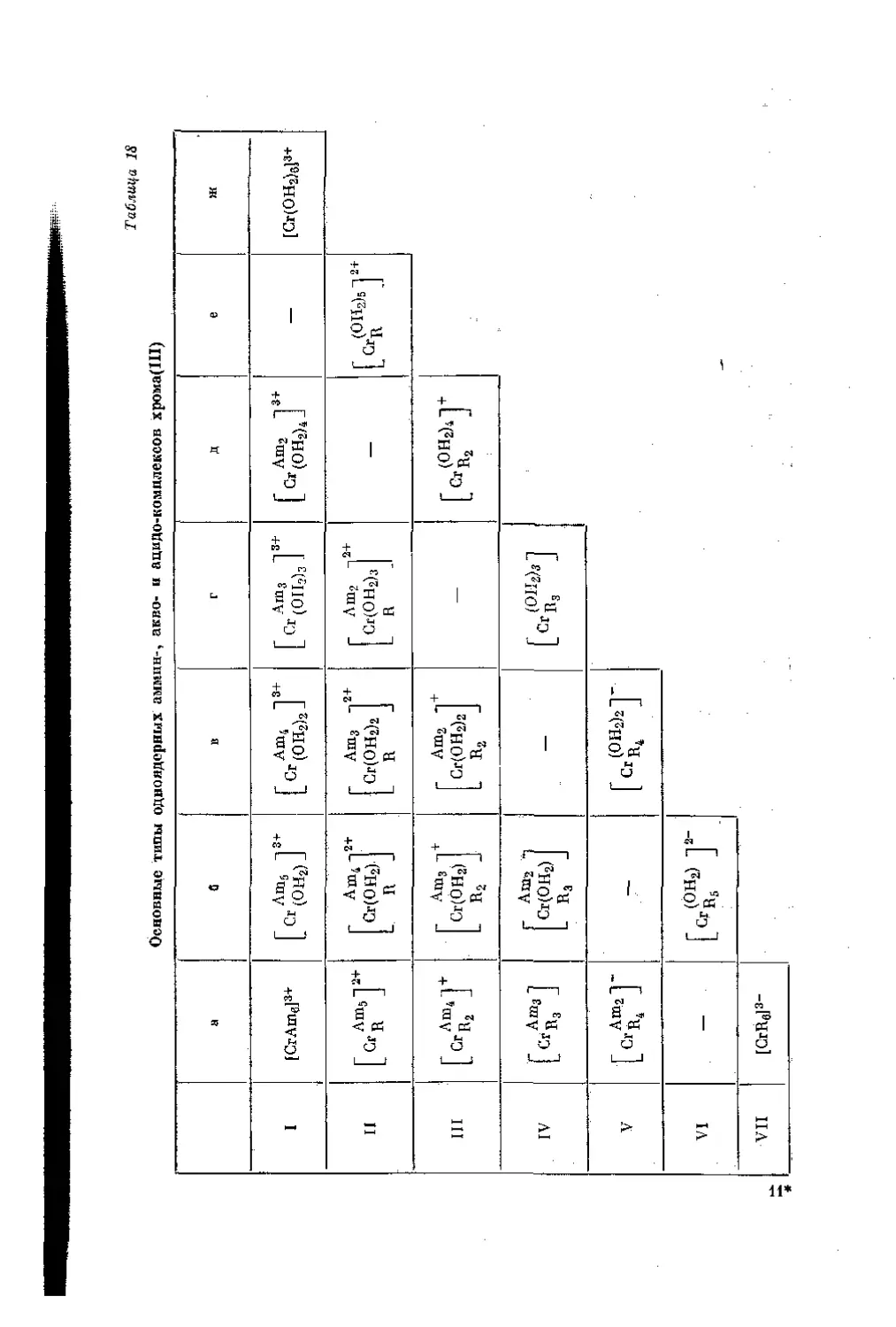
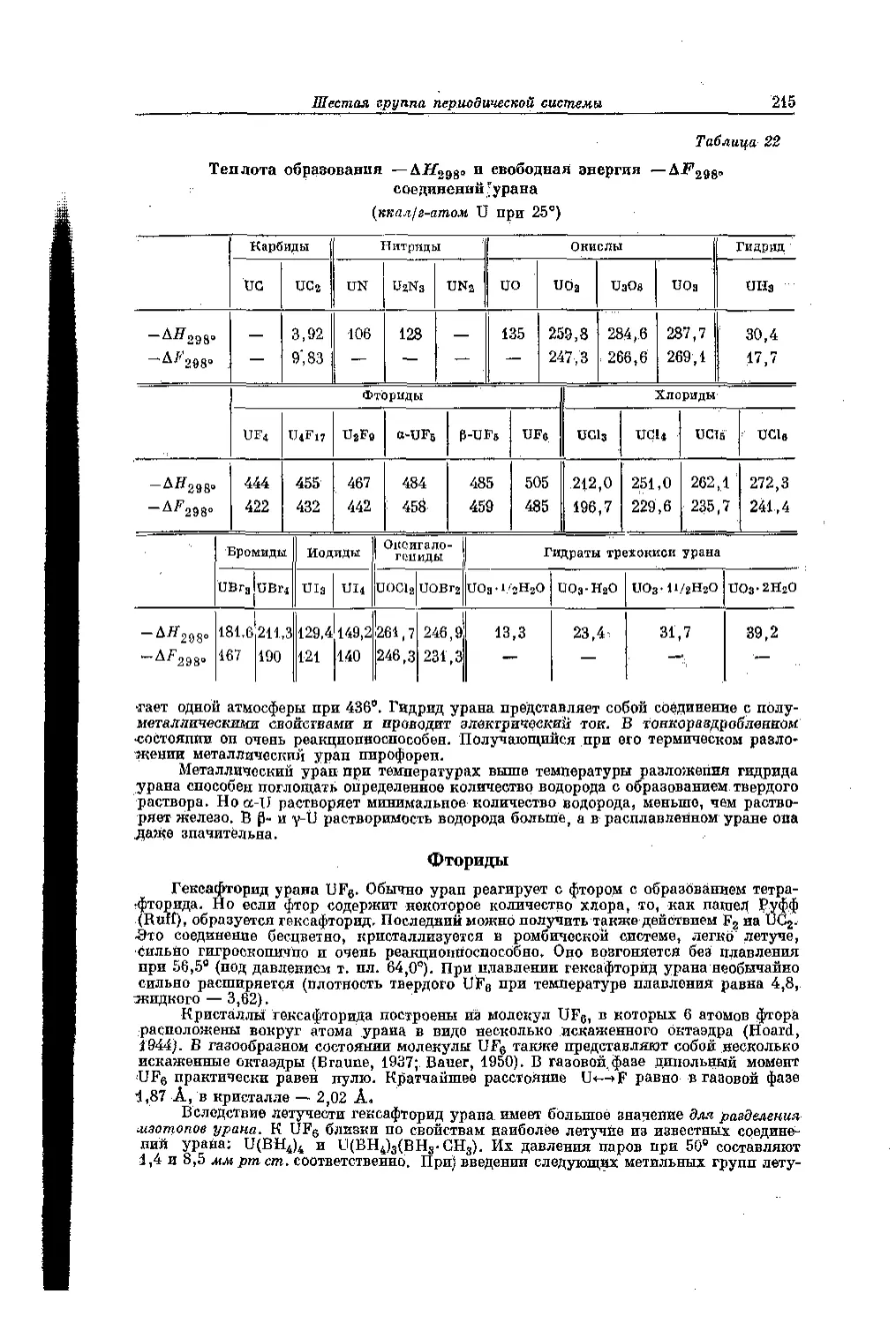
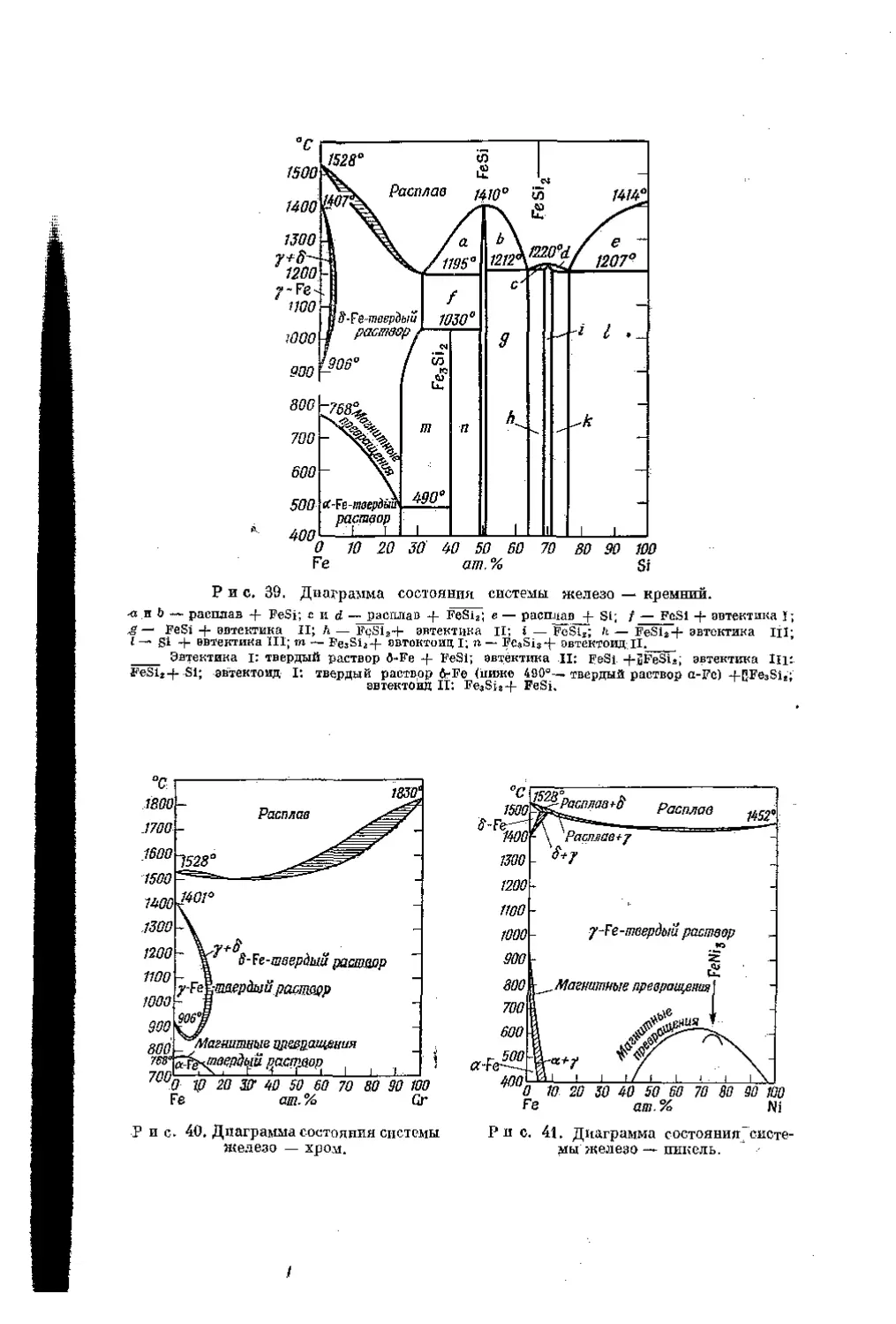
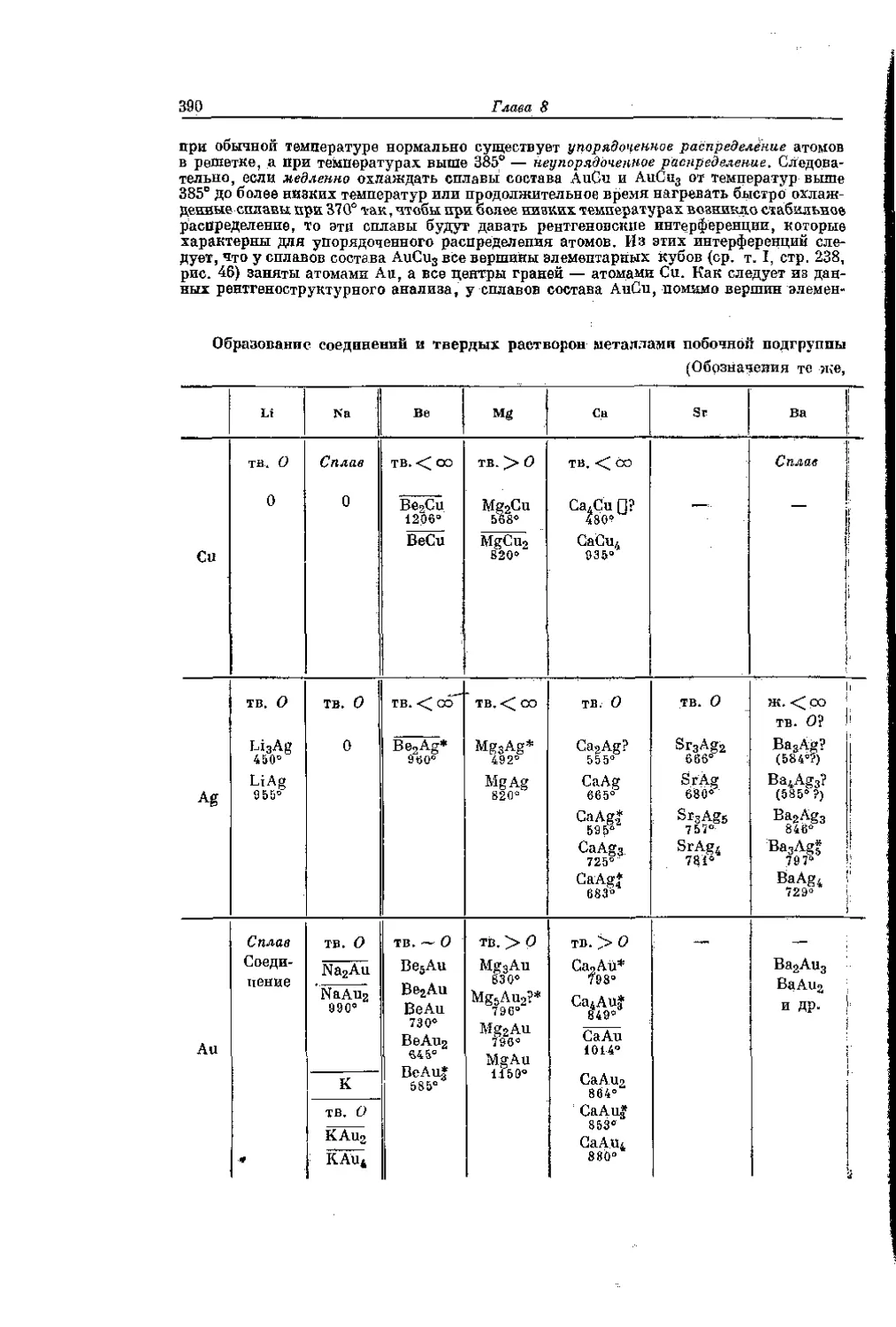
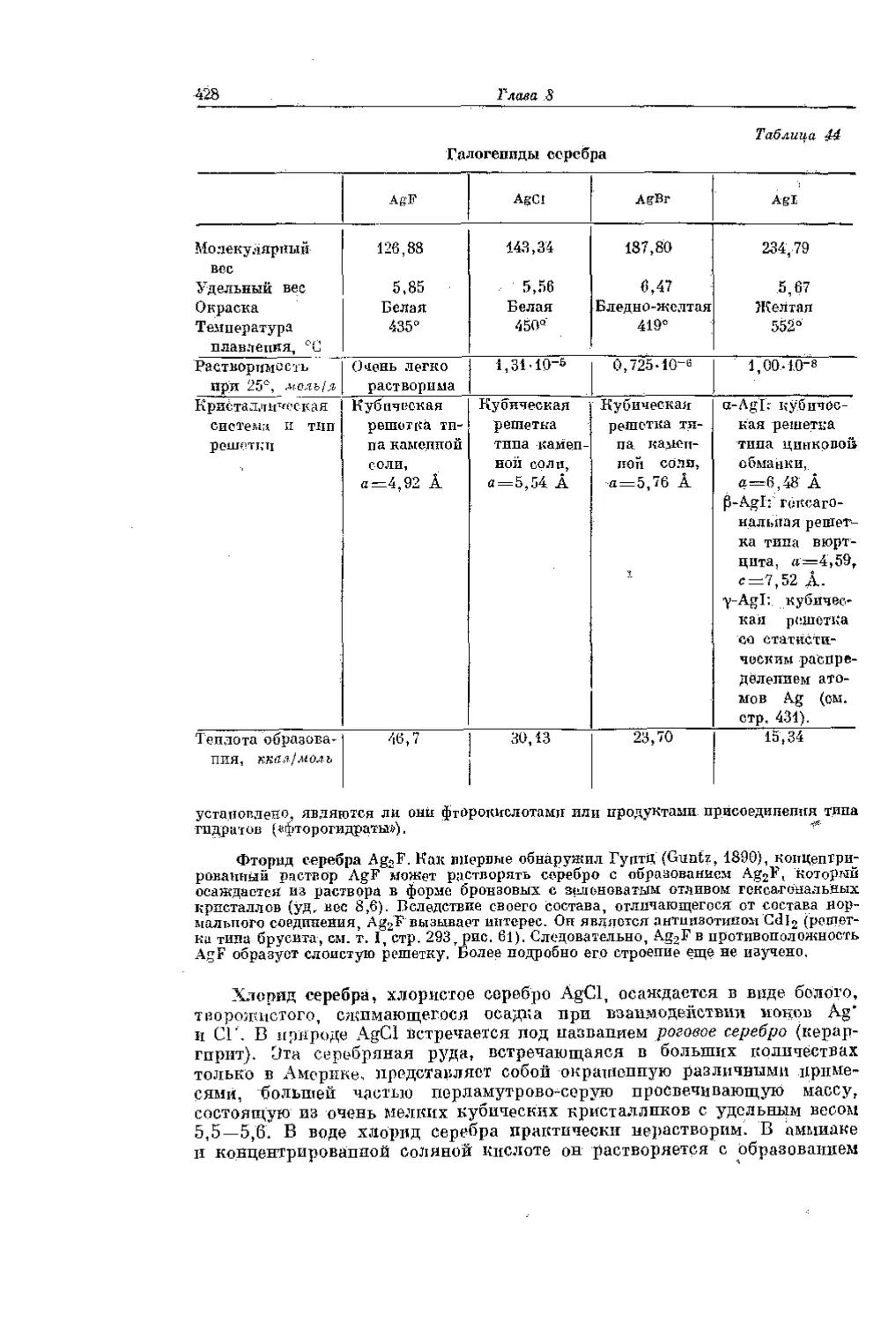
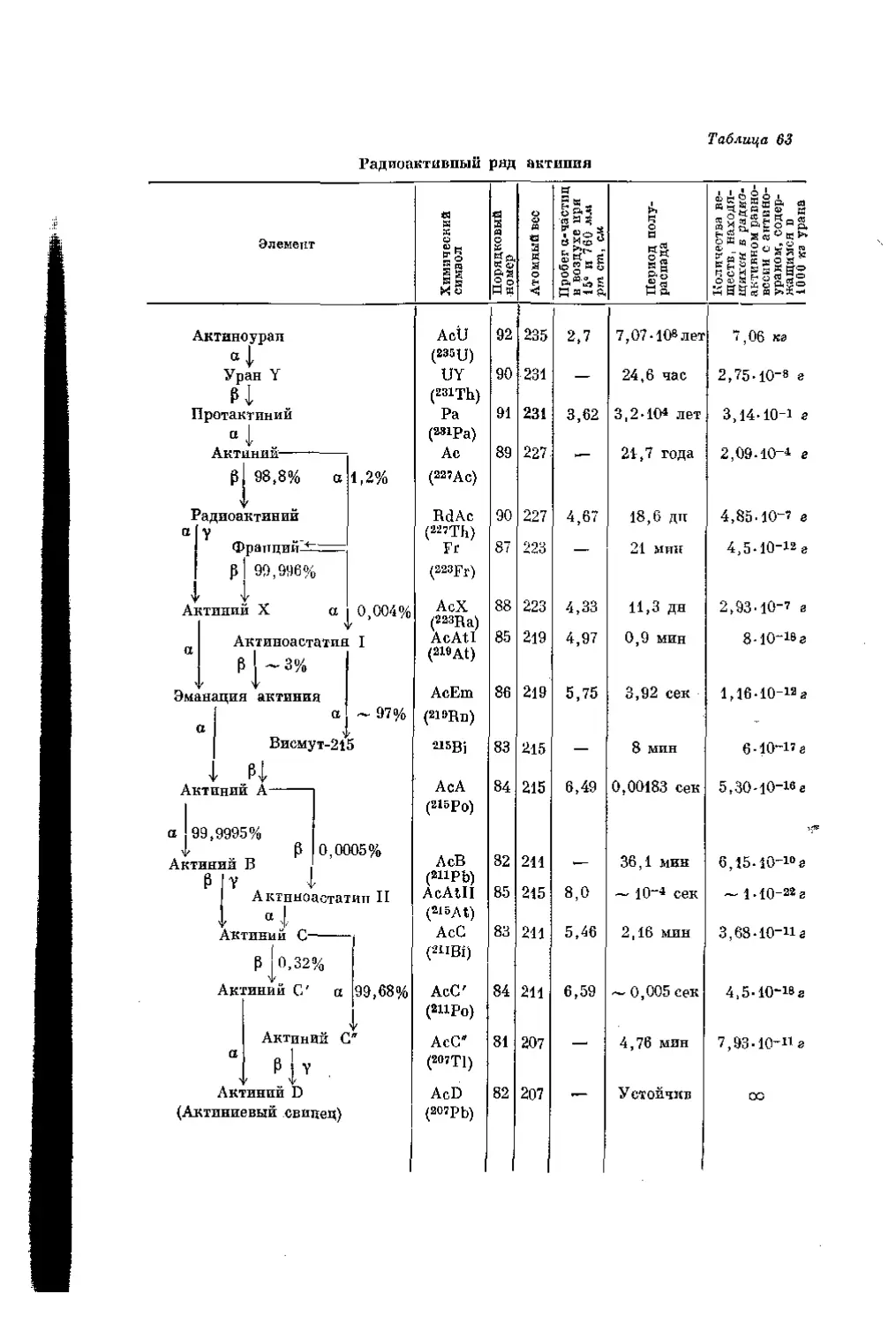
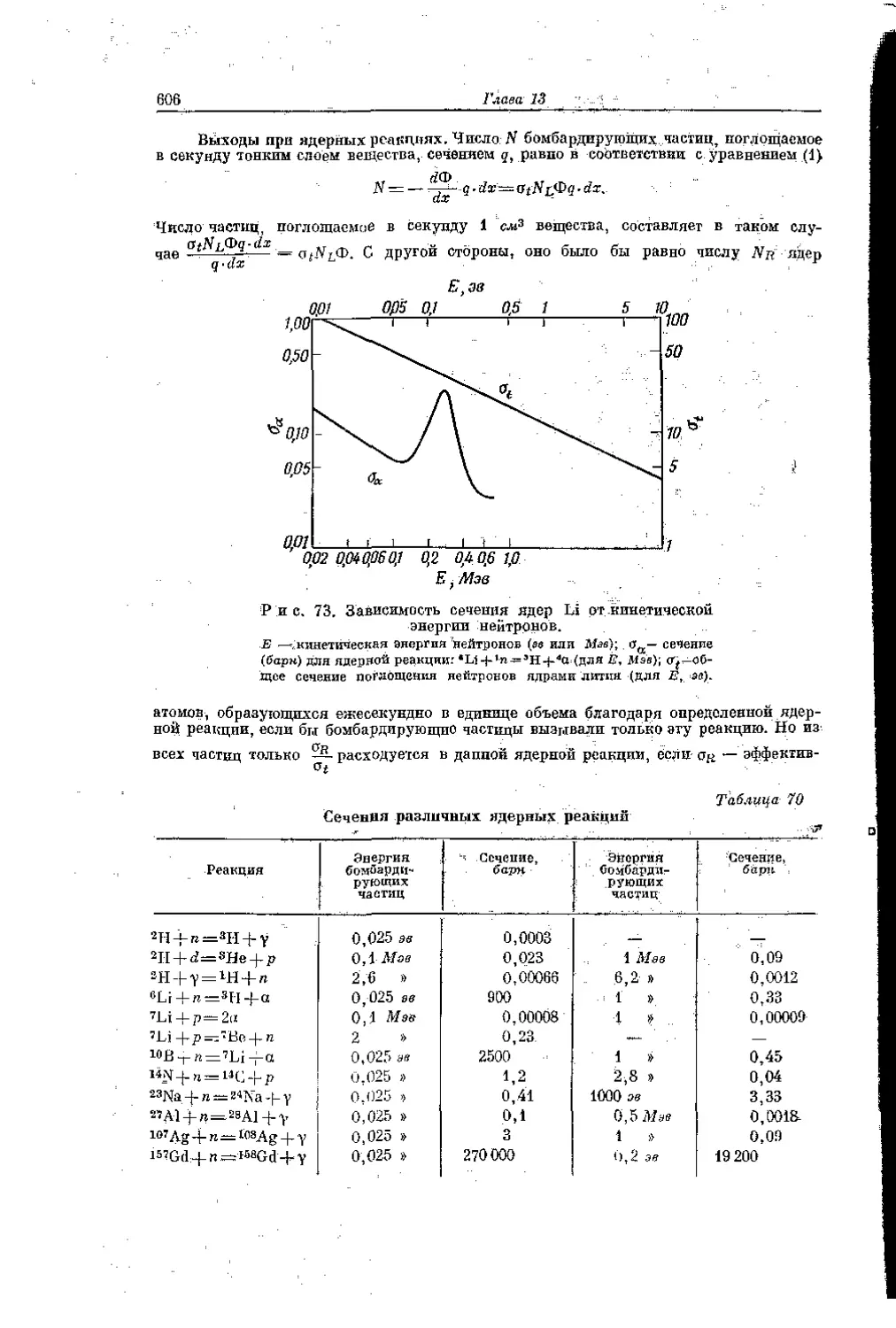
/
























































































































































































































































































































































































































































































































































































































































































































































































































































Текст
Г. РЕМИ
КУРС НЕОРГАНИЧЕСКОЙ ХИМИИ
том
II
ПЕРЕВОД С НЕМЕЦКОГО XI ИЗДАНИЯ канд. хим. наук А. И. Григорьева, канд. хим. наук А. Г. Рыкова, Н. С. Смирновой, канд. хим. наук Н. Я. Туровой
ПОД РЕДАКЦИЕЙ чл.-корр. АН СССР А. В. НОВОСЕЛОВОЙ
*
ИЗДАТЕЛЬСТВО «МИР»
Москва 1966
УДК 546
Книга представляет собой второй том хорошо известного читателю классического «Курса неорганической хймйи» (т. I вышел в 1963 г.). Книга является своего рода энциклопедией, по неорганической химии, одинаково интересной и полезной как начинающему студенту, так и опытному химику. Как п для первого тома, характерны физико-химический подход к изложению рассматриваемого материала, а также внимание к новейшим достижениям неорганической химии.
Книга служит ценным учебным пособием для студентов и преподавателей химических вузов и хорошим справочником для широкого круга химиков, работающих в различных областях.
Редакция литературы по химии
Г. Ре м и КУРС НЕОРГАНИЧЕСКОЙ ХИМИИ Том II Редактор Г. М, Мануйлова. Художник И. А. Литвишко Художественный редактор Е. И. Бескова * Технический редактор ЛГ. 17. Грибова
Сдано в производства ,20/1 (066 г. подписано я печати 1/УШ 1066 т. Бумага 70.x iO8Vis= =26,13 бум. л., 73,15 печ. л., +1 вкл. Уч.-изд л. 79,3s. Изд. XI 3/2463. Цена 5 р. 02 к.
Зак. 56 (Темплав 1666: г. Изд-ва «Мир», пор ту? 96) ¥
ИЗДАТЕЛЬСТВО iMHPo Москва, 1-й Рижский пер., 2 *
Московская типография К» 16 Главполпграфпрома Комитета по печати при Совете Министров СССР.
Москва, Трехпрудный пер., 9
ПРЕДИСЛОВИЕ
К ОДИННАДЦАТОМУ ИЗДАНИЮ
Настоящее издание вновь переработано н дополнено. Многие рисунки заменены для приведения в соответствие с новыми данными или для улучшения в ином отношении. Учитывая, что вскоре произойдет унификация «химических» и «физических» атомных весов на основе углерода 12С = 12, в приложении приведена таблица атомных весов, вычисленных относительно О = 16 и относительно 1SC—12. Помимо этого; приложение дополнено сопоставлением энергий ионизации атомов.
Приношу благодарность проф. д-ру К. Ф. Яру за его усердную и ценную помощь при составлении раздела «Ванадиевые кислоты и вана-даты(У)». Я благодарю также приват-доцента д-ра Линденберга и д-ра Гледис Тидеман за описание новых рисунков, а фрейлейн Тидеман за ее помощь в переработке именного и предметного указателей и за чтение корректуры.
Г. Реми Гамбург, февраль 1960 г.
ПРЕДИСЛОВИЕ
К ДЕВЯТОМУ И ДЕСЯТОМУ ИЗДАНИЯМ
Настоящий том, так же как и т, I, был полностью заново переработан. Достигнутые за последнее время успехи в неорганической химии не только значительно расширили представления о химических соединениях, они привели к более глубокому пониманню общих причин, определяющих поведение веществ. Некоторые области неорганической химии предстали благодаря этому в совершенно новом аспекте. Так, было показано, что соединения, которые у некоторых элементов не соответствуют степеням окисления и вследствие этого раньше считались «аномальными», ~ отнюдь не являются таковыми. Ойи закономерно получаются при определенных условиях, й способность к образованию такого рода соединении ; является одним из характерных свойств элементов, от которых они производятся. Тот факт, что соединения такого рода встречаются' преимущественно у элементов побочных подгрупп периодической системы, т. е. у элементов, рассматриваемых в этом томе, не случаен,“а обусловлен закономерной связью между валентностью и -строением атома. ;
Я связи с этим было бы недостаточным вставить описайие: новых сое- динений с особо отличающимися свойствами в прежний текст; в общих ; разделах текст должен был быть значительно переработан, а в главах.
£
Предисловие к девятому и десятому изданиям
посвященных описанию отдельных соединений, текст нужно было расположить по-новому. В систематическое описание отдельных веществ были введены элементы актиний, протактиний, технеций, прометий и их {
•соединения. До сих пор эти элементы только коротко описывали в общих '
разделах, посвященных систематическому обсуждению отдельных веществ. Причиной этого до недавнего времени являлись недостаточные экспериментальные сведения о химии этих ранее трудно доступных элементов.
В настоящее время имеются очень надежные и точные данные, с которыми оказалось возможным ознакомить читателя. Трансурановым элементам |
посвящена отдельная глава. *
Учитывая большое значение, которое приобрели изотопы, а также | различные области ядерной химии, материал, описанный ранее в главе i
«Радиоактивность, изотопы и ядерная химия», был расширен и разде-
лен на три главы. Ввиду важности для ядерной химии методов, впервые *
разработанных на естественных радиоактивных элементах, они в расши- ?
рейном объеме подробно описаны в главе о естественной радиоактивности. Расширено и углублено также изложение сведений о естественных радиоактивных элементах и вытекающих отсюда следствий. Они необходимы для понимания ядерной химии и ее практического использования. Ядер-ную химию, а это значит науку и технику процессов, связанных с пре-вращением ядер, в настоящее время часто рассматривают в Германии — А в противоположность ученым других стран — как область фпзпкп. |
Однако реакторная техника, основанная на ядерной химии, требует
разрешения многих химических задач и других проблем. Не говоря уже | о том, что химикам, все чаще использующим радиоактивные изотопы как средство исследования, необходимо ознакомиться с основными результатами исследования и методами работы в области радиоактивности ; й ядерной химии.
После выхода т. I этого издания Международным союзом по теоре- тической и прикладной химии (1UPAC) были разработаны новые правила номенклатуры неорганических соединений. В настоящем томе они цриия- I
ты во внимание. Для этого пришлось переработать ужо полностью подготовленную рукопись. К сожалению, не удалось избежать задержки в издании этого тома. Краткое описание новых правил дано в приложении*.
Вновь я должен поблагодарить многочисленных коллег за хорошие советы и великодушную помощь при переработке этой книги. Благодарю моих ассистентов д-ра Линденберга и д-ра Тидеман за ценное сотрудничество; д-ра Линденберга особенно за изготовление новых рисунков, а фрейлейн Тидеман за ценные предложения, переработку всей рукописи в свете новых правил номенклатуры и за тщательное чтение корректуры.
Гамбург, август 1959 г. „
Г. Реми
* Приложение, касающееся номенклатуры, опущено.—Прим. ред.
ВАЖНЕЙШИЕ ОБЩИЕ КОНСТАНТЫ
Абсолютная температура точки плавления, льда: Tfj°c = 273,16s К.
Атмосфера (нормальное давление): 1 атм s 760 ж* рт ст = 1,013250 X
X 10й дин-см~\
Чисм Авогадро: NA = 6,0247 -103S (физическая шкала) = 6,0231-10аз (химическая шкала).
Константа Больцмана: к = = 1,38042-10"1в эрг-град^1.
Элементарная частица электричества: е = 4,8028-Ю"10 эл.ст.ед.== = 1,60207-10~20 эл.магн.ед.=; 1,60207• 10-1я кулон.
Удельный заряд электрона: ~ = 1,75888-10’ эл. магн.ед/г.
Масса электрона (масса покоя): т = 9,1085-10"23 г; массовое число электрона *= 0,00054876.
Мера энергии:
1 эрг = 10'7 дж (ватт-секунд) = 2,777778-Ю-14 квт-ч (киловатт-часов) == = 0,239006-Ю"7 кал.
1 л-атм (литр-атмосфера) — 1,013278-Ю9 эрг = 101,3278 дж = = 2,81466-10"5 квт~ч = 24,2180 кал.
1 сл3-«шл1 (куб. сантиметр-атмосфера) = = 0,1013250 дж --
= 2,81458-10^ квт-ч = 2,42177-10"® кал.
1 кал (термохимическая грамм-калория) = 4,18400 дж = 1,16222 квт-ч = - 0,041292 л-атм = 41,293 сзг-ат.и.
1 КДЛ1 5° (15°-калорйя) = 4,1855 дж = 1,00036 кал.
1 эе (электрон-вольт) « 1,60207-10"1а эрг == 3,82904-10"®° кал; 1 эв на молекулу соответствует-23,0689 ккал/моль (физическая шкала) = - 23,0626 кх-ал/молъ (химическая шкала).
1 ТЕМ (тысячная единицы массы) соответствует 0,93116-10е за на атом, или 8,9858-1010 дж/г-атом = 21,476- 10е ккал!гратом.
Энергия светового кванта (фотона) с длиной волны & см:
• 1,9862• 10~1в эрг = 4 -1,23977-Ю"4 за, что соответствует Л Л Л,
т- 11,9663 дж/моль, или 4 • 2,86002 кал/моль (физическая шкала) == -Л Л
1 1 -
— у -11,9630 дж/моль, или 2,85923 кал/моль (химическая шкала).
Фарадей (электрический заряд одного грамм-эквивалента) = IF = = 96493,1 кулон, что соответствует 26,8037 а-час.
8 Важнейшие общие константы
Газоеая константа' 7?ф113 = 82,079 емУатм = 0,082076 л-атм = = 8,3166 аж = 1,98772 кал на физический моль.
Яхим=82,056 смя-атм=Л),082054 л-алй=8,3143 дж~=1,98718 кал на химический моль.
Скорость света: с = 2,997929-1010 см- сек"1.
Литр (объем 1 кг воды, не содержащей воздуха, при. максимальной плотности и нормальном давлении): 1 л = 1000,028 см?
Молярный объем идеального газа (по химической шкале при 0эС и 760 мм рт ст) — 22,4140 л — 22414,6 cat3. 1 .
Л/яапгаянхае JJjaw&a /лсяаят Ж — 6,S252-JD~^' ум-ж.
Ускорение силы тяжести, нормальная величина (на уровне моря и шипо-те 45э): g0 — 980,665 см-сек~2.
Фактор ^ми'па— \ъ,уми^ вёс° = 1,000275 (условная величина).
ЧАСТЬ ВТОРАЯ
ПОБОЧНЫЕ ПОДГРУППЫ ПЕРИОДИЧЕСКОЙ СИСТЕМЫ, СЕМЕЙСТВО ЛАНТАНИДОВ Й ТРАНСУРАНОВЫЕ ЭЛЕМЕНТЫ
ВВЕДЕНИЕ
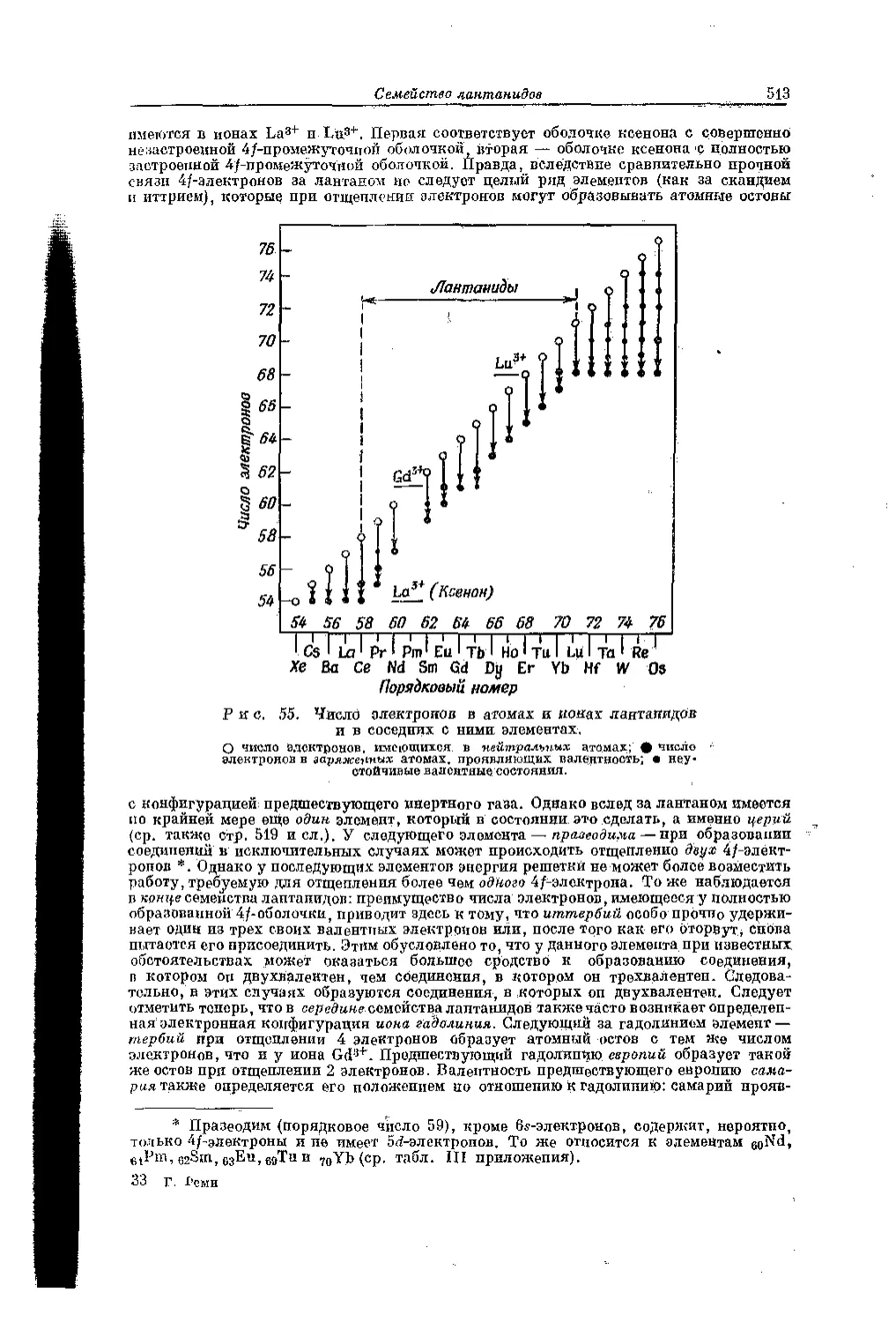
В т. I были рассмотрены все элементы главных подгрупп периодической системы (включая водород), порядковые номера которых на. 1—2 единицы больше или на 5—1 единиц меньше порядкового номера инертного газа. Остальные элементы относятся к Побочным подгруппам периодической системы, да исключением лантана и следующих за ним .14 элементов, которые образуют особое семейство лантанидов, и транс-урановых элементов, следующих за ураном.
Побочные подгруппы периодической системы включают (ср. табл. IV приложения II) элементы с порядковыми номерами 21—30, 39—48, 57, 72—80 и элементы 89—92, Таким образом, они охватывают нижние ряды двух длинных периодов по 10 элементов, а также 10 элементов из третьего длинного периода, в котором содержится 24 элемента (14 элементов этого периода, а именно элементы с порядковыми номерами 58—71, относятся к семейству лантанидов), и, наконец, в побочные подгруппы входят еще первые 4 элемента следующего длинного периода от актиния (порядковый номер 89) до урана (порядковый номер 92). Известные в настоящее время элементы с порядковыми номерами 93—103, следующие за ураном, хотя и составляют продолжение этого периода, однако на основании строения пх атомов и по химическим свойствам их нельзя причислить к элементам побочных подгрупп — они образуют особое семейство подобно лантанидам.
Можно пр одно пожить, что это Подобное лантанидам семейство начинается не с урана, а непосредственно за актинием, так же как в предыдущем длинном Периоде семейство лантанидов возглавляется латаном, аналогом которого является аитипий. С точки зрения строения стома это означало бы, что начинающееся у актиния заполнение (У-уровяя у следующих за ним элементов — тория, протактиния и урана — не продолжается, а сначала происходит заполнение 5/-уровня. Вопрос о том, лак следует рас-поло жить в пер поди ческой системе названные элементы на основании их атомного строения, является спорным, поскольку их строение изучено недостаточно полно. Колее поздние исследования (подробнее см. гл. 14) показывают, однако, что заполнение 5f-yровня начинается весьма вероятно не у тория, а только у трансурановых элементов. Если это так, то элементы торий, протактиний и уран с точки зрения строения атом а должны относиться к побочным подгруппам. На основании химического поведения их прежде рассматривали в качестве аналогов элементов, стоящих в побочных под группах IV, V и VI, групп периодической системы.
Элементы побочных подгрупп по своим свойствам в той или иной степени близки к элементам соответствующих (т. е. относящихся к этому же периоду) главных подгрупп. Это сходство наиболее велико там, где только начинается ответвление побочных подгрупп, а именно в третьем периоде. С возрастанием порядкового номера и номера группы или перио
12
Введение
да это сходство постепенно уменьшается от третьей группы к седьмой, а в восьмой группе совсем исчезает. При дальнейшем возрастании порядкового номера снова появляется побочная подгруппа первой группы, которая при коротком изображении периодов расположена в его пижнем ряду. Здесь снова появляется: Определенное сходство в свойствах, усиливающееся при переходе от первой группы ко второй, где побочные подгруппы примыкают в главным, Чтобы разместить элементы побочных подгрупп в тех же группах, которые занимают элементы главных подгрупп, 3 из 10 элементов ка?кдого нижнего ряда периода, особенно близких по свойствам между собой, а именно Fe, Со, Ni — Ru, Rh, Pd — Os,: Ir, Pt, помещают в одну группу, как ужо было указано в т. I.
В некоторых случаях у элементов побочных подгрупп особенно велико сходство со вторым элементом соответствующей главной подгруппы.. Это неоднократно отмечалось уже в т. I ири характеристике главных подгрупп. При переходе от третьей: группы к седьмой это сходство уменьшается, в восьмой группе исчезает*, а в первой появляется вновь и даже усиливается при переходе от первой группы ко второй. Однако наиболее близки между собой элементы, стоящие друг под другом в одной побочной подгруппе. В середине периодической системы (Ш, IV и V группы) это сходство в общем едва ли меньше, чем у более тяжелых элементов главных подгрупп. В отдельных случаях (цирконий — гафний) оно даже больше, чем между любыми двумя аналогами главных подгрупп. В большинстве случаев, однако, среди элементов одйрй и той же побочной подгруппы наблюдается меньшее совпадение валентности, чем в главных подгруппах. Многие элементы одной побочной подгруппы обладают способностью легко изменять свою валентность. Переменная валентность особенно характерна для, элементов побочной подгруппы:: восьмой группы.
Помимо легкой перемены валентности, элементам побочных подгрупп присущи и некоторые другие особенности. Например, большинство из них образует, как правило, парамагнитные соединения и способны к образованию окрашенных элементарных электролитических ионов, чего не наблюдается у элементов главных подгрупп.
В побочных подгруппах гораздо сильнее, чем в главных, проявляется сходство между рядом стоящими элементами, и иногда оно даже превышает сходство между1 аналогами (т. е. стоящими в периодической системе друг под другом). Например, железо по свойствам, а также по виду и свойствам образуемых им соединений ближе к рядом стоящим, элементам марганцу и кобальту, чем к своим аналогам рутению и осмию. То же характерно для кобальта и никеля. Железо, кобальт и никель расположены в побочной1 подгруппе восьмой группы. Именно в этой и соседних с ней группах сходство по горизонтали оказывается особенно ярко выраженным.
Прежде появление побочных подгрупп могло показаться отклонением от закономерностей периодической системы, В настоящее время очевидно, что оно следует из общего закона распределения электронов в атоме. Существование побочных подгрупп в периодической системе, как показывает изучение спектров, обусловлено тем, что правило, согласно которому в ряду элементов повыв Электропы по мере увеличения заряда ядра, образно Тойорл, «располагаются впе уже полностью сформированной оболочки», не является общим. В определенных местах' рядов элементов электроны располагаются «внутри уже сформированной оболочки». Точнее, в определенных пунктах рядов элементов, несмотря на то что оболочка с главным квантовым числом п завершена, происходит заполнение оболочек с более низкими главными квантовыми числами (л — 1 или п — 2). По спектрографическим данным можно определить,
Введение 13
с какого элемента это начинается *. Теория строения атома позволяет также объяснять, почему это происходит.
При описании главной подгруппы П группы ужо указывалось на одно характерное различие в спектрах однократно ионизированного кальция и нейтрального калия, во всем остальном совершенно аналогичных между собой. Из спектральных термов видно, что у Са+ 34-уровень соответствует пе более высокому, как у К, а брлеенизкбму содержанию энергии атома по сравнению с 4у-уровнем. Из сопоставления уровней .энергии атома калия (рие. 59, т. I, стр. 282) и однократно ионизированного атома, кальция следует, что если бы 34-уровень Са+ находился относительно немного ниже или де-уровень расположился бы относительно немного выше, то 34-уровию соответствовало бы более низкое энергетическое состояние, чем 4з-уровню. При сравнения дугового спектра калия й искрового спектра кальция можно наблюдать сильное возрастание содержания энергии (для Са^ уменьшенное в четыре раза) на 4s-уровне п падение па Зй-уровпе. Поэтому можно ожидать, что у следующего в ряду элемента — скандия значение энергии на 4я-уровпе для двукратно ионизированного атома будет превышать содержание энергии на 34-уровне. Это полностью подтверждается анализом термов спектра скандия. Основным уровнем для 19-го электрона у скандия служит не 4s-уровень, как у калия и кальция, а Зй-уройень. Только 20-й электрон в невоз-бужденпом состоянии занимает 4з-уровень, как в спектре однократно ионизированного скандия. 21-й электрон у скандия также находится на 4s-уровне. Таким образом, создается следующий порядок распределения электронов в девозбуждением атоме скандия:
Is22s^2pe3s23p63d4sa.- л
По мере дальнейшего увеличения заряда ядра, не только 19-й, но и следующие элек-лроны располагаются на 34-уровне, нока чивло их не достигнет максимально возможного для данного уровня (10). Например, у с камбия, как уже упоминалось, 20- и 21~п электроны находятся па 4а-уровне, а у следующего:за ним тиаглмд, как видно нз его спектра, не, только 19-й, но и 20-й электроны размещаются на 34-уровне< И только .21- .и 22-й занимают 45-уровень. У ванадия 19-, 20- и 21-Й электроны занимают 34-уревень и только 22- и 23-я — 4з-уровень **. Сказанное справедливо и для следующих за ванадием элементов, включая никель; все они содержат максимум два электрона на уровне с главным квантовым числом 4; остальные валентные электроны занимают 34-уроветтЬ (ср. табл. Ш и IV приложения). г
Тот факт, что у скандия последний электрон находится па d-, a iie подуровне, как у элементов главной подгруппы III группы, на химические свойства самого скандия влияет очень незначительно. Химические свойства зависят главным образом от того, насколько легко отщепляются электроны^ Особенно это влияет па валентность в гетерополяр пых соединениях. У кальция максимальная электроположительная валентность два, т. е, способность отщеплять только то два электрона, который находятся в невозбужденном атоме на 4з-уровне, но не на Зр-уровне, обусловлена тем, , что последние связаны гораздо прочнее. У скандия же то, что однп электрон в нев избу ж денномсостоя1!ии занимает 34-уровень и связан лишь немного прочнее, чем электроны на 4.у-уровне, мало влияет на его химические свойства. То же справедливо и для'последующих элементов: периодической системы. Большая лабильность, валентности у элементов побочных подгрупп но сравнению с главными подгруппами связала в особым распределением электронов в атомах элементов побочных подгрупп.
Важным следствием распределения электронов на уровне с боле, в низкам главным квантовым числом (во -«(внутренних оболочках») является то, что у железа (порядковый номер 26), стоящего на восьмом месте после аргона, «внешняя оболочкам инертного газа еще ns завершается. Последняя характеризуется наличием в цевозбуж денном атоме двух электронов па s-уровне и шести электронов на /-уровне. Ввиду того что у скандия и следующих за ним элементов всегда часть электронов располагается на 34-уровне, их впачале не хватает для образования слоя нового инертного газа (в данном случае это был бы уровень с двумя электронами на 4s- и шестью па 4р-уров~ иях), И только когда Зд-уровёнь полностью завершен, начинается заполнение уровня •с главным квантовым числом 4, т. е. образование оболочки следующего инертного Таза. Тот факт, что криптон занимает 18-е место после аргона, свидетельствует о том, что па За'-уровне может находиться всего 18 — (2 4- 6) = 10 электронов. Аналогичная
* Еще до того как на основании спектрографических данных было доказано образование промежуточных оболочек, в 1920 г. Ладепбург (Ladenburg) пришел к такому же выводу на основании парамагнитных свойств и окраски ионов элементов побочных лодгрукп периодической системы.
** Сказанное справедливо для нейтрального атома ванадия. В однократно ионизн-Фсвайпом атоме ванадия на 34-уровпе находится и 22-п электрон.
14
Введение
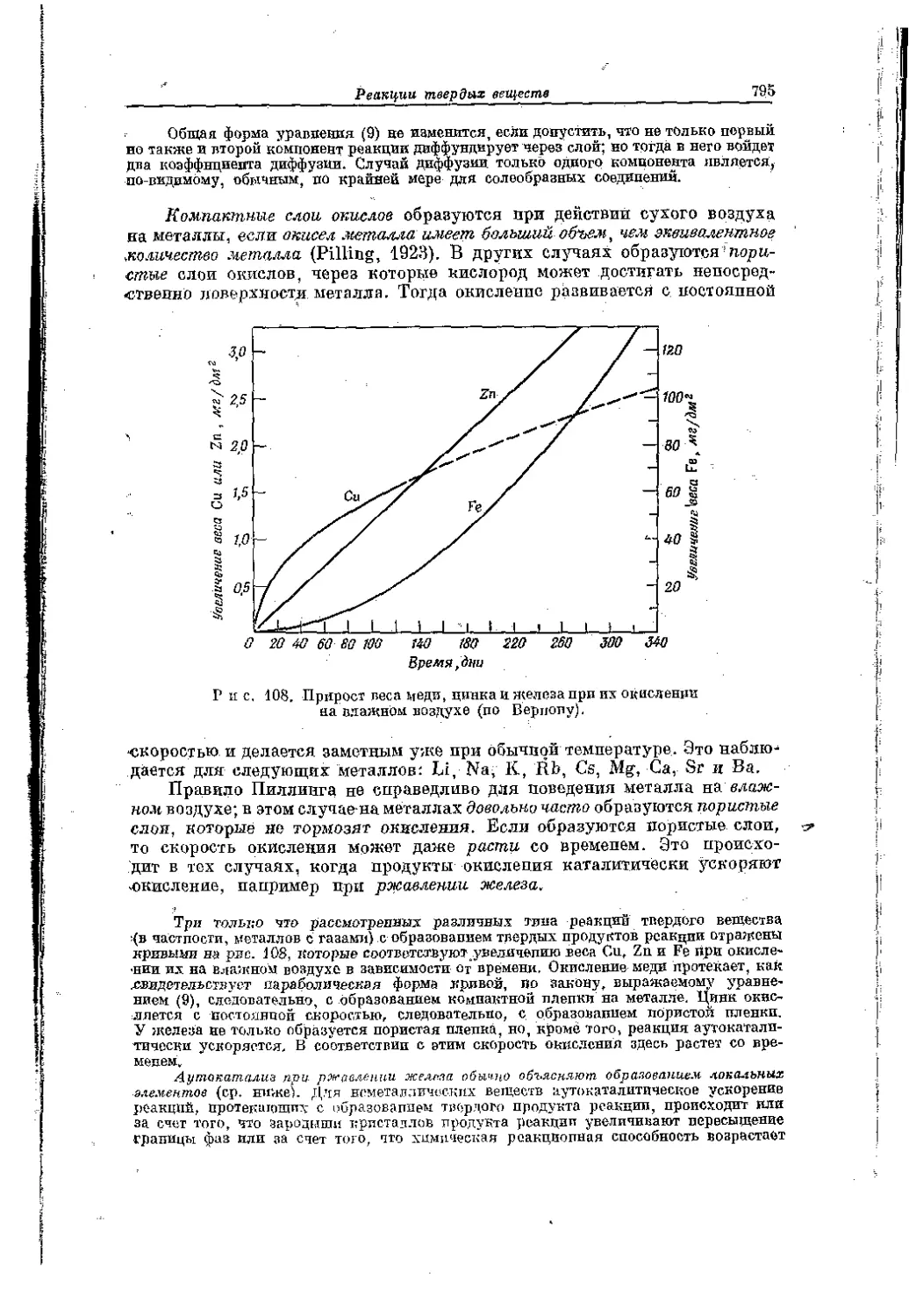
закономерность наблюдается и в следующем ряду; у иттрия, как показывают <~ж?-ральпые термы, вповь начинается образование внутренней оболочки. Следовать гыи начиная с иттрия, электроны распределяются на чй-уровне вместо 5s- или 5р-урахавж пока общее число их не достигнет 10. Таким образом оболочка следующего пи-рр^ЕТП. газа ксенона также завершается через 18 (= 10 4- 2 4- 6) элементов. В следув.. у (восьмом) ряду, начиная е лантапа, снова заполняются внутренние электронные оболочки. В этом случае электронам предоставляются два возможных энергетических уровня с главными квантовыми числами ниже 6, а именно 4) и бй-уровни. Следовательно, оболочка следующего инертного газа (радона) формируется только после завершения обеих) названных «внутренних оболочек# и заполнения бр-уровня. От элементов, у которых происходит образование внутренних оболочек, отличаются так называемые ^переходные элементы», упомянутые а т. /, у которых; происходит заполнение d-уровней;
Наличие в восьмом ряду между лантаном н гафнием, за которым следует ряд элементов побочных подгрупп, особого промежуточного периода элементов с характерными свойствами, очепъ похожих па лантан (семейство лантанидов), указывает на то, что образование обеих оболочек следует друг за друзом. Вначале формируется более глубокая из них, т. е. заполняется 4/-уровень..Это следует из того, что расположенные за лантаном элементы проявляют одинаковую с ним валентность; лишь некоторые из них наряду с валентностью три бывают двух- и четырехвалентнымн. То же подтверждается и анализом термов спектров. Электроны на 4/-уровне связаны настолько прочнее, чем на 5d- и 6з-уровнях, что только в виде исключения входят и число валентных электронов атома. Вследствие этого, пока 4/-уровень не завершится полностью, валентность в общем не изменяется (подробнее см. гл. 10). Можно определять, сколько элементов содержится в группе лантанидов. Следующий инертный газ, радон, у которого по аналогий с остальными инертными газами должно быть два электрона па 6s- и шесть электронов на 6р-уровне удален от предшествующего инертного газа — ксенона на 82 места. Подобно 3d и 4й-уровиям 5й-уровень, отвечающий образованию нижнего ряда элементов, составляющих «побочные подгруппы», должен содержать 10 электродов. Следовательно, число элементов в группе лаптанпдов на основании теории строения атома должно равняться 14 (= 32 —[10 4- 2-}- 6]). Действительно, лантанидный характер проявляют точно 14 элементов, следующих за лантаном; порядковый номер последнего 71. Следующий элемент — гафний (порядковый номер 72), является уже не лантанидом, а подобно самому ланталу обладает свойствами элемента побочном подгруппы. Гафний входит в ряд из 10 элементов побочных подгрупп третьего большого периодо. К атому ряду принадлежат: лантал (порядковый номер 57), гафпий (72) и следующие за ним элементы, нключая ртуть (80).
Наконец, в ряду, возглавляемом радоном, следует ожидать аналогичного заполнения «внутренних оболочек». В то время как у радии (порядковый номер 88) наиболее подйижные электроны в певозбужденпом атоме находятся на 7с-у ровне/ начиная С актипия (порядковый номер 89), наблюдается заподнетше d-уровня, а именно Ш-уровня. Вследствие этого актинии также носит характер переходного элемента, а за ним начинается новый ряд элементов, входящих в побочные подгруппы.
В этом ряду у элементов происходит заполнение 5/-уровня. По данным Сиборга (1949), опо начинается непосредственно за актинием, т. е. у торид (порядковый номер 90). Более поздние исследования *, однако, свидетельствуют о том, что заполнение 5f-уровня начинается, по-видимому, только у нептуния (порядковый помер 93) или у плутония (94). В обоих случаях рио ведет к появлению ещё одной' обособленной группы злелгеитов, подобно тому как наполнение 4/-уровпя привело к появлению семейства лантанидов. Эту группу элементов, характеризующихся заполнением 5/-уровня, называют семейством актинидов **.
* Ср. например: J, К. Dawson, Nucleonics, 10, Mi 9, 39 (1952); Angew; Chem.. 65, 485 (1953).
** Предложенное Сиборгом наименование актиниды, т. е. «элементы, подобные актинию», выражает как сходство элементов этой особой группы с актинием, так и аналогию с элементами семейства лантанидов. Вначале этот термин распространяли на весь ряд следующих за актинием элементов. После того как Дау соном было показано, что заполнение 5/-уровпя начинается только у пептуния или плутония, воз-ивкло сомнение, можно ли в атом смысле элементы горни, протактиний и уран также называть «актинидами». Для всего ряда элементов от актиния до элемента 103 целесообразно употреблять выражение элементы актиниевого ряда. Элемент 103 еще не открыт ***, однако на основании теории строения атома можно предположить, что на нем закончится заполнение 5/-у ровня.
*** В 1961 г. был синтезирован изотоп элемента 103, названного лоуренсием.— Прим. ред.
Введение
15
В табл. III приложения приведен обзор распределения электронов на отдельных уровнях энергии в невозбуждеииом атоме. Данные о распределении электронов основаны главным образом,, па результатах спектральных исследований (включая рентгеновскую область). У элементов, следующих за актинием, они основаны главным образом на магнитных измерениях.
В некоторых случаях распределение электронов с уверенностью не установлено: так, для «Тс распределение внешних электронов возможно не 4d55s2, a 4 Лаю У элементов с порядковыми номерами 58, 59, 61 и с 66 по 68 распределение электронов на /- и ^-уровнях окончательно еще не установлено. То же, как уже упоминалось, справедливо для элементов от 90 до 102 (или 103),
При сопоставлении распределения электронов в приведенной таблице принимается во внимание подразделение энергетических уровней только в соответствии с одним побочным квантовым числом I, т. е. орбитальным квантовым числом. Спектрографические данные позволяют воспроизвести более детальную картину распределения, учитывая еще два побочных квантовых числа т и s (ср. т. I, стр. 122): Это важно, так как по принципу Паули (ср. т. I), исходя из четырех квантовых чисел для каждого заряда, можно определить вероятное число электронов на каждом энергетическом уровне в невозбужденном атоме и, следовательно, вывести важнейшие теоретические общие закономерности периодической системы из одного принципа. В гл. 4 г. I было показано, что на основе принципа Паули можно определить максимальное число электронов па внешней оболочке инертных газов и, следовательно, длину так называемых «коротких периодов». Появление энергетического уровня с орбитальным квантовым числом I = 2 приводит к появлению «длинных периодов» с 18 элементами, так как при 1=2 т принимает значения —2, г—1, 0, +1 и +2 и для каждого из них возможны два значения s; к 8 комбинациям квантовых чисел при i = 0 и ( = 1 добавляются еще 10. При 1 = 3 т = —3, —2, —1, 0, -Н, -J-2, +3; каждому из этих семи квантовых чисел соответствуют значения + и з = — Va, т. е. еще 14 комбина-
ций квантовых чисел, возможных при 1 = 3. Эти 14 комбинаций квантовых чисел обусловливают появление семейства лантанидов и, следовательно, увеличение длины периода с 18 до 18 + 14 = 32 элементов. Таким же образом объясняется и появление семейства актинидов.
Остается выяснить еще вопрос, почему такое своеобразное явлений, как построение «промежуточных оболочек», происходит лить в определенных участках рядов элементов. Ответом являются рассуждения, аналогичные уже приведенным в гл. 4 т. I (стр. 144), о том, что у более тяжелых атомов энергия электронном орбиты, находящейся вне s-уровня, зависит от орбитального квантового числа Z иначе, чем можно было бы ожидать, учитывая только зависимость массы электрона от скорости.
Для наглядности рассмотрим положения, лежащие в основе теории Бора — Зомнерфельда. Из них следует, что, например, у калия 19-й электрон должен быть значительно прочнее связан «а is-уровне, чем на if-уровне. Согласно этой теории, ему соответствует 44- орбит а, т. е. круговая, и на него в случае калия действует аффективный заряд экранированного ядра, равный примерно единице заряда. 4s-ypouHio но той же теории соответствует орбита 4j, т. е. сильно вытянутый эллипс. Электрон па такой орбите связан гораздо прочнее, чем па орбите 44| благодаря тому что у эллиптической орбиты имеется участок, на котором электрон временно подходит очень близко к атомному ядру, заряд которого у калия равен 19, По той же причине 19-й электрон у калия па 4 4-орбите связан прочнее, чем па орбите З3. Однако связь электрона на орбите З3 значительно упрочняется при переходе от калия к кальцию.
Если предположить, что заряд ядра, действующий на электрон на орбите З3 у калия, почти полностью экранирован, за исключением одной элементарной единицы заряда, то заряд экранированного ядра кальция, действующий на электрон, будет равен 2, а у скандия 3.
Энергия связи электрона па орбите З3 в соответствии с уравнением (И) гл. 3 г. I (см. т. I, стр. Ю9) возрастает пропорционально квадрату эффективного заряда
16
Введение
ядра. Гораздо медленнее возрастает энергия связи электрона на 4горбите; в значительной степени это зависит от общего заряда ядра, который при переходе от калия к скандию возрастает от 19 до 21, т, е. в процентном отношении изменяется незначительно. Таким образом, получается, что прочность связи на круговой орбите 3, в конечном итоге превышает прочность связи на сильно вытянутой эллиптической орбите 4j.
В соответствии со сказанным выше, число электронов, необходимое для построения «внутренних оболочек», определяет число элементов побочных подгрупп, т. е. длину нижних рядов. Ответвление побочных подгрупп от главных начинается с элементов, у которых происходит заполнение «внутренних оболочек». Однако конец периода, состоящего из элементов побочных подгрупп, не совпадает с завершением «внутренней оболочки». Например, «внутренние оболочки» меди, серебра п золота, как видно из спектров этих элементовДср. гл. 8), являются завершенными. Однако и эти, и непосредственно следующие за ними элементы (цинк, кадмий и ртуть) по химическим свойствам безусловно принадлежат к элементам побочных подгрупп (а именно к побочным подгруппам первой и второй групп). Таким образом, понятие «элемент побочной, подгруппы» не совпадает с понятием «переходный элемент».
Ввиду того что ответвление побочных подгрупп от главных начинается в третьей группе, изучение элементов побочных подгрупп следует начинать также с элементов побочной подгруппы третьей группы. От них надо перейти к следующим по ряду элементам вплоть до элементов восьмой группы. Затем будут рассмотрены элементы начала чётных рядов длинных периодов, т. е. элементы побочных подгрупп первой и второй групп. На этом обзор элементов побочных подгрупп будет закончен.
Элементы, примыкающие к элементам побочной^ подгруппы, второй группы, относятся к главной подгруппе третьей группы. ;
Элементы, расположённые в восьмом ряду между лантаном и гафнием (14 элементов с порядковыми номерами 58-—71), т. е. непосредственно в том месте ряда, где происходит ответвление побочной подгруппы, по своим свойствам близки лантану й образуют особое семейство. Очень близкие между собой по свойствам эти 14 элементов, окислы которых имеют характер «редких земель», нельзя причислить к побочной подгруппе периодической системы; семейство лантанидов рассматривается в гл. 10-
То же справедливо и для трансурановых элементов, также образующих особое семейство й имеющих большое сходство с лантанидами. Им посвящена специальная глава,< следующая непосредственно за главой о радиоактивности й искусственном делении атома, ввиду того, что эти явления для трансурановых элементов особенно характерны.
Элементы, у которых происходит построение «внутренних оболочек» (т. е. заполнение d~ и /-уровней), как уже упоминалось, названы «переходными элементами». Таким образом, к их числу относятся элементы с порядковыми номерами от 21 до 28 (от Sc до Ni), 39—46 (от Y до Pd), 57—78 (от La до Pt) и все известные в настоящее время элементы (в том числе полученные искусственно) с порядковыми номерами выше 88, т. е. элементы от Ас до U и трансурановые, элементы. Два первых ряда .переходных элементов (от Sc до Ni и от Y До Pd) содержат по 8 элементов. Третий ряд содержит 22 элемента, так как в него входят ещё 14 лантанидов. Последний ряд переходных элементов незавершен, так как у элементов с очень высокими порядковыми номерами устойчивость сильно надаёт по мере возрастания их номера. Ряды переходных элементов начинаются с элемента, занимающего именно- то место в периодической системе,
Введение
17
где происходит ответвление побочных: подгруппОканчивается каждый из них (за исключением последнего незавершенного ряда) элементом; стоящим в периодической системе на два места раньше того пункта, где побочные подгруппы примыкают к главным (Zn, Соответственно Cd, или Hg),
Для побочных подгрупп периодической системы . которые включают переходные элементы, существует правило, что всегда второй элемент более близок третьему в той же подгруппе, чем первому, а в подгруппах, содержащих по четыре элемента, соответственно: третий близок к четвертому.
Это объясняется отчасти так называемым «лантанидным сжатием» (см. стр. 516), благодаря которому атомные и ионные радиусы, второго и третьего элементовкаждой побочной подгруппы мало отличаются друг от друга. Еще нагляднее, чем при Сопоставлении кажущихся атомных и ионных радиусов (рис. 3, т. 1), это видно из рис. 1.
Р,ц е. .1. Радиусы атомов элементов" побочных под-трупп (А).
На.: нем показано изменение кажущихся атомных радиусов элементов побочных подгрупп, расположенная друг под другом. Про этом,, чтобы сделать величины еще более сравнимыми, атомные радиусы тех элементов, которые кристаллизуются: в решетках с координационным числом 12 (как бывает л большинстве случаев), пересчитаны с. учи-.
* В металлическом состоянии (ср. стр. 22 и ел.), шквидимому, не только у элементов третьем группы валентные электроны находятся на «^-уровне, но ну некоторых элементов главной подгруппы второй группы (а именно у Са, Sr, Ва и На). Поэтому при рассмотрении образования и свойств интерметаллических соединений етсмепты главной подгруппы второй группы часто также причисляют к йерехрд-п; । элементам.
2 Г Реми
18
Введение
том величин /KZ, указанных в табл. 42 т. I, на координационное число 12. Из данных, приведенных в табл. 8, 12, 14 и т. д., следует, что сказанное об атомных радиусах справедливо и для иопных радиусов. Из рис. 1 видно, что те аналоги переходных элементов, которые особенно близки по химическим свойствам и свойствам их соединений, имеют близкие атомные радиусы. Из того же рисунка следует, однако, что, помимо аналогичного строения внешней электронной оболочки, обусловленного положением в одной группе, и мало отличающихся атомных (и ионных) радиусов, должны быть и другие обстоятельства, определяющие весьма близкое сходство элементов. Например, наибольшее сходство в поведении и свойствах соединений известно для пары элементов цирконий — гафний. Однако атомные и иопные радиусы у них (ср. также табл. 8 и 12) не так близки, как у пары ниобий — тантал, сходство химических свойств у которых несомненно меньше (подробнее см. стр. 96). У элементов, следующих за переходными элементами, зависим octi, даже обратная. Уже в группе Си — Ag — Аи при сравнении твердых растворов свободных металлов вццпо, Что сходство между 2-м и 3-м элемептами едва ли больше, чем между 1-м и 2-м. В группе Zn — Cd — Hg сходство между 1-м и 2-м даже значительно больше, чем между 2-м и 3-м. Несоответствие в случае пар Zr — Hf, Nb — Та исчезает, как будет показано дальше (см. стр. 99), если принять во внимание различное поляризующее* действие ионов. Следствием этого является небольшой сдвиг пунктирной кривой via рис. 1 вниз. Если учесть это смещение, кривые весьма удовлетворительно передают зависимость сходства между переходными элементами внутри отдельных групп от различия их радиусов. Чтобы устранить несоответствие в побочных подгруппах, следующих за переходными элементами, следовало бы произвести гораздо более сильное смещение пунктирной кривой. Обосновано ли такое сильное смещение различием в поляризующем действии, весьма сомнительно.
Всё элементы побочных подгрупп носят чисто металлический характер. Поскольку понимание различных их свойств возможно только' на основании знания природы металлического состояния, то обсуждению отдельных побочных подгрупп предшествует обзор общих свойств металлов.
Гл а в а 1
МЕТАЛЛЫ И ИНТЕРМЕТАЛЛИЧЕСКИЕ ФАЗЫ
Общие сведения. Металлы отличаются от неметаллов главным образом следующими характерными свойствами. Металлы имеют своеобразный блеск (металлический блеск), который обусловлен их высокой отражательной способностью видимого света. Они обладают незначительной св стоп pony скаем остью и даже в тонких слоях "непрозрачны. Металлы большей частью обладают хорошей ковкостью и поэтому могут быть обработаны при помощи вальцов, пресса, молота и т. д. Однако прежде всего: металлы отличаются высокой теплопроводностью и хорошей электропроводностью, причем их электропроводность возрастает с понижением температуры.
Эти различия гиежду металлами и неметаллами справедливы для твердого и жидкого состояний. В газообразном состоянии они исчезают. Например, пары ртути бесцветны, прозрачны н не проводят электрического тока. Одноатомность в газообразном состоянии также пе является характерным свойством Металлов, так как, с Одной стороны, инертные газы, относящиеся к ярко выраженным неметаллам, одноатомпы, с другой стороны, в парах щелочных металлов имеются в заметном количестве двухатомные молекулы.
Резкой границы между металлами и неметаллами нет. Вещества, которые занимают среднее положение между металлами и неметаллами, называются полуметаллами. Типичные полуметаллы отличаются от металлов, между прочим, тем, что их электропроводность при переходе из твердого в жидкое состояние возрастает, в то время как электропроводность металлов при плавлений падает. Кроме того, полуметаллы отличаются от типичных металлов недостаточной ковкостью. Вещества, которые, за исключением хрупкости, в основном обладают свойствами, подобными свойствам металлов, называют хрупкими металлами.
Появление неметаллического характера у химических элементов-отчетливо зависит от их места в периодической системе (включая водород). Область неметаллов отделена от области металлов в главных подгруппах периодической системы (табл. IV приложения) диагональю, проходящей от бора через кремний, мышьяк и теллур к астатину. Среди элементов, лежащих на этой диагонали, бор и кремний — неметаллы, мышьяк и теллур — полуметаллы. Все элементы главных подгрупп (включая водород), расположенные вправо от диагонали, являются неметаллами. Все элементы, стоящие влево от нее, являются металлами. Те из металлов, которые примыкают к диагонали снизу, а именно германий, сурьма, висмут и, вероятно, также полоний, обладают характером хрупких металлов. Все элементы, принадлежащие к побочным подгруппампериодической системы, как и все элементы семейства лантанидов, а также все трансурановые элементы являются металлами. .
2*
20 Глава 1
Из этого следует, что подавляющее большинство химических элементов (а именно более 4/5 их общего числа) имеет чисто металлический характер.
Химические элементы С металлическим характером, в отличие от сплавов называют «чистыми металлами» *- Сплавы (ср. т. I, гл. 43), помимо металлов, в качестве добавок могут также содержать и неметаллы. Известны сложные химические соединения, состоящие из металлов и неметаллов, которые обнаруживают типичный металлический бцеск и высокую электропроводность, однако наряду с этим они чрезвычайно хрупки; таковы, например, многие нитриды, карбиды и бориды; их не причисляют к металлам (даже к хрупким). Такие соединения в дальнейшем будут рассматриваться как метаялоподобиые соединения.
Некоторые элементы существуют как в металлических, так и в неметаллических модификациях. Например, для фосфора, кроме нескольких неметаллических, известна одна металлическая модификация (черный фосфор); кубическая модификация олова (серое олово) в противоположность обычному тетрагональному олову имеет свойства неметалла. В химии издавпа употребляют названия «металлы» и «неметаллы» (ранее яазывцемые также, «металлоидами») для обозначения дйух различных классов веществ, однако, строго говоря, эти названия обозначают состояния,а:: не вещества.
Получение. Для получения металлов из их соединений применяют главным образом следующие способы:
.7. Химическое восстановление •;
В качестве восстановителя в лаборатории: чаще всего используют водород, в технике — углерод (обычно в форме кокса, реже — древесного угля). При этом обычно исходят и.з окис лов металлов. Сульфиды, переводят в. окислю нагреванием и струе воздуха («обжиг»). Окислы тем легче восстанавливаются, чем ниже их теплоты образования.
Приближеннее представление с: способности металла к в ос ст а поп л ап над дает его положение в ряду напряжений. Окпсльт металлов менее электроположительные, чем цинк, легко восстанавливаются как водородом, так и углем. Для получения более электроне лежите л ютах металлов водород в качестве восстановителя практически не имеет значения. Некоторые из пих, правда, еще удается: получить из окйслов при воздействии водорода,- однако только при очень высокой температуре. Окись ципка еще легко удается восстановить углем; даже щелочные металлы удаётся подучить восстановлением их окйслов .углем. Одиако большинство других сильно электроположительных металлов реагирует с углом с образованием карбида, что препятствует восстановлению бкиёлов до металлов.
Восстановление углем иногда все же достигается, и кар видообразование подавляется, если вместо чистых металлов использовать их сплавы. Для этого, палример, применяют в производстве жолёзо-марганцевые, сплавы (ср, стр. 228).
Для получения (технически) чистых металлов, в тех случаях, когда восстановление углем вследствие карбидообразовапия невозможно, в качестве восстановителя часто применяют алюминий (алюмотермия), реже — магний или кальций.
Для восстановления галогенидов металлов чаще всего применяют- кальций, натрий или калий. Галогениды тяжелых металлов можно, как правило, легко восстановить нагреванием в токе водорода.
При химическом восстановлении в лаборатории металлы чаще всего, получаются сначала в порошкообразном или губчатом состоянии. Сплавление в компактные куски связано иногда со значительными трудностями. Его облегчают-путем присадки способствующих плавлению добавок (цанриМер, буры или хлорида щелочного металла). Эти досылки растворяют мешающие сплавлению загрязнения и, защищая нагретый металл
* Часто па звание металл употребляют в том ;кё смысл?, что и чистый металл i так, например, говорят металлы и ах сплавы.
Металлы и интерметаляическиё фазы 21
от доступа во оду ха, препятствуют образованию окисла или нитрида на поверхности зёрен металла. Действительно, частый металлический порошок легко удается путем нагревания перевести в компактный металл. Дли этого его даже, не Обязательно нагревать до температуры плавления, так как часто уже значительно ниже температуры плавления металлические верна: плотно спекаются вследствие рекристаллизации (см. стр. 783). Па этом основан способ спекания для приготовления;изделий из металлического порошка, который также называют «металлокерамикой».
Электролиз: растворов или, расплавов ' ' ; .
Электролитическое осаждение металлов из водных растворов применяют в технике главным образом для очистки металлов,
Те металлы, которые не разлагают воду, удается выделить из водного раствора чаще всего без большого труда. Препятствие выделению, возникающее вследствие образования окисла на катоде (в случае тех металлов, которые из кислых растворов пе выделяются), можно устранить при помощи комплексообразующих добавок (например, бкеалатов й.чи цианидов). Для выделения благородных металлов также применяют часто такие добавки; так как они способствуют выделению металлов в ком пакт-пой форме.
Все в возрастающих масштабах электролитическое осаждепие металлов из растворов применяют для получения защитного покрытия на предметах, изготовленных из легко корродирующихся металлов (еальватстееия). Электролитическое осаждение применяют также для придания поверхности: металла определенного тиснения, определяемого формой (матрица), на которой происходит осаждение (гальванопластика)
В тех случаях, когда вследствие сильно э л ел троп о лож птельпого характера металла электролитическое осаждение его из водпых растворов невозможно, а путем химических реакции восстановление достигается <• трудом, в Лаборатории используют иногда электролиз неводных растворов, например растворов1 в пиридине.
В технике для выделения сильно электроположительных металлов пользуются большей частью электролитическим выделением из расплавов (электролиз расплавов). Именно так. в технике получают алюминий,, натрий: и кальций и отчасти также магний.
Применяемые для этого расплавы содержат окисли и галогениды соответствующих металлов в смеси с другими соединениями, понижающими.температуру плавления? и повышающими электропроводность и не подвергающимися, конечно, при данных условиях электролитическому разложений.
Потенциалы, требующиеся для выделения металлов из расплавов, нй находятся в простом соотношении с потенциалами разложения, требующимися для водных растворов, так как в последние входят значения энергий гидратаций- (ср; т. I, стр. 181 и ел ).
Как при осаждении из водных растворов наряду с энергиями иы1и;шц1п: определенную роль играют энергии гидратации, та if и цри осаждении из pa сплавов наряду с энергиями ионизации имеют значение силы, которые действуют между разряжающимися ионами и другими состивпыми частями расплава. Поэтому при известных обстоятельствах потенциал, разложения соединения в расплаве можно существенна изменить добавлением других соединений.
Для суждения об основном процессе при ллектроляае расплава .следует также Принять во внимание часто весьма различимо для различных: ионов температурные зависимости потенциалов разложения. Например, ,КаС1 при температуре несколько ниже 600° обладает большим потенциалом разложения, чем CaGlj, а при более высокой : температуре — меньшим; это позволяет получить металлический натрий электролизом расплава смеси поваренной соли и хлорида кальция (ср. т. I, стр. ЦМ к ел.). .
3. Термическое раэлозкение соединений
Термическим разложением подходящих соединений .многие металлы удается получить в особо чистой форме.
Наиболее старым техническим способом, в котором термическое разложений использовали для получения: металла, является получение никеля разложением кар
22
Глава 7
бонила пикеля (см. стр. 331 и сл.). Соответствующим же образом карбонил железа используют для приготовления особо чистого железа (см. стр. 270). В этих двух случаях преимущество метода заключается в том, что большая летучесть названных карбонилов позволяет полностью отделить все примеси.
В других случаях термическое разложение используют, если получение чистых веществ путем химических реакций представляет большие трудности. Так, титан, цирконий и торий при восставоеленпи их соединений химическим путем образуются сначала только в виде порошка.
Вследствие содержащихся в порошке загряз пении его удается в дальнейшем только с трудом сплавить в компактные куски и, даже если это удается, металлы получаются не совсем чистыми. Напротив, эти металлы сразу получаются в компактном состоянии и совершенно чистыми в результате термического разложения их галогенидов (лучше всего иодидов) по способу, разработанному ван Аркелем: и дё Буром.
Равным образом, иа термическом разложении основано получение в чистом состоянии таких металлов, как ванадий, ниобий и тантал. Это осуществляется по способу Болтона нагреванием сильным электрическим током штаб икон из окисла или загрязненного окислом металла (ср. стр. 122 и 128).
Этот способ применим к таким металлам, окпслы которых проводят при высокой температуре электрический ток и которые цри температурах диссоциации их окислов еще не сильно испаряются.
Науку о техническом получении металла из руды называют металлургией. Помимо методов собственно получения металлов, металлургия рассматривает также способы обогащения руд, кроме того, способы очистки, которым должны быть подвергнуты еще сырые металлы, часто получаемые сначала в загрязненном состоянии (рафинирование), а также методы испытания на чистоту.
В качестве загрязняющей примеси в металлах прежде всего надо указать кислород, так как даже незначительное содержание окисла в металле в виде твердого раствора может сильно изменить физические свойства металла (например, электропроводность, ковкость). То же относится часто к азоту. Содержание окисла или нитрида удается часто определить благодаря тому, что металл при нагревании в струе хлора улетучивается в виде хлорида, или благодаря тому, что он растворяется в кислоте, которая не действует на окисел, соответственно нитрид. Для испытания на примеси, особенно носторойпих металлов, наряду с химическими методами полезно пользоваться спектральным' анализом. * ‘ .
Практически большое значение имеет испытание металлов на механические свойства. По результатам этих испытаний удается часто сделать заключение о примесях, так как они могут значительно влиять па механические свойства.
.. -- Теория металлического состояния. Для объяснения высокой электропроводности металлов издавна полагали, что внутри металлов имеются почти свободно движущиеся электроны, которые переносят электричество-Это предположение впоследствии было подтверждено большим числом экспериментальных наблюдений. Оно объясняет, например, почему проводимость тока в металлах не связана в общем случае * с переносом массы, как это имеет место при электролитической проводимости, и почему облучение светом (фотоэффект) или накаливание (эффект Ричардсона) могут обусловить выход электронов из металла. .
Если в металлах имеются свободно движущиеся электроны, то они должны вести себя так же, как молекулы газа в закрытом сосуде.
На основании представления о содержащемся в металлах «электронном? газе» (Riecke, 1898) Друде (Drude, 1902) впервые удалось теоретически обосновать правило Видемана — Франца (Wiedemann — Franz) и дать качественное объяснение термоэлектрическим явлениям.
* При известных обстоятельствах электронная проводимость может перекрываться электролитической проводимостью, см. стр. 28 и сл.
Металла и интерметаллические фаза 23
Правая» Видемана и Франца (1853) ямеег решающее значение для теории металлического состояния, аоскольку оно вскрывает связь между двумя особо значительными для металлов явлениями, а именно высокой электропроводностью и хорошей теплопроводностью. Оно говорит, что электропроводность и теплопроводность метал-лов пропорциональна одна другой или точнее: отношение (л — теплопроводность; и — электропроводность; Т — абсолютная температура) имеет одинаковое значение, независимо от природы металла. Как показал Лоренц (Lorentz, 19QS), теория Друде дает для этого отношения значение примерно на 30% меньше. Только на основании квантовой теории (волновой механики) удалось количественно верно вывести правило Видемапа — Франца из олектронной теории металлов.
G позиций классической теории газов из электронной теории металлов следует один вывод, который находится в явном противоречии с наблюдениями. Теплосодержание одноатомного газа, а также, следовательно, электронного газа в металле по классической теории составляет 3/2/?У, его атомная, соответственно молекулярная, теплоемкость при постоянном объеме, следовательно, равна 3/2Я = 2,98 кал. Так как на каждый атом металла приходится жесть степеней свободы (три для кинетической и три для потенциальной энергии атомных 'колебаний), тепловая теория, оставляя без внимания теплосодержание электронного газа, дает величину е= 5,96 кал. Как показывает опыт (правило Дю лонга и Пти), атомные теплоемкости металлов лежат вблизи 6 Ш *, в то время как они должны были бы составлять приблизительн’о 9 кал, если бы часть подводимого тепла, требуемого классической теорией, приходилась па электронный газ. Это несоответствие было устранено при применении для расчета теплосодержания газа квантовой теории в сочетании с принципом Паули.
Ферми (1926) сделал предположение, что Принцип Паули имеет значение не только для электронов в атоме или молекуле, но вообще для каждой замкнутой системы электронов, следовательно, в частности также и для «электронного газа», содержащегося в металлах **. Отсюда следует, что такой газ при достаточно низкой температуре «вырождается», т. е. имеет другое, а именно более высокое содержание энергии, чем это следовало бы по классической теории. В то время как в соответствии с классической теорией при абсолютном нуле газ должен иметь энергию пуль, согласно принципу Паули, даже при абсолютном пуле энергию равную нулю может иметь только одна частица; все другие частицы должны обладать более высокими различными значениями энергии. Отсюда следует, что внутри области «вырождения» требуемые для определенного подъема температуры затраты энергии меньше, чем в нормальном области. Область температур, в которой газ находится в вырожденном состоянии, тем больше, чем больше число частиц в единице объема и чем меньше масса отдельной частицы. Вследствие чрезвычайно небольших масс частиц электронный газ даже при самых высоких температурах остается еще вырожденным, В результате его удельная теплоемкость практически равна нулю (точнее: В/100 на 1 г-атом металла при комнатной температуре). Таким образом объясняется, почему экспериментально найденные удельные теплоемкости металлов лежат практически не выше, чем значения, рассчитанные из колебаний атомов металла.
Зоммерфельд показал (1927 и сл.), что теория Ферми о распределении энергии по отдельным частицам материи (статистика Ферми) устраняет несоответствия из электронной теории металлов, имеющиеся в её перво-
* В соответствии с правилом Д юл опта и Пти, атомные теплоемкости несколько выше 6 кал. Это происходит от того, что указанное правило относится к атомным теплоемкостям металлов при постоянном давлении. Последние вследствие того, что для расширения требуется работа, несколько больше, чем атомные, теплоемкости пря постоянном объеме (при обычной температуре на 3—10% в зависимости от коэффициента расширения и сжимаемости металла).
** Позднее Дярак показал, что обобщение Ферма можно вывести непосредственно из принципов волповой механики.
24
Глава J
+ Г0-1
О'
-1-
Атом § Метам
1 £
J
начальной форме. Новая теория металлического состояния была заметно расширена и углублена работами других исследователей (например, Nordheim, Bloch, Реierls, Borelius, A. Wilson, Brillouin). На основании этой теории в настоящее время все в большом объеме удается найти и рассчитать величины, характеризующие -различные свойства металлов, и объяснить явления, которые у них наблюдаются. Помимо электро-и теплопроводности это относится, например, к явлению термоэлектричества, контактным Потенциалам (эффект Вольта) и электронной эмиссии с раскаленной проволоки (эффект Ричардсона). Магнитные свойства металла ^могут быть поняты также только на основании этой теории (ср. г. I, стр. 342 и сл.). Все большее значение эта теория имеет также для углубленного понимания: Особенностей химических свойств, связанных с металлическим состоянием; теория металлического состояпия утвсржДает, что в. компактном металле валентные электроны находятся существенно л ином связанном; состоянии, чем в свободных .атомах.::
В то время как в свободном атоме электроны распределены на небольшом числе дискретных уровней, в компактном металле это относится только к тем электронам, которые находятся: так глубоко внутри атома, что на пих не влияют соседние атомы. Напротив, каждый верхний энергетический уровень, занйтый в нормальном состоянии свободного атома электронами, в металле растягивается в энергетическую полосу, которая состоит из множества близко расположенных один к другому энергетических уровней.
А "
Это обусловлено том, что в металлическом состоянии внешние электроны принадлежат в некотором роде всем атомам. Тогда, согласно прип-
Р и с. 2. Энергетические уровни цийу Паули, Два из этих электронов ле. могут меди находиться па одинаковом уровне. На рис. ,2 это
показано Ва примере меди. Слева приведены энергетические уровни/свободного атома меди. При атрм за нуль выбрана энергия однократно ионизированного атома. 4ь-Уросень является . высшим в нормальном состоянии атома меди. В, случае меди он содержит только один электрон (ср. стр. 387 и сл.). Расположенные над пим уровни соответствуют энергетическим состояниям «возбужденного» атома, т. е. это уровни, на которые электроны могут подниматься л результате подвода зпергйи. К самому высшему из них, расстояние до которого от 4т-уровня соответствует энергии ионизации атома (ср. т. I, стр. 136 и ел.), примыкает непрерывная энергетическая полоса, что объясняется, тем, что оторванный электрон, помимо энергии ионизации, может обладать еще кинетической энергией, причем последней — в любом количестве. Как следует из правой части рис. 2, благодаря тому что атомы меди соединяются в решетку металла, чл-уревень растягивается в энергетическую полосу, состоящую из множества близко расположенных уровней. В случае, приведенном на рисунке, то же относится и к Зй-уровню, в то время как более глубоко лежащие уровни пе’изменяются. Отсутствующие в основном состоянии атома энергетические уровни-, которые расположены выше 4s-уровня, также расширяются в полосы, причем последние в случае меди располагаются друг над Другом так, что уже непосредственно выше 4юнолосы Для электронов допустимо практически любое значение анергии.
5
§ -®
о Bbictuuu.
: занятый уровень
----Зр М-
----3s уровень
Е &-100-&
Зр
3s
___2р
~ 2s уровень
2р '2s
-1000-
Я-
,s урмеяь
10000-
is
Металлы. w интерметаллические фазы 25
Расширение энергетических уровней, содержащих валеитпыс электроны, в энергетическую полосу, приводит, между прочим, л, тому, что наименьшая энергия, которая должна быть затрачена, чтобы отщепить электроны от твердого/металла, становится существенно меньше, чем энергия ионизации свободного атома. Эту энергию, так называемую работу выхода, можно определить измерением фотоэффекта или термо-эмпсспопного аффекта. Она составляет для меди 4,3 за, в то время Как работа ионизации, атома меди составляет 7,7 зв. Следовательно, верхний край полосы, содержащей 4«-электроны, в металлической меди лежит на 3,4 эв выше, чем соответствующий уровень в атоме меди.
В нормальном состоянии (при 0° К.) электроны находятся только на таких энергетических полосах, которые соответствуют энергетическим:: уровням, занятым электронами, в нормальном состоянии свободного атома. Если в такой энергетической полосе все уровни заняты электронами, то эти электроны (пока они находятся на полосе) не могут транспор- ; тировать ни электричество, ни теплоту. Несмотря на то что с точки зрения корпускулярной теории электроны считаются подвижными, они не могут ускоряться электрическим полем. В противном случае это означало бы получение энергии. А при получении энергии они должны были бы оод-няться на энергетический уровень, который ужезанят другим электроном (исключая случай, когда добавленной энергии было бы достаточно,. чтобы: перевести их на другую полосу), Это, однако, невозможно но принципу \ Паули.
Если Же только половина- энергетических уровней одной полосы занята электронами, то другая половина может принимать электроны с большей энергией. Тогда, следовательно, практически вес электроны, находящиеся на полосе, могут быть ускорены приложенным напряжением и тем самым они будут способствовать прохождению тока. Если занято более иол овины энергетических уровней, то число электронов, которое может способствовать прохождению тока, меньше чем :в предыдущем слу/ чае. Если занято менее половины энергетических уровней, то это число,: естественно, также меньше. Следовательно, электропроводность металла зависит не от общего числа свободно Движущихся электронов в единице объема металла, а от числа таких электронов, для которых имеются еще незанятые энергетические уровни. Это число называют эффективным электронным числом металла.
Для зависимости удельной электропрдводнбети и от эффективного электродного числа на каждый кубический сантиметр металлов Щфф теория дает следующую формулу: " Г
.::н
где е, т, а и Г — соответственно заряд, масса, скорость и средняя свободная длина пробега электронов. «Средняя свободная длина пробега!) в: смысле корпускул яркой теории означает в волновой механике величину, которая намеряет способность металла образовывать в промежутке между атомами стоячие Электрические Волны. Эта способность уменьшается в результате неупорядоченного внедрения в металл посторонних атомов (ср. стр. 30 и сл.), искажений решетки (см. стр. 36 и сл.), а также в резул/,-тате колебаний атомов около их среднего положения в кристаллической решетке. Так как эти колебаний возрастают с повышением температуры, металлическая проводимость при этом падает. Напротив, уменьшение проводимости, вызываемое посторонними атомами и иными искажениями решеток, не зависит от температуры. Чем сильнее искажена решетка металла, тем более превышается ее нормальное зависящее от температуры сопротивление добавочным сопротивлением, независящим от температуры.
То, что содержащиеся в металле загрязнения повышают его удельное сопротивление за счет независящей от температуры составляющей (причем Даж4тогда, когда речь: идет о небольшом количестве вримеса металла,’ собственная проводимость которого больше, чем проводимость основного металла), было установлено Матиссеном уже в 1864 г. Как позднее было показано, правило Матисс спа верно только в случае таких
26
Гласа 1
примесей, которые образуют твердые растворы, т. е. включаются в металлическую решетку (в общем случае неупорядоченно, ср. стр. 31). Напротив, удельная электропроводность гетерогениоео сплава (т, е. смеси различных кристаллов) по существу аддитивно складывается из удельных электропроводностей составных частей.
Удельная теплопроводность металла X в соответствия с той же теорией выражается формулой
<2>
где Т — абсолютная температура, а А- — так называемая константа Больтцмана, т. е. частное от деления газовой постоянной R на число Авогадро N. Бели разделить уравнение (2) на уравнение (1), то получим
X л5 ( й: V ,,,,
v/-=—UJ' (3)
Это уравнение выражает правило Видемана — Франца. После подстановки числовых значений * в правую часть уравнения (3) получают 2,442-10?. Отсюда при комнатной температуре (Т = 273,15 + 18) оказывается Х/х = 291,15-2,442-108 = 7,110.1010, В качестве среднего значения из найденных для большого числа хорошо проводящих металлов было рассчитано отношение Х/х = 7,11-1010. Следовательно, уравнение (3) для фактора пропорциональности правила Видемана — Франца дает верное значение.
Если удельные электропроводности отдельных элементов умножить на их атомные объемы и полученные таким образом «атомные электропроводности» нанести на систему координат в зависимости от порядковых чисел, то получим при этом периодическую кривую, аналогичную кривой атомных объемов (т. 1), с той разницей; что на вершинах пиков, помимо щелочных металлов, оказываются Си, Ag и Ап. Нашедшая в этой кривой отражение повышенная проводимость металлов обеих подгрупп первой группы периодической системы, согласно электронной теории металлов, объясняется следующим образом: Свободные атомы металлов главной и побочной подгрупп первой группы содержат на внешней оболочке по одному электрону, причем этот электрон имеет побочное квантовое число I = 0. Однако, согласно принципу Паули, оболочка с побочным квантовым числом 1=0 может принять два электрона (ср. т. I, стр. 145 и сл.); следовательно, у названных элементов в атомарном состоянии эта оболочка занята только наполовину, в то время как все нижние оболочки заняты полностью. Квантовомеханический расчет распределения потенциала в решетках этих металлов показывает, что энергетическая полоса, соответствующая этой оболочке в твердом металле, тйкже заполнена только наполовину. Поэтому у этих металлов число способных к переносу тока электронов «.Эфф практически равно числу п валентных электронов, в то время как у других металлов иафф постоянно меньше, чем п.
Полупроводники и изоляторы. Как уже было отмечено, вещества, у которых в твердом состоянии все энергетические полосы полностью запяты,' не могут, вообще говоря, проводить электрический ток. Однако, если немного выше самой верхней энергетической полосы, да которой в нормальном состоянии находятся валентные электроды, лежит другая полоса, незанятая в нормальном состоянии, то электроны
.=l^=1,6o2.w-»; ^у=«(й».101у=2,«2.10,
Металлы и интерметаллические ^а.зы
27
могут в результате подвода тепла переместиться на эту энергетическую полосу, ибо, .котя электронный газ н «вырожден», его способность поглощать тепло не равна нулю, а только лишь очень мала по сравнению с такой способностью нормальных газов. Иона электроны находятся на более высокой энергетической полосе, они могут переносить электрический ток. То же относится и к электронам энергетической полосы, с которой: вышли эти электроны и которая оказывается тогда частично незанятой. Чем выше температура такого вещества, тем больше электронов переходит на более высокий энергетический уровепь и соответственно тем больше его электропроводность. В таком случае говорят о полупроводнике и — если речь идет о простом веществе — о полуметалле. Для последнего характерно, что его проводимость не падает с ростом температуры, как у настоящих металлов, а растет.
G ростом расстояния между обеими указанными энергетическими полосами очень сильно уменьшается число электронов, которые перемещаются на верхнюю полосу в результате возрастания температуры. При большом расстоянии между энергетическими полосами при обычной и умеренно высоких температурах но наблюдают вообще никакой заметной проводимости. В таком случае говорят о непроаоднике (изоляторе), пли, если речь идет об элементарном веществе, о неметалле. Поэтому непроводники отличаются от полупроводник ок только в количественном отношении. Резкой границы между ними провести нельзя (Wilson). Далее следует, что в твердых непроводниках может содержаться также итого я даже весьма много свободно движущихся электронов, как и в хорошо проводящих металлах. Не исключается, конечно, возможность того, что имеются также твердые непроводники (соответственно неметаллы), недостаточная проводимость которых обусловлена отсутствием в решетке свободно движущихся электронов или их исчезающе малым числом.
Чем больше расстояние менаду самым верхним заполненным уровнем и самым нижним незаполненным уровнем в свободном агпоме, тем больше будет в общем случае расстояние в металле между соответствующими опер готическими полосами. Как известно, это можно измерить определением наименьшего напряжения, которое должно быть приложено для «возбуждения.» ат’ома (ср. т. I, стр. 137). Борелнус (Вore-ling, 1939) указывал на то, что все элементы, наименьшее напряжение возбуждения которых лежит существенно выше d as, являются непроводниками. Элементы с наименьшим напряжением возбуждения вблизи б эв являются полупроводниками, и наконец, те элементы, наименьшее напряжение возбуждения которых лежит существенно ниже 6 ав, являются металлами.
Помимо возрастания электропроводности с увеличением температуры, для полупроводников характерно также наблюдаемое иногда очень сильное увеличение проводимости при добавлении незначительных примесей (в этом отношении полупроводники ведут себя противоположно истинным металлам).
Для объяснения этого явления полагают, что посторонние атомы дают дополнительные (дискретные) эвергетичеекяе уровни, которые располагаются между энергетическими полосами, что, естественно, облегчает переход электронов на более высоно-лежащую полосу.
Сверхпроводимость. При очень глубоком охлаждении многих металлов у них неожиданно и в чрезвычайно сильной степени возрастает электропроводность, так что эти металлы в таком состоянии практически не оказывают заметного сопротивления прохождению тока. Это явление называют сверхпроводимостью. В замкнутой цепи, которая образована металлом в состоянии сверхпроводимости возбужденный однажды ток продолжает длительно течь, не требуя при этом никакой электродвижущей силы, чтобы его поддерживать. Сверхпроводимость была открыта в 1911 г. Камер-линг Оннесом (Kamerlingh Onnes) па npimepe ртути, удельная электропроводность которой при 4,2° К скачкообразно падала до неизмеримо малой величины. Позднее это явление наблюдалось также у Ga, In, Tl, Sn, Pb, Ti, Th, Nh, Та и Mo. Сплавление указанных металлов с металлами, которые не проявляют сверхпроводимости, может также способствовать возникновению этого явления. Соединения, обладающие металлической проводимостью (нитриды, карбиды, силициды и бориды некоторых переходных элементов), при низких температурах также могут становиться сверхпроводниками.
Прятана возникновения сверхпроводимости еще не ясна. Многое свидетельствует о том, что в состоянии сверхпроводимости перенос тока обусловлен не теми злек-тронами, которые обычно являются в металле свободно движущимися, а другими, которые в другом случае Не участвуют в движении тока.
Для глубокого понимания природы металлического состояния подробное изучение сверхпроводимости, по-нидимому, может иметь большое значение. Объяснение сверхпроводимости прежде всего очень важно для понимания магнитных свойств металлов; было показано, что между этими свойствами л явлениями, связанными со сверхпроводимостью j имеется тесная связь.
28 Глава J
Возникновение электрической сверхпроводимости но оказывает влияния ла теплопроводность.
Теоретически для идеального металлического монокристалла электропроводность при. 0а К бесконечно велика, а удельное сопротивление, следовательно, равно нулю. У реальных металлов, однако, вследствие имеющихся у них обычно искажении решеток к. сопротивлению, зависящему от температуры, большей частью добавляется сопротивление, которое, согласно правилу Матиссспа (ср. стр. 25), не исчезает нрп . абсолютном нуле (достаточное сопротивление»). Однако в состоянии сверхпроводимости исчезает не только сопротивление, зависящее от температуры, ;цо и остаточное сопротивление; У идеального монокристалла основное различие между сверхпроводимостью и обычной электропроводностью заключается в том, что последняя при абсолютном нуле непрерывно стремится к бесконечности (что не только следует из теории, но и вытекает также из результатов наблюдений, если иа найденного сопротивления вычесть остаточное сопротивление), в то же время сверхпроводимость наступает скачкообразно, так что при охлаждении до нескольких сотых градуса сопротивление-мота.чла падает сразу от заметной до неизмеримо малой величины.
Электролитическая проводимость тока в металлах. Так как валентные электроны могут почти свободно перемещаться между ионами металлический решетки, они, по существу, и обеспечивают перепое тока, однако последнее осуществляется:не исключительно электронами. Определенная часть -токоперенбеа — правда, чрезвычайно-небольшая — может быть также связана с движением ионов металла:: Поэтому при : пропускании тока через сплавы можно иногда наблюдать (когда работают с очень высокими" плотностями тока) концентрационное передвижение составных частей сплава^ т. е. процесс, соответствующий электролизу. Количество металла (е-атол1/фара&ей)г. перемещающееся в направлений положительного тока (к катоду), обозначают как число переноса соответствующего металла в сплаве*. Согласно измерениям Поста. (Jost, 1935—193G) и Зейта (Sei th, 1934), для Сн в сплаве Си-Ли оп ос оставляет, па пример, 7,4- 10-U (при750°), для Pd в сплаве Рй-Лц 1,6- IO"11 (при 900°) и для Ан в сплаве Pt-Aw с небольшим содержанием золота 1,3- 10_i°; (при 180°). Существенно более сильные эффекты наблюдаются в жидких сплавах (Кгыпанп, 1923 и сл.; Schwarz., 1931 и сл.). Это объясняется тем, что в жидиих, соответственно расплавленных, металлах подвижность ионов существенно больше, чем в твердых металлах (что можно показать непосредственно измерением скорости диффузии), в то время как подвижность здект троим с понижением упорядоченности атомов, и прежде всего при переходе из твердого в жидкое состояние, уменьшается.
Ранее часто считали, что движение составных частей Сплава к катоду или аноду зависит От силы их электроположительного характера,. Однако многие экспериментальные результаты трудно объяснить на основании этого предположения; например, при электролизе разбавленной амальгамы и К обогащаются-яа аноде, несмотря на то что они гораздо более электроположительны, чем ртуть. Согласно Вагнеру (Wagner С., 1932—1933), в твердых металлах решающее влияние на движение оказывает, по-видимому, заряд (электрохимическая валентность) составных частей сплава;: наряду с этим существенную роль играют, видимо, также их диффузионные свойства . Для жидких сплавов в хорошем совпадении с опытом находится, пб-видимому, теория1 Шварца (Schwa г г К,, 1933), в соответствии с которой каждый ион металла с большей «плотностью заряда» (отношение заряда к объему) Должен перемещаться к катоду.
ТВЕРДЫЕ РАСТВОРЫ И IIII ТЕРМ ETA.I,; II14 ЕС КИЕ ФАЗЫ
Особой природой металлического состояния определяется и то, что для образования соединений между металлами характерны иные закономерности;, чем для образования соединений, между неметаллами или металлами и неметаллами. Как ужо было отмечено (см. т. I, гл. ijl); интерметаллические соединения часто образуются не в соответствии с простыми стехиометрическими соотношениями и законом постоянных проиорний; их состав часто изменяется внудри в достаточно широких пределах («пс-дальтошгды» или «бертоллиды»). Правда, мея;ду интерметаллическими соединениями и другими типами Соединений нет резкой границы. Даже среди соединений металлов с неметаллами известны такие, состав которых может колебаться. Многие соединения металлов с неметаллами или даже
* Если число переноса относится к металлу, который обогащается на аноде, то оно имеет отрицательный знак.
Металлы w интерметаллические фалы, - •’- - 29
соединения неметаллов между собой по своему составу, структуре, а отчасти также по своим свойствам родственны типичным интерметаллическим соединениям. С другой стороны, встречаются интерметаллические соединения, которые ио своему составу вполне соответствуют солям и солеобразным соединениям, т. е. нормальным валентным соединениям; они могут быть также близки им и по свойствам. Однако в общем для соединений металлов последнее не типично. Это относится прежде всего к сплавам, в образовании которых участвуют металлы побочных подгрупп периодической системы. В этих сплавах часто встречаются вещества, которые так сильно отличаются от типичных химических соединений, что возникает вопрос — законно и целесообразно ли говорись о них как о химических соединениях (см. стр. 3.7 и сл).
В качестве общего обозначения веществ, встречающихся в интерметаллических системах и структурно отличающихся от исходных составных частей системы, а следовательно, также от их твердых растворов, употребляют название интерметаллические фазы. Многие из этих интерметаллических фаз родственны твердым растворам и ранее отчасти рассматривались в качестве последних. Поэтому, прежде, чем подробнее рассматривать интерметаллические фазы, следует сначала более детально обсудить свойства металлических твердых растворов.
ТВЕРДЫЕ РАСТВОРЫ
Твердые растворы называются так потому, что при их образовании в рещетке одного вещества без изменения ее структуры располагаются ; атомы или поны другого вещества, причем внутри границ, в которых происходит образование твердого раствора^ атомы различного вида могут находиться в любых соотношениях.
Например, в кристалле серебра все атомы серебра можно поел вдова телйпо заменить па атомы золота, и структура решетки при этом не изменится.;Когда, наконец, последний атом серебра будет заменея па атом золота, приходят, к чистому золоту, структура решетки которого совпадает со структурой решетки серебра. В этом случае говорят об образовании «неограниченного твердого раствора». Напротив, атомами меди удается заменить только Небольшую часть атомов серебра в кристаллической решётке серебра (при комнатной температуре до 0,2 ат.%), а в решетке меди только нёбольшутр часть атомов меди можно заменить на атомы серебра (до 0,03 ат.%). ;В этом случае имеют дело с образованием «ограниченных твердых растворов»; в интервале концентраций 0,2—99,97 ат.% меди при комнатной температуре существует область несмешивае~ .4!“ н;и .
Смотря по тому, происходит ли включение посторонних атомов и кристаллическую решетку так, что отдельные атомы основной решетки замещаются ими, или так, что посторонние атомы внедряются в пустоты основной решетки, различают твердые растворы замещения * **’тл твердые растворы внедрения (рис. 3, стр. 30).
* Приведенные данные имеют значение для равновесного состояния, С повигш’-нием температуры заметно возрастает взаимная Смешиваемость Си и Ag. (ср. рис. 5, стр. 34). При быстром охлаждении расслаивания не происходят или происходят пспо.т-ностыо,.тогда говорят о ("пересыщенных твердых растворах», которые содержат суще-ствеиио больпщ посторонних атомов я основной решётке, чем это допускается в равновесном состоянии. 'Гакле метастабильпые пересыщенные твердые растворы встречаются в сплавах очень часто (ср. стр. 46 и сл.). Обычно вследствие незпачнтельпой. скорости диффузии оказывается невозможным пблностыо. устранить пересыщение при нормальной или несколько повышенной температуре. .
** Твердые растворы замощения автор называет «сйсшанпыми кристаллами» (Mif-clikrist.aile), а твердые растворы внедрения—«твердыми растворами»,-В переводе введен единый термин «твердый раствор».— Прим. ред.Г
30
Глава, 1
Твердые растворы замещения встречаются гораздо чаще. Образование твердых растворов внедрения наблюдают главным образом при включении в металлическую* решетку атомов неметаллов с небольшими радиусами, таких, как С, N, Я. В обоих
• -Атомы основной решетки
о О-Посторонние атомы
Рис. 3. Схема образования твердых растворов путем внедрения «чужих» атомов, в решетку,
а — основная решетка; б — твердый раствор внедрения; в — твердый раствор замещения; г — с-верхструктурная фаза.
случаях константа решетки изменяется. Изменение происходит непрерывно и пропорционально числу включенных посторонних атомов, а при замещении, кроме того,, пропорционально разности радиусов постороппего и основного атомов (Vegard, 1921).
Обычно посторонние атомы включены в основную решетку полностью-неупорядоченно (ср. рис. 3, бив). В таких случаях удельная электропро-
водность падает с повышением содержания посторонних атомов.
6/
6,0
55
50
ж
С позиций квантовой механики это* объясняется тем, что в результате нерегулярного расположения посторонних атомов-затруднястся образование стоячих волн проводящих электронов металлической решетки, т. е. величина I в уравнении (1) [стр. 25] уменьшается (Nordheim, 1928). Подобно электропроводности при включении, посторонних атомов изменяется растяжимость. Сплавы двух металлов, неограниченно смешивающихся в твердом состоянии, при концентрации 50 ат.% проявляют вследствие этого минимум электропроводности и растяжимости. Сильно уменьшающейся электропроводностью твердые растворы отличаются от гетерогенных смесей. Этовндно-па примере сплава Cu-Ag, прнведеппом на рис. 4. В области образования твердых растворов * добавление меди к серебру или добавление серебра к моди вызывает силь-
ное понижение удельной электропроводности. Напротив, в области гетерогенности электропроводность равномерно возрастает от электропроводности насыщенного твердого раствора меди в серебре до электропроводности насыщенного твердого раствора серебра в меди **. Это происходит в соответствии с тем правилом, что удельная электропроводность гетерогенной смеси кристаллов аддитивно складывается из удельных электропроводностей компонентов (в рассматриваемом примере твердых растворов.
о
Си
Рис. 4.
20 40 60
Серебро, об. %
Электропро водпо си. Cu-Ag,
80
/да
Ад
сплавов
е
I !v
* Область образования твердых растворов, приведенная на рис. существует при 700°, поэтому определение электропроводности (при 0°) в этой области проводили, быстро охлаждая сплав от 700°.
** При температуре измерения электропроводности рассматриваемые твердыв-растворы иересыщепы. См, сноску*,
Металла и интерметаллические фазы
31
Cu-Ag и Ag-Сн) (ср. стр. 26). Поэтому на основании измерений электропроводности удается часто особенно просто и точно установить границы областей образования компонентами сплава твердых растворов.
Для металлов характерна ярко выраженная склонность к образованию твердых растворов. Неограниченная или очень широкая область смешиваемости в твердом состоянии особенно часто наблюдается для металлов побочных подгрупп периодической системы. В бинарных сплавах, оба компонента которых принадлежат к главным подгруппам периодической системы, непрерывные ряды твердых растворов образуются лишь в редких случаях (ср. табл, на стр. 621—623 и 630 т. I).
Как известно, к твердым растворам солеобразных соединений применимо правило Гримма, Которое говорит, что химически аналогичные вещества только тогда могут образовывать твердые растворы (при обычной температуре), когда разница в радиусах взаимно замещающих ионов не слишком велика (согласно опыту пе более 5%); это правило не имеет общего значения для твердых растворов металлов. НапримерСп, радиус атома которой на 10% меньше радиуса Ан, образует с золотом непрерывный ряд твердых растворов, в то время как с Ag, которое имеет почти такой же радиус атома, как Ли, медь только незначительно смешивается.
Сверхструктуры. В некоторых случаях в твердых растворах, состав которых соответствует простому стехиометрическому соотношению или приближается к таковому, при определенных температурах существует упорядоченное распределение обоих типов атомов в узлах кристаллической решетки (см. рис. 3,г), в то время как при более высоких температурах упорядоченное распределение нарушается. Так как упорядоченное размещение различных типов атомов в узлах решетки приводит к появлению дополнительных интерференционных линий в рентгенограммах (ср. стр. 4.2), то в таких случаях говорят об образовании «сверхструктуры».
С образованием сверх структуры связано неожиданное и достаточно сильное возрастание электропроводности. Последнее объясняется тем, что при этом устраняется затруднение для образования электронных волн, вызванное неупорядоченным распределением чужих атомов. Далее, образование сверхструктуры сопровождается в оз-pa ста пнем плотности (правда., только незначительным) (Grube, 1931). Растяжимость при упорядоченном распределении атомов также больше, чем при неупорядоченном.
Сверхструктуры часто имеют более низкую кристаллографическую степень симметрии, чем соответствующие твердые растворы с неупорядоченным распределением атомов. Например, у кристаллов состава АпСн переход иа состояния с неупорядоченным распределением атомов» упорядоченное состояние вызывает сжатие одной из трех Кристаллографических осей, т. е. кубическая гранецентрированпая решетка твердого раствора при образовании сверх структуры превращается в тетрагональную гранецеи-трированную решетку (с0 : а0 = 0,93). Иногда превращение твердого раствора в сверхструктуру приводит к образованию решетки, которая типична для солеобравных соединений. Например, сверхструктура PdCu обладает решеткой типа иодлда цезия (ср. стр. 239 т. I), Она возникает из кубической гранецентрированной решетки соответствующего твердого раствора благодаря тому, что с возникновением упорядоченного распределения атомов ось (.'первоначальной решетки укорачивается на 0,7 но сравнению с осью а.
Сверх структуры занимают промежуточное положение между твердыми растворами и химическими соединениями. Их рассматривают часто как особый случай образования твердого раствора. Однако, так как с переходом из неупорядоченного состояния к состоянию упорядоченно • го распределения атомов связаны скачкообразные изменения свойств, а, кроме того, при возникновении сверхструктуры в принципе наблюдается простое стехиометрическое соотношение компонентов (по меньшей мерс для идеального случая, ср. пиже), сяерхструкгуры рассматривают
32 Глава 1
так же часто как химические соединения. Однако в отличие от собственно химических соединении (ср. стр. 39) их целесообразно обозначить как «сверхструктурные соединения».
Примеры сверхструктур унизаны в табл. 32 и 43 (стр. 264 и .394) , в которых формулы сверхструктур заключены в квадратные скобки.
Помимо сплавов, состав которых соответствует простым числовым соотношениям, определяющимся полностью упорядоченным распределением атомов, возникновение сверхструктур наблюдается также в таких сплавах, состав которых приближается к составу идеальной сверхструктуры. Следовательно, свойства, характерные для сверхструктур, еще остаются (хотя и ослабленные), если в упорядоченной кристаллической решетке часть атомов одного компонента заменить па атомы другого компонента. Однако с увеличением включения атомов другого компонента в решетку (происходит ее раиунорядоченпе) свойства, характерные для сверхструктуры, скоро исчезают.
Границы сопротивляемости твердых растворов. Бинарные твердые растворы, один пз компонентов которых в противоположность другому практически не подвергается действию химических реагентов, обнаруживают, как нашел в 4919 г. Тамман., так называемые границы сопротивляемости. То ость химическое действие имеет место только тогда, когда содержание химически неустойчивого компонента превышает опр идо,генное значение. Это значение зависит от природы химического процесса, однако очень часто оно кратно величине 10(i/8 ат.% (правило c/g моля). Например, сплав Cn-Ag подвергается действию желтого сульфида аммония только в том случае, если он содержит моиео ат.% Ан. С раствором нитрата серебра реагируют с оса-
ждением серебра только такие сплавы Cu-Лп, которые содержат менее 1М?8 ат.% Ап. Причиной возникновения границ сопротивляемости является то, что сплавы построены из однородных кристаллов п полностью гоиогенизованы Достаточно продолжительным нагреванием и, Кроме того, что реакции изучаются при таких температурах, при которых практически пе происходит перемещения атомов в кристаллической решетке (ср. гл. 19).
В характере изменения гальванических потенциалов растворения твердых растворов также проявляются границы сопротивляемости, В то время как для сплавов, состоящих из смеси различных веществ, потенциал растворения не зависит от соотношения количеств Этих веществ в гомогенных сплавах, состоящих из однородных кристаллов, проявляется зависимость потенциала растворения от состава. В последнем случае потенциал растворения часто скачкообразно возрастает прн определенном соотношении концентраций компонентов от значения, соответствующего менее б да г сродни му комноивпту, до значения, соответствующего более благородному компоненту; так, твердый раствор Mu-Cu при концентрации Сп менее 50 ат. % обнаруживает потенциал растворения чистого марганца, а' при концентрации Си более 50 ат.% — потенциал растворения меди. Сплавы Сп-Аа при содержании до 75 ат,% Си имеют потенциал растворения такой же, как у чистого золота; с дальнейшим ростом содержания мед и потенциал растворения изменяется почти линейно вплоть до значения, соответствующего чистой меди. Сплавы Ар-Ап даже с высоким содержанием золота имеют потенциал растворения чистого серебра; однако если перед измерением потенциала сплав погрузить в азотную кислоту, то он даже при высоком содержании серебра Покажет потенциал чистого золота. Из итого следует. что потенциал неблагородных металлов проявляется тогда, когда их атомы находятся на п о вер хпоти твердого раствора. Сплав Ag-An может обладать любым нотеппиалом золота или серебра,— смотря по тому, удалены ли атомы серебра с его поверхности азотной кислотой или. они после прокали-ваппя снова появились на поверхности в результате диффузии. В случае сплавов Cu-Ag достаточно уже их внести при измерении потенциала в раствор, содержащий воздух, чтобы атомы неблагородного компонента были удалены с поверхности То же относится к сплавам Мп-Сн. Объяснить появление скачка потенциала можно тем, что лежащие под поверхностью атомы только тогда теряют активность, когда узлы кристаллической решетки поверхности после удал спил атомов неблагородных компонентов заполнены еще достаточно плотно. Соответственно объясняется также существование границ сопротивляемости по отношению к химическим реагентам
Тамман предположил, что условием защитного действия атомов благородных металлов является их упорадоченное расположение в кристаллической решетке- Вак для твердых растворов Си-Ап. так и в ряде других случаен рентгенографическое нсс.ттё-довапие подтвердило это предположение, обнаружив линии сверхструктур. Однако в ряде твердых растворов, которые также проявляют границы сопротивляемости (например, твердый раствор Ag-Au при 25 и 50 ат. % Ан), рентгенометрически удалось установить отсутствие сверх структур. Таким образом, вопрос о взаимосвязи траппц
Металлы и интерметаллические фазы.
33
сопротивляемости и упорядоченного распределения атомов требует еще дальнейшего выяснения.
1 Гомогенные вещества, у которых не только нет упорядоченного распределения
атомов различного вида в узлах кристаллической решетки, но и вообще отсутствует правильное расположение атомов в пространстве (т. е. стекла), не обнаруживают границ сопротивляемости.
ИНТЕРМЕТАЛЛИЧЕСКИЕ ФАЗЫ
Интерметаллическими фазами называют такие виды встречающихся в сплавах кристаллов, которые отделены от кристаллов составных частей сплавов фазовыми границами *. Следовательно, интерметаллические фазы в структурном отношении нрерывпо отличаются от составных частей сплавов и их твердых растворов. В соответствии с этим они могут появляться только в таких областях концентраций, в которых при данной температуре пет полной смешиваемости между составными частями в твердом состоянии, т. е. там, где твердые растворы не являются стабильными. На диаграммах состояния сплавов интерметаллические фазы отделены большей частью от чистых металлов или их твердых растворов (а также, если образуется несколько интерметаллических фаз, я друг от друга) более или менее широкими «областями гетерогенности», т. е. областями концентраций, в которых затвердевший сплав состоит из смеси кристаллитов различного состава и структуры.
Примерами подобного рода интерметаллических фаз являются прежде всего интерметаллические соединения, приведенные на диаграммах состояния в гл. 13 т. I. В указанных там случаях речь идет всегда об интерметаллических фазах, которые выделяются непосредственно из расплавов. Одпако такие фазы могут образовываться и путем последующих превращений первоначально выделившихся кристаллов, если эти кристаллы, соприкасаясь с расплавом при охлаждении, взаимодействуют с ним с образованием Кристаллов другого состава и структуры («перитектическое превращение») или если превращение с изменением состава и структуры наступает только после окончания затвердевания. Достаточно распространены случаи, когда выделившиеся из расплава фазы при дальнейшем охлаждении претерпевают структурные si изменения без изменений состава. Этот процесс соответствует полиморфным превра-
щевиям обычвых соединений. Это является переходом к тому случаю, когда твердый 3 раствор нри охлаждении, не изменяя своего состава, претерпевает структурное пре-
вращение, приводящее к появлению при более низкой температуре интерметаллической фазы в той области концентраций, в которой при более высокой температуре существуют только твердые растворы.
На диаграммах состояния бинарных сплавов, оба компонента которых принадлежат к главным подгруппам периодической системы, как правило, интерметаллическим фазам соответствуют совсем узкие области гомогенности, отделенные (от областей гомогенности соседних фаз) большей частью широкой гетерогенной областью. В сплавах, в образовании которых участвуют металлы побочных подгрупп, встречаются часто интерметаллические фазы с широкими областями гомогенности, между которыми имеются лишь сравнительно узкие, в отдельных случаях едва заметные гетерогенные области **.
* Общим термином, который можно Применять также для систем с неметаллами, является «нромежгргю'швя фаза».
* * Областью гомогенности фазы называют ту область концентраций и ту область температур, внутри которых гомогенная фаза стабильна. К области гетерогенности, помимо областей концентраций, в которых затвердевший расплав состоит из смеси кристаллов различного состава и структуры, относят, копечпо, также и области, заключенные между кривыми ликвидуса и солидуса, в которых сосуществуют расплав и кристаллы.
3 Г. Реми
34
Глава 1
На рис. 5 приведены области состояния некоторых затвердевших бинарных сплавов при определенных температурах. Области гомогенности отдельных фаз (на рисунке обозначены греческими буквами) с попи-жепием температуры могут расширяться или сужаться. Области гомогенности твердых растворов чистых компонентов с уменьшением температуры большей частью сужаются. Области, гетерогенности на рис. 5 заштрихованы.
Принятый на рис. 5 способ изображения, который дает только соответствующий определенной температуре разрез диаграммы состояния сплавов, упрощает ее рассмотрение. Особенно это удобно при сложных случаях. Например, благодаря различной
а. Си-Ад при 700°
в. Sb-Sn при 200°
О' 10 20 30 40 50 60 70 80 90 100
Sb Sn,am.% Sn
<3. W-Pt при 7600° Гетерогенная область
г» IBS , , z', , | о Ъ io 30 40 SO 00 70 SO SO 100 W Pt, am. % Pt
г. Ni-Zn при 200° $
0 10 20 30 40 50 60 70 80 90 100 №' Zn, am. % Zn
Pae. 5. Интервалы состояний затвердевших бинарных сплавов.
а и б —сплавы с ограниченной смешиваемостью составных частей в твердом состоянии, но без интерметаллических фаз; в —сплав с одной интерметаллической фазой (р); г—сплав с двумя интерметаллическими фазами (0 и у).
ширине области расслаивания становится очень ярко выраженной существенно различная степень образования твёрдых растворов компонентами в системах Сп-Аг И W-Pt.
Интервал: концентраций при определенной температуре в дальнейшем будем называть интервалом состояния. Путем расположения в ряд интервалов состояния сплава при определенной температуре получают, следовательно, разрез диаграммы состояния для данной температуры («разрез состояний#). Если рассматривать приведенные на рис. 5 разрезы состояний в качестве общей схемы, то отсюда можно найти разрезы состояний любых бинарных сплавов, варьируя ширину интервалов состояний. Если, например, на рис. 5,а интервал a-фазы свести к нулю или почти к нулю, то получим тнп сплавов системы Sn-Pi), диаграмма состояния которой показана в т. I на рис. 104. В случае, приведенном на рис. 5, а, интервал гомогенности б-фазы (Zn) равен нулю. Цинк не может в заметном количестве растворить никель с образованием твердого раствора; это видно из того что область гетерогенности у + б (или у + Zn) непосредственно подходит к 100% Zn. Соответственно в тех случаях, когда образуются соединения постоянного состава, т. е. интерметаллические фазы с бесконечно малым интервалом гомогенности, два различных интервала гетерогенности граничат ненос родственно Друг с другом. Их место соприкосновения дает тогда химический состав соединения. Рис. 5, а показывает интервалы состояния твердых сплавов типа Mg-Pb, а рис. 5, г — типа Mg-Cu (ср. т. I, рис. 106 и 107), если в обоих случаях области гомогенности интерметаллических фаз свести к нулю. Естественно, этот способ изображения можно полностью использовать для систем с любым количеством интерметаллических фаз.
Интерметал.тичеекпе фазы в сплавах меди. В качестве примера сплавов, в которых образуется большое число интерметаллических фаз (причем часто с заметными областями гомогепности) на рис. 6 приведены разрезы состояния отдельных сплавов меди, особенно важных в техническом отношении. Фазы различных сплавов, совпадающие в структурном отношении, т. е. в отношении расположения атомов в кристаллической решетке, на рисунке обозначены одними и теми жо греческими буквами без
Металлы и интерметаллические фаза
индексов *. Прежде всего видно, что в этих системах с падением температуры происходят более или менее значительные изменения •*. С изменением температуры не только сдвигаются границы фаз, но с понижением температуры исчезает часть устойчивых при высокой температуре фаз и вместо них возникают новые. Чтобы проследить характер этих изменений в подробностях, следует обратиться к полным диаграммам состояния. Такая диаграмма состояния системы Cu-Zn приведена на рис. 45, стр. 400.
Как показали Вестгрен и Фрагмен, между сплавами, приведенными на рис. 6, имеется близкое родство. Из рисунка следует, что в них имеются несколько структурно
0 10
Си
а. Си-Zn при 580°
яе
Расплав
d. Cu-Zn при 200
20 30 40 SO SO 70 SO 30 100
Zn, вес. % Zn
в. Cu-Sn при 600° de
а>.
О Си
10 20 30 40 50 60-7, Zn , вес.%
г. Си-Sn при 200'
7 е Ул
100 Zn
о да го зо 40 so 60 7о so Hoioo
Си Sn,eec.% Sn
д. Си-Al при 700“ fl Г&з__________ ____________
О 10 20 30 40 50 60 70 SO 90 Ю0
Си Sn,eec.% Sn
е. Си-Al при 200°
«>.
ИН»1
[ 06 ркя ИРасплав , j О 10 20 30 40 50 60 70 80 90 100 Си Al, асе. % Al
О 10 20 30 40 SO 60 70 SO 90 100
Си Al,вес.% Al
Рис. 6, Интервалы состояния в сплавах меди.
Фазы, обозначенные одинаковыми греческими буквами без индекса, имеют совпадающую структуру, а—твердые растворы меди (кубическая гранецентрированная); 0, у, е—фазы типа Юм-Роеери; (j—(кубическая объемноцевтрированиая)—CuZn, или GusSn, или Сиз Al; Р'-сверх-струнтурная форма ₽; у—(кубическая гигантская ячейка, ср. стр. 42)^CusZn3, или CugjStig, или CU3AI4; е—(гексагональная плотнейшая упаковка)=Си2пэ или CujSn.
совпадающих фаз ***. Прежде всего, во всех трех случаях происходит образование твердых растворов меди (a-фаза, кубическая гранецентрированная решетка). При более высоком содержании второго компонента образуется интерметаллическая фаза с кубической объемпоцеитрировэнной решеткой,, в которой оба сорта атомов беспорядочно размещены в решётке ф-фаза). В то время как в системах Си-Al и Cu-Sn эта фаза при низких температурах полностью исчезает, в системе Cu-Zn она изменяется лишь постольку, поскольку ниже 470° в пей устанавливается упорядоченное распределение атомов (р'-фаза). При еще более высоком содержании второго компонента во всех трех системах появляется фаза, решетка которой аналогична рассматриваемой ниже решетке а-марганца (у-фаза). 6-Фазы во всех трех системах не совпадают в структурном отношении; их структуры еще не полностью выяснены. Проявляющиеся в системах Cu-Zn и Cu-Sn в-фазы обладают решеткой с гексагональной плотнейшей упаковкой. в-Фаза, появляющаяся при: высоком температуре в системе С u-А] (на рис. 6 обозначена е3), структурно отличается от предыдущих (кубическая). В системах Cu-Sn
* Поскольку для обозначения структурно различных фаз применяют одинаковые буквы, на рис. 6 их различают при помощи арабских индексов (которые в других случаях не ставят); так, фазы в системе Си-Zn снабжаются индексом 1, в системе Cu-Sn — индексом 2, и в системе Cii-AJ — индексом 3.
** Эти изменения обнаруживаются при условии достаточно медленного охлаждения, когда могут происходить превращения, благодаря которым при более низких температурах образуются стабильные фазы. То, что Путем быстрого охлаждения («закаливания») эти превращения можно задержать и то что стабильные при более высокой температуре фазы можно получить при более низкой температуре (в метастабильном состоянии) имеет большое техническое значение. Это важно также для исследования свойств «высокотемпературных фаз»; закаливанием проб сплавов эти фазы можно часто получить при обычной температуре.
фазы совпадают по пространственному расположению узлов решетки. Распределение различных сортов атомов в узлах решетки может отличаться.
3*
36
Глава 1
и Си-Al при еще более высоком содержании второго компонента появляются интерметаллические фазы , соответственно Т|3 и (+), обладающие совсем узкой областью гомогенности. Им соответствуют формулы CuSn (структура типа красного никелевого колчедана *, СнА1 (ромбическая, структурно родственна у-фазе) и СнА12 (с тетрагональной решеткой). Во всех трех системах способность второго компонента принимать медь в свою решетку незначительна (са-фаза твердых растворов).
В названных системах интерметаллические фазы 1]2, р3 и й, образующиеся при высоких содержаниях второго компонента, на основании их стехиомотрически простого мало изменяющегося состава без оговорок можно рассматривать как химические соединения. Однако и иптерметаллические фазы, появляющиеся при избыточном содержании меди и обладающие растянутым интервалом гомогенности, также можно рассматривать в качестве химических соединений. Наблюдаемые у них сильные колебания в составе можно свести к «дефектам структуры» в решетке соединений. Ниже будет показано, что рассмотрение этих фаз в качестве интерметаллических соединений пе только позволяет изображать их химическими формулами и таким образом облегчает их понимапие, по и дает возможность установить определенные закономерности, от которых зависит появление этих фаз.
ИСКАЖЕНИЯ РЕШЕТОК (ДИСЛОКАЦИИ) И ДЕФЕКТЫ СТРУКТУРЫ
Кристаллические решетки, в которых часть атомов находится на незаконных местах, называются искаженными. Об искажении решеток в узком смысле говорят, если искажены более или менее широкие области решетки вследствие сдвига атомов с их законных мест. Такие искажения
Тип 1
Тип И
Рис. 7. Схема дефектов структур в кристаллической рещетке со едино ния.
Тип I—внедрение имеющегося в избытке компонента в междоузлия; тип II—вакансии в узлах решетки, принадлежащих компоненту, имеющемуся в недостатке; тип III—занятие узлов, принадлежащих одному компоненту, атомами другого компонента, который имеется в избытке.
решеток — дислокации — могут возникать уже при образовании кристалла или в дальнейшем под влиянием внешнего воздействия (например, деформации при механической обработке).
Если в решетке, которая нс искажена, некоторые атомы находятся на «незаконных» местах или часть мест решетки (при неправильном распределении) не заполнена атомами, то говорят о дефектах структуры. В качестве находящихся на «пезакоппых» местах атомов в неискаженной решетке будем обозначать такие атомы, которые либо неправильно расположены в промежутках между узлами решетки, либо, например, в решетках, построенных из двух типов атомов, занимают такие места, которые на основании симметрии решетки должны быть заняты атомами другого типа. На рис. 7 схематично показаны дефекты структуры трех типов. Дефекты структуры первых двух типов можно встретить как
* Структура отклоняется от тина красного никелевого колчедана постольку, поскольку в решетке, соответс-тпующей NiAs, в определенном порядно расположены еще дополнительные атомы Си. Структура п состав (45 ат.% Sri) поэтому лучщо соответствуют формуле CUgSn5;
Металлы и интерметаллические фазы
37
в решетках элементарных веществ, так и в решетках сложных веществ. Дефекты структуры третьего типа возможны только в решетках сложных веществ,
Узлы решетки, па которых при неправильном распределении * отсутствуют атомы называются ^.вакансиями» решетки. Их не следует смешивать с «междоузлиями», т. е. со свободными пространствами, образующимися в упаковке соприкасающихся шарообразных атомов, при обтачном представлении строения кристалла.
Искажения решетки в узком смысле встречаются часто также и у неметаллических веществ (ср., например, стр. 293). У металлов, вследствие их высоких кристаллизационных свойств, искажения как первичные явления встречаются редко. Большей частью они наблюдаются у металлов в качестве вторичных явлений, возникающих в результате деформаций при их обработке. Напротив, значительные дефекты структур типичны для металлов. Это теснейшим образом связано с особой склонностью металлов к образованию твердых растворов. При этом процессе также .происходит замена атомов одного металла па атомы другого или включение атомов в междоузлия. Однако дефекты структур проявляются отнюдь не только в решетках металлов. Многократно удавалось показать, что такие дефекты обычны также и для кристаллов солей. У последних, однако, они большей частью, крайне незначительны **.
Дефекты структур ле влияют на химический состав типично солеобразпых соединений, потому что электростатические силы, которые действуют между их ионами, требуют компенсации противоположных зарядов. Поэтому, если в решетке с о ди АВ Определенная часть ионов А завимаот места, структурно принадлежащие ионам В, то стремление к компенсации зарядов приводит к тому, что соответствующая часть иопов В занимает места ионов А. То же по смыслу относится и к внедрению ионов в междоузлия, а также к появлению вакансии. У интерметаллических соединении, поскольку они очевидно не приближаются по своему строению к солям, это стремление к компенсации зарядов конечно исчезает. Поэтому в решетке интерметаллического соединения АВ атомы В могут занимать часть мест атомов А без соответствующей замены в других местах атомов В на атомы А. То же, по смыслу, относится к включению атомов В в междоузлия или к образованию вакансий в местах, занимаемых атомами А. Каждый из этих процессов ведет к тому, что соединение содержит избыток атомов В, Однако если повысить в соединении содержание компонента А, то можно вытеснить атомы компонента В с занимаемых ими мест, структурно соответствующих атомам А в решетке, и наконец, атомы А могут вытеснить атомы В с соответствующих им мест, так что возникает соединение, которое содержит избыток компонента А. Но следует считать, что в случае стехиометрического состава, соответствующего типу решетки, структура обязательно должна быть свободна от дефектов. Стехиометрический состав может быть обусловлен тем, что различные типы дефектов структуры, например вакансии в местах, соответствующих атомам В, внедрение атомов В в междоузлия, оказываются случайно скомпенсированными.
Рассмотрение птЕтерметалличееких фаз в качестве соединений. Между интерметаллическими фазами с узкими интервалами гомогенности и обычными соединениями не удается провести резкой границы. Поэтому с самого начала *** такие интерметаллические фазы стали считать химическими соединениями. Однако в настоящее время показано, что многие из этих
* Если из решетки удалять атомы в правильной последовательности, то возникнет решетка другого типа. Если, например, удалить атомы из центра и середины ребер каждого элементарного кубика решетки каменной соли (т. е. в соответствии с рис. 44 на стр. 237 т, I—ионы хлора), то последняя перейдет в кубическую гранецептриро-ваппую решетку.
** Число мест с дефектной структурой растет с повышением температуры.
*** Первые систематические исследования способности металлов образовывать ДРУГ с ДРУГОМ соединения были предприняты Г. Тамманом (1906 г. и сл.).
38
Глава 1
интерметаллических фаз, рассматриваемых как типичные соединения, обладают хотя и очень узкими, но все яке заметно протяженными интервалами гомогенности. Это в сочетании с аналогичными наблюдениями, сделанными на соединениях с неметаллами, привело к тому, что постоянство состава перестало быть критерием существования химического соединения .
Почему типичные «соле образные» и «алмазоподобные» соединения проявлятот строго стехиометрический состав, уже было рассмотрено ранее (т, I, гл. 9). Наличие (практическое) стехиометрического состава у типичных интерметаллических соединений обусловлено не столько природой валентности, сколько атомными радиусами компонентов и строением решетки.
Если вследствие различных радиусов атомов, из которых состоит решетка, последняя пе обнаруживает существенных дефектов структры, то вещество должно обладать практически постоянным составом. Если дефекты структуры могут проявляться в значительном числе, то состав соответственно может колебаться. С химической точки зрения это не означает никакого существенного различия, и поэтому нет никакого препятствия для того, чтобы распространить понятие химического соединения на интерметаллические фазы с любой величиной интервала гомогенности, поскольку их удается свести к решетке с определенным соотношением компонентов. Такая «идеальная» решетка, полученная в результате устранения дефектов в расположении атомов, дает тогда химическую формулу соответствующего соединения *.
Если исходя из интерметаллической фазы с упорядоченным распределением атомов все в возрастающей степени менять местами в кристаллической решетке атомы различного вида, то благодаря постоянно увеличивающемуся числу мест с дефектной структурой можно достигнуть, наконец, совершенно беспорядочного распределения атомов в кристаллической решетке. Если не рассматривать случай при высоких температурах, то в кристаллических веществах в полной мере такое явление наблюдается, вероятно, редко, так как в общем с понижением температуры возрастает стремление к упорядочению атомов в кристаллической решетке. Таким образом, не удастся провести границу между интерметаллическими фазами с (практически) полностью разупорядоченным распределением атомов и фазами с сильными дефектами структур. Поэтому целесообразнее всего распространить понятие химических соединений также и на эти фазы.
Правда, для интерметаллических фаз с полностью раз упорядоченным распределением атомов не удается дать «идеальную формулу» па основании структуры решетки. Однако, как пранило, л этом случае можно установить «идеальную формулу» на основании других критериев. Например, имеется интерметаллическая фаза с разупорядоченным распределением атомов, которая обладает очень узким интервалом гомогенности, т. е. практически постоянным составом. В этом случае формула следует ла состава (например, Ag3Al проявляет постоянный состав, однако оба сорта атомов беспорядочно размещены в узлах кристаллической решетки).
Часто интерметаллические фазы с расширенным интервалом гомогенности, поскольку их можно свести к «идеальным формулам», отвечающим химическим соединениям постоянного состава, рассматривают как твердые растворы одного или другого компонента в соответствующем соединении. Однако подобный подход не удается применить к таким случаям, в которых изменяемость состава определяется наличием вакансий в решетке. Это происходит нотому, что соединение, вообще говоря, только тогда стабильно, когда в местах решетки, соответствующих одному из компонентов, имеются вакансии и вследствие этого в сравнении с «идеальной формулой» оно содержит один компонент в избытке. Состав такого соединения может быть (практически) постоянным или заметно варьироваться, однако он должен обязательно отличаться от состава, соответствующего «идеальной структуре». Для стабильности решетки необходимы пе только вакансии, но и другие дефекты структуры. Поэтому формулы интерметаллических соединений даже почти постоянного состава не удается определить чисто аналитически. Это оказывается возможным часто только при помощи структурных определений,
* Как было уже отмечено (т. I, стр, 620), на изменяемость состава будет указывать поперечная черта, постав лепная над формулой. Новыми международными номенклатурными правилами рекомендовано применение значка —, однако из-за того, что он занимает много места, его пе удается поставить над интерметаллическими системами, во многих приведенных в дальнейшем таблицах,
Металлы и интерметаллические фазы
39
Ограничивать понятие химического соединения интерметаллическими фазами с постоянным составом не рекомендуется на том основании, что в связи с непрерывным усовершенствованием методов исследования все время растет число интерметаллических фаз, в которых удается установить заметно протяженные интервалы гомогенности, которые ранее рассматривали как фазы постоянного состава. Ввиду чрезвычайной склонности металлов взаимно обмениваться атомами в своих кристаллических решетках кажется вообще сомнительным, чтобы могли существовать в заметном количестве интерметаллические фазы строго постоянного состава (если не рассматривать случаи при очень низких температурах). Кроме того, оказалось, что интерметаллические фазы с узким интервалом гомогенности (следовательно, соединения в старом смысле) часто в родственных системах так глубоко соответствуют по свойствам структурно аналогичным фазам с широким интервалом гомогенности, что о них следует говорить, как о совервгенно одинаковых веществах.
Например, е-фаза, в системе Cu-Sn, которую вследствие узкого интервала гомогенности всегда рассматривали как соединение CujSn, соответствует е-фазе с широким интервалом гомогенности в системе Cu-Zn. Обе фазы не только имеют одинаковую структуру решетки (гексагональная плотнейшая упаковка), но и родственны также по свойствам; к тому же, как будет показано ниже, между обеими фазами имеется тесная конституционная связь.
Интерметаллические фазы подходят под понятие химического соединения в том случае, если последнее определять следующим образом: химическое соединение — это сложное гомогенное вещество, свойства которого не могут быть непрерывно переведены в свойства одной из его составных частей изменением состава.
Упорядоченное распределение атомов в кристаллической решетке в общем является также мало характеризующим химическое соединение свойством, как и постоянство состава. Имеются химические соединения, которые при высоких температурах обладают неупорядоченным распределением атомов в кристаллической решетке, а при более низких — упорядоченным, причем других изменений в структуре решетки не наблюдается. В таких случаях можно говорить об образовании «сверхструктуры соединения». Например, стабильная при обычной температуре р'-латунь является сверхструктурой, стабильной ири более высокой температуре [J- л ату ни, CuZn.
Если гомогенное вещество, состоящее из компонентов А, В и С удается путем изменения состава непрерывно перевести в компонент С (но не в А или В), то его следует рассматривать как твердый раствор соединеаия А и В в веществе С. Например, как было показано Лавесом (Laves, 1936), соединение AgCd3 образует непрерывный ряд твердых растворов с Mg.
Правило Юм-Розери. Появление фаз, обозначенных па рис. 6 буквами)), у н е, не ограничивается приведенными там сплавами. Структурно аналогичные фазы появляются в большом числе сплавов Си, Ag и Au. Склонность к образованию интерметаллических фаз со структурами р-, у- и е-латупи можно рассматривать как характерное свойство металлов побочной подгруппы первой группы периодической системы. В и п тер металлических фазах типа fj-, у- и s-латуни эти металлы (а также другие металлы, которые могут выполнять их функции, см. ниже) обозначают как «металлы I рода» а металлы, которые образуют с ними эти фазы, обозначают как «металлы II рода». В качестве «металлов II рода» могут функционировать прежде: всего Be, Mg, Al, Ge, Sn, Sb, Zn, Cd и Hg. Для образования названных фаз в 1926 г. Юм-Розери установил правило; структура образующейся определяется отношением общего числа валентных электронов к общему числу атомов. При отношении 21 ; 14 появляется кубическая объемноцентрированная ф-фаза, при отношении 21 : 13 — кубическая ?-фаза и при отношении 21 : 12 — гексагональная е-фаза. В качестве валентных электронов рассматриваются при этом те электроны, которые нельзя отнести ни к одной из замкнутых оболочек. Следовательно, у Си, Ag, An — это один электрон (ср. стр, 387), у Zn, Cd, Hg — это два электрона (ер. стр, 454) и у металлов главных подгрупп столько
40
Глава 1
электронов, снолысо отщепляется нри появлении наивысшей положительной валентности.
Правило Юм-Розери выполняется почти во всех случаях, когда соединения обладают постоянным составом или когда при колеблющемся составе различные атомы правильно расположены в решетке, так что легко можно установить «идеальную формулу» соединения *. В тех случаях, когда наряду с колеблющимся составом
Таблица 1
Соединения, подчиняющиеся правилу Юм-Розери
Число валентных электронов/число атомов
3 -Л жта 21 1 14 21 13 1 ; 4 или 21 ; 12
р-фазы у-фазы е-фаэы
CuBe ChZh CuyAl4 CugZng CuBe3 CuZn3
C^Al Gu31Sns CuaCds CuyGe
Cu5Sn Cu3Sn
Cu£3Sb3
AgMg AgZn Ag6Zns Ag5Al3 AgZn3
AgCd — AgsCde Ag3Sn AgCd3
Ag5Hg8 Agi3Sb3
AuMg AuZn Au5Zns Au5AI3 AuZn3
Au Cd — AusCds AuCd3
имеется неправильное распределение атомов в узлах решеток, составы, требуемые правилом, лежат внутри найденных интервалов гомогенности или очень близко к ним. Следовательно, в этих случаях рассчитанную по правилу формулу можно рассматривать как «идеальную формулу» соединения. В табл. 1 дан обзор ряда соединений, подчиняющихся правилу Юм-Розери.
Как показали Бестгреи и Экманн (Westgren, Ek шапп, 1931), правило Юм-Розери можно распространить на металлы побочной подгруппы восьмой группы и марганец, если принять, что в соединениях с металлами II рода опи имеют число валентных электронов, равное нулю. Например, Мн Al, FeAl, CoAl и NiAl образуют решетки типа fl-фаз, а соединения FeaZn2£, Co6Zn2l, Ni5Zn3J, RhfZn3J, PdGZn21, Pt5Zn2£ образуют решетки типа у-фаа.
Другие правила образования интерметаллических соединений. Часто сильно электроположительные металлы (щелочные и щелочноземельные металлы) с металлами побочных подгрупп образуют соединения, которые по своему составу и структуре аналогичны р-латуни (Zintl, 1933). Однако в случае этих соединений (в которых всегда имеется упорядоченное распределение атомов) правило Юм-Розери не соблюдается; концентрация валентных электронов (отношение общего числа валентных электронов к общему числу атомов) может в пих иметь различные значения.
«Металлы I рода» (Мн, Fe, Со, Ni, платиновые металлы (кроме Rn и Os), Си, Ag, Аи| при высоких температурах образуют друг с другом только твердые растворы; при охлаждении из них образуются часто сверхструктуры (Dehlinger).
По мнению Де линкера (1935), среднее положение между сплавами, которые образуют друг с другом «металлы. I рода», и сплавами сильно электроположительных металлов с металлами I и И рода занимают сплавы, которые образуют друг с другом «металлы II рода?! (прежде всего Zn, Cd, Hg, Mg, Al, Sn). Металлы побочной подгруппы второй группы периодической системы (Zu, C'd, Hg) не образуют соединений между собой и в твердом состоянии проявляют значительную взаимную растворимость только
* Исключение сеет являет появляющаяся в системе Cu-Hg у-фаза"! состава CuIIg. Дал:ее, имеются некоторые соединения, которые, вероятно, имеют требудап.ш правилом состав, но иную структуру. Например, Ag3Al, Лн3А1 и Cu5Si образуют решетку типа ^-марганца (ср. стр. 43).
Металлы и интерметаллические фазы
41
тогда, когда разница между их атомными радиусами незначительна (Cd-Hg). Напротив, Mg при большей разнице атомных радиусов (Mg-Zn) ели только при незначительной химической аналогии элементов (Mg-Hg) образует с цими соединения с упорядочен-ньгм распределением атомов и узкими интервалами гомогенности; при мецьшей разнице атомных радиусов оп образует широкие области твердых растворов, в которых при охлаждении появляются сверхструктуры (ср. табл, 48, стр. 456). Подобно магнию в отношении металлов побочной подгруппы второй группы периодической системы ведут себя А1 и Sn, только в случае последних склонность к образованию соединений выражена еще сильнее, а способ г? ость давать твердые растворы еще более ослаблена. Общим для металлов Mg, Al и Sn ш> сравнению с металлами побочной подгруппы второй группы являются большие атомные радиусы и сильная поляризуемость. По данным Делипгера, в случае перечисленных металлов II рода атомными радиусами п поляризуемостью атомов определяется не только способность к образованию взаимных твердых растворов, но также и способность к образованию соединений. В то же время у сплавов, которые образуют друг с другом металлы I рода, атомные радиусы пе имеют решающего значения даже для образования твердых растворов.
Структуры металлов и интерметаллических соединений. Чистые металлы имеют большей частью очень простые структуры решеток. Они кристаллизуются преимущественно с образованием решеток с плотнейшей упаковкой атомов или решеток, аналогичных решеткам с плотнейшей упаковкой. Из 56 металлов главных и побочных подгрупп периодической системы * 15 при обычной температуре образуют решетку типа магния (с гексагональной плотнейшей упаковкой) и гранецептрировапную кубическую решетку (кубическая плотнейшая упаковка), 13 образуют объем-ноцентрированную кубическую решетку и 2 (Sb и Bi) кристаллизуются по типу сурьмы, решетку которой можно рассматривать как деформированную простую кубическую решетку. Из чисто металлических элементов главных и побочных подгрупп с известной структурой ** только 8 (Мл, Ga, In, fi-Sn, Hg, Po, Pa и U) образуют особые решетки, которые в других случаях у элементарных веществ не наблюдаются, однако иногда встречаются у интерметаллических соединений. Низкотемпературная модификация олова (a-Sn), которая пе проявляет чисто металлического характера, и германий (о котором можно сказать то же самое) обладают структурой алмаза.
У интерметаллических соединений встречаются часто те же решетки, что и у чистых металлов. Только в случаях, когда атомы упорядочеппо распределены в узлах решетки, возникают «сверхструктурные формы» этих решеток. Кристаллографически их рассматривают как особые типы решеток. Однако, если предположить, что устранено различие между разными сортами атомов, сверхструктурная форма переходит в решетку, которая встречается у чистых металлов.
Например, (3-латунь, как уже было сказано, образует кубическую объемяо-центрпрованную решетку, т. е. решетку, которая структурно отличается как от решетки меди (кубическая гранецентрированная), так и от решетки цинка (гексагональная плотнейшая упаковка), однако решетка р-латуни встречается у многих других чистых металлов. Атомы Си и Zh распределены в ней беспорядочно но узлам решетки, поэтому кристаллографическая симметрия решетки такая же, как если бы она была построена из атомов только одного типа. Однако если оба типа атомов упорядоченно распределены в узлах кристаллической решетки (что имеет место в образующейся при охлаждении р'-фазе латуни), то из кубической объемы сцентрированной решетки возникает решетка типа иодида цезия ***. Соединение Сн3А1, также относящееся
* Структура решеток металлов семейства лантанидов см. па стр. 520, трансурановых элементов на стр. 661 и сл., 665 и 678.
** Еще не известны структуры франция и радия.
*** Из рис. 48 т. I видно, что в результате устранения различия между атомами решетка CsI переходит в кубическую объемноцентрированную решетку.
42
Глава 1
к типу fl-латувл, при упорядоченном распределении атомов образует решетку, представленную на рис. 8. Хорошо видно, что эта решетка в результате устранения различия между атомами или в результате неправильного распределения их в узлах решетки также переходит в кубическую объемно центрированную решетку.
Приведенная на рис. 8 сверх структурная форма кубической рбъемноцентриро-ваниой решетки встречается также у соединений Mg3La, Mg3Ce и jV[g3Pr (в. которых атомы всегда упорядоченно распределены в узлах решетки) (Rossi; 1934). Однако эти соединения не относятся к группе р-латуни, так как у лих концентрация валентных электронов не является мерой образовавши соединения.
Структурный тип, впервые установленный ЦинтПем (1932) для соединения NaTl также можно рассматривать как сверхструктуру кубической объемпоцентрирав энной решетки (ср. рис. 9). По этому типу кристаллизуется ряд соединений щелочных металлов (LiZn, LiGd, Li Al, LiGa, Li In, Naln, NaTl). У этих соединений концентрация
Рис. 8. Элементарная ячейка решетки Си3А1 (метастабильная сверхструктурная форма (1-фазы системы Сц-А1, получаемая быстрым охлаждением до 300° стабильной выше 570° P-фазы, обнаруживающей неупорядоченное распределение атомов).
Изотипами NaTl являются: Naln (а— —7.30 A), Ltln (а=е,79А), LiGa (в= sBJjA), L1A1 (а=6,36А), UCd (0= =6,69 A), LIZn (я=6,21 А).
валентных электронов также не определяет образование соединения, и поэтому они не относятся к труппе fl-латуни. В решетке типа NaTl атомы всегда упорядоченно расположены в узлах решетки, а именпо так, что каждый тип атомов порознь образует решетку алмаза.
Решетки, которые, как и приведенные выше, получают'из другой решетки благодаря тому, что в последней часть узлов в определенном порядке занимают атомами другого рода, называют * сверхструктурными типами», потому что в их рентгенограммах появляются дополнительные ляпии («линии сверхструктуры»), помимо линий основной решетки. Линии сверхструктуры появляются потому, что узлы решетки, запятые атомами с различным числом электронов, по-разному отражают (или рассеивают) рентгеновские лучи. Это ведет к появлению дополнительных интерференций па том же основании, на котором включение ионов с равным числом электронов в решетку вместо атомов с различным числом электронов (как показано в т. I стр. 241) приводит к исчезновению линий интерференции.
Из кубической гранецентрированной решетки в качестве основного типа выводится решетка, Приведенная па рис. 114 т. I, относящаяся к CaSn3 и к ряду других Соединений аналогичного состава. Эта решетка проявляется также как св ерх структур а твердых растворов металлов, кристаллизующихся в кубической гранецеятрирован-ной решетке (например: [AuCu3j, [PdCu3l, [PtCn3]).
Решетка у-латуни л соответствующие ей фазы сплавов очень близки решетке модификации марганца (решетка а-маргаица), устойчивой при обычной температуре. Решетка а-марганца родственна кубической объемпоцентрированной решетке. Однако в первой атомы упакованы плотнее и менее симметрично расположены, чем во второй. Элементарная ячейка решетки а-маргаица представляет собой кубик, который содержит не менее 58 атомов («гигантская ячейка»). Такой ящ решеткой обладает, но данным Лавееа (Laves, 1934), соединение Mg17Al12. у-Латунь и остальные приведенные
Металлы и интерметаллические фазы
43
на стр. 40 у-фазы имеют не точно такие же, но близкие элементарные ячейки; они «одержат 52 атома . У большинства из этих у-фаз атомы упорядоченно распределяются в узлах решетки^ Благодаря этому у соединений различного состава могут оказаться различия в степени симметрии, однако если отвлечься от различий в типах атомов, то речь будет идти об аналогичных структурах..
Отдельные соединения, для которых на основании правила Юм-Розери следует ожидать образования структур типа (J-латупи (CnZn), кристаллизуются по типу ^-марганца.. Этохарактерпо для решеток, которые родственны кубической гране-
Щ
Рис. 10. Три простейшие плотнейшие упаковки.
I—г е к са г она льпая л л отн е йша я у па ковк а (тип магния); II—кубическая плотнейшая упаковка (г ране центрированная кубическая решетка типа аргона или меди); III—тип TiNi3.
I — удвоенная элементарная ячейка гексагональной плотнейшей упаковки с длинами ребер а, а, не (ср. т. I, рис. 57). В идеальном случае 'с,~— 1Л6.
-1 и
II — плоские сетки кубической плотнейшей упаковки расположены перпендикулярно лростраяотвеляой диагонали Д гракепеятрировавноЙ кубической ячейки с длиной ребра а (ср. т, I, рис. 4В). III — эту структуру можно рассматривать как смесь гексагональной л кубической плотнейших упаковок. Если обозначить различные плоскости буквами А, В к С, то при гекс аг опальной плотнейшей упаковке будет последовательность: АСАС.... (равнозначно с АВАЛ..., как 'выясняют путем сдвига начала координат на Cj/2). При кубической плотнейшей упаковке будет последовательность АВСАВС... и при гексагональной упаковке типа TiNb— АВАСАВАС.... Элементарная ячейка этого тип.з при одинаковых размерах атомов имеет в ипеальяом случае удвоенную высоту по сравнению е ячейкой обычной гексагональной плотнейшей упаковки.
центрированной решетке и содержат 20 атомов в элементарном кубе. Соединения, кристаллизующиеся по типу p-мар гайца (Ag3Al, Au3Al, Cu5Si, CoZn3), имеют узкие интервалы гомогенности и неупорядоченное раопределенно атомов.
Кубические гигантские ячейки со 112 атомами образуются соединениями NaZn13, KZn13, KCd13, RbCdl3 и CsCd13 (Kctelaar, 1937; Zintl, 1938). Следует отметить исключительно высокое координационное число (24), которое проявляют в этой решетке щелочные металлы в отношении атомов Zn к Cd.
Структурный тип, который представляет собой комбинацию гексагональной плотнейшей упаковки с кубической плотнейшей упаковкой (рис. 10), был обнаружен Лавесом (1939) у соединения T1N13. Нарве. 10доказана не элементарная ячейка кубической плотнейшей упаковки (гранецептрйровэнный куб), а разрез решетки, который долаетТнаглядным ее родство с гексагональной плотнейшей упаковкой. В отличие от последней в случае кубической плотнейшей упаковки, точно друг над другом лежат центры атомов не каждого третьего слоя, а каждого четвертого слоя. Символически это различие можно представить схемой: АСАСАС и АВСАВС, тогда соответствующей схемой для решетки TiNij будет АВАСАВАС.
Интерметаллические соединения общей формулы АВ3 очень часто кристаллизуются в решетке, которая отличается следующими особенностями: каждый атом А одинаково или почти одинаково удален от четырех других атомов А, а каждый атом В — от шести других атомов В; каждый атом А па равном расстоянии (илиприблизительно
44
Глава 1
равном) окрукюн 12 атомами В, а каждый атом В — шестью атомами А. Кристаллические вида, в основе которых нежит подобное строение, называют разами Лавеса, так как они были исследовали главным образом Лавосом (Laves, 1934 и ед.). Известно три типа фаз Лавеса, которые представлены соединениями MgZn2, MgCu,, MgNi2 (ер, табл. 2).
Таблица 2
Фазы Лавеса
Тип MgZna Тип MgClia Тип MgXia
MgZna MnBe2 NaAu3 AgBe2 MgNi2
CaMg2 ReBe3 KBi2 MgZnN i TiCo3
TiMn2 FeBe3 MgCu2 MgZnit3Co0,7 MgAICu
TiFe3 MgAICu CaAl2 MgZnIj2Ag0,s MgSio.jCwi.s
VBo2 PbAu2 W®* **0,2Nii,8 MgZni,gAgo,4
CrBe2 MgSi0,5Culj5 BiAu2 (7’e£i,5BeOj5)Be2 Mg(Zn, Cu)2
MoBe2 Ti-tteg (₽dg,5Bet;ii6)Be2
WBe3 TiCo2 (Сио,<)Веод}Ве2
WFc2 ZrW2 (Ano,5Be0,a)Bo2
Решетку MgZn2 можно вывести из решетки цапка (гексагональная плотнейшая упаковка), если удалить из нее группы из семи атомов цинка, которые образуют два тетраэдра с общей вершиной, и в центр тяжести каждого такого тетраэдра поместить атом Mg. Атомы магния образуют тогда сами по себе решетку типа ртути (или вюртцита). Решетку MgCn2 можно получить аналогичным образом из решетки меди (кубическая плотнейшая упаковка). Для этого следует удалить и о л овину тетраэдров меди, из которых, по-видимОму, построена решетка меди, и в центр тяжести каждого удаленного тетраэдра поместить атом Mg. Атомы магния образуют тогда сами по себе решетку алмаза (или цинковой обманки). Решетку MgNi2 можно рассматривать как комбинацию решетки MgZn2 с решеткой MgCu2. В решетках Лавеса как атомы А, так и атомы В сами по себе образуют взаимосвязанные шаровые упаковки. Однако, если считать атомы шарообразными, то между атомом А и атомом В пи в одном месте нет соприкосновения. Это происходит потому, что сродство между атомами одного вида при образовании решеток Лавеса играет но меньшей мере ту же роль, что и сродство между атомами А и В. Наряду с этим существенную роль играет также отношение атомных радиусов. По данным Делиигера и Шульце (Dehlinger, Schulze, 1939), фазы Лавеса образуются тогда, когда атом А с сильной склонностью к образованию металлической связи встречается с атомом В, предпоследняя электронная оболочка которого не заполнена * и который имеет радиус примерно на 25% меньше, чем радиус атома А.
Структуры металлоподобных соединений неметаллов. Соединения неметаллов В, С, N и Н с переходными металлами имеют большей частые металлоподобный характер (высокую электропроводность, отчасти даже могут проявлять сверхпроводимость). Если отношение атомных радиусов неметалла я металла достаточно мало «0,59), то, как было показано Хеггом (Ilagg, 1931), образуется правильная решетка,которая выводится путем расположения атомов неметалла в пустотах либо решетки трех, чаще всего встречающихся у металлов типов (кубическая грапецентрированная, кубическая объемпоцентрировапная, гексагональная плотнейшая упаковка), либо (реже) ид простой гексагональной решетки с соотношением осей с : а — 1 ; 1 (см. рис. 11), которая не встречается у чистых металлов. Возникшие таким образом соединения почти всегда описываются следующими формулами; М4Х, М2Х, MXOXj. Соединении М4Х и М2Х имеют широкий иитервад гомогенности, соединена я MX.
* Электронной оболочкой считают при этом группу электронов с одинаковым главным квантовым числом (п) и одинаковым орбитальным квантовым числом (I). Следует отметить, что замкнутые в атомарном состоянии внешние оболочки часто при переходе в металлическое состояние расщепляются. Например, внешняя s-оболочка Бе и Mg распадается на s- и р-оболочки, каждая из которых содержит но одному электрону (ср, т. I, стр. 283).
** Приведенные неметаллы обладают в своих соединениях с металлами приблизительно следующими атомными радиусами; Б 0,97 А, С 0,77 A, N 0,71 А, Н 0,45—0,46 А.
Металла и интерметаллические фазы
45
и МХ2 — узкий интервал гомогенности. В решетках соединений МХ2 атомы неметалла X обычно расположены .ее.адр.ио.
Если отношение атомных радиусов X : М^> 0,59, то в пустоты решетки могут внедряться только небольшие количества неметалла. При более высоком содержании неметалла возникают довольно разнообразные типы ре-
шеток, часто со сложной структурой.
На рис. 12 и 13 приведены элементарные ячейки двух соединений этого вида (FeaB и FeSiA Такую же структуру, как Fe2B, имеют Со2В, Ni2B и Al2Cu. Соединение FeSi2 имеет область гомогенности от 68,8 до 72,1 ат. % Si; следовательно, оно всегда обладает высоким содержанием Si, как это и соответствует «идеальной формуле» FeSi2, вытекающей из его структуры. Это объясняют, предполагая, что указанное соединение оказывается стабильным только тогда, когда часть атомов Fe в его решетке замещается (при неправильном распределении) на атомы Si. Более или менее значительным обменом атомов Fe на
Рис. 11. Простейшаягек-
атомы Si объясняется его колеблющийся состав.
Облагораживание сплавов. Как уже было отмечено (т. I, стр. 616 и ел)., механические свойства металлов и сплавов можно улучшить механической обработкой (ковка, вальцовка, протяжка и т. д.). Наряду с повышением прочности сплавов пластической деформацией па холоду (холодная обработка) большое значение имеет также возмож-
с аг опальная решетка. Пример: Wc 50 ат.% С образует простую гексагокаль-яук> решетку (<* 2,90,
=2,83 А.),»которую внедрены атомы G ^вероятно^ в точках а За с \ с координатами , -г-, “5- /» о о л '
иость сильно повышать твердость и прочность определенных сплавов термической обработкой, которая заключается в закалке с последующим
Рис. 12. Элементарная ячейка соединения Fe2B; ао=5,10, с0==4,24 А. Иаотнпами Fe£B являются Со а В (ао=5,01( с0= 4,21 A}, NijB (tt0 = i,98, Со = 4.24А) и АЦСи (<z0 = 6,05, с0 = 4,88 А).
Рис. 13. Элементарная ячейка соединения FcSi3; в0 — 2,68, с0 = 5,12 А.
Состав соединения может колебаться в интервале 68,8—72,1 ат. % 81. Оно содержит, следовательно, постоянно избыток атомов Si. Последние в беспорядке занимают места атомов Ге в решетке.
отпуском при более низкой температуре или старением при обычной температуре. Этот процесс называют облагораживанием пли дисперсионным твердением. Способ твердения был открыт Л. Вильмом в 1911 г. на сплаве алюминия и меди. Он отличается от издавна известного процесса закалки стали (ср. стр. 288) тем, что существенное повышение прочности и твердости происходит не при закаливании, а только при старении или при умеренном нагревании («отпуске») закаленного сплава *.
* Подобным дисперсионному твердению является способ, называемый в сталелитейной яромышлеплосги «облагораживанием».
46
Глава 1
Самым старым и самым известным сплавом, подверженным дисперсионному твердению (старению), является дюралюминий (ср. т. 1, стр. 38G и сл.). Однако в дальнейшем было показано, что имеется очень много сплавов, подверженных дисперсионному твердению. Это обычно такие сплавы, которые при высоких температурах состоят из кристаллов, представляющих собой почти насыщенные твердые растворы, и которые закаливанием можно перевести в состояние пересыщенных твердых растворов. Дисперсионное твердение заключается в том, что сю временем про-
Р ис. 14. Температурная вависиюсть границ насыщения кристаллов алюминия, содержащих медь.
исходит распад этих твердых растворов с выделением кристаллов второго вида в высокодисперсной форме *. Йри-
пято, что повышение твердости и другие связанные с этим изменения свойств обусловлены внутренними напряжениями, " которые вызваны в решетке твердого раствора появлением нового кристаллического вида, образующегося в многочисленных и небольших изолированных областях.
Как уже было сказано, уменьшение взаимного растворения в твердом состоянии с падением температуры наблюдается весьма часто. Особенно ярко выражено такое падение взаимного растворения в системе Al-Си (рис. 14).
Поэтому в случае алюминия, содержащего медь, наблюдается особенно сильное возрастание твердости. То же относится к меди, содержащей бериллий.
Если сплав определенного состава состоит из интерметаллической фазы, устойчивой только при повышенной температуре, которая при охлаждении распадается на две другие фазы и которую, однако, методом закалки можно получить при обычпой температуре в мета стабильном состоянии, то имеются все условия для дисперсионного' твердения. Ибо в атом случае распад можно ировеста таким образом, что продукты распада выделяются в очень высокодисперсном состоянии, так же как при дисперсионном твердении твердого раствора.
Так же как и твердость, дисперсионным твердением можно повысить сопротивление сплава разрыву. Сопротивление разрыву для мелкокристаллического, отожжеиного чистого алюминия составляет около 8 кг/мм2. Холодной обработкой его удается повысить примерно до 25 кг/мм2. Обычный алюминий, содержащий медь (не подвергнутый дисперсионному твердению твердый раствор), имеет прочность до 25 кг/мм2, а после холодной обработки — до 40 кг/мм2.
В результате дисперсионного твердения удается повысить его прочность выше 50 кг/мм2, соответственно после холодной обработки — до 60—70 кг/мм2. Сопротивление разрыву подвергнутого дисперсионному твердению сплава бериллия и меди, содержащего 2,5% Be, составляет даже 130—150 кг/мм2 и имеет, таким образом, значение, которое достигается (или превышается) только у особо прочных сортов стали.
* Видимо, твердость обусловливается не столько собственно распадом твердого-раствора, сколько его начальной стадией. При этом в решетке твердого раствора атомы во многих местах переходят в положения, соответствующие новому кристаллическому виду. Однако сначала это происходит совсем в маленьких областях. Если распад зайдет так далеко, что новый кристаллический вид можно определить рентгенометрически, то твердость снова падает.
Гл а ва 2
ТРЕТЬЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Побочная подгруппа)
СКАНДИИ, ИТТРИЙ, ЛАНТАН И АКТИНИЙ
Порядковый номер Элемент Символ Атомный вес Удельный вес Температура, *>С Удельная теп* лоеыкость Валентность
славления кипении
21 Скандий Sc 44,96 3,20 ~ 1400 —. III
39 Иттрий Y 88,92 4,34 ~ 1500 —— — III
57 Лантан La 138,92 6,18 826 0,045 III
89 Актинии Ac 227 10,07 — — — ш
Общие сведения. Побочная подгруппа третьей группы периодической цистеин включает три редких элемента: скандий, иттрий и лантан и, кроме того, радиоактивный элемент актиний, встречающийся только в очепь незначительных количествах.
Элементы побочной подгруппы третьей группы по своим свойствам тесно примыкают к элементам главной подгруппы второй группы, стоящим в тех же рядах (кальций, стронций, барий и радий). Однако валентность скандия, иттрия и лантана на единицу больше. Й все же элементы побочной подгруппы третьей группы стоят ближе к элементам главной подгруппы второй группы, чем к элементам главной подгруппы третьей группы. Это находится в соответствии с их порядковыми номерами, которые непосредственно следуют за порядковыми номерами элементов главной иод группы второй группы.
Остающиеся после отщепления трех валептных электронов атомные остовы элементов побочной подгруппы третьей группы пе отличаются от атомных остовов щелочноземельных металлов (за исключением на единицу более высокого заряда я более сильного сжатия электронных оболочек, обусловленного более высоким зарядом ядра). Как и ионы щелочноземельных металлов — Саз+, Ба2+ и На3'*' — ионы
Sc3+, Y3+, La34 и Ас3+ обладают электронными оболочками, подобными оболочками инертных газов, в то время как ионы Ga3+, In3+ и Т13+ отличаются от инертных газов, а также от В3+ и AF+, имеющимися у них внешними d-оболочками. Однако, с другой стороны, появление ti-оболочек, сопровождающееся сжатием атомов, приводит к тому, что радиусы ионов Ga3+ и 1н3+ меньше отклоняются от ионного радиуса А!34”, нем ионпые радиусы скандия и его аналогов. У нейтральных атомов электронная конфигурация внешней оболочки в случае галлия, индия и таллия такая же, как в случае бора и алюминия (s'2p), в то время как скандий и его аналоги имеют конфигурацию ds2. Взаимодействие этих различных факторов приводит л тому, что в зависимости от свойств, которые сравниваются, к алюминию оказываются ближе его аналоги, стоящие в главной подгруппе или в побочной.
Как бор и алюминий, элементы побочной подгруппы третьей группы в своих нормальных соединениях всегда трехвалентни. Они в чрезвычайной степени склонны к образованию солеобризных соединений, т. е. сое ди-
48
Глава 2
непнй, в которых они отчетливо электроположительны. Их электроположительный характер непрерывно возрастает от алюминия к лантану и актинию.
Потенциал разложения хлорида лантана в соответствии с данными Неймана, лежит при 800’ па 0,31 в выше, чем потенциал разложения хлорида алюминия т. I, стр. 308),
Вследствие относительно высокого температурного коэффициента у хлорида лантапа это различие ири комнатной температуре было бы еще больше,
Как и все элементы побочных подгрупп, элементы побочной подгруппы третьей группы обладают чисто металлическим характером. Они образуют твердые, очень трудно летучие и очень плохо растворимые окислы. Эти окислы имеют чисто основной характер. В связи с этим окислы элементов побочной подгруппы третьей группы в значительной степени отличаются от типичной амфотерной окиси алюминия. Основной характер у них выражен значительно ярче, чем у окислов аналогов алюминия, относящихся к главной подгруппе третьей группы, из которых окислы галлия и индия также амфотерны. Основной характер отчетливо возрастает при переходе от окиси скандия к окиси лантана. По своему основному характеру окись лантана стоит лишь немного позади окиси кальция.
По аналогии с глиноземом AI0O3 окислы металлов ранее называли вообще землями. Для окислов металлов скандия, иттрия и лантана, которые являются особенно типичными «землями», это название сохранилось. Для металлов, лежащих в основании этих земель, целесообразно употреблять название земельные металлы.
К лантану в периодической системе примыкает ряд элементов, обнаруживающих в своем химическом характере значительную аналогию с ним. Эти элементы называют (по предложению Гольдшмидта) лантанидами. Как уже было сказано, лантаниды образуют в периодической системе особое семейство, которое будет рассмотрено в гл. 10. Окислы скандия, иттрия, лантана и лантанидов объединяют под общим названием редкие земли, так кап в заметных количествах они редко встречаются в природе. Характерным для редких земель является то, что они встречаются в природе всегда вместе. Только скандий содержится в минералах, которые свободны от остальных редких земель или очень бедны ими. Вообще скандий по сравнению с остальными редкоземельными металлами занимает особое место, которое определяется его более близким родством с алюминием.
Электроположительная трехвалентностъ металлов побочной подгруппы третьей группы следует, как и в случае алюминия, из их положения по отношению к инертным газам. В состоянии нейтрального атома они имеют на три электрона больше, чем предшествующий инертный газ. Несмотря на то что один из этих электронов связан иначе, чем у алюминия, а яменпо он находится на d-, а не на р-уровпе, однако этот электрон также сравнительно легко отщепляется, каки оба s-электрона. Например, как показано в табл. 3, в случае скандия энергия, которую требуется затратить, чтобы оторвать только один четвертый электрон, значительно больше, чем сумма энергий ионизации для трех наиболее рыхло связанных валентных электронов.
То, что металлы побил ной под группы III группы нормально только трехвалентны и не встречаются в состояниях с более низкими валентностями, объясняется так же, как и в случае алюминия. Г, т. I было показало, что теоретически пе исключало, что ион AF+ мажет быть перезаряжен в ион А Vм.
Третья группа, периодической системы
49
Таблица 3
Энергии ионизации элементов побочной подгруппы III группы
С ккал/г-ато.и )
Скдядлй ИтгрлЕ Лая та я
I М М1+ + с п М1+-г№Ч« Ш №+-> 154 295 571,8 150 283,7 470 129 262,5 440,1
Сумма 1-рПЦ-Ш 1021 904 832
МЗ+->М*+ + е 1697 — —
Как пола га от Ноддак (Nod dak, 1937), в случае Sc, Y и La (а также лантанидов) при электролитическом осаждении их из водных растворов полной разрядке катиона до металла предшествует перезарядка трехвалентного иона в двухвалентный. Правда, найденные нотенциалы перезарядки лежат у Sc, Y и La лишь немного ниже (0,16, 0,09 и 0,11 в) напряжений осаждения.
Сильно электроположительный характер металлов побочной подгруппы третьей группы проявляется в их высоком сродстве к кислороду и вообще в высоких теплотах образования их солеобразных соединений (см. табл. 4).
Таблица 4,
Теплоты образования соединений элементами побочной подгруппы III группы
( KKEj'l/a-aKflJ
Элементы О кислы фториды Хлориды Бромиды Йодиды Сульфиды Нитриды
Скандий 73,6 63,5 25,0
Иттрий — — 78,3 — 47,7 — 23,8
Лантан 89,8 —1 87,9 — 55,8 49 24,2
В тех случаях, когда теплоты образования известны, они превосходят теплоты образования соответствующих соединений алюминия, причем иногда очень значительно. У лантана сильно электроположительный характер выражается также в его поведении по отношению к водороду, с которым этот металл, подобно щелочноземельным металлам, образует твердый солеобразный гидрид. Правда, гидрид лаптана отличается от гидридов щелочноземельных металлов тем, что содержание водорода в нем зависит от температуры и давления, и формула LaHj соответствует только граничному значению, которое достигается при максимальном поглощении водорода. Таким образом, гидрид лантана представляет собой переход к металлоподобным гидридам со свойствами твердых растворов, которые встречаются в следующих побочных подгруппах периодической системы (относительно гидридов Sc, Y и Ас подробно ничего не известно).
4 г. Реми
50
Глава 2
Карбиды металлов побочной подгруппы третьей группы, получаемые нагреванием окиолов с углем в электрических печах, по своему составу и поведению соответствуют не карбиду алюминия, а карбидам щелочноземельных металлов, т. е. в случае карбидов рассматриваемых элементов речь идет об ацетилидах МС2, в которых металлы электроположительно двухвалентны *.
Склонность к образованию типично вомеополярных соединений, соединений с органическими радикалами, у скандия н его аналогов проявляется незначительно. Взаимодействием хлоридов с реактивом Гриньяра якобы были получены алкильные производные этих металлов в форме продуктов присоединения с эфиром (Pletz, 1938). Ацетилацетонаты, например Sc(C5H7O2)3 (т. пл, 187°, т. возг. в вакууме 200°), также можно, по-видимому, рассматривать как гомеонолярные соединения.
Незначительная способность к образованию типичных металл органических соединений (алкил- и арилнррнзводних), помимо элементов побочной, подгруппы III группы, характерна также для остальных переходных элементов вплоть до эле-ментов побочной подгруппы VIII группы включительно. Такие соединения получаются легко только для хрома (о соответствующих соединениях платины см. стр. 369). В противоположность этому для всех металлов побочных подгрупп I и II групп известны алкильные й арильные производные; в случае металлов побочной подгруппы II группы эти соединения Получаются особенно легко.
Структура кристаллических решеток металлов. В металлическом состоянии элементы побочной подгруппы III группы кристаллизуются в гексагональной плотнейшей упаковке (так же, как элементы побочной подгруппы IV группы). Sc и La, кроме того, могут также кристаллизоваться в кубической плотнейшей упаковке (кубической гране-центрированной). В случае La это устойчивая при высоких температурах модификация. В табл. 5 приведены константы решеток, рентгенометрически измеренные
Таблица 5
Константы решеток, плотности, атомные и иопные радиусы элементов побочной подгруппы Ш группы
a-Sc Гекса-го1ДОн»« пан плотнейшая тиаковна б-Sc Кубическая плотнейшая упаковка Y Гексаго-напъпая плотнейшая упаковка a-La Гексагональная плотней- шая упаковка ₽-La Кубическая, плотнейшая упаковка Ас Кубическая плотнейшая упаковка
Константы решеток, А а=3,309 с =5,273 а = 4,541 а — 3,647 с = 5т731 о=3,770 с =6,079 а= 5,296 а = 5,31
Плотность (рентгенометрическая) 3,02 3,20 4,47 6,19 6,19 10,06
Атомный радиус (rKZ=ia), А 1,641 1,606 1,800 1,877 1,872 1,87
Радиус трехвалентпого иона, А о, 83 1,00 1,21 1,11
плотности, а также кажущиеся атомные радиусы названных элементов. Кроме того, в таблице указаны кажущиеся иоппые радиусы. Пикнометрическй определенные плотности помещены в обзорной таблице на стр. 47.
Сплавы. Поведение металлов побочной подгруппы III группы относительно других металлов хорошо изучено пока только на примере лантана. Последний весьма склонен к образованию сплавов. Почти всегда в них присутствуют не твердые растворы, а соединения, причем часто заметное число их. Принято считать, что остальные металлы побочной подгруппы III группы ведут себя подобно лантану. Сопоставление сплавов лантана и встречающихся в них соединений дано в табл. 57 на стр. 524.
* Экспериментальные данпые имеются, правда, только относительно соединений VC2 и LaCa. Образуется ли также ацетилид скандия или карбид другого типа, еще не установлено.
Третья группа периодической система
51
Среди элементов побочной подгруппы третьей группы особое место занимает актиний, но не из-за химического поведения, а потому, что он вследствие своей особо сильной радиоактивности является короткоживущим и труднодоступным в весомых количествах элементом. Исходя из этого, сначала будут рассмотрены элементы скандий^ иттрий и лантан, актиний же будет описан отдельно.
СКАНДИЙ (SCANDIUM) Sc, ИТТРИЙ (YTTRIUM) Y, ЛАНТАН (LANTHAN) Ьа
Распространение в природе. Скандий, иттрий и лантан наряду с целым рядом других элементов с близким химическим поведением содержатся в отдельных минералах, которые в больших количествах встречаются только в немногих местах, а именно главным образом на Скандинавском полуострове, затем в Европе, на Урале, в Соединенных Штатах, в Южной Америке (преимущественно в Бразилии) и в некоторых областях Австралии, Вместе с аналогичными им элементами скандий, иттрий и лантан образуют группу элементов редких земель.
Окислы этих элементов, редкие земли, подразделяются на две группы. Название одной — группа иттриевых земель — происходит от иттербита (гадолинита) — минерала, найденного первоначально около Иттербй в Швеции, в котором преимущественно встречаются земли этой группы; другая — группа цериевых земель — называется по минералу цериту, в котором содержатся главным образом принадлежащие ей земли. Окислы скандия и иттрия относятся к группе иттриевых земель, в то время как окись лантана — к группе цериевых земель.
Помимо окиси лантана, к группе цериевых земель принадлежат еще окислы церия, празеодима, неодима и самария. К Группе иттриевых земель, помимо окислов скандия и иттрия, принадлежат аелми иттербия, эрбия и тербия. Последние опять-таки являются смесями очень близких земель. Лежащие в их основе элементы, так же как и спутники лантана, относятся к группе лантанидов. Они будут рассмотрены в гл. 10.
Скандий встречается в типичных минералах редких земель большей частью в незначительных количествах. Др сих пор известен единственный богатый скандием минерал — норвежский тортвейтит Sc-JSbO?], который найден также на Мадагаскаре. Цо данным Урбайна (Urbain, 1922), он содержит незначительные количества других редких земель.
В качестве преимущественной составной части, помимо гадолинита (иттербцта) Ц^УЧРеВЕОjo, иттрий содержится еще в ксенотиме (иттриевый шпат) YPO, и в авкаексте, ни об ат е и титанате иттрия. Однако во всех втнх минералах иттрии частично замещен на другие элементы редких земель, в основном из группы иттриевых земель. Лантан содержится прежде всего в церите, ортите и монаците-, правда, он имеется там в меньших количествах, чем церий.
Исторические сведения. В 1788 г. близ Иттербй в Швеции был найден минерал иттербит, названный позднее гадолинитом. В 1794 г. Иоганн Гадолин открыл в нем новую землю, для которой Эккеберг ввел впоследствии название — иттриевая земля. Вскоре после этого (1803)’Клапрот и независимо от нею Берцелиус выделили из другого шведского минерала новую землю, названную цериевой землей. Минерал получил после этого название церит. Впоследствии Мозандеру — ученику Берцелиуса — удалось дальше разделить как цериевую, так и иттриевую земли. Первую он разложил на окислы лантана, церия и дидима, а из второй выделил земли тербия и эрбия, в то время как для главной Составной части оставил название иттриевой земли. Лежащий в основе иттриевой земли элемент —
4*
52
Глава 2
иттрий — был впервые выделен в свободном состоянии (нечистым) Велером в 1828 г. при восстановлении хлорида натрием. Лантан, названный так Мозандером (от греческого Xavftavetv — быть скрытым— потому, что он вследствие отсутствии специфических реакций был обнаружен с большим трудом), был выделен восстановлением хлорида калием; в больших количествах его получили Гиллебранд и Нортон в 1875 г. электролизом расплавленного хлорида.
Приготовление Гиллебраядом и Нортоном больших количеств лантана и других элементов редких земель сделало возможным измерение их удельных теплоемкостей. Это позволило иа основании закона Дюлонга и Пти сделать заключение относительно -атомных весов и валентностей названных элементов. Вначале элементы редких земель вследствие аналогии их соединений с соединениями щелочноземельных металлов и магния считали двухвалентными, их ок ислам приписывали формулу МО.
О диак о рассчитанные из удельных теплоемкостей атомные веса находи л нсь в соответствии только с формулой окисла М2О3.
К тому же заключению, а именно что элементы редких земель следует считать трехвалентными, привели проведенные в то же время исследования Клевеса (Cleves) по изоморфизму соединений редкоземельных элементов с соединениями трехвалентных элементов.
Существование элемента со свойствами скандия было предсказано еще в 1871 г. Менделеевым на основании периодической системы. Он назвал тогда еще неизвестный, но ожидаемый по периодической системе аналог бора и алюминия экабором. Через восемь лот (1879) Нильсоном была выделена из шведского гадолинита и эвксенита новая земля и лежащий в ее основе элемент был назван скандием. Он оказался идентичным предсказанному Менделеевым экабору.
Совпадение свойств, предсказанных для экабора и найденных у скандия, видно из следующего сопоставления:
Эка бор Скандий
Атомный вес — 44
Удельный вес 3
Окись ЕкаО3
Окись, карбонат и фосфат нерастворимы как в воде, так и в щелочах
Сульфат трудно растворим в воде. Он образует двойной сульфат, который не изоморфен квасцам
Атомный вес 44,96
Убельньш вес 3,1
Окись Sc20s
Окись, карбопат и фосфат нерастворимы как в воде, так и в щелочах
Сульфат легко растворим в воде, но трудно растворим в серной кислоте. Оа образует двойной сульфат, который не изоморфен квасцам
Приготовление и свойства. Относительно свойств скандия и иттрия в элементарном состоянии еще имеется мало данных вследствие трудностей получения этих металлов в чистом состоянии.
Лантан впервые был получен в большом количестве и исследован Мутмашюм (Muthmann, 1902).
Мутмани для приготовления лантана (а также других металлов редких земель) использовал электролиз расплава хлорида.
Для плавления соли он соединял угольные электроды топким угольным штабиком, который при прохождении тока раскалялся до интенсивного белого каления. Посла того как соль благодаря этому расплавлялась, пггабик удаляли. Теперь было достаточно проходящего через расплав тока, чтобы поддерживать его в распла-
Третья- группа периодической системы
53
пленном состояния. Позднее этот способ был улучшен главным образом Тромбе (Trombe, 1932) — в основном была понижена температура плавления электролита введением подходящих добавок (КС1 и CaF3). По данным Вейбке (Weibke, 1939), работа при 1000° и достаточно высокой плотности тока дает 50%-ный выход металла по току.
Небольшие количества лантана удается получить методом Гопкинса (Hopkins, 1933) электролизом раствора LaCl3 в этиловом спирте с применением ртутного катода и последующим термическим разложением амальгамы.
Цо сведениям Клемма (1937) небольшие количества лантана можно получить нагреванием его хлорида в атмосфере аргона с жидким металлическим калием.
Более доступен, чем чистый лантан, «смешанный металл». Для его получения используют отходы редких земель, перерабатываемых при производстве калильных колпачков, и избегают трудного отделения лантана от сопутствующих ему элементов. Для многих технических целей «смешанный металл» более подходит, чем чистый лантан, так как он обладает большей реакционной способностью.
По данным Мутманна, чистый лантан обладает способностью к растяжению; его свежеполпрованная поверхность имеет вид белого металла, который на воздухе тотчас тускнеет с окрашиванием. В атмосфере сухого воздуха оп покрывается слоем окисла голубовато-стального цвета, защищающего его от дальнейшего окисления. На влажном воздухе он постепенно превращается в белую гидроокись. Удельный вес лантана равен 6,15; температура плавления, по Кремерсу (Kremers, 1923), лежит при 826°, твердость несколько больше, чем твердость олова. Атомная теплота сгорания больше, чем у алюминия. Расплавленный алюминий энергично соединяется с лантаном, давая прекрасно кристаллизующееся и совершенно устойчивое па воздухе соединенно LaAl4. Лантан очень легко образует сплавы с платиной. Будучи нагрет в струе азота или аммиака, металл образует соединение LaN в виде черной, легко растирающейся массы, которая легко взаимодействует с водой с выделением аммиака. При горении металла в струе воздуха, в основном при сравнительно низкой температуре, наряду с окисью образуется также нитрид лантана. С водородом лантан соединяется очень активно. Согласно Мутманну, лантан при нагревании в токе водорода примерно до 240° раскаляется до красно-желтого свечения, образуя черный продукт, состав которого примерно соответствует формуле LaH3.
По данным Сивертса (Sieverts, 1923), «смешанный металл» (т. е. смесь лантана с другими редкими землями, содержащая около 10% Се) реагирует с водородом уже при комнатной температуре, расслаиваясь при этом на серо-черные листочки. При доступе воздуха продукт реакции не воспламеняется. Теплота его образования была определена Сивертсом (1928) равной примерно 384,6 кал /г «смешанного металла». Это соответствует приблизительно 17,8 ккал/г-акв лантана, если в качестве среднего атомного Веса «смешанного металла» принять атомный вес чистого лантана. Следовательно, теплота образования «гидрида лантана» по порядку величины совпадает с теплотами образования гидридов щелочных и щелочноземельных металлов. Также и объем, занимаемый водородом в гидриде лантана, соответствует объему водорода в гидридах Щелочных и щелочноземельных металлов, на что указал В. Бильтц (Blitz, 1928). Следовательно, гидрид лаптапа в этом отношении примыкает непосредственно к гидридам щелочных и щелочноземельных металлов. Состав продукта, полученного насыщением лантана водородом при комнатной температуре, приближается к составу, определяемому формулой LaH3. Однако Сивертс установил, что содержание водорода в препарате зависит от давления и что при равном содержании водорода давление растет с возрастанием температуры. Поэтому в противоположность Мутманну, Сивертс считал гидрид лантана твердым раствором. Во всяком случае, гидрид лантана представляет собой интересный переход от солеобразпых гидридов, которые образуются щелочными и щелочноземельными металлами, к типичным металлоподобным гидридам со всеми свойствами твердых растворов, какие встречаются у элементов побочных подгрупп периодической системы.
Иттрий был получен Поппом (Рорр, 1864) восстановлением хлорида по методу Велера. Он был описал им как серо-черный металлический порошок, который не окисляется в сухом воздухе, но медленно подвергается действию воды. Иттрий легко раство
54
Глаяа 2
ряется в разбавленных кислотах, однако в концентрированной серпой кислоте — только с трудом. Он не подвергается действию щелочных растворителей. Как установил Кремере, будучи нагрет на воздухе, он загорается при 470°. Кремере получал его электролизом смеси YC13 с NaCl в графитовом тигле, футерованном молибденовой жестью. Иттрий горит не чисто белым светом, как магний и алюминий, а скорее красноватым. В токе хлора он в оси ла меняется уже при 200°.
Металлический скандий первоначально был получен в чистом состоянии Фишером (Fischer W., 1937). Он был выделен электролизом расплавленной смеси КС1, ЫС1 и ScCl3 с применением расплавленного цинка в качестве катода и затем освобожден от цинка вакуумной дистилляцией. Это светло-серый металл с удельным весом 3,1, плавится около 1400° и при атой температуре уже заметно летуч.
СОЕДИНЕНИЯ СКАНДИЯ, ИТТРИЯ И ЛАНТАНА
Соединения скандия, иттрия и лантана имеют общую формулу МШХ3. Следовательно, по своему составу они аналогичны соединениям алюминия, однако их поведение во многих отношениях ближе к поведению соединений щелочноземельных металлов. Их аналогия с последними возрастает при переходе от соединений скандия к соединениям лаитапа. Это проявляется, например, уже у окислов и гидроокисей, которые в противоположность гидроокиси алюминия нерастворимы в растворах щелочей и, несмотря на свою незначительную растворимость в воде, обладают отчетливо выраженным основным характером,
Окисел М|и О3 представляет собой белый порошок, соединяющийся с водой с образованием гидроокиси Мш (ОН)3. Окись лантана соединяется с водой настолько активно, что аналогично негашеной извести ее можно «гасить» водой. Устойчивостью своих карбонатов MJII(G.O3)3 по отношению к воде (при комнатной температуре) названные металлы также больше напоминают щелочноземельные металлы. Правда, при нагревании карбонаты легко разлагаются. Сульфаты М]И(ЗО4)3 также начинают разлагаться при прокаливании. Их давление диссоциации при 900° составляет:
SC2(SO4)3 Y2(SO4)3 La2(SO4)2 11 3 2 ai«w pm cm
Переведение сульфата лантана прокаливанием в окисел без остатка достигается с большим трудом. Нитраты Мш (NO3)3 при прокаливании легко разлагаются.
Хлориды, нитраты и ацетаты скандия, иттрия и лантана легко растворимы. Помимо окисей (соответственно гидроокисей), трудно растворимы фториды, карбонаты, оксалаты и фосфаты. Растворимость сульфатов сильно снижается при переходе от очень легко растворимого сульфата скандия к трудпорастворимому сульфату лантана. Сульфиды скандия, иттрия и лантана, как и сульфиды щелочноземельных металлов, можно получить только сухим способом. При кипячении с водой они разлагаются. Многие из названных солей склонны к образованию двойных (комплексных) солей с солями щелочных металлов. Образуются также й другие продукты присоедипения, например с аммиаком. К воде названные элементы в своих солях также проявляют ярко выраженное сродство. Это подтверждается тем, что не только легкорастворимые, но даже и труднорастворимые простые соли кристаллизуются большей частью со значительным содержанием воды.
Водные растворы солей содержат бесцветные ионы Sc'", Y'" и La"'. Исключение составляет водный раствор сульфата скандия, который содержит преимущественно комплексные ионы, вероятно [Sc(SO4)3]’".
Третья группа, периодической системы
55
С некоторыми неметаллами Sc, Y и La образуют соединения, которые по своему составу соответствуют не обычным валентным соединениям, я скорее приближаются к интерметаллическим соединениям. В качестве примеров таковых следует привести YB0, LaBe, ScSi2, YSi2, LaSi2, LaS2 и LaSe2 (наряду c La2S3 и LaaSe3; для Sc и Y известны только сульфиды и селениды типа M2R3). Среди силицидов LaSis является изотипом ThSiz. YSi2 (а также SmSi2) кристаллизуется по типу, родственному одному из них; напротив, как можно заключить из рентгенограммы, ScSi2 обладает иной структурой решетки (Brauer G., £952).
Окпслы Sc3O3, YaO3 и La2O3 получают в виде белоснежных рыхлых порошков прокаливанием гидроокисей, карбонатов или оксалатов. В то время как полученный прокаливанием Sc203 трудно растворим в холодных разбавленных кислотах, La2O3 даже после сильного прокаливания легко растворим в кислотах. Согласно рентгенометрическим определениям параметров решеток, удельные веса окислов составляют: Sc203 3,09; Y2O3 5,01; La2O3 6,48. Пикнометрические исследования Дают отчасти несколько отклоняющиеся значения, что объясняется, по-видимому, загрязненностью исследуемых препаратов.
Получение чистой окиси скандия (а также и других соединений скандия) лучше всего производить способом В. Фишера [Fischer W., Z. anorg. Chem., 249, 146 (1942)]’. Для этого солянокислый раствор сырой окиси смешивают с NH4SCN и взбалтывают с эфиром. Полученный эфирный раствор скандия и виде роданида упаривают, роданид разлагают азотной кислотой и из разбавленного водой нитратного раствора осаждают аммиаком чистый скандий в форме гидроокиси. Этот способ при соответствующем видоизменении удается также использовать для железосодержащих препаратов скандия.
Окислы Sc2O3 и У2О3 кристаллизуются в кубической системе. Структуру их решетки (тип Sc3O3) удается вывести из структуры решетки СаТ2 (см. т. I, стр. 298, рис. 62), если из последней удалить четвертую часть анионов, а остальные ионы немного
Рис. 15. Элементарная ячейка окиси лантана La2O3 и проекция положений атомов на плоскость ас. а = 3,эи А; с = G.151 А; La_-+0 = 2,43 А.
сдвинуть с их мест. Laz03 образует приведенную на рис, 15 гексагональную слоистую решетку; а = 3,945, с = 6,151 А. То, что речь идет при этом о слоистой решетке, видно из правой фигуры рисунка, которая является проекцией положений атомов на плоскость ас. Каждый атом La окружен почти на равном расстоянии четырьмя
56
Глава 2
атомами О, Расстояние между атомом La, обозначенным па рисунке цифрой 1, и атомом О с цифрой 2 составляет 2,428 А. Расстояние между тем же атомом La и атомами О с цифрами 3,4 к 3 составляет 2,424 А. Расстояние до атомов О с цифрами 4,7 и $ существенно больше (2,697А).
Изотипами La3O3 являются Се2О3, Рт2О3 и Nd2O3, а также интерметаллические соединения Mg3Bi2 и Mg3Sb2.
Относительно магнитной восприимчивости и о парамагнетизме La3O, см. Klemm, Z. anorg. Chom., 256 , 37 (1948).
Гидроокиси Sc(OH)3, Y(OH)3 и La(OH)3 получаются н виде слизистых белых осадков при добавлении к растворам соответствующих солей растворов щелочей или аммиака. Они нерастворимы в избытке осадителей и высыхают на воздухе в фарфороподобные массы состава, соответствующего формулам, приведенным выше. Гидроокись .тантана — достаточно сильное основание. Она жадно притягивает из воздуха СО3 и вытесняет NH3 из солей аммония. Гидроокись иттрия также обладает этими свойствами, хотя и в более слабой степени. Еще слабее проявляется основной характер У гидроокиси скандия. Относительно приготовления Sc(OH)3, Y(OH)3 и La(OH)3 (а также гидроокисей лантанидов) в кристаллическом состоянии и о структурах их решеток см. [Fricke В., Z. anorg. Chem., 254, 107, 116 (1947); 255, 13 (1947/1948); 256, 226 (1948)].
При нагревании гидроокиси переходят сначала в метагидроокиси МО(ОН). Только при более сильном нагревании происходит полное отщепление воды с образованием окиси.
Перекисные соединения. Если соли лантана осаждать щелочью в присутствии перекиси водорода, то образуется соединение, которое можно рассматривать как основную соль лантана и перекиси водорода. Если не учитывать содержание воды, то состав этой соли соответствует формуле La(OH)aOOH. ‘
Разбавленная серная кислота, а также угольная кислота вытесняют из нее перекись водорода. Аналогичное соединение образует также иттрий.
Галогениды. Фториды Sc, Y и La в воде трудно растворимы, остальные галогениды легко растворимы в воде, а также н спирте.
Фториды Sc, Y и La выпадают в осадок при добавлении плавиковой кислоты к растворам соответствующих солей. Фторид скандия очень склонен к образованию двойных фторидов (фтороскандиатов), которые довольно легко растворимы, так что скандий не осаждается из своих растворов нейтральным фторидом, например NH4F.
Фтороскандиаты принадлежат в большинстве случаев к типу MI[ScFel, однако били описаны также соединения типа Mj{ScFs] и M-'-lScFJ. Лсгкорастворамый гекса-фтороскандиат аммония (NH4)3 [ScFe] осаждается в внде октаэдрических кристаллов при упаривании нейтрального раствора соли скандия, содержащего NH4F. Из раствора этой комплексной соли Sc(OH)s не выделяется аммиаком даже при Кипячении. Sc(OH)a, однако, осаждается NaOH или КОН. Гексафтороскандиат аммония разлагается также кислотами. Соль калия растворяется несколько труднее, чем соль аммония. Очень трудно растворима соль натрия.
Фторид скандия ScF3 (d = 2,5) кристаллизуется в ромбоэдрической системе, фторид иттрия YFS (с? = 4,01) — в кубической и фторид лантана LaF3 (d! = 4,49) — в гексагональной. Замечательно, что YF3 образует твердые растворы с CaF2, Хотя оба соединения имеют различный состав и различную структуру решеток. В таком случае говорят об «аномальных твердых растворах». Твердый раствор YF3 в СаК2 обладает решеткой плавикового шяата, в которой избыточные ионы F~ неупорядоченно распределяются в междоузлиях. Высокотемпературная форма диморфного двойного соединения NaYF4 (0-NaYF4, d25 = 3,87) также кристаллизуется но типу плавикового пшата (а =5,45 А); места ионов металла занимают при этом: неупорядоченно распределенные ионы Na+ и Y^L a-NaYF4 (d35 = 3,87) обладает сложным строе-
Третья группа периодической система
57'
наем решетки. Фторохиеъ иттрия YOF также диморфна. Мета стабильная при общиной температуре fl-YOF (d'-b = 5,18) образует решетку плавикового шпата, в которой' ионы О2' и F* статистически распределены в местах анионов (а = 5,363 A.). a-YOF кристаллизуется в родственной, вероятно тетрагональной Искаженной, решетке (Hund, 1950—1951).
Безводные хлориды образуются при нагревании смешанных с углем окйслов в токе хлора. Они представляют собой белые расплывающиеся массы следующего удельного веса: ScCl3 2,39; YC13 2,67; LaCl3 3,818. Их температуры плавления составляют: ScCL 960°, YC13 721°, LaCl3 862°. Из водного раствора кристаллизуются гидраты, а именно: ScCl3 и YCI3 обычно с шестью молекул ами H2O, LaCl3c 7Н2О. Гидролиз в 0,1 и. растворе при 14° для ScCl3 составляет 0,9%, для YC13 0,01%и для LaCI3 0,003%. При обезвоживании гидратов хлоридов нагреванием на воздухе также легко происходит отщепление НС1, особенно в случае хлорида скандия, который переходит при этом в нерастворимый оксихлорид, устойчивый' по отношению к кислотам и основаниям.
Способность к образованию двойных хлоридов с хлоридами щелочных металлов очень незначительна. Из водного раствора Майер (Meyer R.) смог получить двойной хлорид только с хлоридом цезия Cs3LaCle-4H3O, который образуется в виде небольших белых кристаллов.
В то же время соответствующее соединение скандия не было получено в чистом виде вследствие его чрезвычайной растворимости. Двойной хлорид лантаиа и пиридин ия [CjIIsNHlgLaaClg. 2С2Н5ОН бтлл получен из спиртового раствора. Безводный LaCl3 кристаллизуется, как и LaF3, в гексагональной решетке. То же относится и к БаВг3. Напротив, La23 кристаллизуется в ромбической решетке. Бромиды и иодиды имеют следующие температуры плавления:
ScBr3 YBr3 LaBr3 Scl3 YI3 Lal3
960° 904я 763я 945я 1000я 761я
8сВг3 имеет плотность 3,91.
Пптраты. Нитрат скандия Sc(NO3)3-4H2O кристаллизуется в виде бесцветных призматических сильно расплывающихся кристаллов при охлаждении предварительно упаренного на водяной бане раствора Sc(0H)3 в разбавленной азотной кислоте. Нагреванием на водяной бане соль удается обезводить. При несколько более сильном Нагревании происходит ступенчатое отщепление с образованием основных нитратов Sc(OH)(NO3)2. Н3О и ScO(NO3). Уже после слабого прокаливания остается чистая окись. В воде и в спирте нитрат скандия легко растворим. Двойные нитраты не известны.
Нитраты иттрия и ла нт а на кристаллизуются как гекса гидраты Y(NO3)3 • 611,0 и La(NO3)3 6Н2О в виде больших расплывающихся столбиков. Они также легко растворимы в водо и спирте. При нагревании сначала получаются основные соли, затем-Окисли. С нитратами щелочных металлов, а также с нитратом магния нитрат лантана образует хорошо кристаллизующиеся двойные соли состава М|[La(NO3)6]. Двойной нитрат аммония (NH4)2[La(NO3)s] 411,0 очень удобен для очистки лантана фракционированной кристаллизацией.
Сульфаты. Сульфат скандия Sc2(SO4)3 • 6Н2О осаждается в виде бесцветных кристаллов после многочасового стояния на холоду раствора гидроокиси или карбоната скандия в разбавленной серцой кислоте, который предварительно был унарен до состояния сиропа. В абсолютном спирте он нерастворим; напротив, очень легко растворим в воде [по данным Крукса (Crookes), 80 г 8с3(8О4)3 растворяется в 100 г воды при 12°; ио сведениям Бирса (Wirth), 39,9 г в 100 г воды при 25°] и также хорошо растворим в в оду содержащем спирте. В крепкой серной кислоте он мало растворим. При выдерживании на воздухе он отщепляет одну молекулу воды и переходит в Sc2(SO4)3 • 5Н3О. Последний является стабильным при 25° даже при соприкосновении с раствором. Нагреванием до 250° удается получить безводный сульфат. При температуре темнокрасного каления уже происходит отщепление молекулы S03 с образованием основной соли Sc3O(SO4)2. Нагреванием до светло-желтого каления подучают чистую окись.
58
Глава 2
С сульфатами щелочных металлов сульфат скандия образует комплексные соли; M^tSetSOJa], Mj [Sc2(SO4)5] и Mj[Sc(SO4)3]. Раствор нейтрального сульфата также часто содержит скандий в форме комплексных ионов, соответствующих этим солям. Это следует из своеобразных аналитических реакций на скандий: последний лишь частично или очень медленно осаждается из сульфатных растворов под влиянием таких характерных осадителей, как тиосульфат натрия или щавелевая кислота. Сравнительно незначительная электропроводность этих соединений также указывает па ауто-комплексообразование. Исследования переноса ионов показали, что скандий в растворе своего сульфата в значительном количестве перемещается к аноду (Mayer И.). Из растдора сульфата скандия в концентрированной серной кислоте осаждается комплексная кислота H3[Sc(SO4)3J.
Сульфат иттрия, кристаллизуется ИЗ растворов окиси в серной кислоте в виде умеренно растворимых бесцветных моноклинных кристаллов октагидрата Y2(SO4)3- 8Н2О. Растворимость при 1.6° составляет 7,47 г, при 95°—1,99 г Y2(SO4)3 в 100 г воды. Он легко обезвоживается, переходя в безводную соль Y2(SO4)3, представляющую собой белый порошок удельного веса 2,52. Из концентрированного сернокислого раствора кристаллизуется комплексная кислота Ii3|Y(SO4)3]. Из смеси растворов сульфатов иттрия и щелочных металлов осаждаются комплексные соли, например (NII4)4[Y2(SO4)s], K8IY2(SO4)7], K0|Y4(SO4)9].
Сульфат, лантана образует обычно Эннеагидрат La2(SO4)3 9Н2О, который значительно труднее растворим, чем водный сульфат иттрия. Его растворимость при 0° составляет 3,0 при 14—2,6 при 100°—0,7 г La2(SO4)3 в 10Q г воды. Напротив, безводная соль, представляющая собой белый гигроскопический порошок, хорошо растворима в ледяной воде; однако как только раствор несколько нагреется, из пего выпадает трудно растворимый гидрат. Помимо эннеагидрата, получают также еще и другие гидраты сульфата лантана, которые, однако, устойчивы лишь при определенных условиях. С сульфатами щелочных металлов сульфат лантана образует двойною солн (большей частью очень труднорастворимые, особенно в избытке раствора сульфата щелочного металла) преимущественно составов М1 [La(SO4)3} и М| [La(SO4)3]. Была получена также кислота H3[La(SO4)3], соответствующая солям последнего типа.
Карбонаты. Нейтральные карбонаты Sc, Y и La получают из растворов соответствующих солей осаждением карбонатом щелочного металла па холоду. Если осаждение проводят в горячем растворе, то получают осадок, загрязненный основном солью. Нейтральные карбонаты содержат кристаллизационную воду. Карбонат скандия Sc3(CO3)3- 12Н3О сравнительно легко растворим в избытке раствора карбоната щелочного металла. Карбонат иттрия Y2(CO3)S-3H2O также растворим в избытке раствора карбоната щелочного металла. Из раствора получаются двойные соли типа Mi[Y(CO3)2] (+ кристаллизационная вода). Карбонат лантана La2(CO3)3 • 8Н2О, кристаллизующийся в ромбической системе, встречается в природе под названием лантанит. В концентрированных растворах карбонатов щелочных металлов этот карбонат также растворим. При разбавлении растворов осаждаются кристаллы двойного карбоната, например K[La(CO3)2]-6H2O. При нагревании карбонаты отщепляют СО2, переходя сначала в основные карбонаты, а при прокаливании — в окиси, В случае карбоната скандия отщепление СО2 происходит настолько легко, что обезвоживания не удается достигнуть нагреванием без одновременного отщепления СО2.
Ацетаты Sc, Y и La, которые получают действием уксусной кислоты на соответствующие гидроокиси или карбонаты, легко растворимы.
Основные ацетаты осаждаются из растворов аммиаком. Если осаждение вести осторожно, то их можно получить в коллоидной форме в виде гелей или даже золей. Полученный таким образом ацетат лантана окрашивается в синий цвет иодом в результате образования продуктов адсорбции, аналогичных продуктам взаимодействия иода с крахмалом *.
Оксалаты Sc, Y и La состава Sc2(C2O4)3 5Н3О, Y3(C2O4)3 9Н2О и La3(CaO4)3-• ЮН20 выпадают при добавлении щавелевой кислоты к нейтральным или слабокис-
* Для образования этих синих продуктов адсорбции иода решающее значение имеет не только коллоидный характер основных ацетатов лантана и крахмала; их образование, по-видимому, связано с наличием определенных своеобразных атомных группировок у названных и других веществ, которые дают с иодом синее окрашивание. Другие коллоидные основные соли лаптапа не дают окрашивания. То же относится к солям лантана с органически мм кислотами (за исключением пропионата лантана). Так же, как лантан, ведет себя в этом отпошепии соседний с ним празеодим, а также неодим.
Третья группа периодической, системы
59
дым растворам соответствующих веществ в виде творожистых осадков, которые посте* пенно становятся кристаллическими. Их растворимость в воде падает в направлении от соединения скандия к соединению лантана. Для пос.’идиего опа составляет 0,20 .на безводной соли в 100 г воды при 25°. Значительно больше растворимость в разбавленных сильных кислотах; например, 100 мл. 1 н. серной кисло гы при 25° могут растворить 115 мг оксалата скандия или 173 -на оксалата иттрия или 398 мг оксалата Лантана. Значительна также растворимость в горячих концентрированных растворах оксалатов щелочных металлов. Лучше всего в них растворяется оксалат скандия; при охлажден нии растворов осаждаются двойные оксалаты типа M.J[Sc(C2O4)3] . 5Н2О. Значительно меньше в них растворимость оксалата иттрия, который, однако, также образует комплексные соли, например K3[Y(C2O4)3]-9H2O. Для лантана, напротив, не известно никаких оксалатных комплексов.
В соответствии с этим оксалат лантана малорастворим в горячем растворе оксалата аммония и при охлаждении осаждается вновь без изменений. Различная растворимость оксалатов в горячих насыщенных растворах оксалата аммония имеет важное .значение для разделения редких земель.
Аналитические сведения. Скандии, иттрий и лантан удается осадить из сильнокяслых растворов щавелевой кислотой. Они обладают этим свойством, так же как и другие элементы, относящиеся к труппе редких земель (а также торий), однако отличаются в этом отношении от алюминия л щелочноземельных металлов, к ноторым онн в других случаях близки.
Скандий отличается от своих более тяжелых аналогов (и от всех других редкоземельных металлов) способностью осаждаться тиосульфатом; эта способность обусловлена образованием труднорастворимтах основных солей вследствие гидролитического расщепления тиосульфата скандия. Используя растворимость фторида скандия в растворе фторида аммония (вызванную образованием двойного фторида), также можно отделить скандий от других редкоземельных металлов, которые образуют нерастворимые фториды. Однако в соответствии с данными Фишера (Fischer W., 1942) существенно лучше удается провести отделение скандия от других элементов, относящихся к группе редких земель, путем экстракции эфиром на водного раствора роданида аммония.
Иттрий и лантан также довольно легко разделяются благодаря различиям в растворимости их солей. Подобно скандию, их можно идентифицировать, соответственно испытывать па чистоту, определяя удельные веса их окислов, точнее определяя эквивалентные веса. Достаточно характерной реакцией на лантаи является синее окрашивание иодом * поверхпостноактивного основного ацетата лантана.
Однако практически дело идет всегда не о разделении трех названных веществ, а о разделении совокупности всех остальных редких земель и к тому же часто еще об отделении их от тория. 0 применяемых для этого методах речь будет идти в гл. 10.
АКТИНИЙ (ACTINIUM) Ас
Актиний встречается совсем в незначительных количествах в урановых рудах. Содержание актиния в урановой руде составляет приблизительно Usee содержания радия. Актиний был открыт Дебиерном в 1899 г. в отходах от переработки смоляной руды. Его извлекают оттуда в процессе переработки руды на редкие земли, точнее на лантан, к которому примешан актиний и от которого его смогли полностью отделять только в последнее время. Ранее наиболее сильно обогащенные препараты (Lub, 1937; Регеу, 1946) содержали только 1 — 2% Ас2О3; остаток состоял из LasO3. Только в 1950 г. при помощи ионного обмена впервые удалось достигнуть приготовления чистых соединений актиния (в минимальных
* Следует, однако, учесть, что (как уже было упомянуто) еще и другие вещества дают с иодом синее окрашивание, а именно основной ацетат празеодима и различные чисто органические вещества. Основной ацетат неодима дает с иодом фиолетовое окрашивание.
60
Глава 2
количествах). Актиний во многом аналогичен лантану, однако еще ближе по химическому поведению он стоит к кальцию.
Актиний радиоактивен. Он распадается, испуская сначала р-лучи и превращаясь в радиоактиний (RdAc), из которого при а-излучении образуется актиний X (АсХ) — изотоп радия. Последний распадается затем дальше подобно радию, т. е. с образованием первоначально газообразных продуктов распада (эманации актиния. AcEni), из которой затем вновь осаждаются твердые радиоактивные вещества (АсА, АсВ и т. д., подробнее см. гл. 11). Актиний пе является настоящим начальным членом-этого ряда, а образуется в свою очередь в результате радиоактивного-распада изотопа урана, так называемого актиноурана. В качестве промежуточных продуктов выступают при этом уран Y и протактиний. Из последнего благодаря распаду с выбросом а-частиц образуется актиний. Так как протактиний обладает атомным весом 231, для актиния на основаппи теории радиоактивного распада (см. гл. 11) получается атомный вес 227.
Так как актиний испускает ^-лучи очень малой энергии, рекомендуется измерять его радиоактивность, определяя, сколько а-лучей испускается продуктами распада актиния, после того как между ними и актинием1 установится радиоактивное равновесие.
Чистые препараты актиния в настоящее время легче получить искусственно методом ядерных превращений, чем из природного сырья. Если радий подвергнуть нейтронному облучению в атомном реакторе, то образуется актиний по следующей ядерной реакции:
₽
22вца д- Гнойтрон —> 227Ra —> 2afAc
Таким способом Хагемапн (Hageniann, 1950), взяв 1 г радия (в форме RaBr2), получил 1,3 мг чистого актиния. Актиний удалось отделить количественно от радия экстракцией его в форме растворимой в бензоле внутрикомплексной соли, которая образуется с теноилтрифторацетоном-(три фтордикето бутил тиофен F3C-CO -СНа-СО 'C4H3S).
Положение актиния в периодической системе, т. е. его порядиовый номер в настоящее время, так же как и его атомный вес, однозначно определен из генетических отношений актиния к другим элементам, порядковые номера которых и атомные Веса были определены непосредственно. Таким методом оказалось возможным наверняка установить атомный вес и порядковый номер актиния только тогда, когда удалось прочно экспериментально обосновать теорию радиоактивного распада и связанные с ним явления (ср. гл. 11 и 13). Однако место актиния в периодической система было правильно определено уже ранее, причем не только на основании его химической аналогии с лантаном, ио и из его трехвалентности. Трехвалентность была впервые достоверно установлена Хевеши путем определения константы диффузии иона.
По данным Нернста, между константой диффузии соли, электролитической подвижностью ее ионов и их валентностями имеется простая связь. Так как электролитические подвижности элементарных ионов (исключая Н‘ и Li’) колеблются только-внутри достаточно узких границ, то на основании констант диффузии удается сделать. Заключение относительно валентности. Таким методом Хевеши смог впервые определить валентность большого числа радиоактивных элементов.
СОЕДИНЕНИЯ АКТИНИЯ
В последнее время было приготовлено несколько соединений актиния в чистом состоянии (большей частью в количествах нескольких микрограммов) и исследована их структура (Trice!, Hagcnrann, Zachariaseu, 1950). Оказалось, что они являются нзотипами соответствующих соединений лантана. Только безводный фосфат АсРО^ не является изотипом соответствующего соединения лантана (в противоположность-АсРО4-1/гН2О).
Третья группа периодической системы
61
Галогениды. Фторид актиния AcF3 к ристал ли дуется, в гексагональной решетке (а = 4,27, с = 7,53 А.). Помимо LaF3 (а = 4,140, с=7*,336 А), он изотипеп фторидам других редкоземельных металлов, а также UF3, NpF3 и PnF3. То же относится к хлориду актиния АсС13 (а = 7,62, с =4,55 A; LaCl3: а = 7,468, с = 4,336 А) и бромиду актиния АсВг3 (а = 8,06, с =4,68 A; LaBr3; а = 7,951, с = 4,501 А). Оксифторид актиния AcOF кристаллизуется в кубической решетке; он образует решетку типа плавикового шпата (а =5,931 А,; Для LaOF а =5,76 А). Напротив, оксихлорид актиния AcOCl имеет тетрагональную слоистую решетку типа PbFCl. Длины ребер элементарных ячеек (содержащих две формульные единицы) составляют:
PbFCl PbFBr J.aOCl LaOBr AcOCl AcOBr
(2=; 4,09 4,18 4,113 4,147 4,24 4,27 A
С — 7,21 7,59 6,871 7,376 7,07 7,40 A
Халькогениды. Окись актиния АсгО3 кристаллизуется в гексагональной решетке по типу окиси лантана (см, стр. 55), а = ifi"’ с = 6,29 А. Сульфид актиния Ас283 кристаллизуется в кубической решетке (а = 8,97 А) и является изотипом La2S3 (а = 8,706 А) и Ce2S3 (см. стр, 534),
Гл а в а 3
ЧЕТВЕРТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Побочная подгруппа)
ТИТАН, ЦИРКОНИЙ, ГАФНИЙ И ТОРИЙ
ПОРЯДКОВЫЙ номер Элемент Символ Атомный вес У де ль-цый все Температура, °C Удельная теплоемкость Валентность
плавления кипения
22 Титан Ti 47,90 4,49 1670 3260 0,113 II, III, IV
40 Цирконий Zr 91,22 6,53 1857 ~ 3600 0,068 II, III, IV
72 Гафний hi 178,50 13,31 2220 - 5200 0,0341 II, III, IV
90 Торий Th 232,05 11,71 1800 -4200 0,034? II, III, IV
Общие сведения. Элементы побочной подгруппы четвертой группы титан, цирконий, гафний и торий очень близки по свойствам к находящимся в тех яге рядах элементам побочной подгруппы третьей группы (скандий, иттрий, лантан и актиний), но отличаются тем, что обычно они четырехвалентны. Во многих отношениях эти элементы близки также к элементам главной подгруппы четвертой группы, но в отличие от них (как и все элементы побочных подгрупп) никогда не фигурируют в качестве электроотрицательной составной части соединений. За исключением титана, который довольно легко восстанавливается до низших степеней окисления, элементы побочной подгруппы четвертой группы в своих соединениях почти всегда четырехвалентны. В отличие от элементов главной подгруппы четвертой группы у элементов побочной подгруппы с увеличением атомного веса не наблюдается тенденции к переходу в двухвалентное состояние.
Элементы побочной подгруппы четвертой группы образуют твердые, очень трудно летучие и очень трудно растворимые окислы; последние в отличие от соответствующих соединений главной подгруппы весьма легко реагируют с перекисью водорода с образованием перекисей или перекисных соединений, отличающихся сравнительно бодыной устойчивостью.
У титана в состоянии низших степеней окисления впервые во всем этом ряду элементов появляются окрашенные простые электролитические ионы. Это свойство — образовывать окрашенные простые электролитические попы — сохраняется и у элементов последующих побочных подгрупп и исчезает, когда побочные подгруппы смыкаются с главными, а именно в побочной подгруппе второй группы.
Ни один из элементов главных подгрупп периодической системы не может образовывать окрашенные элементарные электролитические ионы.
Четвертая группа периодической системы
63
Своим отношением к водороду элементы побочной подгруппы четвертой группы резко отличаются от элементов главной подгруппы. В этом они более близки элементам побочной подгруппы третьей группы (скандий, иттрий, лантан). Действительно, металлы побочной подгруппы четвертой группы, хотя и способны присоединять значительные количества водорода, но эти количества зависят от температуры и давления.
При комнатной температуре титан и цирконий могут присоединять до двух, а торий — больше трех атомов водорода да каждый грамм-атом металла. С увеличением температуры способность присоединять водород падает. Выше 1000° количество абсорбированного водорода приблизительно пропорционально корню квадратному из давления водорода. Это указывает на то, что в данном случае водород растворен в металле преимущественно в атомарном состоянии.
При пониженной температура характер зависимости количества присоединенного водорода от давления изменяется. Как показывают рентгенометрические исследования (Hagg, 1931), это связано с тем, что только Нри небольших количествах водорода атомы его входят в решетку металла, не изменяя ее; в присутствии большого количества водорода образуются фазы с новой кристаллической структурой, обладающие характером недалыпокадны-л соединений. Тиган, например, образует с водородом твердый раствор, содержащий до 33 ат.%Н. При более высоком содержании водорода появляется новая фаза, гомогенная в области 50 —66,7% И. Она имеет гранецентрированную кубическую решетку, в которой атомы водорода, по-видимому, группируются подобно нонам фтора в решетке флюорита (см. т. I). Узлы решетки, Находящиеся в распоряжении атомов водорода, заняты це все, минимум половина из них. Состав соединения поэтому может колебаться от TiH до TiHa, Кажущийся радиус атома водорода в решетках Гидридов металлов побочной подгруппы четвертой группы составляет 0,45 А,
Ранее многократно дебатировался вопрос о том, являются ли гидриды металлов такого тина твердыми растворами или соединениями. В настоящее время считают, что образование только твердого раствора или соединения зависит от количества Поглощаемого водорода.
Средн окислов металлов побочной подгруппы четвертой группы у двуокиси титана преобладают кислотпые свойства, у двуокиси циркония и гафния -ч- основные, а двуокись тория проявляет только основные свойства. Такое усиление основного характера окислов или соответствующих им гидроокисей с увеличением атомного веса соответствует общему правилу.
Теплота образования двуокисей в побочной подгруппе IV группы иногда значительно выше, чем у двуокисей главной подгруппы (ср. т. 1, стр. 450). В табл. 6 приведены теплоты образования некоторых простых соединений металлов побочной подгруппы IV группы.
Таблица 6
Теплоты образования соединений металлами побочной подгруппы IV группы
(ккал /г-эк в металла)
55,7 [2е02]монокл 64,5 (НЮг1монокл67,9 [ТЬО2]куб 73,13 жидк <5 [TiN] 26,8 [ZrN] 27,4 [Th3NJ 26,0 [TiC] 28? [ZrC] 11,2 [ThC2] 11,4
Из соединений металлов побочной подгруппы IV группы с неметаллами по тину каменной соли кристаллизуются следующие: TiG (а — 4,315 A), TiN (а = 4,225 А), TiO (а = 4,235 A), ZrC (а = 4,687 A), ZrN (а = 4,63 A), HfC (а = 4, 458 А) и, по всей вероятности, также HfN. По типу флюорита кристаллизуется ThO2 (а — 5,57 А), а также ZrO2 и ШО2, если они содержат определенные примеси (см. стр. 87). решетка типа брусита (слоистая решетка) найдена у TiS2 (а — 3,40 А, с — 5,69 A),TiSe2
-64
Глава 3
(а = 3,53 к, с = 6,00 А), ТПе3 (а = 3,79 к, с = 6,45 A), ZrS2 (а - 3,68 А, с = — 5,85 А) и ZrSo3 (а = 3,79 А, е — 6,18 А). Соединения TiSe и TiTe кристаллизуются ио типу NiAs (а — 3,56, с = 6,22 А и а = 3,83, с = 6,39 А), Решетка типа NiAs (рис, 43) может непрерывно переходить в решетку типа брусита (ср, т. I, рис, 61) путем постепенного удаления ионон металла из среднего слоя (наличием которого структура NiAs отличается от структуры брусита), причем удаление ионов металла из кристаллической решетки происходит неупорядоченно статистически. Галогениды металлов побочной подгруппы IV группы образуют большей частью молекулярную решетку.
Титан, особенно в трехвалентном состоянии, в некотором отношении даже более, чем предшествующий ему скандий, сходен с алюминием, стоящим выше в третьей группе. Например, ион Ti’” подобно иону АГ" способен образовывать квасцы. Химические свойства титана в некотором отношении сходны также со свойствами хрома и железа, Четырохвалент-ньтй титан занимает промежуточное положение между алюминием и кремнием. Однако отличается титан от обоих элементов более ярко выраженной тенденцией к образованию двойных или комплексных соединений. Тенденция к комплексообразованию у элементов побочных подгрупп вообще гораздо сильнее, чем у элементов главных подгрунп.
С точки зрения теории Косселя, проявление титаном, цирконием и торием валентности четыре в качестве обычной и вместе с тем максимальной обусловлено их положением относительно предшествующих им инертных газов — аргона, криптона и радона. Отдавая четыре электрона, перечисленные металлы приобретают эдектроп-пую конфигурацию этих инертных гаков. Так как последним соответствуют наиболее стабильные электронные структуры, отщепление больше четырех электронов и, следовательно, проявление более высокой валентности невозможно. В соответствии с теорией Косселя, рассматриваемые элементы пе могут проявлять отрицательную валентность, так как за ними ito следуют инертные газы, электронные оболочки которых могли бы -образоваться путем присоединения нескольких электронов.
Гафний в противоположность упомянутым выше элементам цобочпой подгруппы IV группы удален от предшествующего ему инертного газа ксенона более чем на четыре места. Несмотря на это, его внешняя электронная оболочка аналогична оболочке титана, циркония и тория, и вследствие этого гафпий должен быть четырёхвалентным -Элементом; такой вывод на основе теории строения атома был сделан еще до того, как гафний был открыт (ср. стр. 14 и 95).
Представления о строении атомов титана и циркония, оспованные непосредственно па экспериментальных данных, дают нам анализы спектральных термов. Они позволяют установить вид связи девятнадцатого электрона у титана и тридцать седьмого электрона у циркония в невозбужденных атомах этих элементов. Самый низкий терм у Тг3+ не s-торм (ср, т, I, рис, 59), как в случае кадия или однократно ионизированного кальция, а rf-терм. Это значит (ср. т, I, стр. 282), что электрон, вращающийся вокруг атомного остова Ti4+ (следовательно, девятнадцатый), находится в самом низком энергетическом состоянии на уровне с орбитальным квантовым числом I — 2 (rf-уровепь), а именно па Зй'-уровне. Относительно 20-го электрона в атоме титана на
Энергия ионивации элементов IV группы (ккал/г-атом)
Таблица 7
Главная подгру ива Побочная подгруппа
С Si Ge Sa Р1Э Ti 2г Hf Th
М -> М1+ + е 258,1 186,2 168,4 186,2 170,1 156,9 159,5 — —
М1+-^ MS+4-е 559,1 374,7 366 334,6 344,8 313 322,0 341 —
M3+J-e 1097,9 768,1 786 702,7 735,4 636 553,1 — .—
М3+^М4+ + с 1478,9 1035,2 1049 933 968 992,4 779 — 678
Суммарно:
М М4+-|-4е 3394,0 2364,2 2387 2138 2218 2098 1814 — —
Четвертая группа периодической системы 65
основе анализа термов мы знаем также, что н невозбужденном состоянии он находится на 3cf-уровне. Напротив, 21-й, а также 22-й электроны занимают 45-уровень. Сказанное справедливо для циркония и, вероятно, также для гафния и тория. Вид связи валентных электронов у четырех названных элементов молото передать, следовательно, символами 3d® 4s2, 44s 5sa, 5n!a6s\ 6t/a7sa соответственно. Как видно из табл. 7, энергия, затрачиваемая па отщепление валентных электронов у элементов побочной подгруппы ГУ труппы, угепыпе, чем у элементов главной подгруппы. В соответствии с этим элементы побочной подгруппы обладают более сильным электроположительным характером, чем элементы главной подгруппы,
Структура решетки металла. В элементарном состоянии титан, цирконий и гафний при обычной температуре образуют решётку типа магния (гексагональная плотнейшая упаковка), Длипы ребер элементарных ячеек составляют: для Ti а — =2,9504,0 = 4,6833 А; для Zr а = 3,2312, с=5,1477 А; для Ш а—3. 1946; с = 5,05111. Торий подобно свпггцу кристаллизуется в кубической грапецептрнровапной решетке (кубическая плотнейшая упаковка), а = 5,0843 А.
Выше 885° титан имеет кубическую объёмпоцентрнропаппуго структуру (а — =3,3065 А при 900е). Подобные превращения испытывают циркопий при 862° (а = =3,6090 А при 8639 и торий выше 1400° (а = 4,11 А при 1450°). В табл. 8 приведены кажущиеся атомные и ионные радиусы элементов побочной подгруппы IV группы. Ионные радиусы соответствуют попам с зарядом 4 -|-.
Таблица 8
Кажущиеся атомпьге п ионные радиусы элементов побочной подгруппы IV группы
Элемент Титан Цирконий Гафпий Торий
Атомный радиус (гК2=12), А Ионный радиус, А 1,4615 0,64 1,6025 0,87 1,5805 0,84 1,7976 1,10
Сплавы. В расплавленном состоянии металлы побочной подгруппы четвертой группы, как оказалось, способны растворять другие металлы и образовывать сплавы. Но получение сплавов и металлографическое исследование их очень затруднено высокими температурами плавления этих металлов и повышенной склонностью реагировать при нагревании с азотом и углеродом.
Вследствие этого имеющиеся сведения о сплавах металлов побочной подгруппы четвертой группы (см. табл. 9) еще весьма недостаточны. Во многих случаях известно лишь, что сплавы образуются, но характер их до сих пор не ясен. В других случаях, для которых в табл. 9 на основе металлографических исследований указана растворимость, частично известна диаграмма состояния. На основе полученных до настоящего времени данных создается впечатление, что металлы побочной подгруппы четвертой группы мало склонны к образованию твердых растворов, по весьма охотно образуют химические соединения с другими металлами. При этом характерно, что число соединений, образуемых с определенным металлом, всегда невелико.
Соединения со щелочными и щелочноземельными металлами, по-видимому, по образуются, Это свойство может быть использовано для получения свободных металлов побочпой подгруппы IV группы из нх галогенидов яли окислов. Не образуют они и твердых растворов. Сведения о взаимодействии в другими металлами (насколько исследованы соответствующие системы) приведены в табл. 9.
Дополнительпо следует назвать соединения Ti4Pb, Ti4Bi и Ti3Pt, С металлами V, Nb, Та, Мо и W f-Ti (т. е. высокотемпературная модификация тйтапа) образует неограниченный ряд твердых растворов; напротив, а-титан может только в ограниченной степени включать в свою решетку названные металлы. Титан неограниченно
5 г. Реми
Таблица 9
Образование соединении н твердых растворов металлами побочной подгруппы IV группы с другими металлами и некоторыми неметаллами
Главные подгруппы По Сочные подгруппы
Ее Б Al с si Sn p Sb s Сг Мп Fe Co NI Cu Ag Au Zn Ilg
Т1 Ве2 L i ТВ,>(? TiB TiBa тв. 0? Т1Л1 TiAl3 13 55° тв.>0 Ti2C? TiC 3450° тв,>0 Ti5Si3 2130° TiSi 1540° TiSi* 1700° Сплав ТЦР TiP I'i4Sb TiSb TiSbg Ti2s TiS TljSg ТВ» со Ti2Cr3 [] TiCr3U? Ti2Mn TiMn2 тв.>0 TiaFe TiFe TiFc2 1530° Ti^Co th, > 0 Ti2Ni -шо° tb, >. 0 Ti3Cu Ti2Cu3? Ticu ж. —O tb. 0 0 ж. — 0 tb.^>0 Ti3Au TiAu2 TiZn TiZn, Спляв
TiS2 TiCo TiNi TiCu3 TiAue
TiS3 TiFe3 1387° TiCo2 TiNi, 13’18°
Мой W
— — ТВ, > 0 — — — — — — — tb.>O tb. >. 0 tb, >. 0 tb.^>0
Zr — ZrB Соединение ZrC ZrSi2 — ZrP — ZrS2 ZrCr2 ZrMo3 Zr0Fe3 1040° — ZrNi3 ZrCu3 Соединение Сплав — Сплав
ZrB2 ZtC2? ZtP2 ZrW2 ZrNif ?
— — тв. О? — — —». — — — — — — тв. >> 0 — tb.~O — — ж, >0
— ThB4 ThAlJ 880° ThC £625° ThC2 2655’ ThSi3 — ThP Th3P4 — Ths — — — ТиЖО 1050» ThNi 1200° ThgNij? — ThsAg5 ThAg3 — —' —
Th ThB6 Th2S3 ТКЩ
T113S7 TEN i5 1530° W 13 50»
Примечание.
Твердые растворы: сб — неограниченная растворимости, ж. — -жидкий, та. — твердый, мость неограниченная.
растворимость, < со ограниченная растворимость, > О — очень нс Польша л растворимость, О отсутствие Там, где сделаны указания только для твердого состояния, в жидком состоянии, Как правило, раствори-
Образо ванне соединении, 0 — соединение не образуется, — диаграмма состояния неизвестна, либо известна весьма неполно. Под формулами указаны температуры плавления или разложения соединений. Значкам * обозначены соединения, плавящиеся ипкопгруаитио.
Соединения отмеченные значком П, образуются либо превращением в твердом состоянии, либо (более редкие случаи) отруктуряьгми превращениями происходящими без изменения состава (ср. стр. S4 л 363). Свсрхсщррктурные фазы отмечены квадратными скобками; приведенные для них темпе-ратурь^ от носятсяв! периоду из упорядоченного состоянии в состояние беспорядочного распределения атомов, т. е. соответствуют температуре, при которой соединение теряет собственную интерференцию.
Недальтонидиые соединения отмечены чертой над формулой.
68
Глава 3
смотпттвается также с цирконием или гафнием, но только при температурах, превышающих температуру превращения.
В табл. 9 наряду с металлами указаны некоторые неметаллы, а именно такие, которые с металлами побочной подгруппы IV группы образуют соединили, близкие по характеру к интерметаллическим соединениям. Ярко выраженными металлическими свойствами обладают также нитриды, состав которых в общем соответствует формуле A1N, Они так же, кик бориды типа МВ и карбиды типа МС этой группы, обладают металлической проводимостью, причем у нитридов и боридов она даже значительно
Таблица 10
Удельная электропроводность металлов и металлоподобпых соединений побочных подгрупп IV и V групп (о.м-1 с.ч-1 при комнатной температуре)
Тита и Цирконий Гафний Ванадий Тантал
Металл 1,11-10* 2,24-10* 3,07-10* 0.6-ю* 6,7-10*
Нитриды, MN 4,6-10* 7,4-10* — 1,16-10* 0,74-10*
Бориды, МВ 6,6-10* 10,9-10* 10,0-10* 6,2-10* —
Карбиды, МС 0,52-10* 1,58-10* 0,92-10* 0,64-10*? 1,0-10*
выше, чем у свободных металлов (ем. табл. 10). В табл, 10 указаны также соответствующие соединения элементов побочной подгруппы V группы, обладающие проводимостью, сравнимой с металлической.
ТИТАН (TITANIUM) Тг
Распространение в природе. В природе титан находится главным образом в виде двуокиси TiOa, которая встречается в различных модификациях — обычно рутил, реже анатаз и брукит. В большинстве случаев природная двуокись титана загрязнена в той или ипой степени окисью железа. Железная руда также содержит значительные количества двуокиси титана. Соединения окислен титана и железа встречаются в виде самостоятельного минерала, титанистого железняка или ильменита FeTiO3. Кроме того, следует упомянуть перовскит, титанат кальция CaTiO3 и титанит или сфен, кал ьцийтитанилсиликат CaTiO(SiOi). Двуокись титана часто встречается в соединениях с редкими землями, В небольших количествах она очень распространена, так что почти любая почва содержит заметные количества титана (в среднем выше: ,0,5%).
Исторические сведения. Титан впервые был открыт в виде двуокиси, В 1789 г. англичанин Грегор в Корпвальском железном песке, мена каните, обнаружил окись неизвестного вещества, вначале названного Кнрвапом «мена хин ом». Независимо от пего в 1795 г. Клапрот обнаружил, что рутил представляет собой окисел неизвестного металла, который он назвал титаном. Вскоре после этого стало известно, что Грегор в менаканите открыл металл, идентичный найденному Клапротом. В 1822 г. Волластон в домеппых шлаках обнаружил похожее на металл соединение титана с углеродом и азотом, которое он принял за чистый титан. Это ошибочное представление о титане распространилось и оставалось довольно долго, несмотря па то что уже вскоре после этого, в 1825 г., Берцелиусу удалось восстановлепием натрием двойного фторида калия и титана получить действительно элементарный, по еще недостаточно чистый титан. Поскольку Берцелиус не сомневался в том, что Дпмоппый титан Волластона является чистым кристаллическим титапом, свой продукт он назвал «аморфным тигапом». Однако Ведер в 1849 г. сжиганием в токе хлора доказал, что домеппые кристаллы представляют собой соединение титана с углеродом и азотом состава Ti6CN4.
Получение, Вследствие большого сродства титана не только к таким обычным в металлургии восстановителям, как уголь и алюминий, но так
Четвертая группа периодической системы
69
же и к относительно инертным газам, как азот, получение чистого металла старыми способами не могло быть осуществлено. Чистый титан удалось получить лишь недавно термической диссоциацией тетраиодида тцтана ио методу наращивания па прутках, описанному для циркония (стр. 84). Для получения особо чистого титана и теперь применяют этот способ.
Достаточно чистый титан бил получен впервые X унтер ом (1910) нагреванием смеси тетрахлорида титана и металлического натрия д стальной бомбе:
TiCl4+4Na=4NaCl-|-Ti.
По данным Билли (1921), более подходящим является взаимодействие тетрахлорида титапа и гидрида натрия:
TiCli-|-4NaH=:4NaC14-Ti+2H2.
От абсорбированного водорода освобождаются нагреванием металла в вакууме до 800°. Кроль (1937) описал способ приготовления титана восстановлением окиси титана металлическим кальцием.
Полученный этим способом металл можно подвергать термической обработке, одпако вследствие присутствия TiO on хрупок в холодном состоянии (хладноломок). Позднее Кроль разработал другой способ, который теперь используют в промышленности (см. далее). Шварц в 1956 г. получил технически чистый титап (98—99%-пый) путем превращения двуокиси титана в сульфид нагревапием с HsS й CS2 при 1000э и последующим в ос становлением его металлическим магнием в токе аргона.
В настоящее время в США промышленное производство чистого титана осуществляют главным образом методом Кроля; восстановление тетрахлорида титапа расплавленным магнием проводят в стальпых бомбах при температуре 850° в атмосфере гелия или аргона. Полученную металлическую губку переплавляют в электрической дуговой печи. Таким путем получают компактные блоки длиной 6 м и диаметром 14 см.
Видоизменением процесса Кроля является способ получения титапа, разработап-цып Маддексом (Maddex) и Эствудом (Eastwood) в Battcllo Momoriol Institute (1950). При комбинировании процессов восстановитель Ной п плавильной печей получают сразу компактный титан. Жидкий магний перекачивают сначала в электрически обогреваемую предварительную камеру, из которой он под Давлением аргона наглотается в стальную восстановительную камеру, Из второго сборника туда поступают пары TiCl4. Образующаяся в результате следующей реакции:
2Mgjn + Т i С14 г=TiTE+2MgCl2
смесь жидкого MgCl2 и суспендированного в нем твердого титана непрерывно перетекает в элеь'тродуговую печь, Хлористый магний и та часть магния, которая по прореагировала, испаряются и конденсируются вне печи. Титан сплавляется в компактные блоки, которые по мере накопления выводят через под печи. Одновременно титан служит электродом для создания электродуги между ним и вольфрамовым стержнем.
Для технических целей требуется не очень чистый Металлический титан. Подходящим в этом случае является сплав титана с железом — ферротитан, содержащий 10—25% титана и плавящийся чаще всего ниже 1400°. Ферротитан можно легко получить восстановлением рутила углем в присутствии железа. Если нежелательно присутствие углерода, в качестве восстановителя используют алюминий. Небольшая примесь А1 в ферротитане в большинстве случаев даже выгодна.
Свойства. В компактном состоянии титап похож на сталь; на холоду он обычно твердый, хрупкий и только при температуре красного каления становится ковким. Однако совершенно шитый титан ковок уже на холоду, Чистый титан также отлично полируется, он обладает ббльшим блеском, чем хром. Температура плавленпя Т1 в соответствии с последними
70
Глава 3
данными равна 1670°. В электрической дуге печи Муассана титан возгоняется, По данным зависимости упругости пара от температуры, температура кипения Ti установлена равной примерно 3260°; удельный вес 4,49 (структуру решетки см. стр. 65).
Молярная теплоемкость С.„ металлического титана повышается от 1,278 при —219,7° до 5,880 при 0° (Kelley, 1944) и до 6,507 при 200°, Менаду 200 и 800° она может быть вычислена по уравнению
=4,1964 -|-0,018208; —0,39114-10~®(2 -)-0,29223 • 10" "i3
(Jaeger, Rosenbohm, 1936), Атолла я тепло емкость кубической модификации титана выше температуры превращения составляет 7,525 и пе зависит от температуры. Молярная теплоемкость титана в парах (Gilles, Wheatley, 1951) имеет ярко выраженный максимум (Ср — 6,578) при 135° К и минимум (Ср = 5,096) при 990е К. При температуре кипения титана Ср == 8,034, при 4000° К 8,667 и при 5000° К 9,708. Такая аномальная температурная зависимость связана с легкой возбудимостью атомов титана; то же наблюдается у железа, Теплопроводность титана составляет приблизительно 0,036 кал/см-сек-ерад; удельное сопротивление Q0o = 0,475.10-4 (для очень чистого титана) и удельная магнитная восприимчивость у2ов = + 3,18-10“®.
При низких температурах на воздухе титан довольно устойчив. Выше температуры красного каления в токе кислорода он сгорает до двуокиси (ср. стр. 63). С азотом соединяется при более высокой температуре (выше 800°), образуя нитрид. Поэтому при нагревании титана на воздухе наряду с окисью образуется нитрид. Легче всего титан реагирует с галогенами: с хлором, например, при температуре немного выше 300°, с фтором уже при 150°. При более высокой температуре титан легко соединяется также с другими неметаллами, образуя соединения, отличающиеся устойчивостью по отношению к химическим агентам. Кроме нитрида TiN, здесь прежде всего следует назвать карбид TiC и карбопитрид титапа Ti5CN4, а также образующиеся в виде похожих на серое железо кристалликов силициды Ti2Si и TiSi2.
Порошкообразный титан активно абсорбирует водород: 1 г титана (Sieverts, 1929) при комнатной температуре поглощает 407 мл водорода, т. е. 1,74 г-атома на 1 г-атом титапа (ср. стр. 63), при 1000° только 66 ль®. В результате поглощения водорода титап заметно расширяется (максимум на 15%) и приобретает более светлую окраску, сохраняя, однако, металлический блеск. Теплота образования гидрида титапа, составляющая, по данным Сивертса (1931), 18,0 ккал/г-атом водорода, превосходит, следовательно, теплоту образования гидрида натрия (ср. т. I, стр. 179). Раскаленный в токе водорода титап на воздухе воспламеняется, и абсорбированный водород сгорает бледным пламенем, образуя воду.
Титан та Ki не в значительной степени может растворять кислород, образуя с ним . твердые растворы (до 30 ат.%). Кристаллическая решетка в этом Случае испытывает лишь незначительное расширение (главным образом в направлении оси с), При более сильном поглощении кислорода образуются окислы (см. табл. 9 па стр. 66).
При высокой температуре титан весьма активно сплавляется с железом, образуя соединенно Fe3Ti, Аналогичный состав имеет соединение титапа с алюминием Al3Ti (серебристо-белые квадратные листочки). Его получают методом алюмотермии. Следует отметить, что сплавы титана с металлами еще довольно мало изучены (ср. табл. 9).
В кислотах титан растворяется труднее, чем железо. Например, в разбавлеппой соляной кислоте он растворяется только при нагревании, с образованием фиолетового хлорида титана. Горячая азотная кислота окисляет титан, подобно олову, в нерастворимую ^-титановую кислоту. Превосходным растворителем для титана и его труднорастворимых соединений является плавиковая кислота.
Применение. Титап применяют главным образом в виде сплавов с железом (ферротитан) в качестве добавки к сталям. Титановые стали
Четвертая группа периодической систем**
71
отличаются прочностью и ковкостью. Для достижения этих свойств достаточно содержание титана менее 0,1 %. Такое действие титана обусловлено его способностью препятствовать выделению кислорода и азота при охлаждении, что приводит к образованию пузырьков. В связи с этим титан в виде сплава с алюминием можно рекомендовать в качестве добавки к меди или ее сплавам. Указанное свойство титана аналогично действию фосфора в фосфористой бронзе. Исключительную способность титана связывать азот и кислород используют также для удаления следов газов из эвакуированных объемов, например электроламп.
С тех пор как удалось наладить производство чистого, годного для холодной обработки титана, его применение приобретает возрастающее значение. Сплавы титана, которые по прочности и способности к удлинению весьма близки к нержавеющей стали, но превосходят ее по коррозионной устойчивости и весу, легче, чем сталь, примерно на 40%. Можно ожидать, что титап приобретет большое значение для строительства самолетов, металлических вагонов, судов и судовых механизмов.
Пористый металлический титан и карбид титана можно использовать в качестве катализатора для получения соединений азота с водородом и азота с кислородом. Наряду с высокой активностью они отличаются также большой износостойкостью. Большое значение карбид титана, так же как карбид молибдена, приобретает благодаря применению его для приготовления так называемых твердых металлов или сплавов для режущих инструментов. Их получают спеканием карбидов титана, молибдена или вольфрама с такими металлами, как кобальт или никель. Последние служат в качестве вязкого наполните л я для перечисленных карбидов, отличающихся твердостью, Сплавы титана для режущих инструментов по эффективности значительно превосходят быстрорежущие стали (см. стр, 288). Вследствие высокой прочности на износ эти сплавы можно использовать даже там, где раньше применяли только алмазы.
Соли титана, особенно двойные фториды и оксалаты щелочных металлов, применяют в качестве протравы при крашении тканей и кожи. Раствор хлорида титана(Ш), который легко можно получить растворением ферротитана в соляной кислоте, рекомендуют как безвредное белящее средство для шелка и шерсти. Определенным образом приготовленную и обработанную (в виде тонкого порошка) двуокись титана употребляют в качестве минеральной краски (титановая белая) для покрытий и в ситцепечатании; кроющей способностью она превосходит цинковые белила, а прочностью — свинцовые белила. В соединении с окисью железа и другими окислами двуокись титана служит для производства окрашенных глазурей для фарфора и фаянса (бунцлауэрский глиняный товар). Бруг-гер (Brugger, 1952) разработал способ покрытия стекол тонким слоем TiO2; такое стекло отличается высоким показателем преломления (п = 2,3).
Растворы хлорида титана(Ш) можно использовать в качестве восстановителя в объемном анализе. Однако их применение осложнено тем, что соединения трехвалентного титана склонны к самоокислению и поэтому приходится работать в индифферентной атмосфере. Растворы хлорида титана(Ш) пригодны во многих случаях для препаративных целей.
СОЕДИНЕНИЯ ТИТАНА
В своих соединениях титан преимущественно электроположительно четырехвалентен, но бывает и трсхвалентным, а в некоторых соединениях и двухвалентным; соединения двухвалентного титана, однако, получаются с большим трудом и в водных растворах малоустойчивы.
72
Глаеа 3
Соединения одновалентного титана до сих пор пе получены, хотя некоторые данные свидетельствуют о их существования (например, в системе Ti — S наблюдается максимум летучести для соединения, примерно соответствующего формуле Ti3S).
Четырехвалентпый титан весьма склонен к образованию анионов (ацидо-ионов), например [ТЮ3]" (титанат-нон), [TiFe]" (фторотитанат-ион), [TiClJ" (хлоротитанат-ион), [Ti(SO4)3]" (сульфатотитанат-ион). От трех-валентнОго титана также производятся ацидо-соли. Однако эти комплексы непрочны, их нельзя перекристаллизовать из чистой воды без разложения.
