/





















































































































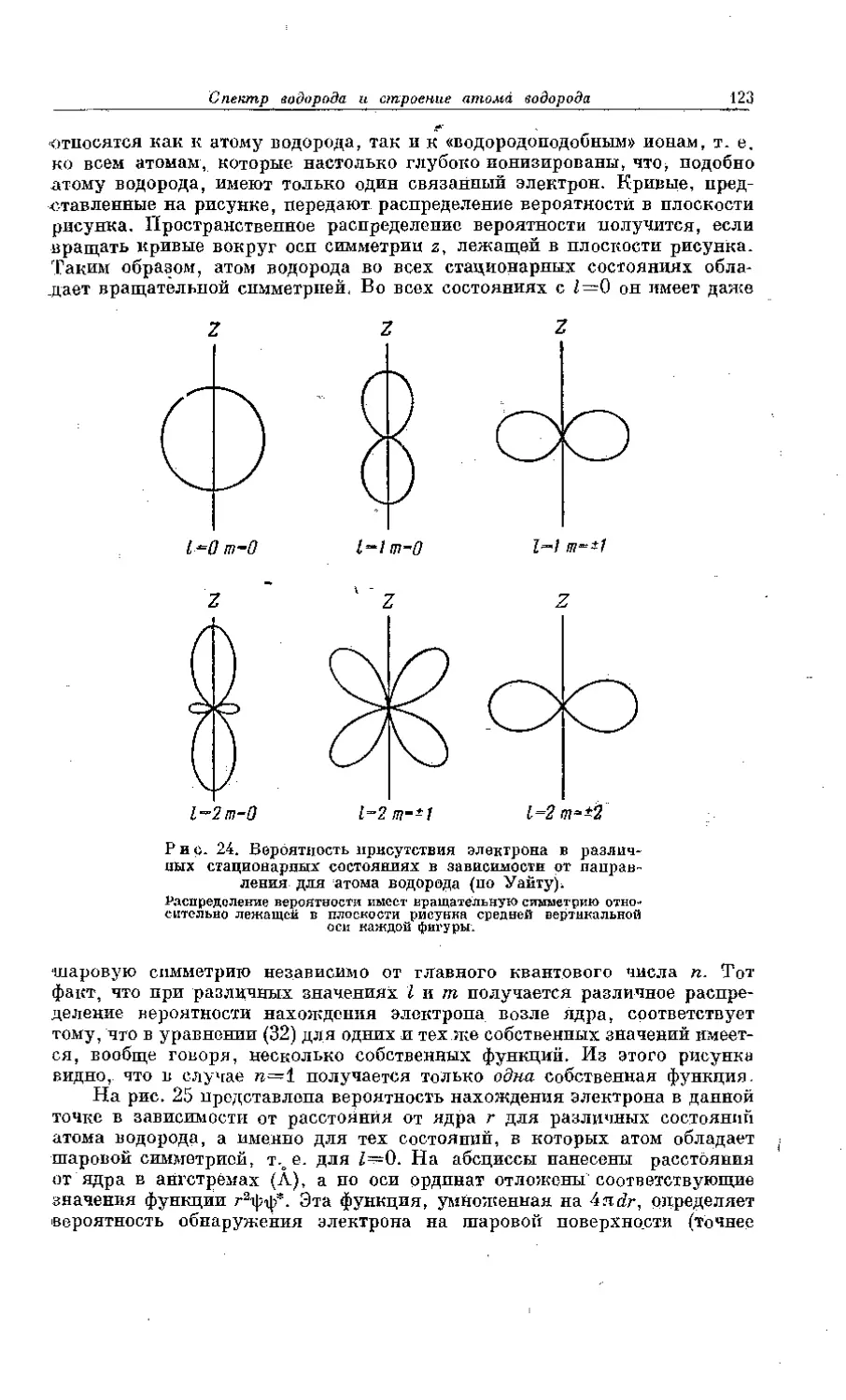










































































































































































































































































































































































































































































































































































































































































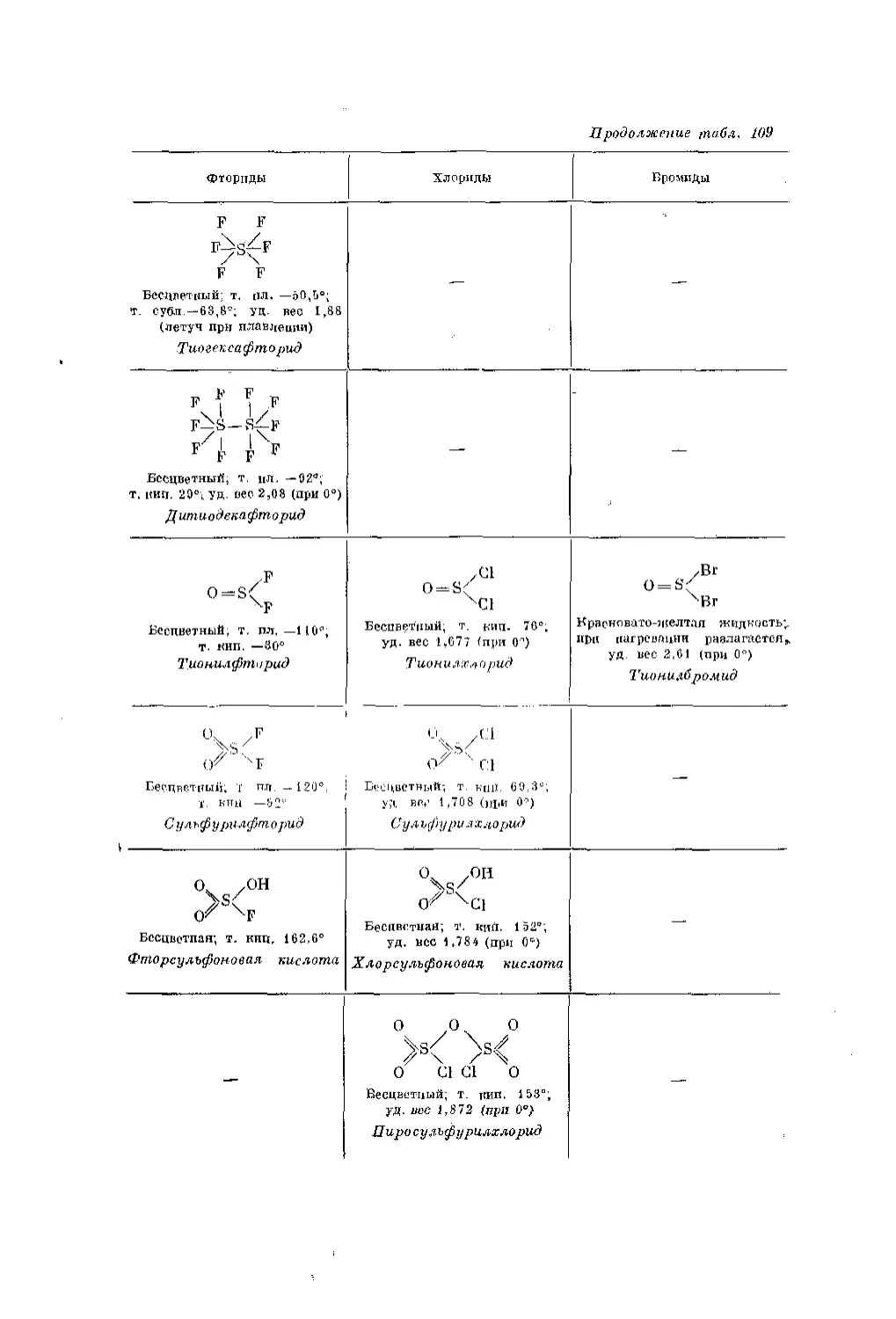















































































































































Текст
Г. РЕМИ
*
КУРС НЕОРГАНИЧЕСКОЙ ХИМИИ
том
ПЕРЕВОД С НЕМЕЦКОГО XI ИЗДАНИЙ
ПОД РЕДАКЦИЕЙ
чл.-корр. АН СССР А. В. НОВОСЕЛОВОЙ
*
ИЗДАТЕЛЬСТВО ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва 196 3
о
Книга представляет собой своего рода энциклопедию по неорганической химии, одинаково интересную и полезную как начинающему студенту, так и опытному химику.
Курс неорганической химии излагается на основе новых представлений о строений вещества. В основу системы расположения материала и всего построения курса положен , периодический закон в современном его освещении. Описание элементов и их соединений, стройно и строго научно систематизировано. Большое внимание уделено кристалло-химической трактовке свойств различных неорганических веществ. Автор часто прибегает к термодинамическим представлениям, обстоятельно излагает координационное учение, современную теорию растворов. Большое внимание уделено вопросам технологии отдельных элементов и их соединений, а также их применению в промышленности. Автор проявляет особый интерес к последним достижениям экспериментальной химии, обстоятельно знакомя читателя с основными результатами, достигнутыми в той или иной области.
Книга является ценным учебным пособием для студентов и преподавателей химических вузов и хорошим справочником для широкого круга химиков, работающих в различных областях.
,Редакция литературы по химии
ПРЕДИСЛОВИЕ
Первое издание книги Г. Реми «Учебник неорганической химии» вышло в свет в 1931 г. (в русском переводе книга вышла в 1934 г.).
Настоящий перевод первого тома этой книги сделан с XI издания.
Со времени первого издания книги в неорганической химии накоплен большой фактический материал, относящийся к химии отдельных элементов, и получены новые данные о строении веществ. В связи с этим автором книги сделано много дополнений почти в каждой главе и написаны новые разделы, в результате чего объем книги по сравнению с первым изданием очень возрос. Книга Реми рассчитана па читателей, уже знакомых с химией из более элементарных курсов, поэтому в ней не рассматриваются основные химические понятия и законы. Опираясь на физико-химические закономерности и теории, автор построил курс неорганической химии на высоком научном уровне и осветил современное состояние неорганической, химии. При изложении некоторых теоретических вопросов автор ие ограничился качественным описанием явлений, как это обычно делается в курсах неорганической химии, но рассматривает и количественную сторону их, используя соответствующие закономерности в математической форме.
В настоящее время неорганическая химия в наших высших учебных заведениях преподается иа первом курсе, когда студенты еще только приступают к изучению высшей математики, поэтому книга Реми не может служить учебником, понятным во всех ее разделах студентам первого курса. Например-, разделы книги «Строение атома водорода с точки зрения волновой механики», «Валентность и сродство» и некоторые другие недоступны для лиц, незнакомых с высшей математикой. Несмотря на это, книга Реми явится прекрасным пособием при изучении неорганической химии и для студентов первого курса,
В книге имеются подробные сведения о свойствах и строении химических элементов и их соединений. Большой фактический материал, относящийся к химии отдельных элементов, излагается очень стройно на основе периодической системы элементов. Хорошо написаны главы, в которых освещаются общие вопросы, например, «Координационная теория», «Сплавы», «Окисление и восстановление» и др..
Очень ценным руководством книга Реми явится для студентов старших курсов, аспирантов и научных работников, которые, зная основы физической химии, могут более глубоко изучать неорганическую химию. Издание книги Реми несомненно будет служить развитию интереса к неорганической химии и более глубокому ее пониманию.
Книга Реми не лишена и некоторых недостатков. Прежде всего объем книги, если ее рассматривать как учебник, слишком велик, и он увели
6
Предисловие
чивался с каждым новым изданием, главным образом, за счёт включения нового материала, появляющегося в литературе в области химии отдельных элементов. К сожалению, в книге почти не нашли отражения работы, выполненные в Советском Союзе. Как это обычно делается при повторном издании учебников, в книге Реми новые сведения в области химии того или другого элемента приведены в большинстве случаев в соответствующих главах в виде многочисленных дополнений петитом. Безусловно, в учебниках для высшей школы должны быть отражены как новые идеи г и теории, так и новые экспериментальные данные, относящиеся к химии отдельных элементов. Но этот новый фактический, материал желательно в большей степени обобщать и частично приводить в виде сводных: таблиц, как это, например, сделано в книге Реми в гл. XIII при рассмотрении интер-. металлических соединений элементов первой — четвертой групп. Таким путем можно было бы сократить объем книги.
Вызывает сожаление, что в книге Реми не нашли отражения проблемы химии полупроводников и очень мало уделено внимания неорганическим и элементорганическйм полимерам. >
При чтении книги следует иметь в виду, что автор иногда использует представления теории резонанса. Мысочли'нёцелесообразным давать примечания в каждом случае, так, как советский читатель, знакомый с критикой этой теории в СССР («Состояние теории химического строения в органической химий», доклад комиссии Отделения химических наук АН СССР, 1954) и за рубежом (В. Хюккелв, К. Коулсон и др.), сам легко составит себе правильное: представление.
При переводе книги Г. Рёми возникли большие затруднения из-за отсутствия общепринятой на русском языке номенклатуры неорганических соединений.
Перевод предисловий и гл. 5, 6, 12, 17 и 18 осуществлён канд. химических наук А. И. Григорьевым, введения и гл. 1 и 2 — капд. хим. наук Й. Д. Колли, гл. 3, 4 и 15 — А, Г. Рыковым, а гл. 7—11,13, 14 и 16 — Канд. хим. наук доц. К. Н. Семененко.
А. Новоселова
ПРЕДИСЛОВИЕ
к десятому и Одиннадцатому изданиям;
Со времени выхода в свет первого издания в январе 1931 г. эта книга неоднократно дополнялась и перерабатывалась в соответствии с постоян- ; ным прогрессом в исследованиях. Вносимые при этом изменения и добавления не ограничивались описанием отдельных веществ. Многократно были переработаны и расширены также главы и разделы общего характера. Однако в конце концов этого стало недостаточно, чтобы отразить те фундаментальные изменения, которые Произошли за последние десятилетия в области неорганической химии. Поэтому для девятого издания книга была полностью переработана. Для следующего десятого издания ее текст был еще раз переработан и дополнен.
В связи с этим были заменены многие 'рисунки. При обновлении текста были также приняты во внимание новые правила номенклатуры неорганической химии, составленные Международным союзом чистой: и прикладной химии (IUPAC). Краткий обзор новых правил номенклатуры дан в приложений ко второму тому.
Общий план, согласно которому книга была первоначально составлена, оставлен неизмененным, так как он вполне оправдал себя. Особенно это относится к методу рассмотрения веществ па основе их места в периодической , системе, как к принципу расположения материала, который использован также в большинстве других учебников неорганической химии. Это касается также включения глав общего характера между главами, которые посвящены описанию отдельных веществ. Трактовка общих закономерностей, определяющих свойства и поведение веществ, с развитием наших познаний приобретает особое значение.
Можно полагать, что в преподавании химии Описание отдельных веществ будет все более уступать место трактовке общих закономерностей.' Однако, принимая во внимание, что эту книгу часто используют: как справочник, автор воздерживался от сокращения разделов, посвященных описанию веществ, наоборот, опи были дополнены многочисленными: новыми интересными соединениями. ’
Некоторые элементы, которые ранее были трудно доступны и только в последнее время стали подробно изучаться, приобрели "между тем зна-чительное научное, а также техническое значение. Это было надлежащим образом учтено. Однако и химия некоторых давно известных элементов была широко развита благодаря новым исследованиям, например химия соединений бора, азота и фосфора. Значительно расширена была уже : обсуждавшаяся в предыдущих изданиях химия водородных соединений' металлов.
Прогресс технических методов (которые следовало учесть в рамках , этой книги) помимо металлов был достигнут, главным образом, для соеди- : нений углерода и кремния.
8 Предисловие к десятому и одиннадцатому изданиям
Чтобы сделать понятной сущность гомеополярной связи, следовало дать представление о строении атома на основе квантовой механики.
Я надеюсь, что мне удалось изложить введение в квантово-мсхани-ческое описание атомного строения, которое должно быть понятно каждому изучающему химию. Оно, по-моему, нс труднее дЛя восприятий, чем основы теории Бора, которая в настоящее время необходима каждому химику.
В качестве первого введения в теорию атомного строения мне представляется подходящей теория Бора — Зоммерфельда в ее первоначальной форме. После того, как была развита квантово-механическая теория атомного строения, представления старой теории стали употреблять только в исключительных случаях.
Знание употребляемых в настоящее время символов теорий атомного строения также необходимо для химика. Кроме того, в современных химических статьях предполагается знание символов термов, применяемых для обозначения состояния атомов и простых молекул в газовом состоянии. Трактовка этих предметов происходит здесь в связи с описанием определенных результатов наблюдений, которые без сомнения ведут к пониманию символов атомного строения и символов термов.
Особое значение приобрели представления и методы, которые обсуждаются в главе «Строение и свойства». Их знание является условием для понимания многих новых результатов исследований почти во всей неорганической химии.
Расположение элементов галлия, индия и таллия в главной подгруппе III группы в качество аналогов бора и алюминия выдержало испытание и поэтому сохраняется. Эти элементы соответствуют бору и алюминию не только своим атомным строением; в результате новых исследований все отчетливее проявляется, что они и в химическом отношении стоят к последним ближе, чем к элементам скандию, иттрию, лаптану и актинию, которые ранее относили к главной подгруппе ИГ группы.
Вновь должен д поблагодарить многочисленных коллег за хорошие советы и великодушную помощь. Особенно благодарю приват-доцента д-ра Линденберга за инициативу в улучшении рисунков и за многочисленные новые схемы. Особенно я должен поблагодарить также моих ассистентом д-ра Фалиуса и д-ра Тидеман: д-ра Фалмуса прежде всего за переработку текста применительно к новым правилам номенклатуры и за переработку предметного указателя; фрейлейн Тидеман за ценные предложения, за тщательную проверку всей рукописи и за чтение корректуры.
Гамбург,, январь 1960 г.
Г. Реми
ИЗ ПРЕДИСЛОВИЯ
К ПЕРВОМУ ИЗДАНИЮ
Задача настоящей книги — дать иодный обзор предмета неорганической химии, включающий рассмотрение фактического материала, закономерностей, связывающих между собой экспериментальные факты, а также следующих из них причинных связей явлений. Для выяснения общих закономерностей и для точного описания веществ и их поведения все возрастающее значение приобретают физические методы исследования. В соответствии с этим в настоящей книге, задачей которой является изложение основ неорганической химия, значительная роль принадлежит физике, как вспомогательной пауке.
В книге сделана попытка па основании подходящих примеров дать изучающим химию доступное введение в атомную физику. При систематическом описании общих разделов неорганической химии но раз использован удобный повод показать, как полученные атомной физикой результаты могут пролить свет на еще нерешенные химические проблемы, могут выявить новые зависимости, дать понять причинную связь уже известных закономерностей й облегчить подход ко многим областям неорганической химии по только для ее исследования, но и изучения.
При пользовании этой книгой предполагается наличие предварительных знаний, которые обычно даются студентам в элементарных курсах; знакомство с важнейшими веществами, их поведением, с понятиями, правилами, закономерностями и теориями, необходимыми для понимания химических реакций, и равным образом, с важнейшими фактами, законами и понятиями физики. Если допустить, что эти понятия читателю , книги (а таковым помимо студента может быть химик-профессионал, желающий ознакомиться с новыми достижениями науки) известны, то все же не следует, что он ими всегда с легкостью может воспользоваться; Поэтому там, где это впервые касается химических проблем, часто приведены определения из элементарного курса физики, которые затем несколько пЬдробпее объяснены.
Однако вывода элементарных физических определений и обоснования простейших физических законов автор принципиально избегает, так как в задачи этой книги не входит избавить химика от основательного изучения учебника экспериментальной физики. То же нужно сказать относительно физической химии, основные начала котором в настоящее время даются изучающему химию уже в первых лекциях, однако к систематической проработке которой он, будучи перегруженным лабораторной 1 работой, приступает сравнительно поздно. В случае важнейших для химика закономерностей, известных ему уже из элементарных курсов, таких как закон действия масс, в этой книге основной упор делается на их применение, а именно они используются в подходящих примерах для числовых расчетов. Эти общие закономерности и теории, с которыми в начале изучения химии знакомятся лишь поверхностно, находят здесь по д
сравнению с обычным описанием их основ в физических и физико-химических учебниках подробное и широкое освещение.
Главы общего содержания расположены между главами, в которых систематически сопоставлены различные вещества, таким образом, что
10 JTa предисловия к первому изданию
каждая -глава общего содержания имеет своим истоком детали, выступающие при описании отдельных свойств. Это отвечает обычному образу мышления, когда стремятся переходить от частного к общему.
Особое значение придается в дальнейшем тому, чтобы в ходе познания полученные выводы общего характера вновь применить к определенным проблемам и сделать их необходимыми для понимания отдельных явлений. Таким образом, приобретается понятие об общих закономерностях наряду с познанием отдельных веществ. Так можно избежать того, о чем еще 30 лет назад Оствальд сказал: «Ученик вынужден поглощать порознь суп и содержащийся в нем. мясной экстракт»?.
Систематическое описание неорганических веществ проведено в книге в соответствии с периодической системой. Это правильный1 принцип расположения материала, рассчитанного на такого читателя, который уже усвоил элементарный фактический материал и может понять основные закономерности периодической системы. Знаменательно, что л Лотар Мейер " и Менделеев пришли к открытию этой системы, пытаясь систематизировать неорганические вещества в целях преподавания. Ни цри каком другой! расположении неорганических веществ не удается в такой'степени, как это позволяет периодическая система, охватить общее, сопоставить аналогичное, позволить отчетливо выявить закономерности. Лучше всего это получается тогда, когда главные и побочные подгруппы периодической системы, будучи разделены, обсуждаются, однако,: в тесной связи, В этом случае помимо аналогии элементов, стоящих в периодической системе друг над другом, и их соединений можно достаточно легко выявить сходство элементов, стоящих рядом.
Данный труд состоит из двух томов, в первом томе рассмотрены воден род и главные подгруппы, во втором — побочные подгруппы, к которым отнесены также лантаниды, ~
В каждой группе периодической системы обсуждению отдельных классов веществ предшествует общий раздел, а в случае каждого класса веществ Опять-таки описанию конкретных веществ обычно предпосылается общий раздел, коротко сопоставляющий свойства этих веществ. Для полного, .понимания этих вводных разделов иногда требуется, правда, пред- <: в зрительное ознакомление с частными свойствами веществ; однако можно предположить, что читатель, еще не знакомый с частными свойствами веществ, после их изучения еще раз вернется к предпосланному их описанию разделу общего содержания.
Те разделы, которые имеют по сравнению с ее главным содержанием второстепенное : значение или5 не одинаково важны для всех читателей, а также включенные иногда в главный текст короткие объяснения — набраны петитом. Набранное: обычным шрифтом составлено; так, что само по себе представляет связное целое, так что при первом чтении работы петит можно пропускать без ущерба для понимания.
При рассмотрении всех важных веществ приведены результаты их структурного анализа, поскольку эти данные в большинстве случаев заменяют теперь представления структурной химии в обычном смысле. ,
На рисунках, чтобы не усложнять их, воспроизведены элементар-. ные ячейки, причем все в одном масштабе (с линейным увеличением в G0 000000 раз). Атомные й ионные радиусы на рисунках также пропбр-циональны кажущимся атомным и ионным радиусам в кристаллах, а именно увеличены в 12 000 000 раз. Гамбург, май 1931 г.
- Г- Реми
ВАЖНЕЙШИЕ ОБЩИЕ КОНСТАНТЫ
Абсолютная температура, точки плавления льда: Та «с = 273,16° К.
Атмосфера (нормальное давление) : 1 атм = 760 лл рт ст ~ 1,013250 X X 10й дн-см~2.
Число Авогадро: Ад = 6,0247-1023 (физическая шкала) ~ 6,0231 X X 10аз (химическая шкала).
Константа Больцмана: k = = 1,38042-10’1й эрг град-1.
1
Элементарная частица электричества: е = 4,8028-10-1в эл. ст. ед. — = 1,60207 10-ао эл. магн. ед. = 1,60207 • 10’1й кулон.
Удельный заряд электрона: = 1,75888- 107 эл. магн.ед/г.
Масса электрона (масса покоя): т — 9,1085-10"38 г; массовое число электрона - 0,00054876.
Мера энергии:
1 эрг = 10-7 дж (ватт-секунд) = 2,777778-10“14 квт-ч (киловатт-часов) —. = 0,239006-10-7 кал.
1 л-атм (литр-атмосфера) = 1,013273-10®эрг = 101,3278 дж = = 2,81466-Ю’6 квт-ч = 24,2180 кал.
1 смР-атм (куб. сантиметр-атмосфера) = = 0,1613250 дж —
, = 2,81458-10 8 квт-ч = 2,42177-10 2 кал- ’
1 кал (термохимическая грамм-калория) s 4,18400 дж — 1,16222 квт-ч — = 0,041292 л-атм = 41,293 смэ-атм.
1 кал[&> (15°-калория) — 4,1855 дж — 1,00036 кал.
1 зв (электрон-вольт) = 1,60207 • 10-1а эрг == 3,82904 -10~ао кал.
1 эв на моль соответствует 23,0689 ккал/моль (физическая шкала) — 23,0026 ккал/моль. (химическая шкала).
1 ТЕМ (тысячная единица массы) соответствует 0,93116 10е эв на атом, или 8,9858-1010 дж/г-атом — 21,476 -10й ккал/г-атом.
Энергия светового кванта (фотона) с длиной волны Л см:
X — 4- 1 >9862 • 10"16 эрг = “ 1,23977 10'4 эв, : что соответствует
•ЛА’ л ।
1 • ' 1
—-11,96633ж/моль или -г--2,86002 кал/моль (физическая шка-Л - Л
1 1
ла) — -г- -11,9630 дж/моль или у-- 2,85923 кал/молъ (химическая Л Л
шкала).
12
Важнейшие общие константы
Фарадей (электрический заря^ одного- грамм-эквивалента) — 1F = 96 493,1 кулон, что соответствует 26,8087 а-час.
Газовая константа: 77ф11Э =82,079 смъ-атм = 0,082076 л-атм — 8,3166 дЖ = 1,98772 кал на физический моль.
7?хим 1= 82,056 c.Hs-aw2.if = 0,082054 л-атм == 8,3143 дж~ = 1,98718 кал на химический моль.
Скорость, света: с = 2,997929-1010 см-сек1.
Литр (объем 1 кг воды, не содержащей воздуха, при максимальной плотности и нормальном давлении) : 1 л — 1000,028 с,и3.
Молярный объем идеального газа (по химической шкале при 0° С и 760 мм рт ст) = 22,4140 л = 22414,6 с.я3.
Постоянная Планка, (квант действия): h = 6,6252 -10“3’ орг-сек.
Ускорение силы тяжести, нормальная величина, (па уровне моря и на широте 45°) : g0 = 980,665 см-сек"~.
Фактор Смита*' = Массовое число 000275 (условная величина).
1 Атомный вес ' '
ЧАСТЬ ПЕРВАЯ
ВОДОРОД И ГЛАВНЫЕ ПОДГРУППЫ ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
ВВЕДЕНИЕ
Хвлыгя :— наука, нанимающаяся изучением распространения, добычи И искусственного приготовления веществ; она изучает также их состав, свойства, превращения и, кроме того, те явления, причины и закономерности, которые находятся в связи с этими превращениями. Разложением встречающихся в природе веществ можно получить строго определенное число основных веществ, из которых построен весь известный нам мир. Эти вещества и являются химическими элементами.
Прежде химическим элементом называли некоторое вещество, которое нельзя разложить на составные части химической реакцией. Однако оказалось, что не всегда можно строго ограничить понятие элемента на основе критерия неразложимости. В настоящее время химическим элементом называют вещество*, все атомы которого обладают одним и тем же зарядом ядра (см, стр. 19).
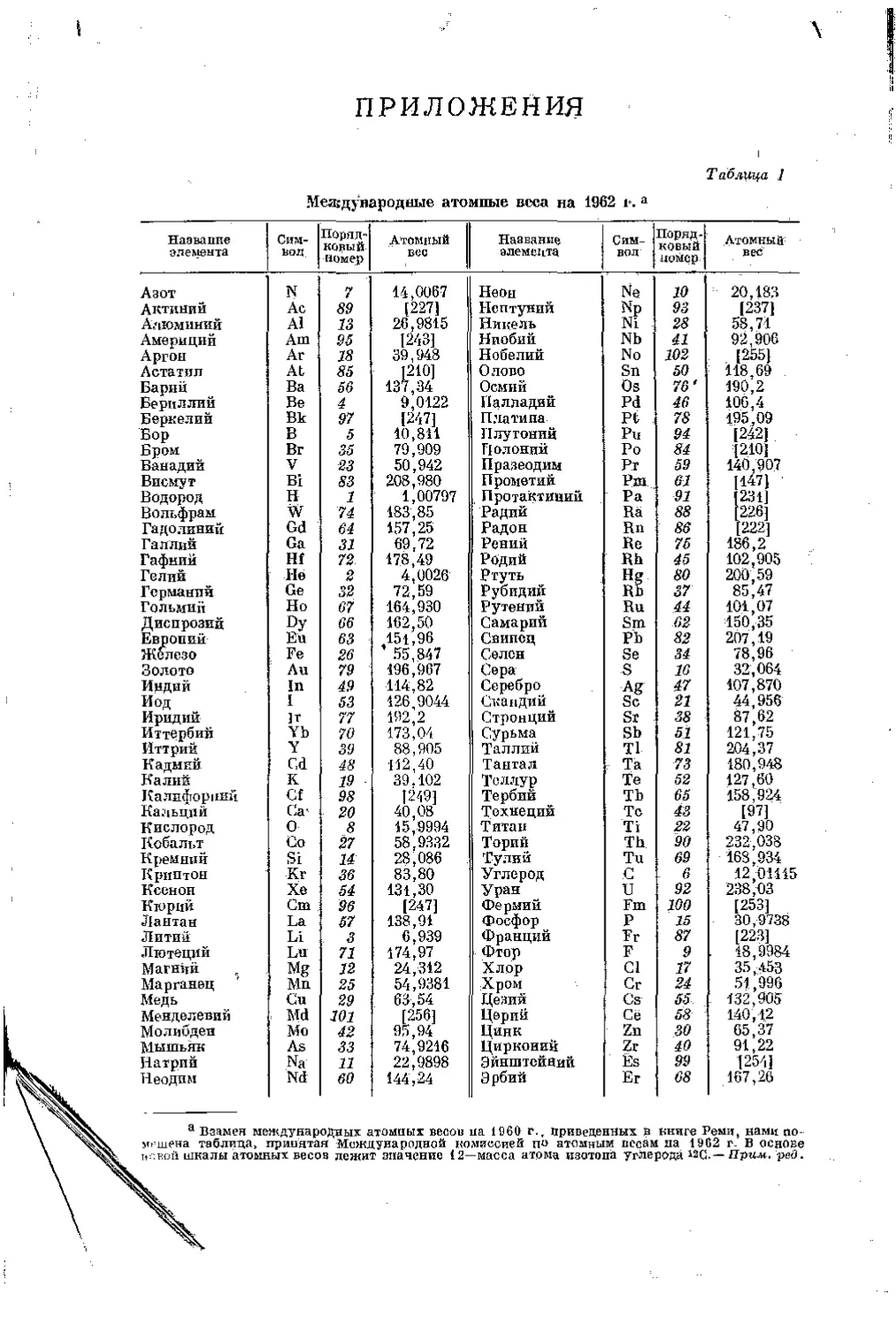
После того как стало возможным менять заряд ядра воздействиями, направленными непосредственно на атомное ядро, и тем самым превращать атомы одного элемента в атомы другого элемента^ появилась возможность получать и такие элементы, которые в природе не встречаются (см. т. II, гл, 13). Включая эти элементы, получаемые лишь искусственным путем, т. е. в результате атомных превращений, в настоящее время насчитывается в общей сложности 103 вида атомов, различающихся зарядом ядра, т. е. известны 103 различных химических элемента (ср. табл. 11 приложения). Из них в природе встречаются только 92.
Одному из этих химических элементов, а именно углероду, принадлежит совершенно исключительная рбль в живой природе. Все живые организмы состоят из соединений углерода, имеющих иногда очень сложной строение. Поэтому раздел химии, специально занимающийся изучением соединений углерода, называется органической химией- В Отличие от этого учение о составе, свойствах, превращениях и т. д. всех веществ вообще, поскольку они не входят в область органической химии, носит название неорганической химии.
Обычно из числа соединений углерода к органическим веществам причисляют те, которые по сво ему составу ,стр о ени ю и свойств ам Лёля ются жи знеинб в ажными у г л ербд-содержащими соединениями. Это справедливо для большинства соединений углерода. Однако существует значительное число углеродсодержащих со единений, которые построены совсем иначе и имеют другой состав по сравнению с упомянутыми выше
* Автор отождествляет понятия химический элемент и простое вещество. Правильнее характеризовать химический элемент как вид атомов, обладающих одинаковым зарядом ядра. Простое вещество образуется путем сочетания атомов одного и того же элемента и является, таким образом, формой существования элемента в свободном состоянии — Прим. ред.
18
Введение
веществами, от которых они поэтому существенно отличаются своими свойствами. Примером могут служить карбиды металлов. Такие соединении углерода относят к неорганическим соединениям. Следовательно понятия соединения углерода и органические соединения не совпадают. Элементарный углерод относится также к области неорганической химии. Простейшие соединения углерода и прежде всего его окись и отвечающая ей кислота, как и простейшие углеводороды, можно отнести как к неоргеническии, так и к органическим соединениям.
Цели и направления работ в области неорганической химии: получение и использование, а также познание специфической природы различных элементов и их соединений (нахождение в природе, получение, свойства и химическое поведение); кроме того, в ее задачи входит изучение общих правил и законов, вытекающих из сопоставления поведения различных веществ, а также тех причин, которыми в конечном счете и обусловливаются свойства и поведение разнообразных веществ.
Специальное изучение свойств и применение тех реакций и методов работы, которыми пользуются для разложения веществ на составные части с целью определения их природы и идентификации, а также для их количественного определения, составляют специальную область химии — аналитическую химию (от aviXixrig — растворение, разделение). Последней также обычно противопоставляли синтетическую химию (от crjv&Ecng — соединение в одно целое), как науку о построении веществ из их составных частей. В настоящее время аналитической химии обычно противопоставляют препаративную химию, в которую синтетическая химия входит в качестве раздела. Препаративная химия (от praeparare — изготовлять, приготовлять) учит приготовлению веществ и рассматривает применяющиеся для этого методы и аппаратуру. В ее задачи входит не только получение сложных веществ из простых, но также и приготовление простых веществ из более сложных; этот последний процесс играет в препаративной неорганической химии далеко не последнюю роль. Методы и аппаратура, применяемые для химического получения веществ специально в технике, а также свойства, методы получения н способы применения этих веществ составляют предмет химической технологии. Выяснением экономического значения встречающегося в природе сырья и получаемых из него химической переработкой продуктов занимается экономическая химия. В задачи последней входит также организация рационального управления химическими производствами, и в этой области, следовательно, ее функции объединяются с задачами химической технологии.
Из других важных областей химии следует указать глектрохимию, которая изучает использование электрического ’ тока для проведении химических процессов; Йотохимию, которая рассматривает влияние света на химические процессы, з других разделов химии следует назвать: коллоидную химию, занимающуюся изучением свойств веществ, когда их частицы находятся в определенных пределах дисперсности, к закономерностей, наблюдающихся для таких состояний веществ (подробнее см. гл. 2); радиохимию, изучающую химическое поведение и важные дня химических исследований свойства радиоактивных веществ, а также атомную. или ядер-ную, химию, которая занимается исследованием превращений атомных ядер и происходящими при этом процессами наряду с изучением свойств и поведения искусственно полученных видов атомов (т. II); дакее, металлографию, применяющую особые методы исследования, которые с течением времени проникни и в другие области химии (гл. 12, т. II); затем кристаллохимию — учение о зависимости между строением кристаллов и их химическим составом (гл. 7), область только что начинающую развиваться, равно как и геохимию — вауку о химическом составе земного шара и о законах распределения в нем различных веществ (т. П).
Здесь нельзя также не упомянуть об истории химии, которая является очань поучительной и важной специальной областью общей химии.
Введение
17
Все названные отрасли химии, поскольку они имеют дело с неорганическими веществами, входят в область неорганической химии. В дальнейшем о них будет идти речь в той мере, в какой это можно сделать в учебнике, охватывающем всю область неорганической химии,
Математика и физика являются важнейшими вспомогательными науками для химии. Для описания веществ необходимо также основательное знакомство с основными понятиями из области кристаллографии. Кроме того, для понимания некоторых отделов химии очень полезно знание основ минералогии, петрографии и геологии, а также общее знакомство с биологическими науками.
Применение в химии физических методов и математических выводов породило в прошлом столетии новую отрасль знания — физическую химию. Основной задачей последней является исследование веществ и реакций физическими методами, выявление общих закономерностей и объяснение поведения веществ на основе важнейших принципов (например, на основе законов термодинамики). Строго разграничить области исследования физической химии, с одной стороны, и физики и химии — с другой, невозможно; особенно этого нельзя сделать в отношении химии, поскольку за последние десятилетия физические методы исследования используются буквально во всех отраслях химии. В наибольшей мере их применяют в неорганической химии, и распространение их в этой области таково, что соответствующее современному уровню знаний изложение неорганической химии, если в нем не принимается во внимание состояние физикохимических исследований, в настоящее время становится уже совершенно немыслимым.
Неорганическая химия охватывает очень широкую область. Помимо более сотни основных веществ, различающихся своими свойствами, ей приходится иметь дело еще с бесчисленным количеством соединений, образующихся в результате взаимных комбинаций основных веществ. Изучение этой обширной области и выделение из ее многообразия наиболее важного фактического материала чрезвычайно облегчается соответствующей классификацией этого материала. Такая классификация химических элементов (а вместе с тем и их характерных соединений) дается периодической системой, основанной на периодичности химических свойств, которая, как будет дальше показано, может претендовать на признание ее естественной системой химических элементов. Поэтому, прежде чем перейти к рассмотрению отдельных элементов и важнейших их соединений, следует остановиться на рассмотрении самой периодической системы. Уже одно ознакомление с ее закономерностями дает краткий обзор некоторых важнейших свойств элементов и их важнейших соединений.
2 г. Реми
Глава 1
ПЕРИОДИЧЕСКАЯ СИСТЕМА ХИМИЧЕСКИХ ЭЛЕМЕНТОВ
Естественная классификация элементов. Периодическая, система химических элементов, предложенная впервые Лотаром Мейером и Менделеевым*, объединяет химические элементы в естественные группы, т. е. в группы попадают отдельные элементы не только по внешнему сходству, но в силу глубокой закономерности, следующей из самой их природы. Поэтому ее называют также естественной системой химических элементов. Периодической же опа называется потому, что основана на следующем факте: химические (и в огромном большинстве также физические) свойства элементов являются периодической функцией некоторой однозначно определимой фундаментальной величины, характерной для каждого элемента. Эта величина изменяется равномерно от элемента к элементу. Если расположить элементы в порядке возрастания этой величины, то через определенные промежутки будут повторяться элементы^ проявляющие в своих свойствах далеко идущую аналогию.
В качество такой величины раньше принимали атомный вес. Одпако если расположить элементы в порядке возрастания атомных весов, то для сохранения периодичности свойств придется все же произвести некоторую перестановку. В настоящее время в периодической системе элементы располагают в соответствии с возрастанием заряда ядра. Зарядом ядра химического элемента называют число, показывающее, во сколько раз положительный заряд ядра атома данного элемента больше заряда ядра атома водорода (подробнее см. гл. 3).
Несмотря па то что каждый встречающийся в природе элемент обладает совершенно определенным атомным весом, который практически не меняется при обычных химических превращениях, все же атомный вес не совсем подходит для однозначного определения понятия «химический элемент». Это является следствием того, что атомпый вес зависит от соотношения в смеси его изотопов (см. т. II), и это соотношение моясет изменяться. На химические свойства это изменение практически пе влияет, даже тогда, когда оно вызывает значительное изменение атомного веса.
Как уже было отмечено, химический элемент однозначно характеризуется зарядом ядра его атома, Каждому элементу отвечает совершенно определенный заряд ядра. Как будет указано в дальнейшем, он определяет число электронов в оболочке электронейтрального атома и тем самым химический характер элемента. Таким образом, периодическая система основана па том, что химические (и большинство физических) свойства элементов являются периодической функцией заряда ядра.
В настоящее времр известно много видов атомов, которые„имся одинаковый заряд ядра, а вследствие этого и одинаковые химические свойства, имеют различные атомные веса (атомы — изотопы)- Точно так же известны многие виды атомов, обладающие
* Приоритет Д. И. Менделеева в Открытии периодического закона окончательно-установлен н науке. —Прим. ред.
20
Глава. 1
при одинаковом атомном весе различный зарядами ядер и в связи с этим характеризую-щиесл различным химическим поведением (атомы — изобары, см, т. И). Отсюда следует, что не существует непосредственной связи между химическим характером и атомным весом. Однако ц новейшие исследования старых законов, полученных на основании эксперимента, показали, что элементы, встречающиеся в природе, проявляют определенные и всегда одинаковые атомные веса*; это те элементы, которые приведены в таблице атомных весов (см. табл. I приложения); Поскольку эти атомные веса при химических превращениях практически не изменяются, их рассматривают как характеристические константы встречающихся в природе элементов**. Их постоянство обусловлено тем, что смеси изотопов встречающиеся в природе (если не учитывать редких исключений) образовались до застывания земной коры***, и тем, что в химических процессах, в которые они с тех пор вступали, соотношение изотопов ио изменяется. Точно так же при химических превращениях, -которым подвергаются эти элементы, соотношение масс изотопов изменяется очень незначительно. Это изменение наблюдается только в случае применения таких химических методов, которые направлены па систематический сдвиг этих соотношений. Об этом будет речь идти в дальнейшем (см. т. II).
Постоянства атомных весов все же недостаточно для объяснения того, что химические свойства элемента хотя и являются в действительности периодической функцией заряда ядра, все же могут быть представлены как периодическаяфункция атомного веса. Здесь важно то обстоятельство, что атомные веса элсментов в рощей возрастают в том же направлении, что и заряды ядер (см. Стр, 261). Вследствие этого одинаковый порядок расположения элементов, за некоторым исключением; получают как в случае распределения их но возрастанию атомного веса, так и по возрастанию заряда. Заряды ядер непрерывно возрастают от 1 до 103 — наивысшего из установленных в настоящее время зарядов. Вместе с тем заряд ядра определяет положение каждого элемента в периодической системе. Пока не были известны заряды ядер и элементы располагали в порядке возрастания атомного веса, последнее обстоятельство не всегда выполнялось. Это происходило потому, что атомные веса изменяются не через равные интервалы. Вследствие этого па основании атомных весов нельзя было с достоверностью решить вопрос, донжон ли между двумя известными элементами находиться еще один неизвестный элемент. В настоящее время установлено, что вплоть до элемента с зарядом ядра 103 ряд элементов является непрерывным.
В то время, когда была предложена периодическая система, ничего не было известно о строении атома и, естественно, о заряде ядра. Вначале элементы располагали в соответствии с их атомным весом, однако со временем было установлено, что этот принцип расположения в некоторых местах нарушается и приходится производить перестановку элементов. Таким образом, появилась необходимость обозначать особыми числами порядок следования элементов в периодической системе. Эти числа назвали порядковыми числами. Позже был найден способ определения, порядковых чисел элементов независимо от периодической системы (ср. стр. 260), и, наконец, оказалось, что порядковые числа идентичны зарядам ядер.
Если элементы, приведенные в таблице атомных весов в алфавитном порядке (см. табл. I приложения), расположить по возрастанию зарядов их ядер Z, начиная от водорода с зарядом ядра Z = 1 до элемента с зарядом ядра Z = 103, то в получившемся ряду элементов периода чески повто
* Это имеет силу, конечно, с известными ограничениями. Например, свинец, который получается из урановой руды или из ториевой руды, обладает иным атомным весом, чем обычный свинец (см. т, II, гл. 11).
** При некоторых обстоятельствах можно наблюдать изменение атомного веса в результате химического процесса. Примером является вода, для которой в течение продолжительного времени в результате процессов, происходящих в аккумуляторах, соотношение обоих изотопов водорода сдвигается й сторону более тяжелого. Содержащийся в такой воде водород имеет значительно больший атомный вес но сравнению с обычным водородом.
*** На основании достижении ядерной физики можно устаповнть'условия, при'кото-рых образуется отдельный эясмсит. Это Происходит потому, что смеси изотопов существовали в природе уже до образования нашей планеты, к такому заключению пришли в результате изучения состава метеоритов; оказалось, чТц элементы, их составляющие, имеют такие же атомные веса, как и у земных элементов.
Периодическая система химических элементов 21
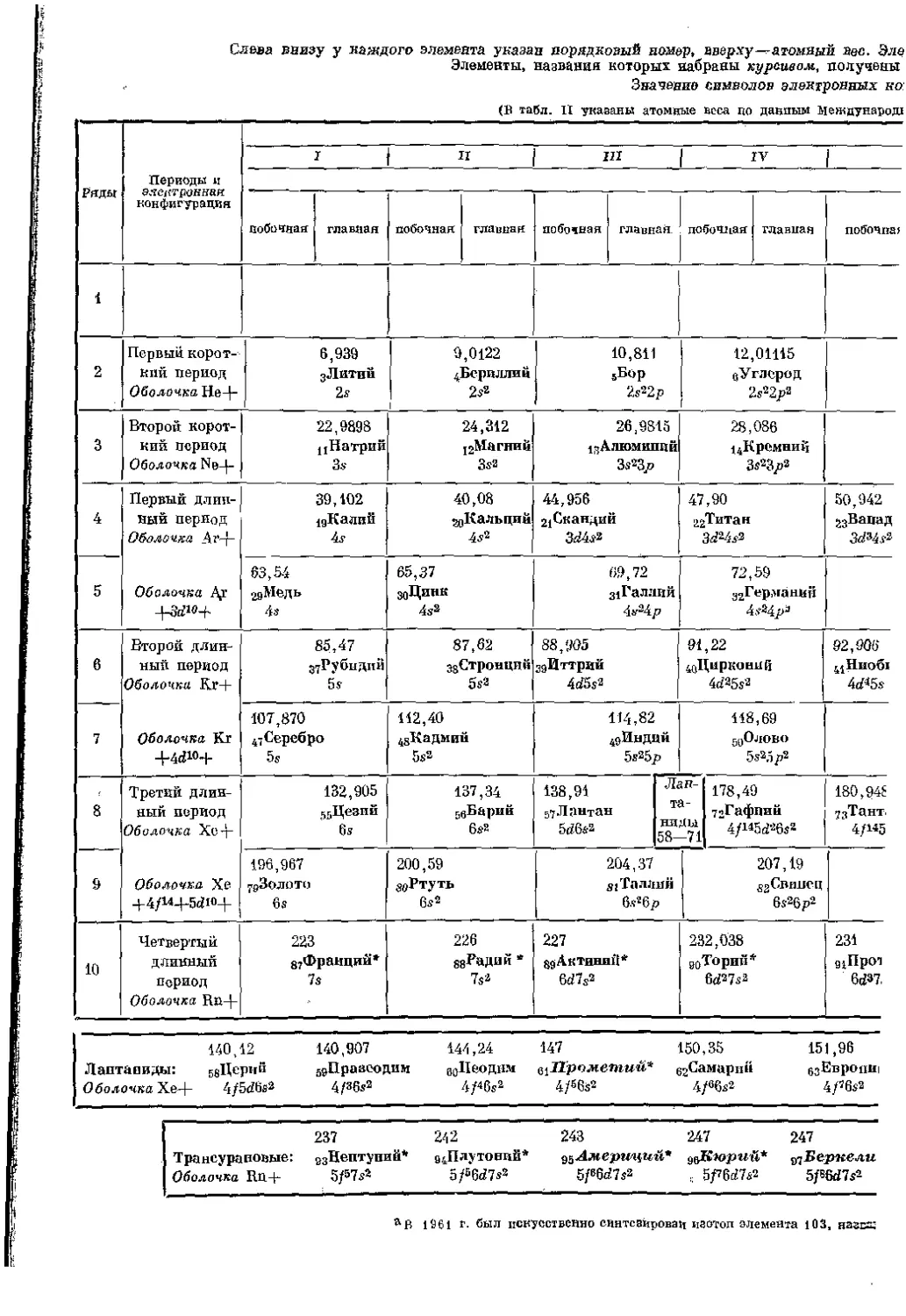
ряются почти одни и те же свойства. Это явление обнаруживается яснее всего, если расположить элементы по рядам друг под другом, как это сделано в табл. 2 приложения. В этой таблице элементы, непосредственно следующие: один за другим по величине заряда ядер, расположены в горизонтальные ряды, однако так, что эти ряды в определенных местах обрываются; получающиеся таким образом отрезки поставлены один под другим. Тогда периодичность свойств выразится в том, что элементы, обнаруживающие значительное сходство, окажутся расположенными один под другим. Такие элементы называют взаимно аналогичными элементами.
Если же расположить, как это и было первоначально, элементы по возрастанию атомных весов, то необходимо произвести перестановку, чтобы только аналогичные элементы находились один под другим. Калий и аргон, следует переставить местами, так как иначе калий попадет в группу инертных газов, а пе в группу щелочшах металлов; далее, более легкий иод приходится поставить после болеё тяжёлого теллура, Равным образом для сохранения химической аналогии никель приходится поместить после кобальта, а протактиний — после тория. Эти несоответствия объясняются, как Мы теперь знаем, тем, что атомные вела еще не представляют абсолютного критерия для естественной классификации элементов. При расположении элементов не по атомным весам, а в порядке возрастания зарядов ядер эти несоответствия исчезают.
Элементы с особо ярко выраженным сходством — строго аналогичные элементы—'следуют один за другим сначала через 8, а затем через 18 мест таблицы. Таким образом, необходимо различать два вида периодов: малые периоды с 8 членами и большие периоды с 18 членами. Последние, однако, можно подразделить таким образом, как это сделано в табл. II приложения, так что входящие в них элементы попадают также п в 8 вертикальных столбцов системы — семейства* (группы) периодической системы; правда, они не строго аналогичны уже находящимся в этих столбцах элементам, но все-таки во многих отношениях обнаруживают с ними родство, особенно в отношении валентности. При расположении таким образом элементов больших периодов обнаруживается иногда также значительное сходство, хотя и не всегда в такой степени, как для элементов малых периодов. Поэтому каждую из 8 групп периодической системы пришлось подразделить на две подгруппы, которые в свою очередь различаются как главные и побочные.
Если таким образом подразделить каждый большой период на два ряда, то в одном из таких рядов на 8 мест (клеток) придется поместить 10 элементов. Чтобы избежать этого, из 10 элементов 3 последних соединяют в одну подгруппу, а именно — в восьмую. Такой прием оправдывается, так как наблюдается очень большое сходство между тремя, рядом стоящими элементами, в то время как в остальных случаях сходство .между элементами, находящимися в вертикальных столбцах, обычно преобладает над сходством между рядом стоящими элементами.
В главной подгруппе восьмой группы находятся инертные газы. Вследствие нулевой валентности их часто совеем исключают из восьмой группы и выделяют в виде «нулевой группы» в начало периодической системы.
Это будет сделано в следующей главе для систематического сравнения элементов и их свойств, причем вслед за водородом будут рассмотрены инертные газы, а затем ужо остальные элементы. Однако нецелесообразно исключать инертные газы из восьмой группы при рассмотрении самой таблицы периодической системы элементов, так как такое исключение затрудняет сопоставление важнейших валентно-химических свойств инертных газов й предшествующих им элементов IV—VH групп. С точки зрения строе-.
* Группы периодической системы автор называет семействами; — Прим. ред.
22
Глава. 1
ния атама также не оправдано 'номсщеппе 'инертных газов в начале периодической системы. Именно на основании закономерностей строения атома (которые будут рассмотрение п дальнейших главах) становится ясным, что каждый период характеризуется образованием новой электронной оболочки, Именно в элементах главной подгруппы I группы каждый раз начинается образования копой оболочки, а у инертных газов опа закапчивается'
В одном ряду элементов, расположенных по возрастанию зарядов ядер, находятся 1.5 элементов, непосредственно следующих один за другим. Для них характерно необычно близкое сходство химических свойств, Особенность этого участка заключается еще и к том, что здесь не обнаруживается характерного изменения валентности при переходе от одного элемента к другому — явление, которое будет подробно обсуждено в дальнейшем. Речь идет б 15 элементах с зарядами ядер от Z == 57 до 2 = 71. Разместить эти элементы в таблице периодической системы обычной формы без натяжки не удается. Раньше иногда помещали их всех в одну клетку таблицы на место, соответствующее лантану (Z = 57). Гораздо правильнее, однако, поступают теперь, выделяя из периодической системы 14 следующих за лантаном элементов и помещая их внизу в качестве особого семейства (семейство лантанидов). Аналогичное особое семейство образуют следующие за ураном элементы — трансураны.
Принимая во внимание строение атома, можно* (как будет видно из дальнейшего), начиная с элемента, стоящего за актинием (тория с Z=SO), рассматривать эти элементы как особую группу. Она располагается аналогично семейству лантани&ов и называется семейством актинидов. Однако первые элементы семейства актинидов, именно торий, протактиний и уран, проявляют далеко идущую аналогию с элементами побочных подгрупп IV, V и VI групп. Поэтому при рассмотрении я:ического поведения этих элементов рекомендуется относить их к вышеуказанным подгруппам. Напротив, элементы, стоящие за ураном — трансураны, по химическому поведению полностью отличаются от элементов побочных подгрупп VII и VIII групп. Таким образом, пр химическим свойствам эти элементы могут быть объединены совсем в особую группу.
Особое положение занимает также водород. Это связано с тем, что он является элементом с самым маленьким зарядом ядра, и поэтому он стоит первым в общем ряду элементов. То обстоятельство, что он предшествует инертному газу (гелию), некоторым образом оправдывает помещение водорода в главную подгруппу VII группы, элементы которой находятся рядом с инертными газами. В табл. 2, приводимой в приложении. ему также отведено это место. Несмотря на многочисленные зависимости. которые существуют между водородом и типичными элементами VII гл явной подгруппы — галогенами, он в общем значительно отличается от них, что обусловлено его специфическим положением первого элемента. Исходя нз этих соображений, рекомендуется выделять его и рассматривать его свойства и поведение прежде других элементов.
Трудности, возникающие при размещении упомянутых элементов в периодической системе обычного вида, зависят от того, что в ёстествен-
* В последнее время считают, что три элемента, стоящие за актинием, т. е. элементы с порядковыми номерами от 90 до 92, не соответствуют по своему строению трем первым элементам семейства лантанидов, а скорее построены аналогично элементам IV—VI побочных подгрупп, Типичная для лантанидов конфигурация электронов проявляется в семействе актинидов, по-видимому, только после нептуния (Z—9-Г'!. Возможно, что четвертый и следующие элементы семейства актинидов по своему строению похожи па четвертый н следующие элементы семейства лантанидов. (Подробнее об этом см, т. II, гл, 14. Ср. также табл, II приложения.) Если эти предположения, установленные на основании данных магнитных измерений, правильны, то элементы тории, протактиний и урап следует поместить впобочные подгруппы IV—VI групп не только по нх химическому поведению, по ц,на основании строения их атомов.
Периодическая система химических алементов
23
ной системе элементов в действнтельност'н имеются четыре вида периодов. Первый из них включает только 2 элемента — водород и гелий; два следующих ряда содержат по 8 элементов; затем идут два периода с 18 членами в каждом; следующий период утке охватывает 32 элелгеита, а именно все элементы от цезия до радона включительно. В этот особенно длинный период п входит в качестве подгруппы семейство лантанидов.
Следующими за радоном элементами открывается другой большой период, Однако этот ряд элементов не закончен. Прежде самым конечным членом ряда был уран. Теперь изучили уже, как указывалось выше, элементы с долее высокими зарядами ядер, чем уран — трансураны (см. табл. 2 приложения).
Общие числа элементов, входящие в названные периоды различной длины, относятся друг к другу лак
2 : 8 : 18 ; 32 или как !
2xV :2Х 2«:2 X 3!:2х 4а
Этот ряд чисел позволяет сделать заключение, что увеличение числа элементов отдельных периодов подчиняется некоторому сравнительно простому закону. В действительности это и имеет место. Основываясь на представлениях Бора о строении атомов, объясним причину увеличения длины этих периодов.
Обычно периодам придают другую нумерацию, чем это рационально было бы делать на основании тех соображений, которые будут приведены в дальнейшем. Дело в том, что закономерность возрастания периодов была установлена сравнительно поздно, по крайней мере ее стали Принимать во внимание совсем недавно. Вследствие этого было принято при нумерации периодов не обращать внимания на первый, состоящий только из двух элементов — водорода и гелия, а начинать счет с периода литий — пеон. Этот период, следовательно, обозначается как первый (1) малый период и т, д., как это изображено в табл, 2 приложения.
В периодиче'ской таблице группы обычно обозначают римскими цифрами, а периоды. и ряды. — арабскими. В дальнейшем для четкого различия главных и побочных подгрупп будем также пользоваться римской нумерацией.
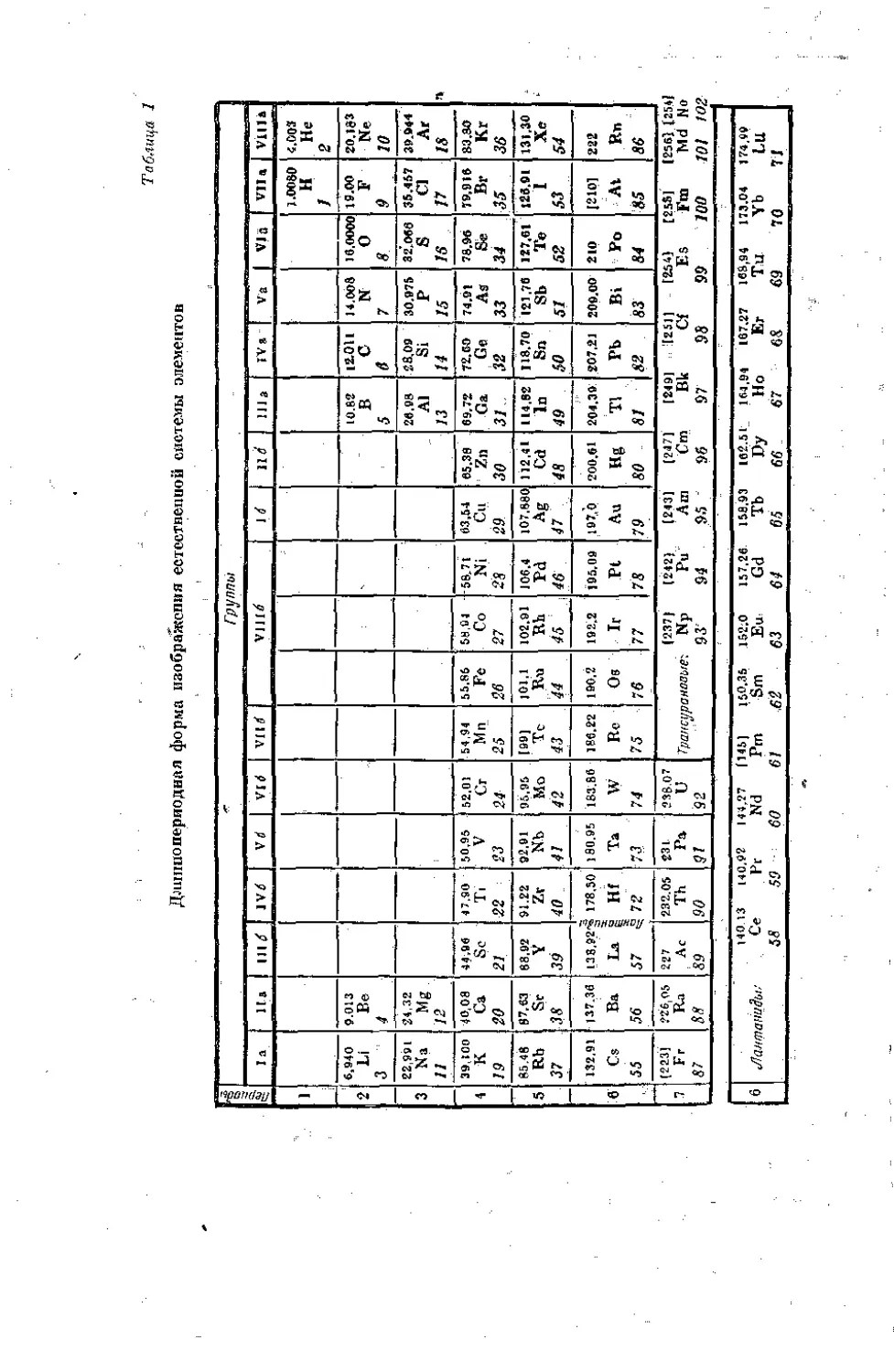
Приведенный в табл. 2 приложения способ представления периодической системы, при котором элементы больших периодов подразделяются на два ряда и, следовательно, горизонтальное протяжение таблицы определяется длиной малых (коротких) периодов, называется короткосгриодной формой в противоположность длгшноперирдноИ форме, когда элементы больших (длинных) периодов располагаются в один непрерывный ряд, В этом случае приходится соответственно раздвигать малые периоды. Уже Менделеев наряду с короткоперподпон применял также и длиннопериодную форму. В табл, 1 периодической системе придан тот,вид (о добавлением открытых с тех нор элементов), который близок применявшемуся Менделеевым.
Длинно, периодная форма системы, при которой избегается разбивка малых периодов на два отрезка, один из которых оказался бы в начале, а другой в копце системы, получается, если расположить элементы так, чтобы они группировались вокруг инертных газов, как это показано в табл. 24 (стр. 149). Комбинация длинно- и короткопер иодной форм получается, если элементы побочных подгрупп выделяют подобно л а нт анид ом. Такая фюрма была предложена Виборгом®. Форма, при которой лантаниды и трансураны располагаются в самой таблице, была уже предложена Томсоном (1895) и Бором (1923) (см, табл. 2), Эта форма расположения, при которой элементы-аналоги связаны черточками, учитывает аналогию, обусловленную строением атома. Рамками отмечены так называемые «переходные элементьы, т. е. элементы, у которых происходит заполнение, rf-оболочек (см. стр, 41 и 352), а также элементы, у которых пополняются /-оболочки,
Химическое поведение элемента главной подгруппы периодической системы (как показано в гл. 5) существенно зависит от положения данного элемента по отношению к инертному газу. На основании этого проводят ра?.деление элементов по главным и побочным подгруппам, а именно: к элементам главных подгрупп (за исключением водорода, который
* Wiberg Е., Z. angew. Chem,, 49, 480 (1936),
Таблица 1
Дли н по периодная форма изображения естсстпешюй системы элементов
Группы
t 1а и. ill 6 IV 6 Vd VI rf VII d v 11M 14 118 UJa IV a va Via Vila Villa
1 J.0080 H 1 4.003 He 2
2 6,940 Li 3 9.013 Be - 10.82 В 5 12.011 c 8 14.008 N 7 le.oooo 0 8 19.00 F 9 20.18» Ne 10
3 22,99 1 Na 11 24,32 Mg . 12 26.98 A) 13 28,09 Si 14 30.975 p 15 32'.O66 S 46 36.457 a 17 39.944 Ar 18
4 39,100 К 79 40,08 Ca 20 44,96 Sc 21 47.90 Tl 22 50,95 V S3 52,01 Cr 24 54,94 Mn 25 55.86 Fe 26 58,04 Co 27 -58.71 Ni 28 63,54 Cll 29 65.38 Zn 30 69,72 Ga 31 72.60 Ge W 74.91 As 33 78.96 Se 34 79,916 Br 35 83.80 Kr 36
5 85,48 Rb 37 87,63 Sr 38 88.92 Y 39 91.22 Zv 40 92.91 N.b J7 95,95 Mo 42 [99] Tc 43 |OI.l Ru & 102.91 Rh 45 : j 106.4 Pd 107.880 Ag 47 112.41 Cd 48 114,82 111 49 118.70 Sn W 121.76 Sb 51 127,01 To 52 126.01 1 33 131,30 Xe 54
132.91 137J6 13 8.92" i 178,50 I SO.95 1B3.S6 1802 190,2 192.2 105.09 197,0 200,61 204.30: 207.21 200.00 210 [210] 222
в Cs Ba La | Hf Ta w Re Os If : Pt Au Hg Tl РЪ Bi Po At Rn
55 56 57 >5 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86
1 (223] Fr 87 226,05 Ra 88 227 Ac 89 23 2. OS Th 90 231 Pa 91 238.07 и 32 Трансурановые^ (237) Np 93 [242] Pu 94 [243] Am 95 ' [247] Cm 96 [249] :: '[251] [254} [255] [256} [254 Bit Cf Es Fm Nd No 97 98 99 100 101 101
HO .13 140.0? 144,27 [145] 15O»3b 152.0 157.26 15B.93 162.51 164,94 167.27 168,94 173,04 174,09
6 Лантаниды/ Ce Hr Nd Pm Sm Eu Gd Tb Dy Ho Er Tu Yb Lu
58 59 6(7 61 .62 63 64 65 6S 67 68 69 70 Tl
Таблица 5
Периодическая система ио Томсену и Бору
’ ’ /- 35 Сз ---------;—— JTFr
-вам
до Д'»
1 1 1 1 1 1 1 1 1 J । ! 1 1 1 .^11 1 90 Th 91 Ра 92 U 4Np
0/ рц
/у Ant
...... ///_ Qfi
/// 07 РЪ
ii. С f
о 0 Г с
/Л. _ J Л Л CrTi
/// Л 1 1/J
1Q2 Np
./
26
1 Лава 1
занимает особое положение) относятся /пе, порядковый номер которых на 1—2 единицы больше или на 5—7 единицу меньше, чем порядковый номер / инертного газа. Остальные же элементы (исключая лантаниды и трансурановые элементы, которые образуют особые группы) принадлежат к побочным подгруппам периодической системы. Таким образом, все ’ элементы распределяются по следующим пяти группам; 1 — водорог), 2 — главные подгруппы периодической системы, 3 — побочные подгруппы периодической системы, 4 — лантаниды, 5 — трансураны.
Аналогия химического поведения и физических свойств отмечается не только для элементов одной главной или одной побочной подгруппы, а в некоторой степени это имеет место и для элементов, принадлежащих к одной группе периодической системы. Более всего это проявляется в III и IV группах периодической системы. Там это настолько сильно выражено, что долгое время нельзя было окончательно решить, какие элементы являются более близкими аналогами бора и алюминия, а также углерода и кремния; таким образом, оставалось неясным, какие из них должны быть помещены в главные подгруппы, а какие — в побочные. На основании предложенного выше определения для гллвпой подгруппы удается получить такое же разделение элементов по главным и побочным подгруппам,'какое получают, основываясь на строении атома (см. стр. 41). Подобное разделение является наиболее целесообразным и общим.
Три фактора, определившие создание периодической системы. Периодическая система в ее современном виде сложилась под влиянием следующих факторов:
1) стремления установить естественную классификацию' элементов;
2) установления существования внутренней связи между характерной для каждого'элемента фундаментальной константой и его свойствами;
3) открытия периодичности в изменении свойств и химического поведения и зависимость их от этой фундаментальной константы.
Эти три фактора, лежащие в основе периодической систешл, развивались независимо один от другого и затем почти одновременно (1869) были использованы для создания периодической системы двумя исследователями — Лотаром Мейером и Менделеевым*.
Исторические сведения. Тот факт, что некоторые вещества (щелочные металлы, щелочноземельные металлы, галогены) образуют естественные группы, был известен еще задолго до установления периодической системы. Попытки систематизировать соответствующим образом и остальные элементы долгое время не удавались, так как тогда еще но было найдено подходящих критериев, на основании которых можно было бы придать и другим, менее близким между собой элементам вполне ясную группировку. Важным моментом в этом направлении явилось установленное в 1829 г. Доберейнсром правило триад.
Добсрейпер заметил, что у родственных элементов, если соединить .их в группы по три (триады), атомный вес среднего элемента оказывается средним арифметическим
* По поводу приоритета Д. И. Менделеева в открытии периодического закона А. Смит (А. Смит, Введение в неорганическую химию. Перевод под редакцией А. В. Раковского, Москва 1028) писал: «Лотар Мейер почти одновременно и независимо от Менделеева открыл периодической изменение ущельных об ьемов простых веществ г. изменением атомного веса»; п далее: «хотя знание удельных объемов простых веществ имеет большое впачевпе Для изучения физико-химических особенностей элементов, однако яспо, что эти удельные объемы нс пригодны для составления системы элементов и что пми во мвогих случаях вельзя руководствоваться для определения места элемента в системе. Вот почему Л. Мейер, открывший периодичность удельных объемов простых веществ, не мог создать периодической системы элементов, что сделал Д, IT. Менделеев, изучивший периодичность не только физических, по и химических свойств»,— Прим. ред.
Периодическая система химических элементов 27
ST из атомных весов крайних членов триады. Таи, атомный вес селена приблизительно равен средней величине из атомных весов серы и теллура;
БД-Те 32.14-127,6 „ - .. _ „ _
—— = —-------у——=/9,8; наид. ат. вес Se=/9,0.
Опираясь па правило триад, Лспсеп в 1857 г. расположил в со известные в то время элементы в закономерную, с точки прейдя этого правила, систему. Система Одлинга, основанная па том же правиле триад и появившаяся в том же году, уже содержит многие элементы в той группировке, в которой они расположены в периодической системе в настоящее время. Однако правило триад, поскольку оно па основании атомного веса допускает группировку в триады также и совершенно несхожих между собой веществ, оставляло в этом отношении широкий простор произволу. Тем нс менее его необходимо •считать крупным шагом вперед благодаря тому, что им впервые установлена возможность положить в основу «сходства» элементов такое соотношение, которое может быть определено количественно. Не менее важным было и то, что здесь впервые была высказала мысль о зависимости между свойствами а атомными весами. Тогда, однако, ёще ис знали методов для точного определения атомного веса. Поэтому для открытия периодического закола решающее значение имело предложение Каницарро (1860) — при определении атомных весов брать за основу молекулярные веса веществ в газообразном л парообразном состоянии так, как это дается теорией Авогадро. Таким образом, была создана довольно строгая основа для* определения атомных весов.
В 1862 г. Бсгье Де Шаикуртуа расположил элементы в порядке возрастания их атомных весов (установленных методом Каницарро) по винтовой линии, вокруг цилиндрической поверхности таким образом, чтобы элементы с близкими свойствами оказались один под другим. Этим впервые была выражена мысль о периодичности-, однако она пе была воспринята должвым образом исследователями того времени, на нее дочти не обратили внимания, и вскоре она оказалась забытой.
Вторично закон периодичности был открыт Дж. Ныолепдсом (1864) который, расположив элементы в порядке возрастания атомных весов, обнаружил, что через каждые 7 элементов (инертине газы в то время еще нс были открыты) свойства их обнаруживают большое сходство. Открытую закономерность он назвал законом октав, •однако иайтн для этого удовлетворительного объяснения оп пе сумел. Только после того как Менделеев в 1869 г. создал свою систему, а в следующем году Лотар Мейер опубликовал набросанную им еще в 1868 г, таблицу, совпадающую в главных чертах с таблицей Менделеева, предположение о внутренней связи между атомным весом и свойствами элементов могло получить широкое признание.
Следует, однако, отметить, что системы Бегье де Шанкуртуа и Дж. Иьюлендса содержали еще много Произвольного и неточного, вследствие чего убедительность идеи, на которой они базировались, в заметной мере ослаблялась. Системы Менделеева и Лотара Мейера почти не содержали такого рода ошибок, хотя в то время избежать их совсем все-такп не удалось ввиду недостаточной ’надежности определения атомных весов.
Во время установления периодической системы число известных элементов было значительно меньше, чем в настоящий момент. В ряду элементов существовало много пробелов, г. с. незаполненных мест в системе, при таком расположении, когда друг под другом стоят элементы-аналоги. Прежде всего незаполненными были клетки, отвечающие аналогам бора, алюминия и кремния с (теперешними) порядковыми номерами 21, 31. и 32. У читывал наличие этих пробелов, Менделеев пришел к заключению о существовании соответствующих этим клеткам, еще не известных тогда элементов — экабора, экаалюминия и экасилиция*, свойства которых он- и предсказал заранее на основании закономерностей периодической системы. Последовавшее вскоре после этого открытие этих элементов (см. ниже: скандий, галлий и германий) явилось поразительным подтверждением представлений, лежащих в основе периодической системы.
Даже после того, как были открыты инертные газы (1894) наряду с новыми элементами из группы редкоземельных металлов, а также после того как открытие радиоактивных элементов — полония, радия, актипия
* Эка (санскрит) — означает одно и то жо.
28
Глава 1
н протактиния значительно расширило число известных элементов^ в периодической системе все же оставались еще пустые места. Эти пустоты определились еще яснее после открытия закона Мозли (см. стр. 253 сл). Этот закон позволил точно определять порядковый номер любого элемента посредством наблюдения его «характеристического рентгеновского излучения» (см. стр. 253). Так, между прочим, оказалось, что еще не запиты места в периодической системе, принадлежащие элементам с порядковыми номерами 72 и 75- Отвечающие им элементы были открыты в 1922 и 1925 гг. и получили названне^я^/шя и рения. Для их открытия имела решающее значение возможность идентификации их на основании характеристического рентгеновского излучения, прежде чем они были отделены от элементов, сопутствующих им в природе.
Долгое время оставались тщетными поиски элементов с порядковыми номерами 6-5 и 67 — называемых до ах открытия экаиодом и зкацезием, несмотря иа то, что их существование было давно предсказано па основе периодической системы. Оба этих элемента, называемые в настоящее время астатин (At) и франций (Ет) относятся к области периодической системы (как видно из табл, 2 приложения), в которой находятся нестабильные легко разлагающиеся радиоактивные элементы. Отсюда можно сделать заключение, что у элементов 85 и S7, по всей вероятности, не Существует стабильных изотонов. Далее можно предсказать, что их нестабильные изотопы должны быть очень короткоживущими. Последнее обстоятельство связано с тем, что в соответствии с опытными данными, элементы с нечетными порядка вымя номерами более ко ротко живу щи е, чем соседние элементы, имеющие четные порядковые номера. В природе радиоактивные элементы с яс очень большим периодом полураспада могут встречаться в значительных Количествах только тогда, когда они Постоянно образуются в результате разложения элемента с большим периодом полураспада. Из таких элементов известны только торий и уран (UI и AcU). Найти элементы 85 и 87 в природе можно было бы только в том случае, если бы они образовывались в результате радиоактивного распада тория или урана. Поэтому само собой разумеется, что все попытки найти их в и ер а дио активных минералах были безуспешными. Эти элементы были найдены среди продуктов радиоактивного распада тория, урана и даже актиноурана. Так, франций был открыт в 1930 г. Маргаритой Иерей (Percy), а астатин — в 1943 г. Карликом (Karlik) и Бернер том (Bernert) (подробнее см. гл. Й и 17).
Элемент астатин, прежде чем оп был найден в природе, был приготовлен иекус-сшвсино (посредством атомных превращений (Se.gr е 194ОД. Два Других свободных места в периодической системе также были заполнены искусственно полученными элементами — 43 и 67. Из правила стабильности атомных ядер (см. т. II, гл. 13) следует, что эти элементы должны быть нестабильны, что й подтверждается наблюдениями. Искусственно полученные элементы 43 и 61 называются теажещгй (Тс) и прометий (Pro). Технеций и прометий не входят в состав естественных радио активных рядов. Скорость распада наиболее долгоживущих изотопов этих элементов много меньше, чем астатина ифрапция; пх распад идет так быстро, что технеций или прометий не могли бы находиться сейчас в земной коре, даже если бы они и образовались в древности. Не исключено» правда, постоянное образование нестабильных элементов в Минимальных количествах под влиянием нейтронов. У технеция это, по-видимому, происходит (подробнее см. т. II).
В настоящее время в ряду элементов от водорода до урана, т. е. от порядкового номера 1 до порядкового номера 92, не только нет свободных мест, но и имеются искусственно полученные элементы (в результате атомных превращений); таким же образом можно теперь получить элементы с более высокими порядковыми номерами, чем уран. Это уже упоминавшиеся трансураны с порядковыми номерами 93, 94, 93, 96, 97, 98, 99, 100, 101, 102 и 103. Они также нестабильны, Подробнее см. т. II.
ПЕРИОДИЧНОСТЬ ХИМИЧЕСКИХ СВОЙСТВ ЭЛЕМЕНТОВ
Периодичность химических свойств особенно ясно обнаруживается составе химических соединений. Состав химических соединений обусловивается еалсягпностя.ми элементов, входящих в данное соединение.
Периодическая система химических элементов 29
Валентность. Валентностью химического элемента называется число, показывающее, сколько водородных атомов может присоединить один атом данного элемента или заместить их в других соединениях. Отвечающую такому определению валентность называют формальной или стехиометрической валентностью в отличие от электрохимической валентности или электровалентности*.
Электрохимическая валентность или электровалентность химического элемента равна числу электрических зарядов**, приходящихся на каждый атом этого элемента в рассматриваемом соединении.
Смотря по тому, заряжены ли й данном соединении атомы рассматриваемого элемента положительно или отрицательно, различают положительную и отрицательную электрохимические валентности. В элементарном состоянии вещества имеют электрохимическую валентность, равную нулю.
В классических химических структурных формулах*** стехиометрической валентности каждого атома соответствует число валентных штрихов, которые от него отходят.
В настоящее время для изображения структурных формул применяют валентные штрихи как символ химической связи., обусловленной электронными парами (см. гл. 5). Для обозначения числа электропПых пар, которыми данный атом обладает вместе с другими атомами, применяют выражение вчисло связей». В органических соединениях число связей в атоме большей частью равно его стехиометрической валентности. В неорганических соединениях это не всегда имеет место; таких примеров известно много.
Стехиометрическая валентность совпадает С электровалентностью в случае тех соединений, для которых применимы оба понятия; в большинстве случаев, если отвлечься от их знака, они численно совпадают. Однако не всегда эти валентности бывают численно равны. Так, в соединении Hg3Cl2 ртуть является стехиометрически Эо-угмлент-ной, а электрохимически опа в нем положительно одновалентна. В обоих структурноизомерных формах азотистой кислоты, ON(OH) и. 02NH, если считать Это соединение тетерополярным, азот является электрохимически положительно тре.гвалептным, а если принять во внимание только формальную (структурную) валентность, то он получается в одном со един сипи трех-, а в другом — пятивалентным.
Во многих случаях можно точно указать формальную валентность, в то время как электрохимическая валентность в точности не известна. Попятие электрохимической валентности к некоторым соединениям возможно и совсем неприложимо. Так, по всей вероятности, совсем не имеет смысла вопрос, какой электр о валентностью обладает углерод в хлороформе СНС13 так как это соединение является гомеополярным, я. е.не состоит из противоположно заряженных ионов. Бывают случаи, когда электрохи-лиическую валентность можно определить точно, а стехиометрическая валентность, наоборот, не поддается определенному выражению. Это наблюдается очепь часто там, где имеются центральные атомы в координационных соединениях (см. гл. 11), Электрохимическая валентность всегда относится к элементу в определенном сто состоянии; стехиометрическая же валентность может быть отнесена вообще к элементу как таковому. Поэтому, например, говорят, что барий но только может выступать как двухвалентный элемент, но что он в полном соответствии с определением валентности является .двухвалентным (стехиометрияески двухвалентным); ^углерод, как правило,—четы-
* Выражение (valenz) валентная сила нередко употребляют также в смысле обозначения той силы, которая взаимно связывает атомы. Поэтому Бильтц, чтобы избежать возможных недоразумений, предложил различать оба эти понятия, применяя термины валентное число и валентная сила.
** За единицу заряда здесь и дальше, поскольку нет особых указаний, принимают заряд вадородного иона. 1
*** Химическая структурная формула (в классическом смысле) касается строения молекулы соединения. Для таких соединений, которые в кристаллическом состоянии построены не из молекул (т. е. из атомных или ионных агрегатов конечной величины; см. гл. 7), классические структурные формулы, строго говоря, пригодны только для газового состояния. Кристаллические соединения, поскольку они состоят нс. из молекул, а непосредственно из атомов или ионов (в принципе из неограниченного количества последних) в отношении структуры причисляют к координационным соединениям {см. гл. 11). . -
30
Глава 1
рехвалептен; сера—Двух-, четырех- и шестивалентна. Такой способ выражения в соответствие с самим определением стехиометрической валентности означает > что один атом бария может, связывать два атома, одни атом углерода — четыре, одни атом серы — два, или четыре, или шесть атомов какого-нибудь равно валентно го водороду элемента. Последний из приведенных примеров одновременно показывает, что валентность элемента может быть переменной.
От стехиометрической и электрохимической валентности падо, кроме того, отличать еще координационную валентность. О ней см. стр. 433.
Понятие, которое в противоположность электрохимической валентности применимо к любым соединениям, — это степень окисления. Степень окисления отдельных элементов, из которых составлено соединение, получается, если заряда атомов распределяются таким образом, что валентные электроны двух неодинаковых партнеров по связи оказываются принадлежащими более электроотрицательному из них. Между равнозначными партнерами по связи валентные электроны распределяются равномерно. Согласно определению, степень окисления пе говорит ничего о фактическом распределении зарядов в соединении; поэтому такое понятие можно прямо применить к гомоополярным соединениям. Например, углерод ж ССН имеет,. степень окпелеиия 4-|-; в СН4 — степень окисления 4 — и в СНС1з — степень окисления 2+. Пр имен сине понятия «степень окисления» оказывается удобным прежде всего при рассмотрении окислительно-восстановительных процессов.
В том случае, когда электроны каждой связи равномерно распределяются между партнерами (независимо от их. электрохимического характера), получаются тан называемые формальные заряды (formal: charges) атома в соответствующем соединении. Так, в каждом из трех соединений СС14, СН4 и СГ1С13 формальный заряд углерода равен пулю. Соответственно в соединении NiCl2 формальный заряд никеля равен нулю, степень окисления его, напротив, равна 2+; в тетракарбониле никеля Ni(CO)4, однако, степень окисления пцксяп равна нулю, а его формальный заряд составляет 4— (ср. т. II, гл. 7).
Для элементов главных подгрупп периодической системы существует правило: наивысшая валентность каждого элемента совпадает с номером группы, в которой он находится. Водород отклоняется от общего правила ввиду того, что ему не отведено особого места в периодической системе, а помещают его в главную подгруппу VII группы периодической системы. В качестве элемента с зарядом ядра 1 водород может отщеплять только один электрон и выступать в качестве положительно одновалентного.
Кислород, фтор и инертные газы не подчиняются этому правилу ввиду того, что они никогда не бывают электроположительными, так как они пе могут отдавать электроны другому элементу (ср. стр. 153).
Если не считать водород, то некоторое отклонение от правила представляет бром, который, как известно, является максим альпо пятивалентным, несмотря на свое по Л о-желне в главной подгруппе VII группы.
В той же мере, в какой элементы могут быть электроположительными*, они проявляют свою наивысшую валентность по отношению к кислороду. (Кроме того, в соединениях с кислородом они могут проявлять также п низшие валентности.)
Ту же валентность, как и по отношению к кислороду, элементы передко проявляют и по отношению к фтору, иногда также по отношению к хлору, сере и другим неме
* Вопрос о том, в какой мере неметаллы в ях кислородных соединениях можно рассматривать в качестве электроположительных составных частой, рассмотрен в тл. 5.
Периодическая система химических элементов 31
таллам. Наоборот, во отношению к водороду большинство элементов обнаруживает более низкую валентность.
Валентность по водороду возрастает в главных подгруппах периодической системы oml к IV группе от одного до четырех и с IV по VIII группу убывает от четырех до нуля. В главных подгруппах IV —VIII групп суммирование валентности но водороду с номером группы всегда дает число восемь.
Характерным свойств ом водородных соединений элементов IV и последующих главных подгрупп является летучеепгъ этих соединений. Валентность, которую проявляют эти элементы, в своих легколетучих водородных соединениях, можно рассматривать как их электроотрицательную валентность (в1 электрохимическом смысле)*. То же можно сказать относительно соединений этих элементов с сильно электроположительными металлами. Эти соедипспия с металлами можно включать в приведенное выше правило, сели придать ему следующую формулировку.
Максимальная электроотрицательная валентность элементов с IV по VIII главную подгруппу при* суммировании с номером группы всегда дает число восемь.
I
Для элементов I, Пи III главных подгрупп периодической системы валентность по. водороду числеппо совпа/даст с наивысшей валентностью по кислороду.
Водородные соединения элементов I и II главных подгрупп, за исключением Bella и MgH2, имеют совершенно иной характер по сравнению с такими же соединениями элементов Щ и последующих главных подгрупп. Эти соединения имеют характер солей, и водород в них является электроотрицательной составной частью.
Состав важнейших соединений, соответствующий положениям элементов в периодической системе, представлен в табл. 3-**
Таблица 3
Важнейшие типы соединений в главных подгруппах пер ио ли ческой системы
Главные подгруппы перводичеевой системы i II ill IV V VI VII VIII
Выгоняй нормальный окисел R,0 " RO RaO3 ro2 B2O5 RO3 r.o, —
Простейшее иодо-родное соединение RH rh2 BHa или RHj RHS rh2 RH —
Элементы побочных подгрупп периодической системы по отношению к кислороду и к неметаллам вообще обнаруживают те же валентности, что и элементы главных подгрупп; однако элементы побочных подгрупп, как правило, в большей степени, чем элементы главных подгрупп, склонны менять свою валентность. В побочной подгруппе I группы одновалентность меди и золота сильно подавлена. В побочной подгруппе VIII группы соответствующая номеру группы йось.имвалентпость была до сих нор обнаружена только для рутения и осмия. !
* Относительно ограничений, которые следует делать, применяя это определение, см,, например, стр. 451.
** В этой таблице R означает любой принадлежащий к данной группе элемент, В дальнейшем буква R в химических формулах будет применяться для обозначения любого элемента или радикала^
32 Глава 1
водородных соединений т5го типа, 4 который дают □лементы главных подгрупп периодической системы, элементы побочных подгрупп обычно не образуют. Для ряда элементов побочных подгрупп существуют, однако, водородные соединения другого характера. Некоторые элементы побочных подгрупп могут поглощать значительные количества водорода, образуя твердые растворы (например, палладий и платина). Больше всего, однако, это свойственно элементам IV и V побочных подгрупп. Они образуют с водородом также и соединения, причем последние могут быть переменного состава. Здесь дело идет о соединениях, которые по своей природе,, по-видимому, близки к интерметаллическим соединениям (см. стр. 67).
В результате изучения линейных спектров в последнее время было доказано существование значительного количества летучих двухатомных соединений водорода (но образующихся в весомых количествах) для металлов побочных подгрупп. Они образуются как элементами главных подгрупп (которые выступают не как одновалентные элементы), так и значительным числом металлов побочных подгрупп. В некоторых случаях их существование можно подтвердить другими мето дамп .“По-видимому, речь идет в данном случае исключительно о соединениях, молекулы которых существуют лйпп, до взаимного столкновения. В связи с этим следует указать, что физики иногда рассматривают в Качестве молекул соединения также и такие агрегаты, которые вследствие очень короткой продолжительности жизни нё могут быть выделены. К ним прежде всего принадлежат те, которые химики называют «радикалы, не существующие в свободном СОСТОЯНИИ».
Электрохимический характер. Те элементы, которые легко образуют электроположительные элементарные ионы, называют электроположительными элементами, тё же, которые обычно образуют только электроотрицательные элементарные ионы, — электроотрицательными. Электроположительными являются металлы, а более или менее выраженный электроотрицательный характер имеют неметаллы. Однако между теми й другими нет резкой разницы. Обычно говорят «о более или менее явно выраженном» электроположительном или электроотрицательном характере в зависимости от того, насколько отчетливо у данного элемента обнаруживается склонность к образованию соответствующих ионов. Склонность к образованию положительных или отрицательных ионов называют электросродством. У некоторых элементов оно настолько незначительно, что для них вообще не удается доказать образования свободных ионов. Одна ко и в этих случаях можно все же обнаружить или по крайней мере устаноВЕГть вероятность того.' что и эти элементы в своих соединениях являются электрически заряженными.
Соединения, состоящие из явно противоположно заряженных составных частой, называют гетерополярными или ионными.', если жо составные части соединения пе обнаруживают явно противополоишых зарядов, то такие соединения называют гомеополярными или атомными соединениями. Из сказанного ясно, что между гетеро- и гомеополярными соединениями нельзя провести резкой границы.
Вместо терминов «тетерополярные» я «гомеопояярные» соединения иногда говорят о полярных и неполярных соединениях. Однако в данной книге автор (по Предложению Дебая) называет полярными, такие молекулы, которые обнаруживают в своем поведении полярность, т.е. действуют на окружающую среду как диполи*. Такое действие могут
* Название «диполь» обычно обозначает систему электрических: зарядов,’ которые распределены так, что центры тяжести положительных й отрицательных зарядов системы не совпадают. Если диполь образуется под влиянием внешних сил, то говорят об индуцированном диполе. Процесс образования диполя называется поляризацией. Единичный атом под влиянием поляризации может также превратиться в диполь (см. стр. 71).
Периодическая система химических элементов 33
оказывать также и гомеополярио построенные молекулы, С Другой стороны, пе каждая гетерополярно построенная молекула должна иметь дипольный характер; так, диполь не проявляется в том случае, если заряды взаимно компенсируются вследствие их симметричного расположения.
В главных подгруппах периодической системы электроположгг,-телькый характер убывает слева направо (т. е. с увеличением номеров групп), а электроотрицательный — соответственно возрастает. Таким образом, в каждом ряду наиболее электроположительный элемент находится в главной подгруппе I группы, а наиболее электроотрицательный (инертные газы ввиду их неспособности образовывать химические соединения исключаются) — в V1L
В пределах каждой главной подгруппы электроположительный характер возрастает параллельно увеличению порядковых номеров (следовательно, в периодической системе по направлению сверху вниз). В том же направлении убывает электроотрицательный характер, В соответствии с этим наиболее электроположительные элементы (цезий и франций) занимают в периодической таблице место слева внизу, а наиболее электроотрицательный (фтор) находится в ней справа вверху. Этой закономерностью обусловливается то, что все неметаллы группируются в верхнем правом углу таблицы. Металлы же, поскольку они стоят в главных подгруппах, располагаются лучами от нижнего левого угла таблицы вверх и в сторону. Побочные подгруппы содержат исключительно металлы, то же относится к семействам лантанидов и трансуранов. Граница между областью металлов и неметаллов в главных подгруппах обозначена элементами: бор — кремпий — мышьяк — теллур — астатин.
Чем более противоположны электрохимически два элемента, тем сильнее в общем их стремление вступать между собой в соединения. Из этого в соответствии со сказанным выше следует, что стремление элементов к образованию соединения тем сильнее, чем дальше в периодической системе Они расположены один от другого.
Тамман установил правило, согласно которому элементы одной и той же подгруппы (главной илп побочной) периодической системы в общем не образуют между собой соединений. Этому правилу, если исключить из пего элементы двух малых периодов, подчиняется огромное большинство элементов как главных, так и побочных подгрупп перио-Д1гческой системы -— особенно металлы.
Основной и кислотный характер. В главных подгруппах периодической системы основной характер гидроокисей увеличивается параллельно возрастанию электроположительного характера образующих их элементов. Наиболее сильные основания являются производными наиболее электроположительных элементов.
Кислотный характер летучих бинарных водородных соединений возрастает для элементов главных подгрупп периодической системы параллельно с увеличением электроотрицательного характера связанных с водородом элементов в каждом ряду слева направо; в каждой группе он, наоборот, возрастает сверху вниз, т. е. в обратном направлении по сравнению с изменением электроотрицательного характера.
О причинах этого явления см. стр. 738,
Сила кислородных кислот находится в несколько более сложной зависимости, чем от положения в периодической системе. Опа зависит от различных факторов. Если сравнивать между собой кислоты аналогичного состава, содержащие кислород, то и в этом случае имеет значение правило, что сила кислот в одном и том же ряду периодической системы возрастает 3 Г. Реми
х
34
Глава 1
слева направо, например H4S4O4, Н3РО4, HeSOj, НСЮд, и убывает в пределах одной и той же труппы сверху впив. ?
Приведенные здесь правила не распространяются на побочные подгруппы периодической системы.
Теплоты образования окислов и хлоридов. Приблизительной мерой способности химических элементов вступать в соединения нередко являются теплоты образования их окислов, хлоридов и аналогичных простых соединений. Эти теплоты также явно зависят от положения элемента в периодической системе.
Рис. 1. Зависимость энергии связей окислов и хлоридов от порядкового' номера элементов III и IV групп периодической системы.
•----• окисли главных подгрупп; • ——- • окирлы побйчпых подгрупп; о —— о хло-
циды главных, подуцт пи; о о хлориды побочкых'цсдурупп; ? приблизительной впа-
дение.
Ордината: теплоты образования, ккал!моль
В качестве примера изменения теплот образования внутри одной подгруппы периодической системы в зависимости от порядкового номера на рис. I приведены теплоты образования окисло в и хлоридов элементов III и IV групп. Кривые наглядно показывают, что как элементы ел а вных подгрупп, таки элемента побочных подгрупп обеих групп проявляют при сравнении со вторым членом главной подгруппы законом ер по а изменение свойств (но сравнению с Al и даже Si). Аналогичный ход кривых наблюдается и в других группах периодической системы (И о t К W, А., Ъ. pliysik. Chirm. (А)> 159 (1932)].
Склонность окислов к гидратации/ Тенденция окислов образовывать с водой соединения в определенных стехиометрических соотношениях резко убывает От I к IV главной подгруппе периодической системы, однако вправо от IV группы эта тенденция опять быстро возрастает, В главной подгруппе IV группы и по соседству с ней гидраты окислов существуют большей частью в виде гелевидпых продуктов переменного состава: вода в пих скорее связана капиллярно или осмотически, нежели химически (подробнее см. гл. 12). >
I ПЕРИОДИЧНОСТЬ ФИЗИЧЕСКИХ свойств
Атомные объемы. Атомным объемом называется произведение атомного веса на удельный объем, или частное от деления атомного веса на ,плот~ ноешь*.
* Плотность в численном выражении равна удельному весу, отнесенному к удельному весу воды при 4Э.
Ввиду того что атомный вес является величиной безразмерной, атомный объем имеет размерность т.н3-г'1.
Периодическая система химических элементов 35
Атомный объем=атомный вес Ху дельный вес = атомпый вес/плотпость
Это выражение называется атомным объемом потому, что опо дает относительную (приблизительную) меру пространства, занимаемого атомами.
Так как удельный объем вещества является объемом, занимаемым I е этого вещества, то атомный объем, как следует из его определения, будет тем объемом, который занимает 1 г-агпом данного вещества. Далее, так как в 1 г-атоме любого вещества содержится одно и тоже число атомов, то если бы атомы всех веществ имели одинаковое расположение и: заполняли одинаковые части занимаемого веществом пространства, атомные объемы различных веществ должны были бы находиться в том же соотношении, как и пространства, реально занимаемые самими атомами этих веществ. Так как это в действительности в строгом смысле не имеет места, то атомные объемы выражают это соотношение лишь приблизительно. ' ?
Атомные объемы являются периодической функцией порядкового номера.
Справедливость этого закона, установленного впервые в 1870 г. Лотаром Мейером (причем им была установлена зависимость между атомным объемом и атомным весом), видна из рассмотрения кривой рис. 2. По оси абсцисс отложены порядковые числа,
Рис. 2. Кривая атомных объемов.
по оси ординат — атомные объемы. Из характера кривой виДпо, что, начиная со щелочных металлов, атомные объемы каждый раз резко убывают, после чего начинают возрастать до следующего щелочного металла (максимума), Однако не только щелочные металлы, но и вообще элементы, входящие в Одну и ту же группу периодической системы, занимают на этой кривой аналогичные места. Наиболее электроположительные щелочные металлы расположены на вершинах кривой, менее электроположительные щелочноземельные металлы все Лежат па ее нисходящих ветвях, но ближе к щелочным металлам, чем к еще менее электроположительному алюминию и его аналогам. На сильно поднятых участках кривой лежат электроотрицательные элементы, причем и здесь аналогичные по периодической системе элементы расположены на соответствующих местах. Важнейшие аналогичные элементы соединены пунктирными линиями.
Сжимаемость, по-видимому, зависит от атомного объема. Поэтому и ее величину можно представить в виде периодическом функции от порядковых: чисел.
3*
36
Глава 1
Кажущиеся атомные и ионные радиусы. Объемы, занимаемые атомами элементарных веществ в кристаллическом состоянии, в настоящее время удается определить значительно точнее, чем это допускает расчет ло упомянутой функции «атомных объемов». Это можно сделать измерениями (о которых подробнее будет сказано ниже) со значительной точностью расстояний между центрами атомов в кристаллах. Если теперь представить себе, что вокруг центров атомов описаны шаровые поверхности так, чтобы они соприкасались друг с другом, то, согласно Брэггу и Гольдшмидту, радиусы этих шаров следует назвать кажущимися атомными радиусами. Это имеет силу для того^случая, когда кристаллы, как, например, кристаллы элементарных веществ, построены из незаряженных атомов. Для веществ, построенных из электрически заряженных атомов (ионов), аналогичным образом получим кажущиеся- ионные радиусы. В последнем случае, определяя расстояние между центрами атомов, сначала получим только сумму кажущихся ионных радиусов. Если, однако, удается каким-либо путем* найти величину одного из этих ионных радиусов, то другой определяется простым вычитанном известной величины из всего расстояния между центрами ионов. Полученную величину можно вновь использовать для определения радиуса какого-нибудь другого элемента, образующего с данным ионом известного радиуса кристаллическое соединение и т. д. В 1926 г. Гольдшмидт доказал, что для некоторых ионов получается в общем лишь с очень небольшими колебаниями одна и та же величина радиуса при определениях ее в кристаллах самых разнообразных соединений. При этом, однако, сравнимы только соединения, кристаллизующиеся в определенных структурных типах, которые Гольдшмидт назвал коммеиэуральными (соизмеримыми) типами. (Подробнее об этом см. т. II.) Для коммензуральных типов, однако, для величины кажущегося радиуса определенного иона независимо от вида соединения всегда получается приблизительно одно и то же значение. Так, величины, получаемые для кажущихся радиусов ионов щелочноземельных металлов, почти (существенно) по зависят от того, вычислены ли они из данных измерений кристаллов фторидов, хлоридов или окислов.
Название «кажущиеся радиусы» атомов и ионов, вычисляемые по данным кристаллографических измерений, принято потому, что эти величины ни в косм случае не означают «действительного протяжения» атомов или ионов; опи только указывают, до каких пределов центры атомов или ионов могут взаимно сближаться при образовании кристаллических соединений. Эти расстояния являются некоторыми величинами, обусловлсч!-ныш1 природой соответствующих атомов или ионов, которые хотя и по строго постоянны**, но, как показывает опыт,могут в первом приближении приниматься за постоянные величины. Другими словами, атомы и ионы в соответствии с опытными данными ведут себя в кристаллах практически как твердые (пе изменяющие своих равмеров) или почти твердые шары; величину радиусов этих шаров и дагот кажущиеся атомные и ионные радиусы. Совершенно такие же величины ионных радиусов, какие получил Гольдшмидт чисто эмпирически на основании измерений межатомных расстояний в кристаллах, рассчитал теоретически Енсеи (1938) па основании принципов волновой механики.
На рис. 3 показано изменение величины гольдшмидтовских кажущихся атомных и ионных радиусов в зависимости от порядкового номера.
* Применяемые в кристаллохимии значения радиусов Для некоторых главных видов иопов (например, О1- = 1,33 A, F’ = 1,32 А) были рассчитаны Вазастьсрна (WasisljBT'na) в 1923 г. из оптических даппых.
** Относительно влияния на радиус нова заряда и числа окружающих его ионов см, гл. 7.
Ппрлвкооын номер
Р и с, 3. Периодичность кажущихся атомных и ионных радиусов.
38 Глава 1
Как видно ин рисунка, кривая “атомных радиусов.аналогична кривой атомных объемов, однако она точнее передает соотношения, так как в ней отсутствуют неправильности, вызываемые различиями в структуре. Подобную же кривую образуют и ионные радиусы. Из рисунка также видно, что радиусы положительных ионов меньше, а отрицательных — больше, чем радиусы нейтральных атомов,
В случае элементов главных подгрупп периодической системы, как показывает рис. 3, происходит параллельное изменение атомных и ионных радиусов. Радиусы положительных попов этих элементов в: общем на 0,7—0,9 А меньше, а отрицательных— на столько же больше, чем радиусы нейтральных атомов.
Оптические спектры. Как известно, некоторые элементы окрашивают пламя бунаеновской горелки в определенные цвета. Характер окрашивания пламепн связан с положением элемента в определенной группе периодической системы. Еще яснее эта зависимость выражается в спектрах излучения в видимой области при исследований пламени с помощью спектроскопа. Оказывается, что спектры элементов, находящихся в одной подгруппе периодической системы, обнаруживают в своем тонком строении чрезвычайно большое сходство. В дальнейшем будет видно, что это явление основано па периодичности атомного строения и обясняется теми же причинами, от которых зависит также и периодический харакер химических свойств. -
Потенциалы ионизации. Периодичность потенциалов ионизации также находится в тесной связи с периодичностью свойств.
Порядковый номер
Рис. 4. Периодичность потенциалов ионизации.
Термин «потенциал ионизации атома» данного элемента соответствует тому напряжению (измеряемому в вольтах), которое должен преодолеть катодный луч, чтобы приобретенная им на этом пути энергия оказа-
Периа&ическая система химических элементов 39
„ :Г
лась достаточном для ионизации атомов данного элемента, с которыми он сталкивается, т. е. чтобы он мог перевести их в электрически заряженное состояние, •
На рис. 4 приведена зависимость потенциалов ионизации первого электрона от порядкового номера важнейших элементов, т. е, величины, напряжения, необходимого для перевода свободных атомов соответствующих элементов в однозарядные положительные свободные ионы. Периодичность проявляется здесь очень явно. Щелочные металлы, как правило, занимают наиболее низкие места на кривой, а инертные газы размещаются на ее вершинах. Почти все элементы-аналоги соединены, как и на рис. 2, пунктирными линиями (см. также стр, 138).
Други© физические свойства. Многие другие физические свойства элементов можно представить как периодические функции порядковых номеров, например точки плавления и кипения элементов, структура кристаллов и мащштная восприимчивость.
Все элементы, обладающие способностью в виде ионов или в виде гете-р аполярных соединений проявлять парамагнитные свойства, находятся в побочных подгруппах периодической системы или среди лантанидов или трансурановых элементов. »
Также и в способности элементов к образованию окрашенных ионов наблюдается периодический характер: все элементы, способные образовывать окрашенные элементарные электролитические ионы, находятся в побочных подгруппах периодической системы или- среди лантанидов или трансуранов. ,
ИЗМЕНЕНИЕ СВОЙСТВ В ГОРИЗОНТАЛЬНЫХ РЯДАХ ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
Тесная связь между элементами наблюдается не только в группах (т. е- с элементами, стоящими выше или ниже в той же группе), но также и с соседними элементами по горизонтальному направлению. Примеры этого явления уже были рассмотрены выше. Так, из кривой атомных объемов сразу видно, что атомный объем магния — элемента, стоящего справа от натрия в том же ряду, настолько же близок к атомному объему Натрия, как и объем стоящего над натрием лития. Та же закономерность наблюдается при рассмотрении и других Кривых, иллюстрирующих периодичность физических свойств для целого ряда случаев. Однако та же закономерность распространяется и па многие свойства, которые обычно принято рассматривать как химические. Например, опа распространяется па электрохимический характер, а также, как было указано выше, й на основной и кислотный характер соединения. Далее, эта закономерность применима к соотношениям растворимости соединений и даже иногда обнаруживается в находящейся обычно в теснейшей зависимости от валентности способности элементов к взаимному замещению в кристалли-, ческих соединениях (изоморфизм).
В дальнейшем условимся ради простоты называть элементы, расположенные в горизонтальных рядах периодической системы непосредственно один около другого, «соседними по горизонтали», а стоящие в одной группе непосредственно один под другим,— «соседними по вертикали».
Особенно сильное сходство соседних по горизонтали элементов наблюдается в побочных подгруппах периодической системы, причем ярче всего оно выражено в VIII группе. Как уже было отмечено, благодаря этому
40
Глава " 1
свойству оправдывается помещение трех соседних по горизонтали элементов этой группы в о Злу и ту же клетку периодической системы.
Совершенно исключительное сходство между соседними по горизонтали элементами наблюдается в семействе лантанидов, т. е. в ряду 14 следующих за лантаном элементов с порядковыми числами от 58 до 71 включительно. Здесь опо настолько велико, что распространяется даже па валентность. Аналогичное, явление имеет место в семействе трансурановых элементов. Причины появления такого сходства рядом стоящих элементов, расположенных в этих областях периодической системы, кроются в строении, атомов этих элементов, Этот вопрос будет рассмотрен в начале гл. 2.
В общем, однако, можно утверждать (за исключением лантанидов и трансурановых элементов), что сходство в химическом отношении, если даже не учитывать валентность, все-таки преобладает в вертикальном направлении. Впрочем, от этого правила более или менее сильно отклоняются элементы первого периода; действительно, большинство, из них оказывается наиболее близкими пе к своим аналогам той же группы и, также хотя ипе в столь сильной степени, к своим непосредственным соседям по горизонтали, но каждый из них обнаруживает особенное сходство по отношению к элементу, стоящему от него справа п ниже в следующем ряду; так, литий близок к магнию, бериллий — к алюминию, бор — к кремнию, кислород — к хлору. Сходство в химическом отношении между бериллием и алюминием настолько глубокое, что долгое время сомневались в двух валентности бериллия и в его аналогии с магнием п щёлочноземельными металлами.
Элементы второго малого периода (натрий и т. д.) обнаруживают другую особенность, а именно из всех элементов главных подгрупп они оказываются в каждом случае наиболее близкими к элементам своих побочных подгрупп; иногда это сходство между ними простирается до такой степени,, что опи в некоторых отношениях подходят ближе к элементам побочной подгруппы, чем к элементам своей главпой подгруппы. Сильнее всего это проявляется примерно в середине периодической системы, а именно в Ш и IV группах.
ПЕРИОДИЧНОСТЬ АТОМНОГО СТРОЕНИЯ
В последнее время удалось объяснить свойства элементов строением их ат,омов. Несмотря на то что эта область исследования, где тесно соприкасаются химия и физика, еще находится в стадии бурного развития, в ней уже достигнуто очень многое, что можно рассматривать в известном отношении как окончательно и твердо установленные факты. Результатам, которые являются наиболее важными для химии, посвящены последующие главы. В настоящий момент уже нет сомнении в том, что многие свойства, которыми различаются элементы, можпо объяснить как:непосредственное следствие строения атомов. Во всех случаях, когда из экспериментальных данных (особенно из спектроскопических) удавалось сделать непосредственное заключение о строении атомов, выяснялось, что атомы тех элементов, которые обнаруживают близкую аналогию в отношении физических и химических свойств (как это, например, в периодической системе можпо сказать в отношении элементов, стоящих один под другим), имеют также и весьма близкое строение. Тот факт, что аналогия между элементами, периодически повторяется, является прямым следствием периодически повторяющейся аналогии в строении атомов.
Периодическая система химических элементов 41
е
В таблице периодической системы, приведенной в конце этого тома, под названиями отдельных элементов определенными символами показано строение их атомов. Значение этих символов разъясняется на стр, 146. При рассмотрении таблицы сразу же обнаруживается, что элементы, стоящие один иод другим, проявляют сходство в строении атомов. Далее выясняются типичные различия в строении между элементами главных и побочных подгрупп периодической системы*. Особенности строения лан- * танидов (появление 4/-оболочкп), которые приводят к тому, что они образуют отдельное семейство, отчетливо отражаются приводимыми в таблице символами. Такая же особенность (образование 5/-оболочки) имеет место и в случае трансурановых элементов. В дальнейшем на многочисленных примерах будет показано, как в общем химическое поведение элементов можно установить по химическому строению и как при этом правило, найденное на основании периодической системы, оказывается обусловленным закономерностями атомного строения.
Близкое ознакомление со строением атомов позволяет установить гораздо более разнообразные соотношения, чем это можно было сделать, основываясь исключительно па положении элементов в таблице периодической системы. То, что представляется исключением, если исходить только из периодической системы, следует с необходимой закономерностъю из строения атомов. В дальнейшем можно убедиться, что не только обсуждавшиеся выше правильности вроде изменения валентности от одной группы к другой или закономерности изменения электрохимического характера, но п кажущиеся с первого взгляда неправильности, как, например, особое положение, занимаемое в периодической системе водородом, своеобразный распад периодической системы на периоды различной длины, поразительно тесная связь редкоземельных элементов, — в действительности являются следствием изменения строения атомов при переходе от одного элемента к другому.
* В самом общем случае различие проявляется при сравнении элементов главных подгрупц с так называемыми «переходными элементами», т. е. с элементами побочных подгрупп III—VIII групп. Чтобы объяснить, почему опи находятся между главными и побочными подгруппами I и II групп, следует рассмотреть значения, приведенные в первой графе таблицы. Можно видеть, что для калия обе внешние оболочки обозначены 3s2 Зр5 4s (оболочки с главными квантовыми числами 3 и 4) для меди они, напротив, обозначаются следующим образом 3s2 Зр8 3<Й° 4s. (Подробнее о влияпии Полектронов на химическое поведение см. т. II.)
*
Гл а в а 2
ВОДОРОД (Hydrogemum) Н
Порядковый помер 1,: атомный вес 1,0080, атомный объем (при температуре кипения) 14,4 с.и3/а, валентность .1.
Общие сведения. Водород наиболее легкий ив всех элементов. По своему атомному весу и порядковому номеру он стоит в самом начале ряда химических элементов и поэтому занимает первое место в периодической системе, В строгом смысле слова его не удается отнести к какой-нибудь определенной группе периодической системы. Его особое положение в периодической системе вызвано тем, что своеобразный первый период системы содержит только два элемента, — водород и гелий, а не так как остальные периоды — 8 и больше элементов. Таким образом, водород объединяет признаки первой й предпоследней {VII) групп. Однако существует большое различие в его отношении к элементам главных подгрупп I и VII групп, т. е. к щелочным металлам и галогенам. Химические свойства, которыми он напоминает щелочные металлы (за исключением его валентности), обусловлены совсем другими обстоятельствами, чём у щелочных металлов. Напротив, свойства, которые определяют ого сродство с галогенами, у водорода объясняются теми же причинами, что н у галогенов. Поэтому водород можно кратко характеризовать следующим образом; водород — это галоген, который вследствие своего особого положения в качестве первого члена в общем ряду элементов проявляет в химическом отношении некоторое внешнее сходство со щелочными металлами.
При особом рассмотрении водорода нельзя не обратить внимания гга его исключительное сходство с галогенами. Несмотря на некоторые различия, он обладает рядом характерных, общих с галогенами свойств. Так же как и галогены, он является неметаллом и, так же как и последние, в элементарном состоянии образует двухатомные молекулы. В этих молекулах, как в случае галогенов, так и в случае водорода, атомы связаны простой связью. Работа, необходимая для разложения молекул на атомы, постепенно убывает в ряду Я— СД—Вг—Р—I. Так же как галогены, водород, может выступать в качестве электроотрицательного иона, т. е. водород аналогично галогенам обладает сродством к электрону. Последнее означает, что в случае присоединения одного электрона к нейтральному атому Н, выделяется энергия. Так же как водород, галогены в соединениях, где они отрицательно заряжены, исключительно о^роеалентны. Соединения водорода с металлами, в которых водород является электроотрицательной составной частью по строению и характеру связи, соответствуют аналогичным сведи нениям галогенов. По своему строению эти вещества подобны солям, и поэтому водород в полном смысле слова можно считать «сил еобр азо вате л см». Точно также и работа, которая должна быть затрачена, чтобы получить положителъно заряженный водород, т. е. атом водорода с отщепленным электроном, является отнюдь по меньшей, чем у галогенов (за исключением фтора). В Этом можно убедиться, сравнив ионизационные потенциалы (см. стр. 140).
Уже одно то обстоятельство, что водород в качестве первого члена в ряду элементов может терять только один электрон, доказывает, что он но многим свойствам сильно отличается от галогенов. Эти свойства необходимо прежде всего выделить при
Водород
43
if ' ч
общем рассмотрении химического поведения водорода, В случае отщеплении от атома водорода электрона остается очень маленькое ядро атома — протон. Это обстоятельство отличает водород от всех остальных элементов*. При соедин спин’такого исключительно маленького водородного ядра с отрицательно заряженным ионом выделяется особенно много энергии. Это относится и к образованию ионов гидроксония (ем.стр. 101) в водных растворах, где ионы водорода соединяются с молекулами воды, и к другим аналогичным реакциям. Поэтому водород, несмотря на высокий ионизационный потенциал, ведет себя как довольно сильно электроположительный элемент. Водород проявляет .склонность к образованию гомеополярных и в соответствии с этим легколетучих соединений**, Вследствие летучести этих соединений равновесия реакций с. их участием смещены в сторону их образования. На этом основана большая восстановительная •способность водорода.
Ранее водород помещали в главную подгруппу первой группы периодической Системы вместе со щелочными металлами вследствие его одновалентности и сильно электро положительно го характера, проявляющегося в значительной восстановительной способности. Щелочные металлы, таг: же как п водород, имеют во внешней оболочке, атома только один электрон. Поэтому опи дают спектры, аналогичные водороду (см. •гл. 6). Они могут быть, как и водород, только одновалентными, но в отличие от него — исключительпо электроположительными. Однако щелочные металлы и водород имеют мало общего, кроме одноположительной валсптпости. Не говоря уже о том, что водород не является металлом, а представляет собой элемент, обладающий в отлпчие от щелочных металлов способностью заряжаться отрицательно, следует также отметить, что •он значительно отличается от щелочных металлов и своим электроположительным характером, У щелочных металлов по сравнению со всеми остальными металлами яаи-более сильно выражен электроположительный характер. Они имеют самые низкие из всех элементов ионизационные потенциалы и находятся левее всех в ряду напряжения (см. стр. 51). Водород же в этом ряду находится значительно правее и имеет даже (юлее •высокий ионизационный потенциал, чем благородные металлы. То, что он стоит левее их в ряду напряжения, следует приписать только более высокой энергии его гидратации, т. с. со верш епп о иной причине, п еж ели та, по которой щелочпые металлы находятся в начале ряда папряжспий. Исключительно малый радиус электроположительного иона водорода (в негидратированпом состоянии) также резко отличает его от ионов щелочных металлов, которые имеют наибольшие радиусы пз всех положительных ионов. Таким образом, помещение водорода в подгруппу щелочных металлов при современном состоянии науки не оправдано..
Распространение в природе. Входя в состав воды и других соединений, водород очень распространен в природе. Его доля участия в строении земной поры (включая гидросферу и атмосферу) оценивается в 0,88 вес,%, или 15,5 ат.%. В свободном состоянии вблизи земной поверхности он встречается редко***. Как случайная составная часть он иногда выделяется в смеси с другими тазами при извержении вулканов, находится среди .газообразных продуктов выделения фумарол, а также присутствует в небольших количествах в виде включений в калийных солях. Наоборот, -атмосфера на очень большой высоте (более 100 км) состоит главным образом из водорода. Кроме того, он присутствует в больших количествах на солпце и на большинстве неподвижных звезд, что доказано анализом их спектров.
ч
Исторические сведения. Водород был открыт в 1766 г. Кавендишем, который установил, что при растворении металлов в разбавленных кислотах выделяется горючий газ. Кавендиш также первый (1781) определил, что вода является продуктом соединения водорода и кислорода. Разлрже-
* Для всех остальных элементов диаметр атомных остовов, полученных после отщепления валентных электронов, лежит в пределах 0,2—3,3 А; величина же водородного ядра составляет лишь 0,00001 А..
** О зависимости между летучестью и характером связи см. гл, 9.
*** Среднее содержание водорода в атмосфере вблизи земной поверхности составляет 5-10~6 %. Относительно содержания в атмосфере дейтерия и трития см. т, II.
44
Глава 2
ине воды раскаленным железом было впервые (1783) произведено Лавуазье (при продувании струи пара через нагретый до красного каления ружейный ствол). Разложение воды электрическим током было осуществлено в 1783 г.
Образование в естественных условиях, получение и .применение. В природе водород образуется главным образом при разложении органических веществ, например целлюлозы или белков некоторыми видами бактерий, большие количества водорода освобождаются при коксовании угля; поэтому светильный и коксовый газы в среднем состоят на 50 об.% из свободного водорода. В последнее время коксовый газ стали технически перерабатывать на водород, применяя для этого способ, аналогичный процессу Липде для получения жидкого воздуха, т. е. отделяя от водорода остальные составные части этог<( газа конденсацией при низкой температуре; водород, как очень трудно конденсирующийся газ, остается нрн этом в газообразном состоянии. Полученный этим способом водород находит применение в процессе «ожижения угля», а отделенные от него другие составные части коксового газа передают по трубам на большие расстояния для снабжения газом городов.
В остальных случаях для получения водорода используют почти исключительно воду; если при этом иногда и пользуются другими исходными продуктами, то последние в большинстве случаев получают 'предварительно из воды.
В настоящем время водород пояучают в огромных количествах. Очень большую часть ого используют при синтезе аммиака, гидрогенизации Жиров и ири гидрировании угля, масел и углеводородов. Далее, водород применяют для синтеза соляной кислоты, метилового спирта и синильной кислоты, при сварке и копке металлов, а также при изготовлении ламп накаливания и драгоценных камней?Водородом наполняют аэростаты и воздушные шары ит. и. В продажу водород поступает в стальных баллонах, где находится под давлением свыше 150 ат.
ТТз числа приводимых ниже методов получении водорода большое техническое значение имеют: получение водорода (или азото-водородной смеси) из водяного газа путем конверсии СО (контактный способ получения водяного газа), из природного газа иля коксового газа в результате ^расщепления метана», из коксового газа или водяного гаао фракционным, снижением, далее злекшроднэом аойы и ж слеза-л проем.и способом. В качестве важнейшего побочного продукта водород получается в процессе электролиза водных растворов хлоридов щелочных металлов и при дуговом способе получения ацетилена. В ограниченном масштабе применяют также способ взаимодействия водяного пара с фосфором (способ Лильенротта) и термическое разложение углеводородов
СН4 —С -|- 2Н2.
В некоторых случаях водород получают в результате каталитического расщепления метанола водяным паром
СНзОП + НгО------> СОг + зн2.
пли в результате каталитического термического разложения а.ммиака
<5 s. по
2NH3— —>N3 + ЗН3.
Однако эти исходные соединения получают в больших масштабах из водорода; между тем получение из них водорода является особенно простым и может быть использовано в таких производствах, которые потребляют его в сравнительно малых количествах (менее 500 м3,'сутки). 1
Водород 45
ВАЖНЕЙШИЕ МЕТОДЫ ПОЛУЧЕНИЯ ВОДОРОДА
1. Растворение цинка в разбавленной соляной кислоте
Zn + 2НС1= ZnCls+Н2.
’ Этот способ чаще всего применяют в лабораториях.
Вместо соляной кислоты можно также использовать разбавленную серную кислоту; однако если концентрация но ел ед ней слишком высока, то выделяющийся водород легко загрязняется SO2 и И2S. Прн пользовании не вполне чистым цинком образуются еще и другие соединения, загрязняющие водород, например AsH3 и РН3. Их присутствием й обусловливается неприятный запах получаемого этим способом водорода.
Для очистки водород пропускают через подкисленный раствор перманганата калия или бихромата калия, а затем через раствор едкого Кали и, кроме того, для освобождения от влаги еще через концентрированную серную кислоту или через слой силикагеля (см. стр. 539). Мельчайшие капельки жидкости, захваченные водородом при его выделении и заключенные в пузырьках газа, не удается удалить пропусканием через промывные жидкости, лучше всего их устранять при помощи фильтра из плотно спрессованной обычной или стеклянной ваты.
Если приходится пользоваться совершенно чистым цинком, то к кислоте необходимо добавить две капли платино хлор пето во до родной кислоты или серпо кислой меди, так как иначе цинк не вступает в реакцию (об этом см. подробнее т. II).
Водород для наполнения воздушных гааров долгое время получали растворением железных опилок е разбавленной серной кислоте (так его получал еще Шарль в 1783 г.). Этот способ использовали вплоть до первой мировой войны.
2. Растворение алюминия или кремния в едкой щелочи
A14-NaOH+3I-I2O=Na[Al(OH)1l+3/2H21
Si4-4NaOH~Na1SiO4+2H2.
Эти реакции применяли раньше для получения водорода в полевых условиях (для наполнения аэростатов). Для получения 1 мэ водорода (при 0° и 760 мм рт ст) требуется только 0,81 кг алюминия или 0,03 кг кремния по сравнению с 2,9 кг цинка или 2,5 кг железа.
Вместо кремния применяют также ферросилиций (кремниевый метод). Смесь ферросилиция и раствора едкого натра, а веденная в употребление незадолго до первой мировой войны во французской армии под названием гидрогенита, обладает свойством, после поджигания тлеть с энергичным выделением водорода по следующей реакции:
Si 4- Са(ОН)2 -J- ДА’аОН = Na2SiO3 + СаО 4- 2Н2.
3. Действие натрия на воду
Na 4- НОН = NaOH 4-»/2Н2.
Ввиду того что чистый натрий реагирует в этом случае слишком энергично, его чаще всего вводят в реакцию в виде амальгамы натрия-, этот способ применяют преимущественно для получения водорода, когда им пользуются для восстановления <dn statu nascendi» (см. стр. 63).
Аналогично натрию с водой реагируют и остальные щелочные и щелочноземельные металлы.
4. Действие гидрида кальция на воду
СаН34- 2Н.0 = Са(ОН)34- 2Н2.
Этот метод является удобным способом получения водорода в полевой обстановке. Для получения 1 лг® водорода теоретически необходимо 0,94 кг СаН2 и, кроме воды, пе требуется пннакпх других реактивов. Отсюда понятно огромное значение гидрида кальция.
4G Глава 2
5. Пропускание водяной пара над раскаленным докрасна железом 4Ha0i-3Fe=Fc3O4-i-4He.
При помощи этой реакции в 1783 г. Лавуазье впервые аналитически-доказал состав воды. Образующуюся при этой реакции окись железа нетрудно восстановить до металлического железа,’ пропуская над ней генераторный газ (см.„стр. 486) так, что продувание водяного пара над одним и тем же железом можно провести произвольное число раз. Этот метод долгое время имел большое промышленное значение, В небольших масштабах его применяют и в настоящее время.
6. Пропускание водяного пара над раскаленным коксом
При температуре выше 1000° реакция идет главным образом по уравнению
Н,о... С -СО-МК.
Вначале получают водяной газ, т. е. смесь водорода й окиси углерода,, с примесью небольших количеств двуокиси углерода и азота (см.. стр. 487). От двуокиси углерода газ легко освобождают промыванием водой под давлением. Окись углерода и азот удаляют при помощи процесса Франка Каро — Линде, т. е, сжижением этих примесей, что достигается охлаждением жидким воздухом до —200°. Следы окиси углерода удаляют, пропуская газ над нагретой натронной известью
CO+NaOH=KaCO?H.
формиат натрия
Этот метод да'ет очень чистый водород, который используют, например,, для гидрогенизации жиров.
Чаще, однако, водяной газ в смеси с парами воды при температуре-4000 пропускают над соответствующими катализаторами, например над. окисью железа или кобальта (контактный способ получения водяного-газа). В этом случае окись углерода реагирует с водой по уравнению-
СО-ЬНоОпар^СОоЦ-Нз («конверсия СО»).
Образующуюся при этом СО2 поглощают водой (под давлением). Остаток окиси углерода (—1 об. %) вымывают аммиачным раствором однохлористой меди. Применяемый в этом способе водяной газ получают пропусканием водяного пара над раскаленным коксом и в последнее время все-больше используют взаимодействие водяного пара с пылевидным углем (превращение угольной пыли в газы). Получаемый этим способом водяной газ содержит обычно большое количество водорода. Выделяемый из водяного газа водород (содержащий азот) применяют главным образом для. синтеза аммиака и гидрирования угля.
7. Фракционированное сжижение коксового газа
Подобно получению из водяного газа водород можно получать фракционированным сжижением коксового газа, основной составной частью которого является элементарный водород (см, стр. 473).
По существу аналогичные методы независимо друг от друга применили в Германии компания Линде и во Франции Клауде (Claude G.). Сначала коксовый газ, из которого предварительно удаляли серу, очищали от СО, промыванием водой под давлением -с последующей обработкой раствором едкого натра,; Затем постепенно освобождали от остальных примесей методом ступенчатой конденсации, которую вели до тех пор,, иона не оставался водород; от последних загрязнений его очищали промыванием сильно охлажденным жидким азотом. Этот метод применяют главным образом для получения водорода для синтеза аммиака,
Водород
47
S. Взаимодействие метана с водяным паром- {разложение метана). Метан взаимодействует с водяным паром в присутствии соответствующих контактных веществ при нагревании (например, при 1100°) по уравнению СИ4-(-Н2Опар-|-48,9 KK<iJi = CO-|-3H, (при постоянном давлении).
Необходимое для реакции тепло следует подводить или извне или применяя «внутреннее сгорание», т. е. подмешивая воздух или кислород таким образом, чтобы часть метана сгорала до двуокиси углерода
CH4-}-2Oa=iCO2-}-2H20nap-|-i91,8 ккал (при постоянном давлении).
При этом соотношение компонентов выбирают с таким расчетом, чтобы реакция в целом была слабо экзотермичной
12CH4-i-5H3OI1ap+5Os=29Hj4-9CO4-3GOS!4-20,4 ккал.
Из окиси углерода посредством «конверсии СО» также получают водород. Удаление двуокиси углерода производят или вымыванием воДой под Давлением или при использовании Жирботол-процесса (см. стр. 483). В качестве исходного вещества в США применяют природный газ (см. стр. 470), в других странах — коксовый газ. Получаемый методом разложения метана водород используют главным образом при синтезе аммиака и гидрировании угля.
9. Взаимодействие водяного пара с фосфором {фиолетовым).
Р 4-4Н,О - Н3РО4 + VaHj.
Обычно работу проводят таким образом, что лары фосфора, получающиеся при восстановлении фосфата кальция в электрической печи (см. стр. 673), пропускают вместе с водяным паром над катализатором при 400—600° (с повышением температуры равновесие приведенной выше реакции сдвигается влево). Разложение образовавшейся вначале Н3РО4 на Н3РО3 и РН3 предотвращают быстрым охлаждением продуктов реакции (закалка). Этот метод применяют прежде вс.его, если водород идет для: синтеза аммиака й последний затем перерабатывают па важное, пе содержащее примесей удобрение — фосфат, аммония.
10. Электролитическое разложение воды.
Ввиду того что чистая йода практически не проводит тока, к ней добавляют электролиты (обычно КОН). При электролизе водород выделяется иа катоде. На аноде выделяется эквивалентное количество кислорода, который, следовательно, в этом методе является побочным продуктом.
Получающийся при электролизе водород очень чцст, если не считать примеси небольших количеств кислорода, который можно легко удалить пропусканием газа над подходящим катализатором, например над слегка нагретым палладированпым асбестом. Поэтому его используют как для гидрогенизации жиров, так и для других процессов каталитического гидрирования. Однако необходимая для электролитического разложения воды энергия довольно велика.:
В качестве побочного продукта водород получается при электролизе хлоридов щелочных металлов (когда его проводят для получения водных растворов щелочей) (см. стр. 208).
Для разрядки 2 а-ионов Н‘, т. е. для получения 1 моля Н2 требуется
2 х 96 500 кулон — 53,6 а-час.
Напряжение разложения составляет 1,23 в (см. стр. 54). Для получения 1 .н3 водорода (18° и 760 .и.« рт ст), следЬватсльно, необходимо (см. табл. 7, стр. 61) теоретически
53,6X1,23X 1000 „
— 22 । Qg еж-ч ~ квт-ч электрической энергии.
48
Глава 2
Практически потребляется значительно больше энергии (4,2—5 кет-ч), что является следствием перенапряжения на электродах (см. стр. 53) и омического сопротивления электролита и диафрагмы. Напряжение па клеммах, как правило, должно быть больше 2 в на ячейку. Разделение анодного и катодного пространства посредством диафрагмы (в большинстве случаев асбестовой) необходимо для предотвращения перемети в алия газов, выделяющихся на обоих электродах, так как для уменьшения падения4 напряжения в электролите электроды располагают возможно ближе.
На рис. 5 представлен поперечный разрез электролизера — именно такого, у которого ячейки подключены одна за другой таким образом, что анод одной ячейки одновременно является катодом следующей (биполярные ячейки). Б результате такого рас-
Р я с. 5. Промышленный метод электролиза воды в биполярных ячейках.
Г — электроды; й — полосой для сбора газа; 3 — гибкие подсоединения;
4 — диафрагма; 5 — окантовка пластины; .6 — изоляция и уплотнение.
положения (введенного О. Шмидтом) падение напряжения при переходе тока от одной ячейки к другой снижается до минимума. Электроды, разделяющие ячейки, изготовлены из жести, никелированной со стороны анодного пространства, и снабжены перфорированными пластинами, на которых происходит преимущественное выделение газа. Таким образом газы быстро выходят из электролизера, Чем предотвращается падение напряжения в электролите.
Снижение проводимости пузырьками газа, взвешенными в электролите, объем которого опи могут уменьшить более чем на четверть, обусловливает значительный перерасход электроэнергии. Поэтому значительный интерес представляют попытки уменьшить объем пузырьков применением высокого давления (100—200 ат) — электролиз под давлением. Если в этом случае выделяющимися газами наполняют стальные баллоны, то получают, следовательно, экономию в работе но сжатию газов. Преимуществом также является незначительный объем ячеек при электролизе под давлением.
В случае биполярных ячеек (которые соединяют до 250) работают при силе тока 10 000 а, в случае униполярных ячеек — даже до 20 000 а. Степень чистоты, подученного таким способом водорода 99,0—99,95%, а получающегося наряду с ним кислорода — 99,6—99,8%.
Водород 49
ЭЛЕКТРОЛИЗ <ВОДЫ; ЭЛЕКТРОХИМИЧЕСКИЙ РЯД НАПРЯЖЕНИЙ
Электролитическое разложение воды, основано на том, что ионы, водорода Н‘ разряжаются на катоде. Нейтральные атомы Н тотчас же соединяются попарно в молекулы На
2Н'-|~2е=Н?,
Конечно, при этом необходимо, чтобы с анода в раствор переходило такое количество положительного электричества, которое равно отрицатель?-ному количеству электричества, переходящему в раствор с катода.
ИОн водорода вводныхрастворах,х.е. гидратированный нопворрубрр (см.стр. 100), называется ионом гидроксония. Обычно его обозначают Н * **. Не следует смешивать его со свободным электроположительно заряженным атомом водорода (ядром водорода, протоном Н+). Свободные ядра водорода (протоны) обладают совсем другим запасом энергии, чем связанные с молекулами Н2О (см. стр. 101), и значительно отличаются от пего своей химической реакционно способностью (см. т. II, гл. 18).
Вместо выражения; «с положительного электрода в раствор поступило два положительных электрических заряда» можно также сказать: «па положительный электрод из раствора перешло два отрицательных электрических заряда». В случае если раствор щелочной, отдача отрицательных зарядов положительному электроду большей.частью идет так, что там. происходит разряжение г и др оксильных ионов — ионов ОН'. Образующиеся при этом нейтральные остатки ОН немедленно вновь соединяются попарно, образуя молекулу воды я отщепляя один атом кислорода. Атомы кислорода также соединяются попарно в молекулы Ог. Таким образом, происходящий на аноде процесс можно представить уравнением
2ОН' - 2е = Н2О Ч- 1/2Ог.
Если раствор содержит соли или кислоты, то иногда на аноде могут вместо гидроксильных ионов разряжаться и другие ионы. Также и на катоде разряжаются исключительно водородные ионы только в том случае, если в растворе отсутствуют какие-либо положительные ионы, которые могли бы разряжаться легче, чем водородные ионы.
Собственный потенциал и потенциал разряжения. Работу, которую необходимо затратить, чтобы разрядить ноны различных веществ, можно точно выразить разностью напряжений (разностью потенциалов) между данным веществом и находящимся в соприкосновении с ним раствором, содержащим ионы этого вещества. Эта разность напряжений Е — собственный потенциал данного вещества — зависит от самого вещества, концентрации раствора и тем,пературы. Существенное значение при этом имеет «действующая концентрация» или «активность» соответствующего иона в растворе. Её получают умножением концентрации с, найденной аналитически (выраженной в молях или трамм-ионах па литр) на коэффициент, так называемый коэ$д5ициент активности /о, который позволяет учесть отклонения от законов, действующих в случае «идеальных разбавленных растворов» (см. стр. 91). Активность а, следовательно, выражается уравнением а = с-/а. Зависимость собственного потенциала от температуры и концентраций можно представить формулой Нернста
J’T
In щ • . (1)
* Знаком е обозначают свободный отрицательный электрический заряд.
** В случае электролитических ионов, .согласно предложению Оствальда, заряд принято обозначать точками и апострофами, например Na’, Zn", Al"1, Cl', SOj\ PO(", Для обозначений электрических зарядов атомов, молекул или радикалов применяют знаки Л- и —.
4 Г. Реми
50
Глава 2
где В — газовая иостояпаая-, Т^— температура, (абсолютная температура) “К; а актпвность ионов соответствующего вещества в растворе; п — число электрических зарядов (в фарадеях), приходящееся на 1 s-аои вещества*. Если Е выражают в вольтах и вместо натуральных логарифмов (In) вводят десятичные логарифмы (log), то ддя 18° (Т = 295,16) получают
,, 0,05777
. £=fe0 -Ь —-— 1ога- (>»>
Так как при а = 1 второй член уравнения становится равным нулю, то Еа соответствует разности потенциалов между веществом в незаряженном состоянии и раствором, содержащим его ионы при активности, равной 1, Обычно Ео называют нормальным или свганЗартным потенциалом соответствующего вещестна. Он зависит от температуры, а в остальном является константой, характерной для данного вещества.
Рис. 6. Цинковый (слева) и водородный’(справа) электроды, соедилеиные в гальванический алемеят.
Непосредственно точное определение разности потенциалов между электродом и раствором соответствующего электролита, т. е. абсолютное значение собственного потенциала, в большинстве случаев затруднительно. Гораздо легче измерить разность потенциалов между двумя электродами^ каждый из которых погружен в раствор соответствующего электролита. Если оба раствора соприкасаются, то они, еслиюставить в стороне небольшой скачок потенциалов на границе соприкосновения (так называемый «диффузионный потенциал»), будут иметь один и тот же потенциал. Таким образом, разность потенциалов между электродами в случае, если активности ионов обоих растворов равны 1, дает непосредственно разность нормальных потенциалов.
В качестве примера рассмотрим опытную установку, показанную на рис. 6. В одном на сосудов (слепа) находится цинковая палочка, погружениая в раствор сернокислого цинка, а в другом — покрытая платиновой чернью платиновая пластинка, погруженная в раствор разбавленной серной кислоты и омываемая струей водорода. Подобный электрод обладает вполне определенным потенциалом, и притом характерным для
* 1 фарадей = 96 493 кулон, (см. стр. 55). Если R выражено также в электро ст а- > тичсских единицах (=8,3143 дж'ц то при 18° получается 2,30259 49з'^~
= 0,057767. При 20° эта величина разпа 0,058164, при 25° — 0,059156.
Водород
51
водорода. Поэтому Он называется водородным электродом. (Простейшая форма водородного электрода, применяющаяся для различных целей, изображена на рис. 7.) Разноси, потенциалов между цинковым и водородным электродами можно легко измерить. Она равна, если пренебречь диффузионным потенциалом (который При надлежащей постановке опыта в большинстве случаев мо;т;по свести практически к ничтожной величине*), разности Е%п—Е™, где —разность потенциалов между цинковой палочкой и раствором Серпо-Кислого цинка, а —разность потенциалов между водородным электродом и раствором серной кислоты. Последнее справедливо ввиду того, что, кроме трех мест соприкосновения (Zn | раствор ZnSO41 раствор Н,5О4 | Н2), в цепи нигде нет скачкообразного падения потенциала. Если условия опыта подобрать так, чтобы активность ионов Za” в растворе сернокислого цинка и активность ионов водорода в растворе серной кислоты равнялась каждая 1, и затем измерить разность потепцяадов между электродами, то окажется, что цинковый электрод будет заряжен на 0,76 в более отрицательно, чем водородный. Это значит, что нормальный потенциал цинка на 0,76 в ниже, чем нормальный потенциал водорода
----0,76 в..
Обычно это кратко выражают так: нормальный потенциал цинка, отнесенный к нормальному водородному электроду, равен —0,76 е.
Соответствующим образом с нормальным потенциалом водорода можно сравнивать нормальные потенциалы любых других металлов. При этом, как уже показывает пример водорода, пет необходимости ограничиваться только чисто металлическими электродами; существенно лишь то, чтобы электрод хорошо проводил электрический ток и чтобы процесс, происходящий на электроде, был точно определенным и обратимым.
В табл. 4 сопоставлены нормальные потенциалы некоторых веществ при 18°, отнесенные к нормальному водородному электроду при той же температуре. Поскольку эти значения относятся к газовым электродам,
они имеют силу только для определенного давления данного газа, равного 1 атм (Нормальные потенциалы для тех веществ, которые не приведены в табл. 4, будут указаны при их описании.)
Р и с. 7. Водородный электрод (простейшая форма).
Таблица 4
Нормальные потепцпалы, отнесенные к нормальному водородному электроду
Катионы 6 Катионы <? Анионы
К |К- —2,9 Ni| Ni- —0,25 F3|F' +2,8
Са|Са" —2,8 Sn|Sn“ —0,16 C,]2| O' +1,36
А] | Л1 — —1,69 Pb|Pb" —0,13 Br, [ Br' +1,08
Мп|Мп" —1,1 H2|H- —0,00 I3|I' +0,58
Zn | Zn‘" —0,76 Cu|Cu- -J-0,35 O3|OR' 4-0,41a
Fe | Fc‘" -,0,51 Hg | Hgy 4-0,793 S I S" —0,51
Cd|Cd" —0,40 Ag | Ag* +0,808
Co|Co- —0,29 An | Au"' 4-1,38
а Следует отметить, что нормальный потенциал кислорода рассчитан для раствора, в котором активность ионов ОН' = 1. В нейтральном растворе со Пет венный потенциал кислорода, вычисленный по отношению в нормальному водородному электроду, равен 4-0,81 в, как это следует из уравнения (1а).
* Относительно точного вычисления диффуэцоппого потенциала см. Н с г-m a n s J, J., Z. physik. Chem., (Л) 176 (1936) 55 и 131.
4*
52
Глава 2
Если теперь расположЙть металлы, включая и водород, “в порядке возрастания их нормальных потенциалов, то получится ряд, в котором наиболее электроположительные металлы займут моста крайние слева, а наименее электроположительные'окажутся справа (электрохимический ряд напряжений, см. табл. 5).
Таблица 5
Электрохимический ряд напряжений
Возрастание нормальных потенциалов
К, Са, А1, Мп, Za, Fc, Cd, Со, Ni, Sn, Pb, Hs, Cu, Hg, Ag, Au <--------- . ~——---------------------—-----,--------
Увеличенье электроположительного характера / :
Вместо того чтобы нормальные потенциалы относить к водороду, 5 находящемуся в контакте с раствором, у которого активность ионов Н' равна единице, нормальный потенциал можно определить каким-либо другим путем. Пернет предложил использовать нормальные водородные электроды. В настоящее время такой способ почти общепринят. Если при измерениях для сравнения потенциалов часто пользуются и другими электродами, главным образом каломельным электродом (см, т, II, гл. 7), ’ то все же найденные величины затем пересчитывают по отношению к нормальному водородному электроду.
В старых работах электролитические, потенциалы отпосели к водородному электроду , у которого водород (тара атмосферном давлении) находится в контакте о раствором, с кажущейся (вычисленной па основапнп электропроводности) концентрацией ионов Н', равцой 1, Такой водородный электрод имеет потенциал на 2,3 ма выше, чем описанный нормальный водородный электрод, т. с. такой электрод, который находится в контакте с раствором, имеющим активность ионов II' — 1. Такими растворами, в которых активность ионов И'составляет.!, являются, например при 25° 1,184 М раствор соляной кислоты и 3,826 М раствор серной кислоты.
Если активность ионов в растворе отклоняется от единицы, то, зная активность ненов а (или концентрацию ионов с и соответствующий ей коэффициент активности) и пользуясь уравнением (Г), можно легко вычислить значение нормального потенциала данного вещества М, отнесенное к нормальному водородному электроду. Эта величина выглядит так: oeh - (EJ1 — ЕИ); ..таким .образом, имеем
osh -bf -Я„н = Е*1—Е«--— in а.
Экспериментальная величина
Если соединить проводом оба электрода цепи, показанной па рис. 6, то от водородного электрода к цинковому потечет электрический ток, причем последний будет непрерывным ввиду того, что между обоими электродами вновь и вновь возникает разность потенциалов. При этом цинк, приобретая положительный заряд, будет переходить в раствор, в то время как на другом электроде в результате разрядки ионов водорода •будет выделяться водород. Если левый сосуд на рис. 6 заменить сосудом , с медным, электродом в растворе сульфата меди, то ток дотечет по проводу, щаоборот, от меди к водородному электроду. Это происходит потому, что л соответствии с данными табл. 4 медь обладает по сравнению с водородом
।
Водород
53
более высоким потенциалом. Таким образом, если в первом случае выделяется водород, а цинк переходит в раствор, во втором, наоборот, в раствор будет переходить:водород, а медь будет выделяться ид раствора. То же явление будет происходить и в том случае, если через раствор пропускать ток, подведенный извне, прилагая к электродам некоторое напряжение (разность потенциалов). При этом всегда будет выделяться то вещество, выделение которого требует приложения наименьшего напряжения^ Очевидно, наименьшая величина того напряжения, которое необходимо приложить для начала выделения данного вещества,— так называемый потенциал выделения или потенциал разряжения,— должна быть равна и противоположна по знаку разности потенциалов между электродом и раствором. Поэтому потенциалы разряжения, которые, конечно, также следует относить к нормальному водородному электроду, равному 0, имеют обратный знак и по величине равны соответствующим собственным потенциалам, отнесенным к нормальному водородному электроду. Отнесенный к нормальному водородному электроду собственный потенциал коротко называют отдельным потенциалом соответствующего вещества.
Перенапряжение. Практически водород и другие газы не всегда начинают выделяться, если приложено теоретически необходимое для этого напряжение. В зависимости от характера электрода, на котором должно происходить выделение, иногда требуется приложить значительно более высокое напряжение. Разность между величиной напряжения, действительно необходимого для выделения, и теоретической величиной напряжения называется перенапряжением. Величина последнего достигает, цапример, в случае выделения водорода на гладком Платиновом электроде (в отлйчие от покрытого платиновой чернью) 0,08 в. Для свинцового электрода эта величина составляет 0,78 в, а для ртути 0,8: в. В случае выделения кислорода на нцкеле, перенапряжение равно около 0,1 в, па железе 0,24 в и на гладкой платине 0,44 в. (Эти значения имеют силу в условиях низких плотностей: тока, см. далее.)
Явление перенапряжения обусловлено тем, что разрядка ионов па поверхности электрода (папрпмер, по уравнению Н3О" + е —> II2O-f-H) происходит с ограниченной скоростью (Smits, 1919; Volmer, Erdey-Gruz, 1930). Первоначально ионы располагаются на поверхности электрода, образуя двойной слой, который ведет себя как конденсатор и повышает скачок потенциала по отношению к. раствору*. Чем выше плотность тока, тем больше собирается у поверхности электрода и ср зарядившихся ионов. Этим и объясняется [что уже в 1900 г. было обнаружено Тафелем (Tafel)] увеличение перенапряжения (ц) с ростом плотности тока. 1. Эту зависимость можно представить уравнением
ц — а -)- Ь log У.
Константа а зависит от природы электрода, в то время как b — только от природы выделяющегося газа. На основе теории Фольмера (Folmer) для водорода при 25° она получается равной 0,118. Такая же величина найдена для большинства металлов. Ней, тральные соли, ионы которых оттесняют от поверхности платины ионы водорода, снижают перенапряжение (Heyrovsky). Другие добавки (например, коллоиды) действуют: аналогичным образом. Перепапряжение падает с ростом температуры н в незначительной мере с повышением давления. Далее, оно уменьшается, если попеременно подводят постоянный и переменный ток. Последнее обстоятельство обусловлено тем, что сначала при пропускании тока перенапряжение не сразу достигает наибольшей величины,
* Не исключены случаи, когда причиной перенапряжения является задержка образования молекул из атомов (Н-рН-» НЕ) (Knorr, 1936). Однако для большинства исследованных случаев это толкование (предложенное Тафелем) опровергается Фолкнером.
54
Глава 2-.
а только постепенно. Временный подъем следует отнести за счет бездействия первоначально существующих активных центров поверхности электрода (Maying, 1936). Перенапряжение находится в подчиняющейся определенным законам зависимости от кра-випны поверхности. На ровной поверхности оно меньше, чем на выпуклой, и больше, чем на вогнутой (Sederholm, 1932). Таким образом, объясняется, почему на металлах е многочисленными шероховатыми частицами (платиновая чернь) перенапряжение минимально и при очень незначительных плотностях тока совсем исчезает,
Потенциал разложения. Величина наименьшего напряжения, которое необходимо приложить для электролитического разложения какого-нибудь вещества {потенциал разложения.}, получается сложением потенциалов выделения соответствующих катионов и анионов. В случае анионов при этом учитывают обратное направление тока. Следует также помнить, что величина п в уравнениях (1), (1а), (16) для анионов отрицательна. Другими словами, можно сказать; чтобы определить потенциал разложения какого-нибудь электролита в численном выражении, следует из собственного потенциала вещества, образующего анионы, вычесть собственный потенциал вещества, образующего катионы. Отдельный потенциал Ес для ^.-валентного иона, имеющего концентрацию с, получают, используя приведенный в табл. 4 нормальный потенциал одинакового иона Ео, по уравнению
„ _ , 0,05777 , ,
, Ес^Еа + -^~^-~ logc/0, (16)
где /0 — коэффициент активности соответствующего иона при концентрации с. В случае вычислений, не требующих большой точности, можно принять /в = 1. Тогда но учитывается различие между аналитической молярной концентрацией с и активностью а.
Для 1 и. в отношении гидроксильпых ионов раствора едкого кали потенциал разложения в соответствии с определением и уравнением (16), если принять /й==1 и концентрацию водородных ионов ви. равной 0\74-10‘14=1 1О‘Ы,1з при комнатной температуре, имеет следующую величину (см. стр, 74):
(Я?+0,058 logcH.)=0,41—0+14,ia-0,058=i,23 е.
Потенциал разложения Z2 (1 И.) в отношении водородных ионов раствора серной кислоты вычисляют из соответствующих отдельных потенциалов (так как cqh,= =1Q-m,13) следующим образом;
Z2^(E0 —0,058 log сон.)—Ь',?^0,414-0,058 14,13—0=1,23 в.
В соответствии с этим потенциал разложения для кислого и щелочного растворов должен выражаться одной и той же величиной. И действительно, разложение происходит при таком напряжении, если в качестве катода исиоиьзовать небольшое платиновое острие, а в качестве анода — большую платинированную платиновую пластинку. Разложение обнаруживается по энергичному выделению водорода на катоде. Однако если, наоборот, взять большой катод и небольшого размера анод, то разложение становится заметным по образованию пузырьков кислорода только при значительно более высоком напряжении. Это зависит от того, что процесс, к которому относится приведенный в табл. 4 нормальный потенциал, заключается в разрядке ионов О"*. Однако их концентрация в кислом растворе столь ничтожна, что при указанных условиях опыта не может происходить заметного выделения кислорода. Если же увеличивать папряже-
* То, что при вычислении собственного потенциала кислородного электрода все же можно примсвять уравнение (16), как если бы дело заключалось в разрядке ионов OIT, объясняется тем, что множитель 2, который вследствие двойного заряда ионов кислорода необходимо ввести для п в уравнение (1), выпадает благодаря пропорциональности концентрации ионов О" квадрату концентрации ионов ОИ'.
Водород 55
ние, то будет достигнут потенциал разряжения гидроксильных ионов, который в растворе с концентрацией водородных ионов равной 1, составляет 1,68 в, Но так как концентрация гидроксильных ионов в кислом растворе также остается незначительной, -го выделение кислорода становится бурным только тогда, когда будет достигнут потенциал разряжения иолов SO), равный 1,9 о.
При вычислении количества потребляемой электроэнергии при техническом осуществлении процесса электролиза к потенциалу разложения следует прибавить сумму перенапряжений на обоих электродах, а также иметь в виду, что напряжение расходуется, кроме того, на преодоление омического сопротивления внутри сосуда и в проводах. Общая потреблявшая энергия определяется как произведение напряжения, силы тока и времени. Произведение силы тока па время определяет количество электричества, пропущенного через раствор. Для разложения 1 г-экв какого-нибудь электролита необходимо пропустить через пего или через его раствор количество электричества, равное 96 493 кулою 1 = 1 фарадею"; или 26,804 а-час.
Условия выделения водорода из растворов, В соответствии со сказанным выше при электролизе водород всегда будет выделяться из раствора в тех случаях, когда последний не содержит никакого другого вещества, выделение которого требует меньшего напряжения, чем выделение водорода. Применяя это положение, следует помнить только, что, согласно уравнению (1), необходимо учитывать концентрацию (соответственно активность) ионов Н' в растворе. На каждое уменьшение показателя степени десяти в числе, показывающем молярную концентрацию (соответственно активность) ионов водорода, ла одну единицу (т, е. при каждом уменьшении этой концентрации в 10 раз) напряжение, которое необходимо приложить для выделения ионов водорода, увеличивается, как это видно из уравнений (1) или (16), на 0,058 в. Так как концентрация ионов Н’ в чистой воде при комнатной температуре равна приблизительно 10"7 й в 1 и. растворах едких щелочей — около 10’1’1, то и для выделения водорода из этих растворов необходимое напряжение на 0,4 или соответственно на 0,8 в выше, чем это требуется для его выделения из нормального раствора ионов водорода.
Если в содержащий водородные ионы раствор — все водные растворы содержат водородные ионы,— внести металл, потенциал разряжения которого выше соответствующего потенциала разряжения водорода в том же растворе, то металл будет переходить в раствор, а водород будет выделяться (в газообразном виде).
На этом основаны способы получения водорода, приведенные выше под номерами 1—3. Этим обстоятельством объясняется не только выделение водорода из соляной кислоты при действии на лее ципка или выделение его из воды при действии патрия, но я разложение раствора едкой щелочи под действием алюминия, так как, несмотря па крайне незначительную концентрацию водородных ионов в последнем растворе, потенциал разряжения водорода в нем все же даже, чем потенциал разряжения алюминия. При этом данный процесс значительно облегчается еще благодаря тому, что алюминий не остается в щелочном растворе в виде иона АГ”, но в большей части переходит в ионы алюмината [AJfOHXJ', что еще повышает потенциал разряжения алюминия. То же объяснение справедливо и в отношении цинка. Нерастворимость металлов вроде алюминия и цинка в чистой воде объясняется вторичным процессом, а именно отложением иа поверхности этих металлов очень тонкого слоя нерастворимых гидроокисей, которые я защищают их от дальнейшего действия воды.
ТЕРМИЧЕСКОЕ РАЗЛОЖЕНИЕ ВОДБГ;
ЗАКОН ДЕЙСТВИЯ МАСС
Приведенные на стр, 46 методы получения водорода основаны в конечном итоге па термическом разложении воды, (следует напомнить в первую очередь разложение водяного пара раскаленным железом или раска-
5й Глава 2
ленным углем). Сущность процесса такова; 5
211,07:211,—07 (2j
j Несмотря па то что разложение воды при температурах, с которыми
приходится иметь дело иа практике, крайне незначительно^ тем не менее
1 л этот процесс все-таки вдет в основном слева направо благодаря тому >
t что выделяющийся кислород тотчас же связывается железом или углем
;• и устраняется из сферы реакции. Что именно это так и.должно происхо-
дить, следует из химического закона действия масс.
Закон действия масс. Закон действия масс в применении к реакции с любым числом веществ гласит:
. Для реакции между веществами А, В, С и т. д., которая является обратимой** и протекает с образованием веществ М, N, Oji т. д. в соот-: ветствии с уравнением
аА-|- ЬВJ с'.: • ... Vi mM-r-uN ; ’!О-| ..
имеет силу следующее равновесие:
|Л]“х(11]ьх|с;-X... Лг
(МГх|:\Г'х|ОГх ..., с‘ ?
'При этом символы, заключенные в квадратные скобки, означают молярные концентрации веществ, т. е. их концентрации, выраженные в молях на литр. Кс —зависящая от температуры констайта («константа равновесия»). Закон действия масс справедлив только для равновесных гомогенных {однородный) систем.
Гомогенной называется такая система, в которой пет поверхностей раздела, кроме "поверхностей молекулярного порядка; Таким образом,: гомогенными являются газы в состояний полного смешения, жидкости (смеси жидкостей — только в том случае, если одна ив них вполне растворена в другой), растворы, твердые тела с однородным со- ; ставом (включая и твердые растворы). Примерами негомогенных систем, являются ; газ, соприкасающийся с жидкостью или твердым телом; несмешивающдеея или не виол- ' ле смешивающиеся соприкасающиеся жидкости; жидкости, соприкасающиеся с твер- ! дыми телами; два или несколько твердых тел различного состава, находящиеся рядом i (за исключением, как уже указано, того случая, когда они полностью образуют твердые ; растворы). Однородные составные части смеси называют также термином :
Таким образом, гомогенные системы являются однофазными.
При применении химического : закона действия масс к реакциям. протекающим в газообразных системах, вместо концентраций веществ, принимающих участие в реакции, можно использовать значение ид .парциальных давлений, так как для газов давления пропорциональны концентрациям. Следует, однако, иметь в виду, что численное выражение константы равновесия станет от этого другим (за исключением случая, когда общее число молекул и вместе с тем суммарное давление при реакции: це изменяется). ,
Если обозначить константу равновесия, относящуюся к давлению, через а константу, выраженную через концентрации Кс, то получим следующее;
A‘f)==(./i/,)2v.A'(, или In A'p=in A'e4-Sv-jnУТ, (4)
* Наряду с этой реакцией происходит также, как это доказал в 3928 г, Б он гофер (Bonhoeffer), термическое разложение пб уравнению 2Н?0 = Н2 Ц- 2011. Однако подробное обсуждение этого процесса при настоящем изложении существенного значения не имеет.
* * Каждую химическую реакцию в принципе можно рассматривать как обратимую.
________________________________Догород____________________________ 57 где Sv :— арифметическая сумма молей, принимающих участие в реакции. При атом число молей по левую сторону от знака равенства считают положительным, а по правую — отрицательным. Например, для реакции 2H2-J-O2=2H2O Sv=3—2=1. Таким образом получается следующая связь между константами равновесия, если принять во внимание уравнения (7) и (8) на стр. 59;
Kv~HT.Kr, '
'Уравнение (4) можно получить па основе газового законаpv^nRT, заменяя в уравнении (3) молярную концентрацию каждого газа к/и через p'RT, причем р означает парциальное давление соответствующего газа. . ;
Условием установления соответствующего уравнению (3) равновесия является, конечно, достаточно быстрое течение реакции. Однако при низких температурах этого обычно не бывает. Системы, не находящийся в равновесий, но в которых вследствие очень малой скорости реакции не замечается превращении, называются метастабилъными.
Закон действия масс был первоначально установлен эмпирически Гульдбергом и Вааге (Goldberg, Waage, 1864). Он получил название «закон действия масс», так как в то время молярные концентрации называли «активными массами». Позднее оказалось, что закон действия масс вытекает также в качестве необходимого следствия из кинетической теории газов и жидкостей. Наконец, его можно вывести также и термодинамически, т. е. показать, что он с необходимостью следует из первого и второго законов термодинамики.
При термодинамическом выводе закона действия масс предполагают, что компоненты реакции подчиняются законам «идеальных газов» и «идеально разбавленных растворов». Вследствие того что реальные газы и растворы отклоняются от этих законов, и тем больше, чем больше их концентрации, то для них закон действия масс имеет силу в указанной выше форме (когда введены молярные концентрации) только как закон, ограниченный и справедливый безоговорочно только для бесконечно разбавленных- растворов. Однако можно добиться того, что этот закон будет действителен с достаточной точностью для любых веществ и при более высоких концентрациях. Для этого аналитические молярные концентрации умножают на определенные коэффициенты, которые называются коэффициентами активности. Произведения аналитических молярных концентраций на коэффициенты активности называют активностями. Активности [введенные в 1907 г. Льюисом (Lewis)], подставленные вместо аналитических молярных концентраций в закон действия масс, делают его справедливым с большой точностью для реальных веществ и не только для бесконечно разбавленного состояния, но и для более высоких концентраций.
Определение активности можно дать термодинамически — независимо от закона действия масс*. Соответственно коэффициенты активности можно определить экспери-
* Например, для растворимого педиссоциирующего вещества активность а при концентрации с определяется как , где Ас — работа, которую необходимо
совершить, чтобы перевести изотермически и обратимо 1 моль растворенного вещества, находящегося в «основном состоянии», в котором его активность aii==l, в состояние с'концентрацией с. В качестве «основного состояния» выбирают такое, в котором вещество следует закону идеальных разбавленных растворов. Строго говоря^ это имеет место только при бесконечно разбавленных растворах (с=0), так что lim а0=с0. с=0
Однако всегда можно указать некоторую конечную концентрацию, при которой а0 соответствует с() в определенных границах точности. Аналогичным путем можно определить активность гйза, пара или растворителя. Относительно определения активности сильных электролитов см. стр. 87.
58
Глава 2
ментально, не подвергая соответствующие вещества химическим превращениям. В табл. В указаны коэффициенты активности некоторых веществ при различных концентрациях. Приведенные значения показывают, что как те коэффициенты активности, которые больше 1, так и те, которые меньше 1, по мере разбавления стремятся к единице. Это должно происходить потому, что, согласно определению, при бесконечном разбавлении исчезает разница между активностью и аналитической молярной концентрацией. Далее, из данных табл. 6 видно, что при умеренных концентрациях (от 0,1 до 1 Л?) для неэлектролитов и слабых электролитов (например, уксусная кислота) отклонения коэффициентов активности От 1 незначительны, а для сильных электролитов — значительны.
Таблица 6 .
Коэффициенты активности водных растворов различных веществ при 25° (т — число молей растворенного вещества в 1000 г растворителя)
тп
i 0,5 0,2 0,1 0,05 0,01 0,005 0,001
3 Метанол 0,80 0,82 0,84 0,87 0,90 0,95
н ф Этанол 0,85 0,87 0,91 0,94 —- — — —
Л цетон 0,935 0,968 0,988 0,994 —•
ф Тростниковый 1,24 1,11 1,05 1,03 1,01 — —
ф tc сахар
Уксусная кислота 0,900 0,960 — -- — — — :
2 j
Е- НС1 0,802 0,750 0,760 . 0,791 0,825 0,902 0,927 0,965
ф H2SO4 0,130 0,154 0,209 0,265 0,304 0,544 0,639' 0,830
NaNO3 0,565 0,633 0,714 0,771 0,820 0,905 0,929 0,966
ВаС1а 0,388 0,392 0,436 0,499 0,564 0,716 0,774 0,881
Са (NO3)2 0,35 0,38 0,42 0,48 0,545 0,71 0,77 0,88
3 MnSO4 0,080 0,110 0,176 0,247 0,333 0,536 0,621 0,780
£ CuSO4 0,041 0,061 0,098 0,149 0,209 0,41 0,53 0,74
о
На основании изложенного выше видно, что в случае газообразных веществ при таких условиях, когда они ведут себя практически как идеальные газы (т. е. при нормальном давлении и не очень низких температурах) в закон действия масс можно, не совершая большой ошибки, вводить аналитические молярные концентрации и соответственно парциальные давления. Если же эти условия не выполняются, то для газов и паров в закон действия масс следует также ввести их активности вместо концентраций.
Зависимость константы равновесия закона действия масс от температуры указывается следующим уравнением, которое было выведено Гиббсом (1879) и Вант-Гоффом;
dip к W .
dT ~РТ2'
или, интегрируя при постоянном значении W, получают
" IV Z I 1 \
(6)
Водород
59
где W — теплота реакций, R — газовая постоянная (выраженная здесь в калориях и равная 1,987 кал)\ 7' — абсолютная температура; К, и К2 константы равновесия при температурах Zj и Г2. Смотря по тому, что принято в jtaaeCTBe констант равновесия К,. или Кр, получают теплоту реакции W или при постоянном объеме Wv или при постоянном давлении H'p.
По предложению Н ер нет а отличают уравнение закона действия масс, выведенное для постоянной температуры [уравнение (3)], названное изотермой реакции, от уравнения (5), выведенного для постоянного объемам названного изохорой реакции (хшоа — объем). Если в уравнении (5) W означает теплоту реакции при постоянном давлении, то уравнение называют изобарой реакции.
Применение закона действия масс к термическому разложению поды. Если теперь применить химический закон действия масс к случаю термической диссоциации воды [уравнение (2) стр. 56], то получим
[На]« х [О81 _ е [НгО]* Кс ' >
или, подставляя вместо концентраций давления рна, Po.2i Рщо, участвующих в реакции газов, получим
Если к этой газовой смеси добавить какое-нибудь вещество, окисел которого обладает при данной температуре меньшим парциальным давлением кислорода, чем вода, то это вещество, образуя окисел, будет до тех пор связывать кислород, пока давление последнего не понизится до величины его парциального давления в окисле. Чтобы при этом Кр оставалась постоянной, необходимо, чтобы частное рн2/рн2о во столько же раз увеличилось, во сколько раз уменьшится давление кислорода ро,г. Поэтому, например, если давление кислорода над окислом добавленного вещества (железа) составляет при данной температуре миллионную часть давления кислорода над водой при той же температуре, то давление находящегося в соприкосновении с ним водорода по отношению к давлению педиссо-циированного водяного пара должно соответственно возрасти в тысячу раз по сравнению с его первоначальной величиной.
। Согласно Эммету (Евине I, 1933) и К any минскому (1934), над смесью Ее и FeO устанавливается следующее равновесное давление:
Температура, ®С 600 700 800 900 1000 1200
?Н=/?н2о 3,° 2>4 2.° 1,fi М М
Таким образом, с понижением температуры равновесие сдвигается в сторону образования водорода. (Это вызвано тем, что с понижением температуры давление кислорода пад FeO снижается быстрее, чем давление водяного пара.) Однако снизить температуру более чем до 600—800° нельзя, так как эта реакция даже в присутствии ускорителей протекает слишком медленно. В случае разложения коксом приходится применять етце более высокие температуры (выше 1000°). В этом случае, впрочем, для повышения давления О2 наряду с равновесием С-)-1/2О2 yi: СО также имеет значение равновесие С0-)~г/202= СО2 (см- стр. 485).
Если в уравнение (8) подставить числовые значения из приводимой ниже таблицы, то получим (ввиду того, что Kp известно из других измерений, см. стр. 75) давление кислорода над FeO при соответствующих температурах. Например, при 1600° К (=727° С) 7Гр=4,5-1О-«1. .Отсюда следует
?сц = К» ()24,5 10- (Ху =0,85-10^1.
Таким образом, при этой температуре давление кислорода над FeO составляет 0,85-• 10*21 ат, или около 6 -10“19 леи рт ст. Этот пример показывает, каким образом дав-
60
Глава 2
левая, которые нельзя непосредственно определить вследствие их очень налой величины, рассчитывают исходя из равновесий, наступающих при взаимодействии. Ниже 570“ Fe3O* обладает еще меньшим давлением кислорода, чем FeO, Повтоиу ниже 570° железо реагирует с парами воды с образованием Fe3Ot.
СВОЙСТВА ВОДОРОДА
Обычный двухатомный водород (Н2)—бесцветный газ* без вкуса и запаха. Зажженный на воздухе он горит голубоватым, очень горячим пламенем. Водород в 14,38 раза легче воздуха и проводит тепло в семь раз лучше последнего.
Что касается коэффициента расширения при нагревании и зависимости объема от давления, то при не слишком низких температурах и не слишком высоких давлениях водород ведет себя почти как «идеальный газ», а именно он хорошо подчиняется законам Бойля—Мариотта и Гей-Люссака, и, следовательно, уравнению газового состояния p-v=nRT, где
Я = 0,082054 л-ат ле.
Так как эффект Джоуля—Томсона, т. е. явление, заключающееся в том, что газ при расширении без совершения внешней работы (т. е. при распространении его в пустоту) охлаждается, наблюдается для водорода только примерно ниже —80°, то это обстоятельство сначала создавало затруднения для его сжижения. Последнее впервые удалось осуществить в 1898 г. Дьюару, который выпускал через узкое отверстие в вакуум сильно сжатый и предварительно охлажденный жидким воздухом до —205° водород. Жидкий водород представляет собой очень легкую, бесцветную, непроводящую электрического тока жидкость, застывающую при кипении под уменьшенным давлением за счет теплоты испарения в твердую массу с удельным весом 0,08.
Важнейшие физические константы водорода сопоставлены в табл. 7.
Вследствие небольшого молекулярного веса в соответствии с законом Грэма и Бунзена (см. т. П) водород обладает наибольшими из всех газов эффузионной и диффузионной способностями.
В воде и в других растворителях водород очень мало растворим и плохо растворим в каучуке. Последнее обстоятельство имеет большое значение для применения водорода в аэростатах наполнения**. Некоторые металлы поглощают его в значительных количествах. При температуре красного каления он диффундирует через них так же, как и через кварц. Наибольшей способностью растворять водород обладает палладий (см. т. II).
Молекулярный водород при обычной температуре химически малоактивен. Только фтор соединяется с ним при обычных условиях***, хлор — при освещении, кислород — только при поджигании смеси. При повышенной температуре водород вступает в соединение со многими элементами, преимущественно с неметаллами, но и с сильно электроположительными металлами (щелочными и щелочноземельными).
* Относительно атомарного водорода см. стр. 63.
*• Ввиду того что водород в каучуке не растворяется, он не может диффундировать в каучуке в обычном смысле, однако склонен к эффузии через поры каучука. Поэтому водород значительно дольше сохраняется в каучуковых резервуарах, нежели углеводороды /светильный газ) и другие газы, которые легко растворяются в каучуке, хотя значительно уступают ему в эффузионной способности.
*** Скорость реакции Hs с F2 сильно зависит от материала реакционного сосуда. В сосуде из магния в темноте и при комнатной температуре реакция практичесии не идет.
Водород
61
Таблица 7
Фазические константы водорода а
Вес 1л водорода при 0°, 760 ллс рт ст и45°геогр. широты = 0,089870г.
Молярный объем 22,4281; плотность по отношению к кислороду, принятому за 1, =0,062893.
Плотность по отношению к воздуху, принятому за 1, =0,06952.
Точка кипения — 252,8° = 20,4° К, уд. вес при этом=0,0700.
Точка плавления —257,3°=15,9° К, уд. вес при —260° = 0,0763.
Критическая температура —239,9°; критическое давление 12,8 атм; критическая плотность 0,0310.
Удельная теплоемкость: ер (при 2О°)=3,306кал; с„ (при 18°)=2,404 кал.
Отношение удельных теплоемкостей cp/cw=l,407 (при 17°), молярная теплоемкость Су—6,847 кал; Су=4,847 кал.
Удельная теплоемкость жидкого водорода при —256° ер = 1,93 кал.
Удельная теплоемкость твердого водорода при —259,8° ср=0,630 кал. Теплота испарения жидкого водорода 108—114 кал/a (при 20,4° К).
Теплота плавления твердого водорода 14 кал/г.
Кристаллическая структура; гексагональная плотная упаковка (из молекул Н5) а = 3,75, c=6,i2A.
Теплопроводность водорода при 0° Л=0,000412 кал/см-сек-а рад, т. е. при градиенте температур 1° через 1 с.и перпендикулярно площади сечения проходит за секунду количество тепла, равное X..
Растворимость в воде: при 0°=0,0215, при 18° = 0,0185 объема водорода на 1 объем воды (взятой при 0°).
Щ-.ислгкула: теплота образования 6 из 2Н= 102,72 ккал/моль Нй (при 0° К), межъядерное расстояние 0,75-10“* с-и, момент инерции 0,467-10'40, дипольный момент равен 0, средняя длина пробега 1,123-10'ь см (при 0° и 1 атм).
Н-атом: ионизационный потенциал 13, 54 в, сродство к электрону 0,715 эв.
а Данные (исключая указанные для молекулы Н;) откосятся к обычному водороду. т.е. содержащему 0,02% Dj (си. стр. 65).
6 Действительно для параводорода (см. стр. 85). В случае смеси п-Н-: о-Нг=-1 :3 получена величина 102,47 кхсл л<оль.
Соединение водорода с кислородом с образованием воды по уравнению
Нг+‘/зОг=Н2О* (9)
сопровождается чрезвычайно большим выделением тепла. Теплота образования еоды из Hs и Os при 25° и постоянном давлении составляет 68,35 ккал на 1 моль жидкой воды или 57,85 ккал на 1 моль газообразной НзО.
Температурную еависимостъ теплоты образования для газообразной Н,0 при постоянном давлении в области от 0 до 2000° можно представить, согласно Льюису (Lewis) и Реиделлу (Bandall), эмпирическим уравнением И7,, —57,444-9,4’10'4 74* 4-1,65-10-’ 72—7,4-НУ10 Т8т где 7 — абсолютная температура; И4,— теплота образовании {ккал/молъ Н2О). Эта величина при 0° равна 57,80, при 100° — 57,98 ккал.
Смесь водорода и кислорода в соотношении, соответствующем уравнению (9) (гремучий газ), особенно сильно взрывает при поджигании.
* В противоположность прежним представлениям установлено, что в процессе реакции не образуется Н5О.> в качестве промежуточного продукта (подробнее см. т. II).
62 Глава 2
В более широком смысле слолгГагремучим газом» иногда насылают также и другие взрывчатые смеси водорода с кислородом или с воздухом. Данные о границах взрывчатости смесей водорода с воздухом несколько противоречивы, В общем можно сказать, что детонации уже пе происходит, если смесь содержит менее 6 или боксе 67 об.% водорода. Для полного сгорания 1 объема водорода требуется 2,39 объема воздуха. Точка воспламенения смеси, образующей гремучий газ, в Значительной мере зависит от сосуда, в котором опа находятся. Чистый гремучий газ взрывает начиная с 500°. В присутствии некоторых катализаторов взаимодействие происходит уже при значительно более низкой температуре; при действии металлов группы платины оно начинается отчасти ужо прн комнатной температуре, при этом иногда без взрыва. Однако большей частью и при этих условиях вследствие выделяющегося при реакции тепла, через короткое время происходит воспламенение смеси, сопровождающееся взрывом.
Безопасное сжигание гремучего газа производится в лоллышя: горелках (кран Даниеля), В таком пламени развивается температура свыше 3100s, Паяльные горелки на гремучем газе применяются в автогенной сварке. Температуру поддерживают такую, при которой получается по возможности меньше шума. При избытке кислорода горелка шумит, а при недостатке — коптит.
Водород соединяется непосредственно также с бромом и серой, а равным образом с иодом, селеном и теллуром. Реакции его с тремя последними элементами доходят до определённого равновесного состояния. В то время как иодистый водород при повышении температуры начинает разлагаться, для селена и теллура равновесное состояние этой реакции с возрастанием температуры, наоборот, смещается в сторону образования соединений. Последние, как и следует ожидать на основании принципа Ле-Шателье, являются эидотермичными соединениями. С азотом водород в сколько-нибудь значительном количестве соединяется только в присутствии катализаторов. Эта реакция экзотермична (подробнее об этом будет сказано при рассмотрении синтеза аммиака).
Присоединение каким-нибудь веществом водорода называют гидрированием*.
Вследствие сильного сродства водорода к кислороду оп очень легко отнимает кислород у других соединений, являясь, таким образом, сильным восстанавливающим средством. Окислы многих тяжелых металлов при нагревании в струе водорода переходят в соответствующие мёталлы.
Некоторые вещества восстанавливаются газообразным водородом уже при обычной температуре — особенно легко хлорид палладия (II) в водном растворе и в гораздо меньшей степени также азотнокислое серебро. Очень многие соединения восстанавливаются водородом (или гидрируются) при обычной температуре в присутствии веществ, «активирующих» водород, к которым, например, относится платиновая чернь или палладий. В присутствии последних водород особенно легко присоединяется к ненасыщенным органическим соединениям [гидрирование по Палю и Скиту]. При повышенной температуре как переносчик водорода действует и никель [гидрирование по Сабатье и Сандерепу].
В последние годы каталитическое гидрирование имеет особенно большое техническое значение, В качестве важнейших процессов следует назвать: гидрирование азота до аммиака, гидрирование угля, нефти и смолы до бензина, гидрировал не окиси углерода до метанола и высших спиртов или до бензола, а также гидрирование ненасыщенных жирных кислот (гидрогенизация жиров).
В лабораториях, особенно органической химии, часто производят гидрирование, «методом встряхивания» при суспендировании в растворителе платиновой черви или палладия. По данным Бизальского (Biesaiski, 1930) эффективным методом является пропускание водорода в раствор, содержащий пенообразователь (черев топкопористый ф а рф ор олый фильтр),
* В органической химии реакции соединения с водородом часто называют также toccmaiмелением. Подробнее см.;гл. 10.
Водород ; 63
— ... “ ч-
Водород особенно активен в момент выделения из своих соединений (in statu nascendi), Это объясняется тем, что в первый момент водород (например, когда он выделяется из серной кислоты при действии цинка или получается путем элекролиза) находится в атомарном состоянии, в то время как в обычном газообразном состоянии атомы водорода соединены попарно в молекулы Н2. Большой реакционной способностью водорода в момент выделения часто пользуются для энергичного восстановления различных веществ. Для этого его получают, растворяя цинк в серной кислоте или в едкой щелочи или внося амальгаму натрия в воду, или, наконец, проводя электролиз. В последнем случае он действует восстанавливающим образом с особой силой, если катод изготовлен из свинца, на котором водород обнаруживает значительное перенапряжение. Таких же результатов можно, согласно Фихтеру (1933), добиться растворением свинцово-натриевого сплава.
Водород при высоком давлении (100—200 ат) и при нагревании (например, 200°), а также в присутствии активирующих веществ может выделять металлы из растворов их солей и тем легче, чем более благородным является металл. Даже такой неблагородный металл, как цинк, может быть вытеснен таким образом из его солей. Одпако гораздо легче это происходит в результате восстановлений связанных с металлами кислотных остатков (например, N Ориона) и выделения металлов в виде гидроокисей (Ипатьев).
Атомарный водород. Если вольфрамовую, платиновую или палладиевую проволоку нагревать в атмосфере сильно разреженного водорода (р менее 0,01 aim pm cm)t то можно доказать образование при этих условиях газообразного атомарного водорода. Он образуется также, как это установил Вуд (Wood, 1922), при пропускании тихих электрических разрядов через водород, находящийся под давлением в несколько десятых миллиметра ртутного столба. Особенно удобен для получения активного водорода способ Вуда в видоизменении Бонхофера. Аппарат, которым пользовался для этого Бонхофер, схематически показан на рис. 8. При: соответствующих условиях опыта можно, пользуясь этим аппаратом, получать активный водород, который, как было доказано, состоит из свободных атомов Н в концентрации до 95 %. Атомарный водород устойчив в точение лишь очень короткого времени (при указанном давлении приблизительно */з—1/s сек)> однако и этого срока достаточно, чтобы пропустить струю (из разрядной трубки) над тем веществом, с которым он должен реагировать. Атомарный водород отличаетей способностью уже при обычной температуре восстанавливать ряд металлических окисло в (например, СпО, РЬО, HgO, AgsO, BiaOs), сульфиды и галогениды. С серой, мышьяком, фосфором он соединяется с образованием соответствующих водородных соединений. С молекулярным кислородом он преимущественно даОт перекись водорода НаОа. Последняя получается, однако, если реакция идет при низких температурах; в противном случае образуется вода. При низких температурах вначале получается изомер НаОа, который при —115° с частичным разложением переходит в нормальную перекись (Harteck, 1932). С ртутью атомарный водород реагирует с образованием устойчивого только при низких температурах твердого гидрида. Олеиповная кислота гидрируется им непосредственно (причем частично полимеризуется), и именно так, что соединяются две молекулы кислоты присоединяя по однцму атому Н на каждую молекулу.
64
Глава 2
На ненасыщенные органические соединения атомарный водород может также действовать дегидрирующим образом, отщепляя Н с образованием На.
Аппарат Бонхофера (рис. 8) состоит из сосуда для электролитического выделения атомарного водорода и разрядного сосуда 4. Последний представляет собой стеклянную трубку длиной 2 м с поперечным сечением 2 см, которой ради экономии места придана S-образная форма, В эту трубку вделаны два цилиндрических электрода из листового алюминия, и она хорошо выдерживает нагрузку 0,4 а постоянного тока при напряжении на клеммах 3000—4000 в. Между точками 3 и 4 вмонтирован металлический вентиль для регулирования (на рисунке заштрихован). Последний регулируют таким образом,
Рис. 8. Прибор для получения атомарного водорода.
чтобьРпри энергичном отсасывании водорода с левого конца аппарата (лучше всего при помощи диффузионного насоса Реде) давление в разрядной трубке установилось в пределах 0,1—1 мм рт ст. Погружая U-образную трубку 1 в жидкий воздух, можно освободить водород от водяных паров, Вещества, отношение которых к действию активного водорода должно быть испытано, помещают в трубку а. Продукты реакции конденсируются в трубке 2 при охлаждении ее жидким воздухом.
Характеризующийся указанными выше реакциями активный водород состоит из атомов водорода Н. Это подтверждается прежде всего тем обстоятельством, что при условиях, благоприятных для образования активного водорода, возрастает интенсивность излучаемого атомами водорода спектра Бальмера (см. гл. 3) по сравнению с поло-' сатым спектром, исходящим от молекулы И2.
Принимая во внимание указанное обстоятельство, следует считать приведенную выше продолжительность существования активного водорода еще сравнительно большой, так как расчет показывает, что между атомами активного водорода должно произойти в среднем до 1 миллиона столкновений, прежде чем они вновь соединятся в молекулу Н2, хотя теплота образования молекулы И2 из атомов довольно значительна (см. табл. 7, стр. 61). Объясняется это следующим образом: чтобы могла произойти рекомбинация, должен быть введен третий партнер по соударениям, который поглощает некоторую часть тепла, выделяющегося при рекомбинации, (Подробнее q механизме реакций см. т. П.) Особенно сильно ускоряется рекомбинация атомарного водорода до Н 2 на поверхности некоторых твёрдых веществ. Ускоряющее действие их можно измерить, помещая соответствующее вещество па шарик термометра и вещая последний в струю газа. Чем больше ускоряющее действие, тем выше поднимается температура вследствие выделяющегося тепла рекомбинации. Помещенные вблизи разрядной трубки небольшие зернышки Pt, ₽d и W раскаляются сами. Это явление, а также реакция с серой (образование H2S) могут быть использованы для обнаружения атомарного водорода.
Водород 65.
Высокая теплота рекомбинации атом ар но fo водорода находит техническое при-мененпе в автогенной сварке особо тугоплавких металлов при помощи факела Ленгмюра. Последний состоит из двух, помещенных в струю водорода вольфрамовых стержней, между которыми возникает электрическая дуга. Мимо них через очень узкие дюзы продувается сильной струей водород. При прохождении через дугу водород частично диссоциирует на атомы, а последние опять, соединяются в молекулы па резко ограниченном участке поверхности металла, на которую направлена струя, В таких условиях достигается локальное повышение температуры до 4000°, Таким образом, при помощи факела Лангмюра могут быть расплавлены даже самые тугоплавкие металлы, например тантал пли вольфрам. Кроме того, атмосфера водорода, в которой происходит плавление, предохраняет металлы от окисления.
Более ранние утверждения некоторых авторов о наличии трехатомного водорода Из не были подтверждены новейшими исследованиями. Иначе обстоит дело с положительно заряженными частичками I1J, которые были обнаружены Томсоном (Thompson) в каналовых лучах. Их существование (в каналовых лучах) было подтверждено Астоном, который произвел исследование их массы.
Тяжелый водород (дейтерий). Обычный водород содержит примесь 0,02 об.% изотопа с атомным весом 2, который называется тяжелым водородом или дейтерием (символ D, подробнее см. т. II). Вследствие незначительного количества дейтерий мало влияет на свойства водорода. Однако при более прецизионных вычислениях следует учитывать, идет ли речь об обычном водороде, т. е. смеси изотопов, или о свободном от дейтерия водороде.
Тритий. Третий изотоп водорода был открыт в 1934 г, Олифантом, Хартеком и Резерфордом), Он обладает атомным весом 3 М называется тритий (символ Т). Тритий нестабилен и претерпевает радиоактивный 0-распад с периодом полураспада 12,5 года. В обычном водороде он не содержится. Его образование н свойства подробно будут описаны в т. II,. гл. 13.
Орто- п пара водород. Удельная теплоемкость газообразного водорода при низких температурах, например при точке кипения жидкого воздуха, значительно меньше, чем ото следует ожидать дия двухатомного газа на основании кинетической теории газов*. Частично это объясняется тем, что при очень низких температурах прекращается вращение молекулы водорода, что может происходить только в том случае, если он ведет себя как одноатомный газ. Одиако полностью ход кривой удельной теплоемкости водорода при очень низких температурах был объяснен только в 1927 г. Гейзенбергом (Heisenberg) и Хундом (Hund), которые теоретически доказали, что обычный водород состоит** из двух модификаций молекулы Н2, которые различаются теПлотами вращения. Их называют орто- и параводород. По химическому поведению они одинаковы, но различаются своими удельными теплоемкостями, а также некоторыми физическими свойствами, Бонхоферу и Хартеку (1929) удалось получить чистый параводород адсорбцией обычного водорода на древесном угле при 20° К. Параводород в чистом состоянии стабилен только при температурах, близких к абсолютному пулю. При соприкосновении с катализирующими веществами (ими могут оказаться даже стенки сосуда) он медленно, даже при комнатной температуре, переходит в ортоводород. Обычный водород состоит из 3 ч. ортоводорода и 1 ч. параводорода. При очень низких температурах стабильны смеси, более богатые параводородом. Все же и в этом случае обычно происходит превращение одной модификации в другую с незначительной скоростью. Эту скорость, однако, можно увеличить применением высокого давления. Таким путем, Эйкену (Eucken, 1912) удалось паблюдать почти одновременно с Бонхо-фером и Хартеком аномальную температурную зависимость удельной теплоемкости водорода при очень низких температурах.
Параводород (p-Hj) обладает несколько большим давлением пара, чем обычный водород (И2), и кипит при—252,871° (обычный водород кипит при—252,754°). Теплота превращения Н2^-р-Н2 при температуре абсолютного нуля составляет 0,337 ккал/молъ.
* В соответствии с кинетической теорией газов молярная теплоемкость одноатомного газа независимо от температуры составляет3/.,/?, т.е. приблизительно 3 кал. Молярная теплоемкость двухатомного газа (при относительно невысоких температурах) независимо от температуры имеет величину не меньше ь/2 Л, т.е. приблизительно 5 кял. Для водорода молярная теплоемкость приближается к 3 кал (при низких температурах).
** Объяснение различия между обеими модификациями Н2 на основе теории строения атома см. примечание па стр. 197.
5 г. Реми
ее
Глава 2
СОЕДИНЕНИЯ ВОДОРОДА
Бинарные соединения водорода называются гидридами. Их можно разделить на четыре класса: 1) газообразные (пли легкой етучие); 2) выси-кополимерные (твердые гидриды, которые построены не по типу солей, но и не обладают металлическим характером); 3) гелеобразные; 4) металлообразные.
Газообразные гидриды, т, е. легколетучи с, нодордд образует с бором, галлием и со всеми элементами главных подеруип IV—VIL групп периодической системы. В той мере, в какой можно ставить вопрос о заряде водорода в этих соединениях, он является <поло$кителъным. Так, галогеноводороды при внесений в воду отщепляют водород в виде положительно одновалентного иона.
Солеобразные гидриды водород образует с большинством элементов главных подгрупп 1 и 11 групп. В солеобразных гидридах воДород является олекпгроотршцателъноИ составной частью (см. гидрид лития стр. 199). При этом он ведет себя как галоген.
В настоящее время для всех элементов гласных подгрупп периодической системы, способных образовывать соединения (т. е. для элементов всех главных подгрупп, исключая инертные годы), известны водородные соединения. Большая заслуга в заполнении имеющихся еще недавно в этой области пробелов принадлежит Вибергу (Wiberg) и Шлезингеру (Schlesinger, 1'140). Приведенный в табл. 8 обзор показывает, что валентность ио отношению к водороду элементов главных подгрупп I—IV групп увеличивается от 1 до 4, а в главных подгруппах [V—VII групп уменьшается от 4 до 1.
Таблица 8
Водородные соединения элементов главных подгрупп периодической системы
Летучие водородные соединения взяты в кружки, а са лео бра ан ы е -- в квадраты >
Увеличивается солеобравпый характер
Увеличивается кислотный характер-
I П 1Ш IV V VI vn
LiH [BeHJ, (Твнл) (chJ) ^fh)
МаН [MgHJ, [AIH^ “) (&h?) (pj) (shJ) (oH^
КН СаН, (fsaHJ^ (GthJ) (AsH^) (SeH^ ^BrH^
RbH SrH, llnHj. (ChJ) CE)
CU{ ЕаНг TIH/) (phhJ) (ah?)
а В йфирпых растворах А1Нз существует в мономерной форме.
0 Известно только в форме двойного соединения, например TlHa-SGallg,
Во главных подгруппах II и III групп имеются твердые .водородные соединения,, которые занимают промежуточное положение между солеобразными гидридами илету-чими водородными соединениями. Они, однако, апачи тел ь по отличаются от солсобраз-ных гидридов и образуют особый класс юысокополимерных гидридов#. Отсутствие, лету
Водород
67
чести, а также ассоциация их простых молекул (например, ВеН2) до агрегатов неограниченной величины, по всей вероятности, основана на образовании водородных мостиков (см. стр. 325) подобно тому, как это происходит при димеризации ВН3 п GaH3 в (ВН3)2 м (GaH3)2.
Из летучих водородных соединений в табл. 8 приведены только простейшие. Наряду с ними образуются также высокомолекулярные летучие водородные соединения, которые получаются вследствие соединения болен простых радикалов. Особенно сильно это проявляется в случае углерода, который, как известно, кроме СН4, образует большое число высших углеводородов. В меиыией степени это характерно для бора и кремния. Для летучих соединений водорода с остальными элементами способность к образованию цепей, если она вообще проявляется, ограничивается объединением двух радикалов. Только для германия известно, кроме GcII4 п Ge2H0! соединение Ge3He (тригерман),,и для серы — соединения с водородом с цепями довольно большой длины (см. полисероводороды).
Некоторые элементы, способные к образованию летучих водородных соединений, могут, кроме того, давать своеобразные нелетучие водородные соединения [например^, пол и силен. (SiH2),., твердый фосфористый водород Р12Ив, твердый мышьяковистый водород (АвН)х]. Природа этих соединений еще недостаточно ясна.
В случае элементов побочных подгрупп периодической системы (если Они склоппы к образованию соединений с водородом) получаются главным образом «металлообразные гидриды». Они часто пе щгеют стехиометрического состава. Иногда водород образует с металлами лишь твердые растворы, входя и кристаллическую решетку без измепепия структуры. Поглощаемые в таких случаях количества водорода в общем пропорциональны корпю квадратному -из давления водорода (Sieverts). В некоторых случаях, прежде всего для элементов побочных подгрупп IV и V групп, происходит структурное превращение, если количества поглощенного водорода превышают определен-шлй предел. Ввиду того что последнее явление связапо со скачкообразным изменением свойств, может идти речь об образовании соединения (см. т. II, гл. 1). Подобные водородные соединения называются металлообразными гидридами. Промежуточное положение между металлообразными и солеобразпыми гидридами занимают гидриды побочной подгруппы III группы, и особенно гидрид лантана..
В результате действия атомарного водорода на элементы побочных подгрупп также образуются соединения типа солеобразпых гидридов (Pietsch, 1933). Одпако среди них не встречаются соединения со стехиометрическим составом. Как показывают исследования линейных спектров, в пламени и в дуге существуют многочисленные двухатомные гидриды. Большинство из них, например CH, NH, ОН, MgH, ZnH, АЩ, АнН, при нормальных условиях пли псуетойчнвы, или нс образуются в весомых количествах.
Важнейшим соединением водорода является вода Н2О. Наряду с ней существует еще одно кислородное соединение водорода, перекись водорода НЙО2. В дайной главе будут рассмотрены только эти соединения водорода; соединения его с остальными элементами будут описаны при обсуждении каждого элемента. Однако здесь придется изложить еще два особо важных класса водородных соединений кислоты и основания — и вместе с тем коснуться природы электролитов, к которым относятся кислоты и основания. Последним в области неорганической химии придается особо важное значение.
ВОДА
Распространение в природе и очистка. Вода—Т12О покрывает большую часть земной поверхности. Она имеет огромное значение в жизни растительного и животного мира. Так, в состав человеческого тела входит по весу больше половины воды. В атмосфере вода содержится в парообразном состоянии и при охлаждении осаж-дается в виде тумана, облакой, дождя, росы, снега и града.
Природная вода никогда не бывает химически чистой. В зависимости от происхождения она содержит различные растворенные, а иногда и суспендированные вещества. Морская вода, как известно, содержит много
5*
68 ’ Глава 2
солон. Дождевая вода часто’"содержит незначительные количества нитрата аммония, а наряду с ним часто и следы других солей. Грунтовые воды и воды источников содержат составные части пород, по которым они протекают. Практическое значение наряду с железными и марганцовыми солями (которые, будучи растворенными в воде в больших количествах, делают ее непригодной для различных целей, например стирки) имеют гидрокарбонаты и сульфаты щелочноземельных металлов ц магния. По содержанию последних определяют жесткость воды. Имеет также значение содержание в воде Тазов, особенно кислорода и двуокиси углерода, присутствий которых оказывает огромное влияние на образование ржавчины при соприкосновении железа с водой.
В зависимости от того, предназначается ли вода для питья или для технических Целей (питание котлов или прачечных), к ней предъявляют различные требования. В качестве пят-ьевой еоЭы обычно более всего пригодна ключевая во»Э«, а при недостатке последней пользуются грунтовой водой. Ее осветляют па водоочистительных станциях большей частью фильтрованием через песок и гальку; Речную воду пор едко приходится подвергать, кроме того, химической очистке. Необходимое в этих случаях обеззараживание производят обработкой хлором или озоном, а в последнее время также дейт станем ультрафиолетовых лучей, Ерли вода предназначается для технических целей, очистка ее заключается главным образом в устранении жесткости. Это осуществляется добавлением химикатов (Са(ОН)а, Na,CO3j, которые осаждают обусловливающие жесткость вещества, или обработкой пермутитом (см. стр. 558), пли же ионообменной смолой (см, стр. 81).. Первая причина непригодности жесткой воды в прачечных заключается в том, что вследствие образования кальциевого мыла (т. е. нерастворимых кальциевых солей жирных кислот, содержащихся в мыле) часть мыла осаждается, что увеличивает затрату мыла. Вторая причина в том, что налЬциевое мыло осаждается на волокнах ткани и придает ей неопрятный вид ц часто дурпой запах.
В химических лабораториях1, а также в некоторых отраслях производства пользуются дистиллированной водой. Последняя практически не содержит солей. Если же такая вода после выпаривания оставляет осадок, то ее необходимо вновь перегнать. Для некоторых промышленных целей готовят воду аналогичной степени чистоты так называемым электроосмотическим методом. В действительности здесь происходит удаление солей вследствие электродиализа, который заключается в том, что в результат те приложенного напряжения ионы постороннего вещества (соли) скапливаются в анодном и катодном пространствах, отделенных одно от другого диафрагмой, и затем удаляются оттуда. Находящиеся в растворе коллоидные вещества (например, кремневая кислота), одиако, таким образом не могут быть удалены. Вошедшие педавно в употребление искусственные ионообменные смолы (см. стр, 81) делают возможным полпое удаление солей из воды. Для этой цели комбинируют ионообменные смолы, предназначенные для катионов и для анионов.
СаЗО44-2Нсмола= H3SO4-)-CaCMOna ),.Кати
NaCl Ц Немела = HCl-f-NacMoaa }
Н2804-Г20Нсиола = 2Нгб4-801сыола) днионит НС1+ОНСмо;1а==Н2О + С1смола /
Под действием ионообменных смол вода очищается также от коллоидно растворенных веществ. Использовав смолы соответствующим образом, можно получить воду, которая по своей чистоте будет близка к дистиллированной и даже обладать особенно незначительной электропроводностью. Одиако в химических лабораториях для аналитических работ, как правило, все еще предпочитают применять дистиллированную воду.
Особенно чистая дистиллированная вода, применяемая дия измерения электропроводности, называется водой для электропроводности^ Одиако она все еще содержит йледы соединений, которые понадают из воздуха, особенно двуокись углерода. Последняя повышает удельную электропроводность от 0,04/10-в для чистой воды при 18° до 1-10"6. Значительные количества так называемой ультрачистой воды, обладающей удельной электропроводностью х=0,05—0,07,10-й, приготовили впервые Вешборн {Washburn) и Виланд (Weiland, 1918) длительным пропусканием воздуха, песо держащего СО2, с последующей дистилляцией из кварцевой колбы. С тех пор было описано много различных методов получения ультрачисгой воды; особенно простой способ принадлежит Тиссену (Thiessen, 1937),
Водород
69
Свойства. Чистая вода не имеет ни запаха, ни вкуса и бесцветна, однако в толстом слое опа имеет голубоватый цвет*. При достаточно сильном охлаждении она замерзает, превращаясь в лед. Температура, при которой лед и вода образуют при нормальном давлении (760 мм рт ст) устойчивую систему, принята за нулевую точку шкалы термометра Цельсия. Температура 100° определяется точкой кипения воды при нормальном давлении. Многие другие физические величины также отнесены к соответствующим величинам для воды. Так, за единицу массы первоначально принимали килограмм — массу одного кубического дециметра воды при 3,98°, т. е. при температуре ее наибольшей плотности. В настоящее время (как возвращение к первоначальному определению килограмма), согласно международному соглашению, в качестве единицы меры объема принят литр, т. е. объем 1 кг не содержащей воздуха воды при максимальной ее плотности и нормальном давлении (760 мм рт ст и 3,98°)**.
Количество тепла, необходимое для нагревания 1 г воды от 14,5 до 15,5°, называют малой калорией (или грамм-калорией) и оно является единицей тепла. В настоящее время единицу тепла определяют соответствующей электрической энергией, а именно в соответствии с термохимическими измерениями калория равна количеству тепла, эквивалентному 4,1840 ватт-секундам (джоуль). Таким образом, 1 кал=4,1840блс. В отличие от «термохимической калории» (кал) единицу, полученную на основе удельной теплоемкости воды при 15°, обозначают как 15°-калория (1 =4,1855 дж). Большей частью теплоты химических реакций
выражают в килограмм-калориях (ккал). 1 ккал =4000 кал.
Таблица 9
Давление пара воды р в интервале между —60 и +350°
J, °C —60 -40 —20 —15 —10 —5 -4 —3 —2 -1
Лед: р, .ч.и рпь ст 0,007 0,09 0,77 1,24 1,95 3,01 3,28 3,57 3,88 4,22
Вода: р, мм рт ст — — — 1,43 2,14 3,16 3,40 3,67 3,95 4,25
t, °с: ; 0 2 4 6 8 10 12 14 16 18 '=
л«-и ргп ст 4,58 5,29 6,10 7,01 8,04 9,21 10,52 11,99 13,63 15,48
t, °C 20 22 24 26 28 30 32 34 36 38
р, мм рт ст 17,53 19,83 22,38 25,21 28,35 31,,82 35,66 39,90 44,56 49,69
t, °C .. 40 42 44 46 48 50 60 70 80 90
р^ мм рт ст 55,32 61,50 68,26 75,65 83,71 92,51 149,38 233,7 355,1 525,8:
(, °C 100 110 120 130 140 150 200 250 300 350
р, лго рт ст 1,00 1,41 1,96 2,67 3,57 4,70 15,35 39,26 84,79 163,20
* О зависимости кривой абсорбции света от степени чистоты см. Lange R., Z. physic. Chem., (А) 159, 303, 1932; 160, 468. ’
** В настоящее время килограмм определяется как масса килограммового sma~ лона (платино-нридиевого цилиндра, хранящегося в международном бюро меры и веса), масса которого соответствует хранящемуся с 1799 г. во французском государственном архиве «первому килограмму». Последний должен был олицетворять массу 1 дм3 воды, при максимальной плотности, однако имеет несколько большую массу. В связи с этим 1 л равен не точно 1000 см3, а 1000,028 с.иэ.
70
Глава 2
Температуры кипения воды при различных давлениях - Таблица 10
/7, b-W..W рт ст 600 650 700 750 755 756 757 758 759 760
1, °C 93,48 95,66 97,71 99,63 99,81 99,85 99,89 99,93 99,96 100,00
рт. ст. 761 762 763 764 765 770 780 790 800 820
1, °C 100,04 100,07 100,11 100,15 100,18 100,39 100,73 101,09 101,45 102,15
Таблица 11
Физические константы воды
Точка замерзания 0,00° с (постоянная точка) =273,16° К Точка кипения 100,00° С (постоянная точка) Плотность льда при 0° =0,9168 Плотность воды при;
при 760 -и.ч рт ст
0° 4° 10°
15° 20°
25°
0,999 868 1,000000 0,999 727 0,999 126 0,998 230 0,997 071а
Плотность ларов воды по сравнению с воздухом (при постоянном давлений) = 1, при 0°=0,624
Вес 1 л воды, насыщенной парами, воды при 100° и 760 •«.« pm ст =0,5974 г.
Критические данные'. Крит, температура 374,1°, Крит, давление 218,5 от.и, Крит, плотность 0,324.
{льда при 0° =0,487 кал; молярная теплоемкость = 8,78 кал воды при 15° = 1,00000хал,5е= 1,00036 кал (ср. стр, 69 н <‘,Л.) паров воды при пост, давлении c{J — 0,462 кал при 100°.
Молярная теплоемкость ср = 8,32 кал', отношение удельных теплоемкостей ср/с,;—1,28.
Теплота плавления льда = 79,40 кал/г, йлн 1,430 ккал/мояъ.
Теплота испарения воды (м.ф); 539,1 при 100°; 583,0 при 25°; 597,3 при 0°.
Поверхностное натяжение (при 20° в соприкосновении с влажным воздухом) =
= 72,7 дин/см.
Вязкость; при 0° =1,789, при 20° 1,002 сантипуаз.
Молярное повышение температуры кннепия=0,513.
Диэлектрическая проницаемость при 0°=87,8, нри 18° 80,1 при 25° 78,3.
Удельная электропроводность х абсолютно чистой воды при;
0°
18°
50°
х=0,01'10-6 0,04-10-* 0,17.10-е
Молекула ГКО: дипольный момент р = 1,84-10“1В; межъядерное расстояние О'—*Н 0,958 А, II+—>Н 1,63 А. Кажущийся радиус в кристаллах 1,7 А. Энергия диссоциации Н2О II -|- ОГТ= 117,6 ккал/моли, ОН —> О-|-Н 100,6 ккал/моли. Валентно-си-
ловая константа для Н2О 7,8-Ю5 дин/см, для ОН 7,7-10^ дин/см.
а Это значение относится к воде, не содержащей воздуха. Для воды, содержащей воздух, при температуре ниже 20° оно уменьшается на несколько единиц в последнем десятичном знаке.
Водород
71
Притяжение Отталкивание
Р и/. 9. Взаимное притяжение динольпых молекул.
При комнатной температуре и даже при 0° йода обладает заметным давлением пара. Это следует учитывать при вычислении объемов газов, собранных пад водой при нормальном давлении (760 л.« рт ст).
Поэтому еслн имеют дело а газами при указанных выше условиях, то от общего давления, под которым находится газ, следует вычитать давление содержащихся в нем водяных паров. Давление пара воды при различных температурах приведено в табл. 9. Температура кипения воды в важнейших областях давлений приведены в табл. 10. В табл. 11 сопоставлены важнейшие физические констапты. Величины, приводимые в табл, 9—11, действительны для воды с нормальным содержанием D20 (о «тяжелой воде» см. т. П).
Как следует из физических исследований, в молекуле воды оба ядра Н образуют с центром атома О угол приблизительно 106°. Из-за асимметричного строения молекула воды обладает резко выращенным дипольным характером. Дипольными или полярными молекулами называют такие, у которых центры тяжести положительных и отрицательных зарядов не совпадают. В случае большого сближения такие молекулы должны испытывать взаимное притяжеиие. Последнее происходит потому, что притяжение центра тяжести положительного заряда центром тяжести отрицательного заряда превалирует над отталкивающим действием одноименных зарядов, так как расстояние между центрами тяжести разноименных зарядов значительно меньше, чем для одноименных (см. рис. 9). В неполярных молекулах под влиянием электростатических сил, исходящих от других молекул, могут возникнуть электрические заряды, в результате чего молекулы могут стать полярными. Такой процесс называется поляризацией, и в этом случае говорят об индуцированном динеле. Отсутствие ди-/ польпого характера указывает на симметричное строение молекулы.
Сильный дипольный характер молекул Н20 .является причиной особой склоппости воды (16-разо вы ватт, продукты присоединения. Они ио. лучаются главным образом в случае построенных
из ионов (солеобразпых) сосдинспйй, однако образуются и с такими соединениями, которые не являются содеобразпыми, а имеют аналогично воде дипольный характер.
Зная вес литра водяного пара при 100° (табл. 11), вычисляют молекулярный вес ГСО 18,31, который немного превосходит даваемый формулой (18,02). Таким образом, вода при обычном давлении в парообразном состоянии ассоциирована совсем а сапа читал ьп о. С увеличением давления ассоциация возрастает. При давлении 4 ат найдена плотность, отвечающая молекулярному весу 19,06. В органических растворителях иода находится полностью в виде димерных молекул (Н2О)2. В ^жидком состоянии она еще сильнее ассоциирован а. Тот факт, что вода при 4ь обладает максимальной плотностью, а при дальнейшем охлаждении опять расширяется, обусловлено увеличением объема, связанного с ассоциацией молекул воды. В связи с тем, что с понижением температуры ассоциации возрастает, вызванное этим увеличение объема превалирует уже пад сжатием, происходящим вследствие охлаждения.
Таммапу (Ташшанн, 1926) удалось впервые приблизительно вычислить степень ассоциации воды па основе изменения объема. Далее в результате сравнения инфракрасных абсорбционных сиектроя льда и воды удалось устаяоннть, что ассоциация молекул в жидкой воде, но крайней мере частично, близка структуре обычного льда. Кроме того, Редлих (Bedlich, 1929) показал, что число «ледяных молекул» с повышением температуры быстро убывает. Точно так же они начинают исчезать при увеличении давления или при добавлении электролита (повышение «внутреннего давления», Tarn-man). Однако даже при 100° жидкая вода еще в значительной степени ассоциирована. Последнее, между прочим, подтверждается сравнением водородных соединений аналогов кислорода с водой, имеющей аномально газ со кую температуру кипения (см. гл. 15). Наконец, Эйкен (Eucken, 1948—1949) показал, что ассоциация воды осуществляется в результате образования ди- и тетрамеров (очевидно, и три- и пептамерон), и, кроме того, вблизи 0° образуются агрегаты из восьми молекул. Можно указать их пр и близи-тельное распределение в зависимости от температуры, Агрегаты из восьми молекул (возможность существования которых резко уменьшается с температурой) являются как раз теми, которые обладают «льдонодобной» структурой. Они занимают наибольший объем, и в основном с их образованием связана термическая аномалия воды.
Глава 2
Рентгенометрические измерения показали', что (обычный) лед обладает структурой тридимита (см, стр, 530), т, с. в и см каждая молекула Н2О окружена тетраэдрически четырьмя другими молекулами. В процессе плавления ив кристаллической решетки удаляются в ост, мич ленные агрегаты й переходят в агрегаты такой величины, при которой теряется регулярная ориентация. Внутри отдельных агрегатов, одпако, вначале расположение молекул остается тем же, что в кристалле (см. рис. 10).
Расширение воды при замерзании связано с тем, что при нерегулярном расположении (или при регулярном только в узких областях) молекулы воды занимают меньший объем, чем при совершенно регулярной ориентации в случае образования, три лимитной,
Р и с. 10. Кристаллическая решетка льда и структура молекулы (НаО)^ («ледяной молекулы»); /г =4,5, е—7,3 А..
•Элементарная ячейка показана жирными линиями.
структуры. Вследствие расширения воды при замерзании (по принципу Ле-Шателье) с увеличением давления температура замерзания понижается. Однако если после замерзания давление превосходит определенную величину, то образуются другие модификации льда, которые плотнее обычного, даже большей частью плотнее жидкой воды. Поэтому разрывающего действия, которое оказывает вода, заключенная в желёзпые сосуды или образования трещин в камнях, при замерзании пе происходит в том случае, если вода перед замерзанием оказывается уже под очень высоким давлением/
Диаграмма состояния воды и правило фаз. На рис. 11 приведена диаграмма состояния воды. Диаграмма состояния — наглядный способ представления областей, существования различных фаз в зависимости от внешних условий (например,давления , И температуры). Согласно правилу фаз Гиббса, для любой системы, находящейся в рав? ’ новесии, имеет силу
I- р = в ;• 2, (Ю)
где Р — число фаз', F — число степеней свободы", В — число компонентов системы, Ввиду того что в данном случае рассматривается система, которая имеет только один компонент — НЕО, то наибольшее число фаз, которые могут одновременно находиться в равновесии, в данном случае равно 3, Число степеней свободы равно 6, т. е. нельзя менять ни давление, ни температуру, чтобы не исчезла пи одна из фаз. Например, три модификации льда 7, II и III существуют вместе (точка G — диаграммы) только при 1=—34,7° и />=2170 кг/с.и2; модификации льда I и III вместе с жидкой водой существуют только при —22,0° и р=2115 «г/с.н2 (точка 7?).( Лед I (обычный лед), жидкая вода и водяной пар могут существовать в равновесии одновременно только при одной температуре п при одном давлении, а именно при р—4,58 hcw рт ст и 1—0,0075° *. Область,
* 0° является температурой, при которой вода в условиях атмосферного давления находится в равновесии со льдом. Повышение давления на 760 мм рт ст снижает температуру плавления Льда па 0,00753°. Понижение давления на (760—4,6) повышает температуру цчавленпя па 0,00753 (760—4,6)/760=0,00748°.
Водород
73
расположенная ниже этой точки, показана па pile. И справа внизу в большом масштабе. Точки сосуществования трех фаз (см. па рис. 11 точки Л; В; С; D; В; В я G) называются тройными точками. Пересекающиеся в тройных точках кривые показывают взаимную зависимость давления и температуры, при которых сосуществуют две фазы. Например, совместное присутствие льда I и льда П или льда I и жидкой воды, а также жидкой воды и водяного пара. В соответствии с уравнением (10) двухфазные системы в даппом случае обладают одной степенью свободы. Это значит, что одно из внешних услов!1Й может изменяться произвольно, второе же изменяется в зависимости От первого. Например, жидкая вода и водяной пар могут находиться в равновесии при^дг.гич-ных температура х (кривая Л//), однако каждойиз этих температур соответствует то ль-ко одно совершенно, определенно с давление пара. Если же при фиксированной температуре изменить давление, то появляется область, где вода стабильна только в жидком
Ловлеиие, ЮНО круг
Рис. 11. Диаграмма состояния воды
состоянии; если менять давление дальше, то уже появится область существования пара. Внутри однофазной, области температура м давление, напротив, могут произвольно и независимо изменяться, так как, согласно уравнению (10), в таком случае система обладает двумя степенями свободы. Затем снова посредством температуры и давления определяют концентрацию. В рассматриваемом случае это значит, что объемопредс; ленного количества взятой воды в однофазной области задается температурой и давлением. ' '
Если кривую давления пара жидкой воды (т. е. кривую НА}, которая разграничивает области существования жидкой и'газообразной фаз, продолжить за точку пересечения се с кривой давления пара льда (кривая 1А\, то будет Достигнута область, в которой устойчив только лед. Это продолжение кривой давления пара жидкой воды показано в пижей правой части рис. 11 пунктирной линией. Несмотря на то что жидкая вода в этой области нестабильна, однако она может временно находиться в этом состоянии вследствие переохлаждения (Мееру (Meyer) удалось наблюдать переохлаждение воды до — 33°J. Измерения давления пара подтвердили, что переохлажденная вода обладает более высоким давлением пара, чем лед при той же температуре (см. табл. 9). Термодинамически можно доказать, что стабильной формой вещества является та, для которой в указанных условиях характерно более низкое давление пара. Нестабильность переохлажденной воды отчетливо проявляется в том, что, как только прекращается задержка кристаллообразования (например, вследствие встряхивания или внесения кристалла льда), вода тотчас же переходит в лед с выделением тепла.
Рассмотренные на примере воды закономерности имеют силу для. диаграммы состояния любой системы с одним компонентом (относительно системы со многими Компонентами см. стр, 9G).
74
Глава 2
Для химика вода имеет особеппо большое значение как растворитель. Кроме того, вода вступает со многими веществами в обменное разложение и является промежуточным соединением во многих реакциях. Это подтверждается тем, что при полном отсутствии воды многие реакции совсем не идут или протекают чрезвычайно медленно.
Электролитическая диссоциация воды. Описанная ныше собственная электропроводность совсем чистой воды возникает потому, что вода способна, хотя и в очень незначительной степени, диссоциировать* с образованием иона гидроксония [НзОГ, и иона гидроксила ОН'
2Н2О7*[ТГ3О]Ч-ОН'. (11)
Константы диссоциации воды для различных температур указаны в табл. 12. При 22° в 10 млн. л воды в диссоциированном состоянии находится 1 моль; при 18° 0,86 моля.
'Габлаца 12
Константы диссоциации воды
Aw=[HsO-]-[OH’]
- Д6,° 17° 18° 19° 20° 21° 22° 23° 24° 25°
0,63 0,68 0,74 0,79 0,86 0,93 1,01 1,10 1,19 l,27x
—log *w= XlO"1*
14,200 14,165 14,130 14,100 14,065 14,030 13 ,.995 13,960 13,925 13,895
30° 35° 40° 50° 60° 70° 80° 90° 99°
i,-89 2,71 3,80 5,95 12,6 21,2 35 53 72 x
x 10*«
В соответствии с законом действия масс для определенной температуры
Уравнение наказывает, что при увеличении конц<:нтрации ионов И?,О' (например, в результата добавления кислоты) концентрация ионов ОН' должна соответственно уменьшаться, и наоборот. Количество педиесоциировапной воды при этом практически будет оставаться без изменения. Действительно, практически совершенно неощутимо, если к 560 000 000 молей воды (1 Л содержит 56 молей Но О) добавить еще 2 моля. Поэтому можно включить концентрацию недиссоциированной воды в значение константы
[I-UOJ х [ОН'] = LHBO]3 X const = (12)
Эту копстапту определяемую уравнением (12), обычно называют константоИ дне-социации воды**. 1
Для чистой воды [Н3О ] = ]^A\V = 0,86-10“т при 18°, так как при диссоциации чистой воды образуются одинаковые количества ионов Н3О" н ОП'. Для [UaO’j = 1 при 18° значение [01Г]=0,7410-11; наоборот, для [ОН'] — 1 значение [ТТ3О]-= — 0,74-IO'14. Ввиду того что практически удобнее производить вычисления с отрицательными логарифмами Л,,-, в табл, 12 приведены эти значения для наиболее употребительного интервала температур. ;
* В предыдущих главах для гидратированного иона водорода (иона гидроксо-ний) применяли укрощенный знак 1Г. Знак [НЯО]’ будет использован только в особых случаях, а имшпто когда нужно показать, что при рассматриваемых процессах идут обмен но-протонные реакции (подробнее см. т- II, гл. 18). Электролитическая диссоциация воды [как видно из уравнения (11)] заключается в протонном обмене между двумя молекулами ILO. Диалогичная ж'е реакция протонногб обмена происходит и при диссоциации кислот (см. стр, 80). То, что в водных растворах, по-видимому, вокруг протона группируется нс одна, а много молекул Н2О (см. стр. 100), не имеет существенного значения для представленнй о протонно-обменных процессах и не должно приниматься во внимание в связи с вышеупомянутым вопросом.
** В советской паучной литературе эту константу называют ионным проиаве-i. _ воды.— Прим. ред.
Водород
75
Термическая диссоциация воды. Ввиду того что реакция, протекающая по уравнению (9) (см. стр. 61), экзотермична. в соответствии с принципом Л е-Шателье при высокой температуре она должна протекать и в обратном направлении. Действительно, как было сказано выше, вода лод действием значительного количества тепла разлагается на О2 и 2На, которые можно получить в весомых количествах.
Степень термическойдиссоциации а, согласно Яернсту и Вартепбергу, для водяного пара при суммарном давлении 1 ат равна:
для Т, °К = 1000= 1200° 1400" 1600=
а, % = 0,00003 0,00081 0,00861 0,051
Хс = 5,49-10’2* 2,73-10’1» 2,78-10-1® 4,96-10"™
Кр = 4,50-10-2г 2,69-10-ja 3,19-10-13 6,51-10-и
для Т, °К = 1800° 2000° 2200° 2400°
а, % = 0,199 0,588 1,42 2,92
Кс = 2,66-10-ч 6,21 -10-‘° 8,ЮТО*9 6,62-10’»
А'р = 3,93-10-» 1,02- Ю-7 1,46-10-* 1,30.10-»
В последних двух строках для различных температур приведены значения кон-•стант равновесия Кс и Кр, получаемые в результате применения закона действия масс к случаю равновесия между 02, Н3 и Н2О, .
Для вычисления константы равновесия Кс при данной температуре из числового -значения установленной экспериментально степени диссоциации а в случае равновесной системы гремучий газ — вода пользуются следующим уравнением:
К Р___________0:3 (J3)
ВТ GH-aHl-a)3 ’ 1 ’
где р — давление {атм)‘, а — степень диссоциации (доли от единицы).
Это уравнение получают из уравнений (2) и (7) следующим образом. Предположим, что при расчетах исходят из чистой воды, пусть ее количество до диссоциации будет 2а моля. После нагревания до той температуры, при которой измеряется диссоциация, количество недиссоциировапной воды, если подверглось диссоциации 2а-а молей, будет, очевидно, 2с(1—а). Так как из каждых двух разложенных термически молекул Н2О получаются 2 молекулы Н2 и 1 молекула О3, то количество присутствующего в равновесном состоянии водорода будет 2аа, а количество кислорода — а-а. Таким образом, общее количество молекул или молей будет теперь
п — 2а(1— а) 4- 2аа аа — а(2 -|- а).
Молярная концентрация каждого рода молекул получится при делепии их количества на объем о, занимаемый всем газом. Таким образом,
1П2О1 = ^-Ц^; |Н2)=-^; [0.]=-^.
Если найденные значения концентрации подставить в уравнение (7), то получим
Па основании этого уравнения можно вычислить Л’с, если, кроме а, известны величины а и ₽.. Однако для ото го удобнее пользоваться уравнением (13), в котором вместо объема подставлена величина р — давление всего газа. Эта величина получается из уравнения (14), если принята, во внимание, что в соответствии с уравнением газового состояния
__ o(2+ct)J?T
”” Р
Так как в данном случае п^а (2 4- a), Kv получается по уравнению 0) умножением Кс на 0,0821 Т.
Перекись водорода IJ2O2 образуется всегда, когда на растворенный в воде молекулярный кислород действует активный водород
76
Глава 2
(например, растворенный в палладии или водород в момент выделения, а также получающийся электролитически). Далее она образуется также-при медленном оКислепии различных органических и неорганических веществ кислородом воздуха, однако в этом случае ЩОа получается в очень незначительных концентрациях.
Техническое получение перекиси водорода или ее водных растворов-н настоящее время ведут преимущественно разложением полученной электролитическим путем надсерной кислоты или ее солей. Наиболее целесообразно сначала получать персульфат аммония, который образуется -с большим выходом, чем надсерная кислота. Из подкисленного-серной кислотой раствора этой соли при соответствующем методе работы при нагревании под уменьшенным давлением можно отогнать Н2О2 (получающуюся в результате гидролиза) точно так же, как это происходит в случае раствора ITaSaO8.
1 Анод Катод
2S0;—2^5,0" | 2Н3О'-|-2е—2Н3ОД-Н2
S-,0^ 4- 4На0=2SO;-J-.2HjO.’ + Н20а
Ввиду того что в результате гидролиза опять получаются ЗО<1"-ионыг в этом процессе^ кроме На, не образуются побочные продукты или продукты разложения.
Следующий технически важный способ получении НаОа основан на образовании ее при окислении (ср. стр.- 822) известных органических соединений, например 2-этилгвдроанТрахинона. Последний реагирует с кислородом воздуха с образованием НаОа и 2-этилантрахинона, из которого каталитическим гидрированием получают вновь исходный продукт.
В незначительной степени применяют еще старый способ, по которому перекись водорода получают, внося влажную перекись иария (см. гл. 8) в приблизительно 20%-пую серную кислоту или в концентрированную фосфорную
BaO2+H2SO4=BaSO4+H3O3; BaO2_pH3pd4=BaHPO1-pH2O2.
Фракционированной дистилляцией под уменьшенным давлением легко получаются сравнительно концентрированные растворы перекиси водорода. В продажу перекись водорода обычно поступает в виде водных растворов с содержанием 3—30 вес. % Н2Оа, причем 30%-ный раствор ее-носит название «пергидролы.
Для промышленных целей И «О а перевозят исключительно в виде-растворов высокой концентрации (30—60%). Такие растворы более устойчивы, чем разбавленные, а кроме того, они не замерзают (30%-ная. перекись водорода замерзает при —30, а 60%-ная при —53°).
Чистая, безводная перекись водорода интересна только для научных целей. Это-сиропообразная, обычно бесцветная, по в очень толстых слоях голубая жидкость. Удельный вес dgft=l,448. Температура плавления —0,46°. Показатель преломления Лр = =1,4067; поверхностное натяжение 75,94 дин! см. Вязкость при 18°=1,307 сантипуаз. Жидкая перекись водорода очень склонна к переохлаждению. Особенно сильно понижает температуру замерз алия вода, с которой Н3О3 образует гидрат, плавящийся нри> —51°, Под уменьшенным давлением перекись водорода перегоняется без разложения, (температура кипения ее при 28 леи ;>т ст равна 69,7°).
Перекись водорода следует рассматривать как чрезвычайно слабую, по все же- -кислоту. В безводном состоянии опа но вызывает покраснения, лакмусовой бумаги, однако ее водные растворы имеют кислую реакцию. Константа диссоциации перекиси
Водород Т1
.водорода — константа равновесия К
112О^И202:
* c<”lst=**
[И-СЬ]
В соответствии сданными Каргина (1929), константа диссоциации цри 20° равна 1,5-10*12. Эта величина приблизительно соответствует константе диссоциации ортофос-форной кислоты в третьей ступени (см, стр, 678), но опа очень низка, например, по сравнению с константой диссоциации угольной кислоты на первой ступени. Тем не менее концентрация ионов Й3О' в1 М водном растворе Н2О2 значительно выше, чем в совершенно чистой воде.
При повышении концентрации ОИ' в растворе по закону действия масс повышается диссоциация Н2О2. При соотношении NaOH/HaO>2 она, согласно данным Кредита (Bredig, 1934), практически проходит полностью.
В качестве натриевой соли перекиси водорода рассмотрим перекись натрия NaOOH, Которую можно выделить на неводных растворов в виде белого кристаллического порошка. Из водных растворов ос также! можно выделить в форме соединения с Н2О и Н2О2.
Перекись водорода обладает столь же высокой диэлектрической постоянной, как и вода. Опа составляет 84,9 при 0tt и повышается в результате добавления воды; максимальное значение 121 получается при ~35% Н2О3.
Об изомере НаО2, существующем только при низких температурах п получающемся действием атомарного Н на О2, см. стр. 63. Эта модификация, по-видимому/обладает таким типом связи, что оба атома водорода связаны с одним и тем же атомом кислорода. .Между тем молекула обычной перекиси водорода, по данным Сезерленда (Sulhorland, 1934) и Симона (Simon, 1935) построена так, что две ОН-грунцы связаны череа кислородные атомы и образуют с осью О—О угол приблизительно 110°, одпако повернуты друг относительно друга на 90°. Расстояние О О=1,47 А, О+- ->11=0,97 Л (Pauling, Giguere, Schomaker, 1943). Аналогично воде перекись водорода в жидком состоянии сильно ассоциирована, в парообразном состоянии, напротив,, мопомолскулярна. Твердая Н202 имеет тетрагональную структуру (а=4,02, с—8,02 А.).
Перекись водорода можно извлечь из ее концентрированного водного раствора встряхиванием с эфиром. С водой она смешивается в любых отношениях. Перекись водорода присоединяется 'к очень мдогим веществам, преимущественно к органическим, а также к некоторым солям, образуя с ними непрочные продукты присоединения. Эту непрочно связанную НаОа называют кристаллизационной перекисью водорода.
Перекись водорода является сильно эндотермическим соединением. ’Несмотря на эго, она практически устойчива как в чистом состоянии, так и в водных растворах. Последнее обстоятельство связано с тем, что скорость ее разложения при обычной температуре практически равна нулю. Однако в присутствии некоторых веществ, особенно платины, серебра или двуокиси марганца, и даже просто при соприкосновении перекиси водорода с шероховатой поверхностью, а также при действии щелочей эта реакция сильно ускоряется, разложение происходит по уравнению
Н4О2— HaO-J-l/aOa-p23,47 ккал*
, Следы тонкоизмеиьченной платины, внесенной в водные растворы перекиси водорода, вызывают бурное выделение кислорода. Очень концентрированные растворы перекиси водорода иногда сильно взрывают при попадании в них пылинок. Медленное разложение перекиси водорода происходит уже под влиянием следов щелочи, которые попадают в рас-твор^вследствие растворения стекла. Поэтому совершенно чистые (без всяких добавок) растворы перекиси водорода сохраняют в парафинированных сосудах. Также и некоторые органические вещества вызывают
* По поводу введения члена. [Н2О]. в константу диссоциации см, стр. 74,
78
Глава 2
разложение перекиси водорода. 'Получающиеся в результате жизнедеятельности организмов вещества, обладающие специфическим свойством разлагать перекись водорода, называются каталазами. Такие каталазы находятся, например, н крови и в слюне.
Каталазы относятся к классу так называемых <{>ермвцтоб. Ови встречаются только в минимальных концентрациях, и неустойчивы. Ферменты играют в жизни организмов такую же роль, как катализаторы в обычной химии. Раньте считали характерной особенностью ферментов их специфичность действия, т..с. действие только на вполне определенные вещества и только в определенном направлении. Однако этот взгляд мощно признать справедливым только с очень большими ограничениями.
Кроме каталазы, существует и другой фермент, оказывающий специфическое действие на перекись водорода, это пероксидаза. Последняя отличается способностью активировать НчО2 таким образом, что опа в присутствии окнсляюпщхся веществ отдает им одни атом кислорода. В отсутствие же окисляющихся веществ пероксидаза не разлагает перекиси водорода.
Кроме того, существуют вещества, которые задерживают каталитическое и ферментативное разложение перекиси водорода, т. е. временна прекращают действие катализаторов или ферментов. К их числу относятся: фосфорная кислота (последняя главным образом применяется для предотвращения разлагающего действия солей марганца и железа) и другие кислоты — преимущественно барбитуровая кислота и мочевая кислота. Чтобы сделать устойчивыми 30 л крепкой перекиси водорода, достаточно 1 а, мочевой кислоты. Таки» небольшие добавки лишь незначительно загрязняют НзОз и в подавляющем большинстве случаев не имеют существенного значения. Поэтому в последнее время их Широко применяют даже* для сохранения высокопроцентных растворов -Н2О2, которые после этого и поступают в продажу в обычных стеклянных бутылях.
Перекись водорода может действовать как окислитель и как восстановитель. Опа окисляет сульфат двухвалентного железа др трсхвалент-ного, сернистую кислоту — до серной, азотистую кислоту — до азотной, мышьяковистую кислоту — до мышьяковой и сернистый свинец — до сернокислого свинца. Из иодистоводородпой .кислоты она выделяет свободный иод и обесцвечивает раствор индиго. Восстанавливающим образом НгОа действует на такие вещества, которые легко отдают свой кислород, например на перманганат калия или на хлорную известь. НзОг восстанавливает также и соединения благородных металлов. Так, при ее действии из растворов солей золота выделяется металлическое золото, окись серебра ею восстанавливается до металлического серебра, окись ртути — до металлической ртути. 1
На способности НаОа легко отдавать активный кислород основано ее прекрасное дезинфицирующее действие, благодаря чему она имеет широкое применение в медицине. Для этих же целей ее выпускают в продажу в виде твердого непрочного соединения с мочевиной под названием «орт и зон». Кроме того, она служит еще в качестве отбеливающего средства, например в косметике (для обесцвечивания волос), однако гораздо в большей степени как отбеливающее средство для технических целей.
г, е V .
Вначале нсрекисг, водорода применяли главным образоьКдля отоелки соломы, перьев, слоновой кости, крахмала, клея, кожи и меховых изделий. Затем ее начали вртт мспять во все увеличивающихся масштабах —• для отбелки волокнистых продуктов (вата, лигнин, шелк, искусственное волокно), и постепенно она все Со ныл е в этом области вытесняет хлорную известь, Н3О2 действует быстрее и меньше разъедает волокно. Большие количества перекиси водорода применяют для отбеливания жиров и масел. В этих случаях применяют по возможности высокопроцентные растворы (др 60%-вых), чтобы как можно меньше вносить воды в обрабатываемое вещество. ’
Водород
79
Значительные количества перекиси’'водорода применяют также для приготовления других отбеливающих веществ, например пероксигидрата тетрабората натрия (см. стр. 379), и для получения органических перекисных соединений. В смеси с. гидразингидратом, метанолом или углевсь дородами она применяется в качестве реактивного горючего и для других подобных целей. Уже перед последней мировой войной ежегодная мировая продукция составляла около 30000 т 30%-ной перекиси водорода.
Аналитически перекись водорода легко обнаруживается по интенсивно желтой окраске, которую она сообщает раствору сульфата титана. Количественное ее определение производят на основании ее реакции с перманганатом калия по уравнению
5H2Os-|-2KMnO1+3HiSO4 = 2MnSOt-(-K3SO4+8H2O Д-5О2. '•
По мере повышения концентрации Н20а скорость этой реакции сначала возрастает, а затем сильно снижается (Riesenfeld, 1934). При исследовании растворов перекиси водорода на присутствие Н3РО/, следует учитывать, что П3О2 окисляет бесцветный иоы MoOJ в интенсивно оранжевый ион MoOJ. Перед испытанием раствора на присутствие Н3РО4 необходимо удалить Н2О2 выпариванием.
Производимо перекиси водорода. В результате замещения в перекиси водорода атомов водорода на другие атомы или радикалы, получаются перекиси, а в результате замещения одного или двух атомов Н на кислотные остатки — надкислоты или перо-ксосали (см., например, стр. 682). Кроме того, перекись водорода образует, как уже указывалось, многочисленные продукты присоединения (пероксогидраты). Относительно различия пероксо гидрате в и пероксосолей (см. стр. 377).
КИСЛОТЫ, ОСНОВАНИЯ и соли
Кислоты, Кислотами называются соединения, которые в результате протонного обмена с водой могут образовать ион гидроксония.
Вещества, которые способны отщеплять протон, называются дис-протидами, те же, которые способны присоединять,— эмпротидами. Вещества, для которых применяются эти обозначения, могут быть как химическими соединениями н обычном смысле, так и. ионами (например, NH‘). Некоторые вещества могут функц иоппрова ть как диспротцды и как эмпротиды смотря по тому, с каким веществом они реагируют*.
Кислоты — это соединения, которые ведут себя по отношению к воде как диспротиды**.
Протонный обмен кислоты с водой заключается в том, что кислота ..способна отщеплять протоны (т.с. электроположительно заряженные атомы И) ц в том, что вода соединяется с ними, образуя [НзО)*-иопы.
TIH-|-H2O R'-|-[H3OJ* (В—кислотный остаток) (15)
Этот процесс называется электролитической диссоциацией кислот. Реакция приводит к равновесию,, которое сдвигается вправо при разбавлении {равновесие диссоциации). В зависимости от степени, с которой различные кислоты диссоциируют в растворах одинаковой эквивалентности, судят о силе кислот (см. стр. 93). Существуют кислоты, которые диссоциируют еще меньше, чем чистая вода. Водные растворы тех кислот, которые диссоциируют сильнее, чем вода, окрашивают лакмус в красный цвет.
* Например, вода ведет себя но отношению к Н.С1 как ампротид (НгО •{- НС1 — = [Н3ОГ -f- СГ, а по отношению и NIT3 — лак диенротид (Н2О -|- NH, = ОН' + +[NH4]‘).
” Не рекомендуется употреблять название «кислота» для всех дпепротидов, так как смысл слова «кислота», которое необходимо при описании химических соеди-пепий, будет тогда полностью изменен. Подробнее см. т, II, гл. 18.
80
Глава 2
13 случае применения закона действия масс к процессу электролитической диссоциации для активностей веществ, находящихся в растворе (ионов и цедиссоциированных молекул), вводят то значение, которое получается, если независимо от концентрации раствора взять активность растворителя, равную 1. Для равновесия диссоциации кислот получается
[R'] х [H3OJ
[HR)x [H3OJ
.=const или
(W)
[R'Jx[II3O ] _
^ThrT^~ :
где X3 — константа диссоциации, кислоты.
Если вместо H3Q’ применяют символ Н‘, то уравнения (15) к (16) получают следующий вид;
HR q± R'-1-н- я
На числовое значение различный способ написания нс оказывает влияния.
Лтом, который, подобно атому Н, в кислоте при соответствующих условиях в растворе может отщепляться с образованием иона, называют ионвгетюсвяганным. Понятие иопогенной связи ничего не говорит о том, был ли полученный ири электролитической диссоциации ион уже заряженным в недиссоции ров айном соединении или он был электрически нейтральным и зарядился только в процессе диссоциации. Особенно сильной способностью вызывать диссоциацию обладает вода. Это связано с ее высокой диэлектрической проницаемостью.
Те кислоты, которые, кроме водорода, содержат только один элемент (например, НС1), называют водородными кислотами, а те, которые содержат также кислород, называют кислородными (цапример, НС1О4, U2SO4); тиокислоты (см, стр. 791); галогенокислоты (см. стр. 852).
Общие способы получения кислот. Кислородные кислоты можно получить;
1. Действием воды на окислы
П р и м е р: серный ангидрид соединяется с водой с образованием серной кислоты. Фосфорный ангидрид при столпим па влажном воздухе образует метафо сфориую кислоту.
- SOs-J-HjO—HzSO^ Р!05-рН8О=2НРО,.
См. также «Получение азотной кислоты из воздуха» (стр. 635, 6И и 646).
Такие окислы, которые, соединяясь с водой, образуют кислоты, называются ангидридами кислот.
2. Гидролитическим разложением таких соединений, обе составные части которых являются кислотообразующими.
При этом способе всегда получаются две кислоты. Поэтому данный метод практически применим лишь в том случае, если есть возможность без особою труда разделить эти кислоты. Часто пользуются гидролитическим разложением хлоридов для получения соответствующих кислот.
Пример:
PCl6-p4H2O=H3BOi+5HCI; PC13-|-3H2O=H3PO3-J-3HCI.
Хлористоводородную кислоту вследствие большой летучести можно, легко удалить выпариванием. В органической химии чаще, чем в неорганической, для приготовления кислот применяют разложение соответствующих хлоридов.
3. Кислородные кислоты получают окислением водных растворов кислотообразующих веществ в отсутствие оснований.
Водород
81
Пример; получение ортофосфорной кислоты окислением фосфора азотной кислотой
3P+5HNO3+2H2O=3H3PO4+5NO.
4. Электролизом водных растворов солей кислородных кислот' на аноде при этом всегда образуется свободная кислородная кислота. Для получения водородных и кислородных кислот применимы следующие методы.
5. Получение кислоты из ее солей в результате действия на них другой кислоты или воды {двойной обмен):
МЛ-(-НВ —> НА -р МВ или МА НОН *—> НА + МОН, Соль Кислота Кислота Соль Соль Вода Кислота Гидроокись
Этот процесс обычно в достаточной степени обратим1 и протекает не до конца. Чтобы он прошел до конца, в данном случае необходимо вывести из реакционной смеси по крайней мере одно из веществ. Это можно произвести следующим образом:
а) образующаяся кислота летуча (нередко только при высоких температурах) или она разлагается на летучий ангидрид и воду.
К этому случаю относится получение наиболее известных кислот, например получение HCI из NaCl и H2SO4 (ср. стр. 844), далее HNO3 из KNO3 или NaNO3 и HaSO4 (см. стр. till); НС1О4 из КСЮ4 и H2SO4 (см. стр. 862); H2S из FeS и HCI (см. стр. 784), а также получение СО2 из СаСО3 и HCI.
б) образующаяся кислота труднорастворима.
Так, кремневая кислота (или гель ее апгидрида) осаждается из подкисленных растворов силикатов палочных металлов; при кипячении растворов титанатов выпадает меГатитановая’кислота. z
в) образующаяся соль труднорастворима.
Этот метод также часто используется. Прежде всего он служит для получения кислот, которые не выдерживают сильного нагревания.
Примеры:
Н2О2 из ВаОз и H2SO4 (стр. 76) ИС1О4 из КС1О4 и H2[SiFeJ. ШО3 из Ва(1О3)2 и H2SO4
г) с другим растворителем.
кислоту или получающуюся при
Н5Ю6 из Ag2H3lO6 и HCI H2SeO4 из BaSeO4 и II2SO4 Н3РО2 из Ва(Н2Р02>2 и H2SO4
этом соль молено извлечь встряхиванием
Приме р—-феррогексациановая кислота, см. т. II.
6. Получение кислот из их солей с применением катионного обмена.
Ионообменные смолы (иониты) — твердые соединения, которые обладают свойством извлекать из растворов определенные вещества, посылая взамен в раствор другие. Это свойство наблюдали первоначально на цеолитах и пермутитах (см. стр. 559). Более или менее ярко выражено это свойство и у некоторых адсорбентов, например у поверхностно активной окиси алюминия (см. стр. 393). Особенно большое значение в качестве ионообменных веществ приобрели в последнее время некоторые искусственные смолы.
Искусственные ионообменные смолы впервые были приготовлены в 1935 г. английскими исследователями Адамсом (Adams) и Холмсом (Holmes). Эти смолы, между прочим, имеют то преимущество, что изменением состава их ионообменную способность можно приспособить для самых различных целей. Например, существуют искусственные смолы, которые обменивают исключительно анионы, и такие смолы, которые обменивают исключительно катионы. У таких искусственных смол: иаипый обмен протекает в-эквивалентные: количествах, чего при ионном обмене с а дсорбентами обычно,нс бывает. В состав искусственных смол, применяемых в качестве ионообменных, входят химические соединения, склонные к солеобразованню, например органические сульфоновые кислоты, которые обладают не адсорбционными свойствами, а вступают в реакции, имеющие характер двойного обмена.
6 Г, Реми
82
Глава 2
В 1949 г. Клемент (Element) показал, что катионеобменную сносе ность таких искусственных смол можно использовать для приготовлена кислот из их солей щелочных металлов. Для этой цели в вертикалы поставленную оттянутую снизу и снабженную краном трубку вводят си циальную смолу, например вофатйт KS*. Промываемый дистиллирование водой слой смолы (нанесенной на фарфоровые пористые пластинки ил кусочки стекла) сначала обрабатывают 5 и. соляной кислотой для нас г щения ионами Н*. Затем промывают дистиллированной водой и надо; няют раствором соли щелочного металла. В результате ионного обмен происходит 'образование кислоты из соли щелочного металла по реакци
NaHsPOa+Н •—ЩРО.,4- Na -...
Поело завершения обмена, который продолжается около 15 мин, раство кислоты спускают через кран. Слой смолы регенерируют, т. е. из пег опять удаляют поглощенные ионы Na*. Для этого сверху вливают 5 т соляную кислоту, давая стекать ей по каплям, и, наконец, промываю смолу дистиллированной водой.
Получение кислоты с применением ионного обмена рекомендуете прежде всего в тех случаях, когда ее получение затруднительно и при ходится применять труднораствбриМые соли тяжелых металлов, которы предварительно должны быть получены из солей щелочных металлов
Описанные реакции обмена могут также иметь аналитическое Значение, например для определения солей ацидиметрическим титрованием (Samuelson О., 1939; Wiesen burger Е., 1942; ср. Klement, Z. analyt. Chcm., 127, 2, 1944; 128, 106, 1948),
Техническое применение ионообменные смолы находят прежде всего при устрапс дни жесткости воды и для аналогичных целей (см. стр. 68). Кроме того, мх применяю и в препаративной имап, папример, для получения нитрата натрия из калийной сели тры и конверсии других солей, дан регенерирования исполЬ за ванных кислот ц основа ний, для удаления электролитов на коллоидных растворов и для удаления следов тяже лых металлов из органических веществ. Например, оказывается, что таким образов можно освободить вино и другие папиткп от следов меди, свинца и мышьяка.
Основания. Основаниями называются такие вещества, которые в водном растворе отщепляют гидроксил-ионы {ОН'}.
Красная лакмусовая бумага под действием оснований, которые диссоциируют сильнее воды, становится синей.
Сильными основаниями прежде всего являются гидроокиси щелочныа и щелочноземельных металлов. Одним из важнейших слабых оснований является гидрат аммиака NHs-HaO (см. также стр. G58), который в водном растворе диссоциирует в незначительной степени по уравнению
NHe- На0 = [NIT*]’ 4- ОН'.
По своему составу основания и кислородные .кислоты значительно не отличаются. Существует много гидроокисей, которые могут функционировать как кислоты и как основания. Их называют амфотерными.
Амфотерность гидроокиси ROH может быть обусловлена двумя различными причинами. Первая заключается в том, что гидроокись в заметпой степени способна как Отдавать протоны молекуле воды, так и отщеплять ионы гидроксила. Вторая — в том, что они могут не только отщеплять ионы ОН', но и присоединять их. Смотря ио тому, какой из этих случаев имеет место в растворах амфотерных гидроокисей, осуществяя-
* Искусственные ионообменные смолы имеют иаавания (с которыми они поступают в продажу) еофатшп, леватит, амберлит, дауэкв с добавками, которые предназначаются для специальных целей.
Водород
83
ются следующие равновесия:
ROH+ НгО qt RO'4-H3O- и ROH qt R‘ + OH'
или
ROH + OH' qt R(OH)J q±: R--t-2OH’.
Применяя закон действия масс, получаем
[Н0']х[И30‘] , [Н(ОН)Й
[R-]XtOH'] =C0PSt’ COOTBel™HO [Н-]х[ОНТ =C0DSt-
Так как в соответствии с уравнением (12) [Н3О"1
iOH'J
то равновесие в обоих
влучаях зависит в равной мере как от концентрации ионов Н30", так и От концентрации ионов ОИ'. Если к раствору добавляют сильную кислоту, т. е. повышают концентрацию ионов Г13О’ (соответственно понижают концентрацию ионов ОН'), то отпоше-, пия [RO']/[R'l и соответственно [R(OII)3]/[R’] должны уменьшиться, т. е. должен про-
текать слева направо процесс
RO'4-ПзО’ qt R’ + OH’-1-HaO, и соответственно R(OH)J q± Й’-|-2О1Г.
Обратное явление произойдет при увеличении в растворе концентрации иопов ОН'. Отсюда следует, что амфотерные вещества должны вести себя по отношению к сильным кислотам как основания, а по отношению к сильным основаниям как кислоты, что "и происходит в действительности.
Общие способы получения оснований. Формально говоря, получение оснований базируется на тех же методах, что и получение кислородных кислот. Одпако в практическом осуществлении и относительном значении отдельных методов существуют характерные особенности. Поэтому целесообразно рассмотреть эти методы и их применение для получения различных оснований.
Метод 1. Получение оснований из окислов, в данном случае, следовательно, из ангидридов основного характера и воды.
1 Несмотря па то что большинство окислов металлов, особенно низших степеней окисления, является основными ангидридами, всо же данный метод получения оснований далеко не имеет того значения, которое он имеет для получения кислот, вследствие того что большинство окислов металлов соединяются с водой крайне медленно.
Большое значение имеет получение этим методом Са(ОН)2 (см. «Гашение извести», стр. 294). Существование равновесия
М20 -|- H2Oqt 2MOHq^2M‘ -р 2ОН'
позволяет применять для тех веществ, гидроокиси которых сами по себе неустойчивы, вместо гидроокисей суспендированные в воде окислы соответствующих металлов; так, вместо гидроокиси серебра, которую как таковую приготовить не удается, пользуются суспендированной в воде окисью, серебра.
Кроме основных ангидридов, функционирующих в качестве оснований, существуют также и другие вещества, которые, присоединяя воду, образуют основания,— например аммиак, который образует с водой гидрат аммиака НИ3-Н2О (См. выше).
По предложению Вернера (Werner А.) вещества такого типа называют анеидро-основанилми (подробнее см. стр. 85).
Метод 2. Гидролитическое расщепление солей.
Этот метод имеет больше аналитическое, чем препаративное значение. В аналитической химии его нередко применяют для изолирования гидроокисей металлов в чистом виде или для отделения трехвалентных металлов от двухвалентных па основании более сильной гидролитической способности первых. Примеры этой реакции см. алюминий, хром, железо.
6*
84
Глава 2
Вода полностью разлагает только соли очень слабых о снов алий с очень слабыми кислотами, например сульфид алюминия Л1233. В большинстве же случаев,* например для хлорида алюминия, гидролиз ведет к равновесию
Л1С13+5Н2О5*ЛЮ(ОН) + 3[Н30р + ЗСГ,
которое, однако, нетрудно сместить полностью вправо. Этого достигают прибавлением к реакционной смеси таких веществ, которые, сами не отщепляя ионов ОН', вступают в реакцию с Н3О'-понами и благодаря этому все время поддерживают концентрацию последних па низком уровне. Чаще всего для этого пользуются карбопатом бария, тиосульфатом натрия или ацетатом натрия. Подобным же образом действует смесь йодида калия с йодатом калия. Подробно об этом см. стр. 868. “
Многие соли (например, карбонат бария) гидролитически расщепляются при обработке их горячим водяным ларом
ВаС03-рН2О^Ва(0Н),-рС02.
Метод 3. Окисление металлов в присутствии воды.
Примеры: железо под действием влажного воздуха образует гидроокись железа 2Ге+з/3о2+т;Н2О= Ре2О3я;Н2О.
Для окисления (щелочных и щелочноземельных металлов достаточно уже действия только воды
‘Na-|-H2O=NaOH+’/2H2.
Метод 4. При электролизе нейтральных растворов солей сильно электроположительных металлов на катоде образуются их гидроокиси.
Этот метод находит широкое применение при техническом получении щелочей.
Метод 5. Получение оснований из солей в результате двойного обмена
Схема; MX ВОН = МОН -рВХ
Соль Основание Основание Соль
Этот метод наиболее технически важен для получения всех оснований, \ за исключением гидроокисей натрия, калия и кальция. Рассмотрим три основных случая применения этого метода:
а) получающееся основание или соответствующий ему ангидрид летучи.
В этом случае основание легко выделяется из реакционной смеси нагреванием. На этом базируется получение аммиака и многочисленных его органических производных.
б) получающееся основание нерастворимо (или труднорастворимо) в воде.
.Ввиду того что гидроокиси всех металлов, за исключением щелочных, нерастворимы в воде или труднорастворимы (как гидроокиси щелочноземельных металлов), этот метод имеет чрезвычайно большое практическое применение. В сущности использованием его является уже каждое осаждение гидроокиси едкой щелочью или раствором аммиака.
в) в некоторых случаях применяют также метод, основанный па образовании труднорастворимой соли.
Технически важцым применением этого метода является каустификация соды (см. стр. 207).
На этом же принципе основан способ приготовления гидроокиси стронция:
SrS -р CuO_^H2O—Sr(OII)2-|-CuS.
(Из целестина)
Соответствующим образом можно получить и едкий барий.
Водород
85
ОБЗОР СПОСОБОВ ПОЛУЧЕНИЯ ОСНОВАНИЙ
Гидроокиси щелочных металлов. Важнейший способ получений — электролиз растворов солей {метод f). Для едкого натра, кроме того, каустификация раствора соды гашеной известью {метод 5е). Получение паи боле с чистого едкого натра осуществляется из натрия и воды (метод 3).
Гидрат аммиака. Водный раствор аммиака получают при пропускании его через воду. В лабораториях аммиак получают и к солей аммония и емкого натра пли гашеной извести (метод За). Относительно технических методов получения аммиака см. стр. 654.
Гидроокись кальция. Гашение жженой извести (метод Г).
Гидроокиси стронция и бария. Из концентрированных растворов хлоридов Действием едкого натра (метод 56) или в соответствии с (5в) или (1).
Остальные основания. Осаждение из растворов их солей действием щелочей (дли аммиака) (метод бб), а также добавлением какой-либо стимулирующей гидролиз соли {метод 2).
Соли. Если к раствору кислоты прилить раствор какого-нибудь основания, то водородные попы кислоты соединяются с гидроксильными ионами основания, образуя воду
Н3О‘4-ОН'=2Н2О.
Если к кислоте прибавить такое количество основания, чтобы в растворе исчезли все ионы ИзО' (т.е. чтобы их концентрация упала примерно до незначительной величины, характерной для чистой воды)^ то такой процесс называется нейтрализацией. В результате в растворе останутся отрицательные ионы кислоты (анионы кислоты) и положительные попы основания (большей частью ионы металлов). Эти противоположно заряженные ионы могут соединяться вследствие существующего между ними электростатического притяжения. Однако этот процесс идет лишь при достаточной концентрации раствора. Образующийся при этом продукт’ называют солью. Иначе соли образуются е результате взаимодействия кислот со щелочами, причем реакция сопровождается выделением воды.
Из кислотных ангидридов и основных ангидридов (окпелов металлов) соли образуются без выделения воды.
Пример-
СОг 4- СаО = СаС03.
Далее ангидрид основания (см. ниже) соединяется с кислотами, образуя сади, также без выделения воды. "Ч
Пример.
NH3_^HC1—> (NH4]C1.
Существуют вещества, которые, соединяясь с основаниями, образуют соля без выделения воды. Они называются ангидрокислотами.
Так же как и электролитическая диссоциация кислоты, образование соло из ан-гиДроосноваппя и кислоты сводится в принципе к протонному обмену
Н2О+НС1= [H3OJ +C1'; NH3+HC1= [NH4]’+CP.
Соответствующий протонный обмен происходит .также между ангидро основанием и Н2О. Зим.и обусловливается щелочная реакция водных растворов ан гидроосп о ваиий
NHs+HzO^t [NHtf+OH'.
Танин образом, анеидроовнованиями называются такие соединения, которые образуют ионы ОН' в результате того, что отщепляют протон от молекул II %О.
Аналогично тому, как ан гидр о основания не отдают ионов ОН’, а присоединяют протон, ангидро киыоты. также нс отщепляют протоны, а присоединяют ионы ОН'. Так ведет себя прежде всего целый ряд амфотерных гидроокисей металлов, являясь антидрокнслотами по отношению к сильным основаниям.
86
Глава 2
Пример. г
Al(Ol-I)s+HOH= [Л1(0Н)4]'+Н-соответственно
А1(ОН)з+ОН'=[Л1(ОН)4]'.
Соли, образованные в результата присоединения основания к ангидро кислоте, называются гидроксосолями.. Примером является К[Л1(0Н)4] — гидр оксо алюмин ат кадия.
Слабые и сильные электролиты. Кислоты, основания и соли являются электролитами, т. с., будучи растворенными в каком-нибудь растворителе с высокой диэлектрической постоянной, и прежде всего, следовательно, в воде, они проводят электрический ток, сами одновременно подвергаясь разложению (электролитическое разложений). Некоторые из названных веществ обнаруживают электропроводность также в расплавленном, а иные даже и в твердом состоянии.
Различают два рода электропроводности: металлическую (электронную), которая, однако, встречается не только у металлов, и электролитическую. Существенное различие между этими видами проводимости заключается в том, что с металлической проводимостью тока не связано цикаких изменений вещества самого проводника, в то время как электролитическая проводимость сопровождается химическим изменением его. Между ними существуют и др у гае отличия. Например, металлическая проводимость уменьшается с повышением температуры, а электролитическая, напротив, с повышением температуры в общем увеличивается.
Для объяснения электропроводности принимают, что в растворах электролитов присутствуют заряженные частицы — электролитические ионы, благодаря передвижению которых от одного электрода к другому и совершается перенос тока, через злек-тролит. Передвижение ионов в растворе можно доказать изменением концентраций, которое наблюдается в тех случаях, когда скорости движения ионов с противоположными зарядами (из которых состоит электролит) неодинаковы, что почти всегда имеет место в действительности. Кроме изменения концентраций, па электродах совершаются химические реакции вследствие того, что на них происходит разрядка ионов. Об этом процессе уже было сказано при рассмотрении электролиза, воды.
Частное от деления удельной электропроводности электролита па его концентрацию, выращенную в молях на кубический сантиметр, называется молярной электропроводностью электролита*. В то время как удельная электропроводность, как правило, быстро уменьшается по мере снижения концентрации электролита, молярная электропроводность в общем возрастает с разведением до постоянного значения, называемого «молярной электропроводностью при бесконечном разведении». На основании зависимости молярной электропроводности от концентрации электролита можно разделить электролиты па два класса — сильные и слабые электролиты. Сильными называют те, которые при больших концентрациях обладают значительной электропроводностью, причем она с разбавлением немного возрастает. Слабыми электролитами называют такие, у которых при больших концентрациях молярная электропроводность незначительна, однако сильно увеличивается с разбавлением. Примерами сильных элект
* Молярная электропроводность р выражается формулой р.. = х/m или р = хх X1000 и, где х — удельная электропроводность, т — концентрация в молях на 1 сл<3, к — объем в литрах, в котором содержится 1 моль электролита. Нередко вместо молярной электропроводности р. приводят значения эквивалентной электропроводности А. Последняя определяется выражением л = х/ц или Л = х-1000 <р, где ц —• концентрация в ераммэквивалентах на 1 с.и3 или <р — обьсм в литрах, в котором содержится 1 s-a.se электролита. Часто электропроводность относят нс к числу молей или грамм-эквпвалентов в литре, а к числу молей или граммэквивалситов в 1000 г растворителя. Применение такой концентрации имеет то преимущество, что она не зависит от'темпе-ратуры.
Водород
87
ролитов являются соляная кислота п^хлористый калий; муравьиная кислота и уксусная кислота — Примеры слабых электролитов (см. рис. 12).
Совершенно очевидно, что электропроводность раствора должна быть тем больше, чем больше число ионов в растворе и чем больше подвижность каждого из этих ионов порознь.
Если предварительно Припять, что подвижность свободных ионов не зависит от концентрации раствора, в котором они находятся, то экспериментально установленный факт, что молярная электропроводность электролитов р (т. е. электропроводность, отнесенная каждый раз
Рис. 12. Увеличение молярной электропроводности для слабых и сильных электролитов с разведением.
и~одному и тому же количеству электролита) возрастает с разведением, можно объяснить увеличением разложения на ионы, т.е. повышением электролитической диссоциации по мере разбавления. Этот процесс идет до тех пор, пока весь электролит не разложится на ноны. Когда Это произойдет, то молярная электропроводность также достигнет наивысшего значения и дальнейшее разведение раствора уже не будет больше оказывать влияния на ее величину. Если через обозначить молярную электропроводность при концентрации с, через — продельное ее значение при бесконечном разбавлении (т. е. при концентраций, равной нулю), то степень диссоциации электролита а, т. е. та его дробная часть от единицы, которая распалась на ионы, выражается следующим образом: а=-^- (а также (17)
Ра \ Ао)
Это становится попятным, если рассматривать электропроводность данного вещества как величину, пропорцноиальпую количеству его ионов. Предположение, что степень диссоциации определяется отношением молярных электропроводностей, было впервые в 1887 г. сделано Аррениусом. Он установил это в результате измерения температур замерзания электролитов. Д именно, как показал в 1886 г. Вант-Гофф, понижение температур
88 Глава 2
замерзания и повышение температур кипения растворов пропорциональны осмотическому давлению растворенного вещества, а оно в свою очередь пропорционально числу молекул, находящихся в определенном объеме, совершенно так же, как это происходит в отношении давления газа газообразных веществ. Поэтому для осмотического давления имеет силу то же Уравнение, как и для газового состояния:
V
где Р — осмотическое давление; п — число молей, находящихся в растворенном состоянии в объеме и; Я — тазовая константа; Т — абсолютная температура.
Для неэлектролитов число п грамм-молекул, содержащихся в определенном объеме раствора, в общем случае равно числу грамм-молекул, которые были растворены в этом объеме (некоторые отклонения могут быть вследствие ассоциации растворенных молекул между собой и с молекулами растворителя). Но для электролитов число растворенных частиц возрастает вследствие того, что растворение в этом случае сопровождается диссоциацией. Соответственно этому и осмотическое давление (а следовательно, и связанное с ним повышение точек кипения и понижение точек замерзания растворов) для растворов электролитов будет больше, чем это теоретически следует из молекулярного веса. Для растворов электролитов в средах,, вызывающих диссоциацию, например в воде, осмотическое давление будет определяться формулой
P^—RT.
V
В соответствии с «классической» теорией электролитической диссоциации (см. ниже) множитель I в этом выражении показывает, в каком отношении возросло число молекул благодаря диссоциации. Его можно также определить как отношение получающегося при криоскопических 0 Других определениях числа молекул к числу внесенных в раствор молекул или как отношение найденных опытным нутом значений понижения точек замерзания или повышений точек кипения к соответствующим значениям, вычисленным на основании закона Рауля, при допущении, что при растворении не происходит изменения числа растворенных частиц.
Согласно закону Рауля, понижение точек замерзания или повышение точек кипения не слишком концеятрированых растворов в данном растворителе пропорционально количеству вещества, растворенного в определенном объеме, и обратно пропорционально молекулярному весу этого вещества
где At — понижение точки замерзания или же повышение точки кипения; р — количество растворенного вещества (в); М — молекулярный вес растворенного вещества; G — некоторая, характерная для каждого растворителя константа, называемая молярным понижением точки замерзания или молярным повышением точки кипения растворителя.
Если через Д£кыч обозначить вычисленное по этой формуле, а через Д(найд — найденное экспериментально понижение точки замерзания или повышение точки кипения, то i определится выражением
Д^найд (18>
Водород 89
Для случая полной диссоциации величина I, согласно «классической теории», будет равняться числу ионов, на которое распадается каждая внесенная в раствор молекула. Для электролита типа хлористого натрия, распадающегося на два одновалентных иона, i при полной диссоциации будет равно 2; при неполной диссоциации t для такого типа электролита определяется выражением
£ = 1+а. (19)
Если через а обозначить степень диссоциации (т.е. ту дробную часть от единицы, которая подверглась диссоциации), то для раствора, в который внесено п молекул вещества, число молекул оставшихся я едиссоцииро ванными, будет равно п(1—а); число же образовавшихся благодаря диссоциации частиц будет в случае электролита, расщепляющегося на два одновалентных иона, равно 2па, а следовательно, общее число частиц (молекул и ионов) будет и(1—а) 2»аи в соответствии с этим отношение числа частиц после диссоциации к числу их до диссоциации определится выражением
ц(1—а)-|-2яа = j п
В общем случае для электролита, распадающегося на х ионов, получим выражение
1= —1) я. (19а)
Аррениус на основании многочисленных экспериментальных наблюдении показал, что для степени диссоциации а числовые значения получаются почти точно совпадающимй как при вычислении их из уравнений (18), (19) и (19а), так и при определении нх на основании изменения молярной электропроводности с разведением [согласно уравнению (17)]. На основании этого он заключил, что значения а, определяемые этими двумя различными методами, действительно представляют собой степень диссоциации электролитов. Такая «классическая» теория электролитической диссоциации оказывается правильной только для слабых электролитов. Как позже было показано, она не оправдывается для сильных электролитов.
Для слабых электролитов степень их диссоциации можно вычислить на основании изменения электропроводности в зависимости от разведения [уравнение (17)] или из криоскопических данных [уравнение (18)]. Если к процессу диссоциации электролита, распадающегося на два одновалентных иона, приложить закон действия масс, то при выражении концентрации через степень диссоциации а получим
—------г~ const или -------const. (20)
v (1—а) 1—а
Принимая во внимание уравнение (17)
Лс'С
т—,. —гл-=const, А, (Ад Ас)
где и (= 1/с) — объем раствора, содержащий грамм-эквивалент электролита [закон разведения Оствальда).
Закон разведения Оствальда приложим к слабым электролитам. Сильные электролиты ему не подчиняются, как это видно из данных табл. 13.
90
Глава 2
Таблица 13
Применимость закона разведения Оствальда для слабых электролитов и непригодность для сильных электролитов
В таблице даны значения
Лас-е
Ло(Ло Лс)
При 18°
Эквивалентная концентрация (с) Слабые электро ли™ Сильные электролиты
уксусная кислота гидроокись аммонии хлористый калий сульфат магния
1 1,40-10'Е 1,40-10-6 2,430 0,0879
0,1 1,70-10-5 1,92-10-6 0,535 0,0333
0,01 1,70.10'5 1,08-10-5 0,151 0,0133
0,001 1,50.10'5 1,56-10-5 0,046 0,0060.
0,0001 1,31Ю5 1,06-10-5 0,013 0,0023
Среднее 1,52-10-5 1,51-10-5
Неприложимость закона разведения Оствальда к сильным электро-' " литам зависит от того, что для сильных электролитов значение а, вычисленное по уравнению (17), пе указывает действительной степени диссоциаций. Лежащее в основе его вычислений предположение,: что подвижность ионов не зависит от их концентрации, приближенно допустимо лишь там, где число ионов в единице объема незначительно. Для слабых электролитов эти условия в какой-то мере выполняются*. Напротив, в растворах сильных электролитов концентрации ионов так высоки, что их подвижность сильно снижается в результате взаимодействия электрических зарядов попов. С разведением растворов это взаимодействие уменьшается и вместе с тем повышается подвижность ионов. Это повышение подвижности ионов объясняется увеличением эквивалентной электропроводности с разведением у сильных электролитов. Таким образом, изменение электропроводности не позволяет для них определить степень диссоциации.
Ввиду того что ионы сильных электролитов ведут себя в основном иначе, чем незаряженные частицы, для определения степени их диссоциации нельзя также применять формулу, в которую входит осмотическое давление. В настоящее время считают, что типичные сильные электролиты, и прежде всего большинство солей, при большом, разбавлении (около 0,01 Л/) полностью диссоциированы, а при более высоких концентрациях почти нацело или в гораздо большей степени диссоциированы, чем это дает классическая теория на основании измерений электропроводности и криоскопических данных. Это положение было развито главным образом Сезерландом, Мильнером, Бьеррумом и Гошем и в 1923 г. точно обосновано Дебаем и ХюкКелем для очень разбавленных растворов**.
* Дри точных вычислениях для слабых электролитов следует также прижимать во внимание действие межионных сил.
** Краткий обзор теории Дебая — Хюккеля дан Mclnnes D, A., J. Franklin Jnst., 225, 661, 1938. Относительно точного расчета констант диссоциации: на основе измерений электропроводности см. там же стр. 678 и сл.
ВоЗород
91
Мильпер (Milner, 1913) впервые покапал, что попы в растворе электролита не распределены совершенно хаоттио (в последнем случае электростатические силы притяжения и отталкивания, оказываемые ими один па другой, должны были ба взаимно уравновешиваться), но что в среднем вследствие электростатического притяжения в соседстве с каждым положительным ионом находится больше отрицательных, чем положительных исков, и, наоборот, вокруг каждого отрицательного иона находится больше положительных ионов, чем отрицательных. Это обстоятельство, однако, независимо от его влияния на электропроводность раствора создает условия, при которых ионы удерживаются в растворе прочнее, чем незаряженные молекулы. Вследствие этого лрп вычислении значения осмотического давления нельзя приравнивать ионы к незаряженным молекулам.
Концентрации иопон в растворах слабых электролитов малы также и в случае высоких концентраций самих электролитов, в связи с чем расстояния между ионами соответственно больше. Поэтому для слабых электролитов влияние сил, действующих между ионами, в значительной мере уменьшается, и практически его следует учитывать только при очень точных расчетах.
Соотношения, существующие в растворах сильных электролитов, значительно осложняются еще и тем, что здесь необходимо учитывать также силы, которые действуют на иопы со Стороны растворителя. Эти силы могут в общем случае вызывать образование сольватов, а в случае воды — гидратов (см. стр. 96 сл.).
Межионные силы оказывают влияние не только па электролитическую проводимость и осмотическое давление, но также и на другие свойства веществ, зависящие от концентрации ионов, например, на потенциалы разряжения и собственные потенциалы, и вообще на все «кажущиеся» и «эффективные» концентрации, в которых вещество принимает участке в равновеском процессе растворения. «Кажущиеся» концентрации, которые соответствуют равновесному состоянию процесса растворения, как уже ранее указывалось, называются активностями. Точно так же как активности неэлектролитов, активности ионов по мере разбавления растворов становятся равными их истинным концентрациям. Однако даже в довольно разбавленных растворах сильных электролитов (с —0,001 моль)Л) еще очень значительны отклонения активностей ионов от истинных концентраций. Напротив, для сильных электролитов истинные концентрации ионов только очень незначительно отклоняются от тех значении, которые получаются из аналитических концентраций с учетом их полной диссоциации. Практически сильные электролиты можно рассматривать (Прежде всего в разбавленных растворах с < 0,1 моль! л) как полностью диссоциированные.
Числа, на которые необходимо умножить истинные концентрации ионов, чтобы получить величины кажущихся концентраций (сначала без учета межионных сил), определяемые измерением активностей [уравнение (1)] или в результате исследования электропроводности [уравнение (17)], а также путем применения осмотического метода [уравнение (18)], называют по предложению Бьеррума коэффициентом активности fa или коэффициентом электропроводности (проводимости) осмотическим коэффициентом /(1 или в общем коэффициентом отклонения.
Коэффициенты отклонения определяются следующим выражением:
Коэффициент, активности-. — , (21)
СН>
где а — активность, т.е. эффективная концентрация; с1У -т- истинная концентрация соответствующего нона.
Р
Осмотический коэффициент: , (Й2)
'о
92
Гласа S
где Р—осмотическое давление, найденное экспериментально; Рс—осмотическое давление, которое обнаружилось бы, если бы ионы вели себя как нейтральные молекулы.
j i и
Коэффициент электропроводности: Ту,—------------, (23)
«ш Но
где аш — истинная (но экспериментальная) степень диссоциации.
Коэффициенты Д, /п, Д, зависят от природы электролита, от разведения и от температуры. В общем случае они не равны, однако при бесконечном разведении все три достигают предельного значения равного 1. Для слабых электролитов в менее разбавленных растворах, однако, они незначительно отклоняются от единицы. Вследствие этого для слабых электролитов можно, не допуская большой ошибки, приравнивать значения концентрации ионов, найденные различными методами.
Таким образом, практически важно следующее:
Для слабых электролитов, особенно при сильном разведении
, , , . . а р 1 ц ,
7а = /о=/я = 1, а также *=----- ^=1.
Для типичных сильных электролитов (большинство солей), при достаточном разведении, наоборот,
аю = 1
и вместе с тем если х по-прежнему означает число ионов, на которые распадается каждая молекула растворенного вещества, то
/<>=— и /ц = 7Г-
В табл. 14 приведены значения коэффициентов отклонения для раствора хлористого калия при различных концентрациях: в последней колонке дапы значения для i—f (=2/rt—1). Эти величины, а также помещенные в предыдущей колонке значения для рассматривались классической теорией пак степени диссоциации. Из данных таблицы
Таблица 14
Значения коэффициентов отклонения для КС1
Молярная концентрация ia /о i — 1
1,0 0,558 0,854 0,755 0,708
0,1 0,762 0,932 0,861 0,864
0,61 0,882 0,969 0,941 0,938
0,001 0,943 0,985 0,979 0,970
видно, что рад деленные двойной чертой величины (в двух последних столбцах) в пределах концентраций 0,1—0,001 обнаруживают очень хорошее совпадение. При современном состоянии теории причину этого указать еще не удается, так как между /„ и / цельзя найти теоретически простой зависимости. Наоборот, между /0 и /я такая зависимость существует, и ее можно вывести термодинамическим путем, что позволяет в случае падобности, зная значение /0 и его зависимость от концентрации, вычислить соответствующее значение для /я.
Коэффициент активности, определяемый уравнением (21), характерен только для определенного вида ионов (например, для СГ-нона). Под коэффициентом активности сильного электролита (например, КС1) понимают среднее геометрическое коэффицивн-
Водород
93
тов активности, его ионое. Коэффициент активности /£х электролита, который распадается на р ионов с коэффициентом активности Д и па q ионов с коэффициентом активности /г, определяется уравнением
1
> /£Х = (М)Р+’.
Примером является коэффициент активности для ВаС12 (где р=1, ?=2)
^аС1г = [/“а”(/^')211/э-
Активностью сильного электролита называют произведение активностей отдельных его ионов. Таким образом, активность ВаС12, который распадается па три иопа, определяется следующим образом:
где с — аналитическая молярная концентрация ВаС12. Ес конечное значение при бесконечном разбавлении, следовательно, равно lim а=4с$.
с=0
К сильным электролитам относятся почти все соли. Для всех солей одного и того же типа, например для солей, расщепляющихся на два одновалентных иона (тип М'Х'), из каких бы веществ они ни состояли,— отношение их молярных электропроводностей рс/р0 или отношение эквивалентных электропроводностей Лс/Ло почти равны между собой; таким образом соли одного типа имеют почти одинаковые кажущиеся степени диссоциации. Это становится понятным в свете новейшей теории. Согласно последней, все соли (за исключением лишь немногих) при растворении полностью распадаются на ионы. Силы взаимодействия, проявляемые находящимися в растворе и отдаленными сравнительно на большие расстояния ионами особенно в сильно разведенных растворах, зависят только от числа зарядов отдельных ионов и лишь в очень незначительной степени от химической природы растворенных веществ. В противоположность этому коэффициенты электропроводности рс/ц0 или Лс/Л0 кислот иногда обнаруживают значительные различия. Однако одноосносиые сильные киСлоты опять обнаруживают далеко идущее сходство, поэтому их можно рассматривать как полностью или почти полностью диссоциированные. Для слабых кислот коэффициент электропроводности с большим приближением указывает на степень диссоциации. Серная, щавелевая, фосфорная и другие подобные кислоты занимают в этом отношении промежуточное положение. Они называются кислотами средней силы (за исключением серной кислоты, которую считают сильной кислотой, так как она в разбавленных растворах па первой стадии полностью диссоциирует), Для сильных оснований отношения коэффициентов электропроводности Лс/Л0 соответствуют значениям отношений коэффициентов электропроводностей для солей.
В табл. 15 и 16 приведены отношения электропроводности Ле/Л0 для важнейших тцпов солон, а также для многих оснований и кислот. Числовые значения в этих таблицах представляют во всех случаях частное от деления эквивалентных электропроводностей при определенных концентрациях Ле па граничные значения эквивалентных электропроводностей Ло при бесконечном разведении (концентрация = 0).
Ступенчатая диссоциация. Многоосновные кислоты, например серпад, щавелевая и фосфорная, не отщепляют сразу всех ионов водорода, способных замещаться на металл. Диссоциация у них идет ступенчато, т. е. сначала отщепляется один ион водорода и только при значительно более сильном разведении происходит отщепление второго ионэ
94.
Глава 2
Таблица 15
Отношение коэффициентов электро про водности Л.с/Лй со .чей 11 оснований (при 18°)
Для солей и сильных оснований отпошениеЛс/А0 указывает кажущуюся степень диссоциации, а для слабых оснований — истинную.
Тин И ример 1 н. эд н.
Соль с одновалентным катионом и одновалентным анионом КС! 0,75 0,86
Соль с одновалентным катионом ид ву хвале нт-f пым аяноцом ! K2SO4 0,54 0,74
Соль с двухвалентным катионом и одно вален г-1 ным апионом СаС12 0,58 0,75
Соль с двухвалентрым катионом н двухвалентным анионом MgSOt 0,25 0,43
Сильное основание с одновалентным катионом кон 0,77 0,89
Сильное основание с двухвалентным катионом Ва(ОН)г — 0,77
Слабое основание NHa-HsO 0,004 0,014
Таблица 16
Отношение коэффициентов электропроводности Л,,/Лв кислот (при 18е)
Для слабых кислот отношение Ле/Лр указывает истинную, а для сильных кислот — кажущуюся степень диссоциации
Сильные кислоты <HNO. I-HG1 1 11230д 1 и. 0,82. 0,79 0,51 0,1 п. 0,93 0,92 0,59
Кислоты средней силы f Н0С004 < щавелевая кислота 0,16 0,31
( Н3РО< 0,06 0,12
Слабые кислоты ( СПдСООН I уксусная кислота 0,004 0,013
1 И3СО3 — 0,001
водорода, а третьего — только при гораздо более сильном разведении или лишь после добавления щелочи и т. д. Поскольку к этим процессам диссоциации приложим закон действия масс, то для каждой стадии диссоциации существует своя константа диссоциации. Этой ступенчатой диссоциацией многоосновных кислот объясняется их способность к образованию1 кислых солей, т.е. таких солей, которые получаются вследствие неполного насыщения кислот основаниями и в которых, следовательно, имеются еще водородные ионы, способные замещаться па металлы.
Ограниченная применимость закона действия масс к сильным электролитам. Неприменимость закона разведения Оствальда к сильным электролитам показывает,
Водород 95
что закон действия масс е его обычной форме неприменим к процессам диссоциации сильных электролитов, а новая теория сильных электролитов объясняет причину этого. Таким образом, следует считать, что при современном состоянии науки утверждение, будто отношение коэффициентов электропроводности ц/ро сильного электролита является истинной степенью диссоциации и что к ним можно применять закон действия масс, является не только приблизительным, но и в основном ошибочным.
Если применяют современную форму закона действия масс, в которую входят концентрации ионов сильных электролитов (например, в случае вычисления произведения растворимости), то необходимо учесть «активности» иопов, При обсуждении вопроса о том, как влияет на равновесие между попами введение сильного электролита, следует прежде всего принять во внимание, что, согласно новейшей теории, активности ионов какого-либо электролита испытывают воздействие любого электролита, а по только того, который имеет с ним общий ион. Таким образом, имеет силу закон* **, говорящий, что коэффициент активности любого электролита зависит от общей ионной силы раствора, а в разбавленных растворах, независимо от природы электролита, является функцией только общей ионной силы J.
Под ионной силой понимают сумму:
•^=1/2(cisJ4-c2s|+ ...),
где сь с2 и т. д. — истинные молярные концентрации,, a zlt ,z2 и т. д. —• заряды различных ионов. Например, в растворе чистого 0,01 М раствора ВаСЬ J составляет i/2 (0,01х 2“-]-0,02х1а)=0,03; В этом растворе коэффициенты активности?* На" и СГ имеют значения соответственно/Ja"=0,50; /д1'—0,86. Если же в раствор ввести только 0,1 моля какого-либо сильного электролита, распадающегося на два иона, то значение J возрастет до величины г/2(0,1-)-0,1)—0,1. Таким образом, общая ионная сила будет равна .7=0,13. Тогда в растворе с такой ионной силон коэффициенты активности будут иметь значения /«a"=0,10 и /а'’=0,79 независимо от того, добавлен ли хлорид калия дли нитрат калия. Туже ионную силу (7=0,13) имеет раствор чистого хлористого бария с концентрацией 0,0433 М; при этом величины /„а” и имеют указанные выше значения. Таким образом, активности отдельных ионов получаются в результате умножения истинных (а для сильных электролитов практических их аналитических) концентраций на соответствующие коэффициенты активности. Например, в 0,01 Л/ растворе чистого ВаС12 активность ионов Ва‘ '=0,01-0,50= 0,0050, а для иопов СГ =0,02-0,86=^ =0,0172. Активность (эффективная концентрация) иопа хлора, таким образом, не является просто удвоенной концентрацией ионов Ва", как это следует из классической теории диссоциации. В случае сильных электролитов для более или менееТрубой,оценки можно пол I. зеваться аналитически определяемыми истинными концентрация мп, которые получают па основании содержания электролита, предполагая полную его диссоциацию и не учитывая коэффициентов активности. Так поступают, например, если необходимо приблизительно рассчитать равновесие смеси сильного и слабого электролитов или если необходимо сделать заключение о влиянии на труднорастворимый электролит незначительных количеств сильного электролита, имеющего одноименный ион. Если же требуется большая точность расчета, то нельзя пренебрегать «активностями». С теоретическом точки зрения для сильных электролитов рационально пользоваться вместо «активностей» кажущимися концентрациями иопов, полученными на основании измерений электропроводности или осмотического давления. Действительно, лишь в редких случаях полученные таким образом значения оказываются более точными, чем полученные непосредственно из аналитических концентраций.
Теоретически закон действия масс применим также и к процессу диссоциации сильных электролитов. А именно в растворе всегда найдутся хоть несколько ионов с противоположными зарядами, расположенными так близко один к другому, что их можно считать вновь соединившимися в ледцссоцнированные молекулы. Однако это число в таком смысле пвди с соци про ванных молекул для сильных электролитов не находится ни в каком определенном отношении к тмгу числу, которое определяется из «кажущейся степени диссоциации».
* Закон о зависимости коэффициента активности от ионной силы был эмпирически памдеп в 1921 г. Льюцсом и Ренделлом. Позднее на основании теории Дебая были получыты подтверждения этого закона.
** Относительно определения коэффициентов активности ионов см. Льюис и Редделл «Термодинамика», Ленинград, 1936.
96
Глава 2
ГИДРАТЫ
Вода обладает свойством соединяться с очень многими веществами, образуя с ними химические соединения. Получающиеся при этом продукты носят названия гидратов, а процесс их образования называется гидратацией. В тех случаях, когда данное вещество связывает не воду как таковую, по когда одновременно со связыванием воды происходит и ее расщепление, так что присоединяются составные части воды, как это, например, имеет место при образовании гидроокиси из окиси:
Н
СаОЦ-.........=Са(ОИ)г,
О-Н
(в этих случаях говорят о, «конституционно» связанной воде) образовавшиеся таким способом соединения в настоящее время обычно уже не называют гидратами,
В соответствии с прежними представлениями такие соединения рассматривали л качестве продуктов присоединения воды, поэтому для них еще удержалось название «гидрат»; так, Са(ОН)2 называют устаревшим термином «гидрат окиси кальция» или «гидрат извести» вместо современного названия гидроокись кальция.
В настоящее время гидратом называют соединения, содержащие, как это следует из их свойств, воду как таковую.
Существуют вещества, для которых еще не известно, связана ли в них вода химически или удерживается механически. Это имеет место в случае всех известных окисло л, которые получаются л виде гидрогелей (см. стр. 394). Поскольку для пнх не установлено, являются ли опи гидроокисями или окислами с механически удерживаемой водой, их называют «оксигидратами».
Диаграмма растворимости гидратов и правило фаз. Мпогие вещества из водных растворов различной концентрации кристаллизуются с различным содержанием воды. Примером тому является СаС12 с 6, 4, 2 и 1 молекулой НаО. Условия, при которых различные гидраты могут существовать в равновесии с водой, можно определить па основании диаграммы растворимости, На рис. 13 приведена растворимость хлорида кальция в воде в зависимости от температуры (кривые растворимости). Из растворов, которые содержат менее 42,5 вес. ч. СаС13 на 100 вес. ч. воды, при охлаждении осаждается прежде всего лед. Тем самым повышается концентрация раствора и одновременно снижается температура замерзания вдоль кривой АВ. В точке В, т. е. при содержании 42,5 вес. ч. СаС12 на 100 вес. ч. Н2О, температура ранца —55% остаток замерзает в виде криогидрата. Из растворов, содержащих более 42,5 и менее 100,6 вес. ч. СаС12, осаждается сначала гексагидрат, хлорида кальция. Концентрация раствора п температура кристаллизации при этом изменяются вдоль кривой СВ. При —55’ остаток опять затвердевает в виде криогидрата.
При значениях концентраций и температур, соответствующих кривым АВ и СВ, в равновесии находятся три фазы, а именно раствор, лед и пары воды пли раствор, гексагидрат и пары воды. Ввиду того что в этом случае приходится иметь дело с трехфазной системой, содержащей два компонента Н2О и СаС12, то в соответствии с правилом фаз [уравнение (10)) эта система имеет одну степень свободы. Поэтому Здесь (в противоположность примеру, приведенному на рис. 11) три фазы могут находиться в равновесии при многих температурах. Каждой температуре, однако, отвечает, в случае одной и той же доппой фазы, определенная концентрация и определенное давление пара раствора, нахоД11Щегося с ним в равновесии. Точно так же для каждой концентрации раствора характерна определенная температура и определенное давление, пара раствора, при котором он находится в равновесии с соответствующей твердой фазой. Точка В является точкой пересечения кривых равновесия льда и гек-сагидрата. Она указывает концентрацию и температуру, при которой обе твердые фазы находятся в равновесии с раствором и его паром. Здесь существуют четыре фазы и, согласно уравнению (10), отсутствуют степени свободы. Такая точка, в которой, как в точке В, одновременно существуют четыре фазы, называется четверной
Водород
.97
точкой. Смесь обеих твердых фаз, являющаяся в данном случае криогидратом*, осаждается таким образом из раствора при совершенно определенной температуре (—55°). Для этого раствора характерен определенный состав (42,5 вес. ч. СаС12 на 100 вес. ч. Н2О) и определенное давление пара (0,015 .и.и рт ст). Криогидрат при температуре выделения имеет такой л;е состав и такое же давление пара, как и раствор. Хотя криогидрат и имеет постоянный состав, все же он является не соединением, а смесью.
Из растворов, состав которых находится между точками Guf (им соответствуют значения абсцисс от 100,6 до 130,2 вес. ч. СаСЬ), должна была бы кристаллизоваться a-модификация тетрагидрата. Однако в результате задержки в кристаллизации этой формы между точками С и D (100,6—112,8 вес. ч. СаСЬ), а иногда и дальше обычно осаждается гексагидраг, а между точками D и Е выделяется нестабильная Р-моди-фикация тетрагидрата**. Из раствора, содержащего 102,7 вес. ч. СаСК па 100 вес. ч.
Р и с. 13. Диаграмма растворимости хлористого кальция.
НгО, т. е. имеющего состав гексагидрита, обычно и осаждается гексагидрат, несмотря на то, что в этой области он нестабилен. Поэтому гексагядрат также плавится при нагревании, как правило, без разложения, а имеппо (в соответствии с рис. 13) нрй 30,2° (по новейшим исследованиям при 29,8°). Ввиду того что из растворов, концентрация которых находится в интервале между 102,7 и 112,8 вес. ч. СаС1г на 100 вес. ч. НаО, выкристаллизовывается сначала гексагидрат, если вносят его затравку, то при охлаждении концентрация раствора растет, пока ие достигнет точки D. При соответствующей этой точке концентрации (112,8 вес. ч. СаСЬ на 100 вес, ч. Н2О) и температуре (29,2°) осаждается смесь кристаллов гексцпщрата и [3-модификации тетрагидрата; при этом состав раствора не изменяется. Таким образом, в точке D, как и в точке В, находятся в равновесии четыре фазы: раствор, пар и обе (нестабильные в данном случае) твердые фазы. Кристаллизующаяся при температуре 29,2° смесь обеих твердых фаз и в атом случае имеет такой же состав, как раствор. Замечательно то, что гексагидрат, как видно из диаграммы, мри одной и той же температуре может
* Название криогидрат, является ошибочным, так как гидратами называют соединения, а не просто смеси. Одпако это пазвапие общепринято.
’* Это соответствует правилу ступеней Оствальда; см. стр. 531. Из растворов, концентрация которых немного ниже значения D (112,8 ч. СаСП на100 ч. воды), осаждается обычно |3-модификация тетрагидрата. Если же внести затравку гексагидрата, то кристаллизуется последний. Стабильный в этой области концентрации а-тетрагид-рат кристаллизуется только тогда, когда вносят его затравку.
7 Г. Реми
08
Гмва 2
находиться в равновесии а двумя различными но концентрации растворами, например при 29,2° в равновесии с раствором, содержащим 112,8 вес. ч., и с раствором, содержащим 94,5 вес. ч. СаС12. В точке, где гидрат соли находится в равновесии с раствором равной с ним концентрации (102,7 ч. СаС12), равновесная температура имеет максимум (29,8е).
Нестабильная модификация тетрагидрата, находящаяся в равновесии с насыщенным раствором (если потереть стенкн сосуда стеклянной палочкой) переходит в стабильную «-модификацию. Превращение происходит мгновенно, если внести натравку «-формы.
Из растворов с содержанием более 127,5 вес. ч., а если внести затравку а-тет-рагидрата, то даже на растворов, содержащих более 436,2 вес. ч. СаС12, осаждается сначала дигидрат; далее, в результате того что концентрация раствора снижается до величины, соответствующей точкам Е и F (127,5 и соответственно 130,2 вес. ч. СаС12), остаток осаждается в виде смеси дигидрата и тетрагидрата. В области от 130,2 до 297 вес. ч. СаС12 па 100 вес. ч. воды стабильным является дигидрат, а при более высоких концентрациях в равновесии с раствором находится моногидрат.
Кривые давления пара; термическая диссоциация гидратов. .Кривые давления пара гидратов СаС12 показаны на рис. 14*. Если кристаллы гексагидрата поместить в эвакуированный сосуд, то они будут разлагаться, образуя тетрагидрат й пары воды до тех пор, пока в сосуде пе будет достигнуто давление пара, отвечающее равновесию.
Рис,. 14, Кривые давления паров гидратов СаСЕ.
В данном случае имеется система из двух компонентов (СаС12 и Н,О) и трех'фаз (гексагидрат; тетрагидрат и пар). Тогда в соответствии с уравнением (4O)^F=1. Система может находиться в таком равновесии при различных температурах, однако каждой температуре соответствует определенное Давление. Соответствующие пары значении давления и температуры, образуют кривую давления вара.
Если жо при определенной температуре, например при 25°, из сосуда откачать водяной пар, то давление будет до тех нор постоянно (р=5,1 .«.и pm cm), пока еще будет иметься в наличии гексагидрат (естественно предположить, что при указанных условиях скорость разложения гидрата достаточна для установления равновесия). Как только исчезнет весь Гоксагидрат, начнут разлагаться тетрагидрат. Вследствие этого давление тотчас падает до значения, соответствующего равновесному давлению тетрагидрата при 25е (р=3,4 льи pm cm, см. рис. 14). Это давление остается постоянным до тех пор, пока тетрагидрат полностью пе перейдет в дигидрат.
Как только исчезнет тетрагидрат, давление скачкообразно снизится до величины, присущей, равновесию дигвдрата (р=2 мм pm. cm) с парами воды и т. д. Если пар откачивают отдельными порциями и каждый раз после этого определяют состав подвергнутого разложению твердого вещества (или по убыли веса, пли намерением откачанных порций пара), то получается приведенная на рис. 15 ступенчатая кривая зависимости давления от состава. Каждая ступень соответствует изменению давления, которое происходит в тот момопт, когда один гидрат исчезает, а цолу-
* Из двух модификаций тетра гидрата рассматривается только стабильная. Кроме того; не обозначена кривая давления пара моногидрата, так как в показанной на рисунке области не были проведены еще точные измерения для итого гидрата.
Нодород
99
чающийся ия пего более бедный водой гидрат еще не начинает разлагаться. Состав каждого гидрата можно установить по кривой разложения.
Вместо разложения при постоянной температуре {изотермическое разложение) можно проводить разложение при нагревании {изобарическое разложение). В таком случае при данном давлении каждый гидрат испытывает разложение при совершенно определенной температуре. Например, в атмосфере с давлением пара воды G .и.ч рт ст гексагидрат хлористого кальция переходит при 28° в тетрагидрат (что сопровождается выделением пара воды); тетрагидрат при 34° переходит в дигидрат, а последний в свою очередь при 49° — в моногидрат. Все это можно хорошо представить, пользуясь рис. 14. В этом случае разложение также происходит ступенчато, т, е. потеря воды длится при постоянной температуре до тех пор, пока не исчезнет соответствующий гидрат. Следующий гидрат начинает разлагаться только тогда, кОгда будет достигнута его температура разложения. В промежуточных температурных областях состав гидратов не меняется.
молекул Н30 на /жлгелулл СаС1е
Рис. 15. Кривая диссоциации СаС1з-6Н2О (изотермическая диссоциация).
Как видно из рис. 13, при одной и той же температуре нестабильная (или мета ста б и льна я) р-модифпнация СаС1г-4НаО легче растворима, чем стабильная a-модификация. При одинаковой температуре она имеет более высокое давление пара, чем стабилвная модификация. Вообще имеет силу следующий закон: при существовании нескольких модификаций одного и того же вещества стабильная при данных условиях модификация обладает наи
меньшей растворимостью и наименьшим давлением пара (или давлением диссоциации). Этот закон является следствием второго закона термодинамики. Далее, термодинамически можно вывести следующее положение, на справедливость которого впервые указал Сакур (Sackur О., 1908). Давление пара гидрата зависит от того, какое более бедное водой вещество из него получается. Например, гекса гидрат хлористого кальция обладает большим давлением пара в том случае, когда он распадается, давая стабильный а-тетрагидрат, по сравнению с разложением, сопровождающимся образованием нестабильпого fj-тетрагидрата. В несколько измененном виде закон Сакура справедлив для термического разложения любого вещества.
Соотношения, приведенные здесь для гидратов, в общем применимы м к другим, находящимся в твердом состоянии веществам, разлагающимся с отщеи.чепием газообразных веществ. Примером являются аммиакаты, как, впрочем, и гидроокиси, окиси, карбонаты и т. д. Однако в этом случае необходимо, чтобы при разложении получалась вторая твердая фаза, иначе система вместо одной будет иметь две степени свободы. Но и в этом случае каждой температуре соответствует не больше одного определенного значения давления щкеоцнзнпп, в го время как последняя зависят от состава твердой фазы. Это явление наблюдалось первоначально в случае гидратов некоторых минералов, цеолитов. Содержание воды в них в зависимости от температуры и давления может колебаться в значительных пределах, не вызывая изменения гомогенности кристаллов. В таких случаях говорят, что имеется «цеолитпосвязанпая» вида. Кроме того, па давление пара может влиять степень раздробления вещества независимо от того, происходит ли это потому, что соответствующие вещества удерживают воду в результате адсорбции из-за сильного развития поверхности, или потому, что вода удерживается в очень топких капиллярах. В обоих случаях давление пара настолько снижается, что эта вода удаляется только е кристаллизационной илн даже с конституционной водой. В связи с тем, что скачкообразное удаление воды перекрывается аффок-
7*
100 L Глава 2
том отдачи адсорбированной или капиллярно удерживаемой воды, ступени на кривой обезвоживании могут быть искривлены или прерывности располагаются неправильно (Ср. стр. .536 а 5о9, а также т. II, гл, 16).
Гидратацип ионов. Если уже в твердом состоянии множество веществ существует в виде гидратов, то последние тем более будут существовать и в водны» растворах, а имеппо при определенных условиях такие гидраты должны быть богаче водой, нем твердые гидраты. В случае ионов стенояь гидратации их в не слишком* разбавленных растворах проще всего определяется измерением электролитического переноса, воды. Последнее оипачает перемещение воды от анода к катоду (или обратно) вследствие различной степени гидратации аниона и катиона. Для электролитического переноса различные исследователи (Buchbock, Washburn, Remy, Baborowsky) получили достаточно хорошо совпадающие значения, на основании которых были вычислены степени гидратации ионов, приведенные в табл 17.
Таблица J“
Гидратация сильно влияет на многие свойства ионов, например на электролитическую подвижность и на потенциал выделения (см. Стр. 181 и далее). Особенно сильно влияет гидратация на ион водорода. Ион гидроксония [НзО]+, полученный в результате присоединения К протону молекулы воды, ведет себя совсем иначе, чем вел бы себя свободный протон. Раньше уже было отмечено, что прочность связи протона с одной молекулой воды особенно велика. В других гидратированных ионах молекулы воды связаны слабее, чем в ионе гидроксония. Их энергии связи -уже не достигают той величины, которая получается если ^разделить теплоту гидратации ионов (значения которой, например, приведены в таол. 28 для щелочных ионов) па число молей связанной с данным ионом воды. Это происходит потому, что, кроме энергии связи, полученной за счет непосредственно присоединенных молекул, в энергию гидратации входит также энергия, выделяющаяся в результате ориентирующего действия иона' на расположенные в отдалений молекулы воды (за счет дипольного характера последних). Ианы гидроксония также оказывают ориентирующее действие ла окружающие молекулы воды. Для них, однако, при наличии большой энергии связи протона эта энергия ориентации численно едва, ли имеет значение.
По данным Вика (Wicke, 1954) ион JH3O]* в водных растворах гидратирован дополнительно, т. е. к нему присоединены еще и другие молекулы воды, прочность связи которых приблизительно соответствует прочности связи молекул воды с другими гидратированными: ионами, панример с ионами щелочных металлов, Согласно этому воззрению, ион гидроксония отличается особенно большой энергией связи молекул воды с протоном. По представлению Вика в водных растворах к нону [НаО]’ присоединяются еще три молекулы воды и образуется комплекс (наличие которого в кристаллизующихся соединениях было утке давно установлено, см. стр, 66,3), имеющий вид пирамиды [HaQJ+. В атом комплексе, по всей вероятности, избыточный протон является очень подвижным, так что он после образования комплекса оказывается не-фиксированным около определенной молекулы воды. К сформированной таким образом «внутренней» гидратной оболочке могут присоединяться еще другие молекулы воды, образуя «внешнюю^ гидратную ободочку. Протон, гидратированный одной молекулой .воды, называют ионом гидроксония.
* По мере разбавления перепое ионами химически связанной воды все сильнее перекрывается течением, которое гидродинамически образует вокруг себя деижущийся ион "(Uli ch, 1933). Уже в 1915 г. Ре шт обратил внимание на то, что имеется возможность гидродинамического переноса части воды.
Водород
101
Как в водных растворах с водой, так и в других средах растворенные вещества часто соединяются с растворителем в более пли менее слабо связанные продукты присоединения. Их называют общим назван нем сольваты (сюда включаются также существующие в водных растворах гидраты). Это явление называется сольватацtieй.
Водородный ион (ион гидроксония). Когда говорят о «водородном ионе» б водном растворе, то под этим всегда следует понимать гидратированный ион. водорода, т. е. ион гидроксония [НзОГ и соответственно |НВО4]+. Из всех электролитических ионов он имеет наибольшее значение. Многочисленные процессы, протекающие в водных растворах, в сильной степени зависят от концентрации в пих водородных ионов (точнее ионов гидроксония). Многие процессы каталитически ускоряются водородными ионами (ионами гидроксония), например, омыление эфира, инверсия тростникового сахара. Некоторые же реакции водородные ионы замедляют (например , упоминавшаяся ранее реакция разложения перекиси водорода). Особенно сильно зависят от концентрации водородных ионов процессы в живых организмах. Ничтожные изменения концентрации водородных ионов жидкостей, входящих в состав живых организмов, в очень сильной степени влияют на функцию ферментов, содержащихся в них, Поэтому в биологии приобрело большое значение определение концентрации водородных ионов. Водородные ионы осаждают многие коллоидные вещества. Это имеет значение также в аналитической химии; так, существует правило, что осаждение веществ, легко образующих коллоидные растворы, следует производить по возможности из кислых растворов.
Электролитическая подвижность водородных ионов далеко превосходит подвижность всех других ионов. Этим определяется высокая электропроводность сильно диссоциированных Кислот. При 18° электролитическая подвижность водородных ионов при бесконечном разбавлении составляет 315. Подвижность остальных ионов в общем лощит в пределах 40—70. Только гидроксил ион ОН' обладает значительной подвижностью, а именно 174. Максимальная подвижность водородных ионов, по-видимому, обусловлена частично тем, что они нс весь путь проходят в виде гидратированных ионов, а соседние молекулы воды обмениваются протонами II\ т. е. поп [НзОГ (соответственно ион [НвО4]*) отдает протон соседней молекуле воды и т. д. Кроме того, большая подвижность водородпых ионов также зависит от незначительной по сравнению с другими ионами их степени гидратации*.
Энергия, которая выделяется в результате присоединения протона ц молекуле воды с образованием иона [Н3О)+ , особенно велика. Она более чем в два раза превышает энергию, которая выделяется нри соединении двух атомов Н в молекулу Н2. Вследствие этого число свободных протонов Н+ в сильнокислых растворах особенно мало. В растворах 1 н. по отношению j< иолам гидроксония (Koltboff, 1930), количество их относится к тисну ионов [Н3О]’ как 1 : low.
Методы определения концентрации водородных ионов. Ввиду того что определение концентрации водородных ионов (концентрации ионов гидроксония) имеет для различных целей совершенно исключительное значение, здесь будут коротко рассмотрены наиболее нажные из этих методов. Все описываемые методы позволяют определить «кажущуюся концент
* Объяснение максимальной подвижности водород1ШХ ионов можно найтн у Wuiff Р., Z. Elektrochem., 47, 858, 1914, Kortum, Euck.cn, Z. Eiektro-chem., 52, 268, 1948, а также Wic.ke E., Z. physikaL Chem., (N.F.)., 1, 340, 1954,;
102
Глава S
рацию водородных ионов, а Йо истинную, поскольку «истинная» отличается от «кажущейся». Согласно сказанному па стр. 91 и далее, числовые значения, подучаемые для кажущейся концентрации, часто зависят от метода, которым они были получены.
Нельзя смешивать методы, применяемые для о пре деления концентра ции водородных ионов, с методами определения содержания «свободной кислоты» в растворе. Последнее определяется титрованием. В том случае когда раствор содержит только свободную кислоту и известна степень ее диссоциации, концентрация свободной кислоты указывает уже на концентрацию водородных ионов. Если же наряду со свободной кислотой присутствуют и ее соли, то концентрацию водородных ионов (соответственно активность ионов) можно в простейшем случае вычислить в результате правильного применения закона действия масс. Большей частью в таком случае производят непосредственное определение концентрации водородных ионов. Последнее особенно относится к случаям сложных смесей, с которыми приходится иметь дело при исследовании жидкостей, входящих в состав организма.
Для определения концентрации водородных ионов применяют главным образом следующие методы.
Методы 1—3 указывают актив, ноешь ионов водорода (точнее иона гидр он с он и я). Таким образом, они применимы к любым водным растворам. Напротив, найденные по методам 4 и 5 кажущиеся концентрации водородных ионов, нельзя просто отождествлять с активностями. Однако в очень разбавленных растворах или в растворах, не содержащих сильных электролитов, отклонения Столь малы, что ими практически можно пренебречь.
1. Потенциометрическое определение концентрации водородных ионов.
Этот метод основан на измерении разности потенциалов между омываемым струей водорода платинированным платиновым электродом, погруженным в раствор с neira-вестпым’ содержанием водородных ионов и «электродом сравнения» (см. рис. 6, стр. 50). Концентрацию водородных ионов (соответственно активность водородных ионов) вычисляют по уравнению (1) (см. стр. 40). Для точных измерений чаще всего применяют этот метод.
2. Колориметрическое определение концентрации водородных ионов (индикаторный метод).
«Индикатором» вообще называют такое вещество, которое дает указание относительно какого-нибудь исследуемого явления, в частности указывает концентрацию водород пых ионов в растворе. Индикаторный метод определения копцептрации водородных ионов оспован па том, что к испытуемому раствору добавляют какое-нибудь органическое красящее вещество, окраска которого изменяется в зависимости От концентрации водородных ионов. Сравнением цвета испытуемой жидкости, содержащей индикатор, с цветом контрольного раствора с тем же индикатором, но с известном концентрацией водородпых ионов, удастся определить концентрацию водородных ионов в испытуемой жидкости при помощи обычных колориметрических приемов. См. также гл. 18.
3. Определение концентрации водородных ионов по каталитическому действию ионов водорода.
Концентрацию водородных ионов в водных растворах можно также определять но каталитическому ускорению некоторых реакций, которое вызывают водородные ионы, К числу таких реакций относятся:
а) расщепление сложных эфиров на свободную кислоту и спирты. Эту реакцию контролируют титриваиием освобождающейся кислоты. Скорость разложения, при прочих рапных условиях, пропорциональна концентрации водородных ионов в растворе; ~
б) инверсия тростникового сахара {сахарозы}. В водпых растворах скорость этой реакции в общем пропорциональна концентрации водородных ионов. Ход инверсии, т, е. гидролитического расщепления тростникового сахара, на глюкозу и фруктозу,
Водород
ЮЗ
можно проследить простым поляриметрическим определением. Этим способом была определена «сила» очень многих кислот;
в) расщепление диазоуксусносо эфира. Эта реакция протекает в водном растворе также со скоростью, в общем пропорциональной концентрации водородных ионов. За ее скоростью легко следить, измеряя количество выделяющегося при этом азота.
На последние три реакции оказывают заметное влияние различные обстоятельства, например добавка к растворам нейтральных солом. Следует отметить, что характер этого влияния до сих пор полностью по выяснен. Поэтому пользоваться полученными результатами следует осторожно. Оказывается, что нейтральные соли и т. д. не оказывают значительного влияния, если учесть, что в Случае каталитического действия водородных ионов существенной является их активность. Последняя в растворах такого рода может сильно отличаться от «кажущейся концентрации», определяемой на основе классической теории (см. также Dubo«x, 1924).
4. Определение концентрации водородных ионов измерением электропроводности.
Удельная электропроводность раствора электролита определяется выражением:
(24)
где 4t, т|г, Т|з> И т. д.— концентрации отдельных видов ионов (в грамм-эквивалентах на 1 е.и3), a kj, [ьдцпт.д,- их электролитические подвижности. Этим соотношением можно пользоваться (введя ограничения, вытекающие из новой теории электролитов) для определения концентрации ионов какого угодпо вида, если известна их подвижность. Однако особенно оно удобно для определения концентрации водородных ионов, подвижность которых очень велпка и благодаря этому оказывает исключительно сильное влияние на электропроводность растворов. Естественно, должны быть известны иодвг/жностя и других ионов. При этом концепт рации этих ионов также должны быть известны, за исключением одной из них, так как требование, чтобы суммарная эквивалентная концентрация положительных ионов равнялась такой же концентрации отрицательных ионон является вторым методом их определения. Если раствор содержит, кроме водородных ионов, только один анион, то, чтобы определить концентрацию водородных ионов при известной подвижности последних, достаточно одного измерения электропроводности. Однако измерение электропроводности чаще применяют Для определения степени диссоциации кислот. Для слабых Кислот степень диссоциации а определяют по уде л т.п ой электропроводности •‘кс при эквивалентной концентрации с (=1000 1]) следующим образом;
где для Ап берется сумма электролитических подвижностей аниона и катиона при бесконечном разведении. Концентрация водородных ионов такого раствора кислоты, выраженная в грамм-эквивалентах па литр, равна с-ц. Таким образом, она получается из удельной электропроводности раствора в виде
100-х,. Ле
что следует также непосредственно из уравнения (24).
5. Определение концентрации водородных ионов из данных определения понижения температур замерзания и повышения температур кипения.
Степень диссоциации а слабых электролитов в соответствии с уравнением (19) или (19а) определяется на основании отклонений понижения температур замерзания и повышения температур кипения от закона Рауля — Вант-Гоффа. Для слабых кислот (принимая во внимав ио общую концентрацию) можно определить и концентрацию водородных иопов.
1
Глава 3
СПЕКТР ВОДОРОДА И СТРОЕНИЕ АТОМА ВОДОРОДА
Спектр водорода. Свечение водорода проще всего вызвать, заключив его под пониженным давлением: в стеклянную трубкуj снабженную электродами (трубку Гейслера), и пропустив через нее электрический ток высокого напряжения (например, при помощи индуктора). В большинстве случаев для этого используют трубки, подобные изображенным на рис. 16, впервые сконструированные Плюккером. Свечение особенно интенсивно в суженной части трубки. Если свет, испускаемый такой водородной трубкой, наблюдать через спектроскоп, то видно, что он состоит из очень небольшого числа резких линий, а именно одной красной, одной зеленовато-синей й двух фиолетовых. Эти линии обозначают буквами На, Up, HY и Нй. Основатели спектрального анализа Кирхгоф и Бунзен в 1860 г. впервые установили, что эти линии являются характеристическими линиями водорода-', Фраунгофер еще в 1814 г. открыл их в солнечном спектре как линии поглощения.
ir.it „ и . Удмми-
Рис. 1G. Трубки Плюккера.
синиц фимвмюи?
"а
7000 6500 6000 5500 5000 4500 4000 , 3500
Лиина волны, Л
Рис- 17. Спектр водорода (серия Бальмера).
Если пользоваться кварцевым спектрографом или, ещё лучше, спектрографом с диффракционной решеткой, то на фотопластинке можно обнаружить еще целый ряд характеристических линий водорода, расположенных в ультрафиолетовой части спектра. В этом случае получается спектр, показанный на рис. 17. Как видно из рисунка, при переходе от длинных волн к коротким отдельные линии все больше сближаются. При этом они ослабевают, пока, наконец, совершенно не исчезают.
Уже при простом рассмотрении этого спектра видно, что частота появления линий Должна подчиняться сравнительно простому закону. Впервые математически сформулировать этот закон удалось Бальмеру в 1885 г. Длииы волн всей совокупности линий можно выразить следующей простой формулой:
|-в-(44)- ”>2' (1)
Спектр водорода и строение атома водорода
105
где /?н — некоторая постоянная, а п — может принимать всецелочисленные значения, начиная от 3. При п~3 получается первая линия видимого спектра водорода Н„, при Пр, при n=z> — Hv и т, д. С увеличением п длина волны X все время уменьшается. Наименьшее предельное значение %, получающееся при и=со, в соответствии с этой формулой равно
Это значение определяет границу последовательности линий со стороны ультрафиолетовой части спектра. Впрочем, ее нельзя непосредственно определить по эмиссионному спектру, ибо по мере уменьшения длины волны линия спектра все более ослабевают и исчезают, прежде чем будет достигнута граница спектра.
Формула Бальмера чрезвычайно точна, как это видно из табл. 18. Длины воли Z выражены в ангстремах (А)-1А=0,1 .u|i=Kr8 с.«. Эмиссионный спектр водорода, получаемый в вакуумных трубках, удалось измерить до 25-й липни, а в спектрах звездпых туманностей — до 33-й.
Таблица 18
Первые линии в спектре водорода
н« И₽ HV н6
п = 3 п=4 п~5 к=6
^яабл> -А- 6562,80 4861,33 4340,47 4101,74
^•вычисл I 6562,80 4861,38 4340,51 4101,78
Константу 2fH, называемую константой, Ридберга, на основании спектроскопических измерений можно вычислить с' исключительной точностью, какой еще вообще не удалось достигнуть ни в одной другой области.
Значение атом постоянной таково*;
Я н = 109 677,581 ±0,007 см*
Серии и их термы. Спектр, который подобно рассмотренному спектру водорода состоит из ряда взаимосвязанных линий, называют серийным спектром, или, короче, серией. Только что рассмотренный спектр называется серией Бальмера.
Кроме серии Бальмера, водород имеет еще несколько серийных спектров; один целиком лежащий в ультрафиолетовой области — серия Лаймана и три в инфракрасной — серия Пашена, серия Брэкетта и серия Пфунда. Эти серии описываются формулам^, совершенно аналогичными формуле Бальмера.
Серил Лаймана т=л» ( < 1 п = 2, 3, 4
Серия Пашена г=л» ( < 1 1 п = 4, 5, 6
ч З2 ’
Серия Б рэкетта т=М < 1 Ч б2 пй ) п = 5, 6 .
Серия Пфунда т=М п2 ) ге = 6.
Константа имеет то же значение, что н для серии Бальмера, четыре серии можно представить одной- формулой
Бее
(3)
nz— целые числа, ni>n2).
* Ср. Birge В. Т., Phys. Rev., [2], 60, 766, 1941.
100
Глава 3
В соответствии с этой формулой длину волны любой: линии в спектре водорода можно представить в виде разности двух величин, имеющих вид (п — целое число). В спектроскопии выражение такого вида называют термом. Длины волн одной серии мощно получить, если в уравнении (3) «а считать равным постоянной величине, а zzi придавать ряд .целочисленных значений.
Поэтому в разности
1 _ЛИ Дн Л М
первый член называют постоянным термом, а второй — переменным. Граница серии определяется постоянны-^ термом.
Линейчатые и полосатые спектры. Кроме линейчатого спектра, водород имеет еще и полосатый спектр. 0s возбуждается при. разряде в трубке Гейслера под небольшим напряжением. Полосатые спектры отличаются от линейчатых тем, что при наблюдении их в спектроскопе с низкой разрешающей способностью опи имеют вид однородных полос. В действительности и эти полосы состоят из отдельных линий, только они очень близко расположены. Однако их расположение принципиально отлично от расположения линий в «линейчатом спектре». Вообще полосатые спектры приписывают двух- или многоатомным молекулам, а линейчатые спектры — свободным атомам. Например, у паров иода можно наблюдать, как при повышении температуры характеристический (абсорбционный) полосатый спектр паров иода исчезает в той же степени, в какой происходит диссоциация молекул па атомы I. Точно так ;ко в случае водорода полосатый спектр приписывают молекулам водорода, а линейчатый — атомам водорода.
СТРОЕНИЕ АТОМА ВОДОРОДА
Теория строения атома Резерфорда.' Уже давно предполагали, что испускаемый атомами свет может дать сведения о строении атома. Электромагнитные волны, к которым относится и. свет, как показывает опыт, всегда являются следствием колебания электрических зарядов. Поэтому логично искать причину испускания света в колебаниях электрических зарядов или по крайней мере в аналогичных или связанных с колебаниями процессах. Электричество, как и материя, имеет дискретную природу. Наименьшие частички свободного отрицательного электричества называются электронами. Отношение массы электрона к массе легчайшего атома, атома водорода, равно 1 : 1837 (ср. стр. 135). В нейтральном атоме должно быть равное число электронов и положительных зарядов. Раньше считали, что положительное электричество распределено по всему объему атома, а электроны, так сказать, «плавают» в нем (томсоновская модель атома). Однако такая гипотеза не-могла удовлетворительно объяснить движения электронов, проявляющееся в испускании света. Эта проблема получила совершенно иное объяснение, когда в 1911 г. Резерфорд, пытаясь истолковать результаты исследования отклонения а-л у чей при прохождении их через вещество, проведенного Гейгером и Марсденом, пришел к выводу, что положительный заряд каждого атома должен быть сконцентрирован в очень малой области внутри атома*. Эта область атома называется ядром. Диаметр ядра атома водорода не превышает 2 -10'13 см, ядра атома золота —не больше ЗЮ"12 см, тогда как диаметр атома, рассчитанный, например, по плотности кристаллических веществ (ср. гл. 7), имеет порядок величины 10-е см.
* Уже в 1903 г. Ленар на основании своих опытов по исследованию прохозкдепия катодных лучей Через вещество пршнсл к выводу, что положительные заряды вс могут быть непрерывно распределены по всему объему атома.
Спектр водорода и строение атома водорода
107
Теория строения атома Резерфорда оспована на том наблюдении, что при прохождении через вещество а-лучи в общем отклоняются очень незначительно, но в отдельных случаях наблюдаются чрезвычайно сильные отклонения. Это можно объяснить только тем, что в отдельных случаях а-лучи проходят в непосредственной близости от центров электрических сил, в которых сосредоточен весь положительный заряд атома. В силу закона Кулона а-чаетицы должны отталкиваться таким положительным зарядом, поскольку они сами заряжены положительно. Необычайно большая сила отталкивания, необходимая длятого, чтобы направить быстро летящие н относительно тяжелые а-чаотицы по совершенно иной траектории, может возникнуть только в том случае, если положительный заряд атома сосредоточен почти в точке. Точным математический расчет, проведенный па основании экспериментальных данных, дает приведенные выше максимальные значения объемов, которые должны занимать положительные заряды, чтобы наблюдаемые сильные отклонения а-частпц можно было объяснить кулоновским отталкиванием.
.Лежащий в основе расчета взаимодействия^ электрических сил закон Кулона гласит: сила, о которой два электрически, заряженных тдла действуют одно на другое., пропорциональна произведению их электрических зарядов и обратно пропорциональна квадрату расстояния между ними. Если электрические заряды представить в Такой системе единиц, чтобы коэффициент пропорциональности был равен единице, то закон Кулона можно выразить следующей формулой:
К=^^, (4)
где К — сила, с которой электрические заряды <?t и действуют один на другой, а г — расстояние между ними. Этот закон был экспериментально найден Кулоном в 1785 г, Легко показать, что если электрический заряд распределен равномерно по поверхности шара, то он действует так, как если бы он был сосредоточен весь в центре шара. Если знаки зарядов одинаковы (заряды одпоимепны), то они отталкиваются, если их знаки противоположны, то заряды притягиваются. Таким образом, К является силой притяжения, если опа отрицательна. Уравнение (4) справедливо в том случае, когда заряды находятся в вакууме. Если заряды расположены в среде с диэлектрической проницаемостью D, то силу взаимодействия надо разделить па D.
Работа А, которую надо выполнить, чтобы в пустоте сблизить из бесконечности электрические заряды и е2 на расстояние г, равна
как это следует из закола Кулона, если проинтегрировать силу по всему и ути. В случае разноименных зарядов работа А будет отрицательной. Это значит, что при сближении заряды выполняют некоторую работу. В каждом случае А означает потенциальную энергию (способность производить работу) системы, состоящей из двух зарядов. Работа, требующаяся, чтобы приблизить единичный заряд к данной точке в поле заряда е, называется потенциальном энергией или потенциалом заряда в в дайной точке.
Теория Бора. Так как поп водорода всегда имеет заряд, равный единице, можно предположить, что заряд его ядра также равен единице. Если это так, то нейтральный атом водорода должен состоять из ядра, имеющего массу 1,008146 по отношению к массе атома кислорода, принятой за 16*, и несущего единичный положительный заряд, и одного электрона (т. е. единицы отрицательного заряда) с массой 0,000549. Согласно закону Кулона, электрон будет притягиваться ядром, и от падения на ядро его должна удерживать некая противоположная сила отталкивания. Примем (Бор, 1913), что электрон вращается вокруг ядра, а атом находится в равновесии благодаря тому, что кулоновскому притяжению противодействует центробежная сила (рис. 18).
Обозначим заряд ядра /?, заряд электрона —е (в данном случае Е =е, но это совершенно пе будем принимать во внимание), тогда, поскольку центробежная сила равна произведению массы т, радиуса г и квад
Точнее, по отношению к массе изотопа кислорода Oia-= 16.
108
Глава 3
Рис. 18. Электрон е, вращающийся вокруг ядра Е.
рата угловой скорости ш, условие равенства центробежной силы и кулоновского отталкивания запишется следующим образом:
тг<й2— -—-=0 или Ё е = т. г3 W2; (6)
Этому условию могло бы удовлетворять любое значение г, определяемое только скоростью вращения электрона “ Но по теории Максвелла ~ _ которой любое изменение скорости или направления
движения электрически заряженной частицы связано с испусканием электромагнитной энергии) такая система не была бы устойчивой. Если бы электрон излучал энергию, то это могло бы происходить только за счет его кинетической и потенциальной энергий, первая из которых определяется квадратом скорости электрона, а вторая—его расстоянием от ядра; это значит, что электрон должен описывать спиральную сужающуюся траекторию и в конце концов должен упасть на ядро. Поэтому Бор отбросил это следствие теории Максвелла и заменил его постулатом: существуют определенные орбиты, на которых электрон может вращаться без потери энергии (н вследствие этого, конечно, без испускания света), и это должны быть такие орбиты, для которых момент вращения равен целому кратному величины h/2n.
Моментом вращения называют произведение .количества движения (рапного произведению массы на скорость и называемого также импульсом вращающегося тела) па ; его расстояние' от центра вращения. Величина h — константа, известная в Квантовой теории под названием «кванта действия» Планка. Ее апачепие равно (5,6252-10"2’ эрг-сек.
Изложенный выше постулат называется первым квантовым условном Бора. Его можно сформулировать так:
h p — ri -д—,
где р —;момент вращения, п — любое целое число — квантовое число орбиты, по которой движется электрон. Если учесть, что
р ~ т v г — т г2 • о, то получим
nh > IS\
—— —mfts. (7)
2л
а если разделить уравнение (6) на уравнение (7)
2цЕе v „
—=™’ (8)
и подставить результат в уравнение (7), то получится
В соответствии с этим уравнением возможны только дискретные орбиты. Радиусы их относятся как квадраты целых чисел, которые, как уже упоминалось, называются квантовыми числами соответствующих орбит. Например, радиус орбиты с квантовым числом п = 3 получается из урав
' Спектр водорода и строение атома водорода 10,9
нения (9), если в него подставить п=? ЗС Согласно теории Бора, электрон, движущийся по этой орбите, ле должен испускать свет. Испускание света должно происходить тогда, когда электрон «перескакивает» с одной орбиты па другую с меньшим квантовым числом. Частота этого света определяется вторым квантовым условием Бора, так называемым частотным уравнением. Это условие формулируется так: разница энергий исходной и конечной орбит, равна произведению кванта действия на частоту
Bi—e2=hvt ’ (10)
где е, — энергия атома в исходном состоянии; е2 — энергия атома в конечном состоянии; v — частота =еА, где — длина волны света, а с скорость света.
В зависимости от того, больше или меньше энергия атома в исходном состоянии ио сравнению с энергией в конечном состоянии, уравнение (10) определяет частоту излученного или поглощенного света. Справедливость этого уравнения была доказана прямым измерением энергии, требующейся для возбуждения атома для определенного излучения (ср. стр. 137).
Энергия вращающегося вокруг ядра электрона складывается из кинетической энергии eJiTI[[ и потенциальной энергии ецот
8 — Ъкпя “г еяот-
Кинетическая энергия равна половине произведения массы электрона т на квадрат его скорости V.
екщ[—1/ум2=4-у--(о (по уравнению (7)].
Подставляя уравнение (9) в (8), получим
8л3я1е?.Е2
«ЭЙЗ ’
гак что
nh 8хдте2Е2 Za^me^E2 (да-= (да-
Потенциальная энергия е!10Т равна (ср. стр. 107)
еЕ Лп2те2Е2 у
еяит=------------_екий.
Таким образом, общая энергия равна
. . _ ... _ 2л2те?£а
е— екин + ьпот— еКИН -----' ‘1г’
Отсюда следует
А ” А3 <4 nlJ' (
В этом уравнении п± представляет собой квантовое число исходного состояния, а иг — квантовое число конечного состояния атома [п в уравнении (9)].
Если ввести число зарядов ядра Z^Eie, а для упрощения выражения величину IE, определяемую уравнением
2 тт
••———=c-Rm (с—скорость света),
но
Глава S
то ^получим, если еще учесть, что v = —. 1
Л
' , (13)
13 случае водорода Е=е, т. е. 2 = 1. Вычислив*
_ 2л®'Пв4 h?,c ,
получим точную формулу, определяющую положение линий в спектре водорода [ср. уравнение (3) на стр. 105J, а именно
Jl = 1,097-105 fX--*\ - (14)
Уравнение (14) совпадает с уравнением (3) не только формально, не теория Бора дает правильное значение и для константы Ридберга с той точностью, с которой известны величины eim, е и h.
При точных вычислениях следовало бы учитывать движение ядра .при вращении вокруг пего олектропа. Строго говоря, так же как и Луна движется не вокруг Земли, находящейся в покос относительно Луны, а Земля и Лупа движутся вокруг общего центра тяжести, так и ядро и электрон в атоме движутся вокруг общего центра тяжести. Расстояние х ядра и у электрона от общего центра тяжести определяется отношением масс ядра и электрона: у : х=М : т. При учете движения ядра в уравнение (11) вместо ш войдет выражение т-ЛТ/ЛГ^-гП-1 и, следовательно, в уравнении (13) д/
вместо 7?ж появится член 7?со — -, так что точпо ато уравнение будет выглядеть так;
1 "~г— т
Т = У Y ' (13а)
л х. »•> л’ / '
Таким образом, значение константы Ридберга в случае водорода Лн будет равно
точно
М по будет
ядра.
37jj+.
выражению Явд--п----------> где Ун+ — масса ядра атома водорода. Чем больше
сравнению е т, Тем меньше величина, вычисленная с учетом движения ядра, отличаться от соответствующей величины, подученной без учета движения ЗТН+
Дли атома водорода -у-—=0,9994557 (ср. стр. 133). Точное значение 7?ср
равно 109 737,309 ± 0,012 сж"1 (Birge 1941; Du Mond u. Cohen, 1953). Отсюда получим: Лп=0,9994557 Лос=109 677,58 сж"1, что превосходно согласуется с величиной, приведенной на стр, 105.
На основании только что развитых представлений Бора возникновение различных серий в спектре водорода объясняется следующим образом.
Электрон, который может вращаться вокруг ядра атома водорода, не испуская света по орбитам, радиусы которых относятся между собой как Is :28:За:и т. д., обычно находится на самой внутренней орбите («=!), поскольку‘в этом случае энергия атома, согласно уравнению (И), минимальна. [Радиус самой глубокой орбиты получается из уравнения (9), если и него подставить п = 1, равным 0,5292' Ю’8с,м.) Если атом получает энергию (за счет удара или излучения), то электрон может подняться на более высокую орбиту. Допустим, что он поднялся па третью орбиту. Если дополнительная энергия сообщается излучением, то атом поглощает
* Значение h приведено на стр. 108; е представляет собой так называемый элементарный квант электричества, равный А,8028-К)-1<> электростатических едиппц (эл. ст. ед.). Кроме того, известно отношение в ; т = 1,7589'10’ж, где с — скорость света (= 2,997ЙЗ-10“‘° СЛ(/йек).
Спектр водорода if- строение атома ео^ороЗд
111
при этом квант света, частота которого^ или длина волны, определяется соответственно уравнением
е2 —В( 1 „ / 1 1 Л
V=^7T- ,fJlu
В состоянии с более высокой энергией, или, как говорят, в «возбужденном» состоянии, атом находится очень короткое время. В среднем уже через 10“8—10“* сек электрон перескакивает, испуская свет, на глубже расположенную орбиту (см, рис. 19). В рассматриваемом случае он может непосредственно возвратиться на первую орбиту. При этом он испускает
Рис. 19. Происхождение различных серий в спектре атома водорода по теории Бора.
свет, соответствующий второй линии в серии Лаймана, длина волны которой определяется выражением
1 /гн /и
X, I2 З2 J
Но электрон может сначала перескочить на вторую орбиту н только после этого на основную орбиту. В этом случае он испустит одну за другой сначала первую линию в серии Бальмера
4=4^) •
а затем первую линию в серии Лаймапа
Отсюда видно, что электронные переходы, которым 'соответствуют линии, принадлежащие к одной и той же серии, всегда, имеют общую конечную орбиту. Например, Длн лини и серии Бальмера это вторая орбита, для линий серии Лаймана — первая, или основная орбита. На рис. 19 представлены переходы электронов с одной орбиты на другую и указаны соответствующие этим переходам эмиссионные линии. При обратных
112
Глава 3
Рис. 20. Электрон на эллиптической орбите (На орбите S»).
переходах возникают соответствующие липни поглощения (абсорбционные липин).
Число п в каждом терме является квантовым числом орбиты, соответствующей этому терму.
Главные и побочные квантовые числа. Для простоты сначала примем, что электроны движутся вокруг ядра по круговым орбитам. Однако форма орбит планет показывает, что движение вокруг центра притяжения может происходить п по эллиптическим орбитам. Теория такого вида движения электрона вокруг ядра атома в 1915 г. была развита Зоммерфе льдом. Если для построения круга нужно задать только одну величину, например радиус, то для построения эллипса необходимо знать два параметра, например большую и малую оси. Вместо одного квантового числа первоначальной теории Бора в теории Зоммерфельда фигурируют два квантовых числа п и называемые главным п_ побочным квантовыми числами. Главное квантовое число п определяет боль
шую полуось эллипса (см. рис. 20) совершенно аналогично тому, как в случае круга это число определяет его радиус [см. уравнение (9)1-Таким образом,
--£7- (15)
Малая полуось эллипса определяется условием
4-А <*»»>.. ; . «в)
При /с=ц и Ь—а, т. е. электрон движется по круговой орбите. При к. < п электрон движется ио эллипсу, эксцентриситет которого тем больше, чем меньше к по сравнению с п. Обычно орбиту, характеризующуюся главным квантовым числом п и побочным квантовым числом к, для краткости обозначают как пк-орбиту, например, показанная на рис. 20 орбита с главным квантовьш числом п=3 ц побочным квантовым числом А=2 обозначается как о2-орбита.
Если на атом не действует внешнее ноле (например, сильное магнитное поле) п пе учитывается зависимость массы электрона от его скорости, что следует из теории относительности (см. в дальнейшем), то энергия атома водорода и по теории Зоммерфельда определяется исключительно главным квантовым числом. Так,что одна и та же линия, наиример красная линия водорода (А=65б2,8 Л) возникает независимо от того, переходит электрон с орбиты З3 на орбиту 22, или с орбиты За на орбиту 21, или с орбиты 31 на орбиту 2а.
Квантовые числа не только определяют в силу уравнения (15) и (10) радиусы орбит, по которым, согласно представлениям, лежащим в основе теории Бора, движутся электроны, но и, как видно из уравнения (11), определяют энергетические состояния, в которых может находиться атом. Энергии этих состояний, как следует из уравнения (11), обратно пропорциональны квадратам соответствующих главных квантовый чисел. Поэтому возникновение различных линий в спектре атома водорода вместо того, чтобы связывать их с переходами электронов с одной орбиты на другую,
Спектр водорода и строение атома водорода 113
как показано на рис, 19, можно объяснить переходами атома из одного энергетического состояния (энергетического уровня) в, другое (эти переходы показаны: на рис. 21). В дальнейшем будем пользоваться исключительно последним способом представления, так как, согласно волновой
Р и с, 21. Энергетические уровни атома водорода.
Энергии атома! водорода, соответствующие различным квантовым числам (и=1, 2, 3 и т. ДО, выражены в плектр он-вольтах. О значении этой единицы энергии и связи квантового числа -п-=г» с ра Сотой ионизации ом. стр. 130 и сл.
механике (см. ниже), понятие об определенных электронных орбитах отброшено, тогда как «энергетические уровни», определяемые уравнением (11), сохраняют свой смысл и в новой теории.
Повышение точности спектроскопических измерений дало возможность показать, что каждая линия серии Бальмера в: действительности состоит из нескольких, по крайней мере двух, линий, длины волн которых, впрочем, различаются чрезвычайно мало (для Яа-.чинии разница составляет только приблизительно 0,05 А, т. е. Менее 0,001% длины волны). Зоммерфельд показал, что теория Бора может в принципе сама объяснить тонкую структуру спектра. А именно,, если учесть (допустив, что Орбиты электронов являются эллипсами) ядваеи-иость массы электрона от его скорости, что следует из теории относительности, то для энергии атома получается уравнение, в которое входит и побочное квантовое число ft. Поэтому энергетические уровни на рис. 21 расщепляются на подуровни. Хотя эти подуровни л лежат чрезвычайно близко один к другому, переходам З3 —> 22, 3, ж>- 2Ь З^ —> 23 и т. д. [на основании уравнения (10)] отвечают теперь уже не одни и те же частоты, или длины волн. Оддако теория Бора — Зоммерфельда не дает количественного объяснения наблюдаемым фактам. Особенно ясно недостаточность количественной стороны этой теории проявляется при попытках приложить ее к более тяжелым атомам.
8 г. Реми
114 Глава 3
Недостаточность теории БорЛ. Волновая механика и квантовая механика. Несмотря на то что теория атома водорода Бора превосходно согласуется с данными спектроскопии для серии Бальмера и аналогичных серий в спектре атома водорода, всё же оказалось, что область применения этой теории ограниченна. Например, она совершенно не может предсказать поведение атома водорода в магнитном поле. Не удалось также объяснить с позиций теории Бора образование молекулы водорода.
Эти затруднения, по-видимому, имеют принципиальный характер. Точно так же как геометрическая оптика, не учитывающая явлений дифракции, принципиально пе в состоянии объяснить существования предела разрешающей способности микроскопа. Указанные выше трудности можно преодолеть с позиций более широкой (универсальной) теории, а именно теории, основанной на теории волн вещества, так называемой волновой механики. Основы волновой механики заложены в 1924 г. де-Бройлем, а вскоре после этого (в 1926 г.) Шредингер использовал ее для построения теории атома водорода. В соответствии с этой теорией движение материальных частиц, например электронов, описывается волновыми уравнениями, совершенно аналогично тому, как в волновой теории света описываются световые лучи*.
Первое квантовое условие Бора, появляющееся в его теории как совершенно произвольная гипотеза, оказывается с точки зрения волновой механики неизбежным следствием общих законов, справедливых для волновых систем. Аналогично и второе квантовое условие Бора является следствием законов волновой механики. Для спектра атома водорода получаются те же уравнения, что и в теории Бора, но, кроме того, волновая механика объясняет симметрию атома водорода, соответствующую его поведению в магнитном поле, даст удовлетворительное объяснение образованию молекулы водорода, и, наконец, рассчитанные с ее помощью потенциалы ионизации тяжелых атомов более согласуются с экспериментальными данными, нежели полученные па основе теории Бора.
Результаты, совершенно аналогичные следствиям волновой механики, получаются при использовании квантов ой механики, основы которой заложены работами Гейзенберга, Борпа и Иордана (1925). В этой теории, к сожалению при отказе От наглядности, анализируются математический уравнения, связывающие непосредственно наблюдаемые в атом ной) физике величины, в частности Частоты и интенсивности характеристических спектральных липий атомов. Удалось показать, что квантовая механика и вол новая механика математически эквивалентны, т. е. уравнения одной теории можпо* непосредственно получить из уравнений другой путем чисто математических преобразований. Но в отличие от квантовой механики волновая механика исходит из более или мепее наглядных представлений. Поэтому и ео выводы о строении атомов можно наглядно интерпретировать.
Но все же наглядность атомпой модели, даваемой волновой механикой, не столь непосредственна, как представления о строении атома Бора — Зомморфсльда. Поэтому в дальнейшем будем по возможности пользоваться очень наглядными представлениями теории Бора. Это можно сделать — если только учитывать границы применимости этой теории — с тем же правом, с каким обычно пользуются представлениями геометрической) оптики для объяснения принципа действия системы линз, папример телескопа и ли* микроскопа. В этих случаях обычно пользуются представлением о «световых лучах» как бесконечно топких линиях, хотя и известно, что тогда нельзя объяснить всех оптических явлений и что (если, например, какую-то роль играют явления интерференции) надо пользоваться представлениями волновой теории света, правда тоже наглядными, но более сложными по сравнению с представлениями геометрической оптики. Так что в дальнейшем будем использовать наглядные представлен ня о строении атома, содержащиеся в теории Бора (и в ее дальнейшем развитии), если только эти представления не противоречат выводам волновой механики.
----- ---------- . \
* С точки зрения волновой маханнкн давний спор между корпускулярной и волновой теориями света не имеет смысла.
Спектр водорода и строение атома водорода 115
Однако н химии встречаются очень важные проблемы, решение которых невозможно в рамках теории Бора — Зоммерфельда. В таких случаях нельзя отказаться от применения результатов волновой механики. Это особенно важно для вопросов, относящихся к проблеме химической связи. Чтобы сделать понятными существо этих проблем и те решения, которые для них найдены, ниже будет дано краткое изложение основных положений волповой механики и ее приложения к теории строения атома.
СТРОЕНИЕ АТОМА ВОДОРОДА С ТОЧКИ ЗРЕНИЯ ВОЛНОВОЙ МЕХАНИКИ
Для линий в спектре атома водорода волновая механика дает те же длины волн, что и теория Бора — Зоммерфельда. И все же представления о строении атома водорода, к которым приходят на основе волновой механики, очень существенно отличаются от представлений теорий Бора— Зоммерфельда. По Бору, в основном состоянии атома электрон движется вокруг ядра по круговой орбите. Поэтому атом должен иметь круговую симметрию. Согласно волновой механике, атом водорода обладает шаровой симметрией, причем пе только в основном состоянии, но и в таких «возбужденных» состояниях, в которых, по Зоммерфельду, электрон движется по эллипсам с больший! эксцентриситетом, а именно по орбитам с побочным квантовым числом fc=l. Эксперимент подтвердил правильность представления о строении атома водорода, созданного на основе волновой механики. В дальнейшем будет показано, что такие представления неизбежно следуют на положений этой теории, а также будет разъяснено, какой смысл надо вкладывать в эти представления.
Основные положения волновой механики. В соответствии с волновой механикой с каждым движущимся электроном (и вообще с каждой движущейся материальной частицей) связан волновой процесс, длина волны которото X определяется соотношением ।
где h — квант действия Планка; т, — масса; и — скорость материальной частицы. Это лежащее в основе волновой механики допущение, высказанное первоначально как гипотеза де-Вройлем, позднее было подтверждено опытами, в которых было показано (впервые Дависсопом и Джермером в 1927 г.), что при прохождении катодных лучей, т- е. движущихся электронов, через вещество возникает дифракционная картина, аналогичная дифракции рентгеновских лучен (ср. гл. 7). Аналогичные дифракционные явления в случае атомных и молекулярных пучков были впервые экспериментально обнаружены Штерном (1929).
«Волны материи» распространяются в пространстве со скоростью в, которая дастся уравнением
ио = еа (18)
где v —т скорость частицы; с — скорость света. 1
Частота v волны вещества связана, как и в случае любой волны, со скоростью распространения н и длиной волны А соотношением
u = vX.
Уравнение колебания и волновое уравнение. В простейшем случае колебание описывается уравнением
1 y = asin(2nvt)« (19)
8*
116
Глава 3
Здесь а — амплитуда колебания, т. е. — если для наглядности принять, что это уравнение описывает колебание некоторой частицы, обладающей массой,— максимальное смещение частицы из положения равновесия в процессе колебания*; у —смещение из положения равновесия в момент времени t иг-частота (число колебаний в секунду).
Уравнение (19) справедливо, например, для колебаний маятника. Но это же уравнение описывает й колебания струны, или периодические изменения плотности воздуха в звуковой волне, или электромагнитные колебания при условии, что рассматриваются простые колебания, т. е, такие, которые происходят с одной частотой -и постоянной амплитудой.
Уравнение (19) справедливо во всех случаях, когда сила, выводящая колеблющуюся частицу из положения равновесии, в каждый момент времени пропорциональна смещению частицы из положения равновесия. Это значит, что в любой момент времени ускорение частицы пропорционально у, т. е. если коэффициент пропорциональности обозначить <н3, справедливо уравнение
d2y
Знак минус появляется в правой части уравнения потому, что сила действует в направлений, противоположном смещению у. Колебания, для которых справедливо уравнение (20), называются гармоническими колебаниями.
(20)
-Р
Р и с. 22. Графическое представление функции у = а sin <at (келе-, \ 2я ,
ванне); со•= — (т — период колебаний).
времени принять момент прохождения частицей
аю • cos tiil,
a®3 sin ati — — <эгу.
Если за начальный момент ] ± _ ^ ... .
пол оженил: равновесия, т. о. считать t—0 при у=0, то решение этого дифференциального уравнения будет иметь .вид
у—a sin. mt. : (21)
Здесь а — некоторая постоянная величина, физический смысл которой будет ясоп из последующего изложения.
То, что уравнение (21) является решением уравнения (20), можно показать, дважды дифференцируя уравнение (21):
dy __ d (sin uii) dt~a dt
<№y d (cos rot) , aa> dt =~
Выражаемая уравнением" (21) связь между у и t графически показана на рис. 22. Из рисунка видно, что колеблющаяся точка проходит положение равновесия (у=0) через определенные равные промежутки времени, причем при этом постоянно меняется направление движения (вверх и вниз). Время между одним прохождением через положение равновесия и ближайшим следующим прохождением через это положение в том же направлении называемся продолжительностью колебания, или периодом колебания. Если обозначить его через г, то из рис. 22 видно, что y=sincot принимает нулевые т
значения при t=0, t=: — , t—т, i —унт. д. Так как в этом случае sin <at=O при <ot=O, cot—л, cot—2л ит. д., если cot измерено .в долях угла, то отсюда следует сот=2л, иди, если заменить т на обратную ей величину — частоту т; то получим w=2jtv. Иод-
* Положением равновесия считают то, которое приняла бы частица, если бы внешние силы не вынуждали со колебаться. В процессе колебания скорость частицы достигает максимального значения в момент прохождения положения равновесия.
Спектр водорода и строение атома водорода 117
ставляя это соотношение в уравнение (21), подучаем уравнение (19). Этим доказывается, что при данных условиях уравнение (19) описывает колебания материальной точки около положения ее равновесия.:
Двукратным дифференцированием уравнения (19) или непосредственной подстановкой 2лу вместо св в уравнение (20) полупим
(22)
Это уравнение называется уравнением колебаний в отличие от рассматриваемого ниже волнового уравнения.
Наибольшее положительное значение у достигает в моменты времени г=1Дт, t = =т-ру4т и т. д. Это непосредственно видно на рис. 22,Аналитически такой результат получается из уравнения (19), если для sin (2nvf) взять наибольшее значение, которое может принимать синус, т. е. 1. Этого значения sin (2лv<) достигает в момент времени ts=i/4t=i/4 v;
sln(2jtv4) = sin-^?t—=sin ~ = i.
' 4v 2
Подставляя этот результат в уравнение (19), получим у=а. Поэтому а представляет' собой наибольшее возможное в данном случае значение у, т. е, амплитуду колебания.
Если колебательное движение может происходить в пространстве, то возникает волна. Длина волны; т, е. кратчайшее расстояние в простран-
*
Р а с. 23. Графическое представление функции
» 2Ла: . .
у = a sui-—r~ (еялна)* А
стве между двумя точками, находящимися в одном и том же состояний колебания, определяется соотношением Х = где и — скорость распространения волны (скорость волны), а у — частота колебаний (число колебаний в секунду). Скорость волны в определепнбм^направлении, например в направлении оси л1, дается уравнением ы== у . Это соотношение справедливо при условии, что скорость волны и постоянна. Подставив в уравнение (19) t— , получим
у = а sin
Это уравнение, которое можно записать также в другом виде
2п
у —a sin -т— х, Л-
описывает распространение колебаний в направлении одной из осей системы ноорди. нат и полностью:совпадает с уравнением протекания колебаний во времени. Графи
118
Глава 3.
чески волна изображается по обе стороны начала координат (рис. 23)*. Этим подчеркивается, что в общем случае колебания могут распространяться но только в одном направлении в пространстве, но одновременно и в противоположном. Как и в урав-нении (19), постоянная а в уравнения (23) обозначает амплитуду.
Двукратным дифференцированием уравнения (23) получаем
ДТ2 \ к
ИЛИ
)^(^-)-(^)‘>
^L+^Ly = <i, (24)
Это уравнение справедливо для линейного распространения коле^ банки. Поэтому' оно называется линейным волновым уравнением.
Можно легко показать, что в любой момент времени анергия (сумма кинетической и потенциальной энергий) колеблющейся материальной точки пропорциональна квадрату амплитуды, ее колебания. Это справедливо, например, для колебаний маятника или струны. В случае колебаний иного типа, например для световых и звуковых волн, квадрат амплитуды является мерой некоторой величины, называемой интенсивностью колебаний.
Общее решение уравнения колебаний. Уравнение (21) не является единственным решением дифференциального уравнения (20). Для других «граничных условий», а именно если положить, что при (=0 смещение у равно не нулю, а у—а, решение имеет вид y=<t cos<at** .Общее решение, т.е. решение, не зависящее от «граничных условий», получим, если Припять у = се“!, где е — основание натуральных логарифмов, а а — первоначально неизвестная постоянная величина. Тогда из уравнения (20) следует
или aeat (a--)J<i)£) =0.
Чтобы это уравнение выполнялось, т.е. чтобы уравнение j/==acfli являлось решением уравнения (20), должно быть
a2^-ti>-=0 или (i = V'r — l).
В этом случае соответствующие решения таковы:
у< = ар>+™\ и y2 = a2e~ial‘.
Согласно известной математической теореме (теореме Муавра), справедливо соотношение eis=cos a+i sia, г. Поэтому можно также написать
T/13sai(cose>^4~i sinwO; у2=а2 (cos wt—i sincot),
где yt и y2~независимые решения дифференциального уравнения (20). Сумма этих решений
у = (а14-а3) cos wt^(af—^Jisin
также является решением уравнения (20). Если учесть, что ы = 2лл’, и считать, что я14~«2==Л1 и i(ai—.аА = А2, то получим-
у—Ai cos 2rtvf А2 sin 2лхЧ. (25)
Эею ураенекие представляет собой общее решение уравнения колебании (S2). Здесь A j и А2 два (в общем случае комплексных) амплитудных множителя, числовые значения которых определяются граничными условиями конкретной физической задачи.
Уравнение колебаний в пространстве, В уравнении (22) у представляет собой переменную, зависящую только от времени t. Однако форма
* Это можно было бы сделать и на рис. 22, так как уравнение (19) не содержит никаких сведений о том, в какой момент времени началось колебание. Точно так же как а: в уравнении (23), t в уравнении (19) могло бы иметь как положительные, так к отрицательные значения.
** В этом легко убедиться, дважды дифференцируя уравнение, представляющее собой решение данного дифференциального уравнения. ;
Спектр водорода и. строение атома водорода 119
уравнения колебаний не изменится, если вместо у ввести некоторую функцию, которую можно обозначить через зависящую не только от t, но и от трех пространственных координат х, ц и г. В таком случае надо только заменить обычную производную частной производной. Уравнение колебаний в пространстве имеет вид
(26)
Его общим решением является 1см. уравнение (25)] уравнение
4r = Aicos2nvf-[-A2sm2nvC (27)
Здесь Ai и Аг представляют собой, как и в уравнении (19), амплитуды, колебаний. Однако они не являются постоянными величинами, а зависят цт х, у и z {или, как говорят еще, являются функциями х, у и я).
Интенсивность колебаний (см. Стр. 118) в этом случае определяется соотношением
|»К|г=тр.»Р% (28)
Здесь [ Т | — «абсолютное значение»у, т. е. величина Чг без учета знака; ф* — величина, комплексно сопряженная с Т *.
Пространственное волновое уравнение. Пространственное волновое уравнение можно получить, если в уравнении (24) переменную у, зависящую только от х, заменить функцией всех трех пространственных коор-, d2y
динат ф, а производную заменить суммой
даф , ааф д3ф ** дх? 1 ду% бг3
Тогда пространственное волновое уравнение будет иметь вид
/29) dz2 dyt Т йгг 3- ц2 т
Уравнение, одновременно выражающее зависимость колеблющейся величины от времени и пространственных координат, можпо получить, если в волновом уравнении функцию ф заменить на’ функцию ф и объединить это уравнение с уравнением колебаний
ч ,/2'г д. <w л_tW 1 —о ; пт
д?2 + ду2 + йз2 да3 ]
В теоретической физике доказывается, что для любой величины Т, зависимость •которой от пространственных координат и времени можно представить таким уравнением, существует возможность волнообразного распространения и что и в этом уравнении представляет собой скорость распространения такого волнового процесса. И наоборот, для каждой величины, которая распространяется в виде волны, зависимость от пространственных координат и времени передается приведенным вылив уравнением. Следует также отметить,что из уравнения (30) можно непосредственно вывести принцип Гюйгенса, лежащий в основ о волновой оптики и вообще объяснения дифракции волн. Для данного случая, однако, важно в связи с последующим изложением указать, что уравнение (30) применимо и к волнам материи, причем физический смысл постоянной и в уравнении (30) и в этом случае заключается в том, что она является
* С комплексной величиной а = х -ф 1у сопряжена величина а* — х — iy. Корень квадратный из их произведения 1/ аа* — f^x2 4- у2 имеет то яге абсолютное значение, что и каждая из этих величии порознь. *
** Эту сумму часто для краткости записывают в виде Дф. Например, уравнение /29) можпо записать в виде Дфф- ф—0. Тоже справедливо и для уравнения (30).
120
Глава 3
скоростью распространения волн материи. Эта скорость не является постоянной в вакууме, как скорость света, а, напротив, зависит от скорости движения в частицы, с которой связана волна [в силу уравнения (18)]. Какой смысл имеет в случае воли материн величина ¥, будет показано позднее.
Уравнение Шредингера. Энергия любой движущейся частицы равна сумме ее кинетической (1/amya) и потенциальной1 (евот) энергий. Так что справедливо равенство Е=1/2тот4-епот. Из уравнения (17) и соотношения u~vX следует, что . Заменив mv2 на 2 (в— 8П0т),,перепишем
это соотношение в виде,
____2?в (в — 8пот) ца - ~~~~№ ’ ’
Подставив этот результат в волновое уравнение (29), получаем уравнение Шредингера, лежащее в основе применения волновой механики к проблеме строения атома
д£2
-та~(е—е^М-
(31)
Строение атома водорода с точки зрения волновой механики. Предпо-
ложим, что атом водорода состоит из ядра с зарядом Е и электрона с зарядом —е. Как й на стр. 107, вначале не будем обращать внимания на то, что в этом случае Е=е, поскольку полученные результаты можно будет тогда распространить на атомы с более высокими зарядами ядер. Если электрон находится на расстоянии г от ядра, то, как ранее было показано, его потенциальная энергия равна епот .== — . Подставив эту
величину в уравнение (31), получим
, сВф. дх- ' ду-
f ,
=о.
(32)
Давно известно, что существуют дифференциальные уравнения, которые имеют решение только при совершенно определенных значениях входящих в эти уравнения «параметров» (т. е. входящих в них произвольных постоянных). Особые значения параметров, при которых можно найти решение дифференциального уравнения, являющееся везде конечным и достаточно быстро убывающим в бесконечности*, называют собственными. значениями такого дифференциального уравнения. Решения, отве- 5 чающие таким параметрам, называются собственными. ^ункфпялш.
Например, дифференциальное
уравнение
d2y .
™Ф=-(а -ая
х^Уу^Л имеет однозначное
решение, если постоянная а. является дельы* нечетным числом. ‘В этом уравнении а является параметром, а числа 1, 3, 5 и т. д. его собственные значения. Для а=1 ретие-ic2 '
ние имеет вид у—е 2 . Выражение е 2 является, таким образом, собственной функ-
цией, соответствующей собственному значению а = 1. Собственным значениям Зи 5 _ -т'-
отвечают собственные функции 2 хе 2 и (4ж1 — 2)е 2 , То, что эти выражения являются собственными функциями привёденпбго выше дифференциального уравнения, соответствующими собственным значениям 1, 3 и 5, можно легко проверить, дважды
дифференцируя уравнения’ у—ё 2 , у—2х-е 2 и т. д. К сожалению, строгое математическое доказательство того, что это дифференциальное уравнение, креме реже-
'* Этими требованиями определяется однозначность решения.
Спектр водорода и строение атома водорода 121
ний, соответствующих нечетным целочисленным значениям параметра а, не имеет никаких других решений, удовлетворяющих требованиям однозначности, довольно сложно,
Шредингер показал, что уравнение (32) имеет, решение при любых положительных значениях е, но лишь при совершенно определенных отрицательных значениях в, а именно при таких значениях, которые удовлетворяют соотношению
г 8=—<33>
где п — любое целое число. Это значит, что электрон, связанный с ядром, атома, может иметь только совершенно определенные значения энергий; поскольку энергия такого связанного электрона е всегда отрицательна*. Состояние, в котором атом может находиться, не теряя электрона, называется стационарным состоянием.
В соответствии с уравнением Шредингера для стационарных состояний атома получаются те же дискретные значения энергии, что и по теории Бора**. Однако, если в теории Бора дискретные значения энергии в получаются из недоказуемого постулата, а именно из предположения, что' электрон может вращаться в атоме без излучения света только по совершенно определенным орбитам, дискретность энергии е является следствием основных положений самой волновой механики и без каких-либо дополнительных предположений неизбежно вытекает из уравнения Шредингера.
Согласно положению волновой механики, стационарными состояниями атома являются такие, в которых волны алектронрв в кулоновском поле ядра не всюду гасятся-ва счет интерференции.
Если электрон, движущийся в силовом поле атомного ядра, не имеет кинетической энергии, достаточной для удаления его из этого поля, он неизбежно должен оставаться вблизи атомного ядра. Но в таком случае его энергия должна отвечать условию, выраженному соотношением (33).
Из законов волновой механики, и соответственно квантовой механики , непосредственно, без какого-либо дополнительного предположения, следует также условие частот Бора Ej— es—hv. В силу уравнения (33) это-соотношение определяет, как было показано на стр. 109, положение линий в спектре атома водорода.
Главные и побочные квантовые числа в волновой механике. Значения е, подучающиеся из уравнения (32) или (33), являются собственными значениями уравнения Шредингера в применении к атому водорода. С каждым из этих собственных значений, характеризующихся главным квантовым числом п, связано несколько: собственных функций. Эти функции отли-
* Это следует из соотношения е = mi13 — -£ . Электрон только тогда удержи-, вается ядром, когда его кинетическая энергия меньше той работы, которая требуете» eEi
для отрыва электрона, т. е. когда £/2 mv'1 <Z~, иначе говоря /когда е <0. Сео водный электрон обладает только кинетической энергией (поскольку г = со). Эта энергия, всегда положительна, и поэтому свободный электрон может иметь любую скорость, (вплоть до скорости света). ь
* * Для точного расчета в уравнение (32) или (33), как и в уравнение (11), вместо.
, • „ , ’
т надо подставить «приведенную массу электрона» р= (см. стр. 110 и сл.).
122
Глава 3
чаются еще двумя квантовыми числами I и т, которые также являются .целыми и называются побочными квантовыми числами.
Побочное квантовое число I волновой механики соответствует побоч-ному квантовому числу к в теории Бора. Однако по своей величине оно не равно последнему, а имеет значение 1~к—1. То, что I отличается от к и его наименьшим значением является пуль, связано с тем, что I является мерой так называемого орбитального момента вращения* атома. Поэтому I называют также квантовым числом орбитального момента вращения или, короче, орбитальным квантовым числом. Из положений волновой механики следует, что орбитальный момент атома может принимать значения У Z (Z+1) ~ . Если атом не имеет орбитального момента, то 1—0.
Побочное квантовое число т учитывает то обстоятельство, что спектральные линии, излучаемые возбужденными атомами, расщепляются в магнитном поле на несколько компонент (эффект Зеемана). Поэтому юго называют магнитным квантовым числом. Если на атом не действует магнитное ''поле, его энергетическое состояние не зависит от т.
Уравнение (33) не отражает влияния побочных квантовых чисел па энергию атома вв его стационарных состояниях. Энергия атома действительно не зависит от побочных квантовых чисел I и т, если справедливы те допущения, которые были сделаны при выводе уравнения (32). Так как влияние магнитного поля не учитывалось, то энергия атома не зависит от побочного квантового числа т, пока атом находится сне какого-либо внешнего электромагнитного поля. Кроме того, при выводе уравнения (32) масса электрона т (а равно, и «приведенная масса электрона» ц) считалась постоянной. Это допущение, как можно показать, не является* точным, если принять во внимание теорию относительности. Поэтому, если учесть следствия из теории относительности, то для различных состояний с одним и тем же главным квантовым числом п получается неболь-шая разница в энергиях, зависящая от значений I, обусловливающая упомянутое па стр. 113 небольшое расщепление спектральных ливши атома водорода**. Значительно больше квантовое число I влияет на величину е тогда, когда вследствие возмущения кулоновского поля между электроном и ядром пе будет уже строгим соотноси
тение е-пот^=——, использованное при выводе уравнения (32). Такое возмущение может быть вызвано внешним электрическим полем (эффект Штарка). Влияние квантового числа I на анергию атома проявляется я в чех случаях, когда атом содержит <более одного электрона. Поэтому у всех атомов с зарядом ядра, большим заряда ядра атома водорода, если эти атомы не потеряли все, кроме одного, пз своих электронов, наблюдается отчетливое, иногда даже довольно сильное влияние орбитального квантового числа I на энергетические уровни атома, а тем самым и па положение его 'спектральных линий.
Смысл ф-фупкции. Функцию ф, входящую в уравнение Шредингера (31) и соответственно в уравнение (32), можно истолковать следующим образом: ф.ф*- do — |ф (Му,(см. стр. 119) определяет вероятность того, что электрон может находиться в окрестности атома в элементе объема dv. Эту вероятность можно представить как функцию расстояния электрона от ядра или как функцию направления в пространстве. Последний случай показан на рис. 24 для различных состояний, характеризующихся указанными значениями квантовых чисел. Кривые, приведенные на рис. 24, имеют следующий смысл: если центр любой из этих кривых соединить прямой с какой-либо точкой на сбответ-•ствующей кривой, то длина отрезка этой прямой будет мерой вероятности присутствия электрона в направлении этой ^прямой- Эти фигуры
* Этот момент вращения называется орбитальным в отличие от момента вращения. связанного с так называемым спином электрона (см. стр. 197 и сл.).
** Наряду с этим “расщепление линий обусловливается также рассматриваемым в следующей главе спиновым квантовым числом.
Спектр водорода и строение атома водорода
123
относятся как к атому водорода, так и к «водородоподобным» ионам, т. е. ко всем атомам, которые, настолько глубоко ионизированы, что, подобно атому водорода, имеют только один связанный электрон. Кривые, представленные на рисунке, передают распределение вероятности в плоскости рисунка. Пространственное распределение вероятности получится, если вращать кривые вокруг осп симметрии г, лежащей в плоскости рисунка. Таким образом, атом водорода во всех стационарных состояниях обла-.дает вращательной симметрией, Во всех состояниях с 1=0 он имеет даже
Рис. 24. Вероятность присутствия электрона в различных стационарных состояниях в зависимости от направления для атома водорода (по Уайту), Распределение вероятности имеет вращательную симметрию относительно лежащей в плоскости рисунка средней вертикальной оси каждой фигуры.
шаровую симметрию независимо от главного квантового числа п. Тот факт, что при различных значениях I и т получается различное распределение вероятности нахождения электропа возле ядра, соответствует тому, что в уравнении (32) для одних и тех же собственных значений имеется, вообще говоря, несколько собственных функций. Из этого рисунка видно, что в случае п~1 получается только одна собственная функция.
На рис. 25 представлопа вероятность нахождения электрона в данной точке в зависимости от расстояния от ядра г для различных состоянии атома водорода, а именно для тех состояний, в которых атом обладает шаровой симметрией, т.о е. для Z~0. На абсциссы нанесены расстояния от ядра в ангстремах (А), а по оси ординат отложены соответствующие значения функции г21);ф*. Эта функция, умноженная на 4 л dr, определяет вероятность обнаружения электрона на шаровой поверхности (точнее
124
Глава 3
в шаровом слое толщины dr) на расстоянии г от центра атома (ядра атома).
Из рисунка видно, что в каждом стационарном состоянии электрон может двигаться не только по одной единственной орбите с определенным радиусом, как это было постулировано Бором. Он не движется и по определенной шаровой поверхности, а может находиться на различных расстояниях от ядра. Но некоторый определенный шаровой слой отличается тем, что-вероятность нахождения электрона внутри этого шарового слоя имеет наибольшее значение. В направлении от этого слоя к периферии атома, вероятность пребывания электрона быстро падает. В направлении внутрь
Рис. 25. Вероятность присутствия электрона в зависимости
от расстояния от ядра (г) для атома водорода.
Выражение 4пгафф*<!г определяет вероятность то?о, что электрон можно обнаружить в шаровом слое толщины dr на расстоянии г от центра. Масштаб по оси ординат раэличеи для разных фигур; он выбран произвольно таи. чтобы максимумы всех кривых имели одинаковую высоту.
атома вероятность пребывания электрона может пройти через другой (однако значительно меньший) гмаксимум. Между этими максимумами лежат шаровые слои, вероятность пребывания электрода в которых равна нулю. Это так называемые «узловые поверхности» стоячих волн, образующихся вокруг атомного ядра. Они соответствуют узловым точкам колеблющейся струны. В некоторых случаях могут возникать также узловые поверхности, проходящие через центр. Конечно^ они не имеют шаровой симметрии, а обладают просто вращательной симметрией. Число таких узловых поверхностей равно I. Общее число узловых поверхностей равно п—1. Следовательно, в волновой механике смысл главного квантового числа п и орбитального квантового: числа I заключается в том, что они определяют число и вид узловых поверхностей.
Пространственное распределение вероятности пребывания электрона можно представить себе в виде облака, окружающего ядро атома. В зависимости от значения I это «электронное облако» обладает вращательной или шаровой симметрией. Можно говорить также и о некоторой (хотя и, пе отчетливой) границе этого облака, поскольку в направлении-бт ядра плотность этого облака после достижения максимума быстро уменьшается. Форма этого облака в некоторой степени передает «форму» атома, отражая его симметрию. Поскольку электронное облако и общем случае имеет наибольшую протяженность к тех направлениях, по которым вероятность пребывания электрона наибольшая, фигуры, показанные на рис. 24, если представить себе, что они вращаются вокруг оси з, дадут приблизительную форму этого облака. Одпако они не дают точной картины — даже отвлекаясь от того, что облако в отличие От образующихся при вращении кривых поверхностей не имеет резкой границы,^ потому что плотность облака, за исключением случая шаровой симметрии, изменяется не по всем направлениям одинаково. Но окружающее ядро атома электронное облако следует рассматривать лишь как наглядное изображение вероятности Пребывания электрона. Как уже было сказано и согласно волновой механике, электрон движется вокруг ядра атома по определенной орбите. Таким образом, в каждый данный момент времени ои находится в одном определенном месте. Одпако это место нельзя точно установить, а можно лишь для
Спектр водорода и строение атома водорода 125
данной точки указать вероятность того, что электрон попадет в нее, или, иными словами, частоту его пребывания в этой точке.
Применение уравнения Шредингера к атомом е несколькими электронами. В принципе уравнения, подобные уравнению (32), можно составить и для атомов с несколькими электронами. Однако уже для двух электронов математическое решение задачи значительно усложняется. В этом случае в уравнение (31) входят вторые частные производные для каждой частицы по трем пространственным координатам, т. е. в общей сложности по шести пространственным координатам; ф будет функцией в шестимерном пространстве. При дальнейшем увеличении числа электронов математические трудности настолько возрастают, что точного решения уравнения Шредингера для многих электронов не имеется. Б таких случаях решение задачи облегчается введением упрощающих предположений. Целый ряд важных для химии результатов был получен при использовании таких допущений на основе волновой'илн квантовой механики.
Глава 4
НУЛЕВАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Главная подгруппа восьмой группы)
ИНЕРТНЫЕ ГАЗЫ
Общие замечания, В группу инертных газов входят то элементы,, которые в обычных услрвиях: вообще нс образуют никаких химических соединений в узком смысле этого слова (т. е. никаких так называемых «валентных соединений»*, см. нитке), а именно келий, неон, аргон, криптон, ксенон и радон.
Порядно-номер / Элемент Символ Атомной веса Вес [ -1, 2 Плотность относительно 1р1 отв ости кислорода^ =i Температура п.'М в ленив, ?с Температура кипения, Ва-лент-пость.
о Гелий • Не 4,003 0,17848 0,12490 -272,16 —268,98 0
J0 Неон No 20,183 0,8990 0,6280 -248,6 —246,03 0
18 Аргон Аг 39,944 1,7837 1,2483 —189,4 —185,87 0
36 Криптон Кг 83,80 3,336 2,614 —156,6 —152,9 О'
54 Ксенон Хе 131,30 ,5,891 4,123 —111,5 —107,1 0
SG Радов Вп 222 9,96 6,97 —71 -65 0
®Атомные веса, приведенные в этой и последующей таблицах, нервы (если только речь-пе идет о чистом элементе} для обычных, встречающихся л природе изотопных смесей и основаны на атомном весе 0 = 1 6,0000 (обычного кислорода). Атомные веса изотопов приведены-в т. II. К природным изотопным смесям относятся и другие величины, приведенные в этой, и последующих таблицах.
° При давлении 25 цтп.ч.
Таким образом, ва.чептноо число ипертных газов равно нулю. Чтобы согласовать, помер группы инертных газов с максимальной положительной валентностью, как это сделано для остальных групп периодической системы, эта группа была названа «нулевой группой и помещена в начале периодической системы. При непрерывном расположении элементов, принятом в табл. 11 (см. приложение), инертные газы попадают в-носьмую группу н качестве ее главной подгруппы. Такое расположение согласуется с закономерностями периодической системы, так как при уменьшении отрицатель пой валентности с возрастанием помер;* группы, начиная с четвертой главной подгруппы, нулевую валентность следует ожидать для элементов восьмой главной подгруппы. Двойственность положения инертных газов соответствует их особому характеру по сравнению с элемента uni остальных главных иодгрувй. Подробнее об этом1 будет сказано в следующей главе.
Отсутствие у инертных газов способности к образованию соединений проявляется также и в том, что инертные газы, в противоположность-другим газообразным веществам при обычных температурах одноатомны*. Иными словами, у инертных газов молекулярный вес равен атомному.
* Относительно наблюдаемых в разрядных трубках чрезвычайно неустойчивых многоатомных молекул инертных тазов см, сноску па стр. 174.
Нулевая группа периодической системы 127
На одноатомность инертных газов указьйает отношение величины теплоемкости при постоянном данлснии к величине теплоемкости при постоянном объеме Ср/с,,. Это отношение у инертных газов равно 5/з=1,666..., т. с. величине, даваемой кинетической теорией газов для одноатомных газов. При низких температурах инертные газы сжижаются, а при дальнейшем довольно незначительном понижении температуры затвердеваю^, Однако низкие температуры кипения указывают на то, что и «вторичные валентные силы», или вандсрваальсовы силы, действующие между свободными атомами инертных газов, очень слабы. Это особенно относится к гелию, температура кипения которого ниже температуры кипения водорода, так что гелий в атомарном состоянии более насыщен, чем водород в состоянии двухатомной молекулы. Температура кипения, а также температура плавления инертного газа тем ниже, чем меньше его атомный вес или соответственно его порядковый номер.
В апдерва а Львовыми силами называют силы, которые обусловливают отклонение поведения газов от законов идеальных газов и переход газов в жидкое состояние. Как показал Лондон (London, 1931), во многих случаях эти силы можно свести к квантово-механическим резонансным силам между электронами различных атомов (см. стр. 155 и сп, и стр, 324). В остальных случаях вандервааЛьсовы силы обусловлены главным' образом поляризационными аффектами (см. стр. 71 и сл.). Дипольный момент инертных газов равен нулю, и они с трудом поляризуются, И все же некоторые из них способны образовывать с определенными веществами, и в первую очередь с водой, неустойчивые продукты присоединения. “
Гидраты инертных газов [Виллард (Willard, 1896), де Форкранд (de Forcrand, 1923 и 1925)] тем устойчивее, чем больше атомный вес инертного газа. Гидраты гелия; и неона не образуются при (У даже при давлении 260 ат. Давление диссоциации гидрата аргона при (Р составляет 105 ат, гидрата криптона — 14,5 ат, а гидрата ксенона — 1,45 ат. Гидрат радона (существование которого было доказано косвенным путем Никитиным в 1936 г.), поскольку его давление диссоциации при 0° меньше 1 ат, должен был бы получаться в кристаллическом состоянии просто при пропускании газа; через воду, охлажденную до 0°, если бы только можно было’работать с большими количествами этого радиоактивного газа. Кристаллические гидраты инертных газов, как. следует из их структуры (см. стр. 248 и сп.), содержат 53Д моля Н2О на моль инерт-пого газа (для гидрата Кг найдено 5,7 моля Н2О) и образуют смешанные Кристаллы, с соответствующим гидратом SO2. В сочетании с различной устойчивостью гидратов инертных газов это можно использовать для разделения этих газов. В соответствии; с данными Виберга (Wiberg, 1948), Аг, Кг н Хе не образуют продуктов присоединения с BF3 и по смешиваются с ним в жидком состоянии. Ксенон в жидком состоянии не смешивается также с SO2, H3S, GH3OH и (СН3)2О; маловероятно также, что он образует с этими веществами продукты присоединения.
Нулевая валентность инертных газов обусловливает очень большое-сходство их «химического характера»; общим свойством для них является; неспособность в обычных условиях образовывать вообще какие-либо «валентные соединения», т. е. соединения, в которых опи были бы связаны обычными валентными силами. Однако было бы преувеличением сказать, что инертпые газы «в химическом отношении вообще не отличаются одни от другого». Имеется много элементов, которые при равной валентности реагируют с веществами с образованием совершенно аналогичных соединений, отличающихся только физическими свойствами (растворимостью и т. д.). Такие характерные различия имеются и у инертных газов — различная летучесть, растворимость, адсорбируемость и т. д.., но только в свободном состоянии.
Исторические сведения. В 1892 г. лорд Релей, исследуя плотность обычных газов (кислорода, водорода и других), установил, что азот, получаемый из воздуха после связывания кислорода, имеет большую
128
Глава 4
плотность, чем азот, получаемый из химических; соединений, таких, как аммиак или нитраты. Рамзай предположил, что различие в плотности объясняется присутствием в воздухе еще одного не открытого тяжелого газа. Ему удалось после удаления кислорода пропусканием воздуха над раскаленной медью связать азот воздуха раскаленным магнием. Оставшийся газ оказался новым химическим элементом с характерным спектром. Одновременно он был выделен лордом Релсем, удалявшим азот старым методом Пристли и Кавендиша (см.стр. G35). Оба исследователя назвали новый элемент аргоном за его химическую инертность (греческое «аргос» ар*р4 — инертный). Рамзай при исследований минерала клевеита, о котором было известно, что при обработке его серной кислотой выделяется газ, похожий: на азот, полагал, что этот газ окажется аргоном. Однако удалось установить, что выделяющийся Из клевеита газ имеет новую спектральную линию, расположенную очень близко к желтой линии натрия, но заметно отличающуюся от последней. Эту желтую спектральную линию уже наблюдали многие астрономы в хромосфере Солнца во время солнечного затмения .в 1868 г. После того как было установлено, что эта линия не идентична желтой линии натрия, Секки первым сделал вывод, что она принадлежит существующему па Солнце и еще не, открытому на Земле элементу, который позднее Локайрр и Фрашгланд назвали гелием (греческое «гелиос» — Солнце). Это название получил й открытый Рамзаем элемент, поскольку его спектр указывал на то, что он идентичен солнечному элементу. Несколько . дней спустя после 'открытия Рамзая и ничего не зная о нем, Лангле в лаборатории Клеве также обнаружил присутствие гелия в клевеите.
В, силу чрезвычайно большого сходства в свойствах и полного отли-чия от всех остальных до сих пор известных элементов едва ли можно было сомневаться, что оба вновь открытых газа следует отнести к одной, и притом новой, группе периодической системы. Но при расположении этих газов в порядке возрастания атомных весов между ними оставалось одно свободное место. По этой причине Рамзай в 1897 г. предсказал существование еще одного инертного газа. Открытию последнего способствовало опубликование незадолго перед тем результатов опытов Хэмпсона и Линде пр сжижению воздуха, позволивших провести сжижение и фракционированную перегонку Аг. Воспользовавшись этим методом, Рамзай буквально через несколько дней открыл даже два новых инертных газа; в на Ча л о более тяжелый, но сравнению с аргоном криптон (греческое «криптос» ярйцто; — скрытый), обнаруженный по двум линиям в Желтой и зеленой частях спектра, а затем предсказанный перед этим более легкий, неон. Последний был назван так по предложению двенадцатилетнего сына Рамзая, который наблюдал новый яркокрасный свет, испускаемый разрядной трубкой и предложил назвать этот элемент «новым» (греческое «то пеон» то' Лоу). Оказалось, что остаток после фракционированной перегонки Аг содержал еще один газ — ксенон (греческое «то ксенон», то qevoy — чужой). Позднее Резерфорд и Содди нашли, что образующиеся из некоторых радиоактивных веществ радиоактивные газы, эманации, по свойствам также должны быть отнесены к группе инертных газов, Рамзай идентифицировал эманацию радия по сё спектру, нашел ее атомный вес по плотности газа и предложил для неё в качестве элемента название «нитон*. (светящийся — от niters'), которое должно было напоминать о фосфоресцирующем свечении сжиженного таза в пробирке. Позднее это название вновь было изменено. В конце концов общепринятым стало название «радон».
Нулевая группа периодической системы 129
Распространение в природе. Атмосферный воздух содержит около 1 об.% инертных газов. Основная доля приходится на аргон, остальные инертные газы присутствуют в очень малых количествах.
В 100 л воздуха содержится 934 мл аргона, 1,82мл неойа, 0,524.«л гелия, 0,114 мл криптона и 0,0087 мл. ксенона (Gltickauf, 1951). Содержание инертных^ газов в воздухе, как и содержание кислорода и азота, практически постоянно. Всюду, куда проникает воздух, можно обнаружить и аргон, например, в крови. По сравнению с распространенностью инертных газов в космосе содержание инертных газов в земпой атмосфере поразительно мало. Объяснение этому См. т. IT, гл. 15.
Газы, содержащие довольно большие количества аргона и особенно гелия, выделяются из многих минеральных вод. Иногда в них находят и большие количества неона. Гелий образуется при всех радиоактивных превращениях, сопровождающихся испусканием а-частид. Поэтому он всегда присутствует в радиоактивных водах и радиоактивных минералах. Из последних гелий выделяют растворением их в кислотах или сильным нагреванием. В некоторых местах Северной Америки на поверхность земли выделяются большие количества газов, относительно богатых гелием.
Содержащие гелий природные источники тазов расположены в США в области, простирающейся от Техаса, гдо находятся самые богатые из них, через Оклахому, Канзас и Огайо до Иью-Иорка. Значительные источники, газов открыты также в Канаде (у Инглвуда, близ Торонто). Содержание гелия в этих газах (основной составной частью которых в некоторых случаях является азот, почаще метан) превышает иногда 1%, Европейские источники гелия значительно беднее. Однако некоторые источники минеральных^ газов содержат Довольно большом процент гелия (Дюргепм 1,8%., Бадеп-Баден 0,85% а Вильдбад 0,71^); однако они могут давать в гоу( в лучшем случае несколько сотен кубических метров гелия. Гелий был найден и в вулканических газах (соффиопи Тосканы 0,26%). Кроме того, небольшие количества его обнаружены во многих местах в каменноугольных газах. Значительные количества гелия присутствуют, как показал спектральный анализ, в атмосфере Солнца, в газообразных туманностях я в некоторых звездах.
Присутствие радона и других радиоактивных эманаций .связано с присутствием радиоактивных элементов, из которых они образуются, поскольку эмапации быстро распадаются. Такой распад, естественно, происходит и тогда; когда эманации находятся вместе с материнскими радиоактивными элементами, однако р этом случае они постоянно вновь образуются. Количество радона, находящегося в равновесии с 1 а радия (так, что в каждый данный момент времени образуется столько же радона, сколько его распадается), составляет 0,653 .«л»® (при 0° и дав Ленин 1 ат.и). Это количество называется кюри, Око служит единицей измерения содержания эманаций в радиоактивных водах*. ч.
Получение п применение. В, Северной Америке начиная с 1917 г. добывают значительные количества гелия из богатых им минеральных газов. В течение долгого времени его применяли для наполнения дирижаблей.
В 1929 г. американский военный флот использовал в этих целях 200 000 м3 гелия. Количество гелия, которое можно было бы получить из североамериканских минеральных газов, оценивается более чем в 1 млн. м3 ежегодно. Примерно 30% этого количества приходятся на долго канадских источников. Гелий в два раза тяжелее
* Кроме меры содержания эманаций, единица кюри служит также общей единицей активности радиоактивных препаратов. В этом случае оно определяется как такое количество радиоактивного вещества, в котором происходит 3,700-1010 ядерных превращений в секунду. Это число приблизительно равно количеству ядерных превращений в 1 к радия в секунду (см. т. II, гл. 13).
9 Г. Реми
130 Глава 4
водорода, однако, поскольку подъемная сила дирижабля определяется разницей между его весом и весом равного объема воздуха, замена водорода па гелий умснь-
' , - " 28,8—2 “408 '
шлет грузоподъемность дирижабля только: в раз.
йо,о—‘A IvU 1
Применяемый в США метод добыта гелия основан на том, что гелий в отлично от других газов очень слабо адсорбируется активированным углем, охлаждаемым жидким воздухом. Этот метод применяли и раньше в .холодильных машинах Линде для получения неона и гелия, из остатков мосле сжижения я ректификации воздуха. Фракционной перегонкой неона и гелия при охлаждении твердым водородом мощно получить практически чистый неон. ОдмаКо Для большинства технических целей (но, конечно, пе для наполнения дирижаблей и воздушных шаров) вполне пригодна смесь неона и гелия, .
В небольших количествах гелии удобнее всего получать по Сивертсу (Sieverts, 1912) нагреванием до 1000—1200° содержащих ого минералов, таких, как клевеит, монацит или тореанит, в закрытой о одного конца фарфоровой трубке. Выделяющийся газ для очистки от образующихся Нй, Н2О и СО2 пропускают через фарфоровую трубку над раскаленной окисью меди и: над -лодочкой с твердой едкой щелочью. От азота освобождаются многократным пропусканием газа над сильно нагретой смесью окиси кальция, магния и натрия, от аргона — цри помощи прокаленного активированного угля.
Кроме применения в качестве: наполнителя для воздушных шаров и т. д., для чего гелий особенно пригоден благодаря своей невоспламеняемости, его используют Для наполнения газовых термометров и в технике низких температур. Неон применяют (часто в смеси с гелием) главным э образом для световых реклам и т. и. (неоновые дуговые лампы, тлеющие лампы). Кроме того, неоновые лампы используют в различных областях электротехники (выпрямители, предохранители, делители Напряжения),: Аргон находит применение главным образом \tak: совершенно индифферентный защитный газ. В смеси с 15% азота его используют для наполнения ламп'накаливания.
В атмосфере чистого аргона легко образуется электрическая дуга. lie образование можно предотвратить добавлением азота, Применение аргона в лампах накаливания наряду с химической инертностью определяется его низкойтепло пр Овод цостью; в настоящее время для этой цели используют также, криптон и ксенон, теплопроводность которых еще пипсе; было предложено при рентгеноскопии дыхательных органов применять криптон и ксенон ввиду их высокой способности поглощать рентгеновские лучи.
В лаборатории аргон* (точнее «сырой» аргон, см. ниже), можно получить, связывая химически азот и кислород воздуха, освобожденного о быч-шым методом, от двуокиси углерода и паров воды.
Кислород обычно удаляют пропусканием воздуха над раскаленной медью. Азот связывают магнием или кальцием или смесью магния, окиси кальция и натрия. Кислород и азот можно удалить одним реагентом, а именно раскаленным карбидом кальция. Еще лучше исходить из содержащего азот кислорода, получепного фракционной перо- ' гонкой жидкого воздуха. В этом случае кислород уже относительно обогащен аргоном (примерно до 3%), поскольку аргон, как й кислород. мепее летуч, чем азот. .
Сначала по методу Общества холодильных машин Линде многократной фракционной перегонкой доводят содержание аргона в жидком воздухе по крайней море до 60%. Затем химическим путём удаляют около 30% ’Кислорода, Остающаяся смесь аргона и азота пригодна для щчполншшя Дамп накаливания..
Получаемый из воздуха аргон содержит Остальные присутствующие ., в воздухе инертные газы- Эти незначительные (составляющие около 0,25 об. %) примеси в обычных случаях применения'аргона несущественны. Очистку «сырого» аргона от сопутствующих инертных гайов производят многократной фракционной перегонкой. Этим методом можно получить ‘ также чистые криптон и ксенон-
Нулевая группа периодической системы 131
Для лабораторного разделения инертных та зов наряду с фракционной перегон* Е кой используют теперь поглощение активированным углем. Поглощаемость инертных газов сильно возрастает с увеличением атомного веса. Фракционным- пог .лощением на. активированном угле при температуре жидкого воздуха можно разделить в зависимости от давления гелии и пеон или неон и аргон. Аргон, криптон н ксенон можно разделять поглощением па активированной угле и последующей фракционной десорбцией методом Петерса (Peters, 1930). Растворенный в жидком воздухе : радон можно полностью поглотить силикагелем.
Разработаны также технические методы, основанные па различной скорости -диффузии инертных газов в атмосферу другого газа, иля методы, использующие для разделения ипертных газов: различную скорость проникновения их через пористые перегородки. Значительные количества крицтопа и ксейона были получены методом Клода (Clotide), который основан на том, что щидкий воздух извлекает ука- • за иные газы из воздуха, охлажденного почти до температуры сжижения/.
Дли получения радона раствор какой-нибудь соли радия оставляют на 4 подели в закрытом сосуде. Это время необходимо, чтобы практически установилось (упомянутое ла стр. 129) равновесие между радием и его эманацией, После зтогр радон мощно удалить из раствора кипяченном или откачиванием в вакууме.
Свойства. Все инертные газы не имеют цвета и запаха. Они относительно трудно сжижаются, но тем легче, чем больше атомный вес. В ряду от гелия до радона температура кипения при атмосферном давлении повышается от—269 до —65°. Температура замерзания радона (при которой давление его паров, еще равно 500 мм рт ст) равна —71°. Поэтому радон конденсируется при весьма малых концентрациях. Если пропускать радон через U-образную трубку, охлаждаемую жидким' воздухом, то он осаждается на стенках трубки, что отчетливо: видно по ярко-зелёной флуоресценции стекла, вызываемой радоном. Если радон находится под очень незначительным давлением, то его конденсация происходит в интервале температур от —152 до -15)“'. Вообще температуры замерзания, а. соответственно и температуры плавления инертных газов лежат пемного ниже температур кипения (см,, таблицу в начале главы). Гелий можно,перевести в твердое состояние только под давлением.
Температура плавления гелия при 25,3 ат равна 1,13° К, при 140 ат — 4,21° К. Гелий образует две жидкие фазы, которые переходят одна в другую при 2,3° К, что сопровождается выделеннем тепла и скачкообразным изменением свойств.
Теплоты плавления ц пспарения инертных газов чрезвычайно Палы. Их значения, а также критические величины, приведены в табл. 19. О кристаллической структуре ипертпых газов в твердом состоянии см. стр. ,240.
Таблица 19 -
Теплоты плавления, теплоты пспарения ц критические величины ипертпых газов
гелий Нее и АргОН Криптон. Ксенон Радон;
Теплота испарения, кнал/молъ ...... 0,022 0,44 1,50' 2,31 : 3,27 " 43
при t, ,°С -270 —246 -186 -151 -109 ‘ -62
Теплота плавления, кг.ал/молъ ...... 0,0033 0,078 0,265 0,36 0,49 0,8
при i, °C ...... . —270,7 -249 -189 -157 : -140 — 71 ;
Критическая температура, °C \ .... -267,9 —228,7 -120 —62,5 -4-16,6: 4^ 104,5
Критическое давление., ат 2,26 26,9 50 54,3 58,2 62,4
Критическая Плотность 0,069 0,4 0,4 0,7 0,9 ' ' 1,2
9*
132
Глава d
В воде растворяются относительно большие количества инертных гадов. Согласно Ланиуигу (Lannung, 1930), в 1 л воды при 20° растворяется 8,8 мл гелия, 10,4 мл пеона, 33,6 мл аргона (объемы газов указаны при О3). Как следует из этих данных, растворимость аргона в воде даже несколько превышает растворимость кислорода. При повышёпий температуры растворимость уменьшается; с увеличением атомного веса ипертного газа растворимость возрастает и достигает у радона примерно 51 об. % при 0°. На стр. 127 уже упоминалось о том, что при высоких давлениях инертные газы образуют кристаллические гидраты. Растворимость инертных газов в органических растворителях в некоторых случаях превышает их растворимость в воде. При низких температурах активированный уголь более или менее энергично поглощает все инертные газы, за исключением гелия (ср. стр. 131). В отличие от водорода гелий не диффундирует через раскаленную платину. Однако при повышенных температурах он (как и водород) диффундирует через кварцевое стекло. Это свойство можно использовать для разделения гелия и неона (Paiieth, 1925).
Инертные газы отличаются Сравнительно хорошей электропроводностью. В этом отношении они значительно превосходят такие газы, как водород, азот и др. Небольшое сопротивление, которое оказывают инертные газы прохождению электрического тока, низкие напряжения, при которых в этих газах возникает светящийся разряд, имеют большое значение для их применения в осветительной технике.,
Инертные газы имеют очень характерные спектры., Которые часто используют для их аналитического определения. Капилляр-трубки Плюк-кера при наполнении его гелием излучает интенсивный желтый свет, при наполнении неоном — яркий кораллово-красный свет. У других инертных газов излучаемый свет зависит до некоторой степени от условий опыта. Обычно излучение аргона имеет красный цвет, криптона — от зеленоватого до лилового, ксенона — фиолетового и радона — яркобелого.
СПЕКТРЫ ИНЕРТНЫХ ГАЗОВ
В табл, 20 приведены наиболее яркие линии спектров инертных газов, расположенные в видимой области. Особенно точно определено положение липин в спектре гелия, поскольку эти линии часто используют для градуирования или настройки спектральной аппаратуры. Хотя длины волн приведены в табл. 20 с точностью до сотых долей, все же в последней цифре могут быть отклонения в несколько единиц.
Капилляр трубки Плюккера, наполненной аргоном под давлением около 3 мм рт ст, излучает красный свет; при меньшем давлении и увеличении напряжения за счет подключения батареи лейденских банок цвет излучения переходит в голубовато-стальной и, наконец, в белый. В «красном спектре» наиболее яркие линии расположены в красной и инфракрасной областях, в «голубом спектре» — в фиолетовой и ультрафиолетовой областях. Для Других инертных газов, поведение которых более или менее подобно поведению аргона, в табл. 20 приведены только обычные спектры, а в них — наиболее яркие линия.
С теоретической точки зрения особый интерес представляет спектр гелия. Еще до появлепия теории Бора было известно, что линии спектра гелия (которых значительно больше, чем приведено в табл, 20, потому что многочисленные линии расположены в ультрафиолетовой области) можно разбить на шести серий. Эти шесть серий образуют две
Таблица 20
Основные спектральные липпп инертных газов в видимой области спектра Длины волн даны в международных ангстремах (А). 1А = 10"8 см
Аргои
Спектральный двет Гелий Неон «красный «голубой Криптон Ксенон Радой
спектр» слентр*
Инфракрасный 8115,3 8112,9 8231,6
'у 8006,2 7854,8 7694,5 7601,5 7587,4 7642,0
7544,0 7535,8
7281,35 7245,2
7065 Д 9 7173,9 6929,5 G965±4
Красный 6678,15 6678,3 6599,0
6506,5 6469,7
Оранжевый 6402,2 6266,5 6163,6 5975,5 6182,4
Желтый 5875,62 : 5881,9 5870,9
5852,5 5400,6 5570,2 5582
5047,74 5037,7 5085
Зеленый 5015,68 4957,0 4979
4921 93 4327,2 4806,0 4817
Синий 471з’14 4715,3 4739,0 4734,1 4766
4671,2 4671,2 4681
4644
4624,3 4614,3 4626
4540.4 4537.8 4510.7 45020 4501,0 4605
Ипдпго 4471,48 4475,0 4463,7
4437,55 4426,0 4453,9 4376,1
4387т9 4300,! 4348,0 4355,5 4350
- 4272,2 . 4277.5 4319,6 4274,0 4307
4266,3 4266,3
4259,4 4200,7 4198,3 4193,5 4203
4143,8 4120,8 4158,6 4104.0 4088,4 4078,8 4167
Фиолетовый 4026,2 4044,4 4057,0 4057,0 3982
3964,7 3949,0 3928,6 3920,1 3951,0 3972
134 Ллава 4
независимые одна от другой групнй, «термы» н^торьгх не удается «комбинировать», как это обычно можно сделать дляхлиний различных серий одного и того же элемента. Ввиду этого долгое время сомневались, является ли гелий простым веществом.