Все растворимые соединения четырехвалентного титана очень сильно гидролизуются. При неполном гидролизе могут получаться оксисоли типа ТЮХа. Так как эти соединения формально (т. е. судя по их составу) можно произвести от радикала ТЮП (пштакил), их рассматривали раньше как соединения тмтанила. Радикал титанил в водных растворах существует в виде иона [Т1О]", но в кристаллических солях, насколько известно, он не сохраняется в качестве самостоятельной структурной группы.
О К целительный потенциал Ti'"/Ti'"' по отношению к нормальному водородному электроду равен —0,04 в. Следовательно, если погрузить платиновую пластинку в раствор, содержащий равные Количества ионов Ti’" н Ti"’*, и соединить ее с нормальным водородным электродом в гальванический элемент, то ток по проволоке, соединяющей оба электрода, пойдет от нормального водородного электрода к платиновой пластинке, погруженной в раствор титановой соли. На нормальном водородном электроде выделяется водород, в то время как на платиновой пластинке ионы Ti’’’ окисляются
Обзор простейших соединений титана
Фториды X ПО РИДЫ Бромиды ИОДИдЫ
TiF3, фиолетовый TiP4, белый TiCI2> черный TiCls, фиолетовый Т£С14, бесцветный ЖИДКИЙ Т1ВгЕ, черный TiBr;!, чорпый TiBr4, я птар по-желтый Til^? коричнево-черный Tilj, темно-фиолетовый Til^, красно-коричневый
Окислы Сульфиды фосфиды Карбиды
TiO, золотнето- Жыгг.ый Ti2O3, фиолетовый ТцО7?, темпо-синий TiO3, белый TiS^j серый TiS, темно-коричневый Ti2S8, зелеповато-черный TiS2, латунно-желтый TiS3, граф»топодоб-пый ti2P? TiP Ti2C? TiC, черный
нитриды
TiN, золотистожелтый
Четвертая группа периодической системы
73
до Ti"“. Ионы Ti‘" являются поэтому настолько сильными восстанови гелями, что при определенных условиях могут выделять водород из кислых растворов.
Окислительный потенциал пары Ti'7Ti'" равен —0,37 в, а пары Ti‘7Ti''’* составляет —0,20 <4
Нормальный потенциал Ti по отношению к раствору соли Ti(II) составляет —1,75 в. Для перехода: Ti 6F' = [TiFej" + 4с он равен —1,24 в; для Ti + 2Н3О = = TiO2 + 41Г + 4 е составляет —0,9а в и для Ti'" 4- Н3О = [TiO]" + 2Н' + е '! равеп —0,1 в.
J Соединения титана(П)
Соединения двухвалентного титана получают энергичным восстановлением соединений титапа(1¥) или (III). Например, из тетрахлорида титана TiCl4 под действием амальгамы натрия образуется хлорид двухвалентного титана TiCl2 в виде черного порошка; который постепенно разлагается водой с выделением водорода. Водные растворы, которые [наряду с хлоридом титапа(Ш)] содержат хлорид титана(П), можно получить растворением TiO в разбавленной соляной кислоте на холоду, При обычной температуре ионы Ti” довольно быстро окисляются водой в ионы Ti'"; при пониженной температуре, однако, устойчивость растворов значительно возрастает. В свободном состояний TiCl2 (уд. вес 3,13) лучше всего: получать методом Руффа (Ruff, 1923) — термическим разложением TiCl3, или методом Клемма (Klemm, 1942) — нагреванием TiCl4 с титановой стружкой, TiBr2 (уд. вес, 4,31) и Tila (уд. вес. 4,99) удобнее всего получать непосредственно из элементов. Обработкой TiCL жидким аммиаком Шумб (Sclmmb, 1933) получил двойное соединение TiCI2-4NH3 в виде if жемчужпо-серого порошка, который подобно безводному TiCI2 реагирует с водой
с выделением Н2.
TiO получают нагреванием смеси Ti и TiO2 (уд. вес 4,93; т. пл. 1750°). Опа изоморфна каменной соли, но в ее решетки (при неупорядоченном распределении) часть i мест не занята. На одной границе гомогенной области пустоты приходятся на долю
[ атомов титана, на другой — атомов кислорода (Ehrlich, 1939), При растворении в раз-
бавленной соляной кислоте происходит частичное окислепие но уравнению: Ti" 4" + Н- = Ti- + 1/3H2.
Соединения титана(Ш)
Соединения титана(Ш) легко получаются в растворах восстановлением растворимых соединений титана(1У) цинком и кислотой или электролитическим способом. Растворы содержат окрашенные в фиолетовый цвет ионы Ti(III). Они весьма склонны переходить в ионы Ti(IV). Восстановительная способность растворов солей титана(1П) значительно больше, чем i солей олова(П) (ср. окислительные потенциалы).
При окислении молекулярным кислородом («сопряженное окисление#) * (т, 1, гл. 16) соединения титаяа(Ш) требуют в присутствии акцептора больше кислорода, чем его нужно для повышения их заряда па одну единицу. Способность трех-валеитпого титана косвенно вызывать окисление кислородом воздуха настолько велика, что н присутствии соединений титана(Ш) в щелочном растворе даже вода может играть роль акцептора. При этом она переходит в перекись водорода. Если перекись водорода сразу после ее образования связывать гидроокисью кальция, то считают, что на один атом ТПП получается х/2 молекулы кислорода (что равно 2 эквивалентам) и образует^ ; ся т/3 молекулы П2О2:
Т;(ОН)з+1/202-|-П2О=Т1(0Н)44-1/2Н2О2.
г В присутствии других веществ, которые могут проявлять свойства акцепторов,
соединения титана(Ш) при окислении молекулярным кислородом требуют два эквивалента последнего на 1 атом Tiili вместо одного; при окислении хромовой кислотой или перманганатом требуется даже три эквивалента кислорода вместо одного.
* В первом томе (гл. 16) термин «Antoxydation» переведен как «самоокисление#. Это неточно, так как в указанных реакциях автор имеет в виду «сопряженное окисление#. (Ср. стр. 759). — Прим. ред.
74
Глава 3
Хлорид титана(Ш); ацидотитанаты(Ш). Безводный хлорид тита-на(Ш) в виде фиолетового порошка получают пропусканием паров тетрахлорида титана в смеси с избытком водорода через раскаленную трубку [лучше всего через «холодно-горячую трубку» по Сант Клер-Девиллю (St. Claire-Deville)]. При нагревании TiCl3 до 700° в токе водорода он распадается на TiCl2 и TiCl4. В растворе хлорид титапа(Ш) получают восстановлением солянокислого раствора соли титана(ТУ) цинком или растворением металлического титана в соляной кислоте.
Из раствора кристаллизуется фиолетовой гексагидрат Т1С13-6Н2О. Если к концентрированному раствору прибавить безводный эфир и насыщать его затем газообразным хлористым водородом, то из зеленого эфирного раствора получается зеленый гексагидрат, т. е. соль того же состава. Фиолетовая соль TiCl3-6H2O имеет строение, подобное фиолетовому хлориду хрома(Ш) (ср. гл. 5), а именно [Ti(OH3)e]Cl3; структура зеленой соли, вероятно, такая же, как и зеленого хлорида хрома(Ш). Следовательно, здесь речь идет о гидратной изомерии (подробнее см. в главе о хроме).
При добавлении раствора хлорида титана (III) к разбавленному раствору золота наблюдается интенсивное фиолетовое окрашивание. Последнее обусловлено адсорбцией коллоидного золота на гидроокиси титана. Образование этого продукта адсорбции совершенно аналогично образованию «кассиевого золотого пурпура» хлоридом олова(П). Реакцией с хлоридом титана(Ш) можно обнаружить до 0,0001 мг золота в 2 мл воды.
Безводный Т1С13 при обработке жидким аммиаком присоединяет 6 молекул NH3, образуя бесцветное двойное соединение, которое легко отщепляет 4 молекулы МН3, однако, прочно удерживает остальные две.
Золеный хлорид титана(Ш) образует комплексные соли типа MlTrGlg Н2О. Еще легче получаются фторсодержащие соли M|TiF6 и M|TiF6. Такие гексаацидо-соли трехвалентного титана существуют даже в таких случаях, когда исходное соединение TiX3 неизвестно, например в случае гексароданотитанатов(Ш) M3Ti(SCN)6-6Н2О.
TiDr3-6H2O образует красновато-фиолетовые кристаллы (т. пл. 115е), легко растворимые в воде, а также в спирте и ацетоне. Безводный TiBr3 существует в двух модификациях. При 400° он распадается в соответствии с обратимой реакцией: 2Т1Вг3
Т1Вг2 4- TiBr4. TiF3 в виде темно-синего порошка получают методом Эрлиха (1951), например при обработке титана или TiCl3 фтористым водородом. Фторид титана химически очень устойчив, в вакууме начинает возгопяться при 900°.
K2T1F5 распадается при возгонке в высоком вакууме. По данным Эрлиха (1952— 1953), при этом наряду с гексафторотитанатом образуется, вероятно, тетрафтороти-таггат по уравнению:
2K2TiF5=K3TiFG-j-KTiF4.
Чистый TiF3 имеет магнитный момент, равный 1,75 магнетонов Бора. Из этого следует, что Tin г имеет не спаренный электрон и, следовательно,существует в неднссоциировэнном соединении в виде иона Т13+.
Сульфат и двойной сульфат титана (III). При электролитическом восстановлении сернокислого раствора сульфата титана(1У) получается сначала похожий на чернила черно-фиолетовый раствор *. Последний при дальнейшем восстановлении спова светлеет и в случае практически полного перехода всего титана в трехвалентное состояние становится прозрачным, окрашенным в чисто фиолетовый цвет. Из этого раствора можно получить кислый сульфат трехвалентного титана ЗТ12(5О4)3-Н2ЗО4-25НгО в виде фиолетового с шелковистым блеском кристаллического порошка. Его можно перевести в безводный нейтральный сульфат титана(Ш) Ti2(SO4)3 выпариванием с концентрированной серной кислотой. При этом образуется темно-синий промежуточный продукт, состав которого еще неизвестен.
* Подобная интенсивная окраска характерна для продуктов, в которых одно и то же вещество присутствует в различных степенях окисления. Такая окраска появляется поэтому часто в качестве промежуточной при окислительно-восстановительных реакциях.
Четвертая группа периодической системы
75
Нейтральный сульфат титана(11Т) представляет собой зеленый кристаллический порошок, нерастворимый в воде, спирте и концентрированной серной кислоте и растворимый в разбавленной серной и в соляной кислотах с образованием фиолетового раствора.
От кислого сульфата титапа(П1) путем замещения водорода кислоты металлом производятся двойные или комплексные соли состава 3Ti2(SO4)3- М|ЗО4 или KpTi^SO^j (с переменным содержанием води), например аммонийная соль (NH4)Ti3(SO4)5-9НЙЬ и рубидиевая HbTi3(SO4)5- 12И2О; обе образуют кристаллы светло-голубого цвета, трудя ора отводимые в воде. Другой вид двойных сульфатов трехвалептпого титана соответствует по составу и форме кристаллов квасцам. Из титановых квасцов, правда, известны только рубидиевые и цезиевые: HbTi(SO4)2-12И3О (красного цвета) и CsTi(SO4)3- 12Н2О (светлые красно-фиолетовые). Эти соли можно перекристаллизовать без разложения из разбавленной серной кислоты, по не из чистой воды. Совсем по другому типу построен двойной сульфат натрия — фиолетовая кристаллическая ыасса'состава NaTi(SO4)2- 2V3H2O.
Подобно двойным солям с сульфатами трехвалеятный титан образует двойные соли с оксалатами MITi(C2O4}2- 2Н3О, где М1 = NH4, К, ВЬ золотисто-желтые кристаллы.
Гидроокись титана(Ш) образуется при действии гидроокисей щелочных металлов на растворы солей титана(Ш) в виде темно-окрашенного осадка, который проявляет весьма энергичные восстановительные свойства и поэтому в чистом виде получается трудно.
Окпеь тптана(Ш), полуторная окись титана Ti2O3 (уд. вес 4,49, т. пл. 1900’) образуется в виде кристаллов ирц нагревании двуокиси титана до 1000s в токе водорода и тетрахлорида титана. Ее кристаллическая решетка такая же, как у корунда; <г0 = = 5,42 А,а = 56’32'.
Нитрид титана TiN. Если соединения тигана восстанавливать при высокой температуре в присутствии азота воздуха, то очень легко образуется нитрид титана, но обычно он содержит примеси. Чистый нитрид титана TiN в виде порошка бронзового цвета получается в результате сильного нагревания тетрахлорида титана или его аммиаката в токе аммиака
ЗТ1С14+ 16NH3=3TiN + 1/EN2+ 12NH4C1.
Очень чистый, компактный нитрид получают методом наращивания подобно нитриду циркония (см. стр. 94). Возможность приготовления нитридов титана и циркония этим методом наращивания указывает на то, что они обладают значительной электропроводностью (ср. стр. 68).
Нитрид титана, несмотря па то что титан в пем трехвалентен, настолько устойчив, что остается неизменным при пропускании хлора даже при 270’. Однако он растворяется в плавиковой кислоте в присутствии сильных окислителей, например перманганата. Оп разлагается также горячей калиевой щелочью: TiN + 2К0Н Н2О = = K2TiO3 + NH3 + 1/2Н2. Под действием перегретого водяного пара нитрид титана разлагается по уравнению: TiN + 2Н2О = ТЮг + NH3 + 1/2Н2. Эту реакцию пытались использовать для получения аммиака.
Нитрид титапа имеет решетку типа каменйой соли (ср. стр. 64). Решетки подобного типа образуют 2rN, VN, NbN и ScN.
С питридом лития в присутствии азота TiN образует двойное соединение Li5TiN3, в котором титан по трехвалентен, как в простом нитриде, а окислен до четырехвалент-лого состояния (Juxa, 1953).
Соединения титана(1¥)
Тетрахлорид титана, хлорид титана TiC]4 получают пропусканием хлора над нагретым титаном, карбидом титана, титаноалюминисм (который образуется при алюмотермии) или над смесью двуокиси титана с углем.
76
1 3
В чистом состоянии он представляет собой бесцветную жидкость (при 0° уд. вес 1,76), кипящую при 136,5° и затвердевающую при —23°. Она имеет резкий запах и Сильно дымит на влажном воздухе. Водой гидролизуется в соответствии с уравнением: TiCl4 + 2НгО = TiO2 + 4ИС1. Если уменьшить гидролиз добавлением кислоты или действовать небольшим количеством воды, то в качестве промежуточных продуктов могут получаться оксихлориды.
Тетрахяорид титана легко образует продукты присоединения, например с аммиаком (TiClr 6NH3 и TiCl4- 8NH3), а также с пиридином, с три- и пентахлоридом фосфора, оксихлоридом фосфора, нитрозялхлоридом, тетрахлоридом серы, хлоридом селена и многими другими соединениями (особенно с кислород-, серу- и азотсодержащими).
Если хлорид титана обрабатывают спиртом и аммиаком, то получают а лк оголять! Ti(OR)4 (где R — алкил или арил). Алкоголяты, как найдено Меервейиом (Meerwein, 1927 —1929), растворяются в спирте с образованием алкоксо-киелот HgtTifOFQJ. Способность алкого.тятов титана присоединять дополнительно OR-группы можно использовать для приготовления высокомолекулярных веществ с органическими полигидроксильпы-ми соединениями (Schmidt, 1952). Подобным образом ведут себя алкоголяты циркония, олова, алюминия и трех валентного железа.
Известны также смешанные алкоксихлориды титана, например TiCl3-OC2H5, который подобно TiCl4 образует двойное соединение с ацетилхлоридом. Это соединение отличается способностью при пониженном давлении перегоняться без разложения (Bradley, 1952).
Хлоротитанаты. Тетрахлорид титана присоединяет ионы хлора, образуя комплексный анион [TiC]6]". Из соответствующих этому аниону солей, хлоротитанатов [точнее: гексахлоротитанатов(IV)] известны аммонийная соль (NH4)2[TiCle] -2Н3О (желтые кристаллы) и несколько солей органических оснований. Соответствующая свободная кислота существует только в водном растворе. О ее образовании при добавлении крепкой соляной кислоты к тетрахлориду титана свидетельствует появляющееся желтое окрашивание. Оно еще интенсивнее в случае бромистоводородной или иодистоводородной кислот.
Фторид титапа и фторотитанаты. Фторид титана TiF4, лучше всего получающийся методом Руффа (Ruff, 1904) при взаимодействии Т1С14 с HF, образует белый рыхлый порошок с удельным весом 2,80. Еще более, чем хлорид, он склонен к образованию ацидо-солей. Последние соответствуют типу M|[TiFfi] [гексафторотитаиаты(1У), или, просто фторотитанаты]. Соли такого типа получены не только со всеми щелочными и щелочноземельными, но также и со многими тяжелыми металлами. Фторотита-яаты калия, рубидия и цезия удобны для определения титана благодаря характерной форме их кристаллов.
Тетрабромид титана Т1Вт4, получаемый аналогично TiCl4 (яптарпо-жолтые октаэдрические кристаллы; уд. вес 3,25, т. пл. 40°, т. кип. 230°), но химическим свойствам весьма похож па хлорид. Он крайне гигроскопичен, очень легко растворяется в спирте (287 г в 100 .и.г’1, умеренно в эфире. ТШг4 кристаллизуется, подобно Sil4 и Gol4 по типу Siil4 (модеку.гиртгая решетка. см. т. I, стр. 576).
Тетраподид титапа Тг14 удобное пг.сго получать взаимодействием Т1С14 с Hl. On кристаллизуется в виде красно-корячневых октаэдров (структура решетки надобна TiBr,, т. пл, l-jiT, т кип. Зб:.3), еэдгыко при хранении переходит в модификацию с более низким порядком симметрии.
Сульфат титана и сульфатотитанаты. При выпаривании тнтаиовой кислоты или двуокиси титана TiO2 с концентрированной серной кислотой образуется оксисулъфат титана (так называемый титанилсулъфат) TiOSO4 в виде белого порошка, растворимого в холодной воде. Горячей
Четвертая группа периодической системы
77
водой он разлагается с выделением гидрата двуокиси титана TiOSO4+IIzO=TiO2+H2SO4.
Кроме оксисульфата, существуют еще и другие сульфаты титана с большим или меныпим содержанием SOg. Относительно существования в свободном состоянии нейтрального сульфата титана Ti(SO4)2 достоверных сведений нет. Правда, известна производная от него двойная соль — сульфата титанат. [трисульфатотитанат(1У)] типа M|[Ti(SO4)g] Оксисульфат титана также образует двойные соли (титанилсульфаты), например (NH4)2[TiO(SO4)2]-H2O.
Другие ацндотитаиаты. Указанные выше двойные соли относятся к типу ацидо-соедппоиий титана, среди которых имеются также соли с другими кислотными радикалами, паиример а?; города пот итак ат ы MjfTiOfSCNjJ, оксодиоксалдтотитаяаты M£lTiO(C2O4)2]. Последние находят применение для мягчения кожи и в качестве протравы при крашении тканей.
Двуокись титана ТЮ2 находится в природе главным образом в виде рутила. Она образует тетрагональные, часто характерно образованные, прозрачные и непрозрачные, красные, иногда желтоватые кристаллы (уд. вес 4,2—4,3). Реже двуокись титана встречается в виде анатаза, кристаллизующегося также в тетрагональной форме, но с другим соотношением осей (уд. вес 3,6—3,95), и ромбического брукита (уд. вес 4,1—4,2). Эдисонит, встречающийся в золотых песках Северной Каролины, представляет собой разновидность рутила.
Стргуктура решетки рутила показана на рис. 63, стр. 298, т. I (а — 4,58, о =2,95, d — = 2,01 А). Каждый атом титана в рутиле окружен двумя атомами кислорода на расстоянии 2,01 А и четырьмя атомами О на расстоянии 1,92 А. Шесть атомов О образуют несколько искаженный октаэдр. Структура анатаза приведена па рис. 16. В анатазе каждый атом титана, также в виде искаженного октаэдра, окружен двумя атомами О на расстоянии d = 1,95 А (на рисунке они соединены двойным плиниями с теми атомами титана, которым они принадлежат) и
стоянии 1,91 А. В то время как структура рутила часто встречается у двуокисей и дифторидов, показанная на рис. 16 решетка известна пока лишь для анатаза. В несколько более сложно построенном ромбическом бруките, поданным Полинга (Pauling, 1928), так же, как и в обеих упомянутых решетках, каждый атом титана окружен в общем шестью (также на разном расстояний) атомами О в виде несколько искаженного октаэдра. Среднее расстояние Ti<—>0 в решетке брукита практически такое же, как в решетках рутила и анатаза.
Различные решетки, в которых кристаллизуется TiO2, занимают промежуточное положение между чисто координационными и молекулярными решетками. Их можно рассматривать как координационные решетки, в которых координационное число титана по отношению к кислороду равно 6. Однако во всех трех решетках два из шести атомов кислорода, окружающих атом титана, отличаются от остальных тем, что они расположены от атома титана дальше четырех других. В решетке анатаза линии связи направлены вдоль с-осен, в решетке рутила — перпендикулярно к ним, а в решетке брукита оба атома кислорода, наиболее удаленные от атома Ti (Ti «—> 0—1,98 А),
78
Глава 3
являются единственными, для которых расстояние до атомов Ti точно одинаково. Расстояния остальных четырех отличаются как от выше названных, так и между собой. Поскольку в этих решетках два атома кислорода располагаются около опреде-ленного атома титана, ее можно рассматривать не как координационную, а как моле-куяярпую решетку, в которой, несмотря на то что атомы кислорода, принадлежащие молекулам TiO2, и оттесняются от титана под действием атомов кислорода соседней молекулы, но все ;ко видно, что они принадлежат определенному атому (ср. описание решетки Л12О3, т. I, стр. 391). Анатав, по данным Шредера (1928), при 642’ претерпевает энантиотропное превращение. Эти модификации обозначаются как а- и р-анатаз. Около 915° анатаз монотропно превращается в рутил. Брукит также монотроппо переходит в рутил. Скорость превращения уменьшается с понижением температуры и ниже 000° становится неизмеримо малой.
TiO2 плавится около 1800°. При более высокой температуре опа начинает отщеплять кислород с образованием Ti2O3. При 2230° давление О2 достигает 1 атм. По равновесию диссоциации теплота образования TiO, из 1/г Ti2O3 и 1/4 О2 составляет примерно 99 ккал (Junker, 1936). Окисыо углерода она восстанавливается до Ti2O3 уже при 800°.
Распространенная в природе двуокись титана редко бывает чистой; как правило, она более или менее сильно загрязнена железом, поэтому всегда окрашена, иногда почти в черный цвет (нигрин). Чистая двуокись титана на холоду бесцветна, при нагревании приобретает желтоватый цвет. Даже в аморфном состоянии она нерастворима в воде и разбавленных кислотах. Медлеппо растворяется в горячей концентрированной серной кислоте, лучше в расплавленных гидросульфатах щелочных металлов. Образующийся при этом сульфат или оксисульфат титана разлагается кипячением с водой, а также в слабокислом растворе. При этом образуется так называемая р-титановая кислота; ее называют так в отличие от обычпой титановой кислоты — а-титановой, получающейся при взаимодействии свежеприготовленного кислого раствора сульфата титана с гидроокисями или карбонатами щелочных металлов или с аммиаком на холоду.
р-Титановая кислота отличается от обычной титановой кислоты, подобно тому как р-оловянная кислота отличается от обычной оловянной. Она гораздо менее реакционноспособна, чем обычная титановая кислота. Например, она почти не растворяется в кислотах, кроме горячей концентрированной серной кислоты. Как между обоими видами оловянной кислоты, так и между титановыми кислотами существует ряд переходных состояний.
Обычная титаповая кислота своеобразно ведет себя при нагревании. Если ее нагревать ио слишком медленно, то при определенной температуре она вдруг раскаляется без изменения веса. У ^-титановой кислоты подобного аффекта не наблюдается. Как показал Бем (J. Bohm) методом добайеграмм, это явление основано на том, что при медленном повышении температуры происходит моментальная кристаллизация аморфного до этого окисла. Внезапное разогревание обусловлено теплом, выделяющимся при кристаллизации. Подобное явление обнаруживают также еще некоторые другие окислы, а именно ZrO3, Nb2O5, Та2О;,, Sc2O3, Сг2О3 и Ге2О3; для многих из них этот эффект наблюдал еще Берцелиус. Предпосылками для появления этого эффекта, кроме большой теплоты кристаллизации, являются повьтгпоггпая способность к кристаллизации, возможность получения аморфной формы и способность последней к перегреву, в результате чего кристаллизация происходит внезапно. Раскаливание лучше всего наблюдать на сильно спрессованных массах, в которых тепло распространяется быстрее, чем в рыхлом порошке.
Обычная титаповая кислота представляет собой слизистое вещество типа гидрогеля. Является лн опа только гелем двуокиси титана или гелем стехиометрия иски определенного гидрата двуокиси — этот вопрос нельзя было решить старыми методами исследования. Позднее, однако, Шварц, применив метод ацетоновой сушки к гелю титановой кислоты, приготовленному гидролизом TiCl3 при 0°, показал с достаточной вероятностью, что в таком геле двуокись титана находится в виде гидрата Ti(OH)4, т. е. ортотшпановой кислоты.
Четвертая, группа периодической системы
79
Коллоидные растворы (гидрозоли) двуокиси титана, как показал Грэм (Graham), легко можно получить пептизацией соляпой кислотой титановой кислоты, осажденной па холоду аммиаком. Значительно более концентрированные гидрозоли удалось Получить Внптгсну (Wintgen, 1936) методом диализа 1—3%-пого раствора TiCl4.
TiO2 подобно SiO2 способна также в некоторой степени переходить в раствор в молекуяяркоднсиерсном состоянии. Ио устойчивость этих растворов значительно ниже, чем молекулярнодисперсных кремневых кислот (Brintzinger, 1931).
Применение двуокиси титапа см. стр. 71.
Титанаты. Тшпанаты щелочных металлов без учета кристаллизационной воды соответствуют главным образом формулам М*ТЮ3 и M|Ti2O5. Мокрым способом их можно получить испарением растворов а-титановой кислоты в крепких щелочах (£-титановая кислота в них не растворяется). В безводном состоянии их получают сплавлением двуокиси титана с карбонатами щелочных металлов. Пирохимическим методом можно получить титанаты других металлов. Некоторые из них образуют при этом соединения типа М^Т1О4(ортотитапаты), другие — типа М|Т1О3 (метатитанаты). Сплавлением получаются также многочисленные полититанаты (соединения, в которых на 1MIQ приходится более 1Т1О2)-
Титанаты щелочноземельных металлов известны следующих типов: MI:lTiO3, Mj^TjOj, M^TiOj. Соединения состава M^ROj Стали известны лишь недавно, в связи с работами Шольдер.т (Beholder, 1953). Последний нашел, что, кроме Ti1^, соединения такого типа способны образовывать также VlV, CrlV, FeIV и Со1^.
Титанат кальция СаТ1О3 существует в природе в виде перовскита. Еще чаще встречающийся в природе ильменит (титанистый железняк) отвечает иногда по составу искусственно получаемому титанату железа (II) FeTiO3. Но чаще он содержит больше ягеле за, чем требуется по этой формуле. Следует отметить его изоморфизм с гематитом Fe3O3. Вследствие этого красный железняк (гематит) содержит часто значительные количества (до 7%) двуокиси титана.
*
Кристаллическая решетка ильменита производится от кристаллической решетка корунда (т. I, рис. 75), которому изоморфен гематит, при замещении атомов А1 атомами Ti и Fe. Метатитанаты Со11, Ni11, Мп11, Cd и Mg почти все кристаллизуются по тину ильменита; напротив, СаТЮ3, SrTiO3 и BaTiO3 кристаллизуются по типу перовскита (см. т. I, стр. 438). И в ильмепите, и в перовските координационное число титана относительно кислорода равно 6. Последнее справедливо для решетки соединения Fe2TiO5, кристаллизующегося в ромбической форме и встречающегося в природе в виде псевдо-брукита. Эта решетка состоит из несколько искаженных октаэдров ТЮ6, которые соединены двумя противоположными углами таким образом, что образуют цени, параллельные оси с. Атомы Fe расположены в пустотах решетки так, что каждый атом связан с четырьмя атомами кислорода почти тетраэдрически.
Титанаты состава MpTiCT кристаллизуются, как показало исследование их структуры (Мп = Mg, Zn, Мн, Со), по тину шпинели (см. т. I, стр. 395). Они содержат радикал [Ti04] С координационно четырехвалентным титаном в качестве структурной группы.
В некоторых минералах радикалы титановой и кремневой кислот могут, в определенных пределах, замещать друг друга. Однако титанит или сфен моноклинный минерал состава CaO-TiO2-SlO2 — нельзя, как предполагали ранее, рассматривать как дисиликат, в котором один атом Si замещен Ti. Учитывая его структуру и возможность частичного замещения титана железом(Ш), лучше всего приписать ему формулу С,.' li*v, Feni)(Q, OH)[SiO4].
80
Г лапа. 3
Рецтгецомотрическпй структурный анализ показал, что в случав титанита речь идет о силикате, построенном из несколько искаженных тетраэдров [SiQ4], в решетках которых атомы Ti расположены так, что каждый ив иих связей с четырьмя тетраэдрами t SiO4], обращенными к нему своими вершинами, и, кроме того, еще с двумя атомами кислорода, которые связывают между собой соседние атомы титана. Атомы титана могут частично замещаться железом(Ш). Одновременно соответствующее число связывающих их атомов кислорода заменяется ОН-группами.
Соединение Li2TrO3 не является собственно титанатом, а представляет собой двойной окисел со структурой каменной соли и статистическим распределением катионов (см. стр. 294), Двойной окисел 2Li2O- 5TiO2 имеет структуру шпинели и рассматривается как £14/зТщ/аО4, т. е. ^З.мсст катионов в решетке заняты Li+ и 6(3 — ионами ТНХ
Пер оке отита нова я кислота и пороке о титана ты. Соли титапа в нейтральном или кислом растворах окрашиваются перекисью водорода в интенсивный оранжево-красный цвет. Из достаточно концентрированных растворов осаждением водным раствором аммиака можно получить пероксотитановую кислоту H4TiO3 в виде коричнево-желтого осадка.
Пер оке отит а новая кислота подобно титановой кислоте образует обычно гель с переменным содержанием воды. Однако Шварцу (Schwarz R.) удалось обезводить эту кислоту путем высушивания ацетоном при 0°. Остаток имел состав Т1О3-2Н3О, в нем па один атом титапа приходился один атом «активного кислорода», т. е. один атом кислорода, эквивалентный по действию на перманганат одной молекуле перекиси водорода в разбавленном растворе серной кислоты. Из этого можно заключить, что соединение содержит одну пороксо-группу —0—0—. То, что речь идет о подлинно пер оке о-кислоте, а не о продукте присоединения Н2О2, Шварц (1935) доказал на основе реакции Ризепфйльда — Лиебхафского (Riesenfeld — Liobhafsky) (ср. т. I, стр. 378). Напротив, ранее считавшееся пероксотитанатом калия соединение K4TiO4- 4Н2О2- 2Н3О является только продуктом присоединения перекиси водорода к ортотитанату калия.
Существуют, однако, подлинно пероксотитанаты, в которых, кроме кислорода, пероксиДпой связью с титаном соединены другие остатки (пороксоацидотитапаты). К ним относятся пор оксо нентафторотитанат аммония
TiF5
и пероксодисульфатотитанат калия
К2
Q
I >Ti(SO4)2
о7
•ЗН2О.
Ион (ОЕ—Ti(SO.)2J" является непрочным комплексом и поэтому в разбавленных растворах распадается с образованием пероксотитанил-ионов по уравнению:
[02=Ti(So4)2r [02=T;]"+2[S04r.
Пер оксо титл пи л-попы образуются, как показал Яр (Jahr, 1950), при добавлении'перекиси водорода к растворам солей титана или титанила любой концентрации, по Сильно подкисленным. Этим пользуются для аналитического определения титана (ср. стр. 82). Между гита пил- и перо кс отита нил-ион а ми в иодных растворах, содержащих перекись водорода, существует равновесие
[TiO]" + H2O2 7^ [Ti=O2T4-H2O;
[TiO-IX [П2О2] [Ti=Oi-] -5-
Из концентрированных пер оксотитан ил-перхлоратных растворов Яр получил кристаллизующееся в пжеигоналыгой форме соединение TiO }С1О4]2-Н2О2 (титаноксодипер-хлорат-перекись водорода), которое очень гигроскопично и в противоположность интенсивно оранжевым растворам солей йе роке отита ни л а бесцветно.
Дисульфид титана TiS2 образуется при пропускании смеси тетрахлорида титана с сероводородом через раскаленную фарфоровую трубку:
TiCl4+2H2S=TiS2+4HCl.
Четвертая группа периодической системы 81
На холоду реакция между тетрахлоридом титана и сероводородом идет иначе, и сероводород в данном случае действует как восстановитель:
TiCl4 + HZS = TiCla+ S + 2HC1.
Дисульфид титана образует латунно-желтые с металлическим блеском чешуйки (о структуре решетки см. стр. 64). При обычной температуре он устойчив на воздухе. При нагревании на воздухе он переходит в Т10Е, при нагревании в токе азота или водорода — в низшие сульфиды: Ti2S3 и TiS. Водой даже при кипячении пе разлагается. Он устойчив также к действию разбавленных серной и соляной кислот и аммиака. Напротив, азотной кислотой н горячей концентрированной серной кислотой дисульфид титана разрушается с выделением серы. В кипящей калиевой щелочи он растворяется, образуя титанат калия и сульфид калия. С сухой двуокисью углерода реагирует при нагревании по уравнению:
Т;Зг+2СО2^ТЮ2-|-2СО + 28й
Как установил Бильтц (Blitz W., 1937), существует также более богатый серой сульфид титана TiS3 и по крайней мере еще одна фаза, отличающаяся по своей структуре от Ti и TiS более низким, чем в моносульфиде, содержанием серы.
Карбид титана TiC входит в состав тнтапсодержащего литья. В чистом виде он впервые получен Муассаном в электрической печи. В небольших количествах его можно получать методом наращивания (ем. стр. 84) в результате реакции, идущей на сильно нагретой проволоке; Т1СЦ + СО ЗТ[2 = TiC -j- Н2О + 4НС1, либо накаливанием угольной нити в парах TiCl4 (Burgers, 1934). Для получения больших количеств чистого компактного карбида применяют метод спекания. Предварительно хорошо перемешанную смесь Ti или ТЮ2 в виде топкого порошка и прокаленной сажи пагревагот в графитовой трубке до 2000° в атмосфере водорода. Образующийся в результате порошкообразный карбид нуд высоким давлением (2000 кг/см2) прессуют в прутки и прокаливают в графитовой трубке при температуре 2500—3000“.
Процесс измельчения и прессования повторяют до тех пор, пока прутка в результате предварительного прессования не станут достаточно плотными и прочными. После этого пропусканием сильного электрического тока через прутки производят «сверхспекание», т. е. нагревание карбнда до температуры, близкой к температуре плавления; в результате этой операции удаляются последние примеси, содержавшиеся в карбиде. Указанный способ можно использовать также для получения других тугоплавких карбидов (например, ZrC, Н£С, NbC, Та С) и нитридов TiN, ZrN.TaN (Agte, Moers, 1931).
Карбид титана по внешнему виду и поведению весьма похож на металлический титан, одпако труднее, чем последний, поддается действию кислот. Температура плавления его 3450°. О структуре решетки см, стр. 63. По данным Бургерса, TiC может связывать в твердые растворы до 0,1 е-атома Ti. Вопрос о том, существует ли другой карбид Ti2C, остается пока еше открытым. TiC благодаря большой прочности применяют для изготовления металлокерамических материалов.
Соединения титана с углеродом и азотом. Известны два соединения титана с углеродом и азотом: дицианид титана (кохранит) Ti(CN)2 — синие, по твердости превосходящие сталь кристаллы — п цианонитрид титана TitoC2Ng — Ti(CN)2-3Ti3N2 — медно-красные кристаллы. Последние образуются в шлаке доменных печей при переработке богатых титаном железных руд; их первоначально принимали за элементарный титан.
Бориды титана. Титан может давать с бором твердые растворы значительной концентрации. Помимо этого, он образует с бором два соединения. TiB (кубический) кристаллизуется по типу ципковой обманки (а = 4,20 A); TiB2 (гексагональный) пзотипен А1В3 (а = 3,03, с = 3,21 А.); по такому же типу кристаллизуются ZrB2, VB2, NbB2, ТаВ2 и СгВ2 (подобным образом кристаллизуется и МоВ2). TiB2no твердости превосходит все известные в настоящее время бориды металлов. Но и бориды других указанных переходных металлов отличаются высокой твердостью, высокими температурами плавления, хорошей электропроводностью и химической устойчивостью. Бориды сплавляются с металлическим железом и их применяют, например, в производстве высокотемпературных материалов и сплавов для изготовления режущих инструмен-ё г. Реми
82
Глава 3
тов. Для технического получения TiB2 ц 2л В, удобно, по данным Киффера [Kiffer, Z. anorg. Chem., 268, 191 (1952)], использовать получаемый в промышленности для шлифовальных целей карбид бора В4С, добавляя к нему В3О3 и Ti либо Zr;
7Ti + ЗВ4С+В2О3 = 7TiB3+ЗСО.
Аналитические сведения. Отличительной особенностью соединений титана является образование интенсивной оранжевой окраски в их подкисленных растворах при добавлении перекиси водорода (см. стр. 80).
Реакция с перекисью водорода очень чувствительна; однако следует учитывать, что ей мешает присутствие плавиковой, к ре инефт ср ист плодородной, азотной и большого количества уксусной кислоты. Титан образует также характерно окрашенные соединения с некоторыми органическими веществами, особенно с алкалоидами, фенолами в некоторыми ок с и-кислотами. Например, хромотроповая кислота (1,8-диоксппаф-талнн-3,6-дпс,ульфокислота) в водном растворе окрашивается в кроваво-красный цвет уже минимальными количествами солей титана.
В окислительном пламени перл фосфорной соли титаном не окрашивается, но при длительном нагревании в восстановительном пламени перл приобретает фиолетовый цвет. Подобную окраску дает перл буры, но только в присутствии больших количеств титана. Для соединений титана(Ш) характерное фиолетовое окрашивание появляется после прибавления цинка, олова и других восстановителей к кислым растворам титановых солей; однако подобное окрашивание наблюдается еще и у некоторых других веществ (соединений вольфрама, ванадия и молибдена).
Все соли титана легко гидролизуются; однако, так как осажденный на холоду и еще не застаревший гидрат двуокиси титана легко растворяется в кислотах без разложения, титан в ходе анализа мокрым путем большей частью понадает в группу, осаждаемую сернистым аммонием. Отделение титана от других веществ этой группы легко осуществляется благодаря его способности выпадать в виде р -титановой кислоты из разбавленного кислого раствора при длительном кипячении.
Лучшим реактивом для переработки нерастворимых соединений титана служит в большинстве случаев гидросульфат калия. Иногда рекомендуется сплавление с содой и поташом. В первом случае, при растворении плава в холодной воде, титан переходит в раствор, во втором — он переходит в титапат щелочных металлов, трудно растворимый в холодной воде и легко растворимый в кислотах.
Количественное определение титана можно производить осаждением: его в виде Ртита нов ой кислоты с последующим взвепщванием в виде TiO3 или, более точно, титр иметр и Чески окислением Ti‘” в Ti”” раствором (NH4)Fc(SO4)3 с K.SCN в качестве ицдикагора. Небольшие количества титана лучше всего определять колориметрически реакцией с Н2О2. Для колориметрического определения еще меньших: количеств, по данным Шенка [Schenk М., Holv. Cliim. Acta, 19, 1127 (1936)], подходит цветная реакция сульфата титана(IV) с салициловой кислотой в концентрированном растворе серной кислоты.
ЦИРКОНИЙ (ZIRCONIUM) Zr
Распространение в природе. Соединения циркония, так же как и титана, рассеяны в небольших количествах по всей земной коре. Крупные месторождения, однако, встречаются редко. Технически наиболее важным минералом (по в отличие от титана не самым распространенным) является двуокись циркония. В виде цирконовой земли (бадделеит) ZrO3 встречается в значительных количествах (главным образом на юге Бразилии и на Цейлоне). Чаще встречается силикат циркония ZrSiO4 — циркон. Но тех-
Четвертая группа, периодической системы
83
ническое значение его меньше, так как он труднее перерабатывается, чем цирконовая земля.
Нередко л природе встречаются силикаты циркония, в которых кремневая кислота частично замещена титановой, ниобиевой или танталовой кислотой. Соединения циркония почти, всегда находятся ташке в минералах, содержащих редкие зелии. Встречающийся в Гренландии и Норвегии эвдиалит наряду с окислами циркония, кремния, железа, кальция и натрия содержит примеси окислов церия п марганца, а также хлор.
По данным Праидтла (Prandll, 1937), минералы циркония почти всегда содержат фосфорную кислоту, хотя количество ее часто настолько мало, что его Нельзя обнаружить, ввиду того что открытию ионов POj” (так ясе как и ЗО^-ионов) мешает значительный избыток ионов [ZrO]”. Присутствие фосфата в двуокиси циркония, однако, можно легко установить благодаря тому, что при прокаливании в токе водорода двуокись темнеет вследствие образования ZrP.
Исторические сведения. Двуокись циркония впервые в 1787 г. была выделена Клапротом из циркона с острова Цейлон. В 1824 г. Берцелиусу путем восстановления калием фтороцирконата калия удалось получить порошкообразный металлический цирконий.
Циркониевые препараты, приготовляемые прежде, всегда в значительной степени были загрязнены гафнием — аналогом циркопия. Получить практически свободные от гафния циркониевые препараты удалось лишь в последние десятилетия.
Получение. Не совсем чистый аморфный металл получают методом Берцелиуса — восстановлением калием или натрием фтороцирконата калия:
K2[ZrF6]-|-4Na^2KF+4Nalf + Zr.
Промышленное получение чпетого циркония подобно титану осуществляют в настоящее время восстановлением хлорида циркопия расплавленным магнием.
По данным Вейса и Неймана (Wei₽, Neumann), методом алюмотермии можно получить расплавленный цирконий, причем сначала вследствие восстановления фтороцирк опа та калия алюминием образуется сплав. Последний представляет собой сплав циркония с 30% алюминия; прежде его называли «кристаллическим цирконием». Если наготовить из этого сплава штифты и пропускать между ними электрическую дугу в атмосфере азота или аммиака под давлением максимум 12 мм рт ап, то цирконий плавится, а большая часть алюминия испаряется. Таким образом, путем повторной плавки можно получить практически чистый компактный металлический цирконий.
Аналитически чистый, во не компактный цирконий получают методом Лели (Lely) и Гамбургера (Hamburger). Смесь безводного тетрахлорнда циркония с избытком натрия нагревают в вакууме до 500°. При этом происходят реакция по уравнению:
ZrCl4-j-4Na=Zr-j-4NaCl.
После выщелачивания продукта реакции вначале водой, затем соляной кислотой цирконий остается в виде порошка, который можно прессовать в прутки. Прутки, однако, но вальцуются, так как этому мешают весьма тонкие, не обнаруживаемые аналитически пленкп окиси или нитрида, покрывающие каждую частичку порошка.
Восстановлением двуокиси циркония кальцием^по методу Кроля (ср. стр. 69) получают компактный и при нагревании пластичный металл; вследствие наличия примеси ZrO такой цирконий хрупок на холоду.
Способ получения совершенно чистого, компактного и ковкого металлического циркония был предложен ван Аркелем и Де Буром (van Arkel, de Boor, 1924) (метод «наращивания» * j. Метод основан на том, что летучее соединение циркония (тетраиодид
* Метод «наращивания» (A ufwachs verfair ген) в литературе принято называть иодидным методом». — Прим, ред,
6*
84
Глава, 3
циркония) подвергается термическому распаду на раскаленной инти в вакууме.
Температура термической диссоциации тстраиодида циркония лежит значительно нии» температуры испарения металлического циркония, так что последний в виде кристаллов нарастает на раскаленной нити, В сосуде из стекла пирекс, приведенном ла рис. 17, через отверстие 1 загружают предварительно полученный одним из указанных выше способов достаточно чистый порошкообразный цирконий, смешанный с иодом. Затем заш, являют отверстие 7, а после создания вакуума в сосуде заправляют отверстие 2. Если теперь, пропуская электрический ток, нагревать вольфрамовую нить 3, натянутую между двумя вольфрамовыми прутками 4 и 5 до 1800°, а вест, сосуд од-5 повременно нагревать в электропечи до 600е, то сначала
образуется Zrl4, который диффундирует к раскаленной нити и на ней распадается с выделением металла; освободившийся иод может перенести новую порцию циркония к нити в виде иодида и т. д. По мере того как нить утолщается за счет выделяющегося металлического циркония, ток увеличивают, например, от1/,; а для нити диаметром 40 р. до 200 л, когда пруток циркония достигнет толщины 5 ж.». Расположенная в центре очень топкая вольфрамовая пить едва ли загрязняет столь толстые прутки. Этим способом можно получить цирконий, пластичный подобно меди, на холоду поддающийся ковке и вальцовке.
Методом наращивания удобно получать и некоторые другие элементы, например бор, кремний, титан, гафний и торий, для которых иные способы не обеспечивают достаточной чистоты. Недавно этот способ стали применять также для приготовления очень чистых соединений указанных элементов (см., например, стр. 75 и 81).
Р и с, 17. Аппарат для получения металлического циркония путем термического разложе-
ния иодида циркония по методу вап Ар коля и де Бура.
Свойства. Чистый компактный цирконий — блестящий похожий па сталь металл. В тонко-
измельченном состоянии он образует порошок черного цвета. Последний легко сгорает па воздухе, в то время как плавленый компактный металл
при накаливании на воздухе лишь тускнеет на поверхности и сгорает только при очень высокой температуре. В порошкообразном состоянии он загорается в токе кислорода уже при красном калении. Тонкие ленты пли проволока могут вспыхивать от пламени спички. Молярная теплота горения Zn превышает эту величину для титана (ср. табл. 6, стр. 63).
Вода и щелочи не действуют на металлический цирконий даже при нагревании. Компактный цирконий не поддается воздействию соляной и азотной кислот любой концентрации как на холоду, так и при нагревании. Однако горячая 60%-ная серная кислота растворяет его. Энергично он взаимодействует с царской водкой. Лучшим растворителем пе только
для металлического циркония, но и для его труднорастворимых соединений — карбида и нитрида —- является плавиковая кислота. По отношению к расплавленным щелочам компактный цирконий весьма устойчив. Газообразный хлористый водород и хлор разрушают его при температуре темно-красного каления. С азотом цирконий, как и титан, энергично соединяется при высоких температурах (около 1000°) с образованием труднорастворимого нитрида ZrN. Очень устойчивы также карбид ZrC и получаемый алюмотермическим сносном или в электрических печах в виде серо-стальных очень блестящих кристалликов силицид ZrSi2. Очень энергично взаимодействует порошкообразный цирконий с водородом, особенно если металл предварительно нагреть в атмосфере водорода выше температуры его превращения (ср. стр. 65) и потом снова медленно охладить до комнатной температуры.
Четвертая группа периодической система
85
Цирконий может содержать в обычной гексагональной решетке свыше 33 ат.% водорода. Напротив, при нагревании уже при сравнительно низком содержании водорода появляется новая фаза, которая имеет структуру, подобную Mn,.N и Fe4N, и в соответствии с этим рассматривается как соединение Zr4H. При содержании 50 ат,% водорода образуется новая фаза (соединение ZrH) и выше 65% — третья фаза, а именно соединение ZrH2 (Hiigg 1931). Теплота образования этого гидрида составляет, по Сивертсу (Sieverts), 20,2 ккал/г-атом.
з; Кислород и азот также способны в определенных количествах гомогенно рас-
I тпоряться в цирконии (кислород до 40 ат.%). При высокой температуре (например,
; 1000°) кислород в решетке циркопия весьма подвижен. Если приложить электрическое
поде, то кислород движется к аноду (de Boer, Fast, 1940). Переход гексагональной модификации (a-Zr) в кубическую (fJ-Zr) у кислород- или азотсодержащего циркония происходит не при определенной температуре, а в некоторой температурной области, в которой, смотря по обстоятельствам, более богатая кислородом a-фаза находится в равновесии с более бедной кислородом (1 -фазой (de Boer, 1936).
Применение. Металлический цирконий, как и титан, вследствие его способности энергично поглощать кислород и азот, применяют в качестве добавки при металлическом литье, особенно сплавов с железом (ферроцирконий). Добавленный к алюминию, он делает его более устойчивым к морской воде. Нить накала из циркония или его сплавов в разрядных трубках отличается высокой эмиссионной способностью и устойчивостью при перегрузке. Благодаря большой коррозионной устойчивости металлический цирконий используют для изготовления тиглей, особенно пригодных для сплавления проб со щелочами. Цирконий дешевле тантала, хотя уступает ему по коррозионной устойчивости. Смесь порошка циркония с нитратом циркония дает вспышку без дыма и запаха.
Широкое применение находит двуокись циркония. Постоянно возрастающее значение она приобретает в производстве огнеупоров, так как проявляет чрезвычайную устойчивость но только по отношению к нагреванию и перепадам температуры, но и к химическим воздействиям при высокой температуре. Целесообразно применять ее в смеси с другими веществами, например с окисью магния (ср. стр. 88). Вследствие тугоплавкости двуокись циркония используют в качестве тепловой изоляции для печен, в которых достигаются высокие температуры. Б смеси с графитом ее применяют для изготовления электрических нагревателей. Двуокись циркония используют также в виде белой краски. В производстве эмали она находит применение в качестве глушителя под названием террар вместо более дорогостоящей двуокиси олова; преимущество ее по сравнению с SiiO2 в большей стойкости к действию кислот. ZrO2 также должна быть хорошим шлифовальным и полировальным материалом. Широко используют ее свойство излучать очень яркий свет в сильно нагретом состоянии (подробнее см. далее).
Благодаря значительной способности поглощать рентгеновские лучи абсолютно безвредная цирконовая земля (двуокись циркония) является весьма подходящим материалом в качестве контрастного препарата пищеварительного тракта при просвечивании рентгеновскими лучами (Polyphos-Ges.).
Вследствие значительной адсорбционной способности гидрат двуокиси циркония применяют для осветления сточных вод.
Различные соединения циркония, например сульфат, оказались пригодными в качестве наполнителя для шелка.
Отличающийся твердостью карбид циркония применяют для резки стекол и в качестве шлифовального материала.
86
Глава J
СОЕДИНЕНИЯ ЦИРКОНИЯ
В своих соединениях цирконий, как правило, положительно четырехвалентен. Подобно титану он может находиться также и в состоянии более низкой валентности, как установил Руфф (Ruff, 1923), которому удалось восстановить бесцветный тстрахлориД циркония ZrCl4 до темного красно-коричневого трихлорида ZrCl3; для этого он нагревал в запаянной эвакуированной трубке до 350° тетрахлорид циркония с порошком А1 в присутствии хлорида алюминия; при дальнейшем нагревании без доступа воздуха ZrCl3 расщеплялся на черный дихлорид и тотрахлорид:
2ZrCI3 ZrCl2 + ZrCl4.
Дихлорид циркония образуется также при нагревании циркония в парах ZrCl4; так же получается ZrBr2(de Boer, 1930). Подробное о низших галогенидах циркония см. стр, 90. Существует также ZrO. Опа растворяется в металлическом цирк опии и делает его хрупким на холоду.
Соединения, производные от четырехвалентного циркония, обычно бесцветны. Это относится и к ионам Zr”“. Но в свободном состоянии опи редко встречаются в больших количествах, так как весьма склонны к образованию анионов. Так, в присутствии ионов F' образуется комплекс [ZrFe]"; в присутствии ионов SOJJ возникают сульфатные или оксосульфат-ныс комплексные ионы, например [OZr(SO4)2]" и т. д. В других случаях ионы Zr"” реагируют с водой;
Zr"“-f-H2O Т4* 2H'-)-[ZrO]” (циркон ил-рад и кал, или циркон ил-ион),
Уг""-)-Й/2ИгО ЗН‘-p/^Zi^CTJ" (дппмрвонил-радллал, или дицир ко п ил-ион),
Zr”"-|-2H2O 4H‘J-ZrO2.
Вследствие этого нейтральные соли циркония в водном растворе в зависимости от концентрации водородных ионов подвергаются более или менее глубоко идущему гидролизу с образованием хорошо кристаллизующихся солей цирконила [ZrO]X2 или дицирконила [Zr2O3jX2*. При более полном гидролизе выпадает двуокись циркония Zr02 или ее гидрат в виде белого осадка. В сильнощелочном растворе последний, присоединяя еще один кислород, переходит в цирконат-ион [ZrO3]"
ZrO2-J-2OH' > |ZrO3)"+ Н2О
Примеры
вполне определенных
соединений цирконила и дицирконила:
ZrOCL, ZrOBr2 ZrOGVOck H2[ZrO(SO4)2]
пиp копилхл о рид цпркоштлбромид ци р конили итрат цщжопклс ер на я кис-
лота (ЦИСА’Льфоок-еоциркиноиан кислота).
ZT9O3GI2 Дициркопнихлорид
Zr2O3Br2 : дицир ко ни л бромид
Zr2O3(NO3)2 дицир кони пнитрат
Zr2O3(SCi\)2-5H2O Диц ирконилроданид
K.JZrpOstSOi)»] дицирнонилсульфат калия (дисульфогрио-
цеодицпрконат калия)
* Возможно, цирконил- и дициркоппл-ионгл в свободном состоянии появляются только в раствор ах соли. У кристаллических соединений речь идет, по-видимому, о солях оксоциркония и триоксодициркония, т. е. о соединениях, в которых ионы О2” являются составной частью решетки анионов, в то время как решетка катионов построена не из [ZiO 1’3+-или [Zr2O3]‘2+-rpynn, а из 2г4+-нонов.
Четвертая группа периодической сиатв.ны
87
Напротив, если рассматриваются продукты переменного состава, выпадающие иногда из растворов солей циркония, например при смешивании их с солями щелочных металлов и слабых кислот, то в большинстве случаев речь идет о продуктах адсорбции кислоты или соли аморфным Гидратом двуокиси циркония,
Двуокись циркония — цирконовая земля ZrO2 — образуется в виде твердого белого, нерастворимого в воде порошка при прокаливании гидрата двуокиси циркония или солей циркония с летучими кислородными кислотами. Температура плавления со чрезвычайно высока (2700°); однако нагреванием ее с веществами (минерализаторами), понижающими точку плавления или растворяющими ZrO2, порошок можно превратить в микроскопические тетрагональные кристаллы. Встречается в природе моноклинная двуокись циркония, которую называют бадделеит (бразилит).
Моноклянна модификация двуокиси циркония при обычной температуре стабильна. Выше 1000°, по данпым Руффа и Эберта (Ruff, Ebert, 1829), она обратимо переходит в тетрагональную модификацию. При нагревании солей циркония до температуры не выше 600° образуется метастабильная тетрагональная окись. Если при прокаливании добавляют определенные окислы, например окись магния, то могут оо разевать с я твердые растворы, обладающие кубической решеткой (типа плавикового шпата), В случае, добавления окиси магпия ряд твердых растворов заканчивается Соединением состава 2MgO-3ZrOa. Моноклинная модификация двуокиси циркония структурно отличается от кубической тем, что решетка полевого шпата в пей незначительно искажена.
Несмотря па то что процесс превращения моноклинной модификации ZrOa в тетрагональную обратим, по происходит он в ту или другую сторону не при одинаковой температуре. Как установил Oxc (Ochs, 1957) методом эманации (см. стр. 578), моноклинная ZrO2 превращается при 1200° в тетрагональную только при условии высокой чистоты. Обратное превращение при охлаждении происходит при температуре 940°. При добавлении 20—45% U3O8 Последняя снижается до 800°.
Кристаллическая двуокись циркония, полученная плавлением, нерастворима в кислотах, за исключением плавиковой. Напротив, слабо прокаленная ZrO2 Довольно легко растворима в минеральных кислотах; после сильного прокаливания, кроме плавиковой кислоты, она растворяется еще только в концентрированной серной кислоте. Сильно прокаленная двуокись циркония легко сплавляется со щелочами или карбонатами щелочных металлов, образуя с ними растворимые в кислотах цирконаты. Удельный вес кристаллической, свободной от гафния двуокиси циркония составляет 5,73.
Будучи нагрета, двуокись циркония излучает интенсивЕЫЙ свет. Благодаря этому свойству ее использовали раньше для проекционных целей пт, и. (цирконовый спет}. Первоначально двуокись циркония применяли в качестве раскаленного тела в ауэровских сетках. Позднее ее использовали в осветительной технике для изготовления стержней накаливания в лампах Периста. В этих лампах имеются прутки (стержни накаливания), которые изготовляют из окиси циркония и окиси иттрия. В нагретом состоянии (разогрев можно производить пламенем спички) масса проводит электрический ток и быстра раскаляется до светлого каления. Лампы Нернста вскоре были вытеснены из осветительной техники лампами с металлическими нитями; для некоторых лабораторных целой (например, для поляриметрии) их еще иногда употребляют как удобный в обращении источник интенсивного света. Проводимость двуокиси циркония является по природе электролитической. У совершенно чистой ZrO2 даже при нагревании она очень мала, по-видимому, еще меньше, чем у кварца.
Как показали Вагнер (Wagner, 1943) и Хунд (Hund, 1952), электропроводность материала штифтов Нернста обусловлена тем, что ZrO2 с У2О3 образует «аномальные твердые растворы», в анионной решетке которых имеются неравномерно распределен ные пустоты, которые могут служить местом временного пребывания ионов О2*, а следовательно, движения их в электрическом поле. Как уже упоминалось, ZrO2 в присутствии некоторых других окислов кристаллизуется По типу плавикового шпата.
88
Глава 3
При этом катионы статистически распределяются в подходящих для них местах решетки; в а ни опп ой решетке образуются пустоты, доступные для кислорода. Например, в твердом растворе состава Иг03- YO1;r, четвертая часть мест в аняоииой решетке (по статистическому распределению) свободна: Твердый раствор ZrO2-YaQ3 имеет гомогенную область от 8,5 до 63 мол.% ¥Oj.Sl Постоянная решетки в этой области возрастает от 5,12 до 5,23 А. В случае образования твердых растворов ZrO2 с MgO и СаО речь идет также об образовании решетки типа плавикового шпата с неполностью заполнеппой авпоппой подрешеткой. Двуокись тория, тоже способна образовывать подобного рода аномальные твердые растворы. Последняя образует решетку плавикового шпата, если она чистая', в пей может содержаться до 30 мол.% YOJjS или до 52 мол.% LaOj.j. Подобпо аномальным твердым растворам ZrO2 при повышенной температуре твердые растворы ThO2 олептропроводны. Зависимость их удельного сопротивления х от абсолютной температуры Т можно представить, по данным Хувда, так: log к = . 3 последнее время стержни Нсрнста, изготовленные из твердых
растворов ThOg-YgOg, применяют для нагревателей электрических печей сопротивления, которые при обычных условиях могут давать температуру до 2000° (Geller, 1943). Изучение зависимости между Электропроводностью н пустотами в анионных решетках имеет значение также для изготовления термостойких изоляторов; так, при высоких температурах хорошими изоляторами могут быть такие вещества, в решетках которых Практически не возникают пустоты.
Коэффициент расширения двуокиси циркония почти так же мал, как и кварцевого стекла. Это свойство в соединении с чрезвычайной устойчивостью к действию кислот и оснований и высокой точкой плавления делает двуокись циркония ценным материалом для изготовления огнеупорных сосудов, прежде всего тиглей для плавления.
Посуда, изготовленная из чистой двуокиси циркония, может трескаться; Руфф1 показал, что причиной этого является превращение модификаций, происходящее при охлаждении после сильного разогрева. Если же к ZrO2 добавить окись магния, то образуются описанные выше кубические твердые растворы, структура которых в процессе охлаждения остается неизмеппой. Поэтому добавление окиси магния, а также других подходящих веществ (например, СаО, YaO3, СеО2), позволяет предотвратить растрескивание изделий из ZrOa.
Для технического получения двуокиси циркония в большинстве случаев исходят и» природной Цирконовой земли. С целью удаления примесей ее обрабатывают кипящей соляной кислотой, после этого выпариванием с концентрированной серной кислотой пли сплавлением с гидросульфатом палия переводят в сульфат циркония; Из полученного раствора аммиаком осаждают гидроокись циркония и прокаливают ее до двуокиси.
Гидрат двуокиси циркония представляет собой белый желатинообразный осадок с переменным содержанием воды. Осажденный на холоду, он растворяется в разбавленных кислотах. Очень легко образует коллоидный раствор. Гель весьма склонен к адсорбции кислот и щелочей и поэтому ими пептизируется. Вследствие способности реагировать со щелочами его называют также «циркониевой кислотой». Если осаждение гидрата окиси вести при нагревании, получается препарат с меньшим при обычной температуре содержанием воды, чем у выделенного на холоду. Такой гидрат трудно растворяется в кислотах и называется р-циркониевой кислотой.
Обычная Циркониевая кислота (а-циркбпиеваякислота), как и обычная титановая кислота, обладает в отличие от fl-цирк оние в ой способностью сильно раскаляться после не слишком мел'генпого нагревания до 300°, когда почти вся вода удаляется. Причиной разогрева здесь также является мгновенная кристаллизация первоначально аморфной окиси.
Цирконаты. Двуокись циркония наряду с основным обнаруживает слабокислый характер. Последний проявляется прежде всего в образо
Четвертая группа периодической системы
89
вании кристаллических цирконатов из расплавов. Несомненно, что ярко выраженная способность коллоидной гидроокиси циркония адсорбировать щелочи связана с кислотным характером окиси.
ч
Способность адсорбировать щелочи у двуокиси Цйркопил настолько сильна, что можно предполагать образование при этом химических соединений. Такого рода солеобразпые продукты, в значительной степени подобные (не принимая во внимание постоянно изменяющийся состав) химическим соединениям, получают, например, прибавлением раствора соли циркония К концентрированным щелочам. Что в данном случае речь идет только о продукте адсорбции щелочи на гидроокиси циркония, образующемся при взаимодействии соли со щелочью, а не о химическом соединении, следует как из изменчивого л большей частью не простого состава осадков, так л из непрерывного отщепления щелочи при его промывании.
Цирконаты, полученные из расплавов, впервые были исследованы Уврардом. Большинство из них соответствует типу M|ZrO3 (метацпрконаты), но наряду с ними встречаются и ортоцирконаты М(ХгО4, Цирконат Кальция CaZrO3 (т. пл. 2350°) изоморфен перовскиту С а'['(Оз и CaSnO-,.
Соединение СаО>ZrO2-SiO3 изоморфно титаниту (ср. стр. 79), и в соответствий с этим его нужно рассматривать как двойной силикат кальция и пири овила CaZrO[SiO4], а пе как смесь цирконата и силиката.
Нероксоциркописвая кислота и перокеоцпрконаты, При Добавлении перекиси водорода к нейтральному раствору сульфата или ацетата циркония получают белый желатине образный осадок, который очень трудно фильтруется и обезвоживается. Шварцу удалосг,, Одпако, показать, что после экстракции жидким аммиаком он обнаруживает состав, аналогичный составу соответствующего титанового соединения, а именно ZrO3-2H2O. Следовательно, речь идет о чисто химическом соединении т. о. об истинно пероксо-соединении — о пероксоциркопиевой кислоте Н—О—О—Zr(OII)3, так как при реакции Ризепфельда — Лиебхафского наблюдается выдел ей н о только иода, но не кислорода. Это соединение образуется также при действии гипохлорита натрия на гидрат двуокиси циркония или при электролизе раствора хлорида натрия, в котором суспендирован гидрат двуокиси циркония. Под действием разбавленной серной кислоты отщепляется перекись водорода. Уже при выдерживали л на теплом воздухе это соединение теряет кислород, по все жо оно более устойчиво, чем пер оке отита новая кислота.
Замена атома кислорода перекисной группой —О—О— или присоединение Н2О2 настолько усиливает кислотный характер двуокиси циркония, что уже в водных растворах со щелочами могут образоваться настоящие соли. Пероксоцирконаты щелочных металлов, так же как и продукты присоединения цирконатов этих металлов с перекисью, растворимы в воде, но не растворяются в спирте. В условиях пизкой температуры и при избытке перекиси водорода Шварц получил соединение K4ZrO4-,4HsO2-2HaO, аналогичное указанному выше продукту присоединения перекиси орто-титанатом калии. Ему удалось также выделить аналогичный соответствующему Соединению титапа и ер оксоцирконил сульфат калия (пероксодисульфатоциркопит калия)
ГСК
Кй | )Zr(SO4)3
Q7
• ЗН2О»
Тетрахлорнд и оксихлорид циркония. Тетрахлорнд цйркония ZrCl4 получают пропусканием хлора над цирконием, карбидом циркония или смесью двуокиси циркония (а также силиката) с углем при нагревании. Это белый кристаллический порошок (удельный вес 2,8), образующий во влажном воздухе облако соляной кислоты и энергично разлагающийся водой с образованием оксихлорида ZrCl4 + Н20 = ZrOCl2 + -|- 2НС1. Гидролиз не уменьшается даже в присутствии большого количества кислоты.
Тетрахлорид циркония образует молекулярную решетку типа Snl4 (см. т. I,. стр. 576). Подобно тетрахлориду титана он весьма склонен к образованию продуктов-присоединепия.' например, присоединяет до восьми молекул аммиака. Присоединяются также некоторые кислородсодержащие органические соединения. С другими соедине-
90
Глава 3
нпями, например с бензойной кислотой, лепием хлористого водорода
тстрахлорид циркония реагирует о выде-
H-j-O-CO • С6н5
i
Н ' - О СО * Об Н s
О-СО С6Н5
О-СО-СаН,
+ 2HCI,
При Присоединении ионов С1- к ZrCl4 образуются слабые комплексные ионы [ZrCl6]". Соответствующие им Соединения типа M|[ZrClej (гексахлороцирконаты) по-луяакугея из спиртовых растворов. Т;щ же как тетрахиорид, они тотчас разлагаются водой.
Образующийся при взаимодействии тетрахлорида с водой оксихлорид циркония (так называемый цирконилхлорид) легко растворяется в воде, а в холодной концентрированной соляной кислоте, напротив, плохо. Он кристаллизуется в форме характерных тетрагональных призм или игл состава ZrOCl2-8H3O, лучше всего при медленном охлаждении сильнокислого раствора хлорида. Образованием цирконилхлорида пользуются для определения, а также для очистки циркония.
При других условиях получается Цирконил хлорид с более низким содержанием воды. Известны также и другие оксихлориды циркония. Например, из спиртового раствора цирконилхлорида ZrOCl2 при действий эфира получают дицирынилхлорид Zr2O3Cl2. Это соединение нерастворимо в воде. Оно медленно осаждается (в течение месяца) также из сильно разбавленных водных растворов цирконилхлорида. Эти и другие оксихлориды определенного состава ио своей природе совершенно отличны от продуктов, образующихся при более глубоком гидролизе или при действии соляной кислоты на гидрат двуокиси циркония, которые ранее (вводя в заблуждение) называли «хлоридами мета цирк они я». В действительности они представляют собой продукты адсорбции хлористого водорода да у окисью циркония, легко переходящие в коллоидный раствор.
О восстановлении тетрахлорида в трихлорид см. стр. 86.
Тетрабромид и тетраиодид циркония, как и их производные, По своим химическим свойствам очепь похожи на тетра хлорид и: его производные. Они близки также по физическим свойствам, как видно из следующей таблицы;
ZrCl4 ZrBr4 Zrl4
Температура плавления (под давлением), °C 437 450 499
Температура возгонки, °C 331 357 431
Теплота сублимации, ккал/моль 26,0 26,5 29,5
Пропусканием смеси ZrBr4 и Н2 иад нагретой до 450° алюминиевой проволокой Юнг (Young, 1931) получил тр'ибромид циркопия ZrBr3; наряду с последним образуется ZrBr2. Образование ZrJ3 и Zrl2 при нагревании Zrl; с избытком Zr наблюдалось Фастом (Fast, 1938). ZrF3 был получен Эрлихом (Elirlich, 1952). В противоположность TiF3, ZrF3 разлагается при попытке возгонки его в высоком вакууме.
Фторид циркония и фтороцирконат циркония. Фторид циркопия ZrF4 образует белые, сильно преломляющие свет, почти перастворимые в воде моноклинные кристаллы с удельным весом 4,6.
Из раствора гидрата двуокиси циркопия в плавиковой кислоте кристаллизуется соединение, которое по составу первоначально можно принять просто за гидрат фторида циркония, но которое, вероятно, следует рассматривать как тетрафтор о ок с оц ирь- опие-вую кислоту H2[ZrOF4)-2H2O, В присутствии фторидов щелочных металлов (или фторидов других элементов) из растворов кристаллизуются двойные соли, большинство которых соответствует типу MJ [ZrF6], Кроме них, наиболее часто встречаются соединения типа Mj[ZrF8J.
Четвертая группа периодической системы
91
Особой устойчивостью отличается гексафтороцирконат калия KsIZcF^J (бесцветные, ромбические призмы). Так как растворимость его в горячей воде значительно больше, чем в холодной, он легко поддается перекристаллизации. Поэтому гексафтороцирконат калия может служить для очистки циркониевых соединений. Он является также подходящим исходным продуктом для получения небольших количеств металлического циркония.
Гексафтороцирконат аммония (NH4)2IZrFe] при умеренней нагревании разлагается на NH3, HF и ZrFt и пригоден поэтому для получения последних. Выше 600° ZrF4 начинает возгоняться. Возгонка фторида представляет удобный метод очистки циркония от железа я других соединений.
Сульфат циркония и сульфатоцирконйевая кислота. Безводный сульфат циркония Zr(SO4)a иолучают упариванием двуокиси циркония или цирконилхлорида с концентрированной серной кислотой в виде белого порошка *. Он растворяется в воде с выделением большого количества тепла. Из водного раствора, содержащего избыток серной кислоты, кристаллизуется соединение, которое по своему составу (ZrO2- 2SO3-‘1H3O) можно принять за гидрат сульфата циркония. В действительности, однако, речь идет о комплексной кислоте, Оксо дисульфат о циркониевой, обычно называемой просто цирконилсерной кислотой:
SO41
Т12 OZr
so.
311,0.
Это следует из того, что в растворе цирконий перемещается не к катоду, а к аноду. Следовательно, он входит в состав аниона и соответственно этому не дает характерных для ионов Zr"’’ или [ZrO]" реакций осаждения.
Цирконил серна я кислота производится заменой SO4-r руппы на кислород в трисулъфатоцирнониевой кислоте Н3[Zr(SO4)3], которая выделяется из сильно-кпелых растворов с тремя молекулами кристаллизационной воды в виде длинных бесцветных игл.
В нейтральном водном растворе цирк опил серна я кислота подвергается гидролитическому расщеплению, причем группа S04 обменивается на атом О или группу ОН. Образующиеся при этом так называемые основные сульфаты циркония представляют собой большой частью мпогоядерпые комплексные соединения, оксо- и гидр оксокислоты. В качестве примера могут быть приведены выделенные Хаузером (Hauser, 1907) соединения тетрацирк онс ер и ой кислоты (окта гидр оксогекса сульфатотетрацирк р-ний кислоты)Н4^г4(ОН)8(ЭО4)в]- 4НгО,аммонийные соли которой были позднее получены РозенгеЙмом (Rosenheiiii, 1919). Эти вещества с большим молекулярным весом образуют характерные кристаллы, по в водном растворе показывают свойства, типичные Зля коллоидов, например они не диффундируют через мембрану из пергамента.
Сульфатоциркоггаты. Сгущением смеси растворов сульфата циркония и Сульфата (или гидросульфата) щелочного металла над серной кислотой при комнатной температуре получают сульфатоцирконаты, наиример K4|Zr(SO4)>j-4H2O **—тетр асу дь-фатоциркопат калия — белые с шелковистым блеском иглы,(ХН4)2[2г(8О4)я1-ЗН2О— трисульф а тоцирконат аммония. Из нейтральных растворов, особенно при нагревании, чаще выделяются оксо- или гидр оке о сульфатоцирконаты. К ним относятся коли уже упоминавшейся тетрацирк онсерной Кислоты.
* Однако получаемый таким способом сульфат в соответствии с данными Хевеши (Ilevesy, 1931) всегда содержит избыток серной кислоты. Полностью удалить ее удается только арн температуре, прп которой наступает уже заметное разложепяе сульфата.
** Содержание воды колеблется.
02
Глава 3
Оксалат цирконила и оксалатоцирконаты. При взаимодействии растворов солей циркопия со щавелевой кислотой получается белый, порошкообразный осадок, состоящий из содержащего воду оксалата цирконила ZrO(C2O4). Горячей водой это соединение гидролизуется.
При избытке щавелевой кислоты цирконий образует оксалатные комплексы. Например, если растворить гидрат двуокиси циркония в растворе кислого оксалата натрия, то через некоторое время кристаллизуется тетраоксалатоцирконат натрия Na4[Zr(C2O4)4];4H2O. Соответствующая солям этого типа кислота в свободном состоянии неизвестна, но известна триовсалаторксоцнрконий кислот аЦоксалатоцирпона л свая) кислота H4IZrO(C3()4)3]. 7Н2О, которую можно получить в кристаллическом состоянии упариванием раствора щавелевой кислоты, насыщенного гидратом двуокиси циркония.
Ацетат циркония. Если безводный тетрахлорид циркония растворить в горячей ледяной уксусной кислоте и затем охладить, то кристаллизуется безводный нейтральный ацетат циркония Zr(C2H3O2)4, Следует отметить, что Цирконий образует именно со слабой уксусной кислотой несомненно нейтральную (нормальную) соль. Относительно других нейтральных по составу, но содержащих воду солей циркопия можно сомневаться: может быть, это действительно оке оаци до-сое дипения, как это доказано в случае сульфата.
Тетраацетат циркония очень неустойчив. Датке при стоянии над серной кислотой он отщепляет уксусную кислоту и переходит в цирконилацегл-ini ZrO(C2H2O2)2. Последний, как и следовало ожидать для соли очень слабого основания и слабой”кислоты, полностью гидролизуется. Образующаяся при этом двуокись циркония остается в растворе в коллоидном состоянии. Очищенный диализом гидрозоль коагулирует при нагревании или прибавлении соляной кислоты, образуя прозрачный студень.
Питраты цирконии. При выпаривании раствора гидрата двуокиси циркония в азотной кислоте получают бесцветные кристаллы состава ZrO(NO3)2- 2Н3О, Это соединение обычно называют нитратом цирконила, хотя вследствие прочной связи его с водой напрашивается предположение, что речь идет об оксонитрато-кислоте. Если это соединение растворить в абсолютном спирте и прибавить эфир, то выпадает нитрат дицирконила Zr2O3(NO3)2-5H2O. Соединение, отвечающее по составу нейтральному нитрату Zr(NO3)4‘5H3O, вероятно, также можно рассматривать как нитрата-кислоту, примерно как оке итетранитратоцирк опий кислоту H2|ZrO(NO3)4] 4п2О. Это соединение выделяется в виде больших прозрачных как вода призм при упаривании в вакууме над пятиокисью фосфора и едким натром при температуре ~ 15° евежеосаждснпого при обычной температуре раствора гидрата двуокиси циркония: в азотной кислоте. Оно чрезвычайно гигроскопично и легко отщепляет азотную кислоту.
Фосфат циркония и фосфатоциркоиаты. Ортофосфат циркония Zr3(PO4)4 должен был бы выделяться при действии на растворы соли циркония ортофосфорпой кислоты или фосфата натрия. Однако в зависимости от условий опыта получают осадки переменного состава. В большинстве случаев они содержат больше фосфорной кислоты, чем соответствует указанной выше формуле. Идет ли при этом речь о кислом фосфате или об адсорбции избыточной фосфорной кислоты, еще не ясно. Во всяком случае, свежеосаждонный гидрат двуокиси циркония обладает чрезвычайно большой способностью адсорбировать фосфорную кислоту подобно гидрату двуокиси олова. Благодаря этому соединения, которые расщепляются водой с выделением гидрата двуокиси циркопия (например, цирко-ннлхлорпд или цирконилнитрат), можно применять в аналитический химии вместо гидрата двуокиси олова или тетрахлорида олова для удаления фосфорпой кислоты.
Фосфат состава 2ZrO2- РаОа выделяется в виде кристаллов из раствора гидрата двуокиси циркония в горячей сиропообразной фосфорной кислоте при стояния. Структура этого соединения еще не известна. Соединение состава ZrO3- Р2О5, образующееся в кристаллическом виде при сплавлении двуокиси циркония со стекловидной фосфорной кислотой, как показал структурный анализ, представляет собой не метафосфат цирконила ZrO(PO3)2l как предполагалось ранее, а пирофосфат Zr[P3O7). Он образует
Четвертая группа периодической системы 93
кубическую решетку (ж = 8,20 А), производную от решетки камеппой соли (см. т. I, стр. 237), путем замепы в ней ионов Na* на Zr4+ и ионов С1_ на группы с центральным атомом кислорода [О3Р—О—РО314-. Эти группы состоят из кислородных тетраэдров, сгруппированных по два около атома Р и имеющих общий угол. Подобные решетки образуют Ш[Р207](а = 8,18 A), Ti[P2O7](a = 7,80 A), Si [Р2О7Ца = 7,46 А ) и Sn[P2O7] (а = 7,89 А).
Двойные соли фосфата циркония с другими фосфатами (фосфатоцирконаты) частично имеют состав, подобный соответствующим образом полученным двойным фосфатам тория, однако не изоморфны им.
Силикат циркония, ZrSiO4 встречается в природе в виде циркона. Циркон является наиболее распространенным минералом циркония. В виде микроскопически маленьких кристалликов он содержится почти во всех изверженных породах. Нередко встречаются также более круцпые кристаллы циркона: тетрагональной формы, большей частью окрашенные примесями в коричневатый цвет, более или менее непрозрачные, они очень красивы. Прозрачные, почти бесцветные и окрашенные, встречающиеся особенно на Цейлоне, в Индии и в Новом Южном Уэльсе, желто-вато-красные (гиацинт) находят применение как драгоценные камни. Они обладают значительной твердостью (7,5 по шкале Мооса) и наибольшим среди благородных камней удельным весом (до 4,8; встречаются также цирконы со значительно меньшим удельным весом, например 4,2).
Циркон це изоморфен, как но лагани раньше, рутилу. Несмотря ад то что они принадлежат к одному классу тетрагональной кристаллической структуры (а именно к голоэдрическому), имеют одинаковое соотношение осей и встречаются обычно
Рис. 18. Решетка циркона.
ил—6,58 ДЕ cQ—5f93 A, St *—*• О = 1,62 А. Отмечены толы?о тс атомы кислорода, которые овнааны с атомами $2Т распоясясеякьпик я левых плоскостях злэдсятарной ячейки., нейоторы^ из этих атомов О лежат вне ^е*
в одинаковых формах, циркон не образует с рутилом твердых растворов и имеет другую структуру решетки.
Структура решетки циркона показана на рис. 18. Там показано прежде всего расположенно атомов Zr и Si: каждый из атомов Si следует представить себе окруженным четырьмя атомами О, рас положенными в форме почти правильного тетраэдра. Расстояние Si-<—>-0 составляет 1,62 А, наименьшие расстояние Zr -<—>- О равно 2,05 А. Одинаковую с цирконом структуру решетки имеют У{РО4], У[ЛзО4] и VfVO4]. При температуре 862° ZrSiO,, обратимо превращается а кубическую модификацию.
94
Глава 3
Кроме ZrSiOj (т. пл. 2430°) в системе ZrO3—SiOo не найдено других соединений. ZrO2 может растворять примерно до 10 мол,% SiO2 с образованием твердых растворов (Жирнова, 1934).
Из двойных силикатов наряду с указанным на стр. 89 соединением CaZrO[SiO4] следует упомянуть найденные д’Ансом (D’Ans, 1930) в тройной системе Na_2O— ZrO2— — SiOo соединения: Xa2ZrO [SiOJ (т. пл. 1477°, плавится HHKonrpygnTHokNa/ZrUSiChls (т. пл. 1540s) и Na2ZrLSi2O7j.
Нитрид циркония. Ввиду того что при высокой температуре цирконий, как и титан, непосредственно соединяется с азотом, металл, полученный восстановлением в присутствии азота, всегда содержит нитрид. Если цирконий получать способом вап Ариеля и де Бура (описанным на стр. 83)— термической диссоциацией иодида в присутствии азота,— то образуется аналогичный по составу нитриду титана нитрид циркония ZrN в виде очень твердых, С металлическим блеском, похожих на серебро, но чуть желтоватых кристаллов (т. пл. 2980°). Это соединение, отличающееся большой устойчивостью, обладает, как и борид ZrB (т. пл. 2990°), довольно значительной металлической проводимостью (ср. табл. 10, стр. 68).
Нитрид циркония, отличающийся по составу от указанного, Получают нагреванием при красном калении тетра хлорида циркония пли его аммиакатов в токе аммиака или водорода. При умеренном нагреваний, если исходить из более высоких аммиакатов, сначала отщепляется аммиак с образованием тетрааммиаката, устойчивого выше 195°. При более сильном нагревании из него путем отщепления НС1 образуется сначала амид циркония: ZrCl4- 4NH3= Zr(NH2)4+ 4IIC1, который устойчив до 260°. Если увеличить температуру до 350°, то получается в соответствии с уравнением: 3Zr(NII2)4 = Zr3N4+ 8NH3 нитрид Zr3N4 (бронзового цвета кристаллики, нерастворимые во всех минеральных кислотах, за исключением плавиковой).
Карбид циркония. ZrC получается вместо чистого металла цри восстановлении соединений циркония углем. Он образует черные с металлическим блеском, очень твердые кристаллы, которые очень хорошо проводят электрический ток и могут быть расплавлены им (т. пл. 3530°) В расплавленном состоянии ZrC растворяет углерод, который снова выделяется в виде графита при охлаждении.
Карбид циркония совсем но так устойчив, как его нитриды. Он растворяется в азотной, серной кислотах и царской водке, хотя на него не действует соляная кислота. Легко растворяется также в расплавленных едких щелочах. Под действием азота при температуре красного каления переходит в нитрид. Еще легче он разлагается хлором, бромом и иодом. Поэтому часто им пользуются как хорошим исходным материалом для получения галоидных соединений циркония.
Бориды циркония. Помимо приведенных в табл, 9, цирконий образует с бором соединение ZrBI2. Это соединение было получено Постом и Глазером (1952) в виде черного морошка. Оно подобно ZrB обладает металлическими свойствами и отличается большой твердостью.
Аналитические сведения. Цирконий дает много реакций, общих с редкоземельными элементами. Однако в отличие от них он не осаждается избытком щавелевой кислоты (благодаря образованию растворимых оксалатов). От элементов цериевой и иттриевой групп и тория он отличается тем, что не осаждается при избытке плавиковой кислоты (образуются растворимые фтор о цирк она ты). Как уже отмечалось, для циркония характерно также образование оксихлоридаКуркумовая бумажка, смоченная раствором соли циркония, подкисленным соляной или серной кислотой, и высушенная на водяной бане, окрашивается в красно-коричневый цвет. Растворы редких земель и тория но дают этой реакции. Мешают, однако, соли титана(1У), так как они дают похожую коричневую окраску; но они легко могут быть восстановлены до солей Т1(Ш). С перекисью водорода в присутствии титана цирконий образует белый осадок.
Четвертая, группа, периодической системы 95
Количественное определение Циркония проводят осаждением его аммиаком в виде гидрата двуокиси или перекисью водорода в виде пероксоцирк они ев ой кислоты и взвешиванием после прокаливания в виде двуокиси.
Микрохимическое определение возможно В виде фтороцирконатов рубидия и цезия. Еще более чувствительна реакция с гидрооксалатом калия (образование тетрагональных пирамид тетра оксалатоцирк она та калия). Определение циркония при сильном разведении можно осуществить капельным, методом при помощи органических реагентов; таких, как р-нитрозо-а-ая фтол, ализарин или п-ди мети л амипоаз оф енил арсоновая кислота, дающих с солями циркония очень трудно растворимые окрашенные осадли.
ГАФНИИ (HAFNIUM) Hf
Гафний был открыт Хевеши и Костерем в 1922 г. и назван в честь города Копенгагена (Нсфиае). Он является постоянным спутником циркония в его минералах. Большинство разновидностей циркона содержит более 1% окиси гафния. То, что гафний, несмотря на это, был открыт лишь недавно, обусловлено чрезвычайно большим сходством химических свойств этих элементов.
Порядковый помер гафния 72. Еще до его открытия из закона Мозли и рентгеноспектральиых данных следовало, что должен быть найден элемент, соответствующий этому номеру. Теория атомного строения Бора указала путь, по которому следовало идти в поисках этого элемента. Опа привела к выводу, что элемент с порядковым номером 72 но должен принадлежать подобно предшествующим ему элементам к группе редких земель, а должен быть аналогом циркония. Следовало ожидать, что он сопровождает цирконий в его природных соединениях; и, действительно, па основании рентгеновских спектров Хевеши и Костер вскоре нашли новый элемент во всех исследованных ими минералах циркония.
Еще в 1895 г. Юлиус Томсен на основе Периодической системы пришел к выводу, что между редкоземельными элементами и танталом должен быть еще элемент, отличающийся ио своим свойствам от редкоземельных металлов и похожий на цирконий. Позднее это предположение высказывалось другими. Когда на основании закона Мозли оказалось, что элемент с порядковым номером 72 еще не найден, он был большинством Причислен it ряду редких земель. В 1911 г. Урбэн (Urbain) полагал, что он нашел новый элемент из ряда редкоземельных металлов, названный им кельтием; Довилье (Dauvillicrs, 1922) считал, что он может па основании, правда, очень слабых рентгеновских линий приписать ему порядковый номер 72, Напротив, Хевеши и Костер ожидали, опираясь па теорию атомпого строения Бора, что элемент 72 следует искать не среди редких земель, а как аналог циркония в циркониевых минералах. Бору незадолго до этого удалось объяснить периодическую систему на основе квантовой теории строения атома. При этом путем рассуждений, указанных па стр. 14, он пришёл к выводу, что элементом 71 ряд редких земель должен заканчиваться. Тогда внешняя электронная ободочка элемента 72 должна быть аналогична внешней электронной оболочке титана, циркония и тория, и, следовательно, в отношении порядкового номера он доджей соответствовать стоящему ниже аналогу циркония. При помощи большого числа линий Е-ссрии Хевеши и Костер легко смогли идентифицировать его в исследованных ими минералах циркония. Ио интенсивности линий оказалась возможной оценка количественного содержания гафния и было показано, что его можно скоп центрировать дробной кристаллизацией двойных фторидов калия и аммония типа MjjZrFg] или Mj[T-IfFjJ. При достаточно большом числе ступеней кристаллизации таким способом можно получить чистый гафний.
Впоследствии оказалось, что гафний постоянно сопровождает цирконий в природе. Особенно богаты им минералы циртолит, альвит, паэгнт и мала»он. Циртолит содержит около 52,4% ZrO^ й 5,5% ПЮ,, Наибольшее содержание гафния но отношению к цирконию в тортвейтите, очень редком минерале, встречающемся в Норвегии и на Мадагаскаре, главной составной частью которого является окись скандия (связанная с кремневой кислотой; ср. стр. 51). Тортвейтит содержит всего 1—2% окиси циркония и почти такое же количество окиси гафния. Минерал, в котором гафпий был бы преобладающей составной частью, Це найден. В среднем минералы, циркония
96
Глава 3
содержат 2—2,5% гафния но отношению к цирконию. Помимо циркония, ни один другой элемент, в том числе и торий, не содержит примеси гафния. Общее содержание гафиия в земной коре (силикатная оболочка) намного превышает содержание, например, ртути, кадмия и висмута (ср. табл. 90, стр. 688)
Сходство между цирконием и гафнием как в свободном состоянии, так и в соединениях больше, нем между какими-нибудь другими элементами-аналогами. Ото обусловлено отчасти тем, что у обоих элементов не только внешние электронные оболочки построены одинаково, но и атомы, а также ионы чрезвычайно близки по размерам (см. табл. 8, стр. 65). Это связано с достройкой внутреннего слоя, ведущей к появлению группы лантанидов. Подробнее об этом см. стр. 516.
Достройка внутреннего слоя, определяющая появление группы лантанидов, привела не только к сокращению атомных к ионных радиусов, но и к ослаблтше поляризующего действия ионов. Ионы гафния, несмотря па несколько меньший радиус, обладают более слабым поляризующим действием по сравнению с ионами циркония. Это видно, например, из того факта, что комплексные соединения гафиия менее устойчивы. Более слабым поляризующим действием иона гафния обусловлено также то, что комплексные соединения гафния в большинстве случаев имеют большие молекулярные объемы, чем комплексные соединения циркопия, в то время как молекулярные объемы простых соединений гафния в соответствии с я ос л одними исследованиями (частично противоречащими прежним) всегда меньше, чем циркония (см. табл. 11, стр. 96).
Таблица 11
Плотности и мольные объемы простых соединений циркония и гафния, у ста но п ленные р он тгепометр ичес ни
Соединение ZrOs (куб ) ШО: (куб ) ZrF4 HtF4 ZrC HIG ZrP2O? HfP2O7
Плотность Мольный объем 6,2" 19,65 10,43 19,17 4,66 35,9 7,13 35,7 6,613 15,61 14,19 13,43 3,195 83,00 4,278 82,41
А цнрконнй вследствие более сильного поляризующего действия ведет себя во многих отношениях так, как если бы он имел радиус меньше, чем следует из простых соединений. Благодаря различному поляризующему действию компенсируется, следователь- i но, различие радиусов между 2т и Hi (часто даже немного сверхкомпенсируется). Таким образом объясняется чрезвычайно большое сходство между соединениями циркония и гафния. Это сходство гораздо больше, чом у соединений ниобия и тантала, хотя атомные (и ионные) радиусы у Nb и Та различаются още меньше, чем у Zr и Hf ; (см. табл. 8 и 12, стр. 65 и 106). Ион ниобия, обладающий более сильным поляризующим действием, чем тантал, обычно ведет себя так, как если бы он имел радиус значительно меньший, чом у тантала. Бели принимать во внимание различное поляризующее действие, то вообще исчезает несоответствие, получающееся дня переходных элементов в том случае, когда о море сходства между соединениями различных аналогов судят по величине атомных или ионных радиусов (ср. стр. 17).
Соединения циркония и гафния очень близки между собой по растворимости, температурам плавления и летучести. Даже у гексафторидов аммония, которые успешно используют для разделения, различия : в растворимости незначительны. Растворимость при 20° составляет для (WH4)z[Zrl^] 1,050 молъ1л, (NHiJalHlFel 1,425 моль; л. Температура плав- I Ленин двуокиси гафния приблизительно на 100° выше, чем двуокиси циркония. Температура плавления карбидов равна для ZrC 3530°, ШС 3890°.
Различие в растворимости возрастает, в случае если переход в раствор сопровождается образованием комплексного соединения с одним из имеющихся в растворе ; веществ. В общем цирконий более склонен к комплексообразованию, чем гафний. На- *
Четвертая группа периодической систем»
97
пример, по данным Хевещц (Hevesy, 1930), в 1 л раствора 13и. НВг при 25° растворяется 0,017 .воль ZrO2 и только 0,001 .чоль Н1О2. Такое относительно большое различие в растворимости обусловлено неодинаковой способностью к образованию комплексов с бромом; это следует из того, что с уменьшением копцептрацин НВг разница в растворимости исчезает. Подобное явление имеет место при растворении окиси в плавиковой и соляной кислотах. Различие в растворимости меньше, а растворимость больше.
Различную устойчивость комплексов циркония и гафния можно с успехом использовать для отделения гафния or циркония. Например, если разбавить водой раствор фосфатов циркония и гафния в концентрированной серной кислоте, то гафпий преимущественно выпадает в осадок в виде фосфата, так как он менее прочно, чем цирконий, удерживается в растворе из-за образования сульфатного комплекса, В этом случае разделению благоприятствует еще то обстоятельство, что фосфат гафния труднее растворяется, чем фосфат циркония; однако фракционированным осаждением фосфата только из водного раствора разделение достигается далеко не так быстро, как указанным выше способом. Можно поступать следующим образом: фосфаты циркония и гафния растворить в плавиковой кислоте и образующиеся при этом фторофосфато-цпрконат и фторофосфатогафпат последовательно разрушить борпой кислотой (образование фтороборат-попов, де Бур, 1927).
Применяющиеся в настоящее время методы разделения циркония и гафния основаны на различной устойчивости комплексов в тих металлов. В части этих методов используют распределение между двумя растворителями, Так Шульц и Ларсен (Schultz, Larsen, 1950) разделяли цирконий и гафпий, встряхивая водный раствор хлоридов обоих металлов с бензольным раствором трифторацетнлацетоин; цирконий преимущественно переходит в органическую фазу благодаря образованию инутрикомллексных солей. Фишер (W, Fischer, 1947) осуществлял разделение этих металлов встряхиванием раствора их сульфатов, насыщенного роданидом аммония, с эфирным раствором родапистоводородной кислоты. В этом случае органическая фаза обогащается гафнием, который переходит в эфирный раствор в виде роданида; цирконии, более склонный к образованию сульфатокомилексов, остается в водном растворе. Другие методы разделения основаны на избирательной адсорбции из растворов, в которых цирконий н гафний существуют в виде комплексных ионов. Можно, например, проводить адсорбцию на силикагеле из концентрированных растворов соляной кислоты в метиловом спирте, при этом преимущественно адсорбируется гафпий (Hansen, 1949/1950; Beyer, 1952). Для выделения из растворов катионов, по связанных в комплекс, или апиопов, присутствующих в виде ацидокомплексов, можно использовать ионный обмен (Seaborg, 1948; Hoffmann, Lilly, 1949). При разделении циркония и гафния этим способом целесообразно поступать следующим образом: раствор, содержащий соли обоих металлов, пропускают через колонну, наполненную адсорбентом или ионообменной смолой, после этого промывают раствором, содержащим подходящий комплексообразователь. Если подобрать достаточно длиппую колонну, то можно за один рабочий цикл добиться весьма четкого разделения.
Естественно, что соединения циркония п гафния различаются иногда по своему удельному весу. Это обстоятельство можно использовать для сравнительно простого определения гафпия в препаратах циркония. Чистая двуокись циркопия, полученная осторожным па г рев апп ем сульфата с последующим прокаливанием при 1000s, имеет при 20° удельный вес 5,73; двуокись гафния, приготовленная аналогичным способом, имеет удельный вес 9,08. Процентное содержание двуокиси гафния х в смеси окислов с удельным весом s при 20° вычисляется по формуле
9,68а; , 5,73(100—ж) 100(^—5,73)
Ч06- + -—loo-----=s пли я=—зд«—‘
Г Реми
98
Глава 3
Гафний, так же как цирконий, с трудом можно перевести б более низкую степень окисления. Однако Шумбу (Schumb, 1947) нагреванием паров ШВг4 с водородом и металлическим алюминием в трубке, в которой поддерживается градиент температуры, удалось получить ШВг3— черно-синее твердое вещество. При нагревании в вакууме выше 300° НГВг3 разлагается на белый HfBr4 и HfBrj глубоко-черного цвета; последний при еще более высокой температуре распадается на ШВг4 и Ilf,
Техническое применение гафния до настоящего времени очень ограниченно. Его используют для изготовления специальных нитей в лампах накаливания и для фотоэлектрических целей, Частично его применяют в рентгеноспектрографии н для изготовления телескопов. Двуокись гафния используют для производства термостойкого стекла,
ТОРИЙ (THORIUM) Th
Распространение в природе. Торий встречается в природе главным образом в виде минерала монацита, в общем не редкого, но скопившегося в немногих местах. Этот минерал представляет собой изоморфную смесь фосфатов редкоземельных металлов и наряду с этим содержит 1—18% окиси тория, преимущественно в виде силиката, а частично, возможно, также в виде фосфата.
Другие минералы редкоземельных элементов также почти всегда содержат торий. Собственно ториевые минералы, напротив, очень редки. Изоморфный циркону силикат тория TliSiO4 встречается в более или менее выветренном состояли и в Норвегии в форме торита и оранжита. Однако эти месторождения почти исчерпаны.Цейлонский ториа~ нит. состоит главным образом из окиси тория ТИО2, но содержит наряду с этим значительные количества окиси урана. Уран почти всегда в более или менее значительных количествах встречается в минералах тория.
Исторические сведения и получение. Торий был открыт в норвежском минерале Берцелиусом в 1828 г. в виде окиси. Элемент получил свое название по имени бога грома Тора, а минерал был назван торитом. Еще Берцелиус пытался выделить металл восстановлением двойного фторида или двойного хлорида калием или натрием. Однако это ему удалось лишь в незначительной степени, ввиду большого сродства тория к кшь породу, азоту и другим элементам, а также в связи с высокой температурой плавления. Позднее разработанные методы получения, основанные большей частью на восстановлении хлорида натрием, также приводили всегда лишь к более или менее чистому металлу. Совершенно чистый торий был получен только недавно — разложением иодида на раскаленной проволоке способом ван Арке ля и де Бура, описанным для циркония.
Свойства. Торий представляет собой металл, в порошкообразном состоянии серый, в компактном — похожий на платину, несмотря на небольшое содержание окисп, мягкий и ковкий (удельный вес и температуру плавления см. на стр. 62). Разбавленные кислоты, включая плавиковую, действуют на него лишь в незначительной степени и очень медленно. Напротив, он хорошо растворяется в кипящей соляной кислоте, а лучще всего — в царской водке. Едкие щелочи на него не действуют. При температуре около 500° торий энергично соединяется с галогенами и серой, а при более высокой температуре также с азотом. С последним он образует коричневый нитрид Th3N4. В токе кислорода торий сгорает с выделением очень большого количества тепла (см. стр. 63) и образует окись ThO2. С водородом порошкообразный торий при температуре красного каления реагирует со вспышкой. При этом образуется продукт, представляющий собой в охлажденном состоянии черный порошок, в котором на 1 атом тория может приходиться более 3 атомов водорода.
Четвертая группа периодической системы 89
С фосфором и серой торий также соединяется непосредственно. Он образует фосфиды ThP и ТЬ3Р4 (Blitz, Meisel, 1938—1939) и сульфиды ThS, Th2S3, ThS2 и Th3S7 (Biltz W., 1941). Об отношении тория к другим металлам еще очень мало известно (ср. табл. 9, стр. 66), При 25° в ртути растворяется только 0,014 ат.% тория (Ратка, 1936).
Применение. Металлический торий до настоящего времени применяют в технике лишь в незначительном объеме *. Окись тория, напротив, имеет огромное значение в производстве калильных сеток. Ее используют также в качестве добавки к вольфраму при изготовлении ламп с металлической нитью, так как небольшие добавки ее увеличивают срок службы нитей. (О применении двуокиси тория при изготовлении монокристаллических нитей из вольфрама см. стр, 193.) Благодаря высокой способности поглощать рентгеновские лучи окись тория используют в рентгенодиагностике в качестве контрастной среды при исследовании пищеварительного тракта. Опа находит также применение в медицине. Последние Годы значительные количества окиси тория используют для получения смешанных катализаторов, применяющихся в производстве бензина методом Фишера и Тропша. Фторид тория в смеси с другими солями пригоден в качестве наполнителя фитильных углей дуговых ламп. Нитрат тория в смеси с порошком магния применяют для мгновенных вспышек.
Вследствие возрастающего вытеснения газового освещения электрическим и поэтому сокращения производства калильных сеток некоторое время наблюдалось перепроизводство тория, что требовало подыскания новых возможностей его применения, Если прежде при переработке монацитового песка основным продуктом были соединения торня, то в настоящее время ввиду значительно возросшего применения соединений церия, лантала, неодима, празеодима и других редкоземельных металлов, соединении тория представляют собой лишь побочный продукт.
Важная новая область применения металлического тория открывается благодаря техническому использованию ядерпой энергии. А именно его; можно применять в так называемых «брутреакторах» при расщеплении уранового изотопа 2ЯЗ U, которого нет в природе (подробнее см. гл. 13).
СОЕДИНЕНИЯ ТОРИЯ
Торий в своих соединениях почти всегда электроположительно четырехвалентен. Соединения тория(1У) обычно бесцветны. Растворимые в воде простые соединения диссоциируют с образованием бесцветных ионов Th"". Гидроокись тория имеет чисто основной характер. Его соли легко гидролизуются при нагревании. Большинство солей тория образует с солями сильноосновных металлов двойные соли (ацидотораты). Особо примечательна способность нитрата тория образовывать многочисленные двойные соли, так как нитраты вообще мало склонны к образованию двойных солей. Растворимость двойных карбонатов тория имеет значение для аналитической и препаративной химии.
Соединения тория с более низкой степенью окисления, а именно Thl3, Thl2, TliBr3, TiiBr2, TliClg и ThCl2 были получены Хейеком (Науек, 1948—1951) и Андерсоном (Anderson, 1949). Их можно получить, например, нагреванием тетра галогенидов с металлическим торием, а также синтезом из элементов, в зависимости от количестаа которых продукт отвечает той или иной формуле.
* Его применяют главпым образом в качестве добавки, снижающей образование окалппы, в сплавах, которые используют для изготовления спиралей в электрических печах, и в качестве поглотителя газов в высоко вакуумной технике.
too
Глава 3
Исходным материалом для получения большинства соединении является нитрат тория или полученная при его прокаливании окись. В технике нитрат тория получают из монацитового песка. Последний представляет собой смесь разнообразных наносных тяжелых минералов, песчаных продуктов выветривания мопацитсодержащих пород. Отложепия огромной мощности встречаются в некоторых местах бразильского побережья, а также в Каро липе (США). Из других месторождений следует назвать прежде всего Индию, Египет, Мадагаскар и Новую Зеландию.
Сырой монацитовый песок сначала сильно обогащают монацитом. Цена его зависит от с одержи пн я окиси тория, которое в обогащенном песке составляет 4—5%. Носок обрабатывают концентрированной серной кислотой при умеренном нагревании; после охлаждения затвердевшую массу растворяют в холодной воде, и торий вместе с редкоземельными металлами осаждается в виде фосфатов после нейтрализации раствора. Фосфаты растворяют в Концентрированной соляной кислоте, осаждают щавелевой кислотой и экстрагпруют хорошо промытеш осадок подогретым раствором соды. При этом основная масса редких земель остается н ера створенной, в то время как тория в виде карбонатного комплекса переходит в раствор. Qi оставшейся Примеси редких земель освобождаются повторной кристаллизацией сульфата ТЬ(ЗО/1)2--ЗТЦО, причем вначале каждый раз осаждают аммиаком гидроокись, а затем ее переводят в сульфат растворением в серной кислоте. Очищенную гидроокись, наконец, растворяют в азотной кислоте и подучают нитрат, используемый в производстве калильных сеток. Если требуется получить его совершенно чистым, следует применить специальные методы.
Окись тория, двуокись тория, торцевая земля. ThO2 образуется при прокаливании гидроокиси тория Th(OH)4 (или гидрата двуокиси, ср. ниже) или солей тория, образованных летучими кислотами в виде белого в зависимости от способа получения плотного или рыхлого порошка. Удельный вес ThO2 9,87. При сплавлении с флюсом, например с бурой, окись переходит в отчетливо кристаллическую форму. Она кристаллизуется в кубической системе (решетка плавикового шпата, a — 5,57 А). Кристаллическая или сильно прокаленная окись тория почти нерастворима в кислотах. Сплавлением с гидросульфатами щелочных металлов или упариванием с концентрированной серной кислотой ее можно легко перевести в сульфат. Расплавленные карбонаты щелочных металлов, напротив, па нее не действуют; так же мало действуют расплавленные едкие щелочи, так как ТЬОЙ в противоположность TiO2, ZrO2 и ИЮ2 не образует солей с основными окислами.
При осторожном прокаливании (500—600’) оксалата окись тория получается в вицо очень рыхлого порошка (гак называемая метаокись горня), который о оглпчие от обычной окиси тория заметно растворяется в кислотах, например в разбавленной соляной кислоте. В действительности при этом идет процесс пептизации подобно мета оловянной кислоте, только гидрозоль окиси тория гораздо б олео устойчив, чем гидрозоль мета оловянной кислоты. Остатки, образующиеся после выпаривания этих гичроюлой, можно снова перевести в коллоидный раствор водой. Золи окиси тория заряжены положительно. (Легкость, с которой окись тория переходит в коллоидное состоя iTiiiy имеет значение для пр имен спи я ее в качестве примеси к вольфраму при изготовлении нитей ламп накаливания.
При смешивании растворов солей тория с аммиаком или гидроокисями щелочных металлов образуется белый, желатипообразный осадок, который обычно называют гидроокисью тория. Действительно ли это гидроокись или только гидрогель другой реакционноспособной формы окиси, пока не известно. По своей структуре осадок аморфный. Только после длительного пагревания в присутствии маточного раствора он дает иа рентгенограмме интерференционные кольца, которые соответствуют
Четвертая группа периодической. системы
101
кристаллической окиси тория. Св еже осажденный осадок легко растворяется в кислотах и жадно поглощает двуокись углерода из воздуха, чем обнаруживает свои сильноосновные свойства. В щелочах ТЬО2не растворяется, но вследствие образования комплексных солей растворяется в растворах карбонатов щелочных металлов.
Чистая двуокись тория в отличие от двуокиси циркония не испускает яркий свет при накаливании. Но если ThO2 содержит небольшие количества окрашенных редких земель, папример окиси церия, то, будучи помещенной в пламя, она светится чрезвычайно ярко. На этом основано введенное Ауэром фон Вельсбахом применение тория для производства газокалильных сеток, имевшее одно время чрезвычайно большое значение. Несмотря на то что газовое освещение в настоящее время в значительной степени вытеснено лампами накаливания, использование его для освещения улиц до сих пор очень распространено.
Исключительная яркость света окиси тория, содержащей некоторое количество окиси церия, обусловлена тем, что окись тория излучает мало тепла и поэтому очень сильпо разогревается в пламени бунзсновской горелки, а окись церия при высокой температуре излучает очень интенсивный свет. Чистая двуокись церия, внесенная в пламя оунзоновской горелки светится очень слабо, потому что наряду со светом одновременно излучает мпого тепла. Так как испускание света сильно возрастает с температурой, а чистая окись церия из-за сильного теплоизлучения мало разогревается в пламепи бунзсновской горелки, ее световая эмиссия 'эффективна только в присутствии излучающей мало света и тепла окиси тория. Только таким путем можно добиться яркого свеч ей и я окиси церия н буцзеновском пламени, требующего в отсутствие окиси тория более высокой температуры. Наиболее употребительна смесь 09% ТйО3 и 1% Се2О3 (ер. ниже). Максимум излучения калильной сетки, изготовленной из этой смеси, лежит в короткопол повой области спектра. Поэтому в противоположность красноватому свету других источников света свет газовых ламп белый с чуть зеленоватым оттепком.
Для практического использования в качестве раскаленных тол имеют значение еще два свойства, отличающие ThO2 от других окйслов (которые по своим термооптическим свойствам также могли бы использоваться), а именно: устойчивость при накаливании и малая летучесть. Малая летучесть существенна также и при использовании окиси церия, поскольку она должна достаточно длительное время удерживаться в смеси.
Для приготовления остова калильной сотки вяжут «чулок» из волокна «рами» и смачивают его смесью нитратов тория и церия, взятых в определенном соотношении. Для обычных кзлильпых сеток используют растворы, содержащие 99,0—99,2% нитрата тория и 1,0—0,8% нитрата церия. Поело высушивания ткань сжигают. Оставшийся скелет из золы, состоящий главным образом из смеси окйслов тория и церия, погружают в раствор коллодия, который в дальнейшем легко сгорает; в таком виде сетки транспортируют.
Окись тория— перекись водорода. Под действием перекисп водорода ив растворов солей тория осаждается белый слизистый осадок, который подобно соответствующим осадкам из растворов солей титана и циркония носит характер гидрогеля и так же трудно обеавояшваетси. После экстракции жидким аммиаком оп имеет состав TI12O7 • 4ИгО. В этом соединении на 2 г-атома тория приходится 3 г-ат.ома активного кислорода, но, как показывает проба Ризенфельда — Лиебхафского, представляет собой не настоящую перекись, а продукт присоединения и ер окиси водорода (и воды) к окиси тория: 2ТйОа-ЗИгО2-Н2О (Schwarz, 1935). Это соединение более устойчиво, чем пероксо-кислоты титана и циркония. В отлнчие от них оно, однако, не реагирует с щелочами с образованием п ер оксо-сол ей. Неспособность тория к образованию подлинных it ер оксо-кислот обусловлена, очевидно, более основным характером окиси тория. Соответственно торий, так же как и цирконий, не образует пероксосульфатов (стр. 91).
Хлорид тория. ThCl4 образует бесцветные иглы (уд. вес 4,59; т. пл. 765°, т. кип. =922°), возгоняющиеся в вакууме уже при 750°. Он получается при пропускании смеси хлора и окиси углерода пли четыреххлористого
102
Глава 3
углерода над нагретой до красного каления окисью тория
ThO24-2CO + 2Cl3=ThC14+2CO2.
В воде хлорид тория растворяется с выделением большого количества тепла. В водном растворе он подвергается глубоко идущему гидролитическому расщеплению. Легко растворяется также и в спирте. Па раствора гидроокиси тория я спиртовой (с небольшим содержанием воды) соляной кислоте кристаллизуются при упаривании гидраты хлорида тория, а при определенных условиях также содержащие воду оксихлориды.
Хлорид тория способен присоединять хлориды других металлов с образованием двойных солей типа 2М[С1-ТЙС14 или M|[ThCle]. Подобно хлоридам титана и циркония, он образует продукты присоединения с аммиаком (ThCi4-6NH3 и ThCl4- 8NH3), с органическими аминами и другими азотсодержащими соединениями, например с пиридином, хинолином, а также со многими кислородсодержащими органическими соединениями. Средн органических соединений некоторые не просто присоединяются к хлориду тория, а реагируют с ним с отщеплением хлористого водорода. Так, бензальдегид реагирует следующим образом;
/С1 H-j-CO-CeH5
ТьЛ + •
Ъс1 H-1-CO-CeHs
^CO-CjHj ^CO-CsHs
+ 2НС1,
Бромид и иодид тория очень близки по свойствам хлориду тория. От них также производятся окепгалогениды. Бромид тория менее, чем хлорид тория, склонен к образованию продуктов присоединения. Дня иодида тория продукты присоединения вообще не известны.
Фторид тория и фторотораты. Безводный фторид тория TliF4 получают пропусканием фтористого водорода над нагретым до 400° безводным хлоридом или бромидом тория в виде белого порошка. При действии плавиковой кислоты на гидроокись тория образуется белый порошок фторида тория, содержащий кристаллизационную воду Он нерастворим в воде и в избытке плавиковой кислоты и поэтому выделяется также в виде студенистого, переходящего затем в зернистый осадка при смешивании растворов солей торн яс плавиковой кислотой или с фторидом, аммония.
Трудной растворимостью фторида тория объясняется тот факт, что известны только немногие из комплексных производных фтористых солей. Концентрация имеющегося в водном растворе фторида тория в общем недостаточна: для образования этих, очевидно-слабых фтороторатных комплексов. И только в случае если они также очень трудно растворимы, их можно получить из водного раствора. Известные до настоящего времени комплексы соответствуют типам М1 [ThF5] и М| [ThFe].
Нитрат тория и нитрат о тора ты. Простой нитрат тория Th(NO3)4 кристаллизуется из растворов гидроокиси тория в азотной кислоте с переменным, в зависимости от условий получения, содержанием воды. На холоду из не очень кислых растворов кристаллизуется двенадцатпводпый гидрат. Он очень трудно растворяется в воде и спирте. Его водный раствор вследствие гидролиза имеет кислую реакцию, и из него постепенно выделяется "основная соль.
Нитрат тория оказался особенно подходящим для изготовления окисного скелета калильных сеток, так как образующаяся из него при прокаливании окись имеет желательную в данном случае особенно тонкую консистенцию. Поэтому нитрат производят в больших количествах. Торговый продукт содержит, как правило, четыре молекулы воды и небольшую примесь сульфата.
Нитрат тория легко образует с нитратами одно- и двухвалентных металлов очень хорошо кристаллизующиеся двойные соли. Двойные нитраты тория (нитратотораты) в большинстве случаев соответствуют, типу
Четвертая, группа периодической системы 103
Mi[Th(NOj)e]. Соли щелочных металлов этого типа безводные, соли щелочноземельных металлов все содержат 8 молекул кристаллизационной воды. Помимо этого существуют и содержащие воду нитратотораты щелочных металлов типа M^fTh^O^s].
Ацетат тория Th(C2H3O2)4 выделяется при упаривании раствора гидроокиси тория и уксусной кислоте в виде бесцветных иголочек. При Кипячении растворов солей тория, смешанных с ацетатом натрия, осаждаются основные ацетаты.
Сульфат тория я сульфатотораты. Безводный сульфат тория Th(SO4)2 представляет собой белый, легко растворимый в воде порошок. При выпаривании смеси двуокиси тория с концентрированной серной кислотой сульфат получается не совсем однородный. Из водных растворов кристаллизуются гидраты. В чистом виде его можно получить, согласно Мейеру (Меуег), осторожным обезвоживанием октагидрата. При сильном прокаливании в пламепи паяльной лампы он переходит в окись.
Гидраты сульфата тория, кристаллизующиеся из водных растворов, содержат в зависимости от условий получения различное количество воды. Так, октагидрат ТЬ(8О4)2-8Н2О получают при упаривании содержащего очень небольшой избыток серной кислоты раствора при температуре 30—35°. Однако он неустойчив. Стабильными являются: ниже 45° нопагид-рат (в соприкосновении с раствором) и при более высоких температурах тетрагидрат.
Прибавление сульфатов щелочных металлов к растворам сульфата тория приводит к образованию двойных солей (в большинстве случаев довольно трудно растворимых) сульф ат оторатов соответствующих типов: M2r[Th(SO4)3f, Mi[Th(SO4)4l, Mf[Th(SO4)5] и Mi[Th(SO4)eJ.
Оксалат тория и оксалатотораты. Оксалат тория Th(C3O4)2-6H2O образуется в виде нерастворимого как в воде, так и в разбавленных кислотах осадка при смешивании растворов солей тория со щавелевой кислотой или оксалатами щелочных металлов. Из раствора, содержащего избыток оксалата щелочного металла, получаются двойные или комплексные соли (оксалатотораты) главным образом типа М][ТЬ(С2О4)4].
Карбонатотораты. Гидроокись тория и многие другие трудно растворимые в воде соединения тория легко растворяются в водных растворах, содержащих карбонаты щелочных металлов или аммония. Это обусловлено образованием комплексных солей — карбонатоторатов М* [Tb(CO3)5J', легко растворимых в воде. Их растворимость используют дня отделения тория от имеющихся в монаците редких земель (ср. стр. 99).
Фосфат тория и силикат тория. Метафосфат тория Tli(PO3)4 получают из расплава. При взаимодействии растворов солей тория с фосфатами щелочных металлов или фосфорной кислотой, по-видимому, но образуются соединения определенного состава. Сплавлением двуокиси тория с кварцем получают силикаты: ThSi20e и ThSiO4. Последние встречаются в природе в виде оранжита (прозрачные с жирным блеском оранжевые кристаллы) или в сильно выветренном состоянии в виде черного непрозрачного торита.
Карбид и силицид тория. Карбид тория ТйС2 образуется при нагревании окиси тория с углем в электрической печи. В чистом виде он образует желтоватые кристаллики. Не только разбавленными кислотами, но даже водой он разлагается с выделением различных углеводородов, главным
104
Глава 3
образом ацетилена, а также водорода. Труднее разлагается силицид тория ThSi2, получаемый в тех же условиях восстановлением окиси кремния,
_ ThSi2 образует, по цаппым Брауэра, тетрагональную решетку с высокой симметрией и правильным расположением атомов. Ему иаотипны LaSi2 и силициды лантанидов Cd, Рг, Nd, а также актинидов USi2, NpSi2 и PuSi2.
Нитрид тория Th3N4 образуется при нагревании металлического тория или смеси ТЬО2 и Mg в атмосфере азота. Он представляет собой устойчивый при нагревании в отсутствие воздуха порошок от желтой до желто-коричневой окраски. При нагревании на воздухе нитрид тория вспыхивает и сгорает до ThO2.
Аналитические сведения. По своим аналитическим реакциям торий похож па цирконий. Так же как последний, он осаждается сернистым аммонием, гидроокисями щелочных металлов или аммония, а также карбонатом бария в виде белой гидроокиси. Для тория, как и для циркония, характерна растворимость полученного при действии карбоната аммония осадка в избытке осадителя. Но по отношению к щавелевой кислоте он отличается от циркония. Оксалат тория в отличие от оксалата цирконила нерастворим в избытке щавелевой кислоты и в разбавленных минеральных кислотах. Правда, он растворяется в оксалате аммопия, но опять выпадает при добавлении соляной кислоты. Далее торий отличается от циркония тем, что оп осаждается из разбавленных растворов плавиковой кислотой, '
От редких земель, вместо с которыми оп осаждается в виде оксалата из кислых растворов, торий может быть отделен благодаря растворимости гидроокиси в растворах карбопатов щелочных металлов или аммония, а также осаждением в виде продукта присоединения его окиси с перекисью водорода, В присутствии редкоземельных металлов можно, по Мейеру и Шпетеру (1911), определить даже очень небольшие количества тория путем осаждения ого из растворов в крепкой азотной кислоте в виде нодата.
Для количественного определения тория обычно его выделяют в вндр гидроокиси или продукта присоединения 2ThO2-3H2O2'H2O, затем прокаливанием при высокой температуре переводят в окись и взвешивают. Для этой цели пригодпо также осаждение йодата тория. При этом рекомендуется проводить «осаждение из гомогенного раствора», так как в противном случае получается слизистый, очень трудно фильтрующийся осадок.
Под осаждением ио гомогенного раствора подразумевается такой процесс, когда осадитель пе вносится как таковой в раствор, а образуется в нем в результате медленно протекающего химического взаимодействия. В рассматриваемом случае ионы 10, получают восстановлением Юр-ионов гликолем. Чтобы обеспечить достаточно медленное протекание реакции, гликоль прибавляют но непосредственно, а дают ему возможность постепенно образовываться, по мере того как протекает гидролиз его мопопцетата (Stine, Gordon, 19531. Этот способ находит разнообразное применение. Большое число веществ, образующихся обычно в виде слизистых, плохо фильтрующихся н сильно адсорбирующих осадков, при осаждении из гомогенных растворов получается в зернистой, хороню фильтрующейся, поверхностно пассивной форме.
Глава 4
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Побочная подгруппа)
ГРУППА ЗЕМЕЛЬНЫХ КИСЛОТ
Порядковый номер Элемент Сим-вод АТОМНЫЙ вес Удель-лый вес Температура, °C Удельная теплоемкость Валентность
плавления кипения
23 Ванадий V 50,95 5,98 1715 -3500 0,1203 п, ni.iv, V
41 Ниобий Nb 92,91 8,56 2470 ~ 5100 0,0645 II, III, IV, V
73 Тантал Та 180,95 16,69 ЗОЮ - 6000 0,0333 II, III, IV, V
91 Протактиний Ра 231 15,37 — - 4200 —• 11,111, IV, V
Общие сведения. Побочная подгруппа пятой группы включает элементы: ванадий, ниобий, тантал и протактиний. Все они соответственно номеру группы преимущественно пятивалентны. Несмотря на то что перечисленные элементы являются металлами, их высшие окислы — пятиокмеи — носят ярко выраженный кислотный характер. Это относится к ванадию, ниобию и танталу, и поэтому пятиокиси этих металлов называют кислыми землями (т. е. кислотообразующими окислами металлов) или «земельными кислотами».
Протактиний принадлежит к радиоактивным элементам. Онг родоначальник актиния (ср. гл. 11). Ра — элемент редкий и трудно получаемый химически, поэтому он еще мало исследован. В соответствии с общим правилом периодической системы он имеет более основной характер, чем первые три элемента группы, которые но могут образовывать простых солей в водном растворе даже с самыми сильными кислотами.
Элементы побочной подгруппы пятой группы, так же как и элементы главной подгруппы, могут быть не только положительно пятивалентными, но существуют и в состоянии более низких степеней окисления. Однако в отличие от главной подгруппы, где тенденция к образованию более низкого положительного заряда возрастает с увеличением атомного веса, в побочной подгруппе эта тенденция убывает в том же направлении. Так, при обработке растворов солей цинком и кислотой ванадий восстанавливается до двухвалентного, ниобий — до. трехвалентного состояния, а тантал при таких условиях вообще не восстанавливается.
В отличие от элементов главной подгруппы V7 группы элементы побочной подгруппы, как и вообще все элементы побочных подгрупп периодической системы, никогда но бывают электроотрицательной составной частью солеобразных соединений. Опп могут, следовательно, отщеплять до пяти электронов, но сами никогда не принимают их; это соответствует тому, что за ними не следуют инертные газы, электронная конфигурация которых отличается особой устойчивостью. С этим связана также неспособ-
106
Глава 4
ность элементов побочной подгруппы пятой группы образовывать газообразные соединения с водородом. Напротив, они дают твердые гидриды, подобные тем, какие образуются в побочной подгруппе четвертой группы.
По данным Сивертса, 1 г ванадия при комнатпой температуре и атмосферном давлении поглощает 157 мл водорода, 1 г ниобия — 104 мл и 1 г тантала — 55,6 ..ил. При500°соответственно 19,9,47,4 и 14,8мл; при 1000°— 2,3, 2,8и 1,4При температурах выше600° количество абсорбированного водорода изменяется почти пропорционально корню квадратному из величины давления. При более низких температурах абсорбция водорода с ун сличением давления возрастает сильнее вследствие образования соединений. При комнатной температуре и атмосферном давлении на 1 ё-атом ванадия приходится 0,71 лт водорода, на 1 г-атом ниобдя — 0,87, тантала — 0,90 ё-атомов водорода (при температурах 500 и 1000° соответственно 0,09, 0,39, 0,24 и 0,01, 0,02, 0,02 г-атолл водорода). Хегг в 1930 г. рентгенометрически установил, что в решетке тантала может содержаться до 12 ат.% водорода. При более высоком содержании водорода сначала образуется соединение Та2Н, имеющее тетрагональную структуру; при еще более высоком — ТаН, решетка Которого подобна решетке цинковой обманки (расстояние Та > Н = 1,901 А) ,
Ванадий, ниобий и тантал образуют кубическую объемноцентрирован-ную решетку; а = 3,0282; 3,3007 и 3,2997 А (ср. т. I, рис, 47, стр. 238), Следовательно, по своему строению они подобны щелочным металлам, но обладают значительно меньшими атомными радиусами. Атомные радиусы Nb и Та в пределах, ошибки измерения совпадают. Ионные радиусы также очень мало различаются (см. табл, 12). Вследствие этого соеди-
Таблица 72
Кажущиеся атомпые и иоппые радиусы элементов побочной подгруппы V группы
Элемент Ванадий Ниобий Тантал Протактиний
Атомный радиус, А Радиус четыре хвален тио го иона, А Радиус пятивалентного иона, А а KZ—координационное чис 1,3.112 (KZa==8) 0,61 0,48 до. 1,4292 (KZ=8) 0,69 0,69 1,4289 (KZ=8) 0,67 0,68 ’ 1 1,6088 (KZ—10) 1,06 ““
пения ниобия и тантала весьма близки по своим свойствам, но это сходство, однако, пе столь велико, как у соединений циркония и гафния (ср. стр. 96 и сл.). Протактиний образует тетрагональную объемноцент-рированиую решетку (а = 3,925, с = 3,238 А).
Элементы побочной подгруппы пятой группы принадлежат к числу редких элементов. К ванадию это относится с некоторой оговоркой, так как он в небольших количествах входит в состав достаточно большого числа минералов; его же собственные минералы встречаются редко. Протактиний, встречающийся в природе в качестве спутника радия,— оба эти элемента образуются при радиоактивном распаде одного и того же элемента, а именно урана или его изотопов (подробнее см. гл. II) — является еще более редким, чем радий (ср. стр, 134 и сл.).
Пятая группа периодической системы
107
Теплоты образования пятиок осей этих элементов составляют (ккал/г-екв металла): fv2O5] 37,3, [NbgOJ 46,3, fTa30j} 49,3. Они, следовательно, значительно ниже, чем у элементов и об очной подгруппы IV группы, если сравнивать одни и те же количества металлов (ср, табл. 6, стр. 63). В обоих случаях теплоты образования возрастают с увеличением атомного веса.
Сплавы. Как следует из табл. 13, сведения относительно сплавов металлов побочной подгруппы пятой группы еще очень недостаточны. Причины этого те же, что в случае сплавов металлов побочном подгруппы четвертой группы. Значительно более точные данные имеются о поведении этих металлов по отношению к некоторым технически более важным металлам побочных подгрупп (Fe, Ni, Си, Ag), а также о системах, похожих на сплавы, в образовании которых участвуют неметаллы.
Тензиметричееднм и рештенометрическим методами Бнльтц (BiJtrz W., 1938) исследовал системы _\b — S и Та — S. Оказалось, что Nh и Та по включают в свои решетки заметных количеств серы, по сульфиды этих металлов по составу могут колебаться в широких пределах. Следовательно, опи обладают характером недалъ-тонидпых соединений. Однако состав большинства этих сульфидов можно представить формулой, так как в большинстве случаев одна из двух граничащих фаз соответствует ей. Например, м о по сульф ид ниобия NbS, не содержащий в своей решетке избыточной серы, может содержать до 1 атома избыт оч пог о ниобия. Полуторный сульфид NbaS3, яанротпв, не может принять избыточный ниобий, по может содержать до 5 атомов избыточной серы (NbS4). Тантал образует трисульфид TaS3, который может содержать избыточную серу (но пе тантал), н дисульфид TaSa, способный принимать серу в таком количестве, что ее содержание превосходит даже TaS3. Далее, Та образует по крайней мере еще более низкий сульфид (Тай?) также с переменным составом.
Следует еще указать, что ванадий, по-видимому, дает сплав с платиной, a Nb -образует аналогичные соединения с Р и As. На основании размеров атомных радиусов можно утверждать, что Nb и Та способны в некотором соотношении образовывать твердые растворы. О системах V — Nb nV — Та пола ничего неизвестно.
ВАНАДИЙ (VANADIUM) V
Распространение в природе. Несмотря на то что ванадий довольно распространен в земной коре, он очень редко встречается в больших количествах. Для технического получения ванадия важнейшим сырьем долгое время служил патронит (сульфид ванадия), мощные месторождения которого находятся главным образом в Перуанских Андах на высоте 5500 м над уровнем моря. В настоящее время большое значение приобрели также ванадиевые руды, встречающиеся в Юго-Западной Африке, Северной Америке и Родезии. Первым из найденных минералов ванадия является ванадинит (ванадиевосвинцован руда) 3Pb3(VO4)2 РЬС1а; он распространен главным образом в Мексике и других южноамериканских штатах, а также в Испании.
В Колорадо и Утахс (на западе США), а также в Южной Австралии встречается карнотит Прежде его добывали
главным образом из-за радия. В настоящее время он является в основном источником урана и в качестве побочного продукта из него получают (наряду с радием) ванадий.
Следует упомянуть далее роскоэлит — силикат сложного состава, содержащий ванадий. Он встречается в Колорадо, Калифорнии, Новом Южном Уэльсе и Южной Австралии главным образом в виде окрашенного в зеленый цвет песчаника (ванадиевый песчаник) и до открытия патронита и карнотита представлял собой важнейшую ванадиевую руду.
Таблица 13
Образование соединений и твердых растворов металлами побочной подгруппы V группы е другими металлами и некоторыми неметаллами
[Обозначения те же, что и в табл. 9 (стр. 66)]
Главные подгруппы Пибичпыс подгруппы
Нс "«1 « AI С N - As S Sc C" Mo w Mrt Fe Cu AS
V Be2V 1 1 VD, 21 i)U° тв. > О V2A1? VAI? VAI,,? тв. >• О v5c V2c V4C3 VC, 2750’ тв, 0 V2Si VSi2 1654° V3N? VZN VN vn2? v3p V2P? V3p2? VP VP2 VAs? VAs2 тв. 0? VS V2S3 VS* VSe VaSe3 VSe2 « — тв. > 0? VMn? ТВ. co ivf;] 1234’ TB.<oo? 8 V a ' e h- ж. ~ 0 тв. 0 0 ж. — 0 тв. 0 0
Nb — — NbB2 >>3U00" NbAl3? Nb2C NbC 3500’ NbS i2 тв, — 0 Nb2N Nl^N.j NbN Kb3N3? NbP NbP2 1 1 тв. 0 NbS Nb2S3 Сплав Сплав 1 1 NbMn2 тв. 0 NbxFey Nb,Fe3 1600’ NbCo2 тв.>О Nb^Nij, NbNi£? 1330’ — 1 1
Та Соединение? Сплав ТаВ2 >3000° TaAJ3? Ta2C TaC 390 9’ 'l’aSi2 TaN 3100’ W TaN2? тв, 0? TaP TaP2 ’—> тв. 0 TaS TaS; TaS^ 1 1 1 1 Сплав TaMn2 тв. > 0 TaFe2 тв. > 0 TaCo2 ТВ, TaNi* 15SQ° TaNh 1 54 5* 1 1 ж. < co тв, О 0
Пятая группа периодической система 109
Небольшие количества ванадия содержатся часто в свинцовых, иногда также в медных и железных рудах.
Исторические сведения. В 1830 г. Зефштрем (Sefstrom) в гибком железе, полученном из руд южношведского Таберга, нашел новый элемент, который по имени скандинавской богини Фрейи, или Ванадис, был назван ванадием.
За 29 лет до этого ванадий бг.гл открыт мексиканским минералогом дель Рио в свинцовой руде, добитой в Зима папе (Мексика), и вначале был назван им благодаря •окрашенным в различные цвета окислам панхромом (от греческого лаv ii'/owpa <<все-цветшлй»), а затем по красивому красному цвету нагретых солей эритронием (еотОоо^— Красный). Однако дгль Рио не удалось установить с достаточной уверенностью, что открытое им вещество новый элемент, Болео того, позднее он сам пришел к выводу, Что это была разновидность хрома. В действительности, однако, добываемая в Зима-пане свинцовая руда представляла собой соединение ванадия, как установил Вёлер сразу после открытии Зефштрема. Ее назвали ванадинитом. Сначала ванадий принимали за аналог хрома, пока в 1867 г. английский химик Роско (Roscoe) точно не доказал принадлежность его к семейству фосфора, т. е. к пятой группе периодической системы.
Получение. Для технического получения ванадия из патронита руду вначале плавят в пламенных печах с добавлением флюсов. При этом тяжелые примеси осаждаются, а ванадий переходит в шлак. Так как производство чистого металлического ванадия связано с большими трудностями, для технических целей обычно получают сплавы ванадия преимущественно с железом, так называемый феррованадий. Получение феррованадия осуществляют в большинстве случаев путем алюмотермии. Для этого шлак, содержащий вападий, смешивают в тиглях с гранулированным алюминием и железом, прибавляют флюсы (плавиковый шпат или буру), нагревают до красного каления и обычным образом доводят до воспламенения. Можно проводить этот процесс и в большем масштабе в шахтных печах.
Получение почти чистого металлического ванадия (98—99%-но г о) осуществлено Руффом и Мартином (Ruff, Martin) ал Юм от ерническим восстановлением трехокиси ванадия в тиглях из магнезии (с добавлением угля) и плавлением подученного королька в вакуумной дуговой печи. Чистый порошкообразный ванадий был получен Дерингом (Doling, 1934) медленным, осторожным нагреванием в токе водорода, в принципе примененным Роско еще в 1870 г. Компактный, совершенно чистый и поэтому ковкий ванадий был получен иодидным методом вак Аркеля в де Бура.
Свойства. Ванадий (в более или менее чистом состоянии) металл серо-стального цвета, чрезвычайно твердый, но поддающийся шлифованию и полированию. Он обладает небольшим удельным весом и очень высокой температурой плавления.
Совершенно чистый ванадий, согласно Руффу, плавится при 1715°, по достаточно небольшой примеси углерода, чтобы температура Плавления его значительно повысилось. Например, для ванадия с примесыо2,7% углерода Руфф нашел температуру плавления равной 2185°, с 10% С —2700°. Температуру плавления повышает также примесь кислорода или окисла. Так как повышение температуры плавления находится и простой зависимости от количества углерода или кислорода, то на основании уста-иоллояия температур плавления различных проб с неодинаковым содеря;анием примесей экстраполированием можно весьма точно определить температуру плавления чистого металла.
110 Глава 4
При обычной температуре в компактном состоянии ванадий не подвержен действию воздуха, воды и щелочей. Он устойчив также и по отношению к не окисляющим кислотам, а а исключением плавиковой кислоты. В сильных кислотах-окислителях — царской водке и азотной кислоте — он растворяется. В сжатом кислороде тонкоизмельченный металл сгорает главным образом до пятиокиси V2O5. Но наряду с этим в качестве продуктов сгорания ванадия образуются и низшие окислы.
Теплоты образования, по данным Симонса и Улиха (Siemons, Ulich, 1940), равны: для V2O3 296, VO2 171, V20s 373 ккал/молъ. Ванадий может поглощать до 0,4 ат, % кислорода, образуя твердый раствор. Его решетка при этом подвергается тетрагональной деформации (подобно решетке н-железа при поглощении углерода, ср. стр. 280) .
При пагревапян металлического ванадия в токе азота образуется нитрид ванадия VN. По-видимому, существуют также другие нитриды (ср. табл. 13). Если VN нагревать с Lr3N в атмосфере азота, образуется, как установлено Юзой (Juza, 1956), двойное соединение LItVIN 4, в котором ванадий в отличие от простого нитрида не трех-, а пятивалентен.
С мышьяком ванадий соединяется при нагревании, образуя VAs и VAs3. При сплавлении ванадия с кремнием в электрических печах в зависимости от условий опыта образуется либо V2Si, либо VSt2, которые различаются твердостью и значительной устойчивостью к химическим воздействиям при низких температурах. Такое же свойстно обнаруживают п карбиды V2C и VC, образующиеся при взаимодействии ванадия с углеродом при более высокой температуре (о других карбидах см. табл. 13).
Применение. Производство стали и желоза — основная область применения ванадия. Ванадий придает стали и чугуну прочность, вязкость, ковкость и устойчивость к толчкам и ударам. Особое значение это имеет в производстве авиационных и автомобильных моторов. Для достижения указанных свойств достаточна примесь 0,1—0,2% ванадия. Эти свойства обусловлены отчасти, как и в случае титана, раскисляющей способностью ванадия, кроме того, qro специфическим влиянием на структуру сплава. Для специальных целей имеют значение сплавы ванадия с другими металлами, например с медью, никелем и алюминием.
Соединения ванадия применяют иногда в качестве переносчиков кислорода. Так их можно использовать для каталитического окисления органических соединений, например для получения анилиновой черной окислением анилина хлоратом или бихроматом калия. Особо важное значение имеют соединения ванадия в производстве фталевого ангидрида путем окисления нафталина, а также в производстве контактным способом серной кислоты (ср. т. I, стр. 759), Их можно выгодно использовать также для получения так называемых «перборатов» для ускорения присоединения кислорода. Ванадиевые кислоты и ванадат натрия находят применение в терапии. Соединения ванадия в небольших количествах используют Для фотографических целен; например, соединения трех- и четырвхвалептпого ванадия служат в качестве проявителей.
СОЕДИНЕНИЯ ВАНАДИЯ
Ванадий образует главным образом четыре окисла:
V O —монокисъ ванадия, окись ванадия(П), черного цвета, уд, вес 5,76;
V 2O3 —полуторная окись ванадия, окись вападия(Щ), черного цвета, уд. вес 4,87;
V O2—двуокись ванадия, окись ванадия(1У), тсмпо-сппего цвета, уд. вес 4,40; я
V2O6—пятиокись ванадия, окись вападия(У), желто-красного цвета, уд. вес 3,34.
Кроме отих окйслов, которые соответствуют всем возможным степеням окисления ванадия, он образует промежуточный окисел V12O2g. Это соединение, подобно промежуточным окислам молибдепа и вольфрама (см. гл. V), изоструктурпо твердым раство
Пятая группа периодической системы 111
рам ZrOv — У2О3, описанным па стр. 88. Как и в них, в анионной решетке У120да имеются пустоты, число которых иногда достигает 30, по распределяются они более правильно, чем в указанных твердых-растворах.
От каждой из степеней окисления, которым соответствуют указанные окислы, производятся другие соединения.
1. Соединения ванадия{11)-, гидроокись ванадия(П) V(OH)g имеет основной характер, ей соответствуют соли типа УХ2. Они образуют двойные соли, т. е. малоустойчивые комплексные соли {ацидованада-игы(П)]. Примеры простых солей ваиадия(П): УС1.а и VSO4-7H2O (изоморфна FeSO4-7H2O). Комплексная соль: K4[V(CN)0]-ЗН2О (изоморфна K4lFe(CN)6J-3H201.
2. Соединения ваиадия(Ш)' соли типа УХ3 и соответствующие комплексные соли \ацидованадаты(111)\. Трехвалеятный ванадий более склонен к образованию ацндо-соединений, чем двухвалентный; но эта склонность слабее, чем у трсхвалентных желоза, хрома и кобальта. Наиболее устойчивой из солей ванадия(III) является сульфат и производная от него сульфатаванадий(III) кислота HV(SO4)2-6H2O.
3. Соединения ванадия(1У): окись четырехвалентного ванадия образует соли [ванадаты(1У)] как с кислотами, так и с основаниями. Их называли ранее еипованадатами. Такие соли, в которых двуокись ванадия выступает в качестве основной составной части, соответствуют (насколько это следует из характера солен, полученных из водного раствора) типу VOX2 (окси-соли ванадия или соли ванадила), а не VX4 (где X — одновалентный кислотный остаток). Положительно двухвалентный радикал [VO]" (ванадил) в этих соединениях играет ту же роль, что и в соответствующих солях двухвалентный тяжелый металл. В водном растворе он имеет светло-сипюю окраску и ведет себя как двухвалентный ион. Ацидо-соли — производные от солей ванадила — по общему правилу наименования координационных соединений (ср. т. I, стр. 445) называются оксоауи-дованадатами(1У). Некоторые соединения ванадия(1У), например тетрафторид ванадия, а также соли щелочных металлов оксосульфатована-дий(1У) кислоты (точнее: диоксотрисульфатодиванадий(1У) кислоты H2[(VO)2(SO4)3]} при нагревании разлагаются па соединения вападия(Ш) и ванадия(У).
4. Соединения ванадия(У): пятиокись ванадия У2О5— кислотообразующий окисел. Соответствующие этой кислоте соли называются ванадатами (точнее: ванадаты(У)]‘ простейшие из них соответствуют формуле Мз [УО4] (ортованадаты), по своему составу они аналогичны солям орто-фосфорной кислоты.
При замощении ионов О2- в ортованадатах па радикал перекиси водорода ОУ получаются пероксоваладаты-. соли кислот H3(VO2(O2)2] и II3(V(O2)41. Кислородный атом в ванадатах может быть замещен серой; так получаются,тиованидаты, например ортотиовападат аммония (NH4)3[VS4). Накопоц, от пятивалентного ванадия производятся также другие гало гемо-соли и ацидо-сопи [ацидованадаты( F)], например 1<2[VO2F3] диоксотрифторованадат(У) калия.
1. Соединения ванадия(П)
Окись ванадия(П), мопоокись вападия VO (47—56 ат.%0) получают восстановлением окситрихлорида вапаДия(У) VOClg или нятпокиси ванадия V2O5, например, сильным прокаливанием последней (при 1700е) в токе водорода. Она представляет собой черный порошок, обладающий в плотном слое металлическим блеском, нерастворимый в воде (с?ПиНН = 5,76, <7реНтг = 6,547). Окись ванадия хорошо проводит электрический ток. В разбавленных кислотах окнсь ванадия растворяется с образованием солей ванадия(П). VO образует решетку типа каменной соли (а=4,081 А). Она устойчива
112
Глава 4
лишь при высоких температурах. При медленном охлаждении окись ванадия разлагается с образованием твердых растворов ванадия и кислорода и более высоких окислов <К!<>татв., 1942).
Гидроокись ванадия V(OH12 выпадает в виде коричневого осадка прн действии щелочи на растворы солеи ванадия(П). Вследствие чрезвычайно сильной способности к окислению, она до настоящего времени еще не получена в чистом виде.
Хлорпд ванадия!!!), дихлоряд ванадия VC1, получается в виде светло-зеленых, похожих на слюду, блестящих Листочков при пропускании смеси тетрахлорида ванадия с водородом через трубку, доведенную до красного каления. VC13 весьма гигроскопичен, Вначале фиолетовый водный раствор быстро становится зеленым, так как двухвалентный ванадий окисляется до трех валентного с выделением водорода. Спиртовой раствор хлорида ваггадия(П) окращои в синий цвет, эфирный — в желто-зеленый. Различное окрашивание растворов свидетельствует об образовании сольватов.
Сульфат ванадия(П) VSCK- 7Н2О образуется при восстановлении сернокислых растворов ванадия амальгамой натрия, цинком или другими металлами. Самый лучший способ получения — электролитическое восстановление на ртутном катоде. Несмотря па то что сульфат вападия(П) склонен к окислению настолько, что постепенно разлагает даже волу, прн определенных условиях его вое же можно выделить в кристаллическом штояпии. Он образует моноклинные, красно-фиолетовые кристаллы VSO4-7П2О, которые способны давать твердые растворы с другими сульфатами типа MnSO4- 7П3О; при этом он является изодимор фи ы и и в зависимости от партнера может быть либо моноклинным, как, например, с же.аезпым купоросом FeSO4-7H3O, либо ромбическим, как в твердых растворах с сульфатом магния MgSOr7H2O.
Сузьфатовападаты(П). Сульфат ванадия(П) с сульфатами щелочных металлов легко образует двойные соли типа VSO4, M|SO4-f)H3O. Они трудно растворимы и немного более устойчивы, чем простой сульфат ваиадНЯ(П), однако обнаруживают еще значительную склонность к окислению.
Гексацп апо ванадат калил(П) кристаллизуется при прибавлении спирта к раствору соли вападия(П), содержащему избыток цианида калия, в виде коричнево-желтых призматических кристаллов K4[V(CN)el-311,0. Это соединение, как и аналогичный по составу гексациапоферрат(П) калин, образует осадки с растворами большинства двухвалентных металлов, по гораздо мопео устойчивые.
2. Соединения ванадия(Ш)
Окись вападия(Ш) — полуторная окись — V3O3 в виде черного, блестящего, очень тугоплавкого кристаллического порошка получают восстановлением при нагревании пяти ок йен ванадия водородом, углем или цианидом калия. Опа имеет структуру корунда: а ~ 5,43® А, а = 53°53', й1)ентР = 5,21. С Сг3О3 и Fe2O3 окись ванадия(Ш) образует непрерывный ряд твердых растворов, а с А12О3— твердые растворы с ограничен ной растворимостью компонентов.
По данным Комаровского и Кнагга (Komarewsky, Knagg, 1951), Va03 является превосходным катализатором в процессах гидрирования. Преимущество его в том, что он не отравляется серусодерждщими органическими соединениями.
V3O3 с FeO образует двойное соединение FeO-V2O3 со структурой шпинели, которое с металлическим железом дает твердые растворы (Alatlieason, 1932). Такие же двойные соединения [шпинели вападия(Ш)) полуторная окись образует с MgU, МнО и СаО; напротив, подобного рода соединении V2O3 с V0 не существуют; однако баз изменения структуры содержание кислорода в соединении может снизиться до 57 ат.% (состав соответствотгпо VO1135),
V2O3 образует с редкими элементами двойные соединения MIIIV03 типа перовскита. При взаимодействии с Lr2O и Na2O получаются LiVO3 или NaVO3 (Rudorff, 1954), постоянные решетки: а. — 2,84; с — 14,7 А или а = 2,86, с — 17,0 А; с/пищ: = = 4,13 пли 4,16; <Х„„тТГ — -4,33 пли 4,36. Эти соединения с щелочными металлами на основе их структуры (тип Na [ИF3]) мэжпо рассматривать как ванадаты(Ш). Известны также в i:i.i и и ; I i 1 • щелочпоземельных металлов.
Гидроокись вападия(Ш) V(OII)3 в вида рыхлого зеленого осадка образуется при действии щелочей или аммиака па растворы содой ванадия(Ш). Она чрезвычайно жадно поглощает кислород.
Хлорид вападпя(Ш), трихлорид ванадия VC13, можно получить нагреванием до 300° в запаянной трубке V2O3 с S2CI2 (Funk, 1940) или термическим разложением
Пятая, группа периодической системы 113
VC14. Он представляет собой блестящие кристаллические листочки, по окраске подобные цветам персикового дерева, похожие па безводный фиолетовый хлорид хрома* Трихлорид ванадия нелетуч и при прокаливании на воздухе переходит в смесь оксихлорида и пятиокиси. В воде (подкисленной во избежание гидролиза) он дает раствор, окрашенный в зеленый цвет. При выпаривании из раствора (если приняты меры, предотвращающие окисление кислородом воздуха) выделяются зеленые кристаллы состава VCI-i-nHoO, как и в случае хрома н железа. С некоторыми ароматическими оксикислотами хлорид вападия(1П) в водном растворе дает интенсивное окрашивание, например с салициловой — аметисто-фиолетовое, с меконовой кислотой — интенсивно краспое. Последнее свойство можно использовать Для Открытия опия (Wo-yaoff, 1934).
Подобно хлориду ведет себя бромид VBr3, ио он значительно менее устойчив, Иодид Vl3 получается непосредственно из элементов при температуре выше 150в в виде коричнево-черного, гигроскопичного кристаллического порошка (уд. вес 4^2). Ом мало растворим в воде, лучше — в абсолютном спирте. При нагревании до 280° разлагается на 12 и VI2. Последний образует слюдообразные листочки с уд. весом 5,0, нерастворимые в абсолютном спирте. Около 800° он возгоняется и только выше 1000° разлагается на 12 и V (Morette, 1938)*
Названные галогениды ванадия(Ш), по-видимому, мало склонны к образованию двойных солей.
Фторид вападня(И1) и фторбванадаты(Ш). Фторид трехвалентного ванадия чрезвычайно склонен к образованию двойных солей. Фторид ваиадия VP3-3H2O получают упариванием раствора гидроокиси ванадин(П1) в плавиковой кислоте в виде темнозеленых ромбических кристаллов.
Двойные фториды трехвалентного ванадия [фтдрованадаты(1П)1 по составу (без учета содержания воды) большей частью соответствуют типу M|VFS. Если они образовапы двухвалентными металлами, то в их состав входят семь молекул кристаллизационной воды, и строение пере дается формулой [МП(ОН2)6] [VF6(OH2)1.
Сульфат ванадия(III), сульфатованадий(Ш) кислота и сульфатованадаты(Ш). Если раствор пятиокиси ванадия восстанавливать в серной кислоте, например, электролитически, то он приобретает зеленую оираску, и при определенной концентрации ванадия и серной кислоты из него выпадают зеленые с шелковистым блеском иглы состава V2(SO4)3‘ H2SO4-12HEO. При осторожном нагревании выше 180° это соединение переходит В нейтральный (по составу) сульфат V3(SO4)3 ~ желтый порошок, нерастворимый в воде, спирте и эфире. Первое из названных соединений, вероятно, можно рассматривать мак сульфатоеая.адий(1Л) кислоту Н !V(SO4)-J-6Н-О, Действительно, от нее производятся соли, такие, как зеленый диеульфатованадатЦЛ) аммония MH4[V(SO4)2b6H2O; кроме того, существуют многочисленные соединения типа i^VtSOJa-lHHaO, т. е. типа квасцов, которые также образуются из раствора указанного соединения и отличаются от упомянутого выше ряда сульфатованадатов(П1) шестью лишними молекулами воды. Двойные соли ванадия типа квасцов в кристаллическом состоянии большей частью окрашены в фиолетовый цвет различных оттенков *. В концентрированном водном растворе, напротив, соли того и другого ряда имеют зеленый цвет. (В разбавленных растворах вследствие гидролиза появляется коричневая и желтая окраска.)
В названных сульфатосолях трехвалентный ванадий чрезвычайно устойчив, Даже в растворе эти соли очень медленно окисляются кислородом воздуха,
Опсалатованадаты(1Ц).Из электролитиче<жи восстановленного раствора пятиокиси ванадия в щавелевой кислоте, содержащего оксалаты щелочных металлов, при упаривании в вакууме выделяются зеленые, моноклинные кристаллы триоксалатоаа-набатов(Ш) щелочных металлов [V(C2O4)3i-3H2O. Это малоустойчивые комплексы; их растворы окисляются на воздухе, в то время как твердые соли на воздухе устойчивы,
Гексацианованадат(Ш) калия K3[V(CN)eJ образуется при взаимодействии раствора соли ванадия(Ш) с избытком цианида калия. Эта соль менее устойчива, чем соответствующие соединении трех валентных хрома, железа и кобальта. Однако водный раствор ее существенно отличается от растворов, содержащих свободные ионы V""* так вместо зеленого он окрашен в винно-красный цвет, приписываемый ионам tV(CN)elm,
* Соли рубидия и цезия существуют в синевато-фиолетовой и красной модификациях*
8 г. Роми
114
Глава 4
Роданованадаты(Ш). Эти соединения типа Mj[V(SCN)e] с различным содержанием кристаллизационной воды являются слабыми комплексами и при растворении в воде быстро разлагаются. Твердые соли7 например Na3[V(SCN)6|- 12Н2О, К3 [V(SCN)eI- 4НеО, (NH4)3[V(SCN)6]-4H2O отличаются яркими красивыми окрасками-и ди- или полихроизмом. Соль аммония, например в плотном слое, зелено-черного-цвета, а в порошкообразном состоянии — красная.
Сульфид вападия(1П), полуторный сульфид ванадия V2S3 получается В виде серо-черного порошка или графитоиодобных листочков при нагревании окиси ванадия(Щ) в токе сероводорода плп пятиокиси ванадия в парах сероуглерода при 7ОС"1. Он весьма устойчив по отношению к не окисляющим кислотам, во растворяется в разбавленной азотной кислоте; в лиццентрпрованпой азотной кислоте он даже воспламеняется.
При прокали папин в токе водорода V2S3 переходит в моносульфид VS. Последний кристаллизуется по типу NiAs (а = 3,34; с — 5,78 А); то же относится к соединениям VSe (а = 3,58; с- = 5,98 А) и VTe (а =л 3,80, с = 6,12 А). Соединения У8ег и VTe2 кристаллизуются по типу брусита.
Встречающийся в природе сульфид ванадия — патронит •— свинцово-серая, в свежем изломе с металлическим блеском руда (уд. вес 2,46 и твердость 3,5), содержит значительно больше серы, чем соответствует составу полуторного сульфида. Согласно данным Клемма (Klemm, 1936) и Бильтца (Blitz W., 1939), соответствующий патрониту высший сульфид ванадия VS4 можно получить препаративным путем.
3. Соединения ванадия(1¥)
Окись вападия(1У), двуокись ванадия VO2, получают умеренным восстановлением? пятиокиси вападия, например сплавлением ее со щавелевой кислотой. Она образует темно-синие, почти черные, блестящие кристаллики (уд. вес 4,654) или, при определенных условиях, темно-зеленый порошок. При нагревании двуокись вападия легко-растворяется в кислотах и щелочах, т. е. является амфотерным окиелом. УО2 пе даст реакции с сипей и красной лакмусовой бумагой. Под действием концентрированной азотной кислоты или при нагревании в токе воздуха двуокись вападия окисляется до пятиокиси.
УО2 (подобно КЬО2) образует решетку типа рутила; а = 4,54, е=2,88 A (NbO2: а = 4,77, с = 2,96 А).
Нагреванием УО2 с окислами щелочноземельных металлов в высоком вакууме-Шальдер (Scbolder, 1953) получил различного тнпа eanaSanw(IV), а именно MjIVO4 и MpVO5. Соединения состава M.JXVO4 в основном имеют структуру шпинели [ванадиевая шпинель(IV)]. Узлы решетки, в обычной шпинели запятые ионами А13+, в данном случае статистически поровну распределены между Ма+-и V4+-ионами. Твердые-растворы, образованные шпинелями трех- и четырехвалентного ванадия, являются полупроводниками, так как между ионами V3+ и У4+ может происходить обмен электронами (Renter, 1957). Этим свойством обладает также полученная Ройт ером (I960)-шпинель лития я вападия(1П,1У) VnlVzv[LiOJ.
Тетрахлорид ванадия и дпхаорид ванадила. Тетрахлорид ванадия, хлорид вана-дия(1У) VC14, представляет собой темную красно-коричневую тяжелую маслянистую-жидкость, затвердевающую лишь при охлаждении жидким воздухом. Температура кипения УС14 154°. Плотность пара соответствует формуле УС14. Однако выше температуры плавления происходит довольно быстрый распад на твердый трихлорид ванадия и хлор. Медленное разложение происходит уже при обычной температуре. При действии воды моментально происходят частичный гидрояиц по уравпепию УС14 Ц-Ц- Н3О = УОС1а 4- 2НС1. Получить чистый тетрахлорид ванадия вследствие этого совсем пе просто. Можно, например, получить его при действии хлора на металлический ванадий, динатрид или дисилицлд вапа дия ним пропусканием смеси трихлорида ванадила VOC13 с избытком хлористой серы через трубку, нагретую до темно-краспого-Каления. От образующихся в обоих случаях побочных продуктов тетрахлорид Отделяют фр акции пир она и пой перегонкой.
Двойные соли тетрахлорида ванадия не известны.
Получающийся при гидролиза тетрахлорида диыорид ванадила [оксихлорид ванадия(1У)| VOGl^ образуется также при взаимодействии концентрированной соляной кислоты с пятиокнсью ванадия
У2О54-6НС1=2у0С124-ЗН2О-рС1г.
Отщепление хлора ускоряется в присутствии мягких восстановителей, подобных спирту или сероводороду. Для получения чистого дпхлорида ванадила самым целесообразным
Пятая, группа периодической система
415
оказался способ нагревания трихлорида ванадила УОС13 с цинком до 400е в толстостенной стеклянной трубке, запаянной с одного конца:
2VOCl3+Zn=2VOCl24-ZnC12.
При этом дихлорид вападила получается в виде травянисто-зеленых блестящих гигроскопических табличен. Водные растворы окрашены в разные цвета, большей частью в синий или коричневый.
Дихлорпд ванадила образует двойные соли с хлоридом пиридина и е хлоридом хинолина состава VOC12-2RC1 (синий) и УОС]2-4ПС1 (зеленый) *.
Дихлориду вападила по поведению аналогичен дпбромид ванадила VOBr2, в безводном состоянии порошок цвета охры. Водный раствор его сипим. Тетрабромид ванадия не известен. Не известны также тетр анод ид и оксидииодйд ванадия,
Тетрафторид ванадия п фторованадаты(1У). Тетрафторид ваиадияЦУ) VF4 образуется в виде очень гигроскопичного желто-коричневого порошка (Ruff) при длительном кипячении тетрахлорида ванадия с безводной плавиковой кислотой. Нагретый в струе азота выше 300°, он разлагается на трифторид и пеятафторид ванадия. В вод-пбм растворе VF4 гидролизуется. В присутствии других фторидов из водных растворов кристаллизуются двойные соли [сксой&поровапаЭйты(1У)1, соответствующие главным образом типу M2T[VOF,(ОИ2)], например (NH4)2(VOF4(OH2)] и [Ni(OH2)e]IVOF4(OH2)b Калиевая соль К2 [VOF4] , однако, кристаллизуется без воды, аммонийная соль состава (NH4)3(VOF5] также является безводной. Сухим путем, по-видимому, получается также соединение К2[УГ6]. Но его существование еще не доказано.
Сульфат ванадила и оксосульфатованадаты(1¥). Безводный сульфат вападила VOSO4 существует в двух модификациях — растворимой и нерастворимой в воде. Последняя получается при пагревапии кислого сульфата ванадила1^ 2VOSO4- H2SO4 с концентрированной серпов кислотой до 260°. При этом образуется серо-зеленый, мелкокристаллический порошок, который при нагревании до 130° с небольшим количеством воды не pi‘ходит в синюю растворимую форму. Получены также различные гидраты сульфата ванадила, окрашенные в цвета от светло- до темно-синего. При длительном выдерживании водных растворов сульфата ванадила из них нслодствие^гпд-ролнза выпадает зеленая двуокись ванадия
V0SO4+H20=V02+H2S04.
Из растворов, содержащих избыток серной кислоты, при упаривании кристаллизуются (светло-синие) гидраты так называемого «кислого вана'дилсулъфатпаЪ 2VOSO4« H2SO4 **. От этого соединения также производятся некоторые двойные соли (с сульфатами щелочных металлов), папример K2SO4-2VOSO4 — светло-синие, микроскопические квадратные пластинки. Если рассматривать эти двойные соли как сульфато-соли типа М2 [У2О2(8О4)2], то «кислый в а на ди л су ль фат» следует рассматривать как ди оке о-трисульфатодивападиЙ(1У) кислоту H2(V2O2(SO4)3]. Другой ряд двойных соединений ванадил сульф ат а (известных также только в виде гидратов соединений со щелочными металлами) имеет состав mIS04‘VOSO4. Эти соединения, которые выделяются в виде темпо-синих кристалликов при прибавлении спирта к концентрированным растворам солей первого ряда, можно рассматривать как оксодисульфатовападаты(1У) MHvO(SO4)2b
Если двойные сульфаты первого типа (ди оксотрисульфатодиванадаты (IV)] упаривать с концентр up ов апл ой серией кислотой, то получаются дисульфатованадаты(Ш) MI[V(S04)2], в то время как эквивалентное количество Vlv переходит в Vv (окислитель но-восстановптелышн процесс). Окисления до Vv не происходит, если исходить из солей аммония, так как NIIJ-ион восстанавливает VV До V1V.
Кипящая концентрированная серная кислота частично переводит соединения VIV в соединения У^ с выделением SO2. При этом устанавливается равновесие между Viv и V . Поэтому при обработке У2О5 кипящей концентрированной серной кислотой происходит частичное восстановление ее до соли V^v с выделением кислорода (Auger, 1921; Sieverts, 1928)^
* Соли кристаллизуются с водой.
** Область существования пеятагидрата простирается примерно до 100’. Тригидрат кристаллизуется при 120°, дигидрат при 150°, полугидрат при 175е, При 190“ кристаллизуется безводное соединение^
8*
116
Глава 4
Вавадилсульфат иногда удобно применять для потенциометрического титрования слабых окислителей (С. del Fresno, 1933).
Сульфитовавадаты(1У). Кроме ок со дисульфитов а па дат он (IV), которые по своему’ тину соответствуют вышеназванным сульфато-солям, существует еще другой ряд сульфито8анадатов(1У) — m|[V3O5(SO3)2J. Известна также соответствующая им кислота H2[V305(S03)2.131/2TI;!0. То что эти соединения, окрашенные подобно большинству еоеданепий оксоваладияЦУ) в синий цвет, содержат ванадий в комплексном анионе, доказано путем наблюдения направления движения ванадия при электролизе растворов (Koppel, 1903).
Океалатовападаты(1¥) легко получаются взаимодействием пятиокиси ванадия со щавелевой кислотой. Известно два ряда такого рода комплексных оксалатов четырех валентного ванадия: mJ [VO(C,O4)2] и Ml [V2O2(C2O4)sj. Они либо синего, либо зеленовато-синего цвета. Щавелевая кислота из растворов этих солей на холоду не выпадает.
Соединение первого порядка между окисью ванадия(1у) и щавелевой кислотой не известно. Подобно щавелевой кислоте другие органические соединения, содержащие группу ОЦ (например, винная кислота, салициловая кислота, пирокатехин), также образуют с радикалом ванадила комплексные соединения такого же типа, как и первый ряд оксалатов. В этих соединениях пе только атомы водорода карбоксильных групп замещспы эквивалентным количеством ванадия, но и атомы водорода спиртовых или фенольных групп.
Род анон апа даты (IV). G роданпд-ионами ионы ванадия(1У) также не образуют соединений первого порядка, а лишь соединения более высокого порядка — охеотет-рараданована9аты(IV) m|(VO(SCN)4]. Тенденция к их образованию должна быть довольно значительна, так как при добавлении к раствору ванадиевой кислоты ионов SON” тотчас появляется характерное синее окрашивание роданованадатов(1У). Для получения этих солей целесообразно восстанавливать У11 до У17 добавлением сернистой кислоты,
Вйнадаты(1У). Как уже упоминалось, двуокись ванадия растворяется в щелочах. Из достаточно концентрированных теплых растворов выпадают соли, содержащие ванадий в анионе и названные вследствие этого eaHadama.nu(IV) (ранее назывались гипованадатами). Кристаллические ванадаты щелочных металлов соответствуют типу Mj[V4OBl-7H2O.
Сульфид ванадия(1У) и тповападаты(1У). При действии сероводорода на растворы ванадагО-в(1 V) щелочных металлов образуются растворимые тш>ванадаигы(1У) еще не известного состава. При добавлении кислоты к их растворам выпадает сначала бурый, а потом черный сульфид, состав которого также ещо точно не установлен.
4. Соединения ванадия (V)
Пятиокись ванадия (диванациппеитоксид), окись ванадия(У) V3OB, можно получить прокаливанием так называемого «метаванадата аммония» NH4VO3 (правильнее: тетраванадата аммония (NH4)4 [V4O12], ср. стр. 118)
2NH4V03=V205+2NH3-|-H20t
в платиновом тигле, или действием воды на трихлорид ванадила (гидролиз) 2УОС13+ЗН2О==У2О5-|-6ИС1.
Пятиокись ванадия представляет собой ядовитый без запаха и вкуса порошок, обладающий от оранжево-желтой до кирпично-коричневой окраской, плавящийся при 660® и при охлаждении затвердевающий в красно-желтые ромбические кристаллические иглы (уд. вес 3,318). Теплота кристаллизации так значительна, что при быстрой кристаллизации затвердевающее соединение под действием сильного сжатия вновь разогревается. Пятиокись ванадия мало растворима в воде (0,07 г в 100 г воды); вода ею окрашивается в светло-желтый цвет; влажная лакмусовая бумага в этом растворе краснеет. Б щелочах V2O5 легко растворяется с образованием ванадатов (см. виже). Пятиокись ванадия растворима
Пятая группа периодической системы
117
также в сильных кислотах; при этом она обнаруживает слабый ио сравнению с кислотным основной характер. При растворении пятиокиси ванадия в соляной кислоте выделяется хлор, причем пятивалентный ванадий частично восстанавливается до четырехвалентного. Если удалять образующийся хлор, процесс протекает до конца.
Если к раствору V3OS в разбавленной серной кислоте (или к раствору ванадатов щелочных металлов, подкисленному серной кислотой) прибавить мышьяковистой кислоты и несколько капель очень разбавленного раствора OsO4 (в качестве катализатора), то сначала реакция не идет. При добавлении раствора хлората калия раствор мгновенно синеет, что свидетельствует о восстановлении V* до Vlv. Этот опыт, описанный
Рис. 19. Плоская сетка, образованная искаженными тетраэдрами У04 в пятиокиси ванадия. Предполагается, что точки пересечения ребер тетраэдров, показанных топкими линиями, лежат выше, а пунктирными— ниже плоскости рисунка. Длины ребер тетраэдров составляют (А): а— 3,13, Ь—3,06, с -- 3,&0, d = d'= 2,72, е = 2,36. Среднее расстояние V -t—> О равно 1,72 А.
Глё (Gleu, 1933), очень наглядно демонстрирует явление, названное Оствальдом «обращенный мир», наблюдаемое чаще в случае индуцированных реакций, когда, восстановление вызывается окислителем (и часто на оборот—о в ислен ие восстановителем).
Пятиокись ванадия легко переходит в коллоидный раствор, она пептизируется, например, азотной или соляной кислотой. Темный кроваво-красный коллоидный раствор в высшей степени устойчив, его можно выпаривать, после чего остается красная бархатистая масса, которая при добавлении воды вновь переходит в кроваво-красный раствор. Частицы в таких гидрозолях пятиокиси ванадия отличаются очень небольшой величиной. Их размеры лежат на границе ультра микроскопической видимости. При старепии гидрозоля пятиокиси вападия можно иногда наблюдать постепенное увеличение ультрамикроскопилеских частичек до микроскопических палочек. Если ориентировать палочки движением жидкости (например, при помешивании золя стеклянной палочкой), золь обнаруживает двойное лучепреломление. При рентгеновском исследовании палочки показывают такую же интерференцию, как и обычная твердая мятиокйеь ванадия, только интерференционные линии расширены вследствие малых размеров кристалликов.
Склонность образовывать микрокристаллы в виде палочек объясняется структурой решетки пятиокиси ванадия. В ее решетке каждый атом V окружен в виде искаженного тетраэдра четырьмя атомами кислорода, три из которых одновременно принадлежат соседним тетраэдрам. Таким образом образуются (зубчатые) дени VO4 неогра-ничеппой длины, которые связаны между собой мостинами из кислородных атомов в сетчатую плоскость (см. рис. 19); одцако для этой цели от каждого У04-тетраэдра используется только ио одному атому О, а для образования цепей — по два. На рис. 19, па котором схематически показана связь у04-тетраэдров, одна из цепей заштрихована,
118
Глава 4
Каждый из тех атомов кислорода, которые образуют вершины тетраэдров, лежащих сверху и снизу от плоскости рисунка, принадлежит только одному из тетраэдров; связь между отдельными сетчатыми плоскостями осуществляется, следовательно, не за спет главных валентностей. Вследствие этого кристаллы пятиокиси ванадия отличпо расщепляются в направлении, параллельном сетчатым плоскостям; опи обладают также отчетливой расщепляем остью в перпендикулярном направлении, а именно вдоль цепей, и в связи с особо плотным расположением атомов в направлении цепей становится понятным преимущественный рост кристаллов в этом направлении.
Ванадиевые кислоты и ванадаты(У). Раствор пятиокиси ванадия в воде реагирует как кислота. Существующая в растворе ванадиевая кислота имеет формулу H6[V10O28], следовательно, соответствует составу 5V2Oa-3H2O. Найцепные Хюттигом (1930) гидраты 2V2O5-H2O и V20j-Н2О существуют только в твердом состоянии. Соли ванадиевой кислоты, ванадаты/1 ), очень разнообразны по составу. Их целесообразно называть в соответствии с соотношением окйслов (М*О : V205). Из слабощелочных растворов кристаллизуются ванадаты с соотношением 4:1, 3:1 (называемые также «ортованадатами») и 2 : 1 («пированадаты»). Из нейтральных растворов получают «мставанадаты» с соотношением 1 : 1. Все они бесцветны. Из кислых растворов кристаллизуются окрашенные (от оранжево-желтых до оранжево-красных) ванадаты; соотношение М.,0 : V2O5 в них может изменяться от 0,8 : 1 до 0,25 : 1.
В водпых растворах, как указывали Яндер и Яр (Tander, Jahr, 1933), Хацедь (Hazel, 1953), Россоти (Rossotti, 1956); Шварцепбах (Schwarzenbach, 1958) и Яр (1959—
I960), существуют следующие равновесия;
2[VO4]’" + 2H- (V2O7j""4-H2O (pH 12—10,6), (1)
2[V2O7]'"'4-41Г [V4O12]'"' + 2I-r3O (pH 9-8,9), (2)
5[V4Oi2]"" + 8H- 2IV10O28]""" + 4H2O (pH 7,0—6,8), (3)
[VyjO^]'""' 4- 6H‘ НДУ1ВО,Й) 5V2O5-|-3H2O (pH 1,6), (4)
ВДУ1о028] + 10П- 10[V02]-+8H20 (pH<l). (5)
Более простые ванадм-ионы склонны, таким образом, переходить, отщепляя воду, в более сложные ионы. Такого рода кислоты, которые образуются из простых кислот (МОиокпслот) путем конденсации, т. е. связыванием двух или болео молекул, сопровождающимся отщеплением воды, называются моли кислотами (ср. стр. 203). Следовательно, в рассматриваемом случае речь идет о поливанадиевых кислотах или лоливанадат-ионах, различающихся числом входящих в них атомов ванадия: например, Э ив а над ат-, т етр «ванадат- и д ек а ва над ат-ионы. Вес этих ионов был установлен криоскопическим методом Яром и Шёппом (Jahr, Schoepp, 1959—1960) (ср. т. I, стр. 336).
Наряду с указанными выше уравнениями приведены значения pH, прп которых в 0,1 М растворах происходят превращения. В промежуточных областях существуют практически только вещества, указанные в левой части уравнений. В промежуточных областях частично имеют место, конечно, еще и такие равновесия диссоциации, которые пе сопровождаются копдепсацией (например, [ViqO3S] 4- Н' [HVJ0O2S]*). Ввиду простоты их здесь гге принимают во внимание.
При добавлении избытка щелочи поливанад ат-ионы в соответствии с приведенными выше равновесиями превращаю теп в коночном итоге в мопованадат-иопы. Переход тетраваггадат-иогпж через ди- в ненова на дат-иопы протекает мгновенно; переход оранжевых дека- в бесцветные тетравападат-ионы происходит медленно по реакции первого порядка. Превращение тетравападат-иояов в д екав ап адат-п оны при подкислении [ур. (3)1 также протекает медленно: спачала образуются темно-красные ионы поливавадиевой кислоты, неустойчивые, но стабилизируемые фосфорной кислотой в «пурпурные фосфовапалат-ионы» (.Tander, Jahr, 1933), которые в противоположвость декаванадисвой кислою мгновенно обесцвечиваются ОН"-ионами. Дек а ванадат-и оны устойчивы в достаточно широком интервале кислотности. Однако с увеличением содержания кислоты растворы становятся неустойчивыми, и постепенно начинается
Пятая группа периодической системы 119
выделение пятиокиси ванадия*. Этот процесс происходит тем медленнее, чем меньше концентрация ванадия; 0,001 М раствор декаванадней ой кислоты устойчив в течение нескольких месяцев. Одновременно в возрастающей степени происходит образование положительных ионов 1VO2]" (Ducret, 1951), т. е. ванадиевая кислота, обладающая амфотерным характером, начинает диссоциировать в данном случае как основание.
Кристаллизующиеся ванадаты являются солями названных ванадиевых кислот. Соли оранжевого цвета — ото декаванадаты; некоторые из них являются декаванадатами оксовападия(У): (0,6 : 1)-ванадат = Mj(VjoOggl, (0,5 : 1)-данадат=М^[НУщОаз], (0,4 : 1)-ванадат = MJ[ГГ2УмО281, (0,33 : 1)~ванадат = М}[VO2]2[У10О28], (0,25)-вана-дат = М J (V О г ] 3 (И Vj о О Е8 ]. О ра г гж евыо (0,8 ; 1) - в анадаты двухвалентных к атиопов про-изводятся, в соответствии с данными Яра (1960), тоже от шестиосп овцой декаванадио-вой кислоты. Они образуют, вероятно, оболочку шара, в которую заключен гидратированный двухвалентный катион, способным отщеплять два протона от своей акво-г-руппы. Таким образом, например, могут образоваться такие соединения, как ВаДУ,0О28. Ва(ОН)2(И2О )4] • нН2о.
Имеющиеся в водпых растворах ионы [У4О12]"" подобно тетраметафосфат-иопам (ср. т. I, стр. 681) имеют кольцеобразное строение. В равновесии с ними в растворах существуют в небольших количествах ионы кислоты Н6 [У4О13[, имеющей цепное строение. Примечательна очень небольшая скорость растворения и кристаллизации «мотаванадатов». Рентгенографически исследованные КУОЭ (Milligan, Vernon, 1952) и КУО.->- НЙО (Christ, Clark, Evans, 1954) содержат бесконечные цепи УО4 или УО5, следовательно, опи производятся не непосредственно от существующих в водном растворе тетраванадат-ионов. Кристаллизующиеся из растворов (2 : 1)- или «пиро»-ванадаты частично являются диваиадатами MIIVjOt], частично кислыми мопованада-тами, например Ag2[HVOJ (Britton, Robinson, 1930). (3:1)- или «орто»ванадаты являются солями монованадисвой кислоты. В случае (4 ; 1)-ванадатов, ио-видимому, речь идет об основных солях MjfOHjlVOJ.
Монованадаты можно получить как сплавлением окислов, так и из водного раствора. Вольшпнстви по ли ванадатов, как показал Лидер, нетрудно получить из водного раствора, если учитывать области существования отдельных поливанадат-иопов и выждать установления равновесия. Из сметав ан ад атов» (тетравападаты) очень легко можно получить чистый, довольно мало растворимым на холоду в противо-лоложтюсть ванадатам щелочных металлов метаванадат аммония NTI4VO3 или IКН4]4(V4OJ2]. Взаимодействием ванадатов щелочных металлов с солями щелочноземельных и тяжелых металлов можно получить соответствующие им ванадаты.
Вападат-ионы весьма склонны к конденсации между собой в более высокомолекулярные ионы, а также к образованию комплексов с ионами некоторых других кислот. Так, с кислотами кремния, олова, фосфора, мышьяка, молибдена и вольфрама образуются комплексные кислоты, которые называют «гетерополикислотами» (подробнее см. стр. 200); при этом роль центрального атома могут играть либо названные элементы, либо ванадий. Солями гетерополикнслот являются, например, указанные выше «пурпуребфосфованадаты».
Ванадинит; структура соединений апатитовой группы. Другой тип комплексных ванадатов представляет ванадинит (ванадиевосвикцовая руда) ЗРЬ3(УО4)2- РЬС12. Эта встречающаяся главным образом в Мексике (Зимднан), Аргентине и Аризопо, а также в Шотлапдаи и Каринтии ванадиевая руда, кристаллизующаяся в виде гексагонально-гомиедрическнх (неполногранпых) столбиков, от желто-коричнев от о до красно-коричневого цвета, изоморфна апатиту, пироморфиту, миметезиту и другим соединениям апатитовой группы.
Соединения апатитовой группы имеют общий состав MIJX2 или
У1Р(ВО4)3Х; М11 — либо Са. либо Pb, R — Р, Аз, либо V н X — С1 либо F. Элементы, соответствующие в общей формуле одному и тому же символу, могут непрерывно замещать один другой. (Вместо CI или Г частично могут входить группа ОН или в эквивалентных количествах О, СО3 или SO4; Са частично замещается Sr.) Соединения апатитовой группы кристаллизуются в гексагональной сингопни, пирамидально-гемиедрически. Опи по только встречаются в виде природных минералов, но могут быть получены и искусственно в кристаллическом состоянии. То, что в данном случае
* Является ли устойчивой У2Оь или V2OS- Н2О в качестве донной фазы в водном растворе, еще не установлено. Некоторые наблюдения свидетельствуют о том, что первоначально выделяется обычно УаО3 и только потом она присоединяет воду, переходя в V2Q5-H2O = [НУО3]«.
120
Глава 4
речь идет пе о двойных соединениях фосфатов, арсенатов или ванадатов с хлоридами^ видно уже из того, что водой из пих пе извлекаются хлориды. Еще Вернер назвал эти соединения координационными соединениями. Он рассматривал их как соединения внедрения типа !CaR^JX2, которые существуют, например, в виде гексагидрата хлорида кальция [Са(ОН3)п]С1. Вернер произвел апатит от этого соединения путем замены двух молекул воды одной молекулой Са3(РО4)3, получив формулу {Са[Са3(РО4)2]3}Х2. Рентгеноструктурным анализом (Naray-Szabd, 1930, Schiebold, 1931) было показано, что в случае соединений апатитовой группы действительно речь идет о координационных соединениях, а именно о типе соединений внедрения. Но предположения Вернера следует поправите, в том отношении, что в апатите в качестве центрального атома комплекса или соответствующей ему в кристалле структурной группы может быть не атом Са, а одповалетный анион X. С этим анионом в первой сфере связаны три группы СаРО4“, а во второй — 12 ионов Са2+ (в двух одинаковых правильных шестиугольниках!, от которых па одинаковом расстоянии находится каждая из шести групп [X(CaPO4)3J4-, так что стехиометрпчески па каждую [Х(СаРО4)3Н--группу приходится -д-==2Са2+-иона, Структуру апатита, таким образом, можно изобразить следующей формулой: '
СаРО4
£
CflafF (С sfcPOfl)® J
или
_СаРО4 СаРО4.
В соответствие с этим общая структурная формула соединений алатитоной группы М2*[Х(МпКО4)я], например для ванадинита Pb2[Cl(PbVO4)3], В некоторых видах апатита анион X- замещается группой, состоящей из нескольких ионов, например, в карбонашоапазпите группой [СО3]а“-Так как эта группа двухвалентна, вторая свободная валентность является центром координации нейтральных молекул (Н2О). В а идро кайл апатите СаЕ(0Ы)(Р04)3 = Ca2((OH)(CaPO4)s] место аниона X- занимает группа ОН-. Структура так называемого оксиапатита Са10О(РО4)6 еще по выяснена. Исследования Трёмеля (Tromel, 1932) вообще ставят под вопрос его существование.
Перокеосоединенйя ванадия(У). При добавлении перекиси водорода к растворам ванадатов щелочных металлов в щелочных шш слабокислых растворах образуются, по данным Яра (1941), желтые дипероксоортозанадат-ношл [V03(O2)2]", а в сильнокислых растворах красно-коричневые катионы пероксованадия(У) [V(O2j]"*. Дипероксо ортов а на дат-иопы и ионы пероксовападия(¥) находятся в равновесий, зависящем от концентрации ионов водорода и перекиси водорода:
[VO2(O2)2]'" + 6H- [V(Oa)]-4-H2O2+2H2O.
Гидрат пятиокиси вападия растворяется в водном растворе перекиси водорода с образованием свободной трехосновной дипероксоортованадиевой кислоты II3 [VO2(O2)2], которая подобно всем пер ок со со единениям ванадия легко разлагается с выделением кислорода. Из солей этой кислоты в настоящее время известны лишь первичная калиевая соль КН3 [VOg(O2)oj-Н,0 (желтые ромбические листочки), и вторичная соль аммония (NH4)2Hiv02(02)2]-ац (желтые иглы), полученные Яром. Приводимые в более ранних работах соли предполагаемой пероксо.истапападиевой кислоты H[VO2(O2)I с легкими и тяжёлыми металлами исследованиями Яра не подтвердились. При температурах около 0я в сильнощелочных растворах, содержащих большое количество перекиси водорода, желтые ди пер оксо ортованадат-ионы постепенно превращаются в сине-сриолетовые- тетрапероксоортоеанадат-моилл (V(O2)4]m. Тетрапероксоорто-ванадаты Mg [V(O,)4J.aqfM1 = Li, Na, NH4, К) кристаллизуются в виде сине-фиолетовых игл, устойчивых том меньше, чем больше радиус катиона.
Пентаеульфцд ванадия и тио папа даты. Имеются сведения, что пентасулъфид ванадия V2SB получается при нагревании окиси ванадия(1П) V2O3 с серой без доступа воздуха до 400". После э к ст р акции продуктов реакций сероуглеродом остается черный порошок, потвидимому, пептасульфида. Одиако, по данным Клемма (Klemm, 1936), существование этого соединения весьма сомнительно. Во всяком случае, оно не было-получено нагреванием V2Ss с избытком серы; более того, при этом образуется указанный на стр. 114 полйсульфид. Продукты, принимаемые за V2Ss, при нагревании на
Пятая группа периодической системы 121
воздухе сгорают до пятиокиси ванадия. Нагретые в инертной атмосфере, они отщепляют серу, переходя в полуторный сульфид. В растворах сульфидов щелочных металлов' или в щелочах эти продукты растворяются с образованием тиосолей или оксотиосолей из которых известны два ряда. Один являются производными от моновападатов, другие — от диванадатов путем замены атомов кислорода серой. В качестве примера можно привести тетратиованадат аммония (NH4)3(VS4], получающийся при унари-ванн и растворов ванадатов, содержащих сульфид аммония, л виде черно-фиолетовых ромбических призм. Как все тио- и оксотиеванадаты щелочных металлов, он легко' растворяется в воде. Тиовападат получают также сплавлением.
Галогениды вападпЩУ) и гадогеновападаты(У). Из бескислородных галогенидов пока известен только пептафторид VFS (твердая белая масса, легко растворимая в воде-н спирте, уд. вес 2.ГП, т. пл. 111,2°, образующаяся при термическом распаде тетрафто-рпда ванадия). Легче образуются кислородсодержащие галогениды VOF3, VOC13; и VOBra, последний из которых даже при умеренном нагревании (медленно уже при обычной температуре) распадается по уравнению УОБг3 = VOBr3 -j- Ч2Ег2. Соединения пятивалентного ванадия с иодом вообще пе получены.
Окситри хлор ид ванадия УОС13 образуется, например, при пропускании сухого хлористого водорода над слабо нагретой пятйокисью ванадия в присутствии пятиокиси фосфора (которая является водопоглощающим средством); V20b -ф- 6НС1 = 2VOC13 -ф--ф ЗН2О. Окситриххорпд ванадия представляет собой желтую подвижную, ио довольно тяжелую жадность с температурой кипения 127° и плотностью пара, соответствующей простой молекулярной формуле. Водой опа гидролитически расщепляется. Таким образом, процесс, протекающий по приведенному выше уравнению, является обратимым. С хлоридом пиридиния окситрихлорид ванадия Образует двойную соль |C5ri&NH] [VOC1J (оксотетр ах лоров ana дат пиридиния). Эту соль следует, получать из спиртовых растворов, так как она гидролизуется.
УЕ3 с фторидами щелочных металлов образует генсафторованадеты M^fVFgj. Они были получены Эмелеусом (Emeleus, 1949) при действии BFa на VC13 и галогениды щелочных металлов. Известно большое число двойных солей пятивалентного ванадия,, производных от оксифторидов. Известны многие представители следующих типов: 3MIF-2VOF3, Sb^F^VOaF и 2MIF-VO2F. Подобно упомянутому выше двойному хлориду пиридиния эти двойные соли молено рассматривать как гало?енрванадатыГ т. е. ванадаты, в которых атомы кислорода частично замещены атомами галогенов. Заместителями, помимо галогенов, могут быть также остатки кислородных кислот,, таких, как серная, иодная, щавелевая. Например, известен океооксалатованадат(У)Т которому приписывают формулу [У02(С204)2],
Аналитические сведения. Ванадий осаждается либо из кислого раствора сероводородом, либо из аммиачных растворов сернистым аммонием. Однако растворы ванадатов щелочных металлов сернистым аммонием окрашиваются в вишнево-красный цвет вследствие образования тиованадатов. При подкислении раствора большая часть ванадия выпадает в виде коричневого пентасульфпда (или полнеульфида? Ср стр. 120). Сероводород окрашивает кислые растворы ванадиевой кислоты в синий рвет вследствие восстановления до солей ванадила(1У). Синее окрашивание получается также под влиянием многих других восстановителей; в некоторых случаях (например, при действии цинка и серной кислоты) синий цврт переходит в зеленый, а затем в фиолетовым, по мере восстановления до соединений вападня(Ш) и далее ванадия(П). Достаточно чувствительна реакция с перекисью водорода. Она окрашивает кислые растворы ванадиевой кислоты в интенсивпый красно-коричневый цвет. Окрашенное соединения не извлекается эфиром, что позволяет открывать ванадий в присутствии хрома (ср. стр. 178). Гораздо чувствительнее, однако, предложенная: Эфраимом [Ephra i m, Helv. chim; Acta, 14, 1266 (1931)] реакция, основанная на восстановлении ионов Fo“* ионами [VO]'* (в щелочном растворе):
Fa— -HVOJ” 4-6ОН’ =Fe" + [VOJ'" +ЗЦ2О.
Если образующиеся при этом ионы Fe" фиксировать при помощи ди метил гл покоима (интенсивное вишнево-красное окрашивание), то этим способом можно открыть до 2,5у ванадия.
Весовым аналитическим методом можно определять ванадий, осаждая его в виде ванадата одновалентной ртути с последующим прокаливанием До пятиокиси ванадия V2O5. Но более удобным является объемный метод: вначале сернистой кислотой восстанавливают пробу до соли ванаднла(ГУ), а затем (после кипячения для удаления избыт
i22
Глава 4
ка ЗО3) титруют раствором перманганата (л сернокислом растворе, нагретом до кипения):
Ю[УОГЧ-2[М11О41' + 7НгО=5УгО5+2Мп“ + 14Н-.
Восстановление Vv до Vй попами Ее" в кислом растворе (процесс, обратный указанному выше в щелочной среде) также пригодно для определения ванадия объемным методом.
НИОБИЙ (NIOBIUM) Nb
Распространение в природе. Минералы ниобия встречаются во многих местах, но редко в значительных количествах. Важнейший минерал ниобия нмогш?», или колумбит, находится главным образом в различных местах США, а также в Гренландии и в Финляндии близ Таммела. В Германии ниобит встречается около Бодеимейса (Нижняя Бавария). Главной составной частью его является няобат железа * Ее(МЪО3)3, который, однако, всегда содержит в более или менее заметных количествах танталит железа Fe(TaO3)2, изоморфный ему.. Если содержание тантала превышает количество ниобия, то минерал называется танталитом.
Ниобий часто встречается вместе с редкими землями. Целый ряд минералов с редкими землями представляет собой ни об а ты или изоморфные смеси ниобатов и та пта лотов. Изоморфная смесь ниобатов и танталатов редких металлов — эвксенит. Близок к нему по составу поликраз. Ниобат кальция со значительным содержанием фторида щелочного металла называется пирохлором ЭТаР-СаО-1\1Ъ2О5 или NaCaNb2O6r.
Исторические сведения. В 1801 г. английский химик Хэтчетг (Hatchett) в полученном из Северной Америки минерале открыл окисел неизвестного металла. Новый элемент он назвал в честь родины минерала колумбием, а сам минерал колумбитом. Позднее этот элемент принимали за тантал, открытым в 1802 г. Экебергом, особенно на основании исследований Волластона и Берцелиуса, Однако, как теперь стало известно, эти исследователи работали но с чистой танталовой кислотой, а со смесью ее с ниобиевой кислотой. Правда, еще Волластон отмечал, что танталиты различного происхождения, так же как и получаемые из них окислы, значительно различаются по удельному весу. Но только Розе (Rose, 1844) пришел к выводу, что это зависит от примеси очень похожего, но отличающегося от него по весу элемента. Он назвал этот элемент ниобием по имени дочери Тантала Ниобеи. Так как Полученная Хатчеттом из колумбита окись ниобия была загрязнена окисью тантала, как и позднее полученная из танталита окись тантала — окисью ппобия, Розе был первым, получившим чистую окись ниобия. Старое название колумбий иногда еще употребляют в английской и американской литературе.
Получение. Для разложения минералов ниобия лучше всего использовать сплавление их с гидросулъфатом калия. При этом окиси ниобии и тантала остаются в виде нерастворимо г о в воде остатка, который, как подробнее описано в главе о тантале, растворяют в плавиковой кислоте и разделяют дробной кристаллизацией двойных фторидов калия. Разделение затрудняется присутствием некоторого количества титана, который в виде фторотитанита остается вместе с ниобием в растворе. Для отделения титана от ниобия (и тантала) рекомендуется отгонять его в виде Т1СЦ, который кипит примерно иа 100° ниже, чем NbCIs и ТаСЦ.
Изящный метод разложения предложен Муассапом (Moissan). Он заключается в том, что измельченный в порошок, смешанный с сахарным углем минерал нагревают в течение нескольких минут в электропечи сильным током (1000 в). В результате окиси ниобия и тантала переходят в растворимые в плавиковой кислоте карбиды, а большая часть примесей других вещие,тв улетучивается.
Техническое получение металлического ниобия осуществляется в настоящее время главным образом обменным разложением карбида ниобия с окисью ниобия при температуре около 2000“ в высоком вакууме.
Старый способ получения металла был предложен Волтоном (W. v. Bolton). При этом лучше всего исходить из чистой пятиокией ниобия Nb2O5. Последнюю либо
* Железо частично вамещеио марганцем (ср. стр. 125).
Пятая группа периодической системы.
123
восстанавливают алюмотермическим способом и образующийся сплав ниобия с алюминием освобождают от алюминия (он при этом испаряется) длительным плавлением в электрической вакуумной печи, либо переводят в двуокись, для чего добавляют парафин (из этой пластичной массы, содержащей Nb2O5, делают нити и прокаливают их с угольным порошком прп температуре белого каления). Двуокись ниобия в отличие от пятиокиси проводит электрический ток и при нагревании сильным переменным током довольно быстро отщепляет весь кислород.
Попытка выделить ниобий электролитическим путем пе привела к удовлетворительным результатам.
Свойства. Ниобий — металл средней твердости, серого цвета, а па полированной поверхности блестяще-белый, немного менее ковкий и значительно ниже плавящийся, чем тантал (ср. стр. 105). Температура кипения его также значительно ниже, чем тантала, и вследствие этого металлический ниобий в отличие от тантала сильно распыляется при накаливании в вакууме.
Ниобий устойчив на воздухе и нагретый в компактном состоянии тускнеет только па поверхности. Одпако в виде опилок при нагревании выше 900° он полностью сгорает до пятиокиси Nb2O5. В токе хлора при красном калении он энергично сгорает с обра- ‘ зовапием пентахлорида NbCls. При нагревании ниобий реагирует также с серой и селеном. С большинством металлов он сплавляется с трудом. С железом, о диак о, Nb образует в разных отношениях твердые растворы. С алюминием образуется соединение NbAl3.
Плавиковая кислота медленно разъедает ниобий. Все другие кислоты, в том числе царская волка, на него нс действуют. Не растворяется он также и в щелочах. Напротив, растворяется в расплавленных гидроокисях щелочных металлов, а в порошкообразном состоянии в расплавленной селитре даже воспламеняется.
Неспособность растворяться в кислотах-окислит елях объясняется тем, что он чрезвычайно сильно пассивируется в растворах окислителей. В соприкосновении с разбавленной серной кислотой он проводит электрический ток только в том случае, если является катодом. На этом основан сконструированный Сименсом и Гальске (Siemens, Halske) выпрямитель переменного тока. Он состоит из двух листов — платинового и ниобиевого — Погруженных в сосуд с 0,1 н. серной кислотой. Через этот прибор проходит только та фаза переменного тока, для которой платина является анодом, а ниобий — Катодом.
СОЕДИНЕНИЯ НИОБИЯ
Подавляющее большинство соединений ниобия являются производными пятивалентного ниобия, несмотря на то что ои может находиться и в более низких степенях окисления, а именно четыре, три и два.
Ниобий четырехвалентен в двуокиси NbO2 и в тетрахлориде NbCl4. Двуокись образуется при нагревании пятиокиси ниобия Nb2Os в токе водорода до температуры около 1000°. NhO, представляет собой черный с голубоватым отливом порошок, нерастворимый в воде и кислотах, который при температуре красного каления опять сгорает на воздухе до пятиокиси.
При дальнейшей потере кислорода двуокись ниобия может перейти в моноокись NbO, которая образует решетку типа каменной соли. Растворимость кислорода в металлическом ниобии составляет около 1,7 ат. %. Тетр ах лорид ниобия в виде коричневых пгл получается при восстановлении NbCI5 ниобием, алюминием, железом или водородом. Он легко разлагается по уравнению
2NbCl4TE^t NbCl3TB+NbCl5ras—28,3 ккал.
По данным Шефера (Schafer, 1952), давление пара NbCk над смесью твердых NbCj4 и NbCl3 при 200° равно 1,2, при 300° — 230 и при 320°—531 .ч.и рт ст. При температуре около 420° и давлении 22 ат в равновесии находятся четыре фазы NbCl3 тв, ЛЬС14ТВ, NbCl3жидк и NbCls газ*
124
Глава 4
Производный от трехвалентиого ниобия трихлорид ниобия NbCl3 представляет собой темные с металлическим блеском листочки. Он получается восстановлением пентахлорида NbCls алюминием при нагревании или при термическом разложении тетрахлорида ниобия NbCJ4.
В растворе соединения ниобия(Ш) получают при обработке соединений нио-бия(У) цинком в кислом растворе. При соблюдении определенных условий восстановление до трехпалентного состояния протекает полностью, что можно проверить обратным титр о метрическим окислением перманганатом до соединений Nb(V), Ионы Nb"*, содержащиеся в восстановленных растворах, окрашены в коричневый цвет1 и весьма склонны вновь к окислению.
Из соединений двухвалентного ниобия, кроме уже упомянутой моноокиси NBO, Шефером получен дихлорид NbCi2.
Соединения ниобия(V)
Пятиокись ниобия Nb2O5 в виде белого нерастворимого в воде порошка (уд. вес 4,46, т. пл. 1460°) получают обезвоживанием ее гидрата — ниобиевой кислоты — или прокаливанием на воздухе солей ниобия; сульфида, нитрида или карбида. Nb2O5 можно перевести в раствор сплавлением как с гидросульфатами, так и с карбонатами или гидроокисями щелочных металлов, что свидетельствует о ее амфотерности, о диак о кислотный характер все же у нее преобладает.
Nbo05 в отличие от Та3О5, существующей в двух модификациях, триморфна (Brauer 1941). Обычно она получается в низкотемпературной форме (изотопной а-Та2Р$) даже при непродолжительном прокаливании. Nb2O5 й Та2О5 образуют твердые растворы; они получаются при непосредственном совместном осаждении из растворов обоих окислов и при последующем обезвоживании геля прокаливанием. При достаточно высоком содержании Nb2O5 (более 76 вес.%) сильно прокаленные твердые растворы имеют структуру решетки Nb2O5, устойчивой выше 850°, н противном случае — низкотемпературной модификации Nb3Os или а-Та2О5. При прокаливании в токе водорода Nb2O6 теряет некоторое количество кислорода, не изменяя при этом решетку. И только при содержании кислорода ниже, чем соответствует формуле NbO2,да, появляется новая фаза, а именно упомянутая выше двуокись NbO2.
Ниобаты и ниобиевая кислота. При сплавлении Nb2O5 с карбонатом натрия пятиокись ниобия вытесняет двуокись углерода и становится на ее место в качестве кислотообразующего окисла. Если на 1 ч. Nb2O5 взять 4 ч. Na2CO3, то выделяется такое количество СО2, которое соответствует образованию ортониобата
Nb205+3Na3C03-2Na3Nb04+3C02.
После выщелачивания сплава водой вместо ожидаемого остатка ортонпо-бата натрия Na3NbO4 получается метаниобат натрия NaNbO3. Он представляет собой бесцветный трудно растворимый в холодной воде мелкокристаллический порошок.
NaNbO3 кристаллизуется по типу перовскита (а = 3,89 А), так же как и KNbO3 (а ~ 4,01 А), в то время как LiNbO3 — по типу ильменита (см. стр. 79). В обоих случаях координационное число ниобия по отношению к кислороду равно 6. Это относится и к пиобатам двухвалентных металлов, Построенных по типу ниобита, например Fe(NbO3)3 и Mn(NbO3)2. Кристаллизующийся в ромбической форме Fe(NhO3)2 состоит из (искаженных) октаэдров FeOe и NbO6, которые в двух измерениях имеют общие углы, а в третьем общие ребра, таким образом каждый атом кислорода одновременно принадлежит трем октаздрам. При атом к октаэдрическим слоям, состоящим из цепей, образованных FeOB,
о О о
/ОХ zO4 : z(\ ; ..-О-..
?Fe:' >Fe<4 >Fe< -
О' j ’О'' ЧО
ООО
Пятая группа, периодической системы
125
с обеих сторон вдоль кристаллографической оси а присоединяются такие же октаэдрические слои из NbOB. Большинство известных в настоящее время ниобатов тяжелых металлов получают из расплавов: так, Mn(NbO3)2, изоморфный Fe(NbO3)2, образуется при нагревании смеси Nh2O5 и MnF2 с RC1 (в качество флюса) до температуры белого каления.
Встречающийся в природе ниобит, или колумбит (черные ромбические кристаллы; структуру см. выше), — изоморфная смесь ниобатов двухвалентного железа и марганца (и соответственно танталитов). Такой же состав имеет (редко встречающийся) тетрагональный моссит.
Из соединений пятиокиси ниобия с основными окислами в других соотношениях следует упомянуть диниобаты. MjbIb2O7. Однако до сих пор но ясно, являются лн получаемые из растворов кристаллогидраты пентаниобатами MjNb6Oj0‘лгН2О иди гек-санйобатами М^МЬ6О19-«Н20, в которых часть воды может замещаться МХОН, как у кристаллогидратов танталатов (ср. стр. 130). Следует отметить, что ниобаты аммония не существуют.
При смешивании растворов ниобатов с серной кислотой выпадает белый, студенистый осадок гидрата пятиокиси ниобия с переменным содержанием воды. Он образуется также при гидролизе солеобразных соединений пятивалентного ниобия, например пентахлорида ниобия или сплава пятиокиси ниобия с гидросульфатом; правда, последний гидролизуется очень медленно и лишь при кипячении — полностью. Гидрат пятиокиси ниобия, называемый обычно ниобиевой кислотой, растворяется как в щелочах, так и в сильных кислотах и в этом отношении подобен оловянной кислоте. Так же как оловянная кислота, он очень легко образует коллоидный раствор, и особенно сильным пептизирующим средством в обоих случаях является соляная кислота.
Существует ли в данном случае определенный стехиометрический гидрат пятиокиси: ниобия и, следовательно, свобод пая ниобиевая кислота как химическое соединение, трудно утверждать, так как содержащие воду препараты Nb2O3 всегда аморфны и при обезвоживании не обнаруживают ярко выраженных ступеней *. Следует отметить очень прочную связь воды в гелях МЬ2О5. Вода содержится в них еще даже около 500°. Некоторые наблюдения (Jander, 1928; Huttig, 1930) свидетельствуют о том, что при разложении растворов ниобатов получается аморфная пентаниобиееая кислота H7Nb5Ole. Но, по-видимому, она не способна кристаллизоваться и образуется в виде типично коллоидного вещества, механически связывающего избыточную воду настолько прочно, что ее невозможно удалить полностью, не удалив при этом часть химически связанной воды.
Ацидониобаты. Заменой в ниобатах атомов кислорода кислотными остатками получают комплексные ниобаты — ацидониобаты. Например, с кислотными остатками щавелевой кислоты — оксалатониобаты — бесцветные кристаллические соединения, большинство из которых соответствует формуле ЗМ^О-Nb2O5-6С2О3, и поэтому их следует рассматривать как оксотр ио кса лат ониобаты М3 [ONb(C2O4)3J. Подобно щавелевой кислоте в кислотный остаток ниобиевой кислоты могут входить, образуя комплексные соединения, кислотные остатки органических оксикислот, а также остатки титановой, фосфорной, мышьяковой, хромовой и вольфрамовой кислот. И наоборот, кислотный остаток ниобиевой кислоты моЖет входить в кислотные остатки перечисленных кислот. Образование таких комплексов наблюдается часто при взаимодействии названных кислот.
К классу ацидониобатов принадлежат также галоеенониобаты, которые будут описаны в дальнейшем; они производятся от ниобатов при замене атомов кислорода галогенами.
Если в ниобатах один или несколько атомов кислорода заменить эквивалентным количествам остатков перекиси водорода —О—О—, то образуются пер оксо ниобаты.
* Автор имеет в виду отсутствие горизонтальных участков па изотермах или изобарах обезвоживания. — Прим. ред.
126
Глаеа 4
Пероксониобаты и псроксопиобнсвая кислота. При действии перекиси водорода на ниобат калия в присутствии избытка щелочи образуется бесцветная или бледно-желтая соль, тетрапероксониобат калия
’02
_о2
о2п Nb
O3J
K3NbOJ = Kj
осаждающаяся из водного раствора спиртом и содержащая ниобий и активный Кислород в отношении 1 : 4. При добавлении разбавленной серной кислоты к концентрированному раствору этого пероксониобата выделяется лимонно-желтый топкий порошок свободной (содержащей воду) перокеониобиевоИ кислоты (точнее, ДЕОксомояопороксо-ниобиевой кислоты!
HNbO4 = H
ГО
Nb03 О
в которой отношение ниобия к активному кислороду равно 1:1. Пероксониобиевая кислота чрезвычайно устойчива; под действием разбавленной серной кислоты она разлагается лшпь при нагревании (на перекись водорода и ниобиевую кислоту).
Пецтахлорид ниобия. NbCls можно получить при нагревании металлического ниобия в токе хлора:
ЫЬ+»/2С12=ХЬС15
или взаимодействием пятиокиси ниобия с некоторыми хлоридами, например СС14:
Nb2Os-)-5CCI4—2NbCl5-|-5COCI2.
Пятиокись ниобия значительно легче реагирует с СС14, чем пятиокись тантала. Однако если смесь обеих пятиокисей с избытком четыреххлористого углерода нагревать до 270° в эвакуированной толстостенной стеклянной трубке, то наряду с пяти-окисью ниобия в реакцию вступает значительная часть пятиокиси тантала, так как Nb2Os служит при этом катализатором. Для получения пентахлорида ниобия описанным выше способом можно использовать углеродсодержащий ниобий, который получается при выплавке металла в электрических печах, а также неполностью восстановленный Продукт, образующийся при прокаливании смссн пятиокиси ниобия'с углем. От оксихлорида NbOUls, примешанного в последнем случае к продукту реакции, пента хлорид можно отделить отгопкой.
Пентахлорид ниобия образует желтые игольчатые кристаллы, плавящиеся мри 204,7°. Температура кипения лежит при 247°. Желтый пар, как свидетельствуют данные определения плотности (при 360°), состоит из молекул NbCjE. В хлороформе, четыреххлористом углероде, а также в S2Cla пентахлорид ниобия растворяется без разложения, а в спирте и эфире с разложением. Водой он полностью гидролизуется с образованием хлористого водорода и ниобиевой кислоты. Если же действовать на него концентрированными сильными кислотами (серной и соляной), то ниобиевая кислота не осаждается. Вероятно, эти сильные кислоты препятствуют гидролизу. Если раствор пентахлорида ниобия прокипятить с концентрированной соляной кислотой, то и после разбавления водой осаждения пе происходит. Образовавшаяся при нагревании в результате гидролиза ниобиевая кислота пептизируется соляной кислотой и остается в коллоидном состоянии в растворе.
Теплота сублимации NbCb, составляет 20,0 ккал/мом (энтропия сублимации = ~ 39,5 нал): теплота испарения расплавленного соединения равна 13,1 ккал./молъ (энтропия испарения = 25,1 кал), ТаС15 и NbCls смешиваются во всех отношениях в жидком и в твердом состояниях. К образованию двойных млей пента хлорид ниобия мало склонен, несмотря на то что известны продукты присоединения с органическими веществами, например с пиперидином NbGl5-6С5ИцП.
* В зависимости от условий опыта соединение получается безводным (Balke, 1908) или е 1/2Н2О (Sieverts, 1928).
Пятая, группа периодической системы
127
Хлорокись ниобия, окситрихлорид ниобия NbOCl3, образует белые с шелковистым блеском иглы, улетучивающиеся при 332°. Плотность пара соответствует формул» NbOClg. Однако при очень высокой температуре продукт разлагается на NhCl5 и Nb3Os. Хлорокись ниобия разлагается также водой:
2NbOCl3+3H2O=Nb2Os+6HGl.
Сероводород при умеренном нагревании переводит NbOCl3 в оксисульфид. Хдорокись пиобия образует двойные соли (оквохлоропиобаты) типа MI[NbOCl4] и M|[NbOCIsJ. Они выделяются проще всего при прибавлении соответствующих хлоридов к растворам ниобиевой кислоты в концентрированной соляной кислого (Weiland).
Подобно пептах л ори ду пиобия ведет себя пурпурно-красный пентабромид. Бролюкиеь ниобия (желтого цвета) менее устойчива, чем хлорокись. Но оиа также образует двойные соли (оксобромониобаты). Лентаиодид Nbl; получают действием паров 13 на раскаленную ниобиевую проволоку (К.бгозу, 1939). Таким жо способом получают Та15, который более устойчив к нагреванию, чем Nbl5.
Фтор он ио б ат и пентафторид ниобия. Склонность к образованию двойных солей наиболее ярко выражена у фтористых соединений ниобия. Если к раствору ниобиевой кислоты в плавиковой кислоте добавить фториды металлов, то в зависимости от количества или концентрации плавиковой кислоты образуются фторониобаты или оксофторониобаты.
Состав простейших фтор ониобатов отвечает в общем виде формулам NbF5-MrF и NbFj-'ZM1? (соответствующую сноску см. на стр. 132); однако большинство этих соединений имеет более сложный состав. Из оксофтор он и оба топ известны соединения состава NbOF3-ЛЬОГд-ЗМ1?, а также и более сложные.
Псптафторид пиобия был получен в 1911 г. Руффом кипячением пентахлорида ниобия в безводной фтористоводородной кислоте с обратным холодильником, охлаждаемым охладительной смесью. Пентафторид ниобия представляет собой бесцветные сильно преломляющие свет кристаллы состава NbFa, плавящиеся в интервале температур 72—73°; температура кипения его 236°.
Нитрид ниобия. Ниобий может содержать в своей решетке весьма незначительные количества азота. Но он образует с азотом несколько соединений (ср. табл. 13, стр. 108). Среди пих NbN, существующий, по дапным Брауэра и Яндера (1952), в трех подробно изученных рентгенографических модификациях, а именно народу с уже давно известной кубической (структура NaCi: а = 4,38 А) в двух различных гексагональных модификациях. Одна из них (а = 2,95, с = 11,25 А) имеет состав, точно отвечаю-пщй формуле NbN, в то время как состав кубической модификации колеблется между NbN и NbNg.gg. Вторая гексагональная модификация (а = 2,93, с = 5,45 А) возникает только при определенных условиях. Соединение Nb4N3 имеет немного искаженную тетрагональную решетку каменной соли (с/а = 0,987) я является гомогенным в области NbN0,J9— NbN0,75, Соединение Nb2N (с областью гомогенности NbNOiSO — NbN0,40) образует гексагональную решетку с плотной упаковкой атомов Nb (а =3,05, с = = 4,96 А) и статистическим распределением атомов N в наибольших пустотах решетки. Все нитриды ниобия очень тверды и хрупки. За исключением серо-желтого кубического NbN, все они имеют чистый серый цвет.
Аналитические сведения. Ниобий осаждается как из кислых, так и из слабощелочных растворов в виде ниобиевой кислоты, которая часто очень трудно фильтруется. Прокаленная пятиокись ниобия нерастворима в кислотах. Ее можно перевести в растворимую форму сплавлением с гидросульфатом или пиросульфатом калия. Цинк и разбавленная серная кислота вследствие восстановления ниобиевой кислоты окрашивают растворы ниобиевой кислоты в грязновато-сипий или коричневый цвет. Танталовая кислота не дает этом реакции.
Об аналитическом разделении пиобия и тантала см. стр. 133.
128
Глава. 4
ТАНТАЛ (TANTALUM) Та
Распространение в природе. Тантал почти всегда сопутствует ниобию я танталитах и ниобитах, которые, как уже было указано, встречаются во всех частях света, по в основном в небольших количествах. Основные европейские месторождения танталита находятся в Финляндии и Скандинавии. Значительно большие залежи имеются в Северной Америке и Австралии.
Танталиты представляют собой мета тантала ты железа (II) Fe(TaO3)2, в которых железо может замещаться маргавцем. Некоторая часть тантала почти всегда замещается ниобием. В ниобитах, как уже отмечалось, ниобия больше, чем тантала. Обычный танталит кристаллизуется в ромбической форме (структура подобна структуре ниобита, см. стр. 124). Но то же соединение может быть тетрагональным, типа рутила (тапиолит). Часто танталовая кислота встречается в соединениях с редкими землями. Так, tpepemetmum является, по существу, ортотантал атом (и ни об ат ом) иттрия Y(TaOi). Иттротанталит- или дитанталат иттрия Y4[T;i2O7]3. Тог же состав, но с большим содержанием ниобия, чем тантала, имеет самарскит (уранотанталит), замечательный благодаря содержанию урана. Микролит в основном содержит дитанталат кальция Са^ТазО?].
Минералов с преимущественным содержанием ниобия известно больше, чем танталовых. В среднем соотношение обоих элементов составляет примерно 1,3 : 1. Встречаются почти чисто ниобиевые минералы, так же как и чисто танталовые в отличие от пары цирконий— гафний, где последний всегда является только примесью. Это объясняется тем, что ниобий и тантал близки но распространенности и обладают меньшим химическим сходством, чем Цирконий и гафний.
Исторические сведения. Тантал был открыт в 1802 г. Экебергом в двух тогда только что найденных минералах, (из Кимнто в Финляндии й из Иттербй в Швеции). Стараясь отметить необычайную для окислов металлов неспособность окиси тантала даже при избытке кислоты присоединять кислотный остаток с образованием соли, Экеберг назвал этот элемент по имени томящегося в аду, согласно греческой легенде, героя — танталом. Оба минерала были названы соответственно танталитом а иттротанталитом.
Получение. Разложение танталовых руд в технике осуществляют нагреванием их с гидросульфатом калия в железных сосудах, выщелачиванием сплава кипящей водой и растворением в HF остающегося порошкообразного остатка танталовой кислоты, загрязненного ниобиевой кислостей. Отделение от ниобия осуществляют обычной фракционированной кристаллизацией фторотанталата калия K2TaF7. Из него восстановлением натрием при нагревании получают металлический тантал:
K3TaF7+5N а -= Та+5NaF+2KF.
Получающийся таким образом в виде черного порошка металл всегда содержит вначале некоторое количество окиси. От нее освобождаются, по Данным Волтона, нагреванием в электрических вакуумных печах; кислород при очень высоких температурах удаляется в виде ТаО или ТаО2 (ср. стр. 129).
Металлический тантал получают также при электролизе Та2О5, растворенного в расплавленном K.2TaF7. Очень чистый металл можно получить методом наращивания в результате термической диссоциации ТаС16 (Burgers, 1934).
Свойства- Тантал представляет собой тяжелый, платиново-серый блестящий металл, довольно твердый, но чрезвычайно ковкий; ковкость его повышается по мере очистки. Он отличается, очень высокой температурой плавления 13027й (Worthing, 1926), 2996° (Malter, 1939)1, а при умеренно высоких температурах исключительной химической стойкостью.
Пятая- группа периодической системы 129
Кроме плавиковой кислоты, на металлический тантал не действуют никакие другие кислоты, даже царская водка. Так же мало влияют на него и щелочи. Расплавленная едкая щелочь также действует на пего слабо; однако при длительном воздействии щелочи его разъедают. При обычной температуре и црн уморенном нагревании компактный тантал устойчив на воздухе; ;в этих условиях он лишь тускнеет и приобретает сине-ваго-черный цвет. При более сильном нагревании на воздухе он превращается в Га2О5. В мелкораздробленном состоянии при нагревании оп сгорает с испусканием яркого света. Раскаленный порошок тантала энергично разлагает воду. При нагревании он также энергично взаимодействует с хлором, а с фтором — уже при обычной температуре. С серой тантал соединяется с образованием пламени. Тантал абсорбирует водород (ср. стр. 106), а при нагревании также азот. При этом он становится хрупким и ломким.
Применение. Так как тантал объединяет превосходные металлические свойства с исключительной химической стойкостью, оп оказался весьма подходящим для изготовления хирургических и зубоврачебных инструментов, например концов пинцетов, игл для инъекций, стрелок м т. д, В некоторых случаях он может заменить платину. Перья из тантала оказываются равноценными золотым перьям с копчиками из иридия, В настоящее время тантал Приобретает все возрастающее значение для изготовления тиглей. Еще недавно применение тантала и его соединений было ограниченным. Но за последнее время оно резко возросло.
СОЕДИНЕНИЯ ТАНТАЛА
Соединения тантала почти все производятся от пятивалентного тантала. Однако известны также вполне определенные соединения тантала с более низкими степенями окисления. Например, при нагревании пентахлорида тантала ТаС15 с алюминием в эвакуированной запаянной стеклянной трубке (лучше всего в присутствии хлорида алюминия) можно получить хлориды ТаС14, ТаС13 и ТаС12. Восстановлёние тантала в водном растворе водородом в момент выделения в отличие от ванадии и ниобия невозможно.
Соединения тантала(У)
Пятиокись тантала. Та2О5 в чистом состояния удобнее всего получать сильным прокаливанием чистого металлического тантала в тоне кислорода. Ее получают также нагреванием на воздухе соединений тантала с летучими или горючими элементами. Большей частью получение пятиокиси тантала осуществляют обезвоживанием «танталовой кислоты». Последняя, однако, часто загрязнена веществами, адсорбируемыми ею при осаждении из раствора, поэтому окись получается не совсем чистой.
Пятиокись тантала—f белый, нерастворимый в воде и кислотах (аа исключением плавиковой) порошок с уд. весом 8,02. Опа не изменяется при прокаливании на воздухе, в атмосфере сероводорода или в парах серы. Однако при температуре выше 100ба Та2О5 взаимодействует с хлором и хлористым водородом. При нагревании в вакууме до 2000° ока отщепляет кислород; по данным Шефера, при этом улетучиваются низшие окислы ТаО и ТаО2. В присутствии углерода отщепление кислорода наступает раньше, так как благодари их взаимодействию давление кислорода понижается до минимума. Однако в этом случае часть углерода реагирует с танталом с образованием карбида (см. стр. 133).
Пятиокись тантала диморфна. При обычной температуре устойчива ее ромбическая модификация (а-Та2О5), изоморфная <z-Nb3O5. Высокотемпературная форма пятиокиси является тетрагональной ф-Та2О5); температура превращения лежит при 1320°.
Чистая пятиокись тантала в отличие от пятиокиси ниобия не восстанавливается до Двуокиси при нагревании в токе водорода, даже еелй ее смешать с порошком цяти-9 Г. Реми
130
Глава 4
окиси ниобия. По-другому ведут себя твердые растворы Та3О5 — Nb2O6. При достаточно высоком содержании Nb2O5, как и ок а аал Шефер, Та2О5 полностью восстанавливается до ТаОа, при этом образуются гомогенные твердые растворы Nb02 — ТаО2,
Танталаты п танталовая кислота. Сплавлением пятиокиси тантала со щелочами или карбонатами щелочных металлов получают танталаты. По своему составу эти двойные соединения пятиокиси тантала (выступающей в качестве кислотного окисла) с основными окислами в общем аналогичны пиобатам. Однако оказалось, что пятиокись тантала еще более, чем пятиокись ниобия, склонна образовывать наряду с нормальными соединениями продукты адсорбции различного состава.
Реакция сплавления пятиокиси тантала с избытком карбоната натрия, протекающая гораздо труднее, чем у пятиокиси ниобия, приводит в конце Концов к образованию ортосоли Ка3ТаО4, о чем свидетельствует количество выделяющегося из расплава СО.. Метатанталаты по своим свойствам весьма близки к метаниобатам, и прежде всего они изоморфны им. В водных, а также в сильнощелочных растворах, как показал Яндер (1925), существуют ноны пентатанта.говой кислоты Н7[Та^О1е). Однако еще но яспо, являются лк кристаллизующиеся с водой танталаты солями этой кислоты, т. е. пеитатанталатами MjTagOщ- тН20, или это гексатанталаты М^ТавОщ-ггН20, в которых часть молекул воды замещена на М^ОН. Калиевую соль, кристаллизующуюся в форме шестигранных призм (в отличие от солей лития и натрия Легко растворимую в воде), можно получить, согласно Виндмейссеру (Windmaisser, 1941), с соотношением К.2О • Та3О5 = 7 : 5 и 8 : 6, причем состав не влияет на структуру кристалла. Танталаты щелочных металлов в растворах сильпо гидролизуются. При значении концентрации попов водорода более 10-а происходит полный гидролиз «с выделением студенистого осадил пятиокиси тантала, так называемой «танталовой кислоты» Выделению осадка препятствуют некоторые кислоты, образующие с пятиокисью тантала комплексные соединения, паприцор мышьяковая кислота, мышьяковистая кислота, но прежде всего органические оксикислоты: вигшая, випоградпая и лимонная. Напротив, координационные соединения типа апатита или ванадинита танталовая кислота пе образует (ср. стр. 110).
Вызывает сомнение, что так называемая танталовая кислота, осажденная из водного раствора, представляет собой определенный гидрат пятиокиси тантала, т. е. кислоту Н,[Та601в], анионы которой, как найдено Яндером, существуют в щелочных растворах. Гель Та2О5 имеет состав, подобный гелю Nb2Os, с той лишь разницей, что он легче отщепляет воду. Кристаллизация наблюдается только после полного обезвоживания. Если гель обезвоживать при не слишком медленном нагревании, то после удаления всей воды внезапно наступает сильное раскаливание. Это явление, наблюдаемое также у пятиокиси ниобия, так же как в случае двуокиси титана, основапо на мгновенной кристаллизации аморфной окиси.
«Танталовая кислота» в отлично от «пиобйевой кислоты» даже свежеприготовленная но растворяется в соляной и азотной кислотах.
В технике «таптадовую кислоту* получают обычно разложением серной кислотой даойного фторида тантала и калия 2KF' ТаР5, который легче всего приготовить в чистом виде:
2K3TaF; + 2H2SO4 -|- 5Ы2О = Та2О5 + 2K2SO4-f- 14HF.
Полученная этим способом танталовая кислота, как уже было указано, всегда в большей или меньшей степени загрязнена адсорбируемыми из раствора веществами.
Псрокеотантя шты и перокеотапталовая кислота. При обработке перекисью водорода растворенного в воде сплава пятиокиси тантала с гидроокисью калия Писаржев-скпй получил пвроксотанталат калия — соединение состава KjTaOg-i^IIjO *. Соль
* При несколько иных условиях опыта получают, так же как для соответствующих соединений ниобия (ср. стр. 126), безводную соль.
Пятая группа периодической системы 131
О, Od
выпадает из водного раствора при добавлении спирта в виде белого мелкокристаллического осадка. Могут быть получены также (не говоря о содержании воды) соответ^ ствующие соли с другими катионами. Эти соединения содержат четыре активных атома кислорода. Структурная формула их имеет вид;
Ml
т. е. они являются производными ортотанталатов при замене п них четырех атомов О I I
четырьмя пероксо-радикалами О—О подобно тому, как псроксониобат калия производится (см. стр. 126) от ортопиобатов.
Свободную пер оке от а нт ал ов у ю кислоту. можно получить, при взаимодействии тетраиероксотанталата калия с разбавленной серной кислотой. Опа (не принимая во внимание содержания воды) имеет состав НТаО4 и содержит один активный атом кислорода. Таким образом, но составу и строению опа аналогична описанной ранее (стр. 126) пероксони обнов ой кислоте и, следовательно, должна быть названа моно-пероксометатанталовой кислотой.
Пептахлорид тантала. ТаС16 образуется при горении тантала (а также карбида, нитрида или сульфида тантала) в хлоре. Его можно также получать взаимодействием пятиокиси тантала с некоторыми хлоридами (например, с пятихлористым фосфором, четыреххлористым углеродом, хлорокисью углерода, хлористой серой, хлоридом алюминия). Это бесцветное, иногда стекловидное, но легко кристаллизующееся вещество с уд. вес. 3,68; т. пл. 216,5° л т. к ни. 233°. Плотность пара соответствует формуле ТаС15. При возгонке в атмосфере, содержащей кислород, оц разлагается, образуя пятиокись тантала. Следует отметить, что при этом в качестве промежуточного продукта нс образуется оксихлорид. Оксихлорид не образуется также при разложении водой; в этом случае сразу выделяются пятиокись тантала (в виде студенистой, содержащей воду, так называемой танталовой кислоты) и хлористый водород. При низкой температуре пептахлорид тантала присоединяет аммиак, Спаку (Spacu, 1937) доказал существование 12-, 10- и 7-аммпакатов. Последний при температуре около 0° разлагается, вероятно, с образованием Ta(NH2)2Cl3-3NII3 (аммонолиз). Двойная соль пентахлорп-да тантала не получена; напротив, пентафторид тантала весьма склонен к образованию двойных солей.
Пентафторид тантала и фторотанталаты. Пентафторид тантала, по данным Руффа, можно получить подобно цептафториду ниобия взаимодействием пептахлорида с жидким фтористым водородом. О и образует бесцветные призмы (т. пл. 96,8°, т. кип. 229°). По Гапу (Hahn О.), его можно также получить прокаливанием двойного фторида тантала и бария 3BaFK-2TaF5 в платиновой трубке при 1000° в медленном токе воздуха, высушенного над пятиокисыо фосфора.
Из растворов пятиокиси тантала в водном растворе плавиковой кислоты пептафто-рид тантала пе выделяется из-за большой склонности этого соединения к гидролизу. При упаривании таких растворов, а также при прокаливании сухого остатка тантал не улетучивается в виде фторида только в том случае, если пятиокись таптала была совершенно свободна от щелочи. Если она содержит щелочь, то улетучиваются значительные количества фторида тантала, так как образующийся в этих условиях фторо-танталат щелочного металла при высокой температуре распадается с образованием относительно легко летучего пентафторяда тантала. Улетучивание можпо практически предотвратить добавлением серной кислоты, которая разлагает фторид тантала.
В отличие от свободных фторидов тантала комплексные соли их, фторотанталаты, можно легко получить из водного раствора пяти-окиси тантала или раствора танталовой кислоты в плавиковой кпелоте. Они выделяются при добавлении к этим растворам соответствующих фторидов металлов. Большое число фторотанталатов соответствует типу ^ABF-TaFg. Существуют также соединения типа MrF-TaF& и натриевая соль состава 3NaF-TaF5. Калиевую соль первого из названных типов
9*
132
Глава 4
2KF-TaF5 *, кристаллизующуюся без воды, вследствие малой растворимости применяют для отделения тантала от ниобия и титана. Эта соль образуется сразу при добавлении ионов калия к раствору иятиокиси тантала в плавиковой кислоте. Так се легко можно перекристаллизовать, при нагревании она значительно более растворима, чем на холоду. Калиевая соль образует тонкие ромбические иголочки, удобные для микроскопического определения тантала. Перекристаллизацию иа горячей воды следует вести в присутствии избытка плавиковой кислоты, так как иначе произойдет гидролитическое разложение.
Приводит ля гидролиз фторотантал атов непосредственно к танталовой кислоте, или в качестве промежуточных продуктов образуются оксофтор отанта латы как определенные соединения, еще не вполне ясно. Во всяком случае, с Клопп ость к образованию оксофторасосдпнений у тантала значительно меньше, чем у ниобия. Из раствора чистой пяти он пси тантала в плавиковой кислоте Гану удалось выделить фтор отанта левую кислоту состава HF- TaF5- 6Н2О в виде перистых игл с температурой плавления 15°.
Соединения тантала с более ппзкими степенями окисления
О низших окислах тантала см. стр. 129 и 130.
Низшие хлориды можно получить, по данным Руффа (1922, 1925), нагреванием до 300° при встряхивании смеси пентахлорпда тантала с порошком алюминия в эвакуированной запаянной стеклянной трубке (в присутствии небольшого количества хлорида алюминия для ускорения реакции). Проще всего таким способом получают трихлорид тантала ТаС1э. По данным Руффа, в расплавленном состоянии он реагирует с избытком ТаС15, образуя ТаС14:
ТаС134-ТаС15 2ТаС14.
Если при температуре 350—400° отогнать пентахлорид, остается трихлорид. При дальнейшем повышении температуры (до 500—600°) трихлорид разлагается, причем снопа образуется пента хлор ид:
ЗТаС13=2ТаС12 ТаС15.
Все хлориды темно-золеного цвета и при обычной температуре твердые. Она различаются отношением к воде. Тетрахлорид разлагается водой по уравнению'.
2ТаС14+5Н2О=ТаС13 + Та(ОН)5+511С1.
Трихлорид в холодной воде растворяется без изменения с образованием зеленой окраски. При добавлении ОН--иопов из раствора выпадает зеленый гидрогель, который растворяется как в кислоте, так и в избытке щелочи и, следовательно, имеет амфотерный характер подобно гидроокиси алюминия. Он весьма энергично окисляется и кипящей водой разлагается с образованием танталовой кислоты:
Та(ОН)3+2НгО=Та(ОН)3 + Н2.
Подобное разложение воды, нагретой до кипения, происходит также под вл пятите трихлорида. Дпхлорид тантала практически нерастворим в воде, но, несмотря на этс„ уже под влиянием холодной воды он Переходит в соединение тряхлалентлого тяптг>~ ла и при этом выделяется водород:
ТаС12 + Н2О = Та- + 2С1- -f- ОН" -f- 1/2Нг.
При упаривании в вакууме раствора хлорида тантала(III), содержащего пзбытс^ соляной кислоты, выпадает наиболее устойчивое комплексное соединение трехвалет-ного тантала с хлором Та3ОС17-ЗН2О, вследствие чего оно было получено раньше простого хлорцда. От него производятся многие соединения, которые также содержу три атома трехвалентпого тантала в молекуле.
* Как показал рентгенометрически при помощи анализа Фурье Хорд (HcarS 1939), структурно это соединение оказалось гептафторотанталвтом К,[TaF-с координационно семивалснтпым танталом (ср. т. I стр. 337). Это справедливо п — аналогичного соединения ниобия (ср. стр. 127). ”
Пятая группа периодической системы 133
Карбиды тантала. Тантал образует Два карбида. Латунно-Желтый TaG кристаллизуется, подобно TiC и ZrC, по типу каменной соли (я = 4,445 А). Подобно этим карбидам ТаС способен растворять определенное количествомоталла (до 10 ат.%), образуя твердым раствор. О проводимости см. стр. 68. Ta2G — серого цвета, имеет решетку, в которой атомы Та образуют гексагональную наиболее плотную упаковку (а — = 3,091, с = 4,93 А).
Нитрид тантала. TaN образуется в виде Серого порошка при нагревании тантала в атмосфере азота. Теплота образования 58,1 ккал/'моль (Neumann, 1934).
Аналитические сведения. С аналитическом отношении тантал чрезвычайно похода на ниобий. Однако из растворов, содержащих большие количества плавиковой кислоты, кристаллизуется фтор отоптал ат калия K2TaF7, значительно труднее растворимый, чем аналогичное соединение пиобия. Последний особенно легко растворяется в теплой воде (образуя соединение K2NbOF5), в то время как фторотантал ат калия при кипячении с водой переходит в очень трудно растворимую основную соль. Используя эту реакцию, можно легко установить, содержит ли тантал примесь ниобия.
Для аналитического разделения тантала и ниобия в основном применяют метод Пауэлла и Шолера [Powell, Schoeller, Z. anorg. Chem., 151, 221 (1926)], основанный на том, что комплексы окса л атотанта латов разрушаются легче, чем оксалатониоба-тов. Образующаяся в результате гидролиза комплекса танталовая кислота выпадает в осадок при добавлении Таллина, с которым она образует бледно-желтый продукт присоединения; осаждающаяся при более низком значении pH ниобиевая кислота образует с тацпипом продукт присоединения ярко-красного цвета.
ПРОТАКТИНИЙ (PROTACTINIUM) Ра
Протактиний был открыт в 1918 г. Отто Ганом и Лизой Мейтнер и независимо от них Соддй и Кранстоном. Поводом для поисков родоначальника Ас служило основанное на законе смещения (см. гл. 11) предположение, что искомое вещество должно быть аналогом тантала. После того как была установлена трехвалентность актиния (в результате диффузионных опытов Ховсши и изучения Флеком его химических свойств) Ра занял свое место в третьей группе периодической системы, и по закону смещения его материнским элементом можно было считать только либр испускающий р -частицы элемент второй группы, либо испускающий а-частицы элемент пятой группы. В первом случае актиний был бы изотопом радия. Но так как радий и актиний оба происходят от урана, обсуждаемый изотоп должен был бы встречаться в природе вместе с радием и, как непременный спутник радия, испускающий р-Частицы, едва ли. мог бы долго оставаться неоткрытым. Правильнее оказалось второе предположение, а именно что искомое вещество принадлежит к пятой группе периодической системы и, следовательно, доляспо быть аналогом тантала *, распадающимся с выделением «-частиц. Для его выделения остатки урановой смоляной руды смешивали с танталовой кислотой, которую потом вновь отделяли. Предположение, что вместе с нею будет выделен искомый аналог тантала, оправдалось. Пятиокись тантала, полученная из остатков урановой смоляной руды, оказалась радиоактивной ii Действительно содержала материнский элемент актиния; это следовало из того, что из нее образовывались короткоживущие продукты распада актиния, идентифицируемые по характеристическим кривым распада. Название протактиний (6 лрюто; — первый в ряду), данное новому элементу Ганом и Мейтнер, должно было, отражать его отношение к актинию как к: непосредственному «потомку».
* Единственный известный до настоящего времени радиоактивный аналог тантала, уран Х2, является р-излучателем.
134
Глава 4
Протактиний иля его пятиокись сравнительно легко можно получить радиоактивно чистыми, т. е. свободными от других радиоактивных веществ. Напротив, чрезвычайно трудно получит г. химически чистую пятиокись, так как опа по химическим свойствам очень близка иятпокшл тантала и отличается от нее лишь немного более основным характером. Помимо этого, весьма затрудняющим обстоятельством являются незначительные количества, в которых протактиний содержится в урановой смоляной руде. На 1 г урана в любом урановом минерале (наряду с 332 мг, радия) приходится только 314 мг протактиния (ср. стр. 569). Однако в 1928 г. Гроссу (Gross) удалось из примерно 500 кг псхо цтого материала выделить 9 мг чистой Ра2О$. Через несколько лет (1934 г.) он получил около 100 мг Ра2О5, к этому времени Грауэ, и Кэдинг (из 5,5 т исходного продукта) получили 500 мг чистого протактиния (в виде KaPaFy), В результате этих препаративных работ было установлено большое сходство протактиния с танталом. Однако оказалось, что Ра2О5 обладает заметно менее кислым характером, чем Та;О5: в атом отношении она ближе к ZrO2. Благодаря возрастающему использованию реакции циркония и тантала, удалось достигнуть дальнейшего обогащения протактиния, позволившего, наконец, из раствора двойного калиевого фторида фракционированной перегонкой выделить чистый K2PaF7. Как показали поздпее Крауз и Мур (Kraus, Moore), особенно хорошо отделяется протактиний от циркония, ниобия и тантала г;ри помощи ионообменных смол. Этим способом легко можно освободиться также от урана и железа.
Наибольшие количества металлического протактиния были получены Гроссом (1934) двумя способами: 1) разложением Окиси иод влиянием каналовых лучей и 2) термическим разложением РаС15 или Ра15 на раскаленной электричеством вольфрамовой проволоке в условиях высокого вакуума. Его получают также восстановлением PaF4 барием при 1400°. Протактиний представляет собой блестящий серо-белый металл, устойчивый па воздухе. Он имеет тетрагональную объемно-центрированную решетку (а = 8,93, с — 3,24 А; <1рснтг — 15,37). При температуре 250° реагирует с водородом, образуя твердый черный гидрид РаГ1э, обладающий кубической структурой (изотипен ПН3), а — 6,648 А. При 800е протактиний реагирует с NH3 с образованием желтого нитрида; при 1400е с углеродом дает карбид (Zacnariasen, 1952—1954).
Генетическая связь протактиния с актинием, а также его химические свойства, указывающие на гомологическое родство с танталом, определяют место Ра в периодической системе (Порядковый номер 91). Экспериментально определенный Гроссом (1935) атомный вес, равный 231, согласуется с положением протактиния в радиоактивном ряду актиния. Конечный продукт — актиний D — это свинец с атомным весом 207 (ср. стр. 5G9); он образуется из протактиния в результате всего шести «-превращений (ср. табл. 63, стр. 569), Следовательно, атомный вес протактиния должен быть на 6 X 4 = 24 единицы больше, чем у актиния D (актиносвинца), т. е. 231, О радиоактивных свойствах протактиния см. стр. 569,
Из тех радиоактивных элементов, которые занимают определенное место в периодической системе, протактиний после радия встречается в природе в самых больших количествах. То, что, несмотря на больший период полураспада, его меньше, чем радия, обусловлено тем, что только небольшая часть радиоактивного урана (4,6%) превращается (через уран Y) в протактиний, а основное количество переходит (через ионий) в радий (ср. стр. 567).
Белее поздние исследования свойств протактиния и его соединений проводили частично па искусственном изотопе 2jaPa. В отличие от естественного а-излучателя он является (J-излучателем (период полураспада 27,4 дня). Его получают действием нейтронов на торий:
В ₽
332Tb (я, У)233Т11 аззра .
СОЕДИНЕНИЯ ПРОТАКТИНИЯ
Протактиний наиболее устойчив в пятивалентном состоянии. Но се может быть также четырех-, трех- и двухвалентным. Последние два состояния встречаются, однако, крайне редко; напротив, переход из пятивалентного в четырехвалентное состояние происходит легко. В водных растворах
Пятая еруппа периодической системы 135
солей пр от актиния (V) имеются, поскольку он не образует анионы, однозарядные катионы [РаО2Г (протактинил-ионы, Welch, 1953).
В водном растворе соли протактиния(У) могут быть восстановлены до солей про-тактиния(1У) при помощи амальгамы цинка (Haissingky, 1951). Фторид, фосфат и ио дат нерастворимы. Сульфат растворяется легко, но образует с K2SO4 труднорастворимую двойную соль. Водные растворы солей протактиния(IV), так же как протактиния(У), бесцветны. Под действием кислорода воздуха ионы Ра“" довольно быстро снова окисляются до [PaO2j '-ионов. При добавлении (N!I4)2CO3 к раствору соли протакт иная (IV) образуется белый осадок, который благодаря образованию комплекса растворяется в избытке осадителя. С виппой и лимонной кислотами также образуются растворимые комплексные соединения.
Энергичным в ос станов лепном в водпых растворах можно получить также ионы Ра"’. Среди пения мротактиттия(Ш) с оос аж даются с соединениями лантана. Потенциал перезаряжения Pa'”/PaOi приблизительно соответствует потенциалу перезаряжения Сг‘7Сг"’ (Andersoa, 1949).
Окислы. Протактиний образует окисли РаО, РаО2 и Ра2О5, соответствующие валентностям II, IV nV, а также промежуточные окислы РаО2,35 или PasOg. Пяти-окись протактиния получают действием аммиака па раствор соли протактиния(У) и нагреванием студенистого осадка па воздухе до 500°. Можно прокаливать также РаО2 в токе кислорода при 1100’. Пятиокись существует в двух модификациях; ромбической изотипной Nb2O5, Та3О5 и USO5 и кубической (а =-5,445 А). Состав последней колеблется между РаО2,5 и РаО3 (Zacharias®, 1950—1954). Двуокись протактиния РаО2 была получена Захариазеном в виде черного порошка при нагревании Ра205 в токе водорода до 1500’. Она также кристаллизуется в кубической сингонии (а = =5,518 А.) и изотипна ThO2, UO2, NpO2, РнО2, АшОа. Если Ра2О5 нагревать в вакууме до 1000’, образуется черный тетрагональный Ра40д (а = 3,835, с = 5,573 А). Выше 1800’ он превращается в другую модификацию с кубической гранецентрированной рещеткой (« = 5,470 Л). Моноокись РаО, имеющая решетку типа NaCl (а = = 4,961 А), образуется в виде пленки па поверхности металлического Ра.
Галогениды. Пентафторид PaF5 был получен Захариазеном (1954) действием BrF3 па Ра2О5 в виде белого твердого вещества, испаряющегося в вакууме при 150°. Водой он гидролизуется. Тетрафторид PaFi, образующийся при взаимодействии РаО2 с Н2 и HF при 600°, окрашен в красновато-коричневый цвет. Пентахлорид РаС15 (бледно-желтые легко возгоняющиеся иглы, т. возг. 301°) легко получается при нагревании Pa2Oj (550°) в токе фосгена:
Ра2О54- бС0С12=2РаС15-|- 5СО2.
Хлорокись РаОС13 бесцветна. При температуре 900° она реагирует со смесью H2S и CS2 с образованием нелетучего светло-желтого оксисульфида протактиния(IV) РаОЗ, изотипного ThOS, UOS и NpOS. Желто-зеленый тетрахлорпд РаСЦ, образующийся цри нагревании РаС15 с Н2 при 800°, изотипеп ThCl4, UC14 и NpCl4. Получены также соединения протактиния с бромом и иодом (Zachariasen, 1950—1954).
Гл а в а 5
ШЕСТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Побочная подгруппа)
ХРОМ, МОЛИБДЕН, ВОЛЬФРАМ И УРАН
Порядковый номер Элемент Символ Атомный все У дель-«ый вес Температура» °C Удельная теплоемкость Валентность
плавления кипения
24 Хром Сг 52,01 7,2 1830 ~ 2300 0,1178 г, II, III, IV, V, VI
42 Молибден Мо 95,95 10,2 2630 — 4800 0,0589 11, III, IV, V, VI
74 Вольфрам W 183,80 : 19,2 3400 -5700 0,0340 II, Ш, IV, V, VI
92 Уран и 238,07 19,0 : ИЗО ~ 3500 0,0274 II, III, IV, V, VI
Общие сведения. В побочную подгруппу шестой группы периодической системы входят металлы хром, молибден, вольфрам и уран. Все эти элементы существуют в нескольких валентных состояниях. В своих нормальных соединениях они максимально шестивалентны соответственно номеру группы. В кислых растворах хром преимущественно трехвалентен, но в присутствии щелочей легко окисляется до шестивалентного. Аналоги хрома в обычных условиях наиболее устойчивы в шестивалентном состоянии. Лишь для урана наряду с шестивалентным относительно устойчиво и четырехвалентное состояние, тогда как остальные валентности этого элемента отступают на второй план. В остальном характерным отличием побочной подгруппы от главной является существование нечетных валентностей наряду с четными. Общим для элементов шестой главной и побочной подгрупп, кроме максимальной валентности шесть, является способность трехокисей образовывать соли с основными окйслами.
Простейшие хроматы, молибдаты, вольфраматы и уранаты по своему составу аналогичны сульфатам MJSO4. Однако в отличие от элементе" главной подгруппы элементы побочной подгруппы обладают ярко выраженной склонностью к образованию соединений, которые на один экве валент основания содержат более одново эквивалента кислотного ангиу рнда. Так, хром, кроме хроматов M*CrO4 = М1О-СгО3 и бихромат:-? М1СггО7 -= М’О-2СгОа (последние по своему составу соответствуй?' дисульфатам M’S2O7 ~ М*О-2ёО3), образует еще трихроматы М|Сг5ОЕ.-:-= М^О-ЗСгОз и тетрахроматы М^Сг4О13 = М*0-4Сг03. В случае ма~ бдатов и вольфраматов число молекул кислотного ангидрида Мс’’ соответственно WO3, приходящееся на одну молекулу основе? окисла ЩО, часто еще выше. Кислоты, которым отвечают соли так::_ типа, называют поликислотами (подробнее об этом см. в разделе, е;к:е щенном вольфраму). Уран обладает меньшей склонностью к обра?:?.
Шестая группа периодической системы 137
нито таких высокомолекулярных поликислот, одиако обычные уранаты имеют формулу, отвечающую не сульфатам, а дисульфатам MiU2O7.
Сила кислот, являющихся производными трохокисей, значительно уменьшается при переходе от хрома к урану. Аналогичное по составу хромовой кислоте Н3СгО4 соединение урана Нгио41пли иОз(ОН)3 представляет собой амфотерную гидроокись, кислотный характер которой сильно подавлен и которая, напротив, отлично образует соли с кислотами — соли уранила [IjO3]X2 (где X — одновалентный кислотный остаток).
Ярко выражен основной характер у гидроокиси четырехвалентного урана; гидроокись хрома(Ш) амфотерна, однако ее основные свойства выражены резче, чем кислотные. Напротив, шестивалентный хром вообще не образует соле образных соединений с кислотами. Хлористый хромил . СгО2С13— типичный х л орангидрид кислоты, а хлористый уранил [UO2|C12— типичная соль.
Правда, молибден и вольфрам образуют соединения с кислотами, однако это почти всегда соединения, в которых соответствующий элемент вместе с кислотными остатками образует более или мопее прочный комплексный аннон. С. точки зрения координационной теории нет принципиального различия между такими аниопама, как, например, образуемыми молибденом л вольфрамом комплексными ионами (Mo(GN)el"" н [W(CN)8]"" и анионами кислородных кислот, например [MoOJ* и [WOJ*.
Как почти во всех побочных подгруппах периодической системы, все элементы шестой побочной подгруппы имеют отчетливо выраженную склоииость к образованию ацидо-соединений. Ст (Ill) наряду со способностью образовывать комплексы с кислотными остатками обладает сильной тенденцией прочно связывать нейтральные молекулы, прежде всего аммиак и его производные. Образующиеся при этом соединения называют аммиинымя комплексами Сг(Ш). Особенностью молибдена и вольфрама является их Способность в форме анионов МоО., или WO) и Мо3О’0 или W3O"0 легко присоединяться к другим элементам (центральным атомам), например к фосфору в фосфорной кислоте, кремнию в кремневой кислоте, бору в борной кислоте. Так образуются комплексные кислоты часто очень сложного состава, однако большей частью относительно простого строения, называемые гетерополикисло-тами. Их наиболее известным представителем является додекамолибдато-фосфорная кислота, аналитический состав которой, без учета связанной ’ воды, отвечает формуле Н3РО4-12МоО3*. Образование ее труднораство-рнмой аммонийной соли при одновременном присутствии в сильно азотнокислом растворе молибдат-, фосфат-иопов и ионов аммония лежит в основе уже упоминавшейся в гл. 14 т.1 реакции на фосфорную кислоту с молибдатом аммония.
Температуры плавления и кипения металлов шестой побочной подгруппы в большинстве случаев очень высоки. От хрома к молибдену и вольфраму они чрезвычайно быстро возрастают, а затем при переходе к урану опять сильно падают. Это об7^ясняется, ло-видимому, тем, что уран имеет отличающуюся от своих аналогов кристаллическую решетку. Хром, молибден и вольфрам кристаллизуются в объемноцентрированной кубической решетке (см. т. I, рис. 47) с длиной ребра а — 2,8846, соот-
0 строении этого соединения см, стр. 201—202.
138
Глава 5
вотственно 3,1468 и 3,1650 А, в то время как уран имеет ромбическую решетку.
Если вольфрам получают электролизом растворов WO3 в расплавленном щелочном фосфате, то образуется другая модификация металлического вольфрама (0-W), которая при температурах выше 600“ мопотроппо переходит в обычную модификацию (a-W). В кубической элементарной ячейке P-W, кроме центра и вершил куба, атомами заняты также и грани (см. рис. 20).
Хром также часто получается при электролизе в виде неустойчивой модификации, иногда с решеткой типа Mg (см. т. I, стр. 279), иногда типа a-Mn (см. стр. 42). И в этом случае ври нагревании происходит мопотроппый переход в обычную модификацию.
Уран иимеет кристаллическую решетку, не встречающуюся пи у одного из металлов. По данным Уоррена (Worrerr, 1937), он образует ромбическую решетку, которую можно рассматривать как искаженную гексагональную упаковку. Но если при такой упаковке каждый атом окружен 12 дру-
Р и с. 20. р-Вольфрам. а = 5,038 А.
гими, находящимися на равном расстоянии атомами, то в решетке урана четыре соседних атома расположены ближе (два на расстоянии 2,786 и два на расстоянии 2,856 А), чем остальные восемь (четыре на расстоянии 3,277 и четыре на расстоянии 3,367 А). Эта модификация (a-U) при температурах выше 660’ переходит в p-U, который при температурах выше 760° в свою очередь переходит в у-U. p-U образует сложную тетрагональную решетку с 30 атомами в элементарной ячейке (Thewlis 1951—1952; Tucker 1951—1953). у-U образует объемноцентрированную кубическую решетку. Определенные примеси (например, хром или молибден) стабилизируют у-форму. так что опа не изменяется при охлаждении до обычной температуры.
Лтожпые радиусы элементов шестой побочной подгруппы, рассчитанные из межатомных расстояний в кристаллических решетках, приведены в табл. 14. Кроме того, таблица содержит
радиусы трех- и четырехкратно положительно заряженных ионов, полученных на основании параметров кристаллических решеток окислов. Для расчета радиусов ионов, имеющих заряд б-f- в настоящее время еще недостаточно экспериментального
материала.
Ранее урап считали элементом с самым большим порядковым номером и наибольшим атомным весом. После того как при помощи ядерных превращений удалось искусственно получить новые элементы, был изучен
Таблица 14
^Кажущиеся атомные и ионные радиусы элементов побочной подгруппы VI группы
Хром Молибден Вольфрам Уран
Атомный радиус, А 1,2491 1,3626 1,3704 1,576
KZ=8a KZ=8 KZ=8 KZ=I2
Радиус трехзарядттого нова, А 0,65 — — 1,06
Радиус четырохзарядного иона, А 0,68 0,68 0,89
а
KZ—координационное число.
Шестая группа периодической системы
139
ряд элементов с еще более высокими, чем у урана, порядковыми номерами и атомными весами, так называемые трансурановые элементы (см. гл. 14). Затем было показано, что по крайней мере два из них (нептуний и плутоний) встречаются в природе, хотя и в очень незначительных количествах.
В табл. 15 приведены величины молярных те плот образования для важнейших окислов металлов побочной подгруппы шестой группы. Переход двуокиси в трех окись связан, за исключением случая вольфрама, с относительно небольшим выделением тепла. И совершенно незначительным является различие между теплотами образования, отнесенными к одному и тому же количеству металла, в случае полуторной окиси и трех окиси хрома. Если при окислении хрома От металла до полуторной окиси выделяется 89,G ккал на 1 г-агпом кислорода, то при окислении полуторной окиси до трехокисн освобождается лишь 1,8 ккал на 1 е-атом кислорода! Впрочем, теплота образования полуторной окиси хрома относительно невелика, если сравнить ее с молярной теплотой образования окиси алюминия (393 ккал). Это обстоятельство важно для описываемого в дальнейшем способа получения хрома.
Таблица 15
Теилоты образования окислов элементов побочной подгруппы VT группы (ккал! моль окисла)
Хром Молибден Вольфрам Уран
Теплоты г полуторной окиси М2О3 268,9 — — —
образования { ДвУ0КЫСи МО3 — 142,8 131,4 270,4
1 трехокисн МО3 137,1 180,4 195,5 303,9
Хром, молибден и вольфрам при нагревании легко реагируют не только- с кислородом, галогенами, фосфором, серой и их аналогами, ио и с углеродом, кремнием, бором и, труднее, с азотом.
Замечательна способность этих металлов давать соединения с окисью углерода — так называемые гексакарбонилы М(СО)6 (бесцветные ромбические кристаллы). Они образуются при действии СО и гриньяровых соединений магпия иа галогениды названных металлов (Job, ср. стр. 380), а карбонилы молибдена и вольфрама получают и непосредственным действием СО на металлы при температурах выше 225° и повышенном давлении (200 ат). Названные карбонилы можно сублимировать без разложения при уменьшенном давлении. Их плотности паров отвечают простым молекулярным формулам. Они нерастворимы в воде, но, напротив, растворяются в индифферентных органических растворителях, например в бензоле. По отношению к химическим реагентам, прежде всего по отношению к кислотам и щелочам, они поразительно устойчивы. Далее о карбонилах металлов см. стр. 375 н сл.
*
Сплавы. О сплавах металлов побочной подгруппы шестой группы (за исключением урана), как видно из данных, приведенных в табл. 16 и 17, имеются довольно разрозненные сведения. В отношении к другим металлам отчетливо проявляется зависимость от положения в периодической системе. В общем склонность к образованию соединении с другими металлами невелика. Образование твердых растворов в широком интервале концентраций наблюдается почти исключительно с переходными элементами.
Таблица J6
Образование соединений и твердых растворов металлами побочной подгруппы VJ группы с элементами главных подгрупп (Обозначения см, в табл. 9, стр. 66)
Главные подгруппы
II Ill 1 IV V VI
Вс i Mg в л1 c SI Sn 1 pl> N 1 p As Sl> Bi S Se Те
Сг Ве2Сг Симе ж. > О . ТВ,? СгВ 1550° СгВ2 18 50’ ж, < ор тв,> О Сг2А1 [] 850’ CrjAlg Q * 890’ CrAl* 1100’ CfaAlfi 1000’ CrAl? 72 5’ : тв, ~ 0 Cr4C* 1 530’ СГ7С3 1670“ Cr3C2 1820’ CrC? тв. >0 Cr3Si Cr2Si CrSi CrSi2 Ж. < CO тв. > 0 0 тв. 0 0 тв. — О cF^n CrN тв, > О CrgP* 1510’ Cr2P CrP CrP3 CrAa Cr2As3? CrAs2? тв. > 0 CeSb 1110° CrSb* 67 5“ ж. О тв. О 0 ж. <оо тв. О (лВ 1565’ Cr2S2 CrSe Cr2Se3 CrTe
Cr2Te3
Но Ве2Мо Сплав Мо2В 1850° МоВа 2250“ тв. 0 MoAIf 735’ тв. > 0 Mo3C 2407’ MoC MoSi2 MogSi^? Сплав тв. — 0 Mo2N MoN M03P MoP MoP2 MoAe2 Сплав Сплае MoS? Mo2S3? MoS2 MoS3 MoS4 MoSe3? MoTe3 MoTe2
W Be2W — W2B 2770’ WB 2860’ W2A1? WA13? WA14 тв. > 0 W2C 2750’ WC* 2600’ тв. > 0 W3Si2 2360° WSi2 2170° — ж. < co тв. 0? 0 WoN тв. 0 W4P? неустойч^ WP WP2 WAs2 Сплав Сплав ws? Was3? ws2 WS3 WSe2 WSe2 Сплав
Таблица It
Образованно соединений я твердых растворов металлами побочной подгруппы VI группы между собой и с металлами следующих за ними побочных подгрупп (Обозначения см. в табл. 9, стр. 66)
Побочные подгруппы
VI VII VIII I 11
Мо W Мп Ro Fe Со N1 Pd Pt Си А4 Au Zn СП Hg
Сг ТВ. со 0 ТВ. со 0 ж. < со ТВ.? СгМп? ТВ- < со 0 ТВ. со [CrFcl 930° ТВ. со Cr3Co2Q 1392° CiCeQ ТВ. —со 0 тв, < ос СГаРсЬ 1398“ ТВ. < со {) ж,< со тв, — О 0 Ж. < СО тв, О 0 ТВ. < со 0 ж. < со 0 ж. < со 0 CrHg? CrHg3?