. Комбинационный принцип, предложенный Ритцем (1908), заключается в том, что методом «комбинации» т. е, сложения или вычитания термов различных серий, можно получить, (обратные) значения длин волн лилий, имеющихся в спектре данного вещества. Если, например, Для спектра водорода рсповпой терм серии Бальмера вычесть из основного терма серии Лаймапа, То получается обратное значение длины волны первой линии серии Лаймапа. В основе этой закономерности лежит тот факт, что, как будет видно из следующей главы, термы определяют энергетические уровни атома, соответствующие его различным стационарным состояниям (ср. стр. 121), Следовательно, комбинационный принцип утверждает, что .атом может переходить из одного стационарного состояния непосредственно в любое другое Стацдоцарнре состояние (за счет поглощения или испускания света). Здесь следует лишь указать^ что если энергетические уровни определяются пе только главными квантовыми числами, а и побочными квантовыми числами, то комбинационный принцип нуждается в некоторых ограничениях (ср. стр. 139). -
.Раньше полагали; что две различные группы серий, характеризующих спектр гелия, следует приписать двум различным элементам, названным ортогелием и парагелием (или астерием). Одпако оказалось, что гелий, вне всякого сомнения, является простым веществом (в смысле химического элемента). Различие спектров объясняется тем, что в случае гелия упомянутые ограничения комбинационного принципа налагают, вообще говоря, запрет на комбинации между обеими Труппами термов. Электронные переходы, Характеризугощиеся.испусканием спектра, приписываемого парагелию, приводят к конечному состоянию, в котором цба .электрона нейтрального атома гелия связаны иначе, чем в конечном состоянии, получающемся при испускании линий спектра ортогелия. Хотя во втором состоянии атом гелия имеет большую энергию, чем в первом, он не может за счет излучения непо средств мню перейти из конечного орто-co стояния в коночное пара-са стояние. Переходы из других состояний ортогелий в состояние парагелия и наоборот за счет излучен ня также практически не происходят, за исключением переходов, сопровождающихся полным отщеплением электрона (ионизацией).
Следует еще отметить, что в 1922 г. Лайман в: дальпей ультрафиолетовой области, открыл еще одну серию гелия, принадлежащую парагелию, так что теперь известны в общей сложности .четыре принадлежащие ему важные сертш. Некоторые другие серии парагелия и ортогелия, ж которым относится лишь очень небольшое число линий, представляют интерес только для специалистов по спектроскопии и атомной физике,
В определенных условиях, если; например, чердз гелий под давлением 1. лш рт ст пропустить искру от больших лейденских бапок, он испускает линии; которые пе принадлежат ни к одной из рассмотренных серий. Аналогичные липни. быЛи также найдены в спектрах зцрзд. Такой спектр, получающийся в разрядной трубке только: за счет искры, называют искровым спектром гелия, в Отличии от обычного дуеового спектра. Линии искрового спектра точно отвечают тем линиям, появления которых еле&увт ожидать как на основании теории Лора, так- и в соответствии в принципами волновой механики для однократно ионизированных атомов гелия, если предположить что они отличаются от атбмов водорода только тем, что ядро атома гелия имеет вдвое больший положительный заряд по сравнению с ядром атома водорода, Линии искрового спектра гелия описываются уравнением (13а) на стр. 110, если принять Z -2 и ;1/=4 (атомный вес гелия). Б полном согласии с теорией для константы Ридберга в слуг чае гелия получается несколько большое значение, чем в случае водорода, а именно по спектр ос коническим измерениям:
ЛЦе=109 722,267 ±0,012:
: Этой величиной можно воспользоваться для очень точного вычисления отношения, массы электрона к. массе ядра атома водорода mlAf^ ио уравнениям
" ДНе_ /1ZIic2-('^н- ••• ДНеГ~^Н ' ...
/'>ц ‘^ir ( I-' Н, 2- "О ;l/H‘ ,. ,, jV/Ht
Ну лева я группа периодической системы 135
Используя приведенные в табл, 69 т. П вел1гчины масс ядер водорода а гелия, для этого .. отношения получаем значение 0,000544628 или 1 ; 1836,12. Отсюда следует, что отно-шение массы протона к массе атома водорода равно
1___99'14557
1,000544628 ’ :
а отношение .насс'Ы электрона к Массе атома водорода
т 0,000544628 , 4 104719
Л/.Г-Пюо544628 =Wo44322 пли 1 .: 1837.12.
Из табл. 21 видно, с какой точностью согласуются значения длин волн линий искрового спектра, гелия, вычисленные по уравнению 2
{--“«(ч—<*>
е экспериментальными данными,
Таблица 21
Длины ноли линий в искровом спектре гелпя
сердя : пй • 1 : Длд =-2' ' 3 4 " 5 6 7. 8 9
Первая се- 1 303,6 256,3
рДЯ ^-выч 303,7 256,25
Вторая се- 2 ^наб.ч 1640,4 1215,2 1085,2 1025,6
рия ^вуч 1640,5 1215,2 1085,0 1025,3
Третья се- 3 А лаб л 1 4685,8 3203,1 2733,3 2511 .,.2 2385,4 2306,2
рия (серия Фаулера) ^ЁЫЧ 4685,9 3203,2 2733,4 251С 3 2385,5 2306,25
Четвертая 4 ?-аабл 6560,1 5411,6 4859,3 4541,6
серия (серия Пикеринга) ^•выч 10123,8' 6560,2: 5411,6 4859,4 4541,7
Так как в соответствии с этими данными искровой спектр гелия, вне всякого сомнения Принадлежащий ившгыировапному гелию Не*, по структуре аналогичен спектру нейтрального атома водорода, то в общем сир аве д либо следующее положение: варактор искрового ейектра (т. е. спектр, испускаемый (однократно) ио пикированными атомами] каждого элемента подобен характеру дугового спектра (т. с. Обычному спектру, испускаемому нейтральными атомами) впереди стоящего е периодической системе элемента (спектроскопический закон смещения ЗоммерфельДа). Спектры, испускаемые нейтральными атомами, называются дуговыми. спектрами, потому что для большинства элементов их получают при помощи электрической дуги,
Спектры других инертных газов еще богаче линиями, чем спектр гелия; например, для пеона.измерены длины волн примерно 900 линий. Структура этих спектров еще сложнее, чем структура спектра гелия. Это i указывает на гсг, что на внешних орбитах атомов остальных инертных газов имеется большее ио сравнению с гелием число электронов;
Ниже будет рассмотрен метод более точного определения числа этих линий. ... '
136 Глава 4
Ионизация н излучение
Для предсказания химической реакционной способности ,6oj интерес представляет, как будет видно в дальнейшем, знание эн (затраты работы), необходимой для перевода атома какого-либо эле в электрически заряженное состояние за счет отрыва электронов. Р; требующаяся для перевода одного атома из нормального состояния (; роп расположен на самой глубоко из доступных для него орбит) в зированпое состояние (электрон полностью оторван) называется pat ионизации для данного атома*. Работу ионизации** можно точно в лить из спектральных данных, даже если ничего определенного не изв о строении атома. Для этого достаточно только предположить, что высший основной терм, получающийся из спектроскопических да] действительно соответствует нормальному состоянию атома. Одна! многих случаях работу ионизаций можно изморить Henocpedcmt производя ионизацию электронным ударом.
Величина энергии, требующейся для ионизации атрма водорода, полу непосредственно из уравнения [11] гл. 3, если принять, что п=1. То же значение гпи получается из обобщенной формулы Бальмера [уравнение (3), гл. 3], ccni тать 1 и пд=со, т.е. из основного терма или соответственно из границы пат коротковолновом серии спектра водорода (серии Лаймана), после умножения и Точно так же йз искрового спектра гелия можно определить работу, необходиму отрыва второго электрона от однократно ионизированного атома, гелия, т. е. pi относящуюся к процессу Не’’ -* Не2+4" е. Эта. работа равна [если принять во л пи уравнение (1) па стр. 135] е=4ЯНс-с-,й.
Однако спектроскопические наблюдения показывают, что основная о с квантовым числом не всегда является нормальным состоянием атома г Для наиболее коротковолновой серии спектра ортогелия получается основной с к=2. Линии, комбинирующиеся с остальными лилиями ортогелия и которые До были бы соответствовать основному терму с л=1, не обнаружены, хоти они и до были бы лежать в области спектра, вполне доступной для измерений. Отсюда в сделать вывод, что в случае ортогелия основной орбитой второго электрона явл орбита с квантовым числом п=2, Напротив, в случае парагелия граница серий зывает на то, что основная орбита имеет квантовое число п— 1. Таким образом, м теперь сказать, что различие между ортогелием и парагелием заключается в что в атомах ортогелия основная орбита второго электрона имеет квантовое число , а в атомах парагелия основная орбита второго электрона, так же как и основна бита первого электрона, характеризуется квантовым числом и=1. Отсутствие ком дующихся линий указывает на то, что переход атомов гелия из одного состояния в гое может происходит!, только путем полной ионизации.
Прямое измерение работы ионизации. Прямое измерение pat ионизации, методом электронного удара было разработано Фра1 (Franck) и Герцем (Hertz) в 1913 г. В основе его лежит следующий дрин в трубке, где находится исследуемый газ, например гелий, полр электроны с совершенно определенной скоростью. Это основано на явлении, что накаленная металлическая нить постоянно испускает эл роны с очень небольшой скоростью. Если эти электроны заставить гаться в электрическом поле, возникающем при подаче .на металл скую нить отрицательного напряжения, а положительного — на расп женную против нити электродную сетку, подобно тому как это дел в радиотехнических усилительных лампах, то такие электроны б] иметь точно известное значение скорости V, а следовательно, н энор
* Точнее, эта работа является работой однократной ионизации. А па лог можпо определить и работу многократной ионизаций.
** В справочной литературе по физико-химическим константам веществ ( употребителен термин «энергия ионизации».—ZZpw.w. ред.
Нулевая группа периодической системы
137
определяемое разностью потенциалов накаленной нити и сетки (lfs mv'~ — =eV, где е — заряд электрона, а. V — разность потенциалов). Обычно скорость электронов выражают через величину напряжения, приложенного для их получения, т, е. в вольтах (е), а энергию, соответствующую этой скорости, в электрон-вольтах (эв). Чтобы перевести значения энергии, выраженной в электрон-вольтах в эрги, надо значение в электронвольтах умножить на 1,602'10'1й, а для перевода в калории — на 3,829-10"20,
1 эи.= 1,602-10-13 зрг = 3,829-10-20 йал. i (2)
Если приложенное напряжение медленно увеличивать, начиная от нуля, то сила тока сначала будет пропорционально возрастать, а затем (если вести опыты с гелием, то при напряжении 19,75 в) сила тока резко упадет до нуля. Сила тока определяется числом и скоростью электронов. Если температуру накала нити поддерживать постоянной, то число испускаемых ею электронов будет также постоянным. Начальное увеличение силы тока объясняется, таким образом, ускорением электронов При увеличении напряжения. Отсюда следует, что электроны, сталкиваясь с атомами гелия, сначала отражаются от них, не претерпевая при этом заметного изменения в скорости, но только- до тех пор, пока их скорость fie достигнет некоторого определенного значения, отвечающего напряжению поля 19,75 в, Как только это произойдет при столкновении с атомами гелия, эти электроны теряют сразу всю скорость, на что указывает внезапное уменьшение силы тока, т. е. теперь они всю свою энергию передают атомам гелия, с которыми сталкиваются] Если и дальше увеличивать напряжение, то сила тока начинает снова возрастать пропорционально разности между приложенным напряжением и величиной 19,75 в, г. е. электроны отдают атомам гелия энергию, точно соответствующую напряжению 19,75 в, Спустя некоторое время (в случае гелия) уже при напряжении 20,55 в сила тока еще раз резко падает до нуля. Далее для гелия такие скачки отмечены при 21,2 и 22,9 в.
Обычно моЖпо наблюдать, что всякий раз, когда происходит резкое уменьшение силы тока, одновременно происходит испускание одной или нёекольких спектральных линий, сохраняющееся и при дальнейшем повышении напряжения. Оказалось, что существует очень простая зависимость между энергией сталкивающегося электрона s=eV в момент появления данной линии (соответственно лилий) и термом, характеризующим эту линию, т. е. возможными значениями энергии атома. Поскольку электрон передает столкнувшемуся с ним атому свою энергию в,| эта энергия е в то же время означает прирост энергии атома за счет соударения с электроном, т. е.
e=s2—ci, где ех — энергия атома до соударения, е, — энергия этого же атома после соударения с электроном. Для атома, перешедшего при столкновении в состояние с энергией за, возможны теперь переходы в другие состояния, энергия которых меньше е? (если только эти переходы ие запрещены «правилани отбора»). Если возможен переход в основное состояние, то длина волпы самой коротковолновой испускаемой линии определяется соотношением
82-6!=-^ = ^. (3)
Это уравнение является не чем иным, как вторым квантовым условием Бора [ср. уравнение (10), гл. 3]. Его справедливость, подтвержденная
138
Глава £
как уже упоминалось, волновой механикой, получает, слёдовательн прямое экспериментальное доказательство.
Наглядно процесс возбуждения свечения атома при электроппс ударе можно представить следующим образом; летящий электрон поди; мает электрон атома, с которым он столкнулся, с основной Орбиты (в смы ле теории Бора) на более' высокую. При возвращении электрона на не.] воначальную орбиту атом испускает в виде света энергию, полученную и при соударении s>. Если энергия сталкивающегося электрона нед< статочпа для перевода электрона атома на более высокую орбиту, то лещ щиц электрон отражается по законам упругого соударения. Напряжена V, требующееся для «возбуждения» атома к испусканию определённо спектральной линии,, называется напряжением возбуждения давно спектральной линии. Напряжение, требующееся для сообщения электрон такой скорости, чтобы он мог вырвать электрон из атома, называете напряжением ионизации Vj. -Работа ионизации равна произведению папрт женин ионизации на заряд электрона :
А/=<+ь : ' -и р
Уравнение (4) дает работу, требующуюся для ионизации свободное атома, или молекулы, в электрон-вольтах. Работу, необходимую для иони зации одного грамм-атома или одной грамм-молекулы, дыражёнпут в килдграмм-калориях, получим, умножая выраженную в калориях работ; ионизации на чйсло Авбгадро* TVa =6,0231 -1023 и разделив, получении; результат на 1000. Так что работа ионизации (ккал/моль) равна работ ионизации (эв/молекула) X23,063. ; : -
В зависимости от того, идет ли речь об отрыве первого электрона о1 нейтрального атома (т. е.о переходе атома во дц ок рати os а ряж е ниьп положительный нон), пли второго электрона (т. о. о переходе однократж заряженного иона в двукратно заряженный), или об отрыве третьей электрона и т. д., различают напряжение первичной, вторичной, третичной и т. д. ионизации. Общую работу, необходимую для многократной ионизации, получим, сложив отдельные работы, требующиеся для ступенчатой ионизации. Например, напряжение первичной ионизации гелия равно 24,48 в, вторичной — 54,14 в. Поэтому для перевода атома гелия в поп Не2* надо затратить работу., равную 24,48+54,14 =78,62 эв. Отсюда, чтобы получить 1 г-uoil II е"+ из 1 моля газообразного гелия,ffnaдо произвести работу, равную 78,62 к 23,063 =1813,2 ккал.
В случае гелия непосредственное спектральное наблюдение света,. испускаемого при возбуждении за счет электронного удара, осложняется тем, что соответствующие ЛИП11И лежат в дальней ультрафиолетовой, области. Однако если сравнить линии, наблюдавшиеся Лайманом в дальней, ультра фиолетовой области серии гелия, с линиями, возникающими при электронном ударе,, то найдем, что наблюдаемые разности потенциалов точно соответствуют разнице энергий, рассчитанных по длинам волн.. Это поясняется рис. 26. На нем. указаны; энергетические уровни, соответствующие напряжениям возбуждения и спектрам. Энергетические уровни обозначены fra рисунке соответствующими квантовыми числами по теория Вора Зоммерфельда. На рисунке не выдержан масштаб, так как иначе основной уровень нрищлось бы расположить слишком далеко. Кроме того, на рисунке не приведены энергетические уровни с кващ
* ’Гнело Авргадро показывает число молекул в грамм-молекуле вещества. В физической литературе это число часто называют числом Лошмидта, хотя раньше делали различие между числом АвоГадро ц числом Лошмидта. Если проводить это различие, то под числом Логпмидта надо понимать число молекул в кубическом миллиметре газа при 0° и атмосферном давлении (= 27 000 биллионов), впервые опр ед еденное Лошмвдтом в 1865 г. Однако обычно под этим числом понимают число молекул газа В кубическом сантиметре (= 27 триллионов).
_ Нулевая группа периодической системы
13!)
товыми числами больше п=5. Не показаны и энергетические уровни, относящиеся только к сериям, расположенным в инфракрасной области спектра, для которых известно лишь очень небольшое число линий. Стрелки, направленные Вниз, соответствуют электронным переходам, отвечающим испусканию соответствующих световых волн.: Раапости энергетических уровней, полученные из длин волн по уравнению (3) и пересчитанные в элейтрон-вольты; названы «вычисленными», а напряжения, соотвётствуюптпё этим разностям энергий (измеренные непосредственно методом электронного удара);— «наблюдаемыми» величинами. Граничная длина волны наиболее коротковолновой серии (Х=502 А) не^измерена непосредственно, а вычислена по формуле серий. Первому минимуму на кривой сила рока — нацряжепре (при 19,75 а) пё соответствует никакая
линия. Этот минимум отвечает энергии, необходимой для перехода стабильного атома парагелия в метастабилглгый атом ортогелия. Энергетический Уровень последнего получается также из основного терма главной серий ортогелия. При этом получается анергия, отличающаяся на 19,77 в от энергии нормального состояния парагелия, что очень хорошо согласуется с наблюдаемым «напряжением превращения».
На рис. 26 Стрелками, соответствующими электронным переходам с одного уровйя ва Другой, показано возникновение линий важнейших серий телия. Тот факт, что термы t парагелия пе комбинируются с термами ортогелия, согласно этой схеме, объясняется у тёц, что никогда электрон сам ио себе не переходит с энергетического уровня парагелия па энергетический уровень ортогелия, и наоборот. Схема поясвяет также, в чем смысл ограничений комбинационного принципа, упомянутых па стр, 134. Как видно, электронные переходы происходят только в тех случаях, когда побочное квантовое число к изменяется лишь на единицу или вообще не .изменяетед. Здесь пе будут подробно описаны другие ограничения комбинационного принципа «правилами отбора»! На примере гелия видно, как, не зная модели атома, сопоставлением результатов л спектральных исследований с результатами измерений методом электронного удара можно найти, даже в сложных случаях, совершенно точное положение энергетических уровней атома.
В табл. 22 приведены значения напряжений первичной .ионизации для элементов, расположенных в порядке периодической таблицы. Индексы, поставленные слева внизу у символов элементов, указывают поряд-
1гмпт'тт1шшш!Н1Я1ИН1ШН11И111
Таблица 32
Потенциалы первичной ионизации элементов (в)
Величины, заключенные л скобки, пе измерены непосредственно, а рассчитаны на основании общих закономерностей изменения экранирования заряда ядра внутренними электронами (Finkе 1 liburg W., Z, Naturforschung., 2А, 16, 1947)
Главные подгруппы периодической системы
Г группл II ГруППЛ III труппа ГУ группа V группа VI группа VII группа VIИ группа
JI 13,54 2Не 24,48СГ1ектр 24,6Пабл
3Li 5,37 ыПа 5,09 19К. 4 ,32 37ИЬ 4,19 55С3 3,86 s-IT (3,97) 4Ве 9,30 i2Mg7,63 2оСа 6,09 38Sr 5,68 йвВа 5,21 sgRa 5,21 5B 8,28 ; IjAI 5,94 S1Ga 5,97 49Ш 5,76 81 Tl 6,08 6С 11,24 i4Si 8,14 32G1) 8 у 10 5qSii 7,34 g2Pb 7,37 7N 14,46 15В 10,43 зз Лв 10,05 wSb 8,35? 83Bi 7,25? 8О 13,57 i6S 10,42 3480 9,75 52Т0 8,89 84Ро(8,3) 9В 17,46 17С1 13,01 35Вг 11,82 531 10,43 35Л1 (9,4) , ifjNe 21,47СПентр igAr 15 >68 одектр 13,94спенТр 54X0 12,08СГ1СК^р l0i69cncltTp 21г5каб.^ 15,4набл 13,Зяабл И^надл
Побочные подгруппы периодической системы
I группа II группа III группа IV группа V группа VI группа VII группа VIII труппа
7 ,(57 4?Ag 7,58 79Л11 9,20 SoZn 9,37 ' 48Cd 8,94 aoHg: 10,41 3iSc 6,7 39T 6,6 g;La 5., 59 8эЛс (5T5) ,2Ti 6,81 t0Zr 6,92 72Hf (7,6) 90Th (5,7) 23V 6,74 itNb (6,9) 73Ta (7,6) alPa (5,7) 24Cr 6,74 42Mo 7,2 74W (7,6) 92U (5,:) 2JMn 7,41 : 43Tc (7,1) 7^Re 7,8 звВе 7,83 2?Go 7,8 cyNi 7,61 HRu 7.5 45Rh 7,7 4ePd 8,3? !0Os 8.7 „1г (8,7) 73Pt 8,8
ЛаНтаниды: ,.0Се 6.54 .„Pi- 5 7R . мл к ai
Нулевая группа периодической системы
141
кбвые номера элементов. Для ипертных'газов в Целях сравнения сопоставлены значения, вычисленные по спектроскопическим данным (индекс «спектр») и найденные методом электронного удара (индекс «набл»). Величины, полученные из спектроскопических данных, имеют большую точность. Как можно видеть, работа, которую надо затратить, чтббы оторвать от атома один электрон, особенно велика для инертных газов (ср. также рис. 4 на стр. 38}., Кроме того, таблица показывает, что в главных подгруппах периодической системы величины работы, требующейся для отрыва одного электрона от нейтрального атома, в общем в каждом ряду увеличиваются слева направо, а в каждой подгруппе уменьшаются сверху вниз. Элементы побочных подгрупп характеризуются некоторыми колебаниями величин работы ионизации, а также нерегулярным изменением их по сравнению с элементами главных подгрупп.
Как будет показано в дальнейшем, величина работы отрыва электронов определяет химическое поведение элемента. Неспособность инертных газов образовывать истинные валентные соединения теснейшим образом связана с высокими значениями их потенциалов ионизации.
Рис. 27. электроном, ( __
(по теории Бора —Зоммерфельда).
Значение побочных квантовых чисел в случае атома гелия. В случае атома водорода, а также однократно ионизированного атома гелия^раз личин между энергетиче-скнми уровнями, соответствующими различным побочным квантовым числам, настолько малы, что опи проявляются только в топкой структуре спектральных линий. Для нейтрального атома гелия эти различия значительно больше. Это объясняется тем:, что для центрального атома гелия, да и вообще для всех атомов, имеющих более одного электрона, энергия орбиты определяется побочным квантовым числом пе только в силу вытекающей из теории относительности зависимости массы электрона от скорости, во и в значительно большей степени по совершенно иной причине. Это легко показать, пользуясь теорией строения атома Бора—Зоммерфельда*, если внимательно рассмотреть орбиты, приведенные на рис. 27. Эта схема приблизительно верпа для ортогелия. Лежат ли орбиты в одной плоскости или нет, имеет второстепенное значение. В любом случае со стороны электрона, вращающегося по круговой ороите
(характеризующейся согласно те'орил Бора — Зоммерфельда квантовыми числами Ц), действует па,, другой электрон, внешний по отпошешпо к первому, отталкивающая сила. Поэтому- притяжение внешнего электрона к положительно заряженному ядру (в рассматриваемом случае ядром с зарядом 2-ф) полностью пе проявляется. Заряд ядра «экранируется» электроном, движущимся цр внутренней круговой орбите. Степень экранирования зависит от расстояния, Электрон, движущийся по эллиптической орбите, характеризующейся, например, квантовыми числами и Zr=l, притягивается Ядром с зарядом 2Ц-, вокруг которого вращается одни электрон, но существу так же, как не экранируемым ядром с зарядом 1Ц-. Но на некотором участке сво.ей
Экранирование заряда ядра вращающимся по орбите 1!
* Квантово-механическая теория строения атома приводит в основном к тем же заключениям о влиянии экранирования на энергетические уровни атома и о связи величины экранирования с главным и побочным квантовыми числами. Поскольку волновая механика отбрасывает представление о движении электронов по определенным орбит юг, даваемое ею объяснение (более точное в отношении вытекающих из него следствий) нельзгг изложить столь же наглядно, как представления, основанные па теории Бора — Зоммерфельда. Конечно, нельзя делать никаких заключений о симметрии атома, используя представления о круговых и эллиптических орбитах (см. стр. 114 и 124 и ел.).
142. I'.iaea 4
орбиты второй электр оппаходптсл ближе-к ядру, чем электрон, движущийся,: пе тоъой орбите i±. На этом участке орбиты на второй электрон действует почта по заряд ядра. В случае электрона, вращающегося пр круговой орбите 2;, экраии ние остается постоянным на'всех участках:орбиТы. Отсюда ясно, что в привода примере энергия электрода на эллиптической орбите 2( будет отличаться] от тип электрона на круговой Орбите 22. Точный расчет показывает, что это етвмтельно так. Поскольку сила, действующая на электрок. из фокуса эллипс постоянна во времени, эллиптическая орбита искажается и движение электрона м приблизительно описать как. движение по эллипсу, большая ось которого в очередь также вращается вокруг фокуса эллипса. Различно энергий эллинтич< и круговой орбит может даже внешне проявиться как. спектральная линия, i;ai видно из рис. 26. Первая циник главной серий как ортогелии, так и парагелия । лбвлена переходами электронов с круговых (2-.) ла эллиптические'(21) орбиты.
Рассматривая внимательно . рис. 26, можно видеть, что линии, отпосяп: к одной и той же серии, характеризуются, "кроме общего основного уровня Jt; и равенством побочных квантовых чисел различных исходных уровней. Это с ведлиео и д.г.ч серий других тяжелых элементов.
У гелия и других элементов различают в основном следующие четыре се главную серию, резкую побочную серию, диффузную побочную серию Н фундаменп пук серию, пли серию Бергмана*. Исходные уровни: этих серий обозначаются только у гелия, но и у других атомов) как г-, rf-и/-орбиты, а энергетические у ни называются соответственно р-, s-, и /-уровнями. Когда термам стали прип вать квантовые числа, то оказалось, что ^орбитам соответствует в теории мерфельда побочное квантовое число 4=2, s-орбидам й=1; 7 орбитам 4=3 орбитам — й=4. Как уже было указано, побочное квантовое число 4 в теории Ёбj Зоммерфельда соответствует побочному квантовому числу I тюдновой механики, к рое на единицу меньше побочного квантового: числа: теории Бора. Таким обра получаем .следующие соотношения:
Название серии Обозначение исходных... уровней для отдельных. лищпГ Побочные кваятовыс числат пряписыоаемый ,йсход-1ШМ :
в теории Борз—3 ом-: жрфёльда Ь ВОЛНОВОЙ ъгекакпке
Главная серия . . . р-уровни ,...' 7=1
Резкая побочная серия .С.. . • ^уровни 4=1 7=0 '
Диффузная побочная серая . .. . Серии Бергмапа (фундаменталь- <4у ровни д=-з/" 7=2
ная) . . /-уровни a—i ' *-з
Различные серий имеют характерные особенности; так что спектроскопист, как п вило, уже по общему характеру серий моя;ст:решить, какие исходные уровни дщ пы быть им приписаны.
Нейтральный атом гелия. «Искровой спектр» гелия показывает, ч в полном соответствии со сказанным ранвезарядядра атома тем йя равен 2-П о этому нейтральный атом гаг ин имеет два электрона. В нормально т. е. наименее богатом энергией, состоянии оба эти электрону связан одинаково (не с читая а нтипа рал. юл i.noro направления' их "/спине ср. стр. 145). Основной терм наиболее коротковолновой серии Тгарарел. показывает, что второму электрону па основном уровне следует прип сать главное квантовое число п=1, т. «^.наименьшее возможное квантов число. Л в соответствии со сказанным выше это должно, быть спранедли
* Название . «фундаментальная: серия» употребляется в. основном в английсЮ й американской литературе. Эта серия назвала так из-за. ев особо простого строени
V
Нулевая группа периодической системы .. ... .143
и для первого электрона, который, конечно, не должен быть связан слабее, чем второй. Система, образуемая этими двумя электронами, отличается особой устойчивостью. Это следует прежде всего из чрезвычайно высокого значения потенциала ионизации гелия. Но еще отчетливее эта особая устойчивость системы электронов в нормальном атоме гелия проявляется при сравнении анергии, требующихся, с одной стороны, Для перевода электрона с уровня 1$ на ближайший более высокий уровень 2s и, с другой стороны, для перехода электрона с уровня, например, 2s па уровень Зр (ср. рис. 26). Первая равна 20,55 эв, а вторая только 2,42 эв. Особая устойчивость системы электронов, имеющейся в нормальном состоянии атома гелия (парагелии), проявляется и в исключительно , большом различий энергии нормального гелия и его метастабилъной формы . (ортогелия). Эта Совершенно исключительная устойчивость определяет химическое поведение не только самого гелия, но и, как будет показано в следующей главе, поведение следующих за гелием элементов. Это утверждение справедливо и для других инертных газов, электронные системы которых, как показывают высокие значения потенциалов ионизации (см. табл. 22), также отличаются исключительной устойчивостью.
Число электронов в оболочках инертных газов. Нейтральный атом гелия имеет, как было показано, два электрона, которые в нормальном состоянии оба находятся на орбитах с главным квантовым числом /д 1. Следующий за гелием элемент, литий, имеет, как будет показано игл. 6. всего три электрона/Один из них, как следует из величины потенциала ионизации, приведенной в табл. 23, связан очень слабо, два же других,
Таблица 33
Потенциалы ионизации эле.ментон от водорода до неона
В колонках с I по X приведены, потенциалы ионизации (в), соответствующие, отрыву первого, второго и т, д. до десятого электрона ,
Элемент I II in j fV V VI VII VII г IX X
1Н 13,54
2Не 24,48 54,14
3Li 5,37 75,20 121,84
4Ве 9,30 18,12 153,11 216,63
г,В 8,28 25,00 37,74 258,03 338,53
еС 11,24 24,26 47,64 64,17 390,02 487,55
-jN 14,46 29,44 47,20 72,04 97,40 549,08 663,73
8О 13,57 34,94 54,63 77,03 113,30 137,42 735,22 867,09
9? 17,46 34,81 62,35 86,72 — 156,37 184,26 — 1097,7
21,47 40,91 63,3 -97 — " — —“ 1355,5
напротив, связаны прочно. Следующий элемент, бериллий, имеет 4 электрона; из них два Связаны относительно слабо, остальные два связаны очень прочно. У следующего элемента имеется 5 электронов, из которых 3 связаны относительно слабее, чем два других, и т. д. (ср. табл. 23),.У каждого следующего за гелием элемента, вплоть до УII главной подгруппы, число электронов, равное номеру соответствующей группы периодической
144
Глава 4
системы, отрывается значительно ''легче, чем последние два электрона. Таким образом, в атоме каждого из этих элементов два электрона (значения потенциалов ионизации которых в табл. 23 выделены жирным шрифтом), так же как и в атоме гелия, связаны особенно прочно. Это позволяет предположить, что имеющаяся в атоме гелия система электронов, отличающаяся особой прочностью, повторяется и в атомах других элементов, так что каждый добавляющийся электрон не входит в эту систему, а связывается вне ее во «второй сфере», как обычно говорят ради наглядности. Данные рентгеноспектрографиц (см. гл; 7) подтверждают это пред- положение. Они показывают, что внутри атомов всех следующих за гелием элементов имеется, как и в атоме гелия, система из двух электронов с главным квантовым числом ?г=1.
Если просмотреть элементы периодической системы вплоть до элемента с порядковым номером 10, то вновь увидим инертный газ — неон. Этот элемент имеет 10 электронов, 8 Из которых находятся во «второй сфере». Поскольку система электронов в атоме неона также отличается особой устойчивостью, то следует, как и в случае системы электронов в атоме гелия, особую устойчивость приписать и такой «вбсьмиэлектрон-ной оболочке». После неона расположены элементы с одним, двумя, тремя нт. д, слабее связанными электронами. Поэтому их нужно отнести в «третью сферу», что опять-таки подтверждают данные рентгепосцектрографиИ, Элемент с порядковым номером 18, аргон, опять является инертным газом. Это значит, что и третья сфера оказывается заполненной, после того как число электронов в ней достигает восьми.
Следующий инертный газ, криптон, имеет на 18 электронов больше, чем аргон. Можно было бы думать, что после образования двух «восьми-элсктронных» образуется оболочка с 18 электронами. В действительности дело обстоит иначе. И в этом случае, т. е. и к «четвертой сфере», вновь образуется восъмиэлектронная оболочка. Однако, чтобы это произошло, требуется 18 электронов, поскольку 10 из них располагаются на «промежуточной ободочке», между третьей и четвертой оболочками. Доказательства в пользу такого заключения будут приведены при рассмотрении побочных подгрупп периодической системы, поскольку их образование теснейшим образом связано с построением промежуточной1 оболочки с 10 электронами. То же повторяется и у следующего инертного газа, ксенона. И он имеет на 18 электронов больше, чем криптон, из них только 8 располагаются на внешней, теперь уже пятой, оболочке, а 10 — на «промежуточной» оболочке между четвертой и пятой оболочками. И, наконец, радон имеет па 32 электрона больше, чем предшествующий инертный газ. Но и в этом случае во внешней оболочке находится только 8 электронов. Все остальные расположены па «промежуточных оболочках», чем, между прочим, и объясняется, как будет показано позднее, появление подгруппы, лантанидов. Итак, можно сделать вывод, что атомы всех инертных газов характеризуются наличием отличающейся особой устойчивостью внешней оболочки, которую в случае гелия образуют два, а у других инертных газов восемь электронов.
Выражение «электроны в атомах расположены на следующих Друг за другом оболочках» следует понимать лишь как образное. Истинный смысл понятия «оболочка» заключен лишь в том, что к одной «оболочке» относятся электроны с одним и тем же главным квантреым числом. Поэтому выражение «электрон принадлежит к третьей ободочке» не означает ничего большего, чем «основной уровень электрона является уровнем с главным квантовым числом и==3». Этим объясняется и то, что при нумерации оболочек в атомах тяжелых инертных газов (криптон и т. д.) опущены «промежуточ
Нулевая группа периодической системы
145
ные» оболочки*. Под «оболочками» можно понимать также л энергетические уровни атома (ср. рис. 55 па стр. 258, на котором представлен разрез энергетических поверхностей в тяжелом атоме). Реальность «оболочек» аналогична реальности поверхностей равного нотепциала вокруг какого-либо электрически Заряженного тела.
Аналогия в строении оболочек инертных газов проявляется еще отчетливее, если наряду с числом электропов сравнивать и их квантовые числа, особенно их побочные квантовые числа. При этом сразу же можно понять, почему у гелия электронная конфигурация, характерная для инертного газа, образуется уже двумя, а не 8 электронами. Для полного описания типа связи электрона в атоме требуется, кроме задания главного квантового числа п, орбитального квантового числа I и магнитного квантового числа т, еще задание четвертого квантового числа, так называемого спинового квантового числа if. Это число учитывает спин, или вращение электрона вокруг собственном оси (ср. стр. 197). Для каждого электрона это число может принимать лишь два значения: -р/з или —1/2 в зависимости от направления вращения. Числовые значения, которые могут принимать все эти побочные квантовые числа, кратко можпо выразить следующими соотношениями: . J
— 1; [ т | 7; s=± 1/г(п, 7, т~~целые числа).
В настоящее время установлено, что атомы инертных газов в основном состоянии никогда не имеют во внешней оболочке электроны, у которых I больше 1. Если 7=1, то, согласно приведенным выше соотношениям, т может иметь значения —1, 0 или ~]-1. Для 1—0 возможно лишь т—0, К каждой из таких возможных пар значений I и т можно присоединить одно из двух значений.?, а именно -р/2 или — J/3. Всего в таком случае возможно ^х4=8 комбинаций кваптовых чисел, и как раз! такое число электронов имеется в оболочках инертных газов. Но для атома гелия возможны лишь две различные комбинации, поскольку л=1, а следовательно, 1=0;
7 = 0, т — 0, и 7 = 0, т = 0, $ = —1/2.
Таким образом, во внешних оболочках инертных газов содержится ровно столько электронов, сколько возможно различных квантовых состояний при условии, что значение I не превосходит единицы.
Принцип Паули. При переходе от инертных газов к элементам с большими зарядами ядер появляющиеся в атомах этих элементов новые электроны располагаются, как уже было отмечено, на других оболочках. Отсюда можно сделать вывод, что оболочка заполнена тогда, когда все возможные для нее квантовые состояния заняты электронами. Паули высказал следующий принцип; в одном и том же атоме не может быть двух электронов, у которых совпадают все четыре квантовых числа (п; 7, т и S').
Из принципа Паули непосредственно следует объяснение различной длины периодов в периодической системе, причем этот принцип объясняет и появление побочных групп, а также семейств лантанидов и актинидов (см. т. II). Этот принцип очень важен и для объяснения природы гомеополяр ной химической связи, как это будет показано в гл. 5.
Символы, используемые для описания строении атома. Если для полного описания типа связи электрона в атоме требуется четыре квантовых числа, то для описания связи между строением атома и химическим поведением элемента в общем достаточно двух квантовых чисел, а именно главного квантового числа п и орбитального квантового числа I. Для указания величины орбитального квантового числа используют следующие краткие обозначения; электрон, находящийся иа энергетическом уровне, характеризующемся 7=0, называют х-электроном, на энергетическом уровне с 7=1—jo-электроном, иа уровне с 7=2 — (7-электроном и с 7—3 — /-электроном.
Эти обозначения связаны с тем, что, как и в случае гелия, линии в спектрах атомов с зарядом ядра, большим заряда ядра атома водорода, можно разбить в основном на четыре серии: главную (prinzipal), резкую (scharfe) побочную, диффузную (diffuse) побочную и фундаментальную (fundamental). Исходные уровни для отдельных _____________________ о
* Подробнее о квантовых числах, характеризующих промежуточные оболочки, см. стр. 259.
10 г. Ремп
146 Глава ; 4
линий этих серий, различающиеся характеризующими их значениями орбитального квантового числа 7, обозначают начальными буквами английских названий этих серий (ср. стр. 142).
Энергетические уровни с орбитальными квантовыми числами больше / = 3 никогда не встречаются в спектрах свободных атомов в нормальном состоянии, даже в случае наиболее тяжелых элементов. Введение сокращенных символов х, р, d и / для числовых- значений I позволяет кратко и точно определить тип связи электронов у атомов всех элементов. Для этого перед символом числового значения орбитального квантового числа ставят значение главного квантового числа, характеризующего данный уровень. Число электронов в атоме, находящихся на уровне, характеризующемся данными ^значениями главного и орбитального квантовых чисел, если оно больше единицы, пишут в виде верхнего индекса у символа орбитального квантового числа. Если электроны расположены на различных энергетических уровнях, то символические обозначения" этих уровней- просто записывают один за другим.
Например, запись «2$-элёктрон» означает, что этот электрон находится на энергетическом уровне с главным квантовым числом л~2 и орбитальным квантовым: числом 7=0. Четыре р-электрона с главным квантовым числом и=2 обозначают символом 2/>4 (читается «два-пэтчетыре»), а символ 2j22/>6 (читается «два-эс-два, два-по-шесть») указывает. что в рассматриваемом атоме имеется два г-электроиа с главным квантовым числом п=2 и шесть р-электронол с главным-квантовым числом /!--2.
Таким образом, электронные конфигураций,'^, е. состояния связи электронов н атомах {водорода, гелия и неона), можно представить следующими символами:
Н - Не Ne lei 1?- " " Ы2а22р«.
С точки зрения строения атомов инертные газы характеризуются тем, что в их «внешних оболочках» все s- й р-уровнй заняты электронами. Под такой «заполненной оболочкой» понимают энергетический уровень атома, в котором все возможные квантовые состояния заняты: электронами. Поскольку в силу принципа Паули на s-уровне- не может быть больше двух, а на /> у ровне больвзе шести электронов, то внешние оболочки инертных газов имеют конфигурации nsznp6, кроме гелия, в атоме которого ввиду того; что при 7i=l Z = 0, имеется всего лишь Два s-электрона и пет р-электронов.
В табл. 2 (в приложении) этими символами указаны: электронные конфигурации всех элементов. Как видно из этой таблицы, особое положение инертных газов среди химических; элементов связано с тем,’ что в атоме инертного газа каждый раз заканчивается заполнение внешней оболочки, а с атома каждого следующего за инертным газом элемента начинается заполнение новой оболочки. .
Эманации. Эманациями (от ётапаге исходить) называют газообразные вещества, которые выделяются из определенных радиоактивных элементов и , сами являются радиоактивными. Известны четыре различные эманации встречающихся в природе’радиоактивных элементов*.
* Кроме того, известны еще три эманации (изотопные эманации природныхрадиоэлементов), которые образуются при радиоактивном распаде не встречающихся в природе искусственных атомов. IIПодробнее см. т. II. -
Нулевая группа периодической системы ИТ
Они названы по именам тех радиоэлементов, в ряду распада которых они образуются: эманации радия (RaEm I и RaEm II), эманация актиния (АсЕт) и эманация тория (Th Em). Первой среди эманаций была открыта (Rutherford, 1900) эманация тория. Уже в следующем году Дорн наблюдал образование эманации радия, а вслед за тем Гизель, а также Дебьерн обнаружили эманацию 'актиния. .Недавно было установлено, что, кроме обычной эманаций радия, образующейся непосредственно из самого радия, имеется еще одна эманация, происходящая от одного' из продуктов распада радия (подробнее см., т. II). Однако она образуется в количествах, значительно меньших, чём первая (отношение количеств при радиоактивном равновесии равно 1 : 1(Г12). Обычная эманация радия представляет собой уже упоминавшийся инертный таз радон.
Оказалось, что не только эманация радия, но и другие эманации являются инертными газами. Все они относятся поэтому к одной и той же группе периодической системы. В тл. 2 т. II будет показано, что теория радиоактивного распада позволяет вычислить атомные веса продуктов распада, Во всех случаях, когда оказалась возможной экспериментальная проверка, атомные веса, предсказанные этой теорией, совпадали с экспет риментально найденными. Такую экспериментальную проверку производили. например, для радона. Рамзай, измеряя плотность таза, получил из многих опытов для атомного веса радона среднюю величину около 223, а: теория распада дает значение 222. Совпадение очень хорошее, если учесть неизбежные ошибки эксперимента при измерении плотности таких малых количеств газа/Теоретическую! величину следует считать'более надежной. Теория распада дает Для второй эманации радия атомный вес 218, для эманации актиния — 219 и для эманации тория — 220. Такие величины атомных весов исключают возможность отнесения эманаций к разным, рядам периодической системы. Таким образом, все они относятся к одному и тому же ряду и к одной и той же группе, но это значит., что все они должны стоять в одной клетке периодической системы,/т.е. все эманации изотопны эманации радия — радону.
Проще — .по с меньшим экспериментальным подтверждением — тот же результат можно получить, если исходить из того факта, что место каждого элемента в периодической системе определяется его порядковым номером. Теория радиоактивного рас-пада(см.:т, II) дает для всех, эмапаций один и тот же порядковый помер 86. Поэтому все они должны стоять., на 86-м месте периодической системы;
Указанные четыре эманаций как изотопные вещества имеют одина^ ковые химические свойства. Ошю Отличаются своими радиоактивными свойствами, которые рассмотрены в т. II.
.Изотопия инертных газов. Несмотря на то что эманация радий (радон) и другие эмапаций. (если они смешаны) нельзя разделить обычными химическими средствами, их легко получить в чистом виде из радиоэлементов, при распаде которых они образуются. Однако остальные инертные газы в природе встречаются ввиде смесей изотопов. Число изотопов в таких, смесях значительно возрастает от гелия к ксенону. Гелий имеет только два изотопа, из которых более легкий, (атомный вес 3) сопутствует более тяжелому (атомный вес 4) в исчезающе: малых количествах (в отношении 1 ; 10е). Неон имеет три изотопа, так же как и аргон. Криптон состоит из шести, а ксенон из девяти изотопов (см. т. II, гл. 12).
1: ,
Глава 5
ВАЛЕНТНОСТЬ И СРОДСТВО
Группировка элементов вокруг инертных газов. Закономерность изменения* некоторых свойств элементов и их соединений проявляется особенно наглядно, если элементы сгруппировать вокруг инертных газов так, как это сделано в табл. 24...
Таблица составлена по принципу периодической системы, однако она- отличается от обычной тем, что группа инертных газов размещена иначе; она расположена так, что при сохранении последовательности элементов, определяемой порядковыми числами, главные подгруппы, даже в больших периодах следуют одна за другой. Таким образом, инертные газы помещены примерно в середину главных подгрупп. При таком расположении инертных газов (как бы па оси, вокруг которой распределены другие элементы главных подгрупп) соотношения элементов в горизонтальном направлении проявляются особенно четко.
Валентность элементов в табл. 24 при переходе от одной группы к другой правильно возрастает как в главных, так и в побочных подгруппах; в последних, правда, это ограничивается тем, что элементы этих подгрупп большем частно легко меняют свою валентность (соответственно заряды) и что валептность, соответствующая номеру группы, там часто уступает место другим валентным состояниям (соответственно зарядам). Сначала в главных подгруппах максимальная положительная валептность возра-. стает от трех до семи, увеличиваясь в каждой группе па единицу. В трм же направлении уменьшается максимальная отрицательная валентность, затем, переходя через пуль, она возрастает до положительного значения. В общем слева направо заряды увеличиваются от 4— до 2-|-, правильно возрастая каждый раз на единицу. Этот процесс продолжается и там, где побочные подгруппы являются продолжением периодов, пока не достигается число восемь. Побочная подгруппа VIII включает зону таких элементов, которым характерны средние значения валентности. Вслед за тем, налипая с 1_|_, происходит новое правильное повышение положительной валентности; опо продолжается у элементов главных подгрупп, где положительная валентность возрастает от трех до семи.
Все элементы, предшествующие инертным газам, образуют пе солеобразные,* а большей частью легколетучие водородные соединения. Те же элементы, которые непосредственно следуют за инертными газами, образуют твердые, солеобразные, водородные соединения. Далее, чем выше в таблице расположены элементы, предшествующие инертным гагам, и чем ближе стоят они к нттрртным газам, тем яснее выражен их неметаллипеский характер. Все илементы, стоящие вправо от инертных газов, являются металлами. Чем ближе расположен элемент в ряду к следующему за ним инертному газу, тем более кислый характер имеет образуемое им водородное соединение. И чем ближе стоит элемент в ряду к предшествующему ему инертному газу, тем более сильное основание он обравует,
В табл. 24 пи один из элементов, стоящих перед инертными газами, п ни один из непосредственно следующих за ними элементов не может образовывать окрашенные элементарные ионы, равным образом ни один из них в ионном состоянии не парамагнитен. Элементами, стоящими непосредственно вслед за инертными газами, считают такие элементы, которые удалены от инертного газа ие более чем на три места.
Таблица 2-i
Группировка элементов вокруг ипертпых гадок
Простейшие водород- Политые соединения мер
Главные подгруппы периодической системы
Побочные подгруппы периодической системы
Основная 4-1И
валентность
Мономер (легролетуний)
Твердый (сомоораз-пшй)
Твердый (долее или менее металлоподобный)
Последний элемент кавюдого ряда ведет себя подобно первому элементу следующего ряда
| Лантаниды'. tliCe, ••’ s«Pr. «Nd. „Рт, .«jSm, бзГи, „Gd, s»fb. «Оу, „Но. мЕг, 65Ти, 10Yb, 71 LU
3Li
uNa
i»K
„Bb
„Fr esRa
jBe
isM8
S18c
J2Ti
23 V „Сг
2зГс „Со EeNi
wCu
Зв^Г 39 Y
tsCs j^Ba „La
»oZr
7;Hf
s»Ac so^h
(i^b 4JMo „Тс
„Ви „Rh „Pd
7flCe 77Ilb fgPt
«Ag
72 A U
(SC<1
^Hg
»1 pa e;U
Трансурановые: MN'p 01Pu (sAm MCna „Bk „Cf (001 m i2|Md|0£
No
Магнитные свойства
Элементарные ионы все диамагнитны
Элементарные ионы частично парамагнитны
Окраска
Образуют толы1,0 нет-ташемые злементар-iibii ипнц
Образуют большей частые окрашенные злементар-
9 ные ионщ- ’
150 Глава, 5
Достаточно приведенных «оотнощёпйй: (которые в дальнейшем буду еще пополнены), чтобы предположить, что многие свойства элементов особенно химические, должны быть связаны с их положением в пер ио диче ской системе относительно инертных газов — другими словами, свойства элементов, и прежде всего их валентность, должны существенно зависеть от того, на сколько единиц его порядковое чиело^ отличается от поряд нового числа инертного таза. Важный шаг, имевший долью поставить эта соотношений в основу учения о валентности, был сделан одновременнс и независимо двумя исследователями •— Косселем (Kosael) в Германии и Льюисом (Lewis) в Америке (обоими в период 1915-* 1'916 гг.).
Теория Косселя. Из элементов, стоящих рядом с инертными газами, рассмотрим прежде всего те, которые способны образовывать элементарные ионы*, т. е, щелочные и щелочноземельные металлы, а также алюминии, скандий и их аналоги, й затем галогены я халькогены (кислород и его аналоги).
Если вычислить общее число электронов, которыми обладагбт эти элементы в форме элементарных ионон, то легко заметить, что это число всегда равно числу электронов в атоме соседнего инертного газа.
Так ион S" имеет 16 2 электрона, ибн СР 171, ион К’ 19—1, ион
Са” 20—2, ион Sc " 21—3 электрона, т, е. каждый на них обладает 18 электронами таким же количеством, как аргон, Вокруг которого эти элементы: сгруппированы. Соответственно иолы F', Na’, Mg" и АГ” обладают 10 электронами, подобие неону, ит. д.
Если сравнить теперь химические соединения, ' образующие при электролитической диссоциации перечисленные ионы, с соединениями других элементов главных подгрупп, которые построены аналогично я обладают аналогичными свойствами, то можно предположить, что в этих соединениях атомы находятся в заряженном состоянии, т. е. в ионной форме, даже и в тех случаях, котда: ионы не содержатся в заметном количестве в свободном состоянии. Существуют соединения, ддя; которых другими путями (например, рентгенометрически, см. гл. 7) можно показать, что в твердом состоянии они также построены из противоположно заряженных ионов. Такое доказательство, например, проведено / для окиси магния Mg О (стр. 242).; Установлено, что она состоит из положительно двухвалентных ионов магния Mg‘~* и отрицательно двухвалентных ионо вскисло рода О2". Путем обобщения Коссель пригнел к выводу, что окислы других металлов также состоят из положительных ионов металла и отрицательных ионов кислорода й что вообще соединения металла с неметаллом всегда содержат металл в положительно заряженном состоянии, а неметалл в отрицательно заряженном состоянии. Далее Коссель предположил, что в соединениях неметаллов, которые построены аналогично гетерополярным соединениям, атомы заряжены противоположными электрическими зарядами, а именно: каждый более электроотрицательный атом должен быть заряжен отрицательно, а менее электроотрицательный атом вследствие этого — положительно. В соответствии с этим слабо электроположительные металлы должны иметь отрицательный ‘заряд,
* Здесь речь идет об ионах в растворах (в общем случае — Водных), Отвосителъ-: j ио'иопов в кристаллах'см. ниже. В канаяовыхлучах и т. д. могут существовать атомы 1 совершенно в аномальных состояниях, которые Не позволяют дслатЬ_заключение о их свойствах и химических соединениях.
Валентность и сродство 151
если они связаны с сильно электроположительным металлом или с водородом. Во всех случаях, поскольку речь идет о простых’молекулах, количество зарядов должно соответствовать формальной валентности._
Еще Абегг (Abegg, 1904) привел многочисленные факты, подтверждающие взгляд, что подавляющее большинство неорганических соединений построено гетеропдляр-но. Однако точных доказательств этого, помимо указанных случаев, пока нет, Поэтому следует учесть, что эти обобщения имеют гипотетический характер, т. е. это рукд--водящая гипотеза, которая ведет к общей теории и которая позволяет попять зависимость свойств, особенно валентности, от положения элемента по отношению к инертным газам. Если эту гипотезу доказать другим путем, то тогда можно судить,, в какой мере эти обобщения фактически обусловливаются теорией, насколько опв впоследствии будут лучше подтверждаться и в какой мере они должны быть подвергнуты сомнению или изменению.
Рассматривая соединения, в которых элементы обнаруживают характерную для их места в периодической системе валентность, в общем как гетерополярные, Коссель рассчитал для первых 57 элементов, до подгруппы лантанидов, количества электронов, которыми они обладают в тех соединениях, где они проявляют высшую отрицательную и высшую положительную валентности. На оси абсцисс рис. 28 элементы расположены „ в соответствии с их порядковыми числами и через равные промежутки; рассчитанное Косселём для каждого элемента число электронов нанесено в качестве ординаты и отмечено черной точкой. Те элементы, которые . могут быть заряжены и отрицательно и положительно,: имеют по две черные точки, которые конечно расположены на одной вертикали однанад другой на расстоянии 8 единиц в соответствии с тем фактом,, что сумма положительных й отрицательных высших валентностей равна 8, на что указал вал еще Аббег. Кружки на рисунке соответствуют числу электронов для элементов в состоянии нейтральных атомов: В то время как эти числа -естественно возрастают от элемента к элементу на одинаковую величину и соответственно этому лежат на прямой, расположенной под утлом 45° к оси абсцисс, черные точки для элементов, расположенных рядом с инертными газами, все лежат на прямых, параллельных оси абсцисс, и находятся от нее на том же расстоянии, как и точка, обозначающая число электронов инертного газа, вокруг которого группируются элементы. Это значит, что число электронов, которыми обладают атомы элементов, стоящих рядом с инертными газами (т. е. элементов главных: подгрупп периодической системы) в своих типичных соединениях, равно числу электронов ближайшего инертного газа. И отсюда следует: «если два элемента, например натрии и фтор, образуют химическое соединение. то один из них отдает , другому такое количество-электронов, что у каждого из них после этого -остается столько электронов, сколько их имеет ближайший инертный газ.
В некоторых местах рис. 28 встречается иное число электронов, чем число электронов у соответствующего инертного газа. Однако это наблюдается^ во-первых, только у тех элементов, которые расположены в побочных подгруппах перйодйчеейой системы, -а именно далеко от предшествующих инертных газов, и, во-вторых, отклонения чисел электронов от соответствующих чисел, электронов инертных газов Образуются только : заснет от дачи электронов и никогда за счет присоединения. Из систем, образованных путем отдачи электронов, известной стабильностью отличаются те, которые имеют 28 -и 46 электронов. :
Исходя из указанного выше положения, Коссель сделал заключение, что системы электронов инертных газов отличаются особой устойчивостью. Устойчивость эта такова, что атомы элементов., непосредственно предшествующих инертным газам, путем присоединения электронов легко
152
Глава И
образуют электронную систему, которая соответствует электронной системе инертного газа. С другой стороны, элементы, следующие за инертным газом, легко отщепляют то избыточное число электронов, которое у них имеется ио сравнению с числом электронов предшествующего инертного газа. Это заключение относительно особой устойчивости электронных
Р и с. 28. Электронные числа в нейтральных и максимально заряженных атомах, проявляющих валентность (цр Кос селю).
систем инертных газов, сделанное Коссьлем на основании химических свойств, вполне подтвердилось впоследствии чисто физическими методами, (потенциалы ионизации), как это было показано в предыдущей главе.
Гетерополярное соединение образуется, по мнению Косселя/ благодаря тому, что противоположно заряженные атомы притягиваются по закону Кулона, При этом они ведут себя в первом приближении как шары, равномерно заряженные по всей поверхности.
Итак, образованно хлорида калия (К.С1) из элементов хлора и калия основано по Косселю на том, что хлор отнимает один электрон от атома.
Валентность и сродство 153
калия. Образовавшиеся таким образом ионы СГ и К.*, следуя, кулоновскому притяжению, соединяются, образуя КС1 (кристалл). Подобный процесс происходит и при образовании MgO из магния и кислорода и т. д.
Совершенно не обязательно, чтобы оба атома принимали электронную конфигурацию одного и того же инертного газа. Вместо калия и хлора равным образом можпо выбрать натрий и хлор или литий и бром. Точно так же число электронов, которое присоединяет каждый атом одного элемента, может быть и не равно числу электронов, которое отдает каждый атом другого элемента, только в целом число присоединяемых и отдаваемых электронов-должно совпадать; Одип атом кремния отдает кислороду четыре электрона; благодаря этому два атома кислорода получают по два отрицательных заряда каждый, и, таким образом, в результате соединения Si1* и2О2* образуется молекула S1O2.
С этой точки зрения можно рассмотреть образование любых других сложных комплексных соединений;
Учитывая сказанное, теорию образования соединении Косселя можно изложить следующим образом: соединяющиеся атомы в результате перехода электронов заряжаются противоположно, а затем сближаются благодаря электростатическому притяжению, обусловленному противоположными зарядами.
Применение теории Косселя. Теория Косселя не только сводят силы химических связей к очень простому принципу, но и позволяет применить этот принцип для объяснения координационных соединений, которые раньше было трудно трактовать в соответствии с представлениями теории валентности. Более подробно об этом см. гл. 11. Далее, теория Косселя значительно превосходит старую неорганическую теорию валентности, потому что без особых вспомогательных данных —по крайней мере в болйе простых случаях — позволяет получить не только состав, но и важнейшие свойства химических соединений. Например, как будет показано в дальнейшем, с позиций теории Косселя можно сразу сказать относительно важнейших гидроокисей, ведут ли опи себя как кислоты или как основания. Впрочем, еще много раз представится случай применить теорию Косселя к конкретным проблемам. Следует здесь только показать на Отдельных, ранее уже упомянутых примерах, как теория Косселя вскрывает, естественные связи и насколько она превосходит старые теории валентности.
Прежде всего па основании представлений Косселя, оказывается, можпо объяснить нулевую валентность инертных газов (точнее, неспособность инертных газов образовывать гетер опои ярные соединения). Если способность элементов вступать во взаимодействии сводится к возникновению противоположных зарядов и в конечном итоге к стремлению принять конфигурацию инертного газа, то, Конечно, у самих, инертных газов нот основания для образования связи. Так как опп ужо обладают стабильной конфигурацией, присоединение электрона, т. е. отрицательного заряда, уже невозможно. Пег оснований также предполагать, что они могут заряжаться положительно (исключая, конечно, трубку Гейслера и экстремальпо высокие температуры). Слишком мало вероятно, чтобы они отдали свои крайне прочно связанные электроны другому элементу только для того, чтобы образовать электронную конфигурацию, которая у них уже имеется. Это заключение полностью подтверждается количественным расчетом апачепий сродства обсуждаемых реакций. ,
Теперь можно объяснить, почему Кислород и фтор никогда не существуют в шести- и соответственно семивалентном состоянии в отличие от своих более тяжелых аналогов. В химических соединениях кислород и фтор вообще не бывают положительно заряжетгпыми, так как у этих элементов электроны связаны особо прочно (ср. табл. 22 и 23). В природе пе существует элемента, который мог бы оторвать электрон от кислорода пли фтора*.
* Почему в гомеополярных соединениях О и F максимально также двух- п соответственно одновалентны, будет объяснено в гл. 15.
154
Глава 5
Йгключшне.ъънре положение ооЭорода в иериодаческой системе легко объясняется да основании теории Косселя, если принять во внимание, что он стоит в таблице до гелия. Так как Н в нейтральном состоянии обладает только одним электроном и поэтому может отдать только один электрон, он, следовательно, в определенном отношении подобен щелочным металлам, которые равным образом отдают только один электрон. Однако Н- сильно .отличается от щелочных металлов и вообще от веек металлов тем, что его электрон сравнительно прочно связан (ср. с табл. 22). Благодаря этому Н оказывается !<смет.а.'1лол1, несмотря па то, что в своих соединениях он большей частью положительно заряжеп. Его расположение перед гелием, инертным газом с двумя электронами приводит к тому, что он может быть также электроотрицательным. Чтобы приобрести конфигурацию инертного газа,, ему нужно присоединить только один электрон. Б ла год аря этому и в согласил с св о йств енным ему н смета л л йческим характером, проявляющимся также в его физических свойствах, водород, несомненно, стоит ближе к галогенам, чем к щелочным металлам..
Теория валентности Льюиса. Опубликованная Дткильбертбм Льюисом в январе 1916 г. теория валентности имеет с.теорией Косселя,: опубликованном в конце декабря 1915 г,, общий принцип, а именно: причиной образования химических соединений является особая стабильность электронных конфигураций, существующих в инертных газах. Однако Льюис в противоположность Косселю выдвигает в своих рассуждениях па пер-вый план гомеополярные соединения, В соответствии с этим он принимает, что отличающиеся особой стабильностью электронные конфигурации возникают не только благодаря полному переходу электронов от одного атома к другому, но очень часто также благодаря тому, что атомы, участвующие в образовании химического соединения, имеют общие электроны.
При создании своей теории Льюис использовал представление о строении атомов, которые теперь устарели и здесь не используются, так как для теории они несущественна. .
В 1919 г. теория Льюиса была развита Лангмюром (Langmuir). Последний показал, что применение ее в некоторых случаях,,приводит к поразительным результатам, что на ее оснований, например, можно объяснить глубокое совпадение физических свойств веществ очень различного химического характера, таких1, как СО и N2, СО2 и N2O, По теории Льюиса — Ленгмюра -именно эти вещества обладают одинаковой внешней системой электронов. Лапгмюр назвал такие вещества иеостерами. Ср. с этим также G о н Ъ е а н A., Nattirwiss., 35, 246, 1948,
Льюис и Лангмюр принимают, что инертные газы (за исключением гелия, обладающего только двумя электронами) содержат на внешних оболочках по 8 электронов. Это предположение совпадает с представлениями о строении атомов, вытекающими из теории Бора. Коссель также придерживался этого положения, не придавая ему, одпакб, в своей теории основного значения, как это делает Льюис. В; теории Льюиса положение о восьми электро иных оболочках1 является существенным. Проявление валентности он сводит к стремлению образовать как можпо больше восьмизлектронных групп (октетов по Лапгмюру). Поэтому Лйнгмюр назвал эту теорию октетной теорией валентности.
Наряду с образованием октетов Льюис и Лангмхор придают также существенное значение стремлению it образованию электронных пар (дублетов), Это представление об образовании электронных пар позднее было принято как наиболее важное, (см. ниже) Приме р:
Н '
Обычный способ изображения CI-C1 О- О Н—CI II — N—Н
Способ изображения но Льюису : CI: С1: : О : .-О : Н : С1.- II: Л : II
1 Н
Помимо способа, предложенного Льюисом, теперь часто применяют также и другие способы изображения:
I i.o-oi : н- сГ| н -х-.н -
’ Н /Я:
Валентность и сродство * 155
Льюис ввел простои способ символического изображения электронов в химиче-•скнх формулах. Те электроны, которые, но его мнению, лежат на внешней оболочке,— л только нх следует принимать во внимание при образовании связи — оп обозначает точками, которые расставляет вокруг соответствующего химического символа. Электроны, являющиеся общими для двух атомов, изображаются точками, стоящими между этими атомами, причем эти точки располагаются ближе к тому химическому символу,... который соответствует более электроотрицательному элементу. Как правило, пара точек, расположенных между символами, соответствует одному валентному штриху в обычном способе изображения. Каждая электронная пара,-^участвующая в'образовании связи, представлена, следовательно, в виде валентного штриха, соединяющего символы, а электронные пары, не участвующие в образовании связи,— в виде •более коротких штрихов, которые располагаются вокруг символа/ ..
Для теории Лыоиса—Лангмюра характерно то, что она в первую очередь занимается гомеополярными соединениями, в то время как теория Косселя,: наоборот, исходит из гетерополярных соединений. Теперь не подлежит никакому сомнению, что для типично гетерополярных соединений взгляды Коссёля по существу оправдываются. В область же менее ярко выраженных гетерополярных соединений они могут быть распространены лишь с осторожностью. Для чисто гомеопоЛярной связи Косселем были развиты представления, которые сродни представлениям Льюиса и Лангмюра, Как оказалось впоследствии, теория Льюиса — -Лангмюра дает весьма плодотворный подход, к пониманию гомеополяр-ной связи. Лежащие в ее основе положения были подтверждены квантово-механической трактовкой химической связи. Одиако нельзя не отметить того, что формулы, основанные исключительно на теории Льюиса — Лангмюра без учета дополнений и углублений, возможных при использовании положений волновой механики, и без использования экспериментального контроля, обладают чисто гипотетическим характером и что выводы из этих формул следует делать с осторожностью.
. i . v
Квантово-механическое объяснение гомеоиолярной связи. Образование гетерополярных соединений без дополнительных объяснений становится понятным с точки зрения теории Косселя, так как сила притяжения противоположно заряженных атомов обусловливается непосредственно законом Кулона. Напротив, не вполне ясно, как атомы, не имеющие, согласно теории Льюиса, противоположных зарядов, могут прочно •соединяться путем совместного обладания электронами. Лишь с использованием представлений волновой механики (квантовой механики): стало возможным удовлетворительно объяснить, как такие не имеющие против воноложных зарядов атомы в определённых случаях могут образовывать прочные связи, в основе которых лежат кулоновские силы.
Уравнение Шредингера (см. стр. 120) для системы, состоящей из двух атомов. Н, принимает следующую форму;
Э2ф . Э2Ф . й2ф , Э3ф . сРф . 8xf + ду1 + + 9у2 + +
, Ыт ( , Г 1 ,11 1 Г 1 И
+ А2 {е е L rab + ri2 ral ra2 rM '
В этом уравнении индексами 1 и 2 обозначены оба электрона, а индексами а в Ь —оба атомных' ядра.
Таким образом, гаъ. —- расстояние между ядрами, г12; — расстояние между электронами, га1 — расстояние: электрона 1 от ядра а и т. д. На основании этого уравнения Гептлером и Лондоном (Heitler, London, 1927) было рассчитано взаимодействие .двух нейтральных атомов водорода, находящихся в основном состоянии. При этом оказалось,
156
Глава 5
что для такого взаимодействия получаются два различных решения. Прй нервом решении вероятность того, что электрон одного атома находится вблизи ядра другого атома, бесконечно мала. При втором решении вероятность этого принимает конечное значение. Первое решение дает положительное значение энергии для любых расстояний между ядрами, т. е. посСпоянное отталкивание. Другое решение дает Отрицательное значение для энергии взаимодействия внутри определенной области расстояний между ядрами, т. е. притяжение. В первом случае оба электрона, принадлежащие к системе, имеют параллельно направленные спины (ff), во втором случае—антипар алл елЬпо (ti). Итак, оказывается, что при сближении обоих ядер потенциальная энергия системы либо возрастает, либо уменьшается, смотря но тому, направлены спины обоих электронов одинаково или противоположно.
Взаимодействие между обоими соединеппымя в молекулу атомами водорода называют кв анто во-механическим резонансом. Это взаимодействие соответствует известному взаимодействию двух связанных (более или менее свободно) а ня логичных маятников. Если один из них заставить качаться, в то время как другой находится в состоянии покоя, то постепенно движение первого передается второму. Наконец, второй начинает колебаться с полной амплитудой и теперь уже в покос первый. Затем процесс идет в обратную сторону и т. Д. Следовательно, между связанными маятниками происходит непрерывный обмен энергией. То же относится к двум атомам Н, связанным посредством одного или двух электронов. Они непрерывно обмениваются своим электроном (в случае иола И$) яле обоими электронами (в случае молекулы Н2).
Н| Н3 и Hi еН2 J H,ciCaH3 и Н^Нг
Поэтому силы кв аптово-мех эпического резонанса называют также обменными вилами. -В случае иона ИJ резонанс приводит к более слабой связи ядер водорода, чем в случае молекулы Н2. В последнем случае результирующее взаимодействие примерно в два раза сильнее, чем связь ядер водорода в ионе На. Для молекулы Н2 расчет дает минимум потенциальной энергии, когда оба ядра находятся па расстоянии 0,74-1O'S см ("Wang, 1928), т. е. на таком расстоянии, которое приписывается им на основании физических измерений (ср. табл. 7). Таким образом, квантовая механика может объяснить образование молекулы Н2 из двух атомов Н. Она дает также правильное значение для работы образования. Джеймс (James, 1935), сделавший наиболее точный расчет, нашел для работы образования значение 4,454±0,013 ев, которое удивительно совпало с надежными экспериментальными определениями (4,45540,001 эв, Beutler).
Следует обратить внимание на то, что взаимное притяжение обоих атомов водо-. рода (хотя спин, т. е. вращательное движение электронов вокруг собственной оси, здесь имеет значение) пе сводится волновом механикой к магнитным силам,'а объясняется электростатическим взаимодействием.
Рис. 29. Электростатическое отталкивание или притяжение в результате действия стоячих электрических волн.
Чтобы понять, наквм образом два тела, не будучи противоположно заряженными, могут путем взаимодействия стоячих электрических волн проявлять одно относительно другого электростатические силы, следует представить себе два параллельно лежащих рядом куска проволоки одинаковой длины, в которых возбуждаются (путем индукции) стоячие электрические волны (см, рис. 29). Если пр о во-л ока нс заряжена, электрические заряды колеблются в пей в разных направлениях. Если к началу периода колебания заряды распределены так, как это показано вытянутыми волновыми линиями на рнс. 29, то после одного нолупериода заряды меняют свои знаки (заштрихованные области), после следующего полупериода снова устанавливается первоначальное распределение зарядов п т. д. Если колебание зарядов в обеих проволоках происходит в одной фазе, как это показано на рисунке слева,
Валентность и сродство
157
io проволоки должны отталкиваться, так кан в этом случае одноименные заряды находятся в ближайшем соседстве. Если заряды колеблются в равных фазах (на рисунке справа), то в ближайшем соседстве находятся разноименные заряды и вследствие этого проволоки должны притягиваться.
Расчеты Гейт л ера Лондона (Heit.ler — London) показали, что мерой гомео полярной валентности атома во многих случаях служит число так называемых неспаренных электронов. Под спаренными электронами понимают два электрона одного и того же атома®, все квантовые числа которых совпадают, за исключением спиновых квантовых чисел (которые у них противоположны). Например, у Пе в нормальном состоянии оба электрона спарены (ср. стр. 145). Так как Не не обладает песпаренными электроламп, оп столь же мало способен образовывать гомоополярные соединения, как и (вследствие своего высокого потенциала ионизации) гетер о полярные соединения. Литий, наоборот, как и Н, обладает неспаренным электроном; поэтому литий может (кроме гетерополярных) образовать также гомеополярные соединения. Так, например, атомы Li ыогут соединяться в молекулу Ы2. Расчет дает, однако, в этом случае (в совпадении с опытом) значительно меньшую величину устойчивости соединения, чем в случае молекулы Н2 (1,14 по сравнению с 4,45 эв}.
Значение теории Гейтлера — Лондона заключается главным образом в том, что она показала принципиальную возможность свести гомеопо-лярнуто связь к известным силам. Область ее применения, однако, ограничена из-за математических трудностей, которые встречаются при обработке более сложных систем. Следует остерегаться Неосторожных обобщений, не подтвержденных точными расчетами. Без сомнения, недопустимо делать из зтой теории вывод, что гомеополярная связь всегда основывается па «спаривании» имеющихся в компонентах соединений неспаренных электронов.
Например, путем строгих квантово-механических расчетов, которые совпали с результатами измерений, Теллер (Toller, 1930) показал, что молекула Н£,_содержащая только один электрон связи в качестве свободного иопа, более стабильна (энергия диссоциации 2,7 эв), чем молекула Li2, в которой связь образуемся Двумя спаренными электронами.
В результате спектрографических и других наблюдений (см. стр. 742 и сл.) установлено, что молекула О2 имеет два песпареппых электрона. Следовательно, при ее образования не происходит спаривания всех электронов, и, несмотря на обладание песпаренными электронами, две молекулы 02 не соединяются с образованием валентного соединения О4 (см. ниже). Кроме того, как следует из спектрографических данных, молекула NO содержит неспареппый электрон, и, несмотря на это, две молекулы NO, насколько известно, не образуют соединения. Уже это небольшое число примеров показывает, что предложение относительно спаривания электронов (так называемое «спиновое насыщение») при образовании атомами гомсополярных соединений следует обсуждать только как рабочую гипотезу, которую нужно применять с осторожностью. Использование этой гипотезы без проверки, если даже получаемые при этом формулы находятся в соответствии с точными результатами измерений (например, спектральными данными), рискованно. Это не лишне подчеркнуть, так как вытекающие отсюда электронные формулы во все возрастающей степени применяют вместо старых структурных формул.
Перенесение точных методов расчета валентного числа и энергии связи, использованных Гсйтлером и Лондоном, на более тяжелые атомы н более сложные со единения потерпело неудачу вследствие встретившихся при этом математических трудностей. Однако Слейтору (Slater, 1931) и Полингу (Pauling, 1931) в результате дальнейшего развития метода Гейтлера — Лондона и Др. удалось квантово-механическим путем рассчитать валентные углы. Так, в случае углерода (ср. стр. 1(53) была получена т<:тра-эдрическая. ориентация четырех валентностей в пространстве, что совпало с существующим до' этого представлением. Другим методом кванте во-механического расчета гомеопоЛярной связи, применяющимся в случае более тяжелых атомов, является метод Хупда (Hund, 1923) и Малликена (Mulliken, 1928). Их результаты качественно совпадают с наблюдениями. Этот метод позднее был развит Герцбергом (Gerzberg) (см. Debye, Leipzig, Vortriige 1931). On основан но существу на том, что эпергети-
* Электроны, общие для двух атомов, считают принадлежащими одному й Другому атому. *
158
Глава 5
Ческие уровни (соответственно квантойло состояния) отдельных электронов в свободных атомах сравниваются: с энергетическими уровнями, найденными при определенных допущениях для тех же электронов в молекулах, образованных из атомов. Смотря но тому, переходят электроны прц соединении атомов па более высокие уровни или нет, следует различать разрыхляющие и связывающие электроны. При этом приближенно действует: простое правило, что стабильная молекула образуется тогда, когда число связывающих электронов больше, чём число разрыхляющих Л исло связывающих электронных пар, уменьшенное па число разрыхляющих электронных пар, дает валентность связи. Например, из вычисленных Хупдом тшантовых состояний следует, что 2 атома азота во внешних оболочках* содержат 8 связывающих и 2, разрыхляющих электрона; в результате получается 8--2=6 связывающих электронов и соответственно 3 связывающие электроппыё пары, так что молекула N2имеет тройную связь. Молекула О2 содержит на 2 электрона больше, чем молекула N3, и именно на 2 разрыхляющих электрона больше. Так как опи должны компенсироваться одной связывающей электронной парой, следовательно, остаются две связывающие электронные пары, в. результате чего получается двойная связь между двумя атомами Кислорода, которая менее прочна, чем тройная связь между двумя атомами азота. Соответствующим образом для, F2 получают еще менее прочную простую связь, так как здесь присоединяются еще 2 разрыхляющих электрона. Два атома Ne вообще пе могут соединяться, так как в результате-добавления еще 2;.следующих разрыхляющих-электронов'теперь все связывающие электронные пары компенсируются разрыхляющими. Как было сказано, молекулы О2 содержат по 2 неспареннЫх электрона. Если бы принцип образования «спаренных электронов» был бы в общем случае решающим дня образования голе о полярных соединений; то следовало бы ожидать, что две молекулы Ог путем спариваний электронов могли соединяться с образованием . валентного соединения О4, которое, согласно опыту, неустойчиво**. В соответствии с теорией Хунда и Малликена образование валентного соединения О4 из двух молекул О2 исключено. Это объясняется тем, что ,2 неспаренных электрона в молекуле О2, как следует из их квантового состояния, являются разрыхляющими электронами. Вследствие Этого соединение со второй молекулой Ог, которая также имеет 2 разрыхляющих электрона, не образуется. Напротив, с одним атомом О, который, согласно изложенному выше, имеет 2 связывающих электрона, может образоваться: соединение (образование 0л). В соответствии с этим Оказывается, что NO хотя и не может соединиться со второй молекулой NO, одпако соединяется с атомом О (образование ..ДО2).
Для проявления гстерополярной валентности решающее значение, согласно изложенному выше, имеют только те электроны, которые расположены вне оболочек инертного газа, так называемые «внешние электроны» атома. По существу то же относится к проявлению и гомеополярной валентности. Вследствие особого значения внешних электронов для про-явления валентности их называют, валентными электронами.
В общем можно сказать, что у элементов главных подгрупп периодической системы максимальная валентность, проявляющаяся в гетеропо-лярных соединениях, определяется общим числом внешних электронов,. а максимальная валентность в гомеополярных соединениях определяется числом неспаренных электронов. Однако следует обратить внимание, что гомеополярные соединения не всегда образуются из атомов в основных состояниях, а в некоторых случаях из атомов: в возбужденном состоянии, в котором число неспарепных электронов больше, чем. в основном состоянии. Например, в первом: периоде системы для электронных: конфигураций в основном состоянии атома, и ...для чисел неспаренных электронов приняты следующие значения;
Li Бе В : с. . : К : О ' Ne.:-
Электронные конфигурации s «а з2р s2p3 s-p3 X2ps j'2pe
Неспареппые электроны 1 0 1 2 - J ... 2. 1 °
* Взаимодействием внутренних оболочек (оболочек гелия) можно пренебречь.
** В сильно сжатом или сжиженном кислороде, .правда, Имеются молекулы (О2)2г однако можно показать, что их образование основано па действии вандёрваадьсовых сил, а не па подлинном проявлении валентности.
Валентность и сродство
15ft
Если бы гомеополярные соединений во всех случаях происходили из основного состояния атома, то углерод должен был бы тогда проявлять максимальную валентность два, а бор — максимальную валентность один; бериллий вообще не был бы способен образовывать гомеополярные соединения. Тот факт, что в гомсополярных соединениях, так же как в гете-. рОполярных, бериллий может быть двухвалентным, бор трех-, а углерод четырбивалентным, показывает, что соответствующие соединения происходят из возбужденных состояний атомов, и именно из таких со- : стояний, в которых все электроны неспарены. Это состояния sp, sp2 и sp3. Как следует из спектрографических данных, требуется, лишь относительно небольшая затрата энергии, чтобы электроны указанных атомов поднять с e-уровня на р-уровень. Требующаяся для этого затрата энер-i гий с избытком^ компенсируется получением энергии, которое связано с: образованием соединения.
Обозначение электронных [конфигураций в молекулах. Для обозначения электронных конфигураций в молекулах могут служить те же сим-: волы, что и для обозначения строения атома (ср. стр. 145), при этом следует только, кроме главного квантового числа п и орбитального квантового числа I, применять еще новое квантовое число К, для которого принимают а ;
(Д как I, постоянно, целочисленно и всегда положительно). Так же как зпачепие I задается буквами s,p, d, j, значение X задают буквами or., л,, Й; ф. Итак, о-электрон это такой электрон, для которого X—0, для я-элек-трона Х=1, для 6-электрона V=2 и для <р-электррна Х=3. В случае молекулы Н2, находящейся в основном состоянии, для обоих электронов п=1, /=0 и, следовательно, Х=0- Итак, конфигурация электронов молекулы Н2 выглядит так: 1 so2, «Внутренние» электроны, т. е. те, которые расположены вокруг отдельных атомных ядер на завершенной оболочке инертного газа, в символах электронных конфигурации молекул не: учитывают.‘ Например, электронная конфигурация существующей в парообразном углероде молекулы С2 передается символом 2so22po2 2рлД То есть молекула Сг содержит (кроме двух оболочек гелия, окружающих ядра углерода) 2 электрона,:для которых /=0и 4=0, 2 электрона, для которых 1, л=0, и 4 электрона, для которых как I, так и Х=1-
Квантовое число X можно наглядно пояснить следующим образом:: энергетическое состонние молекулы зависит от того, имеют, ли облака валентных электронов (посредством которых может быть образно выражена зависимость вероятности пребывания валентных электронов от пространствеппого .направления); свою наибольшую протяженность в направлении линии, соединяющей атомные ядра, или в другом направлении, например перпендикулярном первому. В соответствии с этим sna-чеяие X получают, принимая во внимание пространственное направление орбитально-вращательного импульса, проектируя I на линию, соединяющую .оба. атомных ядра. Если I перпевднкулярно, то проекция имеет значение 0. Если I имеет то же направление, что и линия, соединяющая ядра, то оказывается: Х=/. В случае промежуточных направлений орвитально- вращательного импульса дия К, которое как квантовое число всегда должно быть целочисленным, получают столько различных значений, сколько целых чисел имеется между Он/. Отсюда сразу получается"приведенное выше отношение между >. и
Переходы гетсрополярной связи в гомебполярную. Предположение, что противоположно заряженные атомы, соответственно, ионы, ведут себя как равномерно заряженные электричеством шары (ср. стр. 153),
160
Глава 5
оправдывается лишь приблизительно. Это предположение тем менее допустимо, чем сильнее поляризационное действие, которое оказывают ноны один па другой, когда они сближаются на атомарные расстояния. Поляризацию можпо наглядно представить как деформацию электронных оболочек ,
Электронные оболочки отрицательных ионов деформируются под действием сил положительных ионов, и тем сильнее, чем меньше радиус и чем больше заряд положительных ионов. Далее, отрицательные ионы деформируются- тем сильнее, чем опи больше; например, ион S3* деформируется сильнее, чемиопО3".Ионы с конфигурацией инертного газа проявляют меньшее деформирующее влияние, чем остальные ионы. Поэтому влияние деформации в общем случае становится особенно сильным в соединениях элементов побочных подгрупп.
Известно, что в ряде случаев первый элемент главной подгруппы периодической системы аналогичен в своих свойствах второму элементу следующей главной подгруппы, как, например, Li и Mg, Be и Al, В и Si. Это явление большей частью обусловлено тем, что ионы этих элементов оказывают аналогичное деформирующее действие. Это происходит потому, что оба фактора, оказывающих влияние на величину деформирующего действия — повышение заряда и увеличение атомного радиуса,— здесь приблизительно компенсируются.
С увеличением деформации аниона свойства гетеропоиярпых соединений (ионных соединений) все больше приближаются к свойствам томео-полярных соединений (атомных соединений). Это объясняется том, что с увеличением деформации отрицательного иона его электронная система все более приближается к электронной системе катиона илй даже вовлекается в нее. Благодаря этому все в большей мере становятся определяющими силы взаимодействия между электронами (резонансные силы), к которым квантовая механика сводит гомеополярную связь (атомные соединения). Таким образом объясняется, почему между гетероцолярны'ми и гомеополярпыми соединениями нельзя провести резкой границы; Напротив, здесь можно наблюдать все возможные формы переходов, хотя оба вида связи принципиально различны.
Принципиально между двумя составляющими соединение частями всегда действуют силы, к которым квантовая механика сводят гомеополярную связь. В идеальных ионных соединениях (так называемых чисто гетер о полярных соединениях) этп силы практически не влияют на притяжение ионов с противоположными зарядами. Отталкивание, которое йсходя из квантово-механических представлений появляется в случав, если сближение превышает определенное расстояние, может быть здесь заменено представлением о почти жестких шарах, радиусы которых, а также сжимае~я мость определяются эмпирически*. При умеренной деформации силы гомео по л яркий связи также можяо по принимать в расчет, если вместо них учитывать поляризационный эффект, который получается из эмпирических данных**;
В чисто гемеополярных соединениях действуют одни гомеополярные силы связи. В промежутке лежит широкая область соединений, где для полного особо точпргр количественного описания связи следует принимать во внимание оба вида сил.
Если в принципе в каждой связи существуют кваптово-мехапические силы взаимодействия (притяжение или отталкивание) и лшиь при этом условии на них накладывается прптяжение, возпикающее между противоположно заряженными ионами, то становится ясным, что это дополнительное действие ионной связи только увеличивает прочность связи, по никогда не может понизить ее. Используя представление О таком наложении, Полинг указал метод, при помощи которого для случаев так называемых смешанных гомео- и гетерополярных соединений, расположенных между экстремальными типами соединений, можно приблизительно рассчитать часть энергии
* Как показал Енсеп (Jcacen, 1936), силы отталкивания, возникающие при сильном сближении ионов, согласно кваптово-механическим расчетам, являются основанием для представления о почти жестких шарах.
** Квантово-механическая трактовка связи позволяет в принципе оценивать также поляризуемость ионов.
Валентлюстъ и сродство
161
связи, приходящуюся на ионную связь. При этом делается предположение (правда, не строгое), что чисто гомеополярные энергии связи аддитивны, т. е. что в случае чисто гомеополярных соединений оказывается: А <—> В —1/2 (А <—> А В *—> В), где А»—► В,
А и В »—>В означают эпергни связи молекул АВ, АА и ВВ. Например, энергия связи II2 составляет 4,45,12 — 1,54 и F2 — 2,80 os па молекулу. Отсюда, согласно сде-липпому предположению, рассчитывается энергия, которая приходится на гомеополяр-цую долю связи для HI 3,00, для HF 3,63 эе. Экспериментально найденные значения энергии связи составляют 3,07, соответственно 6,39 эв- В случае Ш рассчитанное значение только немного отклоняется от экспериментально найденного; в этом случае существует, следовательно, почти чистая гомеополярпая связь, о чем свидетельствуют также многие свойства соединения. Напротив, для HF рассчитанное значение энергии связи намного меньше экспериментально найденного. Отсюда Полинг сделал заключение, что в этом случае значительная доля энергии связи (2,76 эв, следовательно, около 43%) приходится на гетер on олярпую часть связи, что также хорошо согласуется со свойствами этого соединения, которые свидетельствуют о его сильно полярном характере.
Аналогичные сравнения Полинг сделал для многочисленных соединений. При этом оказалось, что различные элементы вносят в общую энергию связи тем болъ-. шую долю энергии ионной связи, чем более они различаются по своему электрохимическому характеру. На этом основании элементы можпо расположить в ряд в соответствии с уменьшением, электроположительного характера или возрастанием электроотрицательного характера, который опи проявляют в своих соединениях. Этот ряд представлен в табл. 25.
Таблица 25
Знач ния электроотрицательного характера (электроотрицательности)
элементов no Полингу
Cs 0,7 Ca 1,0 Ti 1,6 Те 2,1 Br 1,8
Rb 0,8 Mg 1,2' Sa 1,7 В 2,1 Cl 3,0
К 0,8 Y 1,3 Ge 1,7 H 2,1 N 3,0
Na 0,9 Sc 1,3 Sb 1,8 Se 2,4 0 3,5
Ba 0,9 Be 1,5 SI 1,8 I 2,4 F 4,0
Li 1,0 Al 1,5 As 2,0 S 2,5
Sr 1,0 Zr 1,6 В 2,0 c ’ '2,5
Семиполярная связь; электровалентность п ковалентность. Особый вид наложения гомео- и гетер о полярной связей представляет семиполярная связь. В качестве простого примера такой связи может быть использована о к ис ь уг лерода (СО), строение которой, правда, еще нельзя считать пполпс доказанным. Вследствие необычайного сходства физических свойств СО и N, уже Лапгмюр в 1$)19 г. сделал предположение, что в молекулах этих веществ имеются совершенно подобные конфигурации олектропов («изо-стеризм», см. стр. 154), т. е. ;С ; СК и соответственно ; N: . Это возможно только в том случае, если атом О отдал атому С один электрон, слсдовательпо, если С заряжен отрицательно, а О положительно. По тогда в соединении СО оба атома связаны как в результате взаимодействия противоположных зарядов, так и вследствие взаимодействия шести спаренных электронов. Итак, здесь имспно тот случай, который изображен на рис. 29 (справа), если»предположить, что в системе на обеих проволоках не только возникают стоячие электрические волны, по проволоки, кроме того, еще заряжены, причем противоположными зарядами. Тогда обе проволоки притягивались бы как под действием постоянных противоположных зарядов, так и вследствие сия, образуемых осциллирующими зарядами.
В отличие от электровалентностей, обусловленных противоположным зарядом атомов или ипов, валептпостн, не обусловленные противоположными зарядами, называют, согласно Сиджвику (Sidgwick), ковалентностями. В случае семиполярных соединений электровалентности в большинстве случаев изображают знаками -ф- и —, ковалентности, поскольку электронные формулы не применяют,— посредством ’штрихов. Изложенные выше представления относительно строения соединения СО, в соответствии с которыми углерод проявляет в нем одну (отрицательную) электровалентность и три ковалентности, можно передать формулами:
С = О или : С :: О :
11 Г. Реми
163
Глава 5
Очень небольшой дипольный момент молекулы СО (см. стр. 483) можно объясни предположив, что существует кваптово-мехаинческий резонанс между атом формой и другой формой СО, представляющей также семиполярное соединение, которое мое быть передано формулой
J -б или : С : 0 :
Подробнее об этом см. стр. 483 и сл.
Дативная связь. При образовании молекулы СО атом О отдаст один электр атому С и благодаря спариванию с этим электроном другого кислородного электро образуется третья связь с атомом С. Это можно выразить также следующим образо оба спаренных электрона, благодаря которым возникает третья связь между С и преЭост.яйляк>вгся атомом О (а не наполовину от атома С и наполовину от атома как это Происходит в случае двух других пар электронов связи в молекуле СО).
Существует много соединений, возникновение которых свободно можно было ( объяснить, предположив, что обусловливающие связь спаренные электроны происх дят не поровну от каждого из связанных атомов, а оба— от одного итого желтом В таких случаях говорят о дативной связи, (авгл.—aovalency). Понятт дативной связи не перекрывается полностью понятием семпполярпой связи, так к; .дативная связь не обязательно должна иметь следствием противоположные заряд атомов, между которыми она образовалась. Если в молекуле СО между обоими атомам имеются три обусловленные электронными парами связи, из которых одна ими характер дативной связи, то эта Молекула пе обязательно должна иметь вследствт этого сильно полярный характер. Ее небольшой дипольный момент, помимо пр па денного выше предположения, можно было бы объяснить также, приняв, что элеы роны, которые вызывают связь, в среднем находятся чаще в окружении ядра О, че ядра С. Благодаря этому различные заряды обоих ядер более или менее компенсирт ются. В результате сомиподярная связь оказывается особым случаем дативмо связи, а именно тем случаем, при котором вероятность нахождения электронов свял вблизи каждого из двух атомных ядер (связь которых они образуют) одинакова нд только немного различается.
Гибридная связь. Образование такого гомеополярного соединения как СН4 (или вообще соединения типа CRJ, согласно октетной теорш валентности, основано на том, что углерод дополняет свою внешнюю оболочку до восьмиэлсктропной путем спаривания своих четырех электроноЕ с электронами того атома, ©которым он образует связь. Такая восьмиэлектронная оболочка имеется в ближайшем, следующим за углеродом инертном газо пеоне. Неон имеет на внешней оболочке электронную конфигурацию Напрашивается предположение, что каждой из четырех связей, образованных углеродом в соединении типа СН4, занята одна из четырех электронных пар', которые имеются в воермиэлектроппоп, оболочке, и, следовательно, что от восьмиэлектронной оболочки с конфигурацией электронов ssjDe (образованной достраиванием четырех электронов) одна связь образуется при участии обоих у-электронов, а каждая из остальных трех связей — с участием двух р-электронои. Однако в действительности это не так. Полипг и др. (1928), применив : квантовую механику, показали, что в каждой из четырех связей участвуют кап .5-, так и р-эдектроны. Следовательно, речь идет при этом о связях, которые не образованы ни х- ни р-элактронами. Следует говорить об особом виде, связи, которая возникла в результате комбинированного действия у-и р-электронов. Поэтому эту связь обозначают по Полипгу как гибридную связь (нем/ Zwit terb in dung, англ, hybrid bond).
Доказательство существования такой гибридной связи основано на том, Что в случае каждой связи при комбинировании волновых функций и р-алоктропов получаются более высокие значения энергии связи, чем если бы для расчета связен применяли эти волновые функции порознь. Если принять прочность связи, образованной одной i-электронпой парой, равной 1, то для связи, образованной р-электропной карой, полу
Валенгпностъ и сродство
163
чают значение приблизительно V3; напротив, путем комбинации волновых функций s-и р-электронов для каждой из четырёх связей получаются равпые значения, а именно 2. Оказывается далее,: что эти четыре связи направлены по углам тетраэдра. Оба эти результата совпадают с представлениями, Полученными уже давно из химии соединений углерода. Для негибрпдпых связей это бы пе имело места.
Гибридная связь имеет значение пе только в соединениях углерода. Как будет показано в гл. 11, понятие об этой связи также важно для выяснения строения других соединении.
Одпоэзектроппая связь. Несмотря на то что в большинстве случаев гомемюляр-ная связь основана па том, что связанные атомы обладают одной или несколькими общими электронными парами, онаможет осуществляться и при помощи одного электрона. В таких случаях также имеется возможность для проявления квантов о-механического резонанса, а следовательно, так называемых’ обменных сил. Это можно но казать уже на примере иона HJ (см. стр. 157). Работа, которую нужно затратить в случае молекулы Hi, для разрыва одно»лектроиной связи (т. е. для перехода Hi —> Н 4г Н*) оказывается равной, согласно спектрографическим данным 2,65 эв. Для разрыва связи, образованной одной электронной парой, в молекуле Г12 нужно затратить почти двойную работу (4,48 эв). Следов атсльпо, молекула Н3 значительно стабильнее, чем яоп Н£, Если же соединяются два неодинаковых атомных ядра, то в случае одпоэлектрон-ной связи обменные силы еще меньше. Этим объясняется, почему по известно соединение, существующее при нормальных условиях, в котором одиоэлектрбнная связь имела бы существенное значение.
Трех элен тропна я связь. Большее значение, чем одноэлектронная связь, имеет трехэлектронная связь. Опа проявляется, по данным Полинга, например, у таких молекул, которые, как NO, NO, и СЮ2, содержат нечетное число электронов. Самым простым примером молекулы, в которой существует трехэлектронная связь, является (спектрографически обнаруженный) иоп HeJ. Из следующей схемы (в которой химические символы соответствуют не атомам, а их ядрам) становится попятным, что при этом, так же как в случае иона Hi, могут проявляться обменные силы:
HqH и Hq Н | HeQ ®Ие и Ilog дНе
Расстояние между ядрами в молекуле Het, полученное из спектрографических данных, совпадает со значением, рассчитанным квантово-механическим путем, и оказывается равным 1,09 А, а работа диссоциация оказывается равной 2,5 зв. Трехзлек-тронная связь поэтому не прочнее, чем одно электронная связь; все же опа имеет значение, так как накладывается ня обьиную го.иеополярную связь (т. в. такую, которая образована одной пли несколькими электронными парами). Так, например, Полинг объясняет образование соединения NO, предполагая, что в нем имеет местрквантрво-мехапический резонанс между следующими формами:
+ +
:N=:: O:, : N :: О :N : О : и : N : б :
Электропы, в результате обмена которых образуется трехэлектронная связь, обозначены при этом более жирными точками, чем остальные. Если бы связь между N и О по становилась бы прочнее в результате наложения трех электро иной связи* на обычную, то следовало бы ожидать, что благодаря димеризации молекул NO в N3O2 (путем спаривания пег,паренных электронов) освобождалось бы так много энергии, что двойные молекулы были бы устойчивы не только в жидком, nd и в газообразном состоянии, В газообразном состоянии, однако, NO существует в форме простых молекул.
Так же как и в случае одноэлекгронной связи, при трехэлектронной связи резонанс проявляется только между такими формами, которые обладают почти рал пой энергией. Этим объясняется, что молекулы или радикалы, для образования которых имеет зпа-
* Чтобы объяснить увеличение прочности связи, достаточно предположения относительно квантово-механического резонанса между двумя первыми формами. Резонанс этих двух форм с третьей принят для объяснения очень небольшого дипольного момента молекулы NO. Одиако тогда следует ожидать, что в резонансе е этими тремя формами окажется также и четвертая форма.
I. Ц*
164
Глава 5
чение трехэтгектронцая связь, получаются лишь благодаря соединению таких атомов, у которых наряды ядер не отличаются или мало отличаются один от другого. Такие соединения будут не раз встречаться в дальнейшем. Трех электронная связь, согласно Полингу, обозначается более простым способом при помощи знака.... Имеющуюся в молекуле Но| резонансную связь обозначим, следовательно, так: Не - • -Не*. Д существующую, например, в нормальном состояния в молекуле О2 связь, состоящую, но Полингу, из обычной простой связи, на которую накладываются две трехэлектронные связи, обозначим следующим образам (ср. гл. 15): О О; или если нужно подчеркнуть, что каждый из двух атомов кислорода содержит пе участвующие в образовании связи электроппые пары, то запишем так.* | О О |.
ХИМИЧЕСКОЕ СРОДСТВО
Чтобы сделать заключение относительно стабильности соединения и условиях его получения, большей частью не требуется знать природу сил связи. Для этого достаточно знать величины сродства расслштриваемых реакций. Сродство можно определить различными методами, которые базируются па измерениях величин, доступных непосредственному наблюдению, определение которых не требует: подробного знания сил связи.
Под сродством химической реакции со времен Вант-Гоффа (1883) понимают максимальную работу (точнее, максимальную полезную работу), которую можно получить при превращениях единицы массы вещества. За единицу массы при этом принимают моль. Обычно сродство реакции относят к числу молей, которое вступает в рассматриваемую реакцию. Сродство имеет размерность: работа/количество вещества. Его величину обычно выражают в калориях.
Максимальна л работа и полезная работа. В соответствии с принципами термодинамики процесс (например, химическая реакция) только в том случае может протекать самостоятельно, т. е. без подвода энергии, если он в состоянии производить внешнюю работу. Процесс дает наибольшую величину впошней работы (максимальную работу), если он протекает изотермически и обратимо. Если процесс идет при постоянном давления р н с этим связано изменение объема ДЕ рассматриваемой системы, то произведенную внешнюю работу мощно разложить на две составляющие
& = £hi + MV. (1)
Вторая составляющая работыуДЕ должна производиться дополнительно, если процесс чает при постоянном давлении; опа не является полезной. Только составляющую Ущ. можно использовать; поэтому ее и называют, согласно Эйкену, полезной работой. Для химических процессов максимальная полезная работа, отнесенная к единице массы реагирующих веществ, служит мерой сродства А/. Следовательно, считается (при отнесении к одному и тому же количеству вещества): Af
Понятие химического сродства было введено в качестве меры стремления веществ соединяться одно с другим. Говорить, о таком стремлении имеет смысл только в том случае, если рассматриваемые вещества реагируют самостоятельно, т. е. без внешнего воздействия (без подвода энергии)*. Логично оценивать это стремление по тому, насколько велики силы противодействия, которые потребовались бы, чтобы не дать возможность веществам образовать Соединение. При обратимом процессе силы противодействия в каждый момент равпы силам, действующим в рассматриваемой системе. Таким образом, работа, которая может совершиться, если реакция протекает обратимо, т. е. максимальная работа оказывается мерой сродства реакции. Так как в большинстве случаев имеют дело с реакциями, которые протекают при постоянном давлении (обычно атмосферном давлении), то целесообразно применять в качестве моры стремления веществ к образованию соединения пе всю внешнюю работу, а только ту ее часть, которая была определена как полезная работа».
* Сюда относятся, конечно, по такие воздействия, которые влияют лишь на скорость реакции, ибо без них реакция также протекала бы, хотя Цри известных обстоятельствах, чрезвычайно долго.
Валентность и сродства
165
Работа образования и сродство образования*. Максимальную полезную работу, которую можно получить при образовании соединения из его составных частей, называют работой образования или свободной энергией образования F данного соединения. Этой величине придают отрицательный знак, если работа совершается процессом**, т. е. если сродство процесса положительно. Работа образования (свободная энергия образования) и сродство равны одно другому с противоположными знаками, если они отнесены к равному количеству вещества. Так как сродство чаще всего относят к таким количествам веществ, которые входят в уравнение реакции (в обычной формулировке), сродство, отнесенное к единице массы вещества (моль), образовавшегося при реакции, называют обычно особым образом; его обозначают как сродство образования данного вещества. Сродство образования обозначается в дальнейшем всегда знаком сродства А/, снаба;енным в качестве индекса формулой образовавшегося вещества.
Например, сродство Л/ реакции Н2 + Cl2= 2НС1 составляет 45,4 ккал (при 25°); сродство образования хлористого водорода в два раза меньше: Л/нс1 == 22,7 ккал/моль (при 25°). Приблизительной мерой сродства реакции часто является теплота реакции W. Благодаря тому что вместо работы обраеоианип часто оперируют сродством образования, удается избежать недостатка, который вызывается большей частью параллельным употреблением величин теплоты реакции и работы реакции с противоположными знаками. Последнее бывает вызвано необходимостью, с одной Стороны, придерживаться обычного в хтши способа обозначения (теплоты решищЙ положительны в .экзотермических реакциях) и, с другой стороны, не нарушать соответствия с обозначениями, принятыми в термодинамике (произведенная работа отрицательна).
Если речь идет просто о сродстве, т. е. если специально пе учитывают зависимость сродства от отношения концентраций участвующих в реакции веществ, то под этим понимают сродство, которым обладает реакция, если все молярные концентрации (или в случае газов давление) участвующих в реакции веществ равны единице (так называемое «нормальное сродство», ср. стр. 168).
Температурвая зависимость сродства. Как следует из термодинамики, зависимость сродства от температуры при постоянном давлении можно представить уравнением z
dT Т ' < ’
где И7 — тепловой эффект реакции (при постоянном давлении). Выраже-
Af—W
нпе —у,—- представляет возрастание энтропии системы при реакции. Следовательно,
At—W—T&.S или :
* Термин «сродство образования» (Bildua^saffinitat) пе является общепринятым; обычно вместо него пользуются термином «свободная энергия образования» со знаком минус. Мерой убыли свободной энергии при образовании соединения является получаемая максимально полезная работа. — Прим. ред.
** В термодинамике условились .величины энергии, подводимой к системе, считать всегда положительными, а величины энергии, отводимой от системы, — отрицательными. Поэтому А обозначает затраченную работу, —А (для этого употребляют ташке знак «$-») — произведенную системой работу. В соответствии с этим Q обозначает подведеппое к системе тепло. Следовательно, IV — — Q, если IV — теплота реакции в обычном в химии обозначении, т. е. тепло, свободно выделяющееся при химической реакции. Соответственно: Af — —Р.
166
Глава. 5
если А5 —возрастание энтропии '‘при реакции, или, короне, энтропия реакции*.
При At > IV энтропия системы возрастает, т. е. система, если реакция протекает изотермически, получает тепло из окружающего пространства. Оно пе может превратиться в работу без понижения температуры окружающего пространства. В случае обратимого процесса тепло снова уходит в окружающее пространство, если реакции) заставить протекать в обратном направлении.
При энтропия системы понижается, т. е. если процесс протекает изотер-
мически, то тепло уходит в окружающее пространство. Равным образом без понижения температуры тепло не может превратиться, в работу. Например, при 18° сродство образования воды на 11,7 кедл/.иоль меньше, чем теплота образования воды (см. стр. 167). Если 18 а гремучего газа при 18° дать обратимо превратиться в воду (это мржно. при4 ближенпо 'осуществить, если процесс проводить в гальваническом элементе из водородного и кислородного электродов), то в качестве энергии, производящей работу, можно использовать лишь величину .4/и,0= 56-7 ккал (в нашем примере в качество электрической энергии), хотя теплота образования составляет П7щ0 = 68,4 ккал. Разница отдается в виде тепла в окружающее пространство. Энтропия воды, следовательно,при данной температуре (Т == 29 Г1 К.) па 11,7/291 = 0,039 ккал меньше, чем энтропия гремучего газа. Для разложения 18 г воды, наоборот, нужно использовать только такое количество электроэнергии, которое соответствует 56,7 ккал. Разница в количестве 11,7 ккил получается из окружающего пространства, если процесс идет изотермически и обратимо (ер. стр. 167 и си)л
Определение величин сродства. Важнейшими методами определения не личин сродства являются следующие;
1. Определение величины сродства из электродвижущих сил
Превращение, протекающее в гальваническом элементе, производит электрическую энергию, величина которой дается произведением напряжения иа клеммах и количеством электричества, перешедшего от одного полюса к другому. Максимальная работа совершается в том случае, если при превращении электричества в тепло не происходит потерь энергии. В соответствии с этим напряжение на клеммах достигает наивысшего значения, если прохождению тока препятствует точно компенсирующая напряжение противодействующая сила, следовательно, для элемента в обесточенном состоянии.
Если допустить, что ие происходит никаких необратимых побочных реакций, до процесс обратим. Итак, максимальная работа и тем самым сродство в случае превращения, происходящего в обратимо работающем гальваническом элементе, задается произведением напряжения на клеммах элемента в обесточенном состоянии и количества электричества, которое было бы перенесено от однрго полюса к другому, если бы превращение не сдерживалось противодействующей силой. Это количество электричества дастся изменением зарядов, участвующих в превращении ионов. Следовательно,
A; —nF!:, (3)
* Энтропия определяется как: U — А = TAS. Здесь U — возрастание общей энергии, любой системы. Эта величина в случае химической реакции, протекающей без выполнения работы, задается теплотой реакции при постоянном объеме (с обратным злаком). Если реакция протекает при постоянном давлении (т.о. с совершением работы), то оказывается: Z7=—W — рДЕ, где IV — теплота.реакции при постоянном давлении. Отсюда следует; Af — W Г AS, если принять во внимание, что, согласно определению, — А — А] рДЕ. .
Валентность и сродство
167
где п — изменение зарядов ионов; F — количество электричества, связанное с 1 г-ионом водорода, и Е — напряжение на клеммах элемента в обесточенном состоянии. Оно количественно равно определенному на стр. 54 напряжению разложения. Если Е измеряют в вольтах, a F выражают в кулонах (LF = 96 493 кулона), то по уравнению (3) подучают сродство в ватт-секундах, соответственно в джоулях. Умножая на 0,23901, получают сродство, выраженное в грамм-калориях на грамм-ион [гкал/г-ион).
Если сродство, отнесенное к 1 иону (вместо 1 г-иа»), обозначить а/, то уравнение (3) переходит в
а/ - п-е-Е, (За)
т. е. произведение измеренной электродвижущей силы на валентность дает непосредственно сродство в электрон-вольтах на ион (зв'ион). Сродство в кило грамм-калориях на грамм-ион (ккал/г-ивн) получают умножением па величину 23,063 (ср. стр. 138). Наоборот, умножением измеренного сродства реакции (ккал/в-ион) на 4,3360-Ю-2 (что соответствует делению на 23,063) и деления иа валентность получают (в количественном измерении) электродвижущую силу гальванического элемента, в котором рассматриваемая реакция используется для получения электрической энергии.
Так как измовепие энергии ирн переходе системы из одного состояния в другое нс зависит от пути перехода, то, следовательно, и максимальная работа, которую можно получить в результате превращения, происходящего в гальваническом элементе, остается та же, если превращение идет иначе, предполагая только, что процесс протекает изотермически и обратимо. Поэтому сродство, рассчитанное из электродвижущей силы ио уравнению (3), относится к определенной реакции как таковой, а не связано с процессом, использованным для ее расчета.
Приме р. Сродство реакции
Zn-J-CuS04=Cu+ZnS04 (4)
можно рассчитать исходя из электродвижущей силы гальванического элемента, образованного цинковой пластинкой, погруженной в раствор ZnS04, и медной пластинкой, погруженной в раствор CuSO4. Если растворы данных ионов 1 Л*, то электродвижущая сила (которая, конечно, может бить непосредственно-измерена) получается из данных табл. 4, стр. 51, Е = 0,35 -ф- 0,76 = 1,11 в (при 18°). Так как изменение заряда ионов составляет 2 (Zn = Zn” 4-2 е; Си" 4- 2 е = Си), то сродство реакции рассчитывается, по уравнению Ai = 2-36'493 1,11 = 214 214 Aw, или 51 200 кал.. Как было найдено, теплю, выделяющееся при реакции (4), составляет 50 НО пал. Следовательно, теплота реакции в этом случае немного меньше, чем сродство**.
Аналогично, принимая во внимание данные, приведенные па стр. 51 и 54, находят значение сродства реакции 2Н2 4~ О3 = 2НаО:
Л/= 4-96 493-1,23-0,2390 = 113 460 кал (при 18°).
Это значение относится к образованию жидкой воды из газов при атмосферном давлении. Теплота образования 2 молей Н2ОЖИДК при 18® сортов ляет 136 740 кал. В этом случае теплота образования заметно больше, чем сродство образования. Из 1 а гремучего газа можно, таким образом, путем соединения составных частей получить значительно больше энергии в форме тепла, чем требуется затратить ее для разложения 1 г воды в форме электроэнергии- При разложении Воды эта разность, составляющая 17% от теплоты образования, отбирается от окружающего пространства в форме тепла ***.
* Поскольку ничего другого не имеется и виду, сродство относят всегда к единице Концентрации; ср. стр. 165 и 168.
** На основании уравнения (2) отсюда следует, Что сродство реакций и в соответствии с этим электродвижущая сила рассмотренного элемента должны возрастать с ростом температуры (правда, лишь очень мало, так как разность Л/ — W ма.чд). Это Подтверждается опытом.
*** Одпако практически наблюдают не охлаждение, а нагревание электролита, так как этот эффект перекрывается обычно эффектом выделения джоулей а тепла током, являющимся следствием необратимого процесса преодоления омического сопротивления.
468
Глава 5
В соответствии Со вторым законом термодинамики невозможно превратить эт часть в энергию, совершающую работу; следователь по, принципиально для полутени работы можно использовать но более 83% теплоты образования воды. В случае угль рода, напротив, теплота его сгорания и сродство образования СО2 имеют при обычно: температуре приблизительно равное значение. Поэтому почти всю теплоту сгорали углерода можно было бы использовать для получения работы, если бы соединение i и О2 пРи обычной температуре удалост, провести обратимо, в то время как при обход ном пути вне тепловых машин неизбежно теряется значительная часть энергии*
2. Определение величины сродства из химического равновесия
Для реакции, которая протекает по уравнению
aA+6B+.,.₽mM+nN+...,
сродство Af дается уравнением
A/ = K-r-2,3G26^Iogbb^j~^-logAc^ , (5,
где. Л — газовая константа (—1,987 кол); Т — абсолютная температура; А’е—константа, полученная при применении к этой реакции закона действия масс- Символы, стоящие в прямоугольных скобках, означают молярныо концентрации (точнее, активности) исходных веществ перед реакцией и полученных продуктов реакции. Надо отметить, что исчезающие при реакции вещества стоят в числителе, а образующиеся — в знаменателе', в соответствии с этим следует поступать при расчете констант равновесия А,;*:‘:.При применении давления вместо концентрации (соответственно активностей) в случае реакций между газами справедливо то лее правило.
Из уравнения (5) видно, что система, находящаяся а химическом равновесии, обладает сродством реакции, равным нулю. Это следует также непосредственно из определения сродства, так как система, находящаяся в химическом равновесии, не меняет самопроизвольно своего состояния, а следовательно, не может Совершать никакой работы,
Из уравнения (5) следует, что сродство реакции зависит от отношения концентраций участвующих в реакции веществ. Если концентрации веществ, стоящих в левой частя уравнения, больше, чем равновесные концентрации, то, согласно уравнению (5), реакция, протекающая слева направо, имеет положительное сродство; в обратном случае положительное сродство имеет, реакция, протекающая справа палево. Значение сродства тем выше, чем больше концентрации, реагирующих веществ отличаются от равновесных концентраций. Если концентрации исчезающих и возникающих веществ все равны единице, то получаем
Л/п = -J?-T-2,3026.1ogAe.
Эту величину Af называют нормальным сродством, реакции.
Пример. Константа равновесия реакции Ь -} Н2 = 2HI при 441° (= 714° К) имеет значение К.. = 0,01984. Поэтому нормальное сродство реакции при давпоп температуре составляет
АЩ = — 1,987-714-2,3026-log0,01984 =-^ 556k кал.
* Даже па самых современных электростанциях в электроэнергию превращается максимально 28% энергии сгорапия угля. Вопрос о непосредственном превращении химической энергии угля пли других горючих веществ в электроэнергию («Brennstoff-ketten») представляет поэтому значительный технический интерес (ср. Elektrochem., 39, 169, 1933; 41, 93 , 405,. 4935). Оствальд назвал решение этого вопроса важнейшей задачей электрохимии.
Если поступают наоборот, то знак соответствующего члена в скобке должен быть измсней.
Валентность и сродство
169
Работу, эквивалентную этому количеств^ тепла, можно получить при превращении 1 моля 12 и 1 моля Н2 с концентрацией 1 М в два моля HI с той же концентрацией и температурой. Если исходить из смеси, в которой концентрация Н2 в 10 раз it ревы-ига ет концентрацию К перед началом реакции и концентрацию III после реакции, то уравнение (5) дает существенно большее значение сродства, соответствующего образованию 2HI.
Af = 1,987-714-2,3026 (log 10— log 0,01984) = + 8828 кал.
В выбранном примере сродство реакции значительно больше, чем ее тепловой эффект, который в случае образования 2 молей Н1 из газообразных продуктов при 441° составляет около 2800 кал.
3. Расчет сродства из теплового эффекта а
Применяя уравнение (2) на стр. 165, можно рассчитать сродство для любых температур из теплового эффекта реакции, если ее сродство известно для одной температуры. Так, сродство превращения одной модификации вещества в другую при некоторой заданной температуре Т можно рассчитать из точки перехода (т. е. из температуры То, при которой обемодцфика- * ции находятся в равновесии и, следовательно, при которой сродство превращения равно нулю) и из теплоты превращения при температуре Т. Чтобы проинтегрировать уравнение (2), следует, правда, знать температурную зависимость теплоты превращения W.
В большинстве рассматриваемых на практике областей температур зависимость разности ДСР молярных, теплоемкостей обеих модификаций от температуры при постоянном давлении можно представить уравнением
ЛСр = a 4- р7' 4- у7'4 (6). .
где о, ₽ и Y — эмпирические константы. Отсюда температурная зависимость теплот превращения при постоянном давлении-задается уравнением
Ж=Ж0—аТ—1/3уГз. : (7)
В этом уравнении 114 означает константу, значение которой можно рассчитать по^уравпопито (7), принимая во внимание уравнение (6), если W известно для какой-нноудь любой температуры [в области применения уравнения (6), т. с. для температур, которые не лежат слишком близко, к абсолютному нулю]. Интегрирование уравнения (2) дает тогда следующее уравнение для расчета сродства:
Af - Wo 4- ОТ In Г ф- 1/2рта ф- 1/6уГз ф. СТ. (8)
Константу Q этого уравнения можпо определить, если вместо Т подставить температуру превращения Тй, так как тогда А} = 0,
Пример, Требуется рассчитать сродство превращении, .моноклинной серы в ромбическую при. 20Р. Зависимость молярных теплоемкостей обеих модификаций от температуры между 0 к 100°, согласно имеющимся измерениям, можно представить уравнениями
сз монокл J 3[62 + о,0072 7'; С* 1,0116 .= 4,12 4- 0,0047 Т.
ЛС-р в у равней пи. (6) означает возрастание молярной теплоемкости, соответственно атомной теплоемкости системы при переходе из состояния 1 н состояние 2.
Следовательно, имеем
Д6’т, = (4,12-3,62)4-(0,0047—0,0072) Т=0,50— 0,00257
И==Ж0—0,507+0,00125712.
Но Льюису и Реп дел л у теплота пре вращении при 0’ (Т = 273) составляет 77,0 кал/мал ь. Отсюда следует
Жо=77,04-0,50.273-0,00125-273^ = 120,3.
Используя это значение и экспериментально установленную температуру превращения
170
Глава 5
To = 368° из уравнения (8), находят значение для С, полагая Af=Q. Тогда
1ЭД 3
С= — ^й^+0,5-2,303 log 368—0,00125-368= - 2,821, ООО
Итак, уравттоппе для расчета сродства превращения серы при любых температурах (внутри области применения уравнения, выражающего температурную зависимость молярной теплоемкости) выглядит следующим образом:
А; = 120,3 + 0,50-Т-2,203. log?’ - 0,00125 Т? — 2,821 Т. (9)
Для сродства превращения моноклинной серы в ромбическую при 25° (Г = 298) это уравнение дает значение Aj — 4-17,6 кал на 1 з-атом S. Положительное значение сродства свидетельствует, что превращение происходит само собой, что также легко можно наблюдать непосредственно. Следовательно, при обычной температуре стабильна ромбическая модификация серы, выше 95°, наоборот, — моноклинная. Например, для 100° (Т = 313°) уравнение (9) дает значение Л/= —1,2 кал; следовательно, в этом случае положительным сродством обладает превращение, идущее в обратном направлении.
Расчет сродства пр уравнению (8) возможен только в. том случае, если для какой-либо температуры сродство уже известно или если известна температура; для которой сродство рассматриваемого процесса равно нулю (точка превращения, точка плавления ц т. д.). Однако, применяя тепловой закон Нернста (ср, стр. 201), можно также вывести уравнения, которые позволяют делать расчет сродства, если только известны тепловой эффект И7 процесса, и его температурный ход*.
Если для определенной температуры известны теплота реакции и изменение энтропии, а удельные теплоемкости начальных и конечных продуктов реакции либо мало различаются, либо их разпость мало зависит от температуры или, накопец, их температурная зависимость в рассматриваемой области известна, то для расчета сродства (и тем самым, согласно стр. 168 и ел. также константы равновесия) можно с успехом использовать метод приближенного расчета, предложенный УлихОм (Ulieh Н., Z. Elektrochem., 45, 521,1939; см. U I i ch Н., Kurzes Lehrbu ch der physikalischea Chemie, neubearb. von Jost W., 9, Au£l. (Darmstadt, 1956), стр. 112 и сл.
4. Расчет сродства из спектроскопических данных
Большое значение с недавнего времени приобрели расчет сродства и расчет химических равновесий из спектроскопических данных.
Из спектроскопических данных узнают прежде всего работу ионизации и работу диссоциации. Работу ионизации получают по уравнению (3), стр. 137, из длины волны X, принадлежащей границе серии. Умножая величину, обратную длине волны (cat'1) на 1,2398-10''*1 получают работу ионизации на молекулу, соответственно на атом в электрон-вольтах. Умножением величии, обратных длинам волн на 2,8592, получают работу ионизации в калориях на моль.
Так же как из атомных спектров (линейных спектров) моткна рассчитать работу ионизации, из молекулярных спектров (полосатых спектров) удастся рассчитать работу диссоциации молекулы. При этом следует обратить внимание, диссоциирует лимолекула на свободные атомы или ионы**.
Спектроскопические данные дают прежде всего величины сродства при абсолютном нуле. Однако, применяя квантово-механические формулы кинетической теории газов, удается также рассчитать и температурную зависимость величин сродства.
* Подробнее’см. учебники термодинамики и физической химии. Для вывода приведенных выше формул см. стр, 261.
** Подробный обзор применения спектроскопических данных для расчета сродства вяэовык редкгщй (и других термодинамических функций газообразных систем) сделал Цейс (Z о i s е Н., Z. Elektrochem., 39, 758, 895, 1933; 40, 662, 885, 1934; 47, 380, 595, 644, 1941; 48, 425, 476, 693, 1942.
Валентность и сродство 171
Применение значений работы ионизации и работы диссоциации для расчета величин сродства химических реакций (соответственно работ образования соединений) здесь следует пояснить на отдельных, совсем простых примерах.
Пример,
1. Расчет работы образования IBr. Величины работ, которые должны быть затрачены для разложения молекул Вг2, 12 и Шт, получают из полосатых спектров
Вг2 = 2Вг — 45, 23 ккал (Gordon, Barnes, 1933),
12 = 21 — 35,40 ккал (Brown, 1931), IBr = I 4- Br — 41,68 ккал (Brown, 1932).
Если сложить два первых уравнения и из суммы вычесть третье, умноженное на 2, то получим
Вг2 12 = 2IBr -f- 2,73 ккал.
СледовательпО, сродство образования <4/ц)г получается из спектроскопических даных равным 1365 кал/моль (для Т — 0°), в то время как Пост (Yost., 1931) измерением равновесий нашел Л/1Вг = 1270 кал/моль (экстраполированное к Т — 0).
Спектроскопически найденные значения работы диссоциации относятся к свободным молекулам. Следовательно, рассчитанное отсюда сродство образования для IBr (так же как значение, экстраполированное из измерений Поста) рассматривается для такого Случая, когда данное вещество при абсолютном нуле остается все еще газообразным. Для перехода к другому агрегатному состоянию следует еще Припять во внимание работы плавления п испарения.
Пример.
2. Расчет работы образования иодида калия. Образование иодида калия из калия и иода можно представить двумя путями: во-первых, непосредственным соединением составных частей, во-вторых, соединением газообразных ионов с образованием кристаллической решетки, благодаря чему освобождается энергия, вычисляемая на основании закона Кулона. Предположив, что все процессы проходят изотермически н обратимо, оба пути можно представить в виде кругового процесса, который, согласно Борну (Born) и Габеру (Haber), можно наглядно изобразить следующим образом:
4-sK
[К1 + П]------—~(К) + (0
Ф +Sr4D£3 I '
+-4/кг ~
[KI] ------ (К+) + (Г)
—GKI
Исходим из 1 г-атома твердого калия|К] и 1 г-атома твердого иода [I]*. Оба вещества переводим прежде всего в состояние свободных атомов. В случае калия для этого следует использовать теплоту сублимации в случае иода, помимо теплоты сублимации iSj, нужно еще учесть теплоту диссоциации отнесенную
к I/, моля 12.
Если осуществить все эти процессы в области, близкой к абсолютному нулю, то, согласно тепловому закону Нернста, тепловые эффекты можно приравнять к затраченной работе. Затем переводим атомы калия в свободные ноны К*. Для этого следует затратить работу ионизации JK. При переводе атомов I в ионы I-, напротив, энергия освобождается. Энергия, освобождающаяся при переводе атомов элемента в, отрицательные ионы (следовательно, путем соединения атома с электронами), обозначается Как электронное сродство Е данного элемента. Свободные ионы К+ и I" можно теперь соединить в Кристаллическую решетку. Освобождающуюся при этом энергию G обозначают как анергия решетки данного соединения. Максимальная полезная работа, которая может быть произведена путем образования 1 г-молекулы KI из элементов (т. е. сродство образования Л/к1), должна быть затрачена, чтобы разложить подид калия на элементы, т. е. чтобы снова восстав овить исходное состояние. В приведенной выше схеме величины затраченной энергии (т. е. энергии, цодведепной к системе) имеют поло-
* Относительно обозначения агрегатного состояния квадратными и круглыми скобками см. стр. 200.
172
Глава о
жителт.гтые знаки, а величины освобождающейся энергии — отрицательные знаки Так как был проведен круговой процесс, общее изменение энергии равно нулю, т. е ^k+^iV^^+J-k’-^-Gki+^k^O. (10
Это уравнение можно использовать для расчета какой-нибудь приведенной в нет величины энергии, если все остальные величины известны. В данном случае употре бим его для расчета А/К1. Требуемый для этого расчета величины энергии, за нсключе нием энергии решетки GK1, известны из экспериментальных данных. Энергию решетк! в случае иоппой решетки можпо рассчитать на основании закона Кулона.
Если рассматривать ионы как жесткие шары с радиусами Г[ и т*2 и зарядами +< и —в, то работа, которую следует затратить, чтобы вплотную сблизить шары, вира зится уравнением
А=-------,— (ем. стр. 107)
?*i -j-Га
Отрицательное значение А показывает, что при образовании молекулы KI из иопог К+ и I- энергия освобождается. Если подставить‘значения иоппых радиусов К+ и Г (см. стр. 180 и 826), то окажется
(4,8028-। £2
(1,33+2,20)’ Р ’
где А — работа образования молекулы К] из газообразных ионов. Умножая эту величину на число Авргадро (Лд = 6,023 1023) и меняя знак на обратный, получим сродство образования в эргах на моль (эра/.иоль). Умножая, далее, на 2,390- 10 s, получим величину сродства образования в калориях на моль (кал/.коль). /
6,534-10-1--6,023-10^-2,390-Ю-3 1= 9,406-101 кал/молъ, Г
ИЛИ, 94,06 ккал/мо.гь. Если прибавить к этому значению, относящемуся /к аеао-образному К1, теплоту сублимации К1 (48,9 ккал/молъ), то получим приближенное значение сродства образования кристаллического иодида калия па газообразных ионов (ири абсолютном нуле), равное 143,0 ккал!моль. Одпако вследствие пренебрежения поляризацией этот расчет должен давать слишком небольшое значение для сродства образования из ионов (соответственно для энергии решетки).
Более точное' значение энергии решетки получают, если закон Кулопа применяют пе к образованию молекулы, а непосредственно к образованию кристаллической решетки (ср. гл. 7) из газообразных ионов. Ибо в таких кристаллических решетках, в которых, как в случае решетки К1 (тина, каменной соли, ср. гл. 7), всё ионы равномерно окружены противоположно заряженными ионами (так насыпаемые «координационные решетки»), поляризующее действие окружающих ионов почти прекращается. В молекулах благодаря одностороннему притяжению поляризация («деформация электронных оболочек») проявляется значительно сильнее. Непосредственный расчет энергии решетки на основании закона Кулопа, при котором было принято во внимание взаимодействие всех ионов, был произведен впервые Маделунгом (Madeking, 1918). Для бинарных соединений расчет можно произвести по формуле* \
л 1 7,-е2
0-=.--—.— AN а. (И)
71 Г
В этой формуле г — кратчайшее расстояние между центрами ионов в кристаллической решетке; Z — валентность иолов; е— элементарное количество электричества; Nд — число Авш адро и Л—константа, величина которой зависит от типа кристаллической решетки [<ад/( каменной соли'. А = 1,748; тип иодида цезия'. А = 1,763; тип цинковой обманки: А — 1,639]. Для решетки плавикового гипата А = 5,039; при Этом для соседи пепий общей формулы М. ГЦ Z = 1, для соединений формулы MlvRj 2 = 2**.
Так называемый «экспонент Отта,а кивания» л учитывает отталкивание ионов, наступающее при большом сближении. Его приближенное значение можно определить
* Вывод формулы, см., например, в монографии В ан-Арке ля (van Arkel) и де Бура (de Воет).
** Простой приближенный метод расчета коцстапт Маделунга А предложил Хенен-дал [II б j е и d a h 1 К., Danske Vidensk. Selsk., mat.-fysiske Medd,, 16, 2, 133, 1938]. Он рассчитал, таким образом, значением для ряда соединений типа иввестковозо -шпата. Вследствие изменения углов между гранями (а, рис. 50) дня этого кристаллического типа значение А у различных веществ не одинаково.
Валентность и сродство
173
X Л
из величины сжимаемости кристалла. Так как 'отталкивание происходит вследствие воздействия электронных оболочек отдельных ионов друг на друга, значение п можно рассчитать также теоретически. По Полингу для ионов с конфигурацией инертных газов п имеет следующее значение (в зависимости от электронной конфигурации):
Электронная конфигурация Не Ne Аг Кг Хе Ин
и= .5 7 9 10 12 44
В случае соединений, состоящих из ионов е различными электронными конфигурациями, например иа К* (конфигурация Лг) ц 1“ (конфигурация Хе), для « Принимают среднее значение (для KL п = 10,5). Как видно из уравнения (11), благодаря отталкивающему действию электронных оболочек энергия решетки уменьшается на дробную часть 1/п*. i
Уравнение (11) дает для энергии решетки K.I (кристаллизирующегося по типу каменной соли, ср. стр. 238) следующее значение '
г 9,5 (4,8028-IO'»)3 л т/й г Леч swots , .
48'6’°23‘10 =6,233‘i0
или 149,0 ккал/молъ. Это значение, как и следовало ожидать, выше значения, рассчитанного первым способом.
Если подставить рассчитанное значение G, а также остальные значения энергии**, известные из экспериментальных данных, в уравнение (10), то получим
Д/и = —21^5—8,03 —17,70—99,65 4-79,34-149,0=81,1 ккал/люль.
Это значение работы образования, Ели сродство, рассчитанное для 1 моля KI (при абсолютном нуле), удовлетворительно совпадает со значением, определенным Итикава (Ishikawa, 1934) при измерении электродвижущей силы (Л/К1 = 77,5 ккал/моль K.I), особенно если принять во внимание, что последнее рассчитано для комнатной температуры.
Значения определения сродства. Так же- как сродство можно определить из данных электродвижущей силы, химического равновесия и т. д., , указанные величины можно рассчитать из сродства. Так, из величин* сродства, найденных по спектроскопическим данным, можно рассчитать константы равновесия и тепловые эффекты реакций. Далее, па основании расчетов сродства молено сделать выводы относительно возможности существования химических соединений. Если для реакции образования кипоте-тического соединения находят слабо отрицательное сродство, то при известных обстоятельствах путем соответствующего измене пин температуры и концентрации можно достигнуть положительного значения сродства [ср. уравнение (5)]. Однако если для рассматриваемой реакции получается сильно отрицательное сродство, то можно суверенностью сказать, что эта реакция не может быть осуществлена ян посредственно.
Таким образом, путем расчета сродства соответствующих реакций удается показать невозможность образования гетерополярных соединений инертных, газов, Например, круговой процесс сродства образования Борна — Габера дает для NeF и NeCl, если принять, что они построены как соли, приблизительно —241, соответственно
* В соответствии с принципами квантовой механики для отталкивания получается более сложное выражение. Однако, поскольку не проявляются существенные поляризационные влияния, уравнение (11) дает энергию решетки большей частью в хорошем приближении. Способы, применяющиеся для более точных расчетов, достаточно сложны (ср. стр. 180).
** Вместо подставлено значение, рассчитанное па основе измерении: Майера (Mayer, 1930) и Биховского и Россини (Bichowsky, Rossini), Остальные значения взяты нз таблиц Ландольта — Бернштейна.
174
Глава 5
—254 ккал}моль. Следовательно, эти соединения ни в коем случае нельзя получить путем химических ^реакций в обычном смысле*.
Однако неспособность к существованию (соответственно нестабильность) соле об-разных соединений инертных газов обусловлен^ но столько высоким потенциалом их ионизации, а скорее тем, что высокая энергия, затрачиваемая для ионизации, не компенсируется соответствующей высокой энергией решетки образующегося соединения, Например, энергия, которая должна быть затрачена для отрыва двух электронов от атома Во (632 ккал/г-атом), значительно больше той энергии, которая должна быть затрачена для отрыва электрона от Ne (495 ккал/г-атом). Однако в случае образования соединений Be энергия решетин соответственно выше; в случае BeF2 она равна примерно 826 ккал/моль, в то время как для NeF рассчитана энергия около181[квал/,иадь. Итак, следует иметь в виду; Эля ооалюжностн образования соединений риаающее значение имеют, не абсолютные величины работ ионизации, а отношение этих величин к величинам: энергии, получаемой в результате образования противоположно заряженными ионами молекул или кристаллов (а при данных условиях также в результате Действия других факторов, например дополнительного присоединения молекул НгО).
Так как в дальнейшем при оценке стабильности соединений для расчета величин сродства инн работ образования часто применяют круговой процесс Борна — Габера, то его следует еще раз пояснить более общей схемой.
Исходные вещества, (тверд., жидк. или газообр.)
+4/
Продукты реакции, (тверд, жпдк. ила газообр.)
+2£iSm+2Qv+SQH
Свободные атомы (газообр") исходных веществ
| —ХЕ
Ионы (газообр.),
гдеQsm—молярная теплота плавления, Qy—молярная теплота испарения; Qs( = Qsm+ +Qv) — молярная теплота сублимации; все величины взяты при Г — О’. (Величины с показателями «а» относятся к исходным веществам, а с показателями «с» к конечным продуктам реакции.) QD—молярная работа диссоциации (для разложения многоатом.-вых молекул газа па атомы), Aj — работа ионизации при образовании положительных иопов, Е — электронное сродство при образовании отрицательных иопов (обе эти величины отнесены к 1 г-атомур, G — молярная энергия рещетки (в случае газов на се месте стоит энергия образования 1 моля газа из ионов); Af— сродство реакции-, исходные вещества -» продукт реакции. Знак суммы 2 свидетельствует о том, что здесь должна быть подставлена сумма рассматриваемых значений для отдельных веществ, при1!ем каждая величина должна быть умножена на коэффициент, который выражает в уравнении реакции число молей, участвующих в превращении. Так как для кругового процесса ;
SQs + SQd-FSAt—S<?4-.4/=0,
то для расчета сродства получим уравнение
Af = SG+ 27? - (2 Aj+ SQd+2<?s)- (12)
В случае газов G заменяют на БмХ; для жидкостей G заменяют па (Вмх -|- Qy) и Qs па
* Это не исключает, конечно, возможность того, что возникшие благодаря электронным ударам или облучению ионы инертного газа или’«возбужденные» атомы инертного газа (т. е. атомы, электроны которых отчасти подняты на более высокие уровни) образуют один с другим или с другими атомами нестабильные короткоживущие многоатомные молекулы или что короткоживущие многоатомные молекулы образуются присоединением нормальных атомов инертного газа к ионизированным атомам других веществ (под действием «вандерваальсолых сил»). Такие, образованные инертными газами, многоатомные молекулы, например Het, ArHg, обнаружены в действительности спектроскопическим путем. Однако в таких случаях дело идет пе о химических соединениях в обычном смысле (ср. стр. 31 и сл.). Продукты присоединения инертных газов характера гидратов газов (гл. 7) также не имеют отношения к изложенному выше.
Валентность и сродство
175
Из табл. 26 по нескольким типичным примерам можно сделать заключенно о влиянии на величину сродства различных входящих и уравнение (12) энергетических величин. Бее приведенные в ней величины энергии относятся к образованию 1 г-же соединения. Энергии решетки рассчитаны по уравнению (11), а стоящие в колонке Л/р;1сСЧ значения сродства—Но уравнению (12). Для сравнения» колонке Л/вкс11 дапы значения сродства, установленные чисто экспериментальными методами, а в колонке И7 — экспе-римептальпо найденные теплоты образования, имеющие, правда, значение для комнатной температуры. Точность расчетов сродства, основанных на круговом процессе Борна — Габора, в общем jotoKa не велика. Однако этот круговой процесс дает превосходный обзор факторов, которые имеют значение для стабильности химических соединений. И» табл. 26 видно, что па величину сродства решающее влияние оказывают главным образом две величины: потенциал ионизации электроположительной составной части соединения л энергия решетки (или в случае газов энергия образования молекул иа ионов), В общем значительно меньшее значение имеет сродство к электрону электроотрицательной составной части. Энергия решетки зависит в соответствии с уравнением (11) главным образом От валентности ионов и расстояния между их центрами в кристалле, т. е. суммы кажущихся ионных радиусов (ср. гл, 7). Суммы радиусов катиона и аниона г = гк ф- гЛ приведены в последней колонке табл. 26. Вследствие особо мало-
ТаблиЦа Зв
Примеры данных, служащих для расчета величии сродства методом кругового процесса Борна—Габера
Соединение ZG ~SMX 2E SAj 2«S "^расся. ^OKCB. IP r
HF 364,2 95,3 312,2 82,7 - — 64,6 64,4
LiH 218,6a —r 16,3 123,8 51,3 39,0 23,7 — 21,6 2,04
NaF 214 —- 95,3 118,0 31,3 26,2 134 .— 136,4 2,31
i/2CaF2 308 — 95,3 206,4 31,3 21,4 144 *—• 144,7 2,36
V2CaO 420 2 —86,5 206,4 29,4 21,4 76 72,7 76,1 2,40
360 — -42 206,4 25,7 28,7 57 '54.9 55,6 2,84
XeF -150 95,3 278,2 31,3 0 64 , — -3,4
V2NaF2 -335 — 95,3 601,3 31,3 12,9 ; 215 — — -2,2
CaF — 183 — 95,3 140,3 31,3 47,8 -59 — — -2,8
а Рассчитано ив активо-механическим методом.
го радиуса ядра водорода последний может образовывать с отрицательными ионами стабильные соединения, аотк работа чоназади« для него значительно больше, чей Зля ксенона, и, кроме того, при образовании иона Н+ для расщепления молекулы Н2 на атомы нужно еще дополнительно затратить 51,3 ккал./z~amo.it.
Малой величиной ядра атома водорода обусловливается его особенно сильное поляризующее действие. Энергия образования молекулы ИР из ионов благодаря эффекту поляризаций почти на 50% превышает значение, рассчитанное без учета этого эффекта. Этим объясняется также то, что все водородные соединения алектроотрица-тельпых элементов легколетучи; это происходит потому, что вследствие силт.ной поляризации энергия образования для свободных молекул здесь больше, чем для ионной решетки.
В нижней части табл. 26 приведены энергии решетки для некоторых неизвестных соединений, которые по заключению Гримма и Герцфельда (Grimm, Hcrzfelcl, 1923) были вычислены при допущении, что эти соединения, если бы они существовали, должны были образовывать такую же решетку, как и аналогичные пи известные соединения. Из полученных таким, образом сильно отрицательных величин сродства образования видно, что такие соединения, как XeF nNaF->, не могут существовать. Соединение СаР, Правда, имеет положительное сродство образования; однако, как можпо заключить пз данных таблицы, при соединении 1 г-атома Р2 с 1/2 г-ато.на Са с образованием
176
Глава. 5
i/a г-атвма CaF, освобождается мпогЬ больше энергии, чем при присоединении 1 и-атома Са к 1 г-атому 1’ с образованием CaF. Итак, хотя CaF и устойчив по сравнению со свободными составляющими, он, однако, не устойчив но сравнению с СаГ2, т. о. CaF должен самопроизвольно разлагаться на Са и CaF2. Одпако следует обратить внимание па то, что с возрастанием температуры равновесие моя;ет сдвинуться.
В общем оказывается, что энергии решетки недостаточно, чтобы произвести работу, необходимую для отрыва электрода, связанного в оболочке инертного газа (в случае самих инертных газов или следующих за ними элементов). Поэтому валентности элементов главных подгрупп определяются их местом по отношению к инертным газам. Энергия решетки уменьшается с возрастанием радиуса как катиона, так и аниона. Поэтому устойчивость аналогично построенных соединений одинакового состава в каждой главной подгруппе периодической системы уменьшается сверху вниз, т. е. с возрастанием радиуса аналогичных ионов. Если сравнивать сродство образования ряда соединений одного и того же катиона в разных валентных состояниях, то в трех первых главных подгруппах периодической системы наблюдается резкий максимум сродства образования у тех соединений, катионы которых отдали все электроны, лежащие впе оболочек инертных газов.
Этим объясняется то, что элементы трех первых главных подгрупп в своих гетер о полярных соединениях почти исключительно проявляют валентности, соответствующие их групповым номерам.
Гл а в а 6
ПЕРВАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Главная подгруппа)
ЩЕЛОЧНЫЕ МЕТАЛЛЫ
порядковый номер Элемент Символ Атомный вес Удсль4 ный вес Температура плавления, °C Температура кипения, °C Удельная теплоемкость Валентность
3 Литий L1 6,940 0,534 179 1340 0,837 I
11 Натрий Na 22,991 0,966 97,8 ! 883 0,295 I
19 Калий К 39,100 0,86 63,5 760 0,177 I
37 Рубидий нь 85,48 1,52 39,0 696 0,080 I
55 Цезий Cs 132,91 1,87 28,45 708 0,048 I
87 Франций Fr 223 а —* —1 — — I
а Этот изотоп является наиболее долгоживущим (период полураспада 21 -чин); образуется в результате а-распада актиняя. Другие изотопы франция можно получить искусственным путем.
Общие заметания. Главная подтруппа группы Периодической системы включает элементы литий, натрий, калий, рубидий и цезий, а также краппе нестабильный элемент франций. Последний встречается в ряду радиоактивного распада актиния (см. т. II). Все эти элементы объединяют под общим названием щелочные металлы, так как гидроокиси главных представителей (натрия и калия) этой подгруппы известны под названием «щелочей». Щелочные металлы очень мягки и обладают весьма низким для металлов удельным несом. Характерна их чрезвычайная легкоплавкость, а также низкие точки кипения.
В химическом отношении щелочные металлы крайне реакционноспособны. Все они разлагают как воду, так и спирт с выделением водорода
Na-f-HOH= №ОН-р/г1Тг, Na 4- ИОС2Н5 = NaOC,H6 -Щ/гН2.
В первом случае при этом возникают растворы с особыми свойствами, которые называют щелочными. Они обусловлены высокой концентрацией ионов гидроксила в растворе. Поэтому растворы с высоким содержанием гидроксильных ионов часто называют «не основными растворами», а «щелочными растворами». Характерным признаком для таких растворов является жгучий, щелочной вкус, а также прежде всего их способность влиять на окраску некоторых органических красителей, например, они окрашивают в сикн.й цвет расгвеор лак.ирей . Концентрированные растворы гидроокисей щелочных металлов сильно разъедают кощу, поэтому их называют также едкими щелочами.
Разложение воды щелочными металлами является необходимым следствием того, что в злектрохимическом ряду напряжений они стоят значительно левее водорода, Нормальные потенциалы щелочных металлов значительно ниже потенциала нормаль-12 г Реми
178
Глива 6
ного водородного электрода в растворах с концентрадией водород пых иопов чистой воды или даже нормального водорб^пого электрода в растворах с 1 и. концентрацией ионов гидроксила. Поэтому щелочные металлы реагируют с водородными ионами даже при такой крайне незначительной концентрации последних, какал наблюдается в сильно щелочных растворах. Сущность реакции мощно представить уравнением
м + н- = м- + ,/2Н2>
где ЭД — атом одного ив щелочных металлов. Щелочной металл, следовательно, отнимает у водородного иопа положительный заряд, вли, вернее, мои водорода отнимает у атома щелочного металла один электрон-
Отдача электрона атомами щелочных металлов происходит настолько легко, что они могут отдать его даже незаряженному водороду, который благодаря этому заряжается отрицательно
М-р/йН2 = м*н-.
Реакция идет при пропускании сухого водорода над слегка нагретым щелочным металлом; еще легче реагируют щелочные металлы с элементами, обладающими ярко выраженным сродством к электрону. Даже наименее реакционноспособный среди них литий тотчас воспламеняется в струе хлора и сгорает в нем, излучая ослепительный белый свет.
На влажном воздухе щелочные металлы тотчас тускнеют и вскоре покрываются толстой коркой гидроокиои. Поэтому их следует хранить под слоем керосина или парафинового масла. При незначительном нагревании на воздухе или в струе кислорода они воспламеняются. Продуктами сгорания большинства щелочных металлов являются не нормальные окислы, а перекиси. Во всех соединениях щелочные металлы проявляют валентность только 1; поскольку соединения можно рассматривать как гетерополярные, щелочные металлы в них всегда несут- один положительный заряд. Щелочные металлы обладают наиболее ярко выраженным электроположительным характером. Правило, что электроположительный характер элементов, т. е. стремление их перейти в электроположительное состояние, внутри одной и той же главной подгруппы периодической системы увеличивается с возрастанием атомного веса, отчетливо выступает у щелочных металлов при рассмотрении их общих химических свойств.
TS некоторых случаях это правило проявляется особенно наглядно. Например, более электроположительный характер калия по сравнению с натрием проявляется в различном отношении этих металлов к воде. Натрий энергично взаимодействует с водой и даже плавится благодаря выделившемуся при реакции теплу; однако при этом водород по воспламеняется даже при доступе воздуха, если только шарик натрия может свободно перемещаться на поверхности воды. Калий же реагирует в водой Столь бурно, что образующийся водород при соприкосновении с воздухом тотчас воспламеняется. Еше отчетливее различие между обоими металлами наблюдается в их отношении к брому и поду. Натрий реагирует с бромом при комнатной температуре только с поверхности; < подом его удается Даже осторожно сплавить без видимого взаимодействия. Если же бросить маленький кусочек калия в жидкий бром, то происходит Сильный взрыв; то же наблюдается при натревцниа калия с иодом.
Сильно электроположительный характер щелочного металла обусловливается, по существу, той незначительной затратой работы, которая требуется, чтобы отнять у атома металла один электрон. Это непосредственно выражается в низких значениях и опиз ац и о иных потенциалов щелочных металлов (см. табл. 22, стр. 140). Слабой связью электронов обусловливается II хорошая электропроводность щелочных металлов, а также и то обстоятельство, что щелочные металлы при облучении их поверхности ультрафиолетовым светом особенно легко отщепляют электроны (фотоэффект). Для химической реакционной способности щелочных металлов легкость, с которой опи переходят в состояние электроположительно заряженных
Первая группа периодической системы
179
атомов, имеет решающее значение. Однако теплоты образования их соединений (см. табл. 27) пе являются непосредственной мерой сродства к эл.ек-
Таблица 27
Теплоты образования соединении щелочных металлов
Приведенные в таблице значения теплот образования даны в кцлограмм-кало-рцях на грамм-атом грелочного металла (при комнатной температуре). Тепловые эффекты отнесены к газообразным неметаллам, за исключением серы, где тепловые эффекты указаны по отношению к ромбической модификации серы
Элемент Окиси Гидро-икисн Гидриды фториды Хлориды Б ромиды Йодиды Сульфиды
Литий 71,5 110 21,6 144,7 98,7 87,1 71,2
Натрий 51,5 102,3 12,8 136,6 97,8 90,4 73,7 44,9
Калий 43,4 101,4 9,8 134,5 104,2 97,5 86,4 43,6
Рубидий 41,7 101,2 — 132,8 105,0 99,2 87,4 43,5
Цезий 41,3 100,3 — 131,5 106,4 101,2 90,4 —
тропу отдельных щелочных металлов, так как они (теплоты) зависят, помимо этого, от ряда других факторов, и прежде всего от энергии решетки.
В табл. 27 сопоставлены теплоты образования нормальных о кисло в М2О в Других солеобразных соединений щелочных металлов, которые мощно рассматривать как приближенную меру сродства, проявляющегося при образовании соответствующих соединений. На первый взгляд можпо думать, что усиление электроположительного характера элементов в ряду литий—цезий должно проявляться также в увеличении сродства образования* (соответственно теплот образования) солеобразцых соединений. Однако табл. 27 показывает, что на самом деле это не совсем так. Это обусловлено тем, что, как было показано в предыдущей главе, сродство образования окисей и т, д. суммируется из нескольких составляющих, которые при Переходе от самого легкого к самому тяжелому элементу группы изменяются различным образом.
Если сравнивать соединение с одинаковыми анионами, то изменение сродства образования в группе щелочных металлов дастся уравнением (12), стр. 174, выражающим разность G — (Aj -ф- Qs). Из приведенных в табл. 28 данных следует, что сумма Aj -|- Qs при переходе от Li К Cs уменьшается от 159,8 до 108,1 ккал/г-атом, т. е. в отношении 3 : 2. Если принять при этом, что энергия решетки G уменьшается в том же соотношении или сильнее, то разность между <7и (.4j Qs) также должна уменьшаться.; следовательно, сродство образования должно уменьшаться от соединений лития к соединениям цезия. Энергия решетки в соответствии с уравнением (11), стр. 172, обратно пропорциональна расстоянию между центрами ионов в кристалле г, соответствующему сумме радиусов ан ио па и катиона. У фторидов, если переходить От LiF к CsF, ионное расстояние возрастает от 2,01 до 3,01 А (ср. табл. 39), таким образом, энергия решетки уменьшается приблизительно в соотношении 3 : 2**.
Итак, сродство образования должно уменьшаться от Li F к CsF. То же имеет место у окислов. Если принять, что G в соотношении существенно меньше, чем (Aj -|- Qg), то направление изменения разности G — {Aj -(- Qg) можно указать лишь в каждом отдельном случае. Для Lil и CsI по уравнению (11), стр. 172, G получается 170,fi, соответственно 130,7 ккал. Таким образом, в Этом случае G уменьшается только в соотношении 4 : 3. Разность G — (Aj Qg) получается из данных табл. 28: для Lil 10,8 и для
* Понятие «сродство образования» соответствует понятию «свободная энергия» со знаком минус. См. прим. ред. па стр. 165.—Прим. ред.
** Энергия решетки уменьшается не точно в том соотношении, в котором возра-п—1
Стает ионное расстояние, так как значение—— в указанном выше ряду повышается на 8% . Константа Маделунга А для всех щелочных фторидов имеет равное значение, так как они обладают кристаллическими решетками аналогичного типа.
12*
180
Глава 6
Таблица 28
Атомные и лонные радиусы, теплоты плавления и испарения, энергия ионизации3* и нормальные потенциалы гцелочпых металлов, а также теплоты гидратации щелочных ионов
Теплоты плавления и испарения дани при температурах плавления и кипения металлов. Теплоты гидратации даны при условии, что теплота гидратация водородного иона равна 250 ккал
Элементы
ЛптиЙ Натрий Калий Рубидий Цезий
Атомный радиус, А 1,56 1,86 2,33 2,43 2,62
Иопный радиус, А 0,78 0,98 1,33 1,49 1,65
Теплота плавления, ккал/е-апюм 0,76 0,63 0,57 0,53 0,50
Теплота испарения, ккал/е.-атом 32,25 23,12: 18,92 18,06 16,32
Теплота сублимации при 0°аес . . . 35,96 26,20 21,85 20,58 18,74
Энергии ионизации, ( М—>М++е 123,80 118,00 99,65 95,90 89,40
ккал/г-атом ( М+—>М2++е 1736,86 1084,5 730,59 631,56 538,7
Нормальный потен- Г набл. . . . -2,96 -2,71 ’ —2,92 —2,92 —
циал, в рассч. . . -2,95 —2,71 -2,90 -2,99 -2,92
Теплота гидратации, ккал/е-ион . . 112 99 82 77 71
В оригинале вместо «аггерпш иояЕгзацгпг» г «работы ионизации».—Приз*. ре th гриведеш числеЕГИ© Ивкиа ей
CsI 22,6 ккал. У иоДйДов в соответствии с этим сродство о бра зов алия возрастает вместе с электроположительным характером щелочного металла, точно так же как у бромида. '
Таким образом, изменение тсплот об разовая ня соединении, сопоставленных в табл. 27, объясняется наложением обоих влияний: увеличением электроположительного характера элементов от лития к цезию и уменьшением энергии решетки в том же направлении вследствие возрастания ионных радиусов. Последнее влияние в случае одного и того же катиона имеет тем меньшее значение, чем меньше заряд и чем больше ионный радиус аниона; поэтому это влияние уже значительно ослаблено у хлоридов, по еще более у бромидов и у иодидов, у которых теплоты образования вследствие этого растут пар алл епьпо росту электроположительного характера элементов.
Радиусы щелочных ионов приведены в табл. 28, радиусы ионов галогенов даны па стр. 826. Для расчета энергии решетки лучше, однако, пользоваться расстояниями между центрами иопов, взятыми непосредственно из размеров решетки, так как правило постоянства радиусов имеет только приблизительное значение. Расстояния между центрами ионов в галогенидах щелочных металлов могут быть взяты из табл. 39 и 40 а
(см. стр. 238 и 239), если принять во внимание. Что для каменной соли г = —и дня
кристаллов типа йодистого цезия г =
В табл. 29 сопоставлены значения энергии решеток галогенидов'Li, Na, К я Rb, рассчитанные де Буром (de Boer, 1936), и значения энергии решеток галогенидов Сз И NHj по данник Майера (Maier, 1932). Эти значения точнее, чем полученные ранее по уравнению (11) стр. 172, так как при их расчете было более точно учтено действие сил, накладывающихся на кулоновское ирнтяжепие; правда, при этом расчет существенно усложняется. Расчеты по "уравнению (11) оказываются достаточными, если не
Первая группа периодической системы 181
требуется значительно большая точность отдельных значений, а нужно только их приблизительное определение для оценки стабильности соединения или для получения представления о влиянии ионных радиусов (соответственно расстояний между центрами ионов в кристаллической решетке) па энергию решетки и на вытекающее отсюда сродство образования соединений.
Таблица 29
Энергия решеток галогенидов щелочных металлов* и аммония (при 0"aQc , ккал/моль)
U Na К вь Gs nh4
Фториды . . 237 213 188 480 174 191
Хлориды . . 195 180 а 164 158 152 162
Бромиды . . 185 172 157 1525 (46 (54
Иодиды . . . а Найдена 181- 6 Найдено 151. в Найдено 154. г Найдено 111. 171 160 149 о 143 139 г 145
«Найдено*—значения, полученные из экспериментально установленных срод-
ства, работы ионизации и т» д. на основании уравнения (12), стр. 174.
Следует отметить незначительную величину теплоты образования у окислов щелочных металлов по сравнению с теплотой образования галогенидов при равном количестве металла. Это обусловлено не исключительно малой энергией решетки окислов, а тем, Что для процесса О + 2 е = 0г“ должна быть затрачена работа, в то время как у галогенов благодаря присоединению одного электрона к свободному атому анергия освобождается (ср. табл. 26).
Несколько неправильная последовательность нормальных потенциалов (см. табл. 23) также обусловлена влиянием различных факторов. Нормальный потенциал является мерой энергетического различия между данным металлом и 1 п: раствором его ионов. Это энергетическое различие определяется не просто суммой теплоты сублимации и работы ионизации. Следует еще учитывать достаточно заметную энергию, освобождающуюся благодаря тому, что во круг-щелочных ионов создается оболочка цз молекул воды (гидратация, ср. стр. 100 и сл.). Выпадающий из общего ряда высокий нормальный потенциал лития обусловлен особенно плотной водной оболочкой иона лития (ср. табл. 17). В ряду от иона лития к иону цезия водная оболочка уменьшается. В гой же последовательности возрастает электролитическая подвижность (см, табл. 30). Хотя ион лптия и является наименьшим среди щелочных ионов, в водных растворах вследствие» наличия самой большой водной оболочки он наименее подвижен.
Таблица 30
Электро литическая подвижность щелочных по нов в растворах при бесконечном разбавлении при 18°
Ът+ Na + К* РЬ* CS*
33,5 43,5 64,6 67,5 68
Энергия, освобождающаяся при гидратации попов, измеряется теплотой гидратации. Суммы и разпостп энергий гидратации можно рассчитать из тепл от растворения*. Если пересчитать приведенные в табл. 28 теплоты гидратации в электрон-воль
* Ср. сноску на стр. 841. Сушил и разности теилот гидратации ионов известны точнее, чем их абсолютные значения. Абсолютное значение теплоты гидратации водородного иона лежит между 250 и 265 хкил!г-ион.
182
Глава б
тах и результат прибавить к нормальным потенциалам, данным в тон же таблице, то можпо получить нормальные потенциалы, которые проявляли бы щелочные ионы, если бы они не были гидратированы: Li* 1,9; Na+ 1,6; К+О,6; Rb+ 0,4; Cs* 0,2 е. Действительно, эти потенциалы равномерно убывают от Li к Cs, как это и следовало ожидать па основании увеличения электроположительного характера атомов при переходе от L1 к Cs и если учесть, что потенциал раствора тем ниже, чем сильнее Стремление данного вещества перейти в ионное состояние.
Принимая во внимание теплоты гидратации, можно рассчитать нормальный потенциал из теплот сублимации металлов и потенциалов иопизации свободных атомов, используя круговой процесс, описанный на стр. 174 и сл.
Исходными веществами являются при этом металлы, продуктами реакции — гидратированные ионы; энергию гидратации используют вместо энергии решетки. Для точного расчета следует, конечно, учесть различие между тепловыми эффектами и изменениями свободной, энергии, а также температурку то зависимость. Макитима [Ма-kishiroa,1Z. fclektrochem., 41,697, 1935] сделал точный расчет для большого числа металлов и получил значения, хорошо совпадающие с непосредственны мн измерениями. В табл. 28, помимо измеренных нормальных потенциалов щелочных металлов, приведены также потенциалы, рассчитанные Макишима.
Соли щелочных металлов бесцветны (если они не содержат окрашенных анионов). Они почти все легко растворимы.-, только литий образует несколько большее число довольно трудно растворимых солей. Водные растворы солей содержат бесцветные, положительно одновалентные щелочные ионы, которые в разбавленных растворах в большей или меньшей степени гидратированы (ср. табл. 17). Соли более легких щелочных металлов в кристаллическом состоянии часто содержат значительное количество воды. Помимо поды, некоторые из этих солей, особенно соли лития, могут кристаллизоваться со спиртом. Однако, за исключением солей лития, большинство щелочных солей не растворимы в спирте или только мало растворимы. В водном растворе щелочные соли практически полностью диссоциированы. То же можно сказать о гидроокисях, которые'поэтому являются самыми сильными основаниями.
Соли щелочных металлов и их гидроокиси в водном растворе не только полностью диссоциированы, но и силы взаимного притяжения свободных попов там также сравнительно незначительны. Это обусловлено, во-первых, циаким зарядом, по, кроме того, большими иоппымн радиусами. Влияние последних становится особенно заметным в концентрированных растворах. У вел и чей и см ионных радиусов объясняется возрастающая сила оснований, т. е. возрастающая «кажущаяся диссоциация» гидроокисей (верпсе рост коэффициента электропроводности/ц) в ряду от гидроокиси лития к гидроокиси цезия. Согласно Xласко (Hlasko, 1935), в 0,031 п. растворах при 25° имеет следующее значение для
ЫОН NaOH КОН Rl)0H CsOH
/ 0,918 0,935 0,938 0,044 0,955
Некоторые другие свойства соединений щелочных металлов, например сравнительно большая летучесть хлоридов, также обусловлены большими радиусами щелочных иолов.
Соли щелочных металлов со слабыми кислотами, например карбонаты, вследствие гидролиза проявляют сильно основную реакцию.
Характерным свойством щелочных металлов является легкость, с которой возбуждается световое излучение их атомов. Если по слишком труднолетучие соединения щелочных металлов внести в пламя бунзеновской горелки, то оно окрашивается. При спектроскопическом исследовании в видимой области появляется несколько характерных линий. Как будет показано в разделе «Спектры щелочных металлов», легкость, с которой возбуждается световое излучение, и простота строения спектров находятся в тесной связи с сильно электроположительным характером щелочных металлов.
Первая группа периодической системы
183
Из теории Косселя следует, что характеристические свойства щелочных металлов определяются их местом в периодической системе. Они находятся в группе, непосредственно расположенной за инертными газами. Их нейтральные атомы содержат в соответствии с этим на один электрон больше, чем атомы предшествующих им инертных газов. Как показывают спектроскопические измерения работы ионизации, этот электрон легко отрывается, в то время как на отрыв второго электрона нужно затратить несравненно большую работу (ср. табл. 28), для чего энергии образования решетки далеко нс достаточно. Этим объясняется, почему щелочные металлы в своих гетерополярных соединениях всегда положительно одновалентны. После отрыва одного электрона оставшиеся атомные остовы, т. е. одновалентные ионы щелочного металла', имеют не только такое же число электронов, как у непосредственно предшествующих инертных газов, но и такую же электронную конфигурацию, т. е. электроны находятся здесь в тех же квантовых состояниях, как и в непосредственно предшествующих инертных газах. Невозможность получения соединений щелочных металлов, в которых они были бы отрицательно заряжены, объясняется слишком большим расстоянием каждого щелочного металла от следующего инертного газа.
Из того факта, что (ср. стр. 195 и сл.) основными термами спектров поглощения атомов щелочных металлов являются s-термы, следует заключить, что каждый атом обладает одпим электроном, который в нормальном состоянии атома находится на энергетическом уровне с побочным квантовым числом I == 0. Этот электрон лежит каждый раз вне электронной оболочки предшествующего инертного газа, т. е. главные квантовые числа соответствующих основных орбит каждый раз на единицу больше, чем у предшествующих инертных газов. Таким образом, основные орбиты у внешних электронов щелочных металлов обозначаются следующими квантовыми числами:
Ы Na К ВЬ Св
\п = 2, (=0 п=3, (=0 п=4, /=0 п = 5, 1—0 п=6, Z=0.
Вследствие значительного экранирования центрального заряда электронными оболочками инертных газов внешние электроны в атомах щелочпых металлов связаны слабо. Связь тем слабее, чем выше главное квантовое число. Так объясняется сильно электроположительный характер щелочных металлов и его возрастание в направлении от Лития к цезию. Этим же объясняется большая величина атомных радиусов щелочных металлов и значительная разница между атомными и ионными радиусами (ср.табл. 28). Последние относятся к атомным остовам, которые остаются при отрыве внешнего Электрона, От лития к цезию атомные и ионные радиусы значительно возрастают в соответствии с положением, что протяженность «электронного облака» (которое в случае ( = 0 обладает шаровой симметрией) сростом главного квантового числа сильно увеличивается (как видно из рис. 25 на стр. 124).
В соответствии с теорией Гситлера и Лондона (ср. стр. 155 и сл.) щелочные металлы должны образовывай., правда с мепьшей стабильностью, также гомеополярные соединения, в которых Они равным образом одновалентны. Это подтверждается опытом. Как ноказынают определения плотности пара, в парах щелочных металлов несколько выше точки кипения паряду с одноатомными находятся также двухатомные молекулы (ср, стр. 192). Алкильные соедимения щелочных металлов, например NaCH3— метилнатрий, также, пожалуй, следует рассматривать как гомеополярные соединения, хотя их растворы в других алкилах металлов, например в диэтилцинке, как нашел Хейн (Hein, 1922), проявляют электролитическую проводимость. Алкилы щелочных металлов были впервые выделены Шленком (Schlenk, 1917). Это бесцветные, нерастворимые в большинстве индеферентных растворителей порошки, которые разлагаются при нагревании без плавления и воспламеняются па воздухе.
Первый элемент к ряду щелочных металлов, литий, по сравнению с другими щелочными металлами занимает во многих отношениях особое место. Он образует, например, различные соединения, которые никакими другими щелочными металлами не образуются. В некотором отношений его поведение больше напоминает щелочноземельные металлы. Подобно
184
Глава 6
последним, некоторые соли лития трудно растворимы, например фосфат, карбонат, фторид. Б олео резко выраженная, чем у остальных щелочных металлов, способность к образованию двойных солей с остальными представителями группы также приближает его к элементам второй группы.
Натрий также проявляет в некотором отношении отклонение от поведения, типичного для остальных членов его группы, но в значительно меньшей степени, чем литий. То обстоятельство, что только калий, третий элемент после лития и натрия, оказывается вполне типичным представителем щелочной группы и подтверждает существующее в периодической системе Правило, согласно которому только у второго или третьего элемента главной подгруппы полностью проявляются характерные свойства группы, в то время как первый и в: меньшей степени ипогДа также второй элемент обнаруживают отклонение от этих характерных свойств. Первый элемент часто является при этом переходным по своему поведению к следующей славной подгруппе. Второй элемент напоминает иногда соединения побочной подгруппы, принадлежащей той же группе.
Натрий и цезий являются чистыми элементами. Остальные щелочные металлы при исследовании при помощи масс-спектрографа оказались смешанными элементами.
Как било установлено Кэмпбелом (Campbell, 4907), калий радиоактивен, хотя и крайне слабо: активность составляет приблизительно i/l000 активности урана. То же явление замечено для рубидия. Распад рубидия сопровождается только p-из л учением, в то время как у калия, кроме p-излучения, отчасти идёт атомное превращение путем электронного захвата (ср. т. II). Наибольшая скорость р-лучей калия, как заключают по их проникающей способности, составляет—2,5-КРо&я/сем, а р-лучей рубидия — примерно 1,8- Ю10 см рек.
Согласно спектрографическим данным, калий представляет собой смесь трех изотопов с атомными весами 39, 40 и 41. Их количества относятся как 93,260 : 0,119 : : 6,729*. Из этих трех изотопов радиоактивным является только изотоп с массовым числом 40. В 1 г калия за 1 сек происходят в среднем 31,6 атомных превращений. Из них 28,3 превращения происходят с испусканием р-частиц, а 3,3 — путем электронного захвата. Отсюда для изотопа калия 40К период полураспада равен 1,18 10й года. Благодаря ради о активному распаду 40К постоянно образуется аргон (точнее, 41)Ат), а именно ежегодно по 3,6- 10’ia „а 1 г калия. И а этом основан калий-аргоновый метод геологического определения возраста минералов (Russel, 1954; Wasserburg, 1955; Gentner, 1955; см. также т. II).
То, что явление радиоактивности связано с одним из наиболее тяжелых изотопов, уже в 1927 г.: обнаружил Хевеши, частично раздели в изотопную смесь методом «идеальной дистилляции» (см. т. II). Оказалось, что та часть, которая была обогащена более тяжелыми изотопами, обладала большей радиоактивностью. Но радиоактивность была только на 4,4% выше радиоактивности обычного калия, в то время как изотоп 41К, как показало определение атомного веса, был обогащен более чем на 10% по сравнению со своей первоначальной концентрацией. Это показало, что свойством радиоактивности обладает не 41К, а40К; именно для 40К при «идеальной дистилляции» следует ожидать наименьшего обогащения. То, что 4ХК не может обусловить радиоактивность калия, вытекает далее из следующего. При p-распаде 41К в соответствии с правилом радиоактивных сдвигов должен образовываться 41Са. Тогда последний должен был бы обнаруживаться в значительном количестве в геологически старых калиевых минералах при условии, что 41К был радиоактивным. Исследование посредством масс-спектрографпи показало, одпако, полное отсутствие 41Са (Hevesy, Aston, 1935). Позднее Смит (Smythe, 1937) измерением активности масс-спектр о скопически выделенного-изотопа 40К смог непосредственно показать, что оп является носителем радиоактивности.
Радиоактивность рубидия определяется распадом изотопа a,Rb, примешанного к обычному 85Rb (в количестве 27,85%). Период полураспада 8,Rh равен 4,6 Юю года (Strassmann, 1956). При этом происходит испускание р-лучей (с максимальной энер
* Это изотопное соотношение считается точным для солей калия из морской воды. У минералов для соотношения 3:9К:41 К найдены значения, которые различаются до 3%; в золе растений обнаружены колебания боле о чем 15%.
Первая группа периодической системы 18 5-
гией 0,132 Мэе) и превращение Rb в стабильный изотоп стронция S7Sr. На этом осно- , ван стронциевый метод определения геологического возраста (ср. т. II). Радиоактивный изотоп цезия в природе не встречается (Hahn, Maltauch, 1942).
Исходя из Периодичсско йсистемы, предполагали существование еще одного ще-лочпого металла е порядковым числом 87 (окацевий). Долгое время все попытки обнаружить этот элемент были безуспешными. Выводы Аллисона (Allison, 1930 и сл.), сделанные изданных магнетооптических измерений, были бездоказательны.Данные Папнша (Papish, 1931), который полагал, что им обнаружены рентгеновские линии элемента 87, также оказались ошибочными (Noddak, 1934), но затем существование экаде а ия в природе было установлено благодаря работам М. Перей (1939), которая нашла, что этот элемент встречается как продует двоякого распада актиния, и назвала его сначала актинием К, позднее францием (Fr). Как уже ранее предполагали исходя из его положения в периодической системе и правил о стабильности ядер, франций неустойчив^ (радиоактивен). Подробнее см. Т. П.
Распространение в природе. Благодаря чрезвычайной способности к окислению щелочные металлы никогда не встречаются в весомых количествах в свободном состоянии, а исключительно в форме своих соединений. Однако в таком виде натрий и калий принадлежат кнаиболее распространенным элементам. Содержание их в земном коре составляет приблизительно 2,6—2,4%. Б общем они встречаются примерно одинаково часто, хотя соединения калия менее распространены, чем соединения натрия. Нали.е^ вый полевой цепат К [AJSi308] и калиевая слюда KAla[A]SisO10](OH, Р)А являются составными частями многих распространенных пород, в частности гранитов*. Наиболее распространенными минералами натрия являются олигоклаз (известково-натриевый полевой шпат) и альбит (натриевый полевой пшат) Na[AlSi308J, образующие аналогичные породы. Хлористый натрий NaCl встречается в виде каменной соли, залегающей во многих местах в виде мощных пластов, которые образовались благодаря высыханию участков морей или колоссальных озер. Иногда каменная соль имеет прослойку солен калия и магния, так называемых «отбросовых солей», как, например, па севере Германии около Стассфурта. В морской воде растворены значительные количества хлористого натрия. Содержание его в воде океана колеблется незначительно и составляет там, так же как в Северном море, приблизительно 2,5%. Воды Балтийского моря, напротив, содержат трлько 0,6—1,7 % хлористого натрия, в то время как в водах Средиземного-моря найдено до 3%, а в водах Красного моря даже до 3,5% этой соли. В неимеющих стока внутренних морях содержание этой соли гораздо больше. Так, воды Мертвого моря содержат примерно 20% хлористого натрия наряду с большими количествами других? солей. По сравнению с содержанием хлористого натрия содержание в морской воде хлористого-калия очень незначительно, оно составляет примерно только V40 содержания хлористого натрия. Это объясняется тем, что в противоположность соединениям натрия земная поверхность сильно адсорбирует соединения калия, так что они не достигают моря. Из земной поверхности калий попадает в растения. Поэтому он содержится в их золе в виде карбоната (поташа). Наоборот, содержание натрия в золе материковых растений незначительно. Только прибрежные и морские растения содержат большие количества натрия, главным образом в виде органических соединений. Отдельные растения содержат также заметные количества лития, например-некоторые сорта табака.
* Гранит, представляет собой горную породу, которая состоит из минералов ортоклаза, слюды и кварца, содержащихся в Нем в виде грубозернистой смеси. Горной породой такого же состава, по другой структуры (сланцевой) является столь же широко распространенный гнейс.
186
Глава в
В человеческих и животных тканях и в жидкостях организма содержится небольшое количество хлористого калия наряду с большими количествами хлористого натрия. Употребляемый для физиологических опытов с животными рингер о некий раствор (аналогичный по составу сыворотке крови), который, например, может поддерживать деятельность изолированного сердца, на 100 ч. 0,6%-пого раствора NaCl содержит по 1 ч. 1%-ного раствора МаПСО3, 1%-ного раствора CaCIg и 0,75%-ного раствора КС1. Значительные количества калия содержатся в поту овечьей шерсти.
В качестве технически важных соединений натрия следует указать еще имеющийся в Гренландии криолит Na3AlF6 и встречающуюся в мощных залежах на побережье Чили чилийскую силитру NaNO3. Однако значение этих минералов определяется не содержанием в них натрия; чилийская силитра, содержащая связанный азот, служит как удобрение, а криолит применяют для производства алюминия.
Литий в небольших количествах сопровождает калий и натрий, главным образом в горных породах и некоторых минеральных источниках. Имеются, однако, также породы, в которых он содержится в значительно больших количествах, напркмер в амбливоните LiAI[PO4]F, трифилине (LI, Na) (Fe, Мн)[РО4], сподумене (трифан) LiAl[Sl2O6], лепидолите. (литиевая слюда) К(А12[Л1513О1(|], Li2[AlSi3Oe(OH,F)4])x X (ОН, F)3 и петалите (Li, Na) AlSi4Oi0. В друзах гранита с острова Эльба вместе с петалитом встречается аналогичный по составу цезиевый минерал CsiAljSigOje'H^O. Поэтому эти минералы называют также «кастор и поллукс», В большинстве же случаев оба щелочных металла — р>убидий и цезий—встречаются только в сопровождении других щелочных металлов лишь в очень незначительных количествах. Однако в незначительных концентрациях они широко распространены. Сравнительно много рубидия находится в лепидолите (более 1%). Много меньше рубидия (примерно 0,015%) наряду со следами цезия содержится в карналите. Методы получения рубидия из кар-налита разработал Яндер (lander, 1929—1930). Обзор щелочных минералов приведен в табл. 31 (стр. 187).
История. Уже в книгах Ветхого завета упоминалось вещество neter, которое служило в качестве моющего средства. То же вещество, которое было известно еще в древнем Египте, встречается и у греческих писателей (Аристотель, Диоскорид) под названием vlrpov, а у древнеримских (Плинии) под названием nitrum. Все эти названия следует отнести к соде* и отчасти к поташу, который тогда еще не могли отличать от соды. Из названия nitpum у арабских алхимиков постепенно возникло слово natron. Кроме того, в рукописях, приписываемых алхимику Геберу (Geber, XIV "и XV вв.), в том же значении встречается название alkali** наряду с употребляемым там впервые названием «.года». Однако у алхимиков более употребительными были названия, указывающие на происхождение соответствующего вещества; например, поташ, полученный ив винного камня, называли sal tartari, а полученный из золы растений — sal vegeiabile. Примерно с 1600 г. для обозначения углекислой щелочи употребляли обозначение sal lixivtosum, из которого затем возникло немецкое слово «Laugensalz» — щелочная соль.
Различие «натра», т. е. основания, образующего поваренную соль, и «кали» (который тогда обычно получали в виде карбоната из золы растений) было впервые отмечено в 1702 г. химиком Шталем (Stahl, 1660—1734). Однако экспериментально различие обоих элементов было доказано только в 1736 г. Дюамелем де Монсо (Duhamel de Monceau, 1700—1781). В 1758 г. Маркграф установил индивидуальность обоих элементов по окрашиванию пламени. Клапрот (Klaproth, 1797) впервые показал, что калии вопреки употреблявшемуся тогда , названию alcali vegetabile встречается также и в минералах. Получить свободные металлы впервые удалось Деви (Davy, 1807). Он подвергал электролизу кусок влажного едкого натра (соответственно едкого кали), лежавшего в платиновой чашке, служившей одновременно катодом.
* Обозначение nitrum для селитры появляется лишь в конце XVI в.
** Вероятно, от арабского слова qualjan — зола растений.
Таблица, 31
Щелочные минералы
Простые соли Двойные и смешанные соли
Галогени Каменная соль NaCl Сильвин КС1 Тенардит NaaSO4 Арканит K2SO4 Глауберова соль Na3SO4’10HaO Чилийская селитра (натриевая селитра) NaNO3 Калийная селитра .KNO3 ды, сульфаты и нитраты Карналлит Дугласит KCl.MgCl2-6H2O 2KCl-FeCl2-2H2O Каинит Эритросидерит КС1 MgSOi- ЗН2О 2КС1 • FeCl3- Н2О Глауберит NaaSO4-CaSO4 Сингенит KaSO4-CaSO4-H2O Глазерит Na2SO4-3K2SO4 Бледит (астраканит) Na2SO4-MgSO4-4H2O Левом Na2S04'MgS04-2V2H20 Вантгоффит 3NaaSO4.MgSO4 Полис алит К 2S 0 4 М gS 0 4 2CaSO4 2112О Шепит K2SO4-MgSO4-6H2O Лангбейнит K2S04-2MgS04 > Квасцовый камень Ярозит (алунит) ш К(АЮ)з(8О4)2-ЗН2О K(FeO)3(SO4)2-3H2O Натриевые квасцы (сол- Натрояро знт фатарит) jjj NaAl(SO4)r12H2O Na(FeO)3(SO4)2.3H2O Калиевые квасцы (кали- нит) KA1(SO4)2-12H3O Дарапскит NaNO3-Na2SO4-HaO Криолит NagAIFg Хнолит 5N'aF-3AlF3 Гиератит KaSiF6 Пахнолит (томееполит) NaCaAlFo-H3O Сульфогалит NaF-NaC1.2Na2SO4
Сода №аСО3-10Ц2О Трана Na2COs'NaHCO3'2li2O Карбонаты Пирссонит Nа2СО3 СаСОз 2НчО Натрокальцит Nа2С03 СаС03 5Н2О Даусонит Na2CO3[AI(OH)2]aCO3
Бораты
Бура (типкад) Боронатрокальцит (тинкальцит)
Na2B407.10II20 КаСаВ5О9-6Н2О Франкландит Na.aCaBeOir7H2O
Фосфаты и арсенаты
Трифил ин LiFe[PO4]
Амблигонит LiAlF[PO4]
Дурапгнт NaA!F[AsO4]
Продолжение табл. 31
Простые соли
Двойные и смешанные соли
Силикаты
(о структуре силикатов см. стр. 541 и сл.)
Полевые шпаты
Натриевый полевой шпат.
(альбит) Na[AlSi3Os]
Калиевый полевой шпат (ортоклаз) K[AlSi3Os]
Натриевый ортоклаз, твердый раствор ортоклаза й альбита
Известк ово-натриевые полевые шпаты ср. стр. 272, твердый раствор альбита и анортита
Нофелвп Na[AlSiO4);
Лейцит К[AlSi2Oa]
Содалит Na3[Al3Si3O12]-NaCl
Новеет,
Na3(Al3Si3O12).Na2[SO4]
Га и пн
Na3[Al3Si3Oi2].(Na2,Ca)(SOJ
Лазурит (ультрамарип) Na3[Al3Si3O12]-Na2S2
Скаполит, твердый раствор
Sa3[AI3Si9O24].NaCl и
CaJAl6Sie-O24]Ca[SO4,CO3]
Слюды
Мусковит (калиевая слюда) KAl.,[AlSi3OI0](OH, F)a
Биотит (магниевая слюда)
K(Mg, WAlSi3Oio](OH, F)2
Флогопит (магниевая слюда)
KMg3IAlSi3O10](OH, F)3
Лепидолит (литиевая слюда) твердый раствор
KAi2[AlSi3O10](OII, F)2 и
KLi3[AlSi3Oe(OII, F)41(OH, F)2
(часто содержит также hb и Cs)
Цинвальдит (литиево-Железная слюда) твердый раствор
K(Mg, Fe)3[AlSi3OI0](OH, F)2 и
KLi2[AISi306(0H, F)4](OH, F)2
Парагонит (натриевая слюда)
NaAl2[AlSi3Oi0](OH, F)a
Щелочные авгцты:
Сподумен (трифан)
LiAl[Si2Oe]
Жадеит
NaAl[Si2O6]
Эгирин (акмит)
NaFe(Si3Oe]
Петалит (кастор)
(Li, Na) AlSi4O!0
Поллуцит (поллуке.)
Cs4Al4Si3O2e. HSO
Цеолит ы:
Натролит Иа3[АЦН13О13]2Н3О
Анальцим Na[AlSi2O3]-Н^О
Апофиллит
KCa4F[Si802(J-8H2O
Хабавит
(Na2, Ca)[Ai2Si4OI2|-6H2O
Томсонит (комнгонит)
NaCa2[AlsSi502o]6H20
Первая группа периодической системы
189
Продолжение табл. 31
Простые сопи Двойные и смешанные соли
Щелочные роговые обманка: .Рибекит Na2FeyFe*n[SieO3al(OH)2 Р лаукофан Nas(Mg,Fe)3 (Al,Fe>2 [SfsO22](OH)2 Десмин (Na2Ca)[AlaSieOie]‘6H2O Филлипсит (К2, Ca)[Al2Si4Oi3]-4H2O Гармотом (К2, Ва)[А12Щ5О14]-5Н2О
-A pifieede о нит (Na3, К2, Ca)(Mg, Fe)3(Al, Fe)3 (Si8Oa2](OH)2 Содержанке воды в цеолитах сильно колеблется.
Литий был открыт в 1817 г. Арфведсопом (Arfvedson), учеником Берцелиуса, сначала в петалите, вскоре затем также в сподумене и лепидолите. Свое название он получил на основании содержания в камнях (WOog— камень). Аналогия между соединениями лития и соединениями щелочей -была уже известна исследователю. Присущее литию красное окрашивание пламени впервые наблюдал Гмелин (1818). Получйть свободный-металл удалось впервые Буизену и Маттиссену в 1855 г. путем электролиза расплавленного хлорида.
Рубидий и цезий были открыты Бунзеном в воде Дюркгаймерского минерального источника на основании присущих им спектров. В соответствии с этим они были также и названы.1 открытый в I860 г. цезий — по двум характерным синим линиям спектра {caesius — сине-серый), и рубидий, открытый в 1861 г. по двум характерным линиям в красной части •спектра (rubldus — темно-красный). Получение металлического цезия удалось впервые осуществить Сеттербергу (Setterberg, 1882) электролизом расплавленной смеси цианидов цезия и бария. К этому времени Бунзеном уже была получена амальгама, цезия. Рубидий был получен Бунзеном в металлическом состоянии электролизом расплавленного!, хлорида^
Получение. Получение технически важных щелочных металлов почти всегда проводят электр олнвшческп. Правда, щелочные металлы можно выделить из их соединений в свободном состоянии чисто химическим путем при помощи таких сильных восстановителей, как углерод, карбид кальция^ кар-. бид железа. Однако в случае более легких щелочных металлов эти способы связаны со значительно большими техническими трудностями, чем электролиз. В условиях крупного производства литий и натрин стали получать исключительпо электролитическим путем. Ранее натрий изготовляли электролизом расплавленной гидроокиси. Но так как в дальнейшем и гидроокись стали получать электролизом хлорида, этот метод получения потребовал в общем значительно большего расхода электроэнергии, чем непосредственный электролиз хлорида. Однако электролиз гидроокиси техни- г чески проще осуществлялся, так как опа обладает более низкой точкой плавления^ чем хдорид. После того как научились достаточно понижать точку плавления хлорида введением подходящих добавок, электролиз хлорида стал все более вытеспять электролиз едкого патра.
190
Глава 6
Способы, основанные на электролизе расплавленного едкого натра, была предложены Кастнером (Caslner, 1890), который пользовался аппаратом, показанным парно. 30. Он состоит из обогреваемого в верхней части железного сосуда, который содержит расплав. В сосуд снизу вставлен железный стержень, служащий катодом, в то время как анодом является окружающий катод железный цилиндр. Кроме того, в сосуд введен еще короткий более узкий железный цилиндр, к которому присоединен цилиндр из проволочной сейш, окружающий верхнюю утолщенную часть катода. Последний должен предотвращать проникновение к аноду выделяющегося на
Рис. 30. Электролиз расплавленного е.дйог о натра (способ Кастнера).
Рис. 31. Даупс-камера для электролиза расплава поваренной соли.
катоде металлического натрия. Натрий из-за своего малого удельного веса собирается поверх плава и его можно оттуда легко удалить. Па апо де вследствие разрядки ионов ОН' выделяются кислород и вода по уравнению
2ОН' = 1/гОг + НгО + 2е.
Большая часть воды испаряется. Однако отчасти она разлагается током, так что на катоде, помимо патрля, всегда выделяется видород, который, выходя из-под, крышки внутреннего цилиндра, воспламеняется. Болес тяжелые примеси расплава собираются в нижней суженной части сосуда. В самом низу сосуд закупорен слоем .застывшего едкого патра. Важно, как указывал уже Кастнер, чтобы температура бани возможно меньше превышала температуру плавления натрия, так как в противном случае металлический натрий смешивается с расплавом и окисляется кислородом, Который при более высокой температуре также в значительном количестве растворяется в расплаве.
Вследствие распределения металлического натрия в расплаве при высоких температурах вначале не удавались попытки выделить натрий непосредственно при электролизе расплавленной поваренной соли, Однако оказалось, что добавкой хлористого-кальция точку плавления можно значительно понизить, так что электролиз такой смеси можно провести при температурах незначительно выше б00s. Па этом основан способ Кибы (Ciba, 1910), который осуществил заводское получение натрия электролизом, хлорида. Очень удобная камера для проведения этого процесса была сконструирована Даунсом (Downs). Даунс-камера (рис. 31) состоит из каменного сосуда, в который вставлены снизу графитовый стержень А, служащий анодом, и сбоку железные катоды К. Анод покрыт железным кожухом 7, на котором укреплена проволочная сетка 2, разделяющая анодлое и катодное пространства. Смеет, хлоридов, которая поддерживается в расплавленном состоянии теплом электрического тока, с поверхности покрыта твер
Первая группа периодической системы
191
дой коркой соли, предохраняющей расплав бт доступа воздуха и одновременно от больших потерь тепла. Выделяющийся на катоде патрий поднимается и течет вдоль по согнутому желобом краю 3 кожуха 7 к Железкой отводной трубке 4, по которой он перетекает в приемник б. Таким образом, жидкий натрий совершенно защищен от влияния воздуха; одновременно край кожуха, почти целиком закрывающий нижнюю часть сосуда, препятствует попаданию поваренной соли в пространство между электродами, прежде чем она совершенно не обезводится и не расплавится.
Исходя из приведенных в табл. 27 и 48 теплот образования NaCl и СаС12, можно заключить, что при электролизе смеси NaCl и СаС12 должен выделяться не натрий, а кальций, так как теплота образования 1/г моля СаС12 меньше, чем теплота образова-иия одного моля NaCI. Это относится, одпако, только к тсплотам образования при обычной температуре. С ростом температуры теплота образования NaCl уменьшается сильнее, чем теплота образования C.aCL- При 620° она составляет для NaCl 90,5 «клл и для */г СаС12 — 90,9 «вал. Таким образом, при этой температуре NaCl имеет меньшую теплоту образования. И поскольку исходя из теплот образования можно сделать заключение относительно сродства, следует ожидать, что NaCl при 620° показывает более низкое напряжение разложения, чем СаС1,*. В соответствии с этим опыт пока' зыкает, что металлический натрий, полученный электролизом расплавленной смеси, только незначительно загрязнен кальцием (в общем менее чем иа 1%). Если дать металлу медленно охлаждаться, то кальций почти полностью отделяется (до нескольких сотых процента), так как с падением температуры растворимость кальция к расплавленном натрии сильно уменьшается. Поэтому способами Кибы и Даунса непосредственно получают патрий требуемой для технического применения чистоты.
Получение калия ведут также отчасти электролизом расплава (в последнее время путем электролитического разложения кислородных солей калия, растворенных в расплавленных галогенидах кадия; при этом на графитовом аноде выделяется СО2). Однако наряду с этим для получения калия существенное значение имеют также чисто-химические способы, и прежде всего карбидные способы (взаимодействие KF с СаС3).
Приготовление металлического лития можно осуществить электролизом расплавленного чистого хлорида, так как последний плавится значительно ниже, чем хлористый натрий. В технике для этого применяют более легкоштанкие смеси, а именно чаще всего смесь LiCI и KCI. В лаборатории для этого можпо также использовать хлористый литий, растворенный п пиридине.
В случае рубидия и прежде всего в случае цезия электролитический метод менее пригоден. Здесь получение лучше вести химическим путем: нагреванием гидроокисей с, металлическим магнием в токе водорода или с металлическим кальцием в вакууме. Согласно дс Буру (de Воет, 1930) , в качестве восстановителя особенно подходит цирконий. Небольшие количества Rb или Cs, согласно де Буру (1927), удобно получать нагреванием их хлоридов в смеси с азидом бария в высоком вакууме. Образующийся при распаде азида барий восстанавливает щелочные металлы нз их хлоридов. Они испаряются и оседают на холодных стенках сосуда.
Свойства. Чистая поверхность щелочных металлов имеет серебристобелый металлический блеск, за исключением цезия, который по данным Костеану (Costeany) в чистом состоянии — золотисто-желтый. Однако блеск этих металлов можно наблюдать только на препаратах, находящихся в вакууме, или на свежем срезе. На воздухе щелочные металлы тотчас тускнеют. Все щелочные металлы чрезвычайно мягки и легко поддаются сжатию. Наиболее твердый из них — литий обладает твердостью
* Данные, приведенные в табд. 37 и 47, основанные па измерениях Неймана (Neumann), показывают непосредственно, что напряжение разложения NaCl лежит при высоких температурах ниже, а при обычпых выше, чем напряжение разложения СаС12. Для 25°, Исходя из приведенных температурных коэффициентов, получается ZbTaCi ~ ж77 и 20аС12 = 3,26 в. Однако по данным этих таблиц напряжение разложения СаС12 превышает напряжение разложения NaCl только при температурах выше 650°, в то время как, согласно техническим данным, это должно происходить уже ниже 620°. Если рассчитать разницу между потенциалами выделения Na и Са При 620°, учитывая, что разность теплот образования равна разности сродства в соответствии с уравнением (3) [стр. 166], то оказывается, что эта разница составляет только 0,02 е. Этот результат лежит пиже ошибок значений, приведенных в табл. 37 и 47. Одпако повышение напряжения разложения СаСЦ2 при добавлении NaCl к расплаву можно объяснить комплексообразованием.
"192
Глава 6
*0,6 по шкале Мооса, следовательно, он значительно мягче талька (твердость талька принята а а единицу шкалы твердости Мооса). С возрастанием атомного веса твердость еще более уменьшается. Натрий обладает приблизительно твердостью белого фосфора, а цезий мягок, как воск.
Сжимаемость, возрастающая отлития к цезию, показана в табл. 32, которая содержит данные, измеренные при комнатной температуре. Значения таблицы дают уменьшение объема в дробных частях объема, отнесенные к давлению в 1 бар, т. е. 1 млн. •дин на 1 поверхности или 75 см рт ст.
! Таблица 32
Сжимаемость щелочных металлов
Металлы Литий Натрий Калий Рубидий Пеняй
-Сжимаемость при 1 баре 8,8- 10-в 15,4-IO-0 31,5-10“® л 40-10“® 61-10“®
Щелочные металлы обладают высокой электропроводностью. При -0° удельная электропроводность лития в 10,9 раза, натрия в 22 раза, калия в 15 раз, рубидия в 8 раз и цезия в 5,2 раза больше электропроводности ртути при той же температуре. Правда, щелочные металлы уступают в этом отношении лучшему проводнику среди металлов — серебру; лучший проводник из щелочных металлов —. натрий —- обладает сопротивлением, почти в три раза превышающим сопротивление серебра.
Щелочные металлы — чрезвычайно легкие металлы. Калий и натрий легче воды; литий плавает в керосине; литий самый легкий среди всех твердых при обычной температуре элементов. Точки плавления и кипения щелочных металлов лежат очень низко (ср. обзорную таблицу).
Щелочные металлы в парообразном состояния в основном одноатомны. Это было сначала установлено путем определения отношения удельных теплоемкостей Ср/ci,. Однако более поздние определения плотности пар а показали, что в основном у самых легких щелочных металлов происходит заметная ассоциация атомов в двухатомные молекулы (существование их впервые было установлено спектроскопически). Например, Родебуш (Rodebu.-jch, 1930) определением плотности пара установил степень ассоциации паров натрия при 5703 и 13,5 мм рт ст равной 13% (что совпало со значением, которое было вычислено из работы диссоциации молекул Na2, установленной спектроскопически). Для паров натрия при атмосферном давлении и температуре кипения патрия степень ассоциации составляет 16% (Ladenburg, Thiele, 1930). •Спектроскопически найденные величины работы диссоциации составляют:
1л2 Na2 К2 Rb2 Cs2
26,3 17,5 11,8 10,8 10,4 - ккал/моль
Пары щелочных металлов интенсивно окрашены. Пары натрия обладают пурпурной окраской, похожей па окраску разбавленного раствора перманганата. Калий в парах окрашен в сине-зеленый цвет, а рубидии — в зеленовато-синий. Аналогично окрашены коллоидные растворы щелочных металлов, которые получают электрическим распыленней металлов в ипдеферентном растворителе, например этиловом эфире. Эти растворы окрашены следующим образом:
Металл
Цвет коллоидных растворов
L1
Коричневый
Na
Пурпурно-фио лотовый до синего
К
Синий до сине-зеле ного
йь
Зеленоватосиний до зеленоватого
Са
Сине-зеленый до зеленовато-серого
По коричневой окраске эфирного золя литий близок к магнию, эфирный коллоидный раствор которого также окрашен в коричневый цвет.
В жидком аммиаке щелочные металлы растворяются, давая темио-•синее окрашивание. В большинстве случаев считают, что эти растворы также имеют коллоидную природу.
!
Первая гр угг па периодической системы 193
Все щелочные металлы разлагают воду (стр. 177). Литий реагирует с водой без плавления, натрий при этом плавится, калий воснламеняется. Еще более активно реагируют рубидий и цезий. Они тотчас воспламеняются при доступе кислорода, даже в отсутствие воды, в то время как остальные щелочные металлы на сухом воздухе или в токе кислорода воспламеняются только при умеренном нагревании. Литий, сгорая, образует окись LiaO, которая загрязнена только небольшими примесями перекиси, Натрий сгорает с образованием перекиси NaaOa. Остальные щелочные металлы дают при сгорании надперекиси (ср. стр. 202). Реакция щелочных металлов со спиртом с образованием алкоголятов уже была упомянута. G безводным эфиром, парафином или керосином никаких реакций не происходит. Желтоватые (соответственно голубовато-серые) корки, которыми постепенно покрываются натрий и калий при хранении в керосине, образуются в результате реакций с кислородными соединениями, содержащимися в керосине.
Будучи слабо нагретыми в атмосфере водорода, щелочные металлы образуют гидриды. С молекулярным азотом соединяется только литий, с образованием нитрида Li3N. Реакция идет особенно энергично при температуре темно-красного каления. Литий медленно реагирует с влажным азотом уже на холоду. Однако тепло, выделяющееся при реакции
3Li -|— ^/^N^ Li3N—| ’ -1 i ,2 ккал, меньше, чем теплота образования гидрида лития (ср. стр. 179), если выделяющееся тепло в обоих случаях отнести к одинаковому количеству лития.
С азотом, возбужденным электрическим тлеющим разрядом, могут соединиться также остальные щелочные металлы. Смотря по условиям опыта, при этом возникают нитриды M3N или азиды MN3 (Wattcnberg, 1930). Нитриды Na и К. можно получить также термическим разложением азидов. Нитриды интенсивно окрашены (при комнатной температуре в красный цвет), устойчивы в сухом воздухе, ио тотчас разлагаются водой или спиртом с образованием аммиака. С водородом Na3N реагирует при 120’ по уравнению
Na3N4-3H2=3NaH+NH3.
При пропускании над расплавленными щелочными металлами газообразного аммиака образуются амиды, например:
Na+NHa^NaNH^+Valh.
Эта реакция, так же как и другие рассмотренные реакции, основана па том, что щелочные металлы легко приобретают электроположительный заряд. Гетерополярный характер щелочных амидов с соответствующей формулой соединений JVrlNlIaF подтверждается хорошей электропроводностью их расплавов и пх растворов в жидком аммиаке.
С углеродом соединяется непосредственно только литий, образуя карбид Li2C2, который, однако, при более высокой температуре снова разлагается на элементы; карбиды остальных щелочных металлов удается получить только косвенным путем. Литий также является единственным щелочным металлом, который соединяется с кремнием. Эти элементы соединяются непосредственно при нагревании, образуя силицид Li6Sl3 в виде темно-фиолетовых гигроскопичных кристаллов, которые весьма реакционноспособны.
Спектры щелочных металлов. Щелочные металлы или их не слишком труднолетучие соединения окрашивают пламя бунзеновской горелки в характерный цвет, а именно; литий — в карминово-красный, натрий — 13 Г. Реии
<94
Глава 6
в желтый, калий, рубидий и цезий — в фиолетовый цвет. При рассмотрении окрашенного пламени: в спектроскоп видны отдельные яркие линии, положение которых настолько характерно, что на этом основании щелочные металлы можно легко идентифицировать.
На рис.. 32 линии, характерные для щелочных металлов, нанесены так, как они видпы при рассмотрении окрашенного пламени в небольшой ручной спектроскоп. Обозначенная буквой D желтая линия натрия появляется уже в присутствии ничтожно малых следов натрия. Она имеется в каждом спектре пламени, подученном в химической лаборатории, если только
Красный
L' П ГТ
Синий Фиолетовый
Na Г) линия
К
Rb
Л 800 700 000 500 4 00.мр
Рис. 32. Сшжтры щелочных металлов.
работу пе ведут с особыми предосторожностями. Так как эта линия удобна для ориентировки других линий спектра, то се наносят пупктиром и в спектрах других металлов. Различная толщина линий на рисунке должна дать приблизительное представление об интенсивности спектральных линий. Обычно наиболее сильных линий в спектре уже достаточно для идентификации. Если исследуемый металл присутствует в небольших количествах, более слабые линии в ручной спектроскоп не видны. Часто также мадо заметна и лежащая далеко вправо фиолетовая линия калия.
В табл. 33 приведены длины волн спектральных линии щелочных металлов, лежащих в видимой части спектра, выраженные в миллимикронах (тр. — миллионных частях миллиметра). Эта единица длин волн является наиболее пригодной для обычного спектрального анализа, так как употребляемые при этом спектроскопы нс в состоянии измерить десятые доли миллимикрона. Следует учесть, что коротковолновый свет при прохождении через призму разлагается сильнее, чем длинноволновый свет. Так как на рис. 32 липни представлены в таком виде, как они наблюдаются при разложении призматическим спектроскопом, красная часть спектра по сравнению с фиолетовой оказывается сильно сжатой. В табл. 33 ради полноты приведены также некоторые линии, которые обычно не видны, если излучение света возбуждается внесением изучаемого соединения
Первая группа периодической системы 185
Таблица 33
Липни щелочных металлов в видимой части спектра
Длина волн (wift); линии, соединенные фигурными скобками, неразличимы в спектроскопах с малой разрешающей способностью
.Читай Натрий Калий Рубидий Цезий ~
670,8 616,1 769,9 | 794,8 1 704,4
610,4 589,3 766,5 ( 780,0 1 760,0
460,3 568,5 В9-3, 1 775,8 ' 697,3
413, г 488,1 691,1 761,9 672,3
301.6 Г,S3,2 740,8 621,3
031,2 629,8 455,5
580.2 620,6 459,3
578,3 421,6
404,5 420,2
н пламя бунзеновской тореики; они становятся видимыми только тогда, когда применяется более высокая температура, которая, например, дости-гается возбуждением небольшой вольтовой дуги между раствором, содержащим испытуемые соли, и находящимся непосредственно над ним острием иридиевой проволоки.
Для получения спокойно горящей дуги положительный ток (в световом шнуре при включении подходящего промежуточного сопротивления) должен течь в направлении от жидкости к острию иглы иридиевой проволоки. При этих условиях получают, например, кромежелтой линии натрия D, еще и вторую линию натрия оранжевого цвета (при Л=(»16 тр). В противоположность первой она видна только тогда, когда натрий присутствует в весомых количествах. Различия в интенсивностях линий на рис. 32 представлены разной толщиной штрихов, а в табл. 33 — разным шрифтом. При небольших концентрациях веществ видны только линии, нанесенные жирным шрифтом.
Спектры щелочных металлов подобно спектру водорода и пскровому спектру гелия построены крайне просто. Поэтому их изучение дало основу для нахождения общих закономерностей в строении спектров. На рис. 33 приведен абсорбционный спектр паров натрия. Этот спектр очень близок к бальмеровской серии водородного спектра
W 550 500 450 400 J5O 500 250
Рис. 33. Спектр поглощения илроп натрия.
(рис, 17). Фактически этот спектр удается воспроизвести, используя формулу, которая выглядит аналогично формуле серии Бальмера. Эта дю серия имеется также в спектре излучении паров натрия и является там наиболее сильной наряду с некоторыми другими, которые не проявляются в абсорбционном спектре. Ее считают поэтому главной серией натрия. Было измеряло по менее 57 линий этой серин. Непрерывный спектр, прилегающий к коротковолновому концу серии, пе наблюдается в эмиссионном спек-13*-
196
Глава б
тре. Кроме главной серии, натрий обладает еще двумя побочными сериями, и так называемой серией Бергмана. Так, например, упомянутая выше оранжевая линия натрия (Л = 616 «iii) принадлежит к так называемой «резкой побочной серии# натрия. То же относится и к другим щелочным металлам. Главные серии являются в каждом случае наиболее сильными, поэтому опн и были открыты в первую очередь, Серии Бергмана были названы в честь ученого Бергмана (Bergmann), который в 1907 г. впервые нашел серии этого типа, и именно у щелочных металлов. Помимо интенсив но ст ей линий (которые у всех серий сильно уменьшаются к концу), названные типы серий различаются также еще и по другим признакам. Однако это интересно только Для спектроскопистов, которые на основании этих признаков распределяют линия в серил. Д.тя химиков серии являются достаточно определенными уже Потому, что под ними понимают совокупность линий, которые мри помощи формул удается представить в правильном последовательности. Они имеют большое значение, потому что позволяют точно предсказать характер связи внешнего электрона в атоме щелочного металла.
На основании изложенного на стр. 111 относительно поглощения энергии мри абсорбции света легко понять, что исходным уровнем для всех линий абсорбционной серии ;ш.тяется основной уровень внешнего электрона в атоме. Липин главной серии в эмиссионном спектре совпадают с линиями абсорбционной серии. Таким образом, основной уровень является конечным уровнем для всех лилий главной серии. Их энергия, полученная из положения границ серий, находится в хорошем соответствии с результатами измерений потенциалов ионизации. Соответствующее основному уровню главное квантовое число получают исходя из того, что уровни, соответствующие инертным газам, заняты*.
У гелия заняты уровни с главным квантовым числом п = 1; для лития поэтому п должно равняться 2. Побочное квантовое число находят для лития на основании теории Бора — Зоммерфельда из следующих соображений, Постоянный терм серии задается энергией основной орбиты. Если ом электрон в нормальном состоянии атома находился бы на 22-орбите, т. е. на круговой орбите, охватывающей па довольно значительном расстоянии 1 к а антовые орбиты (ср. с рис. 27, стр. 141, в котором, как можно представить, заряд ядра 2 заменен зарядом 3 и вместо одного электрона На 1 [-орбитах вращаются два электрона), то следовало бы ожидать, что для постоянного терма «эффективный заряд ядра» мог бы обозначаться числом 1, так как из трех зарядов, которыми обладает ядро лития, два были бы постоянно экранированы. Такой терм можно коротко назвать «водородоподобны». Однако оказывается, что для постоянного терма главной серии определяющий эффективный заряд ядра существенно больше 1. Таким образом, на основании сказанного при объяснении рис. 27 можно заключить, что речь идет об орбите, которая временами приближает электрон к ядру,— следовательно, о сильно эксцентрической орбите. В качество такой орбиты у лития можно рассматривать только одну 21-орбиту как основную орбиту.
Аналогичным образом можно показать, что у остальных щелочных металлов их основные орбиты должны обладать побочпьгм квантовым числом А == 1 (.г-орбиты). С позиций волновой мехапйки (в соответствии с которой представления об «орбитах» в смысле теории Бора — Зоммерфельда, конечно, не могут оставаться в силе) приходят к тому же результату, если принимают во внимание поляризующее действие электрона внешней оболочки на остов атома. Это влияние поляризации на энергию связи электрона можно, между прочим, использовать для расчета из спектроскопических данных поляризуемости щелочных ионон.
Основные термы так называемых побочных серий являются белее «подрродонсдоб-ными», чем основные термы главных серий. Еще более это относится к сериям Бергмана. Вследствие особо ярко выраженного «водородного характера» постоянных термов эти серии, как было указано ранее, также обозначаются как «основные серии». Чем более «водородоподобным» является основной терм серии, тем менее эксцентричной в смысле атомной модели Бора — Зоммерфельда является основная орбита, соответствующая серии, а
Известная желтая линия натрия, так называемая D-линия, является первой линией главной сурии натрия. Обратная величина ее длины волны получается в виде разности основного терма главной серии и основного терма Первой побочной серии. То есть линия проявляется л эмиссионном спектре благодаря тому, Что находящийся на За-уровне в основном состоянии атома электрон, после того как он был поднят с этой орбиты (например, благодаря сильному соударению атомов при температуре пламени) па соседний Зр-уролень, снова возвращается па первоначальный 3 s-уровень. Очевидно, это происходит особегвдо часто, чем объясняется, что Д-липия является
* Главное квантовое число удается также непосредственно определить из спектроскопических данных иа основании рассчитанного из них экранирования заряда ядра.
Первая группа периодический системы
197
самой яркой линией натриевого спектра. Вся последовательность линий главной серии определяется следующими переходами:
Зр —3s, 4р —> 3s, 5р 3s, 6р —> Зя, 7р —> 3s и т. Д.
Обратным переходам соответствуют абсорбционные линии. Совершенно аналогичным образом образуются спектры остальных щелочных металлов,
При более сильном спектральном разложении D-линия оказывается двойной. Она образует так называемый дублет. Объяснение этому дали в 1925 г. Уленбек (Uhlenbeck) и Гоудсмит (Goudsmit) на основании предположения, что электрон ведет себя так, как будто он при своем движении вокруг атомного ядра одновременно вращается вокруг собственной оси. Это вращение может происходить в сторону орбитального движения или в обратную сторону.
Вращение электрона вокруг собственной оса в отличие от вращения вокруг атомного ядра обозначают как dr all (нем.) или spin (англ.). Оно определяется квантовым числом, уже упомянутым на стр. 145 и называемым спиновым квантовым числом в. Вращеппе электрона вокруг собственной оси вносят свою долю в магнитный момент атома, так как вращение электрически заряженного шарика вокруг собственной оси оказывает такое же действие, как электрический круговой ток*. Правда, влияние спинового квантового числа s па магпитньш момент атома, так же как влияние магнитного квантового числа т, обусловленного орбитальным моментом, проявляется только тогда, когда на атом действует внешнее магнмтпов поле. Однако, с другой стороны, вращение электрона вокруг собственной оси оказывает также влияние на вращательный импульс атома. Вследствие этого общий вращательный импульс атома и таким образом его ппергсигтескоо состояние зависит нс только от орбитального квантового числа /, но также и от спинового квантового числа я. Из обоих чисел образуется так называемое внутреннее квантовое число j. Последнее всегда имеет положительное значение, а именно для I — 0 опо имеет только одно значение (j == 1/э), а для каждого I > 0 два значения, например j = 3/з и j = 1/2 для 1 = 1. С позиций волновой механики также можно обосновать спиновое квантовое число s и его комбинацию с I, дающую квантовое число ;, хотя объяснение спинового квантового числа s здесь несколько иное. Так как у щелочных металлов все Z-уровпи, за исключением тех, для которых I = 0, делятся на два энергетических уровня, все линии в спектрах щелочных металлов, которые образуются за счет перехода на основной уровень / — 0, должны давать дублеггил. Это и наблюдается в действительности. Расстояние между линиями дублета сильно возрастает с увеличением атомного веса, У желтой натриевой линии оно так мало (разница в длине волн 5,97 А.), что для разделения этих составляющих требуется хороший спектроскоп. У цезия расстояние, однако, так велико, что обе синие липин цезия различаются даже при довольно слабой дисперсии (разница в длине волн составляет здесь 37,94 А; для лежащего в инфракрасной области дублета первого члена главной cepiin цезия опа составляет даже 422,4А). При переходах иа более высокие уровни, чем основной, в эмиссионном спектре могут появиться более чем две липин, так как в этом случае не только исходный, но и конечный уровень разделяется па два уровня. В таких случаях говорят о «сложных дублетах».
Нужно еще указать, что щелочным металлам, кроме описанных дуговых спектров, которые следует приписать нейтральным атомам (спектры, получаемые в пламени, появляются также л в электрической дуге), свойственны еще также искровые спектры. Последние крайне трудно получить, они очень богаты" линиями и имеЮт такой же характер, как спектры инертных газов. Их приписывают поэтому однозарядно ионизированным атомам М+. Их образование соответствует приведенному на стр. 135 и сл. закону «спектроскопического смещения*. Это является дальнейшим подтверждением представления, что строение атомного остова каждого щелочного металла, образующегося при отщеплении одного электрона, полностью соответствует строению предшествующего инертного газа.
Применение щелочных металлов и их соединений. Металлический натрий, с тех пор кап его изготовление стало дешевим, находит широкое техническое применение. Его используют в качестве исходного продукта
* Пе только электрон обладает связанным с его спипом магнитным моментом; последний был экспериментально установлен также для ряда атомных ядер. Э(о привело к тому, что им также придают спиц (спин ядра). У ортоводорода, например, ядер-ные спины и, таким образом, магнитные моменты обоих находящихся в одной моле-куло протонов параллельны, у параводорода, напротив, антппараллельпы.
198
Глава 6
при производстве перекиси, натрия (моющее средство), а также амида натрия и натрийцианамида. Его используют также в больших количествах в органических синтезах (например, в красильном производстве). В осветительной технике его применяют в натриевых газоразрядных лампах. В лабораториях натрий используют в качестве восстановителя. Для этого обычно вместо чистого металла употребляют мягко действующую амальгаму. Металлический калий также иногда употребляют в лаборатории. Кроме того, калий и прежде всего цезий применяют в фотоэлементах. Помимо этого, рубидий и цезий в свободном состоянии мало применимы. Металлический литий, напротив, приобрел большое техническое значение. Его используют во все возрастающих количествах в сплавах, так как небольшие добавки этого металла существенно улучшают свойства многих сплавов. Преимущественно литий (наряду с натрием и кальцием) применяют для свинцово-подшипниковых сплавов (см. стр. 588) и при производстве склерона (см. стр. 386). Кроме того, он служит в качестве раскисляющего средства для меди и при рафинировании серу содержащего никеля.
Из соединений щелочных металлов в больших количествах используется перекись натрия при производстве моющих средств. В лаборатории она служит как энергичный окислитель как в водном растворе, так и в расплаве. Гидроокиси натрия и калия чрезвычайно часто применяют в лаборатории и в технике в качестве сильных оснований.
Окись и гидроокись лития вследствие их небольшой гигроскопичности используют для приготовления фотографических проявителей в виде порошков.
Исключительно широкое применение находят соли щелочных металлов, особенно натрия и калия.
Если требуется ввести в реакцию какой-либо кислотный остаток, то его применяют большей частью в виде соответствующей соли щелочного металла (натрия или калия). Это предпочтение щелочных солей для реакций объясняется отчасти их легкой растворимостью и плавкостью, а также и тем, что вследствие легкой растворимости большинства щелочных солей они не вызывают нежелательных побочных реакций. Дли технических целей, однако, иногда предпочитают другие соединения, поскольку они оказываются дешевле. Подробнее относительно применения различных щелочных солей будет сказано при рассмотрении соответствующих соединений. Только относительно солей лития здесь следует ещо отметить, что некоторые нз них, например карбонат, салицилат, а также цитрат, применяют как лечебное средство от подагры. Употребляемые для борьбы с этой болезнью минеральные воды в качестве действующей составной части содержат соли лития.
СОЕДИНЕНИЯ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Соединения щелочных металлов соответствуют общему типу MX, где М — означает щелочной металл, а X — одновалентный атом или радикал или же эквивалентное количество многовалентного атома. В приведенной ниже таблице дан обзор важнейших соединений щелочных металлов.
В дальнейшем будет более подробно сказано о гидридах, окисях и гидроокисях, перекисях, а также о приведенных в таблице классах солей. Данные об остальных соединениях можно памти при описании тех веществ, производными которых они являются.
Первая группа периодической системы
199
Обзор важнейших типов соединений щелочных металлов
Гидриды. МН
Окиси Мг0
Перекиси М2О2 и падперекисп МО2 Гидроокиси МОН
Нитриды M3N (см. стр. (93) Карбиды, соответственно ацетилиды MjCj, (см. стр. 507 и 476) Металлические соединения см. табл. 91 (стр. 620); табл.
92 (с.тр. 621); табл. 93 (сгр. 622) к табл. 98 (стр,
Соли, например МС! хлориды
MNO3 нитраты
М2СО3 карбонаты
M2SO4 сульфаты
Двойные соли, например
Na,SО4 3К 3SО4 глазерит Труднорастворимые соли щелочных металлов, например КС1О4
и Na[Sb(OH)c]_
630).
Гидриды. Наиболее устойчивым среди гидридов щелочных металлов является гидрид лития LiH. В этом отношении он близок гидридам щелочноземельных металлов, которые значительно более устойчивы, чем гидриды щелочных металлов. Гидрид лития легко получается при пропускании водорода над слабо нагретым литием, находящимся в железной лодочке. При этом образуется белая твердая, состоящая из правильных кристаллов масса с точкой плавления 680°.
В то время как при высокой температуре гидрид лития чрезвычайно реакционноспособен (диалогично свободным щелочным металлам), при комнатной температуре он исключительно устойчив. При обычной температуре он нс реагирует с сухими газами, например с Оз, С1® и НС1. Водой, напротив, энергично разлагается: LiH-|-HOH=LiOH+I-I2. Выделение водорода вследствие термической диссоциации по уравнению LiH =Ы2-+1IS Н2 начинает становиться заметным в вакууме при 450°.
Как впервые установили Нерпст и Моерс (Nernst, Moers, 1920), расплавленный гидрид лития проводит электрический ток, разлагаясь при этом иа литий и водород. Последний выделяется на аподе и является, таким образом, в соединении электроотрицательной составной частью.
Наиболее ясным дока,ытеяъством того, что разложение, происходящее при пропускании электрического тока через вещество, является электролитическим разложением, служит закон Фарадея, в соответствии с которым равпые количества электричества при электролизе выделяют эквивалентные количества вещества. При пропускании такого количества электричества, которое осаждает 107,88 .иа серебра, должно, согласно этому, выделиться */2 миллимоля Нй(= 11,2 мл Н2). По предложению Неряста Петерс (Peters, 1923) доказал, что это соотионюаие действительно оправдывается при электролизе гидрида лития (для предотвращения конвекционных токов опыты были проведены не с расплавленным, а с твердым гидридом: лития при температурах несколько ниже точки плавления, при которых электропроводность уже значительна).
Остальные гццриды щелочных металлов, так же .как и гцдрцд лития, можно получить непосредственным соединением составных частей; в чистом состояния опи получаются, однако, труднее, чем гидрид лития. По своей природе и по поведению они соответствуют гидриду лития, хотя значительно менее устойчивы. Их температуры диссоциации по данным Ефраима (Ephraim, 1921) лежат много нище, чем температуры диссоциации гидридов щелочноземельных металлов и лития; температуры диссоциации их лежат близко одна к другой.
По-видимому, устойчивость гидридов в направлении от цезия к натрию несколько возрастает. Теплоты образования гидридов щелочных металлов приведены в табл. 27.
200
Глава б
Гидриды щелочных металлов кристаллизуются по тину каменной соли (см. стр. 238). Длины ребер а элементарного куба и рентгенометрически определенные плотности d составляют (Zintl, 1931)
для LtH NaH КН ньн Call
аА 4,085 4,880 5,700 6,037 : 6,376
^рент. 0,77 1,36 1,43 2,59 3,41
^нзмер. 0,82 1,38 1,47 2,60 3,42
В последней строке даны непосредственно измеренные различными исследователями значения d. Ионный радиус Н-, рассчитанный из приведенных выше значений, оказывается равным 1,54 А.
Косвенное определение теплот реакции. Теплоты образования таких соединений, у которых, как в случае гидридов щелочных металлов, прямо их определить невозможно, устанавливают косвенно па основании «закона постоянной суммы тепловых эффектов» Гесса и закона Лавуазье — Лапласа о «равенстве теплот ы образования и теплоты разложения соединения». Например, Сивертс (Sieverts, 1930) для реакций с водой Na ti NaH установил следующие тепловые аффекты:
[Na] H2O 4 aq - NaOH-aq 4- Ч2(На) -Ф 44,4 ккал (1)
[NaH] 4 П2О 4- aq = NaOH-aq 4- (Н2) 4- 31,6 ккал (2)
[Na] 4 i/z (Н2) = [NaH] “ 4 12,8 (3)
Уравнение (3) получается в результате вычитания уравнения (2) из уравнения (1). В соответствии с обоими приведенными выше законами уравнение (3) дает теплоту, которая освобождается, если 1 г-атом твердого Na соединяется j непосредственно с 1/2 моля Н2, давая твердый NaH.
В приведенном выше уравнении NaOH-aq означает 1 моль гидроокиси натрия, растворенный в избытке воды. Вообще в подобных уравнениях символом «aq* обозначают неопределенное количество воды — растворителя, и именно такое количество воды, что дальнейшее увеличение ее не влияет иа тепловой эффект. Теплоту растворения нельзя пе учитывать в термохимических формулах; она составляет, например, для гидроокиси натрия 10 ккал/молъ. Так как изменение агрегатных состояний, а также превращение модификаций связаны с термическим аффектом, то агрегатные состояния (и соответственно модификации) должны указываться в термохимических уравнениях, если они не понятны сами собой. Твердое состояние обозначают тогда квадратными, а газообразное — круглыми скобками, в то время как формулы веществ, участвующих в реакции в жидком или растворенном состоянии, пишут без скобок. Следует еще указать, что тепловые аффекты в термохимических уравнениях приводят обычно при постоянном давлении. Если наблюдения проводятся при постоянном объеме, то их перечисляют на постоянное давление, для чего от теплоты реакции, изморенной при постоянном объеме, вычитают количество тепла, которое соответствует работе, затраченной при расширении для преодоления давления.
Расчет тепловых эффектов из равновесий. Для расчета тепловых эффектов из измеренных равновесных значений температура — давление иногда применяют приближенную формулу Периста. Для часто встречающегося случая, когда твердое вещество образует один газообразный продукт Диссоциации, она имеет вид
Q = (1,75 log Т + С — log р) 4,571 Т, (4)
где О — число грамм-калорий (кал), которые должны быть затрачены при комнатной температуре для отрыва 1 моля газообразного продукта диссоциации; р — давление диссоциации в атмосферах (атм), измеренное прн абсолютной температуре Т; С — условная химическая константа соответствующего газа. Опа составляет, папример, для Н2 1,6; О2 2,8; N2 2,6; С12 3,1; Вг3 3,2; 12 3,9; СО 3,5; СО3 3,2; NH3 3,3; П20 3,6.
Уравнение (4) можно вывести из изохоры реакции (стр. 59) путем введения некоторых приближенных упрощений па основании теплового закона Нернста. Если тепловой эффект известен, то он также может служить для приближенного расчета кривой температура — давление; ранее ото было даже главной областью его применения*.
* Теперь для этого применяют, если возможно, приближенную формулу Улиха (L'lich, ср. стр. 170 и сл.). Опа дает значительно более точные значения; правда, для ее применения нужно, кроме теплоты реакций, знать также значения энтропии и удельные теплоемкости веществ, участвующих в реакции.
Первая, группа г периодической системы
201
Тепловой закон. Нернста, гласит, что измолепне максимальной работы, которая совершается в результате реакции, в зависимости от температуры при приближении к абсолютному нулю также приближается к нулю:
rfA
11ш-^-=0 при Т=0 (А—максимальная работа).
Для давления диссоцпацци гидрида натрия Хюттиг (Hilttig) и Вродкорб (Brodkorb) нашли, например, при 270° (7’ = 543°) 10 .и.и рт ст (= 0,013 атм). Если эти значения подставить в уравнение (4), получим Q = 20 500 кал, или па 1 г-атом натрия, т, е, на !/г моля Н2, 10,25 ккал. Это значение приблизительно совпадает с значением, найденным Сивертсом (Sieverts, см, выше).
Значение приближенной формулы Нернста для расчета тепловых аффектов заключается в том, что для се применения нужпо знать только одну пару значений температура — давление. Если известно две или несколько точек (но слишком отдаленных одна от Другой), то получают более точные значения, используя уравнение изохоры, реакции в интегральной форме. [Уравнение (6), гл. 2]. Подробное объяснение можпо найти в учебниках физической химии.
Окиси. Нормальные окиси МаО щелочных металлов имеют лишь второстепенное значение. Соединение лития здесь также оказывается наиболее прочным. Это единственная окись, которая образуется в качестве основного продукта при сгорании металла па воздухе. Остальные щелочные металлы при достаточном доступе воздуха сгорают с образованием перекисей и надперекисей.
Полученный при горении окисел Li2O также содержит примесь перекиси Li2O2. В чистом виде окись лития получают нагреванием гидроокиси лития, нитрата или карбоната в токе водорода до 800°. Окись лития также слабо восстанавливается углем или окисью углерода, как и водородом. Li2O образует пористую белую массу с удельным весом 2,02. С водой она соединяется лишь медленно, образуя гидроокись, в то время как остальные окиси щелочных металлов реагируют с водой энергично, а иногда даже крайпе бурно (разогревание).
Окись натрия Na2O содержится обычно в виде примеси в перекиси, образующейся при горении натрия. В чистом состоянии она получается, если перекись или гидроокись натрия обрабатывать при нагревании металлическим патрием:
Na2O3 A 2Na = 2Na2O, (5)
NaOII ф Na - Na2O А г/,Н2. (6)
NaaO образуется также при горении натрия в недостаточном количестве кислорода; очевидно, при этом избыточный натрий реагирует с первоначально образующейся перекисью по уравнению (о). Для получения окиси удобна реакция натрия [или по ЦинтЛю (Zintl, 1931) азида натрия] с нитритом натрия
NaN02 + 3^а = 2Na2O A V2N2 [соответственно 3NaN3 A NaNO3 = 2Na2O A 5N2].
Подобно окиси натрия можно получить окиси остальных щелочных металлов. Они имеют желтую окраску, усиливающуюся с возрастанием атомного веса. Окись натрия, так же как окись лития, имеет чисто белый цвет, однако окись калия уже желтобелая, окись рубидия светло-желтая и окись цезия оранжевая. В той же последовательности увеличивается летучесть окисей.
Окиси рубидия и цезия реагируют с водородом при слабом нагревании с образованием гидроокиси и гидрида
ВЬ2О А Н2 = НЬОН А ньн.
Окиси щелочных металлов образуют кристаллическую решетку типа плавикового шпата (см. стр. 298 и сл.), за исключением Сз2О, которая кристаллизуется по типу хлористого кадмия (см. т, II). Большое число сульфидов, селенидов и теллуридов щелочных металлов также кристаллизуется по типу плавикового шпата; только Cs2S, как и Bb2Sc, Cs2Se, ВЬ2Те и Cs2Te, имеют более сложное Строение решетки. В табл. 34 сопоставлены постоянные решеток халькогенидов щелочных металлов, кристаллизующихся по типу плавикового шпата (а — длина ребра элементарного куба в ангстремах). Кроме того, там приведены плотности всех халькогенидов щелочных металлов.
202
Глава 6
Таблица 34
Постоянные решеток и плотности халькогенидов щелочных металлов
<1 Плотность J a Плотность
рейтreпомет риче-ClUJH пикнометриче- ская 1 рентгенометрическая пикнометрическая
ЬЦО 4,62 2,01 2,01 LigSe 6,00 2,8.3 2,91
Na2O 6,55 2,39 2,27 Na2Se 6,81 2,61 2,58
К2О 6,44 2,33 , 1 K2Se 7,68 2,29 —
Rb2O 6,74 4,05 Rb2Se — 3,57
Cs»O — — 4,69 Cs2Se — — 4,39
Li3S 5,71 1,63 1,63 Li2Te 6,50 3,39 3,24
Na2S 6,53 1,85 1,86 Aa2Te 7,31 2,93 2,90
K2s 7,39 1,80 1,81 K2Te 8,15 2,51 2,52
Rb2S 7,65 2,99 Rb2Te — — 3,62
Cs2S — — 4,15 Cs2Te .— — 4,30
Для цезия известен также ряд низших окисло в (недокись) Cs7O, Cs4O (?), Cs7O2t CfjgO и Cs2O. Существование этих своеобразных соединений было установлено уже в 1909 г. Репгаде (Rengade) и позднее подтверждено Брауэром (Вганег, 1947).
Перекиси и па дне рек йен. При горении щелочных металлов в токе кислорода образуются следующие соединения:
Ка2О2 КО2 RbO2 CsO2
Светло-желтая Оранжевая Темно-коричневая Желтая
Соединения типа М^Ог называют перекисями, типа МтОз — надпере-кисями (пшеро концами). Надпере к иен щелочных металлов достаточно легкоплавки. При сильном нагревании надиерекись цезия СзОа отщепляет кислород, переходя в черную Cs2Q3. Нов общем перекиси щелочных металлов, а также надперекисн достаточно устойчивы при нагревании, если только они пе содержат способные к окислению вещества. Однако последним опи легко отдают свой кислород при нагревании, притом даже таким веществам, которые в других условиях окисляются лишь с трудом. Поэтому перекиси щелочных металлов, особенно перекись натрия, применяют в лаборатории для перевода в растворимое состояние способных к окислению веществ, нерастворимых в кислотах.
Перекись литая I,i2O2 образуется в небольшом количестве при горении металла bJtoko кислорода. Ее приготовляют добавлением перокиси водорода к раствору гидроокиси лития; затем при добавлении спирта выделяется кристаллический осадок состава Li2O2' U2O2‘3[I2O, который обезвоживают в течение недели над пятиокисью фосфора.
По существу те же соединения получают действием Кислорода па растворы щелочных металлов в жидком аммиаке. Однако при атом возникают еще и другие более высокие окисли, например Na2O3, К2О2, К-2О3. Цезий образует в этих условиях ряд: Cs2O2, Cs2Og и, наконец, CsO2; последнее соединение подобно тому, которое образуется при более высокой температуре*.
* При действии озона на твердые гидроокиси щелочных металлов образуются озониды, растворимые в жидком аммиаке. (II. А. Казарновский 1948 и ел.). Озониды представляют собой соли. Например, озонид калия КД содержит ионы К.4 и О3,— Прим. ред.
Первая группа периодической системы
203
Перекиси, па пр им ер перекись натрия Na «О а, можно считать непосредственными производными перекиси водорода (НгОа), ибо они количественно взаимодействуют с кислотами или даже с водой с образованием перекиси водорода:
M2O24-H2SO4=M2SO44-II2O3;
М2О2 -ф2Н2О = 2МОИ+Н2О3.
Взаимодействие перекисей с водой совершенно аналогично гидролитическому разложению солей. Поэтому соединения этого типа можно назвать солями перекиси водорода. Из подобия перекисей щелочных металлов с перекисью водорода следует, что палочные металлы в перекисях также одновалентны.
Надперекиси, т. е. соединения типа МОа, наоборот, при разложении наряду с одной молекулой ШОа образуют одну молекулу Оа
2МО3+ H2SO4= М28О4Ц-ПйО2-}- Оа;
2МО,-|-2И2О = 2МОНЦ- Н2О2+О2.
I
Ранее надперекиси щелочных металлов М*О2 рассматривали как тетраокиси MjO4. Однако Касаточкип (1936) и Клемм (Klemm, 1939) установили рентгенометрически, что они имеют такую же структуру, как и перекиси щелочноземельных металлов типа ВаО2 (см. стр. 297) и ацетилиды типа ВаС2 (см. стр. 507). Решетку КО2 можно получить из решетки КС1 (см. стр. 238), если в последней па место каждого атома хлора поместить О2-группу (ее центр тяжести) с расстоянием между центрами атомов 2,08 А. Все липин, соединяющие каждую вторую такую группу, образованную атомами кислорода, проходят параллельно оси с. Последняя растянута ио сравнению с обеими другими осями (с/а 1,178), так что вместо кубической возникает тетрагональная решетка (см. рис. 88, стр. 507). О J‘-группа парамагнитна, так как она имеет одип псспаренпый электрон. Полинг предположил, что в пой имеется одна простая связь, которая совмещена с одном трехэлектронпон связью [| О—О |р“.
Перекись натрия. NaaOa технически получают сжиганием металлического пйтрия в алюминиевом сосуде. Она представляет собой бледно-желтый порошок, который плавится почти без разложения и самопроизвольно не взрывается (теплота образования 124,04 ккал/моль NaaOa. Roth 1947—1948). Напротив, с веществами, способными окисляться, например с хлопком, опилками, соломой, углем, а также с порошкообразным алюминием, она реагирует настолько энергично, что при соприкосновении с ними может произойти сильный взрыв. Если незначительное количество перекиси натрия облить эфиром, уксусной кислотой, нитробензолом или содержащим воду глицерином, то произойдет крайне энергичная вспышка. С серой перепись натрия также реагирует с образованием пламени. Слабее реагирует она с окисью углерода, образуя при этом карбонат натрия (7). С двуокисью углерода образуется карбонат с выделением кислорода (8):
Na2O2+СО = Na2CO3. (7)
№202+С02=№ыСОа-р/202. 1(8)
Последнюю реакцию используют в дыхательных аппаратах, применяемых пожарными и водолазами, а также для обновления воздуха в закрытых помещениях, например на подводных лодках. Для этой цели в продаже имеются препараты под названием оксон, содержащие перекись натрия.
С водой перекись натрия также реагирует весьма энергично с значительным выделением тепла. Последнее основано на образовании гидрата NaaOa-8HaO, который легко можно выделить из водного раствора в виде хорошо плавящихся кристаллических табличек или листочков. Известны
204
Глава 6
также моно- и дигидрат. В разбавленном растворе перекись натрия разлагается с образованием едкого натра и перекиси водорода по уравнению Na2O24-2H2O—2Na‘ -|-2ОН' 4-И2Ог.
Если растворением безводной перекиси натрия требутся приготовить раствор смеси едкого патра и перекиси водорода, то его следует энергично охлаждать, так как иначе пОд влиянием теплоты растворения происходит энергичное выделение кислорода.
Перекись натрия находит все более широкое применение для приготовления отбеливающих ванн для щелка, шерсти, полушелка, соломы, перьев, волос, щетины, губок, древесины, рога, костей и слоновой кости.
Для приготовления отбеливающей ванны перекись натрия растворяют в холодной воде, которую предварительно смешивают с таким количеством серной кислоты, которое необходимо, чтобы нейтрализовать образующийся едкий натр. Остающийся после реакции небольшой избыток серной кислоты нейтрализуют аммиаком. Или же и воде добавляют предварительно сульфат магния, так что освобождающиеся при растворении перекиси натрия ноны гидроксила связываются в виде гидроокиси магния. В продажу поступает стиральный порошок, содержащий, кроме мыльного порошка, перекись натрия.
Гидроокиси. Гидроокиси щелочных металлов, называемые также едкими щелочами, представляют собой бесцветные, очень едкие, довольно легкоплавкие массы. Следует отметить, что едкие щелочи в расплавленном состоянии сильно действуют не только на стеклянную и фарфоровую посуду, но при доступе воздуха также и па платину. Поэтому плавление едких щелочей нельзя проводить в платиновых тиглях. Для этого в лаборатории применяют чаще серебряные тигли; в технике используются железные сосуды.
Гидроокиси щелочных металлов очень хорошо растворимы в воде, причем растворение сопровождается значительным выделением тепла. Так, теплота растворения для NaOH составляет примерно 10 ккал!.\юлъ. Гидроокиси щелочных металлов в водных растворах обычной концентрации почти полностью распадаются на ионы по уравнению МОН =М’-|-О1Г, хотя коэффициенты электропроводности /ц = р/р0 для мало разбавленных растворов вследствие действия межионных сил значительно пиже единицы.
Пример. Для гидроокиси калия в 0,01 0,1 1 М растворе
/и(при 18°) =0,96 0,89 0,77
Аналогичные величины получаются и для остальных гидроокисей щелочных металлов (ср. также стр. 182).
Концентрации (активных) гидроксильных ионов в растворах гидроокисей щелочных металлов больше, чем в растворах всех других гидроокисей металлов при том же эквивалентном содержании; гидроокиси щелочных металлов являются поэтому наиболее сильными основаниями ио сравнению с другими гидроокисями металлов.
Увеличение отавного характера гидроокисей щелочных металлов от гидроокиси лития к гидроокиси цезия йаходится в связи с возрастающим ослаблением связи гидро-ксил-иопов в этом ряду*.
* В соответствии с теорией сильных электролитов в умеренно разбавленных растворах, даже если допустить наличие полной диссоциации, величина ионных радиусов оказывает все же заметное влияние на межионные Силы, так как, чем концентрированнее растворы, тем чаще ионы ударяются один о другой вследствие теплового движения. Работа, которую нужно затратить, чтобы снова разделить ионы, по закону Кулона зависит от величины ионных радиусов.
Первая еруппа периодической системы
205
В качестве меры прочности этой связи'можно рассматривать работу, которую требуется затратить, чтобы оторвать в молекуле гидроокиси щелочного металла ион тидроксила от нона металла. Если рассматривать ионы как жесткие шары а пренебречь влиянием поляризации, то по закону Купона эта работа оказывается равной
D V + Oj г -|-2г0У
где D — диэлектрическая постоянная, г — радиус иона щелочного металла и г0 — радиус иона кислорода. Если в качестве радиусов взять значения Гольдшмидта, то для работы диссоциации* получим величины, указанные" на нижней кривой рис. 34, Гидроокиси щелочных металлов расположены через равные интервалы па оси абсцисс рис. 34, а относящиеся к ним величины А, которые получены подстановкой значений Гольдшмидта вместо радиусов ионов, после деления на (е2/В) • 108 использованы как ординаты. Полученные таким образом точки соединены жирной кривой, которая свидетельствует о том, что рассчитанная работа диссоциации уменьшается от гидроокиси лития к гидроокиси цезия. На рисунке в том же масштабе нанесены также значения работы А', которая затрачивается при отрыве одного иопаН",т. е. при диссоциации по уравнению МОН = [МО]' + Н‘.
Ее рассчитывают по формуле
1 Л
А'
с0
Рис, 34. Работа диссоциации гидроокисей щелочных металлов.
щелочшдх металлов будут давать
Значения этих работ, соединенные светлой кривой на рис, 34, существенно больше, чем величины работ, которые требуются для отрыва ионов ОН'**. Поэтому практически гидроокиси исключительно ионы ОН'. Для сравнения па рис. 34 указана также работа, требуемая для диссоциации молекулы воды. Ее значение представлено ординатой пунктирной линия.
Точки плавления гидроокисей щелочных металлов
LiOH NaOH КОН НЬОН CsOH
Т. пл., ° С = 445 318,5 360,5 301 272,3
Такое же измепепие точек плавления, как у гидроокисей щелочных металлов, наблюдается у л ладное щелочных металлов (Juz а, 1937). Теплоты образования гидро, окисей щелочных металлов из элементов приведены в табл. 27, стр. 179,
Помимо воды, гидроокиси щелочных металлов очень легко растворяются в спирте. Только гидроокись лития умеренно растворима как в воде, так и в спирте и образует, таким образом, переход к гидроокисям щелочноземельных металлов. Опа также менее гигроскопична, чем гидроокиси других щелочных металлов, но кристаллизуется из водного раствора в виде гидрата ГлОН‘Н2О.
При прокаливании гидроокиси щелочных металлов проявляют заметную летучесть, которая возрастает от гидроокиси лития к гидроокиси цезия.
* В справочной литературе по физико-химическим константам веществ обычпо применяют термин «энергия диссоциации» вместо термина «работа диссоциации».— Прим., ред.
** Если учесть влияние поляризации, то для отрыва ядра Н+ получаются существенно более высокие значения работы. Однако значительное количество энергии освобождается при образовании [Н3О]+ из Н+ и Н2О; эта энергия также пе принимается во внимание. Ошибки, допущенные пренебрежением этими двумя факторами, приблизительно компенсируются. ’
2()6
Глава 6
Для получения гидроокисей щелочных металлов можно применять в принципе все методы получения оснований, указанные па стр. 83, Но практически для гидроокисей щелочных металлов имеют значение только четвертый и пятый методы; эле к угр о л из хлоридов и двойной обмен между солью щелочного металла и гидроокисью щелочноземельного металла. Последний метод — каустификация соды едкой известью [Са(ОН)а] — наряду с электролизом служит для технического получения гидроокиси натрия. Указанный метод пригоден также для получения гидроокисей рубидия и цезия. В этом случае исходят из сульфатов, которые взаимодействуют с гидроокисью бария
Rb2SO4+Ba(OH)2=2RbOH-|-BaSO4.
Иногда для получения очень чистых гидроокисей используют взаимодействие щелочного металла с водяным паром (третий метод).
Структура гидроокисей щелочных металлов. Гидроокиси щелочпых металлов (кроме LiOTI, структура решетки которой дана на стр. 245) при повышенной температуре испытывают полиморфпое мревращеппе. Точка превращения для NaOH лежит при 299,6°, для КОН — при 248, дли НЬОН — прп 245 п для CsOH — при 223°. Высокотемпературные модификации Г)аОН и КОН кристаллизуются по типу камоппой соли (а = 5,00 соответственно 5,78 А яри 300°); те же превращения возможны для RbOH (W est С. D,, J, physic. Cliern., 39,, 493, 1935; К.1 о in и» W,, Z. an.org. Chem., 243, 138, 1939—1940].
Структура низкотемпературных модификаций этих гидроокисей еще пе известна. Радиус иона гидроксила уменьшается в направлении КОН, NaOH, LiOH (1,53; 1,50; 1,45 А) в соответствии с ростом поляризации вследствие уменьшения радиуса катиона.
Таблица 35
Структуры решеток гпдро сульфидов п гпдроселепидов щелочных металлов
JJ Na К Rb Cs
М8П Нс несло-довап а-Формл: ромбоэдрическая а=-3,99 А а=96’2Г Р-Ф о р м а: тип NaCl а = 6,07 А. (при 200°) а-Фпрма'. ромбоэдрическая я.=6,61 А а=А)7°,<.ИГ fi-Форма: тин NaCl а = 6,67 А (при 200°) а-Фо р.иа; ромбоэдрическая а = 6,89 А а = 97°,13' Р-Форма тип NaCl л = 6,97 А (при 200°) Tim Csl а = 4,30 А (прп 20’)
MSeH Не исследован । а-Форма ромбоэдрическая 0 = 6,24 А а = 96°27' Р-Фор.и.а;' тип NaCl а — 6,30 А (при 150°) а-форма: ромбоэдриче- ская а=6,83 А а=97'21' Р-Форля: тип NaCl а = 6,92 А (при 180°) а-Форма: ромбоэдриче- ская а —1,12 А п=98°07' Р-Фсрлм: тип NaCl а = 7,21 А (прп 180°) Тип Csl а = 4,43 А (при 20°)
В табл. За указаны структуры гидуосулыридов и гидроселенидов щелочных металлов (a-форма соответствует модификации, устойчивой мри обычной температуре, Р-форма — высокотемпературной модификации).
Первая группа периодической системы 207
Гидроокись иатрия. Едкий натр —"NaOH представляет собой белую, непрозрачную, игол ь та го-к ристал л плоскую, хрупкую, сильно глгроско-пичную массу с удельным весом 2,13. В продажу, как правило, выпускают в виде палочек или пластинок, При температуре красного каления гидроокись натрия плавится; при более высоких температурах начинает испаряться. Из сильно щелочного (ср. стр. 177) водного раствора, называемого натровым щелоком, соединение кристаллизуется с водой. Были обнаружены различные гидраты, в которых содержится от одной до семи молекул воды. При плавлении гидраты обезвоживаются. Растворение гидроокиси натрия в воде сопровождается значительным выделением тепла, которое обусловлено сильной гидратацией попов натрия (ср. табл, 17, стр. 100).
Растворимость гидроокиси натрия равна:
а* 20“ tP0‘
42 109 342 г МаОН /100 г Н2О
В метиловом и этиловом спиртах гидроокись натрия также заметно растворима (спиртовый натровый щелотО.Отпоеителъно плотности и вязкости натрового щелока при температурах до 70° см. Krings W., Z. anorg, Chem,, 255, 294, 1948. Очень чистая гидроокись натрия плавится при 328е, Одпако практически вследствие примесей воды и карбоната точка плавления NaOH лежит большей частью примерно па 10° ниже. При температуре красного калепия гидроокись натрия начинает заметно испаряться.
Техническое получение гидроокиси натрия осуществляют либо электролизом, описанным для гидроокиси калия, либо взаимодействием раствора соды с гашеной известью'.
Na2CO34- Са(ОН)2=2iVaOH4- СаОО3.
Будучи лишь слабощелочной, сода благодаря зтой обработке становится едкой, каустической (от [хагптьхбе — жгучий, едкий). Поэтому полученную таким путем гидроокись натрия называют также каустической содой.
Уже Плипий указывал, что в Египте соду делали более активной, обрабатывая ее жженой известью. Арабы и западные алхимики свободно приготовляли «каустическое кали» при помощи извести.
Едкий натр находит широкое техническое применение прежде всего в мыловаренной промышленности и при производстве красок, где он требуется для окислительного плавления, например при получении ализарина из аптрахинонсульфокислоты. Сплавление с едким натром является также замечательным средством для вскрытия образцов в анализе (В г ц л с k, Н о 1 t j е, Angow. Chem., 45, 331, 1932). При кипячении соломы или древесины с натровой щелочью получают целлюлозу. В результате обработки хлопчатобумажной ткани сильным натровым щелоком (мерсеризация) повышается ее способность к окрашиванию. Если растянутые хлопчатобумажные ткани подвергнуть такой обработке, то они приобретают шелковистый блеск и одновременно повышается их прочность. Натровый щелок применяют также для очистки от жиров, масел и керосина. МаОН часто используемый реактив в аналитической химии.
Гидроокись калня. Едкое кали — КОН представляет собой твердую, белую, хрупкую, плавящуюся при температуре красного каления массу с лучистым строением, вполне подобную гидроокиси натрия, с удельным весом 2,04. Ее теплота растворения равна 12,5 ккал/моль. Как и гидроокись натрия, гидроокись калия сильно гигроскопична и с водой образует гидраты, среди которых известны в твердом состоянии моно-, ди- и тетрагидраты. Дигидрат устойчив при обычной температуре. Он растворяется
208
Глава 6
в воде с отрицательным тепловым эффектом ( — 0,03 ккал! моль). В соответствии с этим растворимость его с повышением температуры повышается, она равна при
0° 20= 100°
97 112 178 г КОН в 100 г НзО.
Водный раствор гидроокиси калия называют калиевым щелоком. Гидроокись калия легко растворяется также в метиловом и этиловом спиртах. Спиртовые растворы КОН в отличие от обычного (водного) калиевого щелока называют (метиловый, этиловый) спиртовый калиевый щелок.
Получение гидроокиси калия можно осуществить взаимодействием карбоната калия с едкой известью (как и в случае гидроокиси натрия). Одиако в технике ее получают почти исключительно электролизом растворов хлористого калил. Большей частью гидроокись калия транспортируется в виде концентрированных растворов (примерно 50%-ных). Выпариванием растворов можно получить твердый едкий кали. Но едкий кали гораздо труднее отдает последние остатки воды, чем едкий натр. Гидроокись калия — калиевый щелок, применяют главным образом в производстве мыла. Вследствие своей гигроскопичности гидроокись калия служит в качестве осушающего средства, далее, как средство, поглощающее двуокись углерода, а также для щелочного плавления. В хирургии едкое кали применяют для прижиганий.
Электролиз хлоридов щелочных металлов. Если через водный раствор хлорида щелочного металла пропустить электрический ток, то на аноде выделяется хлор
Cl'-e=i/scl2. (9)
На катоде выделяется водород, так как надряжение выделения водородных иопов, несмотря на их крайне низкую концентрацию, все же значительно меньше, чем напряжение выделения ионов щелочного металла. Таким образом, разряжаются ионы водорода. Благодаря этому освобождается соответствующее число гидроксильных ионов
Н'-ье = 1/£н2 (10)
Н2О=Н' + ОН' (11)
Н^О+е=1/2нТдЬн', (12)
Общий катодный процесс представлен уравнением (12). Кроме
выделения водорода, па катоде происходит образование щелочи, так как для определенного количества ионов ОН' в растворе имеется эквивалентное количество ионов щелочного металла, освободившихся в результате исчезновения из раствора эквивалентного количества ионов С1' (вследствие разрядки на аноде). Теперь нужно предотвратить взаимодействие накапливающейся около катода щелочи с выделившимся на аноде хлором, иначе может произойти реакция с хлором по уравнению
2ОН'-|-С1г=С1'-|-С1О'4-Н2О,.
т. е. вместо требуемой гидроокиси получают гипохлорит нри одновременном образовании хлорида. Для предотвращения этой реакции были разработаны различные способы: способ с диафрагмой, с колоколом и ртутный способ. Из них преимущественно используется способ с диафрагмой, причем в Европе — главным образом с применением камеры Сименса — Бил-литера.
При способе с диафрагмой анодный и катодный растворы разделены пористой перегородкой — диафрагмой. На рис. 35 схематически представлена камера с диафраг-
Первая группа периодической системы 209
sioil, Электрический ток поступает в камеру через анод А (изготовленный из ретортного угля, графита Ачесона пли плавленого магнетита), выделяет там хлор, который удаляется но трубке 1. Затем, пройдя через диафрагму, ток достигает катодного пространства, где у отрицательного электрода К (представляющего собой большей частью железную сетку) оя выделяет водород, который улетучивается через отвод 2. Одновременно образующийся калийный щелок стекает в отверстие 3, На практике большей частью работают так, что катод расположен очень близко к Диафрагме, и катодное пространство содержит очень мало раствора. Брауеру (Brauer, 1884) первому удалось изготовить подходящую диафрагму; оп применил для этого цемент, смешанный с мелкораз-дроблеяной поваренной солью, которую после затвердения извлекали из цемента растворением. Недостатком метода является большое сопротивление, которое оказывает
Рис. 35. Способ с диафрагмой.
Рис. 36. Способ с колоколом.
диафрагма прохождению тока, а также ее ограниченная прочность, Однако в современных вариантах (см, стр, 210) эти недостатки в значительной степени устранены.
В способе с 'колоколом диафрагму не применяют, Для разделения растворов используется то обстоятельство, что щелочпой катодный раствор обладает большим удельным весом, чем анодпый раствор. Колокол Л (рис, 36) состоит из продолговатой узкой керамической ваипы, представленной на рисунке в разрезе. Сквозь дно этой ванны пропущен угольный иля графитовый анод А, Жидкостью наполняют керамическую ванну 4, на дне которой собирается более тяжелый калиевый щелок, образующийся па железных катодах К. В конце концов он заполняет все катодное пространство. Его заставляют стекать сверху, в то время как анодное пространство непрерывно наполняют свежим насыщенным раствором хлористого калия. Таким образом, предотвращается сдвиг границы м.ежду щолоком и аподпым раствором хлорида к аноду вследствие движения ноиов ОН' в направленна отрицательного тока. Скорость притока раствора хлорида в анодное пространство должна быть отрегулировала так, чтобы движение попов ОН' к аноду компенсировалось притоком раствора электролита.
Ртутный способ основал на том, что потенциалы выделения щелочных металлов значительно понижаются при применении ртутных катодов вследствие большой тенденции щелочпых металлов сплавляться со ртутью; в то же время напряжение, требуемое для выделения водорода, вследствие заметного перенапряжения, которым обладает водород при выделении на ртути, значительно увеличивается. Таким образом, оказывается, что при электролизе концентрированного раствора хлорида щелочного металла с применением ртутного катода водород не выделяется, а напротив, разряжаются попы щелочного металла. Сплав, который щелочпой металл образует с ртутью, в особой камере разлагается водой с образованием щелочи. Разложению сильно разбавленной амальгамы препятствует перенапряжение водорода при его выделении на ртути. Поэтому во второй электролит, камере ртутная амальгама служит анодом, в качестве катода используют железо, на котором водород в щелочном растворе выделяется почти без перенапряжения. Такпм образом, получают установку, схематически представленную 14 г. Реми
210
Глава 6
на рис. 37. Banna 1 разделена в середине почти до самого дна плотной перегородкой. Направление электрического тока указано тремя короткими стрелками. Насыщенный раствор хлорида подвергается электролизу в левой половине между графитовым анодом А и находящимся па дне слоем ртути, действующим в этой части сосуда в качестве катода. Образующаяся там амальгама в результате качающегося движения ванны, вызываемого эксцентриком 2, достигает Правой половины, в которую помещен связанный с отрицательным источником тока железный электрод К. В этой части ванны ртуть,
соответственно ртутная амальгама, служит анодом: она снова отдает при разрядке содержащийся в пей металл в раствор, в то время как на катоде К выделяется эквивалентное количество водорода. Таким образом, в правой части ванны образуется щелочь, которая при этом способе получения отличается особой чистотой (свободная от хлоридов и сульфатов).
Ртутным способом получают не только очень чистые щелочи, но и значительно более концентрированные растворы, чем другими методами, (Подогреванием Пространства, где вдет разложение амальгамы, удается получить щелочные растворы с концентрацией 50—85 %.) Однако это преимущество (помимо более высоких издержек на установку, обусловленных стоимостью ртути) снижается еще и тем, что требуемое напряжение здесь более высокое, чем в современных вариантах установок с диафрагмой. Работающую с наименьшей потерей напряжения (теоретически) установку с колоколом мало применяют, так как опа надежна только при очень тщательном уходе, В настоящее время чаще всего используют способ с диафрагмой. Он был улучшен прежде всего В Я Л литером, В камере Сименса — Биа-литера диафрагма расположена горизонтально. Раствор электролита непрерывно подается через диафрагму и сетчатый
электрод, который лежит непосредственно на пей, в наиолщщпую только водородом донную часть камеры, из которой вытекает уже щелочной раствор. В камере Сименса— Песталоцца электроды (профилированные железные стержни, обернутые асбестом); также расположены горизонтально. Способы с вертикально расположенной диафрагмой усовершенствовались главным образом в США (например, камера Гиббса — Ворсе, камера Нельсона, камера Хукера). Хорошие диафрагменные камеры работают сейчас с выходом по току 95% при напряжении 4—5 е- Этому соответствует выработка 41—52% электроэнергии от теоретически необходимой для разложения*.
Современное мировое потребление электроэнергии для электролиза |хпоридов щелочных металлов оценивается в 2г/г—31млрд. квт-ч ежегодно.
СОЛИ ЩЕЛОЧНЫХ МЕТАЛЛОВ
Общая характеристика солей щелочных металлов приведена на стр, 182, В табл, 36 дан обзор растворимости важнейших солей щелочных металлов. При получении солей щелочных металлов в случае натрия и калия обычно-исходят из встречающихся в природе хлоридов**. В случае лития из природных соединений, как правило, изготовляют прежде всего карбонат, который сравнительно легко получается в чистом виде вследствие своей пло-
* Рассчитанное из значения собственного потенциала напряжение разложения равно 2,2 в (ср. стр. 51 и сл.).
** Относительно получения растворимых солей калия из калиевого полевого гапата см. Т о m u 1 а Е, 8., Апл. Асан. Sci Fenn., Serio Л, 49, № 5, 1938; 59, JV« 12, 1943.
Растворимость важнейших солей щелочных металлов (н солей аммония) в вода (г безводной соли в 100 я раствора)
Таблица 36
ойщан Литий Натрий Калий Рубидий Цезий Аммоний
формуле а °C 9 “С е °C 2 СС & °C 9 °C
Фторид i.i MF 0,26 18 4,22 18 48,0 18 Легко раствор. — Легко раствор. — Расплывается —
Хлориды MCI 44,7 20 26,4 20 25,5 20 47,66 20 65,0 20 27,1 20
Хлораты М fCWal 75,4 18 49,7 20 6,78 20 5,09 19,8 5,91 19,8 Легко раствор. —
Перхлораты М [C1OJ 37,85 25 65,6 15 1,52 15 0,76 14 1,17 14 20,0 25
Перманганаты М [MnOJ 29 16 Расплывается — 5,96 19,8 1,06 19 0,23 19 — —
Нитраты М [NO,] 41,2 20 46,8 20 24,1 20 34,8 20 18,7 20 64,6 16,9
Карбонаты М2 [СО3] 1,31 20 17,6 20 53,2 25 Легко раствор. — Легко раствор. Легко раствор. —
Сульфаты М2 [SOJ 25,7 20 16,0 20 10,07 20 32,5 20 64,1 20 43,0 20
Хроматы ма [Ci-OJ 52,6 18 44,6 21,2 38,6 20 42,4 20 Легко раствор. — Легко раствор.
Г? и хрома ты М2[Сг„0.] 56,6 30 64,3 20 11,6 20 5,0 18 Умеренно раствор. — 23,9 16
Гсксахлоро-платяпаты Ms[PtClJ Легко раствор. — Легко раствор. — 1,09 20 0,028 20 0,009 20 0,67 20
Фосфаты мэ [POJ 0,039 18 11 20 Легко раствор. — Легко раствор* 1— Легко раствор. — Легко раствор. —
Квасцы MAI [SO4]>12H2O — — 16,8 13 4,80 15 1,25 15 0,35 15 6,19 20
Бйтартраты МН [С4Н,0 J Легко раствор. — 6,34 18 0,49 20 1,17 25 8,9 25 2,14 15
212
Глава, 6
кой растворимости; разложением* кислотами из него без труда можно получить большинство других солей. При получении солей рубидия и цезия главная задача заключается в отделении их от калия, так как в этом случае используются главным образом остаточные щелока от переработки калийных солей. Щелок сначала обогащают рубидием и цезием в результате кристаллизации карналлита (ср. стр. 216), затем очистку производят большей частью через стадию выделения квасцов. Отделение рубидия от цезия можно производить, используя различную растворимость их карбонатов в спирте. '
Соли люпия занимают во многих отношениях особое положение среди других солей щелочных металлов. Кроме того что некоторые из пих трудно растворимы в воде (что было отмечено), различные литиевые соли обнаруживают сравнительно большую растворимость в неводных растворителях. В этих неводных растворителях опи в основном сильно диссоциированы на ионы. Понижение температуры замерзания и повышение температуры кипения водных растворов литиевых солей нередко превышают теоретические значения, вычисленные при предположении подпой диссоциации. Это объясняется значительной гидратацией ионов Лития, вследствие чего происходит Заметное уменьшение количества воды, являющейся растворителем.
В результате особой тенденции ионов лития к гидратации почти все соли лития кристаллизуются с водой; так, хлорид обычно содержит 1 молекулу воды, хлорат, а также нитрит 1/а НаО, перхлорат 3 Н2О; то же количество ЩО имеют нитрат, тиосульфат и двтиопат; с одной молекулой Н2О кристаллизуется бромат, сульфат, селенит, сульфит и сслепит.
Особо хорошей растворимостью Отличается хлорат лития LiCIOg, on является, пожалуй, наиболее растворимой неорганической солью. В 100 а воды при 18й растворяется 313,5 a LiCIOj. Соли Лития не образуют, как правило, смешанных кристаллов о другими солями щелочных металлов. Наоборот, они образуют смешанные л двойные соли. Примеры см. па стр. 227.
Большую аналогию с солями щелочных металлов обнаруживают соли аммонал, являющиеся производными однозарядного положительного радикала [NHil* (аммония). Они особенно близки солям калия. Поэтому в табл. 36 для сравнения приведена также растворимость некоторых солей аммония [NHdX.
Аналогия свойств иона аммония и ионов щелочных металлов обусловлена тем, что ион аммония также положительно одновалентен и обладает таким Же ионным объемом, кай и ион калия. О ионе аммония еще будет сказано в связи с аммиаком \Т13, от которого он производится присоединением протона. Наряду с большим сходством соединений аммония и щелочных металлов между ними существуют также и характерные различия, которые обусловлены близким родством радикала аммония с аммиаком. Поэтому, несмотря на аналогию между солями щелочных металлов и ам-мопия, рассмотрение последних следует связать с аммиаком. Кроме радикала аммония известны также еще другие положительные одновалентные радикалы, которые ведут себя аналогично ионам щемчпых металлов.
Соли трех щелочных металлов, находящихся в наиболее тесном родстве, калия, рубидия, цезия очень часто изоморфны. Обычно изоморфизм распространяется также па соли аммония.
ХЛОРИДЫ
Встречающиеся в природе в больших количествах хлориды натрия и калия редко бывают совсем чистыми. Хлористый калий большей частью не только механически загрязнен, но встречается главным образом в виде -двойных соединений е солями магния. Однако при растворении в во де они распадаются,так что безразлично, идет ли речь об отделении механических примесей пли о выделении из двойных солей, — путь очистки одинаков, а именно фракционированная кристаллизация.
Первая группа периодической системы
213
Получение хлоридов рубидия, цезия и лития происходит большей частью путем взаимодействия карбонатов с соляной кислотой.
Точки пллелепия и кипения хлоридов щелочных металлов, а также потенциалы разложения расплавленных солей (последние по измерениям Neumann, 1925) сопоставлены в табл. 37. Из таблицы видно, что потенциалы выделения щелочных металлов из расплавленных солей правильно возрастают от Li к Cs в соответствии с увеличением электроположительного характера металлов в этом ряду. Отсутствие порядка в ряду потенциалов выделения из водных растворов, обусловленное влиянием гидратации (ср. стр. 181 и сл.), здесь не наблюдается.
Таблица 37
Температуры плавления и кипепия хлоридов щелочных металлов; потенциалы разложения расплавленных хлоридов щелочных металлов
Соединения LiCl NaCl КС1 RDC1 CsCl
Точка плавления, °C Точка кипепия, °C 606 1350 * 801 1440 ' 768 1411 717 1385 638 1300
Потенциал разложения при 800°, в Температурный коэффициент ах1&з а Уменьшение потенциала разложен! Так как в случае хлоридОЕ сильных оснований и сильных к тральной реакцией. Однако, как щелочных металлов заметно г паром. 2,38 1,35 гя при пов> щелочь ислот, и; показал идролии 2,62 1,49 дшевив тек сых мета £ водные I Брииё{ дчески 2,77 1,51 шературы ЛЛОВ ре раствор (Впиег разлагаю 2,91 1,50 на 1°. чь идет Ы облад; , 1948), >тся пер 3,04 1,545 о солях 1ют ней-Елориды егретым
Хлорид натрия. Поваренная еолъ — NaCl встречается в природе в морской воде, содержащей в среднем около 2,7 % NaCl, и в виде каменной соли в больших залежах мощностью 1000 и более метров. Такие залёжи находятся в Северо-Германской низменности и около Велички в Галиции.
Северо-германские соляные залежи возникли при высыхании большого внутреннего 'моря, простиравшегося в первобытные времена (в конце среднего цехштейна*) от Урала в глубину теперешней Франции и включающего теперешнее Северное море, которое была тогл,а. отрезано от Атлантического океана. Оно простиралось па юг почти до теперешней долины Дуная, При очень жарком в то время климате, особенно летом, происходило быстрое испарение воды. Это приводило к выделению растворенных в морской воде солей в соответствии с их концентрациями, растворимостью и разницей температур воды летом и зимой. Прежде всего выпадал трудно растворимый в воде карбонат кальция, который лежит поэтому в виде «цех клейновского известняка» под собственно солевыми залежами. Затем выделялись другие соли, а именно лотом преимущественно гипс, ангидрит и полигалит, а зимон каменная соль («сезонные слои»). Наконец происходило осаждепие калийной соли. Высохшие участки моря вскоре покрывались массами песка, позднее частично снова затоплялись, так что в некоторых местах многие залежи находятся друг пад другом. Верхние слои, содержащие калийные соли, были позднее снова смыты. Они сохранялись только в немногих местах благодари наслоению водонепроницаемой глины и имеют теперь большое значение в качестве залежей калийных солей.
* Под цехштейиом в геологии понимают вторую часть так называемого диассовоге или пермского периода, т. е. эпоху, непосредственно следовавшую за каменноугольным периодом.
214
Глава 6
Добыча поваренной соли осуществляется главным образом тремя способами: 1) горнопромышленной разработкой каменной соли, 2) растворением каменной соли под землей и выпаривания полученного рассола, отчасти также выпариванием природных рассолов; 3) из морской воды. испарением в так называемых «соляных садках», а в условиях холодного климата — вымораживанием.
В Германии применяемую в технике каменную соль добывают главным образом путем горнопромышленной разработки, как правило, в виде побочного продукта при добывании солей калия. Самостоятельная разработка каменной соли становится выгодной только при содержании в ней 98—99% NaCi. Более загрязненную соль не добывают, а оставляют в шахте, где она служит*для заполнения выработанных проходов в пластах калийной соли.
Камепвая соль бывает загрязнена главным образом сульфатами кальция н магния. Определенная очистка ее достигается уже ручным отбором кусков ангидрита и гипса после грубого измельчения соли. Дальнейшая очистка осуществляется плавлением или обработкой боЛее чистыми рассолами.
Приготовление пищевой соли, к которой в смысле чистоты предъявляются наибольшие требования, производят главным образом выпариванием естественных или искусственно полученных (путем растворения под землей) водных солевых растворов, так называемых рассолов.
Полученная таким путем соль называется выварочной солью. Перед выпариванием, которое ведут в плоских противнях, рассолы доводят до насыщения. Ранее это производили испарением на воздухе, С этой целью рассол заставляли стекать по стене, сложенной из связок хвороста (градирня). Теперь в большинстве’случаев в рассоле растворяют до насыщения добытую каменную соль.|
Перед выпариванием рассол очищают добавлением хлорида кальция (для удаления сульфатов) и одной извести (для удаления магния). Выделяющаяся гидроокись магния, действуя как адсорбирующее вещество, увлекает одновременно^ собой и органические примеси.
Чистый хлорид натрия не гигроскопичен. Известное увлажнение поваренной соли па влажном воздухе объясняется содержанием в ней примесей. Хлорид натрия кристаллизуется в виде бесцветных правильных кубов* удельного веса 2,17. При температуре плавления (801°) он уже заметно летуч, однако в меньшей степени, чем хлорид калия. (Давление пара NaCl но данным Хориба (Horiba) при 800° равно 1 мм рт ст, в, то время как у K.G1 — мри 800° давление пара равно 4,5 и при^700° 1,5 мм рт ст). Плотность пара соответствует формуле NaCl.
В природе иногда встречается каменная соль, окрашенная в синий цвет, [Эту окраску можно также вызвать искусственно (действием паров натрия или действием катодных лучей иля лучей радия с последующим нагреванием). Поэтому считали, что сипяя окраска природной каменной соли обусловлена находящимся в коллоидной форме металлическим натрием, который является причиной и искусственного окрашивания. Однако, согласно новейшим исследованиям, природная синяя каменная соль не содержит коллоидно растворенного натрия, ибо изменение, которое вызывается в хлориде щелочного металла облучением (р-лучи, у-лучи, рентгеновские лучи), носит другой характер, чем изменение, которое может быть вызвано действием паров металлического натрия. Л именно вызванные в обоих случаях окраски, будучи идентичными в видимой части спектра, различны, одпако, в ультрафиолетовой части. Окрас-
* Иногда хлорид натрия кристаллизуется в форме правильных октаэдров, например, из концентрированной мочи.
Первая группа периодической системы 215
на, вызванная облучением (а также окраска природной синей каменвой соли), о б у слов* лена наличием в кристаллической решетке свободных электронов. Они расположены в определенных свободных (вследствие «нарушений порядка») (ср. т. II, гл. 1) местах решетки, образуемой ионами галогена (S о i t z К, Rev. mod. Physics, 18, 384, 1946).
Растворимость хлорида (см. табл. 38, стр. 216) мало изменяется с температурой. В соответствии с этим (отрицательная) теплота растворения хлорида натрия лишь незначительна (—1,2 ккал/моль), Полученные испарением растворов кристаллы при нагревании растрескиваются в связи с- тем, Что включенный маточный раствор испаряется я кристаллическая корка лопается. Растрескивание каменной соли из залежей В «лички при растворении в воде происходит по другой причине. Это обусловлено выделением заключенных в этой соли сжатых газов (согласно Тамману, главным образом и О2), которые разрывают кристаллическую корку, как только она становится при растворении достаточно тонкой.
Попаренная соль необходима живому организму, особенно при растительном характере пищи. Поэтому се добавляют в корма для скота. Ее применяют при консервировании мяса н рыбы (соление), В технике хлористый натрий является исходным сырьем для получения почти всех других соединений натрия.
Каменная соль является основным исходным материалом при производстве соляной кислоты и сульфата, получения соды, хлора и едкого натра. Кроме того, она служит для многих других промышленных и промысловых целей, например для высаливания мыла и органических красителей; для «хлорирующего обжига» в некоторых металлургических процессах; в кожевенном производстве для соления кож; для глазурования глиняных изделии, Для ускорения таяния снега и приготовления охладительных смесей и т. д.
При низких температурах хлористый натрий Кристаллизуется из водных растворов в форме гексагональных табличек состава NaCl-2 IIгО. При 4-0,15° как дигидрат, так и безводная соль устойчивы при соприкосиовепив с насыщенным раствором. Насыщенный раствор хлорида натрия кипят при 109,7° и содержит 40,4 a NaCl на 100 е воды. Растворимость NaCl в воде сильно снижается при добавлении? НС1. При 18° 1 и. раствор НС1 насыщается хлористым патрием, если Последнего содержится 20,6 г па 100 г раствора, а 3 в. раствор НС1 насыщается хлористым натрием при содержании его 10,6 г на 100 г раствора.
При взаимодействии органических соединений натрия с хлор содержащими органическими соединениями в безводном бензольном растворе получается хлорид натрия в кплмч/днаи состоянии. Он образует с бензолом желтый или желто-красный золь, довольно устойчивый в отсутствие воды. Бромид натрия, но не подид патрия, может образовать аналогичный, однако менее устойчивый органозоль.
Хлорид калия. KCI встречается в природе (в местах залегания калийных солей) в виде сильвина. Последний является наиболее ценным калиевым минералом, так как после размола его непосредственно можно использовать в качестве удобрения. Однако он часто сильно загрязнен примесями хлористого натрия. Если последний образует главнуюУсоставную часть, то продукт называют сильвинитом.
Важнейшие ааледяt калийных солей Германии находятся в Северо-Германской низменности, особеппо в. окрестностях Стассфурта. Кроме того, месторождения калийных солей имеются в Эльзасе. Эти месторождения пе являются непосредственно морскими отложениями (ср. стр. 213), а возникли они в результате вторичных процессов благодаря растворению первоначальных отложений калийных солей и их последующего выделения.
Главной составной частью месторождений калийных солей!и поэтому важнейшим исходным продуктом для получения солей’ калия является двойная соль хлорида калия и хлорида магния, карналлит KCbMgCla-6H2O.
216
Глава
Наряду с этим еще имеют значение: «твердая соль» (смесь 35—70% — NaCl, 10—48% кизерита, MgSO4-H2O, 12—23% КС! с переменным содержанием других солеи калия), затем каинит KCl-MgS0v3H,0 и уже упоминавшийся сильвинит. Эти соли называют «отбросными салями», так как раньше, чтобы достигнуть лежащего под пими слоя каменной соли, их отбрасывали и свозили в отвал. В 1865 г. Либих указал на большую ценность этих солей в качестве удобрений. В настоящее время подавляющее количество солей калия (свыше 95%) применяют для производства удобрений. Для этой цели их часто употребляют уже непосредственно после грубого размола.
Для промышленного получения хлорида калия из карналлита последний растворяют в воде. При этом он распадается на составные части КС1 и MgCl в; при упаривании этого раствора вследствие небольшой растворимости первым кристаллизуется хлорид калия.'
В технике для растворения применяют не чистую воду, а умеренно концентрированный раствор хлорида магния, так как в этом случае примеси (MgSOt и NaCl), содержащиеся в сыром карналлите, в основном остаются пер а створенными.
После кристаллизации в основном хлорида калия маточный раствор содержит значительный избыток хлорида магния, из которого на холоду снова выкристаллизовывается двойная соль КС1 -MgCls' 6Н2О (искусственный карналлит). При обработке водой из него снова можно выделить хлорид калия.
Аналогичным образом производят переработку твердой соли и сильвинита на хлорид калия. Эта переработка основана на том, что при температурах, близких к температуре кипепия воды, хлористый калий растворим лучше, чем хлорид натрия, а на холоду, наоборот, менее растворим (см. табл. 38). Поэтому хлорид калия можно выделить из смеси с хлоридом натрия, действуя па эту смесь горячей водой или, еще лучше, горячим раствором хлорида натрия (маточный раствор после кристаллизации КС1).
Таблица 38
Растворимость NaCl и КС1
Растворимость, г/100 а воды
1J 10е 20° 30° 50“ 70° эо 100°
NaCl 35,6 35,7 35,8 36,0 36,7 37,5 38,5 39,1
КС1 28,5 32,0 34,7 37,4 42,8 48,3 53,8 56,6
Чистый хлорид калия бесцветен, кристаллизуется в виде правильных кубов (часто в комбинации с октаэдрами). Он начинает заметно улетучиваться уже ниже своей точки плавления (768°). Плотность его пара при 2000° соответствует формуле КС).
Хлориды рубидия и цезия получают взаимодействием карбонатов с со ляп ой кислотой или прокаливанием хлороплатинатов. Оба хлорида кристаллизуются в виде кубов. Хлорид цезия заметпо ядовит. И од ид рубидия применяют в медицине, так как он безвреднее, чем иодид камня. Здесь следует указать, что рубидий и цезий склонна к образованию .
В аналитической и препаративной химии хлориды рубидия и цезия применяют иногда для получения чистых двойных хлоридов с хлоридами тяжелых металлов; полученные таким образом двойные соли отличаются большей частые плохой растворимостью и способностью хорошо кристаллизоваться.
Хлорид^лптия LiCl — бесцветная соль, заметно летучая уже при температуре красного каления; при белом калении в струе хлористого водорода может полностью
Первая группа периодической системы
217
испаряться. В противоположность хлоридам Других щелочных металлов расплывается', растворим в спирте, а также в смеси спирта с эфиром. Помимо этилового спирта, растворяется также в метаноле, амиловом спирте и других спиртах, в том числе и в мп о го атомных, например в глицерине, Оп растворяется ив других кислородсодержащих органических растворителях, таких, как альдегиды, ацетон, муравьиная кислота и др. В некоторых случаях он образует с этими растворителями определенные соединения, которые можпо выделить. Из водного раствора безводный хлорид лития кристаллизуется только выше 98°, при более низкой температуре он кристаллизуется с водой, причем в зависимости от температуры содержит 1, 2 или 3 молекулы Н3О,
Хлорид лития (а также и другие галогениды лития) адсорбирует в растворе и в сухом состоянии аммиак. Сухие соли в зависимости от температуры поглощают 1—4 молекулы 1W3. Получепы также многочисленные продукты присоединения органических аминов к хлориду лития. Как правило, к хлориду лития присоединяется только до-3 молекул, а иногда только 1 молекула органического амина.
Хлорид лития получают большей частью действием соляной кислоты на карбопат лития, который более доступен в чистом состоянии, чем другие соли:
Ы2СО3 -J- 2НС1 = 2LiCl + СО2 + НЙО.
Испареппе воды следует вести нагреванием в струе хлористого водорода, так как иначе произойдет гидролитическое разложение.
Бромид и иодид лития во многом аналогичны хлориду. Они также образуют в твердом состоянии гидраты с 1, 2 или 3 молекулами Н20.
Фторид лит ля. Вследствие очень низкой растворимости LiF представляет особый интерес. Он получается в виде зернистого порошка при выпаривании раствора карбоната лития в плавиковой кислоте. При перекристаллизации из расплавленного хлорида калия или бифторида калия образуются правильные октаэдры. Удельпый вес его около 2,6. Незначительная растворимость в воде (в 100 а воды при 18° растворяется 0,27 a LiF) еще более уменьшается при добавлении спирта. В плавиковом кислоте оп легче растворим вследствие образования бифторида LillF2, который мощно выделить в кристаллическом состоянии. Штокбаргер (Slockbarger, 1936) описал получение больших монокристаллов LiF (до 7 см в диаметре). Так как эти монокристаллы превосходят полевой шпат в отношении пропускания света в дальней ультрафиолетовой области, вакуумные спектрографы, предназначенные для исследований в коротковолновой области спектра (до 110 тр), стали снабжать с тех пор линзами и призмами ив фтористого лития, заменившего полевой шпат.
НИТРАТЫ
Нитрат натрия. Натриевая селитра. NaNOa — бесцветная соль, кристаллизующаяся в ромбоэдрах (ромбоэдры принимали ранее за кубы, поэтому в старой литературе натриевую селитру часто называли «кубической селитрой»). Удельный вес нитрата натрия 2,26, точка плавления 311°; при 380° начинается его разложение. Нитрат натрия обладает охлаждающим, горьковатым вкусом, он легко растворим в воде. При растворении происходит сильное понижение температуры и соответственно значительно увеличивается растворимость при повышении температуры. А именно при 0° в 100 е воды растворяется 73 г NaNOa, а при 100° — 175,6 г NaNOa. В природе нитрат натрия встречается в нескольких местах. В очень больших количествах оп находится на чилийском побережье Тихого океана (чилийская селитра). Небольшие месторождения имеются в Египте, за Каспием и в Колумбии Происхождение чилийских залежей еще не вполне объяснено. Чаще всего их объясняют разложением веществ животного или (вероятнее) растительного происхождения, особенно водорослей, под влиянием бактерий. Новейшие исследователи (Perroni, Stoklasa идр.) отдают, однако, предпочтение гипотезе вулканического происхождения чилийской селитры. При вулканических извержениях часто образуются большие количества аммиака, которые в дальнейшем могут превратиться в селитру. Сырая чилийская селитра (называемая caliche) большей частью сильно загрязнена песком и глиной, а также различными солями. Очистку производят рас
;218
Глава 6
творением в горячей воде и перекристаллизацией на холоду. При этот; методе не удаляется, однако, ядовитый для растений перхлорат калил,, КСЮ1. Если он содержится в количестве более 0,5%, то его следует удалять особым способом. Кроме того, в чилийской селитре содержится ещь иодат натрия NalOa, однако он не ядовит, а действует скорее как возбудитель роста растении.
В настоящее время в Германии нитрат натрия получают преимущественно взаимодействием соды с синтетической азотной кислотой. Лишь небольшие количества поступают в виде чилийской селитры. Перед первой мировой войной Германия ввозила ежегодно приблизительно 800 000 т чилийской селитры. Нитрат натрия служит главным образом в качестве азотистого удобрения. Однако в настоящее время он все более вытесняется другими синтетическими азотистыми удобрениями.
Нитрат калия. Селитра, калийная селитра KNOa — бесцветная, «.диморфная» соль, т. е. кристаллизующаяся в двух различных формах. Из кислых растворов она кристаллизуется, как и нитрат натрия, в форме ромбоэдров, в других случаях в форме ромбических призм. Удельный вес ромбической селитры2,105. При 128°происходит превращение в тригонально-ромбоэдрическую модификацию (метастабильную при обычных температурах). Точка плавления 339°. При нагревании выше этой температуры нитрат, выделяя кислород, переходит в нитрит. (Шееде этим методом впервые получил чистый кислород.) Вкус селитры охлаждающий, горький. Селитра легко растворима в воде. Наступающее при растворении понижение температуры еще отчетливее, чем в случае нитрата натрия, в соответствии с еще большим положительным температурным коэффициентом растворимости. Растворимость нитрата калия равна;
Ври 0“ 20° 100=
13,27 31,59 246 г KNO3 в 100 г Н2О
Образование селитры постоянно происходит при гниении животных отбросов в присутствии гидроокиси или карбоната калия. Этим путем она образуется часто в природе, столетиями ее получали так искусственно. Б настоящее время селитру получают либо взаимодействием нитрата натрия с хлоридом калия («конверсионная селитра»), либо — в самое последнее время — разложением карбоната калия или нейтрализацией гидроокиси калия азотной кислотой.
Конверсионный способ основан на том, что при добавлении некоторого эквивалентного количества КС1 к горячему насыщенному раствору NaNO3 выделяется NaCl, в То время как из оставшегося после отсасывания NaCl маточного раствора па холоду кристаллизуется ,KNO$. При нагревании процесс протекает но уравнению]
NaNOs + KCI у»- NaCl 4- KNO3
слева направо, так как выпадает NaCl. На холоду процесс протекает еще дальше слева направо, так как в дальнейшем выпадает KNO3.
Две пары солей, которые могут взаимно превращаться одна в другую, называют «обратимыми парами солей».
Растворимость перечисленных выше солей равна
NaNO3 КС1 NaCl KNO3
при 100° 175 56,6 39,1 246 г соли в 100 г Н2О
при 0э 73 28,5 35,6 13 г соли в 100 г НйО
Нитрат калия служит для получения пороха. В противоположность нитрату натрия он пригоден для этой цели, так: как не гигроскопичен. Далее
Первая еруппа. периодической системы
21
нитрат калия применяют для получения нитрита калия, который (наряду с нитритом натрия, см. стр. 649) используют в производство красителей (для диазотирования). Нитрат калия (так же как нитрат натрия) кристаллизуется всегда без воды. Однако он может присоединять азотную кислоту с образованием кислых нитратов: KNCh-HNOs и KNOa-2HNOa.
Нитрат калия образует с нитратом натрия при обычной температуре смешанные кристаллы лишь весьма ограниченного состава (отсутствие смешиваемости проявляется в интервале содержания NaftOg 0,5—99,9 вес.%). Выше 175°, однако, наступает неограниченная смешиваемость. Смесь обеих солей при содержании 45 вес.% NaNO3 обладает ярко выраженным минимумом температуры плавления (225°). Отсутствие смешиваемости в твердом состоянии можно часто наблюдать в бинарных системах с минимумом точки плавления (J S н е с k с Е., Z. Elektrochem., 48, 45(4, 1942).
Нитраты рубидия и цезия. RbNOs и CsNO3 изоморфны нитрату калия и, Кроме того, очень похожи на него. Растворимость иитрата рубидия также очень сильно возрастает с температурой. У нитрата цезия этого свойства в такой степени не наблюдается. С азотной кислотой образуются продукты присоединения такого же состава, как и в случае иитрата калия.
Нитрат лития. LiNOj получают обычно разложением карбоната азотной кислотой. Он представляет собой бесцветную расплывающуюся соль. Из водного раствора кристаллизуется при обычной температуре с 3 молекулами Н2О. Безводный нитрат лития, несмотря на сильное увеличение растворимости с повышением температуры, не обладает соответствующей этому сильно отрицательной теплотой растворения. Наоборот, вследствие значительной теплоты гидратации, обусловленной присоединением воды, он обладает даже слабо положительном теплотой растворения. LiiNOj не образует смешанных кристаллов с NaNO3 и KNO3.
КАРБОНАТЫ
От угольной кислоты НаСОз происходит два ряда карбонатов щелочных металлов. При полном насыщении угольной кислоты щелочью получаются нейтральные или вторичные карбонаты М|СОз, в то время как при замещении лишь одного из двух водородных атомов образуются кислые или первичные карбонаты МЩСОз. Последние называют также гидрокарбонатами, так как они еще содержат водород, который может быть замещен на металл*. Их название — «бикарбонаты» (так как они образуются в результате взаимодействия одного эквивалента щелочи с двумя эквивалентами угольной кислоты) — устарело. Как «нейтральные», так и «кислые» карбонаты щелочных металлов (гидрокарбонаты) обнаруживают в водном растворе щелочную реакцию, так как вследствие слабости угольной кислоты они подвергаются частичному гидролизу:
МаСОз+Н2О Ь^ОН+Ь^НСОз я
М^СОз+НгО М^Н+НзСОз-
Однако гидрокарбонаты вмегот в растворе более слабую щелочную реакцию, чем нейтральные карбонаты. Щелочную реакцию последних можно обнаружить лакмусом п фенолфталеином; гидролиз бикарбонатов не удается обнаружить при помощи мепее чувствительного к ионам ОН' фенолфталеина.
За исключением карбоната лития, все нейтральные карбопаты щелочных металлов легко растворимы в воде. Гидрокарбонаты также легко растворимы; лишь гйдрокарбоцат натрия обладает сравнительно незначительной растворимостью. Как будет видно в дальнейшем, що имеет большое техническое значение.
* Согласно международной номенклатуре, кислые соли называются еидросолями.
220
Глава S
Прочность связывания СО2 заметно возрастает от Li к К, но затем снова naj^r к Cs. Это иллюстрирует рис. 38, иа котором в соответствии с измерениями Лобо (ЕеЬкщ указаны давления диссоциации (соответствующие равновесию М1СО3 М.О -|-в зависимости от температуры.
Карбонаты К и Rb с соответствующими фторидами образуют конгруептно илю-^ щиеся двойные соединения состава К2СОз*Кй н RbaCOg-RbK. В противоположного этому в системах Li3CO3/LгГ biaaCO3/NaF и CsjCOj/CsF не обрг зуется ни двойных соединений, гз: смешанных кристаллов. О влияипз радиуса катиона па энергию свшо таких соединений см. Schmits Dumont, Z. anorg. Chcni., 2EL 49 (1949).
Рис. 38.
Давление диссоциации карбонатов щелочных металлов.
Карбонат натрия. С осе. МааСОз в безводном состояние представляет собой белый порошок удельного веса 2,4—2,5,. который плавится около 85CF.. В воде сода легко растворяется, причем вследствие образования гидратов растворение сопровождается разогреванием. Важнейший из гидратов, получаемы! в твердом состоянии, кристаллическая, сода, NaaCOa-lOHaO кристаллизуется из водных растворов при температуре ниже 32° в виде больших бесцветных моноклинных кристаллов удель
ного веса 1,45, которые плавятся в своей же кристаллизационной воде при 32°. Водные растворы соды обнаруживают ярко выраженную щелочную реакцию, так как вследствие слабости угольной кислоты соль подвергается далеко идущему гидролитическому расщеплению.
Помимо декагидрата, существует ромбический гентагидрат, устойчивый при соприкосновении с раствором в температурном интервале 32,017—35,3°, а также ромбический моногидрат, который, согласно Вальдеку (Waldeck, 1932), находясь под раствором при 112,5° и давлении 1,27 атм, переходит в безводную соль. Гептагидрат существует также еще в другой модификации, которая при соприкосновении, с водным раствором не устойчива ни при какой температуре.
Сода встречается иногда в природе в водах озер, например в озере Оуэнс в шт. Калифорния, общее содержание соды в котором достигает 100 млн. т\ сода, правда достаточно грязная, добывается из этого озера в результате испарения воды на солнце. Содовые озера наряду с нейтральным карбонатом содержат прежде всего гидрокарбонат. В некоторых местах осаждается двойное соединение гидрокарбоната с нормальным карбонатом Na^COa'NaHCOn, называемое троной (trona или urao). В водах целебных щелочных источников, например в Карловых Варах, также содержатся NaaCOs и Nal-ICOs.
Карбонат натрия содержится в золе некоторых морских водорослей. 100 лет назад соду добывали главным образом из золы растений.
Теперь соду получают почти исключительно способом Солъве {аммиачный способ получения соды). Более старый способ Леблана теперь, по крайней мере в Германии, совершенно пе используют. Производство соды пу
Первая группа периодической системы
221
тем карбонизации полученной электролизом натровой щелочи в противоположность осуществляемому таким путем производству поташа имеет ограниченное значение. Как было указано, едкий натр, наоборот, часто получают каустификацией соды. В США соду отчасти получают из криолита.
По способу Лсблапа каменную соль обрабатывали сначала концентрированной серной кислотой,' получая сульфат натрия (называемый обычно в технике коротко сульфат) и в качестве важнейшего побочного продукта соляную кислоту
2NaCl + H2SO4 = Na2SO4 + 2НС1. ’ (13)
Затем для получения соды сульфат смешивали с карбонатом кальция (известняк) я углем и сплавляли в пламенной печи. При Этом происходили следующие реакции:
Na2SO4 + 2С = Na2S + 2СО2; (14)
Na2S + СаСО3 = Na3CO3 4- CaS. (15)
Соду извлекали из охлажденного сплава выщелачиванием водой, в то время как нерастворимый CaS оставался в качестве малоценного отброса. Способ был разработан Лебланом в 1701 г. на премию Французской академии. Вскоре после этого, сначала в Англии,затем в Германии и Франции, развилась содовая промышленность, которая до 1870 г. основывалась исключительно на процессе Леблана. Только в последнее время процесс Леблана был вытеснен рентабельным способом Сольве.
Способ получения соды по Сольво, или аммиачный способ, основан на образовании сравнительно трудно растворимого гидрокарбоната натрия NaHCOa взаимодействием хлорида натрия с гидрокарбонатом аммония в водном растворе
NaCl + NH4HCO3= NaHCO3-|- NH4CI.
В технике почти в насыщенный раствор поваренной соли пропускают сначала аммиак, затем двуокись углерода. Образующийся NaHCOa отфильтровывают и нагреванием (кальцинирование) переводят в NaaCOs {кальцинированная сода)
2УаНС03=Ка2СОз+С03+Н20.
При этом выделяется половина первоначально взятой двуокиси углерода, и ее снова направляют в процесс. Чтобы обратно получить NH3, в маточный раствор, из которого осаждали гидрокарбонат, пропускают аммиак и водяной пар. Благодаря этому содержащийся там гидрокарбонат аммония переходит сначала в нейтральный карбонат (16) и последний при температурах выше 58° разлагается па двуокись углерода, воду и аммиак (17).
NH4HC03+NH3 = (NH4)2C03; ’ (16)
(NH4)2C03=C02+H20+2NH3. (17)
Аммиак, содержащийся в маточном растворе в виде NH4CI (около 75% общего количества), выделяется оттуда при добавлении известкового молока
2NH4Cl+Ca(OH)2=CaCl2+2H20+2NH3.
гак что наряду с пепрореагировавшим хлоридом натрия единственным отходом является хлорид кальция, который обычно спускают в реки.
Аммиачный способ получения соды был разработан в техническом отношения в 1883 г. бельгийцем Сольве. При этом способе получается очень чистая сода. Аммиачный способ более эффективен, чем способ Леблана прежде в сото потому, что в Этом случае расходуется меньше топлива.
222
Глава. f>
Производство соды из криолита применяют в США, правда в небольших размерах. Способ основан на том, что криолит Na3AlFe при патренапии до красного каления с известняком разлагается по реакции
Na3AlF6 4- ЗСаСО5 = Na3A103 4- 3CaF 4- ЗСО2.
Полученный таким образом алюминат натрия Na3A103 разлагают затем водвЭ двуокисью углерода
2NasA103 ЗН2О 4- ЗСО2 = 3Na2GOs 4- 2А1(ОН)3.
Сода, полученная из Криолита, отличается особой чистотой.
Сода является одним из важнейших продуктов химической промышленности. 13 больших количествах ее используют в стекольном и мыловаренном производствах. Она является также исходным продуктом для получения многих других важных соединений натрия, таких, как едкий натр, бура, фосфат натрия, растворимое стекло и др. Большое количество соды употребляют, кроме того, в прачечных, на бумажных фабриках, в красильном производстве, а также для смягчения воды паровых котлов.. В домашнем хозяйстве сода применяется как средство для чистки.
Гидрокарбонат натрия, кислый карбонат натрия — двууглекислый натрий NaHCOs представляет собой белый, устойчивый в сухом воздухе порошок со щелочным вкусом (удельный вес 2,2). Его получают пропусканием двуокиси углерода через холодный насыщенный раствор NaaCO3.
Na2CO3-]-CO24-H3O = 2NaHCO3. (18)
Гидрокарбопат натрия получают как промежуточный продукт в способе Сольве. Чтобы получать чистый гидрокарбонат натрия, продукт, загрязненный гидрокарбопа-том аммония, растворяют в теплой воде; при охлаждении осаждается чистый тидро-карбонат натрия. Реакцию по уравнению (18) используют в лабораториях для очистки карбоната натрия. Нагреванием примерно до 300° гидро кар б о пат можпо Легко снова перевести в карбонат
2NaHCO3 = Na2CO3 4- СОг 4- Н2О.
В водном растворе (или даже во влажном состоянии) уже при комнатной температуре гидрокарбонат натрия медленно выделяет СО2. Выше 65° выделение СОа из раствора становится энергичным. Растворимость гидрокарбоната в воде, насыщенной СО2, при атмосферном давлении, по данным Федотьева, равна:
при 0° 15* 30* 45*
6,9 8,8 11,02 13,86 a NaHGO3 в 100 е Н?О.
Вследствие гидролитического разложения раствор проявляет щелочную реакцию, правда очень слабую. Она обнаруживается лакмусом и метиловым оранжевым, но не обнаруживается (при 0°) фенолфталеином. Гидрокарбонат натрия применяют главным образом в качестве заменителя дрожжей, как медицинское средство для нейтрализации желудочной кислоты (соль Бульриха), а также для приготовления шипучих порошков. 1
Карбонат калия. — поташ — КаСОз. Белая гигроскопичная масса удельного веса 2,30 с температурой плавления 894°, легко растворяется в воде, причем растворение сопровождается разогреванием (9,5 ккал! моль). Б 100 г воды при 0° растворяется 105, при 25° 113,5, при 100° 156 г КаСОз. Среди получающихся в твердом состоянии гидратов при обычной, температуре устойчив дигидрат КаСО3-2НаО.
Первая группа периодической системы
223-
Техническое получение поташа ведут преимущественно двумя путями: 1) карбонизацией калийного щелока, 2) непосредственно из калиевых солей либо Стассфуртским способом, либо, в последнее время, формиатным способом. Наряду с этим поташ получают также из золы мелассы и из золы овечьей шерсти, а в странах, богатых лесом, из древесной золы.
Еще в древпнс времена было известно моющее действие золы растений, которое связано с присутствием в ней карбоната калия. Получение карбоната калия из древесной золы было известно уже древним грекам и римлянам, однако они пе отличали полученную таким образом соль от встречающейся в природе соды. Алхимики готовили чистый карбопат калия прокаливанием виннокаменной соли КНС4И4О6, а позднее путем сжигания ее с селитрой.
Для получения поташа Леблан разработал способ, совершенно аналогичный, его способу получения соды. Долгое время этот способ имел большое техническое значение.
Карбонизация калийного щелока происходит непосредственно вслед за его электролитическим получением при пропускании через него двуокиси углерода
2KOH-f- СО2 = K3CO3-f- Н20.
По предложению Харгревса (Hargreaves) двуокись углерода вводят непосредственно в камеру для электролиза, так что там тотчас образуется К2СО3; однако в этом случае поташ трудно отделить от хлорида.
Стассфуртский способ [способ Энгела (Engel) и Прсхта (Preehf)f основан па плохой растворимости двойной соли гидрокарбоната калил и карбоната магния. По этому способу двуокись углерода пропускают в раствор хлористого калия, в котором суспендирован МдСОз'ЗНзО;
2KCl-]-3MgC02-3H20-]-C02 = KHG02-2MgC03.4H20-}-MgCla.
Выпадающая трудпорастворимая двойная соль разлагается в воде при температуре около 60° с выделением двуокиси углерода
K.PICO3’ 2MgCO3- 4Н2О = К.2^О2-)~2М^ЕОз" ЗН2О-|- С024“ЗН20.
По формиатному способу (способ Гольдшмидта (Goldschmidt)] поташ получают через формпат калия КИСО2, для чего раствор сульфата смешивают в молярном отношении с известковым молоком, и эту смесь насыщают окисью углерода при повышенной температуре (около 220°) и давлении 30 ат. Реакция идет по уравнению
K2SO4 + Са(ОН)г Ц- 2СО = CaSO4 + 2КНСОг -}- 7 ккал, в результате чего образуется раствор формиата калия, который после отделения гипса упаривают досуха. Методом окислительного] кальцинирования получают затем карбонат по уравнению]
2КНС02 4- О2 = К2СО3 СО2 Н2О 4- 106,2 ккал.
Карбопат калия применяют в мыловаренном и стекольном производствах, при крашении, белении и отмывке шерсти. Его используют также для получения цианида калия, а в препаративной химии — часто в качестве в оду отнимающего средства.
Гндрокарбопат калия. Кислый или первичный карбонат калия KFICO3 несколько более трудно растворим, чем нейтральный карбонат калия, но существенно легче растворим, чем гидрокарбопат натрия (а именно 36,1 г КНСО3 в 100 г воды При 26°). Он имеет бесцветные мотто кликни о кристаллы удельного веса 2,17. При нагревании он распадается так же, как гидрокарбонат натрия:
2КНСОв=КвСОд-)-СО24-НгО.
Карбонаты рубидия и цезия. Rb2CO8 и Cs2CO3 удобнее всего получать взаимодействием сульфатов с гидроокисью бария и последующим упариванием с карбонатом ам-
224
Г лава 6
иония. Обезвоженные прокаливанием соли расплываются на воздухе и растворяются в воде со значительным выделением тепла (8,75, соответственно 11,84 ккал). В спирте карбонат рубидия, так же как другие карбопаты щелочных металлов, незначительно растворим, карбонат цезия, напротив, легко растворим. В 100 г спирта при 19° растворяется 0,74 г Rb2CO3n 11,1 г Cs2CO3, Это используют для разделения рубидия и. цезия. Гидро карбонаты рубидия и цезия еще лучше растворимы, чем гидрокарбонат калия.
Карбонат ;1ития. Ы2СО3 выделяется при смешивании растворов солей лития с растворами, содержащими ионыСО^ в виде труднорастворимой в воде и нерастворимой в спирте белой кристаллической соли. Удельный нес его 2,11. Точка плавления 618°.
При техническом получении карбопата лития большей частью исходят из амблигонита LiAl (POjF, встречающегося в виде больших заложен в Америке. Его растворяют в горячей концентрированной Серной кислоте. Примеси алюминия и железа осаждают добавлением аммиака и сульфида аммония и водную вытяжку смешивают с карбонатом натрия. Выделившийся при этом карбопат лития очищают растворением в соляной кислоте, осаждением примесей серной кислоты хлоридом бария и повторным осаждением соли в виде карбоната. Получение лития из Других минералов проводят аналогично, с той только разницей, что вскрытие производят по-другому — например, в случае сподумена ГлА1 [Si2Oe] путем сплавления с содой. Силикаты можно вскрывать также прокаливанием с СаСО3 или с СаО. Для вскрытия трифилина лучше всего применять концентрированную Серную кислоту с небольшим добавлением ааотпой кислоты, Отделение лития от других щелочных металлов можно производить, используя плохую растворимость фосфата в воде или определенную растворимость хлорида в смеси спирта с эфиром.
Карбопат лития лучше растворим в холодной воде, чем в горячей. При нагревании от 0 до 100° растворимость падает от 1,54 до 0,73 ч, Li2CO3 на 100 ч. воды. Растворимость значительно увеличивается при добавлепип в воду СО2. Этим иногда пользуются для очистки карбоната. Можно предположить, что в воде, содержащей СО2 образуется гидро кар б он ат, так же как в случае щелочноземельных металлов. В твердом состоянии гидрокарбонат лития не получен.
Карбонат лития является исходным продуктом для получения большинства других солей лития. Ранее его применяли для лечения подагры. Теперь для этой цели используют органические соли лития, такие, как цитрат лития или салицилат лития.
СУЛЬФАТЫ
Серная кислота как двухосновная кислота производит два ряда солей — сульфатов щелочных металлов. Нейтральные сульфаты растворяются в воде, давая нейтральную реакцию; кислые сульфаты или гидро-сульфаты* М'НЗОт при растворении дают кислую реакцию. Все сульфаты щелочных металлов легко растворимы в воде.
За исключением сульфата лития, сульфаты щелочных металлов способны образовывать с соответствующими фторидами копгруентпо плавяш.исся двойные соединения тина MjjSOj-MF (Schmitz-Dumont, 1949). Относительно эффективного объема SO4-группы в сульфатах щелочных металлов (и в BaSO4), а также о влиянии природы катиона па связь S—О см. G о н Ь е a u J., Z. anorg. Chem., 255, 309, 1948,
Сульфат натрия. NasSCh получают в больших количествах в качестве побочного продукта при производстве соляной кислоты из хлорида натрия и серной кислоты. Его также получают как побочный продукт из остаточных растворов при производстве хлорида калия. Эти остатки содержат NaCl и MgSO<j. Из остаточного раствора на холоду кристаллизуется содержащая воду соль Na2SO4.10ELO
2NaCl + MgSO4 7+ MgCl2-f-Na2SO4.
Сульфат натрия выделил еще Глаубер в 1658 г. при получении соляной кислоты из хлорида натрия и серной кислоты. В природе сульфат патрпя встречается в многочисленных минеральных водах; безводный сульфат встречается в виде тенардита.
* Название «бисульфаты» устарело.
Нервах группа периодической системы
225
В залежах солей калия он встречается главным образом в виде двойных солей таких как глауберит Na2SO4' CaSO4, астракаиит. Na2SO4’MgSO4-4НгО, лецешп Na2SO4-MgSOfSVzHaO, еантгоффит 3Na2SO4-MgSO4, глазерит lSa2SO4-3K2SO4.
Сульфат натрия кристаллизуется из водных растворов ниже 32,383° в виде содержащих воду больших бесцветных моноклинных призм состава NazSOi-iOHsO, которые па воздухе постепенно выветриваются, отдавая воду. При нагревании выше 32° опи плавятся в собственной кристаллизационной воде с образованием безводной соли. При соприкосновении с раствором выше 32,383° устойчива только безводная соль.
Сульфат натрия легко образует пересыщенные растворы. Содержащий кристаллическую воду сульфат натрия обычно называют глауберовой солью.
Растворимость сульфата натрия достигает максимума при 32,383°. Опа равна
при 0° 10° 20° ЗС° 32,38° 33° 40’ 50* 100*
4,5 8,24 16,1 28,9 33,2 33,1 32,5 31,8 29,8 г Na2SO4
в 100 г раствора
Температуру, при которой безводный рульфаг натрия находится в равновесии с декагидратом, насыщенным раствором и его парами (32,383°), можно использовать в качестве фиксированной температурной точки (Richards, 1903). Ее удается воспроизвести почти без труда, так же как температуру плавления железа. Колебания в содержании DjO, которая обычно имеется в воде, не влияют заметно на положение фиксированной точки, так как если даже НгО совсем заменить на D2O, то точка превращения повысится только на 2,10 (Taylor, 1934).
Точка перехода глауберовой соли в безводный сульфат натрия понижается растворенными в расплаве примесями. То же относится и к другим гидратам солей. Если концентрация примесей не слишком велика, то понижение точки перехода подчиняется тому же закону, какому поднимется понижение точки замерзания раствора (закон Рауля — В ант-Гофф а). В случае глауберовой соли молярное пониженно точки перехода составляет 3,25°. «Солевую криоскопию» можно использовать для определения ионных весов (ср. J a h г К. F., Angew. Chem., 63, 220, 1951). Безводный сульфат натрия, полученный нз водных растворов, образует ромбические бипирампдальные кристаллы удельного веса 2,68 с точкой плавления 884°. В воде Na2SO4 растворяется со слабым разогреванием.
Содержащий воду кристаллический сульфат натрия (глауберова соль) растворяется в воде с сильным охлаждением (—18,76 ккал/моль). Его применяют иногда для охлаждения; в медицине его используют как слабительное. В промышленности NaaSOi применяют при крашении и в аппретурах хлопчатобумажных тканей.
Безводный сульфат натрия, называемый в технике чаще просто сульфатом, в больших количествах паходит применение в стекольном производстве и для получения ультрамарина.
Смесь 1 моля глауберовой соли с 1 молем поваренной соли имеет точку перехода при 17,9°. Превращение (дегидратация глауберовой соли) протекает так медленно, что температура часами остается постоянной. Поэтому эта смесь удобна для получения точно определенной «нормальной комнатной» температуры в сосуде.
Гидросульфит натрия. Кислый сульфат натрия NaHSOr — бесцветная легкорастворимая соль, образуется при умеренном нагревании поваренной соли с концентрированной серной кислотой
H2SO4+NaCl = NaHSOi-f-HCI. i
При более сильном нагревании с поваренной солью гцдросульфат переходит в нейтральный сульфат
NaHS04-}-NaCl = Na2S04-f- НС1.
15 г. Реми
226
Глава в
Нагретый гидросульфат отщепляет воду с образованием тиосульфата натрия NasSaO?; при еще более сильном нагревании последний также разлагается с выделением трехокиси серы
2NaIISO4 = Na2S2O7 -f- H2G; Tsa2S2O7 Na2SO44“SO3.
Гидросульфат натрия и пиросульфат натрия применяют в химическом анализе для вскрытия труднорастворимых соединений. Их используют также для очистки платиновых тиглей.
Сульфат калия. K2SO4 кристаллизуется лишь в безводном состоянии в форме ромбических кристаллов. При 587° он превращается в гексагональную модификацию. Удельный вес при 18° равен 2,67, точка плавления 1074°. Растворимость в воде равна
при 0° 10* 20’ 30’ 40’ 50’ 100’ 170’
7,35 9,22 11,11 12,97 14,76 16,50 24,1 32,9 г K2SO4b 100 г Н3О
В спирте сульфат’калия нерастворим.
В промышленности сульфат калия используют преимущественно для получения стекла и квасцов. Кроме того, он представляет весьма ценное удобрение. Для его технического получения наряду с хлоридом калия в качестве исходного продукта служит встречающийся в месторождениях калийных солей сульфат магния (кизерит), а также его двойные соли.
В водпом ‘растворе сульфат магния сначала реагирует с хлоридом калия по уравнению
MgSO4 -f- 2КС1K2SO4 -ф MgCl2.
Получающийся при атом K.2SO4 дает с MgSO4 трудно растворимую двойную соль (называемую в технике (калийной магнезией)
K2SO4 4- MgSO4 4- 6НаО K2SO*.MgSO4-6H2O.
После фильтрования и промывйи последнюю снова обрабатывают раствором хлорида калия; при этом происходит дальнейшее разложение по уравнению
K2SO4-MgSO4-GlT2O 4- 2кС1 = 2K2SO4 4- MgCl2.6H2O.
Прежде сульфат калия получали разложением хлорида калия или нитрата калия серной кислотой. Таким способом сульфат калия получал уже Глаубер, и еще в XIV в. было известно получение этой соли при действии нагреваемого железного купороса (который выделяет при этом серную кислоту) на калийную селитру. Сульфат калия относится к одному из первых химических соединений, для которых был установлен состав (Glauber, Tachenius, Boyle).
Гидросульфат калия. Кислый сульфат калия KHSO4 получали ранен как побочный продукт при производстве азотной кислоты обработкой селитры концентрированной серной кислотой (ср. стр. 644). В настоящее время его приготовляют растворением нейтрального сульфата калия в избытке разбавленной серной кислоты
KaSO44-H2SO4=2KHSO4.
Из водного раствора гидросульфат калия кристаллизуется с водой. Безводное соединение плавится около 200°. При более сильном нагревании, так же как соответствующее соединение натрия, он переходит сначала с отщеплением воды в пиросульфат, а затем в нейтральный сульфат. Применяется так же, как гидросульфат натрия.
Чистый гидросулъфат калия не гигроскопичен. В чистом состоянии его проще всего получить растворением пиросульфата калия K2S2O5 в водо, затем упаривая раствор досуха и нагревая остаток при 120° до постоянного веса.
Первая группа периодической системы 221
Чистый пиросулъфат калия в лабораторий удобнее всего получить термическим разложением персульфата калия K2S2O8. Последний при нагревании в течение получаса при 290° отщепляет точно один атом кислорода, .
Сульфаты рубидия и цезия. BbsSO-i и CsaSOa образуют ромбический кристаллы, изоморфные K2SOr (при более высокой температуре, а именно выше 657° RbeSOt образует еще другую модификацию). Удельный вес их прп 20° 3,61 и 4,24 соответственно; точка плавления 1074 и 1019° соответственно. Оба соединения отличаются легкостью, с которой они образуют с сульфатом алюминия, сульфатом железа (III) и с сульфатами двухвалентных металлов очень хорошо кристаллизующиеся двойные соли (или смешанные соли). Образование цезиевых квасцов CsA^SO^a-12НвО применяется для обнаружения алюминия.
Сульфат лития. Li2SO4 кристаллизуется из горячих растворов без воды в форме, ио-видимому, ромбических игл. При обычной температуре кристаллизуется в виде гидрата Li2SO4-H2O в топких моноклиппых табличках. (О системе Li2SO4—П2О см.: С a tn р bell Л. N., J. Am. Chem. Soc., 65, 2268, 1943.) Безводная соль плавится при 859°, удельный вес ее 2,21. Растворимость
при 0" 20° 100’
35,5 34,5 29,5 г Li2SO4 в 100 г Н2О
Растворимость, таким образом, весьма значительна, хотя она и пе столь велика как растворимость хлорида и нитрата. Летучесть сульфата лития мпого меньше, чем летучесть хлорида.
Сульфат лития, как и другие соли лития, пе образует смешанных кристаллов с другими солями щелочных металлов. Однако существует целый ряд смешанных п двойных солей'.
NaLiSO4
Na3Li(SO4)2-6H2O
Na4LU(SO4)3-9H2O
Na2Lis(SO4)5-5H2O
KLiSO4
K4Li2(SO4)3
K2Li8(SO4)5-5H2O
Li(NH4)SO4
Сульфит лития Li2SO3 также склонен к образованию подобных двойных солей:
Na2Li i2(SO з) т 6Н2О К 2Li2( S О 3) 2 • Н2О.
У остальных солей лития склонность к образованию'двойных солей с другими солями щелочных металлов далеко не так ясно выражена, как у сульфата лития.
В дальнейшем будет еще сказано о некоторых труднорастворимых солях гцелоч-пых металлов, имеющих значение главным образом в аналитической химии.
Труднорастворимые соли щелочных металлов. Из простых солей натрия трудной растворимостью отличается прежде всего гидрокеоантимонат натрия (ранееназываемый «кислый пироантимонат натрия») Na[Sb (ОН)61. Он выпадает в виде белого, зернистого осадка, состоящего из микроскопических кристалликов характерной формы при добавлении соответствующей соли калия к не слишком разбавленному нейтральному или слабо основному раствору соли натрия
К [Sb(OH)6] -|-NaCl = Na[Sb(OH)6)4- KCI.
Эту реакцию используют в качественном анализе для определения натрия. Употребляемый для осаждения гидроксоантимонат калия (получение см. стр. 719 и сл.) в холодной воде также достаточно плохо растворим, но он: легко растворяется при 40—50°, в то время как для растворения 1 ч. этой соли натрия требуется 350 ч. кипящей воды.
15*
228
Глава 6
Из труднораетворимых солей калия кислый тартрат калия (гидротартрат калия K.HC4H4OJ с давних пор известен под названием винного камня (cremor tartari). Растворимость его при 203 равна 0,57 г соли и при 100° 6,9 г соли в 100 г воды. Растворение значительно уменьшается при добавлении спирта. Нейтральный тартрат калия (кристаллизующийся с водой К2С4Н4О6-*/гНаО), напротив, легко растворим (1 ч. в 0,66 ч. воды при 20°). Легко растворима также смешанная соль KNa |С4И4О1;]-4НгО (сегпетова соль).
Довольно трудно растворимы калийные соли хлорной иплатинохлорп-стоводородной кислот [гексахлoponnaTHHo(IV)кислота]. Значения их растворимостей даны в табл. 36. В воде с большим содержанием спирта они практически нерастворимы. Гексахлороплатинат калия Кг [PtClJ образует желтые небольшие правильные октаэдры. При нагревании соль разлагается на хлорид калия и металлическую платину. Растворимость перхлората калия КСЮг сильно зависит от температуры; опа возрастает от 0,70 е в 100 г воды при 0° до 18,7 г при 100°. Еще труднее, чем перечисленные соли калия, растворяются соответствующие соли рубидия и цезия..
Из трудно растворимых солей- лития уже упоминались фторид LiF и карбонат Гл2СОэ. Фосфат лития LisPO4 образуется в виде трудно растворимо го белого, кристаллического осадка при добавлении к растворам солей лития раствора, содержащего ионы РО^ ; 3 Li' -ф- РО7 = Li3PO4. Этот осадок не плавится в пламени булзепов-ской горелки. Если для осаждения применяют кислые фосфаты, то, чтобы осаждение было полным, добавляется NaOH; иначе образуется кислый фосфат лития LiH2PO4, который растворяется значительно легче. Последний образует с фосфорной кислотой продукт присоединения Ь1НгРО4-Н3РО4-Н2О в виде больших расплывающихся кристаллов.
Аналитические сведения. Открытие щелочных металлов проще всего осуществляется на основании характеристических спектров [12]. Окраска пламени также дает указание. Однако при этом следует иметь в виду, что желтое натриевое пламя может быть вызвано уже следами натрия. Фиолетовое пламя калия скрыто в присутствии натрия, однако его можно видеть и в этом случае, если смотреть на пламя через кобальтовое стекло (т. е. через стекло, окрашенное окисью кобальта в синий цвет), не пропускающее при достаточной плотности желтый свет.
В качестве реакции, применяемой для определения натрия, служит образование гидроксоантимоната натрия Na[Sb(OH)6]. Очень хорошо также пользоваться для этой цели образованием двойной соли NaaCsHsOa. [иОзНСаНзОф и хотя опа лишь умеренно труднорастворима, однако кристаллизуется в очень характерной форме (желтые микроскопические тетраэдры). Для образования этой соли к одной капле раствора, предварительно досуха упаренпой па часовом или предметном стекле, добавляют каплю насыщенного раствора уранилацетата [UOa^CsHaOs^ в разбавленной уксусной кислоте.
Калий можно определить, осаждая его в виде перхлората КСЮ*. Другие упомянутые выше трудиорастворимыё соли также используют Для его определения. Более чувствительна реакция с гекеанитрокобалътатом натрия Na3[Co(NOa)6] (ср. т. II). В этом случае выпадает желтый кристаллический осадок гексанитрокобалътата калия К3 [Со (NO2)e], растворимый в воде в отношении 1 : 1000. При проведении реакций на присутствие калия следует иметь в виду, что большинство из ник характерны и для солей аммония (исключая реакцию с надхлорной кислотой).
Соли рубидия и цезия при использовании реакций осаждения с трудом отличаются от солей калия. Для их обнаружения всегда пользуются спек-
Первая группа периодической системы 229
троскопом; равным образом обнаружение лития надежнее и проще всего можно провести спектроскопически.
Для весового определения натрия и калия их большей частью переводят в хлориды. или в сульфаты и взвешивают в виде этих солей. Вследствие летучести, особенно хырида калия, при нагревании следует соблюдать осторожность. Отделение палия от натрия можно провести, осадив калий в виде перхлората. Из фильтрата натрий целесообразно осадить в форме натриймаенийуранилацетата и взвешивать в виде этой соли (Кogler, 1935). Солп щелочных металлов летучих кислот по Тредвеллу (Trod well, 1933) можно легко перевести во фторсиликаты при упаривании с SiO2 и плавиковой кислотой я взвешивать в виде этих солей. Смеси двух солей щелочных металлов можно определить взвешиванием в виде фторсиликатов и титрованием содержания ионов F’. Кели (Caley, 1931) предложил метод колориметрического определения калия в виде пикрата.
Литий отделяют от других щелочных металлов при обработке смеси хлоридов либо амиловым спиртом, либо смесью равных частей абсолютного спирта и эфира, насыщенной хлористым водородом; при этом ЫС1 переходит в раствор. Для взвешивания его переводят в сульфат.
Гл а в а 7
СТРОЕНИЕ КРИСТАЛЛОВ И РЕНТГЕНОВСКИЕ ЛУЧИ
Молекула и кристалл. При соединении водородного иона с ионом хлора образуется молекула хлористого водорода. Определение плотности пара позволило на основании закона Авогадро сделать вывод, что газообразный хлористый водород состоит из отдельных молекул HCL При сильном охлаждении газа он становится жидким и в конце концов твердым. Наиболее естественным представляется допущение, что и образовавшиеся в затвердевшем веществе кристаллы также состоят из молекул НС1.
Взаимодействие ионов щелочных металлов с ионами хлора носит совершенно иной характер. Прежде всего не удается установить присутствия недиссоциир о ванных молекул в водных растворах; уменьшение молекулярной электропроводности может быть, как мы видели выше, объяснено и иным путем. При упаривании раствора газ не выделяется, как в случае хлористого водорода, а остается соль в кристаллическом состоянии, и требуются сравнительно очень высокие температуры, чтобы перевести ее в газообразное состояние. Хотя в этом и заключается характерное различие между кристаллами типа хлористого натрия и кристаллами затвердевшего хлористого водорода и других газообразных или легко летучих при обычной температуре соединений, но все же ранее по аналогии предполагали, что кристаллы типа NaCl построены из молекул соответствующих соединений. Недостатком этого представления было то, что на его основе нельзя было объяснить характерное различие в летучестях этих веществ. В дальнейшем эти представления были опровергнуты определением строения кристаллов рентгенографическим способом, показавшим, что кристаллы хлоридов щелочных металлов, так же как и других типичных солей, построены не из молекул, а непосредственно из ионов, образующих эти соединения.
Дифракция рентгеновских лучей при прохождении через кристаллы. Известно, что свет при прохождении через узкую щель испытывает дифракцию. Лучи, претерпевшие дифракцию, интерферируют между собой, что и обусловливает в случае монохроматического света появление системы светлых и темных полос. Интерференционные полосы возникают, при прохождении луча через стеклянную пластинку (так называемую дифракционную решетку), на которую нанесены тесно прилегающие друг к другу полосы, или при отражении от соответствующей металлической решетки. Между длиной волны света и соответствующим углом дифракции и постоянной дифракционной решетки (т. е. расстоянием между соседними черточками решетки) существует очень простое соотношение, которое, как известно, и обусловливает образование интерференционных спектров
Строение кристаллов и рентгеновские лучи 231
и позволяет производить измерение длин волн. Для того чтобы интерференция могла возникнуть, постоянная решетки де должна быть меньше половины длины волны света.
Правильная форма образующихся кристаллов свидетельствует о том, что структурные элементы, которыми могут быть, атомы или молекулы, расположены в определенном порядке. Упорядоченное расположение точек называется точечной решеткой.. Интерференционные явления должны возникать, как это следует из волновой теории, при прохождении света через такую точечную решетку или при отражении от густо усеянных точками плоскостей решетки пли плоскостной сетки в том случае, если константа решётки или в данном случае расстояние между соседними плоскостями решетки находятся в определенном соотношении с длиной вопи проходящего света. Нетрудно показать, что при прохождении обычного света через кристаллы не происходит дифракционных явлений. Используя число Авогадро Nд, можно определить число атомов, содержащихся в одном кубическом сантиметре вещества с известной плотностью. На основании этого можно определить и константу решетки.
Плотность поваренной соли (NaCl) d = 2,17, молекулярный вес 58,45. Значение числа Авогадро Na = 6,023-10аз; поэтому в 1 см? кристаллической поваренной соли содержится
24.10-Об,4а
молекул, или вдеое больше атомов. Если представить себе совершенно равномерное их распределение, то па отрезке 1 cat должно поместиться 3,55-107 атомов, что будет соответствовать кратчайшему межатомному расстоянию 2,8 Л, т. в. величине, более чем в 1000 раз мепьшей, чем длина волны наиболее коротковолновой видимой части спектра, и слишком ничтожной, чтобы обусловить дифракцию даже наиболее жестких ультрафиолетовых лучей с длиной волпы около 100 А.
В 1912 г. Лауз сумел показать, что при прохождении рентгеновских лучей через кристаллы происходит их интерференция, вызванная дифракцией. Тем самым было доказано, что кристаллы в соответствии с развивавшимися ранее представлениями обладают решетчатой тонкой структурой. Одновременно было окончательно установлено, что рентгеновские лучи имеют такую же природу, как и световые волны, и отличаются от последних только значительно меньшими длинами волн.
Длины волн обычно применяемых рентгеновских Лучей имеют порядок 0,1—1,0 А, Рентгеновское излучение, содержащее лучи с непрерывным рядом различных длин волн, называют по аналогии с солнечным светом белым рентгеновским излучением. Гомогенное рентгеновское излучение содержит лучи только с определенной длиной волны. О методах его получения будет сказано в дальнейшем.
Лауз, направляя белое рентгеновское излучение на кристалл цинковой обманки ZiiS перпендикулярно к грани куба, т. е. в направлении оси симметрии четвертого порядка, но лучи л па фотографической пластинке, помещенной позади кристалла, интерференционное изображение, приведенное па рис. 39. Это изображение отражает симметрию кристалла; каждом}-’ темному пятну на плоскости соответствует плоскость в кристалле с особенно плотным расположением атомов. Этими плоскостями часто являются грани, ограничивающие кристаллы данного вида.
Можно показать, что дифракция рентгеновских лучей при прохождении их через трехмерную решетку кристалла, происходит совершенно
232
Глава 7
Pat. 39. Лауэтраыма.
Цинковал обманка ZiiS, снято в лучах, перпендикулярных грани куба.
таким же образом, как если бы рентгеновские лучи отражались от п. скостей этой решетки. Теория рентгеновских интерференций для пос.’ него случая может быть развита наиболее простым способом.
1
Теория интерференции рентгеновских лучей, Иптерференция^ренг: новских лучей) как и световых лучей, происходит в том случае, если трсз место взаимное усиление или ослабление в зависимости от угла рассеяит соседних лучей после их отражения от плоскостей решетки. Условие, г обходимое для усиления рентгеновских лучей, отраженных от различие равноотстоящих^одна от другой плоскостей решетки, можно легко/ наг: по рис. 40.
Рис, 40. Возникновение реитгеповских интерференции.
Рассмотрим сначала два параллельных луча 5i и 5з, отражении от соседних плоскостей рснютки. Эти лучи падают па плоскости, решетке по отношению к которым рассматривается их отражение, под углом (угол отблеска). Согласно закону отражения, они отражаются под рас ними углами. При выходе из кристалла лучи будут иметь разность хода определяемую длиной отрезка АВ ф- ВС, на который второй луч отстае
Стрсвнссв кристаллов и рентгеновские лучи 23S
от первого. Взаимное усиление обоих лучей при их попадании на какой-нибудь экран произойдет только в том случае, если разность их хода АВ + ВС будет равна целому кратному длины волны X используемого' рентгеновского излучения. Отсюда имеем следующее условие усиления лучей:
АВС-ВС-гА, где в—целое число, или „ , . . . 2dsina
2asina = M‘. Л=----——— . , (1)
Но АВ = ВС = d sin а, если d — расстояние между обеими плоскостями решетки. Очевидно, и взаимное усиление лучей 5з и S« произойдет y** 2d sin сб
при таких же условиях* Если же —— не будет разняться целому числу», то отраженные лучи будут взаимодействовать между собой и произойдет взаимное погашение; так, если —= 1/а (или 1х/а, 2 /3 и т. д.), то произойдет взаимное погашение отраженных от соседних плоскостей лучей 51 и 5а; в случае 2^^гп?=1/4 (или 3/4, 6/4 и т. д.) будут гаситься лучи Si и S3, а также 5а и Si и т. д. Если пучок отраженных лучей направить на фотографическую пластинку или пленку, то она почернеет только и тех местах, куда попадут лучи, удовлетворяющие условию (1).
Если, как это сделал Лауз, направить на кристалл белое рентгеновское излучение, то в этом пучке всегда найдутся лучи с такими длинами волн, для которых будет удовлетворяться условие (1), и именно для расстояний между различными плоскостями решетки, например в кубической решетке — для ее плоскостей, параллельных граням куба, параллельных граням октаэдра, параллельных граням ромбододекаэдра и т. д. Так как отражение может происходить только от атомов*, то естественно, что от данной плоскости получается тем более интенсивное отражение, чем большее число атомов расположено на ней. Это число оказывается достаточно большим, чтобы вызвать заметное почернение фотографической пластинки в местах, соответствующих только наиболее важным в кристаллографическом отношении плоскостям кристалла. Поэтому на пластинке получаются пятна лишь в тех местах, которые соответствуют именно этим плоскостям. По относительной интенсив пости пятен можно: затем определить, о какой из этих плоскостей в данном случае идет речь.
Рентгенографические методы исследования. Наиболее важными из современных способов получения рентгеновских интерференций являются следующие:
1. Метод Лауэ.
2. Метод Брэгга и его видоизменения (метод вращающегося кристалла Зеемана и Шибольда).
3. Метод Дебая — Шеррера.
Метод Лауэ был уже описан. Расположение приборов при работе этим способом схематически показано на рис. 41. Первоначальный вариант метода «вращающегося кристалла» был разработан в 1913 г. Брэггами (отцом и сыном). По этому методу используют монохроматизированное рентгеновское излучение (рис. 42) пучок, которого направляется па совершенно произвольно ориентированный кристалл. В этом случае условие уравнения (1) остается в общем невыполненным, и поэтому вначале не получается дифракционной картины. Однако при медленном вращении
* Строга говоря, отражение происходит от электронов атомов (см. ниже).
234
Глава 7
кристалла в какой-нибудь момент окажется достигнутым угол отражения,, отвечающий уравнению (1), и именно в этот момент и будет наблюдаться такое отклонение рентгеновских лучей, какое показано на рис. 40. При дальнейшем вращении отраженный луч исчезает, пока направленный ни к ристал лпучок лучей опять нс встретится с плоскостью решетки под
Рис. 41. Метод Лауз.
А — антикатод; К — кристалл; П — фотографическая пластинка; , Б —место на пластинке, соответствующее попаданию первичного не отклоненного пучка*
Рис. 42. Метод Брэгга.
А — антикатод; К — крист а ли; И — фит оттенка; Б — мр.сто на плйкке, соответствующее попаданию первичного пучка»
надлежащим углом. В этом методе, следовательно, на рентгенограмме фиксируется каждая возможная для кристалла плоскость, которая не обязательно должна существовать в действительности в качестве граней данного кристалла, вследствие отклонения рентгеновского луча, строго соответствующего этой плоскости. Плоскости решетки, преимущественно отра
Строение кристаллов и рентгеновские лучи
235
жающие рентгеновские Лучи, как бы вспыхивают при вращении кристалла, подобно зеркалу, вращающемуся в солнечных лучах.
Для определения направления отраженных лучей первоначально Брэгг использовал свойство рентгеновских лучей превращать газы в проводники электричества, т. е. «ионизировать» их. В дальнейшем ионизационная камера Брэгга была заменена куском фотографической пленки, расположенной на некотором расстоянии вокруг кристалла; те места, на которые попадают отраженные рентгеновские лучи на этой пленке, оказываются после проявления почерневшими.
Комбинацией методов Брэгга и Лауэ является разработанный независимо друг от друга Зееманом и Шибольдом метод вращающегося кристалла, известный также под названием метода спектрограммы вращения или метода полной диаграммы.
Рис. 43. Метод Добая — Шеррера.
А — антикатод; К — кристаллический порошок; Я — пленка;
И —место ня пленке, соответствующее попаданию первичного лучка
Метод Дебая — Шеррера (рис. 43) основан на том же принципе, что и метод Брэгга. Дебай и Шеррер (1916) вместо одного вращающегося кристалла применяли мелкий кристаллический порошок. Так как различные частички такого порошка ориентированы в пространстве во всевозможных направлениях, то в совокупности они ведут себя приблизительно так же, как и отдельная частица (монокристалл), приводимая в различные положения относительно рентгеновского луча.
Применения рентгеновских лучей. Интерференция рентгеновских лучей за последнее время получила широкое применение для определения структуры кристаллов (структурный анализ). Постоянную кристаллической решетки, т. о. расстояние между ее главными плоскостями, можно легко определить для кристаллов, имеющих простое строение, путем измерения всего лишь нескольких углов отражения. Вычисление ее производят па основании уравнения (1). Но и для веществ с очень сложным строением можно лишь па основании существования рентгеновских интерференций судить по крайней мере о том, являются ли эти вещества кристаллическими, т. о. построенными упорядоченно или нет, так как только при кристаллическом строении, как Это следует из теории, имеются условия
236
Глава 7
для возникновения четких интерференционных полос или колец. Б случае очень мелких кристалликов но методу Дебая — Шеррера получаются только расплывчатые, т. е. более или менее широкие интерференционные-кольца, причем эти кольца тем шире, чем мельче кристаллики, так что по ширине интерференционных колец можно судить о величине кристалликов. Если диаметр кристалликов значительно меньше Imp, то отчетливых интерференционных колец вообще не удается наблюдать.
Заметное расширение интерференционных колец, получаемых по методу Дебая — Шеррера, происходит в том случае, если размеры частиц лежат между 0,5—0,2 р. Расширение колец закономерно возрастает с дальнейшим уменьшением размера частиц, что и делает возможным определение их величины. Это имеет место в том случае, если речь идет о кристалликах с совершенно правильным расположением в них атомов. Не вполне упорядоченное расположение атомов, т. е. существование так называемых «искажений решетки» (см. т. II, гл. 1), может также привести к расширению интерференционных колец и в случае значительно более крупных кристалликов. Будет ли это происходить, зависит от характера искажений. Существуют и такие искажения, которые обусловливают по расширение, но только уменьшение интенсивностей интерференционных колец. Более подробно см. F г i k е, Z. Electrochem., 44, 29, 1938. Так как в твердых аморфных, а также в жидких и газообразных веществах атомы располагаются не совершенно неупорядоченно, а определенные межатомные расстояния встречаются чаще других, то и для таких веществ наблюдаются более язи мепее отчетливые максимумы в почернениях фотографической пленки. Из положений этих максимумов можпо сделать заключение о строении молекул. Для исследования молекулярной структуры таких веществ, и прежде всего газов, в настоящее время служат не только рентгеновские лучи, по и электронные лучи, которые при прохождении через газы преломляются и испытывают интерференции таким же образом, как и рентгеновские лучи.
Из у равно пия (1) следует, что, если рентгеновское излучение проходит через кристалл, константа решетки которого известна, можно, измерив угол отблеска а, определить длину его волны. Это использование интерференции рентгеновских лучей для измерения длины их воли (рентгено-спектрографии} также получило широкое применение. Особенно важным его результатом явилось открытое Мозли соотношение между длиной волны характеристического рентгеновского излучения элемента и его порядковым числом.
Прежде чем подробнее останавливаться на этом законе, необходимо кратко ознакомиться с результатами определений кристаллических структур при помощи рентгеновских лучей (структурный анализ), поскольку эти определения относятся к ранее уже рассмотренным веществам.
СТРУКТУРНЫЙ АНАЛИЗ (РЕНТГЕНОМЕТРИЯ)
Структура кристаллов. Первые определения структуры кристаллов были проведены в 1913 г, В. Л. Брэггом, в то время студентом Кембриджского университета, исследовавшим по методу Лауэ строение каменной соли (Nad), снльиипа (КС1) и бромистого калия (КВг). Из распределения интенсивностей Брэгг одновременно устанавливал структуру этих кристаллов и находил длины волн рентгеновских лучей, соответствующих каждой дифракционной картине. В настоящее время строение таких кристаллов определяется гораздо проще, а именно путем облучения кристаллов рентгеновскими лучами известной длины волны, так как теперь измерение длин волн рентгеновских лучей производится очень просто, с использованием для этой цели кристаллических дифракционных решеток с известными межплоскостпыми расстояниями. Брэгг установил, что решетки кристаллов каменной соли, сильвина и бромистого калия имеют
Строение кристаллов и рентгеновские лучи
237
строение, приведенное па рис. 44. Отдельные точки (узлы) в таких решетках поочередно заняты атомами (точнее — ионами, ибо атомы, как будет видно из дальнейшего, заряжены) щелочного металла и галогена. Несмотря на то что три упомянутых вещества изоморфны п в соответствии с рентгепоструктурными данными имеют одинаковое строение, распределение интенсивностей в интерференционных картинах NaCl и КВг все же заметно отличается от распределения интенсивностей для KCL Согласно Брэггу, интенсивность отраженных лучей зависит не только от количества узлов решетки, по и от атомного веса частиц, занимающих эти узлы. Брэгг установил, что эта интенсивность пропорциональна квадрату атомного веса. Так как атомные веса К и С1 близки между собой, то кристалл КС1 практически ведет себя так, как если бы его решетка была построена
Рас. 44. Решетка каменной соли. Р и с. 45. Простая кубиче-
а=5,028 А. екая решетка.
исключительно из однородных атомов; он дает рентгеновскую картину простой кубической решетки (рис. 45). Очевидно, приведенная на рис. 44 решетка уже пе будет соответствовать этому типу, если образующие ее лтомы будут различными. Поэтому для NaCl и КВт рентгеновская кар-типа не будет у ясе больше обнаруживать интерференций, соответствующих простой кубической решетке, а будет обладать интерференцией, отвечающей двум вставленным одна в другую гранецентрированным кубическим. решеткам.
Наименьшая часть пространственной решетки, которой присущи все свойства симметрии решетки в целом, называется элементарным параллелепипедом или элементарной ячейкой. Если этот'параллелепипед является кубом, то и соответствующая ему решетка будет кубической. Простой решеткой называется такая, в которой заняты лишь вершины её элементарных параллелепипедов (рис. 45). Гранецентрированной решеткой называется решетка, у которой в элементарных параллелепипедах заняты материальными частичками также и центры граней (рис. 46). Решетка, у которой в элементарном параллелепипеде, кроме вершин, занят и центр, называется объемноцентрированиой (рис. 47). Решетки, которые могут быть образованы, помещением одна в другую двух или нескольких простых решеток, называются сложными. При этом вовсе не обязательно, чтобы отдельные решетки, из которых построена сложная решетка, состояли бы из различных атомов, как это имеет место в случае каменной соли. Решет-
238
Глава 7
на каменной соли ирн заполнений всех ее узлов одинаковыми а тома нт превратится в простую решетку. Грапецентрироврнную решетку можно рассматривать как составленную из четырех помещенных одна в другую простых решеток. Необходимо отметить, что неизвестно ни одного вещества, образующего простую кубическую решетку (рис. 45).
Рис. 46. Гранецептрироваштая куби- Рис. 47. Объемноцентриро-
ческая решетка (плотнейшая кубиче- ванная кубическая решетка,
ская упаковка). Пример: натрий (а—4,30 А).
Пример: аргон (а—5,42 А).
Структурный тип каменной соли. По типу каменной соли построено большинство галогенидов щелочных металлов. Следует отметить, что гидриды, щелочных металлов обладают таким же типом строения (см. стр. 200).
Галогениды щелочных металлов, образующие решетку типа каменной соли, сопоставлены в табл. 39, в которой приведены также длины ребер
Таблица. 39
Галогспиды щелочных металлов, принадлежащие к типу камепиой соли
Фториды a, A Хло риды a, A Бромиды a, A Йодиды a, A
L1F 4,02 LiCl 5,14 LiBr 5,49 Lil 6,00
NaF 4,62 NaCl 5,628 NaBr 5,96 Nal 6,46
KF 5,33 KC1 6,277 KBr 6,58 KI 7,05
RbF 5,63 RbCl 6,54 RbBr 6,85 ВЫ 7,32
CsF 6,01 p-CsCl 7,02 — — — —
— (l-NH, Cl 6,53 ₽-NlI4Br 6,90 P-nh4i 7,24
ребра
отдельных веществ. Длина
а элемен-
элсмептарных ячеек а для
тарной ячейки каменной соли, установленная с очень большой точностью, служит нередко зталоном для других рентгенометрических измерений. Приведенное в таблице числовое значение а относится к природной каменной соли. Так как последняя обычно несколько загрязнена небольшим количеством включенного в решетку хлористого калия, то константа решетки природной каменной соли в действительности приблизительно
Строение кристаллов и рентгеновские лучи
239
на 0,001 А больше по сравнению с константой решетки совершенно чистого хлористого натрия, что необходимо учитывать при очень точных определениях. Межионное расстояние в решетке типа каменной соли равно, как это следует из рис. 44, половине длины ребра куба, т. е. ~.
Структурный тип иодида цезия. Галогениды цезия, такие, как CsBr и CsI, а также и CsCl, кристаллизуются ио другому типу (по сравнению с каменной солью). Они образуют решетку, как бы состоящую из двух простых кубических решеток, расположенных друг относительно друга
так, что один атом одного рода находится в центре элементарного куба, образованного 8 атомами другого рода {тип иодида цезия, рис. 48). Если представить себе, что в решетке типа иодида цезия все узлы заняты только одинаковыми атомами, то она перейдет в объ-емноцентрированную кубическую решетку (рис. 47).
Элементарный куб решетки йодистого цезия содержит, как это следует из химической формулы соединения, одинаковое число атомов Cs и атомов I, хотя с первого взгляда это, быть может, и не вполне очевидно. Каждый не находящихся в его углах
Р и с. 48. Решетка иодида цезия. (<3=4,562 А.)
8 атомов цезия одновременно принадлежит и другим 8 соседним элементарным кубам П, таким образом, входит в состав рассматриваемого куба только своей 1/8 частью. Таким образом, общее число атомов цезия в приведенном на рис. 48ткубе будет 8 х г/а= 1 И, следовательно, равно числу находящихся в нем атомов иода. В качестве центра координат можпо взять но атом Cs, а какой-либо из атомов 1. Элементарный куб решетки каменной соли (рве. 44) содержит 1 -f- i атома хлора и в/2+ ®/в = 4 атома натрия. Межатомное или межионное расстояние в решетке типа иодида цезия равно половине объемной диагонали куба, т. е. (а72))/3.
Галогениды аммония также могут кристаллизоваться и действительно кристаллизуются при низких температурах по типу йодистого цезия. При нагревании решетка типа йодистого цезия у них перегруппировывается в решетку типа каменной соли. Йодистый аммоний NH4I, кристаллизующийся при обычной температуре по типу каменкой соли, при сильном охлаждении образует решетку типа подпетого цезия. Такая же перегруппировка установлена и для хлористого цезия (ср. табл. 39 и 40). При —190° ВЬС1 также может кристаллизоваться по типу иодида цезия (а = 3,74 А при —190°). В табл. 39 и 40 модификации, устойчивые при низких температурах, назва-
Таблица 40
Галогениды щелочных металлов, принадлежащие к типу иодида цезии
Хлориды яТ А Температура превращения. °C Бромиды , а, А Температура превращения. Йодиды a, А Температура превращения. °C
а-CsCl 4,11 415 СвВг 4,29 Сз! 4,56
а-NfI* Cl 3,86 184,3 a-NH4Br 4,05 137,8 a-NHJ 4,37 —17,6
По совершенно другому типу, а именно по типу вюртцита (рис. Й0. стр. 290), кристалли »уетсп фтористый аммоний; а=4,39 А, с=7,02 А.
240
Глава 7
пы «-модификациями; модификации, устойчивые при высоких температурах,— р-мо~ фикациями. Константы решеток а для модификаций, неустойчивых при комнатсс. температурах, относятся к температурам вблизи точек превращения.
Структура решетки щелочных металлов. Щелочные металлы кристам л и дуются в объемноцентрированных кубических решетках (рис. 47). Длпеп,. ребер элементарных ячеек а соответственно равны:
Li Na К Ей Cs
а, А 3,50 4,29 5,51 5,66 6,13
при —10’ при —10°
Однако для К, Rb и Cs это наблюдается только при низких температурах; пр. обычной температуре калий образует тетрагональные монокристаллы (см. стр. 60S
Структура решетки инертных газов. Исследованные до сих пор инертные газы кристаллизуются в кубических гранецеитрированнъгх решетках (рис. 46). Длины ребер элементарных ячеек соответственно равны:
No лг Кс Хе
а, А 4,52 5,42 5,59 6,18
Структура решетки п координационное число. Между строением решеток щелочных -металлов и их галогенидов, как видно, не существует никаких принципиальных различий. В обоих случаях узлы решетки заняты отдельными атомами*. В отиошепни металлов это обстоятельство не является неожиданным, поскольку они в газообразном состоянии одпоатомны; напротив, для их содей химик старой школы должен был бы ожидать в соответствии с их химическими формулами другого строения. Формула NaCl выражает, что 1 атом натрия связывает имени о 1 атом хлора, тем самым в смысле старых валентных представлений исчерпывается способность атома образовывать химическую связь, и он поэтому уже не в состоянии так же прочно удерживать -еще и другие атомы хлора. Однако кристаллическое строение NaCl показывает, что каждый атом натрия окружен шестью атомами хлора, отстоящими от пего па одинаковых расстояниях; равным образом н каждый атом хлора в этом соединении окружен шестью равноотстоящими от пего атомами натрия. Если бы связь между каждыми двумя атомами натрия и хлора чем-нибудь отличалась от других связей, то это должно было бы отразиться и на их расстояниях один от другого. Поскольку в действительности этого не наблюдается, можно заключить, что не имеет никакого смысла представлять себе атомы в it ристал ле NaCl расположенными попарно. Каждый атом п атрия действует па шесть атомов хлора с одинаковой силой. Этим примером подтверждается превосходство более новой теории (электровалентности) по сравнению с прежними представлениями. По электровалентной .теории валентность в соединениях, имеющих тете ров о ляр ное строение, просто обозначает электрический зар.чд. Определение: «хлористый натрий состоит из электрохимически одновалентных противоположно заряженных** атомов (или ионов)о — еще ничего по говорит о структуре соединения, образующегося при взаимном сближении этих ионов. Эду структуру определяют, исходя из общих законов для сил, действующих между электрически заряженными, челами, которые вовсе не требуют, чтобы частицы, обладающие равными и противоположными зарядами, обязательно соединялись попарно. Можно показать, что если притяжение обусловлено в основном только кулоновскими силами, то освобождающаяся f при образовании ионной решетки энергия оказывается больше той энергии, которая бы освободилась при образовании молекулярной решетки, соответствующей соединению атомов попарно. Молекулярные решетки возникают в тех случаях, когда энергия их образования больше, чем энергия образования соответствующих ионных решеток. Это, панрпмер, может произойти, если парной (в более общем случае — групповой) ассоциации атомов иля ионов благоприятствуют лакле-нибудь особые обстоятельства (сильная поляризация).
Пониманию геометрии построения кристаллических решеток в значительной степени способствовало перенесение Пфеифером в эту область понятия о координационном числе, заимствованного из химии комплексных соединений.
* Различие, заключающееся в том, что в одном случае атомы пе заряжены, а в другом заряжены, пока не принимается во внимание.
** Доказательство существования противоположных зарядов в кристаллических соединениях см. в следующих разделах.
Строение кристаллов и рентгеновские лучи
241
Координационным числом в химии комплексных соединений называют число, доказывающее, сколько атомов или групп непосредственно связано с центральным атомом данного комплекса (подробнее см. гл. 11).
В соответствии с представлениями Цфейфера кристаллы рассматриваются как координационные соединения в широком смысле этого слова. Координационное число атома или иона в кристалле показывает, сколько атомов или ионов непосредственно с пим связано и, следовательно, находится па ближайшем от пего расстоянии. В кристалле хлористого патрия, как следует из рис. 44, координационное число хлора равно 6, так как атом хлора здесь окружен 6 атомами натрия, находящимися от него
Д о
на одинаковых расстояниях, равных = 2,814 А. Таким же координационным числом в NaCl обладает и натрий. Это имеет место во всех соединениях типа каменной соли. В иодистом цезии иод, находящийся в центре куба, окружен 8 атомами цезия, расположенными в вершинах этого куба; поэтому под в этом соединении имеет координационное число 8; то же относится, как это следует из соображений симметрии, и к атому цезия. Координационное число 8 присуще вообще всем щелочным металлам в элементарном состоянии, поскольку они образуют объемно центрированную решетку (рис. 47). Еще более высоким координационным числом обладает apron, а именно 12 (рис. 46). Гольдшмидт (1927) показал, что различные структурные типы кристаллических решеток можпо очень четко классифицировать на основании координационных чисел.
Электроны как причина рассеяния рентгеновских лучей; ионпые решетки. Установленная Брэггом зависимость (см. стр. 236) интенсивности рентгеновских лучей, отраженных от плоскостей сетки, занятой одинаковыми атомами, выражающаяся в пропорциональности квадрату их атомного веса, имеет силу только для небольших углов отблеска, да и то лишь приближенно. Эта зависимость находится также в противоречии с установленной рапее Баркла закономерностью, в соответствии с которой интенсивность испускаемого каким-нибудь веществом рентгеновского излучения прямо пропорциональна атомному весу. Это противоречие было устранено Дебаем (1918), показавшим, что Дифракция рентгеновских лучей при прохождении их через кристаллы или при отражении от плоскостей решетки кристаллов основана ~ совершенно так же, как и преломление или отражение обычного света,— па том, что свет, как видимый, так и рентгеновский, попадая на очень мелкую частичку, испытывает рассеяние. При этом такая частичка, на которую падает свет, ведет себя как точка, обладающая собственным свечением*, от которой исходит сферическая световая волпа. Поэтому яспо, что отражение рентгеновских лучей от какой-нибудь заполненной определенным количеством материальных точек плоскости решетки будет тем сильнее, чем значительнее рассеивающая способность отдельных частичек. Дебай, опираясь на принципы классической электродинамики, установил, что интенсивность рассеяния, а вместе с тем, следовательно, и отражения рентгеновских лучей должна быть пропорциональна количеству рассеивающих электронов. Именно электроны и обусловливают в действительности рассеяние рентгеновских лучей. Поэтому распределение интенсивностей рассеянного излучения и дает нам непосредственную меру количества и расположения электронов. Но так как в нейтральных атомах число электронов равно порядковому номеру и так как ему же приблизительно пропорционален и атомный вес**, то отсюда и следует в общем случае приблизительная пропорциональность между интенсивностью рассеянного излучения и атомным весом, т. е., другими словами, справедливость закона Баркла. Однако, как показал Дебай, для малых углов отблеска, согласно теории, получается пропорциональность интенсивности квадрату количества электронов, что подтверждает и приближенный закон Брэгга.
В случае хлористого калия, исследованного Брэггом, числа электронов оказываются равными, если принять, что узлы решетка заняты ионами, и, наоборот, неравными, если считать, что узды решетки заняты атомами. Однако точность измерений, достигнутая Брэггом, была недостаточна, чтобы выявить получающуюся цри этом крайне незначительную разницу в числах электронов. Это различие должно обнаруживаться значительно более отчетливо у фтористого натрия] действительно, нейтральный атом натрия содержит 11, а атом фтора — 9 электронов, а иопы этих элементов содержат каждый по 10 электронов. Из рис. 44 видно, что если в кристалле хлористого или фтористого натрия выбрать плоскости решетки в направлении граней октаэдра, то каждая такая плоскость окажется заполненной атомами только одного рода. Представим себе, что в проведенных (на рис. 44) плоскостях заполнена только поло
* Как известно, это явление используют в ультрамикроскопии,
*• Подробнее об этом см. конец главы.
16 г. Реми
242
Глава 7
вина, скалам, ионами яатрия, и на экране, куда попадают после рассеяния рентгеновские лучи, отметим какое-нибудь определенное место, где лучи взаимно пе погашаются.: тогда для этого случая должно иметь силу условие, выражепноо уравнением (1) на стр. 233, л котором а соответствует расстоянию между плоскостями, ааиолнопиыап ионами Na+. Теперь представим себе, что между этими плоскостями вдвинуты плоскости, заполненные ионами фтора. Тогда отмеченное на экране место окажется освещенным только в том случае, если в уравнении (1) п случайно окажется четным числом: в противном случае это место будет па экране темным, Taj; как если уменьшить d наполовину, то, оставляя остальные входящие в уравнение (1) величины неизмененными, придется уменьшить в два раза и и. Но если п было нечетным, то его половила уже больше пе окажется целым числом и, следовательно, не будет выполнено условие освещенности. Таким образом, в общей сложности половина интерференций, вызываемых рассматриваемыми плоскостями отражения, погасягся. Совершенно другая картина должна получиться, если предположит!,, что эти плоскости заполнены попеременно атомами натрия и фтора, которые преломляют рентгеновские лучи одинаково сильно. В этом случаи в соответствующих местах уже пе может получиться полного затухания, а только ослабление интенсивности света. Дебай нашел, что одпа определенная, неизменно встречающаяся для данного вида решетки Линия совершенно Отсутствует в спектре решетки фтористого натрия, и сделал из этого заключеггие, что узлы этой решетя'н зап о л попы ионами. То же было обнаружено Герлахом и Паули в случае окиси магния.
Можно принять, что в такого рода решетках, особенно в таких, которые образованы веществами, диссоциирующими в водных растворах на цопы, атомы всегда присутствуют в электрически заряженном состоянии*.
То обстоятельство, что рассеяние рентгеновских лучей производится электронами, приводит к тому, что положительные водородные ионы (ядра атома водорода — протоны} вообще не дают интеференций, и поэтому их положение в решетке пе может быть определено непосредствен по. Кроме того, не удается установить положение атомов элементов с небольшим числом электронов вблизи от атомов элементов с большим числом элект]юнок, так как интерференции, вызываемые ими, слишком слабы. Иепо-средствсино определить пололсепис протонов в кристаллической решетке позволяет нейтронография (см. т. II), так как йейтрипы рассеивать должны не электроны, а атомные ядра.
Зависимость кристаллического типа от радиусов ионов. Как было показано Гольдшмидтом, во многих случаях для ионных рошеток можно заранее предсказать их кристаллический тип па оснований следующего правила: каждый ион вследствие электростатического притяжения стремится притягивать возможно большее число ионов с противоположным зарядом, на возможно более близкое расстояние. При этом предполагается, что ноиы представляют собой жесткие и практически недеформирующпеся шары с определенными (по Голтдшмидту) воличцпамй кажущихся радиусов (см, стр. 24). Однако попы ведут себя как не вполне жесткие шары, поскольку их кажущиеся радиусы несколько зависят от типа кристалла. Так, с переходом от типа CsI (координационное число 8) к типу NaCl (координационное число 6) связано уменьшение расстояния между нонами на —3%, а при переходе от типа NaCl к типу с координационным числом 4 межиопное расстояние уменьшается на 5—7%. На ионные радиусы незначительное влияние оказывают уменьшение температуры и давления. В остальном ноша ведут себя как жесткие шары. Поэтому, например, ион хлора С1_ имеет один и тот же кажущийся радиус, независимо от того, входит ли оп в состав решетки вместе с Г Г, К+, N«+ или Ш)+; в пределах сделанных выше ограничений кажущийся ионный радиус С1_ сохраняется И в его соединениях с Cs+ или, например, с 8г-ъ.
Расстояние между центрами ио по в в кристалле можно поэтому приблизительно предсказывать, суммируя тольдишидтовские кажущиеся радиусы соответствующих ионов. Большой точностью такого рода расчеты пе обладают, так как в них не принимается во внимание упомянутая выше зависимость кажущихся радиусов от координационного числа. Кроме того, при атом пе учитывается также влияние па кажущийся радиус определенного иона специфических особенностей других ионов, с которыми он образует соединение. Том не менее такое влияние, как это следует its теории, должно существовать (Полинг, 1927). Например, радиус рои a Na11 должен быть больше в том случае, есла он окружен одаовалептпыми, а не двухвалентными отрицательными попами.
* В качестве общего обозначения зарядов атомов или радикалов в дальнейшем будут использованы значки -|- и —; если же речь будет идти о гопах электролитов в водном растворе, то для обозначения зарядов будут применяться Значки ’ и ‘.
Строение кристаллов и рентгеновские лучи
243
Приведенные ионные радиусы. Межионные расстояния в кристаллах бинарных соединений можно более точно рассчитать при помощи таи навиваемых «приведенных к заряду 1 и координационному числу в ионных радиусов» (Zachariasen, Z. Kris-tall., 80, 137, 1931). Эти радиусы указаны в табл. 41. Межатомное расстояние г рассчитывается по формуле
И+Ф/kz
f2>
|/ Z^Z2
В этой формуле г1} и г" — приведенные радиусы, значения которых взяты из табл. 41, Zi и Z2 — валентности катионов и анионов; п и /KZ— коэффициенты, учитывающие
Таблица 41
Приведенные ионные радиусы по Захариасену (г0, А)
С4" 2,49 Н1* 1,36 Li1* 0,68 Be2* 0,55 В3* 0,42 G4* 0,38 №* 0,35
N3- 2,02 02- 1,76 pi- 1,33 Na1* 0,98 Mg3* 0,89 Al3* 0,79 Si4* 0,69 P3* 0,66 se* 0,64 Cl’* 0,63
S14- 2,97 рз-2,56 S2' 2,20 С11* 1,81 К.1* 1,33 Cas* 1,17 Sc3* 1,03 TH* 0,88 0,82 Gr3* 0,70 Mn’* 0,68
As3* 2,62 Se2- 2,29 Bri-1,96 Rb1* 1,48 Sr2* 1,34 Y3* 1,19 Zri* 1,07 Nb3* 0,98 Mo3* 0,90
Sb3* 2,77 Те3* 2,47 Р* 2,19 Cs1* 1,67 Ba-’* 1,49 La3* 1,30 Th4* 1,24
Ge4* 1,14
отталкивание между ионами, возникающее при большом сближении попов л обусловливающее зависимость межионного расстояния от координационного числа (KZ); п — так называемый показатель отталкивания, зависящий от электронной конфигурации (см. стр. 173). Значение /KZ в .зависимости от п и KZ приведено в табл. 42. Для KZ = 6 /KZ положено равным 1. Координационное число в значительной степени определяется соотношением приведенных ионных радиусов катиона и аниона. В общем
Таблица 42
Значение /Kz по Захариасену
Ti
KZ 5 6 7 8 9 10 И 12
2 0,793 0,834 0,857 0,877 0,890 0,902 0,911 0,919
3 0,862 0,889 0,906 0,919 0,928 0,936 0,942 0,947
4 0,920 0,936 0,946 0,954 0,959 0,963 0,967 0,970
8 1,067 1,053 1,044 1,038 1,033 1,029 1,026 1,024
12 1,160 1,126 1,104 1,088 1,077 1,008 1,061 1 056
244
Глава 7
имеет место следующее соответствие:
для =0,15—0,23 0,23—0,41 0,41—0,73 0,73-1,00 > 1,00
анчои
KZ= 3 4 6 8 12
несколько влияет на межионное расстояние. Вш
Соотношение радиусов также несколько влияет на межионное расстояние. Вш же этим влиянием практически можно пренебречь, за исключением тех случаев, пог^ анионы почти или полностью касаются один другого, что приводит к заметному расширению решетки. В этих случаях уравнение (2) не применимо для расчета межионных расстояний. При помощи приведенных мои пых радиусов удается, конечно, не непосредственно, а с учетом содержащихся в уравнении (2) расчетных факторов определять пространственные характеристики отдельных ионов в кристаллах. Расчет иеж-иониых расстоянии с использованием уравнения (2) можно проиллюстрировать на промере CaF3. Из табл. 41 следует: rj = 1,17, гЙ = 1,33; далее, имеем г; = 2, г3 = 1; для Саа+ (конфигурация аргопа) (см. стр. 173) п ~ 9, для F- (конфигурация неона 117
п = 7. Среднее значение; п — 8. Для соотношения 3 = -4= =0,88, приведенного ! 7*2 1»ОО
выше, находим координационное число, равное 8. Для координационного числа 8 и п = 8 до табл. 42 находим /KZ = 1,038. Межъядер яое расстояние соответственно (1,17+1,33)1,038 „
этому получается равным г = ------ ' — —2,35А. Это значение хорошо согла-
суется с экспериментально найденным г — (а/4)}^3 = 2,36 А (см. стр. 297). Сумма гольдшмидтовских кажущихся радиусов равна г = 1,06 + 1,33 = 2,39 А, т. е. получается значительно более сильное отклонение от экспериментально найденной величины. Однако гольдшмидтовские радиусы имеют определенное значение, так как они дают непосредственную картину пространственного расположения ионов в кристаллах, в то время как приведенные радиусы по Захариасену (за исключением случая одновалентных ионов в соединениях с координационным числом 6) позволяют получать лишь цифровые значения.
Поляризация. Значительные и не проста рассчитываемые изменения (кажущихся) ионных радиусов могут быть вызваны поляризацией. Рассмотренные до сих пор случав все откосятся к слабо поляризующимся ионам, т.е. к ионам с оболочками типа инертных газов.
Как уже было указано, под поляризацией атома или иона подразумевают вызываемое действием внешнего электрического поля, например поля соседнего иона, взаимное смещение положительных ядер и электронных оболочек. Под влиянием поляризации могут создаваться диполи или, если диполи уже существовали ранее, они могут усиливаться (см. также стр. 71),
Поляризация, по Гольдшмидту, приводит к образованию радикалов, например [NH4p, [АО3]-, [SOJ2“ и т. д., которые ведут себя в кристалле как обособленные структурные группы. В этих радикалах атомы или соответственно иопы особенно тесно связаны один с другим. Если радикалы образованы иолами с оболочками инертных газов, то межионные расстояния в них с удовлетворительной точностью можно рассчитать при помощи приведенных радиусов. В других случаях поляризация приводит к образованию слоистых решеток (см. ниже). Под влиянием более сильной поляризации возникают молекулярные решетки. Высркосимметричяые решетки, подобные упоминавшимся до сих пор {координационные решетки по Хунду), образуются в тех случаях, когда влияние поляризации имеет лишь подчиненное значение. В общем можно сказать, что поляризация понижает степень симметрии.
Радиусы и соотношения радиусов атомов и ионов, их зарядов и поляризус-мостей сущщтвенно ле только для структур, но и для Других свойств кристаллов. Этим занимается, как уже упоминалось, кристаллохимия.
Слоистые решетки. Слоистыми решетками называют такие, в которых атомы или ноны расположены слоями и часто так, что отдельные слои содержат только атомы или ионы одного характера, следовательно, они построены, например, попеременно из исключительно положительных и исключительно отрицательных ионов. Типичную слоистую решетку образует гидроокись лития, элементарная ячейка которой представлена па рис. 49, а. Для более ясного представления слоистой структуры на рис. 49, 6 нанесены узлы решетки нижпей половины элементарной ячейки, к которым добавлены узлы решетки, принадлежащие соседним ячейкам. Из рнсупКа видно, что каждый слой ионов Li* расположен между двумя слоями ионов ОН-, каждый из кото
Строение кристаллов w рентгеновские лучи
245
рых заполнен вдвое мепее плотно, чем слой ионов Li*, так что в общем на каждый нон LP, как это и следует из формулы, приходится один ион ОН~. Кислородные ионы в соединении LiOH так сильно поляризованы, что не только располагаются слоями без непосредственного взаимодействия с ионами лития, по даже два слоя, образован ные ионами кислорода или гидроксила, располагаются один за другим. Так как возникновение слоистых решеток связано с сильной поляризацией, то такие структуры образуются главным образом у соединений, в которых катион не обладает строением инертного газа. Они присущи пре имуществен по соединениям элементов побочных подгрупп периодической системы, ио встречаются и в главных подгруппах (папример,
Рис. 49,а и Л. Структура решетки гидроокиси лития (типичная слоистая решетка).
а=3:55А: с=4.33 А; ct=O,807 А.
LiOH) чаще всего в тех случаях, когда отрицательный ион сильно поляризуется вследствие особо малого радиуса положительного иона (в приведенном выше примере главным образом ионами Н*). Вещества со слоистыми структурами обычно обладают значительной спайностью, потому что силы притяжения, действующие между слоями в перпендикулярных к ним направлениях, мпого слабее сил притяжении, действующих впутри каждого слоя. В случае гидроокиси лития одни за другим располагаются два слоя одинаково заряжеппых ОН'-иоаов, которые, если бы пе сильная поляризация, должны были бы даже взаимно отталкиваться. В других случаях, например в случав решетки графита (см. стр. 463), можно заключить о существовании слабых сил притяжения в направлении, перпендикулярном слоям, из того, что в этом направлении межатомные расстояния значительно больше, чем в плоскости слоев.
Особенности строения кристаллических гидроокисей. Отчетливо выраженной слоистой структурой, Вроме LiOH, обладают многие другие гидроокиси, например Mg(OH)2, Са(ОН)2, a-Ze(OH)3, C’d(OH)2, Ni(OH)j, Со(ОН)2, Fe(OH)2 и Mn(OH)2, кристаллизующиеся по типу бруцита, а также и гидраргиллит А1(0Н)3. Одпако известен целый ряд гидроокисей, образующих ие слоистую, а явно координационную решетку, например высокотемпературные модификации NaOH и КОН (тип каменной соли), e-Zti(OH)2 (cm. т. II), далее — изотипные, кристаллизующиеся кубические соединения Sc(OH)j и 1п(0Н)3, в которых каждый ион металла, октаэдрически окружен шестью ионами 0Н~; гидроокиси Y(O]J)3, La(OH)5 л гидроокиси лантанидов, в решетках которых ионы металла обладают относительно ионов ОН" координационным числом 9. В решетках типа Sc(OH)5 12 ионов ОН' (из которых каждые 3 принадлежа г к одному к тому же Мш(ОН)((-октаадру) занимают тройные углы икосаэдра. То обстоятельство, что в двух последних кристаллических типах определенное число ионов OFI" образует ограниченные группы, указывает на то, что в пих между ионами ОН- действуют силы, приводящие к образованию водородных мостиковых связей (см. стр. 32б) (Fricke R., Z. Naturforschg., За, 62, 1948).
В табл. 43 приведены значения «эффективного радиуса» иона гидроксила в кристаллических гидроокисях двух- и трехвалентпых элементов. Эффективным радиусом считают половину расстояния между центрами двух соседних ОН'-ионов, координированных различными ионами металлов. Эффективный радиус в общем уменьшается с уменьшением основного и возрастает с увеличением кислотного характера гидро окиси. Уменьшение эффективного радиуса гидроксила объясняется отчасти поляризующим действием катиона и, по-впдимому, влиянием водородных мостиковых связей.
246
Глава 7
Габлица 43
Эффективный радиус иола гидроксила в соединениях типа М(ОН)2, М(ОН)3, МО(ОН)
Гидроокись Эффективный радиус йена ОН-, А Гидроокись Эффективный радиус иона ОН , А
Са(ОН)2 1,68 Р-Вс(0Н)2 1,43
Mg(OH)2 1,61 e-Zn(OH)s 1,41
Ni(OH)2 1,55 у-А1(ОН)3 1,39
Cd(OH)-, 1,49 В(ОН)3 1,35
У(ОП)3 1,45 y-FeO(OII) 1,35
La(OH)3 1,45 a-FeO(OH) 1,34
i\d(OH)3 1,45 a-AlO(OH) 1,33
Это подтверждается, в частности, наблюдением Глейзера (Naturwiss., 40, 199, 1953 . что адсорбционные максимумы О1Т-колебапия в инфракрасной области спектра с уменьшением эффективного радиуса гидроксила пепрерывЕ смещцготся в сторону больших длин волн.
Яс
4
Я» 2
Строение решетки нитрата натрия. В качестве примера того, каким образом радикалы* могут выступать в кристаллах в качестве структурных групп, рассмотрим строение решетке нитрата, натрия NaNOs. Это соединение, подобно известковому шпату, кристаллизуется в тригональных ромбоэдрах. Кристаллическая решетка его приведена на рис. 50. Из рисунка видно, что кислородные атомы тесно группируются вокруг атомов азота, образуя с последними структурные единицы (группы). Этот тип строения, обнаруженный впервые при исследовании известкового шпата, называется поэтому обычно «типом известкового шпата» (кальцита).
Радикалы, выступающие в качестве особых структурных групп кристаллов, например МО3-ионы, могут, если опи обладают достаточно малым моментом инерции, вращаться вокруг своих осей даже при температурах, весьма далеких от температуры плавления. Это следует как из изменений удельной теплоемкости в зависимости от температуры^ так и из рентгенографпче-сйих данных. Явление вращения наблюдается как для радикалов, так и для молекул, включенных в решетку в качестве ограниченных структурных единиц. В NaNO3 ион йО3 при 150° начинает колебаться около своей оси (рис. 50 — вертикальная ось). При 215° колебательное движение становится столь сильным, что атомам О исходя из рептгеиоструктурных данных уже
невозможно приписать определенные положения в решетке, а при 275й все ионы находятся уже в полном вращении, Для радикалов с меньшими моментами инерции, например NHJ, вращение начинается л прц более низких температурах. Например, в галогенидах аммопия попы вращаются уже при температурах намного ниже 0°. Нитрат калия KNOj образует слоистую решетку типа арагонита. В ней каждый слой из ионов К* расположен между двумя слоями (вдвое менее густо заполненными)
© Na
[’ и с. 50. та натрия вого 6,065 A, а=Д7*14-'
п—A; m=l:dl.i.
Решетка яитра-(тип ипвестко-шпата j.
* Подробнее О понятии радикал и структурная группа см, гл» Н»
Строение кристаллов и рентгеновские лучи
247
ионов ТЧОз таким образом, что над NOj-cnoeM располагается на кратчайшем расстоянии К'-слой, затем опять пи кратчайшем расстоянии — еще один IN Од-слой, а затем на значительно большем расстоянии — один NO 3-слой, над которым непосредственно располагается ближайший К '-слой и т, д. NO^-радикады и в этой решетке, очевидно, ведут себя как структурные единицы.
Двойные соли. Если из раствора шт расплава кристаллизуются две соли в простом стехиометрическом соотношении, образуя при атом особую решетку, то говорят о двойной соли.
В качестве примера строения такой соли укажем двойной сульфат калия и лития
KIJ<SO4) (рис. 51). И в атом случае в кая группа, а именно группа [SO4]'i-.
Двойной Сульфат калия н лития относится к так называемым сме-шанн-ым еемм: Этим термином называли соли, которые образуются при одновременном насыщении многоос-печшой кислоты несколькими основаниями или много основного основания несколькими кислотами. В структурном отношении эти соединения не" отличаются от обычных двойных солей, наирпмер от солей типа каряалита (ср. стр. 216).
Образование смешанных кристаллов л изоморфизм*. Если из какого-либо раствора пли расплава кристаллизуется два вещества с образованием общей кристаллической решетки в со-отношенин, непрерывно изменяющемся в определенных пределах, то в таксы! случае гово
решетка самстаяте.чьно существует структур-
Р ис. 51. Решетка двойного сульфата лития и калия.
о=&,13 A. с=8,60 А.
рят о возникновении смешанных кристаллов. Образование различными вещее тиа дги смеша н-
ных кристаллов, как правило, наблюдается только для таких веществ, которые и порознь кристаллизуются в одинаковых формах. Поэтому рассматриваемое явление наз!.пзают изоморфизмом (too; — одинаковый, роофг; — форма). Сущность изоморфизма и образования смешанных крисяаллоп заключается в том, что изоморфные вещества могут в кристаллической решетке взаимно замещать одно другое. Там, в кристалле хлористого калия ион хлора может быть постепенно замещен ионом брома, причем устойчивость кристаллической решетки от этого не изменяется; свойства хлористого калия в образующихся таким образом смешанных кристаллах непрерывно переходят в свойства бромистого калия. Совершенно неограниченная смешиваемость, представляющая собой наиболее высокую степень изоморфизма, встречается, однако, редко, в большинстве случаев наблюдаются более пли менее значительные разрывы смешиваемости. Особенно часто
это наблюдается в том случае, когда два смешивающихся вещества кристал-
лизуются каждое отдельно в различно построенных решетках или когда
параметры их решеток существенно отличаются.
* В русской литературе вместо термина «смешанные кристаллы» более применим термин «твердые растворы».—Ира л. ред.
248
Глава ?
Главные условия для 'образовация Тдвумяfвеществами смешанных кристаллов в значительном интервале составов следующие: 1) химическое сходство, 2) одинаковая валентность, 3) приблизительно одинаковые атомные или ионные радиусы.
При выполнен и и этих условий возможно дакже и замещение элементарных ионов радикалами.
Явления изоморфизма и образования смешанных кристаллов не вполне соответ ствуют одно другому. Обычно понятию изоморфизма придают более широкое вначепгг по сравнению с понятием образования смешанных кристаллов, считая изоморфными также и такие кристаллы, которые, как, например, известковый шпат и нитрат натрия, обнаруживают столь значительную аналогию в Строении, что кристаллы одного пл этих соединений могут продолжать ориентированный рост в насыщенном раствор:? другого, пе будучи, однако, в состоянии образовывать с ним смешанных кристаллов.. С другой стороны, обычно но считают изоморфными такие вещества, которые, мак, например, NaCl и КС1, несмотря на совершенно одинаковое строение кристаллических решеток, вследствие различия размеров этих решеток очень ограниченно образуют смешанные кристаллы; еще с меньшим основанием можно считать изоморфными такие вещества, которые (например, NaCl и Mg О) обнаруживают только сходное строение кристаллов.
В противоположность двойным солям смешанные кристаллы, как правило, не обладают собственным, свойственным им и отличным от составляющих их компонентов типом решетки. Однако известны также и такие случаи, когда это явление имеет место.
Изотппияи антянзотипня. Если два кристаллических вещества, напрймер NaCf. и MgO, кристаллографически построены совершенно одинаково, т. е. образуют кристаллическую решетку одного типа, ио не могут быть признаны изоморфными в обычном смысле этого слова, то их называют (по предложению Ринне) йзотиппыми.
HsomunWMu. вещеснгвдлш называются такие вещества , которые и.иеюгн. аналогичный состав и одинаковую кристаллическую структуру, но не- способны — или способны в очень ограниченной степени — к образованию смешанные кристаллов.
Вещества, имеющие одинаковую кристаллическую структуру, но в которых анионы и катионы как бы «обменены местами», называются антии^отипными. Например, окисел щелочного металла Ме2О антиизотицеп плавиковому шпату CaF2, т. е. обладает решеткой типа решетки CaF2, в которой на месте отрицатель пых ионов фтора расположены положительные ионы щелочного металла, а на месте положительных ионов кальция — отрицательные ионы кислорода.
Пзотипня и антлизотипия у бинарных соединений очень распространены.
Структура гидратон’газов. «Гидратами газов» называют кристаллические двойные соединения с водой, образованные химически насыщенными и потому находящимися при обычных условиях в газообразном состоянии молекулами — Аг, Кг, Хе, Rn, СН4, СН3С1, С02, N2O, SO21 H2S, С1а, Вг2. Речь идет об очень неустойчивых соединениях, существующих благодаря действию ванде рв аал ьсовых сил (см. стр. 127). Хотя вацдерваальсовы силы сами по себе не способны вызвать агрегацию молекул в определенных числовых соотношениях, гидраты газов имеют все же определенный стехиометрический Состав, который обусловлен пространственными соотношениями, в кристаллических решетках гидратов газов. Структура гидратов газов была объяснена главным образом Штакельбергом (1949—1952). В их кристаллических решетках молекулы Н2О располагаются так, что между пимп появляются закономерно расположенные пустоты, в которые могут внедряться молекулы других веществ, например-Ат или Кт.
В зависимости от размера молекул внедрившегося вещества возникают два типа-решетки. В одном кубическая элементарная ячейка (а =12 А) содержит 46 молекул Н2О, которые окружают 6 больших и 2 маленькие пустоты (диаметр 5,9 и 5,2 А). Элементарная ячейка другого типа (а = 17,2 А) содержит 136 молекул Н2О с 8 большими и 16 значительно меньшими пустотами (диаметр 6,8 и 4,8 А). Какой состав получается для гидратов газов в зависимости от того, только ли большие или все пустоты оказываются занятыми, показывают следующие примеры: 6Вг2-46Н2О или Вг2-•72/,Н20; 8Кг-46Н2О или Кг Н2О; 8СНС13• 136Н2О или СНС13.17Н2О; 8CHCV • 16Н28 13бНао или CHC13-2H2S-17H2O (так называемый двойной гидрат). Состав-гидратов газов, определяемый па основании их структур, в пределах ошибок применявшихся методов совпадает с составом, установленным ранее аналитическим путем.
Строение кристаллов и рентгеновские лучи 249*
Гидраты небольших молекул кристаллизуются по первому типу (гидраты газов в узком смысле итого слова), гидраты больших пип тяжелых молекул — по второму типу (гидраты жидкостей, так как вещества, построенные из больших или тяжелых молекул при нормальных условиях, — обычно жидкости). В обоих структурных типах каждая молекула, включенная в пустоты, окружена большим числом молекул Н2О (28, 24 или 20 в за ввей мости от величины пустоты). С устойчивостью этого чрезвычайно высокого по отношению к ПЕО координационного числа связан тот факт, что гидраты газов этого типа образуются только такими веществами, молекулы которых либо еовсе не обладают поляризующим действием,либо поляризуют очень слабо. Сильно поляризующие молекулы, например NH3 и HCf, образуют гидраты совсем другого типа (см. стр. 658 и 844). Их состав зависит от индивидуального характера соответствующих веществ, в то время как состав гидратов газов указанных типов зависит только от величины молекул и летучести вещества-гидратообразователя и одинаков, для химически совершенно различных веществ (подробнее об условиях образования гидратов газок см. Stackei berg, Naturwiss., 36, 327, 1949).
Большинство гидратов газов устойчиво только при низких температурах или при высоком давлении (см. табл. 44). Замечательно, что их стабильность повышается посторонними газами, находящимися под высоким давлением, например О, и И2. В 1897 г. это паб л го да л еще Виллард, который на этом основании предложил способ получения гидрата иода. Если нет никакого постороннего газа, тот этот гидрат вообще не образуется. Наоборот, под давлением кислорода 330 ат он устойчив вплоть до -}-8о. Нак установил Штакельберг, гидраты газов, например гидрат брома, могут включать в решетку заметные количества О2 и N2. Водород поглощается в мепьшей степени. Так, гидрат брома, распадающийся иод давлением собственных паров при 6,2°, устойчив под давлением кислорода 150 ат еще при 20°, а под давлением водорода 200 ат — только при 9°,
Пространственная химия вегпеств, построенных не пз ионон, В кристаллах с коордппацнонной решеткой, построенной из ионов с конфигурацией инертных газов, в большинстве случаев с удовлетворительной точностью можно рассчитать межионные расстояния, используя приведенные ионпые радиусы по Захариасепу, так нак в агах сзунанх, как правило, поляризационные воздействия не так сильны, чтобы существенно влиять на мсткиоппые расстояния. Но чем значительнее оказывается влияние поляризации, тем мепее удовлетворительным становится этот метод расчета. Межатомное расстояние в слоистых решетках*, называемых Гольдшмидтом координационными решетками несоизмеримого типа (см. стр. 26), можно с хорошим приближением рассчитан, из атомных радиусов, определяемых из атомных решеток свободных элементов. Так как до спх пор нет теоретического обоснования этого эмпирического способа определения, пе следует слишком распространять область его применения. Еще меньшее значение имеют данные, выражаемые формулами для установления соотношений межатомных расстояний в чисто гомеополяр пых соединениях и радиусов атомов элементов, из которых построены эти соединения, в атомных п полных решетках. Для такого рода предположений большое значение могут иметь удельные пли мольные объемы соответствующих соединений в твердом состоянии.
Мольный объем кристаллического вещества с известной Структурой решетки получают при отнесении объема элементарной ячейки к одному атому (или иону) и умножении па общее число атомов (или попов), содержащихся в молекуле этого соединения, ина число Авогадро ДГ t**. Поскольку величина элементарной ячейки и число атомов в ней предсказывается при помощи ионных радиусов ***, а тот метод приводит к наиболее точным значениям мольных объемов. Область применения этого метода,
* В слоистых решетках, построенных из ионов с электронными конфигурациями, подобными конфигурациям инертных газов, расчет межатомных расстояний при помощи приведенных радиусов дает еще удовлетворительные результаты, например расчет для ВеО (см. стр. 290) дает Be «—> О = 1,64 А; найдено 1,65 А.
** Например, элементарная ячейка NaCl, объем которой а3= 5,6283-К)-24 си3, содержит 4 4 = 8 ионов (см. стр. 238). Общее число ионов в молекуле NaCl равно 2.
5 628s-10-24.2-6 (РЗ-Ю23
Следовательно, мольный объем равен —---------—-----—-----— = 26,85 слЮ.
О
*** Основываясь на соотношения rj/r3, можно с большой вероятностью предсказать координационное число, а часто также и тип решетки. Значение предсказания межиопиых расстояний при помощи приведенных ионных радиусов не только в возможности предсказания мольных объемов (которые для подобных соединений в большинстве случаев известны пз измеренных плотностей). Оно особенно важно для облегчения рентген о структур пых определений — предсказания ожидаемых межплоскост-ных расстояний, гак называемых параметрон решетки.
250
Глава, 7
Таблица 44
Теплоты образования, температуры разложения п давление разложения гидратов газов
Теп ..io та образования отнесена к 0,= и к реакции 1 моля газообразного вещества — гидратсюбразователя с жидкой водой
Гидрат
к- Тур.1ЫЙ TJ1H число молрй Н»О
рас:-чд-тя ио из Структур найдено
тешюта a<i-разоваш-тяп ккал'иоль темпера-тууя. У £13“ лощения при I <imr °C даглеиис разложения при (]°
Темоера-.Гипраиюбра- ; тури низа ват ель иениа, °C
Ат - 1SG Тип 1 53/4
сн4 -164 ft ft %4
Кг -153 ft ft 53/4
СГ4 -128 ft 4'4
Хе -107 ft ft 5®/4
с2н4 -104 ft ft 53/4
N.,0 -89 ft 5+.
с,н6 -88 ft ft 5+4
РПз -87 » ft 5g/4
с2н2 -81 » ft 53/4
СО г -79 * ft 5Я/,
СЯ3Р -78 ft 4*
п,я -61 ft •4'4
АзН3 53/4
С,Н8 —45 ft ft 53A
Н2йе -42 ft ft 53/4
«2 -34 ft ft 53/4
c2H0F -32 ?> ft 53/,
СПХ1 - 24 i) ft •44
so2 - 10 ft ft 55/4
СНяВг +4 ft ft 7а/з
С 10а + 10 ft ft
c2il,ci + 13 Тип II 17
(.щНкПг + 38 ft ft 17
СН..С1а +42 ъ ft 17
епд +43 ft ft 17
ch,chgj2 +57 ft ft 17
Bru +59 ft I 7^/3
СИСЬ +61 ft 11 17
-6 13,3 -42,8 105- am
-6 14,5 -29,0 26 >i
5,7 13,9 —27,8 14,5 »
—6 16, 7 —3,4 1,5 »
-6 15,3 -13,4 5,5 »
-6 14,8 -19,3 10,0 »
5,8±0,5 15,0 -15,8 5,2 »
5,9 16,4 -6,4 1,6 »
5,7 15,2 -15,4 5,7 »
-6 14,6 -24 12,3 »
~6 — — 2,1 4
5,7 16,3 +0,35 731 .w.k
— — + 1,8 ‘ 613 »
— — 0 760 i>
5,9 16,8 +8 3i6 »
5,9+0,3 16,2 +9.6 252 »
-6 20,1 +3,7 530 i>
-6 18,1 +7,5 311 »
6,1+0,6 16,6 + 7,0 297 »
-8 19,5 + 11,1 187 »
-8 — + 15,0 ~160 »
-16 31,9 — 201 я
— — — —155 »
— 29 — 116 »
Л7 31,4 — 74 »
— — — —56 »
7,9±О35 19,6 — 45 »
— 30 — —45 »
однако, ограниченна. Он не применим как раз в таких областях, в которых предсказание удельных объемов соединений и смесей представляет для химика особый интерес, например в ендовах и в стеклах. Одпако для атпх веществ объемы можно рассчитывал, С хорошим ирнблтпкеиием па основе закона Бидьтца об at+umueuocmu объе.иос. Этот закон в первую йчещищ, применим для атомных и молекулярных агрегатов. Атомными и молекулярными агрегатами называют такие вещества, которые построены не из противоположно заряженных ионое (по крайней мере непосредственно), а из атомов (или положительных ионов и электродов) или молекул. Примерами атомных агрегатов в этом смысле являются металлы п их сплавы. К молекулярным агрегатам относятся жидкости, расплавы, переохлажденные жидкости и расплавы, стекла, а также молекулярные кристаллы.
Исследования Бильтца и его сотрудников показали, что мольные объемы атомных и молекулярных агрегатов при абсолютном нуле в общем с хорошим приближением можно рассчитать суммированием величин, характеризующих пространственные
Строение кристаллов и рентгеновские лучи 251
свойства отдельных атомов, названных Бильтнем инкрементами объема. Еще Копп (1855) пашел, что мольный объем жидких углеводородов вблизи температуры кипения можно рассчитать суммированием значений объемов, характерных для их отдельных составных частей. Эти значения объемов во многих случаях совпадали с атомпыми объемами, рассчитаптпами из удельных весов соответствующих элементов, по не всегда, так как па пространственные характеристики влияет тип связи.
Бильтц установил, что объемы твердых веществ, приведенные к абсолютному нулю (как неорганических, так и органических), можпо также рассчитать суммированием приписанных онр ед ел енпг>ш атомам или ионам объемных щшрементон. Объемные инкременты зависят от типа связи, т. е., другими словами, от типа соединения. Поэтому для кап от о-либо металла, например, они различны в его соединениях с другими металлами и с неметаллами. Поэтому (ши в общем также нс равны атомным объемам, вычисленным из удельных весов элементов. Объемные инкременты сильно зависят от зарядов соответствующих атомов. Например, атом К имеет инкремент 48,4, а ион К" — инкремент 16. Принимая во внимание влияние зарядов на сумму объемных инкрементов, можпо, как показал Бильтц, приближенно рассчитать мольные объемы таких соединений, которые образу хот типичххые ионные кристаллические решетки. Но все язе расчет с использованием объемных инкремептов в этих случаях приводит к значительно большим отклонениям от наблюдаемых величин, чем к случае атомных или молекулярных агрегатов. Поэтому в этой области для расчетов предпочтительнее применение ионных радиусов.
Есин рассматривать попы как совершенно жесткие- шары, то закон Голх.дпхмидта (см. стр. 36) об аддитивности радиусов будет выполняться совершенно строго. С другой стороны, положение об аддитивности объемов выполнялось бы строго, если бы атомы или ионы вели себя как совершенно пластичные частицы, т. е. могли бы соприкасаться своими поверхностям!! без каких-либо зазоров между нимп. В первом случае действие закона об аддитивности объемов было бы совершенно исключено*, ибо, хюли объем пропорционален (щ г2)3, он пе может бытх. одновременно пропорционален rj -|-Но гак как даже ионы е электронными конфигурациями инертных газон пе ведут себя как совершенно жесткие шары, действительные соотношения оказываются, средними между обойми аддитивными правилами, т. с. действительные мольные объемы лежат, как правило, между объемами, которые рассчитаны па основе законов об аддитивности радиусов и аддитивности объемов. Для соединений, кристаллическая решетка которых построена из слабо поляризующихся ионов, наблюдаемые значения объемов приближают х'я к значениям, вычисляемым на основе закона об аддитивности радиусов. Чем большую роль играет поляризация, тем больше отклоняются рассчитанные по этому закону значения от наблюдаемых, и опыт показывает, что с ростом поляризации они приближаются к тем, которые рассчитаны на основании закона об аддитивности объемов, т. е. суммированием инкрементов объема. Расчет молекулярных объемов из пространственных инкрементов становится ясным па следующих примерах. (Таблицы инкрементов объема см. БильтЦ, а также Z. anorg. Chem., 223, 321, 1935 и 234, 253, 1937.)
Пример 1. Мольный объем- интерметаллических соединений. С точки зрения пространственного строения интерметаллические соединения подразделяются па два класса; в Первом инкременты объема металлов равны или близки к атомным объемам; во втором образование соединения происходит со значительным сжатием, в результате которого у компонентов только часть их атомных объемов играет роль объемных инкрементов. К первому классу принадлежит со единен Xie Mg2Pb. Атомный объем (при 0° К) Mg составляет 13,8, РЬ — 17,9; так как в этом случае объемный инкремент мощно принять равным атомному объему, молярный объем при 0° К равен 2-13,8 Ц- 17,9 = = 45,а (н ай депо 46,5). Ко второму классу принадлежат главным образом соединения, содержащие щелочные или щелочноземельные металлы, например NaPl)g. В соединениях Этого типа свинец имеет объемный инкремент 17,0; натрий — также 17,0. Мольный объем NaPb3 при 0° К равен 17,0 -L 3-17,0 = 68,0 (найдено 68,3).
Пример 2. Мольный объем- силиката. Пространственные инкременты Na 4 и Ог“ составляют по Бнльтцу приблизительно 6,5 и 11. Si14 имеет объемный инкремент 0. Мольный объем Na2Si2O5 при 0р К равен 2-6,5' -|- 5-11 = 68 (пайдено 72).
Пример 3. У дельный объем стекла. Положим аналитический состав стекла 6Na2O-13 SiOj. По Бильтпу О3- имеет в SiO2 значительно больший объемный инкремент, чем в других соединениях (включая силикаты), а именно 13,6. Для определения мольного объема силикатного стекла необходимо учесть, сколько в нем содержатся избыточного SiO2. Наиболее богатым кремневой кислотой определенным соедине-
* Если радиусы мало различаются, можно все же использовать закоп как приближенный.
252
Глава 7
нием Между Na2O и SfO2 является Na2Si2O5. Если вычесть на указанного выше состав-стек л а 0Na2Si2O5l остается 1 избыточная молекула SiO2. Рассчитанный мольный объе— оказывается равным 2-13,6 -|- 12-6,5 30' 11 = 435. Поделив его па paccaniannuij
молекулярный вес (1152,8), получают значение удельного объема при 0° К равным 0,377 Пересчет на комнатную температуру дает значение 0,383 (найдено 0,406). Нампой лучшее совпадение получают, если исходят не из объемных инкрементов отдельна ионов, а из эмпирических «пространственных факторов» для Na2Si2O5 (стеклообразного) и SiO2 (стеклообразного) — 73,1 и 27,2. В этом случае получают удельный объех
для ^Стекла —— 0,404 (чайдепо 0,406).
11о2,о
РЕНТГЕНОС11ЕКТРОГРАФИЯ
Тормозное излучение и собственное излучение. Как известно, рентгеновские лучи возникают, когда быстрые катодные лучи, т. е. электроны с очень большой скоростью движения, встречая на своем пути твердые вещества (антикатод в рентгеновской трубке), внезапно задерживаются, как бы тормозятся. Максимальная энергия испускаемого при этолг отдельным электроном луча определяется соотношением
е=еГ=Лу, 1 (3>
где в — энергия; е — заряд частицы; V — потенциал разряда рентгеновской трубки;. Л — постоянная Планка; v — волновое число (частота) рентгеновского излучения..
Так как в общем случае частицы катодных лучей не отдают всей своей энергии в одном акте торможения*, то для большей части излучаемой
Рис. 52. Распределение энергии в спектре «белого» рентгеновского излучения.