Мо ТВ, со 0 Сплав —. ТВ. < со MogFe,* 1540“ Мо2Ге3* 1480° тв, < со MogCo2 1559° МоСо* 150 0° тв. < ос MoNi* 1345“ MoNiaD 935“ — Ж. <00? ТВ. < со ж. — О тв. О 0 ж. < со тв. 0 0 ж. < со тв. 0 0 — — тв. .~ 0 Сплав
W — тв. О W2Be3 3007“ тв. >• О WeFe? 1640“ WFe2 Q 1040° ТВ. < со WeCo? 1620“ wco3 а 1100“ тв.< со WNie 1525“ —1 ТВ, < со 0 Ж. <ОО 0> ЭЙ> <00 0 Ж. < со 0 — —. тв. -- 0 Сплав
142
Глава 5
Соединения с тяжелыми металлами главных подгрупп образуются в исключительных случаях, а твердые растворы с этими металлами или не образуются вовсе или лишь в ограниченной области. Твердые растворы, насколько известно, не образуются и с легкими металлами. Соединения с последними плавятся большей частью инкон-груонтно, что указывает на небольшую силу связи в таких соединениях. Соединения металлов побочной иод группы шестой группы со щелочными и щелочпоземельпыми металлами не известны. Однако точных металлографических исследований этих систем да настоящего времени не проводили вследствие значительной разницы в температурах плавления и агрессивности паров щелочных и щелочноземельных металлов.
Соединепия металлов побочной подгруппы VI группы с неметаллами главных подгрупп III—VI групп но своей структуре и составу очень близки к интерметаллическим соединениим и поэтому также включены в табл. 16, В отдельных случаях с этими неметаллами образуются недалътониды. Вез изменения кристаллической решетки металлы побочной подгруппы VI группы поглощают незначительные количества неметаллов, за исключением водорода, который обладает повышенной растворимостью в хроме (ср. стр. 116).
От г гонге пне к металлам побочных подгрупп совершенно отлично независимо от того, принадлежат ли они к переходным элементам или нет. Со следующими за ними переходными элементами *, т. о. с металлами побочных подгрупп VII и VIII групп, металлы побочной подгруппы VI группы образуют твердые растворы, в большинстве случаев в широкой области концентраций. В жидком состоянии здесь всегда (за исключением системы Mo — Pt) наблюдается неограниченная растворимость. Отчетливо выраженная способ и ость к образованию сплавов с этими металлами, особеппо с металлами подгруппы железа, имеет исключительное техническое значение (см. стр. 288 и сл.). Во многих случаях соединения металлов побочной подгруппы у! группы с металлами подгруппы железа образуются из первоначально выделяющихся при охла?кдспив твердых растворов, часто образующихся без изменения состава, т. е. характеризующихся только изменением структуры (ср. стр. 34 и 363; такие соединения обозначены в табл. 17 значком П) или это происходит при охлаждении фаз с нор входной структурой (формулы последних заключены в квадратные скобки).
Между собой металлы побочной подгруппы VI группы соединений пе образуют. Напротив, хром, молибден и вольфрам образуют друг с другом пеограниченный ряд твердых растворов.
С металлами побочных подгрупп, которые следуют за переходными элементами, т. е. с металлами побочпых подгрупп I и II групп, металлы побочной подгруппы VI группы неограниченно смешиваются не только в расплавленном состоянии. Единственное твердо установленное исключение представляет собой система Сг — Ан, впрочем, и в этой системе наблюдается заметная растворимость уже в твердом состоянии (золото способно включать в свою решетку до 40 ат. % хрома). До сих пор достоверно не установлено образование соединений между металлами побочной подгруппы VI группы и металлами побочных подгрупп I и II грунп.
ХРОМ (CHROMIUM) Сг
Распространение в природе. Важнейшей хромовой рудой является хромистый железняк, двойное соединение окиси железа (II) и окиси хрома(Ш) FeO-Cr2O.3. В значительных количествах он встречается па Урале, в Южной Африке, в Новой Каледонии, Греции, Малой Азии, на Скандинавском полуострове, в Венгрии и Боснии, в небольших количествах также в Верхней Силезии, Моравии и Штирии. Наряду с хромистым железняком техническое значение имеет встречающаяся в основном в Бразилии и на Урале красная свинцовая руда РЬСгО4.
Нередко хром встречается в алюминиевых минералах как заместитель алюминия. Тан, известны хромовая шпинель, хромовый турмалин, хромовый гранат, хромистая слюда, хромистый хлорит * **.
» Об отпогпштпп металлов побочной подгруппы VI группы к предшествующим ям переходным элементам известно очень мало (см. табл. 9 и 13).
** В мнш: рал огни г г ед хлоритами по ним а тот определенные, подобные слюде минералы (но пе содержащие в отлично от слюд щелочных металлов), которые встречаются во многих сланцевых породах (хлоритных сланцах).
Шестая группа периодической системы
143
Смарагд (природный) представляет собой окрашенный в глубокий темно-зеленый цвет берилл, в котором алюминий замещен хромом.
Исторические сведения. Хром был открыт Вокелепом (Vauquelin) в 1797 г. в сибирском минерале (красной свинцовой руде) и назван так за прекрасный цвет которым характеризуются его соединения — цвет). Вскоре он был обнаружен и в других минералах, прежде всего в хромистом железняке (Tassaert, 1799). Восстановление углем до металла удалось уже Вокелену. Начав с но пыток получения металлического хрома, Ганс Гольдшмидт в 1894 г. разработал алюмотермический метод, который он распространил вскоре па получение других тугоплавких металлов и создание высоких температур в небольшом пространстве.
Получение. Исходным сырьем для технического получения хрома и его соединений почти всегда служит хромистый железняк. Если требуется получить не чистый хром, а только его сплав с железом (содержащий также углерод), так называемый «феррохром», то руду можно непосредственно восстанавливать углем в печах Сименса — Мартена или в электрических печах:
Fed • Ст2О3+4 С=Fo + 2Сг + 4СО.
Напротив, для получения чистого хрома необходимо сначала из хромистого железняка получить возможно более чистую окись хрома, а из нее свободный от углерода хром получают алюмотермическим восстановлением:
Ст3О3-|-2А1 = AJ2Oj-|- 2Сг-f- 130 ккал.
Чистую Окись хргвта получают через хроматы или бихроматы щелочных металлов. Вначале хромистый железняк расплавляют на полу пламенной печи с добавлением гидроокиси или карбоната щелочного металла и жженой извести. При этом хром иод окисляющим действием обогащенного кислородом пламени печи переходит в хромат. Выщелоченный водой хромат под влиянием добавленной кислоты переходит в бихромат. Последний после удаления . труд нерастворимых соединений осаждают зз подходящего раствора в довольно чистом состоянии. В результате восстановления бихромата углем (вместо древесного угля можно использовать опилки иди крахмал) получают частую окись крома(Ш)’.
Na2Cr2O7 -J- 2С=Сг2О3 + Na2CO3-|- СО.
Для алюмотармического получения металлического хрома из окиси хрома последнюю смешивают с алюминиевым порошком и нагревают до вспышки. Для равномерного сгорания к смеси долее ообразао добавить некоторое коаичестдо хромата. Иногда можно добавить и окись хрома, предварительно прокаленную с присадкой щелочи (при этом образуется хромат). Чтобы получить металлический хром, по возможности свободный от алюмипия, для восстановления берут алюминий в количестве, несколько мепьшем теоретического. Практическое преимущество алюмотермического способа получения хрома по сравнению с другими обусловлено частично тем, что получающаяся яри ал гипотермическом вщ ста давлении в качестве побочного продукта плавленая окись алюминия (искусственный корунд) находит применение в качестве шлифующего н полирующего средства. Однако получаемый алюмотермическим способом хром lie является совершенно чистым. Совершенно чистый хром, который в отличие от обычного хрома куется в нагретом состоянии, хотя при комнатной температуре остается хрупким, можно подучить пбез гащиванием у до кт рол ити чески полученного хрома нагреванием в высоком вакууме иди восстановлением СгС13 или Сг2О3 металлическим кальцием в расплаве СаС12 или BaCI2 (Kroll, 1935).
Электролитическое осаждение хрома используют для получения защитных покрытий па других металлах. При этом хром часто осаждают пз теплого раствора хромовой кислоты, к которому добавлен сульфат хрома(Ш). В соответствии с положением хрома в электрохимическом ряду напряжений, его выделение в виде металла происходит только при
144
Глава 5
больших плотностях тока, при которых одновременно выделяется значительное количество водорода. Поскольку Щ частично растворяется в выделяющемся металле, делая ого пористым и растрескавшимся, его следует в дальнейшем удалить нагреванием. При этом одновременно происходит превращение часто первоначально образующейся неустойчивой модификации хрома в стабильную модификацию (см. стр. 138), что приводит к значительному увеличению твердости (Макарьева, 1935).
Основные осабенпоетп процессов, происходящих при электролитическом хромировании, техника которого разработана Форстером, выяснены, хотя и не во всех подробностях, главным образом в работах Sargent, 1920; Liebreich, 1921; Mailer Е., 1926 сл. [см. Z. Е lektroehem., 40, 326 (1934); 43, 361 (1937)]. .Если через чистый раствор хромовой кислоты между двумя инертными электродами пропустить достаточно сильный ток, то после начавшегося в небольшом объеме восстановления НСгО( до ионов Сг’" па катоде наблюдается сильное выделение водорода. То, что выделение водорода происходит, хотя напряжение, требующееся для разрядки попов И’ на 1,3 в выше, чем напряжение, требующееся дня восстановления ионов IlCi'O'. (см. т. I, стр. 821)), обусловлено тем, что последние изолируются на катоде образующейся непроницаемой дня ионов Н СЮ,, пленкой основного хромата хрома(Щ) Сг(ОН)[СгО4]. По предположению Мюллера, в такой пленке все группы Сг(ОН)2+ ориентированы в сторону катода, а все группы [СсО4]а- — в сторону апода, так как в ином случае основной хромат хрома(Ш) растворялся бы в хромовой кислоте, как и в остальных сильных кислотах. Плепка проницаема как для ионов Н‘, так и для некоторых других ионов, например HSO4 и F'. Поэтому иопы Н‘ могут проходить через такую плёнку и разряжаться на катоде. Если.в растворе одновременно содержатся посторонние ионы, например HSOJ или F', то некоторая часть их проходит через пленку вместе с ионами Н‘. Кислота, достигшая катодной стороны пленки, разрушает ее, так что она становится пористой. Проникающие в поры ионы HCrOJ, которые тотчас л;е восстанавливаются на катоде до [Сг(ОН)]а+, закупоривают их вновь, а проникающая кислота дополнительно растворяет пленку и так далее. Так на поверхности катода скапливаются ионы Сг‘“, а иопы Н' расходуются в реакции с группами [Сг(ОП)р+. В результате при достаточно высокой плотности тока наряду с разрядкой ионов ГГ происходят одновременная разрядка ионов Сг'”, приводящая к выделению металлического хрома. Впрочем, условием для этого является не слишком высокое содержание кислоты в растворе: в противном случае происходит только выделение водорода . Поэтому к хромовой кислоте добавляют обычно не серную кислоту, а сульфат хрома. Получить компактные и прочные хромовые покрытия электролизом свободных от хромовой кислоты растворов сульфата хрома значительно труднее, чем электролизом растворов, содержащих хромовую кислоту. Образование пленки хромата хрома, которая затрудняет выделение хрома в отсутствие посторонних ионов (HSO4 или F'), в присутствии таких ионов приводит к образованию особо мелкокрпсгалличоского и поэтому плотного, прочного и блестящего храмового покрытия, поскольку покрытие катода плёнкой снижает скорость роста отдельных кристаллов и, напротив, способствует образованию центроз кристаллизации.
Свойства. Хром представляет собой белый, блестящий, твердый J но хрупкий металл удельного веса 7,2. Он имеет высокую температуру плавления (около 1830") и относительно низкую по сравнению с температурой плавления температуру кипения (примерно 2300° при атмосферном давлении).
При обычных температурах хром химически чрезвычайно устойчив. Он почти пе окисляется на воздухе даже в присутствии влаги. При нагревании окисление протекает только на поверхности. В токе кислорода он сгорает, разбрасывая искры, так же как и при нагревании с хлоратом калии или селитрой. Хром непосредственно соединяется также с галогенами,, серой, азотом, углеродом, кремнием, бором и некоторыми металлами, однако только при нагревании. Содержащий углерод хром еще более ctoehl и тверд, чем чистый металл. Хром растворяется в водных рястворят соляной или серной кислот, причем при соблюдении некоторых условны очень легко._Напротнв, азотная кислота — концентрированная или раэ-
Шестая группа периодической системы
145
бавленная — и царская водка на холоду на хром вообще не действуют. При кипячении хрома с этими кислотами наблюдается очень слабое взаимодействие, причем реакция тотчас прекращается, как только прекращается нагревание.
Своеобразная устойчивость хрома по отношению к этим кислотам объясняется тем, что они переводят металл в состояние особой нереак-ционноспособности, называемое состоянием пассивности.
Явление пассивирования окне ап о уже для других металлов. В случае хрома оно было обстоятельно изучено. Изменение химического характера хрома при обработке названными веществами отчетливо проявляется в том, что после такой обработки он более не растворяется в разбавленной соляной иля серной кислоте, даже если его кипятить с этими кислотами. Если кипячение вести достаточно долго, то можно наблюдать переход пассивного состояния в активное. В большинстве случаев такой переход происходит внезапно: вначале выделение водорода еле заметно, но вдруг оно начинается с такой силой, что вся жидкость вспенивается. Если в этот момент горячую кислоту заменить холодной, обильное выделение водорода продолжается. Но если соляную или серную кислоту заменить разбавленной или концентрированной азотной кислотой, выделение водорода сразу прекращается: хром вновь становится пассивным. Этот процесс можно повторять любое число раз.
В активном состоянии хром, отдавая электроны, легко переходит в раствор в виде положительного иона. В электрохимическом ряду напряжении хром стоит между цинком и железом. (Нормальный потенциал полностью активного хрома в контакте с раствором соли хрома(П) равен по отношению к нормальному водородному электроду —0,56 в.) Поэтому хром способен вытеснять многие металлы, например медь, олово, никель, из растворов их солей. Он способен также вытеснять водород, на чем основана растворимость хрома в нормальном состоянии в кислотах. Явление пассивирования сдвигает хром в ряду напряжений значительно правее, так что сильно пассивированный хром по своей упругости растворения оказывается позади золота и платины* **. Следовательно, в пассивном состоянии он ведет себя как типичный благородный металл. Однако обычно приходится иметь дело не с чисто активным или пассивным хромом, так что практически упругость растворения хрома представляет собой очень мало определенную величину.
Кроме азотной кислоты и царской водки, пассивирование хрома вызывается также всеми другими сильными окислителями. Разные по силе окислители пассивируют с разной силой. Небольшое пассивирование вызывается уже кислородом воздуха.. Поэтому очень трудно сохранить металл в чистом активном состоянии. А ктивирующим действием обладают выделяющие водород кислоты, например соляная кислота. В меньшей степени активируют хром растворы многих солей, некоторые расплавленные соли и многие органические вещества. Хром сильно активируется, если на нем электролитически выделяется водород или если его выделяют из растворов солей электролитическим способом.
Существенное различие между активным и пассивным хромом проявляется при анодном растворении под действием электрического тока. Активный хром переходит в раствор в виде ионов Ст", т. е. в низшем валентном состоянии, в котором он может существовать пассивный хром при растворении образует ноны CrOJ, т. е. находит-:
* Для сильно пассивированного хрома нормальный потенциал был найден равным 4-1,2 в по отношению к нормальному водородному электроду. При сравнении с соответствующими значениями для золота и платины следует учесть, что у этих металлов упругость растворения повышена за счет склонности к образованию комплексных ионов. Поэтому в контакте с аналитически эквимолярными растворами золото и платина заряжаются менее положительно, чем сильно пассивированный хром, Хотя их потенциалы, отнесспиые к концентрациям свободных элементарных ионов, все еще выше, чем потенциал хрома.
** Плотность тока должна быть очень небольшой, так как если на аноде хотя бы и в ничтожной мере начнется выделение кислорода, то немедленно произойдет пассивирование.
10 Г. Реми
146
Глава 5
ся в вметаем присущем ему патентном состоянии. За исключением химического, соответственно электрохимического, поведения, активный и пассивный хром заметно не различаются. Различие, возможно, наблюдается в отражении света; однако это еще окончательно пе установлено.
О теориях, объясняющих явление пассивирования, см. на стр. 790 и сл. Хром, выделенный электрохимически из водных растворов его солей, способен поглощать значительные количества водорода (при обычной температуре до 60, при —50’ до 300 об. Н2 на 1 об. хрома). Возможно, что при этом образуется своеобразный пересыщенный твердый раствор водорода в хроме. Если вести нагревание в вакууме, то при температуре около 60’ основное количество водорода необратимо выделяется. Чтобы достигнуть полной дегазации, хром следует нагревать до 600°. При растворении водорода тип кристал лической решетки хрома пе изменяется — решетка только расширяется (Huttig, 192 а). Гидрид стехиометрического состава СгН3 можно получить, согласно Вайхс.ель^ельдеру (Weichselfelder), действием водорода на эфирный раствор трихлорида Сп',13, содержащий фенплмагнийбромид. До сих пор еще пе доказано, что в данном случае действительно образуется истинное химическое соединение.
Хром в очень топко измельченном состоянии, полученный, например, ив амальгамы отгонкой ртути в вакууме, самовозгорается на воздухе. Коллоидный, хром можно получить электролитическим распылением под изобутиловым спиртом или попеременной обработкой очень тонко измельченного металла разбавленными кислотами и щелочами.
Применение хрома и его соединений. Металлический хром применяют в основном в сталелитейной промышленности. Хромовые стали отличаются твердостью и прочностью, и поэтому их употребляют для изготовления инструмента и частей машин, несущих особо сильную нагрузку, например шариков в шарикоподшипниках. Поскольку слишком высокое содержание углерода для таких сталей вредно, для получения наилучшей продукции нельзя применять феррохром, выделяемый из хромистого железняка восстановлением углем (поскольку обычно он содержит большой процент углерода), а следует использовать хром или феррохром, получаемые алюмотермическим способом. Часто для дальнейшего улучшения качества хромовых сталей в них вводят третий металл, например никель или вольфрам.
И у многих других сплавов, например бронзы, латуни, во много раз возрастает твердость при добавлении к ним хрома. Сплав железа, никеля и хрома в виде проволоки («нихромовая проволока») используют для нагревательных элементов электрических печей.
Поскольку хром обладает наивысшей твердостью из всех используемых в промышленности металлов, он более всего подходит для получения защитных покрытий. Электролитическое хромирование быстро нашло применение, прежде всего в автомобильной и велосипедной промышленности, а также при изготовлении медицинского инструментария. Но и во многих других случаях хромирование все в большей степени вытесняет применявшееся ранее лужение или никелирование.
Еще большее применение, чем металлический хром, находят соединения хрома. Соли хрома(Ш) и хроматы используют в красильной промышленности как протравы. В кожевенном производстве они служат для изготовления прочной хромовой кожи. Соли хрома(П) применяют при кубовом крашении, так как они являются отличными восстановителями. Хроматы и бихроматы относятся к наиболее распространенным сильным окислителям. Их применяют в больших количествах прежде всего при производстве органических краеок, а также при фотопроцессе, так как они обладают свойством переводить желатину и гуммиарабик, подвергшиеся действию света, в нерастворимое в воде состояние (пигментное печатание, гумми печатание). Многочисленные соединения хрома находят применение в качестве минеральных красок. Достаточно упомянуть
Шестая группа периодической система
147
только хромовую желтую (хромат свинца), хромовую красную (основной хромат свинца), хромовую зеленую [окись хрома(III)], цинковую желтую (двойная соль хромата цинка и бихромата калия, 3ZnCrO4'K2CraO?), цинковую зеленую (смесь цинковой желтой и парижской голубой).
Хромовые краски отличаются красотой, устойчивостью к свету (выцветание^ и устойчивостью на воздухе, а часто и очень высокой кроющей способностью. Их применяют не только как кроющие краски пли в живописи, но й для раскраски обоев, в печатании, далее при производстве клеенки и линолеума, а также для раскраски фарфора. При производстве и употреблении хромовых красок следует иметь в виду их ядовитость (ср. стр. 170 и 173).
Для изготовления хромовых красок и хромовых дубителей необходимы соли хрома, совершенно не содержащие ванадия. Обычно это условие не выполняется, так как применяющийся как исходное сырье хромистый железняк, как правило, содержит ванадий. Из хроматных растворов, полученных вскрытием хромистого железняка, ванадий удаляют, по Перрону (Perrin, 1952), перемешиванием со шламом сульфата свинца. При этом ванадий выпадает в осадок в виде ванадата свинца (вместе с некоторым количеством хромата свппца). Осадок можно дальше перерабатывать для получения ванадия.
СОЕДИНЕНИЯ ХРОМА
Соединения двухвалентного хрома малоустойчивы. Наиболее устойчивы производные трехвалентного хрома. Очень устойчивые соединения образует и шестивалентный хром. Впрочем, эти соединения восстанавливаются до соединений трехвалептного хрома довольно слабыми восстановителями, такими, как иодистый водород, сероводород, спирт. При нагревании до красного каления они большей частью разлагаются с выделением кислорода. Лишь в очень немногих соединениях хром является четырех- и пятивалентным, а еще реже — одновалентным.
Трехвалентный хром имеет очень большую склонность к образованию комплексов. Связанные в них атомы или радикалы часто не отщепляются в растворе, так что растворы соединений хрома(Ш) не всегда дают реакции, характерные для ионов хрома(Ш). Сами ионы хрома(III) всегда существуют в водных растворах в гидратированной форме. Шесть молекул воды при гидратированном ионе особенно прочно связаны с хромом, так что они переходят в соли, кристаллизующиеся из таких растворов. Обычные соли хрома(Ш), т. е. соли общего состава Сг(ОНй)0Х3, как и их растворы, фиолетового цвета. Если же соли хрома или их растворы имеют окраску, существенно отличную от фиолетовой, то это всегда является признаком того, что это не чистые аквокомплексы хрома(Ш), а комплексы, в образовании которых принимают участие (в данном случае наряду с водой) другие лиганды.
Обычные гидратированные соли хрома(Ш) очень похожи, за исключением цвета, на соли алюминия.
Например, хром образует двойные сульфаты типа квасцов. Как и соли алюминия, соли хрома(Ш) в водтшх растворах подвергаются значительному гидролизу, причем даже сильнее, чем соли алюминия (о протекании гидролиза см. на стр. 304 и сл.). Сходство распространяется также на другие соединения хрома(Ш) и алюминия. Так, окись хрома(П1) изоморфна корунду и образует с ояислами двухвалентных элементов двойные соединения, относящиеся к группе шпинелей. Сходство между соединениями хрома(П1) и алюминия, выражающееся также в совместном распространении обоих элементов в природе (ср. стр. 142), обусловлено тем, что хром(ПГ) подобен алюминию не только по валентности, но и имеет близкий по величине кажущийся ионный радиус (гСгз+ 0,65 А, Гд!з+ = 0,57 А).
Но хром(Ш) имеет гораздо большую, чем алюминий, склони ость к образованию комплексных соединений. Почти во всех таких соединениях
10*
148
Г лапа 5
координационное число хрома равно 6. Комплексные соединения очень разнообразны, часто ярко окрашены.
Простые соединения хрома, нерастворимые в воде, в большинстве случаев обладают интенсивно!! окраской. Так, окись хрома Сг2О3 интенсивно зеленого цвета; сульфид хрома Cr2S3 черный, нерастворимый в воде; безводный трихлорид хрома СгС13 красно-фиолетового цвета и т. д.
Окись хрома(ТП) Сг2О3 амфотерна, и соли, которые она образует с кислотами, очень склонны к гидролизу; соли, которые она образует с сильными щелочами, гидроксохроматы{111), менее устойчивы, чем соответствующие соли алюминия, гидроксоалюминаты. Окись xpoMa(VI) СгО3 является типичным ангидридом кислоты. Соответствующая ей (известная лишь в растворе) хромовая кислота [точнее, хромовая(У1) кислота] Н2СгО4 очень склонна к полимеризации. За счет удаления воды могут образовываться дихромовая (двухромовая) Н2СгаО7, трихромовая Н2Сг3О10 и тетрахромовая H2Cr4OJ3 кислоты. Отвечающие названным кислотам соли [хроматы(У1), называемые просто хроматами], и прежде всего (момо-) хроматы MJ,[CrO4] и бихроматы М|[Сг2С7], в отсутствие восстановителей и при обычных температурах, а иногда и при небольшом нагревании очень устойчивы. Однако до отношению к веществам, которые легко могут отдавать электроны или принимать кислород, эти соли, особенно в кислом растворе, являются энергичными окислителями, причем сами они переходят в соединения хрома(Ш) (ср.вт. I, стр. 820 окислительный потенциал ионов HCrOj) Из солей хромовых кислот бихромат калия К2Сг3О7 отличается своей исключительной способностью к кристаллизации.
Для хрома известен также «промежуточный окисел». Этот окисел имеет состав Сг3О8 (ср. стр. 171), аналогичный составу закиси-окиси урана tl3O8, и его, по-видимому, можно рассматривать как двойное соединение СгО3 и Сг2Ой (см. стр. 219). Раньше промежуточным окислом считали соединение СгО2 — его принимали за соединение окисей хрОма(Ш) и хрома(У1) Сг2О3-СгО3. Одиако, как оказалось, это соединение представляет собой простую окись хрома(1У) (подробнее см. пиже).
Из соединений пятивалентного хрома в первую очередь следует упомянуть пят и окись хрома Сг2О5 (ср. стр. 171), огненно-красный, легко летучий пентафторид CrF5 (Wartenberg, 1941), дионсифгорид CrO2F (Olason, 1924) и полученные Шарпом и Вольфом (Sharpe, Woolf, 1951) оксофторохроматы(У) KJCrOFJ и AgfCrOFJ. Еще раньше Вейплапд (Welaland, 1905 г. и последующие года) действием очень концентрированной соляной кислоты при низкой температуре на трехокись хрома в присутствии хлоридов щелочных металлов получил оксохлорохроматы(У) М| [CrOGLJ. Цезиевая соль этого ряда образует смешанные кристаллы е оксохлор ониобатом цезия Cr2[NbOCl5]. Оксохлорохроматы(У) типа M^CrOClJ (М1—пиридиний или хинолиннй) были получены уже в 1899 г. Мейером (Meyer R. Г.). Соединениями хрома(V) являются и «красные пер оксохроматы» (см. стр. 177). В соответствии с данными Клемма (Klemm, 1936), полученные Генном (Hein, 1919 и сл.) реакцией Гриньяра хромфенильные соединения, например [Cr(CeH5)3H]X (X — одновалентный кислотный остаток), также следует рассматривать как соединения хрома(У). Термическим разложением хроматов (VI) в присутствии основных окислов металлов Шольдер ((Scholder, 1953) получил такие хроматы(У), как Li3CrO., Sr3(CrO4)2 и Ва3(СгО4)2. Им были получены также хроматы(У) типа гидроксил апатита, например Sr5(piI)(CrO4)3. Нагреванием щелочного бихромата под давлением с СгО3 до 350* в атмосфере кислорода Фанкухен и Уорд (Fankuchen, Ward, 1952) получили соединения состава М1Сг3О3. Некоторые их свойства свидетельствуют о том, что эти соединения следует рассматривать как трихрома-ты(У) щелочных металлов.
Из соединений четы/ютвалептпого хрома известны, кроме полученного Гейном «свободного радикала» Сг(СеНй)4 (ср. стр. 772), фторид CrF4 и хлорид СгС14. Последний, впрочем, может существовать тол [.ко в газообразном состоянии в присутствии избытка Cl2 (Doerner, 1937; v. Wartenberg, 1942). Электроположительный четырсхвалептпып хром присутствует также в окисле СгОа, как это следует из структуры (см. стр. 171) и магнитных свойств соединения (Bhatnagar, 1939). Этому окислу соответствуют
Шестая группа периодической, системы
149
а:/)О.квтм(/У) гцелочноземелъных металлов (например, ВаСсО4), полученные разнообразными методами Шодьдером (1952—1953). Действием F2 на смесь CrGl3 и КС1 Клемм (Klemm, 1950) получил соединение K2(CrFG], которое также содержит xpOM(IV), поскольку опо имеет такую же рентгенограмму, как К2[МпРе].
Одновалентный хром присутствует в полученном Гейпом (1952) соединений [CrDipy3|C104, где Dipy — eta'-дипир и ди л. Оно было получено восстановлением (CrDipy3](C104)2 в водном растворе в виде черно-синего быстро окисляющегося на воздухе порошка. Это соединение нерастворимо в воде, но легко растворяется во многих органических растворителях, например в метиловом и этиловом спиртах, ацетоне, дйоксапе н пиридине. То, что хром в этом соединении существует в: одновалентном состоянии, следует, кроме ого состава, также из величины магнитного момента. Полученное значение (2 магнетона Бора) показывает, что в этом случае мы имеем дело с соединением, представляющим собой комплекс внедрения. В соединении [CrDipy3j(C104)a также имеется комплекс внедрения (магнитный момент: экспер. 2,9, рассч. 2,8 магнетона Бора).
Хля хрома известны также металлорганические соединения типа R3CrX, R4CrX и RjCrX (R — фенил, X — ОН, галоген или 1 экв. кислотного радикала), В соответствии с данными Гейна, эти соединения легко получаются взаимодействием СгС13 с фопилма гний бромидом. Гидроокиси три-, тетра- и пентафенилхрома имеют сильный основной характер. Соответствующие им соли, как и тетрагидрат гидроокиси пеп-тафени.тхрома (СеН5)йСгОН.4НаО, легко получаются в кристаллическом виде.
В щелочном растворе соли ггролм(1П) легко окисляются до хроматов [хроматов(У1)], например хлором, бромом, гипохлоритами, перекисью водорода, а кроме того, двуокисью свинца или свежеосаждениым гидратом двуокиси марганца. В кислом растворе окисление происходит под действием смеси азотной кислоты и хлората калия, или персульфата калия (в присутствии соли серебра как катализатора), или при электролизе.
Хроматы, [хроматы(У1)] в кислом растворе восстанавливаются до солей хрома(Ш) такими восстановителями, как сернистая кислота, сероводород, иоднетый водород, щавелевая кислота, спирт, а при нагревании также бромистым водородом или концентрированной соляной кислотой. При действии перекиси водорода в кислом растворе также происходит восстановление (при этом в качеств промежуточного продукта образуется перекись хрома, ср. стр. 177).
Соединения хрома(П)
При растворении в кислотах хром сначала переходит преимущественно в двухвалентное состояние:
Сг+2Н-^Сг"4-Н2.
Но поскольку ионы хрома(П), приобретая еще один положительный заряд, легко переходят в ионы хрома(1П), то при растворении хрома в кислотах с доступом кислорода получают соли трехвалентного хрома,
И в отсутствие кислорода воздуха некоторая часть иопов хрома (II) получает дополнительный положительный заряд от иопов водорода, причем устанавливается равновесие:
сгч-н- ;±Сг"-+1/2н2. (1)
Положение равновесия, кроме концентрации ионов водорода и температуры, зависит также от природы присутствующих в растворе анионов. В большинстве случаев оно значительно сдвинуто вправо. Поэтому при растворении хрома в кислотах не так-то легко получить растворы солей хрома(П).
Чистые соли хрома(П) можно получить восстановлением цинком в кислом растворе солей хрома(Ш) * при условии, что происходит обиль
* Фиолетовые соли хрома(Ш) легче восстанавливаются до солей хрома(Н), чем зеленые. Вместо солей хрома(Ш) можно исходить из солей хрома(у!) (хроматы и бпхроматы).
150
Глава S
ное выделение водорода. В отсутствие катализаторов взаимодействие ионов Ст'* с ионами Н' по уравнению (1) протекает медленно. Растворы солей хрома(П) можно получить также электролитическим восстановлением солей хрома(Ш) в водном растворе на свинцовом катоде, окруженном глиняным цилиндром.
Безводные соли хрома(П) удобнее всего получать непосредственным соединением компонентов. Хлорид и бромид целесообразнее всего получать действием газообразного хлористого или бромистого водорода на нагретый металл.
Растворы солей хрома(П) окрашены в небесно-голубой цвет. Содержащие воду соли хрома(П) и сильных кислот в большинстве случаев также голубого цвета, тогда как безводные соли и соли слабых кислот окрашены в разнообразные цвета.
Реакции солей хрома(11) очень похожи на реакции солей железа(П). Соли хрома(П) осаждаются при действии тех же реактивов, что и соли железа(П). Как и в случае солей железа(П), выпадению осадка, начинающемуся при добавлении аммиака, мешают соли аммония.
Растворы солей хрома(П), как и растворы солей железа(П), поглощают окись азота, однако не просто присоединяют ее, а восстанавливают в кислом растворе до гидр окси л амина, а в нейтральном до аммиака. Вообще соли хрома(П) отличаются от солей железа своим сильным восстанавливающим действием. Для хрома склонность к переходу из двухвалентного состояния в трехвалентное выражена гораздо сильнее, чем для железа.
Мерой тенденции иопов хрома(П) переходить в ионы хрома(Ш) служит их о»ис-лителъно-восстановителъный потенциал. Он составляет приблизительно —0,4 в. Это значит, что платиновый электрод, погруженный в раствор, содержащий оба вида ионов в концентрации 1 М, приобретает потенциал примерно на 0,4 е меньший, чем потенциал нормального водородного электрода. В замкнутой цепи ток идет от водородного электрода по соединяющей проволоке к электроду, погруженному в раствор хромовых солей. Здесь иолы Сг" заряжаются до ионов Сг‘", в то время как на первом электроде разряжаются ионы Н*. В противоположность этому платиновый электрод, погруженный в раствор ионов Fe"H Fe‘", имеет потенциал белее высокий, даже значительно более высокий (ср. т. I, стр. 820), чем нормальный водородный электрод.
Двухвалентный хром (в противоположность двухвалентному железу) способен образовывать карбонатные соли. Одной из наиболее устойчивых солей хрома(П) является оксалат хрома(И) СгС304- Н2О, получающийся в виде желтого кристаллического порошка при кипячении ацетата хрома(П) с раствором щавелевой кислоты. Известен и еще ряд солей хрома(П) с органическими кислотами. Из них для препаративных целей важен красный ацетат хрома(П).
Окись хрома(П) СгО образуется при свободном окислении хрома, растворенного в ртути, на воздухе. Если СгО нагревать при доступе воздуха, то опа, самораскаляясь, переходит в окись хрома(Ш) Сг2О3. При сильном нагревании в токе водорода она в пр отнв оп о ложность Сг2О3 восстанавливается до металла.
Сульфид хрома(П) CrS получается непосредственно из компонентов с содержанием серы в пределах 50—54 ат.%. Он имеет структуру, аналогичную структуре NiAs, однако прн составе, отвечающем формуле CrS, в сульфиде хрома(П) не все узлы решетки заняты атомами. При занятии атомами всех узлов S-решетки (при составе, отвечающем 54 ат.% S) соединение становится ферромагнитным (Haraldsen, 1937). CrSc* и СгТе также кристаллизуются по типу NiAs
Хлорид хрома(П) СгС12, соответственно Сг2С14, лучше всего получать пропусканием газообразного хлористого водорода над металлическим хромом, нагретым до ярко-красного каления [уравнение (2)], или пропуская водород над нагретым до красного каления безводным хлоридом хрома(Ш) [(уравнение (3)1
Сг 4- 2ИС1=СгС124- И2. (2)
СгС13-|-1/аН2 = СгС12+НС1. (3)
Шестая группа периодической системы
151
Он представляет собой белые иглы с шелковистым блеском, уд. веса 2,88. В парообразном состоянии даже при температурах выше 1500° в значительной степени образует двойные молекулы Сг2С14. В сухом воздухе хлорид хрома (II) довольно устойчив. Напротив, в присутствии влаги жадно поглощает кислород, образуя Сг2ОС14. Хлорид xpoMa(Ll) гигроскопичен. Оп растворяется в воде со значительным выделением тепла (18,6 ккал). Из васильково-голубых растворов, которые проще всего получать введением цинка в сильнокислый раствор х.торида хрома(Ш), можно получить кристаллический гидрат, состав которого в наиболее богатой водой форме отвечает формуле СгС13-6Н2О. Этот гидрат кристаллизуется при 0° и имеет голубой цвет. При комнатной температуре кристаллизуется голубой тетра гидрат, при температурах около 50°— темно-зеленый гидрат, изомерный первому. Обезвоживанием темпо-голубого тетра-гидрата можно получить светло-голубой тригидрат и светло-зеленый дигидрат. Последний при температурах выше ИЗ’ переходит в бесцветную безводную соль. '
С аммиаком Хлорид хрома(П) образует соединения CrCl2-6NII3 (темно-голубой), СгС12-5NHs (фиолетовый), CrCl2-3NH3 (светло-голубой), CrC12-2NH3 (светло-зеленый) и CrCl2-NH3 (светло-зеленый) (Schlesinger, 1933; Ephraim, 1934). Отщепление последней молекулы аммиака происходит при температуре —400“. Необычной устойчивостью отличается соединение хлорида хрома (II) с гидразином СгС12* 2N2II4, которое выделяется в виде осадка сиреневого цвета при добавлении водного раствора гиДразинги-драта к раствору хлорида хроиа(Л) [или к раствору ацетата хрома(11) в разбавленной соляной кислоте (ТгапЬе, 1913)).
Аналогично хлориду хрома(П) можно приготовить зеленый фторид (т. пл. 1100°) и белый бромид (уд. вес. 4,36). Красно-коричневый иодид (уд. вес. 5,02) получается, согласно Гейну (Hein, 1931/1943 г.), при непосредственном соединении Сг с 12 около 800°. Эти галогепады двухвалентного хрома, за исключением фторида, легко растворимы в воде.
Сульфат хрома(П) п двойные сульфщты. Из приготовленных без доступа воздуха растворов хрома в разбавленной сер пой кис лоте пли из восстановленного цинком раствора сульфата хрома(П1) при испарении кристаллизуется голубой гептагидрат сульфата хрома(11| CrSO4-7HaO, который изоморфен железному купоросу. При электролитическом восстановлении сильно серпокислых растворов на катоде кристалли-вуется бесцветный моногидрат CrSO4-H2O.
С сульфатами щолочпых металлов сульфат хрома(П) подобно сульфату железа(П) образует двойные сульфаты типа MjCr(SO4)3-6H2O.
По даппым Стоуна (Stone, 1936), раствор сульфата хрома(Н) в разбавленной серной кислоте является превосходным поглотителем кислорода.
Ацетат хрома(II) Сг(С2Н3О2)2 лучше всего получать, вливая раствор хлорида хрома(II) в достаточно концентрированный, насыщенный двуокисью углерода раствор ацетата натрия. При этом он выпадает в виде красного осадка, мало растворимого в холодной воде, но легко растворяющегося в разбавленных сильных кислотах. Из-за простоты его способа полу-чення ацетат хрома(Н) является удобным исходным материалом дли приготовления других солей хрома (И),
Комплексные соединения хрома(П). Ион хрома(П) стабилизуется при присоединении нейтральных аддеидов. Например, полученные Гейном (1943—1952) соединения [Gr(N2H4)n)I2 и [GrDipy3](C104)2 (Dipy = аа'-дипиридил) в противоположность простым солям хрома(П) в сухом состоянии совершенно устойчивы па воздухе. Чернофиолетовый перхлорат [Сг Dipy3](G104)2, кроме того, негигроскопичен. Его виппо-красные растворы, как и все растворенные в воде соли хрома(11), легко поглощают кислород, причем хром(И) переходит в хром(Ш). С другой стороны, перхлорат в водном растворе легко восстанавливается до соответствующей соли хрома(1), причем цвет раствора переходит в глубокий темно-синий (см. стр. 149). Образование ионов (Or Dipy3i’ из ионов (Сг Dipy3(’' может происходить также за счет окислительно-восстановительной: реакции:
2 [Сг 01ру8)"=[Сг Dipy3]*4-[Cr DipyJ—.
Эта реакция протекает до конца при повышении концентрации ионов ОН', Образующиеся в результате ное ионы [Сг Dipy3|"‘ тотчас же переходят в красные иопы [Cr(OH)(OH2)Dipy2l" (см, стр. 165).
Соединения хрома(Ш)
Окись хрома(Ш), окись хрома, полуторная окись хрома СггО3 в обычной форме представляет собой нерастворимый в воде зеленый порошок.
152
Глава 5
Кристаллическая окись хрома(1П), получающаяся При повышенных температурах, черного цвета, имеет металлический блеск, в большинстве случаев с цветным отливом на поверхности. При растирании она переходит в зеленый порошок. Кристаллическая окись хрома(Ш) изоморфна корунду (см. т. I, рис. 75); а0 = 5,33 А, а = 55°17', dpenTT = 5,21,
Окись хрома(Ш) можно получить многими способами. Обычно при ее получении исходят из легко доступных в чистом состоянии хроматов или бихроматов. В большинстве случаев их восстанавливают прокаливанием с древесным углем (см. стр. 143) пли серой:
K2Cr2O7+S=Cr2O54-KaSOil,
Восстановление происходит и при прокаливании с нашатырем,, поскольку образующийся в этом случае за счет двойного обмена бпхромаг аммония разлагается до окиси хрома(П1).
Кроме уже упоминавшегося применения окиси хррма(Ш) для получения металлического хрома, ее широко используют для получения мае л fl-пых и акварельных красок (хромовая зеленая). Так как эти краски обладают термо- и влагоустойчивостыо, окопные ставни, заборы, садовые скамьи и части станков и машин часто окрашивают хромовой зеленой. Хромовую зеленую часто используют в книгопечатании^ литографии, стекольной * и гончарной промышленности, а также для живописи по фарфору.
Прокаленная окись хрома(Ш) с большим трудом растворяется в кислотах. Однако она легко переходит н раствор при нагревании с раствором щелочного бромата. При этом идёт реакция 5СгаО3 + BBrOi Д- 2НаО = 5Gr3O, -j- 41Г -Ь ЗВ1\. Образующиеся ионы Н‘ каталитически ускоряют реакцию. Растворение в присутствий хлоратов щелочных металлов происходит значительно медленнее (Lyden, 19.35).
Если окись хрома(Ш) получают обезвоживанием гидрата, то при отщеплении последних количеств воды наблюдается саморавогрев (на 1 моль СгаОа выделяется 23 ккал). На основании снятой дебаеграммы Бом (Bohm) с.мог показать, что и основе этого явления лежит уменьшение поверхности при превращении аморфной окта в кристаллическую, подобное таким же процессам, происходящим в случае окислов титана и циркония.
Гидрат окиси хрома(Ш) Сг2О3-.гН2О, содержащий переменное количество воды (обычно называемый гидроокисью. Хрома), выпадает при добавлении Понов ОН' к растворам солей хрома(Ш). Это очень мало-растворимый в воде осадок в свежем состоянии серовато-голубого цвета, имеет типичные свойства геля и легко образует коллоидные растворы, например, при пептизации хлоридом хрома.
Как и Другие гели, Гидрат окиси Хрома обладает сальной поверх постной активностью. Поэтому при осаждении из щелочного раствора он обычно адсорбирует значительные количества щелочи. Чтобы получить его в чистом виде, осаждение следует производить такими веществами, которые не образуют сильнощелочиых растворов или снижают концентрацию попов водорода в растворе лишь настолько, чтобы нарушилось равновесие гидролиза соли хрома;
2С1гХ3-|-ЗНОН Сг2О3 + 61Г-|-6Х',
Удобны для этого, например, нитрит аммония, карбонат цинка, сульфид цинка или смесь иодида и йодата цалия (ср. т. I, стр. 868—869). Осаждение аммиаком мепее пригодно, так нац при его избытке легко образуются комплексные аммиакаты, не осаждающиеся из раствора,особенно ед ля в растворе содерлщгея много солей аммония.
* Окись хрома окрашивает стекло в прекрасный изумрудно-зеленый цвет. Если в стекле содержится избыток окиси хрома, то при охлаждении она выделяется в виде микроскопических зеленых кристалликов (хромо-авентуриповое стекло).
Шестая группа периодической систему
153
Как и все гели, получаемые осаждением, гели окиси хрома(Ш) обладают свойством «стареть», т. е. переходить в менее поверхностно активную и менее реакционноспособную форму. Старение происходит уже при соприкосновении с водой. Точно также стареют коллоидные растворы окиси хрома, особенно в присутствии щелочи. Поэтому при нагревании из таких растворов очень скоро выпадает большая часть гидрата окиси хрома. Застаревший гидрат окиси хрома значительно труднее, чем свежий, растворяется э кислотах и особенно в щелочах.
Гидрат окиси хрома растворяется в кислотах с образованием обычных солей хрома, в щелочных растворах — с образованием хроматов(НТ) (ранее называвшихся «хромитами»), т. е. солей, в которых трехвалентиый хром входит в состав аниона. Следовательно, гидрат окиси хрома, как и гидрат окиси алюминия, амфотерен.
Как показали многочисленные исследования (Muller, 1922; Fricke, 1924, п Schoider, 1934), в концентрированных растворах щелочен существуют гидроксохро-мат(Ш)-ноны [Сг(ОИ)е]'"", [Cr(OH)7]"", [Сг(ОП)е]'". Соли этих анионов можно получить в кристаллическом виДе. В то время как гидр оке охр оматы(Ш) щелочноземельных металлов устойчивы по отношению к воде, гидроксохроматы(Ш) щелочных металлов легко разлагаются водой с выделением гидрата окиси хрбма(Ш). Поэтому при разбавлении растворов эти соли распадаются. В разбавленных щелочных растворах, как было показано Герцем (Herz W.) и Фишером (Pisher Н. W.), гидрат окиси хрома^!!) присутствует в коллоидной форме." он яе диффундирует черев пергаментную мембрану, а электрические свойства таких растворов заметно пе меняются, даже если хром осажден из пих нагреванием.
Гидрокеохромат(Ш) бария Ва3[Сг(ОН)о]2 при нагревании его в смеси с гидроокисью бария в токе ларов воды при температуре —-800° окисляется до хрома та (IV) бария Ва2СгО4 соответственно до двойной окиси бария и Хрома(1У) 2ВаО-СгОг (Schoider, 1952). То. что в соединении Ва2СгО4 хром находится в положительно чстырет-валентном состоянии, было подтверждено Клеимом нри помощи магистохимических исследований. Образование двойного соединения 2ВаО-СгО2 — производного «аномальной валентности хрома» — очевидно, возможно только потому., что СтО2 и ВаО образуют кристаллическую решетку, отличающуюся особой устойчивостью; Шольдер показал, что при надлежащем выборе партнеров реакции можно получить двойные окисли других элементов (например, Мп, Fe, Со), в которых эти элементы существуют в «аномальней валентности». Получаемые таким способом двойные окисли всегда Отличаются особой устойчивостью.
Сплавлением окиси хрома(Ш) с окислами более основных металлов (в большинстве случаев двухвалентных) получают ряд хорошо кристаллизующихся двойных соединении, как правило, состава МПО Сг3О3. Раньше эти соединения принимали за хроматы(Ш) и приписывали им формулу Мп|СгОг/г. С кристаллографической точки зрения все эти соединения принадлежат к классу шпинелей. Так как по строению они полностью аналогичны шпинели MgO-Al2O3, то рассматривать их в качестве «хроматовЩ!)», т. е. солей трехвалентного хрома, в которых хром входит в состав аниона, так же мало оснований, как и считать обычную шпинель алюминатом (см. т. I, стр. 395—396). Важнейшим из двойных соединений окиси хрома(Ш) типа шпинели является встречающийся в природе в больших количествах хромит *, или хромистый железняк FeO-Cr2O3.
Истинными безводными хроматами(Ш) являются кристаллизующиеся по тппу Na(HF2) соединения LiCrOo и 1хаСгОг, полученные Рюдорфси (Rudorff, 1954) прокаливанием смеси Сг2О5 с 1я2С03 иди №2СО3, в виде светло-зелёных порошков. Постоянные решетки; а =2,88, с = 14,6 А; соответственно а=2,96, с=15,9 А; йПИКЕ =4,09, соответственно 4,36, г/гентг = 4,29, соответственно 4,39.
ж Минералогическое название хромит нс имеет ничего общего с устаревшим химическим названием «хромит» для хромата(111). ;
154
Глава -5
Ггогпетова зелень. Сплаваеп и ем бихромата щелочного металла с борной кислотой а выщелачиванием аилу ленные расплава водой получают исключительно красивый зеленый гидрат окиси хрома(Ш), называемый во имени впервые описавшего его приготовление Гюгнета зюгиетовой зеленью или изумрудной зеленой. Его можно получить также нагреванием обычного серовато-голубого гидрата окиси хрома(Ш) в автоклавах при 250° в присутствии веществ, вызывающих конгломерацию. От обычного гидрата окиси хрома(Ш) он отличается тем, что состоит из более крупных частиц (Wohler, 1926), по так ;ке амфотерен, как и первый. Гюгнетову зелень под названием «хромовой зеленой» используют в живописи. Для увеличения ее небольшой кроющей способности ее смешивают с сульфатом бария. Смесь гюгнетовой зелени, сульфата бария и цинковой желтой (см, стр. 147), называемой «зеленая Виктории», применяют для печатания обоев вместо швейнфуртсцой зелени. Аналогичными смесями являются «прочная зеленая», «нюрнбергская зеленая» и «средняя зеленая».
Сульф)ид хрома(Ш) и тиохроматы(Ш). Сульфид хрома(Ш) Cr2S3 нельзя получить из водного раствора, однако он легко получается сухим способом, например при пропускании сероводорода над нагретым до красного каления хлоридом хрома(Ш): 2СгС13 3H2S=Cr3S3 -f- 6НС1 или непосредственно из компонентов. Оп образует черные блестящие листочки, довольно устойчивые к действию кислот, но легко разлагающиеся при действии окислителей (азотной кислоты или расплава селитры).
При непосредственном взаимодействии компонентов получают фазу переменного состава (55—60 ат,% S) и моноклинной структуры, которая при возрастающем содержании серы непрерывно переходит в гексагональную (Haraldsen, 1937).
Для сульфида хрома(Ш) известны хорошо выраженные кристаллические соединения с сульфидами щелочных металлов: тиохроматы(111) щелочных металлов. Например, если сплавлять бихромат натрия с карбонатом натрия в присутствии серы и без доступа воздуха, а затем охлажденный расплав выщелочить водой, то получается тиохромат(Ш) натрия Na[CrSa], имеющий- вид красных гексагональных табличек. Аналогично получают и тиотетрахромат(1П) калия К^Сг^]. Эти соединения исследованы в основном Шнейдером (Schneider, 1897). Кипячением тиохроматов(Ш) щелочных металлов с растворами солей тяжелых металлов Шнейдеру удалось получить также тиохроматы(1П) тяжелых металлов. Это черные или черно-серые порошкообразные, нерастворимые в воде и соляной кислоте вещества, которые, однакб, разлагаются азотной кислотой или царской водкой и тлеют при нагревании на воздухе.
Хлорид хрома(Ш) и комплексы хлорида хрома(Ш). Хлорид хлора{111), трихлорид хрома СгС13 получают в безводном состоянии при пропускании сухого хлора’ пад нагретым до красного каления металлическим хромом (4) или над раскаленной смесью окиси хрома(Ш) и угля (5). Хлорид хрома можно получить также нагреванием окиси хрома(1П) с четыреххлористым углеродом (6) или дитио хлорид ом:
Сг+з/3С!г=СгС134-133 ккал, (4)
Сгг034-ЗС4-ЗС1г=2СгС1з4-ЗСО, (5)
Сг2О34-ЗСС14=2СгС1з4-ЗСОС12, (б)
Безводный хлорид хрома(Ш) образует красно-фиолетовые (окраски «цветущего персика») блестящие листочки. При разогревании до красного каления в токе хлора хлорид хрома(Ш) возгоняется. Если его нагревать в отсутствие хлора, то происходит частичный распад на хлорид хрома(П) и хлор:
СгС13 СгС134-1/2С!2.
При прокаливании на воздухе хлорид хрома(Ш) легко переходит в зеленую окись хрома(Ш):
2СгС134-3/2О2=СггО34-ЗС1г.
Прн действии соответствующих водородных соединений хлор в хлориде хрома(1П) легко замещается серой, азотом или фосфором:
2СгС13+ 3H2S=Cr2S34- 6НС1,
CtC134-NH3=CtN4-3HC1, f СгС1з4-Р1Т3=СтР4-ЗНС1<
Шестая, группа периодической системы
155
В воде и в спирте на холоду безводный хлорид хрома(Ш) не растворяется; даже при кипячении растворение идет чрезвычайно медленно. Но если ввести очень незначительное количество хлорида хрома(И), то происходит быстрое растворение при интенсивном выделении тепла (21,3 ккал/моль СгС13). Аналогичное действие оказывает и добавление таких веществ, которые способны легко восстановить часть хлорида хроца(Ш) до хлорида хрома(ТГ). Получаемый таким способом темнозеленый раствор хлорида хрома(1П) идентичен по своим реакциям тем растворам, которые получаются при окислении воздухом растворов хлорида хрома(П), содержащих избыточную соляную кислоту, а также растворам, получаемым при растворении металлического хрома в соляной кислоте в присутствии воздуха. Такие растворы имеют сладковатый вкус. Если такой раствор охладить после упаривания, то из него выделяются изумрудно-зеленые расплывающиеся кристаллы, обычно содержащие шесть молекул воды на 1 моль СгС13 *. Из свежеосажденного раствора такой зеленой соли после подкисления азотной кислотой и добавления нитрата серебра выпадает только одна треть содержащегося в нем хлора, следовательно, остальные две трети должны быть комплексно связаны. Комплексно связаны, кроме того, четыре молекулы воды, так как Они -остаются в составе соли при высушивании ее в эксикаторе над серной кислотой. Поэтому темпо-зеленому гексагидрату хлорида хрома(Ш) следует приписать формулу** (СгС12(ОН2)4]С1-2НаО {дигидрат дихлоро-тетрааквохром(1 II]хлорида] ***.
В этом соединении хром проявляет координационное число 6, которое характерно для большинства комплексных соединений трехвалептного крома.
Известны еще два других гексагидрата хлорида хрома. В одном из них комплексно связан только одна ион хлора, два других нова освобождаются при электролитической диссоциации. Это открытый Вьеррумом в 1906 г. светло-зеленый гексагидрат. Этой соли, выделить которую па раствора можно только при соблюдении особых условий, приписывают формулу [СгС1(ОН2)5]С12‘Н2О гидрат хлоропонтааквохром(Ш) хлорида. Из ее растворов нитрат серебра тотчас ж® осаждает две трети присутствующего хлора.
Кроме того, существует еще серо-голубой гексагидрат хлорида хрома. Эта соль, из сине-фиолетового раствора которой тотчас же осаждается весь хлор и формула которой, следовательно, [Сг(ОН2)в1С13 [гсксаакво-хром(Ш)], уже давно известна. Ее можно получить из темно-зеленой соли, если долго кипятить разбавленный раствор последней и проводить кристаллизацию при температуре ниже 0° и насыщении раствора хлористым водородом. Эта соль выделяется и из растворов других солей хрома, которые содержат фиолетовые ионы хрома(Ш), т. е. ионы (Сг(ОН2)6]"', например из растворов нитрата хрома(Ш) или хромовых квасцов, если эти растворы насыщать на холоду хлористым водородом. Из трех гекса-гидратов хлорида хрома эта соль наиболее легко кристаллизуется. В серо
* Если раствор концентрируют при температурах ниже 6°, то из него кристаллизуется СгС.13-10Н2О.
** В таких соединениях координационно связанные группы обычно называют в следующем порядке: вначале па звание (неорганического или орг эпического) кислотного остатка, затем нейтральной молекулы, папример Н2О (акво) или NH3 (аммин). Органические нейтральные молекулы называют в последнюю очередь, непосредственно перед названием центрального атома.
*** В названиях координационных соединений часто Опускается одна из рядом-стоящнх одинаковых гласных букв, например дигидрат дихлоротетра«ао(1П)хло-рид.— IfpuMt ред.
156
Глава 5
голубом гексагидрате хлорида хрома все шесть молекул воды связаны координационно. При выдерживании над серной кислотой эта соль пе теряет ни одной молекулы воды.
Три рассмотренных выше соединения, имеющие один и тот же аналитический состав, но различное строение, являются примером того вида изомерии, с которым-наиболее часто приходится иметь дело в химии комплексных соединений. Этот вид изомерий носит название гидратной изомерии. Гидратная изомерия наблюдается у тех координационных соединений, изомерия которых, как в рассмотренном выше случае, объясняется различным типом связи молекул воды.
В растворах устанавливается равновесие между тремя изомерными гидратами хлорида хрома, соответственно между их ионами, причем положение равновесия зависит как от температуры, так и от концентрация и состава растворов. При температуре кипения равновесие устанавливается через несколько минут, тогда как при обычных температурах в некоторых случаях для установления равновесия требуются месяцы. Па холоду и в разбавленных растворах в основном присутствует серо-голубой (в растворе сине-фиолетовый) хлорид, в очень концентрированных растворах — зеленый. При нагревании равновесие также сдвигается в сторону последнего. Однако в разбавленных растворах и при повышенных температурах преобладает фиолетовая форма.
Из серо-голубого, в растворе — сине-фиолетового, хлорида хрома
[Сг(ОН2)в]С13
заменой ионов хлора на другие кислотные остатки образуются многие фиолетовые соли хрома(Ш):
Пон о генно связанные атомы хлора можпо заместить на другие кислотные остатки в светло-зеленом хлориде хрома [СтС1(ОН2)5]С12-Н2О, и это но сопровождается существенным изменением характера соли. Например, заменой этих атомов на остатки хлороплатиновой кислоты [гексахлороплатиновой(1У) кислоты) получают соединение [СтС1(ОН2)5] [PtClgj-SIIgO. Из солей, образующихся при замене в темпо-зеленом хлориде хрома |СгС12(ОН2)4]С1-2Н20 иопогеппосвязаппого атома хлора на другие кислотные остатки, следует упомянуть: [СгС12(ОН3)4]Вг, зеленые легко расплывающиеся кристаллы, и (СгС12(ОН2)4] [SbClg]'6Н2О, зеленые кристаллы.
Комплексы, лежащие в основе обоих зеленых хлоридов хрома [СгС1(ОН2)5р* и [СтС12(ОН2)4)+, получаются из гексааквохром(Ш)-комплекса [Сг(ОН3)6]3+ заменой одной или двух молекул воды па ионы С1_. Возникает вопрос, нельзя ли продолжить такую замену, пока три или болсо атомов хлора не войдут в комплекс вместо Молекул воды. Правда, я кво соединение хрома(Ш) с тремя неионогенносв язаппыми атомами хлора до сих пор неизвестно, но, по-видимому, соответствующую структуру следует приписать красному соединению СгС13-ЗС2Нг;ОИ, полученному Коппелем (Koppel) растворепием металлического хрома в спиртовом растворе соляной кислоты:
г Cl HOC2H6-i
С1 Ст НОС2Н5 .
- С1 НОСгН5 -
В соответствии с такой формулой, это соединение следует называть хлоридом трихлоротриэтанолхрома(111).
Известны многочисленные соединения, в основе которых лежит комплекс с пятью атомами хлора, ней она ген ноев я ванными с хромом. Поскольку в этом случае сумма отрицательных зарядов хлора (пять) больше суммы положительных зарядов хрома (три), комплекс имеет характер аниона. Согласно Вернеру, этот апиоп присутствует в красных двойных хлоридах хгю-на общей формулы СтС13- 2MICL Н3О, которые кристаллизуются из раствора компонентов при упаривании их в токе хлористого водорода. Приписывание этим соединениям формулы пентахлороаквохроматов(1П):
гС1 С1 л
Cl Cr Cl Mj (MT = Li, К, РЬ, Cs, NH4, Т1, »/гВа, i/2Mg)
-Cl OH2J
основано на том наблюдении, что легкорастворимая литиевая соль Li2|CrCls(0H3)] пе дает сразу осадна с солями серебра. Отсюда с.чедует, что в этом соединении весь хлор связан координационно ,и. поскольку хром обычно имеет координационное число 6, падо печатать, что в состав комплекса входит также одна молекула воды.
Шестая, вруппа периодической системы
457
Б водном растворе красные двойные хлориды быстро переходят в зеленые двойные хлориды, которые также можно выделить в кристаллическом виде. Из азотнокислых растворов этих зеленых двойных хлоридов при добавлении нитрата серебра тотчас же выпадает три пятых содержащегося в них хлора. Отсюда следует вывод, что в этих солях 3 атома хлора связаны ионогенно. Вернер приписывает им структурную формулу, в соответствии с которой с центральным атомом хрома координационно связаны, кроме четырех молекул воды, еще два эквивалента хлорида щелочных металлов:
Н2О
Н2О
-Н2О
Ст
он2 -
С1М1
СШ1.
С13
(MT=Li, Rb, Cs, NH4).
Такие соединения Перпер называет соединениями внедрения.
Эти зеленые двойные хлориды можно считать производными серо-голубого хлорида хрома, получающимися заменой двух молекул воды двумя молекулами щелочного хлорида.
Соответственно из светло-зеленого хлорида Бьеррума можно получить желтокоричневые иглы — двойную соль, которую Пфейфер приготовил смешиванием растворов хлорида хрома и хлорида пиридиния:
гН2О
Н3О Сг
-Н2О
Cl 1
С1[руН] ClIpyHjJ
С13,
где [руН] — радикал пиридиний |Q5IISNHJ. Из раствора этой соли осаждаются, как и требуется формулой, только две пятых хлора.
Заменой всех тести молекул воды в катионе гексаанвохрома(Ш) на хлор получают гоксахлорохррматы(Ш), соединения типа м|[СгС10]. Калиевая и натриевая соли этого типа были получены Берцелиусом в виде розово-красных кристаллических порошков.
Различно окрашенные комплексные соединения, являющиеся производными хлорида хрома(Ш), являются прекрасным примером того, как сильно может изменяться цвет комплексных соединений в зависимости от их состава.
Фториды, и бромиды, трехвалентного хрома существуют, вероятно, в формах, подобных или похожих на хлориды, однако они пе столь детально исследованы как последние. Хром в трехвалентном состоянии, по-видимому, не способен к образованию комплексных иодидов. Даже безводный иодид хрома(Ш) до сих пор еще не получен в чистом виде, а только его гидрат состава Сг13-9Н3О. Это соединение образует черно-фи о лотовые кристаллы, превращающиеся при растирании в зеленый порошок. Весь иод в этом соединении связан ионными связями, как это можно заключить по данным температуры замерзания его растворов.
Цианид хрома(Ш) и цианохроматы] Ш). Если к раствору соли хрома(III) добавить цианид калия, то получается осадок цианида хрома(Ш) Cr{CN)3, который растворяется в избытке осадителя и в кислотах. Из растворов, содержащих избыточное количество щелочпого цианида, можно получить гексацианозромат,ы(111) Мз(Сг(СИ)6]. Светло-желтая калиевая соль K3(Cr(CN)6] изоморфна красной кровяной соли (гексацианоферрат(1П) калия] K3[Fe(CN)6].
Роданид хрома(Ш) и родапохроматыЦП). Гидрат окиси хрома растворяется •в роданистоводородной кислоте с образованием роданида хрома(Ш), который, однако, очень трудно получить в кристаллическом состоянии. Это соединение является неэлектролитом. Если к раствору добавить щелочного роданида, то при упаривании из пего кристаллизуются гексароданохроматпы{111) [Cr(SCN)6]. Эти соединения являются прочными комплексными темно-красными солями, свежие винно-красные растворы которых не дают характерных реакций ни на ион хрома(Ш), ни на роданид-ион, а, напротив, обнаруживают электропроводность и изменение электропроводности .при разбавлении, характерные для солей, содержащих одно- и трехвалентные ионы. При длительном выдерживании цвет растворов изменяется до зеленого, при этом происходит распад комплексных содей.
158
Глава 5
Оксалат хрома(ПГ) и оксалатохрознаты(Ш). При добавлении щелочного оксалата к растворам солей хрома(Ш) можно получить нерастворимый, кристаллизующийся с большим количеством воды фиолетовый оксалат хрома(Ш) Ст2(С2О4)3. Это соединение неустойчиво и легко переходит, отщепляя воду, в растворимый теми о-зеленый, аморфный продукт. Из растворов, содержащих избыток щелочного оксалата, выделяются щелочные триокеалатохроматы(И1) М| [Сг(С2О4)3], представляющие собой темно-голубые кристаллы*. Из них методом двойного обмена можно получить соответствующие оксалат о хром а ты щелочноземельных и тяжелых металлов.
В растворах три окса л атохроматов(Ш) обнаруживается прочный комплексный иоп [Ст(СгО4)3]я. Исходя из координационной шестивалептаости хрома можно сразу сказать, что в этом гтогге каждый из трех радикалов
О=С—О—
0=С-0—
занимает два координационных места. Если это действительно так, то у триоксалато-хроматоЩП!) надо ожидать существование оптической изомерии, поскольку радикалы щавелевой кислоты могут располагаться вокруг иона хрома двумя способами,
Рис. 21. Зеркальная изомерия
тряокс а л ато хр оматов( III).
схематически показанными на рис. 21. Приведенные на этом рисунке структурные формулы относятся друг к другу, как предмет к его зеркальному изображению. И действительно, в 1912 г. Вернеру удалось доказать, что триоксалатохроматы(Ш) могут быть разложены на оптически активные компоненты и тем самым подтвердить структуру, предложенную на основании принципов координационной химии.
От триоксаяатохроматов(П1) при замене остатков щавелевой кислоты на НгО, ОН и другие кислотные остатки начинаются многочисленные ряды комплексных окса-латохроматов(Ш), например:
диоксалатодиаквохроматы(Ш) диги др оксо диокса л атохр оматы( III) для Цетато ди оке ал атох р оматы (111) гидроксодиоксалатоаквохроматы(Ш)
М1 [Ст(С2О4)3(ОН2)2]‘, М3 [Ст(ОН)2(С2О4)2]; М3 [Сг(С2НзО2)2(С2О4)21;
Mj[Cr(OH)(C2O4)2(OH2)].
Для всех названных рядов соединений характерно явление цио-трано-иоомерии, о котором подробнее будет сказано ниже.
Ацетат хрома(Ш) п соли ацетатотрихрома(Ш). Нейтральный ацетат хрома(Ш) (Сг(ОН2)е](СаН3О2)3 можно получить растворением свежеосажденной гидроокиси хрома в уксусной кислоте. Эта соль кристаллизуется с шестью молекулами воды в виде сине-фиолетовых игл.
Если к растворам солей хрома(Ш) добавить ацетат натрия, то образуются комплексные соединения, отличительной особенностью которых являются катионы ацета-тотрихрома(Ш). В чистом состоянии они лучше получаются иным путем. Вернер,
* Щелочные трнокса.чатохроматы(Ш) почти всегда плеохроичны. В зависимости от направления, Под которым их рассматривают, они кажутся то темпо-васильковыми, то зеленовато-голубыми, то краевыми.
Шестая группа периодической система
159
а также Вайнланд приготовили а чистом состоянии соли ацетатотри хрома (III) следующих типов (Ас — остаток уксусной кислоты, СН3СОО; X — любой отрицательный одновалентный Кислотный остаток):
[Сг3(ОН)оАс6]Х [соли диеидроксоеекеаацетатотрихромаЩ/)] [Сг3(ОН)Аса(ОНг)]Х3 [соли еидрокеогексаацетатоанвотрихрома^/Ш)] И [Сг3Аса(ОН^]Х3 [соли гексаат1етатодиикеотрихрома(///)].
Аналогичные соли можпо получить и с остатками других органических кислот. Следует отметить, что в этих комплексах всегда содержится три атома хрома. Следовательно, они представляют собой мпогоядерные комплексные соединения. В качестве мостиков, связывающих атомы хрома, в этих соединениях но могут функционировать группы ОН, как это часто наблюдается, так как, присоединив ионы ГР и но изменяя строения соли, группы ОII можно перевести в молекулы ОН2, которые никак не могут быть мостиками, вследствие того, что координационное число кислорода райпо трем. Следовательно, мостиками могут служить только ацетатные группы.
Бринтцпнгер (Briutzinger, 1936) на основании коэффициентов диффузии, полученных методом диализа, " пришел к выводу, что ионы ди гидр оксогекса ацетатотрихрома]!!!) [а также аналогичные комплексные ионы железа(Ш) и кобальта (III)] в водном растворе могут присоединять определенные анионы с образованием двумерных комплексных ионов типа
[[Cr3(OH)2Aca]X4]"""' (X=SOJ-, HPOJ-, НАзОГ).
Такие двусфер ные комплексные новы, образующиеся благодаря присоединению во второй сфере кислотных остатков к катиону, координационно насыщенному в Первой сфере, согласно Бринтцингеру, часто встречаются в растворах. Однако их существование оспаривается [ср. Spandau H.,Z. an erg. Chem., 246, 100 (1941)].
Вследствие образования легкорастворимых ацетатных комплексных солей в основном вышеуказанного типа из растворов солей хрома в присутствии ацетата натрия при кипячении пе выпадает гидрат окиси хрома, несмотря на то что этого можно было бы ожидать по аналогии с алюминием, В холодных растворах также никакие реактивы на хром пе приводят к образованию осадков.
Нитрат хрома(Ш) Cr(NO3)3 образуется при растворении гидрата окиси хрома в азотной кислоте. Раствор в отраженном свете голубоватофиолетовый, в проходящем — красный. При нагревании он становится зеленым, при охлаждении фиолетовая окраска быстро восстанавливается. Соль, кристаллизующаяся из такого раствора, имеет переменное содержание воды, зависящее от условий получения. Ее применяют в ситцепечатании в качество протравы.
Сульфат хрома(Ш) и соли сульфатохрома(Ш). Кроме безводного сульфата хрома(III) Cr2(SO4)3 (представляющего собой порошок цвета цветущего персика, растворяющийся в воде и кислотах только в присутствии восстановителей, как и безводный трихлорид хрома), известен и водный фиолетовый сульфат жролыЦШ) Cr2(SO4)3-18Н2О и несколько содержащих воду зеленых сульфатов хрома, которые образуются при нагревании фиолетового хрома, например при длительном кипячении.
Содержащий воду фиолетовый сульфат хрома — который в зависимости от условий получения может содержать И на несколько молекул воды меньше, чем в приведенной выше формуле,— легко растворяется в воде и обнаруживает в растворе нормальные реакции па ионы хроиа(П1) и сульфат-ионы. Поэтому ему приписывают формулу [Cr(OH2)aj2(SO4)3- пН2О («=2—6). Фиолетовый раствор этой соли при нагревании становится зеленым. Зеленые сульфаты хрома пе только содержат меньше воды и труднее растворяются, чем фиолетовый, но и построены иначе, чем последний: в них сульфатные радикалы, как следует из реакций их растворов, связаны с хромом неионной связью. Поэтому зеленые соли следует считать солями сульфатохрома(Ш). Среди них встречаются продукты гидролитического расщепления, в которых наряду с Н2О и SO4 присутствуют ней оногеп нос вяза иные группы ОН. Вследствие этого превращение фиолетового сульфата в зеленый, как правило, связано с существенным увеличением электропроводности раствора — это обусловлено отщеплением ионол !Г. На основании
160
Глава 5
данных кондуктометрического титрования едким натром Эрдман (Erdmann, 1952) пришел к выводу, что в водных растворах сульфата хрома устанавливаются следующие равновесия:
(010!
(HsOhCr Сг(ОН>)5 (SOJ
+ 2(80!]"
f(HtO)j (OH)S (OHi)i
Or Сг
1(SO4) (W (SOP
Г/
+ H1O.
<1П)
Гексааков-ди-р^сиАроксо -р-сульфато - Лисулыратотетраакво - ди-и-гидрокси р- сильрсисв-
дахром '(т) иоя ' дихромат (ш)-ион
Равновесие (I) имеет существенное значение только в сильно разбавленных растворах сульфата хрома. В умеренно разбавленных растворах доминирует равновесие (II). Оба равновесия при повышении температуры сдвигаются вправо. Реакции (I) и (IJ) протекают быстро слева направо, в обратном же направлении — очень медленно. Это показывает, что для разрушения двуядерногб сульфатного комплекса требуется высокая энергия активации. Равновесие (III) зависит от концентрации. Оно быстро сдвигается в обоих направлениях.
В твердом состоянии зеленый сульфат хрома был впервые получен Этардом (Etard, 1877) и Рекурой (Несонга, 1892). В зависимости от метода приготовления это соединение получают с различным содержанием воды. Уже Рекура показал, что свежеприготовленный раствор зеленого сульфата хрома(Ш) не дает реакций ин на ион хро-ма(Ш), ни на сульфат-ион, следовательно, в этом соединении все сульфатные остатки неиопогенно связаны с хромом. Выпариванием с серной кислотой Рекуре удалось получить из этого соединения целый ряд кислот сульфатохрома (III) (всс в безводной форме), а именно дисульфатохром(П1) кислоту Н [Cr(SO4)J, пентасульфатодихром(1П) кислоту H4|(SO4)2Cr(SO4)Cr(SO4)2] и трисульфатохром(Ш) кислоту Н3[Ст(ЗО4)3]. Эти кислоты легко растворяются в воде, их растворы красивого зеленого цвета. Свежие растворы не дают осадков пн с фосфатом натрия [реактив на иопы хрома(Ш)], ни с хлоридом бария (реактив на сульфа т-ионы). Это относится и к растворам солей этих кислот, сульфатохроматам(Ш).
Присоединять серную кислоту способен и водный фиолещовый сульфат хрома. Получающийся при этом кислый сульфат хрома{111) Н [Сг(ОНч)в](8О4)2'ZTIjO * образует фиолетовые кристаллы, из раствора которых ионы Ва" тотчас же осаждают все ноны S04,
По данным Крауса (Кгагф, 1929), восемнадцативодный фиолетовый сульфат при нагревании сначала переходит без изменения цвета в девятиводный гидрат, затем, изменяя цвет на зеленый, в трехводный гидрат и наконец в безводную соль. Последние три молекулы воды непрерывно отщепляются при повышении температуры (ярн 10 .ил рта ст в температурном интервале 100—325°).
Хромовые квасцы. Из смешанного раствора фиолетового сульфата хрома(Ш) и сульфата калия кристаллизуется двойная соль KCr(SOJ2-12Н2О, которая по составу и кристаллической структуре полностью
* В зависимости от способа получения это соединение кристаллизуется и с ббль-авим числом молекул воды.
Шестая группа периодической система
161
соответствует квасцам КА1(8О4)3-12Н2О и кристаллы которой могут расти и в растворах квасцов. Эту двойную соль называют хромовыми квасцами.
Хромовые квасцы кристаллизуются в виде больших темно-фиолетовых октаэдров удельного веса 1,828. Растворимость в воде при 25° составляет около 24,4 г на 100 -г воды. При нагревании фиолетовый раствор становится зеленым, причем в нем образуются рассмотренные выше зелёные сульфатные комплексные соединения, большинство из которых в отличие от хромовых квасцов очень мало склонно к кристаллизации. Из вновь охла?кдеипо! о раствора хромовые квасцы начинают кристаллизоваться только после многопедельного стояния.
В технике хромовые квасцы получают как побочный продукт при многих процессах, в которых в качестве окислителя используют бцхромат калия. Его применяют главным образом при дублении, в красильной промышленности и ситцепечатании.
Кроме обычных хромовых квасцов (калиево-хромовых квасцов) KCr(SO4)2-12H2O, известны также имеющие аналогичный состав двойные соли фиолетового сульфата хрома(1П) и других сульфатов одновалентных металлов и радикалов. Эти хромовые квасцы в широком смысле слова, которые совершенно аналогичны обычным хромовым квасцам не только по своей общей формуле APCrfSOJ..- 12Н,0 (где М1 — Na, Rb, Cs, Т11, аммоний, гидроксил аммоний, радикал гидразиний), no и по кристаллической форме, называют в зависимости от присутствующего в пих металла или радикала патриево-хромовыми квасцами, аммоииево-хромовыми квасцами и т. д.
По данным реитгеиоструктурного анализа, как уже указывалось в т. I, в квасцах как оЗновалентный атом (соответственно радикал), так и трежвалепт-иый атом окружены шестью молекулами воды. Следовательно в хромовых квасцах присутствует как раз тот самый гексааквохром(П1)-ион, который входит в состав других фиолетовых солей хрома(Ш). Длина ребра элементарного куба калиево-хромовых квасцов, в которых узлы структурной решетки заняты (в таком же порядке, как ив решетке каменной соли) попеременно атомами калия и хрома (см. т. I, стр. 402), очень мало отличается от длины ребра калиево-алгоминисвых квасцов. Опа составляет 12,17 А . Очень близкие значения диины ребра Элементарного куба найдены и для других квасцов. Очень близкими размерами решетки объясняется исключительная способность квасцов к образованию смешанных кристаллов.
При нагревании калиево-хромовые квасцы, согласно Краусу (1929), ведут себя иначе, чем кал и ев о-алюминиевые квасцы, поскольку при обезвоживании последних (равно как и рубидиево- и цезиево-алюминиевых квасцов) в качестве промежуточного продукта образуется только трпгидрат, тогда как при обезвоживании калиевохромовых квасцов в качестве промежуточных продуктов образуются гекса- и дигидраты.
АММИПНЫЕ КОМПЛЕКСЫ ХРОМА(Ш)
Ярко выраженная способность трехвалентного хрома к образованию комплексных соединений особенно отчетливо проявляется в его различных комплексных продуктах присоединения аммиака.
Одноядерные комплексы, В соответствии со своим максимальным координационным числом 6 ион хрома(Ш) может координационно связать шесть молекул аммиака. При этом образуется комплексный ион [Cr(NH3)6]3+ [ион гексаамминхрома(Ш)], заряд которого совпадает с зарядом хрома, фигурирующего в качестве центрального атома, поскольку молекулы аммиака не заряжены. Несмотря на то что молекулы И Г. Реми
162 Глада а
аммиака связаны с хромом лишь координационно, они очень прочно удер-живаются около него. Очень мало элементов (это прежде всего Со и Pt), ионы которых столь я;е прочно, как и ионы хрома(Ш), связывали бы нейтральные молекулы. Особенно прочные связи в этих комплексах объясняются, как это можпо показать (см. т. I, гл. 11), тем, что нейтральные группы связаны с центральным атомом не обычными силами Ван-дёр-Ваальса, а атомными связями, т. е. они владеют электронами совместно с центральным атомом. Как уже указывалось ранее, такие комплексы в отличие от «нормальных» называют «комплексами внедрения». Отрицательные ионы — которые обычно связаны с центральным атомом электростатическими силами— могут, как и нейтральные молекулы, образовывать атомные связи с центральным атомом.
Вследствие прочного связывания молекул аммиака при растворении в воде соединений, содержащих комплекс [Cr(NH3)al3\ даже в отсутствие аммиака не происходит моментального распада комплекса — он существует в растворе как ион и лишь постепенно происходит замещение молекул аммиака молекулами воды. При таком замещении могут образов ваться соединения, в которых 1, 2, 3 и т. д. и, наконец, все молекулы NH3 замещены молекулами Н2О. Существует непрерывный ряд акво-производ-ных иона гексаамминхрома(Ш) (см. табл. 18). В конце ряда * находится ион гексааквохрома(ПГ) [Сг(ОН2)а]3+, исключительная устойчивость которого по сравнению с другими гидратированными ионами хрома уже отмечена была ранее. При замещении молекул аммиака молекулами воды заряд комплекса не изменяется. Существует также и ряд комплексов, который можно представить себе нак следствие постепенного замещения молекул аммиака па отрицательные радикалы (кислотные остатки или группу ОН). В этом случае (положительный) заряд комплекса в зависимости от числа введенных кислотных остатков сначала уменьшается до нуля, а затем вновь увеличивается, но уже с отрицательным знаком. Если представить себе, что все шесть молекул аммиака замещены одновалентными кислотными остатками R, то получим отрицательный трехвалентный комплексный ион гексаацидохромага(111) [СгВ6}3~.
Другой ряд комплексов можно представить себе как следствие замещения в ионе гексаамминхрома(Ш) части молекул NH3 молекулами Н20, а другой части — кислотными остатками В. Вся совокупность получающихся таким образом аммин-, акво-и ацидо-комплексов приведена в табл. 18. Все типы представленных в этой таблице комплексов известны: в форме соединении, в некоторых случаях очень многочисленных. В этих комплексах не только отрицательные радикалы В могут представлять собой разнообразные кислотные остатки или группу ОН, но часто вместо аммиака (Ат) могут выступать другие азотсодержащие соединения, например гидроксил а мин, органические амины, пиридин и т. д. Кроме того, во многих случаях вместо молекул воды могут быть координационно связаны другие кислородсодержащие соединения (спирты, фенолы, эфиры и т. д.), так что от большинства приведенных в табл. 18 типов можно произвести целый ряд подтипов. От каждого из образующихся таким образом комплексных катионов или анионов происходит ряд отдельных соединений, получающихся при соединении комплексного иона с различными другими, ионами противоположного заряда.
* Конечно, соединения гексааквохрома(Ш) не относятся к амминпым комплексам. Однако их можно рассматривать как производные последних. Это же относится и к гексаацидо- и а цидоак во-со единениям.
Основные типы одноядерных аммин-, акво- и а цидо-комплексов хрома(Ш)
Таблица 18
а а В Г д е ж
I [СгАт6]3+ ГсгЛта Г L (ОНг) J Г Ат4 13+ 1ьг(он2)г J Г Ат3 Г+ L Сг(0П,)3 .1 — [Сг(ОН2)0)3+
II КГГ ~ Ат4-Сг(ОН2). - П - 2+ - Аш3 -Сг(ОН2)2 _ R 2+ г Лтг -12+ Сг(ОН2)3 L R —
III [<*г - Ат3 -л Сг(ОН2) L Ra - - Ат3 -Сг(ОН2)2 - + — мщ‘]+
IV [<"”] - Ат2 ъ Сг(ОНг) L R3 J —
V КгГ — [с,(0ВДг]-
VI — [сг№> ]-.
VII [CrRep-
164
Глава 5
Основные типы соединений, являющиеся производными комплексов, приведенных в табл. 18, заслуживают отдельного краткого рассмотрения.
1а) Соединения гекеаамминхрома(ПГ), соединения общей формулы [Сг(Лт)е)Х^.
. „,-,т en pn* Dipy -г
где Ат — NH3, -% , ~~ , (Dipy—а,а -дипиридил). Во внешней сфере комплекса
могут находиться С1_, Вг_, I-, NOj, SO^~, РО|_, а также остатки комплексных кислот, например [PtCl6]2-, [Fe(CN)als- и др. Эти соединения желтого, коричнево-желтого и оранжево-желтого цветов. Поэтому раньше (они были открыты Иергенсепом в 1880 г.) их, как и аналогично построенные амминные комплексные соли кобальта, называли лутео-солями (iuteus — оранжево-желтый).
16) Соединения акаопентаамминхромаЦ! I)
Аль," Сг
Х3 OH2J
ранее называли poaeo-солями, так как соли кобальта аналогичного состава розового цвета. Соединения хрома этого ряда в большинстве случаев окрашены в желтый или оранжево-желтый цвета и очень похожи на гексаамминные соединения по способам получения, кристаллической форме, блеску, растворимости и своим химическим реакциям. Однако в отличие от гексааммиияых соединений, разбавленным водным аммиаком, они не разлагаются постепенно, образуя осадок, а переходят в легкорастворимые гидроксопептааммннные соли
Amf Сг
ОН _
Хг
называемые раньше 1в) Соединения
«основными розео-солями». диаке от етр аамминхрома(11 Г)
Сг
Лт4 (ОВДг.
Х3,
Am — NH-> или тг . 6 2
где
По своему химическому характеру эти соединения близки
рассмотренным выше. Их окраска меняется от оранжевой до кирпично-красной. Соединения этилепдиамипа этого типа известны в двух ожидаемых по теории изомерных формах (ifttc-mpагинизомерия, см. ниже).
1г) Соединения п1риаквотриамминхрома(Ш)
Аш3 1 Сг Х3
L (ОН2)3 J
известны в небольшом числе. Светло-красные соединения, по своему поведению они очень напоминают более богатые аммиаком соединения.
1д) Соединения тетраакводиамминхрома(111)
Сг
Аш2
(ОН2)4_
Х3
окрашены в цвета от красного до красно-фиолетового и по свойствам очень близки к чистым аквосоедипепиям. Опи сильно склонны к гидролизу; их гидроокись [Сг(ОНа)4Аш2](ОН)3 нерастворима в воде в отличие от легкорастворимых гидроокисей гексааммиц- и ацвнентаамминхрома(1П).
* еп и рп — сокращения для втилендиамина
СН2 —NH2
I
сн2—nh2
и (асимметричного) пропилендиамииа
СНч — СН—NH2
I
СН2—NH3
Эти соединения (как и дипиридил) содержат две группы, которые могут замещать ГШЭ. Поэтому они способны занимать два координационных места.
Шестая группа периодической системы
165
Те) Соединения пентаакво-иоиоамминхрома(П1) неизвестны. То, что во всех рядах табл. 18 всегда отсутствуют моноамминные соединения, и отсутствуют только они, указывает на существование какой-то закономерности, проявляющейся в неспособности к существованию моноамминпых соединений.
1ж) Соединения гексааквохрома{111) [Сг(ОН2)в]Хэ. Соединения этого типа окрашены, прежде всего в растворах, в фиолетовый цвет. Сюда относятся уже описанные обычные содержащие воду соли хрома, например обычный гексагидрат хлорида хрома, нитрат, ацетат, сульфат, хромовыо квасцы и другие.
На) Соединения ацидопеитаамминхрома(11I)
Хг,
Апц" Сг L и J
где Ат — NH3, СН3-1Ш2,™и т. д.; R — С1, Вт, I, SCN, NO3, N02, ОН. Эти соеди-
нения генетически близки р оз ео-соединениям (аквопентааммин-соединениям). Реакция
Г Атя-]‘"
-f.R’= Сг
Лт5“[’'
+ Н2О
Сг ohJ
R J
часто необратима. Хлорсодержащие соединения этого типа в большинстве случаев темпо-краспого цвета, поэтому раньше их называли пурпурео-соединениями. , Пб) Соединения
ацидоаквотетраамминхрома(111) Лт4-
Сг ОН2 Х2, R J
где
Am—NH3, 3, 2
Ив) Соединения
где
Am — NH3; R — Пг) Соединения
21РУ ; R — С1, Вг, I, ОН.
ацидодиаквотриамминхрома(111) tAm3 Сг (ОН2)2 R
С1, Вг, ОН, NO3.
a if а й отр и в кво Эн а л(.к ияхр ола (/7/)
Ат3
Сг (ОН2)3 R
Х2,
Х2,
где Ат — NH3, Ру — пиридин; R ~ ОН.
Пд) См. 1е!
Пе) Соединения ацидопентаакеохрома(П1)
(ОИг)5-1
К этому типу относится светло-зеленый хлорид хрома(Ш) Бьер рума.
Ша) Соединения диацидотетраамминхрома(111)
Аш4'
Ст X
L Вг J
Из соединений этого типа известны главным образом соединения этилендиамина. Они интересны ввиду наблюдающегося у них явления изомерии (стереоизомерии).
При октаэдрическом расположении комплексно связанных групп А и В вокруг центрального атома комплекс с общей формулой [ZA4B2] может существовать в двух формах, как это показано на рис. 22. Эти формы различаются тем, что в одном случае обе группы В находятся по одну сторону от центрального атома, по соседству друг с другом, так что прямая, соединяющая их, совпадает с ребром октаэдра (п, следовательно, с прямой, соединяющей концевые точки двух координатных осей на рис. 22), тогда как в другом случае эти группы расположены по разные стороны центрального
166
Глава 5
атома, так что соединяющая их прямая проходит через центральный атом. Первая форма называется цис-формой, вторая — транс-формой, а изомерия такого рода — ц uc-m ра нс-и зом ерией.
Какому из двух экспериментально подученных соединений, находящихся между собой в отношении цис-/пр я ис-изомерии, следует приписать цис-, а какому — трансформу, можно решить, определив, в какой из двух форм можно заменить обе группы В одной координационно-двухвалентной группой (т. е. группой, которая способна
Р п с. 22. Дис-травс-изомерия комплексных соединений.
запять два координационных места). Это соединение будет цис-соединением. В случав диэтилепдиамшшых соединений различие заключается еще и в том, что в противоположность транс-соединению цне-соедипенио представляет собой смесь двух оптически активных форм, поскольку, как видно из рис. 23, в атом случае могут существовать две цис-формы, относящиеся друг к другу, как предмет к его зеркальному отражению. Доказать наличие цис-транс-изомерин у диацид одна тилеидиа минных соедипепий
Рис. 23. Цис-транс- п зеркальная изомерия диэтилендиаминовых соединений.
удалось в 1904т. Пфейферу. В 1911 г. Вернер расщепил на оптически активные компоненты комплекс цыс-дихлородиэтилепдиамипхрома (III)
Г еп2ч+
Сг
L C1J
приготовив его соль с оптически актив и ой а-бромкамфор сульфонов ой кислотой. Goes 1-дихлороднэтиленД!1амипого комплекса с анионом d-a-бромкамфорсульфояовой кислоты (1,</-соль) в отличие от фц-соли малорастворима. Поэтому легко осуществить их разделение. После этого заменой остатка бромкамфорсульфоновой кислоты па другие кислотные остатки X легко получить другие соли цис-ряда типа
Сг
X
в оптически активных формах.
Шестая группа периодической системы
167
Шб) Соединения диацидоахвотриамминзромаЦН) АШзП
Сг 0Н2 X
В2 J
где Ага — NH3; R — Cl и ствование трех изомеров получил хлорид
Вт. Теоретически в этом случае можно предполагать суще-(см, рис, 24). И действительно, Ри Зенфельд (Riesenfeld)
(NH3)3 *|
ОН2 Cl
Cl2
Р и е, 24.
Изомерия соединений типа
Аш3" Сг ОН2 .
В2 J
в трех различных формах (красно-фиолетовой, серой и теми о-зеленой).
Ills) Соединения диацидодиакводиамминхрома{111)
Ат2 -
Сг (ОН2)2 X
L В2 J
С о
где Am—NH3, Ру; R — С1, Вг, ОН, —. Теоретически в этом случае можно предвидеть существование многочисленных пространственных изомеров, которые, однако, до сих пор еще пе получены. По своим свойствам эти соединения очень близки к соединениям диацидотетрааквохрома(Ш).
Шг) См. Те!
Шд) Соединения диацидотетрааквохрома(Ш)
г (ОН2)41
X
Сг
L н2
Наиболее известное из соединений этого типа темпо-зеленый хлорид хрома(III) (ср. стр. 155).
IVa) Соединения триацидотриа.мминхрома(1 Г1)
Аш3-Сг
L R3 J
где Am — NII3, Ру; R — F, Cl, SCN. Соединения этого типа — неэлектролиты.
IV6) Соединения триацидоакводиамминхрома(ГЛ)
Апь*
Сг ОН2
R3 Ф
168
Глава 5
где Am — NH3; R — SCN, ОН; Am — Ру, R — F, С1, ОН. Эти соединения также неэлектролиты.
IVb) См. 1е!
I Vr) Соединения триац.идотриакеохрома(П1)
Правда, соединения общей формулы
(онг)3-
Сг
*3 ,
в чистом состоянии не известны, однако получены производные этого типа, в которых И2О заменена другими кислород- или серусодержащими группами, например ранее уже упоминавшееся соединение
Г (С2Н5О11)3-
Сг
С13
Va) Тетраацидодиаммиихроматы(Ш)
Сг
М1
R4
где Ат — NH3, Ру, ™ ; R—С1, Бг, SCN, * Поскольку в состав комплекса входят
четыре отрицательных эквивалента, он представляет собой анион и образует соли с металлами (М),
К этому классу соединений принадлежит так называемая соль Рей пеке NH4[Cr(SCN)4(NH3)2]-НаО, анион которой часто успешно применяют для получения солей с органическими катионами в чистом состоянии. Б соответствии с данными Мара [Mahr С., Z. anorg. Chem., 225, 386 (1935)) соль Рейнеке удобно использовать для количествен пог о определения меди, так как можно легко провести осаждение последней в форме Cu[Cr(SCN)4(NH3)2], не удаляя из раствора другие металлы (кроме Ag, Hg и Т1). Реакция с солью Рейнеке может служить также и как очень чувствительная качественная проба на медь.
В отношении соединения Cr(SCN)4NO(NH3)2, являющегося производным соли Рейнеке, до сих пор еще точно не установлено, следует ли его считать *нитро шлрейнеке-атом», [NO]+ [Cr(SGN)4(NH3)2]“ или соединением типа IVa: [Cr(SCN)3(NO-SCN)(NH3)2].
V6) См. le!
Vb) Тетраацидодиаквохроматы(1П)
(OH2)21
M1
К этому типу относятся уже упоминавшиеся диоксалатодиаквохроматы(Ц[), Via) См. 1е!
VJ6) Пентаацидоаквохроматы(Ш)
Сг
ОН2“
Rs
Mj
Примером соединений этого типа могут служить уже рассмотренные выше пен-тахлороаквохроматы(Ш). К этому типу относятся и упомянутые па стр. 158 гидро-КСодиокс;1латоаквохроматы(Ш).
VII) Гексааридг>хромать1(ПГ) Ы3[СгП6]. Из соединений этого типа уже были рассмотрены хлоро-, циано-, родано-, оксалате- и сульфатохроматы(Ш).
Многоядерные комплексы. Многоядерные амминные комплексы хро-ма(Ш) содержат несколько центральных атомов хрома. В большинстве случаев они связаны друг с другом ОН-группами *.
* Атомы и группа, которые связывают между собой два центральных атома, обозначают буквой «ц», ставящейся перед их названием. Раньше группу ц-ОН называли «ол»-группой.
Шестая группа периодической системы
169
24-часовое нагревание при 100“
Важнейшими представителями двухъядерных комплексов являются так'называемые рода- и притрохромозые соли я соля ди-р-гидроксоднхрома(1П). Четырехъядер-иый комплекс, по-видимому, входит в состав так называемых родоа0хромоеых солей Иергенсена и описанных Пфейфером этилендиа минных соединений аналогичного состава.
Открытые Иергепсеном в 1882 г. родохромовые соли образуются при окислении кислородом воздуха аммиачных растворов солей хрома(Н). Их легко можпо перевести в так называемые эрптрохромовые соли. Красные нейтральные (нормальные) родохро-мовые соли растворяются в водном аммиаке пли в натриевой щелочи с образованием синих растворов так называемых основных рвдохромовых солей. При добавлении кислот вновь образуются нормальные родохромовые соли. Однако при стоянии голубые растворы через некоторое время приобретают малиново-красный цвет, и (при добавлении спирта) ИЗ них можпо осадить Красные «основные» вритро-соли, которые При добавлении кислоты переходят в нормальные (также Красные) apumpo-соли. При 24-часовом нагревании при 100° последние вновь перегруппировываются в родо-соли:
тг Основания л
Нормальныеродо-соли —.------—> «Основные» родо-соли
(красные) ~ (голубые)
Выдерживание раствора
Нормальные эритро-соли Кислоты «Основные» эритро-соли (красные) * (красные)
Эти взаимные переходы и основные реакции солей объясняются следующими структурными формулами (Jensen, 1937):
+NaOH
[(NH3)sCr-OH Сг(1ЧН3)5]Х5 ----------> [(N1Tg)5Cr-O-Cr(NH3)5]X4-(-NaX-(-HO
Нормальные родо-соли «Основные* родо-соли
[соли дека а м ми м-н -г и др ок со- [с о ли дек нам ми и-д-оксо д их р а ма( ИХ)]
дихрома(1П)]
t I
[(NH3)5Cr-NH2->Crg™=)4]x5 [(ЫНзЬСг-ИНг^Ст^з)*] х4
Нормальные эритро-соли а Основные» эритро-соли
[соли аквоноиааммин-р-амино- [соли гидроксононааммин-ц-амино-
дихрома(1П)3 дихрома(Ш)]
В пользу этих формул, кроме химических реакций, свидетельствует еще тот факт, что родо- и эритро-соли дают совершенно идентичные рентгенограммы. Комплексные катионы нормальных эритро-солей образуются из катионов нормальных родо-солей при замене одной ОН-группы (число электронов 9) на одну группу NHZ (число электронов 9), а одной молекулы NH3 (число электронов 10) — на одну молекулу Н2О (число электронов 10). Поскольку при такой замене число электронов в отдельных узлах кристаллической решетки пе изменяется, остается неизменной и способность к рассеиванию рентгеновских лучен. «Основные» эритро-соли образуются из «основных» родо-солей при замене одного атома О (число электронов 8) на группу 1ЧН2 (число электронов 9) и одной молекулы NH3 (число электронов 10) на группу ОН (число электронов 9). И в этом случае можно ожидать, при одной и той же структуре кристаллической решетки, что рентгенограммы будут Практически одинаковыми.
Соли дн-р-гидрокеодихрома(Ш) представляют собой, как о том говорит их название, комплексные соли хрома(Ш), в которых два атома хрома связаны двумя группами ОН. Этот класс соединений представлен главным образом солями ди-р-гид-роксотетраэтилендиаииядлхрома(1П) [Сг2(ОН)2еп4 (Х4, которые были получены Пфейфером при отщеплепии воды (нагревание до 100—120°) от солей цне-гидроксоак-водиэтилендиаминхрома(Ш) *;
2
еп ОН '
Сг еп ОНя
Х2 -
2Н3О =
еп /ОНХ еп
Crif /Сг еп еп
Х4
* В отличие От цис-солей соответствующие транс-соли не могут превратиться г л и-р-ги др ок со-соли без Структурных изменений. Поэтому в этих условиях они не претерпевают изменений.
170
Глава, 5
Соли этого ряда, из которых известны хлорид, бромид, иодид, роданид, нитрат, тиосульфат и хромат, сине-фиолетового цвета.
Формально комплекс ди-р-гядроксодихрома(ПГ)
еп Н0Н"|3+ Сг еп НОН. введена занимающая два координационных места группа
производным типа
еп ОН (
Сг Сг _еп ОН еп
(>П"|4+
’ можно считать
(тип I в табл. 18) в котором вместо двух трупп НОН
Г НО.
>Сг
еп “1+
еп
Точно также эту группу можно три раза ввести в комплекс и получить четырехъядерный комплекс с зарядом 6+:
en\ "16+ еп/з.
Сульфат последнего был получен Пфейфером при обработке частично обезвоженных хромовых квасцов хлоридом этилендиаммония. Двойным замещением из этой соли был получен целый ряд других солей этого типа. Эти соли гекса-р.-гидроксогекса-этилендиаминтетрахрома(Щ) окрашены в великолепный красный цвет. К этому же типу относятся, вероятно, малиново-красные так называемые родоаохромоеые соли Иергенсепа, которые имеют аналогичный состав, только вместо этилендиамина содержат координационно эквивалентное количество аммиака. Следовательно, эти соли представляют собой, вероятно, соли гекса-ц-еидроксододекааммингпетрахрома(Ш):
Сг
НО.
>Сг (NHS)4
HOZ
Хе
Соединения xpoina(VI)
Трехокись хрома, окись .т/>ол(а(1П), СгО3 ангидрид хромовой кислоты выпадает из концентрированных растворов щелочных хроматов или бихроматов при добавлении большого количества концентрированной серной кислоты. Опа представляет собой темно-красные кристаллические иглы, которые можно промыть чистой (свободной от окислов азота) очень концентрированной азотной кислотой и высушить на пористой керамической пластинке примерно при 70°. Она не имеет запаха, кисловяжущего вкуса и сильно ядовита. (Трехокись хрома прежде всего действует на почки; доза 0,6 а смертельна.) Она ясадпо притягивает влагу из воздуха и очень легко растворяется в воде (166 г в 100 з при 15°) с образованием желто-красных, известных только в растворе хромовой Н2СгО4 и дихромовой (двухромовой) Н2Сг2О7 кислот.
В растворе устанавливается равновесие между хромовой и дихромовой кислотами:
2П2СгО4 Н2Сг2О7+Н2О.
При увеличивающемся разбавления это равновесие сдвигается в сторону хромовой кислоты (монохромовой кислоты). Как хромовая, так и дпхромовая кислоты практически полностью диссоциированы по первой ступени, а по второй ступени, напротив, п очень незначительной степени.
В кристаллическом состояния трехокись хрома образует построенную пз октаэдров СгОо пространственную сетку (см. ниже). В растворе координационное насыщена.* хрома осуществляется благодаря присоединению к СгО3 молекулы Н3О. Водороднал атомы последней отталкиваются положительно шестизарядным хромом, так что ozz
Шестая группа периодической системы
171
отщепляются л виде попов:
О Г °
О Сг + ОН2= О Ст О
О L О
-Р2Н-.
Координационное насыщение СгО3 может Происходить и за счет соединения в растворе уже образовавшихся ионов [CrOJ" (хромат-лонов) о СгО3. Тан образуется бихромат-ион [Ст3О7]":
- О О -г О Сг О Ст О
. О О -
0 Г ° 1’
О Ст+ О Сг О
О L О
Аналогично могут образовываться также ноны, которые содержат еще больше групп СгО3: трихро.нявг-ионы (Сг3Ощ]" и тетрахромат-иопы (Сг^О^)". Такие поли-хромат-ионы образуются тем. в большей степени, чем концентрированнее и чем богаче попами Н‘ растворы хромовой кислоты.
Рис. 25. Элементарная ячейка трехокиси хрома, о == 8,46, Ь = 4,77, с = 5,70 А.
То, что в хромат- и полихромат-ионах координационное число хрома равно только четырем, тогда как в кристаллической трехокиси хрома оно равно шести, объясняется, очевидно, отталкиванием между ионами О2-. D кристаллической решетке трех окиси хрома это отталкивание частично скомпенсировано влиянием шести ионов Сг0*, которые окружают во второй сфере каждый ион Сг04”.
Трехокись хрома кристаллизуется в ромбической системе. В ее кристаллической решетке (см. рис. 25) каждый атом хрома окружен шестью атомами кислорода, расположенными в вершинах несколько искаженного октаэдра, так что каждый атом кислорода принадлежит одновременно двум октаэдрам, причем, однако, нигде два октаэдра не имеют более одного общего атома кислорода (Wyckoff, 1933; Megaw, 1935). Следовательно, октаэдры СгО6 образуют охватывающую весь кристалл пространственную решетку подобно тетраэдрам SiO4 в кристаллической двуокиси кремния.
Удельный вес трех окиси хрома равен 2,80; она плавится .около 190° и уже при незначительном повышении температуры начинает заметно возгоняться. Одновременно начинается отщепление кислорода. Термическое разложение протекает в несколько ступеней: СгО3 —> Сг3Ок^-Сг2О6СгО3—>-Сгг03. Соединения Сг3О8 и Сг2Оа можно получить в чистом виде, если проводить разложение CrOs в атмосфере кислорода при соответствующих давлениях. То, что при термическом разложении образуются только указанные выше соединепия, подтверждено рентгенометрически (Ward, 1952). СгО2 ферромагнитна (Michel, Bcnard, 1935) и имеет структуру рутила.
Трехокись хрома является чрезвычайно энергичным окислителем. Со многими окисляющимися веществами она реагирует со взрывом.
172
Глава 5
Особенно сильно действует она па органические вещества. Часто эта реакция сопровождается воспламенением, например при действии на спирт. Напротив, с уксусной кислотой трехокись хрома можно даже кипятить, и никакого взаимодействия не происходит.
Трехокись хрома находит применение в препаративной химии как окислитель, в медицине — в качество разъедающего средства. Ее разбавленные растворы используют как фиксирующее средство для микроскопических препаратов.
Хроматы. Соли, получающиеся при взаимодействии трехокиси хрома с ангидридами оснований, называются хроматами [точнее хрома-тами(У1)]. Простейшим хроматам, монохроматам, отвечает общая формула М*[СтО4]. Все они, если только входящий в их состав катион не влияет па окраску, желтого цвета. Однако при подкислении желтых' растворов хроматов цвет их изменяется в оранжево-желтый цвет растворов бихроматов Mj(Cr2O7).
Еще более глубокую окраску имеют получаемые из очень кислых и содержащих избыточную трехокись хрома растворов трихроматы М* [Cr3Ow] (темно-красные) и тетрахроматы М*[Сг4О13] (коричнево-красные).
Таблица 19
Растворимость в воде некоторых хроматов и бихроматов (г безводной соля/100 г воды)
Ка.чай Натрий Магний Кальций Стронций Барий
Хромат 62,3 76,6 72 2,3 a 0,123 0,00035
Бихромат 12,7 180 — Легкорастворим, расплывается Легкораст- £10 РИМ Легкорастворим
Температура, °C 20 20 18 19 15 18
а Это значение относится к устойчивой безводной соли. Содержащая воду соль нестабильна и понтону легче растворима. При повышении температуры растворимость хромата кальция уменьшается: при 100° опа составляет только 6,42 е в 100 г воды.
Щелочные хроматы легко растворяются в воде. Растворимость хроматов щелочноземельных металлов сильно уменьшается от магния к барию (ср. табл. 19). Из хроматов тяжелых металлов как практически нерастворимые можно назвать в первую очередь хроматы свинца, висмута, серебра и ртути (в обоих состояниях окисления). Хроматы трех последних металлов темно-красные. Если существуют соответствующие бихроматы, то они в большинстве случаев лсгкорастноримы. Трудпорастворимым является прежде всего тоже темно-красный бихромат серебра Ag2[Cr3O7].
Если к раствору бпхропата добавить соль металла, который образует легкорастворимый бихромат, по ма нерастворимый хромат, например хлорид бария, то в осадок выпадает хромат этого металла, в рассматриваемом случае хромат бария ВаСгО4-Это объясняется тем, что между хромат- и бихромат-ионами устанавливается мгновенно сдвигающееся равновесие:
2CrOJ-|-2ir 7^ Сг2о; + Н2о. (7)
Шестая, группа периодической системы 173
Несмотря на то что в кислых растворах концентрация би хромат-ионов значительно превосходит концентрацию хромат-ионов, последние всо ж о присутствуют в концентрации, при которой достигается произведение растворимости труднорастворимого хромата. Правда, произведение растворимости хромата бария не достигается в сильнокислом растворе, но в слабокислом, например в уксуснокислом, достижимо при условии присутствия в растворе достаточного количества ионов бария. Если кислотность уксуснокислого раствора понизить добавлением ацетата натрия, то из такого раствора можпо количественно осадить шестивалентный хром в виде хромата, поскольку участвующие в равновесии хромат-иопы благодаря образованию осадка до тех пор удаляются из раствора, пока в нем практически не остается бихромат-иопов, из которых могли бы образоваться хромат-иопы.
Если бихроматы в водном растворе имеют кислую реакцию в соответствии с равновесием (7), то хроматы имеют щелочную. Это объясняется тем, что ионы СгО4 вследствие уже упоминавшейся незначительной диссоциации хромовой кислоты по второй ступени частично взаимодействуют с водой с образованием ионов НСгО4:
CrOJ + H2O НСгО£-|-ОН\ (8)
Соли общей формулы М1НСгО4, т. е. кислые хроматы в собственном смысле слова (раньше «кислыми хроматами» называли также и бихроматы) пе известим. В условиях, в которых можпо было бы ожидать их образования, всегда получаются бихроматы.
Нагреванием хромата бария [хромата(У1) бария] ВаСгО4 с ВаС03 в токе азота при 1200° можно, как показал Шольдер (1952), получить хромат(У) бария, Ва3(СгО4)2
2ВаСгО4+ВаСО3=Ва3(СгО4)2-|-СО2+1/иО2.
Шольдер получил также соединение Вай(ОН)(СгО4)3 со структурой апатита. Присутствие в атом соединении положительно пятивалентного хрома было установлено Клеммой благодаря магнетохимическим измерениям. Кажущийся радиус иона Сг5+ составляет около 0,46 А.
Хроматы и бихроматы имеют большое техническое значение. Их используют прежде всего в качестве окислителей, например при получении антрахинона, бензойной кислоты, хинона, искусственной .камфоры и т. д. Вследствие сильной окислительной способности их можно использовать также как отбеливающее средство для масел, воска и древесного уксуса. Они служат для приготовления воспламеняющих и взрывчатых веществ. Растворы бихромата, содержащие серную кислоту, используют для обезжиривания стеклянной посуды. Поскольку восстановители переводят хроматы в соли хрома(Ш), часто исходят из хроматов при получении соединений хрома(III), Кроме того, их применяют в кожевенной промышленности и в производстве чернил. Их растворы служат для фиксации и консервирования анатомических препаратов, в качестве протравы при ситцепечатании, в фотографии для обработки желатины или гуммиарабика, которые под влиянием раствора хромата становятся нерастворимыми в воде в тех местах, где они подверглись действию света (пигментная печать). При техническом получении хроматов исходят из хромистого железняка. Его вскрывают плавлением со щелочью при доступе воздуха («окислительное сплавление»). Если раньше из этого сплава в большинстве случаев получали отличающийся особенно хорошей кристаллизуе-мостью бихромат калия, то сейчас получают в основном бихромат натрия, приготовление которого обходится дешевле и который также легко можпо очистить перекристаллизацией.
Подобно хромовой кислоте, хроматы также чрезвычайно ядовиты. Они могут, особенно при попадании на пораненные места кожи, вызывать глубокие язвы. Это их свойство следует учитывать прежде всего при получении и употреблении хромовых красок.
174
Глава 5
Хромат и бихромат натрия. Хромат натрия NasCrO4 кристаллизуется из водного раствора обычно с кристаллизационной водой (при температурах 19,5° с 10, в интервале 19,5—25,9° — с 6, между 25,9 и 62,8s — с 4 молекулами воды, при температурах выше 62,8° — без воды). Он образует желтые кристаллы, которые, если кристаллизация идет из растворов, содержащих сульфат натрия, всегда загрязняются этой солью. Поэтому чистый хромат натрия получают из очищенного перекристаллизацией бихромата натрия, нейтрализуя его карбонатом натрия:
Na2Cr2O7+Na2CO3=2Na2CrO4+COE. (9)
Бихромат, натрия обычно кристаллизуется в виде дигйдрата, NaECr2O7-2H2O, но при температурах выше 83° — без воды. Он еще более легко растворим, чем хромат. Бихромат легко очищается перекристаллизацией, поскольку1 его растворимость сильно зависит от температуры (приО’в ЮОаводы растворяется 163г,при 98° 433г соли). Безводная соль, сильно расплывающаяся при обычных температурах и значительно растворимая в спирте, плавится при 320°. Около 400° она разлагается, причем выделяется кислород.
В технике хромат натрия получают в больших масштабах нагреванием смеси тонкоразмолотого хромистого железняка с содой и гашеной известью при доступе воздуха. Основная реакция протекает по уравнению
2FeO-Cr2O3+4Na2CO3+’/2O2=4Na2GrO4-f-Fe2O3+4COE.
Смысл добавления извести состоит в том, чтобы создать хорошую пористость расплава и обеспечить свободный доступ воздуха. Сплавление проводят обычно в температурном интервале 1000 —1300°. Поело охлаждения из расплава выщелачивают водой хромат натрия. Обычно его переводят в технически более ценный благодаря большему содержанию хрома бихромат натрия, добавляя к полученному раствору сорную кислоту. Из такого раствора сначала выпадает безводный сульфат натрия. Отделенный от него раствор оставляют после дальнейшего упаривания для кристаллизации при обычной температуре (в этом случае из него выпадает дигидрат) или его нагревают до полного иейарепия воды, после чего вся масса по охлаждении застывает в виде безводной соли. Если получают безводную соль, то обычно после подкисления вновь добавляют некоторое количество едкого натра, так что получают смесь хромата и бихромата натрия с содержанием хрома около 35%. Это делается потому, что получаемый далее бихромат калия имеет такое же содержание хрома.
Если в раствор хромата натрия вводить под давлением двуокись углерода, то образуется бихромат:
2№2СтО4+2СО2+Н2О Na2Cr2OT+2NaHCO3. (10)
Равновесие тем сильнее сдвигается в сторону образования бихромата, чем июне температура, чем выше давление и чем выше концентрация хромата в ксхоцпом ра.стио-ре (Agde, 1934). Образованию бихромата сильно благоприятствует то, что основная часть образующегося бикарбоната выделяется в твердом состоянии. Было предложено использовать эту реакцию для технического получения бихромата патрия, поскольку при этом способе можно заменить серную кислоту значительно более дешевой двуокисью углерода.
Кроме моно- и бихроматов в случае натрия известны, как и в случае других щелочпых и щелочноземельных металлов, также три- и тетрахроматы. Кроме того, для натрия известен также хромат состава Na4CrO5- 13Н2О. Эта соль, кристаллизующаяся в виде желтых больших ромбоэдров, может быть перекристаллизована из воды без разложения. Но если к насыщенному раствору прибавить едкий натр—при Этом можно было бы ожидать стабилизация значительно более основного по сравнению с обычным хроматом соединения,— то из раствора выпадает обычный хромат натрия Na2CrO4.
Хромат и бихромат калия. Хромат калия К2СгО4 всегда кристаллизуется в виде безводных, л им они о-желтых кристаллов, изоморфных сульфату калия KaSO4. При температуре несколько выше 670° он переходит
Шестая группа периодической системы
175
в красную модификацию, плавящуюся при 970—-980°, Хромат калия легко растворяется в воде. Его растворимость мало зависит от температуры.
Бихромат калия K2Cr20j из водных растворов кристаллизуется также в виде безводных крупных оранжевых триклинных пластинок. При обычных температурах его растворимость сравнительно мала и очень сильно увеличивается с ростом температуры (от 4,6 а на 100 г воды при 0° до 94,1 г при 100° и 263 г при 180°). Поэтому соль очень легко очистить перекристаллизацией. Бихромат калия не растворяется в спирте. При нагревании он плавится при 396°. При остывании расплава из пего кристаллизуется неустойчивая при обычных температурах и потому претерпевающая изменение при 240° (сопровождающееся значительным изменением объема) модификация. Бихромат калия теряет кислород только при сильном нагревании. При обычных температурах он вполне устойчив на воздухе и не расплывается. Поскольку он к тому же легко получается в очень чистом состоянии, его используют в количественном анализе для установления титра растворов тиосульфата, применяющихся в иодометрии.
Если к раствору иодида калия прибавить бихромат калия, одновременно добавив разбавленную сорную кислоту, то происходит количественная реакция
СггО; + 6Г + 14Н*=2Ст“- 4-312 + 7Н2О. (11)
Следовательно, на каждый бихромат-ион освобождается 6 атомов иода.
Техническое получение бихромата калия частично аналогично получению бихромата натрия. Только в этом случае при окислительном плавлении хромистого железняка вместо соды применяют поташ. Но так как вследствие большой температуры расплава теряются значительные количества относительно дорогой детучей окиси калия, то в настоящее время предпочитают получать бихромат калия из бихромата натрия, добавляя к раствору последнего хлорид калия:
Na2Cr2O7-|-2KCl = K2Cr2O7—p2NaCl.
Хромат аммония (NH4)2CrO4 и бихромат аммония (NH4)aCr3O7 кристаллизуются, как и соответствующие соли калия, без воды. Однако они значительно мепее устойчивы, чем последние. При нагревании до 200° они энергично сгорают, причем остается зеленая окись хрома в очень тонкоизмельченном состоянии.
Хромат свинца РЬСгО4 встречается в природе в виде красной свин-цовой руды (крокоита). Оп практически не растворяется в воде и выпадает в виде желтого осадка при добавлении к раствору хроматов или бихроматов растворов солей свинца. (Только из растворов с большим избытком СтО3 выпадает бихромат свинца.)
Обычно хромат свиттца образует моноклинные кристаллы. Но, кроме того, существует еще ромбическая модификация (устойчивая в интервале температур от 707 до 783°) и тетрагональная (устойчивая при температурах выше 783°). Ромбическая модификация часто образуется (в соответствии с правилом ступеней Оствальда) при выпадении из водных растворов, по уже нри небольшом нагревании переходит в моноклинную модификацию, устойчивую при обычных температурах. Как ромбическая, так и моноклинная модификации хромата свинца способны образовывать сметанные кристаллы с сульфатом свинца.
Хромат свинца относительно растворим в растворах щелочей, так как щелочи вызывают образование гидроксоплюмбатов(11). Но если добавить очень небольшое количество щелочи, то хромат свинца переходит в очень мало растворимый красный основной хромат свинца РЬСгО4-РЬ(ОН)2
176
Глава 5
(моноклинные пластинки). Еще более богатый свинцом рубиново-красный хромат свинца состава РЬСгО^-ЗРЬО встречается в природе в виде минерала меланохроита (фённцита).
При нагревании хромат свинца легко отдает кислород органическим веществам. Поэтому его применяют как окислитель в элементарном анализе.
Большие количества хромата свинца используют как краску под названием «свинцовая желтая». Краской служит и основной хромат свинца («свинцовая красная»).
Галогенозамещенные хроматы
Если в кислотном радикале хромата
ГО
О 12-
Сг
О О
один отрицательно двухзарядный атом кислорода заменить на отрицательно однозарядный атом хлора, то получим отрицательно однозарядный радикал
"О СГ-
Ст
О О
Соли, являющиеся производными этого радикала М1[СгО3С1(, называются хлорхраматами. Если хлором заменить два атома кислорода, то получим электропейтральную молекулу хлористого хромила
'О СП
Сг
.О С1.
Соединения типа М1[СгО3Х] известны и для других галогенов. Напротив, бром и иод не образуют соединений, аналогичных хлористому хромилу.
Хлорохромат калия К(СгО3С1] получают добавлением соляной кислоты к раствору эквимолярных количеств хлорида калия и трех окиси хрома (12) или кипячением раствора бихромата калия с избытком соляной кислоты (13):
СгО3+КС1=К[СтО3С1], (12)
К3 {Cr2OJ+2 НС) =2К(СгО3СЧ + Н2О. (13)
При охлаждении раствора из него выделяется хлорохромат в виде оранжевых прямоугольных призм удельного веса 2,50. При растворении в воде соль распадается на бихромат кгугия и соляную кислоту, так как реакция (13) обратима. Однако эту соль можно перекристаллизовать из воды, подкисленной соляной или уксусной кислотой.
Фторохромат калия KfCrO3F( (рубиново-красные октаэдры), бромо-хромат калил KjCrCXBr] (темно-коричневые кристаллы) и иодохромат калия К[СгО;Д1 (гранатово-красные кристаллы) получают так же, как и хлорохромат калия.
Хлористый хромил СгО,С1о представляет собой темно-красную, выделяющую красно-коричневые пары жидкость с температурой кипения 117°, застывающую при сильном охлаждении в светло-красные иглы с температурой плавления —96,5°. Удельный вес ее при 15° равен 1,935. Это
Шестая группа периодической системы 171
соединение образуется при действии газообразного хлористого водорода на сухую трехокись хрома:
2НСГ+Сг03==СгОгС124-Н20.|
Его можно получить также действием пентахлорида фосфора, хлористого ацетила, хлористого пиросульфурила или хлорсульфоновой кислоты HSO3C1 на трехокись хрома. Проще всего оно получается при нагревании хромата или бихромата с хлоридом щелочного металла и концентрированной серией кислотой:
К2Сг2О7+4КС14-ЗН28О4=2СгОгС1г4-ЗКгЗО4+ЗН2О<
Хлористый хромил, если он защищен от действия света, довольно устойчив. С окисляющимися органическими соединениями он реагирует в неразбавленном состоянии часто очень энергично, иногда с воспламенением. В некоторых веществах, например в четыреххлористом углероде, сероуглероде, хлорокиси фосфора, он растворяется без разложения. В этих условиях, а также в парах он имеет молекулярный вес, отвечающий формуле СгО2С12. Напротив, водой оп разлагается с сильным выделением тепла (около 17 ккал/моль):
СгОгС1г+2НгО=НгСгО4+2НСЬ
Аналогично он реагирует с едкими щелочами с образованием хроматов.
Для обнаружения хлора в хлоридах хромилхлоридпой реакцией испытуемое вещество нагревают с тонкойзмельчеиным бихроматом калия и серпов кислотой. Выделяющиеся пары поглощают очень разбавленным едким натром. Если в образующемся растворе обнаруживается хромат, то исследуемое вещество содержит хлор, поскольку бромиды и йодиды не дают летучих соединений с хромом, а при этих условиях окисляются до элементарных брома и иода. При проведении пробы следует учитывать то, что хлористый хромил образуют только те хлориды, при действии на которые серной кислоты выделяется свободный хлористый водород. Очень мало диссоциированные и нерастворимые хлориды, прежде всего хлорид ртути(П), хлорид ртути(1) и хлорид серебра, но образуют хлористого хромила. Поскольку фтор образует с хромом соединение, совершенно аналогичное по своим свойствам хлористому хромилу, при проведении хромилхлорцдном пробы следует удалить фториды.
Фтористый хромил CrO2F2 получают взаимодействием' СгО2С13 с F2 или СгО3 с безводным фтористым водородом. Оп представляет собой красно-коричневый газ, который легко конденсируется в темные фиолетово-красные кристаллические иглы (т. субл. 30®, т. пл. 31,6е, давление паров в тройной точке 873 мм рт ст, теплота возгонки 14,6 ккал/моль). Это соединение, если оно сохраняется в темноте, устойчиво. При освещении происходит постепенное превращение в грязно-белую трудно летучую модификацию (Wactenberg, 1941, Grosse, 1951).
Пероксохроматы и перекись хрома
При действии па растворы хроматов перекиси водорода в зависимости от условий опыта (концентрации ионов водорода, содержания H2O2, температуры) образуются различные пероксохроматы, ийи темно-синяя перекись хрома СгО0.
Известно два ряда пероксохроматов: синие пероксохроматы общей формулы М2 |Сг2О12] и красные пероксохроматы общей формулы М*[СтОв]. Кислоты, соответствующие пероксохроматам, в свободном состоянии неустойчивы. В кислых растворах получается не способная к образованию солей перекись хрома СгО6. Она получается в виде растворов и в форме кристаллических продуктов присоединения. В свободном состоянии она, однако, не существует.
1 с I Ремп
i78
Глава 5
Образованием перекиси CrOg объясняется синее окрашивание, полу-чающееся при действии перекиси водорода на подкисленные растворы хроматов. Вследствие распада перекиси с образованием соли хрома(Ш) синяя окраска раствора постепенно сменяется зеленой. Однако если сразу после подкисления раствор встряхивать с эфиром, то перекись хрома переходит в эфирный слой и сохраняется там длительное время. Поскольку синяя окраска перекиси хрома особенно интенсивна, этой реакцией можно обнаружить очень небольшие количества растворенных хроматов («хромпероксидная реакция»).
При действии водного раствора аммиака на офирпый раствор синей перекиси хрома [пятиокиси хрома (VI)! СгО5 можно получить аммиакат другой перекиси хрома — четырехошии хрома СгО,(. Эта перекись известна только в форме продуктов присоединения. При постепенном нагревании с цианистым калием аммиакат CrO^SNHj переходит в обладающий солеобразным характером продукт присоединения СгСЦ 3KCN. Водные растворы соединений четырехокиси хрома Окрашены в коричневый цвет.
Соединения четырехокиси хрома уже Вернером рассматривались как координационные соединения, содержащие пер ок co-гр у пну с электрохимически и координационно шестивалентным хромом
О\ /NH3‘
Oa=Cr*-NH3
Сг
’ CX /CN oACr-CN
To, что пятиокисъ хрома и синие и красные пероксохроматы следует считать истинными с ио.«’со-соединениями (производными перекиси водорода), впервые было доказано Р и Зенфельдом (1908), который установил, что при гидролизе названных соединений образуется перекись водорода. Но так как в кислом растворе опа тотчас же каталитически разлагается хромперокси-соодипениямн, то ее образование нельзя установить непосредственно. Се промежуточное образование доказывается восстановлением перманганата калия. Ризенфельд считал пер оксохрома ты соединениями с сежавалепт-•яым хромом. Однако позднее Шварц (1932 и сл.) показал, что содержание перекисного кислорода в этих соединениях выше, чем нашел Ризенфельд, в опытах которого часть освобождающейся при гидролизе перекиси водорода вследствие саморазложения по вступила в реакцию с перманганатом. Шварц избежал этого, каталитически ускоряя реакцию перекиси водорода с перманганатом кадия добавлением небольших количеств молибдата. Так он нашел, что в синих пер оксохроматах па каждый атом хрома содержится 2t/3 пероксогрунпы. Поэтому им следует приписать формулу двуядерных соединений с жест «валентным хромом. Наблюдение Бема (1926), что К3СгП8 образует смешанные кристаллы с KjNbOs и КуГаОя (ср. стр. 126 и 130), послужило основой предположения, что хром в красных пероксохроматах пятивалентен, так же как ниобий в пероксониобатах и тантал в пероксотапталатах. Окончательно это было установлено Тяббесом (Tjabbos, 1932) и Клеимом (1933), показавшими, что магнитные свойства красных пероксохроматов соответствуют тем, которые Можно ожидать для пятивалентного хрома (<;р. т. I, стр. 341). Напротив, магнитные свойства синих пероксохроматов и продуктов присоединения пиридина к пятиокиси хрома подобны свойствам хроматов и бихроматов, т. е. соединений с шестивалентным хромом. Пятиокнсе хрома содержит, как следует из расхода перманганата, две перекисные группы. Следовательно, назван ным соединениям отвечают следующие структурные формулы:
СХ /0-0“ | ;сг$о-о~ ох ХО~О
м+ м+ м+
Синяя перекись хрома [Пятиоким хрома (¥1)}
Синие пероксахроматы [Пероксохроматы (%)]
Красные пероксохроматы [ Пероксохроматы (¥)]
Установление строения этих соединений устранило противоречие, существозаЕ-шее между прежним предположением о том, Что хром в них семиваленген, ц совремеп-пыми представлениями о зависимости между валентностью и строением атома
Шестая группа периодической системы
179
В соответствии с последними хром не может быть семивалентен, так как в этом случае он должен был бы отдать электрон из аргонной оболочки, что трудно представить ввиду прочности электронной конфигурации в оболочках инертных газов.
Спя я л перекись хрома [пятиокись хрома(УГ)] СтО5 известна только в растворах и в форме продуктов при соединения. Последним отвечает общая формула СгО6'Аш, где Ат означает нейтральную молекулу (пиридин, анилин, хинолин или эфир). Неустойчивость сипей перекиси хрома в свободном состоянии можпо объяснить тем, что ее надо рассматривать как координационно ненасыщенное соединение, которое для стабилизации требует координационного насыщения за счет присоединения нейтралы ной молекулы. Легче всего получается продукт присоединения пиридина к синей перекиси хрома. Его состав выражается формулой СгО5- руг, оп кристаллизуется в виде темно-синих листочков. В воде пе растворяется. Продукт присоединения метилового эфира СгО5-(С11.,)2О, который раньше ошибочно считали кислотой Н,СгО8, получают действием Г12Оа на СгО3 в растворе метилового эфира при низких температурах. Он представляет собой темно-сипне иглы, распадающиеся со взрывом при температуре около —30°.
Синий пероксо хромат кал пн получают осторожным прибавлением 30 %-пой перекиси водорода к раствору бйхромата калия, охлажденному до 0°. Он образует сине-фиолетовые (обладающие дихроизмом) призмы состава КгСг->О13-2Н2О, Которые при нагревании, ударе или при соприкосновении с концентрированной серной кислотой взрываются. При комнатной температуре пероксохромат постепенно разлагается, образуя бихромат калия.
Синий пероксохромат аммония кристаллизуется, как и пероксохромат калия, с двумя молекулами воды, а соль таллия Т12Сг2О12 кристаллизуется без воды.
Красный пе роке ох р ом ат калия К3СгО8 получается при действии 30%-ной перекиси водорода на i ил ыющел очной раствор хромата калия. Он кристаллизуется в темных красно-коричневых призмах. При обычных температурах К3СтО8 довольно устойчив, по при температурах выше 17и° претерпевает разложение, которое нри дальнейшем повышении температуры может привести к взрыву.
В водном растворе при обычных температурах пероксохроматы неустойчивы. В щелочном растворе они распадаются, давая вновь хроматы
[Сг2012]*4’20Н' = 2[Сг04]"4-Н2О-[-57гО2, „
2[СгО8]"' + Н2О =2[CrOJ’ 4- 20 Н' + 7/2О2.
В кислом растворе пероксохроматы переходят в соединения хрома(Ш) [Сг2О I2]’ 4-ЗН’=2Сг—-р 4Н2О+4О2,
[СтО8Г' -j- 6Н‘=Сг— 4-ЗН2О 4- 5/2О2.
Если к растворам добавить перекись водорода, то она каталитически разлагается (при обычной температуре). Поэтому получение пероксохроматов должно проводиться при низких температурах (ниже 0ь).
Аналитические сведения. Аналитически трех валентный хром ведет себя, как алюминий: оп осаждается из водного раствора сернистым аммонием и, точно так же как алюминий, не в виде сульфида, а как гидрат окиси.
Хроматы постепенно восстанавливаются сернистым аммонием до солей хрома(Ш); быстрее протекает восстановление сероводородом. И наоборот, соединения хрома (Ш) легко переводятся в хроматы сплавлением с содой л селитрой (окислительное сплавление). Соединения хрома(Ш) легко обнаруживаются описанной выше «хромпероксидной» реакцией.
В качестве микрореакций может служить осаждение бихромата серебра Ag2 [Сг20,] из азотнокислых растворов нитратом серебра или осаждение хромата бензидиния. Для обнаружения небольших количеств хрсма(Ш) при значительном оазпедении (и в каче
12*
180
Глава 5
стве капельной реакции) очень удобна цветная реакция ионов Ст2О' с дифенил карбазидом (фиолетовое окрашивание). Для этой реакции соли хрома(Ш) следует сначала окислить персульфатом калия с добавлением нитрата серебра как Катализатора (в кислом растворе) [F е i g 1, Angew. Chem., 44, 741 (1931)].
Сопи хрома окрашивают фосфатные перлы в темно-зеленый цвет как в окислительном, так и в восстановительном пламени, поскольку в присутствии щелочен наиболее устойчивым окислительным состоянием хрома является трехвалентное (к которому относится зеленый фосфат хрома).
Для количественного определения хром осаждают из растворов солей хрома(Ш) (удобнее всего при добавлении нитрита аммония или других веществ, нарушающих равновесие гидролиза) и взвешивают после прокаливания в виде окиси Сг2О,3. Если хром находится в виде хромата (или бихромата), удобнее всего определять его иодометрически. Для весового анализа хром осаждают в виде хромата бария ВаСтО4 или хромата ртутиД); последнее соединение переводят прокаливанием в окись хрома(Ш):
2Hg2CrO4=Cr2O3-f-4Hg4-5/2Qa.
Хром можно определять электролитически, выделяя на ртутном катоде. Для этого преимущественно употребляют вращающийся ртутный электрод [Т в t und-1 i б, Z. anorg. Chom., 202, 297 (1931); 215, 19 (1933)].
МОЛИБДЕН (MOLYBDAENUM) Mo
Распространение в природе. Молибден встречается в природе преимущественно в виде молибденового блеска (молибденита) MoS3 и желтой свинцовой руды (молибденово-свинцового шпата, вульфенита) РЬМоСД. Молибденовый блеск находят во многих местах, но редко в больших количествах. Желтая свинцовая руда присутствует в свинцовых месторождениях как вторичный минерал, по довольно редко.
Молибденовый блеск иногда сопровождается другими молибдатами, такими, как повеллит СаЧ,эО4, пцтераит СоМо04, белонооит MgMo04. Чаще, ио всегда в очень Небольших количествах встречается молибденовая охра (молибдит) Мо03.
В незначительных концентрациях молибден очень распространен в природе. Поданным тер Йойлена (II. ter Aleulea, 1931), следы Мо присутствуют во всех ранте-ниях. Молибден необходим для жизнедеятельности а в от об а кте р и й ей гоос ос с нт, способных связывать азот воздуха (Bortels, 1930). Следы молибдена найдены также в тканях животных и человека, прежде всего в селезенке и печени*
Исторические сведения. Древние греки и римляне называли «молибденом» (jidWpSaiva, тоlybdaena) свинцовый блеск, а также некоторые другие свипцовые руды (по-гречески piAn^Sos — свинец). Позднее это название было распространено на другие минералы (которые подобно свинцовому блеску окрашены в черный цвет), прежде всего па графит и минерал, называемый теперь молибденовым блеском. Графит и молибденовый блеск, которые очень близки по внешнему виду, долгов время считала одншд и тем же минералом. Позднее их обычно называли «рисовальный свинец»* и «водный свинец» (латинское plumbago). Впервые их различие обнаружил Шеелен 1778 г. Еиу удалось разложить молибденовый блеск («водный свинец») азотной кислотой, причет он получил белую окись (трехокись молибдена МоО3), которую назвал мояибденонх! кислотой (uctdiim molybdaenae). Металл, являющийся основой этого окисла,— молибден — был выделен в свободном состшшял Хельмом (Hjelm, 1782),
Получение. Основным исходным продуктом для получения молибден служит молибденовый блеск. Поскольку природный молибденовый блеет обычно очень сильно загрязнен (большинство перерабатываемых рут;
Шестая группа периодической системы
481
содержит всего лишь несколько десятых процента чистого сульфида MoS2), его следует подвергать предварительному обогащению. В настоящее время для этого используют в основном флотационные процессы.
В настоящее время флотационный процесс почти везде является основным процессом обогащения руд (особенно сульфидных). Флотация основана на том, что некоторые вещества плохо смачиваются водой, но хорошо бензином, нефтью и другими минеральными Маслами. Если вещества, которые тонут в воде, в топко измельченном состоянии тщательно перемешать с водой и маслом, то вокруг отдельных зерен, плохо смачиваемых водой, возникнет пленка масла и они приобретут способность плавать, так как подъемная сила капелек масла будет больше веса окруженных ими мелких зерен. Если руда наряду с большим количеством легко смачиваемых водой составных частей содержит небольшое количество плохо смачиваемых водой, но хороню смачиваемых маслом веществ (одним из таких веществ является, например, сульфид молибдена), то при флотации эти вещества остайутся в пене, а остальная часть руды осядет па дно. Вместо масла для флотации можно использовать некоторые пепообразующие добавки или барботацито газа. В этой форме флотационные процессы в настоящее время нашли повсеместное применение. Образование пены достигается барботированием воздуха или созданием разряжения. Те вещества, которые смачиваются водой, можно сделать не смачивающимися или трудно смачивающимися, используя определенные добавки, которые адсорбируются поверхностью частиц. Действие Этих добавок (называемых «собирателями», так как они способствуют собиранию в пене извлекаемых частиц минерала) основано на том, что они образуют на поверхности частиц минерала пленку, на которой пузырьки воздуха удерживаются за счет капиллярных сил. Чтобы стали ясны современные методы проведения флотационного процесса, следует, по Оствальду (1932), прежде всего учесть значительные капиллярные силы, действующие в местах соприкосновения трех фаз (в данном случае: воздуха, воды и частицы минерала или покрывающей се п.тепки масла). Эти представления, согласно которым частицы минерала-в значительно меньше» степени выносятся на поверхность за счет подъемной силы связанных с ними нунырыюп воздуха, чем втягиваются в пену мод действием сил па границе трех фаз, хорошо согласуются с тем наблюдением, что всплывание минеральных частиц часто одновременно стабилизирует попу. Введением так наэываемых-«регу лиру тощих всплывание средств» можно изменять действие вызывающих образованно пены добавок («вспенивателей») и «собирателей». Благодаря этому становится возможным последовательно извлекать из одной руды различные минералы, например сульфид свинца и сульфид цника (селективная флотация).
Комбинируя сильнодействующие «собиратели» с подходящими «регуляторами», можно отделять флотационным методом даже окисные руды (окислы, карбонаты и т. д., которые сама по себе прекрасно смачиваются водой) от окисных же примесей (жильных пород). Однако чаще предпочитают при помощи соответствующей обработки получить на поверхности частиц таких минералов сульфидный слой, в результате чего они ведут себя при флотации подобно сульфидным рудам.
Молибденовую руду, обогащенную до и более 70% MoS2, переводят в трехокись молибдена обжигом [уравнение (1)] или сплавлением с содой в пламенной печи и разложением получающегося молибдата концентрированной серной кислотой [уравнения (2) и (3)1. Металл получают из трех-окиси восстановлением водородом [уравнение (4)1 или углем (или угле-родсодержащимн веществами, например канифолью);
MoS2 + ’/s02=Mo03-f-2S03, (1)
МоЯг+ЗКа2СО3+’/гО2=КагМоО4+2Ка28О44-ЗСОг, (2)
Na2MoO44-2HCl=Mt>O3-f-H2O.4-2NaCl, (3)
Мо03 + ЗН2=Мо+ЗИ2О. (4)
Вследствие высокой температуры плавления молибден сначала получается в виде порошка. Затем, прессуя порошок в стержни и нагревая
их в атмосфере водорода переменным током низкого напряжения почти до плавления, можно получить компактный ковкий молибден.
182
Глава 5
Компактный молибден получают также при электролитическом выделении его из эвтектического расплава КС1 — NaCl с содержанием 4 мол.% КэМоС16. Применяя в качестве защиты от окисления аргон, получают 99,9 %-ный молибден.
Коллоидный молибден можно получить, как и коллоидный хром, электрическим распылением под слоем изобутилового спирта или поочередной обработкой топкоизмельченного молибдена разбавленной сол.япой кислотой и разбавленным едким натром. Молибденовый порошок, легко пептизирующийся при такой обработке, получают нагреванием МоО3 или _Х1оО2 с цинковой пылью.
Свойства. Порошкообразный молибден более или менее темного серого цвета, без блеска. Напротив, компактный металл — серебристобелый и блестящий. Оп довольно тверд, по полируется, а при высоких температурах куется н сваривается. Удельный вес молибдена 10,2. Температуря плавления около 2630°. Температура кипения исключительно высокая. Лапгмюр определил скорость испарения молибдена в вакууме при температурах вплоть до температуры плавления ц рассчитал из этих данных соответствующие давления паров. На основании полученных им данных можно вычислить, что температура кипения молибдена при атмосферном давлении составляет примерно 4800°. Молибден хорошо проводит электрический ток; удельная электропроводность при 0° составляет около 34% от удельной электропроводности серебра.
При обычных температурах молибден довольно устойчив па воздухе. При температуре красного каления даже компактный металл довольно быстро окисляется до трехокиси МоОз. При повышенных температурах он реагирует также с хлором и бромом, а с фтором — уже на холоду. Напротив, молибден не реагирует с иодом даже в раскаленном состоянии. Водород очень мало поглощается даже очень тонко измельченным молибденом и уже при температуре 300° полностью удаляется. С углеродом же молибден реагирует при нагревании, образуя карбид. Молибден способен непосредственно реагировать с окисью углерода, если действовать сю при высоком давлении на тонкий порошок молибдена. При этом образуется гексакарбопил Мо(СО)в (сильно преломляющие свет, легко летучие кристаллы удельного веса 1,96). Элементарный азот с трудом связывается молибденом. Нагреванием молибденового порошка в газообразном аммиаке получают нитриды MoaN и MoN.
Согласно Нейману (Neumann, 1934), теплота образования Mo2N составляет 16,6 ккал/молъ. Выше 600° образуется, по Хэггу (1930), еще и третий нитрид, содержащий 28 ат.% азота. Небольшие количества азота молибден может связывать в виде твердого раствора (Sieverts, 1936).
При взаимодействии молибдена с углеродом, как правило, образуется карбид Мо2С, изоструктурный карбиду Та2С н W2C. Кроме этого соединения, существует еще один более богатый углеродом карбид, возможно, состава МоС. Это соединение, по-види-мому, имеет кубическую структуру. Но может также образовывать твердые растворы с гексагональным (вероятно, структурно родственным каменной соли) карбидом вольфрама WC и в этой форме получается гораздо легче, чем в свободном состоянии (Weiss, 1946—19i8; Lauder, Gernier, 1947; Nowotny, Kieffer, 1952).
Разбавленные кислоты не действуют на молибден. Из концентрированных кислот па него действует только концентрированная азотная кислота. Но так как последняя, как и другие окислители, пассивирует молибден, то окисление ею происходит очень медленно. (Умеренно концентрированная азотная кислота реагирует с молибденом энергичнее, чем очень концентрированная.) Молибден окисляется также нагретой почти до кипения концентрированной серной кислотой. Энергичнее дей
Шестая группа периодической системы
183
ствуют царская водка, а также смеси азотной кислоты с фтористоводородной или серной кислотами. В щелочах молибден практически нерастворим. Очень медленно реагирует он и с расплавленными едкими щелочами. Напротив, быстрое окисление происходит при сплавлении с нитратом калия, хлоратом калия или перекисью натрия.
Применение. Молибден применяют в основном при производстве специальных сталей для оружейных стволов, танковой брони, валов и т. д. Уже при очень небольшой добавке молибдена сталь приобретает большую прочность и вязкость. Наряду с хромом, никелем, кобальтом и ванадием молибден используют для получепия быстрорежущих сталей. Его применяют также при производстве магнитных сталей и кислотостойких сплавов. Во всех этих случаях молибден обычно используют в форме ферромолибдена, т. е. сплава железа с высоким содержанием молибдена, который легче плавится и труднее окисляется, чем чистый молибден.
Молибден вследствие высокого сродства к кислороду нельзя применять в качестве раскисляющего средства, так как он удерживает образующийся окисел в твердом растворе. Поэтому, если молибден входит в состав легированной стали, следует принять специальные меры для лучшего раскисления, что достигается добавлением титана или ванадия.
В качестве материала для нитей ламп накаливания молибден менее пригоден, чем вольфрам, так как он скорее распыляется. Напротив, молибден широко применяют для изготовления держателей для нитей ламп накаливания, так как он легко впаивается в стекло и такие спаи не пропускают газ.
Из соединений молибдена паиболее широко применяют молибдат аммония, главным образом для определения фосфорной кислоты (например, в искусственных удобрениях).
СОЕДИНЕНИЯ МОЛИБДЕНА
Молибден образует соединения в различных состояниях окисления. Максимальная валентность молибдена, как и хрома, равна шести. Исключая минерал MoS2, мол беден в тех его со единениях, которые имеют практическое значение как таковые (а именно трехокись молибдена МоО3 и ее производные молибдаты M2IMoOJ), шестивалентен.
Обзор
Хлориды Фториды Окислы
— MoFe M0S3 MoO3
— — — МоэО23
— — — MogOja
МоС15 —' -—•- Mo2Oj ?
МоС14 MoF4? MoSB и Mo(S2)2 Мо02
МоС13 MoF3? Mo2S3 ? МоэО3?
МоСЬ или Мо3С10 — — —
Хлориды. Нагреванием порошка молибдена в токе хлора получают пентахлорид молибдена МоС15 в виде темно-красных паров, конденсирующихся в темно-зол епые, почти Черные кристаллы. Т. пл. 194°, т. кип. 268°. По данным Дебрея (Debray), плотность паров (при 350°) соответствует формуле МоСЦ (9,5 по отношению к плотности воздуха). Лентахмрид молибдена тте проводит электрического тока даже в расилавлея-вом состоянии. С водой он реагирует с обильным выделением тевла, образуя оксихлорид:
MoCls+ Н20 =МоОС134-2нС1.
184
Глава. 5
Тетрахлорид молибдена МпС14 удобнее всего получать пагревапием двуокиси молибдена с растворенным и четыреххлористом углероде хлором (при 250°). Он получается в виде коричневого порошка, который легко возгоняется, образуя интенсивно желтые пары. При нагревании в заплавленной ампуле МиС1; частично распадается на МоС15 н МоС13.
Трихлорид молибдена МоС1г получают, пропуская пептахлорид пад нагретым молибденом (или нагрела и пом в токе водорода). Оп образует темно-красны и кристаллический порошок, нерастворимый в воде и в соляной кислоте. Пурпур но-красные водные растворы трихлорнда молибдена можно получить электролитическим восстановлением на ртутном катоде растворенной в концентрированной соляной кислоте трех-окиси молибдена, В таких растворах трихлорид молибдена существует в виде хлоро-молибдат(1П)-ионив. При добавлении щелочей в зависимости от соотношения концентраций выделяются гексахлоромолибдатьДЩ) М3{МоС1с,] пли пептахлороаквомо-Либдаты(Ш) | MoCls(OH2)]. Органические дмишл обычно присоединяются к трихлориду молибдена МоС13 с образованием неэлектролитов [МоС13Ат3]. Возможно присоединение нейтральных молекул с образованием солей с комплексными молибден(Ш)-иатиопами, ш, пример (MoCl2(NH3)4]Cl (Rosenheim, 1931). Безводный трихлорид молибдена при нагревании до слабого красного каления в токе сухой двуокиси углерода распадается па МоС14 и MoCL.
Датлорид молибдена MoCI, или Мо3С1в представляет собой желтый, нерастворимый в воде порошок. Однако он растворяется в спирте и эфире и имеет в этих растворах молекулярный вес, отвечающий формуле Мо3С16. При нагревании в токе водорода он восстанавливается до металлического молибдена.
Известен также гидрат дихлорида молибдена. Его состав выражается формулой Мо3С1е.4НгО. Как доказал рентгенометрически Броссе (Drosset, 1946), это соединение имеет структуру [МовС18]С1,й-8Н2О. Следовательно, в нем имеется шестнядериый хлорокомплекс, в образовании которого, однако, принимает участие только 2/3 содержащихся в соединении иопов хлора. Ионы хлора, пе связанные в комплекс, легко могут быть заменены другими отрицательными ионами, например ионами ОН-, причем получается кристаллизующееся с 14 молекулами воды соединение [ МоеС18](ОН)4, структура которого также определена рентгенометрически. То, что в дихлориде молибдена (а также в дибром идо) часть атомов галогенов легко обменивается па другие кислотные остатки, было отмечено уже Бломстрапдом (Biomstrand, 1859).
Фториды, Если по отношению к Хлору молибден может быть максимально пятивалентным, то с фтором он образует гексафторид MoF0, который был получен Руффом (при непосредственном соединении элементов) в виде очень гигроскопичной белой ктш-сталлической массы, которая при 17“ плавится в бесцветную жидкость и кипит при 35е. Это соединение очень реакционноспособно и особенно чувствительно к влаге. Более устойчивы (также бесцветные) оксифториды MoOF4 и MoCbFo- Ив их водных растворов при добавлении фторидов щелочных металлов кристаллизуются оксофторомолибда-ты(¥1), например K2[Mo02Fi]- Н2О—бесцветные блестящие чешуйки. Сплавлением МоО3 с щелочными фторидами получают он со фторомолибдаты типа MjlMoOsF3). Только LiF реагирует е МоО3 при нагревании с образованием не фторомодибдата, a MoO2F2. Это объясняется тем, что для устойчивости комплексной соли существенна, кроме энергии образования комплексного иона, также энергия, выделяющаяся при образовании кристаллической решетка из комплексных и простых ионов. Последняя, как это можно рассчитать, в системе LiF— МоО3 меньше, чем сумма энергий решеток простых соединений, несмотря па положительную энергию образования комплексного иона [MoOjFjJ3". В случае триоксофторомолибдатов других Щелочных металлов соответствующие расчеты указывают^ па увеличивающуюся устойчивость в направлении Na Cs (Schmitz-Du moot, 1952). Щелочные триоксотрйфторомолвбдаты кристаллизуются в кубической системе и, по-видимо му, иэоструктурпы кристаллизующейся иэ водного раствора аммонийной соли (NH4)3[MoO3F3).
Известны также оксофториды пятивалентного молибдена, например K2IMOOF5]--Н2О — небесно-голубые блестящие листочки. Лежащий в их основе простой оксифторид до сих пор не выделен в свободном виде, как и пентафторад молибдена. Достоверно не установлено и существование тетрафторида и трифторида молибдена, Одпако для последнего известны двойные соли [фторомолибдаты(Щ)], например K[MoFt]-H2O — светло-фиолетовые кристаллы.
Сульфиды, Для молибдена известны два нормальных сульфида. Т дисульфид MoS3 осаждается сероводородом из подкисленных растворов молибдатов. Дисульфид MoS2 встречается в природе в виде минерала «молибденового блеска».
Шестая группа периодической системы 185
По старым данным, сухим путем можно получить также полуторный сульфид Mo2S3 (серо-стальные иглы). Однако, согласно новейшим исследованиям, существование этого соединения сомнительно.
Кроме нормальных сульфидов, существует еще высший сульфид молибдена состава Л1о84. По-видимому, оп представляет собой полноульфад Mo(S3}2, так как известны его производные соли, которые рассматривают как политиомолибдаты, т. е. соединения, которые образуются из молибдатов [вероятно, не из молибдатов(У1), а ив молибдатов(1У)[ при замене ионов О3- па нолисульфид-ионы S|”.
Трисульфид молибдена и тиомолибдаты. Трисулъфнд молибдена получают разложением кислотами растворов тиомолибдатов или длительным пропусканием сероводорода в теплые подкисленные соляной кислотой растворы молибдатов.
На холоду происходит неполное осаждепио сероводородом, так как сероводород частично восстанавливает молибдаты до молибденовой сини (см. ниже), которая образует коллоидные растворы. Трисульфид также легко образует коллоидные растворы. Для препаративных целей пригоден только первый из приведенных выше методов получения трисульфида молибдена, так как даже при нагревании выпадающий под действием сероводорода осадок не является чистым сульфидом молибдена.
Трисульфид молибдена представляет собой темно-коричневый осадок, легко растворяющийся в сернистом аммонии (а также в щелочных сульфидах) и царской водке. При нагревании без доступа воздуха он переходит, отщепляя серу, в дисульфид молибдена.
Растворимость трисульфида молибдена в сернистом аммонии объясняется образованием тиомолибдатов. Большинству из них отвечает общая формула М|[Мо84]. Однако известны некоторые тиомолйодаты сложного состава. Нормальные тномолибда-ты щелочных и щелочноземельных металлов легко растворяются в воде и в большинстве случаев хорошо кристаллизуются. Их окраска интенсивно красная. Кислоты разлагают их с выделением трисульфида молибдена:
(NH4)2[MoS4] + 2НС1=MoS3+2NH4C1+ H2S.
Дисульфид молибдена MoS2 встречается в природе как минерал молибденовый блеск. Большинство разновидностей этого минерала представляет собой плоские, тонкие, легко гнущиеся, жирные на ощупь и оставляющие серый след па бумаге, похожие на графит листочки удельного веса 4,7—4,8.
Молибденовый блеск (молибденит) очень редко встречается в виде хорошо образованных кристаллов. Он относится к гексагональной системе. Его кристаллическая, решетка приведена на рис. 26. Атомы Мо, как и атомы 8, расположены в плоскостях, перпендикулярных оси с, таким образом, что каждый атом Мо с обеих сторон сопровождается еще одним атомом 8 (на расстоянии с/8 = 1,54 А), Следовательно, как и в графите (ср. т. I, стр. 462), в данном случае имеется слоистая решетка. Этим объясняется исключительная легкость скалывания молибденового блеска перпендикулярно оси с и его преимущественная пластинчатая форма, похожая на форму графита. Расстояния между центрами атомов составляют:
Мб <----> S=2,35 А; МО < Мо = а=3,15 А; 8^ >S = -^-=3,08 А.
4
В европейских Странах молибденовый блеск встречается во многих местах, по в большинстве случаев в очень небольших количествах. Значительные месторождения находятся в Норвегии. Основные залежи открыты в Австралии и Северной Америке,
Дисульфид молибдена можно получить искусственным путем: например, он образуется при нагревании двуокиси молибдена, трехокиси молибдена или молибдата аммония в токе сероводорода. При прокаливании без доступа воздуха это соединение теряет часть своей серы только при очень высоких температурах (в печах Муассана), образуя предположительно полуторный сульфид Mo2S3. Напротив, на воздухе MoS2 легко сгорает До трехокиси молибдена МоО3.
186
Глава 5
Окислы. Наиболее устойчивым окислом молибдена является трех-
окись МоО3, которая поэтому получается как конечный продукт при окислении молибдена кислородом воздуха. Легко получается и двуокись молибдена МоОа. Есть указания на существование окислов Мо305 и Мо2Оз. Интересно существование промежуточных окислов МовО2в
и Мо8Оаз, которые, несмотря на их сложный состав, представляют собой
Рис, 26. Элементарная ячейка молибденового блеска.
а = 3,1 &, с = 12.30 А.
определенные соединения с характерной кристаллической структурой.
Трехокись молибдена МоО3 получается при прокаливании на воздухе молибдена, сульфидов молибдена и других соединений молибдена или при действии на эти соединения азотной кислоты. Обычно ее получают длительным прокаливанием молибдата аммония (см. ниже) на воздухе или многократным выпариванием последнего ,с азотной кислотой и экстрагированием получающегося нитрата аммония водой. Трехокись молибдена представляет собой мягкий белый порошок удельного веса 4,5. При нагревании она желтеет. Трехокись плавится без разложения при 791°. При температурах, близких к температуре плавления, она начинает возгоняться. Прокаленная трехокись молибдена очень мало растворима в воде. Она не растворяется и в большинстве обычных кислот, но растворяется в плавиковой и концентрированной серяой кислотах. МоО3 легко растворяется в щелочах, даже в водном аммиаке и в растворах карбонатов, образуя молибдаты — соли общей формулы M*[MoOJ. Трехокись молибдена можно считать ангидридом молибденовой кислоты Н2МоОь лишь с большими оговорками, так как хотя она легко получается при нагревании последней, но
при простой обработке водой не превращаетсн вновь в молибденовую кислоту.
Если трехокись молибдена нагревать в токе сухого хлористого водорода до 150— 200°, то образуется сублимат, состоящий нз бледно-желтых игл состава МоО3-2НС1, который легко растворяется в воде, спирте, эфире, уксусной кислоте и ацетоне. При упаривании водного раствора остается трехокись молибдена. Эта реакция позволяет проводить легкое отделение молибдена от таких веществ, которые не образуют летучих окислов. С фторидами щелочных металлов (кроме LiF) трехокись молибдена образует бесцветные двойные соединения общей формулы М3 [Mo03F3|, кристаллизующиеся в кубической решетке (ср. стр. 184).
Пятиокись молибдена Мо206 должна получаться в виде черпо-фиолетового порошка при нагревании МоО(ОН)3 в токе двуокиси углерода. МоО(ОН)3 выпадает прп добавлении аммиака к растворам солей молибдена(V) в виде красно-коричневого осадка.
Промежуточные окпелы Mo3O2fi и Мо8О2з, в которых молибден существует в среднем валентном состоя пни соответственно 5,78 и 5,75, имеют голубовато-черный цпед и интересны в связи с нх отношением к молибденовой сини. Их получение и выяснес~? структуры было выполнено в работах Хэгга и Магцели (Hagg, Magneli, 1944—1948).
Двуокись молибдена. МоО2 образуется как промежуточный продукт окпслге~е при осторожном нагревании молибдена на воздухе. Ее получают также ира пропугшт-нии паров воды над раскаленным молибденом или при нагревании в токе водорЕдг нагретой до 470° МоО3, а также при восстановлении молибдатов, палример прн пх сееез-
Шестая группа периодической системы
187
лепип с цинком. Двуокись молибдена представляет собой коричнево-фиолетовый с медлим отливом порошок, хорошо проводящий электрический ток. В противоположность трехокиси молибдена опа пе растворяется в щелочах. Не растворяется она и в кислотах. Азотной кислотой двуокись молибдена окисляется до трехокиси. МоО2 окисляется и аммиачными растворами солея серебра, яз которых она выявляет серебро. С хлором двуокись молибдена образует соединение МоО2С12 и в противоположность трехокиси не образует продуктов присоединения с хлористым водородом. Кристаллическая решетка двуокиси молибдена относится к тину рутила (см. т. I, рис. 63); а — 4,86 А., с = 2,79 А.
Гидратированная форма двуокиси молибдена (в виде черпо-коричневого шлама) была получена Паа леи (Раа!) при восстановлении подного раствора молибдата аммония водородом, в присутствии коллоидного палладия. После очень осторожного высушивания содержание води в этом шламе приблизительно отвечало формуле МоО(ОН)г. Высушивание при нагревании приводило к почти безводной двуокиси. Двуокись молибдена можно получить в виде обратимого коллоида, если в качестве защитного коллоида использовать и ротор б инов о кислый патрий. При дальнейшем воздействии активированного водорода (при нагревании и небольшом избыточном давлении) образуется гель полуторной окиси Мо2О3 (черные, отливающие голубым чешуйки).
Производными двуокиси молибдена являются молибдаты(1У), полученные [Польдером (Scholder, 1963) в форме соединений щелочноземельных металлов типа M^MoOj и М^МоО4 ПРИ восстановлении молибдатов(VI) водородом.
Можно получить и молибдаты(У); Шольдер получил их При «компропорциониро-вапии» молибдатов(IV) и молпбдатов(У1),
Молибденовая кислота и молибдаты
Как уже было сказано, трехокись молибдена легко растворяется в щелочах, образуя молибдаты [точнее молибдаты(У1)[. Состав простейших молибдатов отвечает формуле MjfMoOJ. Однако эти соединения можно получить только из растворов, содержащих большой избыток гидроокиси щелочного металла. Большинство молибдатов содержит несколько групп MoOg на одну группу М2О. Это так называемые полимолибдаты. Торговый «молибдат аммония» также представляет собой полимолибдат.
Из азотнокислых растворов молибдатов при длительном стоянии часто выделяется плохо растворимая в водо молибденовая кисЛота. Опа представляет собой капа реечно-желтые моноклинные кристаллы состава Н2МоО4- Н20. Она легко образует коллоидные растворы, которые получал уже Грэм. При незначительном нагревании в водном растворе это соединение переходит в безводную молибденовую кислоту НгМоО4. Последняя кристаллизуется в виде мелких белых игл. Она существует в двух модификациях, которые мало различаются по внешнему виду, но переходят в ангидрид при совершенно различных температурах, и, кроме того, одна из них образует с водой подобную молоку, не разделяющуюся фильтрованием суспензию, тогда как для другой это совсем не характерно.
В соответствии с данными Япдера (lander) и Яра (Jahr, 1930), а также Бая (Вуе, 1942) и Карпени (Carpeni, 1947), в растворах молибдатов существуют в зависимости от концентрации ионов водорода различные виды молибдат-иопов. Кроме присутствующих в щелочных растворах ионов |MoO4j", это главным образом попы |НМо6О2(]""', [MotO^I""", [Mo802G1"", H7[Mo(204Jm*, Н7[Мо24О78] и Нв[ 1\1о,л0781’". Между этими ионами устанавливаются соответствующие, зависящий от концентрации ионов водорода равновесия, которые аналогичны равновесиям, приведенным для поливанадат-ионов (см. стр. 118). Несмотря на то что этим равновесиям соответствует только сравнительно небольшое число типов молибдатов, а именно (исключая редко выделяющиеся тримолкбдаты **) только гекса-, гепта-, окта-, додека- и икоздтетрамолибдаты, из раст
* Возможно, что этот иок является иоаом додекамолибдатодиводород пой кислоты H3[H2MoI204olw (см. стр. 202).
** В качестве примера тримолибдата можно привести (безводную кристаллическую) соль ЬцМоаОц. Ион [Мо3О11|'"', в соответствии з данными Яддора, и Яра, можно рассматривать как промежуточный продукт конденсации мопо- в гексамоллбдат-ионы. Однако в растворе этот ном- неустойчив.
188
Глава 5
воров могут выкристаллизовываться соли различного состава, так как в лежащих в их основе поликислОтах на атомы металлов можно заместить различное число атомов водорода. Так, известны Ш’п та натрий-, тетранатрий- и трннатрийгексамолибдаты (NaJHMoaOsiM8H20, Na.HJHMo6O2Jl- 13Н2О и Na3H2[HMo6OsJ.7«/2Н2О). Вследствие совершенно различного молекулярного отношения Na2O : Мои3 в этих трех солях (5 : 12, 1 : 3 и 1:4) па первый взгляд совершенно невозможно установить какую-либо связь между ними, Однако, как показывают приведенные выше формулы, это три соли одном и той же кислоты. Точно так же многочисленные другие иолимолибда-ты, аналитический состав которых часто очень сложен, можно свести к нескольким Приведенным выше тинам.
Молибдат аммония. Торговый молибдат аммония, азотнокислый раствор которого используют как: реактив на фосфорную кислоту, получают выпариванием раствора трехокиси молибдена в водном аммиаке. Он выделяется в виде больших бесцветных моноклинных призм. Раньше Это соединение считали тригидратом пента аммоний водород гексамолибдатом (NH4)5H [Мо0О2(1 -ЗН2О. Однако новейшие исследования (Garelli, 1930, Sturdivant, 1937, Lundqvist, 1950) показали,, что соединение является тетрагидратом гсксааммонийгептамолибдата (NH4)0(Mo7O24| -4Н2О.
Нормальная соль (NH4)2[M<jO4] получается при растворении трехокиси молибдена в теплом аммиаке. В отличие от этого соединения торговый молибдат аммония называют также парамолабдатом аммония. Из концентрированных растворов последнего, если они при стоянии на воздухе теряют аммиак, может выделиться соединение (WIf4)4H2/Mc?et}211.5НеО — пелгагидрат тетрааммоннйдиводорадгексаиолибдага. Известен и иолимолибдат аммония, содержащий до 10 ЙоО3 па 1 (МН4)2О,
Если азотнокислый раствор молибдата аммония добавить в большом избытке к подкисленному азотной кислотой раствору фосфата, то выпадает молибдатофосфат аммония в виде красивого желтого осадка состава (NH4)jPO4-12MoO3-6H2O *. Эта реакция очень удобна для обнаружешиц и определения фосфорной кислоты. Однако следует учитывать, что аналогично реагируют с молибдатом аммония мышьяковая, а в некоторых условиях и кремневая кислоты.
Молибденовая синь. Если раствор молибденовой кислоты или подкисленный раствор молибдата обработать восстановителем (например,, двуокисью серы, сероводородом, иодистым водородом, гидразином, виноградным сахаром, цинком, металлическим молибденом), то возникает темпо-голубая интенсивная окраска, вызванная образованием (в большинстве случаев коллоидного) раствора молибденовой сини. Эту реакцкн: используют для аналитического обнаружения молибдена.
Продукты, называемые молибденовой синью, представляют собой смесь разнообразных веществ. Кроме общего для них тем но-гоя убог о цвета, их объединяет г—" и то, что все они содержат молибден в среднем валентном состояний, которое меньег б, гго не меньше 5. При осаждении из водных растворов всегда получаются рентгелз-аморфные продукты с переменным содержанием воды, которые более или менее лете, образуют коллоидные растворы. Можно получить молибденовую сиггъ и в крйеталлл-ческом состоянии, а именно в форме уже упоминавшихся Промежуточных окиелгл Мо0О2(; и i\logO23 также в гидратирован в он форме. Глейзеру удалось получить в кристаллической форме синие соединения MogO^-8tJ2O, Мо4Ои-НгО и Мо2О5-Н2О и охарактеризовать нх (аналитически, рентгенографически и методом изобарного раз.тозшг-ния) как индивидуальные соединения. Вода в этих соединениях связана конституционно, и поэтому Глемзер рассматривал этн соединения как гидроокиси:
MogOjjfOHJia, Мо4Ою(ОН)2 и Мо2О4(ОН)2.
* О строышЕ этого соедвпевия см. на стр. 202.
Шестая группа периодической систем»
189
Они устойчивы к действию едких щелочей и аммиака и отличаются от аморфных синих препаратов, например MosOya;HaO(Audrieth, 1942) и Мо4О(5-х[120 (Glemser, 1951). Соединения Мо40(о (ОН)г и Мо2О4 (0Н)а можно получить также действием атомарного водорода на MoOs. При этом, а также при действии Е1А1Щ на МоО3 получается, кроме того, бордов о-красное соединение MojOifOHJg. Его существование доказано, кроме данных анализа, также рентгенографически и то нзиметри чески (Glemser, 1952). Соединение Mo5O7(DH)s образуется при действии атомарного водорода иа MofK не подо средств с и но, а за счет отщепления На от первоначально образующегося олив ков о-зеленого соединения аналитического состава МоО(ОН)2, которое является, как заключил Г л ем э ер па основании его поведения, не оксогидроокисью молибдена (IV), а оксогидро-ксогидридом Н2Мо5О7(ОП)8. Легко растворяющаяся в воде молибденовая синь MosOg-OH была получена Тредвеллом (Treadwell,. 1946) электролитическим восстановлением парамолибдата аммония в сорнокислом растворе. Она является одноосновной кислотой и имеет молекулярный вес, отвечающий приведенной выше формуле. Присутствует ли в соединениях, относящихся к классу молибденовых синей, наряду с шестивалентным молибденом также нити- или четыреквалеятный, еще следует выяснить.
Коллоидная молибденовая синь легко адсорбируется поверх и ост по активны ми веществами, особенно растительными и жилетными волокнами. Поэтому она является типичным красителем и находит применение для крашения щелка.
Пероксомолибденовая кислота и пер оксомолибдаты. Кислые растворы молибдатов при добавлении к ним перекиси водорода окрашиваются в желтый или оранжево-красный цвет. Из этих растворов можно выделить точно так же окрашенные соединения, которые отличаются от молибдатов большим содержанием кислорода и дают реакции, типичные для перекиси Водорода (обесцвечивание, перманганата, сииее окрашивание с хромовой кислотой, и г. д.). Следовательно, опи являются производными молибдатов, в которых атомы'кислорода замещены перекисной группой —О—О—, и их называют пероксомолибдатами. Простейшим пероксомолибдатам отвечают формулы MiHMo06-aq (бледно-желтые) и M’MoOs-aq (красные, взрывающиеся). Аналогично молибдатам,, однако, известны и пероксомолибдаты гораздо более сложного состава.
В соответствии с данными Яра (1941) в растворе устанавливаются равновесия:
Мео;+2нао3 нмоо;4-он'-|-нго,
МоО;+4Н2Оа MoOJ+4H2O.
(6)
Если ионы ОН', образующиеся по уравнению (5), связывать кислотой, то происходит количественное образование водороддипероксомолибдат-иопов IIMoOJ. При дальнейшем увеличении кислотности происходит распад последних по уравнению HMoOj —> МоОд + Н' 4- Os и конденсация образующихся ионов МоОд в гексамолиб-Дат-иопы НМовОа1""':
6MoOJ4-7H- НМо60;'1"' + ЗН20.
Ионы гексамолибдата могут вновь реагировать с избыточной перекисью водорода, образуя очень устойчивые перокеогексамолибдапг-ионы, существование которых было доказано нзмсфенйом. скоростей диффузии. Соединения, в состав которых входят эти иоггы, окрашены в цвета от интепенвцо желтого до оранжевого и в большинстве случаев имеют очепь сложный состав.
Свободна я перокс-омолибденовая кислота H3MoOi5‘ Представляет собой
аморфный желтый порошок. Это соединение в противоположность синей перекиси хрома СгО5 не экстрагируется эфиром.
Образование желтой пор оксомолибденовой кислоты следует учитывать при испытании перекиси водорода па присутствие фосфорной кислоты при помощи молибдато-фосфатпой реакции. Пород проведением такого испытания перекись водорода следует удалить выпариванием.
190
Глава 5
Соединения молибдена с кислотами
С кислотами молибден не образует простых соединений (если не считать галогенидов и сульфидов, которые не имеют ярко выраженного солообразного характера), а образует многочисленные комплексные соединения (ацидосоли).
Ацидосоли шестивалентного молибдена получают главным образом растворением трехокиси молибдена в кислотах и добавлением соответствующей соли щелочного металла, а также растворением молибдатов в кислотах.
Если серную кислоту насытить нри нагревании трехокисыо молибдена, а затем дать такому раствору остыть, то из него кристаллизуется ди<>ксосульфат молибдена МоО2ЗО4 — бесцветные, расплывающиеся, блестящие шестигранные призмы. Растворением этого соединения в растворах щелочных сульфатов и добавлением избытка серной кислоты к растворам молибдатов щелочных металлов Вайнлапд (Weinland) получил окепеу-1ьфатомолибдаты(У 1} типа M.J |Mo2Oe(SOJ] и М| (Klo2O4(SO4)3J.
Более просты по составу оксофторомолибдаты(У 1), которые получаются растворением молибдатов в плавиковой Кислоте, добавленном фторидов к растворам трехокиси молибдена в плавиковой кислоте или сплавлением трехокиси молибдена с щелочными фторидами (см. также Стр. 184). Известны следующие типы этих соединений:
М’
Г °Л
Мо
-
Г °-Л
Мо
L f2J
м1
Г °21
Мо
L F3J
Ог.сохлоромолибдаты{У1), преимущественно типа М| (МоО2С14], были получены Вайнландом,
К оксогалогеновым солям относятся и простейшие ацидосоли пятивалентного молибдена. Фторо-соли уже упоминались на стр, 184. Хлоро-соли общей формулы М.! | МоОС15] получаются при добавлении щелочных хлоридов к растворам молибденпеи-тахлорида, а также из восстановленных иодистым водородом растворов трехокиси молибдена или молибдатов в соляной кислоте. Опи окрашены в яркий зеленый цвет, хорошо кристаллизуются и устойчивы в сухом воздухе. Бромо-conn аналогичного состава бывают темно-красные и оливково-зеленые. Известны также бромо-соли типа M^MoOBrJ. Они имеют такой же цвет. Пурпурно-красные в растворе родано-соли пятивалентного молибдена по своему составу аналогичны галогено-солям, тогда как состав известных к настоящему времени оксалато-солей значительно более сложен.
Из ацидо-солей четырехвалентного молибдена в первую очередь следует назвать октацчаномолибдаты(ГХг) Mj|Mo(CN)e). Они представляют собой отличающиеся устойчивостью комплексные соединения, которые получаются при действии большого избытка концентрированного раствора цианистого калия па соединения как трех-, так и пятивалентного молибдена. Растворимая в воде калиевая соль K4[Mo(CK)s]-• 2Н2О образует желтые ромбические пластинки. Из концентрированных растворов этой соли при добавлении дымящей соляной кислоты выделяется свободная кислота. Она кристаллизуется в тонких желтых иглах состава H4|Mo(CN)g]-6H2O. Октацнапо-молибдаты(1¥) особенно интересны тем, что в них молибден совершенно определенно имеет редко встречающееся у комплексных соединений координационное число 8. Если же добавить небольшой избыток цианистого калия, то образуютсяфиолетово-красные диохсотетрацианомолибдаты(ГУ) Mj(Mo02(CN)4]<
Из ацидо-солей трехваленткого молибдена наиболее известны, кроме уже упоминавшихся фторо- и хлоро-солей, роданомолибдаты(1И). Им отвечает общая формула М* |Mo(SCN)e], цвет их желтый или красный, и часть из них изоморфна родапо-хроматам(Ш). Это, например, справедливо для ромбической аммонийной соли (NII4)3|Mo(SCN)sp4H2O, которую проще всего получить электролитическим восстановлением па гладком платиновом катоде раствора молибдата аммония в концентрированной соляной кислоте в присутствии большого избытка роданида аммония. Это относится также к калиевой и натриевой солям. Соединением, в котором молибден, возможно, имеет очень редко встречающееся координационное число 7, является полученный Юнгом (.1 oiirig. 1932) цпапомолибдат(ТП) К4 |Мо(СК)7)-2Н2О.
Как ацвдо-голц "двухвалентного молибдена следует рассматривать полученные, впервые Бломсграггдом (В loin st. га nd, 1859) желтые продукты присоединения щелочных хлоридов к дих.т ер иду молибдена. Их состав выражается формулой APCi-MojCIg. В основе таких соединений лежит, вероятно, комплекс, аналогичный комплексу в упоминавшемся на,стр. 184 гидрате дихлорида молибдена. Известны также соответствующие соединения брома и иода.
Шестая группа периодической системы
191
Аналитические сведения. При систематическом разделении катионов присутствие молибдена можно обнаружить по темно-синему окрашиванию фильтрата от сероводородного осадка. При пропускании сероводорода молибден осаждается в виде сульфида, который при обработке сульфидом аммония переходит в раствор.
Если соединение молибдена нагревать в открытой с обоих концов трубке, то обнаруживается сублимат трехокиси молибдена. Последняя легко растворяется в аммиаке с образованием молибдата аммония, который после упаривапия аммиака н при последующем растворении его в соляной или серной кислоте Дает реакцию на молибденовую синь при восстановлении цинком или хлоридом олова(Н). Синее окрашивание появляется, если соединение молибдена упаривать почти досуха с концентрированной серной кислотой и дать остатку постоять на воздухе. Если соединения молибдена восстанавливать в пламени панльпой трубки, то получается интенсивно синий палет, представляющий собой соединение типа молибденовой сини. По мнению Глемзера (195i), это кристаллическая гидроокись.
Микроаналитическое обнаружение молибдена основано па появлении интенсивного Красновато-голубого окрашивания при добавлении к раствору молибдатаэтил-кса пт о ген а та калия SCfOCoHjSK. Эта реакция позволяет обнаружить!),04у молибдена при разбавлгчши 1 : 1 000 000. Столь же чувствительна и реакция молибдата с роданидом калия и хлоридом олова(П) (красное окрашивание вследствие образования К31Мo(SCN)e]). Последнюю реакцию целесообразнее всего проводить в качестве капельной пробы.
Для количественного определения молибден можно осадить из молибдатных растворов нитратом ртути(1) и виде молибдата ртути(1) или в виде сульфида при насыщении сероводородом кислых молибдатных растворов. Оба соединения при нагревании на воздухе переходит в трехокись молибдена, которую можно взвешивать. Удобнее осаждать молибден 8-оксихиноЛгглом из уксуснокислых молибдатных растворов в форме соединения JVlo()2(C.jlIeON)2 (Balanescii, 1930).
ВОЛЬФРАМ (WOLFRAMIUM) W
Распространение в природе. В природе вольфрам встречается в основном в виде вольфраматов, а именно вольфрамита (изоморфная смесь FeW04 ti M11WO4), шеелита (шпат Шееле. тунгстейн), CaWO4 и штольцита (свинцовая руда Шееле) PbWO4. Иногда встречается свободная трехокись вольфрама WO3 в виде вольфрамовой охры (тунгстита).
Чистый вольфрамат железа встречается в тетрагональной форме как рейнит и в моноклинной как ферберит. Чистый вольфрамат маргапца MnW04 — только в моноклппной форме как гюбнерит. Изоморфная смесь FeWO4 и MnWO4 всегда кристаллизуется в моноклинной форме. Распитом называется моноклинная модификация вольфрамата свинца, который обычно кристаллизуется в тетрагональной форме штольцита, изоморфного шеелиту.
Наиболее крупные месторождения вольфрама находятся в Китае, Корее, Индокитае, Бирме и Сцаме, па Малайском полуострове, в США, Боливии, Аргентине и Австралии. Из европейских месторождений важнейшие находятся в Португалии.
Исторические с не депи я. Вольфрам, вернее его окись WO3, был otic рыт Шееле в 1781 Г. в минерале тупгетейне, который теперь обычно называют шеелитом. Вскоре после этого два испанских химика, братья д'Эльхуяр (d'EUiuyar), установили, что этот же окисел содержится в вольфрамите, где он, однако, связан не с окисью кальция, как в тупгетейпе, а с окисламп железа а марганца. Вследствие содержания в тупгетей-яе и в называвшемся в то время «вольфрамом* вольфрамите * металл и получил свое название. От первого происходит и применяющееся еще и теперь французами и англичанами название вольфрама tangsteae соответственно tungsten.
* Уже у Агриколы этот минерал называется iupi spuma («волчья пена»). Про-исхождеиие этого названия связано с тем, что этот минерал, часто встречающийся вместо с оловянными рудами, затрудняет выплавку олова, переводя его в пену шлаков, «сжирая его*.
192
Глава S
Получение. Вольфрамовые руды (большей частью после предварительного механического или электромагнитного обогащения) плавят с содой в пламенных печах [уравнение (1)]. Получающийся при этом вольфрамат натрия Na2WO4 выщелачивают водой и разлагают концентрированной горячей соляной кислотой [уравнение (2)1. Образующуюся вольфрамовую кислоту переводят при нагревании в трехокись вольфрама [(уравнение (3)], а последнюю восстанавливают до металла нагреванием с углем или в токе водорода [уравнение (4)]. При этом вследствие очень высокой температуры плавления вольфрама его получают в виде порошка.
2FoWO4 -j- 2Na2CO3 4- 1/2О2=2Na2WO4 -[- Fe2O3 4" 2CO2, (1)
Na2WO4-|- 2HC1 = H2WO4 + 2NaCl, (2)
h2wo4=wo34-h2o, (3)
WO3 4- 3HZ=W 4- 3H8O. (4)
Спеканием и ковкой порошкообразный вольфрам можно превратить в компактный металл (ср. стр. 193). Расплавленный вольфрам получается при восстановлении его окиси углем или при действии окиси кальция на сульфид вольфрама в электрической дуговой печи. Коллоидный вольфрам получается аналогично коллоидному молибдену (см. стр. 182).
Свойства. Вольфрамовый порошок матово-серый, плавленый вольфрам — белый и блестящий. Удельный вес его 19,2, твердость около 7. Температура плавления вольфрама чрезвычайно высокая (примерно 3400°). Температура кипения, рассчитанная как и для молибдена,) равна примерно 5700°.
Удельная теплоемкость вольфрама в соответствии с его высоким атомным весом — согласно закону Дюлоига и Пти — очень мала (0,0340 при обычной температуре). Электропроводность при 0° составляет около 28,3% электропровод пости серебра при той же температуре. При нагревания до 2000’ сопротивление [возрастает примерно в четырнадцать раз.
При обычных температурах вольфрам устойчив на воздухе. При нагревании он окисляется до трехокиси \VO3. Пары воды переводят раскаленный вольфрам в двуокись. Элементарный азот заметно не реагирует с вольфрамом даже при 1500°. Водород также поглощается в очень незначительных количествах. Из галогенов энергично реагирует фтор с порошкообразным вольфрамом уже при обычных температурах, хлор реагирует только при температуре красного каления.
С азотом вольфрам образует педальтонидпое соединение , которое получается прп нагревании порошка вольфрама с газообразным аммиаком. Структурно оно соответствует соединению молибдена Mo3N, но может быть получено только с 18,2 аг.% N (вместо 33,3% при заполнении всех позиций N-решотки) (H.igg, 1930).
Вольфрам очень устойчив по отношению к кислотам. Это частично обусловлено легкой пассивируемостью вольфрама. Компактный вольфраи даже с концентрированной азотной кислотой и царской водкой взаимодействует только с поверхности. Оп медленно растворяется в смеси азотной и плавиковой кислот. Напротив, па вольфрам энергично действует сплавление с содой и селитрой и другими щелочными окислителями.
Применение. Вольфрам широко применяют для получения социальных сталей, отличающихся особой твердостью, эластичностью и проч-
Шестая группа, периодической система
193
ностыо. Добавленный вместе с хромом к железу, оп дает так называемые быстрорежущие стали, которые сохраняют свою твердость и заточку даже в накаленном состоянии. Для этих целей вольфрам обычно используется в виде ферровольфрама, силана вольфрама с железом, содержащим, как правило, 81—83% вольфрама. Этот сплав получают при восстановлении вольфрамита углем в электрических печах.
Обычно быстрорежущие стали содержат 19% W, 4% Ст, 0,5% V и часто некоторое количество Со. В них присутствует двойкой карбид Fe3W3C, решетка Которого образована октаэдрами Fe и W. Около трети атомов вольфрама могут быть замещены на атомы железа. Поэтому состав насыщенного щелевом карбида отвечает формуле Fe4W2C. Кобальт и никель образуют с вольфрамом аналогичные двойные карбиды. Однако их устойчивость уменьшается в направлении Fe —> Ni (Westgren, 1933),
Вольфрам имеет большое значение и в электроламповой промышленности. Вследствие высокой температуры плавления, малой летучести при одновременно небольшой теплоемкости, вольфрам, с тех пор как из него научились изготовлять нити накала, вытеснил все остальные металлы.
К вольфраму, используемому для изготовления нитей накала, предъявляются высокие требования в отношении чистоты. Поэтому его обычно получают восстановлением трехокиси вольфрама водородом. При этом получают порошкообразный вольфрам, который очепь хрупок и трудно обрабатывается. Поэтому прежде нити накала изготовляли из этого материала «шприцеванием»: тонкоизмоньчепный металл, смешанный с подходящим связующим материалом в пластическую массу, продавливали через алмазную фильеру с очепь маленьким отверстием. Однако получаемые таким способом нити обладали столь низкими механическими свойствами, что были малопригодны к употреблению. Позднее научились превращать порошкообразный вольфрам в Прокатывающийся металл, из которого волочением можно изготовлять нити. Для этого сначала из порошка прессованием, и спеканием изготовляют штифты, вначале очень ломкие. Однако при попеременном нагревании и очень интенсивной ковке через достаточно длительное время опи превращаются в способные к прокатке штабикн. Если затем штабнки осторожно протягивать в проволоку, то тягучесть их возрастает, так что при дальнейшем протягивании можно получить очепь гибкие и прочные нити диаметром до 0,01 л1.к.
Однако из «шприцов аппых» иитей также можно получить гибкие вольфрамовые нити, так называемые монокристаллические нити. Если к массе, из которой «шприцуются» пити, добавить немного окиси тория * и пронести первоначально хрупкие нити через короткую зону с температурой 2400—2600° (которую получают, накаливая вольфрамовую спираль в атмосфере водорода), то такие пити превращаются в монокристаллы, часто длиной в несколько метров. Конечно, при этом скорость проведения пити через «формирующую зону» не должна превышать скорости роста кристалла. Монокристаллические нити, как и протянутые, хорошо гнутся, прочны на излом и не теряют этих свойств при нагревании до высоких температур, тогда как протянутые пити при пагревапии становятся вследствие рекристаллизации хрупкими и ломкими.
Соединения вольфрама иногда применяют в качестве красок: например ослепительно белый и обладающий исключительной кроющей способностью вольфрамат барпя, а также белый вольфрамат Цинка. В керамике используют вольфрамовую кислоту для изготовления желтых плавящихся красок Вольфрамат натрия используют для пропитывания горючих веществ и тканей, чтобы придать им свойства невоспламеняемости. Волъфраматофосфаты применяют для изготовления защитных стекол, но пропускающих рентгеновского и у-иэлучения. Таг; называемые «вольфрамовые брон-зы» (см. стр. 198) в настоящее время используют мало. Карбид вольфрама W2C является главной составляющей твердого сплава «видна» (см. стр., 313), который применяют для изготовления режущих инструментов и буров, а также вместо алмаза при волочепии нитей для ламп накаливания.
* Чем объясняется благоприятное действие окиси тория на образование монокристаллов вольфрама, еще пе выяснено. Однако установлено, что при прокаливании часть окиси тория восстанавливается до металла.
13 г. Реми
194
Глава 5
СОЕДИНЕНИЯ ВОЛЬФРАМА
Вольфрам подобно молибдену образует соединения во всех валентных состояниях (от 2 до 6). Однако, он в еще большей степени, чем молибден, склонен проявлять максимальную швстивалентностъ. Это выражается и в том, что в отлично от молибдена вольфрам образует, кроме гексафторида, и гексахлорид, а также — правда, мало устойчивый—гексабромид. Важнейшими соединениями вольфрама являются вольфраматы,
лежащие в их основе вольфрамовые кислоты и op их ангидрид, трехокись
валъфража* Обз
Фторид Хлориды Бромиды Йодиды Сульфиды Окислы
WF6 WC16 WBr0 WSs wo3
— — — — — w10o39
—“ WC15 WBr5 — — w40n
— WClj -— WI4 wsz wo2
WC13 (только в форме двойных хлоридов) —
'— WC12 WBr2 Wl2 —
Галогениды, При температуре красного каления свежевосстановленный водородом вольфрам соединяется с хлором в гексахлорид вольфрама WCle. При этом Происходит самора аогревание. Это соединение образует темно-фиолетовые или голубоваточерные кристаллы удельного веса 3,52, плавящиеся при 275°. Температура кипения лежит при 347°. В холодной воде гекеахлорид вольфрама практически нерастворим (теплой водой он разлагается), но растворяется в спирте, эфире и других органических растворителях. Он очень склонен к образованию оксихлоридов, таких, как WOCl^ (красный, т. пл, 211°, Т. кип. 228е) и WO2C12 (желтый, т. пл. 265°, летуч при нагревании). Гексахдорид вольфрама, содержащий такие оксихлориды, тотчас же разлагается водой на вольфрамовую кислоту и хлорисил! водород.
Гексабромид WBi'g диалогичен гексахлприду, но разлагается легче. Очень реакционноспособен и газообразный гексафторид WF6 (т. пл. 2,5°, т. кип. 19,5°).
Молекула WF0 нс имеет, как и SFe, правильной октаэдрической,структуры. Атомы F в пей расположены, вероятно, около атомов W в форме ромбической бипирамиды. Кратчайшее расстояние W <—+ F равно 1,64 А. Это же справедливо и для MoFe (Brau-пе, 1937).
Пентахлорид вольфрама WCl5 образуется при термическом разложении г оксихлорида, Это разложение начинается, как показывают измерения упругости паров, уже чуть выше температуры кипения (еще легче разлагается бромид). Для получения неятахлорида гексахлорид вольфрама повторно возгоняют в токе водорода. Пента-хлорид образует блестящие черно-золеные иглы с т. пл, 248° и г. кип, 276°, Плотность зеленовато-желтых паров соответствует формуле WC15. В кислороде WClb сгорает до WOCI4. В воде оп растворяется с частичным разложением. Аналогичные свойства проявляет и плавящийся при 276° и кипящий при 333° (при частичном разложений пентабромид WBrj.
При возгонке гексахл ори да вольфрама в токе водорода, особенно если температура повышена, наряду с пентахлоридом образуются также тетра- а дихлориды. В противоположность ныешцм хлоридам эти соединения не летучи. Тетрахлорид вольфраме WCI4 образует ерро-коричпевуго, рыхлую, гигроскопическую, кристаллическую массу. Под вляяпш'М воды он гидролитически расщепляется. Аналогично ведет себя и черный тетраиодцд WL,, который получается при нагревании в ампуле гексахлорида вольфрама с жидким йодистым водородом.
Трихлорид вольфрама в свободном состоянии неизвестен. Но хорошо известна, его двойные соединения типа M|W2C19 (М* =• К, NH4, Rb, Cs, Tl). Epocce (ВгоззсД 1935) рентгеиоструктурпым анализом доказал, что эти соединения построены из групп ()VaCigp-, образующих гексагональную плотнейшую упаковку. Попы одновалентных металлов располагаются в промежутках между этими группами.
Шестая, группа периодической системы
195
Дихлорид вольфрама- WC13 получают восстановлением г окса хлорида вольфрама водородом (при пе слишком высоких температурах, так как иначе может произойти восстановление до металла), а также при термической диссоциации тетра хлорид а вольфрама. Он получается в виде серой неустойчивой на воздухе массы. При взаимодействии с водой происходит выделение водорода, а на другие соединения дихлорид вольфрама действует как сильный восстановитель. Аналогично ведет себя дибромид WBr2. Коричневый дииодид вольфрама Vvl2, получающийся при пропускании паров пода над свежев осстанов лонным раскаленным вольфрамом, в холодной воде не растворяется. Тёплой водой он разлагается с окислением и одновременным гидролизом.
Сульфиды. Если в раствор вольфраматов пропускать сероводород, то обрадуются — сначала при частичном, а затем и при полном замещении атомов кислорода в вольфрамовой кислоте атомами серы — оксошно-
вольфраматы
М2
"О О’
W
О S.
’О S' W
S S :
и, наконец, тиовольфраматы
Mi
“S S’ w .
-S s
Из растворов последних при добавлении кислоты выделяется шоколадно-коричневый трисульфид вольфрама WS3.
Трисульфид вольфрама очень склонен к образованию коллоидных растворов, например, уже при промывании водой. При нагревании на воздухе он переходит в трехокись вольфрама. Если же его нагреть без доступа воздуха, то, отщепляя серу, он переходит в дисульфид WSa. Последний образуется также при сплавлении трехокиси вольфрама с карбонатом калия и серой. Дисульфид вольфрама представляет собой нерастворимый в воде мягкий бесцветный порошок уд. веса 7,5. Дальнейшее отщепление серы, приводящее к выделению металла, происходит без доступа воздуха только при очень высокой температуре. Но при нагревании па воздухе дисульфид вольфрама легко сгорает до трехокиси. Он реагирует также с фтором, хлором и бромом, образуя соответ-сгвующие гекса галогениды. Кристаллическая решетка дисульфида вольфрама совершенно аналогична решетке дисульфида молибдепа (ср. стр. 185): а=3,18, с = 12,5 А; W*->S = 2,48 А.
Окислы. Как и в случае молибдепа, конечным продуктом окисления металлического вольфрама или его соединений является желтая трех-окись. При действии восстановителей из нее образуется коричневая двуокись.
Как рентгенометрически установил Глсмзер (1943), кроме WOS и W02, существуют еще два окисла. Оп подучил эти соединения, как и Wo2, восстановлением водородом WOy, а также взаимодействием WO3 с металлическим вольфрамом. Один из пих W10O^ (область гомогенности WO2 e2 —WO2d88) голубовато-фиолетового цвета. Другой W4Otl (область гомотопности от 4V02,7ft до W02vM) красно-фиолетовый. Двуокись WO3 (область гомотопности WO2,03 — WO2d00) коричневая.
Трехокись вольфрама W03 образует лимонно-желтый хрупкий порошок, который при нагревании становится оранжевым. При сильном прокаливании, а также при сплавлении с бурой он становится отчетливо кристаллическим. Очень блестящие винно-желтые кристаллы имеют удельный вес 7,23 и принадлежат к ромбической системе. Трехокись вольфрама плавится при 1473° и начинает возгоняться только при температурах выше 1750°. Однако в токе хлористого водорода полное испарение происходит уже при температуре примерно 500°, вероятно в форме окси-13*
196
Г.мва 5
хлорида, В воде трехокясь вольфрама практически не растворяется. Напротив, едкие щелочи растворяют ее с образованием вольфраматов, солей вольфрамовой кислоты H2WO4;, ангидридом которой трехокись вольфрама можно считать только в ограниченном смысле. Она получается из кислоты при прокаливании, однако не может быть переведена в кислоту непосредственно (т. с. присоединением воды).
Кристаллическая структура W03 очень похожа па структуру ВеО5 (см. стр. 254). Только ионы О*- расположены не точно в середине между ионами W6+. Вследствие этого симметрия решетки понижена, так что кубическая в случае Но03 структура переходит в ромбическую. W03 может терять часть кислорода (до WOa95), не претерпевая существенных изменений. в кристаллической решетке. Но самое незначительное изменение содержания кислорода приводит к и вменению цвета. При составе WO2i28 это соединение уже имеет отчетливо синий цвет.
Двуокись вольфрама W02 получается при восстановлении трехокиси вольфрама, например при слабом прокаливании последней в токе водорода. Двуокись вольфрама образует коричневый порошок уд. веса 12,1, который легко вновь окисляется до трехокиси'. Он плавится примерно при 1300°. Более продолжительное по слишком слабое нагревание в токе водорода приводит к восстановлению до металлического вольфрама. Кристаллическая решетка двуокиси вольфрама аналогична решетке двуокиси молиб-депа (тип рутила): «=4,86; е = 2,77 А.
В качестве промеж уточных продуктов восстановления водородом трехокиси вольфрама до двуокиси образуются уже упоминавшиеся окисли п W4O11.
Промежуточный окисел W4O5J образуется и при восстановлении WO3 другими способами, например электролитическим восстановленном в водном растворе. Прежние представления, в соответствии с которыми наблюдаемое при этом синее окрашивание объяснялось образованием окисла Vv2O81 который Должен также Получаться при восстановлении WO3 водородом, опровергнуты исследованиями вал Лиемпта и др. (van Liempt, 1931; Ebert, 1935; Glemser, 1943).
Вольфрамовая кислота и вольфраматы
Как и трехокись молибдена, трсхокись вольфрама может соединяться с окислами основного характера в весьма различных соотношениях. Такие соединения называются вольфраматами. Простейшим из них, так называемым нормальным вольфраматам, отвечает общая формула М*О-WOj = = Так называемые метавольфраматы, содержат 4WO3 па
1 М^О. Кроме того, они всегда содержат воду, часть которой, как следует из ее поведения при обезвоживании, связана конституционно. Еще более сложен состав паравольфраматоа (также всегда содержащих воду), в которых содержится 12WO3 на 5М*О.
«Паравольфраматы» с отношением М|о : WO3 = 5 : 12 по своему составу и структуре отличны от «парамолибдатов», в которых отношение М.]О : Мо03 равно 8:7. В водных растворах маравольфраматовь в соответствии с данными Лидера (1927) присутствуют ионы гексавольфрамата, точнее водородгексавольфрамата [IIWe Оа।]''7". (Включение Н в квадратные скобки указывает па то, что водород входит в состав комплекса.) В кристаллическом состоянии эти соединения, по Линдквисту (1950), содержат не гекса вольфрамат-, а доРекявольфрамат-ион [II2Wн>-.
(1 : 4'i-liа:ыфр,тматы, которые вследстпяе их аналитического состава называют «Метаволыфраматы», частично также являются гексааолъфраматами: это могут быть дважды кислые монсеодородгексавольфраматы М^Н2 [Н W5O21) или нейтральные триеодо-родгексавольфраматчл Я i [ II3 WBO2 j ]. Но свободная кристаллическая метавольфрамовая кислота является нс гексввольфрамовой кислотой, а ЗойекавольфрамовоЙ, однако ее апион отличается от апнопд, который, согласно Линдквисту, присутствует в кристаллических иаравольфраматах. Ее структура соответствует не изополи-, а гетерополикислоте (см. стр. 202). Сопи этой кислоты имеют общую формулу Mj[HaW12O40], Поскольку все (1 : 4)-вольфраматы содержат кристаллизационную воду, на основании только ана
Шестая группа, периодической системы
197
литического состава нельзя решить, отвечает ли их строение формулам: MJ[H2W1204o], или M3H2IHWaO21], или Mj[H3WeO2j], Одпако для одного ряда кристаллических (1 : 4)-вольфраматов установлено, что "они изоморфны солям известных гетерополикислот (см. стр. 202). Те (1 : ^-вольфраматы, для которых это справедливо, без сомнения, являются солями так называемой метаволъфраг^овой. кислоты, т. е. их общая формула Ms [H2W12O40]. Но существуют и (1 : 4)-вольфраматы другого типа. Возможно, что в этих случаях речь идет о солях гексавояъфрамовой кислоты (моно- или триводородгек-савольфрамовой кислоты): М|Н2[HWft031] и Mj [H3W6O2I]. Однако можно предложить и другие структурные формулы. Например, аммонийная соль состава (NH4)2O-4Мо03-•21/3Н2О, как доказал рентгенометрически Линдквист (1950), имеет структуру октамолибдата (NH4)4[MOgO2e[-5H2O. Возможно, что существуют и (1 : 4)-вольфраматы, которые имеют аналогичное строение.
В щелочных растворах вольфраматов при значениях pH до 8 существуют моно-вольфрамат-ионы (ионы |WO4]'' в равновесии с ионами H[W04]')- В области значений pH 8—6 начинается образование гексаволъфрамат-ионов по уравнению
6fwo4r-i-7ir 6hiwo4]'+h- 7» [HW6oBi]’""+3H2o.
В растворах, pH которых лежит между 6 и 4, присутствуют практически только водородгекса воиьфрамат-попы [nW0O3I]'"". При дальнейшем увеличении концентрации ионов водорода может происходить либо прел ращение маков од ородгексав ол ьфрамат-иояов в m/шводородгексавольфрамат-ионщ но уравнению
[HW60ar' + 2H’ Ti [HsWeO21]w,
либо образование додекавольфрамат-ионов:-
2Гт¥6Оа1Г"-<4Н- [H2W12O40rw+2H2O.
Первое равновесно доминирует в сильно разбавленных, второе — в концентрированных растворах (Jander, 1925, 1940; ср. также Спнцьти и Пирогова, 1957). Комплексный анион [HW6O2I)"'" может существовать в двух отличающихся поведением формах с различным содержанием водорода. При возрастающей концентрации ионов водорода только одна из форм превращается в ион [H3WgO21]"' (также содержащий конституционную воду), так называемый nceei) омет а волъфр ам am-ион. Это превращение происходит через дважды кислый кодородовольфрамат-ион в качестве промежуточного продукта:
21Г + [HWaO2J'"" Ti H2[HWeOaI” [H3WeO21]w,
как это было доказано измерениями электропроводности и поглощения света (Sonc-hay, 1943—1944; Jander, 1951).
Интересно отличие конденсации воиъфрамат-ионов, при которой ионово.чъфра-: мат-ионы без каких-либо устойчивых промежуточных продуктов переходят сразу в гексавольфрамат-ионы, от конденсации хромат-ионов, при которой образуются ди-, три- и тетра хромат-иопггь Конденсация молибдат-попов происходит совершенно аналогично конденсации вольфрамат-иопов. Точно так же, как при конденсации молиб-дат-ионов, наблюдается появление в качестве промежуточных продуктов (неустойчивых) тримолибдат-ионов. конденсация вольфрамат-ионов происходит через образование трнвольфрамат-ионов (Anderson, 1949). Но последние еще мепсо устойчивы, чем т рим ол иб дат-и опы.
Кроме мономоиибдатов и моиовольфраматов, из расплавов можно получить также димолибдаты и дивольфраматы (Ноегшацп, 1928). Na2Mo04 и Na3WO4 имеют, как и Ag2MoO4, структуру шпинели, в то время как 1л2МоО4 и LiaWO4 изоморфны фенакиту Bc2SiO4. Б соединениях 1\а2Мо2СЬ и NaaW2O7 ионы Мо2О7 и W2O^- образуют бесконечные цепи. Каждая из таких цепей построена из октаэдров МоО6 или WOa, в которых два рядом стоящих октаэдра имеют общую вершину с присоединенными тетраэдрами Мо04 wihJWQ4.
Вольфрамовая кислота, Из растворов большинства вольфраматов, но только не метавольфраматрв, при нагревании и добавлении сильных кислот выпадает желтая вольфрамовая кислота, практически нерастворимая в воде и в большинстве кислот.
Еслн достаточный избыток кислоты прибавлять к раствору вольфрамата на холоду, то выпадает белый объемистый осадок, так называемая белая вольфрамовая кислота. С ос та в и свойства этого осадка свидетельствуют о том, что это гель трехокиси вольфра-
198
Глава 5
Рис. 27. Шеелит CaWO4. а = < 11,38 А.
Шеелиту изотопны: BaW)4, PbW04,
СаМоО< ВаМиСЬ. ГЬМ.:>0, кроле таги, NaI04, KI04, NH.IO., Agl04t а также ККеЫ4 и КОзО.яК
ма с переменным содержанием воды. Под действием кислот, особенно при нагревании, «белая вольфрамовая кислота» переходит в обычную желтую. Белая вольфрамовая кислота заметно растворяется в воде, причем такие растворы имеют кислую реакцию. Она очень склонна к образованию коллоидов, особенно если при ее получении совсем не было или был очень небольшой избыток кислоты. По ее гидрозоль менее устойчив, чем гидрозоль молибденовой кислоты. При большой концентрации ионов водороде происходит быстрая коагуляция.
Если добавлять кислоту к растворам так называемых метаволъфраматов, то вообще не выделяется никакого осадка. Образующаяся метаволъфрамовая кислота легко растворяется в воде. Ее можно получить действием серной кислоты на метавольфрамат бария «ли сероводорода на метавольфрамат свинца. Она кристаллизуется в больших ромбоэдрах. Состав метаводьфрамовой кислоты отвечает формуле 4WOS- 9НЙО. Ее строение будет рассмотрено в связи со строением вольфрамат окремнев ой кислоты.
Вольфрамат натрия Na2WO4, техническое получение которого уже рассмотрено на стр. 192, является основным исходным продуктом для получения соединений вольфрама и чистого металла. Эта соль легко растворяется в воде (73,2 г в 100г воды при 21°) и кристаллизуется в виде бесцветных тонких блестящих листочков ромбической системы состава Na2WO4-2H2O *. При нагревании на воздухе или при стоянии пад серной кислотой вольфрамат натрия теряет кристаллизационную воду. При кипячении с сильными кислотами происходит разложение с выделением желтой вольфрамовой кислоты.
Кроме обычного вольфрамата натрия, известим поливолъфраматы натрия. Из них следует отмстить мета воль фра,кат натрия, аналитический состав которого выражается формулой Na2O-4WO3- 1бН3О. Из раствора этой соли под действием кислот пе выделяется осадок, так как кислота, производным которой является эта соль, так называемая метаволъфрамввая кислота, легко растворяется.» воде.
Вольфрамат кальция Са[\У04] встречается в природе в виде минерала шеелита, часто в сопровождении оловянного камни. Он кристаллизуется в тетрагональной системе (желтые пирамиды уд. веса 6,0). Кристаллическая решетка шеелита приведена на рис. 27. Такую же структуру имеют штольцит PbjWOJ и вульфенит РЬ[МоО4].
Ферберит Fe[WO4], гюбперит Mn[W04], вольфрамит (Fe, Мп) [WO4], а также ряд других вольфраматов двухвалентных металлов (Co[WO4|, Ni[WO4], Zn[WO4], Mg(WOJ), кристаллизующихся в моноклпнпой системе, имеют кристаллическую решетку особого типа (типа вольфрамита). Такой тип решетки наблюдается только у вольфраматов.
Вольфрамовые броязы. «Вольфрамовыми бронзами» называют разнообразно окрашенные (голубые, пурпурно-краспые, золотист о-желтые и т. д.), химически чрезвычайно устойчивые и прекрасно кристаллизующиеся соединения, имеющие металлический блеск и металлическую электропроводность, которые получают действием восстановителей на иге л очные или щелочноземельные вольфраматы. Аналитически эти соединения содержат эквивалентные количества окисла щелочного или щелочыо-
* При температурах ниже 6° кристаллизуется декагидрат.
Шестая группа периодической системы 199
земельного металла и W2O5l а кроме того, больший или меньший избыток трехокиси вольфрама. Аналогичные соединения образует и молибден («молибденовые бронзы»). Природу молибденовых и вольфрамовых бронз удалось установить только па основании рентгенографических исследований (de Jong, 1932; IJagg, 1935). Натриево-вояь-фрамовые бронзы, если они кристаллизуются в кубической системе, образую! кристаллическую решетку типа перовскита, в которой, однако, обычно заняты пе все места , принадлежащие ионам натрия. Состав этих бронз колеблется в зависимости от числа вакантных мест в решетке в пределах от Nao,3W03 до NaWO3. При составе NaWO3 все узлы решетки заняты; в этом случае вольфрам только положительно пятивалентен. NaW03 золотисто-желтого цвета. При уменьшении содержания натрия цвет изменяется через красный л фиолетовым к темному сине-фиолетовому. Это углубление цвета связано с тем, что в соответствии с уменьшением числа ионов натрия все возрастает число .ионов W5+, окисляющихся до ионов We+. Существуют также я атриев о-в ольфрамовые бронзы, содержащие еще меньше натрия, чсм соответствует формуле Na0,3WO3, Эти соединения синего цвета и кристаллизуются в тетрагональной системе, так как при дальнейшем увеличении пустот структура перовскита становится нестабильной. Кубические патриево-вольфрамовые бронзы являются электронными полупроводниками с высокой проводимостью (и = 1 до 10 о .в-1 цри комнатной температуре).
Вольфрамовая синь. При слабом, восстановлении трехокиси вольфрама или вольфрамовой кислоты в водном растворе или при соприкосновении их с водой получаются интенсивно синие продукты, как правило, переменного состава. В их состав могут входить в зависимости от способа получения различные соединения, которые называют общим именем «вольфрамовая синь». Кроме синего цвета, для этих соединений общим является го, что все они содержат вольфрам в среднем валентном состоянии, которое меньше 6, но больше 5. Следовательно, во многих отношениях вольфрамовая синь соответствует молибденовой сипи. Под водой трехокись вольфрама синеет уже при действии солнечного света. Образование вольфрамовой сини при добавлении цинка или хлорида олова(П) к подкисленному соляной кислотой раствору вольфрамата является очень чувствительной реакцией па вольфраматы.
Осадок, выпадающий цз раствора вольфрамата при действии водорода в момент образования (Zn -f- ПС1), устойчив на воздухе. Напротив, осадок, получающийся при действии SaGla и IICI, на воздухе постепенно бледнеет, превращаясь в желтую вольфрамовую кислоту. Уже такое различное поведение указывает па то, что это две различные формы вольфрамовой сипи. По данным Глемзера (1943—1951), в первом случае получаются водородные аналоги вольфрамовых бронз, т. е. соединения состава HKW03 (х = 0,1—0,5) (Glemser, 1952); кристаллическое соединение H0HWOs получается также при действии на WO3 атомарного водорода или ЫА1Н4. В другом случае образуется соединение, структурно аналогичное вольфрамовой кислоте H2W04 и отличающееся от последней только мсныппм содержанием кислорода и переменным содержанием водорода. Так Же как и молибденовая синь, вольфрамовая синь часто получается не в кристаллической, а в рептгеноаморфнон форме. Вольфрамовая синь, Как и ее молибденовый аналог, склочна к образованию коллоидных растворов.
Пероксовольфрамовая кислота и пероксовольфраматы. Вольфрамовая кислота, а также трехокись вольфрама растворяются в нагретом до кипения растворе перекиси водорода, образуя соединение, которое рассматривают как пероксовольфрамовую кислоту H3WO5*. Действием перекиси водорода на вольфраматы можно получить пероксоволъфраматы.
Из нейтральных или слабощелочных растворов вольфраматов щелочных металлов в присутствии избытка перекиси водорода кристаллизуются желтые тетрапероксо-моновояъфраматы M|[W(O2)4] **.
* Еще не доказано, чтО/Это действительно пер оксо-кислота, а но двойное соединение WO3’H2O2.
** (Оф—перекисная группа. Эти соединения кристаллизуются с водой.
200
Глава 5
Из слабокислых или слабощелочных растворов вольфраматов щелочных металлов, содержащих мало перекиси водорода, кристаллизуются бесцветные тетрапёрбксо-дивольфраматы щелочных металлов Mj [W2O3(02)4], Зависимость образования обоих типов соединений от концентрации ионов водорода и перекиси водорода объясняется том, что в растворах, как доказал Яр (1938—1952), устанавливаются равновесия:
2[WO4]" + 4НгО2 IW203(02)J " 4- 20 И' + ЗН2О,
[W203(02)4r + 20II'4-4H202 2[W(O2)4r + 5H2O,
[HW6O21]...+ 12Нг0г 3[WaO3(O2)4]’ + H’ + 12H2O.
Если при достаточно большом избытке перекиси водорода происходит превращение существующих в кислом растворе гексавольфрамат-иопои [HWe021]'"" в тетрапероксо див о ль фр амат-ноны [W203(02)J", то в присутствии незначительных количеств перекиси водорода в кислом растворе образуются пероксополиволъфраматы, т. е. соединения, являющиеся производными поливольфрамовых кислот.
Кремневольфрамоная кислота. Гетеро поликиелоты, Как впервые наблюдал Мари-ньяк (Marignac, 1862), в горячем растворе кислых вольфраматов щелочных металле» растворяется кремневая кислота. При этом образуются очень хорошо кристаллизующиеся соли, характерной особенностью которых является то, что они на lSiO2 наряду с окисью щелочного металла и водой содержат еще большое число (до 12) групп WO3. Раньше эти соли называли кремневолъфраматами. Теперь их называют поливолъфрама-тоеиликатами (короче, вольфраматосиликатами), поскольку они являются производными кремневой кислоты или ее солей, образующихся присоединением остатков вольфрамовой кислоты (вольфраматных групп). Наиболее богатая вольфрамом калиевая соль этой группы соединений имеет аналитический состав SiO2-12WO3-8К.ОН-ЮН20 или H4SiO4-12\¥О3-8К.ОН-8Н2О. При действии на вольфраматосиликаты кислот отщепляется только щелочь, а отношение SiO2 ; W03 остается неизменным. Напротив,, щелочи полностью разлагают эти соединения.
Как оказалось, вольфрамовая кислота образует соединения, совершенно аналогичные вольфрамат о силикатам, еще с целым рядом кислот, и прежде всего с борной кислотой (воль фра мат оборот ы), фосфорной кислотой [рольфраматофосфаты) и Мышьяковой кислотой (волъфраматоарсенаты).
Примеры:
H3BO3‘12WO3-5K.OH-14H2O—додекавольфраматоборат калия
Н3Г’О4 12WO3 -ЗКОН 21/2Н2О—- додекавольфраматофосфат калия
H3AsO4 - 9WO3 -ЗкО II • 4Н2О—нона вол ъфр а мат оар сенат калия.
Как видно из последнего примера, число радикалов WO3, приходящееся на одну молекулу другой кислоты, может быть и менее 12. Отношение 1 : 12 является верхней границей, которая никогда пе нарушается.
Для комплексных кислот, лежащих в основе приведенных выше солей, характерно то, что они содержат несколько (по крайней мере два) различных кислотных остатка, причем обычно один содержится в значительном избытке. Комплексные кислоты такого типа объединяют под названием гетерополикислот.
Рапьше часто считали, что входящие в состав гетерополикислот многочисленные кислотные остатки связаны друг с другом в цепи. Однако с такой цепной структурой с трудом согласуется значительная устойчивость таких высокомолекулярных соединений. Кроме того, такие формулы пе объясняли существование верхней границы молекулярного отношения разных кислотных остатков. Более удовлетворит ель нах; теория гетерополикпелот была выдвинута Миолати (Miolati, 1908) на основании координационного учения Вернера. Розепгейм (Rosenheim) развил дальше представления Миолати и дал им дальнейшее экспериментальное обоснование. И, па конец, строение этих соединений было установлено рентген ост рукт у рпым анализом (Keggin, 4933, Signer, 1934; Kraus, 1936). Основные положения теории Миолати и Розенгсйма былп Подтверждены, но потребовались также некоторые незначительные изменения вытекающих из этой теории выводов.
Миолати рассматривал граничные вольфраматосиликаты, т. е. такие, в которых отношение SiO2 ; WO3 составляет 1 ; 12, как производные гипотетической кремневая кислоты, в которой атом кремния окружен шестью атомами кислорода в соответспихт с координационным числом 6, играющим исключительную роль в теории Вернере Если представить себе, что каждый из этих шести атомов кислорода замещен остатке::
Шестая группа периодической системы
201
W20J( то исходя из гипотетической кислоты И8[SiO6] (гексаоксок ремнев ой кислоты^ для лежащей в основе граничных вольфраматосилйкатов вольфраматов ремнев ой. кислоты получим следующую формулу;
H8[Si(W2O7)6J.
В соответствии с такой формулой валъфраматокремневая. кислота должна считаться гексаокеокремневоа кислотой, в которой атомы кислорода замещены остатками W2O7. Аналогично иа кислоты Н9|ВОЙ] можпо получить вольфраматоборпую кислоту, а из кислоты HtIPOk] — водьфраматофосфорную. Для них получим формулы
TIJBfWaOyJd и H3IP(W2O,)6].
Аналогичные формулы получаются и для гетерополикислот, содержащих ‘остатки молподеполой кислота,. Конечно, мз теории Миолатл следовало, что должны существо
вать и кислоты, образующиеся при замощении остатками W2O7 и т. д, только части атомов кислорода. Эти кислоты называют ненасыщенными кислотами граничного рядаЛ
Поразительной казалась высокая основность этих кислот, следующая из теории Миолаги: написаппые вне скобок атомы водорода должны быть связаны ионно и, следовательно, обладать способностью замещаться атомами металл он. И когда были получены соли, состав которых отвечал этой высокой основности, в этом стали усматривать существенное подтверждение теории. Действительно, уже упоминавшийся вольфраматоси-ликат калия содержит 8 а том о а калия на 1 атом кремния, как этого н требует теория. Розонгейм гтавнеч, что вольфраматофосфорная кислота (а также молибдато-фосфорпая), которая по теории Миолати должна быть семиосновной, действительно образует соединение с семью молекулами гуанидина CN^ri^:
ICN3H5.H]7 [P(W2O7)6J-
Рис. 28. Расположение октаэдров WOg вокруг центрального атома в гетерополиннслотах типа додекаволь-фраматофосфорной кислоты.
На рисунке я*? видна расположенная сзади четвертая группа.
В случае вольфраматоборнои кислоты была получена соль ртути(1), отвечающая требуемом теории основности. Состав |Этой соли выражается формулой Hgg[B(W2O7)6]’12Н3О (Cop-aux, 1909).
По данным рентгеноструктурного анализа, гетер о/голнкнелоты типа додеканоль-
фраматокремпевой кислоты в кристаллическом состояния построены следующим образом: вокруг центрального атома Si тетраэдрически расноложеаы четыре атома О, каждый из которых является общей вершиной трех октаэдров WO(; (состоящих из шести атомов О, октаэдрически расположенных вокруг атома W) (см. рис, 28). Каждый из этих трех октаэдров имеет с двумя соседними октаэдрами той же группы еще но одному общему атому кислорода, так что каждый октаэдр имеет с двумя соседними октаэдрами общее ребро. Кроме того, каждый октаэдр имеет еще по" одному общему атому О с одним из октаэдров из двух соседних групп, расположенных у того же атома Si. Шестой атом О не связан с другими октаэдрами. Следовательно, па каждую группу, построенную из трех октаэдров WOg, приходится в общем 3 + J —
=10 атомов О. Поэтому каждая из четырех групп, расположенных около центрального-атома Si, имеет состав W901(i,H структура комплексного иона додекавольфрамато-кремпевой кислоты приблизительно передается формулой [Si(W301o)4]4-. Следовательно, со структурной точки зрения додекавольфраматокремневую кислоту надо Считать-тетратривольфрамотокремневой кислотой H4[Si(W30j0)4[. Аналогично построены
До деканол ьфр а мато бо р ная кислота
До де к а вольфр амат ог ерман левая икс лота Д одека вольфрама тофосфорная кислота Д о декав о л ьфрам а т омышьяко ва я кислота
H5[B(W3Ol0)4] H4[Ge(W3Ol0)4] H5IP(WsO10)J H3[As(W3OI(1)4)
202
Глава 5
ж соответствующие молибдато-кислОты. И у солей этих кислот были найдены при репт-геноструктурных исследованиях те же комплексные структурные группы. Например, рассматриваемый па стр. 137 и 188 молибдатофосфат аммония, играющий суще-•ственяую роль при определении фосфорной кислоты, является нормальной Сопью додекамолибдатофосфорной кислоты
(NH4)3[P(Mo3Ow)t].6H3O.
Гетеропояикислоты (и, как правило, нх соли) всегда кристаллизуются с несколькими молекулами воды, число которых зависит от условий получения, В крястцллп-ческих решетках различных гидратов молекулы ноды расположены по-разному. Поэтому 5-гидраты кристаллизуются в кубической, 14-гидраты — триклинной, а 24-тидраты — в тригональной системе. Но строение комплексной анионной группы при этом не изменяется. Интересно различие кристаллических структур 29-гйдрата (куои--ческая) и 3 0-гидр ата (тетрагональная). Такое относительно небольшое изменение содержания воды (Называет существенное влияние на строение решетки * **, тогда как при -одинаковом содержании воды даже изменение центрального атома анионного комплекса не меняет кристаллической решетки. Поэтому одинаковые гидраты всех названных тег ор о поликислот изоморфны друг другу.
Существенное отличие структурных формул, установленных рентгенометрическими измерениями, от предсказанных Миолати заключается в том, что центральный атом анионного комплекса имеет другое координационное число, чом ему приписывалось в прежних теориях. Рассмотренные гетерополикислоты являются производными не гсксгаоксоцислот (координационное число 6), а тетраоксо кислот (координационное число 4). Соответственно этому они содержат не -жесть дивольфрамат о-, а четыре три-вольфрама то-(или тримолибдато-) группы *й. То, что гетерополикислоты являются производными тетра оксокислот, т. е. кислот с мепьщей основностью, чем считали раньше, и, следовательно, сами должны проявлять основность меньше, чем указывается прежними формулами, представляет еще одну трудность для объяснения существования таких соединений, которые подобно приведенным на стр. 200 содержат больше эквивалентов основного металла, чем содержится атомов водорода в свободной кислоте. Рентгенометрические исследования (Kraus, 1939) указывают, по-видимому, на то, что эти соединения представляют собой разновидность .основных солей, образующихся из гидратов нормальных солей при замещении молекул НОН гидроокисью основного ^металла. Какие гидроокиси могут быть связаны таким образом и в каком количестве, будет зависеть от строения кристаллической решетки и от индивидуальных свойств ^включаемых в решетку ионов. Объяснение, вероятно, можно искать и в том, что такие соединения, состав которых отвечает большей основности, чем в замощенной кислоте, получаются только в исключительных случаях и всегда плохо кристаллизуются.
Так называемая метавольфрамовал кислота ПО своей структуре должна быть также отнесена к гетерополикислотам. На основании изоморфизма, существующего между метавольфрамовои кислотой и ее солями и соответствующими соединениями типичных гетерополикислот, уже Kono (Сораих) установил структуру этих соединении. Для метавольфраматов и метавольфрамовой кислоты получаются следующие структурные формулы:
Ke[H3(W3Oi0)4] I8H9O изоморфен (гексагональный) K4[Si(W3O10)J 18НаО
Метавольфрамат калия вольфраматосиликат.у
калия
Ba3[H2(W3Ol0)4]-2711^0 изоморфен (тетрагональный) Ba2,s[B(W3O10)4]-27H2O
Мета вольфрамат барин вольфрамате бора ту Сарая
Н6[Н2(\У3О10)4]-24Н2О изоморфна (тригональная) HB(P(Wa0I0)4j-24H2O
Мета в о Ль фрамо ва я кис л ота в о льфрама то фо сфо р по й
кйслоте
Рентгенометрическими исследованиями установлено, что так называемая метаволь фрамо в а и кислота структурно совершенно аналогична т етратрив о л ьфраматокр синевой, -биркой и -фосфорной кислотам. В мета вольфрамовой кислоте оба атома во до -
* Недостаточным учетом этого влияния объясняются часто встречающиеся в литературе противоречия в кристаллографических дапвых для гетерополикис лот или их солей.
** Уже до того, как были проведены рептгеноструктурные исследования, Яидер на основании того, что в растворе никогда не удавалось установить существование „димолибдато- или див ольфрамя то-ион ов, отклонил гипотезу о том, что они принимают участие в построении комплексных гетерополйкислот.
Шестая группа периодической системы
203:
рода, написанные внутри скобок, играют формально ту же роль, что и атом кремния в вольфраматокремневой, атом бора в вольфраматоборной и атом фосфора в вольфрамате фосф пр пой кислотах. Следовательно, формально оба атома водорода можно считать центральными атомами комплекса метавольфрамовой кислоты, и структурно мёта-вольфрамовую кислоту следует называть вольфраматодиводородной (точнее тетратривольфрамат одиводор одной) кислотой. II метавольфраматы, если они изоморфны воль-фраиато(или молибдат о-) силикатам и т.следует рассматривать как гетерополисолн, например, упомянутым на стр. 198 мот а вольфрамат натрия должен иметь формулу Na6IH2(W3O10)4].29H2O.
Те гетер оно ликис лоты, которые содержат на 1 молекулу кислоты менее 12 групп WQ3, МоО3 й т. Д-*, так называемые ненасыщенные гетероноликислоты, вероятно, построены аналогично рассмотренным выше насыщенным гетер ополи кислотам. Ничего более определенного об их структуре в настоящее время, к сожалению, сказать нельзя. Для гетер оночисолей, которые получаются присоединением WO3 к теплуратам Al|[TeO6} и перподатам сначала казались очевидными при-
нятые большинством исследователей формулы гексамоновольфраматотел луратов Me[Te(WO4)0i и гексамопотюльфраматоиодатов Mf[I(WO4)6]. В соответствии с этими структурами формулами названные гетерополисолн образуются из простых солей нри замещении атомов О ла группы WO4. Однако, как установил Япдер, из растворов, в которых присутствуют только иопы WOj, соли такого типа получить пе удастся. Опи подучаются чаще из кислых растворов, т. е. таких растворов, в которых присутствуют в основном иопы [HWeOai]". Рептгено структурные исследования, проведенные Линдквистом (Lindqvist, 1948) с соответствующими молибдатотеллуратами, показали, что в них присутствует группа [Мо6О24] в форме построенного из октаэдров МоОй шестичленного кольца, в центре которого расположен атом теллура. Структура такого типа была предложена и ранее Андерсоном (Anderson, 1937) как наиболее вероятная с точки зрения размера ионных радиусов. Из щелочных растворов кристаллизуются -соли типа MlTeO4' M|W04 или которые, вероятно, следует рассматри-
вать как обычные двойные соли.
Изоподгикислоты. Если в кислородную кислоту вместо атомов О ввести радикалы той хе кислоты, например в П2СгО4 ввести один или несколько радикалов СгО4, образуются соединения, которые в отличие от гетерополикислот (образующихся при введении в остаток одной кислоты радикалов других кислот) называют иеополикиелотами. Часто из оп о лики слоты кратко называют просто поликислотами, когда це требуется подчеркнуть их отличие от гетерополикислот. Поликислоты (изополикислоты) можно .рассматривать как такие кислаты, которые образуются иг простых кислот, когда две или несколько молекул последних, отщепляя воду, дают новое соединение. Например,
2Н2СгО4—Н30 — НгСгзОу; ЗН2СгО4-—2И2О = H2Cr3Oj[, и т. Д.
(Мово-)хромовая Дихромовая Трихромовая
кислота кислота кислота
Ионы изополикислот мояспо рассматривать как производные ионов простых кислот, образующихся при взаимодействии последних с отщеплепием ионов О3-:
2[СгО^~ — ОВ-=[Сг2О7р-; 3[СгО4]2-—2Оэ-=[Сг3О10]«-
(Моно-) хромат-ион Дпхромат-ион Трихромат-лоп
(Бихромат-ион)
П ри этом отщеп л яющийся и он О 2 з аменяет ся другим и оном кис лоты:
’ О 12-
' О
О Ст О
О
0 Сг О
О
О О
О Ст О Сг О
О О
Н-О3"..
Реакции такого типа, которые приводят к образованию депей из радикалов с отщеплением молекул Н3О или ионов О3- (а также молекул или ионов H2S, NH3, S2~ И т. Д-), носят общее название реакций конденсации.
По склонности к реакциям конденсации ионы кислородных кислот можно разбить на пять групп:..
1. Иопы, которые сообща никогда не вступают в реакции конденсации между собой.
Примеры:
[СО3]2-, [NO3]-, [С1О4Г, [МпО4]-.
* Присоединяться к кислотам с образованием гетерополикислот может также V-,O6.
204
Глава б
2. Ионы, которые могут конденсироваться в ограниченном, но в пределах этого максимального значения в любом числе.
Примеры: [СгО413- и [SOJ2". При конденсации эти ионы образуют только анионные радикалы ограниченной величины: ([СгоО?]3', [Ст3О1(|Р-, [Cr4O13]3~; [SoO?]3", [85О10]2'). Устойчивость таких радикалов падает с увеличением степени конденсации. Для большинства иопов, относящихся к этой группе, способность к конденсации-не превышает первую ступень (образование иопов Эикислот).
3. Ионы, которые конденсируются между собой как в ограниченном, так и в неограниченном числе.
Пример: [SiOJ4". При конденсации ограниченного чнсла ионов [SlOJi- С отщеплением ионов О2- образуются анионные радикалы: [SigOj]®-, [Si3O0le f [Si40j3]10-, [Si50fg]13~, |SiBOlsl13"*. При конденсации неограниченного числа иопов fSiO4]1- образуются цепи, связки, плоскости или пространственные решетки (в одно, двух или всех трех пространственных намерениях) неограниченной протяженности (см. т. I, стр. 541 и сл.).
4. Ионы, которые конденсируются между собой только в неограниченном числе.
Пример; |SbO3]ll-i Пространственную анионную решетку безводного ацтпмона-та(¥) следует рассматривать как продукт конденсации (с отщеплением ионов О3-)-! неограниченного («бесконечно большого») числа ионов [SbOgl11- (см. т. I, стр. 719). В некопдонсированном состоянии ион [SbOg]11- не существует, равно как по существует-оп и в формо продуктов конденсации ограниченного (2, 3, 4 и т. д.) числа ионов.
5. Поны, которые при ограниченной конденсации могут конденсироваться ниже верхней границы не в любых, а только в определенных отношениях.
Примеры: [¥О4]3-, [Мо04]3-, [WO4J2" (см. стр. 118, 187 и сл. 196 и сл.), а также-[GeO3P~ (см. т. I, стр. 567).
Различно между способностью к конденсации ионов групп 1 и 2 то.чько количественное. У иопов кислот, относящихся к группе 2, способность к конденсации в общем-уменьшается ** по мере увеличения заряда и уменьшения радиуса центрального атома. Под влиянием обоих факторов способность к конденсации может быть сведена н; пулю-(группа 1). Отношение группы 3 к группе 2 противоположно отношению группы 1: в группе 3 совокупное влияние заряда и радиуса приводит к появлению максимальной способности к конденсации. Считают также, что исключительная способность к конденсации иопов [StO4[-1“ обусловлена и пространственными факторами. Пространственные факторы могут привести к тому, что ион, например Sb5+, будет стремиться окружить себя большим числом ионов О3-, чем это можно установить по величине его положительного заряда. Группа, к образованию которой стремится такой ион, в данном случае группа [SbOs], может существовать только тогда, когда она имеет возможно большее число общих ионов с соседними группами, т. е. когда происходит возможно-большая конденсация ***. Это осуществляется тогда, когда неограниченное число групп [SbOg]-!! образует одну общую анионную пространственную сетку, как это установлено для кристаллической структуры аптимонатов(У). Ионы группы 4 существуют только в «форме состоящих из них прострапствениых сеток, проявляющихся в кристаллических решетках. Формально они диаметрально противоположны ионам группы 1, которые существуют только в некопденеированном состоянии. Но силы, которые в одном случае препятствуют конденсации, а в другом вызывают ее в самой высокой степени, по-' существу одни и те же.
Ионы группы б занимают особое положение по отношению к нонам остальных четырех групп. Для пих наблюдается своеобразное явление изменения более или менее резкими скачками числа связанных между собой кислотных остатков. Ланример, в случае вольфраматов конденсация в водпом растворе приводит сразу от .илновольфра-матов к гексавольфраматам (через менее устойчивые тривольфраматы), а от пих — к до-дскавольфраматам (ср. стр. 196 и сл.). Эта особенность не может быть объяснена пи одной из структур, пайдеппых для продуктов конденсации ионов 2,3 или 4-й групп.Что к группе 5 относятся как раз те ионы, которые способам образовывать комплексные гете-рополикиюяоты, указывает на то, что комплексные изополикпелоты этой группы структурно очень близки к г стер ополии ис лотам. Действительно, по своей структуре До дека-воль (фр аматный комплекс следует рассматривать как гетерополикомплекс (см. стр. 203),
* Не известно, существуют ли такие радикалы, фигурирующие в качестве структурных групп в кристаллах, также и в растворах.
** Сильное поляризующее действие центрального атома (элементы побочных подгрупп!) может компенсировать и даже превышать влияние заряда на способность-к конденсации.
*** Также пространственно возможная группа [SbOe) становится устойчивой только при уменьшении взаимного отталкивания иопов кислорода: превращение в водных растворах ионов О3~ в ионы ОН", образование [Sb(OH)e]_.
Шестая группа периодической системы
205
Выпадение додекавольфрамовон кислоты из общего ряда изополикислот перестает быть исключительным случаем, если принять, что вообще конденсированные кислотные ионы группы 5 имеют структуру, присущую гетер ополи кислотам. Рент ген ост руктур-пые исследования указывают на то, что действительно наблюдается такое положение. Например, структуру гептамолибдата аммония (NH4)e[Mo7024l-4Ha0 можно получить из структуры гексамолибдатотеллурата аммония (NH4)6|TeMo6OaJ ЛНаО простой заменой атома теллура ла атом молибдена.
В группу 5 долткпы быть включены также попы [NbO6]J~ и [TaOoF-. Эти ноны Относятся к названным попам группы 5 так же, как пошл группы 4 — к нонам предшествующих групп. Ови по известны в неконденсировэнной форме, а конденсируются только в неограниченном числе, образуя простирающиеся на весь кристалл анионы, как в упоминавшихся на стр, 124 безводных ниобатах. Или они конденсируются в ограниченном числе, но ири этом не в любом небольшом числе, а, минуя низшие степени конденсации * **, сразу образуют иогщ [Nb5OI617- и (Та5О10!7- или (NbGO19F' и [Та6О19Р" фсм. стр. 130).
Анионные пространственные сетки (образующиеся при конденсации неограниченною числа ионов) прсусгцрайлциеея через весь кристалл, обычна не относят к анионам поликпелот. Впрочем, по аналитическому составу оксо-соли, которые образуют как раз такие апнойные пр остра ист венные решетки, часто рассматривают как соли поликпелот Для отличия от таких солей кислоты или соли, образующиеся при конденсации оксо-кислот или их солей, анионы которых имеют ограниченную протяженность, можно назвать изополикислотами (или солями) в угком смысле слоеа. Тогда класс иаополнкислот и изополисолей распадется на две группы, представителями одной сбудут полихроматы, а другой — поливанадаты, полимолибдаты и поливольфраматы.
Соединения вольфрама с кислотами
Кроме галогенидов (включая оксттгалогспиды) и сульфидов, соединения вольфрама с кислотами известны только в форме ацидо-соединений. По и их число, если по учитывать галогениды и оксигалогениды, довольно мало.
Для шестивалентного вольфрама известно несколько оксогалогено-солей. Оксо-фторовольфрамат1а(У1) — в общем случае бесцветные, часто хорошо кристаллизующиеся соли, преимущественно типа Mi [W02FJ. Их получают растворением вольфраматов в водном растворе плавиковой кислоты. Ромбическая соль меди(П) Cu[WO2FJ-4Г12О изоморфна соли ()u[TiFe]-4H3O. Соль (NH,)3WO3F3 йзоморфпа (NFQ3ZtF- “и (NH4)aNl)OF6. Известно несколько двойных солеи и Для оксихлорида WO2Cl2,
Раетворстгпем щелочных вольфраматов в концентрированном кислом растворе щелочных оксалатов и восстановлением оловом (станиоль) Олесон (Olsson) получил красивые красные ркеллато-еокп пятивалентною вольфрама общей формулы M|[WO2(C2O4)2]. Эти соединения растворяются в концентрированной соляной кислоте с темпо-синим окрашиванием. Насыщение раствора хлористым водородом приводит к образованию красивых оксохлоровольфраматов(У): M|[WOC1S] (зеленые), M1[WOC14(OH2)] (светло-голубые) я М1 [WOCIJ (коричнево-желтые). Выли получены также соответствующие бромо-соли. Для пятивалентного вольфрама известны и комплексные роданиды.
Важнейшими представителями ацидо-со единений четырехеалентного вольфрама являются октациановольфрама ты (TV) ,.М4[ W(CN)8], аналоги рассмотренных ранее <жтациа1гомолибдлтов(11'). Их можно получить различными способами. Они образуют желтые, в большинстве случаев устойчивые на воздухе кристаллы, водные растворы которых дают нейтральную реакцию па лакмус. Известна и свободная кислота. Она образует желтые расплывающиеся кристаллические иглы состава 6Н3О.
Перманганат калия (в сернокислом растворе) окисляет оКтациановольфраматы(1У) в (желтые же) отГгациановолъфраматы(У) М3 [W (CN)S1, тогда как этот окислитель всегда окисляет вольфрам в других соединениях до шестивалентного ***. Свободная кислота Н3 [ W(CN)g] СН2О образует оранж ево-желтые неустойчивые на в озду х е Kjp иста л ли-ческйе иглы. Из раствора вольфрамата калия в концентрированной соляной кислоте,
* Конечно пившие неустойчивые промежуточные продукты копДенсации не рассматриваются.
** Например, соединения, которые содержат несколько эквивалентов SiO2 на 1М2О, обычно называют полисиликатами независимо от того, имеются ли в них замкнутые анионные радикалы или ленты, сетки и т. д. неограниченной длины.
*** Это относится и к соответствующим соединениям молибдена.
200
Глава 5
приготовленного опр еде лепным образом (по методу Олесона или РозейгеЙма), восстановлением оловом можно получить гидроксопентахлоровольфрамат(1У) калия K2[W(OII)C1>;]. Это соединение представляет собой темно-зеленым кристаллический порошок, который в проходящем свете и в растворе имеет красный цвет. Если продолжать восстановиопне, то в конце концов получаются хлоро-соли трехвалентного вольфрама типа M^W2G19], например K3[W2C191—желто-зеленые гексагональные кристаллические таблички.
Аналитические снедения. При кипячении с сильными кислотами из растворов вольфраматов выпадает осадок желтой вольфрамовой кислоты. Из растворов метавольфраматов такого осаждения не происходит совсем или оно начинается только после продолжительного кипячения (при этом в конце концов выделяется метавольфрамовая кислота). Сероводород также пе образует осадка. Но если сероводородом насыщать щелочной раствор вольфрамата, а затем его подкислить, то образуется светло-коричневый осадок сульфида вольфрама WS3, который нерастворим в кислотах, но легко растворяется в сернистом аммонии.
Если к подкисленному соляной кислотой раствору вольфрамата добавить цинк,, то первоначально выпадающая слизистая белая вольфрамовая кислота вследствие восстановления окрашивается в Красивый синий цвет. При нагревании с хлоридом оло-ва(П) и соляной кислотой происходит в зависимости от концентрации или синее окрашивание раствора, или выпадение синего осадка (вольфрамовая синь, см. стр. 199). Вольфрамовая синь образуется также при выпаривании с концентрированной сорной кислотой соединений вольфрама илн металлического вольфрама. Следует иметь в виду,-что аналогичные реакции дает молибдеп. Иногда в сипийцвет окрашивается и NbjO5, если на нее действовать водородом в момент образования (см, стр. 128),
Фосфатные перлы, бесцветные в окислительном пламени, становятся синими в восстановительном, а при добавлении небольших количеств сульфата железа(Н) — кроваво-красными.
В весовом анализе вольфрам, как правило, определяют в виде трехокиси вольфрама после осаждения желтой вольфрамовой кислоты или вольфрамата ртути(1) Hg2WO4. Отделение От молибдена можно осуществить отгонкой последнего в вида МоО3-2ИС1 в токе хлористого водорода или, воспользовавшись нерастворимостью греховней вольфрама в противоположность трехокисимолибдена, в 50%-помрастворе серпом кислоты, Бепт.чор и Иаттотт (Belcher, Nut ten, 1951) для отделения вольфрама от молибдена рекомендуют ттснользопатт. 1-амино-4-га-аминофенилнафталин. Этот реактив из растворов, содержащих достаточное количество соляной кислоты, осаждает только вольфрам. Мо.тнбден при атом яе захватывается. Очень небольшие количества вольфрама (10у и менее) можно, по данным Фейгля [F е i g 1, Angew. Chem., 45, 674 (1932)], еще с достаточной точностью определять по интенсивному желтому окрашиванию, возиикающему-при добавлении к слабощелочному раствору вольфрамата хлорида олова(П) в присутствии роданида калия.
УРАЛ (URANIUM) U
Распространение в природе. В природе уран встречается в основном в виде урановой смоляной руды. Этот минерал представляет собой двойное соединение окиси у рана (IV) или урана (V) с окисью урана(У1) (см. стр. 219) , в основном отвечающее формуле U3O8. Почти всегда минерал содержит небольшие количества окислов других металлов (железа, свинца, кальция, тория и редких земель). Первыми разработанными месторождениями являются рудники «Иоахимсталь» в Чехии. Этот минерал встречается и в других районах чешских и саксонских Рудных гор и в Кркоше, а также в небольших количествах в Португалии, Англии (Корнуэлл)., Норвегии и Швеции. Самые крупные месторождения расположены в Канаде (у Большого Медвежьего озера) и в Конго (Леопольдвиль)^
Шестая группа периодической система
207
Залежи меньших размеров имеются в США (шт. Колорадо и Юта). Но там, в большом количестве присутствует карнотит K.2(UO2)2 [VOJ2‘3H2O, являющийся наряду со смоляной рудой важнейшей рудой урана.
Минералом, который встречается не во вторичных осадочных отложениях, как урановая смоляная руда, а в некоторых основных породах, так называемых пегматитах, яляется уранинит UO2. Этот минерал всегда содержит окись тория, так как UO2> и ThO2 изоморфны. Некоторые разновидности уранинита содержат наряду с торием значительные количества редких земель, папример встречающиеся в Норвегии минералы клевеит, броггерит и ншепит. Смесью окисей урана и тория, в которой преобладает ThO2, является торианит. Почти всегда содержит окись урана и торит ThSiO4, по, как правило, в очепь небольших количествах. Кроме того, следует упомянуть и ауту нит (уранит, кальдиевоурановая слюда) Ca(UO2)2[PO4]a. 8Н2О; уранопирит (уранит бария) BafUCblolPOJ,-8Н3О; халъколит (торбернит, уранит меди) Cu(UO2)2|PO4J2-8Г13О и цеанерит Cu(UOa)2[Лз0412. Всегда содержат уран и минералы эвксенит и поликрас.
Исторические сведения, Уран был открыт Клапротом в урановой, смоляной руде в 1789 г. Свое название он получил в честь планеты Уран„ открытой за несколько лет до этого Гершелем.
Восстановлением гидрата окиси урана(VI) Клапрот получил двуокись урана, которую долгое время принимали за металлический уран, пока в 1841 г. Пелиго не получил настоящий металлический уран. Он выделил его восстановлением UCI4 металлическим калием. Этот метод в принципе является в настоящее время наиболее удобным методом получения чистого металла.
Прежде техническую переработку урановых руд производили в основном для получения содержащегося в них радия. Окись урапа получали в качество побочного продукта. В настоящее время, наоборот, радяй является побочным продуктом добычи урана.
Горные разработки урановой смоляной руды начались в середине XIX столетия в шахтах «Поахимиаля» в чешских Рудных горах, после того как пришла в упадок вследствие истощения запасов руды добыча серебра, процветавшая там с XVI до начала XIX века *. Одновременно закончились и разработки свинцовых руд. Соединения урапа, добываемые из смоляной руды, первое время паходили очень ограниченное-применение. Их использовали в основном для приготовления красивых флуоресцирующих стекол и керамических красок. Существенное значение урановые руды приобрели только после открытия радия (1898, см. гл. И), для добычи которого вскоре началась их переработка в больших масштабах цочтивовсех крупных странах,и прежде всего в США. Но и тогда применение соединений урана было ограниченным, а в самом металле вообще не было никакой потребности. Но в настоящее время металлический уран имеет огромное значение вследствие использования его для получения плутония и в атомных реакторах, что в конечном счете основано на открытом Ганом (1939) явлении деления атомов.
Получение. Урановую руду вначале выщелачивают серной, азотной или соляной кислотой. Для осаждения алюминия, кобальта, железа-и марганца к раствору добавляют Са(ОН)2 и избыток Na3CO3. Образующийся при этом карбопатоуранат разлагают соляной кислотой и из полученного раствора солей уранила, пропуская аммиак, осаждают уран в виде (NH4)2U2O7— ураната аммония. Прокаливанием осадок переводят в окись U3O8.
Если перерабатываются руды, содержащие медь или мышьяк, то из подкисленного соляной кислотой раствора карбоватоураната уран осаждают NaOII в виде Ха2и2О7. Эту соль вновь растворяют в соляной кислоте и в раствор пропускают H2S. При этом-выпадают в осадок CuS и As2S3. Из фильтрата, освобогкдеяного от H2S кипячением, выделяют затем аммиаком уран в виде (NH4)2U2O7.
* Иоахиметальские серебряные монеты сначала кратко назывались «иоахимста-лерамл», позднее просто «талерами*, а в Америко это название было превращено-в «доллар».
208
Влава б
Если исходной рудой является карнотит, то следует провести некоторые операции для отделения урана от ванадия и фосфорвой кислоты.
Для переработки бедных ураном руд используют особые способы, например нагревание в токе хлора или с другими хлорирующими агента»! (SCIo^-SOCl^, СОС1г, CCJ4). При этом уран отгоняется в виде UC14.
Восстановление и3О3 до металла можно проводить, как нашел Му ас-сан (1883 г.), нагреванием с углем в электрической дуговой печи. Однако получаемый таким способом металл содержит карбиды. Получение чистого металлического урана затруднено (кроме его способности образовывать карбиды) также его высоким сродством к кислороду и азоту.
Для получения чистого металла в лабораторных условиях наиболее пригодным является метод вцп Ариеля и де Бура (van Arkel, de Boer), если его использовать в варианте, предложенном Футом (Foote, 1944—1945; см. также Prescott, Holmes, 1944). Наиболее удобным техническим способом является способ Вестингауза, состоящий в электролитическом разложении UF4 или KUFS, раствореппых в расплаве СаС12- NaCl. Чистый урап можно получить в промышленных масштабах восстановлением UC14 металлическим кальцием (James, 1926) или металлическим калием (полученным взаимодействием СаС3 с КС1; Lautie, 1947):
UCli+2Ca=U-|-2CaCl2 4-131 ккал, UC144-4K=U-|-4KC14-167 ккал.
Обычный метод получения чистого металлического урана для атомных реакторов заключается в восстановлении тетрафторида урапа металлическим кальцием:
UF4-|-2Ca=U4-2CaF24-134 ккал.
При этом исходят из триураноктокепда U3OS, полученного пагреваниом(МН4)2и207 Этот окисел переводят в tJOa, например нагревая его в токе водорода. Обрабатывая двуокись урана 40% -пой плавиковой кислотой я обезвоживая первоначально образую* щийся гидрат нагреванием под уменьшенным давлением, получают тетрафторид урана. Его можно получить и непосредственным воздействием газообразного фтора па двуокись урапа при повышенных температурах. Чистый металлический кальции, требующийся для проведения этого процесса, получают взаимодействием окиси if а льдин с алюминием и последующей возгонкой в вакууме. При реакции тетр а фтор и да урана с металлическим кальцием уран получают в расплавленном состоянии. Для застывания его выливают в формы из плавленого флюорита, затем расплавляют в вакууме в тиглях из того же материала и получают металл с содержанием 99,98%,
Свойства. Уран в чистом состоянии представляет собой серебристо-белый блестяедин металл, который постепенно тускнеет па воздухе. Он не очень тверд, куется и прокатывается при обычных температурах. При нагревании он сначала становится хрупким, по при дальнейшем повышении температуры — очень пластичным. Причиной такого 660“ 760°
изменения свойств являются фазовые переходы ct-U—> |3-U—>y-U (см. стр. 138). Плотность чистого урана составляет 19,05, удельная теплоемкость (при комнатной температуре) равна 0,0274. Температура плавления урана по сравнению с температурами плавления его аналогов относительно низка (1130°). Теплота плавлении равна 2,8 ккал/г-атом.
Температурную зависимость атомной теплоемкости Ср a-урана выше 0° мощпо Представ пт ь формулой Ср = 3,15 -|- 8,44- 10-3Т 4- 0,80- IO5?"2, где Т — абсолютная Температура (Moore, Kelley, 1947). Теплоемкости f>- и y_U по зависят от температуры (10,38 я 9,1 i.i соответственно). При высоких температурах урав значительно более летуч, Чем молибден п вольфрам. Ия зависимости упругости его паров от температуры (0,026 .ад рт ст при 1900J и 2,1 рт ет при 2300°) для температуры кипения получается величика около 3500°, Уделыыя электропроводность чистого металла составляет 4’104 лк-1-с.и-1, а теплопроводность приблизите ль по 65-10-3 кал-см~1, сект1-град~1 Дцри обычных температурах). Уран слабо парамагнитен. Его удельная магнитная ^восприимчивость равна х = 2,6'10~в при 18° (Owen, 1912).
Шестая группа периодической системы
209
При высоких температурах уран является очень реакционноспособным металлом. Уже при незначительном нагревании на воздухе оп сгорает (выбрасывая искры) до триураноктоксида U3O8- Уран очень легко взаимодействует и с галогенами; в атмосфере фтора он тотчас же воспламеняется. С сухим хлористым водородом уран соединяется при температуре темнокрасного каления 250°. При температуре 500° в парах серы происходит энергичное воспламенение. С азотом уран реагирует при 450°. С фосфором, мышьяком и углеродом энергичная реакция начинается только около 1000°. С бором уран взаимодействует только при температуре электрической дуги.
Уран соединяется и с водородом. Гидрид, образующийся при умеренном нагревании, при сильном нагревании вновь распадается. Кроме неметаллов, уран образует соединения со многими металлами.
В разбавленных кислотах уран легко растворяется с выделением водорода и с образованием солей ypana(IV). В топконзмельченном состоянии уже при обычных температурах он реагирует с водой. Напротив, азотная кислота очень медленно действует на компактный металл, очевидно вследствие пассивирования. Порошкообразный уран энергично реагирует и с азотной кислотой, причем выделяется окись азота.
Уран радиоактивен. Он является тем самым элементом, на примере которого впервые наблюдалось явление радиоактивного излучения (см. гл. 11). При радиоактивном распаде урана из него через промежуточные продукты, указанные в табл. 62, образуется радий. Постоянно присутствующий в обычном уране изотоп актиноуран является начальным членом радиоактивного семейства актиния (см. табл. 63).
Применение. Раньте металлический уран пе находил применения. Теперь же он приобрел большое значение, так как его используют в атомных реакторах для получения энергии за счет атомного распада и для выделения плутония (см. гл. 13).
Многие соединения урана имеют определенное значение при получении чистого урана или при разделении его изотопов. В остальном применение соединений урана весьма ограничено. В фотографии соли урана используют Для усиления негативов, а также л качество виражей. Урапат натрия под названием урановой желтой используют Для окраски стекол и глазурей. Уран, содержащий карбид урапа, используют в качестве катализатора Для синтеза аммиака по методу Габера.
СОЕДИНЕНИЯ УРАНА
Уран, как молибден и вольфрам, способен образовывать соединения во всех валентных состояниях от II до VI. Двухвалентным уран является в моноксиде урапа UO. Трехвалентен он прежде всего в очень устойчивых безводных тригалогенидах UX3. Четырехвалентный уран образует очень много соединений. К ним относятся, кроме тстрагалогепидов UX4 и образуемых ими двойных солей, также двуокись урана UOa и соли, образуемые ею с кислородными кислотами. Соединениями, в которых уран определенно пятивалентен, являются пентагалогениды UF5 и UC15. Соединениями шестивалентного урана, кроме обоих гексагалогенидов UF6 и UCle п трехокиси урана IJOg, являются также многочисленные соединения уранила, т. е. соединения типа UO2X3, и уранаты, т. е. соли кислотного ангидрида UO3.
В водном растворе существуют, за пемпогими исключениями, только соединения ypana(IV) и ypana(VI). В общем в водных растворах наиболее 14 г. Реми
210
Глава 5
устойчивы соединения ypana(VI). Ионы урана(1У), если они не стабилизированы комплексообразованием, имеют склонность окисляться с образованием ионов уранила UO2‘‘ или уранатов UQ4" или U2O^'.
Тенденцию ионов U"" переходить в иопы уранила UO" количественно можно выразить величиной окислительного потенциала. Он составляет (при условиях, указанных, в табл. 112 т. I) + 0,41 в для реакции U“” -j- 2Н2О = UOj* Т* 4Н‘ -J- 2е.
Как показывает сравнение с величинами, приведенными в названной таблице, ион U"” в 1 и. растворе по концентрации ионов водорода имеет большую тенденцию к окислению, чем ион Fe", но меньшую, чем иои SH". Если концентрация ионов водорода в растворе уменьшается, то склонность ионов U."" к окислению значительно возрастает. Кроме того, окислительный потенциал растворов содей урана зависит от освещения. приведенные выше значешгя справедливы для темноты. При освещении окислительный потенциал падает, т. о. стремление ионов U’"' к окислению при,освещении возрастает. Точно так же можно часто наблюдать „что восстановлению солей ура-пила способствует солнечное освещение. Но это явление основано на совсем другом эффекте, а имопвО каталитическом ускорении некоторых реакций светом,
В четырехвалентном состоянии уран в кислом растворе существует в виде простого иона U"""" (иона урана(1У)У, в шестивалентном состоянии он присоединяет два иона О2', образуя уранил-ионы [UO2]“. Шестивалентный уран образует также уранаты, например уранат натрия (точнее диурапат натрия) Na2U2O7. Но это соединение в воде не растворяется.
Многие соединения ypana(IV) изоморфны соответствующим соединениям тория, например UO3 изоморфен Thp2f U5P4 изоморфен Th3P4, UP изоморфен ThP. Кроме того, гидраты U(SO4)a (см. стр. 222) изоморфны соответствующим гидратам Th(SO4)2.
Соединения урана придают литому стеклу в большинстве случаев желтый цвет с красивой желто-зеленой флуоресценцией. Растворимые соединения урана сильно ядовиты.
Из органических соединений урана интерес представляют только такие, которые летучи; и поэтому могут служить для разделения изотопов урана. Следует упомянуть тетраизопропилато алюминат урана(1У) U[aI(OC3H7)4]4 — зеленое масло ст,кип,270в.