энергий частота колебаний оказывается более низкой по сравнению с рассчитанной по формуле (3). Поэтому для совокупности лучей максимум интенсивности излучения находится несколько ниже коротковолновой границы, получаемой из этого уравнения. Благодаря этому обстоятельству распределение интенсивностей представляется в виде кривой, приведенной на рис. 52. Эта кривая близка к кривой распределения энергии в спектре света, излучаемого раскаленным твердым телом; одиако в отличие от последней кривая рис. 52 имеет в соответствии с уравнением (3) в коротковолновой области (области высокой частоты) резкую границу. Эта граница, а вместе с ней и максимум
интенсивности, как это непосредственно следует из уравнения (3), с возрастанием потенциала разряда сдвигается в направлении коротковолновой области. Распределение интенсивности тормозного излучения, таким образом, не зависит (в основном) от вещества, из которого сделан антикатод; оно определяется разрядным потенциалом трубки.
Если, постепенно повышая потенциал разряда, смещать границу все более в область высоких частот, то в некоторый вполне определенный момент (в зависимости от материала антикатода) наблюдается появление чрезвычайно сильного излучения. Интенсивность последнего резко отличается от обычного хода кривой распределения интенсивностей и сохра-
* Помимо этого, значительная часть энергии катодных лучен превращается в теплоту.
Строение кристаллов и рентгеновские лучи
253
яяется также и после того, как максимум интенсивности уже перешел через длину волны, соответствующую этому излучению. Такое излучение называют характеристическим рентгеновским излучением, или собственным излучением. Это название указывает на то, что длина волны этого излучения определяется материалом антикатода (собственно говоря, его поверхности).
Баркла наблюдал это явление еще до открытия явления интерференции рентгеновских лучей. Он установил (1905), что частота собственного излучения пе изменяется с возрастанием разрядного напряжения. Это заключение относительно частоты оц сделал ва основании проницаемости, т. е. жесткости рентгеновских лучей, принимая ее пропорциональной частоте. Возможность более точных определений длин воли при помощи интерференции рентгеновских лучей очень скоро привела к важному результату, а имелпо Мозли в 1913 г. открыл простую зависимость между частотой собственного излучения элементов и их порядковыми номерами.
Закон Мозли. При спектральном разложении характеристического
рентгеновского излучения
состоящая из очень небольшого числа линий различной интенсивности. Эти линии обозначают (большей частью в порядке их интенсивностей) буквами греческого алфавита: а, £, у и т. д. Наиболее интенсивная линия а соответствует наибольшей длине волны (точнее говоря, a-линия состоит из двух линий <х и а'; другие линии так же можно разложить дальше). Большинству элементов соответствует несколько определенных серий, которые, однако, лежат в совершенно различных областях спектра. Их обозначают буквами К, L, М ит. д.; Отдельные же линии соответственно обоз-
при помощи кристалла получается серия,
//-> era-
II III г.» 11 ' ~ - —
I i I । I । '__________1 ___1- I____: 3___।
1,2 1.3 1,4 1.5 1,6 1,7 1.8 1,9 2,0 2,1 2.2 2.3 2.4 2,5 2,6 2.7 2.8 Л.)
Рис. 53. Рентгеновские спектры элементов с порядковым номером от 22 до 30 (линий Я-серии).
начаются Ка, La, L& и т. д. Серии различаются’ расположением в них линий (структурой). Наоборот, одна и та же серия имеет для различных элементов аналогичную структуру (рис. 53). Для большинства элементов в области спектра, доступной для измерения длины волн путем дифракции на кристаллах, обнаруживаются только одна или две серии, а именно для наиболее легких элементов (начиная с натрия) серия К, для более тяжелых, кроме того, еще серия L. Для элементов с очень высокими атомными весами появляются еще другие серии (серия М, N, О и Р). Для одного и того же элемента Л'-серия всегда лежит в области более высоких частот, чем Л-серия; линии Z-сериц соответствуют более высоким частотам по сравнению с линиями Af-серии и т. д.
В 1913 г. Мозли установил, что для аналогичных линии, например для АД-лишш, при переходе от одного элемента к другому частоты возрастают с увеличением порядкового номера совершенно одинаково.
254
Глава. 7
Для аналогичных линий характеристических рентгеновских спектр., тстота возрастает пропорционально квадрату порядкового номер.: уменьшенного на некоторую постоянную величину (закон Мозли).
Имеем, следовательно, v r y"v
--^-^const, или М~-а
где v — частота собственного излучения элемента с порядковым номером Z, а — нек... тор ан величина, примерно одинаковая для аналогичных линий различных элемента
const,
Рис. 54. Зависимость частоты собственного излучения от порядкового номера (закон Моэли).
Рис. 54 иллюстрирует, с какой точностью выполняется эта зависимость для ff-серии и даже для каждой липни этой серии. На рис. 54 порядковые номера Z нанесены по оси абсцисс; ординатами взяты вместо корпсп квадратных из частот корни квадратные из обратных значений длин. волн. Обратные значения длин волн называют волновыми числами. Таким образом, например, волновое число vr линии с длиной , , i v
волны А или соответственно с частотой v определяется выражением v = — = — , л с где с — скорость света. В соответствии с уравнением (4) точки пересечения ординат и абсцисс на рис. 54 должны лежать на одной прямой, что фактически и выполняется с очень большой точностью. Незначительное искривление этих линий зависит от того, что число а в уравнении Мозли не является в точности постоянным.
Строение кристаллов w рентгеновские луча 255
Закон Мозли является прежде всего-блестящим подтверждением правильности установленных на основании, периодической системы порядковых номеров. Далее, па его основания представилась возможность проверить в сомнительных случаях, правильно ли, например, были классифицированы трудно различимые между собой химические элементы группы лантанидов и имеются ли достаточные основания признавать некоторые из них самостоятельными элементами.
На основании характеристического рентгеновского спектра удается сравнительно легко и надежно идентифицировать вещество и в таких случаях, когда вследствие слишком больших трудностей, связанных с его получением в чистом виде, методы химического анализа не дают результата, а оптические спектры слишком сложны. На этом основан рентгеноспектральный анализ, к обсуждению которого теперь следует перейти. В дальнейшем изложении, в последнем разделе, еще будет рассмотрен закон Мозли, сыгравший столь важную роль в развитии периодической системы и вскрывший сущность зависимости между атомными весами и порядковыми номерами. Однако прежде необходимо предварительно ознакомиться с теми выводами относительно строения атомов, к которым приводит изучений рентгеновских спектров и которые являются основ ан г, ед г для правильного понимания соотношений между атомным весом и порядковым номером-t- >
Рентгеноспектральный анализ. Для рентген о спектрального анализа чрезвычайно важно, что характеристическое рентгеновское излучение (приближенно) не зависит от того, в каком виде находится данный элемент ~ в свободном состоянии или в виде какого-нибудь соединения. В отношении обычного спектрального анализа ото условие выполняется лишь постольку, поскольку при высокой температуре пламени или электрической дуги соединения разлагаются. Но оптические спектры элементов совершенно отличны от спектров соединений, образуемых этими элементами.
Для получения характеристических рентгеновских спектров достаточно нанести на охлаждаемый водой антикатод незначительное количество исследуемого вещества, поместить антикатод в специально сконструированную рентгеновскую трубку, наложить высокое напряжение и сфотографировать спектр. Рентгеновский анализ не только качественно указывает па присутствие или отсутствие данного вещества, но дает возможность на основании сравнения интенсивностей приблизительно определить его количественно.
Установленная Мозли зависимость позволила также рассчитать рентгеновские спектры еще пе открытых элементов. Это привело к тому, что вслед за открытием закона Мозли последовало открытие новых химических элементов. Основываясь на законе Мозли, Ховеши (Hevesy) и Костер (Coster) в 1922 г. открыли элемент гафний, а в 1925 г. Ноддак (Noddack) и Текк (Таске) — элемент рений (подробнее см. т. II).
Но главным образом и совершенно неожиданно рентгеноспектроско-пия расширила и углубила сведения о строении химических атомов н об уровнях энергии в них.
Собственное излучение и строение атомов. Длины волн собственного излучения элементов, относящегося к рентгеновской области, несравненно меньше длины волн обычпого оптического спектра. Поэтому для их измерения пользуются специальными единицами, называемыми Х-едини-
256
Глаеа 1
щами. (Рентген, открывший рейтгеповские лучи, предложил для пт-^ названые Х-лучей, удержавшееся до настоящего времени в иностранна литературе.) 1Х-единица (Х-Е) = 1,00202-Ю-11 см (1/сХ=1,00202 А). Д~: наиболее длинноволновой A-рентгеновской линии калия, Ка„ длина соответ ствующей волны % = 3737,1 Х-единиц, т. е. эта волна более чем в тысячу раз короче, чем волна линии калия, лежащей в фиолетовой части спектра С возрастанием порядковых чисел длины волн рентгеновских линий пр.: должают, согласно закону Мозли, быстро убывать.
Причина возникновения линий рентгеновского спектра та же, чт. и причина, вызывающая образование обычных световых волн: ею может быть только «перескок» электронов с более высокого уровня энергии ет. более низкий. Каким образом можно истолковать возникновение характеристического рентгеновского излучения, можно понять, если применить формулу Мозли к отдельным линиям в несколько уточненном вид:; что и было проделано Мозли. Согласно закону Мозли, для линии Ас имеем:
v = Ra>(Z—1)а.з/4 (J?m—константа Ридберга),
1)3(1/^—V23). - С
Эта формула вполне соответствует формуле Бора [уравнение (13) в гл. 3|* только в ней величина Z заменена на Z — 1. Это следует понимать в тог; смысле, что на электрон действует не .весь заряд ядра Z — Е/е, но. толькс часть его, в то время как остальная часть заряда ядра как бы экранируется действием другого, уже связанного электрона. Поэтому постоянную а в уравнении Мозли (в приведенном случае а =? 1) называют константой экранирования, Уменьшенный на константу экранирования заряд ядра называют эффективным зарядом.
Из того что все без исключения элементы, у которых соответствующие области спектров доступны исследованию, обнаруживают присутствие К а-линии, непосредственно следует, что все элементы содержат определенный электрон, связанный в них одинаковым образом, и, следовательно, внутреннее строение в пределах сферы, соответствующей этому электрону, у всех элементов одинаково.
Из незначительной величины константы экранирования а для Аа-ли-нии в уравнении (4) следует, что этот электрон, двигаясь по своей внутренней орбите, непосредственно вращается вокруг ядра. Излучение Ка-линии, как показывает уравнение (5), в соответствии с теорией Бора возникает при перескоке электрона со второй на первую квантованную орбиту.
Для £а-линии М.озли установил следующую зависимость:
(as7,4i.
По аналогии со сказанным следует сделать вывод, что эта линия излучается электроном, перескакивающим с третьей на вторую квантованную орбиту, и что на него действует заряд ядра, экранированный приблизительно на 7,4 единицы. Этот электрон и соответствующая ему сфера также являются общими для всех элементов, за исключением самых легких.
* Так как
Ai
то величина йгав приведенной выше формуле должна быть равна
с X в уравнении (13), приведенном в гл. 3
Строение кристаллов и рентгеновские лучи
257
Рентгеновское излучение указывает, таким образом, что атомы тяжелых элементов обладают общей для них всех последовательностью уровней энергии. Их обозначают как К-, L- и т. д. уровни, смотря по той линии (А’а, La и т. д.), которая излучается при перескоке электрона непосредственно с более высокого уровня энергии на данный уровень. Остальные (более высокочастотные) линии данной серии возникают, когда электрон перескакивает не с непосредственно соседнего высшего уровня, а с какого-нибудь более далекого, обладающего еще большей энергией. Этому соответствует и сравнительно меныпая интенсивность этих линий, поскольку такие перескоки электронов происходят реже.
Естественно сопоставить слоистое строение атомов, содержащих дискретные, повторяющиеся электронные оболочки, с их положением в периодической таблице по отношению к инертным газам и с их оптическими спектрами. Спектры и химические свойства щелочных металлов указывают на то, что у каждого последующего иа них имеется новая «оболочка», или новый уровень энергии. Таким образом, предшествующий уровень энергии завершается каждый раз конфигурацией соответствующего инертного газа. В соответствии с таким представлением конфигурации каждого инертного газа следует приписать определенный уровень энергии: конфигурация гелия соответствует, следовательно, низшему уровню энергии, т. е. уровню К; конфигурация: неона — уровню L; конфигурация аргопа — уровню М и т. д. Таким образом, из рентгеновских спектров, если [как это сделано в уравнении (5)] представить их в виде термов для различных уровнем энергии, получаются следующие (главные) квантовые числа:
гелия
К-уро вонь п = 1
Оболочка неона
£-уровень
п = 2
Оболочка аргона
М-уровень
и==,-!
Оболочка криптона
Лг-уровепь
ft = 4
Оболочка ксенона
О-Уровень
п—5
Оболочка радона /'-уровень
На рис. 55 уровни энергии представлены в соответствий с рис. 19 в виде окружностей, а переходы электронов обозначены стрелками. Разница между рис. 19 и 55 заключается лишь в том, что окружности иа рис. 19 соответствуют возможным уровням, на которые электрон, обычно вращающимся вокруг ядра но внутренней орбите, может вообще подниматься, причем эти внешние уровни обычно остаются незаполненными; на рис. 55, наоборот, высшие уро ими, как’ правило, заполнены электронами. Окружности на рис. 55 не представляют самих орбит; по которым вращаются электроны; они только изображают соответствующие этим орбитам уровни энергии. Расстояния между ними па рис. 55 нанесены произвольно.
Собственное излучение возникает вследствие воздействия какого-то внешнего импульса; например, под влиянием ударов катодных лучей электрон сначала выбивается с внутренней орбиты. При этом он полностью отрывается от атома, так как высшие орбиты все уже заполнены Необходимая для этого энергия определяется выражением
где е — энергия уровня, с которого вырывается электрон. Образовавшаяся в оболочке, из которой был удален электрон, «дырка» тотчас заполняется каким-нибудь другим электроном, перескакивающим сюда с одной из соседних внешних оболочек. Если первый электрон был вырван из АГ-уровня, то образовавшаяся на его месте «дырка», как правило, запол-
17 Г. Реми
258
Глава 7
няется другим электроном из соседнего 4-уровня; при этом возникает Ка~линия. Однако вырванный электрон может быть замещен также и электроном из уровня М — тогда возникает А^-линия — или он замещаетсс электроном ив уровня N, с образованием Ку-линий. Последнее не может произойти у тех атомов, у которых па уровне АГ еще ие содержится электронов, т. е. (если отождествление рентгеновских уровней с оболрчкамп инертных газов правильно) у элементов с порядковыми номерами меньшими, чем у калия. Однако такие перескоки электронов не будут происходить в заметном количестве и в тех случаях, если на отдаленных
Р и с. 55. Уровни энергия атома и возникновение рентгеновских спектров (схема).
высших уровнях находится лишь незначительное число электронов. И действительно, А\-линия с достоверностью установлена для элементов только начиная с ванадия, имеющего 5 электронов на уровне IV.
Если переход происходит не с самого высокого уровня, но с какого-нибудь уровня, расположенного под ним, то в последнем в свою Очередь образуется «дырка», при заполнении которой произойдет перескок электрона с более высокого уровня. Поэтому с возникновением А'а-дннии связано возникновение не только других линий К-спектра, но и всех остальных L~, М-, А’- и т, д. спектров, появление которых связано с порядковым номером атома. Однако в то время как линии К$, Ку й т. д.^воз-никают всегда лишь вместе с линией Ка, спектр 4 может быть возбужден и помимо спектра К, спектр М — помимо спектров К и 4 и т. д., но 4-спектр не может возникнуть без одновременного появления спектров М, N и т. д. Эти закономерности были установлены уже вскоре после открытия рентгеноспектр оси опии и в 1914 г. истолкованы Косселем в том именно смысле, как они здесь изложены.
Строение кристаллов и рентгеновские лучи
259
Из изложенной выше теории в полном согласии с опытом следует, что для возбуждения рентгеновского излучения, соответствующего какой-ппбудь линии, недостаточно в отлично От периферийных оптических спектров сообщения атому извне такого количества энергии, которое бы отвечало частоте данной линии, но требуется большая энергия. Так, для возбуждения Ко-линии с частотой va недостаточно сообщить энергию ca = Ava, так как эта энергия была бы в состоянии перенести электрон только до Z-yровня, между тем как для возникновения этого излучения электрон необходимо поднять по крайней мере до периферии атома. Если требующуюся для этого энергию обоаначить через sA-, а напряжение трубки, которое необходимо приложить, чтобы сообщить отдельным частидам катодных лучей энергию efi- — через V^, то эту энергию можно определить из соотношения
^k = Ek = avk. (6)
Атому можно сообщить энергию не только при помощи катодных лучей, но и действуя напето рентгеновскими лучами. Если облучать атом «белым» рентгеновским светом, то он поглотит те лучи, энергия которых hv достаточна для поднятия 1 электрона с внутренней орбиты до периферии атома. Но для этих лучей имеет силу соотношение
s^e.K=tivK.
Те лучи, энергия которых меньше не будут при этом абсорбированы. Таким образом, получается граница абсорбции, частота которой определяется уравнением (6). Из уравпеаия (6) следует, что потенциал возбуждения линии Ка, или — что то же самое — К-серии определяется границей абсорбции соответствующей К~серии и наоборот. Поэтому энергию основной орбиты для А'-серии можно вычислить кай из потенциала ее возбуждения, так и из положения границы абсорбции. То же можно сказать и относительно других серий.
Как ужо было отмечено, при более точном исследовании оказалось, что линия Ка состоит нз двух линий. Это объясняется тем, что, строго говоря, существует нс один. L-уровень, а два, несколько различающихся своей энергией. В зависимости от того, с которого из этих двух уровней электрон перескакивает в образовавшуюся «дырку>> в К-уровне, образуются Ка1 или Ка^-линии. Аналогично уровням энергии, которые можно вывести из оптических спектров, и здесь каждому уровню, кроме главного квантового числа п. необходимо приписать еще и два побочных квантовых числа I и s или возникающее в результате их комбинирования внутреннее квантовое число /. Только таким образом удается представить все множество рентгеновских линий в виде термов. Точный анализ рентгеновских спектров показывает, что L-уровень может расщепляться до 3, ^-уровень — до 5, А-уровень — до 7, О-уровепь — до 5 и Р-уровень — до 3 «подуровней». Как показывает изучение рентгепоспектрограмм, в атомах всех элементов, следующих^ за радоном, встречаются следующие уровни энергии, общие всем этим атомам и характеризующиеся квантовыми числами, приводимыми в табл. 45.
У более легких’атомов заполнены, конечно, еще не все уровни. Как происходит детально заполнение уровней, будет рассмотрено в дальнейшем.
Слоистое в энергетическом отношении строение атомов, обнаруживающееся в структуре рентгеновских спектров, как уже упоминалось, очень важно для правильного понимания природы главных групп периодической системы. Знакомство с этим строением в той мере, в какой оно отражается в тонкой структуре рентгеновских спектров, обнаруживающей подразделение отдельных электронных оболочек, позволяет понять также и особенности Строения побочных подгрупп периодической системы, включая и группы лантанидов и актинидов.
17*
260
Г лае a 7
Таблица AZ
Энергетические уровни атомов, следующих за радоном
к-уро-веиь L -уровень 2И-уровснь
|| Л=1 п — 2 п=2 п=2 л = 3 п — 3 м = 3
Z=0 Z=0 l=i 1=\ Z = 0 Z = 1 1 = 1 Z = 2
i-s/2 /=Va } = ^2. 7=а/а /-3/2 } = :- •
Д'-уровень
сч о II II II Й я il II II . * 1-^ . t-J оГ Я OJ м II II II Z C-J Ift' .11 II II if" ' К Т и (:Г|
О-уровень Р-урОВСНь
TH О -уч II II |г сл ю 1 Т Т ii 1-^ Сд '-ы. 3 II II II Ю С1 .5^ л I.Q ич" II JI ,11, II 1Г || О Ct-гл"1 ' n Т 7,1 'ГГ i7 ft > н* о 1 :Ц
Порядковым номер и число зарядов ядра. Из уравнения Мозли (4*; для каждого элемента получаем некоторое число Z, которое, как уже было показано, идентично порядковому номеру, определяемому последовательностью элементов в периодической системе. Значение порядковых номеров, находимых из уравнения Мозли, заключается прежде всего в том. что они позволяют совершенно однозначно расположить химические элементы на основании их рентгеновских спектров. Так как расположение по порядковым номерам во всех случаях совпадает с расположением их в периодической системе, в основу которого в свою очередь положены химические свойства элементов, то определение порядкового номера на основании закона Мозли позволяет совершенно бесспорно указывать положение в периодической системе также и таких элементов, для которых иа основании остальных свойств и атомного веса этого прежде сделать не удавалось.
Определение порядковых номеров на основании закона Мозли ограничено лить в том смысле, что до сих нор пе удалось измерить характеристическое рентгеновское излучение для инертных газов и дм всех элементов с атомным весом ниже натрия. В отношений инертных газов это объясняется исключительно самой техникой определения; вещества, у которых должно быть возбуждено собственное излучение, должны сами быть в твердом состоянии, либо должны быть переведены в твердые нелетучие соединения, чтобы их можно было нанести на антикатод. Для инертных газов последнее совершенно исключено, а исследованию их в замороженном состоянии препятствует стьтьнос разогревание атгтнкатода бомбардирующими его катодными лучами. Для элементов с атомным весом ниже натрия (соответственно неона) измерение невозможно вследствие того, что соответственные частоты у этих элементов настолько низки (согласно уравнению Мозли, они быстро убывают с уменьшением порядковых номеров), что те кристаллические решетки, которые имеются в пашем распоряжении, уже не позволяют произвести измерение длин соответствующих волн. Тем не менее дня элементов От Li до О удалось все же измерить потенциалы возбуждения серии К, основы-
Строение кристаллов и рентгеновские лучи 261
ваясь на которых можно по уравнению (5) вычислить частоту границы абсорбции К-сер ни.
Для этих элементов Хольтсмарк (Holtsmark, 1923) обнаружил интересное дтл&онение от закона Мозли. Частоты границ абсорбции этих элементов распситожеии, согласно закону Мозли, на продолжении прямой, соединяющей аналогичные частоты для более тяжелых элементов, только в тех случаях (если частоты были определены экспериментально для соединений этих элементов), когда атомы этих легких элементов можно рассматривать в качестве отрицательно заряженных, например кислород в окиси меди. Нейтральные хае элементы с атомным весом ниже неона обладают слишком низким потенциалом возбуждения для К-спектра. Это объясняется тем, что условие возбуждения серии А", оказывающееся для элементов с высоким атомным весом всегда выполненным (а именно чтобы орбиты ближайших высших уровней анергии были полностью заполнены), в данном случае выполняется только тогда, когда атом заряжен, отрицательно. Если следующий более высокий уровень энергии (л = 2) еще заполнен не до конца, то, конечно, перескок электрона, вращающегося непосредственно вокруг ядра, на уровень с п = 2 возмояген и работа, соответствующая этому процессу, или отвечающая ей частота пе могут быть определены по закону Мозли. Это исключение из общего правила подтверждает закономерность и вместе с тем основанную иа этой закономерности теорию,
В то время как для более тяжелых элементов вплоть до лития потенциалы возбуждения соответствуют только границам абсорбции (следовательно, тем частотам излучения, которые атом может абсорбировать, ио которые он, согласно изложенному в предыдущем разделе, не может излучать сам), примыкающие абсорбционные границы спектров гелия и водорода (расположенные в ультрафиолетовой части спектра) соответствуют не только частотам абсорбируемого, по также и частотам испускаемого излучения. Следовательно, у этих элементов непосредственно вращающиеся вокруг атомных ядер электроны не находятся уже во внутренней сфере; они больше пе окружены другими электронами, находящимися на более высоких уровнях энергии. Таким образом, для этих двух атомов зависимость Мозли, Которая здесь опять замечательным образом оправдывается, распространяется н на область оптических спектров.
В предыдущем разделе (стр. 256 и сл.) было показало, что под числом Z в уравнении Мозли следует понимать число зарядов ядра' это число определяет число положительных зарядов, связанных с атомным ядром. Таким образом, зависимость, найденная Мозли, показывает, что число зарядов ядра последовательно возрастает от элемента к элементу, а именно на единицу при переходе от одного элемента к другому.
В нейтральном атоме положительный заряд ядра должен быть скомпенсирован таким же числом отрицательных зарядов, т. е. электронов, находящихся вне ядра. Число электронов в оболочках нейтрального атома равно заряду его ядра. Следовательно, для каждого атома имеет место соотношение: порядковый номер* = заряд ядра = число электронов в оболочках нейтрального атома.
Под «оболочками» атома понимают вес множество вращающихся около атомного ядра электронов. Их не следует смешивать с электронами так называемой внешней сферы атома, которые обозначаются как валентгияе электроны, так как благодаря им осуществляются валентные связи.
Порядковые номера и атомные веса. Атомный вес возрастает в общем приблизительно пропорционально порядковому номеру.
Это положение подтверждает то, что свойства элементов, являющиеся в действительности периодической функцией порядкового номера, приближенно кажутся периодической функцией атомного веса, В какой мере существует пропорциональность между порядковым номером и атомным весом, следует из рис. 56. Видно, что у легких элементов, включая Са, атомный вес приблизительно равен удвоенному порядковому номеру (за исключением водорода, атомный вес Которого равен порядковому номеру). После Са половили атомного веса возрастает быстрее, Чем порядковый номер. Возрастание атомного веса в отличие от возрастания порядкового номера происходит цеупоря*
* Порядковый номер также называют атомным номером.
262
Глава 7
доченно. В этом, следователи!о, причина неправильностей (ошибок), возникаем— при попытках расположить элементы в периодической системе по атомным
Порядковые номера или заряды ядер, а также атомные веса являюи атомными константами, не обладающими периодическим характером. Д^.
них весов элементов нанесены на оси ординат и отмечен и крестиками; Если бы поло вины атомных весов были бы точно равны порядко-вымйномерам, то они попадали бы на начерченную ломаную линию.
них существенно только ядро атома. Периодически изменяющиеся свойства связаны с оболочками атомов, и в первую очередь с наиболее внешними оболочками, занимаемыми валентными электронами. Поэтому периодичность наиболее отчетливо проявляется в валентности элементов.
Гл а в а 8
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Главная подгруппа)
ЩЕЛОЧНОЗЕМЕЛЬНЫЕ МЕТАЛЛЫ
Порядковый номер Элемент Символ Атомный вес Удельный вес Теркеря-тура плавления, °C Температура кипения, *С Удельная тейло^ы-кость валентность
4 Бериллий Be 9,013 1,86 1285 2970 0,540 II
12 Магний Mg 24,32 1,74 650 1100 0,242 П
20 Кальций Са 40,08 1,54 845 1439 0,168 II
38 Стронций Sr 87,63 2,60 757 1366 —' И
56 Барий Ba 137,36 3,65 710 1696 0,068? II
88 Радий Ra 226,05 ~6 -700 — — II
Общие данные. Главная подгруппа второй группы периодической системы охватывает элементы: бериллий, магний, кальций, стронций, барий и радий. По главным представителям этой подгруппы — кальцию, стронцию й бар и го,— известных под общим названием щелочноземельных металлов, вся главная подгруппа второй группы называется также подгруппой щелочноземельных металлов.
Название «щелочноземельные» эти металлы (иногда к ним присоединяют и магний) получили потому, что Их окислы по своим химическим свойствам являются промежуточными, с одной стороны, между щелочами, т. е. окислами или гидроокисями щелочных металлов и, с другой стороны, «землями», т. е. окислами таких элементов, типичным представителем которых является алюминий — гласная составная часть глин. Вследствие такого промежуточного положения окислам кальция, стронция и бария и дали название «щелочные земли».
Первый элемент этой подгруппы, бериллий (если не принимать во внимание его валентность), по своим свойствам горазда ближе к алюминию, чем к высшим аналогам топ группы, в которую он входит. Второй элемент этой группы, магний, также в некоторых отношениях значительно отличается от щелочноземельных металлов в узком значении этого термина. Некоторые реакции сближают его с элементами побочной подгруппы второй группы, особенно с Цинком; так, сульфаты магния и цинка в противоположность сульфатам щелочноземельных металлов легко растворимы, изоморфны друг другу и образуют аналогичные по составу двойные соли. В гл. 1 было указано правило, согласно которому первый элемент обнаруживает свойства, переходные к следующей главной подгруппе, второй — к побочной подгруппе той же группы; и обычно характерными для группы свойствами обладает только третий элемент; это правило особенно наглцдно проявляется в группе щелочноземельных металлов.
Самый тяжелый из элементов второй группы — радий —по своим химическим свойствам, безусловно, соответствует типичным представите
264
Глава 8
лям щелочноземельных металлов, Тем не менее обычно его не пр~ вплючать в группу щелочноземельных металлов в более узком синг.,.. В связи с особенностями его распространения в природе, а также ех ствие наиболее характерного его свойства — радиоактивности (ое; и произошло его название) целесообразнее отвести ему особое место.. Г Sl обсуждении общих свойств элементов этой подгруппы радий не будет р смотрен, поскольку соответствующие физико-химические свойства / до сих пор исследованы недостаточно.
За исключением радия, все элементы щелочноземельной нодгрущ , отпосятся к легким металлам. Легкими называют металлы, удельщ, вес которых не превышает 5. По своей твердости металлы главной щ группы II группы значительно превосходят щелочные. Самый мягкий них, барий (свойства которого наиболее близки к щелочным металла обладает приблизительно твердостью свинца. Точки плавления металл этой группы лежат значительно выше, чем у щелочных металлов.
Величины эффективных атомных и ионных радиусов элементов группы щелочг земельных металлов, полученные на основании исследований кристаллически веще ста, сопоставлены в табл. 46. У каждого из этих элементов атомный и ион^
Таблица ГГ
Атомные и ионные радиусы, теплоты плавления п испарения; эпергия попнзащ— п нормальные потенциалы элементов главной подгруппы второй группы; теплоты гидратации ионов; энергия кристаллической решетки соединений Теплоты плавления и испарения при температурах плавления и испарения, теплоты гидратации (теплота гидратации иопа водорода принята равной 250 мал энергия кристаллической решетки по данным Шермана (Sherman, 1932) и де Бура (de Boer, 1936)
Элемент Есрл.т-лнй Ма гяий Кальций Стро»’ ЦИЙ Барий
Атомный радиус, А. Ионный радиус, А, 1,05 0,34 1,62 0,78 1,97 1,06 2,13 1,27 2,17 1,43
Теплота плавления, ккал^г-апмм Теплота испарения, ккал[ъ-атам Теплота сублимации прп 0” К 2.5 53,5 1,70 32,52 36,56 3,14 36,58 42,82 33,61 49,2 35,66
Энергия ионияа- ( — " е J уг„. м2+ -1- в ции, г,кал!в-ато^1 „ ( мг+-»м®+ + е. 213,7 417,6 3532,7 175,4 344,8 1837,7 140,3 272,4 1173 130,7 253,0 986 119,6 229,3 818
Нормальный потен- ( наблюд, циал, а ф вычисл. Теплота гидратации, йка.т/г-иен —1,69 -1,73 570 —2,40 -2,51 437 -2,76 —2,83 361 —2(87 324 —2,92 297
Эпергия кристаллине- ( cKOit решетки, 1 хлорид ккал/моль ( окисел 713 1080 595 936 537 830 508 784. 488 740
а См. примечание к табл. £8, стр, 180,—Лри.и
Вторая группа периодической системы
265
радиусы несколько меньше, чем у предшествующего щелочного металла. Это сжатие атомов объясняется увеличением зарядов их ядер на единицу по сравнению с соответствующими щелочными металлами.
Общим для всех элементов главной подгруппы II группы является их свойство проявлять в своих соединениях положительную валентность 2 и только в совершенно исключительных случаях они бывают положительно одновалентны. Типичная для них валентность 2-J-, а также порядковые номера элементов заставляют, бесспорно, отнести эти металлы к главной подгруппе второй группы. Кроме того, все они обнаруживают сильно электроположительный характер, который определяется их положением в левой части электрохимического ряда напряжений, а также сильным сродством к электроотрицательным элементам.
В соответствии с величиной нормальных потенциалов элементов главной подгруппы второй группы (см. табл. 46) все перечисленные металлы разлагают воду; одпако действие бериллия и магния на воду Протекает очень медленно вследствие малой растворимости гидроокисей, получающихся в результате этой реакции, папример для магния;
Mg-|-2HOH = Mg (ОН)а -ь Н3.
Образовавшись на поверхности металла, гидроокиси Бе и Mg затрудняют дальнейшее течение реакции. Поэтому даже мелкие опилки магния приходится выдерживать при обычной температуре в соприкосновении с водой в течение нескольких суток, прежде чем они полностью превратятся в гидроокись магния. Остальные щелочноземельные металлы реагируют с водой значительно энергичнее, что объясняется лучшей растворимостью их гидроокисей. Гидроокись бария растворяется легче всего; нормальный потенциал Ва имеет наиболее низкое значение по сравнению с другими элементами группы, поэтому он реагирует с водой, а также со спиртом очень энергично. Устойчивость щелочноземельных металлов к действию воздуха убывает по направлению от магния к барию. В соответствии с положением в ряду напряжений названные металлы вытесняют все тяжелые металлы из растворов их солей.
Нормальные потенциалы, определяющие положение элементов в ряду напряжений, установлены еще пе для всех металлов главной подгруппы II групиы при помощи непосредственных измерений. В табл. 46, кроме измеренных нормальных потенциалов, приведены значения, вычисленные Макишима (Makjshima, ср, стр. 182). Из Данных таблицы видно, что элементы главной подгруппы II группы по сиде своего электроположительного характера значительно приближаются к щелочным металлам. Последовательность нормальных потенциалов здесь та же, что л последовательность потенциалов ионизации. Эффект. гидратации попов, оказывающий в ряду щелочных металлов сильное влияние па величины их нормальных потенциалов, в ряду щелочноземельных металлов отступает па второй план. Это связано с тем, что разности. между теплотами гидратации в энергиями иопизации (см. табл. 46) в группе щелочноземельных металлов значительно больше, чем в группе щелочпых металлов. В обеих группах теплоты гидратации попов меньше, чем энергии ионизации, и значительно меньше, чем сумма последних и теплот сублимация. Металлы обеих групп вытесняют водород пз воды и кислот и в соответствии с этим обнаруживают отрицательный потенциал по отношению К водородному электроду. Причина этого заключается нс в «стремлении указанных металлов перейти в раствор в виде nd л сжито льны х ионов», как это часто полагают, а в стремлении ионов водорода к разрядке с образованием молекулы IIj, т, е. свободная энергия реакции (1)
П+.лд4-е=’/2(Нг)+«? (Б
больше, чем реакции (2а);
М+‘<щ4-е = [М] + <гд (2а)
ИДИ
1/аЬР+.^ + г = 1/2[МЬНя (26)
266
Глава. 8
Нормальный потенциал, отнесенный к нормальному водородному электроду, в сас^ ветствии с уравненном (3) гл, 5, является непосредственной мерой разности стандарт1 пых свободных энергий обеих реакций, описываемых уравнениями (I) и (2а) или (25
Из приведенных в табл. 46 значений нормальных потенциалов (с учетом нормам него потенциала хлора) вычислены приведенные в табл. 47 потенциалы разложеЕгтг хлоридов группы щелочноземельных металлов, относящиеся к водным раствора Последовательность их изменения та же, что и потенциалов разложения (Neumann 1925); значения, полученные Нейманом, а также температурные коэффициенты потенциалов разложения приведены в табл. 47, (Измерения проводили с расплавами сиесеС КС1 и L1C1, которые затвердевали ниже 750'5)
Таблица 47
Температуры плавления и потенциалы разложения хлоридов
1 щелочноземельных металлов
Соединение Beds MgCli CaCls SrCls ВаС! а
Температура плавления, сС 405 715 780. 872 960
Потенциал разложения в водном растворе при IS", е , 3,05 ! 3,96 4,12 4,23 4,28
Потенциал разложения в расплаве при 750°, в . . 1,46 2,25 2,74 2,90 2,99
Температурный коэффициент потенциала разложения в расплаве * . 0,965-10-3 0,712- 10-s 0,714-Ю-з 0,714-Ю-з 0,71’4-Ю-з
В качестве продуктов горения щелочноземельных металлов всегда получаются нормальные окислы МПО. Перекиси щелочноземельных металлов гораздо менее устойчивы, чем в ряду щелочных металлов.
Теплоты образования окислов и других соединений элементов главной подгруппы II группы сопоставлены в табл. 48. Окислы металлов этой группы имеют наибольшие тепло ты образования. О связи этого явления с электроположительным характером элементов этой группы можно повторить то же, что было сказано в гл. 6 относительно данных табл. 27. Для Других приведенных в табл. 48 теплот образования наблюдаются совершенно такие же соотношевтш, как в соответствующих колонках табл. 27; однако в данном случае уже не только у бромидов и иодидов, по и у хлоридов теплоты образования для всех элементов группы возрастают параллельно с увеличением электроположительного сродства,
1 Таблица 48
Теплоты образования соединений бериллия, магпия и щелочноземельных металлов
Значения теплот образования даны в больших калориях на грамм-эквивалент металла (ккал/г-же)
Элементы О в целы Гидриды Фториды Хлориды Бромиды И о ди* ды Сульфиды Нитриды
Бериллий .... 68,7 „— 120,6а 56,3 — 28,1 22,3
Магпий 72,9 — 130,7 75,5 60,8 42,4 41,9 19,2
Кальции .... 76,0 23,0 144,7 95,5 81,0 64,0 57,1 17,1
Стронций .... 70,6 21,1 144,6 98,8 85,4 68,0 55,1 15,2
Барий 66,7 20,4 143,1 102,5 90,1 77,2 51,2 15,0
а Для р-кристобалитной модификации BeF, по данным В. II. Колесова, М, М. Попова, С. М. Скуратова (Ж. неорг, хим., 4, 1233, 1959).—Прим. ред.
Вторая группа периодической системы 267
С водой окисли щелочноземельных "металлов соединяются, образуя гидроокиси, причем энергия этой реакции очень заметно возрастает но направлению от ВеО к ВаО (см. табл. 53). Растворимость гидроокисей также сильно увеличивается от гидроокиси бериллия к гидроокиси бария; по даже растворимость последней при нормальной температуре очень невелика. В том же порядке возрастает и основной характер этих соединений от амфотерной гидроокиси бериллия до сильно основного едкого бария.
Интересно отметить сильное сродство элементов главной подгруппы второй группы к азоту. Склонность к образованию соединений с азотом возрастает у этих элементов с увеличением атомного веса (несмотря на то, что теплоты образования нитридов в этом направлении убывают); у собственно щелочноземельных металлов тенденция к образованию нитридов настолько велика, что последние медленно соединяются с азотом уже при обычной температуре.
Щелочноземельные металлы подобно щелочным металлам соединяются с водородом, образуя гидриды, например
Са-|-Иг = СаН2.
Эти гидриды также имеют солеобразный характер, и поэтому следует считать, что в них, как и в гидридах щелочных металлов, водород является электроотрицательной составной частью.
Труднее получить непосредственно из элементов MgHa, a Bella синтезировать таким путем вообще не удалось. MgHa и ВеНа твердые и нелетучие соединения, как и гидриды щелочноземельных металлов, но в отличие от последних они не обладают ярко выраженных! солеобразным характером.
Все элементы главной подгруппы второй группы образуют бесцветные ионы, имеющие положительный заряд 2; Be", Mg*', Са", 8г", Ва“, На". Бериллий образует, кроме того, бесцветные анионы [ВеОаГ и [Be(OH)<i]". Бесцветны й все соли МпХа указанных элементов, если они не являются производными окрашенных анионов.
Соли радия сами ио себе тоже бесцветны. Однако некоторые из пйх, например хлорид и бромид радия, постеиешю окрашиваются под действием излучения содержащегося в Них радия и, наконец, приобретают окраску от коричневой до черной. При перекристаллизации они вновь становятся белыми.
Многие соли щелочноземельных металлов трудно растворимы в воде. В изменении растворимости этих солей часто обнаруживается определённая закономерность: так, у сульфатов растворимость быстро уменьшается с возрастанием атомного веса щелочноземельного металла. Приблизительно так же изменяется и растворимость хроматов. Большинство солей, образуемых щелочноземельными металлами со слабыми кислотами и с кислотами средней силы, растворяется с трудом, например фосфаты, оксалаты и карбонаты-, некоторые из них, однако, легко растворимы; к последним относятся сульфиды, цианиды, роданиды и ацетаты. Вследствие ослабления основного характера гидроокисей при переходе от Ва к Be, в этой же последовательности возрастает степень гидролиза их карбонатов. В том яге направлении изменяется и их термическая устойчивость: в то время как карбонат бария даже при температуре белого каления разлагается далеко пе полностью, карбонат кальция можно полностью разложить на СаО и СО3уже при сравнительно слабом прокаливании, а карбонат магния разлагается еще легче.
268
Глава 8
С точки зрения теории Косселя причиной двухвалентности элементен щелочноземельной группы является то обстоятельство, что в периодической системе они все удалены от соответствующих инертных газов е; 2 элемента, поэтому каждый из пих имеет на 2 электрона больше, чем предшествующим инертный газ. Вследствие стремйения атомов принять кс~-фигурацию инертных газов у элементов щелочноземельной группы и происходит легкое отщепление двух электронов, но не больше, так как дальнейшее отщепление вызвало бы уже разрушение конфигурации инертных газов.
Это представление можно углубить, если принять во внимание спектр о скоипча-скне данные. Спектры (см. стр. 280 и сл.) показывают, что у атомов каждого элементе этой группы 2 электрона связаны особенно непрочно но сравнению с остальными, с именно на s-уровне стемиже главными квантовыми числами, что и у соседних щелочных металлов, При отщеплении только одного электрона спектр оставшегося электрона находится в том же соотношении к спектру атома предшествующего щелочного Металла совершенно так же, как спектр однократно ионизированного гелия к спектру атома водорода. Однако в соответствии с более высоким главным квантовым числом связь Щ данном случае оказывается далеко не такой прочной, как у гелия. Таким образом, сильно электроположительным характер элементов главной подгруппы II группы объясняется строением их атомов аналогично тому, как Это было сделано для щелочных металлов. Однако из строения атома следует, что электроположительный характер элементов главной подгруппы II группы должен быть в среднем несколько слабее, чем у щелочных металлов. Поэтому у последних па внешней оболочке связь оказывается еще более слабой, чем у элементов главной подгруппы II группы. Справедливость этого положения подтверждается сравнением Потенциалов ионизации (табл. 46), полученных из спектроскопических данных, с данными табл, 28 (стр. 180), Связь электронов па внешней оболочке у металлов щелочноземельной группы прочнее, чем у щелочных металлов, так как атомы последних имеют более высокий «эффективный заряд ядра» (ср. стр. 256 и сл.)
Из приведенных в табл. 46 потенциалов, ионизации (полученных на спектроскопических данных) следует, что для отщепления третьего электрона, принадлежащего оболочке инертного газа, необходимо затратить значительно большую работу, чем нрп отщеплении обоих электронов, находящихся вне оболочки инертного газа. Эта работа значительно больше той, которая выделяется при образовании кристаллической рещеткн. Поэтому невозможно химическим путем получить гетерополярные соединения щелочноземельных металлов, в которых они проявляли бы валентность, большую двух,
Для отщепления двух электронов у щелочноземельного металла необходимо затратить значительно большую работу, чем для отщепления одного электрода. Но эта рнаяица не так велика, чтобы опа могла быть покрыта за счет энергии, освобождающейся при внедрении ионов в кристаллическую решетку при компатлой температуре. Па примере CaF2, приведенном в табя, 26, видно, что энергия образования решетки значительно превосходит эту разницу. Как отмечалось выше, это обстоятельство является причиной отсутствия у щелочноземельных металлов валентности одни в соединениях, стабильных при обычной температуре.
Теоретически не исключено, что с повышением температуры соотношения меняются. Как следует из данных табл. 26, CaF не существует при обычных условиях, так как при низких температурах он менее стабилен, чем CaF.2. Возможно, что с повышением температуры можно достигнуть области, где CaF примерно также устойчив, как CaF2, и, следовательно, может существовать в равновесии с ним в присутствии избытка кальция. Действительно, молекулы CaF, CaCl и т. д, были обнаружены методами молекулярной спектроскопии (Mecke, Johnson). В газообразной форме они существуют в равновесии с днгалогепидами. Однако вопрос об области их термической устойчивости в жидком или твердом состоянии (ср. стр. 177) теоретически еще не решен. Более определенно об этом можно будет судить только тогда, когда при расчетах свободных энергий образования можно будет учитывать зависимость их от температуры и энергетическое различие между кристаллическими, расплавленными и газообразными формами.
Теория Гейтлера и Лондона в применении к щелочноземельным металлам приводит прежде всего к выводу о том, что их атомы я нормальном сост оянии не могут образовывать гомеополярных соединений (так как они пе имеют песпарепных электронов). Поскольку переход одного из двух электронов внешней сферы с s-уровня на соседний ^-уровень связан с небольшой затратой энергии (для Mg 62,2 для Са 43,1 ккал!г-атвм),
Вторая группа. периодической системы
269
теоретически можно предположить, что при переходе атома в «в о и бу ж денное» состояние образуются гомеополярные соединения. Однако в настоящее время еще нельзя провести точный расчет устойчивости атомных связей, образующихся за счет ^-электронов. Опыт показывает, что чисто гомеополярные соединения щелочноземельных металлов, например соединения с органическими радикалами, мало устойчивы. Интересна особая способность магния к образованию соединений тина MgXR (X — галоген, R—органический радикал). Вследствие большого практического значения последних (см. ниже) объяснение природы существующих в них связей и особенно энергетических соотношении представляли бы для химии значительный интерес.
Распространение в природе. Соединения элементов главной подгруппы II группы, за исключением бериллия и радия, широко распространены в природе. Кальций и магний относятся к числу наиболее распространенных элементов (кальция в земной коре содержится 3,4%, а магния 2,0%). Однако благодаря большой химической активности элементы щелочноземельной группы никогда не встречаются в свободном состоянии, а всегда в виде соединений.
Карбонаты кальция и магния в виде известняка и мела (СаСОз), а также доломита (СаСОз-MgCO3) образуют целые горные кряжи. Хотя и не в таких огромных массах, как только что названный двойной карбонат, по местами также в виде мощных залежей встречается простой карбонат магния MgCOa, магнезит, называемый также горьким шпатом, или тальковым шпатом. В Европе оц находится главным образом в Штенер-марке и на острове Эвбее, а из внеевропейских стран — в Калифорнии, Кападе и в китайской части Манчжурии. Местами встречается в больших количествах мрамор, состоящий почти из совершенно чистого карбоната кальция. Меньшее распространение имеют карбонаты тяжелых щелочноземельных металлов: стронцианит SrCO3 и витерит ВаСОз.
Мощные залежи образует гипс CaSCK-SHaO (называемый также селенитом). Разновидностью его является алебастр. Безводный сульфат кальция CaSO$ — ангидрит — наряду с кизеритом MgSCh-HaO является почти постоянным спутником каменной соли. Но и помимо этого, нередко встречаются слоистые залежи ангидрита. О нахождении его вместе с различными двойными солями магния в месторождениях калийных солей уже было указано на стр. 214 и 216. Из двух сульфатов стронция и бария — целестина (SrSCU) и тяжелого шпата (BaSOa) — особо широко распространен последний.
Кз силикатов магния следует указать оливин (Mg,Fe)a [SlOa ], энстатит Mgs[SiaOe], а также содержащие воду силикаты — серпентин, асбест, тальк и морскую пенку. Чрезвычайным многообразием отличаются двойные силикаты магния и особенно кальция (см. табл. 49). К числу соединений кальция относятся следующие минералы: фосфорит (см. стр. 672), апатит ЗСаз(РО4)а-Са(Р,С1)з и плавиковый шпат (флюорит СаГз). Кроме того, следует еще упомянуть встречающуюся в небольших количествах, но довольно распространенную шпинель MgO-ABOa, некоторые разновидности которой ценятся как драгоценные камни.
В качестве продуктов выветривания минералов соединения кальция и магния всегда содержатся в почве, а также в большинстве природных вод, «жесткость» которых и обусловливается содержанием этих солей. В; органической природе кальций и магний встречаются почти всюду: магний входит в качестве составной части в зеленое красящее вещество листьев (хлорофилл); кальций же в виде фосфата образует твердое вещество костей, а в виде апатита — еще более твердую часть зубной эмали. Яичная скорлупа, раковины и кораллы также состоят из карбоната кальция.
Таблица £3
Минералы щелочноземельных элементов
Хлориды Нитраты И йодаты Окис ЛЬ£ Н ГИДРО 01ЕИСЧ1
Бишофит MgCl2-6H2O Карналлит KGl-MgC]z.6H2O Тахгидрит CaGl2.2MgC]2-12H2O Магниевая селитра (нитромагнезит) МдЧМ\-.):>-п2о Известковая селитра (нитрокальцит) Са (NO3)rH2O Бариевая селитра Ba (NO3)2 Лаутарит Са (Юз)2 Брсммелит ВеО Периклаз MgO Вручит Mg(OH)2
Сульфаты
Кизерит MgSO4‘H3O Г орекам соль (эпсомит) MgSOv7H2O Ангидрит CaSO4 Гипс (селенит) CaSO4-2H2O Целестин SrSO4 Тяжёлый шпат BaSO4 Каинит KC'lMgSO4-3HaO Щёнит KoSOv MgSO4‘6H3Q Лангбейнит KaSO4'2MgSO4 Леонит K3SO4- MgSO4 4TIs0 Астра канит (бледит) Na2SO4-MgSO4'4H2O Лёвеит Na2SO4-Mg8O4-2H2O Полигалит K3S04-MgS04.2CaS04.2H20 Сингенит KaSO4-CaSO4-HaO Глауберит Na3S04 CaSO4 Фаузёрит (Mg. Mn)SO4-7HaO Магниевые квасцы (бозез-мапит) Mg [Al (SO4)2]3-24H3O Кеетенит MgO- FeaO3- 3S0,- 12Н3О Ботриоген 2MgO-FesO3-4S03-15H20 Молибдаты и вольфраматы Белоногим MgMoO4 (очень редкий) Паееллит СаМо04 (очень редкий) Шеелит (щееленекий шпат, тунгштейн) CaWO4
Карбонат»
Магнезит (тальковый шпат, горький шпат, гиобертит) MgGO3 Доломит (бурый шпат) CaCO3-MgCO3 Известковый шпат (кальцит) Арагонит J CaG03 Стронцианит SrGO3 Алстанит (Са, Ва) С03 Витерит ВаСОд Тарноеитцит (Са, Г’Ь) С0:! Тидромагнегит Mg (OH)2-3MgCO3-3H2O Патрокальцит Na^COg-CaCOj-SHgO Пирссонит ГСъСОз-СаСОз-ЗНдО Ураноталлит 2СаСОэ. U02 (С03) • 10Н2О
Продолжение табл. 40
Фосфаты, арсенаты и т. д.
Gmpyeum NH4MgP04-6И2О Вагнерит Mg3 (POJ2- MgF3 Апатит ЗСа3 (P04)2-Ca (F, CI)2 Свабит 3Ca3(PO4)2-La (F, Cl, 0H)2 Фосфорит см, стр.. 672 Гердерит CaBe(F, OTI)PO4 (редкий) Сванбергит, смешанный фосфат и сульфат кальция й алюминия Вобиерит Mg3 (РО4)2-8Г1аО Монит Са3(РО4)2'112О Монетит СаНРО4 В рушит СаНР04-11/2П20 Ныобереит М«НРО4.ЗН3О Изоклаз Са3(ОН)Р04-2НаО Цирролит ЗСаО-А]303.Р205.11/2Н20 Таеистокит 3CaO.AiaO3.P2O6-3H2O Гойацит ЗСаО. А]Й03.Р205:9Н20 Верце лит (Mg, Са)3 (AsO4)2 Атопит Саг^Ь^О? Льюисит 5CaO-3Sb205- •ЗТ(О2 Гёрнезит Mg3 (As04)3'8H20 Гайдингерит GaIJAsO4-H2O Фармаколит СаГ1АзО4 - 2Нй0 Вапплерит (Са, Mg) НА8О.(-31/гН20 Ремерит MgHЛяО4.1/2Н2О Арсениосидерит бСаО Микролит, главным образом Ca2Ta2O; Rannum, i-павным образом Ca2Nh2O7 Самарскит, главным образом, щюбат и танталат Са, Fe, Y и Ег (содержит уран) Гелъмит, главным образом танталит Са, Fo, Мп Содержащими железо и марганец фосфатами кальция являются; месселит, таманит (анапаит), ред-дицгит, филлоаит, дис-кинсстит и файерфилдит Известковый у ранит- является двойным фосфатом уранила и кальция; ба-рийу ранит (урандци- ршп) — двойным фосфат том урацила и бария 3As2O3 - 4Fe3O3 • 9Н2О
Бораты
Пинноит Mg (В02)3 - ЗН3О
Ашарит 3Mg3B2Os-2Н2О
Боромагнезит MgaВ4Ojj 2г/3Н30
Пандермит, п ри цеит^С а2 В60 j t 3 Н20
Гидроборацит CaMgBe0ji-6H3O
Колеманит Са3В(!Оц- 5Н3О
Порокалъцит, бсхилит СцВ4О;'4Н3О
Франила!idum Na2CaB(sOIr 7Н2О
Боронатрокальцит (улексит, тппкальцит)
KaCaBsO9-6I-I2O
Суссекси-т (Mg, Мо)3В3О5. JJ30
Сульфоборит iiigzBzO$ -MgS0^А1,'Л120
Люнебургит Mg (ВО->)3-2MgHPO4- 7ИаО
Людвиеит SMgO-Fe.jO^-BgO^
Ворацит 2MggBsOi5 MgCl3
Продолжение табл.
Шпинели
Шпинель MgO-A]2O3
Магнезиоферрит MgO-Fe2O3
Железистая шпинель (плеонаст, цейлонит, черная шпинель)
(Mg, Fe) О (ЛЬ Fe)2O3
Хромистая шпинель (пикотит) (Mg, Fe)O-(Al, Сг, Fe)2O3
Силикаты
Оливин, перидот (Mg.Fe)2 [ЗЮ4] Энстатит Mg2 [Si2O6] Вронцит (Mg,Fe)3ISi2Oe] Гиперстен MgFe[Si2Oe] Хризотил (волокнистый серпентин) Mgs [SiaO1±] • 3Mg(OH)2 •Н2О Антиго рит (листовой серпентин) Mg3[Si4O10(OH)2l-3Mg(OH)2 Морская пенка Mg3 [Si6O15]-Mg(OH)a.3H,O Тальк, стеатит Mg2 [Si4Ow]Mg(OH)2 Асбест см. стр. 285 и сл. Кордиерит Mg3Al3[A]Si5Olsj Г г/мит 3Mg2 [SiOj-Mg (OH,F)2 Волластонит, листовой шпат Са2 [Si2O6] Диопсид CaMg [Si2O6] Геденбергит CaFe[Si2O6]Ga2Al3[SiO4k[OHJ ромбический: цоизит моноклинный: клиноцои- зит ЭЛ14С?О?П * Са2(А1, Fe)3[OH] [SiO4]3 Монтичеллит CaMg [SiOJ Анортит (кальциевый нолевой ганат) СаДЛ1мЯ;йО8] П лагиоклаз (ка а ьциев□ - натриевый, половой шпат) (СаА], NaSi) AlSi2O8 Лучистый камень Ca3(Mg, Fc)B [ОН]2 [SisO22] Датолит Са (ОН) BSiO4 Гомилит FeCa2B2Si20i( Данбурит СаВ2312О8 Аксинит Ca3Al2BSi40j5 (ОН) Варилит Ba4A]4Si7O24 Турмалин см. стр. 359 Берилл Ве3А12 [SigOigj Эвклаз ВеА1 (ОН) SiO4 Фенакит Ве2 [SiO4] Лейкофан (?(а, Са)2 [B6Si2O5 (F,O)2]3 Гельвцн (Fe, Mn)4 [Be3Si3O13l S
Сюда относятся роговая бб-
манкв, скаполит, некоторые ifeo.tumw, слюды, хлориты и подобные им минералы
Везувиан
Ca10Mg2A]4 [Si0,5]& (Si207]2 (0H)4
Магниевоглинистый гранат, : пироп
Mg3Ai2Sj3O12
Известковоглинастъш гранат^ Са3Д]231зО£2
Известковая ромовый гранат, уваровит
Ca3Cr2Si30i2
Иавестковожелезис-тый гранат (ап.лом)
Ca3Fe2Si3OI3
Птс.рпя группа периодической системы
273
Соли магния и кальция содержатся в-довольно заметных количествах в морской воде. Последняя (кроме приблизительно 2,9% хлоридов щелочных металлов) содержит в среднем около 0,30% MgCls, 0,04% MgBr?, 0,18% MgSOi к 0,16% CaSOi. Вода некоторых «горькосоленых» минеральных источников содержит значительные количества сульфата магния.
Бериллий встречается в природе в виде некоторых минералов, не имеющих особенного распространения. Чаще всего из пих встречается берилл Be.sAlslSi^OjgJ, крупные месторождения которого известны в Бразилии, Северной Америке, Африке, Индии, Англии, Норвегии, Испании и в СССР (на Урале). Из других содержащих бериллий минералов следует упомянуть (очень редкие): эвклаз BeA!(OH)SiO4, гадолинит BeiFcYaSisOjg, хризоберилл ВеО АНОз или Ah [В еОг] (последний не изоморфен шпинели и кристаллизуется в ромбической спстомо), наконец, очень редкий минерал фенакит BealSiOd.
Другими редкими минералами, содержащими бериллий (иногда лишь в незначительных количествах), являются: гельвин (Fe, Mn)1[Be3Sij()I2]S, дана лат (Fe, Zn)4 [B^Si3Of2]S, лейхопы (Na,Ca)2(BeSi2(O,F)7], мелшюфан (Ca, Na)2fBis(Sl, A1)2(O,F)T], пгримерит MiiBeSiCC и бертрандит Bes(OH); [Si,Og] [SiOJ,.
Некоторые содержащие бериллий минералы иногда встречаются в виде очень красивых экземпляров и считаются поэтому драгоценными камнями. Так, смарагд и аквамарин — разновидности берилла, а александрит— разновидность хризоберилла. Эвклаз и фенакит тоже считаются драгоценными камнями.
Радий в совершенно ничтожных количествах содержится в рудах урана, из которого он образуется путем радиоактивного распада. Отношение радия к урану в этих рудах практически постоянно и равняется 1 : 3 000 000. Главный минерал, содержащий урап, — урановая смоляная руда — содержит в среднем 0,14 г радия на 1 т (1000 кг). Еще ничтожнее содержание радия в других урановых рудах, например в ау-тупите, карнотите и в других подобных минералах.
Карнотит распространен главным образом л Колорадо и Юте (Северная Америка). Основным месторождением урановой смоляной руды (смоляной обманки) раньше был Св. Иохнмсталь (Богемия). В иоследствии много более значительных месторождений было найдено в районе Катанги (Конго).
Исторические данные, Негашеную известь СаО, получающуюся при обжиге известняка или мрамора, уя;е с давних времен применяли после гашения в качестве цементирующего раствора в строительстве. С топ же целью пользовались в старину и гипсом. Диоскорид, живший в I в.п.э., для обозначения окиси кальция уже употребляет удержавшееся и поныне в строительном деле название «негашеная известь». Сильные щелочные свойства едкой извести были известны очень давно. Позднее вошло в употребление название для окиси кальция известковая земля и вообще для Оки слов металлов название «земли». Так, окись магния, известная с начала XVIII в,, получила название горькая земля. Ужо в конце XVII в. сульфат магния (горькая соль) применяли как лечебное средство в Англии, где его добывали из воды эпсомских минеральных источников; отсюда и произошли старинные названия горькой соли — sal anglicam иля эпсомская соль. Блэк в 1755 г. впервые установил разницу между известковой и горькой землями, указав на различную растворимость обоих окислов и получающихся из них сульфатов.
18 г Реми
274
Глава 8
Тяжелыйшпат BaSO4 впервые стал известен (1602 г.) благодаря открытию одного болонского сапожника, заметившего, что после прокаливангд-с органическими веществами это вещество приобретает способность фосфоресцировать (болонские светящиеся камни), В 1774 г. Шее ле (Scheek открыл окись бария как новую до того времени неизвестную землю, однаю сначала он не обратил внимания на то, что имел дело с новой землей, лежащей в основе тяжелого шпата. Это обстоятельство установил впервые Гап (Gahn), а затем уже оно было подтверждено и Шееле. Вновь открытый окисел получил название барита (от рссрбе — тяжелый), В 1782 г„ Витерингом (Withering) был открыт природный карбонат бария, который позднее в его честь был назвал витеритом. Вскоре после этого oko.tl Стронциана в Шотландии был обнаружен похожий на витерит минерал,, названный стронцианитом. В 1793 г. Клапрот (Klaproth) доказал, чтс последнему соответствует новая, неизвестная до тех пор земля. Вскоре поело этого (1797) была также получена и окись бериллия — Вокеленогл (Vauquelin), который При анализе берилла и смарагда установил присутствие в них некоторой новой «земли», во многих отношениях похожей на глинозем, но в противоположность последнему не способной давать квасцы. Она получила название бериллиевой земли, а металл — бериллия. Во французской литературе он известен под названием glucinium, которое должно было указывать на сладкий вкус бериллиевых солей.
Радий был открыт в 1898 г. супругами Кюри и Бемоном (Curie, Bemoiit). Этот элемент был открыт на основании наблюдаемого ими явления, а имепно, что существуют урановые минералы, гораздо более активные, чем это соответствует содержанию в них урана. На этом основании Кюри предположила, что аномально высокая радиоактивность этих минералов вызывается тем обстоятельством, что в них, кроме урана, содержится еще одно или несколько радиоактивных веществ. Благодаря этому супругам Кюри удалось выделить из урановой смоляной обманки сначала новый элемент — полоний (см. стр. 808) и вскоре и второй новый элемент — радий.
Щелочноземельные металлы в свободном, состоянии впервые получил в 1808 г. Деви (Davy) электролизом с ртутным катодом влажных гидроокисей. В виде амальгам они впервые были получены Борце л кусом (в связи с открытием Деви щелочных металлов). Позже подобным же способом М. Кюри и Дебиерн (Debierne, 1910) получили металлический радий. Металлический бериллий был впервые получен Велером (Wohler, 1828) путем восстановления его хлорида металлическим калием; в чистом состоянии его впервые получил Лебо (Lebeau, 1898) при электролизе фторо-бе рил лата натрия. Металлический магний был получен также электролизом (Davy, 1808). Бюсси (Bussy, 1830) впервые получил его химическим путем (действием паров калия на безводный хлористый магпий). Получение магния электролизом расплавов было впервые осуществлено Бугь зеном (Bunsen, 1852). Пистор (Pistor, 1908) положил начало промышленному применению сплавов с большим содержанием магния.
Получение металлов. Из металлов главной подгруппы Л группы технически наиболее важным является магний. Обычно его получают электролизом чистого обезвоженного и расплавленного карналлита MgCla-KCl или смеси соответствующих солей при температуре, превышающей точку плавления магния, применяя анод из графита Ачесона и железпыи катод. Жидкий магний при электролизе поднимается на поверхность, откуда его извлекают черпаками.
Вторая группа периодической системы 215
Так как при электролизе расплава происходит разложение только MgCl2, его необходимо непрерывно добавлять, MgC]3 получают из магнезита или жженого доломита по реакции
MgO + СО + d2 = MgCI2+ СОг, для которой используют хлор, выделяющийся ла аноде.
В последнее время для производства магния технологическое значение приобрели так называемые термические методы. Важнейшие из них — восстановление MgO карбидом кальция или углем (карботермические методы} и восстановление MgO кремнием (силикотермические методы}.
При производстве магния карботермическими методами ведут восстановление MgO углеродом в электро дуговых печах. Реакция MgO 4 С — Mg 4 СО идет слева направо только при температуре выше 2000°. Чтобы в улетучивающейся смеси паров магния и СО вновь не образовалась MgO, их следует очень быстро охлаждать. Вначале это достигали добавлением водорода (Hansgirg, 1931).
В США, где в основном применяют карботермический метод, быстрое охлаждение реакционных газов осуществляется при введении природных газов.
Более простым в технологическом отношении, чем восстановление MgO углеродом, является восстановление карбидом кальция (Matignon, Thierry, 1915). Восстановление ведут в вакууме в реторте, нагреваемой снаружи почти до 1200°; реакция идет по уравнению
MgO + СаС2 = Mg + СаО Ц- 2С.
Этот метод (применяемый в основном в Англии и во Франции) экономически выгоден только при наличии дешевого карбида кальция.
Силикотермический метод основал на том, что сильно экзотермическая реакция
SiO2 -В 2Mg = Si Ц- 2MgO
при повышении температуры может пойти в обратную сторону. При этом исходят обычно не из чистой окиси магния, а из жженого доломита. Последний в смеси с кремнием или ферросилицием нагревают до 1200—1300° в стальной реторте или в высоко-вакуумцой печи, обогреваемой электричеством. При этом происходит следующая реакция:
2MgO 4- 2СаО + Si = Ca2SiO4 + 2Mg.
Магний (с чистотой 98—99%) отгоняется и осаждается в приемниках, охлаждаемых воздухом.
Техническое получение кальция осуществляют электролизом расплавленного хлористого кальция в смеси с фтористым кальцием и хлористым калием. При этом оказалось целесообразным такое устройство катода, при котором последний едва касается поверхности расплавленной массы; по мере выделения кальция на катоде последний поднимают вверх. Нагревание расплава производят лишь в такой степени, чтобы он оставался жидким. Если вести электролиз при температуре более высокой, чем точка и лав ления кальция, то значительная часть расплавленного кальция переходит в результате диффузии в плав и теряется вследствие вторичного окисления. Если же электролиз вести ниже температуры плавления кальция, то металлический калений выделяется в виде губчатой, сильно загрязненном массы. При описанном устройстве катода на нем вследствие местного разогрева, вызываемого очень высокой плотностью тока, выделяется кальций, причем заметной диффузии его в более холодный плав уже пе происходит. По мере подъема катода па нем застывает выделившийся кальций, так что образуется все более длинный стержень из чистого металла, поверхность которого защищена от окисления коркой из застывшей расплавленной массы.
Кальций высокой чистоты получают в техвике по Хакшпиллю (Hackspill, 1951) при нагревании хлористого кальция с металлическим алюминием. Образующийся при этом хлористый алюминий испаряется, а кальций можно подвергнуть дальнейшей очистке перегонкой в вакууме.
Стронций, так же как и кальций, можно подучить электролитически, в то же время получение бария этим методом сопряжено с большими трудностями. Гораздо лучше по методу Гувтца (Guntz, 1906) и Матиньока (Matignon, 1913) удается восстановление в вакууме окиси 6apim алюминием или кремнием при 1200°, протекающее по реакциям: )
ЗВаО 4. 2А1 = А12О3 + ЗВа; ЗВаО Ц-Si = BaSiO342Ba.
18*=
276
Глава S
Металлический бериллий, согласно Фихтеру (Fichter), проще всего получать в лаборатории электролизом расплавленной смеси BeF, н NaF в молярном отношение 2 : 1. Расплавленную массу при этом помещают в никелевый тигель, служащий одновременно катодом, в то время как анод — толстый угольный стержень — погружаю-в плав. Во избежание образования сплава бериллия с никелем не следует допускать слишком сильного нагревания.
Технический способ получения бериллия, разработанный Штоком и Гольдшмидтом (Stock, 1921, Goldschmidt, 1925), основан на электролизе смеси NaBeF3 и Ba(BeFj}„ или других подходящих смесей, состав которых подбирают так, чтобы они отличались возможно более высокой электропроводностью наряду с возможно меньшей летучестью при температурах выше точки плавления бериллия. При этом, так же как прп получении кальция, применяют охлаждаемые контактные катоды, одпако температуре расплава значительно выше (около 1400°). Бериллии высокой чистоты получают прп перегонке в вакууме (Sloman, Kroll, 1932, 1934—1935). Для этого металл загружают в тигель из ВеО и нагревают током высокой частоты.
Для получения щелочноземельных металлов в очень небольших количествах используют разложение азидов (ср. стр. 651).
Свойства, Элементы главной подгруппы второй группы представляют собой серые или белые металлы (блестящие на свежем разрезе или изломе), которые, однако, па воздухе быстро тускнеют. Их твердость и прочность на разрыв уменьшаются от бериллия к барию.
Таблица йО
Твердость, сопротивление па разрыв н электропроводность металлов главной подгруппы второй группы
Металл Бериллий Магний Кальций
Твердость (по Ритцу) Между 6 п 7 2,6 2,2-2,5
Сопротивление ла разрыв, ке/мм* — 20 5
Электропроводность кубика в 1 cat1, о .и"1 5,41-10* при 20° 23,2-10* при 0° 21,8-10* при 20°
В табл. 50 приведена твердость по Рятцу (по шкале Мосса). В качестве сопротивления на разрыв указан вес нагрузки {кг), которую выдерживает проволока пли стержень с поперечным сечояием 1 млР.
Указанные в сводной таблице в начале этой главы точки кипения щелочноземельных металлов обнаруживают совершенно незакономерный ход, в то время как в группе щелочных металлов точки кипения правильно убывают в направлении от наиболее легкого к наиболее тяжелому металлу. То же можно сказать и относительно точек плавления элементов главной подгруппы II группы. Такое незакономерное изменение точек плавления, вероятно, связано с тем, что при переходе от магния к кальцию и от стронция к барию происходит изменение структуры кристаллических решеток (см. ниже).
Среди металлов главной подгруппы II группы практически наиболее важным является магний, отличающийся серебристым блеском, который Па воздухе вскоре принимает матово-белый оттенок. Магний отличается средней твердостью и довольно тягуч, так что при вальцевании из него удается получать топкие листы и проволоку; однако Эти его свойства значительно изменяются под влиянием очень небольших примесей. Электропроводность магния составляем ЭД5 электропроводности меди и 12/19 эле-цтропроводпости алюминия, а теплопроводность равна 5/13 теплопровод-
Вторая группа периодической системы
277
пости меди и'5/D соответствующей величины для алюминия. Холодная вода действует на магний крайне медленно; тем не менее при действии воды на обезжиренные стружки магния (промытые в эфире) по поднимающимся пузырькам газа можно судить о выделении водорода. С кипящей водой эта реакция протекает гораздо энергичнее.
При нагревании магний реагирует н со спиртом. Если магний предварительно протравить иодом, то реакция протекает почти так же быстро, как и с водой. Магнии растворяется в разбавлепных кислотах с бурным выделением водорода. Амальгама магния очень энергично реагирует с водой уже при обычной температуре. Магний в виде ленты или порошка, зажженный на воздухе, горит ослепительно белым пламенем с выделением белого дыма, состоящего из MgO. Свет, испускаемый магнием при горепии, богат фотохимически активными лучами. Этим пользуются в фотографии (моментальные съемки при свете магния). Во влажном хлоре магний самопроизвольно загорается, сгорая и в этом случае с энергичным выделением света. Магний обнаруживает сильное сродство по отношению к другим неметаллам. Так, при нагревании он легко соединяется с азотом, образуя нитрид состава MgaNs. Последний получается в значительных количествах вместе *с окислом MgO при обжиге магния в условиях недостаточного доступа воздуха. Магний способен отнимать у многих других соединений их электроотрицательную составную часть; так, реакция его с некоторыми окислами или гидроокисями щелочных металлов протекает даже со взрывом. Со многими металлами он образует сплавы; однако лишь некоторые из них имеют значение в технике, так как в большинстве случаев они ломки и слишком легко окисляются. С органическими иодсодержащимя соединениями в эфирном растворе магний образует ’магнййалкилиодиды (Гриньяр).
Метилмагнийипдид
Подобным же образом магний реагирует и со многими органическими бромидами и хлоридами (главным образом с алифатическими); однако в этих случаях реакция протекает лучше в присутствии следов иода или при условии, что магний был предварительно «активирован» иодом.
Магнийалкилгалоидные соединения имеют большое значение в препаративной органической химии (реакция Гриньяра).
Щелочноземельные металлы в узком смысле этого термина — кальций, стронций и барий — по своим свойствам гораздо ближе к щелочным металлам, чем магний. Они значительно мягче магния, хотя их точки плавления выше. На воздухе они окисляются не так быстро, как щелочные металлы. Подобно щелочным металлам, их приходится хранить под керосином. Они легко воспламеняются: барий загорается на воздухе уже при простом раздавливании. В качестве продуктов горения при этом наряду с окислом получается и нитрид. Последний медленно образуется и при обычной температуре. При нагревании в струе азота при температуре красного каления образование нитридов протекает легко и полностью*.
* Кальций реагирует с азотом энергичнее всего около 450s, так как в связи с. полиморфным превращением, происходящим при этой температуре (см. стр. 279), происходит разрыхление кристаллической решетки Са. (У Sr и Ва этого явления пе наблюдается,) Небольшие количества Na на поверхности металла активируют этот процесс, так как Na препятствует образованию пленки нитрида.
278
Глаза 8
Щелочноземельные металлы энергично вступают в реакции и с другие неметаллами, отнимая кислород-от многих онислов. С водой кальцпь. при обычной температуре реагирует довольно вяло, стронций нескольк: энергичнее, а барий очень энергично. Со спиртом они реагируют с выделением водорода. Разбавленные кислоты растворяют щелочноземельные металлы, причем бурно выделяется водород по реакции
Са+2Н‘ = Са" + Н2.
С жидким аммиаком щелочноземельные металлы образуют темпо-синие растворы. После отгонки из этих растворов аммиака остаются твердые продукты, имеющие медный до золотистого блеск и представляющие собой аммиакаты, т. е. продукты присоединения аммиака к названным элементам. Аммиакаты имеют строго определенный состав, а именно [Ca(NHs)e], [Si(Nlia)6] и EBa(NHs)eL Бильтц (Biltz, 1920) установил, что существуют только гексааммиакаты, других аммиакатов эти металлы не образуют.
В присутствии катализаторов (платины) продукты присоединения аммиака постепенно разлагаются с образованием амидов, например
Са (NHj)e = Са (NH2)2 + 4Г<Н3 + И2.
Амиды при нагревании в вакууме могут переходить в желтые имиды. Последние кристаллизуются и кубической грапсцентрированной решетке с ребром куба (я) и плотностью (Д):
CaNH SrNH BaKH
я —5,16А, d = 2,66 | а = 5,45А, rf=4,18 | а = 5,84А, й=5,07
Из имидон при дальнейшем нагревании в высоком вакууме могут быть подучены пернитриды; например Sr3N4 — перпитрид стронция — красновато-коричневый кристаллический порошок, который разлагается разбавленными кислотами с выделением N2:
Sr3N4 + 8НС1 = 3SrCl2 + 2NH4C1 4- N2.
(Hartmann, Frohlich, Ebert, 1934).
При нагревании щелочноземельные металлы реагируют с аммиаком с образованием нитридов и гидридов. Сродство их к водороду настолько велико, что, например, кальций при нагревании в струе водорода образует гидрид, причем эта реакция сопровождается воспламенением
ftr Са-p И2=СаН2.
Щелочноземельные металлы образуют соединения со многими другими металлами; по крайней мере для кальция, который в этом отношении исследован лучше всего, известно большое число интерметаллических соединений.
Металлический радий очень похож па барий, однако значительно более летуч и чрезвычайно неустойчив на воздухе.
Металлический бериллий отличается серо-стальным цветом и значительной твердостью, так что царапает стекло. При обычной температуре он ломок и не выдерживает ковки. Одпако при температуре красного каления Бе становится ковким. Электропроводность бериллия равна около Vi2 электропроводности меди*. В сухой атмосфере он сохраняет блестящую поверхность. При соприкосновении с водой Бе покрывается тонкой пленкой окисла, которая и предохраняет его от дальнейшей коррозии; разбавленные кислоты энергично его растворяют. Концентрированная азотная кислота на холоду пе оказывает па пего заметного действия, а реакция его с разбавленной (2 н.)
* Электропроводность бериллия составляет 40% электропроводности меди. («Бериллий», стр. 275, ИЛ, I960.)—Прим, ред
Вторая группа периодической системы
279
азотном нлслотой на холоду вскоре прекращается. При нагревании эти кислоты быстро его растворяют. От остальных металлов главной подгруппы II группы бериллий резко отличается своей растворимостью в водных растворах щелочей. Впрочем, разбавленное едкое кали растворяет его только при нагревании, но 50%-пый раствор КОИ действует на бериллий уже при комнатной температуре.
Р и с. 57. Гексагональная плотнейшая упаковка.
Пример: МёЦа—3,22; t;=5,23aJ при этом нередко наблюдаются
Строение кристаллнческой решетки. Бериллий и магний в элементарном состоя* нип кристаллизуются по типу гексагональной решетки, приведенной на рис. 57. Элементарная ячейка этой структуры обозначена черными кружками, соединенными на рисунке жирными линиями; кроме того, на рисунке для наглядного представления гексагональной симметрии добавлены еще некоторые атомы, лежащие вне элементарной ячейки (они заполнены точками и Соединены светлыми линиями). Представ лепную на рис. 57 элементарную ячейку можно разделить на Две тригональные призмы. Кроме восьми угловых точек элементарной ячейки, атом бериллия занимает и Центр одной из этих двух призм. В общем элементарная ячейка содержит 2 атома Be, так как каждый угловой атом принадлежит одновременно 8 ячейкам. Эта решетка представляет собой тип гексагональной плотнейшей упаковки. Если представить себе, что атомы — твердые шары и что очень большое их количество размещено один над другим таким образом, чтобы возможно полнее использовать имеющееся пространство (т. е. чтобы между атомами оказался возможно меньший свободный объем), то получим расположение, приведенное на рис, 57. Плотность заполнения при этом достигает 74,05%. Однако существует и другая возможность размещения шаров, при которой пространство окажется также заполненным на 74,05%. Это размещение соответствует структуре, показанной на рис. 46 стр. 238, т. е. гранецентрированной кубической решетке. В этой структурной форме кристаллизуется (ниже 300“) металлический кальций. Оба типа решеток, из которых каждый представляет собой плотнейшую упаковку шарм, встречаются чаще всего у элементарных вещеет.е\ впрочем,
небольшие отклонения от гексагональной плотнейшей упаковки и отношение высоты с элементарной ячейки к обоим (равным друг Другу) ребрам а ее основания пе соответствует точпо теоретической величине_ с/а ~ 1,633.
Для Be а = 2,268 А, с = 3,594 А, с/а= 1,585. Каждым атом Be окружен 12 соседними атомами, причем 6 из них находятся в плоскости основания на расстоянии а, и ко 3 в верхней и нижней плоскостях на расстоянии 2,232 А. Для Mg а — 3,22 А, с = 5,23 А, с/а = 1,624. Каждый атом Mg окружен 12 соседними атомами, находящимися от него на одинаковом расстоянии а = 3,22 А.
Для модификации кальция, имеющей кубическую гранецеитрированную ячейку (а-Са), а — 5,56 А. Каждый атом Са окружен 12 соседними атомами на равных расстояниях (а /2) '1^2 = 3,94 А. При 300° Са претерпевает аллотропное превращение с образованием менее симметричной кристаллической решетки (p-Са), При 450“ эта модификация превращается в третью (у-Са) с гексагональной решеткой (подобна Mg). Кальций, содержащим нитрид, кристаллизуется выше 450° в кубической объемно-центрированной решетке (подобно Ва); однако для чистого кальция этот тип решетки не обнаружен.
Стронций, подобно кальцию, кристаллизуется в кубической грапецентрированной решетке, а= 6,05 А.. Расстояние Sr«—>-Sr=4,27 А.
Барий кристаллизуется по типу кубической объемно центрированной (рис. 47 на стр. 238) решетки; а = 5,01 А. Плотность заполнения Пространства при таком расположении атомов равна 68,02%. Каждый атом В а окружен 8 соседними атомами, находящимися от него на одинаковых расстояниях (а/2)фл3 = 4,34 А.
Кальций и стронции образуют непрерывный ряд твердых растворов. Аналогичным образом Са и Ва, а также Sr и Ва могут давать твердые растворы в Широком интервале концентраций; однако в этих системах в соответствии с изменением типа репгегки появляются области расслаивания (в пределах 4—6 ат.%) (Klemm, 1941). Mg и Са не образуют между собой твердых растворов.
280
Глава 8
Окрашивание пламени и спектры. Кальций, стронций, барий в hsz летучих соединений (например, в.виде хлоридов) вызывают характерна окрашивание пламени бунзепокской горелки. Соли кальция окрашиваггу пламя в кирпично-красный цвет. Соли стронция — в карминово-красныС а соли бария — в желтовато-зеленый. (Соли радия окрашивают плане в карминово-красный цвет.) При рассматривании окрашенного йламеиц в спектроскоп видны линии или группы линий, приведенные на рис. 5S.
-----*- синий
Фиолетовый
Р и с, 58. Спектры пламени щелочноземельных металлов.
Спектры пламепн щелочноземельных металлов не связаны, как это имело место для щелочных металлов, со свободными атомами. При более значительной разрешающей силе спектроскопа оказывается, что многие «линии» спектров пламени щелочноземельных металлов, отличающиеся значительной шириной и отсутствием резкой границы, в действительности состоят из большого числа очень близких линий, из так называемых полов. Ранее уже было отмечено, что полосатые спектры приписывают молекулам. Поэтому в зависимости от того, исследуется ли спектр фторида, хлорида пли окисла щелочноземельного металла, получают совершенно различные полосы. Если в пламя на платановой проволоке внести каплю солянокислого раствора какого-нибудь щелочноземельного металла, то в первый момент возникает спектр хлорида, который, однако, тотчас же переходят в спектр окисла, наряду с которым одновременно появляются также и липин свободного металла. Если вновь смочить платиновую-проволоку соляной кислотой, то опять появляется спектр хлорида и т. д. Несмотря на изменяющийся вид спектров пламени щелочноземельных металлов, ими все же-можно пользоваться для открытия этих металлов; при этом следует обращать внимание главным образом, на характерные, особенно отчетливо про являющиеся линии или соответственно полосы, которые приведены в табл. 51. Указанные в этой таблнце-длины волн относятся к серединам этих полос (поскольку речь идет не о резких линиях, а о полосах).
Таблица 51
Характерные липин спектров (х.ц) пламени щелочноземельных металлов, ненользуемые при аналитическом определении
КалЬцни Стронций Барий
Красная 622,0 Оранжевая 605,0 Зеленая 524,2
Зеленая 553,5 Синяя 460,8 » 513,7
Вморич группа периоди-чесной системы
281
Если значительно повысить температур/, т, е, вызвать описанным на стр. 195 способом появление эмиссионных спектров, то получаются резкие линии, соответствующие исключительно излучению свободных атомов. В этом случае подучаются также линии и в спектрах бериллия и магния. Эти два металла при внесении их в пламя обычной бунзеновскои горелки совершенно не окрашивают его. Линии эмиссионных спектров элементов щелочноземельно}] группы, служащие для их аналитического определения указаны в табл, 52.
Таблица 52
Характерные линии (-мд) амнсекоппых спектров металлов Главной подгруппы второй группы
Ее рилпий Магний Кальций Стронций барий Радий
667,5 516,7 612,2 460,7 493,4 482,6
436,2 518,4 422,7 421,6 455,4 468,2
В табл. 52 приведено только несколько характерных, линий из числа образующихся в сильном дуговом спектре (например, между двумя угольными стержнями, смоченными растворами солей соответствующих элементов, а не между раствором соли п острием иридиевой проволоки, как это описано на стр. 195). При утих условиях для кальция, например, можно наблюдать в видимой части спектра по крайней мере 35 линий, образование которых зависит исключительно от процессов, совершающихся в атомах Са. Кроме этих линий, в ультрафиолетовой и инфракрасной областях появляется еще много других. Вообще все спектры элементов плавной подгруппы II группы гораздо сложнее, ном спектры щелочных металлов. Тем ие менее И здесь удалось распределить их по сериям. Прп этом оказалось, что в то время как серии щелочных металлов состоят исключительно из двойных линий (дублетов), которые переходят в одиночные только тогда, когда разность их длин волн настолько мала, что их уже не удается разложить, металлы щелочноземельной группы имеют три различного рода серии, а именно: состоящие из ряда отдельных линий (синглеты), натем да ряда двойных липин (дублеты.) и, пакотц, из одних только тройных линий (триплеты); отсюда взаимосвязь синглетных и триплетных серий: они образуют взаимные комбинации, но не могут комбинироваться с дублетными сериями. Дублетные серии элементов щелочноземельной подгруппы обнаруживают глубокую аналогию С сериями щелочных металлов. Их можно выразить формулами, совершенно аналогичными тем, которыми выражаются серны щелочных металлов. Разница заключается в том, что в случае щелочноземельных металлов константу, зависящую от эффективного заряда ядра (которая здесь соответствует выражению для водорода и однократно ионизированного гелия), принимают в четыре раза большей, чем в случае щелочпых металлов. Указанная константа пропорциональна квадрату «эффективного заряда ядра», г, е. примерно нропорцяоналыга квадрату заряда атомного остова. Таким образом, д ля заряда атомного остова получается число 2. Это значит, что дублетные серии щелоч-ноземвяъной группы образуются благодаря перескоку электрона-, вращающегося вокруг атомного остова с зарядом 2-|-. Следовательно, дублетные серин относятся не к нейтральным, а к однократно положительно заряженным атомам, т. о., согласно данному на стр, J35 определению, они являются «искровыми спектрами*. Действательио, Пашен нашел, что дублетные системы щелочноземельных металлов при искровом возбуждении получаются значительно более явными, чем при дуговом возбуждении. Дублетные серии щелочноземельных металлов являются сериями искровых спектров, т. е. их носители— уже однократно ионизированные атомы; это подтверждается еще й тем обстоятельством, что вычисленшде из измерений границ главных дублетных серий работы «освобождения» электрона соответствуют по потенциалам ионизации процесса М = М+ + а впачительпо большим потенциалам иопизацнн, относящимся к процессу ЙЕ — М2’ — о. Так, потенциал ионизации магния (установленный Футом (Foote), Меггерсом (Meggers) и Молером (Mohler) методом «электронного удара»], соответствующий образованию Mga+, оказался равным 15 в в соответствии с величиной его, полученной из серийной границы. В то же время образованию однократно заряженного атома соответствует потенциал ионизации 7,6 в. Таким образом, дублетные системы щелочноземельных металлов подтверждают закон Зоммерфельда о спектроскопическом смещении (ем. стр. 135 и ел,).
282
Глава 8
Полная аналогия между эмиссионными спектрами щелочных металлов и искровыми спектрами элементов щелочноземельной группы обнаруживается при рассмотрении рис, 59. На рисунке уровни энергии, соответствующие отдельным спектральным термам, обозначены горизонтальными линиями, так же как и на рис, 21. Отдельные уровни энергии обозначены обычными символами, при этом цифры соответствуют главным квантовым числам; буквы s, р, d и / относятся к побочным квантовым числам соответствующих уровней энергии (см. стр. 142). За пулевое значение энергия (верхняя граница) здесь принята энергия атома яри полном отщеплении соответствующего электрона. Ступенчатое уменьшение энергии по мере приближения электрона к ядру нанесено в направлении сверху вниз. Уровни энергии, находящиеся с левого края рисунка, являются уровнями энергии водородного атома для главных квантовых чисел л — 2, 3, 4 и т. д. Уровень энергии, соответствующий и = 1, находится гораздо
Рис. 59. Сравнение энергетических уровней щелочных и щелочно-земел г.ийх* металлов (по данным искровых спектров).
ниже рамки рисунка. Для уровней энергии однократно ионизированных щелочноземельных атомов масштаб принят вчетверо меньший, чем для нейтральных атомов щелочных металлов, Если отвлечься от фактора 4 (который на чертеже элиминирован путем введения различных масштабов), обусловлепного у щелочноземельных элементов двойным зарядом атомного остова, то, как видно, уровни энергии однократно ионизированных атомов щелочных земель всюду соответствуют уровням нейтральных атомов щелочных металлов. Однако при более внимательном рассмотрении можно заметить и некоторое существенное различие между ними. Действительно, в то время как у Mg+, так же как и у Na, уровень 3d расположен выше уровня Зр, у Са* в отличие от К он лежит чиже уровня 4р. В дальнейшем будет видпо, что это различие, Кажущееся с первого взгляда совершенно несущественным, па самом деле имеет важное значение. Тот эффект, который у кальция выра.жается в смещении ли-шъ одного только терма искрового спектра, при продвижении в ряду олементов только на одно место приобретает столь важное влияние на химические свойства элементов, что благодаря ему, начиная е III группы, происходит ответвление побочных подгрупп,
В противоположность дублетным сериям, синглетные и триплетные серии относятся К нейтральным атомам, Такой вывод следует уже из значительно мепьших эффективных зарядов ядра для этих серий. Однако для главной синглетной серии это яснее всего следует из численного значения границы серии, которое соответствует работе отрыва одного электрона от нейтрального атома и величину которого, например для магния, удалось подтвердить эксперимент а л г. но — измерением этой работы методом «электронного удара» (Vj найдено равным 7,75 в, вычислено ив границы серии 7,61 в). «Искровые линии» металлов щелочноземельной группы появляются уже в дуговом спектре, хотя они здесь и менее интенсивны, чем в искровом. Это объясняется тем обстоятельством, что однократно ионизированные атомы металлов этой груцпы сравнительно легко отдают еще одни электрон — явление, которое, как уже было указано, в свою очередь обусловливает почти исключительную двухвалентность этих элементов.
Вторая группа периодической системы
283
Нейтральные атомы щелочноземельных металлов дают синглетные и триплетные серии, но пе дают дублетных, как их однократно ионизированные атомы пли атомы щелочных металлов. Это явление объясняется следующим образом. Энергетическое состояние атома зависит от его общего вращательного импульса. Если атом имеет на внешней сфере только один электрон, то общий вращательный импульс равен вращательному импульсу этого электрона. В этом случае для вращательного импульса данной орбиты (т. е. для определенного значения I) общий вращательный импульс может иметь только два значения, соответствующие положительному и отрицательному спинам электрона. Как было показано (стр. 197 и сл.), это приводит к появлению дублета в спектрах щелочных металлов. Одпако если атом имеет несколько электронов на внешней сфере, то общий вращателышй импульс составляется из соответствующих орбитальных импульсов и спипов всех электронов. Квантовое число, характеризующее общий вращательный импульс атома, обозначают через J. Квантовое число, относящееся к орбитальному вращательному импульсу всего атома, который слагается из орбитальных импульсов отделышх электронов,— через L, и квантовое число, характеризующее общий спин атома (полученное н результате сложения отдельных спиновых квантовых чисел с учетом их знака) обозначается через ,5'. Но чтобы определить значение J, необходимо Припять во внимание орбитальные и спиновые квантовые чий-ла, только тех электронов, которые находятся па незаполненных уровнях, так как для заполненных уровней как Z, так и S равны нулю.
Для атома металла щелочноземельной группы, имеющего на внешней сфере 2 электрона, 5 может иметь два значения; S — 0 и S = 1 (кроме того случая, когда L 0; тогда 5 может быть равно только 0);. поэтому два электронных спина могут или компенсировать один другой или складываться, Если 5 = 0, то Z= тем самым каждому значению L отвечает только один уровень энергия. Переходы электронов между этими уровнями дают только отдельные линии (синглеты). Но если 5 = 1, то J, как можно показать, имеет три различных значения для каждого L*; например, для В = 1 значения 0, 1 и 2. Тогда, следовательно, каждый Z-уровень расщепляется на три энергетических уровня, и это приводит к тому, что в спектре такого атома, кроме синглета, появляется триплет.
Мульти плотность. Количество уровней энергии, на которые расщепляется уровень, соответствующий определенному спектральному терму, называется мультиплет-ностъю терма. Мультишютность связана с квантовым числом 5 общего спина очень простым соотношением
Мул ьтиплетп ость=25 4*1. (3)
В связи с этим данные о му лътпп лети ости оказываются важными для точной характеристики (нормального или возбужденного) состояния атома.
Обозначения термов. Для точной характеристики состояния атома применяют так называемые символы термов. Их пишут следующим образом; орбитальный вращательный импульс всего атома, результирующий соответствующие импульсы отдельных электронов, обозначают одной из букв: S, Р, D или F. Они соответствуют буквам s, р_ d и / для орбитальных вращательных импульсов отдельных электронов (Ср. стр. 145). Следовательно, 5 обозначает состояние атома, для которого L = 0**, Р — состояние, для которого В — 1, D — состояние, где L = 2 и F — где L = 3. Цифра, стоящая слова вверху перед символом орбитального вращательного импульса атома, указывает мулшлиплетностъ, а внизу справа — квантовое число для общего вращательного импульса атома. Например, (читается: дублетS половина) означает состояние атома с мультиплетностью 2, орбитальным вращательпым импульсом L = 0 п общим вращательным импульсом J = Цг, Это основное состояние для атомов водорода и щелочных металлов, Основному состоянию атомов щелочноземельных металлов, так же как атома гелия, соответствует синглет-терм 150. Другим инертным газам соответствует основное состояние с тем же символом терма *50, так как у пих все электроны
* Эти значения получаются потому, чт0 в данном случае есть три возможности составить векторы орбитального вращательного импульса и спина атома так, чтобы „ , h
суммы общих результирующих импульсов б wait целочисленными кратными .
** Применение той же буквы (5) для различных обозначений; с одной стороны, как символ определенного состояния атома (L = 0), с Другой — как главное квантовое число общего спина оболочки атома,— может привести к недоразумениям; тем не менее такое обозначение общепринято.
284
Глава 8
находятся на заполненных оболочках; в этом случае L = 0 (что выражается спмво-лом 5) н 6’ = О (здесь S означает квантовое число спина) и поэтому J = 0. Иног^с перед символом терма пишут еще главное квантовое число, относящееся к соответствующему состоянию атома.
В то время как основному состоянию элементов II главной подгруппы соответствует с-шюле/пный терм, их возбужденному состоянию может отвечать как синглетный, тан и триплетный термы, То же относится к инертным газам. Наоборот, у водорода и щелочных металлов дублетный терм отвечает не только основному, но и всем возбужденным состояниям.
Для объяснения химических свойств в большинстве случаев не требуется характеризовать состояние атома символами термов. Особенно это относится к установлению связи между валентностями, существующими в гетерополярных соединениях, и строением атома. Для этого в общем случае достаточно данных о числе электронов и характере их связи в оболочке атома, которые обозначаются символами, описанными на стр. 145. Однако символы термов имеют значение не только для спектроскопистов и специалистов по атомной физике, по и для химиков. Для установления некоторых химических свойств на основании строения атома (например, для вычисления магнитных моментов атомов и иопов) в более сложных случаях необходимо знать величины — производные символов термов. Особое значение символы термов имеют для определения валентностей, существующих в гомеополярных соединениях, так как они указывают, сколько неспаренных электронов существует в атомах. В основном состоянии атома число неспаренных электронов на единицу меньше мулътиплетности термов основного состояния. Это следует из уравнения (3) в пр ед положении, что не происходит компенсаций спинов двух электронов, имеющих разные орбитальные импульсные квантовые числа. Опыт показал, что это предположение всегда Справедливо для одного атома в основном состоянии (для возбужденных атомов это не всегда справедливо). Поэтому приведенные на стр. 158 числа для нес паренных электронов в атомах элементов Li—Ne можно обозначить следующими символами термов.
1Л Be в N О F Ne
Строение атома - S- ifip i‘-p2 S2p3 s-pT
Символ терма 1lS 2рУг зр2 грз>2
Эти символы термов получены из спектральных данных и относятся к основному состоянию атома. В каждой главной подгруппе периодической системы все элеметы—аналоги имеют один символ терма.
Для полного описания строения атома необходимы данные как о распределении внешних электронов по s-,p-, d- и /-уровням, так и о символах термов. Например, основное состояние атома О описывается так: s2p4 3Р2.
Соответствующие символы термов используются для характеристики состояния атома, а также для получения данных о состоянии газообразных молекул. Вместо букв 5Г, Р, Dr F для символов термов молекул применяют буквы S, П, А, Ф. Для некоторых элементов побочных подгрупп квантовые числа для общего вращательного импульса атома пе только в возбужденных, по и в основном состоянии предполагаются большими, чем L = 3. Тогда для L = 4 вводят символ G, для L = 5 — символ II я т. д. ji алфавитном порядке.
Применение. Единственным элементом главной подгруппы II группы, применяемым в сравнительно больших количествах в металлическом состоянии, является магний. Вследствие интенсивного выделения света при горении им часто пользуются в пиротехнике, а также в фотографии. Очень распространенные «порошки для моментальных снимков» состоят из смеси порошкообразного металлического магния с такими веществами, как хлорат калия, перманганат калия, пиролюзит или нитраты редкоземельных элементов (которые при сильном нагревании легко отдают свой кислород). Подожженные при помощи фитиля из бумаги, пропитанной селитрой, или других приспособлений для зажигания, эти смеси сгорают почти моментально. Вместо них можно пользоваться также и порошком металлического магния, быстро продувая его в струе воздуха через пламя. Указанное применение магния основано на присутствии в его пламени
Вторая группа периодической системы
285
именно тех лучей, к которым фотографическая пластинка особенно чувствительна. Вообще количество энергии, которое при горении магния переходит в свет, чрезвычайно велико. Отношение части энергии, переходящей в свет, к общему ее количеству, освобождающемуся при горении, для пламепи магния приблизительно в 50 раз больше, чем для газового пламени, Чтобы при горении стеариновых свечей получить то же количество света, которое образуется при горении проволоки магния, пришлось бы сжечь в 140 раз больше по весу стеарина.
Еще большее количество света, чем в случае магния, выделяется при горении циркония и тантала. По ван Лимиту (van Licmpt, 1934 и сл.) металлы при горении в чистом кислороде выделяют следующее количество света:
Mg Al С Zr Се Та Mo W
28 26 1,9 36 9,3 32 8,7 J 9,0 лм/вт.
В препаративной органической химии магнием пользуются для реакций Гриньяра. В металлургии его применяют для освобождения металлов от растворенных в них окислов и сульфидов; магний присоединяет кислород или серу, образуя с ними нерастворимые в расплавленном металле соединения.
В настоящее время все более широкое применение находят сплавы магния. Они отличаются чрезвычайно низким удельным весом и их используют в первую очередь в самолета- н автомобилестроении, а также в машиностроительной промышленности, при производстве оптических и других инструментов.
Так, магналий (сплав магния и алюминия, содержащий 10—30% магния) имеет удельный вес 2,5—2; удельный вес «электрон-металла» Грисгеймера (сплава, состоящего на 90% из магния, с небольшим количеством других металлов — Al, Zn, Си, Мн и для особых целей Si) лишь немного больше удельного веса чистого магния (1,74). Несмотря на сильно выраженный электроположительный характер магния, все эти сплавы устойчивы па воздухе вследствие образования окисных пленок. Для продолжительного соприкосновения с водой; эти сплавы пе пригодны. Однако в последнее время удалось создать сплавы магния, практически устойчивые и к воде (например, ив 2% Мп и 98% Mg).
Злектрон-металл в противоположность алюминию не реагирует с растворами щелочей и плавиковой кислоты. От коррозии па воздухе металл можно предохранить травлением поверхности азотнокислым раствором бихромата щелочного металла. Этот сплав используют в первую очередь на отливки, но при более высоких температурах его можно вальцевать и ковать. Сопротивление на сжатие составляет около 30, сопротивление на разрыв (в зависвмостН от состава сплава) — 10—20 кг.'л/м2. Эту прочность отливки можно повышать прессованием иод давлением 25—40 кг/.ам-. Экономия веса, которую можно достигнуть при использовании электрон-мета л ла, Составляет более 80% по сравнению с железом, 20—40% с дюралюминием — и более 40% — по сравнению с деревом.
Из соединений магния применяют главным образом его карбонат, окись и сульфат (более подробные сведения о них даны при описании отдельных соединений), многие из природных силикатов, Особенно асбест. Последний находит широкое применение в качестве несгораемого, мягкого волокнистого материала. Асбестом называют разновидности роговой обманки или серпентина (хризотила), состоящие из переплетенных волокон. Из плотного серпентина, имеющего окраску от темпо-зеленой до корич
286 Глава 8
невой, изготовляют различного рода орнаменты, плитки и сосуды. Блпт-кий по составу к серпентину жировик служит для изготовления различит предметов обихода, например вставок для газовых горелок [в частностп для горелок в виде рыбвего хвоста и ацетиленовых горелок]; он заменяв также мелки в портновском деле; для приготовления последних приценяется также тальк, Однако талик в основном используют в качеств; порошка и аппретурного средства, а также его включают в состав разннг мазей и косметических средств. Другой содержащий воду силикат, «морская пенка», служит для изготовления курительных трубок.
Особенно разнообразное применение находят соединения кальцин. Известняк чрезвычайно важен в строительном деле. Гидрат изготовляемой из пего едкой извести, т. с. гидроокись кальция, является наиболее часто употребляемым в технике основанием. Гипс применяют для штукатурных работ, для отливок и форм. Равным образом имеют большое значение в технике хлорид, фторид и нитрат кальция. Карбид и цианамид кальция будут рассмотрены в гл. 12, в разделе, посвященном соединениям углерода. Большинство стекол, которые будут подробно описаны в той же главе, относятся к соединениям кальция.
Металлический кальций применяют при получении сплавов свинца. Так, например, на железных дорогах для изготовления подшипников широко применяют сплав свинца с 0,7% кальция, 0,6% натрия и 0,04% лития.
Из соединений стронция наибольшее практическое значение имеет его нитрат, применяемый в пиротехнике', кроме того, в сахароваренной промышленности пользуются карбонатом стронция и его окисью.
Из соединений бария технически важными являются его окись, гидроокись, перекись и сульфат. В качестве химического реактива широко применяют его хлорид, а в пиротехнике — нитрат и хлорат. Особое внимание следует обращать иа ядовитость соединений бария.
Из соединений бериллия наибольшее применение имеет его нитрат. Окись бериллия приобретает все большее значение как материал для изготовления огнеупорной посуды.
Металлический бериллий находит применение для изготовления окон в рентгеновских установках. Оп поглощает рентгеновские лучи в 17 раз слабее, чем алюминий*. Твердость тяжелых металлов при сплавлении их с бериллием увеличивается в значительной степени; так, медпо-бернлллевый сплав, содержащий около 6,3% бериллия, отличается твердостью стали и при этом приобретает чрезвычайно высокую устойчивость по отношению к механическим и химическим воздействиям, При вЫплав-ке меди большое техническое значение имеют очень малые добавки бериллия, обладающие раскисляющим действием (ср. стр. 386). «Электропроводная медь», получаемая при добавках 0,01—0,02% бериллия, обладает значительно лучшей электропроводностью, чем фосфорная бронза (сплавы меди с оловом, к которым при литье добавлен в качестве раскисляющего агента фосфор). Бериллий и его соединения при вдыхании их в небольших количествах в пылевидном состоянии вызывают медленно развивающееся тяжелое заболевание легких [о Противодействующих средствах см.: Schubert J., Chem. Engng. Progr., 48, 26, 1952].
Применение соединений радия основано па их радиоактивности; чаще всего используют хлорид, бромид, карбонат и сульфат радия. Все эти соединения применяют в медицине, а также для научных исследований.
* В последнее время он находит применение в атомной технике в качестве отражателя нейтронов.
Вторая группа периодической системы
287
СОЕДИНЕНИЯ ЭЛЕМЕНТОВ ГЛАВНОЙ ПОДГРУППЫ ВТОРОЙ ГРУППЫ
Представление о составе всех относящихся сюда соединений дает следующая таблица:
Гидриды М^Нг
О кислы МПО, гидроокиси М11(ОН)а
Перикиси — МпОг, надперекиси MOQi
Нитридь!
Карбиды или ацетилиды МпСз
Интерметаллические соединения, см. табл. 91—93 (стр. 620—622) и табл. 98 (стр. 630).
Соли:
галогениды М1]1Ха карбонаты Мп(СОз)
нитраты Мп(КОз)2 сульфаты MII(SO)4
Двойные соли (см. табл. 49).
Соединения магния и кальция получают в самых различных отраслях техники в виде побочных продуктов — отходов производства. Для их полупения исходят преимущественно из природных карбонатови сульфатов. Главным исходным материалом для получения соединений бария является тяжелый шпат, для соединений стронция — целестин. Для технического получения соединений бериллия большей частью исходят из берилла.
Чтобы разложить берилл, его сплавляют с карбопатом калия; растворением плава в разбавленной серной кислоте с последующим нагреванием осаждают кремневую кислоту, затем главную массу алюминия выделяют из фильтрата в виде калиевых квасцов, а остаток его осаждают углекислым аммонием, причем одновременно выпадает и железо, присутствующее в берилле в виде примеси. Из фильтрата, подкисленного соляной кислотой, после удаления из него двуокиси углерода бериллий осаждается аммиаком в виде гидроокиси- Для полного удаления примеси алюминия обработку раствора углокислым аммонием необходимо производить несколько раз. Очистку бериллия можпо также осуществить перекристаллизацией его оксиацетата из хлороформа. Более полного отделения примесеи, согласно Штоку (Stock, 1925), можно достигнуть при перегонке основного ацетата бериллия.
Соединения радия здесь не будут подробно рассмотрены. Насколько известно в настоящее время, они вполне аналогичны соответствующнлг соединениям бария.
ГИДРИДЫ
Все металлы главной подгруппы второй группы способны образовы-вать соединения с водородом, в которых они, как обычно, двухвалентны*. Устойчивость водородных соединений повышается от Be к Са, а затем опять падает от Са к Ва. Водородные соединения щелочноземельных металлов имеют ярко выраженный солеобразный характер, как и гидриды щелочных металлов. Это не относится к водородным соединениям Бе и Mg, продукты их гидролиза свидетельствуют о том, что п в них металлы имеют более сильный электроположительный характер, чем водород. Все водородные соединения элементов главной подгруппы II группы (состав М^Н.в) представляют собой бесцветные, твердые, нелетучие вещества.
* При изучении полосатых спектров установлено существование молекул ВеН, MgH, СаН, Последние существуют, однако, только в газообразном состоянии в'равпе-весии со свободными атомами, и их нельзя выделить в весовых количествах.
288
Глава 8
Гидрид бериллия. ВеН2 был но лучён Вибергом (Wiberg, 1951) обменной реакцией BeCl, с LjTJ в (эфирном растворе) и одновременно Шлезингером (Schlesinger, J95F при взаимодействии Ве(СН3)2 с алюмогидридом лития LiAlII4 или А1(СН3)2Т1:
ВеС12 4-2LiH = BeHs+2LiCl; 2Be(CH3)24-LiAlH4 = 2ВеН24- ЫА1(СН,)4
Be (CHs)2-}-2A1 (СН3)2Н = ВеН2+2А1(СП5)3.
Гидрид бериллия представляет собой белое, твердое, нелетучее вещество, по свойствам подобное гидриду алюминия, по в противоположность последнему не растворимое в эфире. Водой и метиловым спиртом ВеН2 разлагается с выделением водорода:
ВоНа4-2НОН = Ве(ОН)2-J-2IJ2; BeH2-|-2CH3OJf = Be (ОСН3)г -)-2Нг.
Гпдрпд магния. MgH2 получают синтезом из элементов (нагревание под давлением в присутствии MgJ2; В ибер г, 1951). Впервые это соединение было получено Виборгом (1950) термическим разложением диэтилмагния в высоком вакууме: Mg(C2H5)4 = = AfgH2-!-2С2Н4 (при 175°).
MgH2 получается также при замещении магпийдиалкилов в эфнриом растворе диборанйм В2Не (Выборг, 1950) или алюмогядрядом лития LiAlH4 (Шлезингер, 1951).. Гидрид магния представляет собой белое, твердое, нелетучее соединение, слабо растворимое в эфире; п топко измельченном состоянии Mgll2 самовозгорается, но в лиде компактной массы он устойчив па воздухе. При нагревании в вакууме MgH2 отщепляет водород только выше 280’, в то время как Ве112 распадается на элементы уже при 125", a A1HS в зависимости от Степени полимеризации — между НО и 160°. С водой и метиловым спиртом MgH2 реагирует аналогично ВеН2.
Гидриды щелочноземельных металлов.—Гидриды Са, Sr и Ва по внешнему виду и свойствам совершенно аналогичны щелочным гидридам. Давление их разложения, по Эфраиму и Михелю (Ephraim, Michel, 1921), лежит при более высоких температурах, чем гидридов типичных щелочных металлов, но ниже, чем гидрида лития. Устойчивость уменьшается от гидрида кальция к гидриду бария. Металл, образующийся при разложении гидрида, дает твердый раствор с оставшимся гидридом, в связи с чем значительно снижается давление его разложения. Этого явления у щелочных гидридов не обнаружено.
В кристаллической решетке гидридов щелочноземельных металлов, по Цинтлю (Zin 11, 1935), воны металлов расположены приблизительно в гексагональной плотнейшей упаковке (рис. 57). Попы Н_, вероятно, расположены в наибольших пустотах между ионами металлов таким образом, что каждый поя металла окружен 7 попами Н_, рас положе иными z Па различных расстояниях. Ниже приведены плотности гидридов:
дли СаНй SrHn Ball 2
d 1,90 3,2“ 4,15
ОКИСЛЫ И ГИДРООКИСИ
Нормальные окислы элементов щелочноземельной группы получают (помимо сожжения соответствующих металлов (см. стр. 266)] главным образом термическим разложением их кислородсодержащих солей, например карбонатов и нитратов. Опп представляют собой белые, трудно плавящиеся и рыхлые (если были получены при не слишком высокой температуре) массы. Соединяясь с водой, окислы переходят в гидроокиси, теплоты образования которых и растворимость приведены в табл. 53.
Теплоты образования, приведенные в табл. 53, свидетельствуют об образовании стабильно кристаллизующихся гидроокисей (за исключением Са(ОН)2) из стабильной модификации окисла и жидкой воды мри постоянном давлении. Гидроокиси встречаются также отчасти п в метастабильион кристаллической форме, теплота образования которой пи же, чем стабильной. Степень измельчения окисла также оказывает существенное влияние на теплоту образования гидроокиси.
Вторая группа, периодической системы
289
Т ad лица S3
Теплоты образовании гидроокисей из о кислой и растворам ость гидроокисей
Гидроокись Вс(0П)2 Mg(OH)S Са(0Ц)2 Sr(OH)£ Ba(OH)s
i Теплота образования, одгл/лсл* , . t Растворимость, г окиси/100 г раствора 2,7 2.J0-* при 20° 7,3 2-10-» при 18° 15,2 6,1 при 20° 14,7 0,7 при 20° 17,3 3,6 при 20°
Изменение химических свойств в ряду гидроокисей щелочноземельных металлов является хорошей иллюстрацией правила, согласно которому основной характер гидроокисей в пределах одной группы периодической системы возрастает с увеличением атомного веса. Гидроокись бериллия имеет амфотерный характер: она может функционировать в качестве кислоты и в качестве основания. Гидроокись магния обнаруживает лишь слабо щелочной характер. Гидроокиси кальция и стронция уже являются довольно сильными основаниями, а гидроокись бария — настолько сильное основание, что приближается в этом отношении к гидроокисям щелочных .металлов.
Усиление основного характера в направлении от Mg(OH)2 к Ва(О11)2 сказывается также па коэффициентах электропроводности / Последние, по данным Хласко
(Hlasko, 1935), при 25° равны: MgfOHja Са(ОН)з Sr(OH)s Ва(0Н)2
в 0,031 и. растворе / - 0,703 0,737 0,831
в 0,00006 н. растворе 0,882 0,962 0,967 0,979
Наблюдаемую здесь закономерность в измененни свойств гидро окисей можно объяснить с точки зрения представлений Косселя, согласно которым прочность связи между двумя противоположив заряженными атомами в молекуле в первую очередь определяется их электростатическим притяжением, следующим из закона Кулона. Если но аналогии е гидроокисями щелочных металлов для гидроокисей элементов щелочноземельной группы вычислить значения эпергии, которая необходима, с одной стороны, для отщепления одного иона ОН', а с другой — одного пола Н‘, и полученные величины нанести на график (как это было сделано па рис. 34, стр. 205), то получим две кривые, подобные Кривой рпс. 34. Различие между кривыми заключается в том, что в данном случае они будут лежать ближе одна к Другой и подниматься круче; таким образом, amt взаимно пересекутся вблизи гидроокиси бериллия. Здесь, следовательно, разница между энергиями, необходимыми для отщепления ионов ОН' и ионов Н', уже невелика, В соответствии с этим гидроокись бериллия может диссоциировать вс только в качестве основания в соответствии с уравнением
Ве(0Н)2— Ве”+2ОН', (3)
но и в качестве кислоты по уравнению
Ве(ОН)2=ВеО£+2Н-. (4)
Приведенный способ вычисления слишком приблизителен, чтобы дать указание относительно того, какой из этих Процессов в каждом данном случае будет преобладать. Поведение гидроокиси бериллия показывает, что преобладающим является процесс (3). В чисто водных растворах в равновесии
Ве"+2ОН' Bc(K-J-2H’ (5)
будут, следовательно, преобладать ионы, находящиеся в левой части уравнения. Если же концентрацию водородных ионов уменьшить до очень небольшого значения, другими словами, если сделать раствор сильно щелочпым, то, согласно закону действия масс, процесс должен будет протекать слева направо, т. е. в сильно ще.точпом растворе 19 Г. Реми
290
Глава 6
будут образовываться бериллат-ионы [ВеО2[". Соответствующие соли типа М{[ВеОг|» так называемые бериллаты, в которых окись бериллия входит в состав аниона, нельзп выделить из водных растворов, но они хорошо выделяются из спиртового раствора,.
Кислотные свойства гидроокисей могут проявляться также и в том, что спи способны присоединять ионы ОН'. Па основании закона Кулона можпо показать, что прп этом также может выделяться энергия (см. гл, 11). Вероятно, у большинства гидроокисей металлов в водных растворах присоединение ионов ОН' происходит легче, чем отщепление ионов ЕГ; таким образом, амфотерные гидроокиси по отношению к сильным основаниям в водных растворах функционируют главным образом не как собственно кислоты, а как ангидр окисло ты. Поэтому, вероятно, Ве(ОН)2 в щелочных водных растворах диссоциирует не в соответствии с уравнением (4) с отщеплением ионов Н‘, а, наоборот, При присоединении иопов ОН' образует так называемые еидроксо-иоиы.
Be (ОН)з-|-О1-Г = [Бе (ОН)3]' (тригидр оксоберилл ат-ио и), Be (0Н)а-}-20Н' = [Be (OH)J" (тетр агидроксо бе рил лат-ион).
Применение закона действия масс к уравнению
Ве”4-4ОН'±^ [Be(OH)J" (6>
приводит к тому же выводу относительно зависимости концентрации ионов бериллия, ионов бериллата и соответственно гидр оксобериллата от концентрации ионов водорода раствора, что и применение этого закона к процессу, описываемому уравнением (5).
В соответствии со сказанным выше можно ожидать, что и сравнительно сильные основания могут функционировать в качестве кислот или ап гидр окис лот, если только-
Рис. 60. Кристаллическая решетка типа вюртцита.
Пример- ВеО А. е=4,37А,
p=J;gC=i!e5'>A).
удается сдвинуть достаточно вправо равновесие, соответствующее уравнениям (5) или (б), за счет повышения концентрации ОН'. Действительно, Ш о л деру (Sch older, 1935) удалось показать, что даже очень сильное основание — гидрат описи бария — в отношении концентрированного раствора едкого натра ведет себя как амфотерное соединение, Растворимость Ва(ОН)а в воде значительно понижается при прибавлении небольших количеств NaOH, по она опять быстро возрастает если концентрация NaOH превышает 10 молъ!л. Следовательно, в данном случае но отношению к концентрированным растворам едкого натра проявляются те же-свойства, что и в случае типично амфотерных гидроокисей, таких, как Ве(ОН)2 или А1(ОН)3, по отношению к разбавленным растворам едкого натра,
Кристаллическая решетка окислов Mg, Са, 8г и Ба аналогична решетке хлористого натрия (см, рис. 44, стр. 237) с тем только-отличием, что узлы ее заполнены не одновалентными, а двухвалентными
ионами. Указанные окисли имеют тот же тип решетки, что NaCl (и многие другие щелочные хлориды), Герлах (Gerlach) и Паули (Pauli) на основании рентгенографического исследования окиси магния (см. стр. 242) показали, что в основе кристаллической решетки окислов щелочноземельных металлов лежат ионы, а не атомы. Длина’ребер куба а здесь следующая г
MgO СаО SrO ВаО
щА 4,20 4,80 5,15 5,53
Кристаллическая решетка ВеО находится в таком Же отношении к решеткам MgO, СаО и т. д,, как решетка элементарных Be и Mg к решетке элементарного Са, В решетке поваренной соли каждый вид ионов образует гранецентрироваппую кубическую ячейку, т.е. кубическую плотнейшую упаковку. Таким же образом и в решетке ОеШсп бериллия (рис. 60) каждый из двух видов атомов образует ячейку с гексагональной плотнейшей упаковкой. Показанный на рис. GO тип решетки получается, если вставить одну в другую обе эти ячейки. Этот тип решетки обычно называют типом вюртцита, так как он впервые был установлен для вюртцита (гексагональная модификация ZnS). Для ВеО а = 2,69 А, с = 4,37 А. Каждой атом Be окружен здесь четырьмя атомами О, расположенными вокруг него в виде тетраэдра, а каждый атом О тетраэдрически окружен четырьмя атомами Be. Расстояние Be «—+ О = р = 1,65 А.
В соответствии с различием в типах структур ВеО не образует твердых растворов с MgO и СаО. Однако MgO также не дает твердых, растворов с СаО вследствие слишком
Вторая еруппа. периодической системы 291
большой разницы в величинах ионных радиусов. Температуры плавления эвтектик достигают для BeO/MgO 1955°, для ВеО/СаО 1475° и для MgO/CaO 2360" (Ruff, 1933). Эти температуры оказываются значительно ниже^ очень высоких температур плавления чистых окис лов
ВеО MgO CaO SrO ВаО
Температура плавления, °C 2530 2800 2576 2430 1923
Окись бериллия. ВеО (бериллиевая земля, сладкая земля) образуется при прокаливании гидроокиси бериллия или бериллиевых солей, например карбоната или сульфата. Это белый рыхлый порошок, плавящийся и одновременно испаряющийся в электрической иечи, Однако ВеО удается перевести в кристаллическое состояние, прокаливая ее с некоторыми веществами, так называемыми минерализаторами (понижающими точку плавления), или другим способом, способствующим кристаллизации. В воде ВеО растворяется чрезвычайно слабо, являясь наиболее труднорастворимой окисью из всей группы щелочноземельных металлов (см. табл. 53). Щелочные металлы не восстанавливают ВеО. При прокаливании в электрической печи с углеродом ВеО переходит в карбид ВегС, подобный карбиду алюминия.
Гидроокись бериллия. Ве(ОН)а выпадает в виде белого, студенистого осадка, растворимого как в кислотах, так и в едких щелочах при действии на растворы солей бериллия ионов ОН'. Одпако щелочные растворы Ве(ОН)а постепенно снова разлагаются; этот процесс ускоряется при нагревании. При этом в осадок выпадает более трудно растворимая модификация гидроокиси. Ве(ОН)а выдерживает высушивание при 100°; при более сильном нагревании Ве(ОН)з, отщепляя воду, переходит в окись бериллия. Св еже оса ж де иная гидроокись бериллия легко растворяется в кислотах и сильных щелочах, а также в водном растворе карбоната аммония. Своей растворимостью в последнем она отличается от гидроокиси алюминия; другое различие между этими гидроокисями заключается в том, что Ве(ОН)а нерастворима в этиламине. Растворимость Ве(ОН)а в едких щелочах связана с образованием бериллатов. Бериллат калим, КаВеОа, и бериллат натрия, NaaBeOa, были получены из спиртовых растворов. Вода гидролитически расщепляет бериллаты, например:
К2ВеО24-2Н2О = Вс(ОН)г+2К'+2ОН'
и поэтому их пе удается выделить из водных растворов.
Гидроокись бериллия, полученная из берилла, служит исходным материалом для приготовления всех остальных соединений бериллия.
Окись магпия. Магнезия, горькая земля, MgO в виде белого, рыхлого, трудно плавящегося порошка (жженая магнезия, magnesia usta) образуется при горении магния па воздухе или при прокаливании гидроокиси, карбоната, нитрата или других кислородсодержащих солей магния (при сильном прокаливании полностью разрушается и сульфат). В электрической печи MgO сублимируется и затем вновь осаждается в виде кристаллов. Кристаллическую MgO легче получить при прокаливании с минерализаторами, например с боратом кальция, или сильно нагревая ее в струе хлористого водорода. В природе кристаллическая окись магния встречается в виде очень мелких правильных октаэдров и кубов, образуя минерал периклаз (твердость 6, удельный вес 3,7). Окраска периклаза благодаря содержанию в нем железа колеблется от серо-зеленой до томно-зеленой. В то время как вода оказывает на кристаллическую MgO еле замет
19*
292
Глава 8
ное действие, а кислоты реагируют с ней лишь с трудом, порошкообра-ная окись магния легко растворяется в кислотах. Вода медленно пр? вра/цает ее я гидроокись.
Техническое применение окиси магния основано главным образам на ее тугоплавкости. Поэтому MgO используют главным образом дет изготовления тугоплавких приборов-, кроме того, из нее приготовляю^ магнезиальный цемент и ксилолит; в медицине ею пользуются как протс воядием при отравлении кислотами и в некоторых других случаях. В технике окись магния получают прокаливанием магнезита или гидр о окис? магния (образующейся в рассолах, остающихся при производство калиг ных солей), содержащих хлористый магний. Для этого к рассолам доба вляют немного известкового молока (для осаждения железа в виде гидре окиси) и ионов сульфата (в виде сульфата кальция). Последующее добавление известкового молока вызывает осаждение MgO. Другой споссГ получения окиси магния заключается в обработке хлористого магнит перегретым водяным паром
MgCJ2 4- НЯО=MgO 4- 2HG1-
В этом случае в качестве побочного продукта получается соляная кислота; однако этот способ требует больших затрат топлива, так кат разложение MgO а происходит полностью только при температур'-' около 500°.
Окись и гидроокись магния в настоящее время получают (в основном в США) в большом количестве из морской воды. При этом обычно сначала известковым молоком осаждают ионы НС02 в виде СаСОз, затем осадог отфильтровывают и повой порцией известкового молока осаждают понте Mg” в форме Mg(OH)2
Са(ПС03)3+Са(0Н)г = 2СаС034-2НаО;
MgCl2+Ca(OH)3=Mg(OH)24-CaCI2.
Главная трудность заключается в том, что гидроокись магния плохо осаждается из морской воды и плохо фильтруется. Это можно преодолеть, если промытый тляп Mg(0H)2 обработать СО2; при этом получается лучше фильтрующийся гидрат карбоната, который при прокаливании переходит в MgO;
Mg (ОН)2+СО24-2И2О = MgCO3.3H2O,
MgCO3 ЗН2О -> MgO4-CO24-3H2O.
Кроме того, при разложении уплотненного шлама Mg(OH)2 соляной кислотой получают непосредственно MgCl3, который используют для получения металлического магния электролизом его расплава.
«Горькие воды», выпадающие в «соляных садах» (см. стр. 213), раньше шли в отход, теперь их тоже перерабатывают на окись магния. При этом (после удаления брома и осаждения ионов SO" в виде гипса) Mg(OH)2 осаждается юже известковым молоком.
Гидроокись магния. Mg(OH)2 образуется при действии воды на порошкообразную окись магния или на стружки магния и, кроме того, при реакции между гидроксильпыми ионами и ионами магния
Mg"4-2OII' = Mg(OH)2. (7)
Свойства гидроокиси магпия несколько зависят от способа ее приготовления. В воде гидроокись магния плохо растворима. Так как произведение растворимости Пр = IMg”]x [ОН'Р довольно велико, гидроокись магния осаждается водным раствором аммиака неполностью, и осаждения
Вторая группа периодической системы
293
ее но происходит вообще в присутствии больших количеств солей аммония, которые подавляют электролитическую диссоциацию аммиака*. При 100° гидроокись магния не разлагается. При слабо-красном калении она переходит в окись.
В природе гидроокись магния встречается в виде минерала бруцита. Последний образует тонкие, гибкие, блестящие, бесцветные или зеленые листочки, имеющие на изломе перламутровый блеск. Они состоят из кри-
сталлов гексагональноп системы, имеют твердость 2 и удельный вес 2,3—2,4.
Элементарная ячейка бруцита приведена аа рис. 61. Такая слоистая решетка часто встречается у гидроокисей двухвалентных элементов, а кроме того, у иодидов (например, Cdl3, MgJ2, Gala), у MgBA, я у сульфидов, например SnS2.
Окись кальция. Едкая известь, жженая известь, СаО образуется так же, как и окись магния. Получают ее исключительно прокаливанием карбонатов. Для технического получения обычно исходят из известняка, который нагревают в шахтных печах, а в более крупном масштабе — в кольцевых печах, до 800° («обжигают»). Чтобы разложение
СаСО3 = СаО4-СО3
Рис. 61. Решетка бруцита. o=t>=3.t2; с=4.73; d=t,05 А,
могло происходить при указанной температуре, необходимо отсасывать выделяющуюся двуокись углерода, так как давление COs превышает атмосферное давление только при температуре, значительно превышающей 800° (см. табл. 57, стр. 312). Часто, однако, бывает достаточно тяги горячих газов, чтобы вызвать необходимое снижение давления. Обжиг при слишком высокой температуре, помимо лишней затраты топлива, вызывает спекание извести, если известняк содержал примесь Глинистых веществ, что сильно затрудняет последующее гашение такой извести («пережог»). Совершенно чистая окись кальция плавится чрезвычайно трудно. Окись кальция, получающаяся при прокаливании ие содержащего примесей углекислого кальция (мрамора), представляет собой рыхлый аморфный, довольно легко реагирующий с вОдой порошок даже в том случае, когда нагревание производили до очень высокой температуры. Реакция жженой извести с водой известна под названием «гашения» извести- Ока сопровождается выделением значительного количества тепла (см. табл. 55 стр. 289). При внесении окиси кальция в пламя гремучего газа она дает очень яркий свет («друммондов свет»). Расплавленная в электрической печи окись кальция при застывании образует кристаллы удельного веса 3,40. При нагревании до высокой температуры в присутствии угля СаО образует карбид, который широко применяется в технике (см. стр. 478). Кроме приготовления известкового раствора (гидроокиси кальция), СаО применяют как материал для футеровки печей, в качестве
* Как иск аз ал Фредгольм (Fredholm, 1934), значительное повышение растворимости труд нерастворимых солей магния при добавлении аммиака является признаком того, что ион Mg- спо собе к присоединить NH3 с образованием нестойкого комплекса. Однако это комплексообразование оказывает заметпое влияли с на равновесие (7) только при очень высоких концентрациях аммиака.
294
Глава 8
основной добавки при плавке металлов, в стекольном производстве и кат удобрение. Иногда ею пользуются также и в качестве осушителя, одновременно поглощающего СО а. Чистая окись кальция, полученная при обжига мрамора, находит применение в медицине, в частности как средство п.тг уничтожения бородавок.
Гидроокись кальция. Гашеная известь Са(ОН)г— продукт присоедЕ-пения воды к окиси кальция, образует белого цвета пылеобразный аморфный порошок удельного веса 2,08. Са(ОН)з теряет воду только при температуре выше 100°. Давление пара НаО пад гидроокисью кальция достигает 1 атм лишь при 450°. В воде Са(ОН)а растворяется довольно трудно, с небольшим положительным тепловым эффектом (2,8 ккал). Присутствие солей щелочных металлов, и особенно хлористого аммония, несколько повышает ее растворимость. Водный раствор гидроокиси кальция (известковая вода) имеет сильно щелочную реакцию, но все же более слабую, чем эквимолярный раствор гидроокиси калия. При реакции с тростниковым сахаром Са(ОН)а образует сахараты, которые могут содержать 1—6 молекул СаО на 1 молекулу тростникового сахара. Поэтому гидроокись кальция растворяется в растворах тростникового сахара гораздо лучше, чем в чистой воде: так, в 100 е.и3 8%-ного раствора тростникового сахара растворяется 22,4 г СаО. Растворимость же ее в чистой воде составляет:
при 0а Т'й” ТОЙ
0,131 0,126 0,098 0,060 г СаО в 100 а Н2О
Основное применение гидроокись кальция находит в строительном деле в виде известкового раствора. В сахароваренном производстве ею пользуются для выделения сахара из мелассы, черной патоки, остающейся после выделения сахарного песка. Избыток гидроокиси кальция осаждает часть сахара в виде сахарата кальция. Далее, Са(ОН)з применяют для изготовления хлорной извести, а также используют в технике в качестве дешевого основания в очень многих производствах, например для выделения аммиака, для каустификации соды; кроме того, ее применяют в качестве едкого вытравливающего средства, например в кожевенном производстве для удаления волос со шкур или для обезвреживания гниющих органических веществ. В виде известковой воды Са(ОН)г применяют также в медицине, например как средство против ожогов или противоядие при отравлениях серной или щавелевой кислотами. Известковое молоко, которым белят потолки, представляет собой суспензию гидроокиси кальция в известковой воде.
Кристаллическая гидроокись кальция выделяется в виде гексагональных пластинок при упарнвапш ее водного раствора (структурный тип бруцита; а — 3,58 А, с = 4,90 Л); эта фирма Са(ОН)2 гораздо менее реакционноспособна, чем гидрогель, полученный обычным образом — гашением жженой извести. Последний содержит поэтому всегда больше воды, чем соответствует формуле Са(ОН)2, и даже при сильном понижении давлоппя пе отделяет всю воду, прочно удерживаемую за счет абсорбцци. Коллоидный характер оказывает определенное влияние па реакционную способность Са(ОН)2 и в связи с этим на качество полученного из нее цемента.
Окись и гидроокись стронция. Окись стронция (стронциан). SrO, как и окись кальция, обычно получают прокаливанием карбоната. Однако ввиду того что давление диссоциации у карбоната стронция ниже, чем у карбоната калвция (оно достигает 1 атм лишв при 1100°), карбонат стронция приходится прокаливать значительно сильнее. Окись стронция
Вторая группа периодической системы 295
образует белую аморфную массу удельного веса 3,93—4,61. Ее можно получить также в кристаллическом состоянии. Аморфная окись стронция соединяется с водой, выделяя значительное количество тепла и образуя гидроокись Sr(0H)2, которая с избытком воды дает гидраты; наиболее богатым водой является октогидрат 8г(ОН)2-8НгО, который может переходить в моногидрат. Ок та гидрат растворяется в воде с отрицательным тепловым эффектом (—14,6 ккал), в то время как при растворении в воде безводной гидроокиси выделяется 11,6 ккал тепла. Растворимость Sr(OH)a в воде (в противоположность гидроокиси кальция) сильно возрастает с повышением температуры и при 100° довольно значительна. Насыщенный раствор Sr(OH)s содержит в 100 з
при 0" 20" 50" 100"
0,35 0,68 2,13 18,60г SrO.
Гидроокись стронция — сильное основание. С тростниковым сахаром она образует сахарат. Сахарат стронция CiaH22Oii-2SrO растворяется труднее, чем сахарат кальция, и поэтому более пригоден для выделения сахара из мелассы. Кроме указанного применения при сахароварении, окись стронция служит исходным материалом для получения других соединений стронция.
Окись бария. ВаО (едкий барит, баритовая земля) в чистом состоянии удобнее всего получать при прокаливании нитрата иди йодата бария. Однако препарат, образующийся при недостаточно сильном прокаливании, содержит перекись. В технике ВаО большей частью получают прокаливанием карбоната бария, смешанного с углем. Вместо угля можно использовать также карбид бария ВаСа. Указанные вещества вводят для связывания выделяющейся СО а и перевода ее в СО, в результате чего разложение карбоната бария происходит полностьюj несмотря па крайне низкое давление СОг над ВаСОз. Это давление достигает 1 атм лишь при ярком белом калении, когда окись бария уже заметно спекается. Чистая окись бария представляет собой белый порошок удельного веса 4,7—5,8. Техническая ВаО бывает большей частью загрязнена примесью угля и поэтому окрашена в серый цвет. ВаО плавится труднее, чем SrO и СаО. Из расплава опа застывает в виде кристаллов. С водой ВаО образует гидроокись бария, причем эта реакция сопровождается сильным выделением тепла (17,3 ккал). При умеренном прокаливании на воздухе (около 500°) ВаО переходит в перекись.
ВаОф-1/аО2 = ВаОаф-16,2 ккал.
ВаО применяют главным образом для получения^ Ва (ОН) а и ВаОз.
Гидроокись бария. Ва(ОН)г— продукт присоединения воды к окиси бария — в безводном состоянии представляет собой белый аморфный порошок удельного веса 4,5; она плавится, не разлагаясь. В воде Ва(0Н)2 растворяется значительно лучше, чем гидроокиси остальных щелочноземельных металлов. Растворимость ее составляет:
ари С," 20" 50" 80’
1,5 3,84 11,75 90,8 г ВаО в 100 г Н2О
Таким образом, растворимость Ва(ОН)’ заметно повыпшется с температурой, и горячая вода растворяет ее в довольно значительных количествах. Присутствие нейтральных солей приводит к некоторому увеличению ее растворимости, а гидроокисей щелочных .металлов, наоборот,—
296
Глава 8
к сильному понижению. Водный раствор Ва(0Н)з имеет сильно щелочнуа; реакцию; он является чувствительным реактивом на СОа (образуется белый осадок ВаСОз). При упаривании водных растворов Ва(ОН)а выпадает в виде кристаллов, содержащих воду. При обычной температуре и вплоть до 100° и несколько выше в равновесии с раствором устойчивых? является октагидрат Ва(О1-1)2.8НяО. Он может переходить в моногидрат. Октагидрат образует бесцветные тетрагональные кристаллы удельного веса 1,66, которые при 78° начинают плавиться в своей кристаллизационной воде. Растворяется октагидрат в воде с отрицательным тепловым эффектом (—15,2 ккал), а растворение безводной Ва(ОН)а сопровождается положительным тепловым эффектом (-]-12,3 ккал). Спирт высаливает из водного раствора гидроокиси бария значительные количества Ва(ОН)а.. Насыщенный при обычной температуре раствор гидроокиси (баритовая вода) часто применяют в лабораторной практике,
ПЕРЕКИСИ И НАДПЕРЕКИСИ
Щелочноземельные металлы, подобно щелочным металлам, образуют белые перекиси — МпОа и желтые надперекиси — МНО4. Соединения первого типа при осторожной обработке кислотами дают только перекись-водорода, в то время как надперекиси выделяют, кроме того, молекулярный кислород:
СаОЕф- H2SO4= CaO4-(-H2SO4™ CaSO4-(-H2O2-(“ О2.
Белые перекиси, как и перекись водорода и перекиси щелочных металлов, содержат группу Оа_, желтые надперекиси, подобно надперекисям щелочных металлов — группу О’- (Ehrlich, 1944).
В воде как перекиси, так и надперекиси щелочноземельных металлов, растворяются с трудом. Вследствие гидролиза их водные растворы имеют сильно щелочную реакцию
МпО2+2Н2О Мп(ОН)24-Н2О2.
Мокрым путем перекиси щелочноземельных металлов можно получить, действуя перекисью водорода на их гидроокиси, например:
Са(ОН)2ф- НаО2 = СаОг4-2Н2О.
Этот метод синтеза соответствует нейтрализации основания (двухосновной) кислотой и вместе с тем подтверждает (стр. 203) вывод о том, что перекиси являются солями перекиси водорода.
Устойчивость перекисей возрастает параллельно усилению электроположительного характера образующих их металлов. Для бериллия перекиси вообще неизвестны; для магния известны только гидраты, перекисей; для кальция его безводное перекисное соединение СаО2 удается получить лишь обезвоживанием соответствующего октагидрата СаОа.8НаО; перекись стронция ЯгОз можно получить уже непосредственным действием: кислорода па окись стронция, однако лишь при высоком давлении; перекись бария, напротив, легко получается при простом продувании струи воздуха над нагретой окисью ,бария. [ВаОз была известна рапыпе других перекисных соединений,
Надперекиси щелочноземельных металлов до сих лор не удалось выделить в чистом виде, так как о пи всегда получаются в смеси с обычными перекисями. Эти смеси (с приблизительным содержанием 8—9% надперекисеЙ) представляют собой порошки желто-зеленого цвета, устойчивые it умеренному нагреванию, по при более высоких
Вторая группа периодической системы
297
температурах разлагающиеся с образованием йерекисей. При еще более сильном нагревании разлагаются я сами перекиси. Однако для перекиси бария давление разложения достигает 1 атм только при температуре около 800°.
Перекись бария. ВаОз образует белый, довольно трудно растворимый в воде порошок, совершенно нерастворимый в спирте и эфире. С водой перекись бария даст гидрат ВаОз-8НйО; с НаОа также дает соединение ВаОа-НаОа. Водный раствор перекиси бария действует на соли двухвалентного железа как окислитель, а на гексацианоферрат(Ш)калия K3lFe(CN)e] и также на многие другие соли тяжелых металлов, наоборот, как восстановитель. Так, с хлоридом ртути (сулемой) она реагирует по уравнению
HgCl 2 -|- ВаО2=Hg BaCU-f- О2.
При действии разведенных кислот на перекись бария образуется перекись водорода, производной которой является перекись бария. Перекись водорода, как очень слабая кислота (см. стр. 75 и сл.), вытесняется из своих солей более сильными кислотами:
ВаОг4-2И’ = Ва" 4-И2Оа.
При нагревании выше 500° в вакууме или выше 700° на воздухе перекись бария отщепляет кислород
ВаО2=ВаО-|-1/2О2.
Соответствующие величины давления диссоциации р'равны:
при 525* 720“ 790“
р= 20 210 670 л.и рт ст}
Техническую ВаОз получают нагреванием пористой окиси бария до температуры около 500° в струе воздуха. При этой температуре давление кислорода над ВаО а еще настолько незначительно, что окись бария переходит на воздухе в перекись.
Перекись бария применяют для беления шелка, растительных волокон и соломы, а также — в больших масштабах —• для получения перекиси водорода, Ее используют для приготовления перкарбоната бария, для обесцвечивания свинцовых стекол и, кроме того, в качестве дезинфицирующего средства. Применяемые в алюмотермии заиалъные шарики состоят из смеси перекиси бария и магниевого порошка.
Получение кислорода во методу Брина основывалось На образовании перекиси бария при умеренном нагревании ВаО в струе воздуха и отщеплении кислорода При понижении давления или повышении температуры. Однако в настоящее время этот процесс вытеснен другим методом, а именно получением кислорода путем сжижения воздуха (см. стр. 740).
Кристаллическую решетку ВаО3 (Beraal, 1935) можно рассматривать как производную от решетки ВаО (если в последней заменить ионы О2- ионами О|_). Все линии, соединяющие два атома О группы О|“ (расстояние между их средними точками составляет 1,37 А), проходят параллельно оси с, вытянутой по сравнению с двумя другими осями (сfa = 1,27); в результате вместо кубической образуется тетрагональная ячейка (а —5,34 А, с=6,77 А). Аналогичную структуру имеет Sr02 (в = 5,02 А, с — 6,55 А). По этому же типу кристаллизуется большинство ацетилидов (тип карбида бария, си. рис. 88, стр. 507).
Перекись стропиля SrO2 и перекись кальция СаО2 по своему практическому значению уступают перекиси бария. Эти соединения в виде гидратов 8гО2-8НаО и СаО3-8Н2О (изоморфных соответствующему соединению бария) получают действием перекиси водорода или перекиси натрия на раствор гидроокиси стронция или известковую воду. Безводные СаО2 н SrO2 образуются при умеренном нагревании (до 100—130°) их октагидратов или при непосредственном осаждении из горячих растворов. Кислород они выделяют лишь при значительно более сильном нагревании
298
Глава 8
(перекись стронция — лишь ври нагревании выше точки плавления — выше температуры красного каления). Б воде обе перекиси растворяются с трудом. Кислоты разлагают их, как и перекись бария.
Теплоты образования этих перекисей из окислов равны: дан CaOa SrO 2 ВаО 2
5,4 13,5 16,2 ккал/холь
Перекись магния. Техническую перекись магния (содержащую воду и окись магния) получают различными методами (например, при действии Н2О2 на MgO).. Ее применяют в медицине.
Галогениды
Среди галогенидов главной подгруппы II группы фториды занимают особое положение, поскольку они, за исключением фтористого бериллия, трудно растворимы в воде. В то же время растворимость остальных галогенидов этой подгруппы довольно велика, а некоторые даже расплываются на воздухе. Галогениды бериллия в водном растворе в значительной степени гидролизуются. У растворимых галогенидов магния и щелочноземельных металлов склонность к гидролизу проявляется только при более высоких температурах. Галогениды магния и бериллия легко образуют двойные соли со щелочными галогенидами*.
Безводные галогениды щелочноземельных металлов кристаллизуются, как известно, главным образом по типу флюорита (рис. 62). В кристаллах флюорита ионы
Рис. 62 Решетка флюорита. Дли СаКг а=5,45 А,
Рис. 63. Кристаллическая решетка типа рутила.
Примера MgFa (а—4,62; с=3,06; d=2,05 А.)
Са2+ образуют гранецентрироваппую кубическую решетку; F'-ионы занимают середину восьми малых кубов, на которые можно разложить элементарный куб. По этому типу построены;
СаКг SrF2 BaFj RaFa SrCb
5,45 5,78 6,19 6,37 6,97 A.
Тип флюорита вообще часто встречается у соединений общей формулы АВ2, Расстояние между центрами ионов в решетках этого типа составляет (а/4)] 37
Кристаллы MgF2 построены по типу рутила (рис. 63). К этому типу относятся многие двуокиси, и в первую очередь сам рутил TiO2. Элементарная ячейка
* О составе двойных солей щелочных галогенидов с галогенидами магния, которые могут быть нолучепы из растворов, см. Klemm W. Z. anorg. chem., 256, 35, 1948.
Вторая группа периодической системы 299
этого типа состоит Из тетрагональной призмы, углы и центры которой заняты атомами или иояами одного сорта, например ионами Mg2+, в то время кал атомы или ионы другого сорта, например ионы F-, лежат на диагоналях на расстоянии d от углов и соответственно центра (рис. 63).
При охлаждении расплава EeFs (как и Si О,) в большинство случаев образуется стекло. Стеклообразный BeF2, но Баррену (Warren, 1934), имеет структуру, совершенно аналогичную спликатным стеклам (см. стр. 532). Структуры кристаллических BeF2 я SiO2 также близки*; выше 516° BeF2 кристаллизуется по типу ^-кристобалита, ниже 430°— а-кварца. Как и соответствующие модификации SiO2 (см. рис. 94), кварцев о добпый BeF2 плавится шике (540°), чем к ристоб алитоподобный (543°).
Структура СаС12 является искаженным типом структуры рутила (ромбическая ячейка). Каждый С а2* окружен 6 ионами С1_, которые лежат в углах деформированных октаэдров, 2 иона на расстоянии 2,70 А и 4 иона на расстоянии 2,76 A. MgCl2, подобно Mg(0H)2, образует слоистую структуру; расстояние Mg > С1 = 2,54 А, С1 «—* Cl между соседними слоями = 3,59 А.
MgBr2, Mg К в Са12, так же как Mg(0H)2, кристаллизуются по типу бруцита (см. ррс. 61) MgBr2: а = 3,81; с = 6,26 A; d = с/4; Mgla: а = 4,14; с = 6,88 А; d = сЦ\ Са12 : а = 4,48; с = 6,96 A, d = с/4.
Фториды
Фторид бериллия. BeF2, содержащий воду, получают выпариванием раствора гидроокиси бериллия в водной плавиковой кислоте. Для отщепления воды его нагревают в токе фтористого водорода, так как в противном случае происходит гидролиз. Проще получить безводный ВеГ2 ври нагревании фторобериллата аммония [NH4]2[BeFJ в токе С03. BeF2— стекловидная, гигроскопичная масса, легко растворимая в воде. Со щелочными фторидами образует хорошо кристаллизующиеся двойные соли (фторо бе рил латы) типа Ml[BeF3), Ma[Be2F7] и М2 [BeFJ**. Последние легко образуются при выпаривании растворов, содержащих оба фторида. При нагревании на воздухе фтористый бериллий разлагается с образованием оксифторида. Щелочными металлами и магнием BeF2 восстанавливается до металла.
Фторобериллаты обнаруживают структурную аналогию с силикатами. Например: Li[BeF3) или Li2{Be2Fe) изоструктурен энстатиту Mg2[Si206J; LiofBeF^J кристаллизуется по тину виллемита Zn2[SiO4J; у Na2[BeF4] такой же тип структуры, как у форстерита Mg2[SiO4] и всех модификаций Ca2[SiO4]; Са[ВеГ4] кристаллизуется по типу циркона Zr[SiO4]; Na2 [LiBegF?) по типу акерманита Ca2[MgSi2Or] и геленита Ca2|AlSiA107] и т. д. Между диаграммами плавкости систем фтор обе рил латов и силикатов часто имеется полное соответствие. Поэтому фторобериллаты полезно бывает использовать в качестве моделей при изучении силикатов. Талой метод удобен, так как вследствие меньших зарядов ионов фторо бе рил Латы имеют значительно более низкие температуры плавления, чем силикаты (Goldschmidt, 1926; Zachariasen, 1926; O’Daniel, 1941 и сл.; Thilo, 1949 и сл.; Hahn, 1953).
Фторид магния. MgF2 осаждается При реакции иоиа F' с растворами магниевых солей. В воде очень мало растворим (87 мг/л при 18°). Плавится при 1265° (т. кип. 2260°) и при охлаждении образует тетрагональные кристаллы. Редкий минерал — селлаит (бесцветный, со стеклянным блеском, прозрачный, твердость 5, удельный вес 2,97) представляет собой природный кристаллический MgF2. Со щелочными фтори-
* Данные Войту релло (Venturellos, 1941) о том, что BeF2 образует с MgF2 сплошной ряд твердых растворов, нуждаются в проверке, так как они получены в предположении, что BeF2 способен кристаллизоваться по типу рутила. Однако это предположение До сих пор еще не доказано и представляется мало вероятным. Установленная Бентурелло гексагональная модификация BeF2t возможно, имеет структуру, аналогичную тридимиту.
(В настоите в время известпы следующие модификации фтористого бериллия: BeF3(<z = 6,60; о= 6,74 А), подобный -кристобалиту, при 120° превращается в модификацию, подобную а-кристобалиту (а = 6,78 А). Кристобаяитоподобный BeF2 плавится около 780—800° (точно не устало вл ено); BeF2, подобный а-кварцу (а = 4,72; с = 5,80 А), при 220° превращается в модификацию, подобную кварцу (а = 4,77; с = 5,24 А); чистый кварцеподобный BeF2 плавится около 580°. (Новоселова А. В., Успехи химии, 28, 33, 1959].— Л/шм. перес.}
** Кроме указанных типов фторобериллатов щелочных металлов, известпы еще M^BeFj и1 M^BOjFg.— Прим. ред.
300
Глава 8
дамп MgFa образует двойные соли состава MfF-MgF2 и 2M'F-MgF2. Последние получаются при прибавлении MgF2 или MgO к расплавленным щелочным фторидам.
Фторид кальция. Кристаллический CaFs получают при нейтрализации карбоната кальция разбавленной плавиковой кислотой. При действии ионов F' на раствор соли кальция CaFa выпадает в виде студенистого осадка. CaFa очень трудно растворим в воде (16 мг/л при 18°), однако легко образует коллоидные растворы. С фтористым водородом дает легко растворимую кислую соль CaFa-2HF-6HaO. Некоторые другие вещества также повышают его растворимость. Безводный фтористый кальций представляет собой порошок, плавящийся без разложения при 1403° (т. кип. 2500°). При нагревании с концентрированной серной кислотой выделяется фтористый водород в соответствии с уравнением
CaF2+H2SO4=CaSO4+2HF.
В разбавленных сильных кислотах фтористый кальций почти ие растворим. В природе он распространен в значительных количествах в виде плавикового шпата (флюорита). Кроме производства плавиковой кислоты и травления стекла, фтористый кальций находит применение в эмалевой промышленности в качестве средства для глушения эмалей (придания им непрозрачности), а также как антисептик. В небольших дозах он применяется вместе с другими солями кальция для лечения костных заболеваний.
Фтористый кальций имеет особо высокую теплоту образования) (см. табл. 48) и поэтому даже при высоких температурах оп очень устойчив к действию восстановителей, ипрсжде всего расплавленных металлов. Это одно из немногих веществ, которое не разъедается расплавленным ураном. Поэтому тигли и формы для получения металлического урана для атомных реакторов изготовляют из шлакующегося фторида кальция («фторидного шлака»). Посуду из фторидпого шлака изготовляют следующим образом; фторид кальция обрабатывают крахмалом или метил целлюлозой до тестообразного состояния. Тесто формуют и после высушивания при 800—900° шлакуют. Шлакование удается провести только в случае высокой чистоты фтористого кальция (Eichner Ch., 1951).
Фторид стронция. StF2 можпо получить в виде белого осадка при нейтрализации гидроокиси или карбоната плавиковой кислотой или при осаждении иона 8г“ ионом F'. Растворимость в воде 117 мг/л при 18°. При более высокой температуре ои выделяется в кристаллическом состоянии (в виде правильных октаэдров). Т. пл. около 902°, т. кип. 2460®.
Фторид бария. BaF2 получается аналогично SrF2. В воде он тоже трудно растворим (1,63 г/л при 18°). BaF2 образует мелкие прозрачные кристаллы удельного веса 4,83. Т. пл. 1353°, т. кип. 2260°.
Хлориды, бромиды и подиды
Хлорид бериллия. Безводный ВсС12 получают при нагревании металлического бериллия в токе сухого хлора или хлористого водорода. Это вещество представляет собой белоснежные кристаллы, низко плавящиеся (т. пл. 405°) и легко возгоняющиеся (т. кип. 488°). В воде оно растворимо со значительным выделением тепла. Раствор имеет сильно кислую реакцию, так как хлорид в нем частично гидролизовал (при 40° в 0,1 п. растворе гидролиз проходит примерно на 2%). Если подавить гидролиз прибавлением солявой кислоты, то при выпаривании из раствора кристаллизуется гидрат ВеС.12-4Н2О в виде моноклинных табличек, расплывающихся в воздухе. В противном случае выделяется основной хлорид. При нагревании кристаллогидрат отщепляет НС1. Безводный хлористый бериллий легко растворим в спирте и эфире. С последним он образует Продукт присоединения ВеС12-2(С2Н5)2О. Вообще хлористый бериллий очень склонен к образованию продуктов присоединения с органическими молекулами, некоторые из них, вероятно, являются многословными комплексами [см. Fricke R., Z. anorg. Chemie, 39, 317 (1926); Z. anorg. Chem., 170, 25 (1928); 253, 173 (1947)].
Р-то роя группа периодической системы
301
Бромид бериллия. Методы получения и свойства ВеВга аналогичны хлориду. Он возгоняется еще легче, чем хлорид (при 473°). .
Иодпд бериллия. Ве!3 лучше всего получается при нагреваний Ве5С в токо HI. Он образует мелкие гигроскопичные кристаллы. Т. пл. 480°, т. кип. 488°. При нагреваний в токе воздуха Ве12 переходит в окись. Щелочные металлы восстанавливают его. Аммиак присоединяется вплоть до образования соединения BeI->-3NH3*. Это соединение энергично реагирует со Многими органическими веществами.
Хлорид магния. MgCh в больших количествах находится в морской воде. В месторождениях калийных солей он встречается в виде минерала бишофита MgCla-GHsO; в значительно больших количествах — в виде двойной соли карналлита KCI-MgCla-GHsO. Техническое получение осуществляют из раствора, остающегося после переработки карналлита на К.С1. При выпаривании этого раствора в зависимости от температуры кристаллизуется стабильный при обычных условиях гексагидрат MgCls.-бНаО или при быстром выпаривании — продукт, содержащий меньшее количество молекул воды. Только небольшая часть этого раствора перерабатывается на хлористый магний, так как основное количество уходит в виде сливных вод (о получении MgClB из морской воды см. стр. 292). Кроме гексагидрата, MgCj2 образует также гидраты с 2, 4, 8 и 12 молекулами НгО. Гексагидрат имеет, однако, наибольшую область существования, а именно от —3,4 до 116,7°. Он образует расплывающиеся на воздухе моноклинные кристаллы, горькие на вкус, с удельным весом 1,56. Воду из хлорида магния нельзя полностью удалить без разложения соли, так как при нагревании отщепляется хлористый водород и образуется основной хлорид (оксихлорид) переменного состава, например ’
2М IpI2O Mg2OCl2-|-2HCi.
В воде хлорид магния растворяется очень легко. Его раствор содержит:
При -33,6° о° 20° 100°
(Криогидратная точка)
26 .53 54,5 73 128 г MgCl2 на 100 а Н2О
Водный раствор MgCh имеет нейтральную реакцию. Если в концентрированный раствор MgCls внести сильно прокаленную окись магния, то получившееся тесто через несколько часов застывает в твердую массу, образуя так называемый магнезиальный цемент (цемент Сореля), причем происходит соединение оКисла с хлоридом с образованием основных хлоридов.
Для приготовления цемента Сореля лучше всего смешать 10 вес; ч, MgO с 5 вес. ч. безводного MgCl2. Подобные реакции Происходят между MgO и другими солями магния, например MgSO4, Mg(NO3),, МёГ(С2НзОг)2, однако образующиеся к результате цементы по твердости и крепости уступают цементу Сореля. При смешивании цемента Сореля с древесными опилками, отбросами пробкового производства и т. п. получается довольно устойчивая по отношению к выветриванию масса, так называемый ксилолит, применяемый для пастила полов, лабораторных столов и г. и.
С хлоридами щелочных металлов MgCE образует двойные соли. К ним относится природный карналлит KCl-MgCh-GHaO; к тому же типу солей принадлежит встречающийся также в месторождениях калийных содей
* По данным Вплыла и Мессеркпехга (Z. aaorg. Chem., 148, 157, 1925) подпетый бериллий образует продукты Присоединения вплоть до состава Прим, черев.
302 Гл^а. 8
тахгидрит CaCh-2MgCh-12HsO. Соответствующая двойная соль MgCJ* с хлоридом аммония NHsCl-MgCla, 6НаО (которую можно синтезировать из компонентов) при нагревании разлагается, не подвергаясь гидролизу. Поэтому ею можно пользоваться для получения безводного хлорида магния.
Кроме уже упомянутого производства магнезиального цемента й ксилолита, хлорид магния используют для проклейки основы в ткацком деле (которая сообщает мягкость тканям), для пропитки дерева; MgCla также примешивают к воде при поливке улиц; кроме того, MgCla является исходным материалом для получения окиси магния и других его соединений. Из MgCla можно также получать соляную кислоту и хлор. Значительные количества хлорида магния, содержащиеся в конечных щелоках, остающихся при производстве К.С1, во избежание загрязнения рек сбрасывают обратно в шахты. В медицине хлорид магния используют как слабительное.
Бромид магния. Точка плавления MgBr2 711°; по своим свойствам очень близок MgCl2. Подобно последнему он кристаллизуется из водных растворов при обычной температуре в виде расплывающихся кристаллов гексагидрата. MgBr2-6H2O изоморфен гексагидрату хлорида и также содержится в небольших количествах в минералах бишофите и карналлите. Кроме гексагидрата, MgBr2 образует еще декагидрат MgBr210H20. Составу карналлита соответствует двойная соль KBr MgBr2-6Н2О. При кипячении концентрированного раствора бромида магния с окисью магния получается двойное соединение MgBr2-3Mg (ОН)2-9Н2О, кристаллизующееся в бесцветных иглах.
Иодид магния. Mgl2 из водных растворов при обычной температуре кристаллизуется в виде о кт а гидрата и в виде декагидрата. Mgl2— белый, расплывающийся на воздухе, очень хорошо растворимый в воде порошок; применяется в медицине (против сифилиса, золотухи, ревматизма). .
Хлорид кальция. Cad г в безводном состоянии образует белую, чрезвычайно гигроскопичную массу, плавящуюся при 780° и возгоняющуюся при температуре белого каления. Удельный вес плавленого CaCla равен 2,2. Безводный CaCla получают из содержащего воду хлорида кальция при нагревании выше 260°. Обезвоживание следует вести осторожно, так как при слишком быстром нагревании происходит частичный гидролиз, сопровождающийся отщеплением НС1*. Растворение безводного CaCla в воде сопровождается значительным выделением тепла (17,41 ккал), связанным с его гидратацией. Наиболее известный из гидратов — гексагидрат СаСЬ-бНйО — кристаллизуется при испарении раствора хлористого, кальция при комнатной температуре. Он образует гексагональные призмы удельного веса 1,65. При растворении его в воде происходит сильное поглощение тепла (его молярная теплота растворения равна —4,31 ккал), и позтому им часто пользуются для приготовления охладительных смесей. Смешивая этот гексагидрат со снегом в весовом отношении 1,44 : 1, можно получить охлаждение до температуры, соответствующей криогидратной точке, равной —54,9°. Ниже приведена растворимость CaCla в воде:
при -54,9= 9° 4-Ю» 20° 40° 60° 100° 260°
42,5 60,0 65,0 74,5 115 137 159 347 г СаС12 щ 100 г Н2О.
Хлорид кальция легко образует пересыщенные растворы. Кроме гексагидрата, существуют еще два тетрагидрата, один ди- и один моногидрат (см. стр. 97 и сл.)
* О гидролизе СаС13 перегретым водяпым паром и об использовании этой реакции в технике для получения IIC1 см.; Briner Е., Belv. chjm. Acta, 31, 556, 1948.
Вторая группа периодической систелт
303
Растворы хлорида кальция образуются в качестве отходов производства при многих химических процессах, главным образом при получении соды по методу Сельве. Для получения технического хлорида кальция эти растворы очищают кипячением с едкой известью. Чистый хлорид кальция получают при растворении чистого карбоната кальция (мрамора) в соляной кислоте.
Растворы хлорида кальция нередко применяют в качестве бань для лабораторного нагрева, в качестве жидкости для затворов и для приготовления охлаждающих смесей. Температуры кипения и замерзания этих растворов приведены в табл. 54 и 55. Кроме того, растворы СаС1з применяют для пропитывания дерева, тканей и других материалов, которые благодаря этому становятся негорючими. Было предложено также пользоваться ими для тушения огня. Витринные стекла, смоченные раствором хлорида кальция, не запотевают.
4 Таблица 54
Температуры кипения растворов хлорида кальция (Gerlach, Schlamp)
СаС^ в 100 г Г12О, г 6,0 11,5 16,5 21,0 25,0 41,5 69,0
Т. кип., °C 101 102 103 104 105 110 120
СаСЦ в 100г Н2О/ г 101 137,5 178 222 268 292 305
Т. кип., °C 130 140 150 160 170 175 178
Таблица 55
Температуры замерзания растворов хлорида кальция (Pickering, Loomis, Rodebusch)
СаС13 в 100 а раствора, г 0,1 1 5 10 15 20 25 30 32,5
Т. пл., °C —0,051 —0,46 —2,44 -5,89 —10,06 —18,6 —29,9 —48,0 -51,0
Хлорид кальция легко растворим в безводном этилоном спирте (а также в высших спиртах, например в пропиловом, изобутиловом и амиловом; с двумя последними он образует продукты присоединения)*. Из растворов, содержащих, помимо хлорида кальция, гидроокись кальция [например, растворы, получающиеся после переработки на аммиак NELCl и Са(ОН)а] кристаллизуется двойное соединение СаС1а-ЗСа(ОН)я.12НаО в виде тонких длинных игл.
Безводный CaCl-i связывает аммиак, что следует иметь в виду при использовании его в качестве осушителя- Применение CaCla для осушки
* CaCL образует кристаллоалкоголяты состава CaCL.-SROH со всеми спиртами алифатического ряда. [Меншуткип Б. И., ЖРФХО, 38, 1010, 1906]. — Прим, перев.
304
Глава 8
основано па его сильной гигроскопичности. Для этой цели его применяют большей частью в виде пористых кусков, получаемых неполным обезвоживанием (при этом не следует допускать его плавления). Чистый хлорид кальция применяют в медицине.
Вром ид кальция. СаВг2 в безводном состоянии представляет собой белую массу с т. пл. 760° и т. кип. 806—812°; по своим свойствам сильно напоминает хлорид, Однако для него достоверно известен только один гидрат, а именно гексагидрат СаВг2-6Н2О (т. пл. 38,2°), образующий иглы с шелковистым блеском, которые, однако, кристаллизуются с трудом. В воде и спирте бромид кальция легко растворим. Эту соль применяют в фотографии и в медицине,
Иодид кальция. Са1г, имеющий удельный вес 4,9, т. пл. 740° и т. кип. 708—719°, во многих отношениях аналогичен бромиду; из водных растворов он кристаллизуется в виде гекса гидрата Са12.6НаО. Т ас сил и (Tassiiy) описал двойное соединение Са12-ЗСа(ОИ)2] содержащее кристаллизационную воду.
Хлорид стронция. SrCk легче всего получается при растворении карбоната стронция в соляной кислоте. Из раствора при температуре ниже 60° он кристаллизуется в виде гексагидрата SrCU-бНЮ, представляющего собой гексагональные иглы, расплывающиеся на воздухе с удельным весом 1,96; SrCh-бНаО изоморфен соответствующей соли кальция. Выше 60° кристаллизуется дигидрат в виде прямоугольных табличек. При нагревании дигидрата выше 100° получается безводная соль; последняя "плавится при 872°. Во влажном состоянии ее плавление, как и в случае хлористого кальция, сопровождается частичным гидролизом. В воде SrCl-j растворяется очень -легко; такие растворы имеют острый, горький вкус. Ниже приведена растворимость SrCI3 в воде.
при О’ зо°
44,2 53,9 101,9г SrCl2 в 100а Н2О,
Насыщенный раствор SrCla кипит при температуре около 117°. В отличие от хлорида кальция ц этиловом спирте SrClaGHaO растворяется с трудом, а при нагревании еще труднее, чем на холоду. Подобно CaCU, SrCla присоединяет аммиак.
Бромид стронция SrBfj (т. ил. 643°) и иодид стронция Srl2 (т.‘ Ш. 507°) и противоположность хлориду стронция легко растворяются в спирте. Обе соли применяют в медицине- Молярные растворимости галоидных соединений стронция в воде возрастают в ряду: 8гС1й, SrBr2, Srl2.
Хлорид бария. ВаС1з образует в безводном состоянии белую массу, плавящуюся при 878° и имеющую удельный вес 3,86; в воде он растворяется легко, а в абсолютном этиловом спирте — с трудом. В метиловом спирте его растворимость невелика: при 15,5° в 100 ч. СНзОН растворяются 2,18 ч. BaCls. В воде ВаС1з имеет следующую растворимость:
при 1)” 10° 20° 50° ТОО" 104,1
31,5 33 36 43,5 59 60 г ВаС12 в 100 г И20.
Из водного раствора хлорид бария при обычной температуре кристаллизуется в виде дмгпдрата ВаС1<>-2Н2О, представляющего собой бесцвет> ные плоские ромбические пластинки удельного веса 3,06, устойчивые на воздухе. При нагревании они сначала отщепляют одну молекулу воды, а при более сильном нагревании — вторую молекулу.
В технике хлорид бария применяют для «смягчения» содержащей гицс воды, предназначаемой для питания котлов; кроме того, из него получают другие соединения бария, особенно белую баритовую краску.
Вторая группа периодической системы
305
В лабораториях его применяют как реактив, главным образом для качественного и количественного определения серной кислоты. В медицине jBaCh находит применение для различных целей. Хлорид бария отличается неприятным горьким вкусом и сильно ядовит*. Мера противоядия сротив отравления BaCh: после очищения желудка' л ромы ванио 1%-ным раствором глауберовой соли.
При техническом получении BaCla исходят из витерита или из т.яже-шпата. Витерит растворяют в соляной кислоте, а тяжелый пшат для переработки в хлорид нагревают с углем и хлористым кальцием (получающимся в виде отхода при производстве соды по методу Со льве). При этом уголь восстанавливает сульфат бария в сульфид, а последний реагирует t CaCla
BaSO4+4C = BaS-|-4CO; BaS+CaCl2=CaS-j-BaCl,.
Из полученного плава ВаСЗа выщелачивают горячей водой, а небольшое количество непрореагировавщего и перешедшего в раствор сульфида бария разлагают, пропуская через раствор ток двуокиси углерода; образующийся при этом карбонат бария обработкой соляной кислотой переводят в хлорид и затем упаривают раствор до кристаллизации.
Бромид бария. ВаВг2 получается легче всего при нейтрализации Ва(ОН)2 пли ВаСОз водным раствором бромистоводородиой кислоты. В безводном состоянии ВаВт2 представляет собой белую массу, плавящуюся при 847° п имеющую удельный вес 4,79. В воде ВдВг2 хорошо растворим (в 100 г воды растворяются: ири 0° 98, при 2СР 104, при 100° 149 г ВаВг2). Из водных растворов выкристаллизовывается днгпдрат ВаВг2- 2Н2О, изоморфный соответствующему хлориду. Отщепление воды от ВаВг2-2Н2О происходит несколько труднее, чем от хлорида (первая молекула воды отщепляется при 75°, вторая — выше 100°). Бромид бария очень хорошо растворим в метиловом спирте; ппачитещяю растворим он и в этиловом спирте (около 3,1 г ВаВг2 в 100 г этилового скпрта). Двуокись углерода в присутствии кислорода воздуха разлагает водные растворы ВаВг2, причем происходит выдел сипе карбоната бария
; BaBr2+СО2+ V А=ВаСО3_у Вг2.
Иодпд бария. Ва12 легко получается при обработке Ва(ОН)2 или ВаСО3 иодистым водородом или иодом в присутствии какого-нибудь восстановителя. В безводном состоянии оп образует белую массу с уд. весом 4,92. В воде Ва12 очень хорошо растворим (в 190 г воды растворяются: при 0° 170, при 20° 200, при 100° 270 s Ва12), в этиловом спирте растворимость его также значительна. Из воДпых растворов Ва12 криста.тли-зустся в виде гидратов различного состава с содержанием 1—7 молекул воды, расплывающихся на воздухе. Кристаллизующийся в ромбической системе дигидрат Ра12-2Н2О изоморфен соответствующим бромиду и хлориду.
Моно галогениды щелбч поземельных металлов. По данным Вёлера (Wohler, 1909) при'нагревании галогенидов кальция с металлическим кальцием и при быстром охлаждении расплава можно получить мо по галогениды CaF, CaCl и Cal — соединения, неустойчивые при обычпон температуре. ГунтЦ и Бенуа (Guntz, Benoit, 1923—1924) предполагали подтвердить эти наблюдения и получить этим же методом моногалогениды Si и Ва, По данным этих авторов, моно галогениды щелочноземельных металлов представляют собой интенсивно окрашенные соединения; разлагающие воду с выделением водорода. Их образования можно было ожидать при электролизе расплавов дигалоге-пндов. Тем не менее впоследствии существование этих соединении неоднократно подвергалось сомнению, в-частности Бишовским и Россини (Bischowsky, Rossini, 1936), па том основании, что данные Бенуа по теп лотам растворения моно галогенидов незначительно отличаются от теплот растворения смесей нормальных хлоридов и свободных металлов. Попытки обнаружить образование м отстало генидов щелочноземельных металлов рентгенографическим путем дали отрицательный результат. При изучении диаграмм состояния ряда систем дигалогенид Щелочноземельного металла — Щелочноземельный металл Кубиккиоти (Cubicciotti D. D., 1949) пе получил никаких доказательств существования твердых монога логе видов; более того, автор обнаружил, что в системе образуются исключительно твердые растворы. Однако растворимость их ограничена как в твердом, так и в жидком состоянии. 111ефер (Schafer, 1952) при 26 1'. Реми,.
306
Глава. 8
повторном исследовании системы ВаС12— Ва установил, что неограниченная растворимость обоих компонентов в жидком состоянии появляется только выше 1016“. Растворимость резко падает с понижением: температуры. Однако выше 800° даже в твердом состоянии ВаС12 может содержать еще известное количество Ва. Наоборот, металлический В а может растворять ВаС12 тол г, ко в расплавленном состоянии. В точке плавления ВаС12 область расслаивания лежит в интервале 15—93 мол,% Ва. Относительно Ва20, описанного впервые Гуптцем и Бенуа, на основании опытов Шрнля (Schriel, 1937) уже вполне достоверно известно, что он представляет собой только раствор Ва в ВаО.
Смешанныещи дрпды-галоген иды щелочноземельных металлов. Эрлих (Ehrlich., 1953—1955), пытаясь синтезировать моногалогепиды щелочноземельных металлов, не получил доказательств их существования в твердом состоянии. Однако он обнару* жил, что при нагревании смеси щелочноземельного металла и его галогенида в атмосфере водорода при 900’ можно получить смешанныегиДриды-галогениды М11НХ(Х=С1, Вг или 1). Эти соединения представляют собой бесцветные тетрагональные листочки, внешне напоминающие слюду. Водой они разлагаются со значительным выделенном тепла. Строение их молекул аналогично BiOCI, ВЮВг, BiOl (см. стр, 730 л ел.). Гнд-рид-иопы в кристаллической решетке занимают то же положение, что и ионы кислорода в слоистой структуре перечисленных окси галогенидов (рис. 120, б). Вначале зтот структурный тин был обнаружен у соединений PbCIF и PbBrF (Nieuwenkamp, 1931). В соответствии с этим рассматриваемые соединения относят к структурному типу PbClF. В той же структуре кристаллизуются и соответствующие окси галогениды лантана.
Нитраты
Нитраты элементов главной подгруппы II группы, подобно всем /нитратам, хорошо растворимы в воде; за исключением нитратов стронция и бария, они хорошо растворяются и в спирте. Нитрат бериллия в водных растворах подвергается гидролизу (на 1,8% в 0,1 н. растворе при 40°). Прп нагревании нитраты металлов главной подгруппы II группы легко переходят в окислы. Склонности к образованию двойных солей с нитратами щелочных металлов у нитратов главной подгруппы II группы не наблюдается. Такие двойные соли известны только для нитратов магпия и бария (K2Mg[NOsh и KJBalNOsb).
В четверной системе КЛО<;— NaNO3— Ca(NO3)2— Mg(NO3)2 при затвердении расплава, кроме двойной соли KaMg(NO3)4, обнаружены лишь два ряда твердых растворов (К, _Xa)NO3 и (Са, Mg)(NO3)2. (J a n е с к о Е., Z. Elektrochem., 48, 453, 1942). Ив того факта, что безводный Mg(NO3)2 образует с Са(МО3)2 непрерывный ряд твердых растворов, следует, что ом обладают одинаковой структурой решетки1.
Рентгенографическое определение показало, что структуры Ca(NOs)2, Sr(NOa)a и Ba(NOa)2 имеют (если отвлечься от расположения атомов в структурной группе NOa) некоторое сходство с решеткой флюорита (рис. 62, стр. 298). Ионы металлов в каждом случае образуют гранецепт-рированную кубическую решетку. Группы NOa расположены на пространственных диагоналях восьми малых кубов, но не в их серединах, как ионы F" на рис. 62.
Пптрат бериллия. Be(NO3)2 можно легко получить двойным обменом между BeSO4 и Ba(NO3), в водном растворе
BeSO4+ Ba(NO3)2= Be(NO3)a+ BaSO4.
В техники BuNO3 получают растворен нем гидроокиси бериллия в азотной кислоте; из водных растворов он выделяется в виде расплывающихся, кристаллов состава Ве^О^-ЗН^О*, При температуре около 60э последние плавятся в своей кристалли-
* Из водного раствора (при 0 и 20’) кристаллизуется Be(NO3)2 4Н2О. 11 з раствора, содержащего при этих температурах около 50% HNO3, кристаллизуется Be(NO3)2-3H2O. При нагревании выше 60’ происходит плавление, сопровождающееся постепенной потерей воды и окислов азота, подпое удаление которых достигается лишь около 1000s.— Прим. ред.
Вторая группа периодической системы 307
зационяой воде, при 100-' начинается отщепление аяотпой кислоты л, наконец, при. нагревании приблизительно до 200° получается окись бериллия.
Нитрат магппя из водных растворов кристаллизуется в виде гексагидрата: Мл(КО3)2-6112О, образующего расплывающиеся во влажном воздухе ромбические столбики и иглы, которые при 89,1° распадаются с образованием дигидрата, плавящегося при 129,5°. Одновременно начинается отщепление азотной кислоты. При прокат ливапии нитрата магния получается чистая окись магния. Ниже ‘—17,1° устойчивым, является нонагидрат Mg(NOj)2-0H2O. Кроме того; существует еще (мотаста б ильный) тетрагидрат (т. пл. 52°).
Нитрат кальция — известковая селитра С а (NO а) а кристаллизуется в безводном состоянии в виде правильных октаэдров (точка плавления около 561°); однако из водных растворов он выделяется в виде тетрагидрата Са(МОз)2-4НгО, образующего моноклинные призмы удельного веса 1,82, плавящиеся при температуре несколько выше 40° в своей кристаллизационной воде; При охлаждении таких растворов обычно наблюдается значительное замедление кристаллизации. Кроме тетрагидрата, вероятно, существуют и низшие гидраты. При нагревании тетр а гидр ат а выше 100° он легко переходит в безводную соль. Эта соль очень гигроскопична и чрезвычайно легко растворяется как в воде (при 18° растворяется 121 з Са(КОз)г в 100 г воды), так и в спирте. s
Нитрат кальция нередко образуется при гниении азотсодержащих органических соединений в присутствии извести; так, он образует белые выцветы на стенах конюшен (стенная селитра). Прежде нитрат кальция получали, этим способом искусственно в так называемых «селитряницах», ’ куда забрасывали всякого рода животные отбросы, смешивали их со строительным щебнем, известкой и т. п. и время от времени поливали эту массу навозной жижей до тех пор, пока в пей не образовывалось в результате разложения органических веществ достаточного количества нитрата кальция и других азотнокислых солей, которые затем подвергали выщелачиванию. В настоящее время нитрат кальция получают нейтрализацией его карбоната (известняка) или гидроокиси технической азотной кислотой. Полученную этим методом известковую селитру, которую прежде изготовляли главным образом в Норвегии («норвежская селитра»), широко применяют в качестве удобрения. Гигроскопичность норвежской селитры, сильно препятствующая ее равномерному распределению в почве, можно, почти полностью устранить добавлением едкой извести (при этом образуется основная соль).
При нагревании выше точки плавления нитрат кальция сначала выделяет кислород, и только при более сильном прокаливании Са(КОз)а разлагается на окислы азота и окись кальция. Растворы нитрата кальция сильнее абсорбируют аммиак, чем чистая вода. Однако1 твердые продукты. присоединения аммиака к нитрату кальция до сих пор не известны.
•в
Нитрат стронция. Sr(NOa)2 получается проще всего при действии азотной кислоты на карбонат стронция. Его можно также приготовить двойным обменом между хлоридом стронция и нитратом натрия. Последний метод основан на том, что в равновесии
SrC]2-|-2NaNO3 5* Sr(N03)2-f-2NaCl
соли, находящиеся в правой его части, менее растворимы, чем SrCla и NaNOa. Растворимость нитрата стронция составляет при: 0° 39,5; 20° 70,8; 100° 101 г Sr(NO3)a в 100 г воды. На холоду из водных растворов
20*
308
Глава 8
а
Sr(WO3}3 кристаллизуется его моноклинный тотрагидрат Зг(МОз)2-4Н2О с удельным весом 2,25. Тетрагидрат ири нагревании до температур выше 100° легко теряет всю воду. При высоких температурах нитрат стронция кристаллизуется в безводном состоянии в виде октаэдров и комбинации из октаэдров и кубов удельного веса 2,93, имеющих точку плавления 645°. При более высокой температуре сначала отщепляется кислород, образуется нитрит и только ири сильном прокаливании происходит переход последнего в окисел.
В абсолютном спирте нитрат стронция растворяется с трудом (1 : 8500)* и еще труднее в смеси равных объемов спирта и эфира (1 : 60 000). Этим пользуются для отделения стронция от кальция. Нитрат стронция применяют для приготовления красных бенгальских огней.
Нитрат бария. Ва(МОз)г (баритовую селитру) в технике получают различными способами. Проще всего для этого растворять витерит ВаСОз в азотной кислоте. Вместо ВаСОз можно исходить из BaS, получаемого восстановлением тяжелого шпата BaSOr; в последнем случае выделяющийся при реакции сероводород улавливают, пропуская его через едкий натр. Другие способы основаны на обменных реакциях между хлоридом бария и нитратом натрия/чилийской селитрой) или карбонатом бария и нитратом кальция
BaCi2-j-2NaNO3 Ba(NO3)2-|-2NaCl;
BaCO3-[-Ca(NO3)2 Ba(NO3)24-CaCO3.
В обоих случаях происходит образование «обратимых» пар солей (ср. «конверсионная селитра»). ।
Нитрат бария растворим в воде гораздо труднее, чем нитраты остальных Hie л очноземельных металлов. В спирте он практически нерастворим. Его растворимость в воде составляет при: 10° 7; 20° 9,2; 100° 32,2 в Ba(NOa)a в 100 г воды. Ниже 12° устойчивым в соприкосновении с раствором является дигидрат Ва(ПОз)2-2НаО. При обычной температуре нитрат бария кристаллизуется в безводном Состоянии, образуя правильные октаэдры удельного веса 3,24, плавящиеся при 575°. При более сильном нагревании Ва(ПОз)а сначала отщепляет кислород, переходя в нитрит, и только при энергичном прокаливании происходит образование окиси.
Из нитрата бария получают чистые окись и гидроокись бария; его используют также для получения взрывчатых веществ, а в пиротехнике для производства зеленых бенгальских огней.
Двойная соль K2Ba(NO3)4 была описана Уолл бриджем (Wollbridg, 1903). Она образует микроскопические кристаллы в форме тетраэдров. С CsNO3 соответствующей двойной соки нитрат бария не образует; не удалось получить двойного соединения и иа смесей KNO3 и Sr(KO3)2.
Карбонаты
Нормальные или нейтральные карбонаты М[1СОз щелочноземельной группы трудно растворимы в воде. В избытке угольной кислоты карбонаты Са, Sr и Ва переходят в легкорастворимые кислые карбонаты (гидрокарбонаты, бикарбонаты) №(НСОз)а; нейтральные карбонаты бериллия и магния могут существовать в соприкосновения с водой только в присутствии избытка угольной кислоты; в ином случае происходит их гидролиз, «сопровождающийся образованием трудно растворимых основных карбона
Вторая группа периодической системы 309
тов. Кислоты, в том числе уксусная, разлагают карбонаты щелочноземельных металлов, как и другие карбонаты, с выделением СО а. Растворимость карбонатов приведена в табл. 56.
Таблица 56
Растворимость карбонатов щелочноземельных металлон в воде
100 г воды нрп 18е растворяют:
9,4 мг MgG03 1,3 мг СаСОэ 1,0 мг SrCOs 1,72 мг ВаСО3
Карбонат бери л лил. Нормальная соль ВеСО3 образуется из водного раствора только в том Случае, если раствор содержит избыток угольной кислоты. ВеСО3 кристаллизуется из раствора в виде тетрагидрата BeCOjAH^O, который при 100° теряет всю воду, а при несколько более сильном нагревании отщепляет и С02.
Кароонаты щелочных металлов осаждают из растворов солСй бериллия основные карбонаты (переменного состава). Последние, как и нейтральный карбонат бериллия, легко растворяются в избытке карбоната щелочного металла, особенно в концентрированном растворе карбопата аммония, причем образуются двойные соли (довольно легко разлагающиеся водой).
Карбонат магния. MgCOa и естественном состоянии встречается в виде магнезита (тальковый шпат, горький шпат).
Еще более распространена его двойная соль с карбопатом кальция MgCOa-CaCOa — доломит. Оба эти соединения кристаллизуются гексагонально; однако в природе они встречаются в большинстве случаев в виде плотной массы, кристаллическое строение которой нельзя определить невооруженным глазом. Это особенно относится к магнезиту, Доломит часто бывает окрашен различными примесями в более или менее темные цвета. Магнезит обычно бывает белым, нередко со слабым желтоватым оттенком. Твердость его равна 3—5. Приготовленный искусственно карбонат магния образует белый порошок удельного веса 3,04. Из водных растворов нормальный карбонат образуется только тогда, когда раствор содержит значительный избыток угольной кислоты. При температуре ниже 16° карбонат магния кристаллизуется из раствора с 5 молекулами воды, при более высоких температурах выпадает тригидрат, который может переходить в моногидрат. Нейтральный карбонат при кипячении с водой также постепенно гидролизуется, переходя в основные карбонаты.
При прибавлении к растворам солей магния карбонатов щелочных металлов осаждаются так называемые основные карбонаты переменного состава, которые, как правило, не являются определенными соединениями.
По данным Менделя (Menzel, 1930) из водных растворов осаждается только один основной карбонат магния, имеющий определенный состав и структуру, а именно 4MgCO3-Mg(OH)2- 5Н3О. Это соединение при высушивании сравнительно легко отщепляет 1 молекулу НаО, не изменяя своей структуры, Мепцель придает пентагидрату следующую структуру;
г П2О • MgC03, ,CO3Mg- ОН,
>Mg<
. Н3О • MgCO3Z ''CO3Mg- он2.
(ОН),<И2О
Продукты иного состава представляют собой твердые растворы, а часто и просто механическую смесь этого соединения е нормальным карбонатом или с гидроокисью магния.
В природе изредка встречается также основной карбонат магния в кристаллическом состоянии в виде гидромагнезщпа, имеющего состав 4М§СОз-Мд(ОН)-2-4НаО. Аналогичный состав большей частью имеет
310
Глава 8
также и искусственно приготовленный основной карбонат, называемый magnesia alba. Основные карбонаты, как и нейтральный карбонат, отщепляют СОа уже при сравнительно умеренном нагревании; так, для получения из магнезита практически чистой окиси достаточно нагреть его до темно-красного каления (ср. также табл. 57, стр. 3"12). В воде карбонат магния растворяется с трудом (ср. табл. 56), однако лёгче, чем гидроокись магния. Растворимость его зависит от содержания в воде угольной кислоты/ причем присутствие ее повышает растворимость MgCOa; в водном растворе угольной кислоты концентрация ионов СО" очень незначительна вследствие того, что угольная кислота диссоциирует главным образом по уравнению
Н2СОЯ H4-HCOJ. (8)
Так как ионы СО", находящиеся в равновесии с осадком MgCOa,
MgC03 5* Mg"+COJ (9)
соединяются с ионами Н', образуя ионы НСО', равновесие -(9) нарушается и для его восстановления необходимо, растворение такого количества осадка МдСОз, чтобы произведение растворимости
сохранило то числовое значение, которое оно имело в чистом водном растворе, а это Может произойти только в том случае, если уменьшение концентрации ионов СО" компенсируется соответствующим увеличением концентрации ионов Mg".
Магнезит и доломит широко применяют для изготовления огнеупорных камней («обжигом» до окислов). Такне камни идут, например, на обкладку конвертеров, используемых в томас риском процессе производства стали (см. т. II). Полу обожженный доломит* (смесь MgO и СаСОз) используют для изготовления строительных плит и как средство для очистки воды. С малой термической устойчивостью магнезита связано его применение для получения чистой двуокиси углерода, идущей на приготовление минеральных вод.
Особый вид очень плотного магнезита, встречающийся в Боснии и известный под названием «боснийской морской пены», идет для: изготовления курительных трубок. Магнезит наряду с содержащимися в отходах после выработки К.С1 соединениями магния также является важнейшим исходным материалом для получения других соединений магния — главным образом горькой соли.
Искусственно приготовленный основной карбонат магния, является исходным материалом для приготовления многих других солей магния (он растворяется в кислотах гораздо быстрее, чем магнезит); кроме того, его применяют как составную часть пудры, зубных порошков и порошков для чистки, как наполнитель для красок, бумаги и каучука. В ваде magnesia alba его применяют в медицине.
Карбонат магния образует с карбонатами щелочныхметаллов, а также с бикарбонатами двойные соли типа M3Mg(CO3)2, например: KaMg(CO3)2-4HaO — ромбические призмы, KMgH(CO3)a-4HaO — белый порошок; Йа2М§(СО3Б — мелкие тетр агональные кристаллы.
Карбопат кальция. СаСОз в природе встречается в виде известняка, мела и мрамора. Кристаллизующийся в гексагональной системе карбонат
* О механизме термического разложения доломита см., например, Noll W., Angew. Chem., 62, 567, 1950; Haul R<, J. appl. Chem., 2, 298, 1952.
Вторая группа периодической системы . 311
кальция называется кальцитом или известковым шпатом. Последний иногда встречается в природе в виде хорошо образованных кристаллов (большей частью ромбоэдров). Иногда попадаются очень крупные Кристаллы известкового шпата. Известковый шпат обнаруживает двойное лучепреломление, что особенно заметно на больших и совершенно прозрачных ромбоэдрических кристаллах, встречающихся в Исландии (исландский шпат). Удельный вес известкового шпата равен 2,72. Реже встречается в природе арагонит—неустойчивая * ромбическая модификация: кристаллического карбоната кальция (удельный вес 2,93). При 970° кал71ЦИг переходит в другую моднфмкатрпо, которая также относится к гексагональной системе. Арагонит при высокой температуре испытывает аналогичное превращение. Другой неустойчивой модификацией СаСОз является ватерит.
Кристаллическая структура иаеесткоеого шпата И магнезита аналогична нитрату натрия (рис. 50, стр, 246), Арагонит, как и нитрат калия, имеет ромбическую слоистую структуру (см. стр. 247), ватерит. — гексагональную. Решетку доломита можно получить из решетки известкового шпата путем замещения каждой второй плоскости основания, занятой ионами Са2*, плоскостью, состоящей из ионон Mg‘2+.
В подо карбонат кальция мало растворим, поэтому при одновременном присутствии в водных растворах ионов Са" и СО" он выпадает в виде белого осадка. При осаященйи из горячих разбавленных растворов образующийся осадок сначала состоит из очень мелких кристаллов арагонита, которые на холоду медленно переходят в кристаллы кальцита. Из холодных растворов карбонат кальция выпадает в виде аморфной массы, которая, находясь в соприкосновении с раствором, также постепенно переходит в кристаллы кальцита. Известняк и мрамор тоже состоят из болей или менее мелких кристаллов кальцита. Чистый карбонат кальция — белое или бесцветное соединение. Пестрый мрамор обычно содержит примеси — главным образом окисли железа. Желтоватая или серая окраска известняка обусловлена присутствием в нем примесей, среди которых главную роль играет глина. Известняки, содержащие значительные количества глины, называются мергелями; в зависимости: от содержания глины их подразделяют на глинистые и известковые мергеля. Мел Представляет собой мягкую модификацию известняка. Он состоит главным образом из остатков оболочек микроскопических моллюсков древних геологических формаций и из раковин.
Растворимость карбоната кальция в воде, равная при 25° '—1,4 мг СaCO-S в 100 г воды, довольно значительно возрастает при добавлении солей аммония. При кипячении с раствором хлорида аммония карбонат кальция полностью разлагается
СаСО3+ 2NH4CI = CaCi3+2NII3-j- НаО +СО2.
Соли щелочных металлов не приводят к подобному разложению СаСОя. Они также пе повышают его растворимости в воде. СаСОз легко Соединяется с избытком угольноц кислоты и переходит в довольно легко растворимый бикарбонат
СаСО3-}-Н2СО3=Са(НСО3)2. (10)
Поэтому он в значительных количествах растворим в иоде, содержащей угольную кислоту. От содержания в воде бикарбоната кальция зависит ее временная (устранимая) жесткость. Если такую воду прокипятить, то из нее выделяется двуокись углерода и реакция (10) проходит справа налево, а нейтральный карбонат кальция выпадает в осадок. Подобное осаждение
312
Глава 8
центрального карбоната кальция происходит и процессе испарения раствора при обычной температуре. На этом основано образование сталактитов в природе.
При низких температурах карбонат кальция выделяется из водного раствора в виде гексагидрата. Это соединение образует мелкие ромбические кристаллы удельного веса 1,77, которые при обычной температуре очень быстро выветриваются па воздухе с образованием безводной соли, Краус (К гаи В, 1930) разложением гидрата при постоянном давлении (р>= 7 мм рт ст) получил в тензиэвдиометре неустойчивый моногидрат.
При прокаливании карбонат кальция отщепляет СОз
CaCO3s=CaO-f-COs,
однако, как видно из табл. 57, двуокись углерода в нем связана значительно прочнее, чем в карбонате магния. При 897° давление СО а над карбонатом кальция достигает атмосферного*.
Таблица 57
Давление разложения карбонатов щелочноземельных металлов
Давление, м-н рт ста
Карбонат 400“ 450° 500° 540° 700° 900’ 1000’ 1100’ 1200’ 1 300°
MgCO3 0,1 6,8 100 747
СаСО3 0 0 0,1 0,3 22,2 793 2942 . 8739 21800 —
ВаСОа 0 0 0 0 0 0,2 2,7 17,7 92 382
Термическое разложение карбопата кальция (обжиг извести) осуществляется в технике в больших масштабах для получения окиси кальция,.
которая служит для приготовления строительной извести, а также для других целей. Обжигом известняков в смеси с глиной (или естественных известняков, содержащих большой процент глины) изготовляют цемент. Для использования в технике (для приготовления штукатурного раствора — белом меловой краски или школьного мела) природный мел обычно подвергают очистке отмучиванием. Отмученный мел идет также для приготовления зубного порошка, замазки и различных порошков для чистки. Чистый карбонат кальция, получаемый в виде очень тонкого порошка при осаждении его пз| водного раствора, применяют в медицине, например против изжоги. Его используют также для понижения излишней кислотности вина.
Карбопат стронция. Sr СО а встречается в природе в виде ромбического стронцианита, изоморфного арагониту и витериту, в связи с чем миперал обычно содержит примесь карбоната кальция, а иногда и карбоната бария. В технике его перерабатывают главным образом на окись, и гидроокись стронция, которые применяют для извлечения сахара из мелассы. Так как природного стронцианита для этой цели пе хватает т
* Распад карбоната кальция и доломита может ускоряться каталитическим действием посторонних газов (ср. Huttig G., Z. anorg. Chem., 255, 223, 1948) , Энергия активации процесса, происходящего на поверхности при термическом распадо доломита, составляет на воздухе 45 и 55 ккал/молъ в атмосфере СО2. Энергия активаций термического распада кальцита на воздухе равна 48 ккал/моль, а магнезита — 42 ккал/моль (Bischoff, 1950; No]], 1950).
Вторая группа периодической системы 51?»*
то карбонат стронция получают также из целестина SrSOi сплавлением» последнего в револьверных пенах с кальцинированной содой
SrSO4+ Na2GO3=SrCO3+Na2SO4.
Чистый карбонат стронция получается при осаждении растворов солей: стронция карбонатом аммония. Карбонат стронция очень трудно растворим в воде. Его раствор вследствие частичного гидролиза-имеет щелочную* реакцию. Присутствие в воде угольной кислоты значительно повышает его-растворимость. При прокаливании карбонат стронция разлагается труднее, чем карбонат кальция. Поданным Конроя (Conroy) при ИОО°ЗтСОз полностью отщепляет СОг.
При 929° ромбический SrCOa переходит в гексагональный. Если предотвратить отщепление СОа, то эта гексагональная модификация плавится около 4500е.
Карбонат бария. ВаСОз в природе встречается в виде витерита главным образом в некоторых месторождениях свинцового блеска северо-западной Англии. Обычно витерит кристаллизуется в ромбической системе и изоморфен арагониту и стронцианиту (выше 811° стабильной является гексагональная, а выше 982°— кубическая модификация). Из водных растворов ири одновременном присутствии ионов Ва” и СО" выпадает белый осадок карбоната бария. Исходным продуктом для технического» получения ВаСОз является тяжелый шпат BaSOa, который при нагревании до 600—800° в присутствии угля восстанавливается до сульфида бария»
BaS04+2C=BaS-|-2CO2.
В водный раствор сульфида пропускают двуокись углерода, в результате чего образуется карбонат бария, по реакции
BaS+СО24-Н2О = ВаСОз-f- HsS.
Можно также при повышенном давлении нагревать топко измельченный тяжелый шпат с концентрированным раствором карбоната калия; при этом происходит реакция
BaSO4-}- К2СО3 = ВаС03-р K2SO4.
Карбонат бария отщепляет COs при значительно более высокой температуре, чем карбонаты других щелочноземельных металлов; только-при 1400° давление СОа над карбонатом бария достигает атмосферного. Так как карбонат бария еще ниже этой температуры начинает спекаться и вследствие этого теряет пористость, то полностью разложить его только-нагреванием очень трудно (ср. такжо табл. 57).
Карбонат бард я, помимо получения из него других соединений бария, применяют для изготовления специальных сортов стекол (легкоплавких, тяжелых и отличающихся высоким коэффициентом преломления), а также в керамическом производстве. Для получения ВаО — важного исходпого-материала для приготовления ВаОа— особенно пригоден природный витерит, который для этого прокаливают с углем, в то время как для других целей предпочитают искусственно приготовленный карбонат бария как более дешевый.
В воде карбонат бария растворим несколько лучше, чем карбонаты кальция и стронция. Его насыщенный водный раствор в результате гид
314
Глава 5
ролиза имеет щелочную реакцию. С солями, растворы которых вследствие гидролиза имеют сильно кислую реакцию [соли алюминия, железа(П1) или хрома], взмученный в воде карбонат бария вступает в реакцию, в результате которой происходит выделение СОз и выпадение в осадок гидроокисей соответствующих металлов. Этим иногда пользуются в 'аналитической химии для отделения трехналентных металлов группы сернистого аммония от двухвалентных. Растворимость карбоната бария, подобно растворимости остальных карбонатов щелочноземельной группы, несколько повышается в присутствии солей аммония. Довольно значительное количество его растворяется в воде, содержащей угольную кислоту (вследствие образования бикарбоната).
Сульфаты
Сульфаты бериллия и магния легко растворимы, сульфаты щелочноземельных металлов растворяются с Трудом, причем растворимость их сильно убывает не только при переходе от сульфата магния к сульфату кальция, но и дальше вплоть до сульфата бария. В 100 г воды при 18° растворяются: 35,6 г MgSOr; 0,202 г CaSOs; 0,014 з SrSCU и 0,00022 г BaSOi.
Сульфаты элементов главной подгруппы II группы, за исключением сульфата бария, образуют с сульфатами щелочных металлов двойные соли
типа M|Mn(SO4)a.
Сульфат бериллия. При упаривании раствора окиси или гидроокиси бериллия в избытке разбавленной серной кислоты выпадает тетрагидрат сульфата бериллия ВеЗО4-4Нч() в виде бесцветных октаэдров. Кроме тетрагидрата, существуют также ди- и гексагидраты. В воде растворимость сульфата бериллия велика, по н абсолютном спирте он нерастворим, R водном растворе BeSCp несколько гидролизовав, однако в меньшей степени, чем НеСЬ (гидролиз BeSO4 при 403 в 0,1 н. растворе составляет 0,56%). При упаривании раствора BeSO4, но содержащего избытка серной кислоты, в осадок выпадают основные соединении переменного состава. При нагревании гидратов приблизительно до 220 получается безводный ВсЙО*. При более сильном пагре-в а ни п последний начинает отщеплять SOs. После длительного прокаливания при температуре белого каления остается чистая окись бериллия.
С сульфатами щелочных металлов ВсЗО4 образует двойные соли типа M2Be(SO4)2, например хорошо кристаллизующуюся соль К2Ве(8О4)2'2НаО.
О соотношении между двойными сульфатами бёрпллия или щелочноземельных металлов и тех металлов, которые образуют купоросы, см. Schroder W., Z. aiiorg. Chem., 228 и 23!) и сл.
Сульфат магния. MgSCU в безводном состоянии представляет собой белый порошок удельного веса 2,65. С водой оп образует гидраты, важнейшими из которых являются моногидрат (кизерит) и гептагидрат (горькая соль).
Кроме названных гидратов, сульфат магния образует и другие.. Для пего известны соединения с 1, 2, 4, 5, 6 , 7 и 12 молекулами воды; нз них гидраты с 2, 4 и 5 молекулами Н2О нестабильны, а для остальных гидратов области их устойчивого существования следующие:
От -3,9 до 1,3“
MgSO4- 12J-L.0
От 1,8 до 48,3°
MgSO4-7H2O
От 48,3 до 68° Выше S8’
Ромбический
MgSO4.6I-I2O MgSO4.H2O
МОВОИЛИННЫЙ МОНОКЛИННЫЙ
Нестойкая тетрагональная модификация гекс а гидр ат а получается при внесении в пересыщенный раствор сульфата магния затравки из смеси сульфатов цинка и меди. Теплота растворения безводного MgSO4 равна 20,3 ккал, его моногидрата —• 13,3 и гёптагидрата — 3,8 ккал.
Вторая группа периодической системы г 315
Растворимость MgSO4 равна:
при 0° 1С“ 20° 30° 40“
26,9 31,5 30,2 40,9 45,6г MgSO* в 100® И2О
Сульфат магния легко образует двойные соли с сульфатами щелочных металлов. Из двойных солей сульфата магния в калийных месторождениях встречаются следующие:
KCl-MgSO4-3H2O— каинит 3Na2SO4-MgSO4—вантгоффит
К2Й О4 М gS О 4 4Н2О—леонит Ка8 О 4 MgS О 4 • 2€aS О 4 2Н2 О—по л ига л ит
K2SO4 MgSOi 6Н2О—тпёнит K2SO4 -MgSO4 4CaSO4.2H2O—крутит
K2SO4-2MgSO4—лангбейнит Na2S04-MgS04-21/.,IJ20—лёнсит
Na2S О4 MgSO4 4Н3О - астраканит
Двойную соль K2SO4‘MgSO4-6H2O приготовляют также искусственно и под названием калимаенезил выпускают в продажу как удобрительный тук, который особенно ценен для растепий, чувствительных к иопам хлора, ;
Кизерит. MgS04-Ha0 в больших количествах: встречается в месторождениях калийных солей в Германии, главным образом как составная часть так называемой «твердой соли» (см. стр. 216), но также и в виде примеси к сырому карналлиту и другим калийным солям. Несмотря на значительную растворимость сульфата магния (см. выше), его моногидрат, кизерит, лишь очень медленно переходит в раствор и поэтому при выщелачивании калийных солей остается в виде похожей па песок массы, которая при хранении совершенно затвердевает, а захваченная вода связывается благодаря образованию высших гидратов. Если мокрый остаток после выработки калийных солей выложить в формы, то он примерно через сутки затвердевает, и эти твердые глыбы либо перерабатывают на месте па горькую соль, либо отправляют как сырой продукт для приготовления горькой соли на соответствующие заводы.
Чпстый кизерит образует бесцветные молоклинныр кристаллы, имеющие удельный вес 2,57 и твердость 3,8, В то время как моногидрат в виде компактной массы или твердых глыб очень медленно растворим в воде, «кальцинированный кизерит» (обезвоженный путем прокаливания) растворяется в воде довольно быстро. Последний применяют для очистки воды.
Горькая соль. MgSO4-7 Н30 в природе содержится в осадочных породах, остающихся после высыхания озер, а также в растворенном состоянии в воде многих источников (горькие минеральные источники). В месторождениях калийных солей горькая соль образуется в качестве продукта превращения кизерита (в этом случае опа носит название рейхардита}.
Горькая соль кристаллизуется из водных растворов при обычной температуре в виде четырехгранных ромбических призм удельного веса 1,68, изоморфных цинковым и никелевым купоросам. Она устойчива во влажном воздухе, а в сухом подвергается выветриванию; она имеет неприятный горький вкус. Раньше горькую соль получали из минеральных’ вод (в первый раз она была получена англичанином Неемиасом Грью в 1695 г. из эпсомского минерального источника). В настоящее время ее добывают главным образом из кизерита, являющегося отходом при переработке калийных солей. Для этого кизерит растворяют в горячей воде и осветляют растворы отстаиванием в ящиках с хорошей теплоизоляцией; из этих
316
Глава 8
растворов при охлаждении кристаллизуется горькая соль в виде блестящих игл. Ее получают также растворением магнезита в серной кислоте.
Горькую соль применяют в технике для утяжеления хлопка и шелка, для пропитки марли —для снижения ее горючести; в крашении горькую-соль используют как протраву; ее применяют как наполнитель для бумаги и в медицине — как слабительное.
Сульфат кальция широко распространен в природе в виде дигидрата — гипса (селепит) CaS04 2Н2О и в безводном состоянии в виде ангидрита' (карстонит, муриацит).
В питьевой воде сульфат кальция передко содержится в растворенном состоянии и обусловливает поэтому постоянную или неустранимую жесткость водь| (т. е. ту жесткость, которая не исчезает после кипячения). Однако растворимость сульфата кальция в воде все же невелика. При 18° опа составляет 202 мг в 100 г воды и лишь незпачительно изменяется с температурой. Кривая его растворимости имеет плоский максимум между 30 и 40°. Присутствие других сульфатов понижает растворимость CaSO4, однако наличие в воде других солей, а также кислот, не исключая и серной кислоты, наоборот, довольпо значительно повышает растворимость щульфата кальция. С серной кислотой CaS04 довольпо легко образует растворимые в воде продукты присоединения, например CaS04 H2S04 и CaSO4-3H2SO4, которые были выделены в свободном состоянии. С сульфатами щелочных металлов CaSO4 образует трудиорастворимые двойные соли, встречающиеся также в природе, например глауберит Na2SO4 CaSO4 и сингенит K2SO4-CaSO4-H3O.
Из водных растворов при температуре ниже 66° сульфат кальция всегда кристаллизуется в виде дигидрата CaSO4-2H2O (гипс), образующего шестигранные моноклипные призмы удельного веса 2,32. Кристаллы гипса имеют заметную склонность к образованию двойников (в форме ласточкиного хвоста). Гипс распространен в природе в очень больших количествах; иногда встречаются большие, красивые, правильные кристаллы, а чаще — порода, состоящая из мелких и мельчайших кристаллов и имеющая волокнистое, зернистое пли совершенно плотное строение. Гипсовые породы встречаются во всех геологических формациях, ио главным образом они распространены в пермской формации или диасе, в триасе и четвертичной формации, иногда образуя мощные залегания и штоки. Гипс легко отличить ло его незначительной твердости (1,5—2) и прекрасно выраженной способности раскалываться (спайности). Подобно-всем минералам, кристаллизующимся в моноклинной системе, он обладает двойным лучепреломлением. Разновидностями гипса являются мариенглас, пли фрауенглас, и алебастр. Последний очень похож на белый мрамор, но вследствие незначительной теплопроводности не дает при прикосновении, подобно мрамору, ощущения холода. Чистый гипс 6ecj цветеи, или, если он представляет кристаллический агрегат, имеет белый цвет. Различные примеси иногда сообщают ему серую, желтоватую, коричневатую или красноватую, а иногда даже почти черную окраску. z-
При нагревании до 100° гипс отщепляет 3/4 своей кристаллизационной воды и переходит в метастабильный семигидрат (полугидрат} CaSO4 • */аН2О, При обычной температуре последний снова поглощает воду с заметным разогреванием. Если его замесить с водой в виде жидкого теста то он довольно скоро застывает, образуя твердую массу, состоящую из тонковолокнистых, переплетен пых между собой кристаллов гипса. На этом свойстве основано применение гипса в строительном деле, а также при
Вторая группа периодической системы 317
изготовлении скульптур (для отливок). Применяемый в этих случаях жженый гипс («штукатурный») обычно содержит еще меньше воды, чем' полугидрат; однако он не должен быть полностью обезвожен. Если гипс настолько сильно обжечь, что он отдаст всю воду, то он теряет способность в дальнейшем «схватываться», т. е. присоединять воду. Такой гипс называют «пережженым». Природный безводный сульфат кальция — ангидрит — также не способен «схватываться». Однако при очень длительном выдерживании в присутствии воды ангидрит все-таки переходит в гипс. Значительная часть встречающегося в природе гипса образовалась таким путем. Иногда, наоборот, природный ангидрит образуется из гипса. Из водных растворов ангидрит кристаллизуется при температуре выше 66°, Однако если раствор содержит одновременно и другие соли, то ангидрит может выделяться и при значительно более низких температурах. Так, из раствора, который одновременно насыщен хлористым натрием, сульфат кальция выделяется в виде ангидрита ужевыше 30°. Кроме ангидрита, существует еще одна модификация безводного сульфата кальция. Она растворима лучше, чем ангидрит, и поэтому неустойчива.
Природный ангидрит встречается в виде прослоек в залежах каменной соли, а иногда составляет промежуточный слой между залежами каменной соли н калийных солей. Он чрезвычайно распространен и встречается почти в каждой геологической формации, большей частью в смеси с гипсом, который образовался из пего. Ангидрит кристаллизуется в ромбической системе, хорошо раскалывается, однако не в такой степени, как гипс. Он превосходит гипс по твердости (3—3,5) и плотности. Его удельный вес равен 2,8—3. В чистом состоянии он бесцветен, однако нередко бывает •окрашен примесями в синеватый, синевато-серый и другие цвета.
Если гипс или ангидрит нагреть выше 1000°, то они начинают выделять дрехокись серы. Получающийся продукт (твердый раствор СаО в CaSOJ -отличается способностью поглощать воду; при замешивании с небольшим количеством воды он скорее, чем раствор из извести и песка, образует -очень твердую, плотную массу, устойчивую к выветриванию. На этом свойстве основано использование гипса, обожженного при высоких температурах (1300°), для изготовления цементирующих растворов (гипс для строительных растворов, гипс для затирки каменных полов),^которые были известпы еще древним египтянам. Кроме того, «штукатурный гипс» широко применяют для изготовления форм для керамических изделий, л цменно для литья фарфора (для чего он особешю удобен благодаря своей пористости). Тонко размолотый необожженный гипс служит добавкой \ к минеральным краскам (в обойном производстве и в бумажной промышленности).
Давление диссоциации кристаллического CaSO4 (ангидрита) при 1200° равно -3 -U.M рт ст, при 1360°—40,5 мм рт ст. Обезвоженный гипс при высоких температурах •(более 1000°) переходит в ангидрит. До окончания этого превращения CaSO4 имеет уже значительно большее давление диссоциации, чем ангидрит (Zawadski, 1932).
Гиле склонен к образованию пересыщенных растворов. Растворимость его в значительной степени зависит от величины зерен. Это свойство гипса следует учитывать при применения насыщенного раствора гипса для калибрования ячеек, предназначенных для измерения электропроводности. Удельная электропроводность х насыщенного раствора гипса (за вычетом электропроводности воды) при 18* составляет 0,1)01867 ^Melcher), а при 25°— 0,002206 (Hulett). Эти величины относятся к растворам, диаметр частиц гипса в которых ие менее 2ц. При меньшем размере частиц электропроводность насыщенных растворов гипса практически больше не меняется. Более мелкие частицы гипса можно удалить повторным растворением: при неоднократном прнливашш воды 'частицы в течение примерно трех суток переходят в раствор, Что сопровождается одновременным ростом более крупных частиц.
318
Глава 8
Сульфат стронция. SrSO4 в природе встречается в виде целестина. Последний используется в технике для приготовления большинства, сосди-нений стронция. Целестин, хотя и не столь распространен в природе, как тяжелый шпат, все же встречается довольно часто, главным образом в вид& зернистых, слоистых или плотных агрегатов, а иногда в виде хорошо образованных отдельных кристаллов, бесцветных в чистом состоянии пли окрашенных примесями. Твердость его 3—3,5, удельный вес 3,9—4, раствори^ мость 11,4 мг в 100 г воды при 18°. Как и у сульфата кальция, его растворимость повышается в присутствии ряда посторонних веществ, С сульфатами щелочных металлов, например с K.2SO4 и (NH4)2SO4, сульфат стронция образует труди «растворимые двойные соли. При нагревании выше-11520 обычная ромбическая модификация сульфата стронция переходит в Другую, вероятно моноклинную модификацию. Сульфат стронция плавится при температуре белого каления, но еще до этого он начинает терять SO3,
Сульфат бария. BaSO4 широко распространен в виде минералов (тяжелый шпат, барит). Иногда тяжелый шпат встречается в виде отдельных хорошо образованных кристаллов, которые принадлежат к ромбической системе и отличаются большим разнообразием форм (твердость тяжелого пшата 3—3,5, удельный вес 4,48), Одиако обычно он образует волокнистые, зернистые или плотные агрегаты. Сам по себе тяжелый шпат бесцветен, но иногда бывает окрашен примесями. Нередко он содержит значительные количества изоморфного ему сульфата стронция. В некоторых месторождениях он встречается с примесью Сульфата кальция (известковый барит из Фрейберга и Дербишира).
Сульфат бария образует ромбическую решетку, построенную из ионов Ва2+ и [SOJ2-. Такую же структуру имеют сульфаты стронция и свинца. По тому же типу («тип тяжелого пшата») кристаллизуется ряд перхлоратов {М[С1О4], М = К, Fib, Cs, NH4, фтороборатов (M[DF4], M = Rb, NH4), а также перманганат калия-
К[МпО4).
В подо сульфат бария почти нерастворим; при 18° в 100 г воды раствор ряются лишь 0,22 мг BaSO4. Из растворов солен бария при действии ионов. SO" выпадает белый осадок BaSO4, а из растворов, содержащих серную-кислоту или сульфаты,при добавлении к ним ионов Ва". Присутствие сильных кислот значительно повышает его растворимость. В концентрированной серной кислоте его растворимость даже довольно значительна — до 10—12%, что объясняется образованием комплексного-соединения.
Сульфат бария благодаря его крайне низкой растворимости используют для аналитического определения как ионов S04, так и попов Ва". При этом необходимо учитывать, что BaSO4 может увлекать с собой в значительных количествах и другие вещества, находящиеся в: растворе, особенно многовалентные ионы, например: сульфаты трех валентного железа, трехвалситного хрома и алюминия. До сих пор еще не установлено, обусловливается лц это явление адсорбцией пли оно связано с образованием твердых растворов.
По данным Гримма (Grimm, 1924), BaSO4 может давать твердые растворы с очень значительными количествами (до 60 мол.%) КМпО4 (см. также К а г а о g 1 am о w:, Z. auorg. Chem., 222, 249, 1935). Отсюда следует, что для образования твердых растворов, решающим является ве ctojh.ko «химическое сходство», сколько вы пол пение следующих условий: 1) аналогия химических формул (например, Ba(SO4] — KJMnOJ,
Вторая группа периодической, системы
31»
CafCOjJ-^-NajNOg]), 2) совпадение тина кристаллической решетки*, 3) близость расстояний между центрами ионов в решетке (различие расстояний не должно превышать. 5% при комнатной температуре).
Сульфат бария отличается абсолютной устойчивостью по отношению к воздуху, повышению температуры и к действию вредных газов, содержащихся в воздухе (особенно к сероводороду). Поэтому его применяют в качестве минеральной белой краски (перманентная белая — blanc fire), В качестве краски без добавок его не применяют ввиду слишком слабой кроющей способности. Зато его часто используют в качестве примеси к минеральным краскам и к органическим лакам. BaS04 находит широкое применение в качестве добавок к бумаге (обоям, картону, цветной, а также фотографической бумаге). Сульфат бария сообщает бумаге особую гладкость. Для всех указанных, целей сульфат бария следует применять в очень тонко измельченном состоянии. Однако для некоторых целей оказывается недостаточным даже самый тонкий помол природного тяжелого пшата. Поэтому BaS04 приготовляют в значительных количествах также искусственным путем. При этом исходят из витерита ВаСО3 или из природного тяжелого шпата. Последний переводят сначала в сульфид прокаливанием с углем. Полученный таким путем сульфид или природный карбонат бария растворяют затем в соляной кислоте и, добавляя к раствору сульфат натрия или магния, осаждают BaS04. Таким образом полученный BaS04 поступает в продажу в виде пасты с содержанием. 15—20% воды, так как при полном обезвоживании он вновь теряет свою кроющую способность. В качестве побочного продукта сульфат бария образуется прп получении перекиси водорода разложением перекиси бария серной кислотой. В медицине сульфат бария находит применение благодаря своей непроницаемости для рентгеновских лучей, что используют для получения отчетливых изо-бражений при просвечивании желудочно-кишечного тракта. Вследствие исключительно малой растворимости BaSO4 безвреден для организма.
Аналитические данные. При обычном ведении анализа кальций, стронций и барий после предварительного удаления всех тяжелых металлов осаждают в виде карбонатов при обработке, раствора карбонатом аммония в присутствий хлорида аммония. Разделение щелочноземельных элементов производят, используя различную растворимость их нитратов и хлоридов в эфире и спирте. В смеси сппрта и эфира хорошо растворим только нитрат кальция, а из хлоридов в абсолютном спирте нерастворим только хлорид бария. Для отделения бария можно также воспользоваться тем, что из уксуснокислого раствора бпхромат калия осаждает только барий (в виде хромата). Растворимость хромата бария составляет прибди> зительно 1 : 300 000. Хотя хромат стронция тоже'очень мало растворим (около 1 : 800), ио для него произведение растворимости Пр=[$г"]Х X |СгО4] настолько больше, чем для хромата бария, что той незначительной концентрации ионов СгО4, которые могут находиться в равновесии с ионами Сг2О? в присутствии уксусной кислоты (подробнее об этом см. в гл., о хроме т. Л), оказывается уже недостаточно, чтобы произошло осаждение хромата стронция. Растворы солен бария и стронция образуют с гипсовой водой (насыщенный раствор сульфата кальция) осадки; в связи с тем, что произведения растворимости сульфатов бария и стронция значительно
* Относительно образования «аномальных твердых растворов» .соединениями, не имеющими аналогии пи в составе, ни в кристаллической структуре, например СаР2 и YF3, см. т- II.
320
Глава- 8
ниже, чем сульфата кальция, концентрация ионов SO” в гипсовой воде оказывается достаточной для осаждения ионов Ва" даже в том случае, если концентрация последних сравнительно невелика. Для идентификации щелочноземельных металлов наблюдают окрашивание пламени, однако лучшие результаты получают при использовании спектроскопа (см. стр. 280 и ел.)
Магний в ходе качественного анализа осаждают баритовой водой в виде гидроокиси из фильтрата после осаждения карбонатов щелочных металлов. Эту операцию осуществляют после предварительного удаления осторожным нрокаливапием солей аммония. Для Открытия магний удобнее всего перевести в магнпйаммоннйфосфат Mg(NH4)P04>6H30, образующий кристаллы характерной формы.
В последнее время для открытия магния часто применяют также органические реактивы — хинализарин или дифенил карбазид. Последние придают осаждающейся в их присутствии гидроокиси магния голубую или соответственно красную окраску, адсорбируясь па ней; это проявляется особенно ярко прп осаждении из очень разбавленных растворов.
Бериллий нри обычном ходе анализа сопровождает алюминий, от которого его довольно трудно отличить. Несмотря на то что сулЕ,фат бериллия ине образует квасцов, •с сульфатами щелочных металлов он дает двойные соли (см. стр, 314). В отличие от алюминия бериллий не осаждается на холоду избытком концентрированного раствора карбоната аммония. При кипячении такого раствора бериллий-осаждается в виде основного карбоната. Для отделения бери.алия от алюминия можно также использовать растворимость хлорида бериллии в смеси равных объемов насыщенных водного и эфирного растворов хлористого водорода. В такой смеси гидрат хлорида алюминия нерастворим. Количественное определение бериллия производят аналогично определению алюминия (осаждением гидроокиси и взвешипанием в виде окиси).
Количественное определение магния обычно производят осаждением его в виде двойного фосфата магния и аммония, который затем прокаливанием переводится в пирофосфат магния MgaP2O7, являющийся его весовой формой. При количественном анализе кальций обычно отделяют в виде оксалата СаС2Ог ЩО, так как последний наиболее труднорастворимая соль кальция. Высушенный при 110° оксалат кальция взвешивают непо^ ^родственно в виде соли или я виде окиси, которую получают после прокаливания соли в окислительном пламени. Стронций иногда осаждают также в виде оксалата, но лучше его осаждать в виде карбоната; взвещиванме в обоих случаях производят в виде окиси. Стронций можно также определять весовым путем в виде сульфата. Барий обычно определяют в виде сульфата.
Определение магния можно проводить весовым путем, осаждая его в виде магний-аммоняйарсената (NHJMgAsO4; последний растворяется в соляной кислоте, а мышьяковую кивлоту определяют йодометрически (Daubner, 1935). От ионов щелочных и (в присутствии солей аммония) щелочноземельных металле!) Mg можно Отделить по Вергу (Berg, 1927) осаждением о-оксихинол и ном. После соответствующего высушивания осадок взвешивают или титруют бр омом Щр и чески. Кальций определяют весовым методом осаждением в виде оксалата СаС2О^Н2О и титрованием перманганатом щавелевой кислоты, выделившейся из пего при деистами разбавленной серной кислоты (ср. стр. 493). Очень небольшие количества стронция в присутствии кальция мои;но определить нан-более точно спектральным методом в вольтовой дуге (ср. стр. 195, 280 и сл.) (R. u t-hardt К., Z. anorg. Chem., 195, 15, 1931).
Для разделения щелочноземельных металлов пользуются темп же методами, что и в качественном анализе; однако, чтобы добиться полного •отделения зтих трех близких между собой элементов, как этого требует 'количественный анализ, необходимо соблюдать особые предписания.
Гл а в а 9
СТРОЕНИЕ И СВОЙСТВА ХИМИЧЕСКИХ СОЕДИНЕНИЙ
Соединения рассмотренных до сих пор элементов построены, как правило, очень просто. Металлы главных подгрупп I и И групп периодической системы образуют преимущественно соли или солеобразные соединения,., т. е. соединения, по своей структуре соответствующие солям, Подлинные соли построены из положительных и отрицательных ионов. Поскольку при этом речь идет об ионах, по своему строению подобных атомам инертных газов, т. е. обладающих такими же электронными конфигурациями, как и инертные газы, то образованные ими соединения состава АВ или АВ2, как было показано на ряде примеров, обладают большей частью очень простой структурой. Все рассмотренные до сих пор соли и солеобразные соединения очень близки по свойствам, если только пе учитывать их различную растворимость,Соединения металлов главных подгрупп I и II групп, не имеющих солеобразпого характера, т, е. их гомеополярные соединения, также построены очень просто и состоят из двух- или трехатомных молекул — АВ или АВ2, — в которых атомы связаны между собой парой валентных электронов. Соединения этого типа мало различаются по своим свойствам и поведению. Из рассмотренных до сих пор соединений только в отдельных случаях встречались такие, например, гидриды бериллия и магния, которые по своим свойствам и поведению не укладывались полностью в эту простую схему,
В то же время соединения, рассмотрению которых будут посвящены следующие главы, проявляют в своих свойствах и поведении чрезвычайное многообразие. Этим веществам присущи, помимо уже ранее рассмотренных, также и другие типы связи, причем следует указать, что их свойства и поведение определяются не только составом, по и в значительной степени строением. Поэтому, прежде чем приступить к рассмотрению элементов следующих групп периодической системы и их соединений, необходимо остановиться на зависимости между строением и свойствами веществ. Исследование этой зависимости является одной из важнейших задач химии, ибо познание ее позволяет варьировать желаемым образом свойства , веществ, изменяя их состав, или каким-либо иным образом предсказывать существовапне, поведение и условия, прн которых можно получить не известные еще соединения и путем систематического синтеза создавать вещества с желаемыми свойствами. Б то время как в органической химии такие задачи успешно разрешаются уже в течение многих лет, в неорганической химия лишь начинаются исследования, направленные к достижению этой цели.
Классические методы органической химии, позволяющие уста лавливать структуру веществ, в неорганической химии применимы только в отдельных, большей частью 21 Г. Реми
322
Глава О
достаточно узких областях*, и потому основанные па этих методах чисто эмпирические Представления связи между Строением и свойствами нельзя безоговорочно применить [i неорганическим веществам. При исследованиях в этой области, проводящихся с использованием новейших теоретических и экспериментальных методов физики, следует также иметь в виду, что соотношения в органических соединениях представляются зачастую значительно более простыми, чем в неорганических.
То обстоятельство, что старые методы определения строения веществ, основанные главным образом па изучении химических свойств, большей частью совершенно неприменимы для неорганических соединении, объясняется следующим образом: почти все органические вещества построены цз молекул, содержащих ограниченное число атомов и способных переходитт, в газообразное состояние или в раствор, не испытывая при этом существенных Структурных изменений- Напротив, неорганические вещества в твердом состоянии в подавляющем большинстве построены иа неограниченного числа атомов или ионов, При испарении или растворении таких веществ разрушаются силовые поля, в которых находились атомы или ионы в твердом состоянии, и тем самым становится невозможным непосредственное изучение существовавшего прежде типа строения. Кроме того, в органических соединениях почти всегда осуществляется только один тип связи. Не существует принципиальных отличий пи между углерод-углеродными связями (простая и кратная связь, ароматическая связь), ни между углерод-угиорбд-ными и другими связями, возникающими между углеродными и другими атомами в органических соединениях. В неорганических соединениях следует различать многие принципиально отличные типы связей, между которыми существуют многочисленные переходы, которые еще более осложняют положение. Открытое Лауэ цреломлениё рентгеновских лучей при прохождении через кристалл впервые позволило изучить структуру^ веществ, построенных из неограниченного числа атомов или ионов. Принципиальные различия между типами связи, присущими неорганическим веществам, становятся понятными па основе теории строения атома и квантово-механических представлений.
Типы связей. Различают следующие типы связей:
1) гетерополя риал (ионная, электровалентная),
2) т оме он о лярв а Я- (атомная, ковалентная),
3) металлическая,
4) вандерваальсовая (межмолекулярная), г
5) водородная,
Гетерополярная связь обусловлена силами притяжения, которые действуют между противоположно заряженными ионами. Величина этих сил, если рассматривать иолы как тиары, на поверхности которых равномерно распределены заряды, определяется по закону Кулона (ср. стр; 107). Эти силы могут значительно возрастать при искажениях; сдвигах в распределении зарядов (деформация, поляризация, стр. 159 й 244).
При определенном расстоянии между центрами ионов, равном сумме ионных радиусов; притяжение компенсируется отталкиванием, которое очень быстро возрастает при дальнейшем уменьшений расстояния и, наоборот, быстро падает до практический исчезающе малой величины приувеличении этого расстояния. Кривая Г рис. 64 представляет собой рассчитанную таким образом зависимость потенциальной энергии двух противоположно заряженных ионов от расстояния между ними. Положение минимума на потенциальной кривой соответствует расстоянию между центрами ионов п образованной ими молекуле. Если бы отталкивания по было, то потенциальная кривая дальше шла бы так, как указано пунктиром в левой пижией части рисунка.
Квантовая механика утверждают (ср. стр. 155 ц ел.), что могут возникать силы притяжения между; незаряженными атомами, и притом настоль-
*; Примером успешного определения строения веществ в "большой и важной;, области неорганической химии при помощи методов, применяющихся в органической химии, может служить объяснение пространственного строения л^очныо: комплексных соединений, например хрома, кобальта и т, д., данное Вернером на основе явления изомерии и оптической актив гости (см. т. II, гл. 5). Речь в этом случае идет о комплексах внедрения, в которых тян связи приближается к типу связи в органических соедине-" ниях (см, стр. 443)..
Строение и свойства химических соединений
323
но же прочные, пак и между против он оложнозаряженными ионами или зачастую еще более сильные. Если взаимодействие обусловливается этими действующими между незаряженными атомами силами, то говорят б гомео-полярной связи.
Р и с- 64. Зависимость потенциальной энергии от иежъядерного расстояния для ионов Na+-f- CI" (кривая Г) и атомов Na 4* Cl (кривая 11).
На рис. 65 кривая II показывает зависимость потенциальной энергии двух атомов Н, связанных между собой гомеополнрной связью, от расстояния между ними, рассчитанного на основе квантово-механических пред став лений. Для сравнения на рисунке приведена кривая I, отражающая изменение потенциальном энергии молекулы На, построенной из противоположно заряженных ионов Н+ и И". Кривая рассчитана па основе закона Кулона с учетом сил отталкивания между ионами, проявляющихся црн очень сильном сближении. Из рис, 65 видно, что минимум энергии на этой кривой лежит намного выпге, чем минимум анергии на кривой II, так что стабильной оказывается молекула На, построенная из атомов, а не из ноной. Как уже было отмечено ранее (см. стр. 244 и сл,), гомеополярная связь осуществляется путем спаривания электронов. Притяжение между двумя атомами водорода возникает только в том случае, если их^электроны обладают противоположными спинами. Атомы водорода с электронами, обладающими одинаковыми спинами, взаимно отталкиваются. В соответствии с этим между атомами инертных газов действуют по силы гомеопо-лярпоп связи, а только силы отталкивания, так как эти атомы не содержат неспаренных электронов. То же имеет место ।для ионов с конфигурацией инертного газа, например Ма+ н С1". Они отталкивались бы, если бы между ними не действовали силы кулоновского притяжения. Отталкиваются также и свободные атомы Ыаи СГ, как это следует из хода кривой II на рис. 64. Из
рисунка видно, что для молекулы NaCl стабильна гетерополярная связь. Кривые I. и II рис. 64 и 65 заметно различаются вследствие того, .что потенциальные кривые для
• 21*
Р и с, 65. Зависимость потенциальной энергии от межъядерного расстояния для Н+ И " (кривая I) и Н+• Н
(кривая II).
324
Глава 9
ионов (кривая Г) сравнительно медленно стремятся к предельному значению, которое соответствует потенциальной энергии системы при бесконечно большом расстоянии менаду ионами, (Эти значения характеризуются пунктирными прямыми для Na* 4’Cl-или соответственно Н+4- Н“.) Потенциальные кривые для атомов достигают соответ* ствующпх предельных значений, (на обоих рисунках, обозначенных пунктирными прямыми для Na -j- С1 и Н Н) гораздо быстрее. Электростатическое притяжение, следовательно, действует на значительно больших расстояниях, чем силы гомёополярпой связи. .Характерно, далее, что расстояние между предельными значениями для кривых I и //на рис. 64 и 65 сильно различаются. Это расстояние, представляющее собой разницу энергий, между состоянием из свободных ионое А* 4- В- и состоят!ем из свободных атомов А 4" В в первом случае равно 1,39, во втором 12,88 ав. Его можно выразить через разность ЕП1 где JА— работа ионизации атома А й £в— сродство к электрону
атома В. Следует учесть, что всегда, если Jд—£в мало, то образование молекулы происходит из ионое, если УА велико, то образование молекулы происходит из атомов.
Из теории металлического состояния известно (см, т, II, гл. 1), что в веществах с металлическим типом связи между положительно заряженными атомами более или менее подвижно располагаются оторвавшиеся от них электроны,
Вандерваалъсовым называют такой тип связи, который основан па силах, вызывающих отклонение газов от законов идеального состояния и их ожижение при достаточном понижении температуры, Так как эти силы действуют между валептпонасыщенпыми в обычном смысле молекулами, то такие связи называют также «межмолекулярньши связями».
Вандерваальсовы силы, как уже указывалось ранее (стр, 127), могут быть сведены отчасти к поляризационным взаимодействиям, отчасти к кваптово-механическим резонансным силам, В последнем случае говорят также о «дисперсионных силах», так как их возникновение объясняют теми же причинами, которые вызывают дисперсию света®. Эти силы отличаются от резонансных, проявляющихся в гомеополяриой связи, значительно большей сферой действия. Вандерваальсовы силы в общем намного слабее, чем обычные валентные силы. Поэтому на потенциальной кривой, представляющей их действие, присутствует обычно только плоский минимум. Так как вандерваальсовы силы проявляются еще па таких расстояниях, на которых исчезает действие обычных, вызывающих гомеополярную связь, резонансных сил, то они могут вызывать притяжение между атомами, которые под действием обычных резонансных сил отталкиваются, например между двумя атомами инертного газа или двумя атомами Н* с параллельными электронными спинами. Последние не могут соединяться в молекулу, так как при небольших расстояниях между ядрами они взаимно отталкиваются; однако на больших расстояниях между атомами такого рода существует слабое притяжение, вызываемое ванде рваа Львовыми силами.
В химических соединениях перечисленные выше типы связей часто проявляются не в чистом виде; появляются промежуточные формы, которые не соответствуют строго ни одному из этих типов связи.
Если, например, потенциальная кривая (см, рис. 65) для ионной молекулы А+В" лежит немного выше потенциальной кривой для атомной молекулы А : В, то, как это следует из квантовой механики, возможность перехода в ионное состояние значительно повышает стабильность атомной молекулы, т. е. упрочняет связь**. Если, наоборот, ионная связь стабильпее, чем атомная, то возможность перехода молекулы в атомное состояние также будет способствовать ее упрочению. Долю, которую ионное состояние
* Дисперсией в оптике первоначально называли разложение (dispersio) света при прохождении его ч.ерез призму. Этим же термином обозначают явление, на котором основано это разложение, а именно изменение показателя преломления вещества в зависимости от длины волны света.
** Упрочение связи можно паглядпо объяснить предположением, что резонирующие электроны большую часть времени находятся около ядра атома В, чем у ядра атома А. Вследствие этого оба атома взаимно удерживаются ие только силами электронного резонанса, во также еще и вследствие того, что они часть времени оказываются противоположно заряженными.
Строение и свойства химических соединений
325
вносит в общую энергию связи, обозначают, согласно Малликену (1936), как ионность соответствующей связи.
В качестве чисто гетерополярных следует рассматривать такие связи, ионность которых практически составляет 100%, а в качестве чисто гомеопо.тарных — связи ионность которых близка к нулю. Все молекулы, обладающие дипольными моментами, заметно отличающимися от пуля (см. стр. 347), имеют и заметно отличающуюся от нуля ионность связи.
Промежуточные формы существуют не только между гетсрополярной и гомеопоЛярной связями, но и между указанными связями и металлическим типом связи.
Вандсрваальсовы силы в принципе существуют всегда. Но если одновременно с ними действуют другие валентные силы, то они, как правило, полностью перекрывают вандерваальсовы, которые поэтому обычно не обнаруживаются. Целый ряд водородсодержащих соединений '—прежде всего такие, которые имеют водород, связанный с кислородом или азотом, — проявляют особенно сильную склонность к ассоциации, приводящей К тому, что эти соединения ассоциированы в значительнойj степени нё только в жидком состоянии, но также и в растворе в индифферентном растворителе и даже в газовом состоянии. Связь, на которой основано объединение простых молекул этих соединений в ассоциированные молекулы — так называемые «сверхмолекулы», обозначается как связь, обусловливаемая водородными мостиками или просто водородная связь. Водородные мостики могут связывать не только одинаковые, но и различные молекулы. На этом основано, например, взаимодействие Н2О и NHa, а также, вероятно, связывание воды щавелевой кислотой в гидрат щавелевой кислоты.
Предположение о том, что через водород способны осуществляться связи между насыщенными в смысле классической теории валентности молекулами, было высказано еще Ворнером, который приписывал водороду способность проявлять побочную валентность (си. стр. 431), т. е. выступать в качестве координационно двухвалентного атома. Понижение реакционной способности фенольного водорода в некоторых органических соединениях было объяснено Пфейфером (Pfeiffer, 1913) в духе вернеровской координационной теории .на основе предположения о проявлении водородом внутримолекулярных побочных валентностей, которые вызывают, так же как, например, побочные валентности никеля в никельдимети л глиоксиме (т. II, гл. 7), замыкание цикла. (Так называемое «клешнеобразование», иди «хелатообразование», от ~~ звериные когти,
клешни краба.) Термин «водородная связь», или «водородный мостик», был введен Хиггенсом, Латимером и Родебушем (1919—1920). Предложенное ими аа основе теории валентности Льюиса (см. стр. 154) объяснение водородной связи первоначально хотя и четко отличало ее от так называемой «онйеной» связи (которая осуществляется вследствие того, что нейтральная молекула приобретает положительный заряд из-за присоединения иона II * и вместе с тем способность удерживать отрицательный йоп, например {NHJ’C]-), но лее же без дополнительных предположений не находилось я согласии с принципом Паули (ср. стр. 145 сл.). Недостаточным также оказывается, как было показано Бриглебом (Briegieb, 1943), неоднократно высказывавшееся позднее толкование водородной связи как изменение положения атома Н вследствие эффекта мезомерии. Бриглеб указывает на следующее существенное обстоятельство: сравнительно прочная связь, характерная для водородных мостиков, проявляется только между такими молекулами, которые имеют сильно дипольный характер или содержат сильно полярные группы (п а пример, —ОН, — 1ХН2); кроме того, атомы или ионы, участвующие в образовании водородных мостиков, обладают малыми радиусами. Поэтому под действием водородных связей полимеризуются молекулы Н2О и HF, но пе НС1. Но взаимное притяжение дипольных молекул создает только предварительное условие для возникновения водородных связей; для того чтобы связи обладали достаточной прочностью, характерной для водородных мостиков, в образовании их должны участвовать еще и другие .силы. Эти силы следует искать, ио всей вероятности, во взаимодействии между электронными системами атомов, участвующих в образовании связи', природа подобных сил еще не может быть истолкована, по для них, несомненно, существенным является значительное сближение атомов, участвующих в образовании Связи. Эти силы, обусловливающие, помимо электростатического притяжения между дп полями, возникшие-
326
Глава 9
иве водородных связей, обладают очепь ограниченной сферой действия. Таким «бравом, только большая прочность отличает водородную связь от вандерваальсрвой связи, к которой она в остальном: очень близка.
Мезомерия. Так же, как атомные и ионные состояния, между собой могут взаимодействовать и различные энергетически равные или близкие состояния молекул, в которых гомеополярные связи распределяются между атомами различным образом. Известнейшим примером этого является^ бензольное ядро. Среди следующих формул
две первые (формулы Кек у ле) энергетически равноценны. Три-другие (формулы Дьюара) тоже равноценны между собой, но соответствуют более высокому энергетическому уровню, хотя и незначительно отличающемуся от первого состояния. По представлениям квантовой механики различные состояния бензольной молекулы, изображаемые пятью формулами, комбинируются в одно промежуточное, которое энергетически беднее, чём каждое из приведенных состоянии. Этим объясняется особая устойчивость (стабильность) бензольного ядра, В таких случаях говорят о мезомерии-, мезомерия существенно отличается от таутомерии, как и вообще от любого вида изомерии, Изомерные вещества (включая таутомеры) различаются структурно' В случае мезомерии взаимное расположение всех атомных ядер остается неизменным. Свойства мезомерного вещества, согласно сказанному выше, не слагаются аддитивно из свойств его возможных форм, как это имеет место для смеси изомеров. Фактически эти 'формы вовсе не существуют, ими лишь заменяется промежуточпя форма, которую иначе нельзя наглядно представить. Далее, в случае таутомерий не существовало бы различия между обеими формулами Кекуле до тех пор, пока в бензол не введен заместитель. Не было бы также различия в этом случае и между тремя формулами Дыоара. В действительности тип связи, существующий: в бензоле, возникает в результате комбинации всея пяти написанных выше формул.
Упрочение химической связи, возникающее вследствие мезомерии, основано на действии обменных силМИя. ИПачо па квантово-механическом резонансе (так же как и вообще гомеополярная связь) (см. стр. 156), Чтобы этот резонанс мог проявиться в форме мезомерии, должны Соблюдаться следующие условия.
1. Различные состояния молекулы могут резонировать между собой лишь в том случае, если они отличаются только расположением электронов. Расположение атомных ядер в них должно быть практически одинаковым,
2. Различные состояния, если их мысленно представить себе существующими; порознь, должны быть энергетически равшщенпы или почти равноценны.
3. Молекула в своих различных состояниях должна содержать одинаковое число неспаренных электронов. В большинстве случаев это число равно пулю. Это означает, что соединения, в которых имеет место мезомерия, содержат исключительно спаренные электроны.
Из того, что явление мезомерии было первоначально рассмотрено на примере бензола, не следует делать заключение^ что оно присуще главным образом органическим соединениям. Мезомерия очень часто встречается и среди неорганических веществ, за исключением чисто солеобразных.
Строение и свойства химических соединений ...... .327
В Ткачестве примера явления мезомерий в неорганических соединениях можпо
Формула II соответствует классической формуле этого иона с двумя двойными и двумя простыми связями. Электронное расположение, соответствующее формуле II, может существовать в шести мезомерных формах в зависимости от того, как распределяются две двойные и две простые связи между четырьмя атомами кислорода:
Кроме этих шести мезомерных форм, соответствующих формуле II, й Двум, соответствующим формулам,1 и III, необходимо учитывать еще четыре; промежуточные формы между I и II с одной двойной связью и тремя простыми связями и четыре, промежуточные-между II и III с тремя двойными связями и одной простой. Таким образом,-для [ЗО1]'2'-иона всего получается 16 форм, мезомерию между которыми: следует рассматривать к ак веро ятную. К это му до бав ляотся ещё возможность резоиансамёждуформами счисто гомеополярными связями,т. е. такими формами, в которых валентные электроны равномерно распределены между связанными атомами и формами, в которых атомы в известной степени противоположно заряжены, так как электроны, посредством:кото-рых осуществляется связь, теснее связаны с одними атомами, чем с другими.
Таблица 68
Межатомные расстояния в ионах некоторых кислородных кислот (Pauling)
Ион [Sto4]4' 3-[Р041 -2- [SO4] . [СЮ1?"
Расстояние R<—»0 наблюдаемое, А . . . 1,60 1,55 : 1,51 : 1,48
Расстояние Н+--+0, рассчйтан-, пос для простой связи, А. , . 1,83 1,76 : 1,70 ' 1,75
Разница, А —0,23 —0,21 —0,19 : - (1,17
Какие на этих возможных случаев мезомерии [ЗО^-з-иона в действительности приводят к упрочению связей вследствие резонанса*, указать нельзя, так как не существует расчетов энергетических уровней различных принимаемых во внимание резонирующих форм. Но что действительно имеет место такое упрочение связей вследствие резонанса, па это указывает укорочение межатомного расстояния S<—>0, во всяком случав, псклю-
* Автор пользуется представлениями теории резонанса, в основе которой лежит - положение о Том, что строение и свойства молекул, определяются «резонансом структур», т.::е. наложением друг па друга многих электронных состояний или структур, формально возможных по правилам валентности. Согласно этой теории, такое наложение приводит к выигрышу энергии и к устойчивости соединения.
. Теория резонанса была подвергнута в советской литературе критике, и было показано, что понятие -энергии резонанса и электронного резонанса является результатом неправильной трактовки приближенных кваптово-мёханичсских методов расчета молекул и не отражает их реального строения.
(«Состояние теории химического строения в органической хцмии», Доклад комиссии Отделения химических наук АН СССР, М., 1954).— Прим. ред.
328
Глава &
чающее возможность того, что только одна формула I, выведенная на основании октет-ной теории валентности, отражает истинную структуру иона [SO4]a“ (см. табл. 58).
На табл. 58 видно, что положение, наблюдаемое для иона [SO4]3_, справедливо и для ионов других кислородных кислот элементов второго ряда периодической системы. Так как не известно, какие из чисто формально (т. о. без учета присущих им энергетических уровней) возможных мезомерпых состояний в действительности принимают участие в образовании связей, то Полппг рекомендовал сохранить классические формулы строения этих ионов, тем более что в них, как это уже было показано выше на примере иопа,
io. ,ор-
заложены удовлетворительные возможности для выражения различных резонирующих состояний. Только» случае SiOj~-iiona, в котором при классическом способе представления пет двойных связей, возможность мезомерных состояний но следует из классической формулы без дополнительных предположений. Но так как в [SiO4|4 -iione, помимо гомеололярной, существует значительная доля гетеро полярной связи (см. стр.542), то при объяснений укорочения межатомных расстояний и вызванного тем самым упрочения связей удовлетворительным оказывается предположение, что здесь ионный тип связи резонирует с чисто атомным.
Но не во всех случаях классические формулы оказываются приемлемыми с точки зрения новейших результатов в определения Строения молекул. Например, строение соединения ВГ3 соответствует не классической формуле
F :F;
F—В—F или а, как это будет показано в следующей главе, формуле
(три левомерные формы)
Структура и строение. Свойства веществ определяются массами и зарядами ядер атомов, из которых они построены, пространственным расположением атомных ядер к типом связи, существующим между атомами в молекулах этого вещества. Массы атомов (за исключением водорода — дейтерия) существенны только для очень немногих свойств веществ, в первую очередь для удельных весов. Большинство свойств зависит в основном только от пространственного расположения атомных ядер и от числа и состояний электронов, которые в конечном счете опять-таки определяются нарядами ядер. Расположение атомных ядер в веществе считают его структурой. От структурных данных отличают данные, относящиеся к электронным состояниям молекул (распределение электронов у различных атомов, характер их взаимодействия или тип связи, пространственное расположение связей); эти результаты рассматривают как данные о строении молекул.
Сведения о строении, если они являются исчерпывающими, всегда содержат сведения о структуре. По во многих случаях в неорганической химии данным о пространственном расположении, атомов или ионов, участвующих в образовании соединения, придается подчиненное значение по сравнению с данными о характере связи между его отдельными частями. Поэтому часто удовлетворяются неполными сведениями о строении, отражающими только взаимное расположение составных частей молекулы, как, папример, в обычных формулах строения комплексных соединений. Формула K3[Ni(CN)/J свидетельствует лишь о том, что четыре группы CN связаны с никелем и что образовавшийся таким образом комплекс электровалентно связан с двумя ионами
Строение и- свойства химических соединений 329
калия. Какова природа связей между Ni и CN—гетеро полярная она или гомсополярпая, как группы CN в комплексе расположены около Ni (тетраэдрически или плоско), как ионы К+ в кристалле этого соединения располагаются между попами [Ni(CN)4]'2-—на все эти вопросы такая формула не дает ответа*. Вообще в случае комплексных соединений в огромном болыШ!яство случаев полные структуры их еще не известны. Однако для многих неорганических соединений известны их структуры, хотя их строение полностью еще не выяснено.
Влияние структуры па свойства веществ проявляется, например, при полиморфных превращениях вследствие зависимости свойств веществ от особенностей модификаций, в которых они существуют. Особенно отчетливо выражено это влияние в случае алмаза — графита. Здесь изменение структуры сопровождается существенным изменением типа связи (см. ниже). Значительное влияние типа связи на свойства иллюстрируется примером иодид цезия — (J'-латунь (ZnCu). Оба соединения обладают совершенно одинаковой структурой, но в свойствах у них нет ничего, общего. Сравнительно простые соотношения между структурой, и свойствами могут быть установлены для силикатов (ср. стр, 546).
Вещества, обладающие одинаковым составом и структурой и одинаковым числом электронов, но различающиеся расположением электронов, называют «электромерами» (сокращение от электроизомеры). Обычно из различных мыслимых элецтромерных форм существуют только наиболее стабильные. Если имеются различные формы, энергетически не отличающиеся или мало отличающиеся От наиб о л ее ( стабильной, то тогда наиболее стабильным оказывается промежуточное состояние, возникающее вследствие резонанса между отдельными формами. Следовательно, возникает явление мезомерии.
Имеется, однако, много случаев, когда вещество существует в различных формах, не резонирующих между собой и не склонных к превращению одна в другую (ср. т. II, гл, 18).
Классификация веществ по их строению. Все вещества Можно подразделить в зависимости от их строения на шесть классов, приведенных в табл. 59. При сравнении свойств, характерных для типичных представителей этих классов веществ, отчетливой становится связь свойств со строением вещества. Солеобразными соединениями называют вещества, построенные подобно типичным солям (например, хлорид натрия) и образующие ионную, большей частью так называемую координационную решетку (см. стр. 242), К ним принадлежат также и окислы, такие, как СаО и другие ионные соединения с координационными решетками. Напротив, к ионным соединениям не относятся такие вещества, как Hg2CI2 й HgCl2, которые хотя н являются с точки зрения их происхождения солями, но образуют отчетливо выраженные молекулярные решетки. Солеобразный характер могут проявлять только сложные вещества, в то время как к каждому из остальных, приведенных в таблице классов веществ, принадлежат н-простые вещества. Типичные солеобразные соединения образуются из таких элементов, ионы которых обладают конфигурацией инертных газов. Между ними в качестве сил притяжения действуют практически только кулоновские силы. Так как кулоновские силы обладают сферической симметрией, каждый ион стремится окружить себя возможно большим числом противо-положнозаряжепиых ионов. Этим объясняется тот факт, что при кристаллизации твердого вещества ионы объединяются не в ограниченном числе с образованием молекул, а в неограниченном числе с образованием координационных решеток.
* Структуру неорганических соединений, образующих атомную или иоппую решетку, вообще трудно передать формулой: вместо структурной формулы здесь описывают элементарную ячейку.
Классификация веществ по их строению
Таблица 59
Класс : веществ Тип СВЯЗИ- Строение Характерные свойства Примеры
Соле об равные соединения Гетеропо- Ляркая Ионная решетка Трудно летучи, в раствор сипом и расплавленном состоянии— э л е ктр о л иты ,-р ас тво-рнцугся преимущественно в растворите^ лях, вызывающих диссоциацию - NaCl, КВг, СаР2, СаО
Атомные молекулы Гомеополярная Свободные молекулы; в твердом состоянии имеют молекулярную решет-ку Газообразны или легко летучи, не проводят . электрический т ок, в большинстве случаев хорошо растворим ы, прёиму ще ст-вённо в индифферентных растворителях Н2, ()2, С12, СН4, NH3, N,0
Алмазо подобные непуст- i ва Гомеопо-ляркая Атомная решетка Трудно летучи, не проводят электрический ток, нерастворимы, часто очень тверды Алмаз, SiC, в4с Г
Металлы и ж;-таляоподобные соединения Металлическая Металлическая решетка Металлическая проводимость, чрезвычайная склонность к образе ванн ю сме шац*-пых кристаллов Cu, CnZ,:i, Gl Ctlg^Slig
Ва.и дор вааль-совые соеди^ ненпя Вдндер-ваальсо-вая Молекулярные агрегат ты Легко р а спа да юте я при повышения температуры -: Гидраты инертных газов, ассоциированные молекулы жидкостей
Продукты ассоциации и ириса единения сильно полярных водородсо-. держащих соединений Водородная Молекулярные агрегаты Существуют в твердом и жидком СОСТОЯНИИ, в растворах, иногда даже в газообразном состояния (НН2. (IIF)3, : (HF),, (HF)e> (Н2О)а, (fl3O)4s (Н3О)8, (BH-j* |A1!MS -• (М1.(.Ц,0)
Строение и свойства химических соединений 331
: Равномерная структура силового поля вокруг каждого иона может- быть иска-жепа.пбд влиянием поляризации, которая:Приводит к: возникновению слоистых рететок (см, стр. 244), наиболее часто встречающихся среди таких соло об разный соединений, катионы которых принадлежат к побочным подгруппам периодической системы. Среди соединений олемёптев главных подгрупп периодической системы слоистые решетки наиболее часто встречаются среди гидроокисей (ср. стр. 244 и 292), так как ионы гидроксила обладают большим дипольным моментом. Слоистые решетки встречаются также у веществ с преимущественно г оме о полярным характером связи, поэтому, зная лишь, что- вещество образует слоистую решетку, нельзя делать непосредственного заключения о типе связи в нем.
Малая летучесть содеобразных соединений объясняется тем, что в их свободных-молекулах валентные силы энергетически насыщены не полностью. То , что все же стехиометрический состав в твердом состоянии совпадает с составом в газообразном состоянии и что вообще состав чисто соле-о бр а зных соединений всегдапо ст оя иен, объясни етсязаконом электроне й-тральпости: во всяком соединений число .положительных ионов должно быть эквивалентно числу отрицательных.
Соде об разные вещества в расплавленном или растворенном состоянии обладают сильной электропроводностью. Это легко объяснить их,ионным строением.
,, То, что вещества, построенные из атомных молекул, в твердом состоянии иё обладают электропроводностью, без труда объясняется также их строением. В растворе при подходящих условиях эти вещества могут становиться электролитами, по только в том случае, если они под действием растворителя испытывают (в связи с сольватацией) существенные изменения в строений. В диссоциированном состоянии такие соединения, учитывая сольватацию их ионов, отличаются от исходных также и своим составом.
Гомеополярные связи в отличие от электровалентных могут быть описаны как направленные силы, которые следует показывать валентными штрихами. Так как эти силы обладают, кроме того, только небольшой сферой действия (см. стр. 323), то их влияние не распространяется на непосредственно не связанные атомы. Это приводит к образованию четко ограниченных, построенных из конечного числа атомов молекул. Между этими молекулами в основном действуют только вандерваальсовы силы. Этими силами они удерживаются в твердом состоянии. Так как. вандерваальсовы силы относительно слабы, то они могут быть легко преодолены за счёт тепловой энергии; этим объясняется легкая летучесть веществ, построенных из атомных молекул.
Для веществ, построенных из атомных молекул, существует простое сортцоптение между летучестью и молярным объемом в жидком состоянии. ЕщеВщгуГофф высказал предположение, что температуры кипения жидкостей должны быть пропорциональны их молярным объемам; однако эта точка зрения пе была подтверждена экспериментально. По мпопито Билдига (Billig, 1931), существует все же пропорциональность между температурой кипения и молярным объемом в жидком состоянии, если только учитывать ассоциации.'.
Молярные объемы гомеополярно построенных веществ, особенно соединений углерода, составляются приблизительно аддитивно из значений объемов, характерных для отдельных их составных частей (закон Коппа). Разновидности простых гомеополярных связей (многократный связи) следует при этом учитывать суммированием дополнительных числовых значений (инкременты связей). Далее надо сравнивать молярные объемы При одних и тех же состояниях жидкостей (например при температурах кице-пия). Для гомеополярных соединений в жидком'состоянии бб неприменимым является положение об аддитивности произведения из молярного объема и корня четвертой степени из поверхностного натяжения, которое, цо Сегдеиу (Sugden, 1924), обозначается
332
Глава 9
как «парахор» (правило Коппа), В этом случае также следует учитывать типы: связи путем введения инкрементов. С некоторой осторожностью из парахора можно* делать заключения о характере связи в молекуле.
Возникновение гомеополярной связи не всегда приводит к образованию молекул, построенных из ограниченного*числа атомов. Сцепления атомов могут распространяться произвольно далеко ио всем направлениям в пространстве, и это приводит к образованию атомной решетки*. Так как типичным примером веществ, образующих атомную решетку, является углерод в форме алмаза, то такие вещества называются также алмазо-подобными. Вследствие прочности сил, связывающих атомы в этой решетке, вещества, имеющие такую структуру, характеризуются большей частью чрезвычайно трудной летучестью, практически абсолютной нерастворимостью, часто особенно большой твердостью. Гомеополярные соединения, как с молекулярной, так и с атомной кристаллической структурой, имеют, как и ионные соединения, всегда постоянный состав, который у этих соединений обусловлен тем, что для однородных связей всегда требуется равное число электронов. Для интерметаллических и металлоподобных соединений в первую очередь характерна металлическая электропроводность** ***.
То, что с последней связан особый тип связи, следует из теории металлического состояния (см. т. Ц, гл. 1).
Так как ионы, удерживаемые вместе электронным газом, заряжены однозначно, то различные вещества в случае металлической связи могут в определенных пределах замещать друг друга в любых непрерывно изменяющихся соотношениях. &
Границы этих областей фактически определяются геометрическими соотношениями**®. Металлические ионы стремятся под действием распределенного между ними электронного газа образовать возможно более плотную и возможно более симметричную упаковку. Если радиусы иопов значительно различаются, то такая упаковка возможна, как правило, только при определенных соотношениях количеств ионов. Иногда очевидным становится стремление к определенным электронным плотностям, т. е. к числовым соотношениям между электронами и ионами металлов. На это указывает правило Юма — Розери, Но для, всеобщей применимости закона постоянных отношений к веществам с металлической связью нет оснований.
Вандерваальсовы силы также по приводят к агрегации молекул в определенных числовых соотношениях. Но также и здесь, при определенных пространственных условиях, можно прийти к образованию соединений с постоянным и стехиометрически простым составом, например, вследствие того, что с одной молекулой в непосредственное соприкосновение входит только определенное число других молекул. Если под действием вандер-ваальсовых сил в решетку внедряются чужие молекулы, то можно определить их число, зная число мест, имеющихся в решетке и пригодных для подобного внедрения. Значение распределения веществ па классы, указанные в табл. 59, особенно в простых случаях, заключается в том, что таким образом ясно выражается зависимость ряда характерных свойств от строения. Но такая зависимость никоим образом пе осуществляется для
* К атомным решеткам в этом смысле не относят, конечно, решетки металлов и мёталлододобных веществ.
** Это следует понимать л том смысле, что всегда, если существует металлическая электропроводность, должны существовать также металлические связи. И наоборот, если существует металлическая свяэь, с ней обязательно должна быть связана металлическая электропроводность, как это следует из теории непроводников (см. т. II),
*** Об определенном влиянии геометрических соотношений на стехиометрический состав см. Laves Г., Z. anorg. Сйещ., 250, НО, 1942.
Строение и свойства химических соединений 333
всех соединении „ однозначно. Поэтому в классы веществ, приведенные в табл. 59, не удается поместить все вещества на основе приводимых свойств. Уже то обстоятельство, что между различными типами связи существуют переходы, приводит к стиранию границ между этими классами, так что свойства, которые для различных классов веществ типичны, могут выступать и смешанными*. Далее, в одном и том же веществе в различных направлениях могут действовать различные силы связи.
В графите, например, атомы углерода в плоскостях шестиугольников связаны в основном атомными связями. В перпендикулярном к ним направлении действуют, вероятно, вапдорваальсовые силы. В этом направлении связи намного слабее, как это следует из межатомных расстояний (см. рис. 84, стр. 463), а также из топохимических реакций графита (см. т. II, гл. 19). Кроме того, в графите существует также еще некоторая доля металлической связи, ибо оп обладает металлической электропроводностью. Существование различных типов связи в различных пространственных направлениях принимается во внимание при дальнейшем подразделений классов веществ [см. Grimni, Angew. Chem., 47. 1934], но тогда схема без дальнейших дополнений оказывается применимой только к бинарным соединениям. У веществ, построенных более чем из двух элементов, между различными видами атомов существуют часто связи совершенно различного характера. Например, в щелочных сб'лях органических кислот щелочный ион связан гетеро полярно, в то время как между остальными атомами существует гомеополярная связь.
Особенно важный класс соединений, в которых часто сосуществуют различные типы связи, образуют соединения высших гаоряЭков/Поскольку при этом речь идет о комплексных соединениях, в тех из них, которые имеют характер электролитов, между составными частями, переходящими в раствор в виде ионов (без распада комплексов), большей частью также и в твердом состоянии существует ионная .связь. Но внутри комплекса, т. е. между центральным атомом и его лигандами,.могут быть связи совершенно иного рода. Они могут быть обусловлены электровалентными, ковалентными или вапдерваальсовыми силами (подробнее см. гл. 11). В неводных растворах (например, в жидком аммиаке) комплексные ионы имеют даже зачастую металлический характер, (см. стр. 619).
В двойных соединениях солей с электронейтральными составными частями, существующих только в виде очень слабых комплексов или в виде соединений, в которых нейтральные молекулы внедрены в кристаллическую решетку, пе будучи координированными около определенных центральных атомов, связи этих нейтральных молекул, как правило, Осуществляются за счет вандерваальсовых сил. Известнейший пример такого рода соединений — кристаллогидраты. Так как вандерваадьсовы силы сами по себе не приводят к соединению стехиометрического состава, кристаллизационная вода (в тех случаях, когда структура кристаллической решетки не обусловлена определенным положением молекул воды) при повышении температуры или понижении давления непрерывно теряется. К этому же типу соединений, т, е. к типу кристаллогидратов принадлежат легко разлагающиеся аммиакаты, Двойные соли, разлагающиеся в водных растворах на составные части, не отличаются резко по типу связи от простых солей.
* Этим объясняется то, что среди соединений, которые считают обычно валентными, т. е. такими, которые построены гетеро-или гомеополярно, существуют соединения, которые обладают пе строго постоянным составом. Это возможно только в том случае, если для строения существенны, помимо электровалентных или ковалентных, еще и другие связи (металлические связи или вапдерваальсовы силы). В качестве примера можно указать, что в, кубическом сульфиде одновалентной меди (см. т. II, гл. 8) существует тип связи, который имеет частично металлический характер.
334 Глава. 9
ОПРЕДЕЛЕНИЕ СТРОЕНИЯ НЕОРГАНИЧЕСКИХ, ВЕЩЕСТВ
Свойства,-рассмотренные выше, й особенно такие, как летучесть, растворимость и электропроводность, позволяют сделать для целого ряда соединений предположения о природе существующих в них химических связей. Однако в общем эти свойства не позволяют количественно судить о прочности сил связи п не дают каких-либо определенных сведений, пр которым можно было бы четко определить строение. Прочность связей определяется либо из термохимических данных, либо измерением равновесий (например, определением давления разложения). Для изучения свойств, характеризующих строение неорганических веществ, имеется целый ряд специальных физических методов, важнейшие из которых следует здесь коротко рассмотреть.
Пак уже упоминалось, для определения строения неорганических веществ;.методы классической органической химий, основанные главным образом на изучении химических превращений, в; большинстве' случаев неприменимы. Они применимы только к таким веществам, которые можпо перевести в газообразное или растворенное состоя-, ние без изменения их молекулярного состава* и » которых, кроме того, в момент пере- , хода не изменяется взаимное расположение связей. При превращениях неорганических веществ оба эти условия осуществляются одновременно сравнительно редко, Однако имеются, конечно, определенные атомные группы (радикалы), которые при химических превращениях остаются неизменными и существование которых известно уже давно. Все же строение таких групп, .если не считать прочных комплексов, можно определить па основе их химических превращений только в исключительных случаях (см. примечание па стр. 322).
При изучении строения легко летучих или растворимых без электролитической диссоциации веществ определяют молекулярные веса в газе-образном или растворенном состоянии, так как, согласно сказанному выше, можпо в общем предположить, что у этих веществ молекулы, присутствующие в газовой фазе или в растворе, идентичны тем, из которых построено твердое вещество. В веществах, которые при растворении: испытывают электролитическую диссоциацию, часто присутствуют радикалы, состав которых, если они переходят в раствор, не изменяется. Для них имеет значение определение ионных весов. Только в том случае, если речь идет об истипшах радикалах, т. е. о группах, которые остаются неизменными при химических превращениях, на основании присутствия в растворе в виде ионов определенных групп можно судить с уверенностью о существовании их в твердом соединении. Действительно, часто в растворе существуют иные группы, чем в твёрдых сооДицопиях (например, тиосульфат серебра, см. т, II, гл. 8). Но даже и в таких случаях определение ионного веса в растворе представляет значительный интерес, ибо оно часто позволяет сделать заключенно о силах, действующих в растворе между ионами:
Другой ваяжой задачей является определение структуры. Для твердых веществ оно проводится, как правило, рентгенографическим нулем. Для определения молекулярной структуры жидкости или газов большое значение имеет также целый ряд других методов (см. далее). Важнейшие сведения о специфических типах связи и их проявлениях получают прежде всего (не считая изучения полосатых спектров, важных для объяснения строения просто построенных молекул) в результате электрических и магнитных измерений-
* В тех случаях, если это происходит, строение органических соединений можно определить при помощи новейших физических методов (например, целлюлоза). ’
Строение и свойства, химических соединений ’ 335 .
Молекулярный вес. Молекулярные веса газообразных или испаряющихся без разложения веществ определяют, как известно, по плотности пара или газа на Основе закона Авогадро. Молекулярные веса растворимых веществ рассчитывают в большинстве случаев но понижению температуры замерзания1 или по повышению температуры кипения растворов.
Для растворов, температуры кипения которых лежат ниже комнатной, лучше измерять разницы давления паров непосредственно раствора илистого растворителя. Этим методом, например, Иоапни (Joannis, 1892) и Краус (Kraus, 1908) определяли молекулярные веса щелочных металлов, растворённых в жидком аммиаке, а Шток (Stock, 1925) — бороводородов, растворенных в жидком аммиаке. На определении разницы давления паров раствора и чистого растворителя основан также метод определения молекулярного веса путем измерения изотермической дистилляции, который впервые применил^ Барджер (Barger, 1904). Этот метод позволяет работать при обычных температурах и при. помощи* указанного Ульманом (Ulmann, 1931 --1933). усовершенствования проводить точные определения молекулярных весов при очень незначительных молярных концентрациях растворенных веществ, Последнее обстоятельство делает его пригодным для определения молекулярных весов высокомолекулярных веществ, для которых неприменимы методы, основанные на определений понижения температуры замерзания и повышения температуры кипения. Для высокомолекулярных веществ можно также часто предпочесть непосредственное определение осмотического давления с использованием ячейки с избирательно полупроницаемой мембраной. В пользу прямого определения осмотического давления говорит также намного большая точность измерения, чем при косвенных методах. Например, в 0,5%-пом водном растворе вещество с молекулярным весом 250 вызывает понижение температуры замерзания на 0,038':' и повышение температуры кипепия На 0,0104°,. в то время как осмотическое давление изменяется па 497,10 ем вод. ст. Для вещества с молекулярным весом 10 000 соответствующие числа равны 0,00095°, 0,00026° й 12,43 e.w вод. ст. Но непосредственное измерение осмотического давления осуществимо только тогда, когда имеется мембрана, которая проницаема для растворите л я, и.совершенно непроницаема для растворенного вещества. Это условие легче выполнимо для высокомолекулярных веществ, чем для пивкомолекулярных^об определении осмотического давления коллоидных веществ см. т. II, гл. 16).
При определении молекулярных весов или размеров частиц высокомолекулярных веществ также должна оказаться очень полезной улътрацентрифуга.;
Ионные веса. Для определения молекулярных весов.„растворенных веществ можно использовать также измерения скоростей диффузии. В настоящее время этот метод применяется в первую очередь для определения ионных весов. Наиболее доступен расчет молекулярного или ионного веса по скорости «свободной диффузии» (см. т. П, гл. 19), т,. е. диффузии без промежуточной мембраны. Этот метод определения ионных весов был успешно применен ЯндерЬм (Jander, 1925) для объяснения равновесия гидролиза изополикислот и основных солей (пример см, т„ II). Замечательных результатов добился также Бринтзингер (Brintzinger, 1928), измерявший не свободную диффузию, а проницание, т. е. диффузию,, происходящую через промежуточную мембрану. Бринтзингер назвал этот метод «методом диализа», так как он использовал в своей работе аппарат, подобный грэ-хемовскому диализатору. Эмпирически найдено, что у веществ, не сильно различающихся по структуре или строению, коэффициенты диализа (правильнее коэффициенты проницания), т. е. коэффициенты диффузии через мембрану, обратно пропорциональны, так Же как и при свободной диффузии*, корню квадратному из молекулярного или ионного веса. Следует
* Япдер позднее установил (1939), что на нс пользованных Бринтзипгеррм мембранах с очень топкими парами (целлофан и купрофан) эта пропорциональность не осуществляется для частиц с большими молекулярными и ионными весами, но остается в силе и для них при применении так называемых «целлофпльтров» со значительно большим диаметром пор. Скорость диффузии через целлофильтр, так же как й цри свободной диффузии, обратно пропорциональна вязкости раствора. Это закономерное соотношение но имеет места для целлофановых и купрофаповых мембран.:
,336
Глава. 9
обратить внимание на то, что и тех случаях, когда ионы образуют с молекулами растворителя продукты присоединения, по скоростям диффузии можно рассчитать относительные веса этих соединений, в то время как методы, основанные на измерении осмотического давления, дают веса нссольватйрованпых ионов.
Во я ногах случаях весьма применимым способом для определения полных весов окалывается способ «солевой криоскопии». Он основан на том, что температуры плавления (например, СаС1а-6Н2О) и превращения гидратов солей (Na^SO^ 10Н20)и эвтектпт ческие температуры криогидратных смесей (например, лед—KN03) понижаются в присутствии посторонних веществ, растворенных в жидкой фазе. Температурное пониженно соответствует вакопу Рауля. При определении ионных весов солевая криоскопия обладает рядом преимуществ перед обычной криоскопией, так как только такие иопы постороннего вещества вызывают понижение, которые не являются общими с растворенной солью. Для солевой криоскопии применяют до сих пор главным образом следующие вещества (2'11р — температура превращения; Та — криогидратная температура; Е — молярное понижение, отнесенное к 1000 г растворителя).
Na2SO4- IOHjO
7пр 32,38°, Е 3,21s (LGwenherz R., 1895, Darmois Е., 1923),
CaCl2-fiH2O 7пр 30,2°, Е 4,13е (Morgan J. L., 1907, Darmois Е-, 1930—1932),
Лед—KNO:, 7\—2,84°, Е 1,76° (Согпес Е., Muller Н, J., 1932),
Лед—NaNOB П—18,5°, Е 1,55° (Jahr К. F., 1952).
Для солевой криоскопия пригодны также следующие вещества L1NO3-3H2O, Na2S20s-5H20, Na2CrO«- 10Н2О, Ca(NO3)j-4HsO, Zn(NO3).,-3H2O, Mn(NO3)3-3H!!q (Morgan .1. L., 1907, Boutaric A., 1911)
Определение структуры. Определение структур твердых веществ рентгенографическим методом уже было рассмотрено в гл. 7. Так же как ii в твердых веществах, в жидкостях и газах расположение атомных ядер (за исключением ядер водорода) в молекулах можно определить рентгенографически. По положению максимумов почернений на интерференционной картине можно, как зто было показано Дебаем (1929),/рассчитывать межатомные расстояния. Вместо рентгеновских лучей для определения структур газообразных молекул в настоящее время часто применяют электронные (катодные) лучи, которые интерферируют (согласно квантовой механике) совершенно таким же образом, как и рентгеновские лучи.
Определение типа связи. Часто уже на основании свойств, приведенных в табл. 59, можно сделать достаточно достоверные предположения о типе связи. Например, если легкорастворимое вещество растворяется с малой теплотой гидратации с образованием электролитических ионов, то весьма обосновано предположение, что оно также и в твердом состоянии построено из ионов; Но если оно труднорастворимо или его теплота гидратации существенна, то это заключение нуждается в дальнейшем подтверждении.
Водородные ионы, отличающиеся особенно высокой теплотой гидратации, в безводных кислотах, по крайпей мере в большинстве случаев, если пе всегда, связаны более гомоополярно, чем гетеро полярно, и эта гомеополярная связь имеет заметно отличающуюся от нуля ионность (см. стр. 324).
Часто с большой достоверностью характер связи можно определить рентгенографически. Этот метод, определений, который намного обстоятельнее других, прежде всего используют в тех случаях, когда отсутствуют
Строение и свойства химических соединений 33”
приведенные ранее критерии. Рентгенографическое определение типа связи или, иначе, распределение электронов между отдельными типами атомов, основано па том. что рассенваппе рентгеновских лучей вызывается электронами. Как па этой основе можно отличить ионную решетку от атомной, особенно в простом случае, когда существуют два рода атомов, которые в ионной форме имеют одинаковое число электронов, было уже рассмотрено (см. стр. 241). Но если этот простой случай нс имеет места, все равно путем очень точных измерений интенсивности линий можпо рассчитать
распределение электронов между различными видами атомов. При этом часто получают числа, отклоняющиеся от значений, вычисленных и предположении,что заряды ионов равны их валентностям, а именно в том имысле, что рентгенографически определяемые заряды (часто дробные числа) меньше соответствующих валентностей. В таких случаях существует не чисто гстерополярная
связь.
В результате дальнейшего уточнения этого метода (примети;, вне фурье-анализа*) при оценке измеренных интенсивно стой (D нале W.,1915; Havighurst К., 1915: Bragg W., 1933; Grimm. Н., Brill R., Hermann C., Peters H., 1939) электронную плотность в кристалле во всей области между атомными ядрами удается выразить как. функцию координат,
Па рис. 66 представ депо распределение электронной плотности, соответствующее различ
ным типам связи, полученное описанным выше способом. Эти связи в значительной степени соответствуют тому, что можно было бы ожидать на основе представлений, которые уже были выска-
Р и е, 66. Схема распределения электронной плотности для различных типов связи (опреде-ляемого рентгенографически).
Кривые представляют соПой зависимость электронной плотности (d) от расстояния О между центр a bin ядер вдоль направлен ни сияв и
ваны о сущности этих типов связи. Приводимые па рисунке кривые показывают распределение Электронной плотности вдоль линии, соединяющей центры ядер. Электронная плотность представлепа в плоскости (пренебрегая более тонкими оттенками). Иидпо, что в случае цонпой связи электронная плотность в пространстве между иолами падает почти до нуля и, следовательно, каждый ион владеет своими собственными электронными оболочками и не имеет общих электронов с соседними. То же имеет мести в случае ваидорвапльсовой связи. Здесь, однако, остается срав-
* Под «фурье-апализом» понимают метод математической физики, основанной па открытом Фурье законе, что любую функцию /(г) можно представить тригонометрическим рядом
<r0-)-ui sin cos x-J-n^sia 2х-|-ЬЕ cos 2г-}- .. -
Краткий обзор по основам этого метода и его применение при определении электронной плотности см. Sommerfeld A., Naturwiss., 28, 769, 1940.
22 г. Реми
338
Глава 3
нитеяьио Широкое свободное пространство между электронными оболочками, в то время как электронные оболочки ионов касаются. В случае атомной связи электронная плотность вдоль Линии, соединяющей ядра, практически никогда не падает до нуля. Электронные оболочки соседних атомов «вливаются» одна в Другую, одпако только в области очень малых мсжъядерпых расстояний, следовательно — в отчетливо локализованных местах («электронные мостики»). В металлическом типе связи связующие электроны равномерно распределены в пространстве между ионами металла.
Фурье-анализ кристалла, если его ведут со всеми тонкостями, труден и требует много времени. По Оп дает, однако, совершенную картину распределения электронов. Этим методом обнаруживаются такие тонкости в структуре связей, которые па основе существующих теорий евнзи не могут быть предсказаны. Например, оказывается, что между молекулами, которые в обычном смысле рассматривают как насыщенные; помимо вандерваальсовых сил, могут существовать также слабые гомеополярные связи, отчетливо обнаруживаемые благодаря «электронным мостикам». Этот метод позволяет уди-, вительпо тонко устанавливать положения водородных атомов (на основе влияния их на пространственное распределение электроппой плотности), что рентгенографическим путем сделать невозможно.
Магнетохимия. Часто заключение о природе связи, о валентности it других вопросах строения можно сделать сравнительно простым путем на основе магнитных измерений. За последнее время магнитные^мстоды
Рис. 67. Диамагнитные и парамагнитные пластинки в магнитном поле.
очень плодотворно применяют различные исследователи (в Германии .главным образом Klemm W.) для выяснения строения многих веществ. В зависимости от поведения в магнитном поле различают вещества диамагнитные, парамагнитные и ферромагнитные. Мерой магнитных свойств веществ служит магнитная восприимчивость, которая определяется восприимчивостью к магнитным силовым линиям. Вещества с отрицательной восприимчивостью, т. с. такие, которые оказывают большее сопротивление магнитным силовым линиям, чем пустое пространство, называются диамаг-> потными; вещества с положительной восприимчивостью (т. е. хорошо проводящие магнитные силовые линии) — парамагнитными. Пом еще иные между полюсами сильного магнита, первые ориентируются перпендикулярно, вторые — вдоль силовых линий (см. рис. 67). Вещества с особо высокой восприимчивостью (например, железо) называются ферромагнитными.
Магнитная восприимчивость к выражается как « — где 3 — интенсивность намагничивания, т. о. магнитный момент па единицу объема, а б - сила магнитного поля. Мавпипшым моментам называют вращательный момент, который действует в Магнитном попе на магнитик в виде палочки пли па виток, проводящий ток. Единица силы магнитного поля (1 гаусс) есть сила поля, действующего па единицу заряда с силой в 1 дн. Единица заряда есть такой заряд, который взаимодействует с равным ему зарядом яа расстоянии 1 см с силой 1 дн. Часто магнитную восприимчивость относят не к единице объема, а к единице массы. Отнесенная к единице массы восприимчивость равна где d — плотность. Умножая X на молекулярный
вес (или х па молярный объем), получают молярную восприимчивость (Хмоль).
У диамагнитных веществ восприимчивость не зависит от силы ноля и, как правило, пе зависит также и от температуры. Восприимчивость парамагнитных веществ равным образом по зависит от силы поля, но значительно зависит от температуры.
Строение и свойства химических соединений 339
Для многих парамагнитных веществ она обратно пропорциональна абсолютной температуре (закон Кюри) х — C'lT, где С константа, а Т — абсолютнаи температура. Восприимчивость ферромагнитных веществ при незначительной силе поля возрастает с увеличением последнего и падает при его дальнейшем возрастании. При достаточно большой силе поля для ферромагнитных веществ произведение силы поля па восприимчивость, т. е. интенсивность намагничивания Д, практически не зависят от силы поля и, следовательно, восприимчивость обратно пропорциональна сило поля. При возрастании температуры восприимчивость ферромагнитных веществ уменьшается вначале постепенно, а затем при определенной температуре (точка Кюри) внезапно резко падает. При температуре точки Кюри ферромагнетизм переходит в парамагнетизм.
О веществах, подчиняющихся закону Кюри, говорят как о «нормальных парамагнитных веществах». Чаще имеет’ место подчинение закону Вейса q. (В этой формуле 6 означает постоянную, которая для ферромагнитных веществ равна температуре Кюри (°К), но для некоторых веществ 6 имеет отрицательное значение. В последнем случае говорят об «а пт иферр о магнетизме». Температурная зависимость парамагнетизма объясняется тем, что тепловое движенце против о действует внешнему магнитному полю, стремящемуся упорядочить расположение электронных: магнитиков (см. ниже). Чтобы получить истиппые моменты отдельных атомов, надо, следовательно, освободиться от влияния теплового движения. Магнитный момент, отпесеппый к Г о-атому, равен [1— У"ЗВС, где Д — газовая постоянная, С — константа Кюри, отнесенная К 1 г-атому.
Еще наиболее ранкме, в основе своей восходящие к Амперу, теории магнетизма объясняли магнитные свойства предположением о существовании элементарных магнитиков, которые обусловлены электрическим «молекулярным током», т. е. круговым движением электричества внутри атомов. Это предположение удастся уточнить при помощи атомной теории таким образом, что «молекулярные токи» задаются движением электронов по орбитам и их вращением (спином). Поля электронов, принадлежащих одному атому, по своему действию могут либо усиливать друг друга, либо уничтожать, смотря по тому, направлены ли их собственные моменты в одну пли в пр оти но п о ложные стороны. Если они уничтожают действие друг Друга (взаимно компенсируют), то вещество оказывается диамагнитным, в другом случае — пара- (или в особых условиях) ферромагнитным.
Если отдельные моменты электронов взаимно компенсируются и, следовательно, общий момент атома оказывается равным нулю, то njin внесении в магнитное поле в атоме индуцируется противоположно направленный момент соответственно ТОму, как при внесении в магнитное поле проволочного витка последний приобретает противоположно направленный по отношению к нолю магнитный момент из-за возникновения в нем индукционного тока. В то время как в проволоке индукционный тоц из-за омического сопротивления быстро падает, в атоме индуцированный момент сохраняется. Однако вследствие противоположной индукции при извлечении из магнитного ноля этот момент опять уничтожается. В отличие от «индуцированного момента» магнитный момент, существующий независимо от внешнего ноля, называют «постоянным моментом». В парамагнитных веществах отдельные атомы или молекулы имеют по стояцные моменты .»В диамагнмтных веществах постоянные моменты равщ>гиужо' В парамагнитных веществах, у которых вследствие одинаковой (более или менее зависящей от температуры) ориентировки постоянных отдельных моментов при внесений в магнитное поле возникает одинаково направленный с ним общий момент, зависящий от температуры, магнитная, индукция вызывает одновременно в каждом атоме магнитный момент, направленный противоположно внешнему полю и накладывающийся на постоянный момент каждого отдельного атома. Следовательно, в парамагнитных веществах измеряют собственно разницу между его парамагнетизмом и его диамагнетизмом. Но последний в большинство случаев оказывается относительно столь незначительным, что ин можно пренебречь. ,jSg
Из ныптесказанпого следует, что все свободные атомы или ионы с «завершенными электронными оболочками» должны быть диамагнитными, ибо в них взаимно уничтожаются как орбитальные, так и спиновые моменты. s-Э лек тропы, согласно квантовой механике, имеют только спиновые, 22*-
340
Глава 9
по по /.рбиталълые моменты. Если они спарены, то их спиновые моменты взаимно компенсируются. Поэтому атомы или ионы, имеющие, помимо завершенных электронных слоев, только такие электронные пары, также должны быть диамагнитными. Все остальные ионы, если только они суще- ствуют в виде свободных ионов. парамагнитны.
Диамагнитными являются; ’
1. Все инертные газы и все попы с конфигурацией инертных газов. ’
2. Все ионы, которые, помимо оболочек типа инертных газов, содержат только ; полностью завершенные оболочки с <7- или /-электронами, следовательно, например, Ag+, Gds\ In8’, Sa4+ и т. д., далее, ДО’, Та< w«’, Ко7’.
3. Все атомы или ионы, содержащие, помимо завершепшдх слоев, еще только 2я-электрона, например Be,Zn, Ga' , Pb2’, Р3+, Set+. Парамагнитны ионы редких земель (кроме La8’, Се4’, Yha ’ и Lu8’, в которых все электроны образуют закопченные оболочки) и в ряде случаев ионы элементов побочных подгрупп периодический системы, если они проявляют валентность, отклоняющуюся от номера их групп, и если орбиты их электронов не искажены сильно силовыми полями соседних атомов пли ионов.
Величины магнитных моментов р свободных ионов можно определить на. основе ’ теории магнетизма*. Для магнитного орбитального момента электрона, связанного _ - .. .._______________________________________________________________ д.
с орбитой с побочным квантовым числом I, теория дает значение щ = у I (7-(-4) ,
где е — заряд электрона в электромагнитной системе единиц, А — квант действия Плап-„ eh _
ка, т — масса электрона. Величина ^— используется в магнетохимии в качестве единицы, в которой выражают магнитные моменты. Ее называют магнетоном Бора. Она имеет значение У,26’7О-г) гаусс-см3, или, огнесеппая к i молгр, 5580 -гаусс-см3.
с-- eh
Магнитный спиновый момент, составляет для каждого электрона ps = у 3 , сле-
довательно, 1,73 магнетона Бора. По данным орбитальных и спиновых моментов отдельных электропов можно рассчитать общий магнитный момент атома или нова.
В качестве примера в табл. G0 приведены магнитные моменты, которые получены для
ионов редкоземельных элементов в предположении, что в них 4/-уровень заполняется вначале исключительно электронами с параллельными спинами и затем, если пригодные для этого энергетические уровни полностью заполнены электронами с противоположными спинами*®. Совпадение рассчитанных и измеренных значений показывает, что ото пр ед положение соответствует действительности (ср. т. [I, гл. 10).
* Экспериментально постоянный магнитный момепт получается из константы Кюри (см. стр. 338).
Положение о том, что электроны с противоположными спицами всегда стремятся спариться, имеет значение только для s-электронов, но не для электронов с более высоким побочным квантовым числом.
Строение и свойства химических соединений ' 341
Из данных табл. 60 видно, что ионы, имеющие одинаковое общее число электронов, как, например, La3” и Се4+, Со3* и Рг*+, обладают одинаковым магнитным моментом. Это соответствует общему правилу: магнитный момент иона равен магнитному моменту иона предшествующего в периодической таблице элемента, который несет меньший положительный или больший отрицательный наряд и равен моменту иона следующего элемента, обладающего большим положительным или меньшим отрицательным зарядом (закон сдвига Косселя).
Соединения, построенные исключительно из диамагнитных ионон, как в растворе, так и в кристаллическом состоянии, диамагнитны. Соединения, в построении которых принимают участие парамагнитные иопы, проявляют парамагнитные свойства, если в этих соединениях существует чисто ионная решетка. Одпако вследствие образования атомных связей парамагнетизм может полностью исчезнуть.
Парамагнетизм уничтожается прежде всего в тех случаях, если гомеоноляриая связь возникает благодаря спариванию s-электронов, так как последние всегда имеют противоположные спины и соответственно этому противоположно направленные и равные магнитные иомептъу.
Для молекул с атомными связями выполняется в общем правило Лъюса: при четном общем числе электронов общий магнитный момент молекулы равен нулю, при нечетном числе электронов момбпт равен 1,73 магнетона, т. е. соответствует магнитному снивовому моменту электрона. (Исключение из этого правила представляют молекулы Ог п NO). В кристаллических решетках, построенных из атомов или сильно деформированных ионов, соотношения оказываются более сложными. Обнаруживающиеся в пих влияния на парамагнетизм еще не выяснены окончательно. Лредположение о том, что явления ферромагнетизма и антиферромагнетизма определяются взаимным «магнитным Сопряжением» атомов, обусловленным атомными связями, простирающимися через всю кристаллическую решетку, кажется хорошо обоснованным. Ферромагнетизм проявляется, если существуют атомные связи с параллельными электронными спинами (в противоположность обычному случаю'). Проходящие через весь кристалл атомные связи с антипараллельными спинами обусловливают антиферромагнетизм. Во многих случаях па основании изучения магнитных свойств оказывается возможным сделать однозначнее заключение о строении. Это следует показать па нескольких примерах.
а) Валентность. Голубые пер оксохроматы (ср. т. II, гл. 5) проявляют, так же как и продукты присоединения пиридина к голубой перекиси хрома, слабый, не зависящий от температуры парамагнетизм, похожий на парамагнетизм иона ICrOiP-*.
Красные мерок сох рома ты, напротив, имеют нормальный, зависящий от температуры парамагнетизм, соответствующий моменту 1,37 магнетона, который следовало бы ожидать для соединения с нечетным числом электронов. По аналитическим данным как красные, так и голубые пероксо хроматы следует рассматривать либо как двухъядерные с шееишвалонтным, либо как одноядерные с пятивалентным атомом хрома. Из величия магнитных моментов следует, что из этих возможностей для голубых пероксихроматов подходит первая, а для красных вторая.
Магпптнымп измерениями, указывавшими, что соединение диамагнитно,, была также надежно установлена формула П4Р2ОЙ для фосфорповатой кислоты. Тем самым была исключена формула Н2РОз, содержащая почетное число электронов. Исследование магнитных свойств позволило также твердо у ст ап о вить двухвалентность серебра в некоторых комплексных солях (см. т. II). Если бы серебро в них было одновалентным, то эти соли были бы диамагнитны, так как ион Ag+ обладает закопченной электронной оболочкой. Для двухвалентного серебра с нечетным числом электронов можпо было бы ожидать парамагшпязм, как и у двухвалентной меди; для серебра измерениями (Sugden, 1331, Klemn, 1931) были определены такие же значения магнитной восприимчивости, как и для содей двухвалентной меди.
б) Превращение к твердом состоянии. При 130 и 31,5° соединение FeS испытывает превращения с неотчетливо проявляющимися термическими эффектами. Эти превращения, однако, очень четко видны па кривой томператур-
* Парамагнетизм, не зависящий от температуры (большей частью только в незначительной степени), часто наблюдается у соединений, в которых имеются попы переходных металлов с оболочками тина инертных газов, например К2СгО/„ КМиО4, UFn. Он объясняется влиянием поляризации (см. стр. 349).
Глава 9
состояниях может достигать значении,
ной зависимости парамагнетизма, как ото следует из рис. 68. В других случаях, вкото-рых превращения связаны, с отчетливыми изменениями магнитных свойств, магнитные измерения также могут быть с успехом использованы для точного установления темп о р ату jlJU рев ращения. Многие вещества, когда они находятся в «активном состоянии» (ср. т. II, гл. 19), проявляют чрезвычайно сильное повышение магнитной восприимчивости. Восприимчивость в анти которые наблюдаются только у ферромагнитных веществ (см. рис. 69).
Магпетяам металлов. И металлов
магнетизм содержащегося в них элект-ронпого газа налагается на магнетизм
Ферромагнитные 390 °
0,8 0,6 (?4 ,0,2 0
Содёрлсаниеводы, моли
Р и с. 68. Температурная зависимость магнитной восприимчивости FeS (Haraldsen). ''
Р и с. 69. Магнетизм препарата а-ГегОз-ПаО в зависимости от степени обезвоживания (Huttig),
На ось ординат нанесена восприимчивость, изморенная .при комнатной температуре для препарата, который предварительно нагревали до указанной па рисунке температуры: На ось абсцисс нанесено содержание в препарате воды {моль НгО/мо.ть УееОз).
ионов й атомных остовов (см. т. II, гл< 1). Та часть магнетизма металлов, которая связапа с электронным газом, практически не вавшпып от me-Mnepainy-ры, так как магнитное поле может ориентировать л ешь такое число электронов в направлении поля, которое вследствие поглощения энергии может быть поднято па незанятые еще энергетические уровня. Это число пропорционально силе поля, но приближенно не зависит от температуры (Pauli, 1927), В простейшем случае магнетизм металла можно просто рассчитывать, находя алгебраическую сумму пара-ыагиетайма, вызываемого электронами проводимости, и магнетизма атомных остовов. При этом для металлов может получаться положительная восприимчивость, хотя их ионы и обладают законченными электронными оболочками и, следовательно, отрицательной восприимчивостью. Так, щелочные и щелочноземельные металлы парамагнитны, хотя их ноны диамагнитны.
В тех рядах периодической системы, в которых присутствуют «переходные элементы», парамагнетизм металлов возрастает с ростом порядкового номера. Например, найдены следующие значения атомной восприимчивости Хд (произведение из удельной восприимчивости п атомного веса) в ряду эдеме пт о в от 19 До 28. Значения приведены для комнатной температуры);
is К? rol-a si 8 с j/ri 23 V --4СГ ^fjFeafCosaNi
Хд 10,= —20 -}-22 — -)-59 -)-100 Ц-156 -|-5Э5 Ферромагнитны
Появление ферромагнетизма у металлов группы железа оказывается закономерным следствием сильного роста магнетизма, который наблюдается уже в ряду предшествующих металлов, ив которых Сг и Мп в сплавах также уже могут оказываться ферромагнитными (см. т. II, гл, 6).
Элементы с порядковыми номерами, следу тощими за переходные, диамагнитны, В большинстве случаев в каждом из рядов, образованных этими элементами, атомная восприимчивость падает с возрастанием порядкового номера. Например, Хд падает в ряду а9Си— 34Se от —5,4-1О'Й до — 25-10-в. Таким же образом в каждой группе таких элементов атомная восприимчивость в общем падает с ростом порядкового помера, например в группе меди от— 5,4-10'е (Си) через—22-l()-s (Ag) до — 27,3-10~в (Ан).
Строение и свойства химических соединений
343
Некоторые отклонения от закономерности в ходе значений для восприимчивости объясняются различиями кристаллических структур. Действительно, на магнетизм существенно влияет тип решетки, как показывает пример олова, кубическая модификация которого диамагнитна (ХА=—30- Ю-”), а тетрагона л киля модификация парамагнитна {Хд=+2,440-6). Чрезвычайно низким значением атомной восприимчивости характеризуется висмут. (Хд110®= —-219 в направлении осп си —311 перпендикулярно оси с). Самой высокой атомной восприимчивостью среди неферромагнитных металлов обладает палладий (Х-i 0е=4-572 при комнатной температуре). В сравнении С ним гомологи железа поразительно слабо парамагнитны ("Ха* 10s—43,5 для Ru, 9,5 для Os)..Восприимчивость этих двух металлов возрастает с ростом температуры, в то время как восприимчивость палладия с ростом температуры падает. В смешанных кристаллах с золотом Pd диамагнитен (Vogt Е., 1932); из этого можно заключить, что Pd заполняет спою dd-оболочку электронами золота. Такого рода наблюдения над сплавами указывают на то, что металлы VII и VIII побочных подгрупп стрОмятСя заполнить свои внешние оболочки электронами других металлов. Например, металлы МП, Fe, Со и N1 только в том случае укладываются в правило Юма — Розери, рассматриваемое в дальнейшем (см.т. II, гл. 1), если им приписать в сплавах с «металлами II рода» число валецтпых электронов, равное нулю. Сильный парамагнетизм переходных элементов V—VIII побочных подгрупп объясняется, вероятно, главным образом тбм, что решетка их построена из ионов с только частично заполненными внешними оболочками, парамагнетизм которых налагается на парамагнетизм электронного газа.
В случае фёрромаенетизма речь идет, как теперь считают согласно Вейсу (Weiss, 1907), конечно, только об особом случае парамагнетизма, обусловленном тем, что в ферромагнитных веществах одинаковое направление элементарных магнитиков вызывается особенно сильным молекулярным полем, Это молекулярное поле по Гейзенбергу (Heisenberg, 1928) создается вследствие квацтово-механического резонанса между свободно подвижными злектрона5-£И металла. Гейзенберг показал, что, хотя в общем для них стабильным является аптинараллельное расположение, в особых случаях при не очень высоких температурах для определенных групп электронов стабильным может быть и параллельное расположение. Этот случай осуществляется в ферромагнитных металлах п в сплавах. «Точка Кюри» при этом характеризуется том, что в ней уничтожается обусловливающая ферромагнетизм взаимная связь электронов. По Эоммерфельду и Бёте (Sommerfeld, Войте, 1933} ферромагнетизм может проявиться только в случае наличия незавершенных оболочек с высоким побочным квантовым числом и большого среднего расстояния между атомными ядрами в решетке по сравнен пию с радиусами этих оболочек. Эти условия, кроме Fe, Сои N1, выполняются у некоторых редкоземельных металлов; и действительно, яри достаточно низких температурах у них может наблюдаться ферромагнетизм (см. т. II, гл. 10). Ферромагнетизм сплавов Хейслера связан, вероятно, с тем, что расстояние Mn-i—►Мий Сг+—*Сгв них увеличивается из-за внедрения в решетку посторонних атомов.
Строение молекул и полосатые спектры. Одним из важнейших вспомогательных средств для "исследования строения, в первую очередь строения простых по составу молекул, является изучение полосатых спектров. Энергия сложных молекул, не считая энергия их поступательного движения, состоит из вращательной и колебательной энергпи атомов я энергии связей электронов. Изменение к ал; дог о из этих трех видов энергии связано с испусканием или поглощением световых волп (в широком смысле). Поэтому из полосатых спектров можно определять как моменты инерций, по которым рассчитывают межатомные расстояния*, так и получать сведения о прочности отдельных связей (по колебательным частотам). Пэ полосатых спектров молекул, так же как из линейных спектров свободных атомов, можно рассчитывать энергпи связей "электронов в нормальном и возбужденном состоянии молекулы.
Так как полосатые спектры проявляются в видимом и ультрафиолетовой областях, вознпкповетше их связано с изменением всех упомянутых выше трех лидов энергии. Точные намерения этих спектров представляют часто значительные трудности и относятся к особому разделу физики. -
Намного проще построены спектры, возникающие вследствие изменения только вращательной анергии. Многие из внх можно наблюдать в длинноволновой инфра
* Например, для двухатомного газа момент инерции 1а получается из расстояния Дт двух соседних линий полосы вращательного спектра на основе следующего уравнения: Дм = ’ , в котором h — постоянная Планка. Расстояние г между
, «ЯА/ 0
обоими атомами с массами т± и т% рассчитывают по уравнению -г?.
зм
Глава 9
красной области (см. стр. 348). Но для выяснения строения химических соединений служат преимущественно колебателыо-вращателъные спектры, расположенные в относительно коротковолновой инфракрасной области (Х<2Ор). Они служат важнейшим вспомогательным средством для решения структурных вопросов в первую очередь в органической химии. Кроме того, Для выяснения структуры имеют особое-значение спектры комбинационного рассеяния. В неорганической химии их применяют в первую очередь.
Спектры комбинационного рассеяния. Если на газ или жидкость-падает свет, то часть его оказывается рассеянной во все стороны (рслеевскоц рассеяние). Главная часть этого рассеянного света (интенсивность его обратно пропорциональна четвертой степени длины волны) имеет ту же частоту, что и падающий свет. Меньшая часть отраженного света, как это было предположено Смекалом (Smekal, 1923) н экспериментально установлено Раманом (Raman, 1928), имеет частоту иную, чем частота падающего света, по той причине, что часть световой энергии может превращаться в колебательную энергию молекул. Последняя изменяется при этом всегда на один квапт, (vg— частота атомных колебаний). При этом имеет место
ДА
vo—vR= (1>
где vQ— частота облучающего света; тл — частота Раман-света; ДА' — изменение энергии колебания атомов в молекуле; h — постоянная Планка; vR — частота атомных колебаний («собственных колебаний») в молекуле.
Если, следовательно, па вещество направить пучок мо но хроматического света, то спектрографически (при достаточно долгом освещении) можно, помимо линий облучающего света, наблюдать еще линии, частоты которых отличаются от частоты облучающего света и пе зависят от нее-и определяются только собственной частотой атомных колебаний облучаемой молекулы. Если существуют различные колебательные частоты, то получается много линий комбинационного рассеяния. Зная расстояния их от линии облучающего света, непосредственно получают «собственные» частоты различных колебаний, ио которым можно сделать заключение как о прочности связей, так и о структуре. Точнее, для каждого собственного колебания получается не одна, а дне линии комбинационного рассеяния, так как ДА- в уравнении (1) может быть по только положительным, но и отрицательным. (Это значит, что молекула может пе только воспринимать, но и отдавать энергию облучающему свету.) Вторая линия комбинационного рассеяния, которая имеет такое же смещение частоты в фиолетовую область, как первая в красную, большей частью намного слабее первой. Далее, помимо колебательной энергий, со световой энергией может обмениваться и вращательная энергия. Вследствие этого при достаточно большом спектральном разрешении наблюдаются пе отдельные линии, но система из расположенных на небольшом.и одинаковом расстоянии линий, соответствующих различным вращательным квантовым числам. Поэтому спектры комбинационного рассеяния, совершенно так же как и инфракрасные сиектры, можно применять для определения моментов нперции и прочности связи. Экспериментально этот путь часто оказывается более простым.
Для двухатомных молекул частота атомных колебаний vB выражается следующим образом:
v.,= J- l/^ , (2>
Строение и свойства химических соединений
345
где kf — валентно-силовая константа:————|------, где т, и т2 — массы обеих час-
' и т< 1
тиц, колеблющихся около общего центра тяжести. Валентно-силовая константа к, является мерой прочности химической связи. Если v выражено в [сек_1] и масса частиц — в граммах, то kf получается в динах па сантиметр (дн/см). Соответственно этому к? можно определить как силу, которая стремится возвратить частицы в положение равновесия, если они находятся на расстоянии 1 см от положения равновесия, при условии, что вплоть до этого расстояния возвращающая сила пропорциональна удалений от положения равновесия.
В многоатомных молекулах, помимо колебаний в направлении валентных сил, проявляются также так называемые деформационные колебания, т. е. колебания, происходящие под действием сил, противодействующих изменению валентных углов. В нелинейных молекулах, т. е. в таких, в которых один или более валентных углов отклоняются от 180=, могут проявляться к тому же деформационные колебания второго типа, происходящие под действием сил, противодействующих выводу валентной связи из плоскости, в которой она обычно находится. Соответственно Этому в формуле для частот комбинационного рассеяния многоатомных молекул, помимо мпогих валентносиловых констант (для каждой связи одно значение kj), появляются еще другие, так называемые деформационные :константы. С увеличением числа валентно-силовых и деформационных констант в колебательной формуле комбинационного спектра сильно возрастает число линий. Поэтому спектры комбинационного рассеяния молекул, построенных из большого числа атомов, были бы очень сложны, если бы не вносилось упрощение, связанное с тем, что в радикалах, таких, как ОМ, NH2, СН3, колеблются не только отдельные атомы, по и весь он, как целое.
Валептпо-силовые константы, характерные для определенных связей, в большинстве случаев очень мало зависят от соседних связей. Между валентно-силовой константой к/ и средним межатомным расстоянием г0, относящимся к соответствующей связи в положении равновесия, существует соотношение, для которого Бэджер (Badger,' 1934, 1935) нашел следующую эмпирическую формулу;
л/ “Г
'0=1/ г+В-
(3)
В этом соотношении А и В — константы, значение которых зависит от положения в периодической системе элементов, образующих связь. Определение межатомных расстояний таким методом проще, чем их вывод через моменты инерции из вращательных линии, проявляющихся в спектрах комбинационного рассеяния, так как эти линии в последних в бопыпнпстве случаев трудно наблюдаемы.
Точные значения межатомных расстояний получают из электро но графических данных или путем расчета промеров полосатых спектров.
Точность спектрографически определяемых валентно-силовых констапт такая же, как и вычисляемых по данным полосатых спектров. В табл. 61 сопоставлены валентно-силовые копстаиты, определенные спектрографически, для ряда химических связей. Межатомные расстояния, приводимые в таблице, рассчитаны частью по пим, частью получены другим путем. Из молекул, построенных более чем из двух атомов, приведены только такие, которые полностью симметричны. В таких молекулах существуют только однородные химические связи. Поэтому присущие им валецтпо-Силовые константы и межатомные расстояния имеют все одинаковые значения.
Рассеяние света молекулой как в форме релеевского рассеяния, так и в форма излучения комбинационного рассеяния основано па том, .что колеблющееся электрическое поле падающего светового луча, воздействуя на электроны, вызывает периодически изменяющийся электрический момент молекулы. Амплитуда колебания этого электретеского момента тем больше, чем больше поляризуемость облучаемой молекулы. Более точная теория показывает, что интенсивность обычного рассеянного света зависит, помимо интенсивности облучающего света, только от поляризуемости облучаемой молекулы, а па интенсивность излучения комбинационного рассеяния, кроме интенсивности облучающего спета, влияет изменение, которое испытывает поляризуемость вследствие непостоянства расстояний между атомными ядрами. Если на поляризуемость практически не влияют колебания ядер, так как электронное облако, окружающее одно ядро, только О’цчп, слабо воздействует на другое, то излучение комбинационного рассеяния может по обладать заметной интенсивностью. Сильное взаимное влияние электронных облаков всегда проявляется в тех случаях, когда атомы, участвующие в создании молекулы, имеют общие злектроны. Поэтому спектры комбина-
346 Глава У
Таблица 01
Валентно-силовые константы Ilf н межатомные расстояния г0(А) для двух- и многоатомных молекул
Молекула 7<T IO® Молекула kf. IO-5 го-Ю8 Молекула rq. 108
Н5 5,71 0,74 SH2 4,00 1,35 SiCl4 2,68 2,i
HD 5,70 0,74 ScH2 3,12 — GeCl4 2,42 2,08
Ds 5,69 0,74 NH3 7,09 1,02 SnCl4 2,31 2;3
HF 9,55 0,92 PH3 3,36 — TiCl4 2,41 2,3
НС1 5,06 1,28 AsH3 2,80 — CBr4 1,38 1,942
НВт 4,06 1,41 PF3 4,56 1,55 SiBr4 1,74
HI 3,14 1,60 AsF3 3,90 1,72 SnBr4 1,83.
ch 3,32 1,98 pq3 2,11 2,03 SFq 3,98 1,57 ".
O2 41,7 1,20. AsCl3 2,01 2,21. SeF„ 4,67 1,68 ::
22,9 1,09 SbCl3 1,75 2,30 TeFB 4,95 1,83
co 18,9 1,13 BiCl3 1,17 2,46 [Ciosr 5,65 1,48:
NO 45,9 1,15 CH4 4,83 1,09 [BrOsJ- .5,15 1,73
2 w i 11,7 — SiH4 2,90 1,54 5,25 -Д-
C02 15,3 1,16 cf4 ; 5,40 1,36 [Cio4r 6,22 1,48 ‘
CS2 6,88 1,54 SiF4 5,50 1,54 [SOJ2- 6,03 : 1,51 ;
0H2 7,54 0,958 CC14 1,83 1,76 [po4r 5,62 1,55
диодного рассеянна в первую очередь получаются для гомеопояярных веществ*. Для появления эффекта комбинационного рассеяния достаточно, чтобы существовали связи, которые имели бы в значительной степени гомеополярный характер- В чисто ионных соединениях электронные облака партнеров по связи практически не влияют друг па друга. В чисто евпгерополярных соединениях комбинационного рассеяния не наблюдается.
Излучение комбинационного рассеяния присуще как твердым, так и жидким и газообразным веществам. Лучше всего его можно наблюдать на кристаллах и жидкостях, в которых на это излучение пе так сильно налагается рёлеевское рассеянное излучение, как в газах. Возможность непосредственного применения спектров комбинационного рассеяния для разрешения химических проблем видно из следующего: так как «собственные колебания» различаются в зависимости от рода непосредственно связанных атомов и характера связи и на них мало влияют соседние атомы, то можпо устанавливать существование в соединениях определенных группировок атомов па основании характерных для них линий комбинационного рассеяния. Можно использовать эти спектры для идентификации различных типов связи, например для различия обычных комплексов, и комплексов внедрения. Далее, возможным оказывается вывод структуры, молекулы из дацпых спектра комбинационного рассеяния на основе зависимости числа возможных колебательных состояний от свойств симметрии молекулы.
---_--------, ....... ’ ' *
* В большинстве случаев отдельные липни комбинационного рассеяния у них могут поэтому выпадать, в результате чего для определенных связей поляризуемость а da при нормальном расстоянии между ядрами г0 имеет максимум и при этом Станг новится равной пулю.
Строение и свойства химических соединений 347
Спектры комбинационного рассеяния подтверждают, например, предположение Гаича (Hantzsch) о существовании в безводной азотной кислоте «эфнроформы» 02N —ОН (см. стр. 4152 и сл.) Это следует из того, что безводная азотная кислота дает мшш комбинационного рассеяния, характерные как для ОН-, так и для КО2-грулп. Таким же образом удается показать, что безводная серная кислота обладает строением гидр окси л содержащего соединения O2S(OH)2, ибо в ее спектре комбинационного рассеяния присутствуют линии, характерные как для ОН-, так и SО2-групп. Даже хлорная кислота в безводном состоянии является гидроксилсодержащим соединением 03С1 — ОН, как это следует из наличия в се спектре ОН-полос. Так как спектр комбинационного рассеяния этого соединения, помимо ОН-полос, содержит еще шесть линий, возможным становится заключение, что в педиссоцимровэнной хлорной кислоте атомы кислорода располагаются в виде высокосимметрцчной Пирамиды с осями третьего порядка, с атомом С1 в центре тяжести пирамиды и атомом Н в вершине у одного из кислородов. Можно показать, что такой форме присуще упомянутое выше число колебательных состоянии. Для тетраэдрического расположения атомов кислорода получается четыре различных колебательных, состояния, и действительно, водные растворы, в которых, существует тетраэдрически построенный ClOj-иоп, дают спектр комбинационного рассеяния с четырьмя линиями, которые сдвинуты ио сравнению с линиями в спектре свободной кислоты.
Дипольный момент и поляризация. Для определения строения моле-куп существенное значение имеет также измерение электрических дипольных моментов. Для диполя с зарядами 4-е и —е, расположенными на расстоянии I (см. рис. 9, стр- 71), дипольный момент p=e-Z.
Дипольный момент позволяет сделать заключение как о характере связи, так и о структуре.. В молекулах с дипольным моментом*, значительно отличающимся от нуля, не могут существовать исключительно связи, которым можно было бы приписать чисто гомеополярный характер (см. стр. 324). Однако с определенностью можно сказать, что в асимметрично построенных молекулах, — к которым принадлежат все двухатомные молекулы, состоящие из атомов с различными зарядами ядер, — не могут существовать ионные связи, если они только ие имеют соответственно высокого дипольного момента. Симметрично построенные молекулы не обладают дипольным моментом и в том случае, если в них существуют чисто ионные связи. Отсюда следует, что молекулы, имеющие постоянный дипольный момент, всегда должны быть построены асимметрично. Например, молекула АВ3, даже если она состоит из ионов, не имеет дипольного ли мента, если ионы соответственно структурной формуле В-——В’ расположены на одной прямой, ибо в этой случае действие двух противоположных диполей В'—А+ и А+—В“, из которых может быть мысленно построена молекула, взаимно уравновешивается (в таких случаях говорят о: «квадруполе»). Но если обе связи образуют между собой угол, то действие обоих Диполей уничтожается только частично. Такая молекула имеет вследствие этого дипольный характер. Слабый дипольный момент имеют асимметричные молекулы, построенные из различных атомов, даже в том случае, если в них существуют чистые атомные связи, так как электронная плотность около двух атомных ядер с различным порядковым номером никогда пе будет совершенно одинаковой.
Из данных измерения дипольных моментов следует, например, что молекулы СОа и CS2 линейны, в то время как в молекулах Н20, H2S и SO2 атомные ядра занимают вершины треугольника. Подобным же образом измерения дипольного момента молекул NH3, Рн3 и AsJT3 позволяют сделать заключение, что в этих молекулах три атома II образуют основание трехгранкой пирамиды, в вершине которой располагаются атомы N, Р или As**.
* Асимметричные молекулы всегда имеют слабый дипольный момент (см. ниже).
** Более точно расположение атомных ядер (углы и расстояния) можно рассчитать путем комбинирования измерений дипольных моментов с определениями моментов инерции. Независимо От этого углы, как Это было покапано выше, часто рассчитывают такще непосредственно из данных спектров комбинационного рассеяния.
348
Глава 9
Измерение дипольных моментов производят путем определения диэлектрической проницаемости. При этом.следует иметь в виду, что постоянный дипольный момент полярно построенной молекулы в электрическом поле налагается на «наведанный» дипольный момент, вызываемый поляризацией, т. е. сдвигом электрических зарядов в молекуле под действием поля. Этот наведенный дипольный момент возникает также и в неполярных молекулах (т. е, н молекулах с постоянным дипольным моментом, ранным нулю). Он пропорционален силе поля: где Щ—наведенный Дипольный
момент, ®—сила поля. Множитель а служит мерой поляризуемости.
Явление электрической поляризации полностью соответствует индукции магнитного момента магнитным полом (см. стр. 338) с той только разницей, что индуцированный магнитный момент направлен противоположно нолю, а наведенный электрический момент ориентирован по полю. Так же, как и в случае магнитного момента, наведенный момент нс зависит от температуры, то время как постоянный электрический момент, т. е. дипольный момент в обычном смысле, зависит от температуры*; характер зависимости тот же, что и для постоянного магнитного момента. На этом основании можно различать оба эффекта.
Наведенный электрический момент, т. е. момент, для которого существенна поляризуемость а, можно также определить отдельно по уравнению, выведенному Лорептцем (Lorentz) нз электромагнитной теории света:
3 я2— 1
“~4лМл п2 + 2
(4)
дЗ — 1
Выражение ' V называют молярной реф-
где Г/а — число Авогадро, V — молярный объем, л — показатель преломления света с бесконечно большой данной волны. ракцией.
Измерения показывают, что для атомов инертных газов и для свободных ионов с оболочками типа инертных газов молярная рефракция, л также и поляризуемость соответственно уравнению (4) заметно возрастает с увеличением числа электронных: оболочек и что при одинаковых электронных оболочках, например в ряду О2-, F~, No, Na+, Mg2*, опа сильно падает с возрастанием заряда ядра. На поляризуемость ионов существенно влияет образование соединений.
Молярная рефракция ионных соединений поэтому не аддитивна но отношению к рефракции составных частей, как это имеет место в случае органических соединений. Например, нон С1_ в свободном состоянии и в водном растворе имеет молярную рефракцию 9,00, а в соединениях NaCl, Li Cl и НС1 —соответственно только 8,0, 7,4 и 0,67. Молярная рефракция свободных Вг“ и 1“ ионов составляет 12,67ти 19,24 по сравнению с 0,14 п 13,74 в соединениях НВт н Hl. Молярная рефракция свободного иона О2- равна 7, а в соединениях или в сложных ионах она составляет:
GaO MgO ВпО !()? SOi’ СЮ7
6,1 4,2 3,2 4,05 3,62 3,3
Как показывают эти примеры, поляризуемость элементарного аниона вследствие образования соединения с положительно заряженным ионом понижается тем сильное, чем меньше радиус и чем выше положительный заряд последнего и чем сильнее свободный анпоп поляризуем. Чем более сильное поляризующее действие оказывает положительный ион на связанный с ним аппоп, тем меньшему дополнительному влиянию этот анион подвергается со стороны другого электрического поля.
Микроволновые спектры. Исследования дипольных моментов (электрических моментов), а также магнитных моментов можно проводить посредством измерении высокочастотных электромагнитных колебаний. Высокочастотные колебания в области 1010—-1011 герц (длина волны от 3 см до 3 мм) можно также с успехом использовать для определения моментов инерции, межъядерных расстояний и других факторов, обусловливающих структуру молекул. В то время как в инфракрасной области вращательные
* Конечно, это имеет место для постоянного электрического момента, отнесенного к единице объема скопления молекул (газ, жидкость или кристалл). Для каждой отдельной молекулы как постоянный электрический, так и постоянный магнитный момент не зависят от температуры.
Строение и свойства химических соединений
349
спектры в общем налагаются иа липин колебательных спектров н чистые вращательные спектры в инфракрасной области можно получить только для молекул с особенно малыми моментами инерции, имеется большое число молекул, дающих чистые вращательные спектры в области электрических микроволн. Это и.иеет место, например, для л/олеьчглы Н.,О. Далее, в электрическую микроволновую область попадает также так называемый инверсионный спектр молекулы NH3 (см. стр. 646). Микроволновая абсорбционная спектрография является одним из точнейших и наиболее доступных методов для определения структур молекул газа. С ее помощью Вильсону (Wilson, 1950) удалось окончательно установить приводимую . на стр. 365 структуру диборана ВаНв.
ЦВЕТ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЯ
Вещества кажутся окрашенными, если они обладают свойством абсорбировать видимый свет* (весь или определенную область длин волн). Абсорбция видимого света может происходить только в том случае, если в веществе существуют электроны, которые путем поглощения энергии могут быть подняты на вышине уровни таким образом. что волновое число v “ из уравнения е3—s1=/jv (см. стр. 109) попадает в область волновых чисел видимого света. В бесцветных веществах для этого необходима в общем случае большая затрата энергии (соответствующая волновым числам ультрафиолетового света). Поэтому электроны, которые вследствие абсорб- , ции видимого света могут перескочить на высшие энергетические уровни, часто обозначаются как «рыхлоевязайпые» электроны. В действительности для световой абсорбции существенны не прочность связи, но разница в прочностях связей на различных орбитах.
Большинство веществ, построенных из ионов с оболочками типа, инертного газа, бесцветны н окрашенными оказываются только в том случае, если они содержат электроположительные атомы с особенно сильными поляризационными свойствами. Так, сложные поен,? СгО^“ и МпО~ окрашены, хотя они (поскольку связи в них рассматриваются как гетеро-полярные) и построены из ионов с оболочками типа инертных газов. Так как способность к поляризации возрастает с увеличением заряда и падает с увеличением ионного радиуса, то ионам или атомным остовам Сг6+ и Мп7”, характеризующимся высоким зарядом и одновременно малым радиусом, слерует приписать особенно сильную поляризационную способность.
Соединения, в построении которых принимают участие попы с незавершенными электронными оболочками, оказываются почти всегда окрашенными. Так, соединения, образованные элементами побочных подгрупп периодической системы, валентность которых отличается от номера группы, почти все без исключения окрашены. Окрашены также и соединения, в которых элементы главных подгрупп периодической системы проявляют электроположительную валентность, отличающуюся от номера групп, если только они содержат легкополяризуемые анионы.
Общим оказывается то, что окрашенность соединений «углубляется» с ростом поляризуемости аниону, т. е. что абсорбционная область сдвигается в красную область спектра.
* Выражение «окрашенное» используется здесь в аротпиоподажнасть «бесцветио-чу», следоватсльпо, имеется в виду татоке я черный цвет. Окрашенными в узком смысле слова («пестрыми») являются вещества, поглощающие впдпмыйсвет «селективно»
350
Глава ,9
Соответствующие примеры приведены в табл. 62.
Таблица 62
Цвет безводных соединений N1 и Сн
Г Ct Вг I О S Sc । SP4 С104
N111 Желтоватый Желто-вато-коричневый Темно-коричневый Черный Темпо-зеленый Черный Серо-го.чу-бой Желтый Желтый
Си11 Бесцветный То же Корнч-пево-черпый Черный Темно-голубой до черного Зеленоватый до черного Бесцветный
У ионов галогенов поляризуемость возрастает от F- к I-. Соответственно йокраска усиливается от фторидов к иодидам. Тб же имеет место в ряду халькогенидов. В сульфатах и перхлоратах поляризуемость кислородных ионов уменьшается вследствие нх связи с серой или хлором (см. стр. 348). Поэтому приведенные в таблице сульфаты и перхлораты окрашены слабо или совсем не окрашены.
Указанные в табл-62 цвета относятся к безводным солям - То обстоятельство, что их окраска тем глубже, чем сильнее поляризуется присутствующий в них отрицательный ион, и что окраска вообще не появляется, если этот отрицательный ион в значительной степени пе поляризуем, позволяет предположить, что цветность этих соединений вызывается световой абсорбцией ие только катиона, но и аниона. Если это предположение справедливо, то окраски, характерные для многих катионов и проявляющиеся в водных растворах или в содержащих воду солях, возникают вследствие абсорбции света не только капищами, но и молекулами ИаО, Окраска только косвенно обусловлена катионами. В действительности вследствие своего поляризующего действия они только таким образом сдвигают энергетические уровни электронов в молекулах НаО, что эти уровни сдвигаются и область абсорбции этой молекулы попадает в зону видимого света. То же имеет место и для аммиакатов.
На основе этих представлений можно легко истолковать многие трудно объяснимые явления: так, наблюдение, что многие элементы главных подгрупп периодической системы способны давать безводные окрашенные соединения еслеобразнбго характера (папример, PbO, PbS, Bi2O31 BiaSg), хотя образующие их катионы в водных растворах по окрашены. Так как поляризуемость йопа О3' при образовании связи значительно уменьшается (ср. стр. 348), то, очевидно, эти катйопы обладаютноляризацион-иой способностью, достаточной, чтобы вызвать сдвиг абсорбции в область видимого света у ионов О3' и у еще более сильно поляризующихся ионов S2’, но не у значительно слабее поляризующихся молекул НаО.
Ионы элементов побочных подгрупп периодической системы в большинстве случаев имеют значительно мепыпие ионные радиусы, чем ионы элементов главных подтруни. Поэтому они иодяривуют значительно сильнее, если только они обладают неполностью завершенными электро иными оболочками, чем это способны делать при одинаковых условиях ионы главных подгрупп. Таким образом, становится понятым, что ионы элементов побочных подгрупп, если их валентности отклоняются от соответствующей номеру труня и отвечающей полностью заполненным электронами слоям, обладают окраской даже и в водных растворах, т. е., следовательно, вызывают абсорбцию света раополож(щпыми около них молекулами Н3О.
4
Глава 10
ТРЕТЬЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Главная подгруппа)
ГРУППА БОР — АЛЮМИНИЙ
Порядковый номер Элемент Символ Атомный вес Удельный вес Температура плавления» °C Температура itn пения, °C Удельная теплоемкость Валент-яость
5 Бор в 10,82 2,34 —2300 — 0,252 J, п,ш
13 Алюминий А1 26,98 2,70 660,2 2270 0,214 I,II, III
31 Галлий Ga 69,72 5,9 29,78 -2000 0,0918 I, II, 1П
49 Индии In 114,82 7,31 156,4 -2300: 0,0570 1,И,Ш
81 Таллий Tl 204,39 11,83 302,5 —1450 0,0316 I, III
Общие сведения. К главной подгруппе П1 группы периодической системы принадлежат два очень распространенных элемента — бор и алюминий, и три редких — га.игий, индий й таллий. Последние относятся к таким , элементам, которые хотя и распространены во всей земной коре; но присутствуют всегда только в очень малых концентрациях в качестве изоморфных примесей в определенных минералах. В этом смысле о них можно-говорить как о редких, а о галлии и индии даже как об очень редких элементах. Они были открыты методом спектрального анализа.
Элементы главной подгруппы III группы в соединениях, в которых их валентность соответствует номеру группы, максимально трехвалентиы. Бор и алюминий образуют только очень ограниченное число соединении, в которых они проявляют низшие степени окисления. Напротив, галлий, индий и таллий очень легко могут быть переведены в низшие валентные состояния. Но в этом состоянии галлий и индий, однако, менее устойчивы, чем в трехвалентиом. Таллий же чаще встречается в одновалентном состоянии, чем в трехвалентиом. Кроме того, таллий в трехвалентиом состоянии в противоположность галлию и индию имеет лишь очень небольшое сходство со вторым элементом главной подгруппы III группы алюминием, к которому галлин и индий во многих отношениях очень близки. Их гидроокиси, так же как и гидроокись алюминия, амфотерны. Их соли, так же как и соли алюминия, в водных растворах частично гйдролизованы. Как и алюминий, галлий и индий образуют квасцы, т. е. Двойные сульфаты типа М-Мш (SOi)^- 12НаО (конечно, не в такой степени, как алюминий). Трехвалентный таллий образует двойные сульфаты другого типа, а именно типа М1МШ (SO4)s-4HsO. Ио и алюминий образует двойные сульфаты этого типа, и их кристаллизацию при определенных условиях, в которых они вообще способны существовать, можно вызвать добавлением соответствующего двойного сульфата таллия. Помимо способности легко переходить в низшие
352
Глава 10
\ степени окисления, галлий, индиц и таллий отличаются от бора и алюминия мсиыпей теплотой образования их окислов и легкой восстанавливаемостью их до металла. В металлическом состоянии вследствие мягкости и низких температур плавления они очень сильно отличаются от алюминия и особенно от бора.
Бор имеет отчетливо выраженный неметаллический характер. Однако его аналоги — типичные металлы. Соответственно своему неметаллическому характеру бор проявляет склонность к образованию гомеополярных соединений. Аналоги бора образуют преимущественно гетерополярные соединения. Все они способны существовать в водных растворах в виде свободных, если не принимать во внимание оболочку из молекул воды, положительных трехзарядных ионов. Бор таких ионов не образует. Все же в соединениях с сильно электроотрицательными элементами бор также можпо рассматривать как положительно трехвалептпый; необходимо только учесть, что свойства этих соединений вследствие сильного поляризующего действия иопа В3+, обусловленного сочетанием небольшого радиуса с. относительно большим зарядом, в значительной степени приближаются к свойствам гомеополярных соединений.
Электроположительный характер элемептов изменяется с ростом порядкового номера в главной подгруппе III группы не так закономерно, как это наблюдается у элемептов главных подгрупп 1 и II групп. Виа чале он очень сильно возрастает от бора к алюминию, по затем при переходе от алюминия к галлию падает и снова возрастает, правда незначительно, при переходе от галлия к таллию. Столь незакономерное изменение электроположительного характера связано с тем, что в отличие от бора и алюминия элементы галлий, индий й таллий расположены не непосредственно за । цел очно земельны ми элементами, а относятся к так называемым переходным элементам, т. с. к элементам, у которых происходит заполнение d-уровпей (см. табл. 2 в приложении). В то время как при переходе от бериллия к бору и от магния к алюминию заряд ядра возрастает только на единицу, от кальция к галлию оп повышается на целых 11 единиц. Так как при этом главное квантовое число остается неизменным, связь внешних электронов с ядром в атоме галлия сильно возрастает и тем самым электроположительный характер элемента значительно ослабляется- Для индия и таллия наблюдается то же самое.
Бор является отчетливо выраженным кислотообразующим элементом. Окисел алюминия по отношению к сильным основаниям может еще проявлять свойства кислотного окисла, однако в обычных условиях он ведет себя как основной окисел. Окислы галлия (III) и индия (III) также обладают амфотерными свойствами с преимущественно основным характером. Амфотерный характер окисла таллия (ПТ) вследствие его исключительно низкой растворимости проявляется в меньшей степени. Однако основной характер у него выражен не сильнее, чем у окислов алюминия, галлия (III) и индия (III)- Напротив, окисел таллия (Z) имеет сильно основной характер. Получающаяся нв него гидроокись похожа в этом отношении па гидроокиси щелочных металлов.
Сально основной характер гидроокиси таллия (1) соответствует общему правилу, что окислы и гидроокиси, отвечающие низшим степеням окислопия элементов, всегда имеют более основной (или мипю кислый) характер, чем соответствующие высшим степеням окисления. Уменьшение кислотных свойств при переходе от борного ангидрида к окиси алюминия и связанное с этим увеличение основного характера объясняются теми же причинами, что и возрастание основного характера при переходе от гидроокиси лития к гидроокиси цезия и от гидроокиси бериллия к гидроокиси бария. Йоны элементов тлавпой подгруппы III группы с зарядом ЗЦ- в своих гидроокисях сильно
Третья группа периодической системы
353
притягивают кислородные ионы и сравнительно столь же сильно отталкивают ионы водорода. Притяжение водородных ионов к ионам содержавшего кислород остатка сильно ослабляется, если центр полол™ тельного трехзаридного иопа расположен сравнительно близко к иону Н'. Это п происходит в том случае, если объем центрального иона невелик. Благодаря этому у гидроокиси, образованной ионом с наименьшим в данной иод группе объемом, а именно у гидроокиси бора, ионы Н’ отщепляются значительно легче, чем попы ОН'. Отталкивающим действием трех заря дно го положительного попа бора на ионы водорода объясняется также неспособность бора к существованию в водном растворе в виде свободного положительного иона. Так называемые элементарные ноны в действ!|тельпости всегда существуют в водных растворах в гидратированной форме (гидраты попов). Подобный гидрат, например [М11 1 (OII2)3J"в случае бора тотчас же распался бы па В(ОН)3 (борпую кислоту) и 3 попа Н', Радиус пона АБ+ значительно больше, чем иона В3+, поэтому иоп алюминия отталкивает иоиы Н' слабее и удерживает ионы ОН' значительно менее прочно. Этим и объясняется менее кислотный и более основной характер Л1(ОТ1)3. То же справедливо и для гидроокисей аналогов алюминия.
Амфотерный характер гидроокиси алюшшия, т. е. ее способность давать соли пе только с кислотами, по и с основаниями! ие обязательно должен основываться на том, что при ее диссоциации, помимо ионов ОН', могут еще образовываться и ионы Н‘. Образование солей с основаниями может обусловливаться также способностью А1(ОН)3 присоединять еще дополнительные ионы ОН'. Можно показать, что при реакции Al(OH)3-f- ОН'= |A1(OH)J' освобождается энергия (см, гл, 11),. Из опытных данных следует, что происходит именно эта реакция, а пе реакция AI(OH)3=[A1O(OH)2]' -f- 1Г, которая идет только в том случае, если водный раствор гидроокиси алюминия обрабатывать сильными щелочами. Эта значат, что А1(ОН)3 в водно-спиртовых растворах ведет себя пе как кислота, по как ангидро кислот а и образует гидроксосоли, например К(А1(ОН)4], гидр оксо алюминат калия, С другой стороны, иоиы Н‘ способны отрывать ионы ОН' от иона А1(ОН)(, так что устанавливается зависящее от концентрации ионов 1Г равновесие;
[А1(ОН)4]'+1Г Tt А1(0Н)3+Н20, (1а)
А1(ОН)3-|-ЗН- АГ” + ЗНгО. (16)
Эти равновесия характеризуют амфотерные свойства гидроокиси алюминия. Для равновесия, соответствующего (1а), имеем
tA1(0H)il [j£J = 2. Ю-u или --------[АК9Ч)*1----= 2, IO3.
[А'1(ОН)з1 ИЛИ [А1(ОИ)3] (OH'J
Электролитическая подвижность попа [А1(ОН)4]', по Боде (Bode II., 1952), приблизительно равна подвижности гидратированного иона лития. Коллоидных частиц или макромолекулярных агрегатов, состоящих из ионов, согласно данным-Боде, в растворе гндрооксоалюмпната натрия ие существует.
Из водородных соединений элементов главной подгруппы III группы гидриды В и Ga легко летучи. По другим своим свойствам они также соответствуют водородным соединениям элементов, стоящих правое их в периодической системе. Гидриды алюминия и индия — полимерные твердые вещества. Они подобны гидридам бериллия и магния и не обладают, следовательно, солсобранным характером гидридов щелочных и щелочноземельных металлов. Таллий является единственным элементом главных подгрупп периодической системы (не считая инертных газов), для которого не может быть получено в свободном состоянии соединение с водородом*, В виде двойных соединений гидрид Т1 все же получен. Общим для всех гидридов элементов главной подгруппы III группы является то, что в свободном состоянии они всегда полимеризованы (например, (ВН3)2, [А1Н3]Ж). Эта полимеризация основана па сцеплении мономерных молекул посредством водородных мостиковых связей.
* Питч (Pietsch, 1933) нашел, что таллий взаимодействует с атомарным еодородом, с образованием газообразного гидрида, который, однако, очень неустойчив и тотчас разлагается прп соприкосновении со стенками сосуда. Галлий и индий соединяются с атомарным водородом с образованием твердых гидридов. '
23 г. Реми
.354' „ Блаев 10
Главная подгруппа III группы может служить характерным примером того правила, что первый элемент главной подгруппы по свойствам ближе к следующей главной подгруппе, а второй—к побочной подгруппе этой же группы. Бор, если не считать его валентность, по свойствам имеет очень мало общего со своими более тяжелыми аналогами. Как кислотообразующий элемент, он стоит гораздо ближе к соседним углероду и кремпию. У алюминия общего с элементами побочной подгруппы третьей, группы значительно больше, чем у бора; Он близок им не менее, чем тяжелым аналогам главной подгруппы, Во многих отношениях он занимает отчетливое промежуточное положение между бором и элементами побочной подгруппы, а не между борой и элементами главной подгруппы. Например, электроположительный характер правильно возрастает от бора через алюминий к лантану, в то время как в ряду бор — алюминий — галлий — ипдип — таллий, как уже указывалось, такое возрастание отсутствует. Теплоты образования хлоридов и окислов закономерно возрастают от бора и алюминия к лантану, в то время как от алюминия к таллию они Падают (см. рис. 1, стр. 34), Сходство алюминия с его тяжелыми'аналогами из главной подгруппы особенно проявляется в одинаковом строении водородных соединений. С галлием и индием алюминий объединяет также такое характерное для этих элементов свойство, как способность к образованию квасцов.
По теории Косселя положительная трехвалентностъ элементов III главной подгруппы следует из их положения-в периодической системе по отношению к предшествующим инертным газам; в нейтральном-состоянии каждый из них обладает тремя липшими электронами по сравнению с предшествующим инертным газом. Эти три электрона сравнительно легко отщепляются и легко передаются атомам таких элементов, как кислород, хлор и т, д., обладающих большой электроотрицательностью. Образующиеся при этом ионы, обладающие зарядом 3-Ь, соединяются с эквивалентным количеством одновременно образующихся отрицательных ионов кислорода, хлора и т. д.
О строении атомов бора и алюминия можно судить с достаточной точностью на основания спектральных данных. ;
Айал из термов искровых спектров бора и алюмишия доказывает, что Первый электрон, захватываемый атомными остовами В®* и АР’, связывается ими совершенно так же, кая он связан в атоме лития или натрия. Связь, однако, оказывается приблизительно в 9 раз более прочной в соответствии с втрое большим зарядом- ядра, Отсюда можно заключит!,, что атомные остовы В3’ и АР’, несмотря на более высокий положительный заряд их ядер, по расположению в нпх электронов соответствуют атом- -ным остовам Li+ и Na' и тем самым атомам инертных газов Не ii Ne. Разумеется, что орбиты электронов в этих атомных остовах все более сжимаются в соответствии с увеличением ядер по го заряда. Это следует из уравнения. (9) гл, 3 р отчетливо выражено в уменьшении радиусов в рядах Не — 1л+ — Вс2’ В3+ и Ne — Na’ — Mg3’ — AF* (см, рис. 3, стр. 37). А,
, Из рис. 70 видно, насколько полна аналогия между системами спектральных термов Na, Mg+ и АР*. Энергетические уровни, соответствующие спектральным термам, представлены здесь таким ate образом, как и раньше (см, стр. 282). Числа, стоящие паД отдельными энергетическими уровнями, означают квантовые числа пъ по Бору^Зом-мерфельду (так же, как и на рис. 59)7 Шкала на. левой части схемы означает уровни энергии для ге==2, 3 и т. д. водородного атома. Чтобы исключить влияние зарядов ядер, масштабы для уровней энергии в ины для Mg’ в 4, а для А1а+ в 9 роз меньше но сравнению с масштабами для уровней энергии Na. На рис, 70 представлены также уровни энергии атомного остова Si с одним вращающимся вокруг него электродом в масштабе, умейщпеином до Vie- Этот спектр будет еще рассмотрен в гл. 12,
На рве. 71 представлены (в том же масштабе, как и для натрия на рис. 70) уровни энергии, соответствующие различным и iro-разному проявляющимся в излучении света , типам связи третьего удерживаемо го ионом АР’ электрона, относящиеся, следовательно, к нейтральному атому алюминия. Для сравнения приведены также величины урон-
Третья группа периодической системы
Рис. 70. Сравнение энергетических уровней натрия, соответствующих дуговым спектрам с энергетическими уровнями магния, алюминия и крем-
ния, соответствующих искровым спектрам. Все спектры относятся it атомам или атомным остовам с одним и тем жо числом т одиннадцатью электронами.
ней энергии, получаемые из искрового спектра однократно ионизированного атома углерода, о чем подробнее будет сказано ниже. Видпо, что оба этих спектра построены существенно иначе, чем спектры, показанные на рис. 70. В ионе Л12+ электрон находится в нормальном состоянии, т. е. в состоянии прочной связи па 3s-уровне, в нейтральном же атоме А1 он находится на Зр-уровне. Уровень энергии 3s, как видпо: из рис. 71, отсутствует среди спектральных термов нейтрального атома А1. Это значит, что последний присоединяемый алюмиппем электрон пе в состоянии спу-; ститься глубже Зр-уровпя. Уровни 3s,. и а которые мог бы попасть третий электрон*, оказываются уже заняты обоими ранее захваченными электронами (второй из них, как это следует из системы термов А1+, связан таким же образом, как и у щелочноземельных металлов).: Следовательно, три внешних электрона алюмипия (относительно бора можпо Сказать то же самое) связаны не одним способом', два из них находятся на Зя-уровиях, а третий на Зр-урбвпе,
Значение, которое все это имеет для образования гомеополярных соединений бора ji алюминия, будет рассмотрено в дальнейшем. Для образования гетерополярных соединений нс существенно, что различный тип связи (s- или /.-электроны) может в значительной степени влиять на прочность связи электронов с атомным остовом, что, кстати, пе имеет места в случае бора и алюминия. Работа, необходимая для от-
щепления электрона при переходе в ионизированное состояние, много больше, чем. разница в:энергиях связи s- и р-электронов в атоме. Строение атомных спектров галлия,,
* То, что в атоме па каждом s-уровне не может быть больше двух электро но в р следует, из принципа Паули (ср. стр. 145),
п.-5-
.4 -
3-
Al s р d
. С + .
s р d f
6s
5s
4 s
2-
5д
J/s
5d id
3d
Sf
4f
5з
4s
3s
5p
4p
5d id
3d
3f if
sc
E
Р ис. 71. Энергетические уровни;: соответствующие дуговым спектрам алюминия и искровым спектрам углерода.
23»
356
Глава 10
индия и таллия в значите л иной степени соответствует спектру алюминия. Из этого можпо заключить, что в свободных атомах этих элементов внешние электроны в прин-цине располагаются так же, как у бора и алюминия, а именно два электрона па .«-уровне и вднп электрон па /j-уровпе.
Как следует из спектральных термов, максимальная положительная трехеалент-ность рассматриваемых элементов объясняется, таким образом, тем, что эти и только эти три электрона могут отщепляться относительно легко. Общая работа, затрачиваемая па отрыв от атома элемента главной подгруппы Ш группы трех электронов, меньше им, табл. 63) работы, которую нужно затратить Для отрыва одного следующего элект-
Габлица 63
Энергия ионизации9 элементов главной подгруппы Ш группы (кка л/г-« то м)
Бор Алюминий Галлий Индий ТаллиЯ
1М-тМ*~Н ПМ*н-М!++« 1П№*->М’Ч' 190,3 576,1 869,7 137,4 431,8 652,4 137,7 435,9 ,705,3 132,8 387,5 643,7 140,2 468,2 685,0
Сумма 1+ 11+ III 1636,1 ’ 1221,8 1278,9 1164 1293
М3+ -ет- М4+ + е 5946,5 2751,4 1474 1222
а См. примечание к табл. 28, стр. 180. — При.и peg.
рОна. За счет сил, обусловливающих образование ионной решетки, может быть выполнена работа отрыва трех электронов, по не четырех электронов; ибо эта работа возрастает в значительно большей степени, чем энергия решетки, образованной из Соответствующих попов, Рассчитанная по уравнению (11), стр. 172, энергия решетки для соединений четырех валентно го алюминия, изотипных соединениям трехвалентпого алюминия и обладающих одни ано вы мн размерами решетки, превышает всего лишь в 1,8 раза энергию решетки соединений трехвалентного алюминия, Тогда как соответствующие работы ионизации находятся в отношении 3,25 ; 1. Таким образом, становится понятным, почему в гетеро полярных соединениях алюминий всегда бывает только трехвалентпым, а не четырехвалеитпым. Все это справедливо и для других элементов этой группы. При помощи рассмотренного на стр. 174 кругового процесса можно далее показать, что в общем нельзя ожидать, что бор и алюминий при обычной температуре будут образовывать устойчивые в твердом состоянии гетсрополирные соединения, в которых они бы обладали валентностью, меныией чем 3*. Чтобы сделать до сто верша в выводы, необходимо провести расчет стабильности для каждого отдельного случая. Нельзя также, исходя из нестабильности соединения в твердом состоянии, сразу же делать заключение о нсет&билъпостп его также и в растворе. Стабильность в водном растворе в значительной степени определяется теплотами гидратации понос. Насколько сильно зависит от пих стабильность соединения, показывает следующий пример,
Работу, которую следует затратить, чтобы 1 г-атом металлического А! перевести в гидратированный ион АГ", можпо рассчитать по уравнению (12), стр. 174, из теилоты сублимации (57 ккал/г-атом, стр. 358), работы ионизации (см. табл. 63) и теплоты гидратации. Теплота гидратации получается из теплоты растворения алюминиевых солей, равной 1065 вкал/г-ион АГ", если принять теплоту гидратации иона Н‘, равной 250**. Таким же образом получают работу, необходимую для образования гцдра-
* При высоких температурах имеются газообразные соединения, в которых В д А1 существуют в низших валентных состояниях (стопенях окисления) (см. стр, 362 да 387).
** Так как теплоты гидратации ион а 1Г при расчете взаимно уничтожаются, то неточность этой величины не оказывает влияния па конечный результат.
Третья группа периодической системы
357
тированного иона ЛГ" вплоть до неизвестной величины QA1.. теплоты гидратации этого иона:
+57 + 1222 -1065
[АЦ-------> (АЗ) ----> (АР+)-------> At"’,
+ 57 +569 -Qa1—
[A1J------> (Al) ----> (Al3*) —----> Al",
При этом еапграчиеаемая работа борется со знаком плюс. Используя круговой процесс (см. стр. 174), получают отсюда сродство образования *
[А1]=АГ’‘ + Зе —г 214 ккал, (2)
[Л1]=А1“ 2е -f- (QA1.. — 626) ккал. (3)
Далее, имеем
з/^Н^ + !-2(С12)=Н- + С1'+ 31,4 ккал. (4)
Сродство этой реакции получают непосредственно из нормального потенциала хлора (1,36 а), используя уравнение (3) па стр. 166. Из данных, приведенных в табл.7на стр. 61, следует
+51,4 +312,2 -250
V2(H2)!—> (Н) ---------> (Н*)------> Н-,
и поэтому
й(Н2) = Н‘ 4-е — 113,6 ккал. (5)
ур. (2)4-3х [ур,(4)_ур.(5)] : [A1J4-S/2C12 = АГ” д_ ЗСГ 4- 221 ккал, (6)
УР- (3)4-2х (Ур.(4)~уР.(5)) . [A1J4-(C12)=A1- -f- 2СР 4- (QAl.. - 336) ккал, (7)
УР- (6)—Ур-(7) АГ" + />СЬ = АГ" 4-С1'4- (557 — QA1,,) ккал. (8)
Из уравнения (6) Получают, используя уравнение (3) на стр. 166, потенциал разложения Л1С12, отнесенный к 1 37 по АГ" и СГ раствору, равным Z5= 3,2 в, который удовлетворительно согласуется со 'значением, полученным сложением нормальных потенциалов (табл. 4) 3,1 в**. Сродство реакции (8), выраженное в электрон-вольтах, т. е. численно равное потенциалу разложения в вольтах (Z7), для перехода А1С13 в Л1С1г и /4С1г составляет
ZT=24,2— 4,34-10-2-QAl.. .
Ион'Л12*, не имеющий оболочки типа инертного газа, обладает большим- поляризующим действием и соответственно, вероятно, большей теплотой гидратации, чем ион Mg2+. Предполагай, что его теплота гидратации равна теплоте гидратации иона AIg-+(Q-—-437 ккал)’* получаем Z7=Ц-5,2 е. Так как это значение лежит выше, чем напряжение Z3, которое должно быть приложено, чтобы выделить А1 в виде металла, то при предположении такой теплоты гидратации исключается возможность образования иона АГ". Если предположить, что теплота гидратации попа А12+ равна теплоте гидратации иона Вс2* (Q=570 ккал), то получаем Z,=—0,5 в, т. е. реакция, представленная уравнением (8), могла бы сама по себе протекать справа налево: раствор AJ.CI3 должен был бы разлагаться с образованием A1G13 и выделением С12! Отсюда видно, какое значительное влияние на стабильность попа ЛГ‘ оказывает значение теплоты его " гидратации.
Так как теплота гидратации этого иона, по всей вероятности, лежит" между теп-лотами гидратации ионов Mg2 * и Ве2+, то можно считать с известной вероятностью, что Z7 хотя и не отрицательна, по все же меньше, чем Zj. В этом случае, прилагая возрастающее напряжение к раствору А1С13т достигают напряжения Z7 прежде, чем Z5, т. с, при этом происходит вначале восстановление иопов Аг" до АГ’ и лишь при более высоком напряжении, значение которого в этом случае определяется уравнением i (7), происходит выделение алюминия в виде металла.
При образовании гомеополярных соединений существенную роль играет характер связи электронов в атоме. Элементы III главной подгруппы, согласно спектральным данным, в нормальном состоянии имеют в атоме только один псспареяный электрон. (В атомах каждого пз этих элементов термом, соответствующим нормальному состоя-
* Относительно термина «сродство образования» см. сноску на стр. 165. — Прим. ред.
** Расхоящепне объясняется тем, что нс была принята во внимание температурная зависимость величин, использованных для этого расчета. Если это учесть, то получается совпадение до нескольких сотых вольта,
358
Глава 10
вию атома, является гГ’(д-торм,) Ло теории Гейтлера и Лондона в гомеополярных соединениях следовало бы поэтому ожидать одновалентность Этих элементов. И действительно, н полосатых спектрах проявляется существование молекулВН, А1Н, InH и Т1Н; кроме того, для всех элементов III главной подгруппы могут быть обнаружены молекулы мои от ало гсп идо в. Однако при нормальных условиях эти молекулы не стабильны. Например, для молекул ВН н A11I сродство образования пз свободных атомов составляет соответственно 79,Г> и 71,0 ккал!.моль. Для образования из кристаллического бора пли алюминия и молекулы Н2 получаются всё же сильно отрицательные значения сродства (—87 и —35 ккал/молъ). Речь, следовательно^ идет в случае молекул мопогндридов о нестабильных в химическом смысле соединениях. Из. спектральных данных следует, далее, что необходимо затратить очень небольшую работу для столь глубокого «возбуждения» атомов В и А1, чтобы s-электрон перешел бы в соседнее p-со стояние (0,04 и соответственно 0,32 ккал/г-атом). Такие возбужденные атомы с электронной конфигурацией spa содержат три цеспаренпых электрона и поэтому могут вести себя как трехвалептпые. Все же квантово-механический расчет стабильности соединений, производимых от этого состояния, еще затруднителен. Химическое исследование показывает, что гомеополярные соединения элементов III главной подгруппы по сравнению с большим числом гетеронолярпых относительно менее стабильны. Теплота образования ВjHfl, по данным Рота (Polh, 1937), составляет 44 ккал/молъ. т. е, только Vs теплоты образования В2О3. Все же у бора по сравнению с элементами I и II главных подгрупп способность к образованию гомеополярных соединений значительно возрастает. У алюминия она уже значительно меньше и продолжает уменьшаться в группе по направлению к галлию.
Уменьшение склонности к .образованию гомеополярных соединений в направлении Al—> Т1 проявляется, например, в ряду металлерванических соединении в том, что Al, Ga и In при взаимодействий их хлоридов c;MgRGl (реакция Гриньяра) образуют триалкилы MR3, в то время как для Т1 таким путем': межи о получить только Дианки дьноё соединение TlRaCl (см. стр. 427), В случае айияьиых производных реакция Гриньяра для In, так же как и для Т1, приводит, к образованию соединения InRaCl, в то время как для АГ и Ga этим методом получаются триарильные соединения. Способность к образованию перегоняющихся без разложения двойных соединений с эфиром у перечисленных металлов изменяется таким же образом. Известны эфираты различных триалиилов AJ и Ga,. Для In образуется только один, а именно эфпрат триметил-индня, а дли Т1 не существует ни оДпого.
Соединения присоединения металлов главной подгруппы П1 группы с эфиром имеют состав Либо МНз^С-Нг,)./). либо 2МВ3-(С2Н5)2О. Их образование, вероятно, осповапо па том, что эфирный кислород обеими своими еще неиспользованными в химических связях электронными парами занимает вакантные места около атомов металла, дополняя их электронные оболочки до октетов. При этом, следовательно, возникает дативная связь в
H5CZ .AkClhh : (С2Н5)3О4-2Л1(СП3)з= 20;С ;
Н5С/ А1(СН3)3.
Такие же соединегнгя с эфиром известны также у ал кил ьпых производных металлов III побочной подгруппы, по только лишь у Sc и ¥t, а не у; La и Ас.
В табл. 64 приведены теплоты образа вари я соединений В, Al, Ga, In и Т], в которых эти элементы трехналептны.
Таблица 64
Теплоты образований соединений элементов главной подгруппы третьей группы ’ ; (-ккал/г-же)
Элемент Окттсел Фторид . Хлорид Бромид Иодчд Сульфид Нитрид
Бор 58,2 99,0 40,7 4b? , ' — 1и >12
Алюминий ; 66,41 125,3 55,8 40,3 24,4 ... — 21,3
Гаплий 42,9 — ; 41,7 ; 30,8 17,0 — 8,3
Индий 37,1 — i 42л8 32,4; 48,8 — 1,5
Таллий 20?. — 26,7 : — —.
Третья группа периодической системы 359
Теплоты образования соединений элементов главной Подгруппы III группы, если их отнести к эквивалентным количествам, лежат значительно ниже теплот образования соединений элементов главных подгрупп I и 11 групп. Отчасти это обусловлено ина- г чителько возросшей работой отрыва электронов (см. табл. 83). Однако ее повышение у соединений бора и алюминия приблизительно компенсируется увеличением энергии взаимодействия ионов в кристаллической решетке. Для уменьшения теплоты образования, приходящейся на 1 а-эке, в рядах Li — Вс—В иДа^М?-Л1 существенное значение имеет значительное повышение в этом же направлений теплоты сублимации. Последняя, однако ( для большинства этих элементов непосредственно еще нс измерена. То, что от Li и В и От Na к Л1 она существенно возрастает, следует на основании правила Трутона из значительного повышения температур кипения, Правило Трутона гласит, что для еысококипящих веществ молярная теплота испарения X изменяется: приблизительно так же, как и абсолютная температура кипения Ts. Отношение МТ» (константа Трутона) составляет обычно около 21,5. Можно поэтому получать приблизительные значения теплот испарения веществ в калориях путетй умножения абсолютной температуры кипения на 21,5. Для алюминия рассчитанная таким образом теплота испарения равна 25-43 х 55 000 кал1в-атом, Для алюминия непосред-
ственно измерена п теплота плавления', опа составляет 92 мл/г = 2500 кал/а-атом. Сложением теплот плавления и испарения моящо получить приблизительное значение теплоты сублимации*.
БОР (BORON) В
Распространение в природе. Бор никогда не встречается в пр пр 6 де-в свободном состоянии, он всегда оказывается связанным с кислородом. В этой форме оп присутствует в борной кислоте Н3ВО3, которая содержится" в воде горячих источников вулканических местностей, например в Сассо в Тоскане (отсюда название сассолин). Кроме того, в природе распространены многочисленные соли борной кислоты,, месторождения которых, . однако, довольно ограниченны. Из этих солей наиболее известна бура или тинкал Na^BjO^lOHjjO. Техническое значение имеют, кроме того, борацит 2Mg3BgOjs.MgCl/(6o.nbTOHe количества его. имеются, папример, в стассфуртских соляных отложениях), пандермит: Са2В^О11 -ЗНаО> колеманит Са8В6Оп 5Н2О, а также в первую очередь кернит Na.iB/)-. >4НаО, открытый в огромных количествах в 1928 г. в Калифорнии й ставший с тех нор важнейшим сырьем для мировой промышленности буры и борной кислоты.
Из других минералов, являющихся производными борной кислоты, необходимо указать следующие: борокалкцит CaB^Oy-iHgO; боронатрокальцит NaCaR^Og-GHgfJ; франкланд ит Na2C аВ6О < < 7 Н 20; гидроборацит М gCaB^O 4 j - 6IJ2O; боромаг! iesum 2Mg5B4O,r3nsO; ашарит SAIgjB^Oj-.SHnO; пинноит MgB2O4^3H2O; лардереллит (i\H4)2BgU13-4Il2O: лагонизп 2РеВзОв-ЗН2О; сулъфоборит 2Mg2B265-2MgSO4-9НКО; лК'Небурент MgB2O4 <2MgHPO4.7НгО и еингалшп №gAlI3O4. - ‘
Предполагают, что при образовании бор содержащих минералов во многих случаях большую роль играла летучесть борной кислоты с водяными парами. Возникает вопрос, могла ли эта летучесть при определенных условиях привести к тому, что соотношение количеств обоих/ изотонов бора 11В и 1ов оказывалось в разных местах различным. В этой связи интересны определения атомпрго веса В, которые произвел Бриско (Briscoe, 1925—1927) на образцах, выделенных из минералов различного происхождении. Он нашел для бора из калифорнийского колеманита заметно более высокий атомный вес, чем для бора из тосканского сассолина или малоазиатского борацита. (
Радикал борной кислоты встречается в соединении с кремниевой кислотой, например в виде датолита CaBSi()4(OII) и аксинита
* Обычно указывают теплоту'плавления для температуры плавления,1 теплоту : : испарения — для температуры кипения. Для уточненных расчетов теплот сублимации обе Эти величины перед сложением необходимо пересчитать на температуру,' При которой производится определение теплоты сублимации (чаще всего в градусах Кельвина).
360 Глава 10 ,
Ca3Al2BSi4OJg(OH), а также в турмалинах, которые, по-видимому, являются изоморфными смесями боратов и силикатов,
Исторические сведения. Издавна известна бура, о которой упоминается (как о флюсе) уже в сочинениях, приписываемых Геберу. В 1702 г. Гомберг (Homberg) нагреванием буры с серной кислотой выделил из пее свободную борпую кислоту, которую вскоре под названием «-sal sedavltunv» стали применять .в медицине. Элементарный бор (нечистый) был впервые получен в 1808 г. Гей-Люссаком и Тенаром восстановлением борного ангидрида калием, а вслед за тем Дэви получил его и электролитически.
Чистый кристаллический бор был получен впервые Вейнтраубом (Weintraub, 1909) при плавлении «аморфного бора» в вакууме.
Получение. Чистый; кристаллический бор непосредственно получают методом Ban Аркеля и де Бура, подробно описанным в т. II в разделе о цирконии, а также, по Хскшпиллу (Hackspill, 1933), разложением ВС1з в присутствии На в высокочастотном разряде между вольфрамовыми электродами. Восстановлением ВаОз металлическим натрием или магнием получают так называемый аморфный бор в виде коричневого порошка. От примесей его освобождают кипячением вначале с разбавленной соляной кислотой и затем обработкой плавиковой кислотой. Однако весьма сомнительно, можно ли элемент таким образом получить в совершенно чистом состоянии. £-
Сплавлением с алюминием можно получить из «аморфного бора» кристаллическое , вещество, называвшееся ранее тетрагональным бором. Его можно получить непосредственным восстановлением В2О3 алюминием. Ио «тетрагональный бор» всегда содержит алюминий, а при определенных условиях, кроме того, л углерод. Видимо, в этом случае речь идет лишь о соединении A1Bj2, с которым механически смешано отделяемое от него у гл сродсо Держащее вещество, вероятно двойное соединение ЗА1В12-2В^С (Мйгау-Szabd, 1936),
Свойства. Так называемый «аморфный бор» — коричневый порошок без вкуса и запаха, с у дельным весом 1,73. Чистый кристаллический бор имеет серовато-черную окраску, обладает твердостью 9 и удельным весом 2,34 (Richards, Hackspill, Laves). Он кристаллизуется в иглах (вероятно, мопоклипиых) или гексагональных пластинках, чрезвычайно тверд, непрозрачен и имеет очень незначительную электропроводность, повышающуюся с ростом температуры (Laubengayer, 1943). Чистый кристаллический бор химически очень устойчив, в том числе и по отношению к окислителям, Температура плавления бора лежит очень высоко. В электрической дуге бор начинает испаряться. Средняя удельная теплоемкость бора в интервале 0—100° равна 0,252; таким образом, для атомной теплоемкости помучается значение 2,73, сильно отклоняющееся от соответствующего закону Дюлон-га и Пти. С понижением температуры атомная теплоемкость бора понижается еще больше. Напротив, с повышением температуры она все более приближается к величине, требуемой законом Дю лонга и Пти, к которой, по данным. Магнуса (Magnus) и Данца (Вани), при 900° ее значение 5,5 подходит уже довольно близко,
Устойчивый по отношению к воздуху при обычной температуре бор (аморфный) при нагревании па воздухе до 700° загорается и горит красноватых! пламенем; если же его сжигать в струе кислорода, то достигается более высокая температура; бор в незначительной степени улетучивается и окрашивает пламя в зеленый цвет. При нагревании бор соединяется непосредственно с хлором, бромом и серой, но заметно пе реагирует
Третья группа периодической, сметами 36f
с водородом. Начиная с температуры 900°, он соединяется также и с азо* том/ образуя нитрид BN, который поэтому также образуется в качестве побочного продукта при горении бора иа воздухе. Равным образом и с аммиаком при высокой температуре образуется нитрид бора BN.
Концентрированной азотной кислотой или царской водкой, а также при сплавлении со щелочами В окисляется с образованием бор ной кислоты или боратов щелочных металлов; однако расплавленная селитра при 400° на него еще не оказывает заметного действия. Концентрированная серная кислота действует на бор лишь при 250°; фосфорная кислота восстанавливается им до свободного фосфора только при 800°. Водяным паром при температуре красного каления бор окисляется с выделением свободного водорода. С окисью азота бор взаимодействует при температуре красного каления, образуя грехокись и нитрид бора. При очень высоких, температурах бор оказывается в состоянии восстанавливать также окись углерода и двуокись кремния. Благодаря своему сильному сродству н кислороду и к другим электроотрицательным элементам бор может выделять в свободном состоянии металлы из их окислов, сульфидов и хлоридов. Теплоты образования простейших соединений бора приведены в табл. 64 на стр. 358.
Применение. Элементарный бор применяют в ограниченных коли- ‘ чествах в качестве антиокислителя при медпом литье. Он находит также применение в металлургии стали (большей частью в виде ферробора, т. е. сплава с железом с 10—20% В). Добавка уже 0,001—0,003% бора значительно повышает твердость стали. «Тетрагональный бор» вследствие исключительной твердости, почти равной твердости алмаза, был предложен в качестве шлифовального материала. Обширное применение паходят борная кислота и ее соединения, прежде всего .бура. Пероксигидраты боратов играют большую роль в промышленности в качестве отбеливающих средств.
СОЕДИНЕНИЯ БОРА
Обычные соединения бора — галогениды, окисел, нитрид и сульфид, а также бороводороды. — всегда содержат бор в трехвалентном состоянии. Первые из упомянутых соединений следует рассматривать ка к построенные гетерополярно, й бор поэтому в них характеризуется электрохимически положительной трехвааемтностъю: однако следует учесть, что эти соединения вследствие сильного поляризующего действия бора весьма близки к гомеополярным соединениям, особенно по своим физическим свойствам,
В галогенидах BFj и ВСЦ трехеалентностъ бора, принимая во внимание его атомный вес, следует непосредственно из найденных значений упругости паров. Гете-рополярлый характер соединений проявляется в их склонности к гидролизу, который обусловливается наличием большого положительного заряда у бора. Вообще отчетливая склонность бора к присоединению кислородных ионов объясняется его большим положительным зарядом в сочетании с малым ионным радиусом, Сильнее чем ион Os~ удерживается бором песуодий еще больший отрицательпъ:й заряд нон
Вследствие значительно возросшей по сравнению с элементами 1 и II “главных подгрупп склонности бора ц образованию гомеопопярньтх соединений, т. е, к осуществлению ковалентных связей, бор способен одновременно соединяться с электроположительными й электроотрицательными элементами, например с Н и О (аидроксо-бораны и бористые кислоты), но той же причине ол способен образовывать цели, в которых непосредственно связаны атомы В {бороводорады и випоборные кислоты). Химия этих соединений особенно близка химии углерода.
Как уже было указано, бор способен также образовывать соединения, в которых он одновалентен. Существование этих соединений, доказанное анализом полосатых спектров, возможно только при высоких температурах. В табл. 65 приведены опре
362
Глава 10
деленные Шретье (Chretien, 1950) для нескольких таких соединений5 валентно-силовые константы, межатомные расстояния и энергия' диссоциации на свободные атомы. Для сравнения указаны также значения этих величин для обычных соединений бора.
Таблица 65
Валент но-силовые конставты к,, межатомные расстояния гв и энергия диссоциации11 D галогенидов бора
Соединение бин/сл -105 V Л D, СоБДияеяие дин/см 105 Д. Л
BF 7,73 1,26 3,5 I ВГ3 6,69 1,30 6,4
ВС1 3,39 1,72. 5,4 BCIS 3,30 1,73, 4,2
ВВг 2,61 1,89 ‘2 BBrs 2,50 1,87 . 3,1
а См. примечание на стр. 205. — ТГрил. рсО,
Двухвалентен бор в моноокиси ВО. Существование этого соединения доказано как спектрографически (полосатые спектры), так и путем химических экспериментов (см. стр. 373). Но так же как и моно галогениды бора, мопоокись; существует только в газообразном состоянии при; высокой температуре. 1
Технически важным соединением бора является борная кислота Н3ВО3 й ее соли, в первую очередь буря. Борные кислоты обнаруживают большую склонность к образованию комплексных или координационных соединений (см. гл. 11). Простейшими координационными соединениями бора являются борефторйетоводородная (фтороборная) кислота: Н [ВЕЛ и производные от нее соли (фторо^ораты).
Координационные соединения, полностью соответствующие фтороборатам, образует бор со многими органическими радикалами. К ним относятся получаемые по методу Германа (Herman) дибренцкатехинобораты
и подучаемые по Еёзекепу (Boseken) соединения типа [BRjM1, Эти соединения Считали «соединениями пятивалентного бора». Но бор внихвее же, так же как и во фторобор атах, электрохимически. трехвалентен, координационно же чётырехоалемтен. Четвертой валентностью здесь считается дативная связь (см- стр. 162). Свою «од,>>-динационную четырсхваленгбностъ бор осуществляет также в трифенйлборе (CgH5)3B, ; который способен присоединять один атом натрия.
Нормальные бориды металлов, например MgsB2, можно рассматривать как соле-образные соединения, и которых бор является электроотрицательной составной частью. Однако одень часто бориды имеют состав, пе соответствующий нормальным валентно* стнм, и обладают, как, например, А1В, и СаВ0, своеобразной структурой. Так, CaBfi (а также SrВе, ВаВ6, LaB6, СеВ6) образует решетку, которую мо ж до получить из решетки CsI (рис. 48), если в элементарной ячейке атомы: Cs заменить атомами Са, а атомы I — октаэдрами, образованными шестью тесно взаймосвязаннйми атомами В (SiackCIberg, 1931).
Обзор главнейших типов соединений бора приведен в табл. 66.
БОРОВОДОРОДЫ (ВОРОГИДРИДЫ, БОРАНЫ) '
При нагревании металлического магния с борным ангидридом и обработке полученного таким образом вещества, так называемого «борида магния», кислотой получают газ, состоящий главным образом из водорода и содержащий в виде примеси, о чем свидетельствует характерный заиах,
Трешъя группа периодической системы- 363
, - Таблица, 66
Обзор важнейших соединений бора
Бороводороды. В2Не, В4Н10 и др. Соли боранов Соли гидр оксоборанов М1[В3И1(ОН)21 Металлоборо-гидриды (борапаты) А1(ВН4)а, Li[BH4J, Na[BH4] Борореаниче- с кие соединения Алкилы ВН3 Алкоксиды 0B,f Бориды, металлов а) солсобразные Mg3B3 б) нссолеоб- разные СаВ6 Галогениды, BF3 H[BFJ BC]S BBr3 BI3 Кислородные соединения В2О3, во Борные кислоты Н3В0а, ”. нво3 Бораты, например бура Ка3В4ОгЮН3р.; t Нитрид BN Сульфид BoS3 Бо рофо сферт етра-оксид ВРО4 Циклические соединения бора, например боразол B3N3H0 и Н В : hn/Vxh Hb|J!bH Н v 11 Бороксоды ; Борсульфолы
еще и другое летучее вещество — бороводород. Шток (Sluck А., 1912) показал, что в действительности в этом случае речь идет о смеси различных бороводеродов. Ему и его сотрудникам удалось впервые выделить из'этой смеси чистые соединения. Они названы общим термином бораны и различаются по числу атомов В, содержащихся в молекуле — диборан, тетраборан и т. д. Важнейшие из найденных Штоком борбводороды приведены в табл. 67. Формулы ид Точно установлены анализами и определением плотности пара.
Таблица 67
Бороводороды : (борапы) \
ВгНе В1Н.10 ВьИц В5Н9 Bl0Hj4
Температура кипения, °C -92,5“ 18° —. .. -21.Г
Упругость пара пркб’С, 388 57 66 : 7,2 -Г-
м.ч рт ст
Температура ил а в ле- — 165,5° . -120 — 128,6 —45,6 -65,1 99,7
НИИ, °C .
Экспериментально уста- 13,5 2g,7 32,1 32 : 37,25 61,0
новлеппая плотность пара (водород — 1)
Плотность в жидком 0,447 0,59 — 0,61 0,70 0,78.
состоянии (ири — 112°) (при' -70°) (при 0°) (при 0°) (црй 100°)
364
Глава 10
Химические свойства бороводородов были исследованы главным образом Штоком и его учениками — в основном Виборгом — и с успехом использованы для объяснения строения этих соединений, первоначально представлявших значительные затруднения для теории валентности.
Получение и работа с бор о водородами чрезвычайно осложняется вследствие большой чувствительности их к воде, которая их разлагает (через ряд промеж уточных продуктов) па борную кислоту и водород. Известные трудности представляет отделение бороводородов от силанов, которые всегда примешаны к сырому газу вследствие хотя и незначительных, но постоянно присутствующих загрязнений магния кремнием. Йх появления можно избежать, если заменить борид магния боридом бериллия. Работы с бороводородами следует1 вести п особой аппаратуре, которая полностью бы исключала доступ воздуха, влаги и паров, неизбежных при применении обычных вакуумных смазок.
Основным веществом, образующимся при разложении борида магния кислотами (лучше всего 8 и. фосфорной кислотой), является В4Н10 (тетраборан), конденсирующийся уже при 18° газ с своеобразным, вызывающим тошноту запахом и тотчас же загорающийся в жидком состоянии на воздухе. Это соединение также неустойчиво; постепенно оно разлагается с образованием главным образом В3Не (дпборан). Кроме этого, образуются менее летучие бороводороды, например В5Нв.
ВаНв, простейший бороводород, можно получить в чистом виде при нагревании В4Н10 при 100° в течение нескольких часов. Это газообразное (при температурах много ниже комнатной) вещество совершенно устойчиво в течение многих месяцев, на воздухе в газообразном состоянии не воспламеняется. В2Не не реагирует с сероводородом, но еще более чувствителен в воде, чем В4Н10.
Значительные количества B2Ha можно получить проще всего методом Шлезингера (Schlesinger, 1931) действием электрического разряда на смесь И 2 и ВС13 или еще лучше ВВг, (Stock, 1934). Под влиянием электрического разряда образуется вначале по уравнению 2ВВт34- 5Н2= В,НйВг Ц- 5ИВт .«онобро.иЭнбо^пн, который затем диспро-порционирует; 6В2П5ВГ = 5В2Н6-}_ 2ВВг3. В5Нц, ВйН9иВвН10 — бесцветные жидкости, значительно более устойчивые к действию воды, чём ранее указанные бороводороды.
При разложении В(П10 получается еще целый ряд твердых соединений, например Bf0Hf4, бесцветные кристаллы, летучие без разложения, нерастворимые в воде, но растворимые! в сероуглероде, а также пслстучие, твердые желтые соединения бора с водородом. Последние образуются в чистом состоянии под действием тихого электрического разряда на (Stock, 1936). Они нерастворимы в не разрушающих их растворителях (CS2, CCI5 и т. д.), имеют состав [ВЙ]Х и по свойствам и поведению близки нелетучим гидридам следующих за бором элементов: С, Si и Go (ср. стр. 450).
Строение и свойства бороводородов. Бороводороды можно разделить па две группы- с общей формулой ВпИп+4 (В2ИЙ, В4Н3, ВаЙ14, Bt(lITt4) и с общей формулой В„НП,6(В4Н 1(1, BDH||,B6Hf2?). Бороводороды второй группы менее устойчивы, чем первой. Общими для бороводородов обеих групп являются следующие свойства:
1) большая склонность к окислению (т. е. к замене водорода электроотрицательными элементами);
2) непасыщешплй характер: в противоположность бор ал кипам BR3 простейший бороводород имеет молекулярный вес, отвечающий формуле В2Не, а не BHS. В соответствии с этим в твердом состоянии он имеет в сущности такую же кристаллическую структуру, кат; л этан С2Щ (Mark, 1926),'однако В2Иа отличается от С2На некоторыми деталями своего строения, особенно в электронной конфигурации. Строение молекулы В2На и других бороводородов стало ясным после исследований главным образом Питцера, Прайса и Рендлля (Pitzer,. J, Am. chem. Soc., 67, 1126, 1945; Price, J. chem. Physics, 15, 614, 1947; 16, 894, 1948; Rundle, J. Am. chem. Soc., 69, 1327. 1947) и других (Dibeler, Mohler, J. Am. chem. Soc., 70, 987, 1948; S i 1 b i g с r, Bauer, 1. Am. chem. Soc., 70, 115, 1948).
Существование ВГ13 в димерной форме В2Пв объясняется тем, что в соединении В2Ые между двумя атомами бора существуют особого рода двойные водородные мостики, отличающиеся, однако, от обычных водородных мостиков своим неполярным харак-
Третья группа периодической системы 365
тором. Эту идею высказал еще Зеел (Seel, Z. Naturforsch., 1, 146, 1946). По его представлениям диборан существует в мезомериом состоянии
н .н И ИНН Н. .Н
в' в; в 'в; V V н' Н' Н н'“'н 'Н HZ 'ц/ \н
la 16 II
Формула II, имеющая с позиций современной теории валентности то же значение, что и 1а н 16, но являющийся упрощенной формой записи, была предложена еще в 1921г. Днлтоем (Dilthey). Шесть атомов Н образуют два несколько искаженных тетраэдра с одним общим ребром. Атомы бора, расположенные внутри тетраэдров, удалены одни от другого на 1,79 А, От наружных атомов водорода — на 1,18 А. и от мостиковых атомов Н — на 1,39 А. Наружные атомы н образуют с Относящимися к ним атомами Вугол, равный 120°, а с мостиковыми атомами Н, расположенными в плоскостях, перпендикулярных этому треугольнику,— угол 100° (Price, 1948).
Для В4Н10 п В5Нц можно предложить такие же формулы:*
Тетраборан В4НЩ, содержащий В—В-связь, можно получить, как это установил еще в 1926 г. Шток, реакцией, соответствующей синтезу Вюртца:
B2H5I 4- 2Na 4- 1В2Н5-^ВгН5 - В2И5’+ 2NaL
B5Hg, B^Hjg и Bj0Ht4. имеют, вероятно, циклическую структуру.
В2Нв способ ей присоединять NH3 с образованием B2H6-2NH3. llo-вндимому, речь идет об образовании продукта присоединения, а не аммониевой соли, ибо, как показал Бург (Burg, 1947), обмена протонами между В2Нв и тяжелым аммиаком ND3 ле происходит (см. т. II). В2Нв, следовательно, не имеет характера кислоты,. В4Н19 с NH3 образует Продукт Присоединения B4H10-4NH3. Термическое разложение соединений B2H0-2NH3 и ВлНю-йЙНз происходит с отщеплением Н2 и приводит к образованию Соединения B3N3He {боразол, см. дальше).
С диметил амином NH(CH2)2 при 200° В2Не взаимодействует количественно с образованием соединения BHo-NfCHg),, которое при комнатной температуре димерно (бесцветные, нерастворимые в воде, легко сублимирующиеся кристаллы, т. ил. 73,5°), но при нагревании полностью расщепляется на молекулы BH2-N(CH3)2 (Wiberg). Ненасыщенный характер диборана проявляется, например, в его способности при соединять щелочные металлы (которые целесообразно применять в вцде амальгамы). При этом, по Вцбергу, возникают солеобразпые соединения—борановые соли, например
В2Н0 2К=К2[ВгН6].
Если вместо щелочного металла подействовать гидроокисью щелочного или щелочноземельного металла, то с отщеплением водорода образуются дигидроксоборановые соли (ранее называвшиеся «гипобор атам и»)
В2И6 + 2МОН=М1[В2Н4(ОН)21+Нг.
В этом случае происходит не присоединение, а замещение двух электроне игральных атомов И двумя отрицательно заряженными группами ОН. Галоидоводороды также действуют па бороводороды замещающим образом
В2Н6 4- HC1=B3HSC1 4- Н2; В3Н5С1 4- НС1 = В2Н4С12 4- Н2,
Свободные галогены действу гот на бор ов одо роды вначале замещающим образом, цо при избытке галогенов все же протекает реакция полного окисления
В2Н5 -j- 6С12-> 2BC1s4- 6I1C1.
* Структурные формулы бор о водороде в, удовлетворительно описывающие в то время имеющийся экспериментальный материал, были предложены в 1940 г. Б. В. Некрасовым, Современные представления о строении бороводородов см. Липеном и др. Успехи химии, XXV, Яг 10, 1249, 1956— Прим, пеуев.
366
Глава 10
Частично галоидозамещенныё бороводороды легко дйспропорционируют с обратны» восстановлением бороводород а и образованием галогенида бора, например
6В2Н6С1 - 5В2Н6 -I- 2ВС13.
БОРОГИДРИДЫ МЕТАЛЛОВ (БОРОПАТЫ)
Во много раз большее значение, чем Упомянутые выше борановые соли, имеют металлоиорогидриды другого типа, для которого Вибергом было предложено название боронатов. Борогидриды этого типа были впервые получены Шлезингером (1940) при взаимодействии В2Нв с алкилами металлов, например
А12(СН3)в+4ВгЦ6 = 2В(СН3)3Н-2А1(ВН4)3. .
Таким же образом получаются другие металлоборогидриды этого типа. Кроме А1, они известны для Li, К, Na, Be, Mg, Zn, Cd,: Cu, Ag, Ga, Ti, Zr, HI, Th, U, Pu и Np. Большинство из них имеет солеобразную, но некоторые й не солеобразную структуру.
IIесолеобразную структуру имеют прежде всего соединения А1(ВН4)3 й Ве(ВН4)2, Молекула Ве(ВЫ4)2, но электронографическим данным Прайса (Р г i с е, J. Chem, Phys,, 17, 217, 1949) и Бауера (В ап е г, J. Am. chem. Soc.,68, 312,1946; 72, 622,,1864
3
Be Q а в н
Рис. 72. Структура молекулы Be(BH4)2, Расстояния: 1 ч—► в=1,71 А;
1 ч-+ 3=1,83 А; 2 ч—> 4=1,22 А;
2 ч—> 3=1,28 А; 3 ч—+ 4=1,03 А.
ф Д1 ©В ОН
Рис. 73. Структура молекулы А1(ВН4)з.
Расстояния: i ч—* 2=2,15 А; !•—>-3=2,10 Л:
2 ч—, г_1,28 А; 2 ч—+ 4=1,21 А;
3 ч—> 4=1,93 А..
1950), построена из двух искаженных тетраэдров ВН4, связанных с бериллием по: всей вероятности водородными мостиками (см/стр. 325, рис. 72).; А1(ВН4)3 имеет аналогичную структуру по только в пем с атомом металла связаны не две, а три группы ВН4 (рис. 73).
Гелеобразную структуру по рентгенографическим данным следует считать у станов л одной для ХаВН4 н LiBH4. В NaBH4 ионы Na+ n BHj расположены так же, как и ионы в решетке NaCln = 6,15 A (Soldate, J. Am. chem. Sec., 69, 987, 1947). Соединение исключительно устойчиво — в вакууме при: 400° ещё не-происходит его разложения. Li(BH4) кристаллизуется ромбически (с?=0,66), Каждый иОн Li в нем ркру-жеп четырьмя ионами [ВИД (Harris, М е i b oh ш, J. Am. chem. Soc., 69, 1231, 1947). По рентгенографическим данным Захариасепа солеобраЗную структуру следует считать установленной также и для U[BH4J4. '
Бороглдрид лития, боронат лития 1лВН4 — белое твердое нелетучее растворимое в воде й эфире вещество (т. ня. 279е). Его можно получить различными способами, лучше всего при действии гидрида лития на фтористый бор (Wintemitz, 1950; Wittig, 1951) или .эфиры борной кислоты (Schlesinger, 1950) согласно уравнению
4I.il!-! B.X3-l.iBH4-3LiX (X=F u.ni OR).
Третья группа периодической системы
’367
Бора лат лития гидрирует слабее, чем алапат лития. Его Применяют главным образом для приготовления боранатов других металлов
МС1„4-«LiBH4=M(BH*)n+nLiCL
С его Помощью удается также провести особенно простой синтез боразона B3N3He. 3LiBH44-3iXH4Cl -- BsNBHo ч- 0П24-3UC1.
(Schaeffer, Anderson, Schlesinger, 1949—1951).
При действии HCN на L1BH4 в эфирном растворе Виттиг (Wittig, 1951) полу* чил достаточно устойчивый циандборрнат лития Li [BH3(CN)J; Лёгко растворимый в воде н диоксане в кристаллизующийся из последнего с двумя молекулами растворителя.
Ворогидрнд натрия, боранат натрия Na[BH4], так же как и борапат лития,— твердое белое кристаллическое солеобразное вещество, образующееся при нагревании гидрида натрия с борным ангидридом
4NaH 4- 2ВЭО3 —3NaBO34*NaBH4. .
В чистом состоянии его удобнее получать обработкой тоиковзмельченпого гидрида натрия борномети.човым эфиром при 250’:
4NaH (- B(OCII3)3 -> 3Na/OCH34*NaBH4.
Эта реакция протекает так, что вначале' образуется натрийтриметоксибрранат NaHВ(ОСН3)я—г Ка[ВН(ОСЦ3)3] — белое твердое вещество,4 дйспропорциони-рутощее при 231г, с образованием б орана та и тетраметоксл борапата 5
4Na{BH(OCH3)3J=3NatB(OCHs)4]+Na[BH4}.
Более подробно об. этом соединении и о его применении в качестве восстанавливающего средства и для получения дйборапа, а также р других методах получения диборана и металлебороводородов см,: Schlesinger, J. Am. chem. Soc., 75, 186, 1953.
Борогпдрид магния, боранат магния Mg(BH4)3! ио дапным Виберга и Бауэра (Wiberg, Ваяет, 1950) получается при действии избытка боррводорода на диэтилмаг-пин в эфирном растворе
4BJI,.-! 3Mg(C2llE)2^3Mg(Bll4)2-r 2В(С2Н5)3.
Белое твердое вещество, нерастворимое в эфире, взаимодействующее с НС1 с образованием диборана Mg(BH4)24- 2HCl=MgCl24- 2Н24- В2На, разлагающееся метиловым спиртом с образованием твердого белого метокси производно го MgfB(PCH^)4)2, которое при сильном нагревании распадается па метилат магния и борнометилбвын эфир
Mg[B(dCH3)4]a=Mg(OCH3)2+2В(ОСНд)3.
Борогпдрид алгомппия А1(ВН4)3 впервые был получен: Шлезингером (1940) при действии ВаНв на Л1(СН3)3 по реакции
2бгне4-А1(СН3)3=Л1(ВН4)34-В(СН3)3. ;
Это соединение можпо получить также непосредственным взаимодействием АШ3 и В2Н);: 2А IHj 4- ЗВ2Н6 ~ 2А1(ВН4)3 (Wiberg, 1942) пли, что значительно удобнее, реакцией между галогенидом алюминия и борогидридом лития (Schlesinger, 1952)
AlX3-!-3l.i|BH4|=Al(BH4)3-b31 дХ. , - •
Борогпдрид алюминия — ле гко летуча я бесцветная жидкость; dJ=O,569, т. пл. —64,5Р, т. кип,. 44,5°, упругость пара при О’равна 119,5 лки pin. ст. Это самое летучее из всех' известных соединений алюминия) на воздухе самовоспламеняется и имеет чрезвычайно высокую теплоту горения (989 ккал]моль). Поэтому прп сгорании алюмииийборогцдрида выделяется 3750 ккал тепла:на 1кг смеси (А1В3Н424-4-6Ог), в то время как при сгорании углеводорода пентана С5Н12 практически с таким же молекулярным весом, с том же содержанием водорода и с такой же плотностью,.— только 25й0 ккал на 1 кг смеси (С5Н124-иО2). Это подсказывает возможность использования жидкого борогпдрида алюминия в качестве высококалорийного горючего, например в реактивных двигателях. С водой алюмпнийборогидрид энергично реагирует по уравнению
А1(ВН4)3 ф- 12НгО= К A120s3H2O4-3B(OH)s4-12H2 4- 192 ккал. ;;
Это соединение имеет не солеобразный характер. Структуру его см. на. стр. 366.
368 ’
Глава 10
БОРАЗОЛ, БОРОКСОЛ И БОРСУЛЬФОЛ
Боразол, При действии ЬН3 па В3Нв Виберг получил соединение B3N3H6 — боразол:
3BSII6-| 6NII3 = 2B3N3H6+12H2 г
—бесцветную подвижную жидкость с ароматическим запахом (т. кип. 55°, т. пл. —58,0°), горючую и растворяющую жиры, по физическим свойствам чрезвычайно похожую на бензол СсН6. Можно предположить поэтому, что боразол является ияостером бензола
Н н
А
/ ч %
НС СН HN NH
Е :l Е :1
НС СН НВ ВН
н н
Бензол j Боразол х
Знак----служит символом мезомерного промежуточного
состояния между простой и двойной связями.
Расстояние С ч—> С= 1,40 А, расстояние В ч—> N = 1,44 А.
То же имеет место и в случае аналогичных друг Другу продуктов замещения боразола и бензола. Например, метилаамещенные но В или N боразолы B3N3(CH3)3 по физическим свойствам чрезвычайно похожи на мезитилен (1,3,5-триметнлбензол), а гекса-метилборазол В31Ч3(СН3)е полностью соответствует мел лито лу (гекс аметилбе изолу). Но в химическом отношении боразол и его производные отличаются от бензола и его производных аначителвио большей реакционной способностью (W i b е г g Е., Вег., 73, 209, 1940; Z, anorg. сЬеш., 255, 141, 1947; 256, 177, 1948). Свойства, характерные для соединений с двойной связью, в боразоле выражены слабее, чем в бензоле. Степень двоесвязности составляет для боразола примерно 30% по сравнению с 50% в бензоле (Spur г, Chang. J. Chem. Pkys., 19, 518, 1951).
Боразапы, боразоны н бораинны. При получении боразола указанными выше способами в качество промежуточных продуктов образуются следующие соединения:
Н3В ч- NH3 H3B NIT2 НВ ^;NH
Боразан Боравев Боразин
Приводимые названия должны, по Вибергу, отражать ожидаемый изостеризм этих соединений с углеводородами ИВС — СН3 (этан), Н2С = СН2 (этен) и НС = СН (этип). Известны также метил замещенные боразапы и т. д. При исследовании спектров комбинационного рассеяния (Goubeau, 1952) удалось показать,, что в ряду боразан — боразол — боразан прочность связей В — N возрастает совершенно так же, как и прочность связей С — Св ряду этан — бензол — этилен. Этот факт можно рассматривать как подтверждение изостеризма обоих рядов соединений.
Боразоны имеют такой же ненасыщенный характер, как и олефины (алкепы). Но боразоны отличаются от олефинов, например, тем, что, присоединяя Г1С1, НВт и Н20, они пе присоединяют Вг2. Непасыщеипый характер бор азе лов проявляется также в их склонности (правда, только незначительной) к Димеризации, например
Н3СЧ Н3С сня
2 413 NH2 BZ В'
н/'7 н3с/ 4NZ >СН3
Но
Бороксол, Бороксолом называют соединения с общей формулой В3О3В3, содержащие, как и боразол, соответствующую циклическую систему. Основное вещество б про кс о ла В3О3Н3 пе известно, одйако различными путями можно прийти и его производным, Например, из: В2О3 и В(СНд)3 при нагревании под давлением до 300—330°
Третья группа периодической системы
369
(Goubeau, 1953) получают тримегпилбороксол В3О3(СН3)3 — бесцветную жидкость с т. кнп. 80° и т. ил.—38°. । Это же соединение можно получить из мегилборной кислоты СН3.В(ОН)2 путем отщепления воды (Snyder, 1938).
О зн3с-е—он — зн2о=н3с—bZ Хв-сн. ОН О (i
,« ХВ—С1Т3
Стабильность бороксолов падает с увеличением электроотрицательного характера групп, связанных с кольцом; в то время как алкилбороксолы относительно устойчивы, соединение В3О3 (У (СГГз)2]з (т. пл, 64°, т. кип. 224°) во влащном воздухе быстро гидролизуется: В3О3(ОСН3)3 (т. пл. ~ 10°) пе может быть перегнан без разложения; еще менее устойчивы галогениды B3O3F3 или В3ОвС13 (см. ниже).
В бороксоловом кольце, вероятно, осуществляется мезомеряое промежуточное состояние между следующими формами:
Для триметплбороксола В3О3(СН3)3 бороксоловая циклическая структура доказана Бауэром и Бичем (Bauer, Beach, 1940) электроне графически. Тацаки (Tazaki, 1940) сумел рентгенографически показать, что метаборпая кислота НВ03 или H3B5Ofi также обладает бороксоловой структурой. То же, но данным Губо (Goubeau, 1951), имеет место и для Соединения В3О3С13, называемого поэтому три хлор б о роке о.дом. Вероятно, эту же структуру имеет полученный Баумгартеном (Baumgarten, 1940) B3O3FB (трифтор бор оксол). Соединения В3ОЙС13 и B3O3F3 образуются из ВЙО3 и ВС13 или BF3 при нагревания (но устойчивы только при высоких температурах) по уравнению В2О34-
ВХ3;Й; BsO3X3. Вследствие этого они не .могут быть получены частичным гидролизом галогенидов бора.
Борсульфол. Если мысленно заменить в бор оксоле атомы О на S, то получится циклическое соединение с общей формулой B3S3RB, которое Впберг назвал борсульфол. Основное вещество борсульфола В383Н3, так же как и бороксола, не известно. Производными его должна, ло-видимому, являться метатиопорная кислота B3Sj(SH)5 и некоторые другие соединения,, полученные Вибергом (1953), - формулы которых приведены ниже. Метатиоборную кислоту удается получить простейшим образом при действии избытка сероводорода на бромид бора:
ЗВВМ- 6H2S=B3Ss(SH)34-9HBr.
П/ иотатдоборпой кислоты при взаимодействии с бромидом или хлоридом бора получаются соответствующие галогепоборосульфолы. При взаимодействии с метиловым эфиром борпой кислоты получается триметоксиборсульфол. Этим же способом удается iipiriiTii к аминос седине ни ю, имеющему характер ангидрооснования и образующему с ИВг прекрасно кристаллизуюпщеся соли. Водой борсульфолы гидролизуются, по опи растворимы в индиферептных растворителях, Таких, как сероуглерод или^беидаг. По иояпжопггк) температур замерзания растворов вычислены молекулярные веса, соответствующие следующим формулам:
।
8Н
I GI
S HaCOBZ ВОСНз
4 s
I ОСНп
Метатиоборпан кислота Трих.торборсуль- Триметонсиборсульфол .. фол
(HaChNB BN(CH3)3 I I s s
чвх I N (CHs)a T ридиметиламипоборсульфол
24 г. Petal
370
Глава 10
ГАЛОГЕНИДЫ БОРА
Фторид бора и фтороборпая кислота. Фторид бора BF3 — бесцветный,_ удушливо пахнущий гав. Удельный нос 2,37 (воздух принят за 1), вес 1 л-равен 3,07. Т. кип. —101°, т. пл. —128°. Фтористый бор не горюч, но разлагается водой (см, ниже) и поэтому дымит во влажпом воздухе. Его получают при нагревании борного ангидрида с плавиковым пшатом и концентрированной серной кислотой (1). В чистом состоянии его лучше всего готовить методом Хелльригеля (Hellrjgel, 1937) нагреванием фторобората калия КВ F4 с борным ангидридом (2);
В2О3-ЬЗСаГ2+ЗНг5О4=2ВГ34-ЗСа5О4+ЗНгО1 (1>
K[BF4)+2BaOa=K[B4O6F] + BF3. (2)>
При разложении фтористого бора водой образуются борная и фторобор-нал кислоты
4BF3 -И ЗН2О = Н3ВО3 -р ЗИВК4. (3).