Классификация соединений урана
Соединения урана можно разделить на
I. Соединения урана несолевого характера.
II. Образуемые ураном соли.
Соли, которые образует уран, распадаются на две группы:
1. Уранаты и пероксоуранаты.
2. Соединения урана с кислотами:
а) соли урана(У1). Это почти всегда соли уранила, т. е. соли радикала ШО21
б) соли урана(1Уу„
в) соли урана(П1).
К соединениям урапа несолеобразного характера относится преобладающее число бинарных соединений урана.
Нельзя провести резкой грапнцы между бинарными соединениями урана песо ле-образного характера и такими же соединениями соле образ но го характера. Поэтому последние будут рассмотрены вместе с остальными бинарными соединениями урана. При этом будут исключены соединения более чем Двух элементов, если только речь пе идет о соединениях с ярко выраженным характером солей.
Уранаты являются производными трехокиси урана UO3, образующимися при ее взаимодействии с основными окислами. Простейшим урана-
»
Шестая группа периодической системы 211
там отвечает общая формула Mj [UOJ. Но большинство соединений т.рех-окиси урана с основными окислами является диуранатами MHU2O7}_ Пероксоуранаты образуются из уранатов при замене иона Оа~ на перекисную группу Оа“. Пероксоуранатами являются, например, соединения урана с формулой MjlUOJ или M4[0e = U0J.
Соли уранила также являются производными трехокиси урана. Они образуются при взаимодействии трехокиси урана с кислотами. При этом в большинстве случаев на кислотные остатки замещается только один ион Оа” и образуются соли радикала: уранила ШО2]а+:
иОз+гпх^ио^Хг+НзО.
Соли урана(1У) являются производными двуокиси урана U02, имеющей характер основного ангидрида. Им отвечает общая формула UIVX4. Как уже было отмечено, в водных растворах они менее устойчивы, чем соли уранила.
Очень мало устойчивыми в водном растворе являются соли урана(Ш), т. е. соли типа UniX3. Относительно устойчивой простой солью урана(Ш) является иодид урана{ПГ) Щ3. Комплексным соединением, производным соли урана(Ш), является дисулъфатоуран(1¥У) кислота H[U(SO4)2J (ср. стр. 220).
БИНАРНЫЕ СОЕДИНЕНИЯ УРАНА
Обзор бинарных соединений урана с металлами и неметаллами дан в табл. 20.
Приведенные в таблице данные о растворимости в твердом состоянии относятся к «-урану. (J-Модификация несколько лучше растворяет некоторые металлы. Еще более это справедливо для у-урана. Эта модификация способна принимать в свою решетку до 5 ат.% алюминия, 3—4 хрома или марганца, около 2 железа или никеля, а молибдена даже 36 ат.% . Ниобий, который растворяется в а-уранё только до 0,25 ат.% , при 1350° в у-урапе может растворяться до 85 ат.% (правда, при 660е растворимость ниобия составляет только 3,6 ат.%). Возможно также внедрение урана в кристаллические рещетки других металлов, но, насколько известно в настоящее время, только в ограниченной степени. Например, висмут, хром и медь вообще не образуют твердых растворов с ураном, растворимость урана в молибдене, вольфраме и тории также очень мала. Ртуть при 25“ растворяет менее 0,01 ат.% Урана, при 350°—1,25 ат.%.
Из интерметаллических соединений урана UBi изотипен каменной соли. UCo имеет кубическую объемноцеятрированную сложную решетку. IJA12, UMn2, UFe2 и UCo2 образуют фазы Лавеса типа MgCu2; UNi2 дает такую же фазу типа MgZn3 (см. стр. 44). UHg3 кристаллизуется, как и Uwi2, в гексагональной системе, но дает соединения другого типа. UAJ3 и USn3 кристаллизуются в кубической системе по типу Са8н3 (ср. т. I). UHg3 образует гексагональную, VHg4 — довольно сложную, но родственную кубической объемноцевтрированнон решетку. UNi3 и UCus (изотипны РаВе3 и АнВе5) имеют кубическую гранецентрированную решетку, родственную решеткам типа MgCu2. Соединения UgMn, UeFe, U6Co и UeNi кристаллизуются в тетрагональной объемпСцентрированной решетке и изотипны друг другу.
Обзор внутреннего строения бинарных соединений урана с неметаллами дан в табл. 21. Для UO3, кроме модификации, приведенной в табл. 21, известны также другие, но их структура еще не определена. Тетрагональный силицид a-IJSi2 изотипен ThSi2. В табл. 22 приведены теплоты образования и величины свободной анергии для некоторых соединений урана. Эти величины относятся к образованию соединений из элементов в их нормальном состояния при 25е. Только для гидратов трехокиси урана приведены теплоты образования из UO3 и Н2Огазообраан.
Гидрид урана UH3. Если порошок урапа нагревать в токе водорода при 225°, то образуется, как впервые установил Дриггс (Driggp, 1929), гидрид UH3 — соединение с постоянным составом и вполне определенным давлением разложения, которое при 200° равно 0,6 льиpm cm, при 300s—24,8 .ил*рт ст, при 400°—345 .и.и рт ст и достН-
14»
Таблица SO
Образование соединений и твердых растворов ураном с элементами главных и побочных подгрупп
Главные подгруппы
ш IV N V 1 Vl VII
в ЛЬ G S1 Sn p As Sb Bi 1 0 8 Se Те F Cl Br I
— тв. О тн. >0 тв. ~ О TD. 0 тв. — 0 —. •1— тв. 0 ТВ. 0 ——b — —- — __ —
ив2 UA12 1590° ис 25 5 0° и^а U5Sn4 1500° UN UP U2As U3Sb3? UBi uo US U2Se3 UTc L:F3 UC13 UBr3 UI3
ив4 UAliJ Щсг3 стабилен только 2000° U3Si2 16S5’ U$n£ iaae° u2n3 UAs U4O7? u2s3 USe2 U2Te3 UF4 UC14 UBr4 Uh
UN3 UaAs4? uo2 us2 UTe2? kr17 UC15
ив1г UAl* 7 а 0° UC, 2400° USi* 1586= U2o5 US3 tJ2F9 UC16
USi2 ~И00° u3o8 uf5
USi3 1510° uo3 UFe
Побочные подгруппы
IV v VI VII 1 VIII I II
Ti Zr Th V Nb Ta Cr Mo w Mil re Co Ni Cu Ag Au Zn Hg
— — тв. 0 тв. 0 tb.>O тв, 0 тв. о тв. 0 a V о -!-!•* тв. 0 — — — ТВ. 0 tb.>O — тв. 0
UeMn* 720® UMn, 4120° U6Fe* 215° UFe3 1235° U6Co UCo UCo2 UeNi* 754“ UNi? 810° UNi5 1295° у Qi* 1052“ UHg? 450° UHgs* 390° UHg? 360°
Таблица 21
Кристаллическая структура бинарных соединенна урана с неметаллами
Соединения Структура Длина ребра элементарной ячейки, А Число формульных единиц в элементарной ячейке dpОНТг
а ь С
Бориды ив2 Гексагона льная, типа A1B2 3,136 — 3,988 1 12,69
ив, Тетр а т она л ьная 7,075 — 3,979 4 9,38
IJBI3 Кубическая 7,473 — — 4 5,825
Карбида UG Кубическая, типа NaCl 4,961 — — 4 13,63
ис2 Тетрагональная, гра-нецептрир ев апн ая, родствен пая типу СаС2 3,524 5,999 2 11,68
Силициды U3Si Те траго на ль нал, об ъ е м н оцеп три р о-вавная 6,020 — 8,696 4 15,60
L3Si2 Тетрагональная 7,330 — 3,900 2 12,21
USi Ромбическая 5,66 7,67 3,91 4 10,40
g-USte Тетрагональная, объемноцентр про-ванная 3,98 — 13,74 4 8,98
₽-USi2 Гексагональная 3,86 — 4,07 1 9,25
IJSi3 Кубическая 4,035 — 1 8,298
Нитриды UN Кубическая, типа NaCl 4,890 — — 4 14,32
. u2n3 Кубическая, типа Sc2O3 10,700 — — 16 11,24
un2 Кубическая, типа СаР2 5,32 — — 4 11,73
Фосфиды и арсениды UP Кубическая, типа NaCl 5,600 — — 4 10,23
UgPi Кубическая, типа Th3P4 8,214 — — 4 10,04
UP2 Тетрагональная, типа Cu2Sb 3,800 — 7,762 2 8,89
U As Кубическая, типа NaCl 5,767 — — 4 10,77
U3AS4 Кубическая, типа Th3P4 8,507 — — 4 10,94
UAs2 Тетрагональная, типа Cu2Sb 3,954 — 8,116 2 10,15
Продолжение табл, 21
Соединения Структура Длина ребра алеиен-тарпой ячейки, А Число формульных единиц в елемен-тарцай ячейке Бренте
а 6 С
Окислы ио Кубическая, типа NaCi 4,93 "— — 4 14,1
со2 Кубическая, типа CaF3 5,469 — — 4 10,97
С’2О5 Ромбическая 8,29 31,71 6,73 16 8,35
с3оа 6,717 11,967 4,149 2 8,39
а-1Юз Гексагональная 3,971 — 4,168 1 8,34
Сульфиды US Кубическая, типа NaCl 5,484 —- 4 10,87
U2S3 Ромбическая, типа Sb3S3 10,65 10,41 3,89 4 8,78
a-US2 Тетрагональная 10,25 'Ч— 6,30 10 7,54
₽-us3 Ромбическая, изоморфная Th S3 4,22 7,08 8,45 4 7,90
Гидрид UH3 Кубическая, распо-ложение атомов U, как в p~W 6,631 — — 8 10,92
Фториды UF3 Гексагональная, типа LaF3 4,138 — 7,333 2 8,95
uf4 Триклинная (псевдо-моноклинная), изоморфная ThF4 12,79 а 126° 10,72 8,39 12 6,70
a-UFs Тетра гола льва я, ц е-почечнаяструктура 6,512 4,463 2 5,81
№ Тетрагональная, пространственная сетка 11,450 — 5,198 8 6,45
UFe Ромбическая, молекулярная решетка 9,900 8,962 5,207 4 5,06
Хлориды UC13 Гексагональная, типа La(OH)3 7,428 — 4,312 '2 5,51
UCJ4 Тетрагональная 8,30 — 7,49 4 4,87
UCle Гексагональная, молекулярная решетка 10,90 — 6,03 3 3,59
Бромиды и иодиды UBr3 Гексагональная, тала La(OH)3 7,926 — 4,432 ; 2 6,53
UI3 Ромбическая, : типа Lal3 13,98 4,33 9,99 4 6,76
Шестая группа периодической системы
215
Таблица S2
Теплота образования — Aff298, и свободная энергия — Д_РТ298, соединеннй'урана
(ккал/г-атом U при 25°)
Карбиды Нитриды 1 Окислы Гидрид
UC UCS UN USN3 UNa UO иоа UaOa НОз ии3
8 S ttq щ <1 <1 1 I — 3,92 9', 83 106 128 . 1 1 135 259,8 247,3 284,6 : 266,6 287,7 269,1 30,4 17,7
Фториды Хлориды
иг4 U4Fi7 UsFe a-UFB ₽-UF6 UFB UG13 UCI* ГСТв < UCle
— А^298° 444 422 455 432 467 442 484 458 485 459 505 485 212,0 196,7 251,0 229,6 262,1 235,7 272,3 241,4
Бромиды Иодпди Оксигало-r eu иды Гидраты трехокиси урана
иВгэ |иВг4 UIs ш4 JOClo UOBr2 UOg • 1 -;Н2О U0a-H2O иО3-11/2Н2О U0s-2H20
! 1 > о “ч Ц; OP Ч °? 181,6 211,3 167 190 129,4 121 149,2 140 261,7 246,3 246,9 231,3 13,3 23,4 31,7 39,2
тает одной атмосферы при 436°. Гидрид урана представляет собой соединение с полуметаллическими свойствами и проводит электрический ток. В тонкораздробленном •соСтояпии он очень реакционноспособен. Получающийся при его термическом разложении металлический уран пирофореп.
Металлический уран при температурах выше температуры разложения гидрида урана способен поглощать определенное количество водорода с образованием твердого раствора. Но а-U растворяет минимальное количество водорода, меньше, чем растворяет железо. В р- и у-П растворимость водорода больше, а в расплавленном уране опа даже значительна.
Фториды
Гексафторид урана UF6. Обычно урап реагирует с фтором с образованием тетра-гфторида. Но если фтор содержит некоторое количество хлора, то, как пашел Руфф (Ruff), образуется гексафторид. Последний можно получить также действием F2 на iJCj. Это соединение бесцветно, кристаллизуется в ромбической системе, легко летуче, сильно гигроскопично и очень реакционноспособно. Оно возгоняется без плавления при 56,5° (под давлением т. пл. 64,0°). При плавлении гексафторид урана необычайно сильно расширяется (плотность твердого UFe при температуре плавления равна 4,8, жидкого — 3,62).
Кристаллы гексафторида построены из молекул UFe, в которых 6 атомов фтора расположены вокруг атома урана в виде несколько искаженного октаэдра (Hoard., 1944). В газообразном состоянии молекулы UF6 также представляют собой несколько искаженные октаэдры (Вгапне, 1937; Bauer, 1950). В газовой.фазе дипольный момент UF6 практически равен пулю. Кратчайшее расстояние U<—»F равно в газовой фазе 1,87 А, в кристалле — 2,02 Л,
Вследствие летучести гексафторид урана имеет большое значение для разделения изотопов урана. К UF6 близки по свойствам наиболее летучие из известных соединений урана; U(BH4)4 и U(BH4)3(BHS-СН3). Их давления паров при 50° составляют 1,4 и 8,5 мм рт ст. соответственно. При) введении следующих метильных групп лету
216
Глава 5
честь с седин няня падает. Давление паров U'(BH3-СН3)4 (т. пл. 73°) при 50° равпо 0,4 .«.и рт. cm. ISchlesingor, J. Am. Chem. Soc., 75, 219 и 222 (1953)].
Пентафторнд урана UFj, который был впервые получек Руффом (1911), получается в двух модификациях, причем обе они тетрагональны (см. табл. 21). Обычно это соединение описывают как окрашенное (красно-коричневое, зеленое или серое), но в совершенно чистом состоянии оно бесцветно. Это соединение можно получить действием безводного HF на UC15, F2 на UF4 или взаимодействием VFg с UF4. В последнем случае часто образуются в качестве промежуточных продуктов фториды U2F9. и U4F17, дающие рентсенограммы, отличающиеся от рентгенограмм других фторидов урана. Оба соединения черного цвета.
Тетра-и трнфторгщы урапа. Tempa/flmopud урана UF4 получается при действии фтора на металлический уран или при пропускании смеси HF и NH3 над UO3 при температуре около S00Q. UF4 можно получить также осторожным обезвоживанием гидрата UF^VjHoO. Последний осаждаетсяввидо зеленого осадка из растворов солей урана(1У) при действии плавиковой кислоты. При 100° он переходит в моногидрат tiF4-H2O. Безводный UF4 представляет собой зеленый трудно растворимый в поде порошок (т. пл. 96(F). При нагревании на воздухе он переходит в U3Oe. Нагревание в токе водорода прекращает теграфторид в трид/торид (Ruff). Чистый трифторид урана получается этим способом только тогда, когда используемый для его получения тетрафторид совершенно но содержит кислорода (Skinner, 1944). Трифторид VF3 можно получить такжа нагреванием тоикоизмельчепного порошка урана с Ш’4 при 105LF (Glandrow, Cbiotti, 1944). Трифторид урана UF3 образует темные фиолетово-красные кристаллы (структуру см. табл. 21), нерастворимые в воде и в неокисляющих разбавленных кислотах. При нагревании он распадается по уравнению 4UF3 3UF4 + U. Такой распад при температурах выше 1200’ становится значительным. О двойпых солях трифторида урана см. егр. 222.
Хлориды, бромиды и иодиды
При сгорании урана в токе хлора получается смесь тетрахлорида урана (UC]4) и пентахлорида урана. (ПС15). Последний, полученпый в чистом состоянии Руффом (длинные, в проходящем свете ираспые с зеленым отливом иглы), малоустойчив. Уже при комнатном температуре оп медленно распадается на хлор и тетрахлррид урана. Но он может распадаться и иследегвие окпслительно-ЕОсстаяовительпой реакции. В этом случае наряду с тетрахлоридом урана образуется гексахмрид урана UC16. Обе реакции в значительной степени обратимы:
2UClr, rj 2I.1C14+G12 и 2UC15 UC14+(JC16.
Избыток хлора способствует образованию гексахлорида урана.
Гессахлорид урана образует мелкие темно-зеленые или черные кристаллы (Лрикн^3 = 3,36, т. пл. 177,5е). 1.1,1); очень чувствителен к влаге п реагирует с водой, образуя-ПО2С12. По в СС14п в других индифферентных растворителях он растворяется беа разложения. С безводным IJF гекеахлорид реагирует при комнатной температуре, образуя пепгцфторид:
UCle4-5HF=UF5+5HCl+i/2G12.
Татрааялорид урана получают, кроме разложения понтахлорида, л другими способами, например нагреванием в токе хлора до температуры красного каления, смешанного с углем U3OS. Оп образует темно-зеленые кристаллы с металлическим блеском, которые возгоняются нри температуре красного каления (т. пл. 589°, т. кип. 792е). Плотность паров соответствует формуле UC14. В воде тетрахлорид урана растворяется с большим выделением тепла, его растворы окрашены в зеленый цвет. В растворах UC]4 в значительной степени гидролитически расщепляется. При низких температурах UC]4 может присоединять до 12 молекул ЙН3. При раз поженим этого соединения амми-ак отщепляется ступенчато. Прн комнатной температуре устойчив пентааммиакат. Свойствами, аналогичными свойствам тетрахлорида, обладают тетрабромид урана ПВг4 (т. ид, 518е) и тетраиодид урана П14 (т. пл. 506е), которые, однако, менее устойчивы.
Трихлорид урана ПС13 (т. пл. 842°) получают нагреванием тетрахлорида урана в тбке водорода. Он образует темно-краспые, очень гигроскопичные иглы, водный раствор которых вскоре окрашивается в зеленый цвет и при этом выделяется водород. Это относится и К трибромиду UBr3 (т. пл. 730°). Трииодид Ш3 (т. ня. 506’, т. кип. 680°) растворяется в воде с глубоко-красным окрашиванием. Свежеприготовленный триио-дид энергично реагирует с водой, отщепляя иод.
Шестая группа периодической системы
217
Сульфиды. При непосредственном соединении урапа с серой или при действии H,S па UC14 получается дисульфид урана US2 — серый (до черного) блестящий кристаллический порошок (уд. вес 7,90). Вода действует на него очепь медленно, соляная кислота быстрее, а азотная — очень быстро. При нагревании дисульфида урана с избытком серы под давлением образуется трисулъфид урана US3 (уд. вес 5,81) (Biltz, 1940). При взаимодействии UBr3 с H3S илп при нагревании US3 в токе водорода получают диурактрисулъфид U2S3 (уд. вес 8,80). Кроме того, существует еще более бедный серой сульфид состава US. Рентгена структурные исследования показали, что это соединение представляет собой истинный моноеульфид урана. Оп имеет решетку типа NaCl и изоморфен ThS ti CeS. Как US, так и ий83 получаются при прямом взаимодействии элементов. Но если сера взята в избытке, то образуется USg. Последний пе теряет заметно серу даже при 1000°, тогда как уже при умеренном нагревании переходит в US2.
Нитриды урана. С азотом уран образует соединения UN, U2N3 и UN3. Наиболее устойчив мопонитрид урана (светло-серый порошок). Его можпо нагреть в вакууме до 1900° без разложения. U2N3 в вакууме теряет азот уже при 750’, a UNa получается только под давлением. В UN атомы урапа образуют гранедептрироваяпую кубическую решетку. Это относится, вероятно, и к атомам азота, так что в результате получается решетка типа NaCl. I aN3 кристаллизуется по типу Sc-,O3, a UN2 — по типу CaF2. Полуторный питрид может, присоединяя азот, непрерывно перейти в дипитрид. Это легко попять, принимая во) внимание родство типов Sc2O3 и CaF2 (см. стр. 55 и сл.). Значение постоянных решетки см. в табл. 21.
Окислы
По отношению к кислороду уран выступает н основном как четырех-или шестипал еттный. Оп образует черно-коричневую двуокись UCU и оранжево-желтую трехокисъ UO3. Однако более устойчивым, чем эти простые окислы, является зеленовато-черный триураноктоксид U3O8, который считается двойным окислом UO3 с UO2(U3O8 = UO2-2UO3) или UO3 с U2OS(U3O8 = U2O5-UO3) [см. стр. 219]. В природе уран встречается во вторичных отложениях главным образом в форме двойного окисла.
Рентгенометрически достоверпо установлено существование монокиси UO, кристаллизующейся по типу каменной соли. По это соединение в чистом виде получается с трудом. Оно образуется на поверхности металлического урана при его осторожном отделении. Двуокись (JO2 может содержать избыток кислорода. Ее состав может меняться без изменения кристаллической решетки от U02,oo доио2,30. При увеличений содержания кислорода плотность снижается. Это происходит потому, что избыточный кислород располагается в пустотах решетки Жестко (Biltz, 1927) или подвижно (Huttig, 1924). При повышении температуры U02 распадается (если она содержит более двух атомов D на один атом U) на совершенно структурно отличный от нее промежуточный окисел и4Ой и (в зависимости от начального содержания кислорода) U021oo или U3OB. Окислением LOa можпо получить еще два промежуточных окисла — и16О37 и UJ6O38, оба с тетрагональными решетками (Periо, 1952; Anderson, 1953). Еще не выяснено, представляет ли наблюдавшаяся Захариазепом (Zachariasen, 1945) кристаллическая структура состава U0bj5 повое, отличное от UOg соединение U4O7 (соответствующее соединению Th4S7) или в кислородной решетке UO2 могут непрерывно образовываться пустоты, так что область ее гомогенности может простираться как в сторону большего, так и меньшего содержания кислорода. Фазу UO2i!;0 следует, по данным Рапдля (Rundl, 1944), рассматривать как особое соединение U2O5, хотя ее кристаллическая структура очень мало отличается от структуры U3O8 и она, присоединяя кислород, может непрерывно перейти в U3O8.
Трехокись урана UO3 получают осторожным прокаливанием питрат а уранила, аммонийу ранил карбоната или ураната аммония *. Это соединенно представляет собой оранжевый или кирпичпо-красный порошок.
* Совершенно чистую UO3 лучше всего получать нагревали ем дигидрата перекиси урапа.
218
Глава 5
При сильном нагревании опо переходит в U3O8. Трехокись урана имеет характер амфотерного окисла: с основными окислами она соединяется с образованием уранатов, с кислотами образует соли уранила.
При кипячении с водой трехокись урана переходит в гидрат UO3- H2O (ромбические пластинки, не растворяющиеся в воде). Возможно, что вода в этом соединении связана химически. Поэтому это соединение обычно рассматривают как гидроокись уранила UO2(OH): пли урановую кцслоту. Известен также гидрат трехокиси урана состава UO3-2lT2O (лимонно-желтый порошок). При изобарном разложении этих двух гидратов в качестве промежуточных продуктов образуются гидраты UO3- и UO3-
• V2H2O (Hiittig, 1922). иО3-2НЕО при нагревании сначала отщепляет воду непрерывно, пока не достигнет состава UO3- 11/гН2О. Затем при постоянной температуре отщепляется еще 1/2Н2О. При дальнейшем повышении температуры гидрат UO3-H2O вновь непрерывно отщепляет воду до состава а полугидрат при постоянной темпера-
туре переходит в безводную трехокись.
Двуокись урана. UO2 образуется при нагревании трехокиси урапа или триураноктоксида в токе водорода. Она представляет собой черно-коричневый порошок с уд. весом 11,0 и т. пл. 2500—2600°, 11Оа испаряется без разложения. Ее давление паров при 160(? составляет 7.10-1> мм рт ст, при 1800°—а- 10-3 .и.« рт. ст и при 200(г— 0,07 мм рт ст. Двуокись урана [практически не растворяется в воде и едких щелочах. В кислотах (за исключением азотной кислоты, которая окисляет ее с образованием нитрата уранила) двуокись урана растворяется с трудом, образуя соли урана(IV). При нагревании на воздухе, а также в парах воды до красного калепия UO2 переходит в U3O8.
UO2 кристаллизуется по типу CaF2 (см. табл. 21) и во всех отношениях образует смешанные кристаллы с ТЬО2- Магнитные свойства IJO2 (Haraldsen, 1940) указывают на то, что в двуокиси металл существует в виде иопа Е4+я
Триураноктоксид U3O8 образуется как из двуокиси урана, так и из трехокиси при сильном прокаливании этих соединений на воздухе. Ц3О3 образуется также при прокаливании ураната аммония иди урановых полей летучих кислот. При высоких температурах (на воздухе при 900°, в вакууме уже ниже 600°) начинается отщепление кислорода и частичный переход в UO2, с которой U3O8 образует твердые растворы. Поэтому трудно получить чистую двуокись урана термической диссоциацией U3O8. Нагреванием в токе водорода U3Og легко переходит в чистую двуокись урана.
Цвет U3O8 изменяется от зеленого до смолисто-черного. Его удельный вес равен 8,30. Удельный вес U3O8, встречающегося в природе в виде урановой смоляной руды, сильно колеблется в зависимости от его чистоты.
Триуранокт оксид имеет довольно сложную кристаллическую структуру. Икге-роспо, что д его кристаллической решетке все атомы урана структурно эквивалентны, тогда как Ц4 атомов кислорода расположена иначе, чем остальные (Zachariasen, 1944; Rundle, 1944—1945; Grenvold, 1948). Встречающаяся в природе урановая смоляная руда всегда аморфна. Трехокись урана также часто получается в аморфном состоянии. Но можно получить и кристаллическую окись 1Ю3, причем ко крайней мере в трех модификациях, которые различаются не только своими рентгенограммами, но и цветом (оранжевая, кирпично-красная и желтая). Оранжевая модификация кристаллизуется в гексагональной системе (ем. табл. 21) _и образует решетку, в которой в направлении осп с расположены цепи —О—U—О—О—О—L—О— (расстояние О +—* О = 2,08 А). Кроме того, каждый атом урапа окружеп еще шестью атомами кислорода (расстояние U , О =-2.39 А), которые расположены между «ура пи левыми цепями».
Особым свойством трпураноктоксида является образование им смешанных кристаллов с окисью эрбия в области ЕтОь5 : Ъ'О2,3т от 0,27 : 1 до 2,0 : 1. Эти кристаллы имеют структуру плавикового шпата, в которой атомы обоих металлов статистически распределены в "узлах катионной решетки, тогда как анионная решетка может иметь пустоты или избыточные ионы О£_, расположенные в межрешеточном пространстве (Hund, 1952). Образование таких «аномальных» твердых растворов показывает, сколь различными могут быть типы и, при известных обстоятельствах, компоненты твердых растворов, если выполнены другие условия, которые существенны для
Шестая группа периодической системы 219
образования твердых растворов, а именно: возможно меньшие различия атомных или ионных радиусов, близкая поляризуемость и одинаковые координационные числа.
UjOe устойчив по крайней мере до температур 1450я. Но при добавлении Двуокиси циркония давление кислорода над ним возрастает и образуется окись урана с немного измененной кристаллической структурой и с составом в интервале иОй,вз — UO2j43 [Ochs L., Z. Naturforsch., 12b, 215 (1957)],
UO2 имеет характер основного ангидрида. UO3 амфотерна, она растворяется в кислотах с образованием солей уранила [UO2]X2, в едких щелочах — с образованием уранатов M*[UO4] или диуранатов М2 [U2O7]. При растворении в кислотах U3Ob образуется смесь солей урана(1У) и уранила. По этой причине окисел обычно считают смесью окйслов урана(ГУ) и урана(У1); UlVUVI2O8. Однако в соответствии с данными Гаральдсепа (1940) это предположение не согласуется с магнитными свойствами U3Ob. Для этого соединения Гаральдсеи нашел магнитный момент 1,39 магнетона Бора, что существенно отличается от значения, вычисленного для и1¥и^тО8 (1,63), но практически совпадает с величиной 1,42, которая отвечает формуле UjUVlOs. По этой причине соединение U3O8 следовало бы рассматривать как окись урана (V, V1).
Уранаты
Соединения трехокиси урана с основными окислами называются уранатами. Простейшим уранатам (моноуранатам) отвечает формула M’lUOJ. Однако чаще образуются диуранаты, онап&гнчяыо по своему составу бихроматам M*[U2O7] (обычно кратко называемые просто уранатами). Эти соединения образуются, как правило, при действий оснований на растворы солей уранила. Бее уранаты, даже щелочных металлов, нерастворимы в воде.
Моноурапаты получают из расплавов. Известны также три-, тетра-, пента- и гекса-уранаты."
Урапат (или диуранат) натрия выпадает как гексагидрат Na2[U2O7]-6Н2О в форме желтого осадка при добавлении едкого натра к растворам солей уранила. При нагревании оп обезвоживается. В го используют в качестве краски Для стекла и фарфора (урановая желтая).
Если к раствору соли уранила добавить едкого кали, то образуются уранаты сложного состава: K2U7O2a и К4П5О17 (оба соединения содержат воду).
Ураяат (или диуранат) аммония (NHJ2[U2O7] выпадает из растворов солей уранила при добавлении к ним аммиака. Он представляет собой желтый порошкообразный •осадок, практически нерастворимый в воде, но легко растворяющийся в растворе карбоната аммония. Это соединение очень удобно для весового определения урана. Следует только учитывать его склонность к образованию коллоидных растворов при промывании водой.
Дигидрат перекиси урана и пероксоурапаты. Если к концентрированному раствору нитрата или ацетата уранила Добавить перекиси водорода, то выпадает светло-желтый осадок, который после сушки при 100° имеет состав UO4-2H2O. Это соединение разлагается разбавленной серной кислотой, выделяя одну молекулу перекиси водорода на атом урана. Поэтому общепринятым является представление о том, что это соединение — производное гипотетической неизвестной в свободном состоянии четырехоки-си урана U04. Последняя, по-видимому, образуется из трехокиси урана при замене атома кислорода на перекисную группу —О—О—. Однако это еще не доказано. Соединение ПО4-2Н2О можно рассматривать и как U03-II202^ Н2О. Оно не имеет кислого характера, поэтому ранее употреблявшееся название «надурановая кислота» неверно. При действии щелочей это соединение не образует соответствующих ему солей. Но если действовать не только щелочами, но и перекисью водорода, то образуются пероксоуранаты. Эти соединения можно получить и непосредственным действием щелочей и перекиси водорода или перекисей щелочных металлов на водные растворы соединений ура-
220
Глава 5
нала. Так, калиевую соль K4[UO6]-6H2O2 (кристаллизующуюся с переменным содержанием воды), как показал Шварц (Schwarz, 1938), следует рассматривать как монопер-оксоу ранят (VI)-перекись водорода.
иОд-ЗНчО при пагревании переходит, теряя воду и кислород, в U03. Это используют для получения чпстой трехокиси урана. При осторожном нагревании можно достигнуть удаления только половины перекисного кислорода и получить безводную перекись UHO7(=Oa ; С205) (Huttig, 1922; Kraus, 1942). Эту перекись получают также пагревапием в токе кислорода диураната аммония (КИ4)ги2О7 (Kraus, 1944).
СОЕДИНЕНИЯ УРАНА С КИСЛОТАМИ
Известно большое число соединений шестивалентного урана с кислотами. В большинстве случаев они обладают ярко выраженным солеобразным характером. Почти все эти соединения являются производными положительно двухзарядного радикала [UO2]a+ (радикала уранила). Этот радикал ведет себя во многих отношениях подобно элементарному иону двухвалентного металла. Многие соли уранила обладают отчетливой тенденцией к образованию двойных и комплексных солей (аци&о-солей).
Для четырем алентного урана вообще известим только соединения с кислотами вполне определенного состава и по известны соединения со щелочами. Отсюда следует, что гидроокись ypana(IV) в подавляющем большинстве случаев имеет характер основания. Соли ypana(IV) значительно менсо устойчивы, чем соли ypana(VI) или уранила.
Трехвалентный уран присутствует прежде всего в легко получаемых тригалогенидах UX3. Примером соединения трехвалентного урана с кислородными кислотами может служить дисулъфатоуран{111) кислота H[U(SO4)2). Это соединение было получено Розенгеймом при добавлении серной кислоты к электролитически восстановленному при охлаждении льдом раствору хлорида уранила. Оно образует темпо-корич-певые кристаллические пластинки.
Соли уранила
Солям уранила отвечает общая формула UO2X2 (X — одновалентный кислотным остаток). Почти все они хорошо кристаллизуются и легко растворяются в воде. Эти соли обычно желтого цвета с желто-зеленой флуоресценцией. Наиболее употребительными солями уранила являются уранилнитрат UO2(NO3)2-()H3O и уранилацетат и02(С2Нз02)2-2Н20.
Хлорид уранила UO2C12 получается в безводном состоянии при действии газообразного хлора на двуокись урапа (т. пл. 5'78°). Из растворов трехокиси урана в соляной кислоте кристаллизуется тригидрат UO3C]2-ЗНнО в виде желто-зеленых косоугольных призм. При сушке в эксикаторе над Р2О5 образуется моногидрат иО2С1г-Н2О. Эта соль легко растворяется в воде. С хлоридами щелочных металлов и хлоридами органических оснований она образует двойные соли типа M|(UO4C14] [диоксотетрахло-роуранаты(У1)]. Двойные соли образует и фторид уранила UO2F2, причем в этом случае наиболее устойчивыми являются соля типа M|(UO2Fs].
При пизких температурах ПО2С12 способен присоединять до 10 молекул NH3. При разложении этого соединения аммиак отщепляется ступенчато, образуя аммиакаты с 5, 4, 3, 2 и 1 молекулой NH3.
Нитрат уранила получают растворением окислов урана в азотной кислоте. Из растворов кристаллизуется гоксагидрат UOa(NO3)2-6H2O в виде лимонно-желтых призм с желто-зеленой флуоресценцией, которые расплываются во влажном воздухе (т. пл. 59,6°). Нитрат уранила легко растворяется в воде, спирте и эфире. Его легко можно перекристаллизовать. При 21е в 100 в воды растворяется 127 а безводной соли. Если кристаллизацию проводить из концентрированной азотной кислоты, то выпадает дигидрат, плавящийся при 179°. Можно получить и безводный нитрат.
Шестая, группа, периодической системы 221
С нитратами щелочных металлов нитрат уранила образует двойные соли типа Mi[UO2(NO3)3], которые кристаллизуются без воды, но очень гигроскопичны.
Ацетат уранила UO2(C2H3O2)3-2H2O получают растворением гидроокиси уранила иОг(ОН)2 или трехокиси урана UO3 в уксусной кислоте. Но так как трехокись урана получают в основном прокаливанием нитрата уранила, то торговый продукт часто загрязнен нитратом. Из водных растворов ацетат уранила кристаллизуется в виде дигидрата — ромбических призм с сильной флуоресценцией. При нагревании до 110° он обезвоживается, при 275° разлагается с образованием трехокиси урана. Растворимость ацетата уранила относительно мала. В 100 г воды при 15° растворяется 7,69 г дигидрата. При долгом стоянии из водного раствора вследствие гидролиза выделяется основная соль.
Ацетат уранила способен присоединять еще один ацетаТный остаток, переходя в комплексный ион [иОа(СзН3О2)3]'. Поэтому он легко образует двойные соли с другими ацетатами. Такие двойные соли типа и MIMII[UO2(C2H3OS)3]3 большей частью легко кристаллизуются и имеют красивую зелеповато-зкелтую флуоресценцию. Образование кристаллизующейся в виде тетраэдров натриевой соли Na[UO2(C2H3O2)3l часто используют как реакцию на натрий. Аналитическое значение имеют и малорастворимые двойные connNaZn[UO2(C2H3O2)3|3-6Н2О и NaMg[UOa(C2H3O2j3!3-9H20.
Карбонат урапила и карбонатоуранаты. Чистый карбонат уранила. UO2CO3 был найден в Восточной Африке кате продукт превращения урановой смоляной руды (резерфордин!). Осадки, образующиеся при добавлении щелочных карбонатов к водным растворам солен урана, обычно представляют собой продукты неопределенного состава. В противоположность этому двойные или комплексные соли карбоната уранила (карбопатоурапаты) получаются легко.
Если к раствору соли урапила добавить избыток аммиака и карбоната аммония или подействовать раствором карбоната аммония на свежеосаждоппый уранат аммония INН4)2LtJjO?], то получается прозрачный желтый раствор, из которого при упаривании выделяется двойная сонь 2(NH4)2CO3-UO3CO3-2Н,0 (карбонат а имении-уранила) или комплексная соль (NIT4)4 [UO2(CO3)3}-2НгО (диоксотрикарбонатоуранат аммония) в виде желтых моноклинных кристаллов (растворимость около 5 г в 100 г воды при 5°). При длительном кипячении с водой двойная соль разлагается, выделяя уранат аммония. Аналогичные (также легко растворяющиеся) карбонаты уран образует со щелочными металлами. Двойной карбонат урана и кальция Ca2[UO2(CO3)3]-10Н2О встречается в природе в виде минерала урандталлита.
Используя способность урана к образованию легкорастворимых карбонатов, его легко можно отделить от встречающихся вместо с ним в природе редкоземельных, а также других металлов, паиример железа, алюминия и кальция.
Сульфат урапила UO2(SO4).3H2O выделяется в виде желтовато-зеленых кристаллов при выпаривании нитрата урапила с серной кислотой и дальнейшем концентрировании полученного водного раствора. Его кристаллы медленно выветриваются на воздухе, частично теряя воду. Сульфат урапила легко растворяется в воде (в 100 г воды мри 1 з,5е1 растворяется 17,4 а безводной соли). При нагревании до 175° гидрат, выделяющийся из водных растворов, полностью обезвоживается. Безводная соль окрашена в янтарно-желтый цвет и не обладает в противоположность гидратам флуоресценцией. Б растворах сульфата уранила, как показали измерения электропроводности, присутствуют комплексные ионы [UO2(SO4)2]''. При добавлении к таким растворам суль-: фатов щелочных металлов выделяются двойные или комплексные соли в основном типа M1[UO2(SO4)2(OHs)21 и MHUO2(SO4)3].
Сульфид у ранила UO2S выпадает па растворов солей уранила при действии сернистого аммония в виде коричневого- осадка, который растворяется в разбавленных кислотах (даже в уксусной кислоте). На Воздухе во влажном состоянии ои легко раз-па ынстс я, перехода в гидроокись уранила UO2(OH)2. Сульфид уранилаL полученный сухим путем, образует черные с переливами иглы, которые гораздо устойчивее к действию кислот, чем сульфид, осажденный из раствора.
222
Глава 5
Соли ypana(IV)
Сопим урана (IV) отвечает общая формула UX4. Почти все они зеленого цвета и в большинстве случаев легко растворяются в воде. Практически нерастворимым, даже в кислотах, является оксалат у рана (IV). Последний также довольно устойчив, тогда как растворимые соли урана(1Е), которые в растворах сильно гидролизуются очень склонны к окислению. Едкие щелочи или аммиак осаждают из растворов солеи урана (IV) объемистый красно-коричневый осадок, гидроокиси урана(IV), которая очень легко окисляется.
Для приготовления солей урапа(IV) можно исходить из соответствующих солей уранила [U(V])], восстанавливая их, например, водородом в момент образования иди электролитически. При действии соответствующих кислот на U02 соли урана(IV) образуются только в исключительных случаях. Этим способом можно получить фторид урана(IV) [Ш'4], ио не остальные галогениды U(IV), Последние, правда, легко получить сухим путем, например UC14 при действии CCJ4 на UO2 при 450° но уравнению" U02 СС14 = ПС14 -|- С02.
Галогениды урана{1У) легко растворяются в воде и очень гигроскопичны. Только-фторид довольно плохо растворяется. Если фторид получают растворением UO2. в водной плавиковой кислоте, то сначала получают гидрат UF4-2I/2H2O, Который легко переходит в безводную соль при нагревании (см. стр. 246).
UF4 чрезвычайно склонен к образованию двойных солей. Кроме двойных солей-типа MIIUFe, получающихся осаждением из водиого раствора (например, BaUFe, PbUFg) и кристаллизующихся по типу LaFg, известны многочисленные двойные соли, UF4 с фторидами щелочных металлов. Например, сплавлением UF4 с KF можно получить следующие соединения, существование которых установлено рентгеноструктур-ными измерениями (Zach a ria sen, 1948): K3UF7 (диморфный), K2UFe (триморфный), KUF5, KU2Fs, KU3Fi3, KUeF25. K3UF6 может кристаллизоваться как по типу CaF2, так и по типу A1BS (а кроме того, и но третьему типу с более низкой симметрией). Это же справедливо и для Na2UFe. Тетрахлорид урана. UC14 (см. стр. 216) образует в основном двойные соли типаМ}иС1си MzIUC16j но с ВаС1а, например, также двойную’ соль Ba3UCl8.
Соли ypana(IV) значительно менее склонны (чем соли уранила) к образованию’ истинных комплексных солей. Соединением ypana(IV), легко образующим комплексные-соли, является оксалат урана(1У).
Сульфат ypana(IV). Если к раствору U3O8 в крепкой серной кислоте добавить спирт и выдержать его на солнечном свету, то через некоторое время выделяется сульфат урана(1 V), U(SO4)2-4H2O. Оп кристаллизуется в виде темно-.зеленых ромбических пластинок. При растворении этого сульфата в воде сначала образуется прозрачный: раствор, по через некоторое время он мутнеет вследствие выделения оснрвпого сульфата. Часто из водного раствора вместо тетрагидрата кристаллизуется октагидрат. Сульфат урана (IV) устойчив только при температурах ниже 19,5”. Растворимость тетр а гидрата при 24° составляет ДО,9 г , при 63° — 6,7 за окта гидрата — при 18° — 11,3 е, при 62° (при этой температуре он метастабилен) — 58,2 г безводной соли на 100 г воды. Получен также нонагидрат U(SO4)2- 9Н2О. Эти гидраты изоморфны соответствующим гидратам сульфата тория.
Оксалат ypana(IV) получают восстановлением(солей уранила на солнечном-свету щавелевой кислотой или осаждением пз растворов солей ypana(IV) щавелевой кислотой. Он кристаллизуется в виде темно-зеленых кубиков или призм, не растворяется в воде и в разбавленных кислотах, но легко растворяется в щелочных оксалатах. При этом образуются комплексные соли [оксалатоурапаты(1¥)|, сильно отличающиеся по цвету от других солей ypariailV). Легко растворяющийся в воде тетр а око а лат о-уранат калия K4[U(C2O4)4]-5H2O кристаллизуется в виде серых гексагональных табличек. Бариевая сольВа2[и(С2О4)4[-6Н2О образует^красно-фиолотовые кристаллы.
Аналитические сведения. При обычном систематическом разделении катионов уран сопровождает железо. Как и Fe, уран осаждается сульфидом аммония в виде растворяющегося’в кислотах сульфида UO2S. Если к раствору солей уранила добавить щелочь или аммиак, то Уран выпадает в виде ураната аммония или щелочного металла, который вследствие растворимости в карбонате аммония легко отделяется от других, гидратов окисей, осаждающихся этими же реагентами.
Шестая группа периодической системы 223
G гексациан оф ер р ат ом( II) калия К4 [Fe(CN)e] соли уранила образуют коричневый осадок гексацианофоррата(П) уранила (U02)a[Fe(CN)ej. Эта реакция очень чувствительна. Ее используют при капельном титровании гбксацианоферратом(И) калия.
Перлы, фосфорных солей окрашиваются ураном в окислительном пламени в желтый (после охлаждения бледный желто-зеленый), в восстановительном — в зеленый цвет.
Для микроаналитических определений используют двойную соль ацетата уранила и ацетата натрия Na [UO2(C2H3O2)3],
Для весового определения уран обычно осаждают аммиаком в виде ураната аммония (КН4)Й1152О7} и переводят его сильным прокаливанием в U3O3 или прокаливанием в токе водорода в UO2.
Глава 6
СЕДЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Побочная подгруппа)
МАРГАНЕЦ, ТЕХНЕЦИЙ, РЕНИЙ
Порцц-копый ИОМЩ1 Опсмент Опцию л Атомный вес Удельный вес Температура, °C Удельная .теплоемкость Валентность
плавления кипения
25 Марганец Мп 54,94 7,21 1247 2030 0,1214 I, II, III, IV, V, VI, VII
43 Технеций (нестабильн.) Тс 98,91« 11,50 — — IV, VI, VII
75 Рений Be 186,22 21,03 3180 -5500 0,0327 I, П, III, IV, V, VI, VII
а Этот атомный вес относится к долгоживущему изотопу технеция (ср. стр. 251).
Общие сведения. В побочную подгруппу седьмой группы входят элементы марганец, технеций и рений. В отличие от предыдущих побочных подгрупп эту подгруппу составляют нс четыре, а три элемента. Это объясняется тем, что вслед за четвертым элементом предыдущей побочной подгруппы следуют трансурановые элементы, которые, как уже упоминалось, подобно лантанидам образуют особое семейство.
Главным представителем побочной подгруппы седьмой группы является марганец. Его аналог с порядковым номером 43 неустойчив и встречается е природе в очень небольших количествах (ср. стр. 249 и 252). Но искусственно оп может быть получен относительно легко (стр. 251). Так как новый элемент был впервые получен искусственно, Сегре (Segre, 1937) его назвал^ технецием (от Te^vr] — искусство). Аналог марганца с порядковым номером 75 — рений (открытый в 1925 г. Ноддаком и Такке), хотя и стабилен, но принадлежит к числу самых редких элементов. По химическому поведению оба аналога марганца более близки к своим соседям по ряду (молибдену и рутению или вольфраму и осмию) и только но валентному состоянию подобны марганцу.
Марганец соответственно номеру группы максимально семивалептен, но может существовать также во всех более пизких степенях окисления. Основные валентности марганца два, четыре и семь. Однако легко можно получить н соединения трех-, пяти- и шестивалентного марганца. Марганец образует соединения нормального состава, как правило, только с веществами, имеющими сильно электроотрицательный характер. Поэтому можно предположить, что в нормальных соединениях он заряжен положительно. Очевидно, у марганца с увеличением положительного
Седьмая группа периодической система 225
заряда атома уменьшается основной характер гидроокиси и усиливается кислотный (ср. стр. 230). Почти все соединения марганца окрашены; двухвалентного в бледный цвет (бледно-розовый), остальные в большинстве случаев ярко, некоторые даже чрезвычайно интенсивно (см. перманганаты).
Сходство марганца с галогенами — элементами главной подгруппы седьмой группы — очень невелико. Наиболее сильно оно проявляется в соединениях с высшей степенью окисления. Так окись марганца(УП) Ми3О7 можно сопоставить с окисью семивалентного хлора С12О7, а марганцовую кислоту НМпО4— с хлорной НС1О4. Больше марганец похож на соседние с ним по ряду элементы — хром и железо. Подобно им он образует соли типа М:;НЛ1О4 (хроматы(У1) М^СгО4, манганаты(У1) М*МпО4, ферраты(УТ) MjFeOJ. Так же как Ст и Fe, марганец образует в низших степенях окисления труднорастворимые окислы. Ближе всего по свойствам марганец стоит к своему правому соседу — железу. Это сходство проявляется как в аналитических реакциях, так и в том, что оба элемента почти всегда встречаются в природе вмёсте.
G точки зрения теории Косселя, максимальная валентность марганца, равная 7, обусловлена тем, что в периодической системе он занимает седьмое место после одного из инертных газов (аргона). И так как поблизости за пим пет инертного газа, электронную систему которого он приобрел бы, приняв малое число электронов, он проявляет только положительную валентность.
! t 1 =
Рений, несмотря па то что в общем имеет те же степени окисления, что и марганец, отличается от него ярко выраженной склонностью к проявлению валентности 7. Это соответствует общему правилу, что в побочных подгруппах, образованных переходными элементами, тяжелые элементы более склонны к проявлению более высокой валентности, чем легкие. Тенденция рения быть семивалентным так велика, что она накладывает характерный отпечаток на химию этого элемента.
По положению технеция в периодической системе предполагали, что оп по своим химическим свойствам сходен с марганцем и рением. Экспериментально установлено, что последнему он более близок.
' г Теплоты образования некоторых простых соединений марганца и рения сопоставлены в табл. 23.
Таблица S3
Теплоты образования соединений марганца и рения
МпО М113О4 Мп2О3 МпО 2 Мп2О7 ReO3 Re^Oy
93,1 330,5 232,7 125,4 - 165 82,5 297,5 ккал/моль соединения
93,1 112,2 110,4 125,4 82 82,5 148,7 ккал/г-атом металла
M31C12 Мп12 MnS Мпйе Mn6N2 RcS2 Be2S7
112,7 49,8 44,6 26,3 57,8 70,5 — ккал/молъ соединения
112,7 49,8 44,6 20,3 11,6 70,5 — ккал/г-атом металла
13 табл. 24 приведены атомные и ионные радиусы элементов побочной подгруппы седьмой группы.
1-3 Г 1’еуи
225
Глава 6
Таблица 24
Кажущиеся атомные и ионные радиусы элементен побочкой подеру ним VII группы
Атомные радиусы, А Ионные радиусы, А
Мп Тс Во Мп®+ МцЗ+ Мн4+ Мп5+
1,295 \ 1,359 1,375 (KZ = 12) IKZ = 12) (KZ=12) 0,91 0,69 0,52 0,45
Сплавы. Краткий обзор поведения марганца по отношению к металлам и неметаллам, образующим системы, подобные сплавам, содержится в табл. 25. Данные о рении не приведены, поскольку иа указанных в табл. 17 (стр. 141) сплавов пока исследованы лишь немногие.
Как следует из сравнения данных табл. 25 с данными табл. 16 и 17, марганец по отношению к другим металлам (если ограничиться грубым приближением) ведет себя подобно хрому. Это сходство проявляется не только в отношении растворимости и способности к образованию соединений, но часто И в аналогии состава большинства соединений, образующихся в соответствующих системах. Частично это относится и к соединениям с неметаллами (С и Si). Однако, с другой стороны, при сравнении таблиц яспо видно также различие в поведении хрома я марганца по отношению к большинству тяжелых металлов главных подгрупп периодической системы, например марганец в отличие от хрома образует с ними соединения. Еще заметнее это различие при сравнении отдельных сплавов хрома и марганца, если наблюдать на диаграммах состояния поведение составных частей системы в зависимости от температуры. В качестве примера можно указать технически важные системы Ст — Fe и Мд — Fe (ср. рис. 40 и 42). В табл. 17 л 25 для обеих систем указана неограниченная или почти неограниченная растворимость компонентов в твердом состоянии. Эти данные относятся к температурам, близким к температурам плавления. В системе Ст — Fe указанная взаимная растворимость сохраняется и при охлаждении до комнатной температуры. В системе Мл — Fe, напротив, появляются при охлаждении даа твердых раствора; при 500° один из них простирается от 2 до 31 ат.% Мп, другой — от 48 до 63 ат.% Ми. Это происходит потому, что неограниченно смешиваются между собой в (твердом состоянии) только у-модификации марганца и железа. A a-Fe способно образовывать твердые растворы лишь с очень небольшими количествами марганца, так же как a-Mn — с ограниченным количеством железа. Поэтому превращение устойчивой при высоких температурах у-модификации в низкотемпературную «-модификацию влечет за собой появление при низких температурах больших областей разрыва непрерывности (ср. стр. 229 и 281). Вследствие того что температура превращения y-Fe все более понижается с увеличением количества Мн, так что y-Fe при содержании 31 ат.% и более устойчиво'еще при 500°, между областью существования твердых растворов a-Fe, насыщенных Мп(2%Мп), и областью существования твердых растворов a-Mn, насыщенных Железом (37% Fe или 63% Мп), Появляется еще третья область гомогенности, в которой существуют твердые растворы марганца в y-Fe (гм. рис. 42, стр. 286). При 500° эта область простирается от 31 до —48% Мп — границы возможной растворимости Мд в y-Fe при этой температуре. Так как способность образовывать твердые растворы существенно зависит от структуры кристалла, не удивительно, что наличие у марганца трех устойчивых при различных температурах модификаций по сравнению с одной стабильной модификацией у хрома вызывает существенное различие в отношении способности образования твердых растворов в зависимости от температуры.
МАРГАНЕЦ (MANGANIUM) Мп
Распространение в природе. После железа марганец самый распространенный из тяжелых металлов. В небольших количествах он встречается почти всюду; очень распространены также залежи марганцовых
Таблица 25
Образование соединении и твердых растворов между марганцем и элементами главных и побочных подгрупп (Обозначения см. табл. 9, стр. 66)
Главные подгруппы
II III IV V
Be Mg Са В Л1 Ga In TI c SI Ge Sn Pb N p As Sb Bl
МпВе2 ТВ. < со Сплав МпВ МпВ2 тв. > О М пА1* 1180° MnAls* W° MuAl.f 820° ' МпЛК 710° — Mn3In ж. <co тв. > 0 0 tB.<co Mn4C ; MH3G 1 1245° тв. 0 Mn3Si* 1084° Mn5Si3 ISO 0° MnSi 1270° MnSi* 1144° Mn13Ge* 000° Mn5Go2 520° MasGe3 832° Mn3Ce2 74 5° тв, > 0 Mn4Sn 030° Mn2Sn* 657° MnSti*? 648° Ж,<СО тв. О 0 тв, <co M1I4N Mn5N2 Mn3N3 тв. 0 М114Р Mn2P 1300° MnP MnP3 тв. > 0 M n> As 1023° Mn As тв. 0 Mn2Sb 94 8° Mn3Sb*, 872° MnSb -809° ж.< do? MnBi?
.Gl
Побочные подгруппы
VI VII I 11
s Se те Fe Co : Ni «и Rh Pd Ofi Ir Pt Cu Ag Au Zn Cd ng
HC<OO тв, 0 — ТВ, — cc ТВ, co ТВ. СО — — JB, — со — — — ТВ. co ж. <; co ТВ. < co < CO ТВ. <co тв. <; co 0 A я Ж. < co
MnS 1580° MnS2 MnSe MnSe2 1 Male MnTe2 0 0 0 Mn2Pd3Q 1175° MnPd 1515° Сплав 0 0 MH3A11Q Mn.Au. 1237° MnAu2 [] MaAu3 0 MnZnif MnZn?
228
Глава 6
руд. Наиболее важная из них пиролюзит МиО2. В Германии он имеется главным образом в Тюрингии, Гарце, Рудных горах, в горах Фихтель, в Зигерланде и Вестфалии. В огромных количествах залегает он в СССР (Кавказ), Индии, Западной Африке (Гана), Южной Африке и Южной Америке (Бразилия л Чили). Другие руды, образованные окислами марганца — браунит Мп2О3, манганит МиО(ОН) и гаусманнит Мп3О4. Важное значение имеет также карбонат МнСОэ— марганцовый шпат. Руды соседнего с марганцем по периодической системе элемента железа почти всегда содержат более или менее значительные количества марганца. Прежде всего им довольно богаты железный шпат и бурый железняк. Главным источником марганца, добываемого в Германии, служат как раз богатые марганцем железные руды.
В незначительных количествах марганец содержится в растениях п животных. Он каталитически воздействует на химические процессы, протекающие в клетках растепли п живых организмов.
Исторические сведения. Двуокись марганца — пиролюзит была известна еще в древности: известно было и ее применение в качестве «мыла стекловаров». Ее считали разновидностью магнитного железняка (magnes). В отличие от коричневого магнитного железняка Плиний назвал черный немагнитный пиролюзит «женским магнитом». В средние века различали magnes или magnesias lapis — магнитный железняк и magnesia или pseudomagnes (фальшивый магнит) — пиролюзит. Название пиролюзит (Ьгапп-stein) было дапо алхимиком Василием Валентином, который назвал его так потому, что этот минерал (в большинстве случаев черно-серого цвета) давал коричневую глазурь на глиняных изделиях. Стеклоделы же называли пиролюзит благодаря его свойству обесцвечивать железосодержащие стекла «стекольным мылом» (Glasseije) и изменили в соответствии с греческим словом fcavyav<£e tv (очищать) старое название magnes на manganes или lapis manganensis.
Представление о том, что пиролюзит является железной рудой, просуществовало до середины XVIII в. Наконец пришли к заключению, что в нем должен содержаться другой, еще неизвестный металл. Точное доказательство того, что это действительно так, было дано Шееле в 1774г. в его исследовании о пиролюзите, представленном Стокгольмской академии паук. В этом же году Гану удалось получить королек этого металла (bra tins tern me tall) прокаливанием смеси пиролюзита с углем. Его назвали mangane-sium (поэтому по-французски еще и теперь его называют manganese). Позднее этот металл, чтобы не было путаницы с открытым в то же время магнием (magnasiuni), был переименован в марганец (латинское manganlum).
Получение. Металлический марганец проще всего получают из пиролюзита способом алюмотермии. Вследствие очень бурного взаимодействия алюминия с пиролюзитом последний переводят сначала прокаливанием в закись-окись (красного цвета) Мп3О4:
ЗМпОг=Мп304-|-Оа.
В достаточной ли степени удален кислород, устанавливают по потере веса. Полученную закись-акись смешивают с алюминием и поджигают:
ЗМп304 + 8Л1=9Мн+4А1г03-|-602 ккал.
Целесообразно смешивать окись марганца с несколько меньшим по сравнению с теоретическим г.олкчестном алюминия. Шлакование избыточной окиси марганца облегчается добавкой небольшого количества плавикового шпата.
Старые способы выработки марганца путем нагревания окиси с углем в настоящее время уже но имеют технического значения, так как при этом марганец получается загрязненным углеродом, что Делает его непригодным для многих целей. Но очень часто производят железомарганцевые сплавы (ферромарганец и зеркальный чугун), для которых примесь углерода не вредна. В этом случае следует, конечно, примешивать марганцовую руду к железной в соответствующем количестве.
Седъмая группа периодической системы 229
Свойства. Марганец по внешнему виду похож на железо, однако в отличие от него более твердый и хрупкий. Подобно железу марганец в чистом состоянии серебристо-белый, от примеси углерода он становится серым, как чугун. Удельный вес (7,2) его также близок к удельному весу железа, в то время как температура плавления (1247°) значительно ниже, чем у железа. Металл, полученный алюмотермическим способом на воздухе, покрывается пестрыми пятнами, но больше не окисляется даже при нагревании. В мелкораздробленном состоянии марганец окисляется легко, а при некоторых условиях становится пирофорным.
Тонкой змельчепный марганец реагирует с водой, особенно если в ней растворен хлорид аммопия, препятствующий образованию нерастворимого Мн(ОН)2. Нормальный потенциал марганца (по отношению к раствору соли двухвалентного марганца) равен 1,1 в. Следовательно, марганец после щелочных и щелочноземельных металлов, алюминия и его аналогов является наиболее сильным электроположительным металлом.
Марганец, полученный алюмотермическим способом, состоит, как правило, из двух модификаций а- и /5-Мп, первая из которых (уд. вес 7,21) устойчива при обычной температуре, вторая (уд. вес 7,29) — при более высокой (между 742 и 1070°). Обе модификации твердые и хрупкие и имеют сложную кубическую решетку (см. стр. 43). Электролитически осажденный марганец, напротив, довольно мягкий и ковкий O'-Мп, уд. вес 7,21, устойчив между 1070 и 1130°); он образует гранецентрированную кубическую решетку, а = 3,862 А, Мн ► Мп 2,731 (при 1095s). Постепенно он превращается в хрупкий а-Mn. Выше 1130°, как показал Грубо (Grube, 1938), появляется четвертая модификация — й-мар ганец с объем я оцоитрир ованной кубической решеткой; а = 3,081 А, Мн Мп 2,668 А (при 1134°).
В соответствии со своим положением в электрохимическом ряду напряжений (далеко влево от водорода) марганец легко растворяется в разбавленных кислотах с образованием двухзарядного иона Мп" и с выделением водорода:
Мп-|-2Н- = Мп" + Нг.
В концентрированной серной кислоте * он растворяется с выделением сернистого газа, а в азотной кислоте — окислов азота.
В токе хлора марганец сгорает в дихлорид МпС12. Непосредственно он соединяется также с бромом и иодом (образуются МпВтг и Мп12). Очень энергично реагирует с фтором (образование MnFa и MnFs). В азоте Ми воспламеняется при температуре немного выше 1200° и сгорает в нем сильно дымящим пламенем, переходя в нитрид Mn3Na. (Помимо этого марганец образует еще два других нитрида; ср. табл. 25, стр. 227.) С фосфором реакция также сопровождается появлением пламени. Непосредственно соединяется также с серой, углеродом, кремнием и бором; с водородом Мп не соединяется. С окисью углерода марганец непосредственно пе соединяется, однако карбонил марганца Мпа(СО)ю может быть получен при действии СО на смесь дииодида марганца с металлическим магнием под высоким давлением (Brimm и сотр., 1954).
Сплавы Гейслера. Марганец образует, как показал Гейслер (1898), сплавы с некоторыми металлами, например с алюминием, оловом пли сурьмой, отличающиеся ферромагнитными. свойствами, хотя они не содержат ферромагнитных компонентов. При добавлении меди ферромагнитные свойства этих сплавов еще более усиливаются. Это свойство связано с наличием в таких сплавах интерметаллических соединений, гг максимально оно проявляется, когда эти соединения находятся не в чистом виде, а в форме твердых растворов. У хрома также известны сплавы, обладающие ферромагнитными свойствами.
* На холоду концентрированная серпая кислота реагирует с компактным-марганцем очень медленно, при нагревании — быстро.
230
Глава 6
Применение марганца и его соединений. Прежде всего марганец пспользуют для раскисления железа и стали. Для этой цели применяют главным образом сплавы марганца с железом (ферромарганец и зеркальный чугун). Но и в других сплавах, особенно в бронзах, он также служит в качестве раскисляющей примеси (марганцовые бронзы). Известны сплавы марганца с медью. Манганин — сплав, состоящий из 84 ч. Си, 12 ч. Мн й 4 ч. Ni, вследствие незначительной зависимости электропроводности этого сплава от температуры его применяют для изготовления очень точных сопротивлений.
Потребность сталелитейной промышленности в марганцовых рудах (и богатых марганцем железных рудах) чрезвычайно велика. Применение марганцовых руд в других отраслях промышленности имеет меньшее значение, хотя оно также довольно значительно.
Пиролюзит применяют в производстве стекла в качестве осветляющего средства (стекольное мыло); его также используют для окрашивания в фиолетовый^цвет стекол, в коричневый цвет глазурей для гончарных изделий а и т. д. В элементах Лекланше и сухих элементах оц служит деполяризатором. Благодаря способности ускорять окисление льняного масла кислородом воздуха его употребляют для приготовления олиф. При производстве хлора (см. способ Велдона, т, I, стр. 833) пиролюзит играет теперь незначительную роль.
Из других соединений марганца технически наиболее важными являются перманганат, калия КМнО4 (большей частью называемый просто перманганатом), применяющийся как окисляющее, отбеливающее и дезинфицирующее средство, далее хлорид марганца(Н) МпС12 и сульфат марганца^!) MnSO4. Эти, а также и другие соли марганца находят применение в производстве красителей, при ситцепечатании, в качестве протравы для семян (стимуляторы роста). По-видимому, соединения марганца имеют Способность усиливать действие важных растительных ферментов. Различные соединения марганца, прежде всего хлорид Ми(И) и двуокись марганца, находят применение в производстве сиккативов для олиф. Некоторые соединения Мп употребляют в качестве красок, особенно природную умбру; из искусственно получаемых — марганцовая коричневая (минеральный пигмент), марганцовая белая МпСО3, марганцовая зеленая (касселева зеленая) и марганцовая фиолетовая (прочная фиолетовая).
СОЕДИНЕНИЯ МАРГАНЦА
В своих соединениях марганец электроположительно 1-, 2-, 3- 4-, 5-, 6- и 7-валеятен *. Большинству из этих степеней окисления соответствуют окислы.
Низший окисел МпО имеет ярко выраженный основной характер. Следующий за ним полуторный окисел Мв2О3 еще также основной. Двуокись марганца МпО2, напротив, амфотерна. Существование трехокиси марганца МпО3 в свободном состоянии сомнительно. Ее производные — манганаты(¥1) М* [МпО4] являются солями, в которых марганец входит в Состав кислотного остатки. То же относился к манганатам(¥), т. е. солям
* Известны также соединения марганца; в которых он электрохимически нейтрален (нулевая валентность). Это свойство характерно для хрома, железа и многих других переходных элементов. Примером подобных соединений являются карбонилы металлов (ср. стр. 375).
Седьмая группа периодической системы
231
с общей формулой М/[MnOj. Высшему окислу марганца Мп 2О7 соответствует марганцовая кислота НМпО4, принадлежащая к числу наиболее сильных из известных кислот. Таким образом, общее правило, в соответствии с которым у элемента с увеличением валентности понижаются основные и повышаются кислотные свойства, в случае окйслов марганца проявляется особенно ярко.
Различные окислы и гидроокиси марганца в дальнейшем описываются сначала все вместе, а затем рассматриваются отдельно с соответствующими им соединениями.
Соединения марганца с S, So и Те, насколько известно, цо своему составу соответствуют низшим окислам марганца. Напротив, соединения с неметаллами главных подгрупп Hl, IV и V групп имеют состав, непохожий ни на окислы марганца, ни на еолеобразные соединения. В табл. 25, стр. 227 эти соединения приведены вместе с интерметаллическими соединениями марганца, к которым они близки по составу.
Важнейшие соединения марганца являются производными Мп11, МШУ и Mnvn. Соединения Мп111 и MnVl также легко можно получить. В настоящее время не представляет трудностей и приготовление чистых соединений Mnv. Из производных Мп1 известны циано-соли Mf[Mu(CN)6].
1. Окислы и гидроокиси
Для марганца известны следующие простые окислы:
МпО — моноокись марганца (окись марганца), окись марганца (II), зеленая окись марганца;
Мп2О3 — полуторная окись марганца, окись марганца(Ш), черная окись марганца;
МпОа — двуокись марганца, окись марганца(1У), серая окись марганца (пиролюзит);
МпО3 — трехокись марганца, окись марганца(У1). Существование этого соединения в свободном состоянии сомнительно;
Мп2О7 — семиокись марганца, окись марганца(VII).
Известен еще промежуточный окисел Мп3О4, названный по своей окраске, красной окисью марганца. Он кристаллизуется по типу шпинели (ср. г. I, стр. 395), нооиз-за искажения имеет тетрагональную решетку («о = 5,75, е0 = 9,42 А). На основе строения его рассматривают как манг'анат(/У) марганца(Н) Мп3п [MnIVO4]. В природе Mn3O4 встречается в виде гаусманнита (блестящий пиролюзит), в большинстве случаев вместе с браунитом, но. как правило, в больших количествах, чем последний. По внешнему виду они почти неразличимы.
Из двух возможных в соответствии с состав ом формул и MnVfMa^OJ
правильной, как показали Вервей и де Бур (Verwey, de Воет, 1936) путем измерения рассеяния рентгеновских лучей ионами марганца, расположенными внутри и впе радикала МнО4, является вторая (ср. т. I, стр. 241). На основании наблюдений, что Мп3О4 подобно Мн2О3 растворяется в некоторых концентрированных кислотах (например, в фосфорной кислоте) с фиолетовым окрашиванием, характерным для попов Мп"\ в то время как МпО2 в них нерастворим, ранее был сделан вывод, что Мв3О4 следует рассматривать не как соединение 2МиО с 1МнО3, а как 1МпО с 1Мн2О3. Однако образование иопов Мп'" объясняется -гем, что при растворении происходит восстановление ионов Мц"” ионами Мл”, и нерастворимость МлОг не препятствует, следовательно, растворимости производного от нее соединения с другой структурой.
МпО имеет кубическую решетку типа каменной соли (а = 4,43 А), МнОа — тетрагональную типа рутила (ар == 4,40, е0 = 2,87 А), Мн3О3 кристаллизуется обычно
232
Глава, S
в кубической сингонии по типу окиси скандия (ср. стр. 55). Помимо стабильной кубической модификации (а-МпаО3), известна также неустойчивая тетрагональная модификация (подробнее см. стр. 233). В решетках МнО, а-Мп2О3 и МпО2 координационное число Йп по отношению к кислороду равно S. Расстояние Мн <—> О в решетке МнО составляет 2,21 А, в а-МпаО3 — от 2,00 до 2,03 А, в МцО2-1,89 А. Оно умень-
шается, следовательно, с ростом заряда.
МнО, Мп304 и Мл3О3 способны поглощать избыточное количество кислорода с образованием твердых растворов. При этом МнО приобретает состав MnO4lI3, Мп3О4 — Мп30412(; и Мп2О3 — Mn2O3,13. МпО2 в отличие от РЬО2 не растворяет кислорода без изменения решетки (Le В lane, 1934).
Окись маргапца(П) МпО получают восстановлением водородом или окисью углерода более высоких окислов марганца, а также прокаливанием карбоната двухвалентного марганца в токе водорода или азота. Это порошок от зеленовато-сер ого до травянисто-зеленого цвета. В тонко-измельченном состояний МнО легко окисляется. Наоборот, восстановление до металла протекает с трудом, например, в токе водорода и то только при очень высокой температуре. В воде окись марганца(П) практически нерастворима. В природе встречается (редко) в виде манганозита.
Гидроокись марганца(П) Мп(ОН)2 выпадает при взаимодействии растворов солей Мн(П) со щелочами в виде белого, на воздухе быстро приобретающего коричневый цвет осадка. Водным раствором аммиака Мп(ОН)2 осаждается неполностью, а в присутствии солей аммония вообще не выпадает. Причина этого заключается, как и в случае гидроокиси магния, в очень незначительной концентрации гидроксильных ионов по сравнению с той, которая необходима для достижения произведения растворимости (L ~ [Мн"] [ОН']2 = —1-10-12). А в данном случае в силу образования комплексных соединений с аммиаком значительно понижается также концентрация ионов марганца (Weitz, 1925).
В природе гидроокись марганца(И) встречается иногда в виде белых прозрачных листочков пирохроита. Этот минерал нод действием света постепенно становится коричневым. Коричневая окраска — следствие окисления, ведущего через промежуточные окислы или гидроокиси к конечному продукту днуокиси марганца МпОа.
Испрстггы миогоч(ылсН1Тые продукты сиэгслешгя Мй(ОН)2, различные по составу и структуре. Большинство п.ч пих недальтовидные соединения. Например, в Мп(ОИ)г часть ионов ОН- может замещаться ионами Оа- без изменения Структуры. При образовании различного рода продуктов окпепенпя речь идет исключительно о топохимических процессах (Feitknecht, 1944).
По строению кристаллической решетки гидроокись марганца(П) подобна гидроокиси магния (брусит): а == 3,34, с = 4,68, Мн +--* О = 2,30 А. Теплота образования Мп(ОН)г из МдО и Н20)к составляет ~4,2 ккол/молъ.
Окись марганца(Ш) — полуторная окись марганца Мп2О3 — встречается в природе в виде браунита, образующего коричнево-черные скопления, в большинстве случаев в виде примеси к другим марганцовым рудам. Искусственно черный аморфный порошок Мц2О3: получают нагреванием на воздухе двуокиси марганца до 530—940° или же прокаливанием солей марганца(П) при доступе кислорода или воздуха.
При нагревании до температур более 940° на воздухе или выше 1090° в токе кислорода Мн2О3 отщепляет кислород и переходит в более бедную кислородом окись IV) или закисъ-окисъ Мн3О4. Последняя, если
ее достаточно долго прокаливать, при охлаждении не присоединяет вновь кислород. Это важно для весовых определений марганца.
Если Мн2О3 приготовляют осторожным нагреванием оса ледени ой МпО (или Мп304 не очень сильно прокаленной) в токе кислорода (Le Blanc, 1934) или обезвожи-
Седьмая группа периодической системы
233
вапием МцО(ОН) в вакууме при 250° (Dubois, 1934), id сначала получается нестабильная тетрагональная модификация. При более длительном нагревании она мопотропно переходит в устойчивую кубическую модификацию (а-МпгО3), При получении Мо2О3 термическим разложением МпО2 кубическая модификация .образуется непосредственно. Кубическая а-Мп2О3, как уже было указано на стр. 232, изотипна Sc2O3, Длина ребра ее элементарной ячейки, содержащей 16Мп2О3 (что^ соответствует кубу, построенному из 8 элементарных ячеек типа плавикового шпата), составляет 9,41 А. Решетку тетрагональной Д-Ма3О3 можно представить, по Данным Вервея и де Бура (1936), как решетку Мп3О4, в которой появились пустоты в количестве, соответствующем уменьшению содержания марганца. Однако в отличие от Мп3О4 ионы марганца в такой решетке либо трегзарядны, либо, в случае если сосуществуют ионы MiTi+ и Мп4+, они совершенно беспорядочно распределены по всей решетке.
При нагревании в токе водорода окись марганца(Ш) при температуре около 230° переходит сначала в окись марганца(П,1 V) Мп3О4, а при температуре выше 300° восстанавливается до моноокиси зеленого цвета.
При растворении Mn2Og в кислотах в зависимости от выбранной кислоты и температуры образуются либо соли трехвалентного марганца [ср. сульфат и хлорид марганца(Ш)], либо идет окислительно-восстановительный процесс с образованием соли двухвалентного марганца и двуокиси марганца.
Гидрат окиси марганца(Ш) Мп20з-Н20 или метагидроокись марганца МнО(ОН) встречается в природе в виде манганита (бурая марганцовая руда). Она черно-бурого цвета и иногда образует отличные кристаллы, изоморфные гетиту FeO(OH) и диаспору АЮ(ОН). Осажденные из водных растворов и высушенные при 100° гидраты по составу соответствуют манганиту.
Марганцовая коричневая. Искусственно полученную гидроокись марганца(Ш) применяют в качестве черно-бурой краски (марганцовая коричневая, минеральный пигмепт). Получают ее взаимодействием растворов хлорида марганца(П) с хлорной известью и известковой водой. Марганцовую коричневую применяют также для окраски тканей и в ситцепечатании; в этом случае она образуется прямо па волокне в результате окисления ткани, пропитанной хлоридом марганца(П). Малярная краска каштанового цвета, приготовленная из природных смесей гидроокиси марганца с гидроокисями железа и алюминия путем номола и прокаливания, называется умбра.
Двуокись марганца МнО3 — пиролюзит (серый марганец) является важнейшим минералом марганца. Структуру решетки см. стр. 232.
В зависимости от кристаллографической формы различают: полианит (четко Кристаллизующийся пиролюзит); пиролюзит, или мягкая марганцовая руда (пиролю-зить образовавшийся из других минералов марганца с сохранением их кристаллической структуры); серый пиролюзит (мелкозернистый или совсем плотный); псиломелан, или черная стеклянная голова (пиролюзит в форме комковатых образований, большей частью сильно загрязненный примесями); вад, или марганцовая пена (тоже комковатая, но рыхлая МпО2) и марганцовая черная (очень тонкий порошок, похожий на сажу).
Искусственно полученная дауокись марганца имеет три модификации: а-, Д-и p-MnO2 (Dubois, 1936; Glemser, 1939; Walkley и Wadsloy, 1949). /J-MnO3 по структуре соответствует природному пиролюзиту (полианит). В у-МпО2 часть ионов О3- обычно замещена ионами ОН- (одновременно ноны Мп4+ заменяются ионами Мпз+). Препараты ;’-МпО2 часто содержат другие металлы, которые частично замещают ионы Мп4+ (Feitknecbt, 1944).
Все разновидности природного пиролюзита, так же как искусственная безводная двуокись марганца (встречающаяся в марганцовых глинах), полученная, например, умеренным нагреванием нитрата Mn(II), имеют цвета от серого до серо-черного. Исключение составляет вад, он иногда окрашен в коричневые топа. При нагревании на воздухе до температуры выше 530° двуокись марганца начинает отдавать кислород. В вакууме,
234
Глава 6
а также при нагревании в присутствии восстановителей отщепление кислорода происходит при значительно более низкой температуре.
При действии сильных восстановителей в присутствии разбавленных кислот, несмотря на* чрезвычайно малую растворимость в воде, двуокись марганца легко вступает в реакции. G сернистой кислотой она образует дитионат маргапца(П) (1), с перекисью водорода и серной кислотой —• сульфат маргаяца(П) и кислород (2):
МпО2 -{- 2H2SO2=MnS2Og“j- 2На0, (1)
AiaOg-p Н2О2 -j- H2SO4^MnSO4-|- 2Н2О (2)’
В отсутствие кислот перекись водорода также разлагается, однако кислорода в этом случае выделяется вдвое меньше по сравнению с кислыми растворами, так как МнО? не восстанавливается, а действует лишь каталитически:
игог=нгО4-1/гог.
Двуокись марганца каталитически ускоряет также отщепление кислорода при разложении хлората калия (ср. г. I, стр. 860).
На способности МпОг легко отдавать кислород другим веществам основано ее применение в качестве деполяризатора в элементе Лекланше и в сухих элементах. В смеси с окисью меди двуокись марганца применяют для каталитического окисления окиси углерода, например в противогазах (ср. т. I, стр. 484).
При действии на пиролюзит концентрированной соляной кислоты, особенно при нагревании, выделяется хлор. Эту реакцию часто используют для получения хлора в лаборатории.
Реакция протекает таким образом, что сначала образуется тетрахлорид марганца:
МпО2-|-4НС1=МпС14+2НгО,
который очень легко распадается на трихлорид марганца и хлор. При нагревании разлагается также и трихлорид:
МвС1,, -:МпС13 + 1/2С1а; МпС1з=МаС124-1/гС12.
Холодная концентрированная серная кислота не действует на МнО2; при нагревании сначала образуется сульфат марганца(Ш) и выделяется кислород (при 110s); при более высокой температуре сульфат Мн(1П) переходит в сульфат Мп(Ц) и также выделяется кислород.
Гидрат двуокиси марганца образуется большей частью в виде бурого, иногда черноватого, осадка при окислении солей марганца(П), а также при восстановлении в щелочном растворе манганатов (VI) или перманганатов [мангаиатов(УП)]. Окисление солей марганца(П) до гидрата двуокиси марганца может происходить в щелочном растворе, например, под действием хлорной извести или кислорода воздуха. Окисление в кислой среде ср. стр. 236. При электролизе растворов, содержащих достаточное количество азотной кислоты, марганец выделяется на аноде в виде гидрата двуокиси марганца.
Полученные из щелочных растворов продукты всегда содержат примесь щелочей (или гидратов щелочноземельных металлов). Поэтому при необходимости получения чистого гидрата двуокиси марганца следует исходить из кислых или нейтральных растворов, по в таких случаях содержание кислорода в гидрате двуокиси марганца большей частью не соответствует теоретическому. Сомнительно, чтобы гидрат двуокиси марганца содержал определенное количество воды, например ш соответствии с формулой Мп(0Н)4.
Седьмая группа периодической системы 235
Бурый гидрат двуокиси марганца имеет большее значение, нежели серая двуокись марганца, в связи с его значительно большей реакционной способностью; поэтому в качестве окислителя его применяют гораздо чаще, чем природный пиролюзит (например, в производстве синтетических красителей и в лакокрасочной промышленности).
Гидроокиси щелочных металлов и другие основания не только адсорбируются гидратом двуокиси марганца, но и химически связываются им, В виде хороших кристаллов получены соединения МиО2 с основными окислами, отвечающие определенным стехиометрическим соотношениям. Следовательно, гидрат двуокиси марганца обнаруживает В Этих случаях свойства кислоты (правда, слабой). Соли ее называют манга-тталш или,„лучше, манганатами(1У). [О составе и структуре манганатов(IV) и зависимости состава и структуры от способа получения см. Feitknecht, Helv. Chim.. Acta, 28, 129, 149 (1945).] С другой стороны, гидрат двуокиси марганца может давать соли также с сильными кислотами, рдиако эти соли в силу стремления марганца в кислых растворах переходить в двухвалентное состояние очень неустойчивы. В общем двуокись марганца имеет амфотерный характер. Одиако вследствие плохой растворимости двуокиси марганца ни кислый, ни щелочной характер ее не выражены ясно.
Семиокись марганца и марганцовая кислота. Семиокись марганца Мп..От можно получить действием концентрированной серной кислоты на перманганат калия:
2КМпО4+Н28О4=Кг8О44-Мп2О7 + НгО.
Она представляет собой темное, зеленовато-бурое, блестящее, тяжелое масло (уд. вес 2,4). При обычной температуре в сухом воздухе оно в течение продолжительного времени устойчиво. При нагревании разлагается со взрывом на двуокись марганца и кислород.
При внесении в небольшое количество воды семиокись марганца разлагается, вследствие значительной теплоты растворения (~12 ккал/молъ МпрОу). В большом же количество холодной воды опа растворяется без разложения. Раствор ее окрашен в фиолетовый цвет и имеет тот же спектр поглощения, что и раствор перманганата (ср. стр. 247). Отсюда можно сделать вывод о существовании в растворе семиокиси марганца ионов перманганата М11О4, которые образуются в соответствии с уравнением
Мп2О7 + И20 — 2MnOj 4- 2Н .
Безводную марганцовую кислоту НМиО4 получить не удалось, в растворе она устойчива лишь до концентрации 20%. Измерение электропроводности показывает, что марганцовая кислота является очепь сильной. В водном растворе она практически полностью диссоциировала. Кажущаяся степень диссоциации, установленная по данным электропроводности, в 0,1 М растворе составляет 93% (при 25°).
Марганцовая кислота образуется также при действии двуокиси свинца на соли марганца(П) в присутствии концентрированной серной или азотной. кислоты:
2Ми" 4- 5РЬО24- 4Н*=2МпО;4- 5РЪ- 4- ?Н2О.
Реакция имеет значение в аналитической химии, так как благодаря интенсивной фиолетовой окраске ионов Мн04 таким образом можно открыть даже следы марганца. В присутствии ионов С1' эта реакция не идет. Это обусловлено тем, что марганцовая кислота восстанавливается соляной кислотой, а последняя окисляется до элементарного хлора.
Йоны Мп" очень легко окисляются до ионов МиО4 под действием пероксомоно-фосфорной кислоты Н3РО6 (Schmidlin, 1910). Если имеется в запасе свободный от перекиси водорода раствор пероксомонофосфорпой кислоты, то определение марганца этой реакцией гораздо удобпее, чем с РЬО2 и H2SO4.
236
Г лава 6
Вообще марганцовая кислота отличается сильными окислительными свойствами. Еще энергичнее реагирует свободная семи окись марганца. Горючие вещества при взаимодействии с ней воспламеняются. Однако в уксусном ангидриде она растворяется с фиолетовой окраской без разложения .
2. Соли марганца(П)
Соли марганца(П) производятся от низшего окисла марганца МиО при замене кислорода кислотными остатками. Эти соли большей частью окрашены в розовый цвет, так же как и их водные растворы, содержащие ионы Мп". В сухом состоянии и в водном растворе в присутствии избытка кислоты соли марганца(П) вполне устойчивы. В щелочном растворе, напротив, образуется .гидроокись марганца(П), которая весьма склонна окисляться до бурого гидрата двуокиси марганца.
Нейтральные. растворы соли марганца(П) при длительном стоянии также приобретают буроватую окраску, и постепенно из пйх выделяются бурые хлопья. Это обусловлено том, что в растворе образуются следы гидроокиси маргапца(П), которая под действием кислорода воздуха окисляется до очень труднорастворимого гидрата двуокиси. Происходит ли образование гидроокиси марганца (II) в результате гидролиза или под действием имеющихся в стекле посуды щелочей, пока еще не установлено.
Очень сильные окислители, например озон и персульфат (пероксосульфат), окисляют соли марганца(П) даже в таких растворах, которые содержат умеренные количества сильных кислот. Способность персульфатов количественно осаждать марганец в виде гидрата двуокиси из слегка подкисленных серной кислотой растворов используют в количественном анализе.
На течение некоторых окислительных процессов, особенно происходящих под действием атмосферного кислорода, соли марганца(И) действуют ускоряющим образом. На этом основано применение соединений марганца в качестве сиккативов. Сиккативы — препараты, которые, будучи растворены или суспендированы в льпяпом масле, сообщают ему свойство быстро высыхать, так как они ускоряют его окисление кислородом воздуха. Льняное масло, содержащее подобный сиккатив, называют олифой. В качестве сиккатива можно использовать прежде всего некоторые органические соли марганца, а именно марганцовую соль смоляной кислоты (резинат марганца) и| марганцовую соль линолевой кислоты (линолеат марганца), так как оци легче растворяются в льняном масле, чем неорганические соединения марганца. В качестве сиккативов применяют также соединения свинца, одиако по силе действия они уступают марганцовым сиккативам. Лучше всего высыхают олифы, содержащие одновременно и Мп и РЬ.
Обычные соли марганца(П) большей частью хорошо растворимы в воде, особенно хлорид, нитрат и сульфат, а также ацетат и роданид. Из труд-пора створимых следует в первую очередь назвать сульфид, фосфат и карбонат.
Существуют также двойные соли маргапца(П) с солями щелочных металлов и т, и. В водпом растворе эти соединения почти полностью распадаются на составные части. Тенденция к образованию ацидо-ионов у двухвалентного марганца значительно меньше, чем у трехвалснтного.
Иои Мп2+ занимает особое положение в ряду ионов от Са2+ до Zn2+, подобно иону GdS* и ряду лантанидов (ср. стр. 527), так как он содержит наполовину недостроенный 3</-уровень. Из десяти возможных для этой оболочки электронов у него имеется только половина. Эта особенность проявляется в различных свойствах соединений двухвалентного марганца по сравнению со свойствами соединений соседних элементов, например в величине молекулярных объемов (Klemm, 1942).
Седьмая группа периодической системы.
237
Хлорид марганца (II), дихлорид марганца, хлорид марганца МпС12 получают в безводном состоянии при действии сухого хлористого водорода на окись марганца, карбонат или металлический марганец при нагревании. Он образуется также при горепии металлического марганца в токе хлора (теплоту образования см. стр. 225). Безводный хлорид образует розового цвета кристаллы в форме листочков, изоморфные хлоридам кальция и кадмия. Он плавится при 650° и в токе НС1 улетучивается уже при температуре красного каления (температура кипения 1190°). Водородом он не восстанавливается, при нагревании в токе кислорода или в парах воды переходит в Мпв03. В воде хлорид марганца очень легко растворим (72,3 г в 100 г воды при 25®).
Из водвого раствора обычно кристаллизуется тетрагидрат МпС13-4Н2О, существующий в двух модификациях. Неустойчивая модификация изоморфна FeCl2-4H2O. Помимо этого хлорид марганца(П) образует гексагидрат (изоморфный СоС13- 6Н.,О) и днгидрат. При нагревании в токе хлористого водорода вся вода отщепляется. Безводный хлорид очень гигроскопичен, и при растворении в большом количестве воды выделяется 16 ккал/.иоаъ. Безводный МпС12 растворяется также в абсолютном спирте, из которого он кристаллизуется с тремя молекулами кристаллизационного спирта.
Безводный хлорид марганца(П) может присоединять 6 молекул NH3, а также и некоторые другие азотистые соединения, например с гидроксиламипом он образует очень устойчивый продукт присоединения MnClB-2NHaOH.
Он может соединяться также с другими хлоридами, прежде всего с хлоридами щелочных металлов. Двойные соли соответствуют главным образом типам: М^СЬМпС^ и 2М1С1-МцС]2. Водой они разлагаются.
Тетрагидрат хлорида марганца МиС12-4Н2О проще всего получать растворением карбоната марганца в соляной кислоте и упариванием полученного раствора (при температуре ниже 58®).
Для технического получения, поскольку для приготовления хлора до сих пор применяют способ В ель дона, используют образующиеся нри этом растворы хлорида марганца. Сначала из них отгоняют избыток хлора и хлористый водород и осаждают важнейшие примеси (железо и алюминий) введением карбоната марганца. Дальней? шая очистка МаС12, кристаллизующегося при упаривании раствора, лучше всего осуществляется через карбонат.
От хлорида двухвалентного марганца производятся, по данным Файткнех-та (1951), три основные соли (с различными структурами): Мп(ОН)С1, Мн^(ОН)3С1 и Мп3(ОН)5С1. Последняя, однако, малоустойчива. Она имеет переменный состав 11 кристаллизуется подобно Мн(ОН)2 по типу брусита (со статистическим распределением иопов ОН- и С1- и значительным расширением решетки в направлении оси с).
Бромид и иадид марго. нца(11) по внешнему виду и свойствам очень похожи на хлорид. Они образуют почти такие же, как хлорид, гидраты, аммиакаты и т. д. Однако способность к образованию двойных солей у бромида значительно ниже, у иодида же совсем отсутствует.
Фторид марганца^Н) — розовые квадратные призмы, отличается от остальных галогенидов значительно меньшей растворимостью в воде (1,06 г в 100 а воды). Он образует малоустойчивый тетрагидрат MuF2.4H2O и очепь легко разлагающийся аммиакат 3MnF2-2NH3, а также двойные соли типа bPfMnFj].
Сульфат марганца (II) MnSO4 — одно из наиболее устойчивых соединений двухвалентного марганца. Он образуется при упаривании с серной кислотой почти всех соединений марганца, если они не содержат трудно--штучих кислот. В безводном состоянии он почти чисто белый. Из водного раствора MnSO4 выпадают кристаллы красивого розового цвета с различным в зависимости от условии содержанием воды (гепта-, пента-, тетра-п моногидрат).
Устойчивый ниже 9° гептагидрат MnSO4-7H2O встречается иногда в виде минерала миллардита. Обычно он кристаллизуется в моноклинной снигонни, по с цинковым купоросом он может образовывать и ромбические твердые растворы.
238
Глава. 6
При обычной температуре (в соприкосновении со своим водным раствором между 9 и 26°) устойчивым является пентагидрат МпЗО^-бНоО, изоморфный медному купоросу. Его называют также .wap гонкое мм купоросом. Обычный продажный сульфат марганца содержит только четыре молекулы воды. Однако стабильный ромбический тетрагидрат устойчив только в очень небольшом интервале температур*. Помимо этого существует еще одна, сама по себе менее устойчивая, но кристаллизующаяся из водного раствора в более широких температурных пределах моноклинная форма тстрагидрата. Опа образуется, например, при упаривании раствора сульфата мэр-гапца(П) в интервале температур 35—45’; это и есть торговый сульфат марганца.
Для технического получения сульфата марганца(Н) исходят из пиролюзита, который либо растворяют в горячей концентрированной серной кислоте (3), либо прокаливают в смеси с безводным железным купоросом (4):
МпО^Ц- H2SO4—MnSO^-ф НаО (3)
2Mn02+2FeS04=2MnS04-|-Fe203-|-i/263. (4}
Сульфат маргаица(П) подобно хлориду служит исходным материалом при получении многих других соединении марганца. Его используют также для окраски тканей. Применяющиеся для этих целей препараты не должны содержать железа.
При хранении на воздухе гидраты сульфата марганца(П) постепенно выветриваются, За исключением моногидрата, который иногда встречается в природе в виде минерала (шмикит, моноклинный). При нагревании, особенно в присутствии концентрированной серией кислоты, отщепляется вода. Безводный сульфат маргапца(П) может присоединять 6 молекул аммиака, моногидрат — 5 молекул.
Из растворов, содержащих сульфат марганца(П) и сульфаты щелочных металлов, кристаллизуются двойные соли с общей формулой MlSOa-MnSOj (как безводные, так и с 2, 4 и 6 молекулами воды).
Карбонат марганца (II) Л1пСО3 встречается в природе в виде марганцового шпата (малиновый шпат) и имеет важное значение для получения зеркального чугуна и ферромарганца. Карбонат марганца кристаллизуется в гексагонально-ромбоэдрической сингонии; он изоморфен известковому шпату, магнезиту и железному шпату. Природный марганцовый шпат очень сильно загрязнен этими примесями и поэтому большей частью окрашен в серый, зеленоватый или буроватый цвет. Чистый карбонат марган-ца(П) имеет малиновый или розово-красный цвет. Обычно марганцовый шпат встречается в виде тонкозернистых или совершенно плотных масс, кристаллическая структура которых выявляется лишь под микроскопом.
При осаждении из водного раствора [взаимодействием солей марганца^!) с растворимыми карбонатами] получают обычно основные карбонаты марганца. Если же вести осаждение кислым карбонатом натрия из раствора, насыщенного двуокисью углерода, то образуется белый осадок моногидрата нейтрального карбоната марганца МпСО3-Н2О. При нагревании с раствором, содержащим СО2, под давлением и в отсутствие кислорода воздуха его можно превратить в безводный карбонат.
Растворимость карбоната марганца в воде очень мала. Понтону прн комнатной температуре он устойчив по отношению к воде, но при кипячении разлагается (гидролиз). Сухой карбонат марганца довольно легко распадается также при нагревании. Уже выше 100° происходит заметная диссоциация:
МпСО3Ц-23,2 ккал 3"^ MiiO-J-COq. 1
* Между 26 и 27°. Выше 27° в соприкосновении с раствором устойчив только моногидрат.
Седьмая группа периодической системы
239'
При более высокой температуре происходит частичное окисление образующейся окиси марганца(П) за счет двуокиси углерода; вследствие этого выше 330° к СО9 бывает всегда примешана СО. Если МнСО3 прокаливают на воздухе, то образуется Мп3О4, а в токе кислорода — Мн2О3.
Искусственно получаемый карбопат марганца(П) применяют иногда в качестве-малярной краски (марганцовая белая). МнСО3 образует решетку известкового шпата (а = 5,84 А, а = 47’2').
Нитрат марганца(П). Из раствора карбоната марганца в разбавленной азотной кислоте кристаллизуется нитрат марганца(П) в виде гексагидрата Mn(NOa)2-6H2O, если упаривание производить при комнатной температуре; выше 25° или из сильнокислых растворов образуются бесцветные моноклинные призмы тригидрата Mn(NO3)2-3H2O. Существует также моногидрат. При слабом нагревании с азотным ангидридом от моногидрата можно полностью отнять воду. Безводная соль присоединяет до 9 молекул аммиака. В воде она растворяется со значительным выделением тепла (12,9 ккал/моль). Растворимость при 18° составляет 134 г Mn(NO3)2 в 100 г воды.
Нитрат двухвалентного марганца легко образует двойные соли с нитратами редкоземельных элементов. Эти соли оказались очень удобными для разделения радио-земельных элементов способом фракционированном кристаллизации.
Ацетат марганца(П). Из раствора карбоната маргапца в упсусной кислоте кристаллизуется тетр а гидр ат ацетата марганца Мп(С2Н3О2)2-4И2О в виде устойчивых на воздухе бледно-красных игл или табличек (растворим в холодной воде в соотношении 1 : 3). Он находит применение в качестве стимулятора роста растений, сиккатива, а также как переносчик кислорода. Взаимодействием нитрата марганца(П) с уксусным ангидридом можно получить безводный ацетат марганца.
Фосфат и арсенат марганца(П). Из нейтральных растворов солей марганца(П) при избытке двузамещенного фосфата натрия выпадает кристаллогидрат ортофосфата марганца Мп3(РО4)2-7Н2О в виде рыхлого белого осадка. В других условиях можно получить другие фосфаты: ди~ и метафосфаты, а также кислые фосфаты. При действии хлорида аммония, раствора фосфата аммония (или же динатрийфосфата) и небольшого количества аммиака на растворы солей марганца(П) образуется прекрасно кристаллизующаяся двойная соль — аммониймарганецфосфат (NH4)MnPO4-Н2О — кристаллики с шелковистым блеском. Эту реакцию используют в весовом анализе. Это соединение подобно аналогичной соли магния при прокаливании переходит в пирофосфат:
2(NH4)MnP04HI20=Mn2P207 + 2NH3+3H20.
Из растворов фосфата двухвалентного марганца, содержащих значительный избыток фосфорной кислоты, кристаллизуется малоустойчивая комплексная кислота Н4]Мв(РО4)3]-ЗН2О [дифосфатомарганец(П) кислота]. Наличие марганца в анионной форме можно доказать измерением подвижности ионов. То же относится к диарее-натомарганец(11) кислоте Н4[Мв(АзО4)2]-ЗН2О (розовые гексагональные иглы). Последняя реагирует с карбонатами щелочных металлов с образованием безводных солей Na4 [Mn(AsO4),] и K4[Mn(AsO4)2], с карбонатом аммония — кислой соли (NH4)2H2(Mn(AsO4)2] (Grube, 1936).
И з пр осты х а рс ен а тов мар ганца с л е ду ет на зв ать М n3( As О4) 2 Н 2О, МпНAsО4• НаО и Mn[H2AsO4]j, из двойных солей — (NH4)MnAsO4-6Н2О. Основной арсенат марганца^ I) или двоимая соль Mn3(AsO4), с Мп(ОТ1)2 встречается в природе (саркинит, гемафибрит, аллактит, арсеноклазит).
Сульфид марганца, моносулъфид марганца(11) MnS. При действии сернистого аммония или растворов сульфидов щелочных металлов йа рас
240
Глава. 8
твор соли марганца(И) выпадает, как правило, осадок телесного цвета. В зависимости от условий осаждения он может быть желтоватым или красноватым. Это сульфид марганца(П) (обычно содержащий воду). При длительном стоянии в контакте с маточным раствором, особенно при нагревании, он переходит в более устойчивую зеленую форму. Это превращение не связано с присутствием солей аммония, но происходит значительно быстрее, если осаждение проведено большим избытком сернистого аммония. Зеленая модификация сульфида марганца встречается также в природе в виде марганцового блеска (алабандин). Это блестящие, черного цвета правильные кристаллы (тетраэдры), образующие при измельчении зеленый порошок. Искусственно довольно легко мшкно подучить хорошо сформированные кристаллики зеленого сульфида марганца. На воздухе сульфид марганца малоустойчив. Особенно реакционноспособна модификация те,.щспого цвета, при доступе воздуха она довольно быстро становится коричневой и распадается (гидролиз и одновременное окисление до гидрата двуокиси).
Структура решетки ларгамцоеоео блеска или зеленого сульфида марганца (a-MnS), так ;ко кик сульфидов щелочноземельных элементов, подобна решетке каменной соли;, а = 5,21 А, Мп<—► S = 2,61 А. Неустойчивый красный сульфид марганца Встречается н двух модификациях: кубической типа цинковой обманки (а = 5,60 А) и гексагональной типа вюртцита (а = 3,98; о = 6,43 А). При осаждении из водного раствора, как правило, образуются обе эти модификации. При нагревании они монотропно превращаются в устойчивую зеленую форму.
Селенид MnSe подобно ct-MnS кристаллизуется ко типу каменной соли. Эго исключительный факт, так как моносульфиды и моноселепиды всех соседних с марганцем элементов (Ст и V, а также Fe, Со и N1) кристаллизуются по типу NiAs. МнТе подобно всем теллуридам от TiTe до NiTe кристаллизуется по типу NiAs.
Дисульфид марганца, ..дисульфид марганца(П) MnS2. Помимо марганцового блеска в природе встречается также дисульфид марганца, марганцовый колчедан (гауэрит) MnS2. По своей структуре он подобен пириту (см, стр. 296); а = 6,097 Si—> S = 2,09, Мп 8 = 2,58 А. Дисульфид может быть получек также из водного раствора, но только при более высокой температуре (около 160е). О валентности марганца н нем см. стр. 296. При нагревании MnS2 очень легко отдает серу. При 304°, но данным Енльтца (Вiltz XV,, 1936), давление пара серы в нем такое же, как у элементарной серы (55 мм рт ст).
Помимо Мн83 ио типу иприта кристаллизуется Также MnSe2 и МнТе2 (в противоположность FeSe2 н FeTez, имеющих решетку марказита).
Бораты марганца(Н) используют в качестве сиккативов. Их получают на водного раствора взаимодействием солей двухвалентного марганца с бурой. Состав приготовленных таким способом препаратов приближенно соответствует содержащему воду тетраборату. Однородные по составу препараты получить из водного раствора трудно, их получают сплавлением окиси марганца(И) или карбоната с борной кислотой пли бурой.
Оксалат маргапца(П). При добавлении горячего раствора щавелевой кислоты к горячему разбавленному раствору соли марганца(II) или к суспензии карбоната марганца в горячей воде образуется оксалат марганца, который при медленном охлаждении выделяется в виде белого кристаллического порошка состава MnCgOy2Н*ЛО. Коли реакцию проводить на холоду, то сначала получают тригидрат МнС2О4-ЗН2О в виде красивых окрашенных в розовый цвет триклинных игл (образование этих игл используют для качественного микро определения марганца). Однако на воздухе тригидрат неустойчив При храпении он вскоре переходит в Днгидрат. Последний при обычной температуре не отщепляет воду даже над серной кислотой. Только выше 100е он превращается в базвздный бледно-розовый оксалат МпС2О4.
Оксалат мартаяца(11) труднорастворим в воде. Существенно повышают растворимость сильные кислоты, а также щавелевая. Из растворов, содержащих оксалаты щелочных металлов, можно получить двойные соли (содержащие воду) с общей формулой М|С3О4-МпС2О4,
Седьмая группа периодической системы
241
Сульфит марганца(П). При взаимодействии карбоната марганца с содержащей S03 водой образуется сульфит маргаица(И) MnSO3. Будучи довольно трудно растворима в воде, эта соль кристаллизуется из раствора в виде розовых игл, легко окисляющихся на воздухе. Ниже 70° образуется тригидрат, при более высокой температуре — моногидрат. MnSO3 с сульфитами щелочных металлов образует двойные сопи преимущественно типа MjSO3- MnSO3.
Цианид марганца(П) и циапоманганаты(П). Из растворов солей двухвалентного марганца при добавлении циапид-ионов выпадает осадок, по-видимому, представляющий собой цианид марганца Мп(СК)2. Однако в чистом виде его получить не удается, так как па воздухе он очень быстро разлагается. Более устойчивы двойные или комплексные соли циапида марганца с другими цианидами {циаш>манганаты(11) типа
(Мп(С?1)в]}; например, текеациапомапталаЦП) калия К^МгЦСП^-ЗНчО — таблички от темно-фиолетового до сине-стальпого цвета; гексацианоманганат(Й) натрия Na4[Mn(CN)6]-8H2O — аметистовые октаэдры; гексацианомапгапат(П) кальция Ca2[.Mn(CN)0] — темно-синие кристаллы. Водные растворы циапоманганатов(П) почти бесцветны. На воздухе цианоманганаты(П) продолжительное время не могут сохраняться, так как они окисляются. В водном растворе они также довольно быстро разлагаются. Однако свежеприготовленные растворы могут вступать в реакцию обмена с солями тяжелых металлов; при этом радикал без разложения переходит
от одпой соли к другой. Из соли свинца при действии сероводорода можно также получить свободную кислоту Н4 [Mn(CN)e]. Она кристаллизуется в виде бесцветных чешуек, легко растворяется в воде и быстро ею разлагается.
При обработке растворов цианомангаиатов(П) в отсутствие воздуха порошком алюмипия или сплавом Деварда Манию (Mancliot, 1926) получил цианоманганаты(1) Mj [Mn(CN)6]. Такие цианосоли одновалентного марганца окрашены в растворе в интенсивно желтый цвет, в твердом состоянии бесцветны. Они чрезвычайно быстро окисляются. Циапоманганатьщ) можно получить также методом электролитического восстановления (Grube, 1927). Они обладают парамагнитными свойствами (Bhatnagar, 1939; Goldenberg, 1910), в то время как по аналогии с K4[Fe(CN)e] можно было ожидать для них диамагнитных свойств.
Роданид маргапца(П) и роданОмангапаты(П). Роданид марганца!,! Г) Mn(SQN)2 кристаллизуется при упаривании раствора карбоната маргапца(П) в водном растворе родаппстоводородной кислоты. Содержание воды в пем зависит от условий опыта. При обычной температуре наиболее устойчив т’етрагидрат. Он образует крупные светло-зеленые кристаллы, очень трудно растворимые в воде. Концентрированные растворы окрашены в зеленый цвет, разбавленные— в розовый. При нагревании до 100° гидраты (помимо тетрагидрата существуют еще три- и днгидрат) превращаются в безводную соль желтого цвета. С роданидами щелочных металлов роданид марганца(П) образует двойные соли типа Mj[Mn(SCN)e)] Iгексароданоманганаты(П)].
Силикаты марганца. Метасиликат марганца MnSiO3 встречается в природе в виде минерала родонита. Чистый силикат — это розовокрасные триклинные иглы (удельный вес 3,5). Природный силикат вследствие примесей обычно имеет вид грубой массы грязно-коричневого или серого цвета.
Гораздо более редким, чем родонит, является тефроит, ортосиликат марганца, Mn2SiO4. Оба силиката часто в виде красивых кристаллов содержатся также в доменных шлаках и т. п.
3. Соли марганца(1П)
Соли трехвалентного марганца производятся от окиси марганца(1П) Мп20з при замещении атомов кислорода кислотными остатками. Они большей частью окрашены в темный цвет и весьма склонны к образованию комплексных солей (ацидо-соединения); чистыми соли Мн(1П) получают часто исключительно в форме ацидо-соединений. Все соли марганца(1П) малоустойчивы. В кислом растворе они легко восстанавливаются до солей марганца(П); в нейтральном растворе простые соли гидролизуются, при-чем вначале образуется гидроокись марганца(Ш), которая на воздухе быстро переходит в гидрат двуокиси марганца.
16 Г Реми
242
Глава 6
Тот факт, что соли марганца (III) производятся от марганца с особой степенью окисления, а не являются смесыо соединений двух- и четырехвалентпого марганца, следует, между прочим, «> определения молекулярного веса ацетил а дет штата мар-гапца(Ш) Мп [СН(СОС113)2]2 (черные блестящие кристаллы е температурой плавления 172й), растворенного в бензоле.
Хлорид марганца(Ш) и хлороманганаты(Ш). Хлорид марганца (III) MnClc, по-виднмому, существует в темно-буром растворе, получающемся при обработке пиролюзита концентрированной соляной кислотой. Выделить ив него простое соединение не удается, однако можно получить ого комплексные производные — двойные соли хлоромангаиаты^Ш). Они кристаллизуются при охлаждении льдом насыщенного хлористым водородом раствора хлорида марганца(Ш), содержащего хлорид щелочного металла. По своему составу хлоромапганаты(П1) соответствуют формуле М2 [МнС15]. Они окрашены в темно-красный цвет и кристаллизуются без воды, за исключением соли аммония, образующей моногидрат.
Бромид и tiодпд трехвалептного марганца, а также соответствующие двойные соли неизвестны. Напротив, фторид марганца(Ш) MnF3 образуется при действии свободного фтора на иодид двухвалентного марганца: 2Мп1г -f- 3F2 = 2MnF3 4* 21->. При нагревании оп распадается на фтор и фторид маргапца(П). При прокаливании в токе кислорода оп переходит в Mn2O3. С небольшим количеством воды образует красно-бурый раствор, быстро разлагающийся при разбавлении. Такой же красно-бурый раствор получают при растворении Мп2Оэ в плавиковом кислоте (5), а также в результате реакции перманганата с солью марганца(П) в присутствии плавиковой кислоты (6)1
Mn2O34-6HF=2MnF34-3H2O, (5)
МаО^ф4Мп"ф45НР=5МиР3ф4Н2Оф 7Н‘. (б)
При упаривании раствора, содержащего избыток плавиковой кислоты, выпадает кристаллогидрат фторида марганца (Ш) MnFa-2H2G — рубиново-красные палочки. В присутствии фторидов щелочных металлов кристаллизуются томйо-крясные двойные соли типа MT[MnF4] пли MjJ[.MnF6],
Сульфат марганца (III), еульфатоманганаты(111) и марганцовые квасцы. Сульфат маргапця(Ш) образуется при растворении окиси или гидроокиси марганца!Ill) в холодной не очень концентрированной серной кислоте *. Из красного раствора при охлаждении кристаллизуется соединение Mn2(SO4)3‘Г12ЙО4-4112О. Его можно также получить при действии концентрированной серной кислоты на перманганат калия. (Образующаяся вначале гептаокись марганца растворяется при умеренном нагревании в серной кислоте с отщеплением кислорода.) При более сильном нагревании красный сульфат маргапца(Ш) переходит в зеленый. Последний, по данным Кариу-са (Garins), образуется также при обработке гидрата двуокиси марганца горячей концентрированной серной кислотой.
Сульфат марганца(ГП) образуется также при электролизе нагретых растворов с у лхт, фа та марганца(11), содержащих большое количество сер пой кислоты. Растворы окрашиваютс я в красный цвет, и из них выделяется еоедниенае Mn2(SO4)3- H2SO4- 4Н2О.
С сульфатами щелочных металлов сульфат марганца(Ш) образует два ряда двойных солей. Соли первого ряда либо содержат небольшое число Молекул кристаллизационной воды, либо совершенно безводны. Их состав соответствует общей формуле М^ёО4-Мн2(ЗО^)г. Они могут быть представлены как дисульфатоманганатыЦ!/'} М1 [Ми(ЗО4)2]. «Крас
* Растворение происходит только л присутствии небольшого количества окиси марганца(П).
Седьмая группа периодической системы
243
ный сульфат маргапца(Ш)», по-видимому, является производным кислоты Н[Мп(ЗО4)2]-2Н2О [дисульфатомарганцовая(Ш) кислота].
Другой ряд двойных сульфатов трехвалентного марганца с сульфатами щелочных металлов но составу и форме кристаллов относится к классу квасцов. Наиболее устойчивыми являются цезиевые квасцы марганца CsMn(SO4)2-12Н2О — гранатово-красные кристаллы. Соответствующее соединение рубидия уя;с при комнатной температуре отщепляет воду.. Еще легче разлагаются калиевые и аммонийные квасцы.
Следует отметить, что существуют также двойные соли сульфата Мп(Ш) с сульфатами желоза(Ш), хрома(Ш) и алюминия.
Фосфат марганца(III). Известно много фосфатов трехвалентного марганца, и среди них ортофосфат МпРО4-Н2О серо-зеленого цвета, метафосфат Мн(РО3)з красного, остальные фиолетового цвета. Все фосфаты нерастворимы в иоде. Образованием фосфата марганца(П1) обусловлено фиолетовое окрашивание перла фосфорной соли марганца в окислительном пламени.
Определенным образом приготовленный фиолетовый фосфат марганца(III) применяют в качестве малярией краски, отличающейся чистотой тона и устойчивостью на воздухе (марганцовая фиолетовая, перманганатная фиолетовая, нюрнбергская фиолетовая).
От ортофосфата маргапца(Ш) производится комплексная кислота Н3[Мп(РО4)2]; соответствующие ей соли типа MTI-I2 [Мл(РО4)2] [дифосф атома пганаты(Ш) в большинстве случаев с ЗН2О] были исследованы Майером (J. Moyer).
Дейс (Delfi, 1925) получил IIs[Mn(AsO4)3J-3II2O триарсепатомаргаиец(111) кислоту, производную арсената м.арганца{111).
Циаиочангапаты(П1). Цианид маргапца(1[1) неизвестен. Одпако легко можно получить производные от него комплексные соли гексацианом.анеаиат,ы{111) Мз[Мп(СМ)в]. Они окрашены в темно-красный цвет и изоморфны тексацианоферра-там(Ш). В сухом воздухе они довольно устойчивы. Получают их либо окислением гексациапомангаиатов(Н), либо более просто взаимодействием ацетата маргапца(Ш) с цианидами.
Существование гскеацианового комплексного иона [Mn(CN)ep- следует иа реакций двойного обмена растворимых гексацианоманганатов(Ш) с солями тяжелых металлов. Они образуют с нами осадки, в состав которых гекса циановый комплекс входит без изменения. Только в реакции с солями железа(И) трехвалептпый марганец замещается двухвалентным железом, так как ион гексациапоферрата(П) более устойчив, чем гексациапоманганата(Ш).
Ацетат марганца(1П) получают окислением ацетата двухвалентного марганца, растворенного в горячей ледяной уксусной кислоте, перманганатом или хлором. При добавлении небольшого количества воды из раствора через некоторое время выделяется дигидрат Mn(C2HsO2)3- 2Н2О — кристаллы цвета корицы с шелковистым блеском. В сухом воздухе он устойчив. Водой разлагается, но может быть перекристаллизован из растворов в ледяной уксусной кислоте. Ацетат марганца(Ш) оказался весьма удобным исходным веществом для получения других соединений т рехвалентпого марганца.
0ксалатомангапаты(1П). Оксалат марганца(Ш) известен только в виде комплексного соединения оксплатомапганата,{111). ГраомалапгомаяганатпЦЛ) калия К3 [Мп(С2О4)з]-ЗН2О образует темно-красные, почта черные, моноклинные призмы. Он изоморфен триокспдатоферрату(Ш) калия. В темноте это со единение довольно устойчиво при обычной температуре. На свету или при нагревании оно разлагается, при этом кислотный остаток уксусной кислоты окисляется до GOa,a MnI!i восстанавливается до Мп11:
2K3[Mn(C2O4)sJ =2МаС2О4-|- ЗК2С204-|-2С02.
4. Соединения марганца(1У)
Простые соли четырехвалентного марганца очень неустойчивы. Большинство из них известно только в виде комплексных солей ацидомангана-
1G*
244
Глава 6
moe(IV'). Все они более или менее легко гидролизуются водой. Еще легче происходит замещение ацидогруппы кислородом в присутствии щелочей. Так, из сщи<?омангаяатов(1¥) образуются оксоманганаты(1¥), называемые просто манганатами(1 V} или манганатами. Однако вследствие того что соответствующая им марганцовая(1У) кислота (марганцеватистая кислота) является очень слабой кислотой, а ее ангидрид — двуокись марганца МиО2 — чрезвычайно трудно растворим, последний (или его гидрат), как правило, и выпадает в осадок; даже если и происходит образование мапганатов(1 V) — при более высоком содержании ионов ОН',— они почти всегда в той или иной степени загрязнены гидратом двуокиси марганца.
Высокая устойчивость двуокиси марганца, проявляющаяся уже в том, что она в больших количествах встречается в природе (пиролюзит), обусловлена в основном ее чрезвычайно небольшой, растворимостью. В растворе соединения марганца(1У) малоустойчивы. В кислом растворе опи весьма склонны восстанавливаться, но в присутствии сильных окислителей могут быть окислены до перманганатов [манганаты(УЦ)]. В расплавленных щелочах уже иод действием кислорода воздуха они окисляются до манганатов(У) или манганатов(VI).
Из ацидоманганат<зв(7Т') —> общей формулы [МцХв] — следует прежде всего назвать хлоро- и фторопроизводные. Известны также соответствующие этому типу соединения с йодноватой кислотой М12[Мп(1О3)е], Сюда относятся также так называемые глицерил ман ганиты |дигллцсринатоманганаты(1¥)], образующиеся при взаимодействии свежеосаждеппой двуокиси марганца с глицерином G3H&(OH)3 и сильными основаниями. Например, диглицеринатомапганат(1У) натрия Na2[Mn(C3H5O3)2] — желтовато-красный порошок; раствор его в смеси спирта с глицерином имеет кроваво-красный цвет.
Хлорид марганца (IV) и хлороманганаты(1 V). Хлорид четырохвалент-ного марганца, по-влдимому, образуется в качестве первичного продукта при растворении пиролюзита в концентрированной соляной кислоте.. Однако он тотчас разлагается с отщеплением хлора (ср. стр. 234) и вследствие зтого не может быть выделен. Более устойчивы хлорожанганаты(/У), образующиеся и результате присоединения хлорида щелочного металла к хлориду маргапца(1У).
Гсксахлоромапганат(1¥) калия К.г[МпС16] — темно-красные кристаллы — лучше всего получать, по дапным Вейпланда, внесением растворов перманганата кальция и хлорида калия в сильно охлажденную 40% -ную соляную кислоту. Таким же способом можно приготовить соли аммония и рубидия. Известны также фтороманга-иаты(1 V), например К.2 [МнКЕ] — золотисто-желтые гексагональные таблички и K.[MnF5] — бледно-розовый порошок.
Сульфат маргапца(1¥). Если сульфат марганца(П), растворенный в серной кислоте, окислять перманганатом при температуре около 50— 00’, то из достаточно концентрированных растворов выпадают при охлаждении черные кристаллы сульфата четырехвалептного марганца Mn(SO4)2. В 50—80%-пой серной кислоте он растворяется, образуя темно-коричневый раствор. В разбавленной кислоте и просто в воде гидролизуется, образуя осадок гидрата двуокиси марганца. Сульфат марганца(1¥) используют в технике как окислитель, оказавшийся особенно подходящим для некоторых окислительных процессов.
Сульфат марганца(1¥) также способен образовывать двойные соли, однако состав ах довольно сложен.
Седьмая группа периодической системы 245
Манганаты(IV) [мангапиты]. Двуокись марганца МпО2 соединяется с окислами других металлов в самых различных соотношениях. Образующиеся соединения называются .канганатами(1У) или мамганипгами.
Например, с окисью кальция образуются следующие соединения:
2СаО-МпОг,. СаО.МпОг, СаО.2МнОг, CaO-3MnO2,
СаО.5МпОг.
Их получают из расплавов. Подобные соединения при определенных условиях получаются с избытком гидроокисей щелочноземельных металлов. Из расплавов гидроокисей щелочных металлов, как правило, образуются продукты, значительно более богатые марганцем. Продукты, выделенные из водных растворов, в большинстве случаев имеют неопределенный стехиометрический состав. Речь идет при этом только о продуктах адсорбции гидратом двуокиси марганца окислов щелочных и щелочи о-земельных металлов. Некоторые природные разное иди ости пиролюзита, и прежде всего псиломелан и вад, часто очень сильно загрязнены адсорбированными окислами металлов. Многочисленные гидраты двуокиси маргапца, содержащие кислород в количестве 3—4 эке па 1 атом Мп, частично можно рассматривать как мапганаты(1¥) двухвалентного маргапца, а частично —- как продукты адсорбции МпО гидратом двуокиси марганца.
Псроксомапгапаты(1¥). Если в концентрированный раствор перекиси водорода, содержащий большое количество К.ОН, осторожно прикапывать раствор мангапата(¥1) калия при температуре в интервале 15—18°, то постепенно выделяется гидроксотрипер-оксоманганат(1¥) калия К.3[Ма(ОН)(Ог)з/ в виде тонких коричнево-черных блестящих пластинок. При соответствующей обработке его можно перевести в трипероксо-аквомапганат(1¥) калия Кг[Мп(О2)3(ОН2)], кристаллизующийся в виде призм, Гетр он ер оксо-сое дипения К.4 [Мн(О2)л] получены только в лиде твердых растворов с гидроксотрцпероксомапгапатом(1¥) калия. Пер оксоманганаты (IV) калия очень неустойчивы. То же относится и к соответствующим солям натрия, но вследствие меньшей растворимости их легче приготовить, чем пероксомапганаты калия. Возможно, что каталитическое действие МпО2 в процессе разложения перекиси водорода сводится к образованию промежуточных нероксосоедипений МпО2, разлагающихся с выделением кислорода (S ch о Ider, 1949),
5. Манганаты(У)
При сплавлении МпО2 с содой и селитрой иногда вместо обычно наблюдаемого зелепого окрашивания появляется синее. Лукс (Lux, 1946) показал, что это обусловлено образованием манганатов(У) щелочных металлов. Ему удалось выделить в чистом виде соединение Na3 [МдО4]-10Н2О, а Клемм нашел, что магнитная восприимчивость этого соединения соответствует ожидаемой для соединений пятивалентного маргапца. Еще в 1904 г. Аугером и Билли (Auger, Billy) было описано соединение, которое по составу можно рассматривать как манганат(¥). Шмаль (Schmal, 1950) наблюдал, что любой окисел маргапца при прокаливании на воздухе с избытком ВаО при температуре около 900“ переходит в соединения маргапца(У). В чистом виде Ва3(МпО4)3 впервые был получен Шольдером. Позднее им был синтезирован ряд других манганатов пятивалентного марганца. Изготовляемая в настоящее время малярная краска — марганцовая синяя,— как установлено Клеммой в результате магнитных измерений, также содержит пятивалентный марганец.
6. Манганаты(У1)
При сплавлении двуокиси марганца с селитрой образуется плав, в котором содержатся манганаты{У1), т. е. соли с общей формулой MHMnOJ. Эту реакцию наблюдал еще Шееле. Вместо селитры можно попользовать другие окислители в смеси со щелочью. Растворы манганатов щелочных металлов содержат иопы [MnOJ" и окрашены в темно-зеленый цвет.
В технике тщательно перемешанную смесь пиролюзита с едким кали прокаливают при доступе воздуха, являющегося окислителем. Процесс следует проводить таким
248
Рлава 8
образом, чтобы образующаяся по реакции МпО2 + 2К.ОИ + 3/2О2 = К2МпО4 + Н2О вода как можно меньше разлагала полученный мангапат(¥1). При определенных условиях выход достигает 60—70%. Больший выход получить не удается, потому что наряду с мангапатом(УТ) всегда образуются манганаты(У) или манганаты(IV).
В чистом состоянии до настоящего времени известны только манганаты^!) пщлоч«ы.т металлов. Они образуют темно-зеленые, почти черные кристаллы. Манганат(У1) калия; K2[MnOJ кристаллизуется без воды. Из манганатов(УГ) натрия получены тетра-, гекса- и декагидраты.
Манганаты(У!) щелочных металлов легко растворяются в разбавленных растворах щелочей, такие растворы окрашены в зеленый цвет. Чистая вода и слабые кислоты сразу разлагают их до двуокиси марганца и ман-ганатов(¥П) (перманганатов);
ЗМпО4 + 2Н2О=МпО2 4- 2МпО;+40 Н'.
По-видимому, этот процесс обусловлен тем, что свободная марганцовистая кислота И2МпО4 неустойчива. Ее разложению способствует очень небольшая растворимость образующейся при этом двуокиси марганца. При кипячении распад происходит даже в щелочном растворе.
При нагревании выше 500° манганат(У1) калия распадается на манга-пат(1У) (так называемый манганит калия) и кислород, который, правда, вследствие образования твердых растворов выделяется не полностью:
КаМпО4—К2МпО3-р/2О2.
Содержащий примеси манганат(VI) бария используют в качестве краски (кас-селева зеленая), В частности, как неядовитую зеленую краску ее рекомендуют для фресковой живописи,
7. Перманганаты [манганаты(уИ)]
Перманганаты являются производными обладающей кислотными свойствами сомиокнси марганца Мп20т. Общая формула их Mx[MnO4], Попы [МпО4[' (пермангаиат-ионы). придают раотворам фиолетовый цвет.
Наиболее важным среди перманганатов является перманганат калил KM1JO4. Перманганат натрия (трпгидрат NaMnO4-3H2O) имеет небольшое значопие в технике, так как вследствие очень большой растворимости он трудно кристаллизуется. Перманганаты других щелочных металлов представляют лишь научный интерес. Растворимость перманганатов щелочных металлов приведена в табл. 26. Из перманганатов щелочноземельных
Таблица 26
Растворимость перманганатов щелочных -металлов в воде (г безводной соли в 100 а воды)
Формула ЫЫпОд-ЗНзО NaMnO4-3H2O КМпО4 ЛЬМдОд СзМпСц
Растворимость, е ~ 71 Легко растворим, расплывается 6,34 1,12 0,23
Температура, ГС 16 20 20 19 19
металлов следует упомянуть хорошо растворимый в воде перманганат кальция Са(МпО4)2-5Н2О. Его применяют для стерилизации питьевой воды. Известны также перманганаты тяжелых металлов.
Седьмая, группа, периодической системы
247
Перманганат калия КМпО4 — наиболее важную соль марганцовой кислоты — производят в технике в большом количестве. В настоящее время его получают электролитическим окислением манганата(УI) калия".
Мп04 — е=МпО;.
Более старые способы окисления хлором:
MnO^ + i/2Cl3=MnOa-l-Cl', т. е.
К2МпО1+1/2С12=КМпО4 + КС1,
или самопроизвольный распад К.2МпО4 в водном растворе; образующаяся при этом щелочь связывается С03:
ЗК2М[1О4 + 2СО2=2КМпО4+МпОг+2К.2СО3.
В последнем случае 1/3 марганца превращается в МпО2. Однако этот способ все же имеет преимущество по сравнению с предыдущим, так как образующийся в качестве побочного продукта карбонат калия можпо использовать дня сплавления с пиролюзитом, в то время как хлорид калия идет в отвал.
Перманганат калия образует темные, пурпурно-красные, почти черные с коричневым отливом призмы. Он изоморфен перхлорату калия и образует с ним непрерывный ряд твердых растворов *. Перманганат калия умеренно растворим в воде (при комнатной температуре немного более 1/3 молъ!л\ ср. табл. 26). Раствор интенсивно красно-фиолетового цвета имеет характерный спектр поглощения. Его применяют как в аналитической, так и в препаративной химии в качестве окислителя.
В кислой среде в присутствии достаточно сильного восстановителя пермангаиат-иои превращается в ион Ми":
МвО1+8Н- + 5е=Ма“-|-4П2О.
С солями железа(П), перекисью водорода, щавелевой, муравьиной, азотистой кислотами эта реакция протекает количественно. На этом основал весьма простой объемный способ определения перечисленных веществ. Так как раствор перманганата даже при очень сильном разбавлении заметно окрашен, а при переходе в соли марганца(П) окраска исчезает, то конечную точку при титровании весьма просто установить по обесцвечиванию, В колнчоствеинон анализе используют, как иравпло, (1,1 я, раствор перманганата (0,02 моля или 3,1605 г К.МпО4 в 1 л). Одни миллилитр такого раствора соответствует 1/10 мэко определяемого восстановителя. Косвенно при помощи перманганата можно определять и окислители. В этом случае приливают избыток подходящего восстановителя, например щавелевой кислоты или сульфата железа(И), и непр среагировавшую часть оттитровьгаают перманганатом. Мангапатометрические методы — так называют титрометрические методы с применением перманганата — иногда применяют даже для определения веществ, не являющихся ни окислителями, ни восстановителями. Например, кальций определяют следующим образом: осаждают его щавелевой кислотой, осадок растворяют в разбавленной серной кислоте и щавелевую кислоту, количество которой эквивалентно количеству кальция, титруют перманганатом.
В нейтральной или слабокислой среде перманганат восстанавливается только до двуокиси марганца:
МнО;-|-4Н- +Зе=МпОг4-2Н2О.
Эту реакцию используют для определения марганца в солях двухвалентного марганца (метод Фольгарда):
2МпО;+ЗМп'Ч-2НгО=5МпОг+4Н-.
* Непрерывный ряд твердых растворов образуют также NH4[MnO4J и NHJC1OJ. Напротив, K[MnO4j и БЬ [МпО/J лишь в ограниченной степени способны смешиваться. NH4[MnO4] получают взаимодействием (NH4)aSO4 с Ва[МпО4]Е в виде игл; они не гигроскопичны и мри нагревании или трении воспламеняются.
248
Глава. 6
Вместо 6,1 н. jfr. е. 0,02 М) раствора перманганата, применяемого для окисления в сильнокислой среде, нри определении марганца по методу Фольгарда требуется 0,06 п. раствор. При манганатометрическом титровании по Фольгарду следует тщательно следить а а тем, чтобы вследствие окисления ионов марганца(П) не образовывался нерастворимый мангаиат(1¥) двухвалентного марганца. Для этого к раствору прибавляют избыток ионов цинка, которые связывают выделяющуюся двуокись марганца, прежде чем опа успеет прореагировать с ионами марганца(П)>
Если к перманганату калия добавить калиевую щелочь, то образуется манганат(У1) и выделяется кислород:
4М»О^-|-4ОН’=4МаО1+Ог-[-2Н2О.
При длительном стоянии распад углубляется —, образуется МнОо. При нагревании сухого перманганата до температуры умеренного красного каления отщепляется кислород. И в этом случае наряду с манганатом(У1) образуется двуокись марганца или манганат(ГУ):
10КМп04 =ЗК2МпО4+2К2О • 7МпО2 Д- 6О2.
Так как ионы MnOi в щелочной среде гораздо более устойчивы, чем в кислой, перманганатом в щелочном растворе можно титровать многие вещества, которые в кислом растворе взаимодействуют очень медленно, например гипофосфит, метанол, эритрит и ряд других органических веществ. Однако если процесс протекает не очень быстро, можно легко допустить ошибку вследствие саморазложения (даже медленного при небольшом содержании щелочи) ионов МвО4 в щелочном растворе. Поэтому дня количественного определения таких веществ целесообразно использовать только первую ступень восстановления ионов МпО4, приводящую к образованию иопов МпО4 (цо-видимому, в соответствии с уравнением 2MnOj + 2ОН' = 2МнО4 Д- Н2О -к О вначале отщепляется атомарный кислород, который затем реагирует с восстановителем), Эта реакция протекает гораздо быстрее, чем восстановление по второй ступени (МпО4 —>- МпО2). Штамм (Stamm, 1934) показал, что в присутствии растворимой соли бария восстановление останавливается на первой ступени (МпО4 -ч- MnOJ), так как ноны МиО4 тотчас образуют труднорастзоримый ВаМпО4. Для ускорения переноса кислорода к восстановителю рекомендуют добавлять катализаторы — соли тяжелых металлов (например, несколько капель раствора Co(NO3)2). В таком варианте титрование щелочным раствором перманганата пригодно не только для определения многих органических веществ, например для анализа питьевой и производственной воды, но и других, а именно нодатов ([1О2]' —> [1О4]'), Йодидов (Г —> [1О4]'), цианидов ([СК]' [OCN]'), роданидов (1SCN}' [OCN]' Д- [SO4]"), фосфитов ([НРО3:]’->
-> JРО4]'") и гипофосфитов (jHoPOj]' [Р04Г).
Перманганат кадия является наиболее употребительным окислителем в препаративной органической химии. Вследствие сильных окислительных свойств его применяют для отбелки шерсти, хлопка, шелка и других прядильных волокон, а также для осветления масел. Он служит также для очистки веществ от органических примесей, например для очистки от примесей сырой уксусной кислоты. Перманганат калия является хорошим дезинфицирующим средством. Раствором перманганата протравливают различные сорта древесины, чтобы придать ей вид орехового дерева. Его используют также в производстве набивных тканей. В лаборатории применяют для промывания Тазов. В технике его используют для очистки двуокиси углерода при приготовлении минеральной воды.
При нормальных условиях перманганат калия гграктическп по реагирует с водородом. Однако в присутствии нитрата серебра раствор перманганата уже при комнатной температуре и атмосферном давлении поглощает водород. На этом основано его применение для определения водорода в смеси с СП4, С2Н6 и N2, так как они с перманганатом не реагируют [Hein F., Chem. Fabrik, 4 {1931)],
Аналитические сведения. При пропускании сероводорода марганец не осаждается, однако под действием сульфата аммония выделяется телесного цвета осадок — сульфид марганца MaS (безводный). Если марганец
Седьмая группа периодической системы
249
присутствует в более высокой степени окисления, то его восстанавливают до солей марганца (II) кипячением с сульфидом аммония или, гораздо быстрее, с сероводородом.
От остальных веществ группы сульфида аммония марганец можно легко отделить. Сульфид маргапца Легко растворяется в разбавленной соляной кислоте, в то время как отфильтрованные на воздухе сульфиды кобальта и никеля нерастворимы (ср. стр. 316). От цинка и алюминия марганец отличается тем, что его окислы МнО, Мп2О3 и МпО2 пе растворяются в едком натре. Железо и хром отделяют от марганца(Ц) осаждением их аммиаком из содержащих хлорид аммония растворов *,
При сплавлении соединений марганца с содой и селитрой (окислительный плав) образуется зеленый манганат(У1), Раствор его при добавлении уксусной кислоты становится фиолетовым и одновременно выделяется бурый осадок гидрата двуокиси марганца. Весьма чувствительной является реакция окисления марганца двуокисью свинца в присутствии концентрированной азотной или серной кислоты до марганцовой кислоты (ср. стр. 235).
В микроанализе марганец открывают в виде оксалата или капельным методом, основ энным па образовании снпего окрашивания бензидина под влиянием Мп(ОН)3. Еще более чувствительной является капельная реакция с перйодатом калия и реактивом Арнольда (чувствительность 0,001у Мн). В кислой среде перйодаты окисляют Ноны Мп” до ионов MnOj. Последние с реагентом Арнольда дают интенсивно синее окрашивание (ср. т. I, стр. 605).
Перлы солей фосфора или буры в окислительном пламени марганец окрашивает в фиолетовый цвет вследствие образования фосфатов или боратов марганца(1П). В восстановительном пламени окраска исчезает, так как образуются соединения двухвалентного марганца.
Для весового определения марганца пригодно осаждение и взвешивание в виде сульфида или осаждение гидрата двуокиси с последующим прокаливанием его до Мп304. Наиболее удобным методом определения марганца в солях двухвалентного марганца является выделение его в виде двойного аммонинмарганецфосфата NHjMnPOj-HaO и взвешивание в виде пирофосфата Мп2Р3О7. Для отделения марганца от других металлов очень удобно электролитическое выделение его на аноде из концентрированного азотнокислого раствора в виде гидрата двуокиси.
Об объемном определении марганца, а также о применении перманганата калия в титрометрии см. стр. 247,
ТЕХНЕЦИЙ (TECHNETIUM) Тс и РЕНИЙ (RHENIUM) Re
Распространение в природе. Рений — чрезвычайно редкий элемент. В этом отношении его можно сравнить, пожалуй, только с сильно радиоактивными элементами. Технеций — элемент неустойчивый, в природе-он не найден, но был искусственно получен в результате ядерных превращений (см. стр. 250).
В доступной части земпой коры содержание рения составляет в среднем -1-10*’%. Но распространен он неравномерно. В некоторых минералах ого относительно много. Как правило, наиболее богаты рением молибденит, а также платиновые руды. Молибденит содержит 6-1СН5—2-10*3% рения, В платиновой рудо Советского
* Железо предварительно переводят в трехвалентное, добавляя небольшое количество концентрированной азотной кислоты, иначе оно выпадает неполностью. Марганец азотной кислотой пе окисляется.
250
Глава 6
Союза его содержание оценивается до10-4% , т. е, в 1 «г руда содержится 1 мг рения. Окисные руды ре дир достигают такого «высокого* содержания этого металла; в колумбите, например, оно равно 0,5-10*5—2-10-5% , Среди сульфидных руд молибденит оказался единственной, в которой его содержание превышает 10~4%. Сравнительно много рения имеется в доменных шлаках мансфель дер ск их медных сланцев (ср. стр, 252) Из них без особого труда можно осуществить производство рении.
Исторические сведения. Рений был открыт Ноддаком (N oddack) и Такке (Таске) в 1925 г. Уже задолго до этого на основе периодической системы было очевидно существование двух еще гщ найденных аналогов марганца По закону Мозли
порядковые иомора 43 и 75 пе могли принадлежать ни одному из известных в то время элементов. Уже тоща пытались найти эти элементы. Неудачи всех подобного рода попыток, а также сравнение множества различных элементов земной коры привели Н одна К а’и Таксе к выводу, что оба « эк ам ар ганца» должны быть чрезвычайно редкими. элементами. Следовательно, они пе могли быть найдены непосредственно в минералах но характеристичным рентгеновским спектрам. Требовалось предварительное обогащении по мгпыпей мере до 0,1% (грапица чувствительности рецтгепоспектрального метода). Чтобы осуществить такое обогащение, Ноддак и Такке^цбпытались предсказать химические свойства экамарганцев на основе периодической системы. Они пред-положили, что свойства элементов в побочной подгруппе VII группы с увеличением порядкового номера изменяются так же, как и в побочных подгруппах соседних групп, т. е. в VI и первом столбце VIII побочной подгруппы. В результате они пришли к выводу, что экамаргапцы должны бы иметь подобно марганцу несколько валентных состояний, причем более высокая валентность у них должна быть гораздо устойчивее^, чем у марганца, подобно тому как возрастает устойчивость соединений с более высокой валентностью при переходе от хрома К вольфраму и от Железа к осмию в соседних подгруппах. Далее, на основе этих гипотетических свойств предстояло найти способ разделения различных -минералов с целью обогащения их искомыми элементами настолько, чтобы можно было открыть их рентгеиоспектрографически. Для этого необходимо было выбрать в первую очередь такие минералы, в которых в соответствии с законом распределения элементов в земной коре (ср. гл, 15) можно было бы ожидать существования (если они действительно существуют) экамарганцев. Сначала была взята платиновая руда. Однако в распоряжении исследователей ее было слишком мало, поэтому выбор пал на окисные минералы, а именно такие, в которых наряду е молибденом и рутением в заметных количествах содержались также вольфрам н осмий. Такими минералами оказались колумбит и танталит. По закону распределения содержание в земной коре элемента 75 должно быть в 500 000 раз меньше, чем ниобия *, Если предположить, что в колумбите именно такое соотношение между элементом 75 и ниобием, то для достижения границы чувствительности рентген оспек-тральпого метода требовалось по меньшей мере 500-кратное обогащение его элементом 7,j. Однако, пол;и анчто фактическое содержание искомого элемента в нем может оказаться еще меньше, обогащение следовало увеличить еще в 10 раз. Таким образом, йз 1 кя минерала было приготовлено 0,2 а концентрата, состав которого подбирали так, чтобы энамаргинец оставался в совокупности с родственными ему соседними элементами Mo, W, Г<ч и Os, Элемент 73 действительно был открыт в таком концентрате рентгеноспектрографическим методом. Новый элемент был назван (в честь Рейна) рением. В табл. 27 приведены его характеристические рентгеновские спектры.
В результате испытания большого числа минералов после предварительного обогащения на содержание в них рения наиболее богатым оказался молибденовый блеск (норвежский). Из в60 кг чистого молибденового блеска Ноддаку и Такие в 1928 г. удалось впервые выделить 1 а почти чистого репия. Вскоре после этого его стали получать в больших количествах, и металл поступил в продажу. В настоящее время он находит довольно широкое техническое применение (см. ниже). Хенигшмид (HSnig-schmid) в 1930 г. определил атомный вес рения равным 1.86,31, Ноддак и Такке на основе периодического закона в 1925 г. предсказали величину между 187 и 188, Что касается химических свойств, то опи в основном совпали с предполагаемыми.
Подла к и Такне полагали, что наряду с элементом 75 им удалось открыть элемент 43, Они назвали его мазурием. Однако достаточно убедительные доказательства в пользу открытия этого элемента приведены не были. Позднее элемент 43 был нолу-
* Ноддак и Такке предложили рабочую гипотезу, в соответствии с которой отношение среднего содержания экамарганца в земной коре к среднему содержанию рутения и осмия равно отношению в ней марганца к железу, т. е. примерно 1 : 50, Как показали более позцпие исследования (табл. 89, стр. 684), Ноддак и Такке пользовались значительно более низкими величинами распространенности платиновых металлов; ср., например, Данные на стр. 690,
Седьмая группа периодической системы
251
Таблица 27
Вычисленные и Измеренные линии характеристического рентгеновского спектра рения (Ь-серпя)
Линии ч La2
Вычисленные длины волн, А 1,431 1,441 1,235 1,204
Измеренные длины волк, А 1,430 1,441 1,235 1,205
чеп в результате ядерных превращений, Сегрэ, получивший его впервые в 1937 г. бомбардировкой молибденовой мишени дейтропамп (ср. стр. 635) и исследовавший его основные химические свойства, назвал новый элемент, как первый из искусственно полученных, технецием (гк/хц— искусство). Как и следовало ожидать в соответствии с правилом об устойчивости ядер (см. Стр. 630), оп оказался нестабильным. Позднее было получено еше несколько искусственных изотонов техпеция (ср. стр. 636). Все опи также неустойчивы. Наиболее долгоживущий изотоп технеция, найденный в 1947 г. среди продуктов распада урана (й9Тс), имеет период полураспада 2-1(>’ лот. Возраст Земли примерно в 10 000 раз больше (ср. стр. 700). Из этого следует, что даже если первоначально технеций и содержался в земной коре, то за это время он должен был бы исчезнуть. Однако Паркеру и Курода недавно (Parker, Kuroda, 1956) удалось доказать, что в природном уране в крайне и о значительных количествах присутствует радиоактивный изотоп молибдена ййМо, который имеет период полураспада 67 час и в результате /1-распада превращается в ойТс. Это умазывало на то, что ээМо непрерывно образуется при спонтанном я дери ом распаде 238 U (ср. Стр. 644). Следовательно, технеций, очевидно, имеется в природе, несмотря на то что до сих пор оп непосредственно еще не обнаружен (ср. также стр. 638).
СВОЙСТВА И ПОВЕДЕНИЕ ТЕХПЕЦИЯ
По химическим свойствам технеций очень сходен с рением. Сходство с рением у него гораздо больше, чем с марганцем, подобно тому как молибден более близок к вольфраму, чем к хрому, а рутений — к осмию, чем к железу. Сульфиды техпеция и рения, например, в отличие от сульфида марганца нерастворимы в разбавленной соляной кислоте. При нагревании в токе кислорода технеций, а также рений переходят в семиокись, в то время как соответствующий окисел марганца непосредственно из элементов не образуется. Разделение технеция и рения осуществляют пропусканием хлористого водорода в раствор их окислов в 80%-ной серной кислоте при температуре 2003. Рений отгоняется в виде хлорида, а технеций остается в растворе.
Во многих реакциях технеций подобен соседнему по периодической системе молибдену. Это обстоятельство используют при работе с ничтожно малыми количествами технеция, в этом случае в качестве носителя используют соответствующие соединения молибдена.
Первоначально химическое поведение технеция изучали па ничтожно малых количествах этого элемента радиохимическими методами (см. гл. 11). За последнее время в ощутимых количествах стал доступен изотоп В9Тс, так как он является одним из продуктов распада урана в атомных реакторах, а также вследствие его слабой радиоактивности ((период полураспада, по данным Фрида и сотр. (Fried, 1951), составляет 2,12- 10s лет). В виде Тей87 его осаждают сероводородом из водного раствора, подкисленного соляной кислотой. Черный осадок сульфида растворяют в аммиачном растворе перекиси водорода и полученное соединение, технетат аммония NH4TcO4,
252
Глада 6
прокаливают в токе водорода при температуре 600°, Бойд (Boyd) с сотр, приготовил таким способом около 0,6 г спектрально чистого металлического технеция. Величины атомного веса, установленные химическим путем и масс-спектрографически (Inghram, 1947), практически совпади [,98,91), Кристаллизуется технеций, по данным Мука (Mooney, 1947), в решетке с гексагональной плотнейшей упаковкой (л = 2,735, с = 4,388 А.) и изоморфен Во, Пн и Os; <jpeHTt. = 11,50, атомный радиус 1,36 А. Технеций металл серебристо-серого цвета,. Он нерастворим пи в соляной кислоте, пи в щелочном растворе перекиси водорода, по легко растворяется в азотной кислоте и в царской водке. При нагревании в токе кислорода сгорает с образованием светло-желтой летучей семиокиси Тс2О7. Она гигроскопична {т. пд. 119,5°); при растворении в воде образует технециевую («пертехнециевую») кислоту (соответствующую технецию (VII)] НТсО4, которую при упаривании раствора можно выделить в виде темнокрасных, продолговатых кристаллов. НТсО4 — сильная одноосновная кислота. Ее темно-красные концентрированные водные растворы при разбавлении быстро обесцвечиваются; 1 ,11 раствор уже совершенно бесцветный. Пертехнетат аммония [техне-тат(УИ) аммония] NH4TcO4 бесцветен и в чистом состоянии не гигроскопичен. [О свето-поглощеыии растворов техпетата(УП) в ультрафиолете см. Boyd G. Е. u. Mitarb., J. Am. Cbem. Soc,, 74, 556 (1952): vgl. anch R и 1 f.s,M в i n k e, J. Am. Cbem. Sec., 74, 235,] Металлический технеций можно легко выделить из кислого раствора электролитически. Нормальный потенциал, соответствующий образованию иона TcOj из металла но реакции: Тс -ф- 4ll2O = ТсО4 ф- 8Н‘ -ф- 7е составляет + 0,41 в (Flagg, Bleidner, 1945). Эта величина лежит между потенциалами образования ионов МиЩ и НеО^, по ближе к последнему. Свободная энергия образования Тс О ^вычисленная из нормального потенциала равна 160 ккал/г-ион Тс О: Mg, Zn, Fe, Ni, Sn, Pb и Си вытесняют технеций из растворов, содержащих нон ГсО4, в виде металла; хлорид олова(П) п соляная кислота восстанавливают ион ТсО4 до положительного иона, соответствующего более низким степеням окисления технеция. Роджерс (Rogers, 1949) показал, что из щелочного раствора электролитически техпеций в принципе можно отделить как от рения, так и от молибдена. При нагревании технеция (а также рения) в токе сухого хлора оп улетучивается в виде хлорида.
Ввиду того что из отходов атомных реакторов можно наладить непрерывное производство технеция, именно его наиболее долгоживущего изотопа иТе, не исключена возможность его технического применения в будущем. Техпеций относится к числу наиболее эффективных поглотителей медленных нейтронов. В связи с этим следует, очевидно, принимать в расчет его использование дня экранирования ядерпых реакторов (см. стр. 647). Количества технеция, получаемого в настоящее время, исчисляются несколькими граммами.
Содержание технеция в продуктах распада урапа в атомных реакторах составляет около 6,2 вес. % . Вследствие чрезвычайно большой радиоачгтпвкости этих продуктов весьма трудно сразу же переработать их на технеции. Однако спустя некоторое время, когда излучение значительно уменьшается, уже пе представляет никакого труда, отделить техпеций от остальных продуктов распада. В небольших количествах (нескольких миллиграммов) его проще всего получать в ядерпом реакторе н^и бомбардировке окцеи молибдена нейтронами: say;о ф- = S9Mo -ф- у; "Мо —> ®8Tcs От окиси молибдена его легко можно отогнать возгонкой в виде окиси Тс£Ог,
ПОЛУЧЕНИЕ Й СВОЙСТВА РЕНИЯ
Получение, В настоящее время для технического получения рения используют некоторые доменные отходы мапсфельдерских медных сланцев, которые сравнительно богаты им. Отхода, содержащие в среднем 0,005% Не, окисляют и затем выщелачивают, При этом рений в виде ионов ВеО4 переходит в раствор вместе с большим количеством сульфатов тяжелых металлов. Основное количество этих сульфатов отделяется при упаривании, затем добавляют хлорид калия, после чего рений осаждается в виде перрепата калия КВеО4. Его снова растворяют, прибавляют калиевую щелочь, для того чтобы освободиться от соосаждеппых тяжелых металлов, и путем повторной кристаллизации получают полностью очищенный продукт. Таким способом теперь ежегодно производят не менее 251) хе поррената калия нлн 160 хе рения.
Металлический рений в виде серого порошка образуется при прокаливании перрената калия в токе водорода. Для удаления едкого кали перренат промывают горячей водой и затем еще раз прокаливают в токе водорода. Если вместо перрената калия взять перренат аммония, то получают сразу чистый рений в виде блестящего металлического зеркала. Монокристаллическую проволоку из рения получают методом наращивания ван Аркеля и де Бура$
Седьмая группа периодической системы 253
Свойства. Рении — металл, похожий па платину. Совершенно чистый компактный рений довольно мягок и очень гибок. Напротив, штабики, полученные спеканием или плавлением серого металлического порошка — продукта восстановления в токо водорода,— умеренно ковкие и очень твердые (твердость по шкале Мооса равна 8). Плавится рений при температуре 3180°. Кристаллизуется в гексагональной плотнейшей упаковке (ср.,т. I, стр. 279, рис. 57), а = 2,76, с = 4,47 А; атомный радиус 1,38 А. Удельный вес компактного рения 21,03, удельное сопротивление 0,20-Ю*4 ом-см (при 20”). На воздухе при обычной температуре он довольно устойчив. Компактный рений окисляется па воздухе только при температуре выше 1000°. Порошкообразный, напротив, уже при слабом калении в токе кислорода постепенно образует летучую окись Re2O7. Во влажном воздухе он постепенно окисляется уже при обычной температуре, продукт окисления — рениевая кислота НКеО4.
С водородом рений пе реагирует. Не известим до сих пор и соединения с азотом. С фосфором оа образует несколько соединений, йс2Р, НеР,ИеР2 и ReP3. С мышьяком получено соединение, приблизительно соответствующее, формуле Re3As7, С кремнием оп реагирует с образованием ReSi2. С вольфрамом рений дает соединенно W2Re3 (ср. табл. 17, стр. 141), С хромом соединение пе образуется, но получаются широкого диапазона твердые растворы. Об отношении рения к другим металлам пока еще мало известно.
В соляной и плавиковой кислотах рений практически нерастворим, напротив, он быстро растворяется в азотной и более медленно в серной кислотах, образуя рениевую кислоту [рений(\П) кислота]. При сплавлении с едким кали в присутствии достаточного количества кислорода рений переходит в соль рениевой кислоты (ср. стр. 255).
Рений является слабым катализатором в реакциях гидрирования и особенно дегидрирования. Для каталитического окисления он малопригоден вследствие легкого образования летучей семиокиси.
Применение. Репин оказался весьма подходящим при изготовлении наконечников перьев для авторучек; небольшие количества его придают высокую прочность и коррозионную устойчивость по отношению к подсыхающим чернилам. Помимо этого его применяют для производства платиновых сплавов, отличающихся механической прочностью, напрнмер для электродов. По твердости и прочности па растяжение нлатннорениевые сплавы (5% Re) превосходят не только чистую платину, но и сплавы платнпы с иридием (10% 1г), ранее применявшиеся для электродов. Применению рения в качестве материала для тиглей препятствует летучесть его при прокаливании на воздухе вследствие образования Ro207. Это обстоятельство следует иметь в виду также При пользовании нлатипо-платинорениевыми термопарами (см. стр. 345); хотя опи имеют в 3—4 раза более высокую термоэлектрод в ищущую силу, чем нлатино-платипородиевые, но летучесть рения при нагрев алии в окислительной атмосфере постепенно приводит к снижению терм о электродвижущей силы.
СОЕДИНЕНИЯ РЕНИЯ
Рений в своих соединениях преимущественно четырех- и семивалентен.
В соединениях с галогенами рений проявляет наивысшую валентность только в том случае, если одновременно связывается кислород. В противном случае максимальная валентность его по отношению к фтору равна шести, к хлору — пяти. Из галогено-солей наиболее легко доступны соли типа [ReXe], в которых рений чстыуагвалептен. Известны также соединения тусмалентного рения. Имеются основания предполагать о существовании соединений деух- и одновалентного рения, однако в чистом виде они до сих пор пе получены,
254
Глава 6
Обзор важнейших типов соединений рения приведен в табл. 28.
Важнейшие типы соединений рения
Таблица 28
Валентность рения Окислы Оксо-соли Сульфиды Фториды Хлориды Хлоро-соли
III Не.,О3.хНгО —. .—> — RGjjCIg M^ReClJ
IV ВсО2 Mi[ReO3] ReSg ReF4 — Mi[RcCl6]
V '— tffReOJ — — HeCl5 Ml[ReOCI5J
VI ReO3 Mi(ReOJ — ReF6 ReOCl4 M2[ReOCl8]
VII | Ве2О7 Не2О8? (п MI[ReO4] срекись?) Re2S7 '— ReOsCl —
Окислы рения
Помимо окислов, указанных в табл. 28, рений, поводимому, образует еще окисел, подобный молибденовой и вольфрамовой сини. При нагревании рения в токе кислорода наряду с желтой окисью Не2О7 и белой окисью (см. ниже) часто образуется синий налет. В процессе получения ReO2 восстановлением Be2O? или ее производных, как правило, образуются промежуточные вещества синего или фиолетового цвета. По своему составу опи находятся между ВеО3 и ReO2 и, возможно, идентичны синему налету, состав которого еще не установлен^
Семиокись рения. ВевО? получают нагреванием порошка рения в токе кислорода. Она образует желтые кристаллы, плавящиеся при 220s, а уже ниже температуры плавления возгоняется. В воде очень легко растворяется с образованием рениевой кислоты (см. сл. стр.); Re2O7 легкорастворима в спирте, но малорастворима в эфире. Теплоту образования см. стр. 225.
При уме реп лом икгрсванигт желтой семиокпеи в токе кислорода, а также при ее шегуннщи иугги сн.пгатпт.ы ренпа в качестве побочного продукта часто образуется ion с,-лгт.ч? цееюа в впд.е трудно осаждающегося тумана. В конденсированном состоянии они, по нс подтвержденным данным, представляет собой белоснежную кри-сталдичест.ут । массу е высоким показателем преломления. Еще не установлено, что &WT нродунт — вторая модификация ссмиоллси ВегО7 или перекись Вег03.
Трехокись рспия Re03 была получена Бильтцем (Biltz W., 1931) нагреванием Ке2О7 с рением в наплавленной капиллярной трубке при 300’. Опа красного цвета; удельный вес равен 7. Рентгенометрически установлено, что ионы Res+ образуют простую кубическую решетку, в которой кислородные ионы расположены в середине каждого из 12 ребер (ср. т. I, стр. 237, рис. 45).
Двуокись рения ReO3 получают нагреванием семиокпсп рения в токе водорода до 306° иди обезвоживанием ее гидрата ReO2-a-H3O в вакууме при температуре около 3U0q. Ола представляет собой коричнево-черпый порошок, сгорающий при нагревании в атмосфере кислорода (и иногда с пламенем) до сеид ок пси. В токе водорода при температуре 8<Ю° она восстанавливается до металла. При сильном прокаливании в вакууме разлагается по уравнению; 7ВеО2 = ЗВе 2Re3O7 (Biltz, 1933). В соляной кислоте растворяете;! с образованием H2(ReCle], раствор” которой окрашен в коричпево-зединый цвет. Соли этой кислоты (о получении их см. далее) гидролизуются до образования черно-коричневого гидрата двуокиси рения Rc2O-asH2O.
Полуторная окись репня Re3O3 до сих пор известна только в виде гидрата Re20s- zII2O. Опа образуется при добавлении щелочей к раствору Ве2С1й и отличается весьма небольшой устойчивостью. Постепенно разлагает воду с выделением водорода (Geilman, 1933),
Седьмая группа периодической системы
255
Оксо-соли рения
Рений образует следующие оксо-соли:
ReVl1
ReVIMlfReO4]
( M.*(ReG3]
Revj MHRe2O7l
( ьЦ[НеО4]
MI(ReO4] (1 : 1)-перропат, цветпый
M^ReOj] (3 . 1) -перренат,
обычный перренат, тетраоксоренат(У//), бес-пентаоксоренат(У//), от желтого до красного
ренат, точнее, ренат(УГ), зеленый
(i : 1)-гипоренат, мегагииоренаг, траок&тренат/У), желтый
(2 ; 1)-гиПОр<'тгат, пирогипоренат, гептаоксодиренат(У)> желтый
(3 : 1)-гнпоренат, ортогипорепат, тетраоксоренат (V), желтый
RelvM*[ReO3] решит, ренат(1У), коричневый.
Наиболее устойчивыми из них являются ренаты(VII). Относительно легко можно получить в чистом виде п ренатыЦУ). Выделение чистых ренатов(УГ) и рена-тое(У) связано с большими трудностями, так как в расплавах они находятся в равновесии с репатами(1У) и репатамп(УП) в зависимости от температуры (ср. ниже).
Рениевая кислота и перренаты. Семиокись рения является ангидридом рениевой кислоты НВеО4.
Реакция Ре2От + Н3О & 2ГГ + 2ReOj в присутствии большого количества воды сопровождается значительным выделением тепла (6,3 к кал/моль HReOJ. При упаривании раствора выделяется пе свободная кислота, а безводная окись Re2O7. Ропиевая кислота — сильная, одноосновная кислота. Пе водные растворы бесцветны. Проще всего ее можпо получить растворением металлического рения в 30%--ной аэотпой кислоте.
При насыщении рениевой кислоты основаниями образуются соли перренаты [ренаты(УП) М1[НеО4]]. Они бесцветны (в случае если катион сам не дает окрашивания) и кристаллизуются, как правило, без воды.
Большинство из пих хорошо растворимы в роде. Многие плавятся без разложения. Перренаты одновалентных металлов общей формулы MI[ReO4] имеют решетку типа шеелита (см. рис. 27, стр. 198).
Перренат калия КВеО4 является исходным продуктом для получения большинства других соединений рения. Он кристаллизуется без воды, в виде бесцветных тетрагональных бипирамид (типа шеелита, а = 6,615, с = 12,50 А). Растворимость при различной температуре приведена ниже.
0“ 10° 20° 25° 50“ 1001
0,474 0,573 0,990 1,235 3,18 10,44 a KReO4 в 100 г
воды
В отличие от перманганата перренат калия совершенно устойчив в сильнощелочных растворах.
Перренаты можпо получать непосредственно при сплавлении порошка рения со щелочами в токе кие.торода. Если количество кислорода достаточно для окисления металла до пер рената, плав приобретает огненпо-краспый цвет. При большом избытке щелочи образуется не обычный бесцветный иорренат (1 : 1) М1 ReO4, а красный (3 :1)-перрепат М|йеО5.
До образования перрената расплав щелочи с повышенным содержанием кислорода вначале имеет темпо-коричневый цвет, затем желтый и, наконец, зеленый. Как показал Ноддак (Noddack, 1933), это связано со ступенчатым процессом окисления через коричневый ренат(1У), желтый ренат(У) и зеленый ренат{УГ). Оксо-соли, соответствующие промежуточным стадиям окисления, выделить из расплава в чистом виде очень трудно, поскольку между ними в зависимости от температуры и содержания
256
Глава 6
кислорода в расплаве существуют равновесия, частично осповаппые на реакции диспропорционирования, панример 2Rev ReIV + ReVI; 2ReTI Rov + Revn.
Чистые ренаты(IV) M.J[ReO3l проще всего удается получить сплавлением ReO3 со щелочами. Например, ренат(1У) натрия Na3[ReO3l образуется по уравнению: НеО2 -j- 2NaOH = Na2[ReO3] + Н2О. Это коричневого цвета порошок, нерастворимый в воде и щелочах.
Соединения рения с серой
В отличие от марганца моносульфид рения не образуется, но известны дисульфид RcS2 (с ченгыреазвалентным рением) и гептасульфид Re2S7. Последний в противоположность весьма устойчивому дисульфиду (теплота образования 40 ккал) — соединение эндотермическое. Вследствие этого при непосредственном соединении элементов образуется только дисульфид, а гептасульфид получается лишь из растворов.
Г сит ас у.тьф ид ренпя Ro2S7 выделяется в виде черного осадка при пропускании HaS в сильнокислые растворы перренатов. При слишком большом или недостаточном содерщаппи кислоты он остается в растворе в коллоидном состоянии, Оптимальная концентрация НС1 12 г в 100 мл раствора. Гептасульфид рения практически нерастворим в соляной кислоте и растворах щелочных металлов. При пагревапии оп необратимо разлагается на серу и дисульфид ReS2 (Biltz, 1931).
Тиоперренаты. При пропускании сероводорода в нейтральный раствор перре-ната или в разбавлеппый раствор рениевой кислоты появляется желтое окрашивание вследствие образования монотиоперренат-понов [ReO3S]'t Мояотиоперренаты щелочных металлов очень легко растворимы в воде; например, 1 ч. калиевой соли (желто-веленые иглы) K[ReOgS] растворяется в 1,5 ч. воды при 20°. Довольно трудно растворим желтый тиопорренат таллия(!) TlIReO3S]. В растворе он постепенно разлагается, пб-видимому образуя более богатые серой соединения, например тетратиоперренат таллия(1) ТЦНё34}, темно-коричневый осадок.
Дисульфид репил ReS2 образуется как при непосредственном соединении элементов, так и при нагревании гептасульфида (например, в токе азота). Он образует черные, тригональные листочки с уд. весом 7,51, нерастворимые в соляной кислоте и в растворах сульфидов щелочных металлов. При более сильном пагревапии (при 1110’давление паров серы составляет 13 ммртст, при 1225°—$9 мм рт ст) он разлагается: ReS3= Re + S2; образование более низких сульфидов в качестве промежуточных продуктов не наблюдается (BilLz). Металлический рений в весьма незначительной степени образует с RcSs твердые растворы. Однако KeS2 способен растворять избыток серы с образованием твердых растворов, Поэтому очень трудно получить продукт, точно соответствующий составу ReS2.
Соединения рения с гало генам п
Обзор фторидов и хлоридов рения дан в табл. 28 на Стр. 254. Большинство галогенидов рения склонны к образованию ацидо-комплексов. А некоторые из них только в такой форме и устойчивы. В первую очередь это относится к бромидам и иодидам, о существовании которых в свободном состоянии еще очень мало известно. Их ацидо-соли по составу соответствуют соединениям хлора.
Фторид рения. Порошок рения реагирует с фтором при температуре около 125° с образованием гексафторида рения ReFs. Он выделяется в виде бесцветного газа, затвердевающего при охлаждении в желтую кристаллическую массу. Т. пл, 18,8’, т. кип. 47,6’. Упругость пара соответствует формуле ReFc. Можно приучить также оксифториды ReOF4 и ReO2Fs. Фторид семивалептного рения получить пе удалось (Ruff, 1932). При пропускании смеси ReFe и Н2 через нагретую до 200° трубку образуется зеленовато-черная масса тетрафторида рения ReF4 (уд. вес 5,38, т. пл. 124,5°). ReF4 легко растворяется в воде, при этом он шипит и разлагается. При растворении в 40%-пой плавиковой кислоте наблюдается темно-зеленое окрашивание; при добавлении KF из этого раствора кристаллизуется гексафтороренат(1У) калия K2[ReFe)4 Проще это соединение можно получить восстановлением KReO4 иодвдом калия в растворе плавиковой кислоты, Этозелопые октаэдрические кристаллы, изотиппыо K2jPtCl6).
Седьмая группа, периодической системы
257
Хлорид рения и хлороренаты. Ронин при нагревании соединяется с хлором с образованием пентахлорида рения НеС15. Наряду С: ним образуется НеС13 или Ве2С1е, от которого йептахлорид легко отделяется возгонкой в высоком вакууме, R0CI5 окрашен в темный коричнево-черный цвет. В высоком вакууме он возгоняется без разложения, в то время как при нагревании в атмосфере азота отщепляет хлор и переходит в RcoGIg. При нагревании в токе кислорода ReCl5 также отщепляет хлор и образуются ВеОС14 и ReO3CL В разбавленной соляной кислоте оп растворяется частично с образованием кислоты HjfReOCls], частично подвергается окислительно-восстановительной реакции [образование Н2 [ Re( !lfJ и H[ReOj]. С к он центр прованн ой соляной кислотой идет реакция ReCl5 + 2НС1’ = H3[ReCle] -j- 1/2С1а (Ceil ma пн, Biltz, 1933).
Кислота Ha[ReOCl5] малоустойчива. Соответствующие ей оксопентахлороре-наты.{У} MJlReOCltJ в растворе легко разлагаются вследствие гидролиза и окисли-тельно-восстановятельных процессов. По внешнему виду они похожи на гексахлоро-ренйты(1 V), однако в отличие от них обладают двойным преломлением и дают другую интерференцию рентгеновских лучей (Jakob, 1933; Holomann, 1937).
НеС14 в свободном состоянии неизвестен. Производная от пего (при присоединении 2HG1) хлоро рениеёая кислота [точнее, гекеахлорорейиееая(/У) кислота] Нг[ВеС1б] весьма близка но своим реакциям гексахлороплатиповой кислоте Н3 [ptCIg], но но сравнению с пей является менее прочным комплексным соединением. В водных растворах или в виде солей хлорорепатов(1У) опа является наиболее доступной из соединений рения с хлором. Ее можно получать различными способами, например растворением Re О 2 в соляной кислоте или исходя непосредственно из перрепатов, восстанавливая их в с ильп окис л ом растворе (НС1) с помощью HI. /Келто-зелепые октаэдрические кристаллы чистого хлорорената калия [гексахлорорсната(1У) калия] Ka[ReCla[ проще всего получить нагреванием тонкой емеси перрепата калия и иодида калия с концентрированной соляной кислотой. Эта соль изотипна K2[PtCJa]. В центральных растворах она гидролизуется. В соляной кислоте растворяется довольно плохо. Хлороренат цёвия Св2[ВеС1е]растворяется еще труднее.
Трихлорид рения H11CI3 или Пе2С1(;, образующийся при термическом распаде ВеС15 (си, выше), имеет красный цвет. Он легко растворим в воде и в нейтральных растворах быстро гидролизуется, выделяя гидрат полуторной окиси RooOj-tIRO. Б солянокислом растворе ReGl3 весьма устойчив и при обычной температуре пе разлагается даже при действии сильных окислителей, например перманганата и хлорной воды. В солянокислых растворах трихлоряд рения существует в виде комплексном кислоты H[ReClJ; при добавлении хлоридов щелочных металлов кристаллизуются хлороренаты(Л1) типа M1[ReCl4], При нагревании эти соединения диспропорциони-руют по схеме <>MTfReCb] = 3M|[ReCIe[ + Re -j- Re2Cl$ (Gcilman, Biltz W., 1932; I.u.W. Noddack, 1933).
Свежеприготовленный водный раствор трихлорида рения не сразу реагирует с нитратом серебра. Следовательно, он находится в растворе в педиссоциировапном состоянии. Поэтому он легко извлекается эфиром. Определение молекулярного веса в ледяной уксусной кислоте дает формулу ReaCiB. По-видимому, это комплексное соединение
С1 С1 СГ Re Re
CI Cl C1J
как предполагает Бильтц (1936), на основании сходства его спектров поглощения со спектрами И [ReClJ. В отличие от других комплексных неэлектролитов оно хорошо растворимо. Это обусловлено тем, что ропий в нем является координационно ненасыщенным и поэтому способен присоединять дополнительно молекулы воды.
Оксихлорид рения. Если на металлический рений подействовать смесью хлора с, кислородом, то образуется оксихлорид рения. Однако чистый дихлорид легче получать другим способом, например нагреваHKeMtReClg ила Не2С1а л токе кислорода либо взаимодействием ReCl2 с ПеДЭ^. Таким образом Б рук лом (Brukl, 1932) впервые был получен чистый триоксихлорид рения ReO3CI (бесцветная жидкость, f. пл. +4,5°, т. кип. 131°). При избытке ИеС15 образуется окситетрахлорид рения ReOCL, коричнев о-краспые лучистые иглы (т. пл. 29,3е', т. кип. 223°). В то время как ReO3Cl расщепляется водой на IIReO4 и НС1, гидролиз НеОС14 сопровождается окислительно-восстановительным процессом, так что наряду с HRe()4 и HG1 выделяется черным ReO2. В концентрированной соляной кислоте ReOCl4 образует комплексную кислоту H„[ReOCl6], соли которой, однако, очень мало устойчивы. Калиевая соль К2[НеОС1а|, синтезированная И. и В. Поддана ми, сразу после выделения ее из солянокислого Г Г Реми
258
Глава, 6
растнора окислилась по уравнению:
3K2[ReOCle] + 5Н2О = 2К[ПеО4] + К^КсСЦ] + 2К.С1 + 10НС1.
С аммиаком ВеОС14 образует ReO(NHa)2Cl2. Выше 400я это соединение разлагается на Re и ReO2. При внесении оксодиамидодихлорида в ледяную воду происходит обмен С1 на ОН с образованием диамидорениевой кислоты H2[ReO3(NH2)2]. В вакууме при 100° она отщепляет воду п переходит в двуокись диамидорения Re(NHa)2O2 (Brukl, 1933).
Карбонилы рения
Рений подобно марганцу и металлам побочных подгрупп VI, VIII и I групп способен давать соединения с окисью углерода (карбонилы). При этом наряду с карбонилами металлов, например пентакарбонилом рения [Re(CO)f,[2, образуются также залоге нокарбонилы металлов, например ReX(CO)5. Последние были впервые получены Гибером (Hieber, 1939).
Псптакарбонил рения [Ве(СО)5]2 получают действием окиси углерода па семиокись рения при температуре 250г и Давлении 300—400 ат:
Re2O7 + 17СО = [Re(CO)5]2 + 7СО2<
Он представляет собой бесцветные псевдокубические кристаллы, труднолетучие и мало растворимые в органических растворителях. При обычной температуре это соединение вполне устойчиво; выше 250° разлагается.
Галогенокарбонилы репин. Хлоро пент а карбонил рения ReCl(CO)j можно получить, папример, нагреванием ВеС15 с СО и Сц или КВеО4 с СО и СС14:
ReCl5+9CO-HCu=ReCl(CO)5-HCuCl(CO),
KRe04+8C0 + CCI4=ReCl(C0)5+C0Cl2-|-3CO2+KCl.
Если вместо ReCls берут К2[ПеВг6] или Кэ[Ве]в], то получают бромо- или иодопентакарбопилы. Галогепонентакарбопилы реп ня образуют бесцветные, мало растворимые в органических растворителях кристаллы. При обычной температуре они вполне устойчивы.
Как в пептакарбоннло рения, так и в га.тогепокарбонилах часть карбонильных групп может замещаться некоторыми органическими азотсодержащими соединениями [пиридином (Руг), о-фенацтролипом (Plilhrl)]. Получены Ко(СО)йРут2, ReX(CO)3Pyr2 (бледного желто-золеного цвета); Re(CO)aPhlhrl, В еХ(СО)й Ph thrl (желтые).
Аналитические сведения. Рений осаждается из сильно к целого раствора сероводородом в виде гептасульфида Re2S7,ne растворимого в сернистом аммонии. Особенно характерно для рения образование летучего окисла ReaO7 при нагревании ренийсодержащих веществ в токе кислорода. Основные окислы, препятствующие выделению семиокиси, вследствие образования лерренатов связывают добавлением Н3РО4 или SjO2. Образующуюся при растворении Rt‘2O7 в воде рениевую кислоту ННеО4 можно открыть в виде солей, имеющих характерные кристаллы.
Для микроаналитического определения рения, помимо пгрреиатов, весьма удобны также гексахлор о ренаты М2 [ReClel (Goilmann, 1937).
Наиболее падежным является определение рения на основании дуговых (аналитические линии 345,2; 346,1 и 346,5 -wp) или рентгеновских снелтроп (ер. стр. 250). Окислительное пламя бунзеповской горелки окрашивается летучими соединениями рения в блелпо-зелоный цвет.
В весовом анализе проще всего осаждать и взвешивать ренин в виде перрената нитрона С2оН16Л4- HReO41 а также осаждая его в виде сульфида с последующим окислением перекисью водорода [G е i 1 in а и n, Z. aaorg. Chem., 19а, 289 (1931)],
От молибдена ренин можно отделить при помощи 8-оксихииолипа. В фильтрате рений можно затем определять в виде перрената нитрона (Geilmann, 1931).
Гл а в а 7
ВОСЬМАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
МЕТАЛЛЫ ГРУППЫ ЖЕЛЕЗА И ПЛАТИНОВЫЕ МЕТАЛЛЫ
I Порядковый 1 | номер | Элемент | Символ Атомный вес Удельный вес Температура, °C Удельная теплоемкость Валентность
плавления кипеня я
26 Железо 1 Металлы Fe 55,85 7,86 1528 2735 0,1077 I, И, III, IV, VI
27 Кобальт | группы Со 58,94 8,83 1490 3100 0,0928 I, П, ш, IV
28 Инке ль J желоза NI 58,71 8,90 1452 2840 0,107 I, II, III, IV
44 Рутений V Легкие Пн 101,1 12,30 -2400 -4200 0,0553 II, III, IV, V, VI VII, VIII
45 Родий платиновые Ют 102,91 12,42 1966 -3900 0,0591 I, II, III, IV. VI,
46 Палладий J ме'£алп1’т Рй 100,4 12,03 1557 3170 0,0543 11, Ш, IV
76 Осмий 1 Тяжелые Os 190,2 22,7 -2700 —4600 0,0311 II, III, IV, VI, VIII
77 Иридий । платиновые Ir 192,2 22,65 2454 -4500 0,0309 I, II, III, IV, VI
78 Платина J металлы Pt 195,09 21,45 1774 -3800 0,0318 I, II, III, IV, VI
Общие сведения. Восьмая группа- включает три группы элементов, в каждую из которых входят по три элемента, расположенных друг за другом в периодической системе: элементы с порядковыми номерами 26—28; железо, кобальт и никель; элементы с порядковыми номерами 44—46’. рутений, родий и палладий; элементы с порядковыми номерами 76—78; осмий, иридий и платина. Три первых из перечисленных элементов чрезвычайно близки между собой; их объединяют в группу железа. Остальные шесть элементов называют платиновыми металлами по названию наиболее важного и распространенного из них элемента — платины, с которой они близки по свойствам и образуют комплексные месторождения. По удельным весам платиновые металлы можно подразделить на легкие и тяжелые (см. таблицу, приведенную выше).
Девять элементов восьмой группы располагаются следующим образом (цифры слева внизу обозначают их порядковые номера):
а б
1. Железо Кобальт
26 27
2. Рутений Родий
44 45
3. Осмий Иридий
76 77
Никель
28
Палладий
46
Платина 78
17*
260
Глава 7
Сходство расположенных друг за другом- элементов едва ли выражено в каком-нибудь другом месте периодической системы столь явно, как в этих трех рядах. Однако сходство элементов в вертикальных столбцах а, бив также очень велико.
Сравнение основных физических и химических свойств этих элементов показывает, что металлы группы железа более сходны между собой, чем с элементами, стоящими под ними; платиновые металлы, наоборот, обнаруживают большую близость по ^вертикальным столбцам. Поэтому при рассмотрении химических свойств этих элементов наиболее целесообразно объединять железо, кобальт и никель в группу железа, а платиновые металлы подразделять па три подгруппы, включающие каждая по два элемента, т. е. на три диады, каждую из которых образуют элементы, расположенные п одном вертикальном столбце. Классификация элементов восьмой группы приведена в табл. 29
Таблица 29
Восьмая группа периодической системы
А. Группа железа '
Железо
Кобальт
Никель
В, Платиновые металлы
'I диада
Рутений Осмий
II диада
III диада
Родий Иридий
Палладий Платина
Из трех металлов восьмой группы, расположенных слева (железо, рутений и осмий). первые два отдаляют от предшествующих им инертных газов (аргона н криптона) 8 элементов, и, как следует из теории строения атома, внешняя электронная оболочка осмия должна иметь строение, в основном аналогичное атомам железа и рутения (см. ниже)- В соответствии с этим максимальной (положительной) валентностью указанных элементов должна быть 8 Ц-. У рутения и осмия эта валентность действительно проявляется, в то время как железо известно максимум в шестивалентном состоянии. Вообще можно сказать, что здесь в каждом вертикальном столбце склонность элементов к проявлению высших валентностей возрастает сверху вниз (ср. данные, приведенные в крайней правой графе таблицы на стр. 259). В каждом из трех горизонтальных рядов тенденция к проявлению высших валентностей убывает слева направо.
Вполне аналогичные соотношения наблюдаются, если сравнить между собой не .wwauttuwiw, а наиболее часто встречающиеся валентности различных элементов восьмой группы: железо проявляет в основном валентности два и три, рутений — преимущественно четыре, между тем осмий имеет явно выраженную тенденцию проявлять валентности шесть и восемь. Кобальт в простых соединениях преимущественно двухвалентен, а и комплексных чаще всего трех валентен; родий преимущественно трёхвалентен и в простых соединениях, в то время как иридий в важнейших соединениях обычно четырех валентен. Никель почти исключительно двухвалентен; у палладия ага валентиость, хотя и является преобладающей, но пе в такой мере, как у никеля; в отличие от них платина в своих важнейших соединениях (гексахлороплатииовой кислоте и ее солях) четырехвалелтпа.
Восьмая группа, периодической системы
261
По вертикалиj как уже было отмечено при рассмотрении побочных подгрупп периодической системы, тенденция к проявлению более высоких валентностей возрастает с увеличением порядкового номера элемента. В побочной подгруппе III группы указанная закономерность проявляется очень слабо (ср, стр. 49); в соответствующих подгруппах IV, V, VI и VII групп, а также в VIII группе эта; тенденция выражается ум;в вполне четко, но и ограничивается подгруппами, содержащими только переходные элементы. В побочных подгруппах I п II групп этой тенденции не наблюдается. Более того, в побочной подгруппе 11 группы имеет место противоположная закономерность — по мере возрастания порядковых номеров элементы проявляют более низкие валентности.
Осмий по своим химическим свойствам, и особенно ио валентности 8+, очень близок рутению, несмотря па то что он в отличие от рутения не отделен 8 элементами от соответствующего инертного газа. Это обстоятельство связано с тем, что между осмием и предшествующим ему инертным газом (ксеноном) находится группа лантанидов. Появление 14 элементов, составляющих группу лаптапндов, сводится к заполнению 14 электронами 4/-оболочки, которая, условно выражаясь, «находится подкее-ноповой внешней электронной оболочкой». Из 22 (14 + 8) электронов, составляющих разницу в количестве электронов Осмия и ксенона, только 8 связаны вне «ксеноновой внешней оболочки» (они находятся на 5t/-yровнях и, вероятно, отчасти на бв-уровнях). Таким образом, общим для осмия и рутения (а также железа) является то, что в их атомах 8 электронов находятся «за внешней оболочкой предшествующего им инертного газа» [или, выражаясь точнее: в состав атомов названных элементов входят 8 электронов, имеющих более высокое главное квантовое число п или при одинаковом главном квантовом числе п более высокое побочное квантовое число I, чем у наиболее высококвантовой орбиты инертного газа (в нормальном состоянии), который в периодической системе предшествует данному элементу].
Общими свойствами всех элементов восьмой группы являются их металлический характер, цвет (от серого до беловато-серого), тугоплавкость, чрезвычайно высокие точки кипения, очень незначительный атомный объем (для элементов восьмой группы оп составляет лишь х/7—части атомного объема калия). Все эти элементы в более или мепее высокой степени обладают свойством окклюдировать водород и активировать его. Кроме того, они все отличаются значительной каталитической активностью. Общим свойством элементов восьмой группы является их способность проявлять в соединениях различные валентности и легко переходить из одного валентного состояния в другое. Общая у них и склонность к образованию комплексных соединений; при этом характерна их способность связывать не только NH3, но и СО и нередко даже NO, а также их исключительное сродство к радикалам CN. Все они образуют окрашенные соединения даже в состоянии свободных ионов (т. е. имеющих только водную оболочку). Гидроокиси элементов восьмой группы обладают слабоосновным, слабокислым или амфотерным характером. Сродство к кислороду в кая-:дом из трех рядов убывает слева направо. Элементы последнего столбца — никель, палладий и платина — по свойствам приближаются к элементам побочной подгруппы I группы периодической системы, обладающим характером благородных металлов. Общим свойством элементов восьмой группы является также их сильное сродство к сере. Оно выражается в образовании ими как простых соединений («соединений с главной валентностью»), так и соединений высшего порядка («соединений с побочной валентностью»), например многочисленных продуктов присоединения солен платины с органическими сульфидами. Сродство к сере в каждом п,з трех рядов возрастает в направлении слева направо. И в этом отношении свойства металлов восьмой группы приближаются к свойствам элементов побочной подгруппы I группы, для которых очень характерна склонность к соединению с серой в противоположность слабому сродству их к кислороду, что особенно справедливо для меди и серебра.
262
Глава 7
Температуры плавления в каждом из трех рядов восьмой группы убывают слева направо; в каждом из трех вертикальных столбцов они возрастают сверху вниз:
Fe Со N1
1528® ; > 1490° > 1452е
Л Л Л
Ru Rh Pd
2400° : > 1966° > 1557'
Л Л Л
Os 1г Pt
2700° : > 2454° > 1774'
Аналогичная закономерность наблюдается в отношении изменения «атомных объемов) и атомных радиусов, вычисленных на основании межатомных расстояний в решетке:
А толстые объемы, см3 Л толстые радиусы, А
Fe Со № Fe Со Ni
7,10 : > 6,67 ; > 6,59 1,2412 < ; 1,2518 > 1,2458
Л Л Л Л Л Д
Ru Rh Pd Ru Rh Pd
8,27 < 8,29 < £ 8,87 1,3390 < z 1,3450 < Z 1,3755
Л Л Л Л Л Л
Os 1г Pt Os 1г Pt
8,38 < 8,53 < ' 9>Ю 1,3527 < z 1,3572 < Z 1,3873
Твердость отдельных металлов восьмой группы характеризуется следующими величинами (по шкале Мооса):
Fe Со 4,5 5,5 Ni Ru Rh Pd Os 3,8 6,5 — 4,8 7,0 Ir Pt 6,5 4,3
Кристаллическая структура металлов восьмой группы Fe Со N i
Кубическая объемыоцеп* Таирова иная решетка * сг. = 2,8664 А Гексагональная плотнейшая упаковка » а = 2,5063, е= 4,0795 А Граиецентрированная кубическая решетка * (кубическая плотнейшая упаковка) а=3,5238 А
Ru Rh Pd
Гексагональная плотнейшая упаковка а = В,7085, с- 4,2816 А Граиецентрированная кубическая решетка (кубическая плотнейшая упаковка) а = 3,8044 А Граиецентрированная кубическая решетка (кубическая плотнейшая упаковка) а — 3,8907 А
Os 1г Pt
Гексагональная плотнейшая упаковка 0=2,7353, с = rLl3IS 1 А Гранецентрированная кубическая решетка (кубическая плотнейшая унакой ка) а= 3.8389 А Гранецентрированная кубическая решетка (кубическая плотнейшая упаковка) а = 3,9239 А
В табл. 30 сопоставлены радиусы ионов различных степеней окисления элементов VIII группы^
* Эти данные относятся к модификации, стабильной при обычной температуре. Структура других модификаций указана на стр. 280 (ср, также рис. 36 стр. 283), стр, 312 и 332;
Восьмая группа периодической системы
263
Таблица 30
Эффективные ионные радиусы элементов VIII группы
Ион Радиус, Л рез+ соа+ №2+ 0,83 0,82 0.78 Ru4+ Rh3+ рфн 0,65 0,68 0,78 (KZ=6) 0s4+ Ir3+ Pt3+ 0,67 — — (KZ=0)
Ион Радиус, А Fe3+ Со3+ Nis+ 0,67 - — Ru»+ №+ Pd«+ — — 0,66 OS8+ Ir4+ Pt4+ 0,54 0,66 0,52 (KZ=6)
Сплавы. Как следует на данных табл. 31 (стр. 264) и 32 (стр. 26'6), металлы VIII группы обнаруживают ярко выраженную склонность к образованию сплавов. Лишь со щелочными и щелочноземельными металлами, насколько известно в настоящее время, опи пе образуют ни твердых растворов, ни соединений, С металлами других главных подгргупп элементы VIII группы образуют многочисленные соединении и обычно постоянного состава. Это связано с тем, что взаимная растворимость в твердом состоянии у металлов VIII группы и элементов указанных главных подгрупп, как правило, очень мала или вообще отсутствует (см. табл. 31). Соединения подобного же типа, что и с металлами главных подгрупп, элементы VIII группы образуют и с рядом неметаллов, в первую очередь с В, С, Si, N, Р, As и отчасти с S.
Об отношении элементов (Ru, Os, Rjli и I с), не приведенных в табя. 31, к металлам главных подгрупп известно еще очспь мало. Согласно даппым Роде (1929), Rh образует с Bi инконгруэптпо плавящиеся соединения НЬВц, PihBi, и RhBi при отсутствии твердых растворов, В свинце Rh и 1т ограниченно растворимы в ;кидком состоянии, а в твердом — вообще пе растворимы.
Взаимная. растворимость элементов VIII группы и металлов побочных подгрупп зависит от того, является ли второй компонент переходным металлом или элементом другой побочной подгруппы. Металлы VIII группы образуют многочисленные твердые растворы по только между собой, но и с другими переходными элементами (ср. табл. 13, 17 и 25). Только в наиболее далеко стоящих в периодической системе переходных металлах (элементах побочных подгрупп III н IV групп, а также iVh и Та) в твердом состоянии они растворимы лишь весьма ограниченно (см. табл. 9, 13 и 57). Аналогичным образом элементы побочных подгрупп I и II группы (см. табя. 32), как правило, не обладают или обладают лишь в очень малой степени способностью к образованию твердых растворов. Исключение из этого правила составляют, правда, элементы побочной подгруппы I группы и, вероятно, цинк по отношению к Ni, Rd и Pt.
Способность металлов VIII группы к образованию соединений с элементами III — VI групп одинакова в главных и побочных подгруппах (за исключением V и Сг). С металлами побочной подгруппы II группы, и в первую очередь с цинком, почти все металлы VIII группы могут давать соединения типа фаз Юм-Розери (ср. стр. 40). С металлами остальных побочных подгрупп, а также с V и Сг, как правило, пе образуется соединений; однако в некоторых случаях в таких сплавах появляются фазы, обладающие сверхструктурой.
Некоторые типы диаграмм состояния бипарных систем, содержащих железо (как наиболее важный металл VIII группы), приведены па стр. 284—286.
А. МЕТАЛЛЫ ГРУППЫ ЖЕЛЕЗА
Железо, кобальт и никель в металлическом состоянии или в виде соединений обнаруживают между собой очень большое сходство. Железо по своим химическим свойствам примыкает к предшествующему ему в периодической системе марганцу, который и в природе встречается в основном вместе с железом, а никель до некоторой степени приближается к следующему за ним в периодической системе элементу — меди. Общим свойством всех элементов группы железа является их положительная двухвалентность в простых солях. Железо, кроме того, бывает в них трехвалентным даже чаще, чем двухвалентным. Кобальт тоже легко переходит
Образование соединений а твердых растворов металлам
(Обозначения те же, чт
Be Mg Ca B Al Go In TI C Si
Fe тв.<Ух> BexFe (t > 5) BesFe Be2Fe me. 0? .и<?.<(со me. 0? Ci me. > 0 Fe2B* 1389° FeB 1540° me.<Cca |Fe3Al] FeAlQ FeA]« Fe2Al5 FeA]| me. > P 0 - ж. O? 0 me, 0 Fe3G Fe2C? Мета-стаб? mei<Z<^ Fe3SU~ 1030°^ FeSi 1410= FeSi2 1220-
Со me. О ВежСп Be Co me. 0? Сплав? Co2B GoB C0B2 me. j> О CoAl 1628° WAI? 1170° Co All 943° — Co2In jk.<Z co 0 me. ~ 0 me. > 0 Co3C Со2С[]? 225° тв.<сх Co3Sr 1210° Co2Si 1327° CosSijT 1214° CoSi 1440s CoSi, 1277° CoSi3 1306°
Ni ma. >. 0 Be21Ni0 1262° BeNi 1472° me. 0 Mg2Ni* 760° MgNi2 1145° me. 0? Ni2B 1230° IN13B2 nW NiB* 10 2i'i° Ni2D3 12 Mj: me. > 0 Ni3Al* 1390° NiAl 1640° NiaAl| 1182° NiAJJ 842° me. <^00 NigGa 1220° NiGa Ni2Ga3 NiGa4 Ni3Tn Ni2In Ni2In3 NilD3 'Hc.<^ca 0 me. ~O Ni3C M e тает аб. me. G Ni3Si* 1163° NijSb 1255° Ni2Si 1290° Ni3S ur 845°“ NiSi 995° NiSi| 1000°
Pd me. — 0 BtiPd 1465° Be4Pd* 1200° BePd| 1090“ BePd? ЭйО'3' - me 0 Pd2Al PdAl PdAl3 —— Pd2Iri Pdln Pd2In5 — me. 0 0 me. 0 Pd2Si -1490° PdSi 9 00°
Pt me, '> 0 Соединение Соединение Соединение Соединение me, О PtA] PtAl2 PtAlf 780“ P tGa Pt2Ga3 PtGa3 Pt2InB Ptln3 Plln3 me. 0 PIT J 685° me. 0 Сплае Pt2Si-PtSij-
Таблица 31
восьмо» группы с элементами главных подгрупп в табл. 9, стр. 66)
Gc Sn PJ) N p As Sb Bi l. ! s Se Te
1 1 1 me. )> 0 Fe?Sn* 900“ FeSnH 800° FeSn* 496» LMC. 0 me. 0 0 me. >> 0 Fe4NQ Fe^K me. 0 Fp3P* li'tifi’ fe2P 136 5 = FeP FeP2 me. > 0 FesAs 919= Fe3As2 FeAs 1030= FeAs2 ms. 0 Fe3Sb2 1010° FeSb FeSb* 728’ rwc. 0 me. 0 0 me. —0 FeS 1208° FeS2 689’ (Распад на F₽S п газо-обр.S) me ,<co FeSe FeSe2 FeTe FeTe2
: Co2Ge : 1200° CoGcf] ! 082° GoGe2[] 842’ me > 0 ж.< co me. 0 0 me. — 0 Co3N Co2N? Co3N2 14C.<OO me. 0 Co2P 1386= GoP CoP3 me. — 0 Co5As* Co2As* Co3AsJ CoAs 1180° me. 0 CoSb 1191 = CoSb£ 897’ JfC.<^OO me. 0 0 me. — 0 C04S* S30= C0gS* 832° CoS Co3S4 CoS2 COSe CoSe2 CoTe СоТод
CoySn2 1151’ CoSn* 943“ CoSn* 525=
— me. '> О. ж-,<, co me. 0 Ni3N Ni3N2? me, 0 Ni3P* 970’ Ni,P2 1175= Ki2P 1110= Ni6Pa? NiP2 NiP3 me. 0 AijAs-j 998= Ni3AsJ? NiAs 970= NiA=a me. ~>Q Ni3SbQ 698’ Ni5Sb2 1162= Ni7Sb3r| 588’-NiSb 1153’ Ni2Sb3g Ni3Sb£ 626° me. — 0 NiBiif 469° NiBi* 655° Ni3S? Ni6S* NiS Ni3S4 NiS2 "— NiTe NiTe2
1 t" i" Ni3Sn2 1264° Ni3Sn4 794’ ms. 7> 0 0 NiSe NiSe2
Вероятно, существует несколько соединений /»«.<co Pd3Pb 1220= Pd3Pb* 830 = PdPb* 596“ PdPb2 454“ me. 0? 0 me. 0 PdgP* 897= P63P 1047 = Pd5Pa* 860° PdP2 -1150= Pd3As2? PdAs2 msZ>0 Pd3Sb 1182= Pd2SbQ 8 50= PdSb 800= PdSb2 680“ Соединение me. 0 Pd4S[] 761» Pd5S2? PdS 970° PdS2 PdSe? PdSe2 PdTo PdTe2
m«.>0 Pt3Sn* 1305’ PtSn 1300= PtSn* 848’ PtSnJ 745 = me. 0 Pt3Pb* 915’ PtPb* 705’ PtPb? 360’ 0? яс.<оо me. 0 P^PyO 590“ PtPa >1500’ me. 0 Pt2As3? PtAs2 me . )> 0 Pl4Sb*? .752= Pt5Sb?? -637 = PtSh* 1050’ PtSb2 1226° me,~0 PtBi PtBi3? PtS Pt2S3? PtS2 PtSe? . PtSe2 PtTc Pt Те 2 -1250’
Таблица 32
Образование соединении и твердых растворов металлами VIII группы между собой и с элементами побочных подгрупп I и II групп (Обоуначепия те же, что и в табл. 9, стр. 66)
Со Ni 1г Pd Pt Си Ag Аи Zn Gd Hg
Fe ТВ. '—-СО 0 ТВ. '—СО [Ni3Fe] 600° Сплав ТВ, ~со [Pd3Fe[ [PdFe] ТВ. со |ТТйс] ТВ. Ъ- 0 0 яс. 0 тв, 0 0 тв, < со Fe3Au Q тв. >• 0 FOjZn^ FeZn? яс. О? тв. 0 0? тв. — 0 0
Со ТВ, со 0 — ТВ. со 0? ТВ* со [PICoj 825° Р1Со3 □? тн. > 0 0 ж. 0 тн. 0 0 тв. >0 б ТВ, < со c?Zn* Co5Zn^ тв. 0? Сплав! Co3Hg10?
Ni —- тв, <х> 0 ТВ, со [Ptm3] ТВ. со 0 ? ° V л С 8 ТВ. со 0 ТВ. < со NiZn* l(j43° N isZn2t 882° Ni2Zn15? тв. 0? Ш5Сс1*г тв, — 0 0
1 Ли Сплав — — тв. 0 тв, ~ 0 Сплав — —
th С/мги IUhIt] — 1 i n 0 1
Rh TB. co 0 TB. co 0 TB, co [Bb3Cw] [RhCuJ [RhCn3]
Ir — TB. co 0 Сплав
Pd ТВ» co 0 tb. co [PdCu] [PdCua]
Pt TB. co (PtC u] 800» [PtCu3] 500»
г». - 0 1 f: si .-j i Г<т 0?
ТВ, > 0 0 ТВ. > 0 * —“
RhgZiigf RhsCd21
ж. 0? Сплав тв. 0 Сплав — Сплав
ТВ. оо 0 ТВ, со 0 — Сплав Сплав
Р dftZngi PdjCdai
тв. <оо ЬЧзАД Q PtAg □ piAg3 а 960° ТВ. оо 0 Р^2пг1 тв. О PtCd2 725° ptcdS (я >4) G15° PtHg2?
268
Глава 7
в трехвалентноё состояние, а для комплексных соединений эта валентность даже наиболее характерна; в простых солях кобальт в противоположность железу проявляет валептпость три очень редко, а николь, по-видимому, этой способностью уже не обладает. Склонность этих элементов к проявлению валентности один увеличивается от железа к никелю, в соответствии с возрастанием их близости к меди, легко образующей однова
лентные соединения.
В то время как железо подобно марганцу сравнительно легко переходит в шестивалентное состояние [ферраты (VI)], максимальная валентность кобальта и никеля — четыре', они образуют двуокиси, легко разлагающиеся с выделением кислорода и образующие с окислами активных металлов малоустойчивые двойные окислы или оксо-соли.
।
Окиалы ЕеО, СоО и NiO кристаллизуются подобно МнО по тину поваренной соли (т. I, стр. 237, рис. 44), а гидроокиси Fo(OH)3, Со(ОН)2 и Н1(ОН.)2 подобно Мп(ОН)2 но тину брусита (т. I, стр. 293, рис. 61):
FeO СоО NiO а = 4,29 4,24 4,17 А
Fe(OH)2 Со(ОН)2 Ni(0H)2 а=3,24 3,19 3,07 А
с =4,47 4,66 4,60 А
Слоистую структуру, аналогичную гидроокисям, имеют хлориды двухвалентных металлов группы железа. Их решетка отличается от решетки брусита только тем, что отрицательно заряженные ионы образуют кубическую плотнейшую упаковку, а не гексагональную, как в брусите. Этот структурный тин (хлористого кадмия) был установлен для FeCl2, €oG12, NiCl3, а также Для MnCl2, ZnCl2, CdCl2 и MgCl2, а кроме того, для NiBr3 и NiJ2. .
Моносулъфиды Fe, Со и N1 (а также мопо се лепиды, мопотеллуриды н моноанта-мониды) кристаллизуются по типу NiAs (см. стр. 337). Значения параметров соответствующих элементарных ячеек сопоставлены в табл, 33. Некоторые соединения, приведенные в табл. 33, встречаются и в других модификациях (ср. стр. 317 и 336),
Таблица 33
Параметры гексагональных ячеек сульфидов, селенидов, аитпмопидов и других элементов группы железа
(d— кратчайше^ расстояние Fc > Я, Со <—> S п т. д.)
FcS р-CoS fl-NiS FeSe (.oSe 6-NiSe FeSb CoSb NiSb NiAs
Л — 3,43 3,37 3,42 3,61 3,59 3,66 4,06 3,87 3,92 3.61A
С — 5,86 5,14 5,30 5,87 5,27 5,33 5,13 5,19 5,11 5,03A
d= 2,45 2,33 2,38 2,55 2,46 2,50 | 2,67 2,58 2,60 2,43 A
Среди дисульфидов следует отметить FeS2, существующий в двух модификациях (пирит я марказит). CoS2 и NiS2, а также диселениды CoSe2 и NiSe2 кристаллизуются но типу пирита, в то время как FeSe2 и FeTe2 имеют структурный тип марказита. NiTe2 кристаллизуете и в ретпетке типа брусита. СоТе2 встречается в виде двух модификаций — тина марказита и брусита. Между моно- и дител л у рядами Ni и Со существует непрерывный переход, аналогичный имеющему место между TiTe и TiTe2 (ср. стр. 64).
Общим свойством железа, кобальта и никеля является их способность абсорбировать в мелкораздробленном состояния значительные количества водорода. Как показали Шленк и Вейхсельфельдор (Schlenk, Weichselfelder), действуя водородом на эфирные раствори FeCL>, СоС12 и NiCE*, содержащие фенилмагпийбромид, можпо получить
* Указанные хлориды в эфире нерастворимы. По-видимому, автор имеет в виду взаимодействие их суспензий с эфирным раствором C6H5MgBr.— Ириму персе.
Восьмая, группа периодической системы
269
гидриды стехиометрического состава: FeH3, СоН2 и NiH6. Если исходить из три хлорида железа вместо д их Дорида, то, по данным Вейхссльфельдорп, образуется еще более богатый водородом гидрид железа FeIItt. Однако существование этих гидридов точно еще не установлено.
Очень интересно проследить связь между стабильностью наиболее устойчивых комплексных соединений металлов группы желоза и степенью окисления центрального атома. В то время как у железа комплексный ион [Fo(CN)6l4~ с положительно двухвалентным центральным атомом является более стабильным, чем [Fe(CN)6]3-, в котором железо имеет заряд ЗЦ- , кобальт как раз в трс^валентнолг состоянии склонен к образованию наиболее прочных комплексов. В присутствии веществ, образующих с ним прочные комплексы, кобальт исключительно легко окисляется до трехвалентного состояния. Никель в форме некоторых прочных комплексных соединений даже при действии таких не особенно сильных окислителей, как Оа и Н20з, сразу же переходит в челшрехвалентной состояние.
Соотношения между степенью окис лепил центрального атома и стабильностью комплексов становится понятными при рассмотрении их с. позиций теории, развитой Полингом для комплексных соединений переходных металлов. В данном случае речь идет о гюмплекеав внедрения. В таком ионе, образованном присоединением шести CN-групи к одному Fe2+ по схеме
Ее8Ц_+ 6CN'—
NCk ZCN‘ NC-*Fe*-CN NCZ XCN.
4'
NC\ /CN' NC-^Fe^CN .NCT XCN.
4-
центральшдп атом лпе оболочки аргона имеет еще 18 электронов (3(i104s24/>6), т. е. электронную конфигурации), аналогичную криптону. Различие появляется лишь вследствие гибридизации в молекуле комплекса трех 4р-уровней и 4®~уровня из окружения центр ал тит ого атома с двумя ЗаУу ровнями (ср. т. I, гл. И). Энергия резонанса, освобождающаяся за счет такой гибридизации, может быть затрачена па спаривание четырех (/-электронов, пеона репных в свободном ионе Fe2*. Сказанное справедливо пе только для комплекса [FefCNJe]’-, являющегося производным Fe11, но и для соответствующих соединений CoJI1, у шести лигандов которых каждая пара электронов обобществлена с центральным атомом. То же относится и к соответствующим комплексам внедрения Ni*\ Ввиду того что помы Fe2+, Со;1+ и Ni4+ имеют одну и ту же электронную конфигурацию, она сохраняется и в их производных — комплексах внедрения (см. табл. .34, стр. 270). Как показал Хибер [Н i е b е г, Z. anorg. Chem., 269,12 (1952)], чедырехвалентный никель образует комплексные соединения лишь с теми лигандами, сродство которых по отношению к никелю достаточно велико. Для окисления Ni11 до NiIV необходима затрата особо большого количества энергии. Это видно из сравнения потенциалов ионизации элементарных ионов. В водном растворе они составляют
Fc” —> Fe"" Со” —•> Со'" Ni” —> Ni*’1’
4-0,77« 4-l,S4e 4-2,1а*
Положительные зпачЬппя потенциалов ионизации свидетельствуют о том, что во всех случаях для перехода элементарного иона, находящегося в растворе, от более низкой к более высокой степени окисления должна быть затрачена энергия. Тогда сраму становится ясной большая стабильность иона |Fe(CN)e]4- по сравпеишо с [Fe(CN)e]3-. Итак, энергия, которая должна бытт, затрачена па освобождение двух Д-уровней центрального атома для присоединения валентных электронов лигапдбв, в обоих случаях практически одинакова. Кац видно из табл. 34, и у Ferl, и у Fe111 для образования
* Значение потенциала ионизации Ni" —Ni”" найдено измерением окиелм-телыго-иоссгановнтельного потенциала реакции Ni" 4- 2ИгЬ ~ КЮ2 4П‘ 4- 2е 4" |- 1,75 л в предположении, что различие между последним и потенций.том ионизации элементарного иона примерно то же, что между соответствующими потенциалами у свинца (ср. табл. 112, т. I).
270
Глава 7
, Таблица 34
Электронная конфигурация металлов группы железа в элементарном состоянии 11 в комплексных нонах
(Электронные епины обозначены стрелками; спины, участвующие в образовании связей, обведены рамкой)
Ион Электронная конфигурация
Fe3+, Со0+, Ni4+ Конфигурация аргдна 4~ 3d*
П ? 1 LLLL
[Fen(CN)4*- Конфигурация аргона + 3d1* 4 b* dp*
ti|n|n)rnHL tl tl Tl|Tl
[Coln(CN)e]3- Конфигурация аргона + 3d” 4e* dp*
tiltl u in LLL tl tl II Я
" S 1 /8 1 >iiv< 1 RZ 1 XR S — Конфигурация аргона -р 3d” de3 4p?
ti ti ti ti ti ti tl Tl n
Fei+ Конфигурация аргона 4 3d*
t |и I т t t
[Fe^’fCNjop” Конфигурация аргона 4- 3d* 4tfl dp*
п|п| г |in n ti npn 111
Co3+ Конфигурация аргона 4 3d?
ti|ti| т 11t U 1
[Cou(CN)ef- Конфигурация аргона 4 3d1® 44* dp* dd
П ti|u|ru|u tl Tl|Ti1tl t
Nii+ Конфигурация аргона 4 3d*
11 n 11T
S S\ I ZS 1 >Ni"( | Rz t 4R S 4— Конфигурация аргона 4 3d” de3 dp* dd*
U ti ti •Ti ti ti tl ti Tl T t
Kr Конфигурация аргона 4 3d” da1 dp*
U it ti tl ti tl Tl Tl n
комплекса впедрепия должны быть спарены два d-электр он а, не спаренные в элементарных ионах. Так как в комплексном ионе [FeII(CN)e]4- имеются только спареяные-1>лектропы, он диамагнитен. Комплекс [FeIli:(GN)e]3_ содержит один неспаренный электрон, и поэтому оп парамагнитен.
В то время как процесс восстановления Fe'"—> Fe” связан с освобождением энергии, в случае окисления Go" —к Со"" энергия поглощается. Эти изменения в содержании энергии тем больше, чем выше значение потенциала ионизации. Аналогичным образом факт значительно большей устойчивости иона [Coni(CN)6]3- но сравнению с ионом [Co^CNJe]4-, по Полингу, обусловлен тем, что у Со11 два d-уровня освобождаются для валентных электронов лигандов, а один электр оп с 3d-у ровня должен подняться на 4й-уровень. У Ni11 для образования комплекса (Ni11 (CN)6]4“ два электрона.
Восьмая группа. периодической системы
271
должны подняться с 3<7- на 4с7-уровень. Поэтому в случае Ni11 может сразу образоваться комплекс внедрения [Nin(CN)4S3-. Благодаря спариванию обоих Зй-электро-нов, не спаренных в ионе Ni2+, освобождается один Зй-уровень и присоединяются два валентных электрона CN_-rpynn. Остальные шесть электронов располагаются па 4s- И ^р-уровнях. Так как в образующемся комплексе (Nt(CN)^]2" восемь валентных электронов имеют конфигурацию Зй24,?24р4, в соответствии со сказанным в гл. 11, т. I, в этом случае осуществляется плоское расположение лигандов, в то время как комплексы, приведенные в табл. 34, построены октаэдрически. Комплекс [Nt(CN)J2-, так же как и [Fe(CN)e]4~ и 1Со(СМ)й]3-, диамагнитен, во всех перечисленных комплексах все электроны спарены.
Этп соотношения справедливы не только для указанных цианокомплексов, но и для других комплексов внедрения металлов группы железа с шестью лигапдамп, все электронные пары которых участвуют в образовании связей. Стабильность различных комплексов зависит, конечно, и от вида лигандов. Например, ионизационный потенциал комплексного иона [Co(CN)6]"' составляет—0,83 s, a [Co(NHs)g5"‘-/—0,1 е. Тенденция к образованию комплексного иона [Со(С№6Г'' так велика, что комплексный цианид кобальта(П) разлагается водой с вы де лепном водорода. Комплексный нон [Co(NH3)6]”, напротив, в отсутствие кислорода воздуха абсолютно устойчив в водном растворе. Однако в токе кислорода он в соответствии с низким значением своего потенциала иопизации сразу переходит в ион [Co(NH3)e]"’.
ЖЕЛЕЗО (FERRUM) Fe
Распространение в природе. Железо является наиболее распространенным на земном шаре тяжелым металлом. Встречается опо, однако, в земной коре почти исключительно в виде соединений. В самородном состоянии в горных породах Fe встречается очень редко; впрочем, в небольших количествах железо иногда вкраплено в базальты. Оно образует главную составную часть многих метеоритов, предполагается, что и ядро земного шара состоит главным образом из металлического железа. В метеоритах железо находится в виде сплавов с никелем, содержание которого в них колеблется в интервале 5 и 20 вес. %. Нередко в метеоритном железе содержится сульфид двухвалентного железа (троилит), фосфид и окислы железа.
Важнейшими промышленными рудами железа являются его окислы: красный железняк Fo203, магнитный железняк Fe30i, бурый железняк Fe2O3H2O, или FeO(OH), а также его карбонат — железный шпат FeCO3.
Эш руды встречаются почти во всех странах, яо не всегда в удобном для переработки виде. Другой разновидностью бурого железняка, также содержащей фосфорную кислоту, является болотная руда, встречающаяся в Швеции, Голландии и л Северо-Германской низменности. В то время как шведская болотная (озерная) руда после высушивания при 100° содержит 46% железа, болотная руда голландского и северогерманского происхождения обычно содержит в значительных количествах песок н органические вещества. Поэтому она малопригодна для переработки на железо; благодаря исключительной абсорбционной способности по отношению к сероводороду эту руду применяют в основном для очистки светильного газа.
Наиболее распространенным в природе соединением является железный колчедан FeS2. Однако из этой руды нельзя выплавлять железо без предварительной обработки вследствие высокого содержания в ней серы, а также и из-за присутствия сульфидов меди И примеси других мешающих тяжелых металлов. Том: не менее обожженный железный колчедан («колчеданные огарки»), остающийся при производстве Сер пой кис-поты, перерабатывается нд железо только после предварительного удаления из него методом рафипировапия некоторых примесей, которые сами пр себе часто представляют значительную ценность (кроме меди и цинка, в ном иногда содержатся также серебро и золото).
Значительные количества железа содержат также многие силикаты. Продукты их выветривания вымываются и уносятся реками, осаждаясь в виде ила и глип, содержащих окись железа и песок.
272
Глава 7
Исторические сведения. Применение железа известно с древнейших времен. По-видимому, его стали употреблять одновременно с бронзой. В древности его выплавляли так называемым кричным способом (народы, находящиеся на низших ступенях развития, используют этот метод и в настоящее время). Железную руду в смеси с большим избытком древесного угля закладывали в плоские ямы; затем уголь зажигали и раздували огонь мехами. Таким путем получались более или менее связные болванки (крицы) колкого железа, которые затем обрабатывали молотом. В средние века ямы иля плоские очаги для получения железа постепенно стали заменять небольшими шахтными печами, из которых и развилась современная доменная Печь. В XIV в. мехи стали приводить в действие силой воды, Достигнутое благодаря атому значительное повышение температуры .печи дало возможность получать железо, более богато' углеродом, т, е. чугун. Несмотря на то что такой чугун и по поддается ковке, вскоре научились превращать его в ковкое железо путем вторичной переплавки при. сильном продувании — «фршпеванпю. Производство железа значительно развилось в конце XVIII в., когда благодаря открытию паровой машины и строительству железных дорог спрос на железо начал чрезвычайно быстро расти. Недостаток в древесном угле обусловил необходимость использования ископаемых углей в виде кокса — горючего и восстановительного агента. Во второй половине XIX в. фришевание подверглось существенным улучшениям: была введена продувка в конвертере (процесс Бессемера, 1855 г.; способ Томаса—Джнлькриста, 1878 г.) и регенеративная плавка (метод Сименса — Мартена, 1865 г.). Впоследствии к этим способам присоединилась еще плавка в электрический печи, применяемая для получения высокосортных сталей (электросталей).
ПОЛУЧЕНИЕ ЖЕЛЕЗА
Химически, чистое железо можно получать восстановлением чистой окиси железа (последнюю для этой цели лучше всего готовить нагреванием на воздухе оксалата железа) водородом или электролизом водных растворов солей двухвалентного железа, например оксалата аммония-же-лёза(П). В технике в настоящее время его получают в основном при термическом разложении нентакарбонила железа.
Так называемое «карбонильное железо*, получаемое этим методом, содержит в первую очередь некоторое количество растворенного углерода и кислорода. Эти примеси можпо устранить соответствующей дополнительной обработкой, s, Р, Си, Мп, Ni, Со, СТ, Mo, Zп п Si не удалось обнаружить в карбонильном железе даже наиболее чувствительным!! аналитическими методами. В зависимости от условий разложения карбонила жглеао образует равномерное зеркало, слой, или получается в виде пористого продукта (аналогичного электролитическому металлу, выделяющемуся из водных растворовI, или, накопсп, плотного, мелкозернистого порошка, из которого прессованием при п,и репанин до IUOiT («спеканием») можно получить исключительно Гомогенные фасонные детали. Наконец, таким путем образуются объемистые хлопья железа, напоминающие вату, 1 л которых весит менее 10 г. В таком виде Железо оказывается настолько реакциоппоспособпым, что вспыхивает даже от искры.
Несмотря на то что химически чистое железо приобретает все большее значение в технике.(ср. стр. 281), несравненно более важным является процесс получения обычного технического железа, представляющего собой не чистый металл, Э: сплав железа с углеродом. Кроме того, техническое железо обычно содержит еще целый ряд других веществ, часть из которых к нему добавляют намеренно, а остальные являются примесями, в большей или меныией степени ухудшающими качество железа. Свойства железа в значительной степени зависят от присутствия углерода и других примесей, несмотря на то что их концентрация в большинстве случаев очень мала. .Кроме того, свойства железа сильно зависят от предварительной обработки соответствующих сплавов.
Процесс получения различных сортов технического железа разделяемся на два этапа-. первый состоит в восстановлении железной руды до металла (чугуна), второй заключается в удалении из железа вредных примесей и добавлении компонентов, изменяющих качества металла, для получения
Восьмая группа периодической системы
273
определенных технических сортов железа, например ковкого железа или стали. Первый из этих процессов — выплавку железа из руды — производят в доменных печах; второй процесс, «фришование» (т. е. переработку чугуна в ковкое железо, сталг, и т. Д.), раньше осуществляли в кричном горне или путем пудлингования; в настоящее время для этого используют главным образом конвертирование и процесс Сименса — Мартена, а для специальных сталей, кроме того, тигельную плавку и плавку в электропечах.
Доменный процесс- Выплавку чугуна обычно осуществляют в домнах*, которые представляют собой шахтные печи высотой 20—25 at, сло
женные из огнеупорного, шамотового или динасового кирпича (схему см. рис. 29). Сверху в доменную печь загружают окисную железную руду, измельченную до некрупных кусков; при загрузке ее послойно перемешивают с коксом; снизу через фурмы под давлением поступает предварительно нагретый воздух, в котором сгорает кокс. Образующаяся при горении кокса окись углерода восстанавливает железо из окислов:
ЗСО 4 У е2О3=2F 6 +ЗСО3+ + 5,7 ижм**. (1)
Реакция (1) обратима. В соответствии с принципом Ле-Ша-тельс, равновесие ео смещается влево при повышении температуры. Поэтому реакция (1) осуществляется преимущественно в верхней, менее нагретой части домны; часть Ре2О3 восстанавливается в этой зоне лишь до окиси желе-за(Н).
Fe2O3+СО =2FeO + СО2-
(Более подробные снедения об этом см. ниже.) В иижней части домны, нагретой до очень высокой температуры, восстановителем является непосредствен до углерод:
FeO + C+34,5 кхал=Ре + СО. (2)
Температура в нижней части печи настолько высока, что железо плавится и стекает вниз. Пространство, освобождающееся в связи с этим, а также вследствие сгорания кокса, непрерывно заполняют сверху свежими загрузками.
* Для производства чугуна вместо обычных домеппых печен с дутьем можно применять и электропечи. В этом случае углерод является восстановителем, а нагревание осуществляется электрическим током. Электропечи целесообразно строить не как доменные, а по образцу пи.чкошахтных печей [ср. Z. Elektrochem., 42, 337 (1936)]. Их применение вообще возможно лишь в страдах, бедных углем, по имеющих гидроэнергетические ресурсы. Выплавка чугуна в электропечах во всем мире составляет мелсе 0,1% общего производства.
* * Тепловые эффекты в этом и следующих уравнениях относятся к обычной тем пературе.
18 г. Ремп
274
Глава 7
Если в руде железо содержится в виде карбопата, то его перед загрузкой переводят обжигом в Fe2O3: 2FeCOs + 1/2О2 *= Ёе2О3 -f- 2СО2. Порошкообразные руды смешивают со связующими веществами и спрессовывают' полученную массу в куски нужного размера (брикетируют). Для удаления пустой породы, почти всегда имеющейся в железных рудах, ее связывают при помощи соответствующих присадок, причем в случае кислых пород (силикаты, алюмосиликаты) добавляют основные вещества (главным образом известняк), а к основным породам (известь и пр,) — кислые присадки (глинистый сланец или гранит). Эти присадки образуют с пустой породой легкоплавкие шлаки, всплывающие на поверхность расплавленного железа. Одновременна шлаки защищают расплавленное железо от вторичного окисления воздухом дутья. Расплавленное железо выпускают через каждые 4—6 час через специальное отверстие у дна печи, называемое очком для спуска железа. Шлаки по мере их накопления спускают через специальное отверстие, проделанное па определенной высоте. Несколько выше вводного отверстия для фурм («уровня фурм») доменная печь расширяется — эта часть ее называется заплечиками,— а затем переходит в длинную сужающуюся вверх часть, так называемую шахту. В соответствии с большим количеством тепла, необходимым для восстановления железа углеродом [см. уравнение (2)], происходящего в нижней части домны, температура в области заплечиков очень быстро понижается снизу вверх. На уровне фурм она выше 1600°, а в распаре, находящемся между заплечиками и шахтой, она понижается до 800°. В шахте дальнейшее падение температуры происходит уже сравнительно медленнее. На протяжении нижних двух третей шахты температура понижается примерно от 800 до 600°, в верхней же части шахты падение температуры происходит опять песколько быстрее, В области верхнего отверстия домпы — колошника—температура выходящих газов все еще достигает примерно 200°. Своеобразная форма домны обусловливается следующими требованиями: во-первых, необходимо достаточное пространство для быстрого расширения газов дутья, происходящего в зоне заплечиков (что обусловлено образованием окиси углерода и нагреванием); во-вторых, необходимо обеспечить беспрепятственное расширение забрасываемой сверху загрузки при опускании ее во все более сильно нагретые части печи. Ниже уровня фурм, в «горне», собирается расплавленное железо. Под печи — ее основание,— постоянно покрытый расплавленным железом, имеет сводчатую форму, Для предотвращения всплывания кирпичей на поверхность жидкого железа вследствие их значительно меньше г о удельного веса.
Забрасываемая через колошник руда нагревается в верхней части шахты горячими колошниковыми газами и одиопременно обезвоживается. Восстановление окиси железа(Ш) окисью углерода начинается уже ниже 400°. Оно сначала ведет К образованию окиси железа(П, III) Гс3О41 а затем в пнжпей половине шахты при температуре выше 700° образуется металлическое железо, сначала в твердом и очень пористом состоянии:
ЗГе2Ой-|- СО=^2ГезО4-|-СО2-|-8,4 ккал*, (3)
Fi>3O4-^4CO =3Fc-|-4CO2-[-413 ккал. (4)
Чтобы восстановление протекало в надлежащее время и с достаточной полнотой, необходим большой избыток окиси углерода. Поэтому выделяющиеся из колошника газы всегда содержат значительные количества окиси углерода. Часть СО в соприкосновении с пористым железом разлагается в соответствии с уравнением
2СО=С4-СО2-}-38,6 ккал, (5)
так как ниже 1000° окись углерода неустойчива (см. т. I, стр. 485). Выделяющийся при разложении твердым углерод осаждается па железе и по мере опускания железа и повышения температуры реагирует с ним. Это обусловливает понижение температуры плавления железа и способствует его переходу в жидкое состояние. Расплавленное железо при просачивании в виде капель через нижний слой кокса растворяет еще некоторое количество углерода.
Одновременно с восстановлением железа протекает реакция между присадка ми и пустой и up одой. Часть FeO, образовавшейся при частичном восстановлении, в виде силиката переходит в шлак, получившийся в результате этой реакции, и вследствие этого удаляется из сферы реакции с окисью углерода, Однако в дальнейшем, при просачивании ш) каплям жидкого шлака через слой кокса, последний восстанавливает железо в силикате [см. уравнении (2), стр. 273], мри этом происходит также восстановление маргапца, фосфора и в небольших количествах даже кремния.
Доменная печь, после того как она была «задута», должна работать непрерывно. Если в силу особых обстоятельств приходится временно приостановить ее работу,
* Ср. примечание ** п стр, 273.
Восьмая, группа периодической системы 275
следует замазать все отверстия печи, так чтобы се содержимое было по возможности предохранено от доступа воздуха п потерь тепла. При этих условиях иногда удается пустить в ход доменную печь даже после месячного перерыва в работе.
Колошниковые газы, содержащие еще до 2/3 сожженного углерода в виде окиси, после очистки от пыли используют для подогрева воздуха, вдуваемого в печь (оп должен иметь температуру около 800°), и для приведения в действие воздуходувок. Шлак, используют для производства плит для мостовой, Цемента («железный портланд-цемент») и строительного камня («шлаковый камень»). Чугун,, образующийся в результате доменного процесса, частично находит применение без дальнейшей переработки. Однако гораздо большая его часть идет в передел на сталь или мягкое (ковкое) железо,
Передел чугуна. Чугун, полученный в доменной печи, наряду с примесями кремпия, марганца, фосфора (и обычно серы) всегда содержит значительные количества углерода. Вследствие этого он хрупок и пе поддается ковке и прокатке; при сильном нагревания чугун плавится сразу в противоположность ковкому железу и стали, которые размягчаются задолго до плавления. Переработку чугуна в ковкое железо называют переделом, или фришеванием.
Передел (фршпсвание) чугуна в крнчпом горле. Старейшим способом получения ковкого железа из чугуна является фришевание в кричном горне, В емкости, выложенной из чугунных плит (горне), плавят чугун на древесном Угле; для усиления горения
Р нс. 3Q. Передел в кричном горне.
Горн s обложен чугунными плитами. Пса г может охлаждаться водой, находящейся в баке Образующиеся при горении горячие газы, проходя над очагом з для предварительного подогрева, нагревают находящийся там чугун, нредкаяначен-пый для следующей садки. Часть отходящих газов перед выходом в дымовую трубу проходит через камеру 4 И там нагревает воздух, который затем продувается над пламенем горна -5.
последнего пользуются дутьем (см. рис. 30), Для такого передела чугуна в ковкое железо требуется трехкраткая его плавка, При первой планке, так называемой «отбелке», сгорают только наиболее легко окисляющиеся прнмесп — кремний и марганец, Окисление кремния, содержащегося в железе в виде силицида, Происходит в основном по уравнению
FeSi4-3/2O2=FeSiO3 (метасилйкат железа, так называемый «бисиликат»). (6)
Наряду с Метасиликатом при этом образуется также ортосиликат железа Fe2SiO4 I -еппгулосиликат») и, если в Железе содержался марганец, силикат марганца. При второй плавке, «сыром переделе», Происходит окисление фосфора, содержащегося
18*
276
Глава 7
в железо примерно в соответствии с уравнением
2Fe,P-i (jO2=.Fe3(PO4h + РезО4. (7)
Одновременно частт, железа окисляется До окиси железа(П, III), а она в свою очередь окисляет часть углерода, растворенного в железе (в основном в виде карбида Fe3C):
3Fe ф* 2O2=Fle3O41 (8)
Fe3O4-f-4Fe3C^15Fe-f-4CO. (9)
Таким образом получается железо, содержащее еще 1^2% углерода, т. е, сталь (см, стр. 287). Для превращения ее в мягкое железо требуется еще одно фришевание. В процессе, называемом «спелым свежеванием^ (или «спелым фришеванном»), железо благодаря значительному снижению содержания углерода уже полностью не ожижается; нначале и в данном случае образуется окись железа (II, III) («спелый шлак»). Последняя в соответствии с реакцией (9) уменьшает содержание углерода в железо до концентрации менее 0,5%. Процесс обезуглероживания обычпо ускоряют добавлением рацее образовавшихся спелых шлаков (Fe3O4).
Сера, содержащаяся в чугуне, при кричном переделе улетает в виде сернистого газа; этот процесс происходит тем полное, чем дольше длится фришевание. Железо, полученное таким способом, очень чистое и содержит сравнительно мало щлаков, Одпако этот метод сохранился только в некоторых странах, очень богатых лесами (Швеция, СССР).
Передел в отражательных печах (пудлингование). Йудлинёовамие отличается от кричного передела, во-первых, тем, что при нем плавление железа производится
Рис. 31. Отражательная ночь для пудлингования.
пе в б беколе спинов ой топке, где оно находится вместе с горючим, а в отражательной печи, приведенной на рис, 31. В ней пламя от топлива (обычпо каменного угля), сгорающего в топочном пространстве, оборудованном колосниками /, проходит над железом, находящимся в плоской корытообразной емкости 2 («горне»). Расплавленное железо непрерывно перемешивается и поэтому приводится в соприкосновение с воздухом. Это способствует обезуглероживанию вплоть до иолучения мягкого железа в течение одной переплавки. Само название этого процесса — пудлингование — произошло от слова «перемешивать» (по-апГлийски — to puddle). Пр мере обезуглероживания железо делается все более вязким и наконец образует твердые сгустки —«болванки», так как температура становится уже недостаточно высокой для поддержания его в расплав лепи ом состоянии, Болванки содержат еще довольно значительное количество шлаков, от которых их затем освобождают горячей ковкой и прокаткой,
Методом пудлингования можно получать и сталь. В этом случав после образования сгустков продолжают нагревание восстановительным пламенем. Чтобы достигнуть полного окисления примесей (кремния и фосфора), нагревание ведут при более высокой температуре и болев продолжительное время, чем npsi фришеванин стали до мягкого железа в кричном горне.
Сварочное и литое я; очей о; плавка в тигельной печи. Железо, образующееся при сплавлении болванок, полученных кричным или пудлинговым методом, называют сварочным железом. Вследствие неполного удаления из пего: шлака оно всегда более или менее неоднородно и нередко имеет трещины. В середине XVIII в. шеффилдский
Восьмая группа периодической системы
277
часовщик Бснджамеп Гантсмап пачал изготовление часовых пружин из более однородной стали, полученной неренлавкой в тиглях, Тигельная плавка поз;ке была введена Круппом в Эссене в промышленном масштабе и иногда применяется в настоящее Время для получения высокосортных сталей. Тигельной плавкой можпо не только сообщать однородность уже переделанному другими способами желозу, но и пользоваться сю одновременно для фршпевапия. Однако в этом случае переделываемый чугун (к нему добавляют окись железа) не должен содержать фосфора и серы,, (так как примеси при этом способе пе удаляются. Плавку ведут в тиглях, изготовленных из смеси огнеупорной глипьт и графита вместимостью приблизительно 50 кг. Двенадцать или более тиглей устанавливают в длинной узкой камере, где их нагревают в регенеративной топко Сименса (см. ниже).
Описываемыми ниже методами фрипгевания получают непосредствен по литое железо или литую сталь.
Методы конвертирования (Бессемера и Томаса — Джилькристэ). Методы конвертирования основаны на продувке воздуха через рас-
плавленное железо при помощи воздуходувок, что способствует выгоранию углерода и других примесей. Наиболее старый из этих способов — бессемерование, открытый в 1855 г. англичанином Генри Бессемером, применяют п до настоящего времени для передела чугуна, очень бедного фосфором. Переплавку осуществляют в сосуде грушевидной формы, выложенном изнутри огнеупорным материалом. Оп называется грушей Бессемера или конвертером (рис. 32); вращаясь вокруг оси, проходящей приблизительно на половине его высоты, сосуд может опрокидываться. В конвертер, установленным в го риз опта льном положении, вливают чугун обычно непосредственно из доменной печи. После наполнения конвертер приводят в вертикальное положение и через отверстия в дне пропускают сквозь расплавленный чугун сжатый воздух. При этом содержащиеся в железе примеси окисляются. Большое количество тепла, выделяющегося
Рис. 32. Груша Бессемера (конвертер).
при их окислении, поддерживает железо во время этого процесса в расплавленном состоянии (несмотря па то что температура плавления железа значительно повышается по мере снижения содержания углерода).
При бессемеровании, как и в кричном переделе, можно в принципе различать три разных процесса, соответствующих отбелке, сырому и спелому свежеванию. Однако при бессемерования: они проявляются далеко не так отчетливо, как при фришевании кричным способом. Отчасти это зависит от высокой температуры, которая способствует окислению углерода, а с другой стороны, от значительно более быстрого протекания процесса продувки в конвертере (обычно он длится 10—20 .чин). Поэтому иногда железо, полученное способом Бессемера, при почти водном отсутствии углерода содержит значительные количества кремния. В некоторых случаях это да?ке целесообразно. Контроль за ходом продувки в конвертере осуществляется путем наблюдения за выходящим из него пламенем.
При бессемеровании в его первоначальном виде можно было с успехом перерабатывать иа ковкое железо п сталь только те сорта чугуна, которые содержали менее 0.1% фосфора. Вследствие постоянного перемешивания шлаков с углеродсодержащнм железом при этом процессе из первично окисленного фосфора опять образуется фосфид железа:
Fe3(PO4)3-f-2Fc3C-f-3Fe 2FesP-|-6FeO-|-2CO.
(10)
Кремнекислота, содержащаяся в футеровке конвертора, ещё более благоприятствует протеканию этой реакции в направлении слева направо, так как она образует с окисью железа(П) силикат. Поэтому бессемеровский процесс в его первоначальной
278
Глава 7
форме, так называемое «кислое» конвертирование, не освобождает железо от фосфора. Между тем присутствие фосфора в количествах более 0,1% является очень нежелательным, так как сообщает железу хрупкость п ломкость на холоду («хладноломкое» железо).
Переработка в конвертере чугуна, содержащего значительные количества фосфора, оказалась возможной после того, как англичане Томас и Джилькрист в 1878 г. применили для футеровки конвертора основные магаериалы. (обожженный доломит). Последний связывает фосфорную кислоту и препятствует ее восстановлению. Действие основной футеровки еще усиливается при добавлении кусковой едкой извести Са(ОН)г. Шлаки, образующиеся в процессе ^основной» продувки (Томаса — Джипькри-ста), богатые фосфором (томасшлак), являются ценным побочным продуктом. В тонко раз молотом виде они поступают в продажу под названием томасовой муки.
Переход фосфора в (плави происходит главным образом после сгорания большей части углерода. Примерное протекание кислого и основного процессов приведено в виде диаграмм на рис. 33 и 34. По осн ординат отложено содержание в процентах
Рис, 33. Обезуглероживание Рис, 34. О бе зу г лерожив а-
II удаление других примесей ние и удаление других при-
в процессе «кис.лого>> фрише- мссой в процессе «основ-
вания. ного» фришевання.
углерода, кремния, марганца и т. д. в железе в зависимости от продолжительности продувки, Из рассмотрения утих .диаграмм следует, что на примере, изображенном на на рис. 33 («кислый* процесс), вначале происходит главным образом окисление кремния и после этого окисляется только углерод, в то время как содержание в железе фосфора и серы почти не меняется. Па рис, 34 виден ход окисления фосфора в «основном* процессе.
Восьмая группа периодической системы
279
Для удаления серы, в «кислом» процессе необходимо добавлять марганец (ср. стр. 295). «Основной» процесс применим лишь длй сортов чугуна, содержащих немногим более 0,15% серы. Следует отметить, что бессемеровский и томасовскнй процессы применимы для чугунов определен пых составов.
В обоих сталеплавильных процессах, и особенно в «основном;», содержание углерода снижается в значительно большей степени, чем ото необходимо для технических целей. Поэтому перед спуском железа из конвертера к жидкому железу добавляют некоторое количество богатого углеродом зеркального чугуна или ферромарганца, которые вновь повышают в нем содержание углерода, Марганец, кроме того, обладает способностью раскислять растворенную окись желоза. Образующаяся при этом окись марганца Легко отходит в виде шлака. Вместо марганца для раскисления применяют и ряд других веществ, главным образом кремнии, иногда алюминий, ванадий, титап и другие металлы, обладающие сильным сродством к кислороду, а в последнее время—и бериллий. Прп томасировании пород добавлением раскислителей необходимо сначала удалить шлаки, содержащие фосфор; это осуществляют осторожным слипанием их из наклоненного конвертера.
Процесс Сименса — Мартена представляет собой по существу процесс передела в отражательных печах, при котором благодаря очень высокой температуре получается не сварочное (как при использовании обычных отражательных печей — при пудлинговании), а литое железо. Возможность получения достаточно высоких температур появилась лишь после создания в 1860 г. братьями Сименс регенеративной топки. Эта печь представляет собой газовую топку, в которой тепло сгорающих газов используется для предварительного подогрева горючих газов и воздуха; для этого горячие газообразные продукты горения проходят через две камеры, выложенные огнеупорным кирпичом (теплосбориики, регенераторы), где происходит теплообмен. Через некоторое время топку переключают таким образом, что через одну из раскаленных камер проходит направляющийся в печь горючий газ, а через другую — воздух для его сгорания, в то время как отходящие газы нагревают две другие камеры; по остывании первых двух камер вновь производят переключение и т. д. Развивающуюся благодаря этому высокую температуру печи братья Мартен во Франции в 1865 г. впервые использовали для получения литой стали.
i'
На рис. 35 показана печь Сименса — Мартепа для получения литого железа и литой стали, Первоначально литую сталь в печи Сименса — Мартена получали сплавлением чугуна с мягким, т. е. почти полностью обезуглероженным железом. Однако вскоре было установлено, что в такой печи можно получать сталь и даже мягкое железо непосредственно из чугуна, С одной стороны, это достигается регулировкой подачи воздуха таким образом, чтобы пламя, проходящее над очагом, где находится железо, содержало избыточный кислород и Действовало как окислитель; с другой стороны, добавкой к чугуну окисных железных руд, железной окалины и ржавого старого железа (лома, скрапа). В зависимости от содержания фосфора л чугуне очаг обкладывают «основною) или «кислой» футеровкой, В нервом Случае, как и при тома-совок ом процессе, добавляют к железу гашеную известь. Образующееся в этом случае обезуглероженное литое железо при выработке стали приходится вновь обогащать углеродом, причем одновременно (как и при конвертировании) происходит раскисление железа.
Применение процесса Снмепса — Мартена в противоположность конвертированию не ограничивается составом чугуна, так как в этом с л уча е нагревапие производят специальной топкой и оно пе связано со сгоранием тех или «пых примесей, содержащихся в железе. Процесс Симонса — Мартена протекает медленнее, чем конвертирование, при этом происходит более полное отделение шлаков, т. е. улучшается качество железа. Некоторым недостатком процесса Сименса — Мартена является трудно предотвращаемое окисление железа газами пламени во время обезуглероживания железа после его фришевания.
Выплавка электростали. В настоящее время для получения высококачественных сталей все более широкое применение находят печи С илектрообогревом. Ими пользуются в первую очередь для улучшения качеств конвертерной стали путем переплавки, а также для науглероживания и раскисления железа, полученного н печах Сименса —
280
Глава 7
Мартена. Несмотря па сравнительно высокую стоимость электрической энергии, ее применение р данном случае окупается тем, что при этом исключается действие газон пламени на расплавленное железо, что позволяет получать сплавы железа определенного необходимого состава,
Отжиг белого чугуна. Небольшие железные изделия (ключи, части замков, гаек и т. и.) обычно отливают из белого чугуна и затем ужо превращают в мягкое железо. Их закладывают в окись железа и затем в течение продолжительного времени (около 8 суток) нагревают прн 850—1000° (т, е. ниже температуры плавления железа). При этом содержащийся в чугупе карбид железа Ее3С разлагается, выделяя мелкодисперсный углерод, который, поднимаясь и проходя через слон окиси железа, сгорает в ней. В атом заключается отжиг белого чугуна, а изготовленные таким образом яред-^ меты являются изделиями из ковкого чугуна.
Цементация, подобно тому как отжигом можно обезуглеродить чугун, мягкой железо удается обогатить углеродом при длительном нагревании его с порошкообразным древесным углем или другими веществами, легко отдающими углерод в отсутствие воздуха. Этот процесс называется цементацией, а полученная таким образом сталь — цементированной. Знаменитую да.чаес-кукз сталь изготовляли цементацией сырого-
Р и с. 35. Печь Сименса—Мартена.
Железо яаходитгн в стяге 1. Регенераторы I и ГУ (меньших размеров) служат для- подогрева генераторного тазд. a Ji и ЫГ (больших раамвров) — для подогрева воздуха. За одну фазу процесса генераторный газ н воздух проходит, не смешиваясь, через рансопат'ретые камеры' I и II, а раска-тенньте газообразные продукты горения проходят через камеры ГII н ту. Затем продукты-сгорания нери<лк1ча1отсл на камеры I и 11, a aosnvx и генераторный таз проходят черев камеры III и IV.
.железа (т.'е. железа, выплавленного непосредственно из руды в сыродугаомторне). В XVIII и XIX в. хорошую инструментальную сталь нередко изготовляли цементацией топких стержней из сварочного железа, которые затем сваривалив бруски («сварочная рафинированная сталь»),
В настоящее время при цементации в большинстве случаев ограничиваются переделом верхних слоев изделий, полученных формованием из ковкого железа. Тактик метод, используемый для придания твердости поверхности стали, называют цементацией с поверхности.
СВОЙСТВА ЧИСТОГО И ТЕХНИЧЕСКОГО ЖЕЛЕЗА
Чистое железо — белый, блестящий металл, сравнительно малой твердости (4,5). Его удельный вес 7,86, температура плавления 1528°. В твердом состоянии железо может существовать в двух модификациях. При обычной температуре, а также выше 14Q1 ° оно образует объелМоцеНтри-рбванную кубическую решетку (а = 2,8664 А при 20Q), а в интервале 906— 1401°— гранецентрированную кубическую решетку (а — 3,6468 А при
Восьмая группа периодической- системы 281
916d). При температурах ниже 768° железо обладает способностью намагничиваться в магнитном поле (ферромагнетизм). Температура, при которой это свойство исчезает (768°)—- точка Кюри железа (ср. т. I, стр. 339) — проявляется в виде остановок на кривых нагревания и охлаждения * наряду с другими температурами, при которых происходят полиморфные превращения железа (906 и 1401е). Поэтому обычно ** a-железо, способное к намагничиванию, отличают от идентичной в структурном отношении формы, не обладающей ферромагнетизмом, которую обозначают как Ь-желего. Гранецентрированную модификацию (также не обладающую способностью намагничиваться) называют у-железом.
Раньше в интервале 768—906° железо обозначали как (^модификацию, Однако различие между р- и 6-формамп все-таки нельзя установить, и на диаграммах состояния сплавов, в которых: у-оолвсть твердых растворов ограничена определенными концентрациями вообще не удается провести границу между р- и б-областями (ср, рис. 39 и 40, стр. 285).
Чистое железо в противоположность металлу, содержащему углерод, теряет свой магнетизм мгновение после исчезновения электрического поля. В связи с Этим оно находит применение в электротехнике, например, для изготовления электромоторов и трансформаторов, в которых требуется быстрое намагничивание и размагничивание Железного сердечника.
Линейный коэффициент расширения железа между 0 и 100° составляет 1,1- К)-5, а его теплопроводность при 18° равна 0,17 кал/см- сек -град. Удельная теплоемкость равняется при 0° 0,1055, при 725° 0,1467, в интервале 785—919° 6,1571°, а в точке плавления 0,1637. Выше точки плавления теплоемкость изменяется незначительно.
Железо обладает большим сродством к кислороду. Во влажном воздухе оно ржавеет, т. е. постепенно, начиная с поверхности, превращается в гидрат окиси. С сухим воздухом компактное железо начинает заметно реагировать лишь выше 150°. При прокаливании на воздухе оно дает промежуточный окисел Fe3O4, который образуется также при ковке раскаленного мягкого железа молотом (((железная окалина»). В очень тонко измельченном состоянии железо (образующееся, например, при прокаливании оксалата в токе водорода) обладает пирофорными свойствами.
Для защиты железных предметов от ржавления применяют покрытия из других металлов (папример, цинка, олова, хрома, никеля) или окраску (суриком). Особенно эффективна защита железа путем превращения его поверхности в фосфат жело-за(11) («фосфатирование»), Это достигается применением водных растворов кислых фосфатов марганца пли цинка [MnfH2PO4)2 или Zn(H2PO4)2 (метод Варкера)]. Соответствующие присадки ускоряют фосфатирование.
В соответствии с положением в электрохимическом ряду напряжений и разбавленных кислотах железо растворяется, вытесняя из кислот водород и образуя сопи двухвалентного железа. Нормальный потенциал желоба в равновесии с раствором солей двухвалентного железа, отнесенный к нормальному водородному электроду, при 25° составляет —0,440 в-(Randall, 1932). Металлическое железо, погруженное в раствор солеи меди, покрывается слоем металлической меди в результате происходящей при этом окислительно-восстановительной реакции Fe 4- Си” =
Fe"4-Cu.
* Ср. т. I, стр. 612.— Прим,, перев.
** У других веществ пе принято обозначать различными буквами состояния нышо п ниже точки Кюри. В соотпетствнй с этим названия а- или 6-железо часто употребляют для объемнсцентрировалиой модификации как ниже, так и выше точка Кюри.
282
Глава. 7
После погружения в дымящую азотную кислоту железо теряет способность раз-ряжать ноны меди и водорода. Оно Не растворяется в концентрированной HNO3, а подобно хрому пассивируется в ней (ср. стр. 144 и сл. И стр. 790 и сл.).
Вода, пе содержащая воздуха, при комнатной температуре почти не корродирует железо, так как на его поверхттости образуется плотный слой гидроокиси двухвалентного железа, обладающий защитным действием, даже при очень небольшой толщине. В присутствии воздуха, наоборот, образуется пористая гидроокись желсза(П1) и Коррозия поэтому прогрессирует, Еще меньше, чем чистая вода, разъедает железо разбавленный раствор едкого натра (в отсутствие воздуха). Это происходит главным образом потому, что попы ГИГ понижают растворимость Fe(OH)a. (Кроме того, с повышением рП раствора всю рте тает потенциал разряжения ионов Н')7 Однако концентрированные растворы едкого натра довольно сильно разъедают железо даже без доступа воздуха, «особенно при повышении температуры, так как с повышенном концентрация ионов ОН; Fh(OH)2 переходит г, раствор с образованием гидроксо-солей (ср. стр. 291). Образование гидр о к сил l-пых попов сопровождается падением До минимума концентрации ионов Fe", поэтому потенциал железа в концентрированном (~40%-пом) растворе едкого натра сильно отрицателен (—0,86 в). При известных условиях потенциал дая;е в таком .растворе может резко меняться за счет пассивирования и приобретать положительное значение (па пример, -J-0,65 <?), Поскольку ото явление имеет место, железо само по-себе не реагирует в данном случае с выделением водорода и образованием гидроксофср-раг( Г Г Ни |цив; реакция происходит лишь под действием приложенного напряжения п тогда в раствор переходят феррат (VI)-ионы,
При высокой температуре железо очепь энергично реагирует с хлором, а также с серой и фосфором, но пе с азотом. Оно обладает большой склонностью к образованию соединений или сплавов с углеродом и с кремнием. Образование этих сплавов оказывает большое влияние па свойства технического железа.
Сплавы железа в углеродом. В затвердевших сплавах железа с углеродом существует целый ряд компонентов, имеющих различную структуру. Важнейшие из них следующие:
1. Феррит — чистое железо (точнее «-железо).
2. Графпт—гексагональная модификация углерода.
3. Цементит — химическое соединение железа и углерода, Fe3C (содержит 6,58% С).
4. Аустенит— твердый раствор углерода в железе (точнее, в у-железе).
5. Мартенсит — мета стабильная фаза, образующаяся при быстром охлаждении аустенита.
6. Ледебурит — эвтектическая смесь цементита и аустенита (насыщенного углеродом,.
‘ - Перлит — эвтектоидная смесь * феррита н цементита,
О б разевайте ауыпенита. основано па том, что в центрах и в серединах ребер элементарных кубов у-желе.ча могут располагаться атомы углерода (см, рис. 36). Если такое внедрение происходит в каждом пз кубов, то образуется соединение FeC, содержащее 56 ат.% С. В действительности, однако, решетка аустенита устойчива только в том случае, когда лишь небольшая часть ее узлов занята атомами С, Эта часть может колебаться от 0 до 8 ат.% С. Атомы С, участвующие в построении решетки, располагаются статистически в центрах кубов или в серединах ребер. Поэтому аустенит обладает характером твердого раствора. При вхождении атомов С в решетку у-железа эле.ментарная ячейка несколько увеличивается в размерах, причем одинаково во всех направлениях,
Мартенсит можпо рассматривать соответственно как (пересыщенный) твердый раствор углерода в a-железе, Он, как и все пересыщенные растворы, мотастабилен. В решетке мартенсита, показанной на рис. 66, также только часть узлов действительно занята дтомамя С. Если бы были заняты все узды, тояп> образовалось бы соединение FeC. Решетка мартеш’нта по сравнению с a-железом тетрагонально искажена; она вытянута в одном направлении **, а в двух других несколько сжата, Решетка цементита FcsC
* Под аетепщоидом имеется в виду смесь, образовавшаяся при распаде твердого раствора, таким же образом, как при охлаждении расплава происходит образование эвтектической смесй,
*’* Кроме обычной тетрагональной формы мартенсита, существует еще и кубическая. Она образуется из тетрагональной при ничтожпо малом изменении положения атомов С.
Восьмая группа периодической система,
283
как видно из рис. 37, имеет в значительной степени комплексный характер (Shimlira, 1931; Westgren, 1932). Кроме карбида состава Fe3C, существует еще один, возможная формула которого FeaC. Он образуется при нагревании Fe с СО при 225°, но разлагается при более высокой температуре. При непосредственном соединении железа с углеродом этот карбид, по-впдпмпму, получить нельзя,
у-Железо Аустенит
а=3,53 А (экстра - а=3,63 А
полировано до кот- (при 8ат.% С) натнои температурь!)
<х-(6-) Железо
(Ферриту а=2,87А
Мартенсит а0-2,8д, с^З.ООА (при бат.КС)
Рис. 36. Структура ал ем гитарных ячеек сплавов железа с углеродом, • — Fe; О — места, предназначенные для атамов С.
Рис. 37. Репгсткц цементита Ее3С.
а = 4,52,. 6 = 5,08, с=0,78 А. 1?С <—> с — 2,01 А. Атомы Fe образуют призму, в центре которой находится этом С. На рисунке показана такая призма (в которую, .помимо атомов железа, принаниежащих элементарной ячейке, входят еще 2 атома Fe).
На рис. 38 приведена диаграмма состояния системы железо — углерод в области до 5 вес.% С. При добавлении углерода к железу температура плавления Fe сначала снижается (ст А до Е), а затем при дальнейшем возрастании содержания углерода вновь повышается. Эвтектическая точка Е соответствует содержанию 4,2% Си температуре примерно 1140°. Расплавленное Железо, содержание 4,2 вес, % С, затвердевает в ей де ледебурита. Из сплавов с меньшим содержанием углерода в первую очередь осаждаются твердые растворы у-жслеза с углеродом (аустенит). В этом случае твердый раствор беднее углеродом, чем расплав. С расплавом, состав которого соответствует точке Ъ па кривой ликвидуса АЕ, находится в равновесии твердый раствор, состав которого опредсляетсяточкой а не кривой, солидуса АС, Вследствие выделения аустенита расплав обогащается углеродом. Одновременно происходит и возрастание концентрации углерода в аустепите * вплоть до на2 сыщония им при концентрации углерода 1,7% в точке с ; расплав в этот момент достигает точки £, и его остаток затвердевает в виде ледебурита. Из сплавов с содержанием до 1,7% углерода, таким образом, образуется исключительно аустенит, а из более богатых углеродом (до 4,2%) наряду с аустенитом выделяется и ледебурит. Однако аустенит устойчив только при высокой температуре. В зависимости от содержания углерода при медленном остывании сплава аустенит превращается при температурах между точками F (точка превращения ^’-железа в б-железо) и Н или D в смесь феррита и перлита (G — температура превращения 6-железа в a-железо). В качестве промежуточного продукта при этом превращении образуется мартенсит, характеризую-
щийся чрезвычайно большой твердостью; после очень быстрого охлаждения (закалки) оп сохраняется и при обычной температуре. На микрошлифах мартенсит имеет вид темных игл, выпелятопптхся на фоно светлого, еще не измененного аустенита.
Если исходить из сплава, содержащего более 4,2% С, при быстром охлаждении сначала выделяется цементит Fe3C, а после того как содержание углерода понизится то 4,2% (в результате выделения Цементита), остаток затвердевает в ледебурит. При медленном охлаждении вместо цементита осаждается преимущественно графит. При температурах пиже точки плавления смесь железа и графита является более устойчивой, чем смесь железа с карбидом железа. Поэтому карбид железа при про-
* Выделившийся аустенит в дальнейшем еще поглощает углерод.
284
Глава 7
дол жите.льном нагревании до 1000° превращается в графит, Графит при таком превраг Щецин, происходящем в твердом сплаве, выделяется в состоянии очень топкого измельчения («углерод отжига»). Это явление, как уже было упомянуто, используют при «переделе» путем отжига.
0$ /7 4,2
Содержание углерода}%
Рас. 38. Диаграмма состояния системы ,же л езо—углерод (схема).
Другие сплавы жсл«?за. В качестве примеров сплавов железа с другими элементами рассмотрим диаграммы состояния дней пых систем с кремнием, хромом, никелем и марганцем, представ л он тпяе на рис. 39—42.
Диаграмма состояния системы железо — кремний (рис. 39) подобна с о отв етств угощен диаграмме «железо— углерод», поскольку в обеих системах происходит образование твердых растворов и соединений. Существенное отличие, однако, связано с влиянием вторых компонентов (Si и С) па области существования у- ив-фаз железа. На диаграмме системы железо"— углерод область твердого раствора у-железа (аустенит) значительно расширена, в то время как твердые растворы 6-же ле за, несмотря па совершенно ничтожное содержание в них углерода, оказываются нестабильными *. На диаграмме системы железо — кремний твердые растворы у-ж слеза ограничены очень узкой областью, и то время как область существования твердых растворов 4-жеяеза здесь значительно расширена.
Еще более отчетливо влияние легирующего компонента проявляется в областях существования у- и 4-фаз на диаграммах состояния систем железа :— хром и железо — никель (рис. 40 и 41), В обеих системах преобладает неограниченная взаимная растворимость н твердом состоянии (очень узкие гетерогенные области существуют лишь на границе между у- и 4- или «-фазамп). В противоположность системе железо — хром, где
* В области FHD на рис. 38 существуют смеси <5- или a-железа, очень бедных углеродом, и аустенит. Только совсем узкая полоса по краю этой области (па рисунке она не нанесена) соответствует гомогенный твердым растворам <?- или а-железа. Ниже точки А па рисунке также расположена очень ограниченная область существования твердых растворов в-железа, не обозначенная на рисунке, аналогичная области твердых растворов d-железа, содержащих никель па рис. 41.
Рис. 39. Диаграмма состояния системы железо — кремний.
-я и Ь расплав 4- FeSi; end — расплав 4- FeSi2; е — расплав 4- St; t — FcSl 4" эвтектика I; 5 — FeSi 4- эвтектика II; Л — FcSls4- эвтектика II; i — FeSlZ; li — FeSi24- эвтектика Щ-I — Si 4- эвтектика III; tn — FeiSi/4- эвтектоид I; a — FesSis4- эвтектоид II.
____ Эвтектика l: твердый раствор O-Fe 4- FeSl; эвтектика II: FeSi -f-JFeSis, эвтектика 1ц: FeSlj-f- SI; эвтектоид I: твердый раствор 6-Fe (Ниже 400“-— твердый раствор a-Fc) 4-Е^ез811;
эвтектоид II: Fe3Sis4- FeSi.
°C 1800
.1700
1600
1500
1400
.1300
1200
1100
1000
900
8дд\_~Магшшшыв превращения
7000 10 20 Iff 40 50 60 70 80 SO 100
Fe asi. % Or
1830'
Расплав
3528'
1401°
S-Fe-mespdiiiu paauaop y-Fе^-ишердшвpaanspp
ТТ^яверд^и ^aci^eop^
P и c. 40. Диаграмма состояния системы железо — хром.