Реакция протекает таким образом, что вначале вследствие гидролиза образуются борпая кислота и фтористый водород (4), который может соединяться с езде неразло-жнвшимся фтористым бором, образуя фториборную кислоту (5). Частично, однако, он реагирует с выделившейся вследствие гидролиза борной кислотой с образованием гидроксофтороборной кислоты (6). Последняя может вследствие реакции с другой-частью фтористого водорода давать в соответствии с уравнением (7) опять-таки фто-роборную кислоту •„
6F3-|-3HOH = B(OH)3+3HF (гидролиз), (4)-
BF3-|-HF=HBF4 (образование фтороборпой кислоты)] (5)и
B(OII)3+3HF-HB(OH)F3-h2H2O (образование гидроксофтороборной кислоты), (6
ПВ(О11)1'з ГНК “i 11ВГ-- 112О. ;< (7>
Последняя реакция приводит к равновесию. Соответствующая ему константа гидролиза ПВГ4 равна 2,3-10 “3 (при 25®). Калиевая соль гидроксо фторо борной кислоты K[B(OH)F3] лучше растворима в воде и значительно быстрее гидролизуется, чем К[ВК4]. Однако ее можно расплавить, не вызывая при этом разложения (Warn-scr Сп. A., J. Am. chem. Soc., 70, 1209, 1948).
Фтороборпая кислота HBF4 является сильной Кислотой, значительно более сильной, чем плавиковая. На холоду или в водном растворе НВF4 па стекло не действует. При нагревании с водой она даже разлагается,., образуя оксифтор сборные кислоты. Большей частью хорошо кристаллизующиеся соли — фторобораты, многие из которых были получены еще Берцелиусом,— можно приготовить либо растворяя соответствующие-окислы металлов, гидроокиси металлов или карбонаты в водном растворе фтороборной кислоты или внося в плавиковую кислоту фториды металлов, и борную кислоту. Фторобораты большей частью легко растворимы за исключением фторобората калия, выпадающего в виде Студенистого осадка при насыщении фтороборной кислоты гидроокисью калия. Фтороборат, калия KBF4 после высушивания представляет собой белый порошокудель-ного веса около 2,5. Его можно перекристаллизовать из воды, однако, находясь долгое время в растворе, он постепенно гидролизуется, о чем свидетельствует кислая реакция водного раствора; При прокаливании фторобораты разлагаются с отщеплением фтористого бора.
Щелочные фторобораты изотипны щелочным перхлоратам (Hoard J. L., J. Am, cheni, Soc., 57, 1985, 1935; К linkonberg L. J., Rec. Trav. ch., PaVs-Bas, 56, 36, 1937). ’To же относится и к гидроксо фторо боратам, например к Na(B(0H)F3J. Соединение BFj -211ч О следует рассматривать, как гидроксофтороборат гидроксония [0H3nB(OH)F3J (Klipkenberg, 1935), соответствующий перхлорату гидр оксон ня (см. стр. 863). г*
Третья группа периодической системы 371:
Помимо фтороборатов щелочпых металлов, известны фторобораты щелочноземельных и тяжелых металлов. Фторобораты двухвалентных металлов в соответствии е опытными данными устойчивы только в виде гидратов, аммиакатов и т, д,: На основе цикла Борна устойчивость их можно рассчитать теоретически (do Boer, van Liempt, 1927), Фтороборат калия был предложен в качествс плавня, Фтор оборвал кислота ядовита и в очеш, разбавленном состоянии даже препятствует брожению, ...
Помимо HF, BF;1 соединяется также с сухим аммиаком. При этом образуется продукт присоединения BF3-NH3 (теплота образования при 0° равна 41,3 хл-ал), растворимый без разложения в воде (растворимость 36,0 г в 100 г воды нри 25s), При нагревапии выше 125; это вещество разлагается по уравнению
4BF3-NH3= 3[NH4HBF4]>BN ' : 1
(L a u b е в g а у е г, I. Аш. chem. Soc., 70, 2274, 1948). Значительно легче, чем это твердое соединение, разлагаются жидкие продукты присоединения аммиака BF3-2NH3 и BFS'.8NH3, Органические амины я окись азота также присоединяются к BF3. Продукт присоединения с РН3 устойчив только пиже компатнрй температуры.
Молекула BF3 построена плоско, так же как и соединения В(СН3)Г2, В(СНэ)аЕ и расстояния В *--* F=l,30 А, В ,—,С=1,56 А (Вгапле, 1937; Brockway;
Cti3
1937; Bauer, 1942). Продукт же присоединения B(CH3)3-.NH3 или НзС — в<-NHS сн3 .
имеет тетраэдрическую структуру. Аммиак не влияет на прочность связи В — С, в то время как при переходе от BF3 к [BF4]“ связи В — F ослабевают. В ионе [BFJ-расстояние В «—>F составляет 1,43 А. Увеличение этого расстояния по сравнению с 1,30 А. в молекуле BFS соответствует уменьшению валентно-силовой константы i*pF от 6,86/105 до 5,28-Ю5 дип/см (Goubeau, 1952). Это значительное уменьшение становится приятным, если предположить, что IBF4]_ изостерен CFy, тогда как в BF3 существует одна двойная связь, мезомерно распределенная по трем В — F-связям
:р; rF:
F: С : F-: :F -В ;FrB::F
; F : ;F-’
L " -J (Три мезпыерные формы)
В пользу этого предположения говорит наблюдение Губо, что в соединениях В(СНЕ)3 и Н3Й.В(СП3)а связи В—С не различаются пр прочности. В В(СЙ3)3 не могут существовать двойные связи, так как радикал СН3 не может предоставить требуемой для этого второй электронной пары. На основе этой гипотезы возможно объяснение димеризации СН3О- ВР2 п неустойчивости (CH3O)2BF.
Фторид бора используют как катализатор для введения HF в ненасыщенные органические соединения. Кроме присоединения Нй\прн этом может происходить также обмен других атомов галогенов на F (Н е n n е, Arnold, J. Am. chem. Soc., 70, 758, 1948).
Хлорид бора. ВС13, получаемый при нагревании бора в струе хлора или сухого хлористого водорода, а также при пропускании хлора над раскаленной смесью борного ангидрида и угля, представляет собой бесцветную, легко подвижную, сильно преломляющую свет, во влажном воздухе дымящую я;идкость. Удельный вес его 1,43, т. Kini.(Mazzetti) 17,5—18,5°, т. пл. —107° (Stock). Экспериментально установленная плотность пара 4,065 (плотность воздуха ~ 1), откуда следует, что ВС13 в парообразном состоянии мономолекулярен (найденный молекулярный вес 117, рассчитанный — 116,0).
Водой хлористый бор разлагается с отщеплением хлористого водорода
£5С13^ ЗНОН=:В(ОН)э + ЗНС1 (гидролиз). (8)
Другие кислороде оде рщ а щио соединения также вызывают обмен хлора па кислород; так, сорный ангидрид при 120° действует по уравнению
2BCl3-4-3SO3= ВгОа + 38ОгС1г.
24*
372
Глава 10
Хлористый бор, так же как и фтористый бор, склонен к образованию продуктов присоединения. Такие соединения получены прежде всего с хлоридами — с ₽CI3, РОС13, NOGL Аммиак действует замещающим- образом, отщепляя FIC1, например:
ВС134-3NHS= B(NH2)3 + 3IICI или BCI3-|-6NHs= B(NII3)3+ 3NIRCI.
Взаимодействие хлористого боря со спиртами приводит к обмепу атомов CI на спиртовые остатки RO— с отщеплением г я.то генов о до рода, например;
BCls+3C2HsOH=B(OC2n5)j + 3HC] (алкоголиз). . (9)
Обмен происходит ступенчатым образом. Так, могут быть полу чета В(0СН3)С12 борметоксидихлорид (метиловый эфир дпхлороборпой .кислоты), т. пл.—15,0°, т. кип. 58,0й. В(ОСНз)2С1, бордиметоксихлорид (диметиловый эфир монохлороборпой кислоты). Т. пл. —87,б3, т. кип. 74,7"; В(ОСН;5)3 — тримотиловый эфир бор пой кислоты, т. йл. — 29,0°, т. кип. —08,7°. Эти соодипония представляют Собой прозрачные жидкости, разлагающиеся водой на И,зВО3 и СН3ОП, а также НС1. Частично алкокси-
’ Лированные хлориды диспропорциопиругот ио уравнениям
2B(OR)C12^±BGIS4-B(OR)2C1, z (10)
2B(OR)2C1«*B(OR)C124- B(OR)3, (11)
если равновесия (10) и (11) сдвигать вправо добавлением веществ, образующих продукты присоединения (Wiberg, 1931 и 1935). В противном случае эти соединения даже при температурах кипения весьма устойчивы. С окисью ртнЛена СдЩО хлористый бор (SchtnelBer, 1952) реагирует с образованием эфиров С12В—ОС2Н4С1, С1В(ОС2Н4С1)3 и В(ОС3Н4С1)3, из которых два первых чрезвычайно легко диспропор-цнониругот. Таким же образом реагируют с окисью этилепа AsGl3 и SiCl4.
Диборгеграхлорпд. В2С14, слито.'пгрм аплы й впервые Штоком (Stock, 1929), легче всего получить действием тихого электрического разряда на нары ВС13. Это жидкое вещество, лары которого обладают плотностью, соответствующей формуле В2С14, При обычных температурах В2С14 разлагается, но только медленно; под действием горячего раствора NaOH разложение происходит мгповеппо но уравнению
ЗВ2С]4-|-18ОН'=2В3Ов +12С1' + 6Н2О+ ЗП2.
В воде ВЧС14 растворим без выделения водорода. Раствор имеет сильные восстапо-витольные свойства, и предполагают, что в пем присутствует образующееся вследствие гидролиза соединение В3(ОН)4,
Бромид бора. ВВт3 — бесцветная, сильно дымящая тяжелая жидкость; уд. вес 2,65, т. кин. 90,53 замерзает при —46е, Теплота образования 43,2 ккал/молъ. По споим свойствам очень близок хлориду. ВВтк, так же как и ВС13 и BF3, построен н.иско, три атома галогоиа обранутог равносторонний треугольник с атомом В в цотттре. Расстояния В*—>С1=1,73 А, В > Вг=Л,87 A, Cl*—>С1 = 2,9ЭА, Вт^Вг-3,25 A (F^F=2,25 А).
Иодид борд. BR — при обычной температуре твердое вещество, Оп образует Пластин чатьте, бесцветные, очень гигроскопичные кристаллы; т. пл. 43°,т. Кин. 210’; плотность в жидком состоянии (при 50й) равна 3,3; растворяется в сероуглероде, четыреххлпр истом углероде и бензоле', в струе кислорода горит Светлым пламенем. Способность к образованию соединений присоединения, так сильно выращенная у оогалг.т.гг галогенидов бора, у подпетого бора проявляется слабо.
* Под действием тихого электрического разряда В13 разлагается. Прн этом образуется главным образом дибортетраиодид Ва14 — бледно-желтое кристаллическое вещество, при обычной температуре медленно разлагающееся па В13 ц черпый, высо-вд пол и мерный мопоиодид бора (В1)х. Водный раствор В13 обладает сильными восстановительными свинствами. Например, прн действии его на азотнокислое серебро, помимо йодистого серебра, выделяется также металлическое серебро. .
Боралкилы. При взаимодействии галогенидов бора с цинкалкплами получаются боралкилы BR3 (R —алкил), например:
2BCl3+3Zn(CJl3)2=2B(CH3)3+3ZnCl2.
Триметилбор В(СН?)Э —бесцветный газ, триэтилбор В(С2Н5)?— жидкость, кипящая при 95°. На осповании определения плотности пара для
Третья группа периодической системы
373
этих соединений установлены мономолскулярныо формулы. Для приготовления их лучше всего, по Э. Краузе, использовать фтористый бор' действуя па него органическими соединениями магния в эфирном растворе, Боралкилы чрезвычайно легко окисляются. Производные низших углеводородов загораются на воздухе. В запаянных ампулах они устойчивы. При медленном доступе воздуха боралкилы окисляются в алкилбороксиды R-ВО, которые растворяются в воде с образованием алкилборных кислот R‘B(OH),2. Последние очень легко кристаллизуются, чрезвычайно летучи с водяным паром, отличаются пряным запахом и сладковатым вкусом. Растворимы в органических растворителях, в том числе и в жирных маслах. Спиртовый раствор азотнокислого серебра эти кислоты восстанавливают только Jiрн нагревании. Аналогичные свойства обнаруживает трифенил-бор В(С6Н5)3. Эфирный раствор его при внесении металлического натрия постепенно опрашивается в интенсивный рубиново-красный цвет и затем выделяется из него в виде желтых кристалликов трифенилборнатрий NaE(CeHs),.
Кислородные соединения бора
Борный ангидрид. Бг03, продукт горения бора, обычно получают при прокаливании борной кислоты. В2О3 — бесцветная, хрупкая, стекловидная, гигроскопичная масса с удельным весом 1,844, плавящаяся или размягчающаяся при красном калении с образованием вязкой, растягивающейся в нити массы. Борный ангидрид очень устойчив к нагреванию, не восстанавливается углем даже при белом калении, одиако разлагается, если одновременно с углем действовать на него веществами (например, хлором или азотом), которые могут замещать кислород. Борный ангидрид не проводит электрического тока, па вкус горьковат. Б воде растворим с сильным разогреванием. 100 г борного ангидрида, внесенные в 125 г воды, доводят ее до кипения. Сильный тепловой эффект зависит от того, что окисел образует с водой борную кислоту
1/21В2б3]4-3/2НгО-|-ад = Н3ВО3-ач-Ь4,0 ккал.
Указанный топленой эффект имеет место ври растворении в большом количестве воды. Если растворить борный ангидрид в небольшом количестве воды, то выделение тепла оказывается значительно большим, так как теплота разведения для борной кислоты сильно отрицательна. Теплота растворения [Н3ВО3/в большом количестве воды составляет—5,1 ккал/моль; теплота образования [Н3ВО3] из V2 В2О3 и 3/г Н2О оказывается, таким образом, равной +9,1 ккал/молъ. НВО2 существует в нескольких модификациях. Для р- [НВО2] теплота образования составляет +4,4, теплота растр оретгия —0,56 ккал/моль. (Stack elb erg, Roth, 1937). *>
Борный ангидрид застывает обычно в виде стекла. В кристаллическом состоянии его можно получить только при отщеплении воды от а-НВ02. Обезвоживанием Н3ВО3 получают обычно стекловидный В2О3, так как это обезвоживание, как правило, проходит через промежуточные стадии образования fl- или у-НВО2 (см. стр. 374). Кристаллический борный ангидрид (d—2,460, т. пл. 450°) оптически од но о сен и имеет ромбоэдрическую решетку. Стеклообразный В2О3, по Рихтеру (Richter, 1953—1954), имеет слоистое строение с расстоянием между слоями 1,85 А. В этих слоях атомы кислорода располагаются главным образом в вершинах равносторонних треугольников, центры которых занимают атомы В. Расстояние В -4—к О составляет 1,45 А. Для элементов Si, Ge, As, Sb и Se в аморфном состоянии, а также и стеклообразного SiO2 оныты но рассеянию рентгене неких лучен также показали замечательно высокую упорядоченность атомных положений.
Моноокись бора. Если элементарный бор нагревать в смеси с двуокисью циркония, то он испаряется, как показал Цинтл (Zintl, 1940), в форме моноокиси ВО (ср. также Klemm, Z. anorg. Chem., 255, 293, 1948). Существование молекул ВО в газовой фазе было установлено еще в 1925 г. Малликеном (Mulliken) при иеследоваили полоса
374
Глава 10
тых спектров. Химическое сродство реакции Вф-О=ВО составляет около 210 ккал/моль, тогда как для: реакции А1~[-О=А1О (установленной также спектроскопически) — только около 87 каал/молъ.
Борная кислота. Обычная борная кислота или ортоборнал кислота Н3ВО3 — чешуйчатые, белые, прозрачные шестигранные пластинки удельного веса 1,46; получается обычно при добавлении соляной или серной кислоты к раствору боратов, чаще всего буры. Она легко кристаллизуется из горячей воды. Растворимость прп 0° составляет 19,47, при 20° — 39,92 и при температуре кипения насыщенного раствора (102°) 291,2 г Н3БО3
на 1 л. Сильное увеличение растворимости в зависимости от температуры объясняется большой отрицательной теплотой растворения (см. выше). С водяными парами борная кислота летуча. Этим объясняется ее присутствие в водяных парах, которые в смеси с разными газами вырываются из земли в вулканических местностях Тосканы и называются там «соффио-- ни» или «фумаролы».
Борная кислота содержится в минеральных водах, а в незначительных количествах — в ягодах, фруктах, хмеле, а иногда и в вине.
Борная кислота очеиь слабая кислота. Удельная электропроводность 0,94 н. раствора составляет И[8=1,1-10"6. Константы диссоциации равны: Кц= 7,3-10-1(|, й?2=: 1,8>.10_и, А3 = 2-10“и (цри 18°). Таккак диссоциация первой ступени очень мала, то гидролизуются даже од поз вмещенные соли борной кислоты. Трех замещенные; соли нельзя получить из водных растворов.
I.
При нагревании борпая кислота, так же как и расплавленный борный ангидрид, легко растворяет окислы металлов с образованием/солей. Летучие кислоты вытесняются ею из солей. С кремневой кислотой она образует комплексные силикаты (боросиликаты), разлагающиеся соляной кислотой. Поэтому ее используют в аналитической химии для определения силикатов. Так как она образует со спиртами легко летучие эфиры [например, с метиловым спиртом СН3ОН — метиловый эфир борной кислоты Б(ОСН3)3[, то ее легко можно удалять из анализируемого вещества.
Эфиры борной кислоты образуют с аммиаком и органическими аминами двойные соединения. Простейшие из пнх относятся к типу R3lV'B(OR)3, и их можпо; считать производными боразаиа (см. стр. 368). Кроме того, образуются также продукты присоединения более сложного состава (Gotibeau, 1951). Если эфир борной кислоты нагревать с триэтаноламином N(C2H4OH)3, то образуется триэтаноловый эфир борной кис-’лоты N(C3H4O)3B — ромбические кристаллы с т. пл. 231°. Это соединение хотя и растворяется в нитробензоле мономолекул ярко, но все же очень склонно к полимеризации. Водой оно частично гидролизуется. По отношению к кислотам и ансольвокислотам •-(например, SnCl4, SbCl3, HgCl2) оно ведет себя как апги др do снование,- по присоединение происходит только к атому азота, в то время как в аналогичном соединении алюминия N(C2H4O)3AI атом алюминия также способен присоединять другие вещества (Hein, 1952).
Большие количества борной кислоты, используют для приготовления буры, которую прежде в основном и получали из борной кислоты. В настоящее время главным потребителем борной кислоты является производство эмалевых изделий. Благодаря дезинфицирующим свойствам борную кислоту применяют в медицине для присыпания ран, для пропитывания перевязочных материалов и как средство против сильной потливости. Кроме (того, борная кислота входит в: состав различных лекарств. При производство свечей ею пользуются для придания жесткости фитилям; наконец, её применяют дця дубления кож и при производстве минеральных красок. „ "
Третья группа, периодической системы ... ...... '‘'316
При нагревании борная кислота теряет воду и переходит в метабор-лую кислоту НВО2. При растворении в воде метаборная кислота сразу же переходит в орт.оборную. Дальнейшее отщепление воды при нагревании приводит к стеклообразному, содержащему воду В2Оа, без образования в качестве промежуточной ступени тетраборной кислоты Н2В4О;, которая известна в основном только в виде своих солей.
В очень незначительных количествах тетраборпая кислота присутствует в равновесии с мопоборной кислотой HSBO3 в водных растворах последней. По Титесену .{ThygeseTi, 1938) в 0,6 М растворе Н3ВО3 молярные концентрации не диссоциированной тетраборной кислоты составляют 3,5-10-6 и иона тетрабората — 8-10 s, в то время как концентрация лона мояобората вследствие чрезвычайно слабой диссоциации моиоборпой кислоты пе превышает 4-10‘в. Однако с разведением равновесие быстро сдвигается в сторону нона монобората. Например, в 0,1 М растворе его концентрация равна 3- 10 е, тогда как концентрация иона тетрабората надает до 3-10~в и недис-•соции ров липой тетрабррной кислоты — до 6 10“в.
Мета борная кислота. НВО2 существует в трех модификациях. Это становится яспым из диаграммы растворимости, изученной Краденом, Мореем и Мервином (Кга-«сек, Morey, Merwin, 1938). Соотношения между модификациями оказываются такими же, как и для СаС1г-4И2О (рис. 13, стр. 97); из раствора соответствующей концентрации при охлаждении обычно выделяется не стабильная а-модификация,, но менее всего ‘Стабильная у-НВО3, которая медленно превращается в JCмодификацию. Превращение последней в a-модификацию происходит при длительном (в течение нескольких . недель) нагревании в автоклаве. Таким образом, получается, что Н3ВО3 копгруентно плавится при 170,9°, хотя, как это видно из рис. 74, расплав этого состава нестабилен и должна была бы выделиться ci-HBOj. а-НВ02 (кубическая, 8=2,486) плавится ко игру епт по при 236°. Четверная точка, в которой находятся в равновесии четыре фазы; а-НВ02, кристаллизующийся В3О3, раствор и водяной лар, лежит при 235° и 1,9 атм. Упругость водяного пара в 1 атм Над <х-1[ВОа достигается при 225°. Для обеих метастабильных модификаций температуры, при которых давление водяного пара над пи.ми достигает 1 атм, случайна лежат вблизи температур илавлення. $-НВО2 (моноклигпгая, Ц=2,044) плавится конгруентно при 200,9°, у-НВО2 (ромбическая, 8—1,78) — также конгруентпа при 176,0°. То, что обе модификации хоти я пестйби.тьш.г, но плавятся конгруентно, весьма удивительно. Это позволяет предположить, что существующее в них расположение атомов при плавлении в значительной степени остается неизменным. С химической точки зрения это должно означать, что различные формы метаборной кислоты следует считать различными сгруктур-поизомерными соединениями. Для них, по-вйдимому, должны рассматриваться такие же соотношения между изомерами, как и установленные7 для мета фосфор ной кислоты -(ср. стр. 681. и сл.). В этой связи замечательно наблюдение Крацека (Kracek) и его сотрудников о том, что у-НВОа в противоположность другим модификациям отличается чрезвычайно большой расщепляемостью (перпендикулярно оси с). Достаточно уже ггебольшбго давления, чтобы расщепить кристалл на тонкие листочки.
С водяным паром летуча только кислота Н3ВО3. Даже ИЗ очень концентрированных растворов, равновесных с твердой НВ021 улетучивание происходит в форме ортокислоты. Чрезвычайно небольшая летучесть НВО2 и В2О3 объясняется тем, что они в противоположность Н3ВО3 в мот го молекулярном состоянии координационно нена-•СЕПцоны (S tack elb erg, 1937).
Борная кислота растворима во многих органических растворителях, особенно в гндроксилсодержащих. Электропроводность водных растворов борной кислоты повышается при добавлении органических веществ, и особенно многоатомных спиртов, таких, как маннит, дульцит, глицерин. 'Сила борной кислоты вследствие таких добавок возрастает настолько, что ее можно титровать в этом случае с применением в качестве индикатора ‘фенолфталеина. Это объясняется образованием комплексов с органическими веществами. Подробнее об этом см. в следующей главе. Органические вещества, и в первую очередь кислотные остатки, также способны присоединиться к борной кислоте с образованием так называемых гетерополикислот *(с.м. т. II).
376
Глава 10
Соли борной кислоты. Борнокислые соли — бораты большей частью производятся не от нормальной борной кислоты или метаборной кислоты, но от других форм с меныним содержанием воды, существование которых в свободном состоянии не мотет быть доказано. Примерами таких солей служат упомянутые в первом разделе минералы бора. Наиболее часто встречающиеся среди них паходят применение для приготовления технически наиболее важного соединения бора — буры.
Саде:'усачив 5;03, аг ал К
Рис. 74. Растворимость в системе ЕЧСЦ—НаО.
Бораты щелочных металлов довольно легко растворимы в воде, причем растворы вследствие происходящего гидролиза обладают сильно щелочной реакцией. Остальные бораты в воде трудно растворимы.
Многие бораты, подобно борному ангидриду, застывают из расплава в виде стекла. ЛитИй-бериллииборатные стекла применяют для изготовления окон рентгеновских трубок и капилляров, для рентгеиоструктурпых определений методом порошка (линде-манонекое стекло). Хорошая проницаемость литий-берилл ийборатных стекол для. рентгеновских лучей объясняется низкими порядковыми номерами входящих в их состав элементов (ср. стр. 241). Систематическое исследование системы Li20—ВеО — — В203 было проведено Менделем (1942). О метаборате натрия и его гидратах см. М е nfz е lj Н., Z. anorg. Chem., 251, 167, 1943.
Бура. Na2B4O7-ЮН2О — натриевая соль тетраборнои кислоты (см. стр. 375) — образует большие, бесцветные, прозрачные моноклинные)кри-сталлы, которые в сухом воздухе с поверхности выветриваются. Удельный вес 1,72. Водный раствор обнаруживает сильно щелочную реакцию-н поглощает на холоду на 1 моль буры 1 моль двуокиси углерода. Нри< нагревании двуокись углерода выделяется. Растворимость буры в воде-
Третья группа периодической системы 31Т составляет;
при 0° 10° 30° u(j° 80° 100° 1
1,23 1,58 3,75 16,7 23,9 34,3 8 Na2B4O, в 100 8 раствора
Бура встречается и Тибете под названием тинкал, а также в Калифорнии. Прежде тибетский тинкал имел бол иное значение. Но в настоящее время основное количество буры получают из других минералов, прежде-всего из кернита (см. стр. 359).
Кроме обычной буры —декагидрата тетрабората натрия, существует еще пентагидрат Na2E4O7-5НаО, называемый октаэдрической или ювелирной бурой. Он кристаллизуется при высокой температуре (выще 60е), тогда как прн комнатной температуре выкристаллизовывается обычная призматическая бура. Na2B4O7 5Н2О кристаллизуется в гексагональных ромбоэдрах и имеет удельный вес 1,81. Пептагидрат тверже обычной буры и пе выветривается.
При обезвоживании бура ведет себя по-разному, смотря иб тому, была ли опа. предварительно умеренно нагрета (до 50°, например, при сушке) или нет. В первом случае она обладает при 20° давлением пара 10 -и.и рт ст и переходит при обезвоживании обратимо в пептагидрат. Последний обратимо теряет воду до дигидрита, из которого затем при дальнейшем удалении воды возникает моногидрат и, наконец,— безводная соль. Если буру предварительно не нагревали, то^давленис пара над ней при, 20° составляет только 1,6 мм рт ап и она, следовательно, в обычных условиях совершенно устойчива иа воздухе. В вакуум-эксикаторе над Р^О5 она очень медленно необратимо переходит в дигидрат. ТеграгидрЯт, встречающийся в виде минерала (кернит)^ не образуется при обезвоживания дека- или пентагидрата, по может быть получен: из растворов. Подробнее об условиях образования тетрагидрата (кернита) и о соотношениях, в которых стабильны гидраты различного состава, см л Menzel И., Z. anorg. Chem., 245, 157, 1940.
Безводный тетраборат натрия Na2B4O7 (удельный вес 2,37) получается при нагреван ии буры до 350—4.00°. Плавления буры в собственной кристаллизационной воде можно избежать при! осторожном нагревании. Тетраборат натрия плавится при 878°, переходя в стекловидную массу. Расплав, способен растворять окислы металлов, что используют в аналитической химии (перлы буры).
Бура находит широкое применение для изготовления легкоплавкой глазури для фаянсовых и фарфоровых изделий и главным образом для железной посуды (эмаль); кроме того, ее ^-применяют в производстве особых, стойких при изменении температуры сортов стекла (например, ламповое стекло) и в производстве оптических стекол. Благодаря способности растворять окислы металлов буру применяют при паянии и сваривании (ювелирная бура). Большие количества се идут для стирки. Различные применения буры основаны на ее консервирующем действии. Большое значение опа имеет в качестве доходного продукта для получения соединений боратов с перекисью водорода (пероксигидратов боратов).
Б лаборатории буру применяют пе только для получения перлов, но н в 'качестве исходного вещества для установления титра кислот, а также при приготовлении буферных растворов (ср. стр. 268).
Г
Пероксобораты и перокепгидраты боратов. Пероксобораты являются производными боратов при замене в них кислорода на содержащуюся в перекиси водорода перекисную группу —О—О—. Пероксобораты ранее-называли перборатами. Большинство соединений, называемых перборатами, в действительности являются нс истинными пероксосолями, но-всего лишь пероксигидратами боратов, т. е. продуктами присоединения:
378
Глава 10
На0» к обычпым боратам. Их получают обычно при действии йа водные растворы боратов перекисями щелочных металлов или щелочными растворами перекиси водорода. Только в отдельных случаях этим способом можно получить истинные пероксобораты.
Так как вследствие гидролиза истинные пероксобораты также распадаются в водном растворе, выделяя Н2О2, отличить их от двойных соединений с перекисью водорода далеко не просто.
В большинстве случаев для этого используют реакцию Рцзепфельда*, позволяющую отличать пероксосоли от пероксигидратов на том основании, что настоящие пероксо со ли в отличие от П2Оа, а также продуктов ее присоединения выделяют 12 из нейтральных растворов К1 уже на холоду. Так как эта реакция может тормозиться возникающими при гидролизе попами ОН', то к исследуемому раствору целесообразно добавлять фосфатпый буфер (Liebhafsky, 1934). Но даже и в этих: условиях реакция Ризенфельда по указанным выше причинам не пригодна для отличия пероксоборатбв от пероксигидратов боратов. Среди типичных пер оксоборатов с достоверностью известны; ХаВОз (полученный Ло Бланом при смешивании Na О~-ОН с Н5ВО3), KBOr JiHsO .и (NH4)BO3- у, Н2О. Последние имеют, вероятно, следующее строение;
М2
ГНОО
ZOOHT
В- () — В/ (М = К, NH4).
Tlo-видпмому, существуют настоящие пер оксобораты состава М1ВО4-НаО и MLBO5: Н2о -(Partington, 1949),
Так как пероксигидраты боратов в водных растворах разлагаются с выделением перекиси водорода, то они оказывают совершенно такое же действие, как и сама Н2О2, — легко отщепляют кислород, передавая его другим веществам. На этом основано применение «перборатов» (пероксн-гидратон боратов) в промышленности в качестве средства для отмывки .и отбеливания шерсти, шелка, соломы, слоновой кости и многих других .веществ. Обширное применение находят они также и в косметике (для обесцвечивания волос), а также в качестве дезинфицирующего средства. Б смеси с веществами, каталитически разлагающими с выделением О2 пербораты или выделяющуюся при их растворении перекись водорода, их используют для получения кислорода. При работе с пероксигидратами -боратов, содержащими более 15% активного кислорода, необходима осторожность, так как они при трении способны разлагаться со взрывом. Одним из самых известных перрксигидратов боратов является пербура Na2B4O--НаО2 9НвО (рациональное название**; тетраборат натрия-перекись водо-рода-9Н2О) — белая кристаллическая соль, которая получается при растворении смеси борной кислоты с перекисью натрия в воде
4Г1зВОзЦ-Ца2О2 — Na2B4O2 *Н2О2~|-5Н2О-
* О других методах отличия пероксосолей й пероксокислот от пер окси гидратов •см.: LeBlanc, Z. Elektrochem.,29, 193, 1923; М en.ze 1, Z. phys. сЪещ., 105, 402, 1923. Некоторые пероксигндраты боратов При растворении в воде пе отщепляют 112О2, которая зачастую остается в растворе комплексно связанной с ионом бората. Тем самым еще более затрудняется отличие пероксигидратов боратов от пер оксо боратов; поэтому в строении отдельных «перборатов» еще много поясного. Целесообразно проводить различия между соединениями, в которых доказано наличие комплексно связан-ной С ионом бората Г12О2 — иероксчеидпатоборатами,— и пор оксигидратами боратов в узком смысле слова, т. е. соединениями, содержащими только «кристаллизационную» перекись водорода, которая при растворении в воде отщепляется.
** Рациональное название соединений присоединения образуется из последов а- . тельпого перечисления названий составных частей. Число этих частей обозначается арабской цифрой.
Третья группа периодической системы
379
При перекристаллизации из воды эта соль переходит в наиболее часто применяемый технический пероксигидрат бората NaBO2-HsO2-3H2O (тетраборат натрия-перекись водорода-ЗНзО):
NagBiO?' П2О2 -р 4Н-0 = НаВО2.Н2О2-]-МаН2)ВОз-4-2НвВОз.
Пероксигидрат бората обычно получают непосредственным добавлением перекиси водорода к раствору бората натрия или сплавлением перекиси натрия с борной кислотой или с боратами. Это — белая, кристаллическая -соль, растворимая при комнатной, температуре приблизительно в 40 ч. воды с отрицательным тепловым эффектом. Технический продукт содержит около 10,4% «активного кислорода» (см. стр. 77), который аналитически -определяется таким ясе образом, как и в перекиси водорода. Средство для мытья и отбеливания «персиль» содержит, кроме мыла и соды, 10% пер-•оксигидрата бората натрия.
Кроме пер окси гидр ат о в щелочных боратов, имеют значение перокси-тидраты боратов двухвалентных металлов. Последние, например перо-кснгидраты боратов щелочноземельных металлов, трудно растворимы в противоположность пер окси гидр атам боратов щелочных металлов и употребляются поэтому в качестве дезинфицирующих порошков и как средство .для чистки зубов.
Гипо бор на я кислота состава Н4В204 получается, по Вибергу (Wiberg, 1937), тгри гидролизе ее эфиров в лиде белого твердого окисляющегося вещества. Эфиры этой кислоты можно получать из эфиров хлор обо рвой кислоты синтезом Вюртца, впервые примененным в химии бора Штоком:
2В(ОВ)гй1 4- 2Na = В(ОВ)г— В(0В)34- 2NaCJ.
Отсюда следует, что рассматриваемая борная кислота имеет отроение
ПО, .Oil L
)В—В<
НО7 ОН
Эфиры пшоборпой кислоты — бесцветные, неприятно пахнущие жидкости. Метиловый эфир плавится при —24° и кипит при 4-93°.
Кислота И3ВНО2 получена в виде метилового эфира BH(OCHS)2 Шлезингером (Schlesinger, 1933) взаимодействием В2Н6 с метиловым спиртом СН3ОН. Этот эфир —- бесцветная жидкость, т. кип. 25,9Ь, разлагается «одой с выделением водорода
ВЦ(ОСН3)2-|-ЗНОН=Н2Ч-В(ОН)3-Ь2СНзОН. .
Другие соединения бора
Сульфид бора. BsSg образуется в виде белой стекловидной массы при прокаливании бора в парах серы или при пропускании паров сероуглерода над нагретой смесью борпого ангидрида и угля. Он несколько растворим и треххлористом фосфоре и кристаллизуется из этого раствора в виде тонких игл; температура плавления 310°; удельный вес 1,55; с аммиаком, а также с галогенидами бора он образует кристаллические соединения присоединения. Водой сульфид бора разлагается с образованием борной кислоты и Сероводорода.
Растворимое в бензоле соединение BaS3 с HaS состава H2B2S4 получается в виде белыхщгл при действии сероводорода па бромистый бор.
Нитрид бора. BN получается при прокаливании бора в атмосфера азота до белого каления в виде белого, жирного на ощупь порошка с удельным весом 2,34. Нитрид
380
Глава 10
бора по изменяется при нагревании на воздухе, в Струе водорода или сероводорода. ВЛ плавится при ЗООО1 под давлением. Водягюй нар разлагает его при красном калении с образованием аммиака; он разлагается также и прн сплавлении с гидроокисями щелочных металлов.
Кристаллическую решетку нитрида бора можно представить, бели в решетке-графита (см. стр. 463) половину атомов С заме пип. атомами В, а половину — атомами N; «=2,51, с=6,69 А.
Борофосфороксид ВРО.,, — двойное соединение В2О3 с Р2О5, получается в виде белого порошка при сплавлении фосфорной кислоты с борном кислотой. В водном растворе фосфорная и борная кнелогьг ив образуют этого соединения, но если они растворены в серной кислоте пли в ледяной уксусной кислоте, то происходит их совместное выделение. Полученное при обычной температуре и обезвоженное, это двойное соединение довольно быстро разлагается водой; прокаленное, почти нерастворимо в воде, но структурно не отличается (Grnner, 1934) от непрокаленного.
ВРОд образует четко выраженные гидраты с 3, 4, 5 и 6 молекулами Н20. Тригпд-рат дает почти такую же рентгеновскую интерференцию, как и Н3ВОЙ. Решетку ВРО4 (и изоморфного ому BAsOJ можно представить на основе решетки кристобалита (см. стр, 531), в которой атомы Si поочередно заменены атомами В и Р (пли Ае) [Schulze, 1934]. ВРО4, следовательно, является но фосфатом В или борила, а лишь двойным окислом.
Аналитические сведения. Бор открывают обычно по зеленому окрашиванию, которое придают его летучие соединения спиртовому или несветящемуся газовому пламени. Для этой цели борную кислоту или ее соли, обрабатывая метиловым спиртом и концентрированной серной кислотой, переводят в метиловый эфир борной кислоты Б(ОСН3)3
В' (ОН)з+ЗН[ОСН3
В(осн3)34-знон.
Серная кислота служит для связывания воды, вследствие чего реакция из-за сдвига равновесия протекает в указанном направлении. Минералы, испытываемые на содержание в них борной кислоты, смешивают с фторидом кальция и серной кислотой и вносятся в пламя (пли лучше подносятся вплотную к нижней части пламени). При этом образуется летучий (фтористый бор, который дает зеленое окрашивание. Сама борная кислота из-за своей летучести также окрашивает пламя в зеленый цвет.
Об открытии бора по окрашиванию пламени см. также: Gei Imanii W., Z. analyt. Cherri., 129, 3, 1949. Свободная борная кислота, а также подкисленные растворы боратов окрашивают желтую куркумовую бумагу в красно-кортгчновый цвет, появли.11.|Щ1>пС11 при высушивании на водяной бане. Если после этого смочить бумагу щелочью, то окраска переходит в черно-сипюю. Нитрат серебра и нитрат бария осаждают из пейтралыплх растворов боратов белые осадки, растворимые в уксусной кислоте. Осадгщ Ва(ВО8)2 образуется большей частью после добавления нескольких капель аммиака. Борат серебра AgBO2 при кипячении раствора окрашиваетен в коричневый цвет вследствие гидролиза. Отличить различные кислоты при помощи качественных реакций невозможно, тик как они легко переходят одна в другую.
Количественное определение борной кислоты производят, переводя: ее в борн о метиловый эфир с последующим омылением этого эфира гидроокисью кальция
2В(О(ЛЬ)з+Са(О1-Т)2+2Н2О = Са(ВО2)3-]-6СНзОН.
Исходя из взвешенного количества СаО по привесу после прокаливания определяют количество присоединенного Б2ОЭ.
Количественное определению борной кислоты можно также проводить и объемным методом. Для этого титрованием по метиловому оранжевому устанавливают общее количество щелочи и затем добавляют соответствующее количество соляной кислоты.
'Третья группа периодической системы 381
Последнее проделывают, чтобы выделить всю борную кислоту. Затем добавлением глицерина или, лучше, маннита или фруктозы (или иггвертированггого сахара) переводят слабую бораую кислоту и более сильную, одноосновную, которую и титруют щелочью по фенолфталеину. Объемный способ оп редел они я борпон кислоты в присутствии мешающих примесей идеи после их удаления см.: Sieverts, Schafer, Z. analyt. Cheni., 121, 161, 170, 1941; см. также Z. anorg. Chem., 246, 149, 1941; 247, 96, 1941.
АЛЮМИНИИ (ALUMINIUM) Al
$
Распространение в природе. По своей распространенности алюминий среди эле ментов занимает третье место, среди металлов — первое. Оп встречается главным образом в виде двойных силикатов, в полевых шпатах и слюдах и в продуктах их выветривания — глинах. В свободном состоянии алюминий никогда не встречается. Окись алюминия AhOs встречается в виде корунда и наждака. Из гидроокисей боксит АЮ(ОН) имеет наи-болыпес техническое значение в качестве основного исходного продукта для получения алюминия. Большое значение имеет также криолит Na3AlF6, встречающийся главным образом в Гренландии.
Из двойных силикатов следует отмстить: калиевый полевой шпат или ортоклаз .К [AISi3OsJ (главная составная часть изверженных пород: гранита, Гнейса, сиенита, порфира, базальта), натриевый полевой шпат или альбит Na[AlSl30g], кальциевый полевой шпат или анортит Са[ Al^Si^O^], плагиоклаз (твердые растворы кальциевого И натриевого полевых шпатов, к которым относятся олигоклаз, андезин и лабрадорит) .далее слюды: биотит, мусковит, циннвальдит И лепидолит, которые также содержатся в изверженных породах. Нефелин NafAlSiOJn лейцит К|А1813ОЙ] близки к полевым шпатам и ииогда заменяют их в породах. Иа кальциевых алюмо силикатов должны быть еще упомянуты: цоизит Са2А13[SiO4]3[ОН], эпидот Ca2(Al,Fe)3[SiO4]3[Ori] и везувиан, Са(,-.МрчАЪ [SiO^L [Й12Ог]2 [ОИ]4. Силикат магния и алюминия кордиерит Alg3Al3 [AlSisOIS), чистые алюмосиликаты — цианит, андалузит и силлиманит — все три состава AkSiCC (структуру см. на Стр. 541 и сл.). Силикат алюмипия, содер-жалцш фтор — топаз, относится к числу драгоценных камней Al2(0H,F)2(SlO,J.
При выветривании половых шпатов образуется каолин (фарфоровая глина), содержащий воду силикат алюминия состава Al3O3-2SiO2-2H2O. Обычным продуктом выветривания пород, содержащих полевые шпаты, однако, является глина (см. стр. 675). Глины, содержащие большое количество углекислых кальция и магния, называются глинистыми мергелями, а сильно загрязненные окисью железа (III) и песком — просто глинами.
Исторические сведения. Название алюминий происходит от слова alu-.men (квасцы), которое в свою очередь возникло, по Исидору (VII в. н. а.), в связи с применением этого соединения в качестве протравы для крашения: quod lumen coloribus praestat tingendls. Плиний описывает квасцы и их применение и находит упоминание о них уда? у Геродота (V в. до и. э.) иод названием елхлтцдщ. Однако в то время квасцы в современном смысле итого слова, т. с. двойной сульфат калия и алюминия КАфЗОгД-^НаО, не отличали от веществ, действующих аналогично им, например от железного купороса. В чистом виде квасцы были получены впервые, по-видимому, алхимиками. Лежащая в основе квасцов земля, т. е. Окисел металла, была выделена впервые в 1754 г. Маргграфом; впоследствии она получила название глиномма. Деви делал тщетные попытки (Davy, 1808— 1810) электролитически выделить из глинозема А12О3 лежащий в основе его металл.
382
Глава 10
Получить алюминий впервые удалось Эрстедту (1825), восстановившему открытый им безводный хлористый алюминий амальгамой калия при нагревании. Этот способ, только при определенных условиях приводящий к цели, был значительно улучшен Вёлером, применившим вместо-амальгамы чистый (1827) металлический калий. Вёлеру же- принадлежит1 первое сравнительно точное описание свойств металла, и поэтому именно-его обычно считают первым исследователем, подучившим металлический алюминий, так как у Эрстедта нет бесспорных доказательств того, что-полученное им вещество действительно било чистым металлом. Заводское-получение алюминия было разработано первоначально на основе способа Вёлера Сен-Клэр-Девилем, которому удалось снизить издержки производства с 2400 марок до 200 марок за килограмм А1, так что на Парижской1 выставке 1855 г. уже демонстрировался большой слиток «серебра из глины». Между тем в 1854 г. Бунзену удалось приготовить алюминий электролитическим путем, а именно электролизом двойного хлорида натрия и алюминия. Этот метод положен в основу прилгеняемого в настоящее время технического способа получения алюминия; только теперь вместо слиш-; ком летучего хлорида используют раствор окиси алюминия в расплавленном криолите.
До 1883 г. в заводском масштабе металлический алюминии получали во Франции, а затем:в США, Германии и Англии. С 1890 г. значительные количества алюмина я производятся в Швеции, которая использует для; электролиза дешевую энергию гидроэлектростанций.
Получение. Бсо применяемые в настоящее время способы промышлепного получения алюминия основаны на электролитическом разложении окиси алюминия, растворенной в расплавленном криолите. Б качестве материала для электродов используют обычно ретортный графит. Содержимое* ванпы поддерживается в жидком состоянии за счет тепла электрического-тока. Температура ваппы не должна превышать 1000°. Выделяющийся' на катоде металлический алюминий собирается в расплавленном состоянии на дне печи. На погруженном сверху в ванну аноде кислород окисляет графит с образованием окиси углерода СО, которая сейчас же сгорает до двуокиси СО-?. Двуокись углерода частично образуется также и непосредственно на аноде.
Потенциалы разложения составляют (прп 950° С): для NaF 4,5, для A1F3 3,5-и для AI3O3 2,18 в. Эти данные имеют значение в том случае, если анод не взаимодействует с выделяющимися веществами. Если применяют угольный электрод, то потенциал раюнякопля Л12О3, теоретически равный 1,00 в (из-за образования СО), повышается при большой силе тока до 2 а, так как образующаяся СО препятствует разрядке иоиов кислорода. Если напряжение на ванне пе слишком большое и содержание окиси-, алюминия в расплаве не слишком мало, разлагается преимущественно только АЬО3. В очень незначительной степени на аноде происходит образование СГд. При правильном проведении электролиза патрий никогда не выделяется. Так как значительная часть напряжения, подаваемого на ванну, тратится па преодоление ее омического сопротивления, на практике напряжение на ванне поддерживается около 5—б в при плотности тока 70—9 Us на. 1 поверхности анода. Как только напряжение начинает заметно возрастать, необходимо добавлять новые порции окиси алюмипия, Все попытки при получении алюминия заменить электролиз расплава терми-чёским восстановлением окиси алюминия до сих пор не имели практического успеха. При использовании н качестве восстановителя углерода происходит образование карбида алюминия А14С3, которое ие удается предотвратить. Его, однако, можно, как недавно указал Кольмейер (Kohlmeyer, Z. anorg. Cliern., 260, 208, 1949),, сильно ограничить, если Д12О3 плавить очень быстро и смешивать лишь со строго опре-деденпым количеством угля. Этим способом в лабораторных опытах удавалось получать 93%-пьпг алюминии. 1
Третья группа периодической системы.
38$
Окись алюминия, пригодная для получения и» нее металла; чрезвычайно редко встречается в природе в достаточно чистом состоянии. Ее получают почти всюду из-боксита, который добывают главным образом во_ Франции, Венгрии, Далмации, Истрии, СССР, Индии, Арканзасе и Гвиане. Употребляемый для растворения криолит частично изготовляют искусственно. Для понижения температуры плавления обычно-прибавляют флюсы, например фтористый кальций. Таким образом, можно без-труда поддерживать находящийся в ванне плав с 20—'30% A12OS в жидком состоянии при 800—900°. При более высокой температуре разница между удельными весами плава и расплавленного алюминия уменьшается, вследствие чего- А1 не опускается больше на дно ванны и, попадая на поверхность, оторвет.
Встречающиеся в природе бокситы всегда более или менее сплъпо загрязнены окислами железа ц кремневой кислотой. Для получения чистой окиси алюминия, пригодной для использования в электрометаллургии, руду вскрывают нагреванием с СаО и Na2COs (сухой способ} или нагреванием с едким натром в автоклавах (способ Бауэра). В обоих случаях окись алюминия в виде алюминатов переходит в раствор, который затем разлагают либо пропусканием через него двуокиси углерода, либо добавлением уже приготовленной заранее гидроокиси алюминия. В первом случае разложение происходит по уравнению
2[А1(ОН)4]ЧСОг=2А1(ОН)а+СОаЧНгО.
Разложение на второму способу основано па том, что раствор алюмината, полученный при нагревании в автоклаве, после охлаждения н разбавления мота стабилен. Добавляемая гидроокпс[, алюминия ускоряет распад алюмината, прячем разложение происходит следующим образом: (Al(OH)Jr= А1(ОН)3+ОН'. Частицы гидроокиси алюмипия служат центрами кристаллизации. Разновидностью способа Байера является способ башенного вскрытия. По атому способу вскрытие производят пе в автоклавах^ а в высоких башнях, в которых едкий натр стекает сквозь слои боксита. Товарная стоимость алюминия определяется следующими расходами: 32% на А12О3, 4% на криолит, 12% на'электродные угли и 25% на электрическую энергию, если считать стоимость 1 квт-ч в 1,2 пфеннига. Цепа боксита составляет только б % общей стоимости алюминиевого производства. Для Германии имеет большое значение вопрос извлечения А1г0э из местного сырья, т. е. из широко распространенных глин. Вследствие высокого содержания в глинах кремневой кислотыщелочные методы вскрытия, как, папример, способ Байера, для них почти пе пригодны. Чтобы уменьшить переход кремневой кислоты в раствор, для вскрытия глин применяют кислоты, и процесс при этом ведут так, чтобы по возможности взбежать растворения окислов железа, ибо-последующее отделение больших количеств железа от алюминия представляет значительные трудности.
По способу Бухнера (Nuvalonverfahren) отделение А12О3 от SiO2, Ге2О« и TiOg достигается нагреванием глины в автоклаве с азотной кислотой, соответствующей концентрации и в необходимом количестве. Нитрат алюминия, переходящий в раствор, загрязнен в основном только нитратами щелочных и щелочноземельных металлов, от которых его можно отделить фракционной кристаллизацией. Азотная кислота выделяется пр» термическом разложении водного нитрата алюминия, и Л12О3 получается ,в очень чистом состоянии. По способу Гольдшмидта (Goldschmidt) для вскрытия используют водный раствор значительно более дешевой сернистой кислоты. При этом в большинстве случаев в раствор переходят значительные количества железа, которое, однако, образует с алюминием хорошо кристаллизующуюся основную соль; до тех пор пока количество железа не слишком велико, возможно сравнительно простое отделение алюмипия путем фракционной кристаллизации основного сульфита.
На совершенно иных принципах ОспОвап метод Хаглунда (Haglund) (первоначально разработанный с целью извлечения А12О3 из бокситов, но в дальнейшем применяемый также со многим глинам). По этому методу нагреванием окисной руды с углем и серным колчеданом в электрической печи алюминий частично переводят в сульфид, который образует с Ai2O3 легкоплавкий шлак, легко отделяемый вследствие-незначительного удел [.кого веса от одновременно образующегося сплава желоза с кремнием. Застывший шлак, содержащий 80% А12О3 и 20% A12S3, обрабатывают соляной кислотой; при этом выделяется Н23, который можно использовать для получения серы (см. стр. 751), кроме того, образуется А1С13 и остается по растворившимся кристаллический Л1гО3.
Для очистки долучепного электролитическим путел! алюмипия его почти всегда переплавляют в пламенной печи. Технический алюминий п большинстве случаев содержит 99,5% алюминия. Примеси состоят пре-имущественно ив кремния и железа, а иногда — из следов меди.
I
4584 Глава 10
Алюминий с чистотой не мевое 99,99% (рафинированный) можпо получить из ^.лектро.читичоского (обычно 99,7%-пт) промышленного алюминия или из алюминиевой крупы путем так называемого «трех ело иного электролиза». Здесь имеется в виду такой электролиз расплава, при котором анод, состоящий из относительно тяжелого алюминиевого сплава (А1 — Си), располагается на дне ячейки и покрывается электролитом, удельный вес которого добавлением BaF2 или ВаС12 регулируют так, чтобы он оказался легче анодного плава н тяжелее чистого жидкого алюминия, который, таким образом, плавает [[а поверхности электролита, защищающего его от смешения с анодным ил ан ом.
Большое значение имеет повторное получение алюминия из алюминиевой крупы (пылн, остатков). Таким путем производят 50% и более заводского продукта. Обычно его переплавляют с добавкой Mg и Zn (которые в дальнейшем отделяют отгонкой в вакууме). Очень проста и эффективна хлор-азотная очистка' при пропускании хлора в расплавленный легкий металл все содержащиеся в нем примеси (кроме Си) переходят в хлориды, которые выделяются па поверхности расплава. Суспендированные в расплаве оклелы, нитриды и карбиды при этом увлекаются хлоридами. Алюминия теряется только 2—3% на образование А1С13. В конце процесса, занимающего около 10 .иин, ток хлора заменяют током азота.
J
Свойства. Алюминий — серебристый металл с удельным весом 2,70" температурой плавления 660,2° и температурой кипения 2270°. Он Крис таллнзуется кубически, гранецентрированно (рис. 46), а = 4,0414 А. Теплопроводность алюмипия X. = 0,5 при обычной температуре в три раза больше, чем для ковкого железа, и вдвое меньше, чем для меди. г Удельная электропроводность для вытянутой алюминиевой проволоки доставляет около 60% электропроводности медной проволоки. Теплоемкость равна 0,23 (при 100°) и сравнительно с другими металлами весьма высока; она приблизительно в 2 !/2 раза больше, чем для меди или для цинка, и вдвое больше, чем для железа. Теплота плавления также весьма высока (см. стр. 359); поэтому алюминий, несмотря па свою более низкую температуру плавления, плавится труднее, чем. медь; по будучи расплавленным, он дольше остается жидким, чем другие металлы. Алюминий очень легко иод дается обработке, из пего можно вытягивать очень тонкую проволоку, прокатывать в тонкуя» жесть и ковать чрезвычайно тонкую фольгу (листовой алюминий). Сопротивление растяжению чистого алюминия почти в четыре раза меньше, чем меди. Его можно, однако, значительно повысить добавлением нескольких процентов меди. При этом, однако, понижается химическая стойкость алюминия.
О свойствах очень чистого алюминия (99,998%) см. Chaudron G., Helv., chim. Acta, 31, 1553, 1948.
На воздухе чистый алюминий хорошо сохраняется, так как он покрыт топким слоем окиси, предохраняющим металл от дальнейшего окисления. По той же причине он индифферентен и к подели даже к водяному пару при высокой температуре. На него не действует также сероводород, по оп растворим в большинстве кислот, а также в щелочах. Разбавленные органические кислоты, например уксусная и лимонная, слабо действуя па алюминий на холоду, хорошо растворяют его прп 100°; растворение ускоряется также прибавлением поваренной соли. Концентрированная уксусная кислота иа него почти не действует; столь же незначительно действуют жиры и жирные кислоты; не действует на холоду азотпая кислота, ни разведенная, ни концентрированная. Нагревание в этом случае вызывает бурную реакцию.
Растворимость алюминия в кислотах основана на том что он в соответствии со своим положением в ряду напряжений (см. стр. 49) способен разряжать водородные JIOIHi
А!+ЗН- = А!-+3/2И2-
Третья группа периодической системы . 385
То обстоятельство, что алюминий ие реагирует с водой и с разбавленными, слабо диссоциирующими кислотами, объясняется обравоваиием чрезвычайно трудно растворимого окисла алюминон, произведение растворимости которого в растворах с низкой концентрацией водородных ионов еще не достигается. Напротив, в щелочах окись амюмипия растворима с образованием гидроксоалюминатов (см. стр. 353).
Так как в щелочных растворах концентрация ионов А1“' чрезвычайно незначительна, то потенциал металлического алюминия испытывает в этих растворах еще больший сдвиг в сторону неблагородных металлов. Этим и объясняется, что щелочи столь энергично действуют на металлический алюминий.
То обстоятельство, что кислоты, легко отдающие свой кислород, например азот-пая, пе действуют на алюминий, наблюдается и для многих других металлов, весьма активных по отпопгепию к кислороду; это явление называется пассированием (подробнее см. т. II).
Образование слоя окиси, предохраняющей алюминий от действия воздуха и воды, можно предотвратить, если на алюминиевую поверхность поместить ртуть. На амальгамированной алюминиевой жести на воздухе в короткое время появляется белый волокнистый налет, состоящий из гидроокиси алюминия, образующейся в результате действия на алюминий влаги воздуха.
Получаемая таким .образом гидроокись алюминия и образующаяся из нее при прокаливании окись чрезвычайно объемисты и очень поверхностноактивны. Такая окись алгомипия была предложена Вислиценусом (Wislicenus) для определения дубпль» пых веществ и постукает я продажу под названием «волокнистого глинозема». Активирование алюминия под действием ртутя (образование амальгамы) применяют, кроме того, в органической химии, если алюминий используют В: качестве восстанавливающего средства. Такой алюминий иСпользуют для этих целей в виде зернекой (измельченной) амальгамы, получаемой действием двухлористой ртути на зерненый алюминий-
Устойчивость, которой обладает алюминий благодаря наличию окисной пленки, чрезвычайно возрастает, если ла поверхности электрохимическим путем создать окисный слой значительно большей толщийы (0,02 .и.н), чем природная окисная пленка (элоксальный способ). Окисный слой, покрывающий обычно алюминий, возникает вследствие реакции металла пе с атмосферным: кислородом, а с водяными парами, присутствующими в атмосфере. Освобождающийся при атом водород частично поглощается алюминием с образованием твердого раствора. Однако большая часть его адсорбируется на поверхности окисной пленки. Водород, растворенный в алюминии, присутствует в металле в атомарной форме, как это' следует из зависимости его растворимости от давления.
Если мелко измельченный алюминий поджечь, то оп сгорит с ослепительным светом, переходя в окись алюминия. При этом на 1 г алюминия выделится 7,47 ккал тепла. Благодаря столь высокому сродству к кислороду алюминий используют для восстановления трудновосстанавливаю-щихся металлов 'из их окислов и для создания высоких температур, например при сваривании железа. Применяемая для этого смесь алюминиевого порошка или крупы с окислами других металлов, например ГезОг, называется термитом. По данным Вартонберга (Wartenberg, 1936), при реакциях с железным термитом достигаются температуры до 2400°. Обоснованный Гольдшмидтом способ получения трудповосстапавливающпхея металлов восстановлением их окислоп при помощи алюминия называется алюмо тер ми е й.
С хлором алюмипий образует соединения с большим выделением тепла. С жидким бромом он реагирует с вспышкой, с иодом соединяется непосредственно, с серой — при сильном разогревании. Алюминий взаимодействует и с азотом, по лишь при очень высокой температуре, получаемой, например, за счет частичного сжигания порошка алюминия в струе кислорода.
С селеном ц теллуром алюминий соединяется при нагреваний со взрывом. При этом образуются соединения АЬЗез и AlsTes.
Применение. С тех пор как алюминий по своей стоимости стал доступен для промышленного использования, он получил широкое распростра-25 г. Ремп
386
Глава 10
пенне. Из него изготовляют аппараты для промышленных целей, а также многочисленные предметы домашнего обихода.
Часто наблюдаемое потускнение алюминиевой посуды; зависит от содержащихся в алюминии примесей. Для чистки этой посуды пользуются имеющейся в продаже пастой из окиси железа с жиром. Изнутри кухонную посуду чистят древесной золой, наждаком или тонким песком или же кипятят в ней разведенный раствор квасцов с винным камнем (1 ч. винного камин, 2 ч. квасцов; одну столовую ложку этой смеси па 1 л воды).
Обрезки алюминиевой жести, перерабатывают в алюминиевый порошок, употребляемый в качестве литографской краски, а также для изготовления взрывчатых веществ, применяемых в пиротехнике. В электрической промышленности алюминий все более вытесняет дорогостоящую медь. Электропроводность алюмипия составляет всего 60% электропроводности меди, однако это понижение с избытком компенсируется меньшим удельным весом алюмипия и возможностью изготовлять алюминиевые провода из более толстой проволоки. При равной электропроводности алюминиевая проволока весит вдвое меньше, чем медная. Благодаря легкости алюминий применяют в виде сплавов в самолете- и в автомобилестроении. К таким сплавам относятся, например, магналий (КФ—30% Mg) и дюралюминий. —очень твердый сплав, содержащий 93—95% алюминия, 2,5—5,5 % меди, 0,5—2,0% магния, 0,5—1,2% марганца и 0,2—1,0% кремния.
Подобными свойствами обладает склерон — сплав алюминия с 12% цинка, 3% меди, 0,6% марганца, 0,5% железа, 0,5% кремния и 0,08% лития. Г ид ропалий — устойчивый к морской воде сплав алюминия с 3—12% магния. В железо- и сталелитейной промышленности алюминий применяют как раскислитель для удаления из расплавленного железа растворенных в нем окислов. Крупнозернистый порошок алюминия применяют для получения металлов, например хрома, марганца и титана по способу Гольдшмидта, и для изготовления термитов для сваривания железнодорожных рельс и пр. Толкую алюминиевую фольгу используют вместо стаипиоля для упаковки шоколада и разных кондитерских изделий.
Алюминиевую фольгу толщиной около 0,5 р применяют в фотографии для получения мгновенной вспышки.
В последнее время все большее распространение налипает приобретать способ покрытия желеэпых изделий слоем алюмипия для предохранения их от ржавления (алюминирование). Вследствие незначительной разницы коэффициентов термического расширения алюминиевые покрытия должны быть более стойкими, чем цчнковые. Быстро расширяется применение алюминия для изготовления зеркал (в первую очередь телескопов). Зеркальный слой наносят конденсацией паров алюмипия на стекло по способу, предложенному Полем (Pohl, 1912). Такие зеркала обладают значительно лучшими отражательными способностями, чем зеркала с серебряным слоем, и показывают к тому же более высокую отражательную способность в ультрафиолетовой области.
Попытки получить алюминий электролитически из водных или неводных растворов его солей пока безуспешны. По даппым Менделя (Menzel, 1940). электролитическое получение все же возможно из ванн, содержащих алюмииийорганичесКие соединения типа А1Н2Х и ДШХ2 (X — галоид).
СОЕДИНЕНИЯ АЛЮМИНИЯ
Все нормальные соединения алюминия производятся от трех валентного положительного иона А13+п соответствуют, следовательно, типу AIXS, где X — одновалентный радикал.
Третья группа периодической системы
387
Иным строением, отличающимся от строения нормальных соединений алюминия, обладает гидрид алюминия; все же и в пом алюминий трехвалентен.
В особых условиях алюминий способен существовать и в низших валентных состояниях. Например, оп испаряется в виде AKS или ALSe, если сильно нагревать металлический алюминий в смеси с АЬ83 или Al2Se3. Об образовании ALO и Al F при соответствующих условиях см. стр. 392 и 397. Установлено также образование A1GI, А! Вт и All при высоких температурах (в равновесии с металлом и тригалогенидом) (Klemm, 1948). Существование при высоких температурах соединения А1О, в котором алюминий Эвуэяягленпген, установлено анализом полосатых спектров. Замечательна незначительная теплота образования моноокиси алюминия в сравнении с моноокисью бора. В то время как при образовании А1О tig атомов выделяется самое большее 87 ккал/моль, соответствующий тепловой эффект Для соединения бора составляет около 210 ккал/моль.
Ниже приведен обзор важнейших соединений алюминия.
Гидрид алюминия |А1Н3]Х Ала паты: а)несолеобразные, например Кислородные соединения Al^Oj Al (OII)3 Алюминаты, например Г алогециды AIFa Фтороалюминаты, па пример На a[AIF6], А1С13, A1I3 - Другие соли А12(5О4)з Сульфид A12S3 Квасцы, например Нитрид KA1(SO4)2- 12 Н, О A1N
MgHs-2AIHa: б) соле об разные, например Li[AlH4] Ha[AI(OH)t] А1Вгэ, AI(NOB)3 Алюминийорга- AI(SCN)3 ничёские сое- А1 (СН3 СО О) а динениз AIRS, AiRjX, A1RX2
Из кислородных соединений алюминия кристаллическая окись замечательна своей твердостью и значительной теплотой образования, а гидроокись — своим амфотерным характером. Соли алюминия кристаллизуются большей частью с большим содержанием кристаллизационной воды. В формулах приведенной таблицы кристаллизационная вода не указана, так как ее количество является переменным и зависит от температуры и давления. Соли алюминия бесцветны. Из них соли сильных кислот легко растворимы в воде. Из солей кислот средней силы и слабых труд-норастворнмы фосфат; борат и силикат. Легко растворим ацетат алюминия. Водные растворы солей алюминия содержат бесцветные ионы АГ", в значительной степени гидратированные. По Бринтзингеру (Brintzin-ger, 1935), в разбавленных растворах каждый ион АГ" связан с 18 молекулами Н’О. Б связи с этим безводные соли алюминия, несмотря на рост растворимости с температурой, обладают сильной положительной теплотой растворения.
Соли алюмипия, в том числе образованные сильными кислотами, в водном растворе гидролиз ованы, что соответствует слабоосновпому характеру гидроокиси алюмипия. А1(ОН)з вообще не реагирует с очень слабыми кислотами, например с HaS. Если получить соответствующее соединение, например AlsSs, сухим путем и внести его в воду, то оно тотчас же полностью гидролизуется. Наоборот, у солей сильных кислот гидролиз приостанавливается после разложения незначительной части; найри-мер, степень гидролиза у хлорида алюминия в 0,1 н. растворе составляет 2,0%, а у сульфата — 1,3% при 25°.
Алюминий проявляет склонность к образованию комплексных ионов с избытком галоидных ионов или ионов SO4. Это выражается не только в кристаллизации двойных 25* ‘
388 Глава 10
солей кз соответствующих растворов, но и в собственном электрохимическом потенциале алюминия, который при эквимолярных концентрациях солей в растворах хлоридов и сульфатов значительно ниже, чем в растворах нитратов.
При получении солей алюминия большей частью исходят из сульфата алюминия, который получается главным образом из гидрата окиси, для которого в свою очередь важнейшим исходным материалом служит боксит. Кроме окиси, являющейся исходным продуктом для получения металлического алюминия, наиболее употребительными соединениями алюминия являются сульфат алюминия AhfSO^s-18НаО и производимые от него квасцы КА1(ЗО4)2-12НгО.
Гидрид алюминия, аланы и алапаты. Подобно тому как бороврдороды и их производные называются боранами, для соединений А1Н3 и его производных, получающихся при замене атомов водорода па атомы галогенов или органические радикалы, применяют, по предложению Виберга, собирательный тсрмип аланы. Двойные соединения гидрида алюминия с гидридами других металлов называют аланатамц.
Гидрид алюминия. (А1Из]х образует белую некристаллическую массу, разлагающуюся выше 105° с отщеплением водорода. Он был получен впервые в 1942 г. Виборгом при действии тлеющего разряда на смесь три-метилалюминия и водорода.
При этом происходит ступенчатый обмен СН3-групп па атомы Н, который приводит в конце концов через ряд промежуточных продуктов (метялаланы) к образованию твердого, нелетучего, высокополимерпого гидрида алюминия [AlHjL. Наряду с этим образуется Димерпый диметилалан [А1(СН3)аИ]2~ бесцветная, труднолетучая, очень вязкая жидкость, застывающая при охлаждении в стекло, чрезвычайно чувствительная к влаге и со взрывом сгорающая на воздухе пурпурно окрашенным пламенем.
Гидрид алюминия не солеобразное соединение. Он легко образует продукты присоединения, например, с В(СНз)з и с N(CHb)b- Образование соединения присоединения А1Н3.2К(СНз)3 имеет большое значение для выделения гидрида алюминия, получаемого указанным выше способом (ср. стр. 400).
Как впервые обнаружил Шлезингер (Schlesinger, J. Am. chem. Soc., 19, 1199, 1947), гидрид алюминия можно получать в эфирном растворе добавлением А1С1я к эфирному раствору LiAlHi. Последнее соединение образуется, если LiH обрабатывать эфирным раствором А1С1з. Эбулиоскопические определения молекулярного веса (Wiberg, 1952) показали, что в свежеприготовленных эфирных растворах АШ.3 существует в виде мономера. Вскоре, одпако, происходит полимеризация и весь гидрид алюминия постепенно выделяется из раствора в твердом виде. Выделение это прекращается, если добавить вещества, образующие с А1Нз относительно устойчивые продукты присоединения, например триметиламин ЭТ(СНз)з. Таким образом можно приготовить устойчивые растворы гидрида алюминия, которые можно применять для гидрирования.
Продукт яри со единения A1H3-2NR3 (где R=CH3) растворяется в бензоле а виде мономера в отличие от соединения AIH3'NR3, растворяющегося в бензоле в виде димера. В эфирных растворах обе оии мономерные Виберг Заключил из этого, что димеризация, как и у В2Н0, обусловливается возникновением водородных мостиков, а эфир препятствует иди по крайней мере замедляет их образовать, так как оп сам может встать па пх место
Н /NR3 Н. н. /Н Нх ZNR3
Н->А1£ R3N ->А1ф ;А1<~ NR3 Н^а!£
, liZ ХМ!3 н/ '-НЛ \Н LiZ ХОВ2
Третья группа периодической системы 389
Этим объясняется так же н то, почему в эфирном растворе А1И3 способен существовать в виде мономера
1U /OIL hVa1k IIх ^or2
В противоположность связи Al — NR3 более рыхлая связь Al — OR2 постепенно разрушается вследствие образования водородных мостиков, ,что н приводит к выделению твердого гидрида алюминия. В то время как у бороводородов образование водородных-мостиков вызывает только димеризацию, у гидрида алюминия в соответствии с более высоким координационным числом атома алюминия оии приводят к сетчатому сцеплению атомами водорода сколь угодно большого числа атомов AI и том самым к образованию твердого вещества. Причина более высокой координационной валентности алюминия лежит в том, что внешняя электронная оболочка А1 (главное квантовое число »=3) способна принять больше электронов для образования ковалентных связей, чем внешняя электронная оболочка бора (главное квантовое число п= 2).
Выпадение гидрида алюминия из его эфирных растворов можно предотвратить не только триметиламином, но и добавлением галогенидов алюминия, образующих галогенпроизводпые гидрида алюминия (залог ен алая и), например
2А1Н3+ A1XS=, ЗА1Н2Х; А1НЭФ 2А1Х3= ЗЛШХ2.
После испарения эфира эти галогеналаиы остаются в виде продуктов присоединения — эфиратов, которые частично можно перегнать в вакууме без разложения (Wiberg, 1951). Так же как и эфирные растворы гидрида алюмипия и аланата лития, эфирные растворы г ало гена ланов пригодны для избирательного гидрирования органических веществ.
Двойные гидриды алюминия (аланаты). Как и бороводород, гидрид алюминия способен образовывать двойные гидриды. По предложению Виберга их называют аланатами. Среди них особое значение благодаря простоте получения и многообразным возможностям применения для гидрирования, особенно в органической химии, имеет аланат лития — LiAlH4.
Так же как и металлбороводороды (боранаты), аланаты подразделяются на два класса — солеобразные И несолеобразкыб а канаты. Аланат лития Li [АШ4] является солеобразным соединением. В качестве примеров несолеобрааны'л аланатов можно назвать полученные Внбергоми Бауэром (Wiberg, Bauer, 1950—1951) соединения MgH2-2AIH3 и БеН,.2А1Н3. Это твердые, белые вещества, растворимые в эфире; так же как и а.чанат лития, их можно применять для гидрирования. Известны также аланаты Ag, Са и 1л.
Аланат лития. LiAlHa был впервые получен Шлезингером (1947) взаимодействием LiH и А1С1з в эфирном растворе
4LiH4-AlCl3eI.i[AJH1J4-3LjCJ.
По данным Виберга (1952) вместо А1С1з лучше применять AlBrs, так как иначе реакция будет тормозиться из-за выделения LiCl на гидриде лития, в отличие от которого LiBr в эфире растворим. LiAlH< — твердое белое вещество, разлагающееся в высоком, вакууме при 125е па LiH, А1 и На.-Соединение растворимо в эфире; н 0,08 М. растворе оно димерно, в 0,8 М — тримерно. Металлической пылью аланат лития каталитически разлагается, следовательно, в растворе он мета стабилен.
Аланат лития в препаративной неорганической химии оказался превосходным вспомогательным средством Для приготовления трудно получаемых в частом состоянии гидридных соединений. Как было найдено Шлезингером, он количественно превращает ВС13 в B2HG, SiCl4 в SiIT4
4BClg-P 3LiAlH4 2B2H0+ 3LIa1C14,
SiCl4+ LiAlH4 SiH44- LiAlCl4.
Аланат лития c Si2ClG образует соответственно Si2HG, с кремний а лки.ч хлоридами (а лк и лх лор силанами) — алии л силаны, с ал килхлор станнанами — алклзировапные
590
Глава 10
Станнаны [например, (CHs)2SaH2 из (CH3)2SnCl2]; германий-и олововодороды также можно получить со сравнительно хорошим выходом при взаимодействии GeCl4 или SnCl4 с L1AITI4. ВеН2, MgH2 А1Н3 и производимые от них двойные гидриды в большинстве случаев (.также можно удобно получить взаимодействием с ЫАШ4. Де йтеушй содержащее соединение, соответствующее аланату лития — LiAlD4, часто можно с успехом использовать для получения других соединений дейтерия 7 например:
LiAlD4^ 4С12= ЫА1С14ф 4DC1; ЫАЮ4ф- SiCl4= ЫА1С14ф- SiD4.
За последнее время алапат лития Нател обширное применение в органической химии в качестве гидрирующего средства. Он переводит в спирты не только альдегиды я кетоны, но и карбоновые кислоты и их производные. Из алкилгалогенидов получаются углеводороды, из амидов кислот и нитрилов — исключительно первичные амины. Алифатические нитро со единения переводятся в амины, ароматические — в азо соединения. В то же время спирты и эфиры аланатоМ лития обычно не восстанавливаются. Из двойных и тройных связей аланатом лития обычно гидрируются только такие, которые нолярны или могут поляризоваться. В этом заложены обширные возможности для селективного гидрирования. Гидрирование ал анатом лития всегда протекает так, что водород присоединяется к углероду, а алюминий—к электроотрицательному гетеро атому двойной связи
,R
LiA1H44-4O=C( -> Li
: 4R
Путем гидролиза получают желаемый продукт реакции
Li[Al(OCHR2)4]-|-4HOH=4HO—CHR2+Li[Al(OH)4],
В определенных случаях аланатом лития гидрируется также двойная связь С=С, Циглер (Ziegler, 1952), папример, пашел, что этилен СН2=СН2 при температуре немного выше 100° легко присоединяется к LiAlH4 с образованием LiAI(C2H5)4, при гидролизе которого получается этан С2Нщ С аланатом лития подобно этилену реагируют н другие олефины, содержащие расположенную в конце молекулы двойную связь. Органические сульфопы восстанавливаются LiAlH4 до сульфидов. Следует также упомянуть, что, но Кри липкому, Джонсону и Кархарту (К г у n i t zк у, J ohnson, Carhart, J. Am. chem. Soo., 70, 686, 1948), L1A1H4 можно применять для определения числа «активных атомов Н» в органических ^соединениях.
Кадьцийалюминийгидрид [алапат кальция). CatAlHrla, получающийся, по Швабу и Винтерсбергу (Schwab, Wintersberger, 1954), взаимодействием СаНз с AJCh в тетрагидрофуране, по своим свойствам похож па ала-нат лития, и его можно применять вместо LiAIHr для реакций восстановления. Его можно использовать также для синтеза гидрида алюминия, так как без доступа воздуха в сухом тетрагидрофуране, в котором он хорошо растворим, почти количественно происходит взаимодействие с хлоридом алюмипия ио уравнению
ЗСа[ А1Н4)2+2А1С13=8А1Н3 ЗСаС12,
В эфире, дноксапе, бензоле аланат кальция практически нерастворим; с водой он реагирует со вспышкой
Ca[AlHJ2+5HaO = Ca(OIl)2+AlsO34-8Ils.
Самовоспламенение может произойти уже во влажном воздухе. Поэтому отбор проб и взвешивание соединения следует вести в атмосфере сухого азота.
При взаимодействии LiAlII4 с MgBr2 или MgH2 с галогенидами алюминия в эфирном растворе можпо получить магнийаяюминийгидрид г (аланат магния) MgH2’2AlH3 (Wiberg, Bauer, 1950—1952). Взаимодействием соединения А1Н3-А1С13 с Т1С1Э в эфирном растворе Вцбергу и Шмидту (Wiberg, Schmidt, 1951) удалось получить соединение Т1С1[А1Н4]2, устойчивое только ниже —95°.
Третья группа периодической системы
391
Кислородные соединения алюминия
Окись алюминия (глинозем). AhOs подучается при горении алюминия и при прокаливании гидроокиси алюминия или солей алюминия с летучими кислотами в виде белого аморфного, нерастворимого в воде, а после сильного прокалив а яия — и в кислотах, порошка. В природе она встре-
чается в кристаллическом состоянии в виде корунда. Этот минерал, отличающийся большой твердостью, имеет обычно пс-.ча содержания примесей мутную, грязноватую окраску. Окраска разно-видпостей корунда — рубина (красная) и сапфира (синяя), считающихся драгоценными камнями, — также вызвана незначптельпьйги примееямй. Различными примесями могут быть обусловлены еще и другие окраски, например зеленая (восточный смарагд), фиолетовая (восточный аметист), желтая (восточный топаз). Корунд дожно получить искусственным путем методом 'Тольдш'иидта; перечисленные выше драгоценные камни также можно получить искусственно яри соответствующем режиме (синтетические драгоценные камни).
Окись алюминия применяют в виде корунда и наждака (мелкокристаллическая разновидность корунда, загрязненная окисью железа и кварцем) для шлифования и полирования. Далее, корунд
Ри с. 75. Корупд(А1аОз).
<11,^5,12 А; а—55’17-;
Al t—>А[=1,ЗС А.
используют для получения чрезвычайно Огнеупорных материалов, например «плавленого глинозема», применяющегося для футеровки цементных печей. Из обезвоженной гидроокиси алюминия в больших количествах
получают металлический алюминий.
Окись алюминия кристаллизуется обычно в гексагональных гемиэдрических ромбоэдрах (а-А1аО3) с уд. весом 3,99. Решетка окиси алюминия представлена на ряс. 75. Эту решетку можпо рассматривать в качестве молекулярной, так как в иен весьма отчетливо выражены молекулы А12О3. Теи не менее каждым атом алюминия окружен шестью атомами кислорода, причем кислородные атомы, принадлежащие соседним молекулам, расположены даже ближе, чем те, которые принадлежат самой молекуле. Это объясняется сильным отталкиванием, проявляющимся внутри молекул между одиоимевно заряженными атомами. Как видно из рис. 75, одна молекула А12О3 ио л костью расположена внутри элементарной ячейки; другие же молекулы, из которых на рисунке полностью показаны только две, находятся в узлах элементарной ячейки, содержащей в целом две молекулы А12О3. Верхний атом алюминия, относящийся к молекуле А1йОа, лежащей впе элементарного параллелепипеда, расположен на 0,14 А ближе к находящемуся от пего слова и выше атому кислорода, принадлежащему другой молекуле А1203, по сравнению с расстоянием от каждого из трех атомов кислорода, принадлежащему той же молекуле. В соответствии с этим каждый атом кислорода в решетке корунда окружен четырьмя атомами алюминия, из которых два атома, ие принадлежащих этой молекуле, расположены ближе к данному атому кислорода.
Другая гексагональная модификация (p-AkG-.J с уд. весом 3(30 образуется при медленном охлаждении расплавленного окисла, но только в присутствии некоторых посторонних веществ, например MgO. По Биверсу (Beevers, Z. Krist.aUogr., 95, 472, 1936), эти посторонние вещества в значительной степени участвуют в построении решетки (J-АЦОд, так что последнюю, собственно, следует рассматривать не как модификацию А12О3, но как двойной окисел, например Й’агО11А1аО3.
Кубическая модификация окиси алюминия (у-Л12О3) с удельным вес. 3,4 получается обейвоживапяем гидраргилита или боксита или при замеренном прокаливании аморфного оксигидрата. у~Л12О3 имеет одинаковую с у-Уе2О3 шппнелеобразную структуру. Атомы О располагаются в ней, как в шпипельвой решетке. Часть мест, "примерно Го, на которых должны были бы находиться атомы металла, остается незанятыми
392
: Глава 10
Причем пустые моста главным образом находятся там, где в решетке шпинели MgAl2O4 располагаются атомы АГ. Совершенно такую же структуру имеет модификация, которую обозначают как у'-А1аО3, Она отличается от у-А12О3 тем, что в ее решетке пустые места располагаются неупорядоченно и в тех положениях, которые в решетке MgAl2O4 заняты атомами Mg И Al (V е г w е у, Z. Kristallogr., (А) 91, 65 и 317, 1935). у'-А120з при 900° превращается в у-Л12О3. Выше 1000° у-А1аОа переходит в а~А13О3 (теплота превращения 20,6 ккал/моль). у-А12О3 в зависимости от метода получения может обладать совершенно различной: величиной частиц и сдаю искаженной решеткой. Это может вызвать различия в тенлотах растворепия до 10 ккал/молъ (Frick е, Z. NaturforSChg., 1,649, 1946), Имеется также еще несколько неустойчивых модификаций, например б-А1а0з (Parravano, 1928) и £-А1аО3 (Barlett, 1932). При 19503 Л12О3 имеет давление пара около 10-s атм и кипит около 3300°. Из паров А1аО3 выделяется в первый момент в виде аморфного стекла, которое, однако, тотчас же кристаллизуется. При этом при известных условиях образуется до сих пор еще пе полученная другим Путем «мгновенно охлажденная модификация» (Warlenberg, 1952). При определенных условиях, например ири самоокислении амальгамированного алюминия, получающаяся окись алюминия оказывается чрезвычайно поверхностно активной. Ее часто применяют поэтому в качестве адсорбента, катализатора или носителя для катализатора. Он обладает способностью такой же, как и у пермутитов, обменивать неорганические катионы. Торговые препараты, предназначенные специально для хро-матографхического анализа (см. т. II), называются окисью плкшинил Брокмана или окисью алюминия Вилъма. ;
Адсорбционная способность поверхностно активной окиси алюмипия возрастает еще и вследствие того, что посторонние ионы располагаются не только на поверхности окиси, но частично поверхностно внедряются в ее решетку, вытесняя ионы алюминия в раствор. Эта способность к ионному обмену обозначается как «нормутоидпоео свойство окиси алюминия, и та часть адсорбции, которая сводится к этому явлению, называется химической адсорбцией. Фрике (Fricke, 1948—1950) показал, что способность поверхностноактивной окиси алюминия к ионному обмену не обусловлена содержанием в последней щелочей, как это первоначально предполагалось. Действительно, при применении А1аОа, содержащей щелочи (иадрииер, марковской А1аОа, полученной по Брокману), щелочные новы принимают участие в обмене. Однако чистая у?А1аОэ, приготовленная по Фрике, обладает более чем вдвое большей (по сравнению с брок-мановской А1аО3) адсорбционной способностью по отношению, например, к ионам Си-, и часть общей адсорбции, приходящаяся на долю химической адсорбций, в обоих случаях практически оказывается одинаковой (около 40%).
Субокись алюминия. При нагревании в вакууме А1аО3 в смеси с бором до 1300° й,чи с алюминиевым порошком до 1450° происходит сравнительно быстрое нх испарение. По данным Цпнтля (Zintl, 1940), это объясняется образованием летучей субокиси. Ее состав, по данным Грубе (Grube, 1949), соответствует формуле А1аО. Грубе установил, что при нагревании Л12О3 с Si в высоком вакууме До 1800° происходит испарение, связанное с образованием моноокиси кремния и субокисй алюминия (I)
А1аОэ 4- 2Si^2SiO -р А1аО.
В своих опытах он получил два разных сублимата, ив которых один соответствовал по составу соединению SiO, а другой — соединению А1аО.До сих пор не вполне ясно, состоит ли устойчивый сублимат из .этих соединений или из соединений, образующихся из них при охлаждении й результате окислительно-восстановительных реакций (2SiO=Si-|- SiO2; ЗА12О=4А14-А12О3). О существовании А1О .в газовой фазе при высоких температурах см. стр. 387.
Гидрат окиси алюминия и гидроокись алюминия. При добавлении водного раствора аммиака к растворам солей алюминия выпадает гелеобразный осадок, состоящий (если осаждение идет на холоду) вначале из гидрогеля аморфной окиси алюминия, который в соприкосновении с раствором медленно (при нагревании значительно быстрее) превращается в кристаллическую гидроокись, а именно в метагидроокисъ алюминия АЮ(ОН). Метагидроокись получают также (обычно в смеси е аморфный гидрогелем окиси алюминия) при кипячении соли алюминия с веществами, способными при гидролизе связывать ионы Н‘, например с карбонатами, ацетатами, тиосульфатами и т. д. С избытком щелочи осадка це образуется,
Третья группа периодической системы
393
так как прн высокой концентрации гидроксильных ионов происходит образование легкорастворимых гидроксоалюминатов (ср. стр. 353).
Осадок, образующийся при осаждении из кислых растворов, содержащий переменное количество воды (см. ниже) и обладающий в большинстве случаев различной природой, называется оксигидратом в противоположность гидроокиси вполне определенного состава — ортогидрбокиси алюминия А1(ОН)з и метагидрдокиси алюминия АЮ(ОН).
Ортогидроокисъ А1(ОН)з получается в виде белого осадка, обладающего пр рентгенографическим данным кристаллическими свойствами в том случае, если оснащение вели из алюминатных растворов, например при пропускании двуокиси углерода
2[А1(ОН)4]'+СОг=2А1(ОН)3+СО^-|-НйО.
В природе А1(0Н)3 существует в виде моноклинного гидраргиллита. Кристаллическая гидроокись (уд, вес 2,42) в отличий от аморфного окси-гидрата в кислотах растворяется с трудом, При многочасовом нагревании до 100° кристаллическая гидроокись алюминия, опять-таки в отличие от аморфного оксигидрата, пе теряет воды. Но при более длительном нагревании, например в течение 14 суток, в запаянной ампуле при 150° ортогидроокись переходит в содержащую меньше воды, но также кристаллическую метагидроокись А1О(ОН), которая в природе встречается в больших количествах в виде боксита, получившего свое название от города Ле-Бо (Les Ванх, Франция), вблизи которого были открыты большие залежи этого минерала. Боксит образуется при разложении алюмосиликатных пород в тропическом климате, тогда как вне тропиков разложение тех же пород приводит главным образом к образованию каолина.. Искусственно приготовленная кристаллическая метагидроокись называется в отличие от минерала боксита бёмитом.
Другой кристаллической модификацией метагидроокиси алюминия А1О(ОН) является ромбический диаспор, который встречается также в виде минерала. При нагревании до 420° диаспор теряет всю воду и переходит в корунд, тогда как бёмит переходит в безводную окись алюминия уже при 300°, превращаясь ие в корунд, а в кубическую у-охпсь алюминии (см. табл, 68), которая только при температуре светлого каления превращается в корунд.
Таблица 68
Обзор различных модификаций кристаллических окислов и гидроокисей алюминия
А1(ОН)а; । Гидраргиллит <— Байерит . — (метастаб ильный)
АЮ(ОН): Бёмит (йенепт) — Диаспор
300° / 420°
АБФг y-AlgOjj —— --------<——> сыА1дОд
~ 1000° (Корунд)
Объемистый студенистый осадок, образующийся при осаждении солей алюмипия водным раствором аммиака, при высушивании теряет переменное количество воды. На этом основании прежде предполагали существование значительного числа гидратов окиси алюминия, преимущественно весьма сложного состава. Ио в 1888 г. ван Бём-мелеп показал, что такого рода гели ие содержат определенных гидратов и могут терять воду непрерывным образом (см. ниже, кремневая кислота). Эти гели пронизаны миоточкеленными взаимосвязанными мельчайшими порами, в которых вода удержи
394
Глава 10
вается под действием капиллярных сил; кривая обезвоживания подобного геля весьма близка кривой обезвоживания химического соединения. Однако капиллярные силы не могут удерживать воду в том случае, если затруднено образование менисков, например, при проведении обезвоживания пе в вакууме, а путем извлечения воды обработкой геля какой-нибудь жидкостью, сильно поглощающей воду. Вильщтеттеру и Крауту (Wills tatter, Kraut, 1923—1924) и Бильтпу (Biltz) в 1928 г. впервые удалось доказать (первые применяли ацетол в качестве воду отнимающего средства, а второй г— жидкий аммиак) присутствие определенных гидратов окисей в гелях, получаемых при действии аммиака на водные растворы солей алюмипия. К подобным же выводам привели рентгенографические исследования Габера и Бёма (Haber, Bohm, 1925), Бильтца и Мейзеля (Moisei, 1928). Если при нагревании на растворы солеи алюмипия действовать водным раствором аммиака, то получаются осадкиj дающие рентгенограммы, типичные для боксита или бёмита. Осадки, выпадающие при нагревании, имеют такой же слизистый характер, как и те, которые образуются при комнатной температуре и для которых в свежевыпавшем состоянии не может быть доказано кристаллического строения. По Хюттигу, последние в свеже осажденном состоянии следует рассматривать как гидрогели аморфной окиси алюминия, постепенно превращающиеся с химическим связыванием части воды и одновременной кристаллизацией в гидраргиллит А1(0Н)3. Если гидрат окиси алюмипия медленно высаживать1 при комнатной температуре из растворов алюмината, то в зависимости от скорости осаждения (ср. табл. 69) образуется байерит или гидраргиллит — Последний при определенных условиях в виде кристалликов, отчётливо видных под микроскопом.
Baiiepum — у-А1(ОН)3 можно получить не только при правильном проведении гидролиза щелочного алюминатного раствора, но также и при старении аморфного гидрата окиси алюмипия или бёмита под водой (которая не должна обладать ни малейшей кислотностью) или под разбавленным щелочным раствором. Удобно и надежно получать его обработкой амальгамированной алюминиевой жести водой «для электропроводности». При этом вначале образуется рентгеноаморфный тидрат окиси алюминия, который затем в течение нескольких суток переходит, судя по рентгенограмме, в очень хорошо закристаллизовавшийся байерит (Fricke, Schniah, Z. Natur-forschg. 1, 323, 1945). Аморфный гель, образовавшийся вначале при этом методе приготовления (этот же гель можно получить при гидролизе этилата алюминия), чрезвычайно поверхностно активен, и его можно применять для хроматографического разделения катионов. Растворимость его составляет (при комнатной температуре) менее 1(1-е молей на 1 л чистейшей воды (Fricke, S с h m a h, Z. iVatuifoiscli., 1, 322, 1945; Z. anorg. Chem., 255, 253, 1048). При обработке амальгамированной алюминиевой жести кипящей водой или водяным паром получается бёмит АЮ(ОН) (Fricke R., Z. Naturforsch., 2b, 2<i4, 1947). О величине первичных и вторичных частиц байерит;1, бёмита и у-А12О3, а также о величине поверхности и пористости такого рода препаратов см.; Fricke, Z. anorg., chem., 265 , 21 и 41, 1950.
Таблица 69
Обзор различных видон осадков гидроокиси алюминия
а) Из кислых растворов аммиаком осаждаются:
на холоду: аморфный гидрат окиси алюминия;
при нагревании: бёмит А1О(ОН), смешанный обычно с аморфным гидратом окиси алюмипия.
б) Из растворов алюминатов выделяются (например, при пропускании СО2):
а) при медленном осаждении: гидраргиллит а-А1(0Н)3;
б) при быстром осаждении: байерит у-А1(0Н)3.
Разные сорта гидроокиси алюминия обладают специфическими различиями в адсорбционной и ионообменной способности. Как показали Хтоттиг (1931) и Фрике (1936), в ряду: аморфный гидрат окиси алюминия —бёмит — байерит — гидраргиллит — у-А1г03 основной характер поверхности снижается, а кислотный возрастает. Это становится особенно ясным при рассмотрении зависимости pH изоэлектрической точки частцц, суспендированных в воде, от природы этих частиц. Фрике нашел (1949) следующие изоэлектрические точки при 20°;
Аморфный гидрат Бёмит Байерит у-А130з
окиси алюминии
pH 9,45 9,45 — 9,40 9,20 8,00
Третья группа периодической системы
385
При исследовании адсорбционном, способности и каталитической активности недостаточно поэтому характеризовать препарат просто как «окись алюминия» или «гидроокись алюминия». Необходимо рентгенографически определять присутствующую модификацию. Необходимо учитывать также, что, помимо указанных здесь химических различий, па йелнчппу поверхностной активности влияет размер частиц и степень упорядоченности (ср. г. II).
Гидроокись алюминия является исходным продуктом для получения других солей алюминия, особенно сульфатов, а также для приготовления чистой окиси алюминия, пригодной для электролитического получения из нее металлического алюминия. Гидроокись алюминия для превращения в окись, пригодную для этой цели, необходимо сильно прокаливать, чтобы предотвратить поглощение ею воды из воздуха при охлаждении. Аморфный гидрат окиси алюминия, осаждающийся на холоду из водных растворов солей алюминия, находит также техническое применение.
Боксит применяют не только для производства алюминия, но и для изготовления огнеупорных бокситовых кирпичей, получаемых обжигом смеси боксита и глины.
Алюминаты. Выше уже было указано, что алюминаты образуются при растворении в щелочах алюминия или гидроокиси алюминия. В алюминатах алюминий проявляет себя как кислотообразующий элемент. По Вернеру, алюминаты, содержащие воду, состоят из анионов [А1(0Н)4]', (А1(0Г1)Б]2" и [А1(О11)е]3’, например:
А1(ОН)3КОН = К[А1(ОН)4],
А1(ОН)3-Ва(ОН)г=Ва(А1(ОН)6], 2А](ОН)д-ЗСа(ОН)2=Са3[А1(ОН)9]2.
Поэтому их можно рассматривать как гидроксоалюминаты. Для солей Саз[А1(0Н)й]г и Sr3 [А1(0Н)а]а это предположение было позднее подтверждено рентгенографическим определением структуры кристаллической решетки (Brand ей berger, 1933).
Помимо гидроксоплюминатое, гидроокись алюминия с некоторыми основаниями, особенно с Mg(OH)2 nC:i(OH)2, способна образовывать двойные гидроокиси, построенные из чередующихся слоев А1(ОН)3 и Mg(0H)2 или Са(ОН)2 (двойная слоистая решетка, см. т. II), Например, при добавлении едкого натра к раствору, содержащему хлориды алюмппнл и магния, выпадает осадок, который по своему тонкому строению подобен зеленой гидроокиси Co(II, III) (см. т. II). В таких двойных слоистых решетках ионы ОЦ- могут частично' замещаться иона мп СГ. Поскольку в них содержатся молекулы П20, они Могут частично замещаться А1(0Н)3. Вследствие этого подобные соединения могут иметь переменный состав (Feitknecht, Helv. chim. Acta, 25, 406 и 121, 1942). Встречающийся в природе еидрахалумит 2Са(ОН)2-А1(ОН)3.2% Н2О обладает, по дашшм Могавн (Megaw, 1924), структурой с двойной слоистой решеткой. С Sr(OJT)2 и Ва(ОН)2 соединение с тйчздй структурой нс образуется.
Путем сплавления можно получить так называемые «безводные алюминаты». Оки соответствуют главным образом типу Mu[AIsO4i. Соединения этого типа встречаются также в кристаллическом состоянии в природе. Их называют шпинелями:' например, Mg [АЦО41 — обычная шпинель; ZnlALOJ — цинковая шпинель (ганит); Fe[Al„O4) — герципит; (Mg, Fe) {(Al, Fe)3OJ — железная шпинель (плеонаст); (Fe, Mg) {(Al, Ст, Fe)2Oj — хромовая шпинель (пикотит). В шпинелях алюминий может, как это видно из формул, частично замещаться трехвалентным железом и хромом.
Как показывает исследование структур, кристаллические безкодпые алюминаты (шпинели) совсем не являются настоящими алюминатами типа М1|(А1О2)2. В качестве
396 Глава. 10
структурной единицы они содержат не группу [А1О2]_, а группу [М11О4]6-Г. В обычной шпинели каждый атом алюминия «г.ружей на рапном расстоянии (2,02 А) шестью кислородными атомами. Каждый из этих кислородных атомов равноудален от трех; атомов алюминия. Поэтому кислородный атом нельзя приписать какому-нибудь определенному атому алюминия. Зато они могут бытт> связаны с определенными атомами магния, ибо для каждого атома ]1ислорода существует .один атом магния, который к этому кислородному атому расположен ближе, чем другие атомы, и каждый атом магния окружен четырьмя соседними, расположенными вблизи атомами кислорода, удаленными от него на расстояние 1,75 А. Поэтому шпинелям придают формулу AI2[MgOJ, Рентгенографически установлено, что строение решетки Шпинели подлостью совпадает со строением решетки солей Ag2[MoO4J, K2[Zn(CN)4] и им подобных. Это и выражается формулой Al2[MgO4j. Атомная или ионная Группировка [MgO4J существует только как структурная единица в кристалле, и ее не следует рассматривать как радикал или комплекс в химическом смысле. Поэтому о шпинелях лучше говорить ие как о солях, а как о двойных окислах. Шпинели имеют кубическую структуру. Элементарная ячейка (а=8,09 А) содержит 8 формульных единиц. MgAl2O4. Атом Mg так расположен между атомами О, что образует решетку типа алмаза (см. стр. 461).
Помимо пшипелей, известны также и другие двойные окислы с Л12О3. Например, с СаО образуются соединения ЗСаО-А12Оэ, 12СаО-7А12О3 (или 9СаО'5А12О3?), СаО-Л12О3, СаО-2А12Оз п ЗСаО-16А12О3, обладающие довольно сложной структурой кристаллической решетки. Четыре аналогичных соединения образуют А1203 с SrO. В системе ВаО — А12О3 до сих пор найдено только соедппепие ВаО-А12О3. Оно изо-типно с так называемым р-А12О3 (см. стр. 392). То же имеет место для соединений 3 СаО.16Д12О3 и 3SrO.16Al2O3 (W е s I g г е и, Z. anorg. Chem., 234, 1, 1937).
Кислоты почти нс действуют на «безводные кристаллические алюминаты» (шпинели). Наоборот, «водные алюминаты» (гидроксоалюмипаты) легко разлагаются кислотами. При разбавлении водных растворов щелочных алюминатов медленно выделяется кристаллическая гидроокись алюминия А1(0Н)з (Bonsdorf). Ес можно получить таким путем в виде кристалликов, различимых под микроскопом, если разложение вести очень медленно (в течение месяцев) (Fricke). Рентгенограмма этой гидроокиси соответствует рентгенограмме встречающегося в природе гидраргиллита.
Галогениды алюмипия
Фторид алюминия и фтороалюмпнаты. Фторид алюмипия A1F3 образуется при пропускании фтористого водорода над алюминием (1) или над. А1а03 (2) при красном калении. Его можно также получить при сплавлении криолита с сульфатом алюминия (3) и с последующей отмывкой подученного при этом сульфата натрия водой
Al-|-3HF=AlF3-|-3/2H2, (1)
A12O3-HHF=2A1F3+3H2O, : (2)
2Na3AlFa A12(SO4)3—4AlF34-3Na2SO4. (3J-
Полученный таким образом безводный фторид представляет собой
белый порошок удельного веса 3,10, нерастворимый: в воде, в кислотах и щелочах. Он имеет техническое значение, так как его добавляют к шихте при электролитическом получении алюмипия. От фторида алюминия производятся путем присоединения фторидов щелочных металлов и других фторидов двойные пли комплексные соли — (фтороалюмикаты, представляющие собой белые кристаллические, трудно растворимые в воде порошки. Известны три типа фтороалюминатов; М1 [AlFi], М* [A1FS], М* [A1FJ. К последнему типу относится технически важный криолит.
Криолит (ледяной камень) NasAlFe в природе встречается почти исключительно в Западной Гренландии, но зато в очень большом количестве.
Третья группа периодической системы
397
В чистом виде он образует белоснежные моноклинные кристаллы в форме кубов, которые прекрасно раскалываются по плоскостям куба. Удельный вес 2,95, твердость 2,5—3. Он отличается сравнительной легкоплавкостью (точка плавления 1000°). Кислоты на него действуют с трудом, но зато он легко разрушается при кипячении со щелочами или с известковым молоком. Поэтому его прежде употребляли для получения чистой окиси алюминия и соды. II теперь в США (Пенсильвания) с этой целью перерабатывают большие количества криолита. В настоящее время большую часть криолита получают синтетически растворением глинозема и соды в водном растворе плавиковой кислоты или по способу Лезекапна. (Loese-fcahn), основанном на том, что фторид кальция при сплавлении с сульфатом калия и углем образует фторид калия (4), который при взаимодействии с сульфатом натрия переходит во фторид натрия с обратным образованием сульфата калия (5). Из фторида натрия взаимодействием с сульфатом алюминия получают криолит (6):
CaF2J-K2SO4-|-4C=CaS4-2KF+6CO, (4) 2KF-J-Na2SO4 = 2NaF+K2SO4, (5)
12NaF+Ai2(SO/i)3=2Na3AJFs4-3Na3SO4. (6) Помимо использования при электролитическом получении алюминия, криолит применяют также при производстве молочного стекла и эмали.
Структура фтороалюмипатов. Во всех фторо алюминатах, для которых произведены рентгенографические структурные определения, алюминий имеет itoop дин анионное число 6, которое присуще ему- также в кристаллическом A1F3. Во фториде алюминия каждый атом F является общим для двух октаэдров AIFe. В соединении T1A1F4 Вро-
ceT(Brosset, 1937) обнаружил: существование таких же октаэдров, образованных шестью атомами F, из которых четыре являются общими с соседними октаэдрами (рис. 76). В T12A1F5 существуют цепи из ока гад ров; два противостоящих атома F каждого октаэдра принадлежат одновременно двум окатэдрам. В фторо алюминатах типа M3AlFe имеются изолированные октаэдры A1FO. Такой же структурой, как и тстрафторо алюминаты M1A1F4, обладает сложнопост роен ное соединение Na5Al3F14 [хиолит). Его структуру можно вывести из структуры тетрафтороалюмината, если из каждых четырех А1Рв-октаэдров один заменить ионом Na+ (Cowley, Scott, 1948).
Р н с. 76. Элемента риал ячейка T1A1F4.
Тетрагональная, «0=3,61 А; со=6,37Й.
Гидроксофторид алюминия, состав которого может колебаться между A1(OH)2F и A1(OII)F2, получается из раствора, содержащего фторид и сульфат алюмипия, при добавлетшм аммиака. В кристаллической рещетке этого соединения (кубическая элементарная ячейка которого содержит 16 формульных единиц гидр оксофторид а и 6 молекул воды) каждый ион А13+ октаэдрически окружен 6 попами F- или 6 ионами ОН-. Такая решетка чрезвычайно устойчива; она сохраняется при нагревании до 500% хотя при атом отщепляется уже пе только вся кристаллизационная вода, по даже значительная часть воды, связанной в форме гидроксильных групп (Cowley, ScotL, 1948).
Монофторид алюмипия. В отличие от других галогенидов алюмипия фторид алюминия очень трудно летуч. При смешивании с алюминиевым порошком его летучесть при высоких температурах значительно возрастает. Как показал Клемм (Klemm, 1943), ото объясняется образованием моиофторида алюминия A1F, существующего только в газообразном состоянии, а при конденсации разлагающегося; на металл и трифторид.
398
Глава 10
Хлорид алюминия. Alda получается в безводном состоянии при нагревании алюминия в токе хлора или хлористого водорода, или при пропускании хлора над нагретой до красного каления смесью окиси алюминия с углем, или при обработке окиси алюминия смесью хлора и окиси углерода в виде бесцветной (в присутствии примеси хлорида железа — желтоватой) кристаллической массы удельного веса 2,44, которая уже при комнатной температуре заметно летуча и возгоняется при 183°. Точки плавления 192,6° можно достигнуть только под давлением- Вблизи температуры сублимации хлорид алюминия в парообразном состоянии бимолекулярен (структуру молекул А1аС]в см. на стр. 399); около 800° он полностью распадается на простые молекулы Aids. Хлорид алюмипия растворим почти во всех органических растворителях. Его молекулярный вес в эфирном растворе соответствует формуле А1С1з. На воздухе хлорид алюминия сильно дымит вследствие гидролиза влагой воздуха
А1С134-ЗНОН=А1(ОН)з+ЗНСЗ.
В воде ои растворяется очепь легко. Водный раствор вследствие гидролиза обнаруживает сильно кислую реакцию. Степень гидролиза в 0,1 и. растворе, как уже было упомянуто, составляет 2,0%, в 0,001 М растворе — около 4,5%. При выпаривании из водного раствора (получаемого проще всего растворением металла или окиси в соляной кислоте) соль кристаллизуется в виде гексагидрата АЮз-бНаО — бесцветных расплывающихся кристаллов. Если газообразный аммиак пропускать над безводным хлоридом алюминия, то последний присоединяет шесть молекул аммиака с образованием А1С1з‘6ХНз. Это соединение представляет собой легкий белый порошок, отщепляющий NHs только при нагревании до 180°. Безводный хлорид алюмипия образует продукты присоединения со многими органическими веществами, например с хлорапгидридами кислот, эфирами, сложными эфирами и т. д. Он, однако, присоединяет большей частью только одну молекулу этих веществ. Водой эти соединения тотчас же разлагаются. То же происходит и с продуктами присоединения к АЮз других веществ, па пример H’S, SO a, SC] 4, PCIs, а также с двойными солями хлорида алюминия и хлоридов щелочных металлов, имеющих такой же состав, как и двойные фториды, но менее устойчивых но сравнению с ними.
Безводный хлорид алюминия применяют в органической химии для проведения реакций Фриде ля—Крафтса, т. с. для присоединения к бензольному ядру алкильных групп, связанных до этого с галогенами с отщеплением галогенов од орода (папример, СНзС1 + СеНв — СсН5СНз -|- НС1).
Кроме того, его используют в реакциях конденсации, в качестве переносчика галогена и как катализатор в различных процессах, например при дегидрировании и расщеплении. В последнее время Alda в очень больших количествах стали производить в технике и использовать в красочной и парфюмерной промышленности, а также при крекинге нефти.
Потенциал разложения хлорида алюминия, растворенного в расплавленном хлориде натрия, составляет, по данным Неймапа, 1,31 в при 800°. При понижении температуры на 1° он повышается па 1 132 м,в. Для LaCl3 соответствующие числа равны 1,62 в и 1,882 .ив.
Чистый расплавленный хлорид алюминия очень плохо проводит электрический ток. Бильтц (Biltz., 1923) установил, что значительно лучше проводит ток твердый, кристаллический хлорид алюминия. При плавлении последнего тотчас л;с понижается электропроводность. Это обстоятельство исключительное, так как обычно расплавленные соли проводят электрический ток значительно лучше, чем кристаллические. В связи с этим Бильтц нолагает, что твердый хлорид алюминия построен иначе, чем
Третья группа периодической системы
399
расплавленный: первый состоит из ионов, второй — из молекул. Этим же объясняется необычное увеличение объема (почти вдвое) хлорида алюминия при плавлении.
Димерные молекулы АКСД обладают структурой, показанной на рис. 77 и установленной электр оно графически (Pauling, Palmer, Elliot, J. Am. chem. Soc., 60, 1852, 1958). Эта же структура имеет место й для молекул А12Вгв и А121о. В А12(СН3)4СЬ и AL(CH3)4Br2 между двумя атомами алюминия татке существует мостик из атомов галогена (Brockway, Davidson, X, Am. chem. Soc., 63, 3287, 1942). Нелена еще структура молекулы А13(С113)6. По предположению Броявея, атомы А1 в этой молекуле непосредственно связаны, так же как атомы Si л Si2(CH3)6. Электр оно графические определения, одпако, согласуются со структурой, предложенной Рапдлем (Р u n d I с, ,1. Am. chem. Soc., 69, 1327, 1947), в которой оба атома AI связаны посредством двух групп СН3 (см. также Pitzer, G u towski, J. Am. chem.
Soc., 68, 2204, 1946). С точки зрения классической валентности непосредственная связь Al — А1 в этом соединении мало правдоподобна. Теплота диссоциации А12(СН3)0 составляет около 20 ккал!моль. Значение межатомных расстояний во всех перечислен-, них соединениях приведено нище (М —А1 или Si; А — противостоящие атомы галогенов или группы СН3, В — мостиковые атомы галогенов илитруппы СН3).
Соединение Межатомные расстояния, А . величина углов, град
и, > А. м+-*_>в 1. As- '>В Ит——>м в—А1—В А—Al—А
А12сц 2,06 2,21 3,56 3,41 80 118
А12Вг6 2,21 2,33 3,78 3,39 87 115
[ Л1216 2,53 2,58 4,24 3,24 — —
А12(СН3)/1С1г 1,85 2,31 3,43 — 89 - 130
А1г(СН3)4Т5гг 1,90 2,42 3,59 — 90 - 125
A12(CHs)6 2,01 2,2 — -3,2 <А-М-М
Si2(CIl3)B 1,90 ! — 2,34 — —109
Mono мерный А1С]Э обладает особенно сильным поляризующим действием (дипольный момент около 5,3-10-iS ед. CGS). Этим и объясняется его большая склонность к образованию двойпых соединений. Каталитическое действие также связано с поляризующим эффектом, так как в молекулах, присоединяемых к А1С1з, химические связи приходят в возбужденное состояние (см. G о u Ь с а и, Z. anorg. Chem., 254, 126, 1947).
400
Глава 10
Основные хлориды алюминия. Если алюминий кипятить с недостаточным для его полного растворения количеством разбавленной соляной кислоты, то он переходит в раствор в виде; основного хлорида AljfOHJjCl, который можпо высадить из раствора добавлением хлористого натрия. Взаимодействием с Na2^O4 его можно перевести в труди о растворимый основной сульфат AI4(OH)10SO4 (Denk, Bauer, 1951). Другой основной хлорид алюминия А1(О11)2С1 образуется при действии на водный раствор хлорида алюминия металлическим алюминием или при. медленном добавлении к водному раствору A1C1S щелочи. При атом выделившийся вначале гидрат окиси алюминия вновь переходит в раствор с образованием основного хлорида®. В качестве примера основной двойной соли хлорида алюминия можно привести соединение 2Са(ОН)2-• А 1( О Н) 2С1 2TI2О. О но кристаллизуется в гексагона л ьных пл астинках и об л а дает, по данным Фейткпсхта (Feilknecht, 1951), двойной слоистой структурой. Ее стровг
нне можно представить формулой Са2(ОН)в А1С1(Н3О)2, в которой
отрицательно заряженные слои, состоящие из ионов 2Са’+ и 6ОН~, распределены между сдоями, содержащими ионы АР*, С1" и молекулы Н2О. Молекулы Н2О можно удалить без изменения структуры.
Бромид алюминия. А1Вг3—бесцветные, блестящие листочки (уд. вес.3,20, т. пл. 97,5°, т. кип, 265°), бурно разлагающиеся при соприкосновении с небольшим количеством воды и сгорающие в токе кислорода со вспышкой. С большим количеством воды он образует гидрат А1Вг3-6Н2О. Известные в настоящее время двойные бромиды соответствуют тцпам М1 [А1Вг4] и M^AljBr?].
Иодид алюмипия. А1Т3 — уд, вес 3,95, т? пл. 191° и т. кип, 382° — весьма похож на бромид. Известны двойные иодиды типа Mi (Al I41.
В отличие от А1С13, А1Вг3 и АП3 не обладают в твердом состоянии электропроводностью. Степень ассоциации возрастает от хлорида к йодиду (см.: Fischer W., Z. anorg. chem., 205, 37, 1932).
Алюминий органические соединения. Если на металлический алюминий при 100° подействовать диалкилртутью, то происходит, образование алюминййалкиланапример
2AI+3Hg(CH3)2==2Al(CH3)3+3Hg. •
Алюмппийалкнлы представляют собой бесцветные жидкости (точка кипения трнметнл алюминия 130°, триэтйлалюминия1 194е),; воспламеняющиеся на воздухе и бурно разлагающиеся водой на гидроокись алюминия и соответствующий углеводород. Измерения температуры затвердевания, определения плотности пара и другие наблюдения показывают, что эти соединения как в растворенном состоянии вблизи температуры кипения, так и в парообразном существуют главным образом в димерной форме (AIRJa, так же как галогениды А1 и частично талогензамещенные алюминий алкилы.
Алюмпппйалкилы можно применять в ступенчатом синтезе высокомолекулярных парафинов и олефинов из этилена, а также при получении из Этилена производных бензола (Z г с g I с г, Angew. Chem., 64, 323, 1952), Они имеют поэтому промышленное значение. Для их промышленного синтеза используются главным образом следующие реакции:
Mg3Al2+ 6C2H5C1=2A1(C2Hs)3+ 3MgCl21;
А1С13+ 3NaII-f-3C2H4= 2A1(C.3H5)3-|- 3NaCl,
2A1 + 3C2H5C1 4- 3NaH 4- 3CZH4= 2Al(C2H5)3-f- 3NaCl.
* О других растворимых основных солях, которые могут образовываться в качестве промежуточных продуктов при гидролизе хлорида алюминия см. К о h I-schiittcr, Z. anorg. chem., 248, 319, 1941.
Третья группа периодической системы
401
Две последние реакции протекают не непосредственно, а осуществляются через ряд промежуточных реакций, лаирнмср
AIC1H- 2AJ(CsH5)3= 3(С2Н5)2А1С1,
3(СД1512Л1С] т-ЗАта1Г=3(С2Н j)2AlH+3NaGl, 3(С.2Н5),Л]Н+ЗС2Н4= ЗА1(С2Н5)3,
Исходя из 2 молей Л1(С2И5)3, используя 1 моль А1С13, 3 моля NaH и 3 моля С2Н4, получают 3 моля А1(СгП5’)3. Неоднократным повторением процесса получают требуемые количества тршпилальмивия. В димерных, алюминина л килах (Л1Н3)2 часть алкильных групп может замениться на водород. Виберг (1939—1942) при действии тлеющего разряда на смеет, триметил алюминия и водорода полущи соединения Л12Н(СН3)5 и Д13Н2(СП3)4—бесцветные, сравнительно трудцолетучие, очень вязкий жидкости. Они легко образуют соединения присоединения с триметилямивом, например AIIJ2(CHa)2-N(GH3)3. Ирм действии ими на хлорид алюминия можно получить соединение Д12Н4СН3С1. Помимо жидких, образуются также и твердые соединения, из смеси которых обработкой триметнл амином можно выделить двойное соединение A1H3-2N(GH3)2 (бесцветный кристаллы с т. пл. около 90°). При нагревании при пониженном давлении тримотц л амин можно отщепить с образованием нелетучего, твердого соединения [А1113)я. Этим методом был впервые получен в свободном состоянии гидрид алюминия.
Сульфат алюминия; квасцы
Сульфат алюминия. A12(SO4)3 в безводном состоянии — белый порошок с удельным весом 2,71. Из водных растворов кристаллизуется в виде кристаллогидрата, содержащего при обычной температуре 18 молекул кристаллизационной воды. Гидрат Ala(SO4)3.18Н.Ю образует бесцветные игольчатые кристаллы с удельным весом 1,62 и кисловатым вяжущим вкусом. Растворимость его в воде равна;
при 0° 10° 20’ 30’ 50’ 100’
31,2 33,5 36,2 40,5 52,2 89,1 а АЦ(8О4)3 в 100 а Н.20
Вследствие частичного гидролиза водный раствор обладает кислой реакцией
A1s(SO4)3-|-6HOH 2A1(OH)34-3H2SO4.
Продукты гидролиза сульфата алюминия можно получить также в кристаллит веском состоянии. Например, основной сульфат [ЛЮ]25О4-9Н2О встречается в природе в кристаллическом состоянии в виде алюмината (вебстерита). Наоборот, из растворов, содержащих большой избыток кислоты, кристаллизуется кислая соль AlH(3O4)s-i’/2H2O.
Основная двойная соль AlE(S04)3'GCa(OH)2-2GH20, кристаллизующаяся в виде гексагона л ьпьгх иголочек, встречается в природе в ввде минерала эттрингита. Она может также образовываться при затвердевании цемента, содержащего сульфаты, зиа-чптелъпо снижая ого тнердостъ. При нагревании из растпора сульфата алюминия кристаллизуется обычно 16-гидрат (ромбические призмы). Кроме того, существуют гидраты с 27,10 и 6Н2О; при нагревании выше 340° происходит нолпое обезвоживание. Около 600° начинается отщепление SOg (KrauS, 1927).
Сульфат алюминия очень широко применяют в промышленности, главным образом в производстве бумаги. Будучи добавлен вместе с хлористым натрием к бумажной массе, он служит для так называемой проклейки бумаги. Образующийся в результате обменной реакции хлористый алюминий склеивает волокна бумаги. Сульфат алюминия, применяющийся в этом производстве, не должен содержать даже следов железа. Это имеет еще большее .-значение в тех случаях, когда его применяют для дубления кож (белое дубление), а также в качество протравы при крашении ткани. Сульфат алюминия служит также исходным продуктом для получения других солей алюминия, которые можно удобно получать из него взаимодействием с соответствующими солями свинца.
2G г. Реми
402
Глава 10
Применение сульфата алюминия в качестве протравы основано на том, что образующаяся вследствие гидролиза в водном растворе чрезвычайно дисперсная гидроокись алюмипия поглощается и прочно удерживается волокнами шерсти. В свою очередь гидроокись алюминия мщкег связывать органические вещества (с образованием так называемых красильных лаков}, Таким же образом действуют л другие легко гидролизующиеся соли, например сульфат хрома, хлорид олова и др. Шерстяные волокла, обработанные (протравленных) такими солями, могут вследствие способности адсорбированных ими гидроокисей металлов поглощать красители, окрашиваться такими веществами, которые иначе го удерживались бы па волокне. Бумажные волокна не могут, подобно шерстяным, непосредственно поглощать гидроокись алюминия из раствора (горячего). Поэтому при крашении бумажных тканей и получают осадок гидроокиси алюминия внутри волокна, пропитывая его сначала раствором сульфата алюминия и затем действуя па пего щелочью (содой и т. п.).
Промышленное получение сульфата алюминия лучше всего вести растворением чистой (без примеси железа) гидроокиси алюминия в горячей концентрированной серной кислоте. Можно также обрабатывать серной кислотой непосредственно боксит или глину; но при этом возникают затруднения, связанные с очисткой полученного сульфата алюминия от железа достаточно простым способом.
Квасцы. Квасцами называют соединения с общей формулой MIMiI1(SO4)3-12H2O, которые обычно кристаллизуются «правильных октаэдрах, а при особых условиях —также и в кубах. Они принадлежат к классу двойных или же смешанных солей. Двойные соли типа квасцов образуются в первую очередь алюминием, по также и некоторыми другими трехвалентными металлами, например железом и хромом. В качестве одновалентного металла чаще всего участвуют калий, рубидий и цезий. Одновалентный металл может быть замещен радикалом аммония или одновалентным таллием. Различные квасцы изоморфны друг другу.
Кристаллическая структура квасцов. Атомы одповалептпого и трехвалептпого металлов образуют решетку типа каменной соли (длина ребра элементарной ячейки в случае палпевО-алюмипиевых квасцов равна а=12,13 А; в каждом из 8 кубиков, па которые можно разложить элементарный куб решетки каменной соли, На объемной диагонали расположен атом серы, тетраэдрически окруженный четырьмя атомами кислорода. Около каждого атома металла, пак одновалентного, так и тре.твдлеятяого, расположены (> молекул воды. Кристаллизационная вода, следовательно, не группируется в квасцах в виде двойных молекул (Н2О)3 вокруг атомов только трех валентно го металла, как это предполагал Вернер.
Обычными квасцами, по имени которых вся эта группа солей и получила свое название, является сульфат калий-алюмииия (калиевые квасцы) K.A1(SOJ.,-12Н3О. Калиевые квасцы образуют бесцветные октаэдры (при известных условиях также и кубы) с кисловатым, вяжущим вкусом, с удельным весом 1,75. При 92,5’ они плавятся в своей кристаллизационной воде. При дальнейшей! нагревании они легко и полностью обезвоживаются (жженые квасцы). В воде квасцы растворяются с отрицательным тепловым эффектом (—10,12 ккал/молъ квасцов KAI;(SO4)2-12H2O] в отличие от сульфата алюминия, который и в том случае, когда содержит кристаллизационную воду, обладает положительной теплотой растворения. Растворимость квасцов в воде при низких температурах незначительна; но с повышением температуры она сильно возрастает:
ярив' 15® ЗИ° в0° 92,5е *00°
2,95 5,04 8,40 24,8 119,5 154 г КА1(ЗО4)., в 100 г НаО
В абсолютном спирте квасцы пе растворимы.
Третья группа периодической системы
403
В настоящее время для получения квасцов исходят из глины, которую прежде всего обезвоживают и затем вскрывают горячей концентрированной серной кислотой. Из сконцентрированного упариванием сернокислого раствора после прибавления сульфата калия (если имеется достаточный избыток серной кислоты, то для осаждения можно употреблять также и другие соли калия, например хлорид) при охлаждении осаждаются квасцы. Чтобы избежать захвата маточного раствора, смесь при кристаллизации энергично перемешивают; при этом образуется мелко-кристалли-ческий порошок (квасцовая мука).
Прежде для получения квасцов исходили из встречающегося в природе основного двойного сульфата—алунита (квасцового камня) K(A10)3(S04)2.3Ha0 или из сланцевых квасцов — смеси глины или мергеля с серным колчеданом FeS2. Обжигом сланцевых квасцов щолучалц продукт, из которого после длительного выдерживания и многократного обжига после прибавления сульфата калия и выделялись квасцы.
Квасцы применяют в промышленности для тех же целей, как и простой сульфат алюмипия, который все более и более вытесняет их, В тех случаях, когда требуется полностью исключить соли железа, иногда еще предпочитают квасцы, например для травления и при дублении.
В медицине квасцы применяют в качестве вяжущего средства, как слабое раздражающее средство, а также для некоторых других целей.
Другие соединения алюминия
Нитрат’алюмпния. A1(NO3)3 получается при растворения гидроокиси алюминия в азотной кислоте или при обменном разложении сульфата алюминия или же квасцов с цитратом свинца. Эту со ль применяют в качестве протравы для ализаринового красного при крашении тканей, а также при производстве калильпых сеток. В водном растворе нитрат алюминия в значительной степени гидролцзован. Из си льп окисло го раствора соль, содержащую кристаллизационную воду, можпо получить упариванием в виде бесцветных гигроскопических кристаллов. Растворимость при 25° равна 63,7 г AI(NO3)3 в 100 г воды.
При действии раствора питрата алюминия на металлический алюминий можпо получить основные нитраты. Основной двойпой ^нитрат 2Ca(OM)2-Al(OH)2NO3'2H2O имеет, как и аналогичный по составу хлорид, двойную слоистую структуру. Вода в нем связана цеолитяо и теряется уже при комнатной температуре над Р2О6 (Mylius, 1033). Алюминий-кальциевая гидроксисоль соответствующего состава способна, как это было установлено Фейткпехтом (FeitknechL, 1051), связывать многочисленные аниопы, а именно: С1“, п NO,, Br“, СЮ;, МпО;, пикрат-иоп, SOf*, СгО^-,
WO-~, S2O|~ и [Fe(CN)6]3“. Все они в принципе имеют однотипное Строение: между главными слоями, построенными из ионов Саа* с расстоянием между ними 3,32 А, располагаются промежуточные слои, расстояние которых От главного слоя зависит ; от величины аниона. В промежутке между слоями находится вода, связанная в большей или меньшей степени цеолитпо. В большинстве случаев некоторая часть кислот-: ных ионов может быть замещена ионами гидроксила. Это приводит к.тому, что состав
соединений может изменяться в определенных пределах.
I * :
, Роданид алюминия, A1(SCN)3, получаемый взаимодействием сульфата алюминия
в водном растворе с роданидом бария или кальция, применяют как протраву в ализариновом печатании. Для этой дели он поступает в продажу в виде водного раствора.
:i При упаривании его в вакууме образуется безводная соль в виде резипосбразпой мас-
сы. Красная окраска указывает па следы железа (вследствие роданидном реакции на железо),
: Ацетат алюминия (уксуснокислый глинозем). А1(С,НЭОЙ)3 получается
в водном растворе прп взаимодействии сульфата алюминия с ацетатом бария или свинца или при растворении гидроокиси алюминия в уксусной, кислоте. Он гидролизуется значительно сильнее, чем другие рассмотренные соли алюминия. Поэтому из водного раствора постепенно выделяется
J . 26*
-404
Глава 10
осадок гидрата окиси алюминия или основного ацетата. При упаривании остается основная соль, причем выделяется уксусная кислота. Электропроводность нейтрального водного раствора ацетата алюминия поразительно мала; в сильно разбавленных растворах она даже меньше, чем электропроводность эквивалентного количества уксусной кислоты. Ацетат алюминия применяют в красильном производстве для крашения бумажных волокон. При развешивании пропитанных им тканей в теплом сыром помещении (воздушное сушение) ацетат разлагается, причем уксусная кислота улетучивается, а гидроокись алюминия остается на волокне. Широко применяемые в медицине, например для дезинфекции как антисептики, растворы уксуснокислого глинозема содержат основной ацетат алюминия.
Сульфид алюминия. ALS3 можно получить прямым соединением элементов при высокой температуре. Кристаллический сульфид обладает удельным весом 2,37 и температурой плавления 1100°. Водой A12S3 полностью'разлагается.
Кристаллическую структуру A13S3 см. па стр. 407.
Более устойчивы, чем Al2S3, двойные сульфиды GuA1S2 и AgAIS2. Они кристаллизуются по типу халькопирита. Халькопирит CuFeS, имеет структуру, подобную структуре цинковой обмапки (см. т. II); каждый атом S в пей тетраэдрически окружен четырьмя атомами металла, каждому из которых одновременно принадлежат два атома серы. Решетка халькопирита отличается от решетки цинковой обманки тем, что содержит два различных металла и несколько сплющена вдоль оси с, в результате чего возникает тетрагональная симметрия. Такой же структурой обладают соединения СпЛ18с2, AgAISc2, СпА1Те2и AgAlTe2, которые были нолучепы Хапом (Hahn, 1951) нагре-ванисм AloSej ц AI2Te3 с Cu2S или AgoS в откачанной кварцевой ампуле. Галлий и индий также образуют двойные халькогениды типа Х2 со структурой халькопирита (X=S, Se, Тс). Для таллия Хан получил только двойной сульфид CuTlS2, тоже обладающий структурой халькопирита. Некоторые из перечисленных выше двойных халькогенидов имеют характер недальтопидпых соединений. У AglnS, при высокой тем-ufeparype структура халькопирита переходит в структуру, похожую на структуру решетки вюртцита (см. стр. 290).
Нитрид алюминия. A1N образуется при нагревании окиси алюминия с углем в атмосфере азота в электрической печп
’ А 1,0 з+ЗС + N2 = 2A1N 4- 3GO.
При нагревании с едким натром в автоклавах он разлагается с образованием алюмината натрия и аммиака. Эту реакцию пытались использовать для получения аммиака из азота воздуха (способ Серпека), но метод этот пе приобрел практического апачепии.
При нагревании под обычным давлением нитрид алгоминпя разлагается ниже своей температуры плавления. Под давлением 4 ат оп плавится при 2200°.
Структура кристаллической решетки A1N тождественна решетке ВеО (тип вюртцита); а=3,11, с=4,98; Al+~+N=l,87 А.
Карбид алюминии. Л14С3 образуется при непосредственном взаимодействии элементов в условиях высокой температуры. Он растворим в жидком алюминии и выде-ляется из него при охлаждении в виде желтых листочков. Выше 2200° А14С3 испаряется с частичным разложением, так что его пары состоят из смеси Л14С3 и AL Около 2400° общее давление достигает I т. Если создать условия, препятствующие испарепйю И ра.чложению А14С3, то его можно расплавить при 2800°. О поведении Л14С3 по отношению к воде и кислотам см. стр. 507.
Борид алюмпппя. С бором алюминий образует соединения Д1В2 и Л1В12. Гексагонально кристаллизующееся соединение А1В2 замечательно своей тонкой структурой (рцс. 78). Опо имеет решетку типа бруцига (рис. 61) с той лишь разницей, что впей все атомы бора расположены не в двух, а в одном слое.
Третья группа периодической, системы
405
Аналитические сведения. При обычном ходе анализа катионов алюминий осаждают в виде гидрата окиси в группе сернистого аммония.
Осаждение гидрата окиси алюминия сернистым аммонием основано на том, что гидролиз солей алюминия, для которого существует равновесие
2А)Х3 4- .3HOJ-4-aq А12О5-ад4-61Г4-бХ',
протекает подлостью слева направо, так как концентрация ионов И* вследствие образования H2S по уравнению
2Н' -j- S" = H2S
понижается до очень малой величины. Таким же образом действуют все вещества, которые способны значительно понижать концентрацию ионов Н’, например тиосульфат натрия, смесь иодида и йодата (Ср. стр. 868) , карбонат бария и, разумеется, карбонаты щелочных металлов, карбонат аммония и аммиак.
При действии щелочей на растворы солеи алюминия вначале выделяется осадок, который снова растворяется в избытке реактива вследствие образования алюмината.
При сильном прокаливании солей алюминия с летучими кислотами обрадуется окись алюминия А1,03; если эту окись алюминия смочить, небольшим количеством раствора нитрата кобальта .и снова прокалить, то опа окрашивается в красивый синий цвет (тенарова. синь}.
Для доказательства присутствия алюминия в цезиевые квасцы СзЛ1(804)2- 12Н„О. Реакция вместе с тем достаточно чувствительна, и сю можно пользоваться для определения присутствия незначительных количеств алюмипия.
О В
Р и с. 78. Элементарная ячейка Л1Вз.
п=3,60 Д; с=3,24 А.
удобна перевести его
Следы алюминия можно определить в растворе позеленой флуоресценции, появляющейся при прибавлении спиртового раствора морина. Эта крайне чувствительная реакция основана на образовании соединения (аналогичного красильным лакам) между постоянно присутствующей в растворах соли алюминия вследствие гидролиза гидратом окиси алюминия и морином*.
В количественном анализе алюминий обычно осаждается] в виде ‘гидрата окиси и взвешивается в виде окиси алюминий Л12О3.
Из-за гигроскопичности у-А120й прокаливание целесообразно проводить выше 1000° для получения негигроскояичного а-А12О3. При осаждении гидрата окиси алюминия аммиаком необходимо создать определенную концентрацию водородных ионов (4<4рН<л), так как в более кислом, а также и в более щелочном растворе гидрат окиси заметно растворим. Но Фрике (Fricke, Z. anorg. chem., 188, 127, 1930), можно удобно и надежно определять алюминий осаждением двуокисью углерода из щелочных растворов.
Объемно-аналитическое определенно алюминия (в присутствии железа) можно вести, по Даубнеру (D a u 1> п о г, Aug. Chem., 48, 589, 1935; 49,137,1936), осаждением в виде арсената A1AsO4 с последующим Йодометрическим титрованием мышьяковой кислоты. Небольшие количества алюмипия можно определять колориметрически, по Альтену (A lien, Ang. Chem., 48, 273,1935), на основе цветной реакции с эрнохром-
* Морин, или мариновал кислота, содержащийся в желтом дерене (дерево Mor us tinctoria), является органическим красителем [тетраоксифлавопол С^Н^О^ОН^], применяющимся для окрашивания шерсти в желтый цвет.
406
Глава 10
цианином. Для определения содержания окиси в металлическом алюминии (анализ имеет важное техническое значение) пригоден метод Лидера (J a n d о г, Z. anorg. Cliein., 199, 48, 1931), основанный ид том, что при пропускании над металлом при 200° сухого хлористого водорода образуется возгоняющийся хлорид А1С13 (и SiCl4), а А 1,О.з (и FeClJ остается,
ГАЛЛИЙ (GALLIUM) Ga, ИНДИЙ (INDIUM) In, ТАЛЛИЙ (THALLIUM) Т1
Галлий, индий и таллий но свойствам и химическому поведению отличаются от обоих своих более легких гомологов значительно сильнее, чем это имеет место для соответствующих элементов первой и второй главных подгрупп. Это становится понятным при рассмотрении строения атома.
Галлий, индий и таллий но своему атомному строению соответствуют бору и алюминию, поскольку у них у всех одинаковая конфигурация внешней электроппой оболочки (два s-электрона и один /7-электрон). От бора и алюминия они, однако, отличаются, поскольку у них вслед за уровнем s2p следует не уровень s2pe (оболочка инертного газа), а уровень d10 (см. табл. 2 в приложении). Эта особенность строения влияет-па свойства и поведение как свободных элементов, так и их соединений.
Галлий, индий и таллий отличаются, например, от алюминия своими особо низкими температурами плавления. Они отличаются от алюминия также и совершенно особой мягкостью. В то время как твердость и температура плавления сильно уменьшаются от бора к галлию, от галлия к таллию они вновь возрастают. В отличие от этого в первой и второй главных подгруппах твердость и температура плавления уменьшаются последе^ вательно от самого легкого элемента к самому тяжелому. Наиболее значительно отличаются галлий, индий и таллий от бора и алюминия благодаря способности легко восстанавливаться до соединений низшей валентности. Это свойство сильнее всего выражено у таллия, который в‘.св о их важнейших соединениях одновалентен. О менее электроположительном характере галлия, индия и таллия по сравнению с алюминием было упомянуто уже в начале главы.
Среди этих трех элементов ближе всего к алюминию стоит галлий, который в периодической системе располагается непосредственно под А1. Это находит свое отражение в том, что гадлий в отличие от индия и таллия почти всегда сопутствует алюминию в его минералах (хотя вследствие своей редкости он присутствует в них только в виде следов). Далее, общим для алюминия и галлия является то, что.'их полуторные сульфиды гидролизуются водой п поэтому их пельзя получить осаждением из водного раствора, в то время как сульфиды индия и таллия можно осадить из водного раствора, и даже не только сернистым аммонием, но исероводорб-дом. В том, что окисел трехвалептного галлия в отличие от соответствующих окислов 'индия и таллия может кристаллизоваться по тину корунда, опять-таки проявляется более близкое родство между галлием и алюминием,
Относительно свойств окислов, сульфидов н других халькогенидов галлия, индия и таллия, и особенно относительно их структуры, имеются вполне определенные сведения благодаря новейшим исследованиям. Для этих металлов известны халькогениды состава М2Х, МХа’и М2ХЭ. Халькогениды состава М2Х (т. е. халькогениды одновалентно
Третья- группа периодической системы
407
го металла) в твердом состоянии г, полной достоверностью известны только для таллия. Ранее предполагавшееся существование 1п2О и ln2S поставлено, под сомнение повей' пшми исследованиями. Т аи;ко сом нит ел т.п о существование в твердом состоянии халькогенидов одновалентного таллия.
Халькогенид it состава MX — кроме окпелов, который неизвестны,— встречаются у всех трех металлов. Па основании изучения магнитных свойств (Klemm, Vogel, 1934) их отнесли к диамагнитным. Следовательно, металлы в них пе могут Обладать положительной двухвалентностью, так как в этом случае они должны были бы иметь песпареннт.тс «-электроны, следствием чего явился бы парамагнетизм соединений.
Дцамагнстлзм можно обт.яслитъ спариванием s-электронов, т. е. уставов лени ем атомной связи между ионами металла, как это имеет место для иона Hg|+. То, что для соединений галлия :>то действительно имеет место, было показано Ханом и франком (Halm, Franck, 1953) в результате изучения структур GaS и GaSe. Оба соединения кристаллизуются в гексагональной слоистой структуре с расположением атомов S Ga Ga S, где каждый атом галлия окружен тетраэдром из одного атома Ga и трех атомов S.
Для соединений T1S, TISe и InTe структурными исследованиями доказано, однако, что диамагнетизм объясняется тем, что в них ноны металла частично одновалентны, а частично третвалеитны. Эти соединения, следовательно, следует отнести к двойным соединениям типа М1МИ1Х2. Соединения GaTe, IaS и InSu, структуры которых еще до сих пор не изучены, по-видимому, представляют собой переход между этими двумя структурными типами.
Халькогениды МаХ3, соответствующие трехи ал ситному состоянию для всех трех металлов, встречаются в виде окислов, Соответствующие сульфиды, селениды, теллуриды образуются только для Ga и In. Для таллия они существуют только в виде двойных соединений: K2S'T12S3, T12S-T12S3 и Tl2Sc.Tl2Sea.
Из окислов Ga3+, 1пЗ+, TP* GaaO3 встречается в неско.тыгнх модификациях (ср. стр. 411). Одна пз этих модификаций изотипна Sc2O3. 1ц2О3 и Т12О3 также кристаллизуются по типу Sc3O3, который можно вывести из решетки CaF, (см. рис. 62, стр. 298)7 •если в пей изъять четвертую часть анионов, а остальные новы немного сдвинуть со своих мест. GasO3 может также кристаллизоватгщя по типу корунда. Переход от типа корунда к типу Sc2O3 в ряду Л13О3 — GaaO3 — In2O3— Т12О3 обусловливается увеличением радиуса катиона от алюминия к таллию.
Из сульфидов Ga3S3 встречается в нескольких модификациях, две из которых нестабильны и соответствуют типу вюртцита и типу сфалерита, и одна стабильная, представляющая сверуструптуру вюртцитного типа с упорядоченным распределением атомов галлия в тетраэдрических пустотах гексагональной плотнейшей упаковки серы.
In2S3, если его приготовить при низкой температуре, имеет решетку типа шпинели со статистическим распределением атомов индия по всем тетраэдрическим и октаэдрическим пустотам плотнейшей кубической упаковки из атомов серы. При более высокой температуре происходит перераспределение атомов индия с образованием собственно решетки шпинели. В обоих соединениях имеется, следовательно, плотнейшая упаковка атомов S, в пустотах которой располагаются атомы металла. Ga распределяется в тетраэдрических пустотах; для In опи слишком малы. Поэтому он в основном располагается в более крупных октаэдрических пустотах.
Из селенидов и теллуридов Ga2Se3, Ga2Te3 и In2TeB имеют структуру решетки, выводящуюся из решетки типа сфалерита, In2Se3 — диморфное соединение и образует решетку особого типа. Для ее высоко темпер ату рпой модификации можно ожидать слоистую структуру, исходя из внешнего вида кристаллов, похожих на таблички. Тетраэдрические пустоты плотнейшем кубической упаковки, достаточные, по-видимому, в случае атомов Se, для расположения больших атомов нпдия недостаточны^; но подобное расположение имеет место в решетке теллурцца при соответствующей упаковке атомов То; этим и объясняется, почему 1п3Те3 в отличие от Ia2Se3 изотипен Ga2Sc3 и Ga2Te3. Ga;Se3 изотнппы также A13S3 и Al3Sc3 (Geiersberger, 1952).
Сульфиды Ga2S3 и In2S3 близка до структуре и свойствам сульфидам цинка и кадмия, В природе они встречаются вместе с этими металлами. В этом, в частности, проявляется соответствие между аналогами алюминия главной подгруппы III группы и элементами побочной подгруппы II группы, непосредственно предшествующим им в периодической системе. Можно привести еще ряд примеров этого. Так, таллий близок ртути по способности образовывать чрезвычайно большое число соединений с щелочными и щелочноземельными металлами. Подобно щелочным и щелочноземельным металлам, галлий, индий и таллий, если их ввести в пламя горел
408 Глава 10
ки Бунзена в виде достаточно летучих соединений, дают характерные спектральные линии, по которым эти редкие элементы можно легко открыть (ср. стр. 428).
Структура решетки металла. Галлий обладает ромбической структурой, неизвестной для прочих веществ. В этой решетке атомы связаны попарно (Ga<—> Ga=2,437 А). Индий изотипеи у-маргаицу (тетрагональная гранецентрпрованная решетца; каждый атом индия окружен четырьмя атомами на расстоянии 3,24 А й восемью другими на расстоянии 3,37 А). Таллий существует в двух модификациях. «-Т1 (устойчивый при обычной температуре) имеет решетку типа магния (гексагональная плотная упаковка <1=3,45, с—5,52 А). Модификация, устойчивая при высокой температуре (при быстром охлаждении до комнатной температуры существует в мстастабильном состоянии), Р-Т1 образует гранецентрироваппую кубическую решетку (кубическая плотная упаковка; а = 4,84 А).
ГАЛЛИЙ, Ga
Распространение в природе. Галлий встречается в природе как спутник цинка во многих обманках, но только в исключительно малых количествах (0,002%: и меньше). В виде следов он встречается почли как постоянный спутник алюминия. Во всех сортах технического А1 его можно открыть спектрально. Самый богатый галлием минерал —гер.маншп из Тсумба в Юго-Восточпой Африке. В нем содержится 0,6—0,7% галлия.
Исторические сведения. Ле к ок де Буабодран в 1875 г. открыл галлий спектроскопически в цинковых рудах Пиерефптского месторождения во Франции, Он назвал элемент галлием в часть своей родины (Галлия).
Еще в 1874 г. Менделеев предсказал па основ аник периодического закона (пеза-иолнеппая клетка в таблице) су лфство ванне тогда еще нс известного аналога алюминия — энаалюминия. Галлий оказался идентичным предсказанному элементу.
Получение, Методы выделения и получения галлия были разработаны самим первооткрывателем и его учеником Юнгфлойшом. Для получения в лабораторных условиях лучше всего сначала осадить галлий в виде цианоферрата(П). Последний при склоном нагревании превращается в смесь GaaOg н FeaOs. Смесь окислов. сплавлением с бисульфатом калия переводят: в растворимое состояние. Затем пз солянокислого раствора действием большого количества едкого кали высаживают железо (окись галлия растворима в щелочи). После этого галлий можно выделить ни щелочного раствора электролитически. Кейл (Keil, 1926) рекомендует сплавлять цпагюфер-рат(11) с твердым едким кали в серебряном тигле, затем растворять плав в воде (при этом железо выделится в виде гидроокиси) и из фильтрата после подкисления соляной кислотой аммиаком осадить галлий в виде гидроокиси. Гидроокись при прокаливании переходит в окись, из которой можпо получить металл, сильно нагревая вещество в токе водорода, Ричардс (Richards, 1923) описал метод, ио которому свинец,, полученный при рафинировании дистилляцией цинка (выделенного из руд, содержащих галлий), перерабатывается на галлий. Иа горманита галлий мощно просто и количественно получить по методике Берга и Кейла (Berg, Keil, 1932), которая основана на легкой растворимости GaClj в эфире (см. стр. 410).
В Леопольдсхолле несколько лет назад стали получать галлий в заводских условиях при переплавке мансфсльдского медпого сланца. Содержанию галлий остатки, в которых имеются также тяжелые металлы и алюминий — преимущественно в виде сульфатов, фосфатов и молибдатов (Feit, 1933),— прежде всего обрабатывают NaOH. Затем отфильтровывают выпадающие гидроокиси тяжелых металлов, а раствор нейтрализуют. Выпадающий при этом осадок, помимо галлия, содержит еще цирк и алюминий в виде фосфатов и сульфатов. Осадок вновь растворяют В серной кислота и раствор разбавляют водой. При этом в результате фракпдгонлого осаждения сероводородом из сернокислого раствора раствор обогащается галлием в результате отделения остатков молибдена и цинка. Затем раствор смешивают с твердым едким натром, причем фосфорная кпелога Отделяется в виде тринатрийфосфата, почти нерастворимого в щелочи. Оставшийся раствор подвергают электролизу. При тщательной работе получается почти всегда чистый галлий.
Третья группа периодической, системы
409
Свойства. Галлий—относительно мягкий, ковкий металл, блестящего серебристого цвета с голубовато-серыми штрихами. Он плавится при 29,78° (теплота плавления 19,16 кал/а). Расплавленный металл при охлаждении не застывает немедленно, если только его не помешивать палочкой; без такого вмешательства он может оставаться жидким месяцами. Жидкий галлий используют для наполнения кварцевых термометров, предназначенных для измерения высоких температур (применение галлия для зубных пломб см. т. IT, гл. 9).
Таблица 70
Свойства галлия в сравнении с предсказанными свойствами эка алюминия
Нкаалюмипий Галлий
Атомный вес — 68 Удельный вес 6,0 Атомный объем 11,5 Удельный вес окисла 5,5 Металл легкоплавкий Металл устойчив па воздухе Металл способен к образованию квасцов Температура кипения хлорида пиже, чем хлорида цинка Металл легко получается восстагго-впеписм Сульфид пс осаждается сероводородом Атомный вес 69,72 Удельный вес 5,9 Атомный объем 11,8 Удельный вес окисла 3,9 Температура плавления металла 29,78° Металл устойчив на воздухе Галлий способен к образованию квасцов Температура кипения GaCh 201°; ZnCl2 730s Металлический галлий получается при нагревании окисла в ток<> водорода или электро-: литическим путем из водного раствора. Если галлий я растворе присутствует одни, то сероводород не осаждает1 сульфида галлия. (Однако сульфид галиия: можно почти количественно со осадить с другими сульфидами, если они осаждаются из щелочного илп уксуснокислого раствора.)
В табл. 70 приведены свойства таллия л для сравнения свойства окаалюминил, предсказанного Менделеевым на основании периодического закона.
Структуру решетки галлия см. на стр. 407. На воздухе галлий устойчив при обычной температуре. Хлор и бром сильно Действуют на металл уже па холоду., С иодом галлий соединяется при нагревании. Из водного раствора галлий легко можно выделить электролитически, по количественно это сделать трудно. Нормальный потенциал галлия относительно нормального водородного электрода равен —0,52 а.
Соединения галлия. В своих соединениях галлии, как правило, проявляет положительную трехвалентностъ. Соли его бесцветны, в водном растворе сильно гидролизовапы, сильнее даже, чем соли алюмипия, на которые они в большинстве случаев похожи по своим свойствам.
Так же как в случае алюминия, из раствора соли галлия под действием веществ, сдвигающих равновесие гидролиза вследствие понижения концентрации ионов водорода, выпадает белый гидрат окиси. Прибавлением винной кислоты можно предотвратить осаждение вследствие комплексообразования (также как и для алюминия).
Из-соединении двухвалентного галлия известны галогениды GaCl2, GaBr2M халъкся гениды GaS, GaS'e и GaTe. Окись двухвалентного галлия* до СпХ пор не .удалось полу
410
Глава 10
чить, Это тем удивительнее, что халькогениды двухвалентного галлия относительно устойчивы. Соединения двухвалентного галлия, имеющие солеобразный характер, менее устойчивы. Они легко окисляются или распадаются при реакциях окисления па соединение трехвалентного галлия и металл. Еще менее устойчивы соединения одновалентного галлия. Из соединений одновалентного галлия существуют, но-видимому, окисел Ga2O, сульфид Ga2S и селенид Ga2Se. Соединения трехвалентного и одновалентного галлия диамагнитны. Интересно, что это справедливо и для приведенных выше соединений двухвалентного галлии, но крайней мере в твердом состоянии. Их диамагнетизм обусловлен тем, что в кристаллической решетке соединений двухвалентного галлия размещены не ионы Ga2’, а ионы GaF. Ионы Ga2+ здесь связаны попарно гомео-полярной связью, благодаря чему происходит насыщение спинов неспаренных электронов. Образование иопа Ga2’ полностью соответствует образованию ионов Hg+ для ртути
I I
Ga2’ Hg’
(см. т. II). Следует отмстить существование гибрида галлия, летучего при обычпой температуре (см. стр. 412).
1. Соединения трехвалентного галлия
Хлорид СаС13, образующий длинные белые кристаллы с уд. весом 2,47, получается при нагревании металла в токе хлора или хлористого водорода; т. пл. 77,9°, т. кип. 201,3е1 (теплота испарения 11,4 ккал/молъ при температуре кипения; теплота плавления при температуре плавления 5,2 ххал/молъ1, теплота сублимации при температуре плавления 17,0 ккал/молъ). Хлорид галлия легко можно очистить возгонкой в атмосфере азота илн в токе хлора. Определения плотности пара показывают, что пар при температуре, близкой к температуре кипепия, почти целиком состоит из удвоенных молекул (GaCl3)a. При 600° плотность пара соответствует мономолекуаярной форме. Прп более высокой температуре можно обнаружить продукты распада, а также молекулы GaCl3. Расплавленный трихлорид почти совсем не проводит электрический ток. Теплота образования GaCl3 равна 125 ккал/моль. С водой GaCl3 реагирует с большим выделением тепла. На влажном воздухе расплывается с гидролитическим отщеплением хлористого водорода. Водный раствор имеет сильнокислую реакцию п легко выделяет гидроокись. GaCl3 легко и без разложения растворяется в эфире. Хлорид экстрагируется эфиром и из солянокислого раствора. Это свойство мощно использовать для отделения галлия от других элементов (Swift, 1924).
Как показал Улих (IJlich, 1941), GaCl3, как и А1С13, является катализатором органического синтеза. При синтезе некоторых углеводородов каталитическое действие хлорида галлия превышает действие хлористого алюминия.
Бромид GaBig (уд. вес 3,69; т. пл. 121,5°; т, кип. 279°, теплота образования 92,4 к кал/м о лъ) и иодид Gal3 (уд. вес 4,15; т. пл. 212°; т. кпп. 346°, теплота образования 51 ккал/молк) бесцветны и в значительной степени похожи на хлорид. Одпако Gal3 в парообразном состоянии, по-видимому, моиомерен. Как у GaCl3, так и у GaBr3 при нагревании степень ассоциации уменьшается, а у Gal3— увеличивается. Это связано с тем, что для иодида в отличие от хлорида и бромида теплота испарения больше, чем теплота диссоциации Ди мерных молекул (Fischer, 1936). Структуры молекул Ga2Clfl и Ga2Bre соответствуют структуре молекулы А1аС16, т. е. опп образуют сдвоенный тетраэдр с общим ребром и с атомом галлия в центре; С1 <—► С1 = 3,53 А, Вг *-.-» Вг= = 3,83 А. Напротив, мономерный Gal3 имеет плоскую структуру — равносторонний треугольник с атомом галлия в центре; I I —4,16 A (Brode, 1940).
Безводный фторид GaF3 (уд. вес 4,47; т. пл. > 1000°; т. возг. ~950°) растворяется в воде и в разбавленной кислоте только частично даже при нагревании. Напротив, GaF3-3H2O, образующийся при растворении гидрата окиси трехвалентного галлия в водном растворе фтористоводородной кислоты и при последующем упаривании, легко растворяется в разбавленной соляной кислоте. При добавлении NH4F к концентрированному раствору GaFj-3IT2O, по-в»димому, выделяется гексафторогаллат аммония (NH4)3|GaF6], кристаллизующийся в октаэдрических кристаллах. Ряд Других фторогалла то в был получен Пагом (Pugh, 1937). Большинство пз них отвечают формуле Ml(GaF5-H2O],
Окисел трехвалепгпого галлия. Ga2O3 легче всего получить, нагревая нитрат или сульфат. Это белый порошок, который при сильном нагревании теряет способность
* GaO был обнаружен на основании изучения полосатых спектров. Теплота его образования из атомов составляет — 58 ккал/молъ. Для InO па основании изучения полосатых спектров теплота образования составляет 25 ккал/молъ. Т1О, по-видимому, пе существует в газообразной форме.
Третья группа периодической системы 411
растворяться в кислотах и щелочах, подобно окиси алюминия. При нагревании в токе водорода сначала происходит восстановление, по-видимому, ДО Ga2O, а Чатем—и До свободного металла.
Ga2Oa встречается в нескольких модификациях, IIpis сильном нагревании геля Ga2O3 или GaO(OH) образуется мет а стабильная a-Ga3O3. Это гексагональные ромбоэдрически-гемиодри чести* кристаллы, изотопные корунду (а = 5,31 А, а == 55°50'). 0-Модпфикацпя GaaOa стабильна от комнатной температуры до температуры плавления. Онаизотипна 0-А12и3: как промежуточный продукт при нагревании геля окисла обра-зуется Y-Ga2O3. Строение его решетки до сих пор еще неясно, по оно, одпако, пе родственно (Ноу, 1952) шпиш-леподибной структуре у-А13О3 (к а К это предполагал Бём, 1940). Другими модификациями окиси галлия являются: б-Са,03, образующийся при умеренном нагревании Ga(NO3)3 и имеющий структуру Sc2O3, а также e-Ga2Oa, структура которого еще ие определена. Все эти модификация при нагревании с водой под давлением до 300° или в сухом состоянии до 600—1000° переходят в ^-модификацию, а- п 0-Модификацин Ga2O3 могут растворять значительное количество А13О3 с образованием смешанных кристаллов. Кроме того, Ga20a образует с А13О3 двойное соединение с формулой GaAlO3. Теплота образования a-Ga2O3 составляет 258 ккал/молъ. Ga2O3 может давать двойные соединения с MgO и ZnO со структурой шпинели.
Гидрат окиси галлия и гидроокись галлия. Из водных растворов солей трехвалентного галляя при действии веществ, понижающих концентрацию ионов водорода, выделяется белый желатиноподобпый осадок, аморфный по рентгенографическим данным и содержащий переменное количество воды (гидрат окиси галлия Ga2O3-aq). Осадок растворяется как в кислотах, так и в сильных основаниях. Кроме этого (в отличие от гидрата окиси алюминия), гидрат окиси галлия растворяется в значительном количестве концентрированного раствора аммиака. Растворимость в щелочи уменьшается при старении осадка. Растворимость, помимо этого, зависит от количества осадка, что является признаком того, что гидрат окиси галлия присутствует в растворе не только в молекулярно-дисперсном состоянии, по частично и в коллоидном (правило Оствальда для осадков; ср. т. И). _ >
При медленном осаждении можно получить микрокристаллический осадок гидроокиси галлия (III) определенного состава, GaO(OH), (метагидроокись галлия), как из кислого, так и из щелочного раствора. Эта форма гидроокиси имеет, как это показал Дж. Ббм (Bohm, 1938), структуру, аналогичную диаспору AIO(OIJ). GaO(OH) может включать в свою решетку значительное количество АЮ(ОН). Точно так же АЮ(ОН), как в форме диаспора, так и ббмитй, может присоединять значительное количество GaO(OII) с образованием смешанных кристаллов.
Сола, присутствующие в щелочном растворе гидроокиси галлия, называются галлатами. Гидроокись галлия имеет более кислотный и ыепее основной характер, чем гидроокись алюмипия. Поэтому соли трехвалентного гал.чня легче гидролизуются, чем соли алюминия. Это используют для препаративного отделения галлия От алюминия (см. стр. 408).
Сульфид. Ga2S3 был впервые получеп непосредственным синтезом из элементов в 1930 г. (Brukl)*. Чтобы взаимодействие прошло полностью, необходимо очень сильное нагревание (выше 1200°). GaaS3 имеет чисто желтую окраску, уд. вес 3,48 и т. пл. около 1255°. Водой он разлагается с выделением H2S.
Нагреванием до 800° л токе водорода Ga3S3 легко восстанавливается в GaS**. GaS бледно-желтого цвета, уд. вес 3,75, т. пл. ~965°. Этот сульфид устойчив к действию воды. При па грев нии в высоком вакууме происходит распад по уравнению: 4GaS = GaaS3 -Ф Ga2S. Образующийся при этом Ga2S имеет серовато-черную окраску и уд. вес 4,22. При нагревании ыоиосульфид распадается при температуре ниже температуры образования па Ga2Sa и Ga.
Сульфат галлия. Ga3(SO4)3 кристаллизуется из водного раствора, как и сульфат алюминия, с 18 молекулами воды в мягких белых пластинках или звездах. Сульфат
* Образовать белого осадка при пропускании HaS в сильйокцелый раствор соли галлия, наблюдавшееся Лек оком де Буабодроном в 1881 г., относительно которого было предположено, что происходит образование сульфида, объясняется (Brukl), по-видимому, тем, что при этом вследствие гидролиза возникает гидрат окиси галлия; действительно, так как сульфид галлия в соприкосновении с водой неустойчив, его нельзя получить из в один х растворов.
** Препаративное приготоплеппе GaS, по Клемму (Klemm, 1934), лучше осуществлять непосредственным синтезом из элементов, применяя вычисленное количество серы.
412 Глава 10
легко растворим в воде. При нагревали и. обезвоженной соли выше 520° она претерпевает разложение с отщеплением. SO3, При высокой температуре сульфат» значительной степени летуч. Поэтому при упаривании с концентр и ров ап ной H2SO4 до появления дыма SO3 он теряет в весе. Давление диссоциации SO3 составляет, по Маршалу (March al, 1926), при 700° 391 .ил» р/п ст, общее давление пара при 690° достигает 1 атм.
С сульфатом аммония сульфат галлия образует двойную соль, относящуюся к классу квасцов NH4Ga(SO4)2-12Н2О. В 1918 г. Деппис (Dennis) получил изоморфный Квасцам двойной селенат галлия и цезия CsGa(SeO4)2‘12Н2О. Среди основных двойных сульфатов (Einecke, 1941) имеется соединение (NH4)(GaO)s(SOi)2-4II2O изотопное минералу алуниту K(A1O)3(SO4)s ..31Т2О.
Нптрат галлня кристаллизуется из азотнокислого раствора в октаэдрах е восемью молекулами воды Ga(NO3)3-8H2O. Это р а сшивающиеся бесцветные призмы, сильно преломляющие свет (т. пл. ~65°). При нагревании в токе воздуха при температуре около 40° происходит переход в безводную соль Ga(NO3)3.
Нитрид галлия. GaN образуется при нагревании металла в атмосфере NH3 при 900—1000°. Это темно-серый порошок с уд. весом 6,10. Большинство кислот па него не-действует. При нагревании л токе кислорода нитрид медленно переходит в Ga^Oj. (Johnson, п 1932), GaN кристаллизуется гексагонально (решетка вюртцита,, а = 3,18,А, с = 5,17 А.); так же кристаллизуется и ШУ (ср. Стр. 416).
2. Соединения одновалентного и двухвалентного галлия
Хлорид двухвалентного галлия. GaCl2 получается в результате неполного сгорания металла в токе хлора или при нагревании хлорида трехвалентного галлия с металлическим галлием. GaCls образует бесцветные, прозрачные кристаллы (т. пл. 170,5°, т. кип. 535°). Расплав можно легко переохладить; при обычной температуре переохлажденный расплав можно сохранять жидким в течение месяцев. В отличие от GaCl3 расплавленный GaCl2 хорошо проводит электрический ток. Плотность пара. GaCG при 1000° соответствует формуле GaCl2; при более высокой температуре наступает диссоциация.
С водой GaCl2 реагирует с энергичным выделением водорода. Это же явление наблюдается при разбавлении раствора галлия соляной кислотой. Вероятно, такой раствор содержит хлорид двухвалентного галлия. GaBr2 по своим свойствам аналогичен GaCl2.
Окись одновалентного галияЛ Ga2O, по Дюнро (Dupre, 1878), образуется при наг ре пани и Ga3O3 в токе водорода до температуры красного каления. Б рук лом (Brukl, 1931) было указано, что в чистом состоянии Ga2O можно получить, нагревая спрессованную смесь Ga2O3 и Ga. GaaO, по Бруклу, образует темпо-коричпевый порошок (уд. вес 4,77), устойчивый при обычной температуре в атмосфере сухого воздуха, При 500° в высоком вакууме возгоняющийся без разложения и только выше 700° разлагающийся на Ga и Ga2O3. Теплота образования Ga2O около 82 ккал/.крль. Так как в результате новых исследований очень сомнительным представляется существование в твердом состоянии 1н2О и Jn2S, возможность приготовления в твердом состоянии Ga2O требует дополнительной проверки.
3. Алкильные соединения галлия и гидрид галлия
Трпэтнлгаллнй был получен Делисом [(1932) взаимодействием диэтйлртути с металлическим галлием; 3Hg(C2H3)2 -ф- 2Ga == 3Hg -ф 2Ga(C2H3)3. Это бесцветная жидкость с неприятным запахом (уд. вес 1,058, т. пл. —82,3°, т. кип. 142,6°), на воздухе воспламеняющаяся и разлагающаяся водой с сильным взрывом. При низкой температуре реакция с водой протекает по уравнению Ga(C2n3)a-|-HOH — Ga(C2II3)2OH-|--|-С2Нв. Более устойчивыми, чем три этил галлий, являются его продукты присоединения с аммиаком и эфиром; Ga(C,2H 5)3 NH3 и Ga(G2H 5)3-(С2Н 3)2О. Подобным же образом, как и три этил галлий, можно приготовить и трпмети л галлий (т. пл, 15,8°). При взаимодействии Ga(CH3)3 с В2Н(; при —45° образуется] диметил гаЛлий бор анат Ga(GH3)2BH4 т, пл. 1,5°, т. кип. 92°). ,
Гидрид галлия. Триметил галлий при взаимодействии с водородом в тлеющем разряде образует тетра метилди га план в виде бесцветной вязкой жидкости..
Это вещество выше 130° распадается на Ga(CH3)3, Ga и Н2 с промежуточным образованием гидрида галлия (дигалл ana) Ga2II3. Если связывать Ga(GH3)3 добавлением три-
Третья группа периодической системы 413 этиламина, с которым он образует кристаллический продукт присоединения, то распад наступает уже при комнаткой температуре и дигаллап можно выделить нераз-ложившимся. Гидрид галлия — легкоподвижная, бесцветная жидкость (т. кип. 139°, т. пл. —21,4°), распадающаяся при температуре выше 130° со значительной скоростью на Ga и Н2.
Из смешанных при комнатной температуре эфирпых растворов хлорида галлия и галланата лития LiGall/,, как это нашли Внберг и Шмидт (Wiberg, Schmidt, 1952), постепенно выпадает белый осадок, который представляет собой ' высокой о лимерпый твердый гидрид галлия (GaH3)x. Эта форма гидриДа галлия по свойствам и способу образования полностью соответствует гидриду алюминия. Виберг предположил, что первоначально это соединение присутствует в растворе в качестве мономера, а затем постепенно полимеризуется. Благодаря полимеризации устойчивость соединения увеличивается во много раз: в то время как в растворе уже при 35° наступает разложение с выделением водорода и металлического галлия, димерный жидкий гидрид галлия разлагается лишь выше 130'-', а твердый начинает отщеплять водород около 140°. При этом, но-видпмому, сначала возникает низший гидрид (GaH)?, поскольку лишь выше 350° наблюдается выделение металлического галлия. Твердый гидрид галлия относительно устойчив по отношению к воде; разбавленными кислотами он, однако, энергично разлагается с выделенном водорода. Литийгаллийгидрид, соответствующий литий-алюминцйгидриду, образуется при взаимодействии хлорида галлия и гидрида лития в эфирном растворе (Schlesinger, 1947; Wiberg, 1951). Галланат серебра AgGaHj, полученный Виборгом и Хенли (Henle, 1952) взаимодействием AgClO^ и LiGaH* в эфирпом растворе при температуре —100°, уже выше —75° разлагается с выделением водорода и серебра, Еще легче разлагается галлогидрид таллия TI [Galich, который был получен Виборгом и Шмидтом в займ о действием TIGlj с LiGaH4 в эфирном растворе при температуре —115°. Это белое, твердое, растворимое в эфире вещество, разлагающееся уже при —90° на таллий, водород и гидрид галлия.
ИНДИЙ, In
Распространение в природе и исторические сведепия. Индий встречается в очень незначительных количествах в форме сульфидов как примесь во многих обманках. Индий был открыт в 1863 г; Райхом (Reich) и Рихтером (Richter) в остатках от переработки цинковой обманки из Фрейбергского месторождения, которую они исследовали спектроскопически на присутствие таллия. Новый элемент был обнаружен пр своеобразной индиго-синей линии п был назван по ее цвету.
Впачале ипдий считали двухвалентным. Однако Менделеев на основании его свойств поставил его на правильное место в периодической системе й установил его трехвалентность. Валентность, равная трем, была вскоре подтверждена определением удельной теплоемкости, исходя из которой по закону Дюлонга и Г1ти определяется атомный вес и из сравнения последпего с молекулярным весом соединения устанавливается валептпость. Валентность три была установлена, кроме того, путем вычисления атомного объема и открытием соответствующих квасцов. В настоящее время, после непосредственного установления ого порядкового числа (49) па основании закона Мозли по характеристическому рентгеновскому спектру, не приходится сомневаться в положении индия в периодической системе.
Получение. В качестве исходпого продукта для получения индия, в первую очередь используются полупродукты от выплавки свинца и цинка из руд, содержащих иидий*.
Переработку производят в зависимости от природы материала. Например, если имеется в паличии цинк е относительно высоким содержанием индия, то ого обрабатывают соляной кислотой в количестве^ недостаточном для полпого растворения. Иидий при этом остается в лиаме, который содержит немпого неблагородных примесей. Из раствора этого шлама большая часть имеющихся тяжелых металлов осаждается сероводородом. Из фильтрата после прибавления аммиака индий выделяется в виде
* В сравнительно большом количестве иидий содержится в отделяемом фракционированной перегонкой сыром цинке по способу Нью-Джерси (см. т. II, гл1. 9). В последнее время из пего было получено в промышленности довольно Значительное количество ипдия (Ensslin, 1940—1941).
414
Глава 10
Гидрата окиси, обычно вместе с железом, которое перед осажденном должно быть окислено в соль трехвалептпого железа. Способ отделения железа от индия зависит от Содержания последнего. Выделение металлического индия из окисла нагреванием в тоне водорода или электролизом кислых растворов не представляет особых. труд-ностей из-за легкой восстанавливаемости соединений индия.
Свойства. Индий — серебристо-белый, обладающий сильным блеском металл. Он очень мягкий, легко режется ножом и плавится при весьма низкой температуре : (температура плавления -'-156,4°). Температура кипения, напротив, довольно высока (—2300°). Удельный вес 7,31. Удельная теплоемкость 0,057. Относительно структуры его кристаллической решетки см. стр. 408.
В атмосфере сухого воздуха индий не теряет блеск, при нагревании Он покрывается пленкой, по сильно окисляться начинает только при температуре выше температуры плавлении. При нагревании в токе хлора индий энергично сгорает. Оп непосредственно соединяется и с другими галогенами, а также с серой. Нормальный потенциал индия по отношению к нормальному водородному электроду составляет (Kangro, 1954) при 25" —0,336 в (температурный коэффициент равен 0,965 на 1°).
Индии слабо радиактивон. Изотоп In115, который является главной составной частью индия (95,77 ат.%), распадается с испусканием р-лучей (энергия распада 0,63 Мэе}. Период полураспада 6- 10й лет. Конечный продукт распада — стабильный изотоп олова Snr13. Вследствие очепь небольшом скорости распада радиоактивность ипдия наблюдалась только в ранний период его существ овация.
СОЕДИНЕНИЯ ИПДИЯ
В своих соединениях индий обычно положительно трехвалентен. Наряду с этим он может быть положительно одно- и двухвалентным, особенно в соединениях с галогенами и халькогенами. Для соединений индия, происходящих от его низших степеней окисления, характерен распад в водной среде на соединения трехвалентного индия и свободный металл.
Соли трехвалептпого индия, как правило, бесцветны. Соли, происходящие от обычных кислот, за исключением оксалатов, фосфатов и сульфидов, легко растворимы в воде. В растворе они сильно гидролпзовапы. В щелочной среде образуются кислород-Содержащие соли, в которых индий входит в состав аниона, называемые индатами. Индий также может образовывать ацидоемдинтия. В водном растворе ипдий пе образует аммиачных комплексов, одпако из щелочного раствора можно получить продукт присоединения пиридина к хлориду индия состава 1пС13.ЗС5ИьМ (длинные бо.’ше£иглы, т. пл. 253"). В табл. 71 приведен обзор важнейших соединений индия.
Соединения одновалентного и двухвалентного индия в общем устойчивее соответствующих соединений галлия. В твердом состоянии соединения двухвалентного индия, также как и соединения одно-и трехвалентного индия, диамагнитны и, следовательно, подобно соединениям двухвалентного галлия, но-видимому, построены из ионов In* и In3*. В парообразном состоянии соединения двухвалентного ипдия, так же как и соединения одновалентного и трехвалентного индия, могут находиться в мономерном состоянии, как это было установлено Робертом (Robert, 1936) в результате измерения давления пара.
Теплоты образования: InCi 44,6, InCl2 86,8, InCl3 128,5, InBr3 97,2, Inl3 56,5,. In2O3 221,9 хкал/молъ. Теплота испарения: InCl 21,2, InCl2 42,1, 1пС13 37,8, InBr 20,7, InBr2 20,5, InBr3 25.9, Ini 21,5 ккал/моль. Растворимость в 100 а воды прп 22°: InF3 8,50 г, 1пС13 183 г, 1пВт3 536 г, 1н13 1090 г; в 100 г абсолютного спирта; 1пС13 114 г, 1пВг3 285 a (Ensslin, 1942). ~
Потенциал пары 1п"71п- при 25° —0,404 в (температурный коэффициент 0,877 .ms на 1°). Теплота образования ионов (при 25° и иоппой активности 1): Iri = In‘" + Зе + + 43,43 ккал; In — In" + е + 12,47 ккал; 1п"‘ -}- 21п 31п‘ — 6,02 ккал (Kangro,
1954).
Третья группа периодической системы
415
Обзор соединений индия
Таблица 71
Г.пл,, °C |1 Окраска || ТЛ1Л., °C Окраска Т*пл Окраска
InF3 1170 Бесцветный ШР2 — Бесцветный __ —
1пС13 586 Бесцветный, в расплаве желтоватый 1пС12 ~ 235 Бесцветный, в расплаве желто-коричневый InCi 225 Лимонно-жел* тый, выше 120° малиново-красный, в расплаве темно-Красный
1пВт3 436 Бесцветный, в расплаве темно-коричневый 1пВг2 Роговидный, почти бесцветный, в расплаве темнев желтый InBr Карминово-красный, в расплаве интенсивно темдо-кра-снътй
Inl3 210 Желтоватый, в расплава темно-коричневый tola Ini — •—
In2O9 1— Желтый ТпО? Бесцветный 1п2О? — Черный?
In 283 1050 Желтый и красный InS 692 Винно-кра-сныц In2S? 653? Коричневочерный?
Соли: например, нитрат индия IntNOjh-41/2Н2О; сульфат индия In»(SO4)3-5ПгО.
Двойные соли (аци досоли): например, KjInCie- IViHaO (гецсахпориндат (Ш) калия); NHiInCSOth* 12ЩО (аммониевые квасцы индия).
1, СОЕДИНЕНИЯ ТРЕХВАЛЕНТИОГО ИНДИЯ
Хлорид индия (III) и хлорнндаты. Хлорид индия (III) 1аС13 образуется при сгорании нагретого ипдия в атмосфере хлора или при нагревании смеси окиси индия с углем в токе хлора. Это легко во вгоняющиеся белые с перламутровым блеском кристаллические таблички (уд. вес .3,46' при 25°). Хлорид иттдия (III) возгоняется при 418", по плавится только при 586°, Плотность пара при температуре —800° соответствует формуле InClg . В воде 1пС13 растворяется с шипением. Неприятный на вкус, подобный чернилам раствор можно непосредственно получить, растворяя металл в соляной кислоте. IriCLj в растворе сильно гидролпзован (константа гидролиза—1,2-10~5). Из раствора кристаллизуется тетрагидрат InCI3.4fI2O. InCL. образует двойные соли с хлоридами Iде.точных металлов и хлоридами органических основании: (NH4)2InCls. •Н3О, K.9lnCVn;H2O, (CH3-NH5)4InClr, [(CH3)2NJI2]4JnG]7, (CpUyNHJjlnCly (хнво-линтептахлориндат).
Бромид тЕпдпи (III) 1пВт3 (уд. вес 4,75, т. субл. 371°) и иодид индия (III) Inls (уд. вес 4,68; в значительной степени похожи на хлорид. Inls существует, помимо указанной в табл. 71 желтой, также и в красной модификации (Enss)in, 1942). InCl3, IiiBfg и Inl3 в парообразном состоянии при умереппо высокой температуре ассоциированы в димерные молекулы, так же как соответствующие галогениды алюминия, GaC]3 и GaEiy; по своему строепци) они также соответствуют у помянутым соединениям; расстояние между атомами галогенов составляет для In,CLj 3,90, для 1п2Вг0 4,07 и для 1и216 4,36 h.
Фторид индия (11 j) безводный получается при нагревании 1и2О3 или (NIIJsIInF0] в токе фтора (Klemm, 1936). Его удельный вес 4,39, т. пл. 1170°. В проги-
416
Глава 10
воположность трехфтористому гадливо трехфтористый индий очень мало летуч; InF3i как и GaFg, очень мало растворим в воде, в то время как тригидрат InF3-3II2O, выделяющийся из раствора гидрата окиси трехвалентного индия в водном растворе плавиковой кислоты, растворяется сравнительно хорошо (ср. стр. 415). С фтористым аммонием InF3 образует довольно хорошо растворимую двойную соль (NH4)3InFfi. Эта соль при нагревании распадается с образованием нитрида индия, который, как и GaN, имеет структуру вюртцита; а = 3,533, е = 5,693 A (Juzа, 1938). Фтороиндат натрия Na3 [InF6] (растворимость 9,1 г па 100 г П2О) был недавно получен Енслином.
Окись трехвалентного ипдия. 1п2О3 образуется при нагревании гидроокиси, сульфата или иитрата. Это светло-желтый порошок (уд. вес 7,28), при нагревании темнеющий, растворимый в кислотах и нерастворимый л щелочах и аммиаке. При длительном сильном нагревании 1п2О3 становится кристаллической. При нагревании в токе водорода или в атмосфере сухого аммиака при 200—300° происходит восстановление до металла. О строении решетки 1п2О3 см. стр. 426. Теплота образования, но Роту (Roth, 1932), 222,5 ккал/моль, по Холлею (Holley, 1952), 221,3 ккал/моль; по Щтубсу (Stubbs, 1952), рассчитанная из измерений равновесий составляет 196,4 ккал/моль.
1п2О3 с СаО и Cd О образует двойные соединения, соответствующие шпинели по составу и, по-видимому, по структуре (Passerini, 1930).
Гидрат окйеи индия (III) In2Os aq выпадает из раствора солей ипдия при добавлении щелочи, аммиака или какого-либо другого вещества, которое в результате гидролиза создает щелочную среду, например нитрита калия. Гидрат окиси ~ белый, студенистый осадок, нерастворимый в разбавленном аммиаке, вследствие пептизации под действием аммиака может легко образовать коллоидный раствор. Чистая вода также действует пептизирующим образом. Это свойство гидрата окиси может препятствовать образованию осадка в процессе осаждения. Осадок легко растворим в кислотах и в избытке щелочей. Следовательно, оп является амфотерным соединением. Однако из щелочного раствори через некоторое время часть вещества опять выпадает, возможно в результате образования кристаллической гидроокиси ипдия с малым пр о я введением растворимости.
Нитрат нидия(ПТ) кристаллизуется ив раствора гидрата окиси в большом избытке азотной кислоты в виде бесцветных столбиков или игл состава 1п(ХОэ)3.4ЙН3О. Из раствора, содержащего соль аммония, кристаллизуется двойная соль.
Сульфат нндпя(1П). При упаривании коя центрированного раствора индия, окиси ипдия тьлп гидрата окиси ипдия в серпой кислоте кристаллизуется соединение In.H(SO4),. 3’ •’ НаО. Из разбавленного раствора выделяется нейтральная соль In2(SO4)3, в зависимости от температуры с 6—12 молекулами кристаллизации и Eton воды. При нагревании нейтральной или кислой соли до 200° образуется безводный сульфат. Осаждая и идя it в виде соли IbH(SO4)2-3*4 II гО, его можно очень хорошо очистить от примесей. особенно от желоза я олова (Ensslin).
Сульфат индия образует двойные соли с сульфатами щелочных металлов. Двойной сульфат аммония и индия кристаллизуется из водного раствора при температуре выше 36:> в виде тетрагидрата (NH.4)In(SO4)2-4H8O; ниже 36° кристаллизуется двойная соль mumi квасцов (NН.)In(SO4)2-12H,O. Такие же квасцы индий образует С рубидием к цезием. Двойные сульфаты с натрием и калием, наоборот, кристаллизуются с' четырьмя молекулами воды. Индий также образует двойные соли с сульфатом замощенного органическими группами аммония (CH3.NII3][In(SO4)2J.2H2O, (С2Н5. NH3] [ln(SO4)2| ЗЧП2О, {С6НьСН2Маз][Ш(8О4)2]..3112О.
.Сульфпд пе1дпя(Ш) ln2S3.' При пропускании сероводорода в слабо кислый раствор сульфата ипдия вы пада ет желтый осадок сульфида ипдия, легко растворимый в азотной кислоте. При длительном кипячении с разбавленной кислотой получается модификация кирпичного цвета, малорастворимая в азотной кислоте. При сплавлении ипдия с серой образуется сульфид киноварного цвета. In2S3 имеет уд. вес 4,90. Выше 850° он заметно летуч вследствие разложения па In2S и S. По данным Штубса, под давлением сульфид плавится прп 1095°.
СульфпД индия in2S;} встречается в а- и (3-модификациях (Hahn, К lingler, 1949). По данным Штубса (Stubbs, 1952). InaSg образует с TnS два двойных соединения, по-ви-димому со структурой, шпинели состава InS- In2S3 и 3lnS- In2S3. Оба соединении плавятся никоягруепттто (при 840 и соответственно 770°). Ниже 370° InS-1п233*распадается на 3InS.In2S3 и In2S3. Выпадающий из раствора сульфид In2S3 можно обезводить, нагревая его в вакууме. Однако ок остается гигроскопичным, если его не перевести в высокотемпературную модификацию. По мнению Хана и Клинглера,
Третья группа периодической системы
обе модификации сульфида индия различаются подобно у’-А12О3 и y-ALOj. При температуре выше 300° происходит монотропное превращение а- в fJ-In2S3. fJ-In2S3(a = 10,72 A; d^’KH= 4,63) — твердое и хрупкое вещество, изотипноё Y-A)2O3,
Если в нейтральный раствор соли ипдия добавить сернистый аммоний или раствор сульфида щелочного металла, то выпадает белый осадок, который при определенных условиях растворяется в избытке осадителя. Этот осадок рассматривают как тио-соль индия. Для белого осадка, выпадающего при стоянии из водной вытяжки после сплавления окиси ипдвя, соды и серы, Шнейдер (Schneider) нашел состав, соответствующий формуле AaInS2> )ЬО. Таким же образом можпо получить соединение KInS2, кристаллизующееся в гиациптово-красных кристаллах.
При сплавлении индия с селен ом или теллуром получаются соединения In2Se3 и 1п2Те3. Селенид индия имеет сложную структуру и обладает в отличие от сульфида и теллурида мягг;остьго графита. Теллурид, который, так же как и сульфид, тверд и хрупок, кристаллизуется подобно Ga2S3, Ga2Se3 и Ga2Te3 в решетке, которая выводится из решетки цинковой обманки таким образом, что в ней остается пез апо л пенными */з мест, принадлежащих атомам металла, причем распределение этих незанятых мест не упорядочено,
2. Соединения одно- и двухвалентного Индия
Хлорид ппдия(П) 1пС12. При нагревании индия в токе хлористого водорода образуется янтарно-желтый расплав, который застывает в бесцветные кристаллы (уд. вес 3,64 при 25°; т. пл. 235°; т. кип. 570°). Плотность пара выше 1300° соответствует формуле }пС12. Вода разлагает ТпС13 на металлический индий и 1пС13. Процесс окисления— восстановления идет в две стадии:
21пС1,= 1лС1 1иС1з, (1)
31пС1 = 21п 1пС13. (2)
Прп разложении хлорида ипдия (II) соль одновалентного ипдия образуется только в очень небольшом количестве в виде промежуточного продукта, в то время как для бромида индия(П) (уд. вес 4,22), если для разложения использовать холодную воду, реакция приостанавливается на первом стадии и только постепенно протекает дальнейшее превращение. В твердом состоянии 1аС12 изоморфен SnCl2. Это диамагнитное соединение. Поскольку для попов 1п2+ можно было бы ожидать проявления парамагнетизма, то Принимают, что в решетке соединения места Катионов заполнены статистически распределенными ионами 1и+ и 1п3+.
Хлорид ипдпя(1). 1пС1 образуется при1 пропускании паров 1пС12 пад нагретым, индием. Это жидкость кроваво-красного цвета, застывающая в красную массу (уд. вес 4,18 при 25°; т. пл. 225°). Плотность пара при 1100° приближается щ значению, отвечающему формуле InCi, по увеличивается с дальнейшим ростом температуры. В молекуле InCi в парах расстояние между центрами атомов составляет 2,42 А (в ТпВг 2,57 и в Ini 2,86 А). Вода тотчас же разлагает InCi на металл и трихлорид [см. уравнение (2)].
Фторид ппдпя(П). InF2 был получен Клеимом (Klemm, 1936) в результате умеренного нагревания InF3 в токе водорода (при более сильном нагреваний происходит восстанет лепие до металла). Со единение бес цветное, окрашивающееся па влажном воздухе постепенно в серый цвет из-за происходящего под действием воды разложения на lnF3 и металл. Уд. вес InF2 около 6,1.
Окись ипдия(П). 1иО. Если нагревать 1п2О3 в высоком вакууме или в восстановительной атмосфере, то его поверхность постепенно окрашивается в белый цвет. Тил (Thiel, 1928) предположил, что это явление, по-видимому, связано с образованием окисла InO. Существование этого соединения, однако, с достоверностью не доказано и в свете новых исследований представляется очень сомнительным (см. ниже).
Окись ппдия(1), In,О. Окись одновалентного индия, поданным Тила, образуется при нагревании 1п2О3 в токе водорода или при термическом разложении 1п2О3 в высоком вакууме. Это летучее соединение, которое осаждается на холодных участках реакционной трубки в виде твердого налета, в топком слое (в проходящем свете) желтое, п толстом слое черное.Согласно Штубсу (Stubbs, 1952), существование 1п2О, точно так же как и InO, сомнительно, так как рентгенографические исследования препаратов, полученных при нагревании 1п2О3в токе водорода показали, что в них наряду 27 г. Реми
418 Глава 10
с 111оО3 содержался лишь, металлический индий. Можно, правда, предположите^ что при охлаждении др комнатной температуры полученные 1п30 и InO разлагаются на 1п2О3 и металл. Однако это опровергается тем фактом, что; свободная энергия образования 1п3О3 из элементов, рассчитанная Ш ту б с ом в предположении равновесия 1н2О3-Ь ЗНа 21п 4-ЗН2О, имеет значение, практически совпадающее со значений ом, вычисленным из теплоты образования и моляр ной энтропии при 298е К (68,4 ысл).
Сульфид ипдия(П) InS, существование которого было предположено па оснований данных термического анализа Тилом (ТЫН, 1928) и недавно подтверждено Штубб-сом (Stubbs, 1952), проще всего получить сплавлением составных частей в количествах, с о ответст ву io щи х формуле. 1и5 имеет уд. вес 5,18 и- винно-красну и, окраску. Прп нагреваilim под давлением вещество плавится инкоигруоптнр при 680°, е
При нагревании In2S3 в токе сероводорода или при нагревании металла с соответствующим количеством серы по прежним представлениям дрлжеЕЕ был бы/ образа-ваться Jn2S, описанный в виде пороппш черного цвета, с уд- весом 5,90 И т; пл. —653°, но внешнему виду и свойствам приближающийся к 1п20 и разлагающийся разбавленными кислотами с выделением красного In2S3. Он должен был бы возгоняться г в токе водорода и осаждаться в виде иголочек, желтых в проходящем свёто. Однако Хаи (Hahn, 1949) не смог подтвердить образование соединения, а Шгубс (1952) пе1 : сумел обнаружить при изучении системы In — In2S3 признаков существования сое3 динения с формулой In2S.
3. Алкильные соединения индия и гпдрпд индия
Триметилйндий. 1п(СНз)з был получен в 1934 г. Деннисом таким же образом^ как и триэтилгаллий (ср. стр. 412). Это бесцветные кристаллы, сильно преломляющие свет (уд. вес 1,568; т. пл. 88,4; т. Кип. 135,8°), легко растворимые в органических растворителях. Тримётилипдий неустойчив на1 воздухе,. С сухим кислородом при пизкой температуре он реагирует по уравнению 2In(CH3)3-j- i/2O2= [(CH3)2In]2O-f--r C2H6.
Вода также разлагает соединение, причем с отщеплением, двух групп СН2; кис-.лбты огщоялягат третью группу СН3. В этом отношении триметллщгдий пё является,, слёдовательно, аналогом триэтил галлия; в отличие от триатилгайлияон не образует соединений присоединения с аммиаком, по образует такие соединения с эфиром., (СН3)31п р(С2Н5)2 — бесцветная жидкость, d -- 1,241 (при 20’); ,т. .пл. —15°;. т. кип. 139°.
Триэтилиндий In(C2Hs)3 (d — 1,200 при 20°; т. пл. —32°; т. кип. 144°) и трипро-пилиндий In(C3II7)3 (.1 1,187 при 20°; т. пл. —5Г; т. кип. 178°), не способны давать
соединений с эфиром, которые можно было бы перегонял, без разложения,
Прп взаимодействии ТпС13 и NaOCH3 в метиловом спирте образуется триметилат индия 1п(()СНз)з. Это бес цвет! пае кристаллы, легко растворяющиеся в метиловом, спирте н бензоле (Runge я сотр., Z. anorg. Chem., 267, 39, 1951),
Трпфеннлипцнй. In(C6H3)3 (т. пл. 208°) образуется при взаимодействии дифенил-., ртути с металлическим индием: 3IIg(CeH6)2+ 2Тн — 21п(СцН6)3+ 3Hg. Трифенй.чип-дий подвергается воздействию воздуха и влаги, а также медленно реагирует с С02 (с образованием бензойной кислоты) . Он легко взаимодействует с органическими соединениями, содержащими карбонильную группу, например с хлористым бензоилом, б ен зал т, де гидом и бепвофепопом, Склонность к взаимодействию с этими соединениями у 1ее(С,,1 I/; j болЕ,ше, чем у соответствующих соединений галлия и таллия/ (Gilman,/ Jones, 1940).
Согласно Гильмаиу, существует следующая закономерность: реакционная способность метаялореанических соединений уменьшается с увеличением потенциала ионизации. Уменьшение реакционной способности в ряду InR3, GaR3, T1R3 является примет ром проявления этой закономерности (ср. табл. 22, стр. 140). Гильман также показал (1936), что это правило справедливо для алкильных соединений щелочных металлов,, Одпако существуют исключения из этого правила. Например, потенциал ионизаций для металлов второй побочной подгруппы возрастает в ряду Си, 7.n. Ilg: реакционная способность соотвётствуюнЦЕХ им металлорганических соединений убывает в такой же-последовательности только для очень ограниченного числа соединений. В болыппнстЕЮ случаев реакционная способность уменьшается в ряду ZnR2,- CdR2, HgR2. В этом смысле примечательно, что Томпсон (Toropson, 1.936) на основании измерения: спектров поглощения в ультрафиолетовой части спектра для диэтильных..производных нашел увеличение прочности связи и направлений Cd—С, Zn— С, Hg— С, г. в. в полном; соответствии с потенциалом ионизации.
Третья группа периодической системы 419
Гидрид индия, [IilHj]^ был получен Виборгом (Wiberg, 1951) взаимодействием; треххлористого индия с гидридом лития в эфирном; растворе при комнатной температуре
1пС13+ ЗИН _ 1пИ3+ 31 iCL
Это белое, твердое, нелетучее, нерастворимое в эфире, высокомлимерпое соединение; распадающееся выше SO па элементы, т. "
При взаимодействии 1пС13 с LiAlH4 в эфирном растворе при —70° по уравнению 1пС134- 3LiAUl4= Iii(AlIl4)3-|- 3LiCi Виборгом был получеп индийалюмогидрид (аланат, индия). Это тоже твердое, белое вещество* нерастворимое в эфире. Этот двойной < гидрид распадается уя;е выше —40° с образованием гидрида алюмипия, индия и водорода. Следует отметить, что образующимся при этом в мономерном состоянии 1пН3 распадается на элементы прежде, чем юп стабилизируется при низкой температуре в результате полимеризации. При взаимодействии InCl3 с LiAlH4 в качестве промежуточного продукта образуются соединения, в. которых не лее атомы .хлора заменены па группы А1Н4; дпхлораяапат индия 1пС1г[А1Н4] устойчив при 100°.
Для таллия известен только один, очень неустойчивый аланат состава Т1С1(А1Н4)2. Это белое, твердое вещество, нерастворимое в эфире, образующееся только при очень низкой температуре й распадающееся уже при —95° (Wiberg, Schmidt, 1951).
ТАЛЛИЙ, TI
; Распространение в природе. Таллий относится к элементам, которые встречаются во многих местах, но почти всёгдав очень незначительных количествах. Таллий часто содержится в обманках и колчеданах цинка, меди и железа. Кроме того, он часто встречается в калийных солях и слюдах. Обе эти области его распространения соответствуют двум сторонам его химического характера, по которым он, с одной стороны, принадлежит к типичным тяжелым металлам, а с другой, по многим реакциям относится к щелочным металлам.
Собственно минералы таллия чрезвычайно редки. В Македонии был обнаружен в реальгаре арсепосульфид таллия TlAsSa (лорандит), В .Скрикеруме в Швеции был найден крокеаит — относительно богатая таллием изоморфная смесь селенидов таллия, меди и серебра. Близкий состав, но со значительно меньшим содержанием таллия1 имеет берцелианит, обнаруженный там же, а также в Лайбахе (Верхний Гарц),
Исторические сведения. Таллий был открыт спектральным методом в 1861 г. Круксом в шламах свинцовых камер сернокислотного завода в Гарце, Название свое элемент получил по-характерным зеленым линиям своего спектра и по зеленой окраске пламени (OaMog—зеленая ветвь).
Получение. Основное” сырь о для получения таллия, о котором может: идти речь, это пыль, образующаяся при обжиге обманок или колчедана, содержащих таллии. Пыль экстрагируется горячей водой, а таллий из раствора осаждается или пинком, или соляной кислотой в виде хлорида. Для очистки таллий снова переводят в сульфат и после повторного (а в случае необходимости — многократного) осаждения в виде хлорида, металл выделяют электролитически из сернокислого раствора. Технический таллий молите освободить от часто содержащегося в нем свинца растворением металла в азотпой кислоте н осаждения свинца из сильнокислого раствора Сероводородом,
Свойства. Свежий срез таллия белый и блестящий, однако на воздухе1 металл тотчас же тускнеет. Таллий металл мягкий; мягче свинца. Уд. вес 11,83, температура плавления 302,5° ; (теплота плавления 1,03 ккал/г-атом), температура кипения около 1450°.
При 232,3° обычный, гексагональный таллий (а-Т1) переходит в кубическую модификацию (р-Т1). Теплота превращения составляет 82 кал/г-атом. При обычной температуре при перс охлаждении можно получить ₽-Т!.. Уд. вес р-Т1 отличается лишь пе-ипбго (11,86) от уд. веса а-Т1; это соответствует тому, что обе модификации пост-? ровны по принципу плотнейшей упаковки (ср. стр. 408).
" 27»
420
Глава 10
Таллий ионик) в о зо гнать в токе водорода. На воздухе он окисляется с поверхности, образуя окись одновалентного н трехвалентного таллия. Озон, а также перекись водорода окисляют металл до Т1аО3. Вода, не содержащая растворенного воздуха, при обычной температуре лишь очень незначительно воздействует па таллий. В спирте, напротив,- металл медленно растворяется с образованием алкоголята и выделением водорода. При достаточном доступе кислорода выделения водорода пе происходит, В этом случае реакция протекает по уравнению
2Т1 + 2С2Н5ОП + Г'.2 02= 2С2Н5ОТ1 4- Н20.
Образующийся при этом алкоголят одновалентного таллия представляет собой тяжелое, желтоватое масло с уд. весом 3,52 (при 20°). Это соединение затвердевает при — 3°, хорошо растворяется в абсолютном спирте или эфире, его водой немедленно разлагается. Распад наступает также при нагревании выше 130°.
Лучший растворитель для таллия — разбавленная азотная кислота. В разбавленной серной кислоте .металл растворяется значительно хуже и еще хуже — в соляной кислоте. Это находится в соответствии с более или менее плохой растворимостью соединений таллия, образованных этими кислотами. В связи с легкой растворимостью фторида и положением металла в ряду напряжения левее водорода интересно отметить очень слабое взаимодействие таллия с фтористоводородной кислотой.
Нормальный потенциал таллия в растворе соли таллия (I) по отношению к нормальному водородному электроду составляет —0,336 е, поэтому в Электрохимическом ряду напряжений таллий стоит между кадмием и кобальтом. Свипец, медь, ртуть, серебро и золото осаждаются металлическая таллием из раствора. Солянокислый раствор ТЦШ) в присутствии металлического таллия неустойчив. В пем практически полностью Проходит превращение ТГ" -й 2Т1 = ЗТГ.
С галогенами таллий реагирует уже при обычной температуре. При нагревании металл взаимодействует также с серой, селеном и теллуром. При сплавлении металл соединяется с мышьяком и сурьмой. Фосфор, напротив, взаимодействует с таллием лишь с поверхности. С бором и кремнием таллий вовсе не взаимодействует. Он не образует с ними ни смешанных кристаллов, пи соединяется при сплавлении. С молекулярным водородом таллий не взаимодействует. Йе реагирует он и с азотом и двуокисью углерода. В жидком аммиаке таллий также пе растворяется.
Таллий и сто сосдипения па ходят весьма ограниченное применение. Изредка их используют в лаборатории для получения монохроматического зеленого света. Относительно применения соединений таллия в медицине см, стр. 421.
СОЕДИНЕНИЯ ТАЛЛИЯ
В своих соединениях таллий проявляет положительную одно- и трехвалентность. Соединения одновалентного таллия в общем наиболее устойчивы. Соединения трехвалентного таллия легко восстанавливаются до соединений одновалентного таллия и являются поэтому весьма сильными окислителями. Соединения одновалентного таллия во многом похожи на соединения щелочных металлов. Так, гидроокись Т1ОН легко растворяется в воде и является сильным основанием. Весьма хорошо растворимый карбопат похож па соду и К2СО3. Как и щелочные металлы, одновалентный таллий образует большое количество прекрасно кристаллизующихся солей. Соли одновалентного таллия в большинстве случаев бесцветны. При нагревании они относительно летучи. Многие из них кристаллизуются без воды, как соли тяжелых щелочных металлов. Водные растворы солей таллия производных слабых кислот вследствие гидролиза обладают щелочной реакцией.
Третья группа периодической. системы
421
По растворимости солей одновалентный .таллий часто стоит в ряду, который ведет от калия к рубидию м цезию и далее ко всё менее растворимым солям. Это справедливо, например, для нитратов, сульфатов и гекс а хлороплатина тол. Таллий подобно щелочным металлам образует хорошо кристаллизующиеся соли с комплексными анионами. Как и щелочные металлы, одновалентный таллий способен образовывать полисульфиды, например TlaS5, и полниодиды, например Т1- 1а.
С другой стороны, одновалентный таллий во многих отношениях близок к серебру, например своей плохой растворимостью и окраской галогенидов, хроматов, сульфидов и окислов^ Ион одновалентного таллия, так же как и ион серебра, обладает своеобразным свойством давать желтоватый осадок с ионами некоторых слабых кислот, например азотистой и ортомышьяковой кислотами. Характерное отличие иона Одновалентного таллия от иона серебра — отсутствие у пего склонности к образованию в водном растворе аммйнных комплексов, столь сильно выраженное у серебра. В этом отношении таллий приближается к своему правому соседу по периодической системе — к свинцу, с которым у него много общего, и прежде всего в металлическом состоянии.
Подвижность иона одновалентного таллия при 18° составляет 66,6, что, следовательно, только немногим больше, чем для иона К‘ (ср, стр. 181), Эквивалентная электропроводность соля одновалентного таллмя в умеренно разбавленном растворе меньше, а в сильно разбавленном растворе больше, чем соответствующие значения для соли калия.
Соединения трехвалентного таллия часто проявляют заметную склонность к комплексообразованию (например, хлорид). Поскольку, однако, в общем случае этого пе наблюдается, то в водном растворе соли обычно сильно гидролизованы, например нитрат и сульфат., которые уже при стоянии на влажном воздухе постепенно выделяют гидрат окиси трехвалентного таллия. Растворы солей одновалентного таллия вследствие гидролиза имеют щелочную реакцию, гидролиз солей трехвалентного таллия вызывает кислую реакцию раствора.
Кёмплексные соли трехвалентного таллия в большинстве случаев Отвечают формуле или Мз(Т1Хе]. Таллий также легко образует соединения, содержащие
металл Т1 в одновалентном и трехи а л епт ном состоянии. Эти соединения часто отличаются плохой растворимостью и более интенсивной окраской в сравнении с простыми соединениями. Потенциал пары ТГ-> ТГ" составляет + 1,21 в: это значит, что платиновая жесть, погруженная в раствор, содержащий одинаковое число ионов одновалентного и трехвалентного таллия, приобретает из-sa тенденции ионов ТГ" к переходу в ионы ТГ положительный заряд-1-1,21 а. В присутствии веществ, образующих комплексные соединения с ионами ТГ", окислительный потенциал снижается. Поэтому легче всего можно окислить соединения одновалентного таллия в соединения трехвалентного в присутствии веществ — крмплексообразователей.
Соединения таллия весьма ядовиты. Помимо воздействия на нервную систему и органы пищеварения, они вызывают- выпадение волос; из-за этого свойства соединения таллия находят применение в качестве средства для удаления волос при грибковых заболеваниях. Иногда их применяют в медицине и для других целей. Соединения трехвалентного таллия в водном растворе легко восстанавливаются до соединений одновалентного таллия: например, хлорная или бромная вода количественно окисляют в растворе соединения одновалентного таллия в трехвалеитный.
422
Глава, 10
По данным Ха да и Клинглера (1949) таллий образует с серой, селеном и теллуром следующие соединения;
TI2S. T14S3 или ТЦ[Т1Ш83]? ns или Ti1 [Tl11’s2|< T1S4
Tl2Sa
Tl2Te
TISe T
—или TlI[TlInSe2]
TI2S кристаллизуется, по данным Кетелаара (Ketelaar, 1939), в искаженной слоистой решетке, типа б рудита; Т12Se не и зотппеи сул 1.фи ду. TISe п о: р еаультатам: структу р пых определений является двойным соединением селенидов одновалентного и: тр едва л битного таллия. Сказанное справедливо для T1S и, вероятно, также для T14S3. Т1Те имеет сложную структуру; состав итого соединения может колебаться в широких пределах, Т1Те, но-виДимому, близок к пптерметаллическим соединениям^
Таллий является единственным элементом главных подгрупп периодической системы, пе образующим с водородом простого соединения, которое можно было бы выделить в свободном состоянии. Гидрйд таллия, если для его получения исходить из условий, приведенных .на стр. 353, получается в форме двойных соединений, например TlfGaHaJs и Т1С1(А1Щ)2. Однако эти двойные соединения одень мало устойчивы (ср, стр. 412 и 390). ••... ' Д . .
1. Соединения одновалентного таллия
Галогениды одновалентного таллпя (ср. также табл. 72). Т1С1 выпадает из раствора соли одновалентного таллия при добавлении ионов GF. Это белый, творожистый: осаДок, После к ристал лизании соединение можно получать в виде маленьких кубов, В токе сухого хлора при нагревании Т1С1 переходит в двойное соединение TlGlj-TlCl; при обработке хлористым водородом T1CI в конце концов окисляется до TIG1S. TlBr
.... Таблица 12
Галогениды одновалентного таллия
Соединение Т1Г T1CI Т1Нг ти:
Цвет Белый Белый Желтова* тый а-Желтый р-Красный
Удельный вес 8,36 7,00 ‘ 7,5 7,29 а
Температура плавления, °C 327 430 456 440 :
Температура кипепия, °C . , Межатомные расстояния в мо- 655 806 815 824
лскуле пара, А Растворимость, .имоль/л при *— 2,54 - 2,07 2,91
18° Теплота образования, Очень легко : растворяется 12,7 1.48 0,17?
ккал/молъ 77,1 48,5 : 41,3- 31,2 а
& Вто значение справедливо для модификации. стабильной при обычной температуре.
имеет желтоватую окраску, а в остальном является полным аналогом Т1С1. ТП существует в Двух модификациях — желтой й красной. Последняя модификация стабильна при температуре выше 168°, однако остается неизменной Долгое время и после охлаждения до комнатной температуры. Т1С1, Т1Вг и красная модификация ТП(Р-'ГП) кристаллизуются по типу иодида цезия (рис, 48, стр, 239). Длина ребра куба а составляет для Т1С1 3,834, для TIE г 3,97 и для 0-T1I 4,18 А- Желтая модификация, втабиль-
Третья группа периодической системы. 423
иая при обычной температуре (а-ТП), образует ромбическую слоистую решетку, построенную из молекул TH. T1F в отличие от других галогенидов одновалентного таллия очень легко растворяется в воде (1 ч. в 1,25 ч:. воды). Водный раствор имеет щелочную реакцию. Из раствора, содержащего избыточную плавиковую кислоту, кристаллизуется в октаэдрах кислеДЙ фторид Я2Т1Р3.-%Н2О,. Фторид таллия" T1F кристаллизуется в-ромбических листочках и образует деформированную решетку типа каменной соли (л == 5,180; Ь 5,495; с = 6,080 А). Деформация кристалла в вертикальном направлении по оси с обьяСЕЕЯется поляризующим действием иона F- па легко поляризуемый пои ТТ (Kctelaar, 1935). Относительно трико Дида одновалентного таллия ср. стр. 426.
Цианид т:ьтлеея(1). T1GN в чистом состоянии лучше всего ролучать взаимодей-'Ствием цианида бария с сульфатом одновалентного таллия
Ba(CN)2+ T12SO4= 2T1CN |- BaSO4.
Это белый кристаллический порошок или блестящие листочки, довольно хорошо растворяющиеся в воде (15,2 г в 100 г НаО при 14°). Водный раствор имеет сильно основную реакцию. С кодом T1CN взаимодействует по уравнению
T1GN 4- I2- TH. • 1CN. .
Роданид таллия. T1SCN выпадает из раствора соли одновалентного, таллия в виде маленьких блестящих листочков, кристаллизующихся в тетрагональной с и ягой нипри добавлении ионов SCN'. Растворимость при 20° равна 0,32 г T1SCN в 100 а воды.
Гидроокись и окись одновалентного таллия (I), Црй взаимодействии сульфата одновалентного таллня в водном растворе с вычисленным количеством гидроокиси бария образуется очень сильно щелочной раствор, по свойствам совершенно апалогйч-- нып раствору щелочей, который содержит легко растворимую в во до й Спирте гидроокись одновалентного таллия TJOH. Гидроокись выделяется при упаривании в гиде желтой, кристаллической массы. При нагревании гидроокиси выше 100° образуется окись таллня (I) ТЬО в виде черного, очень гигроскопичного порошка. Т12О плавится при ЖЮа, сильно действуя при этом на стекло. Окисью углерода Т12О легко восстав навливается до металла. Теплота образования окисла составляет 42,1 к к ял Дне ль. Теплота образования гидроокиси из окисла составляет только 1,6 ккал/моль T1OFL
Сульфид таллия. T13S осаждается сернистым аммонием или (цз очень слабо кислого раствора) сероводородом. Это черный, очень легко окисляющийся осадок. При паг.ревании с раствором сернистого аммония при 150—20(1° осадок становится кристал-личеснйм (уд, вес 8,40). Сульфид одновалентного таллия осаждается из раствора соли трехвалентного таллия прн действии на него сероводородом, ;но в этом случае лишь в смеси с серой:
2Т1 '-г 3H2S T12S -ф- 25 •(- (ИГ.
В чистом состоянии T12S лучше всего можпо приготовить взаимодействием H2S с раствором этилата одно вале нтпого таллия Т(ОСгН5 в абсолютном спирте. Последний легко подучить, действуя на стружки таллия абсолютным спиртом в атмосфере кислорода. Крупнокристаллический T1..S проще всего получить непосредственным взаимодействием Т1 с S b атрмпых соотношениях 2:1.
T12S очень медленно окисляется под действием сухого кцелорода воздуха в моио-окись и тиосульфат: 2Г12S 4- 2Оа = Т12О T12S2O3. При дальнейшем воздействии кислорода на Т13О образуется Т13Оа. T13S,O3 устойчив при обычной Температуре и разлагается только выше 130°. Если Нагревать эквимолярную смесь Т120 и T12S2O3 или T12S до 250° в атмосфере кислорода, то образуется кристаллическая масса от желтого до зеленоватого цвета с составом, отвечающим формуле T12SO2, Вещество кристаллизуется в кубической гранецеатрированиой решетке. Природа этого соединения еще не выяснена. Вода разлагает его по уравнению £
3T12SO2S= 1BS .B 2n,SQ|.
При дальнейшем пагревапии в атмосфере сухого кислорода (воздуха) при 250° T12SO2 переходит в T12SO4 (Heptor, 1949—1952; van Hippel, 1946). При определенных условиях возможно, кат; установил в 1917 г. Кезе (Case), в результате неполного самоокисления T12S получить фотоэлектрически активное вещество с особой чувствительностью в области длинных но ш.
Сульфат таллия (I). Т12ЯО4 кристаллизуется из раствора, образующегос^при рао-творений таллия: в разбавленной Серной кислоте. Это бесцветные, большие; ромбиче-
424
Глава Ю
сжие иризмы (уд. вес 6,7—6,8, т. ил. 632°). TI2SO4 изоморфен сульфатам калия, руби г дня и цезия, В иоде T12SO4 заметно растворяется (4,87 г в 100 г воды при 20°). При температуре выше слабо красного каления эта соль начинает улетучиваться. Из раствора, содержащего избыточную серную кислоту, кристаллизуется кислый сульфат. Сульфат таллия образует с сульфатом алюминия соединение, относящееся к квасцам,— T1A1(SO4)3- 12Н2О— алюмота.члиевые квасцы.
Нитрат таллия (I). T1NO3 кристаллизуется из раствора металла, гидроокиси или карбопата одновалептпого таллия в азотной кислоте при обычной температуре в ромбических призмах. Выше 61° устойчива тригопально-ромбоэдрическая форма, выше 143,5°—кубическая модификация. Температура плавления равна 206°. При температуре выше 300° наступает разложение. Растворимость соли в воде сильно увеличивается с ростом температуры. В 100 г воды при 20° растворяется 9,55 г соли, а при температуре кипения насыщенного раствора (105°) в 100 г воды содержится уже 594 г T1NO3. /
Хлорат тип л ия (1). Т1САО, образуется при растворении металла и хлорноватой кислоте пли в результате двойного обмена между хлоратом бария и сульфатом одновалентного таллия. Из охлажденного раствора выделяются бесцветные, длинные иглы (уд. вес 5,05), Соль образует смешанные кристаллы с хлоратом калия (с содержанием КС1О3 36,3—97,93 мол.%), Растворимость ее сильно увеличивается с ростом температуры (от 2,00 г при 0° до 57,3 г в 100 г воды при 100°), В горячем растворе, однако, оиа постепенно разлагается.
Растворимость бромата 0,35 г Т1ВгО3 в 100 г И3О при 20®.
Растворимость йодата 0,058 а Т11О3 в 100 а Н3О при 20°.
Перхлорат таллия (I). Т1С1О4 образуется подобно хлорату. Соль кристаллизуется в ромбических кристаллах и изоморфна перхлорату калия. Подобно К С1О4 при нагревании Т1С1О4 переходит в кубическую модификацию. (Для КС1О4 температура превращения лежит при 299,5°, для Т1С1О4— при 266°.) Перхлорат одновалентного таллия растворяется еще лучше, чем хлорат. Растворимость увеличивается от 10 г TLC [О 4 в 100 г воды при 15° до 166,6 г в 100 г воды при 100° ,
Ацетат таллия(1). Т1С3П2Оа образует блестящие, как шелк, расплывающиеся иглы (уд. вес. 3,9, т. шг, 110°). Соль легко растворяется в горячем спирте. Приготовляют ее раствореньем карбопата в уксусной кислоте.
Карбо пат тал.тия(Т). Т12СО3 выпадает из раствора гидроокиси одновалентного таллия, насыщенного СО2, при добавлении спирта. В воде соль довольно хорошо растворяется (5,2 г в 100 г НаО при 18°, 22,4 й в 100 в 1120 при 100,8°), Вследствие гидролиза соли раствор имеет сильнощелочпую реакцию, как и растворы карбонатов щелочных металлов. Из раствора соль кристаллизуется в длинных, моноклинных иглах (уд. вес 7,16; т. пл. 272—273е). При сильном нагревании происходит разложение с отщеплением СО2,
Оксалат таллия(1). Т12С2О4 кристаллизуется из раствора карбоната одновалентного таллия в кипящем водном растворе щавелевой кислоты при последующем охлаждении. Соль образует маленькие призмы с перламутровым блеском, нерастворимые в спирте н эфире. В 100 г воды при 15° растворяется 1,44 а соли. Растворимость увеличивается с прибавлением щавелевой кислоты вследствие образования кислого оксалата Т1НС2О4, Растворимость Т1НС2О4 при 15° составляет 5,35 г .в 100 г воды, t
Фосфат таллия(1). Т13РО4 — бесцветные кристаллы,'довольно плохо растворимые в воде (0,497 г в 100 г 1Г2О при 15°). Одно замещенный фосфат, напротив, хорошо растворяется в воде. Т1Н2РО4 образует моноклинные листочки с перламутровым блеском или длинные иглы, плавящиеся около 190°.
2. Соединения трехвалентного таллия
Хлорид талзпя(П1) и хлороталлаты. При обработке хлорида одновалентного таллия хлорной водой образуется раствор, из которого в процессе упаривания при 60° с одновременным пропусканием хлора выкристаллизовывается тетрагидрат хлорида тре хвале нт на го таллия TIClj-4112О. Это бесцветные, большие, шестигранные таблички, разлагающиеся на влажном воздухе. В вакууме над серной кислотой средине-
Третья группа периодической системы 425-
ние сначала теряет 3 .молекулы воды, а затем и всю воду. Безводный Т1С13 плавится уже при 25°. Водный раствор Т1С13 обладает сильнокислой реакцией вследствие гидролиза. При сильном разбавлении из раствора выпадает коричневый гидрат окиси трехвалентного таллия.
С избыточными ионами хлора Т1С13 легко образует комплексные ноны. Так, из смеси растворов хлорид,а трехи ал ентпого таллия и щелочных хлоридов кристаллизуются двойные соли (хлоро^аллаты), как правило типа M|[T1C1BJ и mJ (Т1С15-Hj>0]. Из раствора хлорида, содержащего избыточную соляную кислоту, кристаллизуется: свободная таллийхлор1.н:>> оеодородиал кислота НТ1С14'ЗН2О в виде тончайших игл. К двойным хлорилам или соответственно хлоросолям этого типа относятся также соединения, являющиеся продуктами неполного окисления хлорида одновалентного таллия или неполного восстановления хлорида трехвалептпого таллия — легко образующиеся так называемые «промежуточные хлориды», т. е. двойные соединения хлоридов одновалентного и трехвалептпого таллия. Эти соединения определяются поэтому как хлороталлаты одновалентного таллия. Из них легче всего в чистом состоянии можно получить соединение Т12 lTlriJCleL Это поблескивающие, желтые гексагональные таблички с уд. весом 5,9, очень плохо растворимые в воде (при 25° 0,0033 моль/л). При осторожном нагревании таллия, пли его одновалентного хлорида в токе хлора образуется бледно-желтая масса состава Ti1 [Tl111^]. При более сильном нагревании этй соединение распадается, однако, по уравнению
ЗТ11 [TimClJ=Т1| [TlIIICJel4-2TlIIICIe.
Хлорид трехвалептпого таллия также легко присоединяет нейтральные молекулы, Так, безводная сот. образует в токе аммиака триаммиакат T1C13-3NH3, который также можно получить из спиртового, по не из водного раствора. Из раствора хлорида трехвалентного талия в спирте или эфире нри упаривании выделяются соединения Т1С1з-С2Н5ОН и Т1С13'(СаН5)2О в виде больших кристаллов.
Бромид таллня(ПТ) и бромоталлаты. Т1Вг3 значительно менее устойчив, чем Т1С13. Напротив, двойные соедипоипя трибромида с монобромндом в чистом состоянии получить легче, чем аналогичные соединения для хлорида таллия. Бромид трехвалент-пбго таллия самопроизвольно с отщеплением брома переходит в двойное соединение Т1Вг3Т1Вг, соответственно Т11 [Т1111Вг4] (длинные желтые иглы). При соприкосновении с водой соединение разлагается на T1J [TlI1IBr(s] (красные гексагональные таблички) и Т1Вг3. Как правило, среди бромоталлатов преобладает тип М1[Т1Вт4] (часто с двумя молекулами воды).
При действии брома на хлорид одновалентного таллия или хлора на бромид одновалентного таллия образуется малоустойчивая смешанная соль Т1С1Вг2.4Н2О или Т1ВгС1г-4Н2О. Удалось также получить смешанные хлоробромотал латы.
Структура решетки га логепота Платов. Среди гало гепоталлатов особенно примечательны вследствие своей структуры соединения Са3Т12С1й, Rb3TIBr6-s/7H2O и КзЛСЦ. 2Н2О. В решетке гексагонально кристаллизующегося Cs3Tl2Cl9 присутствуют в качестве структурных групп двухъядерные комплексные ионы [TLClaP" с координационным числом для таллия, равным шести (Hoard, 1935).
Cl Cl Cl । з~ Cl TI Cl TI Cl I
.Cl Cl Cl J
Элементарная ячейка соединения Rb3TlBr6-e/;H2O, кристаллизующегося в тетрагональной структуре, содержит 14 формульных единиц и, следовательно, 16 молекул воды. Среди 42 понов Rb*, содержащихся в элементарной ячейке, только 2 непосредственно связаны с молекулами воды, и притом так, что каждый из этих двух ионов Rb’ окружен S молекулами воды, образующими куб. Каждый ион Т13+ окружен шестью ионами Вт", образующими октаэдр, так же как, например, ион платины Pt4+ в комплексе [PtCl6]2-или ион алюминия А13+ в комплексе [AlP^]3-, в которых ион металла окружен шестью ионами галогена, образующими октаэдр (рис. 79, стр. 437). Следовательно, решетка рассматриваемого бромоталлата построена из комплексных ионов [Т1Вгв]з*, H0H0B[Bb(01T2)sr и пегидратированных ионов КЬ+, причем последние расположены в пустотах решетки. Решетка соединения КаТ1С16-2Н2О, более богатого водой, отличается от только что описанной пё тем, Что в элементарной ячейке этого соединения большее число щелочных ионов окружены водой. Напротив, дополнитесь-
426 Глава 10
иые 12 молекул воды, содержащиеся я элементарной ячейке этого соединения (которая также содержит 14формульных единиц и соответственно 28 молекул воды), располагаются вокруг двух ионов калия, места которых в рёшейсе совпадают с местами ионов [НЬ(ОНг)8]г в решетке соединения Ш|зТ1Нг1Г8/7Н20. Вокруг каждого из этих ионов калия молекулы воды образуют октаэдр. Поэтому оба основания куба, образованного молекулами воды, входящего в поп [ПЬ(ОН2)Я]+, расположенных перпендикулярно одно относительно другого, раздвинуты гак, что вдоль оси с наблюдается приведенная ниже последовательность групп: октаэдр (Т1С1,р-, квадрат (ОН2)$, октаэдр [К(ОН2)81*', квадрат, (ОН2)4 и затем вновь октаэдр (T1C1J;-, квадрат (ОНа)4 и т. д. В остальном структура совпадает со структурой соединения ПЬ3Т1Вг6!8/7Й2О. На примере этих гидратированных галоидных солей видно, что сложная формула отнюдь по всегда влечет за собой Сложную структуру, а тот о г да даже имеет место об ратное., явление,
Ио днд трех в а лент по го таллия Т113. При добавлении йодистого Калия к раствору •солп трехпалентногб таллия выпадает черный садок ТН3. При перекристаллизации получается вещество, кристаллизующееся в ромбических призмах, изоморфное триио--дидам щелочных металлов. Из этого следует, что твердый трииодид таллия долщен определяться как полииодид одновалентного таллия Т1|1-Т2. В соответствии с этим Т113 чрезвычайно легко отщепляет два атома иода; это, например, можно достигнуть обработкой его органическим растворителем. И иаоборот, можно получить ТП3 присоединением иода к моиойодиду.
В Соответствии с данными Абегга при 25° в соприкосновении с раствором, содержащим не более 0,76’10_& моля свободного иода в 1л, T1I еще устойчив. Приболев высокой концентрации иода T1I переходит вначале в черное вещество, кристаллизую- ? щееся в ромбических кристаллах, состава Т1а18 (соответственно ТП3-5ТН) и только при концентрации иода выше 3,3-10_* молъ/лИз раствора образуется трииодид, В растворе устанавливается равновесие между двумя изомерными формами соединения
Щелочные иодйды образуют с ТП3 двойные соли темно-красного цвета, главным образом типа M^TIIJ.B комплексном иопе 1ТН4]“ таллий, как это установил Абегг при изучении равновесий реакции, трехвалентен,
Фторпд трехвалентного таллия, T1F3 образуется при нагревапии Т12О3 в токе фтора (Klemm, 1936). .Это белый порошок с уд. весом 8,36, При нагревании иа воздухе наступает разложение, одиако в атмосфере фтора соль можно нагреть до плавления (т. пл.~550°). Вода уже при обычной температуре полностью разлагает соль, причем в результате гидролиза выделяется гидроокись трехвалептпого таллия.
Гттдрат’оытсптрехвазептпоготаллия выпадает из водного раствора соли трехвалентного тал.тпя при прибавлении к раствору водного раствора аммиака. Это красно-корпчпевьтй объемистый осадок, легко растворяющийся в кислотах. После высушила? идя при комнатной температуре осадок имеет приблизительную формулу Т1203-Н,0, однако при изобарическом ооезвоживапии отщепление воды происходит' постепенно без скачков (Huttig, 1930). Для полного отщепления воды свежеотфильтррванный осадок надо нагреть до 300° под давлением 10 .ил рт ст. Легче достигается отделение воды при выдерживании осадка с раствором в течение продолжительного времени. При нагревании осадка с раствором получается практически безводный окисел. Рентгенограммы как для содержащего воду осадка, так и для окиси одинаковы. Трехвалентный таллий, следовательно, не образует гидроокись определенного состава йлй: определенной структуры. Прочная связь воды в свежеприготовленном гидрате окиси позволяет, однако, усомниться в том, что молекулы воды действительно беспорядочно распределены в решетке окисла. Поэтому гидроокись трехвалентного таллия следовало бы рассматривать кап пересыщенный твердый раствор воды в окиси трехвалентного таллия.
Окись трехвалептпого таллия.:Т12О3 в тонкоизмельченном состоянии, например пртГполученйй из гидрата окнеп нагреванием с раствором, из которого было произведено осаждение, представляет собой порошок, от темно-коричневого до черного цвета. При термическом разложении нитрата Т13О2 получается ;в виде черных листочков . с уд. весом 16,19 (при 22°). Т12О3 кристаллизуется в кубических кристаллах (по типу Sc2O3; см. том 11) и имеет решетку как у 1п2О3; а == 10,57 А (для 1н2О3 а == 10,12 А). При пагревапии Т12О3 распадается на Т12О и О2. Мелко раздробленная окись уже,ниже 100° отщепляет кислород, в то время как в компактном состоянии черная Т12О3 заметно начинает выделять кислород только при температуре красного каления, ,
Трет ъя группа периодической системы
Сульфат трехвалептного таллня, днсульфатоталлисвля кислота и сульфатотал-латы. Из раствора окиси трехиалбитного таллня в разбавленной серной кислоте обычно кристаллизуется «кислый сульфат» HT1(SO4)2-4H3O. Однако это соединение, вероятно, можно охарактеризовать по как кислую соль, а как комплексную диерлъфати-таллйееую кислоту. Во всяком случае, из смеси растворов сульфата таллия и сульфата щелочного металла кристаллизуется иногда с четырьмя: молекулами воды двойная соль М1 |T1(SO4)2 ] (дисулкфатоталлат), происходящая от этой кислоты. Наряду с такими солями можно получить трнсульфо тал латы чипа ,М| [Т1(ЗО4)3]. Соответствующие двойные соединения также можпо приготовить из растворов, в которых содержится i сульфат как одновалентного, так и трехвалентного таллия. Сульфат трехвалентного таллия применяют как яд против грызунов под названием «целио».
Нитрат трехвалентноготаллия, T1(NO3)3 кристаллизуется из раствора окиси трех-валептпого таллия в концентрированной азотной кислоте при упаривании раствора. Это бесцветные, блестящие, расплывающиеся кристаллы состава Т1(КО3)3- ЗИ2О. В присутствии нитратов щелочных металлов или нитрата одновалентного таллия из раствора кристаллизуется двойной нитрат типа М^ТЦМОд)^], например T1J [Т1Ш-(Np3)j]. Это прозрачные, призматические кристаллы, плавящиеся при 15(r.
Ацетат трехвалентного таллпя. Т1(С2Н3Ог)3 кристаллизуется при охлаждении из раствора окиси трехвалентного таллия в кипящей уксуспойкислоте. Это бесцветные, блестящие листочки/ В то время как это соединение разлагается уже в присутствии незначительных следов влаги, двойная соль, соответственно ацн до Соединение I[Т1(С2Н3Ог)4] (аммонийтетраацетоталлат), кристаллизующееся в .блестящих призмах, устойчива в'атмосфере влажного воздуха.
Оксалат трехпал ентиого таллия, ди оксалате таллиевая кислота и оксалат оталла-ты. Из кислого раствора соли трехвалентного таллпя при добавлении щавелевой кислоты выпадает осадок переменного состава. Нейтральный оксалат трехвалентного таллия в совершенно чистом, состоянии еще до сих пор не Получен. Напротив, комплексную кислоту Н [Т1(С2О4)21 удалось получить в чистом состоянии различными способами. Кислота эта кристаллизуется с тремя молекулами воды. Известны соответствуюпще ей соли М1 [Т1(С2О4)2] и Ml [Т1(С2О4)3| (ди-и трйоксалатоталлаты). Из раствора диокса-латоталлиевой кислоты в концентрированном растворе нитрита калия можно выделить неустойчивое в воде соединение K3[T1(C2O4)2(NO2)2]-H2O, которое кристаллизуется в желтых кристаллах.
3. Алкильные соединения таллня
Триалкильные соединения таллия малоустойчивы и их трудно получить. Несколько легче можно получить диалкнльные соединения типа T1R2X(R алкил, X— одновалентный кислотный остаток). Эти соединения, относительно большая:устойчивость которых характерна для таллия, по своим свойствам' как бы не являются соединениями трехвалентного таллия, а значительно ближе относятся к соединениям одновалентного таллия. В бепово алкильных соединений таллия Лежит одновалентный радикал [Т1П2]* с резко выращенным электроположительным характером. Этот радикал может существовать в растворе в виде свободного иона [Т1В2]‘ и образовывать, с различными кислотными остатками соединения, которые по своим свойствам, папример по растворимости, в значительной мере являются аналогами соединений одновалентного таллия и являются весьма устойчивыми; так, они не разлагаются при нагревании с водой или со щелочью. Для приготовления алкильных соединений таллия по методике Мейера л учите всего исходить из соответствующего, алкилмагнийхлорида и трох-хлористого таллпя. Реакцию ведут в эфирном растворе
2Mg(CHs)ClЯ- Т1С13 = Т1(СН3)2С1 ф- 2МцС12.
Из хлоралкильного соединения легко можно получить другие соли обычпой реакцией двойного обмена. При добавлении сернистого аммония образуется нерастворимый в воде кислый,сульфпд fl’lR2)SII в виде белого осадка. "Для получения основания, лежащего в основе дпалкильных соединений таллия, иодид обрабатывают взвесью окиси серебра в кипящей воде. Гидроокись ITIR^JQH но .своим свойствам соответствует гидроокисям щелочных металлов, Триалкильные’ей единен и я таллия удалось получить впервые в 1930 г. (Groll). Трпэтилталлйй был получен взаимодействием растворенного в пет рол ей ном эфире (С2Н5)2Т1С1 с этиллитием: (С2Н5)2Т1С1 + ЫС2Н6= Т1(С2Н5)3-ф -|-LiCl. Это подвижная жидкость (уд. вес 1,97), перегоняющаяся при пониженном давленйи без разложения. Сухой кислород воздуха пе действует на триэтилталлий, но
428
Глава 10
вода быстро разлагает соединение с образованием гидроокиси диэтилталлия T1(C2H6)s-LHO1I = T1(C2H5)2OH+C2H6.
Трифенимпаллмй Т1(С6Н£)3 при обычной температуре значительно менее реакционноспособен, чем трпфонилгаллнн и трифепилипдий. При нагревании в инертном растворителе (например, в ксилоле) можпо, однако, значительно увеличить его реак-ционноспособность, так что она становится больше, чем у трифоаллирдня и может приближаться к реакционной способности фепилмагнийгалогенидов. Существуют предположения, что увеличение реакционной способности связано с образованием свободных радикалов Т1-С6Н5. Последние, одпако, малоустойчивы и немедленно дцспропор-ционируют, вели ие происходит других превращений, по уравнению ЗТЬС^Щ =, Т1(С6Н6)3-|-2Т1.
Ава литические сведения. Определения галлия, индия и таллия обычно производят наиболее простым способом при помощи спектрального анализа (см. ниже). Для весового определения применяются осаждение гидроокиси аммиаком и взвешивание в виде окиси. Таллии осаждается аммиаком только в трехвалентном состоянии. Следовательно, перед осаждением его необходимо окислить до трехвалентного состояния. Однако, используя растворимость гидроокиси одновалентного таллия, можно легко отделить таллий от галлия, ипдия, алюминия и других трехвалентных металлов.
Определение очепь небольших количеств индия можно осуществить пол яр о графически (Takagi, 1928; Ensslin, 1941). Применяя этот метод для определения содержания ипдия в нерафинированном цинке, Рииюкер (Bienacker, 1952) нашел:, что предварительно надо отделить индий от кадмия. Для этого индий осаждают в виде гидроокиси аммиаком из раствора, содержащего аммонийные соли, а гидроокись растворяется затем в 10%-ной винной Кислоте для съемки полярограммы.
Для спектрального определения галлия и его гомологов: обычно используют спектры хлоридов. Они состоят из немногих линий, приведенных в табл. 73. При определении в пламя горелки Бунзена вносят хлорид или
Таблица. 73
Спектры хлоридов галлия, ипдия и таллия
Галлий ?, = 417,1 лф тт 403,1 ж. (обе линии фиолетовые) Индий лф (индпго-снпяя), 410,1 .»щ (фиолетовая)
Таллий >. = 535,1 лф (зеленая)
окись, смоченную соляной кислотой. Еще лучше вызвать небольшую дугу между раствором хлорида и платиновым или иридиевым острием. Спектр хлорида существенно отличается от характеристического спектра, соответствующего свободному атому, который получается при возникновении вольтовой дуги или пропускании искры между электродами из соответствующего металла. Спектр свободного металла значительно богаче линиями, чем спектр хлорида. При внесении соли галлия в пламя горелки Бунзена заметна только слабая линия 417,1 л<д. Напротив, обе линии, приведенные в табл. 73, становятся очень четкими, если создать небольшую дугу или пропустить искру между раствором и платиновым острием. Зеленая линия таллия расположена почти так же; как и полоса бария 534,7 лф. Однако линия таллия от нее легко отличается своей резкостью и интенсивностью.
Для микроаналитического определения Ga и In пригодна цветная реакция, открытая Питчем (Pietsch, 1933 — 1934), которую дает с хинализарином оксигидрат, образующийся прп гидролизе. Границы определения: 0,02 у Ga“’ и соответственно 0,05 у 1п ‘ ’ в 1 мл. Реакцшо можпо вести и в присутствии солей алюминия или цинка (их оксигидраты образуют апалогпчшю цветные лаки), если только удерживать ионы АГ*' и Zn" в растворе при помощи подходящих добавок (этиламин, пиридин);: одпако в этом случае реакция менее чувствительна.
Глава 11
КООРДИНАЦИОННАЯ ТЕОРИЯ
Радикал. Уже давно было установлено, что определенные группировки атомов химических элементов, например NOs,J SCU, СОа, остаются неизменными при различных реакциях и при замещениях или при обменном разложении могут переходить без изменения из одного соединения в другое, затем в третье, в четвертое и т. д. Такие группировки получили название радикалов.
Вместо названия радикал часто употребляют обозначение группа] иногда радикалы называют также остатками, так как они остаются, если в химических формулах образованных ими соединений вычеркнуть те или иные части, способные замещаться на другие вещества.
Радикалы ведут себя в большинстве реакций совершенно так же, как и химические элементы. Следующие примеры, в которых радикалы заключены в квадратные скобки, могут свидетельствовать об этой аналогии:
2IIC1 -b ZnO = ZaCLj-t* И20, (1)
2Н [ХО3] ф- ZnO = Zh[N0312-> Н20,
2NaCl + HglSOJ = 2НС1 Na^SOJ, (2)
2Na[NO3] + H2[SOJ = 2II[NO3] 4- NaJSOj,
Na2[SO4]+ BaCl2== Ba[SOJ-> 2NaCl,
INH4J2ISOJ+BaCl2= BaJSOJ> 2INH1JC1. ? (3)
Большинство неорганических радикалов в водных растворах присутствует в виде свободных ионов.
Некоторые органические радикалы также способны к существованию в виде Свободных ионов, особенно радикалы кислот, например уксусной кислоты Н[СаНзОг], а также радикалы замещепных органических производных аммопйя.
Однако следует обратить внимании на Щ обстоятельство, что в органической химии кислотными радикалами чаще называют остатки молекул кислот, возникающие после отщепления от них группы ОН. Эти радикалы остаются неизменными при некоторых реакциях (например, при образовании хлорангидридов кислот, амидов кислот и т, д.), при которых пе происходит образование кислотных иопов вследствие отщепления протонов прт! растворении кислот в воде. Радикалом уксусной кислоты называют в зависимости от того, какую именно реакцию имеют в виду, либо группу С3Н3О2, либо группу С2Н3О. Т.тк как в органической химии последнее более целесообразно и поэтому общеупотребительно, то группу С2Н3О2 уксусной кислоты в тех случаях, когда она ведет себя как заряженный ион (папример, в водных растворах кислоты или в ее солях), лучше всего называть просто ионом уксусной кислоты или ацетат-ионом. То же справедливо и относительно других органических кислот.
Долгое время оставался спорным вопрос, могут ли существовать радикалы также и в свободном состоянии, пе будучи соединенными с другими веществами. То, что большинство неорганических соединений существует в виде свободных ионов, объяснялось при этом тем, что эти неорганические ионы рассматривали не в качестве свободных радикалов, а в качестве химических «соединений атомов или же радикалов
430
Глава 11
с положительными или отрицательными электрическими зарядами». Такое представ-Л ле и не возможно только формально и по выясняет сущности дела. Присутствующие-в виде свободных иопов радикалы именно потому и стараются7вступить в химическое соединение с другими находящимися также в виде свободных иопов веществами, так как они электрически заряжены. Все же надо учесть, что радикалы, не несущие электрических зарядов, редко встречаются в свободном состоянии. Реакции, в которых можно было бы ожидать образования свободных электрически незаряженных радикалов, почти всегда приводят к образованию веществ, которые приходится рассматривать в виде продуктов соединения в большинстве случаев двух радикалов. Элементарные вещества ведут себя в общем таким же образом: водород, хлор п т. д. при соответствующих реакциях образуют в качестве промежуточных продуктов свободные атомы, которые тотчас же соединяются в двухатомные молекулы. В основном то же происходит и с металлами в области обычпых температур с той только разницей, что • они образуют не молекулы, состоящие из двух пли нескольких атомов, а кристаллические решетки, построенные из многочисленных атомов.
Структурная группа. В то время как понятие радикал связано с понятием о химических реакциях, в гл. 7 определение структурной
группы является понятием кристаллографическим. Но в общем,-можно сказать, что атомы или соответствен по попы, сохраняющиеся при химических реакциях в виде единого целого, так же ведут себя и при построений кристаллов, таи что радикалы в кристаллической,решётке обычно являются структурными групами этой решетки.
Однако группа, обозначаемая в кристаллической решетке в качестве структурной, отнюдь пе должна оставаться неизменной и при растворений кристалла; или при разрушении структуры решетки. Особенно нельзя это с определенностью предсказать в тех случаях, если уже в самом кристалле связь компонентов, образующих структурную группу, не является очень прочной. В таких случаях растворение: кристалла или , соответственно разрушение решетки может вызвать полный распад структурной группы на отдельные компоненты, так что эта структурная группа уже не проявится в виде отдельных иопов в водном растворе и пе перейдет в другие соединения, за исключением тех случаев, если кристаллическая репгстка этих повых соединений снова окажется построенной вполне аналогично исходной.
Комплекс. У некоторых веществ часто наблюдается образование довольно сложных составных атомных группировок, которые, подобно радикалам, пе изменяются при многих химических превращениях и поэтому проявляют специфические реакции, иногда совершенно отличные or реакции компонентов, из которых они построены. Эти сложные образования называются комплексными группами, или комплексами. Устойчивость (т. е. неизменяемость при химических реакциях) комплексных групп часто не столь велика как устойчивость типичных радикалов*, Те комплексы, которые так же устойчивы, как типичные радикалы, называются прочными комплексами; те же, которые легко распадаются на свои составные части, носят название непрочных комплексов. Хотя комплексы построены из иопов, все же прочные комплексы в водном растворе не обнаруживают реакпин, характерных Л для соответствующих ионов, так как они не распадаются на ионы или распадаются на них лишь в незначительной степени. Непрочные комплексы обнаруживают тем сильнее характерные для их компонентов реакции, чем менее; они прочны и’ чем эти реакции чувствительнее по отношению к компонентам.
Полное отсутствие или ослабление типичных для компонентов реакций настолько характерно для комплексообразования, что о пем всегда говорят в тех случаях, когда типичные реакции отсутствуют вследствие образования особых химических соединений.
•На основании приведенного выше определения многие комплексы в болсе-тпнроком смысле можно рассматривать как радикалы.
Координационная теория
431
Т1рй этом не обязательно, чтобы в каждом случае Группа, характеризующая подобный комплекс, оставалась бы неизменной также и прп химических превращениях, как это бывает е радикалами. Так, упомянутое в предыдущей главе свойство алюминия не осаждаться из растворов, содержащих определенные,органические вещества, например : вннную.кислоту, объясняется образованием комплексных соединений солей алюминия.
Предполагают, что ион алюминия образует с соответствующими органическими соединениями сложно построенные попы. Однако при этом вовсе не обязательно, чтобы комплексные попы, образующиеся из растворов сульфата, хлорида, нитрата к т. д., обладали одинаковым составом. Явление, заключающееся в том, что иопы металлов, например. алюмипия, вследствие образования комплексов не обнаруживают больше характерных для гшх реакций, называют жаукцррахой этих ионов.
Соединения высшего порядка. Простые бинарные соединения, как CIH, Olla, NHa, CH4 называют соединениями первого порядка,,. Далее, к соединениям первого порядка причисляют и те, которые могут быть произведены от соединений указанного выше типа, путем простого замещения. Так, от упомянутых соединений путем простого замещения производятся:
Д1 Д1 Н. ,11
I- н s< р4н )Six ' „
’ ХН ХН IIх ХЧ
.CI /С1 С1. ,С1
J—Cl S< Рс-С1 )Six
ХС1 ХС1 С1Х ХС1
Все эти соединения являются соединениями, первого порядка. К этому ; классу веществ относятся почти все. чисто органические соединения.
: Вещества, образовавшиеся путем соединения молекул первого порядка в новое вещество, которое не может быть получено из соединении первого порядка путем простого замещения, называются соединениями высшего порядка,
Соединениями высшего порядка являются прежде всего- гидраты, аммиакаты, продукты присоединения кислот и аналогичные соединения, образовавшиеся вследствие присоединения органических молекул, а также двойные соли, например карналлит KCI-MgCla-6НаО. Карналлит следует считать соединением высшего порядка даже в том случае,,если совер-щённо не учитывать содержащуюся в нём воду, так как он .образуется (формально) в результате взаимодействия молекул К.С1 и MgCls. Наконец , комплексные соли, например К.ВВд = КF~)<ВFs, также принадлсжат к соединениям высшего порядка.
Если вода, аммиак или какое бы то ни было присоединенное вещество связано сравнительно прочно, так что его трудно отщепить, в том числе и при растворений в воде, й если вследствие этого присоединившееся вещество влияет на реакции тех веществ, с которыми опр связано, или на их окраску, или на какие-либо иные характерные .для первоначального соединения свойства, то его рассматривают как вещество, связанное комплексно. Если при этом комплексно связанными молекулами оказываются те, которые диссоциируют в водном растворе, то при диссоциации отщепляется только одна составная часть; другая же остается в комплексно связанном состоянии.
Например, фтороборную кислоту рассматривают Как соединение высшего по- , рядка, так как она образуется путем соединения BF3 и HF. IJ то же время она является комплексным соединением потому, что молекула HF или по крайней мере часть ее, а. именно поп F-, оказывается прочно связанным с BFj. В водном растворе соединение, диссоциирует следующим образом:
HF-BF3 = ГГ < (F-BFJ',
432
Глава J1
т. о. только одна составная часть связанной комплексно молекулы, а именно ион Н\ отпоил яс тс я при электролитической диссоциации.
Фторо борная кислота является примером комплексной кислоты. От комплексных кислот при замещении иона водорода на металл происходят комплексные соли. Соответственно комплексными основаниями называются такие комплексные соединения, которые при растворении в воде отщепляют ионы гидроксила.
Так как комплексные соли обычно более устойчивы, чем соответствующие им кислоты, их следует определять независимо от: последних. Комплексной в указанном выше смысле называют двойную соль в том случае, если она при растворении в воде не распадается или только незначительно распадается на ионы тех содей, из которых она образовалась. Далее, комплексными солями считают соли, образующие при электролитической диссоциации ноны, содержащие прочло соединенные нейтральные части. Вода также может быть связана в комплексное соединение в качестве нейтральной части; гидратированные иопы только в том случае относят к комплексным ионам, когда вода связана особо прочно и в ясно выраженном стехиометрическом соотношении.
От комплексных солей отличаются двойные соли в узком смысле, т. е. те двойные соли, которые при растворении в воде полностью или в значительной степени распадаются на ионы солей, из которых они построены. Но провести резкую границу между двойными и комплексными солями пе удается.
Главные п побочные валентности. Образование соодипений вйсшего порядка прежде объясняли действием особых сил, отличающихся от сил нормальных валентностей; эти силы называли дополнительными валентностями и обозначали в структурных формулах пунктирными линиями, в то время как обычные или главные валентности обозначали сплошными черточками. Существование соединения HF-BF3 объясняли тем, что бор по отношению к фтористому водороду обнаруживает дополнительную валентность.
/Г /Г
B<~F -|-П —Г=Н—Г---Вг-Г ’ (4)
-F F
Лучше это, однако, сформулировать иначе, Сказав, что бор по отношению к ионам фтора, может проявлять больше валентностей, чем три нормальные^
BF3+F- = [BF4]-. (5)
Ута формулировка и представление реакции уравнением (5) не содержит никакого особого предположений о природе связи четвертого иона фтора. В дальнейшем будет показано, что предположение о существовании особой, отличающейся от обусловливающей проявление обычной валентности силы совершенно не нужно. .
Равным образом и в тех случаях, в которых природа сил, обусловливающих образование комплексных соединений, еще не ясна, можно показать, что не существует, во всяком случае в уже образовавшейся молекуле, принципиальной разницы меящу главными и побочными валентностями.
Координационная связь. Особый тип проявления валентных сил, на котором осповано образование соединений высшего порядка, называют координационной сеянью. Можно представить себе, что атомы, способные к комплексообразованию, стремятся «сгруппировать» (координировать) около себя определенное число других атомов, молекул или радикалов, превосходящее число частиц, связанных обычными силами валентности. Такого рода атом комплекса, около которого группируются координацион но связанные атомы, молекулы или радикалы, называется центральным атомом комплекса (о комплексах с несколькими центральными атомами
Каордипацивнная теория
433
см. ниже; о двухслойных комплексных соединениях, в которых комплексный ион занимает место центрально го атома в другом комплексном ионе, см. т. II). ,
Число, показывающее, сколько атомов, молекул или групп координационно связано атомом элемента в определенном соединении, называется координационным числом или координационной валентностью этого элемента в соответствующем соединен ни. Наибольшее число атомов й т. д. определенного вида, которое может быть присоединено к центральному 'атому, называется максимальным координационным числом центрального атома по отношению к атомам п т. д. этого вида. Соединении, в которых центральный атом обладает своим максимальным координационным числом, называются координационно насыщенными.
Во фтороборной кислоте бор является центральным атомом комплекса. Его координационное число равно 4 (или он координационно четырехвалентен), так как он связан координационно с 4 ион aim F'. О наличии координационной связи свидетельствует то обстоятельство, что число иопов F' превосходит валентность, которую атом бора обнаруживает по отношению к электроотрицательным элементам, а также и тот факт, Что соединение H.F BF3 является соединением высшего порядка.
Наиболее часто встречается координационное число 6. Одпако оно характерно лишь для определенных классов некоторых соединений. Наряду с координационным числом 6 чаще всего встречаются координационные числа 4 и 3.
Учение о соединениях высшего порядка называется координационной теорией-, соединения высшего порядка называются также координационными соединениями. Понятие координационных соединений шире, чем понятие комплексных соединений. Кроме комплексных соединений, координационная теория охватывает двойные соли, распадающиеся в водном растворе на составные части, но в твердом состоянии во многих случаях построенные, вероятно, аналогично комплексным соединениям, и неустойчивые продукты присоединения. Как уже было показано в гл. 7, понятие о координационном числе оказалось плодотворным также и вне области собственно координационных соединений, а именно для понимания строения кристаллов.
Исторические сведения. Координационная теория была создана Альфредом Вернером. Она возникла в связи с изучением соединений металлов с аммиаком, состав которых нельзя было объяснить на основании старой теории валентности, т. е. при попытках отнести их к соединениям первого порядка. Вернер показал, что состав этих и многих других соединений можно объяснить без каких-либо вспомогательных допущений, если только за основу принять положение, что атомы после насыщения их обычных валентностей способны проявить еще дополнительные валентности. Это положение в большинстве случаев является непосредственным выводом из наблюдений; гак. в неоднократно упоминавшемся примере трехфторп-стого бора бор присоединяет еще один ион фтора. Для аммиакатов и их производных Вернер сумел также установить существование изомерных соединений с различными конфигурациями и пришел таким образом к установлению понятия о неорганической изомерии и к стереохимии неорганических соединений. Эти стереохимические представления получили поразительное подтверждение благодаря открытию предсказанной на их основе оптической изомерии комплексных соединений, например у комплексных соединений кобальта, хрома, платины (подробнее см. т. II). Основные поло-28 г. Рени
434
Глава 11
жения теории Вернера, как прежде называли координационную теорию, приобрели большое значение во всех областях химии. Среди исследователей, которые развили теорию Вернера в координационное учение, распространившееся в настоящее время на многие области химии, первое место занимают Пфейффср (Pfeiffer Р.) и Вейнланд (Weinland В.).
Толкование многочисленных соединений высшего порядка как координационных соединений даст для них общий систематизирующий принцип, между тем как без него эти соединения составляли бы огромную неразгаданную область. Первоначально значение координационной теории, если не считать объяснения случаев изомерии, в основном и состояло во внесении порядка в эту совершенно необозримую область. Эта задача, блестяще разрешенная координационной теорией, сама цо себе является весьма важной. Кроме rofo, в последнее время все чаще удается исчерпывающим образом' вскрывать причины, обусловливающие в конечном счете образование соединений высшего порядка. Это очень важно, ибо одно лишь введение понятия о координационной связи первоначально служило лишь описанием того факта, что образование соединений высшего порядка происходит, но не давало этому^факту никакого^объяснения.
Классификация координационных соединений. Все координационные соединения, поскольку их структура известна, подразделяются на;
1) соединения с комплексными катионами;
2) соединения с комплексными анионами;
3) неэлектролиты — соединения с электрохимически нульвалёнт-ным комплексом; ।
4) соединения, построенные из комплексных катионов и комплексных анионов (примеры см. т. II).
Прежде комплексные соединения в соответствии с теорией Вернера подразделяли на соединения внедрения и соединения присоединения.
Простейшим примером образования соединения внедрения является образование гидрата При растворении соли в воде:
СаС12 6П2О = [Са(ОНг)6)С12.
Вода внедряется между ионом Са" и ионами СГ, причем последние оттесняются от иона Са".
Одним ив простейших примеров образования соединения присоединения может служить образование фтороборной кислоты из трех фтористого бора й фтористоводородной кислоты:
HF4-BF3= HF-BF3= HfBFJ.
Оттеснения Иона фтора при присоединении фтористого водорода к трехфтористому бору здесь не происходит. Фтористый водород не внедряется между атомами, образующими BF3, а просто присоединяется к нему. Одяако не все комплексные соединения удается однозначно распределить по’этнм двум классам. Например, и одид тетра мети л-аммония |N(CH3)JI можпо рассматривать и как соединение внедрения N(CII3)3 в СН31, и как соединение присоединения СН31 к N(CH3)3.
Установление существования комплексныхУсоединений, Присутствие прочных комплексов можно установить тем же путем, что и присутствие обычных радикалов, т. .е. на основании присущей им способности при химических реакциях переходить без изменения от одного вещества к другому.
Так, во фтороборной кислоте ион водорода может быть замещен ионами калия и Других металлов; группа [BFJ-, которую поэтому мождо* назвать «радикалом фтороборной кислоты», прн этом остается неизменной.
Координационная теория 43 о
Присутствие прочных комплексов можно установить также путем измерения электропроводности растворов их солей. Молярная электропроводность разбавленного раствора соли зависит главным образом только от того, на сколько ионов распадается эта соль при растворении в воде. Природа ионов, из которых состоит соль, не имеет существенного значения для молярной электропроводности нормальной соли в сильно разбавленном водном растворе. Поэтому на основании измерений электропроводности сильно разбавленного водного раствора — обычно выбирают молярную концентрацию А (см. дальше) — можно установить, на сколько именно ионов распадается каждая молекула растворенной соли.
В табл. 74 сопоставлены значения молярной электропроводности р. некоторых типичных солей при 25° и разбавлении а = 1024 (т, е. в растворах, в которых 1 моль соли растворен в 1024 л). Из данных таблицы следует, что в зависимости от характера соли ее молярная электропроводность при данной концентрации лежит в биределенной области. Для солей, распадающихся,на два одновалентных иона, молярная электро-
Таблица 74
t Молярная электропроводность в зависимости от типа соли 'Разбавление о = 1024, температура i=25®
Соль Схема формулы тип Количестяо ионов Молярная электро-проводность
NaCl КС1О3 AB AB Одно-однопалентпые электро- 2 124 136
AgNO3 AB Jill ТЫ 132 •
ВаС12 ab2 Двух-одно валентные электро- 270
Mg(NO3)2 ab2 ЛИТЫ 3 251 :
K2SO4 a2b О дно-двух валентные электро- 298
ЛИТЫ
AlCi3 AB3 Трех-одновалентные электро- 4 384
- LaCl3 AB3 ЛИТЫ 397
л 1 Na4[Fe(CN)eJ a4b Одно-четырехвалентпые элск- 5 514
з K4[Fe(CN)6] a4b тролиты 583
проводность близка к значению 130; для солей, распадающихся на два одновалентных иона и на один двухвалентный поп, — к значению 270; для солей, распадающихся на три одновалентных нона и па один трехвалентный ион, она находится приблизительно около значения 400 п для солей, распадающихся на четыре одновалентных иопа и па один четырехвалсптпый нон, ее значение находится около 550. Таким образом, на основании молярной электропроводности можно сделать заключение р типе соли п, следовательно, для солей неизвестного строения указать, на сколько именно иопов они рас--п а даются при диссоциации в водпом растворе. Отдельные ионы должны быть, конечно, определены на основании других данных, папример химическим взаимодействием этих иопов.
Еще большее совпадение данных наблюдается для солей одного типа в характера изменения молярной электропроводности при разбавлении раствора. Поэтому пытались, произвести ряд измерений в растворах с вдвое увеличивающимся разбавлением. Если 28"
436 Глава 11
исходит!.'из 1 М раствора (р = 1), то прп этом получается ряд разбавлений:
₽ = 2—4—8—16—32—64—128—256—512—1024 и т. д.
Кривая разбавления для растворов прочных комплексных солей вполне подобна кривой разбавления простых солей. Для более слабых комплексов иногда ла основании кривой разбавления можно сделать заключение о типе разложения комплекса.
Дальнейшее обнаружение комплекса, а также исследование (в сочета-яии с другими методами) и самой прпроды этого комплекса основано на том, что при образовании комплекса вещества, содержащиеся в растворе, не обнаруживают уже характерных для них химических реакций.
Так, в растворе фтороборной кислоты и со солей уже нельзя обнаружить типичную реакцию одной из ее составных частей — трехфто ристого- бора, а именно образованно борной кислоты* при взаимодействии с водой.
В растворе не обнаруживается также характерная для ионов фтора реакция — выпадение фторида кальция при прибавлении попов кальция. Следовательно, раствор ужо не содержит в сколько-нибудь заметном количестве ни трехфтористого бора, пп ноиов фтора. Это (в связи с наблюдениями относительно электропроводности) заставляет предположить, что н растворе образовался комплексный ион [BFJ'.
На основании аналогичных данных делают заключение и об образовании комплексных ионов в растворах солей алюминия, содержащих некоторые органические вещества, например винную кислоту, в связи с тем, что некоторые типичные для ионов алюминия реакции в этих растворах отсутствуют или же алюминий осаждается из них не полностью. Однако в этом случае еще до сих пор пе представляется возможным точно установить состав образующихся комплексов.
Равным образом и свойства растворов, зависящие от осмотического давления, например понижение температуры замерзания и повышение температуры кипения, дают основание для заключения об образовании и о составе комплексов в растворах, поскольку это можно сделать определением числа имеющихся в растворе молекул.
Измерения понижения температуры замерзания и повышения температуры кипения часто служили для подтверждения заключений, установленных совершенно независимо но данным измерения электропровод пости. Но область применения двух первых методов гораздо шире, чем область измерения электропроводности, так как они позволяют установить факт образования комплексов из ионов и из нейтральных частиц,— процесс, который нельзя обнаружить нутом измерения электропроводности.
Подобным же образом и другие методы, дающие указания о числе молекул или о концентрации молекул определенного типа в растворе, могут служить для установления образования комплексов, цанример потенциометрические измерении и изучение равновесий.
Образование комплексных ионов может быть установлено измерением чисел электролитического переноса. Если из чисел переноса следует, что вещества, которое могут функционировать в водном растворе только в качестве положительных ионов — например, все металлы,— оказываются перенесенными электрическим током к аноду, то отсюда можно сделать заключение, что они должны были присутствовать в растворе в виде отрицательно заряженных комплексов.
Следует помнить, что сделать заключение о направлении, по которому перекосятся электрическим током составные части электролита, можно только на основании изменений концентрации около анода и катода, но не на основании того, где именно происхо
* Ввиду того что фторобор а тный комплекс не очень прочен, обычно все же происходит его частичный гидролиз водой. Но этот гидролиз совершенно не обнаруживается, если предотвратить расщепление комплекса по уравнению [RF^]' у* BF3-|- F' прибавлением избыточных ионов фтора.
Координационная теория
437
дит. само выделение. Свинец в крепком азотнокислом растворе выделяется на аноде, так как он в этих условиях окисляется на нем с образованием нерастворимом двуокиси, а вовсе не потому, что оп переносится туда в виде комплексного иопа.~
Измерением чисел переноса можно иногда обнаружить очень неустойчивые комплексные ноны.
В последнее время к этим методам присоединился еще структурный анализ кристаллических координационных соединений при помощи рентгеновских лучей. В гл. 7 уже было показано, что поданным структурного анализа химические радикалы в кристаллах функционируют в общем случае в виде отдельных структурных групп, например радикал INOa]“ и радикал [SOd2^ (ср. рис. 50 и 51, стр. 246 сл.).
Каким образом комплексный радикал в узком смысле этого слова можно обнаружить а кристаллической решетке в виде структурной трунпы — показывает рис. 79,
Рис* 79. Рещетка гексафтороалюмината аммония [Г7Н4]з[А1Рв].
П=8,40 Л; А1 ► F=d=i.66 Л.
на котором представлена элементарная ячейка гексафтороалюмината аммония [NH4]3 [AlFfi]. Из рисунка видно, чт(| атомы F (или же соответственно ноны F') сгруппированы таким образом, что (5 атомов фтора окружают в виде октаэдра одни атом алюминия. На рисунке представлены только части каждого из этих октаэдров, при-падлежлтцие к элементарной ячейке; только один октаэдр (внизу справа) дополнен тремя атомами F, лежащими вио адементарнон ячейки. Если из решетки удалить лежащие в середине ребер атомы N и лежащий в центре атом N вместе с прилежащими к нему атомами Г Г, то получается элементарная ячейка гексафторосиликата [NH4]2fSiFe], о котором будет сказало в следующей главе. Подобная решетка, как и решетка только что упомянутого соединения, была обнаружена у многих соединений аналогичного состава, папример у гексахлоронлатината калия K2[PtCleJ.
И а рис. 80 представлена решетка фторида калия-магния K.F-MgF2. Это соединенно представляет собой пример двойной соли, которая в водном растворе практически полностью распадается на составные части, т. о. на ионы, но для которой в кристаллическом состоянии с известным ограничением мощно ещо говорить о комплексообразовании. В пей все ионы фтора сгруппированы определенным образом ближе к ионам магпия, чем к попам калия: каждый ион магпия па расстоянии а /2= 2,00 А окружен
438
Глава 11
шестью ионами фтора, в то время как самое короткое расстояние между ионами калия и попами фтора .равно (а/2) ]^2 = 2,83 А, т. е. равно расстоянию между отдельными иолами фтора. Координационное число магния в кристаллическом соединении равно 6; следует, однако, учесть, что каждый из шести ионов фтора, окружающих ион магния, одновременно принадлежит и второму иону магния, лежащему вне элементарной ячейки, так что стехиометрнчески на каждый ион магния приходится только три иона фтора. Представленный на рис. 80 тин решетки соединений с общей формулой МНХ3 встречается довольно часто, главным образом в целом ряде кислородных солей (MRO3), например у титаната кальция CaTiOs (перовскит). Поэтому его называют <<перовскитным типом».
Рис, 80, Элементарный куб , двойного фторида калия и магния "
KMgFa (тип перовскита).
Так как известны многие случаи, когда трудно растворимые вещества вследствие образования комплексов становятся легко растворимыми, то в общем можно сделать заключение об образовании комплексов, если растворимость при прибавлении определенных солей к раствору повышается. Однако это заключение нельзя сделать без всяких оговорок при незначительном повышении растворимости, так как на основании новой теории сильных электролитов повышение растворимости трудно растворимых солей можпо вызвать также и прибавлением электролитов с одинаковыми ионами, между как, согласно классической теории, подобное повышение в отсутст-комплексообразования возможно только в том случае, если при-
тем вне бавляются электролиты с посторонними ионами.
Иногда можно обнаружить образование комплексов по изменению окраски. Так, безводный, сульфат меди белого цвета, а обычный, содержащий воду сульфат меди окрашен в синий цвет, как и его водный раствор. Поэтому полагают, что ион меди в синей соли содержит комплексно связанную воду.
Воду, содержащуюся в кристаллических соединениях, называют обычпо кристалла вау ионно И водой. Часто бывает трудно решить, в какой море можно считать ее «связанной комплексно». Поскольку при образовании кристаллов в конце концов решающее зпачение имеют те же силы, что и Для образования соединений высшего порядка, почти всегда можно говорить о координационной связи кристаллизационной воды. Но в более узком смысле обозначение «координационное соединение» или адекватное ему в этой области название «комплексное соединение» можно применять к веществу, содержащему кристаллизационную воду в кристалле только в тех случаях, если молекулы воды в кристалле координированы: около строго определенного иона, в соседстве с которым они и расположены, и когда Они вместе с этим ионом образуют более или менее самостоятельную структурную группу в кристалле. В некоторых случаях наличие таких структурных групп может быть установлено ронтгёнеструктурным аиа-дизом. В общем можно ожидать, что в таких структурных группах вода будет связана прочнее, чем при ином расположении молекул воды в решетке. Поэтому воду, которая легко теряется, обычно рассматривают как кристаллизационную воду в узком смысле слова, а ту воду, которая теряется с большим трудом, считают «комплексно связанной». Но установить какую-либо границу только на основании критерия трудной и легкой способности отдавать воду невозможно. Те же соображения можно высказать и относительно таких аммиакатов, которые известны только в твердом состоянии.
Строение комплексов. До Вернера пытались установить структурные формулы для Комплексных соединений в соответствии со старой теорией валентности, т. е. отнести их к соединениям первого порядка. Это привело р представлению о цепеобразных структурах. Такие «цепочечные,формулы»
Координационная теория 439
были предложены Бломстрапдом для аммиакатов, однако доказать их не удалось.
Прежде чем вывести по методу Бернера структуру упомянутого выше соединения, следует кратко просуммировать, какое толкование результатов наблюдений можно дать на о сно в е ионно й тео рии. Приходится констатировать, что фторид бора обнаруживает способность присоединять один ион фтора. Нейтральный трехфтористый бор превращается при этом-в однозарядный электроотрицательный ион IBFd', называемый комплексным попом
BF3+F' = [BF4]',
Образовавшийся таким образом комплексный ион может вступать в соединения с различными катионами, например с Н', К\ [NH-*]’, Ва". Соединение его с водородом — фтороборная кислота — представляет собой более сильную кислоту, чем фтористоводородная.
Так как катион, связанный с комплексным ионом, может быть произвольным и так как в водном растворе можно установить присутствие комплексного иона в свободном состоянии, то следует рассматривать его независимо от связанного с ним катиона.
Как достроен комплексный анион [BFJ'? Ввиду того что предположение о цепной связи ионов фтора исключено вследствие несовместимости такого допущения с одновалентностью фтора, то остается только принять, что все четыре иона фтора связаны с бором. Остается неясным еще вопрос, одинаковым ли образом или различно ионы фтора связаны с бором. Ни в одном случае, если только речь идет об одинаковых атомах или группах, связанных с центральным атомом, нельзя привести сколько-нибудь убедительных доказательств в пользу существования различных связей. Следовательно, приходится заключить, что связи однородны.
* -г
Во времена Вернера валентность рассматривали как неделимую единичную силу, действующую в определенном направлении. Если строго придерживаться этой точки зрения, то при образовании координационных соединений невозможно проявление других валентностей — тан называемых «побочных валентностей» по Вернеру, После того как вместо цепных формул стали применять формулы с центральным строением по Вернеру, не осталось уже сколько-нибудь веских доводов в пользу того, что валентности, проявляющиеся в Простых соединениях, чем-либо отличаются от валентностей в комплексах. Не исключено все же, что расширение экспериментальных исследований могло бы доказать это предположение. Путем исследования всех возможных случаев изомерии на примере аммиакатов и их производных Вернер, одпако, установил, что координационно связанные группы, пространственно одинаково расположённые около центрального атома, связаны с ним одинаково прочно. Ввиду того что в настоящее время гипотеза, рассматривающая валентность как действующую в определенном направлении единичную силу, применима только в определенной области, да и то в ограниченной степени, предположение об одинаковой связи является наиболее правильным и, во всяком случае, оно может быть положено в основу рассуждений во всех тех случаях, когда не имеется опытных данных, Противоречащих такому предположению.
Связанные с комплексом компоненты, т. е. в случае фтороборной кислоты — ион водорода, а в случае фторобората калия — ион калия, при растворении этих соединений в воде отщепляются в виде ионов. Поэтому Вернер называет их ионогенно связанными частями — в противоположность неионогенно связанным атомам, ионам и группам, непосредственно окружающим центральный атом. Относительно ионогенно связанных групп Вернер полагал, что они связаны не с каким-нибудь атомом комплекса, а с самим- комплексом в целом. Это наиболее естественное предположение, основанное на фактическом материале, так как в растворе
440
Глава 11
электрически заряженный комплекс может проявлять свое действие на большое расстояние только в качестве единого целого. Следует ожидать, что комплексный иоп в основном будет вести себя так же и после того, как ионы соединятся в кристаллы. Поскольку в настоящее время существуют данные рентгено структур кого анализа кристаллических прочных комплексных координационных соединении, можно это предположение считать уже вполне подтвержденным. Прочный комплексный нои в кристалле занимает совершенно такое же место в решетке, как и радикалы [NOa]", [SO4]’ или [NHt]+ (ср. рис. 79).
Вернер предположил, что атомы, являющиеся централ ьньши атомами комплексов, стремятся окружить себя совершенно определенным числом других атомов или групп. Подобно тому как структуру простых соединений определяет обычная валентность (формальная валентность), так, согласно Вернеру, для координационных соединений эту структуру будет определять координационная валентность или координационное число.. При этом безразлично, будут ли координационные валентности насыщены простыми атомами или же целыми группами. Различие имеется только постольку, поскольку существуют определенные группы, которые могут занимать два координационных места, что никогда не наблюдается у простых атомов.
Такими группами, способными занимать два координационных моста («группы с координационной валентностью два»), являются прежде о се го гидразин, этилендиамин и оксалатный пои. Формулы этих групп показывают, чом вызвана их способность занимать два координационных места,
HsN~NH2 П2С—СН2 ГОС—СО-1*
i : II
HaN NH2 О О
U. й ’ J
* ' • •
Они имеют два пространственно отдаленных друг от друга места, от которых исходят силы координационных (•••) валентностей.
Группы, занимающие два координационных места, называются двухвалентными. Следовательно, необходимо принимать во внимание, что атом, обладающий координационным числом и, называется координационно «-валентным, а группа, имеющая координационное число п, — к<к)рди1гациоппо «-валентной.
Тот факт, что многие элементы имеют преимущественно координационное число 6, которое обусловливает расположение неионогенно связанных групп в вершинах правильного октаэдра, т. е. их вполне симметричную группировку, привело -Вернера к заключению, что стремление к образованию пространственно возможно наиболее симметричной группировки имеет для образования координационных соединений особое значение. Вопрос о том, как объяснить это стремление к образованию пространственно возможно более симметричной группировки, остается открытым.
Причины образования координационной связи. Существование координационных соединений объясняется предположениями как о существенном значении кулоновского взаимодействия между противоположно -заряжецными атомами, так и о том, что при их образовании важную роль, играют квантово-моханические резонансные силы, действием которых объясняют образование гомеополярных связей. Оказалось, что оба объяснения совместимы. Для многих координационных соединений предположение о том, что их образование вызывается силами притяжения, действую-
Коордииациоииац теория
441-
щимн между противоположно заряженными ионами или между ионами и полярными молекулами, по совсем удовлетворительно объясняет сам факт их существования, но зато свойства и поведение этих соединений соответствуют свойствам, которые можно было бы ожидать на основании этого предположения. Имеется также большое число координационных соединений, образование которых нельзя объяснить подобным образом. Их состав, свойства м поведение указывают на то, что имеющиеся в них химические связи являются связями другого рода, а именно такого же, как в го.меополлрпых соединениях. Для объяснения образования многих координационных соединений применимо как то, так и другое объяснение. К таким соединениям относятся фторобораты, на примере которых й следует рассмотреть обе теории существования координационных соединений.
Рис. 81. Модель трифторида оара.
Применение теории Косселя к объяснению строения иона фторобората. На основании теории Косселя, т. е, на основании положения, что в гете-ронолярных соединениях атомы различного электрохимического характера можно рассматривать в первом приближении как электрически заряженные шары (соответственно их валентности)действующие друг на друга по закону Кулона, нс только может быть объяснено образование комплексов, например комплексов фтороборной кислоты, но существование подобных комплексов становится даже необходимым следствием, вытекающим из теории. Если рассматривать бор как электроположительную составную часть соединения В Fa, то можно показать, что та же сила, которая вызывает образование трехфтористого бора из электроположительного бора и электроотрицательного фтора, обусловливает и способность бора связывать еще один поп фтора и, следовательно, образовывать комплексный нои
Система, состоящая из одного атома с тройным положительным зарядом, окруженного тремя отрицательно заряженными одовалентпыми атомами (рис. 81), ведет себя па больших расстояниях практически как электро-пейтральная система. Одпако на отрицательно заряженные атомы, которые достаточно приближаются к неп,: влияет сила притяжения, величина которой можот быть вычислена на основании закона Кулона (см,
стр. 107), Предположим, что на рис. 81 представлена молекула трехфтористого бора. Радиус иона В3+ настолько мал, Что три иопа F~ могут касаться один другого. Энергия образованиямолекулы трехфтористого бора из В3+ n3F_ получается, если снова пренебречь сжимаемостью и взаимной поляризацией ионов и в качестве диэлектрической проницаемости подставить величину D = 1 (диэлектрическая проницаемость пустого пространства), приняв за основу модель, показанную на рис. 81;
,, л . З-Зе1 ГЗе3 7,27 ,
’ ‘ ^0 Яо /3
где At — энергия, выделившаяся вследствие взаимного притяжения противоположно заряженных ионов; д-т— анергия, .татраионная на преодоление взаимного отталкивания одноименно заряженных ионов; 2?0— межатомное расстояние В +--> F [принимая для радиуса иона фтора значение, соответствующее ому в кристаллическом соединении (1,33 А), чисто геометрически получим Йо — 1,54 А]. Дли энергии образования молекулы BF3 в соответствии с моделью рис, 81 получаем
Ео^ 4,71-108-е8 эра.
Таким же образом можно вычислить анергию образования комплексного лона (BFj)"> полагая, что он построен из В3+ и из четырех тетраэдрически расположенных
442
Глава /1
вокруг пего исков F'. Межатомное расстояние В +—> F обозначим порез опо оказывается несколько большим, чем R,it а именно Я1=1,вЗ А, Для энергии образования комплексного иона [Вр4|- иа В3+ и 4F- получим
4» Зб- 3 1^” 6 8,33
/?1 = —----= * =541 • 10* • е2 эрг;
2f j . £Jii xti
больше чем £0; следовательно, с образованием [ВЕ4]" из BF3 и F- связан еще дополнительный выигрыш энергии. Тем самым становится понятным образование этого комплексного иопа, т, е. причина проявления «побочной валентности». Если вычислять аналогичным образом энергию образования [BF5]a- из В?* и 5F", то получим 4,53-108-е8 эрг, а для энергии образования [BF6]S- из В3* и из 6F~ получим 4,27-108.еа ЭрВ, 7>ак как энергия образования этих комплексных ионов меньше энергии образования иона [BF4]-, то нет, вообще говоря, оснований для образования этих комплексов. Недействительно, их образования ие наблюдается.
Таким же образом объясняется образование гидроксо-ионов, например [АЦОН*)]', [AJ(QH)6]" и [AJ(OH)J"' из А1(0Г1з) и избыточных ОН'-ионов.
Равным образом в определенных случаях присоединение нейтральных, но обладающих сильно дипольным характером молекул, например Н2О и NHa, можно объяснить как следствие исключительно электростатического притяжения. Но это относится только к тем случаям, когда речь идет об относительно непрочном присоединении.
Применение теории атомных связей для объяснения строения фторо-борат-иопа. В 1923 г. Сиджвик первым применил теорию атомных связей к координационным соединениям. Существенным оказывается при этом предположение, что оба спаривающихся электрона, обусловливающих связь двух атомов, вместо того чтобы быть равномерно распределенными между Обоими атомами, тяготеют к одному из них (дативная связь, см, стр, 162), Если принять это предположение за основу, то образования иона BEj из ВЕз и F” следовало бы ожидать также и в том случае, если бы речь шла о связи В—F как о чисто атомной связи. Проще всего удалось бы объяснить возникновение четвертой связи, если бы В Fa обладал строением, соответствующим формуле I, так как именно в этом случае атом бора не имел бы полностью завершенной внешней оболочки, которая легко могла"бы принять еще одну электронную пару. Но в соответствии с изложенным на стр, 371 строение ВЕз лучше отражается формулой II. Принимая эту формулу за основу, можно объяснить присоединение иона F" к молекуле гВЕз ненасыщенным характером последней, следующим из наличия двойной связи:
:F: : F i
; F:B ; F: + ; F: = ; F: В: F - .• F : В : F F: = ; F : В : F г
- : F : , F • ; F :
’ ‘ II ..
По Полингу, связь В — F па 63% имеет гетер столярный характер, В ионах же (SO4p“ и [C10J-, совершенно аналогичных но составу иону связи имеют пре-
имущественно гомеополярный характер. Доля гетерополярпой связи снижается у них до 22 и 7%, Преимущественно гомеонолярное строение этих ионов доказывается физическими методами исследования. Теория атомных связей объясняет .образование ионов [SO4]3- и [С1О4]” таким же образом, Kait и иона [Bf4]“. Приложение теории Косселя приводит к одинаковым результатам. Однако ее использование для объяснения комплексообразования в тех случаях, в которых связи имеют преимущественно гомеополярпый характер, не всегда оправдано.
Соотношение между обычными и координационными валентностями. Различия в проявлении обычных и координационных валентностей, соглас-
Координационная теория
443
ио вышесказанному, заключается в следующем: если речь идет о гетерополярных соединениях, то при образовании из элементов соединений первого порядка имеет место: 1) обмен электронов между атомами, приводящий к появлению на ппх зарядов, 2) взаимодействие между противоположнозаряженными атомами.
При образовании координационных соединений происходит только второй процесс, так как первый уже совершился ранее; несколько атомов или групп взаимно соединяются, если только это возможно, с выигрышем энергии.
То же происходит при образовании кристаллов, только в этом случае энергия образования выделяется не в результате соединения небольшого числа заряженных атомов в комплекс, а вследствие взаимодействия множества ионов с образованием кристаллической решетки.
Если речь идет о гомеополярных соединениях, то образование соединений первого порядка основано на том, что при взаимодействий между атомами, внешние электронные оболочки которых не полностью заполнены, в результате спаривания электронов возникают обменные силы.
В этом случае образование координационных соединений происходит тогда, когда эти обменные силы могут проявляться в большей области нежели при образовании соединений первого: порядка. Это имеет место либо в том случае, когда число электронов во внешнем слое центрального атома еще более возрастает вследствие увеличения числа партнеров по связи j либо когда при неизменном числе этих электронов увеличивается число атомных ядер, обменивающихся электронами с ядром центрального атома (превращение ненасыщенной связи в насыщенную). ,
Комплексы внедрения и нормальные комплексы. Атомные связи часто приводят к образованию особо прочных комплексных соединений. Не только радикалы, по п нейтральные группы, панрнмер NH2, связываются атомными связями с центральным атомом особенно прочно. В отличие от них такие координационные соединения, в которых существуют только ионные связи между сильно полярными молекулами и ионами (без значительной доли гомеополярности), в большинстве случаев обладают только свойствами относительно слабых комплексных соединений. В этом характерное различие мопеду координационными или комплексными соединениями с ионным характером связей и со связями атомного типа. Последние, согласно Бильтцу (Blits), называют комплексами внедрения, та к как в случае атомных связей электронные оболочки центрального атома и лигандов взаимно проникают одна в другую. Наличие атомных связей между центральным атомом и его лигандами ао многих случаях можно непосредственно доказать, маг нит и ii.ii и измерениями (см. ниже). Комплексы, в которых связи электровалентны или обусловлены вандерваальсовыми силами или дипольным взаимодействием, обозначаются в отличие от комплексов внедрения как нормальные комплексы. Комплексами внедрения, вероятно, являются, как Правило, такие комплексные соадлнелия, которые имеют характер неэлектролитов (например, jCi'Gl3(NH3)3J). Помимо различной прочности связей, комплексы внедрения отличаются от нормальных комплексов также и тем, что в них центральный атом имеет почти всегда постоянное координационное число, в то время как в нормальных комплексах координационные числа обычно колеблются и состав этих комплексов зависит от соотношений, в которых брали для их получения составные части.
Но могут существовать также и «нормальные комплексы» с постоянным координационным числом. Об атом, в частности, Свидетельствует приведенный выше — в предположении существовании чисто ионных связей — расчет энергии образования иона
Отличие нормальных комплексов от комплексов внедрения при помощи магнитных измерений. Простые соли двухвалентного железа, например FeCl2 и FeS04, обладают магнитным м ом оптом около 5 магнетонов Бора. То же имеет место для соединений [Fe(OH2)4]С12, [Fe(NH3)2]Cl2, [Fe(NH3)6]Cl2 идругих слабых комплексных Солей железа. Напротив, соединения K4[Fe(CN)fi] и K3[Fe(CN)5CO] имеют момент, практически не отличающийся от нуля. Из этого следует заключить, что в первой группе этих комплексов железо существует в той же форме, что и в простых солях, и что р аспола-
ш
Глава 21
гающиеся около него группы связаны только вандерваальсовыми силами или за счет поляризационных эффектов. В цианидных комплексах, очевидно, существует другой тип свяли. Предполагая, что каждая циано-группа связана с железом атомной связью, можно ожидать, как это и было показано, диамагнетизм для комплекса (Fe(CN)a|4'. Магнитные измерения подтвердили развивавшееся Бильтцем представление о причине чрезвычайно различной устойчивости двух типов комплексов, а именно нормальных комплексов, в которых в зависимости от рода лигапдов существует либо нонпая связь, либо связь, обусловленная вапдерваальсовымп силами, и комплексов внедрения, существующих благодаря наличию общих электронов для лигандов и центрального атома. К нормальным комплексам принадлежит, например, также комплекс [FeFel3~- Оказалось, что комплекс [Fe(CM)a]3- следует рассматривать как комплекс внедрения, вследствие наличия у пего магнитного момента, равного 1,73 магнетона Бора, соответствующего но правилу Льюиса (см. стр. 341) одному и оспоренному электрону. Экспериментально был обнаружен момент около 2 магнетонов. Следовательно, и в этом случае речь идет о комплексе внедрения. Для карбонилов Fe(CO)j, Сг(СО)6 и Ni(CO)/,, предполагая в них существование атомных связен, следует ожидать диамагнетизм, и действительно, эти соединения диамагнитны.
Комплексообразование у переходных элементов. Явное стремление к образованию прочных комплексов наблюдается у многих «переходных элементов» (см. стр. 352). Как указывает Полинг, это можно обосновать квантово-механически. У ионов переходных элементов существуют неполностью занятые d-уровни. Электроны на этих неполностью занятых d-уровнях очень часто частично или даже полностью не спарены. Например, ион Сг3* содержит три, ион Мпй+ пять, пои Fe2' (помимо двух спаренных) четыре неспаренных d-илектропа. Под влиянием окружающих лигандов электроны таких ионов могут спариваться, причем освобождается большее или меньшее число первоначально занятых d-уровтюй. Так как эти d-уровни Лежат лишь немного iinatej-и /j-уровней следующих оболочек, из этих d-,p- и s-уровпей может происходить образование новых общих уровнем, способных заполняться электронами лигапдов. Расход Энергии, затрачиваемый* на спаривание первоначально неспареипых электронов, покрывается за счет «энергии резонанса» при «гибридизации» уровней. Электроны, обусловливающие атомную связь, принадлежат лигандам. Центральный атом представляет для Этих электронов лишь своп вакантные энергетические уровни или образовавшиеся из них промежуточные, гибридизоваппые уровни. Например, в ионе Со3' имеется два спаренных и четыре неспареипых 3d-электрона. Прп их спаривании освобождаются два 3d-ypOBiiH, образующих вместе с «ы-уровнем и тремя 4р-уровнямп промежуточный уровень, на котором могут разместиться 2 у 6=12 электронов. Эти места могут быть, заняты 6 молекулами аммиака, каждая из которых предоставляет одну электронную пару для образования атомпОЙ связи
3 +
Co 4- 6NH3
H3N^ NH, H3N—* Co NH3 HjN"''* ^NH3
H3N^ NH, HjN—Со—NH3
H3N^
При первом способе написания формулы становится понятным, что в образовавшемся комплексном и еще электронные пары, обусловливающие связь, предоставлены азотом NH3 (свободные молекулы NHS имеют одну свободную электронную Пару). При втором способе написания подчеркивается, что существующие в уже образовавшемся комплексном попе атомные связи по своей природе ничем не отличаютвя от каких-либо других атомпвдх связей. Полинг сумел показать, что и здесь, так же как и у углерода, гибридизация приводит к выравниванию пространственно-направленных связей. Прп гибридизации двух- d-орбнТ, одной а- и трех р-орбит возникает шесть связей; направленных к вершинам, правил,нога октаэдра. Этим и объясняется октаэдрическая структура, предложенная Вернером па основе изучения явления изомерий типичных прочных комплексов с координационным числом 6 (см. т. II, гл. 5). При гибридизации трех d-орбпт и одной s-орбиты или (как в случае углерода) одной s-орбиты с тремя р-орбитами образуются'четыре тетраздрическщнаправленные связи-, при гибридизации одной d-орбиты с одной s- н двумя р -орбитами возникают четыре, лежащие в одной плоскости связи, направленные к вершинам квадрата. Подобное плоскостное расположение чоты-ь рех лигандов действительно было установлено для некоторых комплексов, например
* В этом случае необходимо затратить для спаривания энергию, так как соответствующие d-электроны в нормальном состоянии иона иеспареиы (обладают одинаковым СВК!ЮЛ4).
Координационная теория
445
для комплексов никеля, палладия и ил этилы. Подробнее явления изомерии, связал-ные со структурой комплексных соединений, будут рассмотрены к -г. II, в разделах о соединениях, для которых эти явления лучше всего изучены.
Номенклатура. Обзор множества координационных соединений чрезвычайно облегчается применением рационального и простого обозначен Лил их природы, I. о. состава и строения. Такая единая система обозначений была разработана Вернером.
Предпосылкой для рационального наименования координационных соединенна является знакомство в основных чертах с их строением. Поскольку это строение неизвестно, для двойных солей и других продуктов присоединения приходится сохранять обычные способы обозначения.
Чтобы показать, что атом элемента или группа связаны координационно, к их названию прибавляют окончание «о», предшествующее, названию элемента, к которому она относится.
Исключение составляет аммиак, который обозначается как аммин в координационных соединениях (аммин с двумя ш» в отличие от аминогруппе! NHa). Координационно связанная вода обозначается как «акво». Кислород обозначается словом «оксо»; сера — «тио»; связанная одной связью гидроксильная группа ОП обозначается словом «гидроксо», радикал—О—О перекиси водорода «нероксо»; остальные группы — словами: л:лоро-, фторо-, сульфата-, карбоната-, нитрата-, оксалата-, вообще остатки кислот — ацидо-. Часто вводимая в комплекс группа (этнлендиа-мин) HaN—СНа—СИг—NHa обозначается через чен».
Из атомов или групп, координационно связанных в комплексе, в наименовании или в формуле сначала упоминаются отрицательно заряженные, а затем нейтральные и среди них на первом месте Ч2О (акео-) и аммиак (аммин-). Из остальных нейтральных групп или атомов упоминают сначала неорганические, а затем органические. Название нейтральных групп или атомов, за исключением II2О И NHj, оставляют неизменными.
Число координационно связанных групп обозначают греческими, числительными, стоящими перед их названием;.
1234 5 6 7 8 0 10 12
моно ди три тетра пента гекса гепта окта онпеа дека додека
Если в комплексе связано несколько различных групп, то каждая из них упоминается с соответствующим числом.
Названия чисел не нужны, если они очевидны: обычно моно опускается.
Пример:
(Са (OH2)eJCl2—гсксааквокальппйхлорпд
(Са (№Г3)6] —гексаамминкальций
fCrCJ (ОН.,).. (NH3)J 504—хлородиаквотриамминхром(Ш)сульфат (Cr(OU) (OH2)cn2](NO3)2—гидрси;соакводиенхром(III)нитрат.
Отрицательный комплекс в названиях солей всегда получает окончание «ат». Например, К [BFJ — тетрафтороборат калия, или, короче фтороборат калия. Соответствующая кислота называется, согласно номенклатуре Вернера, тетрафтороборной кислотой, или, короче, фторо-борная кислота*. Это название показывает полную аналогию соединения с борной кислотой, а ие с фтористоводородной кислотой.
* Такие сокращения (опускание цифровых обозначений) допустимы в тех случаях, когда речь вдет о хорошо известных соединениях, которые иельэя спутать. Равным образом у элементов, характеризующихся только одной степенью валентности, можно опускать указания об их валентности.
446
Глава, 21
Электрохимическая валентность (электровалентность) центрального атома в комплексных или координационных соединениях обозначается по интернациональным правилам, так же как и в простых соединениях, т. е. римскими цифрами, В катионных комплексах и в анионных комплексах кислот цифра, обозначающая валентность, ставится после названия соответствующего элемента. В анионных комплексах солей она пишется после оканчивающихся на «ата названий .комплексов.
Пример:
[Сг (NH3)6] С13—гексаамминхром(Щ)хлорид
He[PtClc] — гексахлороплатино([У)кислота К2(Р4С16] — гексахлоро ппатинат(ГУ)калия К4 (Fe (CN)e] — гексацианоферрат(П)калия
Прежде валентности обозначали по предложению Вернера окончаниями, присоединяемыми к названию центрального атома, а именно:
Для валентности: 12345678
Окончание: о о и е ан он ин ен
При наименовании комплексных кислотных остатков в солях это Окончание для обозначения валентности вставляется между названием центрального атома и окончанием «ат».
П р и м е р: гексааммипхромихлорид; гексахлороплатекисЛота; гексахлоропла-теат калия, г ей сацп ано ферро ат калия.
Так как по различным причинам эти обозначения оказались Нецелесообразными*, их отбросили, тем более что единая характеристика валентности как для простых, так и комплексных соединений рассматривается в духе Вернера.
Для вполне определенного обозначения состава достаточно данных о валентности
центрального атома наряду с данными о числе и природе координационно связанных групп, так как заряд комплексного иона определяется алгебраической суммой зарядов центрального атома и связанных с ним групп. Заряд комплексного иона определяет вполне однозначно число ионогепно, т. с. внекомплексно связанных; атомов иля групп.
Если валентность центрального атома установлена ненадежно (например, в соединениях, содержащих в комплексе NO), то упоминание о пей опускается и вместо нее указывается число впекомплекспо связанных атомов или групп. Указание о валентности центрального атома оказывается лшппим в случае незаряженных комплексов (неэлектролитов).
Существуют также координацнощше соединения с несколькими центральными атомами. Их называют многоядерными в отличие от простых сдноядерных комплексов. Атомы или группы, связывающие между собой оба центральных атома, называются мостиковыми. Их обозначают буквой р, которая ставится перед их названием. Например, [(NH3)6Co'NII2-Co(NIl3)5]X5 — соль р-амидодекаммИ|1Дикобальта(1П) или, точнее, соль р-амндо-блс-[пентамминкобальта(Ш)]. Это название соответствует (если об этом идет речь) числу групп, в названии которых уже содержится числительное. Тогда число групп выражают не обычным греческим числительным, а сложным (бис-, трис-, тетракис-) Таким путем достигают однотипности в обозначениях с некомплексными соединениями. Например, бифосфаты — соединения, содержащие два монофосфат-ных остатка, отличаются от дифосфатов, солей дифосфорной (пирофосфорнрй) кислоты НД’2о,. Иногда два центральных атома оказываются связанными кесколькжмк мостиковыми атомами или группами. Число однотипных мостиков обозначают тогда греческим числительным, стоящим перед буквой р. Например; ди-р-сульфидосоединение есть соединение, в котором Два центральных атома связаны двумя— S—мостиками. Напротив, р-дисульфид о со единением называется соединение, в котором оба центральных атома связаны между собой одним —S—S—-мостиком. В качестве примера комплексной Соли, в которой два центральных атома связаны двумя ОН-мостиками, можно
привести соединение
еп /ОН. еп
Сг ХСг ей. ^OHZ ’ еп
Од
* Часто приводила к недоразумениям путаница с обозначением высших и низших валентностей металла через и и о. Иногда под к упро со единениями понимали соединения двухвалентной, иногда соединения одновалентной меди. Такие же неясности возникали для ауро-, меркуро-, таило-, стаппи-, плюмби- и других соединений.
Координационная теория
447
Его следует назвать хлорид дн-р-глдроксотетраэтилендиамипдихром(Ш) или, правильнее, как хлорид ди- р-гидроксо-бис-диэтилендиаминхром(Ш).
Два центральных атома могут быть связаны между собой и двумя различными мостиками. Это имеет место ио многих комплексных соединениях кобальта г Например, полученный Вернером (1910) хлорид р-пероксо-ц-амидодихлорогексамип кобальт(П1)кобальт(1У). Это соединение замечательно тем, что оно существует в двух изомерных формах, различающихся положением обоих ноионогенно связанных атомов хлора.
Симметричное
Асимметричное
В таких случаях говорят о координат иеной изомерии положения. V неорганических соединений изомерня положения наблюдалась до сих пор только в очень редких случаях, тогда как в органических веществах она встречается часто. Однако известны многочисленные неорганические комплексные соединения, у которых наблюдается цис-, транс-иространстланная изомерия или зеркальная изомерия. Многочисленные примеры этого приведены в т. И.
л а в а 12
ЧЕТВЕРТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ/ СИСТЕМЫ (Главная подгруппа)
ПОДГРУППА УГЛЕРОД — КРЕМНИЙ
Порядковый номер Элемент Символ Атомный В VC Удельный вес Температура плавлений, ЙС Температура 1П1ПЩ1ЦН» °C Теплота еубЛИЫа-ЦИИ, къал/г-атом Валентность
в У глсрод с 12,011 3,51а — 3850 170 IV, ni,
п
14 Кремний Si 28,09 2,328 1413 — 85 IV, п
32 Германий Gc 72,60 5,323 958,5 — — IV, II
50 Олово Sn 118,70 7,28 231,8 2362 78 IV, 11
82 Свинец гь 207,21 11,34 327,4 1750 47,5 IV, п
а Алмаа; ср. табл. 77» стр. 458,
Общие сведения. В четвертую главную подгруппу периодической системы входят элементы: углерод, кремний, германий, олово и свинец. Эти элементы непосредственно примыкают по своим порядковым номерам к элементам третьем главной подгруппы (ср. табл. II приложения). В соответствии с этим аналоги углерода н кремния обнаруживают некоторое сходство с аналогами бора и алюминия, т. е. с элементами третьей главной подгруппы, расположенными в больших периодах. Здесь, следовательно, имеются иные отношения, чем при сравнении элементов галлия, индия и таллия с элементами главной подгруппы II группы, расположенными в больших периодах, т. е. со щелочноземельными металл лами, с которыми у них не обнаруживается никакого сходства.
Строение атомов элементов главной подгруппы IV группы полностью соответствует друг другу. Но, как в третьей группе периодической системы, элементы, стоящие в побочной подгруппе (скандий, иттрий, лантан и актиний), несмотря на то что строение их атомов отличается от строения атома алюминия, в некоторых отношениях больше похожи на алюминий, чем его более тяжелые аналоги, стоящие в главной подгруппе, строение атомов которых соответствует строению атома алюминия; так и элементы четвертой группы, стоящие в побочной подгруппе (титан, цирконий, гафпнй и торий), в некоторых отношениях более похожи на кремний, чем его аналоги из четвертой главной подгруппы.1 Однако только последние, подобно углероду и кремнию, проявляют четырехвалентность по отношению как к электроположительным, так и к электроотрицательным веществам и образуют с водородом легколетучие соединения. Эта способность особенно характерна для важнейшего представителя главной подгруппы IV группы — углерода. У кремния она проявляется не
Четвертая группа периодической системы
449
в такой мере вследствие его склонности к образованию кислородных соединений, в первую очередь определяющей поведение кремния. Тот факт, что в определенных классах соединений проявляется особенно большое сходство между кремиием и элементами побочной подгруппы., соответствует правилу, которое постоянно отмечалось в предыдущих группах: второй элемент главной подгруппы является переходным к элементам побочной подгруппы.
В главной подгруппе IV группы, так же как п в предшествующих группах, выполняется приняло, согласно которому первый элемент осуществляет Переход к следующей главной подгруппе. Простейшие кисло родните соединения углерода легко летучи, подобно соответствующим соединениям соседнего авота и в противоположность кислородным соединениям аналогов углерода. Углерод в карбонатах, так же как п азот в нитратах, обладает максимальным коор ди надионным числом 3, в то время как кремний в силикатах имеет координационное число 4 (см. стр. 542). Углеводороды, так же как иэотистоподородпыс соединения, водой нс разлагаются, тогда как водородные соединения кремния и его более тяяшлых аналогов расщепляются водой и т. д.
По отношению к кислороду, галогенам и другим электроотрицательным элементам элементы главной подгруппы IV группы проявляют максимальную валентность, равную четырем, что соответствует номеру группы. По наряду с этим они могут выступать и как двухвалентные.Углерод наряду с СОа п CSs образует также СО л CS; правда, последнее соединение очень неустойчиво*. Соединения Si О и SLS также мало устойчивы. Еще большей склонностью проявлять двухвалентность обладает германий, который, кроме двуокиси н дисульфида, образует еще л дцхлорнд. Тенденция выступать в качество двухвалентного элемента растет далее у олова, для которого устойчивости двух- и чстырехвалептнрго состояния примерно равны. Наконец, у свинца двухвалентное состояние преобладает над чстырехвалептным.
Обнаруживающееся во всех группах периодической системы уменьшение кислотных и соответственно увеличение основных свойств при переходе от более легких к более тяжелым элементам еще более усиливается в главной подгруппе IV группы благодаря увеличивающейся и том же направлении устойчивости низших ступеней валентности. Поэтому в общем основные свойства больше проявляются у соединений низшей валентности, а кислотные — у соединений выстцей валентности. Если элемент образует несколько окислов, то всегда высшие окислы более склонны к образованию кислот и соответственно менее склонны к образованию оснований, чем низшие.
Углерод и кремний являются элементами с ярко выраженным неметаллическим характером — кислотообразующими, элементами. Германий — также к исл ото образующий элемент; правда, в двухвалентном состоянии это свойство у пего выражено крайне слабо. Элементарный германий причисляют к металлам. Олово и свинец по своим? физическим свойствам — типичные металлы. В своих соединениях четырехвалентное олово является преимущественно кислотообразующим элементом; двухвалентное олово амфотерно. У сшита и в четырехвалентном состоянии кислотный характер окисла выражен слабо; двухвалентный свинец преимущественно образует основания, хотя способность к кислотообразованию у него еще исчезает нс полностью.
* Высокая устойчивость CO объясняется, вероятно, тем, что в ней С только стеа-ио-метрически двухвалентен, в то время как в действительности он обладает четырьмя валентностями: одной злептр о валентностью и Тремя к он а л сити остями, причем стабильность соединений повышается в результате мезомерии.
29 г. Реми
450
Глава 12
Усиление металлического характера элементов при переходе от углерода и свинцу, проявляется нс только по отношению к неметаллам; опо достаточно ярко выражено-в их поведении относительно металлов, например щелочных металлов. Свинец и олова-образуют с ними типичные интерметаллические соединения (ср/ табл. 93, стр. 079); некоторые из них при определенных условиях могут проявлять солеобразный характер, выражающийся в склонности к образованию «полиаппонпых солей», таких, как. [Ха(КН3)0]4[РЬ9] (см. т. II). Углерод, напротив, образует с щелочцьгми металлами соединения С ярко выраженными свойствами солей, ацетилиды (см. стр. 476). Кроме того, он может реагировать в форме графита со л (елочными металлами, образуя своеобразно построенные соединения ШС8 и М1С1в (о структуре последних см. т. II). Трем-май и германий занимают промежуточное положение. При непосредственном взаимодействии со щелочными металлами они образуют интерметаллические соединения простого состава MX. Как нашли Клемм и Гоман (Klemm, Hohman, 1948), кремний и германий с К, Rb и Cis образуют, кроме того, соединения состава MSie и MGcs, которые являются переходами от соединений МС1в а МСе (М = К, Rb, Cs) к соединениям KSn4 и КРЬ4, приведенным в табл. 93.
Относительно водорода элементы углерод, кремний, германий, олово-и свинец всегда являются четырехвалептными. Простейшие водородные-соединения (общая формула RHa) легколетучи. Их устойчивость чрезвычайно сильно падает уже от углерода к кремнию, а затем уменьшается еще больше у тяжелых элементов.
До сих пор мало известно о природе бедных водородом нелетучих, еидрцдое этой группы: купрене [СИ]д;, полисилеие [SiH2]x и полигерменах [GeH]K и [GeH^]K.
Сопоставление важнейших соединений позволяет установить аналогию .между элементами четвертой главной подгруппы.
Водородное соединение Окисел Сульфид Хлорид Отрицать л ьны с ПОНЫ Положительные ионы
сн4 co co2 GS CS2 - CC14 ССЦ CS5 . ।
SiH4 (SiO) SiO2 S iS SIS2 — SiCl4 SiOs sis; —
GeH4 Gel) GcO-. Gt»S GeS2 GeCI2 GcC14 GeOJ GeSS ?
SnH, SnO SnOn SnS SllS;, SnCl2 SnCIt SnOJ SnS£, Sn(OH)j Sb“ Sn'""
РЬН4 PbO PbOa PbS — P1)C12 PbCl4 PbO'ij — Pb-- Pb----
Pb(OH)S 5
Теплоты образования важнейших простых соединений приведены в табл. 75.
Таблица 75
Теплоты образования соединений элементов главной подгруппы четвертой группы (ги;«л/г-анв)
Тетра- ГПДриД Моноокись Доу- ОКИСЬ Дих.чо-. РИД •TeirjjQ- : хлорид Моно-сульфид Цис у Jib-ФИД
Углерод (алмаз) 4,56 13,42 23,65 — 6,5 -6,5 -3,9
Кремний 2,2 — 52,1 1— 38,5
Германий — — 32,0 — 31,6 • — —
Олово — 33,6 34,5 40,5 ; 32,0 — +9,3
Свмпец — 26,20 16,1 42,8 — +11,6 м—
С точки .зрения теории Касселя свойства рассматриваемых элементов логически? следуют из их положения в периодической системе. Это справедливо прежде всего.
Четвертая группа периодической системы 451
для углерода и кремния. Последние удалены па четыре места как от предшествующего, так и от последующего инертного гам. Следовательно, они имеют возможность перейти в состояние «подобное инертному газу» либо путем Отщепления 4 электронов, либо путем присоединении 4 электронов. Первое имеет место, по Касселю, тогда, когда углерод д его аналоги реагируют с сильно электроотрицательными элементами, например с кислородом или галогенами, в то время как в реакциях с сильно электроположительными металлами, а также с водородом происходит присоединение электронов. Предпосылкой для этих представлений является тот факт, что углерод и кремний в соединениях с кислородом, галогенами и т. д., по крайней мере условно (см. нище), можно рассматривать как электроположительную составную часть, а в соединениях, например со щелочными или щелочноземельными металлами, а также в соединениях с водородом — как электроотрицательную часть,
Большинство соединений углерода, прежде всего углеводороды п пх производные, обладают ярко выраженным характером еомеополярных соединений. Поэтому теорию Косселя можпо применить к ним только с существенными ограничениями. Однако, если учесть, что и у так называемых гомеополярных соединений в общем на одной составной части скапливается больше положительных зарядов, а на другой _больще отрицательных зарядов*, то теорию Косселя можно принять за основу при объяснении образования этих соединений. Например, образование метана СН4 можно тогда объяснить на основании допущения, что атом С вследствие его стремления принять электронную конфигурацию ипертпого гада заряжается четырьмя отрица-, тельными нарядами, отнимая у четырех атомов водорода их электроны, й затем связывает, электростатически положительные водородные: ядра. (Благодаря малым размерам ядра водорода при этом проникают через впыппюю электронную оболочку внутрь атома.) Учитывая свойства соединений, эти представления следует, конечно, ограничивать, по крайней мере в том смысле, что электроны не полностью отнимаются у атомов водорода и что вследствие этого составные части соединения сцеплены не только за счет противоположных зарядов, но здесь проявляются еще и другие силы (резонансные силы в смысле волновой механики), которые способствуют тому, что в этом случае при образовании нечисто гетеропо л яркого (соответственно гомеополярного) соединения выделяется больше энергии, чем при образовании чисто гетерополяр пых соединений; которых прежде всего следовало бы ожидать на основании представлений Косселя. То же можно сказать относительно образования силана Silli, а также водородных соединений других элементов группы.
Рассматривая вопрос с той ;ке точки зрения, образование четыреххлористого углерода СС14 можно свести к тому, что атом С сравнительно легко отщепляет электроны, имеющиеся у него сверх числа электронов предшествующего инертного газа (гелия), и поэтому отдает их четырем атомам хлора, каждый из которых пытается присоединить 1 электрон, чтобы перейти к электронной конфигурации аргона. Возникшие таким образом противоположно заряженные атомы могли бы образовать чисто ионную связь. Но так как четырехлористый углерод обладает свойствами скорее , гомеополярного, чем гетерополярно го соединения**, вероятно, и в этом случае 4 электрона не полностью отщепляются От углерода и речь идет не о чисто ионной связи. То же справедливо для Других соединений углерода с электроотрицательными веществами.
Несмотря па это, на основе теории Косселя получено объяснение тому, что углерод и его аналоги могут проявлять валептпость четыре как относительно отрицательных, так и положительных элементов. Тот же результат в отношении валентности получается, как показывают следующие формулы, на основе теории Льюиса;
Н ' :С1;
•С. -j-4H= Н:С:Н; ,С. -Ь4.С1’. = -.С;С:С1:
Н ” :С1.”
Преимущество этого объяснения в том, что оно делает понятным тот факт, что с углеродом (и отчасти также с его аналогами) одновременно могут быть связаны положительные и отрицательные элементы (пример: СНС13). Кроме того, оно может быть обосновано квантово-механически.
* То же справедливо для соединений, молекулы которых не обладают электрическим моментом. Такими являются, папример, СН4, СС14, СО2.
Если исходить [[3 представлений Полинга (см. стр< 161), то окажется, что на гетеро полярную часть С—Cl-связи приходится 11% общей энергии связи. Простая связь С—О оказывается соответственно на 28%, С—F на 41%, С—Вг на 2% С—I па 0% и связь С—Н па 7% обусловленной стационарными зарядами атомов.
29*
452 Д.чава 12
Прн приложении теории атомной свя.зв к углероду следует иметь в виду, что соединения, в которых он четырехкалентен, могут быть выведены только и я возбужденного состояния атома G. Когда речь идет об основном состоянии атома С, то по данным спектров имеется в виду 2.А2/А 3 Г’„-с о стояние, следовательно, такое состояние, в котором не спарены только 2 электрона. В этом состоянии углерод'Может быть только двух л ал ситным. Низшее возбужденное состояние, при котором все 4 электрона внешней сферы углерода не спарены, соответствует термам 2s2p35l5‘a, В этом состоянии пе только' три р-электрона, по также б-электроп имеют параллельные ептшы. При спаривании 4 эяоктроцов с электронаип атомов, которые образуют связь с углеродом (так как это обсуждалось в гл. 5), происходит гнбрадизац-ия s и yj-y ровней. Следствием этого, помимо повышения прочности связи, является еще и то, что четыре валентности углерода равноценны и направления но углам тетраэдра.
Энергия, которую следует затратить, чтобы перевести атом С из основного в возбужденное состояние с четырьмя поспарышьтми электронами, составляет около 96,4 ккал/г-атом. (3 11 с n s I о и е, Pliys. Rev., 72, 411 (1947)). Теплота сублимации для С (графа! ири 0° К) с образованием: атомов С в основном состоянии, составляет 170 ккалт-отом. Для расчета энергпи диссоциации соединений углерода но спектрографическим данным в общем не имеет значения разница и энергии между основным . состоянием атома С РА,) и ^-состоянием, так как можпо показать, что, хотя соединения углерода возникают прн возбужденном состоянии атома С, их термическая диссоциация ведет к образованию атома G в основном состоянии.
Двуокись углерода СОг, вероятно, тоже можпо рассматривать как преимущественно гомеополярпос сооднпепнв. Все же это соединение, судя по его свойствам, довольно близко и к гстерополярпым. Как уже указывалось рапсе, между гетеро полярными я гомеополярными соединениями нельзя провести резкой границы.
Соединения кремния с электроотрицательными элементами уже ближе к типичным гетеро полярным, чем к типично гомео полярным соединениям. '1'етрахяорид кремния SiClj, который приближается еще к гомеополярным соединениям вследствие своей большой летучести, заметно отличается от четыреххлористого углерода явной тенденцией к гидролитическому расщеплению и примыкает к ряду предшествующих ему по периодической системе гетер оно лярных хлоридов: NaCl, MgCl2, AlClg, которые диссоциируют па ионы в водном растворе и тем легче подвергаются гидролитическому расщеплению, чем больше заряд катиона. Двуокись кремния Si Оу обладает пе только химическими, по и физическими свойствами соединения, имеющего преимущественно гетеро полярный характер.
В рядах периодическом системы, предшествуютцих рядам, в которых находятся германий, олово и евпттец. пет инертных газов. Этим Ge, Sn и РЬ отличаются от С и Si. Однако, как видно из рис. 28 (стр. 153), число электронов, которыми обладают Ge и Sn в чстырехпололштольно -заряженном состоянии, повторяются у целого ряда соседних элементов; то же справедливо для РЬ, пе приведенного па этом рисунке. Поэтому следует предположить, что электронной системе, имеющейся у четырехполо-жгггелг.ио заряжен пых. Ge, Sn н ГЬ, подобно электронной системе инертных газов, свойственна большая устойчивость по сравнению с другими системами.
Таким образом, тот факт, что число 28 электронов встречается последовательно у одновалентной меди, в* ионах пинка и галлия и т. д. вплоть до шестивалентного селена, наводит на мысль, что это число электронов — хотя и lie в свободном, а только в положительно заряженном атоме — соответствует расположьчппо электронов, отличающемуся относительной стабильностью. То же справедливо относительно числа 46 электро [io в, которое характерно для ряда электролитических ионов*: Ag', Cd", 1гг", Sir--, Sli. и затем положительно заряженных атомов вплоть до иода, а также
относительно числа 78 электронов (пе показ а по па рис.. 28), которое имеется у Ап+, Hg2+, Tl3’, PIA, В А. Здесь отношения даже проще, чем для углерода и кремния, поскольку у олова и свипца четырех положительно заряженные атомы легко обнаруживаются в форме попои.
Равным образом электронные числа 30, 48 и 80, свойственные Ge, Sn и РЬ в положительно двухвалентном состоянии, повторяются в ряду следующих друг за другом элементов, например в ряду G;i+, Gcs+ As3+, Se4+, Вт64-, которые, конечно, пе все встречаются как свободные (электролитические) ионы, однако входят в состав соединений с приведенными здесь зарядами. х.
Образование кислотных ионов двуокисям» и дисульфидами элементов четвертой плавной подгруппы па основании представлений Косселя
* Ср. по этому поводу споску на стр. 242.
Четвертая группа периода ческой системы 45(1
объясняется тем, что при присоединения отрицательного иона, такого, как О2- или S2”, к двуокиси или дисульфиду с центральным атомом, заряженным четырьмя положительными зарядами, выделяется некоторое дополнительное количество анергии подобно тому, как это имеет место при присоединении F" к BFa (ер. гл. 11). В некоторых случаях энергия выделяется и при присоединении второго иона О3' (образование [SiOd4'), что зависит от величины центрального атома. Чем больше последний, тем больше ионов может присоединяться при прочих равных условиях с выделением энергии. ‘
Таким образом, к двуокисям олова и свинца при соединяются максимально четыре нона G2- (которые но причинам, обсуждаемым ниже, могут присоединять еще и ионы водорода) сверх того количества попов О2-, которое содержится в нейтральных молекулах. Это положение можно сформулирован, следующим образом: максимальное координационное пиело по отношению к кислороду е ряду от углерода к олову повышается с 3 до 6. Тот факт, что кислородные кислоты и их соли (так же Как тиокислоты И тиосоли) принадлежат к классу координационных соединений, был отмечен уже Бернером.
Рассмотренные сложные отрицательные ионы могут соединяться с водородом, образуя кислоты, или с другими положительными попами, образуя соли. Если для кислоты H4RiVO4 [ее можно рассматривать и как гидроокись И1У(ОН)4) рассчитать Энергию, которая должна затрачиваться на отделение попа водорода или иола гидроксила, методом, соответствующим описанному в гл. 6 для соединений формулы Л3ОН, то и ай дем, что энергия, затраченная на отделение иона водорода, здесь много меньше, Чем энергия, затраченная на отделение иона гидроксила. Это происходит вследствие сильного отталкивания между центральным атомом, заряженным четырьмя положительными зарядами, и одноименно наряженными атомами водорода (водородные ядра). С увеличением радиуса нейтрального атома в соответствии с законом Кулона уменьшается его отталкивающее Действие. Вследствие этого с увеличением радиуса центрального атома иопы водорода отщепляются труднее, а ионы гидроксила — легче. Этим объясняется, во-перюых, кислый характер гидроокиси Л.(ОП}^ е этой еруппе и, во-вторых, ослабление кислотного характера и одновременное появление основных свойств при переходе от болев легких элементов группы (эти элементы имеют меньшие атомные или ионные радиусы) к более тяжелым (обладающим большими ионными радиусами).
Это справедливо для гидроокисей четвертой группы IbTl^Og, соответственно ОВ1у(ОН)г.
Оловянная кислота, состав которой соответствует формуле Sri(OH)4(OH2)2, может относительно легко отщеплять ионы Н+ только до тех нор, пока «та содержит атомы кислорода, с которыми связало более одного атома водорода. В этом случае К отталкивающему действию центрального атома добавляется отталкивающее действие ионов Н+, связанных с одним и тем же попом О1-. Одиако в попе [Sii(OII)6]2” ионы водорода связаны, по-видимому, так прочно, что при дальнейшей диссоциации имеет место только отщепление ионов OIF. Если при этом соединение переходит в форму Sn(OII)4, то для дальнейшей диссоциации справедливо сказанное выше.
Bluet, сильное проявление основных свойств у гидроокисей, соответствующих второй степени окислении, также оказывается необходимым следствием рассмотренного представления о сущности связи.
Одиако ионы Sh2+ и РЬ2* нельзя просто сравнивать с попами щелочноземельных металлов, .которые обладают примерло теми же. размерами. В противоположность последним ионы Sn2+ и Pb‘J+ оказывают сильное поляризующее влияние, что, между прочим, следует из факта сильной окрав|енноСти их окислов, сульфидов и т. д. (ср. гл. 9). Благодаря поляризующему влиянию повышается прочность связи. Поэтому ионы О2" и групны ОН- в Яц(ОН)а и РЬ(ОТТ)2 удерживаются прочное, чем в соответствующих гидроокисях щелочноземельных металлов; вследствие этого Sn(01I)2 и РЪ(0Н)2 являются тл.т ьлщ слабыми основаниями. Работа отрыва ионов Н’ для пих\ и прежде всего для Йп(О1Г)г, немного больше, чем отрыва ионов ОН-. Таким образом, эти гидроокиси, и прежде всего гидроокись олова, могут функционировать как кислоты относительно сильных оснований. ,
В табл. 7в сопоставлены атомные и ионные радиусы элементов четвертой главной подгруппы, рассчитанные по данным для кристаллических соединений.
Некоторые важные в химическом отношении сведения о строении атома моястЮ получить на основании спектроскопических данных.
454
Глава 12
Таблица 76
Атомные и иоппые радиусы элементов главной подгруппы четвертой группы
Заряд С ; Si &е Зп рь
4— 1,98 2,15 2,15
0 0,77 1,18 ; 1,22 1,40 1,74
2-5- —— — • — 1,32
4-г- -0,1 0,39 0,44 0,74 0,84
Из характера искрового спектра трижды иопйвпровапцого атома кремния Si3*', (уровни энергии которого были уже показаны на рис. 70, стр. 355, следует, что остов атома кремния (если не принимать во внимание более высокий1 заряд ядра и обусловленное им искажение электронных орбит) подобен остовам атомов предшествующих элементов и, следовательно, остову атома предшествующего инертного газа— неона.
Такое же соответствие в расположении уровней энергии имеется в ряду Li, Ве+, Ва+, С3+, из чего следует заключить, что остов атома углерода, если не говорить о ядре, соответствует остову атома гелия. Сравнивая схемы уровней, приведенные на рис. 70 и 71, можно заключить, что у алюминия третий присоединяющийся к остову атома А13+ электроп связан иначе, чем два электрона, присоединившиеся до пего. Из правой половины рис. 71 видно, что у ионизированного углерода С* валентный электрон находится в нормальном состояппи^па 2р-уровне. Электронное облако, символизирующее вероятность его нахождения вблизи ядра, имеет не симметрию шара, которой обладают электронные облака двух электронов, присоединяющихся после образования гелиевой оболочки, а только симметрию вращения. Четвертый внешний электрон, по спектральным данным, также находится на 2^-уровпе. Таким образом, из четырех внешних электронов нейтрального атома С два находятся па 2р-уровне и два — на Ss-уровне. Два последних электрона обладают антинараллельиыии спинами. Энергетические уровни внешних электронов, аналогичные уровням углерода, определяются из спектральных термов для кремния: два электрона на 3s- и два .электрона на 3 ^-уровне; это же справедливо и для других аналогов углерода, которые, следовательно, в протплгиюложпость элементам побочной подгруппы IV группы не только химически, по п спектр ос конически аналогичны обоим наиболее легким элементам главной подгруппы.
Работа, которую следует затратить для отрыва пятого электрона, значительно больше, чем работа, затраченная на отщепление четырех электронов (ср. табл. 23, стр. 143). йто справедливо прежде всего для углерода и кремния; однако и для высших аналогов группы эта работа так велика, что не следует ожидать их появления в стабильных^химнческих соединениях с зярядом, более высоким, чем 4*.
УГЛЕРОД (CARBONEUM) С
Распространение в природе. Углерод встречается в минералах, главным образом в форме карбонатов, производных угольной кислоты Н2СО3. Помимо щелочных и щелочноземельных металлов, карбонаты которых уже были описаны в предыдущих главах, некоторые тяжелые металлы также часто встречаются в природе в виде карбонатов, например, железо — в виде железного шпата FeCOs, марганец — в виде марганцового шпата МпСОз, цинк — в виде цинкового шпата ZnCOs и др.
Углерод является главной составной частью животного и растительного мира. Не существует растительных или животных организмов, в построении которых не участвовали бы в качестве существенной составной части соединения углерода. Поэтому соединения углерода называют
Четвертая группа периодической системы
455
также органическими соединениями. Особенно это относится к сложным соединениям, возникновение которых раньше считали связанным с особой силой, действующей только в живых организмах. Органические соединения чрезвычайно разнообразны, в связи с чем число их очень велико. Ветвь химии, которая специально занимается изучением этих соединений,— органическая химия, стала с давних пор наиболее важным разделом химии,
В атмосферном воздухе углерод содержится в незначительных концентрациях (в среднем 0,03 об. %)* в форме двуокиси углерода СО а, которая поглощается зелеными растениями. Последние обладают способностью при помощи содержащегося в них красящего вещества — хлорофилла образовывать из двуокиси углерода и воды с отщеплением кислорода •* углеводы (С6Н10О5)х, например крахмал и целлюлозу. Необходимая для итого энергия доставляется солнечным светом. Углеводы и продукты их превращения затем вновь распадаются в растительных и животных организмах (последние получают их Из растений) на двуокись углерода и воду в результате процесса дыхания, сопровождающегося выделением энергии. Таким образом, двуокись углерода совершает непрерывный кругооборот между атмосферой и органическим миром.
СО2 атмосферы содержит определенную часть радиоактивного углерода С14, который постоянно образуется л атмосфера в результате ядерной реакции
,№4+orii = 6C>4+IHi.
Необходимые для этого нейтроны п возникают под влиянием космических лучей, которые доставляют еяпюепуядно в среднем 1 нейтрон ла 1 ли3 атмосферы. Так как практически каждый из этих нейтронов реагирует с азотом с образованием С14, а период полураспада последнего, как известно, 5300 лет, можно рассчитать, сколько С14 постоянно находится на Земле. При таком расчете получают цифру около 20 т. Из радиоактивных атомов С в атмосфере тотчас образуется СО2, которая вступает таким путем в биологический круговорот. Общее количество углерода, участвующего в круговороте, т. е. содержании G в биосфере и океане, оценивается, примерно в 3-1043 ri-i. Из них 6-10M т содержится в атмосфере (в виде С02). Из приведенных данных можно рассчитать, что примерно 1-Ю 43 углерода, участвующего в биологическом круговороте, радио активна. Примерно такая же величина была найдена экспериментально <А и d о г s о п, Libby, Physic. Rev., 72, 931, 1947). Экспериментальное определение этого крайне небольшого количества осуществляется путем превращения углерода в метай л обогащения последнего С14Н4 прп помощи разделительной трубки Клу-=зиуса (ср. т. П). В веществе, которое уже ие участвует в биологическом круговороте, радиоактивность затухает в соответствии с периодом полураспада С*4. Поэтому на основании Определения содержания С14 в органическом веществе можпо определить его''«возраст», т. е. время, прошедшее с момента его выхода из биологического круговорота. В веществах, которые «старше» 40 000 лет, определение содержания С14, во всяком случае точпое, значительно затруднено, так как в таких веществах произошло сильное затухание радиоактивности. В соответствии с этим в каменном угле С14 лишь едва обнаруживается. Однако для веществ органического происхождения, образовавшихся п более позднее время, определение содержания С14 позволяет достаточно точно указывать их возраст.
По содержанию С14 в углероде продукты, полученные из органических соединений, которые еще не очень давно вышли пз биологического круговорота, резко •отличаются от продуктов синтетически полученных из угля пли кокса. Грамм свежего •биологического углерода испускает в минуту в среднем 15,6 (3-частиц (Anderson,Libby, 1951), в то время как ископаемый углерод практически больше не обладает активностью. Фалтппг (Faiting, 1952) показал, что измерением радиоактивности С14 можпо но только Отличать, например, природный уксус от синтетического, но и определять, причем достаточно точно, состав смеси природных и синтетических веществ. О других применениях изотопов углерода см. т. II.
* Содержанке двуокиси jn-лерода в воздухе заклеит по данным новейших иссле-довапий от атмосферных условий и может колебаться в пределах 0,02—0,04%.
456
Глава 12
В качество продуктов разложения древпих формаций в природе встречаются угли, главной составной частью которых является углерод (название последнего происходит от слова уголь). Нефть, озокерит (горный воск) и асфальт также являются углеродными соединениями, происхождение которых, по-видимому, следует искать в древних живых организмах, при разложении которых они и возникли. Далее следует еще упомянуть об янтаре, ископаемой смоле состава приблизительно С40Н64О4, и медовом камне (меллит) AlgCi-aCJia-I8H3O, алюминиевой соли бензбл-гсксакарбоновой кислоты СДСООН)е, которая названа поэтому меллитовой кислотой.
Соли щавелевой кислоты также иногда встречаются в природе, например вевелит СаСгСМ-НгО и оксалит FeCaOi- Р/пНзО.
В противоположность ялемеитам трех первых главных подгрупп периодической системы, рассмотренным в гл. 6, 8, 10, углерод встречается в природе и в элементарном состоянии в виде алмаза и графита-
Угли. Углями называют черные или коричневые продукты разложения органических веществ, богатые углеродом. Их можно получить искусственно нагреванием растений без доступа воздуха; по в основном они возникают естественным йутем при самопроизвольном длительном разложении древних растений при значительно более низких температурах. Продукты первого типа называют в зависимости от материала, из которого их получают, древесным углем, костяным углем, кровяным углем и т. д. К природным, или минеральным, углям относятся различные виды каменного и бурого угля, к которым относится продукт неполного разложения— торф.
Процесс «обугливания» представляет собой разложение, при котором другие элементы, первоначально связанные с углеродом, например водород, кислород и азот, выделяются в большом количестве, так что остаток нее больше и больше обогащается углеродом. Подобному процессу обугливания при достаточно сильном нагревании подвергается большинство органических веществ, при этом можно наблюдать появление черного окрашивания, которое характерно для тоикоращюльченного углерода, например для сая;ц. В те чение геологических периодов процессам1 обугливания подвергались дреншк Дре г иены ( породы. Так возникли месторождения каменного угля. Ран-тммыюй ироне хождение утли во многих случаях отчетливо у у опознается по их структуре, особенно в случае более молодых углей.
Те минеральною угли, из которых наиболее полно удалены вещества, первоначально связанные с углеродом, называют антрацитом. Последний состоит более чем па 90% из углерода, наряду с которым содержит лишь несколько процентов водорода и кислорода, а также следы азота. Антрацит имеет окраску от глубоко-черной до железно-серой. Он го put только в сильном токе воздуха и образует совсем короткое пламя, так как при его горении выделяется мало газов. Антрацит — теологически самый старый уголь; однако это относится не ко всем антрацитам, так как температура, при которой идет обугливание, тоже имеет значение. Каменные угли. В узком смысле слова обычно геологически более молодые/угли. Они также имеют черную окраску, но в противоположность антрациту отличаются смоляным пли жирным блеском и горят светящимся п коптящим пламенем, что обусловлено высоким содержанием газообразных веществ в продуктах горец ия, Каменные угли содержат примерно 4—6% II. 5—18% О, 0,5—1,5% А ц только 75—90% С. По составу и свойствам каменных углей различают следующие их виды: жирный уголь, тощий уголь, газовый уголь, пламенный уголь, газопламенный уголь, спекающийся уголь, котельный уголь, песчаный уголь и др. Еще отчетливее, чем у каменного угля, структура растений может быть обнаружена у бурого угля, относящегося К третичному периоду. Он обладает коричневым, а иногда черным цветом, однако всегда значительно мягче, чем каменный уголь или антрацит. В то время как твердость угля и антрацита 2—2,5, тпсрдость-бурого угля только 1,1—1,4. Он содержит 65—75% С, 5—6% ТТ, 20—30% О п 1,2% N.
Самый молодой уголь, состоящий из явно различимых, спутанных между собой растительных остатков,— торф, образование которого можно наблюдать еще и сегод-п я. Он Содержит примерло 55—65% С, 5,5—7% И, 30—40% О и i—2% N, т. е.
Четвертая группа периодической системы 457
очс-пь близок ио составу к древесине (примерно 50% С,’6% ГТ, 44% О и 0,1—0,5% N) и другим растительным волокнистым материалам.
Для тех пи теской оценки различных сортов угля большую роль играет его теплотворная способность. При сгорании 1 кг угля выделяется следующее количество тепла (ккал)-.
Антрацит Каменный уголь Бурый уголь Торф Древесина
8—9000 7 — 80(H) 6—7000 5—6000 4000
Приведенные количества тепла, рассчитанные для горения с образованием жидкой поды, иапыпаплся «верхней теплотворной способностью» соответствующего вещества. /Нггжпли теплотворная способность» получается, если из приведенных выше чисел вычесть теплоту конденсации воды, образовавшейся при горении.
Нефть, или минеральное масло, также, вероятно, возникла при разложении орган инее них веществ и главный/ образны остатков живых организмов. Она состоит из смеси углеводородов, характер которых может быть весьма различным, смотря по происхождению нефти. Известная еще в древности кавказская нефть (бакинская нефть) состоит преимущественно из циклических насыщенных углеводородов, так называемых нафтенов, в то время как американская нефть содержит почти исключительно углеводороды жирного ряда. Нефть встречается в больших количествах еще в Южной Америке, особенно в Венесуэле и Мексике, а также в Иране и Румынии. Наконец, важные месторождения имеются в Ираке, Бирме, а также на Борнео и Суматре. Раньше важной областью добычи нефти была Галиция. Небольшие количества нефти были нандепы в Северо-Германской низменности, например около Панне й Витце в Нижией Саксоции.
Добыча нефти осуществляется путем буреппя скважин в породах, в пустотах которых она находится. Часто под давлением сопутствующих газов, нефть с большой силой выбрасывается из буровых скважнп, одиако большей частью ее добывают при помощи насосов. Сырая нефть, Которая ».Советском Союзе называется просто нефтью, представляет собой жидкость от светло-коричневого до черного цвета, иногда довольно густую с удельным весом 0,8—0,9; ее частично используют просто как топливо. Одиако бблыдую часть нефти разделяют фракционированной перегонкой па составные части, используемые для различных целей. Прежде всего из нефти получают: 1) сырой, бензин — погон, выкипающий до 150° с уд. весом 0,7; 2) керосин — но гон с т. кип. 150—300“ и уд. весом 0,75—0,87; 3) среднее масло пли газойль С т. кип. 300—350°. Из остатка, кипящего выше 350°, путем перегопки в вакууме с перегретым паром можно отогнать еще смазочпос масло с уд. весом 0,9. Остается смесь черных смолистых составных частей — асфальт, продукт, который встречается п в природе, где он образуется в качестве остатка при испарения нефти.
Сырой бензин, называемый в Америко нефтью, большей частью после обработки концентрированной серной кислотой и раствором едкого натра подвергается дальнейшей ратшпе. Таким образом иваучают: пеги релейный эфир или гаколин (г. ш. 30— 70°, уд. вес обычно между 0,64 и 0,66), экстракционный или моющий бензин (г. кип. 70—И Оф н тяжелый бензин (т. кин. 100—140°, уд. вес примерно 0,75). Указанные температуры кипения т удельные веса должны дать только приблизительную характеристику соответствующих продуктов. Они значительно колеблются у продажных сортов, и, поскольку речь вдет пе об однородцокипящих продуктах, даже границы ковебапий Пе могут быть установлены точно. Бензины применяют преимущественно как моторное топливо, а также для экстракции и как средство для чистки; средние масла используют для получения масляного газа, для карбюрирования водяного газа и в качестве топлива для газовых двигателей. Применение других нефтяных потопов ясно из их названий. Бспзтттт, газолин и нефть обладают примерно равной теплотворной способностью (—10 ООО кмл/кз). Из смазочных масел При дальнейшей переработке получают еще одни продукт нефтяной промышленности — парафин. Последний кристаллизуется при охлаждении смазочного масла от —5 до —103 в виде крупных пластинок. Парафин — воскообразный белый прозрачный продукт — является, подобно другим составным частям нефти, смесью углеводородов. Различают твердый и мягкий парафин. Твердый парафин, называемый также церезином, плавится при 52—56°. Его используют преимущественно для приготовления свечей. Более нпзкоплавкип
458
Глава 12
мягкий парафин находит применение при производстве спичек, для аппретуры тканей, для приготовления так называемой восковой бумаги, как изолирующий материал в электротехнике, для изготовления фармацевтических мазей. Для тех же целей служит также жидкий при обычной температуре парафин (парафиновое масло) >— прозрачная, бесцветная, но флуоресцирующая маслообразная жидкость, кипящая выше 360°, с уд. весом 0,88.
Свойства и применение углерода. Чистый углерод встречается в двух кристаллических модификациях: кубической, кристаллизирующейся в форме алмаза, и гексагональной, кристаллизующейся в форме графита. В то время как. алмаз бесцветен, а графит серого Цвета, углерод, полученный при термическом разложении его соединений, имеет глубокую черную окраску (например, сажа). Раньше черный углерод считали особой аморфной модификацией. По новейшим исследованиям эта модификация совпадает в основном по своей тонкой структуре с графитом (ср. стр; 463).
В табл. 77 сопоставлены некоторые характерные признаки различных видов углерода. В качестве примеров того вида, который раньше считали аморфным черным углеродом, указаны сажа и блестящий углерод (см. стр. 465). Последние в противоположность большинству других продуктов, которые собраны под названием «аморфный углерод», можпо получить почти свободными'от посторонних примесей.
Таблица 77
Аллш.? Грабит Черный углерод
салеа блестящий углерод
Окраска и прозрачность Удельный вес Теплота сгорания, кал^г С Бесцветный прозрачный 3,514 7873 Серый, непрозрачный 2,22 7856 Черный, н< 1,85 8129 ^прозрачный 1,86—2,07 8148—8051
Свойства черного углерода в значит ельцом степени зависят от метода получения и могут быть изменены последующей обработкой. Прп очень сильном прокаливании черный углерод, внешне нс изменяясь, становится графитоподобным, т. е. его удельный вес возрастает и одновременно уменьшается теплота сгорания.
Гипотеза о том, что черный углерод но структуре идентичен графиту, была высказана сначала Дебаем и Шеррером (Debye, Scherrer, 1917), которые утверждали, что черный углерод Дает линии рентгеновской интерференции, характерные для графита, хотя и сильно размытые.’Сначала эту гипотезу считали несовместимой с тем, что теплота сгорания различных сортов черного углерода значительно превышает теплоту сгорапия графита, л с тем, что черному углероду свойственна сильная поверхностная активность, которая у графита проявляется лишь в незначительной степени. Гофману (Hofmann U., 1931) удалось всё же показать, что эти свойства черного углерода обусловлены только чрезвычайно малыми размерами его кристалликов и поэтому чрезвычайно большим развитием их внешней поверхности. Наличием ненасыщенных валентностей атомов С на поверхности объясняется как более высокая по сравнению с графитом эпергия черного углерода, так и его большая поверхностная активность (адсорбционная способность и каталитическая активность). Однако поверхностные силы могут проявляться только тогда, когда получение препарата осуществляется при таких условиях, что вншпние поверхности лежат свободно. Поэтому, помимо малых размеров кристалликов, для поверхностной активности (по не для теплоты сгорания) имеет значение также их расположение. Активность проявляется только в том случае, если кристаллики не склеиваются в плотные агрегаты (как в случае ретортного графита), а образуют рыхлый порошок или структуру, пересеченную чрезвычайно большим числом каналов. Свойства черного углерода с увеличением величины кристаллов (сажа блестящий уголь —х ретортный графит —> ачесоновскпй
Четвертая группа периодической системы
459
трафит —> природный графит) постепенно переходят в свойства обычного графита. ’Твердость достигает максимума у ретортного графита, так как, с одной стороны, при беспорядочной ориентации кристалликов она растет в зависимости от их величины и, с другой стороны, уменьшается с увеличением степени упорядочения кристалликов,
В парообразном состоянии углерод почти весь одноатомен, хотя энергия диссоциации молекулы С2 (которую определили спектроскопически) составляет 3,6 эв Д83 хкал/дюль), т. е. больше, чем, например, для С12. Одпако при таких температурах, при которых молекула С2 при нормальном давлении должна была бы диссоциирован, -лишь незначительно, давление пара углерода еще минимально,: а температура, при которой давление пара достигает атмосферного давления, лежит уже так высоко, Что при ней термическая энергия достаточна для почти полного расщепления молекул С2. Например, из рассчитанной Цензом [Z е i s с Н., Z. Elcktrochem., 46, 38, 293, 1940] константы равновесия реакции С2 2 2С следует, что степень диссоциации С2 при .2000° К составляла бы лишь 1,7% (при 3000° К 56,0%), если при этих температурах пар углерода мог бы существовать при нормальном давлеппи. Однако в действительности углерод обладает при 2000° К упругостью пара приблизительно 10'® атм, а цри 3000° К 0,940'® атм, и для этих давлений степень диссоциации равна соответственно 100,0 и 99,9%. Для. S2 степень диссоциации при 2000° К, по данным Цензе, составляет 3,70%, для :Se2—12,9%, для Тс2—27,0%.
Углерод независимо от того, в какой модификации он находится, яе имеет ни вкуса, ни запаха, чрезвычайно трудно плавится и улетучивается и не растворяется во всех обычных растворителях. Он хорошо (растворяется во многих расплавленных металлах, например в железе, кобальте, никеле и в платиновых металлах, и вновь кристаллизуется ® форме графита при охлаждении растворов.
При обычной температуре углерод, особенно алмаз и графит, .химически крайне инертен. Некоторые сорта черного углерода воспламеняются в атмосфере кислорода уже при сравнительно незначительном нагревании. Со фтором черный углерод реагирует уже при обычной температуре. При высоких температурах углерод соединяется с многочисленными элементами: водородом, серой, кремнием, бором и многими металлами. Соединения углерода с металлами и с другими электроположительными Относительно углерода элементами называют карбидами. С азотом углерод непосредственно не соединлется, однако взаимодействие происходит в присутствии водорода с образованием цианистого водорода.
Алмаз. Углерод в форме алмаза образует чрезвычайно твердые, в совершенно чистом состоянии бесцветные, абсолютно прозрачные, сильно преломляющие свет, блестящие кристаллы удельного, веса 3,51. Естественными гранями алмаза часто являются грани правильных октаэдров; однако встречаются и другие формы кубической системы, среди них тетраэдр, что указывает на то, что алмаз принадлежит к тетраэдрической темиэдрии кубической системы. Кристаллы алмаза часто обладают искривленными поверхностями, так что выпуклые кристаллы можно считать характерными для алмаза.
Чтобы алмазы служили украшением, па них искусственно шлифуют новые грани, которые подбирают таким образом, чтобы с возможно меньшей потерей материала получить камень с возможно большим блеском и игрой красок. Это в наиболее совершенной -форме достигается бриллнантной шлифовкой (ср. рис. 82), при которой грани размещают таким образом, чтобы возможно большая часть падающего света полностью отражалась. Показатель преломления алмазов составляет для красного света (линия Фраунгофера) я = 2.407, дли фиолетового света (линия И) п — 2,465. Рассеяние света (дисперсия) также значительна, и на этом основана известная игра красок— «огонь» алмазов и [Соответственно бриллиантов.
Алмазы встречаются большей частью в так называемых россыпях, т. е. в наносных породах, которые содержат и другие драгоценные камни и благородные металлы, <в Ост-Индии, на Борнео, в Новом Юздпом Уэлсе, Британской Гвиане, Бразилии,
460
Глава 12
Калифорнии, Мексике, я прежде всего в Конго, на Золотом Береге и в Южной и Юго-Западной Африке. В Южной Африке они встречаются также в первичных месторождениях, а именно в оливинах вулканического происхождения (кимберлитах).
Раньше добычу осуществляли промыванием породы в бочках и извлечением остатка руками. Теперь в главном месте добычи алмазов, в Капе, предварительно подвергнутую выветриванию породу промывают в центрифуге, остаток, содержащий более тяжелые составные части, пропускают черен смазанный жиром вибрационный грохот, который задерживает алмазы, нс смачивающиеся водой, но смачивающиеся
Рис. 82. Огранка бриллианта.
а — сверху; б — снизу; я — сбоку.
жиром. В самом богатом Кпмберлейском руднике Капа из 2 т породы в среднем получают 1 г алмазов; однако разрабатывают и такие породы, которые в двадцать раз беднее алмазами. Вес алмазов обычно выражают в каратах (1 карат = 0,205 а), однако ота единица несколько колеблется в различных странах. Самым большим алмазом является Куллинэи (3024 карата), найденный в Южной Африке. Другие известные большие алмазы: Регент (193 карата), Орлов (193 карата) п Великий Могол (186 карат).
Алмазы, добываемые в Капской области, редко бывают так чисты,, как бразильские или индийские алмазы, и потому часто по годятся для украшения. Тусклые, почти свинцово-серые алмазы носят название борт. Большая часть добываемых алмазов относится к этому сорту. Однако встречаются и совершенно прозрачные цветные алмазы: желтые, коричневые, фиолетовые, бледно-синие и светло-зеленые. В Бразилии встречаются и совершенно черные алмазы — карбонадо. Они содержат часто 2—4% примесей. Теплота горения карбонадо, по данным Рота (Roth, 192,’)), заметно выше теплоты горения обычного алмаза. Совершенно прозрачные алмазы («чистой воды») применяются для украшения. Требуе-мылйг для этого свойствами, однако, обладает только очень малая часть-(около 5%) алмазов. Остальные употребляют для технических целей: бурения и шлифования особо твердых материалов, резания стекла, для изготовления подшипников для точных инструментов, ушков для протягивания проволоки; алмаз годен для этой цели благодаря исключительной твердости. Алмазы превосходят твердостью всякие другие-материалы. Поэтому они шлифуются только своим же порошком; вместе-с тем они очень хрупки и легко растираются в порошок в стальной: ступке.
В форме алмаза углерод обнаруживает большую химическую стойкость, На него не действуют ни кислоты, ни основания, если только они не отщепляют легко кислорода. Вместе с тем алмаз, нагретый в кислороде до температуры выше 800°, сгорает до двуокиси углерода. Если же сильно нагреть алмаз без доступа воздуха, то он превращается в графит.
Искусственное приготовление алмазов удалось осуществить лишь в самое последнее время. Правда, уже л 1897 г. Му асе ап (Moissan) получил мельчайшие кристаллики (которые он считал алмазами) при растворении угля в расплавленном железе,, которое затем быстро охлаждалось в форме капель, так что углерод выделялся-внутри капель под очень большим давлением.
Четвертая группа периодической системы
461
Р п с. 83. Решетка алмаза а = 3,560 А.
Рядом с элементарной ячейкой для наглядности показана тетраэдрическая координация атома углерода, который лежит в правом верхнем заднем октанте, с его четырьмя соседями.
Позднее в этом направлении были предприняты многочисленные попытки, в результате которых иногда удавалось получать крохотные Кристал .чини, которые обнаруживали свойства ал мала. Однако ни разу ие было да по четного доказательства того, что речь идет действ нтедыто об алмазах. Алмазы, надежно идентифицированные, были впервые получены йСюнид] Electric Со» в США в 1956 г. Они были прпготовдечщ л форме кристалликов с весом примерно 1/1 карата при многочасовом нагревании графита до температуры — Зй(Юс' С под давлением 100 000 кг/см2.
Алмаз — одно из первых- веществ1, структура которого была определена методом интерференции рсчгггеиовскттх лучей. Он образует, как установили в 1913 г. Ерэгги (Bragg W, Н., Bragg IV. L.) и подтвердили более поздние повторные исследовании, решетку, показанную из рис. S3. Это — гранецецтрярованная кубическая решетка, в которую заключены еще четыре атома, расположенные тетр аэдрп чески один относительно другого. Эти атомы С принадлежат также грапецен трированной решетке, которая, одиако, сдвинута на 1/,i пространственной диагонали относи-тельно первой решетки. Каждый атом С равноудален от четырех других, которые окружают его тетраэдрически, па расстоянии (а/4)Цй = 1,54 А. Прост ранств онио е заполнение решетки составляет лишь 34,01%.
Графит. Графит встречается в природе в значите..1!ыпш кол п-чсстве во многих местах, особенно па Цейлоне, Мадагаскаре, в США, в Калифорнии, РГовой Зеландии, а также в Сибири, Корее, в Богемско-Моранеких горах. Менее обширные месторождения имеются также в различных местах Германии, например в Пассау, где графит добывали еще в средние нека. Он образует серые, непрозрачные, частью чешуйчатые, частью землистые массы. Хорошо сформированные кристаллы встречаются редко. Графит образует нхестпсторонпие пластинки с трехугольной исчерчеи-ностью, которые хорошо раскалываются по спайности, параллельной основанию. Твердость графита незначительна (1 но Моосу); он гибок, мягок и жирен наощупь, обладает слабым металлическим блеском, отличается маркостью. На бумаге оставляет черту свинцово-серого цвета, благодаря чему его применяют для изготовления карандашей. Удельный нес графита 2,1—2,3. В противоположность алмазу графит хороший проводник тепла и злектричества. Он легче поддается химическому воздействию, чем алмаз; в пламени бупзеноиской горелки он сгорает, хотя и с трудом; в чистом кислороде воспламеняется при температуре выше 690°. Природный графит часто сильно за грязней, так что после его сгорания остается 2О”о и более золы.
При нагревании с хлоратом калия п азотном кислотой графит образует ток называемую apatfiumoaipo кислоту — кислородсодержащий продукт, который при быстром нагревании вспыхивает. При нагревании графита, смененного дымя вря! азотной кислотой, до красного каления, один сорта его вспучиваются, другие остаются неизменными. При обработке кояденгрирцванной серкой кислотой в присутствии окислителей, графит разбухает и одновременно принимает глубоко-синюю окраску. Аналогичное поведение при определенных условиях обнаруживают графитовые аподы, если пх применяют для плектр'>л и за концентрированных растворов II2SO4, НСЮ4, HNO3 или HF (Thiele Н., 1!И4). Так называемый «аморфный» углерод разбухает Под действии! серной кислоты в гораздо меньшей степени. Процестда, аналогичные вышеизложенному, называют топохимической реакцией (см. т. II).
462
Глава 12
Теплота сгорания графита на 0,20 ккал/г-атом ниже, чем теплота сгорания алмаза, откуда следует, что графит является более стабильной модификацией углерода. Поэтому в условиях, при которых возможна кристаллизация углерода, нормальным является образование графита. Так, растворенный в жидком железе углерод при охлаждении частично-выделяется в виде графита. Этим и объясняется серая окраска чугунаt
Способ искусственного приготовления графита в больших количествах был раз- ’ работай в 1896 г. Ачесопом (Acheson), наблюдавшим образование графита яри иагре-вании s электрической ттечи угля с кварцем дни получений, карборувда SiC. В настоящее время этот метод в больших масштабах используется «Acheson Graphite Company»-па Ниагаре. Оп заключается в том, что уголь, содержащий кварц, нагревают в электрических печах током в несколько тысяч ампер п течение 12—24 час. Раньше полагали, что при атом в качестве промежуточного продукта образуется SiC, при распаде-которого углерод кристаллизуется в форме графита. Однако Арндт (Arndt, 1931> считает, что этот процесс в сущности основан на росте кристаллов графита, которые-уже имелись в угле, и поэтому его можно сравнить с перекристаллизацией, металлов, происходящей при нагревании (ср. стр. 616).
Графит используют для изготовления плавильных тиглей; для этой цели он особенно подходит как огнеупорный, хорошо переносящий быструю смену температур, теплопроводящий материал. Тигли или вытачи- / вают из брусков ачесоновского графита, или формуют из чешуйчатого графита, смешанного с глиной и кварцевым песком, а затем обжигают. Графит используют также для изготовления карандашей, твердость которых варьируется добавкой глины. Далее, Трафит применяют как огнестойкую черную краску для печей, а в смеси с льняным маслом — в качестве краски, предохраняющей железо от ржавления. Мелкочешуйчатыи графит служит смазочным материалом. Искусственно приготовляемый для этой цели коллоидный графит носит название даг (Dag — deflocculated Acheson Graphite); так, например, ойлдаг. применяемый для смазывания цилиндров двигателей внутреннего сгорания, представляет собой смазоч- , ное масло, к которому примешан коллоидный графит. Подобными же препаратами являются аквадаг и колдаг — последний подучают из природного графита. Так как графит наряду другими качествами обладает и хорошей электропроводностью, то он находит разнообразное применение в электротехнике; так, в гальванопластике для получения проводящего-слоя им покрывают матрицы, его используют для изготовления щеток в динамо и в качестве микрофонных углей. Из ачесоновского графита делают (или им покрывают по способу Ачесона) некоторые сорта углей для дуговых лам (например, для прожекторов) и электроды (например, для электролиза хлоридов щелочных металлов).
Тонкое строение графита было установлено рентгенографическими исследованиями Дебая и Шеррсра (1917), а также Хасселя и Марка (Hassel, Mark, 1924). Иа рис. 84, а показана элементарная ячейка решетки графита. На рис. 84, б для наглядности, помимо узлов решетки, входящих в элементарную ячейку, указаны-и другие узлы- Из рисунков видно, что речь идет о слоистой решетке. Каждый атом углерода окружен тремя другими атомами С, лежащими в одной плоскости; иа расстоянии .1,45 А. На несколько больших и равных между собой расстояниях (2,46 А) в той же плоскости лежат еще шесть атомов углерода, также окружающих рассматриваемый атом. В кристаллической решетке отчетливо проявляются определенные, плотно заполненные атомами, параллельные плоскости, которые удалены на достаточно большие расстояния — 3,345 А. Они сдвинуты друг относительно Друга так, что над серединой каждого шестиугольника одной плоскости лежит атом С соседней плоскости. Плоскости, находящиеся на двойном расстоянии (6,69 А), обладают-одинаковым расположением уз-’юв.
Четвертая группа периодической системы
463
Для решетки графита в отличие от решетки алмаза характерно расположение атомов С по углам шестиугольников. В то время как углерод в решетке алмаза, а также в алифатических соединениях- обладает четырьмя валентностями, направленными по углам тетраэдра, в решетке графита валентности углерода в основном Ограничены тремя направлениями, лежатцимн (вероятно, лишь приблизительно) в одной плоскости. Точно такое же строение принимают па основании химического поведения для так называемых ароматических соединений, простейшим представителем которых является бензол. Итак, структура двух главных классов органических соединений в известной мере уже прсдонредслепа строением решетки элементарного углерода в его двух модификациях; в алмазе п графите.
Р и с. 84. Решетка графита 0=2,46 А; с=6,69 А.
Графит реагирует с F2 при температуре около 420°; при этом между слоями, построенными атомов С, располагаются слои, построенные из атомов F, и образуется так пазываемтлп мопофторуглерод — серое, твердое вещество, содержащее максимально 1F па 1С (Ruff, 1934). Кри более высоких температурах образуется газообразный фторид (см. стр. 479).
Черный углерод. Если углерод выделяется при умеренно высокой температуре, то он образует массу глубоко-черного цвета, которую, как уже указывалось, раньше считали аморфным углеродом, Свойства черного углерода, обычно называемого просто углем, определяются в основном исходным материалом и способом его получения. Различные виды угля содержат большей частью значительные количества загрязнений, которые часто оказывают решающее влияние на свойства угля и о которых не всегда можпо сказать, насколько их можно рассматривать просто как примеси и насколько — как химически связанные с углем вещества. При образовании природных углей имеющиеся в растениях соединения углерода с водородом, кислородом и азотом могут переходитв в соединения с более высоким содержанием углерода и без одновременного выделения элементарного углерода. Вопрос о том, когда н в каком объеме при старении углей образуется свободный углерод, в настоящее время еще не выяспеп. Аналогично обстоит дело и при искусственном получении угля, например при нагревании древесины и других органических веществ без доступа воздуха. Обычно все эти продукты объединяют под названием «аморфный уголь»..
464
Глава 12
Технически самыми важными сортами «аморфного угля» являются коке, древесный уголь, костяной уголь, животный уголь и сажа.
Кокс. Коксом называют твердый остаток после сильного нагревания каменного угля без доступа воздуха (коксования). Различают газовый кокс и металлургический кокс. Первый подучают при производстве светильного газа па газовых заводах irpu нагревании особо пригодных для этого сортов угля (газовых углей); такой кокс болов рыхлый, с большой зольностью; как правило, его применяют только для отопления. Металлургический кокс приготовляют из снсцшшьиого коксового угля, который дает меньше газа и сильно Спекается, так что подучается сравнительно плотный и твердый кокс, который используют для доменных печей. Подлежащий коксованию угбль помещают в измельченном и увлажнешгом. состоянии в вертикально стоящие реторты, где он сильно нагревается и спекается, отдавая обычно 20—30% веса сухого угля в виде газов (коксовый газ). Таким образом получается кокс в виде темно-серых кусков, пронизанных трещинами, подобно базальту. Оп содержит приблизительно 90% С, 1% Н, 3% О, 1?,5—1% N и 5% «золы», т. е. несгораемых составных частей. Этот кокс торит совершению без копоти коротким, синеватым пламенем, выделяя 7000— 8000 ккал и а 1 кв.
Пр it коксовании (швелеван'ии) бурых углей получается так, называемый буро-угольный кокс. Оп состоит из матово-черных пористых зерен, величиной в несколько миллиметров, содержащих 15—25% золы и обладающих теплотворной способностью 6000—7000 ккал. При нагревании буроугольиый кокс тлеет, т. е. горит без пламени и дыма. Он может быть использован как топливо, а также для фильтрования воды и приготовления черной краски.
Древесный уголь получают при нагревании дерева без доступа воздуха. Прежде это проводили в так называемых «угольиыа: кострах». В настоящее время обугливание проводят в больших железных ретортах, что позволяет улавливать цепные отгоны (ср. табл. 78).
Таблица 78
Продукты разложения при перегонке каменного угля н древесины
Исходный ирод уст Наложный уголь Д7>св11сина
остаток древесный рголь
Выделяющиеся газы Светильный газ, После ТОГО как из него удалены NII3 ц lioS, содержит главным образом 112, СН4, GO, С2Н4т С2П2, С61Тв; теплотворная способность 5500 ккгы/.ч3 (ср. стр. 473) Древесный газ (менее светящийся) содержит главным образом С02, СО, .СН4 и Н2. Теплотворная способность ~3000 клал/лг3
Дистиллят Аммиачная вода и каменноугольный деготь (последний содержит, бензол, фенол, нафталин, антрацен, пиридип п пр.) Древесный спирт (метиловый спирт), древесный уксус (уксусная кислота), древесный деготь
Древесный уголь — рыхлый черный продукт, в котором можно еще установить ячеистую структуру дерева; этот уголь легко воспламеняется, после чего продолжает тлеть. Зольность его очень мала — около 1%. В противоположность коксу он не содержит серы. Поэтому древесный уголь применяют в металлургии, когда и у я; на особая чистота угля, naiipmiep при рафинировании меди. Далее, он служат для очистки спирта от сивушных масел, при лечении рап (в качестве средства, уничтожающего дурной запах) и для других мсдипипских целой. Особо легко воспламеняющийся пористый и мягкий древесный уголь получают при обугливании черемухи, тополя и оЛьхп прп 300—400’; он служит для приготовления черного угольного порошка.
Четвертая группа периодической системы 465
Костяной уголь (костяная чернь) получают прл нагревании л ретортах предвари* тельно обезжиренных-костей. В качестве побочного продукта при этом получается аммиачная вода и деготь (костяное масло Диппеля). Так как кости состоят приблизительно, на две трети из несгораемых веществ, преимущественно из гидроксплкарбонат-апатнтов (см. стр. 734), костяной уголь содержит .много золы (90% и более). Оп обладает замечательной си особ пос гыо обесцвечивать, и поэтому его используют для удаления красителей и других загрязнений из растворов, например при очистке сахарпого сиропа. Костяной уголь в больших количествах применяют в качестве черной краски, особенно для изготовления обувной ваксы, краски для кожи и типографской краски. Для приготовления лучшей краски для живописи обжигают отходы слоновой кости.
Кровяной п Животный уголь получают нагреванием крови и Других животных отбросов с поташом п дальнейшим выщелачиванием продуктов обугливания. Его применяют главным образом в медицине для удаления из кишечника некоторых ядов и больших скоплений газов.
Вее уназаппые виды применения древесного, костяного, кровяного и животного углей основапы главным образом па высокой адсорбционной способности, которую уголь проявляет по отношению ко многим растворенным веществам и газам. Уголь с высокой адсорбционной способностью называют активированным углем*. Активность угля в значительной мере зависит, помимо исходного вещества, также от способа приготовления. Активность можно часто повысить путем особой обработки угля, например нагреванием с некоторыми пеоргаппческими солями. Адсорбционная способность угля относительно различных веществ весьма различна. Газы в общем адсорбируются тем хуже, чем труднее они сжижйютея. Однако нет полной прямой зависимости между степенью адсорбции газон н их точкой кипения или критической температурой. По данным ангора (19'д2), углем большой активности адсорбируется тем большее количество тех или иных газов, чем ниже упругость их пара в жидком состоянии. Адсорбция стильно возрастает при понижении температуры.
Сажа (ламповая копоть) получается при термическом разложении многих газообразных (пли переведенных нагреванием в газы) соединений углерода. Технически сажу получают из дегтя, каменноугольного масла, нафталина, парафинового масла или из природных гцзитт, богатых углеводородами, путем сжигания этих веществ при недостаточном доступе воздуха и одновременном охлаждении пламени мсталличе-скнмп листами, охлаждаемым!! водой. В настоящее время ее получают также из ацетилена. Сажу широко применяют в качестве черной краски. Опа служит в Первую очередь для приготовления типографской краски, туши, черной краски для живописи, лакированной кожи и резиновых изделий, например резиновой обуви, а также для окраски граммофонных пластинок.
Сажа, древесный уголь и другие сорта угля, полученные при сравнительно слабом нагревании органических веществ, отличаются по своей тонкой структуре от графита, помимо малого размера кристалликов (средний диаметр частиц в направлении перпендикулярном оси с в общем составляет 20—60 А), также и тем, что п у них отдельные плоскости, построенные из углеродных шсстичленпых колец, расположены друг относительно друга не так, как ото показано па рис. 84. Хотя расстояние в направлении оси е у них примерно го же, что и в обычном решетке графита, однако плоскости в обоих перпендикулярных направлениях беспорядочно сдвипуты одна относительно другой (ArnfulL, 1932, Bert, 1932—1933, Warren, 1934, Hofmann, см. особо Z. Eli'k trcichom... 42, 504, 1936). Структуры, которые, подобно структурам этих видов угля, занимают среднее положение между кристаллическими и аморфными веществами ^ср. стр. 532), называют мезоморфными.
Клостящий Углерод. Если над пламенем светильного газа или метана поместить гладкую, нагретую выше 650° поверхность, например поверхность глазуроваппого фарфора, то, как установил Гофман, выделяется очень блестящая черная разновидность углерода — блестящий углерод. Он состоит, как показывает рентгеновский структурный анализ, из мельчайших кристалликов графита, определяемых только рентгенографически. По своим свойствам блестящий углерод занимает в общем среднее место между Другими видами черного углерода и графита. Гофман получил его недавно также в мелкодисперсной форме в виде рыхлого порошка. При одинаковых температурах получения мелкодисперсный углерод по величине кристалликов совпадает с плотным блестящим углеродом (30—40 А), однако значительно превосходит последний по
* Активированный уголь, впервые предложенный Н. Д. Зелинским (1915) в ьычметве сродства защиты от отравляющих веществ, является важнейшей состав-ион частью противогаза. — Прим. ред.
30 Г. Рема
466
Глава 12
адсорбционной способности. Это свойство объясняется1 тем, что у плотной формы только относительно малая?часть поверхности кристалликов доступна адсорбентам.
Ретортный графит; Подобно образованию блестящего углерода,: по при гораздо более высокой, температуре, происходит образование так называемого ретортного графита, Это разновидность угля, образующаяся в качестве побочного Продукта при производстве кокса, Коксовые газы, проходя у очень сильно нагретых стенок реторты, частично разлагаются, и выделяющийся углерод оседает па них в виде твердой плотной массы. Ретортный графит отличается от обычного: графита малой:; плотностью (—2), большой твердостью и отсутствием кристаллических1 свойств, устанавливаемых обычными методами. Сближает его с обычным графитом хорошая электропроводность, и химическая стойкость. Поэтому ретортный графит используют дня изготовления угольных штифтов для дуговых лама, а также электродов'для гальванических элементов.
По данным рентгеновского анализа (дебаеграмма) ретортный графит обладает кристаллической решеткой обычного графита. Однако его кристаллики очепь малы (средний диаметр равен примерно 60 А), так что Опп лежат значительно ниже пре-. делов микроскопической видимости. Так как кристаллики ретортного, графита, так же как кристаллика блестящего углерода, ориентированы беспорядочно, эти виды угля не обладают характерной для обычного графита хорошей расщепляемостью, Твердость ретортного графита также объясняется беспорядочной ориентацией его кристалликов (ср. стр. 459). '
СОЕДИНЕНИЯ УГЛЕРОДА
Разнообразие соединений углерода очень велико, хотя почти все они происходят от углерода одной валентности —от четырехвалентного углерода. Трехвалептный углерод встречается лишь в .некоторых сложных соединениях, относящихся к области органической химии. Известно очень большое число соединений двухвалентного углерода. Они, не считая СО, и котором углерод лишь стехиометрцчески двухвалентен, очень неустойчивы.
Большое разнообразно соединений углерода основано, во-первых, На способности углерода одинаково хорошо соединяться как с электроположительными, так и о элек-троотрицателъными веществами, причем часто одновременно с теми и другими, так что в одном и том же соединении, как, например, в СН4 (метан), атомы: водорода? могут замещаться частично или полностью почти любым другим элементом, по преимущественно зЛемонтом электроотрицательного характера. В связи с этим находится и вторая особенность углерода, на которой основано существование большого числа органических соединений, а именно способность углеродных атомов образовывать прочные свази также друг с другом. Благодаря этому существуют так называемые углеродные цепи, содержащие иногда очень большое число звеньев
И Н Н II Н 11
Н3С —СН3 Н,С —С —СН3 Н3С-С—С —СН3 Н3С—С—С—С—СН3 ИТ. д. Н НН Н II II
Этап Пропан Бутан Пентан
Могут образовываться и «разветвленные цепи», папример:
сн3-сн(си3)—cii(cji.,)-ch2—сн3
Метала тип пентан. .
Третий, четвертый, пятый, шестой и т. д, атомы С в цепи могут снова соединяться с первым атомом, в результате чего образуются кольца. К кольцам снова могуч присоединяться цепи и другие кольца. Далее, процесс, который приводит к связыванию Двух, атомов С, если он идет между двумя атомами С разных молекул или непосредственно несвязанных. атомов С одной и той же молекулы (например, отщепление галогена или Тало геноводо рода), может произойти повторно между теми же атомами,’ Ои приводит тогда к образованию двойных и тройных связей менщу соответствующими атомами : ‘углерода. Если еще принять во внимание, что для таких цепей и колец справедливо все: то, что справедливо для простого углеводорода СЩ, т. е, ихводо родные атомы могут быть замещены разнообразными другими «лигандами» .(от Праге связывать)
Четвертая группа периодической системы 467.
и, кроме того, что членами колен и цепей. могут быть многовалентные лиганды N, О, S (например, атом О в эфире СН3—СН2—О—СИ2—СН3), то можно Представить себе огромное разнообразна соединении, существование которых основано па двух названных выше свойствах углерода. Рассмотрение этих соединений составляем -как уже было отмечено, предмет органической химии. -
Здесь будут рассмотрепы только простейшие соединения углерода.- - . Главные типы их приведены в табл. 79.
Таблица 79
Простейшие соединении углерода
Углеводороды Кислородные соединения : Сульфиды
Примеры: СН4 метан С2Н6 этан С2Н4 эти леи С2Н3 ацетилен ,ч) Окисли СО2 Двуокись углерода .СО окись углерода С3О2 недокись углерода ’ б) Кислоты Н2СО3 угольная кислота, соли которой MjfCOgj— карбонаты Н2С2Ов надугольная кислота, соли которой И J (С 2О е] — перкарбопат ы Я2СгО4 щавелевая кислота, соли которой M|(G2O4j— оксалаты НСНО3 муравьиная кислота, соли которой МТ[СНО2] — формиаты ИС2Н3О2 уксусная кислота, соли которой М,[С2Н3О2]-ацетаты CS;. сероуглерод COS сеpooкись углерода C.S ппюкпсь углерода C3S2 тио недокись- у г ле рода
Карбиды !
Галогениды
Примеры: СаС2 1 Ag3C3 J аЧетода : А14С3, разлагается при действии разбавленных кислот SiC 1 не разлагаются В6С ) кислотами
СР4 четырех фтористый уг-.черод CGi4 четыреххлорястый углерод СОС12 фосген СВгд чотырехбромистый .углерод С14 четырехиоднстый углерод
(CN)2 циап; HGN цианистоводородная ’кислота, соли которой M}[CN] —• цианиды (SCN)2 родая; HSCN рода-нистоводородная кислота, соли которой M^fSCN]— роданиды
/ Средине межъядерпыс расстояния, валентно-силовые ко остеиты в энергии связей, т. е. работа, которую следует затратить для разрыва связи (Glockler, 1951) для важнейших надое связей, осуществляемых.углеродом, приведены в табд. 80.
В первой ко лонке таблицы; указаны соединения, на примере которых получены приведенные данные; во второй — теплоты образования этих соединений из свободных атомов. ’ t :
Углеводороды
Простейшими насыщенными углеводородами являются метан С Hi и этан С3Нс; простейшими ненасыщенными этилен СаНа и ацетилен С2ТГ2. Последние называются попас ы щепными потому, что они легко могут присоединять другиц.вещсстна, например галогены, при этом двойная (соответственно тройная) связь превращается в простую,
•" 30*
468
Глава 12
Таблица 80
Теплоты образования соединений углерода и характерные константы для встречающихся в них связей
И?атом—теплота образования из атомов, к.кал/мплъ' т0—межъядерное расстояние, А; Лу—валентно-силовые константы, ди-н/см-, В—энергия связи простых, двойных или тройных связей, кмл/6,023-1023
Соединение W ятам Связь Го. kf -10-5 В
с2н6 665,7 с-с 1,547 4,5 84,6
Алмаз 169,7 с—с 1,545 — 84,9
531,4 с—с 1,353 7,2 125,0
Ст (газообр.) 135,4 с=с 1,316 9,1 135,4
с2н2 388,4 с=с 1,207 15,6 181,5
СН3-.?1На 187,4 С—N 1,469 4,9 81,9
СНз-N.GO 704,1 C™N 1,25 11,1 141,1
сгя2 537,8 C=N 1,16 17,6 208,7
HCN 326,3 C=N 1,157 17,9 222,8
СН3ОН 480,6 С—О 1,428 4,9 80,9
Н2СО 360,2 с=о 1,21 12,3 157,1
со2 380,9 С=О 1,163 15,3 190,5
со 255,5 С=О 1,128 18,9 255,5
CH (газообр. радикал) 92,2 С-Н 1,1305 4,3 92,2
СН2 (газообр. радикал) 190,2 с-н 1,1112 — 95,1
СН3 (газообр. радикал) 291,2 С-Н : 1,0984 — 97,1
СН4 392,2 С-Н 1,092 5,0 98,1
€2Щ 665,7 с-и 1,0995 4,8 96,9
С2Щ 531,4 с—н 1,071 5,3 101,6
с2н2 388,4 С-Н 1,060 5,9 403,4
Насыщенные углеводороды, т. е. метан, этап, пропан и т. д., объединяются под названием алканы’, ненасыщенные, содержащие двойную связь — под названием алкены, а содержащие тройную связь — под на зван нем алкины. Исходя из этих общих наименований, для этил спа употребляют также название этен, а для ацетиле^ иа —
Важнейшие константы этих соединений даны в табл. 81. Приведенные в таблице значения свес литра» соответствуют весу 1 л определенного газа при 0°. Если разделить эти числа на вес литра кислорода (1,4289 г) или на вес литра воздуха (1,293 а), то моа;по получить удельные веса углеводородов, отнесенные к весу кислорода (соответственно воздуха), принимаемому за единицу. В табл. 81 приведены также теплоты Сгорания рассматриваемых углеводородов. Они отнесены к 1 м3 газа при температуре 0° и давлении 760 .«.и рт,ст, т. е. показывают, какое количество тепла (ккал) можно получить при сжигаппи 1 .и3 газа.
Углеводороды с большим числом атомов С («высшие углеводороды»), в противоположность простейшим углеводородам являются жидкими или даже твердыми веществами. Из смесей жидких углеводородов, как уже упоминалось выше, состоят керосин, бензин, смазочные масла и др. Индивидуальным жидким углеводородом является бензол СеН6. Жидкие углеводороды приобрели большое значение прежде всего как горючее для двигателей внутреннего сгорания. В связи с этим важной проблемой стало их искусственное получение при помощи так называемого процесса сжижения угля.
Четвертая группа периодической системы
469
Таблица SJ
Физические константы простейших углеводородов
Метан Этан Этилен Ацетилен
Формула . , сн4 И3С—СНз НаС=СНа НСзСН
Температура кипения, +; —164 -88,3 -103,9 -83,6
Температура затвердевания, "С .... -184 -172,1 -169,4 -81,8
Критическая температура, .... -82,5 4-32,1 + 9,5 +35,9
Критическое давление, атм 45,7 48,8 50,7 61,6
Вес литра, а 0,7168 1,3564 1,2604 1,1708
Теплота сгорания, ккал/м3 9512 16 750 15 510 14 010
100 об. ч. воды при 0° газа растворяют, об. ч . . . , 5,56 9,87 22,6 173
100 об. V. воды при 20'- растворяют (объемы газов приведены к 0°), об. ч. 3,31 4,72 _ . 12,2 103
100 об. ч. спирта при 0° растворяют газа, об. ч 52,3 -150 360 -600
Сжижение угля. Процесс превращения угля в жидкие углеводороды или в их кислородсодержащие производные в результате присоединения водорода (гидрирования) называют «сжижением угля».
Гидрирование можно осуществлять;
а) непосредственно: при действии водорода на уголь при высоком давлений (200 ат) и температуре около 450° без катализаторов [метод Бергиуса (Bergins)] или с катализаторами (метод 7. G. Farbemndustrle):
б) косвенно: каталитическим превращением окиси углерода и водорода в метанол (метод Г. G. Farbenindustrie) или в углеводороды [метод Ф. Фишера (F. Fischer)], а также при каталитическом взаимодействии окиси углерода с водяным паром (метод Rheinpreufien 4.G.).
Превращение угля в жидкие углеводороды впервые удалось осуществить Вертело при действий иодистбго водорода под давлением па уголь. Позднее Бергнус (1911) нашел, что водород непосредственно соединяется с углем при высоком давлении и температуре выше 400°. Этот процесс можно представить себе следующим образом: ненасыщенные углеводороды, из которых состоит уголь, при нагревании расщепляются и гидрируются Водородом. Применение способа Бергиуса в производственных масштабах осложнено тем, что вследствие неполного гидрирования и отсутствия возможности управления реакцией образуются смолистые и асфальтоподобные продукты. В 1925 г. сотрудники I. G. Farbenindustrie нашли, что, применяя подходящие катализаторы, можно ускорять гидрирование (протекающее в иных условиях лишь очень медленно) я благодаря атому гак управлять процессом, что он будет протекать в желательном направлении (например, в сторону образования легколегучих углеводородов).
Промышленное производство искусственного бензила осуществляется следующим образом. Мелкоразмолотый и смешанный с маслом для образования пасты уголь вместе с небольшим количеством катализатора (он теряется с золой) нагревают с водородом при высоком давлении. Полученное при этом первичное масло пропускают затем в виде пара («газовал фаза») через катализатор (например, соединения вольфрама или молибдена в смеси с другими веществами), расположенный в определенном порядке в реакторе. Рабочие условия такие, как дав.тепно и температура реакции, а ташке расположение и сорт катализатора в «пастообразной» и газовой фазах, можно варьировать в широких пределах, благодаря чему можно Получать нс только чистый бензин, но также й смазочное масло, топливное масло, дизельное масло, осветительное масло. Оба указанных выше процесса гидрирования проводят в автоклавах при температуре 400—450° и давлении окоцо 250 ат. Б качестве исходных веществ можно использовать бурый уголь, каменный уголь или другие уг.теродсодержащне вещества,
J
470 Глава 12
такие, как смолы и масла. В С ПТ Л, папрпмср, метод каталитического гидрирования под давленном применяют для получения цеплых смазочных масел из тяжелых фракций пефтн. В Англии? последнее время гидрированном каменного угля п каменноугольной смолы получают бензил. В Германии уже несколько лот бурый уголь и соответствующая смола, а также в небольших количествах цеФть и масла, выделяемые из каменноугольной смолы, превращают путем гидрирования в бепзин. Синтетический бензин впервые поступил в продажу в 1927 г,
,1 . G. Farbenindustrie разработала также метод (Pier, Mittarch, 1923) получения метадола СН3ОН (метиловый спирт) путем гидрирования окиси углерода при температуре примерно 400° и давлении около 20 ат в присутствии катализаторов, содержащих ццнк. Метанол используют как горючее (и смеси с бензином), в качестве растворителя или как исходное вещество для химических синтезов. В зависимости от условий работы и сорта катализатора от им методом можно получить и высшие спирты.
В метода ф. Фишера. для гидрирования окиси углерода: используют катализаторы, которые состоят из элементов группы железа в соединении со ,ще л очами и Другими окисламн. При работе под давлением образуются смеси, состоящие преимущественно из ^кислородсодержащих производных углеводородов,, (синтол процесс); При работе без избыточного давления образуются углеводороды (синтетический бензин Фишера и Тропша). Исходная окись углерода должна быть тщательно очищена; особенно от сернистых соединений, так как катализатор легко отравляется, Наряду с бензином в небольших количествах получаются газойль и парафин, которые используют как таковые или путем крекинга превращают в бензин. Окисление полученных этом методом высокоплавких парафинов дает мирные кислоты, которые позволяют производить синтетическое мыло. , .. .
Жирные кислоты можно получить синтезом непосредственно из олефинов, которые соединяются с СО н Н2О под высоким давлением в присутствии карбонила никеля в качестве катализатора
ВСН=:СН2 ; СО-ILO - llCIL-CHoCO.OM.
Аналогично можно осуществить синтез эфиров жирпых кислот
RjCH-Clb-f- СО -j- RaOH RjCIk-CIK-CO • OR2.
Из олефинов, СР и Т12 синтезом при высоком давлении в присутствии катализатора ТЬО2, содержащего Со, получают альдегиды
ПСП=СН2+ СО + Н2 = ПСИ2- СИ2- СПО.
В табл. S2 приведены соедпнеини, которые мощи о получить из окиси углерода и водорода. Таблица (и которой приведены лишь некоторые из имеющихся в настоящее время методов) показывает, какие разнообразные превращения окиси углерода мощно вызвать путем изменения катализатора и прочих условий опыта.
В 1951 г. Кёльбсль (К olbel) показал, что для гидрирования СО вместо водорода можно использовал, поду. Ему удалось получить с хорошими выходами бензин, парафин и другие углеводороды при взаимодействии СО с Н20 при нормальном давлений и температуре 210—260°, используя в качестве катализатора азотсодержащие Fe, Со пли Mi (с подходящими добавками). Если процесс вести прп-Высоком давлений (например, 1(Ю ат), то наряду с углеводородами образуются спирты (и прежде всего:этиловый Спирт).
Деуокиеъ углерода также можно прогидрировать водородом до (газообразных жидких и твердых) углеводородов. Уже в 1902 г. Сабатье (Sabatier) и Сендёрёцс (Sea-derens) показали, что при пропускании СО2 или СО в смеси с И2 над нагретым, тщательно измельченным никелем получается СЩ.
Метан СНг — газ, без цвета и запаха, горящий слабо светящимся пламенем. Метан часто выделяется в угольных копях, где он, смешиваясь с воздухом, образует рудничный газ. Он образуется при разложении растительных веществ без доступа ноздуха, например в болотах, и потому называется также болотным газом. (В качестве такового он и был открыт Вольта в- 1778 г.) Во многих местах метан в больших, количеств ах. выделяется из трещин в земной коре, особенно вблизи от источников нефти, например в Пенсильвании, Огайо, Индиане (США) и около г. Баку. Его можно получить и при пиролизе древесины, каменного й бурого углей и т. д. (ср. табл. 78).
I
Таблица 82
Зависимость взаимодействия СО с Н3 от условий опыта
Ката л тиатор TcMiioparypa <4?) и давление Прсвращснпе (схематическое суммарное уравнение) Главный продукт реакции • :/
Nj или Со 2C>C—300 Атмосферное давление со-нн2—сн4н-н2о Иетап
Fe 300—400 Атмосферное давление 2СО-5-2Н2 = СН4.4-СО3 Метан
Со, активирован* ими MgOn ThO2 —t 170—200 Атмосферное давление пСО *}- 2лН3—СпН2т1 -ф*" -(-лНоО: нСО-4*(2н-ф- -ф~1) Н2 = СвН2п+з-ф- -[*лНаО Бензин
Fe, активированное Cu, KOH и СаО 225 10 ати Например, 8СО+10Н2= — С'сН j з 2COg 4~ -ф4Н2О; ОСО + ЮНа-СбЙн-Ь ЗСОоД 3H..0 Бснзип
Fe, активированное Cu, A]2O3 и KOH 230 20 ти Зл GO -J* 3i(Н а=2СпН ап 4* -j-riCO2-J*nH2O Олефины
flu, а л Tir Кирова ji-нап K2CO3 180—200 300 ати с) iH ^^2^) Твердые парафины
TilOa, ЯКТЯвИрб* ' ванная А1303 и K2CO3 450 300 ати Наприлгер, 10C0-f-SR2— = CeH14+4CO2-h + 2Н2° s луни Ими цепями
7н0, активированная CH , И: ACC’s 200.-400 200—300 ати СО-г2Н,==СН3ОН Метиловый: спирт .
ZnO, активированная Fe и KOH 450 200 ати, малая скорость мродускаяия пСО ф-2пН2= = СпН2„+1ОН4-(п-1) нг0 «Изобутиленов масло» (Смесь метилового спирта с изобутиловым н другими алифатическими спиртами)
Fe, активированное AKO3 - н K?CO3 2^ 20 ати 17СО ; 20Н2=cyl, j-5CO^-j7. 4-6Н2О > «гСилтола (смесь утл<лю-дэродов с кислород-содержащими соединениями)
472
Глава 12
До сих пор пе известно, какое количество метана, выделяющегося при гниении, а также из недр земли, достигает атмосферы. Это интересно установить, тан как полученные данные позволят точно рассчитать, как относятся процессы перемешивания воздушных масс путем конвекции и разделения их путем диффузии. К настоящему времени известно только, что вблизи земли влияние конвекции значительно превышает влияние диффузии, и можно полагать, что на высоте около 70 к,м отношения начинают изменяться (ср, Н а г L е с k Р. Ansjew. Chem., 63, 1, 1951: Paneth F., J, Chem. Soc., 3651, 1952].
Метан в лаборатории ложно получить двумя путями:
1) нагреванием смеси ацетата патрия с натронной известью (смесь СаО и NaOH)
NaC2H3Oa-]-NaOH = NaaCO3-|-CH4,
2) разложением водой карбида алюмипия
Л14С3+12НгО=4А1(ОИ)3 + ЗСН4.
Метан легче воздуха (удельный вес относительно воздуха 0,558), Он плохо растворим в воде (см. табл. 81), лучше — в спирте. С воздухом или кислородом мотан образует смеси, взрывающиеся при воспламенении, так как при нагревании до температуры выше 670° он соединяется с кислородом, образуя двуокись углерода и воду:
CH4-J-2O2-у СО2-]-2Н2О,«вдк-]-212,3 ккал (при постоянном объеме).
По закону Авогадро из этого уравнения следует, что для полного сгорания 1 об. ч. метана требуются 2 сб. ч. кислорода пли 10 об. ч. воздуха. В соответствии с этим наиболее сильно взрывает смеет, метана с воздухом в соотношении 1 : 10. Однако и при значительно меньшем содержании метана эта смесь взрывоопасна, чем и объясняются взрывы в шахтах, все еще происходящие время от времени, несмотря на многочисленные предохранительные мероприятия.
Далее, из приведенного выше уравнения следует, что при полном сгорании (с добытком воздуха) 1 об. ч. метана образу гаи 1 об.ч, СО2, Следовательно, уменьшение объема смеси равняется удвоенному содержанию в ней метана, если проводить измерения при обычней температуре и пренебречь объемом жидкой воды. При его ранни смеси водорода с воздухом или кислородом уменьшение объема равняется полута рак ратному содержанию водорода. Если воспламенить смеет, водорода и метана в избытке воздуха (при помощи электрической искры или накаленной проволоьтг) и определить содержание-С'О2 в смеси после сгорания (путем поглощения едким кали), то тем самым непосредственно определяется и содержание СН,5. Если удвоить эту величину и вычесть полученное из величины уменьшения объема, вызванного сгоранием, то получится уменьшений объе-ма, которое следует отнести за счет содержания элементарного водорода в первоначальной смеси. Последняя величина, умноженная на г/3, даст содержание водорода.
В присутствии катализаторов (например, Ni, активированного АЬОз) СП4 моЖет частично окисляться в соответствии с уравнением С114—))О2 Ц: СО + 2П24-+ 6 5 1;кая и.тн СН4+ Н2О £ СО ЗН2— 48,9 ккал и СН4-]- 2Н2О СО2-|- 4Нг— —38,5 ккал. Э1 и реакпип имеют техническое значение для получения водорода (ср. стр. 47). Между СН4 и СО2 нрп соприкосновении с Ni (в;качестве катализатора) также устанавливается равновесие
(РеО'Рн^ДРсЩ’ЛЮз) = ^Р-
log Кр= —12,662/7’-]-13,875 (Fischer, 1931).
Структура правильного тетраэдра, которую химики с давних пор придавали молекуле СН4, была под'1веря:дсна электронографически я спектрами комбинационного рассеяния. Из величины момента. инерции 5,328’10_ад г-с.м2 следует, что межъядерное расстояние С. <--> Н равно 1,093 А. Если заменить 4 атома Н атомами дейтерия, то это расстояние сократится до 1,089 А.
Поскольку в СН4 протоны расположены внутри внешних электронных оболочек (ср. стр, 451), он обладает физическими свойствами, подобными свойствам инертных газов. Температуры плавления и кипепия метана близки к соответствующим константам
Четвертая группа периодической системы
473
Аг. С Кг оп образует непрерывный: ряд смешанных кристаллов (Stackclberg, 1936). При 20,4° К лежит топка перехода СН4, которая обусловлена внутримолекулярными превращениями (Clasius, 1930).
Метан — следующая после водорода основная составная часть светильного газа, получаемого при сухой перегонке каменного угля. В чистом светильном газе содержится в среднем 30—33 об.% метана наряду с 50% водорода. Светильный газ содержит, далее, около 4% так называемых «тяжелых углеводородов» (среди них преобладает этилен наряду с некоторым количеством ацетилена и бензола) и, кроме того, около 9% СО, 2% СОй, 1% Na и следы кислорода. Светильный газ такого состава обладает высокой калорийностью — около 5500 ккал/м^. Однако часто к светильному газу примешивается водяной газ (см. стр. 487), в связи с чем калорийность снижается до 4200 ккал/м9.
Для сгорания 1 .и3 чистого светильного газа требуется 5—6 м3 воздуха; для сгорания 1 л3 чистого водяного газа — только 2,4 л3. Можно считать, что при сгорании 1 л3 городского тала используется около 4 воздуха.
Коксовый >',рч содержит около 55% Н2, 25% СН4, 2% тяжелых углеводородов 4—6% СО, 2% СО2 и 10—12% N2. Тот факт, что его состан отличается от состава светильного газа объясняется главным образом тем, что па коксовых заводах уголь прокаливают сильнее, чем па газовых. Чем выше температура прокаливания угля, тем больше водорода в выделяющемся газе по сравнению с метаном и тяжелыми углеводородами. Так как содержание углеводородов существенно влияет на калорийность, то калорийность коксового газа (4000—4500 ккал/м3) значительно ниже калорийности чистого Светильного газа.
Этан С2Нв — газ без цвета и запаха, впервые получен Кольбе в 1848 г. электролизом концентрированного раствора ацетата кадия. Он выделяется при этом на аноде за счет взаимодействия разряжающихся там ацетат-ионов, т. е. радикалов уксусной кислоты, сопровождаемого отщеплением СО а
СН3 J-COO' СН3
: = 4-2СО24-2е.
СН34-СОО' СН3
I
Кольбе первоначально считал, что здесь образуется радикал метана— метил СНз. Однако вес литра этого соединения показывал, что оно обладает удвоенным молекулярным весом С,НЙ. Метил-радикал чрезвычайно мало устойчив, поэтому метильные радикалы тотчас объединяются попарно, образуя соединение СНз—СНз.
Этап можпо получить также действием металлического натрия на йодистый метил CH3J в эфирном растворе (при 100°) или нагреванием иодистото метила с металлическим цинком в запаянной трубке
СИ31 1 — Ma j
1 -1 СНз
= | -|-2NaI;
„ т---...... СН3
СНз 11 ч- м:
СИзИ
I + Zn ) -СНз b_____]
СНз
СНз
4- Znl2.
При этих методах получения также можпо было ожидать образования метила СНЭ, если бы он был устойчив.
Свободные радикалы СГ13 впервые удалось получить Панету в 1929 г. термическим разложением РЬ(СЛ3)4. Ира этом оказалось, что метильпые радикалы так неустойчивы.
il't Глава IS
что, например, d атмосфере водорода при давлении 2 льк рт ст концентрация" свободных метилов уще через 0,006 сек падает вдвое, а через 0.1 сек они практически исчезают; Поздпсе были получены также чрезвычайно малоустойчивые свободные радикалы — этил, пропил и фенил (Paneth, 1931; Purcell, 1935; Polanyi, 1934). <
Еще в .1924 г. Xi.'iui (Hein) сделал вывод о промежуточном обрааовапии; свободной этильной.группы С2Н5 при электролизе этилата натрия NaC2H5, растворенного в этилате цинка Zn(C2Hs)2, на основании того, что при атом на, аноде выделяются эквимолярные количества этапа и этилена ' ,
2С2Н5 —- 2 с =2С2-Н,5 —>- CgHfl “ФС2Н4 ~
(процесс диспропорцииннровацпя).*
В еще.большой мере, чем радикал этил, к диспропорционированию склонен ради кал н пропил (образование GSHS и С3НЯ). Наряду с этим при более высокой температуре происходит распад с образованием радикала метила (CSH7->C2II4-J-С|13).
Технический метод получения этана заключается в гидрировании этилена. Этап находит применение в охладительной технике. В природе он встречается иногда среди газов, выделяющихся из нефтяных скважин. Этан, подобно метану, является насыщенным углеводородом. Он не реагирует с бромной водой, В воде этан мало растворим, зато значительно растворим в спирте (саг. табл. 81, стр, 469). Он, как и метан, горит слабо светящимся пламенем, однако отличается от метана значительно большей сжижаемостью. Его критическая точка лежит выше комнатной температуры, в то время как метан может быть сжижёп лишь после Сильного охлаждения под давлением.
С2Н6 образует с водой «газ гидрат» (ср. стр. 248) состава С2НЯ.53/4Н2О (найдено 5,8 ± 0-5 Н20, Stackclberg, 1949). Давление разложения последнего составляет при 0° 5,2 ат. Соответствующий гидрат (давление разложения 26 ат’при 0“) образует и СН4.
Этилен. С2Н4 содержится в количестве около 4% в светильном газе. Для получения его в лаборатории обычно нагревают до 160° смесь эти-нового спирта с концентрированной серной кислотой, (к которой целесообразно добавлять крупнозернистый песок). Эту реакцию можно представить как отщепление воды под действием серной кислоты
CJI5OI-1 - Н,О = С2Н4.
Если рлссматртгпать процесс детально, то следует отмстить, что серная кислота сначала реагирует со спиртом с образованием сложного эфира, называемого этилсериой кислотой. Последняя затем распадается, поскольку она не устойчива к нагреванию, на этилен и Серную кислоту
С211гО И НО j SO3II=СМГ.OSO31I МЕО,
Этилсернан кислота
C2H3.OSO.pi-C2H.; |-ll>so.. ч с
В технике этилен получают по методу Ипатьева — пропусканием паров-.спирта над аморфной окисью алюминия, нагретой до 360°. Этилен — бесцветный газ со своеобразным сладким й жирным запахом. Он энергично сгорает, давая пламя значительно более сильно светящееся., чем пламя метана и этапа. В отличие от последних этилен поглощается дымящей серной кислотой и бромной водой- При этом образуется этилсерная кислота C2H5OSO3H и соответственно бромистый -этилен СаЩВгд. Первая при нагревании снова распадается на этилен и серную кислоту. Из бромистого этилена моЩно вновь получить этилен при действий цинковой пыли на его спиртовый раствор
С2НйО5О3Н = С2Н44-И^О4, :
С.Н.Вг ’ Zn —С2П4 ZnBr2.
Четвертая группа периодической системы
475
Этилен умеренно растворим и воде, легко растворим в спирте (см. табл. 81). Ч присутствии катализатора (платиновая чернь или никель) этилен может также присоединять водород, превращаясь в этап
C,,ll4-i- 2П = СгН(
Такое присоединение водорода называют гидрированием. Способность этилена к реакции присоединения объясняют существованием двоиных связей между двумя атомами углерода, которые при присоединении соответствующих веществ превращаются в простые свяли
Молекула С2.Н4 пмеет плоское строение. Угол ИСН, определенный по данным инфракрасного спектра, составляет 120°, следовательно, оп значительно отличается от тетраэдрического угла (110°28'), имеющегося в СН4, С2Йв" и Других насыщенных углеводородах, ‘
Этилен — эндотермическое соединение, одпако теплота его разложения мала 2Ссажа Ц- Ю кхвл.
, Название этилен применяют одновременно и для обозначения соответствующего двухвалентного радикала Н2С—СЙ2. Изомерный радикал НС—СЙ3 называют „ и /х -•
т ил нДен-рд днка л ом.
Исходя из этилена и синильной кислоты можпо получать акриловые смолы, кото? рые цашлпширокое применение в качестве промежуточного слоя для приготовления безосколочпого стекла, как электроизолирующий материал, фармацевтический пластырь, искусственная кожа, лаки, клеящий материал и тому подобное.
Ацетилен СзН2 — бесцветный газ, обычно неприятно пахнущий из-за содержащихся в нем примесей; ядовит, зажженный (температура воспламенения 428°) горит сильно светящимся пламенем. Свет становится ослепительно белым, если использовать горелку, которая дает плоское пламя, в котором вследствие обильного притока воздуха происходит полное сгорание частиц углерода, образующихся в пламени в результате процессов разложения. В других условиях происходит сильное выделение сажи.
Ацетилен — сильно эндотермическое соединение. При детонирующем действии гремучей ртути он взрывает. Разложение можно вызвать й электрической искрой; оно протекает в соответствии с уравнением"
=2С(.а,/и-J-Hg-j-54 ккал.