°C 1500 3-Fer-^ 1400
1300 -
1200-
1100-
1000-
900 -
&^Расплав*$
Расплав 1452^
Расплав* у
y-Fe-твердыи раствор
£
Магнитные превращения]
^л^вная I
300 W 600 а-К™.
ипп _____________
О 10 20 30 40 50 60 70 80 30 100 Fe ат.% Ni
Ри с. 41. Диаграмма состояния системы железо — пикель.
I
286
Глава 7
область стабильного существования твердых растворов у-железа очень узка, а твердых растворов <5-железа очень расширена, в системе железо—никель твердые растворы d-железа существуют лишь в совсем небольшой области, в то же время область твердых растворов у-желеаа эдггь занимает почти всю ширину диаграммы.
На диаграмме состояния системы железо — марганец (рис. 42), которая ужо обсуждалась на стр. 227, преобладает область существования твердых растворов у-жслеза. В этой системе заметно значительное снижение склонности обоих компонентов к образованию твердых растворов с понижением температуры. (Гетерогенные области на рис. 40—42 заштрихованы.)
Расширение или сужение области стабильного существования у-желеэа в бинарных системах с элементами, склонными к образованию твердых растворов, зависит
Р и с. 42. Диаграмма состояния системы железо—марганец (Westgren, Ohmann).
от положении соответствующего элемента в периодической системе и, по-видимому, в первую очередь определяется его атомным радиусом (ер. W over F., Naturw., 17, 304
Неметалл/а обычно растворимы в железе в очень небольших количествах (ср. табл. 31, стр, 264 и сл.), исключение составляют только углерод и кремний. Кислород растворяется в жидком железе в виде FeO, часть которого по охлаждении выделяется и виде твердого раствора (б-Fe образует твердые растворы вплоть до содсржанияО, 12, a a-Fe — только до 0,04 вес.% О). Содержание кислорода в железе уменьшает возможность его обработки при температуре каления, приводит к образованию «красноломкого» желоза. Расплавленное железо присоединяет азот из воздуха в минимальных Количествах. Одиако при прокаливании железа в токе аммиака образуется соединение железа с азотом Fe2N, растворимое в больших количествах в твердом железе и придающее ему значительную твердость. На этом основан метод поверхностной закалки изделий из железа путем нагревания их в токе аммиака (процесс азотирования). Кроме того, существует еще одно соединение железа с азотом Fe4N с довольно широкой областью гомогенности. Выше 660° оно превращается без изменения состава в кристаллы соединения Fe2N (в решетке которого моста атомов азота заняты только наполовину),
В оРород также присоединяется к железу при температуре каления, однако в очепг, малых количествах — пропорционально давлению. Большие количества водорода может содержать электролитическое железо, Это обусловливает его твердость и хрупкость. При отжиге водород выделяется и железо становится ковким,
Особенно большие количества водорода или азота (вплоть до 6 молекул Н2 или Ж на один атом Fe) поглощает, по данным Франкенбургсра (Frankenburger, 1931), так называемое атомарное железо. Его получают в конденсате при одновременном испарении железа и «разбавителя» (например, поваренной соли), взятого в таком избытке, чтобы он препятствовал объединению атомов металла в большие агрегаты. Присоединение молекул газа к атомам Ее связано в этом случае, по-виднмому, с деист-
Восьмая группа периодической системы.
287'
вШ'М так называемых вандсрваальсовых сил — по аналогии с образованием гидратов инертных газов (ср. т. I, стр. 248). Соответствующие свойства обнаруживает п атомарный никель. Инертные га вы к атомарным Fe и Ni не присоединяются.
Технические сорта железа. Кроме углерода, техническое железо содержит еще кремний, марганец и другие примеси. Последние могут в более или менее значительной степени изменять свойства углеродных сплавов железа. Так, примесь кремния в железе затрудняет образование цементита и благоприятствует выделению графита, в то время как марганец влияет в противоположном направлении.
Влияние Si, Мп и т. д. на области существования модификаций железа, обсуждавшееся па стр. 284, имеет практическое значение, в случае если эти примеси присутствуют в больших количествах, например в специальных сталях (см. стр. 288 и сл.).
В зависимости от содержания в железе углерода и других веществ различают следующие сорта железа.
Чугун. Чугуном называют железо, содержащее свыше 2,3% (в основном 5—10%) примесей, в том числе 2—5% С. Перед плавлением такое железо не размягчается и поэтому пе поддается ковке и прокатке, но оно очень удобно для литья, так как хорошо заполняет формы. Температура плавления чугуна колеблется в интервале 1100 —1200°. При комнатной температуре чугун хрупок. Обычный серый чугун (чугуп в более узком смысле) содержит углерод в виде графита. Белый чугун содержит углерод главным образом в виде цементита. Вследствие большей твердости и хрупкости, чем у серого чугуна, белый чугун пригоден для отливок (за исключением изготовления чугунных изделий с целью их последующего отжига) и почти исключительно идет на ковкое железо.
Ковкое железо характеризуется более низким содержанием углерода и вообще содержит меньше примесей, чем чугуп. Содержание в нем углерода обычно колеблется между 0,04 и 1,5%. Ковкое железо плавится при более высокой температуре, чем чугун. При нагревании оно размягчается постепенно и поэтому поддается прокатке. В зависимости от способа приготовления различают сварочное железо и литое железо (ср. стр. 276). В зависимости от того, поддается ли железо закалке, различают сталь и ковкую сталь.
Ковкая сталь содержит не более 0,5% углерода. Она тягуча, сравнительно мягка и поэтому особенно хорошо поддается ковке. По свойствам опа приближается к химически чистому железу, но в отличие от последнего размагничивается с более или менее значительным замедлением (гистерезис).
Сталь содержит больше углерода, чем ковкая сталв, обычно 0,5—1 %. По сравнению с последней она труднее поддается ковке и сварке; но она тверже ковкой стали и при обычной температуре не вязка, а упруга. Однако главным свойством стали является ее способность к закалке. При нагревании до белого каления и при быстром охлаждении (погружением в воду или масло) сталь становится чрезвычайно твердой и хрупкой. Устранение хрупкости стали без снижения ее твердости достигается при так называемом ^отпуске», заключающемся в осторожном нагревании ее в течение короткого времени до умеренно высоких температур (260— 300°).
288
Глава 7
Твердение стали основано на том, что железоуглеродные сплавы с содержанием менее 1,7% С могут быть превращены в аустенит, который при очень быстром оз лаж делан («закалке») полностью пли частично переходит в чрезвычайно твердый мартепсит.. Как видно из диаграммы, приведенной на рис. 38 (стр. 284), температура, необходимая для закалки, различна и зависит от содержания углерода в стали. Как правило, она лежит около 900°. При «отпуске» стали устраняются или ослабляются внутренние напряжения, образующиеся при ее закалке.
Если при отпуске применить б олео сильный пагрев, начинает выделяться перлит в виде очень мелких кристаллов; твердость уменьшается, по при этом возрастает прочность. Такой метод обработки с целью получения возможно более высокопрочной стали называется рафинированием. При этом, однако, но следует вести нагрев до особо высоких температур или слишком долго, так как мартенсит может полностью превратиться в перлит; н противном случае закалку и твердение следует повторить еще раз.
Как видно из рпс. 38, между ковким железом и сплавами железа с: углеродом (сталью) пет нрпнциппальпой разницы. Поэтому и ковкое железо обнаруживает способность к закалке, хотя обычно в крайне незначительной степени.. Тем по менее в Отои отношении между сталью и ковким железом нельзя провести отчетливой границы.. Поэтому в последнее время в технике название «сталь» часто применяют ко всем коек ил сплавам железа, включая и собственно ковкое железо.
Специальные стали. Как уже упоминалось, сталь, помимо углерода, содержит кремний и марганец, но последние два элемента — лишь в незначительном количестве (несколько десятых процента). При возрастании содержания кремния л стали выпте 1% (приблизительно до 2,5%) получаются особо твердые и упругие стали, идущие, например, на приготовление пружин (кремнистые стали}. При увеличении содержанпп марганца получаются также очень упругие и отличающиеся чрезвычайной прочностью, марганцовые стали, которые идут на изготовление осей и других ответственных час.-гсА машин. Особенно высококачественные сорта, предназначающиеся для спецпальннд целей, получаются при добавлении к стали никеля, хрома, ванадия, молибдена, вольфрама и некоторых других металлов. Так, сталь с высоким содержанием никеля (36% обладает чрезвычайно малым коэффициентом расширения и поэтому особенно пригодна для изготовления часовых механизмов (инвар). Сталь, содержащая 46% никеле, имеет такой же коэффициент расширения, как стекло и платина. Проволоку из так слетали можно впаивать непосредственно в стекло, как платиновую. Добавлением чительных количеств хрома можно получать сплавы, устойчивые к действию печных газов, водяного пара и т. д. даже при высокой температуре (жаропрочные стали. Стали, содержащие наряду с хромом (обычно и количестве 4%) вольфрам и ванадил, отличаются способностью сохранять твердость при красном калении, поэтому инструментом из таких сталей можно обрабатывать металлы на высоких скоростях (быстр»-режущие стали). При сплавлении стали с 12 — 15% хрома (VM-сталь) или С 18% хрегл и 8% никеля (У2А.-сталЬ, Nirosta) получаются нержавеющие стали, прймсняющп©^ в последнее время пе только в химическом машиностроении, но и для производств предметов широкого потребления, главным образом ножей. Аналогичный сплав е;:г названием «випла-металл* используют в зубоврачебной технике для протезированхх Медь в качестве легирующего компонента повышает устойчивость железа к хпьштх-с к Им воздействиям. Стали, содержащие медь, обладают, кроме того, способности к твердению (Мосле закалки и старения) (ср. стр. 45).
Некоторые свойства специальных сталей обусловлены влиянием содержащих?.: в них добавок па взаимное расположение областей существования различных ф;-железа. Добавки определенных количеств Ni, Мп, Ст или W к сплавам Fe-C прпведг* к тому, что при медленном охлаждений вместо перлита образуется мартепсит, а в к—-сутствии еще больших количеств Легирующих компонентов — аустенит. Так, йуу кристаллизации железа, содержащего 0,4 вес.% С и менее 8 вес.% Ni, образуй-перлит, при концентрации Ni 8—22 нес,% (и том же содержании С)—, мартенкт а при еще более высокой концентрации Ni — а у степ нт. Стали, образующие прн г-~ лонном охлаждении мартенсит, должны быть названы самозакаливающимися, -г— как ояи уже не требуют специальной закалки. К тому же типу относятся и ста,— затвердевающие в аустрнит, так Как структура аустенита в пих стабильна — обычной температуре. Некоторые ж слез о-ник елевые сплавы отличаются выс— магнитной проницаемостью и поэтому находит применение в электромагнитных усгу ствах (.«проницаемые» сплавы]. Особо высокая проницаемость и совершен по ничтотхы гистерезис (малый остаточный магнетизм') свойственны евл.авам, содертнаНЕШ ~ никеля, 15% железа, 5% молибдена и 0,5% марганца [В о о t h Ь у, В о z о г с. appl. Phys., 18, 173(1947)). Преле дрока тки ей свинцом их Необходимо подкупе об ой термической обработке.
Восьмая группа периодической системы 289
Ферросилиций. Сплавы железа с относительно высоким содержанием кремния (15—18 вес.% или 26—30 ат-%) отличаются высокой кислотоупорностью. Поэтому их применяют для изготовления кислотных насосов, трубопроводов и т. д., которые изготовляют методом литья, так как эти сплавы не поддаются вальцеванию и ковке. В таких случаях обычно нагревают смесь железа (бедного углеродом) и кремния примерно до 1200°. Благодаря происходящей при этом реакции смесь плавится. Ферросилиций (термисилид) более тверд, по менее прочен на излом, чем чугун. Теплопроводность ферросилиция составляет —50%, а электропроводность —65% электропроводности чугуна. Как следует из диаграммы состояния (рис. 39, стр. 285), ферросилиций представляет собой в основном твердые растворы кремния в железе. В пем в минимальных количествах содержится силицид железа (а также углерод, выделяющийся в виде графита при охлаждении желоза). Итак, для применения защити ого действия кремния достаточно использовать существование включений этого элемента в решетку железа даже без образования химических связей (в узком смысле этого слова). Кроме того, часто наблюдается образование твердых растворов у элементов с совершенно различными Электрохимиче.сгтими свойствами (свидетельством чего являются хрупкость, возрастающая устойчивость к действию химических веществ, пониженное электрическое сопротивление); все эти свойства считаются характерными' и для образования интерметаллических соединений (ср. т. I, стр, 618). У сплавов ферросилиция устойчивость к действию кислот дополнительно возрастает, вероятно, благодаря образованию на поверхности защитного слоя, состоящего из SiOs. Поэтому такие сплавы, как правило, особенно устойчивы к действию кислот, обладающих окислительными свойствами.
СОЕДИНЕНИЯ ЖЕЛЕЗА
В своих соединениях железо проявляет главным образом валентности 2-ф- и 3-[~. Производными cta/а-валентного железа являются: окисел FeO, гидрат этого окис ла Fe(OH)2, а также соли многочисленных кислот. Наиболее важным пз них является сульфат, кристаллизующийся с 7 молекулами воды FeSO4-7H2() (железный купорос). Производными трехвалент-пого железа являются окисел Fc2O3 и его гидрат Fe(OH)3. Последний также образует солис большинством кислот, например хлорид железа(Ш) FeCl3-6H2O, и железные квасцы NH4Fe(SO4)2-12H2O. В малоустойчивых ферратах MJ[FeOs] железо шестивалентно.
Кроме окса-сот ей шестивалентного железа [форратбв(УТ)], известны аналогичные соединения четырех- и третвалеитного железа [ферраты(ГУ) и ферраты(Ш)]. Фср-раты(1¥) в чистом состоянии были получены впервые в 1952 г. Польдером (Scholder). Фсрраты(Ш) известны ужо давно; раньше в отличие от ферратов [ферратов (VI)] их называли ферритами. Кроме того, как двух-, так и трехвалентное железо способно к образованию г оксо-солей [гидр оксоферрат ы(П) и гидроксоферраты(П1)]. Однако опи малоустойчивы.
О соединениях одновалентного железа см. стр. 290, 310 и 383.
Соединения железа(П) рапыпе называли ферросоединениями, а желеэа(Ш) — феррисоедннониями. От этих названий сейчас отказались в силу их нецелесообразности. Совершенно устарело и название окиси железа(П) —«закись железа».
Соли как двух-, так и трггаалентного железа с кислородными кислотами хлора, серы и азота легко растворимы и воде. 'Го же относится и к галогенидам (за исключением фторида двухвалентного железа). Нерастворимы его фосфаты, карбонат и сульфиды, а также окислы и гидраты окйслов.
Соли железа обеих валентностей образуют двойные соли с солями наиболее активных металлов (ацидо-соли}. Особенно значительную склонность к этому обнаруживают соли железа со слабыми кислотами. Иногда, например в случае цианистых соединений, двойные или комплексные соли можно легко получить в чистом состоянии, в то время как попытки получить образующие их простые соли в чистом состоянии не увенчались успехом.
Некоторые соединения железа обладают также способностью присоединять аммиак. Так, хлориды железа(П) и (III) поглощают в сухом состоянии до 6 молекул NH3. Аммиакаты железа при растворении в воде разлагаются. Тем не менее при насыщении аммиаком концентрированных растворов аммонийных солей удается получить аммиакаты железа(П) в кристаллическом состоянии (Weitz, 1925).
Значительно более устойчивыми в водных растворах, чем аммиакаты, являются комплексы железа с некоторыми органическими соединениями, содержащими еидрок-19 г. Реми
290
Глава 7
сильные группы. Поэтому присутствие в растворах в сравнительно больших концентрациях таких соединений, как сахар, винная или лимонная кислота, затрудняет осаждение из них железа.
В некоторых соединениях железо может присоединять окись углерода. и, еще чаще, окись амта. На способности растворов сульфата железа(Ш) абсорбировать окись азота с появлением темно-бурой окраски раствора основана проба на азотную кислоту, открытая в 1835 г. Дсбассеном де Ричмондом. Растворы хлорида железа(П) и других его простых солем также поглощают окись азота.
Количество окиси азота, поглощаемой растворами солей железа, в значительной степени зависит от температуры и давления; однако оно пе подчиняется закону Генри и при различных условиях всегда приводит к предельному соотношению: 1NO па lFe“. Маншо (Muncliot) удалось получить такие соединения (содержащие NO) в кристаллическом состоянии, несмотря на их неустойчивость: например, [Feii(NO)]SOi [сульфат П11троп11лжелеза(11)]. Окись азота в соединениях этого типа действительно связана о железом; это следует из опытов по определению чисел переноса. Маншо в ряде других исследований экспериментально подтвердил предположение, что окись азота здесь присоединена как нейтральная группа и не изменяет электрохимической валентности железа.
Прочнее, чем в соединениях типа сульфата и.итровилжелеза(Г!), окись азота спязапа в нитропруссидах, имеющих обязую формулу Mj [Fc(CN)s(N О)]. Последние можно легко выделить и перекристаллизовать из горячих растворов. Другими питро-зля-свединекиямн железа с прочно связанной окисью азота являются соли Руссена, известные в виде двух главных типов с общими формулами MI[SFe(NO)a] и М1 [S3Fe4(NO)T] (красные и соответствен я о черные питрози л тиоферраты). И в этих соединениях группа NO, по данным Маншо, электропейтральна. В соответствии с этим оп рассматривает железо в солях Руссена (в черных солях Руссена от 3 до 4 атомов железа) электрохимически одновалентным.
Известны также пруссидиые соединения *, содержащие окись углерода типа Ml [Fe(CN)6GO)] (карбонил прусса дные соединения). Окись углерода при известных условиях может также непосредственно соединяться с то я к орав дробленным железом, образуя пентакарбонил железа Fi.»(GOj5. При обменной реакции этого соединения С окисью азота получается тегиранит.разил железа Fe(NO)4. (Подробное о ж еле зона р-бонцльных и нитрозплып.ъх соединениях см. стр- 3LKJ н сл,}.
О кислы п гидраты окислов
Железо образует окислы FeO, Fe2O3 и Fe3O4. Кроме того, известны в кристаллическом состоянии гидраты окислов Fe(OH)2 и FeO(OH).
Теплота образования FeO равна 64,0, FeaO3 197,6 и Fe3O3 260,7 ккал/молъ.
Перекись или пероксогидроперекисъ железа;! ! !), вероятная формула которой
IJO-O, ,0-011
yFe-O-O-Fe^ ПО/ ХОН
представляет собой красный, очень неустойчивый порошок. По данным Пеллпап и Виланда (Pcllini, 1909; Wfelaad, 1938), она образуется при действии П2Оа и КОН на ГеС12 пли FeCl3 в растворе абсолютного сиирга яри низкой температуре (—7 У3)..
Окись железа(П), монокись железа FeO получается в виде черного пирофорного порошка при нагревании оксалата железа(П) в отсутствие доступа воздуха. Она реагирует с водой, особенно при нагревании. После сильного прокаливания она теряет свою высокую химическую активность.
Кристаллическая 1W (вюстит) имеет плотность 5,9. Структура решетки описана на стр. 268. Это соединение обычпо содержит несколько меньше железа, чем Это соот-
* Определение нруссядиых соединений см. стр. 308.
В осъмая группа периодической системы 291
ьетствует формуле, так как его кристаллическая решетка оказывается стабильной лишь в том случае, если не все ее узлы заняты атомами железа. Устойчивой FeO ста-новится лишь при повышении температуры. При продолжительном охлаждении окись железа дяспропорционирует па металлическое железо и Fe3O4.
Несмотря на то что в FeO кислород связав исключительно прочно, давление кие-порода над этим соединением при высоких температурах можно обнаружить прямым методом — измерением равновесного давления Рщ/РнзО» устанавливающегося при пропускании водорода или водяного пара над нагретой смесью Fe и FeO (ср. т. I, стр. 46). Так, при 1500° К копстапта равновесия термической диссоциации воды А;р=р2Нз-рОа/р2Н2О, по данным Нсрпста и Вартенберга (Nernst, Wartenberg), составляет 8,15- 10-М. При тОй же температуре вода и водород в равновесии со смесью Fe и FeO находятся в соотношении 0,97 •. 1 (общее давление во всех случаях составляет 1 атм). Отсюда следует, что давление кислорода над FeO нри 1500° К р0,:= 8,15 X Х10-и-0,97^ = 7,7-10-ы атм.
Гидроокись железа(II) Fe(OH)2 образуется в виде белого хлопьевидного осадка при действии щелочи на водные растворы солей желсза(П); опа крайне интенсивно ноглощаот кислород, окрашиваясь во все более томный грязновато-зеленый цвет и, наконец, переходя в красно-бурый цвет гидроокиси железа(Ш). Темный промежуточный продукт, образующийся при окислении, наряду с двухвалентным содержит и трехвалентное железо. Интенсивная окраска этого продукта является типичным примером уже упомянутого явления, заключающегося в том, что соединения, содержащие один и тот же элемент в двух различных степенях окисления, обычно бывают интенсивно окрашены.
Аммиак осаждает гидроокись железа(П) неполностью, а в присутствии большого Количества аммонийных солей осаждения вообще пе происходит. В этом случае концентрация понов ОН' в растворе не может выйти за пределы очень незначительной величины. Вследствие образования в растворе железоаммиачных комплексов, обнаруженных Вейцем, одновременно происходит сильное падение концентрации ионов Fe‘‘, поэтому произведение растворимости гидроокиси железа(П), L — [Fe"]x [ОН']2, в этих условиях не достигается.
Кристаллическая Fe(OH)2 имеет плотность 3,40 (структура ее описана на стр. 268). Теплота образования из FeO и Н2О составляет 4,0 ккал!молъ,
Fe(OH)2 растворима в концентрированном одном натре с образованием гидро-ксоферрата(П) патрия Na2[Fe(OH)J, Наиболее удобным методом синтеза этого соединения является непосредственное растворение железа в виде топкого порошка в кипящей 50% -ной щелочи. При охлаждении раствора гидроксоферрат(И) выделяется в виде красивых сине-зеленых кристалликов. При действии Sr(OH)2 или Ba(OII)s яа Fg(QH)2 можно получить соли щелочноземельных металлов Sr2[Fe(OH)6] н Ba3[Fe(OH)6] — зеленовато-белые микрокристаллические осадки.
Окись железа(1П), окись железа Fe2O3 получают при прокаливании гидроокиси железа(Ш) в виде буро-красного порошка или, при более сильном нагревании, в виде темно-серой блестящей кристаллической массы. После сильного прокаливания она становится нерастворимой в кислотах, так же как и соответствующие окислы хрома и алюминия, которым FeaO3 изоморфна. (Она образует решетку типа корунда; см. т. I, стр. 391, рис. 75; а0 = 5,414 А., а = 55°17'.) Удельный вес прокаленной окиси железа(Ш) равняется 5,20 (йрентг = 5,28). В природе Fe2O3 встречается в виде красного железняка (гематита), образующего плотную зернистую, землистую или чешуйчатую массу серо-стального, черного или красно-бурого цвета. Разновидностями природной Fe2O3 являются железистый сланец (образующий листочки), железная сметана, красный (землистый) железняк кровавик (плотный) и железный блеск (спекулярит). Последний имеет отчетливо кристаллическое строение и окраску от серо-стальной
19*
292
Глава 7
до черной, но на неглазурованном фарфоре дает, как и другие разновидности FegOg, красную черту. Красный железняк является важнейшим исходным сырьем для металлургии. Некоторые разновидности природной окиси железа, главным образом красный железняк (красный болюс — сургучная земля), а также искусственно полученные разновидности Fe2O3 (кол-котар, капут мортуум, венецианская красная, английская красная) применяются в качестве красок, а в некоторых случаях — как катализаторы.
Окисел трехва.тептпого железа, как и А12О3, образует еще и вторую модификацию. По-видимому, она нс встречается в природе, но ее можно получить обезвоживанием рубиновой слюды Габер и Вём (Haber, Bohm.), проводившие рентгенографическое исследовании пои модификации, назвали ее у-окисью железа. у-Окись кристаллизуется кубически, Ге решетка является производной магнетита (см. стр. 295), она образуется при удалении из последней г,/9 атомов Fe. -у-Окись железа, так же как и магпетит. ферромагнитна в отличие от парамагнитной a-модификации. При нагревании до температур выше 400° у-форма окиси монотропно переходит в а-форму.
Третья модификация 3-Fc2O3, как и a-форма, является гексагональной, но в отличие от нее ферромагнитной. Она образуется при одновременном гидролизе и окислении солей желсза(П) (Chevallier, 1927; Glemser, 1939). При более длительном пагревапии до 11(Р она превращается в a-Fe2O3.
Гидроокиси железа (Ш) встречаются в природе в виде бурой железной руды (бурый железняк). Наибольшее значение для металлургии имеют следующие разновидности бурого железняка: «бурый волокнистый железняк» (ксаптосидерит), железная смолистая руда, бобовые руды, железный оолит, скандинавская приморская руда и болотные железные руды. Хорошо кристаллизуется гетит (игольчатая железная руда, пнрросидерит) и образующая обычно тонкие Таблички рубиновая слюда; к аморфным формам относится лимонит. Сильно загрязненные болотные железные руды, встречающиеся, например, в Северогерманской низменности, применяют для очистки газа или в качестве катализаторов, например при получении серы методом Чанса — Клауса (см. т. I, стр. 751). Из них получают также красные краски (железный сурик).
Из соединений FeaO3 с определенным содержанием воды в природе встречается только моногидрат окисла Ке2О3-Н2О пли КеО(ОН). Он существует в двух кристаллических модификациях — гетита * и рубиновой слюды, и, кроме Того, в «аморфном» ** состоянии — в виде лимонита. Содержание воды в природных бурых железных рудах всегда несколько выше, чем Это соответствует их формуле, так Кац FeO(OII) поглощает воду с образованием твердого раствора. Окись железа(Ш) (гематит) тоже может поглощать воду (до 8%) с образованием твердого раствора гидрогематита ***; последний дает характерную для гематита красную черту, в то время как остальные разновидности гидрата окиси железа дают более или менее буроватую черту (Posnjak, 191S; Haber, Bohm, 1925; Курнаков, 1929),
Гётит [a-FeO(OH)] и лепидокрокит [y-FeO(OH)] кристаллизуются ромбически. Первый изоморфен диаспору, последний образует ярко выраженную слоистую решетку. По данным Кайзера и Кратки (Weiser, 1935; Kratky, 1938), существует еще третья (метастабильная) модификация [(J-FeO(OH)], образующаяся при медленном гидролизе FeCl3. Другие соли железа(1П) при медленном гидролизе дают a-FeO(OH), а прп быстром — a-Fe2O3.
* Первоначально название «гётит» было предложено для рубиновой слюда (лепидокрокита). Однако впоследствии его стали применять для обозначения игель-чатых железных руд вообще.
** Лимопнт только внешне кажется аморфным. При рентгенографической ^исследовании он обнаруживает слабые, размытые интерференционные кольца, которые по своему положению совпадают с резкими интерферопционными кольцамп тётита.
*** На рентгенограмме гидрогематита обнаруживаются те же липии, что и у гематита.
Восьмая группа периодической систем»,
293
Относительная устойчивость и взаимные превращения различных модификаций окисла и гидроокиси приведены на следующей схеме:
6 ккал y-FeO(OH)--------> a-FeO(OH)
J1 4 ккал 2 ккал
Y-I'e2O3 a-Fe2O3
а КгьЯл
Указанные значения относятся к соединениям с неискаженной структурой решетки и с достаточной зернистостью, Вследствие малой склонности к кристаллизации они получаются часто в настолько мелкозернистом состоянии, что развитие поверхности заметно влияет на величины теплот образования (ср, стр, 720 и сл,). Еще сильнее сказываются в этом отношении искажения в структуре, часто появляющиеся при большом объеме кристалла (ср. стр. 36 и сл.). Так, Фрике (Fricke) удалось получить образцы у-модификации окиси железа(Ш), теплоты образования которых вследствие указанных причин различались почти на 6 ккал/моль. У препаратов a-Fe2O3, полученных обезвоживанием вязкой аморфной гидроокиси железа(Ш) при различных температурах, различия в теплотах образования достигали 13 ккал/моль Fe2O3' (Fricke, 1935), Окислы желез;! в энергетически возбужденном состоянии отличаются особо высокой каталитической активностью, поэтому такое состояние называется «активным» *. Однако и ряд других свойств, например величины давления диссоциации или констант электролитической диссоциации, в значительной степени могут определяться влиянием искажений в структуре и величиной Поверхности. Как видно нз указанных выше значений, теплоты образования образцов одной и той же модификации при различной активности иногда могут различаться больше, чем соответствующие величины для различных модификаций в нормальном состоянии.
При действии аммиака или других осадителей на растворы солей я;елеза(Ш) образуется окись железа(Ш) в виде буро-красного слизистоаморфного осадка, из которого при высыхании образуются гели с различным содержанием воды **, Обладая большой поверхностной активностью, этот осадок легко увлекает с собой из раствора другие вещества. Особенно специфична его адсорбционная способность по отношению к трехокиси мышьяка; поэтому FeO(OH) можно применять как противоядие при остром отравлении мышьяком. Гидрат окиси железа легко образует коллоидные растворы, и в таком виде его применяют в медицине,
Ферраты(Ш) и гидроксоферраты(Ш). Свежеосажденная гидроокись железа(Щ) растворима в разбавленных сильных кислотах. В горячих концентрированных растворах едкого кали или натра она заметно растворима с образованием ферратов/!11) калия и натрия KFeO2 и NaFeO2, Таким образом, гидроокись железа(Ш) обладает амфотерным характером, однако способность к кислотной диссоциации выражена у нее очень слабо.
Ферраты(Ш) (раньше называвшиеся «ферритами») лучше получать не из раствора, а сплавлением окиси железа(Ш) с окислами, гидроокисями или карбонатами щелочных металлов. Кроме ферратов(Ш) состава 1:1, таким образом можно получить п некоторые ферраты(1П) состава 1 : 2, например K2Fe40y. Известны и гидроксофер-
* Активное состояние можно наблюдать и у других соединений, склонных ь образованию дефектных структур (см. стр. 785 и сл.).
** Гидроокись железа(Ш), свежеосажденная из растворов солей, железа, оказывается рёнтг ено аморфной. По мере «старения» осадка на рентгенограмме появляются линии, характерные для гематита или соответственно гидрогематита. Если же осадки, образовавшиеся при осаждении понов железа(П) ионами гидроксила, окислить кислородом воздуха, то на рентгенограммах полученных препаратов обнаруживаются характерные для гётнта или лимонита линии. В соответствии с данными Хюттига (Hiit-tig; 1930) устойчивость гидратов окислов трех валентно го железа в природных условиях возрастает а следующем порядке: гидрогель аморфной гидроокиси железа(Ш) — гематит -> гидрогематит лимонит -> гетит.
294
Глава 7
pamu(III). Натриевая соль Na$ [Fe(OH)sl-5—йН3О была получена Шольдером (Schoi-der, 1936) при окислении раствора гидроксоферрата(П) натрия кислородом воздуха (при обычной температуре). Она представляет собой бесцветные мелкие кристаллы, довольно слабо растворимые в концентрировапиом едком натре. Водой или разбавленным раствором NaOH эта соль моментально разлагается с выделением в осадок гидроокиси желоза(Ш). Если проводить окисление раствора гидроксоферрата(П) при нагревании (до 40—60°), то вместо октагидрокео-сопи выделяется в осадок тоже бесцветная гсптагидроксоакво-соль Na4[Fe(OH)7(OH2)]-l—2Н2О. При еще более высоких температурах (100—130J) уже не образуется гидр оксо-соль, а осаждается безводная оксосоль NaFeOг в виде оливково-зе.геных кристалликов. Эта же соль в виде коричневокрасных кристалликов выделяется при нагревании выше 130° из 60%-ной щелочи. NaFeO3 образуется также при синтезе феррата(П1) натрия первым из приведенных методов. Причина различной окраски зеленой и красной форм фсррата(Ш) натрия пока не известна.
Красный феррат(Ш) натрия кристаллизуется по типу Na[HF2j. (Однако анионы [OFcO]- не проявляются в решетке в виде таких четких структурных групп, как ат о наблюдается с радикалами FHF в решетке Na[HFa],) Аналогичную структуру имеет и Gu[FeO2J. Пространственная решетка К.FeO,, построенная из бесконечного числа тетраэдров FeO4 и ионов К, в структурном отношении полностью соответствует пространственному мотиву Р-крист оба лита (Barth, 1935). Аналогичную структуру, по данным Гильперта (Hilpert, 1933), имеют и KbFcO3 и Pb(FeO2)2, а также другие ферраты(Ш) одпо- и двухвалентных металлов. Однако окислы некоторых двухвалентных металлов вместо ферратов)!!!] образуют с Fe2O3 оксо-соли — двойные окислы со структурой шпинели. Такие двойные окислы, не обладающие солеобразным характером, Ге2Оэ образует, например, с окислами Mg, Zn, Cd, Си11 или Мп11. Аналогично построен и окисел железа(Ц, III) — Fe3O4. Ферромагнетизм, свойственный большинству таких двойных окйслов, неоднозначно определяется их шпинельной структурой; так, шпинель, состоящая из окйслов кадмия и железа, парамагнитна, хотя другая модификация соединения CdO-Fe2O3, не обладающая шпинельной решеткой, ферромагнитна.
В структурном отношении к двойным окислам [а не к ферратам(Ш)] следует отнести и соединен не LiFeO2. Оно кристаллизуется по типу поваренной соли (а = = 4,14 А), причем ионы Li+ и Fe3+ статистически (т, е. совершенно нерегулярно) распределены в «катионных» узлах решетки (Barth, 1931). Аналогичная структура была найдена и у некоторых других дткшных окйслов— Li2TiO3 {а = 4,10 A), Na2CeOs (а = 4,82 А) и Na2PrO3 (а = 4,84 А).
Между железными шпинелями и обычной шпинелью имеется существенное структурное различно. В обычной шпинели ион Mg2+ расположен: в центре кислородного тетраэдра, а каждый нон Als+ — в центре кислородного октаэдра. В шпинели, состоящей из окйслов магпия и железа, в центрах кислородных тетраэдров находятся ионы Fe3+, а не Mg2-; Mg-'1 и оставшаяся половина ионов Fo3+ занимают места с октаодрп-ческой координацией. Указанные различия выражаются в том, что обычную шпинель изображают формулой Ala[MgO4], а же.тозомагниевую — MgFe[FeOJ или, точнее,. Mgi-a Fei+x [MgxFei_aO4j“ где г = 0,1—0,15. По данным Верная (Verwey, 1953), в структуре желеаомагниевой шпинели почти see (по все-таки не все) иопы Mga+ имели октаэдрическую координацию.
Окись желе за (II, III) Ь"е3О4 является двойным соединением FeO и Fe2O3, относящимся к классу шпинелей. Она образуется (в смеси с другими продуктами) при сгорании железных опилок на воздухе. Упоминавшаяся на стр. 281 «железная окалина» состоит главным образом из окисс железа(И, III). Fe3O4 представляет собой черный, нерастворимый в кислотах порошок уд. веса 5,1; т. пл. 1538°. В природе это соединение встречается в виде магнетита (магнитный железняк, черная железная руда) в больг-глии количествах и является важнейшим источником железа. Магпетвт ферромаг питая и обладает довольно высокой электропроводностью. Благодаря этому из него изготовляют электроды, применяемые прежде всегс для электролиза хлоридов щелочных металлов, а также штифтов для дуговых фонарей. В процессе Сименса — Мартена (ср. стр. 279 и сл.' магнетит наряду с железной окалиной применяют в качестве обезуглероживающего агента. Железная окалина, кроме того, используется для получения термита.
Восьмая группа периодической системы
295
Шпинельная структура магнетита была установлена рентгенографическим путем. Однако в его решетке в центрах кислородных тетраэдров вместо ионов железа(И) находится половина ионов железами). Остальные ионы жеаюза(Ш), а также ноны железа(П) занимают (по-видимому, распределяясь нерегулярно) места ионов алюминия в решетке обычной шпинели AI2[MgOJ. Такое строение магнетита передает фор-мула FeIIFeli:f[FeIIIO4]. То, что ионы Fo2+h Fes+, расположенные вне (FeOj-струк-турных групп, легко обмениваются зарядами, объясняет хорошую электропроводность магнетита (Vcrwey, de Boer, 1936). Параметр кубической ячейки магнетита a = 8,41 А. При уморенном нагревании на воздухе порошкообразный магнетит окисляется до Fe2O3~. Этот метод применяли еще в древности для получения красной окиси железа из черной магнетитовой руды. Прокаливание при очень высокой температуре (выше 1400°), наоборот, сопровождается переходом Fe2O3 в Fe3O>; при еще более высоких температурах происходит дальнейшее выделение кислорода с образованием FeO.
Сернистые соединения
При непосредственном взаимодействии серы и железа образуются два соединения:
FeS—сульфид железа, моносульфид, сульфид железа(П),
FeS3 —сульфид, дисульфид железа(П).
На диаграмме состояния системы железо — сера проявляются только эти два соединения; следовательно, они являются единственными продуктами, синтез которых возможен при непосредственном присоединении 8 к Fo. Не исключено, что другим путем (осаждением из водного раствора) можно получить еще один сульфид—трехвалепт-ного железа — ₽e2S3, однако доказательств его существования до сих пор пе получено (ср. стр. 296).
Моносульфнд железа, сульфид железа{11) FeS образуется при сплавлении железа с серой; этим способом его получают и в технике, где FeS применяют для получения сероводорода. Техническое сернистое железо бывает обычно загрязнено избытком металла. Оно поступает в продажу в виде темно-серых кусков или плиток. Чистый кристаллический моносульфид имеет светлый томпаково-коричневый цвет.
Из водных растворов солей железа(П) при действии ионов S" моно-сульфид железа выпадает в виде черного осадка. Этот осадок в воде практически нерастворим, по растворяется в разбавленных кислотах. Во влажном состоянии оп легко подвергается частичному окислению воздухом до сульфата.
В природе сернистое железо встречается в виде магнитного колчедана (пирротина), кристаллизующегося гексагонально. Аналогичное по кристаллической структуре сернистое железо, встречающееся в метеоритах, называется троилитом. Магнитный колчедан почти всегда содержит никель, поэтому его используют для получения никеля. Содержание серы в магнитном колчедане почти всегда на 1—2% выше, чем это соответствует формуле FeS. Избыток серы включается в решетку сернистого железа.
FeS кристаллизуется по типу NiAs (ср. стр. 268), Его способность присоединять некоторый избыток S (до 3,3 ат.%) основывается па том, что в ого кристаллической решетке часть мест, принадлежащих атомам Fe, может оставаться не занятой. Плотность FeS колеблется между 4,5 и 5,0. Теплота образования составляет 22,8 ккал /моль.
FeS смешивается с Fe в расплаве в любых соотношениях. В твердом состоянии, однако, опи практически нерастворимы. Поэтому при затвердевании расплавов желоза даже с очепь пизким содержанием серы выделяется FeS, который (так же как и FeO) делает железо красноломким, т. е. ковка при красном калении сопровождается в этом случае образованием трещин. Однако при добавлении к железу достаточного количества марганца последний полностью присоединяет серу с образованием MnS, очень мало растворимого в железо-марганцовом расплаве. На Этом основан процесс обессери-
296
Глава 7
вапия чугуна с помощью марганца. Другие содержащиеся в чугуае примеси — С, Р и Si,— еще болев значительно понижая растворимость MnS в расплаве, способствуют обессериванию. Недавно были получены сплавы железа и марганца, в которые добавлена сера, так называемые сернистые стали.. При затвердевании таких сплавов вместо хрупкой и^потому вредной примеси (FeS) в виде очень мелких включений выделяется пластичный MnS. Это приводит к тому, что сернистые стала отличаются исключительной твердостью.
Из водных растворов солей железа(Ш) при добавлении ионов Sff выпадает черный, практически нерастворимый в воде осадок, состав которого соответствует формуле Fe2Sg. Он растворим в разбавленных кислотах. Во вл ал,ном состоянии он легко разлагается на воздухе, образуя гидрат окиси железа и серу. Он выделяет серу и при реакции с соляной кислотой ио уравнению:
FeaS3 4- 4НС1=2FeCl2+2HZS Ц- .8.
Однако маловероятно, что этот осадок является индивидуальным соединением — сульфидом железа(Ш). Он рентгеноаморфен и, по-видимому, представляет собой смесь моносульфида железа и серы, образовавшихся в результате реакции
2Fe— + 3S"=2FeS-|-S,
При непосредственном соединении железа с серой, как уже было указано, сульфид трехвалентного железа не образуется. Тем не менее ой встречается в виде кристаллических двойных соединений. В качестве примеров можно привести минералы — медный колчедан Cu-,>S-Fe3S3 и пестрый медный колчедан (борнит) ЗСгиЭ-FegSg.
Дисульфид железа FeSa очень рас пр о ст рай ей в виде пирита (серный или железный колчедан). Наряду с пиритом дисульфид встречается и в виде марказита (водный или двойной колчедан). При высокой температуре оп легко отщепляет серу. При прокаливании на воздухе он сгорает, образуя Fe2O3 и SO2. Легкости образования сернистого газа из пирита обусловливает его применение в качестве главного исходного продукта для производства серной кислоты. Благодаря значительной теплоте окисления иприта и легкости его разложения обжиг в печах, начавшись, протекает дальше уже самопроизвольно. О применении пиритных (колчеданных) огарков, получающихся после обжнга колчедана, уже говорилось вт. I. В радиотехнике кристаллы пирита применяют в качестве детекторов.
Пирит и марказит окрашены1 в желтый цвет латуни л обладают металлическим блеском. Они различаются по кристаллической структуре.. Пирит, нередко встречающийся в виде хорошо образованных крупных кристаллов (болвшей частвю в виде кубов и пентагондрдекаэдров иле комбинации этих форм), относится к пептагонально-гемиэдрическому классу кубической сингонии, кристаллы марказита — к ромбической.
В 'решетке иприта атомы Fe расположены таким же образом, как атомы Na в решетке поваренной соли. Места атомов хлора в данном случае занимают центры тяжести двух атомов S, причем прямые, соединяющие их, расположены наклонно к осям решетки. Расстояние S <—► S составляет 2,14 А. Ребро элементарного куба в ячейке пирита а = 5,40 А. Структура мариалита родственна решетке рутила, однако имеет более низкую симметрию. Расстояние S«—>• S составляет в ней 2,21 А.
Магнитные измерения показали, что в пирите и в других дисульфидах подобного типа (MnS2, CoSй, NiS2) металлы не существуют, в 'ютыде.гвалентпом состоянии (KlemEj, 1935), MnS3, судя по его магнитным свойствам, можно рассматривать как дисульфщ
Восьмая группа периодической система
297
марганпа(I I) Мnа+(So)2“, однако во елабовыраженным гетеренолярным характером. Еще менее гетер опоя яр ны другие перечисленные дисульфиды, причем наименее гетерой олярным является FeS2. Природа связей, существующих в этих веществах, приближается к интерметаллическим соединениям.
Дисульфиды железа образуют с сульфидами некоторых тяжелых металлов двойные сульфиды. Они, однако, не способны к образованию собственно тио-солей и потому нерастворимы в растворах сульфидов щелочных металлов. Тем не менее сера может частично замещать гидр ок с о-группы в гидр оксоферратах (II) и (III) с образованием г и др о квот ио-соле й (Sch older, 1936), например Na3[SFeII(OH)3],2H2O (черно-зеленого цвета) и м_ гНгОрЛи ч к 1И0Н2> 9IT (темно-коричневого). Образованием 8ЦНО)в (OH'JSJ i*12v
такого рода соединений обусловлена зеленая или темно-красная окраска, возникающая при действии NasS на сильпощелочные растворы, содержащие двух- или соответственно трехвалептпое железо.
Соли железа(И)
Растворимые соли железа(П), например ого сульфат или хлорид, удобнее всего получать реакцией металлического железа с соответствующей кислотой. Водные растворы и кристаллы солей железа(П), содержащие кристаллизационную воду, окрашены в бледно-зеленый цвет, который, таким образом, является цветом ионов Fe". Растворы солей двухвалентного железа вследствие гидролиза имеют кислую реакцию. Нерастворимые соли двухвалентного железа получаются из его растворимых солей' путем обменных реакций. Большинство солей железа(Л) склонно к образованию двойных и комплексных солей с солями активных металлов (главным образом щелочных) и аммония. На воздухе соли двухвалентного железа в большинстве случаев не вполне устойчивы, причем чаще двойные соли бывают более прочными, чем простые. При действии сильных окислителей соли железа(П) количественно превращаются в соли железа(Ш).
Следует отметить, что при растворении железа в кислотах, апиопы которых обладают ярко выраженными окислительными свойствами, например в азотной, хлорной, хлорноватой, бромиоватой, йодноватой кислотах, образуются соли двух-, а не трех-валептного железа (предполагается, что кислоты находятся в разбавленных растворах и растворение происходит на холоду). Сами по себе ионы Fe” пе обладают большой склонностью к окислению до ионов Fe"'. Действительно, па платиновой пластинке, погруженной в раствор, содержащий в одинаковых концентрациях ионы Fe” и ионы Fe'“, обнаруживается потенциал па 0,75 в выше потенциала нормального водородного электрода. Таким образом, в замкнутой цепи происходит самопроизвольная перезарядка ионов железа(Ш) до ионов железа(П), в то время как водород переходит в ионное состояние. Сильно выраженная склонность к окислению соединения железа(П) в значительной степени определяется нарушениями окислительных равновесий. Оно обусловлено чрезвычайно трудной растворимостью окиси железа(1П), легко образующейся в результате гидролиза в водных растворах или в присутствии влаги. Поэтому устойчивость солей двухвалентного железа в водных растворах можно значительно повысить добавлением избытка кислоты, затрудняющей гидролиз.
При действии на металлическое Железо водного раствора S02 наряду с сульфитом железа(11) образуется и тиосульфат желева(Л):
2Fe4~3SO2;=FeSO34-FeS2O3.
Интересно образование дитионата желеаа{11) FeS2Og-5H2O при действии сернистой кислоты на разбавленный раствор сульфата трехвалентного железа при 0°. При окислении тиосульфата железа (II) хлоридом жслеза(Ш) получается тетратионат желева(И):
2FeS2O3 2FeCI3 ^FeS^Og 4-3FeCl2.
Галогениды железа(И)
Фторид железа(П) и фтороферраты(П). При упаривании раствора железа в водном растворе плавиковой кислоты выделяется бесцветный фторид двухвалентного'
298
Глава 7
железа FeF2-8H20, содержащий кристаллизационную воду, по мало растворимый в воде. Безводный фторид, также бесцветный, получается при пагревапии водной соли в токе сухого фтористого водорода. Его можно также получить, действуя фтористым водородом на железо при высокой температуре. Из растворов, содержащих фторид железа(Ц) и щелочные фториды, кристаллизуются двойные соли [g5?aepo<feppa-гам(/7)1 типа M4FeF3] п M}[FcF4].
Хлорид железа(Н) и хлороферраты(П). Хлорид железа(Н) FeCb получается в безводном состоянии в виде бесцветной массы при пропускании сухого газообразного хлористого водорода над железными опилками при красном калении, при нагревании хлорида железа(Ш) в токе водорода или при нагревании иодного раствора хлорида железа(11) (см. ниже) без доступа воздуха. При температуре желтого каления он летуч (плотность его пара при 1000° соответствует формуле FeCl2); на воздухе FcCla расплывается и легко растворяется в водо и в спирте, Водные растворы FeCla удобнее всего получать растворением железа в разбавленной соляной кислоте без доступа воздуха (в колбе с вентилем Бунзена). При упаривании их кристаллизуется тетрагидрат FeCl,-4H2O в виде сипевато-зелевых моноклин-пых расплывающихся кристаллов. Его растворимость при 20° составляет 08,5 г- FoC12 в 100 г воды. С хлоридами наиболее электроположительных металлов FeCl2 образует двойные соли, преимущественно типа M^FeClj] [тетралмороферраты(11)}. Безводный хлорид двухвалентного железа энергично присоединяет аммиак.
Бромид железа(П) FeBr2 [в отличие от хлорида железа(П)[ непосредственно образуется при сгорании железа в парах брома в присутствии избытка железа. Из раствора железа в водном растворе бромнетоводородной кислоты при сильном охлаждении выделяется FeBr2- 6И2О в виде светло-зеленого кристаллического осадка. Выше 45° гексагидрат переходит в тетрагидрат.
Иод ид железа(П) Feb можно получить прямым соединением компонентов. В воде, содержащей иод, железо медленно растворяется уже при обычной температуре. При упаривании раствора в эксикаторе из пего выделяется FeL,-4H2O в виде зеленых кристаллов. При более низких температурах образуются его высшие гидраты, Водкые соли и их растворы при нагревании до 56° чернеют, но при остывании вновь приобретают Пе рв Отта чал ь ним цвет.
О продуктах присоединения СО к галогенидам жолеза(П) см. стр. 377 и 383.
Другие соли жедеза(И)
Роданид железа(И) выделяется при упаривании раствора железа в водной роданпегов одеродной кислоте (в огсугсгвие доступа воздуха) в виде зеленых призм состава Fe(SCN)2-3H2O. Известны также роЗано-сми тина N1J (Fe(SCN)a].
Нитрат железа(П) образуется при растворении железа в разбавленной азотной Кислоте на холоду:
8Fe 4- 20HN О:3=8Fe(NO3)s -|- 2NH4NOS+ 6НгО.
Б чистом состоянии его лучше получать обменной реакцией сульфата железа(П) с нитратом свинца. Он кристаллизуется обычно в виде гекса гидра та Fe(NO3)2-БН3О, образуя си ста о-Зе левые ромбические таблички. Ниже —10° устойчивым является нонагнярат Fe(!NO3)2‘ 9Н2О. Водный раствор нитрата железа(П) разлагается при кипячении с образованием основного нитрата железа(Ш).
Перхлорат желеаа(П). В разбавленной хлорной кислоте при низкой температуре железо растворимо с образованием перхлората жедеза(П). Синтез этого соединения, по Лпндштранду (Lindsbrand, 1936), лучше всего вести при растворении FeS в хлорной Кислоте, так как в этом случае можно работать при обычной температуре и с более высокими концентрациями кислоты; креме того, в этих условиях не происходит окисле-
Восьмая группа периодической системы
299
пия до перхлората железа(Ш). Перхлорат двухвалентного железа кристаллизуется из раствора в виде гексагидрата Ге(СЮ4)2-6Н2О— светло-зеленых гексагональных призм, сохраняющихся в атмосфере, осушенной СаС12 (в присутствии влаги воздуха кристаллы расплываются и одновременно окисляются). В воде эта соль исключительно хорошо растворима (При 0е—80,12, при 60°—106,6 г Fe(C104)2 в 100 г воды); тоже относится и к спирту. В присутствии НС,Ю4 растворимость значит ел ьп о понижается.
Сульфат железа (II) кристаллизуется из водных растворов обычно с 7 молекулами воды в виде железного купороса FeSO4-7H2O. Последний является наиболее важной в техническом отношении солыо железа. Уже в XIII в. имеются упоминания об этой соли железа у Альбе ртуса Магнуса, который называет ее зеленым купоросом. Железный купорос образует светло-зеленые моноклинные кристаллы удельного веса 1,88. На воздухе они медленно выветриваются и одновременно окисляются с поверхности, переходя в желтовато-коричневый основной сульфат железа(Ш). При нагревании железный купорос легко теряет 6 молекул воды; отщепление последней молекулы происходит значительно труднее. Безводная соль представляет собой белый порошок удельного веса 3,0. При еще более сильном прокаливании железный купорос разлагается, выделяя сернистый газ:
2FeSO4=(FeO)2SO4 + SO2.
В технике железный купорос получают растворением железа в разбавленной серной кислоте или подвергают выветриванию па воздухе серный колчедан и часто его увлажняют. В качестве побочного продукта железный купорос выделяется при техническом получении хромовых квасцов. Его применяют для производства чернил и берлинской лазури; в красильном дело (для окраски шерсти в черный цвет), для консервирования дерева, для уничтожения сорняков; иногда его используют и в медицине. Для очцетки железный купорос высаливают спиртом из водного раствора.
В 100 г воды растворяется; при 20°— 26,6, при 56° — 54,4, при 64° — 54,9 и при 90° — 37,2 г FeSO4. Из водных растворов выше 56° FeSO4 кристаллизуется с 4, а выше 64° — с 1 молекулой Н2О. Железный купорос, кроме мопоклиппых, образует ромбические или триклинные кристаллы в том случае, когда кристаллизация из пересыщенного раствора вызвана внесением затравки ромбического цинкового купороса или соответственно триклинного медного купороса; в последнем случае кристаллизуется пента г и драт.
В природе железный купорос встречается в незначительных количествах — в виде выцветов на железном колчедане.
С сульфатами щелочных металлов сульфат желез а (II) образует двойные соли, обычно состава MJSO4-FeSO4-6HaO. Наиболее известна из них аммонийная двойная соль (введенная Мором в объемный анализ) (NH4)2SO4-FeSO4-6H2O. Соль Мора на воздухе не выветривается.
Эти двойные еоди следует, вероятно, рассматривать как дисулъфатоферраты(J Mb [Fe(SO4)2]-6Н2О. Лежащая в их основе кислота Н21FefSOiJjjl известна в безводном состоянии и в виде гидратов с 6,5 и ЗН2О. Описаны также двойные сульфаты железа с сульфатами двухвалентных металлов (в первую очередь с Be, Mg, Zn н Cd). С сульфатами двухвалентных хрома и марганца сульфат жслеза(П) образует пе двойные соли, а смешанные кристаллы.
Карбопат железа(П) FeCO3 встречается в природе в виде железного шпата. Последний подобно известковому шпату кристаллизуется в виде ромбоэдров, нерастворимых в воде. Как и карбонат кальция, он раство
300 Глава. 7
ряется в водном растворе угольной кислоты, образуя кислый карбонат железа(П) (гидрокарбопат железа) Бе(НСОз)2. В таком виде он содержится в воде некоторых минеральных источников. Прн соприкосновении с воздухом эти воды вскоре выделяют гидрат окиси железа, причем избыток угольной кислоты улетает, а выделяющийся карбонат подвергается гидролизу и окисляется кислородом воздуха. Белый осадок карбоната двухвалентного железа, образующийся при добавлении растворимых карбонатов к растворам солей двухвалентного железа, прн доступе воздуха быстро темнеет, а затем и буреет.
При нагревании FeCO3 разлагается на FeO и С03, образуя целый ряд промежуточных продуктов. Давление его диссоциации уже при 282° достигает 1 атм. Энергия диссоциации составляет 20,9 ккал.
Оксалат же,чеза(П)п оксаяатоферраты(П). В водном растворе щавелевой кислоты железо растлоряется с выделением водорода. Из раствора оксалат выделяется в виде-лимоипо-Желтых кристаллов 2FeC2O4-3H2O. Из растворов, содержащих одновременно оксалаты щелочных металлов, можно высолить спиртом двойные оксалаты [оксалато-ферраты(10) состава МJ[Fe(C2O4)2], содержащие различное количество воды в зависимости от природы катиона.
Смесь растворов железного купороса и оксалата калия иногда применяют в фотографии в качестве проявителя.
Силикаты железа (II) получают сплавленном окиси двухвалентного железа с кварцевым песком. В природе силикаты желез а (П) очень широко распространены, но почти всегда встречаются в виде изоморфных смесей с другими силикатами. Сюда, например, относятся оливин (Mg, FeJatSiO^l, гиперстен (Fe,Mg)2[Si2Oe], бронзит (Mg, Fe)2)Si2Oel. Примером двойного силиката двухвалентного железа, имеющего стехиометрический состав, является геденбергит CaSiO3- FeSiO3 или CaFe[Si2Oe].
Фосфаты железа (11). Ортофосфат двухвалентного железа Fe5(PO4), •8Н2О, содержащий кристаллизационную воду, встречается в природе в виде вивианита (синяя железная руда); этот минерал, белый на свежих изломах, па воздухе немедленно окрашивается в синий цвет.
При добавлении к растворам солей железа(И) фосфата натрия получаются фосфаты желсза(П) в виде белых, нерастворимых в воде осадков,, имеющих в зависимости от условий осаждения различный состав.
Цианоферраты(II); желтая кровяная соль. Цианид железаЦ!) в чистом состоянии не известен. При реакции между ионами Fe" и CN' немедленно получаются комплексы. В присутствии достаточного количества ионов CN' всегда образуется особо устойчивый комплекс гексацианоферратЦI) [Fe(CN)8J4-. Известно большое число его солей — гексацианоферра-тое{1 1) M}[Fe(CN)e] (кристаллизующихся обычно в виде гидратов). Важнейшая из них гексадианоферрат(П) калия (желтая кровяная соль, см. ниже). Щелочные и щелочноземельные соли этого комплекса, за исключением гексанианоферрата(П) бария, легко растворимы в воде. Существующие в их водных растворах ионы [Fe(CN)el"" весьма прочны и поэтому не обнаруживают обычных реакций на Fe" и CN'. С двухвалентными ионами тяжелых металлов ионы [Fe(CN)e]"" образуют в основном трудно-растворимые осадки; некоторые из них вследствие характерной окраски или кристаллической формы можно применять для открытия соответствующих тяжелых металлов.
Восьмая группа периодической системы
301
По данным Бринтцингера (Brintzinger, 1934 и сл.), Цианоферрат(II)-ионыв водном растворе находятся в сильно гидра тировали ом состоянии. Как следует из данных скоростей .их диффузии, они содержат 12 молекул Н2О в противоположность ццано-феррат(П1)-ионам, скорость диффузии которых соответствует весу пегидратировапных ионов [Fe(CN)e]s-.
H4[Fe(CN)6]—железистосинеродистая кислота, соответствующая гексациапо-ферратам(П), осаждается в виде эфирата из эфирных растворов ее солей при добавлении к ним концентрированной соляной кислоты. При удалении эфира можно получить свободную кислоту. Она представляет собой белый порошок, устойчивый в сухом состоянии, но во влажном воздухе постепенно окрашивающийся в синий цвет. II4[Fe(CN)e) — довольно сильная четырех осн овна я кислота, хорошо растворимая в воде и еще легче — в спирте. Опа обладает способностью присоединять органические кислородсодержащие молекулы (панример, эфиры, спирты, альдегиды, Кетоны, сложные эфиры), образуя оксопиевые соли, й поэтому ее можно в известной мере рассматривать как реактив на «координационно ненасыщенный кислород».
Н4 [Fe(ClN)e] образует своеобразные продукты присоединения с серной кислотой, например H4[Fe(CX)l5]-7 H2SO4 — ромбические таблички; H4.[Fe(CN)6]-.5HaSO4— Иглы. Известны также двойные соли цианоферратов(II) своеобразного состава, напри-мер: (NH4)4[Fe(CX]er2(XH4)CI-3HaO; K(NH4)3|Fe(CN)e]-2(NH4)CI; K2Na2[Fe(CN)6b 4KNO3,
Известны и эфиры железистосинеродистой кислоты.
Циан о феррат (П) калия, точнее гексацианоферрат(11) калия, известен уже с середины XVIII в. Раньше его получали внесением азотсодержащих органических веществ (например, крови животных, копыт, кож и т. и.) вместе с железным ломом в расплав карбоната калия; по остывании расплавленной массы образовавшуюся желтую кровяную соль выщелачивали водой. Отсюда произошло популярное название соединения «желтая кровяная соль».
В настоящее время желтую кровяную соль получают главным образом иа отработанной массы, остающейся после очистки газов на газовых заводах. Эта масса поглощает цианистый водород, образующийся при сухой перегонке каменных углей, и содержит его главным образом в виде берлинской лазури (см. ниже). При нагревании с гашеной известью большую часть берлинской лазури вначале переводят в легкорастворимый цианоферрат(П) кальция; выщелачивают его водой и к раствору добавляют хлорид калия, после чего выпадает двойная соль К2Са [Fe(CN)0]. Эту соль переводят в чистый цианоферрат(П) калия, действуя рассчитанным количеством карбоната калия К2Са [ Fe(CiN)aj -|- К.2СО2 = K4[Fe(CN)a] -|- СаСО3.
Цианоферрат(Ц) калия кристаллизуется из водных растворов в виде гидрата K4[Fe(CN)6]-3H2O, образующего довольно мягкие моноклиппые кристаллы с удельным весом 1,85. При 100° кристаллы теряют воду и переходят в белый гигроскопичный порошок, при более сильном нагревании разлагающийся с отщеплением азота.
В 100 г воды при 0° растворяется 14,5, а ври 98°—74 г безводного цианоферрата(II) калия. Вследствие большой прочности радикала гоксацианоферрата(11) желтая кровяная соль не ядовита. Она может служить противоядием при отравлении медными или железными солями. В аналитической химии ее используют в качестве реактива на соли железа, меди и других тяжелых металлов.
При совместном присутствии в водном растворе цианоферрат(П)-иопов и комплексных катионов хрома(Ш) или кобальта(1П), по данным Бринтцингера (Brintzinger), происходит отщепление воды с образованием двухслойных комплексных ионов (ср. стр, 160), например:
4[Fe(CN)a(OH2)12]4* 4- [Ст еп3)з+ =
[Fe(CN)eJ ч*з-
IFe(CN)e) ICr ens] [Fe(CN)eJ + 48Н2О
[Fe(CN)e] J
По мнению Бринтцингера н О сев а льда (Brintzinger, Osswald, 1935), справедливо положение О том, что анионы, образующие в растворе акво-ионы, теряя воду, могут присоединять комплексные катионы и образовывать двухслойные комплексные ионы (ср. также стр. 328).
302
Глада 7
Соли железа(Ш)
Соли трехвалентного железа получаются при окислении соответствующих солей железа (II) (например, азотной кислотой или перекисью водорода) или при растворении свежеосажденного гидрата окиси железа(П) соответствующими кислотами. В отношении растворимости они очень похожи на соли железа(П). Растворы солей железа(Ш), не содержащие избытка кислоты, окрашены в цвета от желтова то-бу рог о до темно-коричневого. Однако эта окраска не является цветом иопов железа(Ш), а зависит от присутствующих в таких растворах в коллоидном состоянии основных солей ели гидрата окиси железа, образующихся в результате гидролиза. Вследствие гидролиза растворы солей желеэа(Ш) имеют также сильноецелую реакцию. Если подавить гидролиз добавлением кислоты, то обычно образуются ацпдо-комнлексы. В этом случае окраска растворов в присутствии избытка кислоты зависит от природы анионов соли и добавленной кислоты. Так, растворы фторида железа(Ш) при добавлении соляной кислоты окрашиваются в розовый цвет, а растворы хлорида желе-за(1Н) — в желтый. Однако в общем растворы солей трехвалентного железа, содержащие избыточную кислоту, бесцветны. Действуя на соли желе-за(1П) восстановителями, например сероводородом, SnCl2 или цинком в соляной кислоте, можно количественно перевести их: в соли железа(П). Реакция с иодидом калия в кислом растворе также протекает количественно;
Fe- + I'=Fe"4-i/2I2. (11)
Эти реакции лежат в основе объемных методов определения трехва-лентного желоза. Полученный в первом случае путем восстановления раствор соли железа(П) титруют перманганатом, а после реакции Fe“ с йодидом титрованием тиосульфатом определяют иод, выделившийся в соответствии с уравнением (tl).
Галогениды железа(Ш)
Фторид а;< л\;а(Ш| п фтороферраты! И1). Фторид трехеалентного железа вы деля стся из раствора гидроокиси в плавиковой кислоте в виде светло-розовых мелких крист ал аг.-в 4,5-гидрата при обычной температуре (упаривание пад хлористым кальцием} и 3-гидрата — при более высоких температурах (упармваппе па водяной бане). [3-Гидрат, по данным Нильсена (Nielsen, 1940), образует две модификации. 1 Безводное соединение окрашено в зеленоватый цвет. Оно кристаллизуется гексагонально, плотность его 3,52. Из растворов, со,держащих хлориды щелочных металлов или аммония, кристаллизуются двойные или комплексные соли [jfimopoферраты (///)] преимущественно типа MjfFeFjJ (часто содержащие кристаллизационную воду или HF), а также типов Mj[FeFeJ и MI[FeFi] (Remy, 1933).
Хлорид желсза(Ш) п хлороферраты(Ш). Безводный хлорид железа [хлорид железа (Ш), трихлорид железа [ проще всего получать нагреванием металлического железа в текс сухого хлора. Образовавшийся хлорид сублимируется и осаждается в виде темно-коричневых блесток. Его удельным вес 2,90; температура плавления около 300°, температура кипени» 317°. При нагревший в токе воздуха или при пропускании водяногс пара над нагретым хлоридом жслеза(Ш) он переходит в окись желсза(Ш)
2FeCl3 4~3/2О2=Ре2Оз Ц~ ЗС12,
2ГеС13 + ЗН2О=ГегО3+бНС1.
Хлорид железа(Ш) кристаллизуется в виде гекс а г она левых табличек, отшз вающих зеленоватым металлическим блеском, а в проходящем свете имеющих грана
Восьмая группа периодической системы
303
тово-краспый цвет. При нагревании в вакууме выше 500° хлорид железа(Ш) частично разлагается ио уравнению
2FeCl3 2РеС12-фС12. (12)
Как следует из определения плотности пара [при этих измерениях во избежание разложения FeCl3 по уравнению (12) добавляют свободный хлор], хлорид железа(Ш) л парах при сравнительно низких температурах (— 400°) почти полностью димерен, выше 750° оп нацело диссоциирует па молекулы FeCI3. В спиртовом и эфирном растворах, как было установлено эбуллиоскопическими измерениями, FeCl3 находится в мономерном состоянии.
Во влажном воздухе хлорид железа(Ш) жадно притягивает воду. При этом он расплывается и переходит в темно-коричневую жидкость (Oleum martis). В воде и в спирте оп растворяется очень легко, причем происходит заметное разогревание. С водой FcCl3 образует ряд гидратов. Обычный продажный желтый хлорид железа является гексагидратом ВсС1з-6Н2О. Этот гидрат нередко выделяется из растворов в виде характерных образований (сферолитов). В техникеРеС13-6Н2О обычно получагот растворением железа в соляной кислоте, пропуская через раствор ток хлора для окисления хлорида желеаа(П), образующегося наряду с FeCl3, с последующим упариванием в керамических чашах на паровой бане.
Растворы хлорида железа(Ш) вследствие гидролиза имеют сильнокислую реакцию. Они вызывают быструю коагуляцию яичного белка. На этом осповано их применение (а также ваты, смоченной раствором FeClg) в качестве кровоостанавливающего средства. В технике хлорпое железо применяют главным образом как окислитель в производстве органических красителей, а в некоторых случаях оно служит конденсирующим и хлорирующим агентом; кроме того, нм пользуются как протравой при окраске тканой.
Из растворов, содержащих хлориды щелочных металлов или других электроположительных элементов, кристаллизуются двойные соли —хлороферраты'III). Среди последних наиболее распространены типы MI[FeCl4] и MJjFeCls) (Пегау, 1925).
Если гидролиз раствора IeGl% доведен ДО стадии выделения твердого вещества, то при обычной температуре устойчивым конечным продуктом, как показали рентго-яографическпе определения, является всегда a-FeO(OH) (гены). Вначале осаждаются основные хлориды, переходящие затем через стадию у-FeO(ОН) в cx-FeO(OH). Этот переход, как правило, происходит очень медленно. Поэтому осадки, полученные при медленном гидролизе на холоду, часто даже после месячного хранения под раствором, содержат еще основной хлорид железа. Если основные хлориды образуют коллоидные растворы, для полного превращения их в FeO(OH) иногда требуются месяцы. Лидер и Яр (Jander, Jarh) ну тем измерения скорости диффузии, а также спектров поглощения показали, что в растворах, содержащих избыток соляной кислоты пли легко растворимых хлоридов, т. е. в условиях существования хлороферрат(Ш)-ионов, например [FeClJпри постепенном понижении концентрации ионов Н‘ в первую очередь происходит обмен хлора па ОН по схеме:
[Li’cClJ’ + OH' =р* [Ге(ОН)С13]' + С1',
[ГеС13(ОН)Г-|-ОН‘ +* [Fc(OII)2Ctz]' + Cl'.
Этот процесс закапчивается осаждением основных хлоридов или гидроокиси. Однако железо, оставшееся в растворе, существует опять-таки в виде мономерных гид рок сохл оро-попов, находящихся в равновесии непосредствен по с твердой фазой. Итак, в данном случае осаждеппю не предшествует образование в растворе высокомолекулярных продуктов гидролиза, как это имеет место, например, в растворах перхлората и нитрата желоза(1П) (см. ниже).
В результате гидролиза при нагревании образуется a-Fe2O3. Резкое снижение концентрации ионов Н’, наблюдающееся при добавлении избытка щелочи ими аммиака, приводит к осаждению из растворов аморфной гидроокиси железа(1П).
304
Глава 7
Оксихлорид железа | оксихлор ид жалеза(Ш), оспенной хлорид железа] FeOCl в чистом состоянии лучше всего получать нагреванием в запаянной ампуле смеси FegOj п FeCl3 примерно до 350: иля Fe,O3 в токе сухого НС1 до ~ 240°. Он представляет собой легко расщепляющиеся ромбические листочки, обладающие металлическим блеском, в проходящем свете окрашенные в красивый красный цвет. Уд. вес его 3,55. Оксихлорид железа имеет ярко выраженную слоистую структуру, очень похожую па структуру y-FcO(OH).
Бромид железаПН) (триброМид железа), FoBr3 — коричнево-красные таблички; ,этб соединение вполне аналогично хлориду. Йодид желегауП) устойчив только в равновесии с большим избытком иодида железа(П): Frf3 z±: Felg-f-Vz I2.
Другие соли железа (III)
Перхлорат железа(Ш) Fe(CJO4)3 иолучэют реакцией FeCl, с хлорной кислотой в газовой фазе. Из водного раствора при обычной температуре он выделяется в виде .декагидрата Fe(C104)3-ЮНгО — кристаллов, слабо окрашенных в розовый цвет, обладающих сильным двойным лучепреломлением, не теряющих воду при высушивании над хлористым кальцием и очень гигроскопичных; однако над концентрированной сорной кислотой или над Р2О3 этот гидрат теряет 4 молекулы Н30 (Lindstrand, 1936). Выше 42° из водного раствора кристаллизуется понагидрат Ре(СЮ4)3- 9Н2О. Растворимость перхлората железа(Ш) в воде (при ба—96,99 г, при 60°—-142,0 а на 100 г воды) оказывается больше, чем перхлората железа (II). В спирте эта воль тоже очень легко растворима.
Как показал Лидер (Jaader, 1930 а с л.), гидролиз перхлората железа(1П) в водном растворе протекает совершенно иначе, чем в случае хлорида железа(Ш),— одновременно с заменой кислотных остатков группами ОН происходит конденсация, приводящая к образованию высокомолекулярных продуктов, примерно по следующей схеме:
2Fe(C104)34-H20 (C104)2Fe.—О—Fe(C104)2-|-2IIG104,
2(C104)jFe —О —Fe(C104)2-|-H20 (C104)2Fe—О—Fe—О—Fe—О —Fe(ClQ4)2-r
CiO4 С1О4
4-2НСЮ4 и т, х
Уже в относительно кислых растворах продукты гидролиза указанного типа находятся: в равновесии с пор мал китами солями (или их иолами). С уменьшением концеитрацт— попов И' (например, при медленном добавлении едкого патра) равновесие непрерыаа сдвигается (по не скачкообразно, как это происходит при конденсации ионов большинства кислтп.') по мере роста размеров частиц продуктов гидролиза. Верояыу.. частицы вначале растут преимущественно в одном направлении, и только после это?., образуют длинные цепи, присоединяясь к столбикам или листочкам. Частицы, достигающие таким путем молекулярного веса 40 000 и более, как правило, еще осгаютес в растворе (коллоидном) вплоть до перехода в чистую гидроокись FeO(OH), coup осуждающегося отщеплением последних кислотных остатков. Однако, если только растгу-ры достаточно кислые, в конце концов осаждается устойчивый конечный продует гидролиза — гидроокись железа в форме a-FeO(ОН) (гётнта). Иногда устанавливаете такое равновесие, когда основные соли, обладающие достаточно высоким молекулярным весом, оказываются устойчивыми в растворе в присутствии твердой фазы a-FeO(ОЕ В соответствии со сложным характером превращений равновесия гидролиза устанут ливаются в большинстве случаев исключительно медленно, иногда только в тече~. недель или месяцев.
Яндер и Яр (Jander, Jahr) установили соответствующий механизм гидролсу. и для нитрата железа(Ш) (в этом случае в растворах, содержащих избыток аз оглу у кислоты, в равновесии с продуктами гидролиза существуют питрат-ионы). Гидрату перхлората железа(1П), вероятно, типичен для солей железа(Ш) и сильных кпеу— не склонных к комплексообразованию с ионами Fe'" или обладающих этой спсе:у ностью лишь в слабой степени. Гидролиз солей хрома и алюминия в водных раствсру-протекает в основном таким же образом. В этих случаях с понижением концентргуу-ионов Н‘ вместо скачкообразного роста средней величины частиц продуктов геу лиаа, наблюдается их равномерное увеличение. Поэтому и в таких^ растес-у; никогда не существует продуктов гидролиза с частицами какой-либо одной величину, а всегда присутствуют частицы различных размеров, возрастающих по мере углуйу-ния гидролиза.
Восьмая группа периодической системы
305
Нитрат железа (Ш) образуется при растворении железа в 20—30%-ной азотной кислоте. При обычной температуре в зависимости от концентрации и содержания в растворе кислоты он кристаллизуется в виде ночти бесцветных кубцков, имеющих состав Fe(NO3)3-6HaO, или в моноклинных кристаллах состава Fe(NO3)3'9HaO *. В воде нитрат железа(Ш) растворяется, образуя коричневый раствор, что обусловлено гидролизом (ср. выше); окраска исчезает при добавлении азотной кислоты. Нитрат железа(П1) применяют в качестве протравы при окраске тканей; им пользуются и в медицине.
Сульфат железа (Ш) Fe2(SO4)3 применяют в качестве протравы при окраске тканей и для приготовления железных квасцов и берлинской лазури. Его используют и в медицине. Обычпо приготовляют лишь его водный раствор окислением раствора железного купороса азотной кислотой или при растворении окиси железа в концентрированной серной кислоте. Образующуюся в последнем случае безводную соль переводят затем в раствор кипячением с водой.
Сульфат железа(Ш) образует несколько гидратов, которые, однако, трудно получить в чистом кристаллическом состоянии. В природе его гидраты встречаются в виде кокимбита Fe2(S04)3-9HzO (гексагонального) и кеенстедтита Fe2(S04)3- 10Н20 (моноклинного). При осторожном нагревании гидратов образуется безводный сульфат ;келеза(1И) в виде белого порошка, который лишь медленно растворяется в воде; его растворение, впрочем, сильно ускоряется в присутствии небольшого количества сульфата железа(П). При более сильном нагревании сульфата железа(Ш) он разлагается на окись железа(Ш) и серный ангидрид
Fea(S04)3 — FeaO3-j- 3SO3.
В водных растворах Fez(SO4)3 тоже в значительной степени гидролизуется. Из его растворов можно выделить целый ряд основных сульфатов железаЦИ), многие из которых встречаются в природе как, например, гохманнит (амарантит) Feo03-2SO3-7Н2О; глокерит (купоросная охра) 2Fo2O3-SO3-6Н2О; раймондит 2FezO3-3Sd3-7HzO и т. д.
Двойные соли сульфата железа(Ш); железные квасцы. Наиболее важными из двойных солей сульфата железа(Ш) являются соли, относящиеся к классу квасцов и имеющие общую формулу MIFe(SO4)z-12HzO. В технике в качестве протравы при окраске тканей используют главным образом железо-аммонийные квасцы NH4Fe(SO4)z-12HaO и железо-калиевые квасцы KFe(SO4)2-12H2O. Квасцы получают окислением водного раствора железного купороса азотной кислотой и кристаллизацией после добавления сульфата аммония или калия. В совершенно чистом состоянии железные квасцы бесцветны, но обычпо [вследствие присутствия в них следов сульфата марганца(1П)| они бывают окрашены в бледно-фиолетовый цвет.
Двойной сульфат, по-видимому, комплексного характера иногда встречается в природе под названием вольтаита. Его состав приблизительно соответствует формуле K2FeP(Fe1Il:, AI)4(SO4)12-32—36Н2О, Госснер (Gofiner, 1930 и сл.) получил рид двойных солей аналогичного состава, содержащих NH41 Rb или Т11 вместо К и Мн11, Со11, Mg, Zn или Cd вместо Fe11.
Оксалат железа(Ш) и окса.татоферраты(Ш). При добавлении оксалата аммония к растворам солей железа(Ш) получаются бурые осадки различного состава. Из растворов, содержащих избыток оксалата аммония, удается получить двойную соль
* Возможно, что существуют и другие гидраты.
29 г. Геми
306 Глава Z
(NH4)3[Fe(Ca04)3]-3H20 [трвоясалатоферрат(Ш) аммония] в виде зеленых, устойчивых на воздухе кристаллов. Известны также и некоторые другие соли этого типа.
При действии света па раствор триоксалатоферрата(1П) калия в соответствии с количеством поглощенного света трехвалентное железо окисляет щавелевую кислоту в угольную, а само переходит при этом в двухвалентное состояние. Этой реакцией пользуются для измерения количества света.
Силикаты железа'III) встречаются в природе главным образом в виде двойных солей, например акмита (эгирина) WaFc111 fSioOoJ, андрадита (известково-железного граната) Ca3FeJn [SiO4]3, а также нередко в виде изоморфных смесей с двойными солями алюминия, имеющими соответствующий состав (последние обычно преобладают).
Фосфат желова(1П) FePO. выпадает из раствора хлорида желеаа(Ш) при добавлении к нему Ди натрийфосфата Na2HPO4 в виде желтовато-белого осадка, трудно растворимого в коде. При добавлении к желтому раствору хлорида железа(Ш) фосфорной кислоты раствор почти совершенно обесцвечивается вследствие образования комплексных сае;цтнепий фосфатоферрата(Ш), например [F'e(PO4)3F” Факт существования чисто фосфатных комплексов и отсутствия хлорофосфатных был установлен Енсепом (Jensen, 1934) на том основании, что растворимость FePO4 в кислых растворах, содержащих хлориды и фосфаты, растет с концентрацией фосфат-ионов, но пе зависит от содержания хлорид-ионов.
Производные ацетата железа(1П) [соли ацетатотрижелеза(Ш)]. При действии на св еже осажденный гидрат окиси жедеза(Ш) концентрированной уксусной кислоты образуется темно-красный раствор, из которого при упаривании выделяется красная масса. Раньше ее принимали за ацетат трех валентного железа Fe(GIT3COO)3. Однако в действительности, как показал Вейнлапд, продукты этой и подобных ей реакций (в большинстве случаев неоднородные) представляют собой, каки в случае хрома, соединения, содержащие положительный комплексный иоп гехсаацетатодигидроксотри-желем(РРГ). Обычно этот комплекс присутствует в растворе в виде одновалентного кати-она [Feg(OH)2(CH3COO)er. Вейнлаиду удалось получить различные соли, производные этого катиона *, типа [Fe3(OH)2(GH3COO)e[X [соль гексаацетатоднгидроксотрижеле-за(1П)] в кристаллическом состоянии, среди них и ацетат [Ре3(ОН)2(СГ13СОО)е]СН3СОО-Н2О [ацетат гексаацетатодигидроксотрижелеза(Ш)[. Кроваво-красная окраска, возникающая при смешивании растворов солей трех валентного железа с ацетатом натрия, по Вейпланду, связана с образованием соединений, также содержащих комплекс аЦетатотрижелеаа(Ш). При кипячении таких растворов с ацетатом натрия все содержащееся в них железо выпадает в виде красно-бурого осадка, содержащего железо и остаток уксусной кислоты в соотношении 3 : 1 и состоящего, по-видямому, главным образом из нерастворимого соединения
СН3СОО"
Fe, О3 , . (ОН)а _
которое является производным гексаицетатодигвдроксотри!келеза(Ш), полученным при замещении 5СН3СОО па 30 (вследствие чего комплекс теряет свой заряд). Хром образует подобные же соединения, которые, однако, при кипячении пе разлагаются и пе осаждаются. Кроме того, хром и железо могут в втих соединениях частично замещать друг друга**. Поэтому при кипячении с ацетатом натрия в присутствии большего количества хрома часть железа остается в растворе и, наоборот, в присутствии избытка железа часть хрома переходит вместе с ним в осадок.
Роданид железа(1П) и родамоферраты(П1). При добавлении к раствору соли желез а( III) ионов родана раствор окрашивается в кроваво-красный цвет. Прн встряхивании с эфиром окраска переходит в эфирный Слой. Опа связана с образованием роданида железа(1П) Fe(SCN)3. В присутствии избытка иолов родана образуются, кроме того, ионы гексарода-иоферрата(Ш) [Fo(SCN)er". При достаточной концентрации ионов рода-
* Образование этого катиона было подтверждено электрометрическим титрованием (Treadwell, 1930).
** Веннланду удалось выделить индивидуальные соединения с общими формулами [Fe2Cr(OH)2(CH3COO)6]X и [FeCr2(OH)a(CH3COO)e]X.
Восьмая группа периодической системы
307
на в растворе красная окраска появляется даже при ничтожном содержании ионов Fe"', поэтому рассматриваемая реакция является очень чувствительным методом открытия иона трехвалентного железа.
В роданвферратаз:{11Г}, например гексароданоферрате(Ш) калия K3[Fe(SCN)ep 2Н2О, представляющем собой темно-красные гексагональные призмы, расплывающиеся на воздухе, комплексный анион малоустойчив. Если раствор не содержит избытка ионов родапа, то при растворении в воде комплексные ионы сильно диссоциируют. Однако существование комплексного иопа [Рс(8СК)б]'" было доказано опытами по переносу ионов в спиртовом растворе. Гидроокиси щелочных металлов разлагают роданид железа(Ш), как и другие роданиды тяжелых металлов.
Цианоферраты(Ш); красная кровяная соль. При избытке ионов CN' трехвалентпое железо образует очень прочный ион [Fe(CN)6]w. Наиболее важной из солей — производных этого иона, так называемых гексацианоферратов(111) MJ[Fe(CN)eJ,— является красная кровяная соль, гексацианофвррат(11Г') калия Кэ [Fe(CN)eI, которую впервые получил Гмелин. Красная кровяная соль получается при окислении хлором, перманганатом калия или другими сильными окислителями желтой кровяной соли, гексацианоферрата(П) калия, в солянокислом растворе:
[Fe(CN)6]'+ 1/2CI2=[Fe(CN)6]w+Cl'.
Она представляет собой темно-красные моноклинные призмы, которые при растирании превращаются в желтый порошок и образуют водные растворы тоже желтого цвета. Красная кровяная соль менее устойчива, чем желтая кровяная соль, и потому (в противоположность желтой) ядовита. Она обладает, особенно в щелочных растворах, сильными окислительными свойствами; так, при реакции с перекисью водорода, сопровождающейся выделением кислорода, она опять переходит в желтую кровяную соль, устойчивую в щелочных растворах. Нормальный потенциал системы [Fe(CN)6I"'7[Fe(GN)el'" равен + 0,36 в.
Свободную кислоту H3[Feni(CN)eI можно получить при действии дымящей соляной кислоты на чистый концентрированный раствор гекса-цианоферрата(1П) калия. Однако в чистом состоянии она получается значительно труднее, чем кислота Hs[Foin(CN)6].
Берлинская лазурь и турнбуллева синь. При добавлении к раствору солей железа(Ш) раствора желтой кровяной соли образуется темно-синий осадок берлинской лазури. При взаимодействии растворов солей железа(II) и красной кровяной соли выпадает темно-синий осадок, который называется турнбуллевой синью. Обе эти реакции имеют важное аналитическое значение, как для открытия железа, так и для установления его валентности. Берлинская лазурь, известная также под названием прусской лазури, является хорошей краской, поэтому ее получают в больших количествах.
Образование берлинской лазури и турнбуллевой сини можпо схематически представить следующими уравнениями:
Реакция образования берлинской лазури:
4Fe ’ ‘ ‘ + 3[Fen (CN)oj" " = Fe|n [Fe11 (CN)eJ 3:
Реакция образования турнбуллевой сини:
Fe" + (FenI(CN)e]'"=Fe-" + [FeII(CN)eJ"".
4Fe-- + 3(Fen(CN)6]"”^Fe?II[FeII(CN)6l3.
На первой стадии реакции образования турнбуллевой сини происходит обмен валентностями между Fe11, существующим в виде свободного иона, и Fe111, связанным в ком-
20*
308
Глава 7
плене. Доказательством этого может служить появление красной окраски при действии ионов SCN', обусловленной реакцией с нонами Fe"‘ в момент их образования (Simon, 1936). Итак, осадка, называемые «берлинской лазурью» и «ту рибул левой синью», в основном идентичны друг Другу; однако в зависимости от условий осаждения их состав больше или меньше отклоняется от описываемого формулой Fe4 f Fe(CN)6]3. Прежде всего они всегда содержат калий. Если осторожно, по каплям, приливать раствор соли жедеза(Щ) к избытку раствора цианоферрата(II) калия, то можно получить синюю соль, образующую в чистой воде коллоидный раствор и выпадающую в виде хлопьев при добавлении поваренной соли: она называется «растворимая берлинская лазурь», ее состав соответствует формуле KFe[Fe(CN)e). Обычная берлинская лазурь нерастворима в воде и в разбавленных кислотах и растворяется только в щавелевой кислоте, образуя темно-синий раствор; этим свойством ее пользуются для приготовления чернил, берлинская лазурь очень устойчива в разбавленных кислотах, но крайне чувствительна к действию щелочей (даже сильно разбавленных); при действии щелочей происходит выделение гидроокиси железа(П1) и образование гекса-цианоферрата(П) калия
Пруссидные соединения. Комплексные цианиды железа, содержащие только пять групп CN, в то время как шестое координационное место занято другой группой, называются пруссидными соединениями. Наиболее известен из них нитропруссид натрия; его можно получить действием азотной кислоты на желтую кровяную соль. При этом из раствора сначала кристаллизуется нитрат калия, а затем после нейтрализации избытка азотной кислоты карбонатом натрия — нитропруссид натрия Na2[Fe(CN)5(NO)l" 2НИО в виде рубиново-красных ромбических кристаллов, устойчивых на воздухе. Нитропруссид натрия легко растворим в воде и в спирте, но не особенно устойчив в растворах. Известны также и другие нитропруссидные соли, имеющие общую формулу M|(Fe(CN)5(NO)].
Нитропруссид натрия применяют как реактив на ионы S" и SH', с которыми он дает интенсивную фиолетовую окраску (свободный сероводород не дает этой реакций). Нитропруссид натрия реагирует также с ионами SOiJ, окрашивая их растворы и красный цвет, особенно в присутствии сульфата или нитрата цинка. Этой реакцией можно пользоваться для того, чтобы различать сульфиты и тиосульфаты, так как последние ее пе дают.
Вероятно, железо в комплексе с нитропруссидом не четырех-, а трехвалентио и группу NO, также входящую в комплекс, следует рассматривать как нейтральную’ Поскольку валентность железа в этом соединении окончательно еще пе установлена, не рекомендуется называть его пентацианоферратом(Ш). В таких случаях число атомов, находящихся во внешней сфере комплекса, обозначают греческим числительным перед названием соединения: а«натрийпентацианонитрозилферрат;
При действии на нитропруссид натрия концентрированным раствором аммиака группа NO в нем замещается на NH3. Вместе с тем трехвалентное железо восстанавливается до двухвалентного, а азот переходит в азотистую кислоту:
[(CN)sFe(NO)]’ +NH3+ ОН' =f(CN)sFe(NH3)]'" -ф- О =N—ОН.
Получающийся таким путем понтацианоамминфоррат(П) натрия Na3 [Fe11 (CN)5NH3j можно окислить азотистой кислотой в пентацианоамминферрат(Ш) натрия Na^lFe111 (CN)SNH3]-Н2О (темпо-желтый порошок), В качестве примеров других пруссндаых соединений" можно назвать
M|[FeIIT(CN)s(OH2)] Mi[FeU(CN)5(OH2)]
M|(Fenr(CN)5(NO2)] M|[FeH(CN)5(NO2)]
M,*[FeH(GN)s(AsO2)] MftFe^CN^SOs)] Mi(Fen(GN)5(CO)]
Еолыпнпство этих соединений было получено Гофманом (Hofmann). Пентациано-карбонилферрат(11) калия К3[Fe(CN)s(CO)l31/2Н^О, образующийся при действии окиси углерода на теплый водный раствор желтой кровяной соли, в большинстве случаев содержится л качестве примеси в неочищенной желтой кровяной соли. Известна также соответствующая этому соединению свободная кислота.
В связи с изложенным следует также упомянуть и о соединении Na4 [ Fe(CN)5(NO)]T описанном Унгарелли (Ungarelli, 1925). Оно было получено при действии
Восьмая группа периодической системы 309
гппонитрита натрия NasN2O2 на водный раствор пентациапоаквоферрата(Ц) натрия Na3[Fe(CN)5(OH2)]. Как показали криоскопические измерения, это соединение имеет простую молекулярную формулу. Таким образом, оно не содержит, папример, радикала N2O2, а содержит радикал NO. Однако вопрос о том, является ли эта группа псйтральиой или отрицательно заряженной, т. е. вопрос о валентности железа в этом соединении, пока не решен.
Бринтцингер (Brintzinger) нашел, что скорости диффузии анионов пруссидных соединений по аналогии с циавоферратами(Ш) (ср. стр. 301) соответствуют пегвдра-тнрованным ионам. В то же время скорость диффузии цианоферратов, центральным атомом которых является Fe11, позволяет сделать заключение о сильной гидратации комплексных ионов.
Ферраты( VI)
При нагревании железных опилок с селитрой происходит их окисление, сопровождающееся сильным разогреванием смеси. Охлажденный плав с водой образует яркий красно-фиолетовый раствор, при добавлении к которому хлорида бария выпадает красный осадок цвета кармоизи-на *; состав его после высушивания при 100° отвечает формуле BaFeO4-НаО. Полученный продукт относится к классу соединений железа, соответствующих по своему составу хроматам и сульфатам. Они содержат железо в шестивалентном состоянии и называются ферратами [точнее — ферратами(У1)]. Темно-красный феррат(УГ) калия K2FeOi легко растворим в воде и поэтому подучается в чистом состоянии труднее, чем феррат(У1) бария; он изоморфен сульфату калия. Помимо указанного способа, ферраты^!) можно получать также окислением хлором или бромом гидроокиси железа, осажденной и суспендированной в концентрированном растворе едкого кали, или анодным растворением железа (лучше всего чугуна) в теплом КОН или (еще лучше) в растворе едкого натра:
Fe+8ОН'—бе=[FoOJ" -(- 4Н2О.
Ферраты(У1) являются сильными окислителями, превосходя в этом отношении перманганаты; так, они даже на холоду окисляют аммиак до азота. Соответствующая ферратам(у1) кислота не была выделена ни в свободном состоянии, ни в виде окисла. При иодкйслении растворов pamoe(VI) происходит выделение кислорода, а содержащееся в инх желе-ao(VI) переходит в железо(Ш):
2КеО1+ЮН-=2Ге--|-8/гОг+5Н2О.
Некоторые ферраты(У1), например BaFeO4, при обработке концентрированным раствором КОН постепенно переходят в ферраты(1¥) свыделением кислорода. Шоль-деру (Scholder, 1952) удалось получить чистый феррат(ТУ) стронция Sr2FeO4 при нагревании смеси Sr(OH)2 и Sr3[Fe(OH)6)2 в токе кислорода до 500—800°. Послеуда-ления избытка SrO оставался совершенно черный кристаллический порошок соединения Sr2FeO4. На основании дебаеграммы можно заключить, что это соединение кристаллизуется ио типу 81‘2Мп04и Ва2МпО4, в то же время решетка BaaFeO4, также впервые описанного Шольдером, изоструктурна Ва2Т1О4, Ва2СгО4 и Ва2СоО4.
КАРБОНИЛЫ И НИТРОЗИЛЬНЫЕ СОЕДИНЕНИЯ ЖЕЛЕЗА
Карбонилы железа. Мелкораздроблвнное железо при комнатной температуре и обычном давлении медленно, а при высокой температуре д высоком давлении значительно быстрее соединяется с“ окисью углерода, образуя бледно-желтую жидкость с высоким коэффициентом преломления состава Fe(CO)5, так называемый пентакарбонил железа; Fe(CO)5 дмеет удельный вес 1,494 (при 0°), затвердевает при —20° й кипит при £02,7°. Он легко растворяется в бензоле, бензине и эфире, но нерастворим
* Кармоизин красный — кислотный азокраситель.— Прим. перев.
310
Глада 7
в воде. Газообразные HCinHBr, а также HaS на него не действуют, но йодистый водород вступает с ним в реакцию. Однако перечисленные кислоты реагируют с пентакарбонилом железа в спиртовом или в эфирном растворах (но не в водном). В эфирном растворе пентакарбонил вступает в реакцию с азотной и серной кислотами, с последней — гладко по уравнению ;
H2S 04+Fe(CO)5=FeS О4 + Н3+5СО.
В промышленных, масштабах пентакарбонил железа получают при нагревании до ISO—220° тоякораздробленного железа с окисью углерода под давлением (150— 250 атл*). При охлаждении реакционной смеси конденсируется жидкий пентакарбо-нил, а выходящая из автоклава окись углерода снова возвращается в реакцию. Пента-карбонил железа уже давно применяют в качестве добавки к автомобильному бензину для устранения стука в моторе («моталин»). Это обстоятельство впервые способствовало развитию промышленного производства пентакарбонила. В настоящее время его используют главным образом для получения железа высокой чистоты (ср. стр. 272). Кроме того, при сгорания Fe(CO)s образуется окись железа(Ш), используемая в качестве краски и полировочного материала. Ре(СО)5 является нежелательной примесью в светильном газе, так как он вызывает образование окисных налетов на сетках газовых фонарей, которые ослабляют силу света. Поэтому светильный х^з очищают от пентакарбонила железа при пропускании его через раствор хромовой кислоты или через пористый уголь, насыщенный раствором хромовой кислоты.
На солнечном свету пептакарбонял железа постепенно разлагается * с образованием твердого продукта состава Fe2(CO)9 зннеакарббнила железа., Желтых гексагональных листочков (структуру его см. стр. 382). Это соединение очень чувствительно к действию воздуха. При нагревании до 100° в отсутствие воздуха оно разлагается на Fe и СО, причем опять образуется некоторое количество Fe(C0)5. Более устойчивым, чем аанеакарбонил, является так называемый тетракарбонил железа, который правильнее считать дадека/мрбанил<>м железа Fe3(CO)u. Он образуется, в частности, при обработке пентакарбонила Железа алко гол ят ом патрин в спиртовом растворе (о других методах синтеза см. стр. 382 и 385). Fe3(CO)12 образует темно-зеленые моноклинные кристаллы уд. веса 2,0. В обычных растворителях оп растворим очень мало; в пентакарбониле железа оп растворим и, судя по молекулярному весу, существует в растворе в виде Fe3(CO)l2. (Hieber, 1930). Кристаллическая решетка рассматриваемого соединения тоже построена из молекул Fe3(CO)12 (см. стр. 382).
Тетранитрозил железа. Fe(NO)4 был получен в 1929 г. Нашло (Mancfiot) в виде черных кристаллических нгл при нагревании до 44—45“ в автоклавах леиталарбонила железа с окисью азота. Это соединение очень реакционноспособно. Разбавленной серной кислотой опо разлагается с образованием сульфата нитрозилжелоза(П) (Fe(NO))SO4, С гидросульфидом калия тетранитрозил железа образует черную соль Руссена К [Fe4S3(NO)7l (ср. стр. 290). С тиосульфатом калия K2S2O3 он реагирует, образуя соединение (NO)iFe — S — SO3K; с меркаптаном С2И68Н образуется (NO)2Fe—
—S—С2Н5; с ксантогенатом калия К—S—тетранитрозил железа обра-ЧОС2Н5
g
зует (NO)2Fe.—S—t Эти соединения, полученные раньте другими мето-ХОС,Н,
дами Гофманом и Маншо, можпо рассматривать как производные красных солей Руссена (см. стр. 290), и подобно ям, по мнению Манию, эти соединения содержат электрохимически одновалентное железо.
При действии NO на иодид жсиеза(Н) Fel2, по данным Хибера (Hieber), один атом галогена замещается па 2 группы NO г образуется иоВоВимитрозилжелезо FeI(NO)2 — соединение черпо-корич некого цвета, возгоняющееся без разложения, но очень чувствительное к действию воздуха и влаги. Труднее получить соответствующее бримо-соедипоние FeBr(NO)2. Хлоро-соединение состава FeCl(ND)31 отличающееся большей летучестью, получается еще более сложным путем (строение его см. стр. 318). Соответствующее фторо-соединение, по-видимому, вообще не существует.. Дальнейшие сведения о карбонилах и нитрозилах металлов см. стр. 375..
* Разложения на свету не наблюдается у карбонильных соединений других металлов. Это свойство характерно только для карбонилов железа. Гальдане (Haldane, 1897) обнаружил его, например, у комплекса СО с гемоглобином..
Восьмая группа периодической системы
311
Аналитические сведения. В качественном анализе железо относится к группе сернистого аммония. Сернистый аммоний осаждает его в виде черного осадка FeS или Fe2Ss (соответственно смеси S и 2 FeS) в зависимости от валентности, в которой оно находится в растворе. (Если через раствор перед осаждением железа предварительно пропустить сероводород, то железо бывает всегда Йву.твалентным.) От хрома, алюминия и цинка железо отделяется путем осаждения его в виде гидроокиси смесью раствора едкого натра и перекиси водорода, от марганца — осаждением в виде гидроокиси аммиаком в присутствии хлорида аммония и хлорида гидро-ксиламмония (которые препятствуют осаждению марганца); в последнем случае железо должно находиться в растворе в трехвалентном состоянии; поэтому, если оно двухвалентно, то его предварительно окисляют азотной кислотой. Качественной реакцией иа соли желева(Ш) служит обычно взаимодействие его с раствором роданида; появляющаяся при этом красная окраска при встряхивании с эфиром переходит в эфирный слой.
Степень окисления железа в анализируемом веществе определяется реакцией с желтой и красной кровяной солью. Соли железа(П) образуют с дицианоферрат ом (II) железа в первый момент белый осадок, а с цианоферрат ом (III) — темно-синий осадок (турнбуллевой сини). Соли железа(Ш) дают с цианоферратом(П) железа темно-синий осадок (берлинской лазури), а с цианоферрат ом (Ш) железа не образуют осадка. Если при действии обоих реактивов получяогся темно-синий осадок, то это указывает па присутствие желоза обеих валентностей.
Для весового определения железа обычно используют гидроокись, осажденную водным раствором аммиака, и взвешивают ее в виде окиси после прокаливания. Часто железо определяют объемными методами — титрованием перманганатом:
5Fe-- + MnO;+8H-=5Fe-"4-Mn-- + 4H2O,
или иодометрически (см. также стр.: 302):
Fe"- + r=Fe"-p/2l2.
КОБАЛЬТ (COBALTUM) Со
Распространение в природе. В природе кобальт всегда встречается вместе с никелем, главным образом в виде соединений с мышьяком. Важнейшими из минералов кобальта являются кобальтовый шпейс (смальтит) CoAsa и кобальтовый блеск (кобальтит) CoAsS.
В повой Каледонии и Канаде встречаются большие количества марганцовой черни, содержащей кобальт (черный землистый кобальт, или асболан), В чистом состоянии .кобальт (в количествах 0,5—2,5%) 'иногда входит, в состав метеоритного железа.
Исторические сведения. Раньше на рудниках «кобальтами» называли те минералы, которые, несмотря на их внешний металлический вид, пе поддавались переработке па металл. Подобно насмешливым «горным духам»— «кобольдам» («кобольды» рапыпе назывались также «кобальтами») эти минералы дразнили горняков. Позднее так стали называть только те трудной ер ерабатываемые руды, которые окрашивала стекло в синий цвет. Содержащийся в пих металл, называемый в настоящее время кобальтом, был впервые получен в 1735 г. шведским химиком Брандтом и признан новым элементом.
Получение. Исходным материалом для промышленного получения кобальта служат преимущественно «шпейсы», содержащие никель и кобальт в виде арсенидов,
312 Глава 7
образующиеся при переработке никелевых, медных и свинцовых руд, богатых мышьяком. Йз «шпейсов» обычно вырабатывают только окись кобальта, применяемую для приготовления кобальтовых красок; для этой целя она может быть и. не особенно чистой. Однако в настоящее время промышленность во все возрастающих масштабах производит металлический кобальт.
Получение металлического кобальта в чистом состоянии довольно сложно. Особенно трудно при этом полное отделение его от никеля. Для этого шпейсы или другие мышьяковые руды, содержащие кобальт, обжигом при доступе воздуха переводят в смесь окислов и арсенатов, называемую из-за ее красноватого цвета «сафлором» или щаффером». Затем эту смесь растворяют в соляной кислоте и из полученного раствора осаждают сероводородом медь, свинец, висмут и т. д.: после этого, окислив предварительно железо хлором, его вместе с мышьяком осаждают карбонатом кальция в виде арсената кальция и гидроокиси железа, а оставшийся раствор обрабатывают хлорной известью в количестве, достаточном для осаждения кобальта. Большая часть никеля при этом остается в растворе (при условии, что не было избытка хлорной извести). Из окиси кобальта, которую в случав необходимости очищают повторным осаждением, после прокаливания с соответствующими восстановителями получают металлический кобальт.
Свойства. Кобальт представляет собой блестящий 'металл, похожий на железо, с удельным весом 8,8. Температура его плавления несколько ниже, чем у железа. Кобальт очень тягуч. Он обладает большей твердостью и прочностью, чем сталь. Как и железо, оп ферромагнитен и только выше 1000° переходит в модификацию, не обладающую способностью намагничиваться.
Кобальт обычно кристаллизуется по типу магния (гексагональная плотнейшая упаковка), а = 2,5063, с = 4,0795 Л. Однако для порошкообразного кобальта, образующегося при слабом прокаливании окиси кобальта в токе водорода, был установлен тот структурный тип, по которому обычно кристаллизуется никель — гранецёнтри-рованная кубическая решетка (а = 3,5441 Л). Эта модификация устойчива выше ~ 480°, однако после растворения водорода кубический кобальт устойчив и при более низких температурах, Межатомное расстояние Со -t—» Со у обеих модификаций практически одинаково (2,5036 и 2,5060 Л).
Растворимость водорода в металлическом кобальте несколько меньше, чем в железе. По данным Сивертса (Sieverts, 1934), в 100 г кобальта при атмосферном давлении и при 600° растворяются 0,08, а при 1200я — 0,49 мг Н2. Количества абсорбированного водорода растут пропорционально корню квадратному из его давления. Азот вплоть до 1200е практически нерастворим в кобальте.
Вода и воздух при обычной температуре не оказывают действия па компактный кобальт, но в мелкораздробленном состоянии оп, как и железо, обладает пирофорными свойствами. В разбавленных кислотах, например в соляной или серной, кобальт растворяется значительно труднее, чем железо, что соответствует его положению в электрохимическом ряду напряжений справа от железа (его нормальный потенциал равен —0,28 в). Разбавленная азотная кислота легко растворяет кобальт, а при действии концентрированной HNO3 он, как и железо, пассивируется.
При нагревании на воздухе Со окисляется, а при температуре белого каления сгорает до Со3О4. При нагревании кобальт соединяется со многими другими веществами, причем реакция его с S, Р, As, Sb, Sn и Zn нередко сопровождается воспламенением. При сплавлении с кремнием Со образует целый рид соединений (см. табл, 31, стр. 264 и сл.). При высокой температуре он соединяется также с бором, по не реагирует с азотом. Кобальт легко образует соединения с галогенами. С железом и никелем, а также с хромом и марганцем он образует твердые растворы в любых соотношениях. По отношению к углероду кобальт ведет себя так же, как железо; однако при охлаждении углеродсодержащих расплавов никогда не выделяется карбид Со3С (хотя, поданпым4Руффа, существование его в расплаве является вероятным); если содержание углерода выходит за пределы существования твердого раствора, избыток углерода всегда выделяется в виде графита. При действии СН4 или СО па тонкоизмельчеп-
Восьмая группа периодической системы
313
пый металлический кобальт при слабом пагревапии (ниже 225°), по данным Бара (Bahr, 1930), образуется соединение С о-, С, разлагающееся при более высоких температурах. Каталитическое разложение С1{4 и СО под действием кобальта происходит лишь при таких температурах, когда карбид становится неустойчивым (см. стр. 751).
Применение. Раньше кобальт применяли главным образом в виде двойной соли с силикатом калия (кобальтовое стекло), представляющей собой синюю краску (смальту).
Смальта, известная уже древним египтянам и римлянам, получается при сплавлении окиси кобальта (или обожженных кобальтовых руд) с кварцевым песком и поташом. По остывании сплав размалывают, причем получается великолепная синяя краска, применяемая для окраски расплавленного стекла, керамических изделий и эмалей.
Металлический кобальт получил широкое применение лишь в последнее время. Сейчас все большие количества его идут в первую очередь для получения сплавов и некоторых быстрорежущих сталей.
Такие стали, идущие для изготовления режущих инструментов, допускают применение возрастающих скоростей на токарном станке, так как они сохраняют твердость даже при красном калении. Режущие сплавы, например «видна» или «стеллит», отличающиеся особенно высокой твердостью, позволяют достигнуть еще более высоких скоростей работы.
Кроме режущих инструментов, из этих сортов стали можно изготовлять буры для бурения горных пород (вместо алмазных) и т. д. Видиа (название которого происходит от немецких слов «wie Diamant») содержит карбид вольфрама и около 10% кобальта. В состав стеллита входит около 50% Со, 27% Сг, 12% W, 2,5% углерода и до 5% Fe, кроме того, некоторое количество Ми и Si.
СОЕДИНЕНИЯ КОБАЛЬТА
Кобальт в огромном большинстве простых соединений является двухвалентным, а в комплексных — щресталептным. Известны окислы двух-, трех- и четыреагвалентного кобальта, однако окисел четырехвалентного кобальта не получается в чистом состоянии (ср. ниже). Окисел кобальта(ГУ) СоО3 может давать с снльнросповпыми окислами соли [кобальтаты(1У), ранее называвшиеся кобальтигами]; однако большинство из них и сам окисел кобальта(1У) малоустойчивы. Окисел кобальта(Ш) Со2О3 и в еще большей степени окисел кобальта (Ц) — СоО имеют характер основных ангидридов и образуют соли, имеющие общие формулы СоХ3 [соли кобальта(Ш)] и СоХ2 [соли кобальта(П)]. Оба окисла способны образовывать и двойные соли, кристаллизующиеся по типу шпинелей; окись кобальта(III) образует шпинели с окислами двухвалентных, а окись кобальта(П) — с окислами трехвалентных металлов. Следовательно, существует два вида шпинелей кобальта — МпО-Со2О3 и СоО-М|ПО3. (О шпинели, образованной «кислом кобальта(1У), см. стр. 315.)
Число известных простых солей кобальта(ПТ) очень невелико. Почти все простые соли кобальта являются производными окиси кобальта(1Г). Наиболее важными из простых солей являются хлорид СоС12-6Н2О, нитрат Co(NO3)2-6H2O и сульфат CoSO4-7H2O. Все названные соли легко растворимы в воде. То же относится и к ацетату Со(СН3СОО)2-4Н2О. Большинство же простых солей кобальта и слабых кислот трудно растворимо в воде. Кристаллогидраты солей кобальта(П) при обычной температуре, а также их растворы бывают розовыми или красными. При нагревании они становятся темпо-синими.
Строго говоря, указанные пиже соединения, содержащие воду, нельзя рассматривать как простые Соли — они являются акео-солямп, т. е. координационными соединениями. Однако при нагревании из них легко образуются безводные соли (за исклю
314
Глава, 7
чением нитрата, который при этом разлагается). Часто вместо воды эти соли присоединяют аммиак. Последний также связан в солях кобальта(П) непрочно. Почти все соли кобальта(П) обладают также способностью образовывать а цидо-соли., Последние, впрочем, чаще всего являются малоустойчивыми комплексами, т. о. обладают явно выраженным характером двойных солей.
Значительно более устойчивыми комплексами являются аци до-сое дипения трех-валентного кобальта. Сравнительно большой устойчивостью отличаются и комплексные соединения, образующиеся путем присоединения нейтральных групп к кобальту, имеющему заряд 3-1-. Подобное же явление наблюдается и у хрома. Вообще трехва л битный кобальт способен образовывать соли с анионами многих кислот лишь в тех случаях, когда он сам входит в состав таких комплексных ионов, например [Co(NH3)e]"‘, [Со(ОН)(аЧН3)3]“, [Co(N O2)2(NH3)4p и т. д. Известны многочисленные соли — нитраты, нитриты, хлориды, бромиды, иодиды, цианиды, роданиды, карбонаты, оксалаты, ацетаты,— в которых кобальт находится в виде такого рода комплексов, в то время как соответствующие простые соли (в приведенных примерах также акво-соединения [Co(OH2)fijx3) вообще не могут быть получены.
Трехвалентный кобальт в своих комплексных соединениях независимо от природы лигандов имеет почти всегда координационное число 6, в то время как у двухвалентного кобальта координационное число бывает различным в зависимости от природы лигандов и условии образования соединения и нередко принимает более низкие значения чем fl.
Четыр^валентный кобальт существует в окисле СоО, (неизвестном, правда, в свободном состоянии) и в двойных окислах ЗгоСоО4 и Ва2СоО4, производных СоО2, полученных Шмалем и Шольдером (Schmall, 1950; Scholder, 1952). Двойной окисел Со3О4 или 2Со0‘СоО2 содержит положительно четырех- и двухвалентный кобальт (ср. стр. 315), а некоторые комплексные соединения (например, приведенные на стр. 329) — кобальт с валентностями 4+ и 34--
В 1926 г. Маншо (Manchot) получил тиосульфатный комплекс кобальта, содержащий окись азота и приближающийся по своему типу к азотсодержащим комплексным соединениям железа е серой (солям Руссена). В этом соединении, по мнению Маншо, кобальт (как и железо в солях Руссена) является одновалентным. Это соединение кристаллизуется в виде блестящих листочков цвета латуни и образует с водой чернокоричневый раствор, из которого его можно осадить спиртом. Строение его следующее:
’NO, .S—SO3"
'Со'
no' ‘s—S03.
К3
В соединениях CoC1(NO)2, CoBr(NO)3 и CoI(NO)2, полученных Хибером (Hieber, 1939), кобальт, по-виднмому, одновалентен при условии нейтральности групп NO. Комплексные цианокобалыат(1)-ионы можно получить в водном растворе по методу Грубе (Grube, 1026) электролитическим восстановлением гексацианокобальтат(П)-иои ов.
Окислы и гидроокиси. При нагревании гидроокиси или карбоната кобальта(П) в отсутствие воздуха образуется окись кобальта(11) (моноокись кобальта) СоО в виде оливково-зеленого порошка. Гидроокись кобалъта(ГГ) Со(ОН)2 получается в виде синего осадка, окрашивающегося при стоянии в бледно-красный цвет, при действии на растворы солей кобальта(П) раствора едкого кали. На воздухе осадок гидроокиси медленно окисляется в коричневую гидроокись кобалъта(Т1Г) аналогично гидроокиси железа(П). Быстрее это окисление проходит в присутствии сильных окислителей, например NaOCl, С12, Вг2, Н2О2. В этом случае частично образуется черный диоксигидрат кобальта СоО2-хН2О. Гидроокись кобальта(Ш) * обезвоживанием удается перевести в коричневый
* Препараты гидроокиси кобальта(Ш), осажденные из водных растворов, после высушивания имеют состав, почти точно соответствующий формуле Со2О3-ЗН2О. Они обычно очень мелко дисперсны, но па их рентгенограммах обнаруживается структура безводного Со203. Следует отметить, что моногидрат Со203-Н20, индивидуальность которого доказал Хюттиг (Hiittig, 1929) с помощью кривых обезвоживания, пе отличается по структуре от безводного Со?О3 (Natta, 1928). Со3О3-НгО встречается в виде минерала штайниерита.
Восьмая группа периодической системы
315
окисел кобалъта(1 II) Со2О3, лишь соблюдая особые предосторожности. Обычно еще до полного обезвоживания он отщепляет кислород и переходит в черную окись кобальта(11,1V) Со3О4 или GoPJCo^Oj.
С03О4 кристаллизуется по типу шпинели и магнетита (а = 8,07 А). В отличие от Fe3O4, в решетке которого ионы металла двух- и трехвалентны, в Со304 металл обладает валентностями 24- и 4 4-, так же как это происходит в Мп3О4.Иопы Со4+ занимают те же узлы решетки, что попы Mg2+ в обычной шпинели (Verwey, de Boer, 1936),
При очень сильном прокаливании все окислы кобальта переходят в его моноокись СоО. При пагревапии в токе водорода все они восстанавливаются до металлического кобальта.
. СоО кристаллизуется по типу поваренной соли (а = 4,24 А.). Розовая Со(ОН)2 кристаллизуется по типу брусита (а = 3,17;с = 4,64А)и образует твердые растворы с другими гидроокисями, имеющими тот же тип решетки [например с Mg(OH)2, Zn(OH)2, Ni(0II)a]. Синяя гидроокись кобальта(Н) отличается от розовой более высокой степенью дисперсности и значительно меньшей степенью упорядоченности в расположении атомов в решетке. Вопрос о том, является ли синяя гидроокись самостоятельной модификацией с особым типом структуры, пока остается открытым *. В 1936 г. Файткнсхт при рентгенографическом исследовании синей гидроокиси кобальта (II) установил, что опа подобно зеленому основному хлориду (ср. стр. 320 и сл.) образует двухслойную решетку, причем «главные слои» имеют то же строение, что и у розовой гидроокиси кобальта(П). Однако «промежуточные слои» образованы неупорядоченными гидроксильными группами и расположены примерно па расстоянии 8 А и одновременно произвольно смещены друг относительно друга в направлении слоев. Такую структуру символизирует формула |Со(ОН)2)4-Со(ОН)2, из которой следует, что 80 ат. % кобальта расположено в главных слоях и 20% — в промежуточных. При действии кислорода воздуха па свежеосажденпоо соединение сличала окисляется кобальт промежуточных слоев с образованием [Со(ОН)3]4-СоО(ОН), построенного аналогично зеленому основному хлориду кобальта. Значительно медленнее происходит окисление кобальта в главных слоях, конечным продуктом которого является СоО(ОН) или Co20s-Hj>0.
Окраска безводной СоО зависит тоже от степени упорядоченности атомов и дисперсности; кроме олив ков о-зеленого, этот окисел может быть желтым, серым, коричневым, красповатым, сипеватым или черным; однако до сих пор не установлено окончательно, существует ли, кроме кубической, другая модификация.
Теплота образования кристаллической СоО, цо измерениям Рота (Roth, 1931), равна 57,2 ккал/моль. Теплота образования Со(ОН)2 из СоОкрИет и Н2О)К составляет около 6,2 ккал/моль. Прн реакции с водой очень мелко дисперсной («аморфной») СоО может выделиться количество тепла, более чем вдвое превышающее указанную величину, так как, по данным Микст ера (Mixter, 1909), теплосодержание «аморфной» окиси кобальта(П) превышает теплосодержание явно кристаллической на 7 ккал/молъ.
Гидроокись кобальта(П) растворима в кипящей концентрированной щелочи с сине-фиолетовой окраской. В растворе, как показал Шольдер (Scholder, 1933), существуют гидроксокобалътат(11)-ионь1. Шольдеру удалось выделить соединения Na7[Co(OH)4], Sr2[Co(OH)6] и Ва2[Со(ОН)0] — краспо-фиолотовые кристаллы, очень быстро чернеющие на воздухе вследствие окисления. Водой они тотчас разлагаются. Из растворов небольших концентраций Со(ОН)2в горячей щелочи гидроксосоли невыде-ляются, вместо них образуется красный кристаллический осадок Со(ОН)2.
Не ясна еще природа безводных соединений, образующихся при сплавлении основных окислов с окислами кобальта. Состав их достаточно сложеп, и Со они содержат иногда в различных валентностях. Так, по данным Беллуччи (Bellncci), при сплавлении некоторых окислов кобальта с КОН в токе воздуха образуются серо-стальные листочки состава К20-СоО.ЗСо03. Это соединение в воде и в разбавленных растворах соляной кислоты нерастворимо, но водой медленно гидролизуется. Из концентрированных растворов НС1 опо выделяет хлор, причем кобальт восстанавливается от четырех- до дкухвал ситного. Двойные окислы к об альта (IV) или оксо-соли [кобаль-
* Файткпехт (Feitknecht, 1935) считает, что так называемая зелопая гидроокись кобальта в действительности представляет собой основной хлорид кобальта (ср, стр. 320).
316
Глава 7
таты(1У)] * более простого состава — ВаО-СоО2 и ВаО>2СоО2, а также MgO-Co02,— по данным Руссо и Дюфо (Rousseau, Dufau), образуются при сплавлении окиси или хлорида бария или окиси магния с окисью кобальта(Ш) в токе воздуха; существование этих соединений пока пебсзусловно. Однако в последнее время Шольдер (1952) выделил соединение состава 2ВаО-СоО2 или Ва2СоО4 (кобальтат(1У) бария).
Холгерсон и Карлсон (Holgersson, Karlsson, 1929) при слабом нагревании нитрата кобальта с нитратами магния или цинка получили двойные соединения общей формулы М110-Со203, относящиеся, по рентгенографическим данным, к типу шпинелей.
При прокаливании окиси кобальта(П) с кварцевым песком и карбопатом калия образуется синий стекловидный ортосиликат калия и кобальта^!). При сильном нагревании окиси кобальта(П) с“окисью алюминия получается также синее двойное соединение, имеющее характер шпинели. На образовании этого двойного соединения основана реакция Тенара на алюминий (см. г. I, стр. 405). При прокаливании окиси кобальта(И) с окисью цинка или окиси цинка, смоченной раствором нитрата кобальта (реакция образования зелени Ринмана), образуются в зависимости от температуры прокаливания двойные соединения типа шпинелей или только твердые растворы (подробнее см. стр. 466). Окись магния, смоченная раствором нитрата кобальта, при прокаливании окрашивается в бледно-красный цвет. Эта реакция свойственна также некоторым минералам, содержащим окись магния.
Сульфиды. Кобальт образует несколько сульфидов (ср. табл. 31, стр. 264 и сл.). Важнейшим из них является сульфид кобалъта(1 Г) CoS (мопосульфид кобальта). Он выделяется в виде черного аморфного осадка из растворов ..солей кобальта(П) при действии сернистого аммония. Этот осадок практически нерастворим в воде, но в свежеосажденном состоянии растворяется в разбавленных кислотах — даже в уксусной (a-CoS). Из уксуснокислых растворов сероводород осаждает кристаллический сульфид кобальта (р-CoS), практически нерастворимый в разбавленной соляной кислоте. Если сульфид кобальта, осажденный сульфидом аммония из аммиачного раствора (т, е. а-CoS), отфильтровать в токе воздуха, он становится практически нерастворимым в разбавленной соляной кислоте, хотя остается рентгеноаморфным. По мнению Фрике и Дёпгеса (Fricke, Dongcs), факт нерастворимости осадка объясняется тем, что раствор сульфида аммония, захваченный осадком, при взаимодействии с воздухом отдает серу с образованием богатого серой сульфида кобальта, который нерастворим в (холодной) разбавленной соляной кислоте **. Аналогично сульфиду никеля сульфид кобальта обладает резко выраженной склонностью к образованию коллоидных растворов. Золи обычно окрашены в коричневый цвет. При кипячении с уксусной кислотой сульфид кобальта коагулирует.
* Название «кобальтиты» устарело. Оно использовалось раньше пе только для кобальтатов(1У), но и для кобальтатов(П1).
** Госавами (Gosawaml, 1959) считает, что в данном случае образуется соединение CcioS.;. Только сульфид кобальта, осажденный бесцветным сульфидом аммония, (поскольку оп пс подвергся окислению), растворим в разбавленной соляной кислоте. При осаждении раствором полисулъфида сразу образуется сульфид с более высоким содержанием серы. Он практически нерастворим в разбавленной холодной соляной кислоте, хотя и рент г ено аморфен. Если в аморфный CoS, осажденный бесцветным сульфидом аммопия, после промывания без доступа воздуха пропустить ток воздуха, образуется основной сульфид трехвалентного кобальта. В разбавленной холодной соляной кислоте он растворим примерно наполовину, причем происходит выделение серы. Осадок, по являющийся гексагональными кристаллами, оказывается нераство-«имым вследствие обогащения серой. Аналогичные соотношения наблюдаются и у суль-ида никеля (Fricke, Donges, 1946).
Восьмая группа периодической системы.
317
В то время как а-CoS рентгеноаморфен, Р-CoS имеет гексагональную структуру (см. стр. 268). р-CoS проще всегополучать сухим путем методом Шенка (Schenk, 1942) — нагреванием порошка Со в токе H2S до 700° и, наконец, в высоком вакууме при 750— 800°. Соединение CosSB, мало отличающееся но составу от CoS, получается термическим разложением CoS в токе H2S или из расплава; оно кристаллизуется кубически. Атомы S образуют гранецентрироваппую кубическую решетку. Атомы Со располагаются в ней таким образом, что % иа пих тетрадэрически окружены четырьмя атомами серы, а остальные — октаэдрически шестью атомами S.
По данным Вайбке (Weihkc, 1936), сульфид кобальта(II) CoS остается однофазным даже при известном избытке серы (S : Со минимум 1,05 ; 1). Решетка соединения стабильна только в том случае, если узлы решетки, предназначенные для атомов Со, не заняты полностью; это следует из того факта, что рентгенометрически определенная плотность оказывается больше, чем пикнометрическая. Если бы избыточные атомы S вошли в решетку, плотность, вычисленная с учетом параметров ячейки, оказалась бы меньше пикнометрической.
Минералы сульфида кобальта(II) исключительно редки (на полуострове Индостан встречается сиепурит).
Трипобалъттетрасулъфид Co3S4 встречается в природе В виде кобальтового колчедана (линнеита), но может быть получен и искусственным путем (лучше всего по методу Шенка (Schenk) — нагреванием порошка Со в токе H2S при 400°), В минералах обычно часть кобальта замещена никелем (кобальто-никелевый ко.ччедап). Кобальтовый колчедан встречается иногда в виде хорошо образованных кристаллов (правильных октаэдров), обладающих металлическим блеском, окрашенных от крас-новато-серого до медно-красного цвета. По своей структуре оп, как и Со3О4, относится к классу шпинелей (а = 9,41 А). Выше 450°Co3S4 переходит в Co0Se с отщеплением серы.
Дисульфид кобальта CoS2 получается в виде черного порошка при более длительном взаимодействии расплавленной серы и моноокиси кобальта. Нагревание CoS2 при более высоких температурах сопровождается отщеплением серы. Он кристаллизуется по типу пирита (а = 5,64 А).
Арсепиды. С мышьяком кобальт образует несколько соединений. Из диаграммы состояния системы мышьяк — кобальт (исследованной вплоть до 53,5 вес.% кобальта) следует существование соединений Co5As2, Co3As, Со3Аз2 и CoAs, В природе встречаются арсениды CoAs2 и CoAs3. Первый из них является наиболее распространенной кобальтовой рудой (шпейеовый кобальт). Он изоморфен пириту FeS2, но редко встречается в виде хорошо образованных кристаллов, образуя обычно плотные серые глыбы, содержащие арсениды никеля. CoAs2 иногда встречается в виде ромбической модификации (саффлорита). Кобальтовый блеск CoAsS является важной кобальтовой рудой, он изоморфен пириту и производится от кобальтового шпейса при замене одного атома Аз на атом S, Арсенид CoAs3 иногда встречается в ваде мышьяково-кобальтового колчедана (скуттерудита).
Карбонильные и пптрозн.тьныс соединения. При нагревании тонн о раздробленное о кобальта с окисью углерода под давлением (100 атм) до 150—200° образуется тетракарбонил кобальта Со(СО)4, оранжевые кристаллы, которые при температуре несколько выше 50° начинают разлагаться. Это соединение нерастворимо вводе и довольно устойчиво по отношению к кислотам, не являющимся окислителями. Со(СО)4 растворим в органических растворителях — в спирте, эфире, бензоле, сероуглероде. Определение молекулярного веса показало, что в растворах существуют димеры — Co2(CO)g. При отщеплении окиси углерода образуется черный трикарбонил кобальта Со(СО)3 или Со4(СО)12, который можно перекристаллизовать из бензола,
О карбонилгидриде и карбоиилнитрозилё кобальта см. стр. 383 И 384.
При действии NO на галогениды кобальта(П) по аналогии с железом образуются нитрозилгалогениды кобальта CoX(NO)2 (Hieber, 1941). Они более устойчивы, чем питрозпдгалогениды железа. Их стабильность, как и в случае железа, снижается от иодо- к хлоро-производным; фторидные соединения не удалось получить и в данном случае. Иодид плавится при 131°, бромид — при 116°, а хлорид — при 101°. Соединения возгоняются на воздухе без разложения. Водой они разлагаются. Хибер предполагает, что в соединениях этого типа и вообще в комплексах металлов с NO окись азота существует в состоянии связанного иона (NO)+ или (sNHO:)+. Связь этого иона с центральным атомом осуществляется таким образом, что каждый атом N связан одной электронной парой с центральным атомом, т. е. точно так же, как осуществляется связь СО в карбонилах (ср. стр. 381). СО имеет конфигурацию, одинаковую с (NO)+. Следовательно, связь, с Одной стороны, имеет ковалентный характер, свой-
318
Глава 7
ствепный обычным связям лигапдов в «комплексах внедрения»; с другой стороны, принимая во внимание противоположные заряды па (NO)+-rpynne и па центральном атоме (который Хибер считает отрицательно заряженным; ср. ниже), можно считать этот вид связи семиполярным (ср. т. 1, стр. 161 и сл.). Атомы галогенов связаны ковалентно (гомеополярно).
По ряду 2eFe — 27<'о — 28N1 — 2ЭСн количество нытрозильных групп, которые могут замещать галогены, уменьшается. Поэтому удалось получить следующие соединения:
FcCl(NO)3—CoG1(NO)2—NiCl(NO) —CuCl,
Только в первом из них центральный атом имеет заполненную ободочку *:
-- C2-J 1+1
Ci;Fe(:N(;O:)3
•• in
Поэтому рассматриваемый нитрозилгалогенид мопомерен и сравнительно легко летуч, в то время цац в приведенном ряду летучесть соединений уменьшается.
В водных растворах ионы Со", в отличие от ионов Fe", не присоединяют NO даже в присутствии металлов, связывающих галогены (например, серебряного порошка); благодаря этому облегчается реакция безводных галогенидов кобаль-та(И) с NO, которая возможна вообще только в случае хлоридов.
Соли кобальта(П)
Хлорид кобальта (II) СоС12 (двхлорид кобальта, хлористый кобальт) получают в безводном состоянии при сгорании кобальта в токе хлора или при обезвоживании водных хлоридов; он представляет собой бледно-голубой порошок, который сублимируется, не плавясь. СоС13 очень легко растворим в воде — 46,3 г СоС12 в 100 г воды при 0° (Foote). Водный раствор окрашен при обычной температуре в розовый цвет. Безводный хлорид кобальта очень гигроскопичен к поэтому быстро краснеет на воздухе.
Он растворяется также в различных органических растворителях, папример в спирте, ацетоне, хиполинс и бензонитриле, образуя красивые синие растворы; при упаривании или охлаждении растворов хлорид кристаллизуется в виде синих игл. В 100 а абсолютного спирта растворяется 56,2 г СоС12; растворимость .его в эфире очень мала (0,02 а в 100 0).
С водой хдорид кобальта образует различно окрашенные гидраты:
СоС12-НгО
СоС1г.1,5ПгО
СоС1г2НгО
СоС]г4Н2О
СоС12-6НгО
сине-фиолетовый
т емпо -с ине-ф и о ле т о вый р о " о ват о-фи о л ето вый окраска цветов персика розовый
Гексагидрат при умеренном нагревании окрашивается в синий цвет. Водный раствор при нагревании тоже окрашивается в зависимости от концентрации в различные цвета — от фиолетового до синего. При добавлении к такому раствору концентрированной соляной или серной кислот, а также хлоридов наиболее активных металлов (главным образом кальция и магния) тоже появляется синяя окраска.
* Из восьми внешних электронов атома Fe, по мнению Хибера, лишь один электрон (обозначенный в приведенной формуле х) участвует в образовании гомеополяр-ной свяёи с атомом С1, в то время как 3 молекулы NO должны отдать Fe по одному электрону. У атома Fe оказывается, таким, образом, 10 валентных электронов, т. е. на нем находится заряд 2*. К 10 валентным электронам Fe2- присоединяются еще электронные пары N и пара между С1 и Fe, так что в целом образуется оболочка из 18 электронов.
Восьмая группа периодической системы 319'
Эти изменения цвета растворов вызываются главным образом дегидратацией, происходящей при нагревании. Наименее насыщенные продукты присоединения хлорида кобальта(Н) [а также других галогенидов кобальта(П)] окрашены в цвета от фиолетового до синего, в то время как наиболее насыщенные аддукты, содержащие 6 молекул присоединенного вещества, имеют розовую окраску. Помимо гидратов, ото явление наблюдается и у кристаллических продуктов присоединения других кислородсодержащих растворителей, например метилового спирта. Хлорид кобальта(II) может присоединять и азотсодержащие молекулы, образуя с пими кристаллические соединения, как правило розовые, а менее насыщенные окрашены в цвета от фиолетового до синего (Hantzsch, 1927),
Двойные хлориды двухвалентного кобальта и щелочных или других активных металлов — хлорокобалътатД!I) обычно окрашены в темно-синий цвет. В качестве примера таких соединений можно указать полученные Кемпбеллом (Campbell) двойные хлориды кобальта и цезия: CsC1-CoC12‘2H2O, 2CsC1-CoC12 и ЗСвСЬСоС12. Только в исключительных случаях двухвалентный кобальт образует гекса галогенид-п он; примером может служить Li4[СоС!^]-ЮН2О — синие кристаллы (Beneath, 1927).
Смоченная раствором хлорида кобальта тонкая бумага в сухом воздухе окрашивается в синий цвет, а во влажном вновь приобретает розовую-окраску. Такими индикаторами прежде пользовались для приблизительного определения влажности воздуха. Хлорид кобальта применяют главным образом как лабораторный реактив, он служит также исходным» продуктом для синтеза других соединений кобальта.
Бромид кобальта(П) СоВг2 в безводном состоянии образует блестящие зеленые листочки, легко растворимые в воде, расплмпатпгщьч-я на воздухе в красную жидкость. Гексагидрат СоВг2-6Н3О представляет собой красные кристаллы, тоже, легко расплывающиеся. Над к о иц епт ри ров а пи ой серной кислотой он отщепляет воду. Прн 100* он плавится с частичной потерей воды и образованием пурпурно-синего дигидрата СоВг2-2Н2О, а при 130° переходит в безводное соединение. Среди двойных бромидов, по-видимому, преобладает тип Ml [СоВг4(ОН2)2]. Мейер (Meyer, 1935) синтезировал гексагалогепид — Li4[CoBr6]-12H2O — темно-синио, расплывающиеся кристаллы,
Иоднд кобальта(Н) Со12 получается в безводном состоянии в виде серо-зеленой массы при непосредственном взаимодействии компонентов или при нагревании водной соли до 130°. Со1а, кроме воды, растворим в спирте и ацетоне. Его ди- и тетр а гидраты* окрашены в зеленый цвет. Гекеагидрат Со12-6Н2О — темно-красный; его водный раствор уже при 20q приобретает оливково-зеленую окраску, а при 35° становится хромово-зеленым. При очень низких температурах устойчив также его (светло-красный) нонагидрат. Средн двойных иодидов преобладают соединения типа M3IC0I4], например известно Ся2[Со14]. Исключением является образование гексаиодидных солей — м|[Со10].
Фторид кобальтаЩ) выделяется в виде розовато-красных кристаллов CoF,- 2Н3О при унариваппи растворов карбоната кобальта в водной плавиковой кислоте. Безводная соль CoF2 также имеет красноватую окраску. То же можно сказать и о двойных фторидах — фторокобальтатах(П), например K2[CoF4] и (NH4)2[CoF4}.
Изменение окраски от розовой через синюю до зеленой в ряду безводных галогенидов кобальта(П) : CoF2, СоС12, СоВг2 и Со12обусловлено сдвигом максимума поглощения света от сине-зеленого до красного. Предполагается, что этот сдвиг связан с гомеополярным характером связи металл-галоген в направлении от CoFa к Cola-Таким же образом объясняется различие окраски, наблюдаемое при различных степенях гидратации. Чем больше молекул воды (или аналогичных нейтральных моле кул) присоединяется к иону кобальта(Н), тем в большей степени галогенид-ионы оттесняются от него и тем меньше проявляется поляризующее действие иона кобальта на галогенид-ионы (ср. т. 1,стр. 348 и сл.). У других солей кобальта(П) тоже наблюдается изменение окраски от розовой к зеленой, и тем более значительное, чем сильнее поляризующее действие иона кобальта па окружающие его ионы, т. е. чем ближе-характер связи к гомеоцол яркому.
Основные галогениды кобальта. Основные галогениды кобалыа(1П интересны как представители двух групп основных солей, состав и свойства которых можно объяснить лишь их структурой (Feitknecht, 1933 и сл.)-
320
Глава 7
Основные соли одной группы, представителем к вторых может служить «зеленый основной хлорид кобальта» Co(OH)CI-4Co(OH)S'4Н2О, образуют так называемую двухслойную решетку. В составе coittiz обнаруживаются значительные колебания, несмотря на то что в данном случае речь идет о химических соединениях вполне определенных е структурном отношении. Основные соли другой группы, к которым относится «розовый основной хлорид кобальта» Со2(ОН)зС1, образуют однослойную решетку. Соли, у которых обнаруживается такая структура, имеют всегда значительно более простой состав, чем соли, принадлежащие к первой группе, причем их состав или вообще постоянен, или колеблется в очень узких пределах.
Зеленый основной хлорпд кобальта; двухслойная решетка. При разложении раствора хлорида кобальта(П) щелочью в количестве, недостаточном для полного осаждения гидроокиси, выпавшая первоначально синяя Со(ОН)2 при взаимодействии С СоС12, оставшимся в растворе, частично превращается в зеленый основной хлорид кобальта *. В идеальном случае его состав (колеблющийся в известных пределах) соответствует формуле Со(ОН)С1- 4Со(ОН)2- 4Н2О. Решетка этого соединения построена из слоев Со(ОН)2 так же, как у розовой гидроокиси кобальта(П) в кристаллическом состоянии. Между такими слоями Со(ОН)2 («основными») расположены другие слои, построенные из Со(ОН)С1 и НоО («промежуточные»). На каждый основной слой накладывается один промежуточный, затем опять основной и т. д. Благодаря промежуточным слоям основные слои раздвигаются (на 8,2 А в отличие от 4,65 А у гидроокиси) и одновременно смещаются друг относительно друга в направлении слоя так, что ОН-группы, принадлежащие двум различным основным слоям, располагаются вертикально друг над другом. Этого не наблюдается в случае гидроокиси. Составляющие промежуточных слоев, в отличие от составляющих основных слоев, размещаются в предназначенных для пих узлах решетки совершенно незакономерно. Файткнехт по окраске и соответственно спектру поглощения зеленого основного хлорида кобальта предположил, что в промежуточных слоях кобальт связан в значительной степени гомео-полярно, а в основных — гетерополярно. Различие типов связи сказывается и на химических свойствах: в промежуточных слоях кобальт окисляется легче, чем в основных.
Файткнехт, объяснивший основные особенности структуры описанного типа, назвал ее двухслойной. Такие двухслойные решетки, кроме основных солей кобальта, образуют также основные соли других двухвалентных металлов, например никеля, железа(Л), марганца(Л), кадмия н магния. Соли, обладающие такой структурой, кристаллизуются в виде листочков. Опи обладают способностью постепенно терять воду, так как ее молекулы не имеют определенных мест в решетке; это относится и к другим компонентам промежуточных слоев, поэтому, например, в зеленом основном хлориде кобальта они могут содержаться в переменном количестве. Однако у некоторых основных солей, как следует из рентгенографических данных, катионы и анионы расположены в промежуточных слоях закономерно (что в основных слоях имеет место всегда). Однако и в этих случаях отношение гидроксила к соли колеблется в известных границах, причем гидроксилы и анионы кислоты могут находиться в промежуточных слоях друг против друга или при отсутствии промежуточных слоев в кристаллах существуют зопы, в которых много слоев гидроксилов непосредственно следуют один за другим. Вещества, имеющие двухслойную структуру, оказываются исключительно склонными к топохимическим реакциям (ср. гл. 19). При реакциях их с водными растворами веществ происходит взаимодействие только вдоль плоскости слоев, а но в перпендикулярном направлении.
Розовый основном хлорпд кобальта; решетка, построенная из простых слоев. Зеленый основной хлорид кобальта при длительном соприкосновении с не очень разбавленным раствором хлорида кобальта постепенно превращается в розовый основной хлорид кобальта Со2(ОН)3С1. Эта соль образует так называемую решетку ив простых слоев, т. е. построенную не из различных, а только из одинаковых слоев. Решетка такого типа была впервые обнаружена у основного хлорида кадмия Cd(OII)C.l (Hoard, 1934; см- рис. 50, стр. 480). В случае розового основного хлорида кобальта, пр данным Файткпехта, отдельные слои, построенные из ионов кобальта, закономерно размещаются над расположенными с обеих сторон нонами гидроксила и хлора. Вероятно, расколожоние отдельных ионов в слоях совершенно аналогично решетке розового основного хлорида кобальта(П) пли структуре брусита (ср. т. I, стр. 293, рис. 61), но вследствие частичного замещения ионов гидроксила па хлор расстояния между
* Это соединение ранее ошибочно считали одной из модификаций гидроокиси кобальта(П) (ср. стр. 315).
Восьмая группа периодической системы
321
слоями несколько увеличиваются. Окраска соединения, по данным Файткпехта, позволяет сделать заключение о преимущественно гетер о полярном характере связи между кобальтом и ионами хлора.
«Решетка из простых слоев», кроме некоторых основных солей кобальта, найдена и у основных солей никеля и цинка, а также у основных галогенидов кадмия.
Основные бромиды кобальта. Известно четыре основных бромида кобальта: зеленый, изоморфный зеленому основному хлориду; красно-фиолетовый Со2(ОН)3Вг, который, как и хлорид аналогичного состава, образует решетку из простых слоев, однако расположение ионов внутри отдельных слоев отличается от хлорида; сине-фиолетовый (с двухслойной решеткой) и розовый основной бромид, структура которого пока не известна. В то время как в розовом основном хлориде до 1/з хлорид-ионов может замещаться на бромпд-ионы, красно-фиолетовый бромид не обладает способностью замещать Вт па С1,
Цианид кобальта(П) и циапокобальтаты(П). Соли кобальта(П) образуют с ионами CN' осадок, нерастворимый в воде и разбавленных кислотах, по растворимый в водном растворе аммиака или в карбонате аммония с образованием желтого раствора; после высушивания над концентрированной Серной кислотой этот осадок । имеет состав Co(CN)2‘2H2O. При обработке его в свежеосажденном состоянии раствором цианида калия (при этом следует тщательно избегать повышения температуры, так как иначе кобальт переходит в трехвалентпое состояние, что сопровождается выделением водорода) вначале образуется зеленый раствор, из которого иногда выпадает осадок зеленого цвета KaCo[Co(CN)g). При стоянии раствор постепенно окрашивается в красный цвет. При достаточной концентрации раствора из пего выпадают кристаллические листочки гексацианокобалътата(И) калия К4 [Со(СМ)й], которые После отмывания спиртом и эфиром (в которых они нерастворимы) могут сохраняться в атмосфере водорода. В воде это соединение растворяется легко, образуя темно-красный раствор. При кипячении K4[Co(CN\J окисляется в гоксацнапокобальтат(Ш); в отсутствие воздуха окисление осуществляется за счет кислорода воды и сопровождается выделением водорода. В присутствии избытка ионов циана разложение происходит уже при умеренном нагревании. Кроме описанной калиевой соли, было получено также и несколько других гексациапокобальтатов(Ц).
Цианокобальтат(П)-ионы, по данным Вринтцингера, как и цианоферрат(II)-ионы, в водном растворе присоединяют 12 молекул Н3О в отличие от цианокобальтат(П1)-и цнапоферрат(Ш)-попов, но гидратирующихся в водном растворе.
Роданид кобальта(П) и родапокобальтаты(П). Роданид кобалъта{11) ' Co(SCN)2-4H30 образует темные красно-фиолетовые, ромбические, сильно расплывающиеся кристаллы. В небольшом количестве воды эта соль растворяется, образуя темпо-синий раствор, при разбавлении окраска его переходит в красную, а при добавлении ацетона снова становится темно-синей. В спирте роданид нобальта(Н) растворяется, также образуя темно-синий раствор. Чистую соль лучше всего получать насыщением карбоната кобалъта(П) раствором роданистого водорода и концентрированием его над серной кислотой или реакцией между сульфатом кобальта н роданидом бария.
Роданокобалътаты,(И) в большинстве случаев имеют состав MJ [Co(SCN)J. Аммонийная соль, например, получается при добавлении к раствору соли кобальта(Н) роданида аммония; затем раствор извлекают встряхиванием с амиловым спиртом (или смесью равных частей амилового спирта и эфира) и вытяжку упаривают; выделившаяся аммонийная соль представляет собой синие иглы, обладающие сильным двойным лучепреломлением, п может быть перекристаллизована из ацетона. Из водных растворов она кристаллизуется с водой (NH4)2 [Co(SCN)J-4Н3О. Аналогичным образом можно получить и калиевую соль. Известны и другие тетрарода-нокобальтаты(Н), а также некоторые нента- и гексароданокобальтаты(П).
Реакцию солей кобальта е роданидом калия или аммония нередко применяют для открытия кобальта. При этом чаще всего роданид извлекают из красного водного
21 г. Рсмя
322
Глава, 7
раствора встряхиванием со смесью амилового спирта и эфира, Эта реакция становится еще более чувствительной, если посинение вызвать в самом растворе путем добавления к нему ацетона.
Нитрат кобальта(П), нитрат, кобальта Co(N03)2-6H;>0 образует расплывающиеся кармоизиново-красные моноклинные столбики или таблички, легко растворимые в воде (тоже с кармоизиново-красным окрашиванием). Его применяют к аналитической химии в качестве реактива (главным образом в анализе при помощи паяльной трубки), а также при производстве кобальтовых красок и в керамике. Получают его растворением кобальта, окном Со(П) или карбоната Со(П) в разбавленной азотной кислоте. При пагревании гексагидрат сначала теряет 3 молекулы воды, а при более высокой температуре происходит его разложение, сопровождающееся выделением нитрозных паров и образованием темно-коричневого окисла.
При действии небольшого количества едкого натра па раствор нитрата кобальта(И) образуется зеленый основной нитрат кобальта, соединение состава Co(NO3)2-6Со(ОН), (обычно слегка колеблющегося), образующее двухслойную решетку [распределение компонентов в ее основных и промежуточных слоях по аналогии с зеленым основным хлоридом: кобальта можно выразить формулой Cot,25(OH)(N03)4,5-4Co(OH)2]. При соприкосновении с концентрированным раствором нитрата кобальта зеленая соль превращается в розовый основной нитрат Co2(OH)3(N03), кристаллизующийся В решетке, состоящей из простых слоев (Feitknecht, 1936 и сл.).
Нитрит кобальта(П) и двойные нитриты. Нитрит кобальта) II) должен существовать в водных растворах, одпако его до настоящего времени пе удалось выделить.. Двойные нитриты двухвалентного кобальта, как, например, 2KNO2-Co(NOa)2-II2O (желтый порошок), были получены смешиванием горячих концентрированных растворов хлорида кобальта(П) с нитритами щелочных металлов.
Сульфат кобальта(П) кристаллизуется из растворов кобальта, окиси кобальта(П) или его карбопата в разбавленной серной кислоте при комнатной температуре в виде кобальтового купороса CoS04-7H20; он представляет собой устойчивые на воздухе красные моноклинные кристаллы, изоморфные железному куяоросу. В воде эта соль легко растворима (36 я CoSO4 в 100 г воды при 20е), ио она нерастворима в спирте.
При 40—50° из водных растворов кристаллизуется гекса гидрат CoS0c6H20, В присутствия в оду отнимающих средств, например концентрированной серной кислоты, образуются еще более бедные водой гидраты. При кипячении кобальтового купороса е Концентрированной серной кислотой получается безводный сульфат кобальта в виде красного порошка, при кипячении с водой легко переходящего в раствор. Из растворов, содержащих сульфаты щелочных металлов, кристаллизуются двойные сульфаты, например сульфат аммония-кобальта (NH4)as04-CoS04-6H20 — красные кристаллы, изоморфные сульфату аммопия-магния.
Существуют два основных сульфата кобальта(II): синий, состава CoSO^SCofOHla (с переменным содержанием воды), и фиолетовый, состава 2CoS04-ЗСо(ОН)а-6Н26 Обе соли кристаллизуются в двухслойных структурах. Фиолетовый сульфат, как и синий, обладает способностью непрерывно отщеплять воду и вновь присоединять ее. Окраска фиолетовой соли при обезвоживании резко изменяется в синюю, однако, не возвращается при повторном присоединении воды. Как показало рентгенографическое исследование, при обезвоживании нарушается нормальное, закономерное строение промежуточных слоев и уменьшается расстояние между основными слоями [состоящими из Со(О11)2]. При повторном присоединении воды это расстояние восстанавливается до первоначального, но строение промежуточных слоев остается наруЩеипым (Feitknecht, 1934).
В природе кобальтовый купорос встречается в небольших количествах в виде розово-красного налета на кобальтовых рудах (биберит).
Сульфит кобальта)!!) и сульфитокоба.-1ьтаты(11). При пропускании сернистого газа через воду, в которой суспендирована гидроокись кобальта(П), образуются красноватые, зернистые, трудно растворимые в воде кристаллы сульфита кобалъта(П)
Восьмая группа периодической системы
323
Cc.SO3> 5Н2О. При добавлении сульфита палия к раствору сульфита или хлорида кобапь-та(П) и при нагревании образуется бледно-красный кристаллический, малоустойчивый па воздухе осадок дисульфитокобальтата(П) калия K2[Co(SO3l2]. Это соединение можно также получить Кипячением гидроокиси кобальта(II) с раствором бисульфита калия. Аналогично можно получать и другие сульфптокобальтаты(П), наиример натриевую и аммонийную соли.
Карбонат кобальта(И) и карбопатокобальтаты(Т1). При добавлении к растворам солей кобальта(11) щелочных карбонатов из них, как Правило, выпадают сипие осадки основных карбонатов. Одкако, если в реакцию вводить растворы, насыщенные двуокисью углерода, удается получить гексагидрат нейтрального карбоната СоСО3-6Н2О—фиолетовые, микроскопические кристаллы. При нагревании их до 14и° в запаянной трубке образуется безводный карбонат кобальта в виде светло-красного порошка (состоящего из микроскопических ромбоэдров).
Если осадок, образовавшийся при добавлении к раствору солей кобалЬта(П) карбоната аммония, оставить на некоторое время в соприкосновении с избытком осадителя, он становится кристаллическим, причем образуется двойная соль (NH4)2CO3 • СоСО3-4НгО (ярко-красные призмы). Аналогичный состав имеют двойные карбонаты [карбонатокобальтаты(Н)] патрия и калия.
Оксалат кобальта(П) иокеалатокобальтаты(П).(й>с<гл<гт двухвалентного кобальта образуется в виде розовато-красного осадпа при добавлении к растворам Солей кобальта^!) ионов оксалата. Для получения его в чистом состоянии лучше растворять карбонат кобальта(Н) в водном растворе щавелевой кислоты. В атом случае соединение выпадает в виде розовато-красного порошка, имеющего состав СоС3О4-2Н2О. В чистой воде и в водном растворе щавелевой кислоты оно почтя нерастворимо, но растворяется в теплом концентрированном водном растворе аммиака и особенно легко в растворе карбоната аммония. Из растворов, содержащих избыток оксалата щелочного металла, получаются двойные соли [океалатокобвльтаты(П)], например К2С2О4- СоС304’ 6Н2О —-красные ромбические призмы, образующие в воде прозрачный раствор.
Ацетат кобальтцЩ) Со(СН2СОО)2-4Н2О получается при растворении .карбоната кобальта в уксусной кислоте. Он образует красные кристаллы, изоморфные ацетату магния, цинка, марганца и никеля; в воде растворим легко. Его применяют как средство для отбелки и сушки лаков и олиф, а также в качестве реактива.
Аммиакаты солей кобальта(П). Соли кобальта(П) и сильных кислот, например галогениды и сульфат, а также роданид, могут в безводном состоянии присоединять, как правило, до 6 молекул NH3. Так, при пропускании сухого аммиака над безводным сульфатом кобальта образуется сульфат гексаамминкобалъта(1Г) [Co(NH3)6]SO4 в виде красновато-бе лого порошка. Это соединение можпо получить и пропусканием аммиака через водный раствор сульфата кобальта(П). Оно очень хорошо растворимо в водном растворе аммиака. Спирт высаливает его из водных растворов. Сульфат гексаамминкобаиьта(П) разлагается водой, т. е. является неустойчивым комплексным соединением.
Подобным же образом ведут себя и другие аммиакаты кобальта(П). Так, бледный розовато-красный хлорид гексаамминкобалъта(11) [Co(NH3)b]C12 также можно получить пропусканием (большого количества) аммиака в концентрированный раствор хлорида кобальта(П). В разбавленном аммиаке ок очепь легко растворим; значительно слабее — в концентрированном. Чистой водой оп разлагается. [Go(NH3)6]Bt2 и [Co(NH3)B)I2 тоже представляют собой красные порошки. Таким образом, гексааммип-иые соединения двухвалентного кобальта ио окраске похожи па его гексаакво-соеди-пения. Вместо аммиака могут присоединяться или внедряться молекулы гидроксил-амипа, гидразина, а также органические азотсодержащие ангидрооспования. Следует ггце упомянуть о продукте присоединения гидразина к оксалату кобальта(Ц) СоС2О4-•2_\3Н4 (малинового цвета), который в противоположность гидразиновым соединениям хлорида, бромида и сульфата водой заметно не разлагается.
Галогениды и другие соли кобальта(И) с кислотными остатками, обладающими значительной поляризуемостью, могут присоединять обычно 4 нейтральные молекулы, причем кислотные остатки пе вытесняются во внешнюю сферу. Поэтому соединения типа СоХ2-4Аш (X — Cl, Br, I, GN, SCN нт. д.; Ат — органический амин), как
21*
324
Глава 7
правило, пс электролиты [СоХ2Лпц]. Только в исключительных случаях при присоединении 4 нейтральных молекул происходит вытеснение кислотных остатков во внешнюю сферу с образованием соединений [СоАт4]Х2, способных к электролитической диссоциации; так, о-фонилендпамнн CeH4(NH2)2 «внедряется» во внутреннюю сферу СоС12, образуя [Со(С6Н.,,(МН2)2)г]С1г (вероятно, размер такого лиганда создает пространственные затруднения дли одновременного образования связи между галогеном и центральным атомом;.
Соединения типа [СоХ,Ат4] малоустойчивы. Это справедливо вообще для соединений типа [ZXIX.2Am4]. С малой устойчивостью этих соединений связано и то обстоятельство, что до ейх пор пи в одном случае не удалось установить у соединении типа ]ZIXX2Am4J явления ап с-mp а нс-изомер и к [Н i е b р г, Z. Elektrochem., 39, 24 (1933)]. В тех немногих случаях, когда соединение этого типа оказывалось достаточно стабильным для выделения его в свободном состоянии, из двух возможных вариантов расположения лигандов образуется комплекс с энергетически более выгодным, тем более что вследствие слабо комплексного характера этих соединений лиганды могут легко обмениваться местами.
Соли кобальта(1И)
Фторид кобащ.та(Ш) — хромово-зелепый порошок — был впервые получен Варбьери (Barbieri) в виде гидрата состава CoF3-ЗЦЛТзО при электролизе насыщенного раствора фторида кобальта(И) в 40%-пой плавиковой кислоте (при охлаждении платиновой чаши, служащей анодом). Чистая пода его разлагает, причем выделяется гидроокись кобальта(Ш). В безводном состоянии это соединение было получено в 1929 г. Руффом (Run) действием фтора на хлорид кобальта(И). Это соединение представляет собой (в безводном состоянии) светло-коричневый порошок уд. веса 3,88 и образует гексагональные кристаллы. Примерно при 300° в струе СО2 оно разлагается на фторид кобалъта(П) и фтор. Фторокобальтат(ПГ) калия K-;[CoFB] образуется про действия фтора па смесь СоС1-> и КС1; оп является, по мнению Клемма (Klemm, 1953),. единственным известным в настоящее время «нормальным комплексом» Со111. Все остальные комплексные соединения кобальта (III) относятся к так называемым «комплексам внедрения».
Хлорид кобальта(III) до сих пор получить пе удалось. При растворении гидроокиси кобальтаЦП) в соляной кислоте выделяется хлор. Возможно, что при более высокой температуре СоС13 стабилен в еавообравиой форме. По данным Шефера (Schafer^ 1951), СоС12 при температуре около 800° в токе хлора оказывается значительно более летучим, чем в ток о хлористого водорода или азота. Этот факт позволяет предположить образование в атмосфере хлора летучего высшего хлорида.
Сульфат кобальта(Ш) Co2(SO4)3-18H2O получается при электролиз? концентрированного раствора сульфата кобальта(П), содержащего сернут кислоту, с применением диафрагмы и охлаждением анода. Он образует мелкие синие иглы, немедленно разлагающиеся водой с выделением кислорода; однако в разбавленной серной кислоте сульфат растворяется без разложения.
С сульфатами щелочных металлов сульфат кобальта(Ш) образует квасцы. Так, калиево-кобалътовые квасцы КСо(804)ч-12Н20 получаются при смешивании эквивалентных количеств охлажденных растворов сульфата кобальта(Ш) и сульфата кал пл Это темпо-синне октаэдрические кристаллы, разлагающиеся водой при компатне? температуре с выделением кислорода: их можпо промывать ледяной уксусной кислотой, а затем ацетоном. Рубидиево-кобальтовые и цезйево-кобальтовые квасцы растворимы гораздо труднее, чем калиевая соль, они тоже темно-синего цвета.
ОксалатокпблльтатьЦШ). При растворении гидроокиси кобальта(III) в ксз центрированном водном растворе щавелевой кислоты получается зелепый раствсд. дающий с карбонатом кальция осадок состава Са3[Со(С2Од)3]2, в котором, следовательно, надо предположить присутствие кислоты HjfCoj'C,^)^] [три оксалат ок оба at--(III) кислота]. Однако в свободном состоянии пи эта кислота, ни лежащий в ₽: основе оксалат кобальта(Ш) не известны. В растворах триоксалатокобальт(ПГ) ксг-лота тоже малоустойчива; опа разлагается, выделяя оксалат кобальта (Л) СоСгС^ Колее устойчивыми,чем свободная кислота, являются ее соли Mj[Co(C2O4)3] [триокс^-латокобал[>таты(111)], пацрпмер аммонийпая соль (NH4)3[Co(C2O4)3]‘ЗН2О, образующая темно-зеленые моноклинные кристаллы; ее можно получить при лстряхиванст
Восьмая, группа периодической системы 325
раствора оксалатокобальтата(П) аммония с двуокисью свинца при добавлении ледяной уксусной кислоты или электролитическим окислением смеси растворов оксалата аммония и солей кобальта(П). Кроме триоксалатокобальтатов, известно лишь несколько комплексных оксалатов кобальта(Ш) (в отличие от хрома). Надо отметить еще соль Дюрраиа, которую но Вернеру и Спаку (Werner, 1914; Spacn, 1934) следует рассматривать как гидроксодиоксалатоаквокобалътат(11 Г) калия К.2(Со(ОН)(С2О4)3(ОН2)}т
Гекеацианокобальтаты(1П) и гскеацманокобальт(1П) кислота. Прн обработке солей кобальта(П) в водном растворе избытком цианида калия, как уже упоминалось, сначала образуется гекеацианокобальтат(П) калия K4|Co(CN)ej, переходящий при доступе воздуха в соответствующим кобальтат(Ш) (а при полном отсутствии воздуха с выделением водорода). ГексаиианокобалътатЦП) калия K3[Co(CN)0] образует прозрачные бледно-желтые кристаллы, пзморфные кристаллам гексацпано-феррата(Ш) калия. При добавлении к раствору калиевой соли солей меди(П) или свинца(П) выпадают гексациапокобальтаты(Ш) этих тяжелых металлов. При разложении пх Сероводородом получают свободную гексацианокобальт{111} кислоту, а путем нейтрализации последней можно легко получить и другие, очень хорошо растворимые в воде гексациапокобальтаты(Ш), например соли натрия, аммопня, бария и стронция. Гексапнапокобальт(Ш) кислоту можно получить также непосредственно упариванием досуха с азотной или серной кислотой ее калиевой соли с последующим выщелачиванием остатка водой. Опа образует бесцветные, волокнистые кристаллы HMCofCNJeJ-H^O, растворимые в воде и спирте, по не в эфире.
K3[Co(CTN)g] в растворе жидкого аммиака реагирует с калием в соответствии с уравнением К3 |Co(CN)6] + ЭК = 2KCN -f- K4[Co(CN)4] (Hieber, 1952). Комплекс K4[Co(CN)4] получается при этом в виде коричнев о-фиолетового мелкокристаллического пирофорного вещества. Оно энергично реагирует с водой с выделением водорода и даже взаимодействует с СО2. Кобальт в этом соединении имеет валентность нуль, т. е. его следует рассматривать Как тетрациачокобальтат{<У) калия.
Гексанит ре кобальта ты (III)» В то время как нитрит трехвалентного кобальта в чистом состоянии не известен, его двойные или комплексные соли, гексанитрокобалът,аты(111) Mi[Co(NO,)a} получаются легко. Для приготовления гексанитрокобалътата(1Т1) натрия Na3[Co(NO2)eL; применяемого в качестве реактива на попы калия, смешивают растворы нитрата кобальта и нитрита натрия, добавляют уксусную кислоту, пропускают через эту смесь воздух и выдерживают ее некоторое время, чтобы образующиеся вследствие загрязнения гексанитрокобальтаты(Ш) калия или аммония выпали в осадок. Из прозрачного раствора натриевую соль осаждают спиртом. Повторным растворением в воде с последующим осаждением спиртом ее можно перекристаллизовать. Эта соль представляет собой желтый порошок, легко растворимый в воде с образованием желто-коричневого раствора.
Гексанитрокобалътат(1Г1) калия K3[Co(NO2)6] (соль Фишера) выпадает в виде блестящего ярко-желтого порошка при добавлении гексапи-трокобальтата(Ш) натрия к растворам солей калия. K3[Co(NO2)6] состоит из хорошо образованных призм и бипирамид. В холодной воде он растворим очень слабо (1 :1120 при 17°); горячей водой разлагается, отщепляя NO. Эта соль практически совершенно нерастворима в спирте и в эфире. K3[Co(NO2)e] применяют для из г отопления масляных и акварельных красок под названием кобальтовой желтой или индийской желтой вместо натуральной индийской желтой. Им пользуются также иногда для окраски стекол и фарфора, так как в отличие от обычно применяемой окиси кобальта он не содержит никеля и железа и поэтому сообщает силикатным сплавам более чистую синюю окраску.
Гексанитро кобалътат аммония по растворимости аналогичен калиевой соли. Особенно трудно растворимы рубидиевый и цезиевый гексанитрокобальтаты(Ш) (оба кристаллизующиеся с 1 молекулой воды); их растворимости при 17° составляют соответственно 1 19 800 и 1 ; 20 100.
326
Глава 7
Амминные комплексы трсхвалентного кобальта
Трехвалентный кобальт связывает аммиак значительно прочнее, чем двухвалентный. Поэтому аммиачные растворы солей кобальта(П) очень склонны к окислению в алмннные соли кобальта(Ш) даже при действии кислорода воздуха.
При пропускании в течение некоторого времени воздуха через раствор хлорида кобальта(П), полученного взаимодействием хлорида аммония и аммиака, можно выделить соединенно [CuiNH3)elC)3 [хлорид гексаамминкобальта(Л1)]. Оно образует оран-жево-желтыо кристаллы и поэтому прежде называлось лутеохлоридом кобальта. О строении этого соединения можно судить по его аналитическим реакциям (весь содержащийся в нем хлор немедленно осаждается нитратом серебра) и по электропроводности его растворов, которая имеет значения, характерные для соли, распадающейся па 4 иона.
Помимо желтой соли, из раствора можпо получить красную соль, называвшуюся ранее розеохлорилом кобальта. Она имеет состав СоС13-5NH3’Н2О; в растворе все атомы хлора диссоциируют, и при замене хлора на другие кислотные остатки образуются соли, содержащие наряду с 5NH3 по крайней мере 1 молекулу НЙО. Поэтому полагают, что молекула П20 вместе с 5 молекулами NH3 связана с кобальтом координационно — [Co(OIl2)(NH3)3]CI3 [тлориЗ аквопентаамминкобалъта(11 /)].
Из этого раствора можно помучить еще пурпурно-красное безводное соединение, имеющее состав CoCl3-5NH3, называвшееся прежде пурпуреохлоридом кобальта. Как было впервые установлено в 1870 г. Кроком (Кгоск), этот комплекс, кроме 5 молекул NH3t содержит еще одтт координационно связанный атом С1 (из свежеприготовленного водного раствора этого соединения нитратом серебра при обычной температуре сразу осаждается только 2 атома С1). На основании сказанного этому соединению — хлориду хлороиентаамминкобалъта(11 Г) — следует приписать формулу [CoC1(NH3)5]C12.
Кроме того, известно соединение состава СоС13-4NII3, в котором 2 иона С1 связаны координационно, а в подпои растворе не разложившийся комплекс диссоциирует лишь с образованием одного иона С1. Это соединение следует рассматривать как хлорид дихлоротетраамминкобалъта(ПЦ [СоС12(Г\'Н3)4)С1. По аналогии с соединениями хрома того же типа (ср. стр. 165 и сл.) оно существует в двух изомерных формах — цис И транс. Цис-форма окрашена в сине-фиолетовый цвет, транс- —в зеленый. Поэтому первая раньше называлась виолео-, а вторая — празеох л ори дом кобальта.
Приведенные соединения являются примерами первых членов важнейших рядов амминных соединений солей кобальтами I). В табл. 35 указаны известные типы таких одноядерных комплексов. Они так же разнообразны, как и аналогичные соединения хрома *. И в данном случае вместо молекул аммиака здесь часто встречаются молекулы других азотсодержащих оснований. В качестве отрицательно заряженных групп внутри комплексов, помимо галогенов, могут быть радикалы слабых кислот, в то время как группы X, расположенные во внешней сфере, нередко являются анионами сильных кислот.
Ианы аквоамминкобальта(1П) являются кислотами (ср. стр. 762 и сл.), т. о. они обладают способностью отщеплять протоны и переходить в соответствующие гидроксо-ионы, например
[Co(OH2)(NH3)5r--bII20 [Co(OH)(NH3)5J" + [H3O]-.
Положение равновесия между соответствующими друг другу акво- и гидроксо-попамп ([Co(Ori2i(NH3) = " 32 [Co(OH)(NH3)5]",[Co(OH2)2(NH3)41- ;£ [Со(ОН)(ОНг)(КН3)4[-и т. д.) зависит от числа молекул воды в комплексах; чем больше их входит в состав комплексного иоггд, тем сильнее сдвигается равновесие в сторону образования гидроксо-нопов при одинаковой концентрации ионов водорода (Bronsted, 1628). Вместе со способностью отщеплять протоны возрастает склонность к присоединению
* Следует отметить, что как у хрома(Ш), так и у кобальта(111) неизвестны комплексы, содержащие одну молекулу аммиака. В то же время у железа(П и Ill) (ср. стр. 308), а также у ирндия(Ш) образование подобных соединений имеет место.
Таблица 35
Главные типы одноядерных аммин-, акво* и а цидо-комплексов трех на лент кого кобальта
а 6 в г а е «ТГР vtV
I [СоАш.]3* Ams Со L (онг). 3+ Ат4 Со L (ОН,).] 3+ Ат3 Со . (ОН.),. 3+ Г Ат, !а+ Со —
II 9 * г ut я+ Ат4 Со(ОН.) в 2+ Ат3 Со(ОН.)а в 2+ Ат2 Со(ОН2)3 в зн- — ?
ш Ага/ Со L в, + Ат3 Со^ОН.) +• Ат, Со(ОНа)„ в» + — Ат — электронеатрамная молекула (ЫН,, пиридин ит.Ъ.) R — коороинационносвязаниый оататок одноосновной кислоты или эквивалентное количество остатков многоооновных кислот
IV Ат/ Со в, J 9 — ?
V Г Ат*!' Со в4 — -
VI - (ОН.)!’- Со L Ед
VII [СоВ.]»’
328
Глава 7
электродов, г. е. к образ овацию комплексных ионов двухвалентного кобальта. Поэтому гексааквокобальт(Ш)-ион |СоЮН2)6)"‘, по данным Бренстеда, не способен к существованию. Уже тетраакво-ион [Co(OH2)4(NH3)2]“' наряду с тенденцией к образованию гидроксо-иона [Со(ОН)(ОН2>3(М13)2]‘‘ проявляет явную склонность к переходу в соединение кобальта(И),
Средн аммиакатов кобальта наиболее давно известна соль Эрдмана — тетранитро-транс-дпзмяинкобальтат(Ш) аммония Nll4|Co(NOa)4(NH3)2]. Она представляет собой желтый порошок, состоящий из мелких ромбических кристаллов, и получается при действии нитрита натрия, аммиака и хлорида аммония на хлорид кобальта(П) с последующим окисленном кислородом воздуха. Как указывалось па стр. 165 и сл., комплексы типа [ZA4Ba] принципиально могут существовать в двух формах *— цис-и транс-. Соль Эрдмана является щрапс-формой, это стало известно в 1922 г., когда Рнзенфельд и Клемент (Riesenield, Element) установили, что соль Эрдмана при действии этилендиамина превращается в NH4fC(i|NO;)JNH3)en], а не в NH4[Co(NO2)4en], как можно было бы ожидать в случае соседства обеих NH3-rpynn. Уэллс (Wells, 1936) подтвердил этот вывод рентгенографическим определением структуры, правда, не самой соли Эрдмана, а аналогичной серебряной солц Ag[Co(NO2)4(NH3)a] — тетрагональных кристаллов, образующихся при реакции обмена ее в водном растворе с нитратом серебра; хотя онц и не изоморфны соли Эрдмана, можно предполагать, что в описанных условиях замещения катиона нс происходит изменения конфигурации комплексного аниона. Соединения Na[Co(NO2)4(NHs)2], К [Co(N02)4(NH3)2l и Tl[Co(14Oa)4(NH3)2l оказались изоморфны соли Эрдмана.
Комплексные ионы кобальта(Ш), в которых все шесть координационных валентностей насыщены, по данным Бриптцннгера (Brintzinger, 1935), в водном растворе, как правило, существуют в тех же формах, что и в твердом состоянии; только в исключительных случаях они присоединяют молекулы Н2О или происходит ассоциация в растворе (например, к комплексу [Со он3]з+ присоединяется в растворе, по-видимому, 12*молекул Н2О; комплекс [Co(SO3)2(CN)J6-, очевидно, димереп в растворе). Тем не менее комплексные катионы с координационно насыщенным кобальтом, как показал Бринтцингер (ср. стр. 159), могут присоединять избыточные анионы с образованием двухслойных комплексов. Например:
[Co(NII3)0]'"+ 4SO4 = [[Co(NH3)e](SO4)4].....,
[Co(OH2)(NH3)s]-4-4SO^ = [[Co(OH2)(NH3)5](SOt)4J"
[Co(SO4)(NH3)4]‘~|-4F'=[[Co(SO4)(Nri3)4]F4]'".
Однако таким образом могут присоединяться пе любые анионы, а в соответствии с предположением Бринтцингера и Освальда (см. стр. 301) лишь те, которые в растворе образуют акво-ионы, т. е. особо прочно связывают воду. Б ближайшем окружении этих анионов существует, очевидно, весьма сильное электростатическое поле, хотя в случае катионов Бринтцингер нашел, что акво-иоаы (главным образом гекаа а кв о-и оны) образуются такими металлами, которые вследствие небольшого объема их ионов или относительно большого заряда ()> 2) обладают особо сильным электростатическим нолем.
Соединения, в которых существует двухслойный комплексный иоп кобальта, известны и в кристаллическом состоянии, например:
[NO3]
II [N03][Co(NH3)6][N03] [KO3]
Г [SO4] [SO4]
н Н4 [Co(NH3)6][SO4][Co(NH.,)e] . [SO4] [SO4]
полученные Бепратом (Benrath, 1924). Вероятно, к тому же Классу относятся и описанные Графом (Graf, 1932) соединения кобальтгексааммипоктаборной кислоты, например K[[Co(NH3)e]B80s(OH)l2]‘4H2O, Если вслед за Германсом (Hernans, 1925) пред-
Одпа ив двух форм может оказаться нестабильной.
Восьмая группа периодической системы
329
положить, что многочисленные нолиборные кислоты имеют циклическое строение, то представляется вероятным расположение комплекса кобадьтгексааммина в виде ядра в центре кольца октаборной кислоты.
Остаток азотистой кислоты может Присутствовать во внутренней сфере некоторых из этих комплексов в двух формах. Так, известям два ряда соединений типа [GoNO2(NH3)5]X2. Соединения одного ряда — желтые и значительно превосходят по своей устойчивости соли азотистой кислоты, например по отношению к уксусной кислоте; соединения другого ряда — красные, гораздо менее устойчивы, и поэтому их известно сравнительно нсмвого. Полагают, что в желтых соединениях группа ХО2, как ив желтых органических нитросоединениях, например в нитро эта не С2Н5 — NO2, связана;с металлом (кобальтом) через атом азота, в красных же соединениях ояа, как и в нитритах щелочных металлов М1—О—N = O, связана с Со через кислород. Соединении желтого ряда называют нитро-, а соединения красного — нитрите-соединениями, например:
С1В
Нитропента амминкобалып (Ш)xmpui (коричнево-желтый)
HaN\ '
H3N->Co^-NH3
h3nz \o-n=o
Cla.
Нитритопентааммипкобалып (Ш)хм)рид (бархатисто-красный)
Вернер называет это явление солее ой изомерией. В остальном у рассматриваемых соединений кобальта наблюдаются те же случаи изомерии, что и у соответствующих комплексов хрома.
Среди солей дииитротетрааммипкобальта(1П) [Co(NO2)2(NlT3)4]X(a также солей днвитродиэтплендиамипкобалыа(Ш) [Go(N02)2(in2]X) ^uc-соедипепия окрашены в желто-коричневый цвет, а транс- — в светло-желтый или красно-желтый. Поэтому раньше первые называли флаво-, а вторые — кроцео-солями.
Вернер в 1911 г. именно на примере амминных соединений кобальта впервые обнаружил явление зеркальной изомерии неорганических соединений.
В качестве примеров многоядерных амминов кобалъта(Ш) мо?КИо указать два соединения, которые (наряду с другими соединениями подобного рода) образуются в качестве промежуточных продуктов при окислении растворов солей кобальта(Ц). Под действием кислорода воздуха на аммиачиые растворы нитрата кобальта(II) па Первой стадии образуется соединение [(H3N)5IIiCo—О—О—Coii:i:(NH3)53(NO3)4-2H2O [нитрат декааммин-]1-пероксодико6алъта( III)], кристаллизующееся В виде коричневочерных призм. При окислении кислородом воздуха аммиачных растворов хлорида кобальта(II) сначала образуется смесь темного цвета, состоящая из различных много-ядерных аммино- и пер оке о-соединений, так называемый меланохлорид (дёАп?—чёрный, темный). Характерным компонентом этой смеси является «чистый меланохлорид^— трудиорастворимая фиолетово-черная соль, имеющая формулу
"(Н3Ы)3 ni Со
_ С1г
(ВДЯ
III
NI12—Со (ОН3)
С1
а2
Хлорид трихлороаквогексааммин-11-аминодикобалъта(111)
Первая из пих, коричнево-черная пероксо-соль, при сухом разложении или при действии азотной кислоты образует ярко-зеленую соль, которую следует рассматривать как нитрат декааммин-р.-пероксоко6алъта[111, IV)
[(Н3Х)5Сош —О—О—CoIV (NH3)3](NO3)5.
Это соединение интересно в том отношении, что в нем и в ряде других солей, производных того же комплексного катиона, кобальт существует одновременно в трех-и четырехвалентном состоянии. Существование такого иона предполагал Вернер и экспериментально установил Гле (Gleu, 1938), а позже подтвердил Малатеста (Malatosta, 1942) на основании данных магнитных измерений.
330
Глава 7
Комплексными соединениями кобальта, аналогичными описанному типу четы-рехъядерных комплексных соединений хрома, являются соли додека аммингекса-р-гцд-р ок с отетра к оба л ьта (Ш)
НГО \
Со Co(NH3)4 \НО / з
х6
Соли этого типа окрашены в коричнево-черный цвет. Следует отметить, что существует ряд коричнево-красных солей кобальта, имеющих тот же валовой состав, что и названные выше соли, по вдвое меньший молекулярный вес; опи имеют следующее строение:
ГЯЛ^ /ОН\ /№13] B3N-ZCo4-OH-$Co+-NH, X. HSNZ \ОП/ XNHa
з
(соли гексаамминтри-ц-гидроксодикобальта(П1)]. Наблюдающийся в данном случае вид полимерии называется ядерной полимерией.
Аналитические данные. Кобальт принадлежит к аналитической группе сернистого аммония. Сульфид кобальта, осажденный сернистым аммонием, после фильтрования уже больше не растворяется в разбавленной соляной кислоте по причине, указанной на стр. 316. Поэтому ого вместе с никелем, который ведет себя аналогичным образом, можно легко отделить от остальных элементов группы сернистого аммония при перемешивании сульфидных осадков в разбавленной соляной кислоте. От никеля кобальт можно отделить, если перевести его в груднорастворимую соль — гексанитрокобальтат калия K3[Co(NO3)6] или обработкой раствора обоих сульфидов цианидом щелочного металла, щелочью и бромом или хлором. В этих условиях никель осаждается в виде черной гидроокиси, а кобальт остается в растворе в виде прочного комплексного цианида M|[Co(CN)e]. Оба метода позволяют количественно разделить Со и Ni. Лучшим весовым методом определения кобальта является электролитическое осаждение металла из раствора, содержащего его сульфат, сульфат аммония и пересыщепного аммиаком. Однако кобальт можно также осаждать в виде окисла с последующим восстановлением в токе водорода до металлического кобальта, который является весовой формой.
Перл фосфорной кислоты соединениями кобальта окрашивается в красивый синий цвет как в окислительном, так и в восстановительном пламени. Эта реакция позволяет легко обнаружить присутствие кобальта. Микрохимическое определение кобальта производят либо в виде гексанп-трокобальтата калия K.3[Co(NO3)6), либо в форме его двойной соли с роданидом ртути состава Co(SCN)3-Hg(SCN)3, кристаллизующейся в виде сростков игл великолепного синего цвета.
Реакция с роданидом и ацетоном, описанная аа стр. 321, очень чувствительна. Однако ей мешает присутствие ионов Fe"*, так как появляющаяся в этом случае интенсивная красная окраска перекрывает синюю. Кольтгоф рекомендует для устранения влияния ионов Fe*" добавлять фторид аммония, который связывает железо в комплекс (ср. стр. 302). По данным Дитца fD i t z, Z. anorg. Chem., 225, 73 (1935)), фторид аммония уменьшает чувствительность роданидной реакции на кобальт, поэтому лучше осаждать железо карбонатом кальция.
НИКЕЛЬ (NICCOLUM) Ni
Распространение в природе. В природных условиях никель встречается главным образом в виде соединений с серой, мышьяком и сурьмой, например желтого никелевого колчедана (миллерита, волосистого колче
Восьмая группа периодической системы
331
дана) N1S, красного никелевого колчедана (арсенида никеля, «купферникеля») NiAs и брейтгауптита (антимонида никеля) NiSb, в виде белого никелевого колчедана (хлоаитита) NiAs2, изоморфного шнейзовому кобальту. Кроме того, следует еще указать на герсдорфит (мышьяково-никеле-вый блеск) NiAsS и ульманит (сурьмяно-никелевый блеск) NiSbS.
Для добычи никеля большее значение, чем названные выше собственно никелевые минералы, имеет гарниерит — магниевоникелевый силикат переменного состава, образовавшийся в результате выветривания, а также некоторые виды магнитного колчедана, который часто содержит сравнительно большие количества никеля (в среднем 3%) в виде изоморфной примеси.
Самородный никель в виде сплава с железом довольно часто входит в состав метеоритов.
Исторические сведения. Никель был открыт в 1751 г. Кропштедтом. Название его произошло от «купферникеля» (красного никелевого колчедана NiAs). На языке горняков слово никель в то время считалось бранным. «Купферникелем» тогда павы-вали руду, из которой, несмотря па внешнее сходство с медными рудами, никак не удавалось получить медь. Мнение о том, что «купферникель» является медной рудой, было распространено и среди некоторых химиков даже после открытия никеля до тех пор, пока Бергмап (1775) пе описал точнее свойства никеля и способ его получения. Бергман также первый указал на химическую близость никеля и железа.
Получение. Важнейшими никелевыми рудами являются новокаледонский гарниерит и канадский магнитный колчедан. Последний, помимо никеля, содержит значительное количество меди. Кроме того, исходным материалом для добывания никеля нередко служат так называемые шпейсы, остающиеся после выработки меди и свинца.
При добывании никеля из гарниерита пользуются его особенно сильным сродством к сере. Руду сплавляют с веществами, легко отдающими серу. При этом никель переходит в Ni3S2, в то время как большинство примесей переходят в шлак, образуя силикаты. Частичным обжигом, повторным сплавлением с добавлением кварца и затем продуванием в конвертере «роштейп», обычно содержащий еще большое количество железа, переводят в чистый «никельттейи», теперь уже состоящий главным образом из сульфида никеля. Последний при обжиге превращается в окись иикеля(П) ((1)]. Ее смешивают с порошком древесного угля и, добавив немного воды и муки (в качестве связующего средства), формуют из образовавшейся массы кубики; при их прокаливании образуются сырые рыхлые кубики никеля (2):
NijS^-)-^/гОз—3NiO-p2SO2, (1)
NiO + C=Ni-|-CO. (2)
Магнитные колчеданы, содержащие никель, перерабатывают на никель аналогичным способом; только в этом случае не добавляют серу, а наоборот, обжигом уменьшают ее содержание. Кроме того, ввиду содержания в этих рудах меди при описанном выше способе переработки получается не (сырой) никель, а сплав его с медью. Этот сплав можно разделить па компоненты электролизом, однако обычно предпочитают отделять иикель от меди еще до его восстановления. В Кападе для этого используют главным образом плавку, называемую оксфордским методом. Оп был разработан Томпсоном (Thompson) в Оксфорде и состоит в следующем: в шахтной печи «штейн», содержащий никель и медь, сплавляют с гидросульфатом натрия и коксом. Сульфид натрия, образовавшийся при восстановлении гидросульфата', дает двойное соединение с сульфидом меди(1), содержащимся в «штейне». В верхней зопе печи это соединение переходит в общий плав, а сульфид пик ел я опускается вниз. После охлаждения «головка», обогащенная медью, отделяется от «нижнего плава», содержащего никель. Таким образом, повторным проведением процесса получают продукт, содержащий около 70% никеля и только 1—2% меди. Его размалывают, отмывают от сульфида натрия, подвергают обжигу и, наконец, восстанавливают коксом до чернового никеля, содержащего около 95% Ni и 2—3% Си, Черновой никель перерабатывают на чистый металл электролитическим рафипировапием или карбонильным методом.
Карбонильный метод основан на получении Летучего карбонила никеля Ni(CO)t при действии окиси углерода на металл с последующим термическим разложением
332
Глава /
Карбонила. При этой исходят из губчатого чернового никеля, полученного восстановлением водяным газом окиси никеля, содержащей в небольшом количестве медь. Металл обрабатывают окисью углерода при 50“ й нормальном давлении (метод Мон да — Лашера). Одна к о можно получать карбонил никеля и непосредственно из штейна, содержащего пикель и медь, ec-Jiii реакцию с окисью углерода проводить при высоком давлении (200 ат) и температуре 200—250° (метод «I. G. FarbenIndustrie»). Разложение карбонила пикеля проводят при 200° и обычном давлении. Освобождающуюся при этом окись углерода возвращают в производство. Карбонильный метод позволяет получить никель особенно высокой чистоты (99,9—99,99%). Таким образом осуществляется большая часть мирового производства чистого никеля.
Электролитическое рафинирование используют в о снопе ом при переработке чернового никеля, содержащего йлатипуг так как из анодных шламов легко можно выделить платину и другие металлы этой группы.
Свойства. Никель представляет собой с сребрист о-белый металл, обладающий сильным блеском и удельным весом 8,85—8,90; твердость его 3,8 (по шкале Мооса); температура плавления 1452°. Никель чрезвычайно хиршло полируется, очень тягуч; поддается ковке и сварке; вальцеванием можно приготовить листовой металл; он легко вытягивается в проволоку. Нинель ферромагнитен, но в меньшей степени, чем железо. При 18' его электропроводность составляет 13,8% электропроводности серебра и 14,9% меди. Значительно хуже проводят ток некоторые сплавы Никеля, например константан (см. стр. 333). Теплопроводность никеля равна примерно 15% теплопроводности серебра.
По отношению к воде и воздуху компактный никель при обычной температуре очень устойчив. Однако в тонкораздробленном состоянии он при некоторых условиях обладает пирофорными свойствами. В чистом кислороде никелевая проволока сгорает, разбрызгивая искры. Никелевая жесть подобно стали при нагревании на воздухе обнаруживает цвета побежалости. Разбавленные кислоты действуют на никель менее энергично, чем па железо, однако он легко растворим в разбавленной азотной кислоте; концентрированная азотная кислота его пассивирует.
Никель (как палладий и платина) обычно образует гранецентрированную нуби~ чеекую решетку, где а = 3,5238 А. Однако для никеля, распыленного на катоде в атмосфере водорода, Бредит (Bredig, 1927—1931) обнаружил гексагональную плотнейшую упаковку (а — 2,65; с = 4,32 А), т. е. структуру, аналогичную кобальту. Гексагональный. никель, по данным Ле Клерка и Мишеля (Lo Clerc, Michel, 1939), образуется я в том случае, когда обычный мелкора «дробленный металл в течение продолжлгель-Ного времени находится в атмосфереСО. Поэтому рассматриваемая модификация является стабильной при обычной температуре. При 200° она превращается в кубическую модификацию. Гексагональный никель неферромагнитен.
В электрохимическом ряду напряжений никель занимает место справа от кобальта. Его нормальный потенциал равен —0,25 в.
Нагретый мелкораздробленный никель соединяется с хлором и. бромом, причем ла реакция сопровождается появлением пламени. Оп реагирует также с мышьяком, Сурьмой п фосфором (ср. табл. 31, стр. 264 и с л.). Пикель .содержащий большое Количеств о ф осф о р а, хрупок; о днак о н еб о л ьтп о е с о де рж апие ф осф ора (ок рл о 0,3 %.) п евыша ет способность никеля к отливке и ковко (вероятно, вследствие раскисляющего действия фосфора). В расплавленном состоянии никель легко поглощает также углерод (приблизительно до 6,25%). При затвердевании углерод чаще всего вновь выделяется в виде графита, (При 1315“ никель может содержать в виде твердого раствора 0,5 вес. % углерода, а при комнатной температуре — лишь 9,15%.) Соединение никеля с углеродом в твердом состоянии неустойчиво; тем не монее при термическом разложении СО на мелкораздр облени ом никеле можно получить метастабильное соединение Ni3C, индивидуальность которого была установлена рентгенографически (Вайт, 1928). Особенно энергично реагирует никель с алюминием. При нагревании до 1300° экви-атомных количеств пикеля и алюминия образуется соединение AIJNi, причем реакция
Восьмая, группа, периодической системы
333
сопровождается почти взрывом. (Кроме того, существуют соединения Al2Ni и Al3Ni, разлагающиеся при плавлепии.) Ё кобальте никель обладает неограниченной растворимостью в твердом я жидком состояниях, но не образует спим соединений- Аналогично ведет себя никель и по отношению к марганцу, по крайней мере при более высоких температурах. С хромом он также образует непрерывный ряд твердых растворов, но ни одного соединения. В системе Ni — Fe в твердом состоянии взаимная растворимость практически неограниченна; но образуется FeNij-фаза, обладающая «сверхструк-турой» (ср. рис. 41, стр. 285). Об отношении к другим металлам см. табл. 31 (стр. 264 и сл.) и 32 (стр. 266).
Металлический никель при умеренном нагревании разлагает газообразный аммиак^ на водород н азот. С последним он непосредственно не соединяется. Мелкораздробленный никель в значительных количествах поглощает водород, особенно при повышенной температуре. Но даже при обычной температуре, когда ноны водорода разряжаются па поверхности губчатого пикеля, наблюдается значительное поглощение водорода металлом. Вопрос о возможности синтеза гидрида никеля определенного состава (в весомых количествах) не решен до сих пор. На склонности никеля к окклюдированию водорода и активированию его (путем перехода в атомарное состояние) основана способность Ni служить переносчиком водорода при гидрировании непредельных соединений, т. е. его применение в качестве катализатора гидрирования.
Применение. Уже очень давно никель стали применять для чеканки монет. Так, известны бактрийские монеты, изготовленные до нашей эры, которые содержат наряду с 78% меди 21% никеля. Применение никеля в виде сплавов, например нейзильбера, широко используемого для выделки различных предметов обихода, пе ново. Так, уже в XVIII в. в Европу из Китая ввозили различные предметы, изготовленные из таких сплавов, называвшихся пакфонгами. Под названием нейзильбер (а также аргеитан и альпака) известны сплавы, содержащие около 10 — 20% никеля, 40 — 70% меди и 5—40% цинка; они имеют серебристо-белый цвет, устойчивы к химическим воздействиям и хорошо полируются. В настоящее время предметы домашнего обихода и лабораторные приборы нередко изготовляют из чистого никеля (особенно шпатели, щипцы, тигли и т. и.). Но главным образом никель применяют для гальванических покрытий железнЫх изделий для защиты их от ржавления и придания им лучшего внешнего, вида. Большие количества никеля используют также в современной сталелитейной промышленности, так как добавки никеля к стали сообщают ей особую прочность и вязкость (см. стр. 288). Некоторые никелевые сплавы благодаря их незначительной электропроводности применяют для изготовления сопротивлений. К числу таких сплавов относятся: константан, сплав из 40% Ni и 60% Си, электропроводность которого почти пе зависит от температуры; никелин, состоящий приблизительно из 31% Ni, 56% Си и 13% Zn, также отличается высоким электросопротивлением и слабой зависимостью электропроводности от температуры; манганин (4% Ni, 12% Ми, 84% Си) дает по отношению к меди очень малую тсрмо-э.д.с., и поэтому его используют для изготовления прецизионных сопротивлений. Для обмотки электрических печей часто применяют хромоникелевую (нихромовую) проволоку, состоящую из 60% Ni и 40% Сг. Тонкораздробленный никель и окись никеля широко применяют в качестве катализаторов при гидрогенизации жиров. О никеле Репей см. стр. 757 и сл.
Соли никеля применяют главным образом в ваннах для гальванического никелирования. В некоторых случаях ими пользуются также для окрашивания керамических изделий.
СОЕДИНЕНИЯ НИКЕЛЯ
В соединениях никель преимущественно положительно двухвалентен. Все простые соли являются производными двухвалентного никеля. Кристаллогидраты простых солей никеля и их водные растворы окра-
334
Глава 7
шепы в светло-зеленый цвет. Этот цвет, таким образом, свойствен (гидратированным) ионам никеля Ni". Безводные соли имеют обычно другую окраску.
Почти все соли никеля склонны к образованию продуктов присоединения или координационных соединений типа амминов (к которым, в сущности, относятся и «простые» соли, поскольку они содержат воду) или типа ацидо-соединений. Координационные соединения никеля, однако, менее прочны ио сравнению с соответствующими соединениями кобальта, особенно трехвалентного.
Окислы никеля по своему составу и поведению соответствуют окислам кобальта. Монокпсъ никеля NiO имеет основной характер. Высшие окислы Ni2O3 и NiO2 (известные только в виде гидратов) обладают очень слабо выраженным кислым характером, который сказывается в том, что с сильпо-основпымн окислами (например, ВаО) они образуют двойные окислы (причем одновременно никель стабилизируется в более высоких степенях окисления). Гидратированные высшие окислы разлагаются сильными кислотами с образованием солей никеля(П). В водных растворах щелочей они, как и NiO, нерастворимы.
Кроме высших окислов никеля, извести о несколько комплексных соединений, в которых никель имеет валентности три и четыре. Трехвалентный никель существует (как подтвердили магнитные измерения) в комплексах;
[NiBr3{P(C2Hs)3}2] и [NiCl2((As(CH3)2)2CeH4}J+
(Jensen, 1949; Nyholm, 1950), Оба комплекса содержат электронейтральные лиганды е сильно поляризующимися атомами (Р или As), что, очевидно, благоприятствует проявлению никелем высших степеней окисления. Известно и комплексное соединение трехвал битного пике л я более простого состава — K3[NiFe], а также соответствующий комплекс четырехвалентиого никеля K2[NiF0] (Klemm, 1949). Кроме того, никель проявляет валентность четыре ив соединениях Na[NiIOe]-хН2О и K.[NiIOe]-;r Н2О (Ray, 1946). Достаточно сложно построены комплексные молибдаты четырехвалентного никеля (Нау, 1948). Органические комплексы hhkchh(IV) были получены Фейглем и Хибером (Feigl, 1924; Hieber, 1949).
В исключительных случаях никель может проявлять валентность один. Известен двойной циапнд одновалентного никеля NiCN-2KCN — диамагнитное соединение, которое является, очевидно, двухъядерным К41 Nia(CN)e]. При действии СО оно превращается в К2 [Nri(CN)3(CO)j (Nast, Gehring, 1946). Если считать NO нейтральной частицей в соединениях NiCl(NO), NiBr(NO) и Nil(NO), полученных Хибером и Настом в 1940 г., то пике ль оказывается в них одновалентным. Продукты, которые раньше рассматривали как окись и сульфид никеля(1), по данным Лови и Борпемапа (Levi, Вог-пешапн), не являются индивидуальными соединениями.
При действии металлического калия на К2[Ni(CN)4] в растворе жидкого NH3 Истери Барджесс (Easter, Burgess, 1942) получили соединение K.4[Ni(CN)J, в котором содержится нульвалектный никель. Этот комплекс, вероятно, родствен Ni(CO),,. Действительно, Бергу и Дейтопу (Burg, Dayton, 1949) удалось получить соединения, содержащие одновременно CN" и СО, например К[Ni(CN)(СО)3].
Окислы и гидроокиси. Окись никеля^П) (монокись никеля) NiO образуется при прокаливании гидроокиси пикеля(П) или при термическом разложении карбоната или нитрата никеля; она представляет собой зеленый порошок, практически нерастворимый в воде, но легко растворимый в кислотах. Водород легко восстанавливает NiO до металла. При очень сильном прокаливании NiO переходит в серо-черные правильные октаэдры, обладающие металлическим блеском. При этом NiO утрачивает легкую растворимость в кислотах. В природе окись никеля(Н) встречается в виде фисташково-зеленых кристаллов бунзенита, имеющего удельный вес 0,4. Окись иинеля(П) применяют в керамическом производстве для красок и эмалей; ее используют для окраски стекол в серый цвет, а также в качестве катализатора. Теплота образования NiO составляет 58,6 ккал/моль.
Восьмая, группа периодической системы
335
Гидроокись никеля{11) Ni(O'H)2 выпадает из растворов солей никеля в виде объемистого яблочно-зеленого осадка при добавлении к ним щелочи. При длительном соприкосновении с раствором Ni(OH)2 переходит в кристаллический зеленый порошок. В кислотах, а также в аммиаке и в солях аммония гидроокись никсля(П) растворяется легко. При прокаливании она переходит в окись.
Разложение Ni(OH)3 с образованием окисла происходит, как показал Хюттиг (Huttig-, 1930), уже при 230° (при давлении пара ВОДЫ 10 .и.н рт. ст.); однако при этом выделяется лишь небольшая часть общего количества воды, содержащейся в гидроокиси; полное разложение происходит лишь при сильном прокаливании Ni(OH)2,
NiO кристаллизуется по типу поваренной соли (а = 4,17 A), Ni(OH)2 образует слоистую решетку типа брусита (а = 3,08, с = 4,61 А).
Высшие окислы никеля. При осторожном нагревании на воздухе карбоната или нитрата никеля (до 300°) образуются серые или черные порошки, растворяющиеся в соляной кислоте с выделением хлора, а при растворении их в кислородных кислотах выделяется кислород. Ранее считали, что в данном случае образуется окись никеля(Ш) Ni2O3. Однако состав образующихся продуктов подвержен значительным колебаниям, и содержание в них активного кислорода в большинстве случаев бывает гораздо ниже отвечающего формуле Ni2O3 (Le Blanc, 1926). Этого обстоятельства раньше не замечали, так как получающиеся при прокаливании продукты, которые принимали за Ni2O3, всегда содержат воду. С еще большим содержанием воды, но также и с большим содержанием активного кислорода «окись никеля(Ш)» получается при обработке хлором суспендированной в воде гидроокиси пикеля(П) или при электролитическом окислении гидроокиси пикеля(П) в растворе едкого кали.
Электрод из К12О3, погруженный в 2,8 н. раствор едкого кали имеет, по данным-Форстера (Foerster), потенциал +0,48 в. О диак о, если Nt(OH)2 предварительно электролитически окислить, по окончании окисления впачале обнаруживается более высокий потенциал (+0,6 в). Это явление, по мнению Форстера, связало с тем, что при окислении первоначально частично образуется двуокись никеля NiO3, которая затем постепенно (в течение нескольких суток), отщепляя кислород, переходит в Ni2O3. Так как эти соединения образуют между собой твердые растворы, падение потенциала происходит постепенно, а пе скачком, как это имеет место в других случаях. При добавлении к растворам солей никеля щелочи и хлора или брома тоже вначале образуется гидрат двуокиси никеля.
В кристаллическом состоянии соединение N12O3-2h2O было получено Гофманом (Hoffmann) при сожжении металлического кадия на никелевой пластинке. При осаждении из водных растворов, по данным Отта (Ott, 1933), также можно получить соединение Ni2O3-2H2O. Отт показал, что химически несвязанную воду, содержащуюся в осадке, можпо удалить высушиванием над СаС1^. При дальнейшем обезвоживании происходит разложение этого соединения с потерей кислорода. Поэтому Отт придает ему формулу (IIO)2i\i—О—Ni(OH)2.
По Глемзеру (Glemser), существуют следующие фазы, различающиеся по структуре; NiOi,o7^1,22-а;Н2О, Ni3O4-2H2O, Ni2O3-arH2O и NiO2-zH2O, причем в данном случае речь идет не о перекисях, а о соединениях, в которых никель существует в более высоких степенях окисления.
На образовании гидроокиси никеля(Ш) при анодном окислении гидроокиси нике-ля(П) основано действие аккумулятора Эдисона. Одип нз его электродов изготовлен из высокоактивного железного порошка, а другой — из «окиси никеля([П)» (к которому для лучшей проводимости примешаны никелевые опилки); в качестве электролита служит раствор едкого кали. При разрядке аккумулятора гидрат окиси никеля(Ш) восстанавливается до гидроокиси никеля(П), а железо окисляется в основном в гидроокись железа(П). При зарядке аккумулятора идет обратный процесс;
Разрядка
Fc + Ni2O3 + 3H2O -----—> Fe(OH)2+2Ni(OH)2.
Зарядка
336
Глава 7
Железо-никелевый аккумулятор Эдисона в противоположность обычному свинцовому аккумулятору переносит перегрузки и продолжительное незаряженное состояние. Напряжение на его клеммах при разрядке равно примерно 1,3 в, при зарядке — 1,7 в и выше. В соответствии со значительной разницей между зарядным и разрядным напряжениями коэффициент его полезного действия невысок.
При действии концентрированной уксусной кислоты па холоду па свежеприготовленный гидрат окиси пикеля(Ш) образуется раствор черного цвета, из которого едкий натр вновь осаждает гидрат окиси никеля (III). При добавлении к этому раствору восстановителей он ш медленно окрашивается в зеленый цвет. При продолжительном стоянии или при пагревапии он разлагается. Содержится в этом растворе ацетат пикеля(1П) или лишь коллоидины раствор гидрата окиси никеля(Ш), цока еще пе ясно.
Двойные о кислы-, в которых никель проявляет высшие валентности, можно получать при нагревании NrO с ВаО или SrO в атмосфере кислорода. Таким путем Шмаль (Schmahl) синтезировал соединения Ba3NiaInO5 и Sr2NiJnO6. Аналогичным образом Ландер и Воотеп (Lander, Wooten) получили двойной окисел BaNiIV03 — черный порошок (о!г,е111Г = 6,22), нерастворимый в щелочи; в разбавленной соляной кислоте он растворим с выделением хлора. При нагревании до 730° и давлении кислорода 730 рт. era. происходит разложение BaNiO3 с выделением кислорода и образованием повой фазы. При более сильном пагревапии постепенно происходит дальнейшее выделение кислорода, вплоть до состава Ba2Ni2O5. Соединение Ba2Ni2O5 плавится в атмосфере кислорода при 1200° без разложения.
Сульфиды. Сернистый аммоний осаждает из растворов солей никеля черный сульфид никеля(11) (моносульфид никеля), который вначале выпадает в форме, растворимой в кислотах (a-NiS), а затем при доступе воздуха, как и сульфид кобальта(П), быстро переходит в модификацию, нерастворимую в холодной разбавленной соляной кислоте. При осаждении сернистым аммонием никель частично остается в растворе в коллоидном состоянии (что заметно по коричневой окраске фильтрата). При добавлении уксусной кислоты и кипячении он выпадает в виде хлопьев. Из уксуснокислого раствора при действии сероводорода выпадает кристаллический сульфид никеля ф-KiS), плохо растворимый в разбавленной соляной кислоте.
a-NiS — аморфное вещество, {5-NiS — кристаллизуется по типу NiAs (рис. 43, ср. также стр. 268). Третья модификация — y-NiS (образующая молекулярную решетку, Ni -s—>• S = 2,18 А) встречается в Природе н виде желтого никелевого колчедана', при соответствующих условиях ее можно получить и из раствора. Искусственно полученный y-NiS при соприкосновении с раствором пос тепел по переходит в 6-NiS.
Желтый никелевый колчедан (миллерит, волосистый колчедан) образует гексагональные копьеобразные, лучистые, нередко волосовидные тонкие блестящие кристаллы, окрашенные от латуппо-желтого до коричневато-черно г о цвета (уд. вес 5,3— 5,9). Значительные количества сульфида никеля, как уже было указано, содержатся часто в магнитном колчедане. Мйпорал пентландит (NijlfehSg кристаллизуется по тому Же типу, что и Ctf0Ss (см. стр. 317 ц сл.).
Железно-серый по цвету дисульфид никеля NiS2 получается при сильном прокаливании карбоната никеля с карбонатом калия п серой. Строение его кристаллической решетки аналогично CoS2, FeSz и MnS2 (а = 5,74 А). Сульфид состава Ni3S4 встречается в природе в виде полидимита. По структуре Ni3S4 близок к кобальтовому колчедану (линпеиту) и шпинелям (а = 9,5 А). Из расплава никеля и серы, кроме Ni3S4, Кристаллизуются также (инконгруэнтно плавящиеся) Ni3S3 и Ni8S5.
Известны также двойные сульфиды никеля. Так, при сплавлении окиси 11ИКеля(П) или какого-нибудь ее производного с серой и избытком щелочного карбоната при температурах между краевым и белым калением получаются желтые с металлическим блеском листочки состава K2S'3NiS, Аналогичным образом Бедлуччи (Bellucci) получил из хлорида никеля, серы и окиси барпя соединение BaS-4NiS (темпо-краспые кристаллы).
Арсениды и антимониды. Никель легко соединяется не только с серой, но и с мышьяком и сурьмой. Выше ужо было отмечено, что арсениды NiAs и NiAs3 и антимонид NiSb встречаются в виде минералов. В природе встречаются и смешанные при-
Восьмая, группа периодической системы
337
по своей структуре совпадает
с
Рис. 43. Элементарная ячейка красного никелевого колчедана NiAs, а = 3,61; с = 5,03 А.
с галлы NiAs и NiSb (арит). Состав соединений, образующихся из расплавов, приведен в табл. 31 на стр. 264 и сл.
Строение решетки обычного арсенида никеля NiAs (красного никелевого колчедана) приведено па рис. 43. NiSb, а также (i-NrS, fi-NiSe и NiTe имеют такую же структуру (ер. табл. 33, стр. 268), как NiAs. Диарсенид никеля NiAs2 встречается в двух модификациях: обычно в виде кубического белого никелевого колчедана (хлоаитита) и реже в виде ромбического раммельсбергита. Последний с леллингитом FeAs2 и саффлоритом CoAs2. NiAsS (герсдорфит) и NiSbS (ульманит) кристаллизуются по тину кобальтового блеска CoAsS (ср. стр, 317).
Карбонил никеля, тетракарбонил никеля Ni(CO)4, открытий в 1888 г. Лангером (Langer) (ср. стр. 375), образуется при пропускании окиси углерода прн 50—100° над мел-кориздробленным никелем (полученным восстановлением окиси никеля водородом при 4.00°). Это бесцветная жидкость, кипящая при 43°, затвердевающая при —25° и имеющая прн 20° удельный вес 1,310. Критическая том-п,ератураМ1(СО)4лежитоколо200°, а критическое давление равно примерно 30 атм. Плотность пара Ni(CO)4 (6,01 по отношению к воздуху) соответствует молекулярному весу 173 (вычисл. 170,7). Пары карбонила никеля ядовиты.
О применении карбонила никеля в технике для получения чистого никеля см. стр. 331 и сл. (ср. также стр. 21).
Аэ
М
Наиболее удобным лабораторным способом получения карбопила никеля является действие окиси углерода на легко доступный дитиофен ила мидокарбопат пииеля(П) Ni(S-SC-NHC6H5)2 [Н i о b е г, Z. anorg. Chem., 269, 28 (1952)]. Хибер показал, что в основе этого метода лежит окисление — восстановление соединений никеля и некоторых органических тиокиедот под действием СО ио схеме: 2N111 -*• NilV (комплекс) + Ni°(CO)4. Если не существует предпосылок для образования соединения Ni(lV), то образуется только карбонил (ср. стр. 269 и сл. и стр. 342).
В противоположность остальным соединениям пикеля— парамагнитным, тетракарбопил сильно диамагнитен. Он обнаруживает также значительную дисперсию. При хранении в запаянных стеклянных трубках при обычной температуре он вполне устойчив, мо на воздухе постепенно окисляется. На воздухе тетракарбонил никеля горит ярко светящимся пламенем. Смесь его с воздухом взрывчата. В воде он нерастворим; на него нс действуют разбавленные кислоты и щелочи. Ni(CO)4 растворим в эфире, бензоле и хлороформе. При пропускании тетракарбонила никеля через трубку, нагретую до 180—200°, он разлагается на компоненты, образуя блестящее никелевое зеркало.'Сильные окислители, например концентрированная азотная кислота или хлор, разлагают тетракарбонил никеля с образованием солей никеля и двуокиси углерода или иногда хлорокиси углерода. При соприкосновении Ni(C0)4 с концентрированной серной кислотой происходит взрыв.
Жидкий карбонил никеля реагирует с NO с образованием нитроеилъного соединения, которое, одпако, таким путем не может быть получено в частом состоянии (Mond, 1922; Frazer, 1936). В присутствии влаги Ni(CO)4 образует с NO темно-синий гцдроксопитрозил никеля NiOH(NO).
22 Г. Реми
338 Глава 7
Соли никеля
Хлорид пикеля, хлорид никеля(П), хлористый никель NiCl2 получается в безводном состоянии при сгорании никеля в токе хлора или при нагревании его гидрата или аммиаката. О и образует блестящие, золотистожелтые мягкие (иа ощупь похожие на тальк), легковозгопягощиеся кристаллические чешуйки удельного веса2,56 (структуру решетки см. стр. 268). При умеренном прокаливании на воздухе NiCl2 переходит в окись пике-ля(П) (монокись никеля). Он легко растворим в воде и в спирте (в 100 з воды при 20° растворяется 64,0 г, а при 60°—81,2 г NiCl2. Из светло-зеленого водного раствора он кристаллизуется с водой обычно в виде гекса-гидрата NiCl2 ()H2O—травянисто-зеленых зернистых кристаллов в форме моноклинных нризм, изоморфных кристаллам гексагидрата хлорида кобальта. При выдерживании пад концентрированной серной кислотой гексагидрат постепенно переходит в дигидрат. Известен также моногидрат.
Со щелочными и другийи хлоридами хлорид никеля образует да ойпые соли (хлор ок икелаты), например N Н 4С1 N iC 12 6Н2О — зеленые ромбические призмы; CsCl-JNiGl2 — мелкие желтые кристаллы.
Бромид никеля NiBr3 и иодид пикеля Nil2 вполне аналогичны хлориду. Они кристаллизуются из водных растворов тоже обычно в виде гексагидратов. Для бромида, кроме того, известим его три- и нонагидраты. Существуют также двойные бромида.
Фторид пикеля NiF2. Безводную соль легче всего получать термическим разложением двойного фторида никеля и аммония. NiF2 образует длинные призматические кристаллы, в сухом состоянии имеющие окраску от светло-коричневой до зеленой; они очень слабо растворимы в воде и нерастворимы в спирте и в эфире. При упаривании раствора гидроокиси пнкеля(П) в водной плавиковой кислоте образуется тригидрат i\'iF2-3H2O в виде зернистых бледно-зеленых кристаллов. Двойные соля фторида пикеля (фтороникелаты) были получены сплавленном его с соответствующими солями или смешиванием растворов NiF2 с растворами этих солей; примером может служить соль KF-NiF3'1Т2О, образующая осадок, окрашенный в цвет серы.
При действии К2 на смесь NiCi2 и КС1 получаются соединения K3[A’iIIIFfl] hK2[N11vF6]. К2|NiFs] является пока единственной фторидной солью, которая, судя по магнитным свойствам, является «комплексом внедрения». В случао всех остальных фтористых соней речь идет о «нормальных комплексах» (Klemm, 1953).
Основные галогениды иикеля. При неполном осаждении из раствора хлористого пикеля едким натром образуется основ пай хлорид никеля в среднем состава М1С12-• 3Ni(OH)2, однако состав изменяется в довольно широких пределах (при соотношении СI : ОН от 1 : 1,9 до 1 : 4,3). Он образует «простую» слоистую решетку, отличшо-щуюся от структуры гидроокиси никеля том, что в пей ионы ОН- частично замеЩепы па С1- (причем в решетке опи распределены совершенно беспорядочно) и одновременно ячейка значительно расширяется — вдоль оси е (а = 3,18, с = 5,48 А). Такое увеличение параметра объясняется тем, что между структурами гидроокиси и основной соли не существует ненрерывяого перехода. Основной бромид никеля имеет ту же структуру и средний состав, что и основной хлорид. Однако в случае бромида состав варьирует в значительно более узком интервале (Вг : ОН = 1 : 2,8—1 : 3,3). Существует еще одни основной хлорид никеля Ni(OII)Cl постоянного (или почти постоянного) состава. Он образуется при неполном осаждении едким натром из концентрированного раствора хлористого никеля при нагревании. Ni(ОН)С1 также образует «простую слоистую решетку». Она является производной от структуры NiCl2: половина ионов С1- замощена па ОН-, вероятно, так, как это имеет место в решетке Cd(OII)Cl (см. рис. 50, стр. -580). Тогда псе ионы хлора оказываются расположенными на одной стороне слоя, состоящего из ионов никеля, а все гидроксильные ноны — на другой (1'citkneciit, 193(1).
Цпапнд никеля и двойные цианиды. Прн добавлении к растворам никелевых солей Иопов CN' выпадает цианид никеля ъ виде яблони о-зелен ого осадка, содержащего воду. При нагревании до 180—200° оп переходит в желто-коричневую безводную соль Ni(CN)s. Свежеосажденный цианид никеля растворяется в избытке щелочных
Восьмая, группа периодической системы
339
цианидов с золотисто-желтым окрашиванием. Из получоппого раствора при упаривании выделяются двойные соли (цианопик елаты), например Na2(Ni(CN)4J-ЗН2О, в виде длинных желтых шестигранных призматических кристаллов, теряющих воду при умеренном нагревании. Сильные кислоты разлагают двойные цианиды, причем происходит осаждение Ni(CN)2.
При действии на растворы щелочных цианоникелатов водорода в момент выделения они окрашиваются в кропав о-красный прет. Как покапал в 1914 г. Веллуччи (Bellucci), эта реакция основала на восстановлении никеля До одновалентного состояния. Веллуччи удалось изолировать и проанализировать красное соединение KsNi(CN)3. Соединение диамагггшпо не только в кристаллическом состоянии, но и в растворе. Уже это обстоятельство позволяет заключить, что в данном случае речь идет о двухъядерном комплексном соединении. Этот вывод был подтвержден Настом (Nast, 1952) при рентгенографическом, определении структуры. Было показано, что рассматрггваемый комплекс представляет собой тетрациано-Ди-р-цианодиникелат(1) калия:
K^fCNh-Ni^ ^Ni(CN)2-
Роданид пике.тя и двойные родаппды. При растворении-карбоната никеля в водном растворе р о дап ист оводородной кислоты или при реакции между сульфатом никеля и роданидом бария образуется роданид никеля Ni(SCN)2 (обычно содержащий некоторое количество воды) в виде желтовато-коричневого осадка, довольно легко растворимого в воде с появлением зеленой окраски. При упаривании растворов, содержащих щелочные и некоторые другие роданиды, можно получить хорошо кристаллизующиеся двойные роданиды (родан оникс латы), па прим ер Na3[Ni(SCN)d SH2O- светло-зеленые кристаллы и K4[Ni(SCN)e] - 4Н2О — синие кристаллы; эти соли можно без разложения перекристаллизовать из спирта, а некоторые и из воды.
Нитрат никеля кристаллизуется из водных растворов обычно в виде гексагидрата Ni(NO3)3-6H2O, образующего зеленые, цвета смарагда, моноклинные кристаллы, изоморфные соответствующей соли кобальта. Выше 54,0° кристаллизуется тстрагидрат, а выше 85,4°— дигидрат; ниже— 3° устойчивой твердой фазой является нонагндрат (Sieverts, 1934). Нитрат никеля применяют для окраски керамических изделий в коричневый цвет.
Из основных нитратов никеля особый интерес в структурном отношении представляет соль состава Ni(NO3)3- Ni(OH)2- 6Н2О. В отличие от большинства основных солей никеля, обладающих слоистой структурой, в решетке этого нитрата, по данным Файткпехта, по-видимому, существуют комплексные ионы соответственно формуле [Ni(OII2)6][Ni(OH)a(N03)J.
Пнтрпт никеля и двойные ввтритьг. При действии на сульфат никеля нитрита бария и после упаривания раствора получается нитрит никеля Ni(NO2)2 в видекраено-желтых кристаллов, устойчивых на воздухе. Нитрит никеля растворим в воде с образованием зеленого раствора. Из растворов, содержащих нитриты щелочных или щелочноземельных металлов, кристаллизуются двойные нитриты типа (гекса-
нитр оникса аты), например устойчивый па воздухе никель нитрит (гексаннтропикелат) кал ия К4 [ N i (N О 2) 8 ] — гс орпчн в в о-кр а иные окта э дры, не ра ств оримые в с пи рт е; их можн о перекристаллизовать из воды-
Сульфат никеля получается при растворении металлического никеля, его окиси или карбоната в разбавленной серной кислоте. В технике его получают следующим образом: пикель растворяют в разбавленной серной кислоте, содержащей некоторое количество азотной кислоты; раствор нейтрализуют карбонатом никеля и упаривают. При охлаждении из раствора выделяется гексагидрат NiSO4-6M2O; он существует в виде двух модификаций: в интервале температур 31,5—53,3° образуются синевато-зеленые тетрагональные кристаллы, а выше 53,3°— зеленые моноклинные. Последние изоморфны кристаллам гексагидрата сульфата магния. В сухом воздухе они выветриваются, а во влажном — не изменяются. Из водных
22*
340
Глава 7
растворов при обычной температуре кристаллизуется смарагдово-зеленый, ромбический гептагидрат — никелевый купорос NiSO4-7H2O.
Никелевый купорос иногда встречается в природе в виде моренозита. В противоположность железному и кобальтовому купоросам он кристаллизуется ромбически; кристаллы имеют ту же форму, что и кристаллы горькой соли — сульфата магния; опи образуют друг с другом непрерывный ряд твердых растворов. В ромбическом купоросе никель может замещаться Железом только в пределах до 21% , в моноклинном возможно замещение половины атомов железа.
Из растворов, содержащих, помимо сульфата никеля, сульфаты щелочных металлов или сульфат аммония, кристаллизуются двойные соли. Среди них известны соли главным образом состава M|SO4-NiSO4-6H2O, изоморфные двойным солям соответствующего состава железа, кобальта, магния и других двухвалентных металлов. Сине-зеленый аммониевони-келевый сульфат (NH4)2SO4-NiSO4-6H2O применяют при гальваническом никелировании.
Карбонат никеля и двойные карбонаты. При добавлении к растворам никелевых солей щелочных бикарбонатов выпадает бледно-зеленый мелкокристаллический осадок карбоната никеля NiCO’j-6H2O, содержащего кристаллизационную воду. При осаждении никелевых солей нейтральными карбонатами щелочных металлов образуется основной карбонат никеля.
Если осаждение ведут бикарбонатом аммопия, то наряду с NiCO3-6HsO образуется двойная соль (NH4)HCO3-NiCO3-4II2O, а при известных условиях—только эта соль в виде мелких яблочно-зеленых кристаллов. При осаждении в особых условиях, например При нагревании до 150° водного раствора хлорида никеля с карбонатом кальция в запаянной трубке, удается получить карбонат никеля в безводном состоянии. Кроме приведенной выше двойной аммониевой соли, известны и другие двойные карбонаты никеля, например K2CO3-NiCO3-4I'I2O (ябл очи о-зеленые иглы) и Na2C03'NiC03.10IJ30 (микроскопические мелкие ромбоэдры травянистого Цвета).
Карбонат никеля применяют для окрашивания керамических изделий, а также оя является исходным материалом для получения других соединений пик ел я.
Оксалат никеля получается при растворепии гидроокиси иди карбоната никеля в водном растворе щавелевой кислоты (металлический никель в щавелевой кислоте не растворяется). Оксалат осаждается в виде золеновато-бслых хлопьев также при добавлении щавелевой кислоты к растворам солей никели. После высушивания при 100’ это соединение имеет состав NiC2O4-2H3O. В воде оксалат никеля практически нерастворим, но легко растворяется в разбавленных кислотах или в водном растворе аммиака (с которым он образует комплексную соль). В кипящем растворе оксалата калия оксалат никеля слегка растворим; при охлаждении из таких растворов кристаллизуется светло-зеленая труднорастворимая двойная соль состава K2C2O4-NiC2Ot-fiH2O.
Ацетат инке ля Ni(CH3C00)2-4H20 при упаривании па холоду выделяется в виде кристаллов яблочно-зеленого цвета из раствора гидроокиси никеля в уксусной кислоте. В воде ацетат растворяется легко (1г 6), но нерастворим в абсолютном спирте. Его водный раствор имеет сладкий вкус и при пагреванпи быстро гидролизуется с выделением гидроокиси никеля.
Фосфаты никели. Из довольно значительного числа соединений, образуемых никелем с кислотами фосфора, в качество примера следует указать ортофосфат никеля.. При добавлении к растворам солей ннкеля вторичного фосфата натрия выпадает в виде бледного яблоч по-зеленого, нерастворимого в воде, по растворимого в кислотах хлопьевидного осадка, который в воздушно-сухом состоянии имеет примерный состав Ni3(PO4)2-7H2O. При осаждения фосфатом аммония выпадает соединение (NH4)NiPO4, первоначально также в виде хлопьевидного осадка, который при соприкосновения с маточным раствором вскоре переходит в кристаллическое состояние.
Силикаты никеля. При нагревании NiO с SrO2 образуется ортосиликат никеля Ni2[SiO4], кристаллизующийся по типу оливина (ср. т. Г, стр. 542, рис. 95). В Природе
Восьмая группа периодической системы,
341
л виде мелких зеленых кристаллических зерен или листочков встречается конарит — Nr3Si3OB-2H3O—гидрат силиката никеля. Землистая разновидность его называется рёттизитом (по названию местности). Гораздо чаще, чем зги чистые силикаты пикеля (которые залегают лишь в небольших количествах), встречается гарниерит, образовав^ шийся при выветривании олпвиновых горных пород, содержащих никель. Он окрашен от яблочно-зеленого до смарагдового пвета и является гидратом викелево-магниевого силиката, с сильно меняющимся содержанием никеля. В очень больших количествах гарниерит находится в Нумее в Новой Каледонии (поэтому оп называется также нумеаи-том); он разрабатывается та кг, а также в Орегоне в качестве никелевой руды.
Аммиакаты. Большинство солей никеля способно присоединять аммиак при пропускании его в газообразном состоянии или при добавлении к растворам. Так. из хлорида никеля и аммиака образуется гексааммиакат хлорида никеля NiCl3‘6NH3 в виде почты белого порошка со слегка фиолетовым оттенком, растворимого без разложения в воде, содержащей аммиак. При пропускании аммиака пад безводным сульфатом никеля образуется NiSO4-6NH3 в виде бледно-фиолетового порошка. Из раствора никелевого купороса в концентрированном водном аммиаке кристаллизуется NiSO4-4KH3-2H2O в виде темно-синих прямоугольных кристаллических столбиков. Аналогично получается и Ni(N03)3-4NH3'2H20, образующий большие сапфирово-синие кристаллы.
Нессмпеппо, аммиак в этих комплексах связан таким же образом, как и в соответствующих соединениях кобальта, и, следовательно, в указанных аммиакатах существуют пошл [Ni(i\H3)6P-r и [Ni(OI-I2)2(NH3)4]B4. Соединения, являющиеся производными этих и других иопов аммиакатов никеля, легко роста гримы в воде. Образованием этих Комплексов объясняется растворимость в водном растворе аммиака многих соединений пикеля, нерастворимых в чистой воде, например ого гидроокиси и фосфата. Растворы никелевых солей, содержащие аммиак, обычно окрашены в синий цвет, подобный аммиачным растворам солей меди(И). Это обстоятельство приводило к тому, Что прежде многие пикеиевые руды, например красный никелевый колчедан, принимали за медные не только горняки, но даже некоторые химики (ср. стр. 331).
Кроме приведенных типов аммиакатов [ Ni(NH3)6]X2 и [Ni(OH2)2(NH3)4]X2, в которых никель имеет координационное число 6, известно много других аммиакатов, в которых это (максимальное) координационное число не достигается. Существует также много соединений типа аммиакатов никеля, которые содержат вместо аммиака другие азотсодержащие молекулы (анилин, пириднп, хинолин, этилендиамин и т. д.)
По аналогии с солями кобальта(II) некоторые соединения никеля, содержащие такие нейтральные молекулы, могут присоединять еще до 4 молекул (или до 2 в случае бидентатпых лигандов) без вытеснения кислотных остатков во внешнюю сферу. Соединения типа [NiX2AmJ, однако, неустойчивы и склонны к диспропорционированию, так, Хибер (Hieber) обнаружил, что соединения [NrBraen3j и [Ni(SCN)2ea2] разлагаются в водном растворе по уравнению
3(NiX3en2l + f>H;.O=2[Ni еп3]Х3+ [N)(0H,JelX2..
Ио существует и целый ряд соединений состава NiX2-4Am, в которых никель имеет только координационное число 4, т. е. кислотные остатки в пих связаны ионо-генно в соответствии с формулой |NiAiii4]X2. В первую очередь это относится к соединениям со слабо поляризующимися кислотными иопамп.
Никель очень склонен также к образованию внутрикомплексных солей. К ним относятся соли, в которых атом металла, заместившего водород, например никеля, одновременно связан координационной связью с другим кислотным остатком. (Об образовании хелатных соединений см. т. I, стр. 325.) Впутрикомплекспые соли отличаются часто исключительно низкой растворимостью. Поэтому в последнее время опи приобретают все более важное значение в аналитической химии. Одним из наиболее известных представителей этого класса комплексных соединений является никелъдимет алели оке им, широко применяющийся для аналитического определения никеля.
342
Гласа 7
Реакция ионов никеля с д нм етилгл и оксимом (диацетил диоксимом) сопровождается образованием ярко-красного никельдиметилглиоксима и проходит по уравнению
о он
t !
.. ch3-c=noh ch3-c=n: zn=c-ch3
N1 +2 I =*= I >№< [ H-2jr
CH,-C=NOH СНз— XN=C—СНз
I I
Иг'.'иетилглполсши jjq q
В полученном таким образом впутрикомплекспом соединении никель имеет координационное число 4 (ио мнению Пфейфф ера, в нем днметмлглиоксим существует наполовину в .т м и tt ок си-форме). Око осаждается только из аммиачных, нейтральных или слабокислых (уксуснокислых) растворов, так как обладает основным Характером И образует соли с сильными или средней силы кислотами, например если представить никельдиметилглиокспм формулой
| DITNiDH]: [DHNiDH] 4-2НС1 = [DHaNiDII3]Cl2 (темно-синие кристаллы).
Прн других условиях можно получить иные типы соединений с диметил глиоксимом, например [DH2NiCl2] (желто-зеленое),[DNi(NH3)2] и [DNi(OH2)3] (оба карминово-красные). В то время как никель в этих соединениях всегда имеет координационное число 4, кобальт даже в двухвалентном состоянии во всех стабильных соединениях с днмотняглиоксимом координационно шестивалентен. Таким образом Тило (Thilo, 1931) объяснил отсутствие у кобальта соединения, соответствующего никельдиметил глиоксиму [DHN1DH], а также то, что кобальт не осаждается диметил гл и оксим ом.
К внутрикатплексным, солим относится, вероятно, п соединение Ni(S-CeIL‘NH3)2, полученное Хибером в 1952 г. при замещении солей никеля о-амипотиофеяолом в аммиачном растворе. Оно построено, по-видимому, ив бесконечного числа групп Ni(S-C6H4- NH,)2, связанных между собой так, как показано в структурной формуле I. Это соединенно легко окисляется до темно-сипего кристаллического продукта, содержащего четмрр^валеитный никель. На основании исследования состава и свойств этому соединению приписана формула II. При обменной реакций хл opin ст ого никеля в спиртовом растворе с дитиобепзойной кислотой CeH5:CS(SH), растворенной в эфире, Хибер получил синий днтиобенаоат ннкеля(Н) — Ni(CeH5- CS2)Z. Эта соль тоже легко окисляется в темно-фиолетовое комплексное соединение никеля(1¥), строение которого соответствует формуле III. Криоскопическим методом (в бензольном растворе) Хибер'обнаружил, что рассматриваемый комплекс димерен.
Тшрадиишобгнзоато-ди-ц-тиоИаникелъ №)
Восьмая группа периодической системы
343
Аналитические данные. В систематическом ходе анализа никель осаждается вместе с кобальтом сернистым аммонием в виде черного сульфида, который после «старения» уже не растворяется в разбавленной соляной кислоте. От кобальта никель отделяют осаждением щелочью и бромом или хлором из раствора комплексных цианидов. Отделение от кобальта можно производить, пользуясь также способностью Ni осаждаться в виде нерастворимой в воде внутрикомплексной соли с диметилглиокси-мом, введенным Чугаевым в качестве реактива на никель. Кобальт не образует с диметил глиоксимом осадка. Осаждение диметилглиоксимом применимо для открытия никеля и в микроанализе, а также и для количественного определения.
Несмотря на то что кобальт не образует осадна с диметилглиоксимом, большой избыток его в растворе снижает чувствительность реакции на никель, так как реактив расходуется на образование растворимого комплексного соединения кобальта с диметил глиоксим ом. Кроме того, в присутствии растворимого комплекса кобальта повышается растворимость пикельдиметилглиоксима. Трохвалснтный кобальт мешает этой реакции значительно меньше, чем двухвалентный. Поэтому перед добавлением дометил глиоксима в аммиачный раствор рекомендуется окислять кобальт перекисью водорода. В этих условиях возможно определение никеля реактивом Чугаева в присутствий 200-кратного избытка кобальта. Двухвалентное железо образует с дпметил-глноксимом тоже красную, но легкорастворимую комплексную соль. Палладий и платина образуют нерастворимые желтые комплексы, соответствующие по составу я строению соединению никеля.
Для количественного определения никеля, помимо реактива Чугаева, часто пользуются также реактивом Гроссмана (см. далее). Очень хоро-пгие результаты получаются и при электролитическом осаждении никеля из аммиачного раствора. Наконец, Ni можно осаждать при действии едкого кали и бромной воды в виде коричнево-черной «гидроокиси пикеля(Ш)» и затем после прокаливания взвешивать в виде монокиси.
Реакция никеля с реактивом Грессмана также основана на образовании внутри-компленспой соли. Этот реактив имеет состав (C2H6N4O)2-H,SO4 (дициандиамидин-сульфат). С солями пикеля оп реагирует при добавлении раствора едиогр кали, образуя желтый осадок состава
HaN—С-О О—С—NHa
II \ / И
N Nr N
[ / Ч I
HaN—C=N N=C—NHt
НН
Б. ПЛАТИНОВЫЕ МЕТАЛЛЫ
Общие сведения. Платиновые металлы характеризуются своей устойчивостью по отношению к кислотам, тугоплавкостью, а некоторые и большой твердостью. Кроме того, они обладают исключительными каталитическими свойствами.
Соединения рутения и осмия во многом напоминают соответствующие соединения железа, находящегося в периодической системе над этими элементами, а также марганца, расположенного слева от железа. Родий и иридий обнаруживают некоторое сходство с кобальтом; в частности, они, как и кобальт, легко образуют комплексные соединения типа [МшАтв)Х3. Палладий обнаруживает свойства, переходные к серебру,
344
Глава 7
а платина — к золоту, подобно тому как никель является элементом, переходным к меди. Тенденция к проявлению высших валентностей убывает здесь по направлению слева направо, как в ряду более легких, так и в ряду тяжелых платиновых металлов (так же, как и в группе железа). Рутении в общем чаще образует соединения с валентностью четыре, родий —три, апалладпн —два. Осмий имеет ярко выраженную склонность к образованию четырехоки си, в которой он является восьмивалеитпым. В шестивалентном состоянии он тоже значительно устойчивее, чем рутений. Иридий бывает преимущественно трех- и четырехвалентным, а платина — главным образом двух- и четырехвалентной.
Распространение в природе. В природных условиях платиновые металлы встречаются почти всегда вместе и в основном в самородном состоянии, иногда в виде сплавов между собой. В количественном отношении в рудных месторождениях платиновых металлов платина обычно значительно преобладает пад остальными металлами этой группы. Кроме платиновых металлов — спутников платины,— платиновые руды обычно содержат медь, золото и в первую очередь железо.
Примерный состав платиновых руд (по Пехарду)
pt 1Г Rh Pd Осми-рпдий Си Аи Fe Песок
Платиновая 77,5 1,45 2,8 0,85 2,35 2,15 — 9,6 1,0
руда из СССР, % Колумбийская 80,0 1,55 2,50 1,0 1,40 0,65 1,5 7,20 4,35
платиновая руда, %
Нередко платиновые руды сопровождают также титап — железо и хромистый железняк.
Исторические сведения. Первое достоверное упоминание о платине имеется у Антонио де Уллоа, который и 1748 г. написал отчет о французской экспедиции к западному побережью Южной Америки. В этом отчете оп упоминает, что в Колумбии было найдено золото (в смеси с другими металлами); вследствие трудности разработки этих пород золото, однако, извлечь но удалось. Это упоминание относится к породам, содержащим платину. В настоящее время Колумбия является одним из главных источников платины. Вскоре после появления отчета Уллоа платина была описана Уотсоном как новый металл (1750 г.). Ее название является уменьшительным от plata (по-испански — серебро). Позднее возникли сомнения, является ли платина новым элементом, по в 1774 г. Блондо разрешил этот вопрос положительно. После открытия на Урале в начале XIX в. обширных платиновых месторождений были описаны и спутники платины — палладий, родий, иридий и осмий (в короткий промежуток времени 1803—1804 гг.). Рутений был открыт лишь в 1845 г.
Получение. Для извлечения платины и ее спутников из платиновых руд применяют различные методы, которые отчасти сохраняются в тайне. В основном добыча и переработка платппы сводится к следующему: сначала платиновую руду освобождают отмучиванием от песков, в которых она залегает. После возможно тонкого размалывания ее обрабатывают царской водкой, при этом остается нерастворимый «осмиридий», состоя-
Восьмая группа периодической системы. 345
гций главным образом из сплава осмия и иридия. Затем добавлением известкового молока осаждают из раствора железо, медв, иридий, родий и часть палладтгя. Раствор, содержащий после этого главным образом платину п некоторое количество палладия, упаривают досуха и остаток прокаливают; платина при этом превращается в губчатый металл. Его промывают водой, соляной кислотой и затем прессуют при высокой температуре. Однако полученная таким образом платина еще содержит примеси, и поэтому се подвергают ряду операций очистки. В осадке, получившемся при осаждении известковым молоком, содержатся небольшие количества платины. Для выделения их осадок обрабатывают разбавленной серной кислотой и осаждают платину из раствора в виде хлороплатината аммония (NH4)2 [PtClJ («платиновый нашатырь»).
Все большие количества платины и палладия получают сейчас из анодных шламов электролитического рафинирования меди и других металлов (ср. стр. 332и 398).
Применение. Благодаря своим исключительным свойствам, в первую очоредв химической устойчивости, прочности, тугоплавкости и тем не ме-пее способности к механической обработке, благодаря прекрасным каталитическим свойствам — платина находит очепв широкое применение. Она идет на выделку тиглей, чашек, лодочек, шпателей и др. инструментов и химической аппаратуры; ее применяют как материал для производства электродов, проволоки для сопротивлении и обмотки электрических печей. Из нее делают также хирургические инструменты. Иногда платинируют предметы для защиты их от коррозии (например, иглы громоотводов). Из платины изготовляют различные украшения, особенно оправы для драгоценных камней. Очень значительное количество платины, главным образом в виде платинированного асбеста, применяют в: качестве катализаторов, в первую очередь на сернокислотных заводах. В технологии, однако, в настоящее время платину в большинстве случаев вытесняют смешанные катализаторы (см. стр. 756 и сл.), обладающие таким же каталитическим действием, но значительно менее чувствительные к отравлению. В лабораторной практике ее применяют по-прежнему часто. Соединения платины используют в фотографии (платинотипия), рентгенотехнике (экраны, покрытые цианоплатинатом бария), а также для украшения стеклянных и фарфоровых изделий.
Иридий часто сплавляют с платиной, чтобы повысить ее твердость; это необходимо при изготовлении платиновых физических и хирургических инструментов. Сплавы платины с иридием очень широко применяют в электрических контактах. Эталон метра сделан из сплава 90% платины с 10% иридия. Иридий применяют также для окраски фарфора в черный цвет.
Палладий благодаря красивому белому цвету, напоминающему серебро, применяют для выделки украшений *. Он обладает по сравнению с серебром тем преимуществом, что не чернеет при действии сероводорода. Палладии используют также вместо золота при пломбировании зубов. Им пользуются для окраски фарфора в черный цвет. В научных лабораториях палладий применяют для различных целей в виде палладиевой черни, т. е. в очень мелко раздробленном состоянии; лучше использовать
* При выделке украшений чаще, чем чистый палладий, используют сплавы серебра с палладием. Достаточно лишь небольших количеств Pd (17%), чтобы в значи-те.дьпой степени предохранить серебро от почернения при действии IPS .при 30%-пом содержании. Pd достигается полная защита.
346
Глава. 7
его в виде коллоидного палладия; довольно часто он служит катализатором процессов гидрирования (см. стр. 363).
РвдиИ1 преимущественно в виде сплава из 90% Pt и 10%Н1г, применяют для изготовления термоэлементов. Платииа-платипородиевые термоэлементы имеют по сравнению с применявшимися раньше платина-платиноиридиевыми термоэлементами то преимущество, что они более прочны, так как родий монес летуч, чем иридий *. Для измерения высоких температур (1600 — 2000°) используют термоэлементы, один из сплавов которых состоит из иридия, другой — из сплава 40% иридия й 60% родня. В возрастающих масштабах сплавы платины и родия применяют в качестве катализаторов окислеттня аммиака. Из сплавов родия изготовляют части астрономических инструментов. Тигли ив родия можно использовать nj»n очень высоких температурах (1500° и выше), ня, да платила становится совсем мягкой и начинает заметно испаряться; для аналитических целен родиевые тигли не применимы, так как вследствие поверхностного окисления опн не сохраняют постоянного веса. (Три производстве украшений гальванический покрытия из родия находят все более широкое применение вместо серебрения, та,; как родии, почти совершенно не отличаясь от серебра по цвету, не измениется и i,t;h£'[H<! от пего на воздухе, содержащем сероводород. О применении коллоидною родия при гидрировании см. стр. 355.
Оемнв ещи не так давно попользовали как материал для изготовления нитей в пампах накаливания. Важнейшей областью его применения в последнее время является производство наконечников для перьев авторучек; одна к о теперь для этой цели все больше используют рений (ср. стр. 253). Иногда осмий применяют в качестве катализатора, например в хлоратной пипетке Гофмана (ср. стр. 353). О применении четмрсхокиси осмия в технике микроскопии см. стр. 352,
Рутений отчасти используют как составную часть сплавов при выделке украшений, а также в некоторых случаях в качестве катализатора. Рутениевую красную краску, получающуюся при обработке трихлорида рутения воДиым раствором аммиака, применяют в гистологии для окраски сухожилий на препаратах. С ее помощью можно отличать искусственные волокнистые материалы от естественных.
ДИАДА I: РУТЕНИЙ И ОСМИЙ
Общие сведения. Рутений и осмий — твердые, хрупкие и чрезвычайно тугоплавкие металлы. Они очень устойчивы по отношению к кислотам, по сравнительно легко соединяются с кислородом воздуха. Для рутения и осмия характерна способность проявлять валентность восемь и образовывать летучие четырехокиси RuO4 и OsO4. Кроме того, эти два элемента могут проявлять особенно большое число различных валентностей (см, сводную таблицу в начале этой главы). Во многих соединениях они очень легко изменяют свою валентность. Может быть, с этим связана и их исключительно высокая каталитическая активность.
Окислы — производные низших степеней валентности: рутения и осмия имеют основной характер. По мере возрастания заряда металлов увеличивается способность их окислов К образованию кислот, и только высшие окислы — четырехокиси М^11ГО4— почти совершенно но обладают Кислотным характером. Способность мпогих окислов соединяться с водой с образованием кислородных кислот (и соответственно с основными окислами с образованием солен кислородных кислот) с точки зрения координационной теории объясняется предположением, что у атомов металлов насыщены [io все координационные валентности, которые они могут проявлять по отношению к кислороду. Большинство элементов в радикалах (которые в кристаллической решетке представляют собой иопы илп структурные группы) в случае кислородсодержащих лигандов имеет максимальное координационное число 4. Одпако у некоторых элементов, стоящих в пижпих рядах периодической системы, и в первую очередь у теллура и иода, максимальное координационное число по отношению к кислороду при максимальной положительной валептпости равно 6. Из того факта, что чегырехокнев осмия способна присоединять еще основные гидроокиси (см. стр. 352), следует, что 4 атома кислорода не исчерпывают координационных возможностей осмия в восьмивалептпом состоянии. Однако у четырсхокпеи осмия способность к образованию координационных
* Относительно летучести иридия см. стр, 358.
Восьмая группа периодической системы
347
соединений вообще развита очень слабо. Соответствующие комплексы, например K2[Os04(OH)a], прсят характер довольно непрочных продуктов присоединения. Обладает ли рутений способностью проявлять в восьмпвалентпом состоянии более 4 координационных валентностей по отношению к кислороду, точно еще не известно. В остальных отношениях, однако, склонность рутения и осмия к образованию координационных соединений выражена очень сильно — сильнее, чем у железа.
РУТЕНИЙ (RUTHENIUM) Ru|
Рутений (по содержанию в платиновых рудах) * является наиболее редким среди платиновых металлов. Поэтому он был открыт значительно позже других платиновых элементов русским химиком Клаусом в 1845 г. Название ему было дано в честь России (Ruthenia — средневековое латинское название Руси).
Рутений — спутник платины. Содержится оп главным образом в осмиридии — остатке после разделения платиновых руд царской водкой. Очень редко он встречается и в виде самостоятельного минерала — лаурита, сульфида рутения RuS2, содержащего осмий.
Физические свойства. Рутений в зависимости от способа его получения является матов о-серым или серебристо-белым блестящим металлом, обладающим -чрезвычайно большой твердостью; при этом он настолько Хрупок, что его можно легко растереть в порошок. Он очень тугоплавок и плавятся при значительно более высокой температуре, чем платина. В электрической, дуге при плавлении Ru одновременно испаряется. Оп переходит в газовую фазу также при сильном прокаливании на воздухе, но в этом случае летит не металл, а четырехокись, устойчивая мри очень высоких температурах.
После застывания из расплава рутений имеет уд. вес 12,30; удельный вес металла, образующегося при восстановлении его окисла или хлорида при нагревании в токе водорода, может быть значительно и иже. Удельная теплоемкость рутения равна 0,0553. При температуре кипения рутений, по Муассану, растворяет 4—5% углерода. Он способен поглощать значительные количества водорода или кислорода, особенно при выделении этих газов па рутениевом электроде. В мелкораздробленном состоянии рутений обладает ярко выраженной способностью служить переносчиком водорода или кислорода: например, он каталитически ускоряет реакцию между водородом и азотом с образованием аммиака или окисление спирта кислородом воздуха до альдегида и уксусной кислоты. В коллоидном состоянии рутений можно получать восстановлением его солей гидразином в присутствии гуммиарабика (Gutbier) или акролеином (С asloro).
Химические свойства. В отсутствие кислорода воздуха на рутений не действует ни одна кислота, даже царская водка. Однако содержащая воздух или кислород соляная кислота медленно растворяет его при обычной температуре, а при 125° (в запаянной трубке) даже довольно быстро. При нагревании на воздухе рутений чернеет вследствие поверхностного окисления.
В окислительном пламени паяльной трубки металл сгорает, образуя черный дым двуокиси рутения; одновременно появляется характерный запах его чотырехокиси. Порошкообразный рутений при прокаливании в токе кислорода полностью переходит в сине-черную двуокись RuO3. Фтор действует на порошкообразный рутений уже ниже температуры красного падения, а хлор — при красном калении. С серой порошкообразный рутений реагирует лишь при соблюдении особых условий (см. ниже). С фосфором он образует соединения Rup2, RuP и Ru2P (Biltz, 1939); с мышьяком, так же 1,ак и платина, рутений даст диарсенид RuAs2 (Wohler, 1931). Щелочи в присутствии
* В качестве примеси к сульфидным и окисным рудам в ничтожных концентрациях (пе имеющих цока практического значения для его добычи) рутений так же распространен, как й платина. Подробнее об этом см. в гл, 15.
348
Глава, 7
кислорода или веществ, легко отдающих кислород, например смеси КОН с KN03 или К.2СО3 с КС1О3, а также перекисей, например Ма2О2или ВаО2, при высокой температуре энергично действуют на рутений, образуя с ним рутенаты(У Г) M,RuO4. В растворах гипохлоритов щелочных металлов порошкообразный рутений медленно растворяется уже при обычной температуре с образованием чстырсхокиси.
СОЕДИНЕНИЯ РУТЕНИЯ
Окиелы. Продуктом непосредственного взаимодействия рутения с кислородом обычно является сине-черная двуокись RuO2. При более высоких температурах (выше (>00°) образуются также следы четырехокиси рутения RuO4, которая, однако, при охлаждении разлагается, так как при обычной температуре она мета стабильна.
Низшие окислы рутения известны только в форме гидратов. Черная гидроокись рутения(1 И) осаждается из растворов солей рутсния(Ш) при действии щелочи, однако ее трудно получить в чистом состоянии. Гидроокись рутения{Н) при добавлении щелочи к растворам солей рутения(П) выпадает в виде коричневого осадка, по ее но удастся выделить вследствие быстрого окисления. Даже воздух, растворенный в воде, постепенно окисляет Ru{OH)3 с выделением свободного водорода.
Двуокись рутения RuO2 получается в виде сине-черного порошка при нагревании порошкообразного рутения, хлорида или сульфида его в токе кислорода. Она имеет структуру рутила (а = 4,51, с = 3,11 А). Кислоты на нее не действуют. Водородом нри невысокой температуре RuO2 восстанавливается, а при прокаливании па воздухе обычно пе изменяется; однако при очень высоких температурах RuO2 начинает диссоциировать на рутений и кислород. (При 930° давление кислорода над RuO2 достигает около Зв мм рт. ст.) При термическом разложении RuO2 низшие окислы не образуются.
Четырехокпсь рутения RuO4 легче всего получается при пропускании хлора через раствор рутенатов щелочных металлов шеи при добавлении избытка щелочи к растворам солей рутения; опа образует желтые иглы, плавящиеся при 25° в оранжевую жидиость. Растворимость ее в воде довольно значительна (при 20®—20,3 ев 1 л). Уже при комнатной температуре четырехокпсь рутения заметно летуча. Однако при атмосферном давлении опа не закипает, так как уже тике точки кипения (около 108°) RuCn с сильным взрывом разлагается на RuO2 и О2. Плотность пара КнО4 при 100° и давлении 106 мм рт ст соответствует молекулярному весу RuO?l= 165,1 (Debray, Joly). При очень высоких температурах четырехокись рутения стабильна. Опа и е и змеи я стен и при комнатной температуре, если ее сохранять в темноте в совершенно сухом состоянии. RuO4 чрезвычайно энергично реагирует с органическими веществами, окрашиваясь при этом в черный цвет вследствие образования двуокиси. Она также сильно разъедает каучук, а реакция ее со спиртом происходит со взрывом. RuO4 имеет характерный запах, сильно раздражает слизистые оболочки и образует черные пятна па коже. При реакции ее с концентрированной соляной кислотой выделяется хлор. При этом в качестве первичного продукта реакции образуется оксихлорид шестивалентного рутения RhO2CI2, который в присутствии избытка соляной кислоты быстро восстапавливается до RiiC14. Последний даже в концентрированной соляной кислоте постепенно гидролитически расщепляется, образуя Ru(OH)CI3. Однако При известных условиях до наступления гидролиза ПнС14 может также разлагаться на RuCl3 и хлор.
Сульфид рутении, дисульфид рутения RuS2, получается в виде черно-коричпе-вого осадка при осаждении рутения сероводородом из водпого раствора. При нагревании Ru.CI3 в токе H2S сульфид рутения образуется в виде светло-серой кристаллической массы уд. веса 6,14. Синтез можно осуществить и при непосредственном соединении компонентов, если порошок рутения нагревать с серой в эвакуированной кварцевой ампуле так, что один конец ампулы с рутением находится при 1200°, а другой выходящий из печи, поддерживается при температуре 450°, (Такие условия синтеза были впервые предложены Фарадеем.) На сульфид рутения, полученный сухим путем, пе действуют кислоты, даже царская водна. Только выше 1000° начинается термический распад сульфида на металл и серу. Теплота образования RuS2 составляет 77 к кал /мель (Jnza, 1933; Wohler, 1033). При осаждении и а водпого раствора
Восьмая группа периодической системы
349
образуется еще ряд веществ, которые можно принять за сульфиды; однако они не устойчивы (если вообще в данном случае можно говорить о соединениях определенного состава).
Соединения HuSe2 и RuTe2 очень похожи па сульфид рутения, но менее устойчивы (Wohler, 1933).
Сульфиды, селениды и теллуриды рутения и оелшя кристаллизуются по типу пирита. Параметры элементарных кубических ячеек этих соединений следующие:
RuS2 RuSe2 RiiTez OsS2 OsSe2 OsTe2
a=5,57 5,92 6,36 5,61 5,93 C.37 A
Фториды. Нагреванием мел к о раз др об лепного рутения до температурь! — 300* в платиновой трубке в токе фтора Руфф (Ruff, 1924) наряду с пезначительными количествами низших фторидов рутения (а также большим количеством тетрафт ори да платины) получил пентафторид рутения RuFs. Последний образует темно-зеленую прозрачную твердую массу с температурой плавления 101° и температурой кипения выше 250°. Производная от него соль K[RuFe] была позднее получена Клеимом. Это вещество очень интересно в том отношении, что оно является единственным достоверно известным соединением пятивалентного рутения.
Хлориды. Трихлорид рутения RuCl3 представляет собой коричпево-черпые, нерастворимые в воде и в кислотах кристаллические листочки и образуется при непосредственном соединении рутения и хлора при температуре красного каления. Присутствие окиси углерода благоприятствует этой реакции. В растворимом состоянии это соединение можно получить упариванием в токе хлористого водорода раствора, образующегося при действии соляной кислоты па четырех окись рутения (Remy). Продукты, образующиеся из растворов и считавшиеся прежде трихлоридом рутения [хлорид рутепия(Ш)], при бол со тщательном исследовании обычно оказываются более или метгее чистыми препаратами гидрокситрихлорида рутения Ru(OH)Cl3. Тетрахлорид рутения Пи.С14 образуется в виде малоустойчивого промежуточного продукта при действии HCI на ВпО,,; пи легко разлагается, отщепляя хлор. Трихлорид рутения при высокой температуре также разлагается на рутений и хлор; давление его диссоциации, по данным Реми, достигает 1 атм при 850°. В результате его термического разложения низшие хлориды рутения ие образуются. Водкые растворы хлоридов рутепия при действии на них сильных восстановителей (цинк и соляная кислота, амальгама натрия) окрашиваются в томно-синий цвет в результате восстановления рутения до двухвалентного. При нагревании в токе водорода хлорид рутения очень легко переходит в двуокись рутения. Хлориды рутения обнаруживают сильную склонность к образованию ацидо-соединени.й\ важнейшие из них соответствуют типам MjJRuCIq] [гексахлорорутепаты(!У)], [Mj [Ru2OC1i0][ [ деках лор о-р-оксодпрутена -
ты(1¥)] и M1[RuCIs(0H2)J [пентахлороакворутелаты(Ш)].
Координационные соединения рутения. Рутений образует разнообразные координационные соединения. Среди комплексов двухвалентного рутения устойчивостью отличаются гексацианорутенаты(11) м| [Ru(CN)B]. Бесцветная, слабо растворимая в холодной воде калийная соль этого типа K4[Ru(CN)0]‘3H2O изоморфна аналогичному соединению железа — желтой кровяной соли. Для трегвалентного рутения, помимо чистых ацидо-соедипепий типа MJ[RuX5] и Mj[RuXBJ, известны также соединения, которые наряду с кислотными остатками содержат и нейтральные группы, связанные с центральным атомом. К соединениям этого типа относятся уже упоминавшиеся пеитахлороакеорутенаты(111) Мj [ВuC 15(ОН2)], пентахлоронитрозилрутенаты(11 /) M|[RuC15(NO)], а также аммиакаты, например [Ru(OH)(NO)(NH3)4]C12 [хлорид Г[1Дроксонигрозилтетраамминрутепия(Ш)] и др. Рутений образует также комплексы, соответствующие солям гексаацетатотрижелеза(1П) (Martin, 1952). Примеры Координационных соединений четырех валентного рутения уже были приведены. Координационные соединения шестивалентного рутения, помимо полученных Хове (Howe) диоксотетрахлорорутенатов(У 1) M2(RuO2Cl4], представлены главным образом описанными в следующем разделе рутенатами(У/) [точнее, тотраоксорутепатами( VI) ] MjIRuOJ, а координационные соединения сели вал епт по г о рутения, так называемыми „еррутенатами [рутенатами(УП); точное, тстраоксорутепатами(УП) Mt[RiiO4]. Пока еще не ясно, обладает ли восьмивалентный рутений способностью к образованию координационных соединений, или, другими словами, способна ли четырохокись рутения присоединять другие молекулы.
350
Глава ~
А мм инны е комплексы рутения(П) и(III). Рутений по аналогии с хромом и кобальтом образует прочные комплексные аммиакаты (Gleu, 1936 и сл.) Однако в отличие от устойчивых аммиакатов С г и Go, в которых названные элементы всегда трехва-лентпы, рутений, кроме валентности 3, может проявлять валентность 2.
Глё синтезировал соединения следующих рядов (R — координационно связанный кислотный остаток, X — иопогонно связанный кислотный остаток);
Аммины рутения(1П)
[Rн(NIIj 1 е]Xj—сель гексаамминрутения(11Г) («л у т со- е ол Б>>) [Ии(ОН)(МПй,]Х2— соль гидроксопентаамминрут,ения(Г11) («розео*соль») [hnR(NTT3'.5)X,—соль ацидопентаамми.нрутения(1 II) («пурпурёо-соль»)
[RuRjfNH. i:]№—воль диацидотетрааммш1рутеиия{111), цис- и транс-ряды («виолео-» н «праэео-соли»)
[ Rn('l;i( XIJgVj] —трихлоротриамминрутений
К этим классам соединений относится и давно известный упомянутый выше хлорид гмдрсч<_о::итроаялтетрааммяирутснзя(Ш)
Аммины рутепия(П)
[riri(SO„)(NH3)5]X2— соль тиоксопентаамминрутения(11) i R о (SО 2) (ОIГ2) (NИз)д] X 3 — соль тиоксоаквотетраам.1Ы1н.рутения(1 Г) [ВиЙ(8О2)(ГмН3)4]Х— соль ацидотиоксотетраамминрутения(11) [Ptu(SO3)(NH3)5}-2n2O — диеидрат сулъфитопвнтаамми.нрутейия(1I) )Ки(8О3Н)Е(ХН3)4] — бис-(гидросуяъфито)-тётраамминруте11ий{11)
(NH4)2[Ru(SO3)2(NH3)4J-4Н2О — тетрагидрат дисульфитотетраамминруте-ната(11) аммония
Na4[Ru(SO3)2(SO3TI)2(NH3)2J’ 6Н2О — гексагидрат дисульфито-бис-(гидросульф)ито)~ диамминрутенато.(11) натрия
Амминные соединения рутения(П) в большинстве случаев слабо окрашены. Комплексы лутео-, розер- и п урну ре о-p ядов, как правило, бесцветны или окрашены в светло-желтый цвет, С увеличением количества ацидо-групп окраска углубляется (дпхлоро-соли—оранжевые, трихлоротриамминрутений — красный).
Перечисленные аммины рутения(Н) бесцветны, за исключением тшуксо-соеди-нений, т. е. комплексов, содержащих электропейтральныо группы SO2*. Последние обычно окрашены в желтый, коричневый или красный цвета. Комплексы рутения, содержащие сернистый ангидрид, представляют собой очень интересный тип соединений. В то время как известно большое число соединений., в которых ионы сернистой кислоты — SO;-- и HSO; — связаны координационно (сульфите- й гидро сульф ито-комплексы), до сих Пор не известны комплексы, содержащие электронейтральпую молекулу 8О2.
К амминптог соединениям рутения(П1), по-г и дим ому, близка и «рутениевая красная», открытая ЛГоли (Joly, 1802) (ср. стр. 546), состава 2Ru(OH)ClK-7NH3-ЗН2О. Строение этого комплекса пока пе вполне ясно; однако Гле (Glen) показал, что интенсивное окрашивание, характерное для рутениевой красной, возникает при нагревании солей гексааммин- или нентаамминрутепия(1П) в слабощелочных растворах.
Рутепаты(¥1) п рутенаты(УП). При нагревании порошкообразного рутения с гццратом окиси калия при добавлении хлората калия или селитры получается темнозеленый плав, очень гигроскопичный и растворимый & воде с образованием темного оранжев о-красного раствора. Из этого раствора (который при соприкосновении с органическими веществами легко разлагается с выделением черной двуокиси рутения) при упаривании кристаллизуется pymenam{VI) калия К2Ни04-Н20 в виде призм с зеленым отливом и красных — в топких слоях. При пропускании хлора г. раствор этого соединения красная окраска переходит в зеленую, причем образуется рутенат(УП) — «перрутепат». При дальнейшем пропускании хлора образуется четырех-окись рутения. Если, однако, До ее образования ток хлора прекратить, то из раствора, разогретого благодаря экзотермической реакции, при охлаждении выделяется руте-нат(УП) калия KRuOfp, образующий черные мелкие тетрагональные кристаллы. Рутенаты(¥Г11 в противоположность рутепатам(У1) при высокой температуре (выше 200“) неустойчивы. Они разлагаются также щелочами, образуя рутелаты(У1). Рутепаты(У1) ври обработке их разбавленными кислотами переходят в рутенаты(УП)
* Предложенное здесь название координационно связанной молекулы SO2 — «mu оксо s-группа (сокращенное от «тиодиоксов-груипа) при мыкает к названию ътионил» для радикала >80, являющегося производным SO2.
Восьмая группа периодической системы 351
(при одновременном выделении гидрата двуокиси рутения). Рутепаты(У1) й рутепа-ты(УП) ведут себя совершенно так же, как манганаты(¥1) и манганаты(VII) [дерма нгапаты].
Карбонилы ппитрозилы. Рутений подобно железу и никелю способен непосредственно присоединять окись углерода, по только под высоким давлением. Пттакар-бонил рутения Ru(CO)s получают нагреванием металлического порошка с окисью углерода при 200 nm.it и 180°. Легче его получить нагреванием смеси иодида рутения Rul3 с порошком серебра в токе окиси углерода или — по методу Хибера — действием СО под давлением на сульфид рутения. Ни(СО)5 — бесцветная, прозрачная, летучая жидкость, затвердевающая при —22’ в бесцветные кристаллы. Она легко растворима в бензоле, спирте и других органических растворителях, однако в растворе постепенно разлагается с образованием оранжевого нонаяарбонила RiijfCOjo (гексагоналыпде таблички); в расплаве этот процесс идет быстрее. Кроме того, при разложении образуется еще одип твердый зеленоватым карбонил рутения, по-видпмому, состава Ru(CO)t или Ru3(CO))2.
При действии СО на RuE3 первым продуктом реакции является RuE2(CO)2. Известны аналогия тле соединения с бромом и хлором, а также RuBr(CO) (Manchot, 1924—1936). RuI2(CO)2 — полимерен, однако оп может присоединять некоторые органические азотсодержащие основания, а также фосфины, арсяны, стнбцны с образованием одноядерных комплексов типа [RuE2(CO)2Am2] (Am — пиридин, трифенил фосфин н т. д.). Рассматриваемые соединения представляют собой неэлектролиты, т. е. рутений в пих имеет координационное число 6, так же как в комплексной соли K2[RuI2(CN)2(CO)3[ (Hieber, 1956).
При действии NO на R и2(СО)[} прп умореппо высоких температурах (лучше под давлением) образуется, поданным Маншо, рутения в виде красивых красных
кристаллов. Состав его пока точно не известен, предполагают Ru(NO)4 или Ru(NO)s,
ОСМИЙ (OSMIUM) Os
1 Осмий содержится в платиновых рудах в виде сплава с иридием, нерастворимого в царской водке, так называемого осмиридия-, содержание осмия в нем колеблется между 17 и 80%. Содержание оси иридия в платиновых рудах СССР обычно составляет от 0,5—2,5%. В колумбийских рудах оно тоже равно всего лишь нескольким процентам. В Орегоне н в Австралии, однако, открыты руды с содержанием до 37% о см иридия, а в Калифорнии встречается даже почти чистый осмиридий.
Осмий был открыт в 1804 г. Теннантом, использовавшим более ранние наблюдения двух других французских ученых (Fourcroy, Vauquelin). При анализе минералов, содержащих платину, они обнаружили остаток, нерастворимый в царской нодке, который после сплавления с поташом переходил в растворимое состояние. При обработке его азотной кислотой выделялись пары, раздражающие глаза и слизистые оболочки и вызывающие почернение органических веществ. Это были, очевидно, пары четыре х-окиси осмия. Теннант из летучей четырех окиси выделил металл и по характерному запаху назвал его осмием (бсщТ; по-гречески —запах).
Физ ячеек ие'свойетва. Осмий представляет собой синевато-серый металл, в порошкообразном состоянии он имеет сине-черный цвет. Он очень тверд (7,5 по шкале Мооса), хрупок и его можно легко истолочь в порошок. Из всех платиновых металлов осмий обладает самой высокой температурой плавления (около 2700’). По остывании расплава он имеет уд. вес 22,5, рентгенометрически установленная плотность равна 22,7. Каталитическая активность его такая же, как и других платиновых металлов, однако осмиевые катализаторы легко отравляются. Коллоидный осмий можпо получить аналогично коллоидному рутению, однако это осуществить значительно труднее ввиду склонности осмия к окпеленню.
Химические свойства. Для осмия характерна чрезвычайная легкость, с которой он образует четырехокисъ OsO4. Порошок осмия всегда обладает характерным для его четырехокиси запахом, так как окисление его на воз
352
Глава 7
духе до OsO4 протекает, хотя и в очепь слабой степени, уже прп обычпоп температуре. Ввиду того что четырех ок иск осмия восстанавливается следами жиров, пыли и т. д. в черную нелетучую двуокись, стенки и особенно горло сосудов, в которых хранится ОяО4, бывают покрыты черным налетом двуокиси осмия.
При высокой температуре окисление осмия происходит очень энергично. При комнатной температуре осмий в компактном состоянии почти пе изменяется. Вследствие резко выражении!! склонности к окислению порошкообразный осмий растворяется в азотной кислоте (осойеппо концентрированной), а также в горячей концентрированной серной кислоте, расплавленном бисульфате калия и в растворе гипохлорита натрия. При нагрев,!нип он энергично соединяется также с серой. Соляная кислота, свободная от кнглорода, на него не действует, С фтором осмий соединяется при температуре выше с хлором оп также вступает в реакцию при высокой температуре, но не реагирует при этих условиях пи с бромом, ни с иодом. С углеродом, кроме твердого раствора, т'мий образует и соединения. С фосфором: он соединяется гораздо труднее, чем рутений и железо. В противоположность Ru и Fe осмий образует только одно соединение — фосфид состава OsP2, полученный Билт.тцем (Blitz) при нагревании металлического порошка Os о фосфором по «методу Фарадея» (см. стр. 348).
При еп,1>|В,.н.'нии осмия с поташом на воздухе или при добавлении хлората калия, селитры ti других подобных сильных окислителей образуется осмат калия K2OsO4. Пре подкис лепи и раствора, полученного растворением плава, идет [как и в случав растворения плава манганата(VI)! процесс окисления — восстановления с образованием OsU4 и OsO2.
С окисью углерода осмий образует карбонилы, ио составу, свойствам и методам синтеза аналогичные соответствующим соединениям рутения (стр, 354). О карбо ни л-галогенидах осмия см. табл. 36 па стр. 379 и сл.
СОЕДИНЕНИЯ ОСМИЯ
Окислы. Осмий образует бледно-желтую летучую четырехокись OsO4 л коричневую (черную в порошке) двуокись 0s02. Существуют ли окислы осМия низших валентностей, пока не ясно.
Четырехокись осмия OsO4 всегда образуется при окислении осмия или ого соединений кислородом воздуха или при обработке веществами, легко отдающими кислород. Проще всего OsQ, можно получить нагреванием мелкораздробленного осмия в токе кислорода до температуры красного каления. Четырехокись осаждается в приемнике, охлаждаемом льдом, в виде прозрачных, бледно-желтых, почти бесцветных, блестящих моноклинных кристаллов. Она плавится при 40°, образуя маслянистую жидкость, и, но разлагаясь, кипит при 100°. Плотность пара OsO4 (8,88 при 0е и 760 .«.и) соответствует молекулярному весу, отвечающему формуле OsO4. Уже при обычной температуре четырехокись осмия довольно летуча; она обладает проникающим, очень характерным запахом. Ее пары сильно раздражают слизистые оболочки, особенно слизистую глаз, и очень опасны для органов дыхания. Несмотря на то что четырех-окись осмия более устойчива, чем четырехокись рутения, она все же является сильным окислителем, так как легко восстанавливается до двуокиси и даже до металлического осмия. Ее широко используют (часто под устаревшим названием «осмиевой кислоты») для окраски препаратов, исследуемых под микроскопом. Ее водный раствор пе дает кислой реакции на лакмус. Одпако, как впервые было установлено Чугаевым (1918), четырехокись осмия с сильными щелочами может образовывать непрочные продукты присоединения, например OsO4-2KOH и лк соответственно K3[OsO4(OH)2] [тетра(и,тодт1Гцдроксоосмат(У111) калия). Краус (КтаиС, 1925), помимо калиевои соли этого типа, получил соответствующие аммонийную, цезиевую и бариевую соли, а также рубидиевую и цезиевую соли типа MJ[OsO4F2] [тетраоксодифтороосматы(УШ)], в которых ла место обеих групп ОН находятся атомы фтора. Помимо воды, четырехокись осмия растворима такя;е в спирте и в эфпре. Однако эти растворы быстро разлагаются. OsO4 разлагается также соляной кислотой уд. веса > 1,16 (при этом образуется OsClj; менее концентрированная соляная кислота на OsO4 по оказывает действия в противоположность RnO4.
Двуокпсь осмия OsO2 в виде черного, нерастворимого в воде и в кислотах порошка образуется при восстановлении четырехокиси осмия (так как OsO4 восстанавливается
Восьмая группа периодической системы
353
непосредственно до металла только при пагревании в токе водорода). OsO2 можно получить и другим способом, например сплавлением гексахлороосмата(1У) калия К2 [OsClg] с содой. Кристаллы OsO2 — коричневые с желтым блеском. Структура OsO2 аналогична RuO2 (а ~ 4,51, с = 3,19 А). При пагревании на воздухе двуокись осмия переходит в четырехокись. Водород ее очень легко восстанавливает до металлического осмия. Двуокись осмия применяют в газовом анализе для хлоратпой пипетки Г офмана в качестве катализатора окисления элементарного водорода.
Сульфид осмия, дисульфид осмия OsS2 (теплота образования 62 ккал/моль). получается аналогично Нн82; свойства их также вполне аналогичны, однако с дымящей азотной кислотой л царской водкой OsS2 реагирует медленно.
Как и в случае халькогенидов рутения, устойчивость падает в ряду OsS2, OsSe2, OsTe2 с увеличением атомного веса неметаллического элемента. Структура халькогенидов осмия приведена на стр, 349.
Галогецпды. Работы по галогенидам осмия проведены главным образом Руффом (Ruff). Он получил октафторид осмия OsFg взаимодействием осмия и фтора при 25О3. Октафторид осмия собирался в приемнике, охлаждаемом твердой углекислотой, в виде лимоппо-желтого кристаллического возгона (т, пл. 34,4’, т. кип. 47,5s). При этом наряду с OsFg образовывался нелетучий гексафторид осмия OsFg в виде светло-зеленых кристаллов. В противоположность растворимому в воде октафториду гексафторид осмия разлагается водой, причем образуются четырехокись, двуокись осмия и фтористый водород. При неполном фторировании получается также рид осмия OsF4.
При обработке осмия хлором при высокой температуре, как правило, образуется смесь различных хлоридов. Чистый тетрахлорид осмия OsCl4 Руфф получил нагреванием осмия в токе хлора до 650—700° и медленным охлаждением образующихся при этом желто-коричневых газов. Тетрахлорид осмия получается в виде черных, обладающих металлическим блеском корок. При нагревании в"вакууме или в токе хлора он возгоняется не плавясь. В воде ОзС14 растворяется медленно с образованием желтого раствора, из которого в результате гидролиза вскоре выпадает двуокись осмия. При более сильном нагревании в процессе хлорирования осмия и быстром охлаждении образующихся паров' Руфф получил легко растворимый в воде (и в спирте) трихлорид осмия OsCl3 в виде коричнево-черного, гигроскопичного порошка. Темно-коричневый водный раствор трихлорида имеет слабокислую реакцию, но устойчив не только па холоду, ко и при пагревании; на него действуют слабые восстановители, подобные сульфату железа(П), сернистой кислоте или формальдегиду. При продолжительном кипячении с концентрированной азотной кислотой он образует четырехокись осмия. При нагревании трихлорида в вакууме до температуры примерно 500° он переходит в темно-коричнсвыи, нерастворимый в воде и трудно смачиваемый ею дихлорид осмия ОзС13,
С окисью углерода трихлорид осмия реагирует при температуре выше 220" с образовавшем карбояилхлоридов, например OsCl2(CO)3 — бесцветных призм (Ман-chot, 1925).
Координационные соединения осипл. Осмий подобно рутению образует многочисленные координационные соединения, производное двухвалентного осмия— гексацианоосмии(П) кислота Н4[Оз(СТ\)в] была известна еще Клаусу, Ее калиевая соль К4 (Os(CN)6] ЗН3О, образующая бледно-желтые моноклинные таблички, получается, например, при добавлении цианида калия к щелочному раствору четырехокиси осмия. С солями тяжелых металлов она дает реакции, аналогичные реакциям желтой кровяной соли. Как установил Дюфе (Dufet, 1895), она изоморфна желтой кровяной соли я соответствующей соли рутения. В качестве примеров координационных соединений трехвалептиого осмия можно привести пента-нитроосмат(11 Г) калия K2[Os(NO3)5], образующий янтарно-желтые триклинные кристаллы, И гексахлороосмат{ПГ) калия K3[OsCleJ, В солях типа Mj[OsXa(NO)] осмий следует считать трехвалентным. При нагревании смеси осмия и хлорида калия в токе сухого хлора до температуры темно-красного каления или при добавлении хлорида калия и спирта к раствору четырехокиси ОСМИЯ образуется гексахлороосматЦУ) палия K2(OsCl6] — темно-красные октаэдры, изоморфные соответствующей соли платины. Известны также различные сульф'ито-соединеиия четы рсхв ал битного осмия, которые относятся к тому же типу, что и гексахлор о-с о единения, например Na2[0s(S03Na)6], Na2[Os(OH)2(SO3Na)4] и др. Кроме осматов [точнее тетраоксоосма-moff(F/)] MjjOsOJ известны так называемые о см иловые соединения шестивалентного осмия, которые по номенклатуре Вернера правильнее называть диоксдосматами(У1). 22 г. Реми
354
Глава 7
Общая формула ex Mi [OsO3X4]; их можно получить, например, по одной из следующих реакций:
OsO4+4HC1 + 2K.C1=K.2[OsO2C1J4-C12+2H2O,
OsO4+ЗНгОА -j- 2КОН=KafOsO2(CaO4)2J+2СО2+4Н2О,
OsO4J-2NO + 2KNO2=K2[OsO2(NO2)J, г
;к,оно4+4нх ;± к2[Озо2х41+2н2о.
Последняя реакции обратима. Вода разлагает диоксоосматы(У1) в соответствии с реакцией, указаниои нижними стрелками. Катион шестивалентного осмия содержится в соединениях диоксогетрааммипосмия(У1) (ОзО2(ЙН3)41Х2. Взаимодействие ОзО4 с нитритами при юычной температуре приводит к образованию триоксодинитроосматов (VI) Л1а[ОвО3Х21, называвшихся прежде «осмилокси-солями»:
OsO4 4- 3KNO2=Кй[0з0а(К0а)э] + KNO3.
К координационным соединениям шестивалентного осмия относятся и своеобразные азотсодержащие соединения типа M?[OsNXj] —нитридопентаацидоосматы(УГ), В так называемой ос.енл.иооой кислоте OsOjNH и ее солях—осмиаматах—осмий, по теории Вернера, является восьмивалептным. Эти соединения рассматриваются соответственно как триоксонитридоосмиЩУШ) кислота и триоксонит.ридоосматы(У1ГГ). О координационных соединениях восьмивалентного осмия, приязиодныл четырех окиси осмия, уже было сказано на стр. 346 и 352.
Осматы. Осматы [тетраоксоосматы(УГ)! MatOsOJ п° своему составу соответствуют рутенатам(УТ), но они мепее устойчивы. Поглощая кислород, они легко переходят в четырехокись осмия я выделяют OsO4 в заметных количествах, например уже во влажном воздухе. Осмат калия K2[OsO4l-2H2O образуется при восстановлении спиртом или нитритом калия четырехокиси осмия, растворенной в водном растворе едкого кали. Он кристаллизуется в виде бледно-фиолетовых правильных октаэдров. При нагревании в атмосфере индифферентных газов до 200’ осмат калия теряет кристаллизационную воду, а при нагревании на воздухе образуется четырех окись осмия.
Д11А11 Т1‘. РОДИЙ И ИРИДИЙ
Общие сведения. Родий и иридий представляют собой твердые металлы, еще более тугоплавкие, чем платина, чрезвычайно устойчивые по отношению к кислотам, соединяющиеся с кислородом воздуха при высокой температуре. В то время кап родий в соединениях проявляет в основном валентность три, иридий подобно рутению и осмию обнаруживает большую легкость перехода из одной валентности в другую; однако в противоположность Ru и Os — Bh и Ir никогда не проявляют валентность больше шести л очень редко четыре. Родий и иридий в мелкораздробленном состоянии являются очень активными катализаторами. Помимо простых соединений, известно большое число их координационных соединений с комплексным катионом (в противоположность координационным соединениям рутения и осмия).
РОДИИ J RHODIUM) Rh
Родий встречается в платиновых рудах и, кроме того, в некоторых золотых песках Южной Америки. В мексиканском золоте содержится до 43 % родия. По обычно содержание родия даже в богатых им рудах, как правило, незначительно: оно колеблется между 0,5 и 4,5%.
Родий был открыт в 1803 г. Волластоном (одновременно с палладием). Своим названием он обязан розово-красному цвету многих его соединений (poSeog по-гречески розово-красный).
Восьмая группа периодической системы,
355
Физические свойства. Родий представляет собой белый ковкий металл, имеющий удельный вес 12,42 и температуру плавления 1966°. При плавлении иа воздухе он окисляется с поверхности. В электрической печи его можно подвергнуть дистилляции.
Расплавленный родий растворяет до 7% углерода; при охлаждении сплава углерод выделяется в виде графита. Двуокись углерода, азот и водород при температурах 400—1000е в родии в заметных количествах нерастворимы. Восстановлением трихлорида родия гидразином или распылением металла можно перевести родий в коллоидное состояние. Коллоидный родий, по данным Центе лиса (Zengelis, 1938), является исключительно активным катализатором гидрирования. В его присутствии гидрирование может идти при обычном давлении в нейтральном растворе, в то же время коллоидный палладий катализирует эту реакцию только при повышенном давлении и в кислых растворах. Коллоидный родий обладает бактерицидными свойствами, пе оказывая в то же время вредного действия на человеческий организм; в связи с этим его можно было бы применять в некоторых случаях для терапевтических целей. В очепь активной форме родий получается при осаждении из растворов его солей формиатом аммония в присутствии ацетата аммония и аммиака, спиртом или формальдегидом в присутствии карбоната калия. Полученный этим или аналогичным способом родий в очепь тонкодисперсном состоянии (родиевая чернь) при обычной температуре приводит, например, к разложению муравьиной кислоты на двуокись углерода и водород. Замечательно, что его активность пе исчезает сразу, как это обычно происходит с другими катализаторами в присутствии соединений серы, а наоборот, повышается или сохраняется неизменной (Bredig). Спирт в щелочном растворе в присутствии родиевой черни, выделяя водород, превращается в уксусную киСлоту; из хлорной воды или растворов гипохлорита родиевая чернь выделяет кислород, а Оз переходит в ее присутствии в О,,
Химические свойства. Родий при сильном краевом калении окисляется на воздухе, переходя постепенно в окисел Rli2O3, который при значительно более высоких температура'к вновь разлагается, В твердом состоянии родий может присоединять до 2,3 вес.% (13 ат.%) кислорода. Последние его порции выделяются из металла с трудом, лишь в отсутствие газов-восстановителей. Хлор при темно-красном калении превращает родий в трихлорид RhCl3. По отношению к фтору родий, по данным Руффа, чрезвычайно устойчив. Чистый компактный родий не растворяется ни в одной кислоте, в том числе и в царской водке. Родиевая чернь, напротив, растворяется как в царской водке, так и в горячей концентрированной серной кислоте, а при доступе воздуха и в соляной кислоте. Соляная кислота, насыщенная кислородом, ври 150° действует и на компактный металлический родий; последний немного растворим и в расплавленной метафосфорной кислоте. Полностью родий растворяется при красном калении в бисульфате калия, при этом оп превращается в растворимый в воде сульфат родия Rh2(SO4)3. При прокаливании родия с содой и селитрой или с перекисью бария образуются нерастворимые окислы. Соединения родия легко восстанавливаются до металла при прокаливании в токе водорода; однако при этом необходимо охлаждать Rh током двуокиси углерода, так как иначе вследствие активирования водорода металлом может произойти воспламенение и вторичное окисление Rh.
С мышьяком родий реагирует с трудом, RhA^ получают, по Вёлеру (Wohler), тем же методом, что и IrAs2 (см. стр. 359). С фосфором Rh образует соединения Rh2P, Rh5P4, RhP3 и RhP3, полученные Бильтцем (Blitz, 1940) нагреванием компонентов в кварцевой ампуле. Rh2P кристаллизуется по типу 1г2Р (а = 5,53 А), а не-по типу полевого шпата (а = 5,50 А).
Из интерметаллических соединений родия (ср. стр. 266) техническое значение’ имеет RhBi4; на легкой растворимости этого соединения в азотной кислоте основа® простой метод отделения родия от других платиповых Металлон,
23*“
356
Глава 7
СОЕДИНЕНИЯ РОДИЯ
Насколько известно, родий почти всегда бывает в трехвалентном состоянии. Впрочем, в молекуле гидроокиси он, несомненно, четырехвалентен.
По отношению к фтору родий тоже способен проявлять более высокую валентность, чем 3. Однако пока точно еще не установлено, имеет ли высший фторид родня формулу RhF4 или RliF5. (В качестве главного продукта реакции при взаимодействие родия с фтором образуется его трифториц— красный, мелкокристаллический порошок уд. веса 5,38.) Родий, несомненно, четырехвалентен и в комплексном соединении ka[RhFe], ;
О существовании соединений шестивалентного родия в литературе сообщалось неоднократно, однако окончательно это до сих пор пе установлено. Грубе (Grube, 1937) показал, что в растворах могут существовать новы родия(У1). Из таких растворов выделяются ге.чно-епиие осадки, растворимые и щелочи с синим окрашиванием, в то время как из растворов, содержащих ионы родия(1У), осаждаются темпо-зеленые осадки, растворимые в щелочах с образованием зеленых растворов. Из соединений бедова л ентпого родня тепзиметричесии было доказано существование мопокиси RhO. Известна та к aw комплексная сульфит о-со ль двухвалентного родия Na2[Rh(SOs)2]-•2Н3О, полученная Редденом (Reihlen, 1933).
Для одновалентного родия иавестеп только неустойчивый окисел Rh2O.
Оклады. Двуокись родил, окись родия(1У) RhOa, по Вёлеру (Wohler, 1931), не существует в безводном состоянии. Ее гидрат можно получить при действии очень сильных окислителей. При электролизе гексахлорородата натрия Na3[RhClel в щелочном: растворе у анода образуется темно-зеленый осадок RhO2, содержащий переменное количество воды и обычно примесь Rh2O3. При обезвоживании дкоксигидрата родия происходит одновременно отщепление кислорода с образованием безводного окисла Rh2O . Ди оксигидрат родия очень склонен к адсорбции гидроокисей щелочных илп щелочноземельных металлов. Такие продукты адсорбции рассматривали раньше как «соли, содержащие избыточное количество кислотных остатков», например К30.6ВЬО2. Na2O‘8RhO2, BaO-12RhO2; одпако представляется совершенно невероятным, чтобы в данном случае речь шла об индивидуальных соединениях.
Окись родия(Ш) RtbOi? образуется при нагревании па воздухе металлического родия, а также при прокаливании его нитрата. Rh2O3 представляет собой черно-серый порошок, кристаллизующийся по типу корунда (см. т. I, стр, 391, рис. 75); «о = 5,47 А, <х — 55’40'. Из растворов солей родия щелочи осаждают пента гидр ат Rh2O3 5Н2О в виде ппмотгно-желтого осадка, нерастворимого в воде, до легко растворимого в кислотах. При прокаливании Также образуется безводная, нерастворимая в кислотах Rh2O3.
При нагревании в вакууме Ип2О3 может разлагаться до низших окислов fihG и Rh3O; однако они, по-видимому, метастабильны (Wohler, 1925; Schenck, 1937—1938).
Сульфиды. При нагревании RhCi3 с серой в ампуле Вёлер получил п&апасулъфиё родия Rh2S5 — темно-серую кристаллическую массу, нерастворимую в сильных кислотах и даже в царской водке. Аналогичным образом были получены соединения Rh2Sej и НЪ2Те5 '(Wohler, 1933, Biltz, 1937). Эти три пептахалькогенида кристаллизуются в одной структуре типа пирита («псевдопирита»). Кроме того, по данным Бильтца (1935), существуют еще сульфиды состава Rn2S3, Rh3Sj и Rh3S3i
Простые соли родия. Безводный хлорид родия RhCl3, красный порошок, нерастворимый в воде и кислотах, образуется при нагревании металлического родия в токе хлора при красном калении. Выше 440° он диссоциирует на металл и хлор. Очень легко растворимый в воде (а также в спирте) кристаллогидрат хлорида RhCl3‘4HaO образуется в виде красной расплывающейся массы при растворении гидроокиси родия(Ш) в соляной кислоте. При нагревании его до 180° в токе хлористого водорода он отщепляет воду, не теряя при этом способности растворяться в воде. Однако при более сильном нагревании он переходит в нерастворимый в воде трн-хлорид.
Трихлорид родия, содержащий воду, реагирует при сравнительно высоких температурах с окисью углерода (в присутствии безводного хлорида) е образованней ЯЬ2ОС12(СО)3 — рубиново-красных игл с г. пл. 125,5° (Maachot, 1925).
Восьмая группа периодической системы
357
Бромид и иодид родия обладают свойствами, аналогичными хлориду. Помимо галоидоводородных кислот, родий образует простые соли с другими кислотами, например с азотной, серной, сернистой, с сероводородом и цианистоводородной кислотой *. Он образует также квасцы** MIRh(SO4)2‘ •12Н2О, которые являются переходными к собственно координационным соединениям родия.
Координационные соединения родия. Родий образует довольно устойчивые комплексные соединения. Их можно разделить на два класса; соединения с комплексным катионом и соединения с комплексным анионом. Б соединениях первого типа родий имеет координационное число 6.
Аммиппые соли родия, в которых он входит в состав комплексного катиона, были исследованы главным образом Иергенсепом, По своим свойствам, и в первую очередь по значительной прочности, они соответствуют комплексным солям аналогичного состава хрома и кобальта. Известны представители следующих основных типов;
[Rh(NHs)e]X3 [Rhen3]X3 fRh(OH2)(NH3)5]Xs
Соли гексааммиадодияСШ) Сола триэтилевина- Соли аивопептааммивродин(111)
ывнродия(П11
[RhR(NH3)3]X2 (R—одновалентный кислотный остаток
Соли ацидолентаам- или гидр о нс и льна я группа) мипродин(Ш)
[RhCl2Py4]X (Ру—пиридин)
Соли дихлоротетраииридрнродилпИ)
К солям последнего типа следует также отнести и полученные Чугаевым соединения родия с диметилтлиоксимсм, имеющие строение (ср. стр. 342)
О ОН t NHs | ОНз—O=N4 I eN—C—CH,
I >Rh< I
CH3-C=NZ f XN=C—CHj
I NHs 1
НО О
У солей триэтилепдиаминродия(П1), в соответствии с теорией Вернера, возможно существование двух зеркально изомерных форм. Их действительно удались разложить на оба оптически активных компонента. По сравнению с соответствующими соединениями хрома и кобальта они вращают плоскость поляризации в противоположном направлении. Это обстоятельство указывает на то, что для направления вращения плоскости поляризации решающее значение имеет природа центрального атома.
Соли родия с комплексным анионом представлены в основном ацидо-солями типа SO SO С О
Mi[RhXe] (где X — Cl, NO2, CN, -yi и др.). Однако известны и некоторые
соединения типа Mj[RhXs] (X = Cl, Вг). Кроме того, существуют соединения диметил-г ли оксима с общей формулой М1 [R1iC12(C4H6N2O2H)2J. Лежащий в основе комплекс
* Некоторые соединения родия (по аналогии с хромом), кажущиеся по своему валовому составу простыми солями, имеют в действительности сложное строение. Иногда они образуют даже изомеры (если не учитывать в них содержание воды). Так, сульфат родия(Ш) Rh2(SO4)3, по давним Крауса (КгапВ, 1929), существует в двух Нормах — желтой и красной. Первая даст в водных растворах реакции на Rh” и па 04, между тем свежеприготовленные растворы второй модификации этих реакций не дают. Аналогичные соотношения имеют место, как показал Мейер (J. Meyer, 1936), у селопата родия. Хлорид родия существует в двух формах — коричнево-красной и желтой: [RhCl3(OH3)3] (в растворе не диссоциирует па ионы СГ) и [Rh(OH2)3]CJ3 (ио-видимому, существует только в растворе).
** В основе квасцов лежит желтый сульфат родия. Из растворов красного сульфата их получить не удается.
358
Глава 7
является производным от приведенного выше типа соединений (за счет обмена обеих нейтральных групп NHj на ионы G1). Соединения подобного типа известны и у ириЭця.
Существуют я комплексные соли четыр езжал ентно го родия — фториды типа Ml[RhF6J.
О карбонильных соединениях родия (Hieber, 1943—1956) см, табл. 36, стр( 376 и сл.
ИРИДИИ (IRIDIUM) Ir
Иридий всегда встречается в металлическом состоянии в виде сплавов с платиной и отчасти с осмием. Сплавы иридия с осмием («о.смиридий») в большем пли меньшем количестве находятся во всех платиновых рудах в виде очень мелких, а иногда и сравнительно крупных зерен. Кроме того, небольшие количества иридия встречаются в некоторых природных месторождениях золота. Присутствие иридия было в некоторых случаях доказано и в метеоритном железе. Установлено также, что он присутствует в солнечной фотосфере.
Иридий был открыт в 1804 г. Теннантом (одновременно с осмием). Его название (tgiSiog по-гречески радужный) указывает на многоцвет-ность его соединений.
Физические свойства. Иридий представляет собой серебристо-белый очень твердый и довольно ломкий металл, в котором различимы отдельные кристаллы. При температуре красного каления он малоковкий, однако поддается обработке напильником и полировке. Удельный вес иридия равен 22,65. После осмия он обладает наиболее высокой среди платиновых металлов температурой плавления (2454°).
Иридий (после осмия) является наиболее труднолетучим в высоком вакууме платиновым металлом. Однако при пагревапии на воздухе уже при светло-красном калении оп заметно летит. Эта летучесть, связанная с образованием двуокиси иридия, может оказаться помехой при применении приборов из платины, сплавленной с иридием.
Расплавленпый иридий растворяет некоторое количество углерода, не образуя с ним соедипшттгй; при охлаждении С выделяется в виде графита. Порошок чистого иридия может абсорбировать водород в значительных количествах, особенно если его насыщать водородом электролитически. Коллоидный иридий был получен Паалем (Раа!) при смешивании хлорида иридия, лизальбумипа и крепкого раствора соды, а также Гутбиром (Gutbier) восстановлением хлорида иридия гидразином.
Каталитические свойства иридия выражены слабее, чем у платины и палладия. Для технического применения иридия наряду с его химической устойчивостью большое значение имеет и исключительная твердость.
Химические свойства. На иридий пе действуют ни обычные кислоты, ни царская водка. Однако соляная кислота, содержащая кислород, разъедает его при нагревании до 125° в запаянной толстостенной трубке. Нагретый на воздухе или в токе кислорода до температуры красного каления порошок иридия окисляется до двуокиси 1гО3. Однако при более высокой температуре 1гО2 вновь разлагается, и поэтому выше 1140° металл больше пе окисляется. Сравнительно легко действует на иридий хлор при температуре красного калепия, особенно если 1г смешан с поваренной солью, с которой хлорид иридия образует двойные соли.
При высокой температуре в смеси с поваренной солью иридий реагирует такжэ с бромом и иодом, между тем сами по себе бром и иод почти пе оказывают на нис
Восьмая группа периодической системы
359
никакого воздействия. При красном калении иридий энергично вступает в реакцию с фтором. При нагревании 1г соединяется также ссерой. Прикраспом калении иридий образует соединение с фосфором, которое может плавиться и при температуре белого каления снова разлагается. На этом остгонан метод получения расплав ленного иридия в технике, С мышьяком иридий непосредственно реагирует довольно трудно; однако IrAs2 можно получить нагреванием П'СЬ с As в токе водорода (Wohler, 1931). Сфосфо-ром иридий образует соединения 1г2Р (структура его указана на стр. 355) и 1тР2. Расплавленные щелочи на металл не действуют. По данным Клауса (Claus), при внесении порошка иридия в расплав соды и селитры образуется смесь растворимых и нерастворимых Я воде «иридатов» (см. пиже). Расплавленный бисульфат калия дает в этом случае только нерастворимую 1г3О3,
Окислительно-восстановительный потенциал системы [IrCIsI™/[IrCleJ" в 1 М растворе НС1 составляет по сравнению с нормальным водородным электродом, по данным By (Woo, 1931), 1,02 о (при 25“). Ионы иридия(1¥) обладают, следовательно, значительно большой склоппостыо к переходу в низшие валентности, чем, например, ионы желоза(Ш) (ср. т. I, стр. 820, табл. 112). Следует, однако, обратить внимание на то, что в результате комплекс о образования потенциал перезарядки может значительно изменяться,
СОЕДИНЕНИЯ ИРИДИЯ
Окислы. Двуокись иридия, окись иридия(IV) 1гО2 получается в виде черного порошка при нагревании тонкораздробленного иридия на воздухе или в токе кислорода. По данным Вёлера, нагревание лучше всего проводить примерно до 1070°, тан как 1гО2 при более высокой температуре вновь разлагается. При разложении образуется металлический иридий. Так как 1г может также растворять кислород, то установить состав промежуточных продуктов окисления в данном случае не удается. Подобно двуокиси рутения и осмия, двуокись иридия кристаллизуется но типу рутила; а = = 4,49, с — 3,14 А. Гидрат двуокиси?иридия 1гО2-2Н2О или 1г (ОН)4 получается осаждением из раствора хлороиридата(Ш) натрия Na3[IrCl0| * раствором едкого кали при одновременном пропускании кислорода. При этом сначала образуются фиолетовые и синие коллоидные растворы, из которых через некоторое время выделяется гидрат 1гО2, В зависимости от условий осаждения получаются продукты ипдиго-синего, фиолетового, темно-синего и черного цвета. После высушивания над концентрированной серной кислотой количество содержащейся в них воды соответствует приведенной выше формуле. В щелочах гидрат двуокиси иридия почти нерастворим даже в свежеосажденном состоянии, но довольно легко растворяется в кислотах. Безводный гидрат двуокиси иридия нерастворим в серной кислоте, но растворяется в соляной с образованием комплексной кислоты HglrCle (Wohler).
Раньше считалось, что двуокись ирадия может образовывать с сильно основными окислами определенные соединения, известные под названием иридитое. Однако, по-видимому, в этом случае просто образуются продукты адсорбции. По данным Клауса (Clans), иридий обладает кислотообразующими свойствами и в шестивалентном состоянии дает, например, нерастворимый в воде иридат калия К2[1г20т1. Однако эти сведения Клауса нуждаются еще в подтверждении.
Окись иридия(НГ) 1г2О3 можно получить в виде гидрата при добавлении едкого кали к раствору хлороиридата(Ш) натрия; эту реакцию во избежание окисления необходимо вести в атмосфере двуокиси углерода. Если был взят разбавленный раствор едкого кали, то образующийся гидрат окиси окрашен в зеленый цвет; при действии концентрированного раствора едкого кали оп получается черным. При нагревании с концентрированным раствором КОН зеленый гидрат окиси ириция(Ш) также переходит в черный.
* Эта соль кристаллизуется е 12Н2О.
360
Глада 7
Он всегда адсорбирует некоторое количество щелочи, которую не удается удалить промыванием (Wohler).
Сульфиды. Из растворов солей иридия(1У) сероводород осаждает сульфид иридия IrS2, Его можно получить и сухим путем — нагреванием 1гС13 в токе Н28 до 630й (Wohler, 1933). При термическом разложении его, по данным Бильтца (Biltz, 1937), образуется сульфид иридия(НГ) Ir2S3. При дальнейшем пагревании он распадается на металл и серу без образования промежуточных продуктов. Кроме того, существует еще один сульфид, более богатый серой. Соединению Ir3Ss соответствует определенное давление диссоциации, однако значительное количество серы может присоединяться и в виде твердых растворов. Из величины давления диссоциации рассчитаны следующие значения теп,тот образования сульфидов из твердых элементов:
Ir2S3 IrS2 Ir3S8
25,5 30 35 ккал/г-атом Ir
Сульфид Ir3Ss, трисоленид IrSeg и трителлурид 1тТе3 кристаллизуются в решетке «псевдопирита» (ср. стр. 356),
IrSe3 и 1гГе3 при пагревании переходит соответственно в IrSea и 1гТе2 (Wohler). Термическая диссоциация последних приводит непосредственно к образованию металла.
Галогениды. Продуктом прямого взаимодействия иридия с фтором является гексафторид иридия IrFe. Он был получен Руффом (Ruff, 1927) умеренным нагреванием порошкообразного иридия в трубке из плавикового шпата в токе фтора. Гексафторид иридия представляет собой при обычной температуре очень летучую желтую стеклообразную массу, которая при 44° переходит в жидкое состояние. Т. кип, IrFe, вычисленная по кривой давления пара, равна 53э. Это соединение очень активно. Оно не только реагирует с водой по уравнению IrF6 + 5Н3О = Ir(OH)4 + 6HF-p/2O2*, но и восстанавливается даже хлором, причем образуются IrF4 и GIF. Тетрафторид иридия IrF4 проще всего получается при пагревании гексафторида иридия с порошкообразным иридием; IrF4 представляет собой вязкую густую желтовато-коричневую маслянистую жидкость, которая при действии воды немедленно разлагается на гидроокись четырехвалонтпого иридия и плавиковую кислоту.
Хлориды иридия были тщательно исследованы Велером (1913). Легче всего образуется трихлорид 1гС13. Он получается при нагревании порошкообразного иридия в токе хлора при 600—62011 и образует оливково-зеленый порошок, который при 6509 спекается, приобретая при этом светло-зеленый цвет, и кристаллизуется, пе изменяя своего состава. Он устойчив при 763° (т. е. при давлении хлора 1 атм). При отщеплении от пего хлора образуется коричневый дихлорид иридия, область существования которого лежит в интервале 763—773°. Между 773 и 798° и при давлении хлора 1 атм устойчивым является монохлорид 1гС1, образующий медно-красные мелкие кристаллы уд. веса 10,18. Все эти хлориды нерастворимы в воде, а также в кислотах и в растворах едких щелочей. При нагревании трихлорида с хлором под большим давлением медленно образуется тетрахлорид 1гС14. Гидрат его можно также получить из гексахлоро-иридата(1У) аммония (NH4)2 [IrCIel, действуя на него царской водкой. Гидрат тетра-хлорида образует темно-коричневую расплывающуюся массу. Он малоустойчив; при нагревании даже при давлении хлора 1 атм он отщепляет хлор. Известен также гидрат трихлорнда иридия — темно-зеленые кристаллы, растворимые в воде. Описанные растворимые в воде хлориды иридия, вероятно, являются пе простыми солями, а комплексными гидр оксохлор о-соедилениями.
Соли иридия. Хлориды иридия не обладают типично солеобразным характером. Вообще иридий образует мало простых солей. Состав и строение описанных в литературе солей окончательно еще не доказаны. Между тем иридий образует многочисленные комплексные соли. Переходными от простых солей к комплексным соединениям являются характерные квасцы иридия МЧг (SO4)2-12H2O (M1 = NH4, К, Rb, Cs, TI1).
Трехвалентиый иридий, как и родий, образует соли как с комплексным катионом, так и с комплексным анионом, в то время как четырехвалентный иридий, по-видимому, дает исключительно соли с комплексным анионом (ацидо-соли).
* Наряду с О2 при этом образуется также О3»
Восьмая группа периодической системы
361
Соли иридия(Ш) с комплексным катионом по аналогии с соответствующими солями хрома(Ш) и кобальта(Ш) представляют собой прочные комплексные соединения. Известны в основном тс же типы соединений, что и у родия;
(Ir(NH3)6]X3 IIr(OH2)(NH3)s]X3
Сели гексаамминиридпя(ш) Соли аквопедтаамминиридяя(1ц)
[IrK(NH3)5]X3 [ IrR2(NH3)4]X( + 2Нг0).
Сопи ацидопеитааммиииридпя(ш) Соли диацидотстрааммиинридия(111)
Среди солей трехвалентного иридия с ко.иллексны.н анионом встречаются почти исключительно еексаацидоиридаты(Ш), т. е. соединения типа М|[1гХй]. Известны многочислеппыс представители этого типа (X = CI, Вт, I, N02, CN,^/2SO4, ^SOg, — SO3K и т. д.; М1 .= Na, К, NH4, Ag, Hgl, иногда Н). ГехсахаороиридапиДП!) щелочных металлов Na3 [IrClfi]- 12НИ6 и К3 [ 1гС1й[-ЗН2О служат исходным материалом для приготовления других соединений иридия(1П). Их получают восстановлением соответствующиххлороиридатов(1У) (которые в свою очередь синтезируют при слабом нагревании смеси порошкообразного иридия и поваренной соли или хлорида калия в токе хлора), например нагреванием до 400—500е в токе хлористого водорода или обработкой в водном растворе при обычной температуре сероводородом. Пента-хлороамминиридаты(Ш) М| [IrCls(NH3)j были получены Лебединским (1938). О соединениях иридия с диметилглиоксимом см. стр. 357.
Среди комплексных соединений четырехвалентного иридия типа Ы| [1гХе1 [гексаацидоиридаты(1У)] известны главным образом галогениды. Гексахлороиридат(1У) аммония (NH4)2 ЦгС16) (называемый также иридиевым нашатырем) изоморфен гсксахлороллатипату(1У) аммония. Он выпадает при добавлении хлорида аммония к водному раствору гексахлороири-дата(1У) натрия (который получается в свою очередь нагреванием смеси поваренной соли с порошкообразным иридием в токе хлора) в виде темнокрасного осадка, состоящего из мелких октаэдрических кристаллов. В холодной воде он мало растворим, а в горячей — значительно лучше и поэтому легко перекристаллизовывается из водного раствора. При обработке его царской водкой можно получить гидрат тетрахлорида иридия. Нагревание в токе хлора превращает его в трихлорид. При нагревании в токе водорода хлороиридат(1У) аммония легко восстанавливается до металла.
Карбонильные соединения. Карбонилы иридия [1г(СО)4]2 и [1г(С0)3]а- (ср. также табл. 36, стр. 376 и сл.) по своему составу соответствуют карбонилам кобальта. Они легко получаются при действии СО под давлением на галогениды или комплексные галогениды иридия. В качестве промежуточных продуктов образуются карбонил-галогениды иридия. По составу 1гХй(СО)2 и 1гХ(СО)3 отличаются от соответствующих соединений кобальта, Свойства карбопилгалогенидов иридия — окраска и летучесть — определяются содержанием в пих галогена и окиси углерода. Они представляют собой мономерные гомеополярные соединения с координационным числом 1г, равным 4 (Hieber, 1940 и сл.).
, Хибер доказал также существование карбонилгидрида иридия состава Н1г(СО)4.
ДИАДА ИГ. ПАЛЛАДИЙ И ПЛАТИНА
Общие сведения. Палладий и платина представляют собой ковкие, не очень твердые металлы. Температуры их плавления выше, чем у железа, но ниже, чем у других платиновых металлов. Б кислотах платина и особенно палладий растворимы легче, чем остальные платиновые металлы. Сродство к кислороду у палладия и особенно у платины меньше, чем у остальных металлов платиновой группы. У палладия и платины особенно сильно выражена каталитическая активность.
В соединениях оба элемента проявляют в основном валентности два и четыре. Простые соединения встречаются среди них довольно редко. Напротив, способность Pd и Pt к образованию комплексных соединений выражена необычайно сильно.
362
7
ПАЛЛАДИЙ (PALLADIUM) Pd
Чистый металлический палладий встречается в виде отдельных зерен в платиновых рудах, а также в некоторых золотых песках Бразилии, Колумбии и Кавказа. Иногда он встречается в виде сплавов с золотом, серебром или с обоими металлами. Локье (Lockyer) установил присутствие линий палладия в солнечном спектре.
Палладий был открыт в 1803 г. Волластоном и назван им по имени астероида Паллады, открытого незадолго перед этим.
Физические свойства, По внешнему виду палладий занимает промежуточное место между серебром и платиной; его уд. вес равняется 12,03. Из всех платиновых металлов он обладает наиболее низкой температурой плавления (1557°). Перед плавлением палладий размягчается и поэтому поддается ковке и сварке. Его твердость лишь немного больше твердости чистой платины; он обладает также несколько большей вязкостью. Способность к вытягиванию, наоборот, у пего ниже, чем у платины.
Палладий обладает сильно выраженной способностью абсорбировать некоторые газы, особенно водород, в связи с этим он в значительной степени проницаем по отношению к последнему. Уже при 240° проницаемость водорода можно точно измерить, при повышении температуры опа возрастает.
Листовой палладий при толщине 1 .ил при 240° пропускает 42,3 .и.«3 водорода в 1 мин на 1 с.ка поверхности, а при 1060е— 400 .и.и3. При комнатной температуре палладий может поглощать объем водорода в 350—850 раз больше его собственного. При этом он заметно вспучивается, становится ломким и легко образует трещины. Уже при умеренном нагревании в вакууме палладий вновь отдает водород. Водород растворяется в металлическом палладии главным образом в атомарном состоянии. Поэтому палладий сильно активирует водород.
Электропроводность палладия по мере насыщения водородом снижается. Прохождение электрического тока через палладиевую проволоку, насыщенную водородом, способствует движению водорода вдоль проволоки. Отсюда следует, что атомы водорода в палладия по крайней мере частично диссоциируют па протоны и электроны. Таким образом, водород в палладии существует в «металлическом состоянии».
Скорость растворения водорода в палладии в зависимости от предварительной обработки металла пропорциональна величине давления газа или корню квадратному из нее. В некоторых случаях скорость диссоциации па атомы молекул Н2, адсорбированных па поверхности металла, зависит от времени, в других — от скорости проникновения образовавшихся атомов Н внутрь металла (Wagner, 1332).
Рентгенографическое исследование показало, Что поглощение водорода пе приводит к изменению кристаллической структуры палладия. Тем не менее, после того как содержание водорода превысит определенное значение (зависящее от температуры), происходит скачкообразное увеличение параметра решетки (при комнатной температуре от 3,884до 4,020 А). Дальнейшее растворение водорода приводит уже к постепенному расширению элементарной ячейки (до 4,07 А при концентрации 0,80 ат,%Н). С возрастанием параметра связано и скачкообразное изменение других свойств палладия. Поэтому фазу с расширенной решеткой в системе палладий — водород можно рассматривать как самостоятельную, отличающуюся от твердого раствора, пе обнаруживающего расширения решетки, т. е, как соединение, имеющее довольно широкую область гомогенности. Его «идеальная» формула, по-видимому, Pd2H.
В соприкосновении с палладием водород уже при обычной температуре даже в темноте может вступать в реакцию с хлором, бромом, иодом и кислородом. Насыщенный водородом губчатый палладий загорается на воздухе. Окклюдированный палладием газообразный водород восстанавливает также HgCl2 в HgaCl2; FeCl3 в FeCl2; [Fe(CN)e]'" в [Fe(CN)e]”; [AsOJ"' в [AsOs]"'; 1С1О3)' в Cl'; он превращает SO2 в INO3J'
Восьмая группа, периодической системы,
363
BlNO2l' И NH3;CH3NO3 в CH3NH2; CaH5-NO2 в C6H5-NH2 и т. д. В присутствии кислорода и воды палладий, насыщенный водородом, может вступать также в реакции окисления-, оп превращает СО (которая не окисляется озоном даже при температуре его разложения) при обычной температуре в СО2, N2 — в [NHJfNOJ, бензол — в фенол, толуол — в бензойную кислоту.
Особенно каталитически активен палладий в коллоидном состоянии.
Обратимый коллоид палладия, по данным Па а.ня (Paal), можно получить восстановлением хлорида палладия гидразингидратом, причем к хлорпду палладия добавляют пр отальбинов окисный натрий (в качестве защитного коллоида) и едкий натр (в несколько большем количестве, чем требуется для осаждения гидроокиси). После удаления электролитов методом диализа, упаривания при 60—70° и высушивания в вакууме над серпой кислотой получают черпые пластинки коллоидного палладия, При добавлении воды они легко вновь переходят в золь.
Применение коллоидного палладия, приготовленного по Паалю (способность его абсорбировать водород в 3—8 раз больше, чем у обычного палладия), оказалось особенно ценным для гидрирования ненасыщенных органических соединений, когда реакция гидрирования должна быть проведена с особой осторожностью при комнатной температуре, например для установления строения этих соединений.
Химические свойства. Палладий при температуре темно-красного каления окисляется кислородом воздуха и монокись PdO, которая при более сильном нагревании вновь разлагается. Палладий, расплавленный в атмосфере кислорода, подобно серебру, разбрызгивается при затвердевании, так как в расплавленном состоянии он растворяет больший объем кислорода, чем в твердом.
Фтор прн Темно-красном калении образует с палладием дифторид PdF2, а хлор при тех же условиях — дихлорид PdCl2. Сера и селен при несколько более высокой температуре энергично действуют на палладий, причем реакции сопровождаются значительным выделением тепла. Несколько менее энергично реагируют с палладием фосфор и мышьяк, и только при температуре белого каления вступает в реакцию кремний. Углерод, хотя и растворяется в расплавленном палладии, по при остывании вновь выделяется из пего в виде графита. Палладий образует с большинством металлов сплавы. С никелем, а также с медью, серебром и золотом оп образует непрерывный ряд твердых растворов. Дальнейшие сведения об отношении палладия к металлам и неметаллам приведены в табл. 31 (стр. 264 и сл.).
Указанная в табл. 25 система Мп—Pd (Grube, 1936) интересна тем, что в пей существует соединение I)fn2Pd3l образующееся из кристаллизующегося вначале твердого раствора благодаря превращению, протекающему в твердом состоянии без изменения состава. Образование этого соединения, следовательно, характеризуется тем, что пердичный твердый раствор (имеющий структуру Pd) пиже определенной температуры (1175°) претерпевает структурное превращение и образует решетку, соответствующую соединению Mn2Pd3. В этой структуре атомы Ми и Pd вначале распределяются еще совершенно беспорядочно-, только пиже 530° (без изменения структуры решетки) появляется упорядоченность в расположении атомов (образуется фаза, имеющая сворхструктуру). Второму соединению рассматриваемой системы — MnPd—соответствует максимум на кривой температур плавления (1515°). В структурном отношении при температурах выше 608° опо не отличается от Pd и его кубического гранецептрц-рованного твердого раствора. Только ниже 608° обнаруживается изменение структуры е образованием тетрагональной грапецептрировапной решетки и одновременно происходят упорядочение в расположении атомов.
Разбавленная азотная кислота медленно растворяет палладий. В концентрированной азотной кислоте, если она содержит окислы азота, палладий растворяется очень быстро. Лучшим растворителем для пал
364
Глава 7
ладия является царская водка. Соляная кислота, даже концентрированная, если она не содержит растворенного кислорода и свободного хлора, оказывает на компактный палладий лишь едва заметное действие.
При кипячении палладия в концентрированной серной кислоте он растворяется с образованием PdS04 и SO2. Та же реакция происходит и при сплавлении палладия с бисульфатом калия. При сплавлении с селитрой и содой палладий не окисляется, а при нагревании с перскпсыо натрия оп .переходит в ион окись PdO.
Нормальный потенциал палладия равен приблизительно +0,82 в. Таким образом, палладий в электрохимическом ряду напряжений занимает место между серебром и ртутью (ср. т. I, стр. 38).
СОЕДИНЕНИЯ ПАЛЛАДИЯ
Палладий в своих соединениях в основном двухвалентен, реже — четырехвалентен. Для двухвалентного палладия, кроме его монокини PdO и моносульфида PdS, известны некоторые простые соли, к которым следует также отнести и галогениды PdX2, а также большинство координационных соединений. Четырехва лонги ому палладию соответствуют: двуокись PdO2 (известная только в виде гидратов), дисульфид PdSa, а также координационные соединения, главным образом гексагалогено-палладаты, М| [PdX6] (где X = С1 или Вг).
По отношению к фтору палладий обычно бывает тр«валентным. Чистый трифто-рид палладия PdEg получается сравнительно легко при действии фтора па PdCl2 при 200—250° (Huff, 1929). Од представляет собой черный мелкокристаллический гигроскопичный порошок уд. веса 5,06. Дифторид PdF2 сухим путем в чистом состоянии до сих пор получить не удалось.
Окислы. Монокисъ палладия [окись палладия(П)] PdO образуется в виде черного порошка, нерастворимого в кислотах (в том числе и в царской водке), при нагревании порошкообразного палладия в токе кислорода. При более сильном нагревании он омять разлагается на металлический палладий и кислород. Давление кислорода над PdO достигает нри 875° 1 атм. Окись палладия(П), содержащая воду и растворимая в кислотах, получается при осаждении из водных растворов, например при гидролизе нитрата палладия Pd(NO3)2. В воздушно-сухом состоянии гидрат мопокиси имеет коричневый цвет; при высушивании на водяной бане становится черным. Содержание воды в нем колеблется и вплоть до 500—000° PdO еще не вполне теряет воду, несмотря на то что при этой температуре начинает отщеплять кислород. В свежеосажденном состоянии гидрат PdO растворим в разбавленных кислотах, а также в едком натре. По мере обезвоживания он растворяется о большим трудом. Он обладает слабо выраженными окислительными свойствами. Водород вступает в реакцию как с гидратом, так и с безводным окислом уже при обычной температуре, причем окисел восстанавливается в металлический палладий, раскаляясь докрасна за счет тепла реакции.
Двуокись палладия [окись ПаллаДИя(1У)] удается получить только мокрым путем в видо гидрата. При добавлении щелочи к раствору хлоропалладатов(1У) М| [PdGle] она образуется в виде темно-красного осадка, который при высушивании разлагается и чернеет. При 200° опа полностью превращается в мои окись. Уже пра обычной температуре двуокись палладия медленно разлагается, действуя как сильный окислитель. В свежеосаждеином состоянии опа растворима на холоду в разбавленных кислотах, а также в концентрированном растворе едкого натра. Ее растворимость с потерей воды уменьшается
Восьмая группа периодической системы
365
Сульфиды. Сульфиды палладйя были исследованы главным образом в работах Вёлера (Wohler, 1933), Бильтца (Biltz, 1936) и Вайбке (Weibke, 1935) (ср. табл. 31j стр. 264). Моносульфид палладия PdS осаждается сероводородом из растворов солеи налладия(Н). Оп представляет собой черно-коричневое вещество, нерастворимое в разбавленной соляной кислого и сульфида аммония. Сухим путем (при нагревания хлор опали адата (И) аммония с серой пли термическим разложением дисульфида) получаются синеватые или светло-серебристые блестящие твердые кристаллы, нерастворимые даже в азотной кнслоте н царской водке. Дисульфид палладия PdS2 можно получить различными путями, например натр ев а наем PdCl2 с серой до 450— 500Р. В тонко дисперсном состоянии PdS2 — черно-корачневое вещество, в кристаллическом — черно-серое, нерастворимое в сильных кислотах, но легко растворимое в царской водке. Выше 600® PdS2 диссоциирует с образованием S имопосульфида.
Структура PdSa пока но известна. PdSe2 и PdTe2 кристаллизуются в решетке тяня брусита. PdS и PdSe, очевидно, имеют структуру, аналогичную PdTe, а также NiS, NiSc и ШТе, т. е. типа красного никелевого колчедана. Двойные соединения сульфидов палладия и щелочных сульфидов можно получить тоже сухим путем.
Простые соли палладия, например темный красно-коричневый, очень гигроскопичный дихлорид [хлорид палладия(П)] PdCl2-2H2O, красно-коричневый, также расплывающийся сульфат PdSO4'2H2O и кристаллизующийся в виде желто-коричневых расплывающихся призм нитрат Pd(NO3)2, образуются при растворении гидрата окиси палладия(П) в соответствующих кислотах. Безводный хлористый палладий PdCl2 тоже можно получить нагреванием до температуры темно-красного каления губчатого палладия в токе сухого хлора; он очень легко растворим в воде и расплывается на воздухе. Его водный раствор обесцвечивается окисью углерода, этиленом, метаном и другими газами-восстановителями, причем выделяется металлический палладий. Благодаря этому при помощи очень разбавленного раствора хлористого палладия или пропитанной нм фильтровальной бумаги можно обнаружить присутствие даже следов^окиси углерода или светильного газа.
Безводный хлорид палладия при умеренном нагревании присоединяет абсолютно сухую окись углерода (при пропускании ее вместо с парами метанола) с образованием соединения PdCl2(CO). Этот карбонил хлорид разлагается водой с выделением металлического палладия (Manchot, 1926).
В солянокислом растворе хлорид палладия образует комплексные ионы [PdClJ". Из растворов, содержащих наряду с хлоридом палладия хлориды щелочных металлов, выделяются тетрахлоропалладатъ1{11}, т. е. соединения с общей формулой M|[PdCIJ, например тетрахлоро-палладат(Ф/) калия K^PdCJJ, образующий красно-коричневые призмы, обладающие дихроизмом, изоморфные соответствующей сели платины ср. стр. 371).
Комплексные соли палладия. Кроме тетрахлоропалладатов(П), известно также большое число комплексных солей палладия типа M*[PdXJ, устойчивых иногда даже в тех случаях, когда соответствующие простые соли палладия выделить не удается. Кроме того, известны многочисленные комплексные соли двухвалентного палладия, содержащие аммиак. Почти все аммиакаты можно разделить на два типа: [Pd(NH3)JX2 [соли тетраамминпалладия(//)1 и [PdX2(NH3)2J [соединения диацидодиамминпалладия[1Т}}. При пропускании газообразного аммиака над безводными простыми солями палладия (II) обычпо образуются тетрааммины. Их можно также получить из водных растворов. Пуп пропускании небольшого количества аммиака или при обработке тетра минов кислотами чаще всего получаются диаммины.
О конфигурации диаммянов и тетраамминов палладия(П) см. стр. 372.
366
Глава 7
В четырехвалентном состоянии палладий (в противоположность платине) не способен к образованию комплексных катионов’, однако он может давать комплексные анионы. Так, при пропускании хлора через раствор тетрахлоропалладата(П) калия или при добавлении к нему царской водки образуется гексахлоропалладат(/У) калия K2tPdC]6i, имеющий цвет киновари, кристаллизующийся в виде правильных октаэдров, малорастворимый в холодной воде. Эту комплексную соль калия можно получить также растворением палладия в царской водке или в соляной кислоте, содержащей свободный хлор, и последующим упариванием после добавления к раствору хлорида калия. K2[PdCl6] легко, например, уже при кипячении его водного раствора отщепляет хлор, превращаясь при этом в тетрахлоро пал л адат (II). При обработке K2[PdCle] аммиаком он, выделяя азот, переходит в дихлородиамминпалладий(П) pdCl2(NH3)2.
(PdCl^l
Константа равновесия rojoi'n оч i > по Данным Уэллмана (Wellman, (1930), [гаыц-1 L.J 2J
л 1 и. растворе ионов СГ составляет 4160. Нормальный окислительный потенциал системы PdClj/PdClJ в таком растворе при 25° равен +1,29 в.
ПЛАТИНА (PLATINUM) Pt
Платина почти всегда встречается в самородном состоянии главным образом вместе с другими металлами платиновой группы и, кроме того, с железом, свинцом, медью, а нередко с золотом и серебром. В природе встречается соединение платины — сперрилит PtAs2.
В противоположность золоту, которое содержится в кислых (кварцевых) горных породах, платиновые руды встречаются исключительно в основных породах и притом почти всегда во вторичных месторождениях. Первичные месторождения платины (которые почти не разрабатываются) содержат ее в крайне измельченном состоянии в ваде зерен, вкрапленных в силикаты типа оливина или пироксена. Во вторичные месторождения, по которых добывают в настоящее время платину, она была перенесена еще в древние геологические эпохи из первичных месторождений вместе с галькой и песком, образовавшимися при размывании первпчпых месторождений.
Наиболее богатые месторождения платины находятся на Урале. Вне СССР платина встречается в Европе в незначительных количествах — во Франции и в Испании. Следы ее, встречающиеся в Германии, не имеют промышленного значения. Из внеевропейских стран месторождения платины имеются в Канаде, которая в настоящее время является главным источником мировой продукции, в Колумбии, Южной Африке (в основном в виде сперрилита), в Бразилии, на Гаити, на Борнео, в Калифорнии, Новой Зеландии, Новой Каледонии и Лапландии.
Физические свойства. Платина представляет собой серовато-белый блестящий по очень твердый металл;' она довольно ковка и в горячем состоянии легко поддается прокатке и сварке. Ее удельный вес 21,45, температура плавления 1774°; в электрической дуге она испаряется.
Ван-Лпмпт (van Liempt.) путем экстрансляции данных, полученных измерением в вакууме, установил температуру кипения платины при 1 атм, равную3804°. Удельная теплоемкость платины равна 0,0318, удельная электропроводность х = 9,1 1+ (при 0°), удельная теплопроводность /. — 0,165 (при 17°). Коэффициент термического
Восьмая группа периодической системы
3G7
расширения платины (9,1 • 10-в) очень близок к соответствующему коэффициенту стекла. Поэтому платиновую проволоку можно впаивать в стекло. ' i
При красном калении платина заметно проницаема для водорода, но пе пропускает других газов, например О2, N2, Cl2, HCl, С02, СО, СН4, С2Н4, Н2О, HaS и NH3. Последние два газа (H3S и NH3^npn высокой температуре в соприкосновении с платиной разлагаются, и выделяющийся при этом водород диффундирует через платину.
Платина может абсорбировать значительные количества водорода, особенно в топкораздроблокном состоянии (платиновая чернь). В таком виде она поглощает объем водорода, более чем в 100 раз превышающий ее собственный. Легче всего насытить платину водородом, используя ее в качестве катода при электролизе, сопровождающемся выделением Н2. Платина с большим трудом отдает окклюдированный водород, чем палладий. Однако при прокаливании в вакууме он удаляется полностью.
Платина может абсорбировать также небольшие количества гелия и довольно сильно поглощает кислород (платиновая чернь абсорбирует его в количестве, в 100 раз превышающем ее собственный объем). При растворении в платине водород и кислород значительно активизируются. Па этом основано применение платины в качестве переносчика водорода и кислорода. Особо активна коллоидная платина (например, прп гидрировании ненасыщенных органических соединений в водных растворах или других растворителях). Ее можно получить восстановлением гекса-хлороплатиновой(1У) кислоты п щелочном растворе при помощи гидра-зингидрата в присутствии защитного коллоида. Пааль (Baal), разработавший этот способ получения коллоидной платины, в качестве защитного коллоида использовал лизальбипат натрия. По данным Скита (Skita), в качестве защитных коллоидов можно применять также гуммиарабик или очень чистую желатину. Необратимо коллоидную платину без применения защитных коллоидов получал Бредиг (Bredig) распылением металла под водой при помощи электрической дуги. Приготовленные Бредигом платиновые золи получили широкую "известность главным образом вследствие их исключительной каталитической активности при разложении перекиси водорода даже в очень сильно разбавленном состоянии (0,03 мг)л Pt). Следует, впрочем, указать, что каталитическое действие Pt на НгОг ослабляется в присутствии защитных коллоидов, например желатины.
Химические свойства. Платина является типичным благородным металлом. Она практически не соединяется с кислородом, на нее не действуют сильные кислоты, за исключением царской водки, в которой она довольно быстро растворяется, образуя гексахлороплэтиновую кислоту H2PtCl6.
Па данным Вёлера, платина при умеренном нагревании в атмосфере кислорода ire только растворяет последний, но и образует с пим мопокись PtO. Платиновая чернь может частично окисляться ужо при комнатной температуре. Однако выше 560° уже ни одни иа ее окйслов пе является устойчивым. Концентрированная серная кислота отчасти растворяет платину, особенно при повышенной температуре.
Сильное действие при высокой температуре оказывают на платину перекиси щелочных металлов. Так как их следы образуются при плавлении гидроокисей щелочных металлов при доступе воздуха, то едкие щелочи в значительной степени разъедают платину. Поэтому их нельзя плавить в платиновых тиглях. Окись углерода непосредственно не взаимодействует с платиной. Опасность порчи платиновых тиглей газами пламени, содержащими окись углерода и водород (ср. стр. 368), связана, вероятно,
368
Глава 7
с восстановительным действием газов, диффундирующих через раскаленную платину, по отношению к веществам, нагревающимся в тигле.
Выше 250° платина соединяется с сухим хлором, образуя дихлорид PtCl2. Хлористый водород уже при комнатной температуре медленно взаимодействует с платиной. С фтором она соединяется при температуре темно-красного каления, причем получается главным образом тетрафто-рид платины PtF4; наряду с последним в небольшом количестве образуется и дифторид платины PtF2. Сера при определенных условиях действует на платину, но еще легче вступают с ней в реакцию селен, теллур и особенно фосфюр. С фосфором она образует особо легкоплавкие сплавы. Мышьяк, сурьма, висмут, олово, свинец, серебро с трудом сплавляются с платиной.С железом, кобальтом, никелем, медью и золотом опа образует непрерывный ряд твердых растворов; в системе серебро — платина (в отличие от палладия) в твердом состоянии взаимная растворимость ограниченна. Дальнейшие сведения о сплавах платины приведены в табл. 31 (стр. 264. и сл.), 32 (стр. 266), ср. также табл. 17 (стр. 141).
Сплавы платины с хромом с содержанием хрома до 5 вес.% плавятся при более высоких температурах и обладают бблъншя электросопротивлением, тем чистая платина. Поэтому их используют главным образом в качестве обмотки в электропечах (Немилов, 1934).
В электрохимическом ряду напряжений платина стоит правее всех остальных металлов. Однако ввиду сильной склонности к образованию комплексных ионов ее нормальный потенциал определить не удается.
Правила обращения с платиновой аппаратурой. При использовании в лаборатории платиновой аппаратуры необходимо учитывать, что некоторые реактивы разъедают ее. В первую очередь следует обратить внимание па легкую корродируемость платины вследствие склонности к образованию сплавов при высокой температуре. Поэтому платиновые тигли можно ставить при прокаливании только на фарфоровые, кварцевые или платиновые треугольники.
При высокой температуре платина разлагает углеводороды с обра зованием сажи, причем иногда углерод понадает в платину и, сгорая, делает ее шероховатой и ломкой. Ввиду этого нельзя нагревать платиновые тигли светящимся (коптящим) пламенем. При нагревапии в песветящемся пламени бунзеновскоа горелки необходимо следить, чтобы платиновый тигель не соприкасался с сипим внутренним конусом пдамегпт. Вообще платиновые тигли лучше прокаливать ыа паяльной горелке. Если имеется электрическая печь, то нагревание в ней следует предпочесть всякому другому виду прокаливания.
Не следует сильно нагревать в платиновых тиглях не только металлы, но и соединения, легко восстанавливающиеся до металлов, например серебра, меди, свинца, олова и висмута. Кроме того, в них нельзя прокаливать^ бор, бориды; кремний, силициды; фосфор, фосфиды; мышьяк, арсениды; сурьму, антимониды, а также сульфиды, сульфиты и дитиониты.
Фосфаты, арсенаты и антимонаты также нельзя прокаливать в платиновом тигле в пламени горелки вследствие их легкой восстанавливаемости газами пламени (только в электрической печи). При озолении органических веществ, содержащих фосфорную кислоту, платиновые тигли корродируют только в окислительном пламени *.
Нс следует плавить в платиновом тигле перекисей или едких щелочей.
Для чистки платиновых тиглей их лучше всего протирать морским (но не обычным грубозернистым) песком. Можно также прокипятить их с концентрированной соляной или азотной кислотой, но не с.иесью обеих этих кислот (царской водкой). Наконец, можно чистить платиновые тигли плавлением в них бисульфата иди пиросульфата калия.
Изложенные правила обращения с платиновыми тиглями следует, разумеется, Применять и при использовании других платиновых изделий.
* Для этих целей в настоящее время используют относительно устойчивые К фосфору сплавы платины (Pt-Ru-Nb или РЬ-Та).
Восьмая группа периодической системы
369
СОЕДИНЕНИЯ ПЛАТИНЫ
Платина в своих соединениях бывает преимущественно двух- и четы-режвалентной. Она имеет большую склонность к образованию комплексных соединений; в растворах существуют исключительно комплексные, а не простые соединения платины. Как в двух-, так и в четырехвалентном состоянии платина образует ацидо-соединекия. Среди них соединения четырехвалентной платины значительно устойчивее соответствующих соединений четырехвалентного палладия. Платина в противоположность палладию может образовывать колыглексмый катионы не только в двухвалентном, но и в четырехвалентном состоянии.
Известны также некоторые типы комплексных соединений трехвалентной, платины. В монохлориде PtCl, устойчивом лишь в очень узких температурных границах, платина одновалентна, а в крайне легко разлагающейся трехокиси PtO3 опа шестивалентна.
Платина способна также R образованию металлорганических соединений типа B3PtI (где R — алкил).
Окислы и гидроокиси. Платина, как показал Вёлер, при пе слишком высокой температуре может непосредственно соединяться с кислородом воздуха, образуя окислы. Несмотря на это, чистые окислы платины или их гидраты получаются только косвенным путем.
Низшим (нецелом платины является ее монцкись [окись платины)!!)] PtO. Она неизвестна в чистом состоянии, а только в виде твердого раствора с платиной или о двуокисью платины. Однако соответствующая гидроокись Pt(OH)2 получается в виде черного осадка при действии щелочи иа раствор гетрахлороЕлатяната(1Т) калия К2[Р1С14]. Эту реакцию необходимо проводить без доступа воздуха, так как влажная Р1(0Н)2 легко окисляется при высушивании. В токе двуокиси углерода при 120— 150° она имеет состав, соответствующий формуле РЦОНы, а при более сильном нагревании частично разлагается па двуокись платины и металл. Полуторный окисел Р12О3 известен только в виде гидрата. Его молщо нолучить из дисулъфатоплатина-(III) кислоты ИРЦ804)3 *, полученной впервые Блонделем, при осаждении едким натром. Блондель (Blondel) получил эту кислоту восстановлением раствора гидрата двуокиси платины в концентрированной II2SO4 щавелевой кислотой в виде кристаллических оранжево-красных табличек. При умеренном нагревании в атмосфере кислорода легко происходит дальнейшее окисление полуторного окисла, причем оп одновременно выделяет воду. Сравнительно устойчивым окислом является двуокись платины [окись илатины(1 V)] PtO2, которая образуется в гидратированном состоянии в виде красно-коричневого осадка при кипячении водного раствора тетрахлорида платины Р1С14 с содой. Гидрат двуокиси платины растворим как в кислотах, так и в сильных щелочах. После продолжительного нагревания (приблизительно до 200°) гидрат становится нерастворимым в кислотах. При более сильном нагревании он, не выделив еще всей воры, начинает отщеплять кислород. При этом, как показал Велер, он диссоциирует па платину и кислород; таким образом, при термическом разложении двуокиси платины ле образуется низших окйслов. Вследствие образования при этом твердых растворов с одновременным замедлением реакции при термической диссоциации окйслов платины ле удается наблюдать образования определенных продуктов распада; тем пе менее можно считать, что выше 500° в атмосфере кислорода при давлении 1 атм ки одни из окйслов платины уже не является устойчивым. Очепь мало устойчивая трехокиси платины [окись платины! VI)] PtOj была получена Вёлером анодным окислением раствора гидрата двуокиси платины в едком кали при охлаждении солью со льдом. При этом анод покрывался блестящей золотой пленкой, имеющей состав ЗР!.Оа-К2О; при обработке этого соединения разбавленной уксусной кислотой оно легко отщепляло содержащуюся в Нем щелочь. Трех окись платины медленно разлагается уже при комнатной температуре. При умеренном нагревании опа быстра Н полностью отдает кислород и переходит в двуокись. При действии разбавлешгсз соляной кислоты PtO3 интенсивно выделяет хлор, а при реакции с перекисью водорецд при 0° выделения кислорода не происходит.
* Это соединение содержит кристаллизационную воду.
24 г. Реми
370
Глава 7
Сульфиды. М сносу лъфид платины PtS образуется при непосредственном соединении простых веществ (нагреванием тесной смеси порошкообразных платины и серы). Его можно получить также пропусканием сероводорода в раствор хпоройлатинатов(II) или тетрахлороплатино-вой кислоты:
ИгР1С14+ H2S =4НС14-PtS.
В кристаллическом состоянии PtS образует серые иглы, а из растворов выпадает в виде черного осадка. Сульфид платины очень устойчив по отношению к кислотам и щелочам, особенно в кристаллическом состоянии: кристаллический PtS почти нерастворим даже в царской водке. Дисульфид платины PtS2 выпадает в виде черного осадка при пропускании сероводорода через нагретый почти до кипения раствор хлороплати-натов(ТУ) или свободной гоксахлороплатиновой кислоты *:
НгР«31в+2H2S=6НС1 + PtS2.
Дисульфид платины очень устойчив по отношению к кислотам, однако он медленно растворима горячей концентрированной азотной кислоте и в царской водке. В бесцветных растворах сульфидов щелочных металлов PtSг почти нерастворим, если он но содержит примеси других сульфидов, которые растворимы в таких растворах. В растворах поли-сульфидов щелочных металлов он растворим особенно легко в то1" случае, если наряду с пим присутствуют и другие, растворимые в поли-сульфидных растворах сульфиды мышьяка, сурьмы, олова или золота. При подкислении раствора из него осаждается PtS2 наряду с другими перечисленными сульфидами.
Шнейдер (Schneider) при нагревании очень тонко измельченной платины с карбонатами Щелочных металлов и серой получил двойные соединения состава." MjpfgS-. и M|Pt4Se, имеющие характер сульфо-солей. Соответствующая соединениям последнего типа свободная кислота II2FL4S6, по данным Шнейдера, окисляется па воздухе, образуя воду и сульфид платины Pt2S3, представляющий собой порошок серо-сталыюгс цвета уд. веса 5,52. При нагревании на воздухе оп сгорает, как трут, выделяя свободную платину.
С селеном й теллуром платина образует соединения PtSe2n PtTe2, также обладающие значительной устойчивостью по отношению к кислотам. Они, как и РLS3, кристаллизуются по типу брусита.
PtS образует тетра г опальную решетку, в которой атомы Pt занимают место атомов Zn в решетке цинковой обмапки (рис. 47, стр. 407), Каждый атом Pt окружен 4 атомами S, а атом серы в свою очередь координирует вокруг себя 4 атома Pt (расстояние Pt *-•-* S = 2,32 А). Однако в отличие от цинковой обманки атомы сери образуют не тетраэдр, а плоский прямоугольник.
Хлориды. Платина образует четыре хлорида, области существования которых были определены в 1913 г. Вёлером. Монохлорид платины PtCS устойчив только в узком температурном интервале, между 581 и 5833 (в среде хлора при атмосферном давлении). Область существования дихлорида платины PtCl3 простирается между 435 и 581°, а трихлорида платины PtC!3 — от 370 до 435°, в то время как тетрахлорид платины устойчив только ниже 370’.
Дихлорнд платины, хлорид платииы(П) PtCI2 образуется при нагревание губчатой платины в токе хлора примерно до 500° или (лучше) при термическом разложении тетрахлоридд платили (или также гекса хлор оплати нов ой кислоты). Он представляет собой зеленовато-серый (до коричневого) порошок уд. веса 5,87. Оп пераство-
* При этом нередко часть нлатйны восстанавливается до металла.
Восьмая группа периодической системы
371
Рис. 44. Структура K2[PtCl4], На рису яке приведена элементарная ячейка: а, = 0,03, Со = 4,13 A; Fti- Cl = 2,32 А. Атоны хлора, расположенные вне элементарной ячейки, обозначены пунктирной.линией. Аналогичную структуру имеют KsLPdCI3J (ао — ^7,О4, с„ = 4,10 A; Pd С1 = 2,23 А} и (NH,), [PdCIJ
(«„ - 7.21, с„ = 4,26 A; Pd+—> Cl = 2,35 А).
рнм в воде, но растворяется в соляной кислоте, образуя комплексную кислоту H2PtCl4 (тстрахлороплатиновую), причем одновременно происходит его частичное разложение па платину и гексахлор оплатив оную кислоту.
Самая известная соль тетрахлороплатаповой кислоты — тетрахлороплатинат калия K2[PtCl4j. Наиболее удобным методом его синтеза является кипячение раствора гексахлороплатнпата(1¥) калия с оксалатом калия [взятым в теоретически вычисленном количестве, необходимом для восстановления платипы(1У)). Тетрахлороплати-пат(П) калия образует темно-красные тетрагональные призмы, легко растворимые в воде. Структура его, определенная рентгенографически (Dickinson, 1922; Thoilacker, 1937), представлена па рис. 44. Аналогичную структуру имеют Ka[PdCl4] и (NH4)2[PdCI4}. Интересно отметить плоское расположение четырех атомов хлора вокруг центральных атомов Pt или Pd (ср. стр. 372).
PtCi2 при пагревании. присоединяет СО с образованием соединений Р1С12(С0) или Pt2Cl4(CO)2 (£ пл. 194°), PtCI2(C0)2 (т. пл. 142°) и Pt2Cl4(CO)3 (т. пл. 130°).
PtCl2 образует также вполне устойчивые продукты присоединения с трех-хдористым фосфором, аммиаком и Другими азотсодержащими основаниями.
Тетрахлорпдплатины, хлорид пла-типы(1У) PtCI4 лучше всего получать нагреванием гекса хлор оплатив свой кислоты в токе хлора до температуры примерно 300°. Он образует красно
коричневую, слегка гигроскопичную кристаллическую массу и легко растворяется в воде (и в ацетоне), но мало растворим в спирте. Из водных растворов в зависимости от температуры оп кристаллизуется с различным содержанием воды. В растворе он существует не в виде соли, а комплексной кислоты H2Pt(OH)2Cl4 (ди гидр оксотетрахлор оплатил свой), которую можно получить в свободном состоянии при нагревании до 100° гидратов Р1С14, содержащих, большее количество воды. При растворении тетрахлорида платины в соляной кислоте происходит образование гексахлор оплати -новой кислоты II2PtCle.
Остальные галогениды платины очепь похожи па ее хлориды, температура разложения бромидов ниже, чем хлоридов, еще менсо устойчивы иодиды (Wohler, 1925). В противоположность палладию для платины известны также кислота H2PtI6 и даже свободный тетраиодид Ptl4. Это объясняется большей устойчивостью четы рехвал епт-пой платины по сравнению с четырехвалептпым палладием.
Комплексные соедипсния платины. Известно чрезвычайно много комплексных соединений платины, относящихся к небольшому числу типов и отличающихся сравнительно простым строением.
Обзор важнейших типов комплексных соединений платины: (М1 — 1 йкв металла или электроположительного радикала; R и X - одно-
валентные электроотрицательные атомы или радикалы ное число многовалентных атомов или радикалов). или эквивалент-
Соединения платины; II) Соединения платипы( II I) Соединения платины( IV
Ацидо-соединения: M|[PtX4] MlJPtXJ Mi[PtXe]
Лллшиы: [PtAmJX2 [PLRAm3]X [PlR2Am2] MI[PtOXs] Mi[PtO2X2] [PtAme]X4 [PtR2Am4]X2
Как видно из приведенного перечня типов, двухвалентная платина в основном имеет координационное число 4, а четырехвалентная — 6.
24*
372
Глава 7
Гельман удалось получить комплексное соединение четырех валентной платины С шестью различными лигандами — (PtIBrCl(NO2)(NH3)(C5II5N)J.
Наиболее известными из ацидо-соединений являются производные четырехвалентпой платины — гексахлороплатинаты MJ IPtCle] и соответствующая им кислота: гексахлороплатиновая кислота (сокращенно: хлороплатиновая кислота) * H2PtCle. При действии па нее щелочей образуются гексагпдроксонлатинаты M*[Pt(OH)e] щелочных металлов (золотисто-желтые кристаллы). При добавлении кислоты к их растворам выпадает белый осадок гексагидроксоплатиновой кислоты H2Pt(OH)e.
Из ацидо-соединений двухвалентной платины уже была упомянута тетрахлоро-платиновая кпс.отт H2PtCl4. Получать ее восстановлением тексах л ороплатиновоп кислоты сернистым газом удобнее, чем растворением дихлорнда платипы в соляной кислоте. Одного опа не получается в свободном состоянии, а известна только в виде (темно-красного) водного раствора ее солей — тетрахлор о плати патов M|(PtCl4]. К тому же типу относятся Подробно описанные ниже цианоплатинаты.
Комплексные соединения платины, содержащие аммиак (а также другие азотсодержащие основания), известны в очень большом количестве. Раньше всего были описаны два хлорида Рейзе: EPt(NH3)4]Cl2 и [PtGIs(NH3)J. Первый получается кипячением раствора тетрахлоро-платиповой кислоты с большим избытком аммиака. Он кристаллизуется в длинных бесцветных иглах (с 1 молекулой воды, которая при 110° отщепляется). При нагревании до 250й это соединение, теряя 2NH3, переходит в хлорид «второго основания Рейзе»— желтый порошок цвета серы, плохо растворимый в холодной воде и довольно хорошо — в горячей; кристаллизуется он в виде ромбоэдров. Его свежеприготовленный раствор не сразу образует осадок с нитратом серебра; это доказывает, что хлор в этом соединении входит в состав комплексного иона.
Изомерной хлориду «второго основания Рейзе» является «соль Нейроне», образующаяся в виде зеленовато-желтого осадка при добавлении аммиака к холодному раствору тетрахлороплатииовой кислоты. Опа труднее растворима в горячей воде, чем в'холодной. Хлор в пей также связан координационно. По ат ом у изомерию с хлоридом второго основания Рейзе можно объяснить только допущением, что в соединениях этого типа четыре группы, связанные с центральным атомом, расположены в одной-плоскости. В этом случае как раз и может иметь место цис-транс-ивомерия:
HsNx /С1 НзМ\ /С1
;Pt< /Pt<
H3NZ ХС1 СГ XNH3
цис транс
Измерения дипольных моментов (Jensen, 1936) показали, что «соль Пейроне» является цис-с о единен н ем.
Явление цис-трана-изомерйи удалось установить и для других соединений пла-тины(П) типа [PtX2Am2], а также для соединений с двумя различными аминными остатками, например [ Pt(NHa)2Py2]CI2 (диамминдипиридинплатината(П)хлорнд], Для последнего соединения Гантч (Hantzch, 1926) доказал криоскопическим путем, что оба его изомера в фепольных растворах мономерны и поэтому различие в свойствах нельзя объяснить ассоциацией.
Интересна квадратная конфигурация соединений палладия и платины с четырех-коордипационпым центральным атомом (наиболее часто четыре лиганда располагаются вокруг центрального атома тетраэдрически *).
Плоская конфигурация указанных соединений палладия и платины в настоящее время не вызывает сомнения. Во многих случаях опа установлена рент г ей огр афи-
* Есть сведения о том, что и другие металлы VIII группы (в первую очередь Ни и Ni) могут образовывать комплексы с четырьмя лигандами, располагающимися в плоскости; ср. т. I, стр, 444.
Восьмая группа периодической системы
373
чески, например у тетаралыоропалладатов(П) и тетрахлороплатинатов(П) (см. рис. 44, стр. 371). Рентгенографическое исследование (Hertel, 1931; Сох 4932 и сл.) амминных комплексов двухвалентных Платины и палладия позволяет заключить, что плоское расположение лигандов вполне вероятно и в этом случае; однако структуры этих соединений еще не вполне ясны. Если в таком комплексе две различные Группы расположены в трл»е-положеним друг относительно друга, то прочность связи одной из пих с центральным атомом зависит от природы второго лигапда. Эта закономерность была открыта И. И. Черняевым и названа эффектом траяевлияния. Значительным транс- влиянием обладает, например, NO2-rpynna. Поэтому в цис-нитрохлородиам-минплатине связь Pt с NH3, находящимся в транс-положении к NOS, значительно ослаблена по сравнению со связью Pt — С] в тракс-нитрохлородиамминплатине *. Эффект трала-влияния наблюдается и у соединений с октаэдрической конфигурацией лигандов. Им часто пользуются в препаративных целях.
Известно также много соединений, содержащих одновременно комплексные катионы платины и ее комплексные анионы. Примером таких соединений является «соль Магнуса:», не отличающаяся по составу от хлорида второго основания Рейзе. Однако метод синтеза и химические свойства этой соли свидетельствуют о том, что она является тетрахлороплатинатом тетраамминплатины(Н) [Pt(NH3)4][PtClJ. Это соединение было описано в 1828 г. Магнусом и является вообще первым из известных амминных комплексов.
Следует упомянуть о существовании очень многих платиновых соединении, содержащих координационно связанные серусодержащие молекулы (главным образом органические). По своему типу они в общем соответствуют амминам, но вместо азотсодержащих групп (Am) содержат соответствующее число сульфидных групп SRZ,
Хлороплатинован кислота и хлороплатинаты. Гексахлороплатино-вая(1У) кислота II2PtCI6 является одпим из важнейших соединении платины. Обычно ос получают растворением платины в царской водке с избытком соляной кислоты (чтобы по возможности ограничить образование азотистых соединений), например PtCl4-2NOCl. Для их разрушения гексахлороплатиновую кислоту многократно упаривают досуха с соляной кислотой, причем через раствор лучше пропускать хлор [так как иначе может произойти частичное восстановление до тетрахлороплатино-вой(П) кислоты]. Чистую гексахлоро платиновую (IV) кислоту можно также приготовить обработкой губчатой платины соляной кислотой, содержащей свободный хлор. Из желтых водных растворов гексахлоро-платиновая кислота кристаллизуется с 6 молекулами кристаллизационной воды в виде коричнево-красных расплывающихся призм удельного веса 2,43. Она легко растворима в воде, спирте и эфире. Из солей гексахлороплатинатов [PtClJ наиболее известна аммонийная соль (NH4)2 [PtCle], называемая платиновым нашатырем. Она выпадает из раствора солей аммония при добавлении к ним гексахлороплатиновой кислоты в виде лимонно-желтого осадка, состоящего из мелких правильных октаэдрических кристаллов. В воде она слабо растворима (0,67 г в 100 г воды при 15°). В присутствии хлорида аммония ее растворимость значительно снижается, а прц добавлении спирта можно практически свести ее к нулю. Поэтому платину как для аналитических, так и для препаративных целей часто осаждают в виде гексахлороплатината аммония. При прокаливании (NH4)2[PtCI6) полностью разлагается, причем платина выделяется в тонкоизмельченном состоянии (в виде «губчатой платины»). Гексахлороплатинат калия Ka[PtCle] (структуру его см. т. I, стр. 437) изоморфен аммонийной соли и обладает почти такой же
* По-виднмому, автор имел в виду срагпепие прочности связей Pt — Cl в указанных соединениях (резкое ослабление ее и увеличение подвижности С1 в траке-лзомере по сравнению с цис-).— Прим, персе.
374
Глава 7
растворимостью. Натриевая соль кристаллизуется так же, как свободная кислота, с 6 молекулами Н2О и очень легко растворима в воде и спирте. При добавлении к раствору гекса хлор оплати но в ой кислоты нитрата серебра выпадает светло-желтая, практически нерастворимая в воде серебряная соль гексахлороилатиновой кислоты Aga [PtCl6].
Цианоплатиновая кислота и цианоплатинаты. При сильном нагревании губчатой платины с желтой кровяной солью и последующем выщелачивании плава водой получается раствор, из которого выделяется калиевая соль тетртцианоплатиновой кислоты. H2[Pt(CN)J. Описанным способом ее впервые получил Гмелин. Тетрацианоплатинат калия К.г [Pt(GTST)4] >ЗН2О кристаллизуется в виде плеохроирующих (отливающих синим цветом при рассматривании в направлении оси) ромбических игл удельного веса 2,45. В горячей воде он растворим очень легко, а в холодной — значительно труднее, и поэтому его можно легко перекристаллизовать из водпого раствора. Тетрацианоплатинат бария Ba[Pt(CN)4]-4Н2О (раньше называвшийся платино синеродистым барием) также обладает плеохроизмом; при действии на него каналовых, рентгеновских лучей пли радиоактивных излучении обнаруживает яркую флуоресценцию. Для обнаружения этих видов излучения или нри просвечивании рентгеновскими лучами (для определения контуров просвечиваемого объекта) пользуются экранами, покрытыми «платийосинеродистъим» барием.
Тетрацианоплатинсвая кислота известна также н в свободном состоянии. Проще всего ее можно получить при действии сероводорода на ее ртутную или серебряную соль. Она является довольно сильной двухосновной кислотой. Щелочные и щелочноземельные соли тетрациаиоплэтиновой кислоты растворимы в воде и очень хорошо кристаллизуются. При действии па H2Pt(CN)4 сильных кислот не выделяется цианистый водород, что указывает на прочность комплексного аниона.
Аналитические данные. Платиновые соединения легко (но несколько труднее, чем соединения золота) восстанавливаются до металла, который при растворении в царской водке и последующем упаривании с соляной кислотой можно перевести в гексахлороплатиновую кислоту. Последняя при добавлении: хлорида аммония образует труднорастворимый гекса-хлороплатинатаммонйя (NH4)3[PtGl6l, кристаллизующийся в виде характерных кристаллов (желтых октаэдров).
Сероводород осаждает платину из раствора гекса хлороплатинов ой кислоты в виде дисульфида, растворимого в царской водке. Он растворяется также в желтом сернистом аммонии, особенно в присутствии других сульфидов, растворимых в серпистоп аммонии, образуя сульфосоль, которая при действии кислот вновь разлагается.
Гексахлороиридиевая кислота Н21гС1д образует е хлоридом аммония осадок,, совершенно аналогичный гекса хлороплатинату аммония; однако он окрашен в темнокрасный цвет. Поэтому сколько-нибудь значительное содержание иридия в платината обнаруживается по более темному цвету осадка, образующегося при действии хлориде аммония. Палладий из гекса хлор она л ла днеВ ой кислоты также осаждается хлоридап аммония в виде красного гекса хлор опа я Л а дата (IV) аммония. Однако, посколыи? гексахлоропалладиевая кислота при повышении температуры становится неустойчивой, опа не может существовать в растворе, который упаривается с соляной кислотой. Тетрахлоропалладат(П) аммония (NH4)2 [jPdClJ очень легко растворим. Ра&аС тоже не освящается хлоридом аммония. Однако если к не очень разбавление—-раствору хлорида родия, из которого отогнан избыток соляной кислоты, добавив аммиак то через некоторое время выпадает желтый осадок хлорида хлоропент ааымтл> родия(Ш) [RhCl(NH3)51С13 («хлорид иурцуреородия»). Из растворов, содержакщд oc.wuw, прн пагревании с разбавленной азотной кислотой выделяется четырех скЕ-осмия (ее узнают по характерному запаху). Рутений тоже можно отогнать в ш—7-четырехокиси; для этого в пересыщенный щелочью раствор при умеренном натре
Восьмая группа периодической системы
375
вапип Пропускают хлор. Четырех окись рутения растворяется в приемнике, содержащем концентрированную соляпую кислоту, образуя красно-коричневый раствор; при добавлении цияка он тотчас окрашивается в красивый темно-синий цвет.
Родий, иридий, палладий и платину можно обнаружить в минимальных количествах, пользуясь их исключительно сильной каталитической активностью в реакции водорода с кислородом. При наносепии капли раствора, содержащего эти платиновые металлы или один из них, па тонкий листок асбеста смоченное место прокаливают и пускают па «его струю водорода. В присутствии платиновых металлов можно отчетливо наблюдать, как пак ал не тс я пятно (Пайа, 1930). Специфическая и очень чувствительная реакция на палладий основана па том, что Pd каталитически ускоряет восстановление фосфорномолибденотнш кислоты окисью углерода (с образованием молибденовой сини) (Zeiiglielis, 1910; Feigl, 1930).
Открытие и количественное определение платиновых металлов порознь нс представляет трудности. Однако элементы платиновой группы могут в своих реакциях оказывать взаимное влияние. В зависимости от количественных соотиошепии, в которых присутствуют отдельные металлы, их реакции могут видоизменяться, поэтому точный анализ при одновременном присутствии нескольких и тем более всех платиновых металлов иногда становится очень сложным.
КАРБОНИЛЫ МЕТАЛЛОВ
Соединения окиси углерода с тяжелыми металлами — железом, кобальтом, никелем и некоторыми платиновыми элементами — объединяют под общим названием карбонилов металле^. По своим свойствам, молекулярной н кристаллической структуре они совершенно отличаются от соединений, которые окись углерода образует с активными легкими металлами, например с калием. В то время как продукт присоединения СО к металлическому калию (ср. т, I, стр. 486) является производным бензола*, в молекулах кар бон tin ов металлов именно СО-группы связаны с соответствующим металлом. Итак, карбопилы образуют особый класс соединений, представляющий большой интерес с точки зрения теории валентности. Об их технологическом значении уже упоминалось при описапии важнейших представителей этого класса (ср. стр. 309 и 337).
Ио природе валентности карбонилы родственны комплексным ацетилидам металлов. Оии были исследованы в основном в работах Наста [Nasi, 1955 и с л., Angew. Circni., 72, 26 (I960)). В этой связи следует упомянуть и соединения металлов с некоторыми циклическими углеводородами типа бнс-(циклопентадиенил)-железа Ве(С5Н5)3, так называемого «.ферроцена». [Fischer Е. О., Angew. Chem., 67, 475 (1955); ер. также Wilkinson G., Colton F. A,, Progress ia inorganic Chemistry, p. 456, N.-Y. (1959)].
Известные в настоящее время карбонилы металлов сопоставлены в табл. 36 (стр. 376 и сл.) на основе систематики, развитой в работах Хибера (Hieber). Приведенные в таблице «чистые карбонилы», содержащие только металл и СО, являются производными элементов побочных подгрупп Vi, VII и VIII групп. Элементы следующей побочной подгруппы (меди) присоединяют окись углерода, только если они находятся в виде соединений; такой способностью обладают и соединения всех, металлов VIII группы. Метал л ы п обочн ой по дгр у ппы VI групп ы, паск о льк о и зв естн о в настоящее время, кроме «чистых карбонилов», не образуют других соединений, содержащих окись углерода. Металлы побочной подгруппы VII группы, помимо «чистых карбонилов», способны к образованию карбопилгалогепидов. Сравнительно легко получается карбошьп рения. Синтез карбонила марганца Мп2(СО)10 осуществить уже значительно труднее (стр. 229). Он образует золотист о-желтые моноклинные кристаллы, плавящиеся при 155° (под давлением), нерастворимые в воде, но растворимые в различных органических растворителях. При действии галогенов Mn2(CO)i0 превращается в пентакарбглнллгалогенид марганца [MnX(CO)J— мономерное диамагнитное очень Прочное соединение. При действии па бензольный раствор Ми2(СО)10 мелкораздробленных щелочных металлов образуются щелочные соли карбонилгидрида марганца НМп(СО)5. Это соединение было впервые выделено Хибером (1954) в виде бесцветной жидкости, затвердевающей примерно при —20°.
Исторические сведения. В 1888 г. Лангер (Langer) в лаборатории Монда пропускал технический водород над нагретым никелем (для очистки от окиси углерода;
*) В результате рентгенографических исследований (Weiss, Buchner, Sager, Fatladi, 1963) было установке гю, что (СОК.)зс представляет собой алкоголят КОС = СОК (а не производное гексаоксибензола, как это предполагали ранее).— Прим, перев.
376
Обзор известных л настоящее время карбонилов, карбонил
Карбонилы металлов
24Сг 2 'ill 23₽е 27С0 28*^1
Сг(СО)в уд. вес 1,7,, бесцветные
I ромбические крист., легко сублимируется
Fe(CO)5 уд. вес 1,49, желтая жидкость, т. пл. —20°, т. кип. 103°
Ni(CO)4 уд. вес 1,31, бесцветная жидкость, т. пл. —25°, т. кип. 43° :
Второй 18-электронный период Первый 18-электропный период
II Mn2(CO)IU [ЮДОТПСТО-желтые моноклинные кристаллы, т, пл. 155° |Fe2(CO)0l уд. вес 2,09, зо лоти сто-желтые,триклинные (псевдогексаго-нальные) крист., разлагается при 100° Со2(С0)е уд. ВОС 1,73, оранжевые кристаллы, т. пл. 51°
[Fc3(CO)J3] уд, вес 2,00, веление моноклин- [Со4(СО)Ы черные Кристал*
ные призмы, разлагается при 140° лы, разлагается при 60°
42М0 «Те 44RU 4оВЬ 4ePd
I Мо(СО)6 уд. вес 1,96, бесцветные ромбические кристаллы, легко сублимируются Ru(CO)b бссцветпая жидкость, т. пл. —22°
Второй 18-влектропиый период Первый 18-электронный период число молекул СО на 1 атом металла Карбон ил га лог сипды и карбонигщпаниды металлов 1
да в н о to EJ со Я5 м» го п © К5 / 1=5 3 ЕС СЛ й: в
1 1 1 1 i ч И W П О а: р 3 3 о Сл С#' 6
1 1 4» 1 1 1 1т- Ф Ч £ и n to О 3 *' о ф Sr' р * ь> 1 ь W rj О
1 1 1 1 *4 ф Е to ’ П О сЗ" &> 1
гд ?о 'jo 3 ₽ с я tJ гГ И 3$ 8 М tJ -О rt > 62-OEL^ ГС 1—1 1 1 1 1 ч Ьа4 Л £ 3;Г 2, и К Q О О Q О to рТ' м*" J> jT"1 w rtf со 1 Г<5 п о
й 3 о 1 3 я 3 _о з” и*! ЕС 23 3 3 3 о 1—1 S' 3 n О, ° Л ю 3 л СЦ а «1—1 ч ®J Л ” л 3 3 о 8 s’ 1—1 1 ь
гидридов, карбошмгалогеппдов и карбо ни лцпанидов металлов
w
378
42Мо 4зТс 44Ru 45 Rh
Второй 18* электронный период I II — Ru2(CO)9 оранжевые моноклинные призмы г сублимируется Rh2(CO)g оранжевые кристаллы, т. пл. (с разложением) 76°
— — [Ru3(CO)i£] аелепыо иглы [ЙК4(СО)131 верный —
32-электронный период I 74W J5Re 7вОЯ тт!г 78pt
W(CO)e уд. вес 2,65, бесцветные ромбические кристаллы, легко сублимируется Os(CO)5 бесцветная жидкость, т. пл. —15° — —
II -— Re2(CO)w бесцветные моноклинные Кристаллы, т. вл. 177°, сублимируется Ов*»(СО)р светло-желтые кристаллы, т. пл, 224°, сублимируется 1г3(СО)5 зелено-желтые кристаллы, суб- лими- руется
[Ов3(СО)12]? 1Ь(СО)31Х канареечно-желтые тригональные кристаллы, разлагается при 210°
379
Продолжение табл. Зв
Карбон илтало ген иды и карбоаилцианиды металлов
число молекул СО па 1 атом металла 5CO 4CO 3CO 2CO ICO
I Второй 18-электроппый период I 45кь — — — (RhX(CO)2]2 [HhX(CO)2Py2k 1* [RhX(CO)A2]
48Pd — — HPdCl3(CQ) Mr[PdCl3(CO)] e [PdCMCo)]^ [PdCl(CO) en]
47Ag — — */2CO : Ag3(CO)SO4
32-электронный .период 7SRe HeX(CO)5 — — — '—1
jfiOs — OsX2(CO)4 [OsX(CO)4]2 0sX2(C0)3 OsX2(CO)2 —
77Ir — — ItX(CO)3 IrX2(GO)2 —
380
Глава 7
Карбоиилгидриды металлов
Первый 18-электронный | период 1 I 24Сг 25Мп 2eFe 27С0 Второй 18-элек-т ровный период: HRh(CO)4 желтый, устойчив только в твердом состоянии, т. пл* — 11ь
Г1Мп(СО)5 бесцветная жидкость, т. ил. — 20° H2Fe(CO)4 бесцветная жидкость, т. пл. —70° НСо(СО)4 светло-желтая жидкость, т. пл. —26,2°
32-элок тронный период I 74W 75Не 7вОа 771г
• НИе(СО)5? H2Os(CO)4? Н1г(СО)4
1 — одноядерные, легколетучие соединения, нескора створи мые в индифферентных органических растворителях. Центральный атом имеет, вероятно, олектроипую конфигурацию инертного газа.
II — многоядерные соединения, труд но летучие или вообще нелетучие, в индифферентных органически* растворителях мало или совсем нерастворимы.
пользуясь каталитической активностью Ni в реакции 2СО —С + СОа). При этом он наблюдал образование газа, горящего светящимся пламенем. При высоких температурах газ разлагался с выделением металлического никеля. По охлаждении оп конденсировался в жидкость, анализ которой свидетельствовал о том, что опа представляет собой соединение никеля с окисью углерода — тетра карбонил никеля, Ni(CO)4 Вскоре Монд прсдлоя;ил использовать эту реакцию как промышленный метод получения никеля высокой чистоты (ср. стр. 331 и сл.). Поиски аналогичных соединений родственных металлов привели Вертело (BerLlielo) в Париже и независимо от него Мон да и Квипке (Mond, Quincke) в Лондоне в 1891 г. к открытию пентакарбо пила -железа Fe(CO)s. Опыты по получению карбонилов других металлов приводили к успеху только при проведении реакции между топкопзмельченными металлами и окисью углерода при выдоколг. давлении. Таким образом, в 1908 г. Мопду удалось получить карбонилы кобальта и молибдена, а вскоре после этого оп доказал существование соединения окиси углерода с рутением. В чистом виде карбонилы рутения удалось получить впервые в 1936 г. (Mauch ot). В 1926 г. Джоб (Л. Job) синтезировал карбонил хрома Сг(СО)в, действуя окисью углерода на раствор, содержащий безводный хлорид хрома(Ш) и фенилмагнийбромид. Аналогичным образом в 1928 г. оп получил карбонил вольфрама. Одпако впоследствии было показано (I. G. Farbenindustrie, 1931), что его, как и карбонил молибдена, можно получить при высоком давлении непосредственно цз металла. В промышленном масштабе карбонил железа получают с 1924 г. Начиная с 1928 г. значительное число карбонилов и карбопильпых соединений других металлов было получено Хибером. Он предложил целый ряд новых методов синтеза соединений этого класса. Химические свойства карбонилов металлов и их производных исследованы также главным образом в работах Хибера. Продукты присоединения окиси углерода к соединениям. металлов изучались с 1924 г. в первую очередь Маншо, а в последующие годы н Хибером.
Строение и свойства. Карбонилы металлов, по классификации Хибера, можно разделить на две группы (I и П) так, как показано в табл. 36. Соединения, относящиеся к первой группе, без разложения переходят в газовую фазу и легко растворимы
Восьмая группа периодической системы
381
Продолжение табл. 36
Карбопилгалогенидм и it ар бон и л цианиды металлов
число молекул СО на 1 атом металла 5С0 4СО зео 2СО ICO
32-электронпый период 1 78?* — PtX2(CO)2 JPt(CO)2(NH$)2]C]2 [PtX2(CO)]2 JJPtX3(CO) МТ[Р1Х3(СО)] [PtCJ(CO)(NH3)2[Gl
[Pt2Cl4(CO)3JK
! 79 Ан — — [AuCI(CO)J„
Х-Cl, Бг или I,
А — о-фщшле и диамин, пиридин или l/s о-фенантролина, Ру — пиридин, еп — этилендиамин
в индифферентных органических растворителях — бензине, бензоле, эфире, хлороформе. Молекулярный вес их соответствует обычным мономерным формулам. Карбонилы второй группы мало или совсем нерастворимы в индифферентных органических растворителях и почти всегда плавятся с разложением. В данном случае речь идет только о мпогоадсрных соединениях. Для соединений Ре3(СО)12, Со2(С0)8 и Со4(С0)13, по составу которых нельзя заключить о наличии нескольких центральных атомов, это было установлено 1трИ определении молекулярного веса.
Содержание СО в карбонилах первой группы т. о. в одноядерных соединениях, определяется положением металла в периодической системе. Принимая во внимание физические свойства карбонилов, по мнению Хибера, эту закономерность можно объяснить, исходя из предположения, что два электрона атома С, не участвующие в образовании связи с кислородом в молекуле ; С ; J О ; (ср. т. I, стр. 161), создают систему, общую с внешними электроламп атома металла. Эта система во всех случаях состоит из 18 электронов, т. е. имеет оболочку инертного газа, следующего за соответствующим металлом. Для создания внешней оболочки из 18 электронов Сг, Мо и W требуются 12 электронов. Поэтому они присоединяют 6 молекул СО. Железу-и рутению пеобходимо для этого 10 электронов, л оли присоединяют но 5СО. Никелю нужна 8 электронов, и его карбопил содержит 4СО. Кобальту для завершения 18-электрон-ной оболочки необходимо 9 электронов, поэтому он может образовывать лишьмпого-ядерные соединения (ср. табл. 36). Представление о строении карбонилов металлов, согласно которому СО-группа связана с атомом металла ковалентной связью, не только объясняет состав карбонилов, по п находится в хорошем соответствии с их свойствами и структурой.
Кристаллическая решетка гексакарбонилов Сг, Мо и W, по данным Гофмана (Hoffmann, 1935), построена из молекул М(СО)Й, в которых СО-группы октаэдрически расположены вокруг атома металла М таким образом, что атомы С повернуты в сторону металла. Расстояние М-—> С соответствует ожидаемому для ковалентной (атомной) связи. В структуре пентакарбопила железа также имеет место закономерное неплоское расположение СО-групп вокруг атомов Fe, вероятно, в виде тригональной бипирами-
382
Глава 7
СО СО СО
ды. Эннаакарбопяя железа пос троен из молекул СО Fe СО Fe СО ; СО-груплм в СО СО СО
нем расположены в вершинах двух октаэдров, сочлененных гранями. В соединениях Мп2(СО)10 и йе^СО)^ СО-группы образуют Два октаэдра, имеющих общее ребро. В молекуле Co2(CO)g две тригональные бипирамиды связаны общим ребром. В указанных двухъядерных соединениях атомы металла связаны посредством СО-групп, расположенных между ними, и, кроме того, непосредственно—за счет общей электронной пары. Молекулы так называемого тетракарбоиила железа в кристаллическом состоянии и в растворе образуют тримеры (ср. стр, 310). Для гримера Fe3(CO)J2
СО СО
СО СО СО СО
Брилль (Brill, 1931) считает вероятной структуру: Fe Fe Fe . В этом
СО СО СО СО
СО СО
соединении, по-видимому, отсутствуют непосредственные связи металл — металл. Бро-
куэй (Brockway, 1935) электронографическим путем установил, что в молекуле NiiCO)^ СО-группы располагаются в виде правильных тетраэдров вокруг атома N1. Расстояние Ni+- -* С составляет 1,82 А.,т. е. немного меньше суммы атомных радиусов (2, 01 А.).
Исследование спектров комбинационного рассеяния (Dadieu, 1931), спектров-поглощения в парообразном состоянии и в растворе (Schcibe, 1936), а также магнитные измерения (Мыши, 1931) свидетельствуют в пользу предположения о симметричном строении электронных оболочек в карбонилах металлов. [Ср. также работы С a b 1 е, S h с 1 i и е, Chem. Bev., 56, 1, (1956); Cotton, М о п с h a m р, J. Сйещ. Зое., 1960, 1882.]
С другой стороны, предложенная структура подтверждается химическими свойствами карбонилов металла. Особый интерес представляют реакции замещения. Было показано, что СО-группы в молекулах карбонилов могут частично замещаться! как нейтральными молекулами (аминами, спиртами), так и электроотрицательными атомами или радикалами (например, галогеиамн) (Hieier).
При введении нейтральных молекул на место СО-групп образуются соединения типа Мо(СО)3Аш3, Fe2(CO)4Am3, Со2(СО)5Лш4, Ni2(C0)3Am2, (где Am — NH3 или органический амин), отличающиеся тем, что лигандами в них являются только нейтральные молекулы. Хибер называет их wtuemo координационными соединениями». Примерами спиртовых соединений такого типа могут служить Fe(CO)3(CH3OH), Fe2(CO)e(C2H5OH) и Соа(СО)5(СН3ОН). Они легко образуются при нагревании Fe3(CO)12 или Со2(СО)8 с метиловым или этиловым спиртом при тщательной изоляции от кислорода и влаги воздуха. Соединение Fe(CO)3(CH3OH) в водных и бензольных растворах мономерно; Fe2(GO)0(C2H5OH) в воде растворимо с разложением на Fe(CO)3 (известный до сих пор только в растворе) и FefCOJg^HgOH), Карбонилы металлов, частично замещенные аминами или спиртами, разлагаются под действием кислот, причем часть электро нейтральных атомов железа окисляется ионами Н * До Fe‘ например, в соответствии с уравнениями (Ру — пиридин):
6Fe(CO)3Py + 12H,=Fe3(CO)12 + 3Fe- + 3H2+6CO-i-6[PyH]- (1)
и
4Ре(СО)3(СИ8ОИ)4-2И'=Ео3(СО)1г4-Ре”4-Н2-]-4СН3ОН. (2)
Соответствующим образом происходит и окисление кислородом воздуха:
4Fe(C0)3(CH30H)+I/£02=Fea(C0)i2-|-FeO4-4CH30H. (3>
Если на место СО-групп ввести галогены, то образуются соединения в основном, того же типа, что и при замещении СО нейтральными молекулами. У комплексных галогенидов металлов, например типа (Fe(iVH3)e;X2l обычпо наблюдается вытеснение галогенов на внешнюю сферу, что приводит к образованию почти идеальных ионных связей. В отличие от них в молекулах к ар бонн л галогенидов связи атомов галогенов имеют тот же характер, что в безводных галогенидах этих металлов, т. е. приближаются к атомным (ковалентным) связям. На основании магнитных измерений (К1ешш, 1931) можно заключить, что и карбонил галогенид ах, так же как и в самих карбонилах, электронная конфигурация в значительной степени симметрична, т. е., вероятно, существует тоже 18-электронная оболочка. Однако имеется различие между Характером связей СО-групп, содержащих в свободном состоянии лишь одну пару элек
* Наряду с этим происходит и частичное окисление нонами Н’ и другим путем;; см. уравнения (5) и (6), стр. 383.
Восьмая группа периодической системы
383
тронов с насыщенными спинами, и галогенов, у которых в свободном состоянии существует 1 электрон с ненасыщенным спином*. Поэтому па незаполненный энергетический уровень атома галогена Переходит одни электрон металла, с которым связан галоген. Итак, поскольку характер связи в значительной степени приближается к ковалентному, можно говорить все-таки о противоположных зарядах на атомах металла и галогена в молекулах карбонил галогенидов, так же как ив случае простых галогенидов типа Fel2, лежащих в их основе. Примерами карбонил галогенидов металлов могут служить следующие соединения:
Fcl2(CO)4 FeI2(CO)2Py2
FbBt2(CO)4 FeI2(CO)Py2.
Их можно рассматривать как продукты замещения пептакарбонила железа или как продукты присоединения галогенидов желоза(П). Последнему предположению соответствует и метод синтеза карбонил галогенидов. Их можно получить действием галогенов па карбонилы металлов или окиси углерода (под давлением) на галогениды металлов.
Термическим разложением Fe!3(CO)4, где железо электрохимически двухвалентно, Хиберу в 1940 г, удалось получить соединения Fe11I2(CO)3, FeII(CO)2 и Fe1!. Существование двух последних особенно интересно, так как в них железо находится в одновалентном состоянии, наблюдавшемся ранее только в солях Руссена.
К ар бонн л гидриды металлов. В молекулах карбонилов окись углерода легко окисляется — значительно легче, чем в свободном состоянии. При окислений ионами гидроксила (без доступа воздуха) СО превращается в СОЦ, а водород гидроксила гамё-щает СО в молекуле карбонилов с образованием карбонилгидридов металлов. Например, карбонилгидрид железа, образуется в соответствии с реакцией
Fe(CO)5+2OH'=H2Fe(CO),,-|-CO;. (4)
Аналогичным образом при замещении С о2(С0)3 ионами ОН' получается карбонилгидрид кобальта НСо(СО)4. Кроме того, упомянутое выше окисление центрального атома металла ионами Н" происходит таким образом, что при этом образуются и Карбонил-гидриды металлов**.
Например (А — спирт или амин);
2Fe(CO)3A +2Н- — H2Fe(CO)4+Fe" + 2СО+2 А, (5)
Со2(СО)5АЦ-2Н‘ = НСо(СО)4+Со”-р/2Н24-СО-|-А. (6)
Карбонил гидриды металлов значительно менее устойчивы. Уже выше —20° они разлагаются с выделением водорода;
2H2Fc(CO)4=2Hz + Fe(CO)3+Fe(CO)5; (7)
2НСо(СО)4=Н2+Co2(CO)s. (8)
По физическим свойствам кар б они л гидриды занимают промежуточное положение междупентакарбопиломжелезаикарбопилом никели. В ряду Fe(CO)5 H2Fe(CO)4 -о-—>НСо(СО)4 —> Ni(CO)4 наблюдается непрерывное уменьшение полярного характера молекул. Отсюда можно заключить, что водород в этих соединениях связан тоже ковалентно и так, что ядра Н+ должны в значительной степени внедряться в электронную оболочку, существующую в случае 18-алектропной конфигурации. Карбопилгидриды можпо рассматривать просто как «псевдокарбонилы никеля». Если бы оказалось возможным присоединить протоны прямо к ядру атома железа или кобальта, то образовались бы соединения, идентичные карбонилу никеля.
С другой стороны, существует связь между карбоннлгидридами металлов п летучими водородными соединениями элементов главных подгрупп периодической системы, имеющими законченную симметричную электронную оболочку, в которую внедрены ядра Н+ (ср. т. I, стр. 841). Эта аналогия выражается, например, в способности к образованию солей. Так, у карбонил гидридов известна следующая реакция:
2H2Fe(CO)4-)-[Ni(NH3)o-]-‘+2NH3=rNj(NHJ)snHFe(C0)4J2-|-2[NH4J'.
При этом образуются настоящие соди обычно с известными комплексными катионами. Однако Беренсу (Behrens, 1952) при взаимодействии карбонилов или карбонил галогенидов металлов со щелочными металлами, растворенными в жадном аммиаке, удалось получить щелочные карбопилгидриды. При действии кислот карбопилгидриды выделяются из своих солей в свободном состоянии,
* Термин «спиновое насыщение» см. т. I стр. 157.—Прим, ггерее.
** Реакции типа (1) и (5) часто протекают параллельно.
384
Глава 7
Единственным карбонил гидридом, водород которого не может замещаться на металл или комплексный катион, является карбонилгидрид никеля [NiH(CO)3]3, полученный Беренсом в 1954 г. реакцией:
Ni(CO)4+Na + KH3=NaNIl24-CO4-i/2INiH(CO)3]2.
Он растворим в жидком аммиаке с темно-красным окрашиванием и образует аммиакат |Ni[T(CO)3]a-4NH3 цвета киповарп, легко теряющий аммиак. Кислотами карбонил-гидрид никеля разлагается с выделением водорода;
2|jNiJ[(CO)3]2+2H-=3Ni(CO)4-|-Ni" + 3H2.
В растворе жидкого аммиака этот гидрид димерен, как следует из определения его молекулярного веса (по понижению давления пара NH3). В радикале NiH(CO)3 никель имеет то же число электронов, что и кобальт в радикале Со(СО)4. Поэтому и в случае Ni следовало ожидать димеризации.
Кроме соединений, рассматриваемых как соли карбонил гидридов металлов, известны их металлические производные, пе обладающие солеобразным характером — незлектр«,шты. Соединения указанного типа образуются с аммиакатами таких металлов, как Zn, О;1, Си и Ag, т. е. особенно склонных к образованию гомеополярных связей. Примеры; Fe(CO)4Zn(NH3)3 (бесцветные призмы), Fe(CO)4Cd(NH3)2 (бесцветные призмы), Fe(GO)4Cu2(NH3)2 (желтые иглы). Следует предположить, что в этих соединениях атомы водорода карбонилгидридов замещены атомами металла, связанными ковалентными, связями с С()-груипами (Hieber). В соответствии с этим они могут присоединять меньше молекул аммиака *, чем это соответствует максимальному координационному числу металла в попе (Me(NH3)„Jri+.
'Гак называемые «смешанные карбонилы металлов» также следует рассматривать как производные карбонилгидридов. Примеры их образования:
2НСо(СО)4 + IIgCl2 = 2НС1+ Hg[Co(CO)4]2; FefCO^CdfNH^a1^’ Fe(CO)4Cd 4-4- 2NII3; Fe(CO)s -J- HgSO4 + H2O = Fe(CO)4Hg + H2SO4 + CO2 (Hock). Такие соединения кобальта образуются и непосредственно из компонентов в условиях синтезаs осуществляемого при высоком давлении. В состав смешанных карбонилов Fe могут входить металлы побочной нрдгрунпы П группы, смешанных карбонилов Ср — металлы побочных подгрупп H.lIlnlV груки периодической системы (ШеЬет). Смешанные карбонилы железа нерастворимы и не возгоняются; смешанные карбонилы кобальта сублимируются в токе СО и растворимы в индифферентных органических растворителях.. Следует предположить, что в этих соединениях также осуществляются гомеополярньг? связи между атомами различных металлов и группами СО (того же типа, что и в жок-плексах внедрения»).
В известных случаях карбон и л гидриды металлов могут обменивать свой водород па алкильные группы. Первыми представителями образующихся при этом алкилкар-бонилов металлов были метилпентакарбоиил марганца Мн(СО)3СН3 (бесцветное, диамагнитное вещество, т. пл. 95°) и метил тетракарбонил кобальта Со(СО)4СН3 (светло-желтая жидкость, т. пл. около —44°), полученные Хибером в 1957—1958 гг. Данные ИК-спектроскопии позволяют предположить, что в этих соединениях (первое из которых исключительно устойчиво, а второе очепь легко разлагается) СН3-группа, тан. же как и СО-группы, связана непосредственно с атомом металла,
Карбонилнитрозилы и нитрозилы металлов, СО-грукпы в карбонилах могут замещаться и на NO-грукпы. Образующиеся при этом соединения особенно интересны с точки зрения теории валентности. Хибер предположил, что окись азота в них обладает той же конфигурацией, что и окись углерода, т. е. является электроположительной группой NO+, связанной семиполярной связью (ер. стр. 318), Для появление заряда на NO-гр у пне необходимо перемещение по одному электрону от каждой NO-группы к атому металла. Поэтому для заполнения его внешней оболочки электронам!: участвующими в образовании го.пеоп.олярноИ части связи, NO-групи требуется несколько меньше, чем СО-грунп. Так, пента карбонил у железа соответствует к арб они л нитро-зил Fe(C0)2(NO)2. По той же причине радикал Со(СО)4 может стабилизироваться кат за счет присоединения I Н, так и путем замещения ICO на NO [с образованием Co(CO)3(.NO)]. Хибер показал, что, основываясь на его предположении о природе скак, можно ожидать, что в карбонплпитрозплах металлов будут непосредственно загущаться на амины и другие лиганды только СО, но не NO-группы, связанные с метал лом более прочно вследствие образования электровалентной связи (вместо нова л ентигм
* Вместо аммиака эти соединения могут присоединять органические ампаыт например пиридин пли о-фенаитролин.
Восьмая группа периодической системы
385
Тетракарбонилдигалогенидам железа КеХ2(СО)4 соответствуют тоже мономерные тринитрозилмоногалогениды FeX(NO)3. Их прочность п растворимость в индифферентных органических растворителях возрастает в следующем порядке; FeCl(N0)3 —>FeBr(NO^3 —»- FeI(NO)3, так же как это имеет место >у соединений FeX2(CO)4 (Hieber, 19э8). Кобальт образует димерные нитрозилгаловениды (CoX(NO)2]2; в этом случае речь идет о соединениях ди-р-галогепотетранитрозплдикобальта
Г /С1\ 1
(NO)4Co( )Co(NO)2 4C1Z
Никель образует мононитрозилгалогениды, представляющие собой, по-видимому, тетрамеры [NiX(NO)]4.
Не ясны пока соотношения связей в «чистых нитрозилах металлов». Они известны только в кристаллическом состоянии. Примером этого класса соединений могли бы служить тетранитрозил железа Fe(NO)4 и нитрозил рутения (ср. стр. 351).
Снптез карбонилов металлов. Кроме непосредственного присоединения окиси углерода к металлам, в настоящее время известен еще целый ряд реакций образования карбонилов; некоторые из них имеют важное препаративное значение. Иногда предпочитают проводить синтез по методу Джоба (см. стр. 380), используя реактив Гриньяра. По данным Хибера (1935), при этом в качестве промежуточных продуктов образуются неустойчивые^ метал л органические карбонилы, которые в кислом растворе распадаются с образованием чистых карбонилов. Например; 3Cr(CO)2R4 Ц- 6Н* = = Сг(СО)в4~ 2Сг*' * + 12R + ЗН2, где R — органический радикал. Гексакарбонил хрома до сих пор был получен только этим методом.
Уже было указано, что высокомолекулярные карбопилы металлов удается получить разложением низкомолекулярных в соответствующих условиях.
Часто более простым, чем синтез самих карбонилов металлов, оказывается получение их галогеноеамещенных (при реакции галогенида металла и окиси углерода). Тогда галоген можно связать подходящим металлом — медью или серебром — и перейти таким образом к чистому карбонилу металла, Мапшо (1936) получил таким путем карбонил рутения
4-СО (дод давлением) +Ag
Rul3 ----» RuI2(CO), -----> Ru(CO)5.
По данным Хибера, действие СО под давлением па галогепнды металлов, способных к образованию карбонилов, также можно использовать для синтеза карбопилов. Приведенный в качестве примера на стр, 361 карбонил иридия получается так же, как карбонил и карбонилгидрид родия, указанные в табл. Зо.
Другой путь синтеза карбонилов — исходя из серусодержащих соединений, Так, Мапшо показал, что Ni(CO)4 легко получается при действии СО на NiS, суспендированный в растворе щелочи. В присутствии сульфидов почти всегда можно наблюдать ускорение реакции №пси углерода с металлом, способным к образованию карбонила. Наконец, оказалось, что в солях типа [Ni(?iH3)0j [HFe(CO4)]2 аммиак может легко замещаться па СО (Hieber). В этом случае образуется Ni(CO)4, в то время как радикал jHFe(CO)4] диенропорционпрует по реакции
2[IIFe(CO)J=H3Fe(CO)4 + V3Fe3(CO)J2.
При проведении аналогичной реакции с соединением [Co(NH3)6|[HFe(CO)4)2 образуется к ар б опил гидрид кобальта, так как водород переходит к атому кобальта:
[Co(NH3)s] [ HFo(CO)J2+4СО=[Со(СО)4] [HFe(CO)4]2 Ц- 6N Н3, 2[Co(CO)4][IIFe(CO)4]2 -» 2НСо(СО)4Ц-H2Fe(CO)4+Fe3(CO)12,
25 г. Реми
лава 8
ПЕРВАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
(Побочная подгруппа)
МЕДЬ, СЕРЕБРО, ЗОЛОТО
Порядковый номер Элемент С ГОГ-ВОД А томвий вес Удельный вес Температура, 'G (Удельная теплбеи-кость Валентность
плавления кипения
20 Медь Си 63,54 8,92 1083 2350 0,0916 I, II, III
47 Серебро Ag 107,880 10,50 960,5 1980 0,0557 Г, II, 1П
7!) Золото Аи 197,0 19,3 1063 2700 0,0313 I, III, IV?
Общие сведения. Элементы побочной подгруппы первой группы периодической системы медь, серебро и золото по порядковым номерам следуют непосредственно за последними (стоящими правее других) элементами побочной подгруппы VIII группы (никелем, палладием и платиной). По характеру они во многих отн опте пнях напоминают эти элементы. Однако Си, Ag п А и обладают также рядом свойств, благодаря которым обнаруживается их внутренняя связь с элементами главной подгруппы. первой группы — щелочными металлами. Как и последние, они способны в своих соединениях проявлять положительную одновалентность. Правда, в то время как щелочные металлы встречаются в соединениях только в одновалентном состоянии, элементы побочной подгруппы первой группы (подгруппы меди), и прежде всего медь и золото *, могут выступать в своих соединениях в более высоких валентных состояниях, а именно медь главным образом в положительно двухвалентном состоянии и золото — в трехвалентяом. Двухвалентное состояние для меди и трехвалентное состояние для золота в их соединениях (солеобразных) в общем даже оказываются более предпочтительными.
Соединения, в которых медь, серебро и золото одновалентны, отнюдь не близки по своим свойствам соединениям щелочных металлов и большей частью сильно отличаются от последних, Все соединения одновалентной меди и одновалентного золота либо очень трудно растворимы, как, например, хлориды CuCI и АпС1, либо образуют прочные комплексы. В противоположность щелочным металлам, которые вследствие легкой растворимости, устойчивости и сильной диссоциаций большинства их соединений присутствуют в водном растворе почти всегда в виде свободных элементарных ионов, медь и золоте в одновалентном состоянии в растворе в виде элементарных ионов существуют лишь в исчезающе малых количествах. Серебро в своих нерастворимых и комплексных соединениях ведет себя аналогично одновалентным меди я золоту. Однако Ag может
* Относительно соединений двух- н трех валентного серебра см. стр, 437.
Побочная подгруппа первой группы периодической системы 387
также образовывать отдельные легкорастворимые соли некомплексной природы. Некоторые из них, например нитрат и сульфат, изоморфны соответствующим солям натрия.
По электрохимическому характеру элементы главной и побочной подгрупп первой группы резко отличаются друг от друга. В то время как свободные щелочные металлы относятся к химически активным веществам, металлы побочной подгруппы первой группы обладают ярко выраженным характером благородных металлов. Щелочные металлы стоят в начале, а медь, серебро и золото—в конце электрохимического ряда напряжений.
Среди всех элементарных ионов ионы щелочных металлов обнаруживают наименьшую склонность к комплексообразованию; напротив, медь, серебро и золото па всех ступенях окисления являются ярко выраженными комплексообразователями.
Окислы щелочных металлов жадно соединяются с водой с образованием гидроокисей сильно основного характера, которые крайне легко растворяются в воде с большим тепловым эффектом. Окислы меди, серебра и золота в воде мало растворимы. Содержащиеся в растворах лишь в очень незначительных концентрациях гидроокиси металлов подгруппы меди проявляют более или мепее ярко выраженный амфотерный характер.
Щелочные металлы являются легкими металлами. Медь, серебро и золото, напротив,— типичные тяжелые металлы.
Щелочные металлы обладают большим сродством к кислороду. Медь, серебро и золото аналогично металлам побочной подгруппы восьмой группы очень склонны к соединению с серой.
Соки щелочных металл он бесцветны (если они не содержат окрашенных кислотных Остатков). Соли одновалентных медн, серебра и золота только отчасти бесцветны; некоторые из них слабо, но характерно окрашены. У солей, содержащих окрашенные кислотные остатки, часто наблюдают отчетливое углубление окраски. Почти все соли двухвалентной медн имеют ярко выраженную, большей частые синюю или зеленоватую окраску. Соли трехвалентного золота, хотя и слабо, но также окрашены.
Гораздо сильнее, чем в химическом поведении, аналогия между металлами главной и побочной подгрупп первой группы отражена в оптических спектрах.
В дуговых спектрах металлов подгруппы медн, так же как и в спектрах щелочных металлов, р- и d-термы раздваиваются, т. е. линии, образованные этими термами, проявляют достаточно сильное расщепление в виде дублетов подобно, например, желтой липни натрия (ср. т. I, стр, 197). Так Же как в спектрах щелочных металлов, в спектрах меди, серебра и золота основным термом, т. е. численно наивысшим термом, является s-терм.
В соответствии со сказанным на стр. 142 т. I ато значит, что цтаптачй аимстров находится на орбите с побочным квантовым числом I = 0, Так же как атомы щелочных металлов атомы металлов побочной подгруппы I группы имеют только один электрон, который связан аналогичным образом. Отсюда ясна их одновалентность (соединяющаяся, правда, со способностью выступать также и в более высоко валентных состояниях). Однако, как показывают ионизационные потенциалы, рассчитанные до данным границ абсорбционных серий (а отчасти также непосредственно измеренные) [см. т. I, стр. 140, табл. 22], связь здесь существенно прочнее, чем у щелочных металлов. Этим объясняется отчасти значительно более благородная природа металлов подгруппы меди по сравнению со щелочными металлами. Это свойство определяется также очень высокими тепл стами испарения металлов (ср, т. I, стр. 180) и следует, в соответствии с правилом Т ругана, из высоких вначелни температур кипения (ср. обзорную таблицу в начале данной главы).
Распределение электронов в электронных оболочках свободных атомов в основном состоянии показано в табл. 37. В противоположность щелочным металлам у металлов подгруппы медн непосредственно под внешней оболочкой находится не оболочка 25*
388
Гла#а S
Таблица 37
Распределение электронов в атомах щелочных металлов и в атомах меди, серебра и золота
Порядковый номер Элемент Распределение электронов Символ терма
3 Литий is® 2s
и Натрий Is® 2s3j2p® 3s
19 Калий is® 2s® 2ps 3s® 3pe 4s
29 Медь Is® 2s® 3s® Зр® Зг/ю 4s 2^/2
37 Рубидий Is® 2p® 3s® 3jt>° Srfio 4,S3 5s
47 Серебро is2 2s® 2p6 3s® 3p8 3dK>4s® 4p® 4d1® 5s
55 Цезий Is® 2s® 2pe 3s® 3p>e 3di°4s® 4рв 4d10 5s®5pe 6s
79 Золото Is2 2s® 2pe 3s® 3p« 3di04s® 5№5p65dio 6s
с конфигурацией инертных газов, а оболочка, имеющая 10 электронов с побочным квантовым числом 1=2 (d-оболочка).
В этих «промежуточных оболочках» электроны связаны существенно рыхлее, чей в оболочках с конфигурацией инертных газов.
Это следует уже из того факта, что полностью заполненная d-оболочка не может образовываться в атомах с более низким порядковым номером в результате присоединения электронов и перехода атома в свободный отрицательный ион подобно тому, кап это происходит в случае оболочек типа инертных газов. Точнее, непосредственно ив спектров меди, серебра и золота оказывается, что даже при слабом возбуждении, помимо термов, подобных щелочным металлам, имеются также еще другие системы термов.. Опи образуются оттого, что в возбужденных атомах, помимо наиболее рыхло связанного электрона (s-электропа), в возбужденном состоянии находятся также еще одпп или несколько из десяти d-электропов.
Эти системы термов у меди и у золота проявляются существенно ярче, чем у серебра, у которого соответствующих линий мало и сии имеют незначительные интенсивности. Сравнительно рыхлая связь электронов с побочным квантовым числом i =1 (а'-электроны), которая является причиной появления этих систем термов, приводи? к тому, что элементы подгруппы меди могут отщеплять более чем один электрон,. Существенно более сильное проявление «многовалентности» у меди и золота в сравнении с серебром полностью соответствует спектрам этих металлов. Следовательно, атомные модели, построенные на основании спектров, выражают как аналогию элементами главной й побочной подгрупп первой группы, так и их различие, силы?" проявляющееся в их химическом поведении.
То, что в атомах меди, серебра л залога d-оболочки полностью заполнены, следует из символов термов, соответствующих их основному состоянию, т. е. наинизпгему энергетическому уровню. Символы термов приведены в последней колонке табл. 37.. Так же как у щелочных металлов, у металлов подгруппы меди в основном состояшш атома общее орбитальное квантовое число L = 0 и общее квантовое число J = г г (/ — квантовое число, результирующее орбитальные и спиновые моменты) *. Следовательно, все вращательные моменты электронов взаимно компенсируются, за исключением того, который зависит от спина s-эд ектр опа, находящегося па внешней оболочпх (его орбитальное квантовое число 7=0). Вследствие этого мульти плотность равна 2, т е все спектральные линии, которые возникают При переходе на основной уровень внешней оболочки, образуют дублеты, так же как у щелочпых металлов (ср. т. стр. 282 и слэ.
В табл. 38 сопоставлены еще некоторые физические копстанты элементов ecz-групиы меди.
Медь, серебро и золото диамагнитны. Их удельная магнитная восприимчивость j при комнатной температуре составляет соответственно: —0,085 • 10“в,—0,192 10“-—0,143 • 10~е. Отсюда для атомных магнитных восприимчив остей оказываются xzsxz-пня соответственно; —5,40 • IO-6, —20,7 • 10“6, —28,3 10~®.
* См. т. I, стр. 283.— Прим. реЗ.
Побочная, подгруппа первой группы периодической системы 389
Таблица 3S
Важнейшие физические константы элементов подгруппы меди
Элемент Удельная электропроводность при 18Q Теплопроводность при 18°, па л/см- сек град Атомная теплоемкость, кол Теплота плав Денин (при т. пл.) Теплота сублимации при 0° К, ккпл/г-атом
кал/а ккал/з-атполг
Медь 57,2-104 0,989 5,82 42 2,65 81,2
Серебро 61,4-Ю4 1,006 6,01 25 2,66 68,0
Золото 41,3-104 0,700 6,17 16 3,16 -92
Металлы подгруппы меди кристаллизуются не как щелочные металлы — в сбъем-яоцентрировэнной кубической решетке, а в грацецентрированной (т, I, стр. 238, рис, 46). Длины ребер элементарной ячейки а составляют (при 20*):
Си Ag Аи
а=3,6147 4,0361 4,0786 А.
Кажущиеся атомные и ионные радиусы меди, серебра и золота сопоставлены в табл, 39, В то время как серебро и золото, а также медь и золото в любом соотноше*
Таблица 39
Кажущиеся атомные и ионные радиусы элементов побочной подгруппы первой группы (приведенные ионные радиусы относятся К одновалентным ионам)
Медь Серебро Золото
Атомный радиус, А Иоппый радиус, А 1,2780 -1,0 1,4447 1,13 1,4420
нии образуют между собой твердые растворы, медь и серебро способны к образованию твердых растворов только в очень ограниченных соотношениях (подробнее см, стр. 29 и 34),
В твердых растворах меди и золота впервые удалось наблюдать появление -ксверхструктурных фаз», которые были рассмотрены на стр. 31 и сл, В соответствии с результатами рентгеноструктурных определений вблизи составов АпСп и AuGus
Таблица 40
Растворимость некоторых соединений меди, серебра и золота при комнатной температуре
(лоль/л)
Медь (I) Серебро (I) Золото (I) Медь (II) Золото (III)
Хлорид 1,1-Ю-з 1,31-10'6 Разлагается Легко растворима Легко растворимо
Окись Практически перастйо рнма 1,03-10-4 ~0,2-10-ю 6,8-10-s Практически нерастворимо
Сульфид 3,1-10-5 1,8-10-1? <10“5 3,5-10-5 Разлагается
390
Глава 8
при обычной температуре нормально существует упорядоченное распределение атомов в решетке, а при температурах выше 385° — неупорядоченное распределение. Следовательно, если медленно охлаждать сплавы состава АнСп и АнСн3 от температур выше 385° до более низких температур или продолжительное время нагревать быстро охлажденные сплавы при 370° так, чтобы при более низких температурах возникло стабильное распределение, то эти сплавы будут давать рентгеновские интерференции, которые характерны для упорядоченного распределения атомов. Из этих интерференций следует, что у сплавов состава AuCu3 все вершины элементарных кубов (ср, т. I, стр. 238, рис. 46) заняты атомами Au, а все центры граней — атомами Си. Как следует из данных рентгеноструктурного анализа, у сплавов состава АнСн, помимо вершин элемен-
Образование соединений и твердых растворов металлами побочной подгруппы
(Обозначения те же,
Li Ь'а Be Mg Ch Sr Ba
Си тв. О 0 Сплав 0 тв. < оо Ве2Си 1206“ ВеСи тв. О Mg2Cu 568* MgCu2 820* TB, co ca^cuQ? CaCu4 935“ —: Сплав f
Ag тв. О Li3Ag 450° LiAg 9 55° тв. 0 0 ТВ* < оо" Be2Ag* 960° ТВ, < со Mg3Ag* 492° MgAg 820* тв. О Ca3Ag? 555“ CaAg 665“ CaAg* 59 5“ CaAg3 725° CaAg* 683“ тв. 0 Sr3Ag2 6B6“ SrAg 680“ Sr;jAgs 7 57“ SrAg4 781“ Ж, <co тв. О? Ba3Ag? (584“?) Ba4Ag3? (585’?) Ba2Ag3 846“ Ba3Ag£ 79 7“ BaAg4 729°
Сплав Соединение тв. О тв. ~ О тв. > 0 Mg3Au 830“ Mg5Au2?* 796“ Mg2Au 796“ MgAu 1150“ tb. > 0 Ca2Aii* 798“ CaAu 1014“ — Ba2Au3 BaAua и др.
Аи Na2Au NaAu2 990“ Ве5Ац Вег Ап ВеАи 730“ ВеАнг 646“ | BeAuf 586°
К CaAu? 864“
тв. О КАнг KAu4 CaAu£ 853“ CaAu4 880“
Побочная подгруппа, первой, группы периодической системы
391
тарных кубов, атомами Au заняты еще и центры двух противолежащих граней, а остальные центры граней заняты атомами Си (Borelius, 1925—1928). Медленно охлажденные или выдержанные около 385° сплавы золота с медью того же состава дают острый пик на кривой зависимости электропроводности от состава сплава (Курна-ков, 1916). Сплавы, которые были быстро охлаждены или нагреты выше 385° и затем подвергнуты закалке, пе обнаруживают этого явления. Они дают интерференции, характерные для 0 были ой гранецентрированной решетки, т; е. для твердого раствора с совершенно неупорядоченным распределением атомов. В соответствии с представлениями Курникова в случае сплава, отвечающего,формуле АиСи, превращение при 385° проявляется также в появлении участка замедления па кривой охлаждения.
Таблица 41
первой группы с элементами главных подгрупп первых четырех групп
что и в табл. 9, стр. 66)
1 .1 в А1 Gil In Tl s, Ge 8л
о о - * © g g ТВ, < СО CiijAj 1047“ CujAj* 1016“ Си Al* 626° : CuAl* 590’ ТВ, < CO ТВ. < co Ж. < co тв, 0 0 ТВ, <CO CuSi^f 852° тв. < co Cu4Ge? ; 828“ Cu3Ge* 700’ тв, < co Cu^Sn* Gu3iSng Cu3Sn CusSn? 410“ IK; < eo тв. 0 Q
CusGa* 909“ Ch4Jh 715“
Cu9Ga4 837“ Cu9In4 ? 685“ Cu^In* 688“ Cu5si □ 726“ CuSl0 щ 824’
Си । ,3Ga* 467’ CikbsGa* 249°
Cu2ln?* 671“ Cii013ra 310“ Cu45Si4[J? 6i0“ Cu3Si 859’
ж. —О тв. О 0 ТВ. < со тв, < cd Ag3Ga тв. < co 3 интерметалли- ческие фазы тв. > 0 0 тв. > 0 0 тв . > 0 0 ТВ. <00 О : Л О ft
Ag3Al* 772“ Ag0Sn* 724’ Ag3Srt* 480“
Ag5Ga* 619“ Ag2Ga*? 326*
Ag2Al* 7.28’ 698“
тв. > О Аи4А1* Аи2А1 624“ AusAl3?* AuAl* 625“ AuAI2 1060“ тв. > 0 тв. > О тв. 0 0 td о V о тв. 0 0 : та. > 0 тв. О Ап2РЬ* 418“ АпРЬ? 254“
А Из дСа* 352° Aujln* 647“ Au^n* (z>5) AuSn 418’ AuSn? 309“ AuSaf 252“ '
AU2.3Ga[] 286“ AuGa 468“ AuGa2 492“ Ап31п* 488“ Au, I nJ 484“ Auln 506“ Auln2 544“
392
Глава 8
В следствие небольших атомных радиусов ионы металлов побочной подгруппа: первой группы оказывают значительно более сильное поляризующее действие, че~ иопы щелочных металлов. Поэтому их соединения с отрицательными ионами, еслс они безводны, имеют значительно менсо выраженный солеобразпый характер, чет соответствующие соединения щелочных металлов. Многие из пих но своим свойствам и поведению близки к гомеополярным соединениям. Некоторые из них, возможно, даже следует рассматривать как чисто гомеополярные соединения (ср. стр, 407 и сл,].
Так как атомы металлов побочной подгруппы первой группы имеют тодькс один песпарепный электрон, то следует ожидать, что в чисто гомеополярпых соединениях они должны быть преимущественно одновалентными. Они могут осуществлять более чем одну атомную связь только тогда, когда по меньшей мере один электрет переместится с cZ-уровня на следующий вышестоящий р-уровень,; Требуемая для этого работа весьма значительна. Опа намного больше, например, чем работа, которая необходима для возбуждения атома углерода (ср. т. I, стр. 452). В случае меди работа, требуемая для перехода Зй104у -> 3d94s3p, составляет примерно 6.5 аг; в случае серебра для перехода 4d1G5s(21S'1/2) -* 4dB5s 5р(4Р3/ ) — 6,97 sa и в случае золота для перехода 5d108s —»- 5<79 6s 6р — более чем 7 эв, в то время как у углерода для перехода 2s22pa (3Ро) —> 2s2p3 (55а) необходимо только 4,16 ав.
Очень вероятно, что к чисто гомеополярпым соединениям относятся летучие гидриды Cull, AgH и АиН, которые, как было показано спектрографически, образуются (в минимальных количествах) при высоких температурах. Образование гидрида золота было доказало также Фаркасом (Farkas, 1929) на основании того, что в струе водорода при температуре~ 1400s золото летит существенно сильнее чем в струе азота или гелия. Аналогично ведет себя и серебро; однако в этом случае эффект менее заметен.
Твердые гидриды, получаемые действием атомарного водорода на Си, Ag или Ас (ср. стр. 410, 423 и 441), имеют, вероятно, солеобрагиый характер, В противоположность газообразным гидридам, которые при высоких температурах могут находиться в незначительном количество в равновесии с металлом и водородом, твердые гидриды нестабильны (при обычной температуре мета стабильны). Сравнительно большую устойчивость проявляет твердый гидрид серебра, в то время как среди летучих гидридов самым устойчивым является гидрид золота.
Относительно другого, препаративно более доступного гидрида меди состава СиН. ем. на стр. 410. О гидриде серебра, получаемого в форме двойного соединения,, см. на стр. 423.
Растворимости некоторых соединений меди, серебра И золота даны в табл. 40, (стр. 389).
Сплавы. Как следует из данных табл. 41—43, металлы побочной подгруппы I группы отличаются особой склонностью к образованию сплавов. В таких сплавах образуется множество соединений. Эти соединения чаще всего обладают характерец недальтонидиых соединепий, подчас с очень широкими областями гомогеппбети. Многие из таких соединений принадлежат к типу 1Ом-Розерн (ср. стр. 39 и сл.), в которых металлы побочной подгруппы I группы всегда обладают одним валентным электроном. Образуются также и многочисленные другие соединения, для которых закономерности их возникновения еще пе выяснены.
В поведении рассматриваемых металлов не проявляется ярко выраженных особенностей по сравнению с металлами главных подгрупп, с одной стороны, и с металяамп побочных подгрупп II и III труппы, с другой стороны. Однако их поведение отклоняется от поведения металлов побочных подгрупп IV—VIII групп, т. е. переходных элементов (помимо элементов побочной подгруппы Ш группы).
Как следует из табл. 9 (стр. 66), 13 (стр. 108), 17 (стр. 141), 25 (стр. 227) и 32 (стр. 266), е переходными элементами образуются соединения лишь в незначительных случаях. Таблицы содержат лишь четыре примера образования соединений, если не считать сверхструктурные фазы.Значительной взаимной растворимостью в твердом состоянии с металлами Побочной подгруппы 1 группы среди переходных элементов обладают .по-видимому, лишь элементы побочных подгрупп VII и VIII групп. В этом с.тучае растворимость часто даже ио ограничена.
Поведение металлов побочной подгруппы I группы в отношении металлов побочной подгруппы VIII группы напоминает их поведение в отношении друг друга, рассмотренное г уже на стр. 389 и сл. В обоих случаях, несмотря па достаточно сильнее различие атомных радиусов, наблюдается неограниченная растворимость; при этом часто образуются сворхструктурпые фазы, так же как и в сплавах Ан-Си, Проявляющееся здесь сродство между металлами побочных подгрупп VIII и I групп выражается также в их общей способности функционировать в соединениях типа Юм-Розерп в качестве металлов I типа.
Относительно бронз и близких пм сплавов см. стр. 402 и сл., ср. также стр. 35 и сл.
Побочная подгруппа первой группы периодической системы ЗЙЗ>
«
Таблица 42
Образование соединений и твердых растворов металлами побочной подгруппы первой группы с элементами главных подгрупп пятой и шестой групп
(Обозначения те же, что и в табл. 9, стр. 66)
р Ав Sl> ni S Se Те
Си ж. < со тв. > О Си3Р 1018° СиР2 тв. > О CiigAs 83 5“ CujAsJ? 710°“ тв. > О CugSb2 Ц 469° Cu3Sb 681° Cu,Sb* 58Ь° тв. — О 0 ж. <оо тв. О Cu,S 1130° CuS ж. < co тв. О Cu2Sc 1113° CuSe метаста-бильпое Cn2Se3 ме та стабильное CuSe и Cu2S с3 выделяются только из водного раствора ж. < оо тв. О Си3Те* 623° Си2Те 855°
Ag яс, < со тв, — О AgsP метастабильное AgP3 Ag3P5 метает а-бильное AgP3 тв. > О AggAs*? 595° ТВ. < со AgSbJ-le 562& Ag3Sb* 7O5g тв. О 0 ж, < ОС тв, О? Ag2S 842е Ж, < со тв. О? Ag2Se ж, < со тв, О Ag4Te метастабильное AgTe метаста* бильное Ag3TeJ 46 5° Ag2Te Во8°
Ап тв. О Аи2Р3 Сплав Au3As? Au4As3? AuAs? тв. ~ О AuSb£ 460° тв. > О Au2Bi* 373° 1 Au2S AuS Au2S3 Au2Se3 тв. О АпТе, 464°
Исторические сведения. Медь, серебро и золото относятся к очень давно известным металлам. Медь в виде бронзы служила уже в древние времена для изготовления оружия, инструментов и посуды (бронзовый век). Серебро и золото были общеизвестны и ценились в самые древнейшие времена, о которых имеются сведения. Из свода законов египетского царя Мейеса, написанного за 3600 лет до и. э., известно, что в то время
394
Глава S
Таблица 43
Образование соединений и твердых растворов металлами побочной подгруппы первой группы е металлами побочных подгрупп трех первых групп (Обозначения те ate, что и в табл. 9, стр. 66)
Ag Au Zn Cd Hg La
Си тв. > О 0 ТВ. со [AuCu] [AuCu3] ТВ* <^со GuZn* S05° CujZng 833° Cu0j39Zii * 700* CuZn3* 594°, тв. > 0 Cu2Cd* 549= Cu4Cd3 Cu5Cd8 563= CuCd3* 397= ТВ* <Z co CuHg 96,2* и два других соединения тв. О LaCu * 551 = LaCu2; 83 4= LaCu3* 793= LaCu4 902=
Ag ТВ, со 0 ТВ- <co ТВ* < co тв. < ф Ag4Hgs ? * 276= тв. 0 LaAg 885= LaAgu* 864= LaAg3 &5&e
AgZn 710° AgCd * 722=;
Ag5Zn8* 665° Ag5Cd8* 640= Ag5Hg8* 127=
AgZn3 635= AgCd3* 592=
Аи ТВ. < co [Aa3Zn] 425 = тв. < co ТВ* < со тв. 0 La2Au * 665= LaAu 1360= La An a 1214= LaAu3 1204“
[Au3Cd] Au3Hg* 421 = Au2Hg3* 310“ : AuHga* 124=
AuZn 725= AuCd 627°
Au5Zns 644s AllZtlg* .475= AasCd3 AuCd3
относительная ценность серебра к золоту составляла 1 : SVg в пользу серебра. В Ветхом завете, а Также у Гомера имеются упоминания о всех трех металлах.
Финикийцы обладали золотыми приисками на острове Тазос, серебряными рудниками в Испании и медными конями на Кипре (Суретп). Поэтому медь римляне называли cyprium, откуда позднее образовалось cuprum.
МЕДЬ (CUPRUM) Си
Распространение в природе. В виде соединений медь встречается во многих местах и в больших количествах; самородная медь, напротив, в больших количествах встречается редко (например, на Верхнем озере в Северной Америке). Чаще всего Си встречается в виде сульфида. Сульфид меди(П) GuS (ковеллин) имеет, правда, небольшое значение. Напротив, сульфид меди(1) Cu2S (медный блеск) является важным медным мине
Побочная подгруппа первой группы периодической системы
395
ралом. Наиболее распространенной ;и практически важной медной рудой является медный колчедан (халькопирит) CuFeS3. В Америке вместе с колчеданом встречается очень часто борнит Cu3FeS3. Вообще двойные сульфиды меди и других тяжелых металлов в природе очень широко распространены. Так, не последнюю роль играют, например, для американской добычи меди блеклая руда (двойные соединения Cu2S преимущественно с Sb2S3 и As2S3) и бурнонит (Cu2,Pb)3[SbS3]2. Другими иногда также важными рудами являются основные карбонаты: малахит СнСО3- Сн(ОН)2 и азурит 2СиСО3-Сн(ОН)2, а также окись Сн3О (куприт).
Небольшое значение имеют силикаты меди, среди которых следует упомянуть хриеокол CuSiO3 • 2НаО и диоптан CuSiO3-H2O, используемый иногда в качестве драгоценного кампя (о структуре см. т. I).
Энаргит Си3Аз8^, фаматинит Cu3SbS4, тенорит СиО н атакамит СпС12 • • 3Cu(OH)2 Tipедставляют существенный минералогический интерес.
Иногда медные минералы встречаются в виде мельчайших частичек, примешанных к другим породам, как, например, в слоистых мергелях, окрашенных в черный цвет вследствие большого содержания битума (на юго-востоке Гарца).
Следы меди содержатся также в живых организмах и прежде всего в костях и зубах (Tiede, 1934). Содержание меди в вещество зубов составляет ~ 0,001%. Присутствием меди объясняется окраска костной золы в цвета от розового до бледнофиолетового, которую она приобретает после очень сильного прокаливания (Gabriel, 1894). Такое окрашивание появляется в результате выделения в коллоидном состоянии Сп2О, которая образуется выше 800ч при термической диссоциации СиО. Еще не ясен вопрос, играет ли роль катализатора медь при биологических процессах в живых организмах. Относительно ядовитых свойств соединений меди см. стр. 402.
Получение. Для получения меди имеют значение преимущественно сульфидные руды. Их переработка основана на том, что сульфид меди прежде всего переводят в окись и последнюю затем восстанавливают углом до металла. Однако, так как медные руды обычно сильно загрязнены посторонними примесями, этим операциям должно, как правило, предшествовать обогащение меди.
; Окисные руды, которые чаще всего встречаются только как второстепенные спутники сульфидных руд (из которых они возникли в результате превращений), как правило, не перерабатывают отдельно, а включают в процесс переработки других руд после их переведения в окись меди.
Переработка сульфидных руд распадается на следующие процессы: 1. Обжиг
Этот процесс предназначен для удаления части серы. Чем дольше производят обжиг, тем большая часть сульфидов переходит в окислы. Окислы мышьяка и сурьмы улетучиваются при обжиге. Если при обжиге содержание серы понизится так, что в общем в руде на один атом меди останется только еще примерно один атом серы, то далее следует плавка.
2. Плавка на штейн
При этом продукт обжига, который обычно содержит много сульфида железа, сплавляют в шахтных или пламенных печах. Для этого к нему добавляют кокс и, если требуется, образующие шлак флюсы. В этом случае медь, превратившаяся при обжиге в окись, переходит в сульфид меди(1) по уравнению
2GuO + FeS+G+SiO2=Cu2S4-FeSiO3.+ CO*. (1)
Образующаяся при этом одновременно окись железа в виде силиката переходит в шлак, Так же как и окись железа, другие окислы менее бла-
* Это только одна из многочисленных, идущих параллельно реакций.
38fi
Глава 8
городпых металлов образуют силикаты и переходят в шлак. Образующийся сульфид меди(1) Cu2S соединяется с оставшимся сульфидом железа, давая Cu2S-FeS. Это соединение выделяется в виде «медного штейна> под слоем шлака.
Если реакционная смесь содержит еще мышьяк или сурьму, то последние выделяются в форме аналогичных соединений как «шпейза». Если присутствуют никель и кобальт, то они переходят либо в шпейзу (если она образуется), либо в штейн.
Б случае медных руд, богатых пиритом, обжиг и плавку иногда объединяют в один процесс («пиритная плавка»). В этом случае горение содержащегося в руде дисульфида железа дает тепло, требуемое длн нлавни,
3- Переработка медного штейна на черновую медь
Эту операцию производили прежде по немецкому способу — восстановите льнььм обжигом,, а позднее стали пользоваться преимущественно английским способом — обжигом, сопровождающимся реакционной плавкой. Однако в настоящее время оба эти способа едва ли еще применяют. Получение металлической люди из медного штейна производят теперь почти везде в конверторах путем обжига с дутьем. Этот способ называют бессемеровским процессом.
а) Восстановительный обжиг
Медный штейн подвергают обжигу до тех пор, пока сульфиды почти полностью не переходят в окислы (СцО и Fe2O3). Затем после добавления кокса и кварца производят плавление в шахтных печах; при этом медь восстанавливается до металла, а железо до окиси FeO, которая переходит в шлак в виде силиката.
б) Обжиг, сопровождающийся реакционной плавкой
В соответствии с первоначальной формой этого процесса медный штейн медленно сплавляли при доступе воздуха в пламенных печах:; при этом, как и при обычном обжиге, сульфиды переходили в окислы.. Однако пока еще процесс нс завершался, доступ воздуха прекращало. тогда окислы более легко восстанавливающейся меди могли вступать в реакцию с оставшимися сульфидами. В результате отщепления S(K образовывалась металлическая медь, например по уравнению.
2Cn2S-p3O2=2Cti2O+2SO2 (первая фаза: c&wua) ('
2Gu2O+C\i2S=SOi>-)-6Cii (вторая фаза: реакционная плавка) Г~.
Напротив, с большим трудом восстанавливающаяся до металл окись железа соединялась с добавленным кварцем в силикат:
FeO ф- SiO2=FeSiO3.
Повторением процесса получала «черную медь» с содержанием Си примерно 98%.
в) Обжиг с дутьем (бессемеровский процесс)
Применяемый повсеместно в настоящее время обжиг с дутьем (5ё~-сежройский процесс) развился из двух описанных выше способов. П~. обжиге с дутьел! поступают так: медный штейн (так же как в бессемер l ском процессе—сырое жолезо) в расплавленном состоянии помещу в копвертср, выложенный кислой или (если кислые флюсы —
в реакционную массу) основной более устойчивой футеровкой п Щ"
Побочная подгруппа первой, группы, периодической системы
397
пускают сжатый воздух. При этом сначала окисляется FeS, и получающаяся окись железа образует шлак. Затем начинается окисление части сульфида меди. Образующаяся при этом окись тотчас энергично реагирует с еще не претерпевшим изменения сульфидом до тех пор, пока«в соответствии с уравнениями (2) и (3) вся медь не перейдет в металл.
Для использования способа Бессемера в производство меди, существенным было то, что французу Майе (Manh.es) в 1880 г. пришла идея пропускать воздух в реактор не снизу, как при обычном бессемеровском процессе, а сбоку, иа некотором расстоянии от основания. Таким образом воздух поступал непосредственно в реакционную смесь, находящуюся в расплавленном состоянии вследствие протекающих в пей окислительных процессов, минуя охлаждающуюся и легко затвердевающую («замерзающую») медь, которая собиралась па дне. При получении меди путем обжига с дутьем часто вместо обычного грушевидного конвертера, применяемою в производстве железа, используют барабанный конвертер.
Получение меди путемавтогенногоплавления. Из трех описанных процессов, на которые делится производство меди (также и при использовании обжига с дутьем), два, а именно обжиг и конверторное производство, являются экзотермическими; при плавке, напротив, теплота поглощается. Возникла идея использовать теплоту этих экзотермических процессов. Некоторым образом это было достигнуто в случае так называемого способа пиритной плавки, который был применен в некоторых местах на рубеже нашего столетия; однако его применимость ограничена определенными медными рудами. Способом, применимым почти к любым сульфидным медным концентратам и обладающим, видимо, самым эффективным материальным и энергетическим балансам, является ^способ автогенной плав ни», разработанный недавно в Финляндии (Outokumpu Оу).
При способе автогеппой плавки обогащенную флотацией, хорошо высушенную и измельченную в пыль медную руду в смеси с флюсами, образующими шлак, загружают сверху в шахтную печь. Печь оборудована подогревателем, из которого подается горячий воздух. Оп поддерживает пылеобразную шихту продолжительное время во взвешенном состоянии и способствует одновременно частичному освобождению ее от серы, которая сгорает до S0a. Кроме того, практически всегда содержащийся в медных рудах сульфид железа окисляется до FeO, в то время как Cu2S при правильно регулируемой подаче воздуха не подвергается воздействию. От выделяющейся теплоты сгорания частички пыли сплавляются, и окись железа, образуя силикаты, переходят в шлак. Жидкие капли падают и собираются в пижпей части печи, причем в самом пизу оказывается жидкий медный штейп, а над ним жидкий желез о силикатный шлак.
Штейн и шлак периодически спускают. Штейи в расплавленном состоянии поступает в конвертор, где оп перерабатывается па сырую медь. Железосодержащий шлак также в жидком состоянии подается в электропечь. Здесь его покрывают смесью жжеппои извести и кокса, из которых при температуре электрической дуги образуется карбид кальция. Последний реагирует с силикатным шлаком с образованием обедненного углеродом Железа (стали) и основного силиката кальция, применяемого в качестве удобрения. Наряду с этим образуется СО, сгорание которой дает тепло, необходимое для известкового обжига. Горячие обжиговые газы, выходящие из шахтной печи, в которой сплавляется модный штейп, используют для производства тока для электрической печи. Кроме того, они служат для предварительного нагрева воздуха, подаваемого в шахтную Печь. И, наконец, после обеспыливания их применяют для производства серной кислоты.
Получение меди мокрым способом. Из бедных медных руд и из содержащих медь отходов (например., пиритных огарков) медь получают методом выщелачивания. Выщелачивание производят большей частью, обрабатывая сырье разбавленной серной кислотой, причем в случае необходимости после предварительного обжига сырья для переведения сульфида меди в окись и сульфат.
Из щелока медь можно выкристаллизовать в виде медпого купороса или осадить металлическим железом, а иногда также другими реагентами, например H2S. Однако в настоящее время медь в основном осаждают из растворов электролитически.
Очистка (рафинирование) меди. Медь, полученная различными методами, как правило, вначале еще сильно загрязнена. Рафинирование ее можно вести сухим или мокрым путем, в последнем случае электролитически. Электролиз (сам но себе дорогой процесс) выгоден только
398
Глава 3
тогда, когда медь содержит благородные металлы. Однако электролитическая медь получается всегда особо высокой степени чистоты, и поэтому в настоящее время применяют почти исключительно этот способ для получения меди, используемой в электрических машинах и проводах.
Рафинирование меди сухим путем производят следующим образом: медь плавят в пламенных кечах. При этом часть Си окисляется до Сц2О. Окись растворяется в расплавленной меди н отдает свой кислород содержащимся там неблагородным веществам. После этого оставшуюся Сн2О дезоксидируют внесением в расплав дерева или древесного угля («дразнение» меди) и получают рафинированную ~99,5%-ную медь.
Рафинирование мокрым способом производят электролизом. Черновую медь, служащую анодом, помещают в раствор сульфата меди, подкисленный серной кислотой, и подвергают электролизу; при этом медь с анода переходит в раствор и на катоде освящается чистая медь.
Необходимое для электролиза напряжение очень невелико, так как не требуется никакого напряжения разложения; следовательно, напряжение определяется практически только омическим сопротивлением раствора. При стандартных размерах ячейки для этого достаточно несколько десятых вольта при силе тока 1000 а. Поэтому обычно соединяют несколько сот ячеек последовательно.
Если принять, что ток силом 1 а за 1 мин осаждает 19,76 мг меди и что, следовательно, для осаждения 1 кг меди требуется 1000000/19,76 X 60 = 843,4 а-час, то потребление энергии на 1 кг произведенной чистой меди при напряжении на клеммах каждой ячейки 0,3 в оказывается 843,4 X 0,3/1000 = 0,253 кет-час. Потеря напряжения в токоподводящих проводах при этом в расчет не принята.
Металлы, стоящие в ряду напряжений справа от меди, следовательно, прежде всего серебро, золото и платина, при анодном растворении меди в раствор не переходят, а собираются в виде «анодного пглама» на дне. Возможность извлекать из «анодного шлама» указанные элементы имеет большое значение для повышения рентабельности электролитического способа.
Электролитическое рафинирование меди является первым электрохимическим способом, который стали применять в заводских условиях. Первое приложение его было осуществлено Элкингтонои в Англии уже два года спустя после открытия динамомашины.
Электролитический способ имеет также большое значение для регенерации меда из отходов и старых материалов. При этом поступают обычно так же, как при рафинировании сырой меди. Для извлечения меди из покрытого медью железного скрапа Шаарвехтер (Schaarwachter, 1939) разработал способ, который основан на анодном растворении меди в растворе, содержащем цианид натрия, и последующем катодном осаждепии (ср, стр. 401). Железо при этом практически не подвергается воздействию.. Из железного скрапа, пот; рыт ого латунью, удается таким образом непосреДственил извлечь латунь (ср. стр. 402).
Свойства. Медь — металл своеобразного красного цвета, просвечивающий в очень тонких слоях зеленовато-синим светом. Медь кристаллизуется в кубической системе (ср. стр. 389) и встречается в природе иногда в виде отчетливо образованных октаэдров и кубов. Чистая медь достаточно мягка (твердость пр шкале Мооса 3). Она очень вязкая, хорошо тянется. Медь отличается очень высокими электропроводностью и теплопроводностью (ср. табл. 38, стр. 389). Незначительные примеси (повышающие твердость) большей частью сильно снижают проводимость: прежде всего это относится й хрупким металлам мьнйьяку и сурьме, стоящим в периодической системе далеко от меди.
В металловедении имеется правило, говорящее о том, что легирующая до ба нее оказывает тем более сильное действие, чем дальше в периодической системе она удалена от главпой составной части.
Другие физические свойства меди приведены в обзорной таблиц в начале данной главы.
При обычной температуре компактная медь заметно не подвергается воздействию сухого кислорода воздуха. При нагревании медь тускнея?
Побочная подгруппа первой группы, периодической системы 399
в результате образования поверхностного слоя окиси. При более сильном нагревании она, наконец, полностью переходит в окись меди(1), а при более высоком давлении кислорода — в окись меди(П).
Влажный хлор быстро реагирует с медью уже при обычной температуре. С остальными галогенами медь также взаимодействует очень легко. Ярко выраженным сродством медь обладает по отношению к сере, а также к селену *. Напротив, газообразный азот, даже при более высокой температуре, заметно не действует на медь. Однако, если над медью, нагретой до красного каления, пропускать газообразный аммиак, то она образует соединение с азотом. Уже при температуре ниже красного каления медь взаимодействует с окислами азота, а именно NaO й NO, с образованием Сн2О, а с NO3 образует СиО. Тонкоизмельченная свёжевосста-новленная медь при температурах 25—30° связывает NO2 с образованием так называемой нитромеди Cu2NO2 — коричневой, устойчивой на сухом воздухе массы, которая энергично разлагается водой с отщеплением NO. С углеродом.- медь непосредственно не соединяется. Однако в виде своих соединений медь может замещать атомы водорода в ацетилене С2Н2. Смотря по тому, исходят ли при этом из соединений одно- или двухвалентной меди, получают Сп2С2 или СиС2.
G молекулярным водородом медь непосредственно не соединяется, если не принимать во внимание образования летучего гидрида, который в минимальных количествах образуется только при очень высокой температуре и может бЫть обнаружен исключительно спектрографически (ср. стр. 388). Однако медь вступает в реакцию с атомарным водородом (Pietsch, 1931); правда, при этом образуется (очень неустойчивый) твердый гидрид, [Особый твердый гидрид, содержащий воду, получают взаимодействием сульфата меди с фосфорной а тистой кислотой (ср. стр. 410 и сл.).) Способность растворять водород у меди незначительна. Растворенное количество пропорционально корню квадратному пз давления водорода и возрастает с температурой, В соответствии с измерениями Зиверта (Sieverts) 100 г меди при 409° и атмосферном давлении растворяют только 0,006 мг водорода. При температуре плавления (10835) то же количество меди может растворить в твердом состоянии 0,19 мг водорода, в жйдком состоянии ~~ 0,54 мг, а при 1550я—• 1,24 мг.
Медь может давать сплавы со многими металлами. С некоторыми, например с алюминием, медь соединяется со значительными тепловыми эффектами. Как уже было сказало, в сплавах меди встречаются многочисленные соединения.
Диаграммы состояния медных сплавовиногда довольно сложны. Например, в системе медь-алюминий встречается не менее восьми различных интерметаллических фаз. Среди них четыре являются стабильными при обычной температуре (ср, рис, 6, стр, 35). Медь может, не изменяя кристаллической структуры, включить до 10,5 вес,% А1, а алюминий до 0,5 вес. % Си **.
В системе медь-циик имеются четыре (а со сворхструктурной фазой р-латуни пять) интерметаллических фаз. При обычной температуре три из них стабильны. Они все обладают достаточно широкими интервалами гомогенности. Вез изменения структуры медь может включать до 39,5 вес%. цинка. Цинк, без изменения структуры решетки, может включать только 0,5 вес.% меди. На рис. 45 приведены области существования различных фаз в сплавах меди и цинка. Области, в которых существуют совместно различные фазы (области гетерогенности), заштрихованы. В сплавах медъ-олоео имеется 6 соответственно 7 интерметаллических фаз. При обычной температуре три из них стабильны, а именно соединения Си31Зий, Cu3Sa, Сщ;8п6. Все три имеют весьма узкие интервалы гомогенности. Однако последнее заметно меняет свой
* Из трех селенидов, указанных в табл. 42, только один получен непосредственным соединением составных частей; однако все три селенида встречаются в природе: Cu2Se как берцелианит, CuSc как клокманнит и Cu2Se3 как умангит.
** Здесь и далее приводимые границы насыщения ^'относятся к обычной температуре.
400
Глава 8
состав с повышением температуры. Опо плавится инконгруэнтяо. Два других соединения также при более высоких температурах нестабильны и претерпевают превращения в твердом состоянии. Медь может включить до 44,2 вес,% Sn, пе изменяя своей
Рис, 45. Диаграмма состояния сплавов медь—цинк.
а—твердый раствор Cu-Zn (кубический гранецентрированный); 0-CuZn (кубический о бьем но центрированный; 0' тоже с упорядоченным размещением атомов); у—CujZue (кубическая гигантская ячейка); б—выс.о ко температурная фаза с подробно нс изученной структурой (гексагональной); е—CuZti3 гексагональная плотнейшая шаровая упаковка); р—твердый раствор Zn-Cu (гексагональная плотнейшая шаровая упаковка, однако с другим соотношением осей, чем е; в случае q значение с/а падает, а в случае в возрастает с ростом содержания меди); (y+sj—эвтектическая смесь у4-е. образующаяся при распаде б.
структуры. Олово (белое), напротив, ие включает в свою решетку существенных когг?-чеетв меди. Сплав 94,5 вес. % олова и 5,5 вес. % меди затвердевает как эвтектичесЕгг смесь кристаллов Cu,3Sn5 и Sn. Как уже было указано в гл. I (стр. За и сл,), между сплавами моды с оловом, цинком и алюминием имеется близкое родство (см, таннг с,тр. 402), Очень проста диаграмма состояния сплавов меди о никелем. Опа пол посты?, соответствует схеме, приведенной на рис, 109, стр. 614, т. I, т. е. медь и никель нее в твердом, так и в жидком состоянии неограниченно растворяются друг в друге и ~ -образуют химического соединения.
В разбавленной азотной кислоте медь растворяется с выделением окиси азота и образованием нитрата меди(11); с горячей концентрированной серной кислотой образуется сульфат меди(П) *. В соответствии со ск>-
* Реакция протекает главным образом по уравнениям СкЦ- I-J.5SO4=СцО 4- ЗО^Ц-НзО, CuO4-II2SO4=CuSO4+H2O.
Однако эта основная реакция сопровождается побочными реакциями, которзз были сформулированы Фовлссом (Fowles, 1928) следующим образом:
4Сп Ц- SO2=Cu2S 4-2СиО;
CtigS Ц- H9SO4 =CU2SO4-|- H2S;
H2S H2SO4—S034-S -j-2H20)
S -I- 2Нг8О4=2Н2О -f- 3SO,;
CU2SO4 -J- 2H2SO4 - 2CuSO4 -)-2НчО 4-SO2.
Побочная подгруппа первой группы периодической системы
401
им положением в электрохимическом ряду напряжений (ср, т. I, стр, 52) медь не может обычным образом заряжаться водородными ионами. Поэтому медь в отсутствие воздуха пе подвергается воздействию разбавленных серной и соляной кислот, уксусной кислоты и т. д.
Однако при нагревании газообразный хлористый водород действует на медь с образованием хлорида меди(1):
Си. 1-НС1 CuCi-p/2IT2. (5)
В концентрированных растворах щелочных цианидов металлическая медь также растворяется, при этом выделяется водород:
Си + 2CN' + ПОП [Си i(CN)2] ' + ОН' -|- 1/2И2. (6)
Это происходит потому, что в результате склонности к комплексообразованию способность меди заряжаться положительно возрастает.
Можно предположить, что непосредственно вокруг металла создается равновесие
Cu+Н- Сн-+1/2И2. (7)
Однако обычно оно так сильно сдвинуто влево, что заметного растворения меди и выде-[еиия водорода не происходит. Существенный сдвиг равновесия вправо можно вызвать, создавая условия, при которых либо уменьшается концентрация ионов Си’, имеющихся обычно в растворе лини, в минимальных количествах, либо из раствора удаляется содержащийся там (ташке в минимальных количествах) водород. Первое можно достигнут!,, связывай иопы Си’ сильно комплексообразующими веществами, в;щример ионами CN'.
Если для связывания ионов меди применяют галогепид-иопы, которые образуют < Сп’ далеко не такие прочные комплексы, как цианид-иопы, то достаточно сильный сдвиг равновесия (7) вправо происходит только тогда, когда одновременно в очень сильной степени повышают концентрацию попов IT’. В еысококонцентрироеанных: галогеново дородных кислотах (исключая плавпкоауго) медь фактически растворяется г выделением водорода, несмотря на свое положение в ряду напряжений справа от водорода.
Другая возможность для сдвига равновесия (7) вправо (устранение элементарного водорода) — применение окислителей. Однако последние могут также действовать на медь непосредственно, отнимая у йен) электроны и переводя ее таким образом г, ионное состояние.
Растворение меди в разбавленной азотной кислоте сначала идет очень медленно. Однако, после того как в растворе в соответствии с уравнением
Си + 2Н‘ + КОд =Си- ’ + Н2О + NOj (8)
образуется некоторое количество нптрит-иопов, растворение становится бурным. Если с самого начала добавить к разбавленной азотной кислоте некоторое количество нитрита, то реакция сразу идет с полной силой. В таких случаях, когда возникающий при реакции продукт ускоряет реакцию, говорят об аутокатализе (ср. гл. 17).
Менее благородным металлом, например железом, медь вытесняется из ее растворов. Это используют иногда для технического получения меди (ср. стр. 397). Такое выделение называют цементацией, а полученную таким образом порошкообразную и большей частью сильно загрязненную медь называют «цементационной медыой.
В качестве моры благородности меди в электрохимическом смысле был уже ранее артпюден ее нормальный потенциал (см. т. I, стр. 51, табл. 4). ПластИШса меди, помещенная в раствор, который содержит 1 молъ/л ионов меди(1Г), имеет’ па 0,345 в более выгнкий потенциал, чем нормальный водородный электрод. Еще более высокий потенциал, а именно +0,521 в в отношении нормального водородного электрода, имела бы [пастила меди, если бы она была нагружена в раствор с такой же концентрацией Jb С. Реми
402
Глава 8
ионов меди(1). Следовательно, при одинаковой концентрации ионы медиф проявляли бы большую склонность к переходу в металлическое состояние с отдачей заряда, чей ионы меди(П). Таким образом, окислительная способность первых больше, чем вторых! Правда, в действительности ионы меди(1) всегда присутствуют только в минимальных концентрациях, поэтому вследствие зависимости электролитического потенциала от концентрации ионов истинное соотношение окислительных способностей оказывается прямо противоположным; практически всегда соединения меди(П) являются соединениями с более высокими окислительными способностями.
Медь можно электролитически осаждать вместе с существенно более сильными электроположительными металлами, например с цинком, если очень сильно понизить концентрацию ионов меди, используя процесс комплексообразования, что сдвигает ее потенциал разложения [в соответствии с уравнением (16) на стр. 54, т. I] в Сторону неблагородных металлов. Из раствора солей меди и цинка, содержащего большое количество цианида натрия, при электролизе оба металла осаждаются одновременно в виде латуни, так как в таком растворе напряжение осаждения меди значительно больше, чем напряжение осаждения цинка. Это объясняется тем, что комплексные ионы (Cti(CN)2!' прочнее, чем ионы [Zn(CN)s]' или [Zn(CN)4]". Этот факт используют для получения латунных покрытий на железных предметах для защиты их от коррозии.
По отношению к низшим организмам медь действует как сильный яд. Человеческому организму небольшие количества меди не причиняют вреда даже при длительном употреблении (ежедневный прием менее 0,1 г). Большие количества растворимых солен меди могут вызывать тяжелые заболевания; 2 — Зе их, полученные за один прием, действуют смертельно. Отравления продуктами, содержащими медь, очень редки, так как нища в этом случае обладает ярко выраженным неприятным Вкусом, который делает ее несъедобной.
Применение меди н ее соединений. После железа и алюминия медь является, пожалуй, самым важным металлом, несмотря па то что за последние десятилетия пз многих областей медь в значительной степени вытеснена другими, более дешевывд металлами. Однако она находит все еще разнообразное применение в виде ецлаваз для изготовления аппаратуры, частей машин и инструментов всех видов, а такжн художественно-промышленных изделий. В виде жести медь используют для кровли и обшивки кораблей. В основном медь используют в виде проволоки как материал,, хорошо проводящий электрический ток.
Из сплавов меди прежде всего следует отметить бронзы. Бронзы — это спляг-^ меди и олова, к которым часто еще добавляют для удешевления цинк и свинец. Бронха отличаются твердостью и прочностью, лучшие из пих — орудийные и колокольные бронзы. Последние состоят только из меди и олова. Общим свойством бронз являются» хорошие литейные качества. Бронзы, содержащие примерно др 10% олова, крскг того, хорошо куются и штампуются. Такие бронзы, в соответствии со сказанным пс стр. 35, состоят исключительно из твердых растворов Cu-Sn s. Чтобы предотвратить окисление меди и олова при плавлении и литье бронз соответственно, чтобы восстановить образующиеся окислы снова до металла, перед литьем к бронзам добавляют иногда фосфор в качестве раскислителя (в виде фосфористого олова или фосфористой меди . Полученные таким образом фосфористые бронзы вследствие отсутствия в них окнелех Отличаются особой прочностью и сопротивляемостью **. При литье чистой иг—i в качестве восстанавливающего средства часто используют кремний. Полученную: таким образом медь, свободную от окйслов и содержащую следы избыточного кремншх называют обычно кремнистой бронзой, хотя опа и пе является бронзой в собстпопн—т смысле, т. е. сплавом меди и олова (ср. также т. I, стр. 286). Вместо оловянных брп— все в возрастающем количестве применяют алюминиевые бронзы. Они содержат, как правило, 55—75% Си, 22—42% Zn и 2—20% Д1. Многие из них обнаруживав’.?
* Содержание олова в бронзах, используемых для холодной обработки, прапге-чески лежит несколько ниже, чем зто соответствует границе растворимости при обычц;£: температуре (14,2%). Это обусловлено том. что границы растворимости с ростом температуры сдвигаются в область бедных оловом сплавов (при 792° она лежат при 7,5S„ и что у сплавов, сравнительно быстро затвердевающих из расплава, пе достигает?? равновесия, соответствующего обычной температуре.
’* Готовые фосфористые бронзы содержат, как правило, только следы фосф?.?; так как его обычно добавляют в таком количестве, какое требуется для вссстаЕззп--ния окйслов.
Побочная подгруппа первой группы периодической системы
403
при надлежащей обработке свойства, превосходящие свойства обычных бронз. Широко применяемыми металлами являются томпак- и латунь. Опи представляют собой сплавы, состоящие в основном из меди и цинка, часто с добавками олова, свинца и железа. Они более дешевы, мягче и легче обрабатываются, чем бронзы. При содержании цинка менее 18% они обладают еще красноватой окраской (томпак); при большем содержании цинка (до 50%) эти сплавы имеют желтую окраску и называются латунью.
Сорта латуни с содержанием цинка примерно до 45% не только хорошо льются, но и хорошо обрабатываются холодным вальцеванием, прессованием и протяжкой. Сплавы меди и цинка, содержание цинка в которых лежит в интервале 38—45% (монетный металл), удается легко обрабатывать горячей вальцовкой и ковкой. В соответствии с диаграммой состояния сплавов Cu-Zn примерно в этой области концентраций при температуре красного каления появляется интерметаллическая фаза, называемая ^-латунью *. Ниже 470° эта фаза превращается в так называемую р'-латунь, которая отличается от (1-лату пи упорядоченным распределением атомов в узлах решетки **. Очевидно, что наблюдаемые у монетного металла свойства (поддаваться горячей обработке и коваться) находятся в тесной связи с образованием р-лату-ни. Сплав, содержащий примерно 30% меди и 65—70% никеля (монельметалл), с т. пл. 1410°, который получается непосредствен по восстановлением нйкс.тево-медного штейна (см. стр. 331), отличается кислотоустойчивостью, большой прочностью испособ-ностьго хорошо обрабатываться. Для изготовления столовых приборов и более мелких украшений ранее часто применяли нейзильбер (альпака, альфепид) — сплав, содержащий 50—70% меди, 13—25% никеля, 13—25% цинка и иногда небольшие количества свинца, олова или желоза. Следует упомянуть еще монетные сплавы. Большинство медных монет содержат 95% меди, 4% олова и 1% цинка. Никелевые монеты состоят из меди и никеля или из чистого никеля (ср. стр. 333), Сплав Деварда, состоящий из 50% меди, 45% алюминия и 5% цинка, вследствие своей хрупкости легко дает порошок; его часто применяют в лаборатории в качестве восстановителя.
В препаративной неорганической и органической химии, а также в технике медь часто используют как катализатор.
Как металлическая медь, так и ее соединения отличаются каталитическими свойствами. Под их воздействием особенно ускоряются окислительные процессы. Известным примером этого является окисление хлористого водорода кислородом воздуха под влиянием соединений Меди в дикон-процессе. Довольно часто применяют соединения меди в качестве переносчиков кислорода, например для окисления органических красителей. Если над нагретой медной спиралью пропускать метиловый спирт в смеси с воздухом, то спираль остается раскаленной вследствие образования формальдегида со значительным выделением тепла но уравнению
СН3- ОН•+ ч/зОг=СН20 + Н30.
Медь способна переносить кислород воздуха еще на многие другие вещества. Окисление связанным кислородом часто ускоряется также солями меди. Поэтому, например, при определении азота по Кьельдалю, при котором идет окислительное разложение органических веществ сорной кислотой, часто добавляют немного сульфата меди в качестве катализатора. Восстановительные процессы также могут каталитически ускоряться медью.
Следует указать в этой связи многочисленное использование галогене- и цианосоединений меди(1) в препаративной органической химии в реакциях Запдмайера (перевод диавосоеди пений в галогене со единения и т. д.).
Многие соединения меди используют в качестве красок. Измельченный малахит (ср. стр. 416), а также искусственно приготовленные препараты аналогичного состава находят применение как клеевые или водяные краски под названием медной зелени.
Размолотая медная лазурь (см. стр. 416) представляет собой великолепную медную краску, однако малоустойчивую. Чаще употребляют
* При 800я область 0-латуви лежит в пределах 38—53% ципка. При 700° — в пределах 42—49% цинка. С понижением температуры границы области все более и более сужаются.
** Превращение одного типа в другой проявляется на кривой охлаждения и обнаруживается также благодаря наличию аномалий в изменении электропроводио сти, удельного объема и других свойств при охлаждении или пагревапии.
26*
404
Глава. S
бремерблау, представляющую собой объемистую гидроокись меди(П), приготовленную особыми методами. Другой очень хорошей краской является швейнфуртская зелень (см. стр. 417). Однако вследствие содержания мышьяка она очень ядовита и поэтому запрещена, так же как зелень Шееле — арсенит меди(П) переменного состава,
Другие соединения Меди находят применение в медицине и в качестве инсектицидов. Например, для защиты от пероноспороза различных культур и от фцтофтороза картофеля применяют бордосскую жидкость (ср. стр. 414). Против заболеваний злаков, например ржавчины, применяют медноборатную или меднофосфатную жидкость, которые получают растворением медного купороса и буры или фосфата патрия в воде.
Хлорид меди(1), растворенный в концентрированной соляной кислоте или в водном растворе аммиака, служит в газовом анализе для поглощения окиси углерода. Кроме того, его используют для очистки ацетилена. Хлорид меди(П), окись меди(1) и окись меди(П) применяют для окрашивания стекол и эмалей (СнгО окрашивает в красный цвет, СнО — в зеленый иля голубой). Аммиачные растворы окиси меди используют при производстве искусственного шелка. Одной из наиболее часто применяемых солей меди является медный купорос (см. стр. 414).
СОЕДИНЕНИЯ МЕДИ
Практически важные соединения меди производятся отчасти от одно-валентноп меди (первая степень окисления металла), отчасти от двух-валентной меди (вторая степень окисления). Соединения меди(П) в общем устойчивее. Удается также приготовить соединения, в которых медь существует в третьей степени окисления. Однако последние имеют только теоретический интерес.
Соединения одновалентной меди, папримср красная окись мсди(1) Сп2О, серый сульфид меди(1) Cu3S и простые соли СнХ в воде практически нерастворимы, однако часто растворяются в результате комплексообразования.
Комплексные соединения, так же как и простые соли одновалентной меди, большей частью совсем или почти бесцветны. Исключительной способностью растворов солей одновалентной меди является растворение окиси углерода в результате комплексообразования.
Помимо черной окиси СчО и синей гидроокиси Сн(ОН)2, двухвалентная медь образует многочисленные соли CuX2, средн которых соли с сильными кислотами легко растворимы в воде и в разбавленном водном растворе практически полностью диссоциированы, с незначительным гидролитическим расщеплением. Гидратированные ионы Си'* имеют небесно-голубую окраску. Они довольно легко образуют комплексные ионы (большей частью с изменением окраски). Такие комплексы образуются как с отрицательными нонами (ацидо-комплсксы), например:
Со" -- 'iCl' = [CnCJ4]", так и с нейтральными молекулами, и прежде всего с аммиаком <л1‘ЧАХНз=[Си(ШТ3)41”.
Образованием комплексов с органическими веществами объясняется действие глицерина, сахара, винной кислоты, препятствующих многим реакциям осаждения меди. Эти вещества обладают несколькими гидроксильными труп памп, а медь способна замещать атомы водорода в таких гидроксильных Группах
Побочная подгруппа первой группы периодической системы
405
FT;iпример, она может в щелочном растворе взаимодействовать с. попом вицпой кислоты [С4114О6]" по уравнению
2НгО
н о
г----- —; I I!
Си--|—OH-j-H—I—О—С—С—О'
1____ J I
Na-f-OH + H-1-O—С—С—О' ।------—-—। । ц
О Н
'О—с—с—о-:-н + но-!—
I
'О—с—о—0-Г-Н Ч-НО-1-Na и । I--------_---J
ОН но
II I I II
'0—0—С—О—Си—О—О—С—О'
I | 4- 4HSO.
'О—С—С—ONa NaO—С—С—О'
Эти и аналогичные комплексные ишпд связывают медь так прочно, что в таких растворах свободных ионов меди Си” в достаточном количестве для достижения произведения растворимости, например Си(ОН)2, не имеется. Выпадения Сп(ОН)2 поэтому не происходит. Производные от обоих приведенных комплексных ионов соли натрия — Na2|C4H2O6Cu[-211,0 и Na4[CsH4Oj2CuNa2]- 13Н3О — можно изолировать в кристаллической форме. (Правда, их строение еще не изучено во всех деталях.) При смешивании раствора медного купороса с сегпетовой солью KNa [C4H4O(jJ-4Н2О и гидроокисью калия образуется фелингова жидкость, которая служит реагентом на многие органические вещества (папримор, виноградный сахар). Опа содержит комплекс меди с винной кислотой.
Веществами, подобными виноградному сахару, медь фелинговой жидкости восстанавливается до одновалентной меди, которая выделяется в виде окиси меди(1).
Относительно соединений трехе алештюй меди см. стр. 118 и сл.
Со многими неметаллами, например Si, Р, Те, медь образует соединения, которые по своему составу совершенно отличаются от солеобразных соединений и, по-видимому, приближаются к интерметаллическим соединениям (см. табл. 41 и 42 стр. 390 и сл. и 393).
1. Соединения меди(1)
Окись меди(1) Сн2О встречается в природе в виде минерала куприта — плотной массы цвета от красного до черно-коричневого; иногда она имеет кристаллы правильной кубической формы (большей частью октаэдров) с удельным весом 5,75 — 6,09, Искусственно Сп2О получают добавлением натриевой щелочи и не слишком сильного восстановителя, например виноградного сахара, гидразина или гидроксиламина, к раствору сульфата меди(П) или к фелинговой жидкости. При осторожном нагревании сначала появляется желтый осадок, рентгенограмма которого свидетельствует о Том, что это гель тонко дисперсной окиси меди(1). Постепенно,
406
Глава 8
Р й с. 46. Элементарная ячейка куприта.
а=4,2в А.
а при более сильном нагревании значительно быстрее осадок окрашивается в красный цвет вследствие укрупнения частиц.
В воде окись меди(1) практически нерастворима. Она, однако, легко растворяется в водном растворе аммиака и в концентрированных растворах галогеноводородных кислот с образованием бесцветных комплексных соединений {Cu(NTI3)2JOH и соответственно Н [CuXJ (где X галоген).
В растворах щелочей окись меди(1) заметно растворима. Под действием разбавленных галогеноводородных кислот окись меди(1) превращается в галогенид меди(1), также нерастворимый в воде. В разбавленной кислородной кислоте, например серной кислоте, окись меди(1) растворяется, однако при этом распадается на соль меди(П) и металл:
См2О + H,SO4 =CuSO4+ Н2О + Са. (9)
Структура решетки куприта приведена па рис. 46. Атомы кислорода об разу jot объемы оцентри-роваппый куб с длиной ребраа= 4,257 А; атомы меди образуют тетраэдр с длиной ребра Кратчайшее расстояние Си «—у О составляет ^-Д/з=1,85А. Теплота образования окиси меди равна, по данным Вёлера, 43,0 ккал/молъ Си2О.
Окись меди(1) находит применение для
эмалей в красный цвет, а также для покрытия бы с вредителями растений.
окрашивания днищ судов и
техническое стекол и для борь-
Окись меди(1), загрязненная окисью моди(П), отделяется при вальцевании и ковке моди в виде медной окалины. Чистую окись меди(1) в настоящее время в технике получают обычно электролизом. Для этого пропускают электрический ток через раствор щелочного хлорида, в который погружена медная пластинка в качестве анода; медь в форме ионов [СпС12|' переходит в раствор, в котором она взаимодействует с образующимися па катоде конами гидроксила, давая окись меди(1):
2|.СиС12[' 2OII' =Cu2O + Н2О 4-4CI'. (10)
Галогениды мсди(З). Хлорид .wedu(j) можно приготовить нагреванием медных стружек с концентрированной соляной кислотой при добавлении некоторого количества хлората калия. Черный вначале раствор становится под конец совершенно бесцветным. При разведении из него выпадает соль, которая содержалась в нем в виде комплекса (как продукт присоединения ЙС1), представляющая собой белоснежный в чистом состоянии порошок, быстро окрашивающийся на влажном воздухе в зеленый цвет. Приготовление можно также осуществить восстановлением раствора хлорида медц(П) или смеси растворов сульфата меди (II) и хлорида натрпц сернистой кислотой. В воде хлорид меди(1) очень мало растворим (ср., табл. 40, стр. 392), напротив, он легко растворим в горячей концентрп-роваппойсоляной кислоте. СпС1 растворяется в растворах хлоридов щелочных металлов, а также в концентрированном водном растворе аммиака,
Растворы хлорида меди(1) в концентрированной соляной кислоте абсорбируют окись углерода, однако вновь отдают ее при нагревании Продукт присоединения СпСЬСО удается получить в кристаллической форме. Охлаждением насыщенного при нагревании солянокислого раствора СнС! с одновременным пропусканием газообразного НС1 удается осадить соединенно Н[СнС12] в виде перламутровых игл.
Побочная подгруппа первой группы периодической системы 407
Известны также двойные хлориды меди и щелочных металлов, например KCl-CuCI = К[СнС12]. В аммиачных растворах хлорида меди(1) существуют комплексные аммиакаты, например [Cu(NH3)3]CL Однако их трудно выделить в чистом виде. Помимо указанного аммиаката сухим путем можно получить в чистом виде соединения состава 2CuCl-3NH3 и 2CuCbNH3.
Хлорнд меди(1) способен также присоединяться к ацетилену. Из раствора хлорида меди(П) в безводном спирте ацетилен осаящает соединение состава С2Н3 6СнС1. Соединение состава С2Н2 • 2СнС1 выкристаллизовывается из раствора СнС1 в концентрированной НС1 при длительном пропускании ацетилена.
Пары хлорида меди(1) состоят из удвоенных молекул (СпС1)2, Между тем при определении молекулярного веса в некоторых растворителях, папример в пиридине, получаются значения, лежащие ближе к формуле CuCI. Как свидетельствуют микроскопические наблюдения, хлорнд меди(1) кристаллизуется в виде правильных тетраэдров. Рентгенометрическими исследованиями установлено, что в основе структуры CuCI лежит тип кристаллической решетки, который был впервые обнаружен у цинковой обманки и который поэтому называют типом цинковой обманки. Элементарная ячейка этой решетки состоит из гра-нецентрироваппых кубов, которые образованы атомами одного сорта (напрнмер, атомами Си) и в которые вписаны правильные тетраэдры из
Р и с. 47. Элементарная ячейка хлорида меди(1) (тип цинковой обманки). А.
атомов другого сорта (например, атомы С1;
ср. рис. 47). Если в решетке типа цинковой обманки все узлы решетки заместить атомами одного сорта, то получится решетка типа алмаза (рис. 83, стр. 461, т. I).
Бромид и иодид моди(1) построены так же, как хлорид медиН)., Длина ребра элементарного куба а составляет у CuCI 5,41, у СнВг 5,68, у Gul 6,05 А, Кратчайшие межатомные расстояния Д/з ^таковы: Си +—» С12,34, Си <—>Вг 2,46, Си > 12,62 А,
Эти расстояния здесь в отличие от типичных ионных решеток, например решеток галогенидов щелочных металлов, не равны суммам кажущихся ионных радиусов (2,81, 2,96 и 3,20 А соответственно). Это справедливо пе только для названных галогенидов медн(1), но также и для всех соединений, которые кристаллизуются по типу цинковой обманки. Далее это справедливо также и для веществ, кристаллизующихся по типу вюртцита (для которого была рассмотрена в качестве примера на стр. 290, т. I, окись бериллия), а также кристаллизующихся по типу куприта (ср. выше, стр. 406).
Решетки типа цинковой обманки, вюртцита] и куприта «несоизмеримы» (Goldschmidt) с типичными ионными решетками. Между тем они «соизмеримы»между собой и с решетками большинства свободных элементов. Гримм и Зоммерфельд (Grimm, Sommerfeld) выдвинули в 1926 г. предположение о том, что решетки типа цинковой обманки и вюртцита в качестве структурных единиц содержат неионизированные атомы. Если в случае хлорида, бромида и иоднда моди(1) речь идет о неирнизироваппых атомах, то можно ожидать, что (кажущиеся) атомные радиусы 01, Вг и I можно получить, если из кратчайших межатомных расстояний в соответствующих решетках вычесть (кажущийся) радиус атома меди, полученный при Исследовании решетки металла. Таким образом подучим: rci == (2,34—1,27) = 1,07 А;гВг — (2,46—1,27) =• = 1,19 A, = (2,62—1,27) —1,35 А. Так как иодид серебра также способен кристаллизоваться по типу цинковой обмапки, то атомный радиус иода можно рассчитать
также из его решетки. При этом получают п = (2,81—1,44) — 1,37 А, что удовлетворительно согласуется со значением, рассчитахшым на оснований решетки иодида меди(1). Конечно, существуют некоторые аргументы, свидетельствующие против гипотезы о построении сульфида цинка, окиси меди(1), хлорида меди(1) и т. д. из нононизировэнных атомов. Поэтому вопрос о том, можно ли объяснить на этом основании несоизмеримость решеток типа цинковой обмапки и т. п. с типичными ион-
ными решетками, еще является спорным.
408
Глава S
С образованием галогенидами меди(1) решеток типа цинковой обманки, связана также их способность образовывать продукты присоединения е РН3. Фосфин обладает существенно меньшим дипольным моментом, чем NH3, и вследствие этого присоединяется только особенно сильно поляризующими веществами, и в первую очередь такими, которые образуют молекулярную решетку, как, например, галогениды Ti^', Sn1' и Al (Holtje, 1930 и сл.). Далее оп присоединяется также галогенидами, которые Кристаллизуются в решетке типа цииковоп обманки, как, например, CuCl, CuBr, Cui, и осажденным из водного раствора Agl. Соединения CuCl- РН3, CuBr- РН3г CuI-2PH3 и 2AgI • РН3 были получены Шольдером (S ch о Ider, 1934) действием РН~ на спиртовые растворы галогенидов меди, сильно насыщенные галогеноводородом, прп низком температуре. Они легко разлагаются с образованием соответствующего фосфида. Кристаллизующиеся по типу каменной соли галогениды серебра, AgCl и AgBr, РН3 пе присоединяют.
Так же как в кристаллическом строении, в химическом отношении бромид и иоппу меди(1) Вполне соответствуют хлориду меди(1). Бромид меди(Г) образует бледно-аеде-нов а то-желтые кристаллики, которые плавятся примерно при 500°. Иодид меди(1) образует белый порошок, который, однако, большей частью окрашен в слабо-коричневый цвет примесью иода. Его получают нагреванием меди с иодом и горячей концентрированной иодистоводородной кислотой или в результате взаимодействия раствора сульфата меди с иодидом щелочного металла; при атом целесообразно также добавлять восстановитель, такой, как SO2 и Na2S2O3. Подобно хлориду Cu(I), СнВг и Cui также легко образуют комплексы с аммиаком, галогеноводородными кислотами и галогени-дами щелочных металлов, в концентрированных растворах которых они легко растворимы. Их растворимость в ноде еще меньше, чем у CuCl; при комнаткой температуре опа составляет 2,0-10-4 моля у CuBr и 2,2 • 10-в моля у Cui в 1 л. При действо: HF на CuCl при температуре выше 1000е образуется фторид меди(1), имеющий послг-охлаждения вид рубипово-красной прозрачной кристаллической массы, нерастворимой в воде. Как нашел Эберт (Ebert, 1933), CuF кристаллизуется также по типу цинк:: вой обманки: а = 4,255 А, Си > F — 1,84 А<
По химическому поведению галогениды, одновалентной меди теснс примыкают к цианиду и роданиду.
Цианид медп(1) CuCN (о получении см. стр. 416) представляет собой белый, соскх-щий из моноклинных призм порошок, нерастворимый в воде и в разбавленных кислотах. Он растворяется в концентрированных кислотах, кроме того, в водном расгвср;-аммиака и в растворах солей аммония (с образованием комплексных аммиакатог однако особенно легко он растворяется в растворах цианидов щелочных металл™ (с образованием двойных цианидов).
Известно достаточно большое число двойных цианидов, образованных пняпп-т меди(£). В гальваностегии растворы двойных цианидов щелочных металлов и медт-'~ применяют в качество ванн для омеднения.
Роданид меди(1) CuSCN проще всего получают добавлением ионов SCN' к р--твору соли меди(П), содержащему восстановитель. Он представляет собой бельф. порошок, практически нерастворимый в воде, а также па холоду в не окислят—— кислотах. В концентрированных растворах роданидов CuSCN легко растаорЕщ— с образованием двойных солей. Сухой роданид меди(1) абсорбирует на холоду ами—_ с образованием черного порошка состава 2Cu(SCN)- 5NH3, легко вновь отщецляикщг аммиак. Нз водного раствора удается получить Cu(SCN)- NH3 в виде белого крпспы лического порошка, который также очень легко отдает аммиак.
Ацетат меди(1). Если к горячей смеси аммиачного раствора ацетата медтс: и большого избытка ацетата аммония прикипать раствор сульфата гидроксилатчт-т-т— и затем смесь подкислить избытком уксусной кислоты, то выпадет ацетат ые— ’ в виде тонких белых игл. В сухом состоянии соединение достаточно устойчиво ц отношению к воздуху; водой оно разлагается.
Оксалат медп(1). Из раствора хлорида меди(1) в концентрированной соеешА кислоте щавелевой кислотой или оксалатом калия выделяется белый осадоК| нотцте! считают оксалатом одновалентной меди.
Сульфит меди(1) выделяется при пропускании двуокиси серы в сольно уцгу— кислый раствор ацетата меди(П) в виде белого кристаллического порошка, рагтЕц—-
Побочная подгруппа пергой группы периодической системы
409
мого в водном растворе сернистой кислоты. Он имеет состав Cu2SO,' Н2О. С сульфитами щелочных металлов он образует сравнительно легко растворимые в воде малоустойчивые двойные соединения.
Сульфид меди(1) Cu2S получается в виде черновато-свипцово-серого кристаллического порошка почти с количественным* выходом при прокаливании сульфида меди(П) в токе водорода с добавлением некоторого количества серы. В природе он встречается в виде ромбических кристаллов медного блеска', удельный вес его 5,785, температура плавления 1130°. Из расплава Cu2S затвердевает в кубических кристаллах. В этой форме оп встречается также в металлургических продуктах. Сульфид меди(1) достаточно хорошо проводит электрический ток, однако Хуже, чем сульфид меди(П). Полученный плавлением кубический сульфид меди(1) обладает проводимостью значительно меньшей, чем ромбический.
Кубический сульфид моди(1) подобно щелочным сульфидам Li2S и Na2S является антиизотипом плавикового шпата (ср. рис. 62, стр. 298, т. I); а = 5,56 А (при 170°). Однако, по данным Ральфса (Rahlfs, 1936), всегда запята лишь часть узлов решетки, принадлежащих атомам моди, так что кубический сульфид имеет состав Cu(.sS. Ромбический сульфид, напротив, имеет нормальный состав Cu2S. Он образует сложную решетку, которая сохраняется даже при более высоких температурах.
В воде, а также в растворе сернистого аммония сульфид меди(1) почти нерастворим. Однако другим путем можно получить комплексные двойные сульфиды, которые мояшо рассматривать как продукты присоединения щелочных полисульфидов к сульфиду меди(1). Например, прикапыванием аммиачного раствора сульфата меди к желтому сернистому аммонию удается получить соединение [NHJ [CuSJ в виде гранатовокрасных игл.
Среди двойных соединений сульфида меди(1) с сульфидами тяжелых металлов особое значение (медные руды) имеют медный колчедан (халькопирит) CuFeS2 (о структуре см. т. I, стр. 404), борнит Cu3FeS3, а также двойныесоедипепияссульфидамимышьяка(Ш) исурьмы(Ш) (ср. стр. 395).
Нитрид меди Cu3N получается в впде порошка темно-оливково-зеленого цвета при пропускании тока сухого аммиака прн 250“ над осажденной окисью меди(1), или окисью медн(П), или пад фторидом меди. Он имеет уд. вес 5,84 и является антиизотипом ReO3. Теплота образования его равна —17,8 ккал/моль Cu3N (Juza, 1938 и сл.). Выше 300° нитрид меди распадается на медь и азот. Хлором он разлагается с образованием СпС13, а с хлористым водородом образует CuClj
Аммиакаты солей меди(1). Многие соли мсди(1) очень склонны к образованию продуктов присоединения с аммиаком (аммиакатов или амминов). Это было уже показано на различных примерах **. Полученные из водных растворов аммины солей меди(1) соответствуют большей частью типу
* Раньше часто применяли это превращение для определения меди весовым путем. Однако как только добавленная сера полностью израсходуется, сульфид меди(1) начинает отдавать серу. Поэтому таким путем нельзя получить сульфид меди(1) совершенно в чистом состоянии. Чистым он получается, если нагревание вести либо в смеси водорода п сероводорода, либо в токе двуокиси углерода, который вначале следует пропустить через промывную склянку- наполпеппую метиловым спиртом, и затем насытить парами серы простым способом, предложенным Гааном [Hahn F. L., Вит., 63, 1616 (1930)1.
** Различие между аммиакатами и амминами соответствует различию между гидратами и акво-соединеииямп. Под «аммиакатами» понимают вообще продукты присоединения аммиака. Из них к амминам причисляют только такие, в которых NH3-rpyn-пы принадлежат определенным атомам или иенам с образованием (сильных или слабых) комплексов, а не только расположены в пустотах решетки.
410
Глава 8
[Cu(NH3)2]X. Иногда эти комплексные соединения оказываются устойчивее лежащих в их основе простых соединений. Например, растворением окиси меди(1) и сульфата аммония в водном растворе аммиака в отсутствие воздуха или восстановлением гидразинсульфатом раствора сульфата меди, насыщенного аммиаком, получается аммиакат сульфата меди (I) [диал1мйнмедъ(1) сульфат] [Cu(NH3)2]2-SO4. Он хорошо кристаллизуется в виде белых гексагональных табличек, в то время как простой сульфат меди(1) в свободном состоянии нельзя получить.
Если па водный раствор сульфата меди(И) действовать металлической медью или даже другим металлом (например, магнием или железом), то заметная часть имеющейся в растворе меди пер сходит в одновалентное состояние. Несмотря на это, раствор не содержит сколько-либо заметных количеств ионов Отт, так как в нем одновалентная медь почти без остатка содержится в форме апионвого комплекса.
Растворы амминных комплексов солей меди(1) совершенно бесцветны, если медь содержится в них исключительно в одновалентном состоянии.
Как установил Бьеррум (1934), в растворах попы [Cu(NH3)j‘ и [Cu(NH3)3J‘ существуют в равновесии друг с другом. Оба являются существенно более сильными комплексами, чем Йоны амминов меди(П) (ср. стр. 417). Однако в растворах ионы аммипов меди(1), которые бы содержали более чем две молекулы NH3, не встречаются.
Известны также многочисленные комплексные соли вышеприведенного типа, в которых место аммиака занимают другие азотные соединения, например анилин, пиридин, хинолин, фенплгидразин, тиомбчевипа. В качестве примера следует привести соединение с тиомочевиноп [С11(С8(МНг)3)г]Ь(Оз'Н2О; надо отметить, что простой нитрат меди(1) не известен.
Гидрид меди. Если водный раствор, который содержит 2 моля фосфорноватистой кислоты на 1 моль сульфата меди, нагреть до 40—50е; то из пего медленно выделится краспо-бурый осадок, состоящий из очень неустойчивого соединения меди и водорода. Если после отфильтровывания и высушивания его пагреть, то он отщепляет водород. Если нагревание вести на воздухе, то происходит воспламенение, в результате которого водород сгорает. Во влажном состоянии даже па холоду осадок чрезвычайно склонен к окислению. Поэтому лежащее в его основе соединение в чистом состояния таким путем еще не было выделено. Однако уже ранее ему обычно приписывали формулу СиН, которая затем была подтверждена, особенно опытами Хюттига (Н iittig, 1926). По данным Хюттига, соединение обладает кубической гранэ-ценгрироваплой решеткой с длиной ребра ячейки а — 4,33 А, т. е. расширенная решеткой меди. Напротив, по сведениям Брэдли (Bradley, 1926), атомы меди в этап соединении образуют решетку типа магпия (гексагональная плотнейшая упаковка?:; а = 2,89, с — 4,61 А. Хэгг (Hagg, 1931) предположил, что в этой решетке атомы водорода расположены так, как атомы S в решетке вюртцита; тогда для атома Н получаш-г тот же радиус, который он имеет в соединениях TiH, ZrH и ТаН. Твердый гидрид медп нестабилен. Его теплота образования составляет, по данным Сивертса (Sieverts, 192.8?t —5,1 ккал/молъ СнН.
Как указали Виборг и Гепле (Wiberg, Henle; 1952); чистый безводтплй гпдргп меди СиИ можно подучить взаимодействием иодида меди(I) с литийалгоминийглдрцдая в эфириопиридиновом растворе: 4CuI + L1A1H4 == 4CuH-t-LiI-rAII3. Он представляет собой светло-красно-коричневое твердое вещество, которое растворяется в пиридине с, кроваво-красным окрашиванием. Беаводный гидрид меди устойчивее чем содержащий воду. Безводный гидрид меди отличается также своим отношением К бензоилхлорпду. Последний гидрируется. СпН до бензальдегида
/°
С6Н5—С/ -J-CuH = C6H5—C<f -f-CuCl, ХС1 ХН
Побочная подгруппа первой группы периодической системы 411
в то время как в оду содержащим гидридом меди бензоил хлорид гидролизуется с образованием бензойной кислоты:
.0 .0
С6Н5 — C<f -I- Си Н И,О =С6НЬ- C(f + СиС1 + Нг.
ХС1 “ XJH
Гидрирующее действие Cull меньше, чем, например, гидрирующее действие MgH2 и ЛШ3 или LiAlH4, которые гидрируют бонзоилхлорид, минуя образование альдегида до бензилового спирта.
Другой (также неустойчивый) безводный твердый гидрид меди, вероятно солеобразного характера, был Получен Питчем (Pietsch, 1931) действием атомарного водорода на медь, обладающую развитой поверхностью.
2, Соединения меди(П)
Окись мсди(11) СиО встречается в природе в виде черного землистого продукта выветривания медных руд (мелаконит). В лаве Везувия она найдена закристаллизованной в виде черных триклинных табличек (тенорит'). Искусственно окись меди получают нагреванием меди в виде стружек или проволоки на воздухе при температуре красного каления или прокаливанием питрата или карбоната. Полученная таким путем окись меди аморфна и обладает ярко выраженной способностью адсорбировать газы. Удельный вес окиси меди составляет 6,3—6,4. Уже ниже 1000е окись меди обладает некоторым давлением кислорода [которое, правда, заметно снижается вследствие растворимости образующейся при диссоциации окиси меди(1) в окиси меди(И)]. Водородом окись меди уже ниже 250° восстанавливается до меди, еще легче — окисью углерода.
Окись меди используют при производстве стекла и эмалей для придания им зеленой и синей окраски, кроме того, СнО применяют при производстве медно-рубинового стекла. Для приготовления последнего небольшие количества окиси меди растворяют вместе с восстановителем [чаще всего с окисью олова(П)] в расплавленном стекле. Сначала образуется бесцветное стекло, которое при последующем нагревании приобретает красивую красную окраску («медный рубли»). Эта окраска объясняется распределением в стекле в коллоидном форме металлической меди (ср. также «золотой рубин»). В купроновых элементах окись меди(П) служит в качестве деполяризатора. В органическом элементарном анализе ее применяют как окислитель. В медицине она также находит применение, главным образом в форме мазей.
Кристаллическую решетку окиси меди(П) (тенорита) мощно рассматривать как триклиннодеформироваппую решетку каменной соли. Ее можно получать из решетки каменной соли (рис. 44 на стр. 237, т. I), если ребра в пей расположить под косыми углами (а = 85°21', р = 86°25', у = 93°35'), а длипу ребер сделать различной (а = = 3,74 А, b = с — 4,67 А). В противоположность решетке куприта решетка тенорита соизмерима с типичными ионными решетками. Теплота образования СиО составляет 33,0 ккал/молъ (Wohler, 1933).
Гидроокись меди(11) Си(ОН)2 выпадает в виде объемистого синего осадка при смешивании раствора соли меди(И) с раствором щелочи. Обычно его получают в форме геля, который высыхает в порошок с переменным содержанием воды, а при нагревании даже под водным раствором переходит в черную окись меди. Однако гидроокись меди можно получить также в кристаллическом состоянии. В этом случае ее можно сушить при 100° без разложения, и тогда она обладает составом, соответствующим формуле Сп(ОН)г.
412
Глава 8
Свежеосаж денная гидроокись меди заметно растворима в растворах щелочей. При умеренно высокой концентрации щелочного раствора получают фиолетовые растворы, в которых гидроокись меди существует преимущественно в коллоидной форме. При очень высокой концентрации щелочного раствора получают глубоко-синие растворы, которые содержат гидроокись меди в форме гид роксо-анионов в истинно растворенном состоянии. Таким образом, гидроокись меди амфотерна, однако ее кислый характер очень слабо выражен.
Соли, производные от гидроокиси меди, функционирующей в качестве ангидро-кислоты, называют гидроксокупраталшЦ Г\ (ранее куприты}. Соль патрпп Na2(Cu(OH)J впервые была получена в кристаллическом состоянии Мюллером (.Muller Е., 1923). Шольдер (1933) показал, что выше 180° отщепляется одна молекула Н,С с одновременным черным окрашиванием вследствие распада на СиО и 2NaOH. Вторая молекула воды пе отщепляется даже при 500°; ее отдает соединение только при сплавлении с К2Сг2О7 по уравнению K2Cr,O7+ 2NaOH = К2СгО4 + Na2CrO4 + Н2О.. 11 з этого следует, что рассматриваемое соединение представляет собой но дигидрат оксо-соли Na2[CuO2]-2H2O, а гидраксо-солъ. Щелочноземельные гидроксокупраты также были получены Шольдером: Sr[Cu(OH)4J-Н2О (сине-фиолетовый), Sr2(Cu(OH)e' (светло-синий) и Ba3[Cu(OH)e] (небесно-голубой). Кроме того, были получены также гидроксогалогенокупраты, например Na5(Cu(OH)6Cl(OI'I2)]-6Н2О. Гидроксопиро-катехипатокунрат Na3(Cu(OH)(C6H4O2)2(OII2)b6H2O был олисап уже ранее Вейц-.тапдом (Weinland, 1923).
В водном растворе аммиака гидроокись меди растворяется, вызывая глубокую темно-сипгою окраску
Cu(OH)2+4NII3 = [Cu(NH3)4](OH)2. (И):
Этот раствор (реактив Швейцера) способен растворять целлюлозу. Избытком воды или кислотами целлюлоза вновь осаждается из этого раствора. Это явление используют при производстве искусственного волокна.
Проще всего реактив Швейцера получают обработкой меди при доступе воздуха водным раствором аммиака, содержащим некоторое количество хлорида аммонии Концентрированием раствора при подогревании в струе сухого аммиака удается получить соединение [Cu(NH3)4!(OH)2‘3H2O в виде длинных лазурно-синих расплывающихся игл.
Фторид меди(П). Фторид меди(П) осаждается из растворов гидроокиси меди(И. или карбопата меди(П), насыщенных водной плавиковой кислотой, в втфе светлэ-сииих мелких кристаллов состава CuF2 2Н2О. Нагреванием в токе HF воду может, удалить; при этом остается безводный ChF2 — белый кристаллический норошех (г. ил. 950°), который также получается непосредственно сухим путем. Он кристаллизуется по типу плавикового пшата; а — 5,41 А. Его теплота образования из элементов составляет, поданным Вартенберга (Wartenberg, 1939), 129 ккал. В холодной вод? фторид меди(11) мало растворим (4,7 г в 100 г Н2О). Теплой водой Оп разлагаете^ с осаждением основного фторида Cu(OII)F,
Хлорид меди(П) СчС12 в безводном состоянии представляет собой темно-коричпевую расплывающуюся массу с т. пл. 498°. Из воды он кристаллизуется как дигидрат в виде небесно-голубых ромбических пгп (удельный вес 2,51). После удаления следов воды он становится темно-зеленым. Хлорид меди(И) нс только чрезвычайно легко растворим в воде, но и легко растворим во многих органических растворителях, таких, как спирт, ацетон и. пиридин.
По мнению Бенратта (Beneath, 1932 —1934), соединение СаС12 • 2Н2О является единственным стабильным гидратом хлорида меди(П). Однако, возможно, существуют
Побочная, подгруппа первой группы периодической системы
413
еще другие гидраты в виде нестабильных соединений. Растворимость СнС12 при 0е составляет 40,9 г, при 20° 42,8 г, При 25° 43,6 г, При 50° 46,0 г и при 100° 54,6 г в 100 г раствора.
Раствор хлорида меди в очень небольшом количестве воды томно-коричневый; при разбавлении он становится сначала нелепым, при дальнейшем разбавлении бледно-голубым. При добавлении концентрированной соляпой кислоты даже сильно разбавленный раствор снова становится зеленым. Достаточно концентрированный сильно солянокислый раствор хлорида меди(П) при обычной температуре желтов ат о-зеленый, а при нагревании—коричневый. Сильно разбавленные растворы проявляют окраску ионов меди(П) (пебесно-голубую), В концентрированных растворах существуют аутокомплексы, а в растворах, содержащих соляную кислоту, имеются комплексные соединения с хлористоводородной кислотой. Пос Ji одни е можпо выделить в кристаллической форме. Например, Энгель (Engel) получил соединение СпС12-ИСЬ ЗН2О в виде темпо-грапатово-красных игл; Сабатье (Sabatier) получил гиацинтово-красные иглы состава СпС12 • 2TIC1 5П2О. Хлорид меди(П) образует также многочисленные -двойные хлориды, преимущественно типа M^JCuCy и MgfCuClJ, в то время как СпС1 образует двойные соединения преимущественно тина М1 (СпС12) и М|.[СиС131 (Remy, 1933). Хлорокупраты(1) бесцветны. Безводные хлорокупраты(П), как правило, обладают окраской от желтой до коричневой, а содержащие кристаллизационную воду большей частью имеют свогло-сипюю или сине-зеленую окраску *.
Концентрированный водный раствор хлорида меди(П) способен поглотить значительные количества окиси азота, приобретая при этом черно-зеленую окраску. При разбавлении раствор снова бурпо выделяет окись азота.
В значительно большем количестве СиС12 присоединяет аммиак. Вели охладить насыщенный аммиаком водный раствор хлорида меди(Ц) до 0°, то можно получить содержащий кристаллизационную воду пентааммин. Гексааммин CuCla-6NH3 можно приготовить, пропуская хорошо высушенный аммиак в раствор хлорида меди(П) в этилацетате.
Ранее хлорид меди(П) применяли в дикоп-процессо в качестве переносчица кислорода. Теперь его применяют в качестве переносчика кислорода главным образом при приготовлении органических красителей. В пиротехнике оп служит для получения зеленого пламени. Кроме того, хлорид меди(П) применяют в медицине. В технике его приготовляют растворением окиси меди или карбонита меди в соляной кислоте, а иногда взаимодействием медного купороса с хлоридом бария.
Бромпд меди(П) СаВг2 представляет собой в безводном состоянии блестящие почти черные кристаллы. Прп нагревании до температуры красного каления он отдает половину брома. Из водного раствора в зависимости от температуры СнВг2 выделяется с 2 или 4 молекулами воды в виде коричневато-зеленых расплывающихся на воздухе кристаллов. Безводная соль в токе сухого аммиака при температуре ниже 20° иерехо-щт в гекса а ими и С.нВг2-6КГГ3.
Если раствор бромистого водорода или щелочного бромида смешать с бромидом меди(Н), то раствор становится пурпур по-красным. Следовательно, в этом случае также отчетливо проявляется комплексообразование. Вейн лайду удалось получить комплексную кислоту состава СнВг2. НВг- 10И2О в форме черпых блестящих игл. Двойные бромиды типа M’lCiiBrg] и М|[СнВг4] можно получить в кристаллическом I истояивм.
Иодид меди(П). Если смешать раствор сульфата меди с иодидом калия, то сначала в нем образуется йодид меди(П). Одпако он легко распадается на иодид меди(1) и иод
Сп'- + 2Г=Си18 —> Сн1-р/21;. (12)
* Соль лития LiCuCJ3> 2112О, так же как и продукт присоединения ИС1, облазает гранатово-красной окраской.
414
Глава S
Известны двойные иойидл Cul2-2NH4I-2NH3'4H2O — темно-зеленые иглы, нерастворимые в воде.
Сульфат меди(П) CuSO4 в безводном состоянии представляет собой белый порошок (удельного веса 3,64), который, присоединяя воду, становится синим и поэтому служит для обнаружения воды в органических жидкостях, а также как осушающее средство. При нагревании он разлагается на CuO, SO 2 и 1/2О2.
Из водного раствора, сульфат, меди кристаллизуется с 5 молекулами воды, CuSCh-oHnO — медный купорос — в виде лазурно-синих прозрачных триклинных кристаллов, которые на воздухе с поверхности выветриваются. Удельный вес его 2,286. Медный купорос—технически важная соль меди. Его применяют в производстве минеральных красок, для пропитки древесины (для предотвращения гниения), для консервирования птичьих чучел, протравливания злаковых посевов. Кроме того, он служит для приготовления бордосской жидкости, применяемой для борьбы с вредителями растений (смесь раствора медного купороса и известкового молока). Далее, медный купорос используют для приготовления ванн для омеднения, а также в элементах Даниэля и Майдяигера.
В больших количествах применяют его в производстве органических красителей и при окраске кож. В медицине он служит в качестве вяжущего, прижигающего и рвотного средства.
Техническое приготовление медного купороса производят растворением медных отходов в горячей крепкой серной кислоте [уравнение (13)} или, экономичнее, путем обработки медных отходов теплой разбавленной серной кислотой при хорошем доступе воздуха [уравнение (14)], Кроме того, медный купорос часто получают растворением окиси меди (которую вырабатывают обжигом сульфидных руд) в серной кислоте [уравнение (15)1:
Си + 2H2SO4=CuSO4-f- 2Н20 -j- SO3, (13)
Си + H2SO4 + i/2O2=CuSO4 + Нг0, (14)
СиО-f- H2SO4=CuSO4-f- Н2О. (15)
Иногда медный купорос встречается в природе, например в Чили. Природный медный купорос называют халькантитом, Наряду с ним встречается брохантит, — основной сульфат меди(П) CuSO^ 3Cu(OH)2. Растворением этой природной сульфатной руды в разбавленной серной кислоте готовят медный купорос.
При нагревании медный купорос ступенчато теряет воду. Сначала образуется тригидрат, затем моногидрат и только выше 200° происходит полная дегидратация.
В кристаллической решетке медного купороса каждый ион Си2+ окружен 4 молекулами Н20ц2 атомами О, принадлежащими к раэли’нгым SO ^“-группам, Эти молекулы и атомы находятся в углах искаженного октаэдра. Пятая молекула Н2О не связала координационно с ионом меди, а окружена 2 молекулами Н2О и 2 атомами Осульфатных групп (Lipson, 1934).
С сульфатами сильно электроположительных металлов и радикалов сульфат меди образует хорошо кристаллизующиеся двойные соли (сульфато-соли). Большинство из них соответствует тину M2SO4‘CuSO4-6Н2О и окрашено в бяедпо-зеленовато-синий цвет. Существование комплексных сульфато-анионов меди в растворе мои окли и пой двойной калиевой соли K2SO4-CuSO4-6Н2О, которая изоморфна (XH4)2SO4.MgSO4-6H2O, было доказано Ригером (Rieger) в опытах по изучению чисел переноса,
Растворимость в воде сульфата меди составляет при 0° 14,8 а, при 15® 19,3 г, прн 25° 23,05 г, при 50® 33,5 г и при 100“ 73,6 a CuSO4 в 100 г воды. Насыщенный раствор CuSO4 кипит при 104,0®. Примерно выше 105° в соприкосновении с раствором (находящимся под давлением) устойчив тригидрат.
Действием сухого газообразного аммиака на безводный сульфат меди получают пентааммин CuSO4- 5NII3 в форме сипе-фиолетового порошка. Замечательно, что последний содержит столько же молекул NHa, сколько медпыщ купорос имеет молекул воды. На влажном воздухе аммиак ступенчато замещается молекулами воды, прн
Побочная подгруппа первой группы, периодической системы 415
этом сначала образуемся CuS04-4NH3-Н20, а затем CuSO4-2NH3-ЗН2О. При умеренном нагревании пентааммин диссоциирует на CuSO4-4NH3 и NH3, давлепие которого при 90° составляет 1 атм. .
Насыщением водного раствора сульфата меди аммиаком и последующим его испарением получают соединение CuSO4‘4NH3-Н2О. Оно образуется также под действием газообразного аммиака на твердый медный купорос CuS04 • 5НВО. Это соединение представляет собой томные лазурно-синие прозрачные длинные ромбические призмы. В воде оно легко растворимо (1 : 1,5). При более сильном разведении водный раствор неустойчив, из пего постепенно выделяется основной сульфат меди.
Нитрат меди(П) Cn(NO3)2 в воде очень легко растворим. В зависимости от температуры он кристаллизуется из водного раствора с различным содержанием воды. Из сильно концентрированного азотнокислого раствора CufNOgJa осаждается в безводном состоянии в виде белых, чуть зеленоватых мелких кристаллов.
Тригидрат представляет собой глубоко-сипие, устойчивые на воздухе столбики (т. пл. 114,5°). Гексагидрат образует синие таблички. При: нагревании выше 26° Cu(NO3)2- 6Н2О расплывается, отдавая воду. Ниже —20° устойчив нонагидрат.
Из раствора нитрата меди, смешанного с гидроокисью меди, кристаллизуется так называемым основной нитрат — двойное соединение питрата меди ц гидроокиси меди Cu(NO3)2-ЗСп(ОН)2. Он встречается в природе в виде минерала герардтита. Вероятно, эта соль имеет такую же структуру, как и приведенные на стр. 320 и сл. и 338 я сл. основные соли кобальта и никеля (ср. также стр. 417),
Нитрит меди(П). При растворении меди в азотной кислоте сначала образуется сипе-зеленый раствор, окраску которого принимают за окраску питрита меди(П). Однако в свободном состоянии нитрит не получен, Если уваривать раствор, который содержит только ионы Си" и NOa [его можно получить, например, взаимодействием CuSO4 и Pb(NO2)2l, то происходит образование нитрата, сопровождающееся выделением окиси азота, Удается, однако, выделить так называемый основной нитрит меди Cu(NO2)2- ЗСп(ОН)2. Он кристаллизуется в виде красивых иголочек, мало растворимых в воде и в спирте. Также устойчивыми являются нитриты, включающие ионы амминмеди(П), и, кроме того, различные двойные нитриты.
Состав последних соответствует общей формуле 2MINO2-MII(NO2)2-Cu(NO2)2.
Ацетат меди(П). Из растворов окиси меди в уксусной кислоте кристаллизуется обычно моногидрат ацетата меди(П) в виде темно-сине-зеленых моноклинных призм *.
Кроме того, его можно получить перекристаллизацией основной уксуснокислой меди из уксусной кислоты. При низкой температуре кристаллизуется ромбический пентагидрат.
Обычная основная уксуснокислая медь («ярь-медянка») представляет собой смесь основных ацетатов меди. Ее можно приготовить искусственно повторным увлажнением медных пластин уксусом и действием на них воздуха. Смотря по условиям производства, получают зелёную или синюю ярь-медянку. Ее применяют в качестве масляной или водяной краски, а также для приготовления других медных красок; кроме того, ее используют в медицине, для борьбы с ржавчиной злаков й при золочении огневым способом.
С другими ацетатами, например с ацетатом аммония, ацетат меди(Ц) может образовывать легкорастворимые двойные соли.
* В соответствии с последними магнетохимическимя и рентгенов груктуряыми исследованиями гидрат ацетата меди представляет собой димер 2Сп(ОСОСН3)2-2Н2О, образованный за счет связи Си—Си (расстояние Си <—► Си 2,64 А) и мостиковых ба дентатных ацетатных групп.— Прим. ред.
416
Глава 8
Карбонат меди. Простой нейтральный карбонат меди пока пе получен. Одпако известен хорошо кристаллизующийся основной карбонат. Он часто встречается в природе. Широко распространен (правда, редко в больших количествах) малахит — основной карбопат состава СпСОз-Сп(ОН)2. Последний иногда встречается в виде зеленых, моноклинных, игольчатых, большей частью сросшихся кристаллов, но чаще всего в виде плотной или волокнистой зеленой массы. Мопоклиппый синий азурит имеет состав 2СпСО3>Сн(ОН)2. Особенно красивые кристаллы его найдены около Хесси (близ Лиона), и поэтому оп называется также хессилитом. Под названием медной лазури его применяют в качестве малярной краски, а также для получения синего пламени в пиротехнике. На влажном воздухе оп превращается постепенно в зеленый малахит, что препятствует применению его в качестве краски.
Действием Na2CO3 на CuSO. в водном растворе получают основной карбонат состава СиСОа.Сд1(СШ)2-0,5Г12О. Это соединение при нагревании До 200° переходит в малахит, CiiCO3Cu(OH)2. По данным Биндера (Binder), он дает ту же интерференционную картину, что и малахит, и поэтому структурно его можпо рассматривать кан малахит, в решетке которого расположены молекулы Н2О.
Из двойных карбонатов меди следует упомянуть соль калия К2СО3- СпСО3, которая кристаллизуется в зависимости от условий опыта: либо в безводном состоянии в виде темпо-сииего порошка, либо в 1 молекулой воды в виде светло-синих шелковистых игл, либо с 4 молекулами воды в виде зеленовато-сипих больших табличек..
Оксалат мсдп(П) выделяется из растворов солей меди(П) щавелевой кислотсЭ или, лучше, оксалатами щелочных металлов в виде светло-синего осадка составе СиС2О4- Н2О. Он нерастворим в воде, растворим в сильных кислотах, по порастворпи в избытке щавелевой кислоты. В достаточном избытке щелочных оксалатов оксалат меди(П) растворяется с образованием легкорастворимых двойных солей, которые получены также в твердом состоянии. Некоторые из них соответствуют формует
GhC204‘ 2Н2О.
Оксалат медщП) можно растворить в водном растворе аммиака. Если ьодпъет раствор, насыщенный при нагревании NH3 и СнС2О4, охладить, из него кристаллизует ся пента аммиакат CuC2O4-5NH3,
Цианид мсди(П) Cu(CN)2 образуется при взаимодействии растворсс солей медп(П) с небольшим количеством ионов CNr в виде коричнево-желтого осадка. При обычной температуре этот осадок выделяет циан и быстра переходит в цианид 2CuCN-Cu(CN)2, при нагревании он полностью переходит в цианид меди(1):
Cu(CN)3=GuCN+1/2(CN)2. (F
Из растворов, содержащих избыточное количество цианидов щелочных металлов, получаются двойные цианиды, например K2lCu(CN)4] — белые гексагональные кристаллы, очень легко растворимые в подо
Роданид меди(П) Cu(SCN)2 получают в виде бархатисто-черного порошка растсс рением гидроокиси меди(П) или карбоната меди в концентрированной водной роданлетл водородной кислоте или осаждением роданидом калия из концентрированного растварт соли медЩП), Водой, а еще быстрее растворами роданидов щелочных металлов сл быстро разлагается с выделением роданида медн(1). На некоторые органические вещества (например, индиго) он действует в присутствии воды как сильный окислитель В присутствии аммиака растворимость в воде и одновременно устойчивость родаппх меди(1Г) повышается вследствие образования комплексного аммиаката. В завпепмзгхт от температуры ц копцоитрации аммиака при этом образуются [Cu(NH3)2] [SG\J: — достаточно трудно растворимые светло-еппие иглы, или [Cn(NTT3)J [SCN]2— л сет растворимые темпо-сипне таблички. Последнее из указанных соединений дает tcsx.. моногидрат в форме фиолетовых игл. Из жидкого аммиака получают пента а мила пи"
Побочная подгруппа первой группы периодической системы
417
Аммиакаты солей меди(Л). Двухвалентная медь образует аммиакаты (аммины *) в еще большем количестве, мем одновалентная. Чаще всего среди них, встречаются два типа:
[CiifNIIgJJXa и [Cu(NH3)4]X2.
В растворах, содержащих аммиак и хлорид аммония, существует равновесие между ионами ICufNHg)] ", [Cu(NH3)s]“, [Cu(NH3)3]”, [Cu(NH3)4]'" и ICu(NH3)5]'. Как установил Бьеррум (1931) путем определения констант равновесия, исключая случаи очень небольших концентраций аммиака, в этих растворах всегда преобладают тетрааммин-нонш. При высоких концентрациях аммиака наряду с пими присутствуют также пентааммин~ионт. Напротив, гекса аммин-йоны в растворах не образуются 'В цодтвержденио этого Бриптцпнгер определил методом диализа (ср. т. I, стр. 335) в аммиачных растворах большинства медных солей ионный вес катиона, соответствующий формуле (Cu(NH3)J“. Следует отметить, однако, что в аммиачных растворах сульфата меди и селенита меди он нашел ионные веса, соответствующие формулам [Cn2(SO4)(NH3)4]" и [Сиг(Зо04)(МН3)4]'-*.
Вместо аммиака могут присоединяться также органические азотсодержащие соединения, такие, как анилин, пиридин, хинолин и многие другие. Примеры таких аммиакатов были уже приведены, причем даже аммиакатов тех солей, которые в свободном состоянии не получены.
Так называемые гексо,новые соли медп. Двухвалентная 'медь образует целый ряд содей общего состава СиХ2‘ ЗСи(ОН)2. Вернер назвал их «гексоловыми смями». Он рассматривал их как соединения, некоторых шесть связанных с металлами гидроксильных групп («.«-групп) при помощи «побочных^ валентностей присоединены к центральному атому в соответствии с общей формулой
В настоящее время построенные таким образом соединения называют солями гекса-11-еидроксотетрамеди(//). Однако весьма сомнительно, что рассматриваемые основные соли меди обладают структурой, предложенной Вернером. Рентгенометрически их структура еще не установлена; однако, как следует из рептгеноструктурных исследований Других основных солей, которые Верне!» также рассматривал как «гексоловые сони*, например основных галогенидов кооальта н никеля аналогичного состава (ср. стр. 320 и сл. и.338 и сл,), вернеровские представления нуждаются в исправлении. Примерами основных солей меди этого типа являются минералы атакамит (стр« 395), брохантит (стр, 414), герардтит (стр. 415) и лангит [Cu(Cu(OH)2)3]SO4. Н2О, Кроме того, были иск у сети ей по приготовлены основные соли меди соответствующего состава, где X — Вг, [ 1ЧО2], [С1О3], [1О4] или [SaO6]. Все соли, у которых было исследовано кристаллическое строение, обладают ромб о-бипир амидальным габитусом,
По представлениям Вернера, к типу «гексоловых солей» очень близко стоит швейнфуртсу.ая зелень. Это соединение получается добавлением порциями зелени Шееле Са]1АзО3 к кипящей уксусной кислоте. На 1 молекулу ацетата меди опо содержит 3 молекулы арсепита меди. Верпер придал ему формулу, аналогичную «гекселовым солям»;
Эта формула должна быть, вероятно, изменена, как и формула апатита, который Вер-пор причислил к этой же группе (см. стр. 120).
Известны соединений, аналогичные швейяфуртской зелени, в которой оба ацетат-вона заменены на иопы трихлоруксуспой кислоты, проциоповой кислоты или муравьиной кислоты («группа швейнфуртской зелени»).
* Ср. примечание на стр. 409.
Г. Реми
418
Глава 8
Сульфид м<‘ди(П) CuS. При пропускании сероводорода в растворы солей меди(П) CuS выпадает в виде черного осадка. Оп нерастворим в воде и в разбавленных сильных кислотах. В присутствии кислот оп очень легко образует коллоидные растворы, Кроме того, во влажном состоянии на воздухе он довольно легко окисляется до сульфата меди. Поэтому, если осадок требуется отделить фильтрованием от раствора, из которого он выпал, его следует промывать водой, содержащей сероводород и немного уксусной кисл от ы.
В природе сульфид моди(П) встречается в виде компактных, достаточно мягких, (тше-черных кусков ковеллина, из которых после растирания получают нпдиго-симий порошок. Очень редко находят достаточно большие хорошо образованные кристаллики. Они принадлежат к гексагональной системе. Удельный вес 4,68. Структура решетки довольно сложна.
Сульфид меди(П) обладает достаточно хорошей электропроводностью. При сильном прокаливании в отсутствие воздуха или при Слабом прокаливании (ниже 600е) в токе водорода сульфид медн(Н) переходит в сульфид медп(1)*, а при прокаливании на воздухе — в окись меди(П). В горячей умеренно разбавленной азотпой кислоте сульфид меди(П) легко растворим, в кипящей разбавленной серной кислоте, напротив, очень трудно растворим. В растворах сульфида калия и натрия сульфид меди практически нерастворим; напротив, оп несколько растворим в растворе сернистого аммония, еще более — в растворах полисульфидов щелочных металлов.
Полиеульфнды меди, например CuS3 п Cu2S5, получают смешиванием растворов солей меди(П) с растворами полисульфидов Щелочных металлов и другими путями; однако их трудно получить в чистом состоянии.
Растворимость CuS л растворах полисульфвдов щелочных металлов сильно возрастает с содержанием в полису л ьфиде серы. Увеличение концентрации и повышение температуры также способствуют растворимости. Полнсульфцд калия лучщо растворяет CuS, чем соответствующая" соль натрия (Holtje, 1935),
3. Соединения медп(Ш)
Известно только очопь немного соедпвдний, в которых медь находится в третьей степени окисления. Окись ,меби(Ш) Си2О3 была впервые получена Скаглиаринп и Тореллц (Seagliarini, Torolli, 1921), Они получили ее в виде гранатово-красного порошка при обработке си еже осажденной гидроокиси медн(Н) пероксосульфатом калия. Окись меди(1П) выделяет из соляной кислоты хлор, однако с разбавленными кислотами не образует перекиси водорода. Следовательно, она не является производным 1Т2О2 подобно коричнево-черной перекиси медн, полученной Мозером действием И2О2 па тонн о измельченную Си(ОН)2:
/О О — ОН**
СиОв-П2О =Cuf | -Н2О или Сп^
ХО 'ОН
Отсюда Скю’лиариии и Торслли сделали заключение, что медь в соединении Си.3О3 трехВ|Ч..1Ш1Тна. Правильность этого за г:,.печения была подтверждена существованием производных окиси, которые удалось интерпретировать как соединения Cu(lII).. А именно окись CtiJ [ Й), обладает кислым характером и образует со щелочами красные., очень нестойкие соли типа Мт1Сн(ОН)4], т. е, тетраяидроксокупраты{17Г). Позднее Клеимом и Шестьдером (К йчпш, ЯсйоМег, 1951) были приготовлены также екгекучра-ты(Ш], а именно КСиО2 и Ва(СиОв)2-Н2О. Клемму (1949) удалось также получить
* См. примечание па стр. 409,
** Перекисное соединение меди с. еще большим содержанием кислорода НО-О-Си-О-О-Си-О-ОН было получено Виландом (Wieland, 1938).
Побочная подгруппа первой группы периодической системы, 419
фторо-соли с трсхвалснтиой медью K3[CuFflJ. Другими примерами соединений медп(Ш) являются Кт[Си(Юв)2] • 7И2О и Na.r,H4|Cn(TeO6)2} 18Н2О (Malatesla, 1941). За исключением фторо-комплексных голой, все перечислеппыс в конце соединения мели (111) диамагнитны; следовательно, в этих случаях речь идет о соединениях с комплексами внедрения.
Аналитические сведения. Соединения меди при нагревании с содой па угле паяльной трубкой дают красные металлические блестки или красное зерно, растворимое в азотной кислоте. При добавлении аммиака раствор становится глубоко-синим. При действии сероводорода в кислом растворе появляется черный осадок, при действии гексацианоферрата(Н) калия — красно-бурый осадок, который состоит из гексацианоферрата(П) меди(П) Cu2[Fe(CN)ej; реакция эта очепь чувствительна.
В ходе систематического анализа медь оказывается в той части сероводородного осадка, который нерастворим в желтом сернистом аммонии. В небольшом количестве, однако, медь растворима в сернистом аммонии, и прежде всего в желтом.
Перлы фосфорных солей, полученные в окислительном пламени, окрашиваются медью в желтый цвет при нагревании и в зелеиовато-синий на холоду; в восстановительном пламени вследствие образования окиси медв(1) или дюке металлической меди перлы становятся непрозрачными, окрашенными в крас ко-коричневый цвет.
Для микроаналитических определений очень подходит соединение К2РЬСп(КОг)(;, кристаллизующееся в виде черно-коричневых, тетрагональных кубиков (предел чувствительности О,ОЗу Си), Еще чувствительнее капельная реакция с рубеановым подо-S=C — ХН2
родом | , с которым медь образует ииутрикомлексную соль (предел
s=c — nti2
чувствительности 0,00 бу Си). Эта реакция при подходящих способах pa6orbi(Feigl, 1930) позволяет производить непосредственное испытание сплавов на минимальное I. одержание меди.
При весовом аналитическом определении медь либо осаждают в виде сульфида ыеди(П) и после прокаливания в токе водорода и сероводорода или в токе двуокиси углерода, насыщенном парами метилового спирта и серы, взвешивают как сульфид меди(1) (ср. стр. 409) или пос ле .’Прокаливания на воздухе в виде окиси меди(П), либо выделяют электролитически в виде металла и взвешивают. Последний способ определения самый точный. Очень надежным, кроме того, является определение меди в виде роданида меди(1), полученного осаждением роданидом аммония с добавлением сернистой кислоты (ср. стр. 409). Медь удается также определять титрометрически, папример иодометрически, при смешивании раствора сульфата меди с иодидом калия и титровании иода, выделившегося в соответствии с уравнением (12) на стр. 413, тиосульфатом.
Эфраим (Ephraim, 1930) для обнаружения меди, и прежде всего для определения ✓ОН
/ , который
\CIT=NOH
содержания меди, рекомендует салицилальдокепм
позволяет количествеппо осаждать медь в присутствии любого другого металла из кислого раствора. С иопамп Си" в уксуснокислом растворе он дает зслеповато-белый с желтым оттенком осадок Cu(C7 H6‘O2N)2. По млению Эи если на (Ensslin, 1941), для определения очепь небольших количеств удобно колориметрическое определение е ди э ти лдит пока рб а Минатом п атрия. По даппым Ремп и Михаэльс она (Michaelson, 1911), медь можно очень точно определять электрополяриметрически. Относительно обнаружения и определения меди солью Рейнеке см. на стр. 168,
27*
420
Глава &
СЕРЕБРО (ARGENTUM) Ag
Распространение в природе. Серебро встречается в природе преимущественно в виде сульфида, часто вместе с другими сульфидами, например с сульфидами свинца, меди, сурьмы и мышьяка. С двумя последними сульфид серебра образует двойные сульфиды, которые можно рассматривать как тиосоли—пираргирит и прустит. Пираргирит представляет собой тиоантнмопат(П1) серебра Ag3[SbS3J. Прустит, встречающийся в меньших количествах, представляет собой тиоарсонат(Ш) серебра Ag3[AsS3]. Оба кристаллизуются в гексагональной системе.Те же соединения, хотя и редко, кристаллизуются также и в моноклинной системе, образуя соответственно огненную обманку и ксантокон. Другой Моноклинный минерал, рассматриваемый как метатиоантимонат(III) серебра Ag[SbS2] (миаргирит, гипаргирит), также достаточно редок. Важнейшей серебряной рудой, встречающейся главным образом в Мексике и Южной Америке, является серебряный блеск (аргентит) Ag2S. Серебряный блеск всегда содержится в небольших количествах в свинцовом блеске в виде изоморфной примеси (до 1%J. С медным блеском Cu2S серебряный блеск также образует твердые растворы. Последние называют серебрано-медным блеском (штромейеритом), если они бедны серебром, и ялпаитом, если они богаты серебром.
Иногда в серебряных жилах встречается также дискразит (антимонид серебра) AgsSh, кроме того, полибазит (евгеновьш блеск) — смесь сульфидов серебра, меди и сурьмы, В блеклой руде также могут содержаться заметные количества серебра (до 32%) (серебряная блеклая руда), В Средней и Южпой Америко встречается также часто редкое в других местах^ кубическое роговое серебро (кераргирит) AgCI, иногда бромистое серебро (бромаргирит, также кубическое) AgBr и редко гексагональное иод истое серебро Agl. В самородном состоянии серебро также часто встречается в природе, иногда в хорошо образованных кубиках и октаэдрах (обычно содержащих золото).
Получение. В качестве исходного сырья для получения серебра наряду с собственно серебряными рудами в больших количествах используют содержащие серебро свинцовые, цинковые и медные руды, кроме того, ийогда также остатки обжига серного колчедана (пиритные огарки).
Большие количества серебра получают в качестве побочного продукта при переработке содержащего серебро свинцового блеска. Чтобы выделить серебро из этой руды, обычно содержащей небольшие количества серебра, применяют два способа — способ Паттиисона и способ Паркеса.
По способу Паттинсона, разработанному в 1833 г., расплавленный, содержащий серебро свинец медленно охлаждают и выкристаллизовывающийся при этом чистый свинец отделяют от жидкого расплава, Сплав серебра со свинцом обладает эвтектикой, соответствующей 2,6% серебра, с т. пл. 303°, в то время как чистый свинец плавится при 326°. Следовательно, при охлаждении из бедного серебром расплава должен сначала осаждаться свинец, причем теоретически до тех пор, пока содержание серебра в расплаве пе поднимется до 2,6%, Практически без труда удается получить свинец с содержанием серебра более 2%. Последний плавят на трейбтерд. При этом свипид окисляется до глета, которому дают стечь с поверхности, а серебро остается.
По способу, разработанному Карстеном и Паркесом (Karsten, Parkes), в нагретый до температуры плавления цинка бедный серебром свинец вносят цинк. Цпшг обладает большим сродством к серебру, чем свинец. Поэтому оп растворяет серебре (наряду с частью свинца) и выделяется на поверхности расплава в виде пены, которая при незначительном понижении температуры затвердевает н легко отделяется ст расплава. Из полученной цинковой пены сначала отгоняют цинк и затем отделишь серебро от оставшегося свинца купелированием.
Побочная подгруппа первой группы периодической системы
424
Если имеется сравнительно богатый серебром свинец, т. е. свинец с содержанием более чем 0,1% серебра, то его можно купелировать непосредственно,
Купелирование иногда производят в две стадии, В этом случае сначала процесс ведут до образования сплава с содержанием 50^-80% серебра и продолжают его затем в небольших печах. Однако часто купелирование производят в один прием до получения «бликового серебра», содержаще го 90% серебра, или еще больше.
Содержащие серебро медные руды также имеют большое значение для добычи серебра. После их переработки до медного штейна или до черновой меди Си можно навлечь оттуда обработкой умеренно концентрированной горячей серной кислотой, а серебро при этом остается. Однако, как уже было указано, большей частью содержащую серебро черновую медь рафинируют электролитически й серебро вырабатывают из анодного шлама.
На Мансфельдерском металлургическом заводе содержащий Серебро медный штейн с 18-41 г. перерабатывают по способу Цирфогеля (Ziervogel). Содержащийся в нем сульфид серебра осторожным обжигом при 700—800° переводят в сульфат серебра и последний затем выщелачивают водой.
Описанные способы часто применяют для переработки серебряных руд, свободных от свинца и меди, К этим рудам добавляют перед переработкой свинцовый, блеск или медиуто руду. Это делают в основном в случае богатых серебром руд, Напротив, для вскрытия бедных серебром руд, если переработка со свинцовыми или медными рудами по каким-либо причинам недопустима, применяют такие способы, при которых серебро экстрагируют из руды при обычной температуре. В прежние времена для этого служил способ амалъгамиротшиЯ' Э также различные способы, которые оспова-пы на растворении хлорида, серебра тиосулг.фатпыми растворами (сульфид серебра хлорирующим обжигом легко удается перевести в хлорид). В этой связи следует упомянуть также способ Августина (Augustin), который для вскрытия руды использовал растворимость хлорида серебра в концептрироваипом растворе поваренной соли. Однако эти способы в настоящее время почти все вытеснены цианидным, выщелачиванием,
Способ амальгамирования ранее был особенно распространен в Мексике и Южной Америке. Он основан на том, что серебро легко амальгамируется ртутью. Даже если серебро присутствует в виде, хлорида, оно иогко. поглощается ртутью с разложением хлорида. Прочие серебряные руды необходимо вначале перевести в хлорид серебра путем хлорирующего обжига.
Вскрытий бедных серебром руд, не используемых для получения свинца или меди, почти всюду в настоящее время производят методом цианидного выщелачивания. Этот способ удается применять не только к хлориду серебра, но также непосредственно н к сульфиду серебра. Метод основан на том, что соединения серебра обрабатывают цианидами щелочных металлов и в результате образования комплексных цианидов серебро переходит в раствор.
В том случае- когда серебро присутствует в виде сульфида, реакция протекает па уравнению
Ag-jS-HCN' 2fAg(CN)2J' + S". \
Так как при этом устанавливается равновесно, то, чтобы процесс протекал возможно полнее в правую сторону, из реакционной смеси следует удалять ионы S’,. Это достигаете п пропуска пнем воздуха. Тогда ионы S переходят в В20з'-, SO£- и SGN'-попы. Введением цинкового щтабика можно выделать серебро из цианидного раствора в виде иг/талла.