Ацетилен, находящийся под давлением, особенно сжиженный, иногда взрывает под влиянием ничтожных причин. Но он безопасен, если находится под давлением в несколько атмосфер в смеси с другими газами или в растворе ацетона. Одна часть ацетона растворяет при 15° и давлении 1 ат 25 об. я., а при давлении 12 ат ~ 300 об. ч. ацетилена. Растворимость ацетилена в воде также весьма значительна (1:1при комнатной температуре), еще больше растворимость в спирте (ср. табл. 81).
Растворимость ап стилок а в ацетоле используют для его транспортировки. Его выпускают в продажу и стальных баллонах-, содержащих под давлением 15 ат кизель-: гур, пропитанный раствором ацетилена в ацетоне.
В присутствии платиновой чернп ацетилен, гидрируется водородом, превращаясь в этаж С2Н2+ 4И ~ С2ГГс. При этом расходуется 4 атома водорода, т. е. иа 2 атома больше, чем при гидрировании этилена. Считают, нто в ацетилене атомы углерода соединены тройной связью, которая, присоединяя 4 атома Н, превращается в простую
476
Глава IS
связь
н ,Н -И
н-с 11 i н Н- С; !i 'н Н —( хн
+ =
н-с н II и— L-н С.' чн н—< k J— а и
Еще легче, чем водород, присоединяются галогены, причем максимально 4 атома. Однако при подходящих условиях можно осуществить частичное галотспирование, т. е. присоединить только 2 атома галогена. В первом случае образуются галоидные производные этана, например СНСк— <;НС12— тетр ах лор этан, в последнем — галоидные производные этилепа, в которых сщг: имеется двойная связь, например СНС1—СНС1 — дихлорэтйлен. Ацетилен окисляется спиртовым раствором перманганата калия с образованием смеси муравьиной, угольпой и щавелевой кислот; в кислом растворе в основном образуются муравьиная и угольная кислоты.
Теплота сгорания ацетилена составляет 311,5 ккал/молъ при постоянном объеме. Смесь ацетилена с воздухом чрезвычайно взрывчата. Уже 3,5 об. % ацетилена достаточно, чтобы образовать опасную взрывчатую смесь,
С2И2—молекула линейна; момент инерции ее 23,786-10-40 е-см2, межъядерпые расстояния см. табл. 80. Линейное строение доказано также и для некоторых производных ацетилена, папрнмер II—С = С—С1 и Н—С и С—С slj,
Ацетилен обладает способностью реагировать с солями некоторых тяжелых металлов с образованием труднорастворимых соединений; например, из спиртового раствора AgNOa выпадает белый AgaCa, из аммиачного раствора CuCI — краспо-коричневый СнзСа. Этй соединения называют ацетилидами. Их можно рассматривать как соли ацетилена; хотя ацетилен и не обладает в заметной мере кислотным характером в названных ацетилидах атомы водорода ацетилена замещены металлом. Ацетилиды тяжелых металлов в сухом состоянии крайне взрывчаты. Они детонируют уже При легком потирании. Не взрывчаты ацетилиды сильно электроположительных металлов. Однако их можно получать лишь сухим путем, так как вода их тотчас гидролизует. Способность ацетилидов тяжелых металлов выпадать в осадок из водных растворов обусловлена их чрезвычайно малой растворимостью. Аналогичным образом, получается и ацетилид ртути (П) HgCa.
Ацетилиды щелочных металлов можно получить при действии ацетилена на свободные металлы. При этом сначала образуются первичные или кислые ацетилиды, которые при нагревании, отщепляя ацетилен, превращаются в нормальные или нейтральные ацетил и дЫ, например
К+С2Н2=КИС2-}-V3H2; 2КНС2=К2С2-|- С2Н2.
Ацетилиды ZnCa, CdC2, ВеС2 и^ Л12(С2)3 также можно получить пропусканием ацетилена над нагретыми металлами.
Ацетилид кальция, обычно называемый карбидом кальция, получаемый прн нагревании окиси кальция и угля в электрической печи, имеет важное значение для получения ацетилена. Обычный метод получения ацетилена основан на способности карбпда кальция легко разлагаться под действием воды с образованием ацетилена, т, е. на процессе, подобном гидролизу солей других слабых кислот
Са ' С2 • 211 ОН=Сг11г+Са(ОН)2-|-32 ккал.
Ай алогично реагируют с водой и ацетилиды других щелочноземельных металлов. Разложение водой ацетилидов щелочных металлов, между тем, протекает со взрывом такой силы, что освобождающийся ацетилен тотчас разлагается с выделением
Четвертая, группа периодической системы И7
угля. Нормальный гидролич можно все-таки провести, если медленно пропускать над ацетилидом водяной пар
К2С2-|- 2НОН = 2К0Н 4- С2Нг.
Для получения ацетилена п небольших количествах к карбиду кальция добавляют по каплям воду, Скорость выделения газа при этом регулируется скоростью прикапывания воды. На больших Предприятиях используются щелевые аппараты, в которых карбид порциями вносятся в большое количество воды. Благодаря этому предотвращается опасное нагревание газообразного ацетилена, а также его излшпиее последующее выделенке. Иногда разложение осуществляется под действием мелких брызг воды, которые, испаряясь, поглощают тепло реакции. При этом выпадает Са(ОН)2 в виде сухого порошка, который формуют прессом, обжигают и вновь направляют в к ар бедны е печи. Сырой ацетилен загрязнен аммиаком^ сероводородом и фосфористым водородом. Последний при известных обстоятельствах может способствовать самовоспламенению газа. Загрязнения удаляют промыванием газовой смеси раствором хлористого кальция, который поглощает аммиак, и последующей обработкой смесью окиси кальция и хлорпой извести. Эта смесь окисляет сероводород и фосфористый водород до соответствующих кислородсодержащих кислот, которые затем связываются окисью кальция.
В последнее время ацетилен получают в промышленности во все увеличивающемся масштабе термическим расщеплением насыщенных углеводородов, например СН4. Углеводород, который следует расщепить, нагревают, например, в вольтовой дуге до температуры выше 5000°, а затем быстро охлаждают, подобно тому как в методе «сжигания воздуха»*. Можно показать спектроскопически, что при высокой температуре образуются свободные радикалы С=С, которые затем соединяются с атомами И, давая С2Н2.
В настоящее время большое количество ацетилена используют для синтеза органических соединений (см. ниже). Далее, его применяют при автогенной сварке, а также для целей освещения. Следует обратить внимание на уже упоминавшуюся чрезвычайно высокую взрывчатость смеси ацетилена с воздухом. Как следует из уравнения
2С2Н2+5О2=4СО2-|-2Н2О, наиболее опасна смесь 1 об. ч. ацетилена с 21/в об. ч. кислорода или 12Va об. ч. воздуха.
Если пропускать ацетплен через умеренно нагретую 15%-нуто серную кислоту, содержащую сульфат ртути (II), то в соответствии с уравнением C2II2+ TI2О = С2Н4О** образуется ацетальдегид. Он И получающаяся при его окислении уксусная кислота представляют соб<)й исходные вещества для многочисленных, в высшей степени важных и производимых в большом количестве продуктов промышленности органического синтеза, средн которых наряду с красителями и лекарственными препаратами все большее зпачение приобретают лаки, пленки, волокнистые вещества, пластические массы н искусственные материалы. Катя литическим гидрированием ацетальдегида получают этиловый спирт. В настоящее время значительную часть последнего получают этим методом из карбида кальция (карбидный спирт). При взаимодействии С2Н2 с кислотами, спиртами, фенолами и аминами удается получить «икимеые соединения, т. е. производные радикала СН2= СИ—, которые также представляют собой исходные вещества для получения важных продуктов, например разнообразнейших ценных искусегвениых материалов, которые в настоящее время находят обширное применение для получения воду отталкивающих тканей, для защиты от химически агрессивных Сред, в качестве изоляции электроаппаратов, оболочки для кабелей и проводов, заменителей металла и дерева, в качестве лака, негорючих красок, прессмасс- для изготовления пуговиц, коробок, чашек и т. д. Как показал Ньюленд (Nieuwlarid, 1931), при каталитическом действии комплексных соединений Сп(1) из С2Н2 можно получить винилацетилен СН2 = СН—CsCII, а из последнего нутеМ гидрирования — бутадиен СН2= СН — —СГ1=СН,. При полимеризации бутадиена образуется каучук бу на, а прп присоединении к нему НС1 (с использованием СпС1 в качестве катализатора) — хлоропрен СИ2= СИ—GC1 — СН2, полимеризация которого ведет к каучуку дюпрен, получаемому прежде всего в США и СССР. Изделия из каучука буна по некоторым свойствам, таким,
* Для получения окислов азота.— Прим. ред.
** Синтез Кучерова.—Прим. ред.
478 Глава 12
как терм о стон к ость, маслостойкость, износостойкость, превосходят изделия иэнату-рального каучука. Каучук дюпрен отличается прежде всего' масло стойкостью.
Бутадиен, необходимый для синтеза каучука буна, получали вначале (с 1929 г.) «четырехступенчатым методом»:
2С2П2 2СИ3-СНО —> СН3-СН(ОН)-СН2-СНО Л
Ацетальдегид Альдоль
. CIIt-CH(OII) i.lbC|J2OH С1Г2_— сн—сн = сн2
Б утиле и гликоль Бутадиен .
При этом, а также при разработанном позже Ныолепдом «виниловом синтезе» (см. выше) на 1 моль бутадиена расходовалось 2 моля относительно дорогого ацетилена. Если же использовать разработанный в 1937 г. Penile (Перре) алкиноЛьный -синтез, то необходим лишь 1 моль ацетилена. Алкинольным синтезом называют присоединение ацетилена к альдегидам в присутствии ацетилида меди Cu2C2 ^качестве катализатора,' которое в зависим,юти от условий опыта приводит к одно- или двухатомным сцир- ' там с С=С-связью (алкииолам), Так, при присоединении формальдегида (получаемого синтетически из водяного газа) к ацетилену (при температуре около 100° и давлении 5 ати) сначала образуется бутиндисл НО-СИ2~-С==С—СН2-ОН. Последний легко .гидрируется в водном растворе в присутствии пи кол ел ого катализатора дб бутандиола НОСН2—СН2—С1Г2—СН2ОГГ, и;i которого при отщеплении воды (в присутствии фосфорной кислоты и фосфата натрия в качестве катализатора) образуется бутадиен СН2=СП-СН==СН2.
i Из ацетилена? и формальдегида, а также других альдегидов и кетонов аналогия; иым путем через алкинодьный синтез можно легко получить в технике многие другие-органические соединения, которые находят разнообразное применение, например для Производства искусственных материалов, растворителей, вакс, душистых веществ п фармацевтических препаратов. Техническое значение имеет также аналогично протекающий синтез соединений из ацетилена и органических аминов.
Полимеризацией ацетилена в присутствии соединений никеля-в качестве катализатора можно получить циклические углеводороды, в частности циклорктатетраен СsИ8, очень реакционноспособное вещество, которое может служить в качество исходного продукта для производства множества других органических соединений. Этим путем из ацетилена можно непосредственно получить й бициклические углеводороды, например построенный из одного пяти- и одного семичленного кольца темне синий азулен С10Н3.
СО и Н2О также могут непосредственно присоединяться к ацетилену в присутствии кат а ли зато рои (карбонилы металлов). В простейшем случае образуется акриловая кислота (O2I12+ GO Н2О — П2С=СП— СООИ) или, если использовать вместо Н2О Спирты*амины и т. д., соответствующие ее производные. Прн несколько ипыхуслсн виях получают гидрохинон CfiH4(UH)2 или из С2Н2 и только СО (без Н2О) — циклические кетоны, например е.идриндон СцЩО.
Карбид кальция, СаСс - собственно ацетилид кальция (ибо его-можно производить от ацетилена), обычно называемый просто яг/рбиАли, образует в чистом виде бесцветную кристаллическую массу удельного- , веса 2,22, Оп нерастворим пи в одном из обычных растворителей. Водой разлагается с образованием ацетилена. Из окислов металлов оп выделяет при высокой температуре свободные металлы или взаимодействует с ними» с. образованием карбидов соответствующих металлов. и окиси кальция. С азотом карбид кальция при нагревании переходит в цианамид кальция CaCN-r (см- стр. 504). f
Технически карбид получают нагреванием жженой извести с коксом; или. древесным углем в электрических печах. Элёктр’оды состоят из боль-цптх угольных брусков, через которые пропускают ток до 50 000 а.
При получении в технических масштабах на каждый Килограмм 85% -ного карбиДа расходуется1 около 1 ке едкой извести, 0,7 «а кокса и 2,8--3,5 квт-ч электроэнергии.
Для получения карбида в небольших Количествах лучше всего поступать следующим образом: в плавильном тигле средней величины стачивают наждачным кругом дно,, так что образуется отверстие, через которое вставляется уголь дуговой лампы; толщиной 13—15 лея (рве. 85).
Четвертая группа периодической системы 479
Верхнюю часть угля вдвигают приблизительно на 2 .и,и в тнгль, укрепляют его в этом положении при помощи глнпы и соединяют через сопротивление и амперметр с отрицательным полюсом электрической сети. Анодом служит твердый заостренный “уголь длиной 10—15 с.в, толщиной 7 укрепленный зажимом на стеклянном штативе, который для удобства га ;
передвижения угля вверх п вниз целесообразно снабдить а’’"
зубчатым механизмом. Реакционную смесь приготовляют, Ц ___
тщательно перемешивая 15 г тонко измельченного порошка ркиси кальция о 2и г порошка древесного угля. Сначала насыпают очепь немного этой смеси и включают ток силой примерло 1.2—14 <?. Вместо постоянного тока можно пользоваться п переменным. Но время опыта, продолжающегося около 10—15 мин, понемногу подсыпают смесь. Кроме того, тцгель следует закрыть асбестовым листом с проделанных! в нем отверстием. Расстояние между углями, ранное вначале 1 см, во время опыта доводят до 5—6 см. Количество карбида, получаемого при помощи такого простого устройства, достаточно для приготовления нескольких литров ацетилена.
Рис. 85. Маленькая печь для получения карбида кальция.
Помимо получения ацетилена, карбид кальция расходуется прежде: всего для получения цианамида кальция. Кроме того, его используют в качестве: восстановителя в металлургии.
Чистый СаС2 обладает, как показал Франк (Franck, 1937), решёткой ипой структуры, чем загрязненный примесями технический карбид. Последний кристаллизуется,-: образуя тетр агональную гр ан ецентриро ванную решетку, так же как чистый ,SrC2. и ВаСг (см. рис. 88, стр. 507). Карбид кальция,, содержащий CaCNs, образу единую решетку. Чистый СаС2 легко, а технический с большим трудом превращается в CaGNj.
Галогениды
Четырехфторпстый углерод. CFj, бесцветный газ (т. кип. —128,0?, т. пл. —183,6°, вес 1 л 3,94), можно получить непосредственным соединением составных частей или действием AgF на ССД при 300°. Он плохо растворим в воде, чрезвычайно инертен к химическому и термическому воздействию и позтому пригоден в качестве Запирающей жидкости для тензиметров при низких температурах.
Некоторое количество более богатых углеродом фторидов было* недавно иолучоио Саймонсом (Simons) ’i ?
G2Fe C3F, СДю ‘ CeFI2 C7FU
Т. кип., °C -78,2 —38 — ,7 4-23 -|-5f -|-89
Г. пл., °C —100,6 —183 — —-12 : [-58 —"
Удельный вес, теплоту испарения и другие свойства C2Fe (гексафтороэтан) см.: Расе, Aston, J. Am. Chem. Soc., 70, 566, 1948; C6Ff2 (додекафтороциклогексап) ^кристаллизуется кубическп-гранецентрированно (а~ 10,00 А, диаметр : молекулы 7,07 А); ниже—100° он’ претерпевает изменение состояния, которое вызвано, вероятно, ’исчезновением молекулярного вращения [С h г i s toff ё г is , L 1 ng a f el ler, Cady, J. Am. Chem. Soc., 69, 2502,1947].
. Четыреххлорцстый углерод. CCli — бесцветная, сильно светопреломляющая, негорючая жидкость со сладковатым запахом и наркотическим действием. Он обладает удельным весом 1,593, кипит при 76,7° (давление пара при 0е: 33,4 мм ртст) и затвердевает при —22,87°. [При —47,66° он имеет точку перехода, которую Джонстон (1934) рекомендовал использовать для градуировки термометров.] Растворимость ССк в воде очень незначительна (0,08 г в 100 г воды при 20°), диэлсктричрская проницаемость низка (2,3 при 18°), светопреломляющая способность высокая (показатель преломления к—1,463 для желтого спета).
480
Глава 12
При обычной температуре четыреххлористый углерод химически инертен. Он не реагирует ни с основаниями, ни с кислотами, в том числе и с концентрированной серной кислотой. Между тем он заметно взаимодействует с некоторыми металлами, например с железом и алюминием. В присутствии этих металлов он постепенно разлагается водой по уравнению
СС144-2НгО = СОи-р 4HCI, в то время как в ином случае это превращение при обычной температуре в заметной мере не идет.
Четыреххлористый углерод очень хорошо растворяет органические вещества. Со спиртом и другими органическими жидкостями оп смешивается во всех отношениях. Поэтому его часто используют в лаборатории, и особенно в технике, как растворитель жиро а, масел, смол и т. п. Он служит также средством для тушения поя:аров. В медицине его применяют как анестезирующее и глистогонное средство.
Четыреххлористый углерод получают хлорированием сероуглерода CSa в присутствии переносчиков галогена, например хлорида марганца (II)
CS2+3C12=CC14-|-S2C12.
Образующаяся при этом хлористая сера S2C12 в присутствии подходящих катализаторов, например FcS, и при умеренном нагрела ним (примерно до 60°) также реагирует с CS2: 2SECl2-p CS2= СС14+ GS. Процесс можно вести таким образом, что обе реакции будут идти параллельно. Выпавшая сера возвращается в процесс как исходный продукт для получения СВ2. Очистка четырех хлористого углерода осуществляется промыванием калийным щелоком, за которым непосредствешю следует фракционированная разгонка.
Четырехбромистый углерод. СВг4 получают из СС14 и А1Вг3 при 100?. Он выпадает из растворов в виде моноклиппых кристаллов, которые при 47° превращаются в кубические; т. пл. 93,7°, т. кип. 189,5°. В воде он плохо растворим. При нагревании с водой в запаяппой трубке до 200° протекает гидролиз с образованием СО2 и 4НВг.
Четырех подпетый углерод. С14 модою получить взаимодействием СС14 с А113 или Bilg< Темно-красные кубические кристаллы (уд. вес 4,32, т. пл. 171°), сублимирующиеся в вакууме нрп 90—100°. При более сильном нагревании происходит отщепление пода. То ;т;с происходит и па солнечном свету, причем образуется С214.
Расстояние С<—>-1 в молекуле СГ4 равно 2,12 А (о межатомных расстояниях в других галогенидах углерода см. табл. 61, стр. 346). Правильная тетраэдрическая структура CF41 СС14, СВг4 и С14, также как и СН4, подтверждена физическими методами (дифракция электронов, спектры комбинационного рассеяния),
О смешанных еалогенидах CCIFg, GC12F2 н т. Д. см. Ruff, Z. anorg; Chem., 201, 245, 1931; 210, 173, 1933, а также Hauptschci n, J. Am. Chem. Soc., 74, 1347, 1952. CC12F2, который технически получают при действии HF на СС14 в присутствии SbCl3, применяют в холодильных мат и и ах под названием «фрсот или «фри ген».
Другие фтороугле родные соединения также приобретают все возрастающее техническое значение, панример трифторуксуспая кислота,, CF3COOH (бесцветная жидкости, т. кип. 72,5°), техническое иолучецие которой осуществляют электрохимическим фторированном в жидкой HF. «Перфторированные» органические соединения, т. е. соединения, и которых все атомы Н, за исключением связанных с атомами О, замещены на F, часто отличаются большой устойчивостью к термическим и химическим воздействиям. Из производных этилена C2F4 (т. кип. —76е) н C2C1F3 (т. кип. —28°) путем полимеризации в зависимости от условий получают ;кидкйе, иногда воскоподоб-пьте продукты, которые используют как гидравлические масла, смазывающие и смягчающие средства или твердые искусственные материалы (например, хостеф.юн), которые оказались пригодными прежде всего для изготовления химической аппаратуры. Органические красители, которые вместо СН3- содержат СР3-группы, обладают поразительной чистотой тонов. В качестве примеров перфторированных соединений углерода с относительно длинными цепями можно указать перфторглутаровую (НО)ОС’ -GF2-CF2-CF2-CO(OH) и перфтор адипиновую кисло™ (НО)СО CF2'CF2'CF2-CF2- СО(ОП), которые были получены Хендриксом (Hendricks, 1951). Заслуживают упоминания также синтезированные Вигелоу (Bigelow, 1951) перфторированные амины;
Четвертая, группа периодической системы
CF3<CF2-NF2 (перфторэтпламин) и CF2= NF (перфтормстилепимин). Аипщрид перфтору ксу си ой кислоты. (С F3CO)->O (бесцветная жидкость е т. кип. 39’) оказался ценным вспомогательным средством if препаративпоп органической химии.
Кислородные соединения
а) Окислы
Углерод образует два простых окисла. Продуктом полного его сгорания является двуокись углерода СОз. При неполном сгорании образуется окись углерода СО.
Теплоты образования обоих окислов (при 20° и постоянном давлении) составляют
СграФи^+^/гОг^СО 4-26,64 ккал..
СО-}-V2O3=CO2 -|-67,75 ккал.
СграфитЖ Оа = COg^" 94,39 ккал
Следовательно, тепловой эффект образонапия окиси углерода из угля и кислорода много меньше, чем тепловой эффект дальнейшего окисления моноокиси до двуокиси. Это объясняется тем, что для разрыва связи между атомами углерода'» графите требуется затратить значительную энергию. Поэтому из величины энергии, которая освобождается йри объединении атомбв С и О в молекулы СО, вычитается (помимо энергии диссоциации для молекул 00 очень значительная теплота сублимации углерода, которая составляет около 140 ккал/г-атом.
Еще более бедная кислородом недокись углерода С3О2 была получена Дильсом (Diels) при отщеплении воды от малоновой кислоты под действием пятиокисй фосфора.
i и он.
о = с —С,-С = О — 2Ц?О —о = с=с=с==о
___1___I_ Недокись углерода ]f() [j ; (карбо дика рб<> ни л) 4
'М.ллоиовап itticjioTii
Строение этого соединения следует из способа иго получения и характерных ..реакций. Его линейная структура была подтверждена методом электро по графи и н спектрами комбинационного рассеяния (см. гл. 9). Недокись углерода представляет собой бесцветный удушливый газ (т. кип. 7’1; т. пл. —107°), который в неочшценпом состоянии легко полимеризуется, превращаясь в красное аморфное вмце<-,тно.,.В чистом состоянии ома достаточно устойчива. Как показал Клеменс (Klenicns, 1934), при подходящих условиях она разлагается по уравнению С3О3 ^± СО.,Ц-Сг. При этом газовая фаза приобретает карминово-красную окраску, относительно которой ещь не установлено, вызвана ли она промежуточно появляющимся газообразным углеродом (дикарбон С2) или образованием аэрозоля углерода (ср. т. II). На стенках осаждается твердый углерод (чрезвычайно мелкий графит), окрашенный в пурпурно-красный цвет.
Двуокись углерода. СО а — бесцветный, негорючий газ со слабым кисловатым запахом и вкусом. Он значительно тяжелее воздуха (вес 1 л 1,9768 г при 0е и 760 мм pni ст) удельный вес (по отношению к воздуху) 1,529. Поэтому двуокись углерода собирается на дне емкостей, в которых она образуется (бродильпи, колодезные шахты и т. и.). При входе в такие помещения следует соблюдать осторожность, так как двуокись углерода ire поддерживает дыхания. Она легко сжижается при 20° давлением 56,5 образуя бесцветную, недвижную ’жидкость с удельным весом 0,766. Ее критическая температура 31,3е, критическое давление 72,9 ят.м, критическая плотность 0,464. При обычном давлении двуокись углерода сублимируется При —78,48°. Под давлением 5 атм твердая СОа плавится при —56,7°.
31 г. Реми
482
Глава 12
Кристаллическая двуокись углерода образует молекулярную решетку, в которой атомы С заяимагот узлы гр ан спен три ро ванной кубической решетки с длиной ребра а = 5,63 А. Оба атома О каждой молекулы лежат на одной прямой с атомом С, очень близко к последнему (С<—^0=1,05 А) и направлены по пространственным диагоналям. Свободная молекула также вытянута по прямой и построена симметрично: О=С=О. Межъядерное расстояние С^—*0 в ней 1,13 А.
Теплота плавления двуокиси углерода 2,24 хкал/молъ теплота испарения 6,0 ккаа/моль (при —56°). Жидкая двуокись углерода — плохой проводник электричества; она обладает лишь незначительной растворяющей способностью.
Вода способна поглощать значительные количества газообразной двуокиси углерода. В 100 ч. воды растворяется
при 0° 10° 20’ 25® 60° *
171 119 88 75,7 38 об. ч. СО2 при давлении 1 ат
Зависимость моляр пой растворимости СО2 в воде от абсолютной температуры 23§5 73 выражается уравнением Харнеда и Дэвиса (Hamed, Davis, 1943): log £ — -~--
—14,0184-Ь 0,0152642 2'.
Химически двуокись углерода достаточно инертна. Только с сильными основаниями она, как ангидрид угольной кислоты., (см, стр. 489), энергично соединяется, образуя карбонаты.. При высокой температуре она реагирует также с сильно электроположительными металлами, такими, как К, Mg, Zn, отдавая им полностью или частично свой кислород. При пропускании СОз над раскаленным углем образуется окись углерода по уравнению СОз+С^2СО [равновесие Будуара (Bondouard)].
В состоянии равновесия при давлений 1 атм в газовой смеси, с о пр икасаю шел ся
с углем, содержится [ло опытам Будуара и Майера (Меусг)], % । *
при 1=450° 600° 650° 700° 750° 800° ООО® 1000°
СО2 98 77 61,5 42,3 24,7 6,0 2,8 0,7
СО 2 23 38,5 57,7 75,3 94,0 97,2 99,3
Ниже 800° равновесие устанавливается только л присутствии катализаторов, напрп-мер металлг1ЧГ'(ч;иго железа. Содержащиеся в угле примеси также могут действовать каталитически. На основании этого п пз других данных выясняется вависимость энергии активации, необходимой для реакции, от сорта угля [подробнее см. Wic.ke Е., Z, Eleklrochem., 57, 636, 1953, и 60, 774, 1956].
В небольших концентрациях двуокись углерода не ядовита. В крови постоянно содержится СОг, которая образуется в организме в результате процессов окисления; СОа выделяется при дыхании. Содержащаяся в крови двуокись углерода возбуждающе действует па дыхательный центр. Однако длительное пересыщение ею крови вредно. При вдыхании больших количеств двуокиси углерода наступает не только удушье из-за недостатка воздуха, но и целый ряд расстройств в организме, которые даже и после устранения острой опасности могут привести к смерти.
Как уже было указано, двуокись углерода содержится в незначительных концентрациях (3 : 10 000) в .воздухе. В некоторых местностях, особенно часто неподалеку от вулканов, она выделяется из недр земли. Вода многих минеральных источников (углекислые источники или ключи) при обычном давлении так пересыщена двуокисью углерода, что большая часть ее выделяется при выходе на поверхность земли. В небольшом количестве двуокись углерода содержится во всякой природной воде. Двуокись углерода, связанная с окислами металлов, встречается в природе в громадных количествах в форме карбонатов, это прежде всего карбонаты кальция и магния (ср. гл. 8).
Четвертая группа периодической системы 483
Двуокись углерода получают в лаборатории чаще всего- разложением мрамора соляной кислотой в аппарате Киппа, иногда также нагреванием бикарбоната натрия или магнезита.
В технике получают значительные количества двуокиси углерода, являющейся побочным продуктом обжига извести. Получаемый таким образом гав сильно загрязнен азотом. Для получения чистой двуокиси углерода ее пропускают через башни, в которых по кускам кокса стекает раствор карбоната натрия или калия. Последний абсорбирует СО а с образованием бикарбоната и вновь выделяет ее при кипячении. В качестве абсорбента еще лучше использовать раствор этаноламина НО СН-2-СНз• NHa, который при нагревании выделяет СОа, поглощенный при обычной температуре (Жирботол-процесс). Аналогичным способом двуокись углерода можно получить из коксового газа. В некоторых местностях для добычи двуокиси углерода используют'природные источники газа (например, около Герста в Вестфалии и Гёцпингепа на Рейне) или же ключи (в Эйфеле, Вестервальде, Таунусе). Значительное количество двуокиси углерода выделяется при спиртовом брожении: только в Германии — свыше 100 млн. .к3 ежегодно. В некоторых странах углекислый газ брожения также используют.
Двуокись углерода применяют для производства минеральных вод. Для этой цели в торговлю поступает сжиженная двуокись углерода в стальных бомбах. В медицине СОз используют для приготовления углекислых ванн. Ее применяют, далее, при тушении пожаров и непосредственно как средство, гасящее пламя, и как средство, создающее давление для разбрызгивания других тушащих огонь жидкостей. Двуокись углерода1' очень удобна и в качестве нагнетающего газа для перекачки огнеопасных жидкостей.
Двуокись углерода рекомендуют и в качестве удобрения, так как она является тем питательным веществом, которое растения усваивают иа воздуха. Можно показать, что обогащение воздуха двуокисью углерода сильно способствует росту растений. Углекислотные удобрения при подходящих условиях ценны прежде всего для оранжерей. Твердую двуокись углерода все больше применяют в качестве охлаждающего агента — «сухой лед», который развивает на каждую единицу веса вдвое большую «х ладо производительность», чем лед из воды, и обладает еще тем преимуществом, что испаряется без остатка.
Окись углерода СО образуется в процессе сжигания угля при недостаточном притоке воздуха. Это ядовитый газ без цвета и запаха, который не поддерживает горения, однако сам является горючим. Вес 1 л окиси углерода составляет 1,25001 г (при 0° и 700 мм рт ст), уд. вес (относительно воздуха) 0,967. Следовательно, окись углерода несколько легче воздуха. В воде она растворима лишь слабо (3,3 об. ч, СО в 100 ч. воды ири 0°; 2,3 — при 20°), лучше — в спирте (20,4 об. ч. в 100 ч. спирта в интервале 0—25°). При обычной температуре окись углерода нельзя перевести в жидкое состояние. Ее критическая температура —140,2°, критическое давление 34,6 ат, критическая плотность 0,311. При атмосферном давлении окись углерода сжижается при —191,5°, затвердевает прн —204,0°.
СО (как и N2) кристаллизуется, образуя кубическую, объемно-центрированпую молекулярную решетку с «= 5,63 А. Межатомное расстояние С —>• О в твердом состоянии 1,06 А, в газообразном 1,26 А. Дипольный момент 1,2•10"11 ед. CGS. При ^-211,6° кубическая модификация СО превращается в гексагональную. Теплота превращения составляет 0,151, теплота плавления 0,200 и теплота испарения 1,44 ккал/моль.
Раньше считали, что углерод в окиси углерода двухвалентен, и приписывали ей формулу С=О. - Однако прочность связи С=6 я окиси углерода много больше.
31*
48',
Глава. 12
чем этот следовало бы ожидать для двойной: свяви .(ср. табл. 80, стр. 468). Энергия связи превосходит здесь даже энергию тройной связи; то же справедливо относительно валентно-силовой константы. На основании далеко идущих совпадений в физических свойствах CG и N3 Лангмюр уже в 1919 г. предположил, что обе эти молекулы изосте-рпчньт и в соответствии с этим в молекуле СО, так же как в молекуле Sr2, имеется тропная Связь. Отсюда следует, что молекула СО обладает сильно полярным характером (ср. стр. 369),что, одпако, находится к противоречии с ее очень малым дипольным моментом, установленным экспериментально. Это расхождение можно устранить предположением, что в' молекуле СО имеется «ОДново-.иеяаодчссшш резонанс .между двумя 'формами с противоположно направленными, дипольными моментами. Полипе, который сопоставляет молекулу СО с карбонильной группой альдегидов и кетонов и допускает для последней мезомерию одной полярной и одной неполярной форм*, заключает, что в молекуле СО резонируют даже три формы; помимо двух противоположных полярных, рще одна неполярная форма : :
Мсиомерин ветоков
.Мезомерия окиси углерода
IL 1
>С—СП
R _
)С = О R
I С—О I | С=О ic. - op
! Существование полярной формы окиси углерода с простой свинью объясняется сильно электро отрицательным характером кислорода, а формы с тройной связью — увеличивающейся с числом связей энергией связи, В соответствии с этим, следует ожидать, что обе формы обладают приблизительно равными энергиями и что тем самым даются условия для волново-механического резонанса между ними (ср. стр. 326),
Сходство между молекулами СО и N2 проявляется и в полосатых спектрах. Это относится не Только к основному состоянию обеих молекул, ’S - состоянию (в претиво-положность 3— -основному состоянию для молекул SO, и также О2), ио п в эначитедъ-мок степени к энергетическим уровням их возбужденныхсостояний.
Далеко идущая айалотпя между мол скулами СО я N2 лучше всего отражается формулой С=О, При этом не следует только упускать из виду то обстоятельство, что она служит сокращенным символом для мезомерпого состояния, в котором резонируют две, а возможно, дюке три разные форма СО**. ......
Ядовитость окиси углерода объясняется тем, что она соединяется с красящим веществом крови — гемоглобином, которое поэтому теряет способность соединяться с кислородом. Сродство гемоглобина к окиси углерода гораздо больше, чем к кисло-' роду. Из воздуха с содержанием 0,1% СО, в котором, следовательно, СО и О 2 находятся в отношении 1 : 200, кровь поглощает оба газа в равных количествах. Поэтому достаточно самой ничтожной кон шли рации СО, чтобы значительно понизить способность крови воспринимать кислород к Т!?м самым вызвать удушье. Однако прп вдыхаппи чистого воздуха или, еще лучше, чистого кислорода окись углерода постепенно удаляется п,ч крови. Еелщ это сделать своевременно, отравление окисью углерода по оста-,, вляст длительных вредных последствий. Содержанием окиси углерода объясняется' ядовитость светильного газа и других газов, образующихся при горении в условиях недостаточного доступа воздуха.
Прежде предпринимались успешные попытки снизить ядовитость светильного газа удалением из него окиси углерода (каталитическое превращение с водяным паром (ср. стр. 46). Однако в настоящее время иб экономическим соображениям к светильному газу, как уже упоминалось, стали примешивать водяной газ, несмотря пято, что при этом повышается содержание в пем Окиси,углерода. В противогазах (например, для пожарных) для очистки вдыхаемого' воздуха от СО используют окисление его до СО2 при помощи катализатора, называемого гопкалитом и состоящего из окисей марганца и меди.- Этот метод был разработан в США в первую мировую войну. Он позволяеттакже определять очень малые количества СО.', ооразующуюся при этом СО2 либо улавливают , раствором Ва(ОН)2 и титруют, либо измеряют теплоту окисления (чаще всего авто- : магической регистрацией повышения температуры). >
При пропускания СО в смеси с О2 в аммиачный раствор карбоната меди также можно осуществить окцелеттне СО [с образованием (NH4)2COSJ; однако такое окислспио СО протекает очень медленно.
* Полипг сделал вывод, что в альдегидах и кетонах неполярная форма, соответствующая их классическим формулам, находится в резонансном отношении, с полярной формой, на основании высокого Дипольного момента :Этих соединений (пацример, 2,70 ед. CGS для ацетальдегида, 2,85 ед. CGS — для ацетона).
** О резопапсо молекулярных форм см. прим. ред. на стр. ,327.—Прим, ред..
Четвертая группа периодической системы, 48а
Окись углерода не является настоящим ангидридом кислоты. На гидроокиси щелочных металлов опа действует только при повышенной температуре, соединяясь с ними с образованием формиатов, и соответ-ственно с метилатами щелочных металлов с образованием ацетатов, Эти соли в отлично от самой окиси углерода являются производными формально четыре,г валентного углерода.
СО !-NaOH = Na(CO2IJ]r C0-|-^a0CH3= Na[COa.CIT3l •Годин ат натрия Метилат натрия Ацетат натрия
Если окись углерода в смесис водяным паром пропускать через окись кальция, нагретую до 400°, то получается карбонат кальция и выделяется водород СО+СаО+НаО—СаСОз+На. Сильно нагретый карбид кальция также присоединяет окись углерода с образованием карбоната
CaCs+3CO=CaCOs+4C-
На воздухе окись углерода воспламеняется при температуре около 700° й сгорает, образуя двуокись 2CO-J-O2^2COa. Эта реакция, однако, приводит к равновесию, которое при высоких температурах сдвинуто влево,
Константа Кс уравнения закона действии масс Кс = Нернста и Вартсщберга, атакя;с Бьеррумщ
[С0р[021 [W
равна по данным
при i—t205°
Ке= Ю-12Л5 100а = 0,032 для р— 1 атм
2367°
1О-4’51
21;0
2606° 2S43°
51,7 76,1
В последней строке в объемных процентах даны степени диссоциации окиси углерода, рассчитанные на основании экспериментальных данных; исходя: из них можно вычислить константу диссоциации по формуле (13), выведенной па: стр. 75 для аналогичного равновесия. Из приведенных данных следует, что двуокись углерода при атмосферном давлении и температуре шлпп, 2600° уже наполовину диссоциирована. При повышении давления степень диссоциации двуокиси углерода уменьшается, что следует уже из уравнения реакции ца основании принципа Ле Шателье. Зависимость степени: диссоциации от давления ц температуры количественно выражается уравцёнием (13), стр. 75. При температурах выше'5000° Й давлении J/io алии СО2 практически полностью расщеплена на GO и О2.
Соответствующее равновесие устанавливается также при взаимодействии с водяным паром
qtLCG3-(-Ha; Кс = (РавновеСие водяного газа)
К=0,60 0,90 1,0 1,7 2,6 3,45
при.1 = 700® 800° 830° 1000° 1200е 1400°
Посгтгтт.ку эта реакция протекает без изменения объема, К: пе изменится, если вместо: Концентраций подставить давления. При 830° в реакционной смеси содержатся равные количества всех четырех компонентов, участвующих в реакции. При более низких температурах равновесие сдвигается вправо; следовательно,'тогда окись углерода является более сильным восстановителем; при белое. высоких температурах, напротив, более сольным восстановителем является водород.
Окись углерода при нагревании соединяется с серой, образуя серо-окись углерода COS; при облучении или; в присутствии катализаторов (платиновая чернь или животный уголь) она соединяется с хлором с образованием хлорокиси углерода (фосгена) СОСЬ. Пропуская окись углерода, смешанную с водородом,: пад тонкоизмельченным никелем при 250° получают метан CO-J-3H2 = CH4+H2O.
486
Глава 12
При нагревании окись углерода соединяется с некоторыми металлами, прежде всего с Fe, Ni и Со, с образованием карбонилов металлов, например
Ni+4CO = Ni(CO)4. “
Тетракарбонил ни нем
На металлический калия окись углерода действует уже ири 80°, образуя калийную соль гексаоксибензола — и рои вводное бензола, обладающее формулой
КО ОК
.1 f
С6овке = КО—V>c—ок
кА ок
Это белые кристаллы, легко превращающиеся в «трихипон» C(OS, изомерный окиси углерода.
Многие окислы металлов при нагревании с окисью углерода восстанавливаются до металлов. PdCla в водном растворе реагирует с окисью углерода уя$е при комнатной температуре: СО -f- PdCls-f- НзО = СОа+ Pd + -f- 2НС1. Восстановление даже в незначительных количествах обнаруживается по появлению темной окраски, обусловленной выделением металла, и является поэтому чувствительной реакцией для определения СО. Полу-хлористой’медью в концентрированном солянокислом или аммиачном растворе СО связывается с образованием продуктов присоединения.
Образующееся в солянокислом растворе гг легко опять распадающееся соединение имеет состав СиС1‘СО'2НгО. Его впервые выделил Манию (Manciiot). Поглощение окиси углерода аммиачным раствором попу хлористой меди (более полное, чем в случае солянокислого раствора) используют для удаления СО из газовых смесей, например при анализе.
Другие данные о соединениях СО с металлами побочных подгрупп см. т. II.
Для получения окиси углерода в лаборатории обычно прибавляют по каплям муравьиную кислоту к концентрированной серной кислоте, нагретой до 100°: НСОаН — НгО = СО. Другой способ — действие концентрированной серной кислоты на цианистый калий или на тонкоизмель-ченный порошок желтой кровяной соли..
2KCN-|-2H2S044-2H20 = K2S04+(NH4)2S04-|-2CO;
K4IFe(CN)e14-6H2SO4+6H2O=2K2SO44-FeSO4+3(NH4)2SO4 + 6CO.
Окись углерода выделяется в качестве побочного продукта при различных технических реакциях, например при получении водорода частичным окислением метана (ср. стр 47) и при получении карбида кальция (стр. 478). В очень большом количестве СО производится в технике в форме генераторного и водяного газов.
Генераторный газ, называемый также воздушным газом, представляет собой смесь, которая образуется при неполном сгорании каменного угля или кокса в больших печах, так называемых «генераторах». Его примерный состав; 25% СО, 70% N2, 4% СО2 и незначительные количества Н2, СИ4 и О2. Калорийность генераторного газа составляет 800—1100 ккал/м3, смотря по тому, получен он из кокса или иа каменного угля. Воздушный газ, полученный из каменного угля, обладает большей калорийностью вследствие большого содержания в нем водорода и метана.
Четвертая группа периодической системы
487
Состав, подобный составу воздушного газа, имеют и колошниковые газы домен. Их калорийность равна примерно 750 ккал/м«.
Водяпой газ получают, пропуская водяной пар над сильно нагретым коксом или антрацитом. При этом идет реакция
С -г П2О + 28,1 ккал = СО + Н3. (1)
Указанное количество тепла вдет на окисление черного углерода водяным паром. Образующаяся СО взаимодействует частично с водяным паром, давая СО2. Одпакб при высоких температурах равновесие
СО + H3G - СО2+ Н2,
как видно из цифровых значений константы равновесия, приведенных на стр. 485, сильно сдвинуто влево. Кроме того, в связи с тем что идет реакция (1), концентрация водяного пара в смеси исчезающе мала. Поэтому «идеальш.ш» водяной газ содержит лишь следы СО2 и состоит из равных по объему частей СО и Н2- Его калорийность около 2800 ккал!м3. Практически водяной газ состоит в общем примерно из 40 об. ч. СО, 50 об. ч. Н3, 5 об. ч. СО3, 4—5 об. ч. N3 и некоторого количества СН^. Поскольку образование водяного газа связало со значительной затратой тепла, необходимо через несколько минут работы («холодное дутьем) вновь подвергать уголь горячему дутью. Это осуществляют продуванием воздуха, причем в зависимости от скорости, с которой продувается воздух, и высоты коксового слоя получают либо воздушный газ, который улавливают отдельно, либо в результате полного сгорания лишь двуокись углерода, которая улетучивается. Последнее в общем выгоднее, так как вследствие большего выделения тепла при полном сторанйи меньшее количество угля расходуется при горячем дутье. О применении генераторов Винклера для производства водяного газа см. стр, 757.
Генераторный газ часто по „чу чают в технике для производства газового топлива. Его нельзя использовать вместо светильного газа из-за небольшой калорийности. Водяной газ применяют в технике в тех случаях, когда необходимо создать очень горячее пламя. Несмотря на то что водяном гав обладает вдвое меньшей калорийностью, чем светильный газ, его пламя много горячее, чем пламя последнего; папример, в нем плавится платиновая проволока, что невозможно в обычном пламени светильного газа.. Более высокая температура пламени во дни ого газа объясняется тем, что продукты сгорания водяного газа занимают меньший объем, чем продукты сгорания светильного газа. Следовательно, тепло концентрируется на более узком пространстве. Калорийность водяного газа можно значительно поднять методом карбюрирования. В США карбюрированный водяной газ часто используют для целей освещения. В Европе также в настоящее время к светильному газу часто примешивают водяной газ.
Смешанный газ. Оба процесса, необходимые для производства водяного газа — «горячее дутье» н «холодное дутье»,— можпо объединить, продувая одновременно водяной пар и воздух через уголь. Полученный таким образом смешанный газ называют по имени его изобретателя «газом Дауста» или, поскольку он нашел наибольшее при- ' мепеннс в газогенераторных машинах,— «силовым газом». Как правило, оп состоит примерло из 30% СО, 15% Н3, 5% СО2 и 50% N2 и обладает калорийностью около 1300 т.кал/м5.
Чистую окись углерода можпо выделить из генераторного газа путем контактирования его под давлением с солянокислым раствором CuCI. который поглощает СО из газовой смеси и вковь выделяет ее прн снижении давления. Чистую окись углерода подучают в технике также путем пропускания двуокиси углерода пад раскаленным углем.
Помимо использования в целях отопления (в форме светильного, генераторного, водяного или смешанного газа), окись углерода применяют для восстановления руд, рафинирования никеля (см. т. II), получения фосгена и безводных хлоридов металлов, например AICI3. Однако наиболее существенно то, что в последнее время опа является важным исходным продуктом для ряда индустриальных синтезов. Если смесь СО с Н2 пропускать над подходящими катализаторами, то в зависимости от условий образуются различные продукты гидрирования. В то время как при обычном дав лепив гидрирование над пнксяевым катализатором приводит к синтезу метана, при применении других Подходящих катализаторов при обычном давлении могут образоваться смеси жидких углеводородов (синтез бензина), а при повышенном давлении — либо смеси высших спиртов, альдегидов, кетопов и т, д., которые годятся в качестве моторного топлива («сиптол» Ф. фнщера), либо этим путем можно получать метиловый спирт (метапол) (ср. стр. 470).
488
Улава 12.
б) Кислоты и их соли
Простейшими кислородсодержащими кислотами углерода являются; угольная кислота НзСОз, щавелевая кислота Н2С3О4, муравьиная кислота НСНОа и уксусная кислота МС-зПзОз.
Угольная и щавелевая кислоты двухосновны, т. е. они способны нейтрализовать два эквивалента основания. Они содержат два. атома водорода, способных замещаться на металл, и в водном растворе могут отщеплять два иона И*. Муравьиная н уксусная кислоты одноосновны-, они могут отщеплять лишь один нои И*. В их формулах, приведенных выше, ото свойство подчеркнуто тем, что один атом водорода написан отдельно от друг их t
Ниже приведены формулы соответствующих солей.
МЧТСОз гидрокарбонаты МЧТСгОг гидрооксаяаты
(кислые карбонаты) (кислые оксалаты)
МфСОз (нормальные) карбонаты MJC2O4 (нормальные) оксалаты
М^НОз формиаты МФСаНаОг ацетаты
В водных -растворах нормальных солей, а также при достаточно сильном разведении кислых солей и кислот существуют ионы:
[СОз)" карбонат-исн [СгОзК оксааат-ион
(СНОгУ формиатп^ион [СзНзОаГ ацетат-ион
В СО 2 углерод насыщен в смысле стехиометрической валентности, нр по насыщен координационно. Поэтому С02 может присоединит), сще: один ион О2"
ГО 0'1--
С
О
0=:С = 0+02-->
Если у СО, отнять одип положительный заряд, действуя на пего сильно электроположительным металлом, например натрием (или придать С02 один отрицательный заряд), то углерод в образовавшемся таким образом радикале нс будет, больше насыщенным в смысле стехиометрической валентности. Радикал COj, углерод которого обнаруживает ненасыщенную валентность, может либо взаимодействовать с другим подобным радикалом, образуя ьксал-тт-поп.
о=с~о о=с—о-
+ 2е= | =[С2О4р-,
0 = С = 0 0 = С—0-
либо взаимодействовать с нейтральным атомом Н, образуя формпат-иои
Н
.1 ' ,
О = С«О+е+Н->О=С—О- = [НСОгГ-
Если мысленно заменить в формиат-ионе атом И на группу СН3, то получается аце-таж-иоп
О —С—о-
i =[СН5СО2Г.
СП3 . '
Следовательно, щавелевая, муравьиная и уксусная кислоты получаются из двуокиси углерода путем восстановления* (отнятие положительного заряда или прибавление отрицательного заряда); превращение ее в щавелевую кислоту соответствует восстановлению па одпу, а в муравьиную кислоту — на две ступени; в первом случае каждая молекула СО, присоединяет 1 е, во втором — 2е, как это можно видеть, если написать второе из приведенных выше уравнений в следующей форме:
С02+ 2е +П*= [НСОд]-.
Етце легче, чем получением пазваиних кислот из С02 путем восстановлеппя, протекает обратный процесс — превращение их в СО» путем окисления.
* Подробнее об окислении и во сета по вл ей ин см. в гл. 16.
Четвертая группа периодической системы
489
стадии (4а) (46)
(Harned, 1943;
в две
25“
4,45 ...
30°
4,71
40“
5,06
50
: 5,16,10-’
Угольная кислота и карбонаты. Водный раствор двуокиси углерода обладает свойствами слабой кислоты: оп окрашивает (очень слабо) лакмус в красный цвет, Па осиовацнтг этого свойства можно заключить, что в растворе двуокись углерода находится частично в форме угольной кислоты (НгСОз), которая в свою очередь частично диссоциирует на ионы
СО2-} НцО Н2СО3, (За)
н2со3 2нч-со;. (Зб)
Угольная кислота может реагировать с одним или двумя эквивалентами сильного основания, образуя первичные, или кислые Карбонаты {гидрокарбонаты) и вторичные, или нейтральные (нормальные) карбонаты
Н2СО3Ц-МгОн—М3ПСО3Ц-НчО; Н2СО3+2М1ОН=М^СО34-2Н2О.
Первичный карбонат Вторичный карбонат
Угольная кислота, как двухосновная кислота, диссоциирует
НгСО3 H--HICOJ; нсо; н--р.со".
Величины констант диссоциации для обеих стадий составляют Канко, 193ё) при 1Й°:
^^гс-о^н^^4’01-1077’ '
[СОз -|- п.2^(_>3|
[tlLiUgf
При 25°; Я(= 4,45.-10-’; К2= 5,7-10--п.
Другие значения первичной константы диссоцнацпи: при 0° 10“ t&° 20“ ‘
Kt 2,65 3,43 3,80 4,15
Kt Относится к равновесию ионов со всем количеством двуокиси углерода, находящейся' в растворе (как той, которая растворена как таковая, так ц той, которая содержится в форме педиссоцйировашюй угольной кислоты Н2СО3). Подвыражением |СО24* Н2СО3] следует понимать эту общую концентрацию. Количественное соотношение СО2, содержащейся в растворе в форме Н2СО3 и СО2, растворенной как таковая, было определено Тилем и Штроеккером (Thil, Slrohecker) па том основании, что недиссоциирйваггпая Н2СО3, так же как и ее свободные ионы, моментально реагирует со щелочами, в то время как находящаяся в растворе неизмененная СО2 требует времени для этой реакции. Следовательно, если очень быстро смешать водный раствор двуокиси углерода со щелочью, то в первый момент израсходуется только такое количество основания, которое требуется для нейтрализации действительно содержащейся в растворе угольной кислоты (Н2СОз и ее ионов). Если взять щелочи больше, чем необходимо, то пройдет значительно большее время, пока она нейтрализуется. Таким образом, определением количества моментально прореагировавшей щелочи можно установить и содержание угольной кислоты (в диссоциированном и недиссоцнированном состоянии) в растворе. Опп составляет меньше 1 % общего .количества СО2 и «действительная константа диссоциации» К,^ угольной кислоты
х [Н-ЦНСОд}
[Н2СО3]
выражается величиной порядка 5 -10-4, так что опа больше, Чем константа диссоциации муравьиной кислоты, и много больше, чем Kv уксусной кислоты (ср. стр, 495). Однако практически всегда оперируют «кажущейся константой диссоциации» Kt. Равновесие между СО2, Н2О и Г12СО3 в действительности устанавливается так быстро, что во всех реакциях, которые протекают за время большее, чем доли секунды, константа Kt может служить мерой для установления равновесия, Так что практически водный раствор' двуокиси углерода ведет себя как раствор очень слабой кислоты.
Кажущаяся степень диссоциации угольной кислоты в 0,1 н. водном растворе составляет 0,12%.
490
Глава 12
Соли, угольной кислоты, карбонаты, ъ водном растворе гидролитически расщеплены. В их растворах устанавливаются равновесия
М^СО3+П2О -Г М^Н-НЛГСОа;
К^НСОз+ЩО q± М^Н+ЩСОз.
Поэтому карбонаты обнаруживают щелочную реакцию, причем это справедливо ие только для вторичных пли «нейтральных», но и для первичных или «кислых» карбонатов (гидрокарбопатов). Только по отношению к таким индикаторам, для которых, как для фенолфталеина, цветовой переход щелочь -> кислота протекает тогда, когда раствор является еще слабо основным, первичные карбонаты (гидрокарбонаты) реагируют* на холоду (0° и немного выше) как «кислоты» (подробнее об этом см. в тл. 18).
Гидролитическое расщепление (вторичного) карбоната натрия составляет, по данным Аусрбаха (Auerbach), при 18° в 0,1 и. растворе 3,5% в 0,01 и.— 12,4%. В 0,1 и. растворе карбоната натрия концентрация гидроксид-иоцов составляет, следовательно, при 18° 3,5*10~3 молъ/л. В растворе гидрокарбоната натрия она составляет при той же температуре 1,5 10~в молъ/л.
Известны первичные карбонаты (гидрокарбонаты) щелочных, щелочноземельных и некоторых других двухвалентных металлов. Все они легко растворимы в воде. Исключением является гндрокарбонат натрия, на малой растворимости которого основан метод Сольве получения соды. При кипячении растворов гидрокарбопатов происходит превращение их в нормальные карбонаты с отщеплением СОз.
Вторичные или нормальные карбонаты образуются главным образом одно- и двухвалентными металлами. Нормальные карбонаты, за исключением карбонатов щелочных металлов, трудно растворимы в воде.
Помимо карбонатов щелочных металлов, легко растворим также карбонат аммония. Достаточно легко растворим и карбонат одновалентного таллия.
Все карбонаты разлагаются нелетучими кислотами. Очень слабые кислоты (такие, как борпця и кремневая, и соответственно их ангидриды) разлагают карбонаты только при прокаливании.
При повышении концентрации ионов И' в’водном растворе реакции (За) и (36) (стр. 489) up от екают справа палево. Если в связи с этим раствор пересытится СОг, то двуокись выделяется пз раствора, вследствие чего реакция должна полностью протекать справа налево. Если при обычной температуре раствор еще не пересыщен СО г, то пересыщение часто наступает прп нагревании.
Карбонаты щелочных металлов можпо расплавить без разложения. Прочие карбонаты разлагаются при нагревании, отщепляя СОз: М‘СОЭ = = М|О + СОз.
Этому разложению способствует удаление образующейся СО3 (понижение давления) пли устранение окисла Mt. О из смссп. Последнее можно достигнуть добавлением термостойкой кислоты или со ангидрида, например SiO2, которая образует соль с основ-: ным окислом. На этом свойстве основано разложение карбонатов при прокаливании их с ангидридами очень слабых, однако термостойких кислот, таких, как бор пан и кремневая.
Фосген. Как уже было отмечено, окись углерода может соединяться с хлором, образуя оксихлорид углерода (карбонилхлорид) CQCla. Это соединение обычно называют фосгеном (от греческого слова grog — свет и yEvvav—
Четвертая группа периодической системы
491
образовывать), так как его образование впервые наблюдали (Davy, 1812) в смеси окиси углерода и хлора, выставленной на солнечный свет.
Эта реакция идет не только на свету, но и в присутствии катализаторов. В технике для этой цели используют активированный уголь при 125—150°. В темноте и в отсутствие катализатора СО я С12 реагируют только при температуре выше 500°,‘ одиако при этой температуре равновесие СО 4- С12 £ СОС12+ 20,2 ккал уже сильно сдвинуто влево.
Фосген — бесцветный, чрезвычайно ядовитый газ с удушливым запахом. При охлаждении или под давлением он легко сжижается. Температура кипения 7,56°, температура плавления —118,8°, удельный вес при 4° равен 1,41. Критическая температура 182°, критическое давление 56 атм, критическая плотность 0,52. Ои очень легко растворим в бензоле и толуоле. г*
Фосген можно рассматривать как хлорангидрид угольной кислоты. В соответствии с этим он реагирует с водой с образованием угольной и хлористоводородной кислот
СОС1 г 4- 2НОН—НгСО3+2НС).
Однако при обычной температуре эта реакция протекает очень медленно, так как фосген малорастворим в воде. Фосген легко реагирует с органическими гидроксилсодержащими соединениями (а также с амидами), образуя эфиры угольной кислоты (и производные мочевины). Поэтому его широко применяют при. производстве красителей, а также для синтезов в научных лабораториях. В первую мировую войну его применили как боевое отравляющее вещество.
'Оксифторид углерода COF2, получающийся при пропускании СО над AgF, (см. т. II)— бесцветный таз с резким запахом(т. кии.— 83,1 °, т. ня. —114°), энергично разлагается водой с образованием СО2 и 2HF (Ruff, 1934). Молекула COF2 планарна; Оч->С = 1,225 A; C«-»F==i,313 Д; <FCO=HO° (Karie I. L., Karie J., 1950).
Й аду сильна я кислота и пер карбопаты.. При электролизе концентрированных растворов карбонатов щелочных металлов на аноде происходит образование перкарбо-нат-иовов 2COs —2 е = СгО£. Перкарбонаты AllfC^Oj] следует рассматривать как производные перекиси водорода, как это видно из реакции расщепления, которую они претерпевают при обработке кислотами:
Г О— о 1” О—О
ОС СО -4-211*= | | 4-2СО,.
I О О J II н
Распад с образованием перекиси водорода и гидрокарбоната легко наступает уже при растворении в воде: C2Og + 2Н20 — Н2О2+ 2нС0'3. Поэтому водные растворы перкар-бонатов реагирует в общем как смесь перекиси водорода и гидро карбонатов. Свободную надуеол!:,пую кислоту до сих пор не удалось выделить. Помимо электролитического способа, перкарбонаты можно получить различными химическими превращениями. От перо в сп г цтт ратов карбонатов их можно отличить при помощи средств, рассмотренных па стр. 378.
Щавелевая кислота и оксалаты. Щавелевая кислота НзСзОа образуется в виде своей натриевой соли при пропускании двуокиси углерода над металлическим натрием, нагретым до температуры 350°
2CG24-2Na=jNa£CaO4.
Выход кислоты по этой реакции, однако, незначителен. Практически полное превращение достигается, по данным Хаупта (Haupt, 1913), при распылении жидкого натрия в горячей газообразной двуокиси углерода. Если вместо металла использовать <г.к<мьга.«у натрия, то при комнатной температуре 80% патрия превращаются в оксалат
।
492
Глава 12
натрия (Henglern, 1952); остаток в основном реагирует с образованием карбопата. По Хенглейну превращение протекает таким образом, что сначала Na присоединяется О
II
к СО2, образуя радикал NaO—С. Два таких радикала могут либо соединяться один \
с другим в Na2C’2O4, либо каждый из них может дальше реагировать с натрием по ура в-
пению NaO—Д-Na = Na2O -Н СО, после чего Следует образование карбоната .%>_<> + (.(Л Na2CO3.
В технике получение оксалата натрия осуществляется преимущественно нагреванием до 360° синтетического формиата натрия NaCHCh. При этом отщепляется водород и образуется оксалат натрия NaaCaO-r
C0,Na
: =П2+ I
Н—j—C02Na CO2Na
В технике оксалат натрия но переводят непосредственно в свободную щавелевую кислоту, а в целях обратного получения формиата натрия смешивают с известковым
молоком, после чего в смесь вводят генераторный газ При температуре 200“ и давлении 15 ппьм. При этом в соответствии с уравнением
Na2C,O4+ Са(ОН)2+ 2GO = 2NaCHO£+ СаС£О4 получают формиат натрия NaGHO2 и оксалат кальция СаС2О4. Последний выпадает в виде трудпорастворггмого осадка и его переводят затеи обработкой серной кислотой в щавелевую кислоту
СаС2О4-г П£5О4— CaSO4+ T12G2O4.
Ойсалат натрия получают также при нагревании до ~285° древесных опилок с высококонцентрированным раствором едкого натра. Из оксалата и атрия через кальциевую соль (СаС£О4) путем обработки последней серной кислотой получают свободную щавелевую кислоту.
Щавелевая кислота кристаллизуется из водного раствора с двумя молекулами воды НаСзОт-^НаО в виде моноклинных призм с удельным
Четвертая группа периодической системы
493
весом 1,653 и температурой плавления 101,5°. При нагревании до 100° в токе сухого воздуха гидрат обезвоживается. Безводная щавелевая кислота НаСЮт кристаллизуется в виде ромбических бипирамид с температурой плавления 189,5°. При быстром нагревании или при нагревании с концентрированной серной кислотой она разлагается на двуокись углерода, окись углерода и воду
О I'. -О — I!
-.... ; _С(Ь СО М1.,О.
О к О—Н - '
В воде щавелевая кие. юта умеренно растворима па холоду и очень .легко растворима при нагревай нп, поэтому она пр ск росло перекристаллизовывается, и а воды, В 100 а воды растворяется
Яри If 10* 30" 50" 90“
3,6 5,3 10,2 32,1 120,0 г Н2С2О4.
Еще лучше растворяется она в спирте, В эфире щавелевая кислота растворима труднее, чем в воде.
В кристаллическом ди гидрате щавелевой кислоты обнаруживаются ярко выраженные водородные моет иковые связи. Из данных по рентгенографическому исследован ню структуры и распределения электронной плотности [В г i II, II erm а и n, Р eters, Ацп, Physik, 42, 357, 1942—-1943; Dunitz, Ho bertso у, J, Chem. Soc,; 1947, 142] следует, что дигидрат щавелевой кислоты имеет структуру, приведенную ла рис, 86. (Водородные мостики показаны пунктирными линиями). Как видно из рисунка, водородные мостики имеют различную дайну, .Мостики длиной 2,52 А являются самыми короткими из измеренных до сих пор водородных связей между атомами кислорода. При замене водорода па дейтерий кристаллическая решетка дигидрата щавелевой кислоты сильно расширяется (Robertson, Proc, Л оу, Soc., 170А, 221, 241, 1939),
Щавелевая кислота — двухосновная кислота средней силы. Поскольку опа менее летуча, чем хлористоводородная кислота, то при нагревании вытесняет последнюю из ее солей, например из поваренной соли.
Щавелевая кислота как двухосновная диссоциирует ступенчато
Н,С2О4 H’-t-HCgOJ; НС2О; H’-]-C2OJ.
Копстапты диссоциации для обоих равновесий составляют (при 25°);
к _J**4 jHCoOj] =5 9_;10_а, (И 1:1Сг°*Г= б 4.10-5.
4 ]НС2Оу
[ЩС2О4]
Коэффициент электропроводности Л,;/Ло щавелевой кислоты в 1 н> растворе 0,15, в 0,1 н. 0,31. Согласно этому (кажущаяся) степень диссоциации составляет 15 и соответствен по 31%, Числовые значения обеих констант диссоциации щавелевой Кислоты лежат ближе одна г; другой, чем это обычно имеет место у других двухосновных кислот (например, TI2CO3, H„S). Это обстоятельство связано стом, что у щавелевой кислоты способные к диссоциации атомы И связаны с двумя разными пространственно разделен-ними группа.чп (Treadwell, '1928).
Щавелевую кислоту используют в технике как протраву при печатании тканей, как сродство для отбеливания стеарина и соломы, в качестве 'конденсирующего агента в препаративной органической химии и для разных других целей. В производстве калильных сеток опа служит для осаждения редкоземельных элементов. В аналитической .химии щавелевой кислотой также пользуются для осаждения, особенно при качественном и количественном определениях кальция. Кроме того, в количественном анализе се (а также ее натриевую соль) часто применяют для установления титра растворов перманганата.
494
Глава 12
Это применение осповано на том, что в сернокислом растворе количественно протекает взаимодействие между исфман гаиат-ионами MaOJ и оксалат-ионами, которые при этом окисляются до двуокиси углерода, тогда как перманганат-ионы восстанавливаются до ионов марганца (II);
: 2МаО;+ 5С2О J-|- 1GH- = 2Mn" + WC02+8H20.
Щавелевая кислота в больших концентрациях ядовита. Она встречается в некоторых растениях, главным образом (в виде кислой калиевой соли) в кислице лесной (Oxalis acetosella) и в щавеле (Rumex acetosa).
В качестве двухосновной кислоты щавелевая кислоТа образует два ряда солей; первичные или кислые а вторичные или нейтральные оксалаты. Оксалаты, за исключением оксалатов щелочных металлов, трудно растворимы в воде, но большей частью легко растворимы в сильных кислотах. Щавелевая кислота обнаруживает большую склонность к образованию двойных и комплексных солей,
Оксалат натрия NaaCaOi— кристаллизующиеся без воды бесцветные, пегигроскопические кристаллы. Эта соль очень удобна для установления титра растворов перманганата. По сравнению со свободной щавелевом кислотой опа имеет то преимущество, что не поглощает аммиака из воздуха.
Из оксалатов калия, помимо нейтрального оксалата К2С2О4-Н2О (моноклппные призмы) и кислого оксалата (гидро оксалата) КНС2О4.Н2О, известна еще «сверля ислам соль КНС2О4> 2Н2О — прекрасно кристаллизующийся продукт присоеди-
нения одной молекулы щавелевой кислоты к одной молекуле кислого оксалата. Ее используют, между прочим, для удаления чернильных пятен и ржавчины и называют в торговле «кисличной солью», хотя в Кислице содержится не она, а обычный кислый оксалат (гидрооксалат).
Оксалаты щелочноземельных металлов, среди которых аналитическое значение имеет прежде всего трудно растворимый СаС2СЬ (ср, стр. 320), при нагревании необратимо разлагаются по уравнению МПС2О4= Мпсо3+СО (Wohler, 1932). Из водных растворов они кристаллизуются в виде кристаллогидратов. СаС2О4 образует 1 -гидрат (моноклинный), 2У2-гидрат (триклинный) и 3-гидрат (тетрагональный). Их устойчивость растет в направлении: триклинный -+ тетрагональный-* моноклинный (Kohlschiit-ter, 1930),
По данным Бриптзингера (Brintzinger, 1935) С2О J-ион в водном растворе Прочло связывает четыре молекулы Н2О.
Муравьиная кислота и формиаты. Муравьиная кислота НСНОт, названная так потому, что она содержится в муравьях {formica), где ее впервые и обнаружили, достаточно часто встречается в органической природе,' например в волосках крапивы. В настоящее время ее в больших масштабах получают в технике, так как она находит широкое промышленное применение. Более всего ее используют в кожевенной промышленности для обеззоливания кож и в красильно-набивнем производстве, для подкисле-пия ванн, так как она менее разрушает волокно, чем применяемая также для этой цели серная кислота. Далее, муравьиную кислоту используют как дезинфицирующее средство, например, для дезинфекции сосудов для пива и вина, а также как антисептик, например при консервировании фруктовых соков. Применяют ее и в медицине.
Получение, муравьиной кислоты осповано на способности окиси углерода взаимодействовать при повышенной температуре с гидроокисями щелочных металлов с образованием формиатов щелочных металлов. Например, действуют генераторным газом на тонко измельченный едкий натр при хорошем перемешивании, температуре 120—130° и давлении 6—8 атм и из полученного таким образом формиата натрия NaCHOa действием серной кислоты выделяют муравьиную кислоту.
Четвертая группа периодической системы 495
В более новых методах вместо NaOH используют смесь Са(ОН)а п Na2S04 пли при повышенном давлении (60 ат) только Са(ОН)2, Под давлением 100 ат можно осуществить взайме действ пе СО с водным раствором NH3, Полученный при этом (NH4)CHO2 при разложен ни Н2ЗО4 дает в качестве ценного побочного продукта удобрение (NH4)3SO4.
Чистая Муравьиная кислота — бесцветная, резко пахнущая и раздражающая кожу жидкость, которая затвердевает при умеренном охлаждении. Температура плавления 8,43°, температура кипения 100,6°, удельный вес 1,220. С водой она смешивается во всех отношениях. Водныйраствор, содержащий около 75% муравьиной кислоты, кипит при атмосферном давлении, не меняя своего состава.
Муравьиная кислота — сильный восстановитель. Ее пары горючи. Под влиянием мелко измельченного родия она расщепляется при обычной температуре на водород и двуокись углерода НСНОк= На-J- СОз. Из растворов солей благородных металлов она осаждает металлы. С HgCJs в водном растворе она реагирует по уравнению НСЫОг-}- 2HgGla= СОг-|--l-I-IgaCla-f- 2IIC1. По наблюдениям Скала (Skala А.) при избытке HgCla реакция протекает количественно.
Муравьиная кислота—одноосновная кислота средней силы. Ее констапта диссоциации
х= [7нснб~Т~°1>8'№4 (при 18°)о
Степень диссоциации составляет при 18° в ! и. растворе 1,4%, в 0,1 и. растворе — 4,4 %. Электронографически (Schomakcr, 1947) были определены следующие межатомные
расстояния: С=О = 1,21 Л; С—О = 137 1, О«—* О — 2,28 А; <7 =124°.
ХО
Соли муравьиной кислоты AHCHOs, называемые формиатами, легко растворимы в воде. Некоторые из них, например формиат алюминия А1(СНО2)з, получаемый обменным взаимодействием сульфата алюминия и формиата натрия, находят техническое применение.
Уксусная кислота и ацетаты. Уксусная кислота НСзНзОз представляет собой бесцветную жидкость с резким кислым запахом и кислым вкусом даже при большом разбавлении. Совершенно безводная уксусная кислота, называемая «ледяной уксусной кислотой» (уд. вес 1,049), очень легко затвердевает (т. пл. 16,7°) и кипит при 118,1°. Уже при обычной тем1-пературе уксусная кислота заметно летуча. С водой, спиртом, эфиром, хлороформом и глицерином она смешивается во всех отношениях.
Уксусная кислота — слабая кислота. Как слабый электролит, она отлично подчиняется закону разведения Оствальда (стр. 90).
Степень диссоциации уксусной кислоты при 18° составляет 0,45% в 1 и. водном растворе п 1,4 % ц 0,1 н. растворе. Константа ее диссоциации имеет следующие значения: при 18° К = 1,75(М(Г»; при 25° К = 1,754-1О~ь.
Уксусная кислота находит разнообразное применение. Ее 80%-ный водный раствор, «уксусная эссенция», служит для приготовления столового уксуса. Чистая уксусная кислота (ледяная) находит применение в медицине как лекарственное средство. Разбавленную уксусную кислоту часто применяют в качестве реактива. В технике и в промышленности уксусная кислота служит растворителем для целлулоида и коллодия при изготовлении лаков и политур. Далее, она слу-
-496 Глава 12
ж пт для приготовления свинцовых белил, искусственного шелка (ацетатный шелк), синтетических лекарственных препаратов, таких, как антифебрин, аспирин, антипирин, душистых веществ, которые часто являются эфирами уксусной кислоты, а также органических красителей. Уксусную кислоту частично получают, особенно для пищевых целей, путем сбраживания спирта уксуснокислыми бактериями [Bacterium acetiy Для промышленных целей ее получают главным образом из добываемого при сухой перегонке дерева древесного уксуса. Последний, являющийся в основном смесью метилового спирта и уксусноТГкислоты, обрабатывают известковым молоком. При выпаривании сначала удаляется метиловый спирт, который собирается в качестве побочного продукта. Затем выкристаллизовывается неочищенный ацетат кальция, так называемая серая известь, из которой при обработке.сорной кислотой выделяется уксусная кислота. В последнее время значительное количество уксусной кислоты получают на синтетического (см. стр. 477) ацетальдегида путем окисления его кислородом воздуха при каталитическом действии солей марганца:
СНз-СПО + УзОа^СПз—СО(ОН).
Ангидрид уксу!~поИ кислоты (CH3CO)SO, также часто применяемый в препаративной органической химии, технически получают взаимодействием ацетата натрия с хлористым сульфурилом или введением фосгена в уксусную кислоту в Присутствии MgCl2, причем в качестве побочного продукта .образуется TIC1,
Соли уксусной кислоты — ацетаты и большинстве своем легко растворимы в воде. Только ацетаты серебра и ртути (I) растворяются в холодной воде довольно плохо. Уксусная кислота легко* выделяется из ее солей под действием нелетучих кислот.
Благодаря хорошей растворимости и способности легко кристаллизоваться ацетаты находят широкое применение. Особенно часто применяют ацетат натрия, который кристаллизуется из водного раствора с тремя молекулами воды КаСгНзОз-ЗНаО, в виде бесцветных, прозрачных моноклинных призм. Он очень легко растворим в воде, хорошо растворим также [г и спирте, расплывается при 75° в своей кристаллизационной воде, при температуре около 120<; теряет всю воду и плавится в безводном состоянии при 319°. Его применяют в качестве осаждающего реактива в аналитической химии н часто для понижения кислотности уксусной кислоты. Ввиду того что в водных растворах, ацетат патрий практически полностью диссоциирован, прибавление его к растворам;содержащим уксусную кислоту, сильно повышает концентрацию ацетат-ионов. В результате этого концентрации ионов И" должна соответственно понизиться.
Ацетаты, в том числе ацетаты щелочных и щелочноземельных металлов, обнаруживают ярко выраженную склонность к образованию двойных и комплексных соединений. Так, ацетат натрия способен связывать одну или две молекулы уксусной кислоты, образуя так называемые кислые ацетаты. Действительно, эти соединения представляют собой продукты Присоединения уксусной кислоты к нейтральным ацетатам. Так как уксусная кислота — кислота слабая, то водные растворы ацетатов всдед-: ствие гидролиза обнаруживают щелочную реакцию
М1С21Т3О24-НгО=М,4-ОН'4- НСгНэО2.
Из 15 000 молекул NaCaHsOa в 0,1 н. растворе при 18° только одна подвергается гидролитическому разложению.
Четвертая группа периодической системы 497
Уксусная кислота способна образовывать соли пс только с основаниями, но и с кислотами; однако, поскольку эти соли расщепляются водой, для цх получения следует использовать реакции, при которых не выделяется вода. Так, ацетилфторобррат [CHaCOj+ [BF4I~ получают при соединении CHaCOF c BF3. То, что полученное соединение является солью, моя: и о доказать измерением электропровод и ости (в жидком S02) [Seel F., Z, апогд. Cbeiir., 250, 331, 19431-
Уже давно высказан ное предположение о том, что продукт соединения ацетихло-рида с хлористым алюминием можно представить как содеобразпое соединение СН3СОС1 . А1С13= |СП3СО]+[AICIJ-, было подтверждено Файербродзером (Fair-brother F., J. Chem.. Soc., 139, 503, 1937)], который, применяя в качестве индикатора искусственно полученный радиоактивный хлор (ср, т. II, гл. 13), смог показать, что при присоединении Л1С13 к СН3 — С0С1 содержащийся в последнем хлор становится иопогешгым и вступает в обмен с хлором, первоначально имевшимся в Л1С13.
Ацетат-ион склонен к образованию комплексных ионов (ацстато-йонов) с тяжелыми металлами, при этом он образует преимущественно катионные комплексы; например, с двухвалентными металлами часто образуются ионы ^ипа [МЩСвНзОа^]" (Weinland).
Сульфиды
Нормальным сульфидом углерода является двусернистый углерод CSs, обычно называемый просто сероуглеродом. Низшие сульфиды углерода очень неустойчивы.
Моносульфид (тиоокись углерода CS) образуется, по данным Дьюара (Dewar, 1910—1911), при действии тихого электрического разряда на сероуглерод при —185° в виде белого налета, который при —180° со взрывом превращается в коричневый продукт полимеризации [CS]K. Однако, по данным Клеменсал(Кlemenc, 1930), описанный выше белый налет представляет собой смесь обычных и активированных электрическими разрядами молекул CSj, При повышении температуры последние должны распадаться под каталитическим действием стенок сосуда па [CS]х и S. Появление CS в газовой фазе Клеменс считает, на основании своих, опытов, совершенно невероятным. Во венком случае, свободная молекула CS в высшей степени неустойчива. Несколько более устойчива тионедбкисъ углерода C3S3, которая, как это следует 1гз характера ее реакций, построена аналогично недокиси углерода. В соответствии с этим ее формула имеет вид S—C=C=C=S.
Среднее положение между двуокисью углерода и сероуглеродом занимает оксисулъфид (сероокись) углерода COS.
Сероуглерод. CS2 технически получают пропусканием паров серы над раскаленным древесным углем С + 2S.^t CS2. Он представляет собой бесцветную, в чистом виде приятно пахнущую жидкость, которая, однако, благодаря примесям обычно имеет отвратительный запах. Эта жидкость имеет удельный вес 1,262 (при 20°), кипит' при 46,2° (теплота испарения 6,35 ккал/молъ) и затвердевает только прп очень сильном; охлаждении (—111,6е). Давление пара сероуглерода составляет при 0° 127,1 и при 20°— 298 мм рт. ст. Плотность пара соответствует молекулярному весу, определяемому формулой CSa. Пары чрезвычайно легко воспламеняются (температура воспламенения 236°), они могут воспламениться даже при внесении нагретого предмета. Сероуглерод горит (CSa 4- 30а= СОа+ + 280г + 258 ккал) светло-синим пламенем, богатым фотохимически действующими лучами и при этом обладающим такой низкой температурой, что в нем не обугливается бумага.
Сероуглерод — эндотермическое соединение.. При его распаде освобождается около 15,4 ккал на 1 моль парообразного CS3. Если распад вызван детонатором, например гремучей ртутью, то он протекает взрыло образно, однако широко не распростра-32 г. Реми ! '
498
Глава 12
if тяге
пяется. Медленное; разложение протекает лод влиянием темного электрического раз-' ряда или ври освещении, чем и объясняется появление желтой окраски сероуглерода на свету; Так как иод действием тепла равновесие С -j- 2S -Л CS2 сдвигается вправо, то При нагревании до высоких температур протекает липгь неполное разложение. При этом образуются более бедные серой продукты, например тионёдокисъ углерода C:i42— -красная, очейк резко пахнущая, сильно слезоточивая жидкость с уд. весом 1,27; которая затвердевает прн —0,5° и легко полимеризуется, Превращаясь в черный продукт. Для получения C3S2 следует создать дугу между угольными или' графитовыми электродами в парах сероуглерода пли в жидком; сероуглероде, Для лабораторных опытов можно воспользоваться простым прибором, приведенным на рис, 87. 15 широкогорлой колбе 3, стоящей на водяной бапе, нагревается до кипения сероуглерод. Пары кон депсируются в обратном холодильнике 3, В стеклянный: шар 2, в который впаяны четыре короткие трубки, вставлены два угольных : стержня, между которыми создается вольтова дуга. Угольные стержни укреплены в медных Держателях, которые могут перемещаться, будучи плотно вставленными в корковые пробки, за-крывающис боковые трубки. Продукт реакции после фильтрования1 обрабатывают, медными стружками для удаления растворенной серы. . Если затем выдуть сероуглерод струей тша-тел ъно вы суш ей ного в о здух а, то о станется /несколько миллилитров тёмно-красной жидкости, которую на основании се реакций можно иден-тифпцировать как тионедокись углерода,
Сероуглерод ядовит (поражает дыхательные пути и кожу). При нагревании (выше 150’) он разлагается водой по уравнению
С83+ 2Н2О ==СО2.4-2ЩЗ, Еще легче протопает разложение при нагревании под действием баритовой воды. Такими сильными окислителями, как перманганат, сероуглерод разлагается с выделением серы, С сорным ангидридом оп взаимодействует, образуя сероокйсь" углерода COS
CS2+3SOa — COS-I-4SO2-
В атмосфере двуокиси азота сероуглерод способен гореть. С окисью хлора он рearn? рует, образуя фосген V
: CS3 + ЗСЕО —СОС13 ‘ 2SOCI3.
Рис, 87. Получение CaSj. "При нагреваний =С32\ легко обменивает
серу полностью или 'частично на кислород, : Например при нагревании с окисями металлов. При действии хлора сера может быть по ступеням замещена хлором, а при действии сильных восстановителей —водородом.
При повышен пой температуре CS2 реагирует с Н2, образуя U .S. D присутствий : в качестве катализатора MoS2 или Pl превращение В: H2S протекает в соответствии с уравнением
CSj>4-4H2 2I‘IsS4*QH4‘ kp — (PcH4'PH2sWcs2‘PHa2.2'при 600°, 3,0-10-s прп дао0 (Теггёз, 1934).
Четвертая группа периодической системы 499
Аналогично тому, как СОа, присоединяя один ион кислорода, образует ион [СОзГ, CSa может присоединить ион серы и образовать иод [CSs]". Соли — производные этого иона — называют тиокарбопатами,. Последние легко получаются по реакции CS2+ S' = [CSa]". Например, при встряхивании концентрированного раствора сульфида калия с сероуглеродом образуется тиокарбонат калия KsICSs], который используется в качестве средства против филлоксеры. Свободная тиоугольпая кислота H2CS3— маслянистая, растворимая в воде жидкость, которую можно получить из содей при действии на них соляной или серной кислоты.
Существуют также соединения, которые занимают промежуточное положение между обычной тиоуголг.пой кислотой (собственно т/гитиоугрльцой кислотой H2CS3) и угольной кислотой Н2СО3; в этих соединениях кислородные атомы угольной кислоты только частично замещены атомами серы. Сюда относятся дитиоуголъная кислота Н^СОЗч, моноэтиловый эфир которой HC(OC2H6)S2 называют ксантогеноеой кислотой. Если в натриевой соли этого соединения радикал этилового спирта заменить па. радикал целлюлозы и, кроме того, заменить в последнем один атом Н на Na, то получим так называемый ксантогеиат целлюлозы, Na (C(OCeH8OtNa)S2l. Образование последнего дает возможность иеревеоти целлюлозу в раствор и имеет поэтому большое значение при производстве искусственного телка.
Сероуглерод отличается высокой преломляющей способностью и достаточно сильной дисперсией. Показатель преломления для желтого света (Nan) при 20° составляет 1,6276, Он сильно зависит от температуры, примерно в 20 раз сильнее, чем показатель преломления воды.
Сероуглерод является прекрасным растворителем для жиров, масел, восков, смол, каучука, а также для серы, фосфора, иода и некоторых других веществ. Со спиртом, эфиром и хлороформом он смешивается в любых отношениях. Растворимость его в воде, напротив, незначительна, хотя при низкой температуре (ниже —3°) он образует гидрат ЗСЗз-НгО. В 100 г воды растворяется при 0° 0,26, при 20°— 0,10 г CSa.
Сероуглерод широко применяют в технике в качестве растворителя и экстракционного средства, Он служит также средством для борьбы с вредителями й для других целей. Но главную массу его используют для получения вискозного шелка — искусственного шелка, который получают пропитыванием целлюлозы (ив древесины) раствором едкого натра и последующей обработкой сероуглеродом. При этом образуется растворимый в воде ксантогенат целлюлозы. Концентрированный вязкий раствор последнего продавливается через отверстия (фильеры) в солевой раствор, содержащий также серную кислоту. Серная кислота разлагает соединение с выделением целлюлозы, которая при этом получается в виде тонких., блестящих, как шелк, нитей.
Селеноуглерод CSe2— темно-желтая жидкость (уд, вес 2,68, т. кип. 124°, т. пл. —45,5°) нерастворимая в воде, сгорающая с трудом и получаемая (Grimm, 1936) при взаимодействии CCL, с H2Se, при 50Йа является еще более эндотермичным соединением, чем CS2 (т бил от а образования —34 ккал Ли о ль), чрезвычайно склонным к полимеризации (относительно получения и свойств CSe2 ср. также Ives, Pittman, W аг d-1 aw, J. Chem. Soc., (947, 1080). CTe2, для которого следует ожидать еще меиьшей устойчивости, не был получен. CSSe(желтый, уд. вес 1,98, т. пл. —85°, т. кип. 84°) и CSTe (красный, т, пл, —54°) также мало устойчивы (Stock; 1914),
Сероокись углерода — карбонилсулъфид COS — легко воспламеняющийся газ, без цвета и запаха, который сжижается при—50,2° и затвердевает при —138°. Он образуется при пропускании смеси паров серы и окиси углерода через раскаленные трубки S 4- СО в S = C==O.
32*
МО
Глава 12
В чистом виде его лучше всего получать методом Штока действием кислот иа Тиокарбамат аммония. (Тиокарбаминовая кислота IICO(NH2)S производится от моно-тиоугольной кислоты И2СО2Й путем замены группы ОН ла аминогруппу NH2.)
NH4£S C-NH2 j +2HC1=2NH4G1-|-COS.
Сероокись углерода хорошо растворима в сероуглероде, толуоле и в спирте, несколько менее — в воде. В 1 ч. воды растворяется при 20: 0,55 об. ч. COS. В концентрированном растворе поваренной соли сероокись углерода практически нерастворима. Водой сероокись углерода постепенно разлагается с образованием двуокиси углерода и сероводорода, поэтому во влажном виде она пахнет сероводородом COS + НаО~ СОа-р + HsS. От продуктов разложения сероокись углерода можно освободить Промыванием концентрированным раствором едкого натра, которым она очень слабо поглощается. В отсутствие влаги сероокись углерода устойчива. При нагревании она разлагается по уравнению
2C0S CO24-CS2 И
2C0S 2CO+2S.
Тиокарбамат аммония, который легко получается из COS и NH3, при длительном нагревании, отщепляя сероводород, Превращается в мочевину
CO(NH2)(SNH4) = CO(NH2)2-|-HaS.
Эта очень гладко протекающая реакция предложена Клеменсом (Klemenc, 1930) для технического получения мочевины.
Соединения циана
При прокаливании азотсодержащих органических соединений, таких, как шерсть, кожа, рога, без доступа воздуха и особенно в Присутствии щелочей образуются соединения, содержащие одновалентный радикал — — CN. Наиболее давно известной из них является открытая в 1704 г. Диппелем и Дисбахом берлинская лазурь (см. ниже). По ее названию радикал — CN стали называть циан-радикалом (от xcaveog — томно-синий). Простейшую кислоту, являющуюся производным циана HCN, называют цианистоводородной или синильной кислотой; ее соли называются цианидами.
Кроме синильной кислоты и ее солей, известны многие другие производные циана. Однако здесь опи не будут рассмотрены, так как зти соединения относятся к области органической химии.
Радикал «циаш> в свободном состоянии, т. щ когда он не связан с каким-либо другим атомом и не песет электрического заряда, неустойчив. Если отнять у иона CN' электрический заряд, то два таких радикала соединяются за счет освобождающейся валентности углерода, образуя дициан (CN)z. Последний обычно кратко называют цианом.
Циан. Дициан (CN)a— бесцветный ядовитый газ с резким запахом, несколько напоминающим запах горького миндаля. Он горит пламенем цвета цветущего персика с синеватой каймой. Циан легко сжижается при 15° под давлением 3,3 ат, при обычном давлении — при охлаждении до —20,7°. При —34,4й он затвердевает. Плотность пара отвечает молекулярному весу, требуемому формулой (CN)a.
Четвертая группа периодической системы
501
Энергия, которую необходимо затратить для расщепления молекулы (CN)2 на да а CN-радикала, составляет, по данным Глокклера (Glockler, 19-<8), 127,3 ккол/.коль, а энергия, необходимая для разрыва CN-радикала на атомы, составляет 150,4 ккал/моль.
Циан хорошо растворим в воде, спирте, эфире. Однако он очень легко разлагается в растворах с образованием различных продуктов, среди которых имеется и щавелевая кислота
NG - CN -|-4НгО = НО • СО - СО - OH-|-2NH3.
В других условиях диан полимеризуется в коричнево-черную нерастворимую в воде и спирте пористую массу (парациан}, которая при нагревании без доступа воздуха до 8601 вновь превращается в обычный циан (дда<ийн).
Циан можно получить нагреванием цианида ртути Hg(CN),, который целесообразно смешивать с хлоридом ртути, так как при сильном нагревании и качестве побочного продукта образуется парацнан. Распад цианида ртути — эндотермическая реакция, а взаимодействие его с хлоридом ртути — слабо экзотермическая реакция, поэтому без добавления хлорида ртути смесь приходится нагревать сильнее
Hg(CN)2 + 11 ккал = Hg -Ь (CN)2; Hg(CN)a + HgCla = Hg2Cl2 + (CN)a +0,4 ккал.
Наиболее удобным способом получения циана является взаимодействие сульфата меди с цианидом калия в достаточно концентрированных растворах
Cu'4-2CN'=Cit(C,N)2 ChCN+1/2(CN)3.
Чтобы реакция прошла полностью, смесь под конец слегка нагревают.
Выпадающий в осадок цианид меди (I) можно отфильтровать и обработкой раствором хлорида железа перевести в циаи
CtiCN-| Fe— +Cl' = GuCl-|-lV+4a(CN),.
Цианистый водород н цианиды. Цианистый водород (синильная кислота) — бесцветная легко подвижная жидкость (уд. нос 0,697), кипящая при 26,5°, с наркотическим запахом, в большом разведении напоминающим запах горького миндаля. При —15° опа затвердевает, образуя волокнистую кристаллическую массу.
Следует отметить высокий дипольный момент цианистого водорода. Для газообразной молекулы он составляет 2,88 -10-18 ед. GGS.
Синильная кислота чрезвычайно ядовита ( (противоядие — перекись водорода). Уже 50 мг ее достаточно, чтобы убить человека. Смерть может наступить в течение нескольких секунд. Синильную кислоту применяют вследствие се ядовитости для борьбы с вредителями. Часто используют ее также для синтезов органических соединений. Благодаря летучести HCN можно вытеснить из се солей — цианидов другими кислотами. Для получения ее в безводном состоянии смесь равных частей концентрированной серной кислоты и воды прибавляют по каплям к кускам цианида калия.
Незначительные количества синильной кислоты или продуктов, легко расщепляющихся с образованием синильной кислоты, Часто встречаются в растительном мире. Наиболее известен продукт, встречающийся в горьком миндале в форме амигдалина (от ацеубаХц — миндаль) — соединения, легко расщепляющегося на виноградный сахар, бензальдегид (масло горького миндаля) и синильную кислоту под действием также находящегося в горьком миндале энзима — эмульсина. Синильная кислота постоянно содержится также в неочищенном светильном газе (в общем 0,1—0,3 об.%). Ее удаляют оттуда, пропуская "газтильныи газ через «газею чистительную массу» (в основном болотная руда или колчеданные огарки; ср. т. II), при этом образуются комплексные цианиды железа, которые частично перерабатывают па цианиды щелочных металлов. Непосредственное соединение элементов Н2, N2 и С с образованием HCN
502
Глава 12
протекает только при температуре выше 1800°, Легче идет превращение аммиака в HCN, например, при взаимодействии его с СО при 500—700° в присутствии такихкаталнза-торов, как А12О5 или CeO2(NH3+CO = HCN + Н2О), или при частичном окисление смеси СН4 и NH3 кислородом воздуха при пропускании этих гааов над раскаленное сеткой из платиновой проволоки СН4Ц-1ЧН3+ 3/2О2= HCN 4- ЗН2О 4- 115 ккал. Техническое значение имеет также метод получения HCN из окиси углерода и аммиака» которые в растворе метилового спирта под давлением образуют формамид HC0NHSi распадающийся при нагревании до 400° в присутствии катализаторов на HCN и Н26,
С водой, спиртом и эфиром цианистый водород смешивается во всех отношениях, В водных растворах он распадается на ионы: HCNJ Н' + 4-CN', однако в очень незначительной степени. Цианистый водород — очень слабая кислота, значительно слабее угольной,
HCN диссоциирует при 18° в 1 Щ растворе па 0,002%, в 0,1 н.^~ на 0,007%, Константа диссоциации К — 4,8-10-10 (при 18°). В соответствии с этим соли цианистого водорода сильно'гидролизуются; ыапример, цианид калия KCN при 18° в 1 н. растворе гидролизов ан на 0,38%, в 0,1 и. растворе — на 1,20%,
Соли цианистоводородной кислоты — цианиды отвечают общей формуле MJCN. В некоторых реакциях они напоминают хлориды, Например, своим отношением к нитрату серебра. Последний высаживает из растворов цианидов творожистый белый осадок цианида серебра Ag' 4- CN'= AgCN. Этот осадок лишь слабо растворим в разбавленных сильных кислотах, хотя цианистоводородная кислота и является слабой кислотой.
Тот факт, что цианид серебра практически не разрушается при действии сильных кислот, обусловлен его чрезвычайно малой растворимостью. Произведение растворимости AgCN при 17,5°, по данным Моргана, равно A AgCN= [Ag-][CN'l — 10,07-10“i4. В растворах, полученных обработкой цианида серебра кислотой, концентрация ионов серебра выражается формулой
[Ag-] = [HCN]-HCN'J. (5)
Подставляя в (5)
[H-]|CNq_[H-HCN'l 1 |- К ~ 4,8-10-ю
10,07-10-И
и [CN ] =—ь___— f получим
[Ag*p = 2,00-10-4-[Н*] 4-10,07-10-Н,
[Ag*] = 1,45-10-2 /[Н-]4-4,8- 10-ы.
В 100 л»л 2 н. серной кислоты ([Н1]—1) растворяется в соответствии с уравнением менее 0,2 а цианида серебра. Напротив, если обработать цианид серебра соляной кислотой, то оя превращается в хлорид серебра AgCl, так как произведение растворимости последнего (Aakgi=2,0 • 10-io) достаточно мало и достигается при имеющейся в кислом растворе концентрации ионов Ag-, '
Цианиды щелочных и щелочноземельных металлов легко растворимы в воде; цианиды тяжелых металлов большей частью трудно растворимы [за исключением цианида ртути Hg(CN)J. В результате гидролитического расщепления растворы цианидов щелочных и щелочноземельных металлов обладают сильно основной реакцией и пахнут синильной кислотой.
Ионы С1\7' очень склонны к образованию комплексов. Почти все тяжелые металлы, если они вообще образуют цианиды, с избытком циан-ионов образуют комплексные ноны. Наиболее известным примером последних являются цианоферрат{\\)- и цианоферрат(Ш)-иопы: LFe(CN)0]"" и [Fe(CN)e]'". Соответствующие им калиевые соли называются желтой и .красной кровяными солями: K.4[Fe(CN)a] — калийцианоферрат(П)
Четвертая группа периодической системы
503
и K3fFe(CN)sJ — налийцианоферрат(Ш). Они, так же как и соли щелочных металлов других комплексных цианидов, растворимы в воде. Вследствие комплексообразования цианиды тяжелых металлов, даже цианид серебра, растворяются в воде в присутствии избытка циан-ионов, так что при добавлении небо л i алого количества нитрата серебра в раствор, содержащий избыток циан-нонов, осадок не образуется.
Если смешать раствор желтой кровяной соли с раствором соли железа(Ш), то получается темно-синий осадок — берлинская лазурь. От ее названия производится название синильной кислоты и цианидов (ср. стр. 500).
О составе берлинской лазури см. главу о железе (т. II).
Цианид патрия. NaCN получают в технике главным образом по методу Кастнера — Келльнера из амида натрия NaNHa, который в свою очередь получают, пропуская газообразный аммиак над расплавленным натрием NHa+ Na = NaNHarf- У2Н2. При нагревании с углем до 600° амид натрия превращается сначала в цианамид натрия NasNeC. : 2NaNH2-j-С — = NasNsC + 2На, который при более сильном нагревании в присутствии угля превращается в цианид натрия NasNaC + С •= 2NaCN.
Цианамид патрия является производным цианамида — NC-NH2, а последний — производным цианистого водорода, в котором место водорода занимает амидогруппа — —NHS. /
В более новых методах получения цианида натрия исходят из цианамида кальция. С хорошим выходом он также подучается при взаимодействии NH3 и СО с NaapO3 при 600°, протекающем по схеме
2МНз+ЗСО + ЫагСО3 2ХаСК-р2Н2О-}-Н2+2СОг—29 ккал (Hcnglein, 1930).
Цианид натрия образует бесцветные кристаллы, очень хорошо растворимые в воде и спирте. Его применяют главным образом при добыче золота методом цианидного выщелачивания (см. т. II, гл. о золоте). Кроме того, в гальванопластике и гальваностегии он служит для приготовления ванн, особенно для благородных металлов*. NaCN используют в фотографии,
Цпапид калия. KCN кристаллизуется из водных растворов в форме бесцветных октаэдров. Он гигроскопичен, чрезвычайно легко растворим в воде, существенно Труднее в спирте. При получении его исходят главным •образом из железоцианистых соединений, получаемых из отработанных газоочпетительных масс. Кроме того, его добывают из содержащих три-метиламин газов, образующихся при сухой перегонке мелассы — отхода производства свекловичного сахара.
N(CII3)3 -D1_LG2(’° НСМЦ-2СН4; IIGN-pKOH = KCN-}-H2O.
Цианид калия находит в основном такое же применение, как и цианид натрия, которым он в настоящее время все более и более вытесняется.
В торговых продуктах содержание CN' часто выражают в «процентах цианида калия». 100 г безводного цнапиДа патрия соответствуют 132,65 г цианида калия. В соответствии с этим продажный «цианид калия 100%-ный» пе является чистым цианидом калия, а лишь продуктом, соответствующим чистому KCN по содержанию CN', так
* Гоккель (G o с к с 1 II., Z. Elcktrocb., 40, 802, 1934)] для гальванических ванн вместо ядовитых цианидов рекомендует применять неядовитую тиомочевипу CS(NHt) s
504
Глава 12
как снижение содержания CN' вследствие имеющихся в продуктах загрязнений, таких, как карбопат калия, может компенсироваться наличием NaCN. Соответственно марка «дианид натрия, 100%-ный» отвечает не чистому цианиду натрия, а продукту, который содержит столько же CN', сколько чистый KC.N.
Кристаллическая структура цианида калия, по Данным Боцорта (Bozorth, 1922), соответствует структурам КС! и NaCl (рис. 44, стр. 237), только места атомов хлора заняты центрами тяжести радикалов CN. Их продольные оси лежат в направлении пространственных диагоналей восьми слагающих кубов, на которые распадается элементарный куб. Длина ребра элементарного куба д= 6,55 А. Расстояние С<— — 1,15 А. Наиболее короткое расстояние между К и С, а также между К и N равно 3,0 А,
Цианид кальция Ca(CN)2, т. пл. 640°, используется для борьбы с вредителями.
Цианамид кальция (азотистая известь) получают взаимодействием тонко измельченного карбида кальция с азотом при высокой температуре
CaC2-J-N2 CaCN24-C+72 ккал.
Эта реакция была открыта Роте в 1898 г. и применена в промышленном масштабе Франком и Каро. Она приводит к (дивариаитпому) равновесию (Cochet А.), которое с повышением температуры сдвигается в направлении, указываемом пижней стрелкой. Нагревание (до температуры около 1100') необходимо только в начальный момент. Так как реакция сильно экзотррмичпа, она в дальнейшем идет сама по себе. Примесь сухого хлорида кальция ускоряет реакцию. Остаток карбида удаляют, обрабатывая продукт мелко распыленной водой. Цианамид кальция должен содержать по стандарту не более 0,1% карбида.
Цианамид кальция получается в виде серого порошка. Это соединение производится от цианамида NC Nib при замене атомов водорода на кальций: NC’NCa. Перегретым водяным паром соединение разлагается, отщепляя аммиак
GaCNs4-3IIaO = СаСО3+ 2NH3.
Такое же разложение водой постепенно (и через промежуточные продукты) протекает и при обычной температуре под влиянием почвенных бактерий. Поэтому цианамид кальция получил большое значение в качестве азотного удобрения (а также для производства аммиака, ср. стр. 654).
Применению цианамида кальция в качестве удобрения препятствует его способность пылить, а также значительная щелочность, вызываемая наличием в нем окиси кальция. Однако эти свойства удалось в значительной степени подавить прибавлением незначительных количеств масел или жиров. В настоящее время цианамид кальция выпускают в продажу и в виде зерен.
Чистый цпапамид кальция бесцветен и образует ромбоэдрические кристаллы, которые при температуре около 1300° сублимируются без плавления. Его можно подучить превращением
СаО -> 2HCN GaGNa СО + Н2 и,
наоборот, при действии смеси СО и И, па CaCN2 мОЖпо получить HCN (Franck, 1931). Водой па холоду циапамид кальция разлагается с образованием Ca(HCNa),
2CaCNr2 2НОН — Ca(TICN2)2 -р- Са(ОН)2 -у 161,2 ккал.
При действии кислот образуется свободный цпапамид. Поэтому последний попу, чают, обрабатывая цианамид кальция водой и двуокисью углерода при обычной тем. псратуре.
При нагревании раствора цианамида до 70° получают мочевшгу
CaCN24- П2СО3 = СаСО3CK-NH2; CN-KH2 + НаО = CO(NH2)2
Цианамид Мочевина
Эта реакция имела одно время техническое значение для получения мочевины.
Цианамид магпия MgCN2 получают взаимодействием MgO с IICN при температуре красного каления (Franck, 1931; Hartmann, 1951). Это —. белый мелкокристаллический
Четвертая группа периодической системы
505
порошок, d = 2,170, устойчивый в атмосфере сухого вовдухя; во влажном воздухе ов медленно разлагается с образованием NH3. При 1370° он разлагается на С, Mg и N2,
Соединения тиоциаяа (родана)
Соединения, называемые тиорианистыми, : а также роданистыми., содержат одновалентный радикал — SCN (тиоциап- или родан-радикал), который, подобно циан-радикалу, может выступать и как одновалентный отрицательный нон SCN'. От этого иона производятся роданистоводород-ная кислота HSCX и ее соли — роданиды общей формулы MJSCN. Среди роданидов выделяется интенсивной кроваво-красной окраской роданид трехвалентного железа. Отсюда и происходит название родам, (от p56cog — красный) для тиоциан-радикала.
Роданистые соединения содержатся в небольших количествах в организме, и прежде всего в слюне, В небольших количествах они, в противоположность цианистым соединениям, но ядовиты. Считают, что они обладают стерилизующим свойством и содействуют пищеварению,
В незаряженном состоянии родан-радикал не способен существовать. Как я в случае циана, два таких радикала объединяются, образуя диродан NCS—SCN, который, однако, обычно называют просто радоном.
Родам, диродан (SCN)2 был впервые подучен в свободном состояния в 1919 гг. Зёдербекком (Soderback) ври действии брома, растворенного в сероуглероде, на роданид серебра
2AgSCN + Br2= 2AgBr 4- (SCN)2.
Оп устойчив только при пониженной температуре, образует светло-желтые кристаллы с температурой плавления от —2 да — 3°. Расплав вскоре спонтанно разлагается с выделением желтого дыма и с образованием кирцичпо-красного аморфного твердого вещества. Несколько устойчивее он в растворе. В спирте и эфире род ап растворяется чрезвычайно легко, хорошо растворим он также в сероуглероде и чедыреххлорнетом углероде, Вода тотчас разлагает (SCN)2. В органических растворителях также протекает разложение — медленное при пиакнх температурах, моментальное прп комнатной температуре. При этом выделяется желтое аморфное вещество, которое получал еще Либих, пытаясь приготовить свободный родан путем окисления цианидов хлором в водных растворах. В химическом отношении свободный родам очень напоминает иод. Оп также соединяется с металлами и даже обладает почти такой же окислительной способностью, как иоД- Родап способен вытеснять свободный иод из иодидов и сам может быть вытеснен избытком иода. Следовательно, существует отчетливое равновесие
J, -У 2SCN' 21' 4- (ЙСМ)2.
Плотность пара родана трудно установить из-за его неустойчивости; все же Лехер (Lecher, 1919) измерением понижения точки замерзания в бромоформе (СНВг3) установил, что его молекулярный вес отвечает формуле (SCN)2,
Родан взаимодействует с TI2S в эфирном растворе, образуя роданистую серу S(SCN)3— соедиповне, построенное аналогично хлористой сере ЙС12 ')
2(SCN)2 + H2S = S(SCX)2 4- 2HSCN.
Роданистая сера была обнаружена еще в 1828 г. Лассе нем как продукт взаимодействия хлористой серы SCI2 и роданида ртути SCL 4- HgfSCNJs = HgCls S(SCN)2. Аналогично и.ожпо получить S2(SCN)2, соответствующий S2C12, взаимодействием Hg(SCN)2oS2Cl2, растворенной в четыреххлористом углероде или в сероуглероде: 82С12Ч-+ Hg(SCN)2= lJgCl2+ S,(SCN)2- Существует также нитровилроданид NO-SCN, аналогичный нитрозилхдориду NOCI. Это чрезвычайно неустойчивое интенсивно окрашенное в краспо-корпчневый цвет соединение, которое легко расщепляется на окись азота и родан и благодаря атому обнаруживает реакции последних, папример взаимодействует с белым роданидом меди (I), образуй черный роданид меди (II) iNO'SCN -(--j- Св SCN = Cn(SCbJ)a -|- NO. Питр о зил роданид образуется при обработке нитритом щелочного металла концентрированного раствора роданида щелочного металла, подкисленного сорной кис.тощй; ок может быть также получен взаимодействием окиси азота со свободным роданом.
506
Глава 12
Родамиетоводородная кислота и роданиды. Роданистоводородная (тиоциановая) кислота — бесцветная, маслянистая, очень летучая, резко пахнущая, легко затвердевающая жидкость (т. пл. 5°). В чистом состояний она очень неустойчива, и ее можпо хранить только при пониженной температуре (охладительная смесь) или в разбавленном (менее 5?п) растворе. При ее разложении образуется цианистый водород наряду с твердым желтым продуктом, так называемой изопертиоциановой кислотой HaCaNaSa.
С водой роданистоводородная кислота смешивается во всех отношениях. Ее водный раствор легко получить при разложении роданидов кислотами или при пропускании раствора роданида аммония через катиоцообменные смолы (например, леватит), предварительно обработанный HCI. В безводном состоянии это соединение получают, слабо нагревая сухой роданид ртути или свинца в токе сероводорода
Pb(SCN)2_|-H2S=PbS4-2H8CN.
Роданистый водород — сильная кислота. В водном растворе она, подобно хлористоводородной кислоте, практически полностью или по крайней мере почти полностью диссоциирована.
Соли роданистоводородНой кислоты — роданиды (тиоцианаты) легко получаются из цианидов путем присоединения серы. По химическим свойствам они сильно напоминают хлориды. Как и последние, роданиды образуют с нитратом серебра нерастворимый в воде и разбавленных кислотах осадок — роданид серебра AgSCN. Типичной и очень чувствительной реакцией, на роданиды является уже упомянутое выше красное окрашивание, появляющееся вследствие образования роданида желёза(Ш) при взаимодействии ионов F‘" и SCN'. Родан-ионы сами по себе бесцветны, так же как и их соли с бесцветными катионами. Большая часть роданидов хорошо растворима в воде. Нерастворимы роданиды серебра, ртути, меди и золота. Трудно растворим роданид свинца, который разлагается кипящей водой.
Умеренно к он центрированной (1:1) серной кислотой роданиды разлагаются с выделением COS
MJSCN 4- 2Н2&04+ Н2О = COS (NH4)HSO4^>- MtHSO4.
Некоторые роданиды, а также ион SCN' в растворе, присоединяют SO2. Это свойство можно использовать для удаления SO2 (и H2S) из газов и для получения чистого SO2 (Hansen, 1933).
Техническое применение роданиды находят прежде всего При крашении тканей. В технике получают главным образом роданид аммония (NHi)SCN, действуя NHs в водном растворе на CSa под давлением при температуре около 110°: 2NHa + CSa = (NH«)SCN + HaS. Выделение сероводорода можно уменьшить, добавляя в реакционную смесь гашеную известь HaS + Са(ОН)а = CaS -j- 2НаО. Роданид аммония представляет собой бесцветную соль, кристаллизующуюся в виде пластинок или призм с удельным весом 1.31 и температурой плавления 159°,. Он растворяется в воде очень легко и с сильным охлаждейием, В 100 г воды при 0д растворяются 122, при 20°—162 г (NH4)SCN. Легко растворим он также и в спирте. В лабораториях его применяют как реактив на соли жолоза(П1) и для определения серебра по методу Фольгарда.
Роданид калия KSCN кристаллизуется в виде бесцветных призм, удельного веса 1,9. Он плавится при 161°. Расплавленная соль при 430° окрашена в синий цвет, а при остывании снова становится бесцветной.
Четвертая группа, периодической системы
507
В воде растворяется исключительно легко и с сильным охлаждением В 100 г воды при 0° растворяется 177, при 20°— 217 и при 25°— 239 г KSCN. Роданид калия образуется при сплавлении цианида калия с серой или при сплавлении желтой кровяной соли с поташом и серой. Он находит такое же применение, как и роданид аммония.
Очень легко расплывающийся, но в то же время кристаллизующийся без воды в виде бесцветных ромбических табличек роданид натрия NaSCN используется мало. О структуре роданидной группы саг.; G о о й о а в J., Вет., 73, 127, 1940.
Карбиды
Рис. 88. Элементарная ячейка решетки карбида барин.
а=6,22 Л; с=7,06 Л; ei=>l,4 А.
Карбидами в широком- смысле слова называют бинарные соединения углерода вообще. Но обычно название карбиды относят только к соединениям углерода с металлами, а также с бором и кремнием.
Карбиды (в обычном, узком смысле слова) распадаются на; два класса: карбиды, которые разлагаются холодной или умеренно нагретой водой, и карбиды, на которые вода не действует. Карбиды, которые но разлагаются водой, как правило, также очень устойчивы к действию кислот, даже концентрированных и являющихся сильными Окислителями. В противоположность зто-му они легко окисляются при -сплавлении с гидроокисями щелочных металлов на воздухе,образуя карбонаты.
J. Карбиды, разлагаемые водой или разбавленными кислотами
К ним относятся:
а) Карбиды, образующие ацетилен (ацетилиды)
Их следует рассматривать как соли ацетилена (ср. стр. 475); в соответствии с этим их состав отвечает общим формулам М^С5, МпСг и Водой ил
щенляются с образованием ацетилена. Их солеобразный характер был подтвержден электролизом твердого Na2C2 (Антропов, 11)32). Известны также кислые ацетилиды щелочных металлов М1НС2. В кристаллах ацетилидов радикал — С — С — отчетливо проявляется как самостоятельный структурный алвмеят. Большая часть Лцеттнщов кристаллизуется по типу карбида бария (рис. 88). MgC2 кристаллизуется тетрагонально а0= 5,54 A, cfl= 5,02 А) и, вероятно, является изотопом Th (A (Treadwell, 1948). : Ацетилиды образуют все металлы двух первых главных подгрупп периодической системы, далее Al, Zn й Cd (ср. стр. 47G). Редкоземельные металлы и Th в дивалентном состоянии также образуют ацетилиды (YC2, LaC2, СеС2ит. д... ТЬС2). Так как в этих случаях при гидролизе вследствие изменения валентности металла освобождается водород (Л1е2*+ Н4^ + Н), ацетилен пли его радикал частично гидрируется
до СН4 и С2Н4 (Schmidt J., 1934). Поэтому ацетилиды редкоземельных металлов и тория при разложении разбавленными кислотами дают не чистый ацетилен, а загрязненный значительным количеством (20—30%) метана, некоторым количеством водорода и этилена. Это же справедливо и относительно СГС2, который, по-видимому, также принадлежит к этому типу карбидов.
разбавленными кислотами ацетилиды рас-
508
Глава 12
О взрывчатых ацетилидах Cu(I), Ag и Hg(II) см. стр. 476. [Hg(I)-ацетилид Hg2C2-TI2O ле взрывается, но все же он разлагается при 100*].
б) Карбиды, образующие аллилен
Известен единственный представитель этого типа, впервые полученный Новаком (Nov4k), Mg2C3. Он образует, согласно уравнению
Mg2 |СзТ4П1 - 011 - 2Mg(OH)2+C3H4,
чистый аллилен НС~С—СН3. КТ"2Сз, который кристаллизуется, давая гексагональную решетку (а = 7,43 А, с = 10,59 А), образуется при отщеплении углерода от MgC-при температуре выше 50()г'. При температуре выше 600° он постепенно разлагается на элементы.
в) Карбиды, образующие метан
Карбиды Ве2С и А14С3 разлагаются горячей водой или разбавленными кислотами с образованием чистого метана
Ве2С 4- 4НОН = 2Ве(ОН)г+ СН4;
Д]4С3-Ь- 12НОН = 4А1(ОН)3+ ЗСН4.
В кристаллической решетке этих карбидов атомы углерода удалены друг от друга, Ве3С является антиизотипом CaF2. Близкими свойствами, по-видимому, обладает карбид MrijC, который наряду с метаном дает водород
Мн3С 4- 6НОН = ЗМн(ОН)2+ СН4+ Н2.
г) Карбиды, которые образуют смесь углеводородов (без ацетилена}
Сюда относятся прежде всего карбиды группы железа, далее, один из карбидов. Урала — соединение U2C3, отличающееся тем, что Кри гидролизе дает преимущественно жидкие углеводороды. Карбиды этой группы по своей структуре, по-вядимому, близки к интерметаллическим соединениям. Подробнее они будут рассмотрены в т. 11.
Теплоты образования ряда карбидов, разлагаемых водой или разбавленными кислотами, указаны в табл. 83. По данным 1Птаккельберга (Stackclherg, 1930) образование ацетилена при гидролизе карбидов происходит в том случае, когда радиус атома металла превышает 0,8—1 А.
Таблица S3
Теплоты образования карбидов, разлагаемых водой пли разбавленными кислотами (шм/Ж>лъ}
1л2С2 Na3Ca MgCa Mg2C3 CaC2 BaCs А14Сз
-1-13,9 -4,1 -21 -19 + 14,5 4 0 +40
AgaCa ThCz UC2 М»гС РсдС Со3С NiSC
— 83,6 +46 +29 4-3,6 -2,5 ? —5,8 ? -9,1 -9,2
II. Карбиды, не разлагаемые разбавленными .
кислотами
Устойчивые к действию кислот карбиды обладают (так же как .и большая часть неустойчивых) ярко выраженной способностью кристалл изо-
Четвертая группа периодической системы 509
ваться. Часть их чрезвычайно тверда и хрупка; сюда относятся такие соединения, как карбид бора н карбид кремния, по твердости приближающиеся к алмазу.
Только немногие из карбидов этого класса имеют простые и отвечающие нормальным валентностям компонентов формулы, например SiC, TiC, ZrC. Некоторые карбиды, например В4С, Сг4С, Сг^С^, по составу никоим образом не соответствуют нормальным валентностям компонентов, ОЛИ близки К и;1терметаллическим соединениям, о которых пойдет речь в следующей главе. TiC и ZrC обладают, по Данным Клемма (Klemm, 1931) и Штаккельберга (SLackelberg, 1934), характером типичных интерметаллических соединений, в то время как SiC принадлежит к числу так пазываемгях алмаз» подобных соединений, т. е. ковалентных соединений с атомной кристаллической решеткой. TiC, ZrC, VC. NbC и TaC образуют решетку типа каменной соли. Относительно тонкого строения SiC см, дальше.
Получение карбидов осуществляется главным образом при нагревании соответствующих окислов с углеродом. Последний отнимает у окисла кислород и занимает его место. При очень высоких температурах карбиды распадаются на составные части. У некоторых из них такой распад напивается уже при относительно слабом нагревании. Это касается прежде всего карбидов щелочных металлов, которые поэтому, как указывалось ранее, получают другим способом.
Промежуточное положение занимает Li^C,,'для которого температура разложения хотя и достаточно низка (около 600°), одпако превышает температуру образования.
Карбид кремния, карборунд SiC образуется при нагревании смеси кварцевого песка и кокса до температуры около 2000° в электрических печах
SiO2+3C=SiC-|-2CO.
Взаимодействие начинается около 1600°, однако только при температурах выше 1900° оно приводит к образованию кристаллического SiC. Выше 2200° происходит его разложение, причем кремний испаряется, оставляя графит. Получение карборунда осуществляется главным образом на карборундовой фабрике, основанной создателем метода, Ачесоном, на Ниагаре. Для нагревания там используют ток силой до 10 000 а. Теплота образования SiC из элементов составляет (при комнатной температуре) 28 ккал/молъ.
Карбид кремния в чистом состоянии образует бесцветные кристаллы с удельным весом 3,20, которые по твердости приближаются к алмазу. Химически он чрезвычайно устойчив; даже при высоких температурах хлор лишь слегка затрагивает его поверхность, а кислород и сера не действуют вовсе. Сильные кислоты, даже смесь дымящей азотной и плавиковой кислот, в которой растворяется кристаллический кремний, не действуют на карборунд. Напротив, он достаточно легко разлагается при сплавлении с едкими щелочами на воздухе; SiC + 4КОН + 20 я= К 2S i Оз -f- КаС0з+ + 2НаО. При прокаливании с хроматом свинца он также полностью окисляется.
Кристаллическая решетка карбида кремния очень напоминает решетку алмаза и кремния, несмотря па то, что он относится к гексагональной системе, а алмаз и кремний к кубической. Решетку карбида кремния можно подучить, если в соответственно расширенной решетке алмаза заменить Головину атомод С на атомы Si. Расстояние €<—>Si равно 1,90 А и является, следовательно, почти что средним арифметическим из расстояний С<—’Св алмазе и Si <—> Si в, кристаллическом кремпии: Й (1,54+ 2,34) = = 1,94. Имеется также модификация карборунда, кристаллизующаяся в кубической системе (ранее эту форму считали аморфной); она образует решетку типа цинковой •обманки (ср. т. II) с а = 4,37 А; С ч—, Si также равно 1,90 А.
510
Глава 12
Технический карбид кремния (карборунд) имеет темную окраску,, создаваемую содержащимися в нем примесями, Вследствие своей исключительной твердости он находит широкое применение при шлифовании. Его используют также для приготовления огнеупоров,
Силунд и силит, твердые массы, состоящие в основном из карбида кремния, находят применение в качестве огнеупорных материалов и масс для футеровки электрических печей.
Карбид бора. Т34С, получаемый при нагревании бора или трехокиси бора .с углем при температуре приблизительно 2500 , образует черные блестящие кристаллы уд. веса 2,52. Оп обладает такой твердостью, что его порошком можно шлифовать алмазы [однако, по да иным Лео па (Leon, 1937), порошок карбида бора обладает лишь незначительной долей производительности порошка алмаза, если его используют для шлифования алмазов, в то время как он приближается по своему шлифующему действию-к алмазу и значительно превосходит карбид кремния, если его используют для шлифования твердых металлов]. Карбид бора совершенно устойчив по отношению к хлорату калия и азотной кислоте, с хлором и кислородом при температурах нище 1000"он лишь медленно взаимодействует. Раньше карбиду бора приписывали формулу ВвС, По данным новых исследований (Laves, Ridgway, 1934) он имеет состав В4С, что подтверждено определением структуры кристаллической решетки (Жданов и Севастьянов, 1941; Clark, Hoard, 1943). Карбид бора построен из групп, содержащих три линейно расположенных атома С, и групп, содержащих двенадцать атомов В, расположенных в форме почти правильного икосаэдра.
Аналитические сведения, Эле» ентарный углерод в его различных формах легко распознается по физическим свойствам. Химически он и его-соединения просто и надежно обнаруживаются путем превращения их в двуокись углерода. Карбонаты при обработке разбавленными сильными кислотами, вспениваясь, выделяют двуокись углерода. Другие соединения углерода могут быть переведены в двуокись углерода при нагревании с подходящими окислителями — обычно используют окись меди. Двуокись, углерода обнаруживают при помощи, баритовой воды, в которой при пропускании СОз выпадает белый осадок карбоната бария.
Помимо СО2, имеются еще к другие летучие соединения, которые образуют осадки при взаимодействии с баритовой водой,— это SO2 и HCN. Однако их легко можно-отличить от СО2, например, уже по запаху.
Количественное определение СО2 в карбонатах производят косвенно, путем определения потери в весе при прокалийании или разложении кислотами, или прямо, путем поглощения СО2, образовавшейся при разложения кислотами, растворами гидроокисей щелочных металлов или натронной извести. Содержание углерода в других его соединениях определяют сжиганием чаще всего в присутствии окиси меди как окислителя или. платины как переносчика кислорода и взвешиванием образовавшейся COs, поглощенной раствором КОН или натронной известью («элементарный анализ»).
Очепь малые количества углерода, например в пробах стали, можно просто и очень точпо определить следующим образом: образовавшуюся при сжигапип СО3 улавливают очепь разбавленным раствором Ва(рН)2 п потщщпрметрическп титруют выпавший осадок ВаСО$ (О el sen W, и сотр., Angew. Chem.., 63, 557, 1951).
Чувствительным реактивом на окись углерода является раствор хлорида палладия(П). В газовом анализе окись углерода обычно определяют' путем поглощения аммиачным раствором хлорида медй(,1). Метан определяют, сжигая его до СОз и поглощая последнюю (ср. стр. 476). Нена
Четвертая группа периодической системы 511
сыщенные углеводороды можно поглотить дымящей серной кислотой или бромной водой. Ацетилен можно определить на основании его способности давать осадок при пропусканий через спиртовый раствор нитрата серебра или аммиачный раствор хлорида моди(1) (ср, стр, 570 и сл.).
Для определения малых количеств СО (менее 1%) можно использовать ее взаимодействие с пятнокпсыо мода; 5СО + I2O5=s 12-Ь 5СО3. Иод или двуокись углерода, поглощеппую раствором Ва(ОН)2, можно титровать [Pieters, Z, anal. Chem,, 85, 50. 1931]; одпако более удобен для этой доли гоик алитовый метод (ср, стр, 484),
Щавелевую кислоту можно открыть на основании ее способности обесцвечивать горячий подкисленный раствор перманганата с выделением СОа, Взаимодействие с перманганатом очень удобно также и для ее количественного определения. Для муравьиной кислоты характерна способность восстанавливать растворы солей благородных металлов. Для ее определения можно использовать взаимодействие с HgCIa, описанное на стр, 495. Уксусную кислоту определяют по ее запаху и по еще более характерному запаху ее этилового эфира. Еще чувствительнее какодиловая реакция (ср. стр. 711 и сл.).
Для аналитических целей имеют значение также свойства многих, особенно гидроксилсодержащих органических соединений, предотвращать или делать неполным выпадение веерков некоторых солей тяжелых металлов.
Органические вещества можно удалить выпариванием в присутствии концентрированной сорной кислоты.
КРЕМНИЙ (SILICIUM) Si
Распространение в природе. Кремний после кислорода самый распространенный элемент. Однако в природе он никогда не встречается в свободном состоянии, а всегда в виде соединений и почти исключительно в виде производных двуокиси кремния SiOa. Очень часто встречается свободная двуокись кремния. В форме кварца она образует постоянную составную часть гранита и других широкораспространенных горных пород. При их выветривании кварц в форме зерен и комков попадает в реки и моря, откуда вымывается в виде песка снова на сушу.
Две другие составные части гранита — полевой шпат и слюда — представляют собой силикаты, т. е. соединения, также образованные двуокисью кремния, В природе очень распространены силикаты главным образом сильно электроположительных металлов. В табл. 31 на стр. 188 приведены некоторые силикаты.
Из тяжелых металлов в силикатах прежде всего присутствует железо, особенно в часто встречающихся изоморфных смесях силикатов, например в роговой обманке и в авгитах, а также в слюдах, где железо в большей или меньшей степени замещает магний,
В общем зомпая кора, включая моря и воздушную оболочку, состоит приблизительно на f/г своего веса из кремния и на Уз. — из кислорода. Количество кремния в доступной нам части земной коры равно по весу общему количеству всех остальных элементов минус количество кислорода. Для органического мира кремний имеет лишь " второстепенное значение. Правда, почти все растения содержат незначительное количество SiO2. Относительно богаты двуокисью кремния стебли трав, особенно солома хлебных злаков. Двуокись кремния служит, в них материалом, укрепляющим острв
512
Глава 12
растения. В животном организме SiO2 находится прежде всего в соединительных тканях. С возрастом количество кремния, по крайней мере в человеческом теле, уменьшается.
Исторические сведения. Кремень (silex), кварцевый песок, горный хрусталь й другие богатые кремневой кислотой минералы уже в древности использовали для получения стекла. В XVII в. пришли к заключению, что пригодность этих минералов для изготовления стекла объясняется содержанием в пих особого вещества, позднее названного кремнеземом. В элементарном состоянии кремний впервые был получен Берцелиусом (1822) восстановлением фторида кремния SiFa металлическим калием. Берцелиус установил, что при сжигании кремний переходит в кретнезем и, следовательно, он является элементом, лежащим в основе кремнезема. Фтористые соединения кремния —кремнефтористоводородная кислота HaSiF0 и составляющий ее основу фторид кремния Si Fa были обнаружены Шееле уже полстолстия назад при его опытах с плавиковой кислотой,
Получение и применение. Получение кремния основано на восстановлении магнием, алюминием или углем двуокиси кремния или галогенидов кремния при пагревании. Щелочные металлы менее пригодны для этого восстановления, хотя впервые кремний был получен ири помощи калия. В зависимости от условий опыта получают продукты, довольно различные по внешнему виду и по свойствам. Раньше рассматривали их как различные модификации кремния. В действительности их кристаллические решетки одинаковы. Однако они различаются величиной частиц, развитием поверхностей и содержанием примесей (главных! образом SiOa). Относительно чистым и в виде красивых кристаллических пластинок кремний получают алюмотермическим восстановлением кремнефтористого калия K.i(SiFe]
3K2[SiFQ]-l-dAl = 3Si |-2K[A1F?j]4-2K2[A1Fs],
Метод Вёлера заключается к следующем: сиест. 125 а алюминиевых стружок с 40 г К2 [SiF6] уиергнчпо пагреваЬт около получаса до ярко-красного каления л затем медленно охлаждают. Отделенный от шлака п грубо истолчепнын королёк, состоящий из сплава алюминия с преминем, обрабатывают (сначала разбавленной, затем концепт-рцроваппой) соляпой кислотой. В результате кремний остается нерасширенным. Следует брать избыток алюминия,так как оп в расплавленном состоянии действует па кремний как растворитель, из которого*при охлаждении выкристаллизовывается кремний. Так называемый «аморфный» кремний можпо перекристаллизовывать путем сплавления с алюминием.
SiО2 iipii подходящих условиях также можно восстановить алюминием. Согласно патенту Ниже (Kiihne, 1903), смешивают 400 г алюминиевых стружек, 500 е морошка серы и 360 г кварцевого песка; полученную смесь покрывают сверху смесью порошка алюмипия и серы или другой подходящей воспламеняющей смесью и поджигают раскаленной докрасна железкой палочкой.
Очень реакционноспособную форму кремния получают по способу Гаттермана — Винклера восстановлением двуокиси кремния магнием
SiO2+2Mg=Si4-2MgO.
Для предотвращения бурной реакции к рассчитанному по уравнению количеству смеси добавляют четвертую часть окиси магния. Следует избегать избытка металлического магния, так как в атом случае происходит образование силицида. Очень важно, чтобы вещества были чистыми и абсолютно сухими. Глиняный или шамотный тигель с реакционной смесью помещают в заранее нагретую до красного каления печь (печь Роеслера или другая подходящая). Реакция начинается через несколько минут при сильном разогревании массы. Таким способом получают коричневый порошок, который ранее был назван кре.инне.и». Однако по структуре он не отличается
Четвертая группа периодической системы
513
от кристаллического кремния, лишь всегда загрязнен в значительной степени двуокисью кремния Si О, (ср. стр. 514). Действительно, аморфтлш кремний, как показал Шварц, можно получить термическим разложением монохлорида кремния (ср. стр. 521).
В технике кремнии получают восстановлением кварца в электрической печи углем в присутствии железа. Так как кремний с железом дает сплав ферросилиций, образование карбида ограничено (ср. стр. 509); однако этим способом получают не чистый кремний, а сплавы железа с кремнием. Продукт, называемый в технике чистым кремнием или металлическим кремнием, содержит по меньшей мере 2, а обычно 3—5% железа. Чаще получают ферросилиций, содержащей 25% и больше железа. Ферросилиций с содержанием кремния менее 20% можно расплавить в обычной пламенной печи (ср. т. II).
Для получения кремния электротермическим способом используют обычные почи, таге же как и дли получения карбада и карборунда. Иногда к реакционной смеси добавляют еще СцО. В этом случае образуется промежуточный продукт СаС2, который является прекрасным восстановителем и значительно облегчает получение кремния.
Ферросилиций находит применение главным образом в производстве железа и стали. Он понижает способность железа растворять углерод и таким образом увеличивает выделение последнего в ниде графита. Это иногда бывает нужно при кьграбитье чугуна. При бессемеровском процессе ферросилиции применяют как раскислитель для восстановления образующейся при горении окиси железа. Богатые кремнием сплавы железа применяют в большом количестве благодаря их исключительной кислотоустойчивое ти.
Технически чистый кремний пригоден для раскисления медных сплавов (см. т. II). В полевых условиях кремний применяли для получения водорода (ср. стр, 45 и сл.). Было предложено также использовать его в качестве восстановителя вместо алюминия.
Из соединений кремния большое значение в технике имеют прежде всего некоторые силикаты (см. стр. 547 и сл.). Расширяются также области промышленного применения кремнийорганических соединений. Например, эфиры кремневой и поликремневой кислот применяют в качестве связывающих веществ в керамической промышленности, при изготовлении прочно покрывающих лаков для стекла, для получения особого гомконзмельчен-ного силикагеля и для других целей. Некоторые алкоксиаминосиланы, например
(СН3)3С-ОХ
)Sr(NH2)-, (СН^С-О7
вследствие их водуотталкивающего действия применяют для пропитывания тканей, бумаги, керамических изделий и асфальта при строительстве дорог. Однако ц настоящее время большое техническое значение имеют силиконы (см. стр, 519 и сл,) Силиконы отличаются большой термической и химической стойкостью. Особенно они пригодны в качестве изоляционного материала. Цх применяют также как прозрачные, защищающие от влаги покрытия, Некоторые силиконы очень эластичны (силиконовые каучуки); они сохраняют это свойство при относительно низких температурах (до —65°). Другие силиконы Применяют в виде химически- и термостойких искусственных смол (силиконовая резина).
331 г Реми
514
Глава 12
Свойства. Чистый кристаллический кремний образует темно-серые, блестящие, непрозрачные, часто расслаивающиеся на пластинки октаэдры правильной формы. Твердость его значительна (7 по Моосу), он царапает стекло. Но так как оп очень хрупок, его легко можно превратить в порошок. Удельный вес равен 2,33. Точка плавления лежит при 1413°. При температуре электрической дуги он легко плавится. Подобно графиту он обнаруживает электропроводность (возрастающую с повышением температуры). Удельная теплоемкость при обычной температуре равна 0,171. Вычисленная из нее атомная теплоемкость имеет величину 4,80, т. е. она существенно меньше того значения, которое требуется законом Дюлонга и Пти. Однако с повышением температуры теплоемкость все больше и больше приближается к этому значению..
Грубо кристаллический кремний химически не особенно активен.. Он практически не растворим ни в каких кислотах, даже в плавиковой. Загорается он также с трудом. Но при очень высокой температуре (белое Каление) он соединяется с кислородом и азотом, а при температуре электрической дуги — с водородом. При нагревании Si сплавляется с большинством металлов, но из щелочных металлов — только с литием. В некоторых случаях при этом образуются соединения, например с магнием — силицид магния MgaSi; в других случаях кремний просто растворяется в расплавленном металле, например в алюминии. Из алюминия он вновь выделяется при охлаждении. Поэтому кремний можно перекристаллизовать сплавлением с алюминием, а также с серебром.
Из богатого кремнием расплава при медленном охлаждении кремний выкристаллизовывается в вида хорошо образованных октаз дров, при более быстром охлаждения — в виде листочков, похожих по внешнему виду на графит, Из расплавов, соответствующих эвтектическому составу, кремний выделяется в виде очень мелких кристаллов; эвтектическая точка (ср. стр. 609 и сл.) соответствует в приведенных сплавах низкому содержанию кремния. Таким образом получают описанный Муассаном раетваримый в плавиковой кислоте кремний. Еще более реакционноспособным является кремний, получаемый по способу Гаттермана. Это — рыхлый порошок коричневого или Серо-коричневого цвета, легко загорающийся, очень хорошо растворимый в плавиковой кислоте и также легко взаимодействующий с галогенами. Как показали рентгеноструктурные исследования (Gerlach, Debye, Erauenieldcr, Kastner, Remy), все различные виды кремния имеют одну и ту же кристалл личе скую решетку, а именно решетку типа алмаза (рис, 83, стр, 461) с <i= 5,42 А я кратчайшим расстоянием между атомами, равпым2,35 А. Хотя различия в реакционной способности разных видов кремния вызываются второстепенными обстоятельствами (размеры частиц, развитие поверхностей, содержание примесей), а не существованием различных модификаций, одпако, несмотря на это, различные виды кремния имеют большое практическое значение, так как применение препаратов кремния для определенных целей решающим образом зависит от способа их получения.
♦т
Кремний во всех своих формах реагирует очень легко с едкими щелочами, даже с очепь разбавленными растворами. Уже при соприкосновении со стеклом, из которого может быть извлечено некоторое количество щелочи, вода приобретает способность растворять кремний, по крайней мере при кипячении. Реакция обусловлена склонностью кремпия к образованию силикат-ионол
Si 2ОН'+Н2О=Si GJ -}- 2Н2.
Для протекании этого процесса достаточно уже незначительной концентрации щелочи, так как вследствие слабости кремневой кислоты используемые в слабо щелочных растворах ионы ОН' все время образуются в соответствии с уравнением
SiOS+2H2O=H2SiO3+2OH'.
Четвертая группа периодической системы
515
СОЕДИНЕНИЯ КРЕМНИЯ
Наиболее устойчивыми и важнейшими соединениями кремния являются двуокись кремния п многочисленные производные от нее — силикаты. Сродство кремния к кис,городу чрезвычайно велико и оно определяет поведение остальных соединений кремния. Большинство из них разлагается, как только представляется возможность кремнию соединиться с кислородом (например, при взаимодействии с водой). Следует отметить, что, несмотря на большое сродство кремния к кислороду, связь между ним и гомологом кислорода — серой отличается малой устойчивостью.
Соединения кремния с водородом гораздо менее устойчивы, чем углеводороды. Богатые водородом соединения кремния на воздухе загораются и при действии раствора щелочи быстро разлагаются. Это объясняется очень малым по ера в г гению с углеродом сродством кремния к водороду и особенно большим сродством кремния к кислороду (ср. табл. 75).
Значительно устойчивее кремневодородов алкиллроизводные кремния, т, е. соединения типа SiB4, где R — органический радикал. Таких соединений известно значительное число. Огттт гораздо более похожи на соответствующие соединения углерода, чем кремп ев о дор оды, и но воспламеняются иа воздухе. Если все четыре валентности кремния насыщены различными радикалами, то соединения проявляют оптическую активность. Исключительно устойчивым но отношению к химическим реагентам является карбид кремния (пе отличающийся очень высокой теплотой образования) (ср. стр. 509).
Относительной устойчивостью отличаются галогениды кремния. Одпако водой опи гидролитически разлагаются. При действии спиртов возникают алкоксисоедине-пня, при действии реактива Гриньяра — соединения типа SiR4.
Довольно непрочной оказывается связь Si—N. Она легко разрывается большей частью под влиянием воды или г ал о генов одо родов, если атом N связан одновременно еще с другими элементами. Вероятно, сравнительно более прочными должны быть бинарные Si — N-соединения, например St3N41 которые образуются, согласно Вейсу (Wei 13, 1910) и Функу (Funk, 1924), при пропускании азота пад мелкокристаллическим кремнием лри~ 1400°. (Правда, состав этого соединения еще гте установлен, так как оно еще не получено в чистом состоянии. По-вядимому, существуют также еще другие бедные азотом нитриды кремния.) Сравнительно легко реагирует азот с силицидом кальция, причем образуются соединения CaSiNg и Ca(SiN)2, аналогичные цианамиду кальция н цианиду кальция (Wiihler, 1924). С фосфором кремний образует очень устойчивое соединение SiP, с мышьяком — соединения SiAs и SiAs3. Напротив, с сурьмой и висмутом кремний не соединяется.
Атомы кремния далеко пе так легко, как атомы углерода, способны образовывать цепи без включения посторонних атомов. Высокомолекулярные кремневодороды, например Si4H1{), легко распадаются. Водой даже на холоду связь Si—Si в большинстве случаев разрывается.
Напротив, очень устойчива связь Si—О —Si, которая имеется в силоксанах. Соединения кремния очень склонны к конденсации с образованием кислородных мостиков. Это прежде всего проявляется у силикатов (ср. стр. 541).
Силициды. Силицидами называют бипарные соединения кремния с более электроположительными элементами, а именно с металлами. Силициды очень нохгокп па карбиды, однако они еще больше, чем карбиды, приближаются к интерметаллическим соединениям. Большой частью они имеют явно выращенный металлический вид. По кристаллизационной способности они даже превосходят карбиды.
Большинство силицидов устойчиво по отношению к воде и разбавленным кислотам, Разлагаются силициды щелочноземельных металлов (см- далее) и силицид лития Т r6Si2 (Для остальных щелочных металлов сили-33*
516
Глава 12
циды по известны). По своему составу силициды примыкают к интерметаллическим соединениям. Так жо как у большинства карбидов, валентности во многих силицидах не соответствуют тем, которые проявляются в обычных соединениях углерода и кремния.
Силициды получают сплавлением составных частей или восстановлением двуокиси кремния при большом избытке соответствующего металла.
Силицид магния Mg2S| (т. пл. 1070°) кристаллизуется по типу полевого шпата 1а= 6,37 А). Подобную структуру имеют также Mg2Ce (л= 6,38 A), Mg2Sn (а== = 6,77 А) и Mg2Pb 6.8-1 А). Как доказал методом закалки Велер, при высокой Температуре устойчив также MgSi. С кальцием кремний образует соединения Ca2Sa (т. пл. 920’1), Сайг (г. пл, 1220 ) n GaSi2 (т. пл. 1020°). Сплавы кремния с кальцием (большей частью с 30—35 вес.% Ga) применяют в качестве раскислителей в стале.’штой-пой промышленности, прежде всего для нержавеющих сталей.
Кремний с йг и Ва также образует моно- и дисилициды, с Ва, кроме того, BaSi3l и, вороатпо, еще высшие силициды, Ниже приведены теплоты образования (кхал/люль) CaSi 85,3' CaSia 161,5; SrSi 112,8; StSL, 147,4; BaSi 181,5; BaSi3 399,2 (Wohler, 1932).
Некоторые силициды, например силициды магния и марганца, при разложении кислотами наряду с элементарным водородом образуют также и водородные соединения кремния — силаны.
Крем нс водороды (силаны). Образование Кремнев одорода впервые наблюдалось в 1857 г. Вёлером при растворении содержащего кремний алюминия в соляной кислоте. Как обнаружил Вёлер, пригодным для получения силанов является также силицид магния Mg2Si, который можно получить, например, сжиганием смеси магния с тонко распыленным кварцевом песком: 4Mg -f- SrOa=2MgO + MgaSr. Разложение силицида магния кислотами протекает довольно сложно (см. Schwarz R., Z. anorg. Chem., 215, 288, 1933). Силицид магния, подобно бориду магния, наряду с большим количеством водорода образует самовоспламеняющуюся па воздухе смесь различных крем не водородов, которая была исследована подробно Штоком в 1916 г. и в последующие годы. По предложению Штока кремне-водороды называются силанами. В чистом состоянии Шток получил следующие силаны: монасилагс SiHt, дисилан Si.2Hs, трисилан Si3H8, тетра-силан Si4HJ() (см. табл. 84).
Таблица 84 КремиСводороды (силаны)
Sill* SioHjj Si4Hio
Температура ктш&пня, ° С ... —112 -15 +53 ~85
Температуры апютиция, °C , . Пл отпасть гьза, найденная (води- — 184,7 -132,5 — 117 -93,5
род=1) ............ 16,02 (при 19°) 31,7 (при 21е) 46,5 (при 24°) 6:1,0 (при 25°)
Плотность в жидком состоянии 0,68 (при —185°) 0,686 (при -25°) 0.725 (при 0°) 0,79 (при 0°)
Наряду с этим в их смеси, вероятно, находились еще пента- и гексасилан: SiaH13 и Si6H,4. Относительные количества отдельных составных частей быстро уменьшаются с повышением молекулярного веса- Шток разделил различные силаны путем фракционированной перегонки в аппаратуре, которая работала при абсолютном отсутствии воздуха, так как силаны,
Четвертая i-pynmi nepuodn-riei:.Ki>ii сгктем-ы
517
так же как бораны, очень чувствительны по отношению к воздуху: при доступе воздуха они загораются я взрывают с сильпшг треском.
Мопосплап. Sill,— бесцветный, без доступа воздуха вполне устойчивый газ. В разбавленном состоянии оп имеет слабый затхлый запах. На воздухе чистый мои о* силан большей частью воспламеняется и всегда воспламеняется, если содержит примесь высших гомологов. Щелочи разлагают моносплан в соответствии с уравнением SiII4+ 2011'4- Н.;0 = SiO3 -|- 4Н2. Водой ом разлагается аналогично, но значительно медленнее. Вода, ттс содержащая щелочи (в кварцевом сосуде), вообще по разлагает моносилап. Нрп нагревании (выше 400°) SiH4 распадается па Si и Н2. При действии тихого электрического разряда происходит распад с образованием блестящего, как золото, палю а, состоящего из твердых ненасыщенных гидридов кремния (Schwarz В,, 1935).
Дней лай. Si2Hc по свойствам очень похож на моносилан. В противоположность BsIIfl оц начинает заметно распадаться только при температуре около 300’, С водой реагирует медленно по уравнению St2H64- 4Н.,0 = 2StO24- 7Нг. С четырех хлористым углеродом и хлороформом реагирует часто с воспламенением, в противоположность моносилану, который заметно ими не разлагается.
Белыпие количества моно- и дисилана можпо получить по Джонсону (Johnson, 1935), лучше всего при разложении силицида магния растворенным в жидком аммиаке бромидом аммония, который в этом растворителе действует тан же, как кислота (ср. т.П).
Трпсилап. Si3HB и тот расплав Si,tHI0 менее устойчивы, чем низшие силаны. В еще большей степени это справедливо для высших силанов.
Галогепозамешеиные производные силанов. При действии брома иля При слабом нагревании с бромистым водородом в присутствии бромида алюмипия можно заместить По очереди все атомы водорода в силанах бромом. Например, из SiII4 таким образом получаются соединения SiHsBr, SiHaBr2, 5ШВг3 и SiBr4, первое — газ, другие — также довольно лсгколстучие вещества. Соответствующим образом атомы I-J в S1H4 можно заместить па Cl, I и F. Известны также смешанные типы, например SiHF2Cl и SiHFCla. Некоторые свойства гал от ено силанов описаны в табл. 85. Теплоты нс пар е-
Таблица 36
Плотности, температуры плавления н температуры кипения галогепоеиляпов
Плотность, (ври t, *C) Температура плавления, °C Температура кипения, ’С
SiHF, -131,2 — 97,5
S1H3C1 1,145 ( — 113’) — 118 -30
SiH2Cl2 1,42 — 122 +8
( — 120°)
SiHCJs 1,34 -128,2 4-31,5
SiH3Br 1,533 -94 +2
(O’)
SiHoBra 2,17 -77 4-64,0
(0°)
SiHBrs 2,7 — 73,5 111,8
SiH3I 2,035 (14,8°) -57,0 п-4э,4
StH212 2,746 (15,1°) -1 149,5
SiHJ3 3,286 — —220
(23°) f
518
Глава 12
ния SiH-jI и SiH2I2 составляют 7,13 и 8,05 ккал!моль, поверхностное натяжепие (при 15".| 30,50 и 44,1 дин/см, парахор 182,1 и 267. [Е m е 1 6 u S Н. J., J. chem. Soc., 143, 353, 1941].
Подлеплен [SiIIa]x был получен Шварцем (Schwarz R., 1935) При разложении Сайт ледяной уксусной кислотой пли спиртовым раствором соляной кислоты. Подцеплен — светло-коричневое, твердое вещество, воспламеняющееся на воздухе. Водой он разлагается по уравнению 81На4- 2Н2О = S10a-f- ЗН3; силаны прп этом не образуются, однако образование последних происходит при его термическом разложении в высоком вакууме.
Силоксаны. Галогенозамещенныс силаны легко разлагаются водой. Из частично замещенных силанов при этом получают интересные водород-кислородные соединения кремния, в которых атомы кремния связаны между собой через кислородные мостики
— Si — O—Si =
Шток назвал их силоксанами. Так, при взаимодействии броммоно сил а па с водой тотчас возникает дисилоксан (SiHs)2O, Реакция протекает почти количественно по уравнению
Их
И-^Si—В г н/
Их
H-4S1—Вт и/
Их H-^Si Н/
Нх
H-^Si н/
О4-2НВТ
.Цисилоксан — бесцветный, по имеющий запаха газ, не воспламеняется па воздухе, по, будучи зажжен, горит с образованием белого дыма, состоящего из SiOa (т. киц. —15,2°, т. пл, —144°, уд. вес 0,881 прп —80°). При комнатной температуре без доступа кислорода сохраняется без изменения. Только при температуре красного каления он подвергается быстрому разложению. Соприкасаясь с кислородом, он воспламеняется со взрывом, причем сгорает до SiO2 и Н2О. Щелочами быстро разлагается по уравнению
[(SiHa)aO -]- 4ОН'+ Н.2О = 2S10J + 6На.
При действии воды реакция протекает ступенчато, при] этом в качестве промежуточного продукта образуется просилоксан ILSiO — газ, который легко полимеризуется в аморфную твердую белую массу.
Аналогичным образом из Si2II3Br получают жидкий тетрасилоксан (Si-Hs)aO п т. д. Для некоторых из этих веществ известны также органические производные, в которых па месте атомов И находятся алкил- или арилрадикалы,
Силоксен. Другое интересное соединение кремния, кислорода и водорода — это силоксен SbjOjH,., получающийся при обработке Са81а соляной кислотой
3CaSra-f- 6НС1 4- ЗЩО = Si6O3Hc4-3CaCla-|- ЗН3,
Силоксен — твердое, ни в чем не растворимое вещество, в высшей степени поверхностно активно с, чувствительное к воздуху, воде и свету. Оно впервые было исследовано Каутским, В качестве характерных реакций этого соединения приведены следующие:
SisosH64-ijS=si.oJHBi4-ni, (1)
Si60sHc4-HBr=Si6O5HBBr4-Hs, (2)
SiaO3TIe4" ЗВг2=51вОзН3Вгз4" ЗНВг, (3)
Sr6O3H3Brs -т64Н3 = SjeO3II3(NH3)3 4- 3NH4Br. (4)
Все эти соединения твердые вещества, Они нерастворимы в обычных растворителях, более или менее интенсивно окрашены. Например, получающийся при гидролизе трибромеилоксона тригидрокси силоксен SieO;5H3(OH)3— фиолетово-красный, гексагидрокси силоксен Sie03(OII)6—интенсивно черный. Все эти соедипепия при действии света становятся очень активными, многие проявляют яр невыраженную люминесценцию.
Четвертая группа периодической системы
519
На основании этих реакций Каутский предложил следующую формулу строения сило-ксеца:
Длительным хлорированием сило кс ей Можно перевести в гексахл орсил оксан Cl3Si—-О—SiCl3, а взаимодействием со спиртами ROH — в дикрсмпевокислый эфир XRO)3Si— О—S1(OR)3.
Алкнлпроизводиые кремпия. Значительно более устойчивыми, чем кремневодороды, являются алкилпроизводпы-е кремния, т. е. соединения, в которых с кремнием связаны органические радикалы (алкилы). Их общая формула SiR-j. Известны некоторые соединения типа SiaR6. Органические радикалы R могут быть производными углеводородов алифатического или ароматического рядов. Кремний может быть связан также с радикалами того и другого ряда одновременно.
Алкилпроизводные кремния в отличие от кремпеводородов иё воспламеняются на воздухе, они относительно устойчивы. Водой не разлагаются и не растворяются в пей. Чисто алифатические соединения кремния — легкорастворимые в органических растворителях жидкости, чисто ароматические соединения — твердые при обычной температуре вещества и в большинстве случаев хорошо кристаллизуются.
Для получения алкшшроизводных кремния вначале исходили из цинкалкилов ZnR3, которыми действовали па галогениды кремния, например на SiClv Лучше использовать магнийалкилы. Можно применять реакцию, аналогичную синтезу Вюрца илгцФиттига в органической химии, т.е, действовать натрием па смесь кремнии- й алкил галогенидов
SiCl1+^RC-l-|-8Na=SiR4-|-8NaCI.
Эта реакция идет очень легко с ароматическими углеводородами.
уАлкилпроизводные кремния можно получить непосредственно из SiHt и углеводородов. Как показал Фриц (Fritz, 1951), при нагревании до 400° SiHt с этиленом образуются соединения C2H5-SiHs, (С2Щ)3Я1Н2 п (C3H5)3SiH. Наряду с этим получаются также соединения с различными алкильными остатками, например
|Н3С н,сч
>SiH2 i-.j )SiH3 Н5Са^ Н7С/
При продолжительном нагревании образуются трудно летучие, маслянистые продукты и твердое желтое вещество [SiCH3lx. Точно так же, как с этиленом, SiH4 реагирует с насыщенными углеводородами и с винилхлоридом. При взаимодействии SiHj с окисью этилена и с ацетоном получают алкоксисоединения, например C2H3O,SiH3 и C3H-O-SiHa. Если использовать подходящий катализатор, то SiH4 и его производные реагируют с органическими соединениями уже при низких температурах (Nitz-sche, 1951).
Силиконы. При разложении водой5 триалкилгалогенсиланов получают ал кил заме щепные силоксаны, например
2(CHs)sSiCl -f_H2O=(GH3)3Si—О- Si(CH3)3-|-2HCl.
Если исходить из соединений типа SiREXa (гДе R — алкил, X — галоген), то образуются вещества, которые содержат более или менее большое число
520
Глава 12
R
групп — Si—, снизанных между собой через атомы кислорода, В согласии R
с Киппингом (Kipping, 1901 и сл.) их называют силиконами*. Промежуточной стадией является образование силиколей, соединений, в которых атомы галогена замещены на группы ОН, Силиконы, химия и технология которых развилась в период второй мировой войны в США, имеют большое практическое значение. В химическом и термическом отношении они представляют собой довольно устойчивые соединения. Низкомолекулярные силиконы, в которых SiB-20-групиы связаны между собой в цепи или (чаще) в кольца, являются прозрачными легко подвижными жидкостями. Высокомолекулярные соединения — вязкие вещества. Они содержат длинные цепи, разветвленные или перазветвленные, что зависит от способа получения. В зависимости от длины цепи силиконы — маслянистые жидкости или пастообразные вещества. Их применяют в качестве превосходных смазок, так как их вязкость лишь незначительно уменьшается с повышением температуры. При еще более длинных цепях получаются каучукообразные продукты. При подходящих условиях вместо цепеобразно следующих друг за другом групп SiRaO возникают пространственные сетки, аналогичные тем, которые имеются в силикатах с пространственной сетчатой структурой (ср. стр. 546). Такие трехмерные сетки имеются в силиконовых искусственных смолах, применяемых в качестве электрических изоляторов.
При получении силиконов спадала сап тезируют алкил- или арилгалогсчгосилаиы из S1CI4, из тетр аз тилевого эфира кремневой кислоты или исходя из элементарного-кремния. Затем эти соединения гидролизуют для получения соответствующих силико- ° лей. Силиколи соответствующей обработкой (например, нагреванием или добавлением кислоты) конденсируют в силиконы. Добавлением «грмалкилгалогеносилапа к диалкил галогеносилану можно изменить длину цепи, полученной методом конденсации-Например;
SiR8ClB-|-2HaO=SiIl-2 (ОН)а-|-2НС1; SiR3Cl+HsO=SiR3 (ОН)-|-НС1,
R R R R
НО—Si — ОН-| ИО—Si — ОН = ПО—Si—О—Si—ОИ-(-Н2О;
R R ПК
В R R R R R
НО — Si — О—Si — OH-|-2SiR3(OH) = RSi—О— Si—О—Si— О — SrR4-2H20.
R R R R R R
Сп.тазапы. Соединения, в которых атомы Si связаны однп с другим через имидо-груяпы NII, называются силазанами. Если же образуются кольцевые соединения, то это — !jиклоеилазаюл; примером последних следует считать соединения, полученные Ларсоном и Смитом (Larson, Smith, 1949) из NH3 и Si(CHs),H2
Н
N
(CII3)2Si Й1(СИ3)2
HN Nil
(CH3)2Si
HN
H (CH3)2
N—Si
bi
Si(CHs)2
(CH3)2
Гекс амети лциклот p и си л ааая
(CII3)2 H
Оттамегилци г: л отет pa с плавай
* В развитии химии крем неорганических полимеров большое значение имеют работы К. А. Андрианова (см. «Полимеры с неорганическими главными цепями молекул», изд. АН СССР, 1962).—Прим. ред.
Четвертая группа периодическоа- спете.мы
521
Эти соединения совершенно аналогичны паппростейшпм циклическим силиконам. Шварц педаюго (1952) получил соответствующие соодппопия, которые if а месте С Из-группы содержат радикал RO (R — изопропил или фенил). Он установил, что диамнды (R О) 2S r(NII2) 2 и общем значительно более склопцы к конденсации в циклоешмваиы с отщеплением NH., чем мопоамиды (RO)3SiNH2, из которых при отцщплепии NII^ образуются дисилазаны (BO)3Si—NH—Si(OR)3. Шварц также получил соединения типа (RO)3Si—NR 2 и назвал их силазшеами. Например,
zgh2-ci-i2—сн2
(C6H5O)3Si- N< I
ч:;н2—сн2— си2
Трифено в с । щшг логекс и л сииази в
/ И3СЧ .Н X СО — СН2 — СН2
И А'х Sl-N< !
\H,CZ ЧО—/3 4)0—СНо— снг
Т рнигюир оно псиц пил отетраметп лен д н кетоыма вин
При получении этих соединений исходными веществами были гсксамстплендиамгпт или же хлорангидрпд адипиновой кислоты. Под влиянием атома Si происходили за»гы-кание в цикл, а нс образование линейных полимеров, как можно было бы ожидать по опыту органической^ химии.
Галогениды кремния
Простые галогениды кремния, соответствующие общей формуле SiXs, образуются при непосредственном взаимодействии кремния и галогена, а фториды — также при действии HF на SiOa. Эти соединения сами по себе довольно устойчивы, однако водой уже при обычной температуре быстро разлагаются, что объясняется особенно большим Сродством кремния к кислороду. Б= общем устойчивость галогенидов возрастает от иодида к фториду. Фторид способен присоединять дополнительно ионы фтора, образуя комплексные фторосиликатные ионы, и прежде всего гексафторосиликатный ион
SiF,+2F' = [SiF6]"
Этот ион также вполне устойчив в водном растворе, он представляет собой значительно более устойчивый комплекс, чем фтороборатный ион.
Остальные галогениды кремния ие присоединяют дополнительных ионов галогена.
Галогениды кремния можно рассматривать как галогепзамещенные силаны. Б Соответствии с этим, кроме простых галогенидов SjX4, известны и такие, в которых несколько атомов Si образуют цепи, Эти соединения дают интересные продукты гидролиза. То Же относится и к неполностью замещенным силанам, о которых уже частично шла речь при описании кремне водороде в. В дальнейшем коротко следует остановиться еще на тригалогенсиланах SiITX3, получаемых гораздо легче иным путем. Шварцу удалось получить (1937—1947) хлориды кремния с довольно для иными цепями, например Si10Cl22 и Si26ClS2. Первое вещество представляет собой бесцветное вязкое масло,, которое можно перегнать в высоком вакууме прн 215°; па воздухе оно загорается, водой энергично разлагается с образованием Silc(OH)22. Это соединение — белый рыхлый порошок, медленно окисляющийся на воздухе, причем связь Si—Si постепенно разрывается с образованием мостиков Sr—О—Si. Соединение Si2SCI52 — смело образное, пластичное вещество, которое можпо получить из паров SiCl4 в атмосфере азота при температуре ок(>,ао 1250° в трубке, которую затем быстро охлаждают. При несколько других условиях опыта (атмосфера водорода) Шварц получил соединение 31(0С12Г|П2. Из этого вещества оп получил хлориды кремния, и о строенные в форме колец. Сначала при слабом нагревании из Sit0Cl2()H2 с отщеплением НС1 образуется бицикли-'ческое соединение Sr 1()С11Я — прозрачное желтое вазелиноподобпос вещество. При более сильном нагревании образуются смолооб разные хрупкие, как стекло, вещества с многочленными кольцевыми системами. При этом отщепляются низкомолекулярные хлориды кремния (например, Si^CI^, SisCl3, Si4ClI0). Все эти вещества хорошо растворимы в бензоле. При дальнейшей прогрессивной конденсации возникает в конце концов соединение [SiGl j.c (монохлорид кремния) с неограниченным (но не совсем рав-по мерным) образованием плоских сеток. Это соединение рентгене аморфно, при нагревании до 800° (снова с отщеп.тспием низкомолекулярных хлоридов кремния) превращается в темно-серый аморфный элементарный кремний (Schwarz, 1951—1952).
522
Глава 12
В табл. 86 приведен обзор некоторых характертшх'свойотв простейших галоге-•|Гтвдов кремния.
Таблица 86
Галогениды кремния
Свойства SiF4 8ЮЦ 81Вг4 Sil4
Агрегатное состояние при обычной температуре . . . Температура плавления, ’С Температура кипепия, °C . . Теплота образования, ккал/молъ ......... Продукты разложения холод-ной водой Газообразное Возгоняется при—95 360 H2SiO3 и Н3 [SiF6J Жидкое —70 +57,5 154,0 H2SiO3 и НС1 Жидкое +5,2 + 152,8 88,5 H2SiO3 и НВт Твердое + 120,5 287,5 6,7 H,S1O3 и HI
Свойства Sl^Fo SiaCie SiaBre Sins SiaCIs
Агрегатное состояние при обычной температуре . . . Температура плавления, ° С Температура кипения, 11С . . Продукты разложения холодной водой .... Газообразное —18,7 (780 мм рт ст) —19,1 (т. возг.) H2Si2O4, IIF, H,SiO3 Н2 681Г6] и Н2 Жидкое +2,5 + 147 H2Si2O4 и НС1 f, Жидкое -240 H2Si2O4 я НВг Твердое + 250 H2Si2O4 и Ш Жидкое —67 -215 H4Si3OB и НС1
Тетрафторид кремния (’четырехфтористый кремний) Si Fa получают при нагревании смеси измельченного в порошок плавикового шпата и кварцевого песка с концентрированной серной кислотой. Последняя сначала освобождает из плавикового шпата фтористый водород CaFs+ HaSO4 = = CaSOi-f- 2HF, который реагирует затем с кварцем, образуя фторид кремния SiOa-f- 4HF = SiFa-f- 2НгО. В чистом состоянии SiFa удобнее получать сильным нагреванием фторосиликата бария. При нагревании он распадается на фторид бария и фторид кремния Ba[SiFe] = BaFs4-
Фторид кремния — бесцветный газ с резким запахом. Во влажном воздухе образует густой туман. Вес 1 л ранен 4,693 з (на этом основании установлен молекулярный вес, соответствующий формуле SiF-a). При сильном охлаждении и при обычном давлении фторид кремния переходит из газообразного состояния сразу в твердое (температура возгонки равна —95°, теплота сублимации 6,18 ккал/моль). Он молгет быть переведен в жидкость только под давлением (критическая температура —1,5°, критическое давление 50 ат).
Четвертая группа периодической системы
523
Благодаря большой теплоте образования, равной 360 ккал/моль, это соединение очень устойчиво. Под действием электрической искры разложения не происходит. Напротив, калий и патрий сгорают в его парах с образованием фторидов. Сухие карбонаты и бораты щелочных металлов на холоду не взаимодействуют с SiFi. При полном отсутствии влаги по отношению к SiFi устойчиво также и стекло. Наоборот, водой фторид кремния разлагается с выделением геля кремневой кислоты SiFi-f- 2НаО = — SiOa-f- 4HF. Образующаяся при этом плавиковая кислота соединяется с еще неразложившимся фторидом кремния с образованием кремнефтористоводородной кислоты HsSiFe. В присутствии воды немедленно наступает разложение Si Fa, поэтому фторид кремния следует хранить над ртутью.
С некоторыми веществами SiF4 образует нестойкие продукты присоединения, так, с аммиаком он Дает SiF4'2NH3. При 180—190° SiF4 практически количественно реагирует с А1С13 с образованием SiClF3, SiCl2F2, SiCl3F, SiCl4. Повышая температуру, реакцию можно провести до образования исключительно SiCl4. Подобные реакции идут с А1Вг3 и AlJg. В соответствии с данными Шмайсера (SchmeiBer, 1952) при взаимодействии со спиртами S1F4 в противоположность остальным галогенидам кремния нс сразу превращается в эфир кремневой кислоты, однако при добавлении алкогодята алюминия реакция протекает хороню, SiF4 отличается От остальных галогенидов кремния также и другими реакциями. Например, согласно Шмайссру, окись этилена С2Н4О при реакции с фтористым кремнием превращается в диоксаи С4НвО2, в то время как SiCl4 реагирует с окисью этилена с образованием хлористых эфиров кремневой кислоты (см. стр. 526 и сл.).
Кремнефтористоводородная кислота и фторосиликаты. Кремнефтористоводородная кислота H2SiF6 образуется при взаимодействии фтористого водорода с четырех фтористым кремнием 2HF -ф- SiF4= HsSiFe. Ее водный раствор получается обычно при пропускании фтористого кремния в воду
3SiF44-2H2O — Si03—|— 2H2SiF6.
Выделившуюся кремневую кислоту отфильтровывают. В безводном состоянии кремнефтористоводородную кислоту можно получить действием концентр пр о в энной серпой кислоты на ее соли — фторосиликаты, например на фторосиликат бария Ва[SiFe]. Она при этом выделяется в газообразном состоянии, но уже при обычной температуре распадается в значительной степени (больше чем на 50%) на свои компоненты SiFr и (HF)s. Водный раствор, напротив, не содержит заметных количеств плавиковой кислоты и поэтому не разъедает стекла при условии, что соотношение F : Si в этой кислоте не превышает 6. При концентрации 13,3% кремнефтористоводородная кислота может перегоняться без разложения. Из концентрированных водных растворов при охлаждении выделяются гидраты, например H2SiF6-2H2O — бесцветные твердые кристаллы, плавящиеся при 19°.
Кремнефтористоводородная кислота даже в очень разбавленном водном растворе обладает исключительными дезинфицирующими свойствами. Н а этой основе она находит широкое применение в пивоварении для стерилизации медных и латунных сосудов.
Кремнефтористоводородная кислота — одна нз сильных к-иелот. Она значительно сальнее плавиковой кислоты, приблизительно одинакова ио силе с серной кислотой, te кажущаяся степень диссоциации (измеренная по коэффициенту электропроводности) составляет при 25° в 1 н. растворе 53%, в 0,1 н. растворе — 76%; гидроокиси или окиси металлов нейтрализутот ее с образованием фторосиликатов
2MrOH+H2SiFG —Ml [SiFc]-[-2H2O.
524
Глава 12
Фторосиликаты M|[SiF6J можно получить^как разложением карбонатов кремнефтористоводородной кислотой, так и при смешивании растворов компонентов. В последнем случае иногда вместо обычных гексафторосиликатов получают также еще фторосиликаты другого типа, для которого но известны свободные кислоты.
Так, кроме (Nrr4)2(SiFG J, было получено соединение (NH4)3SiF7. Вероятно, существуют также более сложные комплексные фторосиликаты. О строении решетки (NH4)2[SiF#] см. стр. 437, Одинаковые решетки были установлены у гексафторосиликатов К., Rb, Cs и Т1(1$, а также у гекса фторогерман ат а цезия C.s2[GeFe], кроме того, у ыж-сахлороетапиатов NIL,, К, Rb, Cs и Т1(1) и гексахлоро пл комбатов NH4, Rb, Cs. К" показало реитгеиоструктурпое исследование (Hoard, 19421, соединение (NIl4)sSiF-следует рассматривать как двойную соль (NH4)2lSiFe] п NFT4F, т.е. оно состоит с INH4]+- и [SiFsp--групп и отдельных ионов фтора.
Фторосиликаты щелочных металлов (за исключением лития и аммония) довольно труднорастворимы в воде. Еще менее растворим фторосиликат бария (0,0268 г в 100 г воды при 17,5° С). Остальные фторосиликаты большей частью легкорастворимы. Некоторые фторосиликаты образуют характерной формы кристаллы, что иногда используют в анализе.
Органические производные фторосиликата аммония, например [CII3-NH3]2 [SiFe] (бесцветные, растворимые в воде кристаллы с т. пл. 232°) п [CeII5-NH3]E[SiF6j (голу-болато-серые растворимые в воде кристаллы с т. субл. 220°) применяют в качестве ипс ектофунгицид а.
Фторосиликат калия K2[SiFs] выпадает и виде очень мелкозернистого опалесцирующего осадка при нейтрализации раствора кремнефтористоводородной кислоты гидроокисью калия. Растворимость его равна 0,120 г в 100 г воды при 17,5° и значительно возрастает с повышением температуры. В спирте эта соль практически нерастворима.
Фторосиликат натрия NaJSiFJ немного лучше растворим, чем калиевая соль (0,65 г в 100 г воды при 17,5°). В производстве эмалей эту соль часто применяют вместо криолита в качестве глушителя. В некоторых случаях фторосиликат натрия выгодно применять вместо SiF*. Его получают в больших количествах в качестве побочного продукта при производстве суперфосфата, так как применяемые при этом фосфориты, как правило, содержат фтор.
Фторосиликат магния Mg[SiFs|-fiHoO и фторосиликат цинка Zn(SiF6] 6И2О хорошо растворимы в воде; их применяют для придания цементу водонепроницаемости. Их действие основано па том, что они реагируют с содержащейся в цементе окисью кальция е образованием фторида кальция и кремневой кислоты, которые выделяются в мел-кораздробленном состоянии и закупоривают собой поры цемепта.
Дпкремнийгексафторид Si2F<; (вес 1 л равен 7,778 г) был получен Шумбом (Schwab, 1931) нагреванием SiaCl6 с ZnF2. При взаимодействии с С12 или с Вг2 из него можно получить смешанные галогениды SiF3Cl, S1F2C12, SiFgBr и т. д. Гидролиз Si2Fe протекает с выделением водорода, так как первично образующиеся продукты реакции, согласно уравнению
Si2F,+4H2O=H2Si2O4+ 6Н F, в дальнейшем частично взаимодействуют между собой в соответствии с реакцией
H2Si2O44-6HF=n2SiFe+H2SiO3-|-H2O-|-H2.
Тетрахлорпд кремния {четыреххлор истый кремний) SiCl4 получают нагреванием кремния или смеси двуокиси кремния с углем в токе хлора. Он представляет собой бесцветную, подвижную, дымящую па воздухе
Четвертая группа- периодической систем.ы 525
жидкость с удушливым запахом. Удельный вес равен 1,49. Водой SiCl4 гидролизуется: SiCl4+ 2Н2О - SiO2+ 4НС1. Этим объясняется образование им густого тумана на воздухе. Туман становится гуще, если в воздухе имеются пары аммиака (образование тумана из хлористого аммония). Это используют, например, при маскировке военных судов.
Чстыреххляр истый кремний реагирует такжесокпслами металлов с заменой хлора на кислород, например: 3SiCl4-t-2Al3O3=3SiO2-|-4.AICl3. Реакция с окисью алюмипия иногда протекает настолько энергично, что SiCI4 при этом закипает. Но с большинством окислов металлов он реагирует только при длительном нагревании в толстостенной запаянной стеклянной трубке. G Р2О6 образует РОС13; с SOg даст S2O6C12. Со спиртами четырехъ, юрпстый кремний взаимодействует с образованием эфиров кремневой кислоты, но обычно оп довольно устойчив. Со щелочными металлами он реагирует только мри температуре красного каления (с образованием «аморфного» кремния); с сульфидами большей частью совсем нс реагирует, с сероводородом образует SiClgfSH). При нагревании с НВт и ПГ происходит замена хлора на другие галогены. Таким образом можно нонучитт, также смешанные галогениды кремния, например SiCl3Br, SiCI2Br21 SiClBr3, SiCl:jI и другие. Прп обработке SiCl4 трехфторнстой сурьмой (в присутствии SbCl6 в качестве катализатора) можно получить соединения SiFClg, SiF2Cl2, SiF3Cl и SiF4 (Booth, 1935). С аммиаком SiCl4 соединяется, образуя SIC14-6NH3. Он соединяется также с PIT?,, по только при низких температурах. При пропускании SiCI4 в смеси с воздухом через сильно раскаленную трубку образуются оксихлориды Si2OCle, Si404Cls и другие. Это бесцветные летучие малоустойчивые соединения. Некоторые оксихлориды с довольно большими молекулярными весами (например, SieO5Clf4, Si7OeClIe) получил Шуми (1950) при добавлении воды к охлажденному до —78° эфирному раствору SiCl4. Плотность пара четыреххлористого кремния была найдена равной 9,7 (воздух — 1) при 239°. Это значение позволяет вычислить молекулярный вес, соответствующий формуле SiCI4. Согласно данным Троста (Trosi, 1952), SiCI4 ирпсоединяет органические амиша в соотношении 1:2 и 1:4. Последние представляют собой бесцветные продукты, более устойчивые, чем окрашенные со единен ня состава 1 : 2. Спирты, кетоны и другие кислородсодержащие органические вещества также могут присоединяться к SiCl4.
Монохлорид кремния |81С1]Ж был получен Шварцем (1939) прп термическом разложении Si10Gl32 (ср. стр. 521). Это соединение представляет собой твердое желтое вещество в виде листочков, при комнатной температура устойчивое ио отношению к сухому кислороду, по при 98° воспламеняющееся на воздухе. Оно нерастворимо в инднферепт-ных растворителях. В едком кали растворяется с бурным выделением водорода 2SiCi-f-•4-6OH'=2Cr-j-2SiO3-|-3H2. В кислом растворе (при низкой температуре) идет гищю-лнз С образованием гидрокеисоединения кремния 431С1-[-611О11=8!4(ОН)5-|-4.Н.С1-|-П2. Несколько иначе протекает гидролиз моноиадида кремния [Si 1 ]л, полученного в 1942 г. Шварцем. При этом образуется в высшей степени легко окисляющееся Соединение [Si2O3H4bc (названное Шварцем «над кремневая кислота^), которое, вероятно, содержит бесконечные цепи —Si—Si—. При нагревании в вакууме это соединение, имеющее цист слоновой кости, постепенно разлагается па лимонно-желтое соединение [Si2O3H3)x и красно-коричневое [Si3OgII2]^ в снеси с SiO2 н Si.
Тстрабромид кремния SiBr4 получают аналогично хлориду; он представляет собой бесцветную, сильно дымящую на воздухе жидкость (уд. вес 2,789, т. кип. 152,8°, т. пл. 5,2°). В противоположность хлориду он очень энергично реагирует яри нагревании с металлическим калием. В зависимости от условий опыта при реакции с аммиаком он присоединяет четыре или шесть молекул NH?. Известны также продукты присоединения с другими веществами. /
Тетраиодпд кремния S1I4 получают при пропускании паров пода в токе двуокиси углерода над рас кал г иным докрасна кремнием; он образует бесцветные правильные октаэдры, изоморфные С14, очень легко растворимые в сероуглероде. Для плотности пара было найдено значение 19,12 (воздух=1), соответствующее формуле Sil4. Нагретый газообразный тетраподнд кремния воспламеняется па воздухе и горит красным пламенем. Со спиртами иод11д реагирует иначе, чем остальные галогениды: наряду с кремневой кислотой образуется иодhctt.iй этил и иодистый водород,
Прп нагревании тетр а иод,и Да с мелко измомьчеппым серебром при 290—300° получают гексаиодид кремния Si2l«
2SiI4+2Ag=I3Si-SiI3+2Agl.
526
Глава 12
Это соединение можно хорошо перекристаллизовать из горячего раствора сероуглерода, в котором оно в противоположность тетраиодиду лишь умеренно растворимо. Оно образует бесцветные двупрсломляющне шестигранные палочки, которые в вакууме прп 250° возгоняются с частичным разложением. Если на раствор его в сероуглероде подействовать бромом, то последний замещает под С образованием гексабромида
Si2I6-|- 3Br»=Si2Br6+3I2.
Отсюда и заключают, пто формула этого соединения I3Si—Sil3, так как если бы имелся трииодид с трехвалентным кремнием, то при обработке бромом следовало бы ожидать окисления трииодида в тстрагалогенпд. Аналогичным образом при действии хлора на гексаиодид получают гексахлорид С1ЙЙ1—S1C13, бесцветную жидкость с уд. весом 1,5& при 0°. Это соединение образуется также, если пропускать пары SiCl4 над расплавленным кремнием я затем продукт реакции быстро охладить. Указанные гексагалргеппды кремния прн обычной температуре неустойчивы. Не распадаются они лишь потому, что очепь малы скорости реакций, однако распад происходит уже при незначительном нагревании. Так, Si2Cl6 при температуре выше 350 и ниже 1000° разлагается па S1CI4. и Si. Гексагалоген иды кремния чрезвычайно чувствительны к воде. При разложении их водой па холоду образуется так называемая еиликощаве.левая кислота [Si2O4H2].t — очей, неустойчивая белая масса, которая обладает сильно восстанавливающим” действием и растворяется в едких щелочах с выделением водорода Si2O4H2-|-4OH'= =2SiO3-|- 2Н3О+112. Аналогично при гидролизе Si3Gla получается также очень неустойчивая так называемая силикомезощавелевая кислота [813О6Н4]Я,
Трпхлорсплап. Если пропускать сухой хлористый водород над кремнием ниже температуры красного каления (лучше всего при 380°), то образуется соединение SiHClj, которое па звано силикохлореформа я (вследствие аналогии состава с хлороформом). Его можно получить также хлорированием моносилана SiH4. Трихлорсилан — бесцветная, легко подвижная, не проводящая ток и сильно дымящая на влажномвоз-духе жидкость (г. кип. 31,5°, т. пл. —128,2°), Это соединение растворимо без разложения в сероуглероде, в чстыреххлористом углероде, в хлороформе н в бензоле. С абсолютным спиртом реагирует, образуя SiI-I(OG2II5)3. С водой на холоду образует диоксодиси-локеан (так называемый ангидрид кремнемуравъиной кислоты) — белое объе-
мистое вещество, которое при довольно сильном нагревании па воздухе воспламеняется, по тем ие менее действует как сильный восстановитель, например обесцвечивает перманганат, из раствора цитрата серебра выделяет серебро, из сернистой кислоты — серу. Едкими щелочами диоксодисияоксан и соответственно также трихлорсилан разлагаются с выделением водорода
Si2OsH2-|-4OH'=2SiOIH-H,O-4-2H2;
Й1Т1С13+5ОНг^81Оз+ЗС1'+2Н2ОЧ-Н2.
Соединения SiHBr3 п SiIII3 очень похожи па SiHCl3, но они менее устойчивы. SiHF3 (г. кип. —97,5°, т. пл. —131°), впервые полученное Руффом при действии SbF3 на SiHC]3, уже при очень низких температурах постепенно разлагается. Более устойчивыми являются полученные Бутом (Booth, 1934) соединения SiHCl2F и SiHClF,.
Эфиры кремневой кислоты. При взаимодействии галогенидов кремния-со спиртами или фенолами образуются эфиры кремневой кислоты Si(OR)4 (R — органический радикал). Некоторые из них приобрели важное значение .
11s фенольных эфиров кремневой кислоты следует упомянуть изученные [Кварцем (1951) производимо диокепбепиолов. Опп представляют собой смолообразпые вещества, имеющие различное строение в зависимости от положения гидроксильных групп в бен-аолвном кольце. 'Гак, пир о катехиновый эфир имеет циклическое строение, резорциновый эфяр — цепное строение, а гидрохиноновый эфир, если оп получен взаимодействием SiCl4 с гидрохиноном в подходящем молярном отношении (например, 1 : 2 пли 1 : 2,5). имеет ленточную структуру, причем цепи в цен связаны попарно через мостики
При реакции SiF4 со спиртами или фенолами наряду с эфирами кремневой кислоты одновременно образуется кремнефтористоводородная кислота. Ее можно осадить в виде аммонийной соли при пропускании сухого аммиака. Наоборот, SiR2F2 (R — алкил или арил) реагирует со спиртами и фенолами таким же образом, как и SiGl^ (Jenfcner, 1955), однако в этом случае происходит взаимодействие только при пропускания
Четвертая группа периодической системы
527
NIT3, в то время как для реакции SiCl4 со спиртами или фенолами аммиак Полезен, по не обязателеп. Указанные реакции имеют следующий вид:
SiCl4+4ROH=Si(OR)44-4.lJCl,
;;SiF4+4ROII=Si(OR)4+2H2SiFe,
SiR2F,--2R(jll—2XIl3=SiR2(OR)a-[-2NH4F.
Роданид кремния Si(SCN)4 (белые иглы, т. пл. 143°, т. кип. 314°) подучается взаимодействием хлорида кремния и роданида свинца (лучше всего в растворе бензола)
SiCl4+2Pb(SCN)2=Si(SCN)4+2PbCl2.,
Это соединение устойчиво в сухом воздухе, однако при действии Ьоды. кислот п щелочей, распадается е выделением геля кремневой кислоты или же с образованием силикатов.
Сульфиды кремния. При сплавлении «аморфного» кремния с трехкратным количеством серы получают дисульфид кремния SiS2, который после очистки возгонкой при уменьшенном давлении образует бесцветные с шелковистым блеском волокнистые кристаллы. При обычной температуре он устойчив в сухом воздухе, водой разлагается
SiS2+‘2H2O=SiO2-|- 2Вг8.
Кристаллическую структуру дисульфида кремния определил Циптль (Zintl, 1935). Он получил дисульфид, согласно опытам Тпда, при нагревании до 1100° A13S3 с SiO2. Оказалось, что кристаллы SiS2 построены из бесконечных цепей, причем каждый атом Si тетраэдрически окружен четырьмя атомами серы, а каждый атом серы одновременно связан с двумя атомами Si
zsx zs zs S
..< )Si< )Si< >Si< )Si< \sz 4sz 4
Внутри каждой цепи расстояние между центрами Si*—>-S равно 2,14А, а между Si*—*Si2,77 А. Между соседними атомами S тетраэдра расстояние равно 3,62 А. Напротив, расстояние между атомом Si и ближайшим атомом S соседней цепи составляет4,18 А. Одинаковую с SiS2 структуру имеет волокнистый SiSe2*. Как было показано Вайссами (Weiss Al. ц Ar., 1954), при нагревании моноокиси кремпия при 1200—1400° происходит реакция окисления-восстановления 2SiO=SiO2-[-Si, причем образуется волокнистая модификация SiO2 с такой же, как у SiS3, структурой (плотности 1,975; т. пл. около 1420°). Волокнистая модификация SiO2 метастабильца и при длительном нагревании в интервале 200—800° переходят в тридимит (с сохранением волокнистого вида), а при 1300°—в кристобалит. При действии воды образуется метанремневая кислота (H2SiO3)n. Такие замкнутые цени, которые имеются в кристаллах типа SiS3, характерны для веществ с волокнистой структурой. Цепи расположены по направлению волокон. Цепи SiS4 и соответственно также цепи SiO4 волокнистой модификации SiO2 Отличаются от цепей SiO4 волокнистых силикатов (см. стр. 546) тем, что в них тетраэдры имеют общие ребра, в то время как тетраэдры SiO4 волокнистых силикатов всегда имеют только общие углы (ср. стр. 543). Поэтому в цепях SiS4, а ташке в цепях Si04 волокнистой SiO2 в противоположность цепям SiO4 волокнистых силикатов заряды атомов насыщены, так что каждую цепь можно рассматривать как молекулу неограниченной длины. В соответствии <; этим структура SiS2 занимает среднее положение между молекулярной решеткой в обычном смысле, какая имеется у твердой СО2, и координационными решетками, которые образуются силикатами и SiO2. Дителлурид кремния SiTc2 получается при нагревании смеси Si и Те в высоком вакууме, он образует красные пластинки (г/=4,39), кристаллизуется в противоположность SiS2 и SiSc2 по типу иодида кадмия. Возгоняется выше 1200° н одновременно частично разлагается с образованием серовато-белого мопотеллурида кремния SiTe (Weiss Al., Weiss Аг., 1953).
По данным Гем пеня и Гаази SiS2 способен соединяться с сульфидами щелочных металлов с образованием тиосиликатов Mei SiS3, которые еще недостаточно изучены. Согласно Колсону, при очень высоких температурах образуется также моносулъфид SiS, который при растворении в едких щелочах в отличие от дисульфида выделяет водород.
* При быстром охлаждении расплавленного SiSc2 получается стеклообразный SiSt‘2 (плотность 2,95), который химически значительно устойчивее, чем волокнистый SiSe2 (плотность 3,61), Волокнистая модификация SiSe2 образуется при медленном-охлаждении расплава (Weiss Al., Weiss Аг., 1953).
528
Глава 12
Кислородные соединения кремния
Нормальным и единственным практически важным окислом кремния является двуокись кремния SiO2, из нее взаимодействием с основными окислами образуются силикаты разнообразного состава. Из кремневых кислот, образованных химическим взаимодействием SiO3 и Н,О, в определенной форме известны только очень немногие, причем они получены лишь при особых условиях (ср. стр. 537 и сл.). Двуокись кремния очень склонна давать коллоидные растворы и образовывать с водой гели (ср. стр. 536 и сл.). Эти гели называют силикагелями или гидратами двуокиси кремния, при атом по учитывают того, связана ли вода в них частично химически с двуокисью кремния или нет (что часто нельзя правильно установить).
Изучением спектров абсорбции Еопгоффер (Bonhocffer, 1427) доказал, что при сильном патренапии двуокиси кремния с углем образуется газообразная моноокись кремния S1O. Образование последней происходит также при нагревании SiO2 с Si. Вследствие этого происходит довольно быстрое улетучивание, если смесь обоих веществ нагревают в вакууме при 1300—1400° (Bilt z, 1938). Образование летучей Si О можно использовать для освобождения силикатов от кремневой кислоты (например, A]2Si2OT+ —|—2Si—> А12О3Ч—<iSiO) млн прн получения металлов (например, Nb2O 5-|-5Si —2ЯЬ-|-+5SiO) (Zint.1, 1940).
Двуокись кремния. SiOa встречается в природе в трех различных кристаллических формах: в виде кварца, тридимита и кристобалита.
Р и с. 89. Левый кварц.
Рис. 90. Правый кварц.
Кроме того, содор;кащая воду (в форме геля), она существует в виде опала, а также в землистой форме как кизельгур.
Кварц — кристаллическая форма двуокиси кремния, устойчивая при температуре ниже 870°. В интервале температур 870 и 575° кварц принадлежит к трапецоэдрической гемиэдрии гексагональной системы; ниже 575°— к трапецоэдрической гемнэдрии тригональной ^системы. Нередко кварц встречается в форме чрезвычайно хорошо образованных кристаллов, иногда значительной величины. Кварц обладает двойным лучепреломлением. Он встречается в двух формах, из которых одна вращает плоскость веляризации вправо, другая — влево. Поэтому различают левый и правый кварц. Часто эти формы можно отличить также
Четвертая группа периодической системы
529
по положению граней кристалла, так, на примере, представленном на рис. 89 и 90,— по зеркально расположенным заштрихованным граням $.
На рис. 89 и Bfj речь идет о гранях *, являющихся гранями тригональной бипирае лиды. По ним кристаллы леи,эго и правого кварца можпо различить лишь в том случае, если они заштриховали, м па приведенных рисунках. Если в кристалле хорошо выражены грани то левый и правый кварц легло всего различить по ним.
Если отсутствуют такого типа гр а ли, то, помимо определения оптической вращательной способности, левый ч правый кварц можпо различать еще на основании фигур травления, образующихся при обработке кварца плавиковой кислотой,
%
Разновидности кварца: горный хрусталь прозрачный как вода; дымчатый кварц (называемый также дымчатым топазом) темно-коричневый; морион черный; цитрин желтый; розовый кварц' хризопраз цвета зеленого лука; аметист (большей частью) фиолетовый. Как драгоценный камень особенно часто используют аметист в виде очень прозрачных кристаллов. Кварц принадлежит к числу чрезвычайно распространенных минералов. Очень часто он встречается в качестве составной части изверженных горных пород (гранита, порфира, липарита) и кристаллических сланцев (гнейса, слюдяного сланца). Среди осадочных пород из прозрачных зернышек кварца состоят песчаник и кварцит, а также некоторые пески (кварцевый песок).
Как впервые (1899 г.) установил Ле-1П;пелье путем измерений коэффициента расширения, кварц при нагревании выше 575" превращается в другую модификацию, которая принадлежит к трапецоэдро-гсмиэдрическому классу гексагональной системы.
Рис. 91. Элементарная ячейка fi-левого кварца.
Рис. 92. Элементарная ячейка ^-правого
кварца.
<1=5,01 А; с—5,47 Л (при 600°).
При охлаждении снова появляется прежняя форма. Следовательно, здесь речь идет об энантиотропном превращения. Устойчивую при обычной температур с форму называют а-кварцем инн нивкатемпературным кварцем, Другую —ф-кварцем* или высокотемпературным кварцем. Соотношения осей при этом превращении претерпевают лишь очень незначительное изменение, поэтому оно мало заметно по внешнему виду кристалла. Форма кристаллов остается почти неизменной и, как показывают рентгенограммы, расположенпе отдельных атомов также меняется незначительно.
На рис. 91 и 92 представлены элементарные ячейки лево- и правовращающего ф-к варца. В общем каждый атом кремния окружен четырьмя атомами О, которые расположены на раестопнЕИ 1,59,'Л, образуя почти правильный тетраэдр. Атомы располо-
* Способ обозначения буквами аир неоднозначен. То же относится к тридимиту ц кристобалиту.
34 Г Реми
530
Глава IS
жены винтообразно вокруг показанной на рисунке оси. В левовращающем кварце направление винта противоположно направлению его в правовращающем кварце. (Четыре атома О, которые образуют лежащий на оси тетраэдр, находятся на перпендикулярах, восстановленных па точек базисной грани, которые задаются следующими парами координат: а/зя; */ьа, Vja; 3/$а, 4/йа. Прп Этом за начало коор-
динат принимают задний левый угол базисной грани; во всех случаях вначале указана ордипата, направленная вправо. У левовращающего кварца первые Две координаты относятся к проекции самого нижнего, л правовращающем кварце к проекциям самого верхнего атомов О указанного тетраэдра.1
ритах, так ц в горных породах, однако
Рис. 93. Кристаллическая решетка р-три-димита.
Элементарная ячейка выделена жирными линиями а=5,03А; с=8,23А; Sl-t—+0=4,54 Л.
Тридимит является стабильной модификацией кристаллической двуокиси кремпия в интервале температур 870—1470°; однако ои встречается также и при обычной температуре, по в мстастабйльной форме*. Эта форма двуокиси кремпия тоже часто встречается в природе как в метео-в очень небольших количествах.
Но она представляет интерес, поскольку ее присутствие в горных породах может дать сведения об истории их возникновения.
Чтобы кварц превратить в тридимит, в общем недостаточно нагреть его выше 870°. Для этого перехода скорость превращения чрезвычайно мала даже прп высоких температурах. Иначе дело обстоит, если применить соответствующий растворитель, так называемый минерализатор, например расплавленный вольфрамат натрия. При нагревании с ним можно получить тридимит в виде небольших кристаллических пластинок, которые по форме "соответствуют гексагональной системе. При охлаждении тридимит снова превращается в устой чииый ниже 870° кварц только в том случае, селя присутствует саответсс-вутощий растворитель. Одпако и в присутствии растворителя превращение в кварц идет очень медленно. Так, при условии соприиосиовения тридимита с расплавленным вольфраматом натрия при 825° опо протекает в течение примерно трех су
ток. Если гексагональный тридимит охлаждается в отсутствие минерализатора или же в его присутствии, но очень быстро (для предупреждения превращения в кварц), то ниже 117° Он превращается к ромбическую модификацию. Это Превращение не отражается на внешней форме кристаллов. Ромбическую модификацию, являющуюся оптически двуосной (в отличие от оптически одноосной модификации, устойчивой при более высокой температур с), называют а-триДНмнтом. Она устойчива ниже 117° ио сравнению с ^-тридимитом, но неустойчива или метастабпльпа ио сравнению с кварцем, а- и р-Тридимит также являются энантиотропными модификациями. Встречающиеся в природе ромбические кристаллы а-тр и лимита возникли, как это можно установить па основании их строении, из гексагонального $-тридимита. Прп нагреваний выше 117° ояп опять переходят в ^-тридимит.
Гексагональный р-трпдимит имеет, по-видимому, еще другую точку превращения около 103°. Чтобы различить обе гексагональные формы, их можно обозначить как ;
* На основании электролитического исследования и других наблюдений Флсрке (Florke, 195G) сделал вывод [ср. также Buerger М. J., Ain. Mineralogist, 39, 605, 19541 о том, что решетка тридимита стабильна лишь тогда, когда в ней Присутствует известная часть (0,1=-0,5%) посторонних ионов, Одиако, чтобы окончательно выяснить этот вопрос, нужны еще дальнейшие исследования.
Seine ер т.«л группа пермвической системы 531
и р«-тридимит. Однако «стабильный» выше 163° р2~тРнДидшт очень мало отличается от ₽ {-тридимита.
Структура решетки (J-три лимит э. представлена па рис. 93. Она значительно проще структуры решетки квариа. Решетку fj-тридимита можно получить, если в решетке вюртцита (рис. GO, стр. 290), в которой примем а==5,03, с=-8,22 А, занять сначала все у алы решетки атомами Si, а затем в середину между двумя атомами Si поместить один атом кислорода. Таким образом получим решетку, в которой каждой атом Si тетраэдрически окружен четырьмя атомами О, в то время как каждый атом О одинаково удалей от двух атомов Si (So—>0 = 1 ,э4 А).. Таким образом, каждый атом Si тетраэдрически окружен на расстоянии, равном 3,08 А, четырьмя другими атомами Si, с каждым из которых у пего имеется один общий атом кислорода. Как уже указывалось, подобную структуру решетки имеет (обычный) леЭ. Структура решетки а-тридимита сложнее и ио может еще пока считаться вполне установленной.
Кристобалит (названный по имени горы San Cristobal в Мексике) представляет собой форму двуокиси кремния, устойчивую выше 1470° и до температуры плавления. Ниже точки превращения в отсутствие ускоряющих превращение растворителей кристобалит метастабилен. В природе он иногда встречается в виде мелких кристалликов, включенных в лаву. В общем его месторождения подобны месторождениям тридимита-
Как у кварца и тридимита, у кристобалита также следует различать две формы. «Стабильную» при обычной температуре, также обозначают как п-форму*.
0-Кристобалит относится к правильной системе. Его кристаллическая решетка так;це отличается от решеткп тридимита, как решетка цинковой обманки от решетки вюртцита. Атомы кремния образуют решетку типа алмаза (рис. 83, стр- 461) с д=7,12 А; каждый атом кремния окружеп четырьмя атомами кислорода, расположенными л виде почти правильного тетраэдра на Примерном расстоянии 1,63 А. Следовательно .группировка атомов О вокруг атомов Si в кварце, тридимите и кристобалите по существу одна и та же.
Согласно данным Ньювспкамиа (Nieuwenkamp, 1935), я-крпстобалцт образует подобную Же решетку, которая, однако, тетрагональна; в ней тетраэдры SiO4 несколько повернуты по отношению к решетке р-кристобалпта, так что липин связи Si—О—Si являются непрямыми, как в р-ь'р исто б алите, а расположены под углом (Si<-^0—1,59 А). а<—^-Превращение кристобалита при охлаждении протекает всегда при более низкой температуре, чем при нагревании. Температурная область превращения зависит также от предварительной обработки образца. Она находится приблизительно в температурном интервале 200—270°.
Ниже 1470° кристобалит мепее стабилен не только по сравнению с кварцем, но и по сравнению с тридимитом. При продолжительном нагревании До очень высокой температуры кварц даже в отсутствие растворителей начинает постепенно испытывать превращение; а именно нри этом он переходит в кристобалит, причем даже и тогда, Когда превращение протекает ниже 1470°, т. е. в области, в которой устойчивым является не кристобалит, а тридимит. На первый взгляд этот результат кажется парадоксальным, однако он соответствует прашшу ступеней Оствальда. В соответствии с этим правилом, образование вещества, существующего в нескольких модификациях, протекает ступенчато таким образом, что сначала образуется менее устойчивая модификация, которая затем превращается в более устойчивую. Кристобалит как мепее устойчивая модификация образуется в первую очередь и может сохраниться, потому что его превращение в тридимит приобретает заметную скорость только в присутствии растворителя или же мйперамизатора.
Двуокись кремния в форме кристобалита плавится при 1705°, Кварц плавится приблизительно и а 150° ниже. Охлажденный плав остается в стекловидном аморфном состоянии. Стекловидная двуокись кремния при обычной температуре также мета стабильна. При продолжительном и сильном нагревании происходит медленная кристаллизация («расстекловывание»). В связи со сказанным выше вполне понятно, почему в отсутствие минерализаторов кристаллизация приводит к образованию кристобалита.
* Ср. примечание па стр. 529.
34*
532
Глава 18
Области, в которых перечисленные формы двуокиси кремния действительно являются стабильными, ограничиваются в соответствии Со сказанным следующими точками превращений:
576= 870“ 1470° 170Ь°
а-Кварц < > р-К вари, ^-Тридимит <2? > 3-Кристобалит^— Плав
Кроле того, существуют еще другие точки превращений в областях, в которых соответствующие формы ммастгтбяльны
117° а-Трндимит V ? р-Тридимит, — 260“
а-Кристобалит ^-Кристобалит
— 230“
О метает аб и льнов волокнистой модификации двуокиси Кремния см. стр, 527.
Для иллюстрации областей существования различных модификаций SiO2, установление которых проведено Феннером и Витцелем, может служить д пат рампа на рис. 94. Па этой диаграмме на ось абсцисс нанесены температуры, а на ось ординат —давление пара, которое относится к отдельным модификациям в соответствии с их различной
К в с. .94. Диаграмма состояния двуохися хремяяя.
устойчивостью. Правда, числовые значения этих давлений пока неизвестны; однако, как уже указывалось (см. стр. 73 и 99), менее устойчивая из двух форм какого-нибудь вещества всегда должна иметь более высокое давление пара. Наименее устойчивой моди-' фикацией при обычной температуре и выше приблизительно до 1550° является застывший ' в виде стекла плав — кварцевое стекло. Как уже было отмечено, кварцевое стекло постепенно расстекловивается при очень длительном накаливании, особенно в присутствии примесей; ври этом, как правило, образуется кристобалит. Из диаграммы видпо, что это соответствует правилу ступеней Оствальда, так как при обычных температурах накаливания после кварцевого стекла наименее устойчивой модификацией является кристобалит.
Как показали рентгеновские исследования, аморфная застывшая к виде стекла двуокись кремния (кварцевое стекло) построена не из молекул SiOa, а из атомов SI, связанных через атомыО (или же ионы, ср. стр, 541) таким образом, что образуется сетчатая структура. Она состоит из атомов кислорода, расположенных в'виде тетраэдров вокруг каждого атома кремния; как и в кристаллических модификациях, тетраэдры имеют общие
Четвертая группа периодической системы
533
углы. Однако в кварцевом стекле тетраэдры SiO4 расположены не закономерно, как в кристаллических модификациях, а совершенно беспорядочно [Warren, 19331. Кварцевое стекло обладает чрезвычайно малым коэффициентом расширения, который при 100° равен 5,85-10”7, при 500° 6,2-10*7 и при 1000° 5,45-10”’.
Удалось установить, чтобы бинарное соединение могло находиться а стекловидно-аморфном состоянии, должны выполняться следующие условия:
1) электроположительные атомы по отношению к олектроотрицательным должны иметь координационное число 4 или 3;
2) у тетраэдров или треугольников, образованных электроотрицательными атомами, общими должны быть только их углы;
3) каждая из этих атомных групп должна иметь по крайней мере три общих угла с одпой из соседних групп;
4) каледый электро отрицательный атом должен быть связан только с двумя влет: троположь тельными атомами.
Эти условия выполнимы только для соединений состава А2Ва, АВ2 и А2В5, но не для соединений состава АВ, АВ3, А2В7, АВ4 и т, д. Наиболее известными стекловидными образованиями среди бинарных соединений являются В2О3, As2O3, Sb2Oa, BeF2, SiO2, Ge02, P0O5 и AseO5. Они соответствуют указанным условиям; однако не все соединения, состав которых выражается одной из этих формул: А2В3, А Ва и АгВа,— способны находиться в стеклообразном состоянии. По данным Смекала это справедливо только для таких веществ, у которых имеются смешанные виды связей, в результате чего направленные и ненаправленные связи взаимодействуют между собой. Это возможно благодаря тому, что между ионпой и ковалентной связью существует промежуточное состояние. Последнее имеет место всюду у приведенных ранее бинарных соединений. Однако одна и та же составная часть может проявлять различные виды связи по отношению к различным соседям, например ковалентную и металлическую связи. Это наблюдается у некоторых элементарных веществ, которые способны находиться в стекловидном состоянии, например у As, 8Ь И Sc. У стеклообразных органических веществ также имеется смешаппый вид связи, а именно внутримолекулярная ковалентная связь и межмолекулярная вандерваальсова связь.
Обезвоживанием осажденных из водных растворов кремневых гелей (ср. далее по тексту) можно получить аморфную двуокись кремния в виде белого порошка. Однако полное обезвоживание происходит с большим трудом. В природе двуокись кремния встречается в обезвоженной форме в виде кизельгура. Он состоит из остатков кремневых панцирей «инфузории^ (диатомовых водорослей), живших в глубокой древности, и поэтому называется также инфузорной землей. Он обладает исключительной способностью впитывать жидкости и поэтому его применяют в качестве упаковочного материала для баллонов с кислотами. Пропитыванием кизельгура нитроглицерином (причем кизельгур может впитать тройное количество нитроглицерина по сравнению с собственным весом) получают гурдинамит. Ввиду высокой теплоизоляционной способности кизельгур применяют для обкладки паровых труб. Кроме того, его используют в качестве звукоизоляционных покрытий, а также для многих других целей.
В форме компактных ксерогелей (см. стр. 539) двуокись кремния встречается в природе в виде минерала опала. Красивые экземпляры его используют в качестве драгоценных камней.
Осажденная обычным образом двуокись кремния и ее гели в общем не дают рентгеновской интерференции, которая указывала бы на кристаллическую структуру-То же относится и к ощетам, образующимся при обычной температуре при выделении SiO2 из воды. Напротив, образованные из горячих магматических вод опалы в зависимости от их происхождения показывают интерференцию, характерную для кристобалита яля для кварца.
Халцедон — это минерал, возникший в результате старения опала. В соответствии с этим он беднее водой, чем опал (часто совершенно без
534
Глава 22
водный), и уже обычными средствами можно обнаружить его кристаллическую структуру. Под микроскопом, особенно в поляризованном свете3 видно его волокнистое строение. Разновидностями халцедона являются агат, оникс, карнеол, гелиотроп и яшма, используемые в качестве драгоценных камней, а также кремень, который служил в каменном веке для изготовления инструментов и оружия, а позднее для добывания огня, В настоящее время его также применяют, например, в шаровых мельницах и в керамике. Окраска этих минералов вызывается незначительными примесями, присутствующими в них; так, кремень окрашен в черный цвет примесью угля.
Аморфная двуокись кремния (как осажденная в виде порошка, так и стекловидная) полностью переходит в кварц при многодневном нагревании при 400—500° в автоклавах с раствором карбоната натрия. То же относится к тридимиту и кристобалиту,
Двуокись кремния с химической точки зрения представляет собой чрезвычайно устойчивое вещество. Из кислот ее может растворять только плавиковая, вступающая- с двуокисью кремния во взаимодействие с образованием тетрафторида кремния или кремнефтористоводородной кислоты. В воде двуокись кремния практически нерастворима. Являясь ангидридом кремневой кислоты, опа легко переходит в силикат при сплавлении со щелочами
SiO2 4-.2NaOH=Na2SiO3-j-H2O,
Аналогичным образом она реагирует при сплавлении с карбонатами щелочных металлов, выделяя при этом двуокись углерода:
SiO2 Na2CO3—Na2StO3-p СО2.
Аморфную двуокись кремния можно легко перевести в раствор кипячением с растворами гидроокисей или карбонатов щелочных металлов; при этом образуются растворимые в воде силикаты щелочных металлов.
В неипачптелыюй степени двуокись кремния растворяется также, если Она взаимодействует с метанолом прп повышенной температуре н высоком давлении в присутствии метилата натрия
SlO2+4€H3OH=Si(OCH3)4+2H2O.
Очень незначительная скорость этой реакции позволяет заключить, что для разрыва связи Si—О—Si, имеющейся в твердой SiO2, необходима очень высокая энергия активации. Гораздо легче разрывается связь Si—О—Si .в эфирах полив ремневых кислот: так, из (RO)3Si—O—Si(OR)3 (R — изопропил) при действии изопропилового спирта, 'содержащего изо про пил ат натрия, с хорошим выходом получают Si(OR)4 (Kautsky И., Daubach Е., 1950—1953). В этой связи следует указать также па наблюдение Топчиевой и Баллод (1951) о том, что при адсорбции метанола на силикагеле образуется метиловый эфир ортокремневой кислоты.
Двуокись кремния в виде кварцевого песка находит самое широкое применение. В строительном деле ее добавляют в известковый раствор и примешивают к цементу. Самый чистый кварцевый песок применяют в стекольном и фарфоровом производствах. Из спеченного при высокой температуре кварца (при этом перешедшего в кварцевое стекло) изготовляют химическую посуду, исключительно устойчивую к резким изменениям температуры (вследствие очень малого коэффициента расширения кварцевого стекла) и выдерживающую нагревание до очень высоких температур. Еще в большей степени это относится к совершенно прозрачной посуде, изготовленной из полностью расплавленного кварца. Однако при работе с такой посудой следует учитывать чувствительность кварцевого
Четвертая еррппа периодической системы
535
стекла к щелочам. Раньше одна из главных трудностей при работе с кварцевым стеклом заключалась в том, чтобы при тех высоких температурах (вблизи точки плавления кварца), при которых должно проводиться выдувание этих приборов, предотвратить кристаллизацию, вызываемую следами щелочей. Для этого достаточно уж© тех ничтожных количеств, которые можно нанести па материал, дотронувшись до него перед плавлением и выдуванием потной или сальной рукой. Постепенная кристаллизация кварцевого стекла наступает также при работе с кварцевыми изделиями. «Расстекловывание» (помутнение) наступает тем быстрее, чем выше температуры, которым подвергалось кварцевое стекло в процессе работы.
Прозрачный горный хрусталь применяют для изготовления оптических приборов и украшений. Как было указано ранее, для украшений применяют также и некоторые упомянутые выше разновидности кварца.
Кремневая кислота и силикаты. При сплавлении двуокиси кремния с основными о кисла ми получают силикаты. Для их получения вместо основных окислов можно использовать также соли летучих при прокаливании кислот. Вследствие термической устойчивости двуокись кремния вытесняет легколетучие кислоты. Количественные соотношения, в которых SiO2 может взаимодействовать с другими окислами с образованием определенных соединений, многочисленны. Наиболее простыми по своему составу силикатами являются мета- и ортосиликаты, образующиеся при соединении компонентов соответственно уравнениям
Si02-|~M20=M1S103 и 8iO2+21MlO=MlSiO4
Метя силикат Ортосиликат
Тригл :соси,пикет Тетра оке оси ликат
Формула M^SiOf, для метасиликатов указывает лишь их стехиометрический состав. В противоположность простому аниону ортосиликатов анион, имеющийся в мета си Ликатах, всегда является полимерным (подробнее о структуре силикатов см. стр. 541 и сл,).
Такие силикаты, в которых па 1 Mio приходится 2, 3 и т. д, молекул SiO2, называют ди-, три- и т. д. силикатами, и вообще силикаты, которые стехиометрически содержат па 1 Mio большее число радикалов SiO2, называют «полисиликатами!/.
В то время как силикаты образуются в очень разнообразных соотношениях, свободная кремневая кислота до настоящего времени известна лишь в немногих определенных формах. При известных условиях в водном растворе может находиться ортокрвмневая кислота (тетраоксокремневая) H1SiO1 (см. стр: 538 и сл.), однако только в очень малых концентрациях, -Эта мономолекулярная и растворимая в воде форма кремневой кислоты -очень неустойчива и обычно крайне быстро конденсируется в высокополи-мерные, твердые и практически нерастворимые в воде формы. Среди них с определенным составом наиболее легко получаются метакремневая кислота (триоксокрсмневая) H2SiO3 и дикремневая кислота (пентаоксо-кремневая) H.3SbOs.
Кремневая кислота является очень слабой кислотой. Константа ее электролитической диссоциации крайне мала. Также чрезвычайно мала и растворимость для обычной (т. е. находящейся в конденсированной -форме) кислоты, поэтому уже при очень небольшой концентрации водородных ионов создаются условия для ее осаждения из силикатных растворов. Вследствие этого даже самые слабые кислоты вытесняют кремневую кислоту из растворимых силикатов.
536
Глава 12
Из солей кремневой кислоты в воде растворимы только силикаты щелочных, металлов. Из силикатов практически нерастворимых в воде, некоторые могут растворяться в сильных кислотах с разложением (с выделением геля кремневой кислоты). Однако многие силикаты не подвергаются действию даже и этих кислот и разлагаются только плавиковой кислотой. Действие этой кислоты основало на большой склонности кремния образовывать фторид кремния (ср. стр. 522), Все силикаты легко вступают во взаимодействие с расплавленными щелочными карбонатами. При этом образуются силикаты щелочных металлов и обычно выделяется дву окись углерода. Например;
MjjSiXr. — MgCO3-j—IXa2SiO3,
KAlSinOg-pSNa^CO? — 3№аа5Ю3-(-КА1024-ЗС02.
Водные растворы силикатов щелочных металлов обладают сильно щелочной реакцией. Силикаты в этих растворах подвергаются далеко идущему гидролизу.
Гидролиз метасиликата ватрия, идущий по уравнению
2Na2SiO3^H2O=iS“a2Si2O5^2NaOH,
составляет по Шварцу (1927) в 1 п. растворе 14%, в 0,1 и. растворе 28 и в 0,01 я. растворе 32%, а гидролиз дисиликата натрии в 1 и 0,1 и. растворах достигает 2,4 и 6% соответственно. Присутствия других ионов, кроме SiO3 и Si20s или же [Si.02(0H)2]" и [Si2O3(OH)41", образованных путем присоединения воды, в этих растворах до сих пор установить не удалось.
При подкислении растворов силикатов тотчас выделяется в свободном состоянии кремневая кислота. Однако она обычно не сразу выпадает в осадок, а сначала остается в растворе- Только через продолжительное время начинается выпадение ее в виде хлопьев. Это объясняется отчасти тем, что кремневая кислота может находиться в растворимой мономолеку-лярной форме, которая в зависимости от условий быстро или медленно-переходит с выделением воды в высокомолекулярные формы и в конце концов образует практически нерастворимые высокомолекулярные агрегаты. Однако и после того, как кремневая кислота полностью перешла в нерастворимую форму, еще не происходит ее осаждения, так как она может оставаться в растворе в коллоидном состоянии. Склонность кремневой кислоты образовывать коллоидные растворы (кремневые золи) чрезвычайно велика. Кремневая кислота в коллоидной форме устойчива как в кислых, так и в нейтральных и слабощелочных растворах. Добавлением электролита обычно нельзя осадить ее в виде хлопьев, но не слишком разбавленные растворы медленно превращаются при этом целиком в студень. Из разбавленных растворов выпадают слизистые осадки. Быстрое осаждение достигается прежде всего при действии баритовой воды или концентрированного раствора сульфата алюминия. Однако ввиду того, что-большинство других электролитов вызывает медленное осаждение или застудневание, их следует удалять, если требуется сохранить устойчивые коллоидные растворы в течение длительного времени. Это осуществляют диализом, т, е, внесением раствора в сосуд, в котором коллоидный раствор отделен от чистого растворителя мембраной из пергамента, свиного пузыря, коллодия или других подобных перепонок (диализатор). Вещества, находящиеся в растворе в состоянии молекулярного растворения (кристаллоиды), диффундируют через такую мембрану, в то время как коллоидно-растворенные вещества задерживаются или по крайней мере проникают очень медленно (подробнее см. т. II, гл. 16),
Четвертая группа периодической. системы
537
Некоторые слабо разбавленные кремневые золи застывают в студни, уже при продолжительном стоянии, другие — при выпаривании или добавлении электролита. Богатые водой кремневые студии совершенно прозрачны, мягки и довольно эластичны.
Св ежен рп готе пленные кремневые гели могут содержать до 330 молей Н2О на 1 моль SiO2, При продолжительном стоянии (на воздухе, насыщенном водяным паром) она постепенно начинают сморщиваться, выделяя воду; это явление наблюдается также н для других студией и носит плавание синерезиса. Значительного часть воды можно удалить также растиранием в выжиманием. Еще большие количества воды отделяются при стоянии на сухом воздухе. В эксикаторе кремневые студни и вообще кремневые гели*, наконец, высыхают с образованием совершенно твердой и истирающейся в порошок массы, которая, однако, еще может содержать значительные количества воды, например 6 молей Н2О иа 1 моль Si О,. Высыхание сопровождается весьма значительным сжатием геля. Однако, начиная с определенного момента, сжатие внезапно приостанавливается и прозрачный ге.чь мутпоет; он становится белым, как мел. Бан Беммелен, который впервые изучал эти процессы, назвал точку, мри которой наступает это помутнение, точкой перехода. Например, точка перехода может соответствовать содержанию 2 молей Н2О па 1 моль SiO2. Начиная с той же точки, давление паров воды геля часто становится приблизительно постоянным и остается таковым до тех пор, пока не отделятся вся вода (ирн этом происходит второй «переход» и гель внезапно становится снова прозрачным). Поэтому естественно предположить существование определенных химических соединений, соответствующих составу геля в точках перехода. Для химического соединения, разлагающегося с выделением газообразного продукта разложения, характерно то, что опо При определен пой температуре обладает определенным давлением пара, которое сохраняется до тех нор, пока имеется еще пе разложившаяся часть соединения (см. стр. 98 и сл.), Одпако Ван Беммелен пашел, что содержание воды, соответствующее точке перехода, может изменяться в довольно широких пределах. Согласно Ван Беммелепу, этц явлении с тем ;ке успехом и в некоторых отношениях даже лучше можно разъяснить, предположив, что вода здесь связана не химически, а механически и что прерывистость кривой обезвоживания объясняется тем, что вода весьма прочно удерживается в мельчайших пустотах геля капиллярными силами («впитанная вода»). Действительно, Зигмонди показал, что после удаления заполняющей эти пустоты воды гелем может быть поглощена любая другая жидкость, например бензол, и притом в таком количестве, которое соответствует объему (а ие молекулярному соотношении)) удаленной воды. При повторном высушивании и пропитывании другой жидкостью опять-таки наблюдаются характерные для точки перехода явления. Так как для жидкостей типа бензола химическая связь с SiO2 пе предполагается, тем более, что пет и намека иа стехиометрические соотношения, а, наоборот, происходит только впитывание воды («imbibition») в капиллярные пустоты геля, то вообще нет никакого основания предполагать наличие химической связи между гелем и водой.
Одиако при некоторых определенных условиях можно получить препараты, в которых Содержание воды, как доказано, соответствует определенному простому стехиометрическому соотношению; легче всего такие препараты получают при действии довольно концентрированных кислот, например 80 %-ной серной кислоты, на кристаллические силикаты. Шварц показал, что этим путем можно получить кремневые Гидраты, которые после надлежащей сушки (при применении ацето нового метода Билль-штеттера; ср. сказанное по этому поводу относительно гидроокиси алюмипия) обнаруживают стехпометрачески определенный состав. Здесь речь идет о кислотах HzSiO3 (метакремнееои кислоте) и H2Si20g (дикремнеаой кислоте). Первую можно получить, исходя из .иетасиликата MIS1O3, вторую — из дисиликата M^Si3O5, Если исходить яз сплавов, содержащих окиси щелочных металлов и двуокись кремния в других количественных соотшгпггшнях, то получаются смеси обеих кислот или при известных обстоятельствах такие препараты, которые, кроме обеих кислот, будут содержать свободную двуокись кремния. По составу нельзя сразу решить, представляет ли препарат состава 2SiO,-H20 определенную кислоту H2Si2O,5 или смесь H2SiO3 с SiO2. Одпако Бильцу удалось охарактеризуют. обе названные кислоты как определенные соединения па основании их различного отношения к аммиаку. Обе кислоты, будучи обработаны жиц-
* Выражение «гель» используют для общего обозначения объемных осадков, полученных осаждением коллоидов в виде хлопьев, а также для обозначения студией, образующихся при однородном застывании золей.
538
Глава IS
jcuai аммиаком, образуют с ним продукты присоединения. Однако в то время как мети-кремнсвая кислота при нагревании в вакууме отдает аммиак ступенчато, образуя пэ-следовательно несколько продуктов присоединения аммиака простого стехиометрического состава, выделение аммиака, связанного с дикремневон кислотой, протекает непрерывно. По данным рентгенографического анализа дикромпевая кислота представляет собой кристаллическое вещество, а метакремневая кислота — аморфное.. Метакремневая кислота Гораздо мепее прочное соединение, чем дикремневая кислота. Ее можно получить только прп температуре незначительно выше 0°; при комнатной температуре она распадается но уравнению 2H2Si03=H2Si2O5+H3O. При 150° распадается также и дикремневая к ислота с образованием Si О, и П2О,
При обработке силикатов концентрированными кислотами кремневая кислота получается в виде значительно обезвоженных порошкообразных веществ. Последние хотя и имеют природу гелей, однако она в пих выражена гораздо слабее, чем у кремневых гелей, полученных из водных растворов. Тот факт, что одна и та же химическан реакция приводит к образованию продуктов с различными свойствами в зависимости от того, идет опа в растворе или на кристаллических веществах, наблюдается довольно часто. Такне реакции, и которых свойства твердых продуктов взаимодействия существенным образом определяются тем, что превращение происходит на твердом веществе (т. е. связано местом), называются по предложению Кольшюттера топохимическими реакциями (тбяо5 — место).
Применив метод ацетоновой сушки, Шварц смог показать, что при более низких температурах (0°) существуют гели метакремневой кислоты H2SiO3, получаемые разложением SiCli или SiP4, а в гелях, получаемых разложением растворов силикатов, имеется дикремневая кислота lIsSi2O5.
Для доказательства существования определенных кремневых кислот Тиссен избрал другой путь (Thiessen, 1929). Он исходил из предположения о том, что у гелей с достаточно крупными порами не должно быть отклонений, вызываемых влиянием впитанной воды, и что, согласно законам роста кристаллов, получение гелей: с наиболее крупными порами должно достигаться прп возможно более медленном образовании соединения, составляющего их скелет. И действительно, при достаточно медленном гидролизе этилового эфира орто кремневой кислоты Si(OC2H5)4 ему удалось получить препараты, которые при изотермическом обезвоживании (при 13’) отщепляли воду отчетливо ступенчато. Первая точка излома кривых давление — концентрация соответствовала составу ортокремневой кислоты H^SiOj. При дальнейшем обезвоживании наблюдались еще изломы, соответствующие составам 2SiO2-3H2O, SiO2.H,O и 2SiO3.HaO. Однако существование этих Соединений нельзя считать точно установленным. Вайсеру при повторении опыта Тиссена удалось установить на кривой давление — концентрация лишь точку излома, соответствующую метакремневой кислоте H.,SiO3.
Мил ну с и Г р опт уф ф (МуИиз, Groschut I', 1906) показали, что при разложении растворов силикатов щелочных металлов соляной кислотой кремневая кислота вначале выделяется в кристаллоидном состоянии. Свежеосаждеппая кремневая кислота быстро диффундирует через пергаментную мембрану, и ее раствор обнаруживает значительное понижение точки замерзания. Она отличается от коллоидной кремневой кислоты, полученной при старении, своим отношением: к белку, которым опа не осаждается. Старение объясняется укрупнением частиц, которое по данным Вильштеттера происходит так, что первоначально образовавшаяся H4SiO4 прогрессивно конденсируется с отщеплением воды. Внльттеттер установил, что скорость, с которой происходит укрупнение частиц, в значительной степени зависит от концентрации водородных ионов. Медленнее всего идет старение при pH 3,2. Пропусканием паров SiCl4 в воду при 0° и устранением образовавшегося ири гидролизе хлористого водорода (взаимодействием с окисью серебра) можно было, поддерживая наиболее подходящую концентрацию водородных ионов, получить растворы, в которых по криоскопическим данным содержалось по меньшей мере 75—80% Н^ЗтО^ (соответственно SiOs) в мономолекул яр пой форме.
Другая возможность предотвратить осаждение (вызываемое ростом частиц) твердого гидрата двуокиси к ремния из растворов кристаллоидной кремневой кислоты основана на том, чтобы к моменту, когда конденсация зашла еще не слишком далеко, проэтерифициропать по способу Кирка (Kirk, 1946) гидроксильные группы, вызывающие конденсацию. Таким путем получают эфиры поликремпевой кислоты, которые растворимы в органических растворителях и продолжительно устойчивы. Полученные таким образом эфиры полнкремиевон кислоты имеют техническое значение. Испарением растворителя их можно выделить в твердом состоянии, а в случае необходимости снова растворить. Измерение вязкости их растворов (11 е г R. К,, Pinkney Р. S., Ind. Eng. Chem., 39, 1379, 1947) позволяет заключить, что их частицы имеют Приблизительно форму шара.
Гликолевый эфир Ортон ремнев ой кислоты отличается тем, что в спиртовом растворе он имеет кислую реакцию и образует с алкоголятами продукты присоединения.
Четвертая группа периодической системы,
539
в которых кремний, по-лпдимому, проявляет координационное число, равное пяти...
М\ ZR =-
XOZ
СН,—О. <' И1, СН,—О, j ,0 — СН,
| “ X | +MTOR=J xSi< I снг-о/ о--сн, сн2-о/ хо-сна
Константы диссоциации мономолекулярвой кремневой кислоты составляют: К 1=2,2-10*’», К,=2 10*’2. /<а!=1 -КГ12, Кл< 1 10*12. Как показал Ныовенбург (Van Niemvehbui'j?, 1930), кремневая кислота заметно летуча с водяпым паром, нагретым выше критической температуры. Это наблюдение имеет значение для объяснения некоторых геологических явлений.
Кремневые гели, подобно древесному углю, отличаются превосходной адсорбционной способностью, связанной с их сильно развитой внутренней поверхностью. Особенно эта способность проявляется у высушенных гелей. Для остатков после высушивания студней Фрейндлих ввел общий термин «ксерогели» (^Tjgog — сухой). Поэтому кремневые ксерогели за последнее время часто применяют для адсорбции паров, для очистки жидкостей (особенно для очистки потопов сырой нефти от сернистых соединений), а также для каталитических целей. При этом существенно, что таким ксерогелям можно сообщать специфическое сродство к определенным веществам введением в ксерогели определенных реактивов. Кроме того, структура геля не изменяется или изменяется лишь незначительно при умеренном нагревании (слабое прокаливание). Поэтому поглощенные гелем вещества можно иногда удалить простым прокаливанием и гель использовать снова. Перед инфузорной землей, которую также следует считать кремневым ксерогелем, искусственно полученные кремневые ксерогели имеют то преимущество, что на их структуру может оказывать влияние то или иное изменение процесса получения и это можно согласовать с целями их применения. В хирургии и дерматологий порошкообразные кремневые ксерогели, пропитанные жидкостями, например ихтиолом, перуанским бальзамом и т. д., применяют в качестве поглощающих запахи, дезинфицирующих и одновременно высушивающих присыпок. Кремневые ксерогели применяют для обезвреживания табачного дыма. Пропитанный хлоридом кобальта кремневый ксерогель применяют в лаборатории под названием голубого геля в качестве сильно действующе г о осушителя газов. Ослабление высушивающего действия определяют по переходу окраски хлорида кобальта от голубой в розовую. Гель регенерируют нагреванием. В гальванических элементах и в аккумуляторах электролитную жидкость иногда переводят при помощи кремневой кислоты в студнеобразное состояние в целях уменьшения ее разбрызгивания.
Чрезвычайно распространено применение кремневой кислоты в виде силикатов. Основой фарфоровой промышленности является водный силикат алюминия, встречающийся в природе в виде каолина. В стекольном производстве используется свойство многих «расплавленных силикатов затвердевать в виде стекла». Затвердевание цемента и изготовляемого из него строительного раствора основано главным образом на образовании силикатов. Силикатами являются и применяемые для смягчения воды пермутиты, а также широко распространенная ультрамариновая краска. Некоторые природп/ае г.пгнояодобные ксерогели алюмосиликатов в связи с их избирательной адсорбционной способностью служат в качестве так называемых отбеливающих земель (для обесцвечивания минеральных, растительных и животных масел и жиров). Широкое техническое приме-
540
Глава 73
некие имеют также поступающие в продажу под названием растворимой: стекла сплавы силикатов щелочных металлов.
Растворимое стекло получают сплавлением кварцевого песка с поташом или содой или с сульфатом патрия и древесным углем (обычно кг 3—5 молей S1O2 берут 1 моль окиси щелочного металла). Под влиянцек содержащихся в кварцевом песке следов солей железа образуются окрашенные в зелеповаттле и сероватые тона стекловидные куски растворимого стекла. Одпако болынеп частью растворимое стекло поступает в продажу в виде водных растворов, так как его ^растворение сопряжено с трудностями (в технике растворение осуществляют либо обработкой растворимого стекла водяным паром, либо предварительным смачиванием измельченного растворимого стекла небольшим количеством воды; порошок впитывает воду с образованием твердого геля, после чего топко измельченный гель легко растворяется в воде)*. Растворимое стекло применяют для пропитки бумажных ткапей, проклейки бумаги, утяжеления шелка, для придания твердости хирургическим повязкам, изготовления замазок и склеивающих веществ, для приклеивания этикеток па стекле, придания Дереву и тканям огнеупорных свойств, консервирования яиц (4—10%-ный раствор). Порошкообразный гель растворимого стекла используют в качестве наполнителя для мыла. Растворы растворимого стекла обладают ограниченной устойчивостью; со временем они постепенно разлагаются с выделением кремневой кислоты. При этом часто раствор застывает в студень.
Силикат натрия можно получить сплавлением измельченного кварца с карбонатом натрия (5) или взаимодействием свежеосажденного гидрата двуокиси кремния с рассчитанным количеством едкого натра (6).
SiC)24 Ма.Д.0з=Хаг81Ог-гСОг. (5)
S1O2- |-2\aOH^ya2SiO34 НаО. (6)
Из чистого сплава (т- пл 1088“) силикат натрия застывает в кристаллическом виде. (Напротив, сплавы силиката натрия с другими силикатами, например с силикатом кальция, который застывает обычно также в кристаллическом виде, образуют стекла.) Из водного раствора силикат натрия кристаллизуется с водой. Кристаллогидрат силиката натрия поступает в продажу под названием алкавиля. Его применяют для отбелки фарфоровых изделий, а также для тех же целей, что и растворимое стекло.
Кроме обычного силиката натрия — метасиликата Na2SiO3, из сплавов получены соединения [Va4SiO4, NaeSi2O? и Na2Si2Of, (D'Ans J., 1930). Из водных растворов кристаллизуются силикаты натрия, содержащие воду (см, S t а с k е 1 b е г g, Z. anorg. Chem., 256, 273, 1948). Они очень хорошо растворимы. Еще в большей степени это относится к силикатам калия, которые поэтому с трудом кристаллизуются из водного раствора. Из расплава получены K2SiO3, K2Si2O5 и KgSi4O<), Исключительно хорошо кристаллизуются силикаты лития.
Природа во дусо держащих силикатов щелочных и щелочноземельных металлов. Содержащие воду силикаты натрия Тило (Thilo, 1951) называет кислыми. Это представление сложилось главным образом на основании данных изучения химических свойств и определения веса попов, из которых следует, что в растворах этих соединений существуют одноядерные силикатные ионы, а имеппо [HSiO4]'", [H2SiO4]" й [H3SiO4}'. В соответствии с данными Тило [см., например, Ang. Chem., 63, 201, 1951) ионы кальция также способны образовывать кислые силикаты, например Ca[H2SiO4] и
* См. М й 1 ] е г W. J., Z. anorg. Chem., 236, 232, 1938.
Четвертая ipynnri периодической системы
541
NaCa[HSiO4J. Эти данные тем бояре достойны внимания, что до лих еще не было установлено существование кислых силикатов рентгеноструктурным методом. Тило поставил вопрос о том, пе являются ди еще многие другие содержащие воду силикаты, рассматриваемые до сих лор кат; гидраты, также кислыми силикатами, например минералы афвилит (3Ca0-2SiO2 1L O), рпверсидеит (2CaO2SiO2 Н.>0), крестмореит (2СаО-2SiO23H2O), окенит (CaU 2SiO, 2Н2О), гиролит (2CaO-3SiO2-2H;>O), гиллебрандит (2CaO-SiO2'H2O), фосхипп (5СаО 3SiO.2-3H2O), ксополит (4CaO.4SiOa.II2O) и тобер-морнт (4CaO-5SiO241I2il-( Для афвилита это вскоре было подтверждено рентген о структурным определением (М ogaw Н., Acta Crystailogr., 5, 477, 1952). Как показали недавние рентгене структурные исследовании, для риверсидоита п крести open та, вероятно, следует предполагать такое же строение (Taylor Н. F. W., Min. Magazine, 30, 155, 1953).
Дня объяснения структур силикатов щелочных и щелочноземельных металлов химическим путем имеют значение следующие виды взаимодействия:
1. Гидролитическое расщепление тина гидролиза соли, например;
Na2Ca[SiO4] НОН = NaCa[HSiO4] > NaOH или же
[SIOJi-4- ИОН = (Si03(OH)]s"4- ОН".
2. Гидролитическое расщепление анионов поликислот на более простые, например:
(Sf3O7 ПОН = 2[SiO3(OH)J3" и
LSi4O11(OH)3]e-^. 3H0IJ = 4[SiOa(OH>P 4^ 4Н+.
3. Обратный процесс: конденсация кислотных ионов с выделением воды, например:
2[SiO3(OH)]3"= [Si2O7]°“^ Н2О.
4. Перегруппировка анионных агрегатов тетраэдров SiO4, папример:
Si О2-про стр листве иная сетка -+ [813О91-кольцо.
Каждый из этих типов реакций соответствует определенной области температур. Тило установил, что отиошенне наименьших температур (в градусах Кельвина), при которых силикаты кальция претерпевают эти превращения, к тем температурам, при которых происходят соответствующие превращения у фосфатов натрия, является практически постоянной величиной. Радиус иона натрия Лишь немного отличается от радиуса иона кальция. Вероятно, этим и определяется структурное и химическое родство между этими группами соединений.
Перокси гидраты силикатов. Надкремневые кислоты или их соли, вероятно, не существуют. Однако некоторые силикаты образуют с Н2О2 определенные продукты присоединения. Первым среди них получен (Kraup, 1932—1935) дипероксигидрат мета-силиката натрия Na^SiO^-SH^Oj. При выпаривании в вакууме раствора метасиликата натрия, к которому была добавлена Н2ОИ, Краус получил сначала гидрат Na.,SiO3x х2Н2О2’НаО в виде белого мелкокристаллического порошка. Этот гидрат можно обезводить без значительной потери активного кислорода. Пероксигидраты силикатов щелочных металлов хорошо растворимы в воде и устойчивы в отсутствие влаги. Напротив, пороксигидраты дггсиликатов щелочноземельных металлов и пероксигидрат двуокиси кремния SiO2.H2O2 трудно растворимы и малоустойчивы.
КРИСТАЛЛИЧЕСКАЯ СТРУКТУРА СИЛИКАТОВ
Тонкое строение кристаллических силикатов установлено только в последнее время, главным образом путем рентгеновских исследований. Определение структуры при помощи рентгеновских лучей показало, что кристаллические силикаты образуют Координационные решетки, которые в первом приближении можно рассматривать как ионные*. Принцип
* Наличие ионов в решетках силикатов следует как из'данных изучения электропроводности и подвижности ионов, так и из аддитивности показателя преломления и удельной теплоемкости. Однако имеются основания считать, что при образовании особо прочной связи атома О с атомами Si наряду с электровалентностью имеют значение также силы, которые называют «ковалентностью».
542
Глава 12
строения всех исследованных до сих пор кристаллических силикатов odus и тот же, и он основан на образовании кислородных тетраэдров вокруг каждого иона Si4+, причем расстояние Si«—> О равно около 1,60 А. Если на каждый ион Si1+ сто хио метрически приходится меньше четырех цопав О2-, то этот принцип построения сохраняется благодаря тому, что ионы О2- могут быть общн.ии для соседних тетраэдров. Между отрицательными комплексными ионами, состоящими из отдельных тетраэдров Si От или из тетраэдров, соединенных между собой общими ионами О-~, расположены положительные ноны, участвующие в построении кристалла. Эти ионы располагаются аизможно дальше друг от друга, и, как вообще в координационных решетках, группируют вокруг себя как можно ближе и больше отрицательных иопов (в данном случае ионов О2' тетраэдров SiOi) (ср. стр. 242,1- На основании внутреннего строения силикаты подразделяют иа соединештя с островными структурами, с цепочечными и ленточными, структурами, со слоистыми и с пространственными сётча-тыми структурами.
Островные структуры. Об островных структурах говорят в тех случаях, когда в кристаллической решётке имеются так называемые «островки», т. е. группы, построенные из ограниченного числа ионов Si4+ и О2-, например [ё1О4]!“, [Si2O;]3", [Si3Oe]K *,
Рис. 95. Решетка оливина (Mg, Fe)3[SiO*]. «=10,31 .Л; &=4,755А; с=5,98£ А.
Тетраэдры SiO4 могут существовать как самостоятельные, отдельные группы [SiO4]4- только и таких силикатах, в которых ла каждый атом Si стехпометрически приходится по меньшей мере четыре атома кислорода, следовательно, в ортосиликатах, таких, как фенапгет BealSiOi], ^орстерши. MgalSiOjJ, оливин (Mg, Fe)«(SiO4|, циркон ZrfSiOJ. На рис. 95 представлена элементарная ячейка оливина, которая является примером ортоенлтгката с самостоятельными тетраэдрами SiO4. (Ср. также обсуждаемую в т. II решетку циркона.) Соответствующие структуры определены также в Других соединениях типа MJROJ и М[ВО4], Например, фепакиту, кроме других ортосипикатов, изотнипы соединения Li2 [BeF4], Li2[MoO4l и Lia [ЛТО4]; оливину изотшшы (кроме Mg2[SiO4I, Fea[SiO/J, MgCa [SiOJ и т. д.) LiFen[PO4] и А]а[ВеО4]; циркону изотипны, например, Y[PO4] и Ca[CrO,J.
Кроме радикала |SiO4]’", в построении такого твид решеток могут участвовать также еще другие отрицательные ионы, например F~, (ОН]~, Оа (наряду с ионами металлов). Примеры: цианит (диСтен) AI2O[SiO4] и титанит CaTiO [SiO4J, кроме
* Иногда название оапроонь;?- структуры применяют в более узком смысле, т. е. специально для таких силикатов, в которых имеются группы [SiG4V~. От этих силикатов отличаются силикаты с группами, построенными из большего (но ограниченного) числа тетраэдров Si-От,, например [SijO,]3- и [Si^Or,]3'. Их структуру тогда называют групповой структурой.
Четвертая группа периодической системы
54Э
того, топав А13(ОН, F)3 [SiOj4] и гумит 3Mg2 [SiO4]-Mg(OH, F)a. Решетка гумита и родственных минералов образуется при закономерном внедрении между слоями свойственной оливину структуры cjiocb из Mg(OH,F)2 или же из ионов Mg'2+ и ОН’ (любой может быть заменен на ионы Г ). Аналогично может быть построена структура топаза путем внедрения А1(0Н,Г)3-слоев в решетку дианита. В таких случаях говорят об островных структурах внедрения.
Если количество ионов О2 недостаточно для образования исключительно самостоятельных гругпг [SiCZj р , то связь в тетраэдре осуществляется через общие с соседними тетраэдрами ноям о--. Опыт показывает*, что во всех силикатах соседние тетраэдры SiO4 всегда имеют только один общий ион О2’; другими словами, тетраэдры соприкасаются всегда только вергиинами и никогда ребрами или даже гранями. Попарно соединенные тетраэдры образуют группы (Примеры: Na3Ca2|Si3O7],
Са4(СШ), [Si2OJii минерал тортвейтит Sc2[Si2O7]).TpH или большее число тетраэдров часто связываются один с другим с образованием колец. Это символизируют следующие формулы, в которых повернутые один к другому ребра тетраэдров обозначены штрихами.
O2Si — О-— SiO2 oz •"••d
= (Si3O<j}9-,
OaSi
O2Si—О — Si.O2
O3S1—O — SiO3
= [Si8OI8)«'
В такой кольцеобразной связи тетраэдры образуют группы общей формулы ISinOanI1112-Существование таких колец доказано, например, в кристаллических решетках волластонита (а-воЛластонит) Са3 (Si3Os] (ср. также стр. 70), бенитоита BaTi [Si3Ou] и берилла А1аВе3 [SieOiaJ. Встречаются также структуры включения с кольцеобразной связью тетраэдров, например диоптаз Cu8[Si6OIS]-6H2O, образованный, по данным Хейда (Heide, 1954), на колец [Si8O18]ls- н ионов Си3+, между которыми включены молекулы воды.
Если ограниченное число тетраэдров [SiO4] не образует кольца, то возникают структурные группы общем формулы [SinO3n+1 >2-—. Например, пять тетраэдров
могут соединиться так, что’одип из пих имеет с четырьмя другими тетраэдрами по одному общему иону О2". Такая группировка была Доказана в цуниите А1 [Л112О4(ОН, Р)18] [Si6O18]Cl. Структуру этого сложного соединения можно вывести из относительно простой решетки цинковой обманки (см. т. I I), если в ней заменить иоиы Za'i+ и S2- па структурные группы, показанные в предыдущей формуле квадратными скобками. Иопы АН- и С1-, входящие в состав соединения и компенсирующие избыточные заряды, закономерно распределяются в пустотах решетки.
Известны также силикаты, в построении которых принимают участие одновременно силикатные группы различного состава, например везувиан Cai8Mg2AI4 [SiO4l 5 [Si2O7 ]3(ОН)4.
Цепочечные и ленточные структуры. Тетраэдры SiO4 могут соединяться также с-образован) гем «цепей» Й еле нт», различные виды которых представлены на рис. 96, а—е. Так как Цени имеют неограниченную длину, то при цепном расположении на каждый вон SP- стехиометрическа приходится три (а именно 2-‘-2/3) иона О2~ (см. рис. 96, а и б). Следовательно, cn..i ггкаты с цепочечной структурой по своему химическому составу являются метасиликат.ами**. Одпако так как обозначение состава через формулу Ми [SiO3]
* Теоретическое обоснование этого см.: Pauling, J. Am. chem. Soc., 51, 1010, 1929. В обычных модификациях двуокиси кремния, тетраэдры SiO4 всегда соприкаса-ются также только вершинами. Лишь в малоустойчивой волокнистой модификации SiO2, которая не встречается в природе и получена только при особых условиях, тетраэдры SiO4 соприкасаются ребрами (см, стр. 527).
** Силикаты с островной структурой также иногда имеют стехиометрический состав, отвечающий метасиликатам.
544
Глава 12
дает ложное представление о координационном числе кремния в силикате, то обычно силикаты 0 цепочечной структурой выражают через общую формулу М^1 [Si2Oe|. II р н-иоры: знстатит Mg2 (Si2Oft ], диопсид CaMg[Si20e], сподумен Li Al [Si2O6 ], а также псеадоволластоншп (р-вол л истопит) Cas [Si2O6], который образуется из волластонита (си, выше) нри высокой температуре путем ионотропного превращения. Соответствующее превращение претерпевает rrjoii нагревании изоморфный волластониту пектолит (здесь, по данным Тило, речь идет о кислом силикате NaCa, [Si3Os(OII) ].
В соответствии с рис. 96, е при образовании «ленты» из двух цепей от четырех тетраздров иногда отделяется один нон О2". Таким образом из двух цепей [Si2Oe ]4 возникает лепта [Si4On]6- (также гю ограниченной длины). Возможно, что две лепты
Рис. 96. Цепи (а и б), ленты (виг), плоские гексагональные сетка (0) и нлоские тетрагональные сетки (с), Образованные тетраэдрами Si Од.
[Si4Ojj ]6* можно объединить л лепту [Si8O2l]i°~, состоящую соответственно из четырех цепей; этого, однако, еще пе наблюдали. Примеры ленточных структур: тремолит Ca2Mg6 [Si8O22](OH)2, рибеккит Na2FeJIFeatl[SisO22](OH)2; глаукофан Na2(Mg, Fen)3(Al. FeiiI)2 lSisO22](OH)2; такую же структуру имеет роговая обманка. Пример лепточной структуры внедрения: хризотил (волокнистый серпентин) Mg3 [Si4On ]-3Mg(0H)2-
Однако цепи можно соединить таким образом, что один ион О3"" отделяется от каждых двух тетраэдров. Тогда из двух цепей [Si2Oel4“ получается одна лента [ЗцО10|4-
* В приведенных примерах речь идет целиком о решетке, образованной лентами [Si^Ojj]15-. Если из-за целочисленности атомов нужно удвоить формулу ленты, то, естественно, не имеет смысла написание этой формулы с индексом 2, поскольку речь идет но о законченной группе, а о лепте любой длины. Поэтому удваивают числа отдельных атомов внутри скобки.
Четвертая группа периодической системы
545
(см. рис. 96, г). Эта структура имеется в силлиманите Al2 [Al2Si2Ou]; о.дпако в ней цепи образуются пе только из одних тетраэдров SiO4, а па чередующихся между собой тетраэдров SiO4 и ЛЮ*, и именно так, что в двойной цепи тетраэдры Si04 и тетраэдры А1О4 иногда располагаются друг против друга. Среднее положение между этой структурой и структурой цианита (ср. стр. 542) занимает решетка андалузита.
Листовые структуры (слоистая решетка). Бесконечное расширение лепт[Зг4Оп]в-прпведитк образованию «плоской сетки» или «листа» (см. рис. 96, о). В такой структуре каждый ион Si4+ имеет три общих иона Ой“ со своими соседями. Следовательно, иа каждый ион Si4+ стехис, метрически приходится 1—|-3/в|=21/2 иона О2- иопа. Поэтому этой структуре соответствует формула [SiaO6]s'“ (пли любое кратное от нее). Иа таких более или мепее рыхло наложенных слоями плоских сеток или листов построены слюды и подобные им расщепляемые на слои силикаты. Прим ер ы: тальк Mga [Si4Ola ](OTI)2, антигорит (листовой серпентин) Mg3[Sis010J(OH)a-3Mg(OH)a (слоистая решетка внедрения) , каолинит Al2[Si4O10](OH)a-2Al(OH)3 и др.
Между двумя структурными типами серпентина {хризотил: и а нтигор йт), об означенными формулами MgalSliOifJ-SMgfOHJjj HjO и Mg3[Si4Ole|(OH)2-3Mg(OH)2, существуют непрерывные переходы. Они осуществляются .благодаря все большему расширению лент Si40ji при их конденсации, в результате чего в конце концов достигается структура листа Si4O10. Одновременно с этим лепты Mg(O, ОН, Н20) перестраиваются в бруцитовые слои. В данном случае слоистая расщепляемость не связана с появлением листовой структуры, как в других силикатах. Как показал Нолл, в связи с разной величиной отверстий между листом Si4Oln и бр\ цитовым листом, сконцентрированными каждом двойном слое, может быть вызвано такое сильное искривление двойного слоя, которое приведет к образованию капиллярно построенных волокон. В соответствии с электрон ио-микроскопическими данными, полученными Ноллом, такого вида волокна часто встречаются в волокнистом серпентине (хризотиле). Следовательно, волокнистый серпентин (хризотил) может иметь внутреннюю структуру, соответствующую как одной, так и другой формуле, в то время как слоистый серпентин (антигорит) всегда имеет внутреннюю Структуру, отвечающую формуле Mg3 [Si4O10](OH)2-3Mg(OH)B. Гарниерит, который в природе встречается чаще в виде более или менее загрязненного гидро-ксиснликата магния и пикеля, изоморфен хризотилу. В чистом состоянии соответственно формуле Ni3[Si4Oi0 ](0Н)2 3Ni(0II)a оп был получен Ноллом [N oil, Naturwiss., 39, 233, 1952) гидротермальным синтезом, подобно тому как ранее был получен хризотил.
Электронно-микроскопическим исследованием в гарпиерите установлено наличие капиллярных волокон, что можно было ожидать вследствие одинаковых размеров отверстий сеток Ni(OH)a и Mg(OH)2.
Изоморфные минералы монтмориллонит Al2[Si40ln](0H)2-nHa0, гекторит. Mg3 [Si4O10](OH)2-лНг0, и нонтронит (Fop, Fel11) [Si4010](OH)2-nH20 построены из слоев силикатов металлов (о равномерно внедренными в них ионами ОН); между слоями может быть расположено переменное количество молекул Н20. Этим объясняется возможность обмена катионов и необычно большая способность этих минералов к набуханию (ср. стр. 551 и сл.). Смешиванием хлорида магния и растворов силикатов щелочных металлов Гофман получил слизистые осадки, которые при благоприятных условиях получения дают рентгеновскую интерференцию, характерную для силикатных слоев гекторита', одпако, как показал рентгеновский снимок, силикатные слон в них расположены довольно нерегулярно: параллельно они располагаются только в том случае, если осадки нагревают с растворами щелочей определенных концентраций до температуры около 200s и под давлением. При более сильном нагревании с раствором едкого кали (по пе с раствором едкого натра) происходит превращение в слюды. Изменением количественного соотношения смешиваемых растворов можно получить также осадки со структурой антигорита. При гидротермальной обработке (т. е. при сильном нагревании с водой пли же с раствором едкой щелочи под давлением) осадки не претерпевали изменения структуры. [Hofmann U., 2. anorg. Ciiem., 247, 65, 1941].
Как ужо было отмечено в одном примере, ионы Si4* внутри тетраэдра в некоторых структурах частично могут замещаться па ионы А13+ без нарушения строения этих Структур. В связи с этим, конечно, с каждым атомом А1 отрицательный заряд, приходящийся на тетраэдр, возрастает па единицу. Таким образом, из [Si4Olo]4' возникают [AlSi3Olap' п [АЦЗцОщ]0-. Таким образом, от силикатов группы талька, например ппрофилитл AL [^г4О10](ОН)а, образуются, с одной стороны, такие слюды, как мусковит КАК [AlSi3O(G ](ОН)2, с другой стороны, хрупкие слюды, такие, как маргарит СаЛ12 [Al2SiaO 4 D ] (О Н }2 (присоединение иопов К* или же Са3+ соответствует повышению заряда отрицательной группы).
- Можно считать, что плоская гексагональная сетка из тетраэдров, представленная на рис. 96, д, возникла при соединении шестичленных колец, состоящих из тетраэдров. 35 р Реиж
546
Глава 12
Прг соответствующей связи «оеъмичлегшмх колец тетраэдров получают плеску тетрагональную сетку, в которой чередуются четырех- и в осьмпч Лепные кольев (см. рпс. 96, е). Такая сотка имеется в anorflиллите Са4 [81дО20 J [K(OH2)S](OH, F).
Пространственные сетчатые структуры (каркасные структуры). Если каждый тетраэдр Si 04 имеет с соседними тетраэдрами SiO4 общими все четыре иопа О2-, то образуется пространственный каркас или пространственная сетка. Тогда па каждый ион Si4+ приходится 4/,=2ui- попа. Таким образом получается двуокись кремния SiO2, кристаллическая структура которой уже была рассмотрена. Если в одной часто тетраэдра попы Si4+ замещаются попами Al3* (плп другими ионами с небольшими зарядами и малыми размерами, например ионами Ва+ или Be2'), то одновременно в решетку для компенсации заряда должны войти дополнительные положительные иоиы. Впрочем, структура может с охра питься. 'Гак, нефелин Na[A!SiO4] (гексагональный) имеет одинаковым тетраэдрический каркас с тридимитом, а карнегиит Na[AlSiO4] (кубичек ский) — с кристобалитом.
Кроме сеток, указашгых для различных модификаций SiO2, могут быть образованы тлщке pulp другие нрострапетпслные тетраэдрические сотки, если часть копов Si4 + заменять гыпамп А1;, + . Такне сетки имеются в лолсймк шпатах, например вК|А18130й], в силикатах на,пролн1пие,й группы, например в (Na2(OH2)21 [Al2Si3OIe] (натролит), [Ca(OTIa)3](Al2Si3O10] (сколецит), в анальциме (Nat(OT.I2)4.|i Al4Sj3O24] и в скаполите INа4 [Al3Si3O24jCl. Во всех- этих соединениях прп измене А] па Si алюмосиликатные группы, которые в этом случае могут быть ограничены только произвольно, так как речь идет о каркасах с неограниченным протяжением, переходят в группы, кратные SiO2.
В качестве примеров структур внедрения с решетками в виде пространственных сеток могут служить минералы содалгипной. еруппгл, например содалит Na^f AJeSr6Oa4)С1г и Howaz(-Nas [AleSisO24] [SOJ. Если в решетке нозеана группы [SO4]2- заменить па группы SJh, то получится ультрамарин (ср. стр. 561 н сл.).
Между силикатами с островной структурой и пространственной сетчатой структурой могут существовать переходы. Например, в соединении Ara2CaStO4, кристаллизующемся в форме куба, имеется такая же пространственная сетка, как в кристобалите, если по учитывать сильного искаясепия и расширения тетраэдров СаО4. Однако если в этом соединении инны О2- подчинить только нонам Si4+, учитывая, что расстояние Са«—► О (в среднем 2,00 А) значительно больше расстояния Si <—>0(1,58А) я немного меньше наименьшего расстояния Na<—>0 (2,37 А), то соединение следует рассматривать как двойной силикат с островной структурой. Следователи)о, его внутреннее строение можпо представить как формулой Na2(CaSiO4], так и формулой Na2Ca[SiO4J.
Связь между структуре;! и свокетвамп силикатов. Как было указано, обчэЯСпение структуры силикатов позволило сформулировать принцип распределения, на основании которого ясно видна аналогия в химическом составе между структур неродственными веществами. Кроме того, для многих встречающихся в природе силикатов зто объяснение позволило впервые правильно установить химические формулы., отвечающие составу в тех случаях, когда правильная формула нарушалась вследствие колебаний в составе, вызываемых изоморфным замещением. Одпако прежде всего, оно раскрыло важные связи между кристаллическими структурами и физическими и химическими свойствами силикатов.
Несмотря иа то что исследование этпх связей находится еще в начальной стадии, однако уже выяснилось, что некоторые свойства, как твердость, плотность, расщепляемость, термическая устойчивость, кроме того, оптические свойства силикатов определенным образом связаны с нх внутренним строением. Наиболее ярко это проявляется в расщепляемости. У Site бг.г.чо отмечено, что силикаты с листовое структурой, как правило, показывают особенно большую расщепляемость параллельно поверхностям сеток, так что построенные таким образом соединения, например слюды, часто могут быть расщеплены очень легко ва топкие листочки. Соответственно этому у силикатов с цепочечными и ленточными структурами наблюдается очень хорошая расщепляем ость параллельно направлению цепей или лент. В связи с этим часто эти силикаты имеют вид стебельков или волокон, как асбест. Силикаты с островными структурами, как правило, проявляют гораздо менее выраженную расщепляемость. Поскольку, вообще говоря, расщепляемостью они обладают, то проявляется она главным образом параллельно
Четвертая еруппа периодической системы 647
поверхностям, наиболее плотно запитым ионами О2 . Как и следовало ожидать, исходя из структуры, у кристаллов е равномерными решетками в виде пространственных сеток, какие имеются у кварца, тридимита и кристобалита, большей частью отсутствует отчетливо выраженная расщепляем петь. При частичном замещении ионов Si4+ и о наш А1'1+ часто образуются менее равномерно построенные пространстве иные сетки; эти сетки в связи с различной плотностью упаковки в различных направлениях также обладают большей частью отчетливой, часто совершенной расщепляемостью, какая наблюдается, например, в полевых шаатах.
Искусственное приготовление силикатов и двойных силикатов, которые встречаются в природе, сопряжено иногда с трудностями. Они возникают особенно в тех случаях, когда пытаются получить силикаты нс из расплавов, а приближаясь к природный условиям образования, путем превращений в присутствии воды (при высокой температуре и высоком данленпи — гидротермальный синтез). К соединениям, гидротермальный синтез которых вначале не удавался, принадлежит чрезвычайно распространенный в природе калиевый полевой шпат. Однако при нагревании синтетического лейцита К !AlSi2OJ с водным раствором КгСОз и NaaCOa в автоклаве приблизительно до 200° Барреру и Хиндсу в последпее время удалось получить с хорошим выходом маленькие кристаллики с показателем преломления 1,521", которые до данным рентгеновских исследований можно было идентифицировать как кристаллы калиевого полевого шпата.
СИЛИКАТНЫЕ ПРОДУКТЫ ОСОБО ВАЖНОГО ТЕХНИЧЕСКОГО ЗНАЧЕНИЯ
Стекло
Под стеклом в широком смысле слова понимают аморфно (т. е. без кристаллизации) застывший расплав.
Раньше стекла рассматривали как «переохлажденные жидкости». Однако в соответствии с более новыми представлениями между стеклом и переохлажденной жидкостью существуют значительные различия. Температуру, при которой свойства стекла переходят в свойства переохлажден пой жидкости, называют «точкой превращения’ стекла. В интервале нескольких градусов вблизи точки превращения наступает сильное изменение удельной теплоемкости, коэффициента расширения, температурного коэффициента диэлектрической проницаемости и показателя преломления, также как и температурного коэффициента электропроводности (см. рис. 97). Ниже точки превращения типичные стекла совершенно застывают, так что при царапания на них появляются трещины; выше этой точки они пластичны или вязки. Перечисленные свойства стекол, вагретых выше их точки превращения, ври дальнейшем повышении температуры медленно и непрерывно переходят в свойства жидкости, получающейся при плавлении кристаллического вещества; зато пиже точки превращения они большей частью лишь немного отклоняются от свойств, которые проявляют те же вещества в кристаллическом состоянии. Наряду с исследованием изменения свойств ценные сведения для объяснения сущности cTt'H’i Стибри и него состояния дали в настоящее время рентгенографические методы (ср. стр. 53i и 7:зЬ). В соответствии с этим структуру стекол следует рассматривать как непериодические решетки, но обнаруживающие вследствие этого симметрии в широких областях. Только в узко ограниченных областях определенные атомные группировки сохраняют симметричное расположение, причем с темп же координацией нюти числами, как и в кристаллических сое ди не киях, В силикатных и фосфатных стеклах такими ^атомными группировками являются SiO4- и соответственно РО4-те-траэДры, а в стеклообразной трехокиси бора — треугольники ВО3. Последние, как и в кристаллических соединениях с пространственной сетчатой структурой, связаны через вершины углов, однако расположены одпн к другому нерегулярно, так что возникает неупорядоченная пространственная сетка.
У одного и того жо вещества с текло в и дно-аморфное состояние принципиально структурно не отличается от жидкого состояния. Как показали рентгеновские иссле-
35*
548
Глава 12
цоваиия, в жидком состоянии, прежде всего вблизи температуры кристаллизации, нее имеются уже упорядоченные в узкой области атомные группировки. Однако вех? степень упорядочения в стеклообразном состоянии в общем больше, чем п жидст Это было непосредственно доказано Рихтером (Richter, 1951—1952) прп рептгЕ.. структурном исследовании стеклообразных As й Зе, И все же, исходя только из стр;; турных данных, по удается резко разграничивать: стеклообразное и жидкое состолгг Однако к такому резкому разграничению приводит термодинамическое рассмотри—
В термодинамическом отношении стекла отличаются от переохлажденных жездг стей тем, что они в отличие от жидкостей не находятся в состоянии внутреннего тер^ динамического равновесия. Эго значит, что стекла прн дальнейшем охлаждении соху; няют существующш г прп точке превращения беспорядок (энтропию), в то время ею;
Рис. 97. Температурная зависимость удельной теплоемкости вещества в жидком, стекловидном и кристаллическом состояниях.
Ы — точка превращении; is—точка плавления.
у переохлажденных жидкостей с понижением температуры устанавливается все более увеличивающийся порядок атомных групп. У абсолютного нуля (если можно добиться такого переохлаждения) энтропия переохлажденной жидкости достигает значения пуля. Конечно, нельзя пе учитывать и то, что как раз около точки превращения (точнее, в интервале превращения) происходит значительное упорядочение атомных групп.
Свойством застывать из расплава в стекловидно-аморфном состоянии обладают прежде всего некоторые двойные силикаты. Стекло, в узком смысле этого слова, состоит обычно из смеси силикатов. Большей частью в стекле содержатся силикаты натрия или калия в смеси с силикатом кальция.
Так называемое нормальное стекло имеет состав NaaO-CaO-eSiOa-Обычно применяемое стекло, например оконное или бутылочное, в большинстве случаев близко соответствует этому составу. Точка плавления и прочность повышаются, если натрий заменяют калием {калийное стекло, богемское или йенское стекло). Такое стекло применяют, например, для изготовления трубок для сжигания, используемых в органическом элементарном анализе. Если одновременно кальций заменяют свинцом, то получают стекла, отличающиеся сильной светопреломляющей способностью и высоким удельным весом. Такие стекла применяют для изготовления шлифованной посуды и предметов украшения (свинцовое стекло, свинцовый хрусталь). Стекло, богатое свинцом, называют также флинтглас. Его применяют прежде всего для изготовления оптических приборов (линз, призм). Для этого его используют, как правило, в сочетании с менее диспергирующим кронгласом, которое содержит значительное, количество
Четвертая группа периодической системы
549
PSOE (до 70%). Стекла с высоким содержанием свинца обладают сравнительно низкими тоннами плавления и малой устойчивостью но отношению к химическим воздействиям. Стекла, особенно богатые свинцом и приближающиеся к алмазу по своей светопреломляющей способности называются по имени конского ювелира Штрассера штрасеами. Их используют для подделки драгоценных камней, однако они быстро теряют блеск ввиду своей мягкости.
Добавленном пе значительных количеств окислов, которые образуют цветные силикаты, получают окрашенные стекла; так, при добавлении окиси кобальта получают синее, окиси хрома дли окиси моди(П) — зеленое, окиси меди(1) — красное стекло-Силикат жслеза(П) при высоких концентрациях окрашивает стекло в черный цвет, при незначительном содержании — в грязно-зеленый (цвет пивных бутылок). Особенно интенсивно окрашивает стекло задисг.-оквсь железа; чистая окись щелеза(Ш) окрашивает гораздо слабее (от желто-зеленого до коричнево-желтого цвета). Окраска, вызываемая минимальным количеством окиси железа, может исчезнуть при добавлении к расплавленному стеклу двуокиси марганца; последняя, образуя силикат марган-ца(115), вызывает фиолетовую окраску, являющуюся дополнительной к желто-зеленому цвету силиката железа(Ш). Поэтому двуокись марганца раньше называла «стеклянным мылом*. Наоборот, со аначитемнш! количеством окиси железа двуокись марганца вызывает коричневое окрашивание бутылочного стекла.
Многие стекла содержат также окись алюминия (в большинстве случаев лишь несколько процентов); окись бора также иногда вводят в стекло; так, она присутствует в поиском стекле, применяемом для изготовления нормальных термометров, в шоттов-ском стекле для приборов, и стекле пирекс и в других стеклах, иа которых в последнее время изготовляют стеклянную посуду для домашнего хозяйства, позволяющую кипятить и жарить па открытом огне (дюракс, резиста). Применяемые в последнее время в атомной технике защитные стекла наряду с силикатами бора содержат окись кадмия и фториды (для абсорбции медленных нейтронов) или фосфат вольфрама (для абсорбции у-лучей).
Некоторые боросиликатные стекла обладают свойством постепенно выделять щелочной борат при температурах выше точки превращения. Последний обычно остается в массе стекла в виде мельчайших капелек. Процесс можно про вес гл так, что борат щелочного металла образует отдельную фазу, которую можно удалить выщелачиванием остывшего стекла разбавленной кислотой. При этом остается губчатая масса с очень высокдм с одержав кем SiO2 (до 96%). Из псе спеканием примерно при 1200s получают гомогенное стекло, которое по своим свойствам подобно чистому кварцевому стеклу н размягчается только ирн магревадня выше 3500° (стекло викор) (Hood И. Р., Nord-berg). По методике Викора изделию из щелочного боросиликатного стекла придают форму при обычной рабочей температуре. Затем его нагревают, выщелачивают, спекают, благодаря чему возникает значительное сжатие, одна ко первоначальная форма сохраняется.
Метод Викора используют также для получения высоко пористых, используемых в качестве катализаторов веществ, например катализатор для крекинга нефти. В этом случае после выщелачивания его не спекают, а состав исходного плава видоизменяют нужным образом (например, добавлением СяО или NiO).
Получают также светочувствительные- стекла (фотографическое стекло]. Для этих целей расплав стекла должен иметь состав, аналогичный составу меднорубийового Стекла (см. т. II) или же содержать золото вместе с сенсибилизатором, например окисью церия. При ультрафиолетовом облучении такого стекла через обычпый негатив получают скрытие изображение, которое можно проявить нагреванием до 600—700э.
Получение стекла ведут сплавлением двуокиси кремния (кварца, кремня) с карбонатом кальция (в виде известняка, мрамора, известкового «штата), с кальцинированной содой или с сульфатом натрия и углем. Для Калиевых стекол вместо соды применяют поташ, для свинцовых стекол вместо карбоната кальция — окись свинца и т. д. Сплавленце производят в вапнах или в больших огнеупорных тиглях, в «стеклоплавильных горшках». Печи обычно обогревают генераторным газом с применением топок Сименса. В таких топках горячий отходящий газ используют для нагревания камер, выложенных огнеупорным кирпичом; через эти камеры затем пропускают газы перед их сжиганием, так что они попадают
550
Глава 72
в печь, будучи уже нагретыми до высокой температуры. В последнее время в производстве стекла введено также злектроплавление. На современных предприятиях тигли и ванны, в которых выплавляют оптическое стекло (т. е. стекло для оптических приборов), чаще всего покрывают изнутри платиной.
Свойства стекла можно в зпачительной степени видоизменять, варьируя количественные соотношения и составные части стекла. В общем богатые кремневой пнелотой «кислые» стекла обладают наиболее высокой температурой плавления и наибольшей устойчивостью к химическому воздействию. Увеличением содержания составных частей основного характера повышается плавкость стекла, но одновременно понижается химическая устойчивость. Стекла, богатые щелочами, заметно подвергаются действию кипящей воды. Вода, смешанная с фенолфталеином, окрашивается в красный цвет при кипячении ее в об ычпом новом стакане вследствие извлечения щелочи из стекла. Если в результате частого кипячения часть щелочи извлекается с поверхности стекла, то устойчивость его возрастает. Быстрое повышение устойчивости стекла достигают его пропариванием. Еще сильнее, чем под действием воды и кислот, стекло разрушается щелочами. При применении горячих, сравнительно сильно щелочных растворов для прополаскивания и дезинфекции стеклянной посуды (например, молочных бутылок) для снижения их разъедающего действия в раствор добавляют алюминаты или ничтожные количества ВеО, действующие антикаталитически.
В последнее время (особенно в США) стремятся уменьшить содержание щелочей в наиболее употребляемых дешевых сортах стекол (бутылочное стекло, окопное стекло); так как карбонат натрия—самая дорогая составная часть смеси, применяемой для выплавления этпх сортов стекол, то вместо этого, правда только в ограниченных масштабах (папрнмер, для коричневого стекла), мощно применять сульфат патрия. Иногда дешевые минералы бария и фтора добавляют в качестве флюсов и одновременно с содержанием щелочей уменьшают также содержание SiO2.
Наряду с прозрачностью особенно важным свойством стекла является его способность постелей по размягчаться при нагревании до температуры каления задолго до того, как око перейдет г, жидкое состояние. 11а атом оспоеалю формование стекла при нагревании (стеклодувное цолг)!. Менее благоприятным свойством стекла является его чувствительность к быстрым сменам температур, которая нередко приводит к растрескиванию стеклянных изделий. Стекло, нагретое до высокой температуры, следует охлаждать очень медленно, так как плаче воз пн кают внутренние натяжения, вызывающие растрескивание стекла после его охлаждения. В противоположность этому для некоторых боросиликатных стекол (дюракс, резиста) усто1гчивость к сменам температур достигается как раз путем внезапного охлаждения сформованных изделии.
Боратные стекла, включающие редкие элементы, полученные Мореем (Morey, 1937), содержат бораты, танталаты и вольфраматы циркония, тория и лантана. Они отличаются особо высокой светопреломляющей способностью. Стекла с высоким содержанием фторидов (фторидные стекла} наиболее слабо рассеивают свет и имеют низкие показатели преломления.
Как установил БиЛьтц (1937—1938), в оптической промышленности большое значение имеет то, что показатель преломления стекла можно заранее рассчитать с большой точностью из его состава. Подробнее о структурной химии стекла п о влиянии сил полей катионов па то, будут ли образовываться гомогенные стекла или кристаллические соединения или же в жидком состоянии будет происходить расслаивание смеси, кроме того, о влиянии их на точки плавления соединений, а также о значении сил полей положительных ионов на действие минерализатора см.: D i et г cl A., Naturwisa., 29, 537, 1941; 31, 22 и 110, 4943; Z. Electrochem., 48, 9, 1942. При этом под силой 7.-е, поля понимают выражение , где е — элементарное количество электричества.
Четвертая группа периодической системы
551
Z — валентность положительного иона па — расстояние от центра положительного иона до центра соседнего аниона (следовательно, у силикатов — до центра иона 0s-).
При повышенной температуре, прежде всего выше точки превращения, стекло обладает значительной элсктропроаод гостью. Если работать при таких условиях, когда ионы металлов (или ГГ+) в стекле могут передвигаться, то проводимость имеет чисто электролитическую природу, а именно она основана на Подвижности ионов патрия. В калиевых стеклах перепое тока осуществляется попами калия лишь в том случае, если там отсутствуют им натрия. Силикатные ноны по участвуют в переносе электрического тока (Schwarz, i‘332—iQ33; Manegold, 1932— 1935).
Глазури н эмали. Глазурями называют тонкие стеклянные'пскрытия па довольно -огнеупорных изделиях. Эми ли представляют собой непрозрачные глазури, которые становятся такими под действием веществ, вызывающих глушение. Эмалирование применяют прежде всего для железных изделий. Его осуществляют следующим образом: хорошо очищенные изделия погружают в массу, полученную тщательным смешиванием подходящего (содержащего полевой шпат пли борную кислоту) стекла с водой. После высушивания нанесенную глазурную массу плавят при сильном калении. Вместо погружения изделие можпо покрывать глазурной массой нанесением кашицы или надуванием сухого порошка.
На основную глазурь соответствующим образом наплавляют покровную амаль. Покрытие эмалью производят па основу, нанесенную в два (иногда и больше) слоя, так как эмали содержат обычпо SnO, в качестве средства, вызывающего глушение, и если бы SnO2 соприкасалось с железом, то при нагреваппи происходило бы окисление содержащегося в железе углерода до СО, в связи с чем в эмали образовались бы пузыри. О покрытии глазурями фарфоровых изделий см. стр. 555.
ГЛИНА, КАОЛИН И КЕРАМИЧЕСКИЕ ИЗДЕЛИЯ
Глина и каолин. Глиной называют массу, в сухом состоянии землистую, мягкую, прилипающую к языку, а во влажном состоянии более или мепее пластичную; она образуется при разложении горных пород, богатых полевым пшатом. Состав глин разнообразен и зависит от видов горных пород, в результате разрушения которых они образовались. Чисто аналитическим путем (не учитывая тип связи) определены главные составные части глин — А1г0з, SiOs и НаО. Раньше считали, что каолин представляет собой основное вещество глины, что различные глины, следовательно, являются каолином более или менее сильно загрязненным примесями. Однако в соответствии с более поздними данными глина и каолин — это вещества совершенно различного характера, даже в тех случаях, когда опи случайно имеют один и тот же аналитический состав. Ценные керамические глины содержат в значительном количестве примесь каолина; такой каолин был вымыт из своих первичных месторождений, а позднее снова осажден вместе с другими ко ллоиднора определенными минералами, которые, однако, могут являться образователями глин и сами по себе.
В качестве главной составной части каолин содержит^каолинит, который представляет собой (по рентгенографическим данным) кристаллическое вещество состава АЦОз 2SiO2-2HsO. Глины или совсем не содержат этого соединения или содержат его как случайную примесь (ср. выше). Чистый каолин — белого цвета и отличается сравнительно малой пластичностью. Ввиду того что он служит сырьем для изготовления фарфора, его называют фарфоровой землей.
Глины, которые часто значительно превосходят каолин по своим пластическим свойствам, служат для изготовления гончарных изделий, фаянса, керамических изделий и майолики. Большинство сортов глии окращено в желтовато-серые пли синеватые тона, но встречаются также и совершенно белые глины. Глипы, богатые окисью железа, окрашиваются после прокаливания («обжига») в бурые и красные цвета. Из них изгото
552
Глава 12
вляют обычно глиняные горшки и терракотовые* изделия. ФормовочнвС. землей. называют глину, сильно загрязненную окисью железа и песком.. Такая глина служит преимущественно для изготовления кирпича и черепицы. Глины, сильно загрязненные примесями карбонатов кальция и магния, называются мергелями. Они непригодны: в качество сырья для керамических изделий, однако их частично используют при производстве цемента.
Образование глины происходит при выветривании силикатных горных пород, которое связано прежде всего с их значительным механическим раздроблением (превращение в коллоидное состояние). Наряду с этим протекает подчиненный химический процесс, а именно гидролиз более или менее большей части силикатов (прежде всего полевых шпатов) с образованием аморфных глиноземистых кремневых гелей. Последние называются аллофанами и, по-видимому, представляют собой чистые смеси гидратов окиси алюминия и двуокиси кремния идя спрокаолинами» — тоже аморфные, содержащие воду силикаты алюминия. Прокаолин является, вероятно, определенный
--------Ось Ь
Монтмориллонит
{Д12[ЭЧи|(]]{ОН)2}+пН20
Xх К >х К
----Ось b Каолинит flla[Si2Os](OH),
® Д1 ф К ° Si ( л Mip-aoeunie каждый четвертый атом Si замещен Д1)
О О О ОН
-------*- Ось b
уснова т (коленная слюда/ К{Д1г[5г?Д10|0](СНМ
Ось С
Р и с. 98. Схема минералов глины.
К монтмориллоните расстояние в между слоями зависит от количества воды, содержащейся между Спон ми. Л оптрон itm производится от монтмориллонита При валено иопов А1Э + на Ее3*, гег.таорит при замене 2А13* на SMgS*. Третий ион лр* занимает в решетке место, о Goa чаченное через X. Слюбоебрчзные лгинерч-гы мшш отличаются от мусковита незначительным содержанием щелочей. Часто также в них ионы А1 замешены копами Те кли Mg. Места одиой трети ионов Mg л кристаллической структуре обозначены значком х.
химическим соединением состава Al2O3’2SiO2. On содержит переменное количество воды, которая не связана с ним химически, как в каолините, а примешана к пему, как вода в гелях. Раздробленные только механически и поэтому едце кристаллические составные части горпых пород содержатся в большинстве сортов глин также преимущественно в колл сшднораздробленном состоянии (Соггепз, 1936).
Особые свойства глин создаются определенными составными частями, которые имеют слоистую структуру решетки, образованной шестнч л энными кольцами, состоящими из тетраэдров SiO4. Эти составные части, подобно пермутитам (ср. стр. 558 и сл.), отличаются определенной способностью к катионному обмену (ср, т. II, гл. 19). К ним в первую очередь относится каолин и родственные ему вещества (например, галлоиоит А12Оз-201О2-4.Н2О), монтмориллонит (ср. стр. 545) и некоторые слюдоподобные минералы [Hofmann U., Z. Kristallogr. (А), 98, 31, 1937; Chemie, 55, 283, 1942]. Интересно то, что все эти глииеобразующие минералы имеют аналогичные решетки
Четвертая группа. периодической системы
553
.(рис. 98), Аморфные составные части глин (аллофаны), смешанные большей частью с кристаллическими, пре имуще ст вен в о находящимися в коллоиднораздробленном состоянии составными частями, пе имеют существенного значения для свойств глин.
В то время как выветриваппо горных пород с образованием глин может идти в обычных условиях атмосферного выветривания, образование каолина, как впервые показал Шварц (1933 г. и сл.), связано с особыми условиями. Этому существенно благоприятствуют повышенная температура, повышенное давление, присутствие сильных кислот (например, 11G1), но по угольной кислоты, Одпако, согласно Ноллю (1935 г. и сл.), в геологические периоды каолин мог образоваться также при низких температурах. Действие сильных кислот способствует образованию каолина, так как при этом ускоряется гидролиз полевого шпата. Если исходить из продуктов гидролиза полевого шпата, не содержащих щелочи, то можно наблюдать образование каолина в отсутствие кислоты. Так, Нолл смог синтезировать каолин, исходя, например, из смеси аморфной SiO2 с бёмигом или с байеритом при нагревании ее с водой под давлением. Если нагревать смесь в присутствии раствора едкого натра, то образуется монтмориллонит. Очевидно, образование каолина в природе идет в том случае, если щелочные и щелочноземельные элементы полностью выщелочены из исходных горных пород; в противном случае образуется монтмориллонит. Следовательно, образование каолина в Природе ускоряется прежде всего интенсивным вымыванием и хорошей циркуляцией растворов, а также благодаря кислой реакции вымывающих вод.
Образование каолина — чисто химический процесс, который можно передать суммарным уравнением
2К [AlSi3O8l4-7Н2О = Al2 [Si2O5l (OH)4+ffl2SiOa+2KOH, Полевой шпат Каолин
Каолин может образоваться непосредственно из полевого шпата, а также и из прокаолина, первоначально образованного из полевого шпата при обычном выветривании, если его нагревать с водой под высоким давлением. Если каолин нагреть под давлением в слабо щелочной среде (с раствором щелочного карбоната), то Оп превращается в монтмориллонит Al3|Si4Olt.](OH)2wII3O*, в то время как в сильно щелочной среде из него получаются цеолиты.
При нагревании каолинит отщепляет сначала воду (под давлением 10 мм рт ст при 430°). Механизм отщепления воды показывает, что вода в каолипите связапа химически. Обезвоженный каолинит (мотакаолинит) при более сильном нагревании сначала разлагается на А12Оа и SiO2; При еще более высокой температуре из него образуется муллит 3Al2Os-2SiU2 (наряду с тридимитом),
Рептге неструктурным анализом установлено, что каолинит построен из сетчатых плоскостей, образованных ионами [Si2O5]2*, между которыми включено иногда по два сдоя из [А1(ОН)31+. С каолинитом имеют одинаковый состав минералы дикит и накрит, встречающиеся во многих сортах каолина. Они показывают иную, чем каолинит, картину рентгеновской интерференции, но, по-видимому, построены аналогично (Кегг, 1930; Gruner, 1932).
Чистую глину в виде порошка применяют в медицине й называют «Bolas alba» (pw^og—комок земли).
Изделия, получаемые из встречающихся в природе и искусственно приготовленных пластических смесей глины или каолина с другими веществами, называют «керамическими изделиями» (от слова «epapixeg — глиняный, гончарный). Легко формующейся или «пластической» массой называют вязкую массу, которой при незначительном давлении можпо придать
* Монтмориллонит представляет собой важнейшую составную часть бентонитов, большие залежи которых имеются в США. Монтмориллонит применяют для целей косметики и производства мыла, а в последнее время расширяются технические области его применения; непластпческпм веществам монтмориллонит придает способность к формованию, стабилизирует суспензии, так как он отличается особенно большой способностью к набуханию и к' пептизации. Выяснить причину этого удалось Гофману (Z, Eiectroehem., 41, 469, 19.15] при помощи рентгенографического и коллоидно-химического исследовании; па основании результатов этого исследования он превратил аналогично построенные минералы, распространенные в Германии, в продукты, подобные американскому бентониту. (Ср. также Hofmann U., Z. Kristallogr, (А), 98, 299, 1937; 104, 265, 1942,)
554
Глава 12
любую форму, причем эта форма сохраняется и после прекращения давления. Важнейшие керамические изделия и их характерные свойства сопоставлены в табл, 87.
Таблица 87
Обзор важнейших керамических изделий
Фарфор Излом плотный просвечивающий белый
Каменные лия надо- Излом плотный пепросваливающий белый или цветной (серый, желтый, бурый)
Фаянс Малом пористый пепросвечивающий белый или почти белый; незначительная твердость по сравнению с предыдущими
Гончарные лия изде- Излом пористый не просвечивающий цветной
Кирпичи Пористые, сравнительно крупнозернистые, обычно красного цвета
Огнеупорные делил пз- Пористые или плотпые, плавятся не ниже 1600 •
Фарфор. Фарфор был известен в Китае уже в самые древние времена, а в Европе его стали изготовлять фабричным способом впервые в Мейссе-не (с 1710 г.)*. Фарфор был получен ири сильном прокаливании («обжиге») пластичных масс, изготовленных перемешиванием каолина (фарфоровой земли) с порошкообразными полевым шпатом и кварцем с добавлением небольшого количества воды. Если температура обжига не слишком высокая, то форма изделий сохраняется, только объем их сильно уменьшается, так как фарфор «садится» при обжиге. Одновременно масса («черепок») становится плотной (водонепроницаемой) и звонкой.
Для изготовления твердого фарфора обычно применяют около 50% каолина, 25% подового шпата и 25% кварца. При обжиге каолин сначала отдает конституционную воду. Затем он разлагается на Л13О3 и SiOj, которые растворяются и стеклообразно размягченном полевом шпате. При дальнейшем повышении температуры полевой шпат растворяет в возрастающих количествах крупнокристаллический кварц. По мере того как полевой шпат обогащается двуокисью кремпия, из него осаждается муллит, так
* Изобретателем европейского фарфора в большинстве случаев называют алхимика Вёттгера. Однако правильнее было бы приписать это открытие физику В. Эрен-фриду фон Чирнхаузу. Последнему впервые удалось получить фарфор в результате многолетних исследований. Уже в 1703 г. им была спроектировала фабрика для производства фарфора; однако опа была построена только после его смерти под руководством Вёттгера. Алхимик Вёттгер был привлечен только в 1707 г. фоп Чирнхаузепом в качестве помощника для своих опытов с керамикой. После смерти своего учителя (1708 г,) он выдал себя за изобретателя фарфора (уже в то время оспариваемый факт). Па самом деле оп ввел лишь существенные технические усовершенствования и только благодаря этому сделал мейссепский фарфор предметом торговли; с научной точки зрения приоритет следует присудить фон Чирнхаузу,
Четвертая группа периодической системы
556
как с увеличением содержания SiO2 растворяющая способность полевого шпата по отношению к муллиту понижается. Поэтому готовый фарфор состоит из стекловидном основной массы, которая пропиэаиа тесно сплетенными между собой иглами муллита и оставшимися не растворе иным и зернышками кварца (и крохотными пузырьками воздуха). Как правило, обжиг производят дважды. После первого обжига, так называемого «сырого обжига», идущего приблизительно при 900°, на фарфор наносят прозрачную глаеуръ-, полученные после сырого обжига еще пористые черепки быстро погружают в глазурную массу — водную суспензию каолина, глины, нолевого шпата и мрамора. При нагревании иа нее образуется тугоплавкое стекло. Последующим высушиванием (около 1450°) производят окончательный обжиг. Часто фарфор подвергают еще третьему обжигу в муфельной печи при красном калении после нанесения красок, т. е. перетертого со скипидаром мел ко измельчен к о го цветного стекла. Большей стойкостью обладают краски для «острого» Огня илн подглазурные краски, йаносимые па неглазу-рованпый черепок. Однако существует немного красок, для которых этот метод возможет Окончательно отожженный без глазури фарфор называется бисквитным.
Вместо процесса формования фарфоровой массы, основанного на ее пластичности, фарфоровую массу можпо перевести в жидкое состояние добавлением незначительного количества щелочи и отлить в формы из гипса; в результате поглощения воды обожженной гипсовой формой наступает быстрое отвердение фарфорового изделия.
Фарфор Применяют но только Для изготовления домашней посуды и художественных изделий, но также, и притом в весьма широкой степени, для изготовления химической посуды, а благодаря его электроизоляционным свойствам — для изготовления изоляторов.
От обычного или твердого фарфора отличается мягкий фарфор, из которого изготовляют главным образом художественные изделия. В мягком фарфоре содержится меньше каолина и соответственно этому больше «плавней», например полевого шпата, мела. Такой фарфор в соответствии с его легкоплавкостью подвергают обжигу при менее высокой температуре (обычно при 1200—1300°). Поэтому легко удается произвести его многоцветное подглазурное раскрашивание.
Каменные материалы, подобно фарфору, являются плотными звонкими и настолько твердыми, что не царапаются сталью; кроме того, они очень стойки по отношению к химическому воздействию. Поскольку их изготовляют из глины, они требуют более низкой температуры обжига, чем твердый фарфор (1200—1300°), не обладают такой, как фарфор, просвечиваемостью и в большинстве случаев имеют не белый, а серый, желтый или коричневый цвет. Их часто покрывают только топким слоем «солевой» глазури, который образуется при испарении поваренной соли, бросаемой в печь; благодаря этому на поверхности такого изделия осаждается стекловидный двойной силикат патрия. В химической промышленности весьма часто применяют неглазурованные изделия из каменного материала.
Тонкий каменный материал служит для изготовления ваз и других художественных изделий, в архитектуре для изготовления рельефов и украшения фасадов. Из каменного материала выделывали серые, расписанные голубой краской старинные германские сосуды (кубки и т. и.). Примерами изделий из грубого коричневого каменного материала могут служить водопроводные и канализационные трубы, а также «метлахские плитки». В химической промышленности применяют многочисленные сосуды, изготовленные из коричневого каменного материала: туриллы, змеевики для охлаждения, трубы, ванны и др.
Фаянс, как и фарфор, белого или почти белого цвета, однако он мягче,-так что сталь оставляет на нем царапины; оп легче ломается, порист, поэтому в большинстве случаев его необходимо покрывать глазурью. Фаянс получают из смеси глины, кварца, щелочи и сурика, иногда доба
556
Глава J 2
вляют еще окрашивающие окисли. Фаянс обжигают дважды: сначала без глазури при 1200—1300° (сырой обжиг), а затем несколько слабее с глазурью (окончательный обжиг). Из тонкого фаянса делают мойки, ванны Для купания и т. п. Некоторые сорта фаянса часто окращивают титановой Кислотой в бледно-кремовый цвет (умывальники). Примерами пеглазу-рованного фаянса служат глиняные сосуды, глиняные трубки и т. п.
Фаянс имеет грязновато-cepwii пористый излом. Поэтому его покрыв агат глазурью, которая благодаря прибои,ле пи то двуокиси олова имеет белый цвет и непрозрачна. Раньше фаянс часто применяли для изготовления дешевой посуды; однако фаянс, изготовленный Веджвудом в Англии, почти совершенно вытеснил обычный фаянс аз домашнего обихода. .Колее топкий фаянс пригоден для художественной керамики. По своим свойствам к фаянсу приближается майолика, покрываемая цветной глазурью.
Обычные гончарные изделвя, например горшки для цветов, глиняная посуда, также, имеют пористый налом. На них наносят глазурь, содержащую большей частью свинец; окрашена она обычно добавленными к ней окислами металлов. Окись железа дает желтую, а вместе с двуокисью марганца — коричневую окраску; медь окрашивает глазурь в зеленый цвет.
Кирпич, Обожженный кирпич формуют из глины й затем подвергают обжигу. Вследствие содержания в глине окиси железа кнрцичи большей частью окрашены в красный цвет. Кирпич обладает большой пористостью, так как обжиг ведут цри относительно низкой температуре. Сильно обожженный, плотный й очепь крепкий кирпич называют клинкером.
Огнеупорные материалы. «Огнеупорными» называют такие материалы, которые, пе плавясь, выдерживают нагревание при высокой температуре (ио крайней мере 1600°). Наиболее употребляемым огнеупорным материалом является шамот, который состоит из смеси двух сортов глин: обожженной до спекания, возможно более огнеупорной глипы (собственно шамот) ц красной пластичной глины (связывающая глина). Существуют особые месторождения глянец которые в первую очередь идут на изготовление шамота. Шамот, который содержит обычно около 42—45% А12О3 и 50—54% SiO3, прежде всего применяют для футеровки топок, высокотемпературных печей и рекуператоров. Для коксовых, керамических нечей и для печей сталелитейной промышленности (например, печи Сименса — Мартена) не польз уют в большинстве случаев (впервые полученный в Англии) динас, Его приготовляют обжигом грубозернистого кварцевого песка, смешанного с небольшим количеством известковой массы иди глипы. Глинистый динас содержит 15—17% А12О3 и 80—83% SiO2. Он размягчается при 1350°, одиако плавится только выше 16503. По огнеупорным свойствам его превосходит известковый динас или силикатный камень (с содержанием 1,5—4% Саб, 0,3—2% А12О3 й 94—06% S1O2), который плавится только при 1700—1750s. Его и применяют в первую очередь в печах Сименса—Мартена. Еще большей огнеупорностью обладают так называемые силлиманиты, которые получают обжигом прп высокой температуре силлиманита, цйа-нита или андалузита (минералов одного состава ALSiOj, но различного внутреннего строения, ср. стр. 545), вследствие чего образуется муллит, 3AI2O3'2SiO2, который, как уже было отмечено, является составной частью твердого фарфора.
Из огнеупорных веществ, не содержащих SiO2 или содержащих ее в очень небольших количествах, следует назвать боксит, динамидон, магнезит и доломит..Высокими огнеупорными свойствами обладают магнезия, двуокись циркония и главным образом графит (последний — в отсутствие воздуха).
Цемент
Если содержащий глину известняк или известняк, хорошо йеремешан-ный с глиной, подвергнуть сильному обжигу и затем тщательному размолу, то получается продукт, образующий с водой тесто, которое затвердевает в очень крепкую массу, причем даже без доступа двуокиси углерода иа воздуха (необходимого для затвердевания известкового раствора). Про
Четвертая группа периодической, системы
557
дукт этот, твердеющий даже под водой, называют цементом. Цементы применяют для приготовления так называемых гидравлических, т. е. твердеющих даже под водой, известковых растворов (гидравлический цемент). Цемент широко применяют для различных наземных сооружений. Особенно важно его применение для изготовления бетона.
Для подводных сооружений цементный раствор готовят смешиванием цемента с 1—2 ч. песка, а для наземных — с 3 ч. песка или с 6 ч. песка и с i/2 Ч. едкой извести. К этим смесям добавляют воду для придания им тестообразной консистенции. При добавлении в цементный раствор гравия или Щебня получают бетоп; последний обладает, кроме других свойств, очень важным для его использовапия свойством — крепко схватываться с железом. Поэтому бетоп часто применяют вместо с железной арматурой, которая ыце больше увеличивает его прочность (железобетон). При этом железо не только ко разрушается, но бетон даже предохраняет его от коррозия.
Лучший цемент — портланд-цемент. Кроме него, существуют еще пуциолановый цемент, роман'цемент, шлаковый цемент и гидравлическая известь.
Портланд-цемепт представляет собой зеленовато-серый, дающий мелкую пыль порошок, состоящий из смеси сильно основных силикатов, алюминатов и ферратов(Ш) кальция. Его получают обжигом мелко измельченной смеси известняка и глины приблизительно при температуре до 1400°, при этом происходит спекание с образованием плотного клинкера, который затем размалывают в тонкую пыль.
Состав портланд-цемента в среднем приблизительно следующий, %;
СаО MgO SiO2 А12О3 Fe20a
. .... 63 Щелочи ........................0,5
. . .1,5 SO3............................1,5
21 Нерастворимые в конц. НС1 . . . .0,5 . . . ... 7 СОа-[-Н2О (потери при нрокалива-
. . . . . 3 нии) ..........................2,0
Опыт показал, что оптимальное отношение «основных» и «кислых» составных частей в готовом портланд-цементе равно примерно 2 : 1. Это отношение называется гидравлическим модулем цемента, В указанном выше примере он равен 2,0; наименьшая допустимая величина 1,7, Главная основная составная часть — окись кальция; наряду С ней портланд-цемепт содержит часто незначительные количества окиси магния й окислов щелочных металлов, Главная кислая часть цемента — двуокись кремния, затем окись алюминня и обычно еще несколько процентов окиси железа. Затвердевание цемента основайо главным образом на том, что содержащиеся в нем ортосиликат кальция и основной силикат кальция постепенно разлагаются под действием воды; при этом обрадуется моносиликат кальция CaSiO3 (или же кислый силикат кальция Са |SiOa(OH)2] и гидроокись кальция. Одновременно происходит разложение алюминатов и ферратов (Ill). Со временем первоначально выделившаяся гидроокись кальция превращается в карбопат. Важное значение при затвердевании цемента имеет, по-видимому, также Образование соединения состава 2СаО,81Оа.Н3О. В природе это соединение встречается в виде редкого минерала гиллебрандита. На основании различных химических данных, между прочим, на основании того факта, что это соединение отдает свою конституционную воду при нагревании в две Стадии (половину при 350° и другую при 600°), Тило рассматривает его одновременно как основную и как кислую соль Са2(ОН) [SiO3(OH)J. Эта точка зрения подтвердилась рентгеноструктурным исследованием [И el 1 е г L., Acta Crystallogf., 5, 724, 1952|. Продукты разложения и превращения, образующиеся при застывании цемента, выделяются в форме ультрамикроскопическях, тесно переплетенных кристалликов, и, возможно также, отчасти в аморфном состоянии с образованием гелей. Таким образом, со временем в результате роста кристалликов возникает все более затвердив а тоща я структура. Различие между затвердеванием известкового раствора (воздушного цемента) и затвердеванием гидравлического цемента заключается в первую очередь в том, что в последнем определяющим процессом является образование силиката кальция, Тогда как в первом случае — образование карбонате кальция.
658
Глава 12
Названный ранее в качестве составной части цементного клинкера ортосиликат кальция Ca2S104 имеет четыре различные модификации:
p-Ca2StO4
2130*
Расплав < >
1 42Щ a-Ca2SiO4 ; ~
Генса го нал ь-ыая
<x'-Ca2SiO4
Ромбическая (Вред В гит)
Моноклинная (белит)
y-Ca2SiO4
Ромбическая (тип оливина)
а- и a'-Ca2SiO4 ой,задают строением решетки, аналогичным обеим модификациям K2SO4. Превращение |) -> у и у^а' протекают мопотроппо. Несмотря па то что При обычной температуре устойчивой модификацией является у-форма, в цементном клинкере при нормальных условиях Ca2SiO4 существует в [1-форме {белип-^. Однако иногда при охлаягдепии fl-форма самопроизвольно превращается в у-форму. Это свя-вано со значительным увеличением объема и «рассыпанием» цементного клинкера, что является очень нежелательным явлением, так как При этом Ca2SiO4 теряет способность в дальнейшем связываться с водой. Как установил Тило (19.53), превращения Р —> у пе происходит в присутствии небольших количеств щелочи. Полностью освобожденный от окислов щелочных металлов (например, прн сильном прокаливании) Ca2SiO4 ври охлаждении, как правило, превращается в у-моднфикацшо.
Шлаковый цемент. Если извлеченный из доменных печей шлак облить холодной водой, то получается продукт в виде песка, который после мелкого помола также обнаруживает «гидравлические» свойства, т. с. обладает способностью связываться с водой. Для улучшения его качества и с целью приближения его состава к составу портландцемента к шлаковой муке, содержащей вначале 4а—50% СаО, добавляют перемолотый известняк, обжигают, как и портланд-цемент, до спекания и перемалывают в тончайший порошок. Для удешевления добавляют еще 30% шлаковой муки. Продукт поступает в продажу под названием шлакового иди «железного портланд-цемента». Для его изготовления пригодны только те шлаки, которые получаются при выплавке серого чугуна.
Роман-цемент раньше получали обжигом встречающихся в Природе содержащих глину известняков, 13 настоящее время роман-цементом называют и продукты, получаемые при обжиге искусственных смесей, отличающихся от иортлапд-цомепта малым содержанием СаО (мепее 50%); кроме того, обжиг ромап-цемента ведут не до спекания. Цвет его желтовато-коричневый. По прочности Оп уступает портландцементу.
Пуццолановый цемент. Туф, т. е.'быстро охлажденная водой вулканическая лава, часто имеет природные гидравлические свойства. Еще древние римляне перемалывали такие туфы и получали цемент (pulvis Pu.leola.nus) пуццолановая земля — пайдеппая вблизи Иоццуолп. Такой же натуральный цемент представляют собой греческая санто-риновая земля, а также трасс, получаемый при размоле, туфовых! камней, встречающихся вблизи Эйфеля.
Гидравлическая известь. Осторожным обжигом известняка, содержащего лить 10—20% глины, получают продукт, который благодаря большому содержанию СаО можно гасить в кусках, распадающихся при этом в порошок, так что их не нужно молоть. Изготовление таких сортов «гидравлической извести» обходится дешево, однако их применяют обычно в смеси с цементами, богатыми кремневой кислотой, главным образом с цуццолановой землей или с трассом.
ПЕРМУТИТЫ И ЦЕОЛИТЫ
При сплавлении кварца с каолином и карбонатом натрия (например, в соотношении 1:2:4) получается стекловидный продукт, который! после измельчения и обработки водой переходит в желтовато-белую пористую массу в виде листочков; последняя обладает свойством обменивать содер
Четвертая группа периодической системы
559
жащийся в ней натрий па другие металлы при погружении ее в растворы их солей. Поэтому эту массу называют пермутитом {permutare — обменивать), Получение такого же продукта можно осуществить также мокрым путем, а именно прибавлением силиката натрия к нагретому раствору алюмината натрия.
При внесении пермутита в воду, содержащую растворенные соли кальция и магния, ионы кальция или же магния связываются пермутитом, причем в раствор переходит эквивалентное количество ионов натрия. Это свойство пермутита используют для смягчения воды, применяемой в паровых котлах, в прачечных и красильнях.
Для работы прачечных п красилен чрезвычайно вредно не только Содержание в воде навести, но н присутствие даже следов железа и марганца. Чтобы полностью устранить эти вещества, применяют пермутит, содержащий марганец. Перманганатом калия предварительно окисляют содержащийся в таком пермутите марганец. Последний передает свой кислород солям железЖ Н} и маргапца(П), раствореппым в воде, благодаря чему выпадают в осадок гидроокиси железа(ц1) и марганца(Л').
Если па пермутит, связанный с кальцием или же с другими ионами, подействовать раствором хлористого,’ натрия, то ионы кальция и т. д. снова будут замещаться попами натрия. Этот обмен можно повторить любое число раз.
Встречающиеся в тгрыродр кристаллические соединения, обладающие свойствами, аналогичными пермутитам, называются цеолитами. Например, относящийся к классу цеолитов гмелинит (натриевый хабазпт) может замещать свой натрий па магний
при погружении его в раствор хлорида магния. Кроме того, для цеолитов характерна способность непрерывно отдавать воду без разрушения своей кристаллической решетки.
Пермутиты также непрерывно отщепляют воду при пагревапии. При отщеплении воды от пермутитов и цеолитов не наблюдается ступенчатого повышения температуры разложения, характерного для большинства кристаллических гидратов (ср- стр. 98), наоборот, получаются кривые типа, представленного на рис. 99, с перегибом вместо ступеней. Это связано с тем, что у этих соединений все молекулы воды (или по крайней мере часть их) пе занимают прочного места в решетке. Это относится и к ионам щелочных металлов. Непрочно связанные составные части соединения такого типа названы, по предложения) Хюттига, «блуждающими в кристаллической решетке» [ср. Forts ch г, d. Chem. Phys, и Phy-. Chem., 18, 5, 1924]. Для объявления особой способности к обме-
Р и с, 99. Кривая зависимости содержания воды от температуры в ге-улаидите (изобара, р — 1 льч) рт ст) в качестве примера поведения цеолита при отщеплении воды при’па-тревапии.
ну цеолитов и пермутитов, помимо того, что ионы щелочных металлов не занимают прочного места в решетке | пли же для аморфных пермутитов говорят об их ориентации относительно алюмосиликатного остова), имеет значение также своеобразная структура алюмосиликатного ос it .та этих соединений. Как видно из рентгенограмм, тетраэдры SiO4 и А1О4 в них расположены в виде колец, нанпзаппых одно на другое так, что обра-вуются сквозные трубки, проходящие по всему кристаллу. Молекулы воды, попы щелочных и щелочноземельных металлов находятся внутри этих трубок и могут быть
560
Глава 12
поскольку опи не занимают прочного положения, легко оттуда вытеснены*. В пермутитах и и большинстве цеолитов одна часть воды связана прочнее, чем другая, Нацри-иер, из геулаядита СаА12817О(8'6Н2О только часть воды можно удалить нагреванием без разрушения решетки (Rinne, 1924; Wyart, 1933). Экстракцией жидким аммиаком также можно удалить лишь часть воды (Gruner, 1033), Более прочная связь одной частп воды проявляется в виде перегиба на кривой термического разложения геулаи-дита (содержание воды меньше 3'-'2Н2О и около 2*/^ Н2О) (рис, 99). Аналогичные отношения имеются у других цеолитов, а также у пермутитов.
В 1910 г. Зингер установил, что при обработке пермутитов растворами сульфидов в присутствии кислорода воздуха можно получить вещества от синего до голубоватосерого цвета, которые, по-видимому, приближаются к ультрамарину. По данным Грюнера (1932), при обработке пермутитов растворами сульфидов щелочных металлов в отсутствие воздуха сначала происходит обмен ионов ОН- на ионы SH’. Получающиеся так называемые «сульфидные пермутиты» бесцветны, ио при доступе воздуха переходят в окрашенные «подисульфидные пермутиты», причем из каждых двух 8Н"-групп за счет окисления возникает одна полисульфидная S|*-группа и соответствии с уравнением 2SH--f-1/2O2=S|-+H2O. Если реакция идет при более высокой температуре, то образуется настоящий ультрамарин.
УЛЬТРАМАРИН
Ультрамарин представляет собой минеральную краску, которая встречается в природе в виде лазоревого камня, лазурита (lapis lazuri). Искусственно его получают сплавлением тонкоизмсльченной смеси каолина, безводного карбоната натрия и’серы или при сплавлении каолина, сульфата натрия и угля. Лазоревый камень еще до настоящего времени считается ценным украшением из-за его красивого синего цвета. Искусственно полученный ультрамарин имеет различную окраску в зависимости От способа его изготовления. Основное значение имеет синий, ультрамарин, который является одной из наиболее важных минеральных красок. Однако находят применение также ультрамариновая фиолетовая и ультрамариновая красная краски, мепыпее значение имеет в настоящее время ультрамариновая зеленая. Ультрамариновую синюю применяют в качестве малярной известковой и масляной красок, в типографском Деле, в производстве обоев и бумаги. Ввиду ее способности перекрывать желтоватые тона, доводя их до чисто белого цвета, ультрамариновую синюю широко применяют для «подсинивания» белья, бумаги, сахара, крахмала и многих других веществ. Хорошие сорта такой краски абсолютно устойчивы по отношению к воздуху, свету и мылу, Ультрамарин не растворим ни в каких растворителях. Однако он разрушается растворами кислот, даже очень слабых. При этом он обесцвечивается, выделяя сероводород и кремневую кислоту. Поэтому его нельзя использовать в качестве синьки для сахара, употребляемого при изготовлении маринадов. При обычной температуре ультрамарин устойчив к действию разбавленных растворов щелочей.
Если каолин, карбонат натрия и серу слабо прокалить с ограниченным доступом воздуха, то обычно сначала получается зеленый ультрамарин; последний раньше переводили в синий ультрамарин повторным прокаливанием. В настоящее время его получают за один обжиг. Если к реакционной смеси примешать тонкой вне льченный кварц или кизельгур, то можно получить чуть красноватую ультрамариновую сипюю, которая устойчива по отношению к квасцам. Продолжительным умеренным нагрева
* Одиако по данным Глемзора (1944) вода не во всех цеолитах является легко-подвижпой в этом смысле. 11 меются цеолиты (например, патролит й анальцим), в которых она прочно удерживается в кристаллической решетке. Однако вследствие особой структуры алюмосиликатного скелета цеолитов она может быть удалена из решетки и введена в нее снова или же замещена ва другие вещества без существенного изменения самой решетки.
Четвертая группа периодической системы .561
нием богатой кремневой кислотой ультрамариновой синей с NH4C1 или с хлоратом калия и соляной кислотой получают фиолетовый ультрамарин, который при нагревании в токе хлористого водорода переходит в красный ультрамарин.
Искусственное получение ультрамариновой синей впервые, по-видимому, было предложено Гёте (в его «Итальянском путешествии»), а осуществлено оно было впервые Гмелином в 1828 г. мокрым путем. В последующие годы было предпринято фабричное производство его иа мейссегтспях фарфоровых заводах.
Естественный ультрамарин, или лазурит, представляет собой (в соответствии с анализом) двойной силикат алюмипия и натрия, содержащий полисульфид патрия. Состав соответствует приблизительно формуле NasAlBSieO24S3, однако он может значительно колебаться (ср. ниже). Строение ультрамарина было объяснено па основании Данных
Рис. 100. Элементарная ячейка решетки ультрамарина. 0=9,06 Л.
рентгеновского исследования, главным образом Егера (Jaeger, 1927), а также Под-шуса, Гофмана и Летейского (Podschus, Hofmann U., Leschewski, 1936). Согласно этим исследованиям, в ультрамарине имеется алюмосиликатный остов, который состоит из пространственной сетки, образуемой тетраэдрами SiO4 и Л1О4. Тетраэдры расположены в виде исключительно однородной «сотовой» структуры, «соты» которой ограничивают друг друга полиэдрическими плоскостями, образованными кольцеобразно расположенными атомами Si и А1, связанными между собой через атом кислорода. На каждую шарообразную ячейку, представленную на рис, 100, приходится 13/2=6 атомов Si и столько же атомов А1. Вокруг каждого такого атома тетраэдрически расположено по четыре атома кислорода, из которых один иногда находятся вне элементарной ячейки, представленной па рис, 100, и, следовательно, он принадлежит одной из соседних сот. Внутри каждой соты расположено восемь атомов натрия в виде двух пронизывающих друг друга тетраэдров. В цевтре каждой соты группа S2 занимает такое положение, что атомы серы расположены «статистически» по углам окружающего центр соты октаэдра (т. е. занимая совершенно беспорядочно все возможные положения). Такой октаэдр находится в середине элементарной ячейки. Соответствующие октаэдры, которые окружают середины восьми граничащих сот, расположены в углах элементарной ячейки, (В одном из них отмечены атомы Серы, находящиеся вне элементарной ячейки, а также их возможные положения. Эти положения для атомов серы па рис. 100 обозначены двойными пунктирными кружками.) Группы S2 могут быть образованы как нейтральными молекулами 32, так и радикалами дисульфида 8|_. Отдельные ионы S|~ в некоторых сотах могут располагаться в середине, не занимая углов октаэдра.
36 г Реми
562 Глава 12
Поэтому содержание серы в ультрамарине может в известных пределах колебаться» Большей частью ий элементарную ячейку приходится 2—4 атома серы*. Соответственно этому строение ультрамарина можно представить формулой NaB fAleSi6O241S(2_41.. Часто в ультрамарине содержится меньше (однако никогда пе больше) щелочи, чем соответствует этой формуле. В этол случае в нем содержится соответственно меньше отрицательных ионов серы**. Как показал Грюйер (1935), электропёйтральную серу можво удалить из ультрамарина без разрушения его решетки нагреванием в вакууме. Ионы S3“ можпо извлечь из решетки также плавлением ультрамарина с К CN; одпако. пр и этом одновременно извлекается эквивалентное количество ионов Na"1.: 1’ргопор таким путем получил ИЗ ультрамарина состава NasAlBSi6О24S3 бесцветное соединение Ka6AlLiSjt;O,4, в котором (как показали рентгеновские исследования) сохранился без изменения алюмосиликатный остов. Только около 1050° оп превращается в нефелин, имеющий такой же состав (см. стр. 545). Рентгеновские, а также химические исследования показали, (Leschewski, 1932), что окраска ультрамарина не обусловлена наличием в цом коллоидной серы (как часто предполагали прежде). Она, вероятно, связана с тем, что сера в ультрамарине находится в такой же форме, как в полисульфидах: (см, стр. 793). Подобную ультрамарину структуру обнаруживает ряд других минералов, например нозеан (серый или же бесцветный) Nas [Alf;Si6O24l [S04J, гауин (голубой) Na6Ca2 |Al;-SiBO24] [SO4)2 (Machatschki, 1934), содалит, (бесцветный, иногда голубой) Nae f A1(sSt0O24}C1 2 п еелъеин. (желтый, кдк мед) (Мн, Fe)g |BceSieO2S|S2 (Pauling, 1930). Нозеан можно окрасить в голубой цвет нагреванием с парами серы.
Можно получить также ультрамарин, в котором ионы Na* замещены, например, на ионы К* или Ag*. Кроме того, попы Na1 в ультрамарине можно заменить полностью или частично на другие ионы (нагреванием с растворами солей Tl(I), Са, Sr, Ba, Zn И т. д.). Полученные таким образом замещенные ультрамарины окрашены различно в зависимости от природы тех ионов, которые заменили ионы Na. В соответствии с этим, следовательно, на окраску ультрамарина оказывают влияние также положительные ионы, расположенные внутри алюмосиликатного остова.
Аналитические сведения. Обнаружение кремния, а также аналитическое определение его весовым путем почти всегда осуществляют в форме SiO-2. Характерным для SiOa является превращение его при обработке плавиковой кислотой в летучее соединение SiF4, разлагаемое водой с выделением белого продукта.
Наиболее чу ветвите ;тт, г гой реакцией иа кремний является выделение паров SiF4 в маленьком тигле (платиновом или свинцовом), покрытом крышкой с отверстием и смоченной черной фп.п.грошьчыгой бумагой. В результате реакции ,SiF44-2ff2O= = SiO2 । 4I1F выделяется белея гидроокись кремния, оседающая на червой бумаге в виде белого пятна размером, равным отверстию в крышке тигля (Ciltz).
Наряду с си Л и к ага мп кварц можпо определить простым методом <ГальВитица (Talvitic, Analyl- Chemistry, 23, 623, 1951)—нагреванием с 85%-пой фосфорной кислотой при 220п, При этом силикаты переходят в раствор, а кварц остается перастворенным.
ГЕРМАНИЙ (GERMANIUM). Ge
Распространение в природе. Германий встречается в природе в форме таких редких минералов, как аргиродит AAgsS-GeSa (около Фрейберга в Саксонии и в Боливии) и германит (медпо-желёзный тиогерманат). Германит встречается только и одном месте, а именно около Тсумёба в Южной Африке. Однако там он имеется в сравнительно больших количествах, включенный в блеклую руду, в которой ом отчетливо выделяется
* Количество иопов S2" (так называемой «сульфидной ссры>>) почти всегда меньше количества электронейтральпой серы (так называемой «полисульфидной серы»). Его Определяют по количеству сероводорода, выделяющегося при растворении ультрамарина в кислотах.
** Вместо ионов S2- могут находиться ее. только группы S2 и 8|”, а также частично молекулы воды.
Четвертая группа периадичесхпй. системы. 563
розовой окраской (найден в 1920 г, Шнейдер хоном). В качестве незначительной примеси германий содержится еще в некоторых других, также редких, минералах, таких, как самарскит, танталит, гадолинит. Незначительные количества германия встречаются в некоторых североамериканских цинковых рудах,
Германий концентрируется в некоторых остатках от переработки руды, так что, как' установил в 1916 г. Буханен, остатки могут содержать 0,25% н больше GeOa. Германий иногда в значительных количествах содержится и золе малозольных каменных углей.
История. Германий был открыт в 1885 г. Клеменсом Винклером. При анализе вновь найденной около Фрейберга серебряной руды, а именно аргиродита, оказалось, что сумма содержащихся там составных частей постоянно имеет дефицит 6—7%. Он обнаружил^ что это обусловлено содержанием в ней неизвестного еще до того времени'элемента, который он назвал германием.. Впоследствии оказалось, что германий тождествен экасилицию, предсказанному еще в 1871 г. Менделеевым на основании периодической системы.
Совпадение свойств, предсказаппых Менделеевым для экасилцция, со свойствами, найденными для германия, видно в табл. 88.
Таблица 88
Свойства германия в сравнении со свойствами эка си л ппи я. предсказанными Менделеевым
Эвасилиций Германий
Атомный пос 72 72,60
Удельный вес 5,5 5.35
Формула окисла EsO2 GeOa.
Удельный вес окдела 4,7 4,70
Форму'ла х .торида EsC14 GeCl4
Температура кипения хлорида, °C < 100 83
Более 30 лет, прошедших со времени исследований Винклера, германием почти пе занимались ввиду его чрезвычайной редкости. Широкие исследования стали возможны только после открытия этого элемента в остатках от переработки североамериканских цинковых руд и обнаружения германита в Южной Африке. Эти исследования были проведены тогда главным образом Деннисом (Dennis, 1921) л Шварцем (Schwarz, J929).
Получение. Германит можно вскрыть обработкой тонкоивмельчеиного минерала смесью авотной и серной кислот. Германий осаждается при этом большей 4aCTj.ro в виде двуокиси. От загрязняющих примесей его отделяют растворением в 20%-ной водной соляной кислоте п перегонкой в виде тетрахДорида, который поглощается водой. Получающуюся в результате гидролиза чистую двуокись германия после обезвоживания восстанавливают до металла сплавлением с цианидом калил и древесным у г дем или нагреванием в токе водорода. По Джо псов у (Johnson, 1935) для переработки больших количеств руды вскрытие производят сухим методом. Для этого тонкойзмельчен-вый германит нагревают до 800" в токе азота, очищенного от кислорода, благодаря чему удаляются примешанвыо сульфид мышьяка и сера, затем вад остатком пропускают сухой газообразный аммиак при 825°. При этом возгоняется GeS, который можно легко перевести в окись обработкой азотной кислотой. Нагреванием окиси в токе водорода или прокаливанием с углем из пее получают металл.
36*
564
Глава 13
Свойства. Германий очень хрупкий, серовато-белый блестящий металл, Он кристаллизуется в кубической системе. Твердость составляет около 6,5, удельный вес (при 20°) 5,35, точка плавления 958°. На воздухе компактный германий не изменяется. При температуре выше красного каления оп соединяется с кислородом. С водородом он непосредственно не соединяется и пе обладает по отношению к нему также особой растворяющей способностью- Напротив, при нагревании он легко сплавляется с платиной, золотом, серебром, медью и другими металлами. Эвтектический сплав Ge-Au (с 24 ат. % Ge) обнаруживает заметно низкую для сплавов золота точку плавления (359°). В соляной кислоте германий нерастворим, точно так же в разбавленной серной кислоте; напротив, он растворяется с выделением SO 2 в горячей концентрированной серной кислоте. Умеренно концентрированная азотная кислота переводит его в гидрат двуокиси, так же как олово. С разбавленным раствором едкого кали он пе взаимодействует, однако очень легко подвергается воздействию щелочного раствора перекиси водорода. Его также легко можно перевести в раствор анодным окислением (Jirsa, 1952), при этом он переходит непосредственно в четырехвалентное состояние. В щелочных растворах образуются германаты, в кислых растворах — соли германия(1У).
В соответствии с положением между Si n Sn германий по своим свойствам и свойствам своих соединений в некотором отпоптенни близок к кремнию, в то время как по другим свойствам он приближается к олову. Как и кремний, он кристаллизуется по типу алмаза (длина ребра а=5,63 А). В особых условиях (испарение в высоком вакууме) его можно выделить в аморфной форме. В аморфном германии, так же как и в кристаллическом, каждый атом тетраэдрически окружен четырьмя другими атомами (на расстоянии 2,40 A) [Furst, Glocker, Richter, 1949], в то время как в жидком германии каждый атом имеет восемь ближайших атомов (на расстоянии 2,70 A) [Hendus, 1947]. Подобно кремнию, германий образует как летучие, так и сильно ненасыщеппыо твердые водородные соединения, которые в случае германия легче получаются, чем у кремния. Как и кремний, германий также способен выступать в качестве центрального атома в гетерополикислотах (см. т. II), GeO2, аналогично SiO2, может при застывании из расплавов давать стекла. Кристаллизуясь, GeO2 образует обычно (однако ниже 1033° метает,тбпльпи) решетку, подобную кварцу, по наряду с этим двуокись германия, так же как SnO2, может криста тлттзоватт.ся по ттшу рутила. Германаты и фторогерманаты часто изоморфны соответствующим соединениям кремния. С оловом германий сближает способность в водных растворах существовать в качестве положительного иона и образовывать типичные соли, выпадать из водпых растворов в виде сульфида (образующего тиосоли), а также легко восстанавливаться.
СОЕДИНЕНИЯ ГЕРМАНИЯ
В галогенидах, окислах и сульфидах германий бывает в двух- или четырехвалентном состоянии. Четырехвалситность он проявляет, кроме того, по отношению к водороду и органическим радикалам:. Замечательно, что двуокись германия и дисульфид сравнительно легко растворимы. Соединения двухвалентного германия весьма неустойчивы. Они стремятся окислиться до соединений четырсхвалентного германия.
Соединения четьтрехвалентного германия
Гермаповодороды. Если подействовать цинком на серную кислоту, содержащую хлорид германия, то, как нашел Фогелен (Voegelen, 1902), к выделяющемуся водороду примешан германоводород GeHj. При пропускании такой смеси через нагретую до яркокрасного каления стеклянную трубку на ее стенках осаждается германий в виде блестящего красного в прозрачном слое металлического зеркала.
Четвертая группа периодической системы
565
Формула соединения была выведена Фоголеном исходя из состава германида серебра (1), выпадающего при пропускании гермнноводорода черна раствор нитрата серебра, а также из соотношения, в котором образуются сероводород и сульфид германия при пропускании GeII4 над тонко и и мельче иной освещенной серой (2).
GeJV|-4AgNOs=Ag4G₽+4HNOS( (1)
G<!H4+4S=GeSa+2H2S. (2)
Зга формула была подтверждена Панетом в 1922 г. Он отделил гермаповодород от примеси водорода кондепсаплой посредством жидкого воздуха и разложил затем его нагреванием GeIl4“Ge-f-2H2. Из соотношения образующихся при атом водорода и германия следует формула GeIJ4.
Со сравнительно хорошим выходом получают германово до род при разложении серной кислотой сплава состава Mg2Ge, приготовленного сплавлением германия с магнием, Доннис (1624) обнаружил наряду с обычный газообразным германоводородом (моногерман, т. пл. —165°, т. кип. — 90°), образующимся в качестве главного продукта, еще два других, жидких при комнатной температуре гермааоводорода: дигерман Ge3He (т. пл. —109°, г. кип. 29°) и тригерман Ge3HB (т. пл. —105,6’, т. кии. 110,5°). Моногерман теперь удобнее всего получать взаимодействием GeC],; с ИА1Н4
GeCl4+LiAlH4=GcH4+LiAlGl4.
Расстояние между центрами атомов Ge<—>Ы в молекуле GeH4 составляет 1,37 А.
Помимо летучих гидридов, германий образует сильно ненасыщенные твердые гидриды (полигермены)-, [С1еН]ж и [Се1Ь]ж- Первый был получен Деннисом (1930) гидролизом NaGe. Он представляет собой темно-коричневый порошок, который в сухом состоянии вспыхивает при доступе воздуха, окислителями переводится в соединения четырехвалентного торманип Ge(IV), с сухим хлористым водородом, наоборот, не реагирует. Гидрид (СоНг)ж получен Шварцем (Schwarz, 1933) разложением CaGe разбавленной соляной кислотой, а также раствором едкого натра. Он окрашен в желтый цвет, устойчив в сухом состоянии без доступа воздуха, по на воздухе тотчас воспламеняется со взрывом. С конценгрироватгпой соляной кислотой наряду с GeCla и Н2 образует ряд летучих герма нов одо родов GeH4, Ge2H6 и Ge3He. Эта реакция позволяет сделать вывод о механизме образования силанов при взаимодействии Mg2Si с концентрированной соляной кислотой.
Алкильные сосдипения германия. С органическими радикалами германий образует соединения типа GeR4; например, г действием диэтилцинка Zn(C2Hs)2 ва хлорид германия (IV) получают тетразтилгерманий 2Zn(C2H^)2+GeGl4 = Ge(C2H5)4+2Zn.Cl2. Это бесцветная, не смешивающаяся с водой легкая жидкость со слабым запахом лука (уд. вес 0,991, т. кип. 163°). Асимметричное соединение (C2H5)(C3H7)Ge(CeHs)Br аналогичное асимметричным соединениям углерода можно получить в оптически активной форме (Schwarz, 1931).
Галогениды гер мания (IV)
О важнейших физических свойствах галогенидов германия (IV) дает представление следующее сопоставление:
Ok Wita Температура плавления, °C Температура кипения, °C Расстояние между центрами атомов Ge ч—X, А
GeF4 Вес цвет- Сублимируется при —36,6
1Ш.П
GeClj » -49,5 +83,1 2,09
GeBr4 )> +26 +186 2,32
Gol4 Оранжевый J44 Выше 300 2,48
Известны также смешанные галогениды, например GoF3Cl (т. кип. —20,3е),GeF2Cl, (т. кин. —2,8°) и GeFCLj (т. кип. +37,5°).
566
Глава 12
Тетра фторид германия и фторогерманаты. Действием концентрированной плавиковой кислоты па двуокись германия получают прозрачный раствор, из которого при упаривании выделяется тетрафторид германия, содержащий кристаллизационную воду GeFi-ЗНаО, в виде бесцветных, очень гигроскопичных кристаллов. При попытке обезводить соль нагреванием наступает гидролитическое разложение. Одновременно часть германия улетучивается в виде безводного фторида.
Пря обычной температуре безводный фторид является газом (вое 1 л 6,650 г). Прн более сильном охлаждении он конденсируется в виде белой, хлопьевидной массы, которая цри нагревании возгопяется не плавясь.
Из раствора тетрафторпда германия, К которому примешан фторид калия, кристаллизуется фтпорогерманат, калия KsfGeFJ в виде негигроскопичных, белых гексагональных призм или листочков. Он трудно растворим в холодной воде и нерастворим в спирте. При нагревании до температуры красного каления он разлагается.
K2[GeF6]n (NH4)a[GeFe], по-видимому, изоморфны соответствующим фторосиликатам. Для Csa[GeFe] посредством структурного определения доказан изоморфизм со щелочвыми фторосиликатами- Фторогерм а иат гидрокси л аммония [Ь1Н3(ОН)]2 [GeFe] кристаллизуется так ясс, как [NH3(OII)]2 [SiFe] с 2Й2О, в то время как фторо герман ат гидразина [N2Hn][GeF6], как к [ iV2He] [SiFc], кристаллизуется без воды (Dennis, 1933).
Тетрахлорид германия. GeGh образуется при сжигании германия в токе хлора; в чистом состоянии его лучше получать нагреванием двуокиси германия с дымящей соляной кислотой [по Бауэру (1933) Получение целесообразно проводить в сосуде иод давлением]. Он представляет собой подвижную бесцветную жидкость с удельным весом 1,88, температурой кипения 83°, которая затвердевает при —49,5°, Водой он медленно гидролитически разлагается с образованием содержащей воду мелкодисперсной двуокиси германия.
В нонцоптрпроиапцой соляной кислоте GeCl4 существует л форме еерманийхло-ристоводородной кисло>пы. П2 [GeGl(i ]: это следует из того, что при электролизе Ge перемещается к аноду. Как обнаружил Лаубепгайор (I.aubengayer, 1940), хлорогерманат цезия Cs2 IGeCL, |, представляющий собой светло-желтые октаэдрические кристаллы (уд. вес '3,45), является изо типом (NH4)2 |Р1,С1е].
Прп нагревании титрахлорпда германия с германием происходит частичное восстановление до дихлорнда но уравнению GeCl4-|-Ge 2GeCl2.
Нагреванием GeCl2 Шварц (1952) получил соединение, соответствующее монохлориду кремния [GeC] ]ж. Наряду с ним образуется очень мало устойчивое соединение Ge^Clg и в противоположность кремнию также дихлорид GeCl2.
Оксихлорид германия GeOCU получается взаимодействием GeHCl3 (см. стр. 568) с Ag2O в виде бесцветной, затвердевающей при —56° жидкости. При нагревании он разлагается ва С12 и GeO. Последняя получается при этом в лимоппо-желтой форме, которая переходит при 650° в обычную черную окись (Schwarz, 1932), Другой оксихлорид GeaOCio, который разлагается При нагревании с образованием GoO2, был получен Шварцем (1931) действием О2 на ЬеС14 при 950°. Он представляет собой бесцветную затвердевающую при —60° жидкость.
Тетрабромид германия (1оВг4 (бесцветные, правильные октаэдры, уд. вес, 3,13, т. пл, 26°, т. кип. 135,9°) н тетраиодид германия (оранжевые кристаллы, уд, вес 4,32, т. пл. 14-4°; о структуре см. стр, 576) получается так же, как и хлорид. Водой он разлагается гораздо энергичнее, чем хлорид, поэтому сильно дымит на воздухе. Йодид начинает разлагаться уже немного выше точки плавления на Gela и 12.
Двуокись германия, германиевая кислота и германаты. Двуокись германия GeOa получают сильным прокаливанием германия или сульфида германия в токе кислорода дли окислением указанных веществ кон
Четвертая группа периодической системы
567
центрированной азотной кислотой. Опа представляет собой белый порошок, похожий на песок. Постепенно размягчаясь, двуокись германия полностью плавится про 1115° и затвердевает из расплава в виде стекла, которое, однако, легче рае стек ловывается, чем силикатное стекло. Выше 1250° двуокись германия заметно летуча. В воде она лишь умеренно растворима (при 20” в 100 г воды растворяется около 0,4 г, при 100°— около 1 а).
Растворимость двуокиси германия, как показал Шварц (1931), зависит от количества доппоп фазы. Это связано с тем, что двуокись гормапия переходит в раствор не только в молекулярно-дисперсном состоянии, но одновременно также и в коллоидном в соответствии с правилом допной фазы Оствальда (см. т. II). Образованием золя объясняется также то, что при охлаждении насыщенного при нагревании раствора двуокиси германия не происходит помутнения, хотя растворимость двуокиси германия на холоду существенно меньше, чем при нагревании. Растворы реагируют отчетливо кисло и обнаруживают заметную электропроводность. Первичная константа диссоциации содержащейся в них германиевой кислоты составляет по Пугу (Pugh, 1929), а также по Денпису и Гулециану (Giilezian. 1932) около l l(f (при 20°). Методом диализа Шварц показал, что при pH 8,4—8,8 существуют не ионы GeOg, как в сильно щелочных растворах, а ионы GcjO^.
При испарении водного раствора двуокись гормапия осаждается в виде микроскопических: кристалликов. Двуокись германия с трудом растворяется в кислотах и очень легко в растворах щелочей. Следовательно, кислый характер двуокиси германия значительно преобладает по сравнению с основным. Соединения двуокиси германия с более основными окислами металлов называются германитами. Их можно подучить как из водного раствора, так и сплавлением; они показывают интересные соответствия с силикатами (Schwarz R., Angew. Chem., 48, 219, 1935). Методом диаграммы плавкости доказано существование орто-, мета-, ди- и тетра гермйнатов, например Li,iGeO4 (т. пл. 1298°), Li.,GeO? (т. п.п. 1239°), Na3GeO3 (т. пл. 1083°), Na2Ge2O5 (т. пл. 799°), НагОе409 (т. пл. 1052°). Из водпых растворов обычно кристаллизуются мета герм ан а ты Чаще в виде гидратов).
Помимо растворимой гексагональной модификации, которая имеет решетку низкотемпературного кварца (и =_4,97, с=5,65 А), двуокись германия образует также тетрагональную, нерастворимую в воде модификацию, а именно типа рутила (а— 4,39, с=2,86 А). Выше 1033° опа превращается (крайне медленио) в гексагональную модификацию. Превращение гексагональной модификации в тетрагональную может идти еще медленнее. Его можно ускорить добавлением фторида аммония (Schwarz, 1943), Еще большей растворимостью, чем гексагональная, обладает стеклообразная двуоки гъ германия.
Если двуокись германия получают гидролизом, например, тетрахлорида, то она выделяется с содержанием воды в виде геля. Так как GeO2 значительно более, чем SiO2, склонна к кристаллизации, частицы окиси прп выдерживании под водой постепенно укрупняются; когда окись достаточно стареет, она отдает свою воду даже при стоянии па воздухе. Воду опа теряет не ступенчато, откуда следует, что GeO3 не способна образовывать гидраты (Schwarz, J931). Следовательно, германиевая кислота, подобно угольной Кислоте, существует только в водном растворе.
Пероксттгидраты германитов. Действием Н2О2 на растворы терманатов щелочных металлов подучают, как и В случае кремния, пероксигидраты. Шварц (1930) приготовил перокш тпдрзты германатов K2Ge2O5-2H2O2’2Н2О, Na2Ge2O6-2Н.,О2’2Н2О в NasGoOs 2Н3О, 2ПгО в кристаллической форме и доказал (1935), что при этом речь идет о продуктах присоединения перекиси водорода, а не о перокси с о лях.
Дисульфид германия и тпогермацаты. Если в водный раствор двуокиси германия пропускать сероводород, то осадок сначала не образуется. Если затем смешать раствор с достаточным количеством сильной кислоты, выпадает дисульфид германия GeSa в виде белого осадка.
Это явление основано па том, что только в присутствии гораздо большего количества водородных ионов равновесие СеО2ф4Н* — Ge'"* ф2НгО сдвигается настолько далеко вправо, что копцоптрацпя ионов Ge"" становится достаточной для до стяжения произведения растворимости дисульфида германия 7= [Ge"")x IS"}3. В воде осадок дисульфида лишь умеренно растворим (0,455 г в 100 г воды). Водный раствор
568
Глава 12
дисульфида постепенно разлагается, при этом благодаря Гидролизу выделяется сероводород GeS,-|-2H2O £ GeO;>-|-2H,>S. В сернистом аммонии дисульфид легко растворяется с образованием тиогерманат-иоиое GeSa^S" Ji {GeS3]". При добавлении кислоты (превращение S" под влиянием 1Г в H2S) реакция снова протекает справа налево, однако полностью это происходит только при более высокой концентрации ионов Н‘.
Действуя па растворы тио гермапатов щелочных металлов ацетоном, Шварц (1930) получил соединения NaBGr'2S.'9H3O и KeGe2S7-9II2O (длинные игольчатые, очень гигроскопичные кристаллы). Согласно Бриптзипгеру (1934), в растворе, однако, существуют не [Ge2S7]а [GeSjJ''-ионы. Это является примером правила, установленного также на основании поведения других веществ: в кристаллических соединениях радикалы иногда выступают в качестве структурных групп, которые не существуют в заметном количестве в растворах, находящихся в равновесии с кристаллами.
Близки к тиогермз патам встречающиеся в природе двойные сульфиды германия— минералы: германит (ср. стр. 5G2) — окрашенные в розовый Цвет, расположенные лучами иголочки, аргиродит AgsGeSe — правильные голоэдрические кристаллики с металлическим: блеском, канфиельдит. Ags(Ge, Sii)Sa и ультрабазит (двойной сульфид свинца и Германия). Германит образует решетку типа цинковой обманки.
Соединения германия с азотом. GeCl4 взаимодействует с жидким аммиаком с образованием имида германия в соответствии с уравнением GeCl44-6NH3=Ge(NH)34-4-4NII4Cl. Если последний нагреть в атмосфере азота приблизительно до 150°, то он переходит с выделением аммиака в германам GeoNgH. Германам разлагается выше 300° с образованием нитрида Ge3N4, который распадается около 1000° па Ge и N2. Эти соединения менее устойчивы, чем соответствующие Соедипепия кремния. Соединение, соответствующее амиду кремния Si(NH2)4 (которое разлагается уже при 0°), германий не образует (Schwarz, 1930). Напротив, помимо названных, германий образует также соединения е азотом, в которых оп двух валентен, а именно GeNH и Ge3P<2. Первое Соединение разлагается водой с образованием гидратированной окиси германия(И) GeNH^H2O— =GeO^NH3. При нагревании опо переходит в нитрид германия(П) в соответствии с уравнением 3GeNH=Ge3N2-^NH3 (Johnson, 1934).
Сульфат гермапия(1У) Ge(SO4)2 получен (Schwarz, 1934) действием 8О3 на GcCl4 в виде белого порошка с уд. весом 3,92; гидролитически разлагается водой. С раствором едкого патра взаимодействует с шипенпем, образуя Na2GeOs и Na2SO4. Термическое разложение начинается при 200°,
Соединения германия(II)
Хлорид гермаппя(Н) GeCl2 образуется при пропускапии паров GeCl4 над нагретым металлическим германием. Это твердое вещество, очень чувствительное к влаге и кислороду воздуха. С водой опо реагирует по уравнению GeCL-yH2O=GeO-|-2HCt (с образованием гидратированной окиси гормапия(П), см. пиже), с кислородом по уравнению 20еС124г02=.Ое02-^ОеС14. Па холоду GeCla почти бесцветен, при нагревании окрашивается в оранжевый цвет. При болое сильном нагревании распадаетсц по уравнению 2GeCl3 Ji Ge^-GeCl4.
Трпхлоргерман GeHCl3 по составу соответствует хлороформу и поэтому называется также германийхлороформом. Ов аналогичен хлороформу по некоторым физическим свойствам, однако химическая природа у них разная. Это соедииевцо впервые было получено Винклером при пропускании хлористого водорода вад слабо нагретым порошкообразным германием Gc4-3HCl=GeHCl3+H2. Однако лучше его синтез вести по способу Денниса (1926), пропуская пары GeCl4 пад нагретым германием и обрабатывая соляной кислотой образующийся при этом продукт, который состоит главным образом из GeCl3 : GeCl3 ф- НС1 — GeIICl3. Трихлоргормап представляет собой бесцветную, достаточно трудно конденсируемую жидкость, которая при доступе воздуха мутнеет, образуя оксихлорид. Т. кип. 75,2°, т. пл. —71е'. Германий в этом соединении двухвалентен, так как большим количеством воды оно разлагается с выделением желтого гидратированного окисла германпя(П). Иод действует окиСляюще в соответствии с ураввением GeHCl3^la=GelCl34Hl.
Бромпд германня(П) GeBr2 (бесцветные иглы иля пластиночки, т. пл. 122') получают действием НВг па металлический Ge при 400° и восстановлением образующегося при этом одновременно GeHBr3 цинком. Он растворим в спирте и ацетоне, по нерастворим в бензоле. Водой разлагается с выделением гидратированного окисла германия(П) (Brewer, Dennis, 1927).
Четвертая группа периодической системы
569
С НВг бромид герма пня (II) соединяется, давая трибромгерман GeIIBr3, который слова разлагается при более высокой температуре GeBr3-AJIBrZ GeIIBr5. Триброыгер-ман — бесцветная жидкость, затвердевающая при —24,5°.
Действием НВг на GbII4 в присутствии А1Вгй получают GeH3Br (d^°=2,34, т. пл. —32% т. кип. 52°) и GeH2Br, (d§= 2,80, т. пл. —15°, т. кип. 89°), в то время как реакция Ш с GeH4 при равных условиях приводит к Gel2, причем соответствующие промежуточные продукты не обнаружены (Dennis, 1929).
Иодид германпя(П) Ge!2 —желтые гексагональные пластипки (решетка типа бру-цита; а=:4,13, <>=6,79 А) с т. субл. в вакууме 240°. Проще всего ов получается действием концентрированной иодистоводородной кислоты на гидрат окиси германия(П). Пропусканием паров Gels лад нагретым германием получают Gel2, только с низким выходом, так как при нагревании происходит разложение, сопровождаемое реакцией окисления-восстановления по схеме 2GeI2—Ge-rGeI4 (Brewer, Dennis, 1927).
Окпсь германия(П). При разложении хлорида гермавия(П) водой или раствором едкого натра получают желтый осадок, который при кипячении с небольшим количеством раствора едкого натра становится ржаво-красным в результате частичного окисления. Вероятно, желтый осадок представляет собой гидрогель желтой формы окиси германия(П). Гидроокись Ge(OH)2, по-видимому, существует только в растворе. Осадок, который, принимая во внимание неизвестный характер связи воды, обозначают как «гидрат окиси германия!П)», снова растворяется в большом избытке раствора едкого натра. Согласно Гапчу (Hantzsch, 19U2), электропроводность этого раствора значительно меньше, чем электропроводность чистого раствора едкого натра той же концентрации, из чего можно заключить, что при действии щелочи образуются соли. При насыщении едкого натра в этом растворе точно эквивалентным количеством соляной кислоты гидроокись гермапия(Н) не выпадает. Раствор, который наряду с гидроокисью германия^!) содержит только NaCl, проявляет явно кислую реакцию. Из величины его электропроводности следует, что гидроокись германия является отчетливо выраженной кислотой, хотя и более слабой, чем уксусная. При нагревании выпавшего гидрата окиси в токе азота до 650° получают черную кристаллическую окись гермапия(П) GeO (Dennis, 1930). О желтом GeO см. стр. 567.
Сульфид гермппип(П) GeS получается при уморенном нагревании сульфида германия^ V) GeS2 в токе водорода; GeS кристаллизуется в форме красивых листочков, обладающих в отраженном свете серо-черной окраской с металлическим блеском, а в проходящем свете—ярко-красной. Из солйнокислого раствора хлорида германия(П) оп осаждается сероводородом в виде к рас но-бур ого о садка, который растворим в растворе едкого кали и в противоположность сульфиду, приготовленному сухим путем, в горячей концентрированной соляной кислоте, а также в желтом сернистом аммонии. В последнем случае благодаря окисляющему действию растворенной там серы образуются тиогерма-наты с четы р ехва л ентным г срма н иен G е11 S-ф- S " -ф S = [ Go1 VS3 ]". Ии раствора тио герм а-иата при подкислении осаждается белый GeS2, а из раствора едкого кали — неизменив-тоипся краспо-бурый GeS. Последний имеет большущ склонность к образованию коллоидных растворов. Сульфид германия(П) кристаллизуется в ромбической системе. Он образует решетку, пе наблюдавшуюся пока у других соединений: она имеет вид сильно деформированной решетки каменной соли.
Селениды германия. GeSe (темно-коричневый тетрагональный с уд. весом 5,30, т. пл. 667°) и GeSe3 (желтый ромбический с уд. весом 4,56, т. пл. 707°) были приготовлены Ивановым-Омипым (1940). Селепиды германия образуются при осаждении JI2Se из солянокислого раствора или непосредственным соединением составных частей (нагревание до 5lhj°).
Аналитические снедения. Для гермапия характерна белая окраска осаждаемого из сильнокислого раствора дисульфида, который растворяется в сернистом аммонии. При весовом определении, германия в виде двуокиси следует иметь в виду, что при осаждении из сернокислого раствора она увлекает значительные количества серной кислоты. Серную кислоту можно удалить упариванием с концентрированной азотной кислотой, последующим прокаливанием в экстрагированием аммиаком. При соблюдении определенных условий [Schwarz R., Z. anorg. Chem., 229, 146, 1936J удается электролитически количественно осадить германий вместе с оловом.
570
Глава 12
ОЛОВО (STANNUM). Sn
Распространение в природе. Важнейшей и почти единственной оловянной рудой является оловянный камень (касситерит) SnOa. В первичных месторождениях этот р i встречается включенным в другие породы, прежде всего в граппт («горное оловом), а во вторичных месторождениях он существует («оловянное мило») в виде мелких зернышек, которые тесно перемешаны с песком или глиной, причем содержание олова в рудах, имеющих значение для получения металла, часто очень невелико, в то время как чистая двуокись содержит 78,62% олова.
Довольно редкому оловянному камню сопутствует оловянный колчедан (станнин) CiTSS-FeS-SuS2. Очень редко встречаются небольшие количества олова в самородком состояния вместе с золотом.
Основные залежи «оловянных руд», т, е. пород, содержащих окись олова, находятся на Малаккском полуострове (Малакка, Куантап), в Индонезии (Вавка, Биллитон) я в высокомрной части Боливии. Весьма значительные залежи были найдены также в Австралии (Новый Южный Уэльс, Тасмания), в Мексике, Таиланде и Китае. Важным поставщиком олова является в настоящее время также Конго. В Германии олово встречается главным образом в Саксония. Однако эти залежи дают лишь небольшую часть руды для немецких оловянных переработок. Они базируются преимущественно на импорте оловянном руды. Существенное значение Для получения олова, или «вторичного получения», имеют отбросы белой жести.
Исторические сведения. Олово относится к числу наиболее давно известных металлов. По крайней мере в виде бронзы его использовали уже па заре человеческой культуры (бронзовый век).
В четвертой книге Моисея содержится упоминание об олове под названием «МП», как о предмете военной добычи. В древнеиндийской литературе (Веды, Махабхарата) оно известщ) иод именем trupu. Греки (Гомер) называли его xanottEooS (правда, пока пе ясно, по нодразумевз чея ,-тп под этими названиями также и свиней, так как тогда не было еще строгого рю; । юг ня между оловом и свинцом). Цезарь, сообщая о нахождении олова в Британии. тта :ывает его /йшабиш album, в отличие от обычного свинца plumbum Тах же называет его позднее Плпниц. Вплоть до XII в, Англия была единственной (трагцчт в Европе, где добывали олово (оловятпю-рудные залежп Корнуэлла), Затем вплоть до Тридцатилетием войны большое, значение для добычи олова имели разработки в немецких Рудных горах (Саксонии и позднее Богемии), В результате Три-дцатилетпой войны там почти полностью было разорено Торное дело; только отдельные горные Эйводы были позднее восстановлены. В последнее время имеют значение оловян-но-рудные месторождения в Саксонии. Одпако даже и теперь еще в Германии, как и в других европейских странах, перерабатываются преимущественно импортируемые оловянные руды.
Получение. Олово получают восстановлением оловянного камня углем в шахтных или пламенных печах
SnOg Ц-2С = Sn -|- 2С0.
Перед внесением в печь оловянную руду «обогащают», т. е. освобождают от основных загрязнений механически или отчасти химически (уДалятОт серу и мышьяк обжигом).
Из образующихся при восстановлении оловянпого камня шлаков, содержащих, как правило, еще много олова, последнее можно извлечь по способу восстановления, т. е. плавлением в пламенных печах с добавкой угля и извести (1), или по способу осаждения, т. е. сплавлением с железными остатка ми и углем (2):
SnSiOs+CaO+C=Sn+CaSi03+CO, (1)
SnSiO3+Fe=PeSiO3-|-Sn 1 t
SnO,+ 2C=8iH-2CO. J ' (2)
Четвертая группа- периодической системы '' 571
Получающееся при норной, плавко сырое олово еще сельпо загрязнено железом л в незначительной степени другими металлами. Сначала его освобождают от железа переплавкой, заставляя при этом стекать ио наклонной подложке («зейгорсванке»), затеи его очищают от других металлов окислительным плавлением («дразнение»). Первый из этих процессов основан на том, что при нагревании олова, содержащего железо, .немного выше температуры плавления, железо остается в виде тугоплавкого сплава (так называемых *зойгер-шипов»). При дразпёпии в расплавленное олово погружают жердь из спежтсрублсниого дерева. Выделяющиеся из обугливающегося дерева газы энергично перемешивают олово и благодаря атому достигается достаточное соприкосновение его с кислородом воздуха, вследствие чего примеси окисляются и (вместе с окисленной частью самого олова) собираются на поверхности в виде «оловянных огарков».
Для регенерации олова из отбросов белой жести пользуются либо -электролизом, причем отбросы в корзинах из железной проволоки, служащих анодом, погружают в раствор едкого натра, либо применяют способ извлечения олова при помощи хлора, который основан на том, что олово в противоположность железу легко взаимодействует с сухим хлором,
Свойства. Олово представляет собой серебристо-белый блестящий металл, обладающий незначительной твердостью, но большой ковкостью, так что его можно прокатать до очень топких листов (стаиниолъ). Его удельный вес 7,28, точка плавления 231,8°. В значительной мерс оно улетучивается уже при 1200°, хотя точка кипения его лежит только при 2302°. Из расплава олово затвердевает обычно в виде ^бтрагональ-ных кристаллитов. Кристаллическая структура Sn отчетливо проявляется, если его шлиф протравить соляной кислотой (муаровое олово). Хрустящий звук, появляющийся при сгибании оловянной палочки («оловянный крик»), обусловлен трением кристаллитов друг о друга. Выше 161° олово превращается в другую, ромбическую, модификацию. В этой форме оно очень хрупко, так что его можно истолочь в порошок (лучше всего при температуре примерно 200°), а при падений с небольшой высоты оно разбивается на мелкие куски. На этом основано приготовление так называемого зерненого олова. Третья модификация — порошкообразное серое олово (уд. вес при 18° 5,75) — стабильна ниже 13,2°. Превращение в эту ' модификацию и обратный процесс происходят обычно с бесконечно малой скоростью. При переходе олова в серую модификацию оловянные предметы полностью разрушаются. В местах, пораженных превращением, они рассыпаются в порошок («оловянная чуМа»),
При устойчивых сильных холодах в разных местах оловянных предметов начинается егюптаппое превращение олова в серую модификацию. В этом случае па пораженных предметах образуются серые пятна, которые состоят из порошкообразного серого олова. Эти пылинки, попадая па Другие места, действуют как зародыши кристаллизации, так что разрушающее превращение, если оно началось только в одном месте, распространяется как заразное заболевание. Названии для этого явления «оловянная чума» очень характерно. Превращение тем активнее, чем сильнее охлаждено олово ниже температуры 13,2':'. Одпако в силу того, что скорость реакции с понижением температуры уменьшается, имеется определенная температура, при которой скорость превращения достигает своего максимума. Эта температура лежит около —48°. Превращение ускоряется под действием спиртового раствора «розовой соли»; опо замедляется (по данным Фарупа) прежде всего под действием с о .чей висмута и сурьмы. Термическая и механическая обработка металла и число превращений, которые ужо произошли, также весьма значительно влияют на скорость превращения (Cohen, 1935).
Структура решетки серой» олова (a-Sn) такая же, как у алмаза, но с а==™6,46 А. Обычное тетрагональное олово (ф-Sn) образует (пе наблюдающуюся у других соединений) решетку, которая может быть описана как решетка алмаза, сжатая в направлении оси с. Структура ромбического олова (y-Sn) еще не известна.
572
Глава 12
Превращение a-Sn -* |3-Sn сопровождается не только значительным увеличешхлг плотности, по также значительным усилением металлического характера. Оба свойство, тесно связаны одно с другим, так как для металлов характерна особо плотная упакаЕгх атомов (высокие координационные числа) (ср. т. II, гл. 1). Напротив, неметаллы вследствие наличия направленных связей имеют только небольшие координационные числа (4 и менёе) н вследствие этого обладают достаточно объемными структурами. Наступающее при подводе тепла, очевидно вследствие разрыхления направленных связей, сжатою помимо олова, наблюдается также и у других элементов, которые в периодической системе расположены вблизи границы между металлами и неметаллами, например, у Gc... Si, Ge, Bi и Те. Одиако у этих элементов сжатие не проявляется в твердом состоянии., а наступает только ыры плавлении, а в случае Те даже несколько выше температурю плавления.
По отношению к воздуху н воде олово при обычной температуре устойчиво. Однако при более высокой температуре оно окисляется. Пары олова легко и полностью сгорают, образуя двуокись. Со свободными галогенами олово образует тетрагалогениды. С хлором и бромом оно реагирует уже при обычной температуре, с иодом — при слабом нагревании. С фтором оно не реагирует заметно при обычной температуре, при 100°, напротив, очень энергично (с появлением пламени). Так же достаточно энергично оно реагирует при нагревании с серой, селеном и теллуром. С азотом олово непосредственно пе соединяется, а с фосфором реагирует при нагревании.
Разбавленные кислоты лишь медленно действуют на олово, что объясняется незначительной разницей его нормального потенциала и нормального потенциала водорода (ср. стр. 574). Лучше всего оно растворяется в крепкой соляной кислоте Sn -|- 2НС1 — SnCh + На. (Это уравнение выражает процесс только частично, поскольку для растворения в соляной кислоте большое значение имеет также тенденция олова образовывать хлорокомплексы, например [SnCls]'.) Энергично реагирует олово также с концентрированной азотной кислотой. При этом оно превращается в нерастворимый в воде белый порошок, ft-оловянную кислоту (см. стр. 577 и сл.). Гораздо медленнее взаимодействует олово с концентрированной серной Ы!СЛ()тог1. в которой оно растворяется с выделением SO а.
При кипячении с растворами едких щелочей олово переходит в раствор с образованием гидрокеостаннат-нотюв:
Sb (- 4Н2О+2ОН' = [Sn (ОН)6)" 4-2Нг.
Аналогичным образом олово переходит в раствор, если его используют в качестве анода, погрузив в концентрированный раствор едкого натра
Sn—4e-|-6OH' = [Sn (OH)S]".
Если, однако, плотность тока превысит определенную величину, то олово внезапно «пассивируется», т. е. ведет себя как неразрушающийся электрод (ср. т. II).
Важным для применения олова свойством является его способность легко образовывать сплавы с другими металлами.
Металлическое олово как практически нерастворимое вещество не ядовито. Одиако в старых и сильно кислых консервах могут содержаться растворимые соединения олова, вредные для человеческого организма.
Применение. До изобретения фарфора олово было основным материалом для изготовления кухонной посуды, тарелок, кружек и ковшей.
Четвертая группа периодической системы
573
В настоящее время почти половина производимого олова идет для изготовления белой жести.
При изготовлении белой жести листы железа ополаскивают разбавленной серпой кислотой и затем погружают в расплавленное олово. Вследствие незначительного действия кислот па олово оловянный покров защищает железо от коррозии. Но если покров поврежден, то па этом листе происходит более ускоренная коррозия железа, так как образуется гальвапцческий элемент, в котором железо является анодом.
При производстве белой ?кссти для консервных банок следует применять для покрытия только свободное от свинца олово, так как пе только сам свинец вреден, по и способность олова подвергаться действию раэбавлепных кислот (имеющихся в плодах" органических кислот) также повышается в результате содержания в нем свинца. Часто внутреннюю поверхность консервных банок, предназначенных для фруктов, для лучшей защиты покрывают топким слоем лака («вернпровка»), благодаря чему серебристо-белая поверхность становится золотисто-желтой.
Многие важные сплавы содержат олово, например бронзы, британский металл и мягкий припой. Последний содержит 40—70% олова и 60— 30% свинца. Подшипниковые металлы также являются большей частью сплавами олова. Употребляемые в настоящее время «изделия из олова», в частности органные трубы, содержат почти всегда в качестве примеси свинец. Это заметно уже по их темной в сравнении с чистым оловом окраске.
Б ританским металлом называют сплавы, состоящие в основном из олова с не-болвпшми добавками сурьмы (п в большинстве случаев некоторого количества меди), которые исполвзуют для изготовления предметов домашнего обихода, например столовой посуды, путем штампования.
Подшипниковыми металлам,и являются сплавы, из которых изготовляют под-шиппики для движущихся частей машин (валов и т. д.). Различают подшипниковые металлы белого литвя и красного лцтьл. Первые состоят на 50—90% из олова, 7—20% из сурьмы и большей частью (одержат несколько црещецтов меди. Сплавы, бедные оловом, содержат наряду с ним большие количества свинца. В медполитых подшипниковых металлах, преобладает медь (75—90%), олова в среднем 10% и еще меньше цинка иди свшща. После первой мировой войны вместо оловяппых подшипниковых металлов стали применять свинцовые подшипниковые металлы (ср. стр. 588).
О бронзах, которые также являются сплавами меди й олова, будет сказано при рассмотрении медц.
Применявшаяся ранее для упаковки^многих пищевых продуктов етанниолъ состояла из чистого олова. Теперь вместо нее чаще применяют тонкую алюминиевую фольгу.
Из соединений олова находят применение прежде всего двуокись олова, тетрахлорид олова, «розовая соль» (хлоростаннат аммония), «оловянная соль» (дихлорид олова), а также «сусальное золото» (дисульфид олова). Некоторые соли олова и органических кислот применяют при окраске тканей, например ацетат олова Зн(СШзОа)г, и роданид олова Sn(SCN)a, в качестве восстановителя при протравном печатании; оксалат^олова ЗпСзОг применяют как протраву.
СОЕДИНЕНИЯ ОЛОВА
Олово может быть в своих соединениях:
1) отрицательно четырехвалентпым или электронейтральным и формально четырех валентным;
2) положительно четырехвалентным;
3) положительно двухвалентным.
Возможно, отрицательно четырехвалентное олово существует в водородном соединении олова SnIIr. Однако, вероятно, это чисто гомеополяр-
ное соединение. Алкильные соединении олова также, по всей вероятности. построены гомеополярно.
Как и у углерода, у олова известны соединения, которые в разбавленных растворах распадаются на свободные радикалы ЯпКд с т ре хвале нтным оловом. Полагают, чтц в неорганических соединениях, ио крайней мере кратко временно и в незначительном количестве, олово ташке моя:ет присутствовать в трехвалентном состояние (Ball, 1935).
Положительно четырех валентное олово может существовать в виде-элементарного катится Sn"". Однако в этом состояний оно преимущественно образует анионы, например [SnCle]", |Sn(OH)6]", [SnO^]", и [SnSsJ". Эти иопы присутствуют в гексахлоро-, гексагидроксо-, триоксо- и тритио-станнатах(] V). Пос ле дине большей частью ( известны как станнатыЦУ) или тиастаннатыА/У).
Ь положительно двухвалентном состоянии олово существует в солях в основном в качестве катйопа Sn". Комплексные анионы, производимые от двухвалентного олова, образуются преимущественно с галогенами \галогеностаннаты(/1)]. Также возможно присоединение ионов гидроксила к Sn(OH)s с образованием гидроксостаннатов(1Г).
Ионы Sn" и Sn"" и растворе бесцветны, так же как и их соли, если другие составные частя солей не окрашены. Также и комплексные ионы, производные от олова, в общем случае бесцветнее Некоторые бинарные соединения олова окрашены. Так, моноокись — сине-черно г о цвета, моносульфид — коричневого, дисульфид — желтого.
Соединения олова(П) обладают тенденцией переходить в соединения олова(1¥), как в кислом, так и в щелочном растворе. Поэтому их часто применяют в качестве восстановителя.
Стандартный окнслнтсльно-посегагговягелбнгчй нотенврвл Зп-'/За"’’, втиесеиныИ к нормальному водородному электроду, при 25° составляет +0,154 в (Huey, 1934). Следовательно, платиновая пластинка, погруяптгвая в раствор, содержащий равные количества ионов o tona(H) ir олова(1¥), обладает на 0,154 в более высоким потенциалом, чем иормалi.iiводородный электрод. При включении ток идет ио проволоке от э.токтродн, 1Ю1 ружожого в раствор Sn”/Sn"", к водородному Электроду. При этом у водородного электрида водород переходит в раствор в виде ионов Н‘, в то время как у платинового электрода эквивалентное количество ионов Sa...
исчезает, перехода в ионы Su".
Потенциал еа.иого олова, находящегося в соприкосновении с 1 М раствором ионов олова(Н), т. е. нормальный потенциал олова, составляет —0,158 в (Prytz, 1934). В этом Случае ток идет к олову. Опо переходит в раствор в виде двухвалентного йопа, в то время как на другом электроде разряжается водород. Способностью олова разряжать ионы Н‘ объясняется растворимость олова в кислотах. Однако вследствие незначительного отличия его нормального потенциала от нормального водородного потенциала олово обладает лишь весьма слабой тенденцией к растворению. Поэтому оно обычно заметно взаимодействует только с такими Кислотами, которые, как НС.1, могут увеличить эту тенденцию за счет комплексообразован ня.
Соединения олбва(ГУ)
Гидрид олова S11TI4. Существование этого водородного соединения олова было обнаружено в 1919.г. Панетом (Paneth), который вначале получил его разложением сплава олова с магнием (сидерящщого соединение Mg2Sn) 4 н. соляной кислотой. Позднее оп приготовил ото соединяли» с лучшим выходом катодным восстановлением растворов солей олова на электродах с перенапряжением (лучше всего на свинцовых электродах).
В каждом случае выделяется главным образом водород. В иезначгшщьном количестве к нему, однако, примешано водородное соединение олова", проще всего это можно показать, пропустив профильтрованный через вату газ сквозь раскаленную стеклянную трубку. На стенках последней образуется металлическое зеркало, которое на
Четвертая группа периодической системы
575
основании его реакций можно идентифицировать как оловяппое зеркало. Например( мри обработке такого зеркала газообразным хлористым водородом образуется дихло-рнд.. который дает характерные реакции с раствором хлорида золота (образование кассиевого золотого пурпура, ер. стр. 583), а также с хлоридом ртути (выделение кадоме л и), Это зеркало к тому же в противоположность металлическим зеркалам, возникающим при разложении водородных соединений мышьяка, сурьмы и висмута, не растворимо в холодной концентрированной азотной кислоте и напротив, оно легко растворимо в холодной концентрированной соляной кислоте. Позднее Палет конденсацией жидким воздухом собрал несколько большие количества чистого гидрида олова и определил его точку плавления (—150s) и точку кипения (—52°), а также состав (тем же способом, как п в снучае германоводород а). В настоящее время гидрид олова удается довольно просто приготовить взаимодействием SnCl4 и 1дА1Щ.
Гидрид олова — сравнительно устойчивое соединение. Это довольно необычно, если учесть трудность его образования. В чистом стеклянном сосуде при обычной температуре он может сохраняться в течение нескольких суток. Однако следы металлического олова чрезвычайно ускоряют разложение. Оно тотчас наступает при нагревании до 145—150°. Гидрид олова можно пропустить без разложения через растворы щелочей умеренной концентрации, а также через разбавленные кислоты и большинство растворов солей. Однако растворами нитрата серебра и хлорида ртути (II) он разлагается, а также твердыми щелочами, натронной известью и концентрированной серной кислотой. Гидрид олова очень ядовит.
Тера фторид олова и фторостаннаты(Л’). Тетрафторид слова S«F4 можно Приготовить, по данным Руффа, щцюениом тетрахлорида олова в безводную плавиковую кислоту и умеренным нагреванием последней. Чтобы реакция SnCl4 + 4HF фт jT. SbF4 -|- 4HG1 протекала слева направо, необходим избыток плавиковой кислоты. В качестве промежуточного продукта образуется двойное соединение SnF4-SnCl4, которое разлагается в интервале 130—220°, выделяя чистый SnF4.
Тетр а фтор ид олова образует бесцветную, кристаллическую массу л у чистого строения. Уд. вес 1,78, т. кпп. 705°. С водой соединяется с шипением вследствие сильного выделения тепла. Его раствор проще получить растворением свежООсаждепнЦго гидрата окиси олова(1У) в водном растворе плавиковой кислоты. Есин этот раствор испарить, то останется рединообразная масса, содержащая воду. Напротив, из раствора можно получить легко кристаллизующиеся двойные фториды — фтвростанна-ты. Последние были особенно подробно исследованы Мариньяком. Почти все фторо-стаппаты соответствуют типу M^ShFcI (гексафторостаппаты(1Г) Однако с WH4F, помимо топ), образуется соль состава (NH4)4SiiF8. Кроме соединения K^ShF^-H^O, Мариньяк получил е К F из крепкого раствора плавиковой кислоты соединение KgHSnF^ в виде моноклиппых игл.
Тотрахлорид олова и улороетаимэты(1У). Тетрахлорид олова [хлорид олова(/У)] SnCl4 в технике получают главным образом обработкой отбросов белой жести хлором. ЙпС14 представляет собой бесцветную дымящую на воздухе жидкость (Spiriltitt fumans Libavii) удельного веса 2,229 (при 20°), которая застывает при —36° и кипит при 114°. С сероуглеродом SnCl4 смешивается во всех отношениях и может растворять фосфор, серу, иод, трииодид мышьяка и тетраиодид олова.
В иоде тетрахлорид олова растворяется с сильным разогреванием. Из раствора и зависимости от условий кристаллизуются различные гидраты. Пептагндрат SnCl.,-5H2O (устойчив между 19 и 56°) образует белые, непрозрачные расплывающиеся кристаллы; его температура плавления около 60°. В воде легко растворим.
В водном растворе S[iCl4 подвержен сильному гидролизу, который протекает главным образом согласно реакции: SnCl4 + 2Н2О = SnOa4-+ 4HCI. Образующаяся при этом двуокись олова остается в растворе
576
Глава. 12
в коллоидном состоянии. Выделившаяся одновременно соляная кислота присоединяется к частично нер аз ложившемуся тетрахлориду олова с образованием гексахлорооловянной кислоты Нй [SnCle J. Эту кислоту можно осадить, если в концентрированный водный раствор тстрахлорида олова пропустить газообразный хлористый водород. Она кристаллизуется в виде листочков состава НгЗпС10-6Й2О с точкой плавления 19,2°. Соли, которые производятся от этой кислоты, называются гексахлоростаннатами(1У), или, короче, хлоростаннатами. Им соответствует общая формула M*[SnCl6], где М1- — щелочной металл или эквивалентное количество двух- или трех-валептного металла (о строении решеток хлоростаннатов щелочных металлов см. стр. 524). Техническое значение имеет хлоростаннат аммония (NH4)3[SnClfil, бесцветные октаэдры удельного веса 2,387, легко растворимые в воде. Эту соль называют ташке «розовая соль», «пинкзальд», так как ее применяют в красильном дело в качестве протравы. Водный раствор этой соли в противоположность раствору тетрахлорида олова обладает нейтральной реакцией. Достаточно концентрированный раствор ее не разлагается даже при кипячении.
Тетрахлорид олова может присоединять также многие другие вещества, например NH3, РН3, РС1Е, РОС13, SC14, NOC'i, органические сульфиды H2S и эфиры И2О
Как установил Пибергалл (Niebcrgall, 1951), SnCl4 очень быстро и полностью взаимодействует с С3Н51 в присутствии А1С13 в качестве катализатора, образуя SnI4(SnCl4 -У 4С,Н61 = Sal4 4~ 4С2Н5С1). Следует отметить, что в случае СеС14 аналогичное взаимодействие происходит очень медленно, а в случае S1CI4 — вообще по происходит.
Тетрахлорид олова применяют как протраву при крашении тканей. Кроме того, он служит для утяжеления шелка, в качестве катализатора при хлорировании, а также как конденсирующее средство в органической химии. Тетрахлорид олова является также главной составной частью применяемой при крашении тканей «оловянной протравы», которую готовят растворением олова в царской водке.
Тетра бромид олова и бромостан на ты (IV). Тетрабромид олова SnBr4, получаемый щлюсредствепным соединенном олова и брома, представляет собой белоснежную, кристаллическую массу, уд. вес .3,35 (при 35°), г- пл. 33°, т. кип. 203°. Из водного раствора при обычной температуре кристаллизуется в виде тетрагидрата SnBr4-•4НиО. Гексабромооловянная кислота (Л7) [SnBr6}-8HaO образует белые, бесцветные, расплывающиеся призмы. Известны также ее соли гексабромостаццаты(1У) MhSnBre|.
Тетра под ид олова Snl4 получают непосредственным взаимодействием: олова с иодом (который лучше всего растворить в сероуглероде) или осаждением из Концентрированного раствора SnCl4 под действием KI; он образует сильно светопреломляющие октаэдрические Кристаллы, окрашенные от желтого до желто-коричневого цвета. Они нс изоморфны кристаллам тетрабромида олова. Уд. вес 4,46, т. пл 146°, т. кип. 346°. Тетраиодид олова легко растворим в метилениодиде СН212, сероуглероде, спирте, эфире, бензоле и других органических растворителях, а также в трибромиде мышьяка. Насыщенный раствор тстраиодида олова в трехбромистом мышьяке отличается особо высок им удельным весом (3,731 при 15°). Определение точек замерзания таких растворов дает вдвое большое значение депрессии, чем соответствовало бы простому молекулярному весу. Это явление нельзя свести к электролитической диссоциации, так как растворы практически не проводят электрический ток. Поэтому следует предположить, что здесь происходит диссоциация по схеме: Snl4 Z Snb -f-I2-
Вследствие гидролиза тетраиодид олова не образует прозрачного водного раствора. Поэтому иодостаннаты. нельзя получить из водного раствора. Однако Розепгейм выделил их из спиртового раствора. Тетраиодид олова образует молекулярную решетку. Каждый атом Sn тетраэдрически окружен четырьмя атома да I. Такой же структурой обладает Gel4. Расстояния: Sa «—> I равно 2,65 A; Ge «—> I равно 2,57 А- В молекулах газа соответствующие расстояния составляют 2,64 и 2,48 А.
Четвертая группа периодической системы 577
Алкильные соединения олова. Нагреванием олова с иодистыми алки-ламп можно получить дпалкилиодиды олова. Например, с иодистым этилом олово реагирует с образованием диэтцлиодида олова
I. ZC2HS
Snb2C2H5I= /Sn\
I' сан5
Действуя па последний диэтилцинком Zn(CaH5)2, можно получить тетра-этилолово Sn(C,HJ4. Удобнее исходить из галогенидов олова, замещая в них по методу Гриньяра все или часть атомов на органические радикалы. Таким образом можно получить большое число алкилов (а также арилов) олова, которые в некотором отношении имеют большое сходство с соответствующими соединениями кремния (ср. также стр. 573 и сл.).
Алкильные соединения олова проявляют значительно меньшую способность к образованию продуктов присоединения, чьи галогениды олова, причем способность к присоединению тем ыепьше, чем больше атомов галогена замещено па алкилы.
Если смещать вод Но-спиртовый раствор диэтилиодида олова с аммиаком, то выпадает окись дитпиломва (C2H5)2SnO в лиде белого осадка, растворимого в кислотах с образованием солей; следовательно, диэтилоксид олова ведет себя по отношению к сильным кислотам как основание. То л;е относится к гидроокись тривтилолова (C2H6)3SnOH.
Двуокись олова [окись олова(1У)] SnOa встречается в Природе в виде тетрагонально кристаллизующегося оловянного камня (касситерита). Но, кроме того, ЙпО2 может также существовать в ромбической и гексагональной модификациях. Следовательно, она является триморфной. Нагреванием ее гидратов или оксалата олова получают «аморфную» двуокись олова в виде порошка, кристаллическая структура которого не установлена. Чистая двуокись олова имеет белый цвет. Она сублимируется выше 1800°, но плавясь, в воде нерастворима. Кислоты и растворы щелочей па Нее почти нс действуют. Однако ее легко удается перевести в растворимое состояние сплавлением с гидроокисью щелочного металла или со смесью соды с серой. В первом случае она переходит в стан-uam(IV\, в последнем — в тиостаннат{1У):
SnO2+2NaOH=Na2SnO3 -f-HaO. (3)
Sn02+2Na3C03-[-4S«Na2SnS3-|-Na2S04+2C02. (4)
При прокаливании с углем или при нагревании в токе водорода SnOa восстанавливается до металлического олова. При прокаливании в токе хлора образуется тетрахлорпд олова.
Двуокись олова широко применяют в технике для приготовления различного вида белых глазурей и эмалей. Кроме того, ее используют в качестве полировального средства. Получение ее в большинстве случаев ведут е/кигапием нагретого до высокой температуры олова в токе воздуха. Природная двуокись олова (чаще всего окрашенная примесями в коричневый цвет) является исходным материалом для получения металлического олова.
Тетрагональная двуокись олова имеет такую же кристаллическую структуру, как ц фторисп.гй it аг шг it (^структура рутила», ср. рис. 63, стр, 298) a *= 4,72, с = 3,17, d = 2,06 А.
'Оловянные кислоты и станнаты(1У). Из концентрированного водного раствора сплава SnOa и NaOH кристаллизуется легкорастворимая соль состава NaaO-SnOa-ЗИаО( в виде гексагональных табличек. Эта соль под названием препаратной соли находит техническое применение для 37 г. Ремп
578
Глава 12
подготовки тканей к крашению протравными красителями. Беллучч; и Парравапо (Bellucci, Parravano, 1905), учитывая прочную связь вп— в атом соединении, приписали ему строение Na2 (Sn(OH)eJ. В соответствии с этим Эту соль следовало бы обозначить как гексагидроксоста^ Ham{IV) натрия. Помимо натриевой солн, существуют такте еще друге;; соединения этого типа, например К2 [Sn(OH)e], Са [Sn(OH)6J, Sr(Sn(OH)eL, Pb [Sn(OH)e]. Три последние, нерастворимые в воде, получают взаимодействием гексагидро кс о ст а ин ат а калия с растворимыми солями кальциту стронция или свинца. Эти реакции, так Же как и рентгепоструктуршк исследование (Wyckoff, 1928), подтверждают представление, что в основе сгацпагов, полученных из водного раствора, лежит радикал [Sn(OH)6I^„
Кислота П,8п(ОН)6, соответствующая этому радикалу, в свободном состоянии не выделена. При добавлении к растворам щелочных (гйДроксо-J, стапнатов небольших количеств разбавленных сильных кислот или при пропускании в них двуокиси углерода получаются объемистые белые осадки, которые растворяются в избытке сильных кислот, а также в растворах щелочей. Осадки имеют характер еелей и содержат переменные количества воды. Соответствующие осадки получают также при добавлении аммиака или карбоната аммония к растворам тетрахлорида> олова. Так же как и в случае гидратов кремневой кислоты, еще не установлено, содержат ли свежеосажденные, не обнаруживающие кристаллической структуры осадки химически связанную воду или они являются лишь гелями двуокиси олова. Когда в результате старения (после продолжительного стояния в соприкосновении с раствором или при нагревании) становится возможным рентгенографически обнаружить их кристаллическую структуру, они дают дебаеграммы, характерные для двуокиси олова. Одновременно они становятся все более и более нерастворимыми в кислотах. Обычно эти осадки называют «оловянными кислотами», причем растворимые и нерастворимые в кислотах формы различают как а- и р-оло-вянные кислоты, или как «обычная оловянная кислота» и «метаоловян-пая кислота». Последнюю непосредственно получают при обработке металлического олова концентрированной азотной кислотой.
Нюкущссся растворение р-ияовяцпой кислоты происходит в результате ее обработка концентрированном соляной кислотой с последуюпщм сильным разведением. Одиако при этом происходит по нстикпое растворение, а образование коллоидного раствора вследствие пептизации под влиянием соляной кислоты. Под действием концентрированной соляной кислоты (а также других сильных электролитов) p-о л о винная кислота снова выпадает. Напротив, из раствора а-оловянной кислоты в концентрированной соляпой кислоте мо;кпо отогпать тетрахлорид олова.
Помимо различного поведения в отношении кислот, а- и р-оловянные кислоты различаются также следующими реакциями; а-оловяниая кислота легко растворима как в растворе карбоната калия, так и в растворе едкого кали любой концентрации; (Половинная кислота в растворе карбоната калия пе растворима. Она не растворима также в концентрирован гои едком кадя. Если такой растлор разбавить, ^-оловянная кислота переходит (коллопдпо) в раствор. Из этого раствора не удается получить никакой кристаллической соли. Упариванием щелочных растворов fi-оловянной кислоты получают аморфные вещества, которые прежде принимали за соли «метаодовяп-ной кислоты» и называли «метастаппатами», однако в действительности эти вещества являются лишь продуктами адсорбции щелочи водной двуокисью олова. Из солянокислых растворов (соответственно коллоидных’растворов) сульфат ка.чпя или натрия осаждает на холоду только (5-оловянную кислоту, но пе а-ол о ванную кислоту, а-Оловянная кислота в виде геля прочнее удерживает воду в связанном состоянии, чем Р-оловяппая кислота, и обладает большей адсорбирующей способностью, например, по отношению к фосфорной кислоте п органическим красителям, чел/ р-олиляпная Кислота.
Различные свойства обеих модификаций оловянных кислот были обнаружены еще Берцелиусом (1817), который ввел в связи с этим в химию понятие изомерии.
Четвертая группа периодической системы
579
Согласно Мекленбургу (Mecklenburg, 1909), существ мшые различия в поведении а- и р-оловяпных кислот мол;п<> объяснить, если принять, что оба эти вещества являются различными коллоидными модификациями нерастворимой в воде двуокиси олова или ее гидрата/;-, которые различаются величиной своих частиц. Обычная оловянная кислота (а-оловяппяя кислота) состоит я соответствии с этим из сравнительно мелких частиц, Р-оловяипая кислота — из сравнительно крупных частиц (которые все же находятся за пределами разрешающей способности ультрамикроскопа). Такое представление базируется на непрерывности перехода одной формы в другую при старении гелей. В дальнейшем Мекленбург смог показать, что образующаяся при гидролизе сульфата оловаЦУ) при 0° оловянная кислота по своим свойствам приближаете я к а-оло-вянпОй кислоте; одпако при повышении температуры реакции все более и более проявляются типичные свойства Д-оловяппой кислоты. 9 то подтверждает, что р-ол о влипая кислота состоит из более крупных частиц, так как в общем случае осаждение, идущее при нагревании, приводит к образованию более крупных частиц, чем на холоду.
' Однако сомнительно, чтобы различия между а- и р-оловянныыи кислотами обусловливались только различной величиной частиц. Вильштеттсру (Wills Latter, 1924} удалось получить, используя подходящие способы приготовления и высушивания (’высушивание ацетоном, ср. стр. 394), препараты, которые содержат SnO2 и НЙО в простых стехиометрических соотношениях п обнаруживают в широких температурных интервалах однородный состав. Наиболее активная модификация, осаждаемая аммиаком в присутствии хлорида аммония из растворов SnCl4, при высушивании ацетолом в температурном интервале от —35 до —10° имеет приблизительный состав SnO2-3H2O. После обработки ацетоном при более высоких температурах опа отдаст остальную воду достаточно непрерывно. Напротив, у препаратов, которые значительно дольше были в соприкосновении с водой, т. е. уже несколько «постарели» (и их растворимость в кислотах, прежде всего в азотной и серной, уже отчетливо уменьшилась), при высушивании ацетоном было обнаружено постоянство содержания воды в интервале 0—15° при составе, приблизительна соответствующем 2SnCU'сНаО, и во второй раз в интервале 30—56° (температура кипения ацетона) при составе, соответствующем формуле 45лО2-5Н.,О. На основании этого Вильштеттер заключил, что при «старении!) происходит конденсация, т. е. соединение простых молекул л более сложные с отщеплением воды. В соответствии с данными Зигмонди следует считай., что при переходе о-кислоты в р-кислоту имеют значение одновременно оба процесса: конденсация (по Вилыптсттеру) и укрупнение частиц — полимеризация (но Мекленбургу).
Вильштеттер установи,.] далее, что высушенная ацетоном нри комнатной температуре а-оловянная кислота при последующем высушивании в высоком вакууме пад Ш1ТИ0КИСЫ0 фосфора теряет столько воды, что ее состав выражается формулой H2SnO3 и что она сохраняет этот состав в токе сухого воздуха до +65°. Позднее (1931) Тиссен (Thiessen) и Симой (Simon) независимо друг от друга сделали заключение на основании ступеней (правда, не очень ярко выраженных), полученных па изотермах и соответственно изобарах обезвоживания гелей двуокиси олова, о существовании соединения определенного состава — H2SnO3. То, что это соединение так легко разлагается на SnOg и НаО, обусловливается, по мнению Симона, так же как и в случае кремневых кислот, незначительной теплотой образования в нем связи с водой (около 30 ккал/моль). Согласно Тиссену, помимо SnO2'H20, должны существовать также другие гидраты двуокиси олова в кашютве определенных соединений. Рентгенографически ни один цз гидратов двуокиси олова не обнаружен. Их существование поэтому оспаривается Вейзером, (Weiser, 1932).
Из безводных солей оловянной кислоты в структурном отношении исследованы соединении Mg2SnO4, ZnaSnO4 и Co2SnO4. Они образуют решетку шпинели (см. стр. 395 и ел,), и поэтому их можпо считать ортоста/татами, Соответствующие препараты, имеющие состав метастаннатов Мг|8яО3, на основании рентгенограмм оказываются смесями ортсчрп,mu,im.oe с SnOa (Natta, 1929; Taylor, 1930).
Гели оловянной кислоты или двуокиси олова применяют в ситцепечатании, при крашенин ализариновой красной краской, для приготовления оксалата олова, который также используют при крашении тканей.
Дисульфид олова и тиостаннаты(1У). Дисульфид олова, сульфид' олова{1'¥) SnS.. выпадает в виде желтого осадка из не слишком кислых растворов солей олова (IV) при пропускании сероводорода или из растворок тиостаннатов при добавлении кислот. В технике ого получают большей частью сухим путем, например нагреванием смеси оловянной фольги (или,
580
Гаана 12
еще лучше, истолченной оловянной амальгамы) с серным цветом и нашатырем. Таким способом дисульфид получают в виде золотисто-желтых чешуек гексагональной системы, удельного веса около 4,51, Он поступает в продажу под названием «сусального золота» (Aurum mosaicum, т. е. золото для мозаичных работ) и в виде «оловянной бронзы» его применяют для бронзирования.
Дисульфид олова кристаллизуется по тину бруцнта (рис. 61, стр. 293); а ~ ~ 3,62, с = 5,85 A; Sn Su — 2,55 А.
Тогда как дисульфид олова, приготовленный сухим путем, нерастворим в соляной и азотной кислотах, дисульфид, осажденный из водного раствора, растворяется уже в умеренно концентрированной теплой соляной кислоте
ShS24-4HC1 [ГТ SiiC144“2H2S.
' Он легко растворяется в растворах сульфидов щелочных металлов, образуя легкорастворимые тиостаниаты(1У) щелочных металлов, например тиостаннат (IV) аммония (NH4)2 [SnS3]. Образование тиостанпатов (IV) основано па том, что SnS3 может присоединить еще дополнительные ионы S". При присоединении одного иона S" образуются тритиостаннатЦУ)-ионы ISnSg]") при присоединении двух ионов S" образуются тетпратпио-станна,т(1У)-иолъ1 [SnS4]"".
SnS2+S' = [SiiS3r; SiiS2-j-2S'' = [SnS4]"" (5а и 56)
Тио с тапнаты( IV) образуются также при растворении коричневого моносульфида олова в полисульфидах щелочных металлов
SnS-f-S-j-S* = [SnS3]".
Дисульфид олова растворяется также в растворах гидроокисей щелочных металлов. В атом случае наряду с ионами тиостанната(Г^ образуются ионы гидроксостанпата(1Л7)
3SJJ 8,+6011' = 2 [Sn8s]" 4- [Sn(0H)e]". (6)
Процессы, представленные уравнениями (5а и 56) и (6), обратимы. При подкислении растворов тиостаннатов(ТУ) снова выпадает дисульфид. Это становится очевидным, если к указанным уравнениям применить химический закон действия масс, причем в случае уравнения (6) непосредственно, а в случае уравнений (5а и 56) учитывая, что из-за незначительной диссоциации сероводорода концентрация ионов S'' в кислых растворах очень незначительна.
Т1юстаннат1,т(1 V) щелочных металлов кристаллизуются с большим содержанием воды. Джелли (Jolley, 1935) нашел, что одна часть воды ири нагревании удерживается значите л г, но сильнее, чем другая. Отсюда он сделал заключение о комплексном характере связи этой части воды в апионах соли и придал поэтому аниону содержащего воду тритиостипнатаЦУ) формулу
(8и8Е(ОН2)йр’ или [Sa(OH)8(SH)sJ3~,
а аниону содержащего воду тетратиостанпата(ГУ)—[SnS4(OHa)2p- или [Sn(OH)2S2‘ Подобную же формулу вывел Принтйингер (Brin[zinger, 1934) для тетра-тноатаннат(1У)-иона в водном растворе путем определения лоппаго веса методом диализа. Для тетратиоарсенат(У)- п тетратиоаптимопат(V)-попов он также определил ионный вес, соответствующий днакво-иоцам.
Сульфат олова(1У) выделяется из раствора свежее с аждеип ото гидрата двуокиси слова в разбавленной серной кислоте в виде бесцветных игл состава Sn(SO4)2-2IIaO.
Четвертая группа периодической системы 581
Соединения олова(П)
Хлорид олова(П), дихлорид олова, SnCL, в безводном состоянии в виде белой массы с жирным блеском получается при нагревании олова в токе хлористого водорода. Удельный вес его 3,95, температура плавления 247°, температура кипепия 605°. Плотность паров хлорида олова(Н) вблизи его точки кипения указывает па частичную ассоциацию молекул, которая исчезает лишь выше 1100°. Напротив, его раствор в уретане показывает, в соответствии с данными Касторо (Castoro), понижение точки замерзания, отвечающее простой молекулярной формуле.
В воде, а также в спирте, эфире, ацетоне и этилацетате хлорид олова(11) легко растворим. Из водного раствора он кристаллизуется с водой (SaCl2-2H2O) в виде прозрачных, пе расплывающихся, моноклинных призм или бипирамид с температурой плавления 40,5° и удельным весом 2,710. Это и есть торговая оловянная соль. Техническое получение ее осуществляют растворением оловянных стружек в соляной кислоте. Раствор соли в небольшом количестве воды прозрачен, однако при разбавлении раствор мутнеет вследствие выделения основной соли SnCl2 + 4- Н2О = Sn(OH)CI -j- HCI. При нагревании в токе хлористого водорода до температуры красного каления дихлорид олова, содержащий кристаллизационную воду, можно обезводить.
Дихлорид олова — сильный восстановитель. Он осаждает золото и серебро из растворов их солей в виде металлов. SnCl2 может также выделять в виде металла ртуть или, если его количество недостаточно для этого, восстанавливать соли двухвалентной ртути До солей одновалентной ртути. Далее, он восстанавливает соли железа(Ш) до солей железа(П), арсенаты — до арсенитов, хроматы — до солен хрома(П1), перманганаты — до Солей маргапца(П), нитросоединения — до аминов, соли диа-зония — до солен гидразина. В водном растворе оп медленно окисляется кислородом воздуха
5SnCl3-j-V202-LH2O=SiiCl44~2Sn(0H)Cl.
Добавление металлического олова к раствору препятствует этому окислению .
Дихлорид олова находит широкое применение. Помимо качественного и количественного анализа, его используют в технике в качестве восстановителя для органических нитро-, азо- и диазососдинепий, и особенно в ситцепечатании в качестве протравы. Кроме того, он служит для приготовления лаковых красок.
При нагревании в токе аммиака сухой дйхлорид олова присоединяет одну молекулу NM3. Нагреванием в токе хлористого водорода Энгель получил комплексную кислоту HSnCJa,3H2O в виде жидкости, затвердевающей около —27°. Из растворов SnCl2 при добавлении хлоридов щелочных металлов были получены хлоростаннаты(ГГ) (МЕ [SnCl3] и [SnClJ) в кристаллическом состоянии.
Остальные галогениды олова(П) весьма близки к хлориду олова(П). Фторид оловаЦГ) — белые, мыюклйнные призмы, образующие в поде прозрачный раствор. бромид олова!!/} — слегка желтоватая масса; уд. вес 4,92, т. пл. 232°, т. кип. 619е. Иодид «лсвй(/7) — орэнжево-краспые октаэдры; уд. БСС 5,28, т. пл. 320°, т. кип. 720’, Эти галогениды образуют комплексы, соответствующие Комплексам дихлорида; правда, Для иодида пока известны лишь комплексы типа M4SnI3|.
Окись олова(II) и гидроокись олова(II). При добавлении к раствору соли двухвалентного олова карбоната щелочного металла или небольшого
582
Глава 12
количества гидроокиси щелочного металла выпадает гидроокись олова[К Sn(OH)3 в виде белого очень труднорастворимого в воде осадка Sn" — -j- 2ОН' = Sn(OH)2. После осторожного высушивания она обладгцц-составом, соответствующим формуле. При соприкосновении с pactкоряга солей олова(П), или со щелочами этот осадок легко переходит, особенн. при повышенной температуре, в темную безводную окись олова(1Г) (моноокись олова) SnO. Содержащий воду белый осадок растворим как в кислотах, так и в едких щелочах. В первом случае образуются соответствующее соли олова(П), например, при растворении в соляной кислоте — хлорцг олова(П): Sn(OH), !- 2НС1 = SnCl3 + 2Н3О; при этом гидроокись оло-ва(П) ведет себя в отношении кислоты как основание. По отношении: к сильным основаниям она, напротив, ведет себя как ангидрокислота в соответствии с уравнением
8н(ОН)24-ОН' [8п(ОН)31'.
Константа равновесия К для процесса Sn(OH)2 -f- НОН 8д(О11)з + И* по Гольдшмидту
К — *_[Н i = 4 10_ 10
[8п(0НЫ
Соли, производные гидроокиси олова(П), функционирующей в качестве ангидрокислоты, называются гидроксостпаннагпами^П). (Раньше их называли станнитами или гидроксостаннитами.) Растворы этих солей легко разлагаются, причем из них осаждается безводная окись SnO в виде темного порошка. Поэтому только в последнее время из них удалось получить кристаллические соли (Scholdcr, 1933),
Шолдер получил Na[Sn(OH)s), BafSn(OH)3l2, Ba[Sn(OH)3l2-2H,2O и Sr[Sn(OH)3)2-•2Н2О в “виде бесцветных кристаллических соединений. Щелочноземельные тригидрокси станпаты(П)^п р и нагревании до 100—110“ переходят прежде всего в желтые ц-оксотетрагпдроксодиста1гтгаты(1Т) Мп [(I[O)2Sr)—О—Зп(ОН)2]. При более сильном нагревании из них получаются безводные стататы(1 /) MII[SnO2J. р.-0 квот етр аги д-роксоднстаппагы(ТТ) получаются также непосредственно из подогретых растворов. Стипнаты(П) Ca|’SnO2J, Sr[SnOa] и Ba[SnO3( можно приготовить также действием SnO в парообразной форме па нагретые до высокой температуры окисли щелочноземельных мма..1„iob (Tamaru, 1931).
Для приготовления безводной моноокиси олова можно использовать метод Сэндэл-ла (Sarrdall): дихлорпд олова растирают с небольшим избытком карбоната натрия, смесь нагревают при помешивании на песчаной бане, пока она не почернеет, и затем тщательно промывают сине-черный мелкий порошок моноокиси олова горячей водой.
Моноокись олова (а-SnO) имеет такую же структуру решетки, как красная окись свинца (см, стр. 591). Около 550’, если нагревание вести в вакууме, опа превращается в другую модификацию (р-SnO). При нагревании на воздухе до красного каления она сгорает, превращаясь в двуокись олова. Гидроокись олова(П) дает рентгенограмму, отличную от ЗпО. Из итого следует, что Sn(OH)2 — Другое соединение, а не просто гель SnO.
Относительно термического поведения SnO см.; Spandau Н., Z. anorg. Chem., 254, 65, 1947.
Вследствие склонности к переходу в соединения олова(ТУ), гидроокись олова(П) и растворы гидроксостаннатов(11) действуют как сильные восстановители. На этом основано их техническое применение, например при кубовом крашении и протравном печатании.
Сульфид олона(П), моносульфид олова, SnS образуется при непосредственном соединении олова с серой и выпадает в виде темно-коричневого осадка из растворов солей олова(П) при пропускании сероводорода.
Четвертая группа периодической системы
583
Осадок нерастворим в разбавленных сильных кислотах. В бесцветном сернистом аммонии он также не растворяется, однако растворим в желтом сернистом аммопии.
Так как Зеу^валеитгтое олово обладает меньшей склонностью к образованию кислот, чем четпыретвалентное, тио соли от Звугвалоптного олова ие производятся. Поэтому образом пи t> тиосолей и связанное с этим растворение происходит только в том случае, если, помимо иопов S", присутствует нейтральная сера (как в случае желтого сериистого аммония), которая может окислить двухвалентное олово до четырехъ алепт по го. Если подкислить иолучеппый таким образом раствор тиосоли, то из него не выпадает вновь коричневый сульфид олова(П), а вследствие происшедшего окисления выпадает желтый сульфид олова(1У).
В токе водорода моносульфид олова сублимируется без разложения, образуя кристаллы в виде листочков с металлическим блеском удельного веса 5,27, температурой плавления 882°, температурой кипения 1230е.
SnS кристаллизуется в ромбической системе. Его решетку можно рассматривать как сильно деформированную решетку каменной соли. Каждый атом Sn окружен шестью атомами S и каждый атом S шестью атомами Sa. Однако образующийся таким образом вокруг каждого атома октаэдр сильно искажен: три угла его гораздо ближе к расположенному в середине атому, чем три остальные угла. Таким образом, получается слоистое строение, которому соответствует образование кристаллов в виде листочков (Hoffmann, 1935). Аналогичной структурой обладает двойное соединение PbS-SnS (теаялит).
Сульфат олова(П), согласно Кассету, кристаллизуется из раствора олова в смеси 1 об. концентрированной серной кислоты, 2 об. концентрированной соляной кислоты и 3 об. воды в виде белых легко растворимых в воде игл.
Нитрат олова(Н). Действием разбавленной аэотцой кислоты па олово получают смесь нитратов олопа(ТГ) п oJrOBa(IV). Чистый нитрат одова(П) можно получить, по Веберу (Weber), из раствора мопоокиси олова в азотной кислоте уд. веса 1,2 при охлаждении до —20° в виде белых, очень сильна расплывающихся листочков состава Sn(NC>3)2-20H20. Это соединение чрезвычайно легко разлагается. Несколько устойчивее основной нитрат 6тт2О(Г\тОз) 2, который кристаллизуется без воды и может быть пагрет до 100° без разложения. Однако при более сильном нагревании, особенно при быстром нагревании, а также При ударе и растирании он Детонирует с сильным треском.
Аналитические еяеденяя. Так как олово осаждается сероводородом из кислых растворов и его сульфиды растворяются в желтом сернистом аммонии, то оно вместе с мышьяком и сурьмой относится к группе элементов, образующих «.кислые сульфиды», т. е. сульфиды, способные к образованию тиосолей. Степень окисления олова нередко можно обнаружить уже на основании окраски сульфида, осажденного сероводородом (SnS — коричневый, SnS2 — желтый). Кроме того, присутствие солеи олова(П) обнаруживают ио их восстановительным свойствам. Вследствие этого соли олова(П) удается также легко определить количественна, например, при^'помощи перманганата калия или раствора иода.
Очень чувствительной реакцией на соли олова является добавление нескольких капель раствора хлорида золота к слегка кислому раствору соли олова(П). в результате чего появляется темпо-пурпуровое окрашивание (кассиев золотой пурпур).
Эта реакция оскована на том, что двухвалентное олово восстанавливает хлорид золота до металла и одновременно в соответствии с уравнением: .ISu” -|- 2Атг‘- + + 6Н2О = 2Ац -г 3SnO2 -|- Г2Н- образуется двуокись олова (гидрат). Последняя, оставаясь коллоидно растворенной, адсорбирует мелкодисперсное золото, в результате чего образуется золь с пурпурной окраской. Кассиев золотой пурпур является, следовательно, продуктом HiiaiiMijaii адсорбции двух коллоидов. Если осаждение вести из ие слишком разведенного раствора, то он выпадает в виде темного фиолетово-красного
584
Глава 12
осадка, который можно легко слова перевести в коллоидный раствор («пептизировать») обработкой аммиаком и кипячением. Кассиев золотой пурпур находит применение в качестве красивой красной крастш для фарфора и стекла.
Часто испытание па соли олова(П) ведут также, используя раствор хлорида ртути(П); при этом в зависимости от количественных соотношений образуется белый осадок хлорида ртути(1) или черный осадок ртути. Если олово присутствует в четьгрежвалентном состоянии, то для его определения очеиь удобно также пользоваться образованием характерно кристаллизующегося хлоростанната рубидия, Rbj[SnCIe|. Эта микро-реакция,. т. е. реакция, проводимая с каплей раствора на предметном стекле и наблюдаемая под микроскопом, позволяет обнаружить 0,2 у* олова.
Характерным для олова является поведение его соединений при исследовании их в пламени паяльной трубки. В восстановительном пламени легко образуется металлическое олово; однако при доступе воздуха и высокой температуре оно тотчас сгорает, по крайней мере на поверхности, образуя белую двуокись.
В весовом анализе олово большей частью осаждают в виде сульфида, а взвешивают в форме двуокиси SnOr,. Если олово входит в состав сплава, то его обычно выделяют, обрабатывая сплав концентрированной азотной кислотой, в виде «р-оловянноп кислоты», которую также перед взвешиванием переводят в двуокись прокаливанием. Выделение олова можно вести электролитически либо из кислого оксалатного раствора, либо из аммиачного раствора тиостанната. В обоих случаях олово осаждается на катоде в виде металла и может быть определено взвешиванием.
СВИНЕЦ (PLUMBUM) РЬ
Распространение в природе. Наиболее распространенной и важнейшей свинцовой рудой является свинцовый блеск (галенит) PbS. Он встречается в очень многих местах; па территории Германии в Рейнской области близ Штольберга и Мехерниха, в Зигерланде, в Вестфалии, около Клаусталя и Гослара в Гарце, а такзке в Саксонских рудных горах; на территории Австрии он встречается главным образом в Кбрнтене и Тироле, па территории современной Польши — около Вёйтепа. Из внеевропейских месторождений наибольшее значение имеют месторождения Северной Америки и Мексики.
Часто встречаются также продукты разложения свинцового блеска: англезит (свинцовый купорос) PbSO4 и церуссит, (белая сшпщовая руда) РЬСО3; реже — пироморфит (пестрая епшщовая руда, зеленая свинцовая руда) РЬС12-ЗРЪ3(РО4)2 и миметезит, PbCl2 aPbs(A«O4)2. Иа других минералов свинца, которые, однако, не имеют значения для горнорудной промышленности, следует назвать крокоит, .(каллохром,, красная свинцовая руда) Р1?СгО4, вульфенит (молибдепово-свинцовый шпат пли желтая свинцовая руда) РЬМоО4 и штольцит PbWO4.
Исторические сведения. Свинец относится к наиболее давно известным металлам. Установлено, что его знали древние египтяне, а также, очень вероятно, и израильтяне. Греки называли его /itA-iftoi; или pMrpSog.
* 1у — 0,0.01 мг.
Четвертая группа периодической системы
585
Как уже было отмечено в разделе, посвященном олову, в древности свинец нс всегда четко отличали от олова. Во время пунических войн в Испании существовали уже многочисленные свинцовые шахты, которые были заложены греками и финикийцами и разрабатывались позднее римлянами. Последние использовали свинец главным образом для изготовления водопроводных труб. Об укреплении Железных и бронзовых скоб в каменных плитах путем заливки отверстий свинцом упоминает уже Геродот. Такие свинцовые препараты, как свинцовый глет РЪО, свипцо-вый сурик (миниум) Pl>30j, свинцовые белила (основной карбонат свинца), были известны уже древним грекам и римлянам.
Получение. Металлургическое выделение свинца из свинцового блеска осуществляется в основном тремя путями, называемыми: «обжигательно-реакционная плавка», «обжигательно-восстановительная плавка» й «осадительная плавка».
1. Обжигательно-реакционная плавка. При этом способе получения свинца свинцовый блеск подвергают неполному обжигу в пламенных печах при сравнительно небольшом нагревании (500—600°), т. е. нагревают его при доступе воздуха, но‘так, чтобы только часть сульфида свинца перешла в окись и сульфат в соответствии с уравнением (t), в то время как остальная его часть осталась бы неизменной (обжигательная плавка).
PbS + 3/2О2 = РЬО S02. 1 Обжигательная
PbS -j- 2О2 = PbSO4. J плавка (1)
Заключительная часть — «реакционная плавка» состоит в том, что, продолжая нагревание, по возможности устраняют доступ воздуха; прн этом оставшийся, неиро-рсагировавший сульфид свинца взаимодействует по уравнению (2) с окисью п сульфатом, образуя металлический свинец.
PbS 21’ЬО ЗРЪ 4- SO2. 1 Реакционная
PbS + PbSO4 — 2РЪ -j- 2SO2-J плавка (2)
Иногда весь этот процесс ведут в подовых печах. В этом случае реакции (1) и (2) идут одновременно. О равновесии реакции в смеси PbS — РЬО и скоростях реакции см.: Kohlmeyer Е. J., Z. anorg. Chem., 252, 74, 1943.
2. Обжигательно-восстановительная плавка. При этом способе руду сначала обжигают, однако так, чтобы возможно большая часть сульфида свинца перешла в окись свинца. Для этого в настоящее время используют чаще всего метод обжига с дутьем, предложенный Гунтвпгтоном и Гебсрляйном. Этот метод заключается в том, что через смесь свинцового блеска с известняком и Кварцем продувают или просасывают воздух. Окцслёнйе свинцового блеска до окиси свинца существенно ускоряется благодаря присутствию известняка. Образующийся параллельно сульфат свинца (который в результате восстановительного обжига превратился бы в сульфид) в присутствии кварца переходит в силикат (уравнение (Зв)]. в .результате обжига
с дутьем смесь окиси свинца и силиката евппца во второй части процесса восстанавливаете я в шахтных печах (доменных,печах) коксом пли окисью углерода, образующейся после его сгорания. Правда, последняя не действует непосредственно на силикат свинца; однако благодаря взаимодействию с примешанной окисью кальция пз силиката сначала освобождается окись свинца, а последняя ужо взаимодействует с окисью углерода по уравнению (4а). Самые существенные химические превращения при обжп-гательно-восста ион и тельной плавке можно представить следующими уравнениями:
PbS + - РЬО -|- 3SO2. 1 (За)
PbS + 20, — PbSO4. } Обжигательная плавка (36)
PbSOi + SiO2 = PbSiOs + SO2 + J (Обжиг с дутьем) (Зв)
РЬО + СО — РЬ -г СО2. 1 Восстановительная (4а)
PbSiO3 ф- СаО CO — C,iSiO3 + РЬ + СО2. > плавка (46)
Метод обжигательпо-носстагговигсльной плавки применим дли любых свинца-лых руд и особенно пригоден для таких руд, которые, вследствие содержания в пйх примесей не могут быть переработаны методом обжигательно-реакционной плавки.
586
Глава 15
3. Осадительная плавка. Этот способ находит лишь огранйченноо примененной осцовап па том, что свинец может быть освобожден («осажден») непосредствен^: из сульфида в присутствии металлического железа
PbS -f- Fe = Pb + FeS. (7
Правда, в этом процессе не применяют металлическое железо, а нагревают окп<_ железа с коксом, смешанным со свинцовым блеском в шахтных печах. Образующееся при этом железо реагирует с сульфидом свинца. Способ осадительпой плавки имеет' Ют недостаток, что при ггс слишком сильпом нагревапии процесс, представленных; уравнением (5), протекает частично, а при очень сильном нагревании много свингх теряется в результате пена рения.
Из бедных руд свинец после сульфатного обжига можно экстрагировать Яонцец-ТрирОванпъгм раствором поваренной соли и затем осаждать из раствора электролита чески [способ Тайцтопа (Taintoa)].
Рафинирование свинца
Выплавленный обычным способом Сырой свинец загрязнен медью, сурьмой, мышьяком и серой. Большей частью Он содержит также заметные количества серебра.. Так как очистка свинца от серебра имеет большое значение для добычи серебра, используемые в этом случае методы будут рассмотрены в разделе «Серебро». Для удаления остальных примесей проводят переплавку. Поскольку при переплавке имеется доступ воздуха, мышьяк и сурьма окисляются с образованием арсената и антимоната свинца, которые всплывают на поверхность расплава. Медь образует сравнительно тугоилав-кий сплав, содержащий небольшое количество свинца. Этот сплав также отделяется и одновременно захватывает из свинца лею серу. При переплавке часто нагрев ведут на наклонной подложке так, что свинец медленно стекает. Этот способ называется «зейгерованием». Тугоплавкие примеси остаются при этом в виде «зейгер-шипов»,
В 1932 г. Вильямс (William??) разработал способ, который позволяет проводить рафинирование свинца как непрерывный процесс. Этим способом, который дает свинец с содержанием 99,993%, в Порт-Ппри (Австралия) рафинируют 400— 800 т) сутки свинца.
Свинец, содержания, висмут, целесообразно рафинировать влектролитичеоки. При этом он осаждается из содержащего желатину крсммефторнстокислого раствора (способ Беттса). Желатина способствует осаждению в компактной форме.
Этот способ особенно распространен в Северной Америке, а также в Англии (Ньюкасл) и Германии (Гамбург).
Свойства. Свинец представляет собой синевато-белый металл, блестящий на поверхности свежего среза; однако на воздухе он быстро приобретает матовую сине-серую тусклую окраску. Ph самый мягкий среди обычных тяжелых металлов, значительно мягче, чем олово. Его можно резать ножом и даже царапать ногтем. Вследствие незначительной твердости и большой тягучести свинец легко удается прокатывать в листы, однако ввиду незначительной прочности из него нельзя вытянуть слишком тонкую проволоку. Удельный вес свинца 11,34, температура плавления 327,4°, температура кипения 1750° (Fischer J., 1934). В соответствии с данными Вартенберга пары свинца нри 1870° одноатомны. Свинец кристаллизуется в кубической системе. Удельная теплоемкость его при 18° равна 0,0299, атомная теплоемкость 6,2, что находится в соответствии с правилом Дюлснга и Пти. Теплопроводность свинца относительно небольшая, опа состав.ияет лишь 8,5% теплопроводности серебра. Удельная электропроводность при 18° равна к =4,8-10“4, что составляет 7,8% удельной электропроводности серебра.
В отлично от своих более легких аналогов свинец кристаллизуется в грапецоп-трироваппой кубической решетке (ср. рис. 46, стр. 238) с а = 4,91 А. ,
Свинец легко образует сплавы с другими металлами. С рутыо он образует амальгаму, которая при небольшом содержании свинца представляет собой жидкость. В амальгаме свинец также одпоатомеп.
Четвертая группа периодической системы
587
В электрохимическом ряду напряжений свинец стоит непосредственно перед водородом. Нормальный потенциал свинца по отношению к нормальному водородному электроду составляет —0,130 в. Хотя свинец в соответствии с этим немного «менее благороден», чем водород, в разбавленных кислотах оп в общем пе растворяется. Это связано отчасти с тем, что па чистом свинце водород выделяется только при значительном перенапряжении (ср. стр. 33). В некоторых случаях па свинце образуется нерастворимое покрытие, защищающее его от дальнейшего действия кислоты; так, при соприкосновении свинца с серной кислотой образуется сульфат свпнца, с плавиковой кислотой — фторид свинца. Нерастворимость в умеренно концентрироваппой серной кислоте важна для применения свинца в аккумуляторах, а также в сернокислотной промышленности, где получающуюся в камерном процессе разбавленную кислоту упаривают па свинцовых сковородах до концентрации 60° Вс (78 вес. % H2SO4). Правда, приготовленная таким путем кислота содержит примесь свинца. В соляной кислоте свинец также практически не растворяется. В азотной кислоте он легко растворим вследствие своей сильной способности к окислению.
По даггиым Гарре (Garre, 1933) устойчивость свинца по отношению к серной кислоте можно повысить, сплавляя его с небольшим количеством AgCd4.
При доступе кислорода воздуха свинец медленно взаимодействует со всеми кислотами, в том числе и с очень слабыми, и даже с водой:
Pb+V2O2=PbO,
РЬО-|-2Н- = РЬ--|-Н2О.
Следует отметить сравнительно легкую растворимость свинца в содержащей воздух уксусной кислоте. Вероятно, растворимость здесь повышается благодаря комплексообразованию.
В то время как компактный свинец при обычной температуре подвергается действию кислорода воздуха лишь с поверхности, тонкоизмель-ченный свинец пирофорен. При плавлении свинец покрывается сначала серым окисным слоем, так называемой «свинцовой золой»; при более продолжительном нагревании оп переходит сначала в желтый свинцовый глет РЬО, а затем, если его не слишком нагревать при обильном доступе воздуха,— в красный сурик РЬ304. Прп нагревании свинец непосредственно соединяется также с серой, селеном и теллуром, а также и с галогенами.
Соединения свинца крайне ядовиты. Даже следы свинца могут при длительном действии привести к тяжелым заболеваниям и смерти, так как свинец накапливается в организме. Поэтому сплавы с большим содержанием свинца нельзя применять для изготовления посуды. Опасности свинцового отравления особенно подвержены рабочие, которые заняты в свинцовом производстве. Поэтому там применяют особые меры предосторожности.
Симптомами свинцового отравления являются серая кайма на деснах («свинцовая кайма»), бледность лица и губ, запоры, потеря аппетита. В тяжелых случаях появляются сильные боли в области живота («свинцовые колики»), параличи или боли в суставах, наконец, судороги, обмороки или другие симптомы мозгового заболевания. При своевременном применении необходимых мер излечение возможно.
При растворении в кислотах свинец переходит в бег/ажалентное состояние, образуя ионы РЪ”. Действием сильных окислителей, особенно
588
Глава 12
при электролитическом окислении, он может быть переведен в четырех-валеятнос состояние, Нормальный потенциал, соответствующий перезаряжению РЬ" —> РЪ“” (ср. с оловом), составляет по сравнению с нормальным водородным электродом 1,8 в. Следовательно, тенденция к переходу из четырех- в двухвалентное состояние у свинца гораздо больше, чем у олова. В соответствии с этим соли свипца(П) в противоположность солям олрва(П) не являются восстановителями.
Применение. Металлический свинец широко применяют для изготовления трубопроводов, оболочек для кабелей; листовой свинец используют для покрытии п обкладки, например, свинцовых камер в сернокислотном производстве. Далее, он служит для изготовления тиглей, чаш и сковород для уиарнваппя, а также пластин для аккумуляторов. Благодаря большому весу свинец используют для наполнения снарядов, а также ДЛЯ изготовления дроби.
К свипцу, идущему па изготовление ружейной Дроби, добавляют мышьяк (приблизительно 0,3%), что делает его более текучим и легко разбиваемым на капли в расплав ленном состоянии и более твердым после за тверд овання. Из других сплавов свинца следует назвать металл для отливки типографского шрифта (гарт), содержащий 70—90% свинца, сурьму и часто также олово и третник или мягкий припои. Это легкоплавкие сплавы свинца и олова. Наиболее низкой температурой плавления (181°) обладает сплав 64% олова и 36% свинца, Однако часто применяют более богатый свинцом припой; для запаивания тары, служащей для хранепия пищевых продуктов, например консервных банок, следует употреблять припои с содержанием спяпц.ч не более 10%. О содержащих свинец металлах для заливки ггадишпникив было уже сказано п разделе об олове. Свинцовые металлы для заливки подшипников содержат свинец как главную составную часть, к которой для увеличения твердости добавляют либо сурьму (и Часто наряду с ней также некоторые количества олова, меди, мышьяка и т. Д.), Либо незначительные количества щелочных и щелочноземельных металлов. Ко второй группе относится «дорояшым металл», широко применяемый в настоящее время немецким объединением дорог для изготовления подшипников для коленчатых валов. Он состоит из свтлпгр и примерно 0,7% кальция, 0,6 % натрия и 0,04% лития и при температурах ниже 65' превосходит оловянные металлы для заливки подшипников. Свипцово-с.урьмяггые металлы для наливки подшипников содержат обычно 60—80% свипца, наряду с ггпм сурьму или сурьму и олово в равных количествах. Свинцово-сурьмяные сплавы называют твердым свинцом, а в противоположность им обычный чистый свинец — мягким свинцом.
Среди технически важных соединений свипца прежде всего следует назвать свинцовые краски, которые принадлежат к самым древним из минеральных красок. Важнейшими из них являются свинцовые белила (основной карбонат свипца), затем сурик РЬ3О4 и хромовая желтая РЬСгО4. При использовании этих красок следует учитывать ядовитость свинца. После высыхания масляного слоя иа под грунтовке отравления уже можно не опасаться. Из других соединений свинца техническое применение находят прежде всего свинцовый глет [окись свинца(П)], двуокись свипцаПА7), хлорид свинца, нитрат свинца и ацетат свинца (свинцовый сахар). Применение стекол, содвржагцах свинец, для художественных изделий (Хрусталь) и других целей рассмотрено уже па стр. 549.
С органическими кислотами, содержащимися в смоле и льняном масле, свинец образует соединения, значительно ускоряющие высыхание и твердение употребляемых для окраски масел. Препараты, способствующие быстрому высыхапию красок, называют «сиккативами», а их растворы в льняном масле — «олифами». Помимо окиси свинца, для изготовления сиккативов и олиф применяют еще о кислы марганца и цинка.
Четвертая группа периодической системы
589
СОЕДИНЕНИЯ СВИНЦА
В своих соединениях свинец проявляет преимущественно положительную двухвалентность. Но оп, подобно своим более легким аналогам, может быть также и четлярехвалентнымг, однако в этом состоянии оп гораздо менее устойчив, нем его аналоги.
Как и его более легкие аналоги, свинец в четырехвалентном состоянии образует летучее водородное соединение — гидрид свинца РЬН4 и соответственно сложные соединения с органическими радикалами — алкильные соединения свинца РЬК4. Эти соединения можно рассматривать как гомеополярные,.
В своих алкильных соединениях свинец функционирует как э.чектропомжи-телъный атом (если в этих соединениях вообще может идти речь о его заряде); это следует как из механизма их образования (ср. стр. 604) так и из способ пости относящихся к этому классу соединения окислов РЬОВ2 образовывать отчетливо выраженные соли с сильными кислотами.
Как свободный положительный ион чотырехвалептный свинец существует в водных растворах только в минимальных концентрациях. Он имеет тенденцию присоединять избыточные отрицательные ионы с образованием комплексных анионов.
Так от тетрахлорлда < в|ища РЬС14 присоединенном ионов СГ производится хлоро-плюмбат-иоп [РЬС1с]", от двуокиси свинца РЬО2 — [РЬО3]"- и [РЬО4]'"’-иовы, которые входят как структурные элементы в состав три- и тетраоксоплюмбатов(1У); от гидроокиси свинца(1У) p'b(OJT)4 присоединением двух ионов ОН' образуется гекса-гидр оксо пл юмб ат- и он | Pb(UiI)6J".
Тот факт, что свободные ионы РЬ“" не могут существовать в значительных концентрациях, объясняется чрезвычайно легко протекающим взаимодействием их с водой (гидролиз солей свинца(ТУ)] !
РЬ- '^ 2Н3О = РЬ02 4- 4Н'.
От положительно двухвалентного свинца производятся многочислен^ ные соли, в растворах которых имеется бесцветный катион РЬ”. Из солей двухвалентного свинца трудно растворимы сульфат и галогениды. Кроме того, нерастворимы соли свинца(П), образованные с теми кислотами, которые дают нерастворимые соли также с большинством других металлов.
Соли свинца(П) отличаются склонностью присоединять окись или гидроокись свинца с образованием вполне определенных и часто хорошо кристаллизующихся «основных солей». Гидролитическое расщепление солей свинца(П) и сильных кислот в водном растворе, напротив, протекает чрезвычайно слабо, иногда почти незаметно. Среди «основных солей» свинца( I [) следует отметить свинцовые белила РЬ3(ОИ)2(СС)3)2 как технически крайне важный продукт.
По Вернеру <ч>с полные солп» двухвалентного свиица принадлежат к классу «д-гидроксосоединеппй». В соответствии с этим соединения типа РЬ(ОН)2. РЬХ2, напрямер рассмотренный па стр. 596 основной хлорид свинца Pb[<jH),- 1’1)G12, следует представить формулами:
ОП
ОН
О
О
X—РЬ\ ^РЬ —X, или
О
дн
РЬ^ ^РЬ
О
ОН
590
Глава. 12
Ввиду этого следует ожидать, что основные соли jajtoro типа будут диссоциировать в водном растворе по уравнению [РЬ(ОН)2РЬ]Х2 = [РЬ(бН)2РЬ]- -ф 2Х'. Райф (Reiff, 1936) измерением электропроводности основного перхлората свинца fPb(OH)2Pb] [С1О4]2 показал, что это действительно имеет место. Хейк (Hayek, 193G), изучая растворимость окисей и гидроокисей двухвалентных металлов в растворах солей, подтвердил гипотезу, в соответствии с которой в растворах имеются ди-р~гидроксодиметалл-ионы [М11(ОН)гМ11]”. О кристаллической структуре солей этого типа см. т. II.
Бипарные соединения свинца, как правило, окрашены (например, окись и сульфид); напротив, галогениды, за исключением иодида, бесцветны.
В дальнейшем изложении будут рассмотрены вначале окислы свинца, а затем соли свинца(П) — важнейшие после окислов соединения свинца. Менее устойчивые и соответственно значительно менее важные соединения — соли свинца(ТУ), а также некоторые другие малоустойчивые соединения свинца(1У) будут рассмотрены в конце этой главы.
Окислы
Свинец образует два простых окисла и один смешанный:
Простые окислы Смешанный окисел
РЬО, окись свинца, окись свинца(П) РЬ3О4, сурик, окись свин-ца(ПДУ)
РЬО3, двуокись свинца, окись
свинца(1У)
Прежде считали, что, кроме того, существует «с у б окись» РЬ2О. Новейшие рентгенографические исследовании (van. Arkel, Le Blanc, Fricke, Darbyshire) показали, однако, что продукт, который рассматривали как «субокись свинца», является смесью РЬ и РЬО, Часто допускают существование смешанного окисла состава РЬ3О3, но это не доказано. При термическом расщеплении РЬО, пе образуется РЬ2О3.
Смешанный о к н сел РЬ3О4 образуется при взаимодействии простых окирлов; это осуществляется благодаря тому, что окись свинца(П) обладает преимущественно основным характером, а окись свинца(1У), напротив,— преимущественно кислым характером и в соответствии с этим образует соли (плюмбаты). Между солеобразовапием при взаимодействии двух окислов свинца и солеобразовапием при взаимодействии какого-либо основного окисла с кислотным окислом (ангидридом кислоты) не существует принципиальной разницы (ср. также стр. 592 и 594).
2СаО (-SiO2=Ca2ISiO/(]; 2РЬО-фРЬО2= Pb, [PbOJ.
Соли, производные двуокиси свинца, функционирующей как ангидрид кислоты, называют плюмбитами, точнее плюмбитами(IV). Моноокись свинца также способна, правда в незначительной степени, функционировать как кислотный окисел. Соединения, в которые она входит как кислотная составная часть, называют плюмбаты!! Г).
Окись свинца, моноокись свинца, окись свинца!!I) РЬО, называемую в технике «свинцовым глетом», получают окислением расплавленного свинца при пропускании над ним тока воздуха. Так как он обычно получается при «отгонке» свинца от серебра, его называют также «серебряным
Четвертая группа периодической системы
591
глетом». Температура плавления окиси свинца 884°. Если при получении РЪО нагреть выше этой температуры, то при затвердевании она кристаллизуется в виде плотных ромбических чешуек, При осторожном нагревании, а также при термическом расщеплении карбоната или нитрата
свинца ее получают в пиле рыхлого желтого порошка — массикота. Прежде его применяли как краску. В настоящее время его используют
главным образом для получения сурика.
Мокрым путем окись синица получают кипячением гидроокиси свинца с раствором едкого натра, В зависимости от концентрации NaOH образуется желтая или красная окись свинца, причем обе имеют одинаковый
состав. Красная модификация стабильна прп обычной температуре и поэтому труднее растворима, чем желтая. При продолжительном кипячении с водой желтая окись превращается в красную.
Растворимость .и ч; окиси свинца в воде при 20° равна 1,2 на Н Ю г норм, красной—вдвое меньше. Красная окпсь свинца (йреит = 9,36) принадлежит к тетра го нал 1,тюп, желтая (й[>епт=9,66)— к ромбической системе. Точка перехода лежит при 488° (Cohen, 1934). Красная окнет. свинца образует, по даппым Дикинсона (Dickinson, 1924) и Полинга (Pauling, 1941), сложную рыштку, приведенную на рис. 101 (аналогичную структуру имеет SnO; а0= 3,77 А, с(| = 4,82 A, d = 1,13 А). Желтая окнеь свинца образует, по данным Халла (Иal 1а), ромбическую решетку, которая построена из сдвоенных молекул
_С\
Г1< )1’Ъ
Рис. 101. Элементарная ячейка красной окиси свинца «0=3,9S А; <-(,=4,93 Л; Д=1,19_.Л.
Удельный вес обычного свинцового глета приблизительно равен 9,4. Оп заметно летуч уже ниже температуры плавления. Осторожно нагревая окись свинца(П) на воздухе, можно окислить его до сурика РЬ304; однако для этого пригодна только рыхлая, порошкообразная модификация окиси свинца — массикот.
Такими восстановителями, как водород, уголь, окись углерода, цианид калия, при нагревании окись свинца легко восстанавливается до свободного металла.
В кислотах окись свинца легко растворяется, образуя соли. В растворе едкого натра (не очень концентрированном) она растворяется лишь незначительно; все же растворимость окиси свинца в воде существенно повышается при добавлении гидроокиси натрия, что объясняется образованием со щелочью соли, а именно, как показал Шольдер (Scholder, 1934), образованием гидроксиплюмбатов(П). Следовательно, но отношению к сильным основаниям окись свипца(П) функционирует как ангидрид ангидрокислоты, именно ап гидрокислоты РЬ(ОН)2. В соответствии с преимущественно основным характером окиси свипца ее водный раствор окрашивает красную лакмусовую бумажку в синий цвет.
Если высадить щелочью соль свинца(П) из водного раствора, то по уравнению Pb“ -!- 2OII' — Pb(OH)E образуется гидроокись свинца(П) РЬ(ОП)2 в виде белого осадка. В основном осадок имеет гелеобразный характер и содержит, подобно гидроокиси олова(II), значительные количества воды; одпако Хюттиг (1931) методом изобарического обезвояшва-пия, а также рентгенографически показал, что этот продукт содержит
592
Глава 12
РЬ(ОН)2 кап определенное соединение. Если проводить обезвоживать--гидроокиси свинца при 10(0, то образуется красная окись свинца; пр: более низкой температуре — желтая окись (в соответствии с правила."; студеней Оствальда).
Для диссоциации гидроокиси свиица как основания, протекающей на первой, ступени по уравнению РЬ(ОП)2 Д РЬ(ОН?ф ОН', константа диссоциации выглядит следующим образом:
„ [Ph(OHPjx[OH'l . . ,.
L ~ 4 -10-s <PJei0ner, Auerbach).
Константа равновесия для реакция гидроокиси свиица (как ан гидро кислоты), протекающей по уравнению РЪ(ОН)2 + НОН РЬ(ОН)2+Н‘, имеет аначепие
„ [Pb(OH)5]x|HJ . .. , ...
= ~ 1РЬ(бН)21~L ~ Ь1° <Вег1' Airwell).
В соответствии с этими данными гидроокись свинца в качестве основания обладает примерно той же силой, что и аммиак в водном растворе (когда.оп выступает как ангн-др оо снов ан не). Как ангидрокиемнпа окись свинца слабее фенола, кбпетанта диссоциации которого примерно в 100 раз больше, чем константа равновесия для кислой реакции РЬ(ОН)2.
Помимо применения для получения других соединений свинца, окись свинца используют для приготовления свинцовых стекол (хрусталь, флинтглас, штрас) и глазурей, а также при росписи стекла и фарфора в качестве флюса. Кроме того, она служит для изготовления олиф и пластырей.
Двуокись свинца, окись свинца(1Р), РЬО2 получают окислением солей свинца(Ц) хлором, бромом, гипохлоритом или электролитическим путем. В технике се получают окислением ацетата свиица хлорной известью (6) ими обработкой сурика разбавленной азотной кислотой (7), в процессе которой вымывается окись свянца(П):
РЬ(С2Н3Оа)2-|-Са(ОС1)С1+НаО = РЬО2-1-2ИС2Н3О2-]-СаС12 (6)
РЪ3О4 4HNO5=РЪО2 4-2Р1' +2И2О. (7)
В технике осуществляют также электролитический метод получения, например электролиз раствора хлорида натрия, в котором суспендирована окись свинца. Последняя окисляется в двуокись гипохлоритом, образующимся электролитически (ср. стр. 857).
Двуокись свинца представляет собой коричневый порошок, состоящий из мелких чешуек, удельного веса 8,9—9,2 (dpet!T = 9,42). В воде опа практически нерастворима, однако заметно растворима в кислотах. Следовательно, опа обладает слабоосновным характером". Сильное выражен кислый характер двуокиси свинца: при нагревании с сильно основными окислами она соединяется с ними, образуя плюмбаты(1Р) (обычно называемые короче—плюмбаты).
Различают три ряда плюмбатов(1 V): тетраоксаплюмбаты(/Р)М1[РЬ04], триоксоплюмбаты(7УуМ1[РЪОя] и еексагидроксоплюмбаты (ZF) М|[РЬ(@Н)в1.
Тетраоксоплюм1'>ат(1У) кальция Са2[РЬО4] получают нагреванием РЬО2 с GaO или также смеси РЬО и СаО на воздухе. Соединение разлагается ужо очень слабыми кислотами, например угольной
Са2 [РЬО4] 2H2COs - 2СаСО3 > РЬ02 + 2Н2О.
Четвертая группа периодической системы
593
Плтомбат кальция незначительно растворяй при обычной температуре в воде. Обрабатывая его взвесь в воде перекисью натрия, можно получить триоксоплюмбат. (IV) кальция Са[РЬО3]. При сплавлении двуокиси свинца со щелочами образуются плгомбаты тппа М,[РЬ(ОН)е] [гексагидроксоплюмбаты (IV)].
Строение гекса гид рок сои л юнитов, которые на основании их состава вначале считали воду содержащими триоксоплюмбатами MJ, |РЬО3)-ЗИ2О, было установлено Беллучи (Bellucci) и Парравано (Parravano) для калиевой соли К2[РЬ(ОН)6] нй основании ее изоморфизма с гекса гид р оксоплатинатом калия K2[Pt(OH)ej и гексагидро-ксостаннатом калия K2[Sn(OII)((], В порядке дальнейшего подтверждения своих представлений авторы отмечали, что гексагндроксоплюмбат калия нельзя обезводить без полного разложения (отщенлеппе кислорода и образование КОН наряду с РЬО). Позднее Грубе (Grube) нашел, что соответствующую натриевую соль можно обезводить без глубокого разложения. Этот факт был подтвержден Симоном (Simon, 1928), который установил, что при fill- натриевая соль, отщепляя воду, превращается в трио-ксоплюмбат натрия Naa|PbO3], Это, одиако, не опровергает строения, предложенного Беллучи и Парравано даже в с пучаснатриевой соли, так как нагревание может: вызвать изменение структуры.
Двуокись свинца — сильный окислитель, Будучи нагретой с концентрированной серной кислотой, она выделяет кислород^ а при нагревании с концентрированной соляной кислотой — хлор. Уже при небольшом нагревании РЬО., отщепляет кислород. Если ее растирать с легко загорающимися веществами, такими, как сера или красный фосфор, то происходит воспламенение. Па этом свойство основано применение двуокиси свинца в производстве спичек. Г1люмбат(1У) кальция (в смеси с хлоратом калия) также применяют для этой цели. В элементарном анализе по Деннштедту двуокись свинца служит для поглощения NO2, SOa, CIa, HCi, Br3 и НВг, которые количественно удерживаются слабо нагретой РЬОа.
При разложении двуокиси свинца кислотами перекиси водорода не образуется. Следовательно, двуокись свинца не производится от перекиси водорода, как рапсе описанные «перекиси», например перекись бария ВаО3. Строение соединения] соответствует формуле О = РЬ = О*. Свинец здесь четимрехвалептен.
За исключением размеров (а — 4,93; с — 3,37 Л), кристаллическая решетка двуокиси свинца полностью совпадает с решеткой двуокиси олова. В высушенном состоянии двуокись свинца обычно содержит еще небольшие количества воды. Нагреванием на воздухе или в вакууме воду из нее нельзя удалить полностью так, чтобы При этом не началось одновременно отщепление кислорода. Напротив, согласно Крустнпеопсу (Krustinsons, 1934—1937), двуокись свинца можно получить в безводном состоянии высушиванием в токе кислорода при температуре около 250“. Давление кислорода достигает 1 атм тол,,ко при 344е. Это справедливо для сравпительпо грубозернистой двуокиси свинца. В очень мелкозернистом состоянии, например в таком, в каком она получается nps электролитическом осаждении, обнаружено существенно б о.те о высокое давление кислорода.
В Шотландии и Ида го встречается природная двуокись свинца в виде очень мелких кристалликов серо-черного цвета, которые, однако, дают на глине коричневую черту (платтнерит, тяжелая свинцовая руда).
Сурик РЬ<|04 образуется в виде яркого красного порошка при нагревании мелкодисперсной окиси свинца на воздухе до примерно 500°. В смеси с льняным маслом его широко применяют в качестве масляной краски
* Как все решетки типа рутила, решетку РЬОа можно рассматривать как молекулярную решетку (ср. рис. 63, стр- 298). Одиако если ее рассматривать как координационную решетку, то на каждый атом РЬ приходится в/3 несвязанных между собой атома О, откуда для свинца также получается четырех валентность (в электрохимическом смысле).
38 Г. Геми
594
Глава 12
для защиты железа от ржавчины, а кроме того, его используют как замазку для заполнения стыков между листами и трубами. При техническом приготовлении сурика обычно исходят из порошкообразной окиси свинпг (массикот), нагревая ее при непрерывном перемешивании в токе воздуха. Еще более яркий оранжево-окрашенный сурик (парижская красная, сатурповая киноварь) получается, если исходить из окиси, полученной разложением карбопата или нитрата свинца.
В воде сурик мало растворим. Он * растворяется в расплавленной селитре и кристаллизуется из этого раствора в виде маленьких двупре-ломляющих призм. Удельный вес обычного рыхлого сурика лежит в интервале 8,8 и 9,2, При нагревании сурик темнеет, при охлаждении приобретает первоначальную окраску.
Если сурик нагреть в вакууме, то при температуре около 400е оп начинает терять кислород. Около 550е давление кислорода достигает атмосферы, т. е, парциального давления кислорода воздуха. Следовательно, выше этой температуры на воздухе также происходит разложение, Если предотвратить повышением давления диссоциацию, то сурик плавится при 830°.
Разбавленной азотпой кислотой сурик разлагается с образованием нитрата свинца(П) и двуокиси свинца. Сурик, вероятно, следует рассматривать как тетраоксоплюмбат(1У} свинца(П), PbJPhOj. То, что он не является простой смесью РЬО и РЪ03, следует уже из его давления разложения, отличающегося от давления разложения РЪО2, и было подтверждено рентгенографически. О структуре решетки РЬ3О4 см.; Gross S. Т., J. Am. chem. Soc., 65, 1107, 1943.
Согласно рентгенографическим исследованиям Ле Блана (Бе Blanc, 1932), помимо красной, существует также черная модификация сурика. Ее получают как из РЬО и О2 путем непосредственного соединения, так и разложением РЬО2, если оно проводится ниже 389°. При 390° черный сурик превращается (монотроппо) в обычный красный.
Свинцовый аккумулятор. Аккумуляторы (т. е. собиратели) служат для накоплении электрический'энергии. При «заряжении» электрическая эпергия в результате обратимого электрохимического процесса превращается в химическую энергию; при «разряжении», пепольвуя тот же процесс, протекающий только в обратном направлении, вновь накапливается электрическая эпергия.
Важцейшнм аккумулятором является свинцовый аккумулятор. Он состоит в принципе из двух решетчатых свинцовых пластин, погруженных в серную кислоту удельного веса 1,15—1,20. Одна из пластин заполнена двуокисью свинца, другая — губчатым свинцом. Вместо двух часто применяют несколько пластин, которые располагают гьшеремеыно на довольно близком друг от друга расстоянии, прячем няастппы одного вида соединяют между собой. Если разные пластины соединить проволокой, то ток потечет от пластины с двуокисью свинца (коричневая) к свинцовой пластине (серая). Электрический ток возникает благодаря тенденции четырехвалентпого положительного свинца в РЬО2 разряжаться, отдавая положительное электричество, г, е. принимая электроны.
На положительной пластине
РЪ4+ -|- 2Р’= Pbs+ или
PbOo -I 4Н‘ + 2е = PIT- _L 2I-LO
Одновременно на отрицательной пластипе металлический свинец переходит в раствор в виде положительно двухвалентного иона РЬ = РЬ" -р 2е.
Образующиеся в обоих случаях ионы РЬ” связываются с 80''4-иоиами серной кислоты, давая нерастворимый сульфат свинца, который вследствие нерастворимости остается иа месте исчезнувших РЬО2 и РЬ. Следовательно, процессы па обоих электродах^ можно в общем передать уравнениями:
РЬО2 + 4Н- + SO"4 + 2в = РЬ804 + 211,0, РЬ + SO^ = PbSO4 + 2е.
(8)
(У)
Четвертая группа периодической системы
595
Объединив оба уравнения, для всего химического процесса в целом получают
РЬО2 -> РЬ 4 2Н25О4 = 2РЪ8О4 ф 2НгО.
Поддерживаемая описанными процессами разность потенциалов между обеими пластинами составляет около 2 ц. Если к электродам аккумулятора приложить внешнее напряжение, превышающее ату величину в обратном направлении, т. е. положительную пластину соединить с положительным полюсом внешнего источника тока, то процессы у электродов пойдут в обратном направлении. На пластине, соединенной с положительным полюсом внешнего источника тока, ионы РЬ" Снова станут заряжаться до РЬ'"’, которые тотчас взаимодействуют с водой
РЬ"" + 21ЬО = РЬО3 4- 4Н'.
Иа другой пластине в результате нос ста нов ления ионов РЬ" вновь осаждается свинец.
Прн разряжении концентрация кислоты понижается, так как при этом образуется вода; при заряжении аккумулятора концентрация повышается, так как расходуется вада. Следовательно, состояние зарядки аккумулятора можно контролировать, определяя удельный вес кислоты, Гели продолжать зарядку после того, как будет использован весь отложившийся па пластинах сульфат свинца и, следовательно, ужо но будет больше ионов РЬ", то па свинцовом электроде начнет выделяться водород, а на электроде с двуокисью свинца — кислород: аккумулятор «накипит». Так как для этого требуется приложить более высокое напряжение, чем для разряжения, т. е. повышения заряда ионов РЬ*’ (при пор мяльной концентрации последних), то к концу процесса зарядки напряжение н,т клеммах значительно возрастает, Во время разряжения напряжение быстро падает до 2 е я -затем долго остается почти постоянным, причем это Постоянство сохраняется тем дольше, чем меньше сила разрядного тока. При большей силе разрядного тока пространство около положительной пластины обедняется кислотой, так как убыль кислоты пе мошит быть достаточно быстро восполнена в результате се диффузии. С этим евл ипш падение напряжения при разряжении, так как стремление двуокиси свинца к разряжению в сильнокислом растворе больше, чем в слабокислом.
Так как ввиду указанных причин и вследствие потребления электрической энергии на преодоление сопротивлений кислоты (как при заряжении, так и при разряжении) зарядпое напряжение бывает постоянно выше, чем разрядное, то потеря электрической энергии при накоплении энергии посредством аккумуляторов неизбежна. Па практике эту потерю считают равной 20—25%.
Соли свипца(П)
При получении солен свинца(П) большей частью исходят из свинцового глета или металлического свинца. Последний действием азотной кислоты переводят в легкорастворимый нитрат свинца или обработкой парами уксусной кислоты при доступе воздуха и двуокиси углерода — в свинцовые белила (основной карбонат свинца). Легкорастворимые пли только умеренно трудно растворимые соли свинца получают непосредственным растворением свинцового глета или свинцовых белил в соответствующих кислотах. Трудпорастворимые] соли получают из легко-растворимых солей (нитрата свинца, ацетата свинца) двойным обменом.
Хлорид свинца — дихлорид свинца, хлорид свинца(П) — РЪС1а образует белые ромбические кристаллы с шелковистым блеском. Удельный вес 5,9, температура плавления 498°, температура кипения 954°. В воде хлорид свинца умеренно труднорастворим (см. табл. 89). Поэтому он осаждается оз ие слишком разбавленных растворов солей свинца (11) при добавлении к ним донов СП.
В спирте хлорид свинца растворим гораздо труднее, чем в воде. В глицерине оп растворяется легче, чем в воде (2,04 г в 100 а).
Техническое производство хлорида свинца осуществляют либо растворением окиси свинца или основного карбоната свинца (свинцовых белил) в солянвй кислоте (10), либо растворением гранулированного свинца в разбавленной азотной кислоте
38*
596
Глае a 72
Таблица 89
Структур» решетки и растворимость в воде галогенидов свипца(Н)
P13F2 PbCla РЬВг2 РЫ3
Структура решетки Растворимость, а: тип хлорида свинца fk тип плавикового шпата Тип хлорида свипца Тип хлорида свинца Тип бруцп-та
ммаль[л при 25,? Растворимость, 5,5 38,8 26,5 1,6
ммоль!а При 100" — 119 124 9
и последующим осаждением соляной кислотой (11); освободившуюся во второй части процесса азотную кислоту можно вновь использовать для растворения свинца.
РЪО Ч- 2НС1 — РЪС12+ Н2О;
РЪ3(ОИ),(СО3)2 4- 6НС1 - ЗРЬС12 -> 2СО2 4Н2О. ЗРЬ 4- 8HNO3 = ЗРЬ(ХО3)2 4- 2N.0 4^ 4Н2О, Pb(NO3)2A 2НС1= РЬС12 4- 2HNO3.
В токе двуокиси углерода хлорид свинца удается перегонять без разложения. При нагревании на воздухе он претерпевает гидролитическое расщепление под влиянием влаги воздуха. Нагреванием в токе водорода или с углем в присутствии паров воды он восстанавливается до металла.
Расплавленный хлорид свинца застывает, образуя роговидную массу (роговой свинец). В расплавленном состоянии хлорид свинца обладает значительной электропроводностью; равным образом он хороню проводит ток в иодном растворе при обычной температуре. Исходя из данных электропроводности насыщенного раствора при 25°, следует, что (кажущееся) содержание в нем нодиссоциированной соли составляет 6%; остальная часть хлорида свинца примерно наполовину диссоциирована первично (по уравнению РЬС12 — РЪСГ Д- С17) и наполовину вторично (по уравнению РЬС13 = Pb'" + 2CF)- Раствор обнаруживает кислую реакцию на лакмус; однако степень гидролитического расщепления незначительна (по данным Лея, при 100° в 0,01 п. растворе она составляет около 0,6%).
Хлорид свинца образует ромбическую решетку, в которой каждый атом Pb окружен девятью атомами С1; два атома хлора лежат к РЬ ближе других (расстояние 2,67 А), третий атом С1—на расстояния 2,88 А; шесть остальных атомов хлора расположены на расстоянии примерно 3,05—3,29 А.
РЬС12 может присоединять избыточные ионы С1', образуя комплексы, Вследствие этого растворимость хлорида свинца при добавлении больших количеств хлоридов или НС1 повышается (небольшие количества ионов СГ снижают растворимость). Известные в твердом состоянии галогениды свинца(11) [галогемплюмбаты (//)] соответствуют главным образом двум типам: м! РЬС14 и MIРЬС13; наряду с ними встречаются также сони и других типов, например 'MlPbgCIs-
Хлорид свинца легко образует также основные хлориды. Последние можно рассматривать как соединения РЬС12 с РЬО и с РЬ(ОН)2. Некоторые из инх в природе встречаются в кристаллическом состоянии, например 2РЬО-РЬС12 (мендипит), При действии известковой воды па горюши концентрированный раствор хлорида свипца подучают соединение РЬ(ОН)3.РЪС13, которое поступает в продажу в виде малярной
Четвертая группа периодичееяой системы
597
краски (свинцовые белила Наттинсоиа). Относительно строения этого соединения см. стр. 589.
Хлорид свипца образует также двойные соединения с пиридином и тиомбче-ишой.
Бромид свипца. Бромид свинца[П) РЬВг2 получают таи же, как хлорид свинца; он кристаллизуется из горячей воды в виде белых ромбических игл с шелковистым блеском; уд. вое 6,6. При 373° РЬВг2 плавится, образуя красную жидкость, которая при охлаждении вновь застывает в белую роговидную массу; т. кпд. 916°. На свету бромид свинца медленно чернеет вследствие выделения металла.
Бромид свинца образует такие же двойные соли и другие продукты присоединения, как и хлорид свинца. Из крепкого раствора бромистого водорода он кристаллизуется в виде гидрата РЬВт2-ЗНаО; кроме того, он может также присоединять НВг.
Иодид свипца. Иодид свипца(П) РМ2 осаждается из растворов солей свинца при добавлении ионов I' в виде желтого осадка, который при перекристаллизации из горячей воды превращается в красивые листочки с золотистым блеском. Удельный вес 6,2, температура правления 412% температура кипения около 900°. При нагреваний иодид свинца становится сначала кирпично-красным и, наконец, буро-красным; при охлаждении он принимает прежнюю окраску.
Растворимость в воде иодида свинца существенно меньше, чем растворимость хлорида (ср. табл. 89). Водный раствор этого соединения, окрашенного в интенсивно желтый цвет, совершенно бесцветен. Это объясняется тем, что раствор содержит практически нацело диссоциированное соединение, т. е. ионы РЬ"' и I', которые бесцветны. Следовательно, окраска свойственна исключительно педиссоциировэнному соединению. Из растворов, содержащих подпет оводородную кислоту, кристаллизуется продукт присоединения РЫа-Ш-5На0.
Известны также многочисленные красиво кристаллизующиеся двойные соли иодида свинца. Большинство из них содержит воду,
Иодид свинца(П) обладает слоистой структурой (типа бруцита, ср, рис. 61, стр. 293; а - 4,59, с - 6,86 А).
Фторид свипца. Фторид свинуа.(1Г) PbF3 после иодида свинца является наиболее трудно растворимой солью среди галогенидов свинца (11). Ее получают в ВпДе белого порошка уд. веса 8,24 (t/рент = 8,37) действием водной плавиковой кислоты иа карбонат или гидроокись свипца. Т, пл. 818% т. кип. 1292°. Данные о растворимости см, в табл. 89. Теплота образования 159 ккал/молы Фторид свипца Диморфен. Стабильный при обычной температуре, ромбический a-PbF2 выше 220° превращается в кубический 0-PbF3. a-PbF2 (rfpeiiT 8,37) обладает решеткой, подобной РЬС12; P~PbF2 (йреит = 7,68 при 25°) кристаллизуется по типу ила викового шпата (а = 5,93 Л). Взаимное превращение обеих модификаций происходит обычно очепь медленно.
Цианид синица Pb(CN.)a осаждается из растворов солей свипца при добавлении ионов CN' в виде белого, нерастворимого в воде осадка, который в кислотах растворяется с разложением.
Роданид сшшца Pb(SCN)2 образуется в виде белых, очбш, мало растворимых в воде кристалликов при взаимодействии ацетата свинца и роданида калия в водном растворе.
Как цианид, так и родаппД свинца обладают тенденцией к образованию двойных соединении с галогенидами свинца.
Ацетат свинца РЬ(С2113Оа)2 получают растворением окиси свинца в уксусной кислоте. Ацетат свинца очень легко растворим в воде (50 г в 1Q0 г воды при 25° и 200 а — при 100’). При испарении раствора ацетата свинца соль выделяется в форме кристаллогидрата РЬ(СаН3Оа)2> ЗНаО,
598
Глава 12
образуя светлые моноклинные кристаллы удельного веса 2,50 с температурой плавления 75е. Безводная соль имеет удельный вес 3,25 и плавится только при 280е. Из-за сладкого вкус^, который имеет, однако, неприятный металлический привкус, ацетат свинца называется также свинцовым сахаром. Он сильно ядовит.
Ацетат свинца служит часто в промышленности как исходный продукт для приготовления других соединений свинца. Его применяют также при крашенин тканей я и ситцепечатании. Разбавленные растворы ацетата свинца используют в медицине для компрессов и промываний.
Пропитанная ацетатом свинца бумага после поджигания продолжает тлеть как трут. Водные растворы ацетата свинца способны поглощать значительные количества окиси свиица, Приготовленные таким образом растворы называют свинцовым уксусом. Они содержат основные ацетаты, например РЬ(ОН)(СгНаОй) и РЬ3(О1-1)1(СЙН3О2)2. Другие соединения свиица, и прежде всего галогениды свинца, также легко присоединяют ацетат свинца. Кроме того, он образует с другими ацетатами двойные соли, например с ацетатом натрия NaPb(C2H3O2)3'ЗН2О — триацето-плюмбат(П) натрия.
Формиат свинца РЬ(СНО2)2 — блестящие ромбические столбики, легко растворимые в воде (16 г в 100 а при 16°), По своим свойствам он очень похож па ацетат свинца, но отличается от последнего плохой растворимостью в спирте.
Оксалат свиица РЬС^О^ выпадает в виде белого осадка из растворов солей свинца при добавлении попов C2OJ. Растворимость в воде составляет около 2 мг/л при 25°.
Нитрат свинца Pb(NO3)2 получают растворением свинцового глета, свинцовых белил или гранулированного свинца в горячей разбавленной азотной кислоте и последующей кристаллизацией из раствора, содержащего некоторый избыток азотной кислоты (во избежание загрязнения РЬ(КО3)2 основными солями). При приготовлении двуокиси свинца из сурика .РЬ['А()3)2 получается в качестве побочного продукта. Нитрат свинца применяют при производстве спичек, а также он служит исходным продуктом для получения других соединений свинца.
Нитрат свинца выпадает цд кислого раствора в виде крупных прозрачных кристаллов кубической системы с удельным весом 4,53. Растворимость в чистой воде составляет:
при О 20° 46° ‘66 86° 106°
38,8 56,5 75 95 115 139 г в 100 г Н2О
При нагревании нитрат свинца распадается по уравнению
Р Ь( 2, О3)2 = РЬО ~p2iS O2-j-)7202-
Он легко образует продукты присоединения, прежде всего с окисью свинца (основной нитрат). В иодных растворах нитрата свинца при обычной температуре гидролиз обнаружить нельзя.
Водный раствор нитрата свинца сильно корродирует металлический свинец. По данным .Тиля (Thiel, 1920): это явление Ослопало на том, что нитрат свинца, переходя в желтый нитрит, окисляет свинец, при этом он преимущественно действует па загрязненные места поверхности металла, т. е. на так называемое «промежуточное вещество» (по выражению Тамм ала), которое находится между отдельными кристаллитами (ср, стр, 607) и в котором скапливаются загрязнения металла, так как оно состоит из компонентов расплава, затвердевающих в последнюю очередь («эвтектики») (ср, гл. 13).
Четвертая ep'tmna iiepuodnuecKaii системы 599
Карбонат свинца PbGO., встречается в природе в виде церуссита (бе-лал свинцовая руда), изоморфного арагониту, стронцианиту п витериту. Искусственно его получают в виде кристаллического осадка при пропускании двуокиси углерода в разбавленный раствор ацетата свинца или при добавлении ацетата иди нитрата свинца к раствору карбоната аммония на холоду; удклыгый вес его 6,53. Растворимость в воде РЪСО3 чрезвычайно мала. Она составляет практически около 0,3 м<> в 100 г воды и зависит от содержания угольной кислоты в воде.
При нагревании карбонат свинца легко расщепляется на окись свинца и двуокись углерода. Уже при температуре немного выше 300° давление СОЙ достигает 1 атм.
При смешивания водных растворов солей свинца(П) с карбонатами щелочных металлов при нагревании выпадает основной карбонат свинца. Он образуется также из нейтрального карбоната при кипячении последнего с водой и одновременным пропусканием через реакционную смесь чока воздуха, очищенного от СО,,. В чистом состоянии он имеет состав РЬ(ОНЬ-2РЪСО3 и его широко используют как малярную краску, пзве-стную под названием свинцовые белила.
Свинцовые белила. Имеющие няшное значение в качестве .малярной краски, свинцовые белила приготовляют различными методами; их вырабатывают «сухим путем» (голландский и немецкий способ) или «мокрым путем» (французский и электрохимический способ).
1. Голландский способ является самым старым. Однако он широко используется в настоящее время и состоит в следующем, Тонкие, скрученные а спираль свинцовые пластины подвергают действию паров уксусной кислоты, находящейся в горшках, которые заложены в навоз или другое гпиющее вещество. Процесс гниения дает тепло, необходимое для медленного испарении уксусной кислоты, и двуокись углерода, которая требуется для ттревращения первоначально образующегося основного ацетата свинца в основной карбонат. Процессы, имеющие существенное значение для образования свинцовых белил, можно представить следующими уравнениями:
РЬ + 2НСЙН3О2 4* 1/йО2 = РЬ(С2Н3О2), 4 Н2О,
, РЬ(С2Н3О2)2 4* Н2О = РЬ(ОВ)(С2Н3Ог) 4-'*НСйН3О2,
6РЬ(ОН)(С2Н3О2) + 2СОа = РЬ3(ОН)2(СО3)2 4- ЗРЬ(СгН3О2)а 4“ 2П2О.
Свинцовые делила
Эти реакции протекают параллельно до тех иор, пока пе прекратится выделение двуокиси углерода или не израсходуется весь свипец.
2. По немецкому способу свинцовые пластины подвешивают в камерах, в которые пропускают двуокись углерода, образующуюся при сжигании кокса, а также пары уксусной кислоты, Здесь в основном имеют место те же превращения, что и при получении свиттцовых белил голландским способом.
3, Французский способ осуществляется «мокрым путем». В этом случае основной карбопат свинца образуется при пропускании двуокиси углерода в растворы основного ацетата свинца. Последний получают растворением окиси свипца в кипящем растворе а.пцтата спнпца. На том же принципе, что я французский способ, основаны английсп-нР егюсоб и применяющийся В Советском Союзе чдреенееврейский^ способ с теми нзмеценнямп, что в последних работу проводят не с растворами, а с тесто--образтюй массой, содержащей лишь небольшое количество воды.
4, Электрохимические способы: свинцовые пластипии подвешивают в каче-•стве анодов в подходящие растворы электролитов, например в раствор поваренной «соли, смешанной с карбонатом натрия, и одновременно пропускают двуокись угле* рода, Переходящий в раствор свинец выделяется при этом тотчас в виде основного карбоната
РЬ = РЬ" + 2е, 31>Ъ" 2ОН' 4- 2СО"3 = РЬэ(ОН)2(СО3)г.
GOO
Глава 12
Свинцовые белила представляют собой тяжелый, плотный, аморфный порошок, который очень прочно связывается с маслом. Из всех белых красок он: обладает наибольшей кроющей способностью. Недостатком белил является свойство постепенно темнеть на воздухе, содержащем следы сероводорода (в результате образования сульфида свинца). К поступающим в продажу сортам большей частью примешивают сульфат бария. Содержание сульфата бария до 20% пе снижает кроющую способность свинцовых белил, но повышает их красящую способность. Однако для снижения стоимости краски часто в нее вводят значительно большие количества сульфата бария.
‘Хромат свинца РЬСгО4 представляет собой также технически очень важную краску (хромовая желтая).
Промышленное получение ее производят при смешив апии раствора ацетата свинца с раствором бихромата калия, подкисленным серией кислотой при обычной температуре. При отцх условиях хромат свиица осаждается в виде яркого желтого порошка
2РЬ" 4- Сг2о; + Н2О -= 2РЬСгО4 + 21Г.
Если осаждение вести из нейтральных растворов хроматов, то получается краска более-темного тона.
В природе хромат свинца встречается в виде желтовато-красных моноклинных игл или призм удельного веса 5,9—6,0; это — красная свинцовая руда (крокоит, каллохром), В воде хромат свинца крайне трудно-растворим (0,01 мг в 100 а воды), в азотной кислоте он, напротив, легкорастворим итакжо достаточно легко растворим в растворах щелочи. При нагревании он плавится вначале без разложения, по может отдавать часть своего кислорода другим, обычно трудно окисляемым веществам. Поэтому его используют в элементарном анализе для сжигания серусодержащих веществ.
Перемешиванием увлажнен ж по тг топкоизмеяьчепного хромата свинца с окисью-свинца или осаждением из растворов основных соней свинца нейтральными хроматами получают основные ароматы, которые иногда применяют в качестве красок (хромовая красная). Основной хромат свинца Состава 2РЬО-РЬСг04 встречается в природе в виде фепицита (мелапохроита), в форме тецяо-красных кристаллических листочков. Уд. вес 5,75.
Сульфат свинца PbSO4, называемый также «свинцовым купоросом»,, выпадает в виде белого порошкообразного осадка прп добавлении разбавленной серной кислоты к растворам солей свинца. В воде оп труди «растворим (около 40 мг/л). Значительно лучше он растворяется в концентрированных сальных кислотах (соляная, азотная, серная). При разбавлении большая часть его снова выпадает.
Растворы ацетатов натрия и аммония также способны растворять значительные количества сульфата свинца. Растворимостью сульфата свинца в растворах тартрата аммония, содержащих аммиак, пользуются в анализе для его определения и отделения от других нерастворимых в воде* белых порошкообразных веществ, таких, как сульфат бария, двуокись кремния и т. д., которые не растворяются в тартрате аммония.
Повышение растворимости PbSO4 в присутствии названных веществ объясняется образованием с ними более слабо диссоциирующих соединений (например, образованием мало диссоциированного ацетата свинца при добавлении ацетат-ионов) и отчасти* образованием комплексов.
Четвертая группа периодической системы
601
Сульфат свинца растворяется та я же в концентрированных растворах щёлочей; в этом случае с образованием гидроксоплюмбатов{Н) щелочных металлов.
В природе сульфат свинца часто встречается в виде хорошо образованных больших кристаллов ромбической системы удельного веса 6,1 — 6,4; это англезит (купоросная свинцовая руда). В чистом состоянии эти кристаллы прозрачны, как стекло (поэтому их также называют «свинцовым стеклом»)- Однако нередко они содержат окрашивающие примеси.
Помимо ромбической модификации (о структуре см. стр. 318), сульфат свинца существует также в моноклинной. В некоторых месторождениях ов встречается именно в этой форме (например, в Сардинии).
Точка плавления сульфата свинца лежит между 1150—1200°. Точпсо ее определить не удается, так как это соединение начинает разлагаться при 1000°.
Сульфат свинца может присоединять окись свинца с образованием основных солея. Он образует также двойные соли, например (NH4)2Ph(SO4)2 — мелкие, йро-зрачныо, как вода, кристаллы.
Сульфид свинца PbS выпадает в виде черного осадка при пропускании сероводорода в растворы солей свипца. Он получается также прямым соединением составных частей (нагреванием свинца в парах серы). В природе он встречается в больших количествах в виде свинцового блеска (галенит), часто в форме больших, красиво образованных кристаллов (главным образом кубиков и октаэдров) кубической системы, обладающих евпнцо-во-серой окраской, сильным металлическим блеском и прекрасно выраженной спайностью (по плоскостям куба). Его удельный вес равен 7,5В (структура решетки каменной соли; а = 5,93; Pb<—+S = 2,97 А).
При нагревании па воздухе сульфид свинца окисляется до сульфата свинца и окиси свинца. Этим пользуются при «обжиге» для получения свипца. При прокаливании в токе водорода сульфид свинца постепенно восстанавливается до металла. Окись углерода, наоборот, действует па него только слабо. При нагревании с хлором образуются РЬС12 и SCla. Смесь сульфида свинца и нитрата свинца воспламеняется при температуре примерно 50’ Сплавлением с содой прп доступе воздуха из сульфида выделяется. свободный металл; при этом сначала происходит окисление сульфида до сульфата, который затем взаимодействует с другими порциями сульфида с образованием SO;, и РЬ.
Растворимость осажденного сульфида свинца в воде составляет приблизительно 0,8 мг/л. В разбавленной соляной кислоте и серной кислоте оп практически нерастворим, зато легко растворим в разбавленной азотной кислоте, так как последняя окисляет серу. Концентрированная соляная кислота разлагает его с выделением сероводорода, так же как лимонная кислота, вероятно вследствие своей комплексе образ у тощей тенденции.
Сульфид свинца плавится при температуре около 1110’. С сульфидом железа(П), сульфидом медй(1) и сульфидом серебра расплавленный сульфид свипца смешивается во всех отношениях. Эвтектики (ср. стр. 609 и сл.) лежат при 863’ и 30% FeS, при 540° и 51% Cu2S, при 630° и 77% Ag2S.
Соединения свинца (IV)
Соединения свинцщТУ) распадаются па два класса:
1. Соединения, которые можно рассматривать как соли свинца/IV) с кислотами. Важнейшими соединепцямн этого класса являются: галогениды свщща(1У) РЪХ4, сульфат свинца/fV) Pb(SO4)2 и ацетат свинца/IV) Pb(C3H3O2)4. Помимо этих соединений, которые описаны ниже,
602
Глаза 12
известны также и другие соли свиица(1У), например (первичный) фоо фат. хромат, а также соли свинца(1У) с различными органическими кислотами.
2, Соединения свинца (7Г), не имеющие характера солей. К ним относятся гидрид свинца РЬН4 и алнилыше соединения свинца типа PbR4 (В — алкил). Напротив, алкчтльные соединения свинца тина PbR3X или PbR2X, (X — кислотный остаток) обладают ярко выраженным характером соли и относятся к первому классу.
Основанием, от которого производятся соли свлпца(1У) (или его ангидридом), является двуокись свиица РЬО3. В соответствии с этим соли свинца(1У) образуются, если двуокись свинца растворять в кислотах. Для их приготовления часто удобнее использовать электролиз сильно подкисленных серной кислотой растворов соответствующей соли свинца (II).
Водой соли свинца(IV) гидролитически разлагаются с выделением двуокиси свинца.
Соли свинца(IV) проявляют большую склонность к образованию ацидосоединений путем присоединения солей более сильных оснований, V например
PbCl4-)-2K.Cl = 1<2 [РЬС16]—Гексах.чороплюмбат(1У) калия (изоморфен Ks[PtCl6}}.
Дцидоцлюмбаты значительно устойчивее, чем свободные соли свинца (IV), и существуют иногда даже в тех условиях, когда соли свинца (IV) неустойчивы.
Галогениды свинца (IV) и гарогеноплюмбаты. Двуокись свинца рас-творя&гея в концентрированной соляной кислоте с образованием тетра-хлорида свинца, который, однако, очень легко разлагается с отщеплением хлора
ГИМ 1*н.14 7^ РЬС]г+С1г.. .. (12)
Процесс, и . : ; '.4iin.:ii вторым уравнением, обратим, т. е. дихлорид
свинца мол,ст присоединять в водном растворе хлор с образованием тетра* хлорида свинца, Попытки выделить тетрахлорид свинца из раствора были долгое время безуспешны вследствие легкой расщепляемости этого соединения. Получение его в чистом виде удалось осуществить в 1893 г. Фридриху, который из раствора хлорида свинца (IV), полученного действием хлора на хлорид свинца (II), суспендированный в соляной кислоте, высадил сначала комплексную соль (NH4)JPbCle] и затем разложил ее внесением в охлажденную концентрированную серную кислоту (NHJjPbClJ + H2SO4.= (NH4),SOj + 2НС1 + РЬС14. Будучи нерастворим в концентрированной серной кислоте, тстрахлорид свинца осаждается в виде тяжелой, сильно преломляющей свет желтой жидкости.
Тетпахлоряд свинца РЬС14 — при обычной температуре жидкость, удельный вес его при О71 равен 3,18; при температуре около —15° застывает в желтую кристаллическую массу. Он сильно дымят па воздухе, так как водой гидролитически разлагается. Следовательно, и первая из двух реакций (12), приведенных выше, отчетливо обратима.
Значительно более устойчивыми, чем тетрахлорид свинца, являются производящиеся от него путем присоединения других хлоридов хлоро-плюмбаты [гексахлороплюмбат(ГУ)] М|[РЬС16].
Четвертая, группа периодической системы
603
Кроме уже упомянутой аммонийной соли (NH4)2 [РЬСф 1, были изолированы также другие, хорошо кристаллизующиеся соли этого типа; некоторые из них без разложения выносят нагревание до 200°. Однако чистой водой все они разлагаются уже при обычной температуре с выделением РЬО2. Гексахлороплюмба ты щелочных металлов изоморфны гексахлоростаттпдтам и гексахлороплатинатам щелочных металлов (ср. стр. 5/3). Взаимодействием хлоронлюмбатов с бромидом калия и иодидом калия можно получить также бромоплкмбаты. Mi[PbBr6] н иодоплюмбаты М| [Pbloj. Лежащие в их основе галогениды сштпца(1¥) сами по себе неустойчивы.
Тетрафторид свипца PbF4 был получен Вартепбергом (Warteaberg, 1940) пропусканием F2 над PbF2 про температуре выше 250°. PbF4 — бесцветные тетрагональные иглы; уд. вес 6,7: теплота образования (из элементов) 222 ккал/моль, Тетра-фторид свшщаобразует комплексные < о. иг тпггдМ* [PbFe}, а также соли тппаМ.) HPbFg, которые изоморфны ранее упомянутым K<;liSnFs.
Азид свинца(IV) Pb(N5)4 образуется, по данным Мёллера (Moller, 1949), в водном растворе при взаимодействии с РЬ3О4. Одпако красшдй раствор скоро -обесцвечивается с выделением N2 я о г а по щи нем азида свинца(П) Pb(bl3)2. Взаимодействие РЬО2 с протекает в разле1:-тз?|[ггом, без образования азида свйнца(1¥). Взаимодействием (N Н4)2 [PbClfi] с N nN s Mr л л ср получил остро пахнущее масло, из которого при испарении осаждаются тем но-красные игольчатые кристаллы. Эти кристаллы нельзя проанализировать вследствие их склонности к взрывному распаду; вероятно, ото азидошпомбат(1¥) аммония <
Сульфат свиида(1¥). Д^сульфат. свинца Pb(SO4)2 удобнее всего получать (Elbs) электролизе?! ) ппблизительпо 80%-пой серпок кислоты со свинцовыми электродами г использованием в качестве диафрагмы глиняной ячейки.
Электролиз ведут пня ti нпиостп тока 2—6 а.'й.и2, поддерживая температуру ниже 30°, однако в течение последиего часа температуру поднимают до 50“. При охлаждении раствора сульфат свинца (IV) осаждается в виде желтоватого кристаллического морошка. Водой оп разлагается. При этом в качестве промежуточного продукта образуется бесцветное сиедппепио Pb(OII)2SO4. При дальнейшем разведении последнее распадается с выделенном РЬО2. Концентрированной холодной соляной кислотой сульфат свинца разлагается с образованием желтого раствора, из которого при добавлении хлоридов щелочных металлов медленно выделяются гексахлор оилюмбатьЦ IV) щелочных металлов. С ледяной уксусной кислотой PbSO4 взаимодействует с образованием ацетата свинца(1У).
При осторожном добавлении карбоната калия или карбоната аммония к светлому зеленовато-желтому раствору сульфата свинца в серной кислоте из пего выпадают двойные сульфаты свинца (iV) (тр исулъф а то-пл юмбат) Ks[Pb(SO4)3] и (КП1])2[РЬ($О4)3] в виде тонкозернистого, желтого порошка; в холодной концентрированной серной кислоте они практически нерастворимы.
Сульфат с винца (IV) и образуемые им двойные соли обладают еще болос сильными окислительными способностями, чем двуокись свинца.
Ацетат свипца(1¥). Тетраацвтат свинца РЬ(С2Н3О2)4 кристаллизуется из раствора сульфата свпнца(1У) в умеренно нагретой ледяной уксусной кислоте при охлаждении последнего в виде белых игл с точкой плавления 175“
Проще его можно получить обработкой сурика теплой ледяной уксусной кислотой. Образующийся при атом одновременно ацетат свинца(11) при пропускании хлора можно еще частично перевести в ацетат свинца (IV), в то время как остаток выпадает в виде хлорида свпцца(П):
РЬ3О4 + 8Г1С2Н3О2 = РЬ(С2Н3О2)4 4- 2Pb(t2H3O2)2 + 4ГТ20, (13)
2РЬ(С2Н3О2)2 + С12 = РЬ(С2Н3О2)4 + РЬС12. . (14).
604
Глава 12
Аналогичным образом удается получить соли четырсхвалеатпого свинца с другими органическими кислотами, такими, как, пропионовая, масляная, стеариповад и т. д. Они плавятся без разложения, одиако в воде неустойчивы.
Алкильные производные свинца. Алкильные Соединения Свинца(IV) Представляют интерес с точки зрения механизма их образования. А именно: они возникают при действии алкильных производных цинка или магнийорганических соединений па хлорид свияца(П). Возможно, при этом будут происходить ^следующие взаимодействия (R — органический радикал):
РЬ ; С13—f—Ztx ;П2=2пС12+РЬВ2 или
,R
Р1,С1г+ 2Mg- ^MgBrg-j-MgC^+sPbRa.
-Br
Однако вместо алкильного соединения Свипца(П) PbR2 получают алкильное производное с винца (IV), так как каждый атом свинца (II) восстанавливает другой атом до металла, сам окисляясь до четырехвалентного состояния (окисление — восстановление}
2РЫ1 R2 - РЬ -ф PbiVR4.
В качество примера алкильного соединения свинца следует привести тетра-мет.илсеинсц. РЬ(СН3)4 — бесцветная жидкость уд.: веса 2,03, т. кип. 110° (защищенная от света устойчива, при воспламенении, при известных обстоятельствах•„ взрывчата); тетраэтилсвинец РЬ(СаН6)4 — бесцветная жидкость удельного веса 1,652 (при 20ч), кипящая без разложения только при уменьшенном давлении (давление пара: при 0° 0,056, при 20° 0,260, при 90° 19,0, при 152° 290 м.и pm ст), в воде нерастворима и поэтому ею не разлагается; тетрафенилмииец РЬ(С6Н5)4 — белые иглы с точкой плавления 22-iV
Тетраэтилсвинец применяют в качестве антидетонатора для моторпого топлива (добащеа в колнчествв (У,03%). Смешавтги с ним беияцв окрашивают для продажи, чтобы предупредить вредные для здоропья последствия, к которым может привести неправильное использование такого бензина.
С хлористым водородом или аз от пой кислотой алкильпос производное свинца взаимодействует с образованием продуктов замещения, например PbXR3 и PbX2R^ (X — С] иди NO3), которые обладают вполне выраженным солевым характером и для которых отчасти известны соответствующие им основания (или основные окислы).
Гидрид свинца. РЬЫ4 образуется значительно труднее, чем гидрид олова. Он был получен в 1920 г, Панетом, который использовал для этого особые условия. Оп комбинировал выделение водорода па свинцовом катоде при высокой плотности тока с распыленном электрода, причем оба" явления быстро чередовались. Гидрид свинца, как и в случае гидрида олова, осаждался в виде свинцового зеркала при пропусканий продуктов через раскаленную трубку. При этом применяли, очень плотные ватина фильтры, чтобы предотвратить распыление свинца в виде тумана.
По аналогии с гидридом олова следовало было ожидать образования РЬЩ црк разложении кислотой соединения Mg2Pb. Однако точно этого доказать не удалось-Доказательства были получены для изотопа свинца — торил В При разложений ёазбавценпой соляной кислотой магния, легированного с поверхности торием R-этрм случае на поверхности нагретой стеклянной трубки образовывался невидимый металлический надет, который чрезвычайно чувствительными радиоактивными методами идентифицировали как торий В (после затухания активности, происходящей из ТИС).
Позднее Шультце (Scbuh.ze, 1929) наблюдал образование летучего гидрида свинца также при действии атомарного водорода ца свипец. Однако цопучепный таким образом гидрид свинца не удалосг. сгустить в жадность (и противоположность гидриду свинца, пояучепнрму Панетом), так как он без остатка разлагался при попытке конденсации его.
Четвертая группа периодической системы 605
Аналитические сведения. Соединения синица при прокаливании на угле с содой дают белое ковкое металлическое ядро и желтый налет, связанный с образованием окисла. При систематическом разделении катионов в результате осаждения сероводородом свинец находится в группе основных (т. е. нерастворимых в сернистом аммонии) сулъфидов, От сопутствующего сульфида ртути PbS отделяют на основании ого растворимости в теплой, .умеренно разбавленной азотной кислоте (1 ч. азотной кислоты уд. веса 1,4 + 2 ч. воды), от остальных сопровождающих сульфидов — на основании трудной растворимости сульфата свинца. Характерными соединениями свинца являются хлорид свинца(11) и иодид свипца(П).
Для микроаналитических определений очень удобна легко получаемая из компонентов двойная соль СгЩ'ЬСщХОгЬ; при ее образовании можно установить присутствие свинца в количестве 03 у. Если свинец осаждается как РЬО2, то ого нроще всего удается определить реагентом Арнольда {тетраметилдяампаодифенилметан <CH3)2N -СвН4-С11г-СвГ14 _\(СП9)2] (сине-фиолетовая окраска).
Количественное определение свинца производят чаще всего в виде сульфата. PbSO4. Кроме того, его осаждают на аноде при ялектролдзо слабо подкисленного азотной кислотой раствора цитрата свинца в виде РЬ02 и взвешивают* в таком состоянии или же после слабого прокаливания определяют в виде моноокиси.
Очень небольшие количества свинца в водном растворе можно определить коло-рцметричесгт. Испытуемый раствор смешивают с сероводородной водой или с несколькими каплями бесцветного раствора сульфида натрия. Зависящую от концентрации окраску (желтая до буро-желтой) при готовленного таким образом коллоидного раствора сульфида свинца, сравнивают с окраской таким же образом приготовленного раствора с известным содержанием свинца. Очень надежно и просто удается определить минимальные количества свинца (вплоть до 20 у) по методу Шмидта электролитическим осаждением его в виде РЬ02, растворением последнего в ледяной уксусной кислоте, смешанной с реагентом Арнольда, и колориметрированием раствора. По данным Лидера (Jander, 1937), кондуктометрическим титрованием можно достаточно точно определить количество свинца вплоть до 1 у.
* При атом следует учесть, что двуокись свинца полностью обезвоживается с трудом. Лучше всего ее высушивать при пе слишком высокой температуре и учесть содержание воды в двуокиси, умножая ее на эмпирический фактор (0,9978, если высушивание производили прн 200°).
t
Глава 13 СПЛАВЫ
Сплавами называются гомогенные смеси металлов в расплавленном состоянии н продукты их затвердевания. Жидкие сплавы — это преимущественно растворы' металлов один в другом^ Однако в сплавах могут’ содержаться также и химические соединения в расплавленном состоянии. Природа затвердевших сплавов может быть очень разнообразной. Опи могут быть квазигомогенными (см. ниже) или совершенно пегомоген-ными, могут состоять из твердых растворов иля из соединений металлов между собой или из комбинаций двух последних типов. Металлы, образующие сплав, при затвердевании его могут выделяться таким образом, что получается более или менее грубозернистая смесь из отдельных составных частей; выделение металлов из расплава может при затвердевании и пе наступить или наступить лишь частично; металлы при охлаждении иногда могут вступать между собой в такие соединения, которые оказываются неспособными к существованию при более высокой температуре; это может происходить частично пли полностью, подобные соединения могут вновь образовывать твердые растворы и т. д. Наблюдаемое в этой области разнообразие настолько велико, что изучение природы сплавов, их особенностей и свойств, а также свойств чистых металлов выделилось в особую отрасль знания — металлографию*. Для исследования строения металлои и сплавов металлография пользуется главным образом тремя методами: во-первых, термическим анализом, который подробнее будет рассмотрен ниже; этот метод дополняется вторым, вспомогательным — микроскопическим исследованием шлифованных и полированных н затем соответствующими способами протравленных металличе^ ских поверхностей; в последнее время возник третий метод металлографического исследования — рентгеноструктуриый анализ.
В качестве вспомогательных средств металлографии следует упомянуть; определение электропроводности, определение термоэлектродвижущих сил, изучение' поведения образцов в магнитном поле, изучение потенциалов растворения, твердости и измерение расширения при нагревании (дилатометрия). Для технических целей большое значение имеет изучение зависимости практически важных свойств сплавов от их состава и обработки, например испытание прочности на разрыв и при ударе, коррознойпой устойчивости, поведения яри длительных нагрузках и т. д.
Для доказательства существования и определения состава интерметаллических соединении (см. ниже), помимо перечисленных методов, применяют химический анализ. Интерметаллические соединения часто оказываются более стойкими по отношению-к действию химических реагентов (например, кислот), чем их компоненты. При метоле «анализа остатков» необходимо убедиться (например, исс ледованием под микроскопом)
* Вместо металлографии, часто говорят о металловедении; это неправильно, так как под металловедением можно понимать науку о металлах вообще, включая сюда,, кроме металлографии, и металлургию, т. е. пауку о получении металлов иа руд.
Сплавы
607
в том, что анализируемые кристаллы не содержат включений, так как только в этом случае аналитическое определение состава будет правильным.
Р
и с. 102. Протравлепний шлиф металла, ла котором видка форма кристаллитов.
Структура металлов и их сплавов. Твердение металла или металлического сплава всегда является процессом кристаллизации. Металлы или мх сплавы никогда пе образуют' при затвердевании аморфных пли изотропных* ** продуктов, как ото происходит, например, у стекол. Кристал-литная структура многих металлов заметна уже невооруженным глазом, особенно после травления их поверхности кислотами. В других случаях ее можно обнаружить лить микроскопическим исследованием, а иногда только рентгенографически. Так как в отличие от кристаллизации из растворов при кристаллизации из расплава в виде целого куска металла отдельные поверхности кристалликов ие могут свободно раз вина гься*18. у них получаются иные внешние очертания, чсмутакихжс кристаллов при свободной кристаллизации. Каждая отдельная частица испытывает давление соседних частиц. Частицы такого конгломерата, имеющие не характерные для свободного роста поверхности, а «вынужденные!» (вследствие соприкосновения с соседними частицами) поверхности, называются кристаллитами . К риста л л лты, несмотря на то, что их поверхности не являются поверхностями свободного роста кри
сталлов, имеют довольно правильные формы. Это явление обусловлено1 пространственно ориентированными скоростями их роста. На соответствующим образом вытравленных шлифах границы отдельных кристаллитов большей частью отчетливо ГЕроявляются в виде тонких линий (см. рис. 102). В ограниченных этими линиями областях скапливаются в виде ипромежу точное о вещества» содержащиеся в металле загрязнения. Величина кристаллитов зависит от присущей им способности к спонтанной кристаллизации и от скорости роста кристаллов; этй свойства в свою очередь зависят от скорости охлаждения нДи степени переохлаждения расплава.
Мерой способности к спонтанной кристаллизации является скорость образования центров кристаллизации, т. е. число образующихся в единицу времени в е ди лице объема крнсталлгаационпнх центров. Кристаллизационными централси^яазывают
* Вещество называется изотропным, если оно обладает одинаковымитсвойствами но всем направлениям Кристаллы ан изотропны, поскольку их свойства неодинаковы во всех направлениях. Л..|щр0ны„я (а-роофое — нс имеющий формы) называется такое вещество, которое пе может существовать в правильно обраиованпых формах. Понятие аморфное и даотршгшл- вещество для твердого состояния совпадает, поскольку анизотропия всегда вызывает образование правильных форм при условии, если образование их у твердого тела может происходить свободно.
** Если метали пр я застывании образует неоднородную массу, то в пей могут сохраниться включения комггопептов, выделившихся в Виде кристаллов в первую очередь- в этом случае они обладают характерными для свободного роста кристаллическими поверхностями. Но и они могут в дальнейшем подвергаться последующей деформации.
60S
Глава 13
те точки, в которых начинается рост отдельных кристаллов. Чем больше7число криста дли эацио иных центров, образующихся в единицу, времени, и чем меньше скорость, с которой ;про исходит рост кристалла из его центра: пр -всем на правлен и км. тем более мелкозернистым будет строение затвердевшего сплава. Тонкбкристаллическоестроение большинства металлов обусловливается главным образом их большой7склонностью К спонтанной кристаллизации. д. f " :
Моно к рп столам, Если кристаллизацию металлического” расплава производить в д.'иипилх капиллярах,, то можпо достигнуть того, чтобы при очень незначительном переохлаждении кристаллизация начиналась только в одной точке. Таким способом можно получить один кристалл (монокристалл), заполняющий н.ю трубку‘длиной 20 ел! и больше. Огранка такого кристалла, как иобычных кристаллитов, имеет случайный характер, Одпако в то время Как в кристаллитах .грани по отношению к плоскостям их кристаллической решетки ориентированы совершенно с.тучайш», у кристалла всё плоскости решетки расположены в одном направлении. Монокристалл, как об этом говорит его название, представляет собой единственный? кристалл.
Р и с. 103. Плоскости скольжения проволоки. вытянутой из .,ыниК|шета.'1.'1а (c.\e.\ia). "
хотя оп имеет случайные (т. ё. нербусловливаемые его кристаллических! строением или, иначеплоскостями его кристаллической решётки) внешние грани.
: Другой-Метод получения проволоки, состоящей из одного монокристалла длиной в несколько'метров,будет описан в главе о вольфраме (ем. т. II). Он основан на том, что один конец проволоки на небольшом пространстве'недолго нагревают до температуры, близкой ,к температуре плавления. Тогда, по мере продвйжёнияу.Ьоны нагревания кристаллиты проволоки Превращаются в монокристалл. * :
Еще один метод получения монокристалла был уцазап Чрхральским (СтрсЪга!-skj, 1917). Он заключается в том, что кристаллическое зерпб77 вытягивают из расплава с такой скоростью, с которой происходит кристаллизация. ?
Монокристаллы значительных размеров (весом до нескольких килограммов) можно получать методом, описанным впервые Тамманом и заключающимся 'в том, что охлаждение расплава производят медленно, исходя из одной точки. Для Лого расплав помещают я стеклянную ила фарфоровую трубку,, нижняя часть которой вытянута в виде иглы, и медленно вытягивают его вниз из электрической печи [простая аппаратура описана; Sraunjaniw, Z. pliys. Chem. (Л) 147; 163, 1930].
Щелочные,металлы далий, рубидий и цезии “ уже при обычной: температуре настолько близки к своим температурам плавления, что они- всегда образуют мопо-крметаллы. Поэтому опи в противоположность кодгломбратам кристаллитов< не обнаруживают при обычной- температуре рщгггеповс.крй 'цнтерфёрёнции ттрк йсследовапйи методом Дебая.. -' ...
Монокристаллы по сравнению с зернистым конгломератом: кристаллитов того же вещества. отличаются гораздо большей прочностью па излом-. Тйк как в них нет примесей, внедренных между кристаллитами, то их можно пОлудатьвщрезвычайнрчистом "виде. ?;
Одггакр моискриегаллы сравнительно непрочны. Сопротивлений на разрыв аро-аолоя, сосдготаит- ин монокристалла, обычно много ниже, чем:” просолок, ттлученных протягиванием. Йто объясняется тем, что у монокристаллов-плоскости ско;п.жения расположены вдоль их кристаллографических плоскостей. спайности (рис. 103). Поэтому- проволоки, состоящие из монокристаллов, чрезвычайно тягучи. При вытягивании они утончаются неравномерно и делаются плоскими. Получающиеся при их разрыве отдельные куски пе имеют зернистого или волокнистого строения, как у обычных проволок, но гладки и блестящи; если приготовить несколько кус коп такой проволоки, то у всех кусков конечные плоскости будут параллельны между собой. Проволоки, приготовленные из монокристаллов, не обнаруживают при крученииявления, известного под названием упругого последействия,/Заключающегося в том. что по прекращении действия скручивающей силы возвращение в состояние покоя происходит не сразу. Очень важным свойством, далее; является способность таких проволок даже при высоких температурах (ниже температуры плавления) не изменять свою прочность, в то время как тянутая проволока при нагревании ее утрачивает.
С плавы
Затвердевшие сплавы. Вещество, по своему строению подобное агломератам кристаллитов, получающихся при застывании расплава хими-
чески индивидуального металла, принято называть «к ваз игом огешгым». Затвердевшие сплавы редко бывают кваз «гомогенными; они большей частью состоят из агломератов, кристаллитов, имеющих различный состав. Простейший случай — затвердевание сплава, состоящего из двух
металлов, которые в жидком состоянии неограниченно растворимы друг
в друге, в твердом вообще нерастворимы друг в друге и которые, кроме того, не способны образовывать между собой химических соединений. Явления, происходящие в этом случае, будут объяснены на примере сплавов олово — свинец, относящихся к числу наиболее давно и обстоятельно исследованных металлических сплавов.
Сплавы олово — свинец; ,эвтектика, При медленном охлаждении расплава, состоящего из олова к свинца, в случае если он содержит избыток олова, вначале кристаллизуется олово, а если в пем избыток свинца, то кристаллизуется свинец, благодаря чему сплав все более обогащается вторым колшонентом. Одновременно понижается температура затвердевания; температура Плавления и затвердевания олова (232е) понижается из-за растворенного в сплаве свинца, а температура затвердевания свинца (327°) — из-за растворенного олова, причем это понижение тем значительнее, чем больше содержание в сплаве растворенного в ном второго компонента, так же, как это имеет место и в обычных растворах. Зависимость температуры плавления (и соответственно затвердевания) от состава сплава приведена на рис. 104. Состав в
Рис. 104. Диаграмма плавкости сплавов олова со свинцом (Degen, 190!)).
атомных процентах нанесен на
ось абсцисс, а соответствующие им температуры затвердевания — на ось ординат. На диаграмме видно, что обе кривые затвердевания (соответственно плавления) пересекаются в определенной точке, отвечающей содержанию 24,4 ат.% свинца и 75,6 ат.% олова и температуре плавления 181°*. Эта точка называется эвтектической точкой. Сплав, соответ-
ствующий составу эвтектической точки, обладает из всех возможных сплавов между оловом и свинцом наиболее низкой температурой плавления и затвердевания. Его называют эвтектическим сплавом, или, короче, «эвтектикой» (ей TexTixig — хорошо обрабатываемый), а его температуру плавления — эвтектической температурой. Эвтектический сплав затвердевает как одно целое, не изменяя своего состава, и образует смесь большей частью очень мелкозернистую — кристаллитов обоих компонентов.
* Согласно новейшим исследованиям, 26,1% РЬ, 73,9%~ Sn, т. пл. 183,3°.
39 г, Pejnr
610
Глава 13
Из сплава, содержащего избыток олова, при охлаждении кристалл на олово; из сплава, богатого свинцом, — свинец до тех пор, пока не будаг достигнут состав эвтектики. На вытравленных шлифах при микроскопическом исследовании иногда ясно видно, как “образовавшиеся вначаг». как правило, большие кристаллиты одного из компонентов сплава лены в выделившуюся позднее слоистую или мелкозернистую эвтектику
Если кристаллизация чистого металла идет при сравнительно низкой температуре, то часто образуются округленные, иногда почти совсем круглые очертания ярр^д Это явление объясняется тем, что влияние поверхностного натяжения (стремящего®» вызвать образование круглых форм) при высоких температурах превосходит те мех которые обусловливают полиэдрический рост кристаллитов.
КРИВЫЕ ОХЛАЖДЕНИЯ И ДИАГРАММЫ СОСТОЯНИЯ
Наряду с микроскопическим исследованием травленых шлифов наблюдение самого процесса охлаждения сплава дает представление о природе происходящих при остывании сплава изменений и, следовательно, о характере получающихся при за-
Р и с. 105. Кривые охлаждения.
а — с остановкой; б — с перегибом п оста* новкой; в — с двумя перегибами-
твердевапин продуктов. Диаграммы, на которых, как на рис. 105, температуры остывающего сплава нанесены в зависимости от времени, называются кривыми охлаждения. Аналогичные кривые, называемые кривыми нагревания, получаются также при возрастании температуры при равномерном нагревании.
Кривая а на рис-105 характерна для совершающегося без превращений охлаждения сплава, состоящего только из одного вещества — так называемой однокомпонентной системы. В соответствии с законом охлаждения Ньютона скорость охлаждения. какого-нибудь тела пропорциональна разности его температуры с окружающим пространством, так
что температура нагретого тела понижается равномерно, по с непрерывно убывающей скоростью; Однако на кривой а рис. 105 заметно, что для определенного интервала времени (fft — -А^) происходит прекращение охлаждения; в этом случае на кривой обнаруживается так называемая остановка (т. о. весь участок кривой — Нг соответствует одной и той же температуре ts). Прекращение охлаждения на этом участке кривой обусловливается выделяющейся теплотой кристаллизации при переходе в твердое состояние. Кривая нагревания, если в первом случае удалось избегнуть переохлаждения, а во втором — перегрева, обнаружила бы такую же остановку, лежащую при той же температуре н обусловленную поглощаемой при плавлении теплотой (теплота плавления).
Таким же образом, как в случае затвердевания и плавления, и другие превращения вещества, не связанные с изменением его состава (например, переход из одной модификации в Другую), становятся заметными благодаря появлению на привой остановок, по, конечно, лишь в том случае, если тепловые эффекты таких превращений не слишком малы. Эти
Сплавы
fill
превращения могут происходить как выше,^_так и нитке температуры затвердевания.
Иная картина получается для кривой охлаждения в том случае, если сплав изменяет свой состав при охлаждении, как это имеетj место для сплавов олова и свинца, за исключением эвтектического сплава. В этом случае температура затвердевания во время кристаллизации понижается. Поэтому кристаллизация может здесь идти лишь при охлаждении сплава. Однако теплота, выделяющаяся при кристаллизации, уменьшает скорость охлаждения. Вследствие этого кривая охлаждения такого сплава имеет не остановку, а лишь «точку перегиба» (точка К на кривой б рис, 105). Только после того как расплав приобретет эвтектический состав, на кривой охлаждения обозначится. остановка, так как кристаллизация оставшегося еще жидкого расплава будет после этого протекать при постоянной температуре. Длина горизонтального отрезка кривой охлаждения, т. е. продолжительность эвтектической остановки, дает возможность судить о количестве эвтектики в затвердевшем сплаве. Кривая затвердевания сплава, состав которого с самого начала соответствовал эвтектике, имеет вид, подобный кривой затвердевания простого металла.
В нижней части рис. 104 в виде ординат показана продолжительность эвтектической остановки для различных составов сплава. Обращенное вниз острие кривой aeb (пунктирной), соединяющей конечные точки ординат, отнесенное к соответствующей абсциссе, указывает количественное соотношение компонентов эвтектики и таким образом позволяет проверить : состав эвтектики (24,4 ат.% РЬ), устанавливаемый по точке пересечения кривых затвердевания AjE и BE.
В сплавах олово -- свинец в областях, богатых свцпцом, кривая продолжительности эвтектических остановок (см. рис. 104) достигает соответствующей оси абсцисс (а — Ь) уже при содержании 88 ат.% свинца; сплавы, содержащие менее 12 ат.% олова, не выделяют эвтектики. Это зависит от того, что свинец может и в твердом состоянии «растворять» до 12 ат.%, а по новым исследованиям — до 29,7 ат.% олова (образование твердых растворов). Этим и объясняется появление пунктирной кривой ЬВ. Более подробные сведения см. стр. 614.
Диаграммы плавкости, аналогичные приведенной на рис. 104, характерны также и для других бинарных сплавов металлов, которые не образуют между собой ни соединений, ни по крайней мере в сколько-нибудь заметной степени твердых растворов.
Некоторые оловянные и свинцовые сплавы этого рода приведены в табл. 90.
Помимо эвтектических температур, в ней указаны составы эвтектик. В некоторых случаях эвтектики содержат столь незначительные количества вторых компонентов, что их влияггие па температуру плавления чистого металла до сих пор еще пе уста-
Таблица 90
Сплавы олова н свинца, не обладающих (или обладающих очень незначительной) взаимной растворимостью в твердом состоянии
Сплав ...................Sn-Al Sn-Si Sn-Bi Su-Zn Sn-Cd Sn-Hg Sii-Ga Sn-Tl
Эвтектическая температура,
°C ...................... 229 232 137 204 177 -39 20 170 '
Содержание Sn, пес. % . , . 99,5 100 42 89 67 0 8 56,5
Сплав...............РЬ-Ge РЬ-Sn РЬ-As РЬ-Bi РЬ-Sb Pb-Cd РЬ-Hg Pb-Ag;
Эвтектическая температура,
°C........... 327 183,3 288 125 247 248 : -42 304
Содержание РЬ, вес.% . . . 100 38,1 '97 43,5 87 82,5 0,35 97,5
612
Глава 13
новлепо. В качестве примеров сплавов с очень незначительной растворимостью ксгакь-нбптов можно указать сплавы алюминия с кремнием (эвтектическая температура 577°, 11,7 вес.% Si) и с бериллием (эвтектическая температура 644°, 1,4 вес.% Lefu Другие примеры будут приведены л дальнейшем.
Сплавы магния и свинца; образование соединения. Диаграмма плавкости сплавов магния со свинцом приведена на рис. 106 (Grube, 190с ь как пример того случая, когда оба компонента сплава образуют мещдт собой химическое, соединение. Из диаграммы видно, что кривая плавкости
Рис. 106. Диаграмма состояния сплавов магния со свинцом.
в этом'случае составлена из двух кривых типа, показанного на рис. 104. Соответственно этому здесь имеются две эвтектики, в каждой из которых химическое соединение содержится как составная часть, но в одном случае вместе е кристаллитами из чистого магния, а в другом с кристаллитами из чистого свинца.
Состав соединения Mg3Pb следует из положения максимума (Мх) па Кривой плавкости. Кривая охлаждения сплава, соответствующего ему по составу (80,94 вес. ч. свипца и 19,06 вес. ч. магния), обнаруживает только одну остановку, но пе имеет излома (тип кривой а рис. 105), т. е. вещество с таким составом полностью кристаллизуется при одной температуре. Состав этого соединения можно также определить и по приведенной на рис. 106 пунктирной кривой «продолжительности эвтектических остановок», т. е. продолжительности эвтектических кристаллизации, которые п на рцс. 106 показаны в виде направленных вниз ординат. Этот состав, поскольку при нем не образуется эвтектики, должен изображаться точкой, в которой обе кривые «продолжительности эвтектических остановок» пересекаются с осью абсцисс. Совершенно такой же вид имеет диаграмма плавления сплавов Mg-Sn; и в этом случае образуется Соединение Mg2Sn, к соответственно этому получаются две эвтектики.
На диаграмме рис. 106 кривая ASAfxCD, показывающая зависимость температур затвердевания от состава, с одной стороны, а с другой — прямые ab и cd, соединяющие точки эвтектических остановок, а-также ординаты обеих эвтектических точек и максимума ограничивают определенные поля, характеризующие состояние
Сплавы
613
c. 107. Диаграмма состояния сплавов магния с медью (Sahmen).
системы в пределах соответствующих температур и концентраций. Выше кривых плавкости все вещество находится в расплав ле ином состоянии. Между кривыми плавкости и прямыми ab и cd, соединяющими точки остановок, расположены области, в которых с понижением температуры происходят все увеличивающаяся кристаллизация. Б этих областях совместно присутствуют расплав и кристаллы. Ниже тем- Мо4° нератур, определяемых точками остановок, все вещество затвер- Ю50° девает. Такая диаграмма, наглядно изображающая различные со- !QOO° стояния системы, называется диаграммой состояния системы', д$д° другие примеры диаграмм состояния приведены на рис. 107. ' „„„„ 108, 110. 900
В случае образования нескольких соединений на кривой S50" плавкости соответственно получается несколько максимумов, gpp ’ как это показано на рис. 107, для случая образования двух 7^0“ соединений. Рис. 108 — пример того, как по диаграмме плав- _,tf), кости можпо установить образо-ванне четырех соединений.
Иногда максимум, соответ-сгвующнн данному соединению, подучается в скрытом виде; это 600° происходит в том случае, если это соединение плавится с разло- $50° щепием. Тогда обе соответствующие кривые пересекаются до до- уру» стяжения максимума. Состав соединения в этом случае можно определить только по кривым длительности эвтектических оста- . о ионон. Соединении, разлагающие- Л 91 ся при плавлении, называются инконгруентно плавящимися соединениями.
Образование твердых растворов. В одном из приведенных примеров (сплав Na-Pb) при- В и вне длительности эвтектических остановок пересекают ось абсцисс
по всегда в одной точке. Это явление, поскольку отклонение выходит за пределы ошибок опыта, служит указанием па об разевание твердь»; растворав (см. рис. 108).
Здесь следует рассмотреть только простейший случай образования твердых растворов, который имеет место, если существует неограниченная растворимость как в жидком, так И в закристаллизованном состоянии. Можпо ожидать, что в этом случае кривая охлаждения будет аналогичной соответствующей кривой, характеризующей охлаждение простого вещества или смеси, имеющем состав выделяющегося соединения. Однако в действительности, по крайней мере в общем случае, этого ие происходит; это может произойти лишь для некоторых определенных веществ, когда при вполне определенном соотношении компонентов выделяющиеся кристаллы имеют тот же состав, что и расплав. В общем случае состав расплава отличается от состава находящихся с мим в равновесия кристаллов. Поэтому при затвердевании состав сплава изменяется, а вместе с тем происходит и изменение температуры затвердевания. В идеальном случае получается кривая охлаждения типа, показанного на рис. 105, б. Если соединить между собой точки, в которых затвердевапйе расплавленной массы прн различном ее составе начинается (точка КЛ и заканчивается (дочка Кг), то подушится двойная кривая, приведенная ни рис. 109. Верхняя кривая — кривая ликвидуса, указывает температуры затвердевания для составов, определяемых соответствующими абсциссами, Нижняя кривая—кривая. солидуса, соединяет точки, отвечающие температурам затвердевания конечных продуктов и выражает для каждой относящейся к той же температуре точке кривой ликвидуса состав кристаллов, находящихся при этих уело-
614
Глава 13
виях в равновесии с расплавом; Например, с расплавом состава а находятся в равновесии кристаллы твердого раствора состава Ь. В соответствии с этим состав кристалла
Рис. 108. Диаграмма состояния сплавов натрия со свинцом (Mathewson).
а —расплав 4-Ха4РЬ; Ь — та. раств. (NaaPb-Na-Pb); с — та. расти. (Na4Pb-Na2Pb) +эвтек-
тика I; d — расплав + тв. раств. (laajPb-NasPb); е—расплав 4-тв. раств. (Na2Pb-Na4Pb); ; - тв. раств. (Na2Ph-Na4Pb} 4- эвтектика I; ^ — тв. раств. (NaaPb-NaaPb); h — расплав 4-NasPlr i — Na2Pb+эвтектика И, А — КаРЬ + эвтектика II; I-
расплавч- NaPb; тп — расплав 4-NaPb; ti —NaPb +эвтектика III, о — Na2Piis 4- эвтектика III; р — расплав 4- NaoPbj- q — расплав 4-4- Na2PbE, г — NaaPPs + эвтектика IV; в — тв. раств. (Pb-NajPbs)4-эвтектика IV; 1 — раевлав+тн. раств. (pb-NaaPbg); и —тв. раств. (Pb-NajPbs); и — Na4Pb4-Na.
должен был бы с понижением температуры непрерывно изменяться. Но так как в действительности уже образовавшиеся кристаллы при обычной скорости охлаждения
в большинстве случаев существенно не изменяют своего состава, то реальные кривые ликвидуса всегда более или мопсе сильно отклоняются от идеальных. Теоретическое исследование всех соотношений, возможных при образовании твердых растворов из расплава, было произведено в 1899 г. Розебумом.
Ограниченная растворимость. Нередки случаи, когда растворимость в расплавленном или в твердом состоянии неполная. Область концентраций, в которой не происходит полной растворимости, называют разрывом растворимости.
Рис. 109. Идеальная диаграмма плавкости с неограниченной растворимостью в жидком и твердом состояниях. Расплавы и находящиеся с ними в равновесии кристаллы имеют во всем интервале различный состав.
кроме излома, еще и остановку, подобно
Как пример ограниченпой растворимо-сти е твердом состоянии рассмотрим изученную Л ев конь я (Lewkonja) диаграмму плавкости для сплавов Т1-РЪ (рис. 110). До содержания 5% свинца кривые охлаждения сплавов TJ-Pb имеют, подобно кривой б на рис. 105, две точки излома. В соответствии с ; этим на диаграмме плавкости имеется в этой области типичная для образования твердых растворов двойная кривая (кривая ликвидуса и кривая солидуса). В интервале от 5 до 24% свипца кривые охлаждения имеют, как показывает линия ab, 1 , „. ..
линии б на рис. 105. Эта остановка соответствует образованию эвтектики. Таким обрат
Сплавы
615
лом, в этой области концентраций сплав при затвердевании образует негамогенные агломераты кристаллитов. Это подтверждается также микроскопическим исследованием травленых шлвфов. Таким образом, область образования твердых растворов доходит только до содержания свинца 5%. Эвтектические времена остановки нанесены на горизонтальной липни «,6, в виде направленных вниз ординат. Кривая длительности эвтектических остановок пересекает ось абсцисс в точке bit соответствующей содержанию свинца 24%. Здесь, следовательно, кончается область образования эвтек-
тических сплавов. При более высоких конце дения, имеющие характер кривой в, рис. 105: ванне только твердых растворов. Разрыв смешиваемости для сплавов Т1-РЬ, таким образом, простирается от 5 до 24% содержания свинца. В следующей области кривые охлаждения имеют только две точки излома, в соответствии с чем, начиная с этого места, па диаграмме вновь появляются кривая лмендуса и кривая солидуса. Однако эти кривые в рассматриваемом примере имеют я особенность: они соприкасаются между собой в точке В; там же лежит максимум на кривой плавкости. Если Кривая ликвидуса и кривая солидуса соприкасаются, то это, согласно сказанному выше, означает, что при этом соотношения компонентов выделяющиеся кристаллы имеют такой же состав, как и расплав. Кроме начальной и конечной точек диаграммы, где кривые солидуса и ликвидуса должны всегда сливаться в одпу кривую, так как эти точки соответствуют плавлению чистых веществ, обе эти крйвие могут, как показал Розебум, соприкасаться между собой только в точках максимума или перегиба кривых температур плавлеппя. [Перегибом кривой называется точка перехода ее из вогнутой «ветви» (участка) в выпуклую ветвь]. В атом случае, однако, из на-
Р и с. 110. Диаграмма состояния сплавов таллпя со евняцом.
личин максимума нельзя делать вывод,
что здесь обязательно образуется определенное соединение, несмотря на то, что прн концентрации, соответствующей максимуму, наблюдается совершенно такая же остановка на кривой охлаждения, как это имеет место для чистого вещества (кривая а па рис, 105). В разбираемом случае существование химического соединения следует из того обстоятельства, что максимум расположен как раз в таком месте, где соотношение компонентов соответствует простой химической формуле, а именно Т12РЬ.
Один случаи очень ограниченного образования твердых растворов был уже рассмотрен на диаграмме плавкости сплавов Sn — Pb (рис. 104). На этом: рисунке пунктирная кривая ЪВ — кривая солидуса. Разрыв растворимости в этом случае простирается от 0 до 88 ат.% РЬ*.
Если два вещества взаимно не растворяются даже в жидком состоянии и не образуют между собой соединения, то наблюдаются две остановки, соответствующие температурам, при которых происходит выделение из сплава обоего рода кристаллов.
* Новыми исследованиями установлено, что при эвтектических температурах свипец в твердом состоянии растворяет до 29,7 ат.% олова, а олово — до 1,5 ат.% свинца. То, что ранее была установлена сильно ограниченная растворимость, объясняется тем обстоятельством, что уже незначительные колебания температуры выше и ниже эвтектической резко увеличивают область разрыва растворимости и что равновесие, соответствующее этой температуре, устанавливается очень медленно. При 150° способность олова растворяться в свинце падает до 18,9%, а Свинца в олове — до 0. На диаграммах рдс. 10В и 107 видпо, что магний при высоких температурах образует твердый раствор со свинцом и медью в ограниченной степени; а именно свинец прп температуре плавления эвтектики (468й), образованной твердым раствором, богатым магнием п соединением MgzPb, растворяет до 26 вес.% магния я магний при 722° — до 2,8 лес. % меди.
016
Глава. 13
Проведенные примеры "относятся к сплавам, состоящим из двух веществ (ozsujk ные системы). Для тройных, четверных и т. д. систем имеют место аналогичные сада-ношения; только там они^соответственно сложнее.
Влияние механической обработки на свойства металлов н сплавов
Свойства металла обусловливаются не только его природой, а свойства сплава — пе только его составом. Они в значительной степени я я гл-сит также от структуры, металла, т. е. от величины и формы тех кристаллитов, из которых он построен. Структура металла в очень значительней1
Освещение
Линии скопьхееиия
Рис. 111. Возникновение линий скольжения (схема).
степени обусловливается произведенной ня холоду или при умеренном нагревании .Bf-ханической обработкой (холодном обработкой), которой подвергается металл (ковка, прокатка, прессование и в наибольшей степени вытягивание в проволоку). Так, для меди путем соответствующей механической обработки удается повысить ее сопротивление разрыву в 14 раз.
Изменения, которым подвергается металл в результате холодной обработки, определяются изменением ого структуры. При рассмотрении полированных поверхностей под микроскопом заметны линии скольжения, которые образуются при относительном смещении отдельных частей кристаллитов. Линии скольжения заметны, если смещение происходят в направлении наблюдения; тогда па обращенной к наблюдателю гладкой поверхности выступают бороздки (рис. 111). Линии скольжения ни в коем случае не сиге дует смешивать с образующимися при слишком сильной деформации трещинами. 1Плос-
ностц, вдоль которых происходит скольжение, называются плоскостями скольжения; чем больше способность материала образовывать плоскости скольжения, тем значительнее его пиил/честь', чем меньшее число плоскостей скольжения образует магериал, тем он более крулах. В то время как большинство металлов, а также н их твердые растворы^ в общем легко образуют плоскости скольжения, соединения металлов между собой хрупки (за исключением областей температур, близких к их температурам плавления).
ТамМап п другие исследователи установили, что при механической обработке свойства отдельных кристаллитов но изменяются, Твердость и структура их кристаллической решетки но изменяются. Изменяется только их внешняя форма. Считают, что с этим связано уменьшение внутренних напряжений, которые существуют в металле. Обычно тонкозернистые структуры, например быстро охлажденные сплавы или эвтектики, обладают большей твердостью, чем грубозернистые. При холодной обработке происходит измельчение структуры обрабатываемого материала. Если длительным нагреванием (отжигом) дать возможность зернам вновь вырасти, то металл приобретает свои первоиачальныо свойства. Это явленно называется рекристаллизацией. Кроме того, при вытягивании металлической проволоки кристаллиты, которые до этого имели беспорядочную ориентировку, располагаются примерно параллельно друг другу. Благодаря этому проволока приобретает более или менее волокнистое строение (рис. 112). Последнее достигается нередко и при прокатке. Волокнистое строение особенно увеличивает прочность материала. Если проволоку сильно нагреть, она становится ломкой, так как металл при этом вновь.
Сплавы
617
приобретает то зернистое строение, которое ему свойственно и которое он имел до протягивания в проволоку (рис. 143). Таким образом, становится понятным, почему проволока, состоящая из монокристалла, не содер-
Рис. 112. Волокнистая структура Рис. ИЗ. Зернистая структура про-вытянутой проволоки. волоки после рекристаллизации, вы-
званной отжитом.
жащего, очевидно, деформированных кристаллитов,^ не подвергается «рекристаллизации», сохраняя без изменения свои механические свойства также и при высокой температуре.
ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ И ТВЕРДЫЕ РАСТВОРЫ
Определение и четкое ограничение понятия химическое соединение' в отношении металлов, как и в некоторых других случаях, в которых имеют дело с образованием тверды^ растворов, представляет определенную трудность.
Критерий большей устойчивости химических; соединений по сравнению с физическими смесями (трудность разложения) в атом случае, так же как, впрочем, и во многих других, оказывается недостаточным. Определение химических соединений, основанное на законе постоянных и кратных отношений, является, как показывают последние открытия в химии, слишком узким. ,
На основе закона постоянных и кратных отношений можно дать следующее Определение: продукт взаимодействия двух или нескольких веществ следует рассматривать в качестве, химического соединения в том случае, если атомные соотношения составных частей его выражены простыми целыми числами, не изменяющимися непрерывно' при изменении внешних условий. В общем случае это положение сохраняет свое значение; однако имеются такие вещества, которые с точки зрения приведенного выше-определения следует рассматривать как химические соединения; есть также вещества, которым на основе других критериев должны быть также приписаны свойства химических соединений, хотя И в первом и во втором случаях их состав колеблется в определенных пределах. Прежде пытались рассматривать эти вещества как твердые растворы с другими простыми или сложными веществами, но это объяснение невсегда возможно (см. т. И). Поэтому следует пересмотреть представ ле tine, в соответствии с которым химические соединения всегда обладают постоянным составом. Соединения с непостоянным составом, не подчиняющиеся, следовательно, закону, «формулы рота шюму Дальтоном при становлений атомной теории, называются недалъто видными соединениями в отличие от далыпонидных соединений постоянного состава, подчиняющихся закону Дальтона. Иедальтонидные соединения встречаются не только среди сплавов, по и среди других веществ, к притом совсем иередко. Например, можно указать па соединения с «блуждающими в решетке составными частями^ (см. т. II), Но особую роль педальтолидпые соединения играют в системах, построенных только из металлов, и ипервую очередь в таких системах, вобразовапии которых принимают участие металлы побочных подгрупп. Согласия современным прерставле-пиям, соединения, в состав которых входят только металлы главных подгрупп, обладают непостоянным составом только в исключительных случаях. Поэтому существование недальтонидпых соединений и нх отношение к твердым растворам будет> рассмотрено в дальнейшем нрп) обсуждении побочных подгрупп периодической? системы.
Если, однако, отвлечься от этой пограничной области, не имеющей большого значения для силанов, образованных металлами главных под
«18
Глава 13
групп, в которой понятия твердого раствора и кристаллического химического соединения переходят друг в друга* , то и в случае •металлов соединения и твердые растворы обладают характерными различиями. Твердые растворы по своим свойствам в общем случае соответствуют своим составным частям, в то время как свойства соединений могут значительно отличаться от свойств последних. Уже было отмечено, что кристаллы соединений дочти всегда хрупки. Электропроводность интерметаллических соединений также обычно ниже, чем электропроводность их компонентов. Свойства соединений обычно тем больше отличаются от свойств их комйонентов, чем больше последние отличаются друг от друга в отношении своего сродства к электронам. В то же время интерметаллические соединения также и по своему составу приближаются к обычным химическим соединениям. Состав соединений, образованных электрохимически сильно различающимися металлами, большей частью и в количественном отношении соответствует составу солеобразных соединений этих металлов. Другими словами, металлы, сильно различающиеся электрохимическими свойствами, часто проявляют в образуемых ими соединениях те же (формальные) валентности, как и в своих соляхОднако для огромного большинства интерметаллических соединений те валентности, которые соответствующие металлы проявляют в своих солях, не имеют особого значения. Нередки случаи, когда два металла образуют ряд соединений, причем ни одно из них не имеет состава, соответствующего валентностям металлов в их солях. Поэтому вполне естественно усомниться в том, следует ли придавать вообще какое-либо значение тому обстоятельству, если в длинном ряду соединений, образуемых двумя металлами, какое-нибудь одно или два соединения соответствуют требуемому нормальной валентности составу. Даже в том случае, если два вещества образуют только одно соединение и оно ио своему составу как раз соответствует тому, которое следует из валентностей этих металлов в их солях, — даже в этом случае нельзя еще только иа этом основании связывать состав этого соединения с проявлением нормальных валентностей, присущих образующим его металлам в их солях или других соединениях, носящих явно гетерополярпый характер. На основании сказанного выше можно лишь предполагать такую зависимость для металлов, сильно различающихся своим электрохимическим характером, В этом отношении магний, ио-видимому, больше всего обладает склонностью образовывать с сильно электрохимически отличными от него веществами соединения, соответствующие по своему составу его нормальной валентности. Нередко в интерметаллических соединениях наблюдаются атомные соотношения, значительно отклоняющиеся от простых числовых соотношений. Например, встречаются такие соотношения, как 5 : 6, 3 : 8, 4 : 15, 12 : 13, 12?: 17, 8 : 25 и т. д. Для изучения природы интерметаллических соединений большое значение имеет измерение теплот их образования.
Теплоты образования интерметаллических соединений, как правило, тем выше, т. ё. связи в них тем прочнее**, чем менее благородны их составные части (Biltz).
* В таких случаях для решения вопроса, имеют ли дело с химическим соединением или только с твердым раствором, часто прибегают к калориметрическим определелиям; определениям плотности и при некоторых условиях также и к тензиометрическим определениям. Но все же особое значение именно в этой области приобрели рентеепографические методы (см. т. П).
** Так как речь в этом случае идет о твердых веществах, то величины сродства Можно считать приблизительно пропорциональными теплотам образования.
Сплавы
619
Если к соединению, об разе нэп ному двумя металлами, присоединяются еще дополнительно атомы одного из этих металлов, то соответствующие теплоты образования по мере насыщения становятся все менее значительными. Так, для соединений. Ca4Zn, Ca2Zn3, CaZn4 и CaZnI(J, в которых па 4 атома Са Приходится соответственно по 1, 6, 16, 40 атомов Zn, теплоты образования на 1 г-атом ципка соответственно равны 32, 13, 7,4, 4,8 кте.г; таким образом, чем больше цинка уже связано, тем меньше выделяется тепла при его дальнейшем присоединении. Аналогичные соотношения наблюдаются и для других соединений металлов. Здесь, очевидно, существует та же закономерность, которая проявляется и в случае, например, присоединения аммиака к солям. Это обстоятельство замечательным образом сближает интерметаллические соединения с комплексными или с координационными соединениями и подсказывает возможность рассмотрения их с точки зрения координационного учения {Silts, 1924).
Интересны наблюдения Крауса (Kraus, 1907), согласно которым многие интерметаллические соединения, растворенные в жидком аммиаке, ведут себя как электролиты, т. е. диссоциируют на ионы. Этот процесс полностью соответствует диссоциаций при растворении соли в воде. Циптль (Zintl, 1931—1932) сумел обнаружить (в таких растворах солеобразные комплексные соединения типа полисульфидов; г. е. соединений, образованных (возможно, сольватированным) катионом сильно элек-троположительгеого металла и комплексным анионом, возникающим в результате присоединения нейтрального атома металла к отрицательно заряженному атому этого же металла. Такого рода электроотрицательные ионы образуются только металлами главных подгрупп IV—VII периодической системы, т. е. металлами, стоящими на четырех первых местах перед инертными газами. Тем самым было установлено, что эти (и только эти) металлы могут заряжаться отрицательно, т. е. принимать столько отрицательных зарядов-электронов, сколько необходимо им для заполнения своей электронной оболочки до оболочки типа инертного газа. В связи с этим интересно также наблюдение, что при внедрении Сильно электроположительного атома металла, например Na, в решетку свинца происходит не расширение, а сжатие решетки, хотя атом Na л больше атома РЬ. Это наблюдение объясняет увеличение твердости свиица при добавлении к пему незначительных количеств щелочных или щелочноземельных металлов (см. стр. 588).
Многие неметаллы, помимо обладающего полуметаллическим характером мышьяка, в первую очередь фосфор, кремний и сера, а также селен и теллур и иногда бор и. углерод, образуют с металлами соединения, которые по своему характеру полностью соответствуют интерметаллическим соединениям. Такой жо характер имеет большинство соединений, образуемых названными элементами между собой.
Отношение металлов первых четырех групп друг к другу
Табл. 91—94 показывают, в какой мере металлы первых четырех главных подгрупп способны растворяться Друг в друге в расплавленном или твердом состоянии п какие бинарные соединения они образуют. Для сравнения в таблицы включены также частично и неметаллы этих групп.,
В настоящее время имеется щце очень мало данных о сплавах бериллия с. металлами первых четырех главных подгрупп. С алюминием Be ие образует соединения; между обоими металлами в твердом состоянии существует только очень ограниченная растворимость. В системе Be—Sn в жидком состоянии также отсутствует неограниченная растворимость.
Особенностью металлов четырех первых главных подгрупп, заметной уже при беглом ознакомлении с таблицами, является их в общем случае очень незначительная склонность к образованию твердых растворов. Среди исследованных систем нет пи одной, за исключением систем K/Ro, K/Cs,: Rb/Cs. с неограниченной растворимостью, а также Ca)Sr, Са/Ва, Sr/Ba (см. стр. 272), в которой прн обычной температуре существовала бы значительная растворимость. Во многих случаях металлы ограниченно растворимы даже в расплавленном состоянии. Поразительно далее, что щелочные металлы (за исключением Li) вообще не способны, насколько это известно и настоящее время, к образованию соединений со многими металлами II и III главных подгрупп (например, А1), в то время как с Sn и РЬ они образуют особенно большое число соединений. Из таблиц также следует то положение, что металлы в своих
Таблица Sj
Растворимость и образование соединений элементами первых четырех главных подгрупп
Li Na К Rb с$ Mg Са Sr Ba
Li ж. >0 тв. 0 0 ж. 0 тв. 0 0 ж. 0 тв 0 0 ж. 0 тв. 0 0 ж. оо тв, со LiMg2 UCa2 * .225° — ж. co тв. 0 LlBa,• 425’ LiBa^ 523’
Na ж, со тв- 0 KNa2 ж. со тв. 0 0? ж. со тв. 0 CsNa2 ж. < оо тв. 0 0 ж. < со тв, 0 0 ж. оо тв. 0 0 ж. co тв. 0 Na2Ba 96° Na6Ba 97° NaBa 510°
К ж. со* ТВ, оо 0 ж. со тв. со 0 ж. со тв. 0 0 — —
Rb ж. со ТВ, со 0 Mg ж, со тв, — 0 CaMg2 ж. оо тв. ~0 SrMg ? SrMg2 880° SrMg4 * 598° SrMg9 603° Ж, co тв. — 0 BaMg2 807° BaMg4 * 598° BaMgs 707°
Са тв. co 0 ТВ. <co 0
Sr jb. <oo 0
Примечание
Указания курсивом относятся к растворимости, обычным шрифтом—к образованию соединений. Иннонгруентно плавящиеся соединения отмечены значком *, недальтонидные соединении—черточкой над формулой; знак [ ] указывает, что соединение образуется в результате реакций в твердом состоянии. Цифры под формулами—температуры плавлении или разложения, соединений в градусах Цельсия.
Растворимость;
со неограниченная, < оо ограниченная, > О очень ограниченная,
О отсутствие растворимости, .Ж.—ЖПД1ШЙ, тв,—твердый.
Образование соединений:
О пет соединения,
— диаграмма состояния не изучена или научена недостаточно.
Цифры под формулами означают температуры плаеленНя соответствующих соединений .
Таблица 92
Растворимость и образование соединений элементами трех первых главных подгрупп с элементами главной подгруппы III группы
I Обозначения см. и табл. 91. Если указания имеются только относительно твердого состояния, в жидком состоянии, как правило, существует неограниченная растворимость 1
В Al Ga in T1
LJ — ж. co, тв. О Li2Al, LiAl LiGa тв. <co Llln 625° тв. >> 0 Li4Tl* L13T1 381° 447» bigTla 1л2Т1 f ] 448° 381е LiTl 503’
Na NaB2 ж. < oa, tb. >0 0 Соединение Na Tn тв. >0 NaeTi *, Na3Ti * 77,4° 154» NaTl 305°
К — ж. < се, тв. 0 0 — тв. 0 K2T1 * KT1 242° 335°
Mg Mg3B2 MgsB2 ж. оо, тв. >0 MgjjAl12, MgsAl3 MgAl ? тв. ~0 Mg3Ga2, Mg2Ga 456° 441° MgGa MgGa2 378° 285° тв. >0 Mg5In3* MggJn Mg in, Mgln3 TB. < co Mg5Tl2, MgaTl * 413° 392° MgTl 358°
Са СаВ6 ж. оо, тв. — 0 СаА12, СаА13:. — — tb. <00 CaT.1, Ca3TI4* 970° , 557° CaTl3 524°
Sr SrB6 тв. — 0 StAL, StA14 — SrTi
Ва ВиВв тв. ~0 ВаА14 — — BaTl
в АЩ2, А1В12 — — ж, 0, тв. 0 0
А1 тв. > 0 0 — ж. ~0, тв. 0 0
Ga тв. >0 0 —
la ГВ. <00
Таблица 93
Растворимость и образование соединений металлами трех первых главных подгрупп с металлами главной подгруппы IV группы
Обозначения см. в табл. 91. Если указанна имеются только относительно твердого состояния, в жидком состоянии, как правило, существует неограниченная растворимость
Li Na К Mg Ca Sr Ba Al Ga In Tl
Ge — _NaGe KGe KGe4? тв. ~0 Mg2Ge 1115° CaGe — — тв, >0 0 — — —
Sn тв. О Li4Sn Ia7Sn2 IJ3Sn2 Li2Sn LiSn LiSn2 тв. 0 Nai6Sn4 Na2Sn Na4Sn3 NaSn NaSn2 тв. 0 K2Sn KSu KSn2 KSn4 тв- >0 Mg2Sn тв. 0 Ca2Sn CaSn CaSn3 тв. 0? SrSn SrSn3 SrSns ТВ. 0 ? BaSn3 BaSn5 ТВ. 0 0 тв. 0 0 ТВ. CO In3Sn ? InSn15 ? тв* <; co 0
Pb тв, >0 Li4Pb ; Li7Pb2 Li3Pb -1д5РЬ2 LiPb тв. >0 Na^Pb Na,Pb2 Na2Pb NaPb NaPb3 тв. 0 K2Pb ? KPb ? KPb2 KPb4 тв. >0 Mg2Pb тв. ~0 Ca2Pb CaPb СаРЬз SrPb3 тв. ~0 Ba2Pb BaPb* BaPb3 ж. <oo тв. 0 0 ж. <oo тв. 0 0 ж. <co In15Pb * 1'72° 1и13РЬ2* 159° ж. <co T]2Pb 380°
С плави
623-
Таблица 94
Растворимость в образовапие соединений элементами главной подгруппы JV группы (Обозначения си. в табл. 91)
SI Ge Sn РЬ
С А, —; sue, SiC —; 0 ?
Si ж. co, тв. оо; 0 ж. co, тв. 0; 0 ж. 0, тв. 0; 0
Ge ж. оо, тв. 0; О ж. от, тв. 0; О
8п ж. со, тв. j>0; 0
Рис. 114. Структура решетки С aSn$. о=4,732 A; Са = Sa-3,35 А.
лруктуры является соотношение об-
соединениях между собой, как правило, обладают совершенно иными валентностями, чем в солеобразных соединениях. Далее в Таблицах содержатся примеры того, как часто в интерметаллических соединениях наблюдаются относительно сложные атомные числовые соотношения. Состав соединений, в которых имеют место особенно-.сильные отклонения о г простых числовых’ соотношений, впервые был правйльно-’ установлен на основе рентг ено Структурных определений. Так, соединению Mg1;Al13 (Laves, 1934) ранее приписывали состав Mg3Al2 или Mg4Ai3, а соединениям NatjSn4 и Na15Pbt (Zintl, 1938) — состав Na4Sn и Na4Pb. Следует отметить, что и для состава других соединений (например, Li4Sn я Li4Pb) при помощи рентгенадтрунтуртгого анализа были установлены более сложные формулы, чем предполагалось ранее.
Как уже было указано, соединения переменного состава в бинарных системах, образованных только элементами первых четырех главных подгрупп, встречаются весьма редко. Это, вероятно, связано с малой склонностью этих элементов к образованию твердых растворов, У элементов, в значительной степени склонных к образованию твердых растворов — главным образом у элементов побочных подгрупп периодической системы,— интерметаллические соединения переменного состава образуются очень часто. В определенных типах соединений этих элементов существенным для состава и
щего числа электронов к общему числу атомов (правило Юма — Розери, т. II).
Однако у очень большого числа интерметаллических соединений, в том чисяе-и у образованных только элементами главных подгрупп, число валентных электронов никак не влияет на состав, для которого существенны только чисто геометрические факторы (стр. 333). 1
Б качестве примера структуры типичного интерметаллического соединения карие. 114 представлена решетка CaSn3. Если мысленно занять все ее уэловые точки, атомами о di юг о и. того -же рода, то возникнет представленная на рис. 46 кубическая грапецентрированная решетка, очень часто встречающаяся среди свободных металлов. Такую же структуру, как и CaSn3, имеют ’СаРЬ3 (а == 4,89 A), NaPb3(a = 4,87 А), SrPhj (тетрагонально искаженная решетка а = 4,98, е = 5,03 A), TaSa3{a — А),
LaPb3(a = 4,89 А.), а также ряд других аналогичных по составу интерметаллических соединений с лантанидами или с металлами побочных подгрупп (Ziatl, 1931—1933,. Rossi, 1933). Замечательно, что соединения Mg2Ge, Mg2Sn и ftfg2Pb, соответствую--
624
Глава 13
щие по своему составу солеобразным соединениям, обладают также и структуру типичной для солеобразных соединений, а именно Структурой типа полевого ггдт (Mg2Ge ; а = 6,38 A; Mg2Sn ; а А 6,77 A; Mg2Pb: й=6,84 А). Соединения ГдУА. SrMg2 и BaMg2 кристаллизуются по типу MgZn2 (см. т. II, гл. 1). Mg способен зезе-щать в твердом растворе 0,047 ат. % Са, 0,0042 ат.% Sr и 0,0018 ат. % Ва (при эвтектических температурах). Соответствующие эвтектики: -
Sr-SrMgj 438’
65 ат. % Sr
Ba-BaMg2
358°
65 ат. % Ва
SrMg4-SrMg9
587°
15,5 ат.% Sr
BaMg2-BaMgt
587’
27 ат. % Ва
SrMge-Mg 585*
6 ат.% S»
BaMgB-Mg
634°
2,5 ат. % Ва
По Новотному (Novotny, 1939) существует соединение SrMg, а также LaMg, СеМо и PrMg (см. т. II, гл. 10), кристаллизующиеся ио типу CsI. Однако Клемм (Klemm, 1948) не обнаружил никаких доказательств их существования. Соединение SrAl по структуре близко к ^-латуни, SrAl4 иаотипно ВаА14.
До сих пор речь шла только о бинарных соединениях. Но существуют также и тройные интерметаллические соединения, которые, однако, встречаются реже.
Гл а в а 14
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Главная подгруппа)
ГРУППА АЗОТ — ФОСФОР
Порядковый номер Элемент Символ Атомный вес Удельный 1 вес 1 Тестера- 1 тура илавле-лля, °C Температура кипении, лс Теплота плавления, яжо-Д/з*. tlfttOJrt валентность
7 Азот N 14,008 0,96а —210,0 —195,8 0,085 1, И, ш, IV, V
15 Фосфор Р 30,975 1,826 44,16 2806 0,155 I, in, iv, v
33 Мышьяк As 74,91 5.72й 817г 633 5, Г Ш, V
51 Сурьма Sh 121,70 6.69 630,5 1640 4,8 JII, IV, V
83 Висмут Bi 209,00 9,80 271,0 1560 2,64 II, III, V
а Для твердого авота при 1'емиературе плавления
б Белая (вубпЧеонан) модификация.
в Серая металлическая (ромбическая) модификация.
г 1(ри 36 ат.
Общие сведения. Элементы главной подгруппы V группы — азот, фосфор, мышьяк, сурьма и висмут — в своих кислородных соединениях максимально пятивалентны, по отношению же к водороду они бывают исключительно трехва л битными; Большинство этих элементов пятивалентны также и в отношении других электроотрицательных элементов, прежде всего фтора, хлора, брома и серы. Однако наряду с валентностью пять они всегда проявляют по отношению к ним и валентность три. 1
С увеличением атомного веса этих элементов склонность их к образованию трсхвалентных соединений с кислородом и галогенами все больше и больше преобладает над склонностью к. образованию пятивалентных соединений. Одновременно уменьшается устойчивость их водородных соединений.
Это поведение элементов глав ной подгруппы V группы становится особенно наглядным, если рассматривать их атомы в соединениях с кислородом, галогенами и г. д. в качестве положительно заряженных иопов, а в соединениях с водородом— в качество отрицательно заряженных ионов. Тогда имеет место следующее. Элементы главной подгруппы V группы обладают максимальной положительной валентностью нятъ, но они часто проявляют также и положительную валентность три. Склонность к приобретению только трех положительных зарядов увеличивается с возрастанием их порядкового номера. Кроме того, эти элементы могут выступать и кал отрицательно трехва-левдпые. Эта способность заметно убывает по направлению от азота к висмуту.
40 Г. Рейн
626:
Глава 14
Азот, фосфор, мышьяк и сурьма в твердом состоянии могут существовать в виде: нескольких модификаций.
Обычной модификацией мышьяка, сурьмы и висмута является металлическая модификация. Структура такой решетки представлена на рис. Н5. Ёе можно представить себе как состоящую па двух вдвинутых один в другой гране центрированных ромбоэдров. Эту решетку можно охарактеризовать также, как деформированную решетку типа каменной соли. Черный фосфор кристаллизуется пр тому же типу.
Его решетка построена из двойных слоев. Каждый отдельный слой состоит иа лежащих друг над другом зигзагообразных цепей
Р Р \р/\р/\р/ (4=ЭД°; Р Р—2,17 А)
Атомы фосфора цепей, образующих нижний слой, связаны также поочередно ,с атомами фосфора, входящими в состав цепей, расположенных справа п слева вверху (Р 4—> P^2,20 А). Таким образом, цепи связаны накрест между собой в двойной слои с сетчатой структурой. Каждый атом
Рис. 115. Решетка типа сурьмы.
AS SI) Hi
u=5 ;5i) 6,20 6,56 A
а-3^36' №58' 87°34'
d—е-2; b 1 2,87 3,10 A
ti-t-c—3,1 5 3,37 3,47 A
фосфора имеет, следовательно, трех соседей, из которых два принадлежат к той же цепи, а третий — к другой. В парях Р, As и SB при: сравнительно высоких темйср атурах четы/жгатснпгы, Bi -- йвутатомен. Молекулы Sb4 известпы только в газообразном состоянии. Молекулы As4, напротив, могут конденсироваться с образованием твердого желтого мышьяка, который метаста-бплсн и легко превращается в другие модификации (см. стр. 701). Белый фосфор, построенный из молекул Р4 (тетрафосфор), при обычных температурах также метастабилеп, но стабилен при высоких температурах (выше температуры, плавления красного фосфора) (см. стр. 675).
Замечательным для элементов главной подгрупп!,I V группы являются низкие значения их температур плавления: и кипения по сравнению с рядом стоящими слева элементами периодической системы. Это особенно характерно для наиболее лёгких элементов, В то время как углерод чрезвычайно трудно расплавить и испарить,- азот представляет собой трудпосжйжаемып газ.: Для: фосфора и кремния разница в температурах плавления и летучести также чрезвычайно велика.
Элементы мышьяк, сурьма и висмут в водных растворах могут присутствовать в трехвалептном состоянии в виде свободных положительных ионов As"\ ,Sb"‘ и Bi"’. Однако соответствующие этим ионам соли проявляют большую склонность к гидролизу. Для мышьяка существует равновесие
As— • ЫМ) 7* [АяО-Г-МИГ,
отсюда следует, что этот элемент в виде иона As’" может находиться в заметном количестве только в сильно кислых растворах. У сурьмы ,это равновесие Сильно сдвинуто влево и еще более: — у висмута.
Это положение соотлетствует общему правилу, что основной характер гидроокисей в каждой главной подгруппе периодической системы возрастает по направлению
Пятая группа, периодической системы
627
сверху близ. Невозможность обнаружения свободных положительных попов азота и фосфора можно в соответствии с этим правилом объяснить допущением, что в этом случае равновесие сдвинуто еще сильнее вправо, чем для мышьяка.
Вес гидроокиси, образуемые элементами главной подгруппы V.группы в пятивалентном состоянии, имеют характер кислот.
NO2(OH),
Лзотнаи кислота
ГО(ОН)3,
Фосфорпая кислота
AsO(OH)s,
Мышьяковая кислота
H[Sb(QH)6), Bi2O5-H2O?
Сурьмяная Гидрат пнтиогиен висмута, кислота висмутовая кислота?
В соответствии с приведенным выше правилом кислотный характер гидроокисей в каждой главной подгруппе Периодической системы, уменьшается с возрастанием порядкового номера элемента. Это правило оправдывается й В: даппом случае: азотная кислота — очень сильная кислота, фосфорпая =- кислота; средней силы, а остальные гидроокиси — все слабые или Даже очепь слабые кислоты.
Мышьяковая и сурьмяная кислоты в виде соединений с указанным выше составом известны только в растворах или в виде солей (см. стр. 706 и 718) . Является ли гидрат пятиокпен висмута соединением с определенным составом [виСмутовой кислотой BiOa(OH) или BiO(OH)3), точно не установлено, однако известны соли с вполне определенным составом — производные висмутовой кислоты (см. стр. 729). .'
Таблица 95>
Нормальные гжислы элементов главной подгруппы пятой группы и их теплоты образования (ккал/молъ)
n2o Закись азота: газ гюб разная -—19,6 жидкая—15,6 NO Окись азота: газообразная —21,6 n203 Трехокись азота: газообразная —20,0 жидкая —10,6 Л'Оа Двуокись азота: -8,0 n2o4 четырех окись азота: газообразная —2,4 жидкая -1-6,8 n2o5 Пятиокись азота: газообразная —1,2 жидкая 4-3,6 твердая 4-11,9
— — Р2О3 Трехокись фосфора — P2O& Пятибкись фое-: фора 4-370
— As3O3 Трехокись мышьяка 4-156 — АдЕО5 Пятибкись мышьяка 4-219
— 8Ь20з Трехокись сурь- : мы 4-163 Sb20s Пятиокись сурьмы 4-230
— Bi2O3 Трехокись висмута -J-138 — В12О5 Пятиокись висмута
40*.
628 Глава 14
Элементы главной подгруппы V группы могут проявлять не то.тьес трех- и пятив алентность, но также и другие степени валентности. Это прежде всего относится к азоту и фосфору. Азот по отношению к кислороду может вести себя как одно-, Двух-, трех-, четырех- и пятивалентный элемент. У фосфора, хотя его окис .ты и соответствуют нормальным степенны валентности (III и V), известны кислородные кислоты, в которых os проявляет две другие степени валентности (I и IV).
Краткий обзор окислов и кислородных кислот элементов главной подгруппы V группы приведен в табл. 95 и 96, в которые вошли только нормальные окислы и кислоты., т. е. соединения, соответствующие определенным степеням валентности этих элементов. £ таблицы не включены чепгырехакшш, например SbsO4—SbllISbvO4 (ср. стр. 719) , образующиеся при взаимодействии трехоки си и пятиокиси, а также перекисные кислоты. Молекулярные веса в парообразном состоянии приняты во внимание только для тех -соединении, которые при обычпой температуре газообразны или могут быть исследованы в растворах. Под названиями соединений приведены их теплоты образования. Последние даны во всех случаях На 1 моль соединения и везде, где нет специальных указаний, относятся к соединениям в твердом состоянии. Часть кислот, приведенных в табл. 96, известна только в растворах или в виде их солей.
Таблица 96
Кислородные кислоты элементов главной подгруппы пятой группы и теплоты их образования
Указанные в таблице теплоты образования (ккал) относятся к 1 молю твердого соединения
М2о2 Азотноватистая кислота h2no2 Нитролспло- вая кислота hno2 Азотистая кислота —! HN03 Азотная кислота 4-42,2
Н3РО2 Фосфо рпо па-тис тая кислота 4-140 — Н3РО3 Фосфористая кислота 4-228 Н4Р2О6 Фосфорнова-тая'кислота Н3РО4 Фосфорная кислота -|-304
— — НАзО2 Мышьяковистая кислота — Н3АзО4 Мышьяковая кислота
— — H3SbO3 или 8Ь(ОН)3 Сурьмянистая кислота — H[Sb(OH)6] Сурьмяная кислота
— Bi(OH)3 Гидроокись висмута 4-138 — Bi2O&-H2O пли НВЮ3? Висмутовая кислота?
Пятая группа периодической системы,
629
Ниже приведен обзор хлоридов и сульфидов элементов главной подгруппы пятой группы
Хлориды Сульфиды
+С1 — NC13 - — NsS4 — — NsS4 —
— • Р2С14 РС13 PCJ5 F.+3 — P4S5 — PiS, P+i
— — AsCJ3 — AsaSj AS4S4 — As+3 — — As2Si
— — SbCl3 SbCLi —- — 86283 — — 86285
— — ^4^4 — Bi2Sg — — —
Характерным для сернистых соединений является очень' значительное увеличе-ние ях устойчивости в направленна от азота it висмуту. Сернистые соединения азота црн нагревании вспыхивают; сернистые соединения фосфора в отсутствие воздуха перегоняются без разложения, но па воздухе воспламеняются уже при умеренном нагревании. Значительно устойчивее сульфиды мышьяка, сурьмы и висмута, которые вследствие этого нередко встречаются в природе. Сурьма и висмут даже распространены в природе главным образом в виде сульфидов. Сернистые соединения азота при гидролизе выделяют аммиак и образуют кислородные кислоты серы. Напротив, при гидролизе сульфидов фосфора наряду с кислородными кислотами фосфора образуется сероводород, Это показывает, что в сульфидах азота отрицательный заряд имеет азот, в сульфидах же фосфора, наоборот, сора. Как следует из способов образования, последнее справедливо также и дкя сульфидов мышьяка, сурьмы и висмута, которые вследствие их крайней нерастворимости не разлагаются йи водой, ни разбавленными кислотами.
Обзор простейших водородных соединений (гидридов) элементов главной подгруппы V группы и их важнейшие свойства сопоставлены в табл. 97. Устойчивость гидридов очень сильно снижается от NH, к ВЩ3.
Таблица 97
Свойства простейших соединений элементов главной подгруппы пятой группы с водородом
Свойства Формула и мазванле
аммиак PHS фосфин AsH3 арсин St>Hs стибии Bi Н3 ВИСМУ* тистый водород
Температура кипении, °C —33,3 -87,4 -58,5 — 17,0 +22
Температура плавления, °C Теплота плавления, —77,7 —132,5 -111,2 —88,5 —
ккал/молъ 5,65 3,83 4,34 5,08 —
Константа Трутопа , . . Критическая температура, 23,6 20,6 20,3 19,8 —
°C Плотность жидкости при 132,5 52,0 — '— —
температура кипения Поверхностное натяжение 0,681 0,765 1,621 2,204 —
жидкости прп температуре кипения, дип./см 34,25 20,50 21,08 24,19 —
Дипольный момент моле-
кулы в газообразном состоянии, ц (ед. CGS) 1,44'10-18 0,55-10-18 0,15-10-18 — —
Замечательно, что температуры кипения и плавления, теплота плавления и поверхностное натяжение понижаются от NHS к РИ3 и затем снова повышаются при переходе к BiHs. Это происходит оттого, что аммиак в отличие от Своих аналогов в жид-
Таблица 98
Растворимость и образование соединений металлами Главной подгруппы пятой группы между собой и с металлами четырех первых главных подгрупп
Условные обозначения см. в табл. 91. Если имеются указания только о твердом состоянии, то в жидком наблюдается неограниченная растворимость
Li :Na К : Mg Ca “ 1 Al: Go in Tl Ge Sn Pb
Ав Li3As Na^As K3As | KAs2 Mg3As2 Ca3As2 — AiAs GaAs ; -** яс. < co тй. 0 0 TB. <^co TB. <oo Sn-jAsi 596° SnAs 605° о Л о Й
GeAs 737° GeAs., 732°
As
Sb Li3Sb тв, 0 ,Na3Sb 856" NaSb* 405° TB. 0 K3Sb : 812? JKSb 605® тв. 0 Mg3Sb2 1228° Ca3Sb2 ? Эвтектика 583° I I 1 тв. 0 AlSb 1050® GaSb IriSb tb. <^co Ti7Sb3 226° тв. > 0 0 TB. <^oo тв.>0 0 ТВ. GO . 0
snsb* : 425° .
Sb
Bi тв. О 1л3В1 1145° LiBi* 800° тв, 0 Na3B> .775° ; NaBi 446° th . 0 K3B! ' 671° K3B12. 420° KBi2 ; 558“ тв, ~ 0 Mg3Bi2 800° тв. О Ca3Bi2 928“ CaBi3* 507“ : тв, 0 BaBi3* 447° ' g * ; < f • • p- -Д. C 3 ; ж.<со тв. 0 0 Сплав TH; < CO та.—0 9 ; tb.>O .'0 G ТВ. <00 тв. О о : ТВ. со о
Pb2Bi*?
Ti3Bi 301,5°
Tl3Bi [J 90°
Tl3Bi6 21.3
Пятая группа периодической системы
631
ком состоянии в значительной степени ассоциирован. Что это действительно так, следует хотя бы из того, что аммиаку присуще ненормально высокое значение константы Трутона (см. стр. 358).
Молекула NHs координационно неиасищена. Поэтому она может присоединять еще один нон водорода, образуя положительный радикал [NHJ+ (радикал аммония), который способен существовать в растворе в виде свободного нона. В значительно меньшей степени РНз способен образовывать радикал фосфоний [РЩ1+, У высших аналогов этого ряда способность их водородных соединений образовывать такие радикалы исчезает, одиако она сохраняется еще у их алкйльныл производных.
При замещеции’в водородных соединениях, приведенных в’Табл. 97, всех атомов водорода на алкилы (R) образуются соединения
-М13 PR3 All; . SbR3 BiR3
(Триалкил) -амины -фосфины -арсины -стцбины -висмут ины
Все они, за исключением алкилвисмутипов, координационно яепасыщщгы и способны потому присоединять галоидные алкилы, образуя одновалентные положительные радикалы, например:
N(CH.,)., 4- _ CH3J =К(СН3)3-СНЭ1=(М(СН3)4Г1
Трпметилаяпн иодистый мстил Иодид тетраметчламыония
Нитриды, фосфиды, арсениды, антимониды и ей смут и ды общей формулой МзХ, происходящие от перечислен пых выше водородных соединений прп обмене атомов водорода на атомы сильно олоктроиоложнтельного металла, имеют солеобразную структуру. LisN образует отчетливо выраженную полную решетку (Zinil, 1935); в случае LijBi величины межатомных расстояний также указывают па ионную решетку. Ei3Bi кристаллизуется кубически и, ио Цинтлю (1935), аптнизотипен BrFs, решетка которого близка к решетке Известны также структуры магниевых соединений элементов главной подгруппы пятой группы,
Тип ScsO3 (ем. т. П) Тид La^Qa (см. т- П) Тип Zn3P2, (близкий к типу Sc2O3) Другие структурные типы, (полностью еще
Be3N2 Ве3Р2 — — — Zn3P2 Zn3As2 не установлен пые)
Mg3X2 Mg3P2 Mg3As2 Mg3Sb2 Mg3Bl2 Cd3P2 Cd3As2 ' - Sr3N2 ?. ,
а-Са3Аг — — • — — ' —f —* :: ^-Са3ЭТ2 — т...
Изменение структуры при переходе от MgsAss к MgsSba объясняется, так же как и в случае Ya Оз и LasOa (см. т. II), зависимостью типа решетки, т. е., другими словами, координационного числа от соотношения мойных радиусов (см. стр, 242),
Элементы главной подгруппы пятой группы (включая мышьяк), обладающие ме-таллияескимхарактером, не образуют соединений. Наоборот, As и Sbjатакже Sb и Bi не* ограниченно образуют между собой твердые растворы. Аз И Bi ПО способны к образованию твердых растворов. В расплавленном состоянии эта элементы растворяются одцп в другом неограниченно. Также и с металлами предшествующих главных подгрупп период плоской системы в расплавленном состоянии они смешиваются, как правило, во всех отношениях. (Единственное известное исключение представляет система А1—13 г.) В твердом состоянии с металлами двух первых главных подгрупп они образуют не твердые растворы, а соединения (см. табл, 98). Все соединения СО щелочными и щелочноземельными металлами по своему состав)/ соответствуют одному w тому же типу, а именно типу водородных соединений ННз- Их следует рассматривать в качестве «истиппых валентных соединений», приближающихся, по-видимому, к солеобразным соединением.. Это, между прочим, подтверждается и тем, что таким соединениям на диаграммах состояния соответствующих систем отвечают особо четкие : максимумы температур плавленая. Но, кроме того, образуются еще и многочисленные соединения, которые не отвечают обычным соотношениям, допустимым с точки зрения: валентностей соединяющихся элементов. Эти соединения обладают значительно более низкими температурами плавления, в ряде случаев они плавятся пнконгруентнб.
Металлы главных подгрупп III и IV групп менее способны к образованию со-' единений с As, Sb и Bi, чем щелочные и щелочноземельные металлы)" наоборот, они
632
Глава 14
лучше образуют с ними твердые растворы. Из табл. 98 отчетливо видно изменение хг рактора взаимодействия между металлами главной подгруппы V группы. В системе S^-— Sb существует недальтонидное соединение. Опо кристаллизуется в ромбоэдрически педд-женной решетке типа поваренной соли (Ilagg» 1935). Из этого следует для соедпненгт состав, аналогичный NaCl, т. е. SnSb. Одпако часть узлов решетки, в которых должез. былп бы находиться атомы Sb, может быть замещена атомами Sa, и наоборот. Вс.тез-ствие этого состав соединения может колебаться в определённом интервале, а пмене; от 45 до 55 ат. % Sb. Для характеристики соединения, обладающего переменным составом, по предложению Бильтца, пользуются формулами с черточками вверху, которнг указывают «идеальный» состав SnSb. В системе Pb-Bi также существует не дал ьтонпл-ное соединение (67—75 ат. % Pb). Sb-As—твердый растворы с 25—46 ат. % As, г— Тшебетовскому (Trzebiatowski, 1938), ниже 500° испытывают превращение с образованием кубической гранецентрированной решетки. Это превращение можпо трактовать в смысле образования не дальтон и дно го соединения. Об отношении элементов главней подгруппы V группы к Si и Ge см. табл. 99.
По даппым Юза (J п z a, Natnrwiss., 27, 32, 1939), GeaNj имеет такое же строение решетки, как фенакит BeiSiC^.
Таблица 39
Отношение Элементов главной подгруппы пятой группы к кремнию и германию
N p AS Sb Bi
Si Si3N4 SiP ж. co, тв. 0 SiAs 1083= SiAs2 944° ж. оо, тв. 0 0 ж О, тв. О О
Ge GeaN 4 GoP ж. оо, ТВ. > 0 GeAs 737= GoAsa 732° Ж, co, ТВ. > 0 0 Эвтектика 590° ж. оо, тв. ~ О 0 Эвтектика 271=
Основные свойства элементов главной подгруппы пятой группы удовлетворительно объясняются на основе теории валентности. Наивысшая положительная валентность (пятивалептпость) элементов с порядковыми номерами 7, 15, 33, 51 и 83 следует, по теории Косселя, из того, что каждый из них содержит на пять электронов больше по сравнению с особенно устойчивыми конфигурациями в 2, 10, 28, 46 и 78 электронов (см. рис. 28 на стр, 152). Трехвалентность элементов главной подгруппы пятой группы по отношению к электроположительным элементам, например к водороду, также объясняется стремлением их приобрести особенно устойчивые электронные конфигурации, а именно такие, которые имеются у инертных газов, стоящих п периодической системе после элементов главной подгруппы пятой группы.
По теории Косселя, при этом появляется отрицательный заряд у атомов азота, фосфора и т. д., так что трижды отрицательно заряженный атом приобретает возможность связывать эквивалентные количества других электроположительных атомов. Согласно представлениям Льюиса и Лангмюра, образование октета достигается в результате совместного обладания электронами, которое, Но квантово-механической теории атомной связи, возможно вследствие того, что происходит насыщение спинов трех неспареипых р-электронов, существующих по спектральным данным в атомах элементов главной подгруппы пятой группы, спинами такого же
Пятая группа периодической системы 633
числа неспаренных электронов других атомов. И та и другая теории, таким образом, пригодны для объяснения существования и свойств соединений. Появление четвертой координационной валентности, например у азота в аммиаке, также можно удовлетворительно объяснить, ник это будет показано ниже иа стр. 659, как на основе теории гетерополярной связи Косселя, так и на основе выдвинутой Льюисом и подтверящениой квантовой механикой теории гомеополярной связи. При этом оказывается, что в обеих теориях выдвигаются на первый план только различные аспекты одного и того ясс явления. Эти аспекты, как показывает применение квантовой механики к теории химической связи, должны сочетаться.
АЗОТ (NITROGENIUM) N
Распространение в природе. Атмосферный воздух приблизительно па 4/s по объему состоит из свободного азота. В воздухе содержатся также следы аммиака NH3, образующегося вследствие процессов гниения азотсодержащих органических веществ; после гроз — также азотная кислота HNO3. В больших количествах неорганические соединения азота в природе почти не встречаются, если не считать нитрата натрия NaNO3, вероятно являющегося продуктом разложения растительных и животных остатков, образующего мощные залежи во многих местах, особенно по побережью Чили. Азот является неотъемлемой составной частью живых организмов, так как он входит в состав веществ, из которых построены жизненно важные белки
Исторические сведения. Еще во второй половине XVIII в. было установлено, что в воздухе содержится составная часть, которая не способна поддерживать дыхания и горения. Наиболее ясно это было высказано Шее-ле в его труде «Трактат о воздухе и огне», появившемся в 1777 г.; Лавуазье предложил для газа, названного Шеелс «испорченным воздухом», название азот, т. е. удушливый газ, или удушливое вещество (agtoTiKog — не поддерживающий-жизни). После того как было установлено, что азотная кислота производится от азота, Шаптал (Chaptai) предложил для него название нитрогеи (от nitrum и ysvvd'co — образователь селитры), которое и сохранилось в виде его латинского обозначения.
Наиболее важные соединения азота—азотная км слота и аммиак — в виде их солей были известпы еще арабским алхимикам. В рукописях, приписываемых Геберу, приведено описание получения свободной азотной кислоты. Получение свободного аммиака в газообразном состоянии впервые удалось осуществить только Пристли (Priestley, 1774).
Использование атмосферного азота для получения аммиака и азотной кислоты в промышленном масштабе было осуществлено лишь в начале текущего столетия (получение кальцийпааиамяда ио способу Рота — Франка — Каро приблизительно с 1904 г.; получение азотной кислоты но методу Биркеланда и Эйде с 1905 гл получение азотной кислоты каталитическим сжиганием аммиака по Оствальду с 1906 г.; синтез аммиака по методу Габера — Боша с 1009 г.).
Получение. В лаборатории можно получать чистый азот, нагревая до 70° концентрированный водный раствор нитрита аммония или смеси растворов хлорида аммония и нитрита натрия. При этом происходит разложение по уравнению
NHtNO2=N24-2HaO.
634
Глава 14
Для удаления следов окислов азота выделяющийся газ промывают, пропуская его -через смесь бихромата калия и серной кислоты. Особенно чистый азот получавп термическим разложением аммиака, пропускаемого над никелевым поротиком прм 1000е’ с последующим разделением Ns и На вымораживанием (Harteck, 1930).
(Обычно, однако, в лаборатории азот приготовляют пропусканием воздуха над раскаленной медью, которая при этом поглощает весь содержащийся в нем кислород. Ввиду того что в воздухе содержится 1% аргона, полученный этим способом азот содержит аргон и называется «воздушным азотом».. Для большинства целей примесь аргона в азоте ио имеет значения,
Техническое получение азота производят зачастую аналогичным: образом, только в качестве поглощающего кислород средства вместо- меди используют главным образом уголь. Образующуюся вследствие полного сгорания угля двуокись углерода можно затем легко отделить от азота (промыванием водой под давлением или вымораживанием) в виде побочного продукта реакции.
В очень больших количествах в настоящее время получение азота в технике производят сжижением и фракционированной перегонкой воздуха. В качестве ценных побочных продуктов при этом получаются кислород и аргон, а при определенных условиях, кроме того, еще неон и гелий.
Полученный таким путем азот поступает в производство в сжатом виде в стальных баллонах (бомбах).
Чтобы полностью очистить такой азот от примесей кислорода, применяют цо способу Фрике (см. стр. 741) так называемую «медную башню» или используют описанную Каутским (Kautsky, 1926) аппаратуру, в которой раствор NasSsO4, служащий в качестве абсорбента, приводят в соприкосновение с газом, пропуская его в раствор через глиняные фильтровальные свечи.
Свойства. Азот — бесцветный, не имеющий запаха и вкуса газ, более легкий, чем воздух. Вес 1 л чистого азота при 0° и 760 мм рт ст равен 1,2505 з, а вес «воздушного азота», содержащего 1,185 об. % аргона, составляет 1,2567 г; вес 1 л воздуха при тех же условиях равен 1,2928 з. Азот сжижается с трудом (критическая температура — 147,1°, критическое давление 33,5 атм, критическая плотность 0,3110). Температура' кипения жидкого азота равна —195,8°, температура плавления твердого азота — 210,5°. В воде азот менее растворим, чем кислород; 1 л. воды при 0° растворяет 23,6 мл «воздушного азота» или 23,2 мл чистого азота.
Удельная теплоемкость чистого азота составляет при- постоянном объеме с,, = 0,178, при постоянном давлении ср — 0,249 (оба эти значения приведены для комнатной температуры); ср/сТ, = 1,40. Удельная теплоемкость Жидкого азота вблизи от температуры кипения равна ср = 0,46; теплота испарения 47,74 кал/з.
Ниже — 237,7° азот, как это впервые установили Кеезбм и Кэммерлинг Оянес па основании опытов по исследованию его удельной теплоемкости, превращается в другую модификацию. Эта устойчивая при низких температурах модификация азота ярко светятся при освещении катодными лучами, причем спектр этого свечения очень похож, па своеобразный спектр северного сияния. Выше температуры превращения азот трудно заставить светиться, и тогда оп дает другой спектр (Vegard, 1924).
В газообразном состоянии азот состоит из двухатомных молекул N-j (межатомное расстояние 1,0945 А, момент инерции 13,84-10~ЗД г-смР). Даже при 3000° диссоциация этих молекул но достигает еще значительной степени. Уже из этого следует, что работа диссоциации должна быть особенно велика. Спектрографически для нее получают в качество наиболее вероятной величины значение 9,7и4 as (225,2 ккал/моль) (Gaydon, 1944; Glockcr, 1948). Заметное расщепление азота па атомы происходит под влиянием. ' тлеющего электрического разряда под уменьшенным давлением. Это явленна впервые наблюдал Штрутт (Strutt, 1913). Атомарный азот химически чрезвычайно активен. Например, при обычной температуре он взаимодействует со ртутью [пр данным Тиде
Пятая группа периодической системы 635
(Tiede, 1935), с образованием HgaM], а также с серди и фосфором. РекомбииаДня атомов азота сопровождается желтым свечением, продолжающимся еще некоторое время после прекращения электрического разряда.
При обычных условиях азот очень инертный газ. Однако при высокой температуре его активность значительно возрастает, и тогда он взаимодействует со многими веществами, образуя нитриды.
Под влиянием искровых разрядов азот соединяется в незначитель-ной степени с водородом, образуя аммиак NH3. Однако при высоких температурах равновесие
*2-: 7* 2NH3 (1)
полностью смещено в левую сторону. Поэтому образовавшийся аммиак, лишь только его концентрация превзойдет некоторую минимальную величину', вновь разлагается под влиянием электрических разрядов. Можно, однако, получить большие количества аммиака, если проводить это взаимодействие при возможно более низкой температуре, что и происходит, если реакция протекает в присутствии соответствующих катализаторов. Кроме того, можно сдвинуть равновесие (1) слева направо, применяя высокое давление.
Значительное прямое взаимодействие азота с кислородом воздуха можно вызвать, пользуясь Искусственным приемом. Получающееся в Ртом случае соединение — окись азота NO хотя и стабильно при очень высоких температурах, однако равновесие
-Х:, | О2 у! 2NO (2)
при понижении температуры псе более и болёё смещается влево. Это следует из отрицательного значения теплоты образования окиси азота, приведенной в табл. 95. Несмотря на это, существование окиси азота и при обычной температуре объясняется тем, что скорость со распада при этих условиях практически равняется нулю; окись азота метастабильна. При нагревании она распадается еще до того, Как Достигается область температур, при которых равновесие (2) сколько-нибудь заметно сдвигается вправо. Поданным Нернста, при 2000° в равновесии со смесью азота и кислорода, соответствующей по составу атмосферному воздуху находятся 1,2 об. % NO, а при 3000"—5,3 об.% NO. Поэтому, чтобы получить окись азота в достаточных концентрациях, необходимо, с одной стороны, : применять очень высокие температуры, а с другой, чтобы избежать разложения NO при охлаждении, следует образовавшуюся газовую смесь охлаждать мгновенно, т. е. крайне быстро переводить ее из области температур около 3000° до температуры ниже 1000°. Достаточно высокие температуры для этой реакции получают при применении электрической дуги, а мгновенное охлаждение практически достигается, например, тем, что пламя электрической дуги при помощи сильного электромагнита -растягивают в диск, через который продувают газовую смесь (воздух). Достигаемое таким образом «сжигание воздуха» имеет в настоящее время некоторое практическое значение для получения азотной кислоты (процесс Биркеланда и Эйде).
„ Еще Пристли установил, что если через смесь азота и кислорода, собранную Над водой, долгое время пропускать электрические искры, то происходит уменьшение -объема газа. В данном случае сначала образуется окись азота, из которой затем полу- -чается двуокись азота, и, наконец, азотная кислота, образование которой было уставов-: Лено .еще Кавендпптем (1784). Так как об радующаяся при атом окись азота удаляется вэ сферы: реакции, то, пропуская очень долго искры и ведя реакцию при избытке кислорода, можно полностью перевести весь азот в. азотную кислоту. Для практического
636
Глава 14
получения азотной кислоты этот длительный способ, конечно, не может иметь sesw ния; однако нм именно воспользовался Релей для получения аргона (см. Стр. HF
В табл. 100 приведены теплоты образования некоторых соединений азота па я-нг-мои. В таблице также приведены значения межатомных расстояний в этих соеДЕзе-ниях, валентно-силовые константы и величины работы разрыва этих связей.
Таблица ICO
Теплоты образования соединений азота из атомов и характерные константы азотсодержащих связей
И'атом — теплота образования из атомов, ккал/.иоль; г0 — межатомное расстояние, А /с/ — валентно-силовая константа, бни/сщ; В — энергия связи простых, двойных и тройных азотсодержащих связей, кл’ал/6,023 Л023.
Ссели и (Юл я Связь hf 10-6 в
n2h4 452,6 N—N 1,46 3,6 55,1
N3H 467,4 N=N 1,241 9,1 103,8
225,2 N=N 1,095 22,9 225,2
N20 311,1 №N 1,125 18,0 182,2
nh2oh 367,8 N—0 1,46 .4,1 66,7
n2o 311,1 N=O 1,19 14,4 128,0
no2 245,2 N=O 1,215 9,1 122,6
NO 149,8 N=O 1,151 15,9 149,8
NH (газообразный радикал) 96,9 N—H 1,038 6,0 96,9
NH3 (газообразный радикал) 196,5 N—И 1,028 —— 98,2
N2H4 452,6 N—H 1,02 —' 99,4
NH3 387,9 N—H 1,014 6,4 100,2
N3H 467,4 N-H 1,012 — 100,5
Применение. В лабораториях азот иногда применяют как индиффе рентный газ для защиты легко окисляющихся веществ от окисления кислородом воздуха. Для подобных же целей им пользуются и в технике, например прп переливании бензина и других легко воспламеняющихся жидкостей или для 'наполнения капилляров ртутных термометров. Однако в настоящее время азот в основном используют для приготовления кальцийцнанамида, аммиака л азотной кислоты в производстве удобрений.
Связывание атмосферного азота в соединения, используемые в качестве удобрений, в значительных количествах совершается и естественным путем благодаря деятельности усваивающих азот микроорганизмов. Некоторые виды последних, например Azotobakler, находятся в содержащем гумус слое почвы; вычислено, что благодаря деятельности этих бактерий на каждый гектар почвы среднего состава в течение года может быть ассимилировано до 48 ва атмосферного азота.
Важным фактором накопления азота в почве является также деятельность находящихся в корневых клубеньках бобовых растений, например Serradella, клубеньковых бактерий. Бобовые растения нередко применяют для обогащения почвы азотом. Для этого пользуются ими как зеленым удобренном, запахивая их до созревания, или же, снимая урожай, оставляют л земле корневища. На легких почвах при благоприятных климатических условиях культура бобовых может накапливать в почве свыше 200 кг азота в год на i га.
Для жизненные процессов на земле связывание- азота микроорганизмами («биологическое связывание азота») имеет наряду с ассимиляцией углекислоты фундаментальное значение. Исследования в опыты, особенно Виртанена (Virtanen А. Т., 1931, см.
Пятая группа периодической системы 637
также Ang. Chem., 65, 1, 1953), показали, что при планомерном использовании биологической ассимиляции азота плодородие почв и объем сельскохозяйственной продукций во всем мире может значительно возрасти.
Азот попадает в почву с дождевой водой (в форме азотной и азотистой кислот), так как при грозовых разрядах и вследствие фотохимических реакций происходит в известной степени связывание азота в атмосфере. Увеличение содержания азота в почве, происходящее вследствие этих процессов, намного меньше образования содержащих N веществ в результате деятельности микроорганизмов, но больше (относительно поверхности всей Земли) количества азота, вносимого в виде искусственных удобрений.
Кроме приготовления искусственных удобрений, аммиак, азотная кислота и другие соединения азота находят широкое применение и для многих других целей. Подробнее это будет рассмотрено при описании соответствующих соединений. Однако здесь следует еще упомянуть о большом количестве технически важных органических азотсодержащих соединений — красителей и лекарственных средств,
СОЕДИНЕНИЯ АЗОТА
1. Окислы
Обзор окислов см. в табл. 95 на стр. 627 (ср. также стр. 644).
NO н IVОг существуют также в соединениях в виде радикалов. Неорганические соединения, содержащие NO в виде одновалентное!) радикала, называются нитрозилъ-ными соединениями*, например NOC1 — нитразилхлорид, NaNO — гпгтрозилнатрий. Соединения, относительно которых можно предположить, что они содержат N 0 в виде нейтральной группы (связанное, например, за счет вапдорваальсовых сил), называются н итрд за соединениями, па пример [Fe(NO)]SOj — нитрозо сульф ат двухвалентного железа [нитрозосульфатжелезо(П)]. Органические соединения с радикалом NO также называются нитрозо сое ди пениями. Неорганнческие пр о па водные одновалентного радикала NOa с электроотрицательными веществами называются нитрилоСОедннепиями*. Например, NO2Cl — питрилхлорид. Об обозначении радикала NOa в солда, в органических соединениях и в комплексах СМ. стр. 649.
Закись азота N3O, открытую в 1772 г. Пристли, получают нагреванием сухого нитрата аммония
NH4NO3=N2O4-2H2O.
Разложение начинается при 170° и сопровождается выделением тепла, Поэтому, чтобы не дать ему протекать слишком бурно, следует вовремя прекратить нагревание (см. стр. 660.)
Закись азота — бесцветный газ со слабым приятным запахом и сладковатым вкусом. При вдыхании его в незначительном количестве он вызывает состояние легкого опьянения и судорожный смех (отсюда название веселящий газ). При вдыхании в больших количествах он действует как наркотическое средство. Однако при длительном вдыхании к нему необходимо примешивать кислород, так как сам по себе он не в состоянии поддерживать дыхания. Закись азота легко сжижается (температура кипения—89,5", температура плавления —102,4°, критическая температура 36,5°, критическое давление 71,7 атм). В продажу NaO выпускают в сжиженном состоянии в стальных баллонах и применяют главным образом для наркоза.
Вес 1л закиси азота при 0° и 760 -ч-и рт ст равен 1,9804 г. Плотность в жидком состоянии При температуре кипения равна 1,2257, а при критической температуре —
* Окончание яа обычно отбрасывается, например нитрозамид и нитрамид (ср. стр. 649 и 650).
S38 Глава. 14
0,45, В воде N2O растворяется довольно значительно: 1 об, ч. воды поглощает при О’ 1,ЗОо2, а при 25° 0,5962 об. ч. N-jO. Еще лучше растворяется NaO в спирте.
Вещества, обладающие большим сродством к кислороду, например уголь или некоторые металлы, сгорают в закиси азота почти так же энергично, как в кислороде; однако возбудить начало горения в этом случае бывает труднее, так как молекула NaO, хотя й метастабильна, начинает разлагаться лишь при относительно высокой температуре. Смесь равных объемов закиси азота и водорода при соприкосновении с'пламенем дает энергичную вспышку
На= Nj-|-;H2Orae-|-77,5 ккал.
С большой силой взрывается при воспламенении смесь закиси азота с аммиаком
ЗЛбО 2IVH3 = -р—F 210 ккал.
Твердая закись азата имеет такую же кристаллическую решетку^ как и твёрдая двуокись углерода. Длила ребра ее элементарной ячейки (а 5,72 А) также очень близка к соответствующей длине для СОг.
Рентгеноструктурпым анализом показано, что в твердой закиси азота атомы в молекулеЖО расположены па одной примой. Результаты новейших физически хи селе? доваппй (например, Электр орографических) подтверждают вероятность прямо линейно го расположения атомов азота в молекуле газа Ж О. Так как N2O имеет такое же число электронов, как и СО2, и очень похожа па пёе по своим физическим свойствам-, то Лапгмюр в 1919 т. предположил, что электронные конфигурации в обоих соедппепйях одинаковы (изостерия); Предложен па я Лангмюром формула N :: N ;; О, соответствую-щая О С.:: О, подтверждена новейшими наблюдениями (например, спектрами комбинационного рассеяния), но одновременно и дополнена в том смысле, что эта форма находится в состоянии резонанса с другими:
- + + ~
X::N:;6 и :N; ;N^:
Межъядерные расстояния в молекуле N2O равны N-<—> N= 1,12 A,N -—+ 0=1,19 A (Scho-maker, 1942). Молекула 2s гО имеет момент инерции 66,78ЛО-40 г-см2 и дипольный момент 0,14-10-1S ;>л. ст. ед. Очень небольшое значение дипольного момента объясняется тем, что противоположные моменты обоих семпнолярных форм взаимно уравновешиваются вследствие существующего между ними резонанса.
Окись азота (моноокись азота) NO, продукт соединения азота с кислородом при очень высоких темпера турах , —бесцветный : газ, который, однако, при соприкосновении с воздухом вследствие окисления до NO а образует бурые пары. Температура кипения NO равна —151,8°, температура плавления —163°. В воде окись азота незначительно растворима (1 об. воды при О’ растворяет 0,07 об, NO), но практически совершенно нерастворима в насыщенном растворе поваренной соли. NO обычно образуется при действии на азотную или азотистую кислоты восстановителей. На измерении объема окиси азота, образующейся в соответствий с реакцией.
6Hg-J--2IINO., 4-3H2SOi=2JyO+^[щБО,-; 4Н;О при встряхивании ртути с азотной кислотой и концентрированной серной кислотой, основано определение содержания азотной кислоты при помощи нитрометра Лунге. В лабораториях окись азота получают обычно при действии умеренно концентрированной азотной кислоты на медь
8HNOj-L3Cu = 3Cu(NO3)24-4H2O+2NO.
Окись азота"— весьма реакционноспособное вещество. С хлором и бромом она реагирует, образуя нитрозилгалогениды (см. стр. 6G9). С концентри
Пятая группа периодической системы 639
рованной серной кислотой при одновременном доступе кислорода воздуха NO образует нитро зил серную кислоту
2H2SO44~2NO +1/2О2=2[МО][И5°4!+Н2°.
Хромовой кислотой, подкисленным раствором перманганата или хлорноватистой кислотой NO окисляется до азотной кислоты. Двуокись серы в присутствии воды восстанавливает NO до закиси азота, соли двухвалентного хрома в нейтральном растворе — до аммиака, в кислом — до гидроксилампна. Аммиак и гидроксйламин образуются также при восстановлении NO солянокислым раствором SnCla. Вещества, обладающие большим сродством к кислороду, например уголь, фосфор, магний, энергично сгорают в атмосфере окиси азота, но горячая сера в ней потухает. Смесь равных объемов NO и водорода при зажигании даст вспышку. Окись азота в виде нейтральной молекулы может соединяться с солями многих металлов. Так, с водным раствором сульфата двухвалентного железа FeSO4 она образует темно-бурый нитрозосульфат [Fe(NO)]SO4. При нагревании таких растворов из них полностью улетучивается. NO. Подобные жо непрочные соединения окись азота образует и с растворенным в концентрированной серной кислоте сульфатом меди, а также с раствором хлорида меди в спирте. Большинство таких обладающих сравнительно простым составом соединений устойчиво только в растворах. Значительно прочнее некоторые более сложные комплексные нитрозосое-диненйя, в качестве примера которых следует упомянуть здесь нитропруссид натрия NasfFe(CN)sNOj.
С HCI окись азота образует темно-красное двойное соединение NO-11C1, которое, но Родебушу и Интима (Rodebush, Yntema, 1923), обладает солесбразпым характером и соответственно атому должно рассматриваться как хлорид нитроеония [NOH] С1. Образование этого соединения, впервые обнаруженного в 1909 г. Брокером (Bririer), в последнее время довольно часто являлось причиной ошибочного объяснения опытов при работах в вакуумной аппаратуре я привело, например, к так называемому Открытию «галогенидов инертных газов» или «Красной модификации НС1»; это соединение образуется из компонентов только при очень низких температурах и в присутствии по крайней мере следов высших окислов азота. Оно, как правило, устойчиво только в твердом состоянии и может быть расплавлено только иод высоким давлением.
NO.имеет 11 внешних электронов и, как все газы с нечетпым числом электронов парамагнитна. Основное состояние молекулы соответствует терму 2Д, Работа расщепления на атомы составляет 6,495 ж, межатомное расстояние 1,15 А. О строении молекулы NO см. стр. 163/ Дипольный момент, ее составляет, по Ватсону (Watson, 1934), 0,16- 10“>а эл. ст. ед., момент инерции — 16,43- 10~К> г-сл»2.
В жидком состоянии NO полимеризована. Этим и объясняется высокая и сильно зависящая от температуры удельная теплоемкость жидком NO; высокая энтропия пспа-ренпя, а также ее магнитные свойства. Изучая инфракрасные спектры и спектры комбинационного расстояния, Смит, Келлер и Джонстон (Smith, Keller, Johnston, 1951) заключили, что ири ожижении NO полностью димеризуется, вероятно, с образованием изогнутых молекул ONNO. •
Трехсп.'псь азота NaO3, ангидрид азотистой кислот?/, получают приливанием пр каплям азотной кислоты уд. -веса 1,3 к трубоизмельченной стекловидной трех окиси мышьяка или нагреванием азотной кислоты с крахмальной мукой. При этом образуется темно-голубая жидкость, застывающая при сильном охлаждении в бледно-голубую массу. Температура плавления —102°.
В значительной степени трехокись азота распадается уже ниже , О’ по уравнению
N20j NO4-NO2 или 2N2O3 2NO4-NaO4. (3)
640
Глава 14
При 25° и атмосферном давлении, по данным Абеля (Abel, 1929), в смеси, содержащей по анализу равные количества NO и МОз, содержатся 10% не диссоциирований N->03; рассчитанная на значения констант равновесия теплота образований N«Qt из МО и NO а оказывается равной (при комнатной температуре) 9,6 ккал/моль N.Ob-Слободпая энергия образования при 25° составляет, по Верхоеку (Verhoek, 1931), только —0,44 к кал/моль. <
Вследствие легкой обратимости реакции (3) газообразная смесь равных объемов NO и NO, при большинстве химических реакций ведет себя подобно соединению N3Og; так, при взаимодействии со щёлочами в водных растворах образуются нитриты:
N 3О3 4-2Na ОН = 2N’aNO2+Н2О.
С водой вначале образуется азотистая кислота, которая, однако, быстро разлагается с образованием азотной кислоты
N2O34-H2O = 2HNO2; 3HNO3=HNO34-2NO4-H2O. (Ч
Двуокись азота NO2 и четырехокпсь азота NSO4 (азотноватый ангидрид), Как уже было указано, окись азота легко соединяется с кислородом воздуха, образуя при этом двуокись азота
МО4-1/.,О2=гКО2Ц-13,6 ккал.
Технически двуокись азота получают из нитрозных газов, образующихся при каталитическом окислении аммиака. При сильном охлаждении ее легко можно выделить в твердом виде. Ее можно также поглотить концентрированной серной кислотой (образование нитрозилсерной кислоты см, 639)> и вновь выделить при нагревании последней.
В небольших количествах двуокись азота удобнее всего получать нагреванием хорошо высушенного нитрата свинца, к которому для равномерного выделения газа обычно примешивают прокаленный кварцевый песок.
Двуокись азота — буро-красный, сильно ядовитый газ, обладающий характерным запахом. Оп легко сгущается в красно-бурую жидкость (температура кипения 22,4°), которая при охлаждении бледнеет, затем становится совершенно бесцветной и при —10,2° затвердевает, образуя бесцветные кристаллы. Иаоборот, при нагревании газообразной N02 ее окраска усиливается. Это зависит от того, как показали определения плотности пара, что двуокись азота при охлаждении димеризуется, переходя в бесцветную четырехокись. Существует равновесие, в сильной степени зависящее от температуры
2NO2 NaO4-|-14,7 ккал (при 25е).
Ешли газ или газовая смесь находится под давлением 1 ат, то цри
27" за’ (00 125"
20% 40% 89% 98,7%
четырех окис и оказываются расщепленными в двуокись.
Свободная энергия образования N2O4 из 2NOa, по Данным Верхоеки (1931), при 25 составляет —1,15 ккал, при 45° —0,25 ккал, Следовательно, оно быстро уменьшается с ростом температуры, в то время как теплота образования в так же тенте ра турком интервале изменяется только очень мало (примерно на 1%).
NO, парамагнитна, N2Ot диамагнитна. Поэтому весьма вероятно, что соединение двух молекул 2NO3 в N2O4 объясняется стремлением к спариванию неспарепных электронов в NO2. То, что NO, содержит нес паренный электрон, следует из нечетного
Питая группа периодической системы
641
числа электронов в молекуле. По-видим о лгу, и окраска N О2 обусловливается наличием несиаренных электронов. Молекула ХО2 изогнута (N *—> 0 = 1,215 A,<JlONO= 132*). По Полингу, здесь имеет место мезомерия между двумя состояниями:
1 .Л I
и ISV
•01 22I
Так как в обоих случаях простая связь налагается на трехэлектронную, речь идет в общем о резонансе между четырьмя различными состояниями. Тем самым должна вызываться значительная стабилизация молекулы NO,, исходя из чего и следует объяснять небольшое стремление к димеризации молекул КО, в N,0(. Удельная электропроводность жидкой N,04 при 17° составляет 1,3-10"12 олГ диэлектрическая проницаемость 2,42 (при 18°), показатель преломления п^°' — 1,420, молярная рефракция 15,2 с молярпая поляризация 26,5 aw3. ;
Предполагают, что жидкая N,O4 в незначительной степени распадается па ионы N2O4 N0*4-N0g. Этим объясняется не только ее электропроводность, ио и способность к реакциям следующего типа: [(С2И5)2ТП12]С] ф- N2O4 = [(C,H5)2NHj] [NO3)4-+N0C1; Zu -ф 2N2O4 — Zn(NOs)2 + 260 или Zn + 2NO+ = Zi^-j^NO'. Первая реакция аналогична гидролизу, вторая соответствует разрядке ионов Н’ металлом. Нитрат цинка образует с К2О4 соединение, которому придается формула (NO+)2 [Zn(N'O3)4j2". Его можно рассматривать как аналог гидро кс окис лоты, например H2[Zn(OH)4], Твердая N2O4, по Бредли (Broadley) и Робертсону (Robertson, 1949), построена из плоских молекул со структурой
О. ,0
>N-N< ; N<—>0 = 1,17 A, —>N = 1,64A, < ONO = 126\
O':: ^0
Двуокись азота является сильным окислителем. В ней сгорают калий, фосфор, уголь и сера. Смесь двуокиси азота и сероуглерода сильно взрывается. С водородом NO, вступает в реакцию в присутствии катализаторов, например платипы или мелкораздроблепного никеля, образуя аммиак и воду.
С водой двуокись азота образует азотную кислоту и трехокись азота, которая затем уже реагирует по уравнению (4), так что реакцию в целом можно представить следующим образом:
3N02+H2d = 2HN034-NO. (5)
Образующаяся NO в присутствии воздуха немедленно окисляется до N03, которая в этом случае полностью переходит в азотную кислоту.
Едкие щелочи поглощают N03 с образованием смеси нитратов и нитритов (окислительно-восстановительная реакция).
2?JO2-{-2NaOH=NaNO34-NaNO2-{-Il2O. (6)
Пятиокпсь азота N2O6, ангидрид азотной кислоты, получают при обработке азотттой кислоты пятиокисью фосфора (фосфорным ангидридом)
2HNO3-j-P2O5=2HPO3-|-N2Os.
В чистом состоянии N20s образует бесцветные, твердые, расплывающиеся на воздухе кристаллы (ромбические призмы) с уд. весом 1,63, т. пл, 30°, т. кип. 45—50°. N2O5 в твердом состоянии имеет солеобразную структуру [N02]+[N03’]-, подобно РС16 иди P2C1JU (см. стр. 692), Пятиокись азота очень неустойчива и взрывается без сколько-нибудь заметного ввеюнего воздействия, С водой N2O5 жадно взаимодействует с образованном азотной кислоты.
(N2O5] Н2О 2IINO3 -|- 10,4 ккал; HNO3 ад = HNO3-aq -ф- 7,5 ккал (при 2Сг). С BF3, по Ш мопс с е ру (SchmeiGer, 1952), образуется устойчивое прн комнатной температуре двойное соединение N2O6-BF3.
Из ряда наблюдений можно сделать заключение, что азот способен образовывать еще более богатое кислородом соединение, чем N20g. Ему обычно приписывают формулу NQB, Это соединение образуется, как было показано спектрографическими измерениями и изучением кинетики реакции (Schumacher, 1928), в качестве промежуточного продукта при реакции NhOs (или NO2) с О3 и очень быстро распадается по уравнению 41 г. Рейт?
642
Глава. 14
кисного соединения перекись нитрозила
2NOS = 2NO2 Чг 03. Поэтому оно присутствует с постоянной концентрацией в реагирующей газовой смеси, по недоступно в чистом состоянии. Шварц (Schwarz, 1SSE на основании специфических реакций приписывает ему формулу перекиси, т. е. пере-
o=n/ хо
2. Кислородные кислоты азота
Наиболее важной и устойчивой из кислородных кислот азота является азотная кислота HNO3. Ее ангидрид — пятиокись азота N.,O6. Соли азотной кислоты M*[NOg] называются нитратами. Азотистая кислота HNO, очень непрочна и известна только в сильно разбавленных водных растворах. Болес устойчивы се соли M4NO21 — нитриты. Ангидрид азотистой кислоты — трехокись азота N3O3— существует при обычных температурах только в равновесии с NO и NO3.
В азотной кислоте азот можно рассматривать как электро поло житель но пятивалентный, а в азотистой кислоте — как электроположительно трехвалентный. При дальнейшем восстановлении можно перейти к азотноватистой кислоте H2N2O2r в которой азот, если предположить, что оп заряжен, злектропомжитслъно- одновалентен.
В азотноватистой кислоте азот структурно трехвалентен, так как способ образования и свойства ее приводят к формуле И — О — N — N — О — Н. Азотноватистая кислота получена в Свободном виде, но она очень Неустойчива (взрывчата). Ее соли называются зипонйтритами, Промежуточное положение между азотистой и азотноватистой кислотами занимает, очевидно, еще одна ступень окисления азота, известная только в форме солей,— нитроксилъная кислота H4N2O4
П- О. .0—Н
)N- N<
II— (г ЧО И (см. стр. 650)
От азотной кислоты O2N—ОН при замене группы ОН на группу— О—ОН перекиси водорода производится пероксоазотная кислота O3N—О—О И, hjihHNO4. Соответственно рт азотистой кислоты производится перо ксоа вот и стая кислота ON —О—ОН. Это соединение, изомерное азотной кислоте, существует только как промежуточной при некоторых реакциях (см. стр. 650). 11 ероксоазотная кислота очень взрывчата.
Азотная кислота HN03 представляет собой в чистом состоянии бесцветную жидкость удельного веса 1,522, которая при —41,15° затвердевает и кипит при 84°. При этом она, однако, начинает медленно разлагаться. На свету это разложение происходит уже при обычной температуре. Выделяющаяся при разложении
2HNO3=2.\O.,-!-H2O-i-l/2O2 (7)
двуокись азота растворяется в азотной кислоте и окрашивает ее в желтый, а при значительных концентрациях — в красный цвет. С водой азотная кислота смешивается в любых отношениях. Удельные веса и температуры кипения ее водных растворов сопоставлены ниже,
Удельный вес (d4°) 1.150 1,200 1,300 1,400 1,410 1,420 1,480 1,500
Содержание HNO3, вес. % 25,5 32,9 48,4 67,0 69,2 71,6 89,0 98,2
Температура кипения, °C 106,8 108,8 115,5 121,7 121,8 121,2 100,6 86,0
Максиму лг температуры кипения лежит при содержании 69,2% HNO3. Раствор этой концентрации получают, если исходят как из более слабых, так и из более крепких водных растворов HNOS и нагревают их до постоянной температуры кипения. Поэтому растворы HNO3, называемые «концентрированной азотной кислотой», обычно имеют именно эту концентрацию, Более крепкие растворы HNO3 дымят на воздухе, выделяя пятиокись азота, которая образует с влагой воздуха туман.
Пятая группа периодической системы
643
Смеси, которые, подобно 60’ь-пой азотной кислоте, при кипении или дистилляции не изменяют своего состава, называются азеотропными смесями, (geeuv —кипеть, тдолт'] — перемена, изменение). Концентрации, при которых они «постоянно кипят», и соответственно состав пара постоянно кипящей смеси изменяются в зависимости от давления, в то время как состав пара химического соединения, испаряющегося без разложения, не зависит от давления. Помимо водною раствора азотной кислоты, водные растворы спирта и соляной кислоты являются наиболее известными примерами азеотропных смесей. Hi\O3 образует два гидрата; Н1\О;).П2О (т. пл. —37,85е) и H.NO3-ЗН2О (т. пл. —18,5°). Их существование подтверждается тем, что на диаграмме плавкости системы Н.ХОЯ—Н20 появляются максимумы, подобные максимумам в системе Н2О—ЗО3 (см. рис. 123 на стр. 762). Из кривых температур кипения и упругости пара водных растворов HNO3 нельзя: сделать заключение о существовании гидратов, так как они раз.тагатотся в парах. В случае соединения HNO3'H2O речь, вероятно, идет (по данным Симона (Simon, 1938)] о нитрате гидроксопия [Н3О][Л’О3] (ср, стр, 047). Поразительно, правда, как установил Цинтль (Zintl, 1935), что кристаллический моногидрат нс даёт рентгенограммы, подобной соединению [NH4][NO3] (ср. стр. 863). Это наводит па мысль о том, что это соединение можно рассматривать как орто азотную кислоту или тепграоксразот(У)кислогпу Н3ЙО4 пли 01Ч(ОН)3, что, однако, не подтверждается данными изучения спектров комбинационного рассеяния. Соли,, производные от этой кислоты, а именно Na3NO4 (оргониграт натрия), были получены Цинтлем при сплавлении KaNO3 с Na2O. Органическое производное этой кислоты — тригидроктдиацетилавотиая кислота (СН3-СО-О)2?>(ОИ)3 (т. кип. 45° при 15 мм рт-ст) — было известно уже с 1902 г. (Pictet). Это соединение обладает сильно нитрующим действием и образуется в качестве промежуточного продукта (Bacharach, 1931) при нитровании органических соединении нитратами, растворенными в ледяной уксусной кислоте и л и уксусном ангидриде, по Менке. То обстоятельство^ что растворенная в ледяной уксусной кислоте азотная кислота обладает значительно : большим нитрующим, (и окисляющим) действием, чем водный раствор азотной кислоты той же концентрации, было точно установлено в 1935 г. Бринером (Briner) прн измерении скоростей реакций.
Азотная кислота, особенно концентрированная,—сильный окислитель. Она окисляет серу до серной кислоты, фосфор —сначала до фосфористой, а затем до фосфорной кислот. Особенно энергично взаимодействует азотпая кислота с металлами; только золото, -платина и некоторые металлы платиновой группы пе реагируют с ней. Большинство реакций, в которые вступает азотная кислота, сопровождается выделением окиси азота, например:
8HN634-3Cn = 3Cu(NO3)2-{-2NO-]-4H2O. (8)
Азотная кислота сначала действует на медь как окислитель, по затем следующая порция кислоты образует с окисью меди соль
2HNO3+3Cu = 3CaO-|-H2O-|-2NO.. ЗС«О-|-6НМОз=ЗСп(МО3)2-}-ЗН2О.
Железо в разбавленной (например, 0,2 я.) азоттюй кислоте па холоду (0°) растворяется : е образованием преимущественно нитрата железа (II). С более концентрированной азотной кислотой и при нагревании образуется нитрат ящлсза (III).
Замечательно, что некоторые металлы, на которые энергично действует разбавленная азотная кислота, устойчивы по отношению к высококонцентрированной HNO3; к ним относятся, иацример, железо, хром, алюминий и даже кальций. Это объясняется ? уже неоднократно упоминавшимся явлением пассивирования..
Органические сйедппсния под действием концентрированной азотной кислоты или окисляются, или нитруются. В и ос ле дне,м случае остаток азотной кислоты — NO 2, нитрогруппа, замещает в органических соединениях водород. Так, из бензила CSIIG получается нитробензол CaH5NOa. Этими реакциями нитрования и объясняется тот желтый цвет, который приобретают при соприкосновении с концентрированной азотной кислотой многие органические вещества, в том числе и кожа, так как большинство нитро-4?*
644
Глава, 14
соединений имеет желтый цвет. Особенно сильным нитрующим действие обладает смесь из концентрированных азотной и серной кислот, которую поэтому иногда называют нитрующей кислотой. Вследствие «пассивирующего» действия азотной кислоты эту смесь мряшо хранить в железных сосудах.
На обоих упомянутых выше свойствах азотной кислоты — ее окисляющей способности и нитрующем действии — главным образом й основана ее широкое применение в технике. В качестве окислителя, например., ее используют при получении фосфорной кислоты из фосфора,-щавелевой кислоты — из углеводов, серной кислоты—при камерном способе ее приготовления. Нитрующее действие азотной кислоты используют преимущественно в производстве красок. При производстве большей части содержащих азот органических красителей применяют азотную кислоту. Далее? ею пользуются для приготовления нитроглицерина из глицерина, нитроцеллюлозы (бездымный порох и коллодий) — из*клетчатки, пикриновой кислоты, а также вообще почти всех содержащих азот взрывчатых веществ. Кроме того, HNO3 применяют в производстве нитратов и используют в качестве химического растворителя для большинства металлов. Под названием разделительной жидкости ее применяют для отделения золота от серебра.
Золото и платина нерастворимы в азотной кислоте, но они растворяются в смеси концентрированных азотной и соляной кислот (царской водке), Обычно обе кислоты смешивают в отношении 1 ; 3 (по объему). Царская водка выделяет нитрозилхлорид NOC1и свободный хлор
HN03+3HGI = CJ3-|-2H30+NOC1, (9)
под действием которых металлы переходят в хлориды.
Получение азотной кислоты до качала текущего столетия проводили почти иск.чщ-тнтелыго по способу, применявшемуся Глаубером еще в середине XVII в., а именно нагреванием нитратов щелочных металлов с умеренно концеитрироваппоп серной кислотой с последующей отгопкоп образующейся азотпой*кислоты.
aM^Os+HaSOi^SHNO^-hMlSOi.
Глаубер использовал для этой реакции нитрат калия. В дальнейшем в большинстве случаев исходили из более дешевого и легче приготовляемого нитрата натрия.
В технике используют первую стадию реакции
М^Оз+^Н^Оа^ HNOa+ftfHSOi.
протекающую уже ирп умеренном нагревании (^150°), в те время как полное прсвра-щеттие происходит при таких температурах, при которых в значительной степени идет разложение ьзотной Кислоты,
Основное и почти исключительно промышленное применение находит в настоящее время метод получения азотной кислоты, основанный на каталитическом окислении аммиака (способ Оствальда). По этому методу смесь аммиака с воздухом пропускают над нагретой платиной пли плати-на-родием. Аммиак окисляется в соответствии с уравнением
4NH3 -!,-5О2= 4NO-^-6H204-215 ккал, (10)
образуя окись азота вводу. При охлаждении газов избыточным кислородом воздуха NO окисляется до КО2. Смесь нитрозных газов затем пропускают в поглотительные башни, где она соприкасается с водой, и одновременно через эти башйи продувают воздух; в этих условиях, как было уже описано на стр. 641, и происходит образование азотной кислоты. /
Пятая группа периодической системы
645
Каталитическое окисление аммиака, по Бодопштсйву (Bodcnstein, 1941), происходит следующим образом:
NHg+O = NH-(O (И) NH3O-|-O2=HNO2-ll-JbO (12) HN0j02 = HN0j " (13)
HITO4 -> NO + O2+OH (14) 2ОН = НаО4-О, который штовь принимает участие в каталитическом процессе (15)
После реакции (11) могут также, помимо (12)—(15), происходить и другие реакции, ведущие к образованию NZO или N2, например:
NH3O-|-O = H2O-|-HNO; 2HNO = H20-J-N30 п HNO+NH3O = 2HsO+N2.
То, что смесь аммиака с воздухом при пропускании се над нагретой платиной образует окислы азота, было установлено еще в 1839 г. Кульманом (Kuhlman). Однако для технического использования этой реакции решающим Оказалось наблюдение Оствальда о том, что для получения хороших выходов существенно вести этот процесс так, чтобы газовая смесь соприкасалась с катализатором лишь в течение очень короткого времени, (около 1/1000 сек); иначе будет происходить разложение окиси азота, являющейся очень нестойким соединением, и в результате в смеси, выходящей из контактной печи, останется только элементарный азот.
Поэтому контактные аппараты устроены так, что в них газовый ноток с довольно большой скоростью проходит через частую проволочную сетку. Эта сетка после того, как на пей начинается процесс горения, раскаляется за счет тепла реакции. При абсорбции питрозных газов получается 40—,50%-лый раствор HNO3, пригодный для большинства промышленных целей (производство удобрений). Концентрированную кислоту вплоть до 69% можно получить дистилляцией (см, стр. 642), Если необходима кислота еще более высокой концентрации, то используют водуотнимающие средства — концентрированную серную кислоту или пятиокись фосфора. Непосредственно высококонцентрированную азотную кислоту (до 100%) можно получить при действии на воду или разбавленную азотную кислоту смеси чистых joKn—O,,.
В начале этого столетия возник первый синтетический способ «сжигания воздуха^, наиболее старым вариантом которого явился открытый в 1903 г. способ Биркеланда— Эйде, коротко уже рассмотренный выше. Методы сжигания воздуха связаны с очень большими затратами электрической энергии. Поэтому с самого начала их применение ограничивалось странами, которые, кац, например, Норвегия и Швейцария, обладают особо дешевыми водными источниками этой энергии. Но даже и в этих странах позднее этот метод был вытеснен методом, основанным на окислении аммиака.
Азотная кислота принадлежит к числу наиболее сильных кислот. Ес кажущаяся степень диссоциации (измеренная по коэффициентам электропроводности) в 1 н. растворе равна 82%, в 0,1 н. растворе — 93% (при 18а).
Гантч, основываясь на оптических измерениях Шефера (Schaefers), развил представления, согласно которым азотпая кислота существует в двух изомерных формах, От одной из них производятся эфиры, а от другой — соли:
>N-O-C2HS
(Г
Этиловый вфир
азотной кислоты
О
NO О
Нитрат налия
К
Разница между этими формами заключается в том, что в эфире этиловая группа соединена с азотом посредством одного определенного атома кислорода, в то время как в типичных созях азотной кислоты на положительный ион металла действует отрицательный заряд радикала [NO3]" в целом. В разбавленных водных растворах, в которых радикал азотной кислоты существует в виде свободного иона [NO3]', его заряд действует (тоже как одно целое) на свободные ионы водорода или, выражаясь точнее, на иопы гидроксония [Н3О]’. Однако, как это видно из рассмотрения рис, Г16, практически безводная азотная кислота характеризуется кривой абсорбции, значительно более близкой к соответствующей кривой ее эфира, чем к/гой кривой, которая получается для разбавленных водных растворов азотпой кислоты. Последняя же кривая идентична кривой солей азотттой кислоты. Почти такой же вид, как кривая абсорбции абсолютной кислоты, имеет и кривая абсорбции раствора НМО3 в эфире; таге как, по Гантчу, следует, что значительные изменения в абсорбционных’спектрах всегда обусловливаются изменениями в химическом строении веществ (па обосновании этого положения здесь
646
Глава 14
пвнозможио останавливаться), то это значит, что азотная кислота в безводном состпе-нии п кпслота в эфирном растворе обладают иным строением, чем в водных растворах, а именно тем, которое соответствует ее строению л эфирах азотной, кислоты, следоаг-тельтто, O2N—ОН. Ганга называет
Чисм колебании
Р и с... 116, Спектр поглощения азотпой кислоты, ее сояоп и эфиров.
эту форму <тсевдаазотной кислотой».
Однако кривые 2 и 4 рис, 116 несколько отличаются от кривой 3 (кривой абсорбции для-эфира). По теорпп Гантча, это зависит от того, что безводная азотная кислота не является в строгом смысле гомогенной, но в действительности состоит из смеси «нсевдокис-лота» с солью «истинной кислоты», причем катион этой кислоты образуется при присоединении протона к ледис-социированной кислоте
03N(OH)4-H+=[ON(OH)2]+ или 20;>N(0H) [01\’(ОН)2г 4- [КО3Г; (16)
катион [0N(0H)2]+ называется ионом дигидроксоншпрония.
Развивавшиеся Гантчем представления были в основном Подтверждены и одновременно расширены позднейшими исследованиями, особенно исследованиями спектров комбинационного рассеяния (Dadieu, 1931, Mfidard 1933—1934; Simon, 1938). При этом было установлено следующее; устойчивому только при низких температурах твердому гидрату HNO3-II2O следует приписывать формулу нитрата гидроксония [Н3ОР [NO3]*. При плавлении устанавливается равновесие
[ПзО1++(N03r Н2О+О2К (Oil) (17)
Кривая 1 — HNOa и KSOjB разбавленном вол. пом растворе:1г — Ь1О2-ОП вэфпрнпм рас.твг1.е: 3 — OCjnV(этиловый эфор квотой кислотыj
. i — HNOs почте полностью о tea в о же иная.
В спектре комбинационного рассеяния расплавленного соединения присутствуют, помимо линий, характерных для 1N Оз-группы,- также линии или полосы,
характерные для N02- и ОН-групп, которые отсутствует в спектре кристаллического соединения. Полученные Симоном спектры комбинационного рассеяния представлены на рис. 117. Прй добав-
Р и с, 117; Спектры комбинационного рассеяния безводной азотпой кислоты й сю моногидратов в расплавленном я кристаллическом состояниях.
лея ин воды равновесие, соответствующее уравнению (17), сдвигается, конечно, влево.\В разбавленных водных растворах азотпая кислота присутствует практически
Пятая группа периодической системы
647
исключительно в гидратированной форме [Н3О]+- и [N О3] “-ионов. В жидкой безводной азотной кислоте, по данным спектров комбинационного рассеяния, существует почти исключительно молекулярная форма О2Л'(ОН), однако проявляются также и Лилин, характерные для группы NO?,. В отсутствие воды группа NO3 может образовываться двумя путями;. 2O2N(OH) l'oN(OU)a]+ + [NO3f n 3O2N(OH)7>: [N02]+ + (H30)+4-4-2[NO3]“. Инголвд (1950) установил наличие одновременно обоих равновесий диссоциации. Этим объясняется ранее совершенно непонятная высокая электропроводность практически безводной азотной кислоты. У твердой безводной азотной кислоты отсутствуют, как эго поь'взаяо на рис. 117Т линии, характерные для группы NO,. В этом состоянии, очевидно, азотную кислоту можно рассматривать как чистый нитрат дизид-роксопитрония JON(OH)2][NO3).
Подтверждение своего предположения о существовании иона (0N(OH)2]+ в безводной азотной кислоте Гантч усматривал В открытых им кристаллических соединениях между азотной и хлорной кислотами, которые он считал перхлоратами гндроксонит-рония; [ON(OH)2][C1O4] и [N(OH)3] ГС1О4] 2, Позднейшие исследования, особенно Ииголь-да (Ingold, 1950), все же показали, Что при взаимодействий безводной азотной кислоты с безводной хлорной кислотой образуется не ион еидроксонитрония, по ион нитрония [NO2]+ по уравнению ч
0sN(0H)-{-2H0.C103=[N03J*4-(H3O].*4-2IG1O4]\ (18)
Ипгольд предположил, что и пятиокись азота является солью нитропия, а именно нитратом нитрония [NO2[* [NO3j“ (о солях нитрония см. стр. 670). Тот вывод, что безводная кислота содержит группы ОН в той же форме, как и сильные основания, не следует Ограничивать одной лишь азотной кислотой. Исследованиями спектров комбинационного рассеяния показано таки;о для значительного числа другах кислот (Н3ЛяО4, H,SO4, H2SO5, HeTeOe, НС1О4, HIOj), что они н не диссоциированномсостоянии содержат водород в форме гидроксильных групп. Поэтому естественно предположить, что see кислородные кислоты а над ассоциированном состоянии являются гидрр-ксилсодержа-щими соединениями. В отдельных случаях, когда на основании спектров комбинационного рассеяния отсутствует доказательство наличия гидроксильных групп, Яак в случае фосфорной кислоты, оказывается возможным свести это к влиянию водородных мостиковых связей (см. стр. 679).
Нитраты, Соли азотной кислоты, нитраты Mf(NO3), обычно пригеч товляют растворением соответствующих металлов в азотной кислоте или же насыщением азотной кислоты гидроокисями или карбонатами этих металлов, ,
В производстве нитраты получают в большинстве случаев взаимодействием азотной кислоты с гидроокисями или карбонатами в водных растворах.
При действии нитрозных газов, получающихся при каталитическом окислении аммиака, непосредственно на водные растворы щелочей, согласно уравнению (6), помимо нитрата, образуется нитрит, который можно полностью окислить до нитрата тблько сравнительно сложным способом. Нитрат кальция, не содержащий “интриг а, получается, по Бринсру (Briner, 1929), при взаимодействии сухой Двуокиси азота с карбонатом кальция 3NO24-CaCO3 -= Ca(NO3)2 + NO + СО2 4~ 27 вм.ь Са3(₽О4)2, по Брштеру, также взаимодействует с NO, боз образования нитрита даже в присутствии воды
3NO2 + Са3(РО4)2 4- Н3О - О(И0з)г 4- 2СаИРО4 4- NO.
Все нитраты, производные простых неорганических оснований, легко растворимы в воде. Сравнительно трудно растворяются нитраты некоторых комплексных оснований. Практически нерастворимый нитрат образует одно органическое апгндроосновапие, имеющее состав C.,0H16N4, называемое нитроном, которым иногда пользуются для осаждения иоиов NO’ .при весовом анализе. Нитрат нитрона имеет состав C20H1GN4'HNO3.
При нагревании безводные нитраты разлагаются, отщепляя кислород. При этом нитраты щелочных металлов теряют только треть своего киейо-дэода и переходят в питрнты (19) — реакция, которую можно значительно ускорить добавлением слабых восстановителей (например, металлйческо-
648 Глава 14
го свинца). Нитраты тяжелых металлов, поскольку их нитриты при Еаи ,.-кой температуре неустойчивы, претерпевают дальнейшее разложензв сопровождающееся отщеплением двуокиси азота и кислорода . Например,
KNO3 — KNO2-p/aO2, {ми,
Hg(NOs),=HgO4-2NO2+V2O2.
Нитраты благодаря их способности отщеплять кислородХпри ekij* кой температуре являются прекрасными окислителями. Однако их иг™, ные растворы в большинстве случаев не обладают окислительными се'»-ствами; нитраты в них восстанавливаются лишь при действии сил^а восстанавливающих средств, например водорода в момент выделетзи. В этом случае восстановление может привести к образованию аммтатц который можно получать количественно растворением алюминия, ттл^-е-или сплава Деварда в щелочных растворах нитратов,
Разложение щелочных цитратов может происходить пе только по уравнение (19), но и следующим образом: 2KNO3 = К2О 4- N,+ 5/2D2. Обе реакции эндотер мичны. Если нагревание производить в присутствии металлов с большой теплоте^, горения, то могут происходить реакции следующего типа: 3KNO3 4- 5А1 — ЗКА1О2 — 4-А12О3 4“ 3/2N2, которые сильно экзотермичны и могут сопровождаться взрывом, ftps использовании смесей нитратов натрия и калия для улучшения качества металлаз я сплавов посредством термической обработки (см. т. II, гл. I) во избежание неудач пе следует превышать определенную температуру, выше которой происходит сгорание металла (см. Leschewski К., Z. anorg. chem., 239, 17,, 1938).
Ортопитраты (тетраоксонитраты). При нагревании NaNO3 с Na2O Цинтль. (1937) сумел получить ертончтрат- натрия Na3NO4 белый порошок, расплывающийся на воздухе и полностью гидролизующийся водой до NaNO3 и 2NaOH, При нагревании в сухой атмосфере ортойнтрат устойчивее, чём NaNO3. Он обладает также более "высокой температурой плавления.
Таким же образом Цинтль сумел приготовить и целый ряд не полученных до того времспи «ортосолей» — пз водных растворов или сплавлением с NaOH. Например: NaJBeO3, Na3BO;i, Na3A104 и N;>:iNO3 (ортопитрит натрия). Даже для чрезвычайно мало склонного к солсобразовштию Ag,0 удается получить при нагревании с Ма2О"соль вполне определенного состава [диоксоаргентат(1)натрия] Na3AgO2 в) виде светлосерого, кристаллического порошка. Замечательно, что эта соль даже при 540° в вакууме еще пе отщепляет кислород, в то время как Ag2O начинает разлагаться уже при 160*. То обстоятельство, что эти соединения нельзя получить сплавлением с NaOH или Na2CO3, следует, вероятно, объяснять тем, что их энергия образования недостаточна, чтобы компенсировать энергию, необходимую для отщепления воды, освобождающейся при реакции с NaOH (или СО2 при реакции с Na2CO3).
Пероксоазотпая кислота получается, по Шварцу (Schwarz, 1948), при действии 100%-ной Н2О2 па N0O5 при —80®. Соединение, вероятно, имеет формулу IINO4. Оно очень нестабильно и разлагается со взрывом ужо при —30“; все же эквимолярная смесь 70%-пых растворов HNO3 и И2О2, в которой существует это соединение, устойчива в течение продолжительного времени при 20° и вполне безопасна в обращении. При болео сильном разведении происходит гидролиз, который приводит к полному разложению в 20%-ном растворе.
Еще раньше Д’Анс (D’Ans) наблюдал образование резко пахнущего, чрезвычайно легко разлагающегося соединения при действии И2О2 на N2OS, а также при действии фтора на растворы нитритов (Fichter). Исследователи предполагали, что-fl обоих случаях им удалось приготовить пероксоазетнуго кислоту, по состав соединения не был точно установлен. Возможно, его следовало считать яе пероксоазотной кислотой, а пероксоазотистой кислотой (см. стр. 649). Пероксоазотиая кпалотаг вероятно, образуется в качестве промежуточного продукта при каталитическом окислении аммиака (см, стр. 645).
Азотистая кислота и ,нитриты. Как уже было указано, нитриты щелочных металлов образуются при поглощении растворами гидроокисей
Пятая группа периодической системы 649'
щелочных металлов смеси равных частей NO и NO2; кроме того, их можно-получать умеренным восстановлением нитратов; так, нитрит натрия NaNOg образуется при сплавлении нитрата натрия со свинцом
NaN03-|-Pb=NaNOz4-PbO. (21)
Кислоты выделяют из нитритов свободную азотистую кислоту HNO2, устойчивую лишь в холодных разбавленных водных растворах. При нагревании азотистая кислота (в присутствии песка, осколков стекла или других предметов с острыми краями уже на холоду) разлагается ио-уравнению
3HN03=HN034-2N04-H20. ' (22)
Благодаря большой склонности ц распаду азотистая кислота является очень реакционноспособным веществом. Сильные окислители, например перманганат калия, окисляют ее до азотной кислоты. Под действием амальгам щелочных металлов или электролитически азотистая кислота восстанавливается в более бедные кислородом соединения, например-окись азота, закись азота, гидроксил амин, азотноватистую кислоту или аммиак. Особенно важна ее способность диазотировать органические-амины. Поэтому получаемый указанными выше методами нитрит натрия имеет широкое применение при техническом приготовлении азокрасителей .
Нитрит натрия NaNO.a обычно поступает в продажу для лабораторных целей в виде слегка желтоватых палочек. Он весьма гигроскопичен И очень легко растворим е воде (100 г воды при 2.5'' растворяют 85,5 г NaNOa), но лишь слабо растворяется в спирте. Нитрит калия KNO2 расцлывается па воздухе и растворим в воде еще лучше, чем NaNOa (100 г воды при 25° растворяют 314 г KNOa). При добавлении к такому раствору спирта происходит высаливание. Большинство других нитритов также легко растворяется в воде. В холодной воде относительно плохо растворим (1 : 300) часто применяемый в лабораторной практике нитрит серебра AgNO3.
Нитриты щелочных металлов в сухом состоянии плавятся без разложения. Другие нитриты разлагаются при высокой температуре; так, нитрит бария —при температуре выше 220°, нитрит серебра — при 140° и нитрит ртути — при 75°.
Азотистая кислота занимает пр о Meaty точное положение между кислотами средней силы и слабыми. В сильно разбавленных растворах при 18° найдена константа диссоциация К = 4,5-10"4. В 0,1 и. растворе ее степень диссоциации составляет 6,5%. Ее соли в водных растворах вследствие гидролиза малоустойчивы.
Алот-истая кислота, по крайней мере » своих соединениях, существует в двух изомерных формах:
НО—N=O и Н—n/°.
От них^производягся два ряда органических соединений;
RO —N = O (I) R—NC (В—алкил) (Ц)
;О
Эфиры азотистой кислоты Нитросо единения
0Д1пГиз них (I) легко омылятотся с образованием азотистой кислоты и спирта, Прн. действии на (I) водорода в момент выделения образуются спирты и наряду с ними-аммиак. Соединения другого ряда (II) пе омылятотся, а при их восстановлении образуются органические амины B-NH2, следовательно, при этом азот не отщепляется_
'650
Глава 14
Отсюда можно заключить, что в нитро со единениях азот непосредственно связан с ех-килыгым остатком, как ото и отражено в формуле (II).
Изомерия между обеими формами отряда тельного радикала Ж)2 играет нактт?-pyro роль и в тех комплексных соединения», в которые вступает этот радикал. Те соединения, которые содержат остаток азотистой кислоты в форме ONO, называют ншвдш-то соединениями, а те, в которые он входит в форме NO2, — нитро соединениях Нитрокомплексные соли по своей устойчивости значительно превосходят нигригз-ко.мплскспые.
Амид азотистой кислоты — нитрозоамид ON’NH2—был получен Шаардзе (1934) в виде интенсивно красного раствора в жидком аммиаке при очень низкой температуре при действии NHS на N2O3. Это соединение чрезвычайно легко разлагается (2ON-NH2 — [NH4] [NOal + N2). Значительно устойчивее его-органические прогж водные, которые уже давно были известны под названием нитрозоаминол 0N.NR2.
Растворенные в жидком аммиаке нитриты щелочных металлов взаимодействуют со щелочными металлами с образованием нитроксилатое солей неизвестной в свободном состоянии нитро ксилозой кислоты Г1\-. Например, 2NaNO2 + 2Na = = Na4[N2O4] — нитроксилат натрия. Формула для анион а нитро кси лата [О 2N—NO 2]4-, а не (NO2]a~, следует из его магнитных свойств. Щелочные нитрокеялаты имеют ярко-желтую окраску. В отсутствие кислорода воздуха при обычной температуре в твердом состоянии они устойчивы, однако эти соединения л со же очень реакционноспособны. Разложение водой происходит очень энергично й сопровождается окислительно-восстало ните льпой реакцией. В жидком аммиаке они нерастворимы (Maxted, 1917: Zint1, 1928). :
Пероксоазотистая кислота. Смесь перекиси водорода и нитрита при подкислении разбавленной серной кислотой мгповеипо выделяет бром из бромидов. Это наблюдение было сделано ещё Рашитом (Raschig). Перекись водорода, нитрит и нитрат порознь эту реакцию не дают. Она основана, по Байеру (Ваеуег, 1901), Шмидлйпу (Scbmld-lin, 1910), Трифонову (1922) и Глю (Glen, 1929—1935), на промежуточном образовании, пероксоазот истой кислоты О—N—О—О—-II. Это соединение, изомерное азотной кислоте, образуется, по Глю, при Действий озона на водный раствор щелочного азида. Водный раствор его оранжево-красный. Соединение чрезвычайно неустойчиво, и его нельзя получить в чистом виде в растворе. Оно образуется только как промежуточный продукт при указанных выше, а также некоторых других реакциях. Более устойчивы его щелочные соли. Все же и они до сих пор пе были получепы в чистом состоянии.
Азотнопатпетан кислота и гипопитрпты. Свободная азотноватистая кислота H2N2O2 образует белые кристаллы в виде’ листочков, которые в сухом состоянии чрезвычайно взрывчаты. Опа легко растворяется в воде, в спирте и несколько хуже в эфире. Электролитическая диссоциация ее в водных растворах, как Это установлено па основании измерения се электропроводности, незначительна. Согласно криоскопическим данным, азотноватистая кислота имеет молекулярный вес, соответствующий формуле H2N2O2. ,
Азотноватистая кислота в водном рцстворе в присутствии индикатора фенолфталеина нейтрализуется, как и угольная кислота, одним молем щелочи. Однако, кроме получающихся при этом кислых солей — кислых еипонитритов П^Н [N2O2J, она образует также нейтральные гипонитриты M*[N202], которые в свободном состояний устойчивее, чем легко разлагающиеся кислые гипонитрпты. Водой они, однако, очепь сильно гидролизуются. Свободная кислота в водном растворе разлагается главным образом но уравнению H2N2O2 = Н2О -ф- N2O. Этот процесс необратим, и поэтому закись азота N-О нельзя рассматривать как ангидрид азотноватистой кислоты.
Азотноватистая кислота образуется при-восстанонлении солей азотной или азотистой кислоты или окиси азота в водных растворах: амальгамой натрия." Ее моягно также получить и осторожным окислением гидроксил амин а (23) или же конденсацией гидроксил амина с азотистой кислотой (24), одпако с плохими выходами
Ag2Jo H2|NOH
-ф-Ag2 |О...HjjjNOH
N —ОЦ
~ II -|-2H2O-|-4Ag (23)
N—OH
H0N|-H2+ O!N0H=llD-_N;=N-0.H-bH20. (24)
Пятая группа периодической системы.
651
Оба последних способа образования азотноватистой кислоты и приводят к фор~ муле НО—N=N—ОН. Азотноватистой кислоте изомерен открытый в 1895 г. Тиле нитралшд. Это соединение, как показывает его название, обычно рассматривают как амид азотной кислоты O2N=NH2, однако его строение до сих пор окончательно но установлено.
Основываясь на его кислотном характере (константа диссоциации, по Бренстеду, К = 2,6-10"’), Гантч приписывает ему формулу имидоаготной кислоты HN=N0(0Н); в то же время Педерсен (Pedersen, 1934) на основе исследовании его каталитического разложения щелочами в водных, растворах (H2N2O2 —> N2O-p Н2О) высказывает предположение, что между обеими формами, существует равновесие, которое обычно сильно -сдвинуто в сторону амидной формы.
3. Соединения азота с водородом и их производные
От азота производятся следующие водородные соединения:
1) HN3 — азотистоводородная кислота. Она крайне неустойчива (взрывчата). Ее соли называются азидами.
2) NH, —аммиак. Может присоединять один ион водорода, образуя при этом положительный иоп | NHJ*. Соответственно этому аммиак образует с кислотами соли [NH4]X — соли аммония. Водные растворы аммиака благодаря наличию в них равновесия
NIT3 + H0I] [МВД’+ ОН'
имеют щелочную реакцию. Однако сам но собе аммиак является не основанием (так как он но имеет гидроксильной группы), а ангидрооснованием (см. стр. 83 и 85).
3) NsH4—гидразин. Это соединение также способно образовывать и кислотами соли, а именно соли гидразония (см. стр. 664).
А вот ист плодородна я кислота и азиды. Азотистоводороднаякислота — азоимид) HNa представляет собой бесцветную, обладающую острым запахом жидкость с удельным’весом 1,126, кипящую при 35,7° и затвердевающую при —80°. Это сильно эндотермичное соединение; ее ядовитые, разъедающие слизистые оболочки пары при соприкосновении с нагретыми предметами с большой силой взрываются.
2HN3ra3=3N24-H!S4-141,8 ккал (при постоянном объеме).
В водных растворах азотистоводородпая кисдота устойчива. Это слабая кислота, несколько слабее уксусной. Ее степень диссоциации при 2(Р в 0,1 н. растворе около 1 % (константа диссоциации К = 1,2- 10^в). Вследствие летучести ее легко вытесняют из солей — азидов менее летучие кислоты.
Азиды M*N3 по своей растворимости очень похожи на галоидный соли. Большинство их легко, растворимо. Трудиорастворимыми являются прежде всего азиды серебра, двухвалентного свипца и одновалентной ртути. Исходным материалом для приготовления других азидов, а также свободной кислоты обычно является азид натрия NaNs, получаемый пропусканием закиси азота через расплавленный амид натрия
NaN i Н2 + О ; N2 = NaN3-(-H2O.
Эта реакция протекает гладко слева направо, так как образующаяся вода пемед-ленпо удаляется, вступая в реакцию с еще непрореагпровавшим амидом натрия по уравнению
Н ! ОН 4 Na iNHs=NaOH-|-Nrj3.
652
Глава 14
Азид натрия плавится без разложения и лишь при более сильегт нагревании дает вспышку. Аналогично ведут себя и остальные азпд.^ щелочных и щелочноземельных металлов. Однако азиды тяжелых метет лов, например свинца или серебра, прн нагревании, и особенно прп утх ре, сильно взрывают, и взрыв вследствие детонации легко распространяется на другие взрывчатые вещества. Благодаря атому Свойству азид свит— применяют для изготовления запальных приспособлений. Интересез по своему составу азид аммония, N4H4 = [NHJ [N3L
В то время как а зоти ст о во до родная Кислота разлагается иодом (в присутсптс небольшого количества тиосульфата) с выделением азота
2HN34-I2^2HT4-3N2, азид серебра при действии па пего иода образует азид иода
AgN3-|-l2=IN3-j-AgI.
Азид иода IN3—чрезвычайно взрывчатое, в совершенно чистом состоянии, вероятно, бесцветное твердое вещество, обычно окрашенное в желтовато-белый цвет. Ази&-брома BrWj при обычной температуре — жидкость, азид хлора CIW3, образующийся прн действии азида натрия на гипохлорит натрия по реакций
NaN3 + NaOCI + Н2О = C1N3 + 2NaOH, представляет собой бесцветный, сильпо взрывающийся при со пр икр спо вей и и с пламенем газ.
О получении и свойствах C1N3 и хлоразида серебра AgClN3 см. также: Frierson, К гоцт ad, Browne, J. Am. chem. Soc., 65, 1696, 1698, 1943. Азпд фтора FN3, полученный Брауном и Галлером (Browne, Haller, 1942) при действии 1'2 на 1HN3 в атмосфере азота,— желто-зеленый газ (т. пл. —154°, т. кип, —82°), постепенна разлагающийся с образованием N2F2 но уравнению 2FN3 = 2N2-}-W2F2. В жидком состоянии FN3 взрывчат.
Тещгота образования жидкого HN3 составляет —63,0 ккал,’моль (прп постоянном давлении). Теплота испарения 7,29 ккал/моль, теплота растворения 9,73 клалЛиоль (Gunther, 1935—1936). Прп электролизе азидных -растворов ион N3 разряжается на аноде с выделением азота. Потенциал разрядки иона N3 на гладкой платине составляет 1,18а при 18° (Riesciifcld, 1935).
Строение. Физические методы лсследоваивя (спектры комбинационного рассеяния, вращательно-колебательные спектры, намерение моментов инерции, электропо-графнчестпго исследования) указывают на то, что атомы азота в азотпетоводородной кислого расположены прямолинейно. Структура, следовательно, подобна структуре, установленной для N2O, от которой а зоти сто водородная кислота отличается только тем, что вместо атома О в ней содержится группа NH. Расстояния N + N = 1,14 А, N <—* Nil =1,25 A (Schumaker, 1942). Рентгеноструктурным методом доказано линейное расположение атомов N также и для кристаллического азида.
Если HN3 при пониженном давлении пропускать через сильно нагретую кварцевую трубку, то происходит разложение
hn3=n2+nh.
Возникающий при этом свободный радикал NH образуется и при действии активного азота на HN3, а также, вероятно, при фотохимическом разложений HN3. Подробнее о свободном имидорадикале NH и об образующемся в результате конденсации радикалов NH при низких температурах голубом твердом веществе (NH)K см. стр. 663,
АММИАК
Свойства, Аммиак NHa — бесцветный газ с едким запахом и жгучим вкусом. Он значительно легче воздуха (уд. вес 0,5963, отнесенный к воздуху, как к единице). Вес 1 л NHs равен 0,7713 г. При давлении 8,46 атм и 20° аммиак сжижается, образуя легкоподвижную бесцветную, обладающую сильным светопреломлением жидкость, кипящую при —33,4°. Критическая температура NHa 132,4°, критическое давление 112 атм,
Пятая группа периодической системы
653
критическая плотность 0,236. Температура плавления — 77,7°. Теплота плавления 81 кал /г. Теплота испарения при температуре кипения 327 калгг, при 0° 302 кал!г.
Большую теплоту испарения аммиака используют ь холодильных установках. В значительной мере "она об у ело вл Ена тем, что мапомолекулярпыц в парообразном состоянии аммиак при сжижении, подобно поде, полимеризуется и его деполимеризация при испарении требует большой затраты тепла.
Жидкий аммиак по физическим свойствам во многом подобен воде. Он является хорошим раствор пт ел ем для некоторых веществ, в том числе и для многих солей.
Соли при растворении в жидком аммиаке, так же как и при растворении в воде, испытывают электролитическую диссоциацию. Взаимодействие между различными веществами, растворенными в жидком аммиаке, часто протекает иначе, чем в водных растворах, так как некоторый вещества, пе растворимые в воде, растворяются в жидком аммиаке (подробнее см. т. II).
Диэлектрическая проницаемость жидкого аммиака равна 21 — 23 (при —34°), его вязкость Г| — 0,0026. Собственная электропроводность совершенно чистого жидкого аммиака лранне ничтожна. Нарвалло (Carvailo) удалось довести удельную электропроводность жидкого аммиака (при — 15°) до величины 4.10-10.
Аммиак кристаллизуется в воде кубов. 'Структура его кристаллов была рентгенографически определена Марком (Mark). Расположение атомов N в его решетке видно па рис. 118; атомы I! в аммиаке, вероятно, отдают свои электроны азоту, так как соответствующие им интерференции не обнаружива-
ГРис. 118. Кристаллическая'решетка аммиака.
а= 5 А, (Указано только положение атома азота).
;ЮТСЯ.
В молекуле КП3, как это следует из физических измерений (абсорбционные спектры и спектры комбинационного рассеян ня), ядра водорода располагаются в форме равностороннего треугольника со стороной 1,6 А. Ядро атома азота лежит на перпендикуляре к плоскости треугольника, проходящего в 0,38 А от его центра тяжести, и способно занимать положение по обе стороны от этой плоскости. Эта так называемая инверсия молекулы NH3 приводит К появлению сильных абсорбционных линий в его микроволновом спектре. Инн ср сию молекулы аммиака можно использовать для установления частот колебаний в кварцевых часах (аммиачные часы). Молекула NH3 имеет -относительно высокий дипольный момент (ц = 1,5-10~18 эл.ст.сд.). Аммиак диамагнитен, как это и следовало ожидать, исходя из четного общего числа электронов
в молекуле.
Холодная вода чрезвычайно жадно поглощает аммиак; 1 лл воды при ’0° растворяет 1176 мл (=0,907 е), а при 20°—702 мл аммиака. Однако при кипячении аммиак из водных растворов полностью улетучивается. Водные растворы аммиака ведут себя как растворы слабых оснований. Они окрашивают красную лакмусовую бумажку в синий цвет и для нейтрализации требуют вполне определенного кол и честна кислоты. Из насыщенного кислотами раствора аммиака при упаривании кристаллизуются соли — соли аммония (подробнее см. ниже).
Теплота растворения аммиака в большом количестве воды равна (при 20°) -8,4 г.кал/молъ. Коиш-птрпролавпые водные растворы аммиака замерзают лишь при очень низких температурах. В спирте, ацетоне, хлороформе, бензоле и других органических раствор нт елях цм-чиак растворяется легко, но ие тая хорошо, как в воде.
Получение. Лмявди образуется при разложении азотсодержащих органических соединений. Поэтому в
654
Глава 14
аммония (см. ниже) он встречается также в природе, но большей часть®' лишь в очень незначительных количествах. В отдельных местах земн^ж поверхности, например вблизи вулканов, такие соединения скапливают^ в большом количестве. Однако для получения аммиака этиместорожденин по существу, не имеют никакого значения. Важным источником получение аммиака является побочный продукт производства светильного газа-кокса и сухой перегонки бурых углей аммиачная вода, (газовая вода:;. однако получаемое при этом количество аммиака не удовлетворяет •потребности в нем. Поэтому огромное значение имеет получение аммиака непосредственно из атмосферного азота прямым каталитическим соединением азота с водородом по методу Габера —Боша.
Аммиак из газовой воды.. Образующаяся главным образом при сухой перегонке каменных углей на газовых заводах и при коксовании углей газовая вода представляет собой бесцветную, пахнущую аммиаком, дегтем" и сероводородом жидкость, которая содержит аммиак (большей частью 1—3%) в виде различных солей, главным образом карбоната и, кроме того, в виде сульфида, сульфата, тиосульфата, хлорида,; роданида и т. д. Аммиак из этого раствора сначала выделяют кипячением. При этом улетучивается й та часть аммиака, которая находится в растворе в виде солей, так как они при кипячении гидролизуются; остаток аммиака отгоняют при добавлении к раствору известкового молока.
Из паров аммиака, смешанных с водяным паром и другими Летучими веществами, сначала удаляют основные примеси, а именно СО2 и H2S , путем обработан известковым молоком, а затем охлаждением Конденсируют воду; после этого высушенный таким способом аммиак освобождают от органических веществ, пропуская нац древесным углем или же при промывании парафиновым маслом. Очищенный аммиак затем или сжижают компрессором или же поглощают дистиллированной водой, приготовляя растворы, известные под названием нашатырного спирта.
Иногда аммиак получают при перегонке с разложением отходов сахарного производства', в этом случае его получают главным образом в виде карбоната и сульфата аммопиЯ. В прежнее время аммиак и аммонийные соли получали из самых различных, содержащих азот отбросов органического происхождения, главным образом из перегнившей мочи. Нашатырь (хлорид аммония) этим способом приготовляли еще арабские алхимики. Одно время большое значении имел валъцийцианамидный способ приготовления аммиака Рота — Франка — Каро. Синтез аммиака этим способом основан ла разложении кальцийциаиамнда при нагревании водяным паром или водой под давлением. Связывание атмосферного азота сильно нагретым карбидом кальция с образованием кальцийци ап амида и отщепление аммиака от последнего уже было описано на стр. 504. В настоящее время по способу Рота — Франка — Каро кальцийци ап амид производится еще в большом количестве (см. стр. 656), однако он используется почти исключительно в качестве удобрения, а не для производства аммиака.
Синтез аммиака. Синтез аммиака по способу Габера—Боша основан на возможности активировать катализатором реакцию между азотом и водородом при температурах, лежащих значительно ниже той, при которой равновесие
3/^2,3/а112 ^Нз={-11,05 ккал* (25)
практически полностью смещено влево, и, далее, —на использовании давления для смещения этого равновесия вправо в сторону увеличения концентрации аммиака.
Влияние температуры и давления на равн рвесие (25) качественно определяется па основании принципа Ле-П1ательо. В соответствии с этим ирикципом равновесие
* Величина теплоты образования относится к 20а. При 500° теплота образования равна 12,7, при 700° 13,2, ’при 0° 10,95 ккал. Свободная эпергия образования составляет при 0° 3,91 ккал/молъ NH3.
Пятая группа периодической системы
655
при повышении температуры должно смещаться в сторопу, указываемую нижней, стрелкой, а при повышении давления — н сторону, указываемую верхней стрелкой, Количественно зависимость равновесия от парциальных давлений компонентов и от общего суммарного давления для реакции (25) можно представить на основании закона действия масс следующей формулой;
^NHS _ Р’
(26)
где T’Nj’ "hs_ парциальные давления газов NH2,N2 и Н2; Xv— константа равновесия.
Зависимость Кр от температуры выражается изобарой реакции В ант-Гофф аф( см. стр. 58). Пользуясь ею, Габер вычислил значения константы равновесия Кр для различных температур, приведенные в табл. 101. В таблице также приведены равновесные концентрации аммиака при различных давлениях при условии, если исходная смесь содержит 3 об. ч. Н2 и 1 об, ч.
Таблица 101
Содержание аммиака, находящегося в равновесии со смесью из 3 об. ч. водорода й 1 об. ч. азота
Т, сС 15 1 flm Содержание Nils; об. %
30 ат (00 ат 200 ат 100 0 ат
200 0,660 15,3 67,6 80,6 85,8 98,з;2
300 0,070 2,18 31,8 52,1 62,8 92,6
400 0,0138 0,44 10,7 25,1 36,3 79,8
500 0,0040 0,129 3,62 10,4 17,6 57,5
600 0,00151 0,049 1,43 4,47 8,25 31,4
700 0,00069 0,0223 0,66 2,14 4,11 12,9
800 0,00036 0,0117 0,35 1,15 2,24 —
аоо 0,000212 0,0069 0,21 0,68 : 1,34 — ..
1000 0,000136 0,0044 0,13 0,44 0,87 —
Из рассмотрения этой таблицы следует, что выход аммиака зависит не только от давления, йо в значительной мере также и от темпер ату ры’сивтсз а, т. е. от активности катализатора. Особенно активны я этом отношении рутений, осмий и уран.. Они активны уже при температурах, близких к 400°, однако их применению в технике препятствует высокая стоимость. Хорошим катализатором для технических целей оказа^ лось жеяевв, которым пользовался еще Габер в своих опытах. Обычно его каталитическое действие проявляется только при температуре выше 650°, это действие можпо значительно усилить соответствующими добавками.
П качестве катализаторов пригодны также активированные добавками окись и цианид железа. Обычно используют давление 200 ат и температуру 500°.
Бошу удалось преодолеть технические трудности, связанные с. изготовлен и ем специальной аппаратуры, которая должна выдерживать высокую температуру ц значительное давление. Так как при соединении азота с водородом выделяется тепло, то при надлежащем уст ройстве контактных печей необходимая для реакции температура поддерживается за счет теплоты, выделяемой самой реакцией.
Водород для синтеза аммиака получают в большинстве случаев путем взаимодействия водяного пара с СО в присутствии катализатора (см. стр. 46). Стоимость водорода значительно отражается на общей стоимости синтетического аммиака.
Лежащие в основе современного промышленного синтеза аммиака исследования равновесия его распада и образования были выполнены в 1905—1910 гг. в основном Габером. Первая установка для получения синтетического аммиака была пущена, лотом 1913 г. (Оппау). Ее производительность составляла около 4000 Tlaod NH3. В годы первой мировой войны в связи с потребностью в азотном сырье производство аммиака чрезвычайно возросло. В 191 Г, г, было получено уже 40 ОиО т синтетического NH-,. в 1917 — более 60 000 т. После войны и в других странах на этой же основе лачй-
656
Глава. 14
нает развиваться производство аммиака, так, во Франции был разработан метод (Claude'), в котором применяли давление 1000 ат. Достигаемое при этом увеличив. содержания аммиака в газовой смеси, проходящей через катализатор (см.табл. 111 упропщло способ работы. Одпако это преимущество уравновешивалось ут;-лпчением стоимости аппаратуры и т. д. В дальнейшем во Франции и Италии был рату:^-ботан способ Фаузера и Казале (Fanser, Casale), в котором применяют давление 25С— 800 ат. В других методах, например в методе Mown-Цени, используют меньшие лиг ления (90—100 ат) и добиваются удовлетворительного выхода, проводя реакцию s~i возможно бол се низкой температуре (400°). В настоящее время во всем мире подавлег-щес количество' аммиака приготовляют синтетически.
Получение аммиака в лаборатории обычно производится разложение! содой аммония, например хлорида аммония NH4C1, сильными основаниями
2NH4CI+Ca(OH)2=CaCi24-2NH3-}-2H2O.
Если требуется более значительное количество аммиака, то пользуются жидкое аммиаком иа стальных цилиндров, в которых он поступает в продажу. Небольпшг количества чистого и сухого газообразного аммиака удобнее всего получать нагреванием его двойного соединения с хлоридом серебра
AgCl 3NII3=AgCl -J-3NHS
Применение. Главная область применения аммиака — производство искусственных удобрений. В азотсодержащих удобрениях азот большей частью содержится в виде аммонийных солей или солей азотной кислоты; однако HNOs, как уже было отмечено, приготовляют в настоящее время почти исключительно из аммиака.
Приобретающую в последнее время все большее значение в качестве удобрения мочевину CO(NHa)a приготовляют, помимо указанного на стр. 504 способа, из цианамида кальция, а также и из синтетического аммиака.
Аммиак, взаимодействуя г, двуокисью углерода, образует карбаминат аммония NH4[CO2-NH2] (сц. стр. 661), который прп нагревании выше 115° иод давлением превращается в мочевину
2NIJS> СО2 = [NH4][CO2-Nh2]; [М141[СОг-КН2] = C0(NH2)2 ф Н2О.
В настоящее время в промышленности мочевину пр и гот опия гот непосредственным воздействием СО2 на водный раствор аммиака при 130 —140° и давлении 100 ат. Прп конденсации мочевины с формальдегидом, по Поллаку (Poliak, 1921), получают важные искусственные вещества: r-аурит (растворимое в воде клеевидпое вещество), пластопал (заменитель шеллака) й поллопаз (широко применяемая в настоящее время прессовальная масса). Мочевина начинает приобретать значение и в качестве добавки к кормам для скота.;
^Но и помимо этого, аммиак находнт^широкое применение в химической промышленности. Водный раствор аммиака применяют в аналитической химии как очень удобное осаждающее средство. В домашнем обиходе разбавленными водными растворами аммиака под названном нашатырного- спирта пользуются для чистки и выведения пятен. Его применяют также в медицине. Безводный аммиак благодаря своей значительной теплоте испарения играет большую роль в холодильном деле (холодильники, приготовление искусственного льда).
В научных исследованиях сжиженный аммиак играет большую роль как растворитель и как среда для проведения химических реакций (см. т. II), а также как средство для обезвоживания (метод аммиачной экстракции Вильтца, См. стр. 353 и 537).
Химические свойства. Аммиак, хотя и устойчив при обычкой температуре, является все же сравнительно реакционноспособным веществом.
Пятая еруппа периодической Системы 657 !
’ “ 1 ’ ' к
, Lv Электрическая искра разлагает NHa на составные элементы. При соприкосновении с каталитически активными веществами происходит его час- г'
тичпое'разложение уже при сравнительно небольшом нагревании. На воз- 4
духе аммиак при обычных условиях не горит; однако существуют смеси 9
аммиака с воздухом, которые при поджигании загораются. Он сгорает 1 i Н также, если его ввести в горящее на воздухе газовое пламя. В чистом кис-породе аммиак сгорает бледпо-желтым пламенем преимущественно до азота <'
и воды :
2NH3 -| 3/3Ог=N2+ЗИ;,О.
Находящаяся под высоким давлением кислород-аммиачная смесь взрыв- j
чата. , \ ।:
В присутствии катализаторов можно достигнуть сгорания смесей с воздухом уже при относительно низких температурах (300^-500°). j |
В соответствующих условиях при этом образуются исключительно I'
окпелй азота. Вак уже было отмечено, это обстоятельство используется в промышленности для получения азотной кислоты (метод Оствальда, i {
см. стр. 645). t
При каталитическом окислении аммиака происходит, вероятно, Промежуточное I
образование нитрозилгидрида HiXO. Хартеку (Harteck, 1930—1933) удалось получить j!
это неустойчивое в газообразном состоянии соединение в виде твердого светло-желтого i.
вещества при деветвим атомарного кислорода на NH3 или при присоединении атомарного водорода к NO при температуре жидкого воздуха. Нитрозилнатрий NaNO можно получить действием NO 'на растворенный в жидком, аммиаке натрий (Joannis, 1894) или при нагреваний металлического Na До 470—180° в раз бал ленном 4 ч. азота токе NO (Gohlen, 1939) в виде бесцветного скрытокрлсталличесокого соединения. Цлптль (Zintl, 1933) доказал рентгенографически, что это соединение отличается от имеющего с ним одинаковый состав гнпонитрита натрия. При, растворении в воде NaNO бурно выделяет N2O (кроме этого, образуется гипонитрит и гидроокись натрия). Таким же ।
f образом, как NaNO, можно получить К.NO и питрозилы щелочноземельных металлов. 1
Сильные окислители, например перекись водорода, хромовая кислота
i или перманганат калия, окисляют аммпак и в водных растворах. С хлором и бромом аммиак энергично реагирует !
3Cl2-}-8NII3=N2-{-6NH4Cl.
В большинстве случаев аммиак разлагается также и при пропускании его над нагретыми металлами или окислами металлов, причем иногда азот аммиака взаимодействует с металлом, образуя нитрид. Щелочные и щелоч- I
поземельные металлы замещают в аммиаке один атом Н, образуя амиды, например J
i NH34-Na = NaNH2-|-V2HE.
Амид, натрия
, Чрезвычайно велика склонность аммиака к присоединению к другим ) веществам с образованием аммиакатов, например
J AgCi -|-3NH3 = AgCl 3NH3.
Аммиакат хлорида серебра
? Большая склонность и присоединению к другим веществам сближает аммиак
с водой. Эта способность объясняется большими дипольными моментами этих молекул. Молекулы РНЙ и H.,S, дипольные моменты которых значительно мепьше (см. табл. 97 и 106, стр. 629 и ”738), образуют только очень ограниченное число продуктов присоединения: к веществам, обладающим типичной ионной решеткой, они вообще не ,£ способны присоединяться (Witz, 1925: Holtje, 1936).
. 42 р Реми
1 ' >
«58
Глава 34
Наиболее энергично аммиак соединяется с кислотами а образованная соединений, которые по всем свойствам следует рассматривать как настоящие соли. Они содержат положительный радикал [NHJ* (радикал аммонии^ Производящиеся от него соли называются аммонийными солями. Этот радикал присутствует в виде свободного нона (NH,]' (ион аммония) также и в водных растворах.
Образование иона аммония, которое проще всего можно представить уравнением
_\Ня-|-Н-^>!МПГ, (28
происходит также и в водных растворах аммиака. Однако вследствие крайне незначительной диссоциации воды равновесие (28) в атом случае сильно сдвинуто влево. Коатому в водных растворах аммиака присутствует лишь незначительное количество ионов аммония и соответственно этому также лишь немного гидроксильных ионов, так как они. если не принимать во внимание исчезающе малого количества ионов ОН', содержащихся в чистой воде, образуются в результате реакции
NH34-H2O^[NH4r + OII'. (29)
Тем самым объясняется, почему водные растворы аммиака обнаруживают щелочную (но только лишь слабо щелочную) реакцию и ведут себя как растворы слабых оснований.
Процесс (29) можно представить состоящим ив двух стадий:
Жз+НгО *Н3-Н2О + OU'. (29а)
О том, что яри растворении аммиака в воде образуется соединение, можно заключить по очень высокой растворимости и большой положительной теплоте растворения аммиака (см. стр. 653). Это соединение — тдрат аммиака NII3- II2 О — можно выделить при низкой температуре в кристаллическом виде (т. пл. — 79,0°)- Из растворов с особенно высокой концентрацией аммиака кристаллизуется полугпдрат i\TH3.i/2II2O (т. пл. —78,3’). Напротив. н,ед1.иеоции/1ованная гидроокись аммония [ЛН,]0Н в водных растворах в раиновгсшг е ионами |NIL ]‘ и О1Г не существует, ибо соединение [NIJJOH в исдиссоцпнровашюм состоя пин, как потощал Бриглеб (Briegleb, 1942), неустойчиво. Оно отличается от гидрата аммиака своим строением — это продукт присоединения. В нем вода связана с NII3 как единое целое (вероятно, посредством водородных мостиков). Напротив, гидроокись аммония должна была бы быть координационным соединением такого же строения, как соль аммония (NIIJX; т. е. опа должна была бы Состоять из положительного .иона Nil; п отрицательного иона ОН'. В незначительной стеноцн, судя по уравнению (29а), алсктролитическн диссоциирующий гидрат аммиака является слабым основание.». Соответственно этому гидроокись аммония, так как она существует только в полностью Диссоциированием состоянии, следует рассматривать как сильное основание.
Иримегтяя защш действия масс к равновесию, представленному уравнением (29) для достаточно разбавленных растворов, в которых концентрация воды в результате реакции (29) практически по изменяется, получаем
[Nii;.|x[OH']_ [NH3]
(30)
При 18^ = 1,75 • 10“’.
В соответствии с изложениям выше, К можно рассматривать как константу электролитической диссоциации гидрата аммиака. Однако неправильно, как это неоднократно делали рапыпе, считать се за константу ди сер циации гидроокиси аммония.
При 18° 1М водный раствор аммиака содержит 0,4% аммиака в виде диссоциированной на ионы гидроокиси аммония, а 0,1М раствор—1,3%.
.Пятая группа периодической системы.
(559
С позиции теории Косселя процесс, п ре дета влеп тп.тй уравнением (28), объясняется как следствие влияния атома азота, обладающего в аммиаке тройным отрицательным зарядом, па находящиеся в сфере его ,действия положительные попы. Сначала ион №~ присоединяет три ядра атома водорода, от которых оп получает электроны
МЗ- л- зи+ = Nhs.
Одпако на основании закона Кулона нетрудно показать, что при присоединении еще четвертого положительно заряженного иона водорода к несущему тройной отрицательный заряд атому азота должна выделяться Эпергия, и притом в таком количестве, что, если этот четвертый положительный ион водорода находится в водном растворе в виде гидратированного иона (Н30]\ эта энергия окажется достаточной, чтобы «оторвать» положительный ион водорода от гидратированного иона
NH3 Н" [Н3О]’ = {NHJ- + Н2О.
В соответствии с теорией ковалентных связей способность молекулы NH3 присоединять четвертое ядро Н+ основана на том, что в ней атом азота содержит еще неиспользованную в какой-либо связи электронную пару, которая может быть предоставлена ядру атома Н’’ для образования гомеглюлярпоп связи. Образование иона аммония сво-
н I н -L н - 4- г н’’ 1
н-хц-н* = и 1 и , н :n: и и ti+:n;3’h+
н 1. 1. н - н н+
1а „ io п
дится, таким образом, к проявлению дативной свяли (см. стр, 162 и 446), Возникающая гомеополярпая форма иона [NH4]+, представленная формулами 1а и 16, согласно квантово-мехапичссним представлениям, резонирует* с гетерополяр-ной формой П.
Резонанс с ковалентными связями упрочндет ионные связи. В основном эта трактовка сводится к трактовке па основ© теории Косселя, объясняющей упрочнение связей поляризационными эффектами.
Амальга мн аммоппя. Если обработать концентрированный раствор аммоттийной соли амальгамой натрия плп подвергнуть его электролизу с ртутным катодом, то получится тестообразная масса, называемая амальгамой аммония. Химически она ведет себя подобно амальгамам щелочных металлов; одпако при комнатной температуре она относительно быстро разлагается с выделением На п NII3 в объемном отношении 1 : 2, Разложение идет по уравнению 2NH4 = Н2 + 2NH3, Поэтому раньше предполагали, что в этом соединении в виде сплава с ртутью присутствует радикал аммония NH1(. В настоящее время известно, что металлы и их Сплавы построены не из электро нейтральных атомов, а из положительных попов, расположенных среди свободно передвигающихся между пими электронов (см. т. II, гл. 1). Поэтому в амальгаме аммония присутствуют не свободные радикалы ХН4, а ионы [NH4|+. Их образование основано па том, что металлическая ртуть способна поглощать noni>i[NHJ+, если одновременно с этим процессом атомы ртути получают электроны, необходимые для компенсации зарядов ионон
Соли аммония
Аммонийные соли по растворимости и другим свойствам, обладают большим сходством с солями щелочных металлов. Опп отличаются от солей щелочных металлов своей летучестью при нагревании и, кроме того, тем, что сильные основания (гидроокиси щелочных и щелочноземельных металлов) иповь вытесняют из них свободный аммиак [см. уравнение (27), стр. 65(5].
Приготовление аммонийных солей производится большей частью пропусканием газообразного аммиака в растворы соответствующих кислот или смешиванием кислот с растворами аммиака или карбоната аммония.
* Вероятно, чисто гомееполярйая и чисто гетерополярпая формы резонируют с промежуточными формами, в которых часть ядер атома водорода связана атомом азота томеополяряо, а часть — гетерополярно.
&
«60
Глава 14
Хлорид аммония (нашатырь) [NHJC1 получают при насыщении соляной кислотой растворов аммиака или карбоната аммония
NH34-HCI=[NH4]C1
или
[NHJ2CO34-2MCl=2[NHJC14-H20+C02.
Исходным материалом для получения jVH4C1 в технике служит получающаяся а виде отброса при производстве светильного газа и при коксовании газовая веда (см. стр. 654) . Последнюю сначала концентрируют однократной перегонкой. «Концентрированную газовую воду», содержащую 12—25% NH3, смешивают с концентрированной соляной кислотой 11 затем- упаривают образовавшийся при этом раствор нашатыря досуха. Очистку последнего производят перекристаллизацией или же сублимацией.
Хлорид аммония представляет собой бесцветную соль, обладающую горько-соленым вкусом, легко растворяющуюся: в воде, несколько труднее — в спирте; NH4C1 легко сублимируется и заметно летуч уже при нагревании на водяной бане. Пары хлорида аммония отчасти диссоциированы на NH3 и НС1. При осаждении паров NH4C1 образуется волокнисто-кристаллическая масса; из водных растворов NH4C1 кристаллизуется в виде правильных октаэдров; удельный вес 1,52; теплота растворения —3,84 ккал/моль при 18°.
В старых исследованиях указывалось, что хлорид аммония из растворов,; находящихся в деревянных чанах, выделяется в виде красивых больших кристаллов, в то аремя как иначе он обладает склонностью к образованию мелких перистых разветвленных кристалликов. Увеличение размеров кристаллов объясняется действием пек типовых веществ дерева. Поэтому выращивание возможно более крупных кристаллов нашатыря ведут при добавлении к его растворам растворов пектиновых веществ или других соединении, обладающих подобным же действием.
Структура кристаллической решетки хлорида аммония соответствует структуре иодида Цезия (рис. 48, стр. 239), только на место атомов цезия расположены атомы .Л пли радикалы NHj. Длипа ребра элементарного куба составляет а— 3,859 А (при комнатной темиературс). Лыше 184,5° хлорид аммония кристаллизуется но типу NaCl. Около —30° он испытывает другое превращение, которое, одиако, связано не с изменением структуры решетки, а с торможением ниже этой температуры свободного вращения радикала [УН4]Т около своих осей (см. стр. 247).
Растворимость хлорида аммония в воде составляет;
при 0° 20" 60° юо° 115,6°
29,7 37,2 55,2 77,3 87,3 а М14С1 в 100 а НаО.
При обычной температуре водный раствор хлорида аммония имеет нейтральную реакцию. При кипячении такой раствор становится кислым вследствие улетучивания следов аммиака, образующегося в результате гидролиза.
Нашатырь применяют при паянии, так как он реагирует с окислами металлов, образуя с ними летучие хлориды ц очищая таким образом Поверхность спаиваемых металлов от окислов. Хлористый аммоний служит электролитом в элементе Лекланше и в сухих элементах, а также находит разнообразное применение в химической промышленности ив лабораториях. Его применяют также в медицине.
Нитрат аммония. Дмлюиийиая селитра [NI-I4]NO3, получаемая из аммиака и азотной кислоты, представляет собой бесцветную расплывающуюся на влажном воздухе соль, обычно кристаллизующуюся в виде ромбических кристаллов с удельным весом 1,73; кристаллы плавятся при 169,5° и при несколько более высокой: температуре разлагаются на воду
Пятая группа периодической системы
661
и закись азота*. Аммонийную селитру применяют для приготовления закиси азота, но главным образом для приготовления взрывчатых веществ. Смесь аммонийной селитры и карбоната кальция применяют в качестве удобрения.
Нитрат аммония растворяется в воде с сильным понижением температуры (поглощается 6,2 ккал на 1 люль). Его растворимость прн 0° составляет 118, при 25° 214, при 100° 871 г NH4NO3 в 100 г воды.
Нитрат аммония может существовать в шести различных модификациях, некоторых пять устойчивы при обычном давлении. Превращения происходят при следующих температурах: ‘ ,
-i8" 32,3’ 84,3° 135,2“
Гексагональная а~Ромбическая . > р-Ромбическая Тетрагональная Кубическая
Карбонат аммония [NH412C’O3 получается при взаимодействии двуокиси углерода и аммиака в водном растворе (31) илипрп нагревании смеси сульфата аммония с отмученным мелом (32).
С02+2.\Н3-(-И30 = [М14)3Ср3. (31)
[NH4]2SO4+СаСО3 = [NHJ3CO3+CaSO4. (32)
Полученная вторым способом соль после сублимации наряду с нормальным карбонатом аммония (NIT4)SCO3 содержит кислый карбонат аммония (бикарбонат аммония) [NHjHGO3 и, кроме того, карбамипат аммония (карбаминовокпелый аммоний) [NH4] [CO.3(N Нв)1.
Сухой газообразный аммиак ири взаимодействии с сухой двуокисью углерода образует карбамипат аммония, т.п. аммонийную соль карбаминовой кислоты NH2 -СО(ОН). Эта кислота является производной угольной кислоты СО(ОН)2 « получается из нее при замене одной группы ОИ на группу NII2 (аминогруппу). Вода пере-водит карбамат в нейтральный карбонат, однако этот процесс обратим
[NH4][^C(NH2)] -1- Н2О^± [NH4]2[gC0L
Бикарбонат аммония прп добавлении к нему аммиака или при нагревании раствора (яри этом происходит отщепление С02) переходит в нейтральный карбонат
[NH4]HCO3 Ф N.H3 - ENH4]2CO3; 2[NH4]HCO3 = [NH4]2CO3 4 CO2 H2O.
Нейтральный карбонат при стоянии на воздухе, отщепляя аммиак, переходит в бикарбонат. При нагревании приблизительно до 60° он быстро разлагается на аммиак, двуокись углерода и воду.
Торговый продукт содержит главным образом двойное соединение бикарбоната аммония с карбаминатом аммония
[NH4)HC03.[NH4][C02>NH2].
Сульфат аммония, сернокислый- аммоний, [NH4]2SO4 получают в настоящее время в Германии, помимо прямого насыщения серной кислоты газообразным аммиаком, прежде всего по способу I.G. Farbenindustrie (способ гипс—сульфат аммония), основанному на взаимодействии гипса с карбонатом аммония. По этому способу в тонко измельченную водную
* В зависимости от условий опыта, помимо N2O, образуется большее или меньшее количество N2. Так как прн его образов алии по уравнению 5|NH(]NO3 = 4N2.-T-4-9Н2О + 2HNO3 выделяется свободная HNOa, что каталитически ускоряет разложение, то при плавлении бо.чь rm г х количеств NH4NO3 может произойти взрыл. Пр Трамму (Tramm, 1934), предотвратить взрывообразное разложение можно нейтрализацией кислоты, например аммиаком.
662
Глава! 14
суспензию гипса пропускают аммиак и двуокись углерода
CaSO4+2M13 4-СО3 + Н2О-> CaCO3+(NH4)2SO4.
Сульфат аммония образует бесцветные ромбические кристаллы с удельным весом 1,77, легко растворимые в воде (при 0° 71,0, при 20° 7(5,3, щз 100° 97,5 г соли в 100 г воды). Ето применяют главным образом как удобрение. 1
Сульфат аммония можно получить из светильного или коксового газа поликши-натным способом, разработанным I. G. Farboninduslrie. Этот метод основап наташ что NH3 и H2S в водном растворе взаимодействуют с по литпо патами с образование тиосульфата аммония
2(NH4)2[S,Oe] 4 2(NII4)2S3Oti + 6NH3 + 6H2S 7(NH4)2S2Os + 6S -ф 3H2O.
При Спшгапии серы и обработке сернистым газом раствора тиосульфата политио нате регенерируются
2(NH4)2[S2Os) 4 3SO2->(NH4)2[S4O61+ (NH4)2[S30el.
Большую часть раствора, содержащего тиосульфат, нагревают до 120—130° в автоклавах. При этом выделяется сера и образуется сульфат аммония 2(NH4)2IS2O3] — +(NH4)2(S4O0) = 3(NH4)2SO4 4- 5S. Получающуюся при этом серу можно использовать как таковую либо для переработки на серную кислоту.
При нагревании до 357° сульфат аммония разлагается (при атмосферном давлении) с отщеплением аммиака и образованием расплава, который состоит из смеси ноиэ-менившогося нейтрального сульфата аммопия и образовавшегося при отщеплении аммиака кислого сульфата аммония [MH4]HSO4- Чистый кислый сульфат аммония плавится при 251° и кипит при 490° без разложения.
Кислая [NH4jO-OH и нормальная перекись аммония [Л'Н4)3О2, которые можно рассматривать как первичную и вторичную аммонийные сбЛи перекиси водорода, получают пропуск ан и ем аммиака в сил т.п о охлажденный Крепкий эфирный раствор перекиси водорода (D’Ans, 1913). При плавлении или растворении соль [NH4][HO2J превращается в соединение КП3 • Н 202. Это было установлено Симоном (Simon, 1955) па основании из учения спектров комбинационного рассеяния.
ПРОИЗВОДНЫЕ АММИАКА
Если от NIL; отнять одни атом водорода, то остается одновалентная группа —NIL,. В неорганической химии ее называют амидной группой, если же ома является Лигандом в координационном соединении, — то амидогруппой*. Две амидные группы могут соединиться между собой, образуя диамид H2N—NH2 (гидразин). Соединения, в которых группа NH, связана с металлом, называются амидами металлов (например, NaNH2 — амид натрия). В отличие от этого соединения, в которых радикал ГШ2 связан с определенным электроотрицательным атомом или группой, например С1 или ОН, называют аминами. Так, соединение NH.-.OH называют гидроксиламином и соединения (очень неустойчивые) NH2C1 и NHCL—хлорамином и дихлорамином.
Последние два названия (редко применяемые в неорганической химии для обозначения соединений) образованы по принципу замещения, т. е. соединения NH3‘OH, NH2C1 и NHC12 можно рассматривать как продукты замещения аммиака. Слово «амин» в этом случае обозначает не группу —NH3, а молекулу NH3, например в названии триметпламип N(CH3)3.
* В органической химии, как известно, дня группы NH2 применяют пазванне а.ииОогруппа только для амидов кислот; обычно их называют амино группами. Соответственно в органической химии группу =NH называют имин о группой.
Пятая грцппа периодической системы
663
Если заменить группу ОП в кислотал группой NHa, то в зависимости от того, замещены есе группы ОН или только часть их, получаются амиды кислот (например, ацетамид, сульфамид) или амидокислоты (например, амидоссрная кислота). Часто для амидов кислот применяют короткие названия, например ацетамид, сульфамид, фосфамид (для фос-форилтриамида см. стр, 697). Амидосерпую кислоту раньше называли
амидосульфонОВОН КИСЛОТОЙ. ,о С113С< \NHa Адетилампа (ацетамид), амид уксусной КИСЛОТЫ vHa ^NH2 (У \он С ул ь ф урилаыпд Aw идосе рт!а i i (сульфамид), кисло га. аюжюшгд днямид серной серной кислоты кислоты
Двухвалентную группу HN< в неорганической химии назы-
вают имид или и.иис*огруппой. Соединения, в которых эта группа является анионом, называются имидами. Остающуюся после удаления всех трех атомов водорода от аммиака группу N=, называли ранее в некоторых соединениях нитрило группой. В настоящее время для обозначения этой группы применяют названия нитрид- или ишяридогруппа. Двойные соединения азота с электроположительным элементом называют нитридами, например А1А — пнтрнр алюминия. Если представить азот в качестве заместителя О и ОП группы в кислородных кислотах, то получим нптпрпдосоедипения, например N(SO3H)3 — нмтридосериая кислота.
В свободном состоянии радикал имид NH был спектрографически обнаружен как продукт диссоциации, аммиака при высоких температурах (2000°). Оп также образуется (со средней продолжительностью жизпн 1,3-10"3 сек) при пропускании под уменьшенным давлением (па пример, 0,07 ,<«.« рт ст) а зотистов ода родной кислоты через нагретую до 1000° кварцевую трубку (Rice, 1951). Па охлаждаемых жидким азотом Степках трубки радикалы NH кондеи си pyro тс я с образованием голубого твердого вещества, парамагнитного и не электропроводного. Структура этого соединения еще не вполне ясна, возможно, речь идет в атом случае о ди амиде HN=NH. При — 1254 голубое твердое вещество превращается с выделением тепла в бесцветный азид аммония [NH4]N3.
Хлорамин N И,С1 может быть стабилизирован введением в его молекулу органических радикалов. Например, натриевая соль л-толуолсульфохлорамида СН3-СвН4. SO2-NHC1, кристаллизующаяся с тремя молекулами воды, так устойчива, что может быть перекристаллизована из горячей воды без разложения. Это соединение (торговое название «хлорамин») является очень хорошим дезинфицирующим, отбеливающим я растворяющим крахмал сродством. Его можно также применять при онсидлмегрпче-ском титровании (Noll, 1924); см. ташке: Р о е lli k е W., Wolf F-, Z. anorg. chem., 2(;8, 244, 1952. Окислительное действ ire хлорамина основано на том, что в водном растворе происходит частичное его гидролитическое расщепление с образованием гипохлорита
[CIJ3.CeH4.SO3-NCI]' < H2O7tCII3-CgH4.Sd3.NH2 4-: [СЮ)'.
Гидразаи и гидроксиламин, подобно аммиаку, способны присоединять протоны и образовывать соли с кислотами. Нав и У аммиака, эта способность связана с координационной чс-тырехеалентлостъю азота- Гидразин, содержащий Два координационно четырехвалентных атома азота, способен поэтому образовывать два ряда солей (соли гидразиния) например:
TI..X-NH3+ HC1=JH.>N— NH,]Clr
х “ Хлорид гидраятнш
H:N -NH2 + 2НС1 = [H3N—NH3]C12.
Дпхлорид гйДравтШия
В отличие ог этого от гидроксил амина производится только один ряд солей (солп в идро кс шшлг.ио я пя), п апр и 1 г I? р
Н,\О11 + НС1 = [ИзМОЩСГ
-X л ори д улд родояла мчал ли
664 Глае a 14
Гидразин N.,H4 был впервые получен в 1889 г. Курциусом из органических веществ. Но его можпо получить также и из неорганических веществ, например при восстановлении азотноватой кислоты сернистым аммонием или, еще лучше, по Рашиту, при окислении аммиака гипохлоритом натрия в присутствии клея.
Гипохлорит натрия сначала реагирует с аммиаком, образуя хлорамин NH2-.C1
NII3+NaOCl = NaOH + NHrGl. :
Это весьма неустойчивое вещество тотчас же претерпевает дальнейшее превращение, причем обычно выделяется азот
2NH,C1 -i- NaOCl 4- 2NaOH =. N2 3NaCl ф 3H2O.
Но в присутствии, клея или желатины происходит реакция с аммиаком и образуется гцдразни
NH2; Ci’+ H?;NH2-|-NaOH=II2N—NH34-NaCl-(-H2O.
Гидразин можно осадить из раствора в виде сульфата гидр а типа [N2He]SO4 и вновь выделить его крепким раствором едкого кали. При этом вначале получается гидрат гидразина N2H4-.H2O, из которого безводное соединение получают 1! ср сгонкой с едким патром.
Действие клея при получении гидразина по Раш игу ос попало на том, что оп снизывает ионы тяжелых металлов, и прежде всего попы Си, которые в виде следов присутствуют в реагентах и каталитически ускоряют разложение образующегося вначале NII3C1 с выделением N2. Клей можно заменить другими веществами, которые также очець црочпо связывают ионы Cu (Р f о i f f о г Р., Вег., 80, 127, 1947). '
Гидразин, представляет собой бесцветную, сильно дымящую на воздухе жидкость с удельным весом 1,01.1. при 15°; температура плавления 1,4°, температура кипепия 113,5°. С водой и спиртом гидразин смешивается во всех отношениях. Гидрат гидразина N2H4-H.,O ташке представляет собой жидкость при обычной температуре, но оп застывает при более низкой температуре и кплнт прн более высокой, чем безводнре соединение (температура плавления —40', температура кипения 118,5°при 739,5 мм рт ст,, удельный вес 1,030).
Строение молекулы N2H4;
К <-->N = 1,47 A; N —> Н = 1,04 А; < HNN = 108D. [(Giguere, Schomaker, J. Аш. chem. Soc., 65, 2025,1943].
Гндразнп способен соединяться с одним или двумя эквивалентами кислоты, образуя соли гидразиния. Например, с соляной кислотой образуются дна соединения: Ш2ГД]С1 (хлорид гидразиния) и [N3He]Cl3 (ди-хлорид гидразиния). Сульфат [N3He]SO4 в холодной воде растворим плохо, но в горячей растворяется хорошо, и поэтому его легко можно перекристаллизовать. Кристаллизуется в виде бесцветных толстых пластинок или длинных призм; в спирте нерастворим.
Соли гидразина применяют при получении азидов, а также в аналитической химии в качестве восстановителя. Восстановительное действие гидразина выражено очень отчетливо. Для гидразина или для щелочных растворов гидразиииовых солей особенно характерна способность выделять в свободном" виде благородные металлы, из растворов их солей.
Гидрокспламнп NH2-Ori получают но способу Тафеля электролитическим восстановлением азотной кислоты на свинцовом катоде
н—.О—NOa+6H' + 8e=H—О—NH2-|-2H2O,
Пятая, группа, периодической системы
665
В технике для получения гидрокси ламина применяют, кроме этого способа, также старый метод Рашпга, по которому сначала готовят «ги д рокси ла миндпсу л).фо-кис л ьгй натрий*, действуя нитритом натрия на сильно кислый раствор бисульфита натрия при охлаждении льдом (33), затем обменной реакцией с хлоридом калия осаждают трудно растворимую соль калия (34), которую разлагают кипячением с водой, причем образуется сульфат гндроксиламмодин (35).
NaNO3 -f- NaIlSOa -f- 8O2 = HO—N(SO3Na)2. (33)
HO—N(SO3N;t)2 (- 2KC1 =: HO—N(SO5K)24-2WaCl. (34)
2H0-N(SO3K)2 4- 4H2O. = [HO—NH3]2SO4 -fr 2K2SO4 ф- H2SO4. (35)
Обычно приготовляют только соли (соли гидроксиламмония), так как получение свободного ангидроосноваиия затруднительно и иногда не вполне безопасно. При нагревании свободный гидроксиламин распадается сО взрывом; при нагревании открытым огнем в пробирке очень'небольшого его количества происходит оглушительный взрыв. Раствор гидро-ксиламипа имеет сильно щелочную реакцию.
Соли гидроисиламмония [NH3(0H) IX большей частью легко растворимы в воде. Они, как и свободные а пгидр обе новация, являются сильными восстановите л ям и. Кроме того, необходимо подчеркнуть их весьма сильную ядовитость.
В щелочных растворах гидрокси ламин постепенно разлагается, обычно с выделением азота BnJLjOH ЗН2б + NH3 4- К2. При известных обстоятельствах, папрн-мер в присутствии платиновой чорпи, разложение идет с образованием гипрнитрита 4NH2OH 4- 2N.aO.lI = .4Н2О 4- 2N Н3 4- Na2(N2O2). В обоих случаях вначале образуется (рапсе называемый нитрокси.юм)'. 2NH20H = NH3 4~ Н^О ф
4-NOH ,
Гидрид пнтрозйла тут же реагирует дальше -г либо с гидр оксид амином с выделением азота NOH 4- КН2ОП — Х2 “Ь 2Н2О, либо аниоп нитрозилгидрида димеризуется с образованием азот нов а тостом кислоты
NOIT4 oh'=no’-|-H2O; 2риб]'= [:o:n::n:o:j"
Попы NO' могут быть количественно связаны ионами [NifCNJiV с образованием интенсивно окрашенных красно-фиолетовых ионов трициаяопитрозмл никеля [NifCN^l’^ ф-NO' = [Ni(CN)3NO]" 4* CN'. Поэтому ионы [Ni(CN)4r в щелочном растворе являются очень чувствительным реагентом на нитрозилгидрид. При распаде внтрамнда или гипонцтрнта в щелочном растворе в присутствии ионов [Ni(CN)4’J" красно-фиолетового окрашивания не появляется- Отсюда следует, что эта реакция протекает по через образование нитроз ил гидрид a (Nasi, 1948). Другая реакция на нитрозил гидрид, предложенная Антелн — Рнмнин, основа па па том, что NOH взаимодействует с альдегидами с образованием гидроксамовой кислоты Н-СО-НН(ОИ)Л которая при Добавлении раствора ЕеС13 Дает краевое окрашивание.
Соединения, производимые от гидроксилами на заменой обоих, связанных с азо-том атомов Ни содержащие радикал >N—ОН, называются оксимами. Оксимы, например альдоксим RCH ; N0H и кет ок сим R1R2C : NOH, играют существенную роль в органической химии.
ГидрокслламнПсульфоиовые кислоты. При замещении л гидроксил а мине одного или нескольких атомов водорода на радикал серной кислоты — SO2(OH) получаются соединения, обозначаемые как гидроксиламинеульфоновые кислоты [В=(Н0)О23—)
R< R. Нч FL R.
^N—OH ^N—OH /N—OR pN—OR >N — OR
Hz Rz Hz Hz Rz
'Все пять соединений известны в свободном состоянии или в форме щелочных солей. 5 Эти соединения зиачпге гыго различаются по устойчивости в щелочной среде и, как показал Наст (Nast, 1952), механизмом своего гидролитического распада.
Амиды металлов. Амиды металлов имеют общую формулу Амиды щелочных и щелочноземельных металлов образуются непосредственно при действии аммиака на металлы; например, амид натрия Na А Н.,
Й66
Глава 14
получается при пропускании сухого газообразного аммиаканад расплавленным натрием
NH3 Н Na — NaNH2 4- 1/ЕН2.
При продолжительном стоя.imil раствора металлического натрия в жидком аммиаке амид патрия образуется даже на холоду. При его взаимодействии с солями тяже-лих металлов, растворенными в жпдком аммиаке, можно получить амидыэтих металлов В противоположность дов'ольпо устойчивым амидам щелочных й щелоч по земельных металлов амиды тяжелых металлов большей частью взрывчаты.
Г
Чистый амид натрия образует бесцветную кристаллическую массу. ’ кристаллы которой имеют форму лучей; температура плавления 2105. Оп возгоняется в вакууме, но при 500° разлагается (а в вакууме уже около 300°). Расплав амида натрия действует как энергичный восстановитель и вместе с тем как конденсирующее средство вследствие энергичной реакции с водой
NH2Na + ПОН - NaOH 4-.\Н3С
Амид натрия применяют в технике при синтезах органических веществ, особенно при синтезе индиго, при приготовлении цианцда натрия и азида натрия.
О реакциях щелочных амидов с ангидридами кислот см.: Bock G., Z. anorg. Chem., 233, 155, 1947. Амидыщелочноземельных металлов при нагревании переходят в и.ииЭы MnNH, кристаллизующиеся в кубической"грапецептрнрованном решетке. При дальнейшем нагревании в высоком вакууме желтые имиды щелбчно земельных металлов переходят в краспо-желтые или красно-коричневые пернитриды MpNj. При разложении разбавленном кислотой перпитриды выделяют, помимо двух молей NH3, один моль К2, например SrsN4 4- 8НС1 = 3SrCl2 +2NH4C1 4- N2. (Hartmann, Frohlich, Ebert, 1934.)
Нитриды. Пшпридалш называют соединения азота с сильно электроположительными элементами, преимущественно с металлами. Многие металлы при нагревании непосредственно соединяются с азотом. Иногда удобнее получать нитриды в чистом виде — нагреванием металлов или их окислов или хлоридов в струе аммиака. Нитриды щелочноземельных металлов можпо получать также разложением их амидов.
Те нитриды, которые по составу можпо рассматривать как продукты замещения водорода в аммиаке, например Li3N, Мд3М2, CasN3, 1 Sr3Na, Ba,N,, ZnyN2, AIN, в большинство случаев разлагаются водой (ZnsN2 и A1N — лучше раствором едкого кали) с выделением аммиака. Но существуют такие нитриды, которые нельзя считать просто продуктами замещения водорода в аммиаке, например Mn5N2, W2N3,‘ZrN. Эти соединения часто значительно устойчивее к воде. Нитрид хрома CrN не разлагается водой даже прп 220°.
Большинство нитридов отличается большой стойкостью при пагрева-иии. Подобно окис лам, о пн большей частью тугоплавки.
В качестве двойных нитридов следует рассматривать LiMgN, LiZnN, Li3AlN2» Li3GaN2 (Jnza, 1948). Опн кристаллизуются в координационной решетке н обладают солеобразпым характером. То же имеет место для соответствующих соединений гомологов азота; Лакее, Новотный и Юз (Laves, Nowotny, Juz а) получили следующие соединения и изучили их структуру:
LiMgP LiMgSb LiZnP Li3AlP2
LiiMgAs LiMgBi LiZnAs Li3AIAs2
Пятая группа, периодической системы
667
4. Галоидные соединении азота
Известны два класса галоидных соединений азота. Соединения одного из .них имеют общую формулу XN3, а соединения другого — NX3 (X — галоген). Первые являются производными от азотистоводородной кислоты. HN3, и о них уже была речь в связи с этой кислотой. Вторые можно рассматривать в соответствии со способом их образования как продукты замещения водорода аммиака на галоген. Например, можно получить хлористый азот NC13, действуя хлором на хлорид аммония (в концентрированном водном растворе). Фторид азота NF3, бесцветный газ, сжижающийся при —129°, открыт в 1928 г. (Ruff). Это соединение экзотермнчно и чрезвычайно стойко. В отличие от него хлористый азот NC1.3 и подпетый азот NT3 чрезвычайно взрывчаты.
Аналогичное бромное соединение вообще пе может быть выделено и известно только в виде аммиаката NBr3-6NH3, который тем не менее чрезвычайно легко разлагается. Более устойчивы, чем тригалогениды, оксигалогениды азота. Важнейшие свойства этих соединений сопоставлены в табл. 102 (стр. GG8). 1
Трехфтористый азот NF3 был получен Руффом при электролитическом разложении расплавленного безводного [NH4]HF2. Это бесцветный гаа, сжижающийся при Сильном охлаждении в прозрачную, легко подвижную жидкость. В воде и в едком кали трехфтористый азот практически нерастворим, на сухое стекло и ртуть не действует. по .медленно реагирует с ледяным миром при шнркитацяи няектрмческойискроп 2NF3 + ЗН,0 = 6HF |- ХгО3. С водородом чрезвычайно легко реагирует, даже со вспышкой 2NF3 -р 3112 - (ШЕ -г N3. Структура молекулы NF3: тригональная ппра-/ F
мида с атомом N в вершине; расстояние ►F—1,37 A; <tN' = ]0a:i (Schoma-XF
ker, 1947). К трехфторпстому азоту, полученному электролизом (NTIJHFa, помимо большого количества азота, всегда прлмеигано дойного чрезвычайно взрывчатых соединений NH,F, NITF, и N2F4. Их мощно отделить при пропускании над МпОй. N2F4 (т. кип. —125°) произволнтся от N2H4 так же, как NF3 от NH3. Существует соединение N2F2 (бесцветный газ). Оно также очень взрывчато. По электроне гр а фиче с к им данным, опо имеет следующую структуру:
/V Расстояния; N«—»F = 1,4-4 А
.У N *--‘N—1,25 А
г<иоФорма транс- Фирм а
Открытый в 1934 г. Меди (Cady) и обстоятельно исследованный в 1.935 г. Руффом ншпроксщрторид NO3F имеет чрезвычайно интересную структуру. Оп производится ог окиси фтора F—О—F мри замене одного атома фтора на радикал нитрил — NO2. Это ядовитый газ с удушливым запахом, взрывающийся мрн нагревании. В отличие от NF3t NOF ir i\OaF нитроксифторид — эндотермическое соединение.
Хлористый азот, трихлорид азота, NC13 представляет собой темно-желтое летучее масло с удельным весом 1,65, с резким запахом. NC13 сильно действует па слизистую оболочку глаз и носа. Это соединение чрезвычайно бурно взрывается при нагревании до 93° или при соприкосновении с веществами, способными хлорироваться, например со скипидаром, каучуком. Хлористый азот мощно хранить в сероуглероде, бензоле, эфире, хлороформе или четырех хлористом углероде без доступа воздуха. С аммиаком реакция идет по уравнению NC13 -ф 4NHS == N2 + 3NH4C1. Водой хлористый азот медленно гидролизуется NC1S 4- ЗНОЙ = NH.; 3G1OH.
Таблица 102
Трйгалогсппды и оксигалогеппды азота
Свойства NFj NC13 NI, NOF NOaF NO3F NOC1 NOsCl NOBr
Цвет Температура плавления, °C Бесцветный —208,5 Желтый При обычной температуре жидкий Черпни При обычной температуре твердый Бесцветный -132,5 Бесцветный —166 Бесцветный ; —175 \ Красно* желтый -61,5 Бесцветный -145 Черно-коричневый —55,5
Температура кипения, °C -129,0 — — -59,9 -72,4 —45,9 1 -5,8 —15,0 "J-4 9
Теплота образования, ккал/молъ +26 -54,7 Сильно отрицательная Вероятно» положительная Вероятно, положительная Отрицательная, —7,2 . — —16,9
Бес 1л (0°), г 3,17 — — 2,231 2,971 ' — 2,992 2,81 : (при 100е) Л
Плотность при температуре кипения 1,885 1,65 1,326 1,796 1,507 1,592 1,41
Ллпгал еруппа, перподичесхой системы
669
Хлористый азот получают либо используя обратимость этой реакции яри действии гипохлорита иа подлые растворы амшш или аммонийных солей, либо (для демонстрационных целей) производя электролиз насыщенного раствора хлорида аммония под слоем скипидара, чтобы капли взрывчатого NCL, поднимающиеся на поверхность, тотчас же взрывались и не могли бы накапливаться в значительном количестве.
Йодистый азот. При обработке концентрированного раствора нодида аммония иодом получается пе йодистый азот, а полвиодиды аммония. Но если воздействовать иодом па аммиак в водном ели спиртовом растворе, то получится черно-коричневое вещество, которое в сухом состоянии еще более взрывчато, чем хлористый азот, и взрывается даже при легком прикосновении. Его обычно называют йодистым азотом, хотя состав этого вещества подвержен сильным колебаниям; легче получить в чистом виде продукт присоединения аммиака к йодистому азоту. Наиболее полно изучен моноам-миакат NI3.NH3, кристаллизующийся в виде игл медного цвета с уд. весом 3,5, взрывающихся в сухом состоянии при малейшем прикосновении. То, что это соединение действительно является продуктом присоединения аммиака к иодистому азоту Nl3, следует из реакции с диэтил цинком.
NI3. NH3+3Zn(C2H5)2 = NH3+N(C2HS)3+3ZnC2HsI
Диэтнлцинк ... Триэтилаыин Этилдиaiaiодид
Нптрознлхлорид, хлористый нитрозил, NOC1, об образовании которого при различных реакциях уже упоминалось, хотя и является эндотермическим соединением, но в отлйчие от NC13 не взрывчат. Это объясняется тем, что он обычно распадается ие на элементы, а на хлор и окись азота. Образование же его из этих составных частей является экзотермической реакцией: NO 4- I/,C12 NOC1 -j- 14,4 ккал. Нитрозилхлорид представляет собой красно-желтый, легко конденсируемый газ; ниже — 61° он образует кроваво-красные кристаллы. Водой хлористый нитрозил разлагается на азотистую кислоту и хлористый водород: NOC14-HOH = - NO(OH) + ЦС1.
Реакция обратима: в присутствии в оду отнимающих веществ азотистый ангидрид взаимодействует с хлористым водородом с образованием нитрозилхлорида N208 4- 2НС1- 2NOC1 4“Н20. Для получения нитрозилхлорида в чистом виде пригоден способ, при котором в жидкую N3O3 вводят Р2О5 и пропускают ток газообразного НС1. Как показали электротю графические исследования Кетелаара и Пальмера (Ketelaar, Palmer, 1937), молекула ONC1 в газообразном состоянии изогнута 42 ONC1. = 116°, расстояния О N 1,04 A; N <—> С11,95 А. Дипольный момент, по Кетелаару (1943),
составляет р. = 1,83'Ю-18 эл.ст.ед. Для О К Вт получены соответственно 117°, 1,14 А. 2,14 А 1,87'1СГ1В эл. ст. ед. Из высокого значения дипольного момента следует, что обе связи не чисто гомеополярпы. Кетелаар предполоя;ил, что здесь имеет место мезомерия между обеими формами: —С1 и [ON]*C1' или : О :: N: С1: и ।
|:Ск]. Отсюда понятно также увеличепис расстояния N <—-Cl nN-—-Вт но сравнению с ожидавшимися для чисто гомебполярпогб соединения, а также уменьшение расстояния О<—-N. Каки в хлористом водороде, в других кислотах можно также заменить водород на положительный радикал N.O+ при взаимодействии с Х3О3.Так, по реакции N2O34- 2НС1О4 -- 2[NO][C104] -4 Ы2О образуется открытый " Гофманом (Hofmann К. Л., 1909) нипгроаилперхлорат и соответственно ио реакции Й20з “f" .4- 2H2SO4 = 2 [NO 1 [HSO4] 4* H2O — кислый нитроэилеульфат.. Последний чаще называют -«пн грози л серной кислотой», хотя оп и обладает, но Ганчу (1930), так же как и нитрозил перхлорат [NOJICIOJ, со л ео б раз пой структурой. То же имеет местом для нитрозилфгоробората [NO][BF4j (изотцппого [NH4][JBF4] и питрозилхлоростанпата [IW]2[SnCle) (изо тип по го [N Н4)2 [SnCli; |). Водой все эти соединения, так же как и NOCI, тотчас разлагаются. Теплота образования [NO][C1O4] равна 41,8 ккал! моль
При действии О3 на NOCI (Schumacher, 1929) образуется очень малоустойчивым нитрилхларид NO2C1, распадающийся при нагревании NO2C1^±; НО2 + VaCI,
670
Глава 14
Реакция обратима. То, что ири действий С12 и а N02 под’ давлением образуется NO2C1, наблюдалось уже в 1871 г. Хаибпбахом (Haseribach). Им было показано, NOaCl полу чается также при действии х,:ю [(сульфо но вой кислоты на концептрпро Устную азотную кислоту (уд. вес 1,5). Соединение, следовательно,можно приготовить ерз-нмельно удобным способом. Его можно использовать, по Бриптзипгеру (Brief-zinger, Z. anorg. Chem., 255, 325, 1948), для введения в органическое Соедпиннж одновременно групп NO2h Cl. В жидкой двуокиси серы, взятой н качестве растворителя, могут идти реакции двойного обмена с нитроаил- и нитрил со единениями, соЕгу-щепно аналогичные реакциям двойного обмена между солями в водных растворах Например,
(R = СН3) ; [NO] [SbCUJ + [NR4] [SO3*OR ] = [NRJ[SbCle] -ф- [NOHSO3ORJ; [NO3][SbClb] + [NR4][C1O4] = [NR4][SbCle] [NO2][C1O4]
Ни грилперхлорат
Так же получается нитрил фтороборат [NO2][BF4] и дипитр озил сульфат IN0]2[SO4], который сразу же разлагается с образованием динитрозилдисульфатя 2[N0]2[SO4] = |NO]2[S2O7]-h N2O3. Если пропускать NO в жидкий кислород, то (помимо N2O8) образуется желтая динитрояилперекисъ (NO)2O2. Азид тетрамотидаммэ-ния [NR4][N3] в неводных растворителях взаимодействует с нит р о зи л соединениями — даже с такими, которые практически нерастворимы или пе являются алектролитамв (например, NOCI, NOBr, NOSCN, NOOCO(CH3), Cu(N О)С1г), следующим образом; [NR4] [N3] + NO-X ±= [NR4]X-F N2O -ф- N2. В эту реакцию вступает даже четырех-окись азота, которая ведет себя, следовательно, подобно питрозилпитрату NO-NOj. В отличие от этого NO2C1 не является нитро зил гипо хлоритом NO-ОСЬ Это соединение с питрильным радикалом N Ot и в Других реакциях ведет себя как питрильпое соединение, например но отношению к пятихлорпетой сурьме: NO2C1 -ф- SbCl, = [NO2][SbCLJ (Seel, 1951-1952).
ИопыГ\О+ (нитро зил-иол) и NOt (нитрил-ион) изостериы молекулам СО п СОг.
Нитровилфторид NOF получается нри нагревании NO[BF4] с NaF (Balz, Majlander, 1934), нитрилфторид NO2F образуется аналогичным образом из нитрилфторобората NO2[BF4], который в свою очередь может быть приготовлен из KNO3, В2О3 и BrF3 (Emeleus, 1950) или, проще, по Шмейсеру (Schmeifier, 1952), пропусканием HF в раствор N2O5-BF3 (см. стр. 641) в нитрометане; N20j;-BF3 4- HF = HN03 -f-+NO2[BF4L В соответствии с реакцией 2N2O5 + SiF4 4- 2HF = 2HNO3 4-[NO2]2-- [SiF(;| может быть получен нитрилфторо,:или>:а>п. Примерно таким же способом Jle-мап (Lehman, 1951) синтезировал дини>ири,л>присулъфат ’ [NO2]2[S3OI0], При действии SO3 па Sr(NOs)2 он получил вначале’ соединение N2O,,-4SO3, которое прп пагреваппп до 80° в вакууме теряло часть SO3: N2O5-4SO3 [NO2]2LSgO10j 4* SO3. Выше 12()“ динитрилтри''улъф'1>>>. с отщеплением кислорода переходит в нитрилнитро-знл трисульфат [ N О I [ О ] [ S 3 О j п ] • Нитрилгтр'-г.лорат N О 2 [ С104 ] можно получить, помимо указанного пьппе способа, также действием окиси азота и озопа па двуокись хлора (Gordon, 1940) в виде бесцветных иголочек, растворимых в РОС13, по нерастворимых в СС14. Структуру этого соединения рентгенографически исследовал Кокс с сотрудниками (Сох. 1948). Нитрил перхлорат энергично реагирует с водородсодержащими органическими веществами. Прп нагревании до 120° опо быстро разлагается, но без вспышки.
5. Сернистые соединения азота
Азот образует с серой соединения SsN4 и S4N2 (ранее принимаемое за N2S5), а также одновалентный радикал [S4N3J*.
Нитрид серы (азотистая сера) N4S4 получают взаимодействием серы с жидким аммиаком
10S -|- 11)ХП3 6(NH4)2S-j-S4N4.
Реакция протекает слева направо, если образующийся сульфид аммония удаляют соответствующим реактивом
(NH4)2S -L 2AgI = AgaS+2NH4I
Нитрид серы несколько растворим в органических растворителях; например, 1 кг сероуглерода при температуре кипения растворяет 15 а
Пятая группа uepuoduHtvmtij. системы 671
S4N4. Из охлажденного раствора S4Nt кристаллизуется в-виде золотистожелтых кристаллов с температурой плавления 178°. Криоскопические определения дали формулу S.^Nj. Нитрид серы — сильно эндотермическое соединение, при нагревании или при толчке оно распадается со взрывом
S4_\4 = 4S-р 128 ккал.
Нитрид серt'i нерастворим в воде; по вода гидролизует его при продолжительном кипячении с образованием аммиака и кислородных кислот серы. Отсюда следует, что азот в этом соединении является более отрицательной составной частью, и. следовательно, его необходимо рассмотри-вать как нитрид серы.
При сублимации в вакууме S4N4 деполимеризуется с образованием S2X, — бесцветного, кристаллического, растворимого в эфире вещества, устойчивого только при низких температурах. При комнатной температуре S2N2 медленно превращается в смесь S4N4 и твердого, голубого <; металлическим блеском, нерастворимого в воде и в органических растворителях высоконолимерцогб соединения (SN]X. Это соединение значительно устойчивее, чем S2N2 и S4N4, и обладает полуметаллической электропроводностью (Goehring, 1953).
Своеобразная реакция происходят при действии па нитрид серы хлористой серы S2C12, растворенной в хлороформе. При этом образуется соединение состава S4W3CL Л1Ш1 хлора можно заменить на Вг, I или на кислотный остаток, например — SCN, — NO3. В этом соединении, следовательно, присутствует одновалентный радикал — [S4N3 ]+ — тиотритиазил.
При действии элементарного хлора на S4N4, растворенный в четы ре хх л ори стом углероде, получают, как уже было установлено в 1880 г. Демаркеем (Demar^ay), желтый продукт реакции, к о то р о м у щ i и пи с. ы в а ют формулу с о о динен ня SsN 3С13 (М eu w-sen, 1931). Известно также аналогичное бромное соединение (Muthrnan, 1896). При действий AgF2 на S4N4 в четыреххлормстом углероде Глемзер (Glemscr, 1955) получил соединение S4N4F4 в виде бесцветных тетрагональных игд (dj° = 2,326, т. пл. 153°, дипольный момент 0). Как not,азами рентгенографические исследования, оно построено из симметричных колец в которых с каждым атомом S связан атом F. Прп длительном кипячении смеси S4i\4 и AgF2 В четыреххлористом углероде выделяется бесцветный газ (т. ил. — 108т. кип. —11°), состав и молекулярный вес которого соответствуют формуле SN2F2- Кроме того, образуются также и другие соединения серы с азот том и фтором.
Четырехеернистый a3OTS4Na образует темно-красные кристаллы, плавящиеся при 23°. Переохлажденный расплав при 205 имеет Илотпость 1,71. Это соединение получается при действии сероуглерода на нитрид серы при 100° под давлением пли в качестве побочного продукта при разложении обычного нитрида серы. Оно вспыхивает при нагревании и уже при обычной температуре претерпевает медленное разложение. S4N2 растворим во" многих органических растворителях, например в бензоле, эфире, С§2, СС14 и в таких растворах при низких температурах может храниться продолжительное время без разложения. В спирте S4N2 умеренно растворим, в воде не растворим, но постепенно разлагается с выдслопием серы и образованием аммиака.
Уто соединение вначале считали штгисернметым азотом Я-фф; то, что опо в действительности является четырехсернистым ано том $4ЬГ2, впервые было доказано Мей-зевом (Meuwsen, 1947—1951). Помимо указанных выше способов, это соединение можно получить и в результате следующей реакции:
Hg6(NS)8 4 4S2C12 = 4S4N2 -ф 3ITgCla + Hg2Cl2.
() BF3 чотырехсервистый азот образует чрезвычайно устойчивый, твердый, цвета охры продукт присоединения 84№2-2ВР3. Проводя опыты по гидролизу, Горинг (Goehring, 1952) пока зал, что в молекуле S4N2 содержится один положительно четырехва-лептпый, одни положительно двухвалентный и Два нейтральных атома S, в то время как атомы азота песут ।1Гр11ц.1Ти.тьпые заряды; это делает вероятной циклическую структуру’ соединения, с которой хорошо согласуются результаты измерения диамагнетизма молекулы S4N2 п ее абсорбционные спектры.
Аналитические снедения. Из соединений азота наиболее важны для аналитической хшпш плглшак и азотная кислота, а также азотистая кислота.
672
Гладе 14
Аммиак легко выделяется из своих солей (из солей аммония) псд действием едкого кали или едкой извести и легко распознается по характерному запаху и по свойству окрашивать влажную красную лакмусовую бумагу в синий цвет. Очень малые количества аммиака, например в питьевой воде, открывают реактивом Несслера, с которым можно проводить также и колориметрические определения. В других случаях количественные определения аммиака производят ацидиметрически.
Из комплексных аммиакатов NH3 щелочами выделяется не полностью млн дажв вовсе не выделяется. Содержание аммиака в таких соединениях чаще всего определяют методом Лидера (lander, 1951)), для чего добавляют определенное количество кислоты и избыток ее оттцтровывзют кондуктометрически.
Нитраты, будучи смешаны с раствором, сернокислого закисного железа (сульфата железа(П)) и налиты на слой крепкой серной кислоты, образуют в месте соприкосновения с ней бурое кольцо, которое получается в результате восстановления сульфатом железа (II) выделившейся азотной кислоты до окиси азота и присоединения последней к избытку сульфата желе'за (II) с образованием сульфата комплексного) катиона Fe(NO)s+. Humpиты дают бурую окраску при добавлении даже уксусной кислоты. Поэтому, прежде чем проводить испытание на нитрат кипячением с хлоридом аммония или добавлением мочевины, следует разрушить нитриты, которые также могут присутствовать в пробе.
Количественное определение азотной кислоты в нитратах производят осаждением нитроном, ацидиметрически после восстановления азотной кислоты в аммиак сплавом Деварда или же газометрически путем восстановления азотной кислоты ртутью или хлоридом железа(П) до окиси азота.
Последний способ часто применяют и для определения азотистой кислоты. Кроме того, ее можно определять титрованием перманганатом калия. Когда имеют дело с весьма малыми количествами (исследования питьевой воды), то определение ведут колориметрически, па основании диазотирующего действия азотистой кислоты па органические амины, в результате чего образуются интенсивно окрашенные азокрасители. Об определении Х"Ог в воздухе см.: Beyer, Z. anorg. Chem., 250, 321, 1943.
ФОСФОР (PHOSPHORUS) Р
Распространение в природе. Фосфор встречается в природе почти исключительно в виде солей фосфорной кислоты, главным образом фосфорита ЗСа3(РО4)2-Са(ОН)2* и апатита ЗСая(РО4)2-Са(Г,С1)2. Лишь в отдельных местах встречаются фосфаты железа, например вивианит (синяя железная руда) FejPO4)„-8H„O, алюминия, например вавеллит ЗА12О;(-2Р2О6-• 12Н2О, а также редких земель (см. монацит). Соединения фосфорной кислоты составляют существенную часть растительных й животных организмов. Часть фосфорной кислоты связана в них в виде органических соединений, например в желтке яйца и в веществе мозга — в форме лецитинов;
* В условиях, при которых фосфо.риты могли возникнуть в природе, образуется по новейшим исследованиям (см. стр. 686) не Са3(РО4)2, как предполагали ранее, а двойное соединение ЗСаз(РО4)2-Са(ОН)2, так называемый гидроксил апатит. Поэтому не может быть сомнения,что в фосфоритах присутствует это двойное соединение, частично в изоморфной смеси с карбонат апатитом й обычиым апатитом [см. Rathje W., Вег., 74,,. 349, 1941].
Пятая группа пер ио дической с истемы 673
“ -г-г-*- " •• ' ‘
частично же в виде гидроксилапапшта ЗСа3(РО4)2 - Са(ОН)2 или. карбонат-апатита ЗСа3(РО4)2-СаСО3-Н3О опа вводит в состав основного вещества костей. , 5
.Зубная эмаль не содержит, как предполагали ранее;. преимущественно фторапатит 3Ca3(PO4)2-CaF2, по состав ее неорганического вещества, что было показано поздпёш шими исследованиями (в первую очередь К loment, Tromel, 1933), в основном та- . кой же, как и остальных костей; Большая прочность зубной .эмали объясняется тем, что Гидроксил- или карбонатапатит в пей Присутствует в сравнительно более крупнокристаллическом состояний (размер кристалликов 1б-4 си по сравнению с 1О'& — 1O"S см в остальных костях). »
= Э кек ремонты человека,и животных также богаты фосфорной кислотой (около 40% зольного остатка). Экскременты животных минувших,эпох, находимые в окаменелом виде, так называемые копролпты, состоят почти из чистого фосфата кальция (гидр ок сил апатита?). Значительная часть современных не сто рождений фосфоритов, например мощные месторождения Северной Африки, образовались из больших скоплений экскрементов и трупов животных более ранцих эпох, Есть местности, где и в наг стоящее время можно наблюдать образование мощных отложений птичьих экскремен-- тов (гуано), На островах Тихого океана близ берегов Перу находятся особенно большие залежи гуапо. Помимо фосфата кальция, гуано содержит в большем или меныпем количестве азот (в форме аммиака, мочевины и т. д.). Гуано встречается только в мест-постях, очень бедных осадками. По мере того как азотсодержащие составные части вымываются дождем, гуано обогащается фосфоритом,
т ’
Исторические сведения. Фосфор бый открыт в 1669 г. алхимиком Хеннигом Брандом, когда он в поисках философского камня нагревал выпаренную мочу без доступа воздуха. Фосфор получил название вслед- > ствие своей способности светиться в, темноте (фсщдрброс; —светоносный). Лавуазье впервые стал рассматривать его в качестве химического элемента; свою теорию горения оп подтверждал главным образом на фосфоре.
Получение, Фосфор получают неосновном восстановлением пятиокиси- , фосфора. Р2О5 углем при высокой температуре <
Р2О5+5С^5С()-}-2Р. .. ; %.. , %
Отгоняемый фосфор собирают под водой. Для удаления механических примесей его продавливают в расплавленном состоянии через'пористые каменные плиты или через мягкую кожу или же перегоняют еще раз из железных реторт. Фосфор обычно поступает в продажу в виде палочек, толщиной в палец, которые хранят под водой.
Исходным продуктом для технического получения фосфора служит пурехзамещен-ный фосфат кальц-илСа3(РО4)2 (или гидрокси л апатит, см. выше) в форме фосфорита .. или в форме костяной золы, Трехзамещенный фосфат кальция нагревают » смеси с кварцевым ясском Si02 и углем в электрической печи. S102 вытесняет Р2О3, которая затем - восстанавливается углем
paoP^c+Sn!r4GaSi03+PA i Ca3(PO^)2-}-3SiO2-j-5C=3CaSiO3-f-5CO-f-2P.
Принцип метода был предложен уже в. 180S г, Велером, Ио так как этот способ требует, высокой температуры (свыше 1306°), то он стал технически осуществимым лишь при использовании электрической печи.
В настоящее время этим способом производят значительные количества фосфора (» Германии до второй мировой войны ежегодно около 20 000 т). Большую часть получаемого таким образом фосфора сразу же сжигают до РоО,, который затем перераба-тыкают пн фосфорные удобрения.
В настоящие время очень редко применяют процесс Пелетье (Pelletier), основные принципы .. которого были предложены ЦГееле, По этому методу костяную золу сначала переводят в одно замещённый фосфат кальция, действуя на нее серной кислотой средней крепости S:
Са3(РО.).Н> 2Н ,SO4->4H2O_2CaSO.-2ИЙ(НСа(И2РО4)2, : .• •• (1)
43 г. Рсыв
674
Глава 14
а затем, тщательно смешав од незамещенный фосфат с,древесным углом илн с порсшж&-образным коксом, его нагревают выше 1000° в шамотовых ретортах.
ЗСа(Н2РО4)2+ 10С=Са3(РО4)2^- ЮСО 4- 4Р >.6H2O.
Свойства. Фосфор существует во многих модификациях. Обычней бесцветный, или белый, фосфор (тетрафосфор, см, стр. 626) пролстян.^^ собой мягкую, Как воск, прозрачную массу с характерным запахог. с удельным; весом 1,82, температурой плавления 44,1° и температура® кипения 280°, В воде растворяются только следы фосфора, но с водящее® < парами он заметно летуч. В спирте он также мало растворим, лучше растворяется в эфире, бензоле, скипидаре и жирных маслах, а лучше всел; в сероуглероде, хлористой сере и треххлорйстом фосфоре. Из сероуглерода фосфор кристаллизуется в кубической системе обычно в виде ромбододекаэдров.
Белый фосфор чрезвычайно химически реакционноспособен. В тонко-измеЛьчепНбм состоянии, получающемся, например, iipn испарении раствора фосфора в сероуглероде на фильтровальной бумаге, на воздухе пц самопроизвольно воспламеняется. Б компактных кусках белый фосфор загорается при нагревании немного выше 50°; по этой же причине оп воспламеняется: при трении, поэтому его реЖут только под водой. Фосфор сгорает жслтовато-белым пламенем, образуя пятиокись.- 2Р 4- 6/2О2 = = Р2О5 4- 370 ккал.
В темноте на воздухе фосфор светится. Это происходит оттого, что постоянно присутствующие над ним пары (несмотря на малую упругость пара белого фосфора при комнатной температуре) окисляются кислородом воздуха с выделением света. Многие вещества, например спирт, эфир, скипидар, сероводород, двуокись серы, хлор, аммиак, ослабляют или подавляют фосфоресценцию. В чистом кислороде при обычном давлении свечения не бывает, по опо появляется при уменьшении Давления. При медленном окислении фосфора во влажном воздухе образуется главным образом фосфористая кислота и затем фосфорповатистая.
Белый фосфор жадно соединяется также с галогенами; в токе хлора оп загорается: фосфор энергично реагирует с серой и многими металлами. Сильные окислители, например азотная кислота, окисляют его в фосфорную кислоту. В теплом едком кати фосфор растворяется с выделением фосфористого водорода и с образованием гипофосфита калия
4Р4-ЗКОН4-ЗН2О=РН34-ЗКН2РО3,
Фосфор выделяет легко восстанавливающиеся металлы (золото, серебро, медь, свинец) из их солей, причем он одновременно взаимодействует с ними; например, с медью он дает фосфористую медь.
Белый фосфор очень ядовит. Доза 0,1 г смертельна для человека.
При остром отравлении следует прежде всего возможно, скорее удалить яд, вызвав рвоту или промывание желудка. Противоядием служит сильно разбавленный раствор сульфата меди, который обезвреживает фосфор, переводя его в фосфористую медь, и вместе с тем вызывает рвоту. Длительное вдыхание паров фосфора также влечет за собой тяжелые заболелалля, особенно десоя я челюстей (фосфорный некроз). Вследствие этого употребление белого фосфора при производстве спичек запрещено почти во всех странах.
При нагревании в закрытом сосуде приблизительно до 260° белый фосфор превращается в красный фосфор, значительно менее активный химически и совершенно безвредный. Это превращение происходит медленно й при обычной температуре под действием света. Красный фосфор — порошок темно-красного цвета, нерастворим л сероуглероде; в темноте
Пятая группа периодической системы 675
не светится и воспламеняется лишь при нагревании свыше 400°. Его удельный вес 2,2, т. е. значительно больше, чем белого фосфора. Красный фосфор реагирует с галогенами и серой при более высокой температур ер чем белый фосфор, и пе осаждает металлов из растворов их солей. Но азотная кислота легко окисляет его до фосфорной кислоты. При растирании с хлоратом калия он легко воспламеняется.
Более реакционноспособен, чем обычный красный фосфор, светло-красный фосфор Шенка, получающийся при кипячении обычпого фосфора с трехбромиртым фосфором. Как и бесцветный фосфор, он растворяется в едкой щелочи и осаждает медь ив раствора сульфата меди. Его удельный вес 1,88. По-видимому, в этом случае речь вдет всего лишь об очень тонко-измельченном состоянии красного фосфора; так же как и последний, светло-красный фосфор безвреден.
Обычный торговый красный фосфор нельзя рассматривать как однородный продукт, даже если не учитывать Случайных примесей, а также продуктов окисления, образующихся при продолжите;!гном хранении его па воздухе. Модификация, лежащая в основе красного фосфора, в чистом виде известна под названием фиолетового фосфора, называемого также фосфором Гитторфа, так как Гитторф первый получил его кристаллизацией из расплавленного свинца. Шток показал, что ого можно получить из расплавленной смеси фосфора с висмутом. Застывший плав целесообразно переводить в раствор электролитически, в результате чего кристаллики фосфора остаются не растворит гимн. Удельный вес фиолетового фосфора несколько выше, чем удельный вес торгового красного фосфора: огг составляет 2,35. Его кристаллы относятся к моноклинной системе. При нагревании выше температуры плавления, и особенно при испарении, фиолетовый фосфор превращается в белый. Белый фосфор является модификацией, наиболее стабильной при высокой температуре, но при обычной температуре он мет а ста би лен*. То обстоятельство, что фосфор, несмотря на это, обычно выделяется в виде белого, например пр it кристаллизации из растворов при комнатной температуре, соответствует правилу ступеней Оствальда (ср. стр, 531). Вследствие неустойчивости фиолетового фосфора при нагревании у пего нет вполне определенной температуры плавления. Обычно переход в жидкое состояние наступает около 600°. Приблизительно при той же температуре плавится и фосфор Шенка и обычный красный фосфор.
В соответствии с данными Рота, Де Витта и Смита [В о th, D е W i 11, S m i t h, J. Am. chem. Soc., 69, 2881, 1947] красный фосфор может существовать как в аморфном, состоянии, так и по меньшей мере в трех, а быть может, далее в четырех различных кристаллических модификациях. Три из пих, (триклинную, гексагональную*и тетрагональную) можно получить пе только нагреванием аморфного красного фосфора,' но и непосредственно из паров (фосфора.
Третьей вполне определенной модификацией фосфора является черный фосфор, впервые полученный Бриджменом (Bridgman, 1914) нагреванием белого фосфора до 200° пол Давлением 12 000 ат. Удобнее получать его (Krebs, 1954) длительным нагреванием белого фосфора до 380“ в присутствии топ ко измельченной ртути. По химическому поведению он очень похож на красный фосфор, по окисляется во влажном воздухе быстрее, чем красный. Черный фосфор имеет удельный вес 2,69, твердость 2, обладает окраской цвета желоза и металлическим блеском. Он обладает также металлической электропроводностью и хорошей теплопроводностью. Структуру его решетки см. п.ч стр. 626. Ниже 550° черный фосфор является термодинамически наиболее стабильной модификацией этого элемента. Но при его образовании, в случае отсутствия катили затора, должок быть преодолен очень значительный потенциальный барьер (с,м. т. II. гл. 17).
Для белого фосфора также существует другая (тоже метастаби.чьная) модификация; это следует из того, что при сильном охлаждении у пего Наблюдается двойное лучепреломление. Точка перехода по Фбрлендеру (Vorlander) лежит при —681’.
В парах, приблизительно до 800°, фосфор состоит из молекул Р4. При дальнейшем повышении температуры происходит расщепление этих молекул на молекулы Р2. Как следует из электропогрпфических данных, в молекуле Р4 атомы фосфора распой ложены в виде правильных тетраэдров; Р -—> Р=2,21 А.
* Белый фосфор является метастабильном модификацией вплоть до температуры плавления. См. Mellor .1. W., «А с от р rechen si ve treatise on inorganic and theoretical Chemistry», VIII, 744, 1947. — Прим. ped.
43*
676 Глава 14
Поведение фосфора iio отношению к As,, Sb и .Bi. В расплавленном впаяете й сурьме фосфор-растворим только очень ограниченно. Фосфор и мышьяк в жид гм состоянии смешиваются во всех отношениях. Твердые растворы образуются при содзч-жапни As. 0—43 й 87—100 ат. % . Кроме того, Р й As образуют соединение с областью гомогенности 53—74 ат.%. Аз. Оно кристаллизуется в похожих на графит лцсточеее и обладает чрезвычайно высокой электропроводностью [К 1 ешт, F а 1 к о wsкг„ Z. anorg. Chem., 256, 343, 1948].
: Применение. Вследствие ядовитых свойств 'белого фосфора, применение ёйо ограничено; ого используют,, например, как отраву для грызунсз и в фармацевтических препаратах; Красный фосфор применяют в . больших количествах при производстве спичек; светло-красный фосфор Шенка находит такое же применение. Далее, красный фосфор служит исходным продуктом для получения других соединений фосфора, например хл оридов, а также в качестве галогенирующего агента, например при получении бромистоводородной кислоты (см. стр. 846).
Из соединений фосфора наиболее важны фосфаты, прежде всего в качестве удобрений*. Так Как для этих целей вследствие нерастворимости; в воде мало пригоден встречающийся в природе (в виде .гидроксилапа-тцга) фосфат кальция, го его обычно серной кислотой переводят в растворимый однозамещенный фосфат кальция Сгц'НоРО.,)., [см. уравнение (7). стр. 688]. Получающуюся при этом смесь однозамещенного фосфата кальция и гипса называют суперфосфатом. Если для обработки фосфата кальция вместо серной кислоты** используют фосфорную, то в продукте реакции гипс не содержится и получается двойной суперфосфат. Другое важное фосфорное удобрение — томасовая мука получается при тонком помоле томас-шлака — побочного продукта при производстве стали. Томасовая мука содержит фосфорную кислоту, хотя и не в растворимой форме, .однако в такой, которая усваивается: растениями, главным образом в виде двойной соли Са3(РО4)3 и Ca2SiCh (и соответствующих магниевых солей). Содержание усваиваемой растениями фосфорной кислоты в этом удобрении определяется по тому количеству фосфорной кислоты, которое можно экстрагировать из томасовой муки лимонной кислотой (ицитратно-растворимая фосфорная кислота*). Из удобрений, наряду с фосфорной кислотой, содержащих также и азотистые соединения («смешанные удобрения»), кроме гуано, следует упомянуть костяную муку, а также рыбную и мясную муку. Последние получают из отбросов при. производстве рыбных консервов и на бойнях и т. д.
СОЕДИНЕНИЯ ФОСФОРА
1, Кислородные соединения
Обычным продуктом горения фосфора является пятиокись фосфора Р2О5. Если жё горение происходит При недостатке воздуха, то образуется трехокись фосфора Р2О3.
* Кислые фосфаты цинка и марганца находят обширное применение в качестве защиты от Коррозии (фосфатная, защита, см. т. II),
** Попытки использовать вместо серной к полоты угольную не привели к успеху. Даже в воде, насыщенной двуокисью углерода под большим давлением, растворяются только позначительные количества фосфата кальция. Прииспользовании серной кислоты превращению, происходящему в соответствии с уравнением (7) на стр. 688, способствует плохая растворимость гипса; и случае угольной кислоты реакция приводит к равновесию, которое устанавливается, как только очень незначительное количество фосфата переходит в раствор.
Пятая группа периодической системы„ 677
Четырехокисъ РаО4 следует рассматривать как соединение трехокиси с пятиокиськУ (ср. стр. 684). В литературе часто упоминаются «субоки си фосфора», ио являются ли опи о пр еде лепными соединениями, пока не установлено.
Пятиокись фосфора Р2ОЛ или Р4О10 является ангидридом фосфорной кислоты. HSPO4. Трехокись фосфора РгО3 или Р4Оо — ангидрид о фосфористой кислоты, Н3РО3. Кроме того, фосфор образует фосфорноеатую кислоту Н4РгОв и фосфорноватистую кислоту Н3РОа. Ангидриды, соответствующие двум последним кислотам, не известны.
От обычной фосфорной кислоты (ортофорфорной кислоты} НзРОа путем ластичного отщепления воды («конденсация») производятся различные конденсированные фосфорные кислоты. Важнейшими из конденсированных фосфорных кислот являются: пирофдсфорная кислота (дифос-фюрнап кислота) Н4Р„О7 и метафосфорные кислоты, которые имеют аналитический состав НРО3. Однако эти кислоты и их соли полимерии'. известны, в частности, тримет а фосфорная кислота (fTPO.,)3 и тетраМетафос-форная кислота (НРО3)4.
Н3РО4 и Н4РаО7— первые члены ряда полифосфорных кислот, имеющих общую формулу НтигРпОзгиь например П5Р3О10 (трйфосфорная кислота), ИЙР4О13 .(тетрафос-форная кислота). Они имеют, как будет показано пиже, цепочечную структуру, и в виде солей также образуют цепи очень большой длины. Йеотетраметафосфорная кислота изомерна тетраметафосфорной кислоте. О ео структуре см. ниже.
От ортофосфориетой кислоты H3PQS путем частичного 'отщепления воды производится пирофосфористая кислота Н4Р2О5 и метафосфористая кислота НРОа. Для последней так же маловероятен, как и для метафос-форных кислот, молекулярный вес, соответствующий простейшей формуле. Более подробно она не изучена.
Б' фосфорной кислоте фосфор можно рассматривать как положительно пятивалентный, в фосфорноватой. — положительно четмрехеалентный, в фосфористой— : положительно трехвалентный и в фосфорноеатистой — положительно одновалентный. Эти степени валентности определяются при том допущении, что срази фосфора рассматриваются как гетеро полярные, хотя и с некоторыми ограничениями. В этом случае фосфор и водород должны нести вместе вдвое большее Число зарядов, чем имеется кислородных атомов в соединении, потому что кислород в гетерополярных соединениях всегда двухвалентно отрицателен (см. стр, 739).
Ортофосфорная, ортофосфор истая й фосфо рноватистая кислоты содержат одинаковое число атомов водорода. Однако только в бртофосфорной к ис л оте в се тр и атома водорода могут . быть з а м е щены мета л л амп; в фосфористой кислоте замещаются металлами только Лот атома водорода, а в фосфорноватистой кислоте — только один.
Соли фосфорной кислоты называются фосфатами, соли фосфорнова-той — шпофосфатами, соли фосфористой кислоты фосфитами^ соли фосфорповатйстбй — гипофосфитами. Следовательно, имеются:
Фосфаты
а&н овамещенные (или дигидро-) ортофосфаты
m!im>2o7 тхслые пирофосфаты д игядропи рофосфа ты (дифосфаты)
МТРО3 метафосфаты
М|НРО4 двугвамегуегячыв (ИЛИ мопоЕидрр-) ортофосфаты
М1Р04 гп рехеамегуенные (или нейтральные) ортофосфаты
м1р2о7 нейтральные пирофосфаты пирофосфаты (дифосфаты)
(м'ро3)3 (м’рОз).,
триметафосфаты тетрамета-
фосфаты
678
Глава 14
Гипофосфиты
м5н3РgOg кислые гипофосфиты
м1р2о6
нейтральные гипофосфиты
Фосфиты
МТН2РОЯ
одно замещенные
• фосфиты
Пример пирофо офитов:
Метафосфиты: м’РОз
MlllPO.3 ;
двух» ам ещ е ни ы е фосфиты
Na3H2P2O5
Т рехзамещепнк® не существует
Гипофосфиты
MTH2PO2 (двух и трехзамещенные не существуют).
Строение фосфорных кислот
I. Строение ортокислот и их солей. Если рассматривать кислородные кислоты фосфора как гидроксилсодержащие соединения и принять, что те атомы водорода, которые не могут быть замещены на металлы, связаны; иначе, чем те, которые замещаются металлами, то получим следующие структурные формулы:
,он /Н о=рен \он
/ОН О=Р^ОН ^он О=Р<
О=: хон /Н О = Р^-ОН ^он
,он 4 он
Фосфорная Фосфориоватая Фосфористая Фосфорноватиста я
кислота кислота кислота кислота
Если за основу взять эти формулы, то во всех .кислородных кислотах фосфора, несмотря на его последовательно уменьшающуюся электро-валентность от 5 -i-, 4. ф, 3 -ф до 1 ф , фосфору должна быть приписана валентность в структурном значении этого слова, равная, пяти.
В 1937 г. в результате изучения спектров комбинационного рассеяния Симон (Simon) смог подтвердить предположение о том, что в фосфористой и фосфорноватистой кислотах атомы водорода, которые не могут замещаться на металл, Не посредственно связаны с атомом фосфора. Им было найдено, что в спектрах присутствует характеристическая для связей Р — П-линин, которая для кислоты Н3РО2 является даже самой ярком линией спектра. Константы диссоциации кислот (табл, 103), как это было отмечено Шварценбахом [S с h w а г z е д b а с h G., Helv. chim, Acta, 19, 1043, 1936), также подтверждают тип связи атомов водорода, отраженный: в приведенных выше формулах.
Таблица 103
Копетанты диссоциации фосфорных кислот (Измерено при 18°)
Н3Р04 Н4Р20т адов И3Р03 Н3РО2.
Kt 8,1--10*з >10-1 6,4-10;з 8,0-10-з 8,5-Ю-з
к2 6,0-10’8 3,2-10-г 1,5-10’3 2,6-10-т
К3 1,8-10’12 1,7-10-е \ 5,4-10-8 —
К^ — 6,0-10-8 9,4-10-11 —
Фосфорноватую кислоту по Зальцеру (Salzer) (получившему ее впервые в 1877 г.), согласно ее удвоенной формуле, рассматривали как продукт конденсации фосфорной и фосфористой кислот. Тот факт, что фосфорноватой кислоте отвечает удвоенная формула H4P30e, твердо установлен в результате многочисленных исследований (Согиес, Рагга-
Пятая группа периодической системы 679
vano, Rosenheim, Treadwell, Nylon, Stelling, Bell и т. д.). To обстоятельство, что эту кислоту невозможно получить прп конденсации фосфорной и фосфористой кислот, а также ряд других наблюдений позволяют сделать вывод, что ее структура не должна соответствовать формуле I
НОХ /ОН : ИО/ /ОН
Р —О—1’ — О , а только О=Р—Р=О
НО-7 ЧОН НО-7 ЧОН
I II
Это было подтверждено в результате изучения спектров комбинационного рассеяния (Baudler, 1952). Метиловый эфир фосфорноватой кислоты, полученный Бадлером, обладал иными свойствами, чем соединения состава P2O2(OR)4, полученные ранее Другим путем Арбузовым (1931) и Пиленом (1933). В последнем случае, быть может, речь идет об эфирах соединения I — фосфористофосфорной кислоты [дифосфорной (III,V) кислоты]. Она образуется из соединения с формулой II при медленном, идущем уже при комнатной температуре превращении (см. стр. 691). Соединение I мало устойчиво в свободном состоянии, одпако была получена его натриевая соль (Blaser, 1955), Кроме перечисленных кислот, в виде солей получен еще ряд кислородных кислот, а которых фосфор присутствует в низшей степени окисления. Эбулиоскопические измерения Розенгейма (1906—1910) как будто бы показали, что во многих растворителях эфиры фосфорноватистой кислоты распадаются па ненасыщенные молекулы РО(ОК)2, однако Реми и Фалиусом (Remy, Faiius, 1955) было доказано, что этого в действительно стн не происходит.
В основе приведенных выше структурных формул фосфорных кислот лежит предположение, что в недиссоциированном состоянии их можно рассматривать как гидроксилсодержащие соединения. Подобное предположение возможно в результате изучения спектров комбинационного рассеяния многих других кислородных кислот, для которых,.за исключением HNО3, были получены, доказательства того, что водород в пих в не диссоциированной форме связан в гидроксиле (см, стр, 647); поэтому кислота имеет другое строение, чем соответствующая ей соль. На основании этого можно предположить, что все кислородные кислоты в медиесоциироеанной форме имеют гидроксильные группы. В случае фосфорных кислот, однако, это до сих пор еще точно не доказано. Изучение спектров комбинационного рассеяния [Simon, 1937—1939, см, также Z, Elektrochem., 49, 413, 1943; Veakateswaran, 1936—1938, Mathieu, 1942] показало, что линии, обычно характерные для гндроксилсодержащих соединений, здесь либо вовсе не существуют, либо оказываются исключительно слабыми (ср. с рис. 119, стр, 685), Одпако если водород заменить на дейтерий,то происходит отчетливое уменьшение частоты колебания атбма кислорода. Это указывает па то, что атомы водорода (соответственно Дейтерия) принимают участие в колебаниях атома кислорода и, следовательно, водород (дейтерий) непосредственно связан с кислородом. Отсутствие или, в других случаях, значительное ослабление полос ОН, вероятно, следует приписать влиянию «водородных мостиковых» связей. В спектрах комбинационного рассеяния муравьиной и уксусной кислот до сих пор также не найдено частот, соответствующих группе О.Н; склонность этих соединений к образованию двойных молекул в настоящее время принято объяснять возникновением водородных мостиковых связей,
При полной диссоциации,, т. е. в сильно щелочном растворе, ортокислоты фосфора образуют следующие ионы:
[РО4]'", [PaO6]'T", IPOsHJ" и 11*02Н2]’.
Если рассматривать формулы этих ионов с точки зрения координационного учения, то окажется, что фосфор в ионах всех этих кислот, а также и в попе фосфония [РН4]+ имеет одно :и то же координационное число, а именно 4, допуская, что водородные атомы, не способные к диссоциации, связаны в той же сфере, что и атомы кислорода (см. ниже структурные формулы солей). При таком подходе тот факт, что фосфористая кислота только двухосновна, а фосфор новатистая только одноосповпа, может быть объяснен исходя из координациопной валентности фосфора, равной 4. Однако координационное учение не может объяснить, почему для фосфорноватой кислоты имеет место координационное насыщение вследствие объединения двух групп РО3, а не в результате размещения атомов водорода около фосфора. Для объяснения этого факта теория Льюиса — Лангмюра идет дальше. На основе этой теории удалось полу-
680
Глава 14
чить для перечисленных выше ионов следующие электронные формулы.
:б: Т" :6:р:о; " " ;О:"
Фосфат-ион
:б: :о: :о:р:р:о: ”:6: :6:
Гипофо сфа т -ион
н 2 :о:р:о: : :9: " -л.
Фос фит-ион
-; н - -, :о: р: о: н "
Гипофо сфит-иоп
' н 1’
Н :Ё: Н н
Ион фосфонк^
Во всех случаях Валентине электроны образуют вокруг; атомов фосфора и кислорода октет и связь осуществляется при помощи электронной пары,-
Модель, отвечающая этому условию, пе может быть ^предложена ни для иона [РО313~, пи для иона [НРО3]“. Поэтому с точки зрения теории Льюиса -т Лангмюра отсутствие таких ионов вполне попятно. :Ч
Очень может быть (и уже в некоторых случаях доказано структурными определениями), что приведенные выше ионы переходят из водных растворов в соли почти неизмененными как структурные группы. и взаимодействуют с противоположно заряженными ионами, по сути дела, как единое целое, Таким образом, в координационном смысле возможны следующие структурные формулы солей " \
’О О/
Р
-° 0
Фосфат
0 0] ор—ро mJ о о J
Гипофосфат
"О 0 1 •' р м!
.о :
Фосфит
: "О Н' Р _о н. Гипо фосфит
м1
М|
П. Строение конденсированных фосфорных кислот и фосфатов. В кон денсированных фосфорных кислотах и фосфатах'фосфор всегда обладает координационным числом 4. • •
Для строения этих соедппопий, как это следует из всех установленных до ; сих пор фактов, существенны следующие основные положения;
1. Соединения построены исключительно из тетраэдров Р04.
2. иодобио тетраэдрам Si Од и вообще всем до сих пор известным кондепсировап ныи тетраэдрам В04, тетраэдры P(J4 могут соединяться между собой только через один атом кислорода. Тетраэдры, следовательно, соединяются только вершинами, но пе ребрами или гранями.
3. Если вокруг атома, фосфора расположено несколько групп ОН, то первая из них диссоциирует сильно, вторая слабо,, а третья очень слабо. Водные растворы., соответствующих солей щелочных металлов показывают реакцию нейтральную, слабо щелочную или сильно щелочную в зависимости от того, имеется в анионе соли около атома фосфора одпа или несколько гидроксильных групп.
4. ’Всякое кислородное соединение электроположительно пятивалентного фосфо- , ра содержит по меньшей мере один атом кислорода, который связан с фосфором двойной* связью ;и поэтому неспособен к проявлению гл а врой валентностипо отношению к Другим атомам, л
Полифосфаты, т. е. со с ди я енияс общей ф орму лоп М^^,2 [ Рп О зг1+1 ], постро ены . и в цепей Р04
Моно(орто)фосфат Дп( пир о) фосф ат
м*[Р041 Mj[P2o7]
Трифосфат
Пятая группа периодической системы
681
М*
-М'НЧОе,! \
Тетрафосфат * о о о о if" II ' Li II т
НО —Р—О — Р---О —₽— о—р—он
О- О’ о- о
М^РпОзп.КОН).,]; п^16-90
Высокопоммернм фосфат
Структуры этих Соединений изучал главным образом Тило [Thilo Е., 1941; см. Z. Aug.. Chem., 63, 508, 1951; 64, 510, 1952; Z? anorg. Chem., 272, 182, 1953]. Им было показано, например, что при гидролизе трифосфата образуются один мопо-и один дифосфат-ионы, а при гидролизе тетра фосфата — два дифосфат-иона. Примерами лысо ко пол имёр н ыа: фосф/атов являются соли Грэма, МаДрелла и Курроля (Graham, Maddcell, Kuirol, ср, стр. 690), Для выяснения их структур имело большое значение наблюдение Тило о том, что при сплавлении: Na Н2РО4, NaH2AsO4 и флюса ; получался расплав с анионом в виде длинной цепи, в котором тетраэдры АйО4 закономерно внедрялись в цепь РО4, так что прп гидролизе в зависимости от выбранных соотношений Р : As и смотря по условиям опыта образовывались только моно-, ди-, три-И тетрафосф ат-иопы. Все полпфосфорпые кислоты; содержат два слабо диссоциирующих атома, водорода и наряду с ними столько. Сильно диссоциирующих, сколько: в них имеется атомов фосфора. Цепочечная структура соли Курроля была подтверждена рентгенографическими исследованиями Плиса и Вурстера [Р 1 i о t h, W u r s t e r, Z.anorg. Chem., 267, 49, 1952].
Высок on о л и мершие фосфаты часто обладают (в пределах аналитических ошибок) таким же составом, как и метафосфаты, т. е. соединения с общей формулой M‘POS. Но настоящие метафосфаты имеют не цепную, а кольцевую структуру. Известны три- и тетрщметафосфатъи.
Й^РзОу]
Триметафос фат
* 4о— о 1 р
mJ 1 1 О О
0 = о 1 р= Ч- 1 .." о л.-: • . л_
Mj[P4dI2T
ТетраМе тафо сфат
При гидролизе в результате распада колец образуются три- и соответственно тетра фосфаты. Так как кислоты, соответствующие три- и тетра-метафосфатам, содержат одну группу ОН на каждый атом фосфора и поэтому являются сильными кислотами, водные растворы метафосфатов обладают нейтральной реакцией. Структура, предложенная Тило в основном па основании химических свойств, согласуется с результатами рентгене- ; графических определений.
у. ...
В результате педапнего изучения спектров комбинационного рассеяния, проведенных Симоном (1954), было установлено, что в триметафосфатах существует плоское кольцо в противоположность изоэлектррнному.,(ЗОз)3-кольцу у-модификации серного
682
Глава 14
ангидрида, для которого рентгенографически установлена изогнутая, а именно транс-структура (см. стр. 757),
Различие Симон объясняет тем, что в триметафосфат-иопе связь Р — О имеет значительно более сильный гетерополярный характер, чем связь S — О в SO3.
Иаотетраметафосфорная кислота Н4Р4О12 имеет то же число атомов, что и тет-раметафосфорпая кислота, но является такой же циклической системой, как и три-метафосфорпая кислота. Опа пе известна в свободном состояние, и для нее получены только эфиры, образующиеся при обработке Р4О(п сухим эфиром, например этиловым эфиром (С2Н5)2О или изопропиловым эфиром (С3Нт)аО. Напротив, при гидролизе PiOfo образуется изомерная тетраметафоефорная кислота. Образование этих кислот (и соответственно их эфиров) происходит по следующим схемам:
Тетраметафоефорная кислота Н1Р4О[2
+2Н3О
------
Пятиокись фосфора Р4О10
+ 2RjO
>
Д* 2R3O ----—>
Эфир и зо те трактата фосфорной кислоты R4P4O12
Структура изоформы определяется по продуктам, полученным при гидролизе ее эфиров. При гидролизе тетра этил обо го эфира наряду с двумя молекулами моноэтилового эфира фосфорной кислоты образуется одна молекула диэтилового эфира фосфорной кис л опт и одна молекула фосфорной кислоты (Thilo, 1951).
Надкислоты (пероксо кислоты) фосфора. От фосфорной кислоты производятся еще две надкислот ы (перекисные кислоты), лсононадфосфорная исислата (пероксомонофос-форная) HijPOj л наддифосфорная кислота (пероксодифосфорная) HiPjOg. Первая происходит от орто фосфор пои кислоты, а вторая от пирофосфорной в результате замены одного атома кислорода перекисной группой — Q—О—. Таким образом, фосфор в них положительно пятивалентен (как и в фосфорной кислоте) как структурно, так и электрохимически
ОП ОН ОН
0=Р—О-О—Н 0 = Р—О—О —^=6
1 1 1
ОН он 6н
Мононад фос фор над кислота Над дифосф о рван кислота
Соли перекисных кислот фосфора, например вполне устойчивый в твердом состоянии пероксодифосфат калия K4[P2Og], удобнее всего получать электролитическим путем 2P0j"—2e=P30j ". Моноиадфосфорная кислота образуется, например, по уравнениям,
Н4РгО8 ф-НгО=113РО5 Ч-Н3РО4 или Р2О5^ 2Н2О3Ч> Н2О->2Н3РО5.
Пятая группа периодической системы
683
Перфосфаты отличаются от известных в большом количество перокеигидрапгов 'различных фосфатов (например, Аа2ПРО4'Г120й — псрокеигидрат кислого фосфорнокислого натрия) некоторыми характерными для них реакциями, например окислением Мп” до МпО£.
По данным Бокемюллера (Bockemuller, 1934) при ауто окислении гипофосфита патрия в водном растворе наряду с фосфористой и фосфорной кислотами образуется надфосфористом кислота (как промежуточный продукт и в небольшой концентра-цин). От над фосфорной кислоты ока отличается тем, что вызывает, как и Н2О2, желтое окрашивание раствора сульфата титана, однако опа не дает, подобно перекиси водорода, реакции образования надхромовой кислоты.
Пятиокись фосфора РаО5 или Р4О10 — ангидрид фосфорной кислоты технически получают сжиганием белого фосфора в железных барабанах при обильном доступе воздуха. 'Торговый продукт, белая снегоподобная масса, обычно загрязнен низшими окислами, от которых его можно очистить возгонкой в струе кислорода. Пятиокись возгоняется при 358,9°. При нагревании до высокой температуры под давлением она становится стекловидно-аморфной; аморфная модификация плавится при 565,6°. В парах пятиокись фосфора имеет формулу Р<|0ю. Чистая пятиокись фосфора не имеет запаха. Под действием света пятиокись фосфора люминесцирует зеленым светом, интенсивность которого возрастает с понижением температуры. Пятиокись реагирует с водой с шипением (теплота растворения 35 ккол/молъ). При соприкосновении с воздухом пятиокись тотчас же становится влажной и расплывается в сиропообразную жидкость, так как, соединяясь с водой, образует метафосфарную кислоту или, точнее, смесь триметафосфорной и тетраметафосфорной кислот. Вследствие большого сродства к воде пятиокись фосфора используют для осушки газов и жидкостей, а также для отщепления воды от химических соединений или при химических превращениях, особенно в органической химии.
Пятиокись фосфора, помимо стекловидно-аморфной формы, встречается еще в трех кристаллических модификациях. Обычная гексагонально-ромбоэдрическая модификация (т, шг. 422°) при нагревании медленно переходит в ромбическую модификацию (т. пл. —560°). Однако и эта модификация неустойчива и, до-видимому, постепенно переходит в тетрагональную, устойчивую при высокой температуре (т. пл, 580°) [Hi 11,Faust, Hendricks, J. Am. chem. Soc., 65, 794, 1943]. P3O5 в своей нормальной (гекса гона ль но-ромбоэдрической) форме состоит, как установлено рентгенографически, из молекул Р4Ок), каждая из которых построена из четырех тетраэдров РО4, связанных через один общий атом кислорода. Такую же структуру имеют и молекулы P4OIt, в парах, что было доказано в результате электронографических измерений. Опыты Тило, упомянутые па стр. 681 и с л., по гидролизу иэфироливу Р20э подтвердили существование этой структуры чисто химическим путем.
Трехокись фосфора Р303 или Р40в — ангидрид фосфористой кислоты образуется при сжигании фосфора при ограниченном доступе воздуха. Трехокись фосфора можно отделить от образующейся одновременно пятиокиси, используя ее большую летучесть. Трехокись фосфора образует белую воскообразную кристаллическую массу с удельным весом 2,13, температурой плавления 22,5° и температурой кипения 173°. Летучесть расплавленной трехокией фосфора довольно велика. При 70° давление ее паров составляет уже 37,5 лья рт ст. Молекулярный вес в расплавленном состоянии, в растворе и в парах отвечает формуле Р40в. Выше 210° трехокись фосфора распадается с образованием четырех окиси 2Р4О6 = = J/aP4 + ЗР2О4. При нагревании в струе воздуха трехокись переходит в пятиокись. Она медленно соединяется с кислородом даже при обычной температуре. Окисление трехокиси под уменьшенным давлением сопро-
684 Глава 14
вожДаетея свечением. Пары трехокиси фосфора обладают свойством ионизировать воздух. Трехокись фосфора, подобно? фосфору, 'очень ядовита.
С холодной водой трехокись фосфора, очень медленно ;взаимодействует с:образованием фосфористой кислоты. При действии горячей воды происходит бурная реакция с выделением фосфористого водорода и фосфорной кислоты. Трехокись фосфора также весьма энергично реагирует с хлором й бромом, а выше 150° — также и с серой. При взаимодействии с газообразным хлористым водородом наряду с трех хлористым фосфором образуется и фосфористая кислота
РаО3-ННС1-Н3РО3+РС1э. (3)
Образующаяся яри распаде трехокиси фосфора (при нагревании) чёпгырехокисъ фосфора, Р3О4—бесцветные кристаллы с ярк’.'м блеском, растворяющиеся в воде со зпачпте.тьным разогреванием и с образованием эквимолярных количеств фосфористой и фосфорной кислот. Этот процессор он [в всего можно представить как гидролиз соединения, построенного аналогично четырехокиси сурьмы Sbn,SbvO.; (ср. стр. 707)
pHipVoj ЗН3О=Р(ОН)3^ Н3РО4. •
Четырехокись фосфора нельзя рассматривать как ангидрид фосфорноватой кислоты, так как пе удается доказать ее присутствия в водном растворе после гидролиза.
Фосфорная (ортофосфорпая) кислота. Как уже было отмечено, пятиокись фосфораj соединяясь с водой, вначале образует метафосфррную кислоту (НРО3)3. или (НРО3)4. Эта кислота в водном растворе постепенно переходит (быстрее при кипячении) особенно в присутствии ионов И’ в обычную фосфорнуюортофосфорную кислоту П31’О4.
Ортофосфорную кислоту получают не только из пятиокиси фосфора и воды, но и окислением фосфора азотной кислотой. Для технических целей ее приготовляют преимущественно разложением: фосфата кальция серной кислотой средней концентрации. Ч
Получаемая этим методом кислота бывает сильно загрязнена, но пригодна для многих целей, например для производства удобрений. Чистую фосфорную кислоту готовят всегда из фосфора. Для окисления азотной кислотой можно использовать белый или красный фосфор. Концентрация азотной кислоты пе должна превышать 32%, так как иначе реакция идет слишком бурно. Когда реакция заканчивается, избыточную азотную кислоту удаляют выпариванием; если в фосфоре была примесь мышьяка, раствор Снова разбавляют и обрабатывают сероводородом для осаждения мышьяка, .затем опять выпаривают и конец операции ведут в платиновой чашке, так как горячая концентрированная фосфорная кислота сильно разъедает фарфор. Если сироп, полученный при 15(Д, после охлаждения сам це застывает, то в него вносят затравку в виде кристаллика кислоты.
Прц упаривании водных растворов фосфорной кислоты происходит копдепса-;ция с образованием пирофосфорной кислоты, как только- концентрация кислоты окажется выше той, которая соответствует составу [Н3ОНР.0г(ОН)Д. :Копдсщсация: происходит также, если при обычной температуре раствор упаривать в ’вакууме. Поэтому при сильном упаривании хотя и можно получить продукты, по составу соответствующие фосфорной кислоте, одиако это не будет чистое соединение, которое; можно получить, если Отделить фильтрованием выделившиеся кристаллы: прежде, чем закристаллизуется остаток жидкости [S i m o n, Z. anbrg.: Chem., 242, 313, 1939]. Безводная фосфорная кислота чрезвычайно агрессивна. При 400s с ней взаимодействуют даже золото и платина. I й,
Фосфорная кислота кристаллизуется в прозрачных, как вода, -твердых .расплывающихся: на воздухе ромбических призмах с удельным весом 1,88 и температурой плавления 42,Зэ. Она не образует гидраты, но смешивается с водой во всех отношениях. В продажу" обычно ее выпускают в виде сиропообразного, концентрированного водирцо раствора (83^-98%). Чистую кислоту применяют главным образом в фармацевтической промышленности. Ее прибавляют к растворам перекиси водорода (ср.
Пятая группа париодической системы.
685
стр. 78), В Англии и США ею подкисляют лимонад. Техническая ясе фосфорная кислота, помимо производства удобрений, идет для многих других целей, например при окрашивании тканей, при производстве эмалей, замазки для склеивания фарфора, в зубоврачебной технике и др.
Фосфорная кислота трехосновная, средней силы, Каясущаяся степень ее диссоциации (вычисленная на основании данных по электропроводности) составляет 6% в 1 н. и 12% в ОД н. растворе (при 18°).
Значение отдельных констант диссоциации для всех трех ступеней приведено в табл. 103 (стр. 678).
При титровании фосфорной кислоты раствором едкого натра с индикатором метиловым оранжевым изменение цвета происходит, когда истрачен ровно 1 sks NaOH;
ЙСЬОа ЮОзес.,%
НС10А 77ms, %
BjP04 100 вес. "А
HCLO,/H3PO4 безмИная, зквиммяр.
Рис. 119. Снектры комбинационного рассеяния безводной фосфорной кислоты, водного раствора (ионы [C1OJ") хлорной кислоты, безводной хлорной кислоты и эквимолярной смеси обеих безводных кислот (соединение [Р(ОН)4][С1О«] в равновесии со свободными кислотами).
ОН
ОН
т—1—।—I
ЛОО 3200 3500 см-*
при титровании с фенолфталеином переход окраски происходит тогда, когда истрачено около 2 оке NaOH. Следовательно, фосфорная кислота, хотя и содержит три атома водорода, способные замещаться металлами, вследствие ступенчатости своей диссоциации при насыщении щелочью в водном растворе, может вести себя как одно-основивя или как двухосновная кислота в зависимости от интервала перехода применяемого индикатора (подробнее см. гл. 18).
Фосфорная кислота является ангидроосиовэнием по отношению к хлорной кислоте: она образует с ней соединение, которое по данным Симона (1952) следует считать перхлоратом тетразидроксофосфония [Р(ОН)4] [С1О4] (перхлоратом фосфорной кислоты). При плавлении соединение частично распадается на составные части. Поэтому в спектре комбинационного рассеяния расплава эквимолярной смеси H3POt и НС1О* содержатся линии, характерные как для свободных кислот, так и для иона [С1О*]_ (рис. 119).
Фосфаты (ортофосфаты). Ортофосфорная кислота, как трехосновная, образует три ряда фосфатов. Все однозамещенные фосфаты М1НаРО4 растворимы в воде, из деухзамещенных М*НРО4 и трехзамещенных фосфатов М|РО4 растворимы только соли щелочных металлов.
При прокаливании однооамещенные фосфаты, отщепляя воду, переходят в метафосфаты , двухаамещенные — в пирофосфаты, трехоамещенные (иейтрялыткге) при прокаливании не изменяются. Однако это относится только к тем фосфатам, которые ие содержат положительных радикалов, являющихся производными летучих веществ, например [NH*]*. Нейтральный фосфат магния-аммония (NHJMgPO* при прокаливании переходит в пирофосфат магния MgaPaO?, а двухэ вмещенная соль [NH4]NaHPO4 («фосфорная соль») переходит в метафосфат натрия NaPO3.
Большинство фосфатов растворяется в сильных кислотах. Нерастворимы в первую очередь фосфаты висмута, олова, титана, циркония, гафния и тория.
686 . Глава, 14
Фосфаты щелочных металлов готовят обычно, прибавляя соответствующее количество фосфорной кислоты к растворам гидроокисей п-тп карбонатов. Нерастворимые фосфаты получают в большинстве случаев из растворимых путем двойного обмена. Нацример, при добавлении нитрата серебра к раствору ортофосфата выпадает желтый фосфат серебра
2Na2HPO4+3AgNO3 = Ag3PO4+3NaNO3+NaH2PO4. (4)
При прибавлении к нейтральному или слабо щелочному раствору фосфата соли магния образуется слизистый осадок фосфата магния*. Если же добавлять одновременно хлорид аммония и аммиак, то получится кристаллический магнийаммонийфосфат
Na2HPO44-MgCJ2+NH3+6H2O4=(NH4)MgPO4-6H2O-|-2NaCl. (5)
Фосфат аммония. Обычный фосфат аммония — двухзамещеппый (NH4)2HP04. Он образует моноклинные кристаллы с удельным весом 1,62, которые на воздухе выветриваются, теряя аммиак, Соль легко растворяется в воде. Раствор ее часто применяют в качестве реактива прп анализах, а также в фармацевтической промышленности. В технике она идет для аппретуры тканей, чтобы спизить их горючесть, но в основном ее используют в качестве удобрения.
1 Фосфат натрия. Обычный двухзамещенный фосфат натрия (гидрофосфат натрия) NaaHP04- 12Н.,0 образует бесцветные палочки или плитки освежающего солоноватого вкуса. Оп выветривается на воздухе, теряя воду. При хранении тонкого слоя порошка на воздухе соль через 8—14 суток количественно переходит в ди гидр ат Na3HP04-2Ha0. При нагревании додекагидрат плавится при 403, при 100° становится безводным, а при дальнейшем нагревании превращается в пирофосфат Na4PaO7 (т. пл. 980°).Подобно фосфату аммония,он имеет разнообразное применение.
Трёхзамегцепный фосфат. натрия (тринатрийфосфат) кристаллизуется из водного раствора обычно как додека гидрат, однако, как было показало Монцелеи (Menzel, 1937), состав его колеблется в зависимости от концентрации NaOH в растворе от NagPOv’Oy КиОП да N.j.p’C^-V^aOII. NaOH внедряется в кристаллическую решетку соединен ия. Находясь в соприкосновен ин с-раствором при 70®, эта соль выделяет NaOH и переходит в гексагидрат Na3PO4-61120, который обладает постоянным составом, а выше 100° — в полугидрат, состав которого почти постоянен Na2PO4-%Н2О.. В технике поэтому можно непосредственно получить чистый тринатрийфосфат, упариваниемраствора, который содержит составные части в соотношении, отвечающем формуле. Тринатрий фосф ат является наиболее эффективно действующим средством для смягчения воды, используемой в котлах высокого давления.
Фосфорная соль. При добавлении хлорида аммония к раствору фосфата натрия по уравнению
Na2HPO4-[- [NH4]Cl = lNH4]NaHPO4+NaCl (6)
образуется натрийаммони.йфосфат, или так называемая фосфорная соль. Она кристаллизуется с четырьмя молекулами воды INH4JNaHP04-• 4Н2О в виде бесцветных моноклинных кристаллов и применяют ее главным образом в анализе для получения «перлов фосфорной соли».
фосфат калия. Однозамещенпый фосфат калия КНгРО4 производят в большом количестве; его используют как удобрение.
* При продолжительном храпении под осаждающей жидкостью осадок фосфата магния становится кристаллическим и приобретает состав Mgg(PO4)2 22Н2О. 1
Пятая группа периодической системы
687
Для его приготовления сплавляют смесь фосфорной кислоты и хлорида калия при температуре каления. При этом с отщеплением хлористого водорода и: воды образуется метафойфат калия КРО3, который о был по практически пе растворяется в воде п кислотах. Но если его охладить, вылив расплав на холодную поверхность, то он приобретает способность растворяться в воде. Из раствора одно замещенный фосфат кристаллизуется в виде тетрагональных призм. Он довольно легко растворяется в воде, по не в спирте.
Фосфат кальция. Нормальный или трех замещенный фосфат кальция, трикальций фосфат Саэ(РО4)3, в виде двойного соединения с CaF3 и СаС13 — апатита, а также в виде фосфорита (см. стр. 672) составляет главные запасы фосфора в природе. В небольших количествах он содержится всюду в почве. Он необходим для роста растений, а также для жизни животных.
Трехзамещенный фосфат кальция чрезвычайно плохо растворим в воде, но медленно ею разлагается с образованием гидроксилапатита Са5(Р04)а(0Н) или соответственно ЗСа3(РО4)3-Са(ОН)3, растворимость которого также очень мала.
При добавлении к нейтральному раствору соли кальция раствора двухзамещен-' ного фосфата натрия выпадает двухзамещеннъш фосфат кальция (гидрофосфат Кальция) в виде белого осадка. При длительном выдерживании под раствором осадок становится отчетливо кристаллическим, образуя блестящие бесцветные чешуйки состава Са11РО4-• 2Н3О. Двухзамещенный фосфат, как и трехзамещенный, плохо растворяется в воде, но легко растворяется в кислотах. Води постепенно разлагает двухзамещенпый фосфат' с образованием гпдр ок св л а латита и одно замещенного фосфата кальция
7CaH₽04-j-H20 = Ca5(P04)3OH-|-2Ca(.H2P04)2.
СаНРО4, осаждающийся из нейтрального раствора соли кальция под действием Na2HPO4, вследствие гидролиза всегда загрязнен небольшим количеством гидроксилапатита. При кристаллизации двухзамещепного фосфата кальция гидроксил апатит остается в виде слизистых хлопьев. Вели двухзамещенпый фосфат кальция образуется при осаждении Са(П2РО4)2 раствором Na2HPO4, то гидроксилапатит при этом не образуется
Na3HPO4^ Са(Н2РО4)2= CaIIPO4-b 2NaH2PO4.
[G е г i с к о S., Angew. Chem., 65, 59, 1953]
По данным Курмье (Kurmies, 1953) чистый СаНРО4-2Н2О проще всего можно получить, если одно замещенным фосфат кальция Са(Н2РО4)2 в водном растворе обработать двухзамещеппым фосфатом щелочного металла при комнаткой температуре. Взаимодействие других солей кальция споном НРО4 приводит к образованию СаНРО4. 2Н3О только при вполне определенных условиях. Кроме того, помимо обычно получающегося в этих условиях Сай(РО4)3(ОП), образуется часто также и другой фосфат кальция, который при электронно-микроскопическом изучении имеет вид очень тонких, неправильно ограпёппых табличек, ясно отличающихся от четко ограненных кристаллов соединения СаНРО4- 2Н2О и от игольчатых кристаллов гидроксилапатита. Курмье предположил, что в этом случае печь идет о соединении Са4Н(РО4)3, уже описанном ранее Варпнгтоном (Warington, 1873) и Арнольдом (Arnold, 1950), существование которого, одпако, оспаривалось Хайеком (Hayek, 1951).
Если проводить осаждение фосфатом натрия или аммония после прибавления хлорида аммония или аммиака, то выпадает гидроксилапатит Cas(PO4)g(OH). Трехзамещенный фосфат кальция (трпкальцийфосфат) нельзя получить в чистом виде при осаждепип из водного раствора. Продукт осаждения, считавшийся трикальцийфосфатом, является смесью гидроксилапатита и двухзамещоппрго фосфата кальция, состав которого меняется в зависимости от условий осаждения (baneel, 1930; Schlcede, 1932; Tromel, 1932). Смесь соответствующего состава при сильном нагревании можно, однако, превратить в настоящий трикальцийфосфат. При сплавлении Са(РО3)2 и СаО, помимо трехзамещеппого фосфата Са3(РО4)2, можпо получить соединения Са2Р2О7 (пирофосфат кальция, дика.тьцпйдпфосфат) и Са1Р2Ов (тетракальциифосфат). Последний при нагревании в атмосфере влажного воздуха (например, при прокаливании в открытой электропечи) переходит в смесь гидроксилапатита и окиси кальция ЗСа4Р2О9-(- Н2О•= = 2 Са 5( Р О4)3 (О И) 2С а О. Г пдр о ксил апатит ч р е звычайно устойчивоесоедннепие. В чистом состоянии оп теряет воду и начинает распадаться лишь выше 1500° на три- и тетракальцийфосфаты.
688
Глава 14
Однозамещенный фосфат кальция (монокаЛьцийфосфат) Ca(H2POtk хорошо растворим в воде. Он образуется при действии кислот, лаприигу серной, на трех замещенный фосфат кальция или на гидроксйлапатат в количестве, соответствующем уравнению (7): :
Са3(РО4)2 'Г211,50..,- •iH.;0 = Ca(H2P04)2 |-2CaS04-2H20 или
2Ca5(PO4)3OH-|r7H3SO4-|~12H26 = 3Ca(H2PO4)a-^7CaSO4 <2Н2О.
Выше уже указывалось на значение этой реакций при усвоении природного; фосфата й костяной золы, применяемых в. качестве удобрений (суперфосфатного удобрения).
Фосфорит или гидр ок сил апатит можно не обрабатывать серпой или фосфорной кислотами, а использовать как удобрение, предварительно нагрев их до спекания в смеси с подходящими добавками (главным образом кальцинированная сода, 'известь, : силикатная порода) и Затем мелко измельчив. Продукт, называемый ренанййг-фоефатом, содержит обычно наряду с NaCaPO4 в виде существенной части также ренанUrn. В этом Соединении фосфорная кислота находится в той же форме, что и в томас-шлаке (ср. стр. 676), однако в его образовании наряду с Са3(РО4)2 и Ca2SiO4 участвует и Na3PO4. По рентгенографическим данным ренанит обладает структурой, аналогичной апатиту. Подобно апатиту и гидроксилапатиту, ренанит нерастворим в воде, одцако разрушается содержащимися в пей органическими кислотами, выделяемыми корнямщрастений, и таким образом фосфорная кислота, содержащаяся в этих солях, становится доступной для питания растений-
Одинаковую структуру кристаллической решетки с ренанигпом имеют ряд: соединений, которые по своему химическому составу более или мепее отличаются от рена-цита, входящего в состав ренаний-фосфата (натриево-кальциевый ренанит). Например, соединение Cals(PO4)4(SiO4) 5 также имеет структуру ренанита, так же как и высо-котемпературная модификация КСаРО4 (в противоположность NaCaPO4, изотиппому K2SO4). Входящий в состав натриевр-кальциевого ренанита SiО2 может полностью или частично замещаться на С02. Углекислый ренанит, по Франку и Бледиту (F г а н с к, В г е d i g, Z. anorg. Chem, 230, 1, 1936], имеет средний состав Na6Ca4-(РО4)4СО3. В то время как в системе Na2O — СаО — Р2О5 в качество единственного тройного соединения присутствует сметанная соль NaCaPO4/B системе К20 — СаО РаО5, помимо КСаРО4, были найдены еще соединения КСа4(РО4)3 (замечательно, что это соединение имеет структуру апатита!) и К2СаР2О?. Соединение КСаР04 может включать КчСО;, и СаСО3 в свою решетку. Если это соединение содержит эти примеси, то при охлаждении ниже 705° пе происходит обычно претерпеваемого изменения в структуре [Franck, В г е d i g, Z. anorg. Chem., 237, 49, 1938].
Основная область применения фосфатов кальция — это производство искусственных удобрений. Следует также упомянуть об использовании апатита для изготовления эмалей и матовых стекол, о Применении двухзамещенного фосфата в качестве добавки к кормам (кормовой преципитат), а также в фармацевтической промышленности, Смесь двух замещенного и однозамещенного фосфата можно: использовать вместо винного камня (гидротартрата калия) для изготовления пекарного порошка,'
Пирофоефорная кислота и пирофосфаты. Чистая фосфорная кислота начинает отдавать воду немного выше температуры своего плавления. Если долго нагревать ее до 200—300°, она полностью переходит в пирофосфор-ную кислоту (Н4Р.>07). Пирофоефорная кислота образует бесцветную стеклообразную массу, Легко растворимую в воде. С нитратом:серебра раствор образует не желтый осадок, как с фосфорной кислотой, а белый. На холоду под действием воды пирофоефорная кислота медленно переходит обратно в фосфорную кислоту, при кипячении этот переход совершается быстрее, особенно в присутствии азотной кислоты. :: /
Пятая группа периодической системы 689
Пирофосфррпая кислота четырехо сновка я, по образует только Два ряда срлей, а именно кислые McHilACk и нейтральные пирофосфаты MjP^Oy. Кислые пирофосфаты растворяются в воде обычно со слабо кислой реакцией. Из нейтральных пирофос- ; фатов растворимы только соли щелочных металлов; их растворы вследствие, гидролиза обладают щелочпой реакцией. Пирофосфорная кислота является значительно более •сильной кислотой, чем ортофосфорная (см. табл. 103, стр. 678). Отсюда следует обычае Правило, что сим кислот возрастает при конденсации.
Мета фосфорная кислота и метафосфаты. Есйц продолжать нагревание фосфорной кислоты после отщепления одной молекулы воды, то постепенно образуется метафосфорная кислота. (НР0а)п [п=3 или 4], oбpa-; зующая твердую стекловидную массу (Acidum phosphoricum glacials), которая растворяется в воде' с потрескиванием. Раствор метафосфорной кислоты с нитратом серебра, подобно; пирбфосфорной кислоте, дает белый осадок, но в отличие от пирофосфорнбй кислоты метафосфорная кислота обладает свойством свертывать белок в водном растворе. При -продолжительном стояний, быстрее при кипячении,-особенно в присутствии крепких кислот, раствор метафосфорной кислоты превращается в раствор обычной фосфорной тсислош. При прокаливаний метафосфорная кислота заметно летуча.
Соли метафосфорной кислоты — метафосфаты аналитического состава МгРО3 (величину и структуру молекул см. на стр, 681} получаются при прокаливанйи однбза-мещепных. фосфатов. Наиболее важно образование метафосфата натрия при прокат ливааии '«фосфорной соли» (МН4)КаНРО4= NaPO3-(- H2O4-NH3.
Если затем нагрей, метафосфат с окислом металла или с солью кислоты, летучей при нагревании, то окись металла растворится, образуя смешанный фосфат, например NaPOs-J- CuO=NaCuPO4.
Так как фосфаты тяжелых металлов часто бывают характерно окрашены, то эту реакцию применяют при анализах («лср.ты фосфорной соли»}. ,
Как уже было отмечено, метафосфорная кислота и; метафосфаты в'растворе и в расплаве находятся не в мо помер ной форме НРО3 и М’ИД, но в виде полимеров, имеющих кольцевую (циклическую) структуру (три- й тетраметафоефорная кислота или три- и тетраметафосфаты), В водном растворе с течением времени метафосфорная кислота претерпевает превращения. Гидролиз идет главным образом до ортофосфорной кислоты. При осторожном гидролизе можно, однако, добиться простого расщепления , колец с образованием полифосфорных кислот, например:
(ИРО3)3+Н2О = Н5Р3О1Й.
: Tpi-шетафосфорная Трифосфорнан кислота. ' кислота
Три- и тетраметафосфаты образуются при отщеплении воды ют дигядромоиофосфата в результате нагревания. Образуется ли при этом три-или тетраметафосфат, зависит, по-видцмому, от величины и заряда катиона и способа нагревания. Водные растворы три- и тетраметафосфатов имеют нейтральную реакцию, поскольку соответствующие п.ч кислоты являются сильными кислотами.
Из расплава Na₽O3 может выделяться в трех кристаллических формах, а пирофосфат натрия N а4РгОу существует, вероятно, даже в 5 кристаллических модификациях. При б бы чао й температуре, .однако, можно получить лишь одну из них. Сплавляя :Na4P2O7 и NaPO3, можно получить соединение На^Ою, для которого при обычной температуре найдено две кристаллические формы [Р а г t г i d g е, Н i с k s, S m i t h, J, Am. chem. Soc., 63, 454, 1941 J. Как показали Тило и Ратц [Thilo, Rat z, Z. anorg, Chem., 258, 33, 1949], Ма5РзО10 можно получить при расщеплении кольце^ образного три метафосфата натрия Na3[PsO91 под Действием NaOH. На этом, в частности, основано доказательство кольцевой структуры аниона [Р3ОЙ[3-. С присутствием метафосфорной кислоты и метафосфатов в растворе и расплаве в различных полимерных формах связан тот факт, что и в твердом состоянии эти вещества прн одинаковом составе могут существовать в различных формах, которые часто очень сильно различаются по свойствам [см. J ander G., Kolloid-Beihefte, 54, 1—146, 1942; ср, также М al mgr е n, Z. anorg. Chem., 252, 256, 1944; а также работы ТйЛб, указанные на стр. 681].
44 Г. Реми '
Л
690 : Гласа 14
Полпфосфорпые кислоты и полифосфаты. Полпфосфориые кислоты, подос~ мет а фосфорным, образуются при отщеплении воды от фосфорпой кислоты, но прх этом образуются не кольца (никлы), а цепи. Простейшим их представителем является пирофосфорная кислота (дифосфор на я кислота) Н4Р2О7. Тризосферная кислота известна в виде своих солей, в первую очередь NaZn2P3P10‘9%H2O и Na5P3OI0'8H2C„ Образование последнего соединения наблюдалось уже Флейтманом в 1348 г.; однак-только в результате новейших исследований (Huber, 1936; Andress, 1938) удалось окончательно доказать ее существование. О скорости гидролиза три-, пиро-: п метафосфатов см. W a t z ol, Choinio, 65, 356, 1942.
Трифосфат натрия Иа5[Р3О10] был впервые получен Шварцем (Schwarz, 1895) при сплавлении пирофосфата натрия и соли Грэма (см. ниже). Удобнее эту соль получать пр способу Тило (1949), действуя NaOH па тримета фосфат натрия Na^Oij. Трифосфаты щелочных металлов хорошо растворимы в воде, трифосфапл других'металлов мало растворимы. Тетрафосфат натрия ^а6[Р4О1г1 получается при обработке тотраметафосфата натрия Ма4Г4О12 едким натром при 40°. Реакцией двойного обмена из него можпо получить другие тетрафосфаты, однако они малоустойчивы. К поли-фосфатам относятся также соля, получаемые по вполне определенным методикам, с особенно высокомолекулярными анионами, которые раньше рассматривали как метафосфаты, а именно соль Грэма, соль Мадрелла, соль Курроля.
Соль Грома образуется при быстром охлаждении расплава, полученного из одно замещенного фосфата натрия нагреванием, до 600° или выше, в виде прозрачной гигроскопической стекловидной массы. Это соединение, как установили Лидер (1942) и Другие исследователи на основании определения ионных весов, в зависимости от температуры образования содержит от 30 до 90 Групп РО3. По данным Тило эти группы расположены в виде длинных яеразв отв л ецных цепей из тетраэдров РО4. Эта соль хорошо растворима в водо, однако при комнатной температуре растворение происходит медленно. Под названием «калдон» ее применяют в качестве добавки к мылу для предотвращения выделения извести при стирке в жесткой воде, Это, применение основано на том, что все высокомолекулярные щелочные полифосфаты действуют как ионообменные смолы и их анибны поэтому прочнее удерживают катионы с большим зарядом, например Са2\ чем катионы с зарядом, равным единице.
Соли Мадрелла и Курроля имеют строение, подобное: строению соли Грэма. Соль Мадрелла образуется при нагревании одпозамещенного орто-или пирофосфата натрия до 250—500°. Это вещество существует в низкотемпературной форме, которая построена из nejj аз ветвленных ценен, содержащих 16—32 тетраэдра Poj, и в высокотемпературной форме, построенной ия цепей, содержащих 36—72 тетраэдра Р04 (температура перехода 330°). Дебаеграммы и рентгенограммы, снятые методом вращения, показывают, что строение соли Мадрелла весьма близко к волластониту CaSiO3. Волластонит, как показали рентгеноструктурные определения Барника (Barnik, 1936) н Джефрся (Jeffry, 1953), построен или из колец [Si3O9]s-или из спиралей [SiO3]j: с тремя группами SiO3 в витке. Для соли Мадрелла речь может идти только о спиральном расположения тетраэдров РО4, так как она, как показал Тило, имеет не кольцевую, а цепочечную структуру*.
Соль Мадрелла при кипячении с водой переходит преимущественно в тр1Шета-фосфат, т. е. в со единение с кольцевой структурой. Тидо сумел, однако, показать, что арсенатофосфаты, в которых непосредственно связаны три или более тетраэдров РО4, ведут себя таким же образом, хотя опи заведомо имеют цепочечную структуру. Предположение о том, что длинная цепь тетраэдров РО4 может иметь вид опирали (что находится в соответствии с рентгенографическими данными) , позволяет непосредственно объяснить образование трифосфатиых колец при гидролизе.
Соль Курроля, получающаяся из расплава NaPO3 при соблюдении определенного режима наГрёвапия и охлаждения, существует также в двух формах. Из расплава она кристаллизуется сначала в виде пластинок (с!20— 2,85), которые при растирании рассыпаются в тонкие пушистые волокна (d20=2,56). Такие же изменения происходят под действием коды пли прп храпений па влажном воздухе. Помимо плотности, обе формы различаются рептгоноструктурно, а также п пр своим свойствам; Пластинчатая форма при нагревании выше 400° переходит в соль Мадрелла (d2O=2,67). Из волокон в аналогичных условиях (при 400°) образуется триметафосфат натрия (d20—2,52). Соль Курроля, подобно пермутиту (ср, стр. 558), способна к реакциям катионного обмена.
* От полиарсената натрия (КаАзОй)х, имеющего такую же цепочечную кристаллическую структуру, соль Мадрелла и волластонит отличаются дефектами в расположении ионов в кристаллической решетке, не наблюдаемыми в первом случае (Tliilo, 1954). Похожей структурой обладает также упоминавшийся на стр. 541 и с л. и 557 гиллебрандит. ''
Пятая группа периодической системы 691
Фосф ори о вата л кислота н гипофосфиты. Фосфорнооатая кислота H4P20r образуется наряду с фосфор noir и фосфористой кислотами при медленном окислении фосфора во влажном воздухе. Для получения фосффриоватоп кислоты куски белого фос-: фора помещают в стеклянные трубки, открытые с обоих концов, один из которых вытянут в острие; несколько таких трубок устанавливают в стеклянную воронку и в подставленный сосуд собирают постепенно капающий раствор так называемых «фосфатных кислот». При добавлении ацетата натрия из раствора выкристаллизовывается довольпо трудно растворимый кислый фосфорноватокислый натрий Na3H3P3Og- 6Н2О в виде моноклинных табличек. Это соединение образуется также с хорошим выходом (наряду с фосфатами натрия) при действии смеси крепкого раствора едкого натра и перекиси водорода па красный фосфор. Еще; труднее растворяются свинцовая и бариевая фосфориоватые соли. Из них можно получить свободную кислоту ври взаимодействии с сероводородом или с серной кислотой. Фосфор но вата я кислота кристаллизуется в виде дигидрата в расплывающихся больших табличках П4Р2О6-2Н2О с т. пл. 62°. При высушивании в вакууме над пятиокисьЮ фосфора кислота отдает воду. Обезвоженная Н4Р2Ов при комнатной температуре неустойчива; она постепенно превращается в свой изомер — фосфористо фосфорную [дифосфорную (Ш,¥) кислоту] (см. стр. 679) (‘Кету, Fall us, 1957—1958). Фосфо рноватая кислота — более слабый восстановитель, чем фосфористая кислота. Азотная кислота, не содержащая окислов азота, ее не окисляет, я только лишь способствует гидролизу в соответствии с уравнением Н4Р,О6+Н2О=Н3РО:,-|-Нч1РО4. Эта реакция каталитически ускоряется люоыми .кислотами, т. е. ионами Н' (Blaser, 1933). Фосфорноватая кислота также весьма устойчива к действию восстановителей.
Фосфористая кислота и фосфиты Фосфористая кислота Н3РО3 получается проще всего при гидролизе треххлористого фосфора* РС13+ЗНО1Т = ..3HCI4-НРО(ОН)2.
Она образует бесцветные кристаллы, удельного веса 1,65, плавящиеся около 74е; в воде они очень легко створи .мы. Водород в мо.мепт выделения восстанавливает фосфористую кислоту до фосфористого водорода. Однако она легко окисляется до фосфорной кислоты. Поэтому Н3РО3 осаждает благородные металлы из растворов их солей и восстанавливает хлорид. I'Ig(lT) до Hg(I). Следует отметить, что азотной кислотой, по содержащей окислов азота, она не окисляется (Blaser, 1931). При нагревании в сухом состоянии опа распадается на фосфорную кислоту и фосфористый водород 4Н3РО3=ЗН3РО^-)-РН3 (окислительно-восстановительная реакция).
Фосфористая кислота образует только два ряда солей: однозамещец-ные МЩ2РО3 и двухзамещенные М|НРО3 фосфиты. Можно поэтому предположить, что Н3РО3 содержит один атом водорода, непосредственно связанный с фосфором: Фосфиты щелочных металлов к фосфит кальция.легко растворяются в ноде. Другие фосфиты трудно растворимы,
Пирофосфористая кислота НдРтОд (двухосновная) и метафосфористая кислота ПРО, пе могут быть получены при нагревании обычной фосфористой кислоты (орто-фосфорнстой кислоты), так как фосфористая кислота при нагревании разрушается. Обе эти кислоты были синтезированы ипым способом; они не имеют большого значения.
Фосфорнокатмстая кислота и гипофосфиты. Фасфорноватистая кислота Н3РОа образуется наряду с фосфористым водородом в виде своих солей при обработке фосфора раствором едкого кали или едкого натра (см. стр. 674) или раствором гидроокиси бария. Свободную фосфорноватистуго кислоту получают из гипофосфита бария Ва(Н2РО2)2- ]12О (легко очищаемого кристаллизацией) при взаимодействии с серной кислотой или из гипофосфита натрия NaH2PO2, обработкой его обогащенной ионами Н‘ искусственной ионообменной смолой (например, леватитом). Из концентрированного раствора кислота легко кристаллизуется на холоду в виде бесцветных листочков с уд. весом 1,49. Она плавится при 26,5°, а при содержании следов волы — еще при более нивкой температуре. Водород в момент выДеления восстанавливает Изрд2 в водном
* Помимо гидролиза, в незначительной степени идет реакция окисления — восстановления (Blaser, 1935).
44*
692 * Глава 14
растворе до фосфористого водорода. Ио и сама кислота — сильный восстановитель; она выделяет золото и серебро из,растворов их. содей, сама переходя в начале в фосфористую кислоту. Характерно ее поведение по отношению к растворам медных солей; она осаждает из них водородистую медь (см. т. II), тогда как фосфористая кислота осаждает (да и то лишь при кипячении) металлическую медъ.
Фосфорноватистая кислота образует только один ряд солей: гипЬфосфитгл М^НгРО2. Все они легко растворяются в воде, а соли щелочных металлов — и в спирте.
2. Галогениды фосфора
Фосфор образует три хлорида, Низший из них РС12 (или Р2С14?) получается с трудом и представляет лишь теоретический интерес. Обычный продукт горения фосфора в струе хлора трихлорид—треххлористый фосфор РС13. При действии избытка хлора он присоединяет еще одну .молекулу С12, образуя пентахлорид — пятихлористый фосфор PCI л.
Треххлрриспъый фосфор гидролизуется водой до хлористого водорода и фосфористой кислоты: (см, стр. 690).
; Поэтому следует считать фосфор в треххлористом фосфоре, как и в фосфористой кислоте, положительно трехвалентным.
В пят-йя лорнетом фосфоре фосфор в электрохимическом смысле положительно цЯтпвалснтеи, в структурном отношении он также пятивалентен. Это, однако, можно оспаривать и рассматривать пятихлористый фосфор как продукт присоединения хлора к. треххлористому фосфору Cl2‘PCI3. Действительно, в парах выше 30(1° пятихлористый фосфор полностью распадается на треххлористый фосфор и свободный хлор д, хотя равновесие РС|3- • CI,. I.’CI 531 ккал с понижением температуры значительно сдвигается вправо, все же и при комнатной температуре продуктыг распада присутствуют в значительном количестве. Одпако пятихлористый фосфор не только лишь продукт Присоединения, но и нормальное валентное соединение со структурно пятивалентным фосфором, что следует из электронографического определения его структуры (Rouault, 1938). Было показано, 'что в еавообразном пента&лориде пять атомов хлора расположены вокруг атома "фосфора в виде тригональной дипирамиды (расстояние Р -е,-+ С1в есмовавии Д-кд=2,10 А/ Р«—► С1^ всрг1>ине=2,25 А.; С1«—> G1—3,08 А.). Браун (В га ине, 1937) и Б рок пей (Brockway, 1938) нашли, что аналоги иной структурой облач дает и PFS(P <—> F=1,57 А). Такую же структуру имеет и жидкий пяти хлор петый фосфор. В твердом же снстшнши PCL-, построен из тетраэдров [РС14]* и октаэдров (РС1Д- [данные изучения снектров комбинационного рассеяния и рентгенографические данные (Moureu, 1937; Powell, 1940) J. Близкие структуры были найдены Пауэлом (1941) и Ван Дрплем (van Oriel, 1943) для РВг5 и Франсуа (Francois, 1941) для Sb Cl 5 , Эти соединения в твердом состоянии построены из тетраэдров [РВГ4]* и соответст-,: панно (SbCl4]+ и отдельных иопов Вг~ и С1". Пятибромистый фосфор известен в двух: формах; лимонпо-желтой и красной. Приведенная структура соответствует: красной (ромбической) модификации.
Структурные изменения, которые, претерпевает РС16 при переходе из жидкого состояния в .твердое; объясняют ряд поразительных явлений, которые связаны с затвердеванием'и плавлением этого вещества [М о н г е й, С. В., 203. 257, 1936; 205, 545, ' 1937]. Например, РС15 не имеет резко выраженных температур плавления и затвердевания; на кривых плавления и затвердевания в области Ц5° у пего наблюдается явно выраженный излом, соответствующий максимуму теплоемкости при этой температуре. : Далее, электропроводность его в твердом состоянии много больше, чем в жидком. Диэлектрическая^ проницаемость при плавлении уменьшается; но все же не достигает пуля. По-видимому, в жидком пятихлористом фосфоре находятся в смеси обе формы РС15 и [ РС14) [ РС1в ]. Это предположение делает понятным, почему РС15в реакциях е ненасыщенными органическими соединениями в однихслучаях переходит в РС13, отдавая два атома хлора, ;а в других, отдавая один атом хлора, присоединяется л виде группы PCI4 (Bergmann, 1930), например:
Пятая, группа периодической системы
693
РС1а
В то время как группа [PCl(i]~, по-видимому, присутствует кдк структурная единица только лишь в твердом пятих л ори стом фосфоре, известен целый ряд соединений; в которых содержится комплексный ион [PFej“ (гексафторофдсфатгя). Свободная гексафторофосфорная кислота ITPF6 была впервые получена в 1928 г. Ланге (LHuge) при действии фтористоводородной икс доты па Р2О5. Соответствующие' соли образуются не только i при обработке кислоты основаниями, по также и при взаимодействии PCI5 с фторидами металлов. Они более устойчивы, чем соответствующие'соли гексафторомышьяковой и гексафтор о сурьмя ной кислот, полученных Марипьяком еще в 1868 г/Натриевая соль г сксафторо фосфор ной кислоты из водного раствора кристаллизуется с одной молекулой воды в ромбических кристаллах (пЛ111(н_2,39). Безводное соединение N а [ PFB J к р и ст а л л изу ется ку бич ес ки (<1 п лв ==2,51)i, Ионы н атрия и центры тяжести групп [PF6]" образуют рошетау типа NaCl (изотиппые между собой KfPFg] и Tl[PFe]}, структурно отличаются от Na|PF6] ориентировкой октаздров [PFC]~ по отношению к пространственной диагонали элементарной ячейки. (Bodб, 1951—1952.)
Наряду с кислотой H[PFd ири взаимодействий ‘Р2О6 с HF образуются также оксофторофосфорные кислоты H[PO2F2] и Нг [PO3FJ. Диоксодифторофосфорная кислота довольно быстро гидролизуется. Три оксомонофторофосфо риал ’ кислота, напротив, очень устойчива. Реакции иона [PO3Fj" очень. похожи па реакции иона |SO4]".
Фосфор способен связывать более пяти атомов галогена также и в нейтральные молекулы, Таковы, например, соединения состава РВг7, РВг7С1а, РВт4С1й, РВг0С13 И другие. Они, без сомнения, относятся к классу по ли галогенидов (см. стр. 850).
Обзор простых галогенидов фосфора приведен в табл. 104.
Таблица Jr>4
Галогениды фосфора
PF3 —бесцветный газ, вес 1 л 3,926,. т. кип. —95°, т. пл. -160* i PFS —.бесцветный газ, т. кип, —75°, т. пл. —83"
РСЬ (Р2С14)?—бесцветная жидкость; :т. пл. —28°, т. кип. 180° РС13 — бесцветная жидкость; т. пл. —9Р, т. кин. 75,9° РС]5 — бесцветные тетрагональные кристаллы
ГВ?.. ? РВг3 бесцветная жидкость; т. пл- —41,5°, т. кип- 176° РЙг5 — красно-желтые ромбические иглы; т. пл.?; т.кип, 106°
Р214 — светло-оранжевые триклинные призмы; т. пл. 110° Р13 _ красные гексагональные призмы или таблички; т. пл. 01° Р15 — те мн о-красн ыо и риз -мы
Дихлорид фосфора РС12 (или Р2С14?) был получен Бессоном при действий темного электрического разряда на смесь РС13 и водорода. Это вещество образует бесцветную
694
Глава 14
маслянистую жидкость с сильным запахом фосфора (т. кип. ~ 180°, т.' пл. —28°). при обычной температуре соединение медленно разлагается. Разложенце ускордатсе При освещении и нагревании.
Для соответствующего иодида путем определения плотности пара был найдез молекулярный вес, отвечающий формуле P2I4. Вероятно, формулу хлорида следует также удваивать.
Треххлористый фосфор РС13 получается при сжигании белого фосфора в струе сухого хлора. При этом фосфор самопроизвольно воспламеняется и горит бледно-желтым пламенем; образующийся при этом троххлористып фосфор отгоняется. От примеси пятихлористого фосфора РС13 можно очистить, обработав его белым фосфором. Треххлористый фосфор — бесцветная жидкость с удельным весом 1,57, сильно дымящая во влажном воздухе. Он легко присоединяет другие вещества и ведёт себя,следовательно, как весьма ненасыщенное соединение. Вещества, легко отдающие кислород, а также сам молекулярный кислород ino белее медленно) переводят его в оксихлорид фосфора PQC13; с серой образуется сульфохлбрид фосфора PSC13. С хлором треххлористый фосфор образует иятйхлористый фосфор ГСП.
•л.
Молекула РС13 имеет форму тупой тригональной пирамиды с атомом фосфора в вершине. Такое же строение имеют РР3, РВг3 и PI3. Межатомные расстояния; РУ—>Р=1,52; Fi—»F=2,39 А; Р*—>С1=2,03; С] ч—♦ 01 = 3,09 А; Р *-* Вги2,18 А; Вг+—> 1=2,49 А.
Пятихлористый фосфор РС1й получается при. действии избытка хлора на треххлористый фосфбр. Эту реакцию целесообразно проводить так, чтобы треххлористый фосфор медленно, по каплям, стекал в широкосор-лый сосуд, в который одновременно поступал бы хлор и притом так, чтобы последний всегда находился в избытке. Трубка, подводящая хлор,, должна быть довольно широка, чтобы се отверстие не забивалось образующимся пятихлористым фосфором.
В чистом виде пят’ихлористый фосфор представляет собой белую кристаллическую рчассу, обычно имеющую зеленоватый оттепок вследствие частичного разложения на РС13 и свободный хлор. РС15 возгоняется выше 100 е, по плавясь. Относительно разложения его в парах см. стр. 692:
^Прп нагревании под давлением РС15 плавится в интервале 159—160 5°; его т. ирит, равна 372’. Давление пара РС15 (.«.« рт. ст) можно определить по формуле log />=11,034—16 100/4,57 71. По этой формуле температура сублимации, при которой давление сублимации достигает 1 атм, составляет 432° К (159°). С А1С13 ня тих лор истый фосфор образует очень термостойкое д ной по е соединение AIPCL (т. нл. 380°) [F i -seller W., A. anorg. Chora., 23n, 337, 1938].
Небольшое количество воды, превращает PCL сначала в оксихлорид фосфора Р0С13, который затем при дальнейшем действии воды переходит в фосфорную Кислоту
PC!s4-H;!o=poCl34-2HCi, - : (8)
РОС13+ЗНОИ = РО(ОН)3-|-ЗНС1. (9)
Другие соединения, содержащие кислород в форме гидроксильной группы, также взаимодействуют с РС.Ь, с образованием РОС13. Наряду с этим соединением образуются хлористый водород и хлорид соответствующей кислоты, например
СН3.СО-ОН-|-РС15 = РОС134-НС1 + СН3-СО-С1.
Уксусная кислота Ацетил хлорид
SO2(OH)2-p РС15 = POG13+НС1+SO2(QH)C1.
Серная кислота Хлорсульфоновая
। кислота
______________________Дятад группа периодической системы 695
При действии некоторых кислот вместо их хлоридов образуются ангидридыкио лот, например
2В (О11ИтЗР(Л5=ЗРОС13+биС1Н-В2О3.
Оксихлорид фосфора РОС13 — бесцветная, сильно преломляющая свет, дымящая ио влаялюм воздухе жидкость; удельный вес 1,72, температура кипения 108,7', температура плавления 1,3°;. в чистом виде удобнее всего получается при действии пятихлористого фозфора на щавелевую кислоту PCls+H2Ca04 =РОС134-2НС1+СОг+СО. Пятихлористый фосфор взаимодействует также и с пятиокисью фосфора, образуя оксихлорид ЗРС15-!-+P2O5=3pOCls.
Подобно тому как оксихлорид фосфора реагирует с водой, образуя фосфорную кислоту й хлористый водород [уравнение (9)], он взаимодействует и со многими органическими соединениями, содержащими гидроксил
РСН С)3 л К . ОН = 1 'О'ОН/л J. (10)
Он, следовательно, действует хлорирующим образом, подобно пятихлористому фосфору, по менее энергично, что часто бывает необходимо для более спокойного хода реакции.
Другие галогены также способны замещать группы ОН фосфорной кислоты. Удается получить соединения POFi (оксифторид фосфора — бесцветный газ с резким запахом; т. пл, —68°, т. кип. —40s). PODr3 (оксибромяд фосфора — бесцветные кристаллы с уд. весом 2,822, т. пл. 55°, т. кип. 193°). РОС12Вг (т. пл. 11°, т.кйп. 137,6°) и т. д. Известны, кроме того, оксига.тогеппды, производные от мета- и пиррфосфоршлх кислот/Пока еще не установлено, можпо ли получить окси га л ого виды, соответствующие кислородным кислотам, в которых фосфор проявляет валентность ниже 5.
3. Сульфиды фосфора
Сульфиды фосфора получают при сплавлении фосфора с серой в атмосфере двуокиси углерода. Для этой цели используют красный фосфор (так как белый реагирует слишком энергично) или же медленно испаряют,сероуглерод из раствора, содержащего фосфор и серу. В зависимости от состава исходных смесей образуются соединения P3S3, P.jS7 или Рг8й. При освещении раствора P4Ss и S в сероуглероде в присутствии следов иода в качестве катализатора можно подучить P4S5. Все эти соединения можно перекристаллизовать из соответствующих раствс'рителей или же переплавить; в чистом виде они дают хорошо образованные желтые кристаллы.
V- '
Пентт сульфид — пятпеерч истый фосфор P2S5 в сероуглеродном растворе полимеризован в P4S10; напротив, плотность пара отвечает формуле 1*285. Нетвердом состоянии это соединенна существует в двух модификациях, соответствующих обеим формулам. Сульфиды P4S3 и Р48. характеризуются одинаковым молекулярным состодаием как в растворе, так и в парах. Р^З^при высокой температуре -распадается на P4S3 и P4S7. В отсутствие воздуха (например, в атмосфере двуокиси углерода) остальные сульфиды фосфора перегоняются без разложения. Температуры кипения при атмосферном давлении и теплоты образования приведены ниже:
P4S3 P4S7 PsSs
Температура кипения, °C Теплота образования, 498 523 514
ккал'моль 29,4 65,7 —
При обычной температуре в сухом воздухе сульфиды фосфора устойчивы. P4S3 пе: изменяется и при доступе влаги; напротив, Р48- и Р2ЙД медленно разлагаются водой; поэтому во влажном воздухе опп пахнут сероводородом.
Будучи нагреты при доступе воздуха в присутствии веществ, легко отдающих кислород, сульфиды фосфора сгорают со всцыягкой. P4S3 воспламеняется на воздухе ужо при 100°.
696
Глава 14
Тетрафо£фортрисл1.1ъфид P4S3, называемый также «веской сульфидом фосфсрв^ в настоящее время широко применяют для изготовления спичек, загорающихся дв трении о любую поверхность. Благодаря высоким температурам плавления су.тьфадв фосфора пригодны как вещества для нагревательных бань.
4. Соединения фосфора с азотом
Из бинарных соединений азота и фосфора в чистом состоянии нзвестш лишь P3N6. Однако известно большое число соединений- в которых, помимо азота, с фосфором связаны еще и другие отрицательно заряженные ахс-мы или радикалы. Среди ипх большой интерес предстаййяют</кж(/юкитркх-еа.югениды и соответствующие им метафосфимовые кислоты вследствие своей чрезвычайной склонности к полимеризации. МетафОсфимовые кислоты являются FFpOji3Bодними от фосфопитрилгалогенидов (TNX,)., при заменр галогена X на гидроксил.
Расщепляя их, можно получить имидофосфорные кислоты, т. с. соединения, которые являются производными кислородных кислот фосфора прв замене - О па - Nil. При замене группы —ОН на группу —- Х112 из кислородных кислот фосфора получаются алшдофосфорные кислоты. Следует упомянуть о соединений азота с фосфором, строение которого ещё неизвестно, и о фосфаме с формулой [PN^H]^.
Нитрид пятивалентного фосфора P3N5, .по данным Штока (Stock, 1907), можно получить в Чистом состоянии, если обработать P2S5 аммиаком и долго нагревать полученный продукт в токе аммиака ври температуре выше 850°. Это бесцветный порошок, без вкуса и запаха, с уд. весом 2,51, не растворимый в обычных растворителях, устойчивый на воздухе. При нагревании в вакууме выше 760“ происходит распад соединения на элементы; .при нагревании в токе водорода образуются Р4 и МН31 при нагревании в токе хлора или кислорода P3N5 загорается, при нагревании выше 180° под давлением^гидролизуется водой до Н3РО4 и Mi3.
Фосфонптрилгалогениды. Фосфоиитрилхлорид (PNClj),v был получен еще в 1832 г. Либихом й Велером при нагревании вещества, полученного пропусканием аммиака в PClj, в виде красивого кристаллического возгона. Удобнее получать это соединение,, нагревая PCI3 с Т\ Т14С1 в автоклаве при температуре выше 120“ (удаляя образующийся ИС], как только давление превысит 25 ат). Лучше всего получать ’фрсфонитрил-хлорид по методу Шенка (Schenck, 1924) взаимодействием РС13, раствореппого в четырех хлор петом этане, с NH4C1 прп 135“
nPCl5+nNIT4(1 -(PNCL^-f-^JK'l.
Вещество, полученное таким образом, состоит из смеси изомеров (PNC12)j, (PNC12)4> (PNCL);, и т,"д., что впервые было установлено Штоком (1895). Эти изомеры: можно-разделить фракционированной перегонкой. Помимо температур плавления и кипения, они различаются формами своих кристаллов, а также различной растворимостью-в эфире, сероуглероде, хлороформе, бепзоле и т. д. Твердые соединений устойчивы по-отношению к кислотам и щелочам. В эфирном растворе они гидролизуются водой с образованием мётафосфимовых кислот (см. ниже). ;
На основании свойств этих, веществ Шток предложил для них циклические-структуры
- С1, Р —N ЧРС1 ЛР=.}Г
С12
. .PCI,—к :М>С13
— РС1,^
цикло-Трифо сфони три лхлорид цикло-Тттрафосфоиитрилхлоряд
Оба приведенных выше соединения .являются наиболее устойчивыми из ряда циклических полимеров. Их структура была установлена в результате рентгенографических исследований Егера (Jaeger, 1932); одпако пока ие ясно, связаны
Пятая группа периодической системы ' 697
... ли все атомы хлора с фосфором или же атомы хлора равномерно распределены между азотом и фосфором. (См. также Brockway, Bright, J. Am. chem. Soc., 65",. 1551, 1943.) При продолжительном нагревании выше 300Q С тримерный , фосфо нитрил-хлорид переходит в высоконолнмерное, нерастворимое и нелетучее соединение того Же-состава, которое при обычной температуре по механическим свойствам очень похоже-на слабо вулканизованный каучук (фосфонитрилкаучук). Как было установлено рентгенографически Мейером (Meyer), это вещество построено из очень длинных," изогнутых цепей
С1а р р
- /N7 V\\ .к/ ,NZ
\pz^ ; CIa cl2 .: c,:>
которые при растяжении вещества распределяются параллельно. Обстоятельное изучение условий растяжения показало, что при этом действуют те же силы, что и в случае каучука.
К фосфонитрил хлорид у близки по свойствам фосфоиатрилбромиды н фвефо-нитрилфториды. Фтор содержащие соединения не только способны полимеризоваться, переходя в высокомолекулярные каучукоподобные вещества, цо при иных условиях способны деполимеризоваться до низкомолекулярных 'соединений. Это свойство до сих пор неизвестно для соединений, содержащих только хлор или бром. Например, при нагревании под давлением тетрафосфо нитри л дихлбридгекс а фторид P^^CljFy (бесцветная жидкость с уд. весом .1,874 й т. кип. 105,8°) превращается в фосфонитрилкаучук, а при нагревании при атмосферном давлении деполимеризуется, по-видимому, по уравнению P4N4Cl2Fe -> 2P2N2C1F3 (Schmitz-Dumont, 1958). р
Мотафоефимовын кислоты, соединения с общей формулой [PN(OH), образуются при гидролизе циклических фосфонйтрилгалогснидов. Например, из (PNCla)3 получается триметафосфимоеля кислота [PN(OH)2]3, из (PNC12)4— тетраметафос-фймоеая кислота [РК(ОН)2]л и т. д. Метафосфимовые кислоты — очень устойчивые соединения, образующие весьма характерные для них соли. Обработав их щелочью^ можно расщепить кольцо и перевести их в, соединения, которые отличаются от мета-; фосфимовых кислот па одну лишнюю, конституционно связанную молекулу воды. Известны также амиды метафосфимовые кислот, например PKN3(O1I)4(NH2)2, который образуется при встряхивании эфирного раствора Г\ЛЧС14(А11с NapII. Это соединение можно получить, действуя NH3 на (PhJCl2)3. Строение метафо сфймовых кислот и их производных следует из способа их образования.