Очистка серебра. Полученное одпим из описанных методов серебро всегда содержит немного золота, и очень часто оно содержит также медь. Очистку производят либо аффинированием, либо электролитическим рафинированием.
422
Глава 8
Аффинированием называют очистку серебра путем обработки горячей концентрированной серной кислотой. При этом серебро в виде сульфата переходит в раствор, а металлическое золото (порошок) остается. Несмотря на то что медь переводится сор-пой кислотой в сульфат, в т;ониептрировэнном серной кислоте CuSO,, мало растворим. Внесением медных пли железных пластин из раствора осаждается серебро в виде металла.
Чаще применяют электролитическое рафинирование. Для этого подлежащее рафинированию серебро, содержащее только небольшие количества меди и свинца, в форме пластин подвешивают в качестве анода в раствор сильно разбавленной азотной кислоты или нитрата, серебра. При злектрелизе, помимо серебра, и раствор переходят также свинец н медь. Поскольку концентрация менее благородных металлов по сравнению с к описи грацией серебра не слишком высока, то па катоде вновь выделяется только серебро. Золото падает у анода на дпо, пе растворяясь. Полученное таким путем электролитическое серебро содержит 99,95% и более Ag.
Относпк тыто электролитического осаждения серебра из золота, содержащего серебро, см стр. 440.
Свойства. Серебро является благородным металлом с красивым белым блеском. Оно легко полируется. По твердости Ag стоит между медью и золотом. После золота серебро протягивается лучше всех металлов. Среди всех металлов оно обладает наибольшей электро- и теплопроводностью (удельная электропроводность серебра в обратных омах х — 6,73 10й при 0°, 6,14-10г' при 18", 6,07 >10й при 20°. Теплопроводность X = 1,006 кал/см-сек-град при 18е). Серебро кристаллизуется в кубической системе (о структуре решетки см. стр. 389). Как установил Шеррер по дебайеграммам, кристаллическая структура сохраняется даже в небольших частичках коллоидного серебра.
С золотом, а также со своим непосредственным предшественником в ряду элементов — палладием—серебро образует непрерывный ряд твердых растворов. С медью, напротив, в твердом состоянии наблюдается только очепь ограниченная взаимная растворимость (ср, стр. 29). Это связано с тем. что золото и палладий не только имеют одинаковый с серебром тип решетки, по и обладают близкими константами решеток (Ag 4,077, Ап 4,070, Pel 3,88 А), в то время как медь, хотя тг имеет подобный тип рептеттпт, но обладает существенно мепыпетг константой решетки (а = 3,61 А).
В жидком состоянии серебрю с медью, а также со многими другими металлами смешивается в любом соотпотвеппи. Как в жидком, так и в твердом состоянии серебро весьма огршптчеипо смешивается с хромом *, марганцем, никелем; совсем не смешивается с железом и кобальтом. Серебро пе образует также с, этими металлами химических соединений и, насколько известно, вообще пе соединяется с металлами побочных подгрупп IV— VI1I групп, кроме платили.
Врома того, серебро не образует никаких химических соединений со своими аналогами — медью п золотом: однако оно обра^ет довольно много соединений с металлами побочной подгруппы II группы (Почти все переменного состава). С металлами главных подгрупп серебро образует большое число соединений, однако пе образует соединений с Na, Tl, Ge, РЬ н Bi, а также с неметаллами В и Si (ср. с этим табл. 41— 43, стр. 391 и сл.).
С ртутью серебро дает амальгаму. Ес легче всего получить встряхиванием металлической ртути с раствором нитрата серебра, причем ртуть при этом частично переходит в раствор:
Hg-|-Ag'=Ag-|-i/2Hg2.
Выделяющееся из раствора серебро образует с оставшейся металлической ртутью кристаллические соединения якобы состава Ag3Hg4, Ag3Hg2 и Ag3Hg, которые выделяются в виде длинных блестящих игл. Кристалли-
* С хромом в твердом состоянии серебро вообще по смешивается.
Побочная подгруппа. первой группы периодической системы
423
задней из расплавов получают соединения другого состава (ср. табл. 43, стр. 394).
В соответствии со своим характером благородного металла компактное серебро непосредственно с кислородом не соединяется *; однако в расплавленном состоянии оно может растворять заметные количества кислорода. При затвердевании оно снова отдает наибольшую часть кислорода. Поверхность затвердевшего расплава разрывается при этом выделяющимся изнутри газом, причем часто с та кой силой, что серебро разбрызгивается. Это явление — разбрызгивание серебра — известно давно. Получаемая косвенным путем окись серебра Ag3O разлагается уже при уморенном нагревании. Озон, одпако, легко действует непосредственно на серебро, и прежде всего при небольшом пагревании (около 240°). При этом серебро чернеет и образуется окись серебра(П) AgO (см. стр. 438). Эту реакцию применяют для обнаружения озона. Она очень чувствительна, если применять свежеочищепнуго серебряную пластинку (так как остающиеся при этом па поверхности следы окиси железа действуют сильно каталитически).
В расплавленном серебро в большом количестве растворяется также белый фосфор. При затвердевании расплава часть фосфора с разбрызгиванием вновь выделяется в виде паров. Одвако даже твердое серебро может содержать еще существевные количества фосфора.
Из Приведенных в табл. 42 соединений серебра с фосфором стабильны только AgP2 и AgP3. Соединения Ag3P н Ag2P5 подучают двойным обменом в растворах, а но непосредственным соединенней их составных частей.
Сродство серебра к сере очень велико. При действии влажного сероводорода серебро чернеет с образованием Ag2S. С поверхностным образованием этого соединения связано потемнение серебряных предметов при продолжительном выдерживании их на воздухе. Свободные галогены также медленно соединяются с серебром даже при обычной температуре.
По данным Питча (Pietsch, 1931), серебро реагирует с атомарным водородом при обычной температуре с образованием бесцветного твердого солеобразного гидрида AgH. При высокой температуре оно может реагировать даже с обычным водородом с образованием следов газообразного гидрида (см. стр. 392).
Взаимодействием AgClO4 с LiBH4, LtAIH4 или LiGaII4 в эфирном растворе при . низких температурах Виберг и Хошге (Wiberg, Henio, 1952) получили двойные соеди- 1 нения: AgBII4 (белое), AgAlH4 (желтое) и AgGaH4 (оранжевое). Это очень неустойчивые твердые вещества, которые уже гораздо ниже комнатной температуры распадаются на металлы и водород.
Водные растворы не к недородных кислот в отсутствие воздуха не действуют на серебро. Это соответствует его месту в электрохимическом ряду напряжений. Однако, если нагреть серебро в токе хлористого водорода до красного каления, то оно взаимодействует с ним с выделением водорода:
2Ag4-2IICl 7^ 2AgCl + n2 + 17,2 ккал. (1)
® По-видимому, при обычной температуре иа поверхности серебра образуется совсем топкая окисная пленка, которая защищает Металл от дальнейшего окисления. Па компактном Серебре окпепый слой нс может достигать заметной толщины, так |,ак прн обычпоп или немного повышенной температуре скорость диффузии незначительна (см. стр. 794); одпако при более высокой температуре окись серебра неустойчива; тонко из мельченное серебро в интервале 170—180° может поглотить заметпые количества кислорода с образованием окиси.
424
Глава 8
Как следует из уравнения, эта реакция равновесна. Если исходить из хлористого водорода или Водорода, обладающих атмосферным давлением при комнатной температуре, и нагревать смесь в закрытой амцулё, то но данным Жунио (Jounlaux, 1899), при 600° в равновесии с твердыми Ag и AgCi будут находиться 92,8 об,% хлористого водорода и 7,2 об.% водорода. При 700° 95,0 и 5,0 об.% соответственно. Эти значения свидетельствуют о том, что с ростом температуры равновесие (1) сдвигается влево так, как того требует принцип Ле Шателье. По Этому же принципу равновесие уравнения (1) будет смещаться влево при уменьшении давления. Это предположение было подтверждено экспериментально исследованиями Жунио.
При контакте с водой, содержащей воздух, серебро переходит в раствор, правда, лишь и крайне неапаинтельных количествах:
2Ag+II2O+i/2Oa=2Ag- + 2OH'. (2)
Повышение значения pH, обусловленное реакцией (2), можно доказать.
В противоположность золоту серебро легко растворяется окисляюг щими кислотами (азотной или концентрированной серной). Это свойство серебра используют для отделения его от золота (см. стр. 422 и 440). При доступе воздуха или в присутствии перекиси водорода серебро также легко растворяется в растворах щелочных цианидов; однако в противоположность меди его нельзя, в них растворить с выделением водорода.
Расплавленное едкое кали очень незначительно действует на серебро; его действие можно усилить, если к сплаву добавить селитру.
Применение, Металлическое серебро прежде всего применяют для изготовления украшений и различных поделок; его также применяют и для других целей, например для изготовления химической посуды (тиглей для плавления щелочей), кроме того, в электроиндустрии (для изготовления предохранителей) и в производстве медицинских инструментов.
Неприятный запах и привкус, который принимают иногда серебряные предметы после длительного употребления, связан, по мнению Рау ба (ЙапЬ, 1934), с поверхностным образованием органических сернистых соединений серебра. Подобно серебру ведет себя медь и медные сплавы (в отличие от других металлов). Запах и привкус можпо легко устранить кипячением в сильно разбавленной соляной кислоте.
Раньше стоимость валюты определялась стоимостью серебра. Даже теперь значительные количества серебра, может быть, больше половины годовой продукции, применяют для штампования монет. Для производства монет, а также для большинства иных целей серебро сплавляют с другими металлами, большей частью с МеДыо, так как чистое серебро слишком мягкое. Украшения и изделия из серебра содержат большей частью 80% Серебра. Эвтектика серебряпо-медпого сплава соответствует содержанию меди 28% , Эвтектический сплав имеет т. пл. 778°; ои плавится, следовательно, почти па 300° ниже, чем чистое серебро. Примесь меди вызывает заметную окраску только тогда, когда ее содержание превышает 50%. Применяемый для паяния серебра твердый серебряный припой. * содержат наряду с 20—35% меди обычно также еще 1.0—15% цинка, Серебрение изделий из неблагородных металлов происходит либо накатыванием на поверхность в печах, либо гальваническим путем.
Для химической Посуды (папример, тигли для плавления щелочей) прежде чаще всего применяли чистое серебро. Однако, по мнению Фрёлиха (Frohlich, 1939), для этой цела лучше подходит серебро, к которому добавлено небольшое количество-(0,1—i’,2%! пинеля. Этим предотвращается рекристаллизация металла, которая в противном случив приводит к тому, что серебро после долгого прокаливания становится хрупкий и растрескивается. Добавление Ni заметно пе снижает химическую устойчивость серебра.
В большом количестве используют серебряные солиидругие серебряные препараты. Важнейшей солью серебра является нитрат. В большинстве случаев оп служит в качестве: исходного продукта для приготовления
* Твердым припоем его называют потому, что он хорошо куется.
Побочная подгруппа первой, группы периодической системы 42&
других соединений серебра. Для терапевтических целей применяют часто-коллоидное серебро, которое обладает отличным стерилизующим действием при относительной пеядовитости.
Наиболее старым препаратом итого типа является колларгол (Argentum colloidale) — зеленовато- или синевато-черные листочки с металлическим; блеском, которые образуют мутные коллоидные растворы с небольшим: количеством воды и прозрачные, но ташке коллоидные растворы с болыпим-количеством воды. Большое применение находит также протаргол (Argentum proteinicum), представляющий собой желтый или коричневый порошок. Этиколлоидные серебряные препараты в качестве защитного коллоида содержат более или меиее значительные количества органических веществ, большей частью белка или продуктов распада белка.
Антибактериальное действие Йонов серебра (которое не распространяется, однако, на споры) удается наблюдать даже прн чрезвычайно большом (примерно 2- Ю-11 г-ионл/.ц разведении («ол и година мическое действие», открытое в 1893 г. Нагели (V. Naegeli)].
Металлическое серебро проявляет подобное действие только в том случае, если оно в каком-нибудь месте загрязпепо, так что могут образоваться локальные токи, под действием которых следы серебра переходят в раствор. В случае препаратов коллоидного серебра ввиду содержащихся в них примесей это условие всегда удовлетворяется.
СОЕДИНЕНИЯ СЕРЕБРА
Почти все соединения серебра производятся от одновалентного серебра . Соединения серебра нормальной валентности соответствуют, следовательно, общей формуле AgX. Только в исключительных случаях серебро является электрохимически бегства л ентпым и совсем редко трех-валептным (см. стр. 437 и сл.).
1. Соединения серебра(1)
Большое число соединений серебра практически нерастворимо в воде и отчасти также в кислотах (подробнее см. на стр, 392 и 428). Легкорас-творимыми вводе солями серебра являются прежде всего фторид , нитрат, хлорат й перхлорат *. Ацетат и сульфат также еще достаточно хорошо растворимы. То же относится и к нитриту, особенно при нагревании.
Ионы Ag бесцветны. Бесцветны также только что перечисленные соли, за псклю- ' чонием слабо-желтого нитрита. Нередко, однако, ион серебра образует с бесцветными анионами окрашенные соли. Большей частью эти соли желтые, как, например, Ag3PO4, Ag3AsO3, Ag2CO3, Agf. AgBr— светлый зеленовато-желтым, Ag3AsO4— шаколадно-коричневый, Ag2S — черный.
Если попы серебра вступают в соединение с уже окрашенными попами, то часто окраска углубляется; например, Ag2GrO4 -- красно-коричневый (К2СгО4— желтый), AgnC^O? — темно-красный (К2Сг2О7 — оранжевый); окраска перйодата серебра варьируется от желтой (Ag2H3IO6) до черной (Ag6IO6).
Те мп о-коричнев ан окись серебра в воде очень мало растворима, Однако она придает воде отчетливо основную реакцию, так как растворенная часть ее гидратируется до AgOH, которая практически полностью диссоциирует на ионы Ag‘ и ОН'.
Некоторые соли серебра, такие, как нитрат п сульфат, могут с соответствующеми (безводными) солями натрия образовывать твердые, растворы.
s Перхлорат серебра не только легко растворим, но даже гигроскопичен. Напротив, [Ag(pyr)2]C104 [получаемый взаимодействием с NnC104 раствора AgNO3, смешанного с цнрггдппом (руг)) в воде трудно растворим.
'426
Глаза S
Трудно растворимые в воде солн серебра большей частью легнс растворяются при добавлении некоторых веществ, таких, как аммиак, тиосульфат натрия, цианид калия и т. д. Это явление основано на чрезвычайной склонности серебра к образованию комплексных ионов.
Намерением равновесия в водных растворах удалось обнаружить главным образом следующие комплексные ионы серебра:
( I , .. I [Ag(CN)2]' I [Ag(SCN)J' I [45(8203),]'" I [Ag2IJ" t [Ag(NQ2)2]'
32 I [Ag(CN)3]’ I [Ag(SCN)J"’ I [4g{Saa3)3j""' I [A-gl.j'"* | (слабый комплекс)
На основами it состава кристаллизующихся из раствора солей просто определить вид имеющихся в них преимущественно комплексных ионов не удается, так как они определяются совместно с другими ионами, входящими при кристаллизации в решетку вместе с комплексным ионом.
Из комплексного соединения серебра с окси хинол ином, в котором серебро обладает координационным числом 4, Хейну (Flein, 1935) удалось получить две зеркальпо-изомерпые формы. Отсюда удалось заключить, что в комплексных соединениях коор-динациопио четырехвалентного серебра в противоположность соединениям платины, палладия и других металлов побочной подгруппы VIII группы (см. стр. 372) лиганды расположены тетраэдрически.
Окись серебра Ag20 возникает в виде черпо-норичневого осадка при добавлении ионов ОН' к растворам солей серебра. Можно предположить, что при этом сначала образуется гидроокись серебра AgOH и что последняя тотчас переходит в окись серебра, отдавая воду:
Ag’-[-OII'=AgOH, (За)
2AgOH=Ag2O4-H2O. (36)
Осадок прочно удерживает пасть осадителя (гидроокиси щелочного или щелочноземельного металла). Если осадитель не свободен совершенно от галогенов, то с окисью серебра осаждается также и галогенид серебра.
Суспензия окиси серебра в воде обладает отчетливой основной реакцией. Это свидетельствует о том, что процессы, описываемые приведенными выше уравнениями, могут протекать в обратном направлении, хотя и в незначительной мере. Если содержащиеся в водном растворе окиси серебра ионы Ag' связываются ионами галогенов, то для сохранения равновесия новые количества окиси серебра должны переходить в раствор. Это может привести к тому, что раствор, наконец, сильно обогатится ионами гидроксила.
В препаративной неорганической химии часто используют это свойство суспензии окиси серебра обменивать ионы гидроксила на ионы галогенов и другие ноны, которые дают с ионами серебра нерастворимые соединения.
Приготовленную для этого окись серебра обычно хранят под водой, так как после высушивания препарат пе смачивается достаточно хорошо водой. Кроме препаративной химии, окись серебра находит применение в медицине. Ее используют для этого часто в виде коллоидов, которые получают осаждением ее в присутствии защитных коллоидов (белка и т. п.) (саргол).
Раствор «мост i, окиси серебра в воде, в которцы концентрация СО2 находится в равновесии с концентрацией двуокиси углерода в воздухе, составляет, по данным Роми, 1,1-10-4 лг.01.,.ъ .г црц iSJ. В воде, абсолютно свободной от С02, растворимость
* Между [Agjili]" и [А^Г4Г" существуют, вероятно, такие переходные формы, как [Ag3I5]m, [Ag2/6]"” и j. д.
Побочная подгруппа, первой группы периодической системы
427
меньше. При 18° в соответствии с данными Лауэ (Laue, 1927) опа составляет <57• 10-* моль/л. Добавлением незначительных величеств ионов ОН' растворимость подавляется, большими количествами ионов ОН', напротив, повышается.
Повышение растворимости основано, по млению Лауз, па образовании иопов ; AgOJ' в сильнощелочном растворе.
В соответствии с этим окись серебра обладает не только основным, но также И кислым характером, правда, лишь в иезначительной степени. Если принять, что находящаяся в растворе окись серебра практически полностью существует в форме щтссоципроваппой гпдроошгеи AgOH, то из измерений Лауэ следует, что и чистом водном растворе часть ее, диссоциированная как кислота (т. с, с образованием иопов | AgO]'), составляет примерно одну десятитысячную от того количества, которое диссоциировано как основа пне. В растворе, содержащем 1/100 моля гидроокиси щея очного металла, количество гидроокиси серебра, диссоциированное как кислота, примерно равно количеству ее, диссоциированному как основание.
Теплота образования окиси серебра из элементов составляет 7,02 ккал, свободная энергия образования 2,45 ккал/моль при 25° (Benton, 1932). В соответствии с незначительной величиной свободной энергии образования окись серебра уже при небольшом нагревании начинает отщеплять кислород (при 160° в едва заметном количестве; прп 185—190“ давление кислорода достигает 1 атм). При освещении разложение происходит уже при обычной температуре. Водородом окись серебра восстанавливается уже при 100°. С перекисью водорода Ag2O активно реагирует уже при обычной температуре
Ag2O -|- TTgOa^aAg-i-HjO + (>2- (4)
В водном растворе аммиака окись серебра легко растворима с образованием комплексного соединения. Еще Бертолле было обнаружено, что при продолжительном выдерживании из такого раствора осаждается темный осадок, чрезвычайно взрывчатый даже во влажном состоянии,— гремучее серебро Ag3N [не следует смешивать со взрывчатыми соединениями — серебряной солью гремучей кислоты (фульминат серебра) AgONC к азидом серебра AgN3(. Склонность к разложению со взрывом у гремучего серебра так велика, что, как сообщает Рашиг, препарат, который требовалось промыть в чашке декантацией, взорвался, как только несколько капель воды из промывалки попало на него. Если на поверхности аммиачного раствора серебра образуется компактная корка гремучего серебра, то при сливании жидкости взрыв неизбежен.
Кристаллическое строение Ag2O подобно строению Сн3О ; а—4,74 A, Ag <—<) = = 2,05 А.
Низший окисел серебра (по сравнению с Ag2O) пе существует. Согласно рентгенометрическим определениям (Levi, 1924), рассматриваемые ранее в качестве низшего окисла продукты представляют собой смесь Ag3O и Ag.
ГАЛОГЕНИДЫ
Из галогенидов серебра чрезвычайно легко растворим фторид серебра, другие же галогениды трудно растворимы. Менее всего растворим иодид. Обзор важнейших свойств галогенидов приводен в табл. 44.
Фторид серебра AgF образуется при растворении окиси серебра в плавиковой кислоте; AgF очепь легко растворим в воде (1 ч. в 0,55 ч, при 15°). Из раствора оп кристаллизуется в виде гидрата (в зависимости от условий с 1 или 2 молекулами Н2О), II безводном состоянии AgF представляет собой слоистую кристаллическую массу. Оп способен поглотить 844 об. частей аммиака, Из растворов, содержавши избыточную плавиковую кислоту, выделяются двойные соединения AgF-НУ, AgF-3HF. Еще пе
428
Глада 3
Галогениды серебра
Таблица 44
AgF AgCl AgBr Agl
Молекулярный вес Удельный вес Окраска Температура плавления, °C 126,88 5,85 Белая 435° 143,34 5,56 Белая 450° 187,80 6,47 Бледно-желтая 419° 234,79 5,67 Желтая 552°
Растворимость При 25°, .коль!л 0<1ень легко растворима 1,31.16-6 О,725.1(Гв 1,00,10-8
Кристаллическая система и тип решетки Кубическая решетка типа каменной соли, а =4,92 А Кубическая решетка типа каменной соли, а=5,54 А Кубическая решетка типа каменной соля, я = 5,76 А a-Agl; кубическая решетка типа цинковой обманки, п=6,48 А 0-AgI: гексагональная решетка типа вюртцита, «=4,59, с =7,52 А. y-Agl: кубическая решетка со статистическим распределением атомов Ag (ем. стр, 431).
Теплота образования, ккал! моль 46,7 30,13 23,70 15,34
установлено, являются ли они фтороиислстами или продуктами присоединения типа гидратов (ифторогидрать»).
Фторид серебра Ag2F. Как впервые обнаружил ГуптЦ (Guntz, 1890), концентрированный раствор AgF может растворять серебро с образованием Ag2F, который осаждается из раствора в форме бронзовых с зеленоватым отливом гексагональных кристаллов (уд, вес 8,6). Вследствие своего состава, отличающегося от состава нормального соединения, Ag2F вызывает интерес. Оп является антиизотишвд Cdl2 (решетка типа брусита, см. г. J, стр. 293т рис. 61). Следовательно, Ag2F в противоположность AgF образует слоистую решетку. Более подробно его строение еще не изучено.
Хлорид серебра, хлористое серебро AgCl, осаждается в виде белого, творожистого, сжимающегося осадка при взаимодействии ионов Ag' и СГ. В природе AgCl встречается под названием роговое серебро (кераргирит). Эта серебряная руда, встречающаяся в больших количествах только в Америке, представляет собой окрашенную различными примесями, большей частью перламутрово-серую просвечивающую массу, состоящую из очень мелких кубических кристалликов с удельным весом 5,5—5,6. В воде хлорид серебра практически нерастворим. В аммиаке и концентрированной соляной кислоте он растворяется с образованием
Побочггая подгруппа первой группа периодической система
429
комплексных соединений. При упаривании таких растворов AgCl остается в форме очень мелких, одпако хорошо различимых под микроскопом кубиков. Осажденный хлорид серебра плавится при 450° с образованием оранжево-желтой жидкости, которая при охлаждении затвердевает с заметным увеличением объема в рогоподобную массу. Выше 1000° хлорид серебра заметно улетучивается. При этом он не претерпевает разложения. В парообразном состоянии он частично полимеризован. По данным Вар-тепберга, точка кипения его лежит при 1554°.
Сухой хлорид серебра абсорбирует аммиак с образованием соединений AgCl-NH3, 2AgCl-3NH3, AgCl'3NH3, которые также получаются из водного раствора (правда, только в том случае, если кристаллизация осуществляется при более высоком давлении). Как показали измерения равновесий, водно-аммиачные растворы хлорида серебра содержат преимущественно комплексный и он [А§(КНз)2)*. Помимо аммиака, хлорид серебра образует также легко растворимые комплексные соли с тиосульфатом натрия и цианидом калия. Вследствие образования комплексов хлорид серебра заметно растворим также в концентрированных растворах других хлоридов. Растворимость в концентрированной соляной Кислоте основана на образовании комплексных кислот, например HAgCl2.
Хлорид серебра в значительной степени растворяется также в горячей концентрированной азотной кислоте. То же относится л бромиду, нодиду, цианиду и роданиду -серебра. Из растворов кристаллизуются двойные соли: AgNO3- AgCl - бесцветные лризмы, т. пл. 160°; AgNO^-AgBr, т. ял. 182е; AgNO3-AgI, т. пл. 94°; 2AgNO3-AgI, т. ПЛ. 105е1; 2AgNO3-AgCft; 2AgNO3-AgSGN.
Большое практическое значение имеет светочувствительность хлорида серебра. Хлорид серебра окрашивается на свету с отщеплением хлора и выделением мелкодисперсного металлического серебра в лиловый, бледно-фиолетовый и, наконец, сине-зеленый цвет. На светочувствительности хлорида серебра основано его применение в фотографии (главным образом для копировальной бумаги, кроме того, для диапозитивных пластинок).
Если хлорид серебра облить водой или сильно разбавленной сорной кислотой и добавить цинк, то произойдет восстановление в соответствии с уравнением
2AgCl + Zn=2Ag+Zn” + 2Cl'. (5)-
Вислицепус (Wisllcenus) обнаружил, что в результате такого взаимодействия серебро можно получить в очень тонко диспергированном состоянии, так называемое «молекулярное серебро».
Для его приготовления пелесообразно использовать способ, списанный Гомбергом. В батарейный стакан впосят хорошо промытый водой AgCl, ставят па него мели опор нет у ю глиняную ячейку с укрепленной цинковой палочкой и заливают водой, в раствор погружают платиновую пластинку, которую соединяют с цинком: пос ре дот-лом приваренной к ней платиновой проволоки. Для ускорения реакции к находящейся в ячейке воде добавляют несколько капель соляной кисиотыи затем при помощи сифона выравнивают жидкость так, чтобы поверхность ее в ячейке была всегда ниже, чем в батарейном стакане. Это необходимо для того; чтобы, возможно меньше хлорида цинка диффундировало наружу. Полученное таким образом серое, порошкообразное серебро промывают вначале водой, затем (для удаления остатка AgCl) аммиаком, потом снова водой и, наконец, спиртом и эфиром. После высушивания в вакуум-экси-каюро над серной кислотой его еШе нагревают до 150° и в заключение для получения ip я пулометрически однородного состава просеивают.
Бромид серебра, бромистое серебро AgBr, очень похож на хлорид серебра, однако еще мепее растворим, легче восстанавливается и, что особенно важно, обладает еще большей светочувствительностью. В зави
430
Глава 8
симости от условий из водного раствора он выпадает в виде осадка белого или бледно-зеленовато-желтого цвета. Белая форма обладает самым небольшим размером зерен. Поэтому она относительно легко растворима. При выдерживании в соприкосновении с раствором бромида калия эта форма быстро переходит в зеленовато-желт у to труднорастворимую форму. В виде мягких агрегатов мелких кубических кристалликов оливково-зеленого или желтого цвета бромид серебра встречается в природе (бром-аргерит или бромит) главным образом в Мексике, но почти всегда в изоморфной смеси с хлоридом серебра. Бромид серебра находит широкое применение для производства фотопластинок, пленок и бумаги. В технике ого получают взаимодействием бромида калия и нитрата серебра в водном растворе.
В фото промышленности для приготовления бромосеребряной желатиновой эмульсии раствор желатины, содержащий бромид калия, смешивают с раствором нитрата серебра. Бромид серебра образуется вначале в тонкодиспергированном состоянии. Затем, в результате так называемого «созреваниям, т. е. продолжительного выдерживания в тепле с целью укрупнения зорен, он становится белее светочувствительным.
Иодид серебра, подпетое серебро Agl, еще более трудно растворим, чем бромид серебра. Ввиду исключительно низкого значения произведения растворимости Agl даже концентрированным раствором аммиака растворяется в едва заметном количестве.
В сильно аммиачном растворе ионы [Ag(NH3)2]‘ лишь в очень пезначительной степени распадаются во уравнению
|Ag(NH3)2]- Ag'+2NII3. ' (&)
Существующая при этом равновесии концентрация ионов серебра в сильно аммиачном растворе значительно ниже концентраций ионов серебра в водных растворах хлорида и бромида серебра; поэтому в их присутствии процесс, описываемый уравнением (в), протекает справа палево. Напротив, в водном растворе иодида серебра присутствует так мало попов Ад’, что комплексные аммиачносеребряные иопы образуются лить в пезпачитьлыюм количестве. Однако если на иодид серебра действовать реагентами, которые образуют еще более сильные комплексы с попом серебра, чем аммиак, папример цнапттд-иомами, то содержание ионов серебра в растворе уменьшается настолько, что даже становится меньше, чем это соответствует произведению растворимости иодида серебра ([Ag'Jx [I'] == 10-1й).
Таким образом, даже эту особенно трудно растворимую соль можно перевести в раствор.
В концентрированной подпет овод о родной кислоте и в концентрированных растворах подндов щелочных металлов иод ид серебра заметно растворим, особенно при нагревании. Из этих растворов можно получить комплексные соли, например K.[Agl2] и Кг[Лц1з], представляющие собой бесцветные кристаллические соединения.
Осажденный из водною раствора иодид серебра имеет желтый цвет. Под действием света оп постепенно становится зелонолато-серо-черным. В результате обработки концентрированным водным раствором аммиака иодид серебра становится чисто белым. Это объясняется тем, что образуется соединение AgL bpNHj.
Сухой тюдид серебра в зависимости от давления поглощает 0,5; 1; 1,5; 2 илп 3 молекулы NH3, в то время как бромид серебра образует такие же продукты присоединения аммиака, как и хлорид серебра. Устойчивость аммиакатов снижается от хлорида к нодплу.
Йодид серебра существует в трех модификациях (см. табл. 44, стр. 428). При осаждеппи иодида серебра из водного раствора и при избытке ионов Ag- получают устойчивую при обычной температуре a-модификацию * кристаллизующуюся по типу ципковой обманки. Прп 13Т' опа превращается в р-моДификацию, кристаллизующуюся по типу цюртцита. Последняя получается при обычной температуре в мета стабильном
* Иногда устойчивую выше 146° модификацию обозначают как a-Agl, а устойчивую при обычной температуре — как y-Agl.
Побочная подгруппа первой группы периодической систем и 431'
состоянии, если осаждение ведут избытком иопов I'; одттако уже при растирании осадка происходит превращение его в «-модификацию.
у-Модпфпкация стабильна выше 146°. Она образует кубическую решетку, в которой атомы иода занимают углы и центры элементарных ячеек (кубы с длиной ребра у.'13 А),в то время как атомы серебра совершенно неупорядоченно распределены на SJ вакантных местах решетки. Расстояние Ag*—i> ly a-Agl составляет 2,80 А, у [5-Agl — 2,78 А (цри комнатной температуре); у у-Agl оно колеблется между 2,52 и 2,86 А (при 146°).
Устойчивая выше 146° у-м о дификация интересна тем, что, кат; впервые показал I-рупп (Bruni, 1913), опа обладает чисто электролитической проводимостью, причем в значительной степени. Как показал Тубапдт (Tubandt), перенос тока в этом случае осуществляется исключительно попами Ag+: попы 1_,в нем не участвуют. Удельная , лектропроводпость y-Agl составляет при температуре превращения 1,31 обратных ома (лю), н то время как удельная электропроводность P-AgI при той же температуре равна только 0,00034 .но. Для сравнения следует указать, что удельная электропроводность максимально проводящей серной кислоты прп 18° составляет 0,739 .во. При температуре плавления (552°) удельная электропроводность y-Agl равна 2,64 лю и превышает, таким образом, удельную электропроводность расплава прп той же температуре, которая составляет лишь 2,36 .ко. Напротив, у бромида серебра и хлорида серебра, которые также обладают чисто электролитической проводимостью *, удельная электропроводность возрастает при плавлении от 0,53 до 2,76 и от 0,12 до 3,76’ соответственно. Исходя из найденной Штроком (Struck) структуры решетки, становится понятной особенно высокая электропроводность y-Agl. То обстоятельство, что иопы Ag+ пе пметот определенных мест в решетке, должно существенно облегчить их перемещение. Правда, неупорядоченное распределение одного типа попов в кристаллической решетке пе является необходимым условием для появления электролитической проводимости в твердом состояния, так кап она проявляется, хотя и слабее, далее у таких соединения, как AgBr и AgCl, ионы которых занимают определенные места в решетке. Возможность перемощения для таких ионов обусловлена имеющимися в кристаллах искажениями решеток и дефектами структур (см. стр. 36 и сл.).
В фотографии иодид серебра служит для приготовления коллодиево-эмульсиоп-иых пластинок. Их освещают непосредственно после приготовлении ещо во влажном-состоянип, проявляют сульфатом жслсза(П) в присутствии нитрата серебра и фиксируют раствором цианида калия. II од о серебряные коллодисвые пластинки дают особо контрастное изображение, однако вследствие неудобства их использования их мало-применяют. Наиболее старый процесс фотографии с проявлением — дагерротипия— чепованна светочувствительности иодида серебра. Дагерр действовал сначала на. серебряную пластинку парами иода (т, е. получал топкий слой йодида серебра) и затем,’после облучения, подвергал содействию паров ртути, которые конденсировались на этой пластинке преимущественно па облученных местах.
Применение галогенидов серебра в фотографии
Применяемые в настоящее время фотопластинки.и пленки представляют собой стеклянные пластинки ц тонкие целлулоидные или цел лиловые ** полосы, которые покрыты слоем коллоидного галогенида серебра. В большинстве случаев речь идет о коллоидном растворе бромистого серебра в желатине, реже в коллодии Для специальных целей (репродукции чертежей и диапозитивов) применяют хлоро-серебряные желатиновые пластинки или пластинки с хлористым и бромистым серебрим. Опи хотя и менее светочувствительны, чем бромосеребряные пластинки, однако работают контрастнее ('.жестче»). Под влиянием света происходит разложение следов галогенида серебра па освещенных местах. Освобождающийся при этом галоген абсорбируется желатиной. Освобождающееся одновременно металлическое серебро выде-.тю-тся в виде крайне мелких, различимых лишь в ультрамикроскоп частиц. При б идее слабом пли кратковременном освещении число частиц серебра примерло про-порпиональио количеству падающего света. Если теперь обработать слои, который ешс ио обнаруживает никакоговидпмого для глаза изменения )«скрытое изображение»),
* Перенос тока у этих солей также обеспечивается только ионами Ag+. Примером соли, у которой в твердом состоянии ток переносится только аннонами, служит хторггД свинца.
** Целлит [(вторичная) ацетилцеллюлоза] обладает по сравнению с ijc.mу.гоидом, полученным из нитроцеллюлозы и камфоры, тем преимуществом, что труднее восила-мелиотся. Его недостатком является, однако, значительное изменение объема при тв"отжценпи и высыхании.
432
Глава S
восстановителем («проявителем# *), то произойдет восстанем я епие остальной части галогенида серебра до серебра. Имеющиеся уже мелкие частички серебра служат про этом центрами кристаллизации («зародыши») металлического серебра. Они ла столько облегчают выделение серебра, что восстановление сначала происходит почти исключительно в непосредственной близости от этих частиц, т. е. на ранее освещенных Местах.. •Поэтому в этих местах слой чернеет. После достижения необходимой степени почернения оставшуюся часть пеизменившегося галогенида серебра извлекают в «фиксирующей ванне*, так как иначе оно также в конце концов почернеет. Для фиксации применяют обычно тиосульфат натрия Na2S2O3. Однако для этого можно также применять и другие соединения, которые переводят галогениды серебра в растворимые комплексы, например цианид кадия. Полученное вначале изображение называют «негативом*. В нем наиболее почерневшими являются те места, которые поглотили больше света, т. е. те, которые в оригинале были наиболее светлыми.
Чтобы получить «позитив* с правильным распределением освещении х й затемненных мест, следует в принципе процесс повторить, Это достигается путем «копи-ровапия*. Пегатяв кладут па бумагу, покрытую светочувствительным слоем, яле для получения «диапозитива®—iia пластинку или пленку. Если теперь осветить негатив, то зачерненные в негативе места защищают светочувствительную эмульсию си освещения, в то время как незачерпеппые пропускают свет. Таким образом., происходит новое обращение темных и светлых мест, так что их расположение теперь совпадает с оригиналом.
Для получения фотокопировальной бумаги вместо бромида серебра можно использовать хлорид серебра, так как в этом случае светочувствительность должна быть нс слишком велица. Прежде Для этого часто применяли слои, а которых, как, на пример, в целлоидиновой бумаге, почернение наступало уже при освещении, так чтс особого проявления далее но требовалось, и только еще нужно было провести фиксирование. Однако при применении целлоидиновой бумаги наступает нежелательнее изменение окраски рисунка. Поэтому целлоидиновую бумагу следует «тоиировате* перед фиксацией или. в процессе фиксации. Процесс тонирования основан па том что при погружении в ванну с раствором соли золота серебро обменивается на боле; благородное золото: 3Ag -f- Au'" = 3Ag‘ -|- Au. Вместо солей золота для тон и ров анис можно применять также соединения селена. Особенно красивые изображения получают при тоиировапни бумаги солями платины.
В настоящее время для изготовления копий применяют большей частью бумаге,. требующие проявления. Их слой состоит либо из бромосеребряной, либо из хлорэ-н бромосеребряной желатиновой эмульсии,
ДРУГИЕ СОЛИ СЕРЕКРД
Нитрат серебра AgNCu — технически наиболее важное соединена серебра, обычно получают растворением металлического серебра в азотной кислоте. Если металл содержит медь, то его очищают, осаждая рас.твз-ренное серебро поваренной солью в виде хлорида; AgCl восстанавливала до металла цинком в присутствии небольшого количества разбавленнсА серной кислоты или кипятят с виноградным сахаром или формальдегида^-и натровой щелочью. Полученный металл вновь растворяют в взотне^. кислоте.
Нитрат серебра представляет собой бесцветные ромбические нсгнгрг:-скопичные кристаллы; удельный вес его 4,35. Выше 159,6° стабилыд: гексагонально-ромбоэдрическая модификация (удельный вес 4,19). Точка плавления лежит при 208,5°. В воде нитрат серебра крайне легко растворим. Раствор реагирует нейтрально. Он обладает горьким металлическим вкусом: с повышением температуры растворимость нитрата серебр. .значительно возрастает (при 20° в 100 а воды растворяется 222 a, при 100° 952 г AgNOJ.
* В качество проявителя исп«жзую2_бояьшей частью растворы органичеекпх веществ, например гидро?<иттотта НО——ОН, n-амипофепола (<<род1!на.п>
И п
N.CH3 !
Н _1а
НО-
SO4.
или «метола®
Побочная подгруппа первой группы периодической системы
433
Концентрированной азотной кислотой нитрат серебра частично выделяется из своего насыщенного водного раствора. В разбавленном этиловом спирте оп лишь умеренно растворим (3,8 а в 100 г 92.5% -пого спирта прн 15°), то же относится и к метиловому спирту. Еще менее растворяется он в чистом ацетоне (0,35 г в 100 г при 15°) и бензоле.
Многими органическими соединениями, ианример бумагой, виноградным сахаром, винной кислотой, нитрат серебра легко восстанавливается до металла. При действии последних из аммиачного раствора металл осаждается на стекле в виде блестящего зеркала. Это используют как для обнаружения винной кислоты, так и прежде всего в производстве зеркал. Яичный белок коагулируется нитратом серебра. Поэтому нитрат серебра действует на органические ткани разрушающе. На этом основано его дезинфицирующее действие, используемое в медицине («адский камень»). Большие количества нитрата серебра используют в фотографической промышленности для производства светочувствительных пластинок, пленок, бумаги.
В систематическом анализе нитрат серебра находит широкое применение наряду с нитратом бария или хлоридом бария как групповой реагент на анионы кислот.
Если в раствор, содержащий один из ионов С]', Вт', J', CN', SCN', СЮ', 103, [Ее(СМ)й]'", [Fe(CN)el"", S", добавить ионы серебра, то даже в присутствии свободной аготнцй кислоты выделится осадок. IIе из азотнокислого, а из уксуснокислого раствора ионы Ап' осаждают: С4О4, CrOJ (соответственно CraO,), SOg, S2O3*, (POg)„, РгО?'", HPOg, BrOj. Только из нейтрального раствора ионами Ag' осаждаются РО^', АйО4", АзОд'' (соответственно .АзО^), ВОд” (соответственно ВОд), Si Од (соответственно йт20д) и некоторые органические иопы, например Ионы COJ также дают
с Ag' в нейтральном растворе осадок. Поэтому, если, как это обычно бывает, анализ па анионы проводят в содовой вытяжке, угольную кислоту следует сначала удалить путем подкисления и Кипячения. Только затем можно начать испытание на перечисленные в конце а пионы путем добавления нитрата серебра к раствору, предварительно осторожно нейтрализованному аммиаком.
Нитрат серебра служит также реактивом на мышьяковистый водород в способе ГутцеЙта (ср. т. I, стр. 712),
С нитратами калия и таллия(1) нитрат серебра образует двойные соли, а с нитратом натрия — твердые растноры. В то время как область отсутствия растворимости у нитрата натрия и нитрата калия в твердом состоянии при 218° лежит в пределах 15—75 мол.% NaNO3, у нитрата натрия и нитрата серебра в твердом состоянии эта область при той же температуре лежит только в пределах 26—38 мол.% NaNO3. Правда, в обоих случаях границы области отсутствия растворимости расширяются с паде- •-пнем температуры.
Нитрит серебра AgNO3 выделяется в виде бледно-желтого осадка при добавлении ионов NO1 к растворам солей серебра. Растворимость его при 18° равна 0,332 з в 100 г воды. Она заметно возрастает с температурой. При охлаждении горячего раствора кристаллизуется соль в виде почти бесцветных ромбических игл, тонких, как волосы. С избытком нитритов AgNO3 взаимодействует с образованием комплексных солей типа М1 [Ag(NO2)2]. Электрометрическими намерениями Аббег доказал существование в растворах этих комплексных солей ионов [Ag(NO2)3]\
На свету нитрит серебра черпеет. В водном растворе, особенна при кипячении, он также постепенно разлагается по уравнению
2AgNO2 = AgNO3 + NO + Ag.
При пагревании сухой соли происходит распад па серебро и двуокись азота. В качестве промежуточных продуктов при этом сначала образуются AgNO3 и NO.
Нитрит серебра применяют в препаративной органической химии для получения алифатических нитро-соединений и эфиров азотистой кислоты.
* Ионы S2O3 не осаждаются, если опц имеются в избытке по отношению к ионам Ag'.
28 г, Реми
434
Глава S
Сульфат серебра Ag2SO4 образует бесцветные ромбические кристаллики; удельный вес его 5,45; температура плавления 660°, В воде он довольно трудно растворим (0,80 г Ag2SO4 в 100 г воды при 25°), легче растворяется в разбавленной серной кислоте. Из раствора в серной кислоте кристаллизуются кислые сульфаты, например AgHSO4, которые снова разлагаются водой на нормальную соль и серную кислоту. При плавлении в токе сухого хлористого водорода сульфат без остатка переходит в хлорид Ag2SO4 + 2НС1 = 2AgCl + H2SO4. Вследствие малой растворимости хлорида серебра такое же взаимодействие происходит и в водном растворе.
Получение сульфата серебра ведут либо растворением порошкообразного серебра в концентрированной серной кислоте 2Ag + 2HaSO4 = = Ag2SO4 4- SO2 + 2H2O, либо добавлением сульфата щелочного металла или разбавленной серной кислоты к концентрированному раствору нитрата серебра; 2AgNO3 + Na2SO4 = Ag2SO4 + 2NaNO3.
Как наглел Маншо (Mancfaot, 1924), раствор сульфата серебра в концентрированной серной кислоте поглощает значительные количества СО, особенно при низкой температуре. Разбавленный серпокислый раствор не растворяет СО.
Сульфит серебра Ag2SO3 выделяется при добавлении сернистой кислоты или сульфита натрия к раствору нитрата серебра в виде белого, мало растворимого в воде осадка, который при освещении становится пурпурным и наконец черным. При кипя-чении с водой происходит разложениев соответствий с уравнением 2Ag2SOi3 = Ag2SO4 + + SO2 -|- 2Ag. В разбавленных сильных кислотах сульфит серебра растворяется с разложением. В избытке сульфитов щелочных металлов он растворяется с образованием комплексных солей, преимущественно состава Mr[Ag(SO3)].
Тиосульфат серебра Ag2S2O3 выпадает при добавлении тиосульфата натрия к раствору нитрата серебра в виде белого трудно растворимого в воде осадка, который очень легко растворим в избытке осадителя с образованием комплексной соли. После высушивания он представляет собой белый порошок, сладковатый на вкус. Разбавленный сильными кислотами, би разлагается, медленно разлагается даже водой, причел в последнем случае с выделением сульфида серебра Ag2S и с образованием серной кислоты.
Из растворов тиосульфата серебра в избытке растворов тиосульфатов щелочных металлов можно выделить различные по составу комплексные соли (тиосульфатоарген-таты), например:
Na2S2Oa • 3Ag2S2O3 -2Н2О, 5NaBS2O3 • 3Ag3S3O3 4НнО, 3K2S2O3-Ag2S2O3 -2Н2О,
Na2S2Os-Ag3S3O3-2H2O, 5Na2S3O3 • Ag3S3O3-4H2O, 5K2S2O3- 3 A g3S2O3.
За исключением приведенной вначале соли натрия, все они отличаются большой растворимостью в водо. Их образование, которое происходит, даже если труднорастворимые серебряные соли, такие, как хлорид, бромид и иоднд серебра, обработать растворами тиосульфатов щелочных металлов, имеет важное значение для «фиксирования» в фотографическом процессе. Чтобы уменьшить образование названной выше менее растворимой соли натрия, следует использовать не слишком разбавленные растворы тиосульфата. Из перечисленных комплексных солен ни одна пе переходит в раствор без превращения. По данным Шмитц-Дюмопта (Schmitz-Dumont 1940), в их растворах имеются преимущественно тритиоеулъфато-ионы [Ag(S2O3)3]""'.
Карбонат еерсбра Ag3CO3 выделяется в виде светло-желтого, порошкообразного осадка при смегапвапни раствора нитрата серебра с карбонатом иди гид р ока р б опа том щелочных металлов. Если для осаждения используют карбонат, следует избегать его избытка, таг; как в противном случае осадок загрязняется окисью,
Растворимость карбонита серебра в воде очень незначительна. Существенно легче он растворяется в концентрированных растворах карбонатов щелочных металлов, образуя легко растворимые комплексные соли, например K(Ag(CO3)], которые можно выделить даже я кристаллическом состоянии.
Уже при небольшом нагревании карбонат серебра отщепляет двуокись углерода.. (При 132° давление С02 составляет 6 atat рт ст, а при 218° 752 мм рт ст)
Побочная подеруппа первой группы периодической системы 435
Ацетат серебра Л§СгН3О2 кристаллизуется при охлаждении раствора карбоната серебра в горячей уксусной кислоте в виде бесцветных блестящих плоских гибких игл. Он несколько более растворим, чем сульфат (растворимость его при 20° равна 1,04 з, при 80° — 2,52 г в 100 г воды).
Оксалат серебра Ag2C2O4 выпадает при добавлении растворимых оксалатов к растворам нитрата серебра в виде белого, нерастворимого в воде и в разбавленной уксусной кислоте осадна. В избытке осадителя осадок также нерастворим, так как образования комплекса с избыточными иопами С204 нс происходит. Оксалат серебра разлагается при нагревании до 110°; при быстром нагревании происходит детопация.
Растворимость оксалата серебра при 18° составляет 0,004 г в 100 е воды.
Цианид серебра AgCN выпадает в виде белого осадка при добавлении ионов CN' к растворам солей серебра. В воде, а также разбавленных сильных кислотах цианид серебра практически нерастворим. (В 1 л воды растворяется примерно 2-10"® моля.) Напротив, он очень легко растворяется в растворах цианидов щелочных металлов (вследствие образования комплексных солой).
В водном растворе цианид серебра также существует в форме комплексных ионов: 2AgCN = Ag’ + [Ag(CN)2]'. В других вызывающих диссоциацию растворителях (например, в пиридкне, в котором цианид серебра растворяется значительно легче, чем в воде) отчасти имеются мпот оядорныо комплексные попы, например IAg2(CN)3|' и [Ag3(CN)4J',
В горячем концентрированном растворе карбопата калия цианид серебра также существенно легче растворяется, чем в поде. Он осаждается оттуда цри охлаждении в виде тонких игл с уд, весом 3,943. В водном растворе аммиака оп лишь умеренно трудно растворим. (В 100 мл 10%-ного водного раствора аммиака при 18° растворяется 0,52 с AgCN.) При нагревании циаттид серебра отщепляет половицу циана, и остается «парациановое серебром.
Растворимость цианида серебра в растворах цианидов щелочных металлов объясняется образованием легкорастворимых комплексных солей (циапоаргептатов). В качестве примера можно назвать дицианоар-гентат калия K[Ag(CN)2J, который кристаллизуется в виде бесцветных шестиугольных табличек. При 20° 1 ч. его растворяется в 4 ч. воды. Эта соль образуется также, если действовать раствором цианида калия на металлическое серебро при доступе воздуха.. Она является прочным комплексом, еще более прочным, чем комплексная соль с тиосульфатом. Поэтому? концентрация ионов Ag" в растворах комплексных цианидов серебра крайне мала. Это проявляется также в очень значительном повышении напряжения выделения серебра из таких растворов; оно так велико, что серебро не выделяется даже теми металлами, которые в ряду напряжений стоят значительно левее серебра, например цинком. На атом основано применение комплексных соединений цианида серебра с цианидами щелочных металлов при гальваническом серебрении. Если бы на подлежащих серебрению менее благородных металлах, помещенных в раствор нитрата серебра, серебро осаждалось в результате химического вытеснения, это. привело бы к образованию рыхлого непрочного покрытия. Из комплексных: цианидных растворов серебро, напротив, осаждается только под действием достаточно высокого напряжения, в этом случае оно выделяется в компактной форме в виде плотного покрытия.
Цианид серебра кристаллизуется в гексагонально-ромбоэдрической системе. Другой модификации (в противоположность старым представлениям) не существует, однако, по данным Патта (Natta), AgCN может в широком интервале образовывать кубические твердые растворы с бромидом серебра.
28*-
436
Глава 8
Роданид серебра AgSCN выпадает в виде белого творожистого осадка при взаимодействии растворов солей серебра с ионами SCN'. Осадок практически нерастворим в воде, а также в разбавленных сильных кислотах. (Растворимость его при комнатной температуре составляет 0,8 X X Ю"6 молъ/л.) Напротив, он легко растворяется в избытке осадителя, В этом случае также образуются легкорастворимые комплексные соли (роданоаргентаты).
Из рода и од pre итогов калия в кристаллическом состоянии получены следующие! K[Ag(SCN)a], K2[Ag(SCN)3], K3[Ag(SGN)J.
С бромидом серебра роданид серебра может образовывать твердые растворы, правда в очень ограниченном интервале (область отсутствия растворимости 3—90 вес,% AgBr).
Фосфат серебра. Ортофосфат серебра Ag3PO4 осаждается из растворов ортофосфатов при добавлении нитрата серебра в виде желтого трудно растворимого в воде осадка, который, однако, легко растворяется в разбавленных кислотах. Из уксусной кислоты или разбавленной фосфорной кислоты его можно выделить в кубических кристаллах удельного веса 7,32. На свету соединение постепенно чернеет. Ag3PO4 находит применение в медицине.
Из пиро- и метафосфатных растворов нитратом серебра осаждаются белые осадки Ag4P2O7 и Agi?O3. Последний существует в трех модификациях, которые различаются своей растворимостью и поведением при плавлении.
Сульфид серебра, сернистое серебро Ag2S, выпадает в виде черного осадка при пропускании сероводорода в растворы солей серебра. Он образуется также при действии сероводорода или сульфидов на металлическое серебро. Это наиболее труднорастворимая соль серебра. На основании потенциометрических определений Елинек кашел для произведения растворимости L— [Ag*]2 X [S"] значение 5,7-10~et. Теоретическая растворимость сульфида серебра, т. е. растворимость, которую имел бы сульфид серебра в совсем чистой воде, если бы он растворялся в пей при практически полной диссоциации, не подвергаясь гидролизу, оказывается отсюда 1,8-10-17 молъ/л (при 10°).
Пропусканием паров серы над раскаленным докрасна серебром получают сульфид серебра в кристаллическом состоянии. В природе он встречается в виде серебряного блеска (аргентит) в форме темных свинцово-серых кристаллов, в большинстве случаев кубического габитуса. Уд. вес его 7,2—7,4, твердость 2y-2i/2- Встречающаяся иногда остроконечная форма серебряного блеска, называемая акантитом, по внутреннему строению идентична аргентиту.
AgaS кристаллизуется в ромбической системе, однако по своему внутреннему строению, по-видимому, весьма близок кубической Си2О. При нагревании до 180’ AgjS претерпевает превращение с образованием кубической решетки. Теплота образования сульфида серебра составляет, по данным Рота (Roth, 1935), при 20е 6,о ккал 1моль.
Будучи обработан концентрированными растворами сульфидов щелочных металлов, сульфид серебра переходит, по мнению Дитте (Ditte), в кристаллические двойные соли красного цвета, например Na2S-3Ag2S-•2Н2О, K3S-4Ag2S-2H2O. В природе очень распространены двойные соли сульфида серебра с сульфидами кислого характера, прежде всего с сульфидами сурьмы и мышьяка. К ним относятся прустит 3Ag2S-As2S3 (соответственно Ag3[AsS3]; ксантоконит 9Ag2S-2As2S3-As2S5 (встречается
Побочная подгруппа первой группы периодической системы 437
редко); полиаргирит 12Ag2S-Sb2S3; стефанит 5Ag2S-Sb2S3; пираргирит 3Ag2S • Sb^a (соответственно Ag31SbS3l); миаргирит Ag2S • Sb2S3 (соответственно Ag[SbS2I); серебряновисмутовый блеск Ag2S-Bi2S3 (соответ* ственно Ag[BiS2].
Следует обратить внимание па существование соединения [SAg3](NO3], в котором в качестве структурной группы выступает комплексный катион с электроотрицательным центральным атомом и электроположительными лигандамц. Напротив, в случае Ag3SBr и Ag3SI, получеппых Рейтером (Reuter, 1960), речь идет о соединениях с ярко выраженной координационной решеткой (типа «антиперовскита»).
2. Соединения серебра(И)
Из соединений, содержащих двухвалентное серебро, известны дифторид серебра AgF2, кроме того, AgO, получаемая действием озона на металлическое серебро или анодным окислением серебра, а также ряд комплексных соединений типа [AgnAmJX2, где Ат — нейтральная часть, например органическое азотистое ангидрооснование.
Добавлением насыщенного на холоду раствора пер оксосульфата калия к раствору нитрата серебра и пиридина (C5H5N) Барбьери (Barbieri, 1912) получил соединение (AgII(GjH5N)4]SsOs |пероксосу льфат тетрапиридинсеребра(И)] в форме прекрасных оранжевых призм, которые образуют твердые растворы с соответствующим соединением Двухвалентной меди. Позднее (1927) электролитическим окислением раствора, содержащего нитрат серебра и пиридин, Барбьерц смог получить также соответствующий нитрат [AgII(C5H6N)jl][NO3]2, который также образует оранжево-красные кристаллы. Вскоре после этого Хиберу (Hieber, 1928) удалось приготовить ряд солей серебра(П), которые f вместо пиридина содержали азотистое ангидрооснование— о-фенантролин, комплексно связанный с серебром.
где Х=С1О4, С1О3, NO3, HSO4, V2S2Og.
Добавлением пероксосульфата аммония к раствору, содержащему нитрат серебра и о-фенантролин в соотношении 1 ; 2, он получил сначала шоколадно-коричневый нероксосульфат и из него приготовил двойным обменом другие также коричневые соли. С солями двухвалентных меди и кадмия соответствующего состава эти соединения образуют твердые растворы в любом соотношении, следовательно, они должны обладать одинаковым строением и, таким образом, должны содержать серебро в двухвалентном состоянии. Двухвалентность серебра в этих соединениях подтверждена наблюдениями Клемма (Klemm, 1931), который показал, что они обладают таким же парамагнетизмом как и соответствующие соли меди(П).
Простыми соединениями, в которых присутствует двухвалентное серебро являются дифторид серебра и окись серебра(1Л).
Дифторпд серебра, фторид серебра(П) AgFa, образуется, как впервые наблюдал РУФФ (Ruff, 1934), непосредственно, если на металлическое серебро действовать фтором при очень высокой температуре. С компактным серебром фтор реагирует ниже температуры красного каления и только с поверхности; выше 450° образуется монофторид AgF. Однако если действовать фтором на так называемое «молекулярное серебро» (ср. стр. 429), то уже при обычной температуре начинается реакция с образованием дифторида. По данным Рохова (R о с h о w, J. Am. Chem. Soc., 74, 1615 (1952)], AgF2 удобнее получать при пропускании C1F3 над AgCl. Дифторид серебра (уд. вес 4,7; т. пл. 690°) имеет глубокую темно-коричневую окраску и обладает достаточно сильным парамагнетизмом (ниже —110° оп даже ферромагнитен). Водой тотчас гидролизуется
438
Глава 8
причем в качестве промежуточного продукта образуется, по-видимому, Ag(OH)F который, однако, очень быстро распадается с образованием AgF и кислорода, содержащего озон. Дифторид серебра легко отдает фтор другим вещостиам и поэтому является отличным фторирующим агентом. Его теплота образования из элементов составляет, согласно Вартепбергу (\. Wartenberg, 1939), 84,5 ккал/молъ.
Окись ееребра(11) AgO. Если через раствор нитрата серебра пропускать электрический ток, применяя платиновый анод, то на нем осаждаются черпые кристаллы. Это паб люда л Риттер (Ritter) еще в 1799 г. Состав этих кристаллов обычно передается формулой Ag7NOn или AgjO8(NO3), однакщ по мнению Нойеса (Noyes, 1937), ему соответствует формула Agj30j5(NO3)E. При обработке этих кристаллов кипящей водой получают окись серсбра(П) AgO в виде черного порошка. В воде он нерастворим, однако растворяется в сильных кислотах, например в концентрированной хлорной кислоте (со светлой коричнев ат о-жеят ой окраской) в 6 н. HNO3 (с темно-коричпевой окраской, принадлежащей нитрат о-комплексам). Окись серебра (П) можно получить также окислением окиси серебра(Т) пороке одисульф а том (Barbieri, 1950) или действием озона на окись серебра(Т). Черная окраска металлического серебра, появляющаяся при действии па пего озона (ср. стр. 423), которую ранее приписывали образованию перекиси серебра, обусловлена окисью серебра(П). Это подтвердил Шваб (Schwab, 1955) рентгенометрически и сравнением ее каталитической активности с активностью препаратов окиси серебра, полученных другими путями.
3. Соединения ееребра(Ш)
Соединением, в котором серебро электрохимически трехвалентно, является фторо-соль KjAgFJ, полученная Клеммом (Klemm, 1953).Помимо этого известно еще несколько сложных комплексных соединений серебра(Ш). Речь идет о теллурато-и перйодат ос оо ди нениях, таких, как NaeH3[Ag(TeOe)2] и KeH[Ag(I0e)2], которые получил Малатоста (Malatesta, 1941), а также о некоторых полученных Райем (Ray, 1944) органических комплексных солях серебра(Ш), правда, с комплексным катиопом. Соединения обоих типов, как с комплексным анионом, так и с комплексным катионом, диамагнитны; следовательно, эти соединения являются комплексами внедрения.
Уже Нойс (Noyes, 1937) наблюдал, что при электролитическом окислении AgO, растворенной в хлорной кислоте, осаждается соединение серебра(Ш). Одпако оно устойчиво лишь при соприкосновении с раствором и очень быстро разлагается после выдел епия,
Аналитические сведения. Характерным для серебра является его способность осаждаться из растворов его простых солей под действием соляной кислоты в виде белого творожистого хлорида серебра, растворимого в водном растворе аммиака. Кроме того, характерна его легкая восстанавливаемость до металла, например при нагревании его солей па угле с содой при помощи паяльной трубки. Блестящие белые ковкие серебряные зерна удается легко перевести в раствор нагреванием с разбавленной азотной кислотой. Помимо указанного белого осадка с соляной кислотой, раствор солей серебра дает черный осадок с сероводородом. (сульфид серебра), а после предварительной нейтрализации дает красно-коричневый осадок с хроматом калия (хромат серебра AgECrO4) и желтый осадок с кислым фосфатом натрия (фосфат серебра Ag3PO4).
Для микро а на.литических определений, помимо хромата серебра, очень подходит хлорид серебра, кристаллизующийся в характерных формах при упаривании его аммиачного раствора (граница чувствительности 0,1 у Ag). Еще чувствительнее предложенная Фишером (Ft s с It е г Н., Z. Angew. Chem., 42, 1025 (1929)] реакция с дитизоном; CtiTI 5 — N — N — CS — NII — Nil — Cfh II5 (дифепилтиокарбазон); однако она пе применима в присутствии солей ртути и благородных металлов.
В eecoso.w анализе серебро осаждают либо как хлорид, либо электролитически в виде металла и взвешивают. Для осаждения из растворов, в которых оно присутствует в виде комплекса, по данным Шфейгманна (Steigmann, 1923), часто успешно применяют восстановление серебра
Побочная подгруппа первой группы периодической системы
439
до металла дитиопатом натрия. Часто определение серебра производят титрометрически.
При титровании по Гей-Люссаку к аз от в он нс лому раствору серебра добавляют раствор хлорида натрия известной концентрации до тех пор, пока еще выделяется осадок. При титровании по Моору нейтральный раствор нитрата серебра прикапывают к нейтральному раствору хлорида пат р ня (известного содержания, если требуется определить содержание серебра н растворе) и в качестве индикатора для установления конца реакции иегтользутот хромат калия. Растворимый! легче, чем хлорид серебра, красный хромат серебра образуется только после того, как все ионы хлора будут удалены. По методу Фольгарда титровапие ведут роданидом аммония или роданидом калия и в качестве индикатора используют раствор соли железа(Ш).
В исследуемом растворе белый роданид серебра образуется до тех пор, пока в нем имеются еще ионы Ag'. Как только они исчезнут, появляется красное окрашивание вследствие образования роданида железа(Ш).
По данным Яндера (Jander, 1937), небольшие количества серебра удается определить кондуктометрическим титрованием раствором хлорида натрия даже в присутствии большого избытка солей свинца.
ЗОЛОТО (AURUM) Ап
Распространение в природе. В соответствии с характером благородного металла золото встречается в природе большей частью в самородном состоянии, причем в двух формах: ископаемого золота и намывного золота.
Ископаемым золотом называют золото, находящееся в местах его первичного залегания. Оно бывает вкраплено в виде мельчайших блесток в кварц, иногда встречается в качестве примеси также преимущественно в кварцевых породах, в форме тонких включений или более мощных жил, пролизывающих сульфиды (например, серный колчедан, медный колчедан, мышьяковый колчедан, сурьмяный блеск и т. д.). Наконец, оно встречается с перечисленными сульфидами в форме химических соединений, и прежде всего с теллуром". калаверит АнТе2; сильванит AgAuTe^, нагиагит (листообразный теллур), двойной теллурид и сульфид свинца, золота и сурьмы; иногда встречаются также соединения золота с селеном. В местах залегания соединений золота находится и самородное золото (вследствие легкой восстанавливаемости соединений), но всегда в тснкодиснергированном состоянии.
Часть золота в процессе изменения земной коры уносилась с места первичного залегания и вдоль откладывалась в местах вторичного залегания. Находящееся в таких месторождениях россыпное золото — намывное золото — всегда существует в самородном состоянии в виде мелких зерен, а иногда больших самородков.
В древние времена золото добывали исключительно из россыпей. Однако с усовершенствованием методов добычи все большее значение для производства золота стало приобретать ископаемое золото. Уже в 1912 г. этим методом добывали в/10 количества добываемого золота.
Получение. Самым древним способом добычи золота является промывание золота. Он основан на том, что золотоносный песок подвергают процессу «отмучивания», в результате чего он обогащается тяжелыми золотыми зернами.
Технически значительно болое совершенным вариантом этого способа является гидравлическая разработка», при которой с помощью драг несущие золото рыхлые осадочные породы захватываются и выбрасываются мощными струями воды. Для экономически выгодного применения этого способа в больших масштабах его следует сочетать со способом амальгамирования.
Способ амальгамирования (ртутный способ) осповап па способности ртути растворять золото с образованием амальгамы. Из амальгамы золото удается легко выделить отгонкой ртути. Условие для применения этого процесса — пе слишком мелкие частицы золота, так как иначе они недостаточно смачиваются ртутью.
440
Глава 8
Важнейшим в настоящее время способом добычи золота является применяемый с 1886 г. способ «.цианидного выщелачивания». Иногда этот способ применяют наряду с амальгамированием для извлечения остатков золота. Цианидные щелока позволяют экстрагировать из руды мельчайшие, неамальгамирующиеся частицы^ Этот способ пригоден поэтому не только для переработки еще содержащих золото остатков процесса амальгамирования, но и вообще для переработки таких руд, которые содержат золото в очень тонко диспергированном состоянии, т. е. прежде всего для переработки ископаемого золота.
Впервые испытанное в 1886 г. МакАртуром и Форрестом (MacArthur, Forrest) цианидное выщелачивание почти полностью вытеснило способ хлорирования, широко применяемый перед тем, особенно в Америке [разработан Платтнером (Plattner) в 1850 г.]. При этом способе золото переводили в растворимый хлорид путем обработки влажной руды хлором, выщелачивали и выделяли из раствора сульфатом железа(П), сероводородом или даже адсорбировали на древесном угле. Следует указать еще, что для получения золота из некоторых руд пытались использовать также летучесть хлорида золота при нагревании. Для этого руды нагревали в смеся С каменной солью; хлорид золота при этом улетучивался.
При цианидном выщелачивании мелкоизмельчепную золотую руду обрабатывают разбавленным (большей частью 0,1—0,2%-ным) раствором цианида калия или натрия. Под действием содержащегося в растворе кислорода воздуха золото переходит в раствор в виде комплексного цианида. Из раствора золото выделяют внесением цинковых стружек * или электролитически (только в отдельных случаях).
Из осадка, который более или менее сильно загрязнен цинком, цинк выщелачивают разбавленной серной кислотой. Высушенный остаток сплавляют с бурой. К сплавленному таким образом золоту еще в довольно значительном количестве примешано серебро. Отделение его от золота производят в настоящее время на крупных предприятиях большей частью электролитически. На небольших производствах отделение ведут часто путем обработки концентрированной серной кислотой, а иногда азотной кислотой.
При олехтролитическом отделении золота, которое применяют прежде всего для разделения плати но содержащего золота, подлежащее очистке золото погружают в качестве анода в раствор золотохлористоводородной кислоты [тетрахлорозолото(Ш) кислота]. Прн пропускании тока, помимо полота, с анода переходит в раствор также часть сплавленных с ним металлов. Другая часть (Ir, Rh, Ru) опускается без изменения на дно; Ag выпадает как AgCl, a Ph осаждается серной кислотой в виде PbSO<. На катоде, однако, осаждается только золото, так как оно обладает наименьшей тенденцией оставаться в растворе. Платина также остается в растворе, если только предусмотреть, чтобы раствор ею пе слишком обогащался..
Сернокислотное отделение (аффинация) основано на растворимости серебра в кипящей концентрированной серной кислоте и нерастворимости в ней золота. Оно применимо для всех сплавов золота с серебром независимо от содержания в них золота и не подходит для платино содержащих сплавов, так как платина может сделать нерастворимой часть серебра.
Посредством умеренно концентрированной азотной кислоты (лучше всего 60%-ной) также удается провести отделение золота от серебра. Способ этот требует, однако, чтобы сплав состоял примерно на 1/i из золота и па V4 ив серебра. Приготовление такого сплава называют «квартацией» и название это перенесли затем на весь способ. В XV — XIX вв. квартация была обычным способом отделения золота; однако с развитием производства серной кислоты она была вытеснена способом отделения нрн ПОМОЩИ СерНОЙ КИСЛОТЫ.
Сиойетва золота. С древних времен золото считали «царем металлов». Его выделяли среди других металлов за красивый желтый цвет, живой
* Цинк при этом частично переходит в раствор с образованием цианоцинкат-онов, частично выпадает как Zn(CN)2.
Побочная подгруппа первой группы, периодической системы
441
блеск, устойчивость по отношению к воздуху и ковкость. Твердость золота равна только 2,5—3 по шкале Мооса. Его относят к наиболее тяжелым металлам (уд. вес 19,3).
Точка плавления золота (1063°) лежит примерно лишь на 100° выше, чем точка плавления серебра, и ниже точки плавления меди. Точка кипения, напротив, лежит выше, чем у меди, а именно около 2700°, однако при существенно более низких температурах (начиная с 1000°) летучесть золота уже заметна. Содержащая золото платина полностью освобождается от золота при продолжительном плавлении. При плавлении золото сильно расширяется.
Золото обладает очень большой электропроводностью и теплопроводностью. Его удельная электропроводность и при 18° составляет 67% удельной электропроводности серебра; теплопроводность X равна 70% теплопроводности серебра (иАи = 4,13-105 мо, ^Аи = О,7О5 кал 1см-сек-град, обе при 18°).
Золото кристаллизуется в кубической системе (структуру решетки см. иа стр. 389). В природе иногда встречаются кристаллы золота с поверхностями куба, октаэдра и ромбододекаэдра. С платиной, палладием, серебром, а также с медью золото образует твердые растворы в любом соотношении, несмотря на то что константы решеток названных металлов и золота достаточно сильно различаются; ср. стр. 40 и сл.
С атомарным водородом золото соединяется с образованием бесцветного твердого гидрида, вероятно, солеобразного характера (Pietscii, 1931). Однако это соединение неустойчиво. Напротив, при высоких температурах газообразный гидрид золота устойчив (ср. стр. 392).
Как ярко выраженный благородный металл, золото практически * совершенно не изменяется4 па воздухе, даже в соприкосновении с водой. Даже галогены в виде сухих газов при обычной температуре действуют на золото только с поверхности или не действуют совсем. Например, золото соединяется с фтором только при температуре в интервале 300—400“ (при более высокой температуре фторид вновь разлагается). Напротив, водным раствором хлора золото быстро растворяется уже при обычной температуре. Склонность золота соединяться с хлором или заряжаться при помощи хлора
Au 4-3/sCI2=AttCI3 или Ан+%2С1й=Ап‘*‘+ЗСГ,
в этом случае возрастает благодаря тому, что происходит образование комплексных ионов, а именно тетрахлоро аур ат-ионов
АиС13 + СГ = [ЛпС14]',
или, если нет избытка ионов хлора, в результате образования оксотри-хлороаурат-ионов
, Ап + з/аС 12 + Н2О=[ АиОСЦ" 2Н’.
* По данным Мюллера (Muller J., 1935),'-золото па воздухе покрывается тонким слоем окисла. Если допустить ото предположение, то полная устойчивость золота по отношению к воздуху обусловливается пе тем, что оно с кислородом воздуха непосредственно не может соединяться, а тем, что иа нем образуется защитный поверхностный слой. В этом случае золото по своему отношению к кислороду воздуха отличается от других металлов, папример меди и никеля, по существу только тем, что при темне])атурах, при которых поверхностный слой мог бы расти с заметной скоростью (ср. стр. 780 и сл. и стр. 794), окись золота, образующая этот поверхностный слой, распадается. Теплоты образования окйслов золота достоверно еще не известны. Гидро окись золота(11 Г), согласно Томасу, имеет достаточно большую положительную теплоту образования (—100 к кал/моль).
442
Глава 8
Особенно легко золото растворяется в смеси из 3 ч. концентрированной соляной кислоты и 1 ч. концентрированной азотной кислоты благодаря взаимодействию с хлором in statu nascendi *. Вследствие того что эта смесь может растворять «царя металлов», она получила название «царской водки».
Еще более, чем попами С1', растворение золота облегчается ионами CN'. В этих условиях тенденция золота к растворению настолько возрастает, что в присутствии ионов CN' золото может окисляться даже кислородом воздуха **:
2 Л и ++2^2 Н8О -p4CIS'=2{Au(CN)2] ' -р 2ОН\
Этот процесс лежит в основе получения золота цианидным выщелачиванием.
Золото не подвергается действию расплавленных щелочей. Такие кислоты, как серная, соляная, азотная, фосфорная, мышьяковая, также не действуют на золото как в разбавленном, так и в концентрированном состоянии, если они одновременно не содержат очень сильного окислителя.
В копцептрировэнной серной кислоте золото растворяется в присутствии иодной кислоты, азотной кислоты, двуокиси марганца и т. д. В этих случаях получаются желтые растворы, из которых прн разбавлении водой выпадает гидроокись зокота(Ш). Селеновая кислота, которая действует как очень сильный окислитель, может непосредственно растворять золото.
Нерастворимость золота в менее сильно окисляющих кислотах объясняется ого местом в электрохимическом ряду напряжений. Ан стоит в нем далеко справа от водорода и поэтому по только не может перейти в раствор с выделением водорода, но, как следует из значения его нормального потенциала (ср. табл. 4 иа стр. 51, т. I), не может приобрести заряд в водном растворе под влиянием окисляющего действия кислорода воздуха. Поэтому в противоположность, например, меди золото нерастворимо не только в свободных от воздуха, но и в содержащих воздух кислотах.
Золотой электрод в соприкосновении с 1 М раствором хлорида золота(1П) обладает па 1,08 к более высоким потенциалом, чем нормальный водородный электрод. Вследствие происходящего комплексообразования с участием ионов хлора концентрация ионол золота в таких растворах лежит существенно ниже 1 моль! л. Нормальный потенциал золота лежит поэтому еще выше. Он был определен Ирсой и Елинеком (Jirsa, Jelinek) при измерении растворов сульфата и нитрата золота(Ш) по отношению к нормальному водородному электроду (+1,37 и +1,39 в соответственно). Это высокое значение нормального потенциала отражает трудность получения атомом золота заряда и соответственно легкость потери заряда попом золота.
Нормальный потенциал попа золота(!) лежит еще выше, Чем потенциал иона золота(Ш). Однако, так как свободные попы золота(1) содержатся в растворах всегда в минимальных количествах, соотношение практически становится обратным, так же как у меди.
Так как потенциал разложения золота ниже, чем других металлов, то из достаточно крепких растворов хлорида золота электрическим током (при не слишком высокой плотности тока) выделяется только золото, даже если раствор содержит значительные количества других благородных металлов.
* Выделяемый одновременно царской водкой ни грозил хлорид NOC1 не действует па золото даже при 100°.
** Ср. также стр. 447.
Побочная подгруппа первой группы периодической системы
443
Совсем другим оказывается соотношение, если золото находится в форме чрезвычайно прочного циапо-комплекса. В этом случае его ионная концентрация настолько мала, что потенциал растворепия оказывается ниже потенциала растворения серебра и ртути. Поэтому серебро и ртуть не только не могут выделить золото из раствора цианида зцлота(1) л цианидов щелочных металлов, но они сами осаждаются из растворов их двойных солей с цианидами щелочных металлов металлическим золотом, которое при этом переходит в раствор,
Из положения золота в электрохимическом ряду напряжений (далеко справа от водорода) следует, что золото должно вытесняться из своих растворов почти всеми металлами. При этом предполагается, что золото в них не находится в форме прочных комплексных соединений. Опыт подтверждает это.
Следует учесть, что при осаждении благородных металлов из растворов неблагородными иногда получаются не чистые благородные металлы, а твердые растворы их с осаждающим металлом или даже соединения с ним. Например, из растворов солей золота кадмием выделяется соединение AuCd3,
Такими восстанавливающе действующими ионами, как Fe", Sn", so;, NO;, золото также осаждается из хлорида ого раствора. Однако ионами ртути(1) оно восстанавливается чаще всего только до однозарядного состояния
Л a"' -j- ITg^ ' = Лн' -|-2Hg"
Возникающие при этом ионы золота(1) тотчас выделяются в виде гидроокиси золота(1) в результате реакции с водой
Ли- + 1ЮН= АпОП-р Н \
Это происходит еще прежде, чем они образуются в большом количестве, Однако они все же взаимодействуют частично и по уравнению
ЗЛгГ = Ли" ‘ + 2Аи
так, что наряду с гидроокисью золота(1) осаждается и металлическое золото.
Восстановление под влиянием ионов SOg также часто протекает только до одновалентного золота, а именно тогда, когда соотношение концентраций благоприятствует образованию сульфито-комплекса, защищающего одновалентное золото от дальнейшего превращения.
Многие органические вещества также могут осаждать золото в виде металла, например щавелевая кислота, формальдегид, сахар, винная кислота, лимонная кислота, уксусная кислота и их соли. Окись углерода также осаждает металлическое золото из растворов хлорида золота(Ш), равным образом и ацетилен; последний при этом окисляется, по данным Киндлера, до глиоксаля СНО—СНО. Часто в этом случае золото получается в коллоидной форме. Оно образует большей частью интенсивно окрашенные гидрозоли (например, пурпурно-красные, синие, фиолетовые, коричневые или черные), которые в присутствии защитных коллоидов довольно устойчивы.
Условием для их устойчивости является долное отсутствие коэгулирующе действующих веществ, например электролитов. Осаждающее действие электролита определяется природой его катиона, так как коллоидные частички золота несут отрицательные заряды. При действии больших количеств электролитов частицы золота могут ири известных обстоятельствах перезаряжаться. Поэтому большие количества электролита действуют стабилизирующе.
Для получения чистых гидрозолей золота наиболее эффективны два способа, предложенные Зигмонди *. Один основан гы восстановлении золото(Ш) хлористоводородной
* Подробно см. Р. Зигмонди, Коллоидная химия.
444
Глава 8
кислоты НАиС14 формальдегидом в сильно разбавленном растворе, который слабо подщелачивают добавлением карбоната калия. Б другом способе восстановление осуществляют добавлением нескольких капель эфирпого раствора фосфора. Оба способа можно комбинировать. Такие золи е определенными размерами частиц (в красных растворах золота, приготовленных восстановлением фосфором, частицы имеют, как правило, диаметр 1—б-ltr" с.и) применяют в медицине для диагностических целей (например, для исследования спинномозговой жидкости), а также при биологических исследованиях (например, для характеристики белковых тел и продуктов их распада). Такое использование этих золей ослоп апо на том, что различные белковые вещества вызывают разного характера агломерации частиц золя, проявляющиеся в изменении его окраски.
В качестве восстановителей для получения коллоидных растворов золота предлагают также гидразин, окись углерода и перекись водорода. Интересный способ предложил Донау (Donau, 1913). Он заключается в том, что водородное пламя направляют в сильно разбавленный раствор хлорида золота (1 : 50 000 до 1 : 100 000). В местах, па которые попадает пламя, возникают красные полосы коллоидного золота. Правда, как показали Халле и Прибам (Halle, Prrbam), восстанавливающее действие водородного пламени само по себе является только частью причины выделения золота. Существенным оказывается также участие окиси азота, образующейся в пламени.
Применение, Золото в большом количестве используется для изготовления украшений. Так как чистое золото для этих целей слишком мягко, ого сплавляют с другими металлами, главным образом с медью и серебром.
Содержание золота в этих сплавах («проба») обычно дают либо в тысячных долях, либо в каратах. 24 карата соответствуют чистому золоту. 18-каратный сплав содержит 18 вес. ч. золота в 24 ч., т. е. 750 на 1000 ч.; 14-каратное золото содержит 14 ч. золота в 24 ч., или примерно * 585 ч. на 1000,
Значительные количества золота применяют для позолоты изделии из менее благородных металлов, главным образом серебра и меди.
В стоматологии золото применяют для изготовления пломб, мостов, коронок и т.д. Иногда золото используют для изготовления химической аппаратуры (тиглей, реторт, холодильников).
Из соединений золота наиболее употребительна золотохлористоводородная кислота НАиС14-4НгО, называемая «хлорид золота». Ее применяют прежде всего для приготовления ванн гальванического золочения, кроме того, для окраски стекла и фарфора, в фотографии (для тонирующих ванн) и иногда в медицине. Вместо указанной кислоты используют часто ее натриевую соль (хлороаурат натрия) Na[AuCI4J-2H2O, которую обычно называют «золотой солью».
Ценимое вследствие своей глубокой темпо-красной окраски рубиновое стекла содержит металлическое золото в виде коллоидных частиц, которые можно различить в ультрамикроскоп. Его изготовляют, добавляя к стекольной массе немного золота в виде какого-либо соединения. Сначала таким образом получают бледно-желтое, зеленое иля даже бесцветное стекло. При повторном нагревании неожиданно появляется кроваво-красная окраска. Это происходит тогда, когда частицы золота достигают величины, обусловливающей эту окраску золя. При слишком продолжительном нагревании вследствие дальнейшего укруппепия частиц наступает бесцветное помутнение.
Почти половина годовой продукции золота хранится в форме «золотых слитков» в банках различных стран для обеспечения бумажных денег, выпущенных вместо золота в обращение. Однако в результате накопле-
* Точнее 583,333.., на тысячу частей.
Побочная, подгруппа первой группы периодической системы 445 ния золота в банках немногих стран его значение как средства обеспечения денег и основы для оценки других товаров падает.
В тех немногих странах, где золотые монеты еще находятся в обращении, они состоят в большинстве случаев из 900 ч. золота и 100 ч. серебра.
СОЕДИНЕНИЯ ЗОЛОТА
В своих нормальных соедипепиях золото бывает электроположительно трехвалентно или одновалентно. Наиболее устойчивыми являются в общем соединения трехвалентного золота. Однако все соединения золота легко разлагаются. При действии восстановителей часто даже уже при небольшом нагревании они распадаются с выделением золота.
Из соединений одновалентного золота наиболее важными являются сульфид золота(1) Au2S, галогениды АцХ, а также различные комплексные анионы. В водном растворе простые соединения золота(1) неустойчивы, если не принимать во внимание исчезающе малых концентраций их. Поэтому не существует растворимых простых солей золота(1). Даже очень труднорастворимый хлорид золота(1) AuCl разлагается водой на хлорид золота(Ш) и металлическое золото.
Свободные ионы золота(1) могут существовать в водных растворах лишь в таких концентрациях, которые непосредственно даже не могут быть установлены. Напротив, прочные комплексные соединения золота(1), например цианоаураты(1) M1[Au(CN”)2], заметно устойчивы в водном растворе.
Большинство солей трехвалентного золота также обнаруживают большую склонность к образованию комплексных соединений. Прочные комплексные ионы образуются прежде всего с галогенид-, цианид-, И роданид-ионами. Соли кислот, которые не способны к образованию с ионами золота(Ш) ацидо-комнлексов, устойчивы только в концентрированных растворах соответствующих кислот. При добавлении воды они тотчас гидролитически разлагаются с выделением гидроокиси золота(Ш). В чисто водных растворах ноны Ац‘“ в непосредственно измеримых концентрациях пе могут существовать.
К образованию комплексных катионов золото значительно менее способно, чем к образованию комплексных апионов. Вейтцу (Weitz, 1915), однако, удалось приготовить несколько соединений типа [Au(NH3)4]Xs соли тетрааммиизолота(1П), в основе которых лежит комплексный катион [Au(NH3)4|’“. Нитрат [Au(NH3)4] [NO3 получается при действии аммиака на водный раствор золотохлористоводородноп кислоты, насыщенной нитратом аммония. Другие соли, например нитратоксалат |Ah{NH3)4}(NO3)(C2O4) и труднорастворимый хромат [Au(NH3)4]2iCrO4]3, можпо приготовить из нитрата двойным обменом. Аммиак очень прочно связан в комплексе, лежащем я основе этой соли; концентрированными кислотами он не отщепляется. Под влиянием щелочи, наоборот, происходит разложение. При этом распаде образуются азотсодержащие взрывчатые соединения того же типа, какие получаются, например, при смешивании раствора зо.ч от охлористоводор одной кислоты с избытком аммиака или если продолжительно действовать аммиаком или солями аммония на окись золота («гремучее золото»). По мнению Вейтца, при этом речь идет преимущественно о соединениях Au2O3-2NH3 и Au20n-3NH3, смешанных с соединениями диамидоимидозол о-
Х< II /X
та(Ш) —N — Au<f , где X — одновалентный кислотный остаток (напри-
h2nz \nh2
мер, С], Н03). Напротив, при действии аммиака на эолотохлористоводородпуго кислоту в растворе, насыщенном хлоридом аммония, возникает но взрывчатый, а лишь слабо вспыхивающий при нагревании хлорид диамидозолота(Щ) Au(NH2)3Cl.
Если не происходит комплексообразования, гидроокись золотаПП') растворяется только в очень концентрированных кислотах. Однако,
44S
Глаза 8
с другой стороны, она растворяется также и в сильных щелочных растворах. Опа обладает, следовательно, амфотерным характером. Обычно ее называют золотой кислотой.
Известно несколько соединений, которые на основании их состава можно рассматривать как соединения золота(Н), например АпС1Е, АиВгг„ AuO, AuS, AuSO4. Вероятно, при этом речь идет о двойных соединениях одновалентного и трех валентного золота, например АпСЬАиОз или Au1 [AuinC.lJ и т. д.
Имеются сведения о существовании солей фтора с электрохимически четырех валентным золотом (Hoppe, 1950); однако их существование определенно еще не доказано.
1. Соединения золота(1)
Окись золота(1), монокись золота Ао20, как будто должна получаться прн действии гидроксильных ионов на соединения золота(1) или на соединения золота(Ш) в присутствии восстановителей. Однако существование ее находится под вопросом.
По данным Крюсса (Кгйй, 1887), окись золота(1) в чистом виде можно получить разложением дибромоаурата(1) калия К [АнВг2] калиевой щелочью.
Бромоаурат(Т) калия получается при этом осторожным восстановлением тетрабромоаурата(Ш) калия К [AuBrJ сернистой кислотой в водном растворе при 0°. Окись золота(1) выпадает сначала в виде темно-фиолетового осадка, который считают гидроокисью золота(1) или гелем окисп золота(1); после высушивания над пятиокисыо фосфора этот осадок переходит в светлый серо-фиолетовый порошок, который начинает отдавать кислород примерно при 205°. Новые исследования позволяют полагать, что это вещество, рассматриваемое как окись золота(1), представляет собой лишь смесь Ап2О3 с мелкодиспергированным золотом. Возможна что вначале при выделении и возникает окись или гидроокись золотаЦ)', которая затем быстро распадается; во всяком случае, окись золота(1£,. если опа вообще может образоваться, Крайпе неустойчива.
Галогениды золота(1). Галогениды золота(1) удается сравнительна легко получить, однако они достаточно неустойчивы. Гораздо устойчивее производящиеся от них комплексные галогено-соли.
Хлорид золота(1), монохлорид золота АнС1, получается в виде бледно-желтого порошка при умеренном нагревании (до 185°) безводнотс хлорида золота(III)
АпС1з=AuCl -р С12.
Его, однако, трудно получить в чистом виде. При более сильнее нагревании оп распадается на золото и свободный хлор. Водой он быстра разлагается уже при обычной температуре
3AuC1 = AuC13 + 2Au.
В растворах хлоридов щелочных металлов он растворяется с образованием комплексных ионов [AuClJ' [ион хлороаурат(1)]; однпе. и в этих растворах вскоре происходит разложение с выделением метя— и образованием комплексных ионов трсхвалентного золота.
Побочная подгруппа первой группы периодической системы,
447
В твердом состоянии хлороауратЫ(Т) устойчивы. Например, желтый хлороау-рат(1) калия удается получить плавлением хлороаурата(Ш) калия
K[AuC14] =К[ AuCI2J + C12.
С другими веществами AuCl также образует двойные соединения, например с РС13 и в первую очередь с NH3; он может присоединить до 12 молекул NH3 (при обработке жидким аммиаком).
С окисью углерода AuCl соединяется при слабом пагревапии, давая карбонил хлорида аолота(1) AuCl(CO) — бесцветные, сильно светопреломляющие кристаллы в виде табличек. Проще это соединение получается нагреванием АиС13 в токе СО. Следами воды оно тотчас разлагается с выделением золота и образованием СО2 и НС1 (Manchot, 1925),
Бромид золотз(1). монобромид золота АпВг, получается при осторожном нагревании бромида золота(Ш); оп еще легче разлагается, чем монохлорпд. АиВг растворим в растворах бромидов щелочных металлов с образованием комплексного соединения.
Иодид золота(1), мопоиодид золота Aul, возникает при тех реакциях, при которых следовало бы ожидать образования иодида золота(Ш), например при растворении окиси зодота(1П) в иодистоводородпой кислоте иди при добавлении иодид-ионов к растворам солей золота(Ш). Он получается также при непосредственном взаимодействии, золота с иодом при умереппо высокой температуре. Иодид золота(1) кристаллизуется в тетрагональной системе (д = 4,359, с = 13,711 А) в форме бледно-желтых листочков ('+исн — 8,25). Он обладает цепной структурой, и, исходя из межатомных расстояний (Au <—* *1 =2,60 А), его следует рассматривать как г ом еои о л Ирное соединение (Weiss А. и. А., 1956). При пагрнвапни Aul разлагается еще легче, чом AuClnAuBr. Наоборот, водой он разлагается медленнее, чем другие галогениды. Это связапо, очевидно, с его незначительной растворимостью, В растворе иодида калия он растворяется (с комплексообразованием) без разложения только в том случае, если раствор содержит свободный иод (или трииодид калия) в количестве, соответствующем давлению иода над соединением. Он разлагается растворителями, которые легко отбирают под, например хлороформом и сероуглеродом.
Теплота образования, по данным Бильтца, равпа; для АпС] +8,4; для А нВ г +3,4 и для Aul —0,2 ккал.
Цианид золота(1) и цианоаураты(1). Цианид золота(1), мопоцианид золота AuCN,— желтые, микроскопические, шестиугольные таблички, образуются так же, как иодид, часто при реакциях, при которых следовало бы ожидать образования цианида золота(1П). Кроме того, он получается при нагревании комплексных цианидов золота(1) с соляной кислотой до 50°
Na[Au(CN)2] + IIC1 — AuCN + NaCI+ IICN.
Б одой цианид золота (I) не разлагается, очевидно вследствие слишком незначительной растворимости; он также не разлагается разбавленными кислотами и сероводородом. AuCN легко растворяется в растворах цианидов щелочных металлов с образованием прочных комплексных солей типа Mi[Au(CN)2] — цианоауратЦ). Также в результате комплексообразования растворяется AuCN в растворах тиосульфата натрия, сульфида аммония, калиевой щелочи и аммиака.
ЦианоауратЦ) калия К [Au(CN)2] получают растворением золота в растворе цианида калия при доступе воздуха
2Au-p4KCN+H2O+i/2O2=2K[Au(CN)2j + 2KOH.
Как установил Ь’одлеидер (Codlander, 1896), процесс идет с промежуточным образованием перекиси водорода *:
2Au4-4KCN+2MaO4-O2=2K[Au(CN)2]4-2KOH + H2O2;
2Au+4KCN+4!2O2=2K[Au(CN)2] 4-2KOII.
* Также идет растворепие металлического серебра в растворах цианидов щелочных металлов (Simon, 1935).
448
Глава S
Цианоаурат(1) калия очень легко растворим в воде (примерно i/2 моль!л на холоду и около 7 молъ'л при температуре кипения). В спирте он мало растворим, в эфире совсем нерастворим.
Аналогичными свойствами обладает соль натрия, о значении этих соединений при получении золота уже было сказано.
ТиосульфатоауратыЦ), При добавлении спирта к смеси растворов хлорида золота н тиосульфата натрия выпадает дитиосулъфатаауу>агп(Г) натрия Na3 [Au(S3O3)3] •1/гН2О; он представляет собой бесцветные иглообразные кристаллы сладковатого г вкуса, легко растворимые в воде. Такими восстановителями, как хлорид железа(П), хлорид олова(П) и щавелевая кислота, золото не выделяется из его раствора. Так же мало выделяется сиры при Добавлении кислоты. Однако при добавлений иода образуется тетратионат натрия и иодид залота(1). Из соли бария Ва3 |Aii(S3Os)z|3, разбавленной серной|кие.г!.отой, удается выделить свободную кислоту [дитиосульфатозолото(1) кислоту] Нэ[Аи(8гО3)а]Л/гНгО.
Сулъфптоаураты(1). Прикапывая раствор хлорида золота в щелочной раствор сульфита калия, получают дисульфитоаурат(1) калия К3 [Au(SO3)3l- Н3О в форме белых игл. В воде он легко растворим и является прочным комплексом, Сероводородом из его раствора золото не выделяется. Очистку его следует вести через труднорастворимую бариевую соль. Таким же образом получают соответствующую патрйевую соль.
Сульфид золота(1) Au3S. Если в кипящий раствор хлорида золота(Ш) пропускать сероводород, то выделится1 металлическое золото. При работе на холоду выпадает осадок состава AuS, возможно представляющий собой двойное соединение Au,S и AusS3. Сульфид золота)/) Ап38 лучше всего получать насыщением раствора KlAu(CN)3j сероводородом и Добавлением соляной кислоты. Во влажном состоянии он серо-стального цвета, в сухом коричнево-черный, в воде и в разбавленных кислотах практически нерастворим, однако легко образует коллоидные растворы, особенно в присутствии сероводорода. При действии сильных окислителей (хлор, царская водка) он растворяется с разложением. В водных растворах цианидов щелочных металлов он растворяется с образованием цианозуратов (Г). Сульфид золота(I) растворяется также в избытке водных растворов сульфидов щелочных металлов. При этом оп образует с сульфидами щелочных металлов двойные соли тиоаураты(Г), причем как мопотиоаурат М1 [AuS), так и дитиояурат Mj[AuS3J. Такие же тиоаураты получают при растворении более богатых серой сульфидов золота AuS и AiiaSj в бесцветных сульфидах щелочных металлов благодаря тому, что сульфиды золота отдают избытку сульфида щелочного металла серу с образованием полисульфида. Кислотами тиоаураты(1) разлагаются е образованием сульфида золота(Т)
2| AiiS]' -J- 21Г = Au£S -j- II3S.
Соединения золота(Ш)
Окись золота(1П) и гидроокись золота(Ш) (золотая кислота).'^Есле к раствору хлорида золота(Ш) добавить гидроокись щелочного или щелочноземельного металла или кипятить его после добавления щелочного карбоната, то выделится осадок желто-коричневого цвета, который по существу состоит из гидроокиси золота(Ш), однако обычно сильна загрязненной примесью осадителя. При подходящих условиях загрязнение удается устранить путем экстрагирования кислотами.
В результате высушивания над пятиокисью фосфора получается желто-красный или желто-коричневый порошок состава АиО(ОН). Он растворяется в соляной кислоте и в других кислотах, если они достаточна концентрированные, а также в горячем растворе едкого кали, откуда следует, что оп амфотерен. Так как кислотный- характер преобладает, тз гидроокись золота(Ш) называют обычно золотой кислотой. Соли этой кислоты называют ауратами [точнее: ауратами(П1)5, например К[АиОг|-• ЗН3О — аурат калия.
Произведение растворимости гидроокиси залога получено из значений растворимости его в серной и азотной кислотах, определенных Ирсой и Елинеком (Jirs^ Jelinek, 1924). Оно равно L =[Au,”j x [0Н']э примерно 9-10*40.
Побочная подгруппа первой группы, периодической системы 449
Осторожным нагреванием (до 140—150’) гидроокись золота (золотую кислоту) удается обезводить. Однако окись AugOg начинает заметно терять кислород уже при немного более высокой температуре (примерно начиная со 160°).
Хлорид золота(П1) и золотохлориетоводородная кислота (хлорозоло-тая кислота). Хлорид золотел! 11), трихлорид золота АпС13, в безводном состоянии лучше всего получать действием хлора на золотую фольгу пли на сухой порошок золота, полученный восстановлением сульфатом железа(П) при температуре немного выше 200°. АпС13 возгоняется при этой температуре в токе хлора и осаждается в форме красных игл уд. веса 3,9. Давление разложения хлорида золота(Ш) при 2519 достигает 1 атм. Теплота образования, по данным Бильтца, составляет 28,3 кКал/моль. При повышенном давлении хлора хлорид' золота плавится при температуре 287—288°. В воде он растворяется, давая коричнево-красную окраску, с образованием комплексной кислоты НгАпОС13 [оксотрихлоро-золото(Ш) кислота]:
AuGl3+ HgO=HeAuOC13.
Эта кислота образует труднорастворимую желтую серебряную соль Aga[AuOCl3] (оксотрихлороаурат серебра).
Если смешать коричнево-красный раствор хлорида золота(Ш) с соляной кислотой, то он становится лимонно-желтым, что объясняется образованием золотохлористоводородной кислоты [тетрахлорозолото(Ш) кислота] НАиС14
, НгАиОС13 4- НС1 = НАиСЦ 4- НгО.
В продаже под хлоридом золота, как правило, подразумевают золотохлористоводородную кислоту. В старой научной литературе также под хлоридом золота понимали золотохлористоводородную кислоту. Золотохлористоводородная кислота кристаллизуется в виде длинных, светло-желтых, расплывающихся на влажном воздухе игл состава НАиС14-4Н2О. В сухом воздухе отщепляется одна молекула Н3О. Кроме воды, эта кислота также растворима в спирте и эфире. Из спиртового раствора она кристаллизуется в безводном состоянии.
От золотахлористоводор одной кислоты производятся многочисленные соли [тет.рахлороаураты(1П)] MUAuClJ. Важнейшая из них пгетра-х.юроауратСПГ) натрия Na[AuCLI-2H2O, называемая просто «золотой, солью», кристаллизуется в виде больших ромбических столбиков или табличек. Помимо воды, она растворяется также в эфире и кристаллизуется из эфирного раствора также с 2Н2О. При пагревании она теряет свою поду только при тех температурах, при которых происходит уже отделение хлора. Нерастворимый в эфире тетрахлороаурат калия известен в виде дигидрата К|АнС14]-2НЕО, кристаллизующегося из разбавленных солянокислых растворов в виде светло-желтых ромбических табличек, п полугидрата KfAuClJ-ЧгНгО, который выделяется из концентрированных солянокислых растворов в форме маленьких светло-желтых моноклинных игл. Оба полностью отдают свою воду при 100°.
Бромпд золота(111) АиВг3 и тетрабромоаураты М1 [АаВг4] подобны в своих свойствах соответствующим соединениям хлора. Тетрабромоаурат калия К[АиВг4| .2НгО — пурпурно-красные моноклинные кристаллы — получают обработкой золота бромной кодой и бромидом калия. Как показало рентгена структурное определение (Сох, 1936), в этом соединении атомы В г образуют плоений четырехугольник с атомом Ап в центре. В пустотах решетки размещаются атомы К и молекулы Н2О. Последние едва ли колою рассматривать как координационно связанные с атомами Ан, ибо расстояние 29 Г. Реми
450
Глава 8
Ац«—► Н2О составляет 3,57 А, в то время как расстояние Au <—» Вт — только 2,50 или 2,57 А, В соединении КАиС14 атомы CI, по данным Кокса, также, вероятно, расположены в одной плоскости,
Иодид золота(Ш) АпГ3 не получается прямым соединением составных частей или добавлением растворимых иодидов к раствору НАпС14, так как в обоих случаях возникает мопоиодид. Однако если нейтрализованный карбонатом калия раствор НАпС14 постепенно добавлять к водному раствору иодида калия, то сначала образуются комплексные иопы [Ап14]':
[AuC141' + 4I' = [AuI4]'+4CI'.
После того как таким путем все свободные иодид-ионы устранены, при дальнейшем добавлении [AuClJ' происходит образование иодида золота(Ш):
[AuC141'4-3[AuI4]'=4Aul3-|-4Cl'-,
который выделяется в виде темно-зелено го осадка. При высушивании он отщепляет иод и переходит в иодид золота(1). Комплексная кислота ЩАп14] (тетра иодо золота я кислота) получается растворением иодида золота(Ш) в водном растворе иодистово-дор одной кислоты или также действием иодсодержащей подистов од ор одной кислоты на мелкодиспергировэнное золото. От нее производятся комплексные соли тетраио-доаураты MI[Aul4], например К [AuIJ,—тонкие черные столбики. И кислота, и солп устойчивее, чем простой трииодвд золота; однако они также легко отщепляют иод.
Цианид золота(Ш) и цианоаураты(Ш), Если раствор хлорида золо-та(1П) смешать с цианидом калия, то никакого осадка не выпадет. При упаривании раствора кристаллизуется легко растворимый в воде тетра-цианоаурат{Ш) калия К [Au(CN)4] • Й/г Н2О в виде бесцветных табличек. Нагреванием до 200° соединение удается обезводить. При более сильном нагревании отщепляется циан с образованием дицианоаурата(1) калия KIAu(CN)2I.
Помимо соли калия, имеются также еще и другие цианоаураты(Ш), например легко растворимая в спирте (в противоположность первому) соль аммопия (NHJ [Au(CN)4]-H3O.
Однако соответствующая цианоауратам(Ш) свободная кислота не получена. При добавлении сильных кислот выделяется цианистый водород, а из раствора при упаривании над серной кислотой кристаллизуется тригидрат цианида золота(Ш) Au(CN)s-3H2O в виде больших бесцветных листков.
Если па Дицианоаурат(1) подействовать свободным галогеном, то образуется дигалогеподицианоаурат(1П) MJ[AuX2(CN)21, где X = С1, Вт или Е Известны .многочисленные представители этого типа солей.
Роданоаураты(Ш) MI(An(SCN)4|. При добавлении раствора хлорида золота, нейтрализованного карбонатом калия, к избытку холодного раствора роданида калия получают тетрароданоаурат(Ш) калия K[Au(SCN)4] в виде объемистого оранжевокрасного осадка. Таким же образом получаются другие роданоаураты(Ш). Напротив, свободный^роданид золота(Ш) неустойчив^
Соли золота(Ш) и кислородных кислот, например сульфат золота(Ш), нитрат золота(Ш),. устойчивы только в концентрированных растворах соответствующих кислот. При разбавлении водой: тотчас происходит гидролитическое расщепление с выделением гидроокиси золота(1И) (золотой кислоты).
Вероятно, пазваппые соединения существуют в растворах не как простые соли., а н форме комплексных соединений. Упариванием над едким натром раствора гидроокиси золота(Ш) в сильно концентрированной азотной кислоте можпо выделить комплексную кислоту HAu(NO3)4-3HaO [тетранитратоаомто(11Г\ кислота], представляющую Собой октаэдрические кристаллы с уд. весом 2,84 и т. пл. 72—73°. Известны различные соли этой кислоты, например тетрапитратоаурат калия K[Au(NO3),)—
Побочная подгруппа первой группы периодической системы
451
золотисто-желтые блестящие ромбоэдрические кристаллы. Если кристаллизацию вести в присутствии большего количества нитрата щелочного металла, то выделяется кислая соль гексапитратозолото(П1) кислоты, например кислый тексапитратоаурат калия K3H[Au(NO3)6] — кристаллы л форме табличек.
Тетранитратоауратам(Ш) аналогичны ацетатоаураты(111) М1 [Ап(С2Н3Ог)4], которые получают из растворов соответствующих ауратов(Ш) М1{АнОг| в ледяной уксусной кислоте.
К тому же типу относятся сулъфатоауратыЦП), например дисул ьфатоаурат(Ш) калия К [Au(SO4)2j — светло-желтый кристаллический порошок, получаемый растворением гидроокиси золота(Ш) в концентрированной серной кислоте, добавлением к раствору гидросульфата калия с упариванием при 200’.
Упариванием раствора золота в селеновой кислоте получают селенит золбта(11Г) Au2(SeO4)3 в виде мелких желтых кристаллов.
Сульфид золота(Ш) Au3S3. Так как Ан383 разлагается водой, из водных растворов оп не получается, а получается, по данным Антони и Лючеза (Antony, Lucchesi), действием сероводорода на сухой тетрахлороаурат(Ш) лития Li [АиС14]-2Н3О при —10°. Из полученной первоначально смеси LiCl и Ан283 хлорид лития удается извлечь спиртом. В результате остается сульфид золота(1П) Au2S3 в виде черного аморфного порошка. Нагреванием выше 200° он разлагается на золото и серу.
Если сульфид золота(Ш) обработать бесцветным раствором сульфида натрия при 3—4°, то он очень быстро переходит в раствор с красно-коричневой окраской, вероятно, спачала с образованием Na3(AuS3]. Однако этот тиоаурат(ГП) тотчас разлагается с образованием тиоаурата(1) по уравнению
Na3[ Анш83] + NasS = Na^Au^] -ф Na2S2.
Аналитические сведения. Золото обнаруживают большей частью при помощи восстановителен, которые осаждают его в виде металла.
В компактном состоянии золото легко распознается по красивой желтой окраске. Если исаждемио происходит из сильпо разбавленных растворов, то золото остается растворенным в коллоидном состоянии и тогда, при применении подходящих восстановителей [например, сульфата железа(П)], его легко можно обнаружить даже в минимальных количествах по окраске коллоидного раствора. Если восстановление ведут хлоридом олова(П), то получают интенсивно окрашенный особо устойчивый коллоидный раствор золота (кассиев золотой, пурпур). Образующийся при реакции гидрат двуокиси олова действует в отношении частиц золота как защитный коллоид (ср. стр. 720).
Очень чувствительной реакцией па золото является также цветная реакция с дитизоном (ср. стр. 438), а также бензидиновая реакция по Та на на ев у (1930), которую лучше всего проводить капельным методом на фильтровальной бумаге. Эта реакция основана на ок и слепни бензидина H2N— СвН4—С6И4—NH2 ионами золота(Ш) до хянопиминоподобпых красителей: при этом следует иметь в виду, что другие окислители, папример гидрат двуокиси марганца, также могут способствовать образованию бензидиновой синей.
Для количественного определения золото также чаще всего осаждают в виде металла и затем взвешивают его. В этой форме могут быть определены даже чрезвычайно малые количества золота.
Метод, пригодный для определения совсем небольших количеств золота (до 1-Ю-10 г), был разработан Габером. Он основан на том, что золото спачала осаждают вместе со свинцом, который можно применять в очень большом избытке *. Свинец затем вытравляют, а оставшиеся зерна золота сплавляют с бурой в перлы и измеряют под микроскопом. Главная трудность при определении минимальных количеств золота заключается в том, чтобы абсолютно предотвратить попадание золота в пробу с приме и я емыми реактивами.
* Даже если золото присутствует в водном растворе или суспензии в крайней степени разведения, например, когда речь идет об установлении содержания золота в речной или морской воде, его можно количественно определить методом свинцово-сернистого осаждения.
29*
Глава 9
ВТОРАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ (Побочная подгруппа)
ЦИНК, КАДМИЙ, РТУТЬ
Порядковый номер Элемент Символ Атомный вес Удельный вес Температура, °C Удельная теплоемкость Валентность
плавления кипения
30 Цнпк Zn 65,38 7,130 419,4 906 0,0924 II
48 Кадмий Cd 112,41 8,64 320,9 767 0,0553 II
80 Ртуть Hg 200,61 13,546 (при 20°) —38,84 356,95 0,0334 I, II
Общие сведения. В побочную подгруппу второй группы входят элементы цинк, кадмий и ртуть. Первые два элемента в соответствии с номером группы в своих соединениях положительно двухвалентны. Для ртути, кроме того, известны многочисленные соединения, в которых она одновалентна. Однако эти соединения всегда димеризованы, например в хлориде ртути С1 — Hg — Hg — С! атомы ртути связаны с обеих сторон, так что об одновалентности молено говорить только в электрохимическом смысле, формально жо она двувалентна.
Во второй группе сходство между элементами главной и побочной подгрупп больше, чем в первой. Прежде всего цинк и кадмий в некотором отношении близки к магнию. Например, сульфаты этих элементов с равным содержанием воды изоморфны; состав двойных и комплексных солей часто также аналогичен. Двойные сульфаты ртути типа MJSO4-HgSOj-BHaO соответствуют шёниту KaSC^-MgSOvfiHaO. Для элементов побочной подгруппы второй группы, так же как и для магния, характерно образование алкильных соединений.
Цинк и кадмий по типу кристаллической решетки похожи на бериллий и маг-пий (ср. рис. 57, т. I, стр. 279). Для^п а = 2,6649, с = 4,9468 А, для Cd а = 2,9788, с = 5,6167 А. Длина основного ребра а в элементарной ячейке одновременно является кратчайшим расстоянием между центрами атомов. Решетки Zn и Cd отличаются от идеальной плотнейшей гексагональной упаковки тем, что в них ось с несколько более вытянута по отношению к оси д. Для идеальной гексагональной упаковки — = 1,3333...,
для Zn - = 1,8563, для Cd — = 1 8856. а ’ ' а
Структуру решетки ртути см. стр. 487.
В табл. 45 (стр. 453) приведены атомные и.ионные радиусы металлов побочной подгруппы ТГ группы и их кристаллических соединений. Ионные радиусы соответствуют двухвалентным ионам. Атомные радиусы отвечают координационному числу 12, т. е. если их удвоить, то получается среднее из расстояний между центрами двенадцати соседних атомов. В случае идеальной плотнейшей упаковки все 12 расстояний одинаковы.
Вторая группа периодической системы
453
Таблица 4S
Кажущиеся атомные и полные радиусы элементов подгруппы цинка
Цинк Кадмий Ртуть
Атомный радиус г, А 1,3944 1,5680 1,648
(KZ==12) Ионный радиус, А 0,83 1,03 1,12
Общим для элементов главной и побочной подгрупп второй группы являются также сравнительно низкие температуры плавления и относительно хорошая летучесть. Причем в побочной подгруппе эти свойства проявляются ярче, чем в главной. Среди прочих металлов ртуть имеет наиболее низкую температуру плавления.
В соответствии с правилом Трутона (ср. т. I, стр. 359) относительно низким температурам кипения металлов подгруппы цинка соответствуют небольшие значения атомных теплот испарения (см. табл, 46). Опи значительно меньше, чем у металлов главной подгруппы II группы (ср. т. I, стр. 264, табл. 46). И прежде всего опи так же, как величины теплот плавлении, сущестрептп» ниже, чем у металлов подгруппы меди (ср. табл, 38, стр. 216).
Подобно металлам подгруппы меди, металлы подгруппы цинка диамагнитны. Пх магнитная восприимчивость, отнесенная к единице массы, указана в табл. 46. А томная восприимчивость соответственно равна—9,3-10-0, —20,8 -10-6 и —36,1-10-0. В соответствии с правилом, приведенным на стр. 342 (т. I), эти величины меньше, чем для с о седин х элементов побочной подгруппы I группы и так же, как у них, уменьшаются с увеличением порядкового номера.
Таблица 46
Важнейшие физические константы элементов подгруппы цинка
Теплота плавления (при т. пл.) Теплота испарения 1 j Теплота суСли- мацин при 0° К, ; кпаз/г-атом Удельная электропроводность при 18° Удельная магнитная восприимчивость х при 18’
(При т КПП.)
ftj wo ш о-г/т.сям пал/г i = о S ? <4 Je 14
Цинк 26,4 1,73 419 27,4 31,4 16,5-104 - 0,142-10-4
Кадмий 12,9 1,46 212 23,9 27,0 13,2-104 -0,185-10-»
Ртуть 2,74 0,55 69,5 14,0 15,45 1,044-104 -0,180-Ю-0
В отличие от элементов главной подгруппы второй группы металлы побочной подгруппы этой же группы являются тяжелыми металлами. В этом отношении они близки элементам побочной подгруппы первой группы. Электрохимический характер в ряду Zn — Cd — Hg изменяется так же, как в ряду Си — Ag — Ап, т. е. в обратном порядке по сравнению с главной подгруппой. Тенденция к приобретению положительного заряда значительно падает при переходе от цинка к ртути. В электрохимическом ряду напряжений цинк и кадмий стоят левее водорода, ртуть — правее. По своему характеру она уже приближается к благородным металлам.
454
Глава &
Хотя Hg и образует непосредственно с кислородом окись, стоит немного нагреть HgO, как опа разлагается с выделением свободного металла (ср. стр. 495).
Так же как у элементов побочном подгруппы первой группы, окислы металлов побочной подгруппы второй группы окрашены, правда, ZnO только при нагревании. Гидроокиси очень мало растворимы и имеют лишь слабоосновной характер. Гидроокись цинка обладает ярко выраженной амфотерностью *. При добавлении к растворам солем ртути гидроксил-ионов, так же как и в случае серебра, вместо гидроокиси выпадает в осадок окись (ср. стр. 495).
Сульфиды металлов подгруппы цинка в отличие от сульфидов щелочноземельных элементов нерастворимы в воде. В этом отношении они близки сульфидам подгруппы меди. Подобно последним сульфиды ртути (черный) и кадмия (желтый) окрашены; сульфид цинка, напротив, белого цвета.
Все элементы побочной подгруппы втором группы образуют твердые соединения, с водородом МН3. Их получают взаимодействием иодид о в с алапатом лития LiAlH4. Правда, все они метастабильны и устойчивость их понижается от цинка к ртути. ZnH3 разлагается при температуре 90° (медленно уже при комнатной температуре), CdH2 при —20°, a HgH2 даже при —125°.
Весьма значительное сходство между металлами побочной и главной подгрупп II группы, и особенно с магнием, проявляется в характере спектров или в расположении лежащих в их основе энергетических уровней. Как и в случае щелочноземельных металлов^, дуговые спектры цинка, кадмия и ртути состоят из двух систем главной, побочной и бергмановской серии, а именно из синглетных и триплетных термов. Искровые спектры этих элементов, как и у щелочноземельных металлов, аналогичны дуговым спектрам щелочных металлов. Как в искровых, так и в дуговых спектрах основным термом является s-терм, т. е, в невозбужденном атоме последний и предпоследний наиболее слабо связанные электроны находятся на энергетическом уровне с побочным квантовым числом 1=0. Следовательно, положение наиболее
Таблица 47
Распределение электронов в атомах щелочноземельных металлов и элементов побочной подгруппы второй группы
(Электронные оболочки идеальных газов выделены жирным шрифтом)
Порядковый номер Элемспт Распределение электронов Символ *горма
20 Кальций 1«2 2s22;^ 3s»3pe 4sS
30 Цинк is® 2s32pe 3.s23p«3rfi° 4a®
38 Стронций is® 2ss2pe 3s23p«w 4s24pe 5s2 ^0
48 Кадмий Is® 2s32po 3s23;j«3d10 4,s-24pe4di° 5s3
56 Барий Is® 2s®2pe 3s23;A3rZ»> 4.s"4pi'4rfio 5s®5^e 6S2
80 Ртуть Is3 2s®2/je 3s23jj63<Ii0 4s34pe4tZ104/i4 5s35pe,5^io gsa ^0
слабо связанных, т. е, валентных, электронов у элементов как главной, так и побочной подгрупп север гнев по аналогично. В табл. 47 сопоставлено распределение электронов по энергетическим уровням в атомах металлов побочной подгруппы II группы с распределением электронов в щелочноземельных металлах. Как видно из приведенных данных,
* Cd(OH)2 и HgO также в некоторой степени имеют амфотерный характер, однако он значительно слабее, чем у цинка. Амфотерность обнаруживается, папрнмер, в том, что С<1(ОИ)3 и HgO немного легче растворяются в концентрированных щелочах, чем в разбавленных (стр. 481).
Вторая группа периодической системы
4.55
итомы щипка отличаются от атомов кальция тем, что в них между орбитами 4s2 и 3s2 Зр® (оболочка a pro па) имеются еще 10 3d-электронов. У цинка они в отличие от меди настолько прочно связапы, что нс оказывают влияния на его валемтяовтъ, хотя прочность связи обоих валентных электронов зависит от наличия d-орбиты. Увеличение заряда ядра от 20 до 30 вызывает значительное сжатие остова атома. Вследствие этого оба 4 s-электр опа цинка расположены ближе к ядру, чем у кальция, и связаны прочнее. Этим объясняется тот факт, что цинк менее электроположителен, чем кальций. Аналогичное различие наблюдается между атомами кадмия и стронция^ а также ртути и бария. По у ртути по сравнению с барием, помимо промежуточной оболочки с 10 5 d-электронами, имеется еще оболочка с 14 4/-электр она ми. От цинка к кадмию заряд ядра возрастает па 18 единиц, а при перехода от кадмия к ртути — на 32. Следствием этого является то, что при переходе от кадмия к ртути электроположительность, т. е. способность отдавать электроны, понижается гораздо сильнее, чем при переходе от цинка к кадмию.
Наличием электронов па промежуточном d-уровне объясняется гораздо более сильное поляризующее действие ионов элементов побочной подгруппы II группы, чем следовало ожидать, принимая во внимание их нормальные радиусы. Оно проявляется в повышенной склонности к образованию слоистых решеток, а также таких решеток, которые чнесоивмеримыл с обычными ионными решетками (ср. стр. 407). В соединениях, имеющих решетку, соизмеримую с ионной, все металлы побочной подгруппы 11 группы имеют большие ионные радиусы, чем магний (см. рнс. 3, стр. 37, т. I), Напротив, в соединениях, у которых решетки сильно отличаются от истинно ионных, средние расстояния между атомами или иопами значительно ниже, чем вычисленные па основании ионных радиусов, полученных для этих соединений. Сокращение радиусов можно объяснить поляризующим действием. Но так же, как в некоторых соединениях побочной подгруппы I группы (ср. стр. 407), взаимное влияние компонентов здесь так велико, что ионный характер связи полностью исчезает и можно говорить лишь о гомеополярмой связи.
Несомненно, алкильные соединения, характерные для всех металлов побочной подгруппы II группы, следует отнести к гомеополярным соединениям. Среди них легче всего получаются алкилы цинка (см. Стр. 474). Их образование объясняется так же, как в случае металлов главной подгруппы Л группы (см. т. I, стр. 268).
Сплавы. Металлы подгруппы цинка, так же как подгруппы меди, очень легко образуют сплавы. В табл. 48 (стр. 456) приведен обаор характера взаимодействия этих металлов с металлами главных подгрупп. Сплавы с металлами побочных подгрупп,
рассмотренными в предыдущих главах, указаны в приведенных там таблицах. Металлы подгруппы цинка образуют многочисленные соединения с другими металлами. При этом металлы внутри подгруппы не соединяются (табл. 49), пе соединяются они в общем и с металлами главных подгрупп III и IV групп.
Соединения металлов побочной подгруппы Ц. группы с другими металлами можно разделить па четыре группы. К первой относятся соединения с сильно электроположительными металлами (щелочными и щелочноземельными). Как видно из табл. 48, эта группа чрезвычайно многочисленна. Особенно большое число таких соединений образует ртуть с щелочными металлами. Для пих известно правило Билыпца и Вайбке: способность связывать ртуть, определяемая количеством присоединенной ртути и термической устойчивостью соединений, в группе щелочных металлов возрастает от лития к цезию; для более легких щелочных металлов характерно образование более низких меркуридов, для более тяжелых — более высоких меркуридов.
Ко второй группе относятся соединения
Таблица 49
Образование соединений и твердых растворов между мета Ji ламп побочной подгруппы второй группы
Cd Hg
Zn тв. )> О 0 тв. О 0?
Cd ТВ* <^со 0
Hg
металлов подгруппы цинка с элементами, име-
ющими слабо металлический характер, такими, как мышьяк и сурьма. Соединения этой группы в основном являются нормальными валентными соединениями с неметаллами и большей частью соответствуют им по составу. Характерно, что между этими двумя группами соединений металлов побочпой подгруппы II группы с другими метал-
Образование соединений и твердых растворов металлами побочной
(Обозначения те же,
LI Na К Itb Св Mg Ca Sr Ba
Zn тв. 3> О ж. < co тв. 0 NaZn4 нестабЛ NaZn13 557’ ж. <^oo тв. 0 KZnu 585“ ' *44 tb.>O MgZn* 354’ тв. 0 Ca4Zn* 450’ CaZn* 431’ Ca2Zn3 688’ CaZn4 680’ CaZftj3 717“ Сгелав Сплав —
[biZn]?
LT2Z113 520° LtZh2 [] 93°
MgZn2 590’ MgZn? 381“
LiZn|,5 502“
LiZn* 481°
Cd тв. > 0 Li3Cd 272® LiCd 548’ LiCd30 — 370’ далее: <Р-фаза» тв. 0 NaCd2 385’ NaCd5 360“ ж. < co тв. 0 KCd7? KCd13 487’ Ж. <00 R bed 13 Ж. < OO CsCdia tb. <00 [MggCd] 150’ (MgCd] 251“ [MgCd3] 89’ ж. <00? tb. -<co Ca3Cd? 510’ CaCd 685“ CaCd3 612“ ТВ; P ? SrC.d SrCdJ2 Сплав
Hg тв. О LieHg* 164’ LisHg 375’ Li3Hg* 375’ LiHg 590° LiHg? 340’ LiHgJ 235° тв. 0 Na3Hg* 35’ Na5Hg? 66’ Na3Hg? 119“ NaHg 212’ Na7Hg? 222’ NaHg2 354’ NaHgJ 156’ тв. 0 KHg* 178“ KHgz 279’ KHg’ 204’ K2Hgj 173’ KHgJ 70’ тв. 0 Kh7Hgt 157“ Rb3HgJ 170’ RbHg2 256“ Rb2Hg7 197’ RbsHgt, 194’ Hb2HgJ 162“ RbHgS 132“ RbHgJ 67’ тв. 0 Cs2Hg3? * 171’ CsHg2 208’ CsHg4 164’ CsHge 158“ CsHgTe 73’ TB. 0 MgsHg* 509’ Mg5Hg2? Mg2Hg Mg&Hg3 MgHg MgHg2 170’ TB. 0? CaHg3 CaHg? 265’ CaHg*0 84“ Sr2Hgs? SrHgs? SrHg12? Bapgf0 t65“
а Это соединение получено не из раеиаава, а лишь из раствора (в жидком аммиаке).
подгруппы второй группы с элементами главных подгрупп что П в табл. -9, стр. 66)
Таблица 48
В AI Ga in Т1 Sn Pb AS Sb Bi
я;. О? тв. О 0 ТВ. со 0? ТВ. 0 0 тв. 0 0 ж. < со тв. 0 0 тв. 0 0 ж. -<со тв. 0 0 тв. О Zn3As2 1015’ : ZnAsa 771’ тв. 0 Zn3Sb2 566° Zn4Sb| 563° ZnSb* 54.6’ ж. < co тв. ^>0 0
ж. <С оо тв. О 0 Ж, < ОС? тв. 0 0 тв,)>0 0 тв. ~ 0 0 тв. > 0 0? тв. U 0 тв. О ? Cd3 A s2 721’ CdAs2 621’ tB. 0 Cd3Sb2 (нес-таб.) 423’ CdSb 456° тв. О 0
; i । ! тв, >0 0 ж, < со тн. 0 0 — ТВ. <^со Hg5Ti2 14,5’ тв, ~ О HgaSn ? HgSnJ2? тв; со 0 Hg3Asz Hg3Sb2 тв, О 0
458
Глава 9
ламп (с одной стороны, с си л т.п о электроположительными металлами и, с другой — с элементами, носящими электроотрицательный характер) п периодической системе имеется область (главные подгруппы III и IV групп), с элементами которой они почти не образуют соединений.
Третью группу составляют соединения типа Юм-Розери. Как видно из табл. 1 (стр. 40), цинк и кадмий особенно склонны к образованию таких соединений. При этом они всегда играют роль металлов «второго рода». Вследствие этого третью группу соединений могут образовывать металлы лишь вполне определенной области периодической системы, и именно то, которые способны функционировать в качестве металлов «первого рода» (ср. стр. 39).
Металлы побочной подгруппы II группы оказываются малоспособными к взаимодействию с металлами побочных подгрупп IV — VI групп; по крайней мере существование подобною, рода соединений точно не установлено. В случае если бы они существовали, их мояшо было бы причислить к четвертой группе соединений, которая охватывала бы соединения металлов подгруппы цинка с металлами побочных подгрупп VII, VI[I и I групп, и они отличались бы от соединений тина Юм-Розери. Одпако в побочных подгруппах VII, VIII и I групп число подобных соединений невелико и еще но известны закономерности их образования и состава.
С неметаллами металлы побочной подгруппы II группы, насколько опи вообще с итоги соединяются, почти всегда образуют соединеппя, соответствующие обычной валентности, даже с теми неметаллами, которые с металлами описанных выше побочных подгрупп реагируют с образованием соединений, близких по характеру к интерметаллическим. Это соответствует тому факту, что металлы побочной подгруппы II группы очень близки по своим свойствам металлам главной подгруппы II группы, которые, как вообще все металлы, образуют с неметаллами соединения, отвечающие нормальной валентности.
ЦИНК (ZINCUM) Zn
Распространение в природе. Соединения цинка встречаются во многих местах. Наиболее распространенными рудами являются (благородный) голлгей ZnCOj и цинковая обманка ZnS. В качестве цинковых руд имеют также значение и некоторые силикаты цинка, а именно кремневоцинковая руда (кремнекислый галмей, обычный галмей) Zn2SiO4-H2O, виллемит. Zn2SiO4 и троостит (Zn,Mn)2SiO4.
В Америке найдены красная цинковая руда ZnO и франклинит (Zn,Mn)0 Fe2O3. Последнему родственна цинковая шпинель ZnO-Al2O3, встречающаяся в виде хорошо образованных кристаллов, однако в очень небольших количествах и поэтому не имеющая практического значения как руда.
В небольших количествах цинк содержится иногда в железных рудах, В результате доменных плавок он накапливается в пыли колошниковых газов, преимущественно среди самых тонких компонентов. Содержание цинка (в виде ZnO) там может достигать 30% и более, я поэтому отходы доменного производства наряду с цинковыми рудами имеют значение для производства ципка.
Исторические сведения. Металлический цинк стал известен сравнительно поздно, по крайней мере в Европе, вследствие сложности извлечения его из руд. Но в виде сплавов с медью (латунь) он быд известен Гомеру еще в древности. Этот сплав получали сплавлением меди с рудой, известной в Древней Греции под названием хадцыа (у Плиния cadmla). Отсюда и произошло название га.я.игй первого из открытых минералов — карбоната цинка.
В Европе чистый цинк стал известен только к концу средних веков. В больших количествах его стали получать начиная лишь с конца XVIII ст. Прежде ципк ввозили из Индии. По-вндямому, в Индии, так же как м в Китае, он был известен -задолго до этого.
Вторая группа периодической системы
459
Получение. Прежде цинк получали почти исключительно чисто химическим путем (так называемыми способами дистилляции). Одпако за последние несколько лет сильно возросшее значение приобрел электролитический способ, и в настоящее время электролитическое получение цинка, а также меди, считается важнейшим из электрометаллургических мокрых способов. И только в некоторых случаях, когда проведение электролиза связано с большими трудностями, применяют еще дистилляционные способы. Например, при переработке цинковой обманки, которая в качестве жилых ой породы содержит силикаты, последние превращаются в кремневую кислоту и вследствие этого образуют трудно фильтрующиеся растворы.
Получение цинка чисто химическим путем но «.методу дистилляции» основано на способности окиси цинка легко восстанавливаться под действием углерода (кокса):
ZnO + C=Zn-|-CO. (1)
Красную цинковую РУДУ — франклинит, а также безводный силикат цинка можно непосредственно восстанавливать коксом. Галмей же предварительно переводят в окись цинка пагреванием в шахтных печах (2); предварительный обжиг цинковой обмапки осуществляют в пламенных или муфельных печах (3):
2пСО3=7жО + СОг, (2)
ZbS-^3/^Oo = 1.14,5 ккал. (3)
Выделяющийся в результате реакции (3) сернистый газ можно использовать для производства серной кислоты.
Нагревание циаковой руды с целью восстановления в отдельных случаях осуществляют еще старыми муфельными способами. При Этом окись цяпка, смешанную с коксом, помещают в сравнительно небольшие горизонтальные глиняные реторты или муфели, которые в большом числе загружают в печь и прокаливают. Из муфелей цинк испаряется, увлекается током образующейся окиси углерода и переходит в небольшие приемники, где сгущается до жидкого состояния. Не успевшие сконденсироваться пары цинка улавливаются в виде цинковой пыли в Присоединенных к приемникам жестяпых цилиндрах (аллонжах или трубках). Получающийся таким образом цинк-сырец, содержащий большей частью примеси свинца н некоторое количество железа, поступает на переплавку. Рафинат обычно содержит еще свыше 1% свинца. Очищенный цинк О 99,8% Zn) получают перегонкой рафинированного цинка*.
Образующуюся в качестве побочного продукта цинковую пыль нельзя непосредственно переплавлять на цинк, так как ее частицы покрыты пленкой окиси цинка/ Поэтому^ если цинковая пыль не поступает в продажу без переработки (о применении цинковои пыли см. стр. 463 и сл.), ее примешивают к цинковым рудам, поступающим на переработку.
Гораздо более экономичным, чем старый муфельный способ, является метод, разработанный в Нью-Джерси. В отличие от прежнего ои позволяет осуществлять непрерывное производство и получать цинк-сырец уже сравнительно высокой чистоты (99,5—99,8%). Процесс проводят в вертикальных муфелях из карбида кремния, нагреваемых генераторным газом до 1200—1300°. Реакционную смесь, состоящую из размолотой окиси цинка (или обожженной цинковой обмапки), угольного порошка и связующей добавки, слегка нагревают и затем прессуют в брикеты, которые й загружают в муфели. Далее, согласно предложенному в Нью-Джерси способу, собранный в конденсаторе цинк нагревают до 1100’ и подвергают фракционированной перегонке в колоннах (подобно тем, которые применяют для ректификации спирта). В верхней части колонны собирается богатый кадмием цинк, в средней — очищенный <99,995%), а в пижпей остается цинк, который содержит основную часть примесей, имевшихся в сыром продукте (свинец, медь и железо).
* Растворимость свинца в цинке при температуре плавления последнего составляет 1,5 вес.% (Spring, Romanoff). Давление же пара при 1000° у цинка в 2000 раз больше. Это позволяет при перегонке достигать значительно битее топкого разделения этих металлов, чем при переплавке.
460
Глава 9
Для производства цинка из богатых железом руд New Jersey Zinc Company с 1951 г. примепнет усовершенствованный способ Стерлинга, использующий для нагревания пламя электрической дуги. Благодаря этому ZnO восстанавливается углем рапьше, чем успевает расплавиться шлак. И только в конце происходит восстановление Fe3Oa ДО Ее, которое собирается под расплавившимся к этому времени шлаком, и время от времени их вместе спускают. Пары цинка, выходящие через верхнюю часть печи, сжижаются в конденсаторах.
Электролитическое производство цинка осуществляют следующим образом. Цинковую руду предварительно обрабатывают серной кислотой, цинк переходит в сульфатный раствор. Полученный кислый раствор подвергают электролизу при высокой плотности тока.
То, что цинк способен выделяться из кислого раствора, несмотря па то что в ряду напряжений гш стоит гораздо левее водорода, объясняется высоким перенапряжением водорода при катодном выделении цинка. Одпако это наблюдается только в том случае, когда циии по содержит веществ, понижающих перенапряжение. В противном случае па них начинает выделяться водород, а это влечет за собой не только снижение выхода по току, нои возникновение локального элемента (см. стр. 462) и, следовательно, растворение уже выделившегося цинка. Помехи такого рода могут вызвать даже весьма незначительные следы содержащихся в электролите примесей, если они менее активны, чем цинк, и, следовательно, могут быть им вытеснены. Так, наличие следов германия в некоторых ципковых рудах вызывает значительные затруднения. В настоящее время примеси благородных металлов устраняют в большинстве случаев добавлением к электролиту цинковой пыли. Изготовление достаточно стойкого к электрохимическим воздействиям анода вначале бгдло также связано с большими трудностями. Этим объясняется то, что электролитическое производство цинка только начиная примерно с 1930 г. поднялось до уровня большой техники, хотя зэродллось оно еще в 1894 г. (Хёцфиер, в Фюрфурте на реке Лане) и в принципе превосходила дистилляционные методы. В результате преодоления перечисленных трудностей это производство развилось чрезвычайно быстро, И в 1934 г. уже третья часть мировой продукции ципка производилась электролизом сульфатпого раствора, В настоящее время этим способом производит около 40% всей мировой продукции цинка. Благодаря применению сульфатпого электролизного производства в больших масштабах в Германии удалось полностью удовлетворить потребность в цинке за счет местного сырья. Электролитический способ пе только обеспечивает более высокую чистоту продукта; но в большинстве случаев и более полное вскрытие руд по сравнению с дистилляционными способами, в частности со старым муфельным. Помимо этого, его неоспоримое преимущество заключается еще в универсальной применимости независимо От сорта руды. Например, при переработке богатых кремнием или относительно бедных цинком руд оп дает возможность также легко отделять цинк от примесей. Однако рентабельность этого способа определяется стоимостью электроэнергии, так как расход ее довольно велик. Напряжение на ваннах должно быть порядка 3,3—3,7 в, откуда следует, что при условии 90% -кого использования энергии па 1 т цинка расходуется 3000—3400 кет постоянного тока, пе считая потерь в проводах и выпрямителях.
Иногда прибегают также к электролизу расплава (смеси ZnCl2 с KCI), Это выгодно в тех случаях, когда сырье можно прохлорировать и легко отогнать ZnCl2.
Цинк в виде ZnCl2 получают также выщелачиванием содержащей цинк окалины, образовавшейся при хлорировании. Из полученных растворов ZnCl2, содержащих большие количества NaCl, цинк отделяется электролитически амальгамным способом, разработанным в 1937 г. на Дуйсбургском медеплавильном заводе и представляющим собой разновидность электролиза щелочных хлоридов. В качестве анода используют графит, катодом служит ртуть, налитая тонким слоем на дно ваппы. Образующаяся амальгама цинка становится аподом для второй нанны. Если требуется получить цинковые белила, то в качестве электролита используют раствор NaHCO,. Попадающий в раствор цинк осаждается в виде карбоната, который прокаливают до окиси. Если же хотят получить металлический цинк, то в качестве электролита] используют раствор ZnSOt; цинк выделяется на алюминиевых пластинах.
Свойства. Цинк представляет собой металл синевато-белого цвета, обладающий сильным металлическим блеском, который, однако, во влажном воздухе постепенно исчезает, вследствие образования окисной пленки. При обычной температуре циик довольно ломок, но при 100—150° он ста
Вторая группа периодической системы 461
новится тягучим настолько, что его можно прокатывать в листы й вытягивать в проволоку. Выше 200° он, напротив, делается настолько хрупким, что его можно растирать в порошок. Цинк очень легко плавится (т. пл. его 419,4°) и принадлежит к числу наиболее легко летучих металлов (температура кипения 905,7°). Удельный вес очень чистого литого цинка 7,13 (при 18°), для вальцованного цинка он несколько выше. Плотность жидкого цинка при температуре плавления составляет 6,92. Твердость цинка равняется 2,5 по шкале Мооса, Примеси оказывают значительное влияние на механические свойства цинка, например содержание в нем уже 0,12% железа сильно понижает его пропитываемость.
Теплой ров одп ость линка составляет 61—64%, а электропроводность 27% относительно серебра (ср. табл. 46, стр. 453).
Нормальный потенциал цинка по отношению К нормальному водородному электроду равен — 0,762 в (при 25°) (La Мег, 1934). Следовательно, в электрохимическом ряду напряжений цинк стоит значительно левее водорода.
Благодаря довольно сильной склонности заряжаться положительно, которая соответствует месту, занимаемому цинком в ряду напряжений, он легко растворяется в разбавленных кислотах с выделением водорода
Zn4-2H- = Zn"4-H2.
Чистая вода заметного действия на цинк не оказывает. Однако он растворяется в крепких растворах щелочей, а также в водных растворах аммиака и хлорида аммония, особенно при нагревании. Если цинк очень чистый, то растворения не происходит или оно протекает чрезвычайно медленно. Это относится также и к растворению цинка в кислотах. Поэтому ори растворении очень чистого цинка в кислотах обычно для ускорения реакции добавляют несколько капель очень разбавленного раствора сульфата меди. Медь, стоящая правее цинка в электрохимическом ряду напряжений, тотчас вытесняется им в осадок и, загрязняя поверхность цинка, ускоряет его растворение.
Ускоренно растворения цинка в присутствии (в пем или на его поверхности) примесей обусловлено образованием между ципком и загрязняющими его металлами гальванических пар, так что на местах, где нет примесей, цинк переходит в раствор, а на примесях, состоящих в большинстве случаев из менее активных металлов, выделяется водород. Таким образом, в результате переноса зарядов внутри металла возникают местные токи, как показано на рис. 48 (направление тока указано стрелками)*. В отсутствие примесей водород может разряжаться только на поверхности самого цинка. В результате, как обычно полагают, водород в виде чрезвычайно тонкого слоя удерживается на ней, затрудняя тем самым дальнейшее растворение металла и образование водорода в таком количестве, чтобы он мог собираться в пузырьки и выделяться из раствора. Возможно, что из-за высокого перенапряжения, которое необходимо для выделения водорода па чистом цинке, по может происходить разрядка водородпых ионов н соединенно атомов И в молекулу Нг на поверхности совершенно чистого-цинка (ср. т. I, стр. 53). Эффект настолько велик, что совершенно чистый цинк в разбавленных кислотах практически нерастворим.
В результате амальгамирования поверхности цинка действие примесей снижается, гак как на равномерно амальгамированной поверхности нс могут уже образовываться гальванические нары типа, приведенного на рис. 48. И только если в раствор поместить другой металл , например медь, и соединить его с цинком, как это осуществляют в гальванических элементах (рис. 49), ципк начинает растворяться, и притом в той морс, в какой происходит выделение водорода на меди. Поэтому, чтобы сделать используемые в гальванических элементах цинковые пластинки более прочными, их перед употреблением рекомендуется амальгамировать.
*) Образование такого рода «местных токов» играет важную роль при растворении всех металлов в кислотах (ср. стр. 790).
462
Глава 9
В соответствии с формулой Нерпста [уравнение (1), т. I, стр. 49] потенциал выделения водорода из чистой воды только на 0,41 « больше, чем из 1 н. по ионам Н‘ раствора. Следовательно, цинковый электрод даже по отношению к водородному электроду, погруженному в чистую воду (или нейтральный раствор соли), обладает отрицательным потенциалом. Если тем пе менее цпнк все же не растворяется в воде (в заметном кол ячестве),'то это объясняется тем, что образующаяся при взаимодействии с водой гидроокись цинка практически нерастворима и ее тончайшая поверхностная плёнка предохраняет пипк от дальнейшего растворения. Если удалить защитный слой добавлением веществ, растворяющих гидроокись цинка (щелочь или аммиак), то цинк переходит в раствор с выделением водорода. Соли аммония также облегчают растворение цинка, потому что вследствие гидролиза в них всегда присутствует аммиак. На этом основано применение нашатыря в элементах Лекланше и в сухих элементах.
Р и с. 48. Ускорение растворения цинка загрязнением поверхности (схема).
Р н с. 49. Гальванический элемент.
Потенциал растворения водорода в растворах с 1 н. концентрацией гидроксильных цопов (—0,81 а ио отношению к нормальному водородному электроду) ниже нормального потенциала цинка. В сил ьнощел очных растворах потенциал растворение цинка сдвигается значительно дальше в отрицательную сторону, так как в соответствии с равновесней
Zn"-{-3OH' [Zn(OH)3]'
концентрация свободных ионов цинка Zn”, определяющая потенциал растворения, очень мала. По этой причине в сил ьнощел очных растворах потенциал растворенпг, ципка ппже, чем у водорода. Подобное же действие оказывает аммиак, которых" растворяет гидроокись цинка с образованием комплексного соединения гидроокссц тот р аамминци нк а [ Z n (N Н 3)4 ] (О Н) 2, и б л а г ода ря этом у к о ццентр а ци я св об одних и сп~. цинка также остается пивной. Нейтральные сопл аммония являются зпачптелыл-более слабыми комплексообразователями, но в нейтральных растворах и водородпьЁ). потенциал значительно ниже.
Теплота растворения цинка в разбавленной соляной кислоте составляет 18,2 skj, в разбавленной серной —18,9 ккал/г- экв (при постоянном давлении).
Прн растворении цинка в разбавленной азотной кислоте водород но выделяется, оп расходуется а момент выделения на восстановление кислоты. При этом очень ксл: центрированная азотная кислота восстанавливается до окислов азота, в менее келг центрированной наряду с ними образуется аммиак. Из концентрированной сервсл кислоты также выделяется пе водород, а двуокись серы. Водород, выделяющийся с:.: разбавленных растворов серной кислоты, иногда тоже содержит двуокись сер_ и сероводород.
При нагревании на воздухе цинк сгорает ярким голубовато-зеленых: пламенем с образованием окиси ZnO. При температуре красного каление
Вторая группа периодической системы
463
цинк окисляется также под действием водяных паров и двуокиси углерода, которая восстанавливается до окиси углерода. С галогенами при комнатной температуре цинк реагирует довольно медленно и лишь в присутствии влаги. Сероводород действует на него ужо при обычной температуре, однако образующийся на поверхности сульфид защищает цинк от дальнейшего воздействия сероводорода. При нагревании порошкообразный цинк пепосредствеппо соединяется с серой. С азотом даже в парах цинк заметно не взаимодействует. Напротив, с аммиаком при температуре красного каления он образует нитрид Zn3Na. Правда, реакция протекает не полностью даже с порошкообразным цинком.
В расплавленном состоянии цинк неограниченно смешивается со многими металлами, например с Си, Ag, Ли, Cd, Hg, Mg, Са, Мн, Fe, Со, Ni, A], Sn, Sb, а также с Аз. Однако в твердом состоянии он образует гомогенные смеси только в очень ограниченной мерс. В решетке кадмия он может содержаться лишь в количестве —1 вес.% , с магнием также только в очень незначительном степени образует твердые растворы. Помимо этого цинк со многими металлами образует соединения, например с Си, Ag, Ли, Мп, Fe, Со, Ni, Rh, Pd, Pt, As, Sb,. Mg, Ca, Li, Na, К., по пе c Cd, Ga, Tl, Cr, Bi, Sn и Pb (ср. также стр. 455).
Применение. Цинк находит' широкое применение, особенно в виде листового цинка, например для изготовления бидонов, ведер, ванн, желобов на крышах. Листовой цинк употребляют также для кровель, для обшивки степ, а также для выстилки Kopi.iT, ящиков, ледников и т. п. Многие предметы изготовляют из литого цинка. В больших количествах цинк расходуют на гальванические элементы. В лаборатории его применяют главным образом в виде палочек для получения водорода. Цинковую пыль (обычно с большим содержанием окиси) широко используют в технике и в лаборатории в качестве восстановителя. В металлургии цинк служит для извлечения серебра из свинца (ср. стр. 420) и для осаждения золота в цианидном процессе.
Из сплавов наиболее важными являются сплавы цинка с медью (желтая латунь и красная латунь). Эти богатые медью цинковые сплавы удалось для многих целей заменить сплавами, содержащими мало меди либо совсем без нее (прежде всего алюминиевыми сплавами); в таких сплавах цинк часто составляет основную часть. В качестве примера можно указать белую латунь (85% Zn, 5 Al, 10% Си), люменбронзу (88% Zn, 6 Al, 6% Си), сплав целко (83% Zn, 15 AI, 2% Си).
Большие количества цинка расходуются для покрытия других металлов, особенно железа. Этот процесс осуществляют либо наплавлением (горячее цинкование), напылением и действием паров (паровое цинкоеание), либо гальваническим способом.
Некоторые соединения ципка служат красками, особенно литопон (смесь BaS с ZnSO4,, приготовляемая смешиванием ZnS с BaSOJ и цинковые белила (окись цинка ZnO). Очень мелкую цинковую пыль (цинковая серая) или тонкоизмельченную цинковую обманку (цинксульфидная серая) применяют в качестве покрытий, защищающих от ржавчины, например для мостов и деталей машин. Некоторые соединения цинка, прежде всего хлорид и сульфат, употребляют в медицине в качестве антисептических средств. Основной борат цинка служит присыпкой.
Соединения цинка отпосительно мало ядовиты. Однако в больших количествах они могут быть вредными. Поэтому, ввиду легкой растворимости цинка, цинковые сосуды не рекомендуются для храпения продуктов, их употребление в производстве пищевых продуктов запрещено законом.
464
Глава 9
СОЕДИНЕНИЯ ЦИНКА
В соединениях цинк всегда положительно двухвалентен. В водных растворах содержатся бесцветные ионы Zn'*. И соединения цинка бесцветны, если пе окрашена та составная часть, с которой он соединен. Даже сульфид цинка бесцветен, а при обычной температуре и окись (при нагревании она желтеет).
Склонность к комплексообразованию с большинством анионов у Цинка менее выражена, чем у меди. Напротив, оп значительно охотнее связывает избыточные гидроксильные ионы, другими словами, тенденция гидроокиси к образованию солей с сильными щелочами у цинка значительно больше, чем у меди. Подобно гидроокиси алюминия гидроокись цинка имеет ярко выраженный амфотерный характер. В щелочном растворе она действует как ангидро-кислота, т. е. переходит в раствор не в виде аниона, образовавшегося в результате отщепления иона Н‘, а в виде гидроксоцинкат-ионов, за счет присоединения ионов гидроксила, например цинкат-иопа. [Zn(OH)3]':
Za(OH)2+ОН' = [Zn(OH)3]
Соответствующие им соли называются цинкатами (точнее гидроксоцин-катами). Известны три-, тетра- и гексагидроксоцинкаты: MI[Zn(OH)3j, Mj[Zn(OH)4I или Mn[Zn(OH)4I и M“[Zn(OH)eJ.
Предположение о том, что цинкаты, полученные из водных растворов, представляют собой гидрокео-со.ги, было выдвинуто Пфайффером еще в 1908 г. па основание теории Бернера об ангидро-кислотах. Это представление нашло подтверждение в образовании таких гидр оксо-сол ей гидроокисями других металлов (гидр оке остан и аты и гпдроксоплюмбаты, ср. т. I, стр. 582 и 591), Однако для цинкатов такое предположение было с достоверностью установлено только Шольдером (Beholder, 1933), который показал, что входящая в состав такого рода соединений вода не кристаллизационная, а Конституционно связанная. Натриевая соль состава Na2O-ZnO-2H2O, папример„ при температуре J 90—200° отщепляет только одну молекулу воды. Две другие Ирочка удерживаются даже при длительном нагревании при температуре 465°. Лишь пря сплавлении с бихроматом калия вода полностью выделяется. Если бы названное сощенение представляло собой днгидрат Na2ZnO2-2H2O, то естественно было бы ожидать, что полiiaя потеря воды произойдет ужо при умеренном нагревании. Если же речь идет о гндроксо-соли Na3[Zn(0H)J, то опытные данйые легко объясняются предположением, что это соединение немного ниже 200° разлагается с отщеплением одной молекулы воды, так как Zn(OH)2 выше 100° неустойчив; Na2[Zn(OH)4] =<2нО — 4- 2NaOH Н2О. NaOH при сплавлении с К2Ст2О7 выделяет воду по уравнению
2NaOH -j- K2Cr2O7=Na2CrO4+K2CrO4+ Н2О.
При продолжительном нагревании NaOH также не способна соединяться с ZnO с образованием Na2ZnO2 и выделением воды.
Б качестве примера гидр оксоцинкатов следует указать следующие полученные Шольдером соединения; Na[Zn(OH)3), Na[Zn(OH)3]-,3II2O, Na2(Zn(OH)4K Na2[Zn(OI-I)4].2II2O, Sr{Zn(OH)4].H3O, Ba[Zn(OH)4]-H2O, Sr2 [ZnfOHy, Ba2[Zn(OH)6]. Некоторые из этих соединений, к ан видно из приведенных формул, наряду с конституционной, содержат кристаллизационную воду. В большинства случаев последняя очень легко отщепляется, например гидрат тетра гидр оксоцинкатэ бария обезвоживается при 87° а тригидрат три гидр оксоцинката натрия пад серией кислотой в вакууме уже при обычной температуре.
Пипк дает также комплексные катионы. Они образуются при присоединении нейтральных частиц, особенно аммиака, if ионам цинка Zo’\ В результате образования таких комплексных йрнон большинство нерастворимых в воде соединений цинка (например, гидроокись) растворяются в водном растворе аммиака.
Вторая группа периодической системы
465
Несмотря на большую растворимость, удалось выделить в кристаллической форме целый ряд аммиакатов. Большей частью они соответствуют тину [Zn(NH3)2]X2 (соли диаммйпципка). Некоторые из них кристаллизуются с 4 молекулами NH3 (соли тетраамминцинка). Аммиакаты с б Nlls были получены не ИЗ водного раствора, а сухим способом. Из хлоридов известен ряд: [Zn(NH3) }C12, [Zh(NHs),JC13, [Zrt(NH3)JCl2, [Zh(NH3)6]C12, [Zii(NH3)6]C12; нз сульфатов: [2п(ИН3)2]8О4-H20, [ZnfNH^JSOi, [Zn(NH3)5]SO4.
Помимо аммиака, в состав комплексных солей цинка могут входить'’ многие органические азотсодержащие вещества, например анилин, фенил гидр аз ин, хиполин, стрихнин и т. д. Большинство из образующихся соединений соответствует типу [ZiiAm2]X2, т. е. цинк координирует, как правило, две молекулы органического ангидрооснования; известны, однако, и соединения типа [ZnAme]X2. Сюда относится, например, описанный Ворпером цинктрнэтилен диамин бромид [Zn(en)3]Br2, так как каждый «он»— этилендиамин ri2N—СН2—СН2—NH2 занимает два координационных места,
В табл. 50 сопоставлены некоторые данные о важнейших простых соединениях цинка.
Таблица 50
Теплоты образования, плотность, температуры плавления и кипения простых соединений цппка
Вещество Формула Теплота образования. Плотность (при 25ф) Температура, “С
плавления | кипения
Окись цинка ZnO 41,6 5,78 - 2000° (под давлением) Возгоняется при~1725°
Сульфид ZnS 20,7 4,06 1800—1900° (под давлением) Возгоняется при 1180°
Фторид ZnF2 96,4 4,84 872° —•
Хлорид ZnCl2 49,4 2,904 318° 730°
Бромид ZnBr2 —- 4,20 394° 650°
Иодид Znl2 24,9 4,74 446° —
Из простых солей цинка легко растворимы, в воде хлорид (а также другие галогениды, за исключением фторида), нитрат, сульфат кацетат. Из труднорастворимых соединений в первую очередь следует назвать окись и гидроокись, а также сульфид; далее труднорастворимы карбонат, фторид, фосфат, силикат, цианид и циапоферрат(П). Цианоферрат(П) калия и цинка K2Zn3 [Fe(CN)e]2 применяют в аналитической химии (ср. стр. 475),
Для цинка известно большое число основных солей. Большинство из них, по данным Файткнехта (Feitknecht, 1933), образуют «двухслойную решетку» (ср. стр. 320). «Основные» слои состоят из Zn(OH)2; в «промежуточных» слоях ионы соли закономерно располагаются на определенных местах решетки. Однако у некоторых из этих соединений ионы соли не имеют строго определенного места в промежуточных слоях. Это влечет за собой, как у зеленого основного хлорида кобальта (ср. стр. 320), переменный состав соединений,.несмотря на то что все они хорошо кристаллизуются. Лишь в отдельных случаях среди основных солей цинка встречаются соединения с «однослойной решеткой» [например, Zn(OH)Cl], в то время как для соединений кадмия структуры подобного типа являются весьма характерными.
30 Г. Геми
466
Глава 9
Прежде, до теории Вернера, основные соли цинка рассматривали как «ол-соедп--нения», или как р-гидрокво-соединения(ср. стр. 417). В то время как для тмер&ыз солей это предположение было опровергнуто рентгенометрическими исследованиями, для растворов оно в соответствии с последними данными (Hayek, 1934) весьма вероятно.. Таким образом, предполагают, что в растворе молекулы гидроокиси (цинка иле другого металла) ввиду своего дипольного характера группируются вокруг ионе цинка в качестве центрального атома, образуя комплексный ион подобно молекулам воды в акв о-комплексах. Например:
Zn" + 6Н0Н = (Zii(HOH)a]", соответств енн о
Zn'4-3Zu(0H)2=
/ИСК \ I Zn( yZn I
\H0Z /з.
Растворение окисей тяжелых металлов или гидроокисей в pa створах сол ей ципка как par и объясняется образованием такого рода комплексов. На этом основано, папрпмер, примененпе раствора хлорида цинка в качестве «паяльной жидкости» (ср. стр. 471}
Окись цинка ZnO встречается в большом количестве в природе в одном месте, а именно в Нью-Джерси, и виде красной цинковой руд^ (цинкит). Красный цвет минерала определяется содержанием в нем марганца, которое может достигать 9%. Чистая окись цинка при обычней температуре бесцветна, при нагревании желтеет. Она кристаллизуется в гексагональной сингонии (решетка вюртцита, рис. 60, т. I, стр. 2№; а =3,243, с = 5,195А). Удельный вес чистой окиси 5,78, твердость 4—ё по шкале Мооса. Для технических целей окись ципка получают; обычш сжиганием металлического цинка. Таким способом окись цинка получается в виде чистого белого рыхлого порошка (философская шерсть Lana philosophic^ алхимиков). Ее применяют главным образом в качеств краски (цинковые белила, снежные белила, китайские белила). Наряду с этим ее используют в качестве наполнителя мягкой резины, для изготовления замазки и сургуча; се употребляют также как порошок для чистит и для получения других соединений цинка. Совершенно чистая окпгд. ципка, получаемая прокаливанием основного карбоната, в виде пудрьг или пасты находит применение в косметике (например, для грима) и в меде-цине (преимущественно как присыпка и при изготовлении мазей; океге цинка входит также в состав лейкопластыря). Окись цинка (сама по сеА или в смеси с другими окислами) служит катализатором для ейнтегд метанола.
Каталитическое действие окиси цинка существенно зависит от способа ее nszy чепия. Например, осторожный термическим разложением гидроокиси или нарбшЕЁ. получают ZnO с высокими каталитическими свойствами; напротив, при раз.чожеЕЕ нитрата образуется окись с низким каталитическим действием. Приготовленная ее гидроокиси и карбоната окись тем более действенна, чем слабее был нагрев при г? получении. Препараты, синтезированные различными способами, имеют один тдг решетки, одпако различаются степенью ее завершенности (ср. стр. 36). ПоследШЕ следовательно, определяет каталитическую активность ZnO. Наиболее антивЕЗСЕ катализаторами оказываются препараты с так называемым промежуточным состтт яием. Опп отличаются тем, что решетка исходного вещества (например, ZnCO*) разрушена, а решетка продукта реакции (ZnO) еще не завершена. Подобные «актштдд. переходные состояния» известны л немногих других случаях, когда реакции текают в твердом состоянии (ср. стр. 786).
Не только каталитические свойства, но И химическая реакционная споссбюх^" тем выше, чем больше нарушений имеется в решетке. Известно, например (Нй’У : 1937), что полученная разложением основного карбоната окись цинка тем легчг обя зует с СоО зелень Рин май а, чем ниже была температура разложения (ср. стр.. 4Г'' Теплоты растворения препаратов окиси цинка также в значительной степени злее: ' от исходного вещества и способа его получения. Фрпкке (Fricke, 1932) показал ~
Вторая группа периодической системы
467
у активных препаратов они выше на 1,2 ккал/молъ, чем Для «нормальной» ZnO, т. е. не обнаруживающей существенных отклонений в решетке. Рентгенометрическими исследованиями (1933) Фрикке доказал, что различное теплосодержание и неодинаковая активность обусловлены не наличием модификаций или значительной разницей в величине частиц, а лишь незавершенностью решетки.
При высокой температуре 1100® ZnO реагирует с Ба О е образованием двойного окисла или ок со цинката состава BaZnO2. Подобное соединение ВаО образует также с CdO (Scholder, 1953).
В воде окись цинка практически нерастворима. (Растворимость ZnO в воде, находящейся в равновесии с воздухом, содержащим С02, составляет 3,0 мг/л.)
Гидроокись цинка Zn(OH)2 выпадает в виде белого, объемистого осадка (большей частью с примесью адсорбированных ионов) при добавлении щелочи к растворам солей цинка. Опа легко растворима в избытке осадителя, а также в кислотах и в водном растворе аммиака.
Растворимость в кислотах основана на образовании растворимой соли в результате реакции нейтрализации (4), в избытке щелочи — на образовании цинкатов (5), а в водном растворе аммиака — на комплексообразовании * (6).
Например: Zn(OH)24-113804=ZnSO4-J-2II2O, 1
Сульфат цинка (
вообще: 2тцОН)зЧ- 21Т‘ = Zn” J- 2НгО. J
Например: Zn(OIT)2-J-NaOH=NafZn(OFT)3], Цинкат ватрия ,д,
вообще: Zn(OII)24-OH' = [Zn(OH)3]',
Zn(OH)2-|-4NH3=[Zn(NH3)J(OH)2
или Гидроокись теграамиинцинка
Zn(OH)2+4NHs=[Zn(NH3)4]" + 2OH' 1 J
Гидроокись ципма немного растворима и в растворах солей аммония. При этом устанавливается равновесие:
[NHJ- NH3-J-H’.
Обычно в нейтральных растворах такого рода расщепление происходит лишь в незначительной степени. Но в присутствии гидроокиси цинка равновесие в значительной степени сдвигается вправо, так как опа может реагировать как с NH3, так и с Н'-ионами:
+ [Zn(NH,),j"-r 2HSO,
или Zn(OH)a + 2[NH4]‘ [Zn(NHj)sr +2HSO.
Если взаимодействие растворов солей цинка с аммиаком идет на холоду, то вначале получают студенистый, аморфный осадок («оксигидрат цинка»). Через некоторое время он коагулирует в хлопья; дебаеграммы такого осадка свидетельствуют о начавшемся упорядочении частичек, и вначале, по-видимому, образуется окись цинка ZnO. Лишь при более длительном старении наблюдается интерференция рентгеновских лучей, характерная для гидроокиси цинка Zn(OH)2. В присутствии маточного раствора (вода) процесс полного перехода в стабильную кристаллическую гидроокись цинка длится в течение месяца (Fricke, 1924). Если вести осаждение щелочью, то образование кристаллической гидроокиси проис
* Это следует, например, из данных измерения чисел переноса: при электролизе пересыщенных по отношению к аммиаку растворов цинк перемещается к катоду.
30»
468
Глава 9
ходит быстрее. Скорость кристаллизации зависит также от соли, иа раствора которой осаждают цинк, например из хлоридных растворов кристаллическую гидроокись получают значительно быстрее, чем из растворов нитрата.
Za(OH)2 может существовать в пятн различных кристаллических модификациях. Условия их образования и свойства исследованы главным образом Фринке (1924) а Фапткнехтом (1930} В соответствии с уменьшением теплосодержания различают а-, ₽-, V-, в- и ®-Zn(OH)2. Устойчива лишь последняя из них, кристаллизующаяся в ромбической сингонии e-Zn(OH)3. Остальные метастабильны при определенных условиях получения. Частично они образуются в качестве промежуточных продуктов при переходе аморфного гидрата окиси цинка в ромбическую гидроокись. В присутствии воды все они спустя некоторое время превращаются в e-Zn(OH)s. Последняя ниже 39’ устойчива к действию воды (Hiittig, 1933); при более высокой температуре возникает равновесие между ZnO и водой. Но так как равновесие в этой системе устанавливается крайпе медленно, то e-Zn(0H)2 можно продолжительное время нагревать в вакууме при 60° без видимой потери воды. Наименее устойчивая из кристаллических модафи-кадий гидроокиси цинка a-Zn(OH)2 уже при обычной температуре может под действием воды перейти в ZnO. Правда, в конце концов она снова присоединяет воду, но образует ужо стабильную модификацию е.-2н(ОН)3.
В то время как малоустойчивая модификация a-Zri(OH), (Feitknecht, 1938) имеет гексагональную двухслойную решетку (ср. стр. 320), основной слой которой построен подобно Mg(OH)2 и Cd(OH)2 *, стабильная ромбическая модификация з-7п(ОН)3 образует особого типа решетку, не наблюдаемую обычно у гидроокисей. Она имеет вид пространственной сетки, состоящей из тетраэдров Zn(0H)4, подобно тому как одна из модификаций SiO3 состоит из сетки, образованной тетраэдрами SiO4. Тетраэдры Zn(ОН)4 несколько искажены, так что расстояние Zn*—* ОН колеблется между 1.94 и 1,96 А. Наименьшее расстояние равно расстоянию Zu *—* О в решетке окиси цинка (Wyckoff, 1933; Megaw, 1935).
Теплота образования кристаллической 6-Zn(0Hj2 из кристаллической ZnO и Н20 составляет 2,28 ккал/молъ (Fricke, 1932).
Амид цинка и натрид цинка. П.чкЭ цинка Zn(NH2)2, получаемый взаимодействием этилата цинка Zn(C2H5)2 с аммиаком в спиртовом растворе: Zn('C2H5).i -ф 2NH-= = Zn(NH2)2 2С2Нв, образует белый порошок. Водой он разлагается на Zn(0H)j и NHj. Удельный вес 2ДЗ, теплота образования из элементов составляет 35 ккал/.ааль (Juza, 1937). При температуре красного каления амид цинка превращается в нитрид по уравнению 3Zn(NH2), = Zn3N3 + 4NH3. Нитрид цинка представляет собой серый пороаток, устойчивый при нагревании в отсутствие воздуха (уд. вес 6,2). Водой Zn3N2 тотчас гидролизуется; Zn3N2 -j- 6НОН = 3Zn(OH)2 -j- 2NH3. Нитрид цинка образуется также при взаимодействии цинковой пыли с газообразным: аммиаком. Он обладает диамагнитными свойствами. Теплота образования из элементов составляет 5 ккал/моль (Jiiza, 1938).
Сульфид цинка ZnS встречается в природе во многих местах в виде цинковой обманки (обманка, сфалерит), имеющей кубическую решетку., редко в форме вюртцита, кристаллизующегося в гексагональной сингоапв. Чистый сульфид цинка белого цвета. Природный сульфид окрашен в боле? или менее темные тона за счет примесей (главным образом FeS), Из верных растворов солей цинка сульфид цинка выпадает в виде белого аморфного осадка при добавлении сернистого аммония, а в присутствии достаточного количества ацетат-ионов осаждается сероводородом.Свежеосажден-ный ZnS легко растворим в сильных кислотах; при стоянии он постепенЕ' превращается в более трудно растворимую модификацию. Сульфид цшша легко переходит в коллоидный раствор, например при продолжительны.' действии сероводородной воды. Коллоидный раствор получается такж? при пропускании сероводорода через аммиачный раствор цинковой солп..
* Решетка a-Zn(OH)3 устойчиватолько прп условии, если в Еромешутсчнзл елпях, помимо гидроокиси (распределенной неупорядоченно), содержится количество соли, например хлорида цинка или карбоната.
Вторая группа периодической системы
469
При нагревании в сероводородной воде под давлением аморфный осадок сульфида цинка переходит в кристаллический — так образуется цинковая обманка. Если же нагревают сухой сульфид в токе водорода или сероводорода, то образуется вюртцит. Эта модификация устойчива при более высокой температуре. Температура превращения лежит при 1020° (Allen). Если вюртцит тонко растереть в ступке, то, как следует из рентгеноструктурных данных, оп переходит в обманку.
В кристаллической решетке цинковой обманки (ср. рис. 47, Стр.407) длина ребра элементарной ячейки а =5,42 А, кратчайшее расстояние Zn <—► S равно 2,35 А. Вюртцит образует гексагональную решетку (рис. 60, т. I, стр. 290); а = 3,84, с = 6,28 А. Кратчайшее расстояние Zn <—> 8 в решетке вюртцита равно 2,36 А, т. е. практически такое же, как в решетке цинковой обманки. Это свидетельствует о том, что оба типа решеток «соизмеримы» (ср. стр. 407).
ZnSe и ZnTe изотиптты цинковой обманке (а = 5,66 или 6,09 А). То же относится к HgSe, HgTe и CdTe. CdS и CdSe подобно ZnS могут кристаллизоваться как по типу цинковой обманки, так и по типу вюртцита.
Удельный вес кристаллического ZnS составляет 4,09 и твердость 3,5—4. Разница между удельными весами обманки и вюртцита пе превышает ошибки измерения. Растворимость кристаллического сульфида цинка около 7-10-* молъ/л, све-Же осажденного почти в 10 раз больше. По данным Бильтца, ZnS сублимируется при температуре около 1180°. Тиде и Шлееде (Tiede, Schleede) удалось расплавить его при температуре 1800—1900° под давлением 100—150 ат,
Искусственно приготовленный сульфид цинка находит применение прежде всего в качестве белой малярной краски, одпако чистый сульфид применяется редко, а большей частью в виде литопона. Так называют смесь сульфида цинка и сульфата бария, приготовляемую в технике взаимодействием сульфида бария с сульфатом цинка
BaS-|- ZnSOt—ZnS -{-BaSO4.
Литопон гораздо дешевле, чем свинцовые белила, кроме того, он неядовит и устойчив к действию сероводорода. Однако по качеству покрытия и стойкости он уступает и свинцовым белилам и цинковым (ZnO). Но основным недостатком некоторых сортов литопона является малая устойчивость на свету.
Сульфид цинка под действием ультрафиолетового излучения окрашивается в серый цвет вследствие выделепия элементарного цинка. Вюртцит наиболее быстро подвергается этому процессу, цинковая обманка—медленнее. Чувствительность к свету можно регулировать введением добавок. Например, добавка очень небольшого количества соли кобальта к раствору сульфата цинка перед реакцией с BaS позволяет получить литопон с повышенной светоустойчив остью. На чем основано действие солей кобальта, пока еще не ясно.
Сидотова обманка. Переведенный в кристаллическую форму плавлением, или возгонкой, или же нагреванием в присутствии минерализаторов, сульфид цинка, по возможности освобожденный от примесей, но со следами тяжелых металлов, например марганца или меди, обнаруживает свойство ярко светиться после облучения, подобно фосфоресцирующим солям щелочноземельных металлов. В честь химика, впервые наблюдавшего это явление, приготовленный таким способом сульфид цинка называют сидотов ой обманкой. Б отличие от фосфоресцирующих солей щелочноземельных металлов сидотова обманка сильно светится также под действием радиоактивного и рентгеновского излучений. В связи с этим ее используют для рентгеновских экранов, Употребляемая для светящихся циферблатов я т. и. масса содержит около 1 мг радия или мезотория па 10 в сульфида цинка.
Фосфид цинка. Если над расплавленным цинком пропускать ток водорода или азота вместе с парами фосфора, то образуется фосфид цинка Zn3P2 (трицинкдифосфид). Он представляет собой непрозрачные серебристо-белые тетрагональные кристаллы в форме игл или листочков (с уд. весом 4,54), нерастворимые в воде. При избытке фосфора получают оранжевые прозрачные тетрагональные кристаллы дифосфида цинка ZnP2. Zn3P3 образует решетку, похожую на решетку Sc2O3 (Stackelberg, 1935) (см. стр. 55); так же как там атомы кислорода, атомы фосфора в ней образуют плотнейшую
НО
Глава 9
упаковку. Часть пустот в решетке занимают атомы цинка. Каждый атом цинка окружен чытырьмя атомами фосфора, расположенными в виде искаженного тетраэдра, а каждый атом Р — шестью атомами Zn, которые занимают 6 из 8 углов искаженного куба. Подобную структуру имеют Zn3As2, Cd3P2 и Cd3As2.
Хлорид цинка. Хлористый цинк ZnGl2 получают растворением цинковой обманки, окиси цинка или цинковой стружки в соляной кислоте. Если в качестве исходного материала берут металлический цинк, то в растворе не содержится тяжелых металлов, кроме железа (так как электролитическая упругость растворения у соответствующих тяжелых металлов значительно меньше, чем у цинка). После предварительного окисления хлором железо осаждают в виде гидроокиси Fe(III), прибавляя окись цинка. После упаривания содержащего избыток соляной кислоты раствора получается белый зернистый порошок, состоящий из гексаго-нацьно-ромбоздрических кристалликов. Он легко плавится и застывает затем в виде похожей на фарфор прозрачной массы. В продажу он поступает как в виде порошка, так и в виде переплавленных кусков или палочек.
Расплавленный хлорид цинка довольно хорошо проводит электрический ток. Шультц установил, что удельная электропроводность при 400° и « 0,03, при 706° и = 0,4И обратного ома. При прокаливании хлорид цинка улетучивается (т. кип. 730е); его пары конденсируются в виде белых игл. Хлорид цинка чрезвычайно гигроскопичен. Поэтому во влажном воздухе оп расплывается. Вместо с тем его легко получить из водного раствора в виде безводных кристаллов. Выше 28° хлорид цинка кристаллизуется без воды; ниже этой температуры он образует различные гидраты, однако из очень концентрированных растворов ZnCl2 можно получить в безводном состояние даже при 10".
В воде хлорид цинка растворяется с выделением большого количества тепла (15,6 ккал/моль), п вследствие гидролиза раствор его имеет кис лун реакцию. (В 0,005 н. растворе гидролизуется около 0,1%.) Из-за очена высокого сродства к воде хлорид цинка часто используют для отщепления воды от органических соединений, т. С- для проведения реакций конденсации. Помимо препаративной органической химии, хлорид питтг находит применение в медицине, в больших количествах его использует в ситцепечатании, в производстве органических красителей, часто поступающих в продажу в виде двойных солей с хлоридом цинка, а также цг?:~ пропитывания дерева. Оа служит также для травления металлов, пааут мер при паянии. Хлорид цинка имеет неприятный металлический вну? и действует как сильный раздражитель.
В водном растворе хлорид цинка хорошо диссоциирует; однако в копцентргу ванных растворах, как видно из данных измерения чисел вере носа (HHCcz он часто образует аутокомплекс, хотя и в значительно меньшей степени, чем хгг—-кадмия. Помимо воды, хлорид цинка легко растворяется также в абсолютном mfezt вом и этиловом спиртах, а также в эфире, ацетоне, глицерине, уксусном эфире п друтг: кислородсодержащих органических растворителях. Растворяется оп также я в £-?• содержащих соединениях, таких, как анилин и пиридин.
Хлорид цинка склонен к образованию двойных солей, особенно с ег ридами щелочных и щелочноземельных металлов. Большинство пз ~-соответствуют типу М| [Z11CI4] (тетрахлороцинкаты); некоторые ез соединений кристаллизуются с водой. При 0° из раствора хлорида ср—~ насыщенного газообразным хлористым водородом, выделяются ские кристаллы 2ZnCla • НС1 • 2Н2О. Получено также соетг— - ; ZnCl2 • НС1 • 2Н2О (длинные иглы).
Вторая группа периодической системы
471
Двойную (ромбическую) соль аммбнийцинкхлорид (NH4)3[ZnCl4] (уд. вес 1,88; т. пл. 150°) используют как паяльную воль. Она содержится также в жидкости для паяния, которую приготовляют, растворяя обрезки ринка в неочищенной соляной кислоте и прибавляя к раствору нашатырь. В растворе эта соль, как и все двойные хлориды цинка, полностью распадается на составные части. Применение раствора хлорида цинка в качестве «паяльной жидкости» основано на его способ-ности соединяться (только в растворе) с окислами металлов с образованием комп-К лексных соединений (ср, стр. 466).
Основные хлориды цинка. Простейший из известных кристаллических основных хлоридов цинка имеет состав Zn(OH)C) (цинкгидрок сил хлорид). Он кристаллизуется ^В в форме длинных гексагональных призм (уд. вес 4,57) и образует простую слоистую ^В решетку, подобную решетке основного хлорида кадмия (ср. рнс. 50, стр. 480).
^В Другая основная соль с вполне определенным составом ZnCU- 4Zn(OH)2 кристал-
^В лизуется в виде микроскопических гексагональных листочков (уд. вес 3,29) и имеет решетку с двойным слоем, состоящую из слоев гидроокиси, по Структуре подобных a-Zn(0H)2, и расположенных между ними слоев ZnCI2 (с упорядоченным распределе-пием атомов) (Feitknecht, 1930).
Частичным обменом ионов цинка па ионы других металлов удается получить смешанные основные хлориды. Можно осуществить также частичный обмен ионов хлора в слоях, образованных солью, на гидроксил-ионы п, таким образом, получить вещества с повышенным (нсстехиометрическим) содержанием гидроокиси в соли, Однако и рав-повесил с растворами (оно чрезвычайно медленно устанавливается) устойчива только соль с простым стехиометрическим составом.
Структуру, подобную ZnCl3-4Zn(OH)3, имеют также ZnBr2-4Zn(OH)2, Zn(NO3)2(OH2)2-4Zn(0H)3, ZnSO4(Orj2)4-3Zn(OH)2 и некоторые другие основные соли кадмия, кобальта и никеля. Правда, большинства оспенных солей кобальта и никеля образуют либо простую слоистую решетку, либо решетку с двойпым слоем, в которой атомы промежуточных сдоев расположены неупорядоченно. К этому типу (см, стр. 320) относится третий из основных хлоридов цппка, с еще более высоким содержанием гид-роокиси, чем у перечпелеппых выше. В отличие от них он не имеет постоянного сте-х неметрического состава. Его состав и структуру можно описать формулой Zn(OH.Cl),- 4Zn(OH)2. Основные слои, состоящие из Zn(OH)2, имеют такое же строение, как слои CdCl2 в решетке хлорида кадмия (ср. стр. 482). В соприкосновении с сильно разбавлен-ными растворами хлорида цинка (0,035—0,005 М] оп переходит в основной хлорид следующего типа, состав и структура которого еще не совсем установлены.
Другие галогеппды. Бромид и иодид цинка весьма близки хлориду, однако они ^В пе изоморфны ему. Бромид кристаллизуется без воды в ромбической решетке, а иодид
^В в кубической. Последний имеет желтый цвет. Он находит применение в медицине.
^В В концентрированных или насыщенных иодидом щелочного металла растворах иодида
^В цинка существует значительно больше комплексных галогенидов, чем у хлорида цинка.
^В Бромид занимает промежуточное положение. Фторид цинка, который, по-Виднмому,
^В весьма пригоден для консервирования дерева, отличается от остальных галогенидов
^В цинка, помимо своей кристаллической формы (моноклинной), также и неб о ль-
шой растворимостью (~ 5> 10-5 молъ/л). Подобно хлориду л все другие галогениды ^В цинка образуют двойные соли с галогенидами щелочных металлов. По своему составу ^В эти соли соответствуют общим формулам MIZnX3, MjZnX4 и M^ZnXj.
^В Нитрат цинка Zn(NO3)2 подобно Ni(NOs)3 и Mg(NO3)3, помимо без-
подпой соли, образует четыре гидрата (с 2, 4, 6 и 9 молекулами Н2О), из которых гексагидрат Zn(NO3)2-6H2O (уд. вес 2,07) обычно более устой-^В чив. Он кристаллизуется из водного раствора при температуре выше —17,6° и плавится мри +36,5°. Растворимость нитрата ципка составляет при 18° 115 г Zn(NO3)a в 100 г воды.
^В Изосновных нитратов цинка,помимо у?ке упомянутогоZn(N03)2(OH2)2-4Zn(OH)2
^В с постоянным составом, с двухслойной решеткой и упорядоченным распределением
^В атомов, существует еще более основной питрат, имеющий двухслойную решетку
^В и неупорядоченное распределение атомов в промежуточных слоях. Его удалось
^В получить пока только в выеокоднеперепой состоянии; состав его колеблется, по обычно
^В оп содержит около 9Zn(OH)3 на 1Zh(NO3)2 (Feitknecht, 1933—1938). Существует
^В и еще более основной нитрат цинка, но он еще мало исследован.
472
Глава 9
Нитрит и двойной пптрпт цинка. Нитрит цинка Zn(NO2)2 выпадает в оеадсз при добавлении нитрита натрия к растворам солей цинка. Он легко гидролизуется. С нитритами щелочных металлов образует двойные нитриты, например K3Zri(NO2)4- НяО — Желтые гигроскопичные призмы, очень легко разлагающиеся.
Сульфат цинка Z11SO4 в большом количестве получают в технике растворением цинковой стружки в разбавленной серной кислоте (7) и.тп осторожным обжигом цинковой обманки (8):
Zh-4-H2SO4:^ ZnSO^-t-Н2, (7\
ZnS+202==ZnS04*. (t
Для сульфатирующего обжига особенно пригодна цинковая обманка, содержащая железо. Из раствора, полученного после выщелачивания продуктов прокалп-ваяия водой, примеси более благородных металлов переводят в осадок при помог— подвешенной в растворе пластинки цинка. Железо и марганец осаждают хлорной известью в виде гидроокисей.
При упаривании водного раствора из него ниже 39’ выкристаллизовывается цинковым купорос ZnSO4-7H2O в виде бесцветных, блестящих как стекло, ромбических кристаллов, имеющих форму столбиков, изоморфных горькой соли. Растворимость при 0° 41,7, при 18° 52,7, при 100° 78,С г ZnSO4 в 100 г воды. При быстром нагревании цинковый купорос р а створяется в своей кристаллизационной воде. На воздухе оп выветривается теряя воду.
Цинковый купорос образует твердые растворы с горькой солью MgS04-7Hc' и с другими купоросами, например железным, марганцовым, кобальтовым и никелеаьЁ Нос ними он кристаллизуется пе в любых соотношениях, как с горькой солью. С ш~-ным купоросом он образует ряд твердых растворов: от 16 до 33 мол,% Си — б.чедг-? синие моноклинные кристаллы: от 84 до 100% Си — синие триклинные кристалла
Выше 39° из водного раствора кристаллизуется гексагидрат. И в этом слуш он также изоморфен соответствующему гидрату сульфата магния. Выше 70° криста, лизуется моногидрат. Гептагидрат может кристаллизоваться не только в ромбичест.— но и в моноклнниой сингонии. В последнем случае он метастабилеп и значительно бет растворим, чем ромбическая модификация (например, прп 18° в 100 г воды расти -ряется 58,7 г).
При осторожном нагревании цинковый купорос сначала отщепляет 6 моссу? кристаллизационной воды и только выше 240° — седьмую молекулу. При сппъе” прокаливании сульфат цинка распадается на окись цинка, трехокись серы, двус^г, серы и кислород. В присутствии восстановителей, например угля, распад пр от— -дит легче.
Растворение цинкового купороса в воде сопровождается пог лощением тттт« (—4,3 ккал/ммъ при 18е), растворение моногидрата идет с выделением ? _ (5,6 кг.ал/.моль!.
Из раствора в горячей концентрированной серией кислоте криста л л игу-продукт црисосдияепия ZnSO4-H2SO4 в виде шелковистых игл; из растворов, с"— --жащих сульфаты щелочных металлов, кристаллизуется двойная соль MjS04-Zc5 . 6Н2О. В природе сульфат цинка встречается редко; безводный называется ци.н.к<№ j, (изоморфен тяжелому шпату), водный природным цинковым купоросом (белый пут -рос, госларит).
Сульфат цинка имеет наиболее важное значение в технике. Его отменяют при крашении тканей и ситцепечатании, для приготогшесс г литопона, для пропитывания дерева, в гальваностегии для провавцзш.-;» оцинкованных бакои. В медицине в виде сильно разбавленного раетс - « он служит как вяжущее, ослабляющее секрецию и дезиифицирутш — средство мрм промываниях и перевязках, иногда также как рс~ -Сульфат цинка служит исходным веществом для получения больше-: — других соединений цинка, а также для электролитического Ейфуф. ,_.г.к очень чистого цинка.
* Процесс протекает неколичественно.
Вторая группа периодической системы
473
Из основных сульфатов Ц инка более подробно исследован пока только ZnSO4(OH3)4- 3Zn(OH)3 (стр. 471). Существует еще и более основной сульфат, который также имеет решетку с двойным сдоем.
Сульфит цинка ZnSO3 образуется в виде содержащего кристаллизационную воду осадна при внесении сульфита натрия в раствор сульфата цинка. Его можно цолу-нить также при действии SO, на водную суспензию ZnO или ZnCO3. Берглундом (Berglund) описаны двойные сульфаты, некоторые из них довольно сложного состава.
Тиосульфат цинка ZnS,O31 легко растворимый в воде в любых соотношениях, малоустойчив. Его водный раствор разлагается при упаривании. Однако, по данным Розенгейма, из раствора можно получить двойные соли вполне определенного состава: K2Zn(S,O3)2-Н,0 и (KH,;),Zn(S,O3)2-Н2О. О пн имеют вид длинных белых призм.
Цианид цинка Zn(CN)s выпадает в виде белого осадка при добавлении цианидов щелочных металлов к раствору соли цинка. В воде п в спирте он нерастворим, по легко растворяется в избытке осадителя. Цианид цинка можно получить в кристаллическом состоянии в виде ромбических призм. Он склонен к образованию коллоидных растворов. Цианид цинка йе имеет вкуса, но ядовит. Иногда его применяют в медицине.
Растворимость цианида цинка в избытке раствора циапидов щелочных металлов обусловлена образованием легкорастворимых комплексных солей (цианоцинкатов). Большинство из них соответствуют типу Mj[Zn(CN)4], а некоторые — типу, Мт1гп(СМ)з]. Растворы цианида ципка и медн([) в избытке цианида щелочного металла применяют в гальваностегии для получения латунпых покрытий.
Роданид цинка Zn(SC>’)2 получают взаимодействием роданистоводородной кислоты с карбонатом ципка. Оп легко растворяется в виде и в спирте.
Ацетат цинка Zii(C2H3O,)2> образующийся при растворении окиси цинка в уксусной кислоте, кристаллизуется из водного раствора в виде днгидрата, в форме блестящих моноклинных чешуек (уд. вес 4,73). Перекристаллизацией из Ледяной уксусной кислоты его получают безводным, в форме октаэдров (уд, вес 1,84; т. пл. 242°). При пониженном давлении он возгоняется без разложения *. Легко растворяется (около 1 : 3) в воде. В растворе частично гидролизуется. Степень гидролиза в 1 и. растворе составляет около 1% (Lofmann). Ацетат цинка применяют как защитное средство от огня. В медицине его используют для полоскания горла, при кожных заболеваниях, а также для внутреннего приема.
Карбонат цинка ZnC03 встречается в природе в виде минерала галмея (благородного) или цинкового шпата — гексагонально-ромбоэдрические кристаллы. Однако его кристаллы редко бывают крупными, обычно он представляет собой грубые массы, окрашенные примесями в желтоватый, коричневый, серый или зеленоватый цвет. Чистый карбонат цинка имеет чисто белый цвет. Его получают либо действием раствора гидрокарбоната щелочного металла, насыщенного двуокисью углерода, па раствор соли цинка, либо при действии СО3 на взмученную в воде свежеосажденную гидроокись цинка. Удельный вес его 4,44. ZnCO3 образует решетку кальцита: а = 5,62 А, а — 48°2'. В воде он практически нерастворим, но постепенно разлагается ею с образованием основного карбоната цинка, В сухом состоянии ZnCO3 уже при 150° начинает отщеплять СО2.
Основной карбонат цинка образуется в виде белого аморфного осадка при действии карбонатов щелочных металлов на растворы цинковых солей. Состав осадка изменяется в зависимости от условий. Большей частью он приближается к формуле 2ZnCO3-3Zn(OH)2. Основной кар
* При нагревании (250°, в вакууме) среднего ацетата цинка происходит частичное его разложение и образуется основной ацетат Zn4O(CH3COO)6 в виде прозрачных октаэдров, плавящихся при 249—250°. Подобно аналогичному соединению бериллия Ве40(СН3СОО)в основной ацетат цинка нерастворим в воде, но растворим в хлороформе и бензоле [Auger V., Robin I., Compt. rend., 178, 1546 (1924)]. — Прим, ped-
474
Глава 9
бопат цинка, отвечающий этому составу, встречается в природе как продукт выветривания галмея (цинковый цвет, гидроцинкит). Он имеет такое же строение, как и другие основные соли ципка.
Поданным Файткнехта (1952), различают три типа гидроксикарбонатов цинка; ZnCO3- Zn(OH)2> Н20 (неустойчивый), 2ZnCO3-3Zn(OH)a- Н2О (мельчайшие, различимые лишь в электронном микроскопе кристаллические пластинки) и ZnCO3- 3Zn(OH)s-яН2О (очень топкие иголочки или нити). Последние два имеют переменный состав. По своей структуре они являются производными брусита. Если представить себе, что в их гидроксильных слоях часть ионов ОН~ замещена ионами COj~, то слои основного карбоната приобретают избыточный отрицательный заряд. Для компенсации этого заряда между слоями располагаются в соответствующем количестве ионы Zn’-G (вместе с молекулами воды). Прн этом, если каждый четвертый ион ОН- замещен СО|-, то структуру можно представить формулой [Zn(OH2)]2+{Zn4(OH)0(CO3)2P~, которой соответствует аналитический состав 2ZnCO3- 3Zn(OH)2> Н20.
Оксалат цппка (дигидрат) ZnC2O4-2H2O выпадает в виде белого осадка при добавлении окса лат-ионов к растворам солей ципка. В воде он трудно растворим (6,4 .иг ZnC,O4 в 1 .г пр и! 8s), но легко растворяется в избытке раствора оксалата щелочного металла благодаря образованию комцлексяых ионов [Zn(C2O4)a]’ и [Zn(C3O4)3J'.
Силикат цинка. Ортосиликат ZnsSiO4 встречается в природе в виде виллемита и в изоморфной смеси с Mn2SiO4 (минерал троостит). Оба минерала кристаллизуются в гексагонально-ромбоэдрической форме. Силикат цинка можно получить и искусственно, Водный орт о силикат цинка Zn2SiO4-H2O встречается в больших количествах в виде кремнецинковой руды (кремневый или обычный галмей). В большинстве случаев он образует волокнистые или шаровидные агрегаты, состоящие иа ромбических кристаллов (уд. вес 3,3—3,5). Они либо бесцветны, либо окрашены благодаря примесям. Воду теряют лишь при температуре красного каления.
Метасиликат ципка ZnSiO3 был получен искусственно способом сплавления. При внесении силиката щелочного металла в раствор соли цинка образуется слизистый осадок переменного состава.
Алпплы цинка. При комнатной температуре цинк реагирует с метилиодидом с образованием метилципкиодида СИ3—Zn—I. При нагреваний ои диспропорциони-рует па диметилциик Zn(CH3)2 и Znl2:
Zn + СНз — I=CH3—Zn-l; 2CH3—Za—I=Zn(CH3)2+ZnI2.
Это соединение было открыто Франкландом в 1849 г. Оно представляет собой бесцветную, с неприятным запахом жидкость, сильно дымящую па воздухе и легко воспламеняющуюся; уд, вес 1,38; т. кип. 46°. Таким же способом получают диэтилцинк Zn(C2II<,)2 (уд. вес 1,18; т. кип. 118°) и другие алкилы цинка. Алкилы цинка чрезвычайно реакционноспособны, и поэтому их применяют в органических синтезах. Водой они разлагаются на гидроокись цинка и углеводороды, например
2п(СНз)3+2НОН—Zn(OH)2-|-2CH4.
Алкилы кадмия, например диметилкадмий Cd(CHg)2 (т. кип. 104°), по своему поведению весьма близки алкилам цинка. Об алкилах ртути см. стр. 509.
Гпдрпд ципка ZnH2, белое твердое вещество, легко получается при взаимодействии Zal3 (Wiberg, 1951) иди Zn(CH3)2 (Schlesinger, 1951) с 1лАШ4. При наг реванш до 90° он разлагается на Zn и Н2. С водой реагирует по уравнению
ZnH2^2lIOH=Zn(OH)2+2H2,
С бороводородом и диметилом цинка он образует растворимые в спирте двойннг соединения. С гидридом алюминия, напротив, пе реагирует. Соединение Zn(BHA^ при 85° разлагается на элементы:
Zn(BH4)2=Zn+2В+4Н2.
3
Вторая группа, периодической система
475
Ввиду того что диборан (ВН3)2 диссоциирует на элементы лишь при 500°, предполагают, что при разложении Zn(BH4)3 первоначально ^образуется мономер ВН3; по-видвмому, он менее устойчив, чем стабилизированный водородными мостиками S2- От борапата цинка путем замены ВН4-группы хлором производится соединение
ВН4), твердое белое вещество, растворимое в эфире. Оно весьма похоже на бора-пат ципка, но в отличие от него более устойчиво. Взаимодействием Znj2 с ЫН в эфирном растворе получают в зависимости от условий ZnII2 или ZnlH:
ZnI2 + 2LiH=2LiI + ZnH2,
ZnI2+LiH=LiI + ZnIH-
Подобно гидриду цинка получают гидрид кадмия CdH2 и гидрид ртути HgH2 (S elites in ger, 1951; Wiberg, 1951):
CdR2-pLiAin4=CdHa+2AiH3+2LiR (R=CH3 или I);
HgI24-2LiAlH4=HgHK-|-2AlH3+2LiI.
Однако в этом случае синтез следует проводить при значительно более низких температурах, особеппо для IIgH2, который уже при —125° разлагается па составные части. CdH2 и HgH2 подобно ZnH2 твердые белые вещества. Нов отличие от гидрида цинка они значительно более склоппы к разложению.
Аналитические сведения. Так как из аммиачного раствора цинк осаждается сернистым аммонием, но не выделяется при пропускании сероводорода в кислые растворы, аналитически он принадлежит к группе сернистого аммония. От кобальта и никеля он отличается растворимостью его сульфида в разбавленной соляной кислоте, от железа и маргапца — растворимостью гидроокиси в натриевой щелочи, а от алюминия и хрома — легкой растворимостью Zn(OH)2 в водном растворе аммиака. Далее характерным является белый цвет сульфида цинка.
Прокаленный на угле при помощи паяльной трубки, Цинк образует налет, желтый при нагревании и белый на холоду (окись цинка). Если этот налет смочнть раствором нитрата кобальта и вновь прокалить, получится продукт, окрашенный в характерный зеленый цвет (зелень Ринмана).
Зелень Ринмана, как установлено Хедпеллом (Hedvall, 1914—1932) и Натта (NaНа, 1929), в зависимости от условий приготовления имеет различный состав и структуру. Воли ее получать при очень сильном прокаливании (выше 1000°), то образуются твердые растворы окисей цинка и кобальта(11). (Если содержаниеСоО превышает 70% , то смесь имеет но зеленый, а розовый цвет.) При более слабом прокаливании (800— !Ю0°) в присутствии достаточного количества кислорода получают двойное соединение окиси ципка с окисью Со(Ш) типа шпинели ZnO- Со2О3. Образование этого соединения обусловлено реакцией, протекающей в твердом состоянии (ср, гл. 19),— в первоначально возникающей окиси кобальта(П, IV) Со3О4 ионы Co(IV) обмениваются па ионы цинка с одновременным изменением заряда кобальта:
CogI[CoIVO4]ZnO=С,о)11 [ZnO4] + Со1ХО.
Для микро определения удобно переводить цинк в роданид цинкртути(П) HgZn(SCN)4 или в тетраэдрические кристаллы двойного карбоната цинка и натрия 3Na2CO3-8ZnCO3-8Н2О (граница определения 0,01 -у Zn). Для капельного метода удобна цветная реакция с дитизоном (ср. стр. 438). Благодаря интенсивно красному окрашиванию образующейся при этом внутрикомплексной соли можно определить 0,05 у Zn в присутствии в 2000 раз большего количества алюминия.
В весовом анализе цинк осаждают и взвешивают в виде сульфида ZnS или карбоната с последующим прокаливанием и взвешиванием в виде окиси ZnO.
Можно также определять цинк, осаждая его в виде двойного фосфата с аммонием (NH4)ZnPO4 и взвешивая в виде дифосфата Zn2P2O7. Можно определять его и электролитически, при этом металл лучше всего выделять из раствора двойных цианидов
476
Глава 9
с щелочными металлами. Для объем ого определения цинка его осаждают в виде K2Zns|Fe(CN)e];> из разбавленного солянокислого раствора действием раствора циано-феррата^!) калия определенной концентрации. Конечную точку титрования устанавливают по гаятнаш с уранил ацетатом.
КАДМИЙ (CADMIUM) Cd
Распространение в природе. Кадмий часто встречается вместе с цинком-в цинковых рудах, особенно в галмее и цинковой обманке. Оба эти минерала почти всегда содержат то или ипое количество сульфида кадмия.
Чистые кадмиевые соединения в природе очень редки. Иногда встречается сульфид CdS (гринокит), еще реже окись кадмия CdO.
Основной карбонат кадмия найден в Огави (Юго-Западная Л фрика).
Исторические сведения. Кадмий был открыт в 1817 г. ШтромеЙером (Stromеуег) в Геттингене при исследовании продававшегося в аптеках Хильдесхаймера карбоната цинка. После прокаливания он оставлял коричневатую окись, хотя и не содержал железа. Почти одновременно Херман (Hermann) нашел кадмий в окиси цинка, раствор которой давал с сероводородом вместо белого желтый осадок, вследствие чего она была конфискована в Магдебурге из опасения, что в ней содержится мышьяк. Новый элемент был назван кадмием в связи с тем, что он часто встречается в окиси цинка (которая была известна еще древним грекам под названием исфщм).
Подучение. Для получения кадмия раньше почтя всегда пользовались цинковой пылью, образующейся при производстве цинка дистилляционным способом. В настоящее время его выделяют преимущественно электролитическим способом (наряду с электролитическим получением цинка! или повторным фракционированием содержащего кадмий цинка но способу, разработанному в Нью-Джерси (ср. стр. 459). Однако производстве кадмия из цинковой пыли и из других пылевидных отходов, обогащенные кадмием в процессе переработки руд, занимает еще значительное место.
1. Получение из цинковой пыли, В цинковой пыли, образующейся при получении цинка сухим путем, кадмий накапливается вследствие меньшей по сравнению с цинком способности к конденсации. Посгс добавления кокса для восстановления окиси цинковую пыль подвергают повторной фракционированной перегонке, причем кадмий, как болег легколстучий, каждый, раз накапливается в головных фракциях.
Очень небольшая конденсируемость паров кадмия в процессе перегойки част,-приводит к значительным потерям его. Этих потерь можно избежать при получек-кадмия мокрым путем: содержащую кадмии цинковую пыль растворяют в солят:тз или серной кислоте и из раствора выделяют кадмий либо цинком (Cd" -ф 7.п = Cd — + Zn"), либо электролитически. Иногда объедиияют сухой и мокрый способы tst что осажденный из раствора кадмий, обедненный цинком, практически освобождав" перегонкой от Zn приблизительно до содержания 0,1% Zn.
2. Получение электролитическим способом. Растворы сульфата цинка приготовляемые для его электролитического выделения, не богаты кадмием (ср, стр, 476), Однако его легко можно отделить от цинка благодау-значительно меньшему потенцпалу выделения (ср, табл. 4, т. I, стр. 51 При этом первоначально возникали трудности, связанные со склонность17 кадмия выделяться из кислых растворов в виде губки и дендритов. Однан: при определенных условиях работы их можно преодолеть (напртшер прибавлением в ванны таких коллоидов, как желатина или клеи). Б '"ттль. ном процесс подобен электролитическому получению шьнна.
Вторая группа периодической системы
477
Совершенно чистый кадмий (папример, для нормальных элементов) получают дальнейшей электролитической очисткой и возгонкой в токе водорода или в вакууме;
Свойства, Кадмий представляет собой белый блестящий металл. На воздухе он быстро тускнеет благодаря образованию тонкой окисной пленки. Кадмий довольно мягок, его можно резать ножом. Кроме того, оп очень тягуч: его можно прокатывать в листы и вытягивать в проволоку. Твердость чистого кадмия невысока, однако она сильно возрастает в результате сплавления его с цинком. Кадмий очень легко спаивается. Его температура плавления 320,9°, температура кипения 767°. В вакууме он возгоняется уже при 164°.
Для плотности пара при 1040° Девиль (Deville) нашел величину 3,94 (относительно плотности воздуха, равной 1). Молекулярный вес * вычисленный на основе этих данных, равепЗ,94 X 28,96 = 114,1 (атомный вес 112,4). Следовательно, пары кадмия состоят почти полностью из’атомов, подобно большинству других металлов.
По данным Коана (Cohen, 1914), кадмий существует в трех различных модификациях, различающихся коэффициентами расширения и величинами электролитического потенциала растворения (но лишь на тысячные доли вольта). Взаимный переход их происходит чрезвычайно медленно (за исключением случая, когда Cd соприкасается с раствором CdSO4). Вследствие этого обычпо имеют дело со смесью всех трех модификаций. Структурно, как видно из рентгенометрических исследований, они не различаются,
Удельная электропроводность кадмия при 18° равна 21,5%, удельная тепло-проводоость 22,6% по сравнению с серебром. Кадмий имеет гексагональную решетку подобно цийку и магнию (ср. стр. 452). С магнием он смешивается в любых соотношениях, а с ципком нет.
При распылении кадмия в пламени дуги под водой (по Бредигу) образуется его водный коллоидный раствор. Темно-коричневый гидрозоль в отсутствие воздуха весьма устойчив.
Будучи сильно нагрет на воздухе, кадмий сгорает красным пламенем с образованием коричневых паров окиси кадмия CdO **. Теплота сгорания составляет 65,2 ккал/г-атом Cd. При нагревании ои легко соединяется также с галогенами. При обычных условиях он не реагирует ни с азотом, ни с водородом.
В электрохимическом ряду напряжений кадмий стоит левее водорода, однако ближе к нему, чем цинк, поэтому вытесняет Zn из растворов.
Нормальный потенциал кадмия относительно нормального водородного электрода составляет, по данным Л а мера (La Мег, 1934) и Харнеда (Harned, 1936),—0,402 в (при 25°). Подобно цапку кадмий растворяется в разбавленных сильных кислотах (неокислителях) с выделением водорода. Совершенно чистый кадмий, так ясе как и очень чистый цинк, в нсокисляющих кислотах пе растворяется.
Применение. В настоящее время кадмий применяют главным образом в качестве покрытий для защиты металлов от коррозии. Применение кадмия в этой области, разработанной лишь несколько десятилетий назад, привело наряду с усовершенствованием производства путем введения электролитического способа выделения прежде всего к огромному
* Молекулярный вес газообразного вещества равен плотности его пара, отнесенной к плотности кислорода воздуха, равной 32,000. Из плотности пара, отнесенной к воздуху, принятому за 1, молекулярный вес подсчитывается умножением на 28,957 /' 1 293 X 32 000
( = ’ 4289’ ’ 1’293 и 1,4289 — соответственно вес литра воздуха и кислорода.
Число 28,957 называют также «средним молекулярным весом* воздуха^ .
** При этом образуются также следа перекиси CdO2.
478
Глава, 9
увеличению продукции этого металла. В качестве защитного покрытия кадмий применяют главным образом для автомобильных частей, самолетов, деталей машин, электро- и фотоаппаратов, фортепианной проволоки, винтов и прецизионных инструментов. Его нельзя, однако, применять для изготовления предметов, соприкасающихся с продуктам» питания, так как оп легко разрушается кислотами, а растворимые соединения его очень ядовиты.
Защитные покрытия из кадмия в большинстве случаев наносят электролитическим способом и преимущественно выделением из раствора цианистого калия. Иногда применяй}! разбрызгивание расплав ленного металла; ири этом следует учитывать сильную ядовш-'ч-осгпь паров кадмия. Для осуществления защиты металла достаточен уже очень топкий поверхностный слой кадмия. Так, кадмиевое покрытие железа уже при толщине пленки б р дает такой же эффект, как цинковое покрытие толщиной 12 р и никелевое 25 ц. Вследствие мягкости кадмия покрытия из него малоустойчивы к впешпп-'Е м,.хаттическим воздействиям. Однако это свойство дает преимущество, так'как кадмиевое нокрытие не шелушится при нагрузке рабочих частей на растяжение^ сжатие и кручение. Поэтому кадмий выгодно использовать для покрытий таких мотад2 аических изделии, которые подвергаются сильной коррозии, но но пекут существенна механической нагрузки.
Для оценки защитного действия покрытия против коррозии имеет значенкь. сохраняется ли защитное действие кадмия, осажденного на железе, если Поврежде" его поверхность. Относительно покрытий из олова известно, что коррозия железа тягт где покрытие повреждено, уже не только не задерживается, но, напротив, ускоряетсЕ. так кац образуется гальваническая пара, в которой железо служит анодом. Цинкам покрытие сохраняет свои защитные свойства, даже будучи поврежденным, так ст-в этом случае железо является катодом. Учитывая места, занимаемые железом и кед мнем в электрохимическом ряду напряжений, можно ожидать, что кадмий по отнесению к железу должен вести себя подобно олову. Однако опыт многих наблюдеЕгз показал, что в гальваническом элементе, состоящем из пластинок железа и няпмг-г погруженных соответственно в растворы сульфатов Железа и кадмия, железо, ~~ и в паре с цинком, становится положительным электродом. В соответствии с эт~д оказалось, что кадмий, нанесенный на железо, сохраняет защитные свойства ст—-в том случае, если он и результате нагрева полностью продиффупдирует внутрь Же.тс~ так что первоначальное покрытие совершенно исчезает. Каким образом подс£тг_ наблюдения привести в соответствие с потенциалами растворения, пока еще не яег Возможно, это несоответствие связано с легкой пассивируемостью железа; в т&-~ъ случае, поскольку железо в активном состоянии лить немного активнее кадмия, доге:, точно уже совсем небольшой пассивации, чтобы настолько сдвинуть его в ряду няст--. жениЙ, что оно становится инертнее, чем кадмий.
Помимо испольвования в качестве защитных покрытий, метаащст ский Cd применяют главным образом для изготовления легкоплаегт -сплавов, таких, как сплав Вуда и сплав Липовица (ср. т. I, гл. о виежт легкоплавкий припой (третпик, состоящий из 50% Sn, 25% Ph п 15 . Cd; т. пл. 149°), амальгам для зубных пломб и подшипниковых ме--лов. При изготовлении часов используют содержащие кадмий ж£~у платиновые сплавы, отличающиеся чрезвычайно низкими коэффициент: - j расширения. Далее, кадмий применяют для изготовления nopim ных элементов Вестона, аккумуляторов Юнгпера.
Аккумулятор Юнгнерз (щелочной пике.тево-кадмиевый аккумулятор) ляет собой усопершенствоваиную форму аккумулятора Эдисона (ср. стр. 335у железпого оп содержит электрод из металлического кадмия в смеси с окисьш :..iiii (II, III). Прп разрядке С<1 окисляется до CdO, при зарядке снова восстанавлнз:;: В то время как на железном электроде в аккумуляторе Эдисона в процессе хпг:;н. с самого начала происходит выделение водорода, па кадмиевом электроде си пме;—~ --''-.ц лишь н концу. А так как выделение водорода сопровождается значятедьнзд; — качением напряжения на клеммах, то это значительно повышает коэфф птеезт —' т г :u""hi действия элемента.
Вторая группа периодической система 479
Из соединений кадмия главным образом применяют сульфид в качестве малярной краски, сульфат для элементов Вестона, бромид и иодид в фотографии. Соединения кадмия применяют также в медицине.
СОЕДИНЕНИЯ КАДМИЯ
Соединения кадмия вполне аналогичны соединениям ципка. Их общая формула CdX2. Следовательно, кадмий, так же как и цинк, всегда деух-валентен *. В отличие от бесцветных окиси и сульфида цинка окись кадмия имеет коричневый, а сульфид — желтый цвет. Сульфид кадмия в отличие от сульфида цинка нерастворим в разбавленных сильных кислотах.
Соли кадмия, за немногими исключениями, бесцветны (поскольку опи производятся от неокрашенных анионов). Соли сильных кислот легко растворяются в воде. Растворы содержат бесцветные ионы Cd".
Соли кадмия имеют неприятный металлический вкус и, как уже отмечалось, сильно ядовиты. В разбавленных растворах они оказывают вяжущее действие, а при большей концентрации — разъедающее. В качестве рвотного средства они значительно более действенны, чем соли цинка.
У солей кадмия довольно сильпо проявляется склонность к комплексообразованию. Особенно прочной является связь между попами кадмия и галогенид-ионами. Хлорид, бромид и особенно иодид кадмия в противоположность другим солям металлов в водном растворе диссоциируют неполностью. В таких растворах к тому же образуются аутокомплексы, например
3Cdl2=Cd" + 2[CdI3]'.
Если в раствор внести избыток иодид-иопов (например, в виде иодида щелочного металла), то образуются комплексные ионы с шестью атомами галогена. При упаривании кристаллизуются комплексные соли, соответствующие общим формулам M4CdX3], MJ[CdXJ или M|[CdXe], а иногда также MJ[CdX5], Этим формулам соответствуют также и другие комплексные или двойные соли. Состав их зависит от количественного соотношения компонентов в растворе и от других условий опыта.
Образование упомянутых аутокомплексов можно доказать измерением электропроводности. Вследствие этого и в связи с относительно слабой диссоциацией некоторых солеи кадмия электропроводность их растворов мала по сравнению с растворами солей других металл ов.
Ионы Cd" способны связывать также и нейтральные частицы, например NH3 и его органические производные. Образованием такого рода комплексов и объясняется растворимость в водном растворе аммиака большинства тру дно растворимых в воде соединений кадмия. Одпако особенно трудно растворимый в воде сульфид не растворяется и в аммиаке. По-видимому, в аммиачных водных растворах преимущественно образуются ионы [Cd(NH3)4]". Прн пропускании газообразного аммиака сухие соли поглощают значительно больше четырех молекул аммиака, например сульфат до fi, хлорид до 10, а бромид до 12 NH3.
Кадмий образует также большое число основных солей. Большинство из них, как показывают рентген ост рукт у рпые исследования, имеют простую слоистую решетку. В отлично от циика основные соли, образующие двухслойные решетки, мало извест
* Существование соединений кадмня(1) хотя часто и утверждается, одпако с несомненностью не доказано.
480
Глава 9
ны для кадмия. По аналогии с цинком кадмий образует хорошо кристаллизующиеся смешанные основные сол», иапример Cd(NO3)2‘CYi(OH’)2' 4Н2О (зеленые кристаллы) и С<1Вг2-ЗСи(ОН)г (зеленые гексагональные таблички). Вероятно, в твердом состоянии подобно простым основным солям кадмия они представляют собой соединения со слоистыми решетками.
В качестве примере основных солей кадмия можно привести основные хлориды. Простейший из них имеет состав Cd(OH)Cl. Он образует гексагональную слоистую решетку с упорядоченным распределением атомов, изображенную на рис. 50 (Hoard, 1934). Такую^же простую слоистую решетку с упорядоченным распределением атомов образует, по данным Файткнехта (Feitknecht, 1937), основной хлорид, соответствующий формуле CdClj-ZiCdtOIIJg. Еще два основных хлорида, по составу промежуточные
Рис. 50. Структура решетки гидр оксихлорида кадмия. Гексагональная слоистая решетка; а=3,66, с=10,27А. Расстояние между слоями: Д=1,оз, е=2,70 а. Средние иежионкые расстояния: С4 > ОН=/=2,34 A, Cd t—-<-С1=Д= 2,69 А.
между указанными выше, образуют решетку с неупорядоченным распредел?—г;* атомов. Один из них по своей структуре является производным безводного хлитл_.:.1 кадмия (см. стр. 482) путем замены ь/8 ионов хлора гидроксильными. Структура D7 ‘ по существу такая же, как у гидроокиси няб.ч.ия. Он может содержать 2—2,6СС ’ ...
па 1 молекулу CdCl2. Однако его нельзя перевести в гидроокись кадмия непре—i..7.m вытеснением ионов хлора. Гидроокись кадмия способна, правда, присоединять С ... с образованием^ ер дых растворов, но лишь до 0,13 мол.% . Какие из названных г*;.
нений образуются, зависит от концентрации соли и pH растворов, из котсрыл ши кристаллизуются, а также от срока хранения осадка. Часто вначале bhzezes продукты осаждения, неустойчивые в условиях их образования и лишь со пргх?•:.чи превращающиеся в стабильные вещества.
Окись кадмия CdO образуется при горении кадмия в виде нс~~э«~ вого порошка, который при нагревании в токе кислорода можнг. г-пне-вести в темно-красные кубические кристаллы (уд. вес 8,15). Онг "isaiiii найдена и в природе в виде блестящего черного налета, состоящего е: г”»-эдрических кристалликов. При нагревании окись кадмия оное: ~ Ж начинает возгоняться не плавясь. При более сильном нагреваЕХХ :А
Вторая- группа периодической системы
481
отщепляет кислород. Поэтому при нагревании окись кадмия легко восстанавливается, в токе водорода уже при 270—300°, углеродом или окисью углерода при температуре 700°.
Окись кадмия кристаллизуется в решетке типа каменной соли; а = 4,70 А; Cd «—> 0=2,35 А. Следовательно, по своей структуре она подобна окиелам щелочноземельных металлов, а не окиси ципка.
Окраска окиси кадмия сильно зависит от температуры, до которой она была нагрета. Например, прокаливанием гидроокиси Cd(0H)3 при 350° получают зеленожелтую, а при 800’ — сине-черную окись. Последняя образуется также при длительном кипячении Cd(OH)2 с сильно концентрированным раствором едкого кали (Scholder, 1941).
Ранее предполагали, что при сильном прокаливании, а также в результате восстановления Cd О при высокой температуре образуется «суб окись» Cd, О, Рентгенометрическими исследованиями зто не подтвердилось. Не происходит при нагревании и изменения модификации, По данным Фрики о (Fricke, 1940), сине-черная и золен о-желта я формы окиси различаются только величиной частиц. В соответствии с другими данными, в результате сильного прокаливания происходит отщеплепие кислорода, однако, несмотря на беспорядочное распределение образующихся в О-подрешетке пустот, структура решетки пе изменяется.
Нагреванием в токе хлора окпсь кадмии можно перевести в хлорид CdCl2.
Гидроокись кадмия Cd(OH)2 в виде белого осадка выпадает при действии щелочи на растворы солей кадмия. Она практически не растворяется в избытке осадителя, но растворима в водном растворе аммиака.
Структура решетки CJ(OH)., такая же. как у Mg(OH)2 (типа брусита, ср. т. I, стр. 293, рис. 61); а = 3,47, с — 4 .64 А. Подобную же структуру имеет иодид кадмия.
Теплота образования кристаллической гидроокиси кадмия из CdO и И2О равна 5 ккал!моль (Fricke). Удельным вес ее 4,81.
Растворимость гидроокиси кадмия в воде при 25е равна 1,15-10"6 молъ}л. При добавлении NaOH она вначале понижается, а затем по мере увеличения концентрации щелочи возрастает до 9,0-10"® молъ!л в 5 н. растворе NaOH. На основании этого Пиатер (Piater, 1928) сделал заключение об очень слабом амфотерном характере гидроокиси кадмии. Позднее Шояьдеру (1941) удалось выделить еидрвхеакадмиаты, т, е. соли, производные от Cd(OH)a в качестве ангидрокислоты, например Na2[Cd(OH)4], Sr2[Cd(OH)0l, Ba2[Cd(OH)fiI. При нагревании Cd (О Н)2 или CdO с концентрированным едким патром Шольдер получил натриевую соль. Из концентрированных растворов КОН кристаллизуется не гидроксосоль, a Cd(OH)2.
Амид кадмия Cd(NH2)2 получают ла а имо действием Cd(SCN)2 с KNH2 в жидком аммиаке (В chart, 1915), Он представляет собой желтовато-белый порошок (уд. вес 3,05), на воздухе быстро буреющий. Теплота образования из элементов составляет 13 ккал/молъ (Juza, 1937).
Сульфид кадмия CdS выделяется в виде желтого осадка при пропускании сероводорода в растворы солей кадмия. В виде минерала (гринокита) он встречается, правда редко, в виде землистого налета на цинковых рудах, При прокаливании окиси кадмия с серой он получается в виде красивых кристаллов. Удельный вес кристаллического сульфида кадмия, а также природного составляет 4,8. Осажденный из водного раствора CdS немного легче.
Прокаленный сульфид кадмия, так же как и гринокит, имеет структуру вюртцита с а — 4,14, с = 6,72 и расстоянием Cd S==2,52 A. CdS, осажденный из водного раствора, имеет структуру цинковой обманки с а = 5,82, Cd «—> S = 2г52 А.
Сульфид кадмия нерастворим в разбавленной 'соляной кислоте, но растворяется в концентрированных кислотах, в теплой разбавленной азотной кислоте, а также в кипящей разбавленной в отношении 1 : 5 серной кислоте. Окраска сульфида кадмия зависит от условий осаждения 31 Г. Реми
482
Глава 9
и может изменяться от лимонно-желтой до красно-желтой. Его применяют в качестве малярной краски (кадмиевая желтая), отличающейся яркостью л иричностью. В технике его получают либо осаждением из сернокислого раствора, в большинстве случаев при помощи сульфида натрия, либо прокаливанием смеси карбоната кадмия с серным цветом.
Свежеосажденный сульфид кадмия очень мало растворим в растворе сернистого аммония и совсем не растворяется в растворах сульфидов щелочных металлов. Он легко образует коллоидные растворы (красивого желтого цвета) например пентиац-руется сероводородом в отсутствие сильных кислот.
Хлорид кадмия безводный CdCl2 получается нагреванием кадмия или окиси кадмия в токе хлора или обезвоживанием гидрата. Он представляет собой бесцветную прозрачную массу, которую возгонкой можпе перевести в ромбические листочки. Удельный вес 4,05; т. пл. около 565';'; т. кип. 964е.
Хлорид кадмия образует решетку, подобную бруситу. В брусите (а также иоди-= кадмия, ср. ниже) слои, состоящие из отрицательных ионов, расположены один нац другим, как при гексагональной плотнейшей упаковке, а у хлорида кадмия, как npz кубической плотнейшей упаковке (ср. рис. 10, стр. 43).
Б воде хлорид кадмия легко растворим (110,6 г в 100 а воды при 18'^ Теплота растворения:составляет 3,2 ккал.'мольСИСЕ- Из водного раствора,, который обычно получают растворением металлического кадмия в соленой кислоте, кристаллизуются различные в: зависимости от условий опыта гидраты (1—4 Н2О). При нагревании до 120° они легко превращаются в безводную соль.
Хлорид кадмия немпого растворим также в некоторых органических растворит? лях, например в этиловом спирте (1,5 г в 100 г), в метиловом спирте (1,7 г в 100 с а также в этилацетате и ацетоне. В указанных спиртах он заметно диссоциирует. Пр: растворении в уретапе t>n обнаруживает нормальный молекулярный вес.
Об основном хлориде см. стр. 480.
Бромид кадмия CdBr2, образующийся либо прокаливанием кадмия в парах бр— при температуре красного каления, либо обработкой металла кипящей бромной водр во всех отношениях очень близок хлориду. В безводном состоянии он образует бгг._; с перламутровым блеском листочки (уд. вес. 5,2; т. пи. около 570°, т. кип. 863°). В ц— ном растворе он немного более ассоциирован, чем хлорид.
Иодид кадмия Cell-, в отличие от хлорида и бромида известен лишь в безво~л состоянии. Его получают взаимодействием кадмия с иодом в присутствии воды, а тЙег растворением окиси или карбоната кадмия в разбавленной иодистоводор одной кнец—. Кристаллизуется он в виде бесцветных блестящих тригональных табличек (ух х 5,7; т. пл. около 385я, т. кип. 708—719°).
Иодид кадмия, так же как и гидроокись кадмия, образует решетку типа брутх” («=4,24, с = 6,84, Cd и—» 1 = 2,99 А). Теплота растворения отрицатки:~ (—0,96 ккал/моль при 18°) вследствие малой тенденции этого соединения к гпдратгдг: Растворимость иодпда кадмия при 18° равна 85,2 а в 100 г воды и лишь незначптес; _ возрастает с температурой.
Помимо решетки типа брусита (С 6-тип), иодид кадмия способен кристалл ваться ио другому типу (С2/-тип). Последний занимает промежуточное пололд— между решетками типа С6 и С19 (тип хлорида никеля), в которой крпстадпвлут— СпС'12. В решетке типа С27 ионы металла расположены, как и в решетке С6 в Ю: ионы галогенов в каждой сетчатой плоскости связаны с ионами металлов поперт—-как в решетках типа С6 и С19. Решетку типа С27 можно получить, исходя из . Cd(OH)Cl (рис. 50), замощая ионы ОН- и С1~ ионами I".
Фторнд кадмия Cd F3 (уд. вес —6,6; т. пл. 1110°) является трудно л етучшь, В сг г чие от других галогенидов кадмия он малорастворим в воде (0,29 люлъ/л С
Вторая группа периодической системы
483
Образование аутокомплексов галогенидами кадмии. Измерения электропроводности, осмотического давления, потенциала и чисел переноса позволяют сделать заключение, что иодид кадмия л водных растворах находится в значительной степени н виде аутокомплексныт. солей, например Cd[CdI3]2. Так, в растворах, содержащих О', 1 и более моля Cdl3, числа переноса анионов > 1. Это значит^ что кадмий преимущественно перемещается к аноду. А к аноду он может двигаться только в виде комплексных анионов* например ICdI3]'. Благодаря их образованию по уравнению Cd" + ЗГ = [Cdl3]' общее число свободных ионов в растворе, а следовательно, а их проводимость значительно уменьшаются. Действительно, электропроводность растворов иодида кадмия при умеренном разбавлении оказывается Значительно ниже, чем следовало ожидать для растворов электролита, содержащих двухвалентные катионы и одновалентные анионы. В более слабой мере эта особенность наблюдается также у растворов бромида и хлорида кадмия. В табл, 51 приведены величины эквивалентной электропроводности
Таблица 51
Эквивалентная электропроводность Ле и отношение электропроводностей Лс/Ло растворов галогенидов кадмпя в сраппсшш с теми же величинами для растворов нормального электролита (при 18°)
(Лс — эквивалентная электропроводность при нормальной концентрации с)
CdCis свВщ С (Hz MgCla Cd(NO3)2 ZilGls
Л1= 22 4 18,3 15,4 61,5 54,3 55
Л0.1— 50,0 44,0 31,0 83,4 80,8 82
Ла,01 = 83 76,3 05,(5 98,1 96 98
л0= 115 116 120 110,5 112 112
Л1/Л0“ 0,19 0,16 0,13 0,55 0,48 0,49
Ло,1/Ло= 0,43 0,88 0,26 0,75 0,72 0,73
Ле,о1/Ло= 0,72 0,66 0,55 0,89 0,86 0,87
Лс и отношения электропроводностей Лс/Л0 различных солей кадмия в сопоставлении с соответствующими величинами для типичного двух-одповалетного электролита (MgC]2). Из сравнения величин Лс/Лр ясно видно аномальное поведение галогенидов кадмия, в то время как отношения электропроводностей у нитрата кадмпя, а также хлорида цинка почти не отличаются от такого отношения для MgCk.
Криоскопические и эбуллиоскопические измерения подтвердили, что в растворах галогенидов кадмия концентрации ионов относительно низки, и это выражается в низких величинах отношений Лс/А(;. По данным МанБстта (McBain), можно предположить, что В растворах, особенно иодида кадмия, кроме комплексных иопов, в значительном количестве присутствуют нед иссоци иро и а иные молекулы в соответствии с равновесием:
Cd"4-3lr .. СЛз-дИ . ? [Cdl3|\
В связи с этим также интересны наблюдения Брунса (Bruns, 1925), а именно: электропроводность растворов иодида кадмия при внесении элементарного иода повышается, в то время как в растворах других иодидов, например иодида калия, уменьшается. 12 образует с I' менее подвижные ионы Ц. В случае иодида кадмия реакция с иодом
[Cdi3|'+3i2=Cd--+3i;
приводит к значительному увеличению числа ионов ц, следовательно, к значительному повышению электропроводности.
Двойные галогепнды кадмия. Образование .комплексных анионов, происходящее в растворах галогенидов кадмии, естественно, значительно усиливается от прибавления других галогенидов. Из смешанных раство
31*
484
Глава 9
ров галогенидов кадмия с другими галогенидами кристаллизуются соответственно этому двойные или комплексные соли, преимущественно типа МЦСбХд]. Известны также представители типов M|[CdX4] и M*[CdX6I.
Образование комплексных ионов при смешивании растворов галогенидов кадмия с растворами других галогенидов отчетливо проявляется в том, что проводимость таких смешанных растворов поаддитивпа проводимости составляющих. Например, при 18е эквивалентная электропроводность KI в 1 п. растворе равна 103 6, Cdl3 в 1 н. растворе 15,4. Однако в 1 п. растворе одновременно как по КГ, так и по Cdl2, она равна не сумме 103,6 + 15,4 = 110,0, а 82.
Цианид кадмия и двойной цианид. Цианид кадмия Cd(CN)2 выпадает из растворов голой кадмия при действии цианидов щелочных металлов в виде белого осадка, растворимого в сильных кислотах. Оп растворяется также в избытке осадителя вследствие образования комплексных ионов:
C(l- + 2CN'=Cd(CN)2; Cd(CN)2 + 2GN' = [Cd(CN)bl".
Из растворов были получены кристаллические двойные цианиды, общий состав которых большей частью соответствовал формулам МЦС4(СХ)Ц и MHCd(CN)6].
Измерения чисел переноса и других параметров показали, что двойные цианиды кадмия являются еще более прочными комплексами, чег двойные иодидгл. Однако сероводородом кадмий полностью осаждаете^ из них в виде сульфида.
Растворами двойных цианидов удобно пользоваться для электролитического выделения кадмия (ср. стр. 476).
Роданид кадмия и двойные роданиды. Роданид кадмия Cd(SCN)3 получают рг~ творением карбоната кадмия в роданистойодородной кислоте или путем: дпойного обиед между сульфатом кадмия и роданидом бария. Он выделяется в виде бесцветных коро~ при упаривании раствора. В присутствии других роданидов образуются двойку-роданиды. Большинство из них соответствуют типу Ml [Cd(SCN)4(OH2)2|. Все czz хорошо кристаллизуются, K3[Cd(SCN)4(OH2)2l, например, образует правильна октаэдры. Из водных растворов могут также хорошо кристаллизоваться родашдж содержащие ионы аммиаката кадмия, например [Cd(NH3)a](SCN)3 (MeitzendCE В присутствии пиридина (Ру) из растворов солей кадмия при добавлении рода г— > щелочного металла выпадает соединение (CdPy3](SCN)3 (Spacu).
Нитрат кадмия Cd(NO2)2 чистый проще всего получать растворено^ карбоната кадмия в разбавленной азотной кислоте. Из водного раствпр. при обычной температуре он кристаллизуется с 4Н3О (существуют таЩд гидраты с 2 и 9Н20). Тетрагидрат образует расплывающиеся лучисты иглы с удельным весом 2,45, которые при 59,3° плавятся в собствен^;:.: кристаллизационной воде. Растворимость Cd(NO3)2 127 г в 100 г при 18°.
Водные растворы нитрата кадмия обнаруживают приблизите^;-нормалиную электропроводность (ср. табл. 51, стр. 483). Слсдоватеддд они не содержат в сколько-нибудь значительном количестве компленедд,, ионов. В кристаллическом состоянии для нитрата кадмия также не езе,-""' ны двойные соединения с нитратами щелочных и щелочи оземельд_, т металлов.
Известно несколько основных нитратов кадмия. Те из них, которые имеют ссгтд.в Cd(NO3)2- 4Cd (ОИ)2, подобно основному нитрату цинка такого же состава обрастт'-решетку с двойным слоем с упорядоченным распределением атомов.
Нитрит кадмия и двойные нитриты. Нитрит кадмия Gd(N02), в оедд-чие от нитрата кадмия в виде простой соли весьма неустойчив (легко гдг-ролизуется) и, напротив, способен образовывать довольно устойчив,-
Вторая группа периодической системы 485
и хорошо кристаллизующиеся двойные соли. Так, K[Cd(NO3)3J — бледно-желтые или бесцветные кубические кристаллы; К3 [Cd(lNО3)J — красивые бледно-желтые кристаллы.
Сульфат кадмия CdS04 получают растворением кадмия или его карбоната в разбавленной серной кислоте. Он служит исходным веществом лля получения других соединений кадмия, например кадмиевой желтой краски, а также его применяют в медицине и для заполнения нормальных элементов Вестона.
Из водного раствора сульфат кадмия кристаллизуется в виде гидрата CdS04--8/3Н2О — бесцветных моноклинных призм (уд. вес 3,09). При определенных условиях можно получить также гидраты CdSO4-7H3O и CdSO4- Н%0. Последами существует в двух модификациях с температурой превращения 74,5°. При добавлении избытка серной кислоты выделяется безводный сульфат кадмия, ромбические призмы с уд. весом 4,7. Растворимость 76,2 г в 100 г воды при 18е. При повышении температуры растворимость вначале немного возрастает; выше 41,5° (область существования моногидрата) внезапно падает (при 100° в 100 и воды растворяется 60,8 г CdSO,). Теплота растворения составляет дляСй8О4 10,69,дляС<1804- Н2О 6,05идля CdSO4-8,'3H3O 2,54 ккал! моль.
С сульфатами железа(П) и меди сульфат кадмия образует изодиморфный ряд твердых растворов. При преобладающем содержании кадмия содержание воды и форму кристаллов определяет обычный сульфат кадмия CdSO4- 3/3Н3О, при большем содержании железа — железщгй купорос FeSO4‘ 7Н3О, а при большем содержании меди — медный купорос CuSO4-5H2O. В обоих рядах имеются большие разрывы непрерывности от 0,26 до 51,08 мол.% железа и от 0,55 до 98,29 мол. % медп. С сульфатом кобаиь-та(П), помимо твердых растворов, содержащих 7, и 1И3О образуется также твердый раствор с 4Н2О (Bassett, 1934). (J MtiSO4‘ 4Н2б такя;е известны твердые растворы. Напротив, с сульфатом магния образуется двойная соль [Mg(OH2)e]SO4- [Cd(0H2)e]SO4.
С сульфатами щелочных металлов сульфат кадмия также образует двойные соли, большинство из которых имеют общую формулу M|Cd(SO4)2- 6Н9О. Не известны соли и значительно более сложного состава. Так, Бенрас (Benrath. 1932) нашел, что с K2SO4 образуются следующие двойные соли: K2Cd(SO4)2 (с Р/2 и 4Н2О), K3Cd3(SO4)4 (с 2 и ЗН2О) и K4Cd3(SO4)5-Н2О. В водных растворах в значительной степени, хотя и неполностью, эти соли разлагаются па составляющие их компоненты. Ройер (Донуег) установил. что повышение температуры кипения смешанных растворов сульфата аммония п сульфата кадмия не является аддитивной величиной, рассчитанной на основании температур кипения каждого раствора. Отклонение достигает наибольшей величины при молярном соотношении компонентов 1 : 1. Точпа также существование сулъфато-нонов [М.] I(SO4)3]" наблюдается и в смешанных растворах сульфата аммония с сульфатами Fe11, Со*1, Zn, Mg и Сип. Их устойчивость в ряду названных металлов падает от кадмия к меди. >г
В растворах простого сульфата кадмия оказывается также существуют сульфато-попы, хотя и в весьма ограниченном количестве. Об этом свидетельствует величина отношения электропроводностей, которая в растворах сульфата кадмия, как, впрочем, и в растворах сульфатов цинка и меди, значительно меньше, чем в растворах сульфата магния (ср. табл. 52).
Из водных растворов, содержащих аммиак, сульфат кадмия кристаллизуется а виде аммиаката [Cd(OH3)2(NH3)4]SO4. При пропускании газообразного аммиака над безводным сульфатом кадмия удалось получить гексааммиц {Cd(NH3)e]SO4.
При нагревании выше 700° сульфат кадмия начинает возгоняться. Одновременно он частично разлагается. При 1000° общая упругость пара составляет 330, а упругость SO3 равна 14 мм рт. ст.
Основные сульфаты, кадмия еще мало изучены.
Карбонат кадмия CdCO3 выпадает в виде белого осадка, содержащего обычно гидроокись, из растворов солей кадмия ири добавлении ионов СО3. В кристаллическом виде его можно получить нагреванием осадка е раствором карбоната аммония приблизительно до 170" с последующим медленным охлаждением (удельный вес 4,96). Прп нагревании он разлагается на CdO и СОг- Упругость пара СО2 при 357° достигает 1 атм.
486
Глава S
1'аблица 52
Эквивалентная электропроводность Дс и отношение электропроводностей Ac/Afl в растворах днуд-двухвалеитпых электролитов при 18° (Ле—эквивалентная электропроводность при нормальной концентраций <)
СДЙОд znSo4 CuSO4 MgSO4
;Ч-= 23,58 26,6 25,8 28,9
Ao, 1 — 42,21 46,2 45,0 50,1
Ai) ,о1 = 70,34 73,4 72,2 76,7
Ло= 115 114 114 113
Aj/Ao= 0,20 0,23 0,23 0,26
Ао,г/Ао= 0,37 0,41 0,39 0,44
Ao.oi/Ao= 0,61 0,64 6,63 0,68
Оксалат кадмпя CdCaO4 выпадает в виде белого осадка при действии щавелевой кислоты или оксалата аммония на растворы солей кадмия. На холоду он кристаллп-вуется с ЗН2О в форме листочков, при пагревании образуются безводные призматические кристаллы. В воде GdCa04 трудпо растворим (1 : 13 000). Значительно легче оп растворяется в концентрированных растворах оксалатов щелочных металлов. Из таких растворов выделяются двойные оксалаты, например К2[С(1(С.>О4У 2НаО. Оксалат кадмия растворим также в концентрированных растворах хлоридов щелочных металлов с образованием двойных солей, например К4[Сб2С12(С2О4)3]-вН2О.
Аналитические сведения. В ходе анализа кадмий сопровождает медь. При действии сероводорода подобно меди он выделяется из кислого раствора в виде сульфида, нерастворимого в сернистом аммонии, но легко растворимого в теплой умеренно разбавленной азотной кислоте. Так же как медь, кадмий образует комплексы с аммиаком и вследствие этого не осаждается им. Однако в отличие от меди цианидный комплекс его менее устойчив. Если в раствор, содержащий комплексные цианиды медп и кадмия, пропускать сероводород, то выпадает желтый сульфид кадмиг CdS, в то время как медь остается в растворе.
Прокалённый паяльпой трубкой на угле кадмий образует бурый налет оклей Особенно характерной для кадмия является желтая окраска его сульфида в сочетащ™ с нерастворимостью последнего в сернистом аммонии.
Если через раствор комплексного цианида пропускать электрический ток (прл комнатной температуре), то прп определенной концентрации цианида щелочнет; металла и не слишком высоком напряжении из раствора выделяется мота лян чееггг" кадмий. Прн пропускании электрического тока через подкисленный раствор проела ; cdjiii, напротив, выпадает только медь, в то время как кадмий, стоящий в электрохимическом ряду напряжений левее водорода, остается в растворе.
Для макроопределения кадмий удобно переводить в очень хорошо кристалл—-зующийся двойной роданид кадмия и ртути(П) Hg€d(SCA)4. Еще чуъствнтельщ-реакция открытия его в виде двойного хлорида с рубидием ЙЬ^СЙСЦ. В последах случае граница чувствительности составляет 0,01 у Cd.
Количественное определение кадмия осуществляют обычно осаждением его сульфида е последующим взвешиванием в виде сульфата CdSQ^ Можно также электролитически выделять его в виде металла п закд_ взвешивать. Электролитическое определение л у tune всего проводит; из, раствора, содержащего цианид калия.
Srnopax гр/рцпи периодической системы
487
РТУТЬ (MERCURIUS или HYDRARGYRUM) Hg
Распространение в природе. Ртуть встречается в природе главным образом в виде ее сульфида HgS — киновари. В очень небольших количе-* ствах ей сопутствует также самородная -ртуть, вкрапленная в породу в виде капелек. Основными месторождениями киновари являются Альма-ден в Испании, Пдриа в Югославии, Монте Амиата в Тоскане, кроме того, несколько мест в Калифорнии, Мексике, Техасе, Орегоне, Неваде и Никитовна в Донецком бассейне и СССР.
Минералы ртути, не имеющие технического значения: тивманнйт HgSe; колорадоит HgTe; ртутная роговая дуЭйП§2С12, коссинитHgglg. Частично они встречаются в Гарде. Ртутьсодержа±цими являются некоторые блеклые. руды, а также некоторые разновидности цинковой обманки. В увлекаемой газами нылп, образующейся при обжиге этих руд, содержится металлическая ртуть.
Исторические г ведения. Ртуть и ее получение из киновари были известны еще древним гренам и римлянам. Первое подробное известие об этом принадлежит Теофрасту (около 300 лет до и. э.). Римляне разрабатывали рудники киновари Альмадепы в Испании, и поныне не утратившие своего значения. Функционирование второго из старейших европейских рудников (Идриа л Югославии) относится к концу XV в.
Уже около конца VI в. и. э. ртуть использовали для добычи золота из золотых руд. В начале второй половины XV в. л Мексике (Bartholomyus do Medina) ввели амальгамирование серебряной руды. Па некоторое время это привело к поглощению значительной доли ргугпой продукции Альмадепы.
Особое проц почтение ртути отдавали алхимик и, считавшие ее общей составной частью всех металлов. Они полагали, что измснсппем содержания ртути можно осуществить превращение одного металла в другой. Ртутные препараты были введены в медицину главным образрм иатрохимиками.
До этого опасались применять ртуть в медицине вследствие ее ядовитости, хотя лечебные свойства соединений ртути, например киновари, были известны еще в древности.
Получение. Ртуть получают нагреванием сульфида либо в токе воздуха, либо в смеси с железом или Прокаленной известью:
IlgS -ф- 02=5 Ug 4; SOg,
HgS + Fe=Hg + FeS,
4HgS-|- 4CaO=4Tlg4*3CaS -j-CaSO.j.
Отогнаппые пары ртути впачале конденсируют в специальных керамических холодильниках, представляющих собой небольшие, сверху и снизу открытые глиняные сосуды, вставленные одип в другой и образующие длинные цепи. В настоящее время кондеп-вацпю осуществляют в большинстве случаев в сплющенных керамических трубках, охлаждаемых водой. '
Из увлекаемой газами пыли ртуть обычно извлекают отжатием и пз остатка — путем отгопки.
От механических примесей, таких, как небольшие количества пьцди, жира и т. и., ртуть очищают, пропуская ее через бумажные фильтры, имеющие тонкое отверстие. От нрнмеси тяжелых металлов, о присутствии которых свидетельствует серый «след», оставляемый jaa чистой бумаге, ртуть освобождают многократным пропусканием ее в виде топкой струи черев толстый слой разбавленной азотной кислоты или раствора нитрата ртутп(1}. Применяют также л перегонку в вакууме.
Свойства. При о б ь( чно йт ем пер ат у р е рту т ь — ж и дкий сер е бр не то - б е лый с сильным блеском металл. При атмосферном давлении опа кипит при температуре 356,95° и затвердевает при —38,84°.
При затвердевапии ртуть образует правильные па вид октаэдры; одиако структура решетки принадлежит к гексагоиальпой сингонии. Ртуть кристаллизуется по особому типу: каждый атом Hg в решетке окружен 6 другими атомами Hg на расстоянии 3,005 А
488
Глава 9
и еще 6 на расстоянии 3,477 А. В среднем атомный радиус ртути равен 1,621 А. Эти данные относятся к температуре >—46°. Экстраполируя до температуры -j-20°, получают '[KZ= 12] = *,648 -А-
Ртуть и в твердом состоянии мягкая и растяжимая. Удельный вес ее при различных температурах равен
при 0° 15° 18° 20° 100°
13,59546 13,55846 13,55108 13,54616 13,35166
По данным Шесле и Бланкеяштейна, удельный вес ртути в интервале температур 0—100s можно определить по формуле;
__________________13,59546______________
1+ [в,«В (да)+0.ОЯ (да)1]
Расширение ртути при нагревании довольно значительно и между О и 100° почти пропорционально расширению газа. Удельная теплопроводность ртути (при 0s) составляет 2,2% от соответствующей величины для серебра; удельная электропроводность (при 0°) 1,58% от удельной электропроводности серебра.
Электро про водно стъ ртути использовали ранее для определения единицы сопротивления: сопротивление столбика ртути с площадью поперечного сечения 1 льи2 и высотой 10в,3 см принимали равным 1 ом. В на стоящее в р емя ом как международная единица сопротивления определяется следующим образом: проволока имеет сопротивление 1 ол<, если черев нее при напряжении 1 в проходит ток силой 1 а. (Различие между последним, так называемым «абсолютным омом», и определенным ранее на основе проводимости ртути имеет аначеаие лишь в случае прецизионных измерений.)
Ртуть заметно летуча уже при обычной температуре.
Давление пара ее равно
при 0° 20° 100°
0,00021 0,0013 0,279 .wat рт ст.
В 1 с.ня воздуха, насыщенного парами ртути, содержится при 0° 20° 100°
0,002 0,014 2,42 eHg
j.p*
Плотность нара ртути (относительно Н2 — 2) в температурном интервале 1500— 1700° равна примерно 200. Следовательно, пар одноатомеп.
Пары ртути очень ядовиты. При продолжительном воздействии даже в крайне незначительных количествах они могут вызвать тяжелое отравление.
Характерными признаками ртутного отравления являются слюнотечение, своеобразное покраснение десен и размягчение зубов. Появляется тяжелое нервное расстройство: головная боль, нарушение пищеварения, тремор рук и головы. Но прежде чем обнаружатся характерные симптомы хронического отравления ртутью, может пасту пить общее нервное расстройство: подавленное состояние, вялость, дурнею настроение, бессоница, ослабление памяти и т. и. Восприимчивость к отравлению ртутью носит индивидуальный характер. Наиболее чувствительные организмы могут получить тяжелое отравление даже через зубные пломбы, постоянно выделяющие следы ртути, что, по-видимому, часто происходило при использовании пломб из амальгамированной меди. Гораздо чаще отраяяяются парами ртутя, которые скапливаются в рабочем помещении при неосторожном обращении с ртутью; она скапливается в щелях пола, в пазах линолеумовой облицовки и т. п. и медленно испаряется, постоянно отравлял воздух. Испарению весьма благоприятствует то обстоятельство, что ртуть тонким слоек размазывается по полу при натирке полов. На серьезную опасность, угрожающую
Вторая группа периодической системы
48»
здоровью людей, во столпи о работающих в таких комнатах, настойчиво указывал еще Шток (Stock, 1926). Однако па эту опасность часто слишком мало обращают внимания.
Ртуть легко образует эмульсию при растирании с жиром (серая мазь). При сильном встряхивании ее можно превратить в черный порошок. Чистые водные коллоидные растворы ртути сохраняются с трудом. Однако* в присутствии защитных коллоидов такие растворы ртути устойчивы. Коллоидную ртуть используют в медицине для инъекций, так как даже в очень небольших концентрациях она оказывает сильное бактерицидное действие и гораздо менее ядовита, чем ее растворимые соли.
На воздухе при обычной температуре чистая ртуть не окисляется. Нагретая до температуры кинения при доступе воздуха она постепенна
Ряс. 51. Каломельные электроды.
превращается в окись. При более сильном нагревании окись разлагается. Во влажном воздухе или в присутствии примесей ртуть уже при обычной температуре быстро покрывается пленкой окиси. С хлором ртуть весьма энергично взаимодействует даже при комнатной температуре. С серой она соединяется также при комнатной температуре при растирании. С фосфором, напротив, не соединяется, даже прн более высокой температуре; она растворяется только в расплавленном фосфоре. В концентрированной серной кислоте ртуть растворяется при нагревании; в концентрированной и разбавленной азотной кислотах — на холоду. В зависимости от того, что взято в избытке — ртуть или кислота, образуется соль одно- или двухвалентной ртути. В царской водке ртуть растворяется очень легко с образованием хлорида. С соляной кислотой в отсутствие воздуха и с разбавленной серной кислотой она не взаимодействует. Это объясняется тем, что в электрохимическом ряду напряжений ртуть стоит правее водорода и, следовательно, неспособна вытеснять его из кислот. Большинство металлов, даже медь, вытесняют ее из растворов.
Нормальный потенциал ртутя по отношению к нормальному водородному электроду * составляет -J-0,793 е в растворе соли Hg(I) и -)-0,852 в в растворе соли Hg(II). Потенциал превращения Hgi' —2Hg" составляет -j-0,911 в.
Скачок потенциала между ртутью и 0,1 и. раствором хлорида калия, насыщенного хлоридом ртути(1) (каломель), при 18° составляет0,613 s. Такой «каломельный электрод» очень простой в изготовлении (ср. рис. 51) часто применяют вместо водородного электрода для намерения разности потенциалов. Потенциал каломельного электрода, при.
* Нормальный водородный потенциал рассчитан для раствора с активностью-ионов Т1‘, равной 1 (ср. т. I, стр. 49).
490
Г-taea 9
поденного па ряс. 51, на 0,338 в выше, чем нормаль него водородного электрода (нря 18°). Если вместо 0,1 и. использовать 1 н. раствор хлорида гмлия, эта разница уменьшается до 0,286, а в случае насыщенного раствора (~3,5 н.) до 0,254 в (все данные при 18°).
Амальгамы. Со многими металлами ртуть образует сплавы, так называемые амальгамы. Особенно легко амальгамируются натрий, калий, серебро, золото, а также цинк, кадмий, олово и свинец; амальгама меди образуется лишь при тщательном растирании. Совеем не амальгамируются марганец, железо, кобальт и никель.
Для получения амальгам соответствующие металлы в виде мелких кусочков вносят в ртуть (в случае необходимости при нагревании). Металлы, в компактном состоянии плохо растворимые в ртути, например медь, смешивают с ртутью в виде порошка. В некоторых случаях приготовление амальгамы наиболее удобно проводить электролитическим путем. При этом соответствующий металл под действием электрического тока выделяется из водного раствора на ртути, которая тонкой струей протекает через раствор.
Среди других сплавов амальгамы занимают особое положение, поскольку большинство из них даже при обычной температуре находится в жидком ниц тестообразном состоянии. Часто это обстоятельство оказывается важным при их практическом использовании. Так, для пломбирования зубов применяют амальгамы мягкие и пластичные при температуре киие-пия воды, но полностью затвердевающие при температуре человеческого тела.
По химической природе амальгамы совершенно аналогичны другим металлическим сплавам. В одних случаях они представляют собой просто растворы соответствующих металлов в ртути (в зависимости от температуры они могут быть либо жидкими, либо твердыми). В других — химические соединения. Наиболее легко ртуть образует соединения с металлами первой группы периодической системы (ср. табл. 48, стр. 456) Некоторые практически важные амальгамы, например цинка и кадмия, являются «шеями, а не соедццениямщ
Применение. Металлическая ртуть находит применение по многих технических приборах, и прежде всего в научно-исследовательской аппаратуре. Ее используют, например, в кварцевых лампах, ртутно-паровых выпрямителях, в реле, регуляторах давления, для изготовления термометров и барометров, затворов для газов, а также в качество кипящей жидкости в высоковакуумных насосах Фольмера и для другой научной аппаратуры.
В больших количествах ртуть используют для изготовления киновари (для красок). Из нее получают гремучую ртуть для военной промышленности. Ртутную мазь (серая мазь) применяют при кожных заболеваниях. В Медицине применяются в настоящее время и многие соединения ртути. Из солей ртути важное техническое значение имеют нитрат и хлорид. Например, их используют в производстве фетровых шляп для протравы волос; однако ввиду сильной ядовитости ртути предпочитают употребление для этих целей соединений церия (ср. стр. 530).
Некоторые соли ртути имеют ваяшое значение в связи с их кшпалнпш-ческим действием, особенно в реакциях с углеводородами. Так сульфа? ртути(Г) подобно сульфату медн ускоряет окисление органических веществ концентрированной серной кислотой при определении азота по методу Кьельдаля. Сульфат ртути(П) служит катализатором мри техническое получении ацетальдегида из ацетилена (ср. т. I, стр. 477).
А.чальга.ны., ранее применяемые гораздо шире, во многих сяогг?пх утратили свое значение, например при добыче золота н серебра, дшг
Вторая группа периодической системы
491
позолоты в пламени и для покрытия зеркал. Однако еще значительные количества ртути применяют для амальгамирования зубных пломб *. Важное значение имеет также амальгамирование цинковых пластин для гальванических элементов. В лаборатории в качестве восстановителя часто применяют амальгаму натрия, которую удобно получать погружением кусочков натрия в слегка нагретую ртуть.
Нормальный элемент, Спиду того что ртуть легко можно получить химически чистой и что при соирикоспокении с иодными растворами поверхность ее остается совершенно чистой, особенно удобно использовать ее для элементов с определенным напряжением (нормальные элементы). Наиболее важным из них является элемент Вестона. Он состоит из небольшого сосуда Н-образной формы (рис. 52), в одном колене которого находится ртуть, покрытая слоем Hg.,SO4; дно второго коле па покрыто амальгамой кадмия (12,5%); ее наливают горячую в сосуд и затем дают ей настыть. Оба колена запомнены насыщенным раствором CdSO,,. Чтобы раствор оставался насыщенным при изменении температуры, в него помещают кристаллы CdSO4-®/3HaO. Контактами служат платиновые пр оболочки, впаянные в дно сосуда. Электродвижущая сила этого элемента очень мало зависит от температуры. Она составляет
при 0° 10° 15= 16° 17°
1,0187 1,0186 1,0185 1,0184 1,0184 в
при 18° 19Q 20° 25°
1,0184 1,0183 1,0183 1,0181 з
Зависимость э.д.с. от температурю еще меньше, если вместо гаеыщеяного раствора сульфата кадмия, притотоплэнного при каждой из этих температур, использовать
сульфата кадмия, притотсшзэнного при каждой из этих раствор, насыщенный при 4°. У более старого элемента Вларка, который вместо кадмия н сульфата кадмия содержит цинк и раствор сульфата цинка с избыточными Кристаллами соли, зависимость э.д.с. от температуры значительно больше.
5
KillllilhllM'l
MIIIIIIHlllMlj
СОЕДИНЕНИИ РТУТИ
Ртуть образует два ряда нормальных простых соединений. Их эмпирический состав соответствует общим формулам HgR и HgR 2, где R — одновалентный радикал. Насколько в этих соединениях можно говорить о заряде ртути, в солях первого ряда она электроположительно одновалентна, а второго электроположительно двухвалентна. В соответствии с этим рассматриваются соединения ртути(Г) и ртути (ll).
Строение соединений ртути(I). Во всех соединениях ртути(1), строение которых в настоящее
-,CdSO4-paCTJWp
CdS04-
-кридло/дау
Ндгео4 са+йд
«9
Рис. 52. Нормальный элемент Вестона,
время удалось установить, два атома ртути непосредственно.1 связаны между собой. В соответствии с этим вместо эмпирической формулы HgR правильнее употреблять формулу Hg2R2 или R—Hg—Hg—R. Ртуть в этих соединениях хотя и является электрохимически одновалентной, однако формально двухвалентна. И в^расгворах имеются не однозарядные ионы Hg', а двойные двухзарядные ионы Hgj'.
То, что электрохимически одновалентная ртуть в водных растворах существует в виде ионов Hgj •, можно доказать следующим образом; если встряхнуть ртуть с водным раствором нитрата серебра то устапавливается равновесие, для которого в зави-
* Ввиду вредного для здоровья действия пломб из ртутной амальгамы пытались заменить амальгамы другими сплавами с подходящими свойствами, например сплавом Sn, Bi и Ga (внгаметалл). Однако эти попытки не привели еще к совершенно удовлстверительным результатам.
492
Глава 9
а>
(2)
симости оттого, как идет реакция, по (1) или (2) пути Hg+Ag* Xt Ag+Hg', 2Hg + 2Ag- Xt 2Ag + Hgr, ГАе'1
должно оставаться постоянным либо отношение [Ag] (По зак0ну Действия
масс), либо I^gJ— —. Опыты Огга (Ogg, 1898) показали, что это справедливо
[AgJXfJIgrf'*
только для второго отношения, К такому же результату автор пришел и при исследовании равновесия между нитратом ртутн(1), нитратом ртути(П) и металлической ртутью. Действительно, если встряхивать раствор нитрата ртути(Ц) с металлической ртутью, последняя переходит в раствор, а нитрат ртути(П) превращается в нитрат ртути(Т). Одпако до конца эта реакция но идет, в растворе устанавливается равновесие, для которого справедливо выражение либо (3), либо (4):
Hg" + Hg 2Hg‘
I
Hg-4-iig iigr.
(3)
и
Опыты показали, что в присутствии осадка металлической ртутя остается постоянным 0 /130 при 25°) отношение [П^М] ’ а ве ' ^леАсватеяьно, в обоих
случаях справедливо уравнение, которое предполагает существование в растворах электроположительно одновалентной ртути в вире иона Hgj'.
Из других наблюдений, подтверждающих такое предположение, следует отметить, что электропроводность растворов хорошо растворимых солей ртути(1) по своей величине и зависимости концентрации от температуры (если исключить влияние гидролиза) соответствует двух-одновалентным электролитам (например, нитрату бария), а не одно-одновалентным (нитрат калия).
Ранее существовали еще сомнения относительно того, имеет ли место димеризация электрохимически одновалентной ртути и в кристаллических соединениях иди образование ионов присуще только водным растворам. Результаты определения плотности пара хлорида ртути(1) [а также бромида казалось бы говорят в пользу мономолекулярной формы, так как они привели к моцомолекулярному атомному весу.. Однако те же данные можно было объяснить тем, что в процессе испарения, возможно, HgaCl, диссоциирует на HgCI3 и Hg. В этом случае средний молекулярный вес также соответствовал бы формуле HgCl. Действительно, пластинки золота быстрс амальгамируются парами хлорида ртути(1). Однако из этого еще не следует, что пар полностью диссоциирует на HgCl2 и Hg. В настоящее время рентгенолстнритеж^ удалось определить структуру галогенидов ртути (1). Оказалось, что асе они содержи? спаренные атомы ртути в виде цепей X—Hg—Hg—X (ср, стр. 499). После тот,-, как бт.иго установлено существование связи Hg—Hg не только в водных растворзг но и в твердых соединениях, не оставалось сомнений, что электрохимически ойк,-валеитная ртуть опишяаетея своеобразием^ а. именно она постоянно находится в димерной форме Hg|+.
Получение и свойства соединений ртути(Т). Легкорастворимые согг ртути(1) можно получить либо взаимодействием соответствующих с.одец ртути(П) с металлической ртутью, либо растворением карбоната ртути! Е в соответствующих кислотах. Трудно растворимые соли и другие труди:-растворимые соединения получают осаждением из водного раствора легл:-растворнмых солей. При этом часто вместо ожидаемого соединения ожгла лентной ртути образуется смесь соответствующих соединений ртутв5С с металлической ртутью.
Это всегда происходит в тех случаях, когда соответствующе о соединение ртуде является либо сильным компленсообразователем, либо настолько малодпссоцпп^т- -щим, что концентрация иопов Нд"' в растворе исчезающе мала. В таком случае в г ветствии со сказанным выше н концентрадпя иопов Hg.;* настолько мала, что де;_
* Квадратные скобки означают молярные концентрации (точнее, акппдед'де. соответствующих ионов в растворе или серебра в образующейся амальгаме.. [1сде ;,-трациго металлической ртути можно считать постоянной.
Вторая группа периодической системы
493
леденив растворимости нормально диссоциирующего соединения ртути(1) не может быть достигнуто, даже если это соединение труднорасгворнмо. Так, цианид ртутщ'1) не существует, так как Hg(CN)2 чрезвычайно мало диссоциирует и равновесие
Hgr Hg + Hg" или
П-Ц'г" +CN' Hg4-Hg(CN)2 полностью сдвинуто вправо.
Большинство соединений ртути(1) труднорастворимо, лишь немногие из них растворяются легко, это нитрат, хлорат и перхлорат. Эти соли хорошо диссоциируют подобно солям типа нитрата бария. Однако они гидролизуются, хотя и незначительно. Растворы их имеют кислую реакцию. Все соединения ртути(1) являются довольно сильными восстановителями.
Склонность к комплексообразованию у них мала.
Некоторые соединения ртути(1) окрашены. Карбонат имеет желтый цвет, так жи как иодид (очень неустойчивый); арсснат(Ш) также желтоватого цвета, а арсенат(V) оранжево-красный.
Получение и свойства соединений ртути(П). Растворением ртути в избытке крепкой азотной кислоты получают нитрат ртути(П); при сухом прокаливании последнего образуется окись ртути(П). Растворением ее в соответствующих кислотах приготовляют большинство солей ртути(П). Как правило, они получаются также при окислении соответствующих солей одновалентной ртути.
Как уже упоминалось, соединения ртути(Т1) образуются иногда в результате реакций осаждения, для которых следовало бы ожидать образования соединений ртути(1). Но вместо них часто получают смесь соединений двухвалентной ртути с металлической ртутью, например в случае сульфида и цианида. Подобное явление наблюдается и у медн, например при получении иодида одновалентной меди Cui, нместо ожидаемого соединения меди(П) выпадает более трудно растворимое соединение мсдн(1).
Соли ртути(П) с сильными кислотами подобно соответствующим соединениям ртути(1) бесцветны. Однако электрохимически двухвалентная ртуть весьма склонна к образованию труднорастворимых основных солей, большинство которых окрашены в желтый или оранжевый цвет.
Соли ртути с более слабыми кислотами часто также окрашены, например нитрит ртути(П) — светло-желтого, ортоарсенат(У) — лимонно-желтого цвета. Сульфид ртути имеет две модификации— красного и черного цвета.
Большинство солей ртути(П) хорошо растворимы. Нитрат, перхлорат и им подобные нормально диссоциируют по типу, например, нитрата бария. Растворы вследствие гидролиза имеют сильнокислую реакцию. Гидролиз проходит гораздо глубже, чем у соответствующих солей рту-ти(1). При сильном разбавлении, как правило, выпадают основные соли. Не гидролизуются только соли очень сильных кислот, например перхлорат ртути(П).
Помимо нормально диссоциирующих, имеются также хорошо растворимые соли ртути(П), которые диссоциируют очень слабо. К ним относятся цианид, роданид, хлорид и бромид. (Иодид также очень мало диссоциирует, но он и малорастворим.) Соли, отличающиеся незначительной диссоциацией, соответственно слабо гидролизуются. Особенно мало диссоциирующий цианид вообще не подвергается гидролизу.
У солей ртути(П), образованных средней силы органическими кислотами, такими, как уксусная, степень диссоциации такая же, как у самой кислоты, а нс как у типичных ее солей.
494
Глава 9
В отличие от ионов ртути(Г) ионы ртути(11) чрезвычайно склонны к комплексообразованию.
Из труднора&пворимых солей ртути(П) следует назвать иодид, оксалат и фосфат. Весьма трудно растворим сульфид; роданид и сульфат растворяются умеренно.
Многие соли pryти чрезвычайно легко растворимы в органических растворителях, например в спирте, эфире, бензоле и т. д. Присоединение аммиака нехарактерно для соединений ртути: напротив, ртуть, как правило, замещает, водород в аммиаке (или в его производных) с образованием амидосоединений и даже аммонийных соединений, в которых весь водород замещен ртутью. В более слабой мере это явление наблюдается и у соседа ртути слева — золота (ср. стр. 509).
Насколько легко происходит замещение связанного с азотом водорода ртутью (находящий: и в виде соединения), примером может служить растворение окиси ртути в водных растворах органических амидокислот, индифферентных по отношению к прочим основаниям, например:
.МН-СО(СНз)
IIgO4-2lI2N'CO(CH3)=Hg< + Н2О
XNH-CO(CH3)
Ацетамид Ацетамид ртути
Формально ато реакция сол (’образования- Образующееся соединение, которое по своему составу соответствует соли двухвал сити ой ртути, в водном растворе диссоциирует очень слабо.
Ртуть может легко присоединяться также к углероду или органическому радекад у. Одновалентный остаток —Hg—X особенно легко замещает водород в органических соединениях. Например, при взаимодействии окиси;ртути со спиртом в присутствии едкого натра образуется соединение
/Hg. /Hg\
°\ /С------С\
ПО-Hg Hg-OH
(Hofmann). Безводный ацетат ртути при пагровании с бепзопом при 110° в тсчетг< Нескольких часов переходит в ацетат феиилртути С6Н5—Hg—O-CO(CH<j). Анало-гично реагируют толуол и фонол. Подобно ацетату взаимодействуют и другие соедЕА -нля ртути, например сульфат и нитрат (DimroLli). Об органических соединениях рт^тл типа HgHs (алкильные соединения ртути) см. стр. 509.
Как уже было указано, галогениды ртути, за исключением фторида, мало социируют в растворе. С1, 13г и I, следовательно, связаны с ртутью слабой штааа связью. Практически Неионпыми являются радикалы, в которых ртуть связана с < N п S.
Легкорастворимые и хорошо диссоциирующие соединения ртуг_ как одно-, так й двухвалентной, бесцветны. Слабо диссоциирушгд-й нерастворимые имеют довольно интенсивную окраску.
Раиыне- соединения одповаяситцой ртути называли
а двухвалентной — меркурисоединекияхи. Это противоречило основным иояо~ -~_ : номенклатуры, предложенной Вернером, в соответствии с. которой меркуросхл:: ниями следовало называть соединения ртутн(П), и иринодило к ошибкам. Скцда: : » относится к наименованию соединений и ряда Других металлов, Коатому в дальс:;" :: а. Отказались от буквенного обозначения валентности и условились применять —. ложенное Штоком обозначение ее римскими цифрами.
В связи с тем что между соединениями ртути обеих степгнзй -ния существует тесная связь, в дальнейшем они иногдг. - .л,
вместо.
Вторая группи периодической система
495
Окись
Окись ртути(П). HgO выпадает в виде желтого осадка при действии избытка щелочи на раствор хлорида или нитрата ртути(Н):
Hg" + 2OH' = HgO + H2O.
В виде красного кристаллического порошка она образуется при слабом нагревании нитрата ртути(П), а также нитрата ртути(1):
Hg(NO3)2=HgO + 2NOa + i/2O21.
l-lg2(XO3)K=2HgO + 2NO2.
Последний способ получения окиси был известен еще алхимикам, которые называли продукт разложения «красным преципитатом».
Прежде считали, что красная и желтая модификации различаются только величиной частиц. Рентгенограммы их идентичны, за исключением некоторого расширения липин у желтой формы. Однако различия в величине «зерен» очень малы. Турки (Топгку, 1960), вопреки сказанному, обнаружил существенные различия в электропроводности, диэлектрической проницаемости и магнитной восприимчивости этих: модификаций. Он пришел к выводу, что различие в окраске и ширине линий, по существу, вызвано сильными искажениями решетки и дефектами в распределении атомов. В воде желтая форма растворяется немпого лучше (1 ; 19 300), чеМ красная (1 : 11) 500 при 25"). Раствор имеет слабоосновную реакцию:
HgO+H2O ^Ug“ + 2OH’.
Окись ртути(П), как уже было отмечено, образуется при непосредственном взаимодействии ртути с кислородом воздуха. Окисление Hg идет, однако, лишь при температуре, близкой к температуре кипения ртути. При более высокой температуре окись разлагается на простые-Вещества. Давление пара продуктов разложения заметно ужо при 440J, а при 6ПУ оно достигает 1250 мм рт ст. Следовательно, сродство ртути к кислороду невелико. Теплота образования HgO из элементов 21,6 ккал/моль.
Окись ртути кристаллизуется в ромбической сингонии (уд, вес 11,1). Красна» окись ртути встречается в природе в виде минерала монтроидита.
Окись ртути находит широкое применение в препаративной химии, например при получении хлорноватистой кислоты (ср. т. I, стр. ^57), а также в медицине. Опа служит исходным продуктом для получения других соединений ртути.
В присутствии защитных коллоидов, иапрнмор протальбиновой или лизальби,-noBCiii кислоты, окись ртути можпо легко получить в коллоидной форме.
При добавлении тцелочи к раствору соли ртути(1) получают черный осадок. По-видимому, он представляет собой смесь тоикораздроблеппой металлической ртути и окиси ртути(П), которая образуется в результате окислительно-восстановительной реакции в момент осаждения:
Hg; • + 2ОН'=Hg+ HgO+II2O.
Раньше этот осадок считали просто окисью ртутп(1); однако окись одновалентной ртути выделить в свободном виде еще не удалось. Рентгенометрически также ие обнаружены никакие отклонения решетки полученного осадна от решетки окиси ртутн(П> (Fricke, 1933),
-496
Глава 9
Сульфид и тиосоли
Сульфид ртути, сульфид ртути(II) HgS, существует в двух модификациях, красной и черной. Обе встречаются в природе.
Красный сульфид ртути — киноварь является основным минералом ртути. По своей структуре киноварь (уд. вес 8,1) относится к гексагональной сингонии. В виде кристаллов правильной формы она встречается, однако, редко. Напротив, она обычно образует плотные агрегаты от кирпично-красного до темного красно-коричневого цвета, а в присутствии примесей — иногда сине-черного цвета. Искусственно приготовленная киноварь имеет чистый ярко-красный цвет. Ее применяют в виде малярной краски (киноварь, китайская красная, вермилон) и в качестве грима. Для этой цели ее получают нагреванием ртути с концентрированным раствором пентасульфида калия (способ Доберейнера):
Hg-PK2S5=HgS + K2Sb.
На свету киноварь постепенно темнеет. Происходит ли при этом разложение или превращение в черную модификацию, еще не ясно.
Черная модификация сульфида ртути встречается в природе в небольших количествах в виде сопутствующего киновари минерала ртутной черни (метакиноварь). Она представляет собой черный порошок (уд. вес 7,7—7,8), на первый взгляд аморфный, в действительности же состоящий из крохотных правильных тетраэдрических кристалликов.
Кристаллическую решетку красного сульфида ртути {киновари) можно кратко определить как «деформированную решетку каменной соли». Она как бы образована из решетки типа каменной Соли путем замены атомов натрия на атомы ртути, а атомов С1 па атомы S, причем последние пе точно занимают места С1, а немного сдвинуты. Вследствие такого сдвига расстояния соответствуют уже не кубической, а гексагональной сингонии. Кратчайшее расстояние Пр <—► S составляет 2,52 А. Кристаллическая решетка Чернова сульфида ртути имеет структуру, подобную структуре хлорида меди(1) или цинковой обманки (см- рис. 47, стр. 407); а = 5,84 А. Кратчайшее расстояние Hg <—f S в ней равно 2,53 А, т. е. практически такое же, как у киновари.
Черный сульфид ртути всегда образуется при осаждении ртути ns водного раствора сероводородом, а также и при других условиях. Например, в технике его получают взаимодействием ртути с расплавленной серей (голландский способ) или с тонкоизмельченной серой (ирландский способ). Возгонкой при атмосферном давлении или обработкой раствороц полисульфидов щелочных металлов его можно перевести в красную модификацию .
г» Так как растворимость сульфида ртути в воде чрезвычайно мала, все? соединения ртути, в том числе и прочные комплексные, разлагаются сероводородом с образованием сульфида. Из растворов соли ртутпЦ' выпадает смесь сульфида ртути(П) и металлической ртути:
Hg3“+H2S-HgS+Hg-V2ir.
Сульфид ртути нерастворим или растворяется очень медленно да2=1 в концентрированных кислотах; однако он легко растворяется в даренья водке.
Тио-соли. Сульфид ртути не растворяется ни в щелочах, ни в сернистом аммонии; в концентрированных растворах сульфидов щелочной и гидросульфидов щелочноземельных металлов он растворим. При зггх
Вторая группа периодической системы
497
образуются тио-соли. Они получаются также при сплавлении сульфида ртути с серой и щелочами, например K2[HgS2]-5Н2О — блестящие, очень легко расплывающиеся иголочки.
В водном растворе тио-соли ртути устойчивы лишь при избытке гидроокиси щелочного металла. При пропускании сероводорода в раствор тио-соли, приготовленный растворением сульфида ртути в растворе сульфида щелочного металла снова выпадает сульфид ртути; сероводород связывает ионы S" в ионы HS':
S"<HES 2HS', (5)
и вследствие этого равновесие
ITgS + S' [HgS2f (6)
сдвигается влево. Наоборот, подщелачивание раствора способствует растворению сульфида ртути в сульфиде щелочного металла:
НЗ' + О1Г S" + H2O. (7)
Таким офразом объясняется тот факт, что, несмотря па способность к образованию тио-соли, сульфид ртутя псе ясе не растворяется в сернистом аммонии; в растворе сернистого аммония сера практически полностью находится в виде ионов HS' (ср. т, I, стр. 789), Следовательно, реакция (6) в таком растворе почти пе идет слева направо.
Если сероводород, медленно пропускать через раствор соли ртути(П), то можно наблюдать образование белого, желтого или буроватого осадков, состоящих лз двойных солеи, в которых иопы первоначально связанные с ртутью, еще только частично замещены серой. Такне двойные соли особенно легко получать из растворов слабо диссоциирующих солей ртути, например:
3HgCl2-|-2H2S=Hg3S2Cl2-|-4HCl.
Галогениды ртути(1)
Хлорид ртути(1), каломель Hg2Cla иногда встречается в природе в виде мелких тетрагональных кристалликов (роговая ртутная руда); в технике его получают как сухим, так и мокрым способами. Сухой способ заключается в нагревании тонко растертой смеси либо хлорида ртути(П) со ртутью [уравнение (8)], либо сульфата ртути(П), ртути и хлорида натрия [уравнение (9)]. Мокрым способом хлорид ртути(1) получают восстановлением водного раствора хлорида ртути(П) сернистым газом [уравнение (10)] либо взаимодействием раствора нитрата ртути(1) с раз-
бавленной соляной кислотой [уравнение (И)]:
IJgCl2-|-Hg —Hg2C)2; (8)
IIgSO4 -L Hg 4- 2NaCl = Hg2Cl3 + Na2SO4. (9)
2HgCla + SO2 + 2H2O = Hg2Cl2+H2SO4 + 2ИС1. (10)
Hgj*-]-2Cl'=Hg2C)2. (H)
Полученный сухим способом возогнанный хлорид ртути(1) представляет собой белую блестящую кристаллическую массу, осажденный из раствора — белый порошок. Название каломель он получил благодаря тому, что при смачивании аммиаком он приобретает глубокий черный цвет (xaZZv psActg—прекрасный черный).
Обычпо считают, что черная окраска обусловлена образованием смеси хлорида ампдоргути(Ц) [Hg—NH2]C1 (бесцветного) (ср. стр. 506) и тонкоизмелъченной метал-лкчейкои ртути:
IIg2Cl2+2NH3=4HgNH2]Ci + Hg + NIJ4C1.
32 г. Реми
498
Глава 9
В соответствии с другими данными (Th. F. EgiAins, 4936—1938) черная окраска приписывается соединению ртути(1) Hg-2(NH3)C1. Последнее, особенно в присутствии осаждающей жидкости, разлагается, однако на соответствующее соединение ртути(П) и металлическую ртуть:
HgoCl» + 2NH3=Hg2(NH2)Cl + NH4C1;
Hg2(WCl=[Hg(MH2)]Cl-l-Hg.
Липскомбу (Lipscomb, 1951) удалось показать, что взаимодействие HgaCl2 с водным раствором аммиака сводится к образованию двух различных продуктов, причем в обоих случаях черная окраска обусловлена присутствием металлической ртути. Липскомб нашел, что в зависимости от условий образуется либо [Hg(NH3)]Cl, либо хлорид основания Миллона (если раствор аммиака сильно разбавлен):
2Н?гС12 + 4КНз + ИгО=1Н^гМ]СЬНгО + 2Не+3?П-Ц+ЗС1’
(см. стр, 507). Возможно, что это соединение образуется в качестве промежуточного и при действии более крепких растворов аммиака, по оно Сразу же превращается в амидосое Динеине )HgNH2]Cl.
В воде хлорид ртути(1) трудно чительно легче он растворяется в
Рис. 53. Структура решетки хлорида ртути(1) Hg2CJ2.
«0=6,30. С(г=10,88 A; Hg t—rr Hg=di=2,54 А; Hg ч—* 01=42=2,51 А;С1ч—>С!=4з=3,32 A, Соответствующим образом построены Hg-jBrs (во=6,О1, C(i=li,16A> и Hgala —6>35, ->=lt,57 А) Если принять расстояние iTg «—> Hg равным 2.54 Л, то Hg<—>Вг= =3,58, Hg<—> 1=2,68, ВТ +-»Вт=3,46 и
3 <—> 1=8,67 А.
растворим (2,1 .ий в 1 д при 18°). Зна-растворах хлоридов. В спирте, эфире и ацетоне он практически не растворяется. Удельный вес его 7,16. Прп умеренном нагревании (даже при трении) он желтеет. На свету хлорид ртути (I) быстро темнеет, по-видимо-му, вследствие частичного разложении
Рис. 54. Структура решетки хлорида ртути (II).
Ромбическая элементарная ячейка: и=5;367., 5=12,753, с=4,325 А. Расстояние Hg *—> С1= =2,25 А. (Атомы, принадлежащие одинаковым молекулам, соединены двойными лннпя-ми. (Атомы, обведенные вунктирим, лежат вне в.тементарной ячейки.)
на хлорид ртути(П) и металлическую ртуть. При 383° хлорид ртуттгТ возгоняется, но плавясь. В запаянной трубке, по данным Бильтца и Клеима, он плавится при 525° (частично разлагается на Hg и HgCl2), иерехсун в красно-коричневую жидкость.
Каломель применяют главным образом в медицине как слабительЕде
Вторая, группа периодической системы
499
Бромид ртути(1) Hg2Br2 выпадает в виде белого осадка при действии бромида щелочного металла на раствор питрата ртути(1). Перекристаллизацией из горячего раствора нитрата ртути(1) его получают в виде перламутровых тетрагональных листочков.
Иодид ртута(1) HgJ2 выделяется в виде зеленого осадка при действии небольшого количества иодида щелочного металла на растворы нитрата ртути(1). При растирании увлажненной спиртом смеси ртути и иоДа получают зеленовато-желтый порошок Hg2I2, нерастворимый при обычной температуре в воде и в спирте. Его применяют в терапии. Чистый иодид ртута(1); ярко-желтого цвета. Он неустойчив, очень легко разлагается на Hg и Hgl2. При действии раствора иодида щелочного металла па иодид ртути(1) половина его переходит в раствор в виде комплексного иона двухвалентной ртути с иодом, вторая половина восстанавливается до металлической ртути:
Hg2I2+2l'^[Hgi4]" + Hg.
Структура решетки галогенидов ртутн(1). Рентгеноструктурным анализом для галогенидов ртути(1) была установлена решетка, приведенная на рнс. 53 (стр. 498), где показана элементарная ячейка кристаллического хлорида ртути(1). Она представляет собой слоистую тетрагональную решетку, состоящую из молекул Cl—Hg—Hg—Cl. Ее можпо считать производной решетки тина хлорида натрия, в которой месту каждого атома натрия соответствует центр тяжести двух атомов ртути, лежащих на осн е, а месту каждого иопа хлора — центр тяжести двух атомов хлора.
Фторид ртутц(1) Hg2F2 образуется при растворении св еже осажденного карбоната ргути(1) в водном растворе плавиковой кислоты. Он представляет собой кристаллический порошок (тетрагональной сингонии, уд. вес 8,7) желтоватого цвета, на воздухе быстро темнеющий. В воде растворяется легче хлорида и в отличие от него гидролизуется.
Галогениды ртути(П)
Хлорид ртути(П), хлорид ртути, сублимат (сулема) НдСЬ, получают обычно нагреванием смеси сульфата ртути(П) с хлоридом натрия:
IlgSO^-j- 2NaCJ =^HgCl2-|- Na^SO^,
или растворением окиси ртути в соляной кислоте
HgO + 2НС1 = HgCi2 -j- Н2О.
Полученный сухим путем в виде возгона (отсюда название сублимат), оп образует белую прозрачную массу лучистых кристаллов. Из водного раствора хлорид ртути(П) кристаллизуется в форме бесцветных, ромбических бипирамид. Удельный вес 5,44; температура пл явления 277— 280°, температура кипения 302°. Сулема более летуча, чем каломель. В твердом состоянии она устойчива к действию света.
По данным Брикке (Braekken, 1934), хлорид ртути(П) образует типпчиую лозе-кулярную решетку (см. рис. 54). Этим он отличается ог остальных галогенидов рту-ти(П), среди которых HgF2 имеет координационную решетку (типа плавикового шпата* а = 5,54 A.), a HgBr2 и Hgl2 слоистые, причем решетка бромида ближе к молекулярной (см. стр. 502).
В воде хлорид ртути(П) растворяется довольно легко, особенно при нагревании. (В 100 г воды растворяется при 0° 4,3, при 20° 7,4, при 100° 55 г HgCl2.) В присутствии же НС1 или NH4C1 растворимость возрастает. Хлорид ртути(П) очень легко растворяется также в спирте (50,5 г HgCl2 в 100 г абсолютного спирта при 25°). Он растворим и в эфире
32*
500
Глава 9
(6,45 г HgCl2 в 100 г абсолютного эфира при 18°), и во многих другие органических растворителях, например в бензоле (0,5 а в 100 г при 25ч 1,8 г в 100 г при 84°). В разбавленных водных растворах он постепеннс разлагается с образованием каломели. Водные растворы сулемы вследствие гидролиза имеют кислую реакцию.
Сулему широко применяют в медицине как весьма эффективное антисептическое и дезинфицирующее Средство, а также при лечении сифилисе и других инфекционных заболеваний. Сулема—сильный яд, смертельная доза 0,2—0,4 г. Таблетки сулемы, применяемые для приготовления дезинфицирующих растворов, рекомендуется окрашивать в красный цвет эозином во избежание путаницы. Киан (Куап, 1823) предложил способ сохранения древесины (например, телеграфных столбов) от гниент^-путем длительного выдерживания ее в растворе сулемы («кианизация» В фотографии сулему применяют для усиления негативов. В препаративной химии часто используют (о чем подробнее см. ниже) свойство хлорид; ртутц(П) образовывать хорошо кристаллизующиеся двойные хлорп— для открытия или очистки органических оснований.
Молярная электропроводность водных растворов HgCl2 очень мала по eparzz нню с водными растворами других солей, а также самого хлорида в присутствии с.вобст ной соляной кислоты, хотя его растворы и показывают кислую реакцию. Ни-— -проводимость обусловлена очень слабой диссоциацией хлорида ртути(П). Различил-^ методами, в том числе измерениями потенциала, найдепо, что концентрация ионов Нг в насыщенном (т. е. около т/4 М) водном растворе хлорида ртути(Й) nefr* ка 10-8 г-иои/л, т. е. на порядок меньше концентрации ионов водорода в чист. .. воде! Благодаря гидролизу, протекающему, по-видимому, по уравнению:
2HgGl2+Il3O=Hg2OCl2+2H4-2Cl',
концентрация ионов G1', хотя и значительно больше, чем ненов Hg*‘, однако все очень мала *, Следовательно, основная масса хлорида ртути(П) находятся в растс—-в неднссоциированиом состоянии. По величине электропроводности, измеренной (Ley) при 25°, концентрация ионов С1' в г/32 „W водном растворе хлорида ргупг при условии, что они получаются только за счет выделенной в результате гидрегг .яг кислоты (ср. гл. 12), должна бы быть равной 1,6-10~4 г-иои/л. Если же ионы С1' сб-,я-зуются только в результате диссоциации
HgCI2 [HgCIJ-4-Cl',
то в растворо той же концентрации содержание их должно быть порядка 6-10—1 г-г: В действительности же она равна среднему между этими величинами, так как место оба процесса — (12) и (13). Во всяком случае, можно сказать, что в 1/3, If творе хлорида ртути(П) самое большее 0ф % его гидролизов ано и пе бо-чь-— диссоциировано по первой ступени и что диссоциация по второй стукепи
IHgCl]-
ничтожно мала. Наиболее вероятными для указанной концентрации и томнеу.—<- ищ,, по данным Лютера (Luther, 1904), являются величины: 0,47% гидролизуй^"' ....
и 0,14% диссоциирующего по первой ступени хлорида. Для i/512 М раствврт г ветственно 2,7% и 0,5%.
Хлорид ртути(П) является веществом, удобным для демонстрации своесяцггтт г.
поведения веществ, которые мало гидролизуются и мало диссоциируют. Если г -насыщенному раствору хлорида ртути(П) — вследствие гидролиза показынтт—.....
кислую реакцию ио метиловому оранжевому прибавить раствор NaCl пг- -имеющих нейтральную реакцию, то наблюдается изменение окраски 1тнщтта1—... _
* Полагают, что идет также процесс
HgCl2+2Gl' IHgCl4]'.
Однако равновесие вследствие очень небольшой концентрации cbo6cz~jz: х жж С1' значительно сдвинуто влево,
Вторая группа периодической системы
501
среда становится нейтральной. Это вызвано тем, что благодаря введению ионов СГ концентрация ионов 1Г уменьшается в соответствии с уравнением (12) *. Следовательно, раствор ведет себя подобие раствору слабой кислоты, к которому добавлена нейтральная соль этой кислоты; таким образом, раствор хлорида ртути(11) ведет себя так, будто соляная кислота является слабой кислотой. Из уравнения (12) видно также, что увеличение концентрации иопов Н' должно уменьшить концентрацию ионов СГ. Следовательно, из хлорида ртути(П) концентрированная серная кислота пе может выделить хлористый водород даже при кипячении.
Труднорастворимые хлориды, нормально диссоциирующие в растворах хлорида ртути(П), вследствие небольшой концентрации в пих ионов хлора (например, хлорид серебра) легче, чем в воде, растворяются в водных растворах таких солей ртути, л которых концентрация иопов Hg” в растворе велика. К таким солям относится, например, цитрат ртути(П). Так как иопы Hg" связывают ионы С Г в Не диссоциирующие молекулы HgCl2, произведемте растворимости хлорида серебра достигается только после перехода в раствор достаточного количества ионов Ag‘. По этой причине в присутствии соли ртути(П) ионы СГ не полностью осаждают серебро из растворов нитрата и соответственно нитрат серебра не полностью осаждает хлор из растворов хлорида ртути(П), что следует иметь в виду при количественном анализе этих элементов .
Двойные соединения хлорида ртути(П). Хлорид ртути(П) весьма склонен присоединять другие хлориды с образованием двойнях или комплексных солей, преимущественно типов MTHgCl3 и M]HgCl4. Помимо этих, известны также соединения более сложного состава. Выделены и различные продукты ирис о едино пи я НС1 к IIgCl2, например HgCU'^HCl-• 7Н2О. Часто образуются смешанные двойные соли, т. е. соли, образованные хлоридом ртути(П) е солью другой кислоты, папример:
HgCl2.K2Cr2O7 = K2
Hg
C.l2
Сг2О7
— красно-желтые ромбические кристаллы. Несколько кристаллических соединений HgCl2 образует с HgO.
При действии на раствор соли ртути(11) небольшим количеством раствора едкого патра вначале образуется, как правило, черный коллоидный осадок HgGl2-2HgO. В присутствии маточного раствора ол постепенно при определенных условиях превращается в бесцветные тетраэдрические кристаллы 2HgUlz-HgO (плотность 6,39). Это соединение, как показали рентгеноструктурные исследования (Weiss, 1953), представляет собой соль Океания
CIHg О HgCl Hg Cl
Cl
(расстояние О -f-ч- Hg 2,06 А, Hg<—>С1 внутри комплекса 2,39 А), Ионы хлора во внешней сфере могут обмениваться на ионы фтора. Соединение [0(HgCl)3]Cl очень трудно растворимо в холодной воде. Горячей водой оно гидролизуется с образованием соединения IIgCl2-2HgO черного цвета. При сухом нагревании образуется IIgCl2' 2HgO и возгоняется HgCl2. При более сильном нагревании получают сначала черный продукт HgCl2-4HgO, а затем окись ртути красного цвета. Соединение HgCl2-4HgO образует ромбическую слоистую решетку. Такую же структуру имеет соответствующее соединение брома HgBr3-4HgO (состав его колеблется в интервале 3,5—4 HgO) [Weiss, 1954].
Кристаллизующаяся в виде вытянутых ромбических призм двойная соль NaHgCl3 состоят из связанных л виде цепи октаэдров HgCle. Помимо Этого соединения, HgC]2 образует с NaCI двойные соли Ka2IIgCl4 и NaIIg2Cl5, а также NaJIg^OCluj — волокнистые кристаллы.
* Решая совместно уравнение (12) и уравнение, приведенное в примечании на стр. 500, по закону действия масс получают;
[(IIgCL)4a х [IJ2O] [ИЙ2ОС12]Х[И']аХ[СГ]в
- const.
Из этого равенства еще понятнее, насколько концентрация ионов Н‘ зависит от концентрации ионов СГ.
502
Глава 9
При избытке ионов С Г в растворе хлорида ртути(П) в значительном количестве образуются комплексные ионы (HgCl3]f и [HgC]J\ Этим объясняется значительнее повышение растворимости хлорида ртута(П) в присутствии хлористого водорода хлорида аммония и других хлоридов. Как было отмечено ранее, кислая реакцег хлорида ртути(П) исчезает в присутствии хлорида щелочного металла. Из растворса хлорида ртути(П), содержащих хлориды щелочных металлов, при добавлении слабощелочного раствора (например, гидрокарбоната) основная соль не выпадает, кдз из чистого раствора. Это свойство учитывают при изготовлении таблеток сулеихг их готовят пе из чистого HgCl2, а из смеси равных частей HgCl;> и NaCl.
Бромид ртути(П) HgBr2 из водного раствора кристаллизуется в виде серебристиш листочков, а-из спиртового раствора в виде бесцветных ромбических призм. Он трудяг; чем хлорид, растворяется в воде (0,62 г HgBr2 в 100 г воды при 25°) и еще менее, *а-_ последний, диссоциирован в растворе. В остальном бромид чрезвычайно близок хло риду. Так, он образует продукты присоединения с HgO и двойные соли с друтпхп галогенидами
Криета.гл и чеекая решетка бромида ртути(П) занимает промежуточное цоложезг между слоистой и молекулярной решетками. Так же как в слоистых решетках (тггг брусита) хлорида кадмия и иодида ртуги(И), в решетке HgBr2 атомы металла образует: слой, по обе стороны от которого расположены слои, занятые атомами неметалле: Однако решетку бромида ртути(П) отличает то, что по одному атому брома из обете слоев связаны с одним определенным атомом ртути. Таким образом, каждый агль ртути всегда связан с двумя соседними атомами брома (по одному из каждого спех образуя молекулу HgBr2. (Расстояние Hg-<~-»Br = 2,48 А, расстояние от друге: ближайших четырех атомов Вт 3,23 Л.) В чисто слоистой решетке подобное соодипт-отдельных молекул невозможно. Например, н решетке Cdl2 (тина брусита) и C4S-...-(ср. стр. 482) каждый атом Cd одинаково удален от 3 атомов I или CI в каждом z.;' обоих слоев, а в решетке Hgl2 каждый атом Hg находится на одинаковом расстоягдл от двух атомов I каждого из слоев (в целим, следовательно, от 4 атомов I).
Иодид ртути(П). Hgl2 получают в виде красного порошка при раггл рании смеси ртути с иодом, увлажненной небольшим количеств г и спирта, либо осаждают из раствора нитрата ртути(П) небольшим честном иодида щелочного металла. Он кристаллизуется в тетрагонсх^ кой сингонии (слоистая решетка, Hg«—* I 2,78 А). В воде иодид ртути труднорастворим (примерно 4,4 мг в 100 г при 25°). Легче растворгиг г Hgl2 в спирте, особенно при нагревании. Он растворим также во мигге: других органических растворителях; в бензоле, например, раствориЕгг-’ч,, его значительно выше, чем в воде. При нагревании выше 126q образует другая модификация — ромбический иодид желтого цвета. Поспе.~~га образуется также в качестве промежуточного продукта при получен» иодида из раствора хлорида ртути под действием иодида щелстг г: металла. Желтая модификация образуется и довольно долго егг’ь-няется при быстром разбавлении водой спиртового раствора подцдз т" -ти. При тщательном наблюдении можно заметить, что в процессе ?.х-сталлизации иодида ртути(Н) из раствора всегда в первый момент в , Является желтая форма. Это явление находится в соответствии с ирахдг .ж ступеней Оствальда, согласно которому вещество, если оно имеет нж Деко модификаций, вначале часто образуется в наименее устойчивой с... дхз (ср. т. I, стр. 531).
Третью, еще более неустойчивую бесцветную модификацию впервые сгг;:<г» Таммап примерно в 1910 г. Она образуется при быстрой конденсации паров Hgbcs-ж чем за минуту превращается в красную модификацию.
Иодид ртути(П) в виде мази применяют при лечении некотордЕ явных заболеваний.
Иодид ртути(П) диссоциирует еще меньше, чем остальные гг‘гь-; ииды. Из его раствора не осаждаются ни иодид серебра под ДЕГ’ужш; нитрата серебра, ни окись или основная соль ртути при действии с- ь .чкл
Вторая группа периодической системы
503
Так же как хлорид и бромид, иодид очень легко образует двойные галогениды. При нагревании иодида ртути с разбавленной калиевой щелочью был получен желто-коричневый порошок продукта присоединения с окисью ртути состава HgI3-3HgO.
В качестве примера двойных иодидов можно привести иодид ртути и калия К2 [HglJ -2Н2О— светло-желтые кристаллы, хорошо растворимые в воде и спирте. Водный раствор этого соединения в присутствии щелочи является чувствительным реактивом для открытия и определения очень небольших количеств аммиака (реактив Несслера, ср. стр. 509 и т. I, стр. 672).
Двойной иодид ртути(II) с медью — красного цвета, а с серебром — желтого цвета даже при умеренном пагревании изменяет свою окраску; например, соль меди при 70’ становится шоколадно-коричневой. Поэтому эти соли используют в качестве «термопокрытии», позволяющих судить о степени нагрева деталей машин.
Фторид ртути(П), HgF3 безводный, получают нагреванием фторида ртути(1) в токе хлора при температуре 275° (Ruff). Температура плавления его 645°. О структуре решетки см. стр. 499. Из раствора окиси ртути(II) в водном растворе плавиковой кислоты выделяется дигидрат HgF2-2H2O в виде белой массы. Способностью кристаллизоваться с водой фторид отличается от других галогенидов ртути(И). В отличие от них он также подвергается более глубокому гидролизу и нормально диссоциирует по первой ступени. Двойные ссши фторида ртути(Ц) не известны.
Цианид и роданид
Цианид ртути(П), цианид ртути Hg(CN)2, получают действием водного раствора синильной кислоты на окись ртути или водного раствора цианида щелочного металла на соли ртути(Н), например сульфата ртутй(П):
HgO +2IICN =Hg(CN)2+Н2О,
HgSO4+2NaCN=Hg(CN)2 -H Na2SO4, вообще:
Hg"+2CN'=Hg(CN)s.
Если в качестве исходного продукта используют соединения одновалентной ртути, то также образуется цианид ртути(П), но с примесью металлической ртути (ср. стр. 493).
Диссоциация цианида ртути настолько мала, что уже очень иеболыпого количества ионов CN' достаточно, чтобы произошла реакция, соответствующая последнему уравнению. По этому Hg(CN)2 можно получить также нагреванием окиси ртути с водной суспензией берлинской лазури.
Цианид ртути образует бесцветные без запаха кристаллы в виде палочек. Он довольно легко растворим в воде (8,0 г в 100 г при 0°), в спирте — труднее. Поскольку эта соль почти пе диссоциирует, ртуть не осаждается из водного раствора едким кали или иодидом калия, по осаждается сероводородом.
В насыщенном водном растворе Hg(CN)2 растворяется еще значительное количество HgO с образованием соединения Hg(CN2)- HgO. Оно кристаллизуется в виде белых игл. Раствор имеет щелочную реакцию.
Будучи нагрето выше 320°, оно разлагается на ртуть и циан, частично полимеризующийся в дициан.
Цианид ртути весьма склонен к образованию двойных или комплексных цианидов. Большей частью опи соответствуют типам MI[Hg(CN)3] и MI[Hg(CN)i], однако образуются соединения и более сложного состава.
504
Глава 9
Известны многочисленные комплексные соединения со смешанными анионами, главным образом типа М1 [HgX(CN}2]. Кислотным радикалом X в таких соединениях может служить даже слабый комшгексообразова-тель, например ион NO3. В этом случае при растворении в воде комплексная соль почти полностью разлагается.
Цианид ртути лишен запаха, но имеет отвратительный вкус. Он чрезвычайно ядовит. Применяют его в терапии, а также в качестве дезинфицирующего средства. Гораздо большее применение, чем простая соль, находит упомянутое выше двойное соединение с окисью ртути. Смесью цианидов цинка и ртути переменного состава пропитывают перевязочный материал.
Роданид рт; тп(1) Hgg('SCN)2 выпадает в виде трудпорастворимого белого кристаллического осадги при взаимодействии раствора нитрата ртути(1) с роданидом калия.
Роданид ртути(П) Hg(SCN)2 образуется и вида белого кристаллического осадка при добавлении роданида щелочного металла к раствору иитра та ртути(Л). Он довольно трудно растворим (1 : 4440 при 25°). Значительно лучше роданид ртути растворяется в горячей подо, а также в Спирта. Из спиртового раствора он кристаллизуется в виде красивых иголочек. Электропроводность водного раствора ничтожно мала, следовательно, соль в нем почти недиссоциирована. При пагревапии роданид ртути(И) чрезвычайно сильно вспучивается («змея фараона»).
Из растворов, содержащих избыток ионов SCN’, кристаллизуются $ во Инне соли типа M^HgfSCNJj] и Mf[Hg(SCN)4], Первые, как правило, труднорастворимы, вторые растворяются очень легко. Была выделена также и свободная кислота H2Hg(SCN)i; она образует желтые кристаллы.
Ион fHg(SCN)4]" с тяжелыми металлами (Zn", Ni*’-, Со". Fe") образует труднорастворимые осадии. Кристаллизующаяся в форме белых и гл калиевая соль K2 [Hg(SCN)4J очень легко растворяется в воде. В растворе она обнаруживает типичную для одно-двухвалептного электролита молярную электропроводность и изменения ее при разведении (piO24 — Дзг = 20,7).
Нитраты и нитриты
Нитрат ртути(Т) Hg2(NO3)2 удобнее всего получать растворением при нагревании избытка металлической ртути в умеренно разбавленной азотной кислоте. Он образуется также при взаимодействии нитрата рту-ти(П) с металлической ртутью в водном растворе (ср. стр. 493). Из водного раствора кристаллизуется дигидрат Hga(NO3)2‘2H2O — бесцветные... короткие моноклинные палочки. На воздухе он выветривается, терян воду, а при 70° плавится с частичным разложепием. Нитрат ртутп(Ц очень легко растворяется в воде. Вследствие гидролиза раствор приобретает кислую реакцию. При разбавлении па холоду медленно, а при нагревании быстрее выпадает основная соль, например Hgs(OH)(NO3) — масса светло-лимонного цвета. В присутствии достаточного количества свободной азотной кислоты разложение при нагревании уменьшается.
Растворы нитрата ртути(1) являются сильными восстановителями, Для предотвращения частичного окисления кислородом воздуха в раствор добавляют свободную ртуть.
Нитрат ртути(1) — единственное соединение электрохимически одиова ленте ой ртути, которое образует типичные двойные соли, причем преимущественно с нитратами двухвалентных металлов. Опп отвечают составу 2Hg2(NO3)<2- М11:(К03)2.
Нитрат ртути(П) Hg(i\TO3)2 обычно получают растворением ртутп в избытке азотной кислоты. После упаривания раствора выделяются крупные, бесцветные, расплывающиеся кристаллы состава Hg(NO3)2-H2O..
Вторая группа периодической систем.ы
505
Нитрат ртути(П)’ служит исходным продуктом для получения большинства других соединений ртути.
Нитрат ртути(П) устойчив в растворе только в присутствии достаточного количества азотной кислоты, препятствующей гидролизу. В большом количестве воды оп полностью разлагается на окись ртути и азотную кислоту..
Нитриты. Ни тр u m р m I/; ан ( И Н g2 (N О 3) 2 об ра зу стоя при в за инод ей ста и и избытка ртути с разбавленной азотной кислотой (уд. вес 1,04) прп обычной температуре. По отношению к органическим соединениям он ведет себя подобно нитриту серебра.
Нитрит pmymu(II) Hg(MOa)2 образуется, по Даппым Рея (Ray), при взаимодействии хлорида ртути(1Т) е рассчитанным количеством шггрита серебра в водном растворе. При упаривании раствора в аксикаторе над серной кислотой образуются очень не устойчивые расплывающиеся игольчатые кристаллы. Более устойчивы двойные соли с нитритами щелочных митал.ию. Их удобнее всего получать действием раствора нитрита ртути(П) па растворы иптрптоп щелочпых металлов. Из таких солей известим, например, K[Hg(NO,)3] — светло-желтые кристаллы, Na2(Hg(NO2)4J — светло-желтые гигроскопичные^призмы, K3[Hg(NO2)s(OH2)) — желтые, ромбические кристаллы.
Прочие соли ртути
Сульфат ртути(Т) Hg2S04 образует бесцветные моноклинные призмы (уд. вес 7,12), трудно растворимые в воде и в разбавленной серной кислоте. Его получают осаждением из раствора нитрата ртути(1) разбавлеппой серной кислотой или анодным растворением ртути в разбавленной серной кислоте, а также нагреванием ртути с концентрированной серной кислотой. Водой оп постепенно гидролизуется с образованием еще более трудно растворимой зеленовато-желтого цвета основной соли. Hg2SO4 разлагается также на свету, приобретая серую окраску. Сульфат ртути(1) каталитически ускоряет окисление органических веществ дымящей сорной кислотой. В связи с этим его применяют в технике при получении фталевой кислоты из нафталина. О применении для определения азота по методу Кьельдаля см. стр. 490.
Сульфат ртути(П) HgSO4 получают многократным упариванием ртути с избытком концентрированной серной кислоты (предварительно прибавляя азотную кислоту), а также растворением окиси ртути в серной :с кислоте. Он кристаллизуется безводным, в виде белых листочков, группирующихся в звездочки. В присутствии небольшого количества воды образуется моногидрат HgSO4-H2O — бесцветные, ромбические палочки. Большим количеством воды оп разлагается до основного сульфата рту-ти(11). Последний получается также при нагревании раствора нитрата ртути(П) с сульфатом щелочного металла. Он был известен еще алхимику Василию Валентину. Парацельс назвал его Turpethum minerale. В чистом состоянии он имеет состав HgSO4-2H2O. Это порошок яркого лимонножелтого (уд. вес 6,41), а при нагревании красного цвета. В воде он растворяется довольно трудно. Нейтральный сульфат при умеренном нагревании тоже окрашивается сначала в желтый, а затем в красно-коричневый цвет. Прокаливание при температуре красного каления ведет к его разложению па Hg, SO2 и О2.
Сульфат ртути(П) находит применение в технике в качестве катализатора при получении ацетальдегида из ацетилена и воды (ср. т. I, стр. 477), Иногда его используют как исходный продукт для получения других соединений ртути.
506
Глава 9
Сульфат ртути(П) образует двойные соли с сульфатами щелочных металлов, например K2SO4-3HgSO4-2HaO. Он может присоединять также 1 и 2 молекулы НС1 или НВт.
Сульфит ртути(II) и еульфптомеркураты. Сульфит pmyniu(ll) HgSO3 выделяется в виде белого, очень быстро разлагающегося осадка при действии сульфита щелочного металла на раствор нитрата ртути(II). Чистым легче получается не простая сонь, а основной сульфит HgSOvTIgO.
В вентральных растворах сульфитов щелочных металлов наблюдается растворение окиси ртути(1[) за счет взаимодействия с гидросульфит-нояами, образующимися вследствие гидролиза:
HgO-)- 2HSOJ=[Hg(SO3)a]' 4- На0.
При упаривании растворов кристаллизуется прочная комплексная соль M|[Hg(SO3)2] (дисульфит оме р к ураты щелочных металлов). Прежнее ое название «сульфонат ртути и щелочных металлов». По-видимому, в таких соединениях между атомами серы в 8О|--грунпах и ртутью существует гомеополярпая связь. Эти соединения являются прочными комплексами, о чем свидетельствует то, что из их свежеприготовленных растворов ртуть пе осаждается даже такими реагентами, как едвяй кали, карбонат натрия, фосфат натрия и др. Одиако при стоянии, а особенно при нагревании, они разлагаются с выделением металлической ртути: [Hg(SO3)3)w = Hg -f- S02 + SOJ.
Карбонаты. Карбонат, pmymu(I) HgaCO3 выделяется в виде желтого осадка при действии карбоната щелочного металла на раствор соли ртути(1). Он легко разлагается с образованием Hg, HgO и СОЕ. Нейтральный карбонат ртути(11) до сих пор не известен. При добавлении карбоната или гидрокарбоната щелочного металла к раствору нитрата ртути(П) выпадают основные карбонаты, переменного состава.
Оксалат ртуги{11) HgC2O4— труднорастворимый белый порошок, находит практическое применение благодаря способности разлагаться под действием света. Его используют в фотометре Эбера. Раствор Эдера содержит 80 г (NH4)2C2O4 и 50 г HgClE в 3 а воды *. Б таком растворе оксалат находится в виде легкорастворимого двойного соединения с хлоридом ртуги(П). При освещении происходит следующая реакция:
2HgCla+(NH!J2G2O4=Hg2Cla+2CO2+2NH4Cl.
Количество образующейся каломели пропорционально количеству падающег-света (пока концентрацию реагирующих веществ можно считать постоянной). Следовательно, фотометр Эдера позволяет измерять количеств., света на аналитических весах.
Соединения ртути с азотом
Как уже было сказано, некоторые соединения ртути способны ez только присоединять аммиак, но даже замещать а нем водород. Наибсдг ярким примером продуктов присоединения с аммиаком является «плает^С белый преципитат», а продуктов замещения —«неплавкий белый преципитат» и основание Миллона*
Плавкий белый преципитат. При пропускании аммиака через растя” хлорида ртути(П), содержащий много хлорида аммония, выделяетг; белый кристаллический осадок. Благодаря способности этого ссагя. плавиться при нагревании без разложения, его назвали «плавкий преципитат». Аналитический состав этого соединения —
* Кая показал Ролофф (Rolcff), чувствительность к свету можно значдтег_т увеличить добавлением Hg(NO3)3.
Вторая, группа периодической система
507
Его можно получать также взаимодействием хлорида ртути с жидким аммиаком. Оно легко растворяется в разбавленной азотной, серной, а также в уксусной кислотах. Во всех случаях образуется смесь хлорида ртути(П) с аммонийной солью соответствующей кислоты. Водой плавкий белый преципитат разлагается с выделением аммиака. Это соединение можно рассматривать как хлорид диамми.нртути(11) [H3N —> Hg NH3]C12, что подтверждается рентгеноструктурными исследованиями.
Аналогичные соединения образуются и с органическими замещенными аминами, например пиридином, а также с гидразином. Соединение с гидразином, полученное из спиртово-зфпрного раствора, имеет состав GlaHg-N2H4. Следовательно, гидразин замещает две молекулы МН3, что характерно для координационных соединений. Помимо двух атомов хлора, ртуть может связывать и другие отрицательные радикалы. Например, остатки таких кислот, которые образуют с ртутью хорошо диссоциирующие соли двухвалентной ртути, правда,'при условии, что вместо аммиака становится гидразин или пиридин.
Неплавкий белый преципитат. Если аммиак добавлять к раствору хлорида ртути(П), который совсем не содержит хлорида аммония или только небольшие количества его, то в соответствии с уравнением:
HgCl2 + 2NH3==Hg(NH2)Cl +NH4C1
выпадает белый осадок состава HgNHBCl. Благодаря тому что при нагревании он, не плавясь, разлагается на хлорид ртути(1), аммиак и азот, его назвали «неплавким белым преципитатом1». В воде он нерастворим, но легко растворяется в разбавленных сильных кислотах (в присутствии небольшого количества хлорида натрия). Неплавкий белый преципитат представляет собой хлорид амидортутиСГГ) [Hg :— NHJCI.
Известны также его фтор- и бромпроизводные. Вместо амидогруппы NH2 может входить радикал гидразина, тогда образуется соединение [Hg—NH—NH—Hg]CI2.
Тонкая структура «белых преципитатов». Рентгеноструктурные исследования соединений [Hg(NH3)2]Cl2, [Hg(NH3)2]Br2, [HgfNH^lCln [Hg(NH2)]Br(Bijvoeb, 1936; Lipscomb, 1951; Rfldorff, 1952) показала,что по своей структуре они весьма близки между собой, а также хлориду или бромиду аммония. [Hg(NH3)2]Cl2 и [Hg(NH3)2]Br2 кристаллизуются в кубической сингонии и изотинны друг другу (а = 4,06 А или 4, 23 Л). Кристаллическую решетку [Hg(NH3)2]Br2 можпо представить себе как решетку a-[NH4]Br (т. I, стр. 850), в которой ионы NH| замещены молекулами NH3, -а среди них статистически распределены ионы Hg3+, так что образуются линейные комплексы [H3N Hg ч— NH3]a+. Если в a-[NH4]Br ионы NH| заместить группами NHa, а ионы Hga+ распределить среди, них таким образом, чтобы образовались бесконечно длинные цепи ,NHZ—Hg NH2—Hg NHa, получится структура
[Hg(NH2)]Br. От этого соединения, кристаллизующегося в кубической сингонии (а = 4,339 Л) ромбическая решетка хлорида [Hg(NH2)[Cl структурно отличается тем, что в ней цепи ориентируются параллельно друг другу, что не всегда имеет место в решетке бромида. Соединения [Hg(NHg)2]Br2 и [Hg(NH3)]Bг благодаря структурному родству способны образовывать твердые растворы. Получены также твердые растворы [Hg(NH3)2]Br2 с [NHJBt. Расстояние Hg-*—> N составляет у [Hg(NH3)2]Br2 2,11 Л, у [Hg(NH2)]Br 2,17 А;расстояние Hg«—>Вг равно 2,99 или 3,07 А.Из этого следует, что связь между Hg и N является атомной, а бром присутствует в виде ионов.
Основание Миллона. При действии водного раствора аммиака на желтую окись ртути получают микрокристаллический порошок светло-желтого цвета, практически нерастворимый в воде и в других растворителях. Уд. вес его 4,08, аналитический состав Hg2NHsO3. Это хорошо кристаллизующееся соединение обладает способностью образовывать с кислотами соли. Оно известно под названием основания Миллона. По
508
Глава 9
данным Гофмана (Hofmann, 1900), оно имеет структуру
НО—Hg\
>N< ОН
1но—HgZ н]
а ото соли, содержащие на одну молекулу меньше воды, имеют строение
Г /Нй\ ZH1
О< >N< X 1
• XHgZ “Hj ;
Соответствующее этим солям более бедное водой основание
выделяется при стоянии основания Миллона над едким кали в атмосфере аммиака. Это темно-желтый порошок, с удельным весом 7,42. При нагревании в атмосфере аммиака до 125° отщепляется еще одна молекула воды — образуется темно-коричневый, рыхлый, взрывчатый порошок, удельного веса 8,52, которому Гофман приписал формулу
Hg НОН Hg
Такое представление о строении основания Миллона и его производных подтверждается рентгенометрическими исследованиями (Lipscomb, 1951; Riidorff, 1952), а также тем фактом, что эти соединения получаются., как производные солей аммония. Однако они построены не из изолированных групп типа [OHg2NH2R, а образуют пространственную сетку, состоящую из Н^/Д-тетраэдров, подобно пространственным сеткам SiOa в кристобалите и тридимите Д'т. I, стр. 530 и 531); расстояние Hg *—Х,_ вероятно, составляет 2,07 А. Анионы и молекулы Н2О, по-видимому, неупорядоченно распределяются в пустотах такой сетки. Каждый Hg^A-тетраэдр имеет все 4 иона Hg2+ общими с соседними Нд4К-тетраэдра1пл: следовательно, на каждый ион №' приходится 4/а—2Нд-+-иона. Pchuv-ноструктурпые исследования позволяют, следовательно, припиепп-основанию Миллона формулу
м]0Я-2Н20 или проста [Hg2K]OH-2H2O, соответственно более бедное водой основание и производные от него ссгд (Hg2N)OH -Н20 и I HgWNJX - Н3О.
По данным Липскомба, указанные выше соединения, содержащие воду, кргл—-лизуются подобно ₽-кристобалит у в кубической сингонии. Такая же структура найл -, им для некоторых, соединений типа Hg2NX( = [Hg2N]X, где X = I, NO3, С1О4и соединения были получены из разбавленных аммиачных растворов по реакции
2Hg£ 4- 4NH3 + X'=[UggNjX 4- 2IIg + 3NH;.
Соединение {HgaN]Br кристаллизуется в гексагональной сингонии подобно трпдгзт— (Riidorff). Упомянутое выше соединение Hg2NOH, по-видимому, не cz.tt :*г обменивать ОН-группу на кислотные радикалы.
Все перечисленные соединения малоустойчивы. Основание Миллона разлит,.— г на свету, при растирании в ступке потрескивает. Соединение [Hg3N]OH- Н2О •-
чувствительно к свету, а соединение Hg2NOH при толчке или ударе, а также пул с; вании до 130° взрывается с оглушительным треском. При длительной ваашщдхх:”-.; и
Вторая группа периодической системы
509
ртути с влажным аммиаком при доступе кислорода воздуха образуются соединения, отличающиеся от [HgaN]OH'2H2O и [Hg2N]OH'H2O тем, что в них часть молекул воды замещена NII3 (Weber, 1953), Эти соединения чрезвычайно взрывчаты. Подавить их образование можпо растворением в ртути небольшого количества цинка, который связывает кислород воздуха. В отсутствие кислорода воздуха ртуть с аммиаком не реагирует.
Производные основа пи я Миллона соли (X — F, С1, Вт, I, CN, NOg, N03,1/2SO4, 1/2С03 и т. д.), помимо обработки основа пи я соответствующей кислотой (причем происходит не растворение, а только превращение), можно получать и непосредственно из солей ртути. II именно из тех солей, которые хорошо диссоциируют в растворе. В водном растворе такой соли при действии аммиака идет реакция
2Hg" 4- [NO3]' + H2O + 4NH3 = [Hg2N][NOs] НгО + 3fNHb] ‘ (14)
Малодисеоциирующие соли ртути(П) в подобных условиях образуют соединения, аналогичные «белым преципитатам1.'. Одпако при кипячении с водой или щелочью [уравнение (15) и (16)] они превращаются в соли основания Миллона. Последние образуются также при действии аммиака на основные соли ртути [уравнение (17)], а также аммиака и щелочи па комплексные галогениды ртути [уравнение (18)]:
2[Hg(NH3)2]Cl2+Н2О = [Hg2N]Cl • Н2О -f-3[ NHJCJ, (15)
Плавкий белый
преципитат
2[Hg — NHJC1 + Н2О =[Hg2K]Cl -Н2О + [NHJC1, (16)
Неплавкий белый
преципитат
HgC!2.IIgO-|-2NHi!t^[ng2N]Cl..H20 + [NH4]Cl, (17)
2[HcT.,1J"+\Hi3 + 3CiH' — [Hg2l\]J-H2O + 7I'-|-2HaO. (18)
На последней реакции основано открытие аммиака реактивом Несслера.
Алкильные соединения ртути. При взаимодействии хлорида ртути HgCj2 с ал кил галогенидами магпия, например СН3—Mg—-Cj, образуются алкилы ртути, например Hg(CH3)2 — диметилртутъ (уд. вес 3,07, т. кип. 95°). В качестве промежуточных продуктов получаются ал кил галогениды ртути. Алкилы ртути представляют собой бесцветные жидкости со слабым своеобразным запахом. Пары их чрезвычайно ядовиты. При обычной температуре они в отличие от алкилов цинка устойчивы на воздухе н по отношению к воде. Но несмотря па это, в общем они все же менее устойчивы, чем последние; металлический цинк вытесняет из пих ртуть с образованием алкилов цинка:
Hg(CH3)2-f- Zn—Zn(CH3)2 -f-Hg.
При взаимодействии с влажной окисью серебра алкилгалогениды ртути превращаются л гидроокиси алкилртути. Так из метилиодида ртути СН3—Hg—I (блестящие, нерастворимые в воде листочки, т. пл. 149°) получают гидроокись метилртути. СН3—Hg—ОН — тяжелую, в воде и спирте растворимую жида ость. Ее водный" раствор является сильным основанием и образует с кислотами соли.
Аналитические сведения. Аналитически ртуть можно открыть очень легко. При прокаливании в трубке соединений ртути с содой образуются капельки ртути в виде дистиллята серого цвета. Б этом нетрудно убедиться, рассматривая ого под микроскопом. Из разбавленных растворов ртуть можно выделить при помощи полос медной фольги, которые свертывают и помещают в трубки для прокаливания.
Присутствие паров ртути в воздухе обнаруживают следующим способом. Пропускают воздух через тампоп из чистой золотой фольги, который ’затем помещают в газоразрядную трубку, создают вакуум, нагревают и снимают спектр при высоком напряжении. Даже ничтожно малые следы ртути дают в спектре зеленую линию (?. = 546 тц), а при больших концентрациях еще и индиго-синюю (А. = 456 иц). Очень простой способ обнаружения паров ртути в воздухе предложен ПГтиттом [Stitt, Anal. Cfiem., 23, 1098 (1951)]. Этот способ основан на почернении полос фильтровальной бумаги, пропитанных селеном. Шток [Stock, Angew. Cbem., 44, 200 (1931)] разработал способ качественного и количественного определения очень небольших количеств ртути (менее 0,01 у) в растворах. С этой целью металлическую ртуть электро-
510
Глава, 9
яитически осаждают на медной проволоке, отгоняют в особой сиециаиьио изготовленной стеклянной трубке, в охлаждаемой капиллярной части которой Hg конденсируется, а затем производят микрометрическое определённо величины капель ртути. При определении стояв небольших количеств ртути особенно важно тщательно проверить все применяемые реактивы на содержание ртути и предохранять их от возможного загрязнения за счет атмосферы лаборатории.
В ходе систематического анализа электрохимически одновалентная ртуть при добавлении соляной кислоты выпадает в виде каломели и ее можно легко обнаружить по появлению черного окрашивания при добавлении аммиака. Двухвалентная ртуть выпадает при пропускании серо-водрода в разбавленный солянокислый раствор в виде черного сульфида HgS, который отличается от всех выпадающих совместно сульфидов тем, что он нерастворим как в желтом сульфиде аммония, так и в теплой умеренно разведенной азотной кислоте.
Если ртуть или сульфид ртути растворить в царской водке и после упаривали я смешать с нитратом кобальта и родаппдом калия (без сильного разведения), то образуются красивые томно-сипие иголочки двойного роданида ртути(П) и кобальта HgCo(SCN)t. Эта реакция настолько чувствительна, что в микроанализе ею можно определить даже 0,04 у ртути. Еще более чувствительной (лучше всего проводимой в виде капельной реакции) является реакция с дифепилкарбазоном (окрашивание от синего до фиолетового). Эту цветную реакцию мощно использовать также для количественного определения исключительно небольших количеств ртути (менее 0,05 у) колориметрическим или микроколориметряческим методом (Stock, 1928—1929).
При весовом анализе одновалентную ртуть осаждают в виде HgaCI2s а двухвалентную — в виде сульфида ртути HgS. Оба соединения после надлежащего высушивания можно взвешивать. Следует учесть, что ртутные растворы нельзя выпаривать с соляной кислотой, так как при этом могут улетучиться значительные количества ртути. Поэтому часто удобнее ртуть отделять электрохимическим методом в виде металла. Для этого целесообразно использовать небольшую колбу, в дно которой впаяна платиновая проволочка дм подсоединения катода и перед употреблением покрытая слоем ртути.
Увеличение веса колбы будет соответствовать количеству выделенной ртути.
Объемное определение ионов Hg.C возможно при осаждении ртути раствором Ks[Hg(SCN)i] с применением Fea(SO4)3 в качестве индикатора [Burriel-Marti F., Anal. chfm. Acta, 4, 344 (1950)].
Гл а в а 10
СЕМЕЙСТВО ЛАНТАНИДОВ
Порядковый номер Элемент Символ Атомный вес Валентность Порядковый помер Элемент Символ Атомный Дес Валентность
58 ЦериП Се 140,13 Ш, IV 1 65 Тербий ть 158,93 III, IV
59 Празеодим Рг 140,92 П, III, IV 66 Диспрозий Dy 162,51 III
60 Неодим Nd 144,27 ш 67 Гольмий Но 164,94 III
61 Прометий Pm 145 а Ш 1 68 Эрбий Ег 167,27 III
(исстаби- 1 69 Тулий То 168,94 in
ле л) i 70 Иттербий Yb 173,04 и, ш
62 Самарий Вт 150,35 II, HI I 71 лютеций® Lu 174,99 ш
63 Еврояня Ей 152,0 II, III
64 Гадолиний Gd 157,26 HI
а Для наиболее долгоживущего изотопа (стр. 530).
® Элемент лютеций называется также Кассиопеей (ср. стр, 526),
Общие сведения. Семейство лантанидов охватывает элементы, приведенные в таблице с порядковыми числами 58—71, По своему поведению они тесно примыкают к находящемуся в побочной подгруппе третьей группы лантану (порядковому номеру 57). Они занимают промежуточное положение между лантаном и его обоими аналогами: иттрием и скандием. Вместо с этими тремя элементами они образуют группу редкоземельных элементов *.
Из общей группы редких земель приведенные выше элементы .выделяются благодаря их положению в периодической системе, где они образуют особое семейство- это в свою очередь обусловлено характерной особенностью их атомного строения, что было отмечено уже в начале т. II: элементы семейства лантанидов отличаются тем, что в их атомах происходит заполнение 4/-оболочки. Как уже было отмечено, из этого можно заключить, что порядковый номер (заряд ядра) должен возрасти от первого до последнего элемента семейства лантанидов на 14 единиц, В соот
* По химическому поведению к группе редкоземельных металлов следовало бы отнести также более близкий аналог лантана — актиний. Однако обычно этого не делают, так как основное значение такого крайне редкого элемента определяется его радивактиснпетыо. Напротив, к группе редкоземельных металлов ранее относили также торий, который в большинстве случаев в природе встречается с ними. Однако торий следует рассматривать только как ближайшего родственника, по не как члена группы редкоземельных металлов.
512
Глава 10
ветствии с этим семейство лантанидов включает 14 элементов. Однако один из них, а именно элемент с порядковым номером 61, нестабилен и пе встречается в природе — он получен искусственно. Так же как ниже иода и ниже марганца в периодической системе внутри семейства лантанидов имелось пустое место, и было ото до тех мор, пока не научились искусственно получать элементы, которые не встречаются в природе.
Валентность. За исключением церия, все лантаниды обычно бывают в своих соединениях трехвалентными. Только церий, который в качестве первого элемента семейства занимает в ней особое положение, во многих соединениях бывает также и четырехвалентным. В виде исключения некоторые лантаниды могут образовывать соединения, в которых они проявляют другие валентности. Это—празеодим и тербий, которые, кроме трсхвалентных, могут бытв также и четыреявалентными (празеодим, вероятно, также и двухвалентным); самарий, европий и иттербий, кроме трехвалентпых, могут быть двухвалентными *.
Общая дал всех лантамидов валентность три -следующим образом объясняется па основании теории атомного строения. Как следует из спектрографических ланитах, в нейтральном атоме лантана 2 электрона находятся на tis-уровпе и 1 электрон. на 3d-уровне.
У элементов, следующих за лантаном, происходит последовательная застройка 4/-промежуточной оболочки, однако у всех два послед них электрона или по крайней мере один из них остаются расположенными на ба-уровне, и даже третий от конца. электрон расположен часто не па 4/-уровне, а па 5й-уровне (ср. табл. 1П приложения). Следовательно, часть лантанидов имеет на внешней оболочке такую же электронную конфигурацию, как и лантан (э^бя3). Вполне очевидно, что элементы, у которых □то имеет место, подобно лантану должны быть трехвалентными. Однако в отличие от лантана под 5d 6 s2-о б елочкой у них лежит непосредственно но оболочка инертного газа (5s25pe), а 4/-рболочка, из которой еще могут отщепляться электроны. Этим объясняется то, что у лантанидов наряду с третьей степенью окисления встречается еще также четвертая. Это, однако, имеет место лишь в отдельных случаях, так как в общем связь электронов па 4/-обол очках оказывается существенно прочнее, чем на 5е/-оболочках. Особую прочность связи на 4/-оболочке можно объяснить поведением церия. Это первый элемент в общем ряду, у которого заряд достигает такой величины, что для первого электрона, который, связывается с атомным остовом М4+, 4/-уровень лежит глубже, чем 5<?-уровень; связь этого электрона па 4/-обол очке настолько прочнее, чем связь остальных валентных электронов, что перезарядка, сопровождающая внедрение первого электрона па 4/-оболочКу (Со"" ->Со‘") происходит со значительным выделением энергии (ср. стр. 531), в то время как для перезарядки Се"’ —> Се", сопровождаемой вне древнем второго валентного электрона, требуется значительная затрата энергии (ср. стр. 514).
Исследование спектров показало, что не у всех лантанидов третий от конца электрон находится на 5d-уровне. Часто он расположен на 4/-уровне. Однако прочность связи этого электрона на 4/-уровне очень мало отличается от прочности связи его на бй-уровне. Это следует как раз из того, что третий от конца электрон располагается то па if-, то на 5<i-уровне. Следовательно, этот электрон даже тогда легко отщепляется вместе с обоими бк-электропами, когда оп является 4/-элеКтр он ом. Поэтому даже в таксх случае третья степень окисления является нормальной. То, что наряду с этим иногда имеет место вторая степень окисления, объясняется особо слабой связью обоих 6с-электронов.
Способность некоторых лантанидов проявлять в соединениях валентности четыре или два закономерно связана с расположением этих элементов внутри семейстта.. Это отчетливо видно из рис. 55, Там, так же как ина рис. 28, стр. 152, т. I, число электронов, имеющееся в нейтральных атомах и ионах, зависит от порядкового номера. Есле перенести соображения, изложенные на стр. 151—153 т. I, па приведенный здзх= рисунок, то можпо прийти к заключению, что в семействе лантанидов имеется, очевидно, три электронные системы, которые отличаются сравнительно более высез.чг стабильностью, чем остальные. Это прежде всего электронные системы, когерх
* Это относится к валентностям в твердых солеобразных соединениях. Об зовапии ионов лаптанпдов(И) в растворах см. стр. 515.
Семейство лантанидов
513
имеются в ионах La3+ nlu3+. Первая соответствует оболочке ксенона с совершенно незастроенной 4/-промежуточной оболочкой, вторая — оболочке ксенона с полностью застроенной ^-промежуточной оболочкой. Правда, вследствие сравнительно прочной связи 4/-электронов за лантаном но следует целый ряд элементов (как за скандием и иттрием), которые при отщеплении электронов могут образовывать атомные остовы
76
74 -
72 ~
70 -
68 -
1 65-64-
I 62-
О 8 60-& 58-
56-
54 -о
о о
54 56 58 60 62 64 66 68 70 72 74 76
1 Сз । La1 Рт! Рт1 Ен 1 Tb । Но' Ta I Lu I Та 1 Re Хе Во Се Nd Sm Gd Dtj Er Yb Hf W Os Порядковый номер
Рис. 55. Число электронов в атомах к ионах лантанидов и в соседних с ними элементах,
Q число Электронов, имеющихся. в нгйтролъпых атомах; • числе алектронов в заряженных атомах, проявляющих валентность; • неу* стойчивые валентные состояния.
с конфигурацией предшествующего инертного газа. Однако вслед за лантаном имеется по крайней мере еще один элемент, который в состоянии это сделать, а именно церий (ср. также Стр. 519 и с л.). У следующего элемента — празеодима — при образовании соединений в исключительных случаях может происходить отщепление двух 4/-элёкт-ропов *. Однако у последующих элементов энергия решетки не может более возместить работу, требуемую для отщепления более чем одного 4/-электрона. То же наблюдается в концесемейства лантанидов: преимущество числа электронов, имеющееся у полностью образованной 4/-оболочки, приводит здесь к тому, что иттербий особо прочно удерживает один из трех своих валентных электронов или, после того как его оторвут, снова пытается его присоединить. Этим обусловлено то, что у данного элемента при известных обстоятельствах может оказаться большее сродство к образованию соединения, в котором оп двухвалентен, чем соединения, в котором он трехвалентеи. Следовательно, в этих случаях образуются соединения, в которых оп двухвалентен. Следует отметить теперь, что в середине семейства лантанидов также часто возникает определенная электронная конфигурация иона гадолиния. Следующий за гадолинием элемент — тербий при отщеплении 4 электронов образует атомный остов с тем Же числом электронов, что и у иона Gd!i+. Предшествующий гадолипию европий образует такой же остов при отщеплении 2 электронов. Валентность предшествующего европию самария также определяется его положением но отношению к гадолинию: самарий прояв-
* Празеодим (порядковое число 59), кроме Ss-электронов, содержит, вероятно, только 4/-электроны и пе имеет 56-электронов, То же относится к элементам ^Nd, ellJm, егЗш, (jjEu, 6ЙТ11 и 70Yb(cp, табл. III приложения).
33 Г. Геми
514
Глава 10
л нет двухвалентность, при этом число его электронов по крайней мере приближается к числу электронов в ионе гадолиния Gd3*.
Ион еадолииия занимает поэтому особое положение в семействе лантанидов', он (хотя и в значительно метшей мере) соответствует особому положению инертных газов в общей системе элементов. Это особое положение иона гадолиния в семействе лантанидов, на которое впервые указал Клемм (Klemm, 1929)помимо особенностей изменения валентностей, проявляется также еще в изменения других свойств, например спектров поглощения (см. стр. 518 и сл.) и магнитных свойств трехвалентных ионов.
Рис. 56. Восстановительный потенциал трехвалентных ионов Лантанидов (кривая /). потенциал выделения двухвалентных ионов (кривая II) и общий потенциал разрядки трех валентных ионов (кривая III).
Значения ординат, относящихся к кривым I и II, нанесены слева, а значения, относящиеся к кривой III,— справа.
Это удается объяснить на основе атомной теории. А именно магнитные моменты nczxz лантанидов удается вывести из того, что при заполнении 4/-уровня каждый из esrz подуровней, обозначаемых различными магнитными квантовыми числами т (ср. ?.. U стр. 422)., на которые делится 4 (-уровень, заполняется сначала одним электродах только когда все имеющиеся /«-подуровни «наполовину» заполнены, провсхсдг™ внедрение в них второго электрона (с противоположным спином). Следов ателье гадолцнпй, как седьмой элемент в ряду лантанидов, отличается тем, что в ионе Go-’’ gee без исключения имеющиеся т-подуровни заполнены «наполовину». Из хода патгт-циала ионизации в рядах В — Ne, Al — Ат и Ga — Кг (см. т. I, стр. 38, риг,. е видно, что такое «половинное заполнение» m-нодуровтгей (как это имеет место в рядах у атомов N, Р и As) ведет к упрочению электронной связи в атомных оболочдтг То же," следовательно, можно ожидать для иона Gd3+ *.
В водном растворе соединения лантанидов (II) в строгом смысле нес.тапп-гу-так как все ноны лантанидов (II) разлагают воду с выделением водорода. Правда, -Жени о происходит с большей или меньшей задержкой. У соединений еврвпшг 1
* Па основании непосредственных измерений или спектральных данных г ; значений энергии ионизации Ms+ —> М1+ в семействе лантанидов еще пе пзггззт;-: Однако с большой вероятностью удается предсказать, что кривая, которая ns-jss. зависимость энергии ионизации от порядкового номера, л случае Gd5* обпаруЕццд -если не пик, то по крайней мере излом (так же как кривая In — Хе па рпс.. 4 т.Л е ‘Д’ чае Sb).
Семейство л/штаиидпе
515
эта задержка так велика, что их раствори при обычной температуре практически устойчивы. По данным Иоддака (Noddack, 1937), все лантаниды при разрядке их трех-валеитпых ионов до металлов могут существовать в водном растворе в промежуточном состоянии в форме двухвалентных ионов. Под да к обнаружил ато путем съемки полярографических кривых с применением ртутно-капельного электрода. На рис, 56 нанесены измеренные Поддан ом значения восстановительного потенциала (М"' —> М") и потенциала выделения (М” Мамял1г). Разность этих значений, т. е- расстояние между соответствующими точками па кривых 1 и II, является мерой относительной стабильности различных иопоп лантанидов в двухвалентном состоянии. Из рисунка видно, что те лантаниды, относительно которых известно, что они могут существовать в соединениях в двухвалентном состоянии (Eu, Yb и Sin), также ив водном растворе в форме двухвалентных ионов существенно стабильнее; чем остальные лантапиды, у которых восстановительный потенциал и потенциал разрядки очень мало различаются (на 0,065—0,145 а). Если исключить Ен, Yb и Яш, то наибольшая разница между восстановительным потенциалом и потенциалом разрядки наблюдается у гадолиния (0,145 в). Следовательно, последний, находясь в виде электролитического иона в водном растворе, легче восстанавливается из трехпалептпого до двухвалентного состояния, чем большинство других попов лантанидов. Это, одиако, но противоречит представлению, что свободны и Отт Gd3+ отличается большей стабильностью по сравнению с остальными ионами лантапидов(Ш), ибо вследствие влияния тепл от гидратации на свободную энергию образования электролитических ионов их потенциалы перезарядки и выделения изменяются пе всегда одинаково с изменением потенциалов ионизации свободных ионон и атомов (ср. т. I, стр. 181 ц сл,). Впрочем, особое место гадолиния в ряду электро литических попов лантанидов проявляется также, если сравнить значение энергий, требуемых в совокупности для разрядки трехвалеитиых ионов. Они получаются (эв/ион], если к восстановительному потенциалу трехвалентных ионов прибавить удвоенную величину потенциала разрядки двухвалентного иона [ср. т. I, стр. 167, уравнение (За)]. Связывающая нх кривая (III на рис. 56) имеет для гадолиния отчетливый излом.
Основность гидроокисей*. Все гидроокиси лантанидов имеют ярко выраженный основной характер-, они находятся в этом отношении ближе к гидроокисям щелочноземельных металлов, чем гидроокись алюминия. Одиако гидроокиси, т. о. гидратированные окислы лантанидов существенно монее растворимы, чем гидроокиси щелочноземельных металлов, и поэтому в противоположность тем полностью осаждаются щелочами далее в присутствии солей аммония. При добавлении по каплям очень разбавленного раствора щелочи или аммиака происходит последовательное осаждение: (Sc”'), Lu”’, Yb‘”, Tu”', Er"’, Ho‘", Dy*", Tb'", Sm'”, Gd*", Eu’", (Y"‘), Nd'”, Pr*“, Ce'", (La’") (для сравнения в скобках приведены ионы редкоземельных элементов, относящиеся к побочной подгруппе III группы). Несмотря на то что таким путем нельзя достигнуть полного разделения различных веществ, все же удается наблюдать (особенно если подобное фракционированное осаждение повторять несколько раз), что наступает частичное разделение, соответствующее указанной последовательности. Полученный таким образом ряд соответствует ряду с возрастающими значениями произведений растворимости и, следовательно, с возрастающей основностью гидроокисей. Отсюда следует, что гидроокиси лантанидов по своей основности располагаются между гидроокисями скандия и лантана. Наиболее основная среди гидроокисей лантанидов гидроокись церия(Ш) очень мало отличается по своей растворимости (и основности) от гидроокиси лантана. Наименее оенбвпая гидроокись лютеция приближается к гидроокиси скандия, в сравнении с которой она, правда, еще сильно оси овна, так что скандий может быть довольно легко отделен от остальных редкоземельных металлов путем фракционированного осаждения аммиаком.
* Под основностью гидроокисей (Basizitat) автор подразумевает силу оспова-еиё —Прим. ред.
33*
516
Глава. 16
Имеются также еще и другие критерии, на основании которых окисям лантанидов можно расположить по возрастающей основности, например на основании различной способности нитратов разлагаться при нагревании. Чем более основна гидроокись, лежащая в основе нитрата, тем oil устойчивее в общем случае. Этим и другими аналогичными методам путем повторного их применения можно разделить редкие земли с достаточно отличающееся основностью. Поэтому базирующиеся на различной основности методы разделения имели большое значение для открытия редких земель. Однако если теперь потребуется как можно точнее расположить отдельные члены но их основности, не прибегая к разделений, то для соответствующих измерений более подходящими являются другие методы, которые используют земли, уже выделенные в чистое состоянии. Такие измерения были проведены Джемсом (James, 1914 и 1921) и Еадресом (Endres, 1932). Первый измерял количества иода, освобождаемые различными сульфатами в водном растворе пз иодид-иодатиой смеси; он также определяя скорость, с которой сульфатные растворы при температуре кипения выделяли двуокись углерода из карбоната натрия. Таким образом он получил следующий ряде понижающейся
Р н с. 57. Зависимость ионпого радиуса От порядкового номера в ряду лантанидов.
основностью, в котором опять для сравнения приведены металлы земель, припадлежг-Щйе к побочной Подгруппе III группы: (La), Се, Рг, Nd, Sm, Gd, Th, Dy, (Y), £>,. Tu, Yb, - . . (Sc), Таким образом, внутри семейства лантанидов основность пад^г= с возрастанием порядковых номеров, причем точно в той же последовательности.
Это тесно связано с уменьшением ионных радиусов, происходящим в соответстЕ— с рис. 57, точно в той же последовательности. Чем Меньше радиус положительпщ: трохвалентногоиона, тем прочпее должны связываться по закону Кулона ионы гидроксила. I? свод» очередь уменьшение ионных радиусов, называемое по Гольдшмидту «лантанидным сжатием.», обусловлено возрастанием притяжения, действующ—': на окружающие ядро электроны, в связи е ростом заряда ядра. В то время как образование новой внешней оболочки, т. е, застройка электроналш уровня с более высслгхд квантовыми числами, имеет следствием (скачкообразный) рост атомного радпущ в семействе лантанидов, где все элементы принимают электроны на «одну промежущ-пую оболочку», происходит постоянное уменьшение атомного радиуса. Это сад-т к тому, что последние лантаниды обладают даже меньшими атомными и нснеыгт радиусами, чем иттрий, имеющий гораздо более низкий заряд ядра и, следователь’” содержащий значительно меньше электронов. Этим объясняется то, что иттрий по csr^s свойствам, зависящим от ионного радиусами прежде всего по основности, оммапс:;:. срейк лвшнаииЩиз.
Ендрес непосредственно определил отношение произведений раствариилп гидроокисей. Он установил параллельное изменение основности и ионных ращщг Только в случае иттрия было найдено явное отклонение, поскольку для пщреещд г иттрия оказалось несколько большее произведение растворимости, чем для гепц?-.—: г диспрозия, в то время как в соответствии с рис. 57 ионный радиус иттрия еж;:::щ < меньше, чем ионный радиус диспрозия.
Семейство лантанидов
517
Окислы и соли. Окислы лантанидов, как и окислы скандия, иттрия и лантаиа, в воде практически нерастворимы, однако они обладают ярко выраженным основным характером. Как уже отмечалось, основность возрастает с возрастанием порядкового номера так, что окисел последнего элемента семейства лантанидов — окись лютеция — по своей основности лежит примерно посредине между Y2O3 и Sc2O3.
Полуторные окислы элемеитсп, следующих непосредственно за лантаном, как и La2O3> кристаллизуются в гексагональной слоистой решетке (стр. 55, рис. 15), Длины ребер элементарных ячеек составляют
La2O3 СеаО3 Pr2O3 Nd2O3
(2 = 3,94 3,88 3,86 3,85 A
£* — 6.13 ti, 06 8,01 6,02 A
Остальные полуторные окислы, как Sc203 и Y203 (тип Sc2O3 см. стр. 55 и сл.), образтют кубическую решетку с длинами ребер, приведенными в табл. 53 (Zacliariasen, 1928;'Klemm, 1939).
Таблица 53
Длины ребер элементарной ячейки а кубической модификации полуторных окйслов и Кажущиеся радиусы г трехвалентных иопов редкоземельных элементен (Л)
Ho. Ьа5О3 GC0O3 Pr*O;i Nd-зОз Sm2O3
a 9,81 10,801 11,40 j 7 10,99 11,078 10,928 10,864
r 0,83 1,06 1,25 — — 1,17 1,14 1,12
Gd2O3 та3о3 ПО2ОЗ Егй o3 TjqOa YbaO3 Lu3O3
a 10,818 10,71 10,672 10,60 10,526 10,476 10,437 10,396
r 1,11 1,08 1,07 1,06 1,04 1,02 1,00 0,99
Кроме указанных двух типов решетки, существует еще третий, который встречается отчасти наряду с кубической (как форма, стабильная при более высокой температуре), отчасти с гексагональной (как форма, стабильная прп более низкой температуре). При подходящих условиях (например, при осторожном прокаливании нитрата) удается также получить Ьа2О3 и У(12О3 в форме кубической модификации (Loliberg, 1935). Се2О3 и Рг2О3 можно'также закристаллизовать по тину ScaO3 (Klemm, 1939); однако Се203 вследствие легкой окисляемостц нельзя получить в этой форме достаточно ч петым.
В противоположность тем редким землям, которые относятся к побочной подгруппе III группы, окислы лантанидов большей частью окрашены. Растворы обнаруживают в этих случаях характеристичные абсорбционные спектры, отличающиеся сравнительно острыми полосами (ср. табл. 54).
Ионы Се"’, Gd’“, Yb'" и Ltt‘“ бесцветны; другие ионы более шли менее интенсивно окрашены. Церий образует бесцветный ион только в трехвалентном состоянии; четы р е хвал епт лып церий дает в растворе соединения с окраской от оранжевой до итттепсивпо-краспой. Однако двуокись церпя почти бесцветна. Также и другие лаптапнды, образующие окрашенные ионы, имеют белые окислы. Отраженный
518
Глава 76
Таблица 54
Спектры поглощения лаитапидов в водной растворе
[Цифры в таблице соответствуют длинам воли (.ир.). Если границы полос не указаны, эти цифры соответствуют середине полосы или положению самого сильного максимума]
Церий
Празеодим
Неодим
Самарий
Европии
Гадолиний Тербий Диспрозий
Гольмий
Эрбий
Тулий Иттербии Лютеций
Полосы поглощения в видимой области отсутствуют (сильное поглощение в ультрафиолете)
5S7 589 481,9 (острая) 469 (острая) 444 (широкая) 730 087,7 678,6 637 629 саз 594—562 .534—498 480,2 475,5 469,1 461 (диффузная) 433 427,2 418,2 500 499,5 489,2 487—472 464 451 443 (Диффузная) 418 416 415 402 зэо,5 374,6 362 536 525 465,6 465,1 464,7 394,3 385,3 380,9 376,6 374,0 361,7
Полосы поглощения в видимой области отсутствуют 573 523 437,5 370,7—375,2 369,4
753 479—468 453,4 450,1 427,4 398 388 (широкая) 365 641 550—553 485,2 480 473 468 458—443 422 417 з&0 386 361
В67 653 S43 549 5,41,3 523 521 491,5 487,1 453,4 449,7 442,2 407 405 379,4 364 359 356 699 682 658,3 464,2
Полосы поглощения в видимой области отсутствуют й » 0 » » »
от окрашенных окислов свет при спектроскопическом наблюдении обнаруживает те же характеристические абсорбционные полосы, какие наблюдаются в проходящей свете для растворов солей.
Расширение исследований абсорбционного спектра на ультрафиолетовую область покавало (Praudtl, 1934), что в семействе лантанидов абсорбционные полосы закономерно сдвигаются с возрастанием порядкового номера. От празеодима к гадолинию полссх сдвигаются от красной области в ультрафиолетовую, а от гадолиния к тулию вповь сдвигаются из ультрафиолетовой области в красную. Наряду с этими имеются полосы, которые не смещаются или смещаются лишь несущественно, ио которые об—-руживаютту особенность, что их интенсивность постоянно уменьшается от празеодим к европию, у гадолиния опи совсем исчезают, а затем вновь появляются у еле дугах— элементов. По-видимому, по своей способности поглотать свет ионы лантанидов (Кб симметрично группируются вокруг иона гадолиния. Это проявляется также в окрссх ионов:
а7Еа“' (бесцветный)
ssCo"' (бесцветный)
5ЭРг""‘ (желто-зеленый)
60Nd"* (красно-фиолетовый)
61₽т'” (розовый)
62^И1''’ (желтый)
йдЕц • (почти бесцветный)
e4Gil'‘‘ (бесцветный)
ОбТЬ"' (почти бесцветный)
(;(Цу ‘ ‘ (бледао-желто-зепеный)
6т11о’ ’ ’ (коричневато-желтый)
yjEr-” (розовый)
eoTir - (бледно-зеленый)
(бесцветный)
t^Ltt •' (бесцветный)
Семейство лантанидов
519
Характерное различие между лантанидами и редкоземельными элементами побочной подгруппы III группы выражается в их магнитных свойствах. В то время как сояп скандия, иттрия и лантана диамагнитны, лантаниды (кроме лютеция) обнаруживают в своих соединениях (в которых опи трехвалентпы) парамагнетизм (ср. г. I, табл. 60). Некоторые лантаниды очепь сильно парамагнитны и по своей удельной магнитной восприимчивости сильно превосходят парамагнитные соли железа, кобальта, никеля и марганца.
Соли лантанидов, как и соли остальных редкоземельных элементов, кристаллизуются чаще всего с водой. Хлориды, нитраты и сульфаты в воде легкорастворимы. Труднорастворимы оксалаты, фториды, карбонаты и фосфаты. Полученные восстановлением сульфатов сильно окрашенные сульфиды при кипячении разлагаются водой.
Большинство солеи редкоземельных элементов образуют с соответствующими солями щелочных металлов и аммония, а часто также и с солями двухвалентных металлов двойные соли, среди которых многие обладают замечательной кристаллизационной способностью.
Многие из этих двойных солей, и прежде всего двойные нитраты (например, двойной нитрат аммопия и двойной нитрат магния), двойные сульфаты, двойные оксалаты и двойные карбонаты, оказываются весьма удобными Для разделения редких земель путем фракционированной кристаллизации. фракционированную кристаллизацию двойного нитрата аммопия использовал уже Менделеев Для отделения лантана от «дидима» (ср. стр. 525). Ауэр фон Вельсбах (Ап о г von Welsbach) использовал двойные нитраты аммония при разделении дидима на неодим и празеодим, осуществленном им впервые методом фракционированной кристаллизации.
Ранее упоминалось, что первый элемент семейства лантанидов, церий, занимает в этом семействе особое положение. А именно соединения четырехвалентного церия совсем выпадают из ряда типичных для лантанидов соединений. Напротив, они тесно примыкают к соединениям тория. Поэтому рацее церий часто рассматривали как аналог тория и отводили ему то место в периодической системе, которое в настоящее время занимает гафнии.
При сравнении свойств этих соединений четырехвалеитный церий оказывается ближе к торию, чем гафний. Это вполне понятно на основании теории строения атомов. Прежде всего с точки зрения этой теории оказалось совершенно естественным, что первый элемент в ряду лантанидов, кроме трехвалентного состояния, может быть и в четырехвалентпом. Этот элемент содержит только 1 электрон на 4/-уровне, отно- ' сительно которого сразу можно предположить, что он сравнительно не очень прочно связан (например, по аналогии с электроном на ЗФуровне скандия; ср. стр. 13). Однако, поскольку атом церии отдает этот электрон, т. с. поскольку он переходит в состояние положительного четырехвалентного иона*, он теряет признаки, характерные для лантанидов. Следовательно, Се1+ является для семейства лантанидов «в известной мере чужеродным элементом» (Hevesy), Если бы в ряду элементов не происходило застройки 4/-оболочки, то в побочной подгруппе IV группы в качестве аналога тория стоял бы не элемент со свойствами гафния, а элемент, который в электроположительном чотырехвалентном состоянии вел бы себя точно так ясе, как электроположительный четырехвалентпый церий. Он стоял бы к торию по своим свойствам несомненно ближе, чем гафпий. Ибо благодаря «лантанидному сжатию» гафнии но своему атомному и ионному радиусу и, следовательно, по всем своим свойствам, связанным с величиной этих радиусов, стоит ненормально близко к цирконию и соответственно ненормально далеко от тория. Гафний отличается также от тория тем, что в виде положительного четырехвалентного нона он пе обладает, как последний, конфигурацией инертного газа. В то же время церий в этом отношении сходен с торием.
* Перед тем как оторвется (прочный) электрон, связанный с 4/-уровнсм, должны, конечно, сначала отщепиться три более рыхло связанных электрона.
520
Глава 10
Свойства металлов. В форме металлов лантаниды можно получить тем же способом, что и лантан. Приготовляемый часто в технике «смешанный металл» получают из смеси редких земель, обычио цериевой группы. Цериевый смешанный металл содержит в среднем примерно 50% Се, 40% La, 3% других металлов редких земель и 7% Fe. Он «пирофорен» (т. е. опилки воспламеняются и сгорают с выделением света и пламени), и поэтому он пригоден для изготовления кремней (для карманных зажигалок). Для этих целей к нему добавляют еще немного железа, иначе он слишком мягок. Образование искр при трении смешанного металла и его сплавов с железом о рифленую сталь основано па легкой воспламеняемости и высокой теплоте егорапия редкоземельных металлов.
Для церия н цериевого смешанного металла температура воспламенения в чистом кислороде лежит около 150°; для лантана ока равна примерно 450’. Теплоты образования каждого моля окислов равны
для LaaO3 СеОа PrsO3 PrO3 Nd2O3 Sm203
539 233 439 231 435 430 ккал
Следовательно, они лежат иногда существенно выше, чем теплота образования окиси алюминия (402,9 ккал). Высокие значения тепло? образования окислов соответствуют сильно электроположительному характеру металлов редких земель, который проявляется также в их высоких потенциалах выделения (см. рис. 56, стр. 514),
Лантаниды легкоплавки: точка плавления церия лежит, по данный Билли (ВШу, 1931), при 815°.
Примерно двадцать лет назад из лантанидов в металлическом состоянии были известны только церий, неодим и празеодим. В 1935 г. Урбана (Urbain) присоединил к ним гадолиний. Позднее в небольших количествах Клемм (1937) приготовил в металлическом состоянии всё лантаниды и исследовал структуру их решеток, а также их магнитные свойства. Клемм использовал для их получения восстановление хлоридов щелочными металлами, которое он проводил в отсутствие воздуха в стеклянное трубке при умеренном пагревании. По окончании восстановления избыточный щелочной металл отгоняли. Отделения от щелочных хлоридов» образующихся При взаимодействии, не проводили, так как они не мешали измерениям. В табл. 55 приведены структуры решеток и значения плотностей, полученные из параметров элементарных ячеек.
Структура решеток лантанидов
Таблица 55
Элемент Тип решетка Длины ребер элементарной . ячейки, А Плотность Элс-i мсят Тип решетки Длины ребер элементарной ячейке, А Плсг-
а-Се Г ек car он а дт>-и латной ша я уда-копка тО о CM g «Р Q5 ео' 1! К g. у 6,78 | Gd Гексагональная плотнейшая упаковка а=3,636, с=5,783 7 , Sj>TL
|3-Се Кубическая грин (.'центрированная а=5,161 6,810 - Tb . ! То же а =3,601, с = 5,694 8,3^ >4
а-Рг Гексагональная плотнейшая ула-- ковка а=3,672, с=5,918 6,776 i Dy » > а=3,590, «=5,648 8,4=
Семейство лантанидов
521
Продолжение табл. 55
Элемент Тип решетки Дли ны ребер элеме н-тарной , ячейки.А Плотность Элемент Тип решетки Длины ребер элементарной ячейки, А- Плотность
₽-Рт Кубическая грансцентрированная а = 5,161, 6,805 Но Гексагональная плотнейшая упаковка а=3,577, с=5,616 8,764
Nd Гексагональная плотнейшая упаковка <1 = 3,658, с =5,900 7,001 Ег То же а =3,559, с=5,587 9,164
Pm — — Ти & & <1=3,537, с== 5, ииб 9,346
Sm Ромбоэдрическая S а II II о о со съ 7,54 Yb Кубическая гранецентрированная «=5,468 7,010
Eu Кубическая объемно-цептриро-ваяная а = 4,590 5,244 Lu Гексагональная плотнейшая упаковка я=3,503, с = 5,551 9,740-
Мандатные измерения показали, что все лантаниды в металлическом состоянии, парамагнитны. У металлов от Еп до Та ниже определенной температуры («точка Кюри», ср. т. I) парамагнетизм переходит в ферромагнетизм. Ярко выражено это, однако, только у Gd. Ниже приведены температуры Кюри
для Hu Gd Tb Dy Ио Er Ти
15 302 205 150 — 40 10° К
Следовательно, опи закономерно зависят от порядкового номера. Особое положение гадолиния в семействе лантана, как показывают эти данные, подтверждается также и магнитными свойствами.
На рис. 58 приведены атомные объемы лантанидов в зависимости от порядковых номеров. Элементы европий, иттербий и самарий, которые отличаются от остальных лантанидов своей способностью выступать в соединениях в двухвалентном состоянии (см. рис. 56 ыа стр. 514), в металлическом состоянии также значительно отклоняются от остальных лантанидов своими особенно большими атомными объемами. Как видно-из рисунка, два из названных вначале элемента по атомным объемам являются аналогами не лантана, а бария. Фактически из магнитных свойств следует, что решетин европия и иттербия, как и бария, состоят из двукратнопо дожито л ьно заряженных ионов. Самарий, который существенно труднее переходит в двухвалентное состояние, чем европий и иттербий, по своему атомному объему также значительно меньше, чем последние, отклоняется от остальных лантанидов.
Если бы все решетки лантанидов, как и лантана, состояли из трехкратпо-за ряженных ионов, то следовало бы ожидать для кривой атомных объемов примерно такого хода, который показан па рис. 58 начинающейся от лантала пунктирной кривой. Се отклоняется от этой кривой в противоположную сторону по сравнению с Еп и Yb. В соответствии с этим магнитные измерения показывают в его металлической решетке присутствие ионов Се4+, Правда, при обычной температуре ионы Сез+ отчетливо преобладают над Се44-. Поэтому для церия отклонение от равномерной кривой много меньше, чем для Еп и Yb. Еще меньше эти отклонения для Рг и ТЪ. Можно поэтому предположить, что они имеют ту же причину, что и в случае Се. Рг и ТЬ объединяет с Се способность выступать в соединениях в четырехвалентном состоянии, хотя эта способность у пих значительно менее выражена. Замечательно, как глубоко отражается у лантанидов изменение химических свойств в ходе кривой атомных объемов.
522
Глава .10
Если отвлечься от различий, следующих из того, что некоторые лантаниды могут проявлять другие степени окисления, помимо 3, то, как показывающих химические свойства, они ведут себя совершенно аналогично лантану. С водородом они образуют гидриды того же типа, что и гидрид лантана (ср. гл. 2), а с азотом соединяются с образованием нитридов.
Порядковый номер
Рис. 58. Атомные объемы лантанидов и соседних с ними элементов.
Jj’ 1 Теплоты образования некоторых простых соединений лантанидов 'привед^к в табл. '56. Теплоты образования окйслов лантанидов уже были указаны выше.
Обзор поведения, лантанидов по отношению к другим металлам (насколько это исследовано) приведен в табл. 57.
Помимо первых двух элементов ряда лантанидов, в таблицу введен также лагжым. В результате сравнения оказывается, что аналогия между Интернета л л пчесшигд соединениями лантанидов и соединениями лантана очень значительна. ОбразуетгЕ но только большое число соединений аналогичного состава, по и эти соединения съедают также одинаковыми точками плавления. Помимо приведенных в таблице, лазгсг образует еще некоторые другие интерметаллические соединения и прежде всего LaGa LaGa3, LaPb31 La3Ni (т. пл.515°), La3Ni, LaNi (т. пл. 685°) и ЬаМц (т. пл. Вероятно, соответствующие соединения образуют также Се, Рт и другие лаптггтцдк.
Семейство лантанидов
523
Таблица 56
Теплоты образования соединений .чантанндоп(111) (ввал/г-зка)
Се 1>г Nd Sm Gd Dy Но Ег Tit Lu
Хлорпды 86,6 84,8 80,3? 81,7 79,2* 77,6 77,3 76,3 75,9
Йодиды 54,5 53,8 52,8 51,8 49,2 48,1 47,3 46,7 45,9 44,4
Сульфиды — — 43,8 — — с — —► — — —
Нитриды 26,0 — — — — — — — —
Гетдрады 21,1 — — — — —- — —
* Для модификации, устойчивой при обычной температуру.
Одпако эти системы еще пе исследованы. Приводимый в статьях состав соединений Т1 с Се и Рг основан по-видимому, только на аналогии и нуждается еще в экспериментальной проверке. Ибо, как свидетельствуют данные табл. 57, несмотря на хорошие в целом совпадения, в отдельных случаях между соответствующими системами появляются различия. Помимо соединений, найденных термическим анализом и приведенных в таблице, лантан образует с магнием Mg,La. Это соединение обнаружил Лавес рентгенометрически [Laves F., Natiirwiss., 31, 96 (1931)]. Оно кристаллизуется по типу MgCu»(cp. стр. 44) с а = 8,77 А.
С кремнием лаптапидтя образуют соединения типа MSi2. Соединения первых элементов подгруппы лантанидов являются изотилами LaSL. В ряду LaSi2 — CeSia — PrSi2 — NdSij — SmSio происходит регулярное возрастание рентгенометрически установленных плотностей (от 5,41 до 6,26). В этом, так же как и в случае окислов (стр. 517), отчетливо проявляется «лантанидное сжатие» (Вганег, 1952).
Металлоподобные соединения лантаниды образуют также с бором. Они имеют состав MBfi и являются изотипами СаВ0 и LaBe (см. т. I, стр. 362; ThBfi имеет подобную Же структуру). Эти соединения окрашены в сине-стальной цвет, в то время как бориды щелочноземельных металлов имеют цвета от блестящего черного до черно-коричневого. Как все соединения с такой структурой решетки, они обладают значительной металлической проводимостью.
С элементами главной подгруппы V группы лантаниды образуют (исследованы Се, Рг и Nd) среди прочих соединений соединения состава MX со структурой NaCL С Zn a Cd также образуются соединения состава MX, однако со структурой типа CsI (landelli, 1936 и сл.).
Пять лантанидов слабо радиоактивны. Это лантан (который претерпевает превращение при поглощении электрона), неодим (р-излучение), самарий (а-излучение), гадолиний (а-излучение) и лютеций (р-излучение). Их радиоактивность основана на содержании (большей частью незначительном) в этом элементе нестабильного изотопа (подробнее см. стр. 576 и сл.). Атомные ядра 14"Sm и 153Gd, на распаде которых основана радиоактивность самария и гадолиния, наряду с атомным ядром изотопа гафния являются единственными встречающимися в природе * средними (повесу) ядрами, у которых установлен распад с а-излучеиием.
Исторические сведений- О двух первых этапах истории открытия редких земель — о почти одновременном открытии иттриевой и цериевой земель и затем примерно через 40 лет о расщеплении Мозандером каждой из этих земель на три новые земли — уже сообщалось в гл. 2 этой книги. То, что наблюдалось в начальной стадии изучения редких земель, характерно также и для дальнейшего: с совершенствованием методов исследования и разделения удавалось все дальше расщеплять вещества,
* Известно несколько искусственно полученных изотопов лантанидов, которые распадаются с а-излучением.
Образование соединений и твердых растворов лантаном и лантанидами с другими металлами (Обозначения те ясе, что и в табл. 9, стр. 6(5)
Таблица 57
Mg Al TI Si Sn Bl Fe Cu ле Ли Zn Cd Hg
La ж. < по та. О .\TgqU* 662’ М1?-Д.,а* 7 ij6J MgLa MgiXa 50 3° тв. О LaAl* 869’ LaAl 2 1424’ LaAL 1222’ та. 0 La3Tl 1200’ LaTl 1182’ LaTl3 1096’ LaSi2 тв. 0 La2Sn 1420’ La2Sn3 1188° LaSna 1150’ LaSn3 ? LaBi -- та. 0 LaCu* 551’ LaCu2 834’ LaCu-}* 793’ LaCu4 9 02’ тв. О LaAg 885’ LaAg3* 8 64’ LaAg3 9 55’ тв. О La2Au* 665’ LaAu 1300’ LaAu2 • 1214’ LaAu3 1204°' La Zu LaCd LaHg/t
Се тв. О MggCe* ' «22° Mg3Ce 780’ MgCe 740’ ТВ, О Се3А1 014’ Се2Л1* 598° CeAl* 780’ СеА1г 1465’ CeAl4 1250’ Ce2Tl? CeTl? CeTl3? тв. O? CeSi 1530’ CeSi2 тв. 0 CesSn UOO’ Ce2Sn3 1165’ CeSn3 , 1140’ CeSn3 тв. 0 Ce3Bi* 1400’ Co4Bi3 1630’ CeBl* 152 5’ GeB L S68° TB. <00 CeFe-,* 773’ Ce2Fo5* 1090’ тв. 0 CeCu* 515’ CeCu4 820’“ CeCn4* 780“ CeCufi 940° — — Ce4Zn? Ce2ZB? CeZn CeCd тв. O? CeHg4
Рг тв. О 798° MgPr 767° TB. 0 PrAJ* 906’ PM1, 1442’ PrAU 1244’ Pr3TP PrTP PrTl3? PrSLj PrSn3 PrBi тв. 0 PrCu* 503’ PrCu2 Bit’ РгСн4* 824’ PrCuB 962’ тв. 0 PrAg 928° PrAg2* 878’ PrAg3 950’ тв, 0 Pr^Au* 710’ РгАн 1350’ РгЛи2 1210’ PrAuA 1200° PrZn PrCd PrHg4
Таблица S3
Схематический обзор истории открытия элёмептов_ редких земель
Иттриевая земля
(Гадолин, 1794)
. Иттрий
Эрбий (Моз ан дер,
Тербий
1843)
Иттербий (Марипьяк, 1878)
Скандий
(Нильсов, 1879)
Эрбий
Иттербий
~ Кассиопей (Лютеций)
(Ауэр фов Вельс бах, 1905—ISO7; Урбейн, 19 07)
------(11ео-) Эрбий
Гольмий
(Клеве, 1879)
----—Тулий
Гольмий
-Диспрозий
(Ленок де Буайодрап, 1886)
Самарий
Цериевая земля (Клапрот, Берцелиус, 1803): Церий __ Дидим (Мозандер, 1839 сл,) — Лантан Самарий (Ленок до Вуабодрак, 1879) ~ Гадолиний (Марипьяк, 1880) —Дидим Е вропий (Демарса, 1896) — Неодим (Ауар фон Вельсбах, 1885)
Празеодим
526
Глава 10
ранее рассматриваемые как однородные, на новые (как это несколько схематично представлено в табл. 58); в конце концов их число достигло числа, известного в настоящее время, а именно 17 редких земель (включая не встречающуюся в природе окись прометия). Три из этих 17 редких земель, относящиеся к побочной подгруппе III группы, уже сравнительно давно были открыты и выделены в чистом состоянии, в то время как некоторые из земель семейства лантанидов были обнаружены только на исходе прошлого столетия.
Прошло примерно столько же времени, сколько прошло между открытием иттриевой и цериевой вел-ель н их расщеплением на окислы (в общем шести металлов — шптрия, арб «.я, тербия, церия, дидима и лантана), пока пе наступил новый период в истории открытия редких земель (около 1880 г.) За это время Бунзен разработал метод спектрального анализа, ставший важным вспомогательным средством для идентификации и испытания чистоты вещеотв. Кроме того, были обнаружены также новые, легко доступные минералы, из которых можно было получить большие количества редких земель. Таким минералом был, например, самарскит, из которого Лек ок де Буабодрап (Loccoq de Boisbandran) выделил в 1879 г. самарий (и его окисел); уже за год до этого Делафоптен (Delalont&ine) спектрально доказал неоднородность полученной из этого минерала «окиси дидима» (путем сравнения ее спектра поглощения со спектром поглощения препарата дидима другого происхождения). В 1880 г. Мараньян (Marignac) выделил из самарскита, кроме окиси самария, еще один новый окисел — окись гадолиния. Еще за два года до этого Мариньяку удалось выделить из эрбиевой земли, рассматриваемой до того как окисел однородного вещества, бесцветный окисел (в противоположность розовой окиси эрбия), который по своим свойствам стоял между скислом арбия и иттрия и поэтому был назван окиси ом иттербия. В 1879 г. Нильсой (Nilson) выделил новый окисел из старой эрбиевой земли, окись скандия (ср. стр. 52), В том же году Клеве (Cleve) удалось выделить оттуда еще два новых окисла: окислы тулия и гольмия. Последний впоследствии также оказался неоднородным. Оп содержал еще диспрозий характеристический абсорбционный спектр которого был в 1886 г. получен впервые Лекок де Буабодраном.
Ауэру фон Всльсбаху (Auer von Welsbach, 1885) удалось расщепить дидим па две части: неодим и празеодим. Как. уже упоминалось, ему удалось это осуществить путем фракционированной кристаллизации двойных нитратов — редких земель и аммония Метод фракционированной кристаллизации до этого мало разрабатывался. Впоследствии ок приобрел чрезвычайно большое значение для аналитического, а также препаративного разделения редких земель. Примерно в то же время, когда Ауэр фон Всльсбах разрабатывал метод фракционированной кристаллизации, началось применение минералов, дающих редкие земли, и прежде всего монацитового песка (см. Стр. НИЛ в производство газокализьных колпачков.
Дальнейшие годы в основном характеризуются получением и подробным изучением свойств безукоризненно чистых редких земель, нежели открытием новых представителей этой группы. Фактически оставалось даже не слишком много из них открыть, ибо, как известно в настоящее время, иэ 16 стабильных элементов рассматриваемся группы (включая скандий, иттрий и лантан) в 1890 г. были известны уже 14. В 1895 г. Демарса (Dem area у) удалось отделить от самария еще один элемент с характеристическим абсорбционным спектром. Впоследствии оп получил название европий. В 1905 т» Ауэр фон Вельсбах обнаружил спектральную неоднородность иттербия, получепвстс Мариньяком, и выделил из него (1907 г.) методом фракционирован!той кристаллпзац=2 новый элемент (в форме окиси), практически чистый, и назвал его кассиопеем. Псптг в то же время этот же элемент был обнаружен У рб айн ом (Urbana), который дал егг название лютеций.
После открытия в 1913 г. Мозли рентгеновской спектрографии оказалось можным точно установить число еще пе найденных редкоземельных металлов на ocuczz-нин порядковых номеров, рассчитанных из рентгеновских спектров. Оказалось, сг между барием (порядковый помер -Io) н танталом (порядковый номер 73) лежпт dczz-16 элементов и что все известны, кроме двух с порядковыми номерами 61 и 72„ Ez основании теории атомного строения оказалось, однако, чтоэле.мент 72 не более к металлам редких земель; как было отмечено н гл. 3, он был вскоре найден -и Костером в минералах циркония. Как уже упоминалось, не встречаюпщйса □ zzzz роде зяемент 61 (прометий) был получен искусственно (п результате атщшнк ~--вращений). Впервые он был обнаружен в продуктах расщепления ура в a fen. гт-в 19-15 г. (Coryell, Marinsky, Gleadonin). В настоящее время известно утке —~ число изотопов этого элемента (см, стр. 635), однако все они нести
Семейство лантанидов
527
Классификация редких земель. Первые элементы ряда лантанидов по свойствам своих соединений, прежде всего по их растворимости, стоят особенно близко к лантану, в то время как остальные, начиная примерно с европия, ближе стоят к иттрию. Благодаря этому все редкие земли делят на две группы, из которых одна, включающая в качестве технически важной составной ча’сти церий, обозначается как группа цериевых земель, а другая, к которой относится иттрий, называется группой иттриевых земель. Группу иттриевых земель удается еще далее разде-лить на основании более или менее тесной взаимосвязи ее отдельных членов, так что в конце концов приходят к следующей классификации:
I. Цериевые земли II. Иттриевые земли
1. Окись 2. Тербиевые 3. Эрбиевые 4, Иттербие- 5. Окись
Окислы: лантана церия празеодима неодима самария иттрил земли Окисли: европия гадолиния тербия земли Окисли: диспрозия Т0Л1ДП1 я эрбия тулия вые земли Окислы: иттербия лютеции скандия
Этой более ранней классификации, основанной по существу на аналитико-препаративном поведении соединений редких земель, в 1929 г. была противопоставлена классификация Клемма, который исходит из представлений об атомном строении.
С точки зрения атомного строения оказывается прежде всего, что элементы скандий, иттрий и лантан составляют особую группу (или часть особой группы, а именно побочной подгруппы III группы периодической системы). Они противостоят остальным редкоземельным элементам, которые объединяются в отдельную группу, а именно семейство лантанидов. Эта классификация также уже давно общепринята (при этом, конечно, не следует забывать тесные связи между первыми тремя элементами, особенно иттрием и лантаном, и лантанидами, которые выражаются даже в их названиях). На основании особого места, которое гадолиний занимает внутри ряда лантанидов (ср. стр. 514), их подразделяют теперь в соответствии с классификацией Клемма на две подгруппы, каждая из которых содержит элементы, расположенные по их порядковым номерам, причем одна включает элементы от церия до гадолиния, другая — от тербия до лютеция.
Если рассмотреть с этой точки зрения зависимость свойств лантанидов и их ионов (соответственно их соединений) от порядкового номера, то обнаруживаются три различные закономерности.
1. Ярко выраженная периодичность свойств проявляется в том, что изменение свойств в первой подгруппе повторяется во второй подгруппе аналогичным образом. •
Такая периодичность проявляется, например, о изменениях валентностей. Как показывают дальнейшие сопоставления, изменение валентностей, которые проявляются в изолированных соединениях, во второй подгруппе по существу такое же,
528
Глава IQ
как в первой, с той лишь разницей, что способность к проявлению других валентностей, помимо (III), вообще говоря, во второй подгруппе выражена слабее, чем в первой.
I подгруппа ' Валентность i ЭДСМОЦТОВ III, IV II, Ш, IV - III III n, Ш II, III Ш
1 Элемент 58 Се saPf eoNd eiPm G3Sm esEu c«Gd
подгруппа г Валентность । элементов III, IV III III HI III II, Ш III
с Элемент 65ть ввпУ 67Н° esEr etiTu 70Vb 7iLu
Ход кривых I и 11 на рис. 56 (стр. 514) также свидетельствует об отчетливой периодичности, равным образом как и кривая атомных объемов на рис. 58 (стр. 522). Это легко заметить, если сравнить ее с кривой атомных объемов, относящейся ко всем элементам (рис. 2, стр. 35 т. I).
К свойствам, периодически изменяющимся внутри семейства лантанидов, относится, кроме того, магнетизм, причем как магнитные свойства соединений, построенных из ионов, так и магнетизм самих металлов.
2. В семействе лантанидов мощно также наблюдать симметричное изменение свойств, т. е. у элементов, стоящих слева и справа от гадолиния, с ростом порядкового номера свойства изменяются в противоположном направлении.
При этой зависимости свойств от порядкового номера аналогичными оказываются пе те элементы, которые в приведенных выше рядах стоят Друг под другом, а те, которые одинаково удалены от середины семейства лантапидов. Опи оказываются стоящими друг пад другом, если принять способ паплсания, который выбран па стр. 518 для выражения закономерного изменения окраски растворов солей лаитаиидов с изменением порядкового номера. Это изменение окраски, соответственно сдвиг поясе поглощения света с ростом порядкового номера, является типичным примером «симметрично протекающего*- изменения свойств в семействе лантапидов.
3. Далее, имеются свойства, которые изменяются внутри семейства лантанидов непрерывно в одном направлении. Однако в этом случае на кривых, которые показывают зависимость рассматриваемых свойств от порядкового номера, у гадолиния часто наблюдается отчетливый излом.
Примерамн таких кривых является кривая ионпых радиусов (рис. 57, стр. 516J и кривая общих энергий разрядки т рехвал ентных каков (кривая III на рис. 56, стр.514) „ На этих кривых проявляется особенность гадолиния. Она также отчетливо проявляется на кривых тепл от растворения редноземельиых металлов в соляной кислоте и теплят образования безводных хлоридов (Klemm, Bommer, 1941).
В то время как аналитико-препаративпое поведение редких земель, т. е. нх -способность разделяться, основанная на различии растворимости соединений, главным образом * определяется одним свойством ионов, а именно величиной их радиуса., атомное строение в общем является определяющим для химических свойств (а такжг для физических). Поэтому можно считать, что классификация, исходящая из атомного строения, позволяет сделать более точный обзор свойств и значительно облегчает понимание их зависимости, чем старая классификация. Старая классификация редких земель и клеммонская систематика относятся друг к другу примерно так же, как классификация металлов по группам аналитического процесса разделения относится к классификации периодической системы. Отсюда следует одновременно, что для практических целей старая классификация по-прежнему сохраняет свое значение..
* Наряду с радиусами определенную роль играет также различная сила поляризующего действия. Для отделения церия используют его способность переходить св четыреквалентное состояние; это исключительное и важное свойство церия не учитывается в старой групповой классификации.
Семейство лантанидов
529
Распространение в природе. Нахождение редких земель в природе в первую очередь определлетел свойствами, используемыми в аналитцко-препаративной систематике, и вытекающим из лих различным родством отдельных редких земель. (Поэтому эту систематику называют также «минералогической».) Несмотря на то что более или менее постоянно все редкие земли находятся вместе, имеются минералы, в которых преобладают элементы одцрй и той же аналитико-препаративной группы, а в других минералах — другой группы. На этом бегло основано старое разделение на цериевые земли, которые главным образом содержатся в церите, и иттриевые земли, которые главным образом содержатся в иттербите (гадолините), хотя уже Берцелиус установил (1814), что в иттербите в значительно меньшем количестве содержится также церий.
Важнейшими минералами, которые содержат преимущественно цериевые земли, помимо церита (водусодержащий силикат Металлов цериевых земель), являются ортит (двойной силикат металлов цериевых земель, алюминия и кальция) и монацит (фосфат металлов цериевых земель с содержанием в среднем 5% окиси тории).
Минералами, которые содержат преимущественно иттриевые земли, кроме гадолинита или иттербита (основной силикат, окрашенный в черный цвет железом), являются ксенотим (аналогичный монациту фосфат металлов иттриевых земель), иттротанталмт, самарскит и фергузонит (изоморфная смесь ниобатов и танталитов, содержащая металлы иттриевой группы), а также еексенит (содержащий, кроме ниобиевой и танталовой кислот, также титановую кислоту).
Кроме собственно минералов редких земель, имеются также еще многочисленные минералы, которые содержат редкие земли В очень небольших количествах. Это прежде всего фосфаты, арсенаты(У), ванадаты(У), молибдаты и вольфраматы кальция, стронция, железа и свинца. Количественное соотношение отдельных редких земель в них отчасти другое, чем в ранее названных минералах. Так, в некоторых из них имеются смеси редких земель, в которых преобладают окислы 8ш, Ей и (id, которые в собственных минералах содержатся только в незначительных количествах.
Получение и применение. Для технического получения редких земель в качестве исходного материала обычно используют монацитовый песок. В переработанном состоянии оп содержит в среднем наряду с 5% окиси тория примерно 60% цериевых земель и 3—4% иттриевых земель. Первоначально его перерабатывали из-за наличия в нем тория. При получении окиси тория (стр. 100) или нитрата редкие земли отбрасывали как побочный продукт. В настоящее время редкие земли, и прежде всего церий и его соединения, находят техническое применение в столь большом количестве, что они теперь стали главным продуктом переработки монацита.
Обычно редкие земли сначала выделяют из кислого раствора вместе с торием в виде оксалатов. Путем дальнейшей обработки осадка теплым насыщенным раствором оксалата аммония торий удается перевести -в раствор в виде оксалатотората аммония. Остающиеся нерастворснными оксалаты металлов редких земель сначала переводят, чаще всего прокаливанием, в окислы и затем приготовляют из них путем растворения в кислотах и добавления соответствующих солей подходящие двойные соли, которые затем методом фракционированной кристаллизации разделяют на отдельные земли (если это требуется для технического применения).
В то время как церий вследствие особого поведения его четырехвалентных соединений сравнительно легко отделяется от других редких земель, приготовление в чистом виде последних путем фракционированного осаждения или кристаллизации (почти исключительно используемым ранее) очень затруднительно из-за незначительных различий в растворимости их соединений. Некоторые позднее предложенные способы использовали другие методики, напримор фракционированное распределение между двумя песмешпвагощимися жидкостями ( Fischer, 1937). Для разделения и приготовления в чистом виде больших количеств редких земель используют часто в настоящее время ионообменные смолы (ср. т. I, стр. 81). Европий (хуже иттербий) удается, согласно методу Брунла (Brukl, 1936), сравнительно быстро отделить от других редких земель путем электролитического восстановления в присутствии сульф ат-ионов. При этом 3'1 Г, Реми
530
Глава 10
сульфат европия(П), как труднорастворнмое соединение, осаждается. Если европий присутствует в растворе только в незначительной концентрации, так что произведепиа растворимости EuS04 но достигается, то осаждение можно вызвать добавлением SrCl3 в процессе электролиза. При этом выпадающий SrSO4 захватывает EuSO4 с образованием твердого раствора. После прокаливания: (при этом образуется ЕнгО3) легко удаетСя экстрагировать европий изосадна в виде хлорида. Электролиз можно повторить без добавления SrCls, так как SrSO4, хотя сравнительно и: в малых количествах, захватывает в свою решетку сульфаты редких земель(Ш), например Sm3(SO4)3 и Gd^SO^
В то время как для изготовления калильных колпачков требуется чистая окись церия или по мепыпей мере такая, которая свободна от окрашенных окйслов, для многих других технических целей достаточно приготовления смесей, которые содержат редкие земли в естественном соотношении, в каком они присутствуют в монаците. Полученные из этой смеси соединения называют «цериевыми соединениями». Смесь окйслов используют для обесцвечивания стекла.
Нитраты в смеси с порошком магния применяют в качестве порошка для вспышек. Фториды используют в углях дуговых ламп; пропитанные ими угли дают особо яркий чисто белый свет. Фитили углей для дуговых киноламп содержат до 60% фторида церия. Оксалаты применяют в фармацевтических целях (перемезин). Смесь металлов —«цериевый смешанный металл»,— приготавливаемую электролизом расплава хлоридов (подобно тому как это описано в случае лантана), применяют в качестве легирующей добавки или как раскисляющее средство в сплавах легких металлов. Цериевое железо (ср. стр. 520) используют в качестве «пирофорного сплава» при изготовлении кремней для бензиновых зажигалок н т. п.
Чистые соединения церия, празеодима и неодима применяют для окрашивания стекол и эмалей, а такя;е в производстве фарфора. Кроме того, соединения церия используют для травления шерсти при изготовлении фетровых шляп, а также для аналитических целей (стр. 533)„ Соль неодима З-сульфонзоникотиновой кислоты находит терапевтическое применение как средство, препятствующее свертыванию крови (тромбо-дим). Смесь окйслов празеодима и неодима служит под названием «окпсе дидима» для изготовления неофаиового стекла.
Не встречающийся в природе прометий получен только путем атомных л рев ращений. Различные ядерные реакции, в результате которых он образуется, принапри» на стр. 635. Получить его наиболее долгоживущий изотоп I46Pm (период полураспада примерно 30 лет) можно, например, путем облучения самария (в форме Sm2O3) нейтронами. От неизменивше го ся самария его можно отделить при помощи ионообменных смол. Однако более доступен, чем I49Pm, образующийся при расщеплепии; урана 14'Рщ (период полураспада 2,6 года). Атомный реактор мощностью 1000 кет производит ежедневно примерно 16 мг 14;Рш,
ВАЖНЕЙШИЕ СОЕДИНЕНИЯ РЕДКОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
Следующее ниже короткое описание важнейших соединений редких земель базируется па старой («аналитико-препаративной» или «минералогической») классификации. Применяемое при этом подразделение ни небольшие подгруппы облегчает обзор. Важнейшие зависимости, лежащее в основе новой клейновской систематики или вытекающие из нее, утес были рассмотрены в общей части.
Семейство лантанидов
531
I. Цериевые земли
Первый элемент подгруппы цериевых земель, лантан, уже рассмотрен в гл. 2 этой книги. Технически важнейшим элементом является церий. Относительно остальных (празеодима, неодима и самария) следует отметить главным образом, что они в противоположность двум названным выше образуют окрашенные трехвалентные ионы с характеристическими абсорбционными спектрами.
Празеодим получил имя по зеленой окраске своих солей (ngaoaios—луково-зеленый). Соли неодима — фиолетовые, окисел — светло-голубой. Соли самария —‘ топазово-желтые. Электрической дуге самарий сообщает розово-красную окраску.
Празеодим и самарий относятся к таким элементам редких земель, которые в определенных соединениях могут проявлять также и другие валентные состояния^ кроме (III).
Если соли празеодима и летучих кислот прокаливать на воздухе, то можно получить черно-коричневый окисел состава Pr6Olt. Сплавлением с хлоратом натрия его удается перевести в черную двуокись РгО2. Последняя получается также нри нагревании РгеОц в атмосфере кислорода под давлением, в то время как при прокаливании в токе водорода образуется желтая Рг2О3. Двуокись кристаллизуется по тину плавикового шпата (а = 5,36 А). Окись Pr60fl имеет очень близкую к пей структуру решетки (а =5,53 А). При обработке кислотами эти более высокие окислы претерпевают восстановление, в результате которого образуются соли нраэеодима(Ш).
По данным Прапдтла (1938), если смесь Рг,О3 и YaO3 прокаливать в атмосфере кислорода под давлением, то присоединяется гораздо больше кислорода, чем это соответствует образованию РгО2, По этим данным можно было заключить, что празеодим способен окисляться до еще более высоких степеней, чем до четырехвалентного состояния. Однако, согласно Маршу (Матвей, 1946), этого не происходит. Последний смог установить только образованно РгО2 и принял, что наблюдаемое Прандтлом сильное возрастание веса могло обусловливаться поглощением паров воды крайне гигроскопичной y2O3. Напротив, согласно Маршу, празеодим при подходящих условиях, можно восстанавливать до двухвалентного состояния; например, он нашел, что нри сильном нагревании смеси РгаО3 и Th62 в токе водорода желто-зеленый Рг203 переходит в черный РгО.
Нагреванием безводного хлорида самария SmCl3 в токе водорода получают красно-коричневый дихлорид SmCl2. С водой он тотчас взаимодействует с выделением водорода, и самарий при этом спова переходит в трехвалентное состояние. Получен также дииодид самария Sml2.
Промежуточное положение между неодимом и самарием занимает по своему химическому поведению прометий. Соединения РшС13 и Pm(NO3)3 легко растворимы в воде. Трудно растворимы Рт2О3, Рт(ОН)3 и Рта(СаО4)3-10Н20. Соединения прометия окрашены в розовый цвет, за исключением хлорида, который в безводном состоянии желтый. В растворе хлорид прометия тоже окрашен в розовый цвет. Спектр поглощения водпых растворов солей ^прометия аналогичен: спектру растворов солей неодима, с той лишь разницей, что у последнего отдельные полосы сдвинуты на 8 тц в фиолетовую область/
СО ЕДИНЕНИЯ,ЦЕРИЯ
Соединения треявалентного церия подобны соединениям остальных элементов из группы редких земель, особенно соединениям лантана. Как и те, они бесцветны. Соли устойчивы на воздухе. Однако гидрат окиси церия(Ш) проявляет большую склонность к окислению. Поэтому на воздухе он быстро принимает темную окраску и наконец переходит в желтый гидрат окиси церия(1У).
Соли четыреявалентного церия в против оно ложность солям трехвалентного церия в большинстве малоустойчивы. В водном растворе они достаточно сильно гидролизованы, так что при разведении растворов иногда выпадают основные соли. Большинство солей церия(1У) легко
34*
532
Глава 10
претерпевают восстановление до солей церия(Ш). Более устойчивыми, чем простые соли церия(Г^), являются двойные соли, среди которых двойной нитрат MHCc(NO3)Gf отличается хорошей кристаллизационной способностью. Среди простых солей церия(1У) одной ин наиболее устойчивых в растворе является сульфат. Хлорид известен только в форме темно-красного раствора комплексной кислоты НаСеС18 и в форме солей этой кислоты. Он легко переходит с выделением хлора в бесцветный хлорид Церия(Ш).
Потенциал перезарядки Сл"’Кее"" при тех же условиях, при каких он дай для веществ, приведенных в т. I, табл. 112 (стр. 820), составляет -pi,61 в по отпошешпо к нормальному водородному электроду. Следовательно, соли церия(1У) еще более сильные окислители, чем соли зоЛота(Ш) (ср. т. П, стр. 442 и сл.). Поэтому, как и последние, они восстанавливаются уже слабыми восстановителями, например солями железа(П) или даже перекисью водорода.
Окислы п гидраты окислов. Прокаливанием солей церия и летучих кислот па воздухе или сжиганием металла получают вкось церия СеО2 в виде почти белого го слабой желтизной порошка, который при нагревании становится лимонно-желтым. Он кристаллизуется по типу плавикового шпата (а = 5,40 А) и обладает уд. весом 7,2, В соляной и азотной кислотах прокаленная окись церия практически нерастворима. Очень концентрированная серная кислота переводит его в сульфат церия) IV), в то время как менее концентрированная серная кислота действует на него, вызывая частичное восстановление и выделение кислорода. В присутствяивосстанавителей двуокись церия растворяется также в соляной и азотной кислотах. При умеренно сильном прокаливании в токе водорода двуокись церия перехопит в темно-синий промежуточный окисел состава Се4О; *. Если этот окисел нагревать на воздухе, то он снова превращается в двуокись. Растворами щелочей или аммиака из растворов солей церия(1у) осаждается гидрат окиси церия(1¥) в виде желтоватого слизистого осадка. Из растворов солей цсрия(Ш) теми же реагентами выделяется белый гидрат окиси церия(Ш). Однако последний на воздухе быстро окисляется, причем в качестве промежуточного продукта образуется темно-фиолетовый промежуточный окисел. Если ла раствор соли церия(Ш) подействовать перекисью водорода и аммиаком, то выпадет интенсивно окрашенный в красно-коричневый цвет гидрат перекиси церия в виде слизистого осадка. Используя эту реакцию, можно определить даже незначительные количества церия по желтому окрашиванию раствора при добавлении перекиси водорода.
С Na2O двуокись церия образует двойной окисел Na2CeO3, обладающий структурой каменной соли и статистическим распределением катионов (стр. 294). Двойной окисел получается при нагревании Се02 с Na20 (Zintl, 1940) или с NaOH (D’Ans, 1930). Соответствующий двойной окисел прааеадома был получен Циптаем нагреванием смеси Рг203 и Na£O в атмосфере кислорода. Се2О3 является изотипом La20p (ср. стр. 55). Полностью аналогичную структуру имеют соединения La2O2S, Ce2OaS и Pu2OaS. Их решетка производится от решетки La2Oa, если каждый третий атом кислорода заменить на атом S (Zachaciasen, 1949).
Хлориды. Хлорид церия (ИГ) СеС13в безводном состоянии получается нагреванием окнела в токе хлора или хлористого водорода в виде кристаллической расплывающейся массы. Он очень легко растворяется в воде и спирте. Из водного раствора кристаллизуются гидраты, которые при нагревании на воздухе дают оксихлорид церия. С. хлоридами органических оснований хлорид церня(Ш) может давать двойные соли типа Мг[СеС14]. Двойные хлориды, соответственно комплексные соли, известны также для чежырехпалентного церня, например [C5HsNH)a[CeCle] [гексахлороцерат(1У) пиридявия] — ягелтмй кристаллический осадок..
Согласно Клемму (1934), если на безводный СеСЬ или Ce2S3 действовать элементарным фтором, то образуется тетрафторид церия CgF.. Он бесцветен и нерастворим в воде. Его удельный вес составляет 4,77, При пагревапии в вакууме до 400° он пе отщепляет фтора, по восстанавливается при нагревании в токе водорода до 390’, давая CeF3 (т. пн, 14ВДД.
* Прескаливанием в токе водорода при 1250® получают окись церия(Ш) Се£Оа.
Семейство лантанидов
533
Нитраты. Нитрат, церия(1П) Ce(NO3)3-GH2O кристаллизуется из растворов, например карбоната церия в азотной кислоте, в виде бесцветной, лучистой расплывающейся массы. Он легко растворяется в воде и спирте. С нитратами аммония, магния и другими нитратами двухвалентных металлов он образует хорошо кристаллизующиеся двойные соли.
Нитрат церия(1У), помимо двойной соли, известен только как основной нитрат Ce(OH)(NO3)3-3H2O, который получается в виде красных кристаллов при растворении гидрата окиси церия(1У) в концентрированной азотной кислоте и упаривании раствора. В воде он растворяется с желтым окрашиванием и вследствие гидролиза с кислой реакцией. При добавлении азотной кислоты свежеприготовленный раствор становится красным. Аналогичный аффект вызывает добавление нитрата щелочного металла. Из растворов, смешанных с щелочными нитратами, кристаллизуются светло-красные (безводные) нитрато-соли типа [Ce(NO3)6]. С двухвалентными металлами образуются нитрато-соли аналогичного состава; однако эти соли кристаллизуются с 8НЙО, Нитрата if epam(/V) аммония (NH4)2(Ce(NO3)6], который легко растворим В воде, ио достаточно трудно растворим в азотпой кислоте, дает возможность сравнительно просто отделять церий от сопровождающих его земель, двойные нитраты которых существенно легче растворимы в азотной кислоте.
Сульфаты. Сульфат церия(Ш} Ce2(SO4)3, иолучеппый обработкой нитрата или оксалата серной кислотой, представляет собой в безводном состоянии белый гигроскопичный порошок. Так же как безводные сульфаты других редкоземельных металлов, он хорошо растворяется в воде при 0° с образованием сильно пересыщенных растворов по отношению к устойчивому при этой температуре додека гидр ату. Помимо до дека гидрата, существуют еще многие другие гидраты. При нагреванип до 4.00—450° можно получить из них безводный сульфат.
Если сульфат нагреть выше 500°, то он начинает разлагаться, Иагревапием до белого каления удается получить чистую двуокись. С сульфатами щелочных металлов сульфат церия(1 II) образует двойные соли, например K[Ce(SO4)2]-II2O — бесцветный, труднорастворимый кристаллический породит (наряду с ним сосуществует также Ke[Ce4(SO4)9]-8П2О и [Ce(SO4)4[. Легче, чем двойные калиевые соли, растворяются двойные аммонийные соли: (Г\ТН4) [Ce(SO4)2] и (NH4)5 [Cc(SO4)4] (или (NH4)8[Ce2(SO4)7]?). Дисульфатоцсрат(Ш) аммония обычно осаждается в форме метастабильного тетрагидрата; в соприкосновении с раствором стабильными являются (в зависимости от температуры и концентрации) моногидрат и безводная соль (Schroder, 1938).
Сульфат церия(1У) Ce(SO4)2 получается в безводном состоянии нагреванием двуокиси церия с Концентрированной серной кислотой в виде ярко-желтого, нерастворимого в концентрированной серной кислоте кристаллического порошка. В воде он легко растворяется, вызывая коричпевато-желтую окраску. Из водного раствора ниже 3° оп кристаллизуется с 12 молекулами Н2О, между3°и 42,5°— с 8Н2О и выше 42,5°— с4Н2О. При более сильном разведении раствора происходит гидролитическое расщепление, которое приводит К осаждепиго труднорастворимых основных солей. При подходящих условиях удается получать сравнительно устойчивые золи сульфата церия (Janek, 1933), При добавлении серной кислоты свежеприготовленный раствор сульфата церия(1У) окрашивается в темно-красный цвет, Путем исследования чисел переноса Джонс (Jones, 1935) смог установить, что это связано с образованием комплексных кислот. Из растворов, к которым добавлены сульфаты щелочных металлов, кристаллизуются двойные сульфаты |суяь^атоцерашы(/кг)], например K4[Ce(SO4)4I-2ПЙО— очень мало растворимые ор а токе во-желтые моноклинные кристаллы; (ЙН4)е[Се(ЗО4)5]-• 2Н2О —орапжево-краспые, моноклинные кристаллы.
Сульфат церия(1У) часто применяют да я оксндиметрического титрования (Furman, 1928 и сл; Willard, 1928 и сл.). Он паходпт также иногда препаративное применение для окисления органических соединений.
Карбонат церил. Известен карбонат только для шрегвалентного церия. Он выделяется при добавлении карбоната аммония к растворам солицерия(Ш) в виде микрокристаллического осадка состава Се2(СО3)3- 5Н2О, С избытком концентрированного раствора карбоната аммония карбонат церия дает двойную соль NH4[Ce(CO3)2]-3H2O, Карбонаты калия и [[атрия также образуют двойные соли с карбопатом церия.
Оксалат церия. С щавелевой кислотой церий также образует соединения только в трехвалентном состоянии. Четырехвалентный церий тотчас восстанавливается щавелевой кислотой. Оксалат церия Се2(С2О4)3- 10Н2О получают в виде белого кристаллического осадка при Добавлении щавелевой кислоты к раствору соли церия. Как и все оксалаты редкоземельных металлов, оксалат церия не только практически нерастворим в воде, по и очень мало растворим в разбавленных сильных кислотах. В 100 мл
534
Глава 10
воды нри 25° растворяется 0,041 -кг безводной соли, в то время как в 100 мл 1 н, серной кислоты при 20° растворяется 164 мг. Растворимость оксалатов в воде в ряду редких земель возрастает в общем с уменьшением основности земель. Она наибольшая у оксалата скандия, в то время как растворимость в кислотах, наоборот, наибольшая у оксалата лантана.
Сульфиды церня. Церий образует сульфиды CeS, Cc3S4, Ce2S3 и Ce2S4. Ce2S3 получается в результате сильного нагревания безводного хлорида или сульфата в токе сероводорода, Он представляет собой чёрно-фиолетовый порошок уд, веса 5,10. Если вести только умеренное нагревание, то образуется соединение Ce2S4 (темно-коричневый порошок). Как следует из ого химического поведения и парамагнетизма *, последний следует рассматривать как продукт присоединения S к Ce3S3 полисульф идя о го типа. Следовательно, в противоположность СеО2 оннроизводйтся Пе от четырехвалентного, а от ту ^валентно го церия. Примерно при 720° Ce2S4 отщепляет серу и переходит в Ce3S3, Соответствующий нолисульфид образует также лантан: La2St или La2S3-S (золотистый желтовато-коричневый порошок),
Ce2S3 кристаллизуется с образованием кубической объемцоцептрированной решетки (а = 8,617 А). Как установил Захариазен (1949), он обладает интервалом гомогенности, простирающимся от CeSi,5 до CeS j,33. При составе CeS1>S3(= Ce3S4) элементарная ячейка (а = 8,608 А) содержит 12 атомов Со и 16 атомов S; при составе CeSi;6( = Ce2S3) мест металлической решетки не занята. Co2S3 имеет ярко выраженный характер гетерополярпбго соединения. С уменьшением содержания серы все больше убывает ионный характер; это проявляется также в усилении внешнего металлического характера. Изотипами Ce3S3 являются La2S3 {а — 8,706 A),Ac2S3 (а = 8,97 A), Pu2S3 (а = = 8,437 А) и Am2S3 (а = 8,428 А).
II, Иттриевые земли
Среди элементов группы иттриевых земель (ср. стр. 527) наиболее распространенным является иттрий, соединения которого ужо была рассмотрены в гл. 2 этой книги. Среднее содержание окиси иттрия в минералах редких земель (в атомных процентах) примерно в 3 раза больше, чезз содержание тербиевых, эрбиевых и иттербиевых земель, вместе взятых. Скандий, окись которого, как наименее основная среди редких земель^ является как бы переходной от последних к окиси алюминия (глинозему^ также уже была рассмотрена в гл. 2,
Соединения тербиевых, эрбиевых и иттербиевых земель настолько близки основным своим свойствам к соединениям иттрия, Что для их характеристики досТ1Ж> чоя короткий обзор.
Тербиевые земли
К группе тербиевых земель относятся европий, гадолиний и тербий.
Земли, относящиеся к тербиевой группе, хотя и не обладают характеристлякЕЕ-ми абсорбционными спектрами, однако дают весьма характеристические дуговые gee-тры. Гадолиний окрашивает пламя электрической дуги в карминово-краспып еддтг тербий — в желтовато-белый. Европий даже пламя бунзеновской горелки окраин в великолепный красный цвет.
Соли европия обладают слабо-розовой окраской. Их растворы дают лишь ненеег: большей частью весьма слабых, полос поглощения. Соли гадолиния и тербня fcr.;,-?-: ны. Одпако тербий образует темно-коричневый более высокий окисел, coxspsror кислорода в котором примерно соответствует формуле ТЬ4О,; оп обнаруживаем
* Так как ион Се1+ обладает таким же расположением электронов, что п взк—: г то для солеобразпых соединений церия(1У) следовало бы ожидать ---
(см. т. Г, гл. 9). И Действительно, СеО3 диамагнитен. Одпако, как покаки [?--« (1930), Ce3S4 также сильно парамагнитен как Ce2S3, и он имеет практпчзиЕД et i: же молярную восприимчивость, как типичные соли церня(Ш), напршггз -
Семейство лантанидов
535
же структуру решетки, как РгО2 11 Ргвои (а = 5>28 -А.). Его получают^црокаливанием на воздухе солей тербия и летучих кислот. Вследствие иптепсилной окраски этого окисла даже незначительное содержание тербия в других окис л ах этой группы делается заметным. ТЬ2О3, белого цвета, тоже относится к Gd2O3; Eu2O3 слабо-розовый.
Нагреванием ТЬ4О7 в атмосфере кислорода под давлением удается окислить его до ТЪ6Оц. Если одновременно добавить Y3O3, то кислорода поглощается все больше. Таким путем можно приготовить препараты, которые содержат ТЬ и О почти в отношении 1 : 2 (Prandtl, 1938; Marsch, 1946). Однако полного окисления до ТЬО2 пока не удалось достигнуть. Вообще говоря, более высокие окислы тербия менее устойчивы, чем такие же окислы празеодима. Это проявляется в том, что они значительно легче восстанавливаются при пагревапии в токе водорода до ТЬ2О3.
Уснлепие присоединения кислорода окислами Рг2О3 и ТЬ2О3 при добавлении Y2O3 связано, согласно Маршу, с тем, что как пол утора окиси, так й двуокиси Рг и ТЬ способны включать У2О3 в свою решетку с образованием твердых растворов. Благодаря Включению в решетку У2О3 затрудняется сильное сжатие решетки, возникающее при переходе полутораокиси л двуокись. Помимо Y2O3, этого удается достигнуть включением других окислов редкоземельных металлов. Показательно, что присоединение кислорода ими также облегчается, чего нельзя сказать о тех окислах, в случае которых названные условия не выполняются.
Помимо нестабильного прометия, европий является самым^ редким элементом среди редких земель. Он примерно в 500 раз более редок, чем иттрий, и примерно в 150, чем церий.
Хлорид европия ЕпС13, так же как и хлорид самария, удается восстановить до ЕпС12 нагреванием в токе водорода. ЕпС12 бесцветен, он стабильнее, чем SmCl2, и окисляется водой только при нагревании раствора. В воде он легко растворим и кристаллизуется из нее в виде дигидрата ЕиСЬ'2П2О. Водный раствор дихлорида можно получить также непосредственно восстановлением растворенного трихлорида цинком в соляной кислоте, EuCl, является изотппом РЬС121 в то время как EuF2 (получаемый так же, как ЕпС1г, плп кипячением EuSO4 с концентрированным раствором NaF) имеет структуру плавикового шпата (а — 5,796 А). EuBr2 и Ен!2 также могут быть получены (Klemm, 1938); известен и EuS (Nowacki, 1938). Последний, получаемый нагреванием Eti2(SO4)3 в токе H2S в виде коричнево-фиолетового порошка, кристаллизуется по тину камеппой соли (а = 5,957 А). То же относится к ЕпТе и EuSe, которые были приготовлены Клеимом (1939). EuSO4 вследствие малой растворимости дегко получается электролитическим восстановлением водного раствора сульфата евро-пия(Ш) (ср. стр. 5291, Он изоморфен SrSO4 и BaSO4.
Безводный ЕпС12,присоединяет аммиак с образованием октааммина EuCla-8NH3, так же как SrCl2. Другие соединения евроиия(П) также во многом аналогичны соединениям стронция. Это объясняется тем, что радиус иона Еп2+ очень мало отличается от радиуса вона Sri+,
Эрбиевые земли
К группе эрбиевых земель отпосятся диспрозий, гольмий, эрбий ктулий. Все земли этой группы образуют окрашенные ионы и дают абсорбционные спектры, которые частью весьма характеристичны (ср. табл. 54, стр. 518), Растворы солей диспрозия имеют цвет от желтого до зеленовато-желтого, растворы солей гольмия окрашены в коричневато-желтый цвет, растворы солей эрбия — интенсивно розовые, а солей тулия — зеленые. Диспрозий дает в ультрафиолетовой области еще более характеристический абсорбционный спектр, чом в видимой области. Диспрозий и гольмий обладают наибольшим парамагнетизмом из всех элементов редких^ земель в их соединениях. Название гольмий производится от «Стокгольма, тулий от «Туле»— старого названия Скандинавии,
Иттербиевые земли
Иттербиевые -Земли, т. е, окислы иттербия и лютеция, бесцветны, так же как я их соли. Однако они дают характеристические дуговые спектры. Иттербий Окрашивает пламя электрической дуги в зеленый цвет, лютеций—в голубовато-зеленый. Отделение иттербиевых земель от других земель семейства лаптапидов было осуществлено па основании их более слабого основного характера уже в 1878 г. Мариньяком при фракционированном разложении нитратов при нагревании. Однако только в 1907 г. Ауэру фон Вельсбаху удалось разделить два весьма похожих элемента — иттербий и лютеций. Он достиг этого путем длинного ряда фракционированных кристаллизаций двойных оксалатов редких земель и аммония, после того как уже в 1905 г, Марипьяк
536
Глава 10
констатировал разлагаемость старого иттербия в результате наблюдения искрового спектра.
Как показал Клемм (1929), YbCl3 удается легко восстановить до YbCl2 при нагревании его в токе водорода примерно до 550°. Янч (Jantsch, 1931) получил позднее также YbBr2 и Ybl2. Последний кристаллизуется по типу брусита (а=4,48, й = 6,96 А). Халькогениды восстанавливаются до соединений иттсрбия(П) труднее, чем галогениды. В случае Yb2O3 вообще не удается достигнуть восстановления, а в случае Yb2Sg достигается только неполное восстановление. Напротив, путем нагревания Yb2Se3, соответственно Yh,Te;,, в токе водорода Клемм (1939) получил YbSe и, еще легче, YbTe практически в чистом состоянии (черные пор описи, являющиеся изотипаыи NaCl; а — 6,340, соответственно 5,867 А). Как следует из постоянных решеток этих соединений, ион Yh2+ имеет почти тот же радиус, что и Саа+.
Аналитические сведения. В аналитическом отношении характерным для всей группы редких земель является незначительная растворимость их оксалатов в кислотах. Поэтому при систематическом разделении присутствие редких земель обнаруживают в результате осаждения, когда после отделения веществ сероводородной группы солянокислый раствор (концентрация кислоты в котором, однако, не должна быть слишком велика) смешивают с щавелевой кислотой.
Для более точной характеристики тех родиих земель, которые образуют окрашенные соли, удобно проводить спектральное исследование их растворов в проходящем свете. Однако следует нри атом учесть, что наблюдаемые в спектрах поглощения полосы могут подвергаться влиянию таких веществ, которые способны действовать па окрашенные ноны, даже если сами они абсолютно бесцветны. Особые характеристические абсорбционные спектры дают празеодим, неодим и эрбий (ср. табл. 54, стр. 518). Эмие-сцепные спектры (дуговые и искровые) редких земель чрезвычайно богаты линиями. Поэтому при их применении для идентификации частое успехом используют так называемые последние линии. Это как раз те линии, которые при неоднократном разбавлении вещества исчезают самыми последними. Самым простым путем для точной идентификации и особенно двя испытания на чистоту служат рентгеновские спектры. В общем рентгеноспектрографически удается обнаружить еще 0,1% редкой земли. Иногда можно достиг [[уть и существенно более высокой чувствительности, например используя особо высокую силу тока и продолжительную экспозицию.
Глава 11
РАДИОАКТИВНОСТЬ И ИЗОТОПИЯ
Определение радиоактивности. Радиоактивными веществами называются такие вещества, которые имеют свойство самопроизвольно (т. е. бед внешнего возбуждения) длительно излучать энергию. В большинстве случаев лучи, испускаемые радиоактивными препаратами, обладают способностью проникать через непрозрачные для обычного света вещества, например через плотную черную бумагу или металлические пластинки, а также разряжать несущие электрический заряд тела, засвечивать фотопластинки, а часто, как например лучи радия, вызывать свечение люми-несцирующих веществ. Радио активность — это свойство атомов, т, е. излучение, испускаемое радиоактивным элементом, не зависит от того, существует ли он в элементарной форме или в виде какого-либо соединения.
Открытие радиоактивности. Вскоре после того как Рентген в 1895 г, открыл лучи, в дальнейшем названные его именем, Беккерель решил испытать, не обладают ли флуоресцирующие вещества способностью испускать лучи, которые подобно рентгеновским лучам проникают через непрозрачные материалы — ведь трубки Рентгена, кроме рентгеновских лучей, испускают обычное излучение флуоресценции. С этой целью он испытал различные флуоресцирующие вещества и среди пих урановые препараты и нашел, что эти вещества действуют на фотопластинки через черную, непроницаемую для света бумагу. Но другие исследованные Беккерелем флуоресцирующие вещества этой способностью не обладали, В случае же урана это свойство оказалось присуще не только флуоресцирующим солям уранила, но и нефлуоресцирующим уранатам.
В конце концов оказалось, что все соединения урапа, а также сам металлический уран испускают специфическое проникающее излучение. Кроме того, как выяснилось, это излучение в противоположность излучению флуоресценции совершенно не зависит от предварительного освещения или какой-либо иной предварительной обработки препарата. Беккерель открыл также свойство этих лучей разряжать электрически заряженные тела (делая проводящим окружающий воздух), что впоследствии часто использовали для количественного излучения этих лучей. Особенно удивительным было свойство открытого Беккерелем излучения, названного позднее радиоактивным излучением, оставаться длительное время неизменным. Впоследствии это получило объяснение и одновременно ограничение в выдвинутой Резефордом и Содди (1903) теории радиоактивного распада.
Поиски других веществ, обладающих, как и урап, свойством радиоактивности, вскоре привели к открытию радиоактивности тория (Шмидт,
538
Глава 11
1898, и почти одновременно М. Кюри). В том же году супруги Кюри открыли два новых элемента, которые оказались значительно более радиоактивными, чем уран. Эти элементы были названы полонием и радием.
Кая уже было отмечено в т. I, прн исследовании радиоактивности минералов урана (для измерения которой определяли ионизацию воздуха радиоактивными веществами) М. Кюри заметила, что, если дия искусственно полученных соединений урана пх активность всегда соответствовала содержанию в пих урана, минералы урана часто обнаруживали значительно большую активность, чем можно было ожидать, Например, поахимстальская урановая смоляная руда оказалась втрое более радиоактивной, чем металлический уран. Из того факта, что активность чистых урановых препаратов всегда пропорциональна содержанию в них урана независимо от рода соединения, М. Кюри сделала вывод, что радиоактивность является свойством атомов. Если это так, то большую активность встречающихся в природе соединений урана можно было объяснить, только допустив, что эти минералы содержат но крайней мере один элемент, превосходящий по своей активности уран. М. Кюри вместе с мужем 11. Кюри и их сотрудником Бемопом (Beniont) провела химический анализ урановой смоляной руды, контролируя при этом распределение активности, для чего измеряла ионизирующую способность па различных стадиях анализа. Уже в осадке, полученном при осаждении сероводородом (уран выпадает в осадок с металлами группы сернистого аммония), обнаружилось присутствие сильно радиоактивного вещества. Прн дальнейшем анализе это вещество следовало за висмутом. Поскольку сам висмут не испускает столь интенсивного радиоактивного излучения *, то это должен был быть новый элемент, очень близкий висмуту по свойствам. М. Кюри назвала новый элемент полонием в честь своей родины Польши. В Ходе дальнейшего разделении был найден еще один активный элемент — спутник бария. Этот элемент был назван радием вследствие его чрезвычайно высокой радиоактивности.
Вскоре после этого радий был Получен в чистом состоянии. Это было очень трудно осуществить, так как радий содержится в урановых рудах в очень небольших количествах. Даже сравнительно богатая радием иоахимстальская урановая смоляная руса в 5 кг содержит только 1 мг радия. Но М. Кюри получила от австрийского правительства Почти бесплатно два вагона остатков от переработки урановой смоляной руда.. Из этого сырья после длительной работы ей удалось выделить 0,1 g чистой соли радпг (бромида радия).
Очень скоро после открытия полония и радия Дебьерн (1899) нашег в отходах от переработки урановой смоляной руды еще один радиоактивный элемент — актиний (axTivceic; — излучающий). Этот элемент пр~ аналитическом разделении выпадает вместе с железом и редкими земле-ми. Поскольку содержание актиния в урановог! смоляной руде еще значительно меньше, чем содержание радия, то получение весомых количеств: чистого природного актиния чрезвычайно трудно и до сих пор еще ез удалось **.
Общая характеристика радиоактивного излучения. Испускаемое радпгщ и другими радиоактивными элементами проникающее излучение обнаруживается в основном по трем вызываемым им эффектам: 1) по вызываем:^ им люминесценции, т. е, благодаря его свойству вызывать свечение npz обычных температурах некоторых веществ, например сернистого цикле. 2) по ионизирующей способности и 3) по действию на фотопластинку.
После проявления фотопластинки опа чернеет в тех местах, которое подверглись действию радиоактивного вещества. При непосредственна контакте несколько миллиграммов соли радия вызывают такое почернение уже через несколько минут. Но еще более интенсивно ионизирующее
* Радиоактивность висмута (см. т. I, стр. 727) столь мала, что имевшимися в рупору средствами ее вообще нельзя было обнаружить.
** Искусственный актиний уже получен в весомых количествах (несколько чл--лиграммов) по ядер ной реакции: 226Ra[n, yj227Ra Я 225Ас.
Радиоактивность и изотопия
539
действие радия. Оно настолько велико, что достаточно 0,001 мг радия, чтобы почти мгновенно разрядить находящийся вблизи от него электроскоп.
Люминесценция, вызываемая радиоактивными лучами, в общем случае заметна невооруженным глазом только в темноте. Свечение тем сильнее, чем интенсивнее радиоактивное излучение. Чистые соединения радия сами интенсивно люминесцируют; под действием собственного излучения опи светятся голубоватым светом. У расплавленных хлорида или бромида радия такое самопроизвольное свечение настолько сильно, что его видно даже при дневном свете. Но еще отчетливее, чем чистые соединения радия, светятся те препараты радия, которые содержат барий,
Более подробные исследования показали, что у радия наблюдается три различных вида излучений. Однако одновременно было отмечено, что эти три вида излучения не все испускаются непосредственно радием.
1. Лучи первого рода, названные а-лучами, обладают небольшой проникающей способностью. Они отклоняются в электрическом и магнитном полях, по пе очень сильно. Направление отклонения показывает, что они заряжены положительно. Следовательно, они очень напоминают получающиеся в вакуумных трубках каналовые лучи, открытые Гольдштейном (Goldstein) в 1886 г., и состоят, как и последние, из положительно заряженных частиц материи, с большой скоростью выбрасываемых в пространство. Но скорость а-частпц значительно больше, чем скорость частиц обычных каналовых лучей. Кроме того, величина отношения электрического заряда к массе у а-частиц не переменная, как у каналовых лучей, а постоянная и равна 1/3. Как было доказано (см. стр. 546), я-частицы представляют собой дважды положительно заряженные атомы гелия, т. е. атомы гелия, которые потеряли оба электрона — ядра атомов гелия.
2. Второй вид радиоактивных лучей, названных 0-лучами, обладает значительно большей проникающей способностью, чем а-луни. В магнитном и электрическом полях 0-лучи относительно сильно отклоняются и оказываются при этом отрицательно заряженными *. Опи во всех отношениях аналогичны катодным лучам, превосходя их только в скорости. Как и катодные лучи, опи состоят из движущихся с невероятными скоростями элементарных частиц отрицательного электричества (электронов), Если катодные лучи обычно имеют скорость около половины скорости света, то среди 0-лучей известны такие, скорость которых только на 1% меньше скорости света.
3. Лучи третьего рода, у-лучи, обладают чрезвычайно высокой проникающей способностью. Они не отклоняются в магнитном и электрическом полях и, следовательно, не заряжены. Эти лучи наиболее близки рентгеновским лучам, от которых они отличаются только значительно меньшими длинами волн.
ВАЖНЕЙШИЕ ДЕЙСТВИЯ РАДИОАКТИВНЫХ ВЕЩЕСТВ
Прежде чем ближе ознакомиться с этими тремя видами излучения, следует подробное рассмотреть лишь коротко охарактеризованное выше действие излучений радия и других радиоактивных веществ.
* Теперь 0-луча.ми называют не только электронные лучи, по и позитронные лучи (состоящие из положительно заряженных частиц, масса которых равна массе электрона). Если нужно различить аги два лида лучей, то говорят о (i~- и 0+-лучах. Поскольку дальше пойдет речь об излучения естественных радиоактивных веществ, то под 0-лучами всегда будут пониматься 0_-лучи.
ИО
Глава 11
Ионизация газов. Способность ионизировать воздух и другие газы, присущая препаратам радия (а также тория и урана), связана почти исключительно с а-излучением. Это относится к тем случаям, когда препараты нс экранированы, т. е. они не окружены какой-либо оболочкой, непроницаемой для сс-лучеп (см. ниже). В меньшей мере ионизирующая способность характерна и для р-лучей. Наименьшим ионизирующим действием обладают у-лучи.
Такое сравнение справедливо при условии, что мерой ионизирующего действия служит количество ионов, образующееся на равных отрезках пути *. Если соединении радия нанесено топким слоем на две пластинки, расположенные на расстоянии 5 е.«, то вклады в ионизацию находящегося между ними воздуха (мерой которой является его электропроводность), обусловленные действием а-, ₽- и у-лучей, относятся между собой как 10 000 : 100 : 1.
Визуальное обнаружение ионов газов. Ионы газов действуют подобно частицам пыли, являющимся центрами конденсации при образовании капелек воды в пересыщенном влагой воздухе. На этом основан разработанный в 1911 г. Вильсоном метод обнаружения ионов газов, а тем самым и траекторий а-дастиц, вызвавших образование этих ионов (рис. 59),
Как известно, ири расширении сжатого газа вследствие совершаемой им работы по преодолению обычного давления воздуха газ охлаждается. Если при первоначальной температуре газ насытить парами воды, то при его расширении вода, избыточная но сравяе-
" 11 с- 53. Траектории и-частиц пню с упругостью ее паров ври бол со низкой
в камере Вильсона. температуре, выделяется в виде тумана; од-
нако при умеренном охлаждении это произойдет только в том случае, если в газе имеются центры клтдеиеации для отдельных капелек воды, из которых и состоит туман. Обычпо такими центрами служат частички пыли. Но было показано, что аналогичными центрами конденсации могут быть и ионы газов. Если из газа удалить частички п!(Ши (что легко достигается конденсацией на них водяных капелек, благодари чему частички пыли оседают на дно), то образование тумана при постоянном охлаждении (которое легко регулировать, проводя адиабатическое расширение гава) происходит вновь, если газ ионизирован действием рентгеновских лучей или какого-либо радиопренарата **. Можно даже установить, положительные это ионы или отрицательные и в каком соотношении опи содержатся. Обычно для конденсации на положительных ионах требуется большее охлаждение, чем па отрицательных. Так, было показано, что всегда образуются равные количества положительных и отрицательных ионов. С.дуды тумана, оставляемые ионизирующими излучениями, можно сфотографировать, как ото видно из рис. 59.
* Па каждом сантиметре своего пути 0-лучи образуют значительно меньше ионов, чем а-лучи, но в одной я той же среде их путь больше. Общая иопивацпя для п- п 0-лучей одной и той же энергии практически одинакова (в среднем по одной паре пеней на каждые 3-2,5 ав). Это же справедливо и для других частиц матерям, обиадатзпшх высокими скоростями, например для протонных лучей (ср, стр. 553 и 598).
** Препараты, которые содержат (любые) радиоактивные вещества, коротко называют «радпопрепаратами» (пе следует путать с радиевыми препаратами, которые содержат радий).
Радиоактивность и изотопия
541
Пропикающая способность радиоактивных излучений. п-Лучи полностью задерживаются 0,1-ми л ли мет ровен алюминиевой фольгой, листочком слюды или листом плотной писчей бумаги, 0-лучи — только 5-миллиметровой алюминиевой фольгой или свинцовой жестью толщиной 1 -и .и, у-лучи проходят через эти вещества практически не ослабляясь. Поглощенно излучения различными веществами приблизительно пропорционально их удельным весам. Ио вещества с очепь высоким удельным весом поглощают излучение даже лучше, чем следует им этого правила.
Люминесценция, Кристаллический сульфид цинка при иопадании па него радиоактивного излучения ярко светится голубоватым светом, а экрант покрытый циано-платинатом бария, флуоресцирует зеленым светом. Еще сильнее флуоресцирует минерал виллемит (силикат цинка Ztj2SiO4). Его флуоресценции также зеленая. Шеелит (CaWO4) и алмаз, а также большинство флюоритов светятся голубым светом. В меньшей степени радиоактивные излучения вызывают свечение и многих других веществ: кроме минералов, это прежде всего стекла и большое число органических соединений. Флуоресценция возбуждается также внутри глаза человека. Поэтому приближение радиоактивных препаратов отчетливо воспринимается даже с закрытыми глазами,
Люминесцирующее действие почти исключительно связано с а- и 0-лучами, испускаемыми радиоактивными веществами. Напротив, люминесценцию, вызываемую у-лучами, вряд ли следует учитывать. Если а-лучи пе задерживаются, то люминесценция в большинстве случаев обусловлена главным образом их Действием. Однако существуют вещества, люминесценция которых вызывается только 0-лучамп, а а-лучи пе заставляют их светиться. Так ведет себя, папример, минерал кунцит (открытая американцем Купцом разновидность минерала трифана), а также салипирин (салициловокислый антипирин).
У многих веществ свечение исчезает сразу же при удалении радиопрспарата, Другие же обладают и поел есвечеп гам («фосфоресцируют»). Сульфид цинка фосфоресцирует иод действием 0-лучей диффузио п высвечивается медленно. Напротив, свечение этого вещества под в,апяпис-м а-лучеп настолько резко ограничено пространственно и во времени, что его молшо использовать для счета отдельных а-чаетил,, наблюдая вызываемые ими вспышки света.
Сцинтилляция. Если в лупу или микроскоп (лучше всего с увеличе-
нием 50 : 1) рассматривать поверхность экрана, люминесцирующего под действием падающих на него а-частиц, то ясно видно, что светится но вся поверхность равномерно, а нспышки света возникают то в одном, то в другом месте поверхности и сразу же исчезают. Очевидно эти вспышки света, названные сцинтилляциями (от scintilla — искра), вызваны отдельными попадающими на экран «-частицами, и поскольку такие вспышки можно сосчитать, то можно определить, сколько а-частиц попадает па поверхность экрана или
докрытого сульфидом циика
Рис. 60. Спинтарископ.
1 — экран, покрытый сульфилом пинка; пород ним на кончике иглы препарат рал ИЯ. Лучи, попадающие от препарата и а экран, вызывают сцинтилляции, которые можно наблюдать и лупу 3.
испускается радиоактивным препаратом.
Прибор, который позволяет очепь просто наблюдать сцинтилляции, вызываемые радиоактивным препаратом, представляет собой сконструированный Круксом «спинтарископ» (оштф^д — искра, oxonctv — наблюдать), показанный на рис. 60. Сцинтилляция возбуждается также во мно-
гих алмазах.
Почернение фотографических пластинок. Почернение фотопластинок, расположенных непосредственно пад топкими слоями радиопрепаратов, также вызвано в основном действием а-лучей. Но па большом расстоянии иочернение вызывается преимущественно 0-излучением. Относительно высокая проникающая способность последних я их различная поглощаемость веществами различной плотности позволяет, как и в случае рентгеновских лучей, обнаруживать с их помощью на фотопластинке контуры внутренних частей непрозрачных тел. Однако изображения на таких «радиографиях»
5642
Глава 11
более расплывчаты, чем на рентгеновских снимках, вследствие более сильного по сравнению с рентгеновскими лучами рассеяния р-лучей. Улучшения качества изображения можно достигнуть, используя для получения снимков только у-лучн, а-Излучение совершенно непригодно для получения радиографий.
Окрашивание под действием радиоактивных излучений. Стекла; и другие вещества при продолжительном воздействии на них радиоактивного излучения постепенно окрашиваются. Голубой цвет, в который иногда окрашена природная каменная соль (см. т. I, стр. 214), вероятно, обусловлен воздействием радиоактивного излучения. Под действием излучения пустые места в решетке ионов хлора, существующие вследствие «неупорядоченности», занимаются свободными- электронами. Поглощение света такими электронами приводит к окрашиванию. Оно может быть вызвано и искусственно при облучении радиоактивными излучениями. Эльстер и Гейтель (Elster, Geitel) наблюдали, что сульфат калия, искусственно окрашенный в зеленый цвет под действием излучения радия, обладал сильным фотоэлектрическим эффектом, т. е. иод влиянием ультрафиолетового света из него вырывается заметное количество электронов.
Часто окрашивание, вызванное радиоактивным облучением, исчезает после нагревания. Способность некоторых веществ светиться под действием радиоактивных излучений благодаря окрашиванию значительно ослабляется, но вновь восстанавливается, если окрашивание устранено нагреванием.
Если, как это часто бывает у некоторых минералов, особенно у слюд, какое-либо вещество содержит включения частичек радиоактивного элемента, то под действием а-лучей (а они эффективнее других лучей вызывают окрашивание) окрашивается только ближайшее окружение радиоактивной частички, так как в твердых телах пробег а-лучей очень мал. Образующиеся таким образом вокруг каждой радиоактивной частички кольца (которые в поляризованном свете обнаруживают плеохроизм и потому называются «плеохроичными кольцами») позволяют обнаружить присутствие чрезвычайно малых количеств радиоактивного вещества. Окись урана в количество 5 • 10-10 г, в равновесии с которой находится (см. ниже) только 10~1в г радия, в слюдах девонского периода * окружена вполне отчетливым плеохроичпым кольцом, хотя там в среднем испускается 1 а-частица за 10 час. Даже такое количество радия, как 10-1’ г, испускающее одну а-частицу в 4—5 суток, в некоторых случаях обнаруживается по образуемым им плеохроичным кольцам. При этом одновременно доказывается, что другие элементы, часто содержащиеся в слюдяных минералах в значительно больших количествах, не испытывают радиоактивного распада с испусканием ct-частиц, так как даже за геологические эпохи не обнаруживается действие испускаемого ими излучения.
Другие химические действия радиоактивного излучения. По отношению к газам радиоактивное излучение действует подобно тихому электрическому разряду. И в этом случае наиболее действенным является а-излучепие. Супруги Кюри уже в 1899 г. наблюдали озонирующее действие лучей радия. При достаточной активности радиопрепарата образование озона можно обнаружить даже по запаху. Окись углерода, двуокись углерода, аммиак, хлористый водород и другие соединения под действием радиоактивного излучения разлагаются на составные части.
* Девонский период — геологическая эпоха в историн Земли, непосредственно предшествующая каменноугольному периоду.
Радиоактивность и изотопия 543
И наоборот, эти соединения синтезируются из составных частей. Вода разлагается на водород и кислород. Поэтому влажные препараты радия не следует продолжительно хранить в запаянных стеклянных ампулах, так как возникает опасность разрыва ампул под давлением выделяющихся газов. Точно так же при работе с радиоактивными эманациями следует учитывать, что под действием их лучей из смазки кранов выделяется двуокись углерода. Химически изменяются также вазелин и парафин. Кварцевое стекло под воздействием а-активных препаратов постепенно становится хрупким. Изменяются и металлы. Точно так же сами радио-препараты могут претерпевать вторичные химические изменения. Так, бромид радия на воздухе под действием собственного излучения (иди образующегося озона) отщепляет бром и постепенно переходит в карбонат.
Физиологическое действие. В организме радиоактивное излучение вызывает раздражения и воспаления. Поэтому при обращении с сильно радиоактивными веществами следует соблюдать большую осторожность. В некоторых случаях достаточно приложить на несколько минут палец к дну капсулы, содержащей такое вещество, чтобы вызвать болезненное воспаление, не заживающее в течение нескольких месяцев (ср. стр. 629 и сл.). С другой стороны, иногда действие радиоактивных излучений на организм используют в лечебных целях. Хорошие результаты получены прежде всего при лечении рисовых опухолей. В настоящее время многие медицинские институты имеют радий в количество нескольких граммов, т. е. относительно большом.
СВОЙСТВА и природа трех видов радиоактивных излучении
1. Альфа-излучение
Пробег, скорость и ионизирующая способность. а-Лучи проходят через газовую среду по приблизительно прямым траекториям (см. рис. 59). В некоторой точке они внезапно исчезают, т. е. начиная с некоторой точки больше не обнаруживается ионизании или какого-либо другого действия а-частиц. Путь, проделанный а-частицей до исчезновения ее ионизирующего действия, называют пробегом. ct-Лучи, испускаемые однородным в радиоактивном смысле веществом, в общем * имеют практически одинаковый пробег. Поэтому величина пробега а-лучей является, важнейшим средством идентификации радиоактивного вещества.
а-Частицы, испускаемые однородным веществом, обладают одинаковыми началь-иыми скоростями. Их скорость можно определить аналогично скорости катодных и каналовых лучей, наблюдая отклонения в магнитном и электрическом^ елях (см. стр, 580). По мере проникновения в какую-либо среду скорость а-частид уменьшается. Вблизи конца пробега скорость быстро уменьшается и а-частяца резко тормозится. Число а-частиц, вызывающих ионизацию, остается до конца пробега Почти постоянным, а затем быстро падает до нуля. Ионизирующее действие а-частиц обратно пропорционально их скорости, т. е- оно тем больше, чем больше время,требующееся для прохождения через атом.
На рис. 61 показала ионизация газа а-частицами в зависимости от удаления от раднопр опара та и, следовательно, от скорости частиц (по измерениям Гейгера). Измерения относятся к ионизации воздуха при 12° и давлении 760 .«.ч рт. ст,, вызываемой а-частицами продукта распада радия, радия С' (см. ниже). На оси ординат отложено число ионов, образующихся на каждый миллиметр траектории. Как видно, ионизация непрерывно возрастает при увеличении расстояния от источника иэлуче-
* Об этом см. на стр. 554.
544
Глава 11
иия, т. е. при уменьшении скорости, достигая максимальной величины, непосредственно перед точкой торможения, после чего резко падает до нуля. Одпа а-частнца радия С' с пробегом около 7 ем образует па своем пути прпморпо 237 000 ионов, прежде чем она поглотится средой. Резерфорд и Гейгер нашли, что число а-чаетиц, испускаемых в i сек таким количеством радия С', которое находится в равновесии с 1 г радия (см. ниже) составляет 3,4' КМ 1 г радия вместе со своими продуктами распада, включая радий С', испускает, вчетверо больше (Г-частиц (13,6-1010) в секунду, или около 8 биллионов п-частиц в минуту.
Счет частиц осуществляют следующим образом: при помощи чувствительного прибора наблюдают ионизацию воздуха, вызванную одной частицей, прошедшей через
определенное отверстие. Каждая частица, прошедшая через отверстие, вызывает разряд электрометра. Число, найденное методом Резерфорда и Гейгера, согласуется с определенным по счету числом сцинтилляций.
8000
SOOO-
4000
3000
2000*-
1000
7000-%-60вО-Ё: । g
о
§
О 1 2 3 4 6 6 7
Расстояние от источника оЛгуеелця.ои
Р п с. 61. Зависимость ионизирующей способности а-лучей от скорости частиц.
Пробег а-частицы в водороде в четыре раза больше ее пробега в кислороде при равных температуре и давлении. В шестнадцать раз более тяжелый кислород, следовательно, тормозит а-лучи в четыре разг сильнее, нем водород. Как нашяп Брэгг и Клем а ни, в общем справедливо утверждение, что атомная тормозная способность вещества приблизительно пропорциональна корн” квадратному из его атомного весг.
Д.чя сложных веществ молекулярная тормозная способность приблпзс-тельно равна сумме тормозных способностей отдельных входящих в пе
состав элементов.
В каком-либо определенном газе пробег а-лучей пропорционален абсолютной температуре и обратно пропорционален давлению. При пробег в 1,055 раза больше, чем при 0л.
По данным Гейгера, пробег а-лучей приблизительно проиорцпонаггх кубу их начальной скорости. Если через Q обозначить пробег при £ а через v — начальную скорость (с.м/сек), то справедливо уравненг-.
р = 9,25-10-аэ-е3, или V— 1,026-10®. f q.
Относительная тормозная способность и воздушный эквивалент, Отпошс— гОг
— = В, гдер^— пробега-частиц л воздухе (при нормальном давлении), а — пхгтг,-
6s
бег в веществе <7, называется относительной термосной способностью этого
для а-лучеЙ. Например, для алюминия В ~ 1660, для слюды 2000 и для золота 4_-Часто пробег а-лучей в твердом веществе выражают в единицах массы (ле'ея-которую имеет фольга из этого вещества толщиной как раз достаточной для 3a~s“r?: пня а-лучен. Поэтому часто применяют выражение «пробег в мг/см'-г>. Если тан ваемын «пробег в мг/см2» разделить па 1000 где — плотность вещества, то clt-чин собсгвсппо пробег р,< (пробег в обычном смысле слова) в сантиметрах, так как г~-т пластинка толщиной р.ч и площадью q (см2) равпа т = qs- <? rf,s грамм, Cze=“ " телъно, тачая пластат-.а на каждый квадратный сантиметр имеет массу КЖ-г. , йМ-Рч-rl^; ,,
миллиграмм, и —j—г -Д—- = о,ч. У множа я pg па относительную тормознута г-лг.т.п Hjuij
вещества (В), получают пробег соответствующих а-частиц в воздухе (см -
а-лучя, испускаемые (чистым в радиоактивном смысле) торием, па~':~ г ~ ваются алюминиевой фольгой с массой 4,43 мг па каждьгй квадратнкЗ гатгтх'П-тому говорят: «Пробег а-лучей тория в алюминии равен 4,43 ~ ц-
мииия равпа 2,70, Собственно пробег (пробег в обычном смысле)с-с'Ш'-У т. тгг -.а-
Радиоактивность и изотопия
545
ляет, следовательно в алюминии = 1,64-Ю-3 см и в воздухе 1,64- 10"3Х
j Uw’ ZT I и
X 1,66-103 = 2,72 ел. Кроме «пробега в мз/смЧ, часто приводят «воздушный эквивалент» данного вещества, т, е. массу его в мв/см2, тормозная способность которой для а-лучей равна тормозной способности столба воздуха толщиной 1 см. Эта величина составляет, например, для алюминия 1,63 м.г/см2. Если «пробег в «не/с№» разделить па ату величину, то сразу получим пробег в воздухе ^pg|=2,72^) •
Используя соотношения, существующие между определенными выше величинами, можно, например, вычислить пробег а-частиц в веществе или в воздухе, смотря по имеющимся в распоряжении
<•
экспериментальным данным, по следующим формулам *: _ pl-ioWs QS~ #S-B& ’
_р£-^8-Дй
1000-
„ _ ns . _О_1П6_. Qs ~~ 10И,3 ’ Vb &SBS’ n&Bs . _ ns .
eL — 100(Ws’ 'L ’
6Е = В8?8',
Здесь gs— пробег а-лучей в веществе X; g£ — пробег а-лучей в воздухе (обе величины в сантиметрах); В&— относительная тормозная способность вещества б1; dg— его плотность; pg— так называемый «Пробег в .чг/е.ч2» а-лучей в веществе 8 и Оу— воздушный эквивалент вещества X. Следует помнить,что дди тЭднс имеют размерности длипы; напротив fig имеет размерность плотности [з/с.нЗ], a цд— размерность плотности, умноженной на длину.
Отклонения величины пробега. Воздушный эквивалент вещества пе является величиной, совершенно независимой от скорости «-частиц. Это же относится и к относительной тормозной способности. Поэтому для точных измерений пробег определяют
Р и с. 62. Распределение пробегов а-частиц радия С'.
непосредственно в сухом воздухе при попил;еипом давлении, чтобы получить возможно большие величины проходимого а-частнцамй пути. То, что а-частица с определенной начальной скоростью имеет определенный пробег, обтлсняется тем, что при каждом приводящем к ионизации столкновении с атомом теряется вполне определенная часть кинетической опергип. В воздухе эта потеря энергии составляет в среднем 32,5 as на образующуюся пару ионов. Но в отдельных случаях наблюдаются столкновения, при которых сталкивающемуся с частицей атому передается значительно больше энергии [(например, 1000 за). Такие связанные с большой потерей энергии столкновения распределены вдоль пути отдельной частицы статистически, и это приводит к тому, что длина этих путей колеблется около некоторого среднего значения. Рис. 62 иллюстри
* Эти формулы применимы также для протонных, дейтронных и т. д. лучей (см. стр, 599 и сл.).
35 т1, Реми
546
Глава. 11
рует это на примере а-частиц радия С' [по данным И. Кюри и Мерсье (Mercier)], Для -каждого значения пробега, нанесенного на ось абсцисс, по оси ординат отложено в процентах число а-частиц, которые имеют эту величину пробега. При пробеге в воздухе 6,70 сл еще обнаруживаются все первоначально испущенные а-частицы, при пробеге 7,02 сл все опи исчезают. Уменьшение их числа наибольшее в том месте, где находится точка перегиба кривой,т, е. при абсциссе, равной 6,87 см. Однако обычно в качестве величины пробега а-частиц радия С' приводят не эту наиболее часто обнаруживаемую величину, а величину 6,95 йл, которую получают, если продолжить касательную к кривой в точке перегиба до ее пересечения с осью абсцисс (пунктирная линия на рис. 62), Величину пробега, н большинстве случаев очень мало отличающуюся от последней, получают также, если представить число ионов, образующихся на единицу длины пути, как функцию расстояния от источника излучения (см. рис, 61) и продолжить прямолинейно ниспадающую ветвь кривой до ее пересечения с осью абсцисс.
Заряд а-частиц. Резерфорд и Гейгер при помощи чувствительного прибора непосредственно измерили количество электричества, излучаемое радиоактивным препаратом в виде а-лучей. Поскольку они первоначально определили число испускаемых а-частиц, то из данных этих опытов они могли непосредственно найти заряд одной а-частицы, тогда как измерение отклонений в электрическом и магнитном полях позволяет определить только отношение заряда частицы к ее массе. Для заряда а-частицы было получено удвоенное, значение положительного элементарного кванта, т. е. удвоенный заряд иона водорода (или ядра атома водорода). Почти в то же время аналогичные опыты были проведены Регеиером (Regener), который получил те ащ результаты.
Природа а-частиц. Поскольку, как это было установлено из величин отклонений в магнитном и электрическом полях, отношение заряда а-частиц к их массе равно в среднем половине отношения заряда иона водорода к массе атома водорода, а каждая а-частица несет два положительных заряда, то получается, что масса а-частицы в четыре раза больше массы атома водорода. Единственным известным нам элементом с атомным весом 4 является гелий. То, что из а-частиц действительно образуется гелий, Резерфорд доказал, перенеся испускающую а-лучи эманацию радия в стеклянный капилляр, стенки которого были настолько тонки, что пропускали а-излученис. Как оказалось, вокруг капилляра постепенно накапливался гелий, что было доказано при помощи спектроскопа. Специально было проверено, что стенки капилляра не могли пропускать никакой газ. Этот опыт дал прямое экспериментальное подтверждение высказанному, ранее предположению, что а-частицы представляют собой дважды, положительно заряженные атомы гелия. Как следует из спектроскопических данных, приведенных в гл. 4 т. I, гелий, потерявший два электрона, тем самым потерял всю свою электронную оболочку. Осталось только «ядро» атома. Поэтому а-частицы можно рассматривать как испускаемые с большой скоростью ядра атома гелия.
Так как 1 г радия испускает в секупду 13,6-1О10 ядер гелия, то это составлкгз
13,6-lQin
27-1018*
= 5,03-10-» е.«3 гелия в секупду, или 159 .нл3 в год.
2. Бета-лучи
fJ-Лучи в отличие от а-лучей неоднородны даже для одного п того — элемента, т. е. они состоят из электронов с различной скорсстъщ. Неггсс-лее быстрые из них движутся со скоростями, близкими к скоуостп
* Число Лошмидта, см. т. I, стр. 138.
Радиоактивность и изотопия
547
Рис. 63, Радиевые часы.
То, что частицы 0-лучей варяжепм отрицательно* **, следует на направления их отклонения в электрическом или в .магнитном поле. Это мол;но продемонстрировать такте на сконструированных Штруттом (Strutt) «радиевых часах», Опи представляют собой (см. рис. 63) маленькую стеклянную трубку, в которой заключен препарат радия (трубка I), изолированно укрепленную в эвакуированном стеклянном сосуде 2 на кварцевой нити 3. На нижнем копне трубки укреплены два тонких золотых листочка 4, а весь стеклянный сосуд внутри окрапирован станиолем 5, который заземлен. а-Лучи радиевого препарата задерживаются стенками стеклянной трубочки, а 0-лучи достигают с та полевого экрана. То, что 0-лучи песут отрицательное электричество, доказывается появлением положительного заряда на золотых пластинках. Если в приборе находятся около 30 мг бромида радия, то ул;е через 1 мин листочки полностью расходятся, соприкасаясь при этом со станиолевым экраном, и затем сразу опадают. Этот процесс может продолжаться сколько угодно долго н весь прибор представляет собой, так сказать, «часы», которые пе требуют завода.
Определяя отношение заряда к массе для частиц катодных и 0-лучей, Кауфман (Kaufmann, 1901—1906) смог доказать зависимость массы от скорости, следующую из теории относительности.
Максимальная энергия 0-частпц. Кинетическая энергия 0-частиц, испускаемых при каком-либо определенном радиоактивном распаде, может принимать любое значение внутри некоторых известных пределов. Но различные значения энергии рас пр од слепы по 0-частицам совершенно определенным образом. Если графически представить зависимость числа 0-частнц, имеющих некоторое определенное значение энергии, от величины энергии, то получим кривую, форма которой подобна форме такой же кривой для 0-лучей любого радиоактивного вещества. С увеличением энергии число 0-частиц, имеющих эту энергию, сначала растет, проходит через максимум и затем постепенно падает до нуля (см, рис. 66). Энергия, при которой число частиц равно пулю, является максимальной энергией, которую можно обнаружить у 0-частиц, испускаемых даннымвсщестлом. Эту энергию обозначают как £макс- Она является для каждого распадающегося с испусканием 0-частиц элемента характеристической. величиной точно так же, как величина пробега для элементов, распадающихся с испусканием а-частиц.
Ионизирующая способность. Ионизирующая способность 0-частиц на равных отрезках траектории значительно меньше, чем у а-частиц. Как и для а-частиц, способность уменьшается при увеличении скорости частиц
ионизирующая
а-Частицы радия С', скорость которых меньше только скорости а-частиц тория С', на равных отрезках траектории образуют приблизительно в девять раз больше ионов, чем 0-частицы такой же скорости, и в триста раз больше, чем самые быстрые 0-частицы.
В воздухе при атмосферном давлении самые быстрые 0-частицы образуют около 50—100 ионов па сантиметр траектории, а самые быстрые а-частицы — от 20 000 до 40 000,
Увеличение ионизирующей способности с уменьшением скорости достигает своего предела при значении скорости примерно 3 • 103 см/сек. При дальнейшем уменьшении скорости ионизация вновь уменьшается и вскоре совершенно исчезает.
Поглощение 0-лучей. Поглощение 0-лучей происходит по другому закону, нежели поглощение а-лучей.
* См. ссылку па стр. 539.
** Общая ионизация, как и в случае а-лучей, возрастает с увеличением начальной скорости частиц. Она пропорциональна начальной кинетической энергии частиц (ср, примечание на стр, 540).
35*
548
Глава 11
fi-Лучй в отличие от а-лучей не тормозятся внезапно, а постепенно ослабляются, что связано частично с их поглощением, но в большей степени обусловлено их «рассеянием», т. е. изменением траекторий р-частиц.
Если fi-лучи проходят через поглощающее вещество в направлении я (см. рис, 64), то уменьшение интенсивности J их пучка (измеряемой ионизацией при прохождения через определенный слой воздуха) выражается уравнением
Рис. 64,
Коэффициент пропорциональности р называют коэффициентом поглощения р-лучей для данном вещества. Он возрастает с увеличением плотности вещества. Кроме того, ого величина зависит от энергии р-частиц * ** я поэтому различна для р-лучей, испускаемых различными радиоактивными элементами. Например, коэффициент поглощения алюминия для р-лучей НХ2 равен 14, для р-лучей UXf—510, а для р-лучей RaD—5500. Поскольку скорость р-частиц при прохождении через поглощающее вещество уменьшается, то для каждой отдельной частицы ионизация, вызы-** ваемая ею па данном отрезке траектории, увеличивается. Но одновременно благодаря рассеянию значительно уменьшается число частиц, движущихся в направлении х. Конкуренция этих факторов приводят к тому, что, как правило, коэффициент и для пучка Р-лучей сдвппой начальной скоростью остается приблизительно постоянным. Интегрируя уравнение (1) при постоянном р, получаем
(2)
где J.q — интенсивность Р-лучей при их вхождепии в поглощающее вещество; х — пройдеппый лучом путь; е —- основание натуральных логарифмов.
3. Гамма-лучи
Проникающая способность у-лучой не только превышает такову© а- н р-лучей, но и выше, чем проникающая способность рентгеновских лучей. Существование у-лучей впервые было открыто Виллардом (1900) на примере радия. Испускание радием у-лучей легко обнаружить по их действию на фотопластинку или по вызываемой ими флуоресценции экрана, покрытого циан оплат ин ат ом бария или виллемитом. Конечно, при этой необходимо экранировать р-лучи. Промежуточный экран из свинца толщиной 2 мм полностью поглощает р-лучи. Труднее обнаружить у-пзлуче-ние слабо радиоактивных препаратов, например урана или тория **»
у-Йз л учение наблюдается пе у всех радио элементов, а лишь у некоторых из них-у-Излучение сопровождает как а-, так и [J-распады. Его появлояие объясняется освсэ-нымн положениями теории атомного распада следующим образом; после испускания а- пли (5-частицы многие атомные ядра остаются в возбужденном состоянии и отдано-избыток энергии в форме электромагнитных колебаний очепь высокой частоты, т.. е.. я форме у-излучения. В соответствии с их происхождением (первичные) у-л учи *** всегда дают линейчатый спектр, на основании которого можно сделать выводы об энергетических уровнях атомных ядер, аналогично тому как из оптических и реатгеновенгщ спектров можпо определить энергетические уровни атомных оболочек.
Поглощение у-лучей подчиняется, как и поглощение Р-лучей, соответствующшу экспоненциальному закону
* Коэффициент ц тем меньше, чем больше кинетическая энергия. Псзтсму сл'-величину можно использовать для приблизительного определения скорсгт р-чаггсг
** В случае урана и торпя у-лучи испускаются по пспосредствгиЕэ атгл-п ментами, а их продуктами распада (см. табл. 60 и 62).
*** О вторичном у-и ал учении см. на стр. 552.
Радиоактивность и изотопия
549
только в этом случае коэффициент поглощения ц имеет намного меньшие значения. Его величина, кроме природы поглощающего вещества, зависит также от дайны волны у-лучей. Чем меньше длина волпы, тем меньше и коэффициент йог лощения у-лучей, т. е. тем больше их проникающая способность.
ОБЪЯСНЕНИЕ ЯВЛЕНИЯ РАДИОАКТИВНОСТИ НА ОСНОВЕ ТЕОРИИ АТОМНОГО РАСПАДА
В 1903 г. английским исследователям Резерфорду и Содди удалось объяснить радиоактивные процессы на основании их теории атомного распада. Согласно этой теории, сущность явления радиоактивности заключается в самопроизвольном распаде атомов радиоактивных веществ. При распаде атомы испускают частицы, число которых соответствует числу распавшихся атомов. Атомы, образовавшиеся в результате радиоактивного распада, в общем случае в свою очередь являются радиоактивными я за счет дальнейшего распада переходят во все более простые атомы. В конце концов распад приводит к образованию стабильных нера-диоактивных атомов.
Эти представления объясняют длительное испускание энергии радиоактивным веществом без подвода внешней энергии. Излучаемая энергия равна уменьшению потенциальной энергии атома при его распаде. Для некоторых радиоактивных веществ (например, для урана) не удается установить уменьшение интенсивности излучения даже при длительном наблюдении. Но в соответствии с теорией атомного распада в этих случаях постоянство интенсивности излучения только кажущееся. Это как раз те случаи, когда за время наблюдения распадается исчезающе малая доля атомов. Но если за время наблюдения распадается значительная доля атомов, то легко можно обнаружить уменьшение интенсивности излучения. Для многих радиоактивных веществ измерение падения интенсивности излучения является важнейшей их характеристикой.
При атомных распадах образуются элементы, которые отличаются от исходных не только радиоактивными, по и, как было установлено, химическими свойствами. Следовательно, в случае радиоактивного распада мы имеем дело с процессом превращения элементов.
Радиоактивные ряды (ряды распада). В связи с тем что радиоактив- * ный элемент при своем распаде превращается в другой радиоактивный элемент, этот — в третий, следующий — в четвертый и т. д., образуется ряд продуктов распада, совокупность которых называется радиоактивным рядом соответствующего элемента- Встречающиеся в природе радиоэлементы образуют четыре радиоактивных ряда. Но только три из них имеют практическое значение, а именно ряд тория (табл. 60), ряд урапа — радия (табл. 62) и ряд актиния (табл. 63). Четвертый радиоактивный ряд, который имеет только теоретический интерес,— ряд нептуния (об этом радиоактивном ряде см. стр. 680 и 682). Каждым из этих рядов заканчивается неактивным элементом. Тот элемент, из которого непосредственно образуется следующий, называют материнским элементом. Непосредственный продукт распада радиоэлемента называют дочерним элементом.
Период полураспада и константа распада. Как следует из теории атомного распада, интенсивность излучения, испускаемого каким-либо определенным элементом, постоянно уменьшается. Время, за которое
550
Глава 11
интенсивность испускаемого элементом излучения уменьшится до половины ео первоначальной величины, называется периодом полураспада соответствующего радиоэлемента.
Если через No обозначить число атомов радиоэлемента, имеющегося в момент времени i = 0, т. е. в начале наблюдения, то, согласна теории атомного распада, число атомов элемента, оставшихся неизменными к моменту времени I, дается уравнением *
Nt=N0-e-u. (4)
В этом выражении е— основание натуральных логарифмов, X—копстапта, величина которой зависит от природы радиоэлемента. Эту константу называют константой распада. Обратная ей величина называется средней продолжительностью жизни атома данного элемента. Период полураспада Т, т. е. время, за которое число атомов ради о э.дем опта и интенсивность испускаемого им излучения уменьшаются наполовину, связан с константой распада соотношепием
7,=0,69315— , (5)
Л
как это следует из уравнения (4), если в него вместо I подставить период полураспада Т, а вместо Nt — половину начального числа атомов . Тогда получаем
Д“-=^ее~?'т или 1п 2 = — 0,69315.
2 Л Л
Для i=10T отношение *L=±=J^ .
Следовательно, за время, равное десяти периодам полураспада, число атомов радиоэлемента уменьшается примерно в тысячу раз. Поэтому во многих случаях по истеченпп десяти периодов Полураспада можно считать, что излучение, непосредственно испускаемое данным радиоэлементом, практически исчезает.
Определение констант распада. Логарифмируя уравнение (4), получаем
log Nt = log No—0,4343 Л t. (6"
Это выражение является уравнением прямой. Если в системе координат на ось ординат нанести значения log^Vj, отвечающие различным моментам времени t, нанесенным на ось абсцисс, то получим прямую лпгьпт:. наклон которой к оси абсцисс позволит определить константу ряспадд
tg а= 1Qg ЛЪ—tog дг£ =0)4343.k (!7
При определении константы распада этим методом нет пеобходимо&Еп в знании истинного числа радиоактивных частиц в измеряемом препарате Достаточно определить число частиц, испускаемых за достаточно коротнпз интервалы времени. Это число пропорционально общему количеству радиоактивного вещества, а фактор пропорциональности исключается npz вычитании логарифмов. Время, когда начато наблюдение, т. е. t = С может быть произвольным моментом.
Уравнение (6) справедлив0 для того случая, когда имеется однородное радиоакпгз-вое вещество. Если же присутствует смесь атомов различного сорта с раалттчттнпп секстантами распада, то получается не прямая, а некоторая кривая. Однако в этих сау=-
* Вывод формулы см. на стр. 561.
Радиоактивность и изотопия
551
ях, используя математический анализ, можно определить отдельные значения X для различных видов атомов. Конечно, если такую смесь можно разделить химическим путем, то следует использовать именно эту возможность.
(8)
Определение констант распада относительно долгоживущих веществ производится другим методом, так как для определения этих констант по первому методу потребовались бы годы. Если период полураспада равен годам, то количество радиоактивного вещества за несколько часов практически не изменяется. Поэтому в таких случаях в уравнение (И), dN
[стр, 561J вместо отношения дифференциалов можно ввести отношение
„ AN конечных разностей , и тогда получим
AN 1 А----At ' N '
Значение N можно получить из веса и общего состава препарата, массового числа радиоактивного атома и числа Авогадро. Уменьшение ЛГ( = —ДЛГ) за время At равно числу частиц, испускаемых препаратом за это время.
Не для всех радиоактивных элементов можно определить период полураспада, или константу распада, непосредственным измерением, т. е. наблюдая уменьшение интенсивности излучения. Это невозможно прежде всего для особенно долгоживущих элементов, таких, как торий и уран (уран I). Для этих элементов уменьшение интенсивности излучения за практически возможные перподы времени совершенно незаметно. Но в этих случаях период полураспада, или константу распада, можно определить из отношения количеств веществ, находящихся в радиоактивном равновесии.
В случае очень быстро распадающихся веществ часто для приблизительной оценки величины константы распада используют эмпирически найденное Гейгером и Нат-толом (Nuttall, 1911) соотношение между величиной пробега а-частиц, g и константой распада X:
logX=4+Blog q, (9)
где А н В — постоянные, из которых А для трех радиоактивных рядов имеет несколько различные значения. Уравнение Гейгера — Наттола выражает связь двух фундаментальных величин радиоактивного превращения, а именно связь между константой распада и скоростью, с которой а-частицы вылетают из атомного ядра. Объяспить эту связь удалось Гамову в 1928 г. (см. стр. 631).
Кривые Саржента. Связь, аналогичная связи между величиной пробега а-частиц и константой распада, существует, как нашел в 1933 г, канадский физик Саржент, между максимальной анергией Р-лучей Емакс и константой распада. Если для различных естественных радиоактивных 0-излучателей построить график зависимости log X от log Емакс, то окажется, что большинство точек лежит на двух прямых линиях (см, рис. 78). Оказалось что, так называемые «кривые Саржента» можно объяснить на основании теории p-распада, разработанной Ферми (1934) вслед за Паули (1931). В соответствии с этой теорией с хорошим приближением оправдывается уравнение X^fc-Емако, откуда при логарифмировании получаем
log X=log X-J-5 log £мапс. (10)
В этом уравнении к— некоторый параметр, который может принимать различные значения в зависимости от того, высока или мала вероятность связанного с Р-излуче-пием перехода атомного ядра из высшего в низшее энергетическое состояние. Если для X принять два различных значения, то из уравнения (10) получим две прямые линии, которые приблизительно отвечают найденным Саржентом для значений к ">• Р-излучатслей естественных радиоактивных рядов. Теория оставляет возможность для к принимать более чем два значения. По-видимому для Р-излучателей, полученных при искусственных превращениях атомов, наблюдаются другие кривые Саржента.
552
Глава 11
Радиоактивное равновесие. Если разные члены радиоактивного ряда не отделены, а, напротив, каждый из них длительное время соприкасается со своим материнским элементом, то с течением времени меягду отдельными членами ряда устанавливается такого рода равновесие, при котором для каждого элемента количество распадающихся в единицу времени атомов равно количеству образующихся за этот же период. Если начальным членом ряда является «долгоживущий» элемент, как, например, радий или торнн, то суммарная активность ряда в течение длительного времени остается практически постоянной. Подробнее сущность радиоактивного равновесия будет рассмотрена ниже (стр. 560).
Вторичные лучи
На основании теории атомного распада можно было бы ожидать, что после испускания а-лучей атомы-остатки будут иметь удвоенный отрицательный заряд. Однако оказалось, что эти атомы притягиваются отрицательным электродом. Следовательно, они каким-то удивительным образом приобретают положительный заряд. Это свойство можно использовать, чтобы сконцентрировать их в желательном месте, например на поверхности металлической проволоки. Для этого нужно только поднести такую отрицательно заряженную проволоку к раднопрепарату.
Лучи отдачи. Можно показать, что положительный заряд атома-остатка объясняется последующей перезарядкой, которую претерпевают первоначально отрицательно заряженные атомы-остатки. По законам механики атомы после испускания а- иле 0-частиц испытывают отдачу, точно так же как ствол орудия после выстрела. Часть атомов-остатков благодаря этой отдаче вырывается в свободное пространство с необычней скоростью, особенно при испускании а-частицы, В этом случае скорость атома-остатка составляет около 2% скорости а-частицы, или, по порядку величины, около скорости света. Благодаря столь большой скорости атомы отдачи вызывают довольыЕ сильную ионизацию молекул газа, с которыми они сталкиваются. Но прежде веет: они сами теряют при таких столкновениях оба избыточных электрона и иногда еса один дополнительный электрон. Этим и объясняется их положительный заряд. Еетг исключить по в озм ежи осты столкновения с молекулами газа, ла пример проводя измерения в высоком вакууме, то перезарядки не происходит,
В газе с давлением примерно равным атмосферному атомы отдачи теряют скег высокую скорость, а с ною и ионизирующую способность, уже пройдя путь в долю ылг-лнметра. После того как их скорость падает до нормальной скорости молекуд тьет они ведут себя как обычные ионы газа, Атомы-остатки, образующиеся при 0-распад с самого начала не имеют скоростей, превышающих скорость молекул газа прп ет мальных условиях.
Отрицательные вторичные лучи. Лучи отдачи представляют собой пример с~ -ручного излечения, вызываемого радиоактивными процессами. Существуют еще и ?
вторичные излучепия, а именно положительные и отрицательнее, Отрицатель^ -вторичное излучение возникает при попадания а-, 0- или у-лучей па атомы др;," веществ и состоит в основном из электронов, скорость которых значительно екет -скорости 0-частиц. Дж. Дж, Томсон, который впервые наблюдал это излучение в 12. - -назвал его б-лучами, б- Л учи (называемые также медленными 0-лучами) в otx~z « вызываются действием а-лучей. а-Лучи способны также вызывать появление втерт- : у-лучей, впрочем, очень слабых,
Более сильное вторичное у-изл учение возникает при поглощении 0-лучлй. возникновение полностью аналогично возникновению рентгеновского излучелпт -попадании катодных лучей на антикатод. «Вторичное 0-иалученце», соировсч.т“_ь : * 0-распад, состоит в ос и овном из отраженных или рассеянных первичных 0-т г Но при этом могут возникать и собственно вторичные 0-лучи. Последние пмепгт зт , i тер р-лучей.
у-Л учи могут вызывать как вторичные у-л учи, так и вторичные 0-лучп.. 7" ч* наблюдается и для рентгеновских лучей.) Вторичные 0-лучи, возникаю ттт. г --вии у-лучей, по проникающей способности соответствуют первичным р-—-- ?- т зовапие п вызываемая ими ионизация в основном и обусловливают _
ствие у-л у чей.
Радиоактивность и изотопия
553
Положительные вторичные лучи. Особый интерес представляет положительное вторичное излучение, возникающее при столкновении а-лучёй с другими атомами. Если масса сталкивающегося атома примерно равна массе а-частицы или еще меньше, то такие вторичные лучи имеют значительный пробег. Из законов столкновения можно рассчитать, что при столкновении с ядром водорода, последнее в наиболее благоприятном случае, т. е. при центральном ударе *, приобретает скорость в 1,6 раза большую, чем скорость а-частицы. Поскольку пробег пропорционален третьей степени скорости, такие ядра водорода должны обладать примерно в четыре раза большим пробегом, чем а-частицы, с которыми они столкнулись. Что это действительно так, впервые наблюдал в 1914 г. Марсден, сначала при прохождении а-лучей через газообразный водород, а затем при их прохождении через любое содержащее водород вещество.
ОПРЕДЕЛЕНИЕ ЭНЕРГИИ РАСПАДА
Вначале для определения энергии распада пользовались калориметрическими методами, т. е. измеряли тепло, выделяющееся при радиоактивном распаде. Однако область применения калориметрических методов ограничена. В общем случае они пе позволяют также проводить точных измерений энергии отдельных видов излучения (а-, р-, у-и вторичных излучений), а для более глубокого понимания сущности процессов радиоактивного распада большое значение имеет знание величин энергии, связанной с каждым видом излучений. Поэтому в настоящее время используют исключительно такие методы, которые позволяют непосредственно дифференцировать все виды излучений. Для суммы этих величин получают как раз то значение, которое определяется калориметрически.
О методах определения энергии распада по величине связанной с ним потери массы см. стр. 613.
Энергия, освобождающаяся при а-распаде, проявляется как кинетическая энергия продуктов распада. Следовательно, она равна сумме кинетических энергий а-частицы и атома-остатка. Величина кинетической энергии атома-остатка относится к величине кинетической энергии а-частицы, как масса а-частицы к массе атома-остатка. (Это непосредственно следует из закона сохранения количества движения.) Следовательно, чтобы вычислить энергию распада, надо только найти кинетическую энергию,, а-частицы, испускаемой при распаде, и умножить ее на отношение , где М — массовое число (т. е. округленный атомный вес) продукта распада.
Значительно сложнее расчет в случае fj-распада, поскольку р-частицы не обладают одной определенной энергией. Как следует из дальнейшего, .иа.чеи.налъное значение эяергли р-частиц представляет собой как раз энергию, освобождающуюся при fJ-распаде.
* Для центрального упругого удара справедливы следующие законы сохранения*, лцс, -1~т2С2~т^1
Я
где и тг — массы обоих тел; с( и с3— их скорости до столкновения; и — скорости после столкновения, Если положить — 4, т2 —1 и с3 = 0, то получгкг
16
^=УО‘С1-
554
Глава 11
Как а-, так и p-распад часто сопровождаются у-излучением. Последнее может быть связано как с оболочкой атома, и в таком случае имеет характер вторичного излучения, так и с ядром атома. Во втором случае у-излучение вызвано тем, что ядро, испустившее а- или р-частицу, переходит из состояния с большой энергией в состояние с меньшей энергией. Это явление совершенно аналогично явлениям, сопровождающимся испусканием света или рентгеновского излучения, при переходе электронов атомной оболочка из состояний с более высокой в состояния с более низкой энергией, То, что в ядре атома, так и в атомной оболочке, существуют различные дискретные уровни энергии, имеет большое значение для теории ядерных сил, т. е. сил, которые удерживают вместе составные части атомного ядра. Как будет показано в дальнейшем, эти уровни энергии можпо пайти ие только из наблюдений у-излучений, но также из других явлений, наблюдающихся при сс-распаде.
Энергия «-частиц. Энергию а-частиц можно точно определить по их отклонению в магнит нои поло. Более удобно и в общем не менее точно определяется эта энергия из весивши пробегов а-частиц, Поскольку связь между энергией и пробегом а-частиц довольно сложна, лучше всего ее представить в виде кривой, Такие кривые можно найтп в работах Ливингстона и Бете и Халловея (Livingston, В о t h е, Rev. mod. Phys., 9, 266 (1937), H al 1 ov ay Livingston, Phys. Rev., 54, 31 (1938)]. Приведенная на стр. 544 формула Гейгера является лишь приближенной. Даже при незначительных требованиях к точности эту формулу можно применять только к а-ча-стнцам, пробег которых в воздухе составляет3—7 см. При меньших величинах пробега начальная спорость а-частиц существенно больше, при больших — существенно меньше, чем это следует из формулы Гейгера.
В общем а-частицы, испускаемые одним определенным видом радиоактивных атомов, все имеют совершенно одинаковые начальные скорости, или начальные энергии. О диак о встречаются случаи, когда некоторые группы а-частиц испускаются с существенно другой энергией. Например, торий С испускает шесть групп а-чаетиц. Каждая из этих групп имеет различную величину начальной энергии. Эти значения начальной
Таблица 59
Спектр а-частиц тория С
Энергия а-частиц, Af 90 Вероятность, % Энергия распада, Мае Отличие энергии распада от максимальной энергии
6,084 27,15 6,201 -—
6,044 69,78 6,161 0,040
5,762 1,81 5,873 0,328
5,620 0,16 5,728 0,473
5,601 1,09 5,709 0,492
5,480 0,01 5,585 0,616
энергии приведены в первой колонке табл. 59. Цифры, приведенные во второй колонке этой таблицы, показывают долю а-частиц, имеющих некоторое определенное значение начальной энергии. В таких случаях говорят О спектре а-частиц. В приведенном примере существование групп а-частиц с различной энергией объясняется тем, что ядро тория С", образующееся за счет а-распада ядра тория С, частоспачала образуется в св о зб у щ дени ом» состоянии, т. е. в состоянии с большей энергией, чем энергия ядра в основном состоянии. Энергия распада ядра тория С, а следовательно, п энергия испускаемой при этом а-частицы наибольшая в том случае, когда при а-распаде непосредственно образуется ядро тория С" в осноеком состоянии. Но если при распаде образуется возбужденное ядро тор ня С" то энергия распада уменьшается на разницу между энергиями возбужденного и невозбужденного ядер (см. рис. 65). Разница между энергиями возбужденных ядер и ядра в основном состоянии приведена в четвертой колонке табл. 59. Величины энергии распада, приведенные в третьей колонке табл. 59, полу-
Радиоактивность w изотопия
555
208+4 * „ , чепы умножением значении энергии первой колонки на отношение ^д В большинстве случаев ужо спустя неизмеримо малый интервал времени возбужденное ядро излучает избыточную энергию в форме у-квантов и переходит непосредственно или через промежуточные состояния в основное состояние. Следовательно, энергии
Рис. 65. Спектр а-частиц тория С. Связь между энергетическими уровнями ядра тория С" и энергиями у-нвантов, испускаемых при образовании тория С".
Числа на стрелках указывают энергию (Мае), освобождающуюся нри испускаиии этих излучений. Слева у энергетических уровней возбужденных состояний ядра приведены разницы их энергий по сравнению с энергией основного уровня.
Энергетические уромиядра
у-лучей также должны быть равны разницам энергетических уровнен и намерение энергии у-лзлучеиия. дает второй, независимый метод определения положения энергетических уровней в возбужденных ядрах тория С", В правой части рис. 65 приведены расчетные значения энергии таких у-лучей. Они совпадают с наблюдаемыми. Как в и ди о, из рисунка, проявляются пе все возможные переходы между энергетическими уровнями ядра тория С", а только некоторые ив них. Это показывает, что аналогично переходам в атомной оболочке для переходов в ядре также существуют справила отбора», согласно которым некоторые из них «запрещены», т. е. очень мало вероятны,
Спектр «-частиц может быть вызван и тем, что некоторые из ядер, испускающих а-частицы, в момент распада находятся л возбужденном состоянии. В таком случае, кроме нормальных а-частцц, будут испускаться и а-частицы с высокими энергиями. Одиако число таких а-частиц очень мало по сравнению с числом нормальных а-частиц, так как в общем переход возбужденного ядра в основное состояние происходит за счет у-излучения Значительно быстрее, чем распад е испусканием а-частицы. Поэтому «-распад возбужденных ядер наблюдается только для особо короткоживущих «-излучателей.
Энергия у-лучеп. Энергия у-кванта, каки энергия любого кванта света, определяется произведением /iv. Одпако частоту v или соответствующую ей длину волны к можно непосредствен пи определить только в исключительных случаях, Для этого, как и в случае рентгеновской спектроскопии, наблюдают рассеяние на гранях кристалла. Нов случае у-лучей, частота которых значительно больше частоты рентгеновских лучей (а это в первую очередь относится к большинству первичных у-лучей), этот метод
* 208 равно массовому числу тория С' (см. табд. 60).
556
Глава. 11
непригоден. Приблизительной мерой энергии у-излучения может служить его проникающая способность. Чем «жестче^, т, е. чем большей энергией обладают у-лучи, тем менее они поглощаются. Но для точных измерений, однако, постукают иначе; измеряют энергию электронов, которые вырываются у-излучением из оболочки атомов, на которые падает э,то излучение. Возникающие под действием у-лучей «вторичные р-лучи» имеют в отличие от «первичных р-лучей», связанных с атомпым ядром, дискретные значения энергии. Кинетическая энергия электрона, вырванного у-квантом, равпа энергии у-кванта, уменьшенной на первоначальную энергию связи электрона *. Последнюю можно определить на основании характеристического рентгеновского излучения, испускаемого при вырывании электрона с энергетического уровня, на котором он находился в оболочке атома (см, том I, гл. 7). Кинетическую энергию свободного электрона можно определить по его отклонению в магнитном поле. Из того, что вырываемые первичным у-пзлучепием электроны имеют вполне определенные дискретные значения энергии, следует, что такие Же определенные дискретные значения энергии имеют и сами у-л учи. Это в свою очередь может быть обусловлено только тем, что атомное ядро само может находиться только в определенных дискретных энергетических состояниях и при переходе из состояния с более высокой энергией в состояние с более низкой энергией разница энергий излучается в форме у-кванта. Пример такого процесса ужо был рассмотрен выше (рис. 65). При этом речь шла о р-цз л учении, соиро-вождающем «-распад. Точно так нее у-излучедие может сопровождать и p-распад. Следовательно, и Р-ра спад может приводить к образованию как нормальных, так й возбужденных атомных ядер.
Внутренняя конверсия у-лучёй. Часто бывает так, что у-квант вырывает электрон из электронной оболочки того самого ядра, которое испускает у-пЗл учение. При этом возникает рентгеновское излучение, характерное дня дочернего элемента. Этот процесс-называют внутренней конверсией, у-лучей, Как правило, такую внутреннюю конверсию претерпевает только небольшая часть испускаемых при радиоактивном распаде у-кваптов, В общем число таких у-кв антов тем меньше, чем выще энергия у-изл учения, f-Лучи, образующиеся при внутренней конверсии у-лучей, по своему поведению близки к вторичному излучению и имеют подобно у-лучам и в отлично от первичного р-пзлз-чения дискретные значения энергии.
Энергия р-частиц. Первичные р-лучи, т, с', электроны, испускаемые непосредственно радиоактивно распадающимся ядром, не имеют, как уже упоминалось, дискреты^
Рис. 66. Спектр р-частнц радий Е.
значений анергии. Они имеют все возможные скорости от пуля до различного ДЛЛ ЕД;: дого р-язлучатедя максимального значения. Если графически представить завЕкждхд. числа Р-частиц, имеющих некоторое определенное значение энергии, от г-д:”.:;:
* Это справедливо для тек случаев, когда речь идёт о процессах, езддддддд фотоэффектом. Но в некоторых случаях может наблюдаться эффепп при котором у-квант передает электрону только часть своей энергии, а сам —•
по законам упругого удара. При этом изменяется с о ответствен и о уменье—г— -:—
при столкновении частота у-кванта, Из наблюдений эффекта Комптона д.’г:" .де лить первоначальную энергию у-кв анта.
Радиоактивность к изотопия
557
Мзвг13РептутЗ 7,179—i I- I । ft ft J. J if
^ПлЯШТии 0,057™ \ 0.061 7 j 00871Р75 0,205 77 | Г
О.®7-
0,776-
' 0,227
7
Эяергеттеские аровни ядра зз9рц
0,503-
v 0,275 Г
р-тастиц, то получим кривую, приведенную на рис. 66, Эта кривая называется спектром р-частиц. Общая форма кривой для р-частиц одинакова независимо от источника их происхождения. Только коночная точка кривой, отвечающая максимальному значению энергии, наблюдающемуся у исследуемых р-частйц (обозначенная па рис. 66 как #MaKC) различна для р-частиц различного происхождения. Определение этого предельного значения энергии требует тщательного измерения распределения числа частиц по различным значениям Эпергпи, так как кривая приближается к оси абсцисс под довольно острым углом. Максимальные скорости 3-частиц, испускаемых различными Р-излучателями, составляют в общем от 25 до 99% скорости света.(Им отвечают максимальные значения энергии2?макс в пределах 0.625—3,15 Л/м. Чаще всего опи лежат около 1 Мэе.
Для определения скорости или кинетической энергии р-частиц и распределения частиц по величине энергии используют различные методы. Одним из наиболее употребительных является так называемый метод маа-нитной линзы, т. е, проволочной катушки, по которой идет электрический ток, в сочетании со счетной трубкой Гейгера — Мюллера. При прохождении катушки р-частицы отклоняются от их первоначального направления и описывают спираявные траектории. Отклонение различно в зависимости от скорости частиц и напряженности магпптного поля, создаваемого проходящим через катушку электрическим током. При давней силе тока на счетчик попадают только те Дчастйцы, которые имеют вполне определенную скорость. Варьируя силу тока, можпо заставить попадать на счетчик все P-частицы по порядку, начиная с наименьших скоростей и кончая наибольшими, и тем самым можно найти чпслочастиц, имеющих некоторую определенную скорость.
Энергия, освобождающаяся при ₽-распаде, равна но сродней кинетической энергии f>-частиц, а ее .какси-мальному шче«т. Это следует как из измерений потери массы, связан-ИОЙ С , .,г,цич. ic Ji —-',1П лглаии-О! 1'1 juppi ,14 ^.1 . S'SCJ,
так и из других наблюдении (ср, стр, 565). Однако большая часть р-частИГГимеет меньшую энергию, чем освобождается при р-распаде атомного ядра. Чтобы объяснить эту потерю энергии *, была выдвинута гипотеза, что при В-рас па де, кроме электронов (пли позитронов, если речь идет о Р+-распаде, см. стр. 600 и 616), атомное ядро испускает и другие (и притом незаряженные) частицы, которые технически еще не умеют обнаруживать. Во всяком случае, масса таких частиц значительно меньше массы нейтрона, а вероятно, и меньше, чем масса электрона. Возможно, что их масса покоя — как и масса покоя квантов света — равна нулю. Эти гипотетические частицы назвали нейпгрипа.
Как уже упоминалось, при Р-ра спа де, равно как и при а-распаде, часто продукт распада получается в возбужденном состоянии. Если это имеет место, то ядра материнского вещества часто испускают несколько групп Р-частиц, каждая из которых имеет непрерывный спектр, похожий па спектр, приведенный на рис, 66, но с различными значениями максимальной энергии Так как спектры различных р-частиц перекрываются, то часто очень трудно, а иногда и просто невозможно непосредстсгапо определить: максимальные значения энергии различных групп р-частиц. Bnpcssi,
* Если бы при Р-распаде нейтрино не испускалось, то, кроме закона смрзгжддя энергии, был бы нарушен также закон сохранения мбмепта вращения.
У J 7 7
Рис. 67. р-Распад неятупия-239 и у-йзлучение образующихся возбужденных ядер плутония.
Числа на стрелках указывают максимальную энергию различных групп р-частйц или энергии у-лучей (.Mas). Слева у энергетических уровней возбужденных состояний ядра приведены Полученные из энергий р-частнц разницы их энергий по сравнению е энергией основного уровня. Числа в скобках указывают, сколько процентов от общего числа Д-частиц приходится аа данную группу.
0-
пеции иотерп массы, связан-
р-распадом по закону эквивалентности массы и энергий (см. стр. 609)
558
Гласа 11
в большинстве случаев для определения энергии распада из значений энергий 0-частпц достаточно найти максимальную энергию для одной группы 0-частиц, если имеется падежный критерий того, какое из обязательно наблюдавшихся в этих случаях у-излучеаий сопровождает испускание данной группы 0-частиц. На рис. 67 приведен сличай, когда для всех групп испускаемых 0-частиц были определены максимальные значения энергии. Получающиеся при этом энергетические уровни ядра продукта распада полностью согласуются с теми, которые можно рассчитать па энергии наблюдающихся при этом у-лучей *. Схема распада, представленная па рис. 67, относится к элементу нептунию, входящему в группу трансурановых элементов. Она относительно проста и во многом аналогична приведенной на рис. 65 схеме распада с испусканием нескольких групп а-частиц. Две более сложные схемы 0-распада показаны на рис. 81 и 82. Они относятся к радиоактивным превращениям, при которых в различных энергетических состояниях находятся не только ядра, образующиеся в результате 0-распада, но и ядра, испускающие 0-частицы.
ЭМАНАЦИИ И АКТИВНЫЕ ОСАДКИ. РАДИОАКТИВНЫЙ РЯД ТОРИЯ
То, что радиоактивные процессы связаны с превращением элементов, впервые было показано при исследовании эманаций и активных осадков.
Эманациями называют газообразные вещества, которые постоянно выделяются радиоактивными элементами — радием, актинием и торием. и которые в свою очередь являются радиоактивными. Как ужо указыва-лось ранее (см. т. I, стр. 146), эманации имеют характер инертных газов.
Первым открытым радиоактивным газом была этанация тория. Она была обнаружена вскоре после установления радиоактивности тория. [Оуэнс (Owens) н Резерфорд, 1899—1900 гг J Вслед затем были открыты эмли/щин. радия и актиния.
Исследование процессов, связанных с образованием и распадом эманаций, сыграло огромную роль в разработке теории атомного распада. Поэтому следует ближе ознакомиться с закономерностями этих процессов, и это лучше всего сделать на примере радиоактивного ряда тория (табл. 60), так как впервые эти закономерности были установлены для членов этого ряда.
К открытию эманация тория пришли следующим образом. Сначала различные исследователи заметили, что радиоактивность соединений тория иногда сильно колебалась, если эти соединения исследовались в открытых сосудах, а при работе в закрытом сосуде радиоактивность только возрастала до некоторой постоянной конечной величины. Но если после этого через сосуд пропускали ток воздуха, то интенсивность излучения вновь уменьшалась. Как показал Оуэнс, это излучение проходило через пористую бумагу, которая полностью поглощала обычное а-излученпе, Резерфорд объяснил это явление, предположив, что препараты тория выделяют в воздух частицы, которые сами могут служить источником радиоактивного излучения. Он смог показать, что эти частицы вели себя совершенно так же, как молекулы газа; они захватывались током воздуха, проходили вместе с ним через воду, могли проходить через фильтр из ваты и диффундировали через бумагу, но полностью задерживались тонкой пластинкой слюды.
Резерфорд нашел, что все предметы, с которыми соприкасалась эманация, в свою очередь становились радиоактивными. Такая «индуцированная радиоактивность», как показал Резерфорд, связана с тем, что на этих предметах оседали выделяемые эманацией твердые радиоактивные частицы —«активный осадок». Резерфорд показал также, что такой актпЕ-ный осадок должен состоять из нескольких веществ. Форма кривой, выражающей зависимость индуцированной активности от времени, в свод? очередь зависит от времени экспозиции, т. е. от времени, в течение которого
* Конечно, измерение энергии у-лучей позволяет определить не сами гззрзе-тнчсские уровни ядра, а различия в их энергиях.
Радиоактивность и изотопия
559
предмет соприкасался с эманацией (рис. 68). Из формы кривой активности Резерфорд заключил, что активный осадок тория должен состоять по крайней мере из двух продуктов с различными периодами полураспада. В настоящее время эти продукты распада называют торий В и торий С.
Вскоре, пользуясь различной летучестью, удалось разделить эти два вещества: при 1000° тории В испаряется. Остающийся торий С имеет период полураспада 1 час. Из кривой постепенного уменьшения индуцированной активности, полученной дня длительной экспозиции (кривая II на рис. 68), получается период полураспада ~ 11 час. Следовательно, этот период полураспада относится к торию В. Кривые,
Рис. 68. Зависимость уменьшения активности, индуцированной эманацией тория, от времени экспозиции.
Кривая I — короткая экспозиция; кривая II — длительная экспозиция.
Рис. 69. Активность тория, свободного от тория X, увеличивается с такой же скоростью (кривая I), с какой уменьшается активность тория X (кривая II). 5)
Отклонение хода кривых от нормального в первые 2 дня объясняется образованием и исчезновением Других продуктов распада тория (активных осадков).
приведенные па рис. 68, получают простое объяснение, если предположить, что торий G образуется из не имеющего собственного а-излучепия тория В, период полураспада которого равен 10,6 часа. В таком случае при короткой экспозиции сначала имеется только практически торий В. Поэтому а-радиоактивность сначала равна нулю. Она возрастает, по мере того как из тория В образуется торий С. Но так как торий С распадается быстрое, то его количество вновь начинает уменьшаться, как только количество тория В уменьшится настолько, что торий С не может более образовываться с той скоростью, с которой он распадается. Напротив, в случае долгой экспозиции в препарате с момента наблюдения содержится то количество тория С, которое соответствует содержащемуся в осадке количеству тория В, и наблюдается простое уменьшение «-активности в соответствии с кривой II. Это уменьшение a-активности связано с уменьшением образования тория С из тория В, по передает уменьшение количества тория В, хотя пе он, а его продукт распада, торий С, испускает а-частицы.
В 1906 г, Ган на основании того, что торий С испускает а-лучи с двумя различными пробегами, пришел к выводу, что природа этого продукта должна быть еще болео сложной, и в течение последующих Десяти лет разработал теорию двойного распада тория С, в соответствии с которой этот элемент переходит в неактивный изотоп свинца, торий D (ториевый свинец), с одной стороны, путем а-распада с последующим испусканием Р-частиц, а с другой стороны, путем. Р-распада с последующим испусканием а-частиц. При втором пути образуется чрезвычайно короткоживущий элемент, торий С', продолжительность жизни которого была определена пе непосредственно, а оценена при помощи соотношения Гейгера — Паттола между продолжительностью жизни и величиной пробега а-частиц. Опа составляет одпу десятимиллионную секунды. Другой довольно
560
Глава 11
короткоживущий, но, конечно, «долгоживущий» по сравнению с торием С' продукт, торий- Л, был обнаружен впервые в 1911 г. Гейгером и Резерфордом. Эти исследователи пришли к выводу о существовании тория А на основании наблюдения Гейгера и Марсдена, что эманация тория испускает а-лучи с двумя существенно различными пробегами.
Одновременно проводили исследования членов радиоактивного ряда тория, которые предшествуют эманации тория. После того как в 1900 г. сэр Вильям Крукс показал, что от урана химическим путем можно отделить вещество, которое действует на фотопластинку значительно сильнее, чем уран, — это вещество было названо уран X (теперь его называют уран X,)— Резерфорд и Содди нашли, что и от тория можно химически отделить вещество, которое во много тысяч раз более активно, чем торий, из которого оно образуется. Опи назвали его по аналогии с ураном X торием X. Но если сравнить активность тория, от которого был отделен торий X, с активностью последнего, то можно наблюдать своеобразное явление, что в то время как торий X постепенно теряет свою активность, активность пространственно полностью отделенного от него тория вновь возрастает (рис. 69). Более подробное исследование этого явления на примере тория X и урана X привело Резерфорда к формулировке теории атомного распада.
Резерфорд показал, что нетканую в займе за висим ость изменений активности тория и тория X можно понять, если допустить, что торий продолжает в той же мере образовывать новые количества тория X, а активность последнего уменьшается по экспоненциальному закону.
Эту связь наглядно можно пояснить па следующем примере: "возьмем два градуированных сосуда, имеющих в пииспей части отводы и крапы (например, оба измерительных цилиндра колориметра), и отрегулируем краны так, чтобы при одной и той же высоте столба жидкости опа вытекала из них за одно и то же время, Пусть :в начале один сосуд пуст, а во второй из большого сосуда (который настолько широк, что в течение всего опыта высота столба жидкости в ном практически не изменяется, т. е. в простейшем случае водопровод) вливается вода. Отрегулируем скорость наполнения цилиндра водой так, чтобы при некоторой определенной высоте столба жидкости в цилиндре уровень воды в нем оставался постоянным (высота h-,, рис. 70), т. е. сколько воды в него вливается, столько и вытекает. Этот случай аналогичен препарату тория с постоянной активностью, т. е, торшо перед отделением тория X. В зтом случае постоянная активность препарата тория соответствует в нашей модели некоторой постоянной при данных условиях скорости вытекания воды. Но скорость вытекания воды пропорцио-
нальна высоте водяного столба в цилиндре (при условии, что крап ведет себя как узкая трубка, к которой применим закон Пуаза, и можно с достаточной точностью принять, что это так). 13 тот момент, когда прекращается подача воды в цилиндр, высота столба воды в пем. начинает уменьшаться, Это же происходит п с акшвиостыв тория, поело того кап отделен торий X. Пока ториц X находится вметтг с тернем, его количество и активность остаются постоянными. Но как только X сходен от тория, его активность начинает уменьшаться, как показысгист —is, со
Р и с. 70. Модель, демонстрирующая распад радиоактивного вещества и накопление ого в материнском радиоактивном препарате.
Радиоактивность и изотопия
561
тому же закону, что и скорость вытекания жидкости из несвязанного с резервуаром цилиндра А. Но в той же мере, как в от ом сосуде уменьшаются количество и высота столба воды, а тем самым и скорость се вытекания, в сосуде Б, который подставлен под струю в тот момент, когда был отставлен сосуд А, количество и высота столба воды увеличиваются. Если проводить такой опыт, то обнаружится, что высоты столбов воды, /ц и Л2, в обоих сосудах А и Б постоянно дополняют друг друга до Л3, т. е. до высоты столба, отвечающей равновесию. И тому же закону' подчиняется общая активность отделенного и распадающегося тория X и вновь образующегося тория X в старом препарате тория. Ли типи ость отделенного тория X в каждый момент времени уменьшается ровно настолько, насколько увеличивается активность вновь образовавшегося тория, 1г сумма обеих активностей всегда равна первоначальной активности.
Легко вывести закон, по которому изменяется скорость вытекания воды из сосуда А с того момента, как прекращен в пего приток воды из резервуара. Если считать, как было принято, что скор ость вытекания пропорциональна высоте столба воды в сосуде, то скорость вытекаппл ее будет уменьшаться тем скорее, чем скорее будет падать высота столба. Однако высота столба воды в цилиндрическом сосуде пропорциональна количеству воды и уменьшение количества воды прпорционально скорости вытекания. Последняя же, т. е. скорость уменьшения столба воды в сосуде, пропорциональна в кажцдш момент времени общему количеству воды N в new.
ям
(И)
где f — время, а). — некоторая постоянная, зависящая, кроме ряда других факторов, и от ширины отверстия.
Точно такой же закон справедлив и для уменьшения активности тория X. В этом случае IV — количество активного вещества, ал — его константа распада. Интегрируя уравнение (11) в пределах от 0 до £ и обозначив при этом количества активного вещества в моменты времени t = 0 и t как Л'о и Nt соответственно, получаем
— ln^ = U, или Nt=Noe~M.
Это уравнение не что иное, как приведенный на стр. 550 Закон радиоактивного распада. Его применимость не ограничивается только случаем тория X. Этот закон сейчас проверен на огромном числе других радиоактивных веществ, и нет никаких сомнений в том, что каждый радиоактивный распад чистого вещества подчиняется этому закону.
Поскольку в ряду следующих Друг за другом превращений каждое отдельное превращение подчиняется закону, выраженному уравнением (11) — конечно, при этом константа распада X различна для каждого члена ряда,— из этого следует важная связь между количествами отдельных веществ в радиоактивном ряду. Для каждого вещества ряда скорость распада в соответствии с'уравпением (11) пропорциональна его количеству. Но последнее для каждого вещества, присутствующего вместе со своим материнским веществом, зависит от скорости, с которой это вещество образуется из материнского. Следовательно, в конце концов должно наступить состояние равновесия, при котором в каждый момент времени распадается кап раз такое количество атомов данного вещества, сколько их вновь образуется (радиоактивное равновесие). Пока скорость образования больше скорости распада, количество данного вещества JV, а вместе с пим и скорость его распада будут возрастать. Напротив, если скорость образования меньше скорости распада, то количество вещества JV будет уменьшаться до тех пор, пока скорость его распада пе станет равной скорости образования. Следовательно, уравнение (11) позволяет на основании значений констант распада Ха, и т. д. различных веществ радиоактивного ряда рассчитать количества TVi, Nz, Л'з и т. д. этих веществ, которые будут находиться между собой в состоянии радиоактивного равновесия. Поскольку в этом случае скорости распада различных 36 Г Реми
562
Глава 11
веществ равны, то справедливо
XiW1=X2Ws=X3.^3 и т, д. (12)
Если вместо констант распада подставить периоды полураспада Т„ Т2, Т3 л т. д., то для случая радиоактивного равновесия получим
Следует иметь в виду, что Д\, ДД, ZV3n т. д.— это числа атомов. Если ;ке нужно рассчитать количества веществ в граммах, то эти величины следует разделить на атомные веса.
И в этом случае радиоактивный ряд можно сравнить с системой водяных сосудов, которые имеют равный диаметр, но разную ширину отверстий для стока воды. Прн этом эти резервуары расположены так, что вода перетекает из одного в другой. В таком случае н этой системе установится равновесие, при котором высота столба воды л каждом резервуаре будет постоянной, так как для скорости вытекания воды из каждого сосуда справедливо уравнение (11). Единственным условием установления такого равновесия является то, чтобы иервый сосуд имел диаметр отверстия очень небольшой по сравнению с диаметром самого сосуда, т. е., иными словами, скорость вытекания иа пего была практически постоянной. Точно так же и в радиоактивном ряду раинове-оно может установиться только в том случае, когда активность исходного вещества остается практически постоянной. Конечно, если в радиоактивном ряду имеются члены с небольшими константами распада (большими периодами полураспада), то равновессе установится только спустя длительное время.
Количество относительно короткоживущего продукта распада, которое образуется из долгоживущего материнского вещества через определенное время t до установления радиоактивного равновесия, определяется уравнением
^ = У!^(1~е-^). (14
В этом выражении JVj — число атомов материнского вещества; Zj — его констапп: распада; Xj — число атомов продукта распада, имеющееся к моменту времепг I, если в начальный момент продукт распада отсутствовал, а Ц— его констаитЕ распада.
Это уравнение можно получить следующим образом: количество продукта рассада, которое образуется за время dt из материнского вещества, равно количеству материнского вещества, распадающегося за время dt; если в обоих случаях количества веществ выразить через число атомов, то образуется 1Ы1 A^-dt атомов продукта распад? За это же время dt распадается Л2- N-dt атомов продукта распада. Следоватсльг?, за время dt число атомов продукта распада увеличится на (Л1 Л\ — Z2:V) dt, отщ-'-
Если учесть сделанное вначале предположение (Zj Z2), то величину .V( меж?г считать постоянной. Тогда после интегрирования получим In (Xi JVj — X, ;Vf) — lnZ-иУд == — Х2г, что является логарифмической формой уравнения (14),
В результате ряда исследований, из которых большая часть rhg-д выполнена Ганом (1905 исл.), было выяснено, что торий X образуется е непосредственно из тория, а при распаде другого, чрезвычайно спльт радиоактивного вещества — радиотория. Радиоторий был обнарус./ почти одновременно Ганом в остатках от выделения тория из ториаисщ-и Бланком (В)апс) в осадках радиоактивных водных источников. Вс. -следствии оказалось, что радиоторйй по своим химическим свойстсгл настолько близок к торию, что его нельзя отделить от последЕГ химическими методами. То, что его все же удается получить свободнш; * тория, объясняется тем, что оп образуется из вещества, которое мпх 11 отделить от тория — из так называемого мезотория.
Радиоактивность и изотопия
503
Существование последнего промежуточного продукта еще до его отделения от тория было доказало Ганом па основатгйи того, что, как это отмечалось п другими исследователями, часто торговые, т. с., как правило, старые препараты тория, имели другую a-активность, чем те препараты, которые были выделены самими исследователями из минералов. В свежеприготовленном тории радиоторий находится сначала в количествах, отвечающих радиоактивному равновесию. Если бы оп образовывался непосредственно из тория, то его количество и, следовательно,; его активность оставались бы и далее постоянными. В действительности же они сначала уменьшаются, затем снова возрастают и в конце концов достигают уровня, отвечающего радиоактивному равновесию. Это можно объяснить только тем, что между торием и радиоторием находится относительно долгоживущее по сравнению с последним, не испускающее а-частиц вещество, которое отсутствует в свежих препаратах тория н, следовательно, химически отлично от тория. Действительно, вскоре Болтвуду (Boltwood) удалось показать, что мезоторий легко «лдг итется от тория химическим путем. Период полураспада мезотория равен 6,7 года, раднотория — 1,9 года.
В 1908 г. Гаи нашел, что химическим путем ме в от ори и можпо разделить ца два вещества, которые оп назвал мевоторий 1 и мезотории 2 и из которых последний является продуктом распада первого, но очень быстро превращается дальше в радиоторий.
Таким образом, распад тория в неактивный конечный продукт торий D (ториевый свинец) происходит через большое число промежуточных членов ряда. В достаточно «состарившихся» препаратах тория, прежде всего в ториевых минералах, все эти продукты распада находятся в состоянии радиоактивного равновесия.
Вещества, которые последовательно образуются из тория при его радиоактивном распаде, приведены в табл. 60. Указанные в таблице названия этих веществ в большинстве случаев объясняются их историей открытия *. Но. кроме того, эти названия в общем хорошо отражают положение отдельных членов в радиоактивном ряду и позволяют обнаружить некоторые аналогии в других радиоактивных рядах. Это можно видеть при сопоставлении табл. 60 с табл. 62 и 63. В каждой из этих таблиц генетические связи элементов выражены соединяющими их стрелками. Каждая стрелка направлена от материнского вещества к дочернему. Рядом со стрелкой обозначено излучение, испускаемое при данном превращении. Кроме того, указаны величины пробега а-частиц в воздухе, периоды полураспада, а также порядковые номера и атомные веса продуктов распада, найденные по «закону сдвига при радиоактивном распаде» (см. стр. 565 и сл.).
Устойчивым конечным продуктом радиоактивного ряда тория является ториевый свинец. От обычного свинца он отличается атомным весом, что можно было проверить на примере свинца, выделенного из ториевых минералов. Полученная величина хорошо совпадает с предсказанной на основе теории распада (см. стр. 573).
В предпоследней колонке табл. 60 приведены количества отдельных продуктов распада, которые находятся в радиоактивном равновесии с 1000 кг тория**. Значения химических символов, приведенных в последней колонке таблицы и в скобках во второй колонке, будут рассмотрены в дальнейшем (стр. 566 и сл.).
Из продуктов распада тория практическое применение находит мезоторий (смесь MsThj и MsTh^ в радиоактивном равновесии). Как и торий, меэоторий получают из монацитового песка. Поскольку из
* Вместо названий, приведенных в табл. 60, в настоящее время чаще употреб" ляют химический символ, приведет! пый в скобках во второй колонке таблицы, напри" мер «радий-228» вместо «мезоторяй 1»< Эти «рациональные названия» обладают тем преимуществом, что они сразу указывают на химические свойства продукта распада (ср. стр. 568).
** Эти величины получены подстановкой в уравнение (13) значений периодов полураспада, приведенных в шестой колонке таблицы. Для случаев двоякого распада следует учитывать соотношения обеих ветвей распада.
36*
Таблица 60
Радиоактивный ряд тория
Элемент Химический символ Порядковый номер Атомный вес Пробег а-частиц ввоадухспри 15° в 760 Jimi pm cm, CJH Период полураспада Количества веществ, находящихся в* радиоактивном равновесии с 1000 ка тория Изотопы
Торий 4 тъ (ШТЬ) 90 232,05 2,72 1,39-Юю лет 1000 кг | UXj. UY, Io, RdAc, RdTh Ra, AcX, ThX
Мезоторчй 1 4 MsTh, (2S8Ra) 88 228 — 6,7 лет 4,7.10"* г
Мезоторий 2 4? MsTh2 рйАс) 89 228 — 6,13 час 4,94.10-8 3 Ac UX, UY, Io,
Радистории 4? RdTh (228 Th) 90 228 3,95 1,90 лет 1,34-10-* г RdAc, Th,
Торий X а| ТЕХ (2E*Ra) 88 224 4,30 3,64 № 6,92-IO-7 s | Ra, AcX, MsTh, RaEm,
Эмапацпя тория а у ThEm (EE°Rn) 86 220 5,02 54,5 сек 1,18-10-Ю г AtEm, AcEm RaA, AcA,
Торий А а | 99,986% 1 В ThA (216PO) 84 216 5,66 0,158 сек 3,35-10-1зе BaC', AcC'P ThC', RaF RaB, AcB
То ₽ зий В 0,014% Y Ториоастатин , !«• ThB (212pb) ThAt (2ieAt) 82 85 212 216 6,84 10,6 чае ~10-з сек 7,94-10-в г | ~3.10-1» в RaD, AcD TJlD, RaG RaAt,-, AcAtl, AcA til
Торий С : 65% 0. Y 35% ThC (sxsBj) 83 212 4,79 60,5 мип 7,56.1О-э в RaC, AcG, RaE, Bi RaC', AcC%
То зий С' ThC' (212PO) 84 212 8,61 ЗЛО-7 сек 4.10-1»в j RaA, AcA, ThA, RcF
а Торий С’ ₽ V ThC" (29811) 81 208 — 3,1 мип 1,33.-10-Юе { RaC', ACS'. Tl RaD, Ai£c
То (Т< зий D зриовый свинец) ThD (2«ePb) 82 208,0 Устойчив со | ;RaR Ari2:
Радиоактивность и изотопия
565
мезотория очень быстро образуются «-активные продукты распада (RdTh и т. д.), его можно применять для многих целей вместо радия. По сравнению с последним мезоторпв имеет тот недостаток, что его активность через несколько лет значительно уменьшается. Но с другой стороны, в соответствии с более коротким периодом полураспада мезоторий значительно более активен, чем радпй. Поэтому мезоториевые препараты (всвежеприготовленном состоянии) с активностью, равной активности препарата радия, значительно дешевле последних. Ввиду этого мезоторий применяют прежде всего для производства радиоактивных светящихся красок, а также для облучений к медицинских целях.
Как видно из табл. 60, в радиоактивном ряду тория дважды встречается двоякий распад. В таких случаях говорят о разветвлении в радиоактивном ряду. Приведенные в таблице побочные пути распада в обоих случаях вновь вливаются в основной путь. Если через различные промежуточные продукты образуется один и тот же продукт распада, то суммы энергий распада обоими путями должны быть одинаковыми. Например, должна выделяться в сумме одна и та же энергия, как для случая распада тория С через торий С' (т. е. в результате f-распада с последующим «-распадом) в торий D, так и для распада тория С через торий С" (т. е. благодаря а-распаду с последующим f-распадом) в торий D.
Что это действительно так, можно видеть из данных табл. 61. Ядро тория D, Таблица 61
Энергетический баланс для процессов образования тория D из тория С через торий С’ и через торий С"
Образование тория D через торий С' Энергия, Мае Образование тория D через торий С” Энергия, Мэа
f-Частицы ThC а-Частицы ThC' Лучи отдачи 2,256 8,776 0,166 а-Частицы ThC Лучи отдачи f-Частицы ThC" у-Излучение возбужденного ядра ThD 6,084 0,117 1,792 3,204
Сумма 11,198 Сумма 11,197
образующееся нри р-распаде тория С", сначала находится в возбужденном состоянии, из которого опо переходит в основное состояние, испуская у-лучн*. Конечно, при составлении энергетического баланса необходимо учитывать энергию этого у-излучения. Совпадение энергетических величин для распадов этим двумя путями можно получить только в том случае, если для энергии f-распада в обоих случаях принять л а кс и.чал ъ-иое вкаченив кинетической энергиир-частиц а пе ее среднее значение. Это одно из наблюдений, из которых следует, что энергия р-распада равна максимальной энергии р-частиц, а не ее среднему значению (стр. 557).
Химическая природа продуктов распада
Законы смещения. Химическая природа продуктов распада определяется двумя законами радиоактивного смещения, найденными Содди и Фаянсом.
* В общем существует по крайней мере шесть возбужденных состояний, в которых может находиться ядро ThD, образовавшееся за счет f-распада ThC". Но в преобладающем большинстве случаев опо образуется в том состоянии, энергия которого выше па 3,204 Мэв энергии основного состояния.
566
Глава 11
1. После радиоактивного превращения, сопровождающегося испусканием а-частицы, вновь образующийся элемент относится к той группе периодической системы, номер которой на два меньше номера группы материнского вещества.
2. При ^-радиоактивном распаде происходит смещение в следующую группу.
Например, из относящегося к главной подгруппе второй группы периодической системы радия с порядковым номером 88 при а-распаде образуется относящийся к нулевой группе радон (эманация радия) с порядковым номером 86. Из радона, такя-te за счет «-распада, образуется элемент, который по химическому поведению должен быть отнесен к главной подгруппе шестой группы периодической системы (порядковый номер 84). Его называют радием А. Этот элемент также распадается с испусканием «-частиц, образуя элемент, относящийся к главной подгруппе четвертой группы (порядковый номер 82), радий В. Радий В распадается, испуская р-луии, поэтому образующийся при этом элемент, радий О, относится к главной подгруппе пятой группы и имеет порядковый номер 83 и т, д. (см. табл. 62). По этим правилам были определены и порядковые номера приведенных в табл. 60 членов радиоактивного ряда тория.
Законы смещения непосредственно следуют из теории радиоактивного распада, если принять, что а- и 0-частицы испускаются атомными ядрами. Эти законы были установлены при многочисленных химических исследованиях веществ, образующихся при радиоактивном распаде.
Атомные веса продуктов распада. Из теории атомного распада следуют не только законы, определяющие положение продуктов распада в периодической системе, но опа позволяет также определить атомные веса этих веществ. При испускании «-частицы масса ядра, а тем самым и атомный вес элемента должны уменьшиться на 4 единицы, тогда как испускание р-част и цы сопровождается очень незначительным изменением массы. Следовательно, при «-распаде образуются элементы с атомным весом на 4 единицы меньше атомного веса материнского элемента, а при р-распаде образуются элементы практически с тем же, что и у материнского элемента, атомным весом. Обобщая, можно сказать:
Испуская а-частицу, атом превращается в другой атом, порядковый номер (заряд ядра) которого на 2 единицы, а атомный вес на 4 единицы меньше. Напротив, при испускании ft-частицы атом превращается в другой атом с порядковым номером (зарядом ядра} на единицу большим и практически тем же атомным весом.
Об уменьшении массы, связанным в соответствии с принципом эквивалентности массы и энергии с выделением энергии при радиоактивном распаде, см. стр. 613.
Изотопия. Образующиеся при радиоактивных превращениях вещества, носящие характер химических элементов, часто соответствуют тем местам периодической системы, которые уже заняты другими элементами. Они отличаются от этих элементов устойчивостью своих атомов и в большинстве случаев атомными весами. Это явление называется изотопией. Следовательно, изотопами называются такие элементарные вещества, которые по зарядам их ядер (порядковым номерам) должны быть помещены на одно и то же место (ev tw шютблы) в периодической системе. В последней колонке табл. 60 приведены то вещества, которые являются нзотопагш ядер, приведенных в первой колонке. Поскольку заряд ядра определяем
to-о "То ЕЙ "То Ей 'й’й "~Ё5 “"Та И ti Ей to Ей "’Та W’ t* Wq nC ^C1 ьа C Гй "To e
S * '-zjC? Й 5» ё£ пи Й5> ГдО £й ре, Й° аА тер аВ пр аА !18А to -у ti ixg3 £₽> Ed W —1 О н* —| H cz! fcts Id gfx h^t-o 1 *» £*4 « —। Oj
tr о о- 5 - ’ Се ’S' 3-3 « P> * 'E* '
oo te 0o co CO W 00 00 00 00 05 СО OJ 00 сл СО ьо 0о 0s со се Со со С? t§ S0 О СО о ё Порядковый номер
206,0 206 210 о 210 I 210 ьо >£> 218 214 218 214 to со ti 226,05 о WZ Си 234 Ь5 W ' 2380,1 Атомный вес
1 1 3,85 1 | 1 6,95 1 05 4,1 5,53 1 4,67 Ф* О 05 3,30 ei'e 3,22 1 1 1 2,63 Пробег а-частиц в воздухе яри 15° и 760 At.w ртп ст, сл«
Устойчив НИН gs‘? 138,8 ди 4,95 дп 22,3 года 1,32 мин 1,45-Ю-4 сен 1 0,019 сек 19,7 мин секунд Несколько 26,8 мин 1 3,05 мин 3,825 дн i 1590 лет 8,3-10* лет JO S о tn а ч 6,7 час ник 24,10 дн "ci Си О V г? Период полу-распада
8 05 О ! 9 У о Cl 0» : 2,63-10-e г 4,32-Ю-з г г ет-ОРЗбЧ о р А <4 н-Ь. о 09 Cs> 7,4-10-9 г 8 н-01 Т~ "о 5 । ад «А И*. о f «п со 2,147-10-е г 3,32-10-1 г 17,6 г 50,1 г ГЛ ^8? S 1 е о о оо О о П5 4Х К' Л О «R CS> 1 993 кг Количества веществ, находящихся в радиоактивном равновесии с 1000 кг урапа (или 903 кг UI)
Радиоактивный ряд урапа — радия
568
Глава 11
число и расположение электронов в оболочке атома, а это существенно для химического поведения, то изотопические вещества практически не отличаются по химическим свойствам. Поэтому химическими методами изотопы разделить нельзя, если они перемешаны; с химической точки зрения смесь изотопов можно рассматривать как один элемент. Однако положение о невозможности разделения смеси изотонов химическими методами имеет, как будет показано в следующей главе, ограниченное значение. Поэтому в настоящее время химический элемент определяется но как вещество, которое невозможно разложить химическим путем, а как вещество, все атомы которого имеют один и тот же заряд ядра (порядковый номер).
Различные изотопы одного и того же элемента можно обозначать следующим образом: к названию химического элемента добавлять характеризующее его массовое число *, например торий-232 (обычный торий) в отличие от тория-230 (иония) и тория-228 (радиотория). Аналогично обозначают и химические символы, только по мел-: ду нар одному соглашению массовое число пишут слева в виде верхнего индекса (например, mTh). В табл. 60, 62 и 63 для каждого члена соответствующего радиоактивного ряда наряду с традиционным названием приведено в скобках и принятое написание химических символов. Порядковый номер (заряд ядра) для каждого атома определяется уже его химическим символом. Но если хотят особо подчеркнуть его, то пишут слева внизу у символа элемента (например, “®2Th).
Изобарные атомы. Как видно из данных, приведенных в табл. 60, кроме атомов, обладающих различными атомными весами (соответственно массовыми числами) при одном и том же заряде ядра, существуют и такие, которые при различных зарядах ядра имеют равные массовые числа (например, MsThi и МзТЬг, т. е. ^eRa и ед8Ас). Химически различные атомы, имеющие одинаковые массовые числа, называются изобарами.
Правило осаждения Фаянса и правило адсорбции Панета. Чаете случается, что образующийся в результате радиоактивного распада элемент вследствие его неустойчивости можно получить только в чрезвычайно малых количествах, так что одна из важнейших химических реакции — осаждение из раствора — не может служить для концентрирования этстс элемента, так как даже при значительном избытке осадителя невозможна достигнуть произведения растворимости образующегося малорастворпмсгг вещества. Однако фаянс нашел, что справедливо следующее правило:
Радиоэлемент, даже если он находится в невесомых количествам соосаждается вместе с осадком, если этот осадок образуется в та^л: условиях, при которых этот радиоэлемент, если бы он имелся в весазх&х количествах, образовывал бы труднорастворимое соединение.
Это явление объясняется найденным Панетом правилом адсорбции
Твердое вещество адсорбирует те радиоэлементы, соединения которых с электроотрицательной частью (кислотным остатком) адсорбирующее: вещества малорастворимы в данном растворителе.
Впрочем, с течением времени стали известны многочисленные исключения из в~л-го правила. Строго соблюдаются лишь следующие найденные Ганом законы
Закон соосаждения: элемент при сколько угодно малой концентрации в кристаллическим осадком в том случае, если он может входить в крг&смьллсжляс&с
• Массовым числом называют округленный до целого яялт?—я я ::л: l,.-" соответствующей разновидности атомов.
Таблица 63
Радиоактивный ряд актиния
ура:
Актиноуран Act! 92 235 2,7 7,07-108 лет 7,06 кг
а | (235U)
Уран Y UY 90 231 — 24,6 час 2,75-10-8 г
н (231 Th)
Протактиний Pa 91 231 3,62 3,2-IO4 лет 3,14-10-1 е
а 1 (asipa)
Актиний Ac 89 227 H— 21,7 года 2,09.10-4 г
р|э8,8% а 1,2% (22’Ac)
Радиоактиний RdAc 90 227 4,67 18,6 дтг 4,85-10-7 в
а у (22’Th)
Фраиций +- - Fr 87 223 — 21 мин 4,5-10-12 г
Р 99,996% (223FT)
* У , Актинии X а 0,004% AcX 88 223 4,33 Н>3 дн 2,93-10-т а
у (223Ra)
Актиноастатин I Ac At I 85 219 4,97 0,9 мин 8-10-19 0
а 1 Р ~ 3% (21«At)
Эманация актиния AcEm 86 219 5,75 3,92 сек 1,16-10-12г
а - 97% (2i»Rn)
Висмут-215 215B] 83 215 — 8 МИН 6-10-1’0
1 P’l' Актиний А AcA 84 215 6,49 0,00183 сек 5,30-10-1ве
(Si&Po)
а 99,9995%
| Р 0,0005% Актиний В AcB 82 211 36,1 мин 6,15-10-1°е
р у (SHPb)
Актиноастатип II AcAtll 85 215 8,0 — 10“4 сек — 1-10-2*0
а (®15At)
Актиний С i AcC 83 211 5,46 2,16 мин 3,68-10-11г
Р |0,32% (2HBi)
Актиний С' а 99,68% AcC' 84 211 6,59 — 0,005 сек 4,5-10-18г
(2HPo)
Актиний С" AcC' 81 207 — 4,76 мин 7,93-10-11г
а я 1 Р j Y (20JT1)
Актиний D AcD 82 207 Устойчив OG
(Актиниевый свинец) (2O5pb)
570
Глава, 11
решетку осадка. В противном случае он остается в растворе, даже если его соединение с противоположно заряженными нонами этой, решетки сколько угодно мало растворимо в данном растворителе.
При этом следует иметь л виду, что элементы в мипимальпых концентрациях могут входить в состав кристаллической решетки как за счет образования истинных твердых растворов, так и за счет образования «аномальных твердых растворов» или «внутренней адсорбции» (см. стр. 578).
Закон адсорбции' элемент при сколько угодно малой концентрации