При кипячении солей метафосфимовых кислот с 30%-ной уксусной -кислотой происходит их распад с образованием имш)офосформ.ых кислот. .Наиболее простая из пих — имиддметафосформая кислота [OP(NH)OH]3, образующаяся также неяо-средственпр при действий NH3 на Р3Оа. Имидометафрсфориая кислота — бесцветная масса, легко растворимая в воде и в спирте. При кипячении постепенно разлагается на Н3РО4и NI‘I3.
(’Амиды (фосфорных кислот образуются при гидролизе их сложите эфиров,;которые в свою очередь получаются при обработке хлоридов сложных эфиров фосфорной кислоты аммиаком, Например, амидофосфорную кислоту 0=P(0H)2.NH2 можно получить по уравнениям
OP(O.CeHB)2Cl+2NH3=OP(O-GeHB)2MHs4-NH1Cl; " 0P(0-CeH5)3-NH2+K6H+H0H=KE0;,P(0H)-NHs]-}-2C?H50H;
К [О2Р(ОН).КНя]ф-Н' = ОР(ОН)з‘МНа4-К.' (с искусственной- иоаообмеяной-смолой)-
Амидо фосфорная 1 кислота — двухосновная кислота. Оца кристаллизуется в бесцветных призмах, легко растворимых в воде й плохо — в спирте. В растворе она постепенно, а при кипячении довольно быстро гидролизуется с образованием Н3РО4 иф?Н3, т. е. фосфата аммония.
Таг; же, лак н моноамидсфосфоряую кислоту, можно приготовить и диамидофос-г формую кислоту OPfOHKNH,),. Триамидофосфорную кислоту (фосфорокситриамиД) ОР(ЛГН2)3 можно получить, по Клементу (Kiement, 1954), при действия аммиака па раствор оксихлорида фосфора в хлороформе OPCI3-r6NH3=OP(NH2)s-|-3NH4Cl.: Это бесцветные сладковатые иглы, легко растворяющиеся в воде. Соединение это устойчиво в сухом воздухе, но в присутствии влаги теряет аммиак е образованием [NH4] [О2Р(ОН) 'NH2i- При нагревании с разбавленным NaOH образуется Na [OaPiNH^gj (крисгаллнаустся с 4Н2О). При взаимодействии с AgNO3 образуется
*698 Глава 14
весьма устойчивое комплексное соединение [OP(NH2)3Ag]NO3, кристаллизующееся в листочках, блестящих, подобно шелку.
Триамид тиофосфорной кислоты SP(NH2)3, полученный Клементом аналогично триамиду фосфорной кислоты, разлагается при действии влажного воздуха не как три а мид фосфорпой кислоты с отщеплением двух групп NH, в виде NH3, а с отщеплением всех трех групп NH2, переходя при этом в jNH4l2[SPO2(OH)]. Напротив, йрп нагревании с раствором едкого натра SP(NH2)3 ведет себя как OP(NII2)3, образуя Na[SPO(NH?)2].
Фосфам [PN2Hjx образуется при нагревании продукта взаимодействия NH3 с РС15 на воздухе. Это белый рыхлый порошок, нерастворимый в воде. При нагревании на воздухе фосфам медленно окисляется до Р2О5. При взаимодействии с расплав ленным едким калц фосфам разлагается Со вспышкой па аммиак и фосфат калия. Строение фосфама и природа ряда других фосфор азотных соединений, близких к нему по составу мл по способу образования, еще не выяснспы.
5. Соединения фосфора с водородом
Фосфор образует два легко летучих водородных соединения: газообразный при обычной температуре фосфористый водород — фосфин РН3 и жидкий при обычной температуре фосфористый водород.— дифосфин РаИ4. Является ли самостоятельным соединением так называемый «твердый фосфористый водород» примерного состава РаН или Р4аЩ, еще нс установлено (ср. стр. 699).
Фосфин РН3, как и NHj, дает соли с кислотами, о со бе q по с га логеново дородными (соли фосфония), Но эти соли значительно менее устойчивы, чем соли аммония; например, иод ид фосфония, образующийся по уравнению РН3+И1=(РНД)Г -(бесцветные, тетрагональные кристаллы, возгоняющиеся при 80°), разлагается водой. Бромид фосфония [РН4|Вг (т. возг. 3(Р) и хлорид фосфония [РН4]С1 (т, возг.. — 28°), несмотря на низкую температуру сублимации, в парах полностью разлагаются па РИ3 и г ало гено водород.
РгН4 не соединяется с кислотами; твердому" фосфористому водороду Ра11 приписывают способность сказывать вещества основном характера, например пиперидин.
Фосфин (мопофосфии) РН3 в чистом состоянии получают при действии веды или, лучше, раствора едкого кали на иодид фосфония 1РНтН
[PH,,]!-[- Н2О = РП3-НН3ОГ6 1г ИЛИ [РН4]1-|-О.Н' = РН3+Н2О-|-1'.
Фосфин можно получить также при разложении фосфида кальция Са3Ра или фосфида магпия Mg3P2 соляной кислотой. Но в этом случае к нему бывает примешано псболыпое количество дифосфина- То же относится М к фосфористому водороду, получаемому при действии едкого дали иа белый фосфор (см. стр. 674).
Фосфип — бесцветный, ядовитый газ с неприятным чесночным запа--хом. В сжиженном состоянии он кипит при —87,4° и затвердевает при —133°. Фосфин несколг.ко растворим в воде: 0,26 об. в 1 об.- воды. Чистый фосфин воспламеняется иа воздухе при температуре около 150° и сгорает в фосфорную кислоту РН3-(-2Оа = Н3РО4.
Если газ содержит дифосфин РеНц то он самовоспламеняется. Для предотвращения этого дифосфип удаляют, например, конденсируя его При охлаждении льдом.
В водном растворе фосфип РН3 количественно реагирует с HgCl2 по уравнению PH3-(-3HgCl2=P(HgCl)3-i-3riCl. На ацидиметрическом титровании образующейся Яри этом НС1 оспо в ап способ определения РН3, разработанный Уилметом (Wjliaet, 1927). Для определения очень малых количеств более пригодно йодометрическое титрование P(HgCl)3, предложенное Бейером (В е у е г, Z. anorg. Chem., 250, 312, 1943). P(HgCl)3 окисляется подом в растворе бикарбоната до Н3РО4.'В твердом
ГГлтал группа периодической системы . . ~ 899
Состоянии фосфигг образует кубическую гранецеитрпроваинуго молекулярную решетку. Оп претерпевает два полиморфных превращения (при —184 и —243°).
Дпфосфип Р3Н4, ранее называемый «жидким фосфористым водородом», образуется Обычно в качество побочного продукта при получении мопофосфнна. Оя представляет собой бесцветную жидкость с т. кип. 51,7° и т, тв. —99°, самовоспламеняющуюся и распадающуюся, особенно на Свету или в соприкосновении с пористыми телами, па фосфин и «твердый фосфористый водород».
«(Твердый фосфористый водород» Р2Н пли Р12Н6, свет до-желтый, устойчивый на воздухе порошок, образуется при разложении самовоспламеняющегося фосфористого водорода, т. о. смеси моно- и дифосфцноп, например, концентрированной соляной кислотой. Он нерастворим в воде и почти во всех других растворителях, по растворяется с желтым окрашиванием в жидком, фосфористом водороде, а также в расплавленном -фосфоре. Шенн установил, что понижение температуры замерзания его раствора в фосфоре соответствует формуле Р12ИВ. Ройсн (Royen, 1936— 1938) высказал предположение, что твердый фосфористый водород нельзя считать определенным соединенном. Он рассматривает твердый фосфористый водород как продукт сорбции РН3 на аморфном фосфоре и считает соединение, которое до сих пор считали солью Р2Н, как «нолианиолиую соль» Р1Т2, т.е. как соединение Типа [NHi]+[РН2-Рх]“. Это соедините можно получить, как cmv удалось показать, непосредственно из NII3, ;рн3ир.
6. Фосфиды
Фосфидами называют соединения фосфора с элементами сильно электроположительного характера. Их подразделяют на два класса. К первому из них относятся те фосфиды, которые производятся от фюсфнпа РН3 при замене водорода эквивалентным количеством металла. Сюда относятся фосфиды щелочных, щелочноземельных металлов и алюминия. Эти фосфиды разлагаются водой, а еще легче — разбавленными кислотами, с выделением фосфористого водорода, например
(.а 31’2 -f- 6Н2О—ЗСа(рН)2-й 2РИ3.
; Ко второму классу относятся те фосфиды, которые по "составу и внешнему виду соответствуют соединениям, образуемым металлами между7 собой; вода ц разбавленные кислоты большей частью не действуют на эти соединения или действуют слабо. К этому классу относятся фосфиды большинства тяжелых металлов.
Фосфиды получают, обрабатывая металлы, а иногда их окислы или хлориды элементарным фосфором или восстаиап.тивия фосфаты углем или же соогвегствутогцдаг металлом. Например, фосфпД магния получагог при нагревании с магнием какого-нибудь фосфата (или иных соединений фосфора).
Фосфиды большей частью тугоплавки. За исключением фосфидов благородных металлов, они весьма стойки к нагреванию.
Аналитические сведения. Чтобы установить наличие фосфора в : какой бы то пи было форме в исследуемом веществе, это вещество, измельченное в тонкий порошок, нагревают с несколькими кусочками магниевой проволоки. При этом магний соединяется с фосфором, образуя фосфид магния Mg3P3; реакция в большинстве случаев сопровождается вспышкой. При гидролизе образовавшегося фосфида магния выделяется фосфористый водород, который можно узнать по характерному запаху.
Определение элементарного белого фосфора имеет большое значение в судебной химии. Фосфор обнаруживают обычно на основании характерного свечения фосфорных паров, образующихся при перегонке пробы с водяным паром (проба Митчерлиха). Свечение, наблюдаемое в темноте, появляется главным образом в том месте, где водяные нары конденсируются
700
Глава 14
в холодильнике, потому что там пары фосфора впервые соприкасаются с воздухом.
Фосфорную кислоту (ортофосфорную) обнаруживают обычно на основании ее реакции с молибдатом аммония (NH4)2MoO4 (желтый осадок ив сильно кислого азотнокислого раствора). Этот же метод годен и для количественных определений. Можно также проводить осаждение из аммиачного раствора в виде магнийаммонийфосфата (NH4)MgPO4-6HsO, который перед взвешиванием прокаливают и переводят в пирофосфат магния MgaPjOT.
Очень быстро и легко можно определить содержание фосфора в фосфоритах, удобрениях и других торговых продуктах кондуктометрическим титрованием перхлоратом висмутила (J and er G., Angew. Chem., 49, 106, 1936) в растворе хлорной кислоты. Относительно определения фосфора в чугуне и феррованадиевых сплавах см.: Meuwsen A., Z. anorg. Chem., 251, 45, 1943.
Об отличии ортофосфорной кислоты от пиро-и метафосфорной кислот по окраске осадков с нитратом серебра, а также о реакции, характерной для метафосфорной кислоты (коагуляция раствора белка), уже было упомянуто, так же как и о различной восстановительной способности кислот, соответствующих различным степеням окисления фосфора.
МЫШЬЯК (ARSENICUM) As
Распространение в природе. Мышьяк иногда встречается в природе в самородном состоянии, но главным образом в виде соединений. Важнейшие соединения с металлами — арсениды; в большинстве случаев они встречаются в изоморфных смесях с сульфидами. Наиболее всего распространен мышьяковый колчедан (ядовитый колчедан, арсенопирит), изоморфная смесь FeAs2 и FeS2, чаще всего с приблизительным составом FeAsS. Чистое мышьяковистое железо FeAs2 встречается в виде минерала лоллингита.
Из арсенидов следует упомянуть: белый никелевый колчедан NiAs2, красный нике-левый колчедан (мышьяковистый никель) NiAs, шпейсовый кобалып СоАз2, кобальтовый блеск CoAsS, мышъяковоникелевый колчедан (герсдорфит) NiAsS.
К иному типу относятся содержащие мышьяк блеклые руды, например теннантит (светлая блеклая руда 4Cu2S.As2S3) и мышъяковосеребряная обманка (светлая красная руда SAgjS-As^j).
Их следует рассматривать как тиоарсениты (си. стр. 710 и сл.). К тиоарсенатам относится энаргит 3Cu2S.As2S5=Cu3AsS4.
В природе встречаются и сульфиды мышьяка: реальгар As4S4 и аурипигмент AsgSj. Как продукт выветривания мышьяковистых руд встречается и окись мышьяка АзаОз (мышьяковый цвет, арсенолит).
Мышьяк вообще настолько распространен в природе, что металлы, добытые из сернистых руд, почти всегда содержат As в качестве примеси, и довольно трудно полностью удалить его.
Исторические сведения. Природные мышьяковосернистые соединения реальгар и аурипигмент упоминаются уже у Аристотеля и его ученика Теофраста под названиями oavdapa^r] (сайдарах) и agoeviKov. Название auripigmentum, указывающее на золотисто-желтую окраску минерала, встречается впервые у Плиния. Диоско-рнд (I в. в. а.) говорит уже о прокаливании «мышьяка* (т. е. сернистого мышьяка). Алхимики более подробно изучали сульфиды мышьяка. Продукт их прокаливания они называли «бельм* жышъяхом*. Многие из них считали мышьяк основной составной
Пятая группа периодической системы 701
частью всех металлов, подобно сере. Получение металлического мышьяка впервые описано у Альберта Магнуса (XIII в.). Свойство мыиплка окрашивать медь в белый цвет и как бы превращать ее в серебро немало способствовало укреплению характерной для алхимического периода веры в возможность превращения металлов (транс-мутация). Алхимикам была уже известна ядовитость «белого мышьяка», т.е. трех-окисн мышьяка. Применение соединений мышьяка для лечебных целей начал латрохимик Параде..п.с.
Получение. Мышьяк получают главным образом нагреванием мышья-нового колчедана без доступа воздуха в трубках из огнеупорной глины. Мышьяк возгоняется и конденсируется в приемнике
FeAsS = FeS-|-As.
Можно также исходить из мышьяковистого железа. При нагревании без доступа воздуха оно разлагается
FeAs2= FeAs -ф- As.
Образующийся арсенид железа FeAs прокаливают
2FeAs •ф' 302 в РваО^'ф’ AsgOj.
Возгоняющуюся трехопись мышьяка можно восстановить углем до металлического мышьяка. Но обычно этого не делают, так как трехокись мышьяка боа ее употрсбима, чем металлический мышьяк.
Свойства. Подобно фосфору, мышьяк встречается в нескольких модификациях. Обычная форма — металлический или серый мышьяк. Он представляет собой серо-стальную кристаллическую массу с металлическим блеском, хрупкую, с незначительной твердостью (3—4 по шкале Мооса), проводящую электрический ток (удельная электропроводность при 0° равна 4,19% электропроводности серебра). Удельный нес 5,72, магнитная атомная восприимчивость %а=—5,5-10“е.
Мышьяк возгоняется, не плавясь, при 633°. При нагревании под давлением он плавится. Под давлением 36 атм температура плавления лежит около 817°. Плотность пара до 800° соответствует формуле Аз4, выше 1700° — формуле Аз2.
Пары мышьяка бесцветны. При резкам охлаждении паров] получается желтый мышьяк, состоящий из прозрачных, мягких, как воск, пластичных кубических кристалликов удельного веса 1,97, тогда как серый (металлический) мышьяк кристаллизуется гексагон аль но-ромбоэдрически. Желтый мышьяк растворим в сероуглероде, летуч с водяными парами и действует как сильный восстановитель, следовательно, подобен по свойствам белому фосфору, ио значительно менее устойчив, чем последний. Уже при слабом нагревании, а также под влиянием света, он быстро переходит в серый (металлический) мышьяк.
В качестве промежуточного продукта при переходе желтой модификации мышьяка в обычную металлическую часто появляется третья форма мышьяка, так называс-мый черный мышьяк. Эта форма мышьяка образуется также при пропускании мышьяковистого водорода через раскаленную стеклянную трубку в виде налета на ее стенках (мышьяковое зеркало). «Черпый мышьяк» (он может быть также окрашен и в серый цвет) — стекловидно-аморфное вещество. Удельный вес его колеблется^в интервале 4,7—5,1 в зависимости от способа приготовления. Магнитная атомная восприимчивость Ха——23-10_е. При температуре выше 270° происходит монотропное превращение в металлическую модификацию. Четвертая форма, бурый мышьяк (уд. вес 3,7—4,1), получается при восстановлении солянокислых растворов трехокиси мышьяка, например, хлористым оловом (хлоридом олова (11)] или фосфорноватистой кислотой. До сих пор еще не установлено, возникает ли при этом особая модификация или очень тонко-измельченная обычная.
При нагревании на воздухе мышьяк сгорает синеватым пламенем, образуя белый дым трехокиси мышьяка As.,O3. При горении распространяется характерный чесночный запах. Крепкая азотная кислота пли царская водка окисляют мышьяк в мышьяковую кислоту, а разбавленная
702
Глава 14
азотиая или концентрированная серная кислота, а также кипящие щелочи — в мышьяковистую кислоту. С хлором As реагирует со вспышкой.. При высокой температуре As непосредственно соединяется и со многими другими элементами. С тяжелыми металлами, даже с такими, с которыми он не дает соединений, оп очень легко сплавляется (т. е. легко растворяется в расплавленном металле). Застывшие сплавы большею частью хрупки, даже при очень небольшом содержании мышьяка. Например, менее ^юсо мышьяка делает золото хрупким.
Нормальный потенциал мышьяка, отнесенный к нормальному водородномv электроду, составляет около +0,3 а. Следовательно, мышьяк менее электро положите^ лен, чем водород, а такщо чем сурьма и висмут.
Применение. Металлический мышьяк используют главным образом для изготовления ружейной дроби (о составе см. «Свинец»), Вообще же как металл он не имеет большого технического значения. Важнее соединения мышьяка, особенно трехокись и сульфиды. О применении последних будет сказано при их описании.
Следует указать на сильную ядовитость растворимых соединений мышьяка. Они вызывают смерть после нескольких суток тяжелых мучений. Ядовитые свойства соединений мышьяка используют для борьбы с вредителями сельского хозяйства и в медицине.
СОЕДИНЕНИЯ МЫШЬЯКА
1, Окислы и гидроокиси
Мышьяк образует два окисла: трехокись мышьяка АзаОз и пятиокись мышьяка АнгОз. Оба они имеют характер ангидридов кислот. От трехокиси мышьяка производится мышьяковистая кислота [мышьяк (III) кислота] г существующая только в водном растворе; известны, однако, многочисленные ее соли — арсенаты I арсенаты(Ш)]. От пяти окиси мышьяка производится м.ышьякетая кислота [мышьяк(У) кислота]. Ее соли называются арсенат амт I точнее, арсенаты(У)].
T;ii, па-зысалгую As2O4 следует рассматривать как сме-
шанный ангидрид мышьяковистой я мышьяковой кислот, пли двойное сосдинёниё-трехоьнсп мышьяка с пятиокисыо.
7 еелогчы еиранования окислов (из элементов) равны;
AsjOg ЛВ2О4 AsoOj
153,8 174,3 219,4 ккал/.чоль
Трехокпсь мышьяка. Окись мышьяка(Ш) А«2О3 или As4Oe, называемая также белым мышьяком, получается в больших количествах при обжиге мышьякового колчедана или других природных арсенидов.
2FeAsS + 3CL = Fe2O3 + 2SO2 + As3O3.
На многих заводах, перерабатывающих мышьяковые руды, она получается в качестве побочного продукта. Белый дым,состоящий из трехокиси мышьяка и появляющийся при обжиге таких руд, был известен еще в средние века под названием «рудный дым».
Для выделения трехокиси мышьяка газы, образующиеся при обжиге, пропускают через длшшыс каменные каналы. С.обирающуюся в них «ядовитую муку» очищают возгонкой; в зависимости от температуры, прн которой происходит конденсация, трехокись получается в виде рыхлого белого порошка или в виде стеклообразного продукта («мышьяковое стекло»).
Пятая группа периодической системы
70CJ-
Трех окись мышьяка обычно постукает в продажу в виде бесцветной, стеклообразной массы с раковистым изломом. Удельный вес ос 3,70. Но эта масса постепенно становится непрозрачной, как фарфор, и переходит в агломерат октаэдрических кристалликов (уд. вес 3,87).
Непосредственно такие кристаллы получаются при кристаллизации из води ого раствора. Процесс кристаллизации из раствора сопровождается отчетливой люми-яеедетщиг®. Кубпчмшимжтаэдрл веская форма является при обычной температуре' стабильной модификацией трехокиси мышьяка. Ее т. пл. 310°, т. кип. 465°. Существует еще и вторая кристаллическая модификация, а именно моноклинная, стабильная при температурах выше 221". Обе эти формы встречаются также в природе, кубическая — под названием арсенолита (мышьяковый цвет), мопок.'ИШная — под названием клаудетита.
Трех окись мышьяка легко улетучивается при нагревании. Пары конденсируются на стенках сосуда, если температура их выше 310°, образуя мышьяковое стекло; при конденсации ниже этой температуры получается мука, состоящая из октаэдрических кристалликов.
Плотность пара при 800" соответствует формуле As4O6; лишь при 1800°' плотность пара трехокиси мышьяка отвечает формуле АвгО3. Эбулиоскопические определения молекулярного веса в нитробензоле также приводят к удвоенной формуле As406.
Кристаллы арсенолита также построены из молекул As4O0. Центры тяжести: молекул расположены подобно атомам углерода в решетке алмаза (а=11,0й А). В отличие от арсенолита клаудетит, стлбЕльпый при более высокой темпера туре, имеет слоистую структуру. Это противоречит правилу, в соответствии с которым стабильная модификация вещества при более высокой температуре имеет более высокую симметрию кристалла, чем низкотемпературная модификация. Грехокиеь мышьяка, также отклоняется от правила ступеней Оствальда, а также и от правила, гласящего, что однородное вещество имеет резко выраженную температуру плавления. Эта аномалия, по мнению Странскн (Stransici), объясняется тем, что слои в: решетке клаудетита построены из бесконечного числа атомов пли соответственно ионов, образование которых или перестройка молекул As4Ofl в которые требует относительно большей анергии активации (ср. т. II, ГЛ. 17), ибо в обоих случаях должны разрушаться нормальные валентные связи, что не имеет моста при испарении арсенолита, ярл котором должны нарушаться только слабые вопдерваальеовы связи, двйсгвую-щие между молекулами Лз4О6. При наличии следов водяного пара превращение арсенолит^клаудетит ускоряется в обоих направлениях, так как вследствие образования промежуточных соединений с водой сильно снижается необходимая энергия активации. Таким же образом можно объяснить, согласно воззрениям Стрински, аналогичные явления, наблюдаемые для Sb40g, Р2О4 соответственно Р4О10, As4S4₽ Р, Ар и (CN)2. Величина энергии активации испарения, разумеется, не влияет на равновесие между твердой и парообразной фазой. Энергия активации не влияет также на давление пара, но опа может сильно влиять па скорость исвиренБИ. Для значительного большинства веществ энергия активации меньше или приблизительно равна теплоте парообразования. Однако имеются случаи, когда энергия активации парообразования в несколько раз больше теплоты парообразования. Указанные выше вещества являются такими, у которых для одной модификации имеет место первый случай, а для другой — второй. В таком случае скорости испарения обеих модификаций могут различаться на несколько порядков. Можно даже наблюдать парадоксальное явление — ускорение конденсации пара при нагревании. Странскп называет это явление «вынуящмпюм шшщщеацней». Это можно легко продемонстрировать на примере Ак2О3: в запаянном стеклянном сосуде, па которого откачан воздух, содержится небольшое количество арсенолита, в нескольких сантиметрах от которого впаяна платиновая спираль. Сосуд помещают в термостат с температурой 200". Если генеря нагреть сип ради выше 700°, то As2U3 исчезнет со дна сосуда и осядет в значительно более горячей зоне па стенках сосуда против спирали. Этот Процесс объясняется следующим образом. Молекулы Лв4О6, образующиеся при испарении арсенолита, активируются около платиновой спирали, т. е. частично распадаются на мол скул гл As2O3, и поэтому делается возможной их кондеясагщя с образованием клаудетита или клаудетито-подобного стекла, которое г следствие высокой энергии активации, необходимой для
704
Глава 14
испарепия, испаряется Крайне медленно в противоположность арсенолиту, кот;;^4 испаряется быстро, так как его энергия активации меньше, чем теплота испаряли
О полиморфно см. также; Schulman, Sth am Ь, J. Am. ch~ Soc., 65, 878, 1943.
Трехокись мышьяка легко восстанавливается до металлическое, мышьяка. Так, если нагревать АвгО3в маленькой трубке для прокаливание с углем или с цианидом калия, то мышьяк, образовавшийся в результат-, восстановления, осаждается в более холодной части трубки в виде чернот? зеркального кольца (мышьяковое зеркало, проба на мышьяк по Берцелиусу). Из растворов трехокиси мышьяка в большом количестве концентрированной соляной кислоты хлористое олово [хлорид олова(П)] осаждает металлический мышьяк в виде черно-бурого осадка. Эта реакция лежит в основе определения мышьяка по Беттендорфу. В кислом раствора водород в момент выделения восстанавливает трехокись мышьяка до мышьяковистого водорода. Окислители окисляют AsaO3 до мышьяковой кислоты. Окисление идет легче всего в присутствии щелочи.
Раствор трехокиси мышьяка в присутствии бикарбоната натрия связывает такое количество пода, сколько требуется для окисления мышьяка из трех- в пятивалентный. Эту реакцию используют для количественного определения мышьяка в растворах трехокиси мышьяка*.
Трехокись мышьяка умеренно растворима в воде; стекловидно-аморфная форма растворяется значительно лучше кристаллической. Для последней растворимость при 0° составляет 1,2, при25° 2,1 и при 75° 6,0 г AsaO3 в 100 г воды. Это сладковатый раствор с неприятным металлическим привкусом. О сильной ядовитости, трехокиси мышьяка уже упоминалось.
Доза менее 0,1 г трехокиси мышьяка может быть смертельной, если опа введена в желудок и не приняты соответствующие меры; стимуляция рвоты или обезвреживание яда переводам в нерастворимое соединение (например, действием окиси магния) иди адсорбция яда ; в; тв г осажден л ой гидроокисью желоза. Замечательна способность организма до некоторой степени привыкать к этому яду. Привычные потребители мышьяка иногда переносят без вреда значительную дозу, в несколько раз большую, чем та, которая для обычпыо организма была бы смертельна.
Мышьяк широко применяют в борьбе с вреди гелями сельского хозяйства. В стекольном производстве его используют для осветления стекла. Он идет также для изготовления красок, особенно швейнфуртгкой зелени и, кроме того, для медицинских целей.
Мышьяковистая кислота и арсениты. При взаимодействии с водой трехокись мышьяка’ образует мышьяковистую кислоту [мышьяк(Ш) кислоту]. -J
В свободном состоянии мышьяковистая кислота неизвестна, но можно доказать ее присутствие в водных растворах в равновесии с продуктами диссоциаций. Еще не ясен вопрос, какую формулу следует приписывать мышьяковистой кислоте. Ее можно рассматривать не только как ортокис
* Раствор нельзя подщелачивать гидроокисью или карбонатом щелочного металла, так как иод вступает в реакцию с ними, образуя гипоиодит (см. стр. 867). Достаточно той концентрации гидроксильных ионов, которая образуется вследствие гидролиза бикарбоната, чтобы равновесие
АзОу-)-1г-|-2ОН'^А8ОУ+2Г4-ПяО)
пол по стьюА сдвинул ось вправо.
705
Пятая. группа периодической систры
лоту H3As03 или As(OH)3 — это определение представляется предпочтительнее остальных,— но и как метакислоту HAsO2 или АнО(ОН) или, .наконец, как гексагидрокеокислоту H3[As(OH)e] (см. ниже). Мышьяковистая кислота может диссоциировать нс только как кислота (уравнение (1)), по и как основание (уравнение (2)J
As(OIl)s AsO"a4-3H* , (1)
и
Л s{ ОН).,, As' “ 4- ЗОН'. (2)
Отношение содержания ионов As'" к ионам AsO3" очень сильно зависит от концентрации водородных и соответственно гидроксильных ионов в растворе, как это и следует из закона действия масс применительно к равновесию, существующему в растворе. Комбинируя уравнение (1) с уравнением (2) и применяя закон действия масс (учитывая при этом постоянство произведения концентрации водородных ионов на концентрацию гидроксильных ионов), получаем
[As“J .
,-?• г»»~^7.ттчк~ const.
| AsO 'J )Н']6
Лишь в сильно кислых растворах имеются в значительном количестве ионы As" • Мышьяковистая кислота — очень слабая кислота, приблизительно такой же силы, как и бориая, Ещо слабее она диссоциирует как основание. Первые копстапты диссоциации в качестве кислоты и в качестве основания при 25° имеют следующие значения:
_ liMsO'KH'I „ [As(OH);noH'i
л>^ : in - *°0НО6=“[Ацо1ад---------1'10
Некоторые данные свидетельствуют о том, что мышьяковистая кислота в растворе находится, Ц0-В11ДИМ0МУ, в мета-формо HAsO2. В таком случае приведенные константы диссоциации относятся к равновесию AsOiAH' yi HAsO2 и AsO* АОН'
HAsO2. В противоположность этому Вринтзиигер (1935), однако, обнаружил методом диализа, что мольный вес арсенит-ионов соответствует формуле [Аз(ОН)в]"'' или |Н2Ав(.ОН)61’’. Если, принимая это во внимание, мышь жр кистой кислоте приписать формулы Н3Ля(ОН)0 или соответственно Аз(ОИ)3(ОН2)3, то вышеприведенные константы можно отнести к равновесию
{As(OH)4(OH2)J'4- (ОНЫОН,)5,
[Ая(ОН)г(ОН2)4У A OH'^tAs(OH)3(OH2)3-A На0.
Соли мышьяковистой кислоты называются арсенитами [арсенаты(Ш)]. Арсениты щелочных металлов очень легко растворяются в воде.
Труднее растворяются арсениты щелочноземельных металлов. Соединения мышьяковистой кислоты с тяжелыми металлами в воде практически нерастворимы. Слизистые гидроокиси металлов, например гидроокись железа, способны адсорбировать значительные количества мышьяковистой кислоты. Кислоты разлагают арсениты, потому что в кислых растворах равновесие AsO'”-}~6H" As"“+3H2O сдвигается настолько вправо, что
концентрация ионов арсенита оказывается уже недостаточной для достижения произведения растворимости даже труднорастворимых солей.
Большинство солей мышь яко в петой кислоты соответствует типу MIAsO2; они, следовательно, производятся не от орто-кислоты H3AsO:; или соответственно H3[As(OH3)], а от мета-ниспоты ПАААА В этом одна иа причин, позволяющих утверждать суще-
* Наряду с мета- и ортоарсенитами известны также арсениты со сложным составам. Например, по данным Нельсона (1941), арсенит натрия в зависимости от количественного соотношения компонентов в распадре_ кристаллизуется цз водного раствора при 35° в виде 14aAs3O5, NaAsO2, Na4As2O3-7H2O и Na4As2O6.
45 Г. Реми
Глава 14
706
£.
ствование мета-формы мышьяковистой кислоты. Но возможно, что преиму1цк~2 сап образование первичных солей обусловлено только очень слабокислым хар гидроокиси мышьяка, который должен способствовать гидролизу вторичный z тичных солей в водных растворах с образованием первичных солей или нх е. ь например: f
K3AsO3 4- 2Н0Н = ЗК‘ 4- 2ОН'4- H2AsO3
или
K3AsO34r 5НОН = ЗК‘ 4- 2ОН'ф- H2As(0H);.
В качестве примера ортоарсепита можно назвать желтый, трудно растворимый арес-^ серебра Ag3AsO3.
Арсенит меди Сп(АзО2)2 красивого зеленого цвета прежде применяли в ньх-стве краски (зелень Шееле}.
Пятиокись мышьяка, мышьяковая кислота и арсенаты. Пяпшок;.. мышьяка [окись мышьяка(У) 1 As2O5 белая, стекловидная масса с удельным весом 4,1, не может быть получена аналогично пятиок ней фосфора прг окислении трехокиси кислородом воздуха, а только при обезвоживание ее гидрата As,O5-4HsO — мышьяковой кислоты (см. ниже). Во влажно?.' воздухе пятиокись мышьяка расплывается. С водой она вновь соединяется в мышьяковую кислоту.
Мышьяковая кислота [мьцш>як(¥)кислота] H3AsO4 образуется прп окислении мышьяка или трехокиси мышьяка крепкой азотной- кислотой или другими сильными окислителями. Она выделяется из сильно концентрированного водного раствора большей частью в виде очень мелких бесцветных расплывающихся кристаллов состава H^AsO^ 1/2НаО- (Об отношении ее к нагреванию см- ниже.)
Мышьяковая кислота действует как довольно сильный окислитель, но большей частью только в кислом растворе. На этом свойстве основано ее применение в производство органических красителей. Она служит также исходным продуктом для органических мышьяковистых соединений, применяемых в медицине (сальварсан и неосальварсан), >
Растворимость мышьяковой кислоты очень велика. При 20° 100 г воды растворяют 630 г As2O5, а при высокой температуре — еще больше. Мышьяковая кислота является трехосновной кислотой средней силы, но она несколько слабее фосфорной. Ее константы диссоциации (прп 25°) равны: Kj—5’10*3, А2=4‘10“6, Я,3=610"1о.
Соли мышьяков ой’к не л оты — арсенаты [арсенаты(У)] пбдсв сему Составу и растворимости в общем соответствуют фосфатам*. Некоторые’из них изоморфны фосфатам, например магнийаммонийар сенат (NH4)MgAsO4-6Н3О, который получается в тех же условиях, что и магнийаммоний-фосфат, и аналогично ему переходит при прокаливании в пироарсенат магния Mg3As2O7. Это вещество особенно пригодно для весового определения мышьяковой кислоты. Нитрат серебра осаждает из раствора арсе?-натов3 шоколадно-бурый (в отличие от желтого фосфата) осадок арсената серебра Ag3AsO4. Двух замещенный арсенат натрия (_На,НАзО4 (из водного
* Так же, как и тринатрийфосф ат, трин атрий ар сенат, кристаллизуется в виде додекагидрата, и, по данным Мепцелл (Menzel, 1937), его состав меняется в пределах между Na3AsO4 и Na3AsO4A/4NaOH. Различие заключается только в том, что арсенат можно получить из водного раствора также и со стехиометрическим составом, что в случае фосфата невозможно. Относительно обезвоживания фосфата и арсената Натрия при нагревании см.: Menzel, Z. Elektrochem.., 43, 104, 1937; см. также Z. anorg. Chem., 233, 49 и 209, 1937.
Пятая группа периодичесхой система
70,7
раствора соль кристаллизуется с 7НаО*), кроме медицинских целей, часто применяют в большом количестве для опрыскивания фруктовых деревьев.
Другие арсенаты, например Ca3(AsO4)2, также находят применение как инсектициды в сельском и лесном хозяйстве. Арсенат кальция готовят окислением арсенита кислородом воздуха или другими химическими агентами (например, раствором гипохлорита) или электролитическим способом.
Окисление кислородом воздуха можно осуществить не только в растворе, по и сухим путем при нагревании в токе воздуха. Например, арсенит кальция сгорает при нагревании в токе воздуха с образованием арсената. В основе реакции лежит, как показал Рейзаус (Reissaus 1931), окислительп о-в о останови тельный распад арсенита при нагревании 5Са3(АзО3)г=ЗСаз[АаО4!2^-4Аз'4-6СаО. [Эту реакцию можно использовать для получения чистого арсената и мышьяка, если вести нагревание в вакууме, добавляя необходимое количество АзгО3 для связывания СаО;ЗСа3(АзО3)2ф 4-2As2O3=3Ca3(AsO4)2-p4As.] При нагревании в токе воздуха мышьяк снова окисляется до As2O3, который после соединения со свободным окислом металла переходит в арсенит, и вновь распадается в результате окислительно-восстановительной реакции, и так до тех пор, пока весь арсепит по суммарному уравнению Ca3[AsO3]24-O2=Ca3(AsO4)2 не превратится в арсенат. Выяснение механизма этой реакции имело большое значение для металлургии руд, содержащих мышьяк. 11s водного раствора в зависимости от состава раствора осаждаются арсенаты кальция: CaH4(AsO4)2, СаНАаО4, Ca6II2[AsO4]4, Ca3[AsO4]2 и Са4[АбО4]2(ОИ)2; последние четыре1 формы выделяются с кристаллизационной водой (Pearce, 1936; Guerin, 1941). Моногидрат CaHAsO4 встречается в природе в виде гайдингерита, а дигидрат — в дяде фармаколита (см. стр. 272).
Структуру кристаллической, решетки арсенатов см.: Helmholz L., J. Am. chem. Soc., 64, 354,, 1942.
KH2AsO4 кристаллизуется тетрагонально, Ag-3AsO4 — кубически. В обеих солях атомы кислорода групп (AsO4) образутот правильный, лишь слегка искаженный тетраэдр; расстояние As 0=1,74 Л.
Приведенная выше формула мышьяковой кислоты [мышьяк (V) кислота] HaAsO4 основана на формулах арсенатов, вполне аналогичных фосфатам. Однако соединение, которое кристаллизуется из водных растворон пятиокиси мышьяка, по своему составу пе соответствует этой формуле, а отвечает полугидрату мышьяковой кислоты H3AsOt или соответственно тетрагидрату пятиокиси мышьяка Аз2О6-4НгО. При обезвоживании из него сначала образуется соединение состава H5As3010 пли соответственно 3As2O6-5H2O, которое начинает распадаться уже при 120° на пяти-окись мышьяка я воду, ио удерживает последние остатки воды настолько прочно, что для полного обезвоживания нагревание должно продолжаться до 500®. При обез-вояшвании не образуется таких соединений мышьяка, которые цо своему составу соответствовали бы пиро- и метафоефорной кислотам. Однако при более низкой температуре (около —30°) из водного раствора можно получить еще один, более богатый водой, гидрат As3O6’7H2O (Simon 1927). Цока еще ие установлено, следует ли считать последнее соединение дигидратом обычной мышьяковой кислоты H3AsO4-2H2O или одноосновной гидр оксокис йотой Н[Ав(0Н)в].
Тот факт, что при обычной температуре мышьяковая кислота присутствует в водном растворе в виде H3AsO4, хотя кристаллическое соединение такого состава пе известно, был установлен Симоном и Фехером (Simon, Feh£r, 1937) на основании изучения спектров комбинационного рассеяния этих растворов в сравнении со спектром кислой соли. Появление в спектре почти безводной H3AsO4 сильной полосы, соответствующей группе ОН, позволяет сделать заключение, что мышьяковая кислота, подобное отной кислоте, существует в двух разных формах: в «форме сложного эфира»
* Гидраты NaJIAsO,;, по мнению Менцеля, в соприкосновении с водным раство-
—1,14° 4-20,5"
ром [имеют следующие области существования:' Лед <—.—_ Додекагидрат <—— 55,3° 57,4° Э9,5"
Гептагидрат Пентагидрат <-----Моногидрат < Безводная соль. При
тензиометрическом исследовании ступень моногидрата ио улавливается, в случае, если к пентагидрату не добавлено очепь небольшого количества моногидрата в виде затравки.
45*
708
Глава 24
и в «форме соли». Первая форма существует в концентрированном растворе, вторая — в разбавив ином. К такому же заключению пришел рапьшо Лидер (lander, 1934 который установил, что мышьяковая кислота (так же как кремневая и теллуровас. кислоты) в недиссоцип ров айном состоянии иначе адсорбирует ультрафиолетовы! лучи, чем в диссоциированном состоянии или в форме соли. В примерно 1 и. растворе, по Бринтзипгору, арсенат- или соответственно гидр о арсен ат-ион присутствует в следующих формах:
при pH > 13: [AsO4(OH2)12]"' при 7 < рН < 11: [HAsO4(OHa)6r,
при pH <4 6: [H2AsO4(OH2)2]'.
2. Галогениды мышьяка
Обзор галогенидов мышьяка приведен в табл. 105. Меньшая устойчивость мышьяка в пятивалентном состоянии по сравнению с фосфором проявляется здесь в отсутствие пептахлорида, пептабромида и пентаиодида.
Таблица 105
Галогениды мышьяка
AsF3 AsF5
Бесцветная жидкость, уд. вес Бесцветный газ, т. затв.
3,01, т. затв. —8,5°, т- кип. 63° — 79,8а, т.кип. — 52,8°
AsClg
Бесцветная жидкость,’ уд. вес 2,17; т. затв. -16,0°, т. кип. 130,2=
; А^вг,
Бесцветные кристаллы в виде столбиков, т. н.ч. 31,2°, т. кип. 221 °
Asl3 Темпо-красные призмы, т. пл. 124*
Asl3
Красные гексагональные таблички, т. пл. 146°, т. кип-—400°
Бее галогениды мышьяка можпо получить непосредственным взаимодействием элементов. Но иногда бывает выгодно получать их иным путем, особенно фториды. Мышьяк и иод, по данным Моитипье (Montignie, 1941), при содержании мышьяка менее атомного отношения 1 : 2 в жидком состоянии смешиваются только ограниченно. При содержании 88 ат.% I образуется эвтектика (т. пл. 73=). В молекуле парообразного Asl3, как было установлено Хасселем (Hassel, 1941) в результате электронографического исследования, атомы расположены в вершин ах тригональной пирамиды As <—>-1=2,54 А; <£1—As—1=98°30'. Галогениды мышьяка обладают способностью присоединять другие вещества, тешример аммиак. С фторидами металлов, например с фторидом калия, иентафторпд мышьяка дает двойные соединения типа AsF5-MiF и AsF5’2MjF; прочие галогениды мышьяка образуют главным образом соединения типа 2AsX3-3MIX.
Пятая группа периодической системы 709
Трихлорид мышьяка, треххлористый мышьяк, AsCJ3 представляет собой бесцветную, дымящую на воздухе жидкость, образующуюся при сгорании мышьяка в газообразном хлоре. Проще всего AsCls образуется при пропускании сухого хлористого водорода над трехокисью мышьяка при 180—200°. При растворении трехокиси мышьяка в водном растворе соляной кислоты образование этого соединения идет не до конца, так как в растворе устанавливается равновесие
As(OH)3-L3HCl АзС13+ЗН2О,- (3)
Но при достаточном избытке концентрированной соляной кислоты можно отогнать весь мышьяк из раствора в виде треххлористого мышьяка (вместе с нарами соляпой кислоты). На этом основан аналитический метод отделения мышьяка от сурьмы и олова. Это разделение еще лучше у соответствующих бромидов.
Б водном растворе треххлористый мышьяк частично распадается на ионы As’’’-l-ЗСГ; одновременно происходит глубокое гидролитическое расщепление 1уравнеиие(3)1; см. также уравнение (1), стр. 705.
3. Сульфиды н сулъфосолп мышьяка
При сплавления мышьяка с серой можно получить сульфиды As4S3, As4S4, Ag.,S3 и As3Sg. Проще всего трехсернистый мышьяк AsaS3 получается при пропускании сероводорода в солянокислый раствор трехокиси мышьяка
2As* - + 3H2S = As2S3+6Н-.
Пептасульфад мышьяка, пятисерннстый мышьяк, As,S5 можно подучить аналогичным образом из раствора пятивалентного мышьяка
2As... Ф 5II2S=As2S55 + ЮН*.
Но реакция протекает в этом направлении только при соблюдении вполне определенных условий. Сероводород частично восстанавливает пятивалентный мышьяк, так что, как правило, вместе с пятисер нистым выпадает смесь трехсернистого мышьяка (трисульфида мышьяка) и серы:
2AS...4-2H2S=2As--(-2S+-4H* 1 „
2as-S-As s lw J 2AS +5H2S = ASsS3+2S+10H*.
Встречающийся в природе AsjS4 называется реальгаром, а также мышьяковой красной, мышьяковой обманкой, рубиновой серой или сапда-рахом; природный АзгЗз называется аурипигментом (мышьяковая желтая обманка, мышьяковая желтая). Оба соединения получают также синтетически.
Реальгар (чаще приготовляемый искусственно) применяют в живописи и прп изготовлении фейерверков. Он имеет вид красной, стекловидной массы, а будучи размолот,— оранжевого порошка. Оп возгоняется без разложения, и плотность его пара соответствует формуле As4S4. Едкое кали разлагает его па трех серии с тын мвппьяк (переходящий в раствор с образованием тиоарсепита и арсенита) и свободный мышьяк
3As4S4=4As2S3-|-4As.
Трехсерншупый м-ышьяк — красивое^ лимонно-желтое' соединение, которое при 310° плавится в красную жидкоств, а прп 707° кипит, не разлагаясь. Это соединение особенно склонно давать коллоидные растворы.
710
Глава 14
Трехсернистый мышьяк применяют как краску в живописи — в чистом виде под названием королевской желтой, а в неочищенном состоянии., приготовляемом сплавлением трехокиси мышьяка с серой, под названием опермснта. Б последнем веществе содержится пепрореагаровавшая трехокись мышьяка, и оно ядовито в отличие от чистого сульфида.
Трехсернистый мышьяк нерастворим в воде и в кислотах, даже в концентрированной соляпой кислоте. Но он легко растворяется в веществах, обладающих щелочной реакцией, особенно в растворах сульфидов щелочных металлов (в том числе и в сульфиде аммония).
Растворимость в них обусловлена способностью мышьяка образовывать тиосоли As3S3 4 3S" = 2jAsS3]”' (4)
Тиоа рее нит-ион
Этот процесс вполне соответствует образованию яонов арсенита при растворении трехокиси мышьяка в щелочах, но только вследствие ничтожно малой концентрации ионов О" в последнем случае пишут 6ОН' вместо 30'',
АД2О3 ф-: 6ОН' _- 2(AsO3]"f 4. ЗН3О* (5)
Арсснит-ион
Под действием щелочей (в том числе водного раствора аммиака) образуются ионы тиосолей наряду с ионами кислородной соли
As3Ss > 6ОН' = [AsS3]"' + |AsO3]"' 4 ЗН2О, (б)
Таким же образом трехсернистый мышьяк реагирует также и с карбонатами щелочных металлов, включая карбонат аммония
AsKS3 4- ЗСО3" = [AsS3I"' 4 IAsOsJ'" 4- ЗСО2. (7)
Если на трехсернистый мышьяк подействовать желтым сернистым аммонием, то растворенная в ном сера окислит арсенит до тиоарсената, так же как в случае сульфида олова; ср. стр. 581.
As2S3 > 3S" 4- 2S = 2(AsSJ'". (8)
Тцозрсенат-ион
Тиоарсеналил получаются также и при растворении пятисернистого мышьяка в сернистом аммонии, при растворении As3S5 в щелочах, водном растворе аммиака или в растворе карбоната аммония тиоарссчщт получается в смеси с арсенатами или же с оксптйоарсеш|тами. Попы тиоарсеиата, по-видимому, в растворе присутствуют в виде диакво-иопов [А=8.;(ОП3)3]“' (см. стр. 581). При подкислении раствора тиоарсепита или тио арсената вновь выпадает Азг83 или As2S5, так как при добавлении ионов Н* концентрация ионов S" становится исчезающе малой вследствие образования очень мало Диссоциированного H3S. При этом реакция должна протекать в обратном направо Кении пз-за сдвига равновесия влево. Кислоты, соответствующие тиоарсепитам и тиоарсенатам, в свободном состоянии неустойчивы. Однако удалось получить их эфиры (Klement, 1935), папример фениловый эфир тиомыщьяковистой кислоты As(S C6H5)3 (бесцветные иглы, т. пл. 95°); л-толиловый эфир тиомыщьяковистой кислоты As(S-C6H4-CH3)3 (светло-желтые иглы, т. пл. 76°); n-толиловый эфир тио мышьяковой кислоты S—As(S C6H4-CH3)3 (желтые иглы, т. нл. 74°),
4. Мышьяковистый_водород и арсениды
Мышьяковистый водород арсин (моноарсин) AsH3 образуется при действии водорода в момент выделения на растворимые соединения мышьяка, например
[As2O3+6Zn-|-6HsSO1=2AsH34-6ZRSOil4-3H2O.
Для получения больших количеств арсина разлагают арсениды, например арсенид цинка, разбавленной серной кислотой
Zn3As2-)-3H2SO4=2AsH3-|- 3ZnS.O4.
Пятая группа, периодической системы
711
Мышьяковистый водород — бесцветный газ с неприятным чесночным аалах ом. Он чрезвычайно ядовит', вдыхание небольшого количества смертельно. Газ, плотность которого (воздух=1) равна 2,70, что соответствует формуле AsH3, сжижается при —55° и затвердевает при —114°. Подожженный, он сгорает бледно-синим пламенем в трехокись мышьяка и воду. Если охладить пламя, введя в него холодную фарфоровую чашку, то сгорает1 только водород, а мышьяк выделяется в виде черного или бурого пятна (проба Марша). Если мышьяковистый водород пропускать через стеклянную трубку, нагретую до красного каления,то он распадается в соответствии с уравнением
AsH3«=As-yS/BH2-|-44,2 ккал.
Мышьяк осаждается тотчас же за зоной нагрева в виде черного зеркала с металлическим блеском. Однако при смачивании зеркала каплей раствора гипохлорита натрия или смеси перекиси водорода с едким натром оно растворяется с образованием арсената натрия.
Мышьяковистый водород — сильный восстановитель. Из раствора нитрата серебра он осаждает металлическое серебро
AsHs-|-()AgNO3-|-3H2O = GAg+As(OH)3+6HNO3.
Б то же время с твердым нитратом серебра реакция протекает с образованием желтого двойного соединения Ag3As-3AgNO3. Этими реакциями пользуются при определении мышьяка ио Гутцейту.
Испыталяе на мышьяк по Гутцейту производят следующим образом: подлежащее испытанию вещество помещают в пробирку, куда затем приливают разбавленной серной кислоты и бросают крупинку цинка, не содержащего мышьяка. Поело удаления вагой со стенок пробирки капель жидкости пробирку покрывают фильтровальной бумагой и па нее кладут кристалл нитрата серебра. Присутствие мышьяка обнаруживается по пожелтению нитрата серебра или (при увлажнении нитрата) по его почернению. Следует иметь в виду, что подобным же образом реагируют фосфористый водород, а также сурьмянистый водород.
Диарсшт. По данным Наста (Hast, 1948), открывшего диарсин Аз3Н4, он^еще менее устойчив, чем дифоефнн Р2Н4. Уже при —100° As2H4 распадается на АзН3 и красный твердый мъпиъяксеистый водород (AsH JK. Образование последнего наблюдали уже в 1924 г. Мозер и Брукл (Moser, Brukl) при окислении AsH3 солями Sn(IV).
J Органические арсины. Взаимодействием треххлористого'мышьяка"'с дпалвилцлп-ком получаются органические арсины, т. е, алкилпроизводпые мышьяковистого водорода, например триметиларсин As(CH3)3
2AsCl34-3Zn(CH3)-2=2As(CH3)3-^32nC)3.
Это — ядовитые, отвратительно пахнущие жидкости, имеющие характер силько ненасыщенных соединений. Кислород^ сера и галогены окисляют их до соединении типа (CH3)3AsO, (CH3)3AsS, (CH3)3AsC12. С алкилнодидами они соединяются с образованием иодидов тетрамкилареония [R4As)I, которые переводятся влажной окисью серебра в гидроокиси шетраалкиларсои1Ч1[В4Аз](ОН)— кристаллические, весьма гигроСКОПИ-ческие вещества с сильно основным характером. С кислотами они образуют соли тала [R4As]X, к которым принадлежат и указанные выше иодиды тетра алкил арсония.
При нагревании трех окиси мышьяка с ацетатом калия перегоняется жидкость с отвратительным запахом; «дымящая мышьяковистая жидкость» Каде. Главная составная часть ее — окись какодила [(CH3)2As j2O. При действии на иее соляной кислоты получается хлор ид какодила (CH^AsCl, нз которого в свою очередь можно получить цианид какодила (Cns)3AsCN, сульфид какодила [(CH3)2As]2S и т. д. Все эти чрезвычайно реакционноспособные соедяпепия являются производными радикала какодила (CH3)aAs—. Название эта cm получил вследствие очень неприятного запаха, Свойственного его производным (иахшбце—ДУрно пахнущий). Свободный какодил (CH3)aAs—As(CH3)2 представляет собой жидкость (также с отвратительным запахом), жипящую при 17Сг и воспламеняющуюся па воздухе; это соединение можно рассма
712
Глава 14
тривать как ал килпрол вводное диарсина H2As—AsH3, соответствующего гидразину H3N — NH, или дифосфипу НгР—РН2.
Арсениды. Соединения мышьяка с металлами называются арсенидами. Их получают в большинстве случаев сплавлением компонентов, причем реакция часто протекает со значительным выделением тепла. Некоторые арсениды осаждаются из растворов солей металлов мышьяковистым водо-ёодом, например арсенид меди CusAs2 (относительно CuaAs3 см. ниже), оединения мышьяка со щелочными и щелочноземельными металлами, а также некоторые другие, например арсенид цинка Zn3As2, разлагаются водой или кислотами с образованием мышьяковистого водорода, и их можно рассматривать как его производные. Другие арсениды по своему составу и свойствам относятся к классу интерметаллических соединений. Они большей частью очень устойчивы по отношению к кислотам- К арсенидам последнего типа относятся арсениды, встречающиеся в природе (см. стр. 700).
Аналитические сведения. Как переходный элемент между неметаллами и металлами V группы мышьяк обладает характерными признаками тех и других. Это отражается и на его аналитической характеристике. Его находят как при испытании анализируемого вещества на кислоты, так и в ходе разделения на катионы. С нитратом серебра соединения мышьяка образуют желтый осадок арсенита или шоколадно-коричневый осадок арсената. Сероводород осаждает мышьяк в виде лимонно-желтого сульфида; при этом полное осаждение пятивалентного мышьяка возможно из очень сильно кислого раствора. Сульфид мышьяка легко растворяется в сернистом аммонии, а также в отличие от сульфидов сурьмы и олова — в карбонате аммония, но пе растворяется в концентрированной соляной кислоте. !
Для идентификации мышьяка может служить реакция Беттендорфа (см. стр. 704).
Для количественною определения мышьяк осаждают (если он трехва-лентный) большей частью в виде сульфида As2S3, являющегося его весовой формой, Если же мышьяк пятивалентный, то лучше всего его осаждать из аммиачного раствора с магнезиальной смесью* в виде магний-аммонийарсената (NH4)MgAsO4-6H2O и переводить затем для взвешивания это соединение путем прокаливания в пироарсенат магния Mg3As,O7. Трехвалсптный мышьяк можно определять Tait же и титрованием иодом в растворе бикарбоната калия (см. стр. 704).
Б судебной медицине для открытия мыщвяка служит обычно проба. Марша. При умелом выполнении ею можно обнаружить и определить самые ничтожные количества мышьяка**. Для быстрого испытания торговых кислот на мышьяк применяется способ Гутцейта (см. стр. 711). Кроме того, можно также пользоваться для открытия мышьяка способностью меди выделять мышьякиз солянокислых растворов мышьяковистого ангидрида — в форме арсенида меди Cu5Asa (проба Рейна).
* Магнезиальной смесью называют смесь водных растворов хлорида магнии,, хлорида аммония и аммиака.
** При помощн предложенной Лонеманом методики проведепия пробы Марша можно с уверенностью определять 1/10у мышьяка. Столь малые количества мышьяка даже количественно могут определяться с достаточной точностью (1%),если выделившийся в виде зеркала мышьяк перевести в раствор и кондуктометрически оттитровать-в виде арсепита по методу Лидера [Aug. chem., 48, 267, 1935] сильно разбавленным! иодным раствором.
Пятая группа периодической системы
713
СУРЬМА (STIBJL.VJ) Sb
Распространение в природе. Сурьма встречается в природе главным образом в виде трисульфида— трехсернистой сурьмы SbgS3, серой сурьмяной руды. Продув том разложения ее является ЗЬ2О3— белая, сурьмяная руда, сурьмяный цвет. Изредка сурьма встречается и в самородном состоянии, иногда в изоморфной смеси с мышьяком (аллемонтит). Кроме того, подобно мышьяку, она часто содержится в свинцовых, медпых и серебряных рудах.
В качестве примеров, встречающихся в природе соединений сурьмы с металлами, можно указать и а брейтгауптит NiSb (изоморфный вюртциту ZnS), ульманнит NiSbS (изоморфный пириту FeS,)n дискразит Ag2Sb (изоморфный медпому блеску Cu2S). В природе распространены многочисленные двойные сульфиды или тиосоли сурьмы, к которым принадлежат прежде всего гексагональная темная красная руда (сурьмяная или сурьмяиосеребряная обманка, пираргирит) Ag3SbS3 и моноклинная такого же состава огненная обманка', далее, содержащие сурьму блеклые руды, например черная руда (темная блеклая, сурьмяная блеклая руда) 4GuaS-Sb2S3; двойными Сульфидами или тиосолями являются также бурнонит 3(Pb,Cu2)S- Sb2S3; буланжерит 5PbS’2Sb2S3; джемсонит 2PbSf?b2S3; полибазит 9(Ag, Cu)3S- Sb2S3; стефанит (хрупкая стекловидная руда) 5Ag2S-Sb2S3; серебряно-еурьмяцый блеск (миаргирит или гипаргирит) Ag2S-Sb2S3; медно-сурьмяный блеск (халькостибит) Cu2S-Sb2S3; свмуова-сурьмяный блеск (цинкенит) PbSSb2S3.
Исторические сведения. Сурьмяный, блеск был известен еще в древности; его применяли для окраски л черный цвет бровей’ и ресниц. Греки называли его еийрр!, римляне — stibium. Впоследствии ему было дано название (вероятно, заимствованное с арабского) antimonium, которое в дальнейшем стали применять и к самому металлу, получаемому из руды. Живший в XV столетии бенедиктинский монах Василий Валентин подробно описал в своей <еТриумфальной колеснице антимония» приготовление металлической сурьмы, а также бывшие тогда уже в употреблении ее сплавы, например сплав со свинцом для отливки типографского шрифта, и значительное число препаратов сурьмы. Природную сернистую сурьму он назвал сурьмяным стеклом, откуда и возникло название «сурьмяный блеск». В яатрохимический период развития химии препараты сурьмы принадлежали к числу самых распространенных средств лечения, среда них и «вечные» пилюли из металлической сурьмы. В качестве рвотного средства применяли вино, выдержанное некоторое время в чашах из сурьмы. В настоящее время медицина использует сурьмяные препараты только в ограниченном количестве.
Однако недавно Синтезированные органические соединения, содержащие сурьму, приобрели большое значение как специфические средства от некоторых тропических болезней.
Получение. Сурьму получают либо сплавлением сульфида,с железом (метод вытеснения) Sb2S34-3Fe-=2Sb+3FeS, либо обжигом сульфида и восстановлением полученной четырех окиси сурьмы углем (метод обжига — восстановления)
Sb,S3+5O2 = Sh,O4-|-3SO2>
Sb2O4 4- 4G = 2Sb -ф4С'О.
Метод вытеснения годен только для достаточно чистых руд. Однако из руд, содержащих большое количество пустой породы, можно получить предварительно довольно чистый сульфид сурьмы, если руду нагревать до тех пор, пока не расплавится и не стечет относите л г.по легко плавящийся сульфид сурьмы. Полученный таким образом «Зейгерованный» сульфид сурьмы издавна называется «antimoriium erudumv>.
По методу обжига. — восстановления четырехокись сурьмы получают во время обжига только в случае обильного доступа воздуха. Однако обжиг можно производить в условиях умеренного доступа воздуха. Тогда вместо нелетучей четырехокнеи Sb3O4 образуется летучая при нагревании трехокись Sb2O3, которую можно собирать в конденсационные камеры (обжиг с испарением). При восстановлении окиси углем (древесным или коксом) в качество флюса, покрывающего плав, прибавляют, например, глауберову соль или соду, чтобы предотвратить испарение расплавленной сурьмы. Этот
714
Глава И
флюс (плавень) одновременно очищает сурьму от загрязнений (мышьяка, железа, медп переводя их в шлак.
Весьма чистую сурьму (99,6%) можно получить а.гвктролиткческиа* ра<^инирс-ванием. Разработан метод, позволяющий получать электролитическим путем очень чистую сурьму непосредствен но из сульфидных руд.
j Свойства. Сурьма — серебристо-белый с сильным блеском металл,, хрупкий и умеренно твердый (3 по Моосу), легко превращающийся в порошок. Сурьма проводит электрическим ток: удельная электропроводность при 0° составляет 3,76% по сравнению с серебром. Удельный вес сурьмы 6,69. Температура плавления 630,5°, температура кипения 1640°.
Сурьма, как и мышьяк, в парахчетырехатомнаБЬд. В твердом состоянии она встречается в нескольких модификациях. Обычная серая или металлическая сурьма кристаллографически относится к гексагонально-ромбоэдрической системе (см. строение на стр. 628 и сл.). Пропуская кислород в жццяий сурьмянистый водород при —90°, Шток получил желтую сурьму, соответствующую желтой модификации мышьяка и белой модификации фосфора. Желтая сурьма значительно менее устойчива, чем желтый мышьяк. При температуре выше —80° она быстро чернеет — даже в темноте. На солнечном свету почернение наступает гораздо скорее и при еще более низкой температуре. Получающаяся при этом черная сурьма представляет собой, по данным Штока, третью модификацию сурьмы, Она возникает также при действии кислорода или воздуха на жидкий сурьмянистый водород при температурах выше —80°. В чистом виде ее лучше всего получать быстрым охлаждением паров сурьмы. Удельный вес черной сурьмы 5,3, т. е. значительно меньше, чем серой сурьмы. Черная сурьма химически активнее серой. Опа окисляется на воздухе уже при обычной температуре и может даже воспламеняться. При нагревании без доступа воздуха черная модификация превращается в серую.
Четвертая форма — так называемая еврывчатая сурьма — открыта в 1855 г. Горе (Gore). В этой форме сурьма осаждается на катоде при электролизе растворов хлорида, бромида или иодида сурьмы при достаточно высокой плотности тока. Если полученную таким образом сурьму потереть каким-нибудь твердым предметом или быстро нагреть, она превращается в обычную сурьму с выделением тепла (около 20 кал] г) и распылением. Вон (Bohm) доказал рентгенографическим методом, что взрывчатая сурьма аморфна и что сильное разогревание при переходе ее в обычную форму объясняется внезапным выделением тепла кристаллизации.
Колее глубокие исследования структуры аморфной или взрывчатой сурьмы содержатся в э .ле кт poms графических работах (Prins, 1933) и в работах С применением монохроматических рентгеновских лучей (Glocker, 1942). Этими исследованиями и установлено, что'взрывчатая сурьма — стекловидно-аморфное вещество. Каждый атом такой сурьмы окружен четырьмя другими атомами на расстоянии 2,87 А и двумя атомами па расстоянии 3,51 А, в то время какв решетке обычной (серой) сурьмы каждый атом сурьмы окружен тремя атомами на расстоянии 2,87 А и тремя — на расстоянии 3,37 А.
На воздухе при комнатной температуре металлическая сурьма устойчива. Нагретая выше температуры плавления, она на воздухе загорается. При горении образуется главным образом летучая при высокой температуре трехокись Sb3O3, возникающая также и; при действии водяного пара па сурьму при красном калении. С хлором порошкообразная сурьма взаимодействует со вспышкой, образуя при этом пентахлорид—пятихлористую сурьму SbCl5. Так же энергично реагирует она и с другими галогенами. С серой Sb соединяется при сплавлении, так же, как и с фосфором, мышьяком и со многими металлами. При нагревании с нитратами или хлоратами щелочных металлов порошкообразная сурьма со вспышкой образует щелочные соли сурьмяной кислоты.
В азотной кислоте сурьма растворяется с образованием трехокиси или пятиокиси сурьмы в зависимости от ее концентрации. С горячей концентрированной серной кислотой она реагирует с выделением двуокиси
Пятая группа периодической системы 715
серы и с образованием сульфата сурьмы. В соляной кислоте и в разведенной серной кислоте сурьма не растворяется.
Нерастворимость сурьмы в пеокисляющих кислотах соответствует ее месту в электрохимическом ряду напряжений. Ее нормальный потенциал, отнесенный к нормальному водородному электроду, составляет от 4*0,1 до 40,2 в. Таким образом, сурьма более электроположительна, чем мышьяк, и менее электроположительна, чем водород.
Если поместить электрод из сурьмы в какой-либо водный раствор, то оп принимает потенциал, зависящий от концентрации попов водорода в растворе. На этом основано применение сурьмяных электродов при потенциометрических определениях pH, предложенное впервые Улем (Uhl, 1J123) и Кольтгофом (Kolthoff, 1925). Процессы, -определяющие потенциал такого электрода, до сих пор полностью не ясны. Потенциал не всегда воспроизводим непосредственно, а также непостоянен, как правило, во времени. Этим ограничивается возможность применения сурьмяных электродов, которые в других отношениях обладают рядом преимуществ, не нуждаясь, например, в пропускании водорода или в прибавлении посторонних веществ (хингидрона) ц исследуемому раствору.
Применение. Металлическую сурьму применяют главным образом для изготовления сплавов. Она обладает способностью значительно повышать твердость мягких металлов, например олова и свинца. Сплавы свинца с сурьмой называются твердым свинцом. Литеры для печатания отливают из сплава свинца с сурьмой {типографский металл), содержащего 15-^ 25% сурьмы; часто этот сплав содержит также и олово. Сплавы олова с сурьмой (например, 90% олова, 8% сурьмы, 2% меди — британский металл.) служат для изготовления посуды и т. ди другие подобные сплавы применяют для изготовления подшипников (см. стр. 573 и 589).
Соединения сурьмы играли одно время (со времен Парацельса) большую роль в медицине. Некоторые из них применяют и теперь, например пентаеулъфид сурьмы и главным образом рвотный камень (см. стр. 723). В настоящее время сурьма приобретает все большее значение для Приготовления сложных лекарственных синтетических препаратов. Главным потребителем пентасульфида сурьмы является резиновая промышленность (см. стр. 724). Из прочих соединений:сурьмы находят техническое применение трисулъфид сурьмы, трихлорид сурьмы, пентахлорид сурьмы, также антимонат свинца (неаполитанская желтая).
СОЕДИНЕНИЯ СУРЬМЫ
Сурьма в своих соединениях почти исключительно трех- п пятивалентна. Соединения пятивалентной сурьмы проявляют большую склонность к переходу в кислом растворе в соединения трехвалеятной сурьмы и действуют поэтому как окислители! они, например, выделяют иод из растворов иодндов.
Четырех ед лепт пой (электроположительно четырехвалептной) сурьма является в тетр ах л ори,не п его пронзводных-комплексных солях, соответствующих гексахлоро-станнату Мц[ЯпС;.161 и год с ах лор оплатил ату Ml[PtCl6]. Тетрахлорид) сурьмы и его производные отличаются чрезвычайно интенсивной окраской, тогда как соединения трех- и пятивалентной сурьмм с сильно электроотрицательными элементами (за исключением сульфидов) бесцветны или окрашены очень слабо.
1. Окислы и гидроокиси
Производным трехвалентной сурьмы является ее трехокисъ SbaOff или Sb40a, а производным пятивалентной — пятиокись Sb2Oa. Соединением обоих окислов является четырехокисъ сурьмы ЗЬ2О4.
71G
Глава It
Соответствующая трехокиси сурьмы гидроокись Sb(OH)3 Имеет отчетливо выраженный амфотерный характер. Она растворима в кислотах с образованием солей суръмыШ J) SbX3. Чаще, однако, образуются не нормальные соля типа SbX3, а основные — преимущественно соли антимонила. SbOX. В щелочах трехокись сурьмы растворима с образованием антимонитов [антимонат(Ш)] MHSbOsl..
Соли антимонила и антимониты могут производиться и от гидроокиси с меньшим содержанием воды, например SbO(OH). Эту гидроокись (в зависимости от того, какой тип диссоциации имеется в виду) можно называть метагидроокисыо сурьми или сурьмянистой кислотой
SbO(OH) [SbOp + Oir
Sb(OH)3—Н2О = SbO(OH);
Орто гидро-
окись
сурьмы
Метаги дро - А нтимо нпл -
окись ион
сурьмы
зьоон [Sbo2]'4-H-
Метасурьмя- Антимонат (III)-нистая кислота мок
Представляется сомнительным, существует ли гидроокись трехвалептной сурьмы пе только в водном растворе, но и в свободном состоянии. При попытках выделить ее образуется гель с переменным содержанием воды. При старении, а также находясь в контакте с водой гель переходит в кристаллическую Sb2O3. Если гидроокись трех-валентиой сурьмы вообще и существует в твердом состоянии, то она должна быть чрезвычайно неустойчивой, подобно кремневой кислоте.
Гидроокиси, производные пятиокиси сурьмы, имеют ясно выраженный кислотный характер. Они называются сурьмяными кислотами, а их соли — антимонатами(У).
Антимонаты, содержащие кристаллизационную воду, производятся от гексагидро-ксосурьмяной кислоты Н [Sb(OH)e| (ер. стр. 717). Сама кисло га в свободном состоянии не получена, так как иитты' [S.IxOli),;]' уже в водном растворе, как показал Лидер, конденсируются н триант к-ионы [hbjOji Jкак только возрастает концентрация ионов 11 , Jijin ji.Tiепшем ni.i.’jiaCTiiHiiJi концентрации ионов Н’ образуются титраон-тимона.т-^юи.ы ‘:
Окислы и оксигидраты сурьмы в отличие от соответствующих соеди-) пений мышьяка очень плохо растворимы в воде.
Трехокись сурьмы, окись трехвалентноп сурьмы Sb2O3 или Sb40e, образуется при нагревании металлической сурьмы при доступе воздуха, выше температуры плавления в наклонно установленном тигле. При этом она возгоняется и на более холодных частях тигля оседает в виде кристалликов правильной формы (сурьмяный цвет). Для ее получения лучше всего-вести гидролиз ал&аротова порошка Sb4O5Cl2 (кипячение его с раствором карбоната натрия)
Sb4O5Cla+Na2CO3 = Sb4OQ4-2NaC14-CO2.
Получаемая таким образом трехокись сурьмы представляет собой белый, нерастворимый в воде порошок с удельным весом 5,2—5,3 и температурой плавления 656°. Она очень летуча; при нагревании окрашивается в желтый цвет, по охлаждения вновь становится белой. Плотность пара отвечает формуле Sb4Oe.
В кристаллическом состоянии трехокись сурьмы, так же как итрехокись мышьяка, / существует в двух формах; та и другая встречаются в природе. Кристаллы сенармонти
Пятая группа периодической. системы
717
та представляют собой октаэдры кубической системы (уд. вес 5,25), изоморфные арсенолиту. Другая форма — ромбическая (уд. вес 5,7)— валентинит (белый сурьмяный блеск, сурьмяпый цвет).
Ниже 570° стабильной модификацией является кубическая, выше 570° — ромбическая (теплота превращения 3,24 ккал/молъ).
На основании данных рентгеновского анализа можно заключить, что кристаллическая решетка сенармонтита состоит из молекул Sb4O6, центры тяжести которых образуют решетку типа алмаза (см. стр. 463) е данной ребра куба а = 11,14 А. Расстояние между центрами тяжести молекул d = 4,83 А.. Каждая молекула Sb4Oe состоит из кислородного октаэдра, в который «вставлен» тетраэдр, образованный атомами сурьмы. Расстояние О+—> Sb — 2,22 А. Подобную же структуру имеет арсенолит (а = = 11,08; d = 4,78, 0<-->Л$ = 2,01 А).
В разбавленных серной и азотной кислотах трехокись сурьмы нерастворима. Однако опа растворяется в соляной кислоте, а также в растворах некоторых органических кислот, например в винной кислюте. В щелочах SbaO3 растворима с образованием антимонитов [антимонатов(111)]. При прокаливании на воздухе трехокись сурьмы взаимодействует с кислородом с образованием четырех окиси SbaOi. Однако она легко восстанавливается до металла, например, при нагревании в токе водорода, а также при прокаливании с углем или цианистым калием.
Гидрат, трехокиси] сурьмы и антимониты [антимонаты(III)]. Если раствор рвотного камня (см. стр. 723) при пониженной температуре (0°) обработать разбавленной соляной или серной кислотой, то образуется объемистый белый осадок, который обладает свойствами геля и содержит переменное количество воды. Осторожным высушиванием содержание воды в осадке можно довести до такого, которое приблизительно соответствует гидроокиси трсхвалеитной сурьмы Sb(OH)3; однако остается неясным, содержится ли Sb(OH)3 в осадке как индивидуальное соединение. Если вести разложение при более высокой температуре, то образуется осадок, который в воздушно-сухом состоянии содержит меньше воды, чем это соответствует формуле гидроокиси. Гидролиз SbCl3 при температуре выше 40°непосредственно ведет к образованию кристаллического безводного Sb2O3. Гель гидрата трехокиси сурьмы с большой активной поверхностью даже под водой постепенно переходит в кристаллический окисел. Трехокись сурьмы растворяется в кислотах с образованием солей сурьмы(Ш) (см. стр. 723). Если трехокись сурьмы обработать щелочью, то образуются антимониты [антимонаты(111)], т. е. соли метасурь мни истой кислоты, например
Sb(OH)34-NaOH=NaSbO34-2H2O.
Натриевая соль NaSbO2-3H2O, кристаллизующаяся в октаэдрах, плохо растворяется в воде в противоположность очень хорошо растворимой калиевой соли. Раствор антимонита является восстановителем, например, он осаждает металлическое серебро из аммиачного раствора нитрата серебра.
Пятиокиеь сурьмы SbsOa в гидратированном состоянии образуется при окислении сурьмы концентрированной азотной кислотой. Высушенный продукт реакции — желтоватый порошок с удельным весом около 3,8; в воде растворим Очень плохо, но окрашивает влажную синюю лакмусовую бумажку в красный цвет. При нагревании еще до полного удаления воды начинается отщепление кислорода.
По Вестгрену (Westgren, 1937), при нагревании примерно до 800° образуется вначале соединенно SbniSI>2 О6(ОН) (изотипное В1Та,ОйГ). Только после длительного
718
Глава 14
нагревания это соединение переходит в Sb2O^. Область существования последе^ лежит, по данным Симона (Simon, 1927), в интервале 780—920° (при давлении ки!хг> рода 10 лм1 рт. ст).
Сурьмяная кислота и аптимопаты(У). Обычная простая сурьмяная кислота [моносурьмяная(У)кислота] известна только в растворе. является, как это следует из состава и свойств ее солей (см. ниже), гекса-гидроксокислотой HISb(OH)J.
Раньше сурьмяпой кислотой называли продукт осаждения с постоянно изменяющимся содержанием воды, образующийся, например, при гидролизе SbCl5 (дия гидролиза применяют воду, насыщенную хлором, что предотвращает восстановление пятивалентной сурьмы). Поскольку все-таки природа этого соединения еще неизвестна, то еГо лучше называть гидратом пятиокиси сурьмы. Это белый порошок, растворимый как в соляной кислоте, так и в растворе КОН, В воде он практически не растворяется, но, однако, в значительной мере переходит в коллоидный раствор, если обрабатывать, св еже осажденный препарат водой.
Так Называемая «осажденная сурьмяная кислота», 1г. е. гидрат пятиокиси. сурьмы, имеет характер геля. Поэтому содержание в ней воды изменяется в зависимости от условий ее получения, а йОсле обезвоживания обычно не получается веществ с стехио-метрически простым составом. Только при высокой температуре, цак показал Симон (Simon, 1927), получается определенное соединение состава ЗЗЬ2О5‘бН3(\Т' е. H5Sb3Of(): (трисурьмяная кислота, см. стр. 716). Образование этого гидрата происходит, однако, гораздо труднее и медленнее, чем образование соответствующего соединения мышьяка. Сурьмяная кислота папоминает кислоты олова (элемента, расположенного рядом с сурьмой в периодической системе) по своей способности существовать в двух формах, соответствующих а- и ^-оловянной кислоте. Как и у оловянной кислоты, здесь также возможны любые промежуточные формы. Обычная сурьмяная кислота (гексагидро-ксосурьмяпая кислота) является кислотой одноосновной. При комнатной температуре-ее константа диссоциации составляет 4,-10~5. Первая константа диссоциации триеурь-мяной кислоты, равная 5,3-10 *4, является, следовательно, в соответствии с общим правилом более высокой, чем константа диссоциации мопокислбты.
Из солей сурьмяной (V)кислети в качестве реактива на натрин применяют антимонат( V ) калия, (гексагидрокеоантимонат калия) K[Sb(OH)e]r представляющий собой зернистый белый порошок, довольно трудно растворимый в холодной воде и несколько лучше в теплой. Так как растворитель антимоната натрия Na[Sb(OH)e| еще значительно меньше, то при действии раствора соли калия на раствор соли натрия образуется осадок.
Для при гот о влепи я антимоната палия сплавляют пятиокись сурьмы с избытком гидроокиси калия; затем сплав растворяют в небольшом количестве воды и оставляют кристаллизоваться. Осаждающуюся соль очищают непродолжительным нагреванием с несколько раз сменяемой водой.
Гидр оксо антимонат калия прежде почти всегда рассматривали как кислый пиро-антимонат, содержащий кристаллизационную воду K2H3Sb2O,-6Н2О. Однако уже в 1921 г, Томула (Tonnila) на основании данных по измерению электропроводности высказал предположение, что это не кислая соль четырехосяовной, а нейтральная соль одноосновной кислоты. Это мнение было полностью подтверждено в 1926 г. Лидером (Jander) н Брулем (Briill) на основании измерения понижения температуры замерзания и скорости диффузии, а также па основании' определения влияния на растворимость соответствующей натриевой спаи присутствия электролита с одноименным ионом. Хам-мет (Hammet, 1929), по-видимому, первый указал, что образующаяся при осаждений ив водного раствора натриевая соль имеет состав, точно соответствующий формуле Na[Sb(OH)e], Наконец, в 1933 г. Полинг показал, что исходя из соотношений ионных радиусов для рассматриваемой соли может идти речь лишь о формуле гидроксосояи MI[Sb(OH)e]. Ронтгеяоструктурпые определения (Beintema, 1935, Weatgren, 1938) подтвердили формулу Мг(8Ь(ОН)в]. Кроме солей моиосурьмяной(У)кислоты (гекса-гядроксосурьмяпой), известны соли полисурьмяных(У)кислот, например три- и тетра-антимонаты(У) (ср. также стр. 716).^
Безводные антимонаты (V), приготовленные сухим путем, т. е. сплавлением окие-лов, по своему составу являются большей частью солями кислот сурьмы, аналогичных
Пятая группа периодической системы
719
О
дислотам фосфора: H3SbO4 (ортосурьмяпая кислота), H4Sb2O7 (пир о сурьмяная кислота) и HSbOj (метасурьмяная кислота). Как уже было отмечено, существование этих кислот не доказано. Но даже и для солей в тех случаях, когда их точное строение могло быть определено рентгенографически, было установлено, что в структурном -отношении они не являются производными названных кислот. По свойствам эти соли близкй к мета- и полисиликатам. Например, часто образующиеся антимонаты двухвалентных металлов, такие, как Са^8Ь2О7 или Pb2Sb2O7, содержат не конечные радикалы [Sb2O7J1-, а группы [SbjOjP*, образующие, как и группы [AlSi3OB]- в алюмосиликатах типа полевого шпата, пространственную сетку, простирающуюся на весь кристалл. Одиако структура этих солей еще более сложная, чем в алюмосиликатах, так как сурьма в соответствии с большим ионным радиусом имеет более высокое координационное число, чем кремний. Поэтому в этих солях каждый нон 8Ъз+ окружен не четырьмя, как ион Si4+ в силикатах, но по Крайней мере шестью и максимально даже еосыиыо ионами О2-.
Метаантимонат натрия NaSbO3 образует решетку, в которой сурьма, как н в гидроксоантимонатах, обладает координационным числом 6. Эта соль кристаллизуется, как установили Вестгрен и Шревелиус (Westgren, Schrewelius, 1938), по типу ильменита (ср. т. II). Наряду с этим NaSbO3, так же как и AgSbO3, встречается и в кубической модификации.
Антилюнат.(У)свинца, получаемый при сплавлепии нитрата свинца с рвотным намнем и поваренной солью, применяют в качестве краски под названием неаполит-анской желтой, особенно в керамике.
Кислотами антимонаты разлагаются с образованием (в зависимости от условий опыта) трисурьмяной ^кислоты или геля пятиокиси сурьмы.
Четырехокись сурьмы. Оба окисла сурьмы, как трехокись, так и безводная пятиокись, при нагревании на воздухе до 800—900° переходят в четырехокись Sb fi^. Из гидрата пятиокиси иля из образующегося при его нагревании соединения Sb3O6(OH) (см. стр. 717) четырехокись в чистом состоянии получается только в результате длительного прокаливания.
Четырехокись сурьмы образует белый, едва растворимый в воде порошок с удельным весом 6,59, при нагревании делающийся желтым. При очень сильном нагревании происходит отщепление кислорода с образованием трехокиси. При прокаливании с углем или цианистым калием четырехокись легко восстанавливается до металла.
Четырехокись сурьмы, согласно рентгеноструктурным исследованиям (DihLstrom, 1938), соответствует двойному окислу Sb2O3 и Sb2O6 или ортоантимонату трехвалептной сурьмы SbIIISbvO4. Это соединение изотипно SbTaO4. В кристаллических решетках обоих соединений не существует в качестве структурных групп радикалов SbO4 или ТаО4, по в них имеются пространственные сетки, простирающиеся на весь кристалл, образованные чередующимися октаэдрами 8ЬШОВ и SbYo» (соответственно TavOl}). Вокруг ионов Sb5+ и со ответствен ио' TaG + ионы О2* образуют почти правильный октаэдр, в то время как вокруг ионов Sha* ионы О2* образуют сильно искаженный октаэдр.
При сплавлепии четырехокиси сурьмы и щелочи (или щелочного'карбопата) можно получить соодипепия, которым ранее приписывали формулу M(sb2O6. Подобные соединения встречаются также и в природе: ромеит CaSb2Os, аммиояит HgSb2O5. Эти соединения, по данным Пата (Natta, 1933), имеют точно такуюйке структуру,"как и ппроантидюиагы тина AfPsbj Согласно Цедлитцу (Zedlitz, 1931), они, напротив, иаотипны пиро хлору (ем. т. II). В соответствии с этим ромеиту следовало бы прп писать формулу NaCaSb^O^/OH).
^2. Галогениды! (сурьмы
Галогениды сурьмы (кроме"не'существуК1ще«) в виде простого соединения тетрахлорида SbClJ соответствуют общим формулам SbX3 и SbX5. Их можно получить непосредственным взаимодействием сурьмы с галоге
о
720
Глава 14
ном. но обычно для их получения применяют другие методы. Водой галогениды сурьмы в сильной степени гидролизуются — менее всего фториды.. Более стопки по отношению к воде, чем простые галогениды, их двойные соединения с галогенидами металлов, которые надо рассматривать как комплексные соли. Такие комплексные соли, галоидоантимонаты, [галои-доантимонаты(1 II) и галоидоантимонаты(У )] образуются всеми галогенидами сурьмы. Наибольшая склонность к их образованию отмечается у пента галогенидов.
Трифгорпд сурьмы. Трехфтористая сурьма SbF3 выделяется при упаривании раствора трехокиси сурьмы в плавиковой кислоте в виде бесцветных расплывающихся кристаллов с уд. весом: 4,4, растворимых в воде с кислой реакцией. Гидролизуются они в значительно меныпей степени, чем другие галогениды сурьмы. Трехфтористая сурьма легко образует двойные соли с фторидами щелочных металлов фтороaiimимокиты (фторо антимонаты(Ш) 1 преимуществ он во типа MISbF4 и MlSbF5, а также образует двойные соли с хлоридами и сульфатами щелочных металлов. Двойную соль с сульфатом аммония SbFy (NH4)2SO4 [сульфатотрифгороантимонат (III) аммоцня] применяют в качестве протравы при крашении тканей под названием «сурьмяной солш.
Пептафгорид сурьмы — пятифтористая сурьма SbF5, получающаяся в чистом состоянии (по методу Руффа) при продолжительном действии безводного фтористого водорода на пептахлорид сурьмы, представляет собой бесцветную маслянистую жидкость с уд. весом 2,99 и т. кип. 150°. 1
В парообразном состоянии SbF5 сильно полимеризован (в противоположность AsF5, испаряющемуся в виде отдельных молекул). С водой SbF6 реагирует с шипением. С фторидами щелочных металлов SbFs образует двойные соединения, преимущественно тина MI[SbF6] [гексафтороантимонаты (V)]. Кроме того, SbF6 легко образует продукты присоединения, например с SbF3, Р«О6 и Ь. Пятифтористая сурьма растворяется в жидком SO2 с выделением тепла. Она образует с SO2 соединенно SbFf,-SO, (бесцветные кристаллы, т. пл. 57°); cNO, — бесцветное соединение SbFs-NO21 которое при 150° вновь распадается на SbFs и NOK. SbFe может также присоединять S, So и То с образованием Соединений (SbFshS (белое), (SbFs)2Se (желтое) и (SbF5)2Te (желто-v коричневое) (Aynsley, Peacock, Robinson, 1951).
Трихлорид сурьмы — треххлористая сурьма SbGl3 — бесцветная, мягкая, дымящая на воздухе масса tButyr am antimonii, сурьмяное масло) с удельным весом 3,06; температура плавления 73,4й; температура кипения 223". Лучше всего ЗЬС1Я получается при растворении тонкоизмельчеппой серой сурьмяной руды в горячей концентрированной соляной кислоте Sb2S3 -HGН Cl —2SbGl3 + ЗН aS.
Треххлористую сурьму применяют в медицине в качестве раздражающего средства. Она служит также для протравы металлов, например при воронении ружейных стволов.
Треххлористая сурьма — довольно реакционноспособное вещество; например, при взаимодействии с концентрированной серной кислотой она образует сульфат сурьмы и хлористый водород
ЗЙЬСЦ + 3H3SO4 = Sb2(SO4)3 -ф 6ИС1.
Польше всего SbCl3 склонна к образованию продуктов присоединения. С хлоридами щелочных металлов т ре хх.тто ристая сурьма образует двойные соли — хлороанти-монаты(Ш'), большей частью типа M3[SbCl6], но в ряде случаев и более сложного состава.
Оксихлорид сурьмы. Треххлористая сурьма хорошо растворима в небольшом количестве воды; ири разбавлении же раствора вследствие гидролиза выпадают основные хлориды (оксихлориды), например SbOGl хлорид антимонила и Sb..(Or!Gl, — алгаротов порошок. Последний назван так по имени жившего в XVI в. итальянского врача Алгарота, который
Пятая группа периодической системы
721
стал применять его как лекарственный препарат; в настоящее время его почти не применяют.
Пентахлорид сурьмы — п.чтихлористал сурьма SbCJ6 получается: при обработке хлором треххлористой сурьмы. В чистом состоянии SbCl6 представляет собой бесцветную, часто окрашенную в желтоватый цвет жидкость с удельным весом 2,35, которая при охлаждении затвердевает в кристаллы, плавящиеся при 2,8°. При пониженном давлении SbCls перегоняется без разложения; при атмосферном давлении SbCl5 испаряется при 140и, по уже при стой температуре она начинает терять хлор. При взаимодействии с сероводородом выделяется хлористый водород и образуется сульфохлорид сурьмы SbSClg. Пятихлористая сурьма способна отдавать многим органическим веществам часть своего хлора, переходя в трех-хлористую сурьму. Поэтому ее иногда используют в органической химии в качестве хлорирующего агента.
Пятихлористая сурьма проявляет большую склонность к образованию продуктов присоединения. Особенно хорошо известии многочисленные соединения ее с органическими веществами, содержащими кислород, хлор или азот. SbCl5 соединяется также и со многими хлоридами металлов с образованием при этом преимущественно соединений типа М1 [SbCl6] [гексахлороантимонаты, точнее, гексахлороантимонаты(¥)].
Тетра хлорид сур ьмы (четыре х х.ц о ри стая с у рьма) их лороа чти м онаты( IV). Если к солянокислому раствору треххлориетон сурьмы прибавить эквимолярное количество пятихлористой сурьмы, то получается темно-коричневый раствор в отличие от бесцветных в чистом состоянии или почти бесцветных растворов исходных веществ. То же соединение, по-видимому, образуется в качестве промежуточного продукта при окислении хлором треххлористой сурьмы в пяти хлористую. Предполагают, что при этом образуется тетрахлорид сурьмы ЯЪС14, существующий в равновесии с двумя другими хлоридами сурьмы
SbCl3 -ф- SbCl5 £ 28ЬС14.
SbCl4 но удается изолировать, но при прибавлении хлоридов рубидия и цезия легко подучаются производные от нее комплексные соли Rb2[SbCl0] и CsB[SbClg]. Эти соли кристаллизуются в октаэдрах, окрашенных в густой темно-фиолетовый цвет, и изоморфны сочни щелочных металлов гексахлоро свинцовой Н2(РЬС1Я], гексахлоро-оловяпной H2[SnClrJn гексахлороплатиповой (платипохлориСтОводородной) H2[PtCleJ кислот. В этих соединениях сурьма, подобно свинцу, олову и платине в перечисленных трех кислотах, является электроположительно четырехвалентным элементом. Хотя электроположительно нетырехвалеитпая Сурьма обладает несла рентам электроном, соединения типа M2[SbX0] являются диамагнитными. По мнению Йенсена (Jensen, 1944), это, а также цветность соединений, объясняется взаимодействием между группами |SbXsp_, которое ведет к тому, что электроны соседних атомов Sb4+ могут объединяться в пары, причем атомы галогенов при атом фигурируют в качестве «каналов» для перемещающихся электронов.
Трпбромпд сурьмы — трехбромистая сурьма SbBr3 — бесцветные ромбические кристаллы (уд. вес 4,15; т. пл. 96,6°; т. кип. около 280°) получающиеся со вспышкой при внесении порошкообразной сурьмы в жидкий бром. Поэтому целесообразнее получать ее, действуя на сурьму бромом, растворенным в сероуглероде. В химическом отношении St В г, ведет себя вполне аналогично SbCl3 и, подобно ей, образует двойные соединения с бромидами щелочных металлов, но большей частью значительно более сложного соста пц
Пентабромид сурьмы известен только в виде двойных соединений. Вромоанти~ манаты — точнее, бромолнтимонаты (V)— отвечают преимущественно типу М1 (SbBr6], большей частью о ли содержат воду; полувека также свободная кислота HSbBre-3H2O (гекса бромосуръмяная кислота.).
Войнлапд (Weiitland) получил двойную соль, производную от тетрабромида сурьмы — гексабромоантимонат^ТУ^рубидий Rb2[SbBr0J.
Триподпд сурьмы — трехиодиету ю сурьму 8Ы3 удобнее всего получать, внося порошок сурьмы в раствор иода в сероуглероде. Она кристаллизуется из раствора 46 г. Реии
722
Глава 14
в рубиново-красных гексагональных пластинках, изоморфных трниодидам мыт—-- t и висмута (уд. вес 4,85; т. пл, 171°; т. кип. около 401°). Трохиоднстая сурьма суще?^-и в другой модификации — ромбической, желтого цвета с уд. весом 4,77. С подц^лл щелочных металлов она образует двойные соли иодоант,имвнаты(1П). Sbl3 может соединять избыточный иод, образуя твердый раствор. Последний легко удалслт*^ подходящими растворителями, например эфиром; в соответствии с новайг-т-т-^ исследованиями Sbl5 не существует.
3. Сурьмяные соли
Рассмотренные в предыдущем раэдбле галогениды сурьмы можно счх: тать сурьмяными солями галоидоводородных кислот. Однако они отличаются от типичных солей металлов своей большой летучестью и малой дис социацией в водных растворах, У них преобладает гидролитическое разложение над электролитической диссоциацией.
Соли кислородных кислот известны толькодля трбивалентной сурьмы.. Окисел пятивалентной сурьмы обнаруживает столь слабые основные свойства, что он не может образовать солей с азотной, серной и другими кислотами. Однако и соли трехвалентной сурьмы чрезвычайно склонны к гидролизу. В качестве промежуточных продуктов при этом могут образоваться основные сурьмяные соли. Частично, по-видимому, речь идет об оксисолях трехвалентной сурьмы SbOX (ранее называемых «солями антимонилао: ср. стр. 720). Основные соли сурьмы в воде большей частью нерастворимы, но претерпевают все же постепенно дальнейший гидролиз. Относительно большей устойчивостью отличаются сурьмяные соли некоторых органических кислот, например типа рвотного камня (антимонилтартрат калия), в которых сурьма входит в состав прочного комплекса.
Большинство солей сурьмы способно присоединять соли щелочных металлов с. образованием двойных или более или менее прочных комплексных солей. В качестве примеров таких двойных или комплексных солей можно указан, па сулы0ат<1антилс>н(1ты(111) М1 [Sb(SO4)a] и оксала-тоан/онмоннтыС IП') Мr ISfНС,О4)-31 и M|[Sb(C,O4)3L
Сульфат сурьмы. Sli3(SOj3 получается при растворении сурьмы, трехокиси сурьмы, трасу,аьфид’а сурьмы или алгаротова порошка в горячей концентрированной серной кислоте. Он кристаллизуется в бесцветных шелковисто-блестящих расплывающихся иглах с уд. весом 3,62. С небольшим количеством воды он образует гидрат; большим же количеством воды гидролизуется. Прп обработке сульфата сравнительно разбавленной серной кислотой происходит только частичпый гидролиз с образованием сульфата антимонила (SbO)aSO4. Меньшую склонность к разложению по сравнению с нейтральным сульфатом обнаруживают двойные соли, образующиеся при присоединении к нему сульфатов щелочных металлов М1 [Sb(SO^)^]—сульфатоаптимонаты(Ш).
Нитрат сурьмы. При внесении трехокиси сурыпд в холодную дымящую азотную кислоту it разбавлении затем ее водой выделяются белые, с перламутровым блеском кристаллики основного нитрата сурьмы. Нейтральный нитрат иа водного раствора нельзя получить.
Антимонилтартрат калия (рвотный камень). При охлаждении прозрачного раствора смеси трехокиси сурьмы с кислым виннокислым калием (получающегося при кипячения) кристаллизуется двойная виннокислая соль калия и антимонила K[G1H„O4Sb(OH)i!] SG Н..О, обычно называемая рвотным камнем (Tartarus emeticus).
KHC4H4O6-]-V'2Sb3O3 = K[C4H2O6Sb(0Hs)]-i/2H2O.
Пятая группа периооической. системы
723
Это соединение, описанное впервые Адрианом фон Минзихтом, жившим в первой половине XVII Bi, последователем Парацельса, еще и теперь часто применяют в медицине. Кристаллы рвотного камня бесцветны, легко растворимы в воде п нерастворимы в спирте.
Замечательно, что рвотный камень далека не так склонен к гидролизу, как неорганические Соли сурьми. Уже это указывает иа то, что рвотный камень не является антпмонилтартрптом, т. е. солью винной кислоты и радикала [SbO]+, связанного как катион, как это в большинстве случаев предполагали раньше; наоборот, можно считать, что сурьма тесно связана именно с радикалом винной кислоты. Уже Шиф (Schiff, 1863) высказал предположение, что сурьма в винной кислоте, помимо атома водорода карбоксильной группы, замещает также оба атома водорода в спиртовых гидроксильных группах. Это предположение было Позднее основательно экспериментально подтверждено Рейленом (Reihlen, 1931). Для объяснения кислой реакции винного камня в водном растворе полагают, что с сурьмой комплексно связана молекула воды, которая способна отщеплять ион водорода, так что в водном растворе существует следующее равновесие:
'О=с—О\ -
И—С —О— Sb—он
Н—С—о/
О = С—О'
Рвотный камент, поэтому следует рассматривать как калиевую соль аквоеуръмяпв-еинной кислоты И[CiHaOllSb(OHs)]*. Известен еще ряд других солей акяосурьмяно-винпой кислоты. При нагревании эти соединения прежде всего теряют воду, связанную вне комплекса (количество ее изменяется в зависимости от природы катиона). При более сильном нагревании (для натриевой соли в вакууме уже при комнатной температуре) отщепляется также и вода, находящаяся внутри комплекса. Свободная кислота теряет воду уже при кристаллизации.
4. Сульфиды ц тиосолй сурьмы
Сурьма образует два сульфида; SbES3 и Sbr.S5. Они получаются при сплавленяп серы с сурьмой или же пря осаждении сероводородом из подкисленных растворов солей трех- и пятивалентной сурьмы. Оба сульфида сурьмы способны, подобно соответствующим сульфидам мышьяка, соединяться с ионами S", если последние имеются в избытке, с образованием тиосолей; поэтому Они растворимы в сернистом аммонии:
Sb2Ss-f-3S’ 2(SbS?r; SbaSji+aS" 7» 2[SbS4]m
Тиоаитимонат( I i 1)-ион Тиоавтиио
нат(У}-ион
При прибавлении кислоты сульфиды вновь выпадают в осадок: 2[SbS3]"'+6H‘ = Sb2S3+3H2S; 2[SbSi]"'+6H' = Sb2S5-f-3H2S.
Сульфиды сурьмы в отличие от сульфидов мышьяка растворимы в концентрированной соляной кислоте, но нерастворимы в водном растворе карбоната аммония. В растворах щелочей они растворяются так же, как и сульфиды мышьяка.
Тиоантимопаты в подпои растворе, по-видимому, присутствуют в форме тиоди-аквоионов [SbS4(OHa),]'" илив форме гидро ксотиотиоло-ионов [Sb(OH)2S2(SH)2]'" (ср. стр. 580). Прн обработке сульфидов сурьмы раствором щелочи происходит, по-ви-димому, точно так же образование гидроксотиоло-ионов.
* Соединение'которое иногда неправильно называют «сурьмяно-тривиппой кислотой», является настоящей виннокислой солью сурьмы, и его следует рассматривать как кислый антимонтритартрат.
46=»
724
Глава 14
Так же как и у мышьяка, свободные тиокислоты сурьмы в отличие от .-<гг.ищи
неустойчивы (Klement). Эфиры тиокислот сурьмы все же распадаются значит.-.-„ин легче, чем соответствующие соединения мышьяка; например, в против оползни :«—щ. последним ови немедленно гидролизуются водой.
Трисульфид сурьмы — трехсерцистая сурьма Sb2S3 встречается в ст i роде в виде серой сурьмяной руды (важнейшая руда сурьмы), иначе „ ваемой сурьмяным блеском, антимонитом, стибнитом. Она предстагдг -собой серые иглы с металлическим блебком, с удельным весом 4,6, тг^--ратурой плавления около 550°, теплотой образования 38,3 ккпл/'л.-:'? Нагретая на воздухе, она переходит (при достаточном доступе воздух, в четырехокись (см. стр. 713). Тонкоизмельченная порошкообразная сеу. сурьмяная руда растворяется в теплой концентрированной соляной ед лоте с образованием трихлорида сурьмы (см. стр. 720). Серая сурьмяы..., руда и изготовляемый из нее Antimonium crudum (см. стр, 713) нахе-;,' применение в качестве исходного материала для получения сурьмы п t солей, в пиротехнике, а также для приготовления спичек и рубиновс.. стекла.
Трехсернистая сурьма, осажденная из раствора на холоду или при умеренном нагревании, окрашена в красивый оранжево-красный цве-После высушивания она имеет удельный вес 4,15; оранжево-красныйсуп-т-фид сурьмы аморфен. При нагревании без доступа воздуха он превращаете, в кристаллическую серо-черную модификацию. Теплота превращении, составляет 7,5 ккал/моль (Fricke, 1942).
Двойное соединение трехсернистой сурьмы с трехокисыо сурьмы — так называемая сурьмяная киноварь, помимо применения в медицине (под названием «кврлест’:, находит широкое применение в качестве красной краски. Это соединение образуется, например, при обработке SbCl3 тиосульфатом натрия. В природе оно встречается в вид= вишнево-красных, моноклинных, игольчатых кристаллов и называется красной сурьмяной рудой (пиростибит, сурьмяная обманка).
По данным Долге и Фрике (D onges, Fricke, 1945—1947), сурьмяная киноварь представляет собой Sb2S3, который в зависимости от способа приготовления может содержать переменное количество примесей Sb2O3 и S. Красная окраска по сравнению с красно-ораажевым Sb,O3 обусловливается большим: размером частиц.
Пентасульфид сурьмы. Пятисернистую сурьму Sb,S5, называемую также золотистой серой (Sulfur auratum), в технике получают разложением соли Шлиппе [тиоантимонат(У) натрия] разбавленной серной или соляной кислотой
2Na38bS4+ 3HaSO4 = Sb2S5+3Na2SO4+3H2S.
Золотистая сера Sb2S5 — нерастворимый в воде оранжевого цвета порошок. Ее с давних пор применяют в медицине, особенно в ветеринарии, но в первую очередь Sh2S5 используют для вулканизации каучука. Вулканизованные сю резиновые изделия имеют характерный красный цвет.
Тиоантимонат(У) натрия Na3SbS4, называемый обычно солью Шлиппе, получают, внося серу и порошок серой сурьмяной руды в кипящий раствор едкого натра или в смесь известкового молока с содой. Раствор едкого , натра растворяет серу частично с образованием сульфида натрия NaaS, который затем реагирует следующим образом;
3Na2S-|-2S+Sb2S3=2Na3SbS4.
Из охлажденного раствора тиоантимонат(У) натрия кристаллизуется с девятью молекулами воды Na3SbS4-9H2O в виде светло-желтых, легко выветривающихся на воз,духе тетраядро». У рольный нес 1,86.
Пятая группа периодической системы Т25
5. Сурьмянистый водород
Сурьмянистый водород — стибин — SbH3 получается, аналогично мышьяковистому водороду, при действии водорода в момент выделения на растворимые соединения сурьмы.
Для получения его в больших количествах лучше всего использовать метод Штока, заключающийся в следующем: 1 вес. ч. Sb нагревают с 2 вес. ч. Mg и полученный таким образом порошкообразный сплав вносят в холодную разбавленную соляную кислоту. Для очистки от примеси водорода стибин конденсируют, используя жидкий воздух.
Сурьмянистый водород — бесцветный, плохо пахнущий газ, почти не уступающий по ядовитости мышьяковистому водороду; он немного растворим в воде (Va об. в 1 об, воды), лучше в сцирте (15 об. в 1 об.), еще лучше в сероуглероде (250 об. в 1 об. при 0°). 1 л газа весит (при 0° и 760 .431 рт ст) 5,685 г. Сжиженный газ кипит при —18° и затвердевает при —91,5°.
Будучи зажжена, смесь сурьмянистого водорода и водорода горит бледно-зеленым пламенем, оставляющим на холодной фарфоровой чашке черное пятно металлической сурьмы. В отлично от получаемого подобным же образом мышьякового пятна пятно сурьмы не исчезает от смачивания его раствором гипохлорита натрия или смесью едкого натра с перекисью водорода. Таким же свойством отличается и зеркало сурьмы, которое получается при пропускании через раскаленную стеклянную трубку водорода, содержащего сурьмянистый водород.
С нитратом серебра сурьмянистый водород реагирует, подобно мышьяковистому водороду, С сухим нитратом серебра он образует желт о-коричневое двойное соединение которое под действием влаги разлагается с выделением черного
сурьмянистого серебра Ag3Sb.
Подожженный чистый сурьмянистый водород разлагается со взрывом^ при этом выделяется 34 ккал/моль.
Органическое стиблпы. Аналогично производным фосфористого и мышьяковистого водорода — органическим фосфинам и арсинам — От сурьмянистого водорода (сти-бпна) производятся органические стибины SbB3 (В — алкил), Например, при взаимодействии SbC!3 с диметялци пком кол у чается триметидетибин £Ь(СН3)з — самовоспламеняющаяся жидкость 2SbCl3 + 3Zn(CH3)2=3ZnCl2 -|-2Sb(CH3)3.
С соляной кислотой он реагирует с выделением водорода, подобно сильно электроположительному металлу 8Ь(СН3)Й -f- 2НС1 = Sb(CH3)3Ci24 H2. С йодистым метилом три метил стцбип образует соединение, обладающее типичными свойствами Соли, в водном растворе полностью распадающееся па ионы |Sb(CH3)4]’ и Г — йодид тетра-метилетибония Sb(CH3)s СН31 = [Sb(CH3)4]I.
При взаимодействии с влажной окисью серебра из пего можно получить гидро-окисг, т^траметилстибаиия (Sb (СН3)4]ОН, обладающую свойствами сильного основания. Совершенно аналогичны свойства остальных алкилстмбинов,
6. Соединения сурьмы с металлами (антимониды)
Соединения сурьмы с металлами, антимониды, по своим свойствам соответствуют типичным интерметаллическим соединениям. Часто они образованы на счет валентностей, отличающихся от нормальных. Это прежде всего справедливо для соединений сурьмы с металлами побочных подгрупп периодической системы, В соедипшшях с сильно электроположительны мп металлами обеих первых главных подгрупп сурьма, напротив, предпочитает, по-видимому, проявлять валентность такую же, как и по отношению к водороду, т. е. равную 3 (ср. табл. 98, стр. 630). С оловом сурьма образует недальто-нидиое соединение SaSb, со свинцом сурьма не соединяется, образуя лишь твердые растворы только в узко ограниченных областях состава, тогда как в расплавленном состоянии сурьма смешивается с РЬ во всех отношениях. G никелем сурьма образует четыре соединения: iVi2Sb3, NiSh, Ni3Sb2 и А7ц8Ь. Соедипеине 4iSb встречается в при
726
Глава 14
роде под названием брейтгауптита. С серебром сурьма образует два шгкопгруентно плавящихся недальтонидных соединения. Одно из них Ag3Sb (20—25 ат.% Sb) встречается также и в природе (дискразит).
Аналитические сведения. При систематическом ходе анализа сурьма осаждается сероводородом в виде нерастворимых в разбавленных кислотах оранжевых сульфидов (Sb,S3 nSb2S6), которые растворимы, подобно сульфидам мышьяка и олова, в сернистом аммонии с образованием тиосолей. От сульфидов мышьяка они отличаются, кроме своего цвета, нерастворимостью в растворе карбоната аммония и растворимостью в концентрированной соляной кислоте. Из солянокислого раствора сурьма осаждается металлическим цинком в виде черного порошка.
При обработке окислов сурьмы цинком и соляной кислотой получается сурьмянистый водород; последний образует при тех же условиях, как мышьяковистый водород, металлическое зеркало, причем зеркало сурьмы легко отличить от зеркала мышьяка на основании реакций, указанных на стр. 725, и других реакций.
В пламени паяльной горелки соединения сурьмы образуют белый налет (Sbs03) и хрупкий металлический королек. Из кислых растворов солей сурьмы, как правило, при их разбавлении выпадают белые осадки основных соединений сурьмы.
Для количественного определения сурьму осаждают обычно в виде сульфида, который взвешивают после высушивания в струе двуокиси углерода или после окисления его в четырехокись сурьмы.
ВИСМУТ (BISMUTUM) Bi
Распространение в природе. Висмут встречается в природе как в самородном состоянии, гак и в виде соединений. Самые распространенные среди них сульфид Bi2S3 сйсск) и окись Bi2O3 (висмутовая,
охра). В основном висмутовые руды не очень распространены. В Европе встречаются главным образом в Саксонско-Богемских рудных горах. Главнейшие из: месторождения находятся в Южной Америке (Боливия) и в Австралии (Тасмания),
Подобно мышьяку и сурьме, висмут иногда встречается в природе в виде двойных сульфидов, например: сеинцововисмутоеый блеск (галеновисмутит) PbS-Bi2S3; серебря-новисмутоеый блеск (a pre нто висмутит) Ag^S-BljSg (таким же составом обладает пл:енар-гирит) и медное нему товый блеск (эмплектит) Cu2S.Bi2S3; медновисмутовая обманка (виттихенит) 3Cti2S Bi2S3; лилианит 3PbS-Bi2S3 и т, д. Следует упомянуть также О селеновисмутовом блеске В13863 и тетрадимите Bi3Te2S. Содержащей висмут золотой рудой является австралийский мальдонит Au2Bi. Вследствие более основного характера окиси висмута ио сравнению с окислами мышьяка п сурьмы окись висмута встречается в природе и в форме основного карбоната (висмутовый шпат).
Исторические сведения. О висмуте, как о металле, похожем на олово, упоминает впервые Василий Валентин (в XVb.). Но, однако, еще долгое время многое было ыеясио относительно его природы. Только в XVIII в, два химика, последователи теории флогистона, Потт и Бергмаи точнее охарактеризовали висмут как особый элемент с металлическими свойствами. Уже в XVI в, окись висмута Bi2O3 использовали в качестве краски, а основной нитрат ипсмута (испанские белила) — в качестве косметического средства.
Получение. В старину металлический висмут получали из руд, содержащих его в самородном состоянии, нутом выплавки («зейгерования»).
Пятая группа периодической системы .727
В настоящее время его получают'из руд, содержащих окислы, при восстановлении их углем в тиглях или пламенных печах (способ восстановления). Сернистые руды либо предварительно окисляют обжигом, либо сплавляют непосредственно с желез ом для получения висмута (способ вытеснения).
Способ восстановления: В12Оз~(-ЗС. = 2Bi-{-3C.O.
Способ вытеснения; BioSg-J-3Fe=2Bi~(-3FeS.
Получаемый таким образом висмут большей частью загрязнен различными примесями: .мышьяком, сурьмой, свинцом, железом, медью и серой. Иногда оп содержит также серебро и золото. Последнее можно экстрагировать из расплавленного висмута оловом. Для удаления меди предварительно нутом окислительной плавки устраняют все остальные примеси, а затем остаток сплавляют с сульфидом натрия, в результате чего выделяется сернистая медь. Если требуется большая чистота висмута, например для фармацевтических препаратов, то обычно производят еще и рафинирование мокрым путем, напримёр растворением в азотной кислоте и кристаллизацией из нее нитрата. В производстве для получения очень чистого висмута применяют также электролитическое рафинирование.
Свойства. Висмут — белый с красным оттенком блестящий хрупкий металл грубозернистого строения. Он легко раскалывается и превращается в порошок. Удельный вес 9,80. Температура плавления 271,0°, температура кипения 1560°. Твердость, по Моосу, 2,5.
Удельная электропроводность висмута прп 18° равна 1,37% и теплопроводность —1,93% по сравнению с серебром. Удельная теплоёмкость при 18° равна 0,029, атомная теплоемкость — 6,1 кал. В парах висмут образует двухатомные моле-, нули (Вц,).
Встречающийся в природе висмут является чистым элементом (ср. т. II, гл. 12). Он не абсолютно стабилен и обладает исключительно слабой радиоактивностью. Распад происходит с выделением сс-лучей (3,0 Мае) и с образованием стабильного изотопа Т1М5. Период полураспада висмута =2,5 -10Ы лет, т. е. примерно в 20 миллионов раэ больше, чем у тория, и в 50 миллионов раз больше, чем у урана.
При обычной температуре висмут на воздухе устойчив, при температуре красного каления он сгорает с синеватым пламенем в желтую окись BiaOs. С хлором порошкообразный висмут соединяется со вспышкой. При нагревании он взаимодействует с бромом, иодоле, а также с серой, селеном и теллуром. С азотом к фосфором висмут непосредственно не соединяется. Вода при обычной температуре на висмутпе действует, если онаие содержит растворенного кислорода. При прокаливании в атмосфере водяного пара висмут медленно окисляется. В пеокисляющих кислотах висмут нерастворим; не действует на него и холодная концентрированная серная кислота. В горячей концентрированной серной кислоте висмут растворяется с выделением двуокиси серы. Самый лучший растворитель для висмута — азотная кислота.
Нерастворимость висмута в нео квел нищих кислотах обусловливается его положением в электрохимическом ряду напряжений. Нормальный потенциал висмута по отаошевню к нормальному водородному электроду составляет около <0,2 е; следовательно, висмут благороднее водорода.
С металлами висмут легко образует сплавы. Некоторые из них замечательны своей очень низкой температурой плавления. Случаи, когда висмут образует с металлами соединения, довольно редки. Чаще всего такие соединения образуются с металлами с сильно выраженным электроположительным характером (см. табл. 98, стр. 630).
VIS
Глава. 14
Из легкоплавких сплавов висмута наиболее известен сплав Вуда, состоящей з 7—8 вес. ч. висмута, 4 вес. ч. свинца, 2 вес. ч. олова и 1—2 вое. ч. кадмия, Оп плавшгт при 70°, Самый низкоплавкий («эвтектический») сплав из этих металлов плавпг":т при G0’; он состоит из 15 вес. ч, висмута, 8 вес. ч. свинца, 4 вес. ч. олова и 3 вес. ч. тет--мия (сплав Липовица), Такой же эвтектический сплав представляет собой метлгг. Розе, состоящий из 2 вес. ч. висмута, 1 вес. ч. олова и 1 вес. ч. свипца. Температуры-плавления его 94°.
Применение. Металлический висмут применяют для приготовление легкоплавких сплавов, а также иногда в качестве добавки к британскому металлу и металлу для подшипников. Из соединений висмута наиболее употребительны окись и основной нитрат. Окись висмута служит, например, наряду с окисью свинца в стекольном производстве для изготовления оптических стекол с высоким коэффициентом светопреломления, а также для цветных глазурей. Основной нитрат применяют при окраске фарфора для закрепления обжигом позолоты. Главным же образом его используют в косметических и медицинских препаратах, В медицине висмутовые соединения находят постоянное применение: так, основная висмутовая соль галловой кислоты Bi(OH)2-ОСО-С6На(0Н)3 (Bismuium subgallieum) является обычным средством для присыпки ран (порошок «дерматол»); висмутовые соединения применяются также и прн лечении сифилиса.
СОЕДИНЕНИЯ ВИСМУТА
Большинство соединений висмута являются производными трехвалентного висмута и отвечают, таким образом, общей формуле BiX3. Однако висмут бывает также двух- и пятивалентным.
Соответственно отчетливо основному характеру окиси висмута Вь03 соединения висмута с более электроотрицательными атомами или радикалами имеют большей частью характер солей. Висмут, как и его пившие аналоги, образует газообразное соединение с водородом, которое, однако, малоустойчиво:
Соединения висмута с более электроположительными металлами имеют второстепенное значение ic.m, выше).
1 , Окислы и гидроокиси
Трехокись висмута. Обычная окись висмута (трехокись висмута) BiaO3 образуется при сжигании металла, а также при нагревании нитрата или карбоната висмута; это желтый, цвета серы на холоду и краснокоричневый при нагревании порошок с удельным весом 8,76, температурой плавления 820° и теплотой образования 137,8 ккал/моль, Расплавленная окись висмута действует на платину так же сильно, как окись свинца. При высокой температуре окись висмута легко восстанавливается до металла, В кислотах она растворяется с образованием соответствующих солей висмута; в разбавленных щелочах Bi2O3 нерастворима.
Нерастворимость окиси висмута в разбавленных щелочах, отличающая ее от амфотерных трехокисей мышьяка и сурьмы, указывает на ее основной характер. Однако при сплавлении с другими окисламтг металлов BiaO3 может вступать с ними в соединения. Так, с окисью свинца опа образует следующие соединения: 2РЬО-ВЬО3, 2PbO.3Bi2O3, РЬО 4BiaO3.
Bi2O3 образует три модификации. Подробнее см.: Schnmb/ Rittner, J. Am. chem. Soc., 65, 1055, 1943.
Низший по сравнению с трех окис но окисел висмута, согласно новейшим исследованиям, в виде определенного соединения, по-видимому, не существует. То, что
Пятая группа периодической системы
729
раньше называли моноокисью висмута, представляет собой, по Нейсе ер у (Neusser), смесь В13О3 и Bi,
Гидроокись висмута Bi(OH)3 осаждается гидроксильными ионами из растворов солей висмута в виде белой хлопьевидной массы. Ее очень трудно получить в чистом состоянии, так как она, обладая коллоидной природой, легко адсорбирует кислотные ионы, которые прочно ею удерживаются. Очень легко гидроокись висмута получается в виде коллоидного раствора. Если осажденную обычным путем Bi(OH)3 высушить при 100°, то получается соединение приблизительного состава BiO(OH). Гидроокись висмута легко растворима в кислотах с образованием солей. В разбавленных щелочах она нерастворима.
Так как орто гидроокись висмута Bi(OH)3 характеризуется определенной рентгенограммой, то ее следует считать индивидуальным химическим соединением. При изобарическом обезвоживании ортогидроокиси или соответственно три гидрата окиси висмута В12ОЭ-ЗН2О обнаруживается существование еще двух других гидратов: Bi3O3-2H2O и Bi203'H20 [или BiO(HO)] (Hflttig, 1931).
Пл тиокисы висмута, висмутовая кислота и висмутаты. При обработке взвеси гидроокиси висмута в концентрированной щелочи сильным окислителем (например, хлор, Перманганат калия, персульфат калия, циансферрат(Ш) калия) образуются щелочные висмутаты, т. е. щелочные соли висмутовой кислоты HBiOg, однако обычно с примесями. Впервые таким путем удалось получить чистые соли висмутовой кислоты Шолдеру (Beholder, 1941): желтый висмутат, натрия. NaBiOg, коричневый ортовисмутат натрия Na3BiO4, краспо-фнолетовый висмутат калия КВ1О3, а также висмутаты щелочноземельных металлов и тяжелых металлов при взаимодействии щелочных висмутатов с соответствующими солями, Ортовнсмутат натрия, как это установил Цинтль (Zintl, 1940), легко получить сухим путем при нагревании Bi2O3 с Na2O2, в молярном отношении 1 ; 3 (Bi3O3 + 3Na3O3 = 2N;i3BiO4 + ПэСЕ). В этих условиях переход висмута в пятивалентное состояние осуществляется так легко, что даже уже нагревание Bi2O3 с Иа2О в атмосфере сухого воздуха ведет к образованию висмутата. При обработке щелочного висмутата (загрязненного Bi2O3) азотной кислотой можно получить гидратированную пятиокисъ висмута 1И2О5. При этом, однако, легко происходит отщепление кислорода; в большинстве случаев удается получить продукт, представляющий собой смесь пятиокиси и двуокиси висмута BiO2. В определенных условиях последнюю мощно получить в чистом состоянии (Worsley, Robertson, 1920, Scholder, 1941).
2 Соли висмута
Характерные для висмута соли являются производными трехвалеит-наго висмута. Опи большей частью бесцветны; соли, образованные сильными кислотами, легко растворимы в воде. Однако прозрачный растпор получается только при значительном избытке кислоты, так как иначе вследствие гидролиза образуются основные соли, которые выпадают в виде нерастворимых осадков. Основные соли по своему составу соответствуют формуле BiOX (оксисоли висмута).
Ранее соединения BiOX называли солями висмутила} однако оказалось, что опи не содержат в качестве определенной структурной группы «висмутил»-радикал [ВЮГ, То же следует сказать и с, солях сурьмы типа SbOX, которые рапьше назывались солями антимонила, Почти все оксисоли висмута плохо растворимы в воде. Единственной солью, которая легко растворяется в воде и полностью переходит в раствор, не содержащий избытка кислоты, является о ксиперялорат в нему та BiOlClOiJ, который легко образуется при растворении ВьО3 в относительно разбавленной хлорной кислоте (Fichter, 1923). Обычно оксинерхлорат висмута кристаллизуется в виде моногидрата в маленьких ромбоэдрах. Из сильно разбавленного раствора кристаллизуется в виде игл малоустойчивый тригидрат; из концентрированной НС1О4, напротив, по-видимому, выделяется нейтральная соль Bi [С1О4]3 ЗН2О, которая, однако, также неустойчива.
730
Глава 14
Из растворов, содержащих соли висмута и щелочных металлов, часто кристаллизуются двойные соли (ацидосоли). Это указывает на то, что соли висмута обладают склонностью присоединять находящиеся в избытке кислотные ионы с образованием сщидо-ионов, например BiCl34-Cl' = [BiCl4J'. Концентрация свободных ионов висмута Bi"' в растворах солей висмута определяется двумя, одновременно существующими равновесиями
Bi"'+H2O -т± [В1ОГ4-2Н- и -± [BiXt]'
(X' —кислотный ион). Однако эту концентрацию часто бывает трудно оцепить с достаточной точностью.
Галогениды висмута
Трсхфторнетый висмут в фторовнс.чутаты, Трехфтористый висмут, представляющий собой практически нерастворимый в воде серовато-белый кристаллический порошок о уд. весом 5,32, подучается при обработке окиси висмута плавиковой кислотой или при прибавлении фторида калия к возможно менее подкисленному раствору нитрата висмута. Фторид висмута заметно растворим в концентрированном растворе фторида калия вследствие образования комплексной соли. Комплексная соль [NH4|[BiF4] (тетра фторо висмутат аммония) получается, по Гельмольту (Helmolt), в виде прозрачных блестящие кристалликов.
При нагревании BiF3 в токе фтора при температуре 500J образуется, по данным Вартенберга (Wartenberg, 1940), пятифтоуиетый висмут в виде бесцветных ромбических игл (т. пл. 725—730°). ОксифторпД пятивалентного висмута был получен еще в 1908 г, Руфом (Ruff) в виде двойной соли KF-BiOFj.
Хлорид висмута, оксихлорид висмута и хлоровисмутаты. Хлорид висмута BiCl3 был получен еще Бойлем [при нагревании висмута с хлоридом ртути(П)], Для его приготовления лучше всего растворить окись висмута в соляной кислоте или металлический висмут в царской водке и после упаривания перегнать остаток без доступа воздуха. Хлорид висмута представляет собой белоснежную кристаллическую, па влажном воздухе расплывающуюся массу (удельный вес т.В: температура плавления 232°; температура кипения 447 ).
Водой хлорид висмута гидролизуется с образованием оксихлорида висмута BiOCl, Он образует белый кристаллический порошок уд, веса 7,72, который при нагреваний окрашивается от желтого до коричневого цвета, а при охлаждении опять становится почти бесцветным, Под действием света он постепенно темнеет. То же явление наблюдается и для хлорида висмута; но у него коричневая окраска постепенно исчезает в темноте, тогда как у оксихлорида висмута она сохраняется.
С хлоридами щелочных металлов, аммония и т, д. хлорид висмута образует комплексные соли (хлоровисмутаты) преимущественно типа М1 [BiCl4] и M3[BiCl5l, Кроме того, хлорид висмута склонен к образованию продуктов присоединения, например с аммиаком.
Если хлорид висмута долго нагревать в ампуле с металлическим висмутом, то образуется дихлорид висмута В1С12 в виде очень гигроскопической черно-коричневой кристаллической массы. О и образуется также в качестве промежуточного продукта при медленном действии хлора па висмут при обычной температуре. Водой дихлорид разлагается с выделением оксихлорид;г висмута BiOCl,
Структура окси галогенидов висмута. Bi OF образует сложную координационную решетку.'Соединения BiOCl, BiOBr h ’BiOI образуют тетрагональные слоистые решетки. Эти решетки построены из слоев металл — кислород (рис. 120, а), между которыми расположены двойные слои из атомов галогенов (рис. 120,6). [О других соединениях подобного типа (тип смешанного хлорида-фторида свинца) см. стр. 306.) Смешанные оксина логтиды висмута с другими металлами, которые часто обладают очень сложным
Пятая еруппа периодической системы
731
•составом, кристаллизуются в большинстве случаев, как и простые окспгалогсниды висмута, в тетрагональных табличках и близки к ним по строению решеток. Многие из них, например ВаВ1О2С1 и LiBi3O4ClB, построены на слоев кислород — металл, между
которыми расположено по орно.иу слою из атомов галогенов (рис. 120, в). В других смешанных оксигалогенидах между двумя слоями металл — кислород располагаются тройные слои из атомов галогенов, в средней плоскости которых расположен еще один слой из иопов металла (рис. 120, г), который, однако, в большинстве случаев заполнен частично, что и приводит к образованию стехио метрически непростых соединений, например Cab25Bi1>flOaCl3. Три последовательных слоя (на рисунке о, в, е) могут встречаться в различных комбинациях. Это часто приводит к образованию соединений, состав которых может колебаться внутри известных пределов (не-дальтонидные соединения). Отсюда следует, что колебания в составе, обусловленные особыми геометрическими свойствами, могут встречаться не только в интерметаллических, но н в типичных «со.адобрааньиг» (т, е. построенных из ионов) соединениях. Не имеет смысла называть их «твердыми растворами», так как соединения, твердыми растворами которых они должны были быть, сами ио себе часто не существуют, Например, существует оксихлорид •состава Cd2—зхВ1и2хО2С13 (х = 0,20—-0,30), в то время как соединения предельного •состава Cd2BiO2Cl3 (% — 0) и. CdBi(O4Cle (® = !<) не получены [Sil 1 ё n L., Natur-wjss,, 30, 318, 1942; ср. также Z, anorg. Chem., 250, 173, 1942).
Соединения B1SC1, BiSBr, BiSI, a также BiSeBr и BiSei (так же, как SbSBr, SbSI, SbSeBr и SbSel), кристаллизующиеся в ромбических иглах, имеют, по Дёнгесу (Donges, 1950), ленточную структуру. Ленты, например
раснолагаЮтСЯ в направлении оси с, параллельно которой кристалл поэтому легко раскалывается. Соединения BiTeBr и BiTel
Ме - металл X •• галоген
также кристаллизуются в слоистых рёшет-
Рис. 120. C.TOucTiiir структура оксигалогенидов висмута.
Соединения кристаллизуются тетрагонально Ось а параллельна, ось с перпендикулярна плоскости рисунка. Соединение построено из слоев металл—кисло род без промежуточных слоев галогенов, подобно РЮ и SnO (со. стр. 5S0).
нах, но не в тетрагональных, как соответствующие окс)и'ало)'ениды, а в гексагональных. По своим структурам опи, по-впди-мому, похожи. па структуры бруцита ft упорядоченный расположением атомов Те я Вт (Соответственно I) в узлах решетки запятых атомами кислорода.
Бромид висмута BiBr3 и нодчд висмута Bil3 аналогичны хлориду. Премий
висмута образует желтые кристаллы с удельным весом 5,60, плавящиеся при 218° в темно-красную жидкость. Ивдид висмута — блестящие томно-коричневые кристаллические листочки с уд. весом 5,8 и т. пл. 408°. Он значительно менее растворим в воде, чем хлорид, а потому из растворов осаждается ионами иода. Бромид висмута под влиянием воды легко переводится в бесцветный оксибромид, тогда как иодид вследствие
732
Глава 74
своей незначительной растворимости — только прн кипячении. Окышодид впсмугц кирпично-красный тяжелый порошок, применяется в медицине. Как бромид, тц_т и иодид висмута образуют комплексные бромо- и иодовиемутаты, а также и друге-; продукты присоединения.
В нему тоио диет ово до родную кислоту И [Bili] (кристаллизуется с 4П2О) применяют в аналитической химии. Многие алкалоиды (сильно ядовитые, органическЕт азотсодержащие основания, например стрихнин, хинин, кокаин) образуют с пей нер&-створимые осадки. Поэтому ее калийной солью пользуются как групповым реагентом ел алкалоиды. Соль о-оксихиполина с висмутоиодистоводороднон кислотой [С9Н6(ОН)КН” [BilJ удобна вследствие ее чрезвычайно малой растворимости для количественного определения висмута (Berg, 1927).
Роданид висмута и родаповисмутаты. Из раствора гидрата Окиси висмута в рода-иистов ОДО родной кислоте кристаллизуется желтый роданид висмута Bi(SCN)3, который присоединяет роданиды щелочных металлов с образованием родановисмутатов
Нитрат висмута образуется при растворении порошкообразного висмута в азотной кислоте. Из упаренного раствора выделяются большие триклинные кристаллы в виде столбиков состава Bi(NO3)3-5H2O. При нагревании вместе с водой отщепляется и азотная кислота, так что в результате остается океинитратеисмутаВЮ^О^). Последний образуется также и прн растворении нитрата висмута в воде наряду с другими основными нитратами в зависимости от условий опыта. Приготовленный определенным образом основной нитрат висмута М agister ium bismuti или Bismutum subnitri-cum широко применяют в медицине в качестве дезинфицирующего, уничтожающего запахи и одновременно вяжущего средства. Его используют в качестве порошка для присыпки ран, ири желудочных и кишечных заболеваниях и особенно при дизентерии и холере. Наконец, его применяет под названием жемчужных или испанских белил в качестве неядовитого белого грима, О применении его для окраски фарфора уже упоминалось па стр. 728. Нейтральный и основной нитраты висмута обычно используют для приготовления других соединений висмута.
Сульфат висмута — Bi2(SO4)3 кристаллизуется из растворов висмута, а также его окиси или сульфида в концентрированной сер пой кислоте в виде тонких белых гигроскопичных игл, С небольшим количеством воды он образует гидраты; большими количествами воды гидролизуется с образованием основной соли. Сульфат висмута присоединяет сульфаты щелочных металлов с образованием двойных солей [ сульф атови ему-татои(Ш)], например K[Bi(SO4)3] и KsfBi(SO4)3],
Карбонат висмута, Прн действии щелочных карбонатов илй карбоната аммония на растворы солей висмута или его окисла или сульфида осаждается основной карбонат висмута (В1О)2СО5, содержащий воду. (BiO)2CO3 встречается также и в природе (висмутовый шпат).
Сульфид висмута. Обычный сульфид висмута— трисулъфид висмута, трехсернистый висмут BisS3 — выпадает при пропускании сероводорода в раствор какой-нибудь соли висмута в виде темно-коричневого осадка, растворимого только в концентрированных кислотах
2Р. i' * 4- 311, S = В l2S3 -ф 6Н •.
Сульфид висмута в отличие от сульфидов мышьяка и сурьмы нерастворим в сульфидах щелочных металлов.
Если приготовить сульфид висмута сплавлением висмута с серой, то он подучается в виде свинцово-серой лучистой кристаллической массы с удельным весом 6,5. Осаждаемым из водного раствора коричневый аморфный сульфид висмута переходит постепенно (быстрее при высокой температуре)
Пятая еруппа периодической системы 733
в серую кристаллическую форму. Последняя и встречается в природе (висмутовый блеск, висмутин). Природный висмутовый блеск образует длинные призматические, вертикально сильно испещренные кристаллы, цвета от серо-стального до оловянно-белого, большей частью соединенные в древовидные сростки и по внешнему виду очень похожие на сурьмяный блеск. Висмутовый блеск изоморфен сурьмяному блеску и природному сульфиду мышьяка. Несмотря на то что большие месторождения висмутового блеска встречаются довольно редко, тем не менее значение его как висмутовой руды очень велико.
Встречающиеся в природе двойные соединения сульфида висмута с сульфидами других металлов уже были описапы на стр. 726. Соответствующие двойные сульфиды можно получить и сплавлением.
Кроме трисульфида, висмут, по-видимому, образует и моиосулъфид BiS. Это соединение должно образовываться при нагревании Bi О (ранее принимаемого за моноокись висмута) в токе сероводорода в виде грифельно-серого порошка BiO + H2S == = BiS + Н20. Будучи довольно устойчивым при обычной температуре на воздухе, мопосульфид висмута при нагревании до температуры красного каления в атмосфере сухого С03 разлагается на висмут и трехсернистый- висмут.
Соединения Bi,Se3 и Bi2Te9 (им изотиплы As2Se2 и $Ь2Те3) кристаллизуются в ромбоэдрических кристаллах, образуя слоистые решетки. Слои, образованные только из атомов одного рода, располагаются перпендикулярно оси с, и иритом таким образом, что между двумя слоями висмута попеременно располагаются один или два слоя селена (соответственно теллура). Если заменить один слой Тс на спой серы, то образуется тетрадимит. ВьТе23. В нем слои расположены в такой последовательности: —Bi—Те—То—Bi—S-.
Кроме соединения В138е3, висмут образует с селеном еще соединение BiSe (также гексагональное), в то время как в системе Bi—Те присутствует только соединение Bi2Te3 (Рагга van о, Korber, 1930).
3 . Вл см у тис ты й водород
Впсму'пгстый водород с очень незначительным выходом (наряду с большим количеством водорода) образуется при разложении соляной кислотой порошкообразного сплава висмута с магнием. Он малоустойчив, медленно разлагается уже при обычной температуре, а при высокой — моментально. Присутствие его устанавливают но образованию «висмутового зеркала», аналогично тому, как открывают мышьяковистый и сурьмянистый водород. Зеркало висмута отличается от зеркала мышьяка своей нерастворимостью в растворе гипохлорита натрия, а от сурьмяного зеркала —нерастворимостью в желтом сернистом аммонии.
Удобнее всего открывать висмутистый водород реакцией на пламя (реакция Донау). Для этого в водородное пламя вносят В' платиновом ушке небольшое количество окиси кальция, которая в случае содержания в горящем газе висмутистого водорода извлекает из пего висмут. Если затем платановое ушко вынуть ненадолго из пламени так, чтобы окись кальция застыла, н потом опять ввести его в пламя, то плав начинает светиться интенсивным васильковым цветом. Реакция, которую можно повторять любое число раз, позволяет определять присутствие даже 10*10 е висмута. Сурьма вызывает также интенсивное окрашивание до небесно-голубого цвета, а мышьяк вызывает только слабое бледно-зеленое окрашивание.
После тщетных попыток прежних исследователей приготовить висмутпстый водород удалось Панету (Paneth, 1918). Он использовал для своих опытов сначала не обычный висмут, а его уадиоомтиеный изотоп, а именно торий G- (ThC). Благодаря сильной радиоактивности ТЕС можпо было определять несравненно меньшие количества водородного соединения ThC, чем это возможно для обычного висмутистого водорода. Таким образом, Папету удалось по только сравнительно легко установить присутствие искомого вещества, но и определить важяейтиие его свойства и найти наиболее подходящие экспериментальные условия для получения вещества в весомых количествах с применением в качестве исходного продукта обычного в нему га.
734
Глава. 14
Алкильные соединения висмута BiR3 можно рассматривать как производный висмутистого водорода. Они, одиако, значительно превосходят последний по устойчивости. Получаются висмута л килы прп взаимодействии бромида висмута с цинк-ananien.iiOMj
2BiBr3 3ZnH2 = 2BiR3 4* 3ZnBr2
или прп обработке хлорида висмута магнийалкилбромидом. Замечательна их способность присоединять хлор или бром с образованием соединений пятивалентного висмута, например
(С6П5)3Вг + Cla = (C6H5)3B1CJ2.
Аналитические сведения. Висмут осаждается сероводородом из кислого раствора в виде коричневого сульфида BiaS3, не растворимого в сернистом аммонии. Сульфид растворяется в 20%-иой азотной кислотес образованием нитрата. Аммиак осаждает висмут из раствора его солей в виде белой гидроокиси Bi(OH)a.
При разбавлении кислых растворов солей висмута водой обычно выпадает белый осадок, состоящий из основных солей висмута. Это характерная реакция для солен висмута; однако следует учитывать, что и соли сурьмы обладают такой же реакцией.
Для количественного определения висмут выделяют и взвешивают чаще всего в виде сульфида. Можно также осаждать его в виде гидроокиси пли основного карбоната, затем восстанавливать цианистым калием 11 полученный металлический висмут взвешивать.
Малое количество висмута удобно определять по методу Мара [Mahr С., Z. anorg. Chem., 208, 313, 1932; Z. analyt. Chem., 102, 41, 1935], осаждая висмут в виде Bi[Cr(SCN)e]. Об определении висмута в виде тетраиодо висмутата о-оксихиполипа см. стр. 732. О весовых определениях малых количеств висмута см.: StammH.,., Z. analyt. Chem., 115, 1, 1938.
В пламени паяльной горелки соединения висмута дают хрупкий королек металла и светло-желтый налет окисла.
Для микроаналитического определения особенно пригоден характерно крпсталлнзующипся я довольно трудно растворимый пентаиодовисмутат цезия 2Cs2| BiTrJ 5Н2О. Граница определимости при пользовании этой реакцией лежит около 0,1у. Относительно определений самых незначительных следов висмута реакцией на пламя, по Донауг см. выше.
Глава 15
ШЕСТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Главная подгруппа)
ГРУППА КИСЛОРОД - СЕРА
орядкопий номер Элемент Символ Атомный вес Удельный вес Температура плавления, °C Температура кипения, °C Теплота плавления Теплота испарения Валент- TiOCTii
ккал,' г-атпол<.
8 Кислород О 16,0000 1,27 а -218,9 - 182,96 0,053 , 0,814 п
16 Сера S 32,066 6 2,06® 119,0г 444,60 0,35 2,52 II, IV, ¥1
34 Селеп Sc 78,96 4,824 220,2к 688 1,6 4.35 11,1V, VI
32 Теллур Те 127,61 6,25 е 452,0 1390 4,27 11,0 П, IV, VI
81 Полоний Ро 210 9,32 — "— — — И, IV, VI
а Твердый ниглород по!) температуре кипения.
6 Вс ле детине непостоянства отношения ивотопов в естественной смеси изотопов атомный^вес серы может отклоняться от указанного значении па величину ±0,003.
в Ромбическая сера.
г Моноклинная сера. Относительно температуры плавления ромбической серы см. стр. 752,
Л Гексагонально-ромбоэдричесьтгй (серий, металлический; селен.
u Гексагонально-ромбоэдрический теллур.
Общие сведения. В гла вную подгруппу VI группы периодической системы входят элементы кислород, сера, селен, теллур и полоний. Первые четыре элемента, имеющие неметаллический характер, объединяются под названием халькогенов, что значит «образующие руды». Все элементы главной подгруппы VI группы могут давать соединения с водородом и в своих соединениях с сильно электроположительными элементами заряжены отрицательно. Сильнее всего неметаллический характер выражен у кислорода и серы. Селен и теллур занимают промежуточное положение между неметаллами и металлами. Так, к элементарном состоянии селен существует как в неметаллической, так и в металлической модификациях. Для элементарного теллура металлическая модификация является даже наиболее обычной. По по своим химическим свойствам и эти два элемента стоят ближе к неметаллам. Их сходство с металлами в химическом отношении проявляется лишь в том. что селен и теллур могут образовывать соли с сильными кислотами, в которые они входят в качестве электроположительной составной части. Особенно это относится к теллуру, хотя и его соли очень мало устойчивы. У последнего (наиболее тяжелого) элемента группы, радиоактивного и сравнительно короткоживущего полония, металлический характер выражен более ярко. Он способен существовать в водном
736
Глава 15
растворе в виде элементарного положительного иона и по сво им хтгмтгч е№~ ч свойствам близок не только к стоящему пад ним теллуру, но и к соседнее? слева висмуту.
В газообразном состояния даже при самых низких температурам кислород всегда двухатомен. Его аналоги, наоборот, имеют очень большую склонность к ассоциации и образуют двухатомные молекулы тольнм при сравнительно высоких температурах. Работа, требующаяся для расщепления таких молекул па атомы (термической диссоциации), сильна уменьшается с увеличением атомного веса.
Энергия диссоциации, ккал/молъ (при 0°К)
Оз 8г 8вз Тег
117.2 75.6 62 53
Для всех элементов главной подгруппы VI группы характерно проявление в соединениях отрицательной валентности, равной двум. С точки зрения теории Косселя, это объясняется тем, что в периодической системе каждый из этих элементов стоит за два места до инертного газа. Стремление атомов этих элементов приобрести «конфигурацию инертного газа;» приводит к тому, что каждый из пих легко принимает два дополнительных электрона.
Это не следует понимать в том смысле, что при получении атомом такого элемента заряда 2 — выделяется энергия. Это отнюдь не так. Например, в случае кислорода энергия освобождается только тогда, когда к атому присоединяется только один электрон; для присоединения второго электрона необходимо затратит*' энергию, так что в целом процесс О Н- 2с = 0s’ протекает с поглощением энергии. Другими словами: сродство О к электрону при образовании иона 0-~ из атомов О (и, конечно, из молекул 02) отрицательно. Это сродство можно вычислить при помощи описанного на стр. 174 и сл. кругового процесса. Согласно Бригдебу (Biiegleb, 1942), свободная энергия образования О3* из О составляет 150 ккал/г-атом, О- из О —56 ккал/г-атом и Ой- из ОН—45 ккал/г -атом. Аналогично для свободной энергии образования S3-из SaT0M вычислена величина 84 ккал/г-атом.. (Свободная энергия образования S3* из батон в соответствии СО спектрографическими данными равна —65 ккал/г-атом.) Работа, необходимая для образования отрицательно двухзарядных иопов, в случае образования кристалличеекнх соединений прок (водится за счет сил решетки (ср. гл. 5). Если этот процесс происходят в водном, растворе, необходимая энергия покрывается за счет энерглен гидратации.
М‘-;юп свободней!, энергии образовавши электролитических ионов является потенциал растворения. Нормальные потенциалы (при 18’), отвечающие образованию отрицательных деухзарлдных ионов, составляют по отношению К нормальному водородному электроду для
+0,41 -0.51 -0 77 -0,91 в
Величина, приведенная для кислорода, справедлива для 1 М раствора по отношению к ионам ОН , тогда как остальные величины относятся к 1 М растворам по отношению к ионам S", Se" и Те". Эти величины показывают, что легкость, с которой происходит переход в состояние отрицательных электролитических ионов, сильно снижается от кислорода к теллуру. Если кисдородпый электрод соединить с нормальным водородным электродом, то по внешнему проводнику положительный ток потечет от кислородного электрода к водородному. На кислородном электроде кислород будет заряжаться до отрицательно двухзарядных ионов (эти ионы О" тотчас же по образовании будут реагировать с водой, давая 2ОН'), на водородном электроде водород, образуя ноны водорода, будет переходить в раствор. Напротив, если платиновую фольгу, погруженную в сульфидный раствор (так называемый «серный электрод»), соединить с нормальным в о дородным электродом, то во впекшей цепи положительный ток потечет от второго к первому. Ионы S" будут разряжаться; точно так же будут вести себя ионы So* и Те", только их тенденция к разрядке будет еще более ярко выраженной.
Максимальную валентность шесть, соответствующую номеру группы, обнаруживают сера, селен, теллур и, во всяком случае, полоний, по не кислород. В славных валентных соединениях кислород никогда не проявляет
Шестая группа периодической системы
737
валентности выше двух*. По теории Кессел я, появление нормальной валентности шесть только у серы, селена и т. д. объясняется тем, что эти элементы сравнительно легко могут отдать шесть электронов. Хотя и у кислорода (по спектроскопическим данным) шесть электронов расположены на «внешней оболочке» атома (точнее, на энергетическом уровне с главным квантовым числом 2), однако, как показывают высокие значения потенциалов ионизации кислорода (ср. табл. 23 на стр- 143), электроны в атоме кислорода особенно прочно связаны**. Отсюда следует, что не существует такого элемента, который смог бы оторвать электроны от атома кислорода. Поэтому во всех гетеронолярпых соединениях кислород всегда является отрицательно заряженной составной частью и как таковой он обычно является двухвалентным, во всяком случае, никогда не имеет валентности больше двух***.
Аналоги же кислорода способны отдавать до шести электронов, и поэтому в соединениях, которые имеют гетерополярнын характер, они могут являться положительно заряженными составными частями, а именно нести заряд до б-р. Если отщепляется мепее шести электронов, то гомологи кислорода могут проявлять положительную валентность меньше шести. При этом они, как и элементы ранее рассмотренных главных подгрупп, отщепляют электроны парами, так что в гетерополярных (по теории Кос-селя) соединениях эти элементы могут иметь, кроме шести, еще четыре и (однако значительно реже) два положительных заряда.
По отношению к сильно электроотрицательным элементам, таким, как кислород и фтор, сера проявляет валентность 6-(-. Селен и теллур в соединениях со фтором также имеют валентность 6-ф, но в соединениях с кислородом — преимущественно валентность 4-ф.
В гомеополярных соединениях кислород всегда двухвалентен, точнее говоря, имеет две главные валентности, если не учитывать существования описанных на стр. 747 (устойчивых только в растворе) радикалов, которые образуются при диссоциации некоторых органических перекисей.
Тот факт, что кислород и в гомеополярных соединениях никогда не проявляет валентности больше двух, с точки зрения теории атомной связи объясняется следующим образом: в атоме кислорода имеется шесть внешних электронов, находящихся на энергетическом уровне с главным квантовым числом п = 2. Согласно принципу Паули (см. стр. 145 и сл.), на таком уровне может находиться максимум восемь электронов. Так как обычно каждая гомеойоляркая связь образуется парой электронов, для которой каждый из связанных атомов представляет одни электрон, то при образовании двух главных валентностей число внешних электронов атома кислорода доходит до восьми, т. е. до максимально возможного числа. Чтобы образовалось бол ее двух валентных связей, по крайней мере одни электрон должен подняться с уровня с главным квантовым числом 2 на уровень с главным квантовым числом 3, Как следует из спектральных термов кислорода, для этого надо затратить очень большую энергию, а именно около 210 ккал/о-шпом.. В атомах аналогов кислорода, наоборот, ни одному электрону пе надо подниматься ня уровень с большим главным квантовым числом, чтобы стало возможным образование более двух гомеополярных главных валентных связей, так как в силу большего зиачеиид главного квантового числа внешние энергетические уровни этих атомов могут содержать больше восьми электронов. Впрочем, и в атомах гомологов кислорода, чтобы они проявили валентность больше двух, электроны должны быть подняты на более высокий энергетический уровень, но пе е большим гласим.и
* О валентности кислорода в оксон новых соединениях см. стр. 748 и сл.
** Это обусловлено высоким эффективным зарядом ядра в сочетании с малым главным квантовым числом (2).
*** В перекиси водорода и ее производных кислород является электрохимически одновалентным и структурно Овевал сытным. О существующей в щелочных и щелочноземельных перекисях группе Oj см. стр. 747.
47 Р. Рейн
738
Глава 15
квантовым числом, а всего лишь на уровень с большим побочным квантовым числом, а именно па ^-уровень. Если образуются две валентные связи, такого перехода электронов пе происходит, поскольку, как следует из спектров, и у кислорода и у его гомологов основному состоянию атомов соответствует триплетный терм (3Т32). Это значит, что-атомы кислорода и его гомологов в основном состоянии содержат деа неспаренных электрона. Следовательно, они могут проявлять валентность два, не требуя какой-либо-энергии возбуждения, кроме энергии, необходимой для распада молекул на атомы, тогда как для проявления ими высших валентностей такая энергия возбуждения необходима. Отсюда понятно, почему в чисто гомеополярных соединениях и гомологи кислорода проявляют в основном валентность 2.
Халькогены являются двухвалентными в элементарном состоянии и в кристаллической и аморфной формах (см. стр. 754 и 798). При таком строении каждый атом имеет двух ближайших соседей, так что образуются цепи или циклы. Расстояния между атомами, принадлежащими различным цепям или циклам, значительно больше. Этим Самым оправдывается предположение, что между такими цепями или циклами действуют вторичные валентные силы (вандерваальсовы силы). Из отношения межатомных расстояний внутри таких цепей и между ними можпо заключить, что величина вторичных валентных сил возрастает от селепа к теллуру. Это можно рассматривать как признак перехода к металлической связи.
По отношению к сильно электроположительным элементам элементы главной подгруппы VI группы всегда выступают отрицательно двухвалентными. Поэтому их нормальным соединениям с металлами и водородом отвечает общая формула M2R или соответственно H2R. Соединения с водородом (халыюгеноводороды см. табл. 106), как все простые водородные
Таблица 106
Свойства водородных соединений халькогенов
Соединение нео H2S H2Se HST e
Температура плавления, °C ... . 0,00 —85,60 — 60,4 -51*
Температура кипения, °C 100, Oil -60,75 -41,5 -1,8
Критическая темнёр.п урй Lf Теплота плавлеитш, сьч и;.ч |пр<т 500 100,4 137 —
температуре плавления.» Теплота испарения» (при 1,430 ii,5676 — —
температуре кипения) 9,715 4,663 4,75 -5,7
Константа Трут она, 25,9 20,9 20,4 -21
Плотность прн температуре кипения Поверхностное патяжепие при тем- 0,958 0,993 2,004 2,650
пературе кицеипя, дин1см% . . . Теплота образования (прп 20° it но- 58,9 28,7 28,9 30,0
стоянном давлении), ккал/моль Свободная энергия образования +68,35 +4,80 —18,5 -34,2
(при 25°), ккал/молъ Копс танты влек грози ги чес ко и дне- -56,72 —8,42 — •
социацип ири 18"
К, 1,3 -Ю"1®3 0,87-10~7 1,9-10-1 2ДМ0-3
К, — 0,79-10-is — —
Дипольный момент молекул н газо-
вой фазе (п в ед. CGS) ..... 1,85-Ю-is 1,10-Ю-l8
а В качестве константы Эиссоi.cttaui.ii.i во<)ы указывают, Кроме того, ll'J X ГОН'К Оно имеет значение О.74-10-Ы (при 18°), см. стр. 74.
обычно произведение
Шестая группа периодической системы
739
соединения элементов, стоящих в главных подгруппах правой половины периодической системы, легко летучи. Все опи в значительной степени способны электролитически диссоциировать с отщеплением ионов Н'
Летучесть водородных соединении элементов главной подгруппы VI группы сильно увеличивается от 1ГаО к Н38, а затем снова уменьшаетед при переходе к более тяжелым аналогам. Низкая по сравнению с ее аналогами летучесть воды обусловлена сильной ассоциацией ее в жидком состоянии (см, стр. 71 и сл.), которая выражается также в ненормальном значении константы Трутона и п высоком значении доворхпостцого натяжения (см. табл. 106). Как видно из табл. 106, подобно температурам кипения, изменяются и критические температуры и температуры плавления. Теплоты образования сильно уменьшаются нрн переходе от Н2О к Нг'Ге. Степень электролитической диссо циации сильно увеличивается в том я;е направлении.
Увеличение Кислотного характера прп переходе от воды к теллуристому водоро ДУ, с точки зрения теории Косселя, объясняется увеличением радиусов отрицательных ионов от кислорода к теллуру. Чем больше последние, тем меньше, согласно закону Кулона, работа, требующаяся для отрыва положительно заряженного атома водорода, и тем в большей степени, при прочих равных условиях, сдвинуто равновесие в сторону диссоциации. Аналогично объясняется увеличение кислотного характера бинарных летучих водородных соединений при увеличении порядкового номера и в других главных подгруппах периодической системы.
Соединения халькогенов (О, S, Se, Те) с электроположительными элементами называются халькогенидами. Халькогениды сильно электроположительных элементов кристаллизуются в большинстве случаев в типичных ионных решетках. Часто они имеют одинаковую структуру со структурой аналогично построенных: галогенидов; например, халькогениды общей формулы МПХИ часто имеют решетку типа каменной соли, как и галогениды с общей формулой NFX1. Точно так же н гидрохалъкогениды. щелочных металлов имеют типичную кристаллическую решетку. Большинство из них при обычных температурах имеет искаженную, а при повышенных температурах — правильную структуру типа NaCl (см. стр. 206). CsHS и CsHSe образуют решетку типа CsI (West, 1934; Klemm, 1939). Лишь в очень редких случаях образуется решетка слоистого типа (до сих найденная только в случае LiOH, ср. стр. 244 и сл.).
Очень большое сходство между галогенидами щелочных металлов и гидрохалькогенидами этих же металлов проявляется» изменениях теплот образования (рис. 121),
а также теплот растворения й теплот гидратации. Аналогично Изменяется и сродство к протону у ионов НВ- (В — халькоген) и ионов галогенов. Наоборот, па вычис-
47е
740
Глаза 15
лепные величины сродства к электронам свободных радикалов НН влияет протез деформирующий иоп В-. Поэтому эти величины изменяются иначе, как видно с: рис. 122, чем величины сродства к электронам атомов галогенов (Klemm, 1946
Рис. 122. Величины сродства к электрону для иопов галогенов и гидрохалькогон-радикалов.
Аналогия между гидр охал ькогенидамн щелочных металлов М*НВ и галогенидами щелочных из таллов MJX или соответственно между ионамг HR* п Х~ дает превосходный пример прп менимости установленного в 1925 г. Грпммоа закона гидриднаго одвига, который гласит: химические свойства атомов, стоящих е перис дцчссхой системе за четыре места до инерк -ноге газа, изменяются при присоединении -с атомов водорода (а = 1, 2, 3, 4) так, чт%. образующиеся группы атомов ведут себя как впсе.вдоатомън>, аналогичные атомам элементов стоящих в периодической таблице на а групг. правее. В соответствии с этим законом радикалы GH3, NH^ и ОН являются аналогами атомов F. а если опн имеют отрицательный заряд, то ионов F*; радикалы С Ну и NH2' подобны нонам О2*, а нейтральные молекулы СН4, NHj, ОН2 и FH в некоторых отношениях подобны инертному газу Ne,
КИСЛОРОД (OXYGENIUM) О
Распространение в природе. Кислород является важнейшей для жизни, поддерживающей дыхание составной частью атмосферного воздуха. Содержание кислорода в сухом воздухе составляет 20,9 об.% или 23,0 вес.%, причем в открытом пространстве содержание кислорода в воздухе очень мало изменяется (не более чем на 0.1%). Несмотря на то что при дыхании и за счет процессов горения кислород непрерывно расходуется, его количество все время пополняется благодаря процессам фотосинтеза, происходящим в золеном веществе растении на солнечном свету. Вода содержит 88,81 все. % кислорода, мировой океан — около 85,8% и доступная нам часть твердой земной коры — 47,3% (в форме окислов и кислородных солей). Общее содержание кислорода в земной коре, океане и воздухе оценивают примерно в 50 вес.%, т. е. кислород принимает такое участие в строении земной коры (включая атмосферу), как все остальные элементы, вместе взятые,
В результате действия силы тяжести состав воздуха с увеличением высоты должен изменяться в сторону увеличения доли легких составных частей (гелий, водород) по сравнению с тяжелыми (кислород, аргон). Однако этот эффект проявляется только яа очень больших высотах; вблизи Земли он не может проявляться из-за постоянного перемешивания составных частей атмосферы под влиянием конвекции. Оказалось, что конвекция распространяется на значительно большие высоты, чем предполагали ранее. Как нашел Панет, исследуя пробы стратосферы, полученные при помощи ракет [J. atm, terr. Phys., 1, 49, 1950; Nature, 168, 358, 1951 i, процентный состав воздуха вплоть до высоты 60 «л» остается таким же, как и у поверхности Земли. Только па больших высотах заметно вызванное гравитационным разделением изменение соотношения составных частей воздуха.
Кислород, встречающийся и природе, является смесью трех изотопов с массовыми числами 16,17 и 18 (см. т. 1[, гл. 12). Вследствие колебания соотношения изотопов атомный вес атмосферного кислорода на II.00011 атомных единиц больше атомного веса кислорода лз морской воды (Dole М., 1936). Эту незначительную разницу следует принимать во внимание, так как атомный вес кислорода (точнее, атомный вес природной смеси изотопов кислорода) лежит в основе величин атомных весов остальных элементов. (Об атомпом весе изотопа С12 — 12 как основе для шкалы атомных весов см.
Шестая группа периодической системы
УЛ
т, II, гл. 12.) Различие изотопного состава кислорода, зависmn.ee от способа его получения, найдено также Юри (Urey). Он установил, что содержание Ols в водах современных морей примерно на 0,7% ниже, чем и первичных породах (граните, базальте), и в среднем на 0,7% выше, чем в осадочных породах (см. также т. II, гл. 12),
Исторические сведения. До того как был открыт кислород, воздух Считали про стым веществом. Мысль о том, что воздух доджей состоять из двух веществ, из которых только одно может поддерживать дыхаппе и горевие, впервые была ясно выражена Шееле в 1777 г. в его сочинении «Трактат о воздухе и огне». Опыты, которые привели Шесле к такому выводу, были проведены им еще за несколько лет до этого. Впервые Чистый кислород он получил при сильном прокаливании селитры; позднее он получил кислород также при действии концентрированной серной кислоты на пиролюзит (двуокись марганца). Независимо от Шее ле в 1774 г. Пристли получил кислород нагреванием окиси ртути и сурика. Открытие кислорода послужило Лавуазье основой для создания его теории горения. Поскольку Лавуазье считал этот элемент существенной частью кислот, он назвал его principe acidlftant или Оху gene (от — кислый, yevvico— получаю), или по латыни Oxygenium. Несмотря па то, что позднее было установлено, что существуют бескислородные кислоты, название, данное Лавуазье этому элементу, сохранилось.
Получение, В настоящее время в технике кислород получают преимущественно фракционированной перегонкой жидкого воздуха, по методу Линде или фракционированным сжижением воздуха по методу Клоде. Кроме того, значительные количества кислорода л плутают электролизом воды (см. главу о водороде). Получаемый при сжижении воздуха кислород обычно содержит немного (в среднем 3%) аргона, температура кипения которого близка к температуре кипения кислорода.
В продажу кислород поступает в стальных баллонах. В лабораториях в большинстве случаев пользуются этим кислородом. Если его надо получить в лаборатории, то используют электролиз (например, калиевой щелочи с никелевыми электродами) или получают его термическим разложением соединений кислорода, например нагреванием хлората калия (к которому примешивают для ускорения разложения пиролюзит или другие катализаторы) или перманганата калия.
Кислород в точно рассчитанных количествах можно получить, если раствор перманганата калия вливать по каплям в подкис ленный серной кислотой раствор перекиси водорода (ср. стр. 79). Очень удобным во многих случаях методом является взаимодействие хлорной извести (в кубиках) с раствором перекиси водорода, которое можно проводить в аппарате Киппа.
Для до к азате лъства присутствия следов кислорода в газах можно воспользоваться истодом Каутского и Хирша [Kautsky, Н i г s с h, Z. nnorg. Chcin., 222, 126, 1935]. Он основан на том, что фосфоресценция трипафлавипа (акридинового красителя), адсорбированного на Силикагеле, подавляется следами кислорода. Перед анализом пробы надо удалить И2О и NH3, которые также подавляют послесвечение.
Небольшиекодичес.тва кислорода, согласно МакАртуру [McArthur,), appl, Cbeia., 2, 91, 1952], можно легло определить, поглощая кислород раствором СгС1г известной концентрации, окисляя избыток СгС12 железными квасцами н оттлтровывая образующиеся ионы Fe" бихроматом. О потенциометрическом определении следов кислорода в газах пли водных растворах см.: L a i t i п о n, Higuchi, Czuka, J, Am. cbem. Soc., 70, 561, 1948.
Для очистки применяемых в лаборатории защитных газов (водорода, азота, инертных газов) от кислорода пользуются главным образом «медной колонкой» Фрике (Fricke) и Мейера (Meyer, 1939), см. также Fricke, Z. Elektrochern., 53, 76, 1949. Эта колонка состоит из осажденной на кизельгуре активировав noli меди, которая, будучи нагрета до 160°, умепыпает парциальное давление кислорода в газе до менее чем ЗЛО’4 мм рт ст, Следовательно, после прохождения через медную, колонку газы, находящиеся при атмосферном давлении, содержат пе более чем 4Л0*а % кислорода.
Свойства и применение. Важнейшие физические копстапты кислорода приведены в табл. 107 (стр. 742). Обычный элементарный кислород состоит из двухатомных молекул О2 (относительно озона Os см. ниже).
742
Глава 75
Таблица 107
Физические константы кислорода
Вес 1 л при 0°, 760 -«-и рт ст и иа 45° географической широты 4,42895 г.
Плотность (по отношению к плотности воздуха, принятой за 1) 4,4053; удельный вес жидкого кислорода при температуре кипения 4,448; удельпый вес твердого кислорода (при — 2523) 1,126.
Т. кип, —483,00°; т. пл. —218,8°; крит. т. —448,9°; крит. дав ленке 49,7 атм,; крит. плотность 0,430.
Удельные теплоемкости газообразного кислорода (прп 20°); ср—0,2184 кал, ct,= =0,183 кал, Ср/а(, = 1,395. Молярные теплоемкости: Ср = 6,989 кал, С„=5,84 кал.
Удельная теплоемкость жидкого кислорода при —200* ср=0,394 кал.
Удельная теплоемкость твердого кислорода при —222* ср=0,336 кал; при —256° ср = 0,078 кал.
Теплота испарения жидкого кислорода 54,0 кал/г (при —183°).
Теплота плавления твердого кислорода 3,3 кал/а (при —219°).
Теплопроводность кислорода при 0° и 1 атм (ср. табл. 7) \ — 0,0000583 вял-сл»*1-
еек^граЬ^1.
Растворимость кислорода в воде: при 0° = 0,0491 при 20°—0,0311, прп)100°—0,0170 об. (приведенного к 0°) в 1 об. воды.
Магнитная восприимчивость х — 104,4-10"6 (при 20°), парахор 54,0,
Молекула О2‘ дипольный момент 0, момент инерции 19,23-10г-см-, межъядерное расстояние О-<—?-0 = l,207t 10*8 энергия диссоциации 5,084 эв (117,2 хал/
/моль), валентно-силовая констапта 11,7-40s дин!см.
Озон О3: т. кип. —111,5°, т. пл. —251,4°; крит. т. —5°, крит. давление 92,3 атм; крит. плотность 0,54; уд. вес при температуре кипении 1,46; молекулярная теплота испарения; X = 3500 -ф- 3,499Т—0,0581IT2- кал; дипольный момент 0,49-40~18 эл.ст.ед; магнитная восприимчивость: X=0,15-10~s (при —183°); растворимость в воде: при 0° 0,494; при 18° 0,454 об. (приведенного к 0°) в 1 об. воды. Энергия диссоциации О3—><1О; 141,2 хкал/жоль; межъядерное расстояние см. стр. 745; валенгно-силовая константа 7,5-Ю5 дня! см.
Шидь-нп кислород светло-голубого цвета. Оп довольно сильно парамагнитен, э.тектрияссгпа по проводит. Твердый кислород также светло-голубого цвета. При температурах немного ниже температуры затвердевания кислород имеет гексагональную кристаллическую решетку. При более сильном охлаждении (—230°) его кристаллическая структура изменяется. При —255° происходит новый фазовый переход.
Кром:е NO, кислород является единстве иной существующей при обычных температурах в газовой фазе двухатомной молекулой, для которой уетаповлен парамагнетизм. Но тогда как молекула NO содержит нечетное число электронов, в молекуле О2 число их четное. Из наличия у молекулы О2 парамагнетизма можно заключить, что не все ее электроны образуют пары с противоположно направленными спинами (ср. гл. 9). И действительно, как показывают спектроскопические данные (основное состояние 33), молекула О2 содержит два «неспаренпых» электрона (см. также стр. 157 я сл.).
Полинг считает, что в молекуле О2 имеется пё одпа образованная двумя парами электронов двойная связь, а одпа простая связь, окруженная двумя трехалектронными связями. С этой точки зрения строение молекулы О2 будет выглядеть следующим образом 10-гтт- О |. Обычно прочность трехэлектронной связи составляет максимум половину прочности простой связи. Если бы имеющиеся в молекуле | О —-j— О | три связи только складывались, вряд ли получилась бы прочность связи, большая прочности обычной двойной связи. Однако обо трех электродные связи взаимодействуют между собой, результатом чего, как можно показать теоретически, является существенное увеличение устойчивости связи в том случае, если спины двух неси арен пых электронов имеют одинаковое направление. Так, получается, что 32-состояние молекулы О2, которое фактически имеет место, лежит на 0,97 ав ниже, чем ^-состояние, в котором имеется нормальная дврйпая связь между атомами кислорода и результирующий спил которой равен нулю. В свою очередь 1А-состояние лежит на 0,67 эв ниже ^-состояния, в котором, по мнению Полипга, так ;ко как и в 33-состоянни, имеется одна простая связь,
Шестая epynnd периодической системы
743
окруженная двумя трехэлектрон пымн связями, но взаимодействие между песпаренными электронами яе увеличивает, а уменьшает прочность связи, так ван в этом случае спины обоих электронов не складываются, а, как видно из обоаначепия терма, друг .друга компенсируют.
Одной из наиболее характерных особенностей кислорода является -ei'o способность соединяться с болыпзнством элементов с выделением тепла и света. Чтобы вызвать такое соединение, сгорание, часто требуется нагревание до определенной температуры — температуры воспламени ния, так как при обычной температуре кислород является довольно инертным веществом. Однако в присутствии влаги медленное соединение с кислородом (медленное сгорание) происходит уже при обычных температурах. Важнейшим примером такого процесса является дыхание живых организмов. Но и другие протекающие при обычных температурах процессы медленного горения в природе весьма многочисленны (см. также стр, 821 н сл.),
К процессам медленного горения относятся ржавление и потускнение металлов, тление дерева и другие процессы гниения н разложения. Вследствие длительности таких процессов выделяющееся при этом тепло целиком рассеивается в окружающем пространстве. Однако в некоторых условиях такое тепло может скапливаться н привести к воспламенению, т. е. к началу энергичного, протекающего с появлением огня горения. Так происходит «самовоспламенение» влажного сена, соломы, угля и других горючих материалов. Поэтому во влажном состоянии их нельзя хранить в больших количествах. Для лучшего теплоотвода их следует проветривать и постоянно контролировать температуру,
В специально сконструированной горелке за счет сгорания водорода ® кислороде можно получить температуру выше 2000'. Еще более высокую температуру получают в пламени ацетилено-кислородной горелки. Пламеней! таких горелок пользуются для сварки и резки металлов, плавления платины, кварца и других очень тугоплавких материалов. Жидкий кислород пли сильно обогащенный кислородом жидкий воздух часто применяют для изготовления взрывчатых веществ, которые получают смешиванием пористого угля или других горючих составляющих, например нефти, парафина, нафталина, с жидким кислородом или жидким воздухом (оксилнквит). В лабораториях жидкий кислород й особенно жидкий воздух часто применяют для создания низких температур, а также, например, для очистки трудно сжижающихся газов от легко конденсирующихся примесей, таких, как вода, двуокись углерода («вымораживание»). О других применениях кислорода см. Лашин (Laschin) и Каущ (Kausch).
В настоящее время все большее значение приобретает применение кислорода в дыхательны» аппаратах, которые используют в самолетах, подводных лодках, при тушении пожаров и т. д. Эти аппараты представляют собой стальные баллоны, наполненные кислородом под давлением; в других случаях кислород получается в них непосредственно за счет разложения перекиси щелочного металла (см. стр. 204). Применяют аппараты, в которых имеется запас жидкого кислорода. Дыхательные кислородные аппараты имеют дополнительное устройство для поглощения выдыхаемой двуокиси углерода. Кислородные дыхательные аппараты особой конструкции применяют для оказания помощи пострадавшим от удушья, утонувшим или при других случаях потери сознания или клинической смерти. Взрослый человек в спокойном состоянии потребляет около 20 л кие лороj (а в час, при физических нагрузках — в несколько раз больше. Вдыхание чистого кислорода безвредно, если только его давление не превосходит 1 атм.
ОЗОН
Появление озона наблюдается всегда, когда происходит электрический разряд в воздухе, например в помещении, где работает электрическая машина пли искровой генератор. Озон узнают по характерному запаху,
Гла-111 la
от которого он и получил свое название (o£evy — пахнуть). Уже в 1785 г. ван Марум отмстил появление этого запаха при работе электрической машины. Шёнбайн, давший название этому веществу, констатировал (1840), что оно образуется при электролизе разбавленной серной кислоты. Озон получается также при медленном окислении влажного белого фосфора на воздухе, при разложении богатых кислородом веществ (перманганат, бихромат) концентрированной серной кислотой. Кроме того, он образуется при действии фтора на воду и при действии ультрафиолетовых лучей на кислород воздуха. Поэтому запах озона всегда ощущается вблизи кварцевой ртутной лампы.
Раньше озон принимали за высший окисел водорода; однако удалось показать, что он состоит только из кислорода. Его можпо получить из чистого сухого кислорода и вновь перевести в кислород. При этом 2 об. озона дают 3 об. обычного кислорода. Отсюда на основании закона Авогадро для молекулы озона получается формула О3. Правильность этой формулы подтверждена измерением плотности пара.
Долго оставался спорным вопрос о том, содержит ли воздух вблизи земной поверхности, например в горных местностях, озон. Исследования, проведенные химическими методами, указывали на содержание озона порядка нескольких миллионных процента. Так как в таких методах для доказательства присутствия и определения озона использовали его окисляющее действие, не была исключена возможность того, что окисление вызвано другим присутствующим иногда в воздухе в виде следов вещест-; вом (например, NO2), поэтому доказательство присутствия озона, полученное такими методами, позднее оспаривалось. Однако в конце концов Еюисон (Buisson) и Фабри (Fabry, 1930-1931) и Гоц и Ладенбург (Gotz, Laden burg, 1931) доказали независимо Друг от Друга присутствие озона в атмосфере на основании изучения спектра поглощения воздуха в ультрафиолетовой области. Найденные ими значения по порядку величины согласуются со значениями, полученными ранее химическими методами. Содержание озона в атмосферном воздухе объясняется в основном тем, что в верхних слоях атмосферы молекулы кислорода О2 расщепляются ультрафиолетовым излучением па атомы, которые образуют с другими молекулами О3 (при тройном Столкновении) озон ; О2 Ч--ф-0 = О3 4- 24 ккал. Молекулы О 3 частично снова расщепляются светом и вновь образуются при соударениях молекулы О2 с атомами О. За счет одновременного образования 11 разложении озона в атмосфере устанавливается стационарная (зависящая от температуры и давления) концентрация озона, достигающая максимальной величины на высоте около 25 л.«. Общее число молекул озона во всей атмосфере равно числу молекул О, и А 2 в слое поз iv у а то пн дней в тч’коль ко миллиметров (при нормальных условиях). (Вся атмосфера, ег .ш бы ее плотность не уменьшалась е высотой, простиралась бы на 8 пад поверхпоётью Уемтп.! В ультрафиолетовой области спектра озон имеет чрезвычайно большое поглощение. При длипе волны Z _ь 0,25 у озон поглощает сильнее, чем любой металл в видимой части спектра. Поэтому слоя атмосферного озона, несмотря па его чрезвычайно низкую концентрацию, достаточно для полной защиты органической природы от смертоносного действия ультрафиолетовой части солнечного излучения, а именно излучения с длиной волны, меньшей 0,2 р,. Вертикальные потоки воздуха, интенсивность которых сильно колеблется в зависимости от времени года и погоды, приносят часть образующегося в верхних слоях атмосферы озона к поверхности Земли. Но здесь под действием атмосферной пыли нт. д. он быстро разлагается. Поэтому содержание озона близ земной поверхности очень мало. «Озонированный» воздух хвойных лесов — выдумка. Но все же при благоприятных климатических условиях локальными направленными вниз потоками воздуха озон может сильнее, чем где-либо, попадать в пижпие слои атмосферы и уничтожать пыль, запахи и другие загрязнения воздуха. В этом состоит его косвенное оздоровительное действие. Однако никаких доказательств непосредственного физиологического действия воздуха с природным содержанием озона пока пет.
Для получения, озона в настоящее время обычно пользуются действием тихого электрического разряда на кислород в озонаторе Сименса. Основную часть такого озонатора составляют две вставленные одна в другую стеклянные трубки, между которыми пропускают озонируемый, хорошо высушенный кислород (или воздух). Стеклянные трубки покрыты обкладками, через которые проходит переменный ток высокого напряжения; таким образом, через кислород проходят тихие разряды. Под их действием
Шестая группа периодической системы
небольшая часть кислорода превращается в озон. Большего обогащения кислорода озоном можно достигнуть, пропуская его последовательно через несколько озонаторов. Таким путем легко можно получить озон — кислородные смеси с содержанием озона до 10% и более.
Чистый озон (физические конетапш си. в табл. 107) можно выделить из озон-киелородной смеси сжижением. Уже в газообразном состоянии озон окрашен в голубой цвет; в жидком состоянии он темно-синий, почти черный. Как нашел Ризенфельд, при низкие температурам озон пе во всех отношениях смешилаетея с воздухом.
При распаде озона освобождаются значительные количества тепла: О3=3/а 03+ 34,2 ккал (при постоянном объеме). Поэтому чистый озон легко взрывается.
Разбавленный озон при обычных темпера турах распадается очень медленно, Кроме нагревания, сильно ускоряют распад некоторые вещества, например двуокись марганца, окись свинца, натронная известь; распад ускоряется и при облучении ультрафиолетовым светом.
Работа с разбавленным озоном, получаемым в озонаторе, не опасна. Следует только учитывать, что в больших концентрациях озон разъедает слизистую дыхательных путей. Резиновые шланги разрушаются озоном моментально. Поэтому соединения трубок нужно уплотнять ртутью. На органические соединения озон, как правило, действует очень энергично уже при обычных температурах: под действием озона выцветают органические краски, разрушаются микроорганизмы. Многие вещества, такие, как эфир, спирт, светильный газ, смоченная скипидаром вата, при действии озонированного воздуха воспламеняются. При осторожном действии озона ца ненасыщенные органические соединения часто получаются так называемые озониды, причем озон присоединяется по месту двойной связи. Озониды, как правило, очень неустойчивые соединения, легко разлагаются водой. Эту реакцию часто можно использовать для установления строения органического соединения или в препаративных целях.
Озон, как правило, взаимодействует со многими веществами, с которыми кислород прп обычных температурах реагировать не может. При действии озона черный сульфид свинца PbS переходит в белый сульфат PbSO4, белая гидроокись свиица(П) РЬ(ОН)3 переходит в коричневую двуокись РЪОа. Особенно характерным для озона свойством является способность черппть металлическое серебро с образованием перекиси серебра AgO. Для количественного определения озона в большинстве случаев пользуются реакцией с иодидом калия в нейтральном водном растворе О3+ 2KI -f- Н30 = Ia+ 2КОН -р Оа. Выделяющийся иод при и одкнслепип оттитровывают тиосульфатом.
Кроме йспользования в препаративной химии, озон применяют как очищающе!? воздух и стерилизующее средство, например на бойнях, в пивоваренном производстве, холодильниках и для обезвреживания воды. Первые большие установки для озонирования воды были построены в Висбадене ц Падерборне, затем в Ницце, Германштадте, Петербурге, Париже. В настоящее время озонирование воды с целью ее обезвреживания постепенно вытесняется более дешевым хлорированием.
Строение молекулы озона. Как показали злектронографические исследования (Shand, Spun, 1943), в молекуле озона три атома кислорода образуют тупоугольный треугольник с длинами сторон 1,26—1,26—2,26 А м углом 127°. Межъядерные расстояния пе подтверждают предположения
Глава 15
о том, что между атомами О основания треугольника существует химическая связь, т. е. опровергают предполагавшуюся раньше циклическую структуру.
Вероятно, в молекуле озона существует мезомерия между следующими формами:
Две последние формы вносят, по-видимому, значительно меньший вклад в мезомерпое промежуточное состояние, чем две первые, поскольку на основании величины дипольного момепта и межатомных расстояний можно заключить, согласно Мельчеру (Melcher, 1946), что каждая из связей 0—0 только на 10% имеет характер двойной связи.
Атомарный кислород. При тлеющем разряде при пониженных давлениях (менее 1 мм рт ст.) кислород, как и водород (только с меньшим выходом), распадается на атомы. Атомарный кислород, согласно Хартеку (Harteck, 1930 и сл.), обладает сравнительно не Очень высокой реакционной способностью- Например, столкновения с молекулами Н2 только в 0,2% случаев приводят к образованию ЩО. В небольшой степени реагирует атомарный кислород и с СН4 (менее чем на 1%), с HCN (5%) и с СО (5%) и, напротив, на 100% с НВг, HgS, CSs, СН3С1, СН2С1г, СНС13 и многими другими органическими соединениями. Если вести реакцию атомарного кислорода с органическими веществами при очень низкой температуре (температуре кипения жидкого кислорода), то часто получаются — как и в случае низкотемпературных реакций атомарного водорода (ср, стр. 63) — соединения, которые при обычных температурах неустойчивы. С кислородом атомарный кислород реагирует по уравнению О2-[- О = О3. Образующийся за счет этой реакции озон при новом соударепии с атомом кислорода вновь превращается в обычный кислород 03 -р О —2 О2, Этим объясняется то, чТо получаемый при низких давлениях атомарный кислород не содержит больших количеств озона.
Активированный кислород. Реакционную способность кислорода можно повысить, активируя молекулы Оа. Это происходит, как показал Каутский (1931—1933), в том случае, если молекулы О3 привести в контакт с красителями, возбужденными светом до состояния флуоресценции. Ес ли столкновение с молекулой 02произойдет прежде, чем возбужденная квантом света молекула красителя попустит полученную ею энергию в виде флуоресцентного изучения, то энергия возбуждения может перейти от молекулы красителя к молекуле О,. За счет этой энергии такая молекула 02на короткое время переходит в состояние, в котором опа способна вызывать реакции окисления, не идущие под действием нормального кислорода.
СОЕДИНЕНИЯ КИСЛОРОДА
Окиси и перекиси. Продукты соединения кислорода с другими элементами называются окисями, а сам процесс — окислением.
Название окисление, кроме процессов соединения с кислородом, часто употребляют в общем смысле для тех процессов, которые по своей сути аналогичны соединению с кислородом, папримор для процессов соединения с галогенами, серой и другими элементами электроотрицательного характера. Подробнее об) этом см. в следующей главе.
Различают нормальные окиси и перекиси. Нормальные окиси формально являются производными воды как основного вещества, в которых атомы водорода замещены на эквивалентные количества иных атомов (или радикалов). Перекиси формально являются производными перекиси водорода. Они содержат характерную группу — О — О — (перекисную группу) или соответствующий ион [О—О]2-. Каждый из двух кислородных атомов перекисной группы имеет только одну свободную валентность, так как вторая используется на образование связи между атомами кислорода.
Шестая группа Периодической. системы.
747
Таким образом, окиси являются соединениями перекиси — с общей формулой RJO.,.
Примеры нормальных окисей
Н
с общей формулой ЩО,
Перекиси
С1.
НзС\, н3с/
И—о R1—О
I; I; Ва‘П08р-.
R1-
W
Приведенные в качестве примеров окиси соответствуют воде пе только формально, по и структурно, если только они построены иа отдельных молекул. Такие окиси в большинстве случаев газообразны или легко испаряются; опи образуются главным образом неметаллами. Большинство окисей .игталлм трудно летучи и имеют «солеобразный» характер (см, гл. 9). Промежуточное положение между этими двумя классами занимают некоторые окиси, которые, как В308 и SiO3, труднолетучи, но имеют тенденцию образовывать стекла при застывании.
Как в нормальных окисях, так и в перекисях кислород двухвалентен. Очень редко встречаются соединения, в которых кислород одновалентен.
При окислении метилового эфира фенаптрепгидрохинона калийцианоферра-том (Ш) или окисью свинца получается перекись
Само это соединение бесцветно, тогда как его растворы окрашены. На этом основании, а также из результатов определения молекулярного веса, согласно Гольдшмидту (1922), можно сделать вывод, что при растворении происходит частичный распад молекул по связи, обозначенной 4- Поэтому в сильно ненасыщенном продукте диссоциации кислород формально можно рассматривать как одновалентный. Этот случай совершенно аналогичен соединениям с формально трехвалентным углеродом. Долгое время предполагавшееся существование только мономолекулярных органических соединений одновалентного кислорода не подтвердилось. Следует отметить существование одно-валентпой группы 0« , т. е. отрицательного нона с нечетным числом валентных электронов, образованного двумя атомами с общим четным числом валентных электронов, существующем в так называемых «двуокисях щелочных металлов» М[03 и «четырех-окисях щелочноземельных металлов» Мп04. на основании их строения эти соединения называют теперь на ^перекисями щелочных и щелочноземельных металлов (ср. стр. 202 и 298).
Окиси получают главным образом двумя следующими методамй:
1. Непосредственным соединением элементов с кислородом. Кислород непосредственно соединяется с большинством элементов, за исключением благородных металлов (образование минимального количества окиси на поверхности) и галогенов. При непосредственном взаимодействии с кислородом большинство щелочных металлов дает не нормальные окиси, а перекиси или надперекиси. О получении перекисей и надперекисей щелочных металлов см. гл. 8.
2, Нагрева и ием гидроокисей или других легко разлагающихся кислородных соединений. Многие гидроокиси уже при обычных температурах распадаются на окиси и воду, например гидроокиси серебра и ртути. Так же распадается и устойчивая только в водных растворах угольная кислота. Из других легко разлагающихся соединений для получения окисей металлов используют прежде всего карбонаты и нитраты.
Большинство окисей способно соединяться с водой с образованием гидроокисей. Те окиси, которые при этом образуют кислоты, называются
748 Глава 15
ангидридами кислот. Большинство окисей металлов ведет себя как ангидриды оснований. Но многие окиси имеют амфотерный характер, т. е. соответствующие им гидроокиси могут диссоциировать и как основания, и как кислоты. Имеются также окиси металлов, которые являются ярко выраженными ангидридами кислот, например трехокись хрома СгОз, окись марганца(УП) Мп2О7. В этих случаях всегда речь идет об окисях высшего валентного состояния соответствующего металла. Есть также индифферентные окиси, т. е. окиси, которые не являются ни ангидридами оснований, ни ангидридами кислот, например закись азота.
Большинство окисей очень устойчиво к нагреванию. Только окиси благородных металлов разлагаются уже при слабом нагревании. Из перекисей устойчивыми к нагреванию являются только перекиси щелочных металлов (за исключением лития).
Многие окиси при нагревании с водородом или углем восстанавливаются до свободного элемента.
О получении гидроокисей уже говорилось в гл. 2. Гидроокиси большинства тяжелых металлов, а также гидроокиси бериллия и магния в воде-нерастворимы; значительно более растворимыми являются гидроокиси щелочноземельных металлов, а гидроокиси щелочных металлов растворяются легко. В воде легко растворяется и большинство гидроокисей неметаллов (кислородные кислоты).
Оксониевые соединения. Многие окиси (и перекиси) способны образовывать с другими веществами продукты присоединения (соединения высшего порядка). Это свойство присуще в первую очередь воде', оно-выражается в образовании ею многочисленных гидратов. Но и многие другие кислородные соединения проявляют способность к присоединению. Среди неорганических соединений это в первую очередь гидроокиси (и перекись водорода), среди органических — спирты, эфиры, альдегиды и кетопы. Эти кислородные соединения могут присоединяться как к солям, металлов, так и к кислотам. Например:
(СИ0)5О lb 1 = (CHi,)2O.HCl.
Образующиеся при этом двойные соединения называются оксониевыми соединениями. Обычно они имеют солеобразный характер. Их общая формула такова: (R3O)n-MX, где R3O — какая-либо окись, а MX — соединение, состоящее из положительного радикала М (обычно это металл или водород) и отрицательного радикала X (кислотный Остаток или гидроксил). Чйсло п молекул R.,O, которое может приходиться на 1 молекулу МХ„ может быть и больше 1
Образование оксониевых соединений объясняется, по Верперу, тем, что кислород может выступать как координационно трехвалешпный, т. е. за счет координационной связи он может удерживать непосредственно 3 лиганда. При этом третий лиганд может быть нейтральной молекулой. Однако с присоединением к атому кислорода может быть связана диссоциация присоединившейся молекулы, так что непосредственно при соединенным оказывается только электроположительная часть, а электроотрицательный остаток электростатически удерживается в этом случае положительным зарядом образующегося комплекса, пе будучи связанным с каким-либо определенным местом этого комплекса. Первый случай можно представить формулой I, второй — формулой П:
>0 • • мх Rz
FR
0-М R'
II
I
X
Шестая группа периодической системы
749
В каждом случае имеется координационно ’трехвалентный. кислород. Электрохимическая валентность кислорода в оксониепых соединениях такая яге, как и в окисях, производными которых они являются, т. е. также, поскольку кислород в утих соединениях можпо рассматривать как заряженный, она равна 2—. Несмотря на отличающуюся на единицу электровалентность центрального атома и на единицу меньшее координационное число, оксониевые соединения являются полными аналогами аммониевых соединений.
И с точки зрения теории атомных связей соединения оксония будут полными аналогами соединений аммония. Как и в случае последних, присоединение третьего лиганда в оисонневых соединениях происходит, согласно теории атомной связи, за «Чет датиеной свяви
R\ и\ Гйх
[Of 4- И= [О > MX, соответственно (О —> Ы
К/" Lrz
Различие между соединениями оксония и соединениями аммония заключается в том, что в соединениях оксония третьим лигандом у центрального атома обычно является металл иля водород. Однако Меервейпу (1937—1939) удалось получить а «третичные оксониевые соли» [R3O)X, соответствующие четвертичным солям аммония [U4N]X.
Раньше кислород в солях оксопия считали четырехвалентним. Эта точка зрения основывалась главным образом на развивавшихся фот* Байером (я. Ваеуег) и Виллиге-ром (Villiger) представлениях о строении этих соединений, согласно которым обе части соединения MX должны быть непосредственно связаны с кислородом окиси В20. Однако после того, как Вернер показал, что соединения оксония, вне всякого сомнения, являются координационными соединениями, не осталось более никаких причин для допущения необычной четырехвалентпости кислорода. Даваемое координационной теорией объяснение строения оксониевых соединений охватывает и такие случаи, когда на один атом кислотного водорода приходится более одной молекулы окиси. Такие соединения, поскольку их строение нельзя было объяснить на основании теории о четырехвалентпости кислорода, называли «аномальными оксониевыми солями». Существование таких соединений, число которых отнюдь не столь мало, надо было предвидеть с точки зрения теории Вернера, так как пет никаких причин, чтобы водород всегда проявлял только одну побочную валентность (т. е. чтобы его координационное число никогда не было больше двух).
Ион гидроксония. Основную группу оксониевых соединений образуют соединения Н2О’НХ или [Н30]Х. Ион [Н3О]+ называют ионом сидроксония. Последний, например, существует в перхлорате гидроксония {Я3О)+[СЮ4]_ (ср. стр. 863),
Образование иона гидроксоппя [Н30]+, как и вообще образование оксониевых соединений, формально можпо объяснить координационной трехвалснтностыо кислорода- В случае этого простого комплексного иона можно указать причину, почему кислород проявляет именно, координационную валептпость 3. А именно, если вычислить энергию, которая выделяется при присоединении 1, 2, 3 и т. д, протонов к иону кислорода с зарядом 2—, то получим, что эта энергия болоте в случае Присоединения ЗН+ к 10s-, чем при присоединении 211+ к 102~. I
Если для упрощения расчета не принимать во внимание поляризацию и происходящее, но всей вероятности, внедрение протонов через внешнюю Электронную оболочку внутрь атома кислорода, то получим (при условии, что г0—радиус иона кислорода, (А)]:
^2
Ог--|-Н+ = ОН-4-2— 103 зрг; {И
Со
«а |.)Н-4-П+ = ОН2+1,5-5-10« эре; (21
го
е2
0П2 |-H+ = fOH,p 4-0,8 —10s эр»; (3)
го
[0П3]1 4-Н* = [OIIJ2 + 4-0,06 — 10s (41
4
Отсюда видно, что энергия, выделяющаяся прп при со единении протона к молекуле НаО, хотя и меньше, но того яге порядка, что и энергия, выделяющаяся при присоединении
750
Глава 15
протона к нону гидроксила ОН-. Отсюда следует, что процесс (3) может конкурпро- |
вать с процессом (2), что проявляется в частичной электролитической диссоциацЕ? 1
поды. Равновесие диссоциации воды в принципе* можно представить следующие: ]
образом: |
2НгО . ( [Н3О] ++ОН~. (5.1
Равновесие (5) очень сильно сдвинуто влево, но все же диссоциация воды проис- 1
ходят в измеримой степени**. Если присутствуют соединения, которые легче отщепли- ij
ют протоны, чем вода, что имеет место для большинства кислот, то Степень диссоцпа- ;
ции, конечно, будет значительно выше. -
Наоборот, как видно из приведенных выше расчетов, нельзя ожидать образования j
в измеримом количестве ионов [ОТЦр*. До сих нор фактически не получено никаких j
доказательств существования таких ионов. Вообще соединения, в которых кислород
имел бы координационное число большее 3, не известны: координационное число 3 является ,наксшиа.гьны.и координационным числом кислорода***.
С точки зрения теории атомной связи образование иона гидроксония объясняется, как и вообще образование оксониевых соединений, возникновением дативной связи за счет кислорода
н\ Гн\ 1 +
|О|+н+= |О_>ц
HZ Lhz
Тот факт, что не происходит присоединения четвертого протона, хотя в ионе (Н30]* имеется еще одна, пе использованная для образования связи пара электронов, объясняется тем, что эта пара электронов образована s-электронамп, которые при возникновении гомеополярной связи сообщают ей значительно меньшую устойчивость, чем участвующие в образовании связей с тремя протонами р-электроны (ср, стр. 162).
Продукты присоединения с молекулярным [кислородом. Многие кислородные соединения могут образовывать продукты присоединения с молекулярным кислородом. К таким соединениям относятся, по данным Тpayбе (Trauhe, 1916), так называемые-«озоновокислые щелочи». Они образуются при действии озона на гидроокиси щелочных металлов и представляют собой интенсивно окрашенные оранжево-желтые про-дукты, которые при растворении в воде или кислотах по образуют никакой перекиси водорода, а отщепляют только газообразный кислород, вследствие чего Траубе-придисал им формулу (МЮНЬ., .О2.
СЕРА (SULFUR) S
Распространение в природе. В природе часто встречаются значитсяь ные залежи серн (большей частью вблизи вулканов); в Европе она встречается прежде всего в Сицилии. Еще большие ее залежи имеются в Америке (в штатах Луизиана и Техас), а также в Японии. В вулканических местностях часто наблюдается выделение сероводорода H„S как подземного-газа; там же он встречается и в растворенном виде в серных водах. Вулканические газы часто содержат сернистый газ SO.,. Очень распространены сернистые соединения металлов. Наиболее часто встречающиеся сульфиды: железный колчедан (пирит) FeS2, медный колчедан CuFeSa, свинцовый блеск PbS и цинковая обманка ZnS. Еще чаще сера встречается в виде сульфатов, например сульфат кальция (гипс и ангидрит), сульфат магния (горькая соль и кизерит), сульфат бария (тяжелый шпат), сульфат стронция (целестин), сульфат натрия (глауберова соль).
* Следует учесть, одиако, что [Н3О]*-и ОН“-ионы гидратированы, а молекулы-Н20 ассоциированы.
** В качестве энергия, необходимой для диссоциации, используется тепловая энергия.
*** Последнее утверждение автора пе точно. Кислород в ряде соединений обладает координационным числом 4, например в некоторых окислах: ВеО, ZnO и др. —Прим, ред.
Шестая группа периодической системы 751
В доступной части твердой земной коры содержится примерно 0,03 вес.% серы; мировой океан содержит (в виде сульфатов) около 0,09% серы, В метеоритах часто обнаруживают сернистое железо Fей. Считают, что на больших глубинах под силикатной оболочкой (см. т. II) имеется зона, состоящая в основном из сульфидов. Сера относится к элементам, которые необходимы для органического мира, так как опа является существенной составной частью белков. Они содержат 0,8—2,4% химически связанной серы. Растения получают серу из сульфатов, содержащихся в почве. Неприятные запахи, возникающие при гниении трупов яшвотных, объясняются главным образом выделением соединений серы (сероводорода и меркаптанов), образующихся при разложении белков.
Каменные угли содержат в среднем 1 — 1,5% серы, частью в виде органических соединений, частью в виде серного колчедана.
Исторические сведения. Так как сера встречается в природе в самородном состоянии, опа была известна человеку уже в глубокой древности Применение горящей серы для дезинфекции упоминается еще Гомером Диоскорид сообщает о применении серы в медицине. Большое внимание уделяли сере алхимики. Многим из них была уже известна серная кислота. Василий Валентин в XV в. подробно описал ее получение (нагреванием железного купороса). Фабричным способом серпая кислота была подушена впервые в Англии в середине XVIII в. (см. стр. 761).
Получение. Серу получают главным^образом выплавлением ее из горных пород, в которых она содержится в чистом состоянии.
В Сицилии, которая до 1914 г. была основным поставщиком серы, ее получали раньше и частично получают и сейчас способом, при котором содержащие серу породы складывают в большие кучи {calearonty, кучи покрывают глиной или отработанной рудой и проделывают в них вертикальные каналы для воздуха. Затем их сверху поджигают. Сера медленно сгорает, а нес тор евшая сера плавится и стекает вниз в подставленные формы. Выплавка серы в такой куче может длиться, смотря по ее величине, от 1 до 3 мес. Выход сырой серы составляет 50—70% от ее количества, содержавшегося в породе. Значительная часть серы теряется в виде сернистого газа. Вследствие вредного влияния SOa на растения законом запрещено сжигание серных куч в период с августа по декабрь.
Выплавленную в кучах сырую серу очищают дистилляцией из чугунных реторт. Пары отводят в большие, выложенные кирпичом камеры, где они оседают в виде серных цветов (серного цвета)г если температуру в камерах поддерживать ниже 112°. Если температура поднимется выше, то на щ>лу камеры собирается расплавленная сора, которую выливают в деревянные формы и пускают в продажу под названием nepeiinoiioii серы,.
В Луизиане и 'Гехасе добычу серы производят по методу Фраша, который заключается и том, что в буровые скважины под давлением подают перегретую воду, расплавляющую серу под землей, после чего расплав поднимается на поверхность под давлением горячего воздуха. Получаемая таким способом сора очень чистая (99,6%) н поэтому не требует дальнейшей дистилляции.
Производство серы в США началось только в начале текущего столетия, но очень скоро США далеки превзошли все остальные государства.
В Японии содержащие серу породы расплавляют в нагреваемых перегретым паром котлах. В последнее время этот метод стали применять и в Сицилии. То, что в Сицилии сохранились связанное с большими потерями выплавление серы в кучах, объясняется недостатком в этой стране угля.
В Англии раньше значительные количества серы извлекали на остающегося при получении соды по способу Леблана сульфида кальция. Для этого сульфид кальция разлагали по способу Чанса двуокисью углерода и водой CaS + СОг Ч- Н?О*= — СаСО3 + Н23. Образующийся при этом сероводород HaS пропускали в печах Клауса в смеси с воздухом над катализатором (бурый железняк или боксит), в результате чего осаждалась сера
H2S + i/2Os=IbO-| S.
jn2 Глава 15
Метод Чанса.—Клауса сохраняет свое значение в настоящее время Й.1л получения серы в» гипса или. тяжелого шпата. Нагреванием с углем эти породы переводят в сульфид кальция или соответственно бария, которые затем переводят в серу описанным выше способом.
Все увеличивающееся значение приобретает способ выделения серы из светильного или коксового газа, водяного газа и генераторного газа, а также из дымовых газов. В выходящих газах газовых и коксовых батарей сера содержится, как было указано, в основном в виде H2S и удаляется из них массой для очистки газов (см. стр. 501). Из этой массы серу можно извлечь, например, экстракцией подходящими растворителями (сероуглеродом или сернистым аммонием). Однако для получения серы удобнее поглощать сероводород из отходящих газов мокрым путем, как это производят, например, в описанном па стр. 632 методе получении политионатов по Фельду.
Свойства. Сера существует во многих модификациях. Устойчивой при обычных температурах модификацией является ромбическая сера (a-сера). Она имеет хорошо известный желтый цвет, удельный вес 2,06, твердость 2,5, удельную теплоемкость 0,172 (при 16°), обладает очень малыми электро-и теплопроводностью. При трении она сильно отрицательно электризуется. В воде она нерастворима, немного растворима в бензоле, спирте, эфире и других органических растворителях; легко растворяется в сероуглероде; 100 ч. сероуглерода растворяют при 0° 24,0, при 22’ 46,1, при 55° 181,3 ч. серы. Из раствора сера кристаллизуется в той жз модификации.
В пиперидине сера растворяется (независимо от модификации, в которой она существует) с красным окрашиванием. Это можно использовать как качественную реакцию на элементарную серу.
При температурах выше 110'* ромбическая сера расплавляется в подвижную желтую жидкость. Если, медленно охлаждая, расплав, дать части серы застыть, а оставшуюся жидкую серу слить, то внутренность получившегося углубления покрывается игольчатыми, почти бесцветными кристаллами, Это и будет моноклинная модификация серы (ф-сера). Опа имеет удельный вес 1,96 и температуру ила иле пня 119,0'. При обычных температурах эта модификация неустойчива; спустя некоторое время иглы тускнеют и распадаются на мелкие ромбические кристаллы.
Превращение происходит быстрое, если в расплав внести кристаллик ромбической серы, но только при температурах ниже 95,6° С. Выше 95,6°, наоборот, ромбическая •сера переходит в мопоклиппуго, если она соприкасается с последней. Таким образом, при 95,6° лежит точка, перехода между обеими энантиотропными формами. Превращение моноклинной серы в ромбическую сопровождается выделением тепла. Теплота превращения составляет при 0’ 2,4 нал!?, (О свободной энергии превращения см. стр. 169.)
Другую модификацию, также моноклинную, но с иным отношением осей, чем у обычной моноклинной серы, получают в форме имеющих перламутровый блеск желтовато-белых листочков, если медленно охлаждать горячий, почти насыщенный раствор
* В зависимости от условий опыта для температуры плавления ромбической серы получают различные значения. При медленном нагревании происходит превращение ромбической серы в моноклинную. Если ромбическую серу нагревать быстро, то обычно состав расплава неопределенен, так как в нем происходит медленное частичное превращение серыЗ?, в серу S,, (см. стр. 753). При температуре 112,8s ромбическая сера находилась бы в равновесии с расплавом, содержащим только серу S^. Эта температура называется идеальной температурой плавления и затвердевания ромбической серы. Из расплава, в котором находите я в равновесия сера S? п сера S^, ромбическая сера выкристаллизовывается при 110,2’. Если нагревать серу медленно, то в расплаве может установиться равновесие и ромбическая сера будет плавиться при этой температуре при том условии, что она не превратится перед этим в моноклинную серу (естеетсенная температура плавления и затвердевания рсдабнчккой серы).
Шестая группа периодической системы
753
серы в бензоле, скипидаре или спирте (Muthraann, 1890; Neumann, 1934). В этой же форме сера кристаллизуется при ее осаждении на воздухе из раствора полисульфида аммония. В этих случаях, по-видимому, речь идет о модификации, которая неустойчива ври всех температурах. То, что мри оиределепных условиях эта модификация образуется, соответствует правилу ступеней Оствальда (см. стр, 531).
Своеобразные изменения претерпевает сера при дальнейшем нагревании. Выше 160° она становится коричневой и по мере увеличения температуры все более вязкой; при 200° она становится темно-коричневой и вязкой, как смола. Выше 250° ее вязкость снова начинает уменьшаться, и при 400° она опять становится легко подвижной; при 444,60° сера кипит.
Если сильно нагретую сору вливать тонкой струей в холодную воду, то получается коричнево-желтая пластическая и вязко-эластичная масса (пластическая, сера). Эта эластичная сера растворяется в сероуглероде только частично. Остается рыхлый порошок, представляющий собой еще одну модификацию твердой серы. Растворимую часть пластической серы также можно рассматривать как особую модификацию. Нерастворимая в сероуглероде модификация содержится, между прочим, и в серном цвете.
Твердая сера, получаемая быстрым охлаждением жидкой серы или ее паров, всегда ограниченно растворяется в сероуглероде. Количество норастворившогося остатка зависит от температуры, до которой сера была нагрета перед резким охлаждением. Таким образом, в твердой аморфной сере, полученной быстрым переохлаждением, имеются две модификации, которые должны присутствовать и в жидкой сере, так как «пластическая сера» является не чем иным, как переохлажденным расплавом. Растворяющуюся в сероуглероде аморфную модификацию называют 8?л г о растворяющуюся — Sp*. В расплаве обе модификации находятся в равновесии. Резким охлаждением серы ъютао определить положение равновесия при различных температурах. Так, было найдено, что при 114,5°** 3,6%, при 160° 11% и при температуре кипения около 40% серы в расплаве находится в состоянии модификации Sg. При отсутствии катализаторов равновесие устанавливается очень медленно.
Одновременным присутствием в жидкой сере обеих модификаций и можно объяснить своеобразные явления, наблюдающиеся при нагревании расплавленной серы. Постепенный переход желтой окраски в коричневую вызывается, очевидно, тем, что имеет желтый, a темно-коричнево-красный цвет, Свизаипыо с нагреванием изменения вязкости можно объяснить, предположив, что 8^ имеет значительно большую вязкость, чем п что при повышении температуры вязкость 8^ уменьшается, В таком случае увеличение концентрации модификации будет приводить к увеличению вязкости до тех пор, пока, наконец, вязкость 8|л пе будет уменьшаться с температурой быстрее, чем возрастает ее концентрации.
При обычных температурах сера превращается в серу 8?, (правда, при отсутствии катализаторов чрезвычайно медленно), а сера 8?_ переходит в устойчивую модификацию, ромбическую серу.
Величины, полученные для плотности паров серы, и их зависимость от температуры указывают на то, что в парах серы одновременно присутствуют различные молекулы, а именно Sg, Se, S4, Sa. При повышении температуры изменяется их сравнительное содержание. Например, по
* Согласно Атену (Alon, 1912) и Бекманну (Beckmann, 1918), в расплаве можно различить даже три модификации серы—кроме 8?ь й S^, еще Srt. Чтобы пе слишком усложнять этот вопрос, в длиной книге по будет приниматься во внимание различие между S(1 и Sn.
* * Свежерасплаилеппая чистая серп затвердевает в моноклинной форме при 119,0°. Если расплав некоторое время выдерживать при температуре, превышающей температуру затвердев а пил, то точка затвердевания постепенно падает до 114,5°. Это объясняется тем, что в расплаве постепенно образуется отвечающее равновесию количество серы SR, понижающее температуру затвердевания серы Sfi7
48 г. Реми
754
Глава 15
данным Брауне (Вгаипе, 1951), при температуре 450“ и давлении 500 мм рт ст 53,9% общего давления пара приходится на долю молекул S8, 37,0% — молекул Sa, 4,9% — м'олекул S4 и 4,2%,—молекул Sa. При 750“' и таком же суммарном давлений пара отношения этих количеств составляют соответственно 0,1, 0,8, 7,2 и 91,9%. Около 2000° происходит, наконец, распад молекул Ss па отдельные атомы S, Вероятно, с этой диссоциацией связано изменение цвета паров серы, наблюдающееся при нагревании.. Пары серы при температуре примерно около точки кипения оранжевожелтые, при повышении температуры цвет изменяется сначала к красному, затем снова светлеет, так что ири 6503 пары серы приобретают соломенно-желтый цвет.
Быстрым охлаждением паров серы, состоящих из молекул S2, до температуры жидкого азота Райсу (Rice, 1953) удалось получить твердую серу в пурпурно-красной модификации, построенной из молекул S2, Однако эта модификация неустойчива и очень быстро превращается при нагревании до комнатной температуры в желтую серу (энергия активации этого перехода составляет 3,1 ккал’г-ато.к S).
Для молекулярного веса растворенной серы в различных растворителях получены совпадающие значения, отвечающие формуле Sg. Рент-ге ио структурное исследование показало, что и в кристаллическом состоянии сера построена из молекул S8.
Согласно Уоррену (Warren, 1935), ромбическая сера построена из изолированных, не плоских восьмичленных циклов. Это же относится, по данным Беруэлла (Burwell, 1937), и к моноклинной сере, в которой, правда, часть молекул S8 вращается или колеблется около осн, перпендикулярной циклу. Пластическая сера построена, согласно Мейеру (Meyer, 1934), из нерегулярно расположенных зигзагообразных Цепей. Если пластическую серу сильно растянуть, то она приобретает волокнистую структуру, в которой в со зигзагообразные цепи расположены параллельно; одновременно с этим пластическая сера затвердевает. Превращение подвижной желтой жидкой серы в вязкую коричневую жидкую серу объясняется, по Уоррену, вероятно, тем, что циклы, состоящие из молекул Ss, которые сначала преимущественно существуют и в жидкой сере, при дальнейшем нагревании раскрываются в топ или иной степени в цепи. Поэтому можно заключить, чти циклы Ss отвечают сере S? , а цепи из атомов S—сере S . В настоящее в рот г я называю! S,,— иаклооьи1.аеера и — катенополисера.
Тенденция к образованию цепей нац. подаете я по только у элементарной серы, но и у многих соединений серы (например, полит иохлоридол, по л не еро водородов). С эпер-гетиш скии точки зрепня эти понятно, так как, по Полингу, энергия образования простои связи S—S составляет 6,3,8 ке.лл/мол-ь и, следовательно, только на 25% меньше энергии образования простой связи С —С (84,6 ккая/моль, см. табл, 80).
Тепловой эффект реакции Spomg -+ %S2 при постоянном давлении составляет прп пересчете на комнатную температуру 14,45 ккал/г-атом S. Эта суммарная теплота реакции складывается из следующих частей (хмл/г-атом):
0,075 0,33 2,51 0,90 3,52 .7,10
8ромб —> С’манокл --> 8ЖИдК1 —> Ь8 (пары, —> С’б —>_Ь41—» -’З'
Работа диссоциации S2 -> 2SaT0M составляет в зависимости от истолкования спектров поглощения 102,6 или 75,6 ккал/моль S2, или соответственно 51,3 или 37,8 ккал/г-атом S. Меньшая величина согласуется с расчетами Гольдфипгера (Gold finger, 1936), ос нова шшмп на более ранних измерениях давления паров, выполненных фон Вартепбергом (V. Wartenberg), Большая величина лучше согласуется с экстраполяцией энергетических уровней для различных колебательных состояний молекулы S2 [экстраполяция Б ер джа-Споне pa (Birge-Sponer)].
Спектроскопически установлено, что основному состоянию молекулы % отвечает терм 32. Поэтому молекула S2 имеет два песпаренных электрона ц должна, подобно молекуле О3, быть парамагнитна. Полинг считает, что в молекуле S2, как и в молекуле О2, имеются две трех электронные связи, окружающие одну простую связь. Такую же связь он допускает и для молекул SO и Se3, основным состояниям которых также отвечает терм 32. Таким образом, строение этих молекул, согласно Полингу, выражается формулами S гН S, S гН О и So r~Se.
Шестая группа периодической системы
755
Суспендированная и коллоидная сера, /Если раствор полисульфида кальция (полученный длительным кипячением известкового молока с серой) разлагать разбавленной соляной кислотой, то получается белая жидкость, так называемое серное молоко. Опо представляет собой суспензию топкоизмельченной аморфной, растворимой в сероуглероде серы в воде или со ответственно в разбавленном растворе хлорида кальция. Аналогичные суспензии образуются ц при разложении других ноли-сульфидов.
Коллоидный раствор серы получается, если медленно пропускать сероводород в возможно более концентрированный холодный раствор сернистого газа или если разлагать тиосульфат натрия разбавленной серной кислотой по методике, описанной Раффо (Raffo, 1908). Из совершенно прозрачного Коллоидного раствора серу можно осадить добавлением электролита; однако в большинстве случаев происходит только частичное выпадение серы, так как коллоидный раствор серы в противоположность коллоидным растворам металлов мало чувствителен кдобавлению электролитов. Малые количества электролитов, прежде всего кислот, даже стабилизируют золи серы. Сера, выпадающая при добавлении электролитов, может быть обратно переведена в раствор при действии воды.
По данным Фрейндлиха, получаемые таким способом гидрозоли серы содержат в качестве дисперсной фазы серу 8Ц. Устойчивость этих гидрозолей может обусловливаться содержанием в них пентатионовой кислоты H2S5O6, апионы которой адсорбируются на частичках и сообщают им отрицательный заряд. При длительном стоянии из таких золей выкристаллизовывается ромбическая сера, которая утке пе переходит в раствор при действии воды.
Совеем иной характер имеют гидрозоли серы, которые получают вливанием спиртового раствора серыв поду. Они значительно мопее устойчивы; спустя некоторое время мутнеют и при добавления электролитов, прежде всего кислот, необратимо коагулируют. По Фрейндлиху, такие растворы, вероятно, содержат в качестве дисперсной фазы серу 8^.
Устойчивость всех гидрозолей серы можно значительно повысить, добив лян к ним защитные коллоиды. Стабилизированные таким способом препараты выпускаются промышленностью. В виде растворов или паст их используют для борьбы с фиаок-серой (болезнью виноградных лоз), а также в медицине — против экземы, лишаев и пр.
Уже при умеренно повышенных температурах сера является очень реакционноспособным веществом. Она непосредственно соединяется почти со всеми элементами, а с металлами часто даже с сильным выделением тепла. Уже при обычных температурах сера заметно действует на медь и серебро, с ртутью она соединяется при температуре кипения жидкого воздуха.
Применение. Серу используют для получения черного пороха, в пиротехнике и при производстве спичек. В значительных количествах се применяют для опыления виноградников. Общеизвестно применение горящей серы для дезинфекции. «Окуривание серой» основано на действии сернистого газа, образующегося при сгорании серы. Сернистый газ часто используют как отбеливающее средство, например для шерсти, шелка и желатины. Значительные количества серы применяют для .вулканизации каучука и гуттаперчи, а также для получения сероуглерода, ультрамарина, киновари п органических серных красителей. В медицине серные мази и серное молоко применяют для лечепия кожных заболеваний, а серный цвет как внутреннее средство, например, при хронических расстройствах пищеваренья и дистрофии. О применении коллоидных препа-48*
756
Глава 15
ратов для этих целей уже упоминалось выше. Серу используют также для изготовления оттисков, отливок и замазок.
На соединений серы важнейшим является серная кислота. О ее применении, как и о применении других соединений серы, будет сказано в конце разделов, посвященных описанию свойств этих соединений.
СОЕДИНЕНИЯ СЕРЫ
1. Окислы и кислородные кислоты
Сера образует четыре нормальных окисла: трехокись серы SO3; двуокись серы SO2; полуторную окись ее.ры S2Og п моноокись серы SO; кроме того, сера образует две перекиси: S3O; и SO4. Трехокись серы является ангидридом серной кислоты Н^ЙОд, двуокись’серы — ангидридом сернистой кислоты H:(SO3. Отвечающие этим кислотам соли называются сульфатами M*[SO4'j и сульфитами MJS63J.
Кроме того, для серы известны следующие кислородные кислоты и их соли, не имеющие среди окислов серы отвечающих им ангидридов:
Кислоты
II2S2O3 тиосерная кислота (неустойчива)
Соли
Mj[S2O3] тиосульфаты
H2S2O2 тиссернистая кислота* H^S^-Og политштовые кислоты**
HgS^Og дитмоновая кислота
политионаты**
Ma(S2Oe] дитионаты
M|[S2O4] дитиониты
M^[SO2j су.к.фоксилаты,
Полуторную окись серы S2O3 можно рассматривать по ее составу как ангидрид янтнопистой /i W’.7O77>i IcyiiECгвупицен только в виде со.юй) Н^С^; однако эта окись но имеет неП1МТ1('дс-1Т1Г(П|иг1', .iTijoiireiuia г, со 1.яч лптионистой кислоты, днтпонитам. Точно так же ..ifOH.uio.ftci: r-i.< ц.,г шпм <:т»011<‘тви.ч пе является ангидридом сульфоксиловой
кпоюш (си. стр 7~lit.
Из двух и'дет, и; > !< серы ч-ич.ырехон.и<. ъ серы, SO4 не имеет характера ангидрида кислоты. Производны мп другой перекиси серы, семиокиси серы S2O7, являются две пероксокислогы: перЬксояонвсерная кислота {кислота Карр, ранее называвшаяся сульф ом он о Перкислотой) H2SO5 и пероксодисерния кислота H2S30s. Эти пероксокис-лоты можно рассматривать как производные перекиси водорода й серной кислоты; возгону они будут рассмотрены после серной кислоты.
Важнейшими из перечисленных соединений являются трехокись серы и двуокись суры, а также кислоты—их производные. Двуокись серы— нормальный продукт сгорания серы на воздухе;
Трехокись серы — серный ангидрид SO3 — получают в технике, пропуская смеси газов («отходящие газы»), образующиеся при обжиге серного колчедана и других сульфидных руд (медного колчедана, цинковой обманки, свинцового блеска) над катализаторами (прежде платиной, в настоящее время в большинстве случаев ванадиевыми£соединениями). В отходящих газах содержатся (наряду7 со значительными количествами
* Тиоеерниепгая кислота получается только как промежуточный продукт при различных реакциях (си. стр, 772;.
В политионовых кислотах и политиоиатах х может принимать значения 3, 4, 5, 6, а возможно, еще более высокие.
Шестая группа периодическоа системы
757
атмосферного азота) двуокись серы (сернистый газ) и кислород. Последние в присутствии подходящих катализаторов реагируют по: уравнению SO24-i/,O2 SO3(rs3)4-23,2 ккал. (1)
Обжиг серного Колчедана и других сульфидных руд производят в большинстве случаев на длинных решетках пли в многоэтажных печах с механическим перемешиванием или во вращающихся трубчатых печах. Наиболее современными способами являются флейс-процесс (FleuB-ver-fahren), обжиг во взвешенном состоянии и трайл-процесс (Trail-Ver-fahren) специально для обжига цинковой обманки.
Во флейе-процесее (метод вихревого обжига шихты) руда в виде довольно крупных зерен загружается в шахтпую печь н поддерживается во взвешенном состоянии током подаваемого снизу воздуха. За Счет тесного контакта воздуха с частицами руды обжиг происходит быстро и полно. Флейс-процесс применяют также при хлорирующем обжиге и иных процессах хлорирования (получение хлорной извести), кроме того, дли обжига извести и т. и,, а также для каталитического крекипга и синтеза бензина по Фпшеру — Трошпу. В двух последних случаях вводят катализатор в виде зерен, поддерживаемых во взвешенном состоянии реакционной смесью газов. Флейе-нроцесс впервые был разработай для получения водяного газа (Winkler) на Веденском а ин л ино-содовом заводе (1922).
При обжиге во вавешенгюм состоянии, который в основном применяют при производстве серной кислоты и сульфит целлюлозы, обжиг происходит частично в прямом, частично в противоположном потоке. В применяющихся в этом процессе печах Николь-са—Фримана очень .мелкозернистую руду, смешанную с предварительно нагретым воздухом, вводят сверху. Она встречается с идущим навстречу вторым потоком воздуха, которым она и обжигается до копца в средней, сравнительно обедненной SO2 золе печи.
Метод обжига, разработанный компанией «Mining and Smelling Company» в Трейле (Британская Колумбия), так называемый пылевой обжиг, особенно пригоден для обжига пылевидной цинковой обмапкц, которая получается при флотационпом обогащении (см. т, IT). Ципковую обмапку в виде ныли вдувают сжатым воздухом в аижпюю часть печи для обжига. Частицы пылн подхватываются поднимающимся потоком воздуха, обжигаются, проходя через горячую зону печи, и частично уносятся с яыходящпмгг ив верхней части печи газами. Но основная часть после непродолжительного нахождения во взвешенном состоянии, недостаточного для полного обжига, падает па дно печи и там окончательно обжигается.
Реакция, выражаемая уравнением (1), обратима. Поскольку при образовании ЙОй выделяется тепло, равновесие при увеличении температуры сдвигается справа налево, т. е. SO3 распадается. Однако при слишком низких температурах реакция вообще пе идет, В присутствии платины реакция проходит со значительной скоростью уже при 400°; окись железа оказывается эффективной только при температуре 600s.
Если исходить из чистой (свободной от азота) реакционной смеси стехиометрического сноп та. выход SOs составляет 98,1 % при 400'J и 76,3% при 600°. При использовании платпиы в качестве катализатора получается, таким образом, значительно больший выход, чем в случае применения окиси железа. Поэтому в технике прея,де для этого процесса ясиользовалн почти исключительно платану, Б настоящее время почти всегда в качество катализаторов для процесса получения S03 используют соединения ванадия (пятш1кпсь ванадия или сульфат ванадила). Если эти соединения, с- соответствующими добавками, палеетп па подходящий инертный носитель (например, силикагель, цеолит), то по I.волн эффективности они почти Но уступают платине. При этом они значительно дсппев,.н? ц имеют, кроме того, то преимущество, что не отравляются соединениями мышьяка так легко, как платана.
Если к представленному уравнением (1) равновесию применить закон действия масс, то получим
Ры,*Ро1
—г——=Ар
-so3
ЙЯО3 ]
или
758
Глава 15
Как видно, отношение количеств 8О3и SO2 пропорционально корню квадратному пз парциального давления кислорода, и, следовательно, его можно увеличит!,, добавляя к отходящим газам воздух. Если исходить пз отходящего газа состава 84,85% N3, 10,10% SO3 и 5,05% О2„ то выход SO3 при 400° равен 96,2%, при 600° — 59,1%, Если же отходящие газы еще до проведения их над катализатором разбавить четырьмя частями воздуха, так что состав их будет 80% N2, 2% SO2 и 18% О2, то выход 8О3 возрастет до 99,5% прп 400° и 80,5% при 600°.
Получающийся при контактном способе серный ангидрид (трехокись серы) в большинстве случаев пе конденсируют как таковой, а используют для получения серной кислоты. Если же требуется выделить это соединение, то его получают нагреванием дымящей серной кислоты. Этим способом выделяют небольшие количества серного ангидрида в лаборатории. Его можно получить также нагреванием пиросульфата натрия (или других ниросульфатов)
Ь Na2S2O7=Xa2SO4-|-SOs.
Сульфат железа(Ш) при нагревании также отщепляет SO3. На этом основан самый старый способ получения серной кислоты.
При охлаждении трехокись серы конденсируется в бесцветную, прозрачную, похожую на лед, сильно дымящую на воздухе массу, которая плавится при 16,85° и кипит при 44,8° (y-SO3)- Ее удельный вес при 13° равен 1,995, при 20° (расплавленной) — 1,97. Если трехокись серы долго хранить при температуре ниже 25°, то она переходит в другую модификацию (p-SO3), состоящую из белых, шелковистых, сплетающихся в] виде войлока игл. Эта «асбестообразпая» модификация имеет более высокую температуру плавления, чем «льдообразпая». Перегонкой можно снова получить обладающую низшей температурой плавления, «льдообразную» модификацию. Существует и третья, еще более тугоплавкая модификация (a-SO3), которая получается в чистом состоянии только в особых условиях (Sinits, 1981). Торювая трехокись серы представляет собой смесь а-8О3 с P-SOs и,"Т0[13и в большинстве случаев преобладает). Обычно такая треды.'иск серы начинает плавиться около 40°, однако температура плавления (а также давление пара) зависит от отношения количеств этих модификаций. Это отношение изменяется в основном в период плавления и испарения, так как 0-модификацпя быстрее переходит в жидкое состояние и легче испаряется, чем а-модпфпкация.
Смесь а- и р-модификацйй S03 в том случае, когда взаимное превращение в достаточной степени замедленно (при отсутствии влаги), по дет себя как смесь двух различных, взаимно растворимых в твердом состоянии веществ. Поэтому опа не имеет вполне определенной температуры плавления, а плавится в некотором (в иных случаях значительном) температурном интервале. Точно так же прп возгонке наблюдается уменьши ние давления пара. Поэтому эти модификации можно разделить фракционной возгонкой. Аналогичные явления будут наблюдаться и в случае Других встречающихся в различных аллотропических модификациях веществ, например трехокиси мышьяка, хлорида алюшишя и фосфора. Чтобы применить и таким системам правило фаз, надо обо модификации рассматривать как отдельные составные части системы (компоненты). Смите, развивший теорию этих явлений и обосновавший ее экспериментально, называет такие системы, в химическом смысле состоящие из одного вещества, по ведущие себя как системы, состоящие из двух веществ (бинарные системы), <<псевдобипарнымн системами», а модификации, образующие в твердом состоянии единую смешанную фазу,— «псевдо компонентами». См. также стр. 703.
О структуре у-8О3 см.: Westrick, R., Rec. trav. chim., 60, 794 (1941).
Б газовой фазе молекулы 8О3 имеют, согласно Бальмеру (Palmer, 1938), плоскую конфигурацию, т. е. три атома кислорода образуют равносторонний треугольник, в центре которого расположен атом серы (расстояния: S»—>О 1, 43 А; О«—2,48 А).
Шестая группа периодической системы
759
Трехокись серы исключительно энергично соединяется с водой, образуя серную кислоту
SOj 4- JL»O=H2SO4 -У21.28 ккал (при 20°). (2)
Если капля воды попадает на трехокись серы, происходит реакция со взрывом. Вследствие очень сильного сродства к воде трехокись серы часто отнимает воду от таких веществ, которые содержат конституционно связанную воду; поэтому опа обугливает органические соединения, на пример целлюлозу.
Серная кислота H2SQ4 является продуктом соединения трехокиси серы с водой {см. уравнение (2)1. В технике сильно концентрированную серную кислоту (98%-ную) получают в настоящее время почти исключительно контактным методом, менее концентрированную (78%-пую) — более старым камерным методом.
Производство серной кислоты контактным методом заключается в том, что описанным выше способом из отходящих газов обжига сульфидных руд получают трехокись серы и поглощают ее концентрированной серной кислотой. Затем такую поглотившую SO3 серпую кислоту смешивают с более разбавленной, так чтобы концентрация ее оставалась постоянной.
Получающийся прп контактном методе серный ангидрид ве удается просто поглотить водой, так как при этом образуется плотный туман. Вносимые с потоком газов частички SOa жадно поглощают иоду, образуя очень мелкие капли. Эти капельки тумана находятся во взвешенном состоянии в потоке газа. Вследствие их малой подвижности они с трудом приходят в соприкосновение с поглотителем, так что почти невозможно полностью поглотить трехокись серы, если она за счет попадания влаги перешла в тумап. Поэтому выходящий из контактных печей, содержащий 80а газ пропускают через 98%-ную серную кислоту; при этом не образуется никакого, мешающего поглО-щспию тумана.
Контактный метод был разработан в 1831 г. апгличаппном Филипсом (Philipps). Однако практическое значение оп приобрел только 65 лег спустя, после того как в основном работами Винклера (Winkler) и Книча (Knietsch) были выяспопы важнейшие вопросы для эффективного проведения коптактпого процесса (зависимость равновесия НСь—S03 от концентрации и температуры, природа кат а ли За торных ядов н возможность их устранения, поведение SOa при поглощении) и на этой основе были выработаны основные технологические условия процесса. Особеппо важными оказались освобождение отходящих газов обжига от суспендированной в них в виде коллоидной пыли трехокиси мышьяка — сильного катали заторного яда — и правильное регулирование теплового режима для получения необходимой температуры в контактной камере,
Камерный процесс производства серной кислоты, которым еще получают основное количество 78%-ной серной кислоты, используемой промышленностью по производству удобрений, основан на соединении сернистого гозэс кислородом и водой в присутствии окислов азота с образованием серной кислоты. Протекающие в этом случае реакции схематически ( о точной последовательности этих реакций см. ниже) можно представить следующими уравнениями:
NO+V2O3=NO2, (3)
S03+H20=H2S03, 0)
II2SO3+NO2=H2SO4+-X0. (5)
Как следует из реакций, окись азота NO выступает в роли переносчика кислорода. Она окисляется кислородом воздуха до двуокиси азота и вновь переходит в окись азота, отдавая кислород сернистой кислоте,
760
Глава lo
после чего снова может присоединяться кислород и т. д. Таким образом, небольшое количество окиси азота могло бы перенести любые количества кислорода, если бы не происходили в значительной степени побочные реакции, приводящие постепенно к ее расходу на образование закиси азота и самого азота, которые уже по способны переносить кислород. Если не учитывать побочных реакций, то и в случае камерного способа получения серной кислоты, идет каталитический процесс. Название «камерный способ» этот процесс получил потому, что описанпыр реакции обычно протекают в больших покрытых листовым свинцом камерах, которые, как правило, располагают по три, одну за другой.
Прежде чем запустить в первую камеру очищенные от пыли и Подогретые примерно до 300° отходящие газы обжига, их пропускают через башню, в которой навстречу потоку газов стекает по пористому материалу «нитрозная кислота», т. е. насыщенная окислами азота умеренно концентрированная серная кислота (башня Гловера). При атом газы охлаждаются и насыщаются окислами азота, тогда как кислота в свою очередь концентрируется. Затем газы направляют в свинцовые камеры. Одновременно в камеры подают распыленную воду и азотную кислоту в таком количестве, которое необходимо для пополнения происходящих в процессе производства серной кислоты потерь окнслов азота.
Образующаяся в свинцовых камерах умеренно концентрированная серная кислота («камерная кислота», в большинстве случаев ~ 60%-ная) стекает вниз на дно камеры, откуда периодически ее спускают. Выходящие из камеры газы (состоящие в основном из азота воздуха, подаваемого для обжига руды) уносят с собой значительные количества окнслов азота. Чтобы возвратить в производство окислы азота, выходящие газы, прежде чем направить их в вытяжные трубы, пропускают через башни Гей-Люссака (чаще всего две). В этих башнях газы орошаются стекающей навстречу им 80%-вой серной кислотой, которая после насыщения окислами азота смешивается с «камерной кислотой» и возвращается как «нитрозпая кислота» в башню Гловера, где и отдает окислы азота.
Большинство исследователей считает, что образование серной кислоты в свинцовых камерах происходит В основном через цитр озилг идр о сульфат [NOHHSOiJ, ранее называемый «нцтрозилсернон кислотой» (см. стр. 669), который затем разлагается водой с образованием серной кислоты. При недостатке воды питрозилгидросульфат может выделиться в тцн|Сгаллн'1еской форме («камершао кристаллы»). Сам нитрозил-гидросульфат может гиг шь образоваться через иные промежуточные продукты, например;
SO2 4- IEO + АО» = HONO SOs-OH,
Су.иьфоиитроновая кислота
2HOAO SO2 ОН -|- А’О2 = 2HO-S03-ONO + NO 4- Н2О,
Пит рози л гидросульфит
2HO-SOa-ONO + Н3О = 2H2SO4 ф NO3 NO.
Эти представления о механизме процесса развивались в основном Лунге (Lunge, 1885 и сл.) и Верлем (Borl, 1906—1907 и 1931 и сл.). Образование в качестве промежуточного продукта еульфонитрогювой кислоты (называемой им «янтрсзосульфокислотой» а часто «Синей» или «фиолетовой кислотой»*) принималось и Рашигом (1887 и сл.), однако он предполагал иной механизм ее образования и распада. По данным же Мюллера (Muller, 1934) и Абеля (Abel, 1928 и сл.), ни сульфонитро нова я кислота, ни питрозилгидросульфат, ни тем более азотистая кислота HNO3 не могут быть промежуточными продуктами, ускоряющими окисление 80s. В целом До сих пор еще не ясно, какие из рассмотренных механизмов процесса имеют наибольшее значение. Можпб-только считать твердо установленным па основании кинетических исследований Боден-штейпа (Bodenstein, 1918 и 1935 н сл.), что первичным процессом является Окисление NO кислородом воздуха (вероятно, по схеме 2NO"A’sO2; 1Ч2О2 -f- O3-*2NO2) до NO2, а последняя (через промежуточные продукты и, по-видимому, за счет различных протекающих одновременно реакции), переходя вновь в 140, переносит кислород на 80s.-в присутствии воды или Н230з [ср. уравнения (3)—(5) на стр. 759].
* По данным Сила (Seel, 1953) вто соединение следует рассматривать как гид росу льфат окиси азота и нитрозила [ON-ХО]+[HSOJ-.
Шестая группа периодической системы
761
Камерный способ получения серной кислоты был разработан в середине XVIII в. в Англии. Важнейшими этапами на пути его развития были: введение Гей-Люссаком (1827) процесса возврата нитрозных газов и открытие в 1859 г. Гловером метода извлечения окиси азота из питрозной серной кислоты при действии горячих газов, образующихся при обжиге сернистых руд, что позволило вести процесс непрерывно (круговой процесс). В настоящее время вместо свинцовых камер в больших установках применяют башни из кислотоупорного материала, подобные башням Гловера, по крупнее. Между этими камерами включают небольшие ящики, изготовленные из листового свинца, в которых нитрозная кислота распыляется быстро вращающимися вальцами (башенный процесс),
Серпая кислота, получаемая камерным или башенным методами, сначала имеет умеренную концентрацию. Камерная кислота обычно содержит 60—70% H,SO4 (удельный вес 1,50—1,62), башенная — около 78% H2SQ4 (удельный вес 1,71). Последнюю в таком виде и применяют для многих целей, например для получения суперфосфата. Однако камерную кислоту, если только ее не проводят через башню Гловера, необходимо концентрировать упариванием- Упаривание производят в с винцовых чанах до концентрации 78%. Для еще большего концентрирования выпаривание следует проводить в платиновых сосудах, фарфоровых или кварцевых чашах. Однако такое концентрирование в настоящее время производят очень редко, так как для этого эффективнее применить контактный метод,
В продажу поступает главным образом примерно 98%-ная H2SO4 (удельный вес 1,841). Контактным методом получают не только эту сильно концентрированную кислоту, но и дымящую серную'кислоту (олеум), т. е. серпую кислоту, содержащую растворенный избыточный серный ангидрид. Вначале контактным способом получали только такую кислоту.
Концентрацию серной кислоты часто выражают через удельный вес, определяемый при помощи ареометра. Раньше служившие для этого ареометры градуировали в большинстве случаев в градусах Боме. 50° В б соответствуют уд. весу 1,530 (62,53% HeSO4), 60’ В6 —1,710 (78,04% HsSOJ, 65° Be — 1,820 (90,05% H2SO4), 66° Be -1,841(96—98% H2SO4). Однако нри содержании серной кислоты более 90% определения концентрации но удельному весу становятся очень неточными.
Упариванием невозможно получить совершенно безводную серную кислоту. Максимальная температура кипения серной кислоты лежит при 338° и отвечает концентрации 98,3% H2SO4- Если растворением рассчитанных количеств SO3 в концентрированной серной кислоте довести ее стехиометрический состав до точно отвечающего формуле H2SO4, то она затвердевает и плавится при 10,36°; однако при нагревании такая кислота теряет SO3 до тех пор, пока ее состав не будет соответствовать 98,3% H2SO4.
Удельный нес серной кислоты вблизи этой концентрации имеет пологий максимум, как покапывают следующие цифры:
Концентрация 1Г8О4, % 94,60 95,60 96,38 98,20 98,72 99,12 99,31
Уд. вое. при 15° 1,838 1,840 1,841 1,841 1,840 1,839 1,838
Кривая температуры затвердевания растворов серной кислоты имеет отчетливый максимум при составе, отвечающем формуле H2SO4 (рис. 123). Температура плавления 98%-пой серной кислоты лежит при 3,0°, 100%-ной — нри 10,36°, а серной кислоты, содержащей 5%-лый избыток &0з, — ври 3,5°. Электропроводность растворов серной кислоты падает все более резко по мере того, как концентрация серной кислоты начинает превышать 95%, и Снова возрастает, как только превышена концентрация 100% (рис. 124).
762
Глава 15
На основании максимумов, наблюдающихся на кривой зависимости температуры затвердевания растворов серной кислоты от соотношения 80з/На0 (рис. 123), можно сделать вывод, что в таких растворах существуют различные гидраты серной кислоты, а имеппо Н28О4-Н2О (т. мл. 8,5°), H2SO4-21I2O (т. пл. — 38°) и H2SO4-4H2Oi (т. па.
— 27°). Как нашел Бильц (Biltz, 1934), серная кислота образует еще два гид-
0 10 20 50 АО 50 60 70 80 90 100
CaSes.'^ai'-ue 5Г)3, %
Рас. 123. Диаграмма плавкости для системы 80s—НаО.
рата, которые плавятся ипкопгруелтпо, а именно H2SO46H2O (т. пл, —54°) и HaSO4-8H2O (т. пл. —62°). Кроме того, диаграмма температур плавления свидетельствует о существовании соединения H2SO4SO3, или Н2ВзО7—пиросерной кислоты. Эта кислота представляет собой прозрачную кристаллическую массу с температурой плавления 36’. Пиросерная кислота смешивается во всех отношениях как с сорной кислотой, так и с сорным ангидридом, Большинство таких смесей йри обычных температурах жидки, они и являются продажной дымящей серной кислотой (олеумом), Из растворов, содержащих более 18,35% и менее 61,87% избыточного по отношению к формуле II2SO4 серного ангидрида, при охлаждении выкристаллизовывается пиросерная кислота Н28аО7. Кривая изменения давления паров дымящей серной кислоты указывает на существование, кроме H2S20j, еще одного более богатого SO3 соединения, вероятно состава (Remy, 1942).
Чистая концентрированная серная кислота представляет собой маслянистую, тяжелую бесцветную жидкость. Она отличается чрезвычайно
Рис. 124. Зависимость удельной электропроводности и эквивалентной электропроводности серной кислоты от концентрации.
7 _ удельная электропроводность х (при IS*)» 2 — эквивалентная электропроводность А (при 18й).
высоким сродством к воде. При смешивании концентрированной серной кислоты с водой выделяется значительное количество тепла (20,42 ккал!моль H2SO4 при смешивании с избытком воды при 20°). Поэтому смешивать
Шестая группа периодической системы
7Q3
серную кислоту и воду следует с осторожностью, вливая серную кислоту топкой струей в воду, а не наоборот. Концентрированная серная кислота поглощает пары воды и из воздуха. На этом основано со применение как осушителя. Ес часто используют как водуотнимающее средство при химических реакциях. Концентрированная серная кислота обугливает многие органические соединения, которые при этом отщепляют воду. В то же время концентрированная серная кислота является окислителем, особенно при нагревании, причем она, отщепляя кислород, переходит в сернистую кислоту. lie только металлы, но и такие вещества, как сера и уголь, окисляются горячей концентрированной серпой кислотой с выделением сернистого газа. В разбавленной серпой кислоте, напротив, растворяются только те металлы, которые в электрохимическом ряду напряжений стоят Левее водорода.
Еще более сильное водуотнимающее и окисляющее средство — дымящая серная кислота {олеум). Она вызывает сильные и болезненные ожоги Кожи, если ее не удалить тотчас же сухой тряпкой (при попадании влаги ожог усиливается).
В технике большое значение имеет поведение серной кислоты по отношению к желеяу и свинцу. Если в разбавленной и умеренно концентрированной серной кислоте железо чрезвычайно легко растворяется, то оно очень устойчиво по отношению к концентрированной серпой кислоте, что объясняется пассивированием его поверхности (см. стр. 381:.), Серную кислоту с концентрацией выше 93% можно хранить и кипятить в чугунных сосудах. Особенно устойчивым является чугун с большим содержанием кремпия, так что из такою чугуна в настоящее время изготовляют аппараты для упаривания серной кислоты. Tlpit обычных температурах устойчивым по отношению к концентрированной Серной кислоте является и ковкое железо. Поэтому его используют для изготовления железных бочек и цистерн. Иа ковкое железо не действует н олеум, если он содержит более 27% свободного SOa.
Свии.еу подвергается действию разбавленной и умеренно концентрированной серной кислоты только с поверхности, причем образуется сульфат свинца, препятствующий дальнейшему действию серной кислоты. Но в концентрированной серной кислоте сульфат свинца сравнительно легко растворяется, поэтому свинец менее устойчив по отношению к концентрированной серной кислоте, особенно при нагревании.
Серная кислота — двухосновная сильная кислота. Ее кажущаяся степень диссоциации (измеренная по коэффициенту электропроводности Ас/А0) в 1 п. растворе равна 51%, в 0,1 н. растворе —59%.
Серная кислота отчетливо диссоциирует в две ступени. Один йот! водорода в растворах средней концентрация отщепляется практически полностью. Отщенлепие второго пона водорода подчиняется закону действия масс
1^2Д^Д£1 = ^=51129.10-з при 18° (Hamer, 1934). (6)
|HSO4]
Если для 1 п. раствора серной кислоты принять степень диссоциации по цервой ступени равной 100%, то для второй ступени получим значение 2,5*. Отсюда среднее значение стёпепн диссоциации 51,2% удовлетворительно согласуется с велпчппой,
* При вътчттсТёпнц диссоциация серпой кислоты по второй ступени следует учитывать подавление ее за счет диссоциации по первой ступени. Если ц, и нг — степени диссоциации (в долях единицы), то цз с молей HaSO4 аа счет диссоциации по уравнению Г128О4;:Н8О) — И получится по с-а, г-ионоо USOi и IP, а за счет диссоциации по уравнению HSO'tJ:SO) 4- £.Г образуется с-аг-на г-иоиа S04 н еще с-сц-аг г-иона Н*, па что расходуется с и, «-иона ИЗОд. Если с — общее количество серной кислоты (молъ/л), то получим
[SOJl — c a^a,: [Н']=?с-а((1+аг); {IISOJJ = <?• crt (1—а2).
Если эти величины нодставпть в уравнение (6), то для И| = 1 и е== 0,5 получим «2 = 0,025, а при с = 1 «! = 0,013.
764
Глава 15
определенной по коэффициенту электропроводности (51,4%) - Дня 2 н. серной кислоты среднее зпачепне степени диссоциации по первой и второй ступеням подучается равным 50,0%. Поэтому для такого раствора [Н*| = 1,012 и pH = — 0,005.
Удельная, электропроводность серной кислоты при концентрации серной кислоты-около 30% (7,5 н.) имеет отчетливый максимум (zlg = 0,739) (см. рис. 124), тогда как эквавалентная электропроводность в этой области по мере разбавления равномерно увеличивается. Если проследить за изменением электропроводности при разбавлениях менее 10 вес.%, что соответствует 2 г-экв/л, то скорость нарастания электропроводности сначала несколько уменьшается, но при разбавлениях менее 0,2 е-оке/.и снова пронсхо-дпт сильное возрастание электропроводности, что объясняется значительным увеличением в этой области диссоциации по второй ступени. Если на оси абсцисс откладывать корень кубический из эквивалентной концентрации, а по осп ординат — эквивалентную электропроводность, то получим кривую, которая между 0,5 и 0,2 г-акз-H»SO4 имеет отчетливую точку перегиба, отвечающую началу диссоциации по второй ступени.
Серная кислота находит разнообразное применение в лабораториях и различных отраслях промышленности. Главнейшим ее потребителем является промышленность, производящая удобрения, где серпая кислота используется в первую очередь для производства, сульфата аммония (на газовых заводах и коксовых батареях) и суперфосфата. Кроме того, серная кислота используется для очистки растительных масел, жиров и минеральных масел, для получения других кислот, сульфатов, простых и сложных эфиров. В промышленности органического синтеза, кроме-концентрированной серной кислоты, часто используют дымящую серную кислоту (олеум), особенно для сульфирования, т. е. введения в органические соединения вместо атома водорода группы SO3H. Умеренно концентрированную с-ерпую кислоту (72—75% ) используют для получения пергаментной бумаги. Серпая кислота удельного веса 1,15—1,25 (имеющая примерно максимальную электропроводность) используется в аккумуляторных батареях. Разбавленную серную кислоту употребляют и в медицинских целях.
Сульфаты. Как ^.вгхосиовпая, серная кислота образует два ряда солей: кислые сульфаты (гидросульфиты) M1HSO4 и нормальные (нейтральные) сульфаты МН',.
кислые сульфаты известны только для щелочных металлов. Они кристаллизуются из растворов нормальных сульфатов в присутствии избытка серной кислоты и образуются при разложении серной кислотой солен щелочных металлов других кислот при умеренно повышенных температурах.
Пример.
Na2SO4 + H2SO4 = 2NaHSO4 (кислый сернокислый натрий, гидросульфат натрия) бпсульфат натрия,
NaCl-| H2SO4 = NaIISO4+HCJ.
В воде кислые сульфаты очень легко растворяются. Они также легко плавятся. При нагревании выше температуры плавления кислые сульфаты, отщепляя воду, переходят сначала в пиросульфиты M£S2O,, при дальнейшем нагревании — в нормальные сульфаты и серный ангидрид:
2NaHS04 = Il20-|-Na,S207 (пнросульфат натрия),
Na2S2O7 = Na2Sp4+SO3
Некоторые нормальные сульфаты M*SO4 устойчивы при паг рев алии
Шестая группа периодический системы 765
Сульфаты щелочных металлов плавятся без разложения (Na3SO4 плавится при 884°, K2SO4 — при 1074°). Сульфаты щелочноземельных металлов и сульфат свинца можно нагревать до красного каления без разложения. Одиако многие сульфаты начинают разлагаться уже при красном калеяпй. При этом наряду с окисью металла (а в случае благородных металлов — самого металла) образуется серный ангидрид или образуются как продукты его распада сернистый газ и кислород; такой распад происходит тем активнее, чем выше температура разложения сульфата. Особенно легко разлагаются сульфаты трехвалентных элементов. Например, давление диссоциации сульфата железа (III) ЕщфЗОД» достигает заметной величины уже при 500°, а яри 721° равняется .атмосферному давлению. Давление диссоциации сульфата алюминия превышает атмосферное давление при температуре около 750°. Поэтому нагреванием этих веществ можно получать серный ангидрид.
Большинство сульфатов хорошо растворимо в воде. Трудно растворимыми являются сульфаты щелочноземельных металлов (см. гл. 8) и сульфат свинца- Довольно плохо растворяется сульфат серебра. Большинство хорошо растворимых сульфатов кристаллизуются с водой. Многие сульфаты имеют склонность к образованию двойных солей (сулъфатосолей).
В качестве примерев сульфатов, содержащих кристаллизационную воду, можно привести купоросы'. железный купорос FeSO4'7H2O, цинковый купорос ZnSO4-7H2O, медный купорос CuSO4'5H2O. Примерами двойных сульфатов могут служить квасцы. В водных растворах сульфат-доны (так же как н селепат-ионы) удерживают две молекулы воды [SO4(H2O)2]'' (Brlntzinger, 1935).
Сульфаты можно получить следующими способами:
1) растворением металлов в серной кислоте;
2) насыщением серпой кислоты окисями или гидроокисями металлов «(или ангндрооснованиями, например аммиаком);
3) разложением солей летучих кислот серпой кислотой;
4) обменным разложением между’ сульфатом и солью того металла, сульфат которого желательно получить;
5) окислением сульфитов или сульфидов.
Сульфаты находят очень обширное применение. Об этом сказано в разделах, посвященных отдельным сульфатам, или в главах, посвященных металлам, производными которых являются сульфаты.
Действием SO3 па сульфаты можно получить полисулъфаты. Например, из K2SO4 м 50з образуется трисульфат калия, K2S30i? (Thilo, 1938). Может быть, возможно получение и еще более богатых SO, соединений, однако, во всяком случае, менее устойчивых. K.2S3O10 можно нагревать без разложения до —150°. При более сильном нагревании он разлагается по уравнению
K^O^KaS/WSOa.
При температуре около 50° реакция протекает п обратном направлении.
Строение серпой кислоты п сульфатов. Поскольку серная кислота и ее ангидрид, чрехокпсь серы, пе имеют характера производных перекиси водорода и в их молекулах, очевидно, пет перекисной связи, то серу в этих соединениях надо считан, шести-валентной, и пмеппо, если эти соединения можно было бы считать гетеро поляр вы мп (что, впрочем, допустимо лишь с известными ограничениями), положительно шеети-валентной. Тс,, что сера может быть шее тика лоптпой, вне всякого (юмвеипя Подтверждается сущеютвоваццем гексафторида серы, для которого в силу одновалентности фтора отпадает любая другая формула, кроме формулы с шестивалентной серой. Исходя из этих положений для еерпоц киыкфгы. если ее рассматривать как гидропоильное соединение, получается структурная формула I. Если же, напротив, принять, что атомы водорода связаны не с отдельными атомами кислорода, а о радикалом SO4 как целым, то получим формулу И. Такое строение имеют прежде всего типичные соли серной кислоты (формула ill). Рентгенометрические определения показали, что в солях серной кислоты радикал SO4 отчетливо проявляется как структурная группа (ср. рис. 51 стр. 247). С точки зрепия координационного учения сере в сульфатах следует
766
Глава la
приписать координационное число четыре.
0, >он 0 О’ 0 О’
X ; S Иг; S Ml
0х хон .0 0 -0 о
1 И ш
Пероксоссрная кислота и пер оксосульфаты. Если серную кислоту подвергнуть электролизу при пониженных температурах (ниже 30°), применяя гладкий платиповый анод и высокую плотность тока, то получается пероксосерная кислота (точнее, пероксодуссрная кислота) H2S2Os, (ранее называемая надсерной кислотой). Точно так же получают пероксосульфаты (пероксодисульфаты) MJSa08, (ранее называемые персульфатами), если в аналогичных условиях вести электролиз растворов сульфатов.
.Пероксосульфаты образуются с лучшими выходами, чем свободная кислота. Это особенно относится к легко кристаллизующимся пер оксосульфатам аммония, и калия, получающимся без труда в чистом виде. Чтобы предотвратить восстановление па катоде, в электролитической ячейке устанавливают глиняную диафрагму или покрывают катод пленкой гидроокиси хрома, которую очень легко создать, добавляя хромат, восстанавливающийся иа катоде и выделяющийся в виде гидроокиси.
Пероксосерная кислота и пероксосульфаты являются сильными окислителями. Под их действием из растворов солей марганца, свинца, пикеля и кобальта выпадают высшие окислы этих металлов; соли хрома(Ш) переходят в хроматы и т. д. В сухом состоянии пероксосульфаты (пероксодисульфаты) совершенно устойчивы. Все пероксосульфаты растворимы в воде.
Пероксосульфаты, кроме применения при получении Н2О2 (ср. стр. 76), используют как окислители и отбеливатели. Пероксосульфат аммония (NH4)3SaOe применяют в фотографии для ослабления слишком, плотных негативов. Его используют также в аналитической химии, например для разделения марганца и хрома.
То, что порог,с-1-О’. "iL'f :Т;.н iггерикоi.ihlt, .тъФатьГ) имеют формулу M1[S2Os], а не бо;.цт простою фюряулс .4 ' | з-Ср |. пц _1 он но уетапоилепо измерением электропроводности и Tirieii щмерзщтня цх воипьи растворов. Строения пе рок со дисульфатов подтверждается п способом их образоващгя.
О
2 OSO
О
О 0 1’
OSO-OSO о о
В соответствии^ такой точкой зрения не роке о дисульфаты являются производными перекиси водорода. Тогда как растворы ее солей относительно устойчивы, свободная лер оксодисерп а я кислота гидролитически легко распадается с образованием rwp-оксомопосерной кислоты (кислоты Каро), также являющейся производным перекиси водорода.
ОХ ХО
НО. „О
-ох Х0
/0Н ох хон
но, у,о но—ох хо
Н [—- он
Пероксомоносерная кислота
И, наоборот, из пероксомоноссрпой кисло/ы действием хлорсульфоновой кислоты IlSOgCl можно получить обычную нороксосерпую, т. е. пероксоди серную, кислоту
04s/^--+-l^0-°\sz0 = °х/° °V/°+HCi ох хон гюх хо ох хон нох хо
Шестая группа периодической системы
767
Пероксомоносерпая кислота H2SO6 (кислота Каро) получается прп электролизе серной кислоты как продукт распада первоначально образующейся пер оксоди сер пой кислоты, причем тем быстрее, чем концентрированнее серпая кислота. Перо кс с; моно серную кислоту получают еще действием серной кислоты на пероксодисульфаты, а также действием перекиси водорода на холодную концентрированную серную кислоту. Последняя реакция, правда, обратима
H2SO4 4- Н2О2 H2SO5 4- Ц20.
Реакцию, протекающую справа налево—гидролитическое расщепление веройсомо-носерной кислоты, образующейся прп электролизе концентрированной сорной кислоты,— используют в технике для получения перекиси водорода.
В чистом состоянии в виде красивых кристаллов (т. пл. 453) пороксоммюсерпую кислоту мол; по получить взаимодействием хлор супь фо нов ой кислоты HSOgCl с перекисью водорода.
О С| 4 Н -О- О- II 0-0- Н
Х > ' — + ПС]
0^ ^0—В 0^ \о—Н
Приведенный метод получения одновременно указывает и на строение этой кислоты. Пероксомопосерпая кислота является сильной о Дно основной кислотой. В водном растворе опа постепенно разлагается. Серпая кислота в больших концентрациях ускоряет разлоиачию.
Сопи нер оксомоно серп ой кислоты малоустойчивы. В отличие от серной кислоты нп иероксомояо-, пи пероксодисерная кислота пе образуют труднорастворимую бариевую или свинцовую соли. Из растворов йодида казня пероксомопосерпая кислота даже при большом разведении моментально выделяет свободный под. Под действием пероксодисерной кислоты выделение иода происходит значительно медленнее, а перекись водорода выделяет иод в очепь небольшой степени. Три названных вещества довольно сильно различаются и своим окислительным действием, например на органические соединения.
Семионпсь серы S2O7 образует, по данным Вертело, который получал ее действием томного электрического разряда па смесь двуокиси серы и кислорода, маслянистые капли, затвердевадощии при 0“. Ока, по-видимому, образуется также при действии озона па двуокись или трехокись серы. Это соединение легко разлагается с выделением кислорода,, особенно в присутствии воды.
Если работать так, чтобы выделение кислорода было затруднено, то при гидролине семиоки сц серы получается пер ок со ди серпая кислота. Поэтому семиокись серы можно рассматривать как ангидрид дер оксо дисер ной кислоты.
Четырехокпеь серы 804 образуется, как нашел Фихтер (Ei с 11 ter, 1926 и сл.), при действии фтора па попы SOj. В чистом состоянии она впервые получена ТПварцом (Schwarz, 1934) иод действием тлеющего разряда на смесь двуокиси серы и кислорода прп давления 0,5 мм рт ст. Чстырехокись серы представляет собой твердое белое вещество, плавящееся с разложением при -1-3°. В безводной серной кислоте четырех-окись серы растворяется без разложении. Взаимодействуй с водой, S04 образует по H9SO5, а медленно распадается в водном растворе, выделяя кислород. Опа является сильным окислителем, например переводит соли марганца (II) в иермапгапат, а соли меди (11) — в соли меди (III).
Двуокись серы SOa образуется при сгорании серы па воздухе (S 4-+ О, — SO, 4- 70,9 ккал при постоянном давлении и 20°), а также прп нагревании сульфидов в токе воздуха или кислорода. Получение SO2 таким способом при производстве серной кислоты уже было рассмотрено. Для получения двуокиси серы в лаборатории пользуются главным образом восстановлением горячей концентрированной' серной кислоты под действием меди, ртути, угля или серы. Кроме сернокислотного производства, двуокись серы используют в технике во многих других случаях: как отбеливаюш.ее средство, прежде всего для соломы, шерсти и шелка, а также кукурузной муки и сахара; как консервирующее средство; SO2 подавляет брожение. При вдыхании в больших количествах двуокись серы ядовита.
768
Глава. 15
Двуокись серы представляет собой бесцветный, резко пахнущий газ, который пе поддерживает горения и но горит сам. Однако под действием катализаторов она, как уже было отмечено, соединяется с кислородом воздуха, образуя трехокись серы. В водных растворах двуокись серы является сильным восстановителем. Благодаря этим восстановительным свойствам она обесцвечивает многие органические красители. На этом основано действие двуокиси серы как отбеливающего средства.
Двуокись серы применяется и в холодильном деле. Она очень легко конденсируется и имеет большую теплоту испарения. При температуре кипения сжиженного Таза (—10,0°) теплота испарения составляет “96 кал!г. Давление паров SO2 при 0° равно 1,52 атм, при 20° — 3,3 атм; удельный вес жидкой двуокиси серы при —10° равен 1,46. Критическая температура 157,2°, критическое давление 77,7 атм, критическая плотность 0,51. Температура замерзания —72,5°. Плотность в газообразном •состоянии при нормальных условиях составляет 2,2630 по отношению к плотности воздуха, принятой за 1 (вес 1 л газообразной SO2 равен 2,9256 г). Так как двуокись серы легко конденсируется, ее поведение довольно плохо подчиняется законам идеальных газов.
Молекула SO2 имеет изогнутое строение; S«-^O == 1,432 А; <510—8—0 = 119,04° [Су ab 1 е, Smith, J. Chem. Phys., 19, 502, 1951]; дипольный момент ц =!1,61 10’lS.
Сернистая кислота и сульфиты. Двуокись серы очень хорошо растворима в воде (100г воды растворяют при 20° 10,5, при 10° 15,4г SO2; при 0° •один объем воды растворяет более 70 объемов двуокиси серы). Водцый раствор двуокиси серы имеет отчетливо выраженную кислую реакцию. В нем содержится двухосновная сернистая кислота H2SO31 ангидридом которой надо, следовательно, считать двуокись серы. В свободном состоянии сернистая кислота не известна, так как она легко распадается на воду и двуокись серы. Поэтому и водный раствор сернистой кислоты сильно пахнет двуокисью серы,
Сернистая ьш- щтп mirriнтпрует как двухосновная кислота в две ступени. Копстапты ..pi г со ин тцтш равны А'1 = 1,610’’ (при 18°), К? = 1,0-Ю-7 (при 18°). Копстапта диссоциации по цервой ступени A1T представляет собой отношение произведения ионных концентраций [HSOg] х [H'J к общей концентрации, присутствующей в растворе недиссоциировапной двуокиси серы (H2SO3 + SO3). Доля последней, которая присутствует в растворе в виде кислоты H2SO3,'no спектроскопическим измерениям составляет всего несколько процентов.
SO3 образует с водой твердый гидрат, состав которого приблизительно отвечает -формуле SO2 6Н2О. В этом случае, вероятно, следует говорить о «гидрате газа» (см. •стр. 248 и сл.), а не о пептагидрате сернистой кислоты.
Сернистая кислота образует два ряда солей; кислые сульфиты MIHSO3 кнормалыше (нейтральные) сульфиты M|SO3. Оба типа солей разлагаются -сильными кислотами с выделенном двуокиси серы:
2MIHSO3-UHaSO4 = M^SOA+2H2O4-2SO2,
Miso, •i-H2SO4=M^SO4+H2O4-SO2.
Все кислые сульфиты, гидросульфиты (рапес называвшиеся также «бисульфитами»), легко растворяются в воде. Многие из них известны только в водном растворе, например кислый сульфит кальция (гидросульфит кальция) Ca(HSO3)2, раствор которого, сульфитный щелок, используют при получении целлюлозы. Разнообразное применение в технике находит также кислый сульфит натрия (гидросульфит натрия) NaHSO3,
Шестая группа периодической системы
769
который получают пропусканием двуокиси серы через насыщенный па холоду раствор карбоната патрия; NaHSOg кристаллизуется в виде маленьких, бесцветных блестящих призм. Его применяют как восстановитель, например в красильном деле и ситцепечатании, далее, как отбеливающее средство, а также в качестве исходного продукта для получения дитионита натрия Na2S2O4.
При отщеплении воды от гидросульфита натрия 2NaHSOg = Na-jSgO5 4- FI2О или прн пересыщении водного раствора гидросульфита натрия двуокисью серы получается пиросульфит натрия Na2S2O5. Обычное торговое название этого соединения «метабисульфит патрия». Однако чаще попользуют получающийся таким же способом пиросульфит калия K3S3O5 — блестящие, довольно твердые кристаллы, медленно растворяющиеся в воде. Его используют в фотографии для получения кислых ванн, кроме того, в красильном дело и в ситцепечатании.
Нормальные сульфиты М|[80з], за исключением сульфитов щелочных металлов и аммония, мало растворимы в воде, но растворяются в водных растворах сернистой кислоты. Водные растворы нормальных сульфитов вследствие гидролиза имеют щелочную реакцию, но только по метиловому оранжевому или лакмусу, по не по фенолфталеину.
Сульфиты являются очень реакционноспособными соединениями. В водном растворе опп легко окисляются до сульфатов. С другой стороны, они легко восстанавливаются, например, цинковой пылью в водном растворе до дитпоиитов (гипосульфитов) M*S2O4; при нагревании в сухом состоянии с углем, цинком и другими металлами — до сульфидов. При действии серы на водные растворы щелочных сульфитов получаются тиосульфаты M*SaO3; действием оксихлорида фосфора на сухие соли получают тионилхлорид SOC12. Сульфиты, и прежде всего сульфиты тяжелых двухвалентных метал.цов, обладают ярко выряженной склонностью к комплексообразованию (к образованию сульфитосолей).
Сульфит аммония (NH4)aSO3 в водпом растворе кислородом л о я дум медленно окисляется до сульфата. В присутствии комплексных аммиакатов кобальта, как установил Фордендер (Vorlancler, 1930), реакция настолько ускоряется, что ее можно использовать в технике для получения азота из воздуха ц одновременно сульфата аммония.
Обычно сульфиты получают пропусканием двуокиси серы через водные растворы или суспензии гидроокисей или карбонатов или растворением соответствующих металлов в водных растворах сернистой, кислоты. Малорастворимые в воде сульфиты получают двойным обменом между сульфитами щелочных металлов и растворимыми солями других металлов.
Г О 'Г
В растворах сульфитов присутствуют ионы [О S ОJ", в которых серу можпо рассматривать гит: имеющую заряд 4-|-. Можно предположить, что при кристаллизации содей эта itutii.i ciixp.iiiHiorcH в качестве структурных групп. Для свободной серпистой кислоты iiii.bj тииуощть возможность существования ее в двух структурных формах:
,ОН /н
o=s< и он (X ^он
1 I1 1
Симметричная флрма Асимметричная форма
В одной из них сера структурно четырех-, в другой структурно шестивалентна. Органические производные, в которых атомы водорода замещепы алкилами П, известны для обеих форм. Од пгт называются эфирами сернистой кислоты, другие — эфира-49 г. Ремп
770
Глава 15
ми а.пкилсерпой кислоты (раньше «эфиры а лкил сульфокис лот»).
.OR % /И
-o=s< >z
^OR \0R
Диажиловый эфир Алкиловый эфир сернистой кислоты алкилсерной кислоты
То, что в эфирах алкилсерной кислоты одна алкильная группа связана непосредственно с атомом серы, следует из того, что в этих соединениях только одна из двух алкильных групп (т. е. та, Которая связана с кислородом) отщепляется гидролитически («омыляется»). По данным Шварце нбаха (Schwarz ев bach, 1930), па ос пои алии значений констант диссоциации можно сделать вывод, что сернистой кислоте следует приписать «симметричную форму» (I), тогда как для иона гидросульфита возможны обе таутомерные формы Н—SOg и НО—S02.
Сульфит натрия Na2SO3 обычно получают следующим образом; сначала, пропуская двуокись серы через водный раствор 1Ча2С03, карбонат натрия переводят в гидросульфит натрия, а затем добавляют еще такое же количество карбоната натрия. Если упаривание вести при нагревании, то выпадает безводная соль; ниже 37,0" сульфит натрия кристаллизуется с семью молекулами воды. Растворимость в воде составляет при 0° 14,2 а, при 18,2"—25,3 г Na2SO3 в 100 г воды. В спирте соль мало-растворнма. В фотографии сульфит натрия применяют Для прекращения процесса проявления и фиксирования. В медицине он служит антисептиком. Кроме того, его используют для удаления хлора из отбеливавшихся им веществ, а также как консервирующее средство. В Германии запрещено употреблять сульфит натрия для консервирования мяса, так как оп возвращает нормальную окраску испорченному мясу; кроме того, указывается также, что сульфит натрия вреден для здоровья.
Сульфит калия KaSO3 в технике получают пропусканием двуокиси серы в концентрированный раствор едкого кали, пока проба раствора (предварительно разбавленная) пе перестанет окрашивать раствор фенолфталеина. Момент насыщения можно установить также, контролируя удельный вес. Сульфит калия кристаллизуется, как гидрат K2S'O3'H2O, в воде бесцветных, слегка расплывающихся кристаллов горького вкуса. Он с трудом растворяется в спирте, но очень легко — н воде.‘Раствор сульфита калия применяют в ситцепечатании как восстановитель.
Тиосерпая кислота п th осу ль (фаты. Если сульфиты кипятить с тонко-измельчеппой серой, то каждый ион сульфита присоединяет один атом серы, за счегг чего сульфиты переходят в тиосульфаты — соединения общей формулы XnS2O:J.
,\a2SO1-pS = Na2S2Ct3, и общем виде: SOg-|-S = S2O3.
Тиосульфат натрия Тиосульфат-ион
Щелочные тиосульфаты образуются, кроме того, при действии иода на смешанные растворы сулг.фидов и сульфитов щелочных металлов
S'-ySO"-)-Ia=S2O"-^2r.
Техническое значение имеет окисление полисульфидов до тиосульфатов под действием кислорода воздуха, например
Na2S5-{-3/2O3=Na2S2O3-|-3S;
CaS24-3/2O2=CaS203.
Большинство тиосульфатов легко растворяется в воде. Мало растворимы тиосульфаты свинца, серебра и одновалентного таллия. Очень трудно растворим тиосульфат бария. Растворимые тиосульфаты очень хорошо кристаллизуются; как правило, они кристаллизуются с водой. Тиосульфаты тяжелых металлов в большинстве случаев очень склонны к образованию комплексных солей. Так, тиосульфат серебра очень легко растворяется в водном растворе тиосульфата натрия, причем образуются комплексные тритиосулъфат-понъ! [Ag(S2O3)3]""'r (ср. т. II).
Шестая группа периодической системы, 771
ГОГ ГО
OSO OSO
I О
Кислоты разлагают тиосульфаты в водном растворе, при этом осаждается сера и выделяется двуокись серы. Однако осаждение серы происходит вд мгновенно, а спустя некоторое время. Поэтому предполагают, пти в растворе первоначально образуется тиосерная кислота Н2320з, которая, однако, мало устойчива в растворе
S2O"4-2H-^H2S2O3. -> II2O+SO2H-S.
Тиосерная кислота более устойчива в эфирном растворе. Но и в нем она распадается при нагревании, а именно сначала по уравнению H2S3O3 = = H2S -|- SO5. Эта реакция обратима; поэтому в эфирном растворе H2S2O3 можно синтезировать из H2S и SO3. Безводную тиосерпую кислоту можно получить также взаимодействием тиосульфатов с хлористым водородом (Schmidt, 1956).
Из солей тиосерной кислоты известны представители только одного ряда, а именно нейтральные соли, в которых оба атома водорода кислоты заменены на металлы. В отличие от очень неустойчивой кислоты некоторые соли довольно хорошо сохраняются даже в водном растворе.
Способ образования и поведение тиосульфатов показывают, что входящий в них тио су ль фат-ио n (S аО s ]'' мо жнор ассматрнв ать как производное су л ьфат-иона IS О4 ]", в котором од и п атом кислорода заменен па атом серы, или как производное сульфкт-иона fSO.j]”, имеющее дополнительный атом серы:
Г О 1'
OSO
S
Сульфит-ион Сульфат-и о в Тиоеульфат-ион
Раньше тиосерную кислоту считали продуктом восстановления сернистой кислоты и называли «серноватистой кислотой». Однако в соответствии с вышеизложенным переход иона S03 и поп 82О3 не является восстановительным процессом. В известном смысле его можно даже считать окислительным процессом (ср. стр. 810 и сл.). Поэтому тиосерпую кислоту нельзя называть «сор по в ати стой кислотой». Поскольку замена в соединении кислорода на серу в общем случае обозначается приставкой «тио», то название «тиосерная кислота» показывает, что речь идет о соединении, производном серной кислоты, в котором атом кислорода замещен па атом серы. То, что тиосерная кислота имеет строение, отвечающее этому названию, в настоящее время можно считать твердо установленным.
Тиосульфат натрия Na2SaO31 в продаже все еще встречающийся под названием «серноватистокислого натрия», раньте получали в технике главным образом из сульфида кальция, являющегося побочным продуктом при получении соды по методу Леблана. Сульфид кальция при вылеживании па воздухе в значительной степени переходит в тиосульфат кальция, из которого затем взаимодействием с сульфатом натрия можно получить тиосульфат натрия.
GaS2O3-(- Na2SO4= Na2S2O3—J-CaSO^.
В больших количествах тиосульфат натрия получают как побочный продукт прп производстве серных красителей (см. стр. 789). Из водных растворов тиосульфат натрия обычно кристаллизуется в виде пентагпд-рата Na2S2O3-5H2O Пентагидрат представляет собой прозрачные моноклинные кристаллы, легко растворимые в воде, причем при их растворении поглощается значительное количество тепла (И кк а л/л олъ). При 48,5° иентагидрат плавится в своей кристаллизационной воде. Расплав можно сравнительно легко переохладить и сохранить и таком состоянии в течение нескольких суток. Только при встряхивании раствора, при трении стенок стакана стеклянной палочкой или при внесении в виде затравки
49*
772 Глава J5
кристаллина твердой соли происходит мгновенная кристаллизация, сопровождающаяся выделением значительного количества тепла.
Тиосульфат натрия имеет разнообразное применение. В фотографии его используют как фиксирующую соль для удаления яепророагировав-шего бромида серебра с проявленных пластинок и т. д. При отбеливании его используют как ^антихлор», т. с. для удаления хлора из отбеливаемых им тканей
8вОз=|-4С.124-5Н2О = 23О:-|-8С1,Н- ЮН'.
С иодом тиосульфат натрия реагирует с образованием тетратпоната натрия Na2S4Oe
; 2s2o;+i2=s4o;'+2i'.
Реакция протекает количественно и лежит в основе применения тиосульфата натрия в иодометрии. Взаимодействие с бромом, которое в кислых растворах протекает также количественно по уравнению, аналогичному уравнению реакции с хлором, можно использовать в бромометрип UD'A n s, Angew. Chem., 63, 45, 1951],
Су льфацсульфо новые кислоты. Вместо того чтобы рассматривать тиосор ную кислоту как производное серной кислоты, получающееся в результате замены атома кислорода на атом серы, ее можно также считать производным На8 (сульфа; ta, ср. стр. 784), в котором одцп атом водорода заменен на группу SO3IJ, и называть сульфа; (сульфоновой (точнее, сульфан.чи-.о.усульфоковой) кислотой. Т,чк же кал тносериую Кислоту можно получить действием 80 з на H,S, но данным Шмидта (1956), Действием S03 на нолисуль-фапы II3Sti можно получить полисул1.ф>лн(тлъфоновые кислоты. Этим методом можно приготовить как по.тисульфаишаносульфоновые кислоты II—(S)1t—SO3II, так и Нили-су.чьфапДисульфоновые кислоты HSO3—(S)J(—SO3H. Последние идентичны политионовым кислотам. которые рассмотрены ниже. Полисудьфапсульфоновыо кислоты образуют класс соединений, из которых до сих пор, кроме тиосерной кислоты, не был известен ни один другой представитель. Шмидту удалось указанным выше методом получить, кроме кислоты tIsSz<)n, также кислоты IJ2S3O31 H2S4O3 и HaS5O3.
Тцчсерппетая кис юта IbSyj.., нтн S^SiOH),, в водном растворе, вероятно, ВаХИДИТсЯ В /ЧОШйтЧ’; !,li г H_s и J!_.S(f3
-,ll2 - USOHb ~ H50 + S = S(OH)2,
В i BoOopnijM состоянии получить ее He удается. Эфирами. тносернцстоп кислоты большей частьго называют соединения общей формулы йкО^Нг, полученные впервые в 1895 г. Ленгфельдом, прп действии метилата или этилата натрия па растворенную в петролейним. эфире хлористую серу SAllg. Эти соединения представляют собой бесцветные, резко пахнущие жидкости. Их структурной формулой, одиако, является 1 о s в.
не приписывавшаяся цм ранее формула S = S(OB)2, а формула р./ \д/ \q/ > на правильность которой указывают величины их парахора, дипольного момента и данные спектров комбинационного рассеяния (Stamm, 1937; Scliefbe, 1938).
Политионовые кислоты и по литиониты. Если через водный раствор двуокиси серы пропускать сероводород, то получают так называемую жидкость ВАкенродера — раствор, содержащий политионовые кислоты. Под политионовыми кислотами понимают соединения общей формулы Н38гОй, где х может принимать значения 3, 4, 5, 6 и, вероятно, еще более высокие*. В основном жидкость Вакепродера содержит кислоты H3S4O6 (тетратионовую кислоту) и Il2SbOe (пентатионовую кислоту). При действии сернистой кислоты па сероводород в качестве промежуточных продуктов получаются тиосерная кислота Нг$30г
* Дктисиювая пиелита. 112Й2ОС не относится н Еоллтионовым кислотам (ср. стр. 779).
Шестая группа периодической системы
773
и тритноновая кислота H2S3Oe. Однако последняя в присутствии тиосерной кислоты, присоединяя серу, тотчас же переходит в тетра- и пентатионовые кислоты.
Реакции, имеющие практическое значение для получения политионовых кислот, изучали главным образом Дебус (Dehна), Форстер (Foerster), Рпнеифельд (Rie.mmfeld), Басетт (Bassett) и Штамм (Stamm). Долгое время почти общепринятым мнением было, что при взаимодействии H2S с H2SO3 в качестве первого промежуточного продукта получается моноокись серы SO или суль^оксижпая кислота S(OH)2. Однако Шелку (Schenk) удалось показать, после того как он сумел получить SO (ср. стр. 778), что при реакции H2S с SO2 не образуется SO и что последняя реагирует с H2SO3 без образования политионовых кислот. С другой стороны, Ноак (Noack, 1925) и НЕ та мм (1938 и сл.) доказали, что продукт гидролиза соединений S2C12 и S2O2R3, а также соединение H2S2O2* реагируют, с одной стороны, с H2SO3 и с другой — с HaS, образуя как раз те соединения, присутствие которых наблюдается в жидкости Вакенродера. Поэтому в настоящее время считают, что (как уже предсказывал в 1922 г. Форстер) соединение Л<>3202 является первичным продуктом взаимодействия H2S с Н;8О_? и что политионовые кислоты образуются в результате следующего ряда превращений:
Н28+Нг8О3 H2S2O2+H2O, (7)
.H2S2O2+2HsSO3=H2StO64-2H2O, (8)
H2S,(Ob-!-H2SO3 H.2S306+H2S2O3, (9)
П2ЙгО3 = S+H2SO3, (10)
H 2s2o 2 4 H2S - 3S 4- 2H2O, (11)
H2S4Oft-|-S = nzS5Oe.
(12)
Сырая жидкость Вакенродера содержит значительные количества коллоидной серы. Если ее очистить от серы, па пример добавлением небольших количеств хлорида лантана, то образуется бесцветный, не имеющий запаха раствор с сильнокислой реакцией, который можно сконцентрировать выпариванием па водяной бане. Однако в безводном состоянии содержащиеся в нем политионовые кислоты получить пе удается, хотя легко получаются их соли — политионаты М|[8жОе]. Среди них особой устойчивостью отличаются политионаты щелочных металлов. Менее устойчивы политионаты щелочноземельных и тяжелых металлов. Легче всего разлагаются тритионаты, конечными продуктами их распада в водном растворе являются серпая и сернистая кислоты и сера**. Большинство политионатов легко растворяется в воде, по многие не растворяются в спирте.
Таблица 108
Растворимость в воде полигпопатов щелочных металлов при 20° (1 г безводной соли на 100 г раствора, по данным Kurtenacker, 1933—1938)
NaaS^Oa ЗН2О • 2ЁН-/.1 NaaSsOs-2H:O (NHj)2. saoe (NH4)2- -SKhs K2S4OG KaS60s- f 1,'гНгО
52,9 50,4 52,0 56,9 54,4 18,4 23,2 24,8
* В настоящее время еще ire известно, имеет ли это соединняие строение HO-S-S-OH или S—S(OH)2.
** Подробнее о распаде политионатов в водном растворе см.г Goehring М., Z. anorg. Chem., 254, I85, 1947.
774
Глава 15
По данным фон Дейнеса (v. Delises, 1933) и Шейн зона (Meawsen, 1952), политионаты: тяжелых металлов проще всего получать обменной реакцией перхлоратов атих металлов с политионатами щелочных металлов.
Трптионаты, Тритионат калия Кя8зО6 получают действием двуокиси серы на тиосульфат калия в водном растворе*. Тритивнат натрия NajSsOe можпо получить при действии перекиси водорода па сильно охлажденный насыщенный раствор тиосульфата натрия
2Na3S3O3 -ф- 4НоО2 = Na2S3O0 ф- Na2SO4 *ф- 4Н2О.
Свободную тритионовую кислоту H2S3Oe можно получить действием на ее калиевую соль кремнефтористоводородной кислоты, хлорной кислоты или винной кислоты; однако эта кислота устойчива только в водных растворах.
Тетратиопаты. Тетратионат натрия NajS^O,;, как уже упоминалось, образуется при действии иода на тиосульфат натрия. Аналогичным способом можпо получить также тетратионат калия К.3Н4Ов. При действии на него рассчитанного количества винной кислоты выделяется свободная тетратионовая кислота H2S40e. Однако ее также пе удается выделить в безводном состоянии,
Пейта тио паты. Пентатионат калия K2S5Os можно получить из жидкости Вакеп-родера при добавлении в нее ацетата калия. При этом сначала выпадает тетратионат калия в виде призматических кристаллов. Из маточного раствора выделяются пластинчатые кристаллы пентатионата калия. При действии на него винной кислоты выделяется свободная^ пентатионовая кислота IIaS5Oe. Она также устойчива только в водных растворах.
Гексатионаты. Существование гексатионовой кислоты H2SrtO8, присутствие которой в жидкости Вакепродера допускалось еще Добусом (1888), было надежно установлено в 1928 г. Вайтцем (Weitz), которому удалось получить чистые кристаллические соли этой кислоты. Вполне устойчивый в твердом состоянии, но очень легко разлагающийся в водном растворе, особенно в присутствии щелочей, гексатионат калия K2SeOe можно получить при взаимодействии тиосульфата калия с нитритом калия в сильно солянокислом растворе при пониженных температурах (от —10 до —15°), Он выделяется в виде белых кристаллических чешуек. Особенно хорошо кристаллизуется бензидиновая Соль гексатио но пой кислоты [С i2H (2N9H 31 [S60eJ (бесцветные тонкие: кристаллические иглы).
Строение политионовых кислот. Строение тетратионовой кислоты или соответственно тетратионат-ионов следует из того, что она образуется при окислении иодом тиосульф ат-иопов.
Поскольку атомы иода отрывают по одному электрону от двух атомов серы, между этими атомами серы может образоваться связь. Эта реакция вполне аналогична реакции образования пероксоднсульфат-ионов из двух частично потерявших заряд суль-фат-ионов (ср, стр, 766),
Предполагают, что тритионаты и пентатионаты построены аналогично тетратйо-натам, т. е, О О О О
'О —S—S —S —О'; 'О — S—S — S—S — О'
АО О А '
Фритионат-ион неитатионаг-ион
* Механизм этой реакции еще недостаточно ясен. Интересно отметить, что тритионат натрия аналогичным способом получить не удается.
Шестая группа периодической системы
775
С точки зрения формальной теории валентности в полптггоповых кислотах имеются два атома шестивалентной серы, связанных между собой одним, двумя или более атомами Змржалептпой серы. Однако к вопросу о формулах можно подойти и с точки зрения координационного учения, В атом случае политионовые кислоты можно было бы рассматривать как двухъядерные соединения, оба координационных центра которых заняты положительными координационно четырехвалептпыми атомами серы, тогда как связующие мостики образованы частично отрицательными, частично нейтральными координационно двухвалентными атомами серы.
Недавно Шмидту (Schmidt, 1956) удалось получить политионовые кислоты в безводном состоянии, а именно действием SO3 на сульфапмоносульфоиовые кислоты
HSO3—(S)n—Н 4- S03 = HSO3-(S)n-SO3H.
Этим методом можно получить политионовые кислоты с числом атомов серы до 10. Окисление сульфапмоносульфоновых кислот иодом в водном растворе также приводит к образованию политионовых кислот
2HSO3-(S)n-II + I2 = HSOg-tSJzn-SQsH 4- 2HI.
Высокомолекулярные полптионаты. Еще в 1928 г. Вайтц наблюдал, что возможно образование политиопатов с более чем шестью атомами серы. Одпако удается получить только их смесь, вероятно, потому, что высокомолекулярные политионат-ионы имеют тенденцию распадаться иа ионы с меньшим и с еще более высоким молекулярным весом. Все же дробной кристаллизацией, например бензидиновых солей, можно разделить эти смеси. Этим методом Вайтцу удалось позднее получить в кристаллическом состоянии политионаты бензидина, содержащие до 10 атомов серы. Политионаты с особенно высоким молекулярным весом (с 20—40 атомами S) образуются при действии на тиосульфат калия концентрированной соляной кислоты в присутствии S2C13. Эти политиопаты существуют в водном растворе в коллоидной форме. Очевидно, их нитеобразные анионы, подобно анионам в мылах, склонны располагаться рядом, образуя мицеллы (см. т. II). По мнению Вайтца (1952), так называемые гидрофильные золи серы Одена (Odea, 1913) представляют собой не коллоидные растворы серы, а коллоидные растворы политнопатов с очень высокими молекулярными весами (с 40—60 и более атомами серы),
Дитноновая кислота и дитионаты. Дитионовую кислоту H2S2Oe раньше также причисляли к политионовым кислотам. Однако она не имеет к ним непосредственного отношения, а скорее представляет собой промежуточный член между сернистой и серной кислотами. Дитионовую кислоту в виде ее марганцевой соли получают действием двуокиси серы на взмученный в воде гидрат двуокиси марганца
MnO24-2SOa=MnS2OG.
При этом некоторая часть двуокиси марганца реагирует с образованием сульфата марганца по уравнению
МпО2 4 SO2 = MnSO4.
Вместо двуокиси марганца можпо использовать другие окиси тяжелых металлов (в реакционно способной форме, т. е. в форме гидроокиси), обладающие слабыми окислительными свойствами, например гидрат окиси железа (III). В этом случае сначала образуется сульфит железа(Ш), распадающимся затем па сульфит железа(П) и дптио-пат же..1еза(П).‘
Ш П И
Fe2O3 -L 3SO2 = Fc2(SO3)s; Fe2(SO3)3 = FeSO3 + FeS2Oe.
Сулъфит-ионы можно также перевести в дитионат-ионы анодным окислением на электродах из платинированной платипы
2SO3 4- 2е = S2O;.
Однако при этом основным процессом является образование сульфат-иопов по уравнению
so; - 2е > н3о = so; + 2Н-.
Взаимодействием дитионата марганца с гидроокисью бария получают хорошо кристаллизующийся дитионат бария ВаЙаО4-2Н2О, Разложением
776
15
бариевой соли рассчитанным количеством серной кислоты получают свободную днтноновую кислоту Н282ОЙ. Однако она устойчива только в водных растворах. При сильном концентрировании раствора дитионовая кислота распадается на серную кислоту и двуокись серы
П,Р3О6==Нг8О4-|-8О2.
Аналогично распадаются при нагревании и соли дятионовоё кислоты, па приме]» дитионат калия при 258°: K.2S20e = I^zSO4 -f- SO2 — 5,0 ккал (Tammann, 1932; Roth, 1935). О механизме реакции распада дитионовой кислоты см.: Stem тп И., Вег. 67, 726. 1934; S t a m m> G о е h г i n g, Z. phys. Chem., 183 (А), 89, 112, 241, 1938.
Все дитиопаты легко растворимы в воде и в большинстве случаев-хорошо кристаллизуются. Все они, за исключением дитионатов щелочных и щелочноземельных металлов, распадаются при кипячении растворов, а некоторые медленно даже при обычных температурах.
Еще рацьпто большинство авторов принимало для дптионовой кислоты формулу, предложенную Менделеевым;
(HO)O2S-SQ2(OH).
В соответствии с этой, формулой дитпоноваи кислота является продзиодным серной кислоты, получающимся при соединении двух радикалов серной кислоты — SO2(OH), точно так Же как щавелевую кислоту (НО)ОС—СО(ОП) можно рассматривать как продукт соединения двух радикалов угольной кислоты — СО(ОН) (карбоксильных радикалов). Эта форигула для дитвоповой кислоты, как и соответствующая формула для дитионат-иона [OsS — SO3}2*, полностью подтверждена ре пт гон остр у кт у ртп л м определением цезиевой соли Cfsa [320й] (Hagg, 1932) и магнето химическим и исследованиями (см. гл, 9),
Дитиониты, Если дитионаты образуются при окислении'
сульфитов, то дитиониты M2[S2O4.| получаются при их восстановлении'.
р . Ш _Н2О.
Их можно получить, как видно из этого уравнения, также электролитическим восстановлением растворов, содержащих гидросульфит-ионы, но лучше получать восстановлением цинком растворов гидросульфита натрия, содержащих избыток сернистой кислоты:
2NaHSO3-f- H2S03-|-7,n = ZnS034-Na2S204+ 2Н2О.
Дитионит натрия (гипосульфит натрия) выделяется из этих растворов при добавлении спирта или поваренной соли. Он кристаллизуется в виде кристаллогидрата Na2S2O4-2H2O, однако в этой форме он мало устойчив, и поэтому его переводят, удаляя воду, в безводную устойчивую соль. Дитионит применяют как восстановитель при кубовом крашении и вытравном способе печатания, В газовом анализе дитионит натрия используют как поглотитель кислорода. Кроме того, оп служит для получения ронгалита (см, ниже), также применяющегося при кубовом крашении и вытравном печатании. Вследствие очень сильной способности к окислению дитиониты (гипосульфиты) очень неустойчивы в растворе или в виде кристаллогидратов. Даже без доступа воздуха в растворе или во влажном состоянии они постепенно распадаются па тиосульфаты и пиросульфиты
2Na2S2O4= Ха3820з -(-Na3S2O3.
Шестая группа периодической системы
777
При нагревании разложение происходит ио уравнению
= Na^SgOs 4- Na^SOj + SO2 4- 2il,7 ккал*.
Относительно устойчива цинкован соль ZnS,O4. То же относится к ее двойным солям с дитпонитамп щелочпых металлов. В воде дитионнты, за исключением дитио-нита кальция, легко растворимы. Для натриевой соли растворимость составляет 21,8 г Na2S2O4 в 100 г воды при 20°. Соответствующая дитионитам свободная кислота— дитиониетая кислота — нВ существует.
Образование дитаонитов при и ос становлении растворов гидросульфитов впервые-наблюдал Шонбайп (Schonbein) в 1852 г. Твердую соль, а нменпо натриевую соль, впервые выделил Шутцепбергер (Schirtzenberger, 1869). Последний ошибочно приписал ей состав Л а.Н8Ог и дал название гидросульфит натрия. Правильный состав ft'aSOj или NaES2O4 был определен Бернтсеном в 1880 г. Тем не менее неверпое название «гидросульфит» встречается, особенно в технической литературе, и по сей день. Введению названия гипосульфиты или соли серноватистой кислоты, предложенного для соединений MlS2O4 Бернтсеном и другими, препятствовало употребление (хотя и устаревшее) этого названия для тиосульфатов (солей тиосериой кислоты). Против того, чтобы соли состава MaSgO^ называть «гипосульфитами», можно также выдвинуть еще Другое возражение, а именно что это на зван но пе соответствует солям M|S3O4 с точки зрения обычной номенклатуры солей кислородсодержащих кислот, а, строго говоря, опо должно относиться к солям состава M2SO2 (сульфоксилатам). Уже Нойес (Noyes, 1929) указывал на то, что в соответствии с обычными правилами наименования солей кислородсодержащих кислот соли M2Sa()/r должны быть названы дитионитами, так как состояние окисления серы в среднем па один атом серы в этих соединениях на единицу (удвое1щую) меныпе, чем в дитиопатах. Поэтому в настоящее время для солей M2S204 предложено название дитиониты, а для соответствующей им кислоты — дитиопиетая кислота.
Строение дитвояатов или соответственно дитионит-ионов fS^O^]21 до последнего времени оставалось спорным. Химические реакции дитиопитов протекают не столь однозначно, чтобы но ним можно было с достоверностью судить о их строении. Только исследования спектров комбинационного рассеяния [Simo n A., Z. anorg. Chem., 260, 161, 1949] внесли ясность в этот вопрос. Эти исследования показали, что в Дятио-нит ионе [S2O41-1, имеется простая S—S-связь (без свободного вращения) и, по всей вероятности, оп имеет плоское и совершенно симметричное строение. Структурная формула дцтионит-иона, по-видимому, такова;
-/°lT
Id/ ^о|
[О-S—SCG]2* пли
Каящьп) из двух атомов серы в этом иопе имеет три ковалентные связи и песет положительный заряд, равный трем. Способ образования и реакции дитиопитов согласуются с такой формулой.
Сульфокспл<>вал кислота (ронгалит). При действии формальдегида ЯСНО па дитионит натрия получают (применяющийся в красильном деле в качестве восстановителя) ронгалит:
Na3S2O4 4- 2НСНО 4- Н2О = CII2(OII)-SO2Na - CHa(OII)-SQ3Na.
Онспметансульфшпшокис- Онсиметанеульфоньвокпс-лыit натрий (рюяаалмт) пый натрий
Раньше большинство ученых считало ронгалит производным сульфо ксиловоИ кислоты (неустойчивой в свободном состоянии). Однако он скорее всего является производным пе этой кислоты, а и золю р ной ей (также неустойчивой) кислоты HSO (ОН). Нортому его следует называть пптрновой солью оксиметансульфиноеой кислоты CH2(0J1)-lS0(0Hj. Эта точка зрения подтверждается прежде всего наблюдением, что Ка-дублет в рентгеновском спектре ронгалита имеет примерно то же положение, что и слектрс 71-толуилсульфнновой кислоты CH3-CeH4-SO(OII) (Eaessler, Goehring,
Сульфо ксилт-ап. кислот» [гидроокись серы (II)] S(QH)2 образуется в качестве-промежуточного продукта при гидролизе SCL, SfOCoIls)2 и аналогичных соединений. О реакциях с-ульфокспловой кислоты см.: Goehring М., Z. anorg. Ghent., 253, 304, 1947. Вопрос о существовании солеа сульфоксило вой кислоты (сульфокси латов)
* Прп 190° такой распад натриевой соли происходит со взрывом. Поскольку уже поглощение влаги вызывает нагревание, большие количества соли могут самораска-ляться благодаря повышению температуры.
778
Глава 15
-ице не ясен. Шольдер"( Sell older, 1935) полагает, что при осторожном подкислении водно-аммпачного раствора" СоС12 и Na2S2O4 ему удалось получить сулъфоксйлат кобальта СоЗОг-ЗГ^О, При этом сначала должно было произойти гидролитическое расщепление дитионит-пона на сульфит- и сузьфоксилат-ионы:
[S2OJ" + Н2О = [ЗО3р Ч- [5О2Г + 2Н-.
Неустойчивые в водном растворе сульфокеилат-поны [ЗО-г]" улавливаются затем попами кобальта с образованием нерастворимой соли, выделяющейся пз раствора в виде темпо-коричпевого осадка. Однако, согласно Боде (Bode, 1952), доведение в магнитном поле полученного этим методом препарата не соответствует ожидаемому для соединения CoI1SO2. Эфиром сульфоксиловой кислоты, предполагая, что она имеет обычно приписываемое ей строение S(OH)2, считается полученное Мейвзеном (1936) соединение S(OC2H5)2. Од получил это соединение при каталитическом разложении . диэти лтиосу, гьфита
S=S(OC2H5)2 = s+ S(OC3H5)2.
Это бесцветная, резко пахнущая, горючая, пе смешивающаяся с водой жидкость (т. кип. 117° при 733 льи рт. ст.).
Полуторная окись серы S3O3 получается в виде голубоватых кристаллических чешуек при действии серы на совершенно безводную трехокись серы. Избыток трехокиси серы можно удалить декантированием, так как полуторная окись серы в ней очень мало растворима. При попадании влаги полуторная окись серы образует темно-голубой раствор. В дымящей серной кислоте полуторная окись серы растворяется с образованием растворов от голубого до коричневого цвета в зависимости от содержания в серной кислоте SOs. Коричневые растворы содержат коллоидно растворенную серу, образующуюся при распаде очень чувствительной к влаге полуторной окиси серы. Раньше и синие растворы рассматривали как коллоидные растворы серы в трехокиси серы. Однако еще в 1875 г. Вебер установил вполне определенный стехиометрический состав таких растворов, Что было недавно подтверждено Фогелем (Vogel) и Партингтоном (Partington).
По составу полуторную онись серы можно рассматривать как ангидрид дитионис-топ кислоты (гидроссрпш’топ кислоты ( H2S2O4; однако до сих пор нс установлена связь,]между полуторпоп гпщсью серы и этой кислотой или ее солями,
Моноокись серы. SO впервые получена в чистом состоянии в 1933 г. Шенком при действии тлеющего разряда па двуокись серы пли смесь последней с парами серы. Она представляет собой бесцветный газ, распадающийся при обычных температурах в отсутствие катализаторов очень медленно (с выделением соры);однако,если этот газ сконденсировать охлаждением, он уже не испаряется без разложения. При обычных температурах он не реагирует с кислородом воздуха. Водой SO Сначала разлагается до уравнению 3SO -р ЗН2О = H2S 4- 2H2SO3, после чего происходит выделение серы и образование политионовых кислот.
Спектроскопические наблюдения уже в 1929 г. привели Генри (Henri) к мысли, что при действии тлеющего разрида из S03 образуется SO. Еще раньше образование SO в качестве промежуточного продукта предполагали во многих химических реакциях. Образование SO при взаимодействии SO Cis с некоторыми металлами, например Sn и Sb, как и при сгорании серы в избытке ее паров, было спектрографически установлено Шенком. Кроме того, оказалось, что SOC12 при 900°, a SOBr2 уже при 520° в значительной степени начинают распадаться на SO и С12 или соответственно Вг2, тогда как распад 80s на SO и О2 начинается в заметной степени только при значительно более высоких температурах.
2, Соединения серы с галогенами (галогениды серы)
С фтором сера образует соединения SFe, S2F10, SF4, SF2 и S3F2, с хлором — соединения SCI4, SCI3 и S2CI2. Последнее соединение является первым членом класса соединений общей формулы SnCl2, в которых имеются цепи, образованные большим или меньшим числом атомов
Шестая группа периодической системы
779
серы. Эти соединения называют политиохлоридами или дихлорполисулъ-фанами. С бромом сера образует только соединения типа SnBra (политиобромиды или дибромполисульфаны). Соединений серы с иодом выделить не удалось*.
При замещении гидроксила в кислородсодержащих кислотах серы па галоген получаются соединения, из которых в первую очередь следует упомянуть тионилхлорид SOC12, сулъфурилхлорид (хлористый сульфурил) SO2CI2 и хлорсулъфоновую кислоту SO2(OH)C1.
Соединениями тиоиила называются соединения, содержащие двухвалентный радикал >SO, соединениями сульфурила, — содержащие радикал >ЗОа. Соединения, получающиеся пз симметричной формы сернистой кислоты OS(OH)2 заменой одной из гидроксильных групп па другой одновалентный атом или радикал, называются •сульфииовыми кислотами, а аналогичные производные серной кислоты 028(0П)2 называются сульфоновыми кислотами,, пли сульфо кислотами.
В табл. 109 приведены свойства и формулы простейших соединений серы с галогенами.
Таблица 10&
Галогсппды п кислородные кнелотьт серы
Фториды Хлориды Бромиды
! Cl-(S)n-Cl Полшпиохлориды. (д и хлорпол и сул ьф а н ы) см. стр. 782 Вг—(S)n —Вг II о лити обромиды (дибромполисульфаны) см. стр. 784
ZS. Ж р/ Xg/ Бесцветный; т. пп. —130,5°; т. кцп.—38° Дитиофторид / .s. XI си Оранжево-желтый; т. пл. —76,5°, т. кип. +137,1°; уд. вес 1,709 (при 0°) Дитиохло рид вг/Ч/Вг Темтго-краеный; т. пл. —46°; перегоняется без разложения, уд. вес 2,635 (при 30°) Дитиобромид
ZF 8( XF Бесцветный; т, кип. —35° Тиодифторид XI S^ci Гранатова-красный; т. кип. 59°; уд. вес 1,623 (при 15°} Тиодихлорид —
1хр к/ Ч' Бесцветный; т. пл —124°, т. КИП. —40° Тиотетрафторид Cl\s/C1 ClZ Cl Бледно-желтый; плавится с разложением примерно яри —30° Тиотетрахлорид —
* По данным Рао (Вас, 1940), иодистая сера (дцтяолодвд) ,S2I2 образуется в качестве промежуточного продукта при действии КI Иа растворенную в СС14 хлористую серу SjClg. Это же относится и к иодистому тионилу (тионнлиодиду) SOI2.
Продолжение гпабд, 109
Фториды Хлориды Бромиды
F F F F Бесцветный; т. пл. —аО,6°; т. субл — 63,8°; уц. вес 1,88 (летуч при плавиепии) Two г е к са 56 т орид - -
р F F F^S-S<F F М F Бесцветный, т. пл. — 92’; т. кип. 20°; уд. вес 2,08 (при 0°) Д ит иодекафторид — —
/Р O = S< XF Бесцветный; т. пл. —110°, т. кип. —30° Тио нил фт .ipU'J ,С1 O = S( ХС1 Бесцветный; т. кип. 76*; уд. вес 1.С77 (при 0п) TwoHiMir/io/we* /ВГ o=s< хВг К ра снс вато-з । {елтля ж ид к q сть*г ирц наг рева нии р а зла гас тел уд вес 2:61 (при 0°) 7 гио н и лбр о.м ид
(J, /F ""F Беецввтвый; т пл. — 126°. т кттн —82“ Сульфурилфторид о^'4 Cl Бесцветный; т. кцц. 6Э:3“; уд вс,’ 1,70 8 ()Ц,и О’) Сулъфурилх ло piu) —
%S/0H tZ 4F Бесцветная; т. кип, 162.6° Фторсулъфоновал кислота Ох .011 Чд/ О^'УС1 Бе с цветная; т. кип. 152’; уд. вес 1,78 4 (при 0°) Хлореулъфоновая кислота —
— ° 0 0 ^S\ /Ч 0 С1 С1 0 Бесцветный; т. тпш» 158°; уд. пес 17872 (при 0°} Пиросулъфурилхлорид —
Шестая группа периодической системы,
781
Вода гидролитически разлагает все галогепиды серы, за исключением фторидов, причем при разложении низших галогенидов, например S2CIa и SC121 выделяется сера.
Тетрафторид серы (четырехфтористая сера) SF>, может быть получен взаимодействием SC12 с NaF в ацетонитриле прп 70—-SO'2'; SF4 можно использовать в качестве фторирующего агента. Оп реагирует с альдегидами, кетонами и другими карбонильными соединениями, заменяя О на 2F.
Гексафторид серы (шестифтористая сора) SFg образуется прп непосредственном взаимодействии элементов, протекающем с большим выделением тепла. Гексафторид >серы представляет собой бесцветный, лишенный запаха газ с высоким удельным весом (5,107 по отношению к удельному весу воздуха, принятому за единицу; вес 1 л >6,602 г). Этот газ мало растворим в воде, несколько лучше в спирте. Он не горит: при нагревании в токе кислорода или в токе водорода остается без изменений. Он вообще мало активен. Однако при нагревании оп разлагается сероводородом до уравнению
SFe ф- 3H2S = 6IJF ф 48.
Как установили электро по графическим методом Брауне (Вгавпе, 1933) и Брокуэй (Brockway, 1933), в молекуле SFe атомы фтора расположены но вершинам правильно то октаэдра вокруг атома серы. То же справедливо для SeFe и ТеЬ’й. Значения межъядерных расстояний приведены в табл. 61 на стр. 346.
SeCI4 имеет тетраэдрическое строение Se<—»Cl = 2,13 А; ТеС14 имеет неправильную структуру: Те ч—» С1 = 2,33 А. Молекулы TeCI2 и ТеВг2 изогнуты: Те <—>С1= =2,36 W, Тс<-> Вт =г 2,51 А.
Дитнохлорид (дитиодихлорид) SaCI2, называемый также хлористой серой, представляет собой оранжево-желтую, дымящую на влажном воздухе жидкость с неприятным удушливым запахом. Ее получают пропусканием хлора над расплавленной серой и применяют для растворения серы при вулканизации каучука. Ее используют также для приготовления -сероуглерода и других соединений, особенно хлоридов, поскольку она легко теряет хлор.
Хлористую серу применяют часто также при криоскопических или эбулиоскопических определениях молекулярного веса, так как она растворяет очень многие вещества. Молекулярное Понижение температуры замерзания для 1000 г хлористой серы равно 5,36°, повышение температуры кипения 5,02°. Растворимость серы в S2C12 нрн -20“ составляет около 22 г S в 100 г раствора. Вода медленно разлагает хлористую серу с образованием хлористого водорода, двуокиси серы и серы. При этом образуются также тиосерная кислота и и о нити о новые кислоты и в меньших количествах сероводород и серная кислот^. В соответствпп с исследованиями Лоа к a (Noack, 1925) и Штамма (Stamm, 1942 и сл.) механизм реакции можтю представить следующим образом; Сначала при гидролизе хлористой серы но уравнению S2Ch + 2Н0Н = Sa(0II)2 Д- 2НС1 образуется неустойчивая в свободном состоянии /гмосерлистая кислота (или какое-то иное изомерное ей соединение). Первичный продукт гидролиза частично распадается по уравнению H2S2O2 = H2S ф- SO2. Другая часть может дальше реагировать с продул, та \щ распада H2S и SO3 по уравнениям, приведенным на стр. 773. Этими реакциями объясняется появление всех упомянутых вьпне веществ, за исключением серной кислоты, Образование последней становится понятным, если допустить, что S:1C12 в расттшрь частично распадается па хлор и серу. Для паров хлористой серы (прп температуре выше 31’if) такой распад экспериментально доказан (Yost, 1935).
Ни основании установленного уже Карнусом (Саи us) образования хлиристои серы при обработке хлористого тцппила пента сульфидом фосфора
Г - |- [, O^SC12 - Р2О>, 4 5S=SC13
и па основании дуутх наблюдений, например большого сходства спектров поглощения S3Cla и О=ьС12, раньше в качестве вероятной структуры для хлористой серы при-C,i
ним,тли структуру 8^4 . Одпако спектр комбинационного рассеяппя евндетедь-
ЧС1
ствует о наличии одной простои спжш между цЯ&яып атомами серы (Venkates-waran, 1931; Miznsliirna, 1937). Бальмер (Райгцт, 1938) Электр о нографпческимя
782
Глава 15
S Cl измерениями определил, что структура хлористой серы такова: (расстоя-
ния: С1 <—► S 1,99 A, S » S 2,05 А, C1SS 103°). Дипольный момент хлористой серы-(ц = 1,06.10-18 эл. ст, ед., Scheibe, 1938) согласуется как с асимметричной, так и (при
учете возможности свободного вращения вокруг простых связей) с симметричной структурам. Гиакомолло (Giacomello, 1935) да основании химического поведения-хлористой серы пришел к выводу, что обе формы находятся в равновесии.
Дяухлористая сера (днхлорид серы) SC12, Если в хлористую серу пропускать ;
хлор, то она поглощает его, причем окраска ее постепенно темнеет. Раньше пе было- i
достаточной уверенности в том, образуется ли при этом новое соединение или просто- i
происходит растворение хлора. Оказалось, что в действительности происходит и то-и другое. Сначала хлор просто растворяется, по затем частично вступает в реакцию |
с хлористой серой, образуя двухлористую серу. Последняя в чистом состоянии пред- |
ставляетсобой гранатово-красную жидкость (уд. вес 1,62 прн 15°; т. кип, 59°). В обыч- |
ных условиях она в очень сильной степени распадается на хлористую серу и хлор; I
поэтому химически она ведет себя как смесь этих двух веществ, молекула ЗСЬ ноли- 6
нейпа, S С1 - 2,00 А, < = 103°. *
Соединения SC12 и S2C12 можпо рассматривать как первые члены гомологического-- 5
ряда дихло размещенных сероводородов (ср. стр. 784 и сл. и стр. 793). Это относится 3
и к остальным ди галогенидам серы и дисер ога до генидам. !-
Ио.читпохлорпды SnCl2. Сера пе только просто растворяется в хлористой сере, но и внедряется в молекулу последней. При этом образуются в основном соединения S3C12 и 84С12. Руффу (Buff, 1924) удалось выделить-эти вещества в формещродуктов-присоединения А1С1з-283С12 и A1C13-2S4C12 (винно-красные, разлагающиеся жидкости), Политиохлориды с особо высоким содержанием серы были приготовлены недавно Фегером (Feher) и Баудл ер ом (Baudler, 1952) нагреванием S2C12 в токе водорода в трубке для закаливания. При температуре нагревательного стержня 860—875° он получил ,
оранжево-желтые маслянистые продукты состава от S2OC13 до S24C12, которым он на-основании спектров комбинационного рассеяния приписал цепочечную структуру, аналогичную Структуре много сернистых водородов (ср. стр, 793 и сл.). При еще более высоких температурах он получил продукты, затвердевающие при комнатной температуре^ содёржапне серы в которых колебалось от S2SC12 до примерно Si(;oCl2.
Тетрахлорнд серы. Четьтреххлористая сора SC14 в чистом состоянии устойчива, только при низких температурах Руфф получил ее в виде бело-желтых, плавящихся примерно при —Зи° криста т (об действием жидкого хлора па хлористую серу. При нагревании до комнатной температуры четырехх.тористая сера распадается полностью-па SC12 и СБ. Водой это соединение разлагается на НС1 и 8Оа. Значительно устойчивее, чем свободная четыреххлиристои сера, ее двойные соединения с другими хлоридами, например SCl4-SnCLr; ЯС14 SbCl;; SC14 А1С1ч-> и др. Водой н эти соединения быстро-разлагаются.
Хлористый тионил (тионплхлорид) SOC13 образуется при действии пяти хлористого фосфора (пентахлорида фосфора) на двуокись серы
РС15 4- SO2=РОС134- SOC12.
Вместо двуокиси серы”~можно действовать пятихлористым фосфором на сульфиты-2РС15 4- CaSO3 == 2РОС13 + СаС12 + SOCI2, В технике хлористый тионил получают главным образом действием двух лор л стой серы на трехокись серы SC124-8O3=SOC124-4- SO2, От одновременно образующегося при этом пиросульфурилхлорида (хлористого пиросуаьфурила) S20sCl2 хлористый тионил можно отделить фракционной перегонкой, Вместо двух.пористой серы можно использовать хлористую серу, а выделяющуюся в этом случае в соответствии с уравнением S2Cls -|- 8О3 = SOC.l2 -4- SO2 & серу можно перевести действием хлора обратно в хлористую серу.
Хлористый тионил представляет собой бесцветную, сильно дымящую-жидкость. Она кипит при 78° и несколько выше этой температуры начинает распадаться на двуокись серы, хлор и хлористую серу. Водой хлористый тионил разлагается на двуокись серы и хлористый водород
SOC124- Н2О = SO2 4- 2НС1.
Шестая группа периодической системы
783
Основное применение хлористый тиопил находит в органической химии в качестве хлорирующего агента, для введения хлора в органические молекулы на место других атомов или радикалов.
Действием Л На на SOC1, можно получить соединение HN(SONH2)2 (имидоди-сульфинамид). Если это соединение обработать сжиженным НС1, то при этом образуется желтый сулъфипимид (тионилимид) OSNH. Это вещество может перегруппировываться в темпо-красное изомерное соединение, которое следует рассматривать как оксим моноокцси серы S=N—О—II (Goehring, 1951—1952), Изомерия между этими двумя соединениями аналогична изомерии между изоциановой н циановой кислотами.
Хлористый „ сульфурил (сульфурилхлорид) S02CI2 образуется при, непосредственном соединении двуокиси серы с хлором на солнечном свету или в присутствии подходящего катализатора (камфоры или активированного угля) SO2 + С1а = SO2C12, Он получается также при нагревании хлорсульфоновой кислоты
2SOa(OH)Cl = SO,(OH)34- SO2C12,
В чистом состоянии хлористый сульфурил представляет собой бесцветную, резко пахнущую жидкость. При стоянии он желтеет вследствие частичного распада па двуокись серы и хлор. Продукты распада можно удалить, пропуская через жидкость индифферентный газ. При действии большого количества воды хлористый сульфурил разлагается на серную кислоту и хлористый водород
S02C12+2HOH=S02(OH)2+2HC1.
Хлористый сульфурил является хорошим, мало ионизирующим растворителем для многих органических и неорганических веществ. Его применяют в органической химии как энергичный хлорирующий агент.
Фтористый хмрсулъфурил (суд ьфурилхлор фторид) SO2C1F получен Бутом (Booth, 1936) взаимодействием SO2C12 с SbF3 в присутствии SbCi5. Это бесцветный газ; т, пл. —124,7°; т. кип. 7,1°; теплота испарения при температуре кипения 6,34 ккял/лоль; в жидком состоянии (при 0°) имеет уд. вес 1,623 и поверхностное натяжение 17,2 дин!см.
Хлорсульфоновая кислота SO3(OH)Ci образуется прп непосредственном соединении хлористого водорода с трехокисью серы НС1 4- SO3 =• = SOa(OH)Cl или при действии концентрированной серпой кислоты на треххлористый фосфор, хлорокись фосфора или нятихлорнетый фосфор SO2(OH)2 -J- РС15 = SO2(OH)C1 + РОС13 + НС1. Она представляет собой бесцветную, сильно дымящую па влажном воздухе жидкость с резким запахом. С водой хлорсульфоновая кислота реагирует с шнпепием, распадаясь при этом на серпую кислоту и хлористый водород.
С органическими веществами хлорсульфонован кислота реагирует в зависимости от условий с образованием сульфокислот (сульфоновых кислот) или хлоридов сульфокислот:
O2N-C6H5 4- C1SO3H = O2N-C6H4.SO3H + НС1
Ни тро бензол Нитробен зол с улъ фо но ва я
сполота
0%' 4 /| S() II = OaN-.C(!H4-SO2Gi -ф- ИС1 ф 11,30,,
Иитробенаолсульфохлорид
II сотому в ортавичесъ ой химии есошогда используют для и о л у лева я сульфохл ори до в, реже для приготовления сульфокислот, так как последние обычно удобнее получать действием па сульфщу емоо вещество концентрированной иля дымящей серной кислоты.
Фторсульфо по иля кислота SO2(OH)F получается при соединении SO3 с HF, однако лучше ее получать по методу Руффа (Ruff, 1914) нагреванием CaF2 с дымящей серной кислотой (60% SOp или по методу Мейера (Меуег, 1932) действием последней на K.HF-2- Для многих препаративных целей она более пригодна, чем хлорсульфоновая
’784
Глава IS
кислота, так как значительно устойчивее последней; даже при 900° опа еще пе претерпевает никакого разложения. С Nad опи взаимодействует с образокаишчи фторосулъ-фона/па натрия (фторосульфата натрия) Na[SOoFJ. При действии воды фтор сульфоновая Кислота медленно разлагается на H3SO4 и НК.
3. Сероводороды (сульфаны)
С водородом сера образует ряд соединений общей формулы Нг8п. По предложению Фегера, эти соединения называют сероводородами, или сульфонами. Простейшим и наиболее важным представителем этого класса соединений является обычный сероводород (моносероиодо-род, мопосульфан) HaS. От него производятся остальные члены ряда замещением в соединении Н—S—И атома серы на более или менее длинную цепь атомов серы. Так образуются соединения II—S—S—Н (дисероводород, дисульфан), Н—S—S—S—И (трисероводород, трисуль-фан) и т. д.
Обычно эти соединения в отличие от^ простого сероводорода называют полисеуо-водородами пли пояисульфидами водорода' Этим, одпако, выражается только состав соединений, т. е. то, что в них па два атома водорода приходится более одного атома серы. Если же название должно указывать и па структуру соединения, то предпочтительнее номенклатура, предложении!! Фегером, Эта номенклатура основ а аа па том принципе, что лолисероводороды являются производными обычного сероводорода (сульфа на) в том смысле, в каггом пасьпцеяпые углеводороды с прямой цепью являются производными метана. Собирательное название еулъфаны позволяет образовывать названия таких соединений, которые получаются из соединений общей формулы H2Sre замещением водорода на другие атомы и радикалы. Эго название можно распространить, таким образом, на все соединения формулы B2Stl, если только но своей структуре они аналогичны сероводородам, причем внимание обращается па генетические отношения, существующие между соединениями II2Sn п замещенными сульфапамп, например галогепсупьфапамп (SnXJ, сульфанамн щелочных Металлов (M^S71), алкилсуль-фанами (R2S„) и т. д.
Из незамещенных с ульфа ни в II аЯ,, к настоящему времени получены в чистом виде соединения < одной .щчп.ю атомов серы, содержащей до восьми атомов серы включительно, а в форм? iowei'i шипены аналогичные соединения со значительно большей длиной цепь. Its XTopi.A дьф.гииг нзпеигньт сиедпиоипя с одной цепью, включающей до ста атомов серы (гр стр. 78тI. X,торсу льфаны с очень большой длиной цепи можно получить, ка с было у г, аза его нагреванием смеси ЭаС12 и Н2 в трубке для закаливания и, кроме того, валимо действием илп SCla с JIaS пли полисероводородами, например, по уравнению
. 2SaCla -|- TlaS = S5C12-|-2HC1.
При этом вначале образуются смеси различных гомологов, которые можно разделить фракционированной перетопкой при пониженном давлении. В качестве примеров бром-сульфапов можно назвать, кроме дибромдисульфана S2Br2, полученные Фегером (1954) соедянеяпя Й4Вга (дибромтотрасульфан), S5Br2 (дябромпентасульфап) и З^Вг2 (дибром-октасульфаи). Галогеносульфаны (полиеерогаиогениды) с содержанием серы вплоть до 24 атомов па молекулу представляют собой маслянистые жидкости. Тем, что смеси этих га доге его сульфапо в перегонкой при пониженном да в лепки удается разложить на отдельные гомологи, опи отличаются от растворов серы в SaCla или S2Br31 из которых отгоняется только применявшийся в качестве растворителя днеер о ди галогенид.
Сероводород {сулъфатр моносульфан} H2S получается при прямом соединении образующих его элементов (см. ниже). Однако обычно его получают действием кислот на сульфиды, как правило, действием соляной кислоты на сульфид железа FeS
FeS-, 2ITCJ =FeCl3-|-H3S.
От увлекаемых при этом паров соляной кислоты газ очищают, ЕЕроуп.шая его водой. Вместо соляной кислоты для разложения сульфида железа можно использовать разбавленную серную кислоту; однако в йтом случае получающийся сероводород легко
Шестая группа периодической системы
785
загрязняется двуокисью серы. Сульфид железа, получаемый технически сплавлением серы с железом, обычно содержит несвязанное железо. Поэтому приготовляемый нз такого сульфида сероводород содержит также свободный водород, в меньших ьюли-чествах кислород и азот, а также пары воды. Как правило, ати примеси не мешают. Удалить их можно, сжижая газ прп помощи твердой углекислоты ^фракционированной перегонкой. Обычные осушители в случае сероводорода не пригодны, так как он с шиш реагирует.
Для приготовления очень чистого (однако пе совершенно свободного от COj) сероводорода часто пользуются разложением Са8 или BaS разбавленной соляной кислотой. С пол [(ой гарантией совершенно чистый сероводород можно получить синтезом его нз элементов. Лучше всего в этом случае пропускать смесь паров серы п водорода через нагретую до 600° стеклянную трубку (Klemenc, 1932).
В природе сероводород часто присутствует в вулканических газах, а также в газах, выходящих из земли во многих вулканических местностях (сольфатары близ Неаполя). В растворенном состоянии он содержится в воде серных источников. Встречающиеся там «серные бактерии» не производят сероводород (например, восстанавливая сульфаты), а, напротив, он необходим для пх существования, поскольку они используют для своей жизнедеятельности энергию, выделяющуюся при окислении сероводорода до серы или сущ.фатов Но есть микроорганизмы, которые, наоборот, способны восстанавливать сульфаты до сульфидов, т. е. солей сероводородной кислоты.
Сероводород представляет'собой бесцветный газ, который при большом разбавлении неприятно пахнет «тухлыми яйцами». Он сильно ядовит. При вдыхании даже в небольших количествах он вызывает головные боли, головокружение и тошноту. В больших концентрациях он, подобно цианистому водороду, может привести к моментальной смерти. Сероводород тяжелее воздуха (уд. вес 1,1906;'нес 1 л газа 1,5392 а), он легко конденсируется в бесцветную жидкость (см. табл. 106).
Давление пара жидкого сероводорода при 0е' равно 10,3 атм. Крит, темп, 100,4°; крит. давление 89 атм\ крит. плотность 0,31. Жидкий сероводород прекрасно растворяет многие органические соединения, а неорганические, напротив, в нем мало растворимы. Растворенные в сероводороде соединении очень слабо ионизируются, что объясняется его довольно низкой диэлектрической проницаемостью, которая при —60е равна примерно 10, при 01? — около 6.
В твердом состоянии сероводород образует кубическую гранецептрированную молекулярную решетку. При —169,6 и —145,0° он претерпевает превращения, которые, впрочем, связаны не с изменением структуры решетки, а вызнаны свободным вращением молекул IbS вокруг двух осей. Теплоты превращения составляют 0.365 и 0,109 ккал/моль соответственно (Giauque, Clusius, 1936).
При нагревании, сероводород распадается на составные элементы. Реакция S Н2 Н,8 + 4.8 ккал, следовательно, легко обратима. Зажженный на воздухе сероводород сгорает голубым пламенем, и при этом образуются вода и двуокись серы (13), а прн ограниченном доступе воздуха — вода и сера (14):
HsS-l-^/aOg^ НуО-|- (13)
H2S +> /2О3=ТТгО S - (14)
Сероводород легко реагирует с большинством металлов, особенно в присутствии влаги или ири нагревании, образуя соответствующие сульфиды. При нагревании оп взаимодействует также со многими окисламп металлов с образованием сульфидов. Сероводород энергично реагирует с фтором, хлором и бромом, мепее энергично — с иодом. Концентрированная серная кпелота окисляет его с выделением серы и в свою очередь восстанавливается до двуокиси серы: ЩйСЦ 4- H2S S u SO2 : 2НаО. Вообще сероводород является сильным восстановителем,
В воде сероводород очень хорошо растворяется — примерно вдвое лучше, чем двуокись углерода. Одни объем воды может поглотить при 0° 5U Г. Гемп
786
.Глава 15
4,65, при 10° 3,44, при 18° 2,75 и при 20° 2,61 объема сероводорода. Теплота растворения при 20° составляет 4,52 ккал/моль (Roth, 1934). При низких температурах образуется кристаллический гидрат H2S'5-/4HaO (о структуре его см. стр." 248 и сл.) Сероводород образует продукты присоединения и с другими веществами, например с многочисленными галогенидами. Еще лучше, чем в воде, сероводород растворяется в спирте (11,8 об. в 1 об. спирта при 10°). Насыщенный водный раствор сероводорода в лабораториях применяют в качество реактива под названием сероводородной воды. ЕгО нельзя долго хранить, так как в водном растворе H2S медленно окисляется кислородом воздуха с выделением серы.
Сероводород является очень слабой двухосновной кислотой. Как кислота он электролитически диссоциирует в две стадии, Константа диссоциации для первой стадии равна, по данным Тредвелла и Кубли (Treadwell, Kubli, 1946), 0,87310'7 при 20°, а константа диссоциации
для второй стадии составляет, согласно Конопику (Konopik) и Лебёрлю (Leberl, 1949), А'., = 0,79‘10~1:! (при 20°, экстраполирована к нулевой ионной силе). Диссоциация во второй стадии настолько мала, что она не отражается на электропроводности растворов даже при наибольших разбавлениях. Степень диссоциации первой стадии при 18° в 0,1 н. растворе составляет 0,13%.
Сульфиды. Соединения серы с более электроположительными эле-, ментами называются сульфидами. Большинство сульфидов, а именно сульфиды металлов, по способу образования и химическому поведению следует рассматривать как соли, сероводородной кислоты.
Поскольку сероводород является двухосновной кислотой, от него производятся два ряда сульфидов: кислые сульфиды или гидросульфиды М^НЗ и нормальные сульфиды М|3. Все кислые сульфиды очень легко растворимы в воде. Из нормальных сульфидов также легко растворимы сульфиды ще~ лонных металлов. В водном растворе опи очень сильно гидролизуются (в 1 и. растворе примерно на 90%) по уравнению
Na2S4-IIOH ХаОН -:-XaHS или S'+HOH 5* OH'+IIS'.
Поэтому их растворы имеют сильно щелочную реакцию. Нейтральные сульфиды щелочноземельных металлов как таковые в воде не растворяются. Одиако при действии воды они претерпевают гидролитическое расщепление 2CaS + 2НОН = Ca(HS)2 + Са(ОН)2, а образующийся при этом кислый сульфид переходит в раствор. При кипячении раствора он также разлагается Ca(HS)2 -|- 2НОН = Са(ОН)2 -J- 2Нг8. Еще легче гидролизуются сульфиды некоторых многовалентных металлов, например сульфид алюминия А1283, сульфид хрома Cr2S3, сульфид кремния SiS2. Кислоты разлагают все эти сульфиды с выделением сероводорода.
Большинство сульфидов тяжелых металлов настолько мало растворимы в воде, что гидролитического расщепления их не происходит. Некоторые сульфиды разбавленными сильными кислотами не разлагаются. Произведение растворимости этих сульфидов настолько мало, что даже при понижении концентрации ионов S" в растворе за счет прибавления ионов Н' концентрация ионов металла в растворе, находящемся в равновесии с сульфидом (донной фазой), очень незначительна. Поэтому при пропускании сероводорода такие сульфиды будут выпадать в осадок даже из очень кислых растворов.
На том, что одна часть тяжелых металлов осаждается сероводородом из кислого раствора, а другая выпадает в осадок только из аммиачных
Шестая группа периодической системы 187
растворов при действии на них раствора сульфида аммония (ср. стр. 789), основано применение этих реактивов для разделения катионов при еноте-магическом анализе.
Из кислого раствора сероводород осаждает следующие элементы в виде их сульфидов:
а) .мьийьяк, сурьму и олово;
б) серебро, ртуть, свинец, висмут, медь и кадмий.
При действии сульфида аммония осаждаются следующие элементы! цинк, марганец, кобальт, никель, железо, хром и алюминий. Два последних элемента выпадают в виде гидроокисей, так как их сульфиды гидролизуются водой.
Сульфиды элементов, приведенных под рубрикой (а), отличаются тем, что они способны растворяться в (желтом) полисульфиде аммония, образуя при этом тиосоли (см. ниже), тогда как сульфиды элементов группы (б) в этом реактиве ие растворяются.
Приведенный выше перечень элементов ограничен наиболее часто встречающимися-Надо отметить, что при проведении систематического анализа серебро, одновалентную ртуть и некоторую часть свинца до пропускания сероводорода обычно осаждают добавлением соляной кислоты, так что получающийся в ходе анализов сероводородный осадок не содержит сульфида серебра.
IJроигеедение растворимости ряда сульфидов приведено в табл. НО. Эти величины вычислены на основании соотношения
—RT -2,3026- log L,
где i произведение растворимости, а Арп — нормальное сродство реакции 2М’-|-~p_S"=M2S [или соответствующей реакции для многовалентных ионов металлов (ср. Таблица 110
Произведение растворимости кристаллических сульфидов металлов при 25°
[но Goates J. R-, J. Am. chem- Soc., 74, 835, 15)52]
Сеединение П роизведенце растворимости Свободная оиертвя обратования,
сульфида, иона металла, ПК£1Д/г-1Д<1Н
MnS 1 -10-11 —47,6 — 53;4
FoS 5-10-w —23,32 —20,30
NiS 2-10-31 —18,8 -11,1
ZnS 8-i0“25 -47,4 —35,184
CoS 8.10-sa —21,8 -12,3
Co^Sg ~ 10'124 —47,6 29,6
CdS 7.10-27 —33,6 —18,58
PbS 8-10-28 —22,15 -5,81
He;S 3-10-52 —10,22 39,38
CoS 8.10-3S —11,7 15,53
ClhS 1-10'48 —20,6 12,0
Ад-.Й 7-10'50 —9,56 18.430
11 - 7 -10-20 —21,0 — 7,755
Bi-jSg — 10-oe -39,4 ~15
La,S3 2-10'13 —301,2 —172,9
Ce2S3 6-10-11 ' —293,0 -170,5
50*
788
Глава 15
стр. 168)]. Лежащие в ослопе такого расчета величины свободной энергии образования (гидратированных) попов металлов и сульфидов также приведены в этой таблице (свободная энергии образования иона S"' равна 20,64 ккал/г-ион при 25‘?). Приведенные значения справедливы для сульфидов в кристаллическом состоянии. Однако в ходе анализа прн осаждении сульфиды всегда получаются не в кристаллическом виде, а в форме гелеобразных, аморфных, как показывает рентгеновский апалнз, осадков с сильной поверхностной активностью. Для таких осадков произведение растворимости должно иметь значительно большее значение, тал: как для веществ в топкодисперсном состоянии на величину внутренней энергии, а тем самым и на растворимость очень сильно влияют упорядоченность, величина час тип п развитость поверхности (ср. т. 1[, гл. 16).
Для получения сульфидов используют следующие методы:
1. Взаимодействие гидроокисей с сероводородом.
дтпм методом получают в первую очередь растворимые в воде сульфиды, т. е. сульфиды щелочных металлов. Поступают следующим образом: сначала насыщают раствор гидроокиси щелочного металла сероводородом. При этом получается кислый сульфид (гидро сульфид) (15). Последний прибавлением равного количества щелочи переводят в нормальный сульфид (1G).
iXFaOH-j-H2S NaHS-4-H,O. (15)
NaHS !-NaOH = Na3S+H2O. ' (16)
2. Взаимодействие солей в нодполг растворе с сероводородом или сульфидом аммония.
Этим методом получают в первую очередь нерастворимые в воде сульфиды. Об их значении для аналитической химии уже говорилось.
3. Восстановление сульфатов прокаливанием с углем.
В технике сульфид натрия получают главным образом этим методом Na-jSO,, v 4С _= Ха;В 4С0. (17)
Этот же метод испилi.iynrc тля иолу’пчшя. сульфидов щелочноземельных металлов.
4. Непосредствен hi и- соединение элементов.
Гак подучают, как ужо упомийалось. сульфид железа, нагревая железо с серой:
Fi- |-S FeS. (18)
Соединение металлов с серой протекает в большинстве случаев очень легко, часто с большим выделением тепла. Однаь-о оно редко приводит к образованию совершенно чистого продукта.
Сульфиды щелочных и щелочноземельных металлов бесцветны. Сульфиды тяжелых металлов, напротив, в большинстве случаев интенсивно окрашены, часто черные-
Мпоггге сульфиды ири нагревании без доступа воздуха не претерпевают разложения. Но некоторые теряют серу. Таи. например, пирит FeS3 уже при сильном нагревании распадается па сульфид железа(П) и серу; сульфид onoua(IV) распадается прп нагревании па сульфид оло-ва(П) и серу. Устойчивые к нагреванию сульфиды в большинство случаев можно нагревать в токе водорода; при этом они не изменяются. Напротив, при нагревании в токе кислорода или воздуха («обжиге») большинство сульфидов переходит в окисли, и иногда частично и в сульфаты. Сульфиды, выпавшие из водного раствора, уже при обычных температурах в значительной степени подвергаются окислению, если они во влажном состоянии долгое время находятся в контакте с током воздуха. При этом
Шестая группа периодической системы 780
происходит или выделение серы (19) или образование сульфата (20):
Fe2S3+aq+a/aO2=Fe2O3aq-|-3S; (19)
CtiS-|-2O2=CnSO4- (20)
Легко окисляются и растворенные сульфиды; при атом они действуют как сильные восстановители.
Сильное восстановительное действие сероводорода и сульфидов в растворе обусловлено незначительным сродством образования иопов S", В гальваническом зле-’ менте, составленном из нормального водородного электрода и платиновой фольги, погруженной в раствор сульфида, «серный электрод» вследствие тенденции ионов 8" разряжаться (как уже было указано па стр - 736 п с л.) становится отрицательным, а водородный электрод — положительным полюсом.
Сульфид натрия Na2S в технике получают главным -образом восстановлением сульфата натрия углем [ср, уравнение (17)]. При температуре ниже 48° он кристаллизуется из воды с девятью молекулами НаО в виде гигроскопичных, квадратных призм, В чистом состоянии он бесцветен, по часто вследствие присутствия полисульфидов (см. ниже) имеет слегка желтоватую окраску.
Растноримость сульфида натрия прп 18° составляет 18,06 г в 100 г воды. Вследствие гидролиза водный раствор сульфида натрия имеет щелочную реакцию. Кислородом воздуха он легко окисляется до тиосульфата
2Na2S-i-2O24-HaO=Na2S2O3-L2NaOH,
В технике сульфид натрия применяют главным образом как восстановитель для органических нптросоединений, например при производстве серных красителей. При этом сульфид натрия также переходит в тиосульфат. При дублении кож сульфид натрия используют для удаления волос со шкур.
Сульфид калия K2S получают, как правило, описанным на стр, 788 методом, смешивая эквивалентные количества растворов едкого кали и гидросульфида калия (кислого сульфида калия). В чистом состоянии K2S кристаллизуется бесцветным, обычно в виде пента гидрата, K,S-5HgO.
Окрашенный в цвета от желто-зеленого до коричневого торговый сульфид калия, известный также под названием «серкой печени», получаемый сплавлением карбоната калия с серой при доступе воздуха, представляет собой смесь полисульфидов калия, (см. ниже) и тиосульфата калия. Его часто применяют в медицинских целях, например для протираний и ванн при кожных заболеваниях.
Сульфид аммония. Если разбавленный раствор аммиака насыщать сероводородом, то ио уравнению
NH3-|-H2S = NH4HS
образуется гидросульф ад аммония (кислый сульфид аммония). Если к такому раствору прибавить ранное количество аммиака, то оп не будет связываться- Такой раствор, называемый серноаммиачным раствором или, короче,сернистыломмонием и часто применяемый в лабораториях, прежде всего для аналитических целей, не является раствором сульфида аммония [NH4]2S, а представляет собой раствор эквимолярных количеств гидросульфпда аммония [NH4 JHS и аммиаки.
790
Глава 15
Истинный нормальный сульфид аммония [NH.j]sS существует только при низких температурах и в отсутствие воды. Кислая (первичная) соль, гидросулъфид аммония, NII4HS, получаемый в кристаллическом виде охлаждением до O'J смеси газообразного аммиака и сероводорода, очень легко улетучивается, распадаясь При этом на составные части NH3 .ц H2S. Уже при 20° давление диссоциации составляет 355 ль» рт ст. Гидросульфид аммония в значительной степени распадается и в водном растворе, В 1 М растворе гидросульфида аммония около 4% соли находится в форме аммиака и недис-социировапного сероводорода
NHiHS5>NH3-^ H?S.
Степень гидролиза сульфидов калия или натрия при той же концентрации более чем в 100 раз меньше,
В качестве аналитического реактива значительно чаще, чём бесцветный сернистый аммоний, используют так называемый желтый сернистый аммоний, который получают растворением серы в бесцветном сернистом аммонии и который содержит растворенные полисульфиды аммония (ср. ниже).
Сульфиды щелочноземельных металлов. Сульфид кальция CaS (т. пл. —2400°) раньше получали как побочный продукт при производстве соды по методу Леблапа. Теперь его получают главным образом прокаливанием сульфата кальция с углем и применяют при дублении кож я в косметике кагг средство для удаления волос. Для тех же целей используют получаемый прокаливанием окиси кальция с серой полисульфид CaS.. («серн о кальциевая печень»), который находит также разнообразное применение в медицине. «Сернокальциевый отвар», получаемый кипячением известкового молока с серой, также содержит полисульфид кальция, и его применяют как средство борьбы с вредителями.
Сульфид бария В aS получают прокаливанием при температуре 600—800° измельченного в тонкий порошок и тщательно гмешинного с углем тяжелого шпата
BaSO4 л- 2(\ = Ва8 -f- 2СОг*.
Торговый сульфид бария представляет собой серый рыхлый порошок. Он служит для получения литопона, а также, как и сульфид кальция, для удаления волос со ? шкур. Полисульфид бария В aSx представляет собой светло-коричневый порошок (Barium su-lfuralum jl.avum) и служат как средство защиты растений, для уничтожения насекомых и как эффективное средство для удаления волос.
Структура решетки сульфидов Щелочноземельных металлов подобна решетке их окйслов (типа каменной соли):
MgS CaS SrS BaS
а=5,19 5,(58 6,01 6,37
Светящиеся краски. Прп добавлении к сульфидам щелочноземельных металлов следов солей тяжелых металлов я сильном прокаливании в присутствии флюсов они приобретают свойство продолжительно светиться после предварительного освещения. Препараты, Отличающиеся таким свойством, называют фо сферами или светящимися красками. В настоящее время их обычно получают прокаливанием о Kite лов щелочноземельных металлов или их карбонатов со смесью серы и солей щелочных металлов и прп добавлении очень незначительных количеств солей тяжелых металлов. Например, смешивают 40,0 а карбоната стронция, 9,0 г серы, 1 е карбоната лития, 1,0 г трис у л ь-фида мышьяка и 2 .ил раствора нитрата таллия (1 : 200) и прокаливают 3/4 часа при температуре около 1200°, например, в Печи Рослора. При этом получают светло-зеленую массу, способную к послесвечению. Аналогично можно получить голубые, Голубов атозеленые, желтые, оранжево-красные и другие препараты. Открытие светящихся красок связано со случайным наблюдением жившего в середине XVI в, болонского
* Если нагревать сильнее, то наряду в СО2 будет образовываться СО, что приводит к увеличению расхода угля.
Шестая группа периодической системы
791
сапожника Винцениуса Касциаролуса (V in centius Casciarolus), занимавшегося алхи-иней. Оп нашел не известный ему ранее минерал (тяжелый шпат) и прокалил его с углем, после чего обнаружил, что получившийся препарат светится в темноте. Препараты, пригодные для использования в качестве светящихся красок, технически впервые получил в 70-х гг. прошлого столетия англичанин Балмейн (Balmain), после того как- было установлено, что свойство прокаленных сульфидов щелочноземельных металлов светиться зависит От присутствия в них следов солей тяжелых металлов. По мнению Ленарда (Lenard), получение «фосфора» определяется тремя компонентами: 1) сульфидом, 2) флюсом, 3) тяжелым металлом. Флюс следует добавлять в относительно большом количестве, а тяжелый металл — в виде следов (~10-4). В качестве флюса могут служить такие вещества, как хлорид патрия, фторид кальция, фосфат натрия, бура и т. п. Как показали Ти де (Tiede) и III ле еде (Schleede), присутствие флюса излишне, если плавление сульфида вести при более сильном нагревании под давлением.
Тиосоли, Многие сульфиды растворяются в растворах сульфидов щелочных металлов, образуя тиосоли*, например
AszS3+3K£S=2K3 [AsS3],
Тиоарсенит валяя
Этот процесс вполне аналогичен процессу образования солей кислородных кислот при соединении кислого и основного окислов
As2O3+2K2O=2KsIAsO3],
Арсенит кадия
Раньше сульфиды, растворимые в растворах сульфидов щелочных металлов н способные к образованию тиосолей, Часто называли «кислыми сульфидами», а остальные сульфиды — «основными сульфидами». Одпако такая номенклатура в настоящее время пе используется, так как в соответствии с. общими правилами наименования солей «кислыми сульфидами» следует называть кислые соли сероводородной кислоты т. е. гидросульфиды M^IS, а употребление этого названия в другом смысле только приводит к путанице.
Тиосоли можно также рассматривать как соли, аналогичные солям кислородных кислот, но только содержащие вместо кислорода серу.
Образование анионов тиосолей по аналогии с образованием анионов солей кислородных кислот можно представить следующими уравнениями:
As2O3+3O'5t2[AsO3r, или As2O34-6OH' 2 (АаО3Г+ЗН2О; (21
As2S3-f- 35# 2 [AsSj]"7, или At^Sg-f- 3SH' 4-3OIT 2 [AsSgl^'+^ITO; (22)
As2S3+3O’ [AsS3]"-|- [AsO3]”', или Аз^Н-бОН' [AsS3]w-|-[AaO3]"'4-3H2O. (23)
Уравнение (23) показывает, что могут образовываться одновременно апионыкак тио-, так и кислородных кислот, а именно в том случае, когда сульфиды, растворимые в растворах щелочных сульфидов, обрабатывают щелочами (см. также стр. 580 н 710).
При подкислении раствора большинство тиосолей распадается с выделением сероводорода и освобождением исходного сульфида, так как свободные тиокислоты, как правило, неустойчивы.
II в этом отношении не существует принципиального различия между солями тио- п кислородных кислот. Средн последних имеется много таких, соответствующие кислоты которых ие способны к существованию, так что вместо кислот прп подкпе-
* В соответствии с международным правилом номенклатуры те кислоты, которые получаются замещением атомов О на атомы S в кислородных кислотах, должны яаэы-ватьсяпшоккслота.ни, а их соли — тиосолями. Раньше для тиокислот часто употребляли название сульфокислоты, которое, однако, применяли одновременно и для совершенно иного класса соединений, а именно для производных серной кислоты типа В .SO3H (наряду с более старым названием для этого класса соединений «сульфоновые кислоты»).
792
Глава 16
ле ни и растворов выделяются (после отщепления воды) окислы. Разложение тио солен следует рассматривать просто как следствие сдвига равновесий, представленных уравнениями (22) п (23), справа налево за счет увеличения в растворе концентрации иопов водорода.
Способность к образованию тиосолей представляет, как уже указывалось, аналитический интерес, особенно в случае сульфидов мышьяка, сурьмы и олова.
Тиосоди образуют также платина, золото, германий, теллур, молибден, вольфрам, ванадии и углерод. Тппеоли всех этих элементов можно получить обрабеткоГт соответствующих сульфидов раствором сульфида щелочного металла. Еще ряд тиосолей можно приготовить сплавлением, однако относительно полученных таким способом соединений часто остается сомнение, действительно ли имеют дело с настоящими тиосолями, а не с двойными сульфидами (в узком смысле этого слова, т. е. с двойными соединениями сульфидов, пе имеющих характера солей).
Полпсульфиды и полисероводброды. Растворы щелочных сульфидов способны растворять значительные количества серы, и при этом образуются окрашенные в цвета от желтого до коричнево-красного полпсульфиды, т, е. соединения общей формулы MjSn, где п обычно имеет значения от 2. до 5, но в некоторых случаях может принимать и еще большие значения.
Полисулъфиды щелочных металлов образуются также при стоянии растворов, щелочных сульфидов на воздухе вследствие медленного окисления гпдросулъфил-но-пов кислородом воздуха
2HS' Ч- JsO2 =- Н2О 4- So.
Полисульфиды щелочных металлов получают также сплавлением сульфидов щелочных металлов с серой. Кроме того, их можно получить, сплавляя гидроокиси или карбонаты щелочных металлов с серой. Однако в последнем случае получающиеся полисульфиды бывают загрязнены одновременно образующимся тиосульфатом, а при доступе воздуха и сульфатом. Уже давно известная верная печень (hepar sulftiris от длар — печень) и является такой .larpauneitnoir смесью полисульфидов щелочных металлов.
Кроме полщ’.ульфпдов щеючных металлов, известны также полис у.чл,-фиды щелочноземельных металлов. Самыми устойчивыми являются, по-вйднмому, полпсульфиды с четырьмя атомами серы.
Обзор по лученных к настоящему времени по дисульфидов щелочных металлов дая в табл. 111. В то время как с увеличением содержания серы окраска полисульфидов углубляется, полиселениды и яолителлуриды, которые также приведены в этой таблице, всегда серо-черного цвета. О магнитных свойствах и пространственном строении зтцх соединении см.: Klein m, Z. anorg. Client., 2A1, 281, 1939..
Таблица 11/
Полпсульфиды, полнееленпды и пояителлуриды щелочных металлов
Полпсульфиды Полисе лени дм По лит ел лу ри ды
Na2S2 K2s2 Cs2S2 Na2Se2 K2Saj Na2Te2 К2Тс2 НЬ,Те2
— КчЗ.з Rb3S3 Cs2Sfl Ma2Se3 K2Se2 — К2Тсз ВЬ2Твя.
NaaS4 K2S4 BbsS4 Cs2S4 Na2Sc4 K3So4 —
Na2Sr, k2se HbaSb Cs2Hb -• КгЗс5 —
— K3S8 RbflSg Cs2S(; _Na2Sc8 ? Na2Te(j
Шестая группа периодической системы,
793
Гидролитическое расщепление полисульфидов происходит в значительно мепыпей степени, чем обычных сульфидов. Поэтому в отличие от нормального сульфида аммония (NH.s)2S полисульфиды аммония [например, (NH4)2S4, (NH4)2S5-Н20, (NH4)2Sb- 1/2Н2О] при обычных температурах устойчивы. Кислоты разлагают полисульфиды с отщеплением серы
Na2S2+2НС 1 = 2NaCl-фH2S -J- S.
Однако при соответствующем подборе условий можно получить в свободном состоянии отвечающие лолисульфидам кислоты HaSx, называемые полисероводородами или полисулъфидами водорода.
Пол вдул ьфиды водорода образуются также при разложении водных растворов тиосульфата соляной кислотой (v. Deines, 1929). При соблюдении определенных условий эксперимента таким путем получают прозрачное, желтое, вязкое масло, представляющее собой, по всей вероятности, смесь по ли сульфидов водорода с чрезвычайно бол ьшим молекулярным весом (Feher, 1952). Об аналитическом определении серы в полисульфпдах см.: W i n torsberger, Z. anorg, Chem., 236, 369,1938; а также F е 11 ё г, Z. anorg. Chem., 255, 316, 1948 и 260, 273, 1949.
О получении в чистом виде H3Sa, II2S3 и H2S4, об образовании H2S5 и H2Stl при фотохимическом разложении II2S3, а также о получении в чистом виде II2S6 й H2S6 см.: F е 11 ё г F., Z. anorg. Chem., 1947 и сл.
Молярные теплоты образования жидких полпсульфидов водорода (ив Н2 и 8роМб> составляют для H2Sjj 4,2, для H2S3 3,5 и для HaS4 3,4 ккал. Образование газообразного HaS2 на 112? п паров серы (S2) также является экзотермическим процессом (6,4 вкя.тДноль), тогда как аналогичная реакция образования перекиси водорода сильно ондотермичпа (Н3ОГ[Г1 | :JiO2 — НаО2газ — 24,2 ккал).
Структура по-’шеу.тьфпдоп водорода. Дисульфид водорода (дисульфан) H3S2 имеет структуру, в основных чертах аналогичную структуре перекиси водорода, что следует из его спектра комбинационного рассеяния (Feher F., 1941). В нем второй атом, серы не присоединен к молекуле H2S как таковой, а связан тем же способом, что п первый: II—S—S—Н.
Как следует из опытов Фегера, в более богатых серой полисульфпдах водорода, атомы серы связаны в цепи'.
И—S—S—S—Н Н — .8 — S — 8 — Я — Н
Трисульфнд водорода (три су ль фан)
Тетрасульфид водорода (тетрад ульфа и)
Н —Я —S —S-S —S —н
Пен та сульфид водорода (пшггаеульфап)
Кроме спектров комбинационного рассеяния, это подтверждается молярными! объемами и показателями преломления этих соединений. Теплоты образования отдельных нолисульфидов водорода также лучше всего согласуются с цепеобразной структурой. Раньше полагали, что полисероводороды построены «цептрнчно», т. е. в нх молекулах центральный ион окружен координационно связанными атомами серы. Если бы это было так, то следовало ожидать, что соединение H2S5, являющееся в таком случае структурным аналогом кислоты H3SO4, будет отличаться от остальных полисульфидов водорода чрезвычайной устойчивостью, так же как серная кислота является самой устойчивой из кислородных кислот серы. Однако на самом деле этапе так. Кроме того, предполагая, что формулы «центричные», следовало бы также объяснить тит факт, что существуют нолисульфиды водорода с еще большим содержанием серы, чем H.,S- (ср. стр. 784). Го же относится и к ео.гл.и полисульфидов водорода,, среди которых также [[метется соединения, содержание серы в которых значительно-превышает число атомов серы в формуле M[Ss (например, [-VH4]2S<)).
Богатые серой спел пне пня галогенов типа SnX2 следует рассматривать, как уже упоминалось па стр. 784. как гало г ено производные по л нсср о водородов. То, что в этом случае речь идет ш? о растворах серы в галогенидах серы, таких, как S2C12 или S2BrE, удалось показать Фегеру измерением молекулярных весов и частично на оснований спектров комбинационного рассеяния. Эти соединения, подобно сероводородам, образуют гомологические ряды, начальными членами которых следует считать дита-логениды серы и дитиогалогепиды. Структурное родство этих соединений с сероводородами (сульфанани) можно выразить тем, что называть эта соединения галогенсуль-
794
Глава 15
В качестве примера можно привести первые члены гомологического ряда;
-X лорсу ль фанов;
Cl—S—Cl Cl—S—S—Cl Cl—S—S—S—Cl Cl—S—S—S—S—G1
Ди хлор ид серы Дитиах лории Тритиохлорид Тетратиохлорид
(дцхлорсульфан) (дихлор дисульфан) (дихлортрисульфа я) (дихлортетра сульфа и)
Алкилеульфиды и соединения сульфония. При замене одного или Двух атомов водорода в молекуле сероводорода па органические радикалы образуются меркаптаны =
R, fk
или тио спирты и алкилеульфиды. или тиоэфиры ^S. Последние способны нри-
Их Rz
соединять алкилгалогепиды, прежде всего алкилиодиды, с образованием соединений сульфония
R.S + RI = R3SL
При обработке иодидов алкилсульфония влажной окисью серебра получают гидроокиси алкилсульфония— соединения, которые по их химическому поведению следует рассматривать как истинные основания, из которых при подкислении кислотами можно получить соли (соли сульфонил). Содержащийся в соединениях сульфония радикал R3S переходит в раствор как положительно заряженный ион [RjSl". Строение соединений сульфония соответствует, по всей вероятности, строению оксрниевых соединений. Поэтому их следует рассматривать как координационные соединения -с координационно трехвалентной серой, т. о. как соединения, в которых сера, кроме двух нормальных связей, образует одну донорную связь:
[rsr]x пли
R—S —> R
Аналитические сведения. Для самого общего доказательства присутствия серы в каком-либо соединении служит проба Гепара. Она основана на восстановлении соединений серы до сульфида натрия при нагреваний их с содой в восстановительном пламени бунзеновской горелки или трубки Лота и образовании черного сульфида серебра, при нанесении охлажденной и увлажненной смеси на кусочек серебра. Сульфидине руды распознают по выделению дну.)писи серы прп нагревании пробы в открытой с дЪух сторон трубке, наклонно расположенной в пламени, т. е. при нагревании в токе воздуха (запах горящей серы).
Сероводород легко узнают по его неприятному запаху и по реакциям с растворенными в воде солями тяжелых металлов. Для доказательства присутствия следов сероводорода, например в воздухе, часто применяют пропитанную раствором ацетата свинца бумагу (свинцовую бумагу), которая вследствие образования сульфида свинца чернеет. Очень чувствительным реактивом на сульфиды в водном растворе является нитропрус-’Сид натрия, который в присутствии сульфидов дает фиолетовое окрашивание. Значительно менее чувствительной является реакция нитропруссида натрия на сульфаты (красное окрашивание, особенно в присутствии •сульфата или нитрата цинка). При добавлении сильных кислот сульфиты выделяют двуокись серы (распознаваемую по запаху); так же ведут себя тиосульфаты, причем последние одновременно выделяют серу1 (ср. стр. 771). Нейтральный раствор сульфита реагирует с иодом по уравнению
805-Нг-|-Иг0=8О:-|-2Г-|-2Н-.
При этом раствор иода обесцвечивается и реакция его становится кислой. Тиосульфаты также обесцвечивают раствор иода, но без образования кислоты (ср. стр. 772).
Сулъфат~ионы образуют с ионами бария белый, не растворимый -в разбавленных сильных кислотах осадок сульфата бария ВайСЦ. От фто
Шестая еруя па периодической, системы
795
рида бария BaFa и от фторосиликата бария Ba(SiFE] (также мало растворимых в кислотах) его можно отличить при помощи пробы Гепара.
Количественное определение серы обычно производят осаждением и взвешиванием в виде сульфата бария. В органических соединениях серу чаще всего определяют этим методом; для этого ее переводят или в серную кислоту нагреванием с концентрированной азотной кислотой в запаянной ампуле (метод Кариуса), или в сульфат натрия нагреванием с перекисью натрия (метод Вюрцшмитта).
О применении тиосульфата натрия в ио до метрик и бромометрин уже было упомянуто.
СЕЛЕН (SELENIUM) Se И ТЕЛЛУР (TELLURIUM) Те
Распространение в природе. Следы селена присутствуют во многих природных сульфидах, например в железном колчедане, медном колчедане^ Цинковой обманке. В увлекаемой с отходящими газами пыли, образующейся при обжиге таких селеясодержащмх сульфидов, его относительное содержание значительно увеличивается. Шламы, образующиеся в свинцовых камерах при производстве серной: кислоты, часто содержат значительные количества селена.
Поскольку селениды почти всегда изоморфны соответствующим сульфидам, они обычно находятся в смеси с последними. Чистые соленовые минералы очень редки. Можпо упомянуть берцелианит fai^Se, тиеманит HgSe, науманит Ag3Se. Несколько чаще встречаются чистые теллуровые минералы, например изоморфный иауманиту гессит Ag2Te, далее, алтаит РЬ'Ге, колорадоит HgTe, сильванит AgAuTe4 и другие теллуриды серебра и золота (петцит, мутманит, креннерит). Часто встречаются также кислородные соединения теллура, например ТеО2 — теллуровая охра. Иногда теллур встречается в чистом самородном состоянии и вместо с селеном и серой. Красноватая японская теллуристая сера содержит 0,17% То и 0,06% Se.
Теллуриды вместе с сульфидами встречаются реже, чем селениды. Поэтому теллур в общем менео распространен, чем селен; но в отдельных рудах содержание его выше, чем содержание селена.
Одним из наиболее важных минералов теллура является листоватый теллур (нагиагит), изоморфная смесь сульфидов и теллуридов, главным образом свинца, золота, меди, серебра и сурьмы.
Исторические сведения. Теллур был найден в 1782 г. в золотоносных рудах Трансильвании и назван Клапротом, который определил его основные свойства, в честь Земли теллуром (от tellus — Земля). В 1817 г. Берцелиус в шламах свинцовых камер одного из сернокислотных заводов нашел элемент, очень похожий по своим свойствам на теллур, который он назвал в честь Луны соленом (от оеАцут] — Луна).
Получение. Селен получают из пыли, образующейся при обжиге селенсодержащих сульфидов, или из шламов свинцовых камер. Содержащие селен шламы свинцовых камер имеют пе чисто серый, а красновато-серый цвет. После обработки концентрированной серной кислотой с добавлением нитрата патрия селен переходит в раствор. При этом он в основном переходит в селенистую кислоту HaSeO3, а частично в селеновую кислоту H2SeO4. Последняя при нагревании с соляной кислотой
796
Глава 15
также переходит в селенистую кислоту. Если затем в раствор пропустить сернистый газ, то выпадает элементарный селен в виде красного аморфного осадка.
Для очистки селена его сжигают в кислороде, насыщенном парами дымящен азотной кислоты; прн атом сублимируется чистая двуокись селена. Из раствора SeO2 в воде после добавления соляной кислоты можпо вновь осадить Селен, пропуская сернистый газ или добавляя гидра зингндрат.
Из шламов свинцовых камер селен можно выщелочить, обрабатывая их теплым раствором цианистого калия. В этом случае образуется селено циан ат калия К [SeCN ], который разлагается соляной кислотой с выделением селена.
Поскольку в шламах свинцовых камер теллур в больших количествах присутствует лишь в редких случаях, для получения этого элемента используют главным образом листоватый теллур (нагиагит).
Этот минерал можно вскрыть несколькими способами, папример, сначала удалить кипячением с копцептрироваппой соляной кислотой содержащиеся в нем сульфиды, а остающийся теллурид золота разложить азотной кислотой, выпарить раствор досуха, остаток растворить в соляной кислоте и осадить теллур сернистым газом.
Если селен н теллур присутствуют одновременно, то для их разделения можно воспользоваться различным поведением солей селенистой и теллуристой кислот по отношению к серной кислоте. Прн добавлении серпой кислоты выпадает двуокись теллура ТеСЦ, тогда как селенистая кислота остается в растворе.
Свойства. Селен, как и сера, существует во многих модификациях. При восстановлении селенистой кислоты сернистой кислотой или другими восстановителями, а также при быстром охлаждении паров селена получают рыхлый, красивый красный порошок. Он отличается только размерами частиц от стекловидного селена, получающегося при быстром охлаждении расплавленного селена, папример ири выливании его в холодную воду. Стекловидный селен представляет собой очень хрупкую блестящую массу, цвета от красно-коричневого до свшщово-серого, с удельным весом 4,28— 4,30, которая легко рас трасте я в красный порошок. Стекловидный селен неэлектропройоден. В воде он совершенно не растворяется. Однако он несколько растворим в серной кислоте и сероуглероде, давая в первом случае зеленый, во втором — рубипово-красный раствор. При температуре около 50° стекловидный селен начинает размягчаться, при большем нагревании он, выделяя значительное количество тепла, переходит в серый кристаллический селен, который получается также при медленном охлаждении расплавленного селена. Кристаллическим селен значительно менее растворим в сероуглероде, чем красный селен. Кристаллический селен представляет собой серую кристаллическую массу, которая в темноте очень слабо проводит электрический ток. Но если его осветить, то электропроводность возрастает примерно в тысячу раз; в темноте она вновь падает до первоначальной величины.
Это явление объясняется тем, что при освещении связи электронов в селене ослабевают или электроны даже отрываются. Как известно, при облучении металлов светом с очепь короткой дипнон волны они испускают в пространство электроны (фотоэлектрический эффект). Селен также обладает этим свойством, если его облучать ультрафиолетовым светом. Так как свет с большей длиной волны сообщает атому или соответственно его электронам меньшую энергию [в силу уравнения (10) на стр. 109], чем свет с более короткой длиной волны, то можно предположить, что при облучении видимым светом электроны хотя и вырываются из атомов, но со скоростью, недостаточной для выхода в свободное пространство. Этот эффект должен наблюдаться как у селена, так и у обычных металлов; однако в силу значительно большей электропроводности последних наблюдать у них этот эффект не удается.
Шестая -группа периодической системы 797
Светочувствительностью селена пользуются для конструирования так называемых «селеновых ячеек», правильнее «селеновых мостиков» и селеновых фотоэлементов (точнее, селеновых полупроводниковых фотоэлементов). В обоих случаях получаются приборы, преобразующие световые колебания в электрические. Их используют для устройства сигнализационных приборов («электрический глаз») и других приспособлений для автоматического пуска ма<пин (например, эскалаторов), а также используют в звуковом кино, фототелеграфе и телевидении. Селеновые мостики дали первый толчок этим открытиям, но для широкого практического применения оказались мало, чувствительным и и инертными. Практическое применение этих эффектов стало возможным лишь тогда, когда вместо селеновых мостиков были сконструированы фотоэлементы, в основе работы которых лежит фотоэлектрический эффект. В таких фотоэлементах сначала использовали исключительно щелочные металлы.. Изобретенные позднее фотоэлементы с запирающим слоем (Schottky^ Lange, 1930), для которых (наряду с известными полупроводниковыми соединениями, например Сц2О) снова оказался особенно пригоден селен, для многих практических целей значительно превосходят щелочные фотоэлементы.
Селеновый, моетиг, представляет собой две проволоки, намотанные параллельно на изолирующий материал, например фарфор, между которыми наносится тонкий слой расплавленного селена. Застывающий в стекловидную массу селен при длительном нагревании переходит затем в кристаллическую модификацию («сенсибилизируется»). Различают «твердые» гелвповьте мостики, электропроводность которых при освещении медленно, в течение нескольких минут, достигает предельного значения, а в темноте также медленно уменьшается, и «мягкие» селеновые мостики, электропроводность которых уже через несколько секунд после начала освещения достигает максимальной величины, чтобы затем, при продолжении освещения, медленно, а в темиото моментально умепьшптьсн. Электропроводность металлического селена возрастает во времешч и без освещения, если приложить высокое напряжение: кроме того, при высоком напряжении она выше, чем при низком. Аналогично электропроводности при освещении металлического селена увеличивается и его теплопроводность.
Устройство обычного фотоэлемента В основных чертах таково: на части внутренней стенки стеклянного сосуда осажден тонкий слой щелочного металла, соединенный подходящим способом с отрицательным полюсом источника тока; против этого слоя расположен (ипогда кольневой, иногда сетчатый) электрод, который соединен с положительным полюсом. Стеклянный сосуд эвакуирован нлп наполнен инертным газом. При освещении с поверхности щелочного металла вырываются электроны, которые притягиваются положительным электродом и тем самым создают электрический ток. Если внутреннее пространство фоте зле мента заполнено газом, то ионизация гада увеличивает ток. В фотоэлементам: с запирающим слоем пространство между двумя электродами заполнено веществом, из которого при освещении освобождаются электроны, как это имеет место прежде всего у селена (в несколько меньшей степе ип также, например, у Сн2О). Тогда как для перехода электронов из металла в вакуум или газовую фазу даже у щелочных металлов требуется произвести относительно большую работу, переход аледтроиов между двумя соприкасающимися твердыми проводниками происходит очщи. легко. Этим объясняется высокая чувствительность фотоэлементов с запирающим сдоем. В соответствии с данными Ланге (Lange, 1931) и фотоэлементе с запирают дим Слоем пз окиси меди (I) на единицу светового потока бм вспомогательного напряжения возникает ток примерно в 6—12.7 раз большей мощности, чем в обычно применяемых щелочных фотоэлементах, работающих- со вспомогательным напряжением 120—200 : се leriijBbiii фотоэлемент превосходит меднозакисный по чувствительности еще в 1г’’—2и р , ; Лоеш втеку фотоэлементы с запирающим слоем прн освещении работают как самостоятельные источники -тока, они пригодны прежде всего для таких фотоэлектрических приборов, которые должны работать независимо от внешних источников тока, наприме р фотоэкспонометров. Далее их используют для фотоэлектрических выключателей и сигналов (например, автоматические шлагбаумы, сортировочные машины, включение уличного освещения при наступлении сумерек, оптическая блокировка на железных дорогах) п дли различных фотометрических приборов (спектрофотометры, колориметры п т. и.).
798
.Глааа 35
Серый селен кристаллизуется в гексагональной ромбоэдрической системе (физические константы см. на стр. 735). В жидком состоянии селен коричнево-красный. Пары его желтоватого цвета. При температурах выше 900° селен состоит из (парамагнитных) молекул: Sea, при более низких температурах происходит дальнейшая полимеризация. Селен, растворенный в сероуглероде, имеет, согласно криоскопическим измерениям Бекмана и Пфейфера, молекулярный вес, отвечающий формуле Зой.
Вследствие сильно выраженном сцдонностн к переохлаждению серый селен выделяется из расплава только крошечными кристаллами. Однако сублимацией при пониженном давлении можно получить иглы длиной в несколько сантиметров (Strawman is, 1940). Эти иглы имеют хорошо выраженные грани призм, однако на их концах в большинстве случаев вообще нельзя найти правильных поверхностей. Очевидно, это вызвано тем, что отдельные (спиральные) цепи, из которых построен серый селен, растут в направлении осн с довольпо независимо. Слабые связи, которые имеются между ними, недостаточны, чтобы определить рост этих цепей так, чтобы могли образовываться плоские поверхности (de Boer, 1943).
Из растворов и сероуглероде можно выделить н кристаллической форме и красный селен, а именно в двух различных моноклинных модификациях с различным отношением осей: Sea (уд. вес 4,48) и Sep (уд. вес 4,40). а-Селея может образовывать смешанные кристаллы с серой, Как и стекловидный селен, красный к ристал личе сияй селен метастабилен. При нагревания он переходит в металлическую модификацию. Однако при быстром, нагревании его можно расплавить примерно при 18(г без того, чтобы он претерпел фазовое превращение.
Кристаллическая структура. Решетку серого селена можно представить себе как производную от простой кубической решетки (рис. 45, стр. 237), в которой атомы смещены из угловых положений в различных направлениях. Если в простой кубической решетке каждый узел окружен шестью другими, расположенными на равных расстояниях, то каждый атом селена имеет двух ближайших соседей (на расстоянии 2,32 А) и четырех более далеких (на расстоянии 3,46 А). Рядом стоящие атомы образуют бесконечные спиральные цепи. В решетке теллура, аналогичной решетке селена, расстояния между атомами составляют 2,86 и 3,74 А. Кристаллический красный селен в обеих своих формах (а и £) имеет решетку, построенную, как и в случае серы, из молекул Se8.
В стекловидном селене молекулы, как и в кристаллической решетке гексагонального селена, образуют цепи. Эты цепи (в отличие от расплавленного селена) расположены параллельно, но не связаны закономерно, как в кристаллическом состоянии (Prins, 1937; Glocker, 1942; Richter, 1952). Чтобы расположить эти цепи таким закономерным образом, надо разорвать множество связей Se—Se и образовать множество новых. Обусловленная этим значительная энергия активации и объясняет относительную устойчивость стекловидной формы селена (а также и стекловидных модификаций других веществ). Примеси ускоряют переход стекловидного селена в кристаллический. Это и попятно: в связи с тем, что Примеси реагируют с Отдельными атомами селена, цепи во многих местах разрываются, зато должно приводить к облегчению закономерной ориентации.
Различия, ян я логичные различиям между стекловидным и кристаллическим селеном, существуют между аморфным и кристаллическим лышьяяо.и, По мнению Рихтера, появление наряду с кристаллическими стекловидпо-аморфных модификаций с наибольшей вероятностью следует ожидать у таких элементов, которые, как сера, селен, теллур, фосфор, мышьяк я сурьма, в кристаллическом состоянии (по крайней мере в одной из модификаций) образуют цепочечные пли слоистые структуры. Поскольку между цепями или слоями существуют лишь слабые связи, то они не всегда оказываются достаточно эффективными, а это приводит it возникновению аморфного состояния-Если же связи равнозначны до всем направлениям, то повод к образованию аморфных модификаций возникает ЛиШг> в исключительных случаях, а именно тогда, когда в качестве составных частей выступают, как у кремния и германия, целые атомпые группы (правильные тетраэдры), которые вместо правильного относится иго го расположения (кристалл) могут быть связаны беспорядочно (аморфное состояние), В такнх случаях црпмеси способствуют возникновению аморфного состояния, так как они затрудняют правильную ориентацию групп атомов.
Теллур кристаллизуется в гексагонально-ромбоэдрической системе изоморфно серому селену. Он представляет собой серебристо-белое сметал-
Шестая группа, периодической системы
7991
лическим блеском вещество относительно низкой твердости (2,5 по шкале-Мооса) и притом хрупкое, так что он легко растирается в порошок. Его-электропроводность мала — примерно в 100 000 раз меньше, чем у серебра. Она несколько увеличивается при освещении, по медленнее, чем в случае селена. Уде.нжый вес кристаллического теллура 6,25. Если теллур-ос аждать из водного раствора, восстанавливая теллуристую кислоту сернистой кислотой, то он осаждается в форме объемистого коричневого, осадка с удельным весом 6,0. Представляет ли собой этот так называемый аморфный теллур особую модификацию или является просто мелкодисперсной формой кристаллического теллура, еще недостаточно ясно.
Если быстро охлаждать расплавленный теллур, то так называемый аморфный теллур получается в смеси с кристаллическим. Однако если дать свободно окисляться на воздухе раствору теллурида калия, то из такого раствора выпадает теллур в форме тонких игл (гексагонально-ромбоэдрические призмы). Теллур плавится при 452° и кипит при 1390е1 с образованием золотисто-желтых паров, которые примерно до 2000° состоят в основном из молоку л Тс‘2> выше этой темпера туры происходит в значительной степени распад на атомы. Как установил в 1952 г. Клемм, расплавленный теллур при. температуре, несколько выше температуры плавления, имеет максимальную плотность, аналогичную наблюдаемой у воды (ср. стр. 572).
Прп на г ревя нии па воздухе селен и теллур сгорают, первый — чисто голубым, второй — голубым пламенем с зеленоватой каймой, образуя, двуокиси селена SeO2 и теллура ТеО2. При сгорании селена появляется специфический запах (запах «гнидой редьки»), тогда как горящий теллур имеет лишь слегка кисловатый запах. Селен и теллур энергично соединяются и с другими электроотрицательными элементами, например с галогенами, а также со многими металлами. С водородом непосредственно -соединяется только селен, а теллур не реагирует или реагирует в очень-незначительной степени. Энергично происходит соединение с серой, однако при этом образуются не химические соединения, а растворы или, при их затвердевании, смешанные кристаллы.
Кислоты, не являющиеся окислителями, не действуют на селей и теллур. Однако оба элемента растворяются в концентрированной серной < кислоте, а также в азотпой кислоте и в едких щелочах.
Применение. Селен применяют в фотоэлектрических приборах, используемых в звуковом кино, фототелеграфе и телевидении; кроме того, например, для автоматического включения и выключения освещения улиц, для фотоэлектрических измерений и т. д. (см. стр. 797). Чувствительность металлического селена к световому потоку н его колебаниям превосходит во много раз чувствительность человеческого глаза. Далее, селен используют для окрашивания стекол: он дает розово-красные, красные или красно-желтые тона. В остальном селен и теллур, а также их соединения! находят очень ограниченное применение. Следует указать, что в последнее время соединения селена иногда применяют вместо солей золота в тонирующих фотографических составах.
Соединения селена, подобно соединениям мышьяка, сильно ядовиты. Например, селеноводород даже при большом разведении вызывает головную боль и топшоту. При больших концентрациях даже небольшие количества его сильно раздражают слизистую оболочку. При попадании па кожу соединения селена вызывают экзему и болезненные ожоги. В последнее-время органические соединения селена пытались использовать для борьбы с раком.
SOo
Гаана 15
Соединения теллура значительно менее ядовиты, чем соединения селена; так как в организме они очень быстро восстанавливаются до неядовитого элементарного теллура. Последний затем постепенно выводится из организма в форме отвратительно пахнущих органических- соединении теллура.
СОЕДИНЕНИЕ СЕЛЕНА И ТЕДЛУРА
Селен и теллур образуют соединения с большинством элементов. Однако они не соединяются между собой и с серой, а, как уже указывалось, образуют с пей и между собой твердые растворы. Самыми устойчивыми среди соединений селепа н теллура являются, с одной стороны, соединения с кислородом и легкими галогенами (фтором и хлором), а с другой — соединения с такими сильно электроположительными элементами, как щелочные металлы. Например, образование селенида калия K2Se из элементов протекает со взрывом.
По отношению к металлам и водороду селен и теллур, подобно сере, электроотрицательно двухвалентны. Однако, когда они выступают как электроположительные элементы, валентность их равна главным образом четырем. Лишь но отношению к фтору они являются определенно шестивалентными, тогда как из кислородных соединений самыми устойчивыми являются двуокиси и отвечающие им кислоты и соли, т. е. соединения четы-регва.пентных селена и теллура, а пе, как в случае серы, соединения, где она является шестивалентной (трехокись серы, серная кислота, сульфаты).
В общем формальная аналогия между соединениями серы, селепа и теллура довольно велика. Опп проявляется в рассматриваемых подробно ниже соединениях с кислородом и водородом и мепее отчетливо в со единениях с галогенами, а также в следующих соединениях:
AsgSy As2Se3 As3Te3
Sb2S3 Sb.>Se3
Sb.,Te-
CS2 I'SeS ETi'S
(SCN)., [SCNf
(ВеСК) •> [SeCN}'-
[TeCN ['
(SsO6r И т. д.
[SeS4O0]" и т. д,
[TeS4Ofi]" II т. д,
По свойствам mi r"C нншнод re ictta п теллура отличаются от соединений серы (нз которых формаих MijiKHu получить заменой серы на селен пли теллур) главным пбрЯЗОМ .МОНЬШОИ VtTl.<tl4llBOl..Ibl«>.
Из соединений с азотом состав. аналогичный сернистому азоту достоверно установлен только для ;/лешлээии п-зоша ~т,\. (тетраселентетрашгтрнд). Теллуристый а-но/п (пе полчченпып в 'питом виде! имеет, по да ними III трекера (Strecker, 1934), состав ТезУ;.
1. Окислы и кислородные кислоты
Наиболее устойчивыми окислами селепа и теллура являются их двуокиси SeO2 11 ТеО.,. Теллур легко образует трехокись ТеО3, тогда как трехокись селена SeO3 получается с трудом (см. стр. 802).
В газовой фазе при высоких температурах возможно (в равновесии с продуктами распада) образованно моноокисей SeO и ТсО, как показали исследования полосатых спектров этих молекул. Свободные энергии их образования из молекул составляют —81 и —G2,8 ккал!малы Однако, по-видимому, эти молекулы не могут быть изолированы (ср. О 1 с m я о г О., Z. аиогд, Chem., 256, 103, 19-181.
Двуокись селена легко соединяется с водой, образуя селенистую кислоту Hj,ScO3; соли последней называются селенитами.
Двуокись теллура мало растворима в воде. Соответствующая ей теллуристая кислота имеет переменное содержание воды. Отвечающие этой кислоте соли, теллуриты, в простейшем случае имеют общую формулу
Шестая группа Периодическая системы
S01
М£ТеО3. Они, так ?ке как и селениты, по составу аналогичны сульфитам. Такая же аналогия суп (ест в уст между сернистой кислотой, селенистой кислотой и теллуристой кислотой, если не учитывать для последней пере-меппого содержания воды, что объясняется большой склонностью теллуристой кислоты к «полимеризации» (точнее, конденсации).
Точно так же, как при окислении сернистой кислоты Н,йОз получается серная кислота HgSO4, окисление селенистой кислоты HESeO3 приводит к селеновой кислоте II2SeO4. В последнем случае требуются зпачителыю более сильные окислители. Аналогично при действии сильных окислителей на теллуристую кислоту образуется теллуровая кислота. Эта кислота, ангидрид которой соответствует ангидриду серной кислоты, отличается от H2SOA содержанием воды. Это верно по крайней мере для обычной теллуровой кислоты, ортотеллуровой кислоты. НеТеОй. Соли селеновой кислоты, называются селенитами, соли теллуровой кислоты — теллуритами.
IJ
!! Двуокись селена SeO3 образуется при сгорании селена в токе кисло-
9 рода или воздуха. Кислород перед этим лучше пропустить через дымящую
азотную кислоту, так как присутствие окислов азота значительно уско-1 ряет реакцию горения селена. Чистая двуокись селена образует блестящие белые иглы (удельный вес 3,954 при 16°). Она легко сублимируется. При 315° давление возгонки достигает 1 атм.
При нагревании под давлением двуокись солена плавится без разложения. Расплав ее оранжево-желтого цвета. Двуокись Селена заметно окрашена и в газообразном состоянии в желто-зеленый цвет. Двуокись селена легко растворяется в воде, а также в концентрированной серпой кислоте и в спирте. После испарения спиртового раствора остаются большие прозрачные пластинки, представляющие собой моноэтпловыйэфир селенистой кислоты. Сухая двуокись селена соединяется с хлористым водородом, давая соединение S с О -2 ТIС1. Диалогичные двойные сое,циней и я дв у о кис ь се ле на <) б р а зу ет и с другими веществами. В противоположность двуокиси серы двуокись селена легко восстанавливается до элементарного селена. Уже пыль может вызвать частичное восстановление, что придаст красную окраску продукту.
g
Селенистая кислота и селениты. Уже при лежании па влажном воздухе двуокись селена притягивает воду, с которой она соединяется с образованием селенистой кислоты H2SeO3. Последнюю получают обычно растворением измельченного п порошок селена в разбавленной азотной кислоте. Селенистая кислота кристаллизуется в расплывающихся, бесцветных, гексагональных призмах, удельного веса 3,007 при 15*°, которые на воздухе выветриваются, теряя воду.
(л’.Ч!'ппсдая кислота — слабая двухосновная кислота. Ее константы диссоциации равны К\ — 4-Ю-3 ц ЛГ2 — 0,9-10'® (при 25°). Она образует два ряда солей: кислые и /гг ььчу j селениты. Все селениты бесцветны. Водные растворы нейтральных
селг’пптоп вг действие гидролиза имеют щелочную реакцию. Селенит-ионы, а также теллур hi-по i ты ь ццпоч растворе существуют, согласно Брнятципгеру (Brinizmger, 1935), в впде ггдхагп 1.рёксё-1р.п||.ч; [SefOIDiJ" и ГГе(011)е]" соответственно.
По ।'пгишевию к бе,-:iin.piuii хлорной кислоте селенистая кислота ведет себя как ангидроосн1штп!ш юпл рщцкрует с хлорной кислотой, образуя перхлораттригидро-ксоселена(1\ । ЬсСш.Жь — ПС1()4 [Se(OH)3](С1О4[, Это соединение представляет собой бесцветные мщ 10.1гшппые кристаллы (т. пл. 33е), легко растворяется в нитрометане и проводит г рас творе электрический так (Arlman, 1939).
В противоно Ki.binKTb сернистой кислоте селенистая кислота существует только в одной форме — са и ere;ричнеш O-Se(OH)2.
Об этом можно судить, из пример, па основании того, что опа образует только один ряд алкильных С1>|'лпвонпй Be0sR3, которым ввиду пх способности к омылению следует приписать формулу О—Se(OR)a.
51 Г. Реми
802
Глава 75
Селенистая кислота легко восстапавлнвается до элементарного селена. Такое восстановление вызывается, например, сернистым газом, но количественно обычно идет только в присутствии соляной кислоты. Иначе происходит образование селепополитио-Повых кислот, т. е. политионовых кислот, в которых сера частично замешена па селеп. Соляная кислота разрушает эти кпслоты с осаждением селена. Элементарный селей осаждается и другими восстановителями, например дитионитом натрия (гипосульфитом натрия), солями гидразина и гндроксиламлна. С иодистым водородом селенистая кислота реагирует количественно по уравнению H2ScO3 + 4Ш =Se 4- 213 4- ЗН2О. При действии сероводорода выпадает красновато-желтый осадок, состоящий из смеси серы и селена
H2SeO3 + 2H2S=Se + 2S + 3HaO.
Двуокись селена часто успешно применяют в органической химии для проведения окисления в определенном направлении. Например, при действии двуокиси селена СН2-группа, расположенная рядом с кетогрунпой, может быть окислена до СО-группы. Некоторые углеводороды с двойной связью под влиянием 8сО3 переходят в дикетопы (Riley, 1932),
Двуокись теллура ТеО2, как и двуокись селена, образуется при горении теллура. Однако' для ее получения удобнее вести окисление теллура сильно охлажденной концентрированной азотной кислотой. При выпаривании или разбавлении раствора выпадает двуокись теллура в виде бесцветных, тетрагональных, октаэдрических кристаллов с удельным весом 5,7—5,9.
Кристаллическая решетка ТеО2 аналогична кристаллической решетке SaO2, РЬО2 и MgF2 (рис. 63);« = 4,79, с ~ 3,77 А. Этот тип кристаллической решетки (тип рутила) характерен прежде всего для целого ряда окнслов и дифторидов элементов, расположенных в побочных подгруппах периодической системы.
При нагревании двуокись теллура желтеет. При нагревании до красного каления ТеО3 расплавляется и одновременно начинает несколько улетучиваться. Из расплава опа затвердевает и виде ромбических (или моноклинных?) игл уд. веса 5,78 (при 14°). В воде двуокись теллура очень мало растворима (1 : 150 000). Раствор имеет по кислый, а горько-металлический вкус и не дает отчетливого красного окрашивания лакмусовой бумаги. Значительно лучше двуокись теллура растворяется в концентрированных сильных кислотах, прячем Опа образует соли с этими кислотами. В качестве примеров таких солей можно принести Te2G3(OH)NO3, Те2О3(ОН)С1О4, Te2O3SO4 и Te2O3SeO4. В абра.швщиш тнкиго рода солей теллура(1У) проявляется в некоторой етепоия основной ха [(актер pej окпеи теллура. Однако значительно более ярко выражен ос кислоттлй характер, вследствие чего двуокись теллура легко растворяется в едких Щелочах. Получающиеся при этом соли, отвечающие теллуристой кислоте, называются теллуритами.
Теллуриты и теллуристая кислота. Простейшие теллуриты по своему составу соответствуют сульфитам и нормальным селенитам, их общая формула М(ТеО3. Теллуриты щелочных металлов, кроме метода растворения двуокиси теллура в растворах сильных щелочей, получают также сплавлением се с едкими щелочами или карбонатами щелочных металлов. Теллуриты щелочных металлов бесцветны и легко растворяются в воде. Теллуриты остальных металлов плохо растворяются в воде. Отвечающая этим теллуритам теллуристая кислота Н2ТеО3—очень слабая кислота, так как водные растворы теллуритов щелочных металлов частично разлагаются даже угольной кислотой. В чистом состоянии теллуристая кислота до сих пор еще пе подучена, так, как она проявляет большую склонность конденсироваться в высокомолекулярные комплексы, отщепляя воду. При небольшом повышении температуры вся вода отщепляется и образуется ангидрид теллуристой кислоты — двуокись теллура.
Некоторые теллуриты при застывании из расплавов образуют стекловидные мао? сы. Теллуритовые стекла имеют в некотором отношении необычные физические свойства. Многие из них отличаются, как утверждает Стенворт (Stanworth, 1952), особенно высокой прозрачностью в инфракрасной области спектра.
Трехокись селена SeO3. согласно данным Ломана (Lehmann, 1952), получается с хорошим выходом при кипячении K2SeO4 и SO3 с обратным холодильником. При этом образуются два жидких слоя; верхний, пред-
Шестая группа периодической системы 803
ставляющий собой раствор SeO3 в избытке S03, может быть декантирован с нижнего, содержащего полисульфат калия. При испарении SO3 в остатке можно получить чистую SeO3.
Прежние попытки получить Se03 обезвоживанием H.,SeO4 оказались безуспешным и. Только Рейпбольдту (Rfaeinboldl, 1930) удалось получить это соединение прп действии на элементарный селен кислорода, активированного током высокой частоты и тлеющим разрядом. Эта методика была улучшена в 1934—1936 гг. Me лохом (Meloehe), однако получающийся продукт все же сильно загрязнен SeO2.
Трехокись селена бесцветна, плавится без разложения при 118° в сильно преломляющую жидкость с удельным весом 2,75, которая легко переохлаждается, нс затвсрдева^. Трехокись селена легко сублимируется в вакууме. В твердом состоянии опа существует в двух модификациях —> кубической и асбестоподобной игольчатой, аналогичной трехокиси серы. С водой SeO3 соединяется с шипением, образуя H2SeO4. Однако эта реакция протекает не так энергично, как в случае SO3, и без образования -тумана.
Селеновая кислота и селениты. Селеновую кислоту H2SeO4 получают при обработке селенистой кислоты сильными окислителями, иапример хлорноватой кислотой. Ее соли — селениты M.lSeO4 — проще всего полу чаются сплавлением селена, селенидов или двуокиси селена с селитрой, или обработкой растворенных в воде селенитов хлором (1), или электролитическим окислением селенитов (2):
SeO^|-Cl2+H2O = SeO;-f-2H-4-2Cl': (1).
SeOJ-l 2О1Г—2e = Sc0J+H20. (2)
При добавлении к растворам селепатов пи гратов бария или свинца выделяются малорастворимые седоваты бария или свинца соответственно. Если затем эти соли обра ботать серной кислотой или сероводородом, то можно получить чистую селеновую кислоту;
BaSeO4 4- H2SG4 = BaSO4 4 H2SeO4*; (3)
PbSeO4 4 H£S = PbS 4 H2SeO4. (4) ‘
Испарением в вакууме прп температуре около 200° получают безводную кислоту, которая при обычных температурах затвердевает в виде бесцветных, гексагональных призм (т. пл. 57’).
При нагревании селеновая кислота легко отщепляет кислород, поэтому испарять ее без разложения можно только в высоком вакууме. Селеновая кислота довольно энергично соединяется с водой. При этом образуются два соединения; H2Se04-H2O (т. ил.-|-26°) и H2SeO4-4H2O (т. пл. -51,7’).
Даже в умеренно разбавленном растворе селеновая кислота в значите плюй степени электролитически диссоциирует подобно серной кислоте, которой опа в значительной мере аналогична по химическому поведению. Например, концентрированная селеновая кислота, подобно серной кислоте, обугливает органические соединения. Однако по окислительной способности селеновая кислота значительно превосходят серную. Например, из смеси концентрированных селеновой и соляной кислот выделяется хлор, так что эта смесь может растворять золото ц платину,
* Сслепат бария значительно более растворим, чем сульфат бария, поэтому реакция (3) идет до конца. Растворимость селепата бария составляет 8,25 ate в 100 г воды при 25°.
51*
804
Глава IS
как царская водка. Из всего сказанного следует, что реакция, представленная уравнением (1), обратима.
Сили селеновой кислоты — селенита — во многих отношениях аналогичны сульфатам, например своей растворимостью и кристаллическими формами. Онц также образуют двойные соли, подобные квасцам. При нагревании селенаты менее устойчивы, чем сульфаты. Они довольно легко, отщепляют кислород и вспыхивают при нагревании на угле, выделяя свободный солеи.
Селенат калия K2SeO4 лучше всего получать взаимодействием КаСО3 с BaSeO4 по уравнению К2СО3 -f- BaSeO4 K.2SeO4+BaCrO3. Селенат бария BaSeO4 получают по методу Блюменталя (Blumenthal, 1913), прибавляя к раствору Ва(ХО3)2 раствор H2SeO4, приготовленный окислением. SeO2 азотной кислотой и К Вт Оз- Селенат калия К28о04 изоморфен с ул;.фату кал ня K2SO4 it способен, как последний, присоединять SO3, образуя K2SeS2Oi(i, который, подобно K2SsO10, нрн нагревании примерло до 150° переходит, отщепляя SO:;, в K2SeSO7. Это соединение приблизительно около 440° разлагается по уравнению K2SeSO7 = K,SO4+ SeO2+ ОСО (Lehmann, 1952).
О содержании кристаллизационной воды и растворимости ссленатов металлов, образующих купоросы, см.: Klein ЛАли. China., [11], 14, 263, 1940.
Теллуровая кислота и теллуриты. Теллуровую кислоту получают, действуя сильными окислителями на теллуристую кислоту. Например, по Штауделмейеру (Staudcnmeier) ее получают следующим образом: топ к о измельченный теллур растворяют в возможно меньшем количестве горячей разбавленной азотной кислоты и к этому раствору медленно прибавляют хромовую кислоту. Удобнее проводить окисление теллура методом, предложенным Мейером (Meyer, 1930), водным раствором хлорноватой кислоты. При упаривании такого раствора кристаллизуется ортотеллуровая кислота состава НДеОв. Хорнер [Horner Н. J., J. Am. chem. Soc., 74, 3694, 1952] рекомендует получать теллуровую кислоту окислением двуокиси теллура перекисью водорода в аммиачной среде. При этом снэта ia ни тучасгся теллурат аммония, из которого дей-ствнем KOHiH'iiTpHpi.вацпои азотной кислоты освобождается теллуровая гл ini.- Ортшел.)уровня кислота существует в двух различных формах: в кубической с удельным весом 3,033 и в моноклинной с удельным весом 3,071. „Первая прп соприкосновении с раствором постепенно переходит во вторую. При температурах ниже 10е1 из водного раствора кристаллизуется гидрат НеТеОв-4Н2О. Растворимость теллуровой кислоты в воде довольно велика
По сравнению с серной и селеновой кислотами теллуровая кислота очень слабая — настолько слабая, что ее нельзя титровать щелочью. Поэтому электропроводность водных растворов теллуровом кислоты очень мала. II на вкус она пе кислая, а сладковато-металлическая. Коистатгга диссоциации первого иона водорода составляет, по Бланку (Blanc), 6-1СГ'. Поэтому в солях теллуровой кислоты, теллуритам, в большинстве случаев не все атомы водорода теллуровой кислоты замещены на металл. Од па ко известны и такие теллураты, где замещены все атомы водорода, например серебряная соль AggTeOy и ртутаая соль Hg3TeOB. Не выделяющийся из водного раствора нейтральный ортвтеллурат натрия Ма^ТеО^ был получен Ццнтлем (Zintl, 1938) нагреванием NaaTeO4 с перекисью натрия. Na2TeO4 можно получить обезвоживанием Ма2П4ТеО6, кристаллизующегося на водного раствора.
За счет присоединения молекул ангидридов других кислот к атомам кислорода теллуровой кислоты образуются еетервполикислоты, например Щ [Те(О -МоОз)в] и Н6 [Te(O-W03)el *. Теллуровая кислота склонна также л к образованию аутокомплок-сов. При нагревании до кипения в ее растворах происходит настолько сильное укрупнение частиц, что такой раствор приобретает характер коллоидного раствора. При
* Подробное о гетер ополи кислотах см. т. II.
805
Шестая группа периодической- системы охлаждении идет обратный распад па более мелкие частицы и, наконец, па отдельные молекулы.
Прн нагревании орто теллуровая кислота отщепляет воду и переходит в конце концов при температуре выше 300° в желтую трехокись теллура ТеО3. Последняя практически не растворяется в воде и иццпфферептпа по отношению к химическим реагентам, а при нагревании до красного каления отщен.аяет кислород и переходит в двуокись теллура. По данным Л1онтанье [Montlgnie, 1945—1947], кроме обычной модификации ТеСЬ (ц-ТеО3, уд. вес 5,075), существует еще одна модификация (Р>ТеО3, уд. вес 6,21), которая получается при длительном нагревании а-ТеСО в запаянной трубке при температуре 310°. В отличие от желтой аморфной а-ТеОа fJ-ТеОз серая и кристаллическая.
Петти ортотеллуровую кислоту нагревать в запаянной трубке до плавления, то образуется аллотеллурое.ая кислота, бесцветная к лей ко-сир опо о б раз пая масса, которая смешивается с водой во всех отношениях, образуя раствор, отличающийся от растворов ортотеллуровой кислоты одной характерной особенностью: ати растворы в противоположность растворам ортотеллуровой кислоты имеют отчетливую кислую реакцию. Аллотеллуровая кислота в водном растворе постепенно переходит в орто теллуровую кислоту.
2. Соединения с галогенами
Галогениды селена и теллура в общем устойчивее, чем галогениды серы. С фтором се леи л теллур взаимодействуют как четырех- и шести-валептпые элементы. По отношению к остальным галогенам теллур проявляет валентность два и четыре, тогда как селен образует с пими, кроме тетрагалогенндов, соединения типа Se2X2, аналогичные хлористой сере.
1'и.лв<>ениды селепа и теллура
Фториды SeF4 бесцв'; жидкость SeFs бесцв. газ Хлор и д ы Sc2Cl2 коричнево-желтая жидкость ScCl4 бесцв. кристаллы Бромиды Se2Br2 темно-красная жидкость ЯеВт4 желтый порошок Йодиды Se2I2 (?) черный твердый
TeF4 бесцв. ТеС12 черно-зеяе- ТеВг2 черно-зеленый Те12 (?) черный
твердый пый твердый твердый твердый
Ti'F5 бесцв. газ ТеС14 бесцв. ТеВг4 красно- Те14 черные
Te2F10 кристаллы желтые кристаллы кристаллы
По да иным Носта (Yost, 1930—1931), SeCl4 и SeBr4 в газообразном состоянии полностью распадаются на С12 ц SeCl2 или соответственно на Вг2 и SeBr2; но оба ди гало-гонида существуют только в виде газов как продукты диссоциации соответствующих тетрагалогенндов. ТеС14 (т. кип. 390°) пе разлагается. На основании электропографп-ческнх исследований Шомакер (Scliomaker, 1940) ирипгея к выводу, что молекулы ТоС14 в газовой фазе имеют следующее строение: атом Те образует с двумя атомами С1 угол, равный примерно 93° (расстояние; С1 *—* €1 = 3,37 А). Два других атома С1 расположены по обе стороны плоскости, образованной первыми тремя атомами, перпендикулярно к пей, так что они образуют с атомом теллура угол около 170'. Все pact’TiijTfiTjя Те*—► С1 равны 2,33 А. В этом случае можно говорить об искаженной тритона, 1 ы।.!![ бипирамиде, обе вершины которой заняты атомами хлора, тогда как два из трех углов треугольника, перпендикулярного липни, соединяющей первые два атома хлора, заняты атомами хлора, а третий — двумя электронами атома теллура, не участвующим 11 в образовании химических связей.
Аналигиищи.' строение подтверждено [McCullough, Hamburger, J. Аш. die и. Soc., 03, 803, 1941] рентгеновским анализом для молекулы твердого (кристаллизующегося в ромбической системе) дибромида дифепилселепа Бе(С6Н5)2Вг2. В этой молекуле оба атома Вг расположены в вершинах, а две фенильные группы СаН5 — в двух из трех экваториальных углов тригональной бипирамиды (расстояния: Se*— Вг = 2,52 A; Se <—> С = 1,91А; Br—So—Вт = 180“; *£ С—Se—С.—
- 110°). Подобное же строение имеет иоп [1O2F2]“ (ср. стр. 869).
Молекула ТеВь?, как следует из электронографпческих измерений, изогнута. Вг—Те—Br = 98J; piiccci ijiniie Те*—>Br = 2,50 A [Bogers, S р п г г, J. Am. chem? Soc., 69, 2102, 1047].
806
Глава 15
Некоторые из приведенных выше галогенидов способны образовывать комплекс-ные кислоты, присоединяя галогеноводород. Например, тетрабромид селена SeBr4, соединяясь с бромистым водородом, образует гексабромселеновую кислоту H,[SeBre]; при присоединении бромидов щелочных металлов получаются соли этой кислоты. Гексахлорселенаты М, [SeCl0] были получены Петцольдом (Petzold, 1932). Для теллура известны комплексные кислоты Н [ТеС15], Н(ТеВгй] и Н(Telj] (все кислоты кристаллизуются с водой), а также комплексные соли обтцпх формул М2[ТеС1в], MllToBrd и М|[То16].
3. Водородные соединения
Селеноводород H2Se и теллуроводород Н2Те по своему составу соответствуют сероводороду. Они очень похожи ла сероводород и по химическому поведению, однако значительно уступают сероводороду в отношении устойчивости. Их теплоты образования отрицательны (ср. табл. 106, стр. 738).
Селен, подобно сере, легко соединяется непосредственно с водородом. В случае теллура непосредственного соединения элементов не происходит, однако теллуроводород можно получить с хорошим выходом при электролитическом выделении водорода на теллуровом электроде. В очень разбавленном состоянии теллуроводород получают при разложении теллуридов металлов, например теллурида алюмипия А12Те2, водой или кислотами. Селеноводород получают этим методом почти в чистом виде. Его можно получить также, пропуская водород над элементарным селеном при температуре выше 400°.
Селеноводород представляет собой бесцветный газ, который легко сжижается и легко переходит в твердое состояние (см. табл. 106). Вее 1 л селеноводорода при 0° равен 3,672 е. Оп разлагается легче, чем сероводород, и уже при попадании пыли, в присутствии влаги, а также при взаимодействии с кислородом воздуха выделяет элементарпыйселен. Подожженный, он сгорает васильковым пламенем с образованием белого тумана двуокиси селена, а при недостаточном доступе воздуха — с выделением красного селена. При обычных температурах селеноводород метает аби ле и. В случае отсутствия катализаторов оп начинает с а мокро из во лило разлагаться С заметной скоростью только при температурах выше 300’. При более высоких температурах равновесие между селеноводородом и составляющими его элементами сдвигается в значительной степени в стерону образования соединения. Поэтому, как уже упоминалось, пропуская водород над раскаленным селеном, можно получить селеноводород. При этом во время охлаждения газа начинается частичный распад его, так что за нагреваемым местом трубки образуется селеновое кольцо.
Селеноводород еще болев ядовит, чем сероводород. Запах его похож на запах сероводорода, при этом он одновременно вызывает болезненные п долго пе заживающие воспаления слизистой дыхательных путей и глаз («селеновый насморк»),
В воде селеноводород растворяется лучше, чем сероводород. При этом оп также способен образовывать (правда, очень малоустойчивые) гидраты. Концентрация насыщенного водного раствора селеноводорода при атмосферном давлении и комнатной температуре приближается к 0,1 Af. Водный раствор, из которого па воздухе вскоре начинает выделяться красный селен, имеет отчетливо кислую реакцию. Степень электролитической диссоциации составляет в 0,1 М растворе при 25° примерно 4% (величины констант диссоциации приведены в табл. 106). ‘Гак что селеноводород является значительно более сильнойкислотой, чем сероводород. По своей силеон близок к муравьиной кислоте.
Селеноводород образует два ряда солей; кислые селениды MIHSe и нейтральные селениды М(Яе. В растворах селенидов щелочных и щелочноземельных металлов селен легко продолжает растворяться, образуя лолиселепиды M»Sev, точно так же, как в растворах сульфидов щелочных и щелочноземельных металлов образуются полпсульфиды. В большинстве случаев растворы селенидов щелочных металлов, содержащие полиселениды, окрашены в красноватый цвет. В чистом состоянии они, вероятно, бесцветны. Селениды тяжелых металлов, напротив, подобно сульфидам тяжелых металлов, все более или менее окрашены и в воде, а часто и в кислотах, нерастворимы. Кроме обменной реакции между селеноводородом и солями тяжелых металлов, селениды в большинстве случаев можно получить сплавлением компонептов.
Шестая группа периодической системы
807
Теллуроводород представляет собой бесцветный газ с неприятным запахом, напоминающим запах мышьяковистого водорода. Он конденсируется еще легче, чем селеноводород (ср. табл. 100), в жидкость, по-видимому, бесцветную в совершенно чистом состоянии, но вследствие частичного разложения почти всегда желтоватую. Теллуроводород очень легко разлагается и подвержен действию даже самых слабых окислителей. С кислородом воздуха он реагирует уже при обычных температурах с выделением теллура. Зажженный на воздухе, он сгорает голубым пламенем до двуокиси теллура. В воде теллуроводород легко растворяется, но в растворе он неустойчив; па-пример, при доступе воздуха происходит почти мгновенное разложение с выделением теллура. Сипа теллуроводородной кислоты сравнима с силой фосфорной кислоты (см. табл. 108).
Из солей теллуроводорода — теллуридов м1Тс — теллуриды щелочных металлов растворяются в воде и бесцветны. Одиако при доступе воздуха тотчас же начинается покраснение растворов, причем за счет окисления образуются полителлуриды. Теллуриды тяжелых металлов в воде но растворимы и окрашены в темный цвет. Некоторые из них, подобно теллуриду алюминия А12Те3, разлагаются водой, другие же — только кислотами.
Алкпльпыс соединения. При перегонке нейтральных или кислых селенидов с алкил сери о кислыми солями получаются алкилселенидн SeRa (R — алкил) и алкилселе-номеркаптаны. RSeH — соединения, соответствующие а л «и л сульфидам (тиоэфирам) и меркаптанам.
Н а и ри м е р:
K2Se + 2KO.S02.OC2H5 = 2.K2SO4 + (C2H5)2Se
От иле ерш? кислый каппа Дизтвяселснид
KUSe 4- КО 8Ое ОС2Нй = K2SO> -(- G2Hs-ScH
Рт н. i селеноне р каптан
Л на логичным oopli.ii.M можно получить алкилтеялуриды ТеВ2. А лк и лее ле ни ды и алкилтеллуридт.1 являв лея легко кипящими жидкостями с отвратительным занахом. Они легко присовдипяют галогены и кислород, образуя такие соединения, как <C2II;i)2SeCI,. (С2П 5)2ТеС1;;, (СгНй)28еО и т. д. Последнее из указанных соединений имеет основной характер и образует с кислотами соли, например (C2H5)2Se(NO3)2. Так же ведут себя и соединении теллура. Характерна способность а л ки лее лепи до в и ал ки л теплу ридов нрпсоедииять алкнлиодиды, образуя алкилселенониевые и алкилтеллуропиееыв •соединения, аналогичные алкилсульфониевым соединениям:
(C2H5)gSo Ч- С2Н51 == [(C2H,)3So]I, ;
(С2Н5)2Те + C2HSI = [(С2Н5)3ТеЦ.
Действием на эти соединения влажной окиси серебра получают соответствующие гидроокиси, которые, как и гидроокись сульфония, являются сильными основаниями.
Аналитические снедения. Селем просто определяется в его соединениях благодаря тому, что он легко переходит в красный элементарный селен. II для количественного определения селен в большинстве случаен переводят в элементарное состояние, но при этом выпадающую первоначально из растворов красную модификацию длительным нагреванием в соприкосновении с раствором переводят в легче фильтруемую зернистую серую модификацию.
При нагревании на угле в пламени паяльной горелки селеновые соединения распространяют характерный запах (запах «гнилой редьки»). Если в восстановительное пламя горелки ввести какой-либо холодный предмет, то в присутствии селеновых соединений он покрывается красноватым налетом, который растворяется в концентрированной серной кислоте с зеленым окрашиванием в результате реакции Se -ф- H2SO4 SeSO3 4- Н2О. При разбавлении водой вновь выпадает красный селен, так как реакция обратима.
Соединения теллура в этих условиях дают налет от черного до коричневого цвета, однако при этом не выделяется какой-либо характерный
808
Глава 16
запах. Налет теллура растворяется в концентрированной серной кислоте с красным окрашиванием
Te + II2SO4 TcSO3 + H2O.
При добавлении воды выпадает черный осадок, представляющий собой вновь образовавшийся элементарный теллур.
При весовом аналитююском определении в случае теллура поступают так же, как в случае селена: сначала переводят его в теллуристую кислоту, восстанавливают ее подходящим восстановителем (сернистой кислотой, гидразином) до элементарного теллура и взвешивают его.
При систематическом анализе селен и теллур под действием сероводорода выпадают вместе с серой. При обработке сернистым аммонием оба эти элемента, подобно сере, переходят в раствор и снова выпадают при подкислении.
О микро определении селена и теллура см,: Hecht F., Z. anorg. Chem., 251. 14, 1943.
ПОЛОНИЙ (POLONIUM) Ро
Полоний, самый тяжелый элемент подгруппы кислород — сера, принадлежит к радиоактивным элементам. Он относится к тем элементам, которые были открыты только благодаря исследованиям радиоактивности, и является первым повым радиоактивным элементом. Вскоре после того-как па основании свойств урана было открыто явление радиоактивности, а затем оно было найдено и у тория, Поль и Мария Кюри в 1898 г. открыли полоний, когда они анализпровали составные части ураповой смоляной руды, чтобы установить причину ненормально высокой для соединений урана радиоактивности этого минерала.
Полоний является значительно более радиоактивным, чем радий. Поэтому пе р и* i д 1 < л л >. р д с п а л а ш, ж t п и я мал. Актив ность препар ата по линия ежедневно умс11ьш,и:?тт1 нрпблцвптельно на 0,5%. Через 140 дней активность состав1яег ио Ливону первоначальной величины.
Как про ivkt распада разня (радий Е), полоний постоянно присутствует в старых препаратах радия. Однако вследствие его малого периода полураспада содержание полония в таких препаратах не может быть велико. С 1 г радия в равновесии находится только приблизительно Vjooo а полония. Поэтому содержание полония в урановой смоляной руде примерно в 5000 раз меньше содержания в пей радия. Так что для получения 1/2-) г полония надо переработать свыше тысячи тонн урановой смолки или свыше 300 г* бромида радия, сохранявшегося не менее 30 лет в твердом состоянии. Лучшим исходным веществом для получения полония мог бы быть радий D, который, однако, может быть получен тоже только из радия пли эманации радия.
Чаще всего исходным материалом Для получения полония служат остатки от переработки урановой смоляной руды на радий. Из солянокислою раствора таких остатков полоний сначала осаждают сероводородом вместе с Другими осаждающимися сероводородом металлами. Осадок затем перерабатывают для получения висмута. Этот соседний с полонием и аналитически очень похожий на него элемент содержится в ураповой смоляной руде п огромных по сравнению с полонием количествах. По так как сульфид полония менее растворим, чем сульфид висмута, последний можно обогатить сульфп-
* „, 385.8-104
(очное. 226,6-25-2,14
319 г ИаВг2.
Шестая группа периодической системы 809
дом полония методом дробной кристаллизации. Обогащение полонием можно произвести также дробным осаждением основного нитрата висмута при осторожном разбавлении сильно азотнокислого раствори, или дробным осаждением хлорида одо-ва(11) из солянокислого раствора, или осаждением ври погружении в соя я по кислый раствор пластиккя из меди, серебра пли висмута.
Часто полоний удобно осаждать из водного раствора электролитически. При пропуска ни и тока он осаждается на катоде. Лучше всего в качестве катода использовать золотую фольгу, поскольку полоний, осажденный на платиновой фольге, по удаляется с лее полностью дана; прн кипячении с кислотами. Удалить полностью полоний с платиновой фольги можпо только, нагревая се длительное время при 1000 '.
Нормальный потенциал полоппя по отношению к нормальному водородному электроду равен -|-0,0 г (Joliot, 1929; Erbachor, 1933). Полоний более благородный метала, чем серебро, и поэтому вытесняется серебром из раствора. Этим часто пользуются при радиохимических работах. В кислых растворах полоний существует в виде положительного двухзарядного нона Ро".
По своему химическому поведению полоний напоминает, с одной стороны, своего соседа — висмут, с другой — своего ближайшего аналога — теллур. Вследствие легкого гидролитического разложения солей полония его соединения легко образуют коллоидные растворы получающейся при гидролизе гидроокиси полония.
Аналогия между полонием и теллуром была установлена в 1903 г. Марксальдом (Marcwald), который ири погружении в солянокислый раствор остатков от переработки урановой смоляной руды чистой висмутовой пластинки получил вещество, принятое им сначала за новый радиоактивный элемент, который он назвал «радиотеллуром» вследствие большого сходства реакй.пн этого вещества с теллуром. Позднее, определив период полураспада, он убедился в том, что это был ранее открытый полоний. В 1918 г. Панету удалось показать, что полоний, подобно своим более легким аналогам, образует летучие соединения с водородом. Это было доказало тем же методом (см. стр. 733), как и в случае соединения водорода с торием С, т. е. как и в случае впсмутистого водорода. Полонистый водород обнаруживает большое сходство с последним, но он еще мепее устойчив, чем внсмутцстый водород. С другой стороны, полонистый водород обнаруживает аналогию с теллуристым водородом, например в отношении чувствительности к пасыи[<>нной воздухом воде.
Как показал 'Гаммап (Tnmmann, 1932), полоний очень склонен к образованию-смешанных кристаллов с такими металлами, как серебро, медь, цинк, кадмий, олово, свинец, сурьма, висмут, но не дает смешанных кристаллов с теллуром. Последнее объясняется тем, что структура кристаллической решетки полония сильно отличается от структуры решетки теллура. Кристаллическая структура полония была определена Рол i.e (Hollier, 1936) электро ^графическим методом на 0,1 у чистого металлического нолонця. Полоний образует мопоь"лишило кристаллы особого тина. Каждый атом полония окружен шестью Другими, расположенными в вершинах октаэдра, который,, однако, сильно искажен, так что все расстояния между атомами различны. Они изменяются в пределах 2,81—4,13 Д.
В противоположность металлическому полонию его соединения образуют Смеша иные кристаллы с соответствующими соединениями теллура. Соли полония в водном растворе взаимодействуют с дитпокарбаминалами патрия типа NaS-CS NR2 (К — алкил) с образованием ие диссоциированных, растворимых в хлороформе комплексных соедттш'шп!. Вместо с аналогичными комплексными соединениями иикеля(П), кобаль-та(ПГ) п в нс,му та (III) ИЗ раствора с оосаж даются даже следы полония. Однако в качестве <<110Сгг1!?лей>> (см. т. II) для соединений полония чаще всего используют соединения теллура. С ацетилацетоном СН2(СО-СПз)-, полоний образует соединение, которое, вероятно, представляет собой внутреннюю комплексную соль чгшырсхвалеит-пого подойдя, т.н; как опо образует смешанные кристаллы с а цетила цетон атом тория Th (Clip О СП j2]4 и может быть отделено от ацетил ацетопата алюминия А1 [СН(СОСП3)г|?; ее нт оиг> осаждено с ацетил ацето патом тория.
Глава 16
ОКИСЛЕНИЕ И ВОССТАНОВЛЕНИЕ
Окисление, Как уже было указано в предыдущей главе, соединение какого-нибудь вещества с кислородом называется окислением. Однако существует целый ряд процессов, обнаруживающих очень большую аналогию с реакцией соединения с кислородом, например соединение металлов с хлором^ бромом, серой и подобными им элементами, имеющими неметаллический характер. Эта аналогия нередко проявляется уже внешне. Так, сурьма сгорает в атмосфере хлора совершенно так же, как и в воздухе или в кислороде, и большинство других металлов можно заставить гореть не только в кислороде, но и в хлоре, в парах брома, парах серы и т. д. В ряде «случаев соединение с этими элементами происходит даже гораздо энергичнее, чем с..кислородом. В отношении фтора это справедливо даже в большинстве случаев. Образующиеся в результате этих процессов продукты можно путем реакций совершенно иного характера, чем типичные процессы окисления, превратить в те же продукты, которые получаются при непосредственном соединении с кислородом. Так, продукт горения олова в струе хлора, тетрахлорнд олова йпСЦ, можно разложить, действуя на него водой (гидролиз), и затем, высушив или прокалив полученное вещество, получить тот же конечный продукт — двуокись олова SnCU. который образуется прп непосредственном сжигании олова на воздухе. Изучение всех изложенных выше процессов привело к тому, что термину -«о кислей не* в настоящее время придают более широкий смысл, обозначая им пе только соединение с кислородом, но и родственные ему процессы, в частности соединение металлов пли водорода с фтором, хлором, бромом, серой, а также с иодом и другими аналогичными им веществами, вообще с веществами, имеющими электроотрицательный характер.
Сущность процессов окисления, как это впервые было ясно показано 'Оствальдом для таких веществ, которые в водных растворах существуют в виде ионов или, входя в состав соединений, несут на себе электрический заряд, состоит в присоединении ими положительных или в отдаче отрицательных зарядов; точнее, процесс окисления является отщеплением электронов от атомов или радикалов. То обстоятельство, что с равным основанием можно говорить о процессе окисления в том случае, когда свинец, соединяясь с хлором, переходит в хлорид свинца РЬС12, как и в том, когда он, соединяясь с кислородом, превращается в окись свинца PtO, в конечном итоге оправдывается тем, что в обоих случаях свинец из элементарного электронептрального состояния путем отщепления электронов переходит* в электроположительно двухвалентное состояние
Рь+С13=РЬ2+ац- и рь-Н/2о2=рь2+02-.
Относитоиьно обозначения зарядов см. примечание на стр. 242.
Окисление и восстановление SH
) То, что свинец в окиси свипца действительно имеет два положительных заря-
; да., нельзя утверждать с такой же достоверно ст в го, как в случае хлорида свинца,
•:з который как в растворе, так и в расплавленном состоянии сильно диссоциирован на
; ионы РЬ2+ и 2С1 . Одиако, принимая во внимание свойства окиси свинца, едва ли можно
j сомневаться, что и ото соединение имеет гетероиол яркое строение и что свинец в нем
является электроположительной составной частью; то же справедливо и для других окислов металлов. Впрочем, в случае кислородных соединений неметаллов иногда не ясно, можно ли говорить в обычном смысле слова о противоположных [ зарядах на кислороде и других составных частях этих соединений. Это прежде всего
j относится к соединениям углерода. В тес случаях, когда сущность окисления заклю-
“ чается не в приобретении положительного заряда, под окислением понимают просто
i соединение с кислородом (при определенных условиях также и отщепление водорода;
,, см. ниже). Конечно, в таких случаях нельзя в понятие окисления включать также
и процессы присоединения хлора, брома, серы и т. Д-, которые называются тогда хлорированием, бромированием и т. Д- Таким образом, можно прийти к двум, не полностью совпадающим определениям понятия «окисление»: окисление в чисто химическом и окисление в электрохимическом смысле.
Под окислением в чисто химическом смысле подразумевают е первую очередь соединение какого-нибудь вещества с кислородом. Кроме того, окислением в чисто химическом смысле считают и те реакции, при которых происходит отнятие водорода от какого-нибудь вещества. Отнятие водорода называть «окпслеписм* уместно только в том случае, если его можно в принципе представить происходящим под действием кислорода.
Термином чокислениеъ в электрохимическом смысле охватываются все химические (или электрохимические) процессы* при которых происходит отнятие электронов у одного какого-либо вещества при посредстве другого (прямо или косвенно).
Примером окислительной реакции в смысли первого определения может служить юкисдепие азотистой кислоты в азотную
hno2 + О = Н1ЧО3.
Окисление гадр окей л а мина в азотноватистую кислоту можно рассматривать как пример второй части первого определения
НО— N :Н2 НО—N + = Ц +2Н3О.
ПО-И. О Но- N
Наоборот, отщеплете? водорода, происходящее при термическом разложении гидрида кальция
СаН2 Са 4- Нг,
не есть процесс окисления, а, согласно второму он ре делению, является даже в злектро-химическоч сдай: ю процессом, противоположным окислению — это процесс восстановления (СМ. ППЯ'.'е)-
Для лучшего уяснения понятия окисления в электрохимическом смысле можпо привести еще несколько примеров.
Перевод какого-либо вещества из его элементарного состояния в соедпнеппе, в котором (ню является положительно заряженным, Например РЬ в РЬО или в РЬС12, так же как и перевод уже заряженного вещества в более высокую степень зарнжепня, например РЬО в РЬ()Й, РЙС1 <> в РЬС141 PbSO.s в Pb(SO4)2 и т. д., следует рассматривать как окисление. Равным образом окислением считается и перевод вещества, имевшего отрицательный заряд, и элементарное (электронейтральное) или в электроположительное состояние, например из 83- в S или в из Г123 в S или в H4SO4.
Окисление (отщепление электронов) может также происходить при электролизе например 2+ 4 +
PbSO,+Н380л — 2е = РЬ (SOJ3 4- 2Н*.
812
Глава 16
Электроны и в этом случае переносятся (косвенным путем) на другое вещество, так как электролиз, при котором совершается окисление сульфата свинца па аноде,, может протекать лишь при том условии, чтобы одновременно происходило восстановление какого-нибудь вещества па катоде (папример, PbSO4 в РЬ или 2Н* в На; подробнее об этом см. ниже). К процессам окисления в электрохимическом смысле следует далее причислять растворение металлов в кислотах, в том числе и в так называемых «неокисляющих» кислотах*, папример цинка в соляной кислоте
Zn + 2ПС1 = ZnCl2-|- Н2 или Zn 4- 2Н‘ = Zn” + Н,, растворение металлов при соприкосновении их с растворами солей более благородных металлов, например растворение цинка при впссетЕни его в раствор сульфата меди
Zn + CuSO4 ~ ZnSO4 + Си или Zu + Си" = Zu” + Сп.
Переход щолопяых и щелочноземельных металлов л их гидриды, в которых ати металлы заряжены положительно, также следует рассматривать как окислении в электрохимическом смысле
Li + 1-гП2= LPH-.
Восстановление. Восстановление (от лат. reducere) означает, как показывает само название, переход какого-либо окисленного вещества в его первоначальное состояние. Это — процесс, обратный окислению. И в отношении процесса восстановления, как и окисления, следует различать восстановление в чисто химическом смысле, заключающееся в отнятии кислорода или часто также п прнсоедипспии водорода, и восстановление в электрохимическом смысле, сущность которого заключается в присоединении к восстанавливаемому веществу электронов.
Перевод окиси свипца в свинец, например нагреванием с углем
РЬО + С = РЬ 4- СО, (1)
является процессом восстановления. В этом случае говорят, что окись свинца при нагревании с углем восстанавливается до свинца. Но в электрохимическом смысле-и переход хлорида свинца в сцинец также является восстановительным процессом; в общем случае всякий процеш’, при котором положительный заряд какого-нибудь вещества понижается плц его ui.pimare.TiiiiijH заряд повышается, является процессом, восстановления, Е о с ст а подите жиыо пропеесы могут совершаться также и алектролп-тпчегким путем, нричем s.tittt прош ло 1.ят иа катоде, в то время как процессы электролитического ОШ11...1С1Т11 Я irpOTl-riellOT на
Можно кратко сформулировать следующие определения восстановительных процессов: восстановительными процессами являются такие химические или электрохимические процессы, при которых происходит отнятие кислорода от данного вещества или присоединение к нему электронов. Присоединение водорода, поскольку с ним пе связано появление отрицательного заряда на том веществе, к которому он присоединяется,: пе подходит под это определение процесса восстановления. О пем в дальнейшем еще будет идти речь.
Когда при химическом превращении к какому-нибудь веществу присоединяются электроны, должно всегда присутствовать другое вещество, которое эти электроны отдает; и наоборот, вещество может отдавать химическим путем электроны только в том случае, если присутствует другое вещество, присоединяющее эти электроны. Поэтому всякий окислительный, процесс Iвсегда связан с восстановительным процессом, и наоборот. Для
* «Неокисляющими» кислотами обычно называют такие кислоты, растворяющая способность которых во отношению к металлам обусловливается исключительно стремлением водородных понов разрядиться вследствие присоединения электронов; к таким кислотам относятся, например, соляная кислота, разбавленная серная кислота, уксусная кислота.
Окисление и восстановление
813
электролитического окисления и восстановления сказанное справедливо с тем ограничением, что в этом случае оба процесса могут быть пространственно отделены. Электролитическое окисление происходит, как уже было •сказано, па аноде, а восстановление — на катоде; но и в этом случае восстановление на катоде может совершаться только в той море, в которой на аноде происходит окисление, так как с анода в раствор переходит такое же количество положительного электричества, какое отводится из раствора через катод.
Раскисление. Для гетер оно лярпых сое дни опии отщепление кислорода всегда соответствует понижению положительного заряда связанной с кислородом составной части соединения, т. е. с присоединением ею электронов, так как кислород в этих соединениях всегда является отрицательно заряженной составной частью. В гомеопо.т hit-пых соединениях, наоборот, отщепление кислорода ие вызывает обязательного изменения заряда той составной части, которая была первоначале по связана с кислородом. В таких случаях, которые часто встречаются в области органических соединений, процесс отщепления Кислорода нередко называют раскислением.
Гидрирование и дегидрирование. Во многих случаях восстановление соединении согвршаотся водородом. При ртом водород может действовать двояким образом: может отниматв кислород от того вещества, с которым вступает в реакцию, или же присоединяться к восстанавливаемому веществу, отдавая ему электроны, что происходит, папример, при реакции водорода с серой
h24-s=JJ>-.
Наконец, оба процесса могут протекать одновременно, т. с. может происходить отнятие кислорода при одновременном присоединении водорода.
Не всякое присоединение водорода можпо рассматривать как восстановление. Так, соединение щелочных и щелочноземельных металлов с водородом в электрохимическом смысле является не восстановлением, а наоборот, процессом окисления, поскольку водород, соединяясь с этими металлами, отнимает, у них электроны.
Во многих случаях нельзя определенно сказать, возникают ли при взаимодействии с водородом в получившемся соединении разноименные заряды на водороде и на другой составной части соединения. Это прежде всего относится к органическим соединениям. Обычно в органических соединениях водород рассматривают как более электроположительно заряженную составную часть, и с этой точки зрения до некоторой степени оправдывается термин «восстановление» и в этих случаях; когда же пет определенных указаний на то, что присоединение водорода сопровождается отдачей им электронов, т. е. главным образом при присоединении водорода к ненасыщенным органическим соединениям, правильнее называть такие процессы по bi установлением, а гидрированием. Соответственно этому и отнятие водорода, когда оно не сонр'яжено с отдачей электронов, целесообразно называть дегидрированием.
О гйнщшг в первую очередь тогда, когда необходимо подчеркнуть,
что процесс (в шпрот.ом гмысле стопа рассматриваемый как окислительный) протекает не в результате ищищ. и Ш тиня с кислородом, но при отщеплении водорода. Например, окисление спирта и а льде гид есть де гидрнрованне
< II > । II .О|| + О ьл СН3СНО | Н2О;
далее, Виланд считает дегидрированием также процесс окисления альдегида я водном растворе в уксусную кислоту, так как этот процесс, как было установлено, протекает
814
Глава 16
в присутствии воды таким образом, Что образовавшееся вначале путем присоединения воды неустойчивое в свободном состояния соединение СН3СН(ОН)2 отщепляет два атома водорода.
/0Н /ОН
сн3с < - j и+о;=сп3 -с^о + ЩО.
н
Окислители и восстановители. Вещество, которое при химической, реакции отдает кислород или присоединяет электроны, называют окислителем, а вещество, которое присоединяет кислород или отдает электроны,— восстановителем. Обычно, однако, эти термины употребляют в более узком смысле. К окислителям в более узком значении слова обычно относят только такие вещества, которые обладают сильно выраженной склонностью-отдавать кислород или присоединять электроны и проявляют это свойство по отношению ко многим другим веществам. Равным образом к истинным восстановителям обычно относят лишь то вещества, у которых ясно выражено их свойство отнимать от других веществ кислород или передавать, им свои электроны.
Так, в процессе, представленном уравнением (1) на стр. 192, окись свинца действует на углерод в той же мере окисляющим образом, как и углерод на окись свинца, восстанавливающим образом. Поэтому можно сказать, что окись свипца в этом процессе является окислителем, а углерод — восстановителем. Однако окись свипца обычно не называют в общем смысле окислителем ввиду того, что, за исключением содержащих углерод соединений*, число веществ, которым окись свинца отдает свой кислород, сравнительно певелико. Наоборот, углерод называют в самом общем смысле восстановителем, так как его способность отнимать при высокой температуре кислород, от других соединений выражена чрезвычайно сильно.
Те окислители, у которых свойство отдавать кислород или присоединять электроны выражено н очень сильной степени, называются сильными окислителями. Слабые окислители обладают этой способностью в значительно меньшей степени, по все же у них она явно выражена. Таким ?ке: образом различают сильные и слабые восстановители.
Основными окислителями, помимо кислорода воздуха, являются: перекись водорода и другие перекиси, хлор, бром, гипохлориты (главным, образом гипохлорит натрия и хлорпая известь), хромовая кислота, или трехокись хрома, бихромат калия, перманганат калия и концентрированная азотная кислота. Не столь сильно, как перечисленные выше окислители, действуют концентрированная серная и разбавленная азотная кислоты. В качестве примеров слабых окислителей следует упомянуть окись-серебра и при нагревании — окись меди. Очень сильным окисляющим действием при высокой температуре обладают также нитраты и хлораты.
Основными восстановителями при нагревании являются: углерод (его чаще всего применяют в виде кокса или древесного угля), окись углерода и водород. Очень сильным восстанавливающим действием, особенно при высоких температурах, обладают щелочные металлы, магний и щелочноземельные металлы и алюминий. В технике, кроме того, для восстановления часто пользуются железом. Для восстановления в водных растворах па холоду применяют главным образом: хлорид олова(П), сульфат железа(П), сернистую кислоту, щавелевую кислоту, муравь
* Если иметь в виду действие окиси свинца только по отношению к органическим соединениям, то в этой области ео можно назвать окислителем и в общем значении Этого, слова.
Окисление iz восстановление
815
иную кислоту, формальдегид, этиловый спирт, гидроксил амин, гидразин, иодистый водород, сероводород, фосфористую кислоту, элементарный водород в присутствии катализаторов (палладия или платиновой черни), а также водород «в момент выделения», т. е. водород, выделяющийся при действии металлов па кислоты или щелочи (см. стр. 63).
G электрохимическом точки зрения характер действия окислителей и восстановителей можно представить следующими уравнениями;
Ок и слит ели
Кислород воздуха 03 4 4е = 2О2"
Перекись водорода П2024 2е = Н20 4 О3"
Хлор или бром С12 -г 2е = 2С1" или Вг2 4 2е = 2Вт "
Гипохлориты СЮ’ 4" 2е — CL" 4 О2'
Трехокись хрома СтОй 4 Зе = Сг3* 4 ЗО‘2~
Бихромат калия К^СюО- -|- 6е=2К+ 4 2СгЗ+4 7О2’ или С г. О; 4- 6е= 2Cr3*4 7О2-Перманганат калия КМпО44 5с= К* 4 Мп2+4ч02" или МпО; 4 5е — Мп2* 4 4О2~ Азотная кислота Т11\О3 4 Зе = NO 4 Н+ 4 2О2" или NO" 4 Зе = NO 4 2О2" Серная кислота И2вО4 + 2е — Н40з 4 О2" Окись серебра Ag2O 4 2е = 2Ag 4 О2" Окись меди СиО 4 2е = Си. 4 О2*
Нитрат калия KN034 2е = KN024 О2’ или NO’ 4 2е = NOJ 4- О2"
Хлорат калия КСЮ3 4 6с = КС1 4 ЗО2’ или С1О" 4. бе — С1” 4 ЗО--
Восстановители.
Углерод С 4 О2" = СО 4 2с
Окись углерода СО 4 О2- = СО2 4 2е
Водород Н2 =: 2Н+ 4 2е или Н2 4 = Н2О 4 2е
Металлы, папример железо М = М5'- », папример Ге = Fe2* 4 2е
Хлорид олова(И) SjiCU 4 2С1" ~ SnCl4 4 2е или Sn2+ = Sn1* 4 2е
Сульфат жолеза(П) FeSO4 = Fe3* 4 SO;’ 4 е пли Fe2* = Fes* 4 е
Сернистая кислота II2SO3 4 О2' = H2SO4 4 2е или S0|' 4 О2’ = SO;’ 4 2е
Щавелевая кислота НаС2О4 4 О2* = Н2О 4 2СО2 4 2е или С2О;’ — 2СОг 4 2е Муравьиная кислота НСООН 4 О2" ~ Н20 4 СО2 4 2е Формальдегид НСНО 4 О2- = НСООН 4 2е
Этиловый спирт СН3.СН2.ОН 4 О2" = СНЯ-СНО 4 Н2О 4 2с
Г ядра кем ламин 2М12ОН 4 О2- = Na 4 3112О 4 2е
Гпдразгш NZH4 4 2О2’ = N2 4 2Нг0 4 4е
Подпетый водород 2HI = 12 4 2И* 4 2е или 21" = 12 4 2е
Сероводород H2S = S 4 2Н* 4 2е или HaS 4 О2- = S 4 Н2О 4 2е
Фосфористая кислота Н3РО3 4 О2" = Н3РО4 4 2е
Водород в момент выделения Н = Н* — « пли 2Н 4 О2" = Н2О 4 2е
Приведенные у равней и я ясно выражают сущность действия окислителей и восстановителей. заключающуюся в общем свойство первых л рис о единить электроны, а вторых — отдавать электроны.
Тщ; как при химических реакциях всегда совершается только обмен электронами и никогда пе происходит образования свободных электронов®, то для удовлетворения общего уравнения реакции частные уравнения процессов окисления и восстановления, подобные приведенным выше, должны быть всегда связаны между собой таким образом, чтобы в результате их суммирования не оставалось свободных электронов.
Относительно электролитических процессов справедливо сказано на стр. 193.
8J6
Глава 16
Например: ПгО2-!-2е=Н3О+О-2- I Н2С2О4+Оа-=Н2О + 2(Юг+2г /Ч
Н362+Н2С2О/, =2Н2О -Ь 2СО3
С12-!-2> = 2С1” 1
2х I, Г +
Cl2-|-2To2+^2CL- + 2Fe:1*
Количества окислителей и восстановителей, присоединяющие или отдающие одно и то же число электронов, называют эквивалентными количествами. Так, одна молекула перекиси водорода эквивалентна одной молекуле щавелевой кислоты или одной молекуле хлора (так как можно сравнивать окислители также и в отношении их количественного действия). Одна молекула хлора иди перекиси водорода эквивалентна двум попам двухвалентного железа. Два иона железа(П) эквивалентны одной молекуле щавелевой кислоты мт. д. Одним эквивалентом какого-нибудь окислителя или восстановителя называют такое его количество, которое принимает или соответственно отдает 1 электрон. Поскольку один нейтральный атом кислорода в состоянии присоединить 2 электрона, можно также сказать, что 1 эквивалент какого-либо окислителя или восстановителя есть такое его количество, которое соответствует переносу 1/„ атома кислорода. Одни грамм-эквивалент окислителя или восстановителя равен такому ого количеству (в граммах), которое соответствует переносу 8 граммов (электронейтрального) кислорода или, что то же, одной электрохимической единицы заряда*.
«-Нормальным раствором окислителя или восстановителя называется раствор, содержащий в литре «а» грамм-эквивалентов окислителя или восстановителя.
При составлении уравнений реакций окисления п восстановления следует (при пользовании привецоиными пыш,? и аналогичными схемами для частичных реакций) иметь в виду, что и вооо-рчу-ове iio<o iuiiOMhi ирелорода О2- никогда не появляются в свободном с, 'Стоянгш 11 сьотгьи-пцо* ,(1| зачетной концентрации. Они немедленно вступают и сш.'.т11и।",।не I., ирисутстау ющими в реакционной смеси электроположительными составны:ип ч;ц:т!сш ; и г идиом растворе, если раствор кислый, Они соединяются с «линии водорсиж збризул гюд;, и других сдуЧннх — с водой, С образованием ио-ч нов гидроксила
О2 -|-21Т+ Н2О или О2" + Н2О = 2ОН-.
Это обстоятельство необходимо учитывать при составлении уравнений — путем добавления соответствующего количества иопов 11* пли молекул НЙО или, если реакция протекает в отсутствие воды,— нутом введения в уравнения других, электроположительных составных частей, присутствующих в реакционной смеси. До же имеет место и при введении в уравнения тех веществ, которые должны доставить необходимые для реакции заряженные отрицательно атомы кислорода. Несколько примеров поясняют сказанное.
Первый пример. Окисление йодистого водорода бихромат.ом калия в серпокислом растворе:
Сг2О|- + 6е=2СгЗ+4-7О2- 1 , Зх| 2Ь = 1а+2с } +
’ Сг2ОГ +61-^2СгЗ< + 31г4-7О2-'1 703-414Н- =7Н2О J +
Cr^of--L-GT--L14H+= 2Crs+ 4- 312 4- 7Н2О
Общее уравнение: К2Сг2О7 -|- 6HI 4- 4H2SO4 = Cr2(SO4)3 4- K2SO4 4- 3I2 4- 7Н.2О.
* Электрохимической единицей заряда является фарадей, т. е. такое количество электричества, которое выделяется при разрядке 1 г-атома одновалентного элемента. Т фарадей = 96 493 кулона = Кд электронов (где Лд — число Авогадро).
Окисление и восстановление
817
Второй пример. Окисление щавелевой кислоты перманганатом кадия в сернокислом растворе:
2.x I МпО;4-5е=-Мп2» + 4О2- 1
5х ] («О? =2СОаЦ-2е J +
2MnOj +5Сг0Г = 2Мц2<-[- 10С02+802- т
8О2- + 16Н+=8Нг() J +
2МпО^ + 5С2О?"+16[1+ —гМяЗ* + ЮСОг + «Нг0
Общее уравнение:
2КМпО4 + 5Н2С2О4 + 3H2SO4 = 2MnSO4 + Кг80>, + 10С02 -|- 8Н2О.
Третий пример. Окисление сульфата маргапца(П) перманганатом калия в нейтральном растворе:
2х ] MnOj+3e=MnO2+2O2-'|
3 х. | Мн2+20-“ = MnO2 +2е J
2Мп0г + ;Шп2++202-=5МдО2 1
2Н2О = 2О2-+4Н+ J
2MnO,i-|- ЗМп2++2НгО=5МпО3 + 4Н+
Общее уравнение:
2KMnO4 + 3MnSO4 + 2IK0 5МпОг + 2KHSO4 + Н2ЙО4.
Четвертый приае р. Окисление сульфата хрома сплавлением с содой и селитрой с образованием хромата патрия:
Зх I N0,+2c ==К0;+02- I
2х | CfSl+4O2- = CrOr + 3e J +
3N0a+ 2Cr3* + 5О2“ = 3N0J +2СгО;“ 1
50О|“ = 5СО2+502“ J “Г
3NOs+2GrS+ + 5С0|- — 3N0»" +200’-+500г
Общее уравнение:
ЭККО, -1- Сс2(ЙО4)3 + 5Na3CO3 = 3KNO2+ 2Na3CrO4 + 3Na2SO4 + 5C02.
Рассмотрение приведенных выше двух примеров окисления перманганатом калин показывает, что разложение окислителей при okkc.ihmiuh происходит пе всегда одинаково. У пермапгапата калия в нейтральном пли слибокислом .растворе опо протекает иначе, чем в сильнокислом. Равным об pari ом и для других окислителей и восстановителей характер реакции окисления и восстановления иногда изменяется в зависимости от условий их проведения. В некоторых случаях одно п то же вещество в зависимости от условий может даже действовать то как окислитель, то как восстановитель. Интересным примером этого является перекись водорода, реакционная способность которой обусловливается ее стремлением превращаться в воду. Эта реакция может идти как в соответствии с уравнением
Н3О2 + 2с = Н20 + 0““, (2)
так и по схеме
П30, -| Оа“ НаО + 02 + 2е. (3)
Таким образом, перст;пн. водорода в различных реакциях может как присоединять электроны, так й отщеплять их: другими словами, перекись водорода обладает способностью действовать как окислитель или как восстановитель. Как восстановитель Н2О3 ведет себя в реакциях с сильными окислителями, папример с перманганатом калия. Если же реакция протекает в кислом растворе, то образуется соль марганца(П) 52 г. Реми
818
Глава. 16
я выделяется молекулярный кислород:
2 х I МиО4- = Млг-ь + 402- .
5х | II2O2-j-O2~=H2O-|-O24-2e |-|-
30*--] 6Н- = ЗН3О J
2MnOj + SH2O2 + 6II* ^2Мц2*+5О3 4-8Н2О
Кomosнируя уравнения (2) и (3), получаем
2Н2О2 = 2Н3О 4- 02.
Это и есть приведенное в гл. 2 уравнение самопроизвольного разложения перекиси водорода в присутствии катализаторов.
Особенностью разложения перекиси водорода, сопровождающегося окислением или восстановленном, является то, что в обоих случаях при этом образуется одно и то же вещество — вода, причем в одном из них выделяется, кроме того, мол окулярный кислород. Подобное явление, заключающееся в том, что одно и то же вещество, превращаясь в различные продукты, может при этом действовать то как окислитель, то как восстановитель, встречается очень часто. В принципе оно вообще имеет место для всякого вещества, которое может существовать в различных степенях окисления, Например, как уже было отмечено выдщ, окнсь свинца, при высокой температуре действующая по отношению к углероду как окислитель, в ос ста на вливает хлорную известь, которая переводит окись свинца в двуокись.
Написание окислительно-восстановительных уравнений, аналогичных рассмотренным в предыдущих примерах, не представляет никаких трудностей даже в сложных случаях, если только известны продукты реакции. Писать ли частные реакции, как это сделано здесь для краткости и лучшего восприятия, вначале в виде ионных уравнений (поскольку речь идет действительно о ионных реакциях) или отказаться От их использования — имеет лишь второстепенное значение. Необходимо только иметь в виду, что кислород, если он освобождается в соответствии с частными уравнениями, всегда имеет два отрицательных заряда, ес.чн речь идет о химически связанном кислороде, а не о молекуле кислорода. Помимо этого, следует учитывать в то, что число атомов одного и того же рода в обоих частях равенства должно быть равным (что, конечно, должно выполняться для любого химического уравнения) и что общая сумма зарядов в обеих частях равенства также должна быть одинаковой. Недостающие заряды должны быть добавлены в виде свободных электронов.
Следовательно, при написании окислительно-восстановительных реакций нужно исходить прежде всего из конечных иридуктов реакции, во-первых, окислительного и, во-вторых, eoccmiHtociiuie-ienoMi npfi^fri-oc, устанавливая оквивалеиыностъ обоих, т. е. умножая оба частных уравнения на соответствующие числовые множители так, чтобы в во сета щитете льном процессе приобреталось бы столько же электронов, сколько нх терялось в окислительном, и суммируя затем оба уравнения. Прп этом ноны кислорода О2 остаются по обе стороны знака равенства, так что приходится еще учитывать процесс, приводящий к их связыванию и затем вновь суммировать. При некотором опыте третье уравнение (компенсация попов О2-) можно составлять до сложения первых двух и таким образом проводить суммирование только одни раз.
Электролитическое окисление и восстановление. Поскольку сущность электролиза заключается в разложении веществ электрическим током, т. с. в их превращении при присоединении к ним электронов на катоде и отнятии электронов па аноде, то каждый электролитический анодный Процесс является окислением, а катодный восстановлением. Так, сущность электролиза соляной кислоты заключается в том, что па аноде происходит разряжение отрицательных ионов хлора, т. е. окисление до элементарного хлора, а иа катоде совершается разряжение положительных ионов водорода, которые при этом восстанавливаются до элементарного водорода.
Анод: 2С1' = С124-2е. Катод : 2Н‘4*2е=Н3.
Сказанное относительно этих искусственно вызванных, протекающих под влиянием подвода тока извне процессов всецело относится также и к протекающим самопроизвольно и порождающим электрический ток химическим реакциях!, происходящим в гальванических элементах; так, процесс,
Окисление и восстановление
819
дающий электрический ток в известном элементе Даниеля, основан па том, что на одном электроде этого элемента растворяется цинк, отщепляя электроны и переходя в ионное состояние, а на другом осаждается медь, ионы которой, присоединяя электроны, разряжаются. И в этом случае первый процесс является окислением, а второй — восстановлением.
Процесс окисления-, Zn — Zn" -р2е.
Процесс восстановления: Cii"-|-2e=Cu.
Ранным образом и процессы разряжения или перезаряжения ионов окисляющих или восстанавливающих веществ в более узком значении этих терминов, например попов MnO', CIO^, NO, и т. д., путем надлежащего комбинирования их с электродами можно использовать для осуществления процессов, дающих электрический ток. Это особенно важно потому, что возникающие при этом разности потенциалов позволяют количественно определять силу различных окислителей и восстановителей.
Окислительный потенциал. Если в водный раствор, содержащий, папример, ионы двухпольны to го п новы чгтырехвалептпого олова в одинаковых концентрациях, погрузить платиновую пластинку и получившийся таким образом электрод соединить, как показано на рис. 6, стр. 50, для ципкового электрода, с нормальным водородным электродом в одну цепь, то по внешней цепи потечет положительный ток в направлении от погруженного в раствор солей олова электрода к водородному электроду. Вызывающие ток процессы в этом случае следующие'.
Sii"” + 2e=Sn” и Н2=2Н’+2е.
Стремление олова перейти из четырех валентно го в двухвалентное состояние настолько велико, Что опо при соответствующих условиях может переводить водород из элементарного состояния в попы (т.н. окислять его) и, иреще того, производить еще внешнюю работу, так как разность потенциалов между обоими электродами, которую в принципе можно использовать для совершения внешней работы, составляет здесь, как уже было указано в гл. 12, при разомкнутой цепи 0,2 в, Разпость потенциалов между платиновым электродом, погруженным в эквимолярный водный раствор ионов РЬ" и ионов РЬ"", и нормальным водородным электродом равна даже 1,8 в. В этом случае погруженный в раствор платиновый электрод является положительным полюсом. Таким образом, йоны РЬ обладают еще значительно более высоким окислительным потенциалом, чем ионы Sn"". Если шштиповую пластинку, погруженную в раствор ионов Sn" и Sn"", соединить с другой такой же пластинкой, погруженной в раствор ионов РЬ" п РЬ“", то положительный ток от последней пластинки устремится к первой пластинке; ионы РЬ"" станут восстанавливаться, а ионы Sn" — окисляться. Тот потенциал, которым обладает платиновый электрод, по груженный в эквимолярную смесь ионов двух различных степеней окисления данного вещества, по отношению к нормальному водородному электроду, называется окислительным потенциалом более высокой степе-пи окисления этого вещества или также потенциалом перезарядки этих ионов. Некоторые таких окислительных потенциалов или потенциалов перезарядки приведены в табл. 112. Кроме потенциалов, относящихся к простой перезарядке ионов, в этой таблице также приведен ряд окислительных потенциалов, относящихся к реакциям, в которых принимает участие и растворитель — вода, как это, например, происходит при восстановлении иона нитрата
Х’О7Д-3<"=КО-1-2О2- 1
. и.,: -л. N0, з । 4H-=NO-|-2H,O
20^- -, -Ш=2Н2О J -т -г .
илп в случае весе та поп шипя ие свободных ионов РЬ"", а РЬО2 в попы РЬ-- : РЫК-}-2е-}-41-Г =РК‘ 4-2Н2О.
Ввиду того что приведенные в табл. 4 па Стр. 51 потенциалы, согласно сказанному выше, также можпо рассматривать как окислительные потенциалы (в более широком смысле), то значения потенциалов, приведенных в табл. 112, можно непосредственно сравнивать с соответствующими значениями табл. 4. Так, папример, из числовых значений этих потенциалов следует, что если комбинировать цепь из серебряного электрода в 1 М растворе ионов серебра с электродом Sn'7Sn',,,, то получится разность
52®
820
Глава 16
Таблица 112
Нормальные потенциалы окислителен, отнесенные к нормальному водородному электроду
Потенциалы перезарядки e Другие окислительные питеициалы (?
[Co(CN)6]"" -> [Co(CN]e]"' Cn’->Gu" Sn” —> Sn"" [Fe(CN)e]""->|Fe(CN)er" Fe” —> Fe‘" Hg^2Hg- ТГ-^ТГ" Co’-^Co’" Ph" -д- Ph— 2so; -> s2c^ -0,83 e +0,17 +0,2 +0,36 +0,77 +0,91 +1,21 ++84 + 1,8 +1,08 Н2+2ОН' ->2Н,0 КО+2НгО -> NO;,+4H‘ 2НгО —s-O2 + 41T’ Gr- + 4IT2O-> JICrO;-i-7H' Mn" +2HaO -> MnO, +4H-Pb" + 2H2O -> PbO2+4H-CI'-j-3H20^-C10i + 61r MnO2+2H2O^-MnO;+4H-А Г + 2НгО-^ Ni02-| AH’ O2+H2O O3+2H' -0,82 e +0,95 + 1,23 +1,3 +1,35 +1,44 + 1,44 +1,59 -M,75 +2,07
потенциалов 0,6 в, причем ток будет идти во внешней части цепи от первого электрода ко второму, т. е. ионы Sa" будут окислите,ся в попы Sri'"', а попы Ag' будут восстанавливаться до металлического серебра.
Подобно тому как раин ость потенциалов между каким-нибудь металлом и раствором его ионов зависит от концентрации их в растворе (см. стр. 49 и сл.), так и потепци алы перезарядки и окислительные потенциалы в более узком смысле находятся в зависимости от концентрации соответствующих ионов в растворе, причем потенциалы перезарядки зависят от соотношения концентраций обеих стоп о пей окисления, например от Sn""/Sn”, а окислительные потенциалы, относящиеся к таким реакциям, в которых принимает участие вода, кроме того, также п от концентрации в растворе водородных иля соответственно гидро доильных иопов. Окислительные потенциалы, приведенные в табл. 112, относятся к растворам, в которых все участвующие в реакции иопы находятся в 1 М концентра н.пы. Эти потенциалы указывают величину заряда погруженного в соответствующие растворы платинового электрода по сравнению с нормальным водородным электродом. Вместе < том потенциалы, приведенные в таблице, указывают также п величину работы (в воп.тахц которую необходимо совершить, чтобы окислить 1 с-о/.и ,’тайного вещества при одновременной разрядке 1 г-акв ионов водорода, следователи .по, в направлении, обратном указанному стрелкой. Таким образом, процессы, происходящие само произвольно и еыаыеяющис появление тока, протекают, поскольку потенциалы имеют положительные значения, в направлении, обратном показанном у стрелками.
Окислительные реакции и диепропорциопирование. Часто наблюдают, как из вещества, в котором атомы какого-либо элемента находятся в средней степени о кис лини я или электро нейтральны, образуется два других вещества, из которых одно производится От высшей, а другое от низшей степени окисления соответствующего Элемента. Например, хлорат калия при нагревании превращается в перхлорат и хлориД; свободный хлор с едким натром образует гипохлорит тг хлорид, а фосфор — гипофосфит и фосфин:
4КСЮ3 ЗКС1О4 + КС1, (4)
Cl2 + 2NAOH - Na [СЮ] + NaCl + Н2О, (5)
0 1+ , з-
4Р + 3NaOH + ЗН2О = 3Na[HsPOJ + РЦ3. (6)
Такого рода процессы называются окислительно-восстановительными, ибо их можно рассматривать как окисление части атомов элемента за счет восстановления другой части атомов этого ;ке элемента. Это будет видно, если представить общие уравнения
Окисление и восстановление
821
в виде частных ура «пенни так, как это было сделано рапсе:
3.x | СЮз -г 0'2~^С10; 4-2й (окисление)
С! О з -j- бе = СГ + ЗО2' (восстановление)
4С1О; = ЗСЮ; 4-Сг
5'2С1г4-О2- =СЮ'ф-с
Ч2С12+. = С1'
2ОН' = ОЗ-4*НЧО
С.124-2ОН' = С;Ю' + С1'-) Н2О
Зх | P-|-2OH'=H2POi-H P-|-3e-i-3ir==PII3
4Р+ЗОН'-I-ЗН2О = 3H2POS 4* РН3
(окисление)
(восстановление)
(окисление) (восстановление)
Как следует пи приведен пых выше частных уравнений, рассмотрение этих реакций в виде комбинаций окислительных и восстановите явных в электрохимическом смысле процессов но зависит от того, считать ли атомы образующих их элементов в соединениях электрически заряженными или приписывать соединениям чисто гомеополярный характер.
Обласгт. огяе..>птвлг,по-вос«таиовитольных реакций частично налагается ид область реакции диенрот>рцнон.и./>оиаиия. Под диспропорционированием понимают образование простых соединений in более сложных того же типа и с таким же числом атомов и радикалов. Папример:
1Жл13Г:1^5ВгН6-|-2ВС13 (см стр. 364) (7)
.'Tl;;) — 2BGI3-I-B(OCIl3)3 (см. стр- 369) (8)
2СН3-СНО НКОИ = [СН3СОО]К4-С;Н3-СП2'ОН. (9)
Уксусный Ацетат Этиловый
альдегид палия спирт
Реакция (9) является одновременно окислптельно-восстаиочительноп реакцией и реакцией диспропорционирования. Реакция (8) является реакцией диспропорционирования, по не окпслптелт.по-восстаиовительпой. Диспропорционирование, продствлепное реакцией (7), можно только в том случае рассматривать как окисление — восстановление, если предположить, что бор в хлордиборане более электроположителен пли, во всяком случае, мешю электроотрицателен, чем в дпборапе. Это предположение ио подтверждается известными в настоящее время опытными данными. Поэтому в этом и в аналогичных случаях лучше говорить о диспропорционировании, чем об окислении — в ос ст а но в лепив. Наоборот, реакции (4) — (6) являются реакциями окисления — восстановления, а но диспропорционирования.
Самоокисление. Под самоокислением понимают такие окислительные реакции, которые протекают под действием кислорода воздуха при обычной или слегка повышенной температуре. При таких реакциях можно нередко наблюдать, что в период окисления часть кислорода переходит в более активную форму, так что он приобретает способность окислять такие вещества, которые при данных условиях обычно кислородом воздуха пе окпс.ияюгся. Так, арсенит натрия в водном растворе пе окисляется кислородом воздуха, а сульфзит натрия., напротив, медленно окисляется. Если Же струю воздуха пропускать через водный раствор, содержащий одновременно арсенит и сульфит натрия, то наряду с окислением сульфита в сульфат происходит также и окисление арсенита в арсенат, причем при надлежащих условиях на каждую молекулу сульфата, образующегося при окислении сульфита, образуется тоже одна молекула ар сенат а*. В т;шнх случаях говорят о половинном распределении кислорода. Процесс следует представить таким образом: сначала сульфит-пин присоединяет одну молекулу кислорода и образовавшего! прй этом перекисное соединение, в данном случае пон [О2 ; SO3|", немедленно отдает один атом кислорода арсеппту:
[ЗО3Г + О2= fO2.'SO3r,
1.0, ; £O3.I" I- [AsO3]'"= [S04]"4- [AsOJ'".
Теория пропсы:'щ самоокисления, протекающих при промежуточном образовании соединений нирекнепого тина, была намечена Траубе (Trairbe) и подробнее разрабо
* Подобные совмегтио протекающие реакции окисления были изучены Н, Л. Шиловым (1905) и названы, им сопряжениями реакциями. — Прим. ред.
822
Глава If)
тана Энглером (Engler). В то время предполагали, что все реакции самоокисления протекают через стадию образования промежуточных перекисных продуктов*. В настоящее. время известна, что они могут происходить и иными путями, а именно при промежуточном образовании свободных радикалов (см. ншке). Вещества, которые, подобно сульфиту патрия, могут непосредственно окисляться кислородом воздуха, называется, по Энглеру, самрокислшпелями, а те, на которые само окислители переносят кислород,— акцепторами. В присутствии соответствующих самоо кислите лей, например растворенного в палладии водорода, многие вещества, например иодистый водород, окись углерода, аммиак, щавелевая кислота, бензол, белое индиго и целый ряд других органических веществ, могут функционировать как акцепторы. Все Эти вещества сами по себе при обычной температуре не окисляются кислородом или же окисление их протекает при этих условиях настолько медленно, что его не удается заметить. Однако в присутствии растворенного в палладии водорода их окисление происходит быстро: йодистого водорода — до иода, окпец углерода — до двуокиси, аммиака — до азотистой кислоты и т. д.** В этих случаях также можно подобрать такие условия, прн которых кислород распределялся бы поровну между сам о окислите ле м и акцептором. Однако пе всегда нужно, чтобы половина кислорода, необходимого для образования промежуточного неустойчивого перекисного соединений, непременно переносилась затем, на акцептор. Непрочное перекисное соединение (по Энглеру, пазы, ваемое мольокисью), в рассматриваемом примере нон [02 : SO3j” (ре идентичный иону [НО- О' SOs] монопадсерной кислоты), иногда может само реагировать с само окислителем
[O2:SO3]" + [S03]“ = 2[SO4r
или две его молекулы могут реагировать между собой с образованием О2
2JO2 :SO3K = 2[SO4]” + О2.
Наконец, иногда ото перекисное Соединение в присутствии воды гидролизуется с образованием перекиси водорода. При самоокислении в водном растворе нередко наблюдается образование перекиси водорода.
Перекись водорода при само окис лопин может возникать не только благодаря гидролизу первоначально образовавшейся перекиси самоокислителя, но также и другим путем, а именно так, что данное вещество сначала присоединяет что вызывает активизацию водородных атомов последней, благодаря чему они приобретают способность вступать в реакцию с молекулярным кислородом. Так именно обстоит дело в изученном Траубе случае самоокисления цинка в соприкосновении с водой, содержащей воздух:
И
-о — II .......-О ОН Н—о
-1- || = 4- ।
--о— н - о '-он п- о
н
В подобных случаях говорят о непрямом самоокислении, при котором перекись водорода является косвенным еамоохи.слип>елем, а ТО вещество (в приведенном примере цнпк), которое активирует пе непосредственно молекулярный кислород, подобно настоящему самоокис лите лад, но в результате реакции с водой активирует водород, называется пееедосамоокислителем. Тяжелые металлы в водных растворах часто действуют как псевдосамоокпелнтели. К таким реакциям, например, относится наблюдавшееся уже Шс-нбейном (Schonboin) образование перекиси водорода при действии кислорода воздуха на свинец, находящийся в соприкосновении с разбавленной серной кислотой, причем здесь па каждую молекулу сульфата евппца образуется одпа молекула перекиси водорода
РЬ + H2SO4 + О» =? PbS04 + Н2О2.
В присутствии акцептора перекись водорода может переносить на него кислород. Но во всех случаях как прп прямом, так и при косвенном самоокислении па акцептор
* Теория окисления веществ кислородом е образованием промежуточных перекис пых соединений была развита Л. И, Бахом (1897),— Прим, ред,
” Промежуточно образующаяся при этом перекись не вполне идентична обычной перекиси водорода, так как она более активна, чем последняя. В этом случае предполагают образование раствора перекиси водорода в палладии или малоустойчивого соединения палладия с перекисью водорода. Вследствие того что Хартек наблюдал образование устойчивою только при пиукой температура изомера перекиси водорода при действии атомарного водорода па 02 (см. стр. 63), можно допустить промежуточное образование именно этого соединения для рассматриваемой выше реакции.
Окисление и восстановление
823
никогда не может быть перенесено больше половины (иногда и меньше) первоначально присоединенного самооквсяителем кислорода. Этим сам о ок целители отличаются от катализаторов, которые могут передавать теоретически неограниченные количества вещества. Другое отличие заключается в том, что катализаторы могут без подвода энергии извне активировать течение только такого рода процессов, которые не потребляют энергии, в то время как при посредство само ок целителей, без притока энергии извне могут протекать также окислительные процессы, связанные с потреблением энергпи; и этих случаях необходимая энергия доставляется за счет энергии, выделяющейся при окислении самоокисяителя, и только весь процесс в целом: окисление само-, окислителя -j- окисление акцептора — должен сопровождаться выделением. энергии-
Процессы самоокисления играют, по-видимому, большую роль в жизни живых оргонивмцв. Быв. может, этим и объясняется псключптельпая способность организмов производить окисление. Так, например, организм человека и животных в состоянии окислять бепзол в фенол и даже сжигать углеводороды парафинового ряда (вазелины). Некоторые бактерии могут окислять водород в воду, аммиак — в азот и в нитраты, метан — в двуокись углерода. Осуществить перечисленные процессы окисления (да ji то не вес) при обычных температурах в лабораторной обстановке удается только при помощи самых энергичных окислителей. Поэтому Энглер предположил, что в организме в качестве окисляющих веществ действуют первично образующиеся при самоокислении перекиси (мольокцеп).
Новые исследования привели к видоизменению н ограничению этих представлений. В настоящее время известно, что окислительные процессы, происходящие в организме, Часто протекают через многочисленные промежуточные стадии, каждая из которых обладает только низшим о кисните л иным потенциалом. Показано далее, что для эпщгих окислительных процессов большую роль играет промежуточное образование свободных радикалое. Оказалось, что это имеет значение даже для таких реакций, в которых в качестве промежуточных продуктов образуются перекись водорода и Другие перекиси.
Роль свободных радикалов в окислительных и еамоокиелптельных процессах. На стр, 201 было уже отмечено, что самоокисление может протекать через промежуточно образующиеся свободные радикалы. Это наблюдается прежде всего в тех случаях, когда -самоокисление происходит в присутствии катализаторов. Например, ускорение самоокисления сульфита патрия в водном растворе солями медн(П), по Габеру (Haber, 1931), основано на том, что ион Си" вначале окисляет ион SO; до свободного радикала S О J
Си"+so;=си-+so;. (16)
В отсутствие кислорода воздуха образуется дитионат 2SO; = S2O6', но в присутствии растворенного кислорода происходят следующие реакции:
SO's+ Н2О = ИЗОГОН', (11)
hso3 4- о2 н2о ч- so; = 2во; 4- гн' 4- он (is;
н он + so;-i-h-=hso34-oh', (13)
нз которых две последние, связанные в так называемые «цепные реакции» (см. т. II), протекают с образованием иона SO') с затратой Оз столь продолжительно, пока не исчезнут вследствие какой-либо побочной реакции участвующие в них радикалы HSO3 или Oil, Так как ион Си" вновь окисляется кислородом воздуха до Си", добавкой незначительных количеств медной соли можно вызвать самоокисление любых количеств сульфита. Если окислять сульфит в дитионат другим, электролитическим путем, прп котором также необходимо предположить промежуточное образование SO.(- пли HSOa-радикапов, то за время этого окисления избыточный сульфит в значительной степени с ам i ю кис л пется.
Другой пример реакции, протекающей с образованием свободных радикалов, дает активация разложения перекиси водорода солями ягелеза(П). По Габеру и Baij-су (Haber, Weiss, 1034), опа протекает следующим образом:
Fe" -1-Н.гОг=Ре- 4-ОН' 4* ОЦ, (14)
-> ОИ-{-Н2О2=Н2О4-НО2, (1.Э)
НО34-Н2О2=О3 +На0 + 0Н. (16)
Fe" 4- ОП = Fo"'4-Oir. (17)
К реакции образования свободного ОН-радикала по уравнению (14) присоединяются цепные реакции (15) н (18), протекающие до тех лор, пока случайное столкновение
824
Глава 16
постоянно образующихся заново ОН-радикалов с ионами Fe" не вызовет обрыв цепи по уравнению (17) (см. т. II, гл. 17). По новым данным механизм реакции сложнее, чем это предполагал Габер, по в принципе его схема правильно объясняет ускорение разложения перекиси водорода солями железа(П)— появлением свободных радикалов, вызывающих цепную реакцию. Образованием свободных ОН-радикалов удается также объяснит!, чрезвычайную окислительную активность перекиси водорода и в смеси с раствором солей железа(11) (реактив Фентона). Промежуточное образование свободных ОН- ц ЦОг-радикалов при химических реакциях было вначале установлено из наблюдений над протеканием реакций в газообразных системах. Однако нет никаких оснований для сомнений в том, что эти радикалы могут существовать также и в растворах. Вероятно, НО2-ради кал является именно тем радикалом, который во многих случаях должен быть поставлен на место предполагавшейся ранее «мольокиси». В этом случае сущность механпма реакция при самоокислении в принципе сводилась бы к следующим процессам; окисляющееся вещество отдает вначале один электро!! молекуле 02 (уравнение (18)] и пои О; с молекулой II20 образует Н02-радикал [уравнение (19)], который отрывает от окисляющегося вещества второй электрон [уравнение (20)], а образовавшийся таким образам поп 1102 реагирует е Н\О с образованием Н2О2 [уравнение (21)].
024-e=0j; 02 + П2О=НО2 + ОН*; (18) и (19).
HO2-|-e=HOv, ILOj + Н20 = Н202-|- ОН'. (20) и (21)
При таком самоокислении каждая молекула О2 окисляет 2 экв соответствующего велтест-ва и образует одновременно одну молекулу На02. Ilo, кроме того, могут также протекать и другие процессы, например радикал НО» может реагировать не по уравнениям (20) и (21), а следующим образом: 2НО2 = Н2О2 + О2 и 2Т1О2 = Н2О^О3. Неоднократно наблюдавшееся при самоокислении образование следов озона можно объяснить именно этим путем.
Подтверждением промежуточного образования свободных радикалок в неорганических реакциях рассматриваемого типа служит наблюдение Эванса (Evans, 1946), согласно которому полимеризация некоторых виниловых соединений, например стирола, о которой известно, что она вызывается свободными органическими радикаламиг возбуждается также смесью перекиси водорода и солей железа(т1), а также многими другими неорганическими системами, в Которых имеют место окислительные процессы. Этим методом Эванс (1947) сумел доказать, что, например, окисление солеи железа(Ц) свободными галогенами нэп гипобромитом протекает через промажу точное образование свободных радикалов:
Fc”_|_Br, — Fe*'* — Вт' —Hi; Fe ” j-HGBr _ Fe"'-| О1Г-|-Вг. (22) и (23) lie только химическом путем, но тг ,m.i учением годном может быть вызнано промежуточное образов.! min 1|Ю|7иД|1ых радикс юн. Ни пример, самоокисление сульфита натрия ускоряется и[Н| облучении его светом.
Па каждый световой квант hv, который при этом расходуется, нарождая — С отщеплением электронов — свободные радикалы SO) плв HSO3 па попа SO), многие десятки тысяч подов SO) на основе приведенных выше цепных реакций окисляются кислородом до ионов SO) (Biickstrom, 1927). Это наблюдение позволяет понять тот факт, почему часто вещества, вполне устойчрвые к кислороду воздуха в темноте, на свету им ОКИСЛЯЮТ!'я.
Под самооквелением, протекающим под действием света, понимают по только,, как уже было указано, процессы, протекающие сами ио себе, т. е. без затраты энергии. Хотя подводимая извив световая энергия в случаях, подобных приведенному выше, чрезвычайно мала по сравнению с производимым действием, эта энергия все же расходуется; речь идет, следовательно, не о каталитическом ироцессе. Вначале самоокислением считали (как об этом свидетельствует название абтбе от Само, добровольно) только такое окисление кислородом воздуха, которое протекает само но себе. В дальнейшем это ограниченно было оставлено, так как, как показывает пример с самоокислением сульфита, при протекании процесса в значительном масштабе характер возбуждения последнего имеет подминсНпоо значение.
В какой мере практически важнейшие самоакаслительиые процессы, особенно имеющие биологическое значение, протекают через нестабильные перекиси и в какой мере от! являются цоипыми реакциями, протекающими с образованием свободных радикалов, в отдельных случаях еще не вполне яспо. После исследований, в первую очередь Виланда, цепной характер некоторых биологически важных реакций самоокисления можно считать доказанным или, во всяком случае, очень вероятным. В результате работ Габера, Вейса, Эванса и Других исследователем теперь склоняются к тому, чтобы придавать решающее значение свободным радикалам в различных реакциях.
Гл а в а 17
СЕДЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Главная подгруппа)
ГАЛОГЕНЫ
Порядковый номер Элемент Сцм-uii/i Атомный вес Удельный вес Температура плавления, Тестера ту ра кипения, °C Теплота плавления Теплота-испарении Валентность
•ккалА -атолс
9 Фтор г И),(нД 1,108а -223 -187,9 0,17 0,80 1
17 Хлор Cl 35,457 1,57 — 102,4 -34,(1 0,81 2,2 г, 1II, IV, V, VII
35 Бром Вг 79, 916 3,14 —7,3 +58,8 1,76 3,82 I, III, V
53 Иод I 126,91. 4,942 +113,7 +181,5 2,0 7,45 Т, III, V, VII
85 Астат [Hi At 210 s — — — — — 11о-видимому :• как у иода
а -Для жидкого фтора (соответственно хлора) при точье нипенпл.
6 Значение для наиболее долге,живущего ияптопа (Период полураспада S,3 чж-).
Общие сведения. Главная подгруппа VII группы периодической системы включает элементы фтор, хлор, бром, под, а также нестабильный элемент астатин, который в незначительных количествах встречается в качестве промежуточного продукта радиоактивного распада (подробное см. т. Н). Астатин был обнаружен лишь недавно. Астатином назвали долгое время безуспешно отыскиваемый экаиод, т. е. более тяжелый аналог пода, существование которого следовало бы ожидать па основании периодической систедгы. Оказалось, что астатин действительно химически очень близок иоду. Однако точных данных о его свойствах и поведении еще мало.
Элементы фтор, хлор, бром и иод являются веществами с ярко выраженным неметаллическим характером. Фтор в хлор при обычной температуре газообразны, бром — жидкость, а иод — твердое тело. В газообразном состоянии все они образуют двухатомные молекулы и все очень реакционноспособны. Особенно это относится к наиболее легким из них. Первый элемент группы — фтор, вообще говоря, является самым реакционноспособным из известных элементов. Большая реакционная способность этнх элементов связана с тем. что их атомы стремятся перейти в отрицательные однозарядные ионы. Вследствие такой тенденции элементы этой группы соединяются с легкими металлами, у которых они отрывают электроны, образуя соединения с типичным характером солей. Элементы седьмой группы объединяют под названием галогены, т. е. солеобразователи (от oiXg — соль и ytvvav — производить)*. Обнаружившаяся способность
* Пазвапие «галоген» было впервые предложено в 1811 г. Швейгером для хлора вследствие его способности к сплеобразованию.
«26
Глава 17
этих веществ образовывать соли при соединении с металлами в период открытия галогенов была очень необычна; в то время считали, что существенной составной частью кислот, по крайней мере сильных, и типичных солей является кислород. Однако галогены могут образовывать бескислородные соли и бескислородные кислоты, а именно галогеноводородные наело ты., соединения типа НХ (X — галоген). Галогене водородные кислоты (за исключением фтористоводородной кислоты) — очень сильные кислоты, и не только в водном, но также, например, в спиртовом или эфирном растворе.
Соединения галогенов с сильными электроположительными веществами называют галогенидами. В этих соединениях галогены всегда одновалентны.
Помимо склонности к образованию отрицательных ионов (голозе-нид-ионов), для проявления большой реакциопноспособности галогенов имеют также значение сравнительно небольшие величины работ, которые требуется совершить для расщепления их молекул на атомы (см. табл. 113).
Свойства галогенов (ср. также табл, па стр. 825)
Таблица 113
Фтор Хлор йром ИОД
OKpacita в газообразном состоя-
ееПИ Слабо Желтовато- Красновато- Фиолетовая
зОоЧеновато- зеленая коричневая
желтая
Молекулярные объемы (жидк.
при т. кип.), мл 34 45 54 68
Атомный радиуса кристаллах, А — 1,07 1,19 1,36
Иоппый радиус в кристалла?;, А Момсггт инерцЕгЕ! молекул газа 1,33 1,81 1,96 2,20
(а - с.»-} 25,3-10'« ИЗ, 7-10-40 310-10-40 742-10*40
МежъядЕЕрноЕЕ расстояние в молекулах газа, А ....... 'Степень термической диссоциа- 1,135 1,998 2,284 2,67
ции, %
при 1000° К , 4,3 0,035 0,23 2,8
при 2000° К ........ 08,9 37 72,4 89,5
Энергия диссоциации (Х2—>-2Х) при 0 °К, ккал/молъ Сродство к электрону (ХЦ-е = 37,4 57,2 45,4 35,5
= Х"), ккал/г-атом . . . , . Нормальный потенциал, в (огне- 81,2 86,5 81,5 74.2
семный к нормальному водородному электроду при 18° С) +2,8 d-1136 ’ -4-1,08 +0,58
Теплота гидратации отрицательного иопа, ккяд/а-ион .... -Электролитическая подвнжпость 128 96,9 92,2 85,8
отрицательного иона при бесконечном разведении (при 18°) 47,6 66,3 68,2 66,8
Седьмая группа периодической системы
827
Величина сродства к олентрону, т. е. энергия, которая может производить работу благодаря присоединению электрона к свободному атому, имеет положительное значение у всех галогенов. От фтора к хлору она сначала увеличивается и затем слова уменьшается к моду. Значительно сильнее, как показывают даппые табл. ИЗ, уменьшаются в том же направлении величины нормальных потенциалов. Они являются мерой количества энергии, которая освобождается при переходе галогенов из газообразного состояния, т. е. из состояния двухатомных молекул, в состояние однократно отрицательно заряженных электролитических ионов. Отнесенный к нормальному водородному электроду Нормальный потенциал у всех галогенов положителен. Например, «хлорный электрод», если его поместить в раствор с 1 и. концентрацией ионов СГ, облагает на 1,36 е более высоким потенциалом, чем нормальный водородный электрод.
11 ри появлении тока положительное электричество течет от хлорпого электрода но внешнему проводнику к водородному электроду. У этого электрода водород переходит в раствор в виде положительного иона, в то время как у хлорного электрода хлор переходит в раствор в качестве отрицательного иона. Выделяемое таким путем при -образовании раз ба в ле пион соляной кислоты количество энергии составляет в соответствии с уравнением (3) (стр. 166) 31,3 ккал/моль IICI. Это количество равно сумме свободной энергии образования 1ТС1 и свободной энергии растворения НС.1 в воде. Вычитая последнее (8,6 ккал/.коль'), получаЕОт значение свободной энергии образования НС1, равное 22,7 ккал, в то время как спектроскопически было найдено значение 22,76. Значения нормальных потенциалов, приведенные в таблице, были измерены непосред--ствопно. Однако они могут быть рассчитаны также посредством кругового процесса, приведенного Eta стр. 171 и сл., из спектроскопически определенных значений энергий диссоциации ет сродства к электрону. Учитывая температурную зависимость значений энергии, получают, как показал Макишима (Makishima, 1935), хорошее совпадение рассчитанных таким образом величин с наблюдаемыми. При этом оказывается, что, как и в случаях, указанных в гл. 6 и 8, для значений нормальЕЕМХ потенциалов определяющими являются но супщству теплоты, гидратации.
Значения последних, приведенные в табл. 113, рассматриваются в предположении теплоты гидратации для 1 г-иона Н‘, равной 250 ккал (Ср. стр. 181).
Галогены могут образовывать соединения как с электроположительными, так и с илектроотрицателъными. элементами. За исключением фтора, они проявляют в них, помимо валентности единица, также более высокие валентности, а именно хлор и иод могут быть максимально семивалент-ныма, бром, насколько известно до настоящего времени,— пятивалентным.
В соединениях с электроотрицательными элементами галогены наряду с семивалентпым состоянием выступают также в пяти-, трех- и одновалентном состоянии. Хлор, кроме того, образует окисел, в котором он четырех -валентен (двуокись хлора СЮ2). С химической точки зрения реакции галогенов, приводящие к различным Валентным состояниям, а также их соединения могут быть лучше всего поняты, если рассматривать галогены в их соединениях с более электроотрицательными веществами электроположительно заряженными. В этом случае, например, обнаруживается правило, широко определяющее поведение галогенов, заключающееся в том, что последние в общем наиболее устойчивы в крайних, более всего различающихся валентных состояниях, т. е. когда они отрицательно одно- и положительно семнвалентны (бром соответственно пятивалентен). Однако иод в ряде случаев наряду с семивалентным проявляет и пятивалентное состояние-
С точки зрении, кюуии Косселя, отрицательную вазеитшк.ть галогенов, равную единице, н положительную валенткоегь семь можно о6ьясееить, исходя из их положения в таблице еео 11Тггишенлю к инертным газам, иди соотыет с тленно тем, что в их атомах имеется семв. элеЕГгропов с равным главным квантовым числом. Коссель предположил, что апешние оболочки атомов галогенов стремятся заполниться до оболочек инертных газов. Между тем. бы,лее определены величины сродства к электрону галогенов (хотя еще не очепь точно) (Mayor, 19.32). Положптс.иЕЛые значения этих величие (см. табл. 113) свидетелт.ствуЕот, что присоединение электрона к атому, т. е. переход атома ® отрицательный нон, в случае Е'алогепов идет с выделением энергии. Таким образом
828
Глава 17
объясняется особая стабильность соединений, в которых галогены участвуют в виде отрицательных ионов.
Для перевода галогенов в состояние положительных ионов, как и у всех атомов, следует затратить работу. Как показали спектроскопические измерения, эта работа для отЕЦОпления восьмого электрона галогенов значительЕЮ больше, чем работа, затрачиваемая для отщопл'еппя от первого до седьмого электроЕЩ галогена (ср. табл. 23, стр. 143). Так объясняется максимальная электроположительная валентность валогеновг равная семи. В случае фтора работа, требуемая для отщепления одного электрона, ужо очень высока по сравнению с работой, которая может быть произведена в результате электростатического притяжения, действующего между одновалентными ионами (ср. стр. 175 сл.). Отсюда попятно, почему фтор никогда не выступает в качестве электроположительной составгЕой части соединений.
То, что фтор никогда также пе обладает в гомеополярних сшщипепиях большей валеЕЕТпостЬЕЕ), чем единица, объясняется теорией Геймера — Лондона, так же как максимальная двухвалентность кислорода следует из при ее ди и а Паули. Напротив, аналоги фтора, также клее аналоги кислорода, могут проявлять в го мео полярных соединениях более высокие валенщости, так как вследствие более высоких г.чавЕЕЫх квантовых чисел их внешних эл(жтронов на внсепеесй оболочке их атомов могут располагаться болое чем 8 элеЕЕтронов. Правда, при этом часть внешних электроЕЕов должна перейти па энергетЕЕческик уровень с более высоким орбитальным кеейеетовым числом (<2-у ровен ь). Следовательно, соединения, в которых галогены проявляют более высокую валентность, чем единица, производятся не из основного состояния атома, а из возбужден пых состояний. Основное состояние атома соответствует у всех галогенов дублетному терму (3Рз;.,), г- е- в основном состоянии все электроны, кроме отгиото, спарены.
Относительно наиболее сильный электроположительный элемент среди галогенов, иод, мо;кет выступать в соединениях в качестве электроположительного монл, а именно электроположительного одновалентного иона, как в перхлорате пода НС1О4], а также трехвалентного — в последнем случае чаще всего в форме радикала иодила [10]+ (пример: иодилсульфат [l6]aSO4). Вероятно, в водном растворе в очень небольших количествах йод может существовать в качестве положительного нона Г. Его способность к образованию положительных ионов значительно возрастает благодаря соединению с органическими радикалами (образование иодо-нневых соединении, см стр. S'iS) или при присоединении нейтральных молекул (см. стр. 837 и с, ij, Таким путем удалось недавно стабилизировать в кичое’тпс по.пычПтелъногн одпоиалентцого иона также бром (см. стр. 838). В общем, однако, галогены нс выступают и электроположительном состоянии в качестве элементарных электролитических ионов, а соединяются с другими элементами, обычно с кислородом, давая отрицательные радикалы, которые часто очень устойчивы в водных растворах.
Величины энергии, которая должна быть затрачена для перевода молекул газа в свободные, оДЕНшратно положительно заряженные иоееы в соответствии с урав-неЕЕием — Х+ -f- с, в случае галогеЕЕОЕЕ (кроме фтора) существе ее его меньше, чем для водорода. Они были рассчитаны из значений энергий диссоциации и перВЕГЧных по) тенциалов ионизации (см. табл. 7, 22 и 113).
Для И+ F+ СР Вг+ Г
364 411) 327 298 258 ккал.;г-атом
То, что хлор, бром ЕЕ ИОД В ПРОТИВОПОЛОЖНОСТЬ водороду ЕЕ© образуют в водном растворе в значительном количестве положительные ионы, объясняется, ееозможно, сравнительна еесбольшой теплотой гндратацип для положительных ееонов гадогоЕЕов. Вероятно, ых теплоты гидр ат а инее лмеегь немного отличаются от те пл от гидратации Отрицательных шшов галогенов (см- табл. ИЗ), в то время ееэее ееоее водорода обладает очень высокой теплотой ПЕДратацпи. Когда образование соединений происходит в водном растворе, она покрывает большуЕО часть энергии, необходимой для расщепления молеЕ{ул на и ионизации атома Л. у галогенов соответствующая энергия может 6ыте, получена благодаря добавлению других веществ, молекулы которых присоединяются к до сожитель ее ому иону с ббльЕПИм выигрышем энергии, чем молекулы воды.
Седьмая ерунпа периодической системы
829
Как показывает опыт, в случае брома и иода могут быть получены соединения, в которых галогены существуют в положите л ыю одновалентном состоянии в качестве центральных атомов комплексов. Таким образом объясняется стабилизирующее действие комплексообрамоания. Это можно часто наблюдать в аналогичных случаях. Например, одновалентное золото не может также переходить в раствор в виде элементарного иона Ли.’, а только в виде комплексного иона (ср. т. II).
В случае золота энергия, требуемая дли образования свободного одиократноноло-житольного попа (газового иона) из элементов в твердом состоянии, даже 6o.te.ehc, чем для брома и иода. Энергия, требуемая для стабилизации положительного иона, может быть скорее всего получена присоединением к нему отрицательных ионов (в случае золота, папример, присоединенном ионов CN'). В случае галогенов, таким образом, объясняет’ее существенно большая стабильность но сравнении» с их положительными нонами ионов кислородных кислот, которые образуются при присоединении ионов 02“ е> галогенам. Возможно, что стабильность ионов, которые производятся от высших валентных состояний атомов галогенов, например ClOg и CIO.J, попытается за счет упрочения связей благодаря действию сил волпово-механичесЕ;ого резопанеа.
При сравнении между собой галогенов в общем наблюдается очень правильная вакономернпстъ в их поведении. Так, отрицательное электросродство, т. е. стремление заряжаться отрицательно, как уже было сказано, непрерывно уменьшается от фтора к иоду. Как следствие из этого элементарный фтор вытесняет все другие галогены из их соединений с металлами; хлор, напротив, разлагает только Соединения с металлами брома и иода, а бром — только соединения иода. Лежащий в основе этой реакции процесс всогдее заключается в зарядке вещества с более отрицательным электроеродством при посредстве находящегося в ионной форме вещества с менее отрицательным электросродсгпвом.
\.Р£4-С1- = К--!-ьгс1г;
1/гС1г-(-ВГ = С1* >/аВг2 и т. д.
Наоборот, из га л а refio кис л о родных кислот и их солен, т. е. из соединений, в которых галогены положительно заряжены, элементарный иод вытесняет хлор it бром, а элементарный бром вытесняет хлор. Большему отрицательному электро сродству соответствует меньшее стремление заряжаться положительно. Следовательно, уменьшение отрицательного электросродства проявляется у галогенов также и л положительном состоянии, одееяко в этом случае оно менее резко выражено, чем нри переходе из элсктронейтрального состояния в состояние отрицательного иона. Реакция между ялемептарным иодом и х,.юр;п-ионом гладко протекает при подходящих условиях:
13 с лучае рядом стоящих галогенов подобная реакция протекает гораздо реже* Однако существующее в главных подгруппах периодической системы правило о возрастании яле, i / 11 /I о 1игуье.ъы toe о та р а кулдера элсменп i оа с возр а с?на н исм атом ное о веса, песо м-[Eenrrij. проявляется также и в группе галогенов*
Далее, от фтора к иоду происходит правильное возрастание молекулярных объемов, а также атомных и ионных радиусов. Об этом свидетельствуют числа, при-веденные в i.ioa 113. В этом же направлении наблюдается и равномерное возрастание точек н,.гои.ifin 1Я п кипепия галогенов (см. обзорную таблицу в начале этой главы). Это возрастание Е'вялагго нее только с увеличением молекулярного объема, но также, вероятно, с уЕленыпЕЧшем взаимного насыщения атомов внутри молекулы в направлении от фтора к lE'j.ny. С неполным насыщением атомов в молекулах связапа также окраска галоЕёнов свойство абсорбировать видимый свет, благодаря которому появляется окраска, г. общем об пленяется наличием относительно слабо связанных электронов. Иитеневвтпп тг- 1.1гграшивания возрастает от фтора к иоду (ср. табл. ИЗ).
У иода при нысоких температурах можно отчетливо наблюдать расщепление молекул 12 на атомы. _М|)..нч;улы брома и хлора термически значительно труднее расщепляются на атомы; иди ее ко и у них при очень высокой температуре термическая диссоциация достигает эамётгЮи величины (см. табл. 113).
830
Г ла ва 17
Можпо ожидать, что увеличению кажущихся радиусов отрицательных элементарных ионов галогенов в кристаллах должно Соответствовать уменьшению электролитической подвижности, ионов галогенов. Однако в действительности электролитическая подвижность увеличивавшей, от нона фтора к нону брома, и только от последнего--несколько снова уменьшается к иону пода (см. табл. 113).
Причину возрастания электролитической подвижности от попа фтора к попу брома можно объяснить уменьшающейся гидратацией ионов, поскольку при переходе от хлора к брому в случае одного и того же катиона это проявляется в увеличении электролитического переноса воды в направлении к катоду (ср. табл. 17, стр. 100). Так как величина электролитической подвижности обусловливается двумя факторами, действующими в противоположных направлениях (а именно возрастание ионного объема — в направлении снижения, и уменьшение гидратной оболочки — в направлении повышения подвижности), то, вероятно, в этом и есть, причина, что в ряду электролитических подвижностей галогенов у иона брома проявляется максимум.
Как во всех гд явных подгруппах периодической системы, в подгруппе галогенов первый элемент (фтор) занимает особое положение по отношению-к другим элементам группы. Как было уже отмечено, фтор никогда не бывает заряжен положительно. Если сравнить свойства аналогичных по составу соединений галогенов, то особое место фтора опять-таки отчетливое проявляется. Так, фтористый водород отличается от других галогеповодо-родов заметно меньшей электролитической диссоциацией в водном растворе и, далее, своей склонностью к образованию «кислых солей» №НГ2. Фториды часто сильно отличаются от остальных галогенидов своей растворимостью. Хлориды, бромиды и иодиды щелочноземельных металлов — все очень легко растворимы в воде и даже отчасти расплываются. Наоборот, фториды щелочноземельных металлов труднорастворимы. Хлорид, бромид и иодид серебра практически нерастворимы, фторид серебра, наоборот, расплывается.
Следует, одиано, отметить, что даже от мода да серебра к хлориду серебра растворимость возрастает и что также от иодпдов шел очно земельных металлов к хлоридам’ растворимость уменьшается, правда лишь незначительно. Следовательно, при переходе от хлора к фтору во всех случаях наблюдается очень сильное изменение свойств, которое хотя и слабо выражено, олггзко паб издается в там же направлении в ряду аналогов фтора,
Галогены могут образовывать химические соединения между собой. Молекулы этих соединений содержат том больше атомов, чем дальше образующие их элементы отстоят друг от друга в группе галогенов.
Аналог иода с порядковым числом 85, «экаиод», существо в ан [те которого можно-было предполагать на основании периодической системы и который поэтому про до п-жптельпо искали, был обнаружен толь по педавпо.
Ранее считали, что он долзкоп встречаться в природе в качестве спутника иода.
Однако затем из правила стабильности атомных ядер (см. т. II) оказалось, чтО «экаиод» является нестабильным элементом, т. е. он радиоактивен. Такой элемент, если оп обладает незначительной продолжительностью жизни, в природе может встречаться только как член природных радиоактивных рядов распада (ср. т. II). Несколько-лет назад Карлику и Бернерту (Karlik, Bernert, 1 !)43 и сл.) удалось обнаружить, что действительно элемент с порядковым числом 85, т, е, экаиод, встречается как среди продуктов распада тория, так и среди продуктов распада урана (подробнее см. т. II). Этот элемент благодаря своей не устойчив ости получил название астатин (символ At, та dor «тот — неустойчивый). Относительно получения и свойств астатина см.: Johnson, Leininger, Segre, J. chem. Physics., 17, 1, 1949;
Структура решеток. В твердом состоянии га л о тепы* образуют молекулярную решетку" Межатомные расстояния в (двухатомных) молекулах этой решетки состав
* Структура решетки твердого фтора еще не известна.
Седьмая группа периодической системы
831
ляют: С1 <-> С1= 1,82 А (при —185°), Вг<->Вг = 2,27 А (при —150°), If—> 1 = 2,70 А (при комнатной температуре). Решетка 13 приведена па рис. 125. Это явно выраженная слоистая решетка. Таким образом объясняется построение кристаллов иода в виде листочков. Бром образует решетку аналогичного типа. В то время как 12 и Вт2 кристаллизуется в ромбической системе, С12 кристаллизуется в тетрагональной системе, причем особого типа.
Рис. 125. Элементарная ячейка решетки иода.
Распространение в природе. Галогены встречаются в природе в виде» своих соединений почти всюду, однако в очень различных количествах. Более всего распространены соединения хлора. Однако соединения фтора1 также встречаются в виде больших месторождений.
Фтор входит в состав минералов, прежде всего плавикового шпата (флюорита) CaFa, криолита Na3AlFs и апатита Ca5(POj)3(F, С1),. Хлор Содержится в морской воде в виде хлорида натрия NaCl и других щелочных и щелочноземельных хлоридов (ср. стр. 270). В виде таких же соединений находится он в соляных залежах, образовавшихся при высыхании морей. Иногда встречаются также хлориды тяжелых металлов, прежде всего в форме двойных соединении, таких, как атакамит ЗСч(ОН)2-СаС12. В составе земной коры (включая мировой океан) на долю хлора приходится 0,19%. Бром встречается обычно вместо с хлором в виде аналогичных соединений, но в значительно меньших количествах. На 200 вес. ч. хлора в морской воде приходится приблизительно 1 вес. ч. брома и х/ю вес- ч. иода. В аналогичном соотношении названные элементы встречаются в твердой земной коре. Иод находится в морской воде главным образом в форме органических соединений и меньше в форме иодидов. В залежах селитры Чили и Боливии иод присутствует в виде йодата, а иногда в виде перйодата. Элементарный под встречается иногда среди продуктов вулканической деятельности.
СВОБОДНЫЕ ГАЛОГЕНЫ
Исторические сведения. Первым среди галогенов в свободном состоянии был получен хлор. Шееле получил его впервые в 1774 г. окислением соляной кислоты пиролюзитом.
С точки зрения флогистонной теория Шееле правильно назвал его «дефлогисти-роваппой соляной кислотой». На основании того факта, что хлорпая вода выделяет яш
832
Глава 17
солнечном свету кислород с образованием соляной кислоты, Бертолло предположил позднее, что хлор является веществом, содержащим кислород, и дал ему название «acide murlotique axygene» (окисленная соляная кислота*). Однако после напрасных попыток Гей-Люссака и Тенара, а также Деви оторвать от хлора кислород, например при пропускании его иад раскаленным углем, последний в 1810 г. причислил хлор к элементам и предложил для пето название, оспованное па его собственной окраске, chloric gas плп Chlorine (нт WkbqsS — Желто-зеленый). Теперешнее более короткое название дано Гей-Люссаком, который вначале оспаривал мнение Деви, однако в 1813 г. после своих исследований пода присоединился к нему.
Иод был открыт в 1811 г. парижским фабрикантом селитры, но имени Куртуа, в соде, приготовленной из золы прибрежных растений. В 1813 г. он был подробнее исследован Клементом, Дезормом, Гсй-Люссаком и Дени. Гей-Люссак тогда же установил его аналогию с хлором. Он дал ему название но фиолетовой окраске Паров (IcogiSTjg — цвета фиалки).
Бром был открыт Баларом в 1826 г. в маточном растворе при приготовлении морской соли. Балар подробно исследовал его и обнаружил аналогию с хлором и иодом. Оп назвал его по резкому запаху (^gwpog — зловоние).
Вскоре после установления точки зрения Деви на хлор (1810) Ампер сделал предположение о существовании в плавиковой кислоте элемента, аналогичного хлору, т. е. фтора. Попытки многочисленных исследователей изолировать гипотетический элемент долгое время оставались безуспешными вследствие взаимодействия фтора со стенками сосуда, с водой, применяемой в качестве растворителя, и т. Д. Только в 1886 г. Муас-сану удалось получить фтор путем электролиза в аппарате из платины фторида калия, растворенного п безводном, сжиженном фтористом водороде- Элемент был назван по встречающемуся в природе его соединению с кальцием — плавиковому шпату, который в металлургических процессах служит в качестве флюса (fluo).
Получение. В ее попе ио, туче пня галогенов в общем случае лежит процесс
_\ \. 2е (X—галоген),
т. е. получение их происходит при разрядке ионов галогена. Эту разрядку можно осуществить либо электролитическим путем, либо химическим окислением. Иод получают также при восстановлении его кислородных соединении.
Фтор. Вследствие чрезвычайно высокого сродства этого эле Агента к отрицательным зарядам разрядку ионов фтора можно произвести только электролитическим путем. Уже было отмечено, что получение фтора впервые удалось осуществить Муассану электролизом фторида калия, растворенного в безводном, сжиженном фтористом водороде. Теперь пользуются электролизом расплавленного гидрофторида или дигидрофторнда калия.
В соответствии со способом Арго (1910) гидрофторид калия KIIF2 подвергают электролизу прп 240—250° в электрически обогреваемом не дном сосуде, который одновременно служит катодом. В качестве апода используют графитовый гптабик, окружен-
* В смысле данного в предыдущей главе расширенного определения окисления хлор действительно следует рассматривать как продукт окисления соляной кислоты (правильнее — хлористого водорода). Однако Бертой ле понимал под окислением (или оксигеяизацией) присоединили кислорода.
'е&ьмая группа периодической системы
833
ими «диафрагмой» в виде закрытого снизу, по имеющего боковые прорези медного Цилиндра. Диафрагму применяют для предотвращения доступа к аноду выделяющегося у катода водорода. В противном случае фтор снова с ним начнет соединяться со взрывом. На электродах происходят следующие процессы:
Анод: F- = y2F3 + г. Катод: 1Г + е = Ц2Н2.
По Фредеnxareiiy (1’i'edenhagen, 1928) с целью понижения температуры плавления вместо KF-HF целесообразно применять KF'2HF. Вместо сосуда из меди теперь применяют также сосуды из магния или монель-мета л ла. По способу Лебо и Дамье (Lebeau, Damien, 1925) электролизу подвергают KF-3HF при температурах в интервале 50—.100°. В качестве материала для анода применяют никель. По данным Вигелоу (Bigelow, 1936), для получения наиболее чистого фтора лучше всего проводить электролиз КН F2 при 250° в хорошо изолированном никелевом сосуде, применяя в качество материала для электродов графит.
Хлор. Хлор получают в больших количествах в качестве побочного продукта при электролизе хлоридов щелочных металлов (ср. стр. 208).
В лаборатории хлор (если его ие берут из баллона) получают обычно при слабом нагревании крепкой соляной кислоты (или смеси поваренной соли и умеренно концентрированной серпой кислоты) с двуокисью марганца МиО2
4НС1 4-МпОг=МпС1г4-2Н2О4-С12.
В принципе п период реакции здесь происходит окисление хлористого водорода двуокисью марганца. Ес ли этот процесс рассматривать по стадиям, то сначала образуется тстрихлорид марганца, который затем разлагается на хлорид марганца и хлор:
МпО2 -L 4IIC1 МцС14 + 2Н2О; МпСЦ = МпС12 CU
Ранее хлор в большом количестве получали в технике по способу Велдона (предложенному в 1866 г.). В этом способе н качестве окислителя.сначала также попользовали двуокись марганца, а затея се снова регенерировали из растворов хлорида марганца(Ц) (при обработке известковым молоком и продувании воздуха) в виде двойного соединения с окисью кальцпя, например СаО-МпО2. Это соединение йотом снова взаимодействует с соляной кислотой.
МлС12+ 2Са(ОН)2< /2О2 = СаО-МпО2ф- СаС12-ф 2Н2О.
СаО МпО2 ф- 6НС1 = CaCI2-f- МпС12 -ф С12 -ф ЗНаО.
Таким образом, в методе Велдона окислителем является кислород воздуха, как почти и во всех других способах технического получения, кроме электролитического.
Для окисления хлористого водорода можно также непосредственно применять кислород воздуха; этот метод используется в хлорном процессе Дикона (1808 г.). По этому способу смесь воздуха и паров соляной кислоты пропускают при температуре около 450° над глиняными ятарнкамй, пропитанными хлоридом меди СпС12. Хлорид меди действует как катализатор [[роцесса 2HCI Ч~ LO, = Н2О -ф С12. Несмотря иа то что по способу Дикона хлор получается в очень разведенном состоянии, этот метод оказался удобнее для приготовления хлорной извести и Др., чем способ Велдона. Однако теперь этими двумя способами хлор получают очепь редко, так как широкое распространение электролиза щелочных хлоридов временно вызвало даже его нере-iip о из во дет во.
Бром получают при пропускании хлора в маточные растворы, остающиеся прп переработке стассфуртских отбросных солей
2ВгтН-С12—2С1'Н-Вг2.
В США исходят отчасти из содержащих бром остаточных щелоков солевых производств. Там добывают бром также непосредственно из морской вода. Его высвобождают оттуда хлором при pH 3,5 и извлекают воздухом, затем восстанавливают SO2 до НВг, который обогащают методом абсорбции примерно в 4000 раз, после чего бром вновь вытесняют хлором. По аналогии с хлором бром можно также получать электролизом, однако предпочитают вытеснение хлором, так как последний имеется в избытке.
Иод в небольших количествах получают из золы водорослей, в которой он содержится в вице иодида; его получают так же, как и хлор, т. е. 63 г. Реми
834
Глава 17
либо электролизом (1), либо нагреванием с двуокисью марганца и серной кислотой (2).
2I' = Iz+2e (1)
2Ка14-ЗНг5О4+МпО2=2ГСа113О4+Мп8О4+2Н2О+12 (2>
Иод можно также выделять из щелоков при пропускании через них хлора 21' + С12 = 2С1' 4- 12; однако при этом возможны потери иода, так как в случае избытка хлора часть иода окисляется до йодата, который остается в рассоле.
Основное количество иода теперь получают из маточного раствора чилийской селитры, в котором он присутствует в виде йодата натрия. NalO s.
Иодат либо восстанавливают гидросульфитом патрия
2IO;+5HSO^=3HSOl+2SO![+II2O-|-l2, либо поступают рациональнее; сначала добавлением раствора гидросульфита патрня и сульфата меди осаждают иодид мсди(Т) Cui юг,+3hso^=3iiso;+г,
2Г -]~2Cu“+HSO J+H2O = 2Cul + HSO J ф-2Н-,
а затем иод освобождают из Cui нагреванием с серной кислотой и двуокисью марганца или окисью железа(Ш)
2CuI -ф- 6H3SO4 4" 2FeaO3 = 2CnSO4 4" 4FeSO4 4" 6H3O 4* О-
После того как выделившийся иод отфильтровывают, смесь сульфатов меди и железа(П) вновь применяют для осаждения иодида меди(1).
Физические свойства. Фтор — газ, в толстом слое окрашенный в слабый зеленовато-желтый цвет; он обладает резким, одурманивающим запахом, несколько напоминающим запах хлорноватистой кислоты. Вес литра газа составляет 1,71, его удельный вес относительно воздуха, принимаемого за единицу. 1.32. Ниже температуры —187,9° фтор сгущается в бледно-желтую жидкость удельного веса 1,108 со сравнительно высоким показателем преломления; точка затвердевания лежит при —223°.
Энергич. ди-социации моле;, yen F2, т, е. работа, необходимая для расщепления молекулы на .пимы, по новым исследованиям [Wahrhaftig, 1942; Schumacher, 1947; Does с her. 1952; Wise, 1952; Wkke, 1953] оказывается меньше, чем энергия диссоциации С13 и Вг2. Она составляет лишь 37,4 ккал/молъ (при 0° К) и, следовательно, только немного превосходит энергию диссоциации молекулы 12 (ср. табл. 113). Сродство к электрону атома Г также заметно меньше, чем сродство К электрону атома хлора; оно практически равно сродству к электрону атома Вт. Молекула Fa по сравнению с молекулами остальных галогенов и вообще по сравнению со всеми молекулами газов, за исключением молекул Не, Ne и 1Ь, отличается необычно малой поляризуемостью. Это объясняется особой стабильностью электронной оболочки фтора, которая также проявляется в чрезвычайно высоком значения потенциала ионизации атома F (400 ккал/г-атом). Этот потенциал ионизации превышают только потенциалы ионизации Не и No (ср. рис. 4 на стр. 28).
С точки зрения строения атома особенности молекулы F3 объясняются тем, что d'-электро иное состояние не проявляется в имеющейся в этой молекуле связи, в то время как в случае более тяжелых аналогов d-элсктроппое состояние заметно участвует в образования молекулярной связи.
Хлор — желто-зеленый газ с резким запахом, сильно действующий на слизистую дыхательных путей. Вдыхание хлора даже в незначительных количествах может привести к смертельному воспалению легких. Литр хлора весит 3,220 г. Следовательно, он в 21/а раза^тяжелее воздуха. Он легко сжижается при повышенном давлении (давление его паров ирн 20° составляет 6,6 атм). В продажу он поступает поэтому в сжиженной форме
Седьмая группа периодической системы
835
в стальных бомбах и цистернах. Морские перевозки его осуществляются даже в танкерах. При атмосферном давлении точка кипения жидкого хлора —34,0е, точка затвердевания —102,4°, Критические константы: ^ирит= 143,5 , р1(рит—76,1 атм, (4$рит 0, э7.
Бром — темпо-бурая, тяжелая жидкость (уд. вес 3,187 при 0°) с едким неприятным запахом; легко образует красно-бурые пары. Жидкий бром, попавший на кожу, вызывает болезненные раны. Пары брома сильно действуют па слизистую дыхательных путей и глаз и опасны даже в сильно разведенном состоянии. Точка кипения брома +58,8°. При —7,3° происходит затвердевание, в результате чего образуется бурая кристаллическая масса со слабым металлическим блеском.
Иод — твёрдое при обычной температуре вещество. Он образует темно-серые чешуйки с металлическим блеском удельного веса 4,942. Хотя его точка кипения лежит при 184,5°, уже при комнатной температуре он заметно летуч н обладает своеобразным запахом. Пары иода также ядовиты и вызывают сильное катаральное воспаление слизистой носа и глаз («иодный насморк»). Иод плавится при 113,7°. Одиако уже ниже этой температуры летучесть иода столь велика, что он при не слишком быстром нагревании обычно сублимируется без плавления.
Следует отметить (Villard, 1896; Dewar, 1898; Braune, Strassmann, 1929), что давление паров брома и иода в присутствии посторонних газов (например, Ng, О2, СО2) ваше, чем в вакууме.
Химическое поведение. Фтор — чрезвычайно активное в химическом отношении вещество. Будучи смешан с водородом, оп обычно* самопроизвольно воспламеняется (даже в темноте), большей частью с сильным взрывом. Фтор также соединяется уже па холоду с бромом, иодом, серой, фосфором, мышьяком, сурьмой, бором, кремнием, с древесным углем** и, кроме того, со многими металлами — с образованием пламени или с сильным раскаливанием. Некоторые металлы, например медь, па холоду или при небольшом нагревании реагируют только с поверхности, так как образующийся поверхностный слой препятствует продолжению'реакции.Однако при более сильном нагревании с этими металлами|фтор также энергично реагирует и в отдельных случаях, например с цинком, оловом, алюминием, реакция сопровождается сильной вспышкой. При температуре красного каления действию фтора подвергаются также золото и платина. Большинство химических соединений разлагается фтором, в том числе стекло и кварц. С аморфной двуокисью кремния фтор реагирует даже с воспламенением. Он превращает ее в тетрафторид кремния, отщепляя кислород. С сероводородом и аммиаком идет реакция'с образованием пламени. Галогс=иоводороды (кроме фтористого водорода) также энергично разлагаются фтором.
Из поды фтор вытесняет кислород, образуя фтористый"водород, при чем кислород отчасти выделяется в форме озона.
' Все :ип реакции объясняются ярко выраженным стремлением фтора переходить в отрицательно ллряжепттое состояние, в результате чего он способен отрывать электроны почти от всех других элементов. Фтор не реагирует непосредственно только
* См. irpiBieHiHirri..' *** па стр. 60.
** С графитом реакция идет только при краевом калении. Алмаз не реагирует еще и при 700а.
53*
836 Глава 17
с кислородом и с квотой, а с хлором взаимодействует лишь при зажигании. Однако если эти элементы находятся в отрицательно заряженном состоянии, то фтор отрывает у них избыточные электроны и выделяет их в элементарной форме
Na+CI-+J/.F2=Na+F-+72C12,
Si4+O<"+2F3 = 5Н+ГГ+О,.
Соединения фтора характеризуются особенно высокими значениями те плот образования (на грамм-эквивалепт; см., например, табл. 27 и 48). Наряду с высоким значением сродства к электрону определяющими здесь факторами являются небольшие размеры атомных и ионных радиусов. Этим фтор отличается от брома и иода, значения сродства к электрону которых не меньше, чем у фтора, но которые обладают существенно большими атомными и ионными радиусами. Особая агрессивность элементарного фтора объясняется как высоким значением тенлот образования его соединений, так и повышением скорости реакции расщепления молекул F2 на атомы, которое становится заметным уже при умеренно высокой температуре [ср. Wicke Е., Angew. Chem., 66, 701, 1954]j
Хлор также является в высшей степени активным химическим элементом. Однако по своей активности он стоит позади фтора. Так же как и фтор, хлор непосредственно реагирует с большинством элементов, но всегда менее энергично, чем фтор. Он не соединяется непосредственно с азотом и кислородом, а также с углеродом. Однако взаимодействие с этими элементами возможно обходным путем — через другие соединения. С кислородом он образует несколько окислив, правда все они в высшей степени нестойки. В углеводородах хлор замещает водород (частично или полностью). При этом одни атом молекулы хлора С12 соединяется с атомом водорода, а другой — с освободившейся валентностью атома углерода, например СН4 -j- С12 = НС1 + СН3С1. Хлор непосредственно присоединяется к ненасыщенным органическим соединениям, например
И2С = СН2-[-С12 = С1Н,С— СН2С1.
Влажный хлор обесцвечивает большинство органических красителей, так как образующаяся в качестве промежуточного соединения хлорноватистая кислота НОС1, окисляя, разрушает красители.
В воде хлор заметно растворим (2,3 об. в 1 об. воды при 20°). С водой он образует также продукт присоединения, который имеет состав С12-• 5:,/4Н2О (найдено 5,9±0,3 Н2О, Stackelbcrg, 1949). (О структуре см. стр. 248 и сл.) Хлор выделяется в виде зеленовато-желтых кристаллов при пропускании его в охлажденную льдом воду; однако в открытом сосуде эти кристаллы устойчивы только до 9,6°.
Хлор частично взаимодействует с водой с образованием хлорноватистой кислоты и соляной кислоты
С1з4-Н3О ^НОСЦ-НС! или С12-рОН' HOCl-j-Cl'.
[относительно механизма реакции см.: Morris J. С., J. Am. Chem. Soc., 68, 1692, 1946J.
Часть хлора, прореагировавшая таким образом, весьма значительна. При О3 в растворе, содержащем в 1 л 25 ммоль хлора в форме хлорноватистой и соляной кислот, находится 49% хлора, а в растворе, содержащем в 1 л 5 лмюль хлора,— 89%.
Хлорноватистая кислота постепенно распадается на соляную кислоту й кислород
НОС1 = НС1 > у2о3
Под влиянием света эта реакция значительно ускоряется. На прямом солнечном свету можпо поэтому легко наблюдать выделение кислорода из хлорпой воды. Это явление химики наблюдали уже далпо, и оно было причиной того, что многие вначале хлор принимали за соединение кислорода.
Седьмая группа периодической системы
837
Под влиянием электрического разряда при пониженном давлении можно получить хлор е атомарном состоянии (Rodebusb, 1933; Schwab, 1933). Так как хлор ужо в обычпом состоянии очень активное вещество, реакции атомарного хлора мало чем отличаются.
Бром по химическим свойствам аналогичен хлору, однако уступает ему по реакционноспособности. Как в парах, так к в жидком состоянии он соединяется непосредственно с большинством элементов, причем часто также с появлением пламени, например с фосфором, мышьяком, сурьмой, висмутом и оловом (станпиоль). Алюминий также очень активно взаимодействует с бромом. Различным образом ведут себя по отношению к брому золото и платина. В то время как золото легко переводится бромом в трибромид АпВг3) платина вообще с ним нс взаимодействует. Металлический натрий (в противоположность калию, см. стр. 178) также лишь слабо корродирует под действием1 брома даже при 200°.
На органические вещества бром действует так же, как и хлор, вступая в реакции замещения. Отбеливающее действие брома слабее, чем у хлора. С крахмальным клейстером бром образует оранжево-краспый продукт абсорбции.
В воде бром мало растворим (3,55 г в 100 г воды при 20°). НижеТ6,2° он образует с водой, как и хлор, гидрат. По Штакельбергу (1949—1952), этот гидрат обладает составом Вг2.72/3Г12О (о структуре см. стр. 248 и сл.) Легче, чем в воде, бром растворяется в некоторых органических растворителях, например в хлороформе и сероуглероде.
Водпый раствор брома, тотс же как и хлорная вода, выделяет иа прямом солнечном свету кислород
Вг2 Н2О = ИОВт -р НВт; IIО Вт = НВг -р !4О2.
Атомарный бром (монобром), который был получен Швабом (Schwab, 1934), подобно атомарному хлору, обладает особой склонностью к рекомбинации. Оказывается, что при каждом ударе о стенку’сосуда, независимо от ее материала, атомы брома снова соединяются в молекулу Вг2,
Иод гораздо менее активен, чем хлор и бром. Он непосредственно и энергично соединяется с некоторыми элементами, например с серой, фосфором, железом, ртутью; напротив, тенденция к соединению с водородом у него незначительна.
Скорость реакции иода с водородом при обычной температуре не может быть измерена. При повышенной температуре между йодистым водородом и составляющими его элементами устанавливается равновесие Ьд±2Н1 (ср. также табл. 114).
Как и остальные галогены, иод с кислородом непосредственно не соединяется. Косвенным путем он образует, однако, окисел 1гОй, Который в противоположность окислам других галогенов является экзотермическим соединением.
Б воде под мало растворим (1 : 5500 при 10°), он образует растворы со слабой буровато-желтой окраской. Он легко растворяется во многих органических растворителях: в сероуглероде, хлороформе и четыреххлористом углероде с фиолетовой окраской, в бензоле — с красной, а в некоторых кислородсодержащих органических веществах, таких, как спирт, эфир, ацетон,— с бурой окраской. Фиолетовые растворы содержат молекулы 1а. Обычно считают, что в других растворителях существуют соединения иода с молекулами растворителя (сольваты); однако, согласно Кортюму (Kortrim, 1947) н Рису (Rees, 1951), различие в окраске иода
838
Глава 17
в разных растворителях основано на различии сил взаимодействия между молекулами 12 и растворителем; совсем необязательно предполагать при этом образование стехиометрических продуктов присоединения. С крахмальным клейстером*, а также со слизистыми осадками основных ацетатов лантана или празеодима иод образует темно-синие продукты адсорбции.
Растворы иода в некоторых органических растворителях, например в бензофеноне, в котором Он растворяется с фиолетовым окрашиванием, обнаруживают электропроводность. В связи с этим можно заключить, что иод там отчасти расщепляется на ионы I' -f- Г. Па наличие такого расщепления указывают также про исходящие в подобных растворах реакции обмена. Например, иод, растворенный в бензоле или хлороформе, обменивается с перхлоратом серебра по уравнению
I2 + AglClOJ == Agl-М(С104].
Соль иода(1) удается стабилизировать присоединением образующих комплексы нейтральных частей, например пиридина, так что она может быть выделена в кристаллической форме в виде днпиридиниод(Т)перхлората [I(C5H5N)2] [С1О4]. Нитраты я: другие соли с одновалентным положительным иодом также можно выделить в виде таких комплексов. Солевой характер этих соединений доказан измерением электр о провод пости и движения попов. Оказалось, что аналогичные соединения образуются также и бромом. Они также могут проявляться в виде положительных электролитических ионов, если их стабилизировать в виде комплексов с нейтральными частями. Устойчивые на сухом воздухе, эти соли, однако, разлагаются водой. При действии раствора едкого натра соли иода(1) переходят в продукт присоединения с неизвестным в свободном состоянии окислом иода(1); например, из бесцветного II(C5TT5W)a] [N03l возникает оранжево-красный LO(C5H5N)4 (Carlsonn, 1924 и 1932 и сл.; Birckenbach, 1932 и сл.; Ушаков, 1931 и 1935).
Применение. В то время как ранее фтор применяли лишь в редких случаях — в исследовательских работах, а именно для приготовления фторидов, не получающихся другим путем или получающихся с трудом (Ruff), а также в качестве особо сильнодействующего окислителя (Fichteг)— сейчас он все в большей мере находит техническое применение. Прежде всего он служит (правда, болючей частью не в элементарном состоянии, а в форме фторидов металлов с высоким содержанием фтора, таких, как SbFa, BiF5, PbF4, CoF.j) для приготовления соединений, которые полу1-чают из алифатических углеводородов путем замены атомов И на атомы F, Даже если эти соединения состоят только из углерода и фтора, во многих своих свойствах они аналогичны углеводородам, однако они не горят и пе окисляются. Их применяют в настоящее время в качестве смазок, растворителей и жидкостей для холодильных установок (ср- стр. 480 и сл,).
Хлор, бром и иод весьма часто применяют в элементарном состоянии.
Хлор служит для приготовления многочисленных соединений хлора, как органических, так и неорганических, и прежде всего хлорной извести, которую в больших количествах используют для отбелки. В возрастающей степени его применяют для синтетического приготовления соляной кислоты (см. стр. 222). Большие количества хлора нужны, для стерилизация питьевой воды. В Северной Америке, где хлорирование питьевой воды было введено в 1912 г., в настоящее время его применяют уже всюду. Сточные воды также хлорируют для устранения запаха и гнилостных веществ. Все в возрастающей степени хлор применяют для извлечения олова из отходов белой жести, а также для приготовления хлорсодержащих органических соединений, которые во все увеличивающемся
* Кроме крахмального клейстера, известен еще ряд других чисто органических веществ, которые дают с иодом синюю окраску.
Седьмая группа периодической системы
839
объеме используют в качестве растворителей для лака и как синтетические материалы. О применении хлора для получения брома уже было сказано.
Бром применяют для приготовления неорганических и главным образом многочисленных органических соединений брома. В лабораториях вследствие относительного удобства употребления его часто используют вместо хлора в аналитических и препаративных целях. Его применяют также в целях дезинфекции. Бромид и броматжилия применяются в бро-мометрип и броматометрии.
Иод в лаборатории применяют для иодометрии, а также в препаративной органической химии. Далее, он служит для Приготовления неорганических и многочисленных органических соединений иода. Многие такие соединения, так же как элементарный иод, используются в медицине, так как иод обладает исключительным антисептическим н к тому же кровоостанавливающим действием. В то же время он действует раздраягающе на кожу. Большие количества иода, принятые внутрь, действуют отравляющим образом.
СОЕДИНЕНИЯ ГАЛОГЕНОВ
1. Галогеноводороды
Галогеноводороды — бесцветные газы, довольно легко превращаемые в жидкости. Фтористый водород сжижается уже при 19°. Водой галогеноводороды в высшей степени жадно поглощаются. Водные растворы их очень хорошо проводят электрический ток. Хлористый, бромистый и иодистый водород в водных растворах почти нацело диссоциированы
нх Н’-фх'. (3)
В значительно меньшей степени диссоциирована фтористоводород-мая кислота. Это видно уже из приведенных в табл. 114 отношений электропроводностей AcZAe, хотя они дают только кажущуюся степень диссоциации.
Электролитическая диссоциация фтористоводородной кислоты, как кислоты сравнительно слабой, подчиняется химическому закону действия масс. Однако иопы F', образующиеся при диссоциации ио уравнению HF Н’ -J- F', при соединяю тс я частично (особенно в концентрированных растворах) к недиссоциироваиным молекулам HF ; F' -|- HF HFj. Поэтому концентрации ио вок а водных растворах фтористого водорода определяются равновесиями, которые задаются обоими представ ленными уравнениями. Константы этих равновесий еще ие вполне надежно определены. 'Согласно измерениям (Pick, '1912; Hndleston, 1924; Rolh, 1937—1939), в качестве предварительных можно принять следующие значения:
jHlxfF'1 10-4. №]
[HF] 1 [F'J X [HF]
В 1 и. растворе примерно 10% фтористого водорода находятся в форме ионов HF^ и 1%—в форме попов F'. Следовательно, в концентрированных растворах существует преимущественно р, in попсе ин
2HF H4-HFS, (4)
Ранее предполагали, что в по слишком разбавленных водных растворах фтористоводородной кислоты содержатся преимущественно двойные молекулы (HF)2 и что -опа диссоциирует как двухосновная кислота. Однако это предположение пе подтвердит лось. Данные по молярной электропроводности нейтральных фторидов и их завпеи-мость от концентраций показывают, что это соли одноосновной Кислоты, а не двухосновной. Так называемые кислые фториды или гидрофториды, т. е. соединения общей
840
Глава 17
Свойства галогеноводородов
Таблица 114
Соединение HF нет НВг HI
Теплота плавления, °C .... -83,1 -114,8 -86,9 —50,7
Температура каления, °C ... +19,54 -84,9 -66,8 -35,4
Критическая температура, °C +230,2 +51,3 +91,0 +150,5
Теплота плавления, ккал/молъ НХ (при т. нл.) 1,094 0,505 0,575 0,686
Теплота испарения, ккал/моль НХ (при т. кип.) 1,850 3,85 4,210 4,72
Константа Трутона, Х/Лч .... 6,3 20,4 20,4 19,9
Плотность (при т. кип.) .... 0,991 1,187 2,160 2,799
Поверхностное натяжение у (при т- кип.), дип/см ....... __ 23,18 25,40 26,69
Вес 1 л газа (при 0°), а .... .— 1,6391 3,6443 5,7888
Теплота образования (при 20° п пост, давлении), ккал/молъ НХ +64,4 +21,9 + 7,3 +1,32 а
Свободная энергия образования Л/ (при 25°), ккал/мМъ .... —65,0 —22,75 —12,86 —1,85 ®
Степень термической диссоциации (при 1 атлг), % при 300° 0,3-10-а 0,003. 19
при 1000° —' 0,014 0,5 33
Теплота растворения (при 205), ккал/моль -+12,4 + 17,4 +20,5 +19,6
Кажущаяся степень электролитической диссоциации ACAO в 0.1 ы. растворе (при 18°). % 10 92,6 93,5 95
Энергия ионизации (НХ—>П++Х“ в вакууме), ккал^молъ .... +304,2 +328,2 +327,7 -|-316,3
Дипольный момент газовой молекулы, р (в ед. CGS) .... 1,91+0-*® 1,04+0-*® 0,79+0'*® 0,38+0’*»
Момент инерции молекулы газа, а-сж2 1,35-10^0 2,65-IO-™ 3,26-10'« 4,3+10'*»
Межъядерноо расстояние Н.ч—>Х в молекуле таза, А 0,92 1,276 1,4Ю 1,62
Магнитная восприимчивость (при 0°), %мол-10’ —8,6 -22,1 -32,9 -47,7
а Значение рассчитано для образования HI из газообразного иода. Для образования из-тпеербого иода W——£.,23 и Г=вЛб ккалулюль HI £при пост, давлении и 20 или. 26°).
формулы Mi[HF2], можно совершенно удовлетворительно объяснить как координационные соединения М1 [HFg] с водородом в качестве центрального атома комплекса. Тот факт, что ионы фтора легче, чем другие иопы галогенов, образуют с водородом комплексы [HF2]', легко объясняется существенно большей энергией образования, которая соответствует фторидному комплексу, вследствие незначительного радиуса нона фтора по сравнению с радиусами ионов других галогенов.
Седьмая группа периодической системы
841
Процессы, представленные уравнениями (3) и (4), точнее могут быть сформулированы следующим образом
нх+НгО^*[Н3ОГ + X- (а)
в
2hf ч- що^тзог + 1ВД'. (6)
Электролитическая диссоциация галогеноводородов в водном растворе обусловлена тем, что водородные ядра удерживаются элоктронейтральнымп молекулами воды не намного слабее, чем однократно отрицательно заряженными нонами галогена. Л так как, кроме того, выделяется еще энергия при гидратации ионов галогена, то в целом реакция (5) является энергетически выгодным процессом*. То же оказывается справедливым и в случае других сильных кислот. В неводных растворах диссоциация, если опа имеет место, ташке происходит благодаря присоединению в о дородных ядер К молекулам растворителя. Чистые галогеноводороды не просадят электрического тока, и в отсутствие воды при обычной температуре опи также очень мало реакционноспособны.
Поэтому сомнительно, чтобы галогеноводороды были построены как гетерополяр-ные соединения, тем более что другие факты свидетельствуют, по-видимому, о их еомеополярном строении. Во-первых, энергия, необходимая для расщепления кристаллических соединений ла свободные (т. с. бесконечно удаленные) ионы в случае гало-геповодородов (сумма теплоты сублимации и работы ионизации в табл. 114), значительно больше, чем энергия, которая должна быть затрачена для расщепления лонном решетки щелочных галотепвдов («энергия решетки», см. стр. 181), несмотря на то, что молекулярные объемы кристаллических галогеноводородов имеют приблизительно ту же величину, что ц молекулярные объемы щелочных галогенидов. Во-вторых, о го мео-пол ярном строении галогеноводородов свидетельствует, по-видпмому, незначительная величина теплот сублимации по сравнению с величинами работ ионизации. У галогенидов щелочных металлов теплоты сублимации составляют примерно 30% величин энергии решеток, у галогеноводородов — лишь 1—2%. (Это существенное различие выражается также в значительно большей летучести галогеноводородов по сравнению с галогенидами щелочных металлов.) В-третьих, указанно на гомеополярнос строение галогеноводородов усматривают также в большом различии между их точками плавления и точками плавления галогенидов щелочных металлов. Это и другие связанны© с ним свойства не дают, однако, определенного доказательства о преимущественно гомео-полярном характере связи, а следовательно, ц о преобладании действия сил полно в о-механнчеслого резонанса лад притяжением, обуеловлепяым стапвопармямп зарядами; эти свойства можно объяснить также при помощи представления, что у галоееноводородск ядро атома водорода проникло сквозь внешнюю электронную оболочку внутрь отрицательного иона. Об этом свидетельствуют значения, которые получают Для моментов инерции молекул галогеноводородов различными методами (например, спектрографическими измерениями). Вычисленные на этом основании расстояния менаду ядром водорода и ядром отрицательного ио на в молекулах галогеноводородов приведены в табл. 114_ Из данных таблицы следует, что межъядерные расстояния много меньше, чем радиусы ионов галогенов (ср. табл. 113). Путем внедрения ядра водорода внутрь иопа подучают молекулу галогеноводорода, построенную аналогично инертному газу. Однако молекулы галогеноводородов в противоположность атомам инертных газов обладают достаточно высокими дипольными моментами (см. табл. 114). Иа основапнп дипольных моментов газообразных галогеноводородов Смит [Smyth Р., J. Am. Chem. Soc., 68, 171, 1946] заключил, что химическая связь у IIF гетеронолярна на 43%; у НС1 — иа 17%, у ЛВг — на 12% и у HI — на 5%; однако по мнению Робинзона [В о b 1 n s о п J.Chem. Phys., 17, 1022, 1949], по дипольным моментам нельзя сделать заключение
* Например, из данных, приведенных в табл. 113 и!14, а также на стр. 181 (примечание), оказывается
ЦС1 = Н+4-СЬ — 328,6 ккал
1Г-|-НгО-]-ад = [Н3О]‘ aq + 250 ккал
Cl~-\-aq~Cl'&<[-{-98,9 ккал
IIC1 J-II2O-|-aq==[H3O]‘*aq-|-Cr aqJ-18,3 ккал
Экспериментально же, наоборот, теплоты гидратации ионов получают из тецлот растворения и работ ионизации соединений.
842
Глава 17
относительно доли ионной связи в галогене водородах*. ОтноситеЛ1,но дипольного момента HF, НС1 и НВг в растворах cu.:Weith, Hobbs, Gross, J. Am. chem, Soc., 70, 805, 1948.
В твердом состоянии галогеповодороды образуют молекулярную решетку. НС1 и НВт кристаллизуются выше 98°, соответственно 111° К — в кубической гранецентри-.рованнои решетке (как инертные газы, ср. рис. 46), при более низкой температуре — в ромбической решетке (выше 119° К НВг существует в третьей модификации с достаточно сложной решеткой); 1П кристаллизуется в тетрагональной грапецентрировэнной решетке (Simon, 1924 и сл., Natta, 1931 и сл.). Помимо точек превращения, обусловленных изменением структуры решетки, кристаллические галогеноводороды обладают также еще другими точками превращения, которые свидетельствуют о появлении свободного вращения молекул с возрастанием температуры.
В изменении точек плавления, точек кипения, критических температур и т. д. у галогеноводородов проявляются такие же аномалии, как и у халькогеноводородов (ср. стр. 738). Так же как там, эти аномалии обусловлены полимеризацией воды, здесь они обусловлены полимеризацией фтористого водорода в жидком состоянии. Однако константа Трутопа, которая для воды имеет аномально высокое значение, в случае -фтористого водорода имеет аномально низкое значение (см. табл. 114). Это происходит оттого, что фтористый водород при обычпом давлении даже в газообразном состоянии -шшьно полимеризован. При понижении давления происходит расщепление па простые молекулы HF. Поэтому теплота испарения фтористого водорода с уменьшением давления сильно возрастает. При давлении 20 мм рт ст она составляет 390 кал!в или 7,80 ъкал!молъ HF (Fredenhagen, 1934). Отсюда получается ?,/7’s = 26,6, следовательно, аномально высокое значение для константы Тручона, как это и можпо было ожидать вследствие полимеризации в жидком состоянии.
Следует отметить, что в газообразном фтористом водороде наряду с молекулами i(HF)2 имеются также в значительном количестве более высоко ассоциированные молекулы, а именно главным образом молекулы (HF)?, (HF)0 и (HF)8 (Brieglob, 1953). При более низких температурах (и соответственно уменьшенном давлении), например при 4° и 300 ,н.к рт ст, последние преобладают по сравнению е молекулами (HF)3. Они образуются ири посредстве водородных связей и, вероятно, имеют преимущественно вид цепей. Эти соединения представляют единственный известный до сих пор пример ассоциации в газообразном состоянии с образованием цепей посредством водородных связей; С высокой степенью ассоциации молекул фтористого водорода -связано то, что газообразный фтористый водород по наблюдениям Франка (Franck, 1953) обладает При низких температурах сравнительно высокой для газа и сильно зависящей от давления теплопровод г гостью. Прн О'3 и давлении в интервале 100— 200 мм рт ст. она достигает даже тгимопрОЕйДиоети жидкой воды.
Термическая диссоциация галогеноводородов щ> общему уравнению 2НХ^±Н3-|-+ Х2 очень сильно возрасте т от хлористого водорода к подистолту водороду. В то время как для хлористого и бромистого водорода при 300° распад на составные части юще не может быть прямо обнаружен**, для йодистого водорода при этой же температуре оп уже весьма значителен. Подучающееся отсюда сильное уменьшение сродства галогенов к водороду с возрастанием атомного веса проявляется также в значительном уменьшений теплою образования от фтористого водорода к иодистому. Последние в случав галогеноводородов дишьщемного отличаются от велнчип свободной энергии •образования (СМ. табл. 114).
Получение галогеноводородов можно осуществить следующими методами:
а) прямым взаимодействием элементов, папример
С12-рн2=2ИС1. - (7)
Этот в принципе самый простой метод имеет сейчас большое значение в качестве способа получения соляной кислоты (ем. ниже);
* Как показал Ритнер [Rittner Е. S., J. Chem. Phys., 19, 1030, 1951 [, значения дипольных моментов, энергий связи и колебательных частот газообразных молекул галогенидов щелочных металлов хорошо Совпадают с экспериментальными данными, если их рассчитать при предположении чисто ионной связи (принимая во внимание поляризацию); однако доля иошгой связи, рассчитанная по Полингу из измеренных дипольных моментов, должна составлять в среднем только примерно 70%.
** Приведенные в табл. 114 значения вычислены из данных, найденных при более высоких температурах па основании температурной зависимости распада.
Седьмая группа периодической системы
843
б) действием кислот на галогениды.
Это важнейший метод получения плавиковой кислоты, а также все еще самый употребительный метод получения соляной кислоты:
CaF2+H2SO4=CaSO44-2HF, (8)
NaCl + H2SO4=NaHSO4+HC]; (9)
в) гидролитическим разложением галогенидов:
РВг3 4- ЗН2О = Н3РО3+ЗНВг; (10)
г) действие водородных соединений, например H2S, на свободные галогены в водном растворе:
I2-UH2S‘a<I = 2HLaq4-34-17,5 ккал. (11)
Оба последних приведенных метода применяют главным образом для приготовления бромистоводородной и, иодистоводородной кислот. Обмен (11) основан на том, что теплота растворении Н1 существенно больше, чем теплота растворения H2S. В газовом состоянии реакция протекает в обратном направлении
’ 2111 4~ S — I; 4- HsS 4" 5,0 ккал.
Этот процесс можно использовать для получения чистого II2S.
Фтористый водород HF обычно получают нагреванием фторида кальция (плавикового пшата) с концентрированной серной кислотой [см. выше уравнение (8)]. Так как HF действует на стекло, то получение его проводят в платиновом или свинцовом сосуде. В безводном состоянии фтористый водород можно получить нагреванием гидрофторида калия или натрия KHF2 = KF 4- HF.
Ниже 19,54° фтористый водород образует бесцветную, весьма подвижную, сильно дымящую жидкость; выше этой температуры фтористый водород представляет собой бесцветный газ, который при обычной температуре и атмосферном давлении, как уже было отмечено, сильно полимеризован. Однако прй пониженном давлении и более высокой температуре наступает достаточно быстрая диссоциация на простые молекулы HF. При 90° имеются почти исключительно простые молекулы HF.
В ясидком состоянии фтористый "водород равным образом сильно ассоциирован -(см. выше). Безводный Жидкий фтористый водород не проводит электрический ток. Диэлектрическая постоянная /КПДКОГО фтористого водорода, согласно Фреденхагопу (Fredenhagen, 1929), почти так же велика, как у воды. Жидкий фтористый водород можно хранить в серебряной, медной или стальной посуде.
Фтористый водород сильно растворяется в воде (при обычной температуре они смешиваются неограниченно). Раствор, называемый плавиковой кислотой, растворяет большинство металлов с выделением водорода; ои растворяет также стекло, так как влажный фтористый водород реагирует с двуокисью кремния и с силикатами, образуя летучий тетрафторпд кремния S1F-. Совершенно сухой фтористый водород не действует на стекло даже в жидком состоянии и реагирует с большинством металлов только при высокой температуре. На способности,плавиковой кислоты разлагать двуокись кремния и соединения двуокиси кремния основано ее применение для вскрытия силикатов и травления стекла. Обнаружение фторидов при помощи пробы травления (ср. стр. 852) также основано на свойстве плавиковой кислоты разъедать стекло. На золото и платину плавиковая кислота не действует, свинец она разъедает только с поверхности. Поэтому для приготовления плавиковой кислоты или при работе
844
Глава. 17
с ней пользуются посудой из платины, свинца или каучука; однако для многих целей пригодна также парафинированная стеклянная посуда.
Кроме травления стекла, плавиковую кислоту применяют в технике для удаления кремния из труб, а в литейном деле для удаления приставшего к литые песка. Опа служит также для приготовления перекиси водорода из перекиси натрия. Вследствие дезинфицирующего действия ею пользуются в пивоваренном производстве для сохранения дрожжей. Иа этом же основано ее применение для консервирования анатомических препаратов.
Торговая плавиковая кислота чаще всего содержит 40% HF (уд. вес 1,130 при 20°). Она выделяет на воздухе крайне резко пахнущие, очень ядовитые пары. Пары, так же как и сам раствор, сильно разъедают кожу и вызывают весьма болезненные раны.
Если нагреть концентрированную плавиковую кислоту до кипения, то сначала будет выделяться фтористый водород. Одновременно возрастает температура кипения, и при 120’ начинает перегоняться раствор постоянного состава, а именно 35,4%-ная кислота (уд. вес, 1,118). Если исходить из более разбавленного раствора, то в связи с идущим испарением воды удается получить кислоту той же концентрации.
Хлористый водород НС1 и его водный раствор — соляная кислота были получены впервые Глаубером (XVII в.) при нагревании поваренной соли с серной Кислотой. Этот метод получения, как правило, до сих пор еще используют в лаборатории. Его также преимущественно применяют в технике; однако все в большей мере сейчас используют синтетическое приготовление соляной кислоты при сжигании водорода с хлором.
Обменное взаимодействие серпом кислоты с повареппой солью протекает в две стадии. На холоду с выделением хлористого водорода образуется сначала в соответствии с уравнением (9), стр. 843, гидроеульфат натрия NallSOj. При нагревании (до температуры красного каления) реакции идет дальше. При обмене гидросульфата натрия с поваренной солью снова образуется хлористый водород NaCl -Ь NaHSO4 = =Na2SO4 -ф- НС1.
Синтетическим методом со ляпе ю кислоту приготовляют из газов, полученных в качестве побочных продув гон прп электролизе хлоридов щелочных металлов — водорода и хлора. Эти газы сжигают в кварцевой шахте, причем: получают особенно чистую соляную кислоту.
Хлористый водород — бесцветный газ с резким запахом и вкусом (физические константы см. в табл. 114). Плотность газа относительно кислорода равна 1,1471, что соответствует молекулярному весу 36,71, в то время как рассчитанный по формуле НС1 молекулярным вес оказывается равным 36,47. Следовательно, хлористый водород при обычной температуре состоит из простых молекул НС1. Его можно достаточно легко перевести в жидкое состояние. Даже вблизи температуры сжижения плотность ^аза все еще близка к нормальной-
Водой хлористый водород жадно поглощается в больших количествах п с сильным выделением тепла (см. табл. 114). При атмосферном давлении 1 об. воды при комнатпой температуре может растворить около 450 об. хлористого водорода.
При сильном охлаждении в зависимости от состава раствора из растворов кристаллизуются различные гидраты: НС1-ЗН2О (т. пи. —24,9°), HGL 2НгО (т. пл. —17,6°) и НС1-Н2О (т. пл. —15,3°). Правда, из раствора, насыщенного приД° хлористым водородом под давлением 1 атм, может выделиться только тригидрат (с содержанием 40,3% HCI). Остальные гидраты выделяются из растворов, насыщенных хлористым водородом под давлением выше атмосферного. Раствор, насыщенный хлористым водоро
Седьмая еруппапериодической системы 845
дом при атмосферном давлении, при 0° содержит 45,4 вес.%* НС1, а при 15° — 42,7 вес.%. Если такой раствор нагреть, то сначала выделяется хлористый водород, пока при температуре около 110° нс достигается смесь достоянного состава с содержанием хлористого водорода 20,24%. Смесь того же состава можло получить, если исходить из более разбавленных растворов. Однако, как показал в 1859 г. Роско, состав смеси, кипящей при постоянной температуре, зависит от давления, при котором производится перегонка. Следовательно, здесь мы не имеем дело о определенным химическим соединением, как предполагали ранее (ср. стр. 643).
Водные растворы хлористого водорода обычно называют соляной кислотой. Содержание в ней хлористого водорода устанавливают чаще всего посредством ареометра.
Соляная кислота
i удельного веса 1,060 1,124 1,16 1,19
при 15° содержит, %НС1 12,2 24,8 31,5 37,2
Торговую кислоту называют «разбавленной», если она содержит, иапрщгер, 12,2% ИС1; если она содержит 24% и больше НС1, ее называют «концентрированной соляной кислотой». В лаборатории разбавленной соляной кислотой обычно называют 2 и. (7%-пую, уд. вес 1,035). Как показывают приведенные выше данные, достаточно точное содержание раствора хлористого водорода (%) можно получить, если удельный вес, уменьшенный на единицу, умножить на 200,
Техническая торговая соляная кислота окрашена примесями в желтый цвет (главным образом хлоридом Железа). Чистая соляная кислота, напротив, совершенно бесцветна.
Соляная кислота обладает очень высокой электропроводностью. Ее удельная электропроводность и для различных концентраций при 18° имеет следующее значение:
Содержание соляной
кислоты, % 5 10 20 30 40
к 0,395 0,630 0,7615 0,662] 0,515
Таким образом, удельная электропроводность достигает максимального значения в области средних концентраций так же, как и в случае серной кислоты.
По химическим свойствам соляная кислота является типичной сильной кислотой. В качестве сильной кислоты она находит широкое техническое применение, например для получения хлоридов металлов, хлорида аммония и для многих других целей, В лаборатории соляная кислота относится к наиболее употребляемым реактивам; ее применяют также в медицине.
Вследствие сильного сродства к воде газообразный хлористый водород применяют иногда как водуотнимающее средство или как конденсирующее средство. О влиянии HCI на растворимостъ соединений см.: Fischer W., 7.. anorg. Ghem., 247, 384, 1941.
Помимо воды, хлористый водород сильно растворим также в спирте, в эфире н еще во многих других жидкостях. Наоборот, жидкий хлористый водород может служить раствори гелем для спирта, эфира и многих других веществ. Однако на большинство металлов жидкий хлористый водород не действует, оп не реагирует в общем также с окисламп, сульфидами и карбонатами. Газообразный хлористый водород при
* Поскольку в лаборатории высококонцентрированную соляную кислоту приготовляют чаще всего прокус капнем газообразного НС[ в открытый сосуд, охлаждаемый ледяной водой, его значение по лполнп достигается, а получают тогда чаще всего приблизительно 42,5 % -ну ю кислоту.
846
Глава П
температуре каления реагирует с выделением водорода с металлами, причем дако с такими металлами, на которые водная соляная кислота без доступа воздуха не действует, например с медью и серебром. В то время как с фтором хлористый водород немедленно взаимодействует уже при обычпой температуре с образованием пламени, с кислородом воздуха он реагирует только в присутствии катализаторов, причем обычно только при повышенной температуре. Однако равновесие 4НС1 4- Ог5*2НгО-|-: + 2С12 сдвигается при возрастании температуры влево, так что при температуре около-600° коэффициент равновесия по закону действия масс
7;НС1 х Ро2
Риф х р£Л«
достигает единицы. Следовательно, выше этой температуры более сильным окислите- . лем является хлор, а ниже — кислород.
Бромистый водород НВг можно получить, как и хлористый водород, обменным взаимодействием бромида калия с серной кислотой. Однако последняя в этом случае должна быть умеренной концентрации (уд. вес 1,4), так как иначе она частично будет окислять образующийся бромистый водород до брома. Получаемый таким способом водный раствор бромистоводородной кислоты удается с концентрировать путем фракционированной перегонки до содержания в нем 48% НВг.
Во л ее концентрированную кислоту или газообразный бромистый водород можно получать гидролизом трех бромистого фосфора [уравнение (4), стр. 843J, При этом обычно поступают так; сначала в колбу помещают красный фосфор, смачивают его водой, приливают по каплям бром и затем смесь осторожно подогревают, в результате чего выделяется НВт. Удобным способом получения водной б ромист.о во дородной кислоты является также и тот, при котором смесь брома и воды разлагается сульфидом бария 4Вг2 + 4И2О ф- BaS= BaS04 4- 8ИВг. По окончании реакции, которая сопровождается сильным выделением тепла, отгоняется водная кислота. '
Бромистый водород — бесцветный газ. чрезвычайно похожий по своим, свойствам па хлористый водород, но легче его окисляющийся, отчего водный раствор бромистого водорода па свету постепенно окрашивается, в бурый цвет.
Растворимость бромистого водорода в воде очень велика (600 об. в 1 об. воды; при 0°). При низких температурах образуется несколько кристаллогидратов (НВг-4П2О, т. пл. —56°; НВг-ЗН3О, т. пл. —48’, НВю2НЕ0, т. шг. — 11,3° и НВг-Н2О;, последний под давлением 1 пики разлагается при —28,5°).
Йодистый водород HI еще легче окисляется, чем бромистый водород,, так что его можно отнести к яркой ыраженным восстановителям. В остальном он очень похож па бромистый и хлористый водород. Как и другие галогеповодороды, иодистый водород — бесцветный резко пахнущий газ-и в водном растворе является сильной кислотой.
1 об. воды может поглотить при 10° и атмосферном давлении приблизительно 425 об. газообразного HI. Существуют три гидрата: Н1-4НгО, т. пл. —36,5°; Н1-ЗН2О, т. пл. —48° и 1-П-2И2О, т. пл. около —42°. Раствор, насыщенный йодистым водородом при комнатной температуре и атмосферном давлении, содержит приблизительно 70% HI и обладает удельным весом 2,00. При перетопке образуется раствор, кипящий при постоянной температуре, равной 127°, о содержанием 57,0% И1 (уд. вес 1,70). Концентрированная подпето води родная кислота растворяет серебро с выделением водорода. Хотя серебро стоит в ряду напряжений правее водорода, эта реакция происходит благодаря тому, что образуется достаточно прочное комплексное соединение Н [Agl3j, в результате чего концентр а пи я ионов серебра в растворе становится! исчезающе малой. Медь в иодкетоводородной кислоте не растворяется.
Седьмая группа периодической системы 847
Без доступа кислорода воздуха газообразный иодистый водород и его водный раствор ^при обычной температуре совершенно устойчивы даже на свету. При доступе кислорода, напротив, происходит медленное окисление его до иода. Поэтому на воздухе растворы йодистого водорода, особенно концентрированные, быстро буреют. Свет действует при этом ускоряюще. Окрашивание можно предотвратить добавлением медных стружек, которые связывают выделявшийся иод с образованием нерастворимого иодида меди(1) Cui.
При нагревании иодистый водород восстанавливает алкилиодиды и спирты и гидрирует в некоторых случаях ненасыщенные органические соединения; однако обычно он присоединяется последними с образованием иодидов, например:
С2Н5-1 ф- Н1=СгН64-12,
С£и5-ОН -J- 2HI =s С3Не -ф Н,0 >• 1г,
CjIJ;t) "ф" 2HI = CfjHjg -ф- 1£,
С2Н4 + HI = С2Н51,
Йодистый водород получается аналогично бромистому водороду путем гидролиза йодида фосфора.
Обычно поступают следующим образом: к Смоченному водой (10 ч.) поду (100 ч.) прикапывают кашеобразную смесь красного фосфора (5 ч.) я воды (10 ч.), Образующийся вначале пснтаподид фосфора Р15 тотчас гидролизуется с образованием фосфорной кислоты и йодистого водорода
v + 5,м3 = piь; рт5 -Ф- 4Нз° = HSPO4 > 5Ш.
Водные растворы йодистого водорода умеренной Концентрации удобно получать пропусканием сероводорода во взвесь иода в воде (ср. уравнение (11), стр. 843]. Избыток сероводорода удаляют пропусканием двуокиси углерода. Выделившуюся серу отфильтровывают; в случае надобности раствор предварительно кипятят, чтобы сера коагулировала.
2. Галогениды
Гйлогенидам,и называют соединенпя галогенов с более электроположительными веществами. Галогениды металлов в большинстве случаев обладают явно выраженным солевым характером. Они являются солями галогеноводородных кислот. К типичным солям относятся прежде всего галогениды легкшс металлов, Иная природа у галогенидов неметаллов, папример у ССЦ, SiCl4, S2C1£. В отличие от галогенидов металлов опи в большинстве случаев легко летучи и либо нерастворимы в воде, либо ею тотчас полностью разлагаются. Галогениды металлов в воде большей частью легко растворимы и практически полностью диссоциируют (за исключением, например, HgCb, HgBr2 и У галогенидов, производных
Слабых оснований, как и вообще у солей слабых оснований и сильных кислот, диссоциация сопровождается частичным гидролизом, который, однако, в слабо разбавленных растворах в большинстве случаев не идет слишком далеко.
Своей растворимостью фториды несколько отличаются от остальных галогенидов. В то время как из хлоридов, бромидов и иодидов особенно
* Рассчлталпые по отноничтю электропроводностей ЛсЛ4о хам-ущиеся степени диссоциации лежат заметно ниже истиной степени Диссоциации. Особенно это справедливо для галогенидов многовалентных элементов вследствие действия меж ионных сил, как у всех солей мни го валентных металлов. Кроме того, это обусловлено и тем, что часто образуются аутокомплевсы (см. т. II).
848
Глава 17
трудно растворимы соли, серебра, а соли щелочноземельных металлов, наоборот, очень легко растворимы и отчасти даже расплываются па воздухе, фториды щелочноземельных металлов, и прежде всего фторид кальция, трудно растворимы; наоборот, фторид серебра крайне легко растворим и расплывается.
В качестве труднорастсоримых солей еще следует назвать галогениды одновалентной ртути, одновалентной меди и (поскольку они вообще существуют) одновалентного золота, а также фторид свинца. Хлорид свинца, бромид свинца и иодид свинца па холоду также достаточно трудно растворимы, однако при нагревании растворяются значительно легче.
Для получения галогенидов металлов пользуются общими методами приготовления солей, о которых будет сказано в следующей главе. Приготовление галогенидов неметаллов было уже описано при рассмотрении отдельных соединений.
Галогениды принадлежат к числу важнейших соединений отдельных элементов. Особенно это относится к хлоридам. Поэтому (и поскольку последние производятся от рассмотренных элементов) о них было уже сказано. Некоторые другие галогениды отдельных элементов также многократно упоминалисв в предыдущих главах. В дальнейшем следует лишь описать еще фториды, бромиды и иодндьг щелочных металлов и аммония.
Среди органических галогенидов особое место занимают соединения иода, поскольку от них производятся соединения иодония. Это Соединение типа [R2I]X (R — органический радикал, X — одновалентный кислотный остаток или гидроксильная группа). Эти соединения находятся в таком же отношении к простым алкил- (соответственно арил-) галогенидам, как соединения аммония к аммиаку. Соединения иодония обладают солевым характером; в их растворах радикал иодония выступает в качестве свободного положительно го иона [Rai]'. Гидроокись иодония [RaI]OH — сильное основание.
Фто рады
Фторид натрия XaF выпадает в виде белого зернистого осадка при пропускании фтористого водорода в не слишком разбавленный раствор гидроокиси или карбоната патрия. Осадок состоит пз правильных кубов или октаэдров (уд. вес 2,78; т. ил. 988°; т. кип. 1700°).
В техпйке его получают сплавлением криолита с едким патрон: NajAlFe + + 6NaOH = Ха3[Л1(ОН)й]‘т- 6NaF. При выщелачивании сплава алюминат натрид переходит в раствор, фторид же остается не растворенным.
Растворимость фторида натрия в воде довольно незначительна (при 15° 4 ч. NaF растворяются в 100 ч. воды). В спирте он практически нерастворим. Водный раствор имеет щелочную реакцию, так как там частично происходит следующее превращение;
2F'+Н0Н = [НК3]' 4- ОН'.
Из растворов, содержащих избыточную плавиковую кислоту, при упаривании кристаллизуется кислый фторид натрия (гидрофторид патрия) NaHF2 в виде бесцветных ромбоэдров. При нагревании это соединение выделяет фтористый водород и может поэтому с лужитв для его получения (ср. стр. 843).
Фторид натрия применяют главным образом для пропитки древесины в целях защиты ее От гниения, а также как дезинфицирующее средство в технологии брожения. Иногда его применяют также в медицине.
Седьмая группа, периодической системы
849
Фторид калия KF в противоположность фториду натрия очень легко растворим в воде и расплывается да воздухе. Все же его удается осадить добавлением спирта. При этом он выпадает в виде дигидрата KF-2H2O, который плавится ири температуре около 46° в собственной кристаллизационной воде. Безводная соль плавится при 848° и кипит приблизительно при 1500°. Опа кристаллизуется в правильной системе (уд. вес 2,48). Водный раствор, так же как раствор фторида натрия, обладает щелочной реакцией. Из растворов, содержащих избыточную плавиковую кислоту, кристаллизуется кислый фторид калил (гид рофторйд калия) К Н F3 (уд, вес 2,37), который при прокаливании даёт нейтральную соль. Исходя из безводной плавиковой кислоты Муассап получил продукты присоединения с двумя и тремя молекулами HF: KF-2HF и KF-3HF. Кроме того, существуют еще KF-27aHF и KF-4HF (Cady, 1934).
Фторнд аммония NH4F можно получить по способу, предложенному еще Берцелиусом, нагреванием смеси, состоящей из 1 ч. хлорида аммония и 2*/4 фторида натрия. При слабом нагревании фторид аммония сублимируется и осаждается па холодной крышке сосуда. Он очень легко растворим в воде и расплывается на влажном воздухе. В растворе он в заметной степени распадается на аммиак и кислый фторид (гидрофторид).
2|NHJF=[NH4--HHFd'+W
Поэтому его нельзя приготовить упариванием смеси растворов аммиака и фтористого водорода, так как при этом выделяется аммиак и кристаллизуется гидрофторид аммония [NEhJHFa (уд. вес 1,21, т. пл. 124°). Гидрофторид аммония, так же как и плавиковую кислоту, можно применять для травления стекла. Он дает более шероховатое травление, чем плавиковая кислота. В спиртовой промышленности и пивоварении его используют в качестве дезинфицирующего средства, особенно для дезинфекции шлангов и трубопроводов. Нейтральную соль применяют в медицине.
Из безводного жидкого фтористого водорода можно, кроме того, получить продукты присоединения NH4F-2HF, NH<F-3HF hNH4F-5HF, температуры плавления которых лежат очень низко (вблизи 30°) (Ruff, 1933).; NH4F-2HF (в форме по л у гидрата №H4F-2HF- У> Н2О) также получается и из водного раствора (Hassel, 1932).
Бромиды
Бромид натрия NaBr чаще всего получают обменным взаимодействием соды с бромидом железа Fe3Brs
4KaaCO3+Fe3Br8=8NaBr-|-Fei!iO4+4CO24
При этом обычно поступают следующим образом: железные стружки и другие железные отходы обливают водой. Постепенным введением брома железо переводят в растворимый бромид железа(П) FeBr2. Зеленый раствор бромида железа сливают и смешивают дополнительно с бромом (1/з взятого ранее количества): Fe ф- Вг2 = = FeBr2; 3FeBr2 - Вг2 Fe3Brs. Раствор последнего, сконцентрированный упариванием, вводят при хорошем перемешиваний в кипящий раствор соды. Осадок гидрата окиси железа(П, Ilf) отфильтровывают. Из сгущенного упариванием фильтрата кристаллизуется выше 30,7° безводный бромид натрия (уд. вес 3,20). При обычной температуре кристаллизуется дигвдрят NaBr-2H2O (уд. вес 2,18). В ннтервале от —24 до —28° при соприкосновении с раствором устойчив пептагпдрат.
Бромид натрия можно приготовить также введением брома в горячий раствор едкого натра до насыщения; после упаривания раствора остаток, состоящий из смеси бромида и бромата натрия, нагревают 5-4 г. Реми
850
Глава 17
с порошком древесного угля:
3Br2-j- 65аОН=5NaBr Ц- NaBrO3+ЗН2О;
2NaBrO?4- ЗС = 2NaBr-f-3CO2.
Чистый бромид натрия представляет собой бесцветные кристаллы (т. пл. 760°, т. кип. 1393°). В воде оп очень легко растворим и также достаточно легко растворяется в спирте. В 100 г воды при 0° растворяется 79,5 г NaEir, при 20°—90,3 г и при 100°—120,5 г NaBr. Точка кипения насыщенного раствора лежит при температуре 121°. Бромид натрия применяют в терапии в качество успокаивающего средства (Natrium bromatum).
В ланаловых лучах, может также существовать соединение Na2Br. Это было обнаружено масс-спектре графически, М a t t а и с Ъ, Ewald, Hahn, S t r a s -зшапв, Z. Physik, 125, 598, 1943.
Бромид калия КВт получают так же, как бромид натрия, и опи очень похожи по своим свойствам; однако КВт пе образует гидрата. КВт — бесцветные кристаллы кубической системы; уд. вес 2,75; т. пл. 748°; т. кип. 1380°. В 100 г воды при 0° растворяется 54 г КВг, при 20°—65 г, при 100°—105 г; в 100 г абсолютного спирта при 25° растворяется только 0,13 г. Бромид калия находит достаточно широкое применение в терапии {Kalium bromatum) и, кроме того, в фотографической промышленности (ср. т. II).
Бромид а миопия NH4Br можно приготовить так Же, как бромид натрия, — взаимодействием водного раствора аммиака с бромидом железа FejBrg или осторожным прибавлением брома к охлаждаемому льдом крепкому раствору аммиака. Реакция протекает с сильным выделением тепла 3/2Вг2 4- 4NHs = ЗЛ’ЩВг + -|- 105 икал.
Следует избегать избытка брома, так как в соответствии с уравнением К’Н4Вг-рЗВг2 = = 4НВг NBr3 может образоваться очень взрывчатый бромистый азот (ср, стр. 667).
Бромид аммония бесцветен; так же как бромид калил, образует кристаллы кубической системы. С бромидом калия он дает смешанные кристаллы. Уд. вес его 2,39. Он легко сублимируется. В воде лыко растворим: 69,7 - в 100 г воды прп 15°. Применяют его так же, как бромид калия. 1
Йодиды
Иодид натрия Nal получают подобно бромиду натрия. Так же как и бромид, иодид натрия при более высокой температуре (выше 65е) кристаллизуется в безводном состоянии — при обычной температуре в виде . дигцдрата, а ири низкой температуре (между —13,5° и —31,5°) в виде пентагидрата. Иодид патрия находит применение прежде всего в терапии {Natrium jodatum). Безводная со л в образует бесцветные,^ кубические кристаллы удельного веса 3,66 (уд. вес дигйдрата 2,45). Она плавится при температуре 661° и кипит при температуре 1300°. Растворимость при 0° составляет 159 з, при 20°—179 и при 100°—302 г Nal в 100 г воды. Он также .очень легко растворим в абсолютном спирте (43 г в 100 г этилового спирта и 78 а в 100 г метилового спирта при 23°).
Иоднд калия KI получают обоими способами, указанными для бромида натрия. Оп кристаллизуется в виде бесцветных кубов (уд. вес 3,12; т. пл. 677°; т. кип. около 1325°). Растворение в воде сопровождается сильным понижением температуры. Иодид калия не образует гидратов.
Растворимость при 0° составляет 128 г, прн 20’ — 144, при 100° — 209 е в 100 г воды; при 20° 1,75 г иодяда калия растворяется в 100 г абсолютного этилового спирта и 16,5 г в 100 г абсолютного метилового спирта.
Седьмая группа периодической системы
851
Иодид калия находит широкое применение в медицине (Kalium jodatum) несмотря на то, что оп в отличие от иодида натрия вреден для сердца. Его используют также в фотографической промышленности. Кроме того, он служит в качестве исходного продукта для приготовления других соединений иода.
Иодид аммония NH41 получают насыщением иодистоводородной кислоты аммиаком или карбонатом аммония. Он представляет собой белый гигроскопический кристаллический порошок (уд. вес 2,44). В воде и в спирте од очен,. легко растворим (167 а в 100 г воды При 15°). Его применяют также в медицинских целях.
Полигалогеннды. Некоторые соединения более тяжелых газогенов с металлами способны присоединить еще дополнительное количество атомов галогена. Например, иодид калия KI легко присоединяет еще два атома (соответственно 1 молекулу) иода с образованием темного красно-бурого тримодида калия К7Й. .При присоединении еще большего количества иода можно получить наконец эннеаиодид калия. К I9: KI + Ia = KIa; KI + 4- 412 — KI,,, Таким я;е образом многие другие йодиды присоединяют иод с образованием соединений общей формулы где п может быть равно 2—9. Такне соединения называют полииодидами, а вообще соединения типа М:ХП — полигалог-сиидалис.
Помимо полииодплов щелочных металлов, средн которых наибольшую склонность к образованию тон । галогенидов проявляют рубидий и цезий, известны также полииодиды главным образом органических аммониевых оснований. Три- н гоптаиодяды □тих органических оснований обладают в общем к pac.no-бур ой окраской, пента- и эпнеа-иодмды — тем ио-зеленой. Существуют также ди-, тетра- и гексаиодиды главным образом тяжелых металлов (Си и Hg) и алкалоидов. Из поиибро.иьЗоа следует назвать: RbBrj — красные кристаллы; CsBr3— желтовата-красные; CsBr6 — темно-красно о очень неустойчивое соединение.
Полигалогеннды можно подразделять на содержащие галоген только оЗчсго вида (только иод или только бром) и содержащие одновременно различные галогены-. Первые следует рассматривать, вероятно, как непрочные продукты присоединения элементарного галогена к простому галогениду, например К1д = К1-12, Оял достаточно легко отдают избыточно присоединенные молекулы галогена.
По ли галогениды, которые содержат различные галогены, возможно, построены так же, как и те, которые (.'одержат атомы галогена одного вида. Однако часть из них, по-видимому, приближается н гмогеносолям, которые будут рассмотрены в следующем разделе. Это одиосится, например, к полученному Эмелиусом (Emelins, 1949) соединению KF-IF5, которое, оков ярко, следует рассматривать как гекекфтороиод&т К [IFeJ.
Проще всего можно получить полииобиЗм, и среди них наиболее рас-црост раненный трииодид к«лняК13, который первым из полииодидов был приготовлен Джонсоном в 1877 г. й поэтому называется также солью Джонсона. В водном растворе он распадается на ионы К*и [13]’. Аналогичным образом диссоциируют остальные полигалогеннды. В воде многие из инх растворимы, правда отчасти с разложенцем. Однако их электролитическую диссоциацию можно изучать в других подходящих растворителях.
Растворы полииодидов, главным образом раствор полииодида калия, часто применяют. когда требуется получить иод в водной среде в возможно большей концентрации. Для приготовления раствора иолииодида калия нужно не слишком разбавленный раствор иодида калия смешать с иодом. Иод легко растворяется в KI, однако вследствие нестойкости полииодяда так же легко выделяется слова.
852 Глава. 17
Поведение такого раствора, который содержит элементарный, иод, слабо связанный с ионами иода, в большинстве случаев соответствует практически поведению настоящего раствора иода.
Галогеносели и галогене кие лоты. Белый инет во галогенидов многовалентных элементов способно присоединять галогениды низко валентных сильно электроположительных элементов, главным образом галогениды щелочных металлов и часто также галогеноводород. При этом образуются двойные или комплексные соединения, которые в соответствии с номенклатурой Вернера обозначаются как галогеносоли. (соответственно галогена кислоты). Многочисленные примеры таких галогеносолой ,и галогеио-кисло^быди уже приведены. Первыми рассмотренными подробнее соединениями такого типа являются указанные в гл. 10 фторобораты и борофто-ристоводородная кислота.
Как было изложено в гл. 11, образование подобных сведя пений объясняется тем, что при присоединении: отрицательных ионон к электроне игральному в целом образованию, состоящему из многовалентного положительного иона и соответствующего числа одновалентных отрицательных ионов, во многих случаях дополнительно выделяется анергия. Это происходит главным образом тогда, когда отрицательные ионы обладают лишь незначительным диаметром. Благодаря этому среди галогепосолей и галогеио-кислот наиболее устойчивыми являются обычно соединения фтора., а соответствующие соединения иода существуют редко и их устойчивость в большинстве случаев заметно уступает устойчивости других гадоге но соединений.
Двойные галогеносоли играют большую роль в неорганической химии. Однако и в органической химии ими часто пользуются, так как они отличаются хорошей кристаллизационной способностью и поэтому пригодны для получения веществ в чистом состоянии.
Получение галогеносолой и- галогене кислот чаще всего производят простым смешиванием водных растворов компонентов. При упаривании выкристаллизовываются двойные соединения. Устойчивость комплексов, лежащих в основе этих соединений, очень различна. Существуют такие, которые при растворении л воде полностью распадаются на свои компоненты; но имеются также и другие, у которых даже при сильном разведении растворов не удается обнаружить обычными реактивами никаких свободных галогенид-иопов, а обнаруживаются лишь только сложные иоиьг (комплексные ионы). Такие устойчивые комплексные галогеирсоеди-пения образуются главным образом металлами побочных подгрупп периодической системы. Наиболее известными примерами являются илатино-хлористоводородная кислота HalPtClJ и ее соли (гексахлороплатинаты).
Гаяогеносааи можно производить от солей кислородных кислот, если предположить, что каждый отрицательный двухвалентный атом кислорода заменяется двумя отрицательными одновалентными атомами галогена. Так, из мета борной кислоты НВОЭ иоау чается, и ап ример, тетра фторсборная кислота НВу,, и» метакрешгевой кислоты HjSiOs — гекса фторо к рем нов а я кислота H£SiF0 и т. д. Если замещается только часть атомов кислорода на атомы галогена, то образуются ок со гало гоно соединения. Если часть атомов кислорода заместить атомами галогена, а Другую часть -другими отрицательными атомами или радикалами, например ОНТ или УСД, то получаются ги дрок со га логе но соединения, иди нитрогалогеносоедивения и т. д.
Аналитические сведения. В отсутствие кремневой кислоты, для открытия фторидов обычно служит проба, травления (ср. также стр. 843 и сл.). К топкоизмельченному веществу добавляют концентрированную серную кие.юту и осторожно нагревают в платиновом или свинцовом тигле, покрытом часовым стеклом. Освобождающийся фтористый водород обнару-
Седьмая группа периодической системы_____________________853
дай в а ют по травлению стекла. Если же присутствует кремневая кислота (кварц или силикаты), то выделяется фторид кремния, который на стекло не действует. Если в этом случае тигель покрыть крышкой, просверленной в середине, и положить на нее влажный черный бумажный фильтр, то в случае, если исследуемое вещество содержит фторид, на фильтре проявляется вскоре белое пятно, которое объясняется образованием гидрата двуокиси кремния в результате гидролиза фторида кремния.
Следует отметить, что фториды, так же как сульфаты, дают осадок с хлоридом бария*.
Хлориды, бромиды и иодиды с нитратом серебра дают осадки, нерастворимые в разбавленной азотной кислоте. При нагревании смеси растворимого хлорида и бихромата калия с концентрированной серной кислотой выделяется хромилхлорнд CrOjCb, который при поглощении раствором едкого натра образует легко обнаруживающийся хромат (ср. т. 11). Если растворимый бромид смешивать с хлорной водой, выделяется бром. При избытке хлорной воды образуется винно-желтый хлорид брома ВгС1. Еще легче хлором вытесняется под из йодидов, который растворяется в хлороформе или сероуглероде с фиолетовым окрашиванием. Избыток хлора окисляет под в водном растворе до бесцветной йодноватой кислоты. Поэтому, если, кроме иода, в растворе присутствует также бром, добавляя достаточное количество хлора, можно получить окраску, характерную для коследпего.
Количественное определение фтора можно осуществить осаждением и взвешиванием его в виде фторида кальция CaFa.
Если, кроме ионов F', имеются еще другие ионы, которые дают осадки с ночами Са", то нагреванием с кварцевым песком и концентрированной серкой кислотой фтор переводят во фторид кремния. Последний улетучивается и поглощается разбавленным спиртовым раствором хлорида калия, где благодаря следующим реакциям: 3SiF$ 4-Ч-2НаО = SiOa +: 2H2SiFfi; H2SiF$ + 2КС1 = KaSiF0 + 2HC1, — иа каждую их трех молекул SiF4 образуется четыре молекулы НС1. Количеств!) освобождающегося хлористого водорода, I а-аке которого соответствует 3 с-эив ионов фтора, определяется титрованием раствором едкого натра (метод Пеифильда). 'Гак как этот метод дает правильные результаты только при полном отсутствии влажности в перегонной аппаратуре, Виллард (1933) рекомендовал вместо серной кислоты применять водную хлорную кислоту, Тогда фтор перегоняется в виде водной кремнофгггоркетоеадорвдкоа кислоты (ар. етр. 523). Последняя может быть оттитрована раствором нитрата тория с использованием в качестве индикатора смеси иитрата циркония и ализарина [см. Frees J., Z. anaiyt. Chem., 110, 251, 1937]. При Определении фтора но Вилларду целесообразно воспользоваться аппаратурой, описанной Эрлихом [Ehrlich, Р., Angew. Chem., 65, 131, 1953), В технике в большинстве случаев работают ио методу Штарка и Холи (Starck, Hawley), который основал на том, что ионы F' сначала осаждают в виде хлоро-фторида свиица PbCIF, осадок отфильтровывают и растворяют в разбавленной азотной кислоте, после чего в растворе тцгрометрпчески определяют содержание лоиов СГ. [См. Specht F,, Z. anorg, Chem., 231, 181, 1937]. Эрлих [Е h г 1 i с h, Z. analyt. Chem., 133, 84, 1951] рекомендует осаждать фтор в виде бромофторида свинца PbBrF, который, между прочим, имеет то преимущество, что может быть осажден из более кислого раствора.
Для количественного определения остальных галогенов пх обычно осаждают к сиде галогенидов серебра. Последние либо взвешивают непосредственно, .либо тптромстрически определяют количество серебра, необходимое для их осаждения.
* Вследствие существенно большей по сравнению с сульфатом бария растворимостью фторида бария в енлыю разбавленных растворах по происходит образования осадка. Фторид кальция менее растворим, чем фторид бария, однако и его растворимость также в 7 раз больше, чем растворимость сульфата бария.
854
Глава 17
Небольшие количества ионов F'(20 .ив — 20 у) удобно определять (даже в присутствии Cl', NOa, SO4 и SiOg ионов) кондуктометрическим титрованием раствором хлорида алюминия (Jander, 1939). Определение ионов СГ в небольших количествах (при подходящей методике до 1 у) также можно проводить кондуктометрически (титрованием с нитратом серебра (J ander, 1937). Для определения очень небольших количеств СГ весьма рас пр о стр а не юл нефелометрические способы, разработанные Ричардсом (Richards, 1904) и Лэмбом (Lamb, 1920) [см, также Kolthoff, J. Am. chem. Soc,, 55, 1915, 1933],
3. Окислы л к мелородные кислоты галогенов
В кислородных соединениях галогены не обнаруживают между собой столь глубоких аналогий, как в других соединениях.
В то время как соединения между фтором и кислородом были обнаружены только недавно, хлор образует несколько отчасти уже давно известных окнслов, а именно: С12О, С1ОЙ, С12Ое и С1ЙО7*, а также несколько кислородных кислот: НСЮ, НС1О2, ЙСЮ3 и НСЮ4. Часть кислот производится от других валентных состояний хлора, не встречающихся в окис-лах. Получение окнслов брома (ВгаО, ВтОй и ВгОз — все очень неустойчивы) удалось осуществить только в последнее время; из кислородных кислот брома с достоверностью известны только две: НВгО и НВтО3. Иод образует весьма устойчивый, окисел I2OS, который является ангидридом йодноватой кислоты НЮ5. Последняя наряду с иодной кислотой Н5Юв является наиболее устойчивой среди галогене кислородных кислот.
Иодная кислота существует в нескольких формах с различным содержанием воды. В ее растворах и в растворах ее солей между этими формами (или соответствующими им ночами) наступает равновесие.
[10J'-H2H3O [1О6]"'4-2НЧ-Н2О [1О6]"'"-р4Н\
2[IO4]'+H2O qt [12ОЙ]""-|-2Н‘ и т. д.
Кроме того, от электроположительною одновалентного иода производится иодноватистая кислота [ОН, которая, правда, может существовать только непродолжительное время в растворе.
Далее известим еще киелородлые соединения иода состава Ю2 (соответственно 12О4) и W Однако, как можно заключить из их поведения, они не являются окислами в обычном смысле; их следует рассматривать как йодаты трехвалентпого иода, соответственно радикала иодила [Ю|\ являющегося производным трех валентно го пода:
3+- 5+ З* 5*
I4o9= I цо3]3; i2o4=[ioj[io3j.
Соединения фтора, хлора и брома с кислородом зндотермтшы. Для их образования поэтому требуется подвод энергии. Это можно осуществить, если па соответствующую газовую смесь действовать электрических! раз
* Помимо приведенных выше нормальных окислов, хлор образует еще окисел, . который, вероятно, следует рассматривать щтк. перекись. Он был получен в 1923 г, Гомбергом действием иода па перхлорат серебра в эфирном растворе. Он известен только в растворе, одпало можно установить, что иа один С1 в нем содержится четыре О. Если его молекулярный вес (что считает вероятным Гомберг па основании своих опытов) отвечает формуле С12О8, то его образование можно представить следующим образом:
-pAgl.
Седьмая группа периодической системы
855
рядом (ср. получение O2F2 и ВтО2). Однако необходимая энергия может быть также получена в результате другого химического процесса, связанного с образованием окисла. Например, прн взаимодействии HgO с С)2 по уравнению (12), стр. 856, энергия, необходимая для образования С1аО, предоставляется благодаря одновременно происходящему соединению С1г е Hg.
КИСЛОРОДНЫЕ СОЕДИНЕНИЯ ФТОРА
Ди фторид кислорода. В 1927 г. Лебо (Lebeau) и Дамьен (Damiens) нашли, что при электро.тазе расплавленного гидрофторида калия КНГ3, до тех вор вола он еще содержит иоду, в качестве побочного продут;та выделяется кислородное соединение фтора состава ОУ2. Для отделения этого соединения от примешанного к нему элементарного фтора смесь следует пропустить через воду. В воде это соединение ггезначитсль-ио растворимо, в то время как элементарный фтор взаимодействует с водой с выделением кислорода. Как затем вскоре установили Лебо и Дамьеп, лучший выход дифторида кислорода можпо получить при действии фтора на разбавленный раствор едкого натра
2F, ф 2NaOH = 2NaF ф Н30 ф OF».
Получение OF» в большом лтстчестве почти л чистом состоянии удалось осуществить в 1930 г. Руффу (КпЙ). Путем анализа и Определения плотности Руфф установил формулу соединения OF». Теплота образования его составляет около —8 ккал/моль.
Дифторпд кпсдо рода — бесцветный газ с характерным запахом сильно раздражающий дыхательные путь [вес 1 л газа равеи’2,421; т. кип, - 144,8°; т. пл. —223,8; уд. вес (при т. ним.} 1,53). В жидком состоянии он окрашен в интенсивно-шел тип цвет. На холоду он но действует на стекло и вообще ,менек реакционноспособен, чем элементарный фтор. В воде мало растворим (6,8 мл в 100 мл воды прп 0°). На холоду он лишь очень медленно реагирует с водой . Однако, таге как реакция OF3 ф- И2Опар— 02 4-2HF + 74,8 ккал силыю э изотермична, то при поджигании (например, электрической искрой) происходит взрывообразиоо взаимодействие. Если OFa нагреть в сухом состоянии или подвергнуть облучению, то он ггосгепеико распадается па О» и Fa (по-еледппм л стеклянном иди кварцевом сосуде тотчас, образует SiF4 и О2).
Молекула OF2, так же как молекула ОН2, образует угол. Согласно совпадающим данным' электропографических определений (Boerscli, 1935) и исследования инфракрасного спектр» (Fohlmunn, 1935), <5 FOF равен 100,6°. Межъадерпое расстояние О-—>F составляет 1,4) А.
Фторид кислорода O3F2 был получек Руффам к Мешдощм (Rulf, Meaael, 1933) непосредственным соединением О» и F3 под действием тлеющего разряда. В газообразном состоянии он слабо-коричневый, в жидком — вишнево-крастгый, в твердом — -оранжевый (т. пи. 163,5°; т. кип. —57°; уд. вес при г. кип. 1,45; теплота испарения 4,57 кхал/моль). Уже немного выше температуры кппмшя FaOa разлагается на F3 и О2 (Schumacher, 1936).
Помимо фторидов кислорода, известны также еще некоторые другие соединения л которых У связан с кислородом, а именно! NO2(OF), C'.JOa( OF) и CF3(OF) (Cady, 1934 и 1948; Ruff, 1935; Yost, 1935). [Относительно строения NOa(OF) см. Полппг и Брокуэй, а также Hill, В i g о 1 о ту, J. Am. chem. Soc., 59, 13, 2127, 1937),
КИСЛОРОДНЫЕ СОЕДИНЕНИЯ ХЛОРА
(о б з о р)
01 ТТгЛьг Кислоты Салп
fJA'Wi'h zro;.’'7s иbiit таз СЮа, Z.VDp? • зеЛС[1СП1(1Тй->Ьб5Ч- тай гая CJ f ш few, zz jj i? ъ д:,.д L1 pa т темя 11 -кр a g- ная HWAKijCTj> селсцолисъ хлора, боец нот и ан ЖИДКОСТЬ НСЮ, лмарковати-СГПвЯ А*11СЛО?^<2 ’НС 10а, хлористая кислота НСЮ3г хлophовйтая кис лоти НСЮ4т хлорная шша М^СЮ], гипохлорит М1] С1О2], хлориты M^CJOJ, хлораты перхлораты
856
Глава 17
Все окисли, хлора взрывчаты. Из кислот в свободном состоянии получена только хлорная кислота. При нагревании она также взрывает; при разложении ее на хлор, кислород и воду освобождается около 16 ккал!моль тепла. Остальные кислородные кислоты хлора известны только в водном растворе. Соли всех четырех кислот получены в кристал-л инее ком состоянии. Их устойчивость значительно возрастает от гипохлоритов к перхлоратам.
Строение. Возрастание устойчивости и вместе с тем силы кислородных кислот хлора с увеличением числа атомов кислорода не подтверждают предполагаемые ранее «цепочечные формулы», и соответствии с которыми, например, хлорной кислоте приписывали строение Н—О—О—О—О—CI. Поэтому приблизительно с конца прошлого столетия стали считать, что в названных кислотах все кислородные атомы непосредственно связаны с хлором и, следовательно, хлор в своих кислородных кислотах обладает валентностями, ступенчато возрастающими в порядке нечетных чисел: 1, 3, 5, 7, Поэтому кислоты можно представить следующими формулами:
О
1 Ш v Ю Цун
Н—О—CI, Н-Ю-С1=0, И—Q—С1< , 11-О-СЬО.
%
В солях по крайней мере сильно электроположительных металлов с ионами металлов связаны, вероятно, не определенные кислородные атомы, а целые ионы:
' иг •}-
OGIO
ОСЮ о
J »
OV11O' *
С1 о о .
Эти «ядерные формулы», выведенные сначала на основании химических свойств, были цодтверждещ.т в последнее время рентгене структурными определениями солей. Ничто не препятствует считать валентности хлора в приведенных выше нонах (а также в кислотах) по существу обусловленными соответствующим положительным зарядом атома хлора, т. е, отдачей, или по меньшей мере частичной отдачей (переход па более высокие квантовые уровни), соответствующего числа электронов, Следовательно, ноны мощно считать построенными гетер он о лярио или смешанно гомеополярно — гетеро-полярно. 0>:ислы хлора, вероятно, следует рассматривать как чисто гоме,аполярные соединения. Может быть, этим объясняется, что для части кислородных Кислот (а именно дли хлористой кислоты и хлорноватой кислоты) не подучены ангидриды и что, с другой стороны, на некоторых окислов (а именно двуокиси хлора и гекс а окиси хлора) нельзя образовать кислоты. s
Окись хлора, хлорноватистая кислота и гипохлориты
Окись хлора С1гО образуется при пропускании хлора над сухой окисью ртути при 0°
HgO+2Cl2=HgCls4-Cl2O. (12)
Это желто-коричневый, неприятно пахнущий газ, сильно действующий на дыхательные пути; значительно тяжелее воздуха (удельный вес относительно воздуха 3,02). Его легко можно сконденсировать в красно-бурую жидкость (т. кип. -}-3,8о).
Молекула С120 образует угол; межатомное расстояние С1<—»О равно 1,70 А /С1
111в, дипольный момент ц = 1,69 10'18.
Окись хлора является эндотермическим соединением (теплота образования —18,6 ккал при постоянном давлении). С12О легко распадается
Седьмая группа периодической системы
857
со взрывом на составные части, например при нагревании, при переливании в жидком состоянии или при соприкосновении с горючими веществами. Водой С12О легко поглощается, образуя с ней хлорноватистую кислоту С12О + Н3О ~ 2ИОС1. Следовательно, окись хлора является ангидридом хлорноватистой кислоты.
Хлорноватистая кислота НОС1 известна только в водном растворе. Как уже было указано на стр. 836, она образуется наряду с соляной кислотой или ее ионами при взаимодействии хлора с водой
ci24-h2o HOGi-4-нч-ср. (Гз>
Если ионы Н’ и Ci' устранять добавлением окиси ртути
IIgO4-2H4-2Cl' =И^.1в-)-Н20 *,
то процесс (13) полностью протекает слева направо. Поэтому при пропускании хлора в воду, которая содержит суспендированную окись ртути, можно приготовить достаточно концентрированные растворы хлорноватистой кислоты
2иг4-НйО+Н^0^.2НОС14-Н§и2.
Молекула НОС1 образует угол-сН-^О—С1, который равен, ^113°. Межатомное расстояние в пси составляет Н 0 = 0,957 А, О +—> С1 = 1,70 А, Валентпо-силовые постоянные имеют значение 7,3510®, что соответствует 3,86-10® дин/см (Hodberg, Badger, 1951).
Растворы хлорноватистой кислоты окрашены в слабый зеленовато-желтый цвет и обладают своеобразным запахом (похожим на запах хлорной извести). Растворы постепенно разлагаются, особенно иа свету, с выделением кислорода и одновременным образованием хлорноватой кислоты НС1О3. Вероятно, реакция протекает Так, что сначала отщепляется кислород НОС1 = НС1 -J- Ок затем часть его действует в момент выделения гтя еще не разложившуюся хлорноватистую кислоту с образованием хлорноватой кислоты HOG!-f- 2О = НС1О3. Хлорноватистая кислота является очень слабой кислотой (константа диссоциации ее К = 3,6-10*® при 25е). Ее соли называются гипохлоритами.
Гипохлориты — соединения общей формулы М1 [СЮ 1 — образуются вместе с хлоридами при действии хлора на сильные основания, например, 2NaOH + С1г = NaOCl 4- NaCl + Н2О. Растворы гипохлоритов щелочных металлов можно также получить, непосредственно при электролизе хлоридов щелочных металлов. Для этой цели необходимо только устранить указанные на стр. 208 приспособления, которые должны препятствовать смешиванию выделяющегося на аноде хлора со щелочью, образующейся на катоде.
Растворы: гипохлорита калил были получены впервые фабричным способом ПО инициативе Вертолле в 1792 г. в г. Жавеле близ Парижа нри пропускании хлора в раствор кзрбоната калия. Эти растворы были названы «жавелевой водой» («Еаи de Javeilc'»). В 1820 г, Лабаррак (Labarraqne) получил аналогичным способом (пропуская хлор в раствор соды) растворы гипохлорита натрия (Eau de Labarracjue «лабарракова вода>>). Однако приготовленные этим способом белильные растворы малоустойчивы, так как они, как правило, содержат свободную хлорноватистую кислоту
2\я2СОч + С12 + Н20 = NaCl Ц- 2NaIICO3 NaOCl;
NaOCl + С12 -А Н2О - NaCl + 2НОС1.
Растворы гипохлоритов щелочных и щелочноземельных металлов, не содержащие хлоридов, можно получить насыщением растворов или суспензий соответствующих
* Хлорид ртути HgCl2 в противоположность другим солям диссоциирует только в незначительной стешеип.
-858
1
Глава 17
гидроокисей растворами хлорноватистой кислоты. Испаряя растворы при ни□ кой температуре, названные гипохлориты получают в кристаллическом состоянии. Как правило, они содержат кристаллизационную воду.
Гипохлориты отличаются сильными окислительными свойствами. Растворы их применяют главным образом в качестве белильных щелоков. Этому пе мешает присутствие хлоридов в растворах гипохлоритов, приготовленных обычным способом.
, Хлорная известь. При пропускании хлора над гашеной известью образуется двойная кальциевая соль хлорноватистой и хлористоводородной кислот
С1а4-Са(ОН)2 = СаС1(ОС1)+НОП. (14)
Эта двойная соль является главной составной частью хлорной извести. приготовляемой в технике в больших количествах.
Хлорная известь представляет собой белый зерпистый порошок, обладающий своеобразным запахом, который приписывают обычно хлорноватистой кислоте, освобождающейся благодаря действию двуокиси углерода, содержащейся в воздухе. Как все соединения хлорноватистой кислоты, хлорная известь легко отдает кислород и действует поэтому сильно окисляющим образом. Например, она окисляет окись свинца и закись марганца или их соли в щелочном растворе до двуокисей РЬО2 и МпО2. При действии на хлорную известь соляной пли серией кислот выделяется хлор
СаОС1г+2НС1 = Са(Й2+И»О + С12, (15)
CaOCl2 + H2SO4 = CaSO4-|-H2O-|-Cls. (16)
Предполагают, что при этом сначала освобождается хлорноватистая кислота, которая, однако, моментально реагирует с соляной кислотой с выделением хлора:
СаОСЬ + 2НС1 = CaCl, + НОС! + НС1 )
СаОС12 + HjSO,, CaSO4 + НОС! Ч- HC1J
HOC! + НС 1 = Cl 2 • H нон.
На прямом солпечном свету па воздухе (главным образом в присутствии двуокиси углерода) хлорная известь достаточно быстро отщепляет кислород. Такое же разложение происходит, когда растворы хлорной извести нагревают в присутствии некоторых окпелов и гидроокисей, действующих в качестве катализаторов, например окиси меди, окиси железа, гидроокиси пикеля, гидроокиси кобальта.
Эту реакцию можно использовать для получения кислорода в лаборатории.
В противоположность сильно гигроскопичному хлориду кальция СаС12 чистая хлорпая известь не притягивает воду из воздуха. Хлорид кальция не может быть также экстрагирован из пес непродолжительной обработкой спиртом. На этом базируется представление, что хлорная известь является смешанной солью Ca(OCl.)CJ. Это представление первоначально было высказано Одлпнгом (Оtiling). Как показал Нейман (Neumann, 1926), хлор ид-гипо хлорит кальция СаС1(ОС1) существует в хлорной извести в виде комплексного соединения с Са(0Н)2. Смотря по условиям образования, можно получить соединения
[ЗСаСЦОС!) Са(ОН)2|-5Т12О,
[ЗСаС1(ОС1).Са(ОН)2].ЗН2О и [СаС1(ОС1)-Са(ОН)21Н2О
(кристаллизационную воду можно извлечь из соединений в вакууме), Первое из трёх приведенных соединений образуется при условиях, используемых в процессе технического получения хлорной извести. Ойо устойчиво в закрытых сосудах в отсутствие свети. На солнечном свету в закрытом сосуде происходит разложение, являющееся окислительно-восстанови тельным процессом: 6СаОС13 ₽= 5СаС12 + Са(СЮ^)2. На воздухе,
Седьмая группа периодической системы
859
даже в темноте, происходит постепенное разложение с отщеплением кислорода, Распаду чрезвычайно способствует присутствие углекислоты. Поэтому хлорная известь, которая была приготовлена из неполностью обожженной извести или с применением хлора, содержащего двуокись углерода, не устойчива даже в закрытых, светонепроницаемых сосудах.
Хлорную известь все в более широком объеме применяют в качестве отбеливающего средства для целлюлозы, бумаги и тканей, отчасти также еще для стирки и в домашнем хозяйстве; здесь она, однако, все более вытесняется перекисными отбеливающими средствами, обычно менее разрушающими волокна ткани. Часто хлорную известь используют также в качестве дезинфицирующего средства. Значительные количества ее применяют для очистки сырой нефти.
Белильные свойства технической хлорной извести определяются содержанием в ней гипохлорита. Его содержание устанавливают определением количества хлора, который выделяется при добавлении соляной кислоты [ем. уравнение (15), стр, 858 J. Его выражают в процентах от общего веса продукта и обозначают naif содержанке «действующего хлора». Содержание ««действующего хлора» в качественном торговом продукте лежит между 35 и 39%. Илистом соединении [3CaCl(OCV|-Ca(OH)2J-5Il2O оно составляет 39,05% . Часто содержание действующего хлора выражают в градусах. При этом различают онслийсвис градусы и французские, или градусы Гей-Люссака'. Английские градусы означают содержание «действующего хлора» в весовых процентах; французские градусы, наоборот, показывают, сколько) литров га сообразного хлора (при 0° и 760 дни рт. <:<>,.) Ъыдеддет 1 кг продукта.
Получение хлорной । язве сти первоначально было осуществлено в Англин Теннантом (Tennant) в 1799 г. Ес, второй половине прошлого столетия хлорную известь еще в больших количествах выл о мыл » в Германию из Англии.
В настоящее время транспортировку хлорной ялиестп заменяют часто транспортировкой жидкого хлора, из которого уже на месте получают необходимые растворы хлорной извести путем пропускания его через известковое молоко. Недавно вместо хлорной извести все в возрастающем объеме в продажу стал поступать, особенно на экспорт, технический гипохлорит кальция Са(ОС1)2 с содержанием «действующего хлорал до 80% (чистый Са(0С1)2 имеет 99,19% «действующего хлора»].
Хлористая кислота и хлориты
Хлористая кислота НС102 образуется в качестве промежуточного продукта при гидролизе двуокиси хлора. Одни ко о на очень быстро разлагается. Болес устойчивыми в растворе являются ее соли — хлориты М' [С1О3]. Они получаются вместе с хлоратами при действии двуокиси хлора на растворы гидроокисей или карбонатов щелочных металлов
Зою, -р зон- = ciOs + + н3о.
Хлориты, свободные от хлоратов, мп наго получать, смешивая водные растворы двуокиси хлора и переписи натрия (иля перекиси водорода и гидроокиси натрия):
2С1О, + Ха2О, 2Na[C102J + О3,
2С1О2 + НгОе + 2КаОН = 2Na[CIO2J + О3 -ф 2Н2О.
Растворы хлоритов действуют кик сильные окислители. Твердые хлориты, особенно хлориты тяжелых металлов, при нагревании или ударе разлагаются со взрывом, Теплота образования С(0, 17,18 ккал, HCJO2>aq 13,68 ккал/молъ; свободная энер-гпя образования: соатветггвеино 0,23 кхал/маль (t = 25а) [Latimer W. М.,
.Б Лш. ciiirijt. Sue,, 69. 2598, 1947].
Хлорноватая кислота и хлораты
Хлорноватая кислота НСЮ3 образуется, как уже было указано, при разложении хлорноватистой кислоты. Обычно при ее получении исходят из ее солей — хлоратов. Например, ее получают взаимодействием хлората бария с разбавленной серной кислотой
Ва[С1Оз}24-Н38ОА=Ва[ЙО4] %2НСЮ3.
860
Глава 27
Упариванием в вакууме бесцветный раствор можно сконцентрировать без разложения кислоты до содержания 40% НСЮ3. Если упаривать далее, то начнется медленное разложение; разложение становится очень быстрым, когда концентрация превысит 50%, Разбавленные растворы можно нагревать почти до кипения без разложения. Однако веществам, способным окисляться, хлорноватая кислота, особенно в концентрированном растворе, легко отдает свой кислород.
Хлорноватая кислота является сильной одноосновной кислотой. В водном растворе она практически полностью диссоциирует. Кажущаяся. степень диссоциации (найденная из отношения электропроводностей Л/Лф) составляет 79% в 1 и. рацтворе при 18°.
Хлораты. Соли хлорноватой кислоты — хлораты М1[С10з1 — образуются при разложении гипохлоритов в водном растворе при нагревании
3[СЮ]' = [С1О3]'+2С1'.
Поэтому они образуются (вместе с большим количеством хлорида), если пропускать хлор в теплые растворы гидроокисей щелочных или щелочноземельных металлов или если подвергать электролизу без диафрагмы jijnpn нагревании растворы хлоридов щелочных металлов
ЗС1г4-6ОН’ = [СЮзГ + 5СГ+ЗН2О.
Хлораты — бесцветные вещества, в твердом состоянии при обычной температуре совершенно устойчивы, в воде : легко растворимы. Их растворы обладают мепее сильным окислительным действием, чем растворы гипохлоритов. При нагревании хлораты отщепляют кислород. Часто при этом сначала происходит распад на хлорид и перхлорат
4MI(C103] = MICl-f-3MIIC104]. *
При более сильном нагревании перхлорат разлагается затем на хлорид > и кислород
М1[СЮ4( = М|С1 + 2О2.
Хлорат калия (хлорноватокислый калий, Kali ckloricum) K[C103J в технике получают различными способами, среди которых наиболее известным является способ Либиха. Хлор пропускают в нагретое известковое молоко и смешивают с хлоридом калия, после чего при охлаждении выкристаллизовывается хлорат калия,, в то время как хлорид кальция остается в растворе
6Са(ОН)г-|-6С13=Са[С1Оз1!»-|-5СаС1г-|-6НгО, Са[С1ОзГ2+2КС1 = 2К[С1Оя14-СаС12.
Хлорат калия образует бесцветные, блестящие, моноклинные таблички охлаждающего вкуса (уд. вес 2,33; т. пл. 370°). В горячей воде он очень легко растворим, значительно труднее растворим в холодной (при 0° в 100 г воды растворяется 3,3 а КС1О3, при 20°—7,3 г и при 100°—56,0 г КС1О3), В абсолютном спирте К.С1О3 не растворим. Около 400е хлорат калия начинает разлагаться. При этом сначала образуется перхлорат Калия, который затем, при болео сильном нагревании, разлагается с отщеплением кислорода (ср. выше). Примесями, такими, как двуокись марганца, опись железа и т. д., температура разложения значительно снижается и обра-
* Наряду с этим, как правило, происходит также отщепление кислорода с непосредственным образованием хлорида.
Седьмая группа периодической системы 861
зовация перхлората при этих условиях ие происходит, Прй быстром нагревании до температур, лежащих выше его температуры разложения, хлорат калия может с силой взорваться.
Вследствие способности легко отдавать кислород при нагревании хлорат калия часто используют при сплавлении и т. д. в качестве окислителя. В болыпях количествах его применяют в спичечной промышленности.
Появившиеся сначала в Швеции «безопасные, спички», которые загораются только при трепни о специально приготовленную поверхность, в качестве воспламеняющегося вещества содержат смесь, изготовляемую из хлората калия,; серы нлц сернистой сурьмы, раствора декстрина, стеклянного порошка п красителя. Поверхность для трения смазана смесью красного фосфора, раствора декстрина и сернистой сурьмы. Под действием тепла, выделившегося при трении, следы красного фосфора превращаются в белый фосфор; пос ледний, воспламеняясь, поджигает воспламеняющуюся массу спички, а от ное загорается дерево.
Хлорат калия применяют, кроме того, в пиротехнике и для изготовления взрывчатых веществ. Его исиользуюг также в медицине, например разбавленный раствор хлората калия служит для полоскания горла. В больших количествах (более 1 г), как и все хлораты, он ядовит.
Хлорат натрия] Na[CJOa) чрезвычайно легко растворяется в воде (101 г в 100 г воды при 20е) и поэтому в чистом состоянии получается пе так легко, как хлорат калия. Он также расплывается на воздухе, однако кристаллизуется без воды, а именно обычно в кубической системе.
В метастаб.изилюм состоянии бывает также ромбоэдрическим и моноклинным. Применяют его в качестве окислителя, как исходное вещество для получения перхлоратов, а также в большом количестве для борьбы с со ринка ми.
В настоящее время хлорат патрия приготовляют в значительно больших количествах, чем хлорат калия, п притом почти исключительно электролитическим путем.
Двуокись хлора и шестиоквсь хлора
Двуокись хлора С1Ог—зеленовато-желтый взрывчатый газ с резким запахом. Он образуется при действии концентрированной серной кислоты на хлорат калия благодаря тому, что освобождающаяся иа хлората .калия хлорноватая кислота распадается под действием серпой кислоты
КСЮ3-Н128О4==НС.Юз + КНЗО4,
ЗНС1О3=1ТС1О4+Н2О+2С1Ог. ?
Ири действия разбавленной серной кислоты на смесь хлората калия и щавелевой кислоты также получается двуокись хлора
2К.СЮ3-|- ГГ2СЙО4-^- HaSO4= K2SO4+2Ff2O-j-2CO2-(-2C(O2.
Охла?кденисм двуокись хлора легко удается превратить в красно-бурую жидкость. При более сильном охлаждении она затвердевает, образуя оранжево-красные твердые хрупкие кристаллы (т. пл. —76°; т. кип. +9,9s), Плотность пара соответствует формуле С1О2.
Как и молекула CLO. молекула Ci Оа также образует угол; межъядерное расстоя-
ние С1«—> О составляет 1,49Д; равен 116,5° (Hedberg, 1950); дипольный
ЧО
момент р 0,78.16-1».
862
Глава 17
Молекула СЮ3, так же как молекула NO->, содержит нечетное число электронов. Полинг предположил, что у двуокиси хлора мезомерия имеется между двумя состояниями
/О I jO|
[СК- и |С1^_
О | \ |
с простой связью, на которую накладывается трехэлектронпая связь, так же как у NO2 (ср. сур. 641).
Двуокись хлора — сильно эндотермическое соединение (теплота образования —26,3 ккал) и, следовательно, очень неустойчиво. Уже при слабом нагревании или при соприкосновении с горючими веществами происходит распад на кислород и хлор, сопровождающийся сильным взрывом.
При соприкосновении с Р2 также тотчас происходит распад со взрывом. Однако при осторожном проведении опыта получается соединение C1O2F (бесцветный газ, т. кип. —6°, т. пл. —115°), которое, вероятно, следует рассматривать как фторид хлорноватой кислоты (Schumacher, 1942).
В воде двуокись хлора легко растворима, она образует с ней твердый гадрат, который, однако, с трудом получается в чистом состоянии. В темноте при обычной температуре двуокись хлора в водном растворе остается неделями почти без изменений. Лишь очень медленно начинается взаимодействие с водой с образованием соляной и хлорноватой кислот, В качестве промежуточного продукта при этом образуется хлорноватистая кислота, В присутствии значительных количеств ионов хлора гидролиз двуокиси хлора сильно ускоряется. С еще большей скоростью происходит разложение в спиртовых растворах. В этом случае разложение останавливается на стадии образования хлорита
2С1О,+2ОН' = СЮ^-|-С1О'-|-НгО.
Шеетцоквсь хлора С12ОВ была цолучепа в 1925 г. Боденштейном при исследовании разложения жидкой двуокиси хлора под влиянием спота- Опа возникает также при действии о во па па двуокись хлора, и при применений подходящей аппаратуры (не следует применят!, смазанных кранов) может быть таким образом сравнительно легко приготовлена в больших ко.1 ячеств ах.
Шестионись хлора представляем собой темно-красную, дымящую на воздухе жидкость уд. веса 1,65, которая затвердевает при —1".
В чистом состоянии при комнатной температуре она довольно устойчива, оДпако при coupiiKOCiioBiHunj с органическими веществами взрывает с большой силой. С водой при известных условиях она также реагирует со взрывом. Образование вещества со свойствами шестнокиси хлора еще в 1843 г. паб.чгодал Миллон, но ему нс удалось правильно определить его состав. В против оно до ж л ость другим о кис лам хлора шести-окись хлора обладает при обычной температуре лишь незначительным давлением пара (около 1 мм рт ст). Поэтому ее можно сравнительно легко отделить от пих фракционированной перегонкой в вакууме.
Семнокись хлора (ангидрид хлорной кислоты), хлорная кислота и перхлораты
Семнокись хлора С12О7 является ангидридом хлорной кислоты НСЮ4 и может быть получена из нее обезвоживанием при обработке пятиокисью фосфора
2НС1О4-|-Р2О5=С1аО7-|-2НРО3.
Семиокись хлора — бесцветная, весьма летучая (т. кип. 83°), маслянистая жидкость. Она часто взрывает при соприкосновении с пламенем или от удара; с иодом опа также реагирует со взрывом. Однако С12О7 можно вылить па бумагу, дерево и аналогичные материалы и при этом она не вступает с ними в реакцию.
Седьмая группа периодический системы
863
Семиокись хлора медленно растворяется в холодной воде с образованием хлорной кислоты Ci «О, + Н20 = 2НСЮ4.
Хлорная кислота НСЮ4 является самой устойчивой среди кислородных кислот хлора. Ее можно получить в свободном состоянии действием концентрированной серпой кислоты на перхлорат калия КСЮ t + H2SO4 = = KT-ISO4 + НС1О4. .
Реакционную смесь оиорожяо нагревают на масляной бане, медленно повышая температуру от 90 до 160°. JB вакууме, создаваемом водострумвым насосом, перегоняется безводная кислота. Ее можпо очистить повторной перегонкой в вакууме.
Безводная хлорная кислота — бесцветная, весьма подвижная, сильна дымящая на воздухе жидкость, которая затвердевает только при сильном охлаждении (температура плавления —112’'). НСЮ4 можпо перегнать, без разложения только при пониженном давлении. Если ее нагреть, то она в результате разложения окрашивается в коричнево-красный цвети, наконец, взрывает. Медленное разложение происходит также уже на холоду, особенно в присутствии примесей. Веществам, способным окисляться, хлорная кислота легко отдает свой кислород (по пе брому). При соприкосновении с горючими материалами, такими, как бумага, дерево, древесный уголь, она госиламеняет их со взрывом. При попадании на Кожу она вызывает болезненные и с трудом поддающиеся лечению ожоги.
С водой хдорпая кислота смешивается в любых отношениях. Опа образует с ной несколько гидратов, среди которых моногидрат HClOi-HaO отличается сравнительно высокой точкой плавления (4-50°). Концентрированные растворы хлорной кислоты в противоположность безводному соединению обладают маслянистой консистенцией, подобно концентрированной серной кислоте. В водном растворе хлорная кислота гораздо устойчивее, чем в безводной форме. Это относится даже к высококонцентрированным растворам, папример к 72%-пому раствору, который перегоняется с постоянной температурой кипения около 203°, правда при этом, отчасти разлагаясь; еще устойчивее разбавленные растворы НСЮ4, Хлорная кислота является одноосновной кислотой. Она относится к наиболее сильным ив известных кислот. Ее соли М1 [СЮ* ] называются перхлоратами,
Хаорная кпепота образует соли даже с такими по живительными радикалами, которые в других случаях встречаются редко, например радикалом нитро в и я (NO-j]’’ (ср. стр. (>й7). Моногидрат хлорной кислоты моя;но рассматрпгать ваг: солеобраэиои соединенно, а именно как перхлорат' гидроксония [ОН3|+[С1О4]-. Это следует главным образом ns рентгенограммы соединении, снятой Фолкнерам (Volmer, 192<П, которая ока адась почти идентичной рентгенограмме перхлората аммония [N Н4 J+ICIO4]’.
Ггя’ цой Oil на F от хлорной кислоты производится пврхлорилфторпд ClOaF, который был получен Энгельбрехтом (Engelbrecht, 1952) при электролизе перхлората 1тэтр.Ю|, рчгтгмраютати в жидком фтористом водороде. Это бесцветный газ (т. кип. -ДЖ. т п | . —ЮД который разлагается крепким раствором щелочи на перхлорат и фгйрвд ‘.гт'мю., с ци’Ктй практически ие взаимодействует. Изомером итого соединенья является интктапып Еоде (Bode, 1951) хл-орилоксш/ппорид ClO-j(OF).
Перхлораты. Сели хлоркой кислоты — перхлораты являются наиболее устойчивыми кислородными соединениями хлора. Многие из них в твердом состоянии, так же как и в водпом растворе, проявляют почти неограниченную устойчивость. Однако при нагревании происходит разложение, в процессе которого с образованием хлорида выделяется кислород. В случае перхлората калия К.СЮ4 это наступает только выше 400°; легче разла
864
Глава 17
гаются те перхлораты, которые кристаллизуются с водой, а также те, которые содержат органические составные части. Большинство перхлоратов очень легко растворимы в воде. Перхлораты калия, рубидия и цезия давольно трудно растворимы в холодной воде, но существенно легче растворяются в теплой воде (ср. табл. 36, стр. 211). Поэтому они хорошо кристаллизуются и легко приготовляются. Последнее относится также еще к перхлорату аммония, который тоже обладает пе очень высокой растворимостью.
При приготовлении перхлоратов щелочных металлов обычно исходят из соответствующих хлоратов, которые либо разлагают при нагревании (17), либо окисляют электролитически в водном растворе (18)
4KC10s=3KC104-fc-KCI *, (17)
ClOJ-j- Н2О—2е=CJOJ-j- 2Н-. (18)
Остальные перхлораты сбыл но приготовляют растворением соответствующих металлов, гидроокисей или карбонатов в водном растворе хлорной кислоты, который можно получить, папример, взаимодействием перхлората калия с кремнефтористоводородной кислотой. Выпадающую при этом трудпорастворимую соль калия этой кислоты отфильтровывают 2КС1О4 ф- H3SiFe = K.2SiFs ф- 2IICIO4 (о получении сухим путем см. ниже).
Водную хлорную кислоту часто используют й качестве реактива на иопы калия, а также для их количественного определения. Перхлораты, и прежде всего перхлорат аммония NH4CIO4, нечувствительный к ударам, но взрывающийся от инициирующего воспламенения, применяют в промышленности взрывчатых веществ вместо легко разлагающихся и поэтому более опасных хлоратов.
Перхлорат калия встречается в природе в небольших количествах в неочищенной чилийской селитре (CaZze/ze). Так как он является сильным ядом для растений, то его следует удалять, если селитру применяют в качестве удобрения.
Относительно аегнорает варимых перхлоратов чаще всего отмечается, что она расплываются; однако в ряде случаев было установлено (например, NaC104, ГлС1О4, Ва(С1О4)Е], что это происходит только с солями, загрязненными 11СЮ4. Перхлораты щелочноземельных металлов все в возрастающей мере, находят техническое применение, например, для приготовления фейерверков. Условия приготовления этих соединений сухим путем, который удобен тем, Что при этом получаются безводные перхлораты, были исследованы Смитом (Smith G, F., Z. anorg. Chonl., 223, 1, 1935].
Весьма интересным свойством перхлоратов является их очень незначительная склонность к гидролитическому расщеплению. Кроме того, насколько известно, ион СЮ4 не способен фигурировать в качестве составной части комплексов.
Аналитические сведения. Гипохлориты легко распознают по хлорному запаху, который появляется при подкислении их растворов;’кроме того,' их обнаруживают по сильной отбеливающей способности. С нитратом серебра опи дают осадок хлорида серебра AgCl, так как образующийся вначале гипохлорит серебра AgOCl быстро разлагается по уравнению 3AgOCl AgC103‘+ 2AgCl..
Количественное определение содержания гипохлорита в хлорной извести, белильных щелоках и т. д, производят обычно по методу Попо. По этому методу для определе
* Одновременно часть хлората калия разлагается но уравнению 2КС103= 2КС14-Ч-ЗО2.
Седьмая группа периодической системы 865
ния «действующего хлора» пробу титруют раствором трехокиси мышьяка. Мышьяковистая кислота окисляется гипохлоритом до мышь яко гоа кислоты. /Гонец реакции устанавливают по появлению пятен на подкрахмальдай бумажке.
Хлораты и перхлораты не дают осадков с нитратом серебра. Хлораты в отличие от перхлоратов можно восстановить двуокисью серы или в щелочном растворе водородом в момент выделения (который получают при посредстве сплава Деварда), после пего хлорид серебра выпадает. Для перхлоратов характерно образование осадка при добавлении хлорида калия; этот осадок вновь растворяется при нагревании.
КИСЛОРОДНЫЕ СОЕДИНЕНИЯ БРОМА
Окислы брома с трудом получаются и крайне неустойчивы. Поэтому они стали известны только в последнее время.
Шумахер в 1929‘г. действием озона на бром первым получил окисел ВгО3 или ВгаОе, который образует бесцветные иглы и устойчив только пи же—80°. Шумахер придал ему формулу }lr3Os; однако Пфягогмахер (Pflugraacber, 1955) показал позднее, что его состав отвечает формуле ЕгО3. Удвоена ли эта формула по аналогии с шестиокисью хлора С1аО|} нелеп,ствие неустойчивости соединения, установить не удалось. Действием тлеющих разрядов па смесь Вга и О2 Шварцу удалось получить в 1937 г. двуокись брома, Соединение осаждается в средней части разрядной трубки, охлаждаемой жидким воздухом, в виде твердого вещества цвета яичного желтка, которое но плавится, а распадается при 0° па Вг.2 и О2. Наряду с этим при распаде образуется окисел Вг3О. Образование итого соединения было впервые обнаружено Цинтлем в 1930 г. при пропускании паров Вгй над HgO при 50—100°. В чистом состоянии оп был получен только в 1938 г- Шварцем прн разложении двуописи брома в высоком вакууме [подробнее см. J. prakt. Chem., N. F., 152, 157,1939]. Ёг-0 устойчив только июне —40°. Он обладает темно-коричневой окраской, растворяется в CCI4 с интенсивно зеленой окраской и лег>;о взаимодействует с раствором натровой щелочи, давая гипобромит.
Из кислородных кислот брома с достоверностью известны только две кислоты: бромноватистая кислота НБгО и бромноватая кислота НВгО3. Эти соединения и их соли, зипобромиты и броматы, во многом аналогичны соответствующим соединениям хлора.
Бромистая кислота ПВтО3 соответственно бромит-иопы [ВгО2Р, По-видлмому, существуют в качестве промежуточных продуктов (очень мало устойчивых даже в растворе) при некоторых превращениях, например при разложении гипобромит-иопов, которое является о кислите ль по-восстановительным процессом: 2ВгО' = BrOi -ф- Вг'; ВгО» -|- ВгО' == ВгО3 л- Вт' (Ciarans, 1913). Однако об этой реакции еще мало что известно.
Несмотря на то что среди кислородных соединений хлора наиболее устойчивыми являются хлорная кислота и перхлораты, доказать существование аналогичных соединений брома не удалось.
Б ром нова чиста я кислота НВгО и гипобромиты МЧВгО! получаются точно так же. как хлорноватистая кислота и гипохлориты. Как и гипохлориты, они обладают ярко выраженными отбеливающими и окислительными свойствами. При пропускании паров брома над гашеной известью удается получить соединение, аналогичное хлорной извести. Растворы гипобромитов щелочных металлов вследствие удобства их получения применяют иногда в количественном анализе в качестве окислителей. Они имеют соломенно-желтую окраску и обладают своеобразным ароматическим запахом. При нагревании или при подкислении они разлагаются тотчас с образованием бромида (соответственно бромистого водорода) и бромата.
55 г. Реки
866
Глава 17
Растворы гипобромитов медленно разлагаются даже при обычной температуре; это происходит отчасти вследствие окисления — восстановления гипобромит-ионов (ЗВгО'=]?гОз + 2Вг'), отчасти вследствие неустойчивости образующейся при гидролизе бромповатистой кислоты. Константа диссоциации НВгО составляет, по данным Скрабаля (8 к г a b а 1, Z. Elektrochem., 48, 314, 1942], около 2-10~и.
В кристаллическом состоянии гипобромиты впервые были получены Шольдером-[Scholder R., Z. anorg. Chem., 268, 279, 1952], а именно: NaOBr-5H2O, NaOBr- 7H3O и KOBr-3H2O. Эти соединения (желтые иглы, очень легко растворимые в воде) очень неустойчивы, прежде всего это относится к соли капня, которая быстро разлагается уже при О’.
Бромная кислота НБгО3 образуется при пропускании хлора п бромную воду
Вг2Н-5С1г 4-бНгО = 2НВгОз+ 10НС1
или из ее бариевой соли при взаимодействии с разбавленной серной кислотой
Ba[BrO3]2-|-H2SO4=Ba[SO4]-|-2HBrO3.
Эта кислота устойчива только в водном растворе. Раствор бесцветный; при концентрировании происходит разложение.
Броматы. Более устойчивыми, чем бромноватая кислота, являются ее соли—броматы М1[БгО3]. Их получают аналогично хлоратам при прибавлении брома к нагретым растворам гидроокисей щелочных металлов или электролизом растворов бромидов. Как и хлораты, броматы легко-отдают кислород. При нагревании со способными к окислению веществами, такими, как уголь или сера, они дают вспышку и легко распадаются также сами по себе с выделением кислорода.
Бромат калия, КВгО3 — бесцветные, тригональные кристаллы, d — 3,27. Растворимость при 20° составляет 6,9 г, при 100°—49,7 г в 100 г воды. КБгОз применяют прп броматометрическом титровании. При температуре выше 434° эта соль разлагается с выделением кислорода.
Вромтпомвтрия основана на том, что ионы BrOJ могут быть Легко количественно восстановлены в солянокислом растворе, причем, как только в о сста повитель израсходуется, образуется свободный бром (BrOJ + 5Вг' 4- 6Н‘~ЗВг2 + ЗН2О), который, разрушая цветной индикатор (метиловый оранжевый или метиловым красный), указывает на конец титрования.
Растворы, содержащие сеобоЙпый бром, также применяют для титрометрического-определения восстановителей; кроме того, применяют растворы КВт для определения сильных окислителей. В этом случае речь идет о бромометрии. При бромометрий в качестве цветного индикатора обычно используют индигокармин. Ошибки, которые могут произойти вследствие высокого давления пара растворов брома, уменьшаются, если пользоваться аппаратурой, описанной Д’Ансом (D’Ans J., Angew. Chem., 63, 45, 1951].
КИСЛОРОДНЫЕ СОЕДИНЕНИЯ ИОДА
Если воздействовать иодом на гидроокиси щелочных или щелочноземельных металлов или на карбонаты щелочных металлов в водном растворе, то, как и в случае хлора и брома, сначала образуются гипоиодиты — соединения общей формулы МЧЮ]. Однако они настолько неустойчивы, что уже при обычной температуре за очень короткий срок в результате окислительно-восстановительного процесса переходят в йодаты M4IO3L Еще более неустойчива сама иодноватистая кислота, которую можно получить на короткий срок в сильно разбавленном растворе при встряхивании водного раствора иода с окисью ртути. НЮ является очень слабой кисло
Седьмая группа периодической системы
867*
той и присутствует поэтому вследствие гидролитического расщепления также в водных растворах гипоиодитов. Б чистом состоянии гипоиодиты нельзя получить из-за их большой неустойчивости. Водные растворы их обладают запахом шафрана и в отсутствие избытка щелочи окрашивают в синий цвет иодкрахмальную бумажку и проявляют еще более сильное отбеливающее действие, чем растворы гипохлоритов и гипобромитов.
Так называемая иодноватистал кислота ЮН является амфотерной гидроокисью, у которой ио существу основной характер преобладает. Константа диссоциации по кислотному типу в соответствии с уравнением ЮН ГО'4-Н' составляет около 4.10_i3 [Skrabal, Вег., 75, 1570, 1942—1943]; константа диссоциации по типу основания в соответствии с уравнением ЮН Г ОН' составляет 3‘ 10"10 [Murray II., J. Chem. Soc., 127, 882, 1925]. Константа гидролиза для 13 4- НОН IQH -МЧИ' составляет З'Ю-13, а константа диссоциации по уравнению I2 Г 4-1' равна 9,6-10-9.
Гидроокись иода стабилизируется присоединением нейтральной молекулы иода к электро положительному атому йода. Образующееся таким образом соединение 12-ЮН устойчиво продолжительное время в кислом растворе, в то время как ЮН при равных условиях тотчас распадается на иод и подловатую кислоту (Skrabal, 1909).
Пятиокись иода, йодноватая кислотами йодаты. Йодноватая кислота ШОз устойчивое, чем хлорноватая и бромноватая кислоты. Она легко образуется в чистом кристаллическом состоянии и может быть, например, выделена из своих солей — йодатов (йодата натрия) при нагревании с серной кислотой (19). Кроме того, ее можно приготовить окислением иода концентрированной азотной кислотой (20) или окислением иода хлором в водном растворе (21).
Образующуюся в последнем случае одновременно хлористоводородную кислоту устраняют добавлением окиси серебра (22).
NalO3+H2SQ4=HlO3-}-NaHSO4, (19)
5HNO3+3/al2f=3HI03+5NO+H20-<2°)
1,+5С,12+6НэО = 2ШО34-10НС1, (21)
10НС1+5Ag2O = iOAgCl 4- 5Н3О. (22>
Йодноватая кислота образует бесцветные кристаллы со стеклянным блеском и горьковато-кислым вкусом. Она кристаллизуется в двух различных, не переходящих одна в другую формах, принадлежащих к одному и тому же бисфеноидальному классу ромбической системы. Б воде она крайне легко растворима (100 г воды растворяет при 16° 310 в НЮ3). Если ее нагревать, то она отчасти плавится при 110°, превращаясь в более бедную водой форму, так называемую ангидроиодноватую кислоту Hl3Og. Эта кислота кристаллизуется также из водного раствора при температуре выше 110°. Если дать постоять йодноватой кислоте при 30—40° на сухом воздухе, то в результате выветривания происходит переход в более бедную водой форму. При нагревании до 195° отщепляется вся вода и остается пятиокись иода 12О6 в виде белого порошка. Последняя распадается в темноте только выше 300°. Б отличие от окислов остальных галогенов это — экзотермическое соединение (теплота образования 43 ккал/моль HOj). Пятиокись пода легко растворяется в воде с образованием йодноватой кислоты; таким образом, ее следует рассматривать как ангидрид последней..
Соли иодповатой кислоты, йодаты, в большинстве случаев по составу соответствуют общей формуле МЧО3. Однако известны также йодаты состава КРНПССЕ (гидродииодаты) и М1Н2[Ю3]3 (дигидротрииодаты). Последние можно рассматривать как продукты присоединения йодноватой кислоты к нормальным йодатам М11О3. Подобные продукты присоединения уже встречались у различных кислот.
55®
868
.Глава 17
Йодноватая кислота обладает ск дойностью к полимерияацил, правда в концентрированных растворах. Однако она является одноосновной кислотой Н1О3. Это следует прежде всего из проводимости растворов ее нейтральных Солей. Она вполне соответствует проводимости солей типа MX, а не типа (ср. стр. 435), Константа диссоциации йодноватой кислоты составляет, согласно Абелю (Abel, 1934), К — = 0,19 при 18°. /
Относительно структуры кристаллической решетки Н1О3 см.: Rogers, НеЬ mholz, J. Am. chem. Йос., 63, 278, 1941.
Нормальный потенциал электрода AgIO3 составляет 4- 0,358 в [Cavallaro L., Gazz. chim. ItaL, 72, 343, 1942],
Коэффициент активности иона 1O3. по данным Ковалларо, оказывается 0,9104 для с = 0,01; 0,8379 для с = 0,05; 0,7979 для с = 0,1 и 0,7475 для с = 0,2 люль/л.
Йодноватая кислота и йодноватокислые соли склонны к образованию продуктов присоединения с галогенидами щелочных металлов, например NaBr'NaIO3-6H2O, а также к присоединению других кислотных ангидридов, таких, как SO3, SeO3, ТоО3, МоО3 и т. Д. (образование гетерополикислот).
Иодаты много устойчивее, чем хлораты и броматы. Однако они также являются ярко выраженными окислителями, дают вспышку на раскаленном угле (однако не очень сильную) и детонируют в смеси с горючими веществами от удара.
Иодат кальция CaIIO3]s встречается в природе в виде лаутарита. Иодат натрия Na[IOj содержится в незначительных количествах в качестве примеси в чилийской селитре.
Приготовление йодатов можно осуществить окислением иодидов в щелочном растворе. Окисление молено провести электролитическим путем.
Иодаты получают также растворением иода в горячих растворах гидроокисей щелочных или щелочноземельных металлов. Интересным способом получения является способ, основанный на действии водных растворов хлоратов на элементарный иод.
Реакция протекает весьма сложно, так как выделяемый вначале иодом из хлората хлор в дальнейшем вновь реагирует с. образованием хлората. Например:
2КС1О3 + Ь • ;-Н2О = К ГГ[1 о3]г+ С124-К.ОН;
Гидродииодат калия
Gb~i-K0H=KC1 + IJ0C1
При нагревании
ЗН0С1 ----—-------> НС1О3+2НС1;
HG103 4-2НС14- ЗКО Н = К СЮ3Ч- 2КС1+ЗН2О.
Если отбросить другие побочные реакции, то в качество суммарного уравнения оказывается
11КС1О3 4* 612 4- ЗН2О = 6КН[103]2 4 ЗС12 ф 5КС1.
Йодноватая кислота, соответственно подкисленный раствор йодата, реагирует с иодистым водородом с выделением иода
НЮ34-5Ш = 312+ ЗИ3О,
или
10^4-51’-]-6ГГ=3124-ЗН2О.
Это выделение иода в случае йодноватой кислоты происходите существенно большей скоростью, чем в случае хлорноватой и бромноватой кислот, хотя последние в других случаях гораздо менее устойчивы, чем йодноватая кислота.
Если R раствору, который, как, например, раствор сульфата алюминия, вследствие частичного гидролиза дает сильно кислую реакцию, добавить смесь иодида
Седьмая группа периодической системы
863
и йодата калия, то в результате приведенной выше реакции затрачиваются водородные ионы. В связи с этим равновесие гидролиза сдвигается; гидролиз протекает полностью, если удалять освобождающийся иод тиосульфатом. По предложению Штока этим пожауюгся иногда в аналитической химии для осаждения гидроокисей, которые при других условиях легко увлекают вещества из раствора. Растворы йодатов можно применяй, для оксидиметриЧСскоро титрования (иодстгометрия).
От йодатов замещением О на F производятся фтороиодаты.г например фтороиодат калия K[IO2Fa]. По данным Гельмгольца и Роджерса [Helmholz, Rogers, J. Am. chem. Soc., 62, 1537, 1940J ион [1ОгГ2]" обладает такой же структурой, как ТеС14 (см. стр. 805). Межатомное расстояние: I + О = 1,93А; I+-+F = 2,00 А,
< К ~ 100°.
О
Иодная кислота и перйодаты. Многие йодаты, например иодат бария, при прокаливании переходят в перйодаты
5Ва{1О3]г = Вай[1О6]2+«3+9О2. Ио дат барин Перйодат бария
В растворенном состоянии перйодаты получают окислением йодатов хлором в щелочном растворе
[ГО3Г + бд1-Г4-С1г=[1Ов]-">'-|-2С1'4-ЗН2О
или электролитическим путем
]1О3Г 4- 6ОН'—2е=IЮ6]""'+311гО.
Из соли бария взаимодействием с серной кислотой можно получить свободную кислоту
Гу 1 ое), 4-5H2SO4 = 5Ba|SO.J + 2HsIOe.
Ее также можно получить непосредственно, например электролизом раствора йодноватой кислоты.
Иодная кислота Н5Ю6 образует бесцветные призматические, быстро расплывающиеся иа влажном воздухе кристаллы, которые плавятся несколько выше 130°. При немного более высокой температуре иодная кислота уже начинает разлагаться; она переходит при этом в ангидрид йодноватой кислоты, пятнокись иода, выделяя, кроме воды, также кислород. Следовательно, ее собственный апгидрид не удается получить. В воде иодная кислота легко растворима. Она является пятиосновной кислотой, и притом весьма слабой. Ее нормальные соли типа М.|[Ю6] (ортоиериодаты) сильно гидролизовапы в водном растворе.
Известны нейтральные и- кислые орт о перйодаты. Но, кроме тога, существуют также перйодаты, производные от более бедных водой форм иодной кислоты; это прежде Вет о мееопериодаты М* (1OEJ (пентокеопсриодаты),метапериодаты М1 JIOJ (тетра-<п;соп1'рИ1.щаты) ц дипериодаты МдЕ120д] (энпеа оксопери од ат ы).
Пе получающийся из водного раствора нейтральный ортопориодат натри я Na5IOB был приготовлен Циптпем (Zintl, 1938) п аг рев а пи ем NaIO4 с Na2O. Соединение значительно более устойчиво при нагревании, чем метанерподат натрии. Оно образуется также, если Nal нагревать с КаЕОй или Na2O при доступе воздуха. Его можно получить также сплавлением Nal с NaOH; однако в этом случае образование идет очень медленно. Нолес быстрое окисление NaJ до Na5IOe в расплаве NaQH удается достигнуть npii добавлении NaNO3 [Z i п t 1 Е., Z. anorg. Chem., 245, 20, 1940].
В отличье от перхлоратов, а также от свободной иодной кислоты почти все перйодаты лишь мало растворимы в воде. При прокаливании они разлагаются.
В то время как- (нейтральные) ортоиериодаты весьма устойчивы и постоиещго разлагаются (с образованием иодидов) только при сильном прокаливании, метаперио-
8ТО
Глава 17
даты, разлагаются при нагревании, часто со взрывом. Метапериодат аммония может взрываться уже от слабого трения (от прикосновения шпателем). Следует отметить, что некоторые перйодаты при нагревании переходят сначала в йодаты с отщеплением кислорода, в то время как некоторые йодаты, как, папример, иодат бария, при прокаливании, наоборот, дают перйодаты. Как следует из потенциала перезарядки, йодаты в водном растворе устойчивее, чем перйодаты. Платиновый электрод, погруженный в раствор, который содержит ио дат- и периодат-ионы в равной концентрации, обладает на 1,51 в более высоким потенциалом, чем нормальный водородный электрод [Abel, 1932]. Метапер йодаты, например NaIO4, К1О4, ИЫО4, (NH_s)IO4,AgIO4, образуют (в противоположность метаантимонатам, ср. стр. 719) решетки, в которых радикалы 1О4 выступают как законченные структурные группы, причем эти решетки относятся к типу шеелита (см. т. II).
Аналитические сведения. В отличие От хлорат-ионов бромат- и иодат-ионы дают с нитратом серебра труднорастворимые осадни, которые растворяются в водном растворе аммиака. Если на разбавленный аммиачный раствор подействовать двуокисью серы, то выпадает бромид серебра, соответственно иодид серебра. Последние можно отличить друг от друга на беповапип их различной растворимости в концентрированном растворе аммиака или лучше перевести их в раствор действием цинка и серной кислоты или сероводородной воды, а затем после добавления хлорной воды извлечь свободные бром или иод встряхиванием раствора с хлороформом.
4. Соединения галогенов между собой
(Обзор)
C1F, газообразный; ВгС1устойчив только IBr, твердый; ICI, твердый; две
т. пл. — 155,6°; в равновесии с Вг2 и т. ц.ч. около 40°; мо дификации
т. кип. —100,1° С1г BrF, жидкий; т. пл. около —33°; т. кип. около 20° т. кип. 116® а-(стабилвная): ру~ биново-краспые иглы; уд, вес. 3,22; т. пл. 27,2°; р -(метастабиль-
ClFa," газообраз- BrF3, я;идкпй; 1С1а, твердый; ная); коричнево-
ный; т. ПЛ. 8,8°; уд. вое 3,11; суб- красные ромби-
т. пл, —82,6°; т. кип. И ,3° т. КИП. 127° лимкруется ческне таблички; т. па. 13,9°;
ВгЬ’д жидкий; т. пл. — 61,3°; т. кип. 40,5° IF6, жидкий; т. пи. 8,5*; т, кип. 97* IFr, газообразный; т. пл. 4,5°; т. кип. 5,5* т.mm. около 100°
Как видно из приведенного обзора, галогены, как правило, могут образовать между собой соединения тем с большим числом атомов, чем дальше они отстоят один от другого в периодической системе. Галогены, расположенные в периодической системе непосредственно один над другим, способны в общем проявлять друг по отношению к другу только одновалентность; лишь хлор по отношению к фтору может быть также тнреявалентным. Бром по отношению к фтору может быть максимально пятивалентным, а иод — максимально селгивалентным.
Соединения галогенов всегда легко летучи; самое высококипящее из них BrFs кипит при 127°. Большинство из них малоустойчиво; однако ня одно из я тих соединений не взрывчато. Все они экзо тер мичны. Однако
Седьмая группа периодической системы 871
теплоты образования их большей частью лишь незначительны. Теплоты образования составляют: для 1Бг 2,5, для ICI 7,9, для 1С13 21,5, для ВгС1 0,34, для CIF 15,0 и для CIF3 42,0 ккал{молъ.
Относительно ассоциации C1F3 с образованием (CJF3)2, равновесия диссоциации и спектра поглощения C1F3, энергий диссоциации F3, GIF я BrF, а также теплот образования C1F и C1F3 ем. Schumacher Н, J., Z. Natarforschg., 2а, 358, 1947.
Мопофторид хлора C1F был получен Руффом в 1928 г. при прямом соедияеиии составных частей при 250°. Приготовление этого соединения затрудняется чрезрычай-пой агресспапостыо сойдняепия, которое реагирует со стеклом с образованием взрывчатых окисло» хлора, а на металлы, включая мышьяк я сурьму, действует еще энергичнее, чем сам фтор. Монофторид хлора — почти бесцветный газ, который лишь при сильном охлаждении может быть переведен в жидкое и затем в твердое состояние.
Трифторид хлора C1F3 образуется при действии избыточного F3 на C1F. Это легко конденсирующийся гаа (вес 1 л составляет 3,57 я), в жидком состоянии светло-зеленый, в твердом состоянии бесцветный. Следует отметить чрезвычайно большую реакционную способность трифторида хлора, в парах которого моментально затирается стеклянная вата. MgO, CaO, ALO-, и другие довольно устойчивые вещества также вступают в реакцию с трифторидом хлора, которая сопровождается появлением пламепи (Ruff, 1930). В США C1F3 выпускают в настоящее время в продажу в бомбах. Во многих случаях его можно применять вместо F2 для фторирования.
Три фторид брома Вт?.; образуется при непосредственном взаимодействии составных частей. При этом в качестве промежуточного продукта возникает монофторид брома BrF, который диспропорциопирует затем: на BrF3 и Вг2. Трифторид брома представляет собой бесцветную тяжелую жидкость, которая сильно дымит на воздухе и активно действует на кожу. Он также очень реакционноспособен. С Fa соединяется при 200°, образуя пентафтарид брома Bt.F5 (Ruff, 1931—1933).
Пентафторнд подаГГА, — бесцветная, дымящая на воздухе жидкость, пары которой сильно действуют па органы дыхания. Пентафторид иода также очень реакционноспособен. Он получается пропусканием фтора над сухим иодом. Подобно трифториду хлора, еще бо.тее реакционноспособен гептафторад иода IF- (Ruff, 1930—1931).
Как установил Брауне (В г а и и е Н., Z. physika). Chem,, (В), 35, 250, 1937) путем электро но графических исследований, в молекулах газообразного 1₽5 все пять Атомов F удалены на равное расстояние от атома иода (1*—>-F = 2,57 А); по всей вероятности, они расположены в форме тригональной бипирамиды.
Моно хлорид брома ВгС1 был открыт Баларом уже в 1828 г. ирп пропускании газообразного хлора в жидкий бром. Однако достоверно существование соединения было установлено только более поздними исследованиями. Оно устойчиво только в равновесии со своими продуктами распада — Вт2 и С12. Степень термической диссоциации составляет При комнатной температуре 43% деплота образования 0,34 ккал) моль (Brauer, 1935).
Монохлорид иода ГС [образуется при пропускании хлора лад иодом или при кипячении последнего с царской водкой и последующего извлечения хлорида из разведенного раствора эфиром. Он образуется в виде красно-бурого масла, которое переходят при перетопке в б о лес устойчивую при обычной температуре твердую а-модпфпкацпю — ру би но во-красные иглы. Водой он сначала разлагается на хлористый водород ц гидроокись иода (иодноватистую кислоту)
IC1 + НОН = НС1 4 ЮН.
Од па ко последняя очень быстро разлагается, так что в качестве конечных продуктов гидролиза ounoaowTi n хлористый водород, йодноватая кислота и свободный иод
5IC1 -Ф ЗНОН == 5НС1 -ф НЮ3 + 212.
Бромид иода ГВт в значительной степени аналогичен моно хлориду нода. Как можно заключить из данных гидролиза, иод в этом соединении является электроположительной составной частью. Это представление подтверждается также тем, что другие отрицательные радикалы также могут соединяться с одновалентным иодом, например остаток цианистоводородной кислоты может соединяться с ним с образованием цианида-
872
Глава 17
иода ICN, а остаток азотисговодородной кислоты — с образованием анида иода I[N;J (см. ниже).
Трихлорид иода 1С13 получают действием избыточного хлора на иод или обработкой монохлорида хлором. 1С13 — желтые, расплывающиеся па воздухе иглы с крайне резким запахом. При нагревании он распадается на IC1 и С12. В органических растворителях, таких, как спирт, эфир, бензол, он растворяется с темной оранжево-красной окраской. Водой оп разлагается по уравнению
21С13 4 ЗН2О = 5НС1 4 IC1 4 НЮ3.
Добавлением соляной кислоты гидролиз можно сдвинуть в обратную сторону. Солянокислый раствор трихлорида иода очень удобен для вскрытия сульфидных минералов, таких, как пирит, так как это соединение проявляет по отношению к сере исключительные окислительные свойства (Birk, 1928; Wilke-Dorfurt, 1930).}
Трихлорид иода способен присоединять хлориды щелочных металлов, а также хлориды других сильно электроноложнтельных веществ с образованием хорошо кристаллизующихся солей типа
_С1 С1
Эти соли [тетрахлороиодиты, тетрахлороиодаты(Ш) j можно также получить действием па йодаты крепкой соляпой кислоты
К[Ю3] 4 6НС1 = KfIClJ -1- CJ3 4 ЗН2О.
Несмотря па то что трихлорид иода отщепляет хлор при нагревании, его все же пол 1.зя рассматривать только как продукт присоединения СГ3 к IC1, а следует Считать соединением первого порядка, в котором иод трехвалеитеп.
5. Солеобразные соединения с электроположительным галогеном
Как уже было указано на стр. 838, иод и бром способны также образовывать солеобразные соединения, в которых они фигурируют в качестве катионов.
Значительное число подобных соединений известно главным образом для иода, причем в них иод может быть как электроположительно одно-валептным, так и т/ж^валситпым.
(юл собранные соединения ио да (I). С о ли ио да (I) обычных сил ьных кне лот, т акио, как I [С1О31 и 1 [СЮ41, сами по собе неустойчивы; однако они могут быть стабилизированы нутом процесса комплексообразования, папример, путем присоединении пиридина (ср. стр. 838). Кроме того, электроположительный одновалентный иод можно стабилизировать, если дать ему образовать соли с такими кислотными ионами, которые сильно поляризуются (это те кислотные ионы, которые известпы как сильные комплоксо-обравоватспи). Соединения этой группы, папример l[CN], I[0CN], I [SCN], I[N3], получаются в большинстве случаев, так же как комплексные ’* соединении типа [1(CsIT5N)2] [NO.,], при взаимодействии солей серебра с 1, в неводном (например, эфирном) растворе: Ag [OCN [ 4 1г = Agl 4 I [ОСМ ]. Они ие обнаруживают, правда, в своей массе оолеобразного характера, как соединения с комплексным катионом иод(1)..
Относительно иодониевых соединений (соединениях типа [П21]+Х_) см. стр. 848.
Солеобразпые соединения нода(Щ). Из солей электроположительного трехвалепт-ного иода первым был получен ацетат иода(Ш) Г[С31Г3О3|3 (Schiitzenbergor, 18(11), Это бесцветные, гексагональные, устойчивые в темноте кристаллы. Позднее подобные соли были приготовлены в большом числе главным образом Фихтером (Fichlor, 1915): например, перхлорат 1[С104|, 2Н2О (зелено-желтый), сульфат I2 [SOJ3 (светло-желтый) и фосфат 1[РО4] (желтый). Соединения 14О9 и Г2О4 также причисляют it этой группе, так как они, как уже было отмечено, рассматриваются в качестве йодатов 14О9 — = НЮз1з [иодат иоДа(Ш), желтовато-белый порошок]; 12О4 = [10] [1О3] (иодилио-дат, бледно-желтые кристаллики).
Другая группа солсобразных соединений трехвалентного иода производится от подозосоедпнепий. Иодооосоединениями (ио аналогии с нитр о зосо единениями) назы
Седьмая группа периодической системы
8'3.
вают соединения типа R—1 = 0 (R — органический радикал), которые получают, например, окислением определенных аридиодидов дымящей азотной кислотой. Однако известны также алифатические ио дозе соединения, особенно такие, которые производятся от олефипов. И одозосо единения, исследованные главным образом Вильгеродтом (Willgerodl) и Тиле (Thiele, 1909), имеют ярко выраженный основной характер и образуют с кислотами настоящие соли R—IX2 (X — кислотный остаток). Водой эти соли разлагаются с образованием иодоаосоединений,
И о доз ос о единения — светлые серо-желтые вещества со своеобразным пронзительным запалом — разлагаются при нагревании; вз ароматических иодоэосоедине-ний в результате диспропорционирования образуются арилиодид и арилиоддиоксид* ^0
2Н=1=О=И-I-f-Н—
Ч>
Арилиоддиоксид-соединения могут быть произведены из йодноватой кислоты заменой гидроксильной группы на арильную группу. В отличие от иодозосоединений они ае обладают основным характером. Их водные растворы проявляют нейтральную реакцию. В то время как иодоз о соединения действуют сильно окисляющим образом, иодосоединения обладают только слабыми окислительными свойствами. Однако если их нагреть в сухом состоянии, то она распадаются с сильным взрывом.
* И О до соединение.— Прим, порее.
Г л ае а 18
ОБРАЗОВАНИЕ СОЛЕЙ И НЕЙТРАЛИЗАЦИЯ
Нейтральные, кислые и основные соли. Важнейшее и наиболее распространенное галоидное соединение — поваренная соль, называемая в повседневной жизни коротко «солью» —в химическом отношении является типичной солью. Солями в химии называют вещества, которые можно получить взаимодействием кислот и оснований с , выделением воды.
Так, поваренную соль (хлорид натрия) можно получить соединением хлористого водорода с гидроокисью натрия
NaOH+HCl = NaCl+H2O. -
Эта реакция основана на том, что"водород хлористоводородной кислоты, отделившийся в виде иона, соединяется с гидроксильной группой основания — гидроокиси натрия, равным образом существующей в виде иона, образуя при этом воду. Остающиеся после этого ионы Na’ и С1' существуют в водном растворе без изменения. Если раствор испарить, ионы соединяются, образуя кристаллическую решетку, представленную на рис. 44 (стр. 237).
* J То же относится к образованию других солей. В случае мало растворимых солей, например К[С1О4], CaF2, BalSOJ, соединение образующих соль ионов в кристаллическую решетку происходит большей частью уже в разбавленном растворе; свободных ионов в растворе остается лишь столько, сколько соответствует растворимости соли. В очень редких случаях образующие соль ионы соединяются в молекулы уже в растворе; это наблюдается, пожалуйтолько у солей ртути. Почти вСе остальные соли, пока они растворены в воде, практически полностью расщеплены па ионы. На этом основано важнейшее свойство растворимых типичных солей: все типичные соли являются сильными электролитами.
Если все способные к диссоциации атомы водорода кислоты вступают в реакцию с гидроксильными группами основания, то кислоту называют «насыщенной» основанием. Соответственно говорят о насыщении оснований кислотами. Такие количества кислоты и основания, которые необходимы для взаимного насыщения, называют эквивалентными, Соли, которые обра-зуюгсн благодаря взаимному насыщению эквивалентных количеств кислоты и основания, называются нормальными или нейтральными солями.
Как видно из многочисленных примеров, часто в кристаллическом виде образуются соли, которые возникают благодаря тому, что в реакцию вступает несколько эквивалентов кислоты с одним эквивалентом, основания. Такие соли называют кислыми солями или гидросолями.
Образование солей и нейтрализация
875
П рнмеры;
2NaOH+H3SO4=2ri3O4-Nfl2SO4 нейтральный сульфат натрия
Wa.OM-^H>S()4=H2O-(~NaHSO4 кислый сульфат натрия (гидросульфат натрия)
3NaOII-{-H3PO4=3H2O4-Na3PO4 нейтральный фосфат натрия
2NaOJI4-IJ3PO/,=2IJ2O-)-NagIJPO4 кислый двувамеиртный фосфат натрия (гидрофан-фат натрия) .
МаО114-ИзРО4 = Н2О4-МаН2РО4 кислый однозамещенный фосфат натрия (дигидро-фосфат натрия)
Равным образом часто существуют соли, которые содержат па один эквивалент кислоты больше, чем одни эквивалент основания (илиангидрида основания): такие соли называют основными солями или гидрокси- (соответственно окси-) солями
Примеры:
I’bCOs нейтральный карбонат свинца
РЬ3(О11);.(СО3)2 основной карбонат свинца (дигидроксидикарбонаг свинца, свинцовые >бол ил а)
SbCIj хлорид сурьмы
SbOCI основной) хлорид сур [.мы (оксихлорид сурьмы, антимон л лхлорйд)
Общие методы приготовления солей. Для приготовления солей иеполь-.зуют главным образом следующие общие методы:
1. Соединение кислот и оснований с образованием воды.
Лежащая в основе этого метода реакция выражается общим уравнением
НХ+МОН=МХ-|-НОН.
В препаративной химий вместо кислот и оснований часто применяют их ангидриды.
Например, нитрат свинца Pb(Nd3)2 в технике получают растворением окиси свинца (т. е. ангидрида основания—гидроокиси свинца) в азотной кислоте. Для приготовления силикатов применяют почти всегда кварцевый песок, т. е, двуокись кремния, ангидрид кремневой кислоты и т. д.
Если применять для реакции ангидриды кислот и оснований, то Процесс соле-образоваиия формально является простым процессом присоединения, например
СаО -ф- SiO2 = CaSiOs,
в то время как при получении солей из кислот и оснований дело идет о двойном обменном разложении
п
НХф-М(ОН) = МХ-1- нои.
I t
На двойном обменном разложении основан ытакже три последующих приведенных метода образования селей.
2. Обменное разложение между кислотой (или ее ангидридом) и солью
Г|
НХ-|- М¥ = MX -J- Я Y (X и Y—кислотные остатки).
Об обменном разложении было уже сказано в гл. 2 в качестве метода получения кислот. Его также часто используют для приготовления солей, правда больше в ап а литической, чем в препаративной химии. Этот метод применяют прежде всего тогда, когда соль
876
Глава 18
возникающая в результате обмена, труднораствврима (например, осаждение сульфат» бария серной кислотой, осаждение сульфидов тяжелых металлов сероводородом) или когда получающаяся при обменном разложении кислота (или ее ангидрид) легколетуча (например, получение хлоридов, нитратов, сульфатов и т. д. при добавлении, соответствующих кислот к карбонатам). В утих случаях вследствие нарушения равно-веси я реакция полностью протекает слева направо. Это происходит также в том случае, если при обмене выделяется труднорастеоримая кислота', однако последнее имеет лишь небольшое значение для препаративного получения солей.
3, Обменное разложение между основанием (или его ангидридом) и солью
м
M(OH)4-RX = R(OH)4~MX (М и R—металл или положительный радикал).
I t
Это обманное разложение, имеющее важное значение при получении оснований (ср. стр. 84), лишь в исключительных случаях применяют для получения солей, Эют-метод с успехом применяют в том случае, если получаемая соль производится От такой слабой кислоты, что может существовать только в щелочном растворе.
4. Обменное разложение между двумя солями
MY4-RX=MX + RY.
I .. . t
Этот метод часто применяют для приготовления солей. Его применяют всегда тогда, когда соль, возникающая при разложении, существенно отличается от других рассматриваемых здесь солей своей растворимостью пли летучестью.
Если в водном растворе и нет существенного различия в растворимости, то оно может появиться При применении других растворителей. Этот метод построен исключительно на различии растворимости, соответственно летучести. Если имеется достаточная разница в растворимости, то таким образом можно приготовить даже сравнительно-легко растворимые соли; правда, для очистки их надо часто еще перекристаллизовать. Известным примером применения этого метода является получение конверсионной селитры (см. стр. 218).
5. Растворение металлов в кислотах
При зтом либо просто водород кислоты замещается металлом, как,, например, при растворении цинка в серной кислоте (получение цинкового купороса)
Zn4-HaSO4=ZnSO4+H2,
либо часть кислоты одновременно разлагается, окисляя металл, как, например, при растворении свинца в азотной кислоте (получение нитрата свинца)
3Pb+8HNO3=3Pb(NO3)2-pNO-HH2O.
Однако и в первом случае, строго говоря, происходит пе простое замещение, а перемещение зарядов; -эго становится попятным,; если реакцию записать как ионную
Zn 4- 2Н‘ 4- SO4 = Zn'' + SO4+Н2 или
Zn4-2H- = Zn”4-H2.
Следовательно, различие между двумя указанными случаями заключается в основном в том, что металл в первом примере окисляется (заряжается, давая иоп) ионом водорода, во втором — кислотным остатком. Чем правее стоит растворяемый металл л электрохимическом ряду напряжений, том более сильными окисляющими свойствами должна обладать кислота, применяемая для растворения. Так, цинк растворяется уже
Обрааование солей к нейтрализация
877
в разбавленной серной кислоте (окислитель — ион водорода), медь же — только в концентрированной серной кислоте или в азотной кислоте (окислитель л обоих случаях — кислотный остаток) и, наконец, золото растворяется только в царской водке (окислитель — хлор).
Для получения солей имеет значение также обмен зарядами между двумя метал-'лими. Этим пользуются иногда для очистки содей. Например, железный купорос (сульфат железа(1Г)] можпо очищать от меди [и одновременно от соли железа(Ш)! внесением в его раствор небольшого количества чистого железа
Cu”-f-Fe — Fe-'-f-Gu,
2Fe-'-4-F0 = 3Fe-.
Иногда для получения солей используют также перенос зарядов мелсду неметалл ат. Примером этого является указанный в предыдущей главе способ получения ГИД,роди-йодата калия КН[Ю3]2 при действии иода па хлорат калия.
6. Соли водородных кислот получают часто при непосредственном •взаимодействии элементов.
П римеры:
Fe -ps=FeS; Sn -J- 2C12=S nCi 4;
2Sb-(-3h=2SbI3; 2Al-|-3Se=Al2Se3.
Кислые соли получают неполным насыщением кислот или присоединением кислоты к нейтральной соли. При применении второго метода получения солей (вытеснение одпой кислоты другой) часто вначале образуются кислые соли.
Основные соли получают при неполном насыщении оснований. Однако обычно для их получения смешивают растворы нейтральных солей и свободных оснований или частично гидролитически разлагают нейтральную соль, папример вливая ее раствор в горячую воду. (Так получают преяще всего трудпорастворпмые основные соли сурьмы и висмута.) В случае приготовления солей тяжелых металлов и слабых кислот при работе с водными растворами часто вместо нейтральных солей получают основные соли,.
Гидролиз. Под гидролизом или гидролитическим расщеплением соли донимают разложение ее водой с получением вновь нислоты и основания, из которых была образована соль, например:
А1С13+ЗНОН :£ 3IICJ + А1(ОК)з,
Bi(NO,)3-|-3HOH 3HNO3H-Bi(OH)3.
Если читать эти уравнения справа налево, то они будут представлять собой образование солей из кислот и оснований. Следовательно, гидролиз солей является процессом, противоположным процессу образования солей.
Вообще же говоря, определение гидролиз применяют для таких процессов расщепления веществ водой, которые отличаются тем, что составные части воды соединяются с продуктами расщепления. К понятию гидролиз, следовательно, можпо причислить, папример, также расщепление эфира на кислоту и спирт. Многие соединения, а именно галогениды не мега дли в, в качестве продуктов гидролитического расщепления образуют две кислоты; так, например, треххлористый фосфор расщепляется водой па хлористоводородную кислоту и фосфорную кислоту, четыреххлористый кремний — на хлористоводородную кислоту и кремневую кислоту (соответственно гидрат двуокиси кремния)
РС13 + ЗНОН = ЗНС1 + Р(ОП)3,
SiCl4 + ЗНОН = 4НС1 + Н281Ол.
В то время как процессы, подобные только что приведенным, протекают в водном растворе почти всегда полностью, гидролитическое рас-
878
Глава 18
щепдение солей (а также эфиров) часто приводит к установлению равновесия. В таких случаях говорят о частичном гидролизе. Положение равновесия зависит от условии опыта. Гидролизу способствует повышение температуры, разведение раствора и главным образом нарушение равновесия вследствие выделения в виде осадка или улетучивания одного или обоих продуктов-раз л о женин.
Гидролитическое расщепление солей происходит только в том случае, если составные части воды могут образовывать существенное количество недиссоциированных молекул с продуктами электролитической диссоциации солей, т. е. ион водорода — с анионом, а ион гидроксила — с катионом соли (или только один ИЗ ИОНОВ ВОДЫ С одним из ионов соли). Это происходит тогда, когда соединение аниона с водородными ионами представляет собой слабую кислоту или соединение катиона с гидроксильными ионами является слабым основанием. Поэтому все соли, происходящие либо от слабой кислоты, либо от слабого основания (либо от того и другого), претерпевают гидролитическое расщепление тем в большей степени,, чем слабее кислота или основание,
В качестве Примера гидролиза соли слабой кислоты рассмотрим гидролиз цианида натрия NaCN, Последний, как все типичные соли, в водном растворе вначале полностью расщепляется на ионы
NaCN = Na* CN\
Но так как цианистоводородная кислота HCN является слабой кислотой, часть ионов; CN' соединяется с ионами Н' воды, давая недйссоцииро ванную цианистоводородную кислоту (образование которой может быть непосредственно установлено по запаху}
CN' Н* £ HCN.
В связи с этим нарушается равновесие диссоциации воды
НОН £ И‘ 4- ОН',
а так как по химическому закону действия масс [Н’] X [OH']=const, то в соответствии с уменьшением концентрации ионов II* в растворе концентрация ионов ОН' должна возрастать. Так как в этом случае речь идет о соли сильного основания, ионы ОН' остаются свободными в растворе и он обнаруживает поэтому основную реакцию. Это будет понятнее, если оба приведенных выше уравнения объединить в одно
CN' НОП HCN ОН'.
Соответственно при гидролитическом расщеплении соли слабого основания и сильной кислоты в растворе образуются свободные ионы Н*; следовательно, в этом случае раствор обнаруживает кислую реакцию. Есля же сель, подвергающаяся гидролизу, ; происходит от слабой кислоты и слабого основания, то реакция раствора определяется тем, какое из двух соединений более диссоциировано. Если они оба диссоциированье приблизительно одинаково, то раствор остается практически нейтральным. Так, раствор ацетата аммония (\НА)СгНэО2 обнаруживает нейтральную реакцию, хотя эта соль заметно гидролитически расщепляется, В общем иа изложенного выше следуетг
водные растворы солей, происходящих от слабых кислот и сильных оснований, вследствие гидролиза обнаруживают, основные свойства; растворы солей, происходящих от слабых оснований и сильных кислот, вследствие гидролиза обнаруживают кислотные свойства. Вследствие гидролиза основную реакцию могут проявлять не только растворы нейтральных (нормальных) солей, но также и кислых солей. Например, водные растворы кислых сульфидов щелочных металлов (гидросульфиды) М HS вследствие гидролиза проявляют сильно щелочную реакцию.
Часть растворенного количества соли, которая при данных условиях гидролитически расщепляется, т. е. которая взаимодействует с водой, образуя кислоту и основание, обозначается как степень гидролиза соли.
Образование солей и нейтрализация
879
Она выражается в дробных частях от единицы или в процентах. Степень гидролиза зависит от температуры и' разбавления^
Степень гидролиза можно вычислить из силы кислоты и основания, из которых соль образована. И, наоборот, измерение степени гидролиза используют часто для определения силы (точнее: констант диссоциации) слабых кислот и оснований.
Измерение и расчет степени гидролиза. Вообще говоря, В водном растворе соли М1Х можно представить следующие процессы:
A^X^M'-f-X' (диссоциация соли); (1)
М‘-1-НОН -А М0Н4-Н- I (2)
Х'+НОН НХ+ОН' j гидролиза; (3>
МОН М‘+ОН' (равновесие диссоциации основания); (4)
НХ 5* H'-J-X' (равновесие диссоциации кислоты); (5)
НОН Н’Ц-ОН' (равновесие диссоциации воды), (6)
Если речь идет о соли сильной кислоты и сильного основания, то процессы (2)—(5) практически отпадают; концентрация ионов Н’ чистой воды от растворения соли не изменяется.
Если речь идет о соли слабой кислоты)! сильного основания, ТО практически отпадают процессы (2) и (4). Степень гидролиза Pi (в дробных частях единицы) в этом случае в соответствии с определением задается как
(7)
где (НХ] — молярная концентрация возникшей в результате гидролиза Кислоты, а с (аналитическая) молярная концентрация растворенной соли. Количество ионов ОН' в растворе равно сумме ионов, образовавшихся в соответствия с уравнениями (3) и (6). Следовательно,
[0Н'1 = [НХ] + [Н-], или [НХ] = [О1Г]-[1Г]. (8)
Так как (см, стр. 67)
[И‘] X (OH'] = A-W (ftw—-константа диссоциации воды), (9)
то для степени гидролиза получают следующее выражение:
в [0Н'’-7оЙ
pi=----_----
ГОнт-^_^-(нт
ИЛИ ₽1- [ОН']-с ~ IH-J-c *
(10а)
Следовательно, степень гидролиза (для указанного случая) можно экспериментально определить измерением концентрации, водородных ионов или, точнее, активности водородных ионов*-.
* Как следует из сказанного на стр. 91 и сл., в выражение равновесия входят не действительные концентрации, а активности ионов. Если обозначить ан, и аон, активности И* [I ОН'-ионов, то оказывается ан. = /д. X [Н*] и аон, = /од, X (ОН'], где f7Т*2, (ОН'} — действительные концентрации, a /ц., /он,— коэффициенты активностей ионов И* и ОН' соответственно. Если принять во внимание, что ka является произведением активностей, а не произведением действительных концентрации ионон Н* и ОН', то оказывается, что в уравнениях (10а) п (106) Aw должно быть еще поделено па произведение /и.X /од.. Благодаря этому уравнения (11а) и (116) переходят в
й.
ЛН‘ х /ОН' х е
И 02 =
°н-
4 г х
880
Глава 18
Если гидролиз приводит к отчетливо основной реакции раствора ([ОН']^>10”в при комнатной температуре), A'w (~10*14) практически исчезает в сравнении с [ОН']2 и соответственно [H'j2 исчезает в сравнении с Z-w. Уравнение (10а) принимает тогда более простую форму
[ОН'] .. Ч
с [1Г] -с '
(Иа)
Соответственно для степени гидролиза соли слабого основания и сильной кислоты оказывается
[1Г]=—*w [Н-1
Р3=- [ЬГ1.е ~ и, если [1Г]>10Л («б) и (Иб)
Приведенные д предыдущих и последующих разделах формулы, как следует из их вывода, справедливы для солей одновалентных металлов. Однако формулы (11а) и (116) могут иметь также значение для солей многовалентных металлов, если с и них считать не молярной, а эквивалентной концентрацией.
Эти уравнения, однако, не применимы для расчета гидролиза солей слабых оснований и слабых кислот. В этбм случае следует учитывать все .уравнения от (2) до (6), и тогда не получается простого соотношения между концентрацией водородных ионов и степенью гидролиза. Степень гидролиза можно, однако, тогда рассчитать из констант диссоциации кислоты и основания.
К общей формуле для расчета константы и степени гидролиза ив констант диссоциации соответствующих кислот и оснований подойдем путем следующих простых соображений. Прп этом предположим, что реакция идет в гомогенной среде, а именно в водном растворе, и что, следовательно, не происходит никакого выпадения в осадок или улетучивания продуктов реакции. Различие между дойстяптольнымя концентрациями и активностями можно пе принимать во внимание. Из дальнейшего будет видно (см. стр. 882), что значения, рассчитанные из констант диссоциации [в противоположность значениям, найденным из концентраций водородных ионов по уравнению (Юа) или (11а), соответственно (106) или (116)], в большинстве случаев лишь мало от зтого зависят.
а) Гидролиз солей слабых кислот и сильных оснований. Если поддерживать температуру постоянной, то, применяя закон действия масс к уравнению (3), можем получить
К
[НХ]Х[ОН']
[Х'1 х [П3О]
= const.
При обычных концентрациях количество воды благодаря взаимодействию (3) практически не изменяется: можно, следовательно, ее концентрацию ввести в константу и таким образом получить
[ИХ]х[ОН'1 = г .
-- [Х7~---- гидр1 (12)
^гидр называют константой гидролиза. Уравнение (5) в результате применения закона действия масс дает *
— ^НХ (Л’нх—константа диссоциации кислоты, НХ). (13а)
Комбинируя это уравнение с уравнениями (9) и (12), получим
^гйдр = у? (14а)
ЛНХ
Константа гидролиза соли слабой кислоты и сильного основания равна частному от деления константы диссоциации воды на константу диссоциации кислоты.
Подставляя уравнения (13а) и (9) в уравнение (8), получим
В случае слабой и средней степени гидролиза ([5<0,01) уменьшение [X'] в реакции (3)
Образование солей и нейтрализация 881
можно оставить без внимания* и (X'J заменить на аналитическую молярную концентрацию соли с.
Тогда получим
^онч=|/ ^(1+лтЬг)’ (15а)
Подставив это в уравнение (10а), получим
1/Т ,_А _Z .
’ Я'нх^нх+с)
(16а)
б) Гидролиз соли слабого основания и сильной кислоты. Для этого случая аналогичным образом получим уравнения:
=^5пш i7f jrOir—константа диссоциация основания МОН), (136)
И” гидр—7=----- (14о)
ЛМОН
Константа гидролина соли слабого основания и сильной кислоты равна частному от деления константы диссоциации воды на константу диссоциации основания
Ш']= 1/TW( i+Д.
* V Лмон/
|/ Кион (£МОн+й)
(156)
(166)
в) Гидролиз соли слабой- кислоты и слабого основания. В этом случае должны быть приняты во внимание равновесия (2) — (6) и относящиеся к ним уравнения (13а), (136) и (9). Умножая (13а) на (136), получим, Принимая во внимание (9),
{ МОН) х [НХ] Ч-IM J X 1X') &НХ X
(14в)
Прн гидролизе солей, производимых от слабых оснований и слабых кислот, концентрация конов Н" часто претерпевает, как уже было отмечено, совсем незначительное изменение. Это тот случай, когда прп гидролизе образуются практически эквивалентные количества недиссоцниро ванных кислоты п основания. Так как было принято предположение, что они остаются в растворе, то (МОН) == (НХ1 и |М‘] = IX']. Часто можно без большой ошибки считать [ИГ] = [X'] = с. Тогда из уравнения (14в) следует
(МОПР |ПХ]3] = Aw
с- сг й'нх х #МОц ’
(МОН) = [ИХ]=е]/ =-----. (15в)
г Лнх х Лмон
* В случае более сильной степени гидролиза (^>0,01) уменьшение [X') должно быть принято по виштине- f X'! = с — [НХ ]. Напротив, в этом случае в ураввенпп (8) [ 1Г) становятся исчезающе малой величиной в сравнении с [HXJ. Итак, (ОН') = = [НХ] =:: с — (ХД— с — Г-К’нх(OH'pMw); [ОИ'Р ==(ЛттЖнх) •(.'—[ОН']). Отсюда следует
> 1' с( И Г+«/4с — Уа;4с), где а — .
Это же уравнение дает степень гидролиза для силл слабого основания и сильной кислоты (для Р>0,01), если принять а = ^/^мон-
56 г, Реми
882
Глава. 18
| f —.
V А’пх х А’мон
и для степени гидролиза получим В fM0HJ : iHXJ
Рз с с
Как следует из уравнения (16в) для- солей слабых оснований. и слабых кислот, при
сделанных предположениях степень гидролина независим, от концентрации. Для солей си.гъмыл; оснований и слабых кислот или слабы# оснований И сильных кислот., напротив, степень гидролиза в соответствии с уравнениями (16а) и (166) растет с уменьшением концентрации, Во всех случаях, как следует из уравнений, гидролиз тем сильнее, чем Меньше значение констант диссоциации Я^нх и-^МОЮ 01 чем сла(5ее участвующие в образовании соли кислоты и основания. Для солей сильных оснований и слабых кислот или слабых оснований и сильных кислот в табл, 115 сопоставлены рассчитанные при помощи приведенных уравнений степени гидролиза в зависимости от констант диссоциации слабых кислот (Лнх) или слабых основании (Хмон) и от аналитических молярных концентраций солей с. В случае солей сильных кислот и сильных оснований каждое из приведенных выше уравнений для расчета степени гидролиза имеет Р == О, так как в этом случае А'нх = А'МОд=со.
При выводе приведенных выше уравнений, служащих для расчета степени гидролаза, а также при расчете приведенных в табл. 115 значений, различие между актие-
Таблица 115
Степени гидролиза солей (при 22°)
Числа показывают степень гидролиза в дробных частях от 1 при молярной концентрации раствора с для слабой кислоты (или основания) с копстантой диссоциаций: К и сильного основания (или кислоты).
К С—1 4 с=0,1 с=0,01 е—0,001. <:=0,0001
10-1 0,30-10*® 0,71-10'* 0,95-10’* 1,00.10-* 1,00-10’*
10-8 0,10-10-ь 0,30-10’* 0,71-10-s 0,95-10’* 0,995-10’6
10'3 0,32-10-5 0,995-10-5 3,0-10'5 7,1-10”® 9,5-10’*
ю-* 0,10-10’* 0,32-10-1 0,995.10"* 3,0-10'* 7,1.Ю~*
10’® 0,32-10» о,ю-ю-а 0,32-10’® 0,995-IO’® 3,0-40'3
1О-о 0,10-Ю-з 0,32.10® 1,0-40’® 3,2-dO-3 9,95-10'®
10’8 0,10-10-» 0,32- Ю’1 1,0-10'» зд-ю-» 9,5.10'2
10-п 0,10-10-1 0,32-Ю-> 0,95-10-1 2,7-10-" 6,2-10'1
Ю-щ 0,095 0,27 0,62 0,92 1,00
постами и действительными концентрациями не принимали во внимание. Если учесть это различие, то для средних степеней гидролиза оказывается, что значения рассчитанные по уравнению (16а), должны быть умножены еще на фактор V SHa'
чепия 02 [уравнение (166)] — на фактор ^/м ЧН- и значения р3 [уравнение (16в)] следует умножить на‘фактор )-z/x--./м*. Здесь /х', /м-, /оН' и/н-^- коэффициенты активности ионов X', М‘, ОН' и Н* в рассматриваемых растворах, С учетом различия между активностями и действительными концентрациями уравнения (14а) и (146) пе изменяются.
Примеры для расчета степени гидролиза. Если значения, данные на стр, 74 и 495, подставить вместо и Хн;. в уравнение (16а), то для степени гидролиза a if ema -та натрия при 18° в случае с = 1 найдем р = 7^10^""Г~бО ~ ддя
с = 0,1 В = 6,5-10~*; для с = 0,0001 оказывается В = Т/ --------212'*’.12 ---—
’ ’ r V 1.75.10*®-1,175.10’*
=0,19-10-2. Следовательно, в процентах степень гидролиза при данных концентрациях
Образование солей и нейтралигация
883
составляет; 0,0021, 0,0065 и 0,19%. А в учетом коэффициентов активностей получаем значения: 0,0022, 0,0066 и 0,19%.
Используя приведенные па стр. 502 даппые для Анх, получаем значение для константы гидролиза цианида калия
/ 0 74-10“14
₽= У бЗ-Ю-гЦ-Г^О^0’39,10"58 для Ш раствора и
Р = 1,2410"а для 0,Ш раствора*.
Учитывая коэффициенты активностей, получим 0,37-10-1 и 1,245-Ю-2 (экспериментально найдено: 0,38-10-2 и 1,2010-2). Степень гидролиза ацетата аммония можно
(16»): В= 1/ , е° ’7410 У „=6,49 104 Если при-
1 1 н V 1,75-10-®-1,75-10-» F
рассчитать по уравнению
пять во внимание коэффициенты активностей, то в этом случае оказывается существенно более низкое значение, так как У Z, тг.х /л =/„_ . =0,54. Таким образом, для степени гидролиза получают р = 0,54-0,49 10“2 — 0,27-10-*, или 0,27%, Вообще говоря, если учесть различие активностей и действительных концентраций, подлежащих корректировке, значения, рассчитанные по уравнению (16в), существенно изменяются, в то время как значения, рассчитанные по уравнениям (16а) или (166), большей частью изменяются незначительно. Это происходит потому, что в одном случае корректирующий фактор равен корню из произведения, в другом случае ~ корню из частного от деления коэффициентов активностей, а так как последние для одновалентных ионов имеют большей частью мало различающиеся значения, то корень из их частного, как правил!), мало отклоняется от единицы.
Нейтрализация. Раствор называют нейтральным, если он не обнаруживает ни кислой, пи щелочной реакции (neutrum — ни то, ни другое). Следовательно, под нейтрализацией имеют в виду устранение кислой или щелочной реакции**. Кислая реакция, которая обусловливается присутствием избытка водородных ионов в растворе, устраняется добавлением ионов гидроксила, т. е. добавлением основания; обусловленная же присутствием избытка гидроксильных ионов в растворе основная реакция устраняется добавлением водородных ионов, т. е. кислоты. Сущность нейтрализации заключается в соединении ионов водорода и гидроксила с образованием практически недиссоциированиой молекулы воды
НЧ-ОН =Н20. (17)
Если к раствору разбавленной сильной кислоты добавить эквивалентное количество разбавленного сильного основания или наоборот, то, поскольку возникающая благодаря соединению кислоты и основания соль остается растворенной, процесс нейтрализации обусловливается реакцией, представленной уравнением {17). Практически это единственная реакция, происходящая в этом случае,— ведь ионы, составляющие соль, имеются в растворах совершенно диссоциированных кислоты и основания в топ же форме, как п затем в растворе соли. Вследствие этого количество тепла, освобождающееся при взаимном насыщении сильных кислот и оснований (теплота нейтрализации) в разбавленных растворах, почти не зависит от природы кислоты и основания.
Иначе обстоит дело, если в реакции участвуют слабые кислоты или основания. Если раствор слабой кислоты смешать с основанием, то в ре-
* Так как степень гидролиза составляет лишь немногим больше 1%, уравнение-(16а) дает еще достаточно точное значение. Посредством уравнения, приведенного в примечании на стр, 881, получают 0 = 1,233 10~2.
** Как следует отсюда, «нейтрализация» и «насыщение» (ср. стр. 874) являются1 различными понятиями.
56*
884
Глава IS
зультате реакции ионов гидроксила с ионами водорода равновесие диссоциации кислоты нарушится. Диссоциация кислоты будет продолжаться до тех пор, пока эта кислота будет в состоянии давать водородные ионы. То же можно сказать относительно ионов гидроксила, если дело касается слабого основания. Поэтому оказывается, что в случае слабых кислот и оснований теплоты нейтрализации существенно различаются в зависимости от природы соединений. При нейтрализации слабых кислот и оснований равновесие устанавливается таким образом, что концентрация водо-родпых«или гидроксильных ионов не просто зависит от разности смешиваемых количеств кислоты и основания, а определяется также в силой кислоты или основания. Это следует уже из того, что растворы «нейтральных» солей (т. с. образованные эквивалентными количествами кислоты н основания) не обнаруживают, как уже было отмечено, обычно нейтральной реакции, если в образовании солей участвуют слабые кислоты или слабые основания.
Установление точки, в которой наступает взаимное насыщение кислоты и щелочи, так называемой точки эквивалентности, имеет большое значение для титрометрического определения кислот и щелочей. Для этого служат большей частью цветные индикаторы. Их обычно называют коротко индикаторами. Это — вещества, окраска которых зависит от концентрации водородных ионов в растворе. Одним из таких индикаторов является, нанример, краситель лакмус (азолитмин), который в кислом растворе обладает красным цветом, а в основном — синим. Для качественных испытаний на кислую или основную реакцию пользуются большей частью полосками бумаги, пропитанными указанным веществом (лакмусовая бумага). Для титрометрического определения кислот и оснований лакмус применяют редко. Для этой цели обычно используют индикаторы фенол-фталеии и метиловый оранжевый или вместо последнего также метиловый красный, предложенный Руппом (1908). Фенолфталеин в кислом растворе бесцветен, в основном — красного цвета. Метиловый оранжевый и метиловый красный в кислом растворе обладают красным цветом, в основном — оранжево-желтым и соответственно желтым. Важно только иметь в виду, что переход окраски у применяемых индикаторов отнюдь не происходит при концентрации водородных ионов, которая соответствует точке нейтрализации, т. е. лри концентрации водородных ионов чистой воды (IH'J = — 10~7). Изменение окраски, вообще говоря, не происходит резко при определенной концентрации водородных ионов, а распространяется чаще на достаточно широкий интервал концентраций.
В случае лакмуса, например, интервал перехода от красной окраски к синей простирается от [Н] = 10'6 до [Н‘] = 10"8, т. е. в интервале, равном трем единицам степени концентрации водородных ионов. Области перехода других индикаторов приведены в табл. 116. Как видно из этой таблицы, интервал перехода фенолфталеина лежит в более щелочной области, а метилового оранжевого и метилового красного — в более кислой области.
Когда дело касается титрования сильных кислот сильными щелочами, отклонение области перехода указанных индикаторов от собственно точки нейтрализации ([Н‘] = 10“7), так же как и тот факт, что область эта имеет значительную протяженность, пе имеет в общем практического значения, так как в этом случае концентрация водородных ионов вблизи точки нейтрализации внезапно изменяется на очень значительную величину. Реакция раствора внезапно становится из сильно кислой сильно основной, или наоборот.
Изменение окраски индикаторов
Таблица 116
И Л Hl 1 :ЛТ| ф Интервал значений pH Изменения окраски л иола я^>щелочна я Количество индикатора в to лд
Метиловый фпилетоы.ш 0,1-3,2 Желтая 5± синяя 5i фиолетовая 2—8 капель 0,05%-ного водного раствора
Мотаииловый желты й 1,2—2,3 Красная 52 * желтая 3—5 капель 0,1%-иого водного раствора
Тропоолин 00 1,3-3,2 Красная 52 X желтая 1—3 капли 0,1%-пого водного раствора
Диметиловый желтый (мае- 2,9-4,0 Криспа я 52 * желтая 2—5 капель 0,1%-ного спиртового раствора
ляный желтый) Метиловый оранжевый 3,1-4,4 Красная 5: Ц орапжояожелтая 1—3 капли 0,1%-ного водпого раствора
Конго красный 3,0-5,2 Сипе-фиолетовая 5± красная 1—5 капель 0,1%-ного водного раствора
Метиловый красный 4,2-6,3 Красная 51 L желтая 2—4 капли 0,2%-ного водно-сниртового раствора
Лакмоид 4,4-6,4: Красная 5; !; сипя я 1—5 капель 0,2%-пого спиртового раствора
л-Потрофенп.ч 5,0—7,0 Бесцветная 5* желтая 3—20 тиноль 0,4%-ного водпого раствора
Нейтральный красный 6,8-8,0 Красная 5: желтая 2—4 капли 0,1%-пого водно-сниртового раствора
Лакм)С («з о лит мин) о,0-8,0 Красная 5: ?: синяя 10—20 капель 0,2%-цого водного раствора
Розолопая кислота 6,9-8,0 Коричневая 5± красная 1—4 капли 0,1%-ного водно-спиртого раствора
Куркумин 7,8—9,2 Желтая 5± красно-коричпевая 1—5 капель 0,1%-пого водного раствора
Фенолфталеин 8,2-10,0 Бесцветная 5± красная 3—10 капель 0,1%-пого водно-спиртового раствора
Ализариновый желтый 10,1-12,1 Желтая 5± лиловая 5—10 капель 0,1%-ного водного раствора
Нитрамин 11,0-13,0 Бесцветная 5* оранжев о-коричнева я 1—3 капли 0,1%-ного водно-спиртового раствора
886
Глава 18
Рис. 126 дает наглядное представление о зависимости водородного показателя £ от состава смеси, что соответствует процессу изменения концентрации водородных ионов при постепенном добавлении раствора сильного основания к раствору сильной кислоты. На рисунке на оси абсцисс отложено соотношение кислоты и основания в растворе, имея в виду, что соотношение 50% кислоты и 50% основания соответствует образованию нейтральной соли. На ординату нанесены водородные показатели pH, т, е. отрицательные логарифмы концентраций водородных ионов,
красный
желтый
бесцветный
красный
О 10 20 SO 40 50 SO 70 80 90 ЮО Щелочь, %
Р и с. 126. Нейтрализация сильной кислоты сильным основанием.
существующих приразличных составах. Видно, что кривая, соответствующая зависимости водородных показателей от отношения количеств кислоты и основания в растворе, проходит вертикально через точку нейтрализации (рН7), так что почти перпендикулярно пересекает области перехода фенолфталеина и метилового красного, которые отмечены на рисунке штриховкой. Предположим, что смешано 50 мл 0,1 н, соляной кислоты с 49,9 дал 0,1 и. раствора едкого натра. Концентрация водородных ионов в этом растворе, содержащем в 99,9 дал. 0,00001 г-экв избыточной кислоты, составляет 10“4.0 г-экв/л^(рН == 4,0). В растворе еще ясно фиксируется кислая реакция обоими индикаторами. Однако достаточно прибавить еще 0,2 мл 0,1 п. раствора едкого натра, чтобы достигнуть внезапного понижения концентрации водородных ионов до 10“1()0 (pH == 10,0), при которой раствор в отношении обоих указанных индикаторов обнаруживает основную реакцию.
Совсем иной характер имеет кривая водородных показателей, когда дело касается насыщений слабой кислоты сильным основанием или сильной кислоты слабым основанием. Ход кривой водородных показателей в этом случае представлен на рис. 127. Кривая I дана в качестве примера
Образование солей w нейтрализация
881
Кривой насыщения слабой кислоты сильным основанием. Она соответствует ходу кривой нейтрализации при насыщении ОД н. уксусной кислоты ОД я. раствором едкого натра. Нак видно из рисунка, область перехода метилового красного минуется в этом случав весьма медленно. Когда же
переход окраски наконец происходит, раствор не содержит’ еще всего количества основания, эквивалентного кислоте. Это не имеет места даже ц в точке централизации Щ'] = Ю'7, а, как будет показано ниже, достигается только тогда, когда водородный показатель снижается до величины 10 “®\ Если же при титровании приливать ио каплям кислоту к
щелочи, то начало изменения окраски метилового красного происходят уже за точкой насыщения. Следовательно, слабую кислоту, например уксусную, нельзя ти-тросатъ, применяя в качестве индикатора метиловый красный (а также метиловый оранжевый, как видно из табл. 116), однако; как показывает кривая I, слабую кислоту можно титровать с фенолфталеином. В качестве примера соотношений, существующих при смешивании слабого основания с сильной кислотой, приведена кривая II на рис. 127. Она характеризует изменение водородных показателей при титровании 0,1 н. соляной кислоты ОД М водным раствором аммиака или, если рассматривать ее ход справа налево, при титровании водного раствора аммиака соляной кислотой. Как следует из хода кривой, слабое основание нужно титровать не с фенолфталеином, а с метиловым красным.
8 -
а &0оласть перехода
фенолфталеина^
I! -
пролает перехода
г шепиликииясипи. х, §Л'чХЩХ\\\\хх\Хч\'С<х
12 -
/Зк
100 90 80 70 SO SO 40 30 20 10 О
14 \--1-----1--1---i---f----1---1--1----1-1
0 10 20 30 40 50 60 70 80 90 100
«•____1_
ч— кислота г%
Щелочь. %
Рис. 127. Кривая I нейтрализация уксусной кислоты едким натром; кривая II — нейтрализация соляной кислоты водным раствором аммиака.
Приведенные ла ряс. 1213 и 127 в качестве ординат значения водородных показателей можно не только экспериментально определить, их удается также очень просто рассчитать. Для чистой 0,1 и. соляной кислоты [Н‘] = 10-1, pH 1*. Если смешать 55 .и.г кислоты с 45 .ил 0,1 н. раствора едкого натра, тоД5 ли кислоты переходит в форму голи. Остаток 10.«л распределяется да 100мл раствора. Следовательно, концентрация водородных ионов понижается ДО 5/к первоначальной величины и pH равно 2. Соответственно для смеси на 50,5 ч. кислоты и 49,5 ч. основания получают pH 3 и для смеси, из 50,05 ч, кислоты и 49,95 ч. основания — pH 4. Смесь из 50 ч. кислоты л 5й и основания [теоретически**] точно нейтральна, следовательно, pH 7, Смесь из 49,95 мл кислоты и 50,05 м щелочи содержит 0,1 мл 0,1 в. щелочи, распределенной в 100 .и.,, следовательно, [0Л']=Ю“4, pH 10. Соответственно для смеси из 49,5 ч, кислоты и 50,5 ч. щелочи находят pH 11, а для смеси из 45 ч. кислоты и 55 ч. щелочи — pH 12, в то время как для чистой 0,1 я. щелочи — pH 13.
* Активность вэцородиых ионов bJ0,11 и. соляной кислоте равна 10-1>06. Эта ничтожная разница не имеет здесь значения.
** Вследствие неизбежного загрязнения реактивов точка нейтрализации таким путем практически недостижима (см. последний раздел этой главы).
888
Глава 18
Для слабой, кислоты концентрация водородных ионов получается из константы диссоциации Анх кислоты:
[П']=-&^нх, (18)
pH = —log [Н‘] = log (X'] -log [НХ] - log АГПХ. (19)
Для уксусной кислоты (при 183) Лих = 1,75 10“s = Ю’4.76; — log Анх = 4,76. Для чистого раствора кислоты [Н‘| к [X'], следовательно:
[Н-]»=4НХ] X АГнх,
рН= —*/2 log [HX]-V2 log А'нх.
Практически [НХ] можно считать равной общей концентрации кислоты. Таким образом, для 0,1 п. уксусной кислоты
рН = —‘/з log 0,1 4,76=0,54-2,38 = 2,88.
Смесь из 95 ч. 0,1 н. уксусной кислоты и 5 ч. 0,1 н. раствора едкого натра имеет по ацетату натрия 6/100Х 0,1 нормальность и по свободной уксуспой кислоте X 0,1 нормальность. Водородный показатель для этой смеси в соответствии о уравнением (19) будет равен
рН= log --------log wo’ +4,76^3,Д1.
В смеси, состоящей из двух третей уксусной кислоты и одной трети раствора едкого натра, концентрация соли равна концентрации свободной кислоты
[X'] = [НХ], рН=4,76.
Смесь из 55 ч. уксуспой кислоты и 45 ч. раствора едкого патра оказывается по соли
45 X 0,1 „ - 10 X 0,1 ’ „ „
—... нормальной и по своооднои кислоте—тдк—— нормальной. Следовательно, Ivv lULf
pH = log 0,045—log 0,01 4- 4,76 = 5,41.
Для смеси из 51 ч. уксусной кислоты и 49 ч. раствора едкого натра
pH = log 0,049—log 0,002 4- 4,76 = 6,15.
При смешивании 50 ч. 0,1 к. уксусной кислоты с 50 ч. 0,1 н. натровой щелочи получают «нейтральную соль#. Однако вследствие гидролиза она обнаруживает щелочную реакцию. Уравнение для вычисления водородных показателей получают из уравнения (15а), стр. 881. Так как в нашем случае единица исчезающе мала по сравнению с с /К нх, уравнение переходит в
[ОН'] = й^.(с/Хнх)^ плй
Логарифмируя, получим
pH-Vs log 4 —V2 log КHX4-V2 log с.
Этот раствор является 0,05 я. по соли (с = 0,05), следовательно, pH = 7,06 4- 2,38 — 0,65 = 8,79.
Смесь, состоящая нз 49,5 мл уксуспой кислоты и 50,5 мл едкого натра, наряду с 49,5 ч. соли содержит 1 ч. свободного едкого патра. Таким образом, раствор является 1/1оо'О,1 нормальным по ионам ОН': pH 11,0. Аналогичным образом получаются водородные показатели и при дальнейшем добавлении раствора едкого натра; ход кривой с этого момента такой же, как н в первом примере.
Если с водным раствором аммиака смешивать сильную кислоту, то для концентраций гидроксильных ионов, соответствующих различным соотношениям смешанных веществ, практически получают те Же значения, какие были пайдены для смесей уксусной кислоты и едкого патра для концентраций водородных ионов. Это обусловливается тем, что для водного раствора аммиака константа диссоциации имеет почти то же значение, как и в случае уксусной кислоты. Поэтому кривая II на рис. 127 проходит
Образование солей и нейтрализация 889
аналогично кривой /, только значения отрицательных логарифмов концентрации гидроксильных ионов откладываются не от верхнего края диаграммы вина, как водородные показатели, а от пижпего края вверх.
Колориметрическое определение концентрации кодородных ионов. Различное положение областей перехода индикаторов и постепенное изменение окраски в пределах области перехода делают возможным определение концентрации водородных ионов колориметрическим путем. Если, например, находят, что раствор краснеет от прибавления метилового красного, но не краснеет от добавления метилового оранжевого, то из этого следует прежде всего, что водородный показатель меньше 6, но больше 3. Более точное значение водородных показателей можно определить затем, сравнивая окрашивание пробы, смешанной с метиловым красным или с метиловым оранжевым, с окраской растворов известной концентрации водородных попов, к которым добавлен тот же индикатор.
Буферные смеси. Смесь слабой кислоты или слабого основания с соответствующей солью в оп ре де лени ой области концентраций водородных ионов (положе вис которой зависит от силы слабой кислоты или основания) испытывает при прибавлении кислоты или щелочи сравнительно незначительное изменение концентрации водородных ионов. Этим явлением пользуются, если требуется приготовить растворы с низкой, но достаточно определенной Концентрацией водородных или гидроксильных ионов. Получить такие растворы простым разбавлением растворов сильных кислот или оснований нельзя, так как, если их нормальность при разведении опускается ниже или даже 1 /юоо, то, как показывает рис. 126, даже минимальное добавление кислоты или щелочи оказывает огромное влияние иа концентрацию ионов Н‘. Однако при работе с растворами трудно предотвратить доступ небольших количеств кислоты (углекислоты из воздуха) или щелочи (из стекла сосуда). Поэтому растворы с низкой, но достаточно определенной концентрацией водородных или гидроксильных ионов готовят смешиванием слабых кислот или основании с их солями. Вследствие эластичной сопротивляемости этих смесей изменению концентрации водородных ионов их называют буферными смесями. Буферное действие такой смеси уменьшается с разведением. Для данной концентрации это действие будет максимальным, когда смесь содержит кислоту (соответственно основание) и ее соль почти в равных количествах. Сведения относительно приготовления буферных смесей можпо найти в приведенных ниже статьях. Тиил (Thiel А., 7). Elektrochem., 40, 150, 1934] предложил ряд буферных смесей, полезных тем, что их очень удобно готовить из веществ, легко получаемых в чистом состоянии. При их использовании можпо всегда быстро приготовить растворы с точно определенной концентрацией водородных ионов в области между рИ 1,5 и 11,0.
ПРИЛОЖЕНИЯ
I
Таблица ]
Меахдупародные атомные веса на 1962 г. а
Название элемента Символ Порядковый номер Атомный вес Название элемента Символ Порядковый номер Атомный вес
Азот N 7 14,0067 Неон Ne 10 20,183
Актиний Ас 89 [227] Нептуний Np 93 [237]
Алюминий А1 13 26,9815 Никель Ni . 28 58,71
Америций Am 95 1243] Ниобий Nb 41 92,906
Аргон Аг 18 39,948 Нобелий No 102 [255]
Астатин At 85 [210] Олово Sn 50 118,69
Барин Ва 56 137,34 Осмий Os 76' 190,2
Бериллий Бе 4 9,0122 Палладий Pd 46 106,4
Веркелий Вк 97 [247] Платина Pt 78 195,09
Бор В 5 10,811 ] 1лутоний Pu 94 [242]
Бром Вг 35 79,909 Полоний Po 84 [210]
Ванадий V 23 50,942 Празеодим Pr 59 140,907
Висмут Bi 83 208,980 Прометий Pm 61 147]
Водород н 1 1,00797 . Протактиний Pa 91 231]
Вольфрам W 74 183,85 Радий Ra 88 226]
Гадолиний Gd 64 157,25 Радон Rn 86 222]
Галлий Ga 31 69,72 Рений Re 75 186,2
Гафний Hf 72 178,49 Родий Rh 45 102,905
Гелий Не 2 4,0026 Ртуть Hg 80 200,59
Германий Ge 32 72,59 Рубидий Rb 37 85,47
Гольмий Но 67 164,930 Рутений Ru 44 101,07
Диспрозий Dy 66 162,50 Самарий Sm 62 150,35
Европий Ен 63 151,96 Сииисц Ph 82 207,19
Железо Fe 26 55,847 Селен Se 34 78,96
Золото Au 79 196,967 Сера S 16 32,064
Индий In 49 114,82 Серебро Ag 47 107,870
Иод I 53 126 9044 Скандий Sc 21 44,956
Иридий ]T 77 192,2 Стронций Sr 38 87,62
Иттербий Yb 70 173,04 Сурьма Sb 51 121,75
Иттрий Y 39 88,905 Таллий T1 81 204,37
Кадмий Gd 48 112,40 Тантал Ta 73 180,948
Калий К 19 39.102 Теллур Те 52 127,60
Калифорний Cf 98 ]249] Тербий Tb 65 158,924
Кальций Ga* 20 40,08 Технеций Tc 43 [97]
Кислород 0 8 15,9994 Титан Ti 22 47,90
Кобальт Co 27 58,9332 Торий Th 90 232,038
Кремний Si 14 28,086 Тулий Tu 69 168,934
Криптон Kr 36 83,80 Углерод C 6 12,01115
Ксенон Xe 54 131,30 Уран U 92 238,03
Кюрий Cm 96 [247] Фермий Fm 100 [253]
Лантан La 57 138,91 Фосфор P 15 30,9738
Литий Li 3 6,939 Франций Fr 87 [223]
Лютеций Lu 71 174,97 Фтор F 9 18,9984
Магний Mg 12 24,312 Хлор Cl 17 35,453
Марганец Mn 25 54,9381 Хрем Cr 24 51,996
Медь Cu 29 63,54 Цезий Cs 55 132,905
Менделевий : Md 101 [256] Церий Сё 58 140,12
Молибден Mo 42 95,94 Цинк Zn 30 65,37
Мышьяк As 33 74,9216 Цирконий Zr 40 91,22
Натрий Na 11 22,9898 Эйнштейний Es 99 [254]
Неодим Nd 60 144,24 Эрбий Er 68 167,26
а Взамен международных атомных весов на 1060 с.т Приведенных в книге Реми, нами noir пшена таблица» принятая Международной комиссией по атомным весам на 1962 г. В основе TfziEoii шкалы атомных весов лежит значение 12—масса атома изотопа углерода 12С.—При-w. ped.
Слева внизу у каждого элемента указан порядковый номер, вверху—, а томный вес. Эле Элементы, названия которых набраны курсивом, получены
Значение символов электронных ко
(В табл. II указаны атомные веса но данным Международ!
I И 1
Ряды Периоды и аделтроквая конфигурация
побочная главная побочная глаипак побочная главная побочная главная побочна!
1
2 Первый короткий период Оболочка Не-|- 6,939 3ЛИТИЙ 2s 9,0122 ^Бериллий 2s2 । 10,811 5Бор 2s22p 12,01115 «Углерод 2s22p2
3 Второй короткий период Оболочка Ne-f- 22,9898 уНатрий 3s 24,312 ^Магний 3s3 26,9815 ^Алюминий 3s23Jo 28,086 14К ремни и 3s23p3
4 Первый длинный период Обомчха Ar—f— 39,102 igKajinii 4s 40,08 ^Кальций 4s2 44,956 21 Скандий 3fMj'2 47,90 щТитан 3^2 50,942 йзВапад MWS
5 Оболочка Дг -prfw-b 63,54 2вМедь 4s 65,37 зоЦинк 4s2 69,72 з1 Галлий 4sa4p 72,59 32Германип 4ss4pil
6 Второй длинный период Оболочка Кг+ 85,47 57? У би ди Й 5s 87,62 Э8Стронций 5s2 88,905 здИтТрИЙ 4d5s3 91,22 ^Цирконий 4d25s3 92,906 41Hho6i 4rf<5s
7 Оболочка Кг Ц-4<110+ 107,870 47 Серебро 5s 112,40 48Кадмий 5s2 114,82 49Индий 5я25р 118,69 гщОлово 5s2.i р2
8 Третий длинный период Оболочка Хе-у 132,905 .вдЦезий 6s 137,34 звБарий 6«2 138,91 57 Л;1нтан Лантаниды 58-71 178,49 72Гафппй " if«5d26s2 180,945 73Тант. ‘ 4/«5
9 Оболочка Хе +4/U_|-5dio_j_ 196,967 7рЗолото 6s 200,59 й0 Ртуть 6s2 204,37 з/Галлий 6s2 6р j 207,19 язСвииец I 6s26p2
10 Четвертый длинный период Оболочка R&-]- 223 8;Франций* 7s 226 88Радий * 7s3 227 88АктиниЙ* 6d7s2 232,038 в0Торип* 6d27s3 231 81ПрО1 6(137.
140,12 Лантаниды: ^Цернй Оболочка Хе-|- 4/5d6s2 140,907 59П рапсодии 4/36s2 144,24 0ОПеоднм 4/<6®2 147 в 1 Ир о л! еш мй* 4/®6s2 150,35 62Самарпй 4/e6s2 151,96 63Евро11Н| 4/’6s2
237 212 243 247 247
Трансурановые: взНептупий* в4Плутоний* ^Амерыл^ий* &йКюрий* мБеркели
Оболочка R.H+ 5p7sa af^dls3 Sf^dls^ ;; 5f 6rf7s2 5pSdls-
аВ 1861 г. был искусственно синтезирован изотоп элемента 103, нагкд
(ты, у которых вес виды атомов нестабильны (радиоактивны), отмечены звездочками. чусстченно, но существование их в природе не доказано,
(гураций см. стр. 145.
комиссии по атомным весам па 1962 г,- -Jlptui, реОл
Группы
7 VI I VII { VIII
подгруппа
главная побочная главная побочная главная побочная главная Подгруппа (нулевая трудна)
1,00797 ! Водород 1s 4,0026 аГелий IS2
14,0067 1 Азот 2s22/>s 15,9994 3Кислород 2s22/>i 18,9984 ;}ФТ0р 2s22p6 20,183 10Неоп 2s22p®
30,9738 16Фосфор 3«23/>з 32,064 ittCcpa 3s23/>i 35,453 17Хлор 3s’a3p5 39,948 jgApron 3s*3pe
51,996 24Хром 3cf64s 54,9381 ззМарганец 3dfi4s2 55,847 58,9332 58,71 звЖслезо 27 Кобальт ^Никель 3</e4s2 33’4s2 3d84sa
74,9216 33Мышьяк 4sa4p3 78,96 з4Се.теп 4^24уА 79,909 ззБром 4sa4p® 83,80 ззКрпптон 4s34pe
95,91 42Молибдсп 4d®5s' 97 43Технсцин* 4dB5s2 101,07 102,905 106,4 44Рутепий 46Родий 46Палладий 4di5s 4d»5s 4dio
121,75 51Сурьма 5з25рЗ 127,60 заТеллур 5s25/>4 126,9044 53И0Д 5s25pB 131,30 54Кеенон 5sa5p®
J 183,85 т4Вольфрам 4/M5d*6s2 186,2 7 «Рений 4/«5dB6s2 190,2 192,2 195,09 7дОсмий 77 Иридий удПлатнна 4/l45d°6sa 4/H5d76s2 4/l4S3a6s
208,980 ззВисмут* 6s36p3 210 84 Поло ни if* 6.f26p« 210 8 5 Астатин* 6sa6p5 222 ввРадон* бз^бр®
ДИНИ* 238,03 92Уран* 6(/47s3 Трансурановые алемепты (См. ниже)
7,25 Гадолиний 4/75d6s2 158,924 162,50 65Тербий Диспрозий 4/»5d6s2 4/35d6s2 164,930 е7 Гольмии 4/I05d6s2 167,26 168,934 173,04 174,97 в8Эрбий ваТулий 70Иттербий 71 Лютеций 4/«5ti6s2 4/i36s2 4/i46s2 4/i45d6i2
249 254 253 256 255 257
ддКалифорний* ъдЭйншпгей'ний* 100Ферлнй* 101Afew9e.teeww* 1иНобелый* 10за
5/a6d7s2 5iw5d7sV 5fs&?7s2? 5/i26d7»2? 5/i3617s2?
лаурспсием.Вго массовое число предполагается равиыл 157, —Прим, pel).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
абсорбент 483
абсорбция 259
света 349 ,
авгиты 511
квогадро число 138
\втогсипая сварка 477
Агат 534
Агрегаты
атомные 250
молекулярные 250
Адсорбция химическая 392
Азеотропные смеси 643
Азиды 651
Ааот 625—672
аналитические сведения 67!
атмосферный 633, 636
атомарный 634
биологическое связывание 636 галоидные соединения 667—670 двуокись 640
двусерпистый 671
закись 637
иодистый 669
кислородные кислоты 642—647
модификации 634
распространение в природе 633
общие сведения 833
(iKticxu 635, 637—642
окись 638
Получение 633
применение 636
пятиокись 641
свойства 634-т-636
сернистые соединения 670—672
треховне;, 639
фтористый 667
хлористый 667
четырехсот сь 640
Азотистоаодородная кислота 651
Азотная кислота 218, 347, 642—647
амид 650
изомеры 645
перокси 642, 648
синтетическая 218
спектры 646
Азотноватистая кислота 650, 651
изомер 651
Азулен 478
Аквадаг 462
Аквамарин 273
Акво су ръмяи овинная кислота 723
Аккумуляторы 594
Акриловая кислота 478
смола 475
Аксинит 359
Активность 57, 91, 95
ионов 95
Активные состояния веществ 342
Актиниды 22
Акцепторы 822
Алгаротов иоропгок 716, 720
Алебастр 269, 316
Александрит 273
Алка зил в 540
Алканы 468
Алкены 468
Аякилбориые кислоты 373
Ал килборо Кейды 373
Алкяяселеяиды 807
Алкияселеномеркаитаны 807
Алкиясиланы 389
Анкилсульфкды 794
Алкилтеллуриды 807
Алкииольиый сип Tea 478
Алкиды 468
Алкоксааминосиланы 513
Аллемонтит 713
Аллилен 508
Аллотеллуровая кислота 8&5
Аллофаны 552, 553
Алмаз 458, 459, 460
искусственный 460
Алмазонедобтше вещества 330, 332, 50,9
Альбит 185
Альдегиды 470
Альдоль 478
Алкмшяаты 387, 395
Алюминий 263, 351—359, 381—40б
аланаты 387—390
аланы 388
алкилы 358, 400
амальгама 1585
амфотерность 352, 353
аналитические сведения 405
арсенат 405
ацетат 387, 403
ацетилид 476
борат 387
борид 404
борогидрид 367
бромид 400
валентность 351, 354, 356, 387
892
Предметный указатель
гало ген аланы 389
галогениды 396—400
гетеро полярные ср единения 356
гидрид 353, 387, 388
гидроксофторид 397
гидроокись 351, 353, 387, 392—394
иодид 400
карбид 382, 404, 472
квасцы 351, 387
комлексные иопы 387
монофторид 397
нитрат 403
нитрид 404
обнаружение 227
окись 387, 391, 393
— адсорбент 392
— Брохмана 392
— кристаллическая решетка 391, 392
— модификации 391, 393
оксигидрат 388, 393
работа ионинации 356
роданид 403
силикаты 387
соединения 386
спектральные термы 354, 355
сплавы 386
строение атома 354, 355
субокись 392
сульфат 388, 401
сульфид 387, 404
теплота гидратации ионов 356, 357
— образования соединений 358
— плавления 359
тряарилы 358
формиат 495
фосфат 387
фтор алюминаты 396, 397
фторид 396
хлориды 357, 388, 398—400
— димерпые молекулы 399
— основные 400
эфираты 389
Алюминий ер танине ски с соединения 400
Алюмкнироватгне 386
Алюмосиликаты 381
Ал юмота лицевые квасцы 424
Алюмотермия 385
Амблигопит 186
Аметист 391, 529
Амигдалин 501
Амидная группа 662
Амидо фосфорная кислота 697
Амиды 662, 665
Амипы 662
Аммиак 651, 652—666
гидрат 249, 658
гидроокись 658
жидкий 656
инверсия 653
каталитическое окисление 644
получение 653—656
применение 656
производные 662
Аммиакаты 350, 431, 657
Аммоний
азид 652
амальгама 659
бикарбонат 661
бромид 850
галогениды 181, 239, 246
гексафторо алюминат 437
гексафторогаллат 410
гексахлороплюмбат 524
гексахлороСтаннат 524
иодид 851
иоп 212
карбаминат 661
карбопат 490, 661
нитрат 660
перекись 662
пиросульфат 769
роданид 506
селитра 660
сернистый 789
соли 211, 228, 658
сульфат 661
сульфид 789, 790
тетраацетоталлат 427
тиокарбамат 500
фосфат 686
фторид 239, 849
хлорид 660
хлоростанпат 576
Амфотерность 748
Амфотерные вещества 83
Анализ
металлографический 606
рентгенографический 231—235, 336
— метод Брегга 233
— — Дебая — Шеррера 235
— Зеемана и Шибольда 235
— Лауз 231—233
рентгеноструктурный 606
термический 606
Аналитическая химия 16
Апальцим 546
Ангидрид 213, 269, 316, 317
ангидрокислоты 591
Ан гидро кис лота 85, 290, 353, 867
Ангидро основание 83, 85
Англезит 584, 601
Андалузит 381, 545
Аид евин 381
Апяоиы комплексные 439
Анортит 381
Антигорит 545
Антимонаты 716, 718
Антимониды 725
Анти мопн л сульфат 722
Аптимонилтартрат 722
Антимониты 716, 717
Антиферромагнетизм 339 341
Антрацит 456
Апатит 269, 672
Арагопит 311
Аргиродит 562, 563, 568
Ароматические соединения 463
Ареепаты 706, 707
Арсениды 712
Арсепиты 705
Арсеполит 703
II реаметный указатель
893
ApCHUbj 710
органические 711
Асбест 269, 285
Ассоциированные соединения 330
Астатин 28, 830
Асфальт 456
Атом 106—125, 134, 144—146, 158, 159,
174, 255, 259—261, 284
водорода 106—125
возбужденный 158, 159, 174
импульс вращательный 283
— орбитальный 283
квантовое число ’283
мостиковый 446
оболочка 144, 146
основное состояние 158, 284
сник 283
строение 40, 255, 259
тин связи электрона 145
число электронов 261
энергетические состояния ИЗ
— уровни 113, 134, 260
Атомный вес 19—21, 261
константа 262
помер 261
объем 34
плотность 34
радиус 36
решетка 332
Аурипигмент 700
Ацетальдегид 477, 478
Ацетат-ион 488, 497
Ацетатный шелк 496
Ацетаты 485, 488, 496
Ацетилен 475—478, 511
гидрирование 475
определение 511
полимеризация 478
Ацетилиды 450, 476
Ацетилфтороборат 497
Ацидосоли 730
Аэрозоль углерода 481
Байерит 394
Бальмера Серия 104
формула 105
Барий 263, 264, 266, 274, 278, 319
азид 191
аммиакат 278
бромид 266, 305
гидрид 266—267, 288
гидроокись 265—267, 289, 290, 295
тшнд 278
иодпд 266, 305
карбонат 269, 274, 313
кристаллическая решетка 279
падиерокись 296
нитрат 308
нитрид 266
окись 266, 288, 236, 2.91, 235
окрашивание пламени 280
перекись 297
селейат 803
силициды 516
спектр 280
сульфат 269, 318, 795
сульфид 266, 790
фторид 266, 300
фторосиликат 523
хлорид 266, 304
— ди in др ат 304
хромат 319
Баритовая вода 206
Бауэра способ 383
Башепное вскрытие 383
Безопасные спички 861
Белая жесть 572
Белильный щелок 858
Бензин 457, 469, 471
синтетический 470, 487
Бензол 463
Бенитоит 543
Бергмана серии 196
Берилл 273, 543
Бериллаты 290, 291
Бериллий 263, 265, 266, 273—276, 278,
'279, 286
амфотерность 289
анионы 267, 290
ацетилид 476
бромид 303
гидрид 267, 288
гидроокись 265, 267, 289
иодид 301
карбонат 309
кристаллическая решетка 279
минералы 273
нитрат 266
окись 266, 274, 288, 290, 291
перекись 296
сульфат 314
сульфид 266
фторид 299
хлорид 266, 300
Берлинская лазурь 500, 503
Берцилианнт 419
Бемит 393, 394
Биполярные ячейки 48
Бисквитный фарфор 555
Бифосфаты 446
Боксит 381, 383, 393—395
Бокситовые кирничи 395
Болотный газ 470
Бор 351—381
аморфный 360
аналитические сведения 380, 381
атомная теплоемкость 360
бромид 372
валентность 351, 354, 356, 361
галогениды 361—363, 369—372
гидриды 353
гомологи 352
иодид 372
карбяд 510
кислородные соединения 363
ко о р ди нации иные сое дине п ия .362
моноокись 362, 373, 374
нитрид 361, 363, 379
окиелы 361
окрашивание пламени 360
поляризующее действие 352
894
Предметный указатель
применение 361
распространение в природе 359
свойства 360, 361
строение атома 354, 355
сульфид 361, 363
теплота образования соединений 358
тетрагональный 360, 361
триалкилы 358
удельная теплоемкость 360
фторид 328, 369—371, 441, 442
фтороборная кислота 369, 370, 371
присоедипение аммиака 371
хлорид 371, 372
алкоголиз 372
гидролиз 371
продукты присоединения 372
циклические соединения 363
чистый кристаллическим 360
элементарный 360
энергия ионизации 356
Вора
квантовые условия 114, 137
постулаты 108
теория 107, 114
условия частот 121
формула 256
Боразаны 368
Боразены 368
Боразины 368
Боразол 368
БоралкиЛы 372
Борацаты 363, 366, 367
Борановые со ли 363, 365
Борапы 363
Бораты 363, 376
пероксигидраты 361, 377
Борацит 359
Бориды металлов 362, 363
Бористые кислоты 361
Борна — Габера круговой процесс 175
Борная кислота 359, 360, 370, 373, 374
соли 376
эфиры 374
Борный ангидрид 373
Борогидриды 366, 367
Бороводороды 361—365
Бороксол 368
триметил 368, 369
Борорганические соединения 363
Боросиликаты 374
Бррофтористоводородная кислота 362
Борсульфид 379
Борсульфол 3.69
БорфосфороксиД 363, 380
Брейтгауптит 726
Бриллиантовая шлифовка 459
Британский металл 715
Бром 825—855, 865, 866
азид 652
антимонаты 721
атомарный 837
кислородные соединения 865
нахождение в природе 831
получение 832
применение 838
физические свойства 835
фториды 871
хлориды 871
Броматометрия 866
Броматы 866, 870
Бромиды 849, 853
Бромистая кислота 865
Бромистоводородная кислота 843, 846
Бромистый водород 846
Бромная кислота 866
Бромноватая кислота 865
Бромноватистая кислота 865
Бронза 570, 573
Бруцит 293
Бульриха соль 222
Бура 359, 360, 376, 377
ювелирная 377
Бутадиен 477, 478
Бутан 466
Бутиндиол 478
Буферные смеси 889
Вавеллит 672
Бакенродера жидкость 772
Валентная сила 29
Валентное число 29
Валентность 29—33, 148—164, 176, 284,
341, 432, 439
готерополярная ,158
главная 432, 433
координационная 433
максимальная 158
побочная 432, 442
стехиометрическая 29
теория Косселя 150—154
— Лапгмюра 154
— Льюиса 154
— онтетяая 154
— электрохимическая 29
Валентные углы 157
Ванадий 258
Вапдерваальсовые силы 127, 332
соединения 330
Ватерит 311
Вебстерит 401
Бевелит 456
Везувин 381, 543
Вейса закон 339
Вердера теория 433
Вернировка 573
Веселящий гаа 637
Вещество
аморфное 607
изотропное 607
модификации 99 ,
простое 15
Велера метод 512
Вивианит 672
Винилацетилен 477
Виниловые соединения 477, 478
Виннокаменная соль 223
Випяый камень 228
Висмут 343, 726—734
алкилы 734
аналитические сведения 734
ацидосоли 730
Предметный указатель
895
валентность 728
галоидные соединения 730 гидроокись 729--731 карбонат 732 распространение в природе 726 нитрат 732 окислы 728 оксигалогепиды 730, 731 оксихлорид 730 получение 726 применение 728 роданид 732 свойства 727 соединения 728—734 соли 729
— комплексные 730 сульфат 732 сульфид 732 Висмутаты 729 Висмутина соли 729 Висмутовый блеск 726, 733 водород 733 зеркало 733 Кислота 729 охра 726 шпат 726
Витерит 313
Вода 67— 75 , 269 , 273 , 292 , 311, 438, 482, 537, 559, 654, 786 аммиачная 654 газированная 482 давление пара 69 диаграмма Состояния 72 диссоциация 74, 75 дистиллированная 68 жесткость: 68, 269, 311 известковая 294 ионное произведение 74 конституционно связанная 96 кристаллизационная 438 обеззараживание 68 питьевая 68 приходная 67 свойства 69 сероводородная 786 температура кипения 70 термическое разложение 55, 59 ультрачистая 68 цеолитиосвяз ап ная 99 электролиз 49 электролитический перенос 100
Водород 22, 42—125, 156, 157, 225, 265, 784, 793 атомарный 63 валентность 42 выделение из растворов 55 дегидрирующее действие 64 жидкий (Ю ион 101 молекула 156, 157 распространение в природе 43 образование в естественных условиях 44 общие сведения 42 орто 65 пара 65
положение в таблице^Менделеева 22 получение 44—59 потенциал 323 применение 44 свойства 60—65 соединения 66—103 спектр 104 спин 156
сродство к электрону 265 строение атома 106—125 тяжелый 65, 225 физические константы 61
Водородная связь 322, 325, 493 Водородный
Мостик 325
показатель 886
электрод 51
Водяцой газ 485, 487
Воздух, сжигание 635 Волластонит 543, 544 Волновая механика 114—125 Волновое уравнение 118 пространственное 119
Восстановители 814—818
нормальные растворы 816
Восстановление 812 электролитическое 818
Вульфепит 584
Вымораненвание 743
Гадолинит 273 ‘
ГаагиДрат 474
Газойль 457
Газолин 457
Галенит 584
Галлий 351—359, 406-—413, 452
алкилы 412
амфотерность 352
бромид 410
валентность 351, 354, 356, 409
галогениды 410
гидриды 412, 413, 353
гидроокись 351, 411
Двухвалентный 412
квасцы 351, 412
нитрат 412
нитрид 412
одновалентный 412
— окись 412
окись 407, 410, 412
получение 408
работа иоцизации 356
селениды 407
сцектры 356
структура решетки 408
сульфид 44, 407
теллуриды 407
теплота образования соединений 358
триалыилы 358
триарилы 358
фторид 410
халькогенид 406, 407, 410
хлорид 410, 428
Галогениды 826, 847—854
аналитические сведения 852
896
Предметный указатель
Галог еноборосульф о лы 369
Галогеноводородные кислоты 826, 844— 847
Галогеноводороды 839—847
получение 842
строение 841
электролитическая диссоциация 841
Галогепокислоты 852
Галогепосоли 852
Галогене сульфоны 793
Галогены 350, 825—874
валентность 827
кислородные кислоты 854 комплексообразование 829 распространение в природе 831 общие сведении 825—831 окислы 854
окраска 829
подвижность ионов 830
получение 832—834
применение 838
соединения 839—874
структура решетки 830
физические свойства 834
электросродство 829
Гарниерит 545
Гасящее пламя 483
Гаттермана — Винклера способ 513
Гауин 562
Гаусс 338
Гафний 255
Гем тл ера — Лондона теория 157
Гексабромоловяниая кислота 576
Гексагидро к сопл юмбаты 593
Гексаметялциклотрнснлазан 520 Гексатиопаты 774
Гексатиоповая Кислота 774
Гексафторофосфаты 693
Гексафторофосфорпая кислота 693
Гексахлороловянпая Кислота 576
Гекторит 545
Гелий
атом 142
спектр 132
Гелиотроп 534
Гель 537
голубой 539
кремневый 537, 539
Гемоглобин 484
Генераторный газ 486, 487
Генераторы 486
Винклера 487
Геохимия 16
Герман аты 567
Германиевая кислота 567
Германий 448—454, 562-569
аморфпый 564
аналитические сведения 569
валентность 449
гидроокись 453
кристаллический 564
получение 563
свойства 564 и
строение атома 453
Германий(П) 568, 569
бромид 568
гидроокись 569
иодид 569
окксь 569
трибромгерман 569
трихлор герман 568
селенид 569
сульфид 569
хлорид 568
Германий(1У) 564—568
алкилы 565
бромид 566
галогениды 565
двуокись 566
дигерман 565
дисульфид 567
имид 568
модификации 567
моногерман 565
нитрид 568
оксихлорид 566
полигермены 565
сульфат 568
тетраэтил 565
фторид 566
фторогерманаты 566
хлорид 566
хлористоводородная кислота 566
Германит 408, 562, 568
Гермаяовэдороды 564
Гетерополикислоты 375, 804
Гстерояолярпая связь 322, 346
Гетерополярные соединения 32 443
Гиббса — Ворсе камера 210
Гибридизация уровней 452
Гидразин 449, 651, 663, 684
Гидразин гидрат 664
Гидраэиния соли 664
Гидраргиллит 393, 394
Гидратация 96, 100, 212
Гидраты 96—103 431
газов 248—250
давление пара 98
диаграмма растворимости 96
термическая диссоциация 98
Гидридпого сдвига закон 740
Гидриды 66, 67, 287, 388, 389
газообразные 66
Двойные 389
двухатомные 67
металлообразные 67
несолеобразные 388
полимерные 66
получение 389
солеобразные 287
Гидрппдон 478
Гидрирование 62, 390 , 469, 475 , 813
аланатом 390
Бергнуса метод 469
косвенное 469
непосредственное 469
Гидрогенизация жиров 62
Гидрокалумит 395
Гидрокарбонаты 488—490
Гидр окси л амин 662—664
Гидроксил аммония соли 663, 665
Предметный указатель-
897
Гидроксильные ионы, концентрация
888
Гидр оксо алюминаты 353, 395
Гидр оке о бораны 361
соли 363
Гидроксоиопы ,290, 442
Гидроксоплюмбаты(П) 591
Гидроксоний-иоп 100, 101, 749
Гидроксосоли 86, 353
Гидроисостаннаты(11) 582
Гидрокедфторсборная кислота 370
Гидролиз 877—883
копстапта 880
степень 878—883
частичный 878
Гидроокись 331, 747
Гидрооксалаты 488
Гидротермальный синтез 547
Гидрохинон 478
Гиллебрандит 557
Гильмана закономерность 418
Гипобороты 365
Гиноборная кислота 361, 379
эфиры] 379
Гипоиодиты 866
Гипонитрпты 650
Гипофос ф аты 677, 691
Гипофосфиты 677, 692
Гипохлориты 857, 864
Гипс 213, 269, 286, 316, 317
Гипсовая вода 319
Главные подгруппы 829
Глазурь 551
Глауберит 316
Глауберова соль 225
Глина 381, 551—554
керамическая 551
Глинистые мергели 381
Глинозем 381, 385, 391
волокнистый 385
плавленый 391
Гмелинит 559
Гнейс 381, 529
Гомеонолярные связи 321, 331, 332
соединения 32, 183, 357, 443
Гончарные изделия 556
Гопкалит 484
Гонкалптоный метод 514
Горение 743
Горный хрусталь 512, 529
Горькая земля 273
соль 273, 314
Горячее дутье 487
Гранит 381, 511
Графит 245, 333, 456, 458, 461—463
ретортный 466
Графитовая кислота 461
Гремучий газ 61
Гриньяра реакция 277
Грисгеймера сплав 285
Группы 21
Грэма соль 690
Гуапо 676
Гумит 543
Гурдинамит 533
1/а 57 г. Ремп
Даг 462
Давление разложения гидратов газов 250
Дативная связь 358, 442
Датолит 359
Даунс-камера 190
Даусона газ 487
Двойные водородные мостики 364
окислы 396
соединения 333
соли 431, 432
Дегидрирование 813
Дейтерий 65
Деформационные колебания 345
константы 345
Джонсона соль 851
Джоуля — Томсона эффект 60
Диаграмма состояния 610, 613, 614, 615
плавкости 611
Диазоуксусцый эфир 103
Диализ 335, 536
Диализатор 536
Диамагнетизм 407
Диамагнитные свойства 338—340
Диаспор 393
Диборан 364, 365
мезомерия 365
Дибортетраиодид 372
Дибрепцкатехи но бораты 362
Дивариантпое равновесие 504
Дикарбон 481
Дикит 553
Дикремпевая кислота 535, 537, 538
Дикремпийгексафторид 524
Днлтея формула 365
Димотилаланы 388
Диморфная соль 218
Динас 556
Диоксодисил оксан 526
Диопсид 544
Диоптаз 543
Диполь 32
Дипольпый момент 348, 357
Дисилазаи 521
Дисилоксан 518
Дисперсия 324
Диспропорционирование 474, 820
Диснротиды 79
Д песо ниацин 485
Дисульфатоталлиевая кислота 427
Дитнопаты 775
ДитиОпиты 770, 777
Дитиопистая кислота 777
Дитноповая кислота 775, 776
Днтноггольная кислота 499
Дифосфаты 446
Дифосфин 699
Дихлорэтан 476
Дициан 500
Доломит 269, 309, 310
Драгоценные камни синтетические 391
Дразпсчше 571
Древесный уксус 496
Друммондов свет 293
Дубление белое 401
Дублет 154
Дублетные серии 282
898
Предметный указателъ
Дьюара формулы 326
Дюралюминий 386
Единица тепла 69
Железный пшат 454
Железо, комплексные соли 443
Железобетон 557
Жемчужные белила 732
Жженые квасцы 402
Жирботол-процесс 483
Жирные кислоты 470
Жировик 285
Закон
Авогадро 335
Бильтца 250
действия масс 55—60, 94, 485
— — константа 485
постоянных отношений 332
смешения Зоммерфельда 135
Заряды 29, 157, 876
единица 29
Отталкивания 157
перемещения 876
притяжения 157
Зеемана эффект 122
Зейгерование 571
Зелень Шееле 706
Зубная эмаль 673
Известковая земля 273
Известковое молоко 294
Известковый раствор 294
селитра 307
Известняк 269, 293
Известь 207
гашеная 294
едкая 214, 293
жженая 293
негашеная 273
серпая 496
Излучение 136
Изобары 20
реакция 59
Изобарическое разложение 99
Изомерия 433, 447, 578
Изоморфизм 39, 212, 247, 248
Изостеризм 161, 638
Изоетсры 154, 368
Изотерма реакции 59
Изотермическая дистилляция 335
Изотермическое разложение 99
Изотетра мета фосфорная кислота 682
Изотипия 248
Изотопы 20
Имбибцция 537
Имидогруниа 663
Имиды 663
Имипогруппа 662
; Инверсия сахарозы 102
Индий 351—359, 406, 413—419
аланат 419
алкилы 418
алюмогцдрид 419
амфотерность 352
ацидосоли 414, 415
бромиды 414, 415, 417
валентность 351, 354, 356, 413
Гекса хлорин дат 415
гидрат окиси 416
гидрид 419
гидроокись 351
днарилы 358
изотопы 414
под иды 414
квасцы 351, 415, 416
нитрат 416
окись 414—417
получение 413
работа ионизации 356
радиоактивность 414
распространение в природе 413
селенид 407, 417
соединения 414
спектры 356
структура решетки 408
сульфат 416
— двойной 416
сульфид 407, 415—417
теллурид 407, 417
теплота образования соединений 358
тиосоль 417
триалкилы 358
триметил 418
трипропил 418
трифенил 418
триэтил 418
фторид 415—417
халькогенид 406, 407
хлориды 414, 415, 417, 428
эфират триметилиндия 358
Иядий(1) 414, 417
окись 417
хлорид 417
Индий(П) 417, 418
бромид 417
окись 417
сульфид 418
фторид 417
хлорид 417
Индикаторы 102, 884, 885, 887
Индуцированный момент 339
Инертный газы 127, 132—135, 143, 147,
153, 240, 241, 257, 260
валентность 127, 153
гидраты 127
изотопия 147
спектры 132—135
структура решетки 240
уровни апергИи 257
число электронов в оболочках 143
электронные конфигурации 147
Инсектофунгициды 524
Интерметаллические соединения 332, 508
Интерференционные кольца 236
Инфузорная земля 533, 539
Предметный указатель
899
Иод 652, 825—855, 866—870, 872 ацетат 872 бромид 871 валентность 828 гидрат 249 кислородные соединения 866—870 кристаллическая решетка 831 распространение в природе 831 получение 833 применение 838 пятиокись 867 растворы 838 соли 838, 872 физические свойства 835 фториды 871 химические свойства 837 хлорид 871, 872
Иодаты 867, 868, 870
Иодистоводородная кислота 843, 846
Йодистый водород 846
Поди гы 850, 853
Иодпая кислота 854, 869
Йодноватая кислота 854, 867, 868 Иодноватистая кислота 854, 866 Иодоаптицопаты 722 И одоз о соединения 872 Иодометрия 869 Иодониевыо соединения 872 Иодо ста ни а ты 576 Ионизация 136 напряжение 138 потенциал 38, 140, 143 работа 136, 138
Ионная сила 95
Ионные веса 335
Ионообменные смолы 81
Ионы 160, 336, 349, 353, 431, 452-453, 646, 658, 749, 770, 879 активность 879 аммония 658 нодорода 336 гидратированная форма 353 гидроксония 100, 749 Дигидроксонитропия 646 кислотные 452, 453 маскировка 431 нитрония 647 Отрицательные 160 тиогерманат 568, 569 тип оболочки 349 тритиосульфат 770
Ипатьева метод 474
Какодил 711
Какоди.'Ювая реакция 511
Калий 171, 177, 178, 180—193, 199—208, 211, 213, 215, 216, 218,2*19. 224, 226— 229, 238, 353, 370, 415, 486, 499 501 — 503, 506, 524, 649, 686, 718, 729, 769, 774, 789, 817, 848, 850, 851, 857. 866 азид 193 амальгама 209, 210 амцц 193, 205 аптимонат 718 бихромат 211 бромат 866
бромид 850 висмутат 729 галогениды 181, 238 гекса гидр ок состапн ат 524 гексапитрокобальтат 228 гексаоксобензол 486 гсксафторосилшмты 524 гексахлороиндат 415 гексахлороплатинат 211, 228 гидрид 179, 193, 199, 200 гидрокарбопат 219, 223 гид рок со алюминат 353 гидроКеоантимонат 227 гидроокись 179, 182, 204—208 гидросульфат 226 гидротартрат 228 гипохлорит 857 изотопы 184 иодид 171, 850, 851 карбид 193
карбонат 182, 211, 219, 220, 222, 223 карбонилмагнийфторид 437, 438 минералы 185—189 нитрат 211, 218, 219, 247 — кислый 219 нитрид 193 нитрит 649 окись 201, 202 оксалат 494
— сверхкислый 494 перманганат 211, 817 перекиси 193, 202 перхлорат 211, 218, 228 пироаптимонат 769 пяросульфат 226, 227, 769 полигалогепиды 851 политиопаты 774 получение 189, 191 применение 198 радиоактивность 184 роданид 506 селенид 201, 202 спектр 193—197 сульфат 211, 224, 226 сульфид 201, 202, 789 тартрат 228 теллурид 201, 202 теплота образования соединений 179 тиокарбонат 499 ферроцианид 485 фосфат 211, 686 фторид 211, 848 хлорат 211, 860 хлорид 211, 213, 215, 216, 248 хромат 211 цианид 501 циапоферраты 502, 503 фтороборат 370 фторо герма нат 566 фторосиликат 524
Каломельный алектрод 52
Кальций 97, 191, 213, 214, 244, 263, 266— 269, 273, 275—294 296, 300, 302—304, 307, 310—312, 316, 319, 320, 476, 478, 479, 485, 504, 516, 558, 592, 593, 687, 768 , 790 , 853, 859
57*
900
Предметный, указатель
аланат 390
алюминий гидр ид 390 амид 278
аммиакат 278
ацетилид 476, 478
бромид 266, 304
гидрид 266, 267, 278, 288
гидроокись 267, 289, 294
— гидрогель 294
гидросульфит 768
гипохлорит 859
имид 278
иодид 266, 304
карбид 476, 478, 479, 4§5
карбонат 213, 269, 310—312 кристаллическая решетка 279 надперекись 296 нитрат 307 нитрид 266, 277
окись 266, 273, 276, 278, 288, 290, 291, 293
окрашивание пламени 280 оксалат 320 ортосиликат 558 перекись 296
распространение в природе 269 силикаты 269
силицид 516
соединения с металлами 278
спектр 280
сульфат 269, 316
сульфид 266, 790
тетраоксоплюмбат 592 триоксоплюмбат 593 фосфат 687
фторид 244, 266, 268, 300, 853
хлорид 97, 191, 214, 266, 302, 303
цианамид 478, 504 цианид, 504
Каменная соль 185, 213—215, 237, 238 кристаллическая решетка 237, 238
Каменные материалы 555
Каолин 381, 539, 551—554
Каолинит 551
Каолирит 545
Карбаминовая кислота 661
;Карбиды 459, 476, 478, 479, 507, 508
кальция, см. Кальция карбид образующие аллилен 508 — ацетилен 507 — метай 508
— смесь углеводородов 508
устойчивые к кислотам 508
Карбонадо 460
Карбонаты 488—490
гидролиа 490 кислые 489 нейтральные 489 разложение 490
Карбонизация 223
Карбонилсульфид 499
Карбонилы металлов 486
Карборунд 509, 510
Карботермический метод 275
Карналлит 215, 216, 431
Карнегиит 546
Карнеол 534
Каро кислота 767
Касситерит 570, 577
Каталазы 78
Катализаторы 469, 472
Катодные лучи 336
Каурит 656
Каучук 477, 513
силиконовый 513
Квадруполь 347
Квантовая механика 114
Квантовое число 108, 112, 121, 141, 145, 159, 197, 257, 259
К вантово -механический резонанс 326
Кварц 511, 528
дымчатый 529
розовый 529
Кварцевое стекло 532
Кварцевый песок 512, 529, 534
Кварцит 529
Квасцы 211, 212 , 387 , 402, 424, 765
Кекуле формулы 326
Керамические изделия 553, 554
Кермес 724
Корнит 359
Керосин 457
Кизельгур 528, 533
Кизерит 269, 315
Килограмм 69
Киппа аппарат 483
Кирпич 556
Кисличная соль 494
Кислород 735—750, 855
активированный 746
атомарный 746
валентность 736, 737, 749
жидкий 742
координационное число 750
молекула 157
распространение в природе 740
получение 741
применение 741 свойства 741—743 соединения 746—750
фторид 855
Кислота 33, 79—82, 93, 94, 103, 327, 432, 470, 478, 488,489, 493, 495, 647,663, 679, 812, 874
амиды 663
водородная 80
двухосновная 488, 489, 493
диссоциация 93
жирная 470
кислородная 80, 647, 679
' комплексная 432
константа диссоциации 80 межатомные расстояния 327 многоосновная 93 насыщенная основанием 874 пеокнеляющая 812 одноосновная 488, 495 получение 80
сила 33
алектролитическая диссоциация 79, 103
Клаудетит 703
Предметный указатель
901
Клинкер 556
Клоде метод 741
Кобальт, цвет безводных соединений 350
Ковалентность 161
Коксование 464
Коксовый газ 473
Коксы 464
Колдаг 462
Колебания 115—119
амплитуда 116
гармонические 116
интенсивность 116
период 116
простые 116
уравнения 117—119
частота 116
Колчеданы 750
Комплексные
соединения 333
соли 431
Комплексы 430, 434—440, 443, 444
внедрения 443
методы установления существования 434—438
многослойные 300
нормальные 443
переходных элементов 444
строение 438—440
Цианидные 444
Конверсионный способ 218
Конверсия окиси углерода 46
Конденсация 579
вынужденная 703
Константы 11, 56, 58, 105, 108, 110, 134,
345, 346
валоптно-силовая 34а, 346
Планка, см. Планка константа
равновесия 56, 58
Ридберга, см. Ридберга константа
Концентрация
водородных ионов 101—103, 887, 88.9
молярная 56
Координационная теория 429, 433, 445
Координационное число 240, 241 244,
433, 440, 453
Координационные
связи 432, 440, 441
соединения 433, 434, 443, 445, 446
— классификация 434
— многоядерные 446
— одноядерные 446
Коппа правило 331
Кордиерит 381
Королевская желтая 710
Корунд 381, 391
Косселя закон сдвига 341
теория 130—154, 238, 441
Костяная черць 465
Коэффициенты 49, 37, 91, 95, 213, 335
активности 49, 57, 91, 95
диализа 335
осмотические 91
отклонения 91
температурные 213
электропроводности 92
Красильные лаки 402
Краска перманентная белая 319
Крахмал 455
Кремень 512 »
Кремневодороды 515
Кремневый гель 537
золь 536
Кремнезем 512
Кремнефтористоводородная кислота 512, 523, 853
Кремниевая кислота 513, 526—528, 534—
536, 538
ангидрид 534
коллоидная 536, 538
эфиры 513, 526, 538
Кремний 448—454, 509, 511—562
алкилпроийводные 515, 519
аморфный 512, 513, 521, 525
аналитические сведения 562
валентность 449
галогениды 515, 521, 525
гексабромид 526
гексахлорид 526
гидроксисоедипения 525
гидроокись 453
двуокись 452 511, 515, 527—535, 543, 546
дисульфид 527
дителлурид 527
ио Диды 525
карбид 509, 515
металлический 513
модификации 527, 532
моноокись 528
распространение в природе 511
нитриды 515
оксихлориды 525
перекристаллизация 514
получение 512
реакционная способность 514
роданид 527
строение атома 453
сульфиды 527
тетрагалогениДы 452, 525
фториды 512, 521, 522
хлориды 452, 521, 524, 525
Кремний органические соединения 513
Кривые
ликвидуса 613
нагревания 610
охлаждения 610, 611
солидуса 613, 615
Криогидрат 96
Криолит 186, 222, 381, 396
Криоскопия 336
Кристаллизация 607
спонтанная 607
теплота 610
Кристаллиты 607
Кристаллические редпетки 230, 231, 233, 235, 237—240, 241, 242, 244, 245, 329, 331, 482
гранецентрировапные 237
иопные 230, 241, 242
координационные 244, 245
кубические 237
молекулярные 230, 240, 244, 482
902
П редметный указатель
объемноцентрироватшые 237, 240
постоянная 231, 233, 235
сложные 237—239
слоистые 244, 245, 329, 331
тетрагональные 240
точечные 231
элементарные ячейки 237
Кристаллогидраты 333
Кристаллоиды 536
Кристаллохимия 16
Кристаллы 219, 236, 247, 443
образование 443
смешанные 219, 247, 613, 617
структура 236
Кристобалит 527, 523, 531, 546
Кровяная соль 502
Крокезит 419
Крокоит 584
Кронглас 548
Ксаптогепат целлюлозы 499
К сан то генова я кислота 499
Ксерогели 533, 539
Ксилолит 301
Кулона закон 107
Купоросы 765
Купрен 450
Курроля соль 690
Лабрадорит 381
Лазоревый камень 560
Лазурит 561
Лакмус 177, 884, 885
Лаиновая копоть 465
Лантаниды 22, 23, 40, 41, 255
Лаутерит 868
Лауэграмма 231
Леблапа способ получения соды 221
Леватит 506
Лед, решетка 531
Ледяной камепь 396
Лейцит 381
Лепидолит 186
Лиганды 466
Лильенротта способ 44
Линде метод 741
Липарит 529
Литий 179, 481, 182, 184, 185—189, 191, 193—203, 205, 209—213, 216, 217, 219, 220, 224, 227—229, 238, 244, 245, 367, 389, 515
азид 193
аланат 389
амальгама 209, 210
амид 193, 205
бихромат 211
борогидрид 366
бромат 212
бромид 217
галогениды 181, 238
гексахлороплатинат 211
гидрид 179, 193, 199, 200
гидрокарбонат 219
гидроокись 179, 182, 204—206, 244, 245
гидротарграт 211
дитиопат 212
иодид 179, 217
карбид 193
карбонат 182, 184, 211, 219, 220
минералы 185—189
нитрат 211, 219
нитрид 193
нитрит 212
окцсь 193, 201, 202
перекиси 202, 203
перманганат 211
перхлорат 241, 212
получение 189, 191
применение 198
селепат 212
селенид 201, 202
силицид 193, 515
спектр 193—197
сульфат 211, 212, 224, 227
сулвоид 201, 202
сульфит 212
теллурид 201, 202
тиосульфат 212
фосфат 184, 211, 228
фторид 179, 184, 211, 217, 228
хлорат 211, 212
хлорид 211—213, 216, 217
хромат 211
цианоборадат 367
цитрат 224
Литийгаллийгпдрид 413
Лоллипгит 700
Лорандит 419
Льюиса правило 341
Магналий 285, 386
Магнезиальная смесь 712
Магнезиальный цемент, см. Цемент магнезиальный
Магнезит 269, 309
Магнезия 226, 556
Матретиам металлов 342
Магнетоны Бора 340
Магнетохимия 338
Магний 263—295. 298, 301, 302, 307, 309,
314, 315, 320
аланат 390
алкилиодид 277
алгоминийгидрид 390
амальгама 277
аммоний арсен ат 320
аммонийфосфат 320
борогидрид 367
бромид 266, 302
гидрид 267, 288
гидроокись 214, 265, 267, 289, 292
иодид 266, 302
карбонат 269, 309, 310
кристаллическая решетка 279
нитрат 307
нитрид 266, 277
окись 242, 266, 288, 290, 291
оксихлорид 301
перекись 298
распространение в природе 269
силикат 269
Предметный указатель
903
силицид 516
спектральные термы 354, 355
сплавы 285, 632
сульфит 226, 263, 264, 269, 273, 274, 314
сульфид 266
фосфат 320
фторид 266, 299
фторосиликат 524
хлорид 266, 292, 301
цианамид 504
Магнитная восприимчивость 338
Магнитный момент 338—341
Маделунга константа 172, 179
Мадрелла соль 690
Майолика 556
Малоновая кислота 481
Марганцовый шват 454
Маскировка иолов 431
Масло
гидравлическое 480
горького миндаля 501
, дизельное 469
изобутиловое 471
минеральное 457
осветительное 469
смазочное 469
топливное 469
Массикот 591
Медовый камень 456
Медь 452
родапиД 505
Мезомерия 326, 329
кетонов 484
окиси углерода 484
Мезоморфная структура 465
Мел 269
Меласса 503
Мендипит 596
Мергели 311, 552
Меркаптаны 794
Мерсеризация 207
Метаоорная кислота 375
Метакремневая кислота 537, 538
Метадлобороводороды 363
Металлография 606
Металлоподобные соединения 330
Металлы 33, 148, 264, 607, 616 876
Метай 451, 470—473, 485, 510
Метасиликаты 535, 543
Метатиоборяая кислота 369
Метафосфаты 681, 689
Метафосф им овые кислоты 697
Метафосфорнал кислота 677, 689
Метиловый
красный краситель 884, 885
оранжевый краситель 884, 885
спирт 470, 471
Метллэтилпептад 466
Микрореакцци 584
Минерализаторы 291
Мозли закон 28, 253—255, 260, 261
Молекулы
асимметричные 347
ассоциированные 325
атомные 330, 331, 347
поляризация 347, 348
полярные 32, 71 рассеивание света 345 симметричные 347 стабильные 158 строение 328, 329, 346 олектрояные конфигурации 159
Молекулярный вес 335 определение 335, 336
Моль окиси 822
Молярная восприимчивость 338 рефракция 348
Молярный объем 331
Момент вращения 108
Мотюбромди боран 364 Монокристаллы 608 Монофторуглерод 463 Монтмориллонит 545, 553 Морин 405
Морская пенка 286 Мочевина 500, 656 Мрамор 269, 303 Муллит 554, 556 Мультиплети ость терма 283 Муравьиная кислота 494, 495, 511 Мусковит 545 Мыло синтетическое 470 стеклянное 549
Мышьяк 700—712 амфотерность 705 аналитические сведения 712 галогениды 708 модификации 701 окись 702 получение 701 применение 702 пнтиокись 706 распространение в природе 700 свойства 701 соединения 702—712 сульфиды 709 сульфосоли 709 тиосоли 710 трихлорнд 709
Мышьяковая кислота 706 Мышьяковистая кислота 704, 705 Мышьякозое зеркало 704 Мышьяковый колчедан 700 цвет 700
Надперекиси 193, 202
Надугольная кислота 491
Наждак 381, 391
Накрит 553
Напряжение возбуждения 138 иоиизапия 138
Натрий 177—186, 181, 189, 193—229, 238, 241, 246, 248, 249, 291, 354, 355, 367, 485—496, 503, 507, 524, 536, 540, 541, 578 , 6(4, 639, 648, 649, 651, 657, 665, 685, 690, 691, 705— 707, 719, 724, 729, 768, 769—771, 774, 776, 789, 804, 834, 849, 850, 857, 874, 878 азцд 193, 651
904
Приб-четный. утзаглелъ
алюминат 222 амальгама 209, 210 амид 193, 503, 665 антимонат 719 арсенат 706, 707 арсенит 705 ацетат 485, 496 бериллат 291 бихромат 211 борогидрид 367 бромид 849 висмутат 729 галогениДы 181, 238 гексапитрокобальтат 228 гексахлороплатинат 211 гидрид 179, 193, 199, 200 гидрокарбонат 219, 222 гидроксоантимонат 227 гидроокись 179, 182, 204—207 гидросульфат 225, 226 гидросульфит 768 гидротартрат 211 гипосульфит 769, 776 гипохлорит 857 дитионат 776 иодат 218, 834 иодид 850 Карбид 193, 205 карбонат 182, 211, 219—222, 228 Метилат 485 минералы 185—189 нитрат 211, 217 , 218, 246 , 648 нитрид 193 нитрит 649 нитрозил 657 нитропруссид 639 окись 201, 202 оксалат 494 перекиси 193, 202—204 перманганат 211 перхлорат 211 пиросульфат 2 26, 769 пиросульфит 770 по ли тионаты 774 получение 189—191 применение 197, 198 роданид 507 селенид 201, 202 силикат 536, 540, 541 соединения 177—186,198—208,210—229 спектр 193—197 спектральные термы 354, 355 сплавы 614 Станнат 578 сульфат 211, 224, 225 сульфид 201, 202, 789 теллур ат 804 теллурид 201, 202 тетраборат 377 тиоантимонат 724 тиосульфат 771 ферроцианид 485, 492 фосфат 211, 686, 690, 691 фторид 211, 241, 848 фторосиликат 524 хлорат 211, 861
хлорид 179, 191, 211—216, 240, 241,
248, 249т 874
хромат 211
цианамид 503
цианид 503, 878
Нашатырный спирт 654
Нашатырь 660
Нейтрализация 85, 883—889
теплота 883
точка 884
Нельсона камера 210
Неметаллы 148
Неорганическая химия 15, 16
Неорганические вещества 334
Нерпста тепловой закон 201
формула 49
Нефелин 381, 546
Нефть 456, 457
перегонка 457
Никель 350
тетракарбопил 486
Нитраты 642, 643, 647, 648, 672
гидроксопия 643
орто 648
щелочпые 648
Нитридогруппа 663
Нитриде со единения 663
Нитриды 666
двойные 666
Нитрилогруппа 663
Ннтрилоперхлорат 670
Нитрилосоедияении 637, 669, 670
Нитрилофторид 670
Нитрилофторосиликат 670
Ннтрилохлорид 669
Нит ритоеде динеин я 650
Нитриты 642, 648, 672
Нптроген 633
Нитрогрупна 643
Питр о зил гидрид 657
Нитро зиле ери ая кислота 640, 760
Нитрозилперхлорат 669
Нитрозилроданид 505
Нитрозилсульфат 669
Нитрозилфторид 670
Нитрозилхлорид 505, 669
Нитро зил ьпые соединения 637
Нитрозная кислота 760
Нитрозоамины 650
Нитрозонийхлорид 639
Нитрозе со единения 637
Нитр оксида в ая кислота 642, 650
Нитрон 647
Н итр осоед ин епия 650
Нозеан 546, 562
Иоптронит 545
Ньютона закон охлаждения 610
Обесцвечивание органических Красителей 836
Обжиг 554, 556
пылевой 757
Обменные Силы 163
Обогащение 570
Обугливание 456
Предметный указатель
905
Объем
мольный 249—251
твердых веществ 251
удельный 251
Огнеупоры 556
Озокерит 456
Озон 743—746
строение молекулы 745
Озонатор Сименса, см. Сименса озонатор
Озониды 745
О золовок целые щелочи 750
Ойлдаг 462
Окисление 810—812
перманганатом калия 817
электролитическое 818
Окислители 814—818
нормальные растворы 816
Окислы 34, 486, 746—748
амфотерные 748
гидратация 34
индифферентные 748
кислотные 748
металлов 486
основные 748
теплота образования 34
Окрашенные соединен ня 349
Оксалатоталлаты 427
Оксалаты 488, 494
кислые 494
нейтральные 494
Оксигидраты 96
Оксимы 665
Оксониевые соединения 748
Октав закон 27
О ята мстил цикл отетра сил аза п 52 0
Окуривание серой 755
Олеум 761, 762
Олефины 471
Оливин 269, 542
Олигоклаз 185, 381
Олифа 588
Олово 343, 448— 454, 570—584
аналитические сведеиия 583
валентность 449, 573
горное 570
модификации 571
муаровое 571
нормальный потенциал 574
пассивирование 572
получение 570
применение 572
решетка 571
соединения 573
сплавы 573, 609, 611
строение атома 453
Олово(П) 581 —584
бромид 581
гидроокись 453, 581
иодид 581
питрат 583
окись 581
сульфат 583
сульфид 583
фторид 581
хлорид 581
Олово(1У) 574—580
алкилы 577
гидрид 574
двуокись 577
дисульфид 579
ди мил оксид 577
сульфат 580
тетрабромид 576
т'етраиодид 576
тетрафторид 575
тетрах.торид 575
тетраэтил 577
триэтилгидроксил 577
фтор оста ни аты 575
Оловянная
кислота 453, 572, 577
протрава 576
соль 573, 581
чума 571
Оловянное
зеркало 575
мыло 570
Оловянный
камепь 570, 577
колчедан 570
«крик» 571
огарок 571
Ониевая связь 325
Оникс 534
Опал 528
Онермепт 710
Органические вещества, возраст 455
— химия 15, 455
Ортизон 78
Ортосиликаты 535
Ортостаинаты 579
Осмотическое давление 88, 335
Основания 82—85, 93, 94, 432
амфотер[1ые 82
диссоциация 93
комплексные 432
насыщенные кислотой 874
получение 83—85
Оствальда Закон разведения 89
— правило ступеней 531
Отбеливающие
земли 539
средства 768, 838
Отжиг 616
Палладий 343
Пандермит 359
Парамагнетизм 341, 407
Парамагнитные вещества 338, 339
нормальные 339
Парафины 457, 471
Парациан 501
Пассивирование 385, 643
Паули нрипйен 145
Пектолит 544
Пентан 466
Пербура 378
Пергидроль 76
Перекиси 746—749
Перекись водорода 75—79 817
обнаружение 79
58 г, Реми
906
Предлгетный указатель
получение 76
производные 79
Перенапряжение 53
Переходные элементы 352
Периклаз 291
Перйодаты 869
Периодическая система элементов 18—40
группы 21, 23
периоды .21, 23
ряды 3!)
форма изображения 23—25
Перкарбоиаты 491
Пермутиты 539, 558—560
нолисульфцДные 560
сульфидные 560
Пермутотдпое свойство алюминия 392
Пернитриды 278, 666
Пероксидаза 78
Пероксоазотцстая кислота 650
Пероксоазотпая кислота 648
Цероксобораты 377
Персияь 379
Пер Фтор адипиновая кислота 480
Пер фторамины 480
Перфторглутаровая кислота 480
Перфторимины 481
Пор фтору хсусиая кислота 481
Перхлораты 350, 863, 865
Перхлорил фторид 863
Песчаник 529
Петалит^ 186
Петролвйпый эфир 457
Пироморфит 584
Пиросерная кислота 762
Пиросульфаты 764, 765
Пирофосфаты 689
Пирофосфористая кислота 691
Пирофосфорная кислота 677, 688
Питая реакция 428
Плавиковая кислота 843
Плавиковый [плат 300
Плавка
обжигаю льно-восстановительная 385
осадительная 586
реакционная 586
Плагиоклаз 381
Планка константа 108
Пластова л 656
Пл гамбиты 590, 592
Поваренная соль 213, 214, 874
пищевая 214
Подкреипеван кислота 525
Подшипниковый металл 573
Полевой шпат 185, 298, 381, 511
Полигалит 213
Поли галогениды 851
ПоэитгфМеп 450
Полинга метод 160
Полпсероводороды 793
Полисилен 450, 518
Полисил и каты 535
Полисульфнды 792
Политпогалогенпды 779
Полцтиопаты 772
Политионовые кислоты 772
Полпфосфаты 680, 690
Полйфосфорные кислоты 677, 680, 690
Поллопаз 656
Полоний 808, 809
Полосатые спектры 343
Поляризация 32, 71,160, 244, 347, 349, 350
Поляризуемость 348
Порфир 529 ।
Порядковые числа элементов 20
Поташ 186, 222, 223
получение 223
Потенциал
возбуждения 260, 261
выделения 53
гидратации 181
ионизации 38
нормальный 50, 51, 181, 182, 820, 827
образования 179, 200
окислительный 819
отдельный 53
перезарядки 819
разложения 54, 213
разряжения 49, 91, 252
собственный 49, 50, 54, 91
Потенциал ьп а я кривая 322
Правило Гольдшмидта 242
Правило фаз 72
ПреЕгаративная химия 16
Проба травления 843, 852
Прометий 28
Пропап 466
Просилоксан 518
Противогаз 484
Процессы
лосстаповптельцыо 812
окислительные 811, 823
присоединения 875
самоокисления 823, 824
Псевдобпнарныо системы 758
Лсевдокомпоиепты 758
Работа
ионизации 170, 355, 356
максимальная 164
образования 165, 171
полезная 164
Равновесие при нейтрализации 884
Радий 263, 264, 267; 273, 274, 278, 280,
286, 287
радиоактивность 264
соли 267, 273
Радикал
итлцд 663 *
иодил 663
кислотный 429
метил 473
неорганический 429
Епгтропия 863
органический 429
пропил 474
свободный 473, 474, 823
сульфурил 779
гнонил 779
тиотритиа зил 671
уксусной кислоты 429
фепил 474
Предметный указатель
907
циан 500
атил 474
этилен 475
Радиохимия 16
Радиусы
атомные 36
полные 36, 242, 243
кажущиеся 36, 242
приведенные 243
аффективные 245, 246
Разложение двойное обменное 875, 876
Раскисление 813
Расстекловывание 531, 535
Растворимость 614
Растворитель 499
Растворы
водные солей 878
коллоидные 192, 215
рипГеровскис 186
Рауля закон 88
Рвотный камспь 722, 723
Реакции
восстановительные 818, 820
диспрокорпионировапия 821
о кислится ы.тые 811, 820
топохимические 461, 538
Реальгар 709
Редкоземельные элементы 340
Резерфорда теория строения атома Ю6
Резонанс квантого-мсханический i 56
структур 327
Резонансные сцлн 166
Рекриста л лизаци i е 615
Ренаннт 688
Рений 255
Рентгеновские лучи 230—233, 235, 211, 252
возникновение 252
дифракция 230, 231, 241
интенсивность 252
интерференции 232, 233
максимальная энергия 252
применение 235
проницаемость 253
рассеяние 241
Рентгеновский акуоиз 255
Рентгеновское излучение
собственное 252, 253, 25ё, 257
серии 253
тормозное 252, 253
характеристическое 253—256 Рентгенометрия, см. Структурный авали Ре и гге коспектр огр афия 255, 252—262 Решетки
координационные 172
слоистые 462, 543
типы 172, 438
:нторги я 171
Рибеккцт 544
Ридберги константа 105, 11(1, 134
Ривонфсльда реакция 378
Ритца комбинационный принцип 134
Роговая обманка 511, 544
Родаи 505
Роданиды 506
Роданистоводородная кислота 506
Родановпсмутаты 732
Розовая соль 573, 576
Ропгалит 776, 777
Ртуть
амальгама 209, 210
хлорид 329 цианид 501 Рубидий 177—229 азид 193 амальгама 209, 210 амид 193, 205 бихромат 211 галогениды 181, 238 гексафторосиликат 524 гекса хлороплатинат 211, 228 гексахлорошгюмбат 524 гидрид 193, 199, 200 гидрокарбопат 219 гидроокись 179, 182, 204—206 гидротартрпт 211 изотопы 184, 185 подид 179, 180, 216 карбид 193 карбонат 182, 211, 219, 220, 223, 224 минералы 185—189 литрат 211, 219 ЕЕИТрИД 193 окись 201, 202, перекись 193, 202, 203 перманганат 211 перхлорат 211, 228 пели галогениды 216 получение 189, 191 применение 198 радиоактивность 184 селенид 201, 202 спектр 193—197 сульфрт 211, 224, 227 сульфид 201, 202 сульфит 206 теллурид 201, 202 фосфат 211 фторид 179, 211 хлорат 211 хлорид 211, 213, 216 хромат 211
Рубин 391
Рудничный газ 470
Рутил 298
Сажа 458, 465
Самоокисление 821, 823, 824
Самоокнслители 821, 822
Сапфир 391
Сассолин 359
Светильный газ 473
Светящиеся краски 790
Свинец 448—454, 584—605, 719, 811, 812
аналитические сведения 605
,1[»тимонат 719
валентность 710
гексахлсфостаннат 524
гидроокись 453
двуокись 592, 593
колориметрия 605
окислы 590, 811, 812
58*
908
Предметный указатель
получение 585
поляризующее действие 453
применение 588
рафинирование 586
роговой 596
свойства 586—^89
соединения 589—605
сплавы 588, 609, 611, 612, 614
615
строение атома 453
теплота образования соединений 450
Свинец(П) 590—592, 594—599
ангидрокислота 592
ацетат 597
бинарные соединения 590
бромид 597
галогениды 589, 596
гидроокись 591
иодид 597
карбонат 599
нитрат 598
окись 590—592
оксалат 598
основные соли 589, 596, 598
роданид 597
соли 595
сульфат 600
сульфид 601
тетраоксоплюмбат 594
формиат 598
фторид 597
хлорид 595
хромат 600
цианид 597
Свинец(7У)
азид 603
алкилы 602 , 604
ацетат 601, 603
ацидосоедицения 602
галогениды 601, 602
гидрид 589, 602, 604
окись 592
соединения 601
сульфат 603
— двойной 603
тстраметил 604
тетрафенил 604
тетра фторид 603
тетраэтил 604
хлороплюмбаты 602
СвИещовйя
бумага 794
краска 588
руда 584 , 601
Свинцовое стекло 548
Свинцовые белила 496, 588, 589, 599
Свинирвыи
аккумулятор 594
блеск 584, 601
глет 585, 590
Металл 588
сахар 598
сурик 585, 593
уксус 598
хрусталь 548
Свободная Диффузия 335
Связь
атомиая 443
вапдерваальсовая 322, 324,325, 337
водородная 322, 325
гетерополярная 159, 322—324
гибридная 162
гомеополярная 159, 322, 324, 441
дативная 162, 442
двойная 466, 475
ионная 337
ионогенная 439
межмолекуляриая 324
металлическая 337
ценоногенная 439
одноэлектронная 163
ониевая 325
прочность 163
семиполярная 161
трех электронная 163
тройная 466, 475
Сегнетова соль 228
Селен 452—499, 795—808
алкильные соединения 807
аналитические сведения 807
валентность 800
водородные соединения 806
галогениды 805
двуокись 801
Кристаллический 796, 798
листоватый 795
минералы 795
получение 795
применение 799
соединения 800—808
стекловидный 796, 798
трехокись 802
Селениты 804
Селенистая кислота 801
Селенистый аэот 800
Селениты 801, 803, 806
Селеновая кислота 803
Селеноводород 806
Селеновый мостик 797
фотоэлемент 797
Селеноуглерод 499
Селитра
калийная 218
конверсионная 218
чилийская 186, 217
Сенармонтит 716
Сера 735 —740 , 750—795
аналитические сведения 795
галогениды 778—783
двуокись 767
двухлористая 782
кислородные кислоты 756, 779
коллоидная 755
кристаллическая 75й
модификации 752—754
моноклинная 752
моноокись 778
распространение в природе 750
нитрид 670
окислы 756
окись полуторная 778
пластическая 753
f
Предметный указатель
909
пллитиохлориДы 782
получение 751
применение 755
роданистая 505 ромбическая 752 семиокись 767 соединения 756—795 суспендированная 755 трехокись 756—759 фториды 781 хлориды 781, 782 четырехокись 767
Серебро
арсенит 706
галланат 413
роданид 506
Серная кислота 347, 759—765 диссоциация 763 производство 759 электропроводность 764
Серная печень 789
Сернистая кислота 768, 769
афиры 770
Сернистый аммоний 789
Серпокальциевая печень 790
Серпокальциевый отвар 790
Серное молоко 755
Серпый цвет 751
Сероводород 784, 785
Сероокись углерода 485
Сероуглерод 497—499
Серпентин 285
Сжимаемость 35
Сиккатив 588
Снлазипы 520
Силазины 521
Силаны 451, 516, 517
га логе но замещенные 517, 521 Силикагели 528
Спликат-ион мезомерия 328
Силикатный камень 556
Силикаты 381, 511, 515, 535—547
волокиистые 527
двойные 548
кислые 540, 541
кристаллическая структура 541—547 псроксигидраты 541
Силиколи 520
Силиконовые каучуки 513
Силиконовые смолы 520
Силиконы 513, 519
Силикотермцческий метод 275
Силикохлороформ 526
Снликощавелепая кислота 526
Силит 510
Силициды 512, 515
Силлиманиты 381, 545, 536
Силоксаны 515, 518
Сило к сен 518
Силунд 510
Сильвин 187, 213
Сильвипнт 215
Сименса — Бпллитера камора 208, 209
Сименса озонатор 741
толки 549
Сименса — Песталоцца камера 210
Сингенит 316
Сипглет 283
Синглетные серии 282
Синглет-терм 283
Синерезис 537
Синильная кислота 500—502
Синтол 471, 487
процесс 470
Системы
гомо генные 56
мотастабильные 57 пегомогенные 56 Сколецит 546 Скоиалит 546 Сланцевые квасцы 403 Слюды 185, 381, 511, 529, 545 Смарагд восточный 391 Смешанный газ 487 Собственные колебания 346 функции 120
Сода
кальцинированная 221 каустическая 207 кристаллическая 220 получение 220—222
Содалит 546, 562
Соединения алкилселенониевые 807 ар или одо ди оксидные 872 ароматические 463 ассоциированные 330 бинарные 624 внедрения 434 высшего порядка 431 гетеронолярные 152, 155, 158 гомеополяр ны с 154, 155, 158 далиоцидные 617 ипкоцгруеитные 613 интерметаллические 450, 617, 623 иодония 848 иоппые 160 коммензуралъные 36 комплексные 434 координационные 433 педальтопидные 617 неорганические 433 образование 174, 612, 620, 622 оксопиовыо 748 органические 813 первого порядка 431 неременного состава 623 присоединения 434, 658 Семиполярные 161 солеобразпые 321, 329, 330 стабильность 164 сульфоция 794 химические 173, 617
Соли аптимонила 715 ацетилена 476 водородных кислот 877 гидролитическое расщепление 877, 878 двойные 220, 224, 227, 247, 432 диссоциация 93 кислые 94, 874, 877
910
Предметный указатель
комплексные 431, 432
нейтральные 888
основные 875, 877
очистка 877
полианиоццые 450
приготовление 875
смешанные 227, 247
Сольватация 101
Сольваты 101
Сольве способ 221
Соляная кислота 844, 845
активность водородных ионов 887
Спаривание электронов 323
С иектр а л ь еты е серии
Бальмера 105, 111
Брэкетта 105
Лаймана 105, 111
Пфунда 105
Спектрография микроволновая абсорбционная 349
Спектры 38, 106, 134, 230, 259, 344—348 дуговые 134
Интерференционные 230
искровые 134
колебательно-вращательные 344 комбинационного рассеяния 344—347 линейчатые 106
микроволновые 348
оптические 38, 259
полосатые 106, 344
Спин 156
Спиновое квантовое число 197
— насыщение 157
Сплавы 606—624
затвердевшие 609
структура 607
ферромагнитные 343
эвтектические 609
Сподумен 186, 544
Сродство 164—176
нормальное 168 образование 165 определение величины 166—173 температурная зависимость 165 химических реакций 164, 170 , 357 электронное 171
Сталактиты 312
Станнаты 577, 582
Станниоль 571, 573, 837
Старение 579
Стекло 286, 299, 540, 547—551
боратное 550
боросиликатное 549
викор 549
калиевое 548
кислое 550
нормальное 548
окрашенное 549
основное 550
пирекс 549
получение 549
растворимое 540
свинцовое, см. Свинцовое стекло фотографическое 549 фторидное 550 пготтовское 549
Степень окисления 30
Стибины 25
органические 725
Стиральный порошок 204
Строение
комплексов 438—440
молекул 328
Стронцианит 312
Стронций 263, 266, 267, 269, 275, 278, 280, 288, 291, 294, 295, 297, 300, 304, 307, 312, 318, 319
аммиакат 278
бромид 266', 304
гидрид 266, 267, 288
гидроокись 267, 289, 294, 295
имид 278
иодид 266
карбонат 269, 312
кристаллическая решетка 279
нитрат 307
нитрид 266
окись '266, 288, 290, 291, 294, 295
окрашивание пламени 280
перекись 297
распространение в природе 269
спектр 280
сульфат 269, 318
сульфид 266
фторид 266, 300
хлорид 266, 304
— гексагидрат 304
— дпгидпат 304
хромат 319
Структура
групповая 542
it арка спая 546
ленточная 543
листовая 545
мезоморфная 465
неорганических веществ 334, 336
островная 542, 546
силикатов 541—547
сотовая 561
цепная 543
Структурная Группа 430
формула 29
Структурный анализ 236—252
Сульфансульфоновые кислоты 772
Сульфаны 784
Сульфат-ион, мезомерия 327
Сульфатоталлаты 427
Сульфаты 350, 7 64
Сульфиды 341, 786—790
железа 341
Сульфиновые кислоты 779
Сульфирование 764
Сульфятосоли 769
Сульфиты 769, 794
Сульфокислоты 779
Сульфоксиловая кислота 773, 777
эфиры 778
Сульфонил соли 794
Суперфосфат 676, 688
Сурьма 713—726
аналитические сведения 726 валентность 715
Предметный ркааателъ
911
галогениды 719—722 гидроокись 715 минералы 713 модификации 714 нитрат 722 окислы 715—717 оксихлорид 720 получение 713 применение 715 пятиокись 717 соединении 715—726 силаны 715 сульфат 722 сульфиды 723, 724 тиосоли 723 чстыр ех окись 719
Сурьмяная кислота 716, 718 Сурьмяная киноварь 724 руда 713, 724
Сурьмянистая кислота 715 Сурьмяные соли 722 Сурьмяный цвет 713, 716 Сусальное золото 573, 586 Сухой лсд 483 Сайделла метод 582
Таллий 351—359, 406, 408, 419—428, 015
аЛапат 419
алкилы 358, 427
алкоголях 420
арилы 358
валентность 351, 354, 356, 406, 420
гаялогидрид 413
галогепосаллат 425
гексахлоростанпат 324
гидрид 353, 422
гидроокись 352
определение 428
селенида 422
соединения 420—428
спектры 350
сплавы 615
структура решетки 408
сульфиды 422
теплота образования соединений 358
трифешгл 428
халькогенид 406, 407
хлористоводородная кислота 425
Таллий(Г) 420, 422—424
ацетат 424
галогенида 422
гидроокись 420, 423
карбонат 424
нитрат 424
окись 423
оксалат 424
перхлорат 424
сульфат 423
сульфид 423
фосфат 424
хлорат 424
цианид 423
ТаЯлий(Ш) 420, 421, 424—427
ацетат 427
бромид 425
бромталлаты 425
гидроокись 426
иодид 426
комплексные соли 421
нитрат 421, 427
оксалат 427
сульфат 421, 427
фторид 426
хлорид 424, 428
хлороталлаты 424, 425
Тальк 269, 286, 545
Твердые растворы 279, 613—015
Теллур 795—808
алкильные соединения 807
аморфный 799
амфотерность 802
аналитические сведения 807
валентность 800
водородные соединения 806
галогениды 805
двуокись 802
минералы 795
получение 796
пр имя пение 800
соединения 800— 808
Теллуриты 804
Теллуриды 422, 807
Теллуристая кислота 802
Теллуристый азот 800
Теллуритовые стекла 802
Теллуриты 802
Теллуровая кислота 804
Теллуроводороды 807
Температура
воспламенения 743
кипения 331
плавления 213
Тепарова синь 405
Теория реиопапса 327
Теплота
гидратации 357
кристаллизации 610
образования 250, 618
плавления 610
поглощения 225
растворения 21 □
сублимации 359, 452
Теплотворная способность угля 457
Термит 385
Термические методы 275
Термы 106, 137, 259, 283
Тетраборап 364, 365
Тетрабор пая кислота 375
ТетраметилсвииеП 604
Тетраеилоксан 518
Тетраэдров группировки 543
Технеции 28
Тинкал 359
Тпокарбаминовая кислота 500
Тиокпслоты 791
Тнонил иодид 779
Тиогерная кислота 771, 772
Тпосернйстая кислота 781
Тиосиликаты 527
Тносоля 791
Тиостапнаты 577, 579
912
Предметный указатель
Тиосульфаты 770
Тиоугольная кислота 499
Тиоциан 505
Типографский металл 715
Типы связей 322, 329, 336, 337
Титанит 542
Титрование 884, 886—889
Томасовая мука 676
Топаз 381, 391, 543
Топливо моторное 487
Топохимические реакции 461
Тортвейтит 543
Торф 456
Точка эквивалентности 884
Трансураны 22, 23, 28, 40, 41
Триад Правило 26
Тригалогеисиланы 521
Тридиметил амиц о бор сульф о л 369
'Тридимит 527, 528, 530
Триизо пр опоксицик лот етр а м ети лев ди-кетосилазии 521
Триметил бор 372
Триметилиндии 418
Триметидфосфаты 681
Тримет оксибор сульфол 369
Триплетные серии 282
Тритий 65
Трифенилбор 362, 373
Трифенилталлий 428
Триф ен ок си ци к л оге кс и леи лазип 521
Трифилин 186
Трихлороборсульфол 369
Трихлорсилан 526
Три ати л бор 372
Триэтил галлий 412
Тройная точка 72
Тропа 220
Трутона правило 359
Турмалин 360
Тяжелый шпат 318
Углеводороды
высшие 468
я;идкие 468
насыщенные 467, 474
термическое расщепление 477
не па см щепные 467
определение 511
разветвленные 471
тяжелые 473
циклические 478
Углеводы 455
Углерод 15, 157, 454—511
аморфный 458, 461, 463
аналитические сведения 510, 511
блестящий 458, 465
валентность 449
галогениды 479—481
— смешанные 480
гидроокись 453
двуокись 452, 455, 470, 481—483,. 510
— круговорот в природе 455
двухвалентный 466
кислородные соединения 481—497
мопосульфид 497
недокись 481
одноатомный 459
окись 471, 481, 483—487
— взаимодействие с водородом 471
— мезомерия 484
— молекулярная решетка 483
— окисление 484
— продукты присоединения 486
оксисумьфид 497
оксифторид 491
оксихлорид 490
применение 458 радиоактивный 455 распространение в природе 454 сероокись 485, 499 спектры 484
строение атома 453, 454
сульфиды 497
теплота образования соединений 450
тио недокись 497, 498
трехвалептный 466
черный 458, 463 четырехбромистый 480 четырехиодистый 480 четырехфтористый 479 четыреххлористый 451, 479, 480
Уголь активированный 465 антрацит 456 бурый 456 древесный 456, 464 животный 465 каменный 456 костяной 456, 465 кровяной 456, 465 сжижение 468, 469 теплотворная способность 457
Угольная кислота 488—490
Удобрения 218
Уксусная кислота 495—497, 511
ледяная 495
определение 511
степень диссоциации 495
Уксусная эссенция 495
Ультрабазит 568
Ультрамарин 546, 560—562
замещенный 562
строение 561
Ультрацентрифуга 335
Упругое последействие 608
Уравнение
волновое 115
газового состояния 60
ионное 818
колебания 115
Шредингера, см. Шредингера уравнение
Уровня энергии 257
Факел Лангмюра 65
Фарадей 50, 816
Фарадея закон 199
Фарфор 554, 555
Фарфоровая земля 551
Фаянс 555, 556
фенакит 542
фепицит 600
Предметный указатель
913
Фенолфталеин 884, 885
Ферробор 361
Ферромагнетизм 341—343
Ферромагнитные вещества 338, 339
Ферросилиций 513
Физическая химия 17
Фиксирующая соль 772
Фильеры 499
Флейс-процесс 757
Флинтглас 548
Флюсы 383
Формиаты 485, 495
Формовочная земля 552
Форстерит 542
Фосген 485, 490
Фосфам 698
Фосфатная защита 676
Фосфаты 677, 680, 685—688
высокополимерные 681
Фосфиды 699
Фосфин (198
Фосфиты 677, 691
Фосфопитр и л галогениды 696, 697
Фосфор 672—700
аналитические сведения 699
белый 674
галогениды 692—696
красный 674
модификации 674
надкислоты 682
нитриды 696
оксихлорид 695
получение 673
применение 676
пятпокись 683
свойства 674
соединения 676—700
сульфиды 695
трехокись 683
хлориды 693
черный 675
четырехокись 684 '
Фосфористая кислота 677, 678, 691
Фосфористый водород 699
Фосфорит 269, 672, 688
Фосфорная кислота 677, 678, 684, /00 соль 686 — перлы 689
Фосфорноватая кислота 341, 677, 678, 691
метиловый эфир 679
Фосфор нов атиста я кислота 677, 678, 69'1
Фосфорные кислоты 676—684. 688—692, 697
амиды 697
конденсированные 677
строение (>78
Фосфоры 790
Фотохимия 16
Фотоэлектрический эффект 796
Фотоэлементы 79 7
Фракционированная перегонка нефти 457
Франка — Каро — Линде процесс 46
Франций 177, 185
Фреон 480
Фри ген 480
Фриделя — Крафтса реакция£398
Фтор 825—855
валентность 827, 828
кислородные соединения 855
получение 832
применение 838
распространение в природе 831
сродство к электрону 834
физические свойства 834
химические свойства 835
Фтор о алюминаты 387
Фторо бериллаты 299
Фторобораты 362, 370
Фторидный шлак 300
Фториды 839, 848, 853
Фтористоводородная кислота 839
Фтористый водород 842—844
— хлорсульфурил 783
Фторбборная кислота 431, 432, 434, 436,
439
Фторосиликаты 523, 524, 542
Фторостаннаты 575
Фтор сульфонов а я кислота 783
Фумаролы 374
Функция ф 122
Фурье-анализ 337, 338
Хаглунда метод 383
Халцедон 533
Халькогениды 350, 739
Халькогены 738
Халькопирит 404
Химическая технология 16
Хинолингептахлориндат 415
Хиолит 397
Хлор 208, 242, 825—864, 870—873
азид 652
атомарный 837
двуокись 861
кислородные соединения 855—865
о кислы 856
перекись 854
получение 833
применение 838
распространение в природе 831
семиокись 862
физические свойства 834
химические свойства 836
шести окись 862
Хлорамин 663
Хлорантимонаты 721
Хлоратоталлаты 427
Хлораты 859, 860, 865
Хлорв нему тэты 730
Хлориды 34, 853
Хлорилоксифторид 863
Хлористый водород 844, 845
гидраты 844
растворимость 845
сродство к воде 845
Хлористая кислота 859
Хлористая сера 781
Хлористый сульфурил 783
Хлористый тпенпл 782
Хлориты 859
Хлорная известь 838, 858
914
11 редметный указатель
Хлорная кислота 347, 863
Хлорноватая кислота 859
Хлорноватистая кислота 857
Хлорный электрод 827
Хлорокиси 856
Хлоропрей 477
Хлоростаппаты 576, 581
Хлорофилл 455
Хлорсульфоновая кислота 783
X л орс уль фурил фтористый 783
Хризопраз 529
Хризотил 544
Царская водка 644
Цезий 177—229
азид 193
амальгама 209, 210
амид 193, 203
бихромат 211
бромид 239
галогениды 181, 238
гексафгорогерманат 524
гексафторосиликат 524
гексахлороплатинат 211, 228
гексахлоростаннат 524
гидрид 193, 199, 200
гидрокарбонат 219
гидротартрат 211
изотопы 185
иодид 239
карбид 193
карбонат 182, 211, 219, 220, 223, 224
минералы 186—189
нитрат 211, 219
нитрид 193
окись 201, 202
перекиси 193, 202, 203
перманганат 211
перхлорат 211, 228
полигалогеннды 216
получение 189, 191
применение 198
селенид 201, 202
спектр 193—197 ,
сульфат 206, 211, 224, 227
сульфид 201, 202
теллурид 201, 202
фосфат 211
фторид 211
хлорат 211
хлорогерманат 566
хлорид 211, 213, 216, 239
хромат 211
Целестин 318
Целио 427
Целлофильтр 335
Целлюлоза 455
Цемент
гидравлический 557
магнезиальный 292, 301
портланд 557
пуццолановый 558 ,,
роман 558
г (лаковый 558
Центральный атом комплекса 432
Центры кристаллизации 607
Цеолит 553, 559, 560
Церуссит 584
Цехштей нов скин известняк 213:
Циан 500
Цианиды 500, 502
Цианистый водород 501
Цианит 381, 542
Циклические кетоны 478
Цикл о октатетра еп 478
Цикл о сила залы 520, 521
Цинк
ацетилид 476
сульфат 263:
фторосиликат 524
Цинковый пшат 454
Циркон 542
Цирконий, применение 191
Цитрин 529
Цоизит 381
Цуниит 543
Шамот 556
Швейнфуртская зелень 704
ШвеЛевапие 464
Шелк искусственный 499
Шпинель 269, 395
Шредингера уравнение 120, 125
Шрифтовый металл 588
Штольцит 584
ПГтрассы 549
Щавелевая кислота 491—494, 511
— безводная 493
— дигидрат 492
— определение 511
Щелевые аппараты 477
Щелоча о земельные металлы 263
ацетаты 496 ;
валентность 265, 268
гидриды з— галогениды 306
гидроокиси 267
гомеополярные соединения 268, 269 минералы 270—272 мопогалогёпиды 305
нитриды 267
нормальные потенциалы 264, 265
окислы 266, 267
оксалаты 494
перекиси 266
перхлораты 864
получение 274, 275
применение 284—286
радиусы атомные 264
— ионные 264
силициды 515
соединения 286—320 спектры 268 , 280—284 сульфиды 790 фторобораты 370 хлориды 266
цианиды 502
Щелочные земли 263
Щелочные Металлы 177-^198 алкины 183
Предметный указатель
915
ацетаты 496
ацетилиды 476, 507
гидроксоплюмбаты 601
гидроокиси 187
гоиеополярные соединения 183
карбонаты 222, 223
координационное число 241
минералы 187—189
пеДокцси 202
нитраты 217, 218
нормальные потенциалы 177
пероксиглдраты силикатов 541
перхлораты 864
полигалогепиды 851
политионаты 773
получение 189
свойства 191—193, 342
; силикаты 540,, 541
соединения 193, 198—229
спектры 193—197
сульфаты 225—227, 765
теплота образования соединении 179,
181
тпостанпа.тш 580
тиосульфаты 770
Уровни Энергии 257
фторо антимонаты 720
фторобораты 370
электролитическая подвижность 181
Щелок
калиевый 208
натровый 207
спиртовый 207, 208
Эвтектика 609
Эвтектическая температура 609
точка 609
ЭкаалГОминий 27, 408, 409
Экабор 27
Экаиод 28, 830
Экасилиций 27, 563
Экацезии 28
Эквивалент 816
Экранирования константа 256
Экспонент отталкивания 172
Электровалентность 161
Электродиализ 68
Электролиз 208, 811
грехслойяый 384
Электролитическая диссоциация 87, 93
Электролиты 86—9,3, 329
активность 93
Электронные
лучи 236
облака 124
плотности 337
Электроны 121, 157—15(.i
валентные 158, 261
внешние 158
внутренние 159
неспаренвые 157
разрыхляющие 158
связывающие 158
спароппые 157
спаривание 157
Электропроводность 86 металлическая 332 молярная 86, 435 эквивалентная 86 электролитическая 86
Электросродство 32
Электростатическое взаимодействие 156
Электрохимический ряд напряжений 52— 54
Электрохимия 16
Элементарный анализ 510
Элементы 15
аналогичные 21
главной подгруппы 23
кислотообразующие 449
общее число электронов 150
оптические спектры 255
переходные 23, 41
периодичность свойств 28—40 побочной подгруппы 26 порядковый номер 20, 260, 261 свойства 19, 150 электроотрицательность 32, 161 электроположительность 32, 33
Элокса.тгьный способ 385
Эмаль 551
Эманация 146
Эмиссионный спектр 281
Эмнротиды 79
Эмульсии 501
Энантиотропные превращения 529
Энаргит 700
Энергия
диссоциации 170
образования 441
отрицательная 165
положительная 165
потенциальная 107
решетки 171
уровни 257
Энстатит 544
Энтропия реакции 466
Эпидот 381
Этан 466, 473, 474
галоидные производные 476
Этеп 468
Этилен 474j 475
бромистый 474
получение 474
Этилендиамид 440
Этиловый спирт 470
Этилсериал кислота 474
Этин 468
Эттрингит 401
Эффект Джоуля — Толсона, см. Джоуля-Томсона эффект
Ядерная химия 16 Ядро атома 106 заряд 19, 256, 261 притяжепие 156 отталкивание 156
Янтарь 456 Яшма 534
ОГЛАВЛЕНИЕ
Предисловие ............................................................ 5
Предисловие к десятому и одиннадцатому изданиям.......................... 7
Из предисловия к первому изданию...................... 9
Важнейшие общие константы ......................,.................... И
Введение ................................................................ 15
Глава 1. Периодическая система химических элементов...................... 19
Периодичность химических свойств алементов . . , .............. 28
Периодичность физических свойств.................'............ 34
Изменение свойств в горизонтальных рядах периодической системы 39
Периодичность атомного строения ............................... 40
Глава 2. Водород .................................................. 42
Важнейшие методы получения водорода............................ 45
Электролиз воды; электрохимический рид напряжений.............. 49
Термическое разложение воды; закон действия масс............... 55
Свойства водорода ... ......................................... 60
Соединения водорода ...................................... . 66
Вода .................................................... . 67
Кислоты, основания и сопи...................................... 79
Обзор способов получения оснований ........................... 85
Гидраты . ....................................... 96
Глава 3. Спектр недорода и строение атома водорода..................... 104
Строение атома водорода ............................. 106
Строение атома водорода с точки зрения волновой механики .... 115
Глава 4. Нулевая группа периодической системы (Главная подгруппа восьмой группы).............................................................. 126
Инертные газы ............................................... 126
Спектры инертных газов....................................... 132
Ионизация и излучение .............................. . 136
Глава 5. Валентность н сродство . .................................... 148
Химическое сродство
164
Оглавление
917
Глава 6. Первая группа периодической системы (Главная подгруппа) . . . 177
Щелочные металлы ............................................ 177
Соединения щелочных металлов................................. 198
Соли щелочных металлов . . . ........................... 210
Хлориды ..................................................... 212
Нитраты ..................................................... 217
Карбонаты .................................................. 219
Сульфаты.................................................... 224
Глада 7. Строение кристаллов и рентгеновские лучи . .................. 230
Структурный анализ (рентгенометрия)...................... . 236
Рентгеноснектрография ....................................... 252
Глава 8. Вторая группа периодической системы (Главная подгруппа) . . . 263
Щелочноземельные металлы................................... 263
Соединения элементов главной подгруппы второй группы .... 287
Гидриды ..................................................... 287
Окислы и гидроокиси.......................................... 288
Перекиси и падперекиси ...................................... 296
Галогениды............................,.................. 298
Фториды ................................................. 2^3
Хлориды, бромиды и иодиды................................ 300
Нитраты ................................................ 306
Карбопаты ................ , ......... 308
Сульфаты................................................. 314
Глава 9. Строение и свойства химических соединений ................... 321
Определение строения неорганических веществ.................. 334
Цвет неоргавических соединений............................... 349
Глава 10. Третья группа периодической системы (Главная подгруппа) . . 351
Группа бор — алюминий...................................... 351
Вор.......................................................... 359
Соединения бора............................................. 361
Вороводороды (борогидриды, борашя)........................... 362
Вор о гидриды металлов (боранам)......................... 366
Воразол, бороксол и борсульфод............................... 368
Галогениды бора.............................................. 370
Кислородные соединения бора.............................. 373
Другие соединения бора................................... 379
Алюминий . .................................................. 381
Соединения алюминия.......................................... 386
Кислородные соединения алюминия...................... . 391
Галогениды алюминия ................................... 396
Сульфат алюминия; квасцы ................................ 401
Другие соединения алюминия .............................. 403
918
Оглавление
Галлий, индий, таллии....................................;........ 406
Галлий ..........................., .................... 408
1. Соединения трехвалентного таллия ..................... 410
2. Соединения одновалентного и двухвалентного галлия .... 412
3. Алкильные соединения галлия и гидрид галлия....... , 412
Индий .................................................. 413
Соединения индия......................................... 414
1. Соединения трехвалентного индия ..................... 415
2. Соединения одно- и двухвалентного индия .......... 417
3, Алкильные соединения индия и гидрид индия............. 418
Таллий............................................. . 419
Соединения таллня..................................... " 420
1. Соединения одновалентного таллня.................. 422
2. Соединения трехвалентного таллия ................ 424
3. Алкильные соединения таллия....................... 427
Глава 11. Координационная теория............................... 429
Глава 12. Четвертая группа периодической системы (Главная подгруппа) 448
Подгруппа углерод — кремний ............................. 448
Углерод...................................................... 454
Соединения углерода .................................... 466
Углеводороды.......................................... 467
Галогециды ......................................... 479
Кислородные соединения.............................. 481
Сульфиды ................................... ........ 497
Соединения циана..................................... 500
Соединения тиоциаца (родаца) , ... .................. 505
Карбиды .............. ......... ..................... 507
Кремний ........................................................ 5Г1
Соединения кремния...................................... 515
Галогениды нремпия ............................... • 521
Кислородные соединения нремпия ...................... 528
Кристаллическая структура силикатов...................... 541
Силикатные продукты особо сажного технического значения . . . 547
Стекло ........................................... . . 547
Глина, каолин и керамические цзделля.................... 551
Цемент............................................. 556
Пермутиты и цеолиты ................................... 558
Ультрамарин . ; . . . ............................... 560
Германий.........,............................................ 562
Соединения германия .................................. 564
Соединения чотырехвалентнего германия................. 564
Галогениды германия(1У)............................. 565
Соединения гормания(П) ................ 568
Олово ................................................ .......... 570
Соединения олова . ..................................... 573
Соединения олова (IV, , ................ . . ........ 574
Соединения олова(П) . ............................... 581
Оглаелеиие 919
Свинец ......... .............................................. . 5S4
Соединения свипЦй . ............................. . . -589
Окисли ..... . 590
Соли свипца(И)................................... ... 595
Соединения свнниа(1\г) ............................. 601
Глава 1,3* Сплавы ............. - ............................606
Кривые охлаждения и диаграммы состояния.................. 610
Влияние механической обработки на свойства металлов и сплавов 616
Интерметаллические соединения и твердые растворы ........ 617
Отношение металлов первых четырех групп Друг к другу . . . 619
Глава 14. Пятая группа периодической системы (Главная подгруппа) . . 625
Группа азот — фосфор........................*........... 625
Азот ........................................................ 633
Соединения азота ...................................... . 637
(1. Окислы............................................ 637
2. Кислородные кислоты азота......................... 642
3. Соединения азота с водородом и их производные.... 651
Аммиак . ................................................ 652
Соли аммония . . . ,.................................. 659
Производные аммиака . ............• *.................. 662
4. Галоидные соединения азота ....................... 667
5. Сернистые соединения азота........................ 670
Фосфор ........................ ....................... . - - - 672
Соединения фосфора...................................... 676
1. Кислородные соединения ............................ 676
Строение фосфорных кислот................................ 678
2. Галогениды фосфора ...............,............... 692
3. Сульфиды фосфора ................................. 695
4. Соединения фосфора с азотом....................... 696
5. Соединения фосфора еводородом ............. 698
6. Фосфиды........................................ . 699
Мышьяк.......................................................... 700
Соединения мышьяка ...................................... 702
1. Окислы и гидроокиси.............. , ............ 702
2. Галогениды мышьяка .................. ........... 708
3. С ул вфиды и сульфосоли мышья ка................. 709
4, Мышьяковистый водород и арсенидь!................ 710
Сурьма ....................................................... 713
Соединения с урьмы.................................... 715
1. Окислы и гидроокиси............................... 715
2. Галогеппды сурьмы.................................. 719
3. Сурьмяные гили ................................. 722
4. Сульфиды и тиосоли сурьмы.......................... 723
5. Сурьмянистый водород ............. . . . ......... 725
6. Соединения сурьмы с металлами (антимониды)»...... 725
Висмут.................................. . ................ 726
Соединения висмута...................................... 728
1. Окислы и гидроокиси.............................. 728
2. Соли висмута...................................... 729
3. Висмутистый водород............................... 733
Глава 15. Шестая группа периодической системы (Главная подгруппа) . . 735
Группа кислород — сера 735
920
Оглавление
Кислород.............................................................. 740
Озоя ........................................................ 743
Соединения кислорода......................................... 746
Сера ..............................,.................................. 750
Соединения серы.............................................. 758
1. Окислы и кислородные кислоты........................ 756
2. Соединения серы с галогенами (галогениды серы) ...... 778
3. Сероводороды (сульфаны) .............................. 784
Селен и теллур , ... ............................................... 795
Соединения селена я теллура................................ 80G
1. Окислы и кислородные кислоты ......................... 800
2. Соединения с галогенами.......................... 805
3. Водородные соединения...........,.................... 80(4
Полоний ............................................................ 888
« Г л а в'а 16. Окисление н восстановление............................. 810
Глава 17. Седьмая группа периодической системы (Главная подгруппа) 825
Галогены..................,................................. 825
Свободные галогены.......................................... .831
Соединения галогенов ................................... . 839
1. Гало генов одор оды................................... 839
2. Галогениды.......................................... 847
3. Окислы и кислородные кислоты галогенов................ 854
Кислородные соединения фтора.....................,........... 855
Кислородные соединения хлора................................. 855
Окись хлора, хлорноватистая кислота и гипохлориты........ 856
Хлористая кислота и хлориты.............................. 859
Хлорноватая кислота и хлораты ........................... 859
Двуокись хлора и шестиокись хлора........................ 861
Семиокись хлора (ангидрид хлор поп кислоты), хлорная кислота и перхлораты........................................... <862
Кислородные соединения брома................................. 865
Кислородные соединения иода . . ............................
4. Соединения галогенов между собой................. . , 870
5. Солеобразные соединения с электроположительным галогеном 872
Глава 18. Образование содей и нейтрализация........................ 874
Приложения........................................................... 890
Предметный указатель................................................. 89J
Г. Реми
КУРС НЕОРГАНИЧЕСКОЙ ХИМИИ
& Редактор Г, М. Мануйлова
Художник II. A. JlumsutUKP Технический редактор АГ. П. Грибова
Сдано в производство 9/1 Г 9(53 г. Подии сане к печати 12/VI Г 963 г. Бумага JCX1 ОЭЫГ<; = =ЙЗ.В бум, л 79,1 иеч. и., в т/ч. Г вкл. Уч.-ивд. л, S4.6. Изд. Й/5Й56. Иена 6 Р- 12 к.
Зак. 605.
*
Издательство иностранной литературы. Москва, 1-й Рижский пер., д. 2
*
Московская типография М 5 Мое гор совнархоз а, Москва. Трехпрудный пер., 9
4' СПИСОК ЗАМЕЧЕННЫХ ОПЕЧАТОК I i , 1 Fj i" i
Страница Строка Напечатано Следует читать
6 34 14 снизу 15 сверху теории резонанса внутри одной подгруппы теории pesoнанса внутри одной группы 1 1 ;
34 S5 S5 20 сверху 14 сверху 18 снизу (пи сравнению с А1 и даже Si) 96 493 кулою ангидрид основания (по сравнению с Д1 или Si) 96 493 кулон ангиврооонования 1 i
417 13 сверху t — '4iX = '4iy 14 4v 1!
128 16 снизу и ой л гос ноилто^ i
163 16 снизу Дг:6; и :N:O: :Х;б: п ;Х:О: ' i
177 Я сверху Главная подгруппа труппы Главная подгруппа первой I
328 328 451 675 22 сверху 24 сверху 11 снизу 2 снизу .ДА 11ЛИ .-F.-Biii: "«‘I •ifiiirF: н С-_Г4Н = Н:С-И; Н ;С1: •Cr+4:Ck=:C:CGi: \са «Л comprehensive treatise Группы :F; ИЛЙ :F:B;F: :F; или ... :F:B:F: Н ЧУ4-4Н==Н:С:Н; Й ;С1; •С- 4-4;С1: = :С1:С:Й1:. -.СП «А с от proh e»s ivc; treatise 1 i
Зек. 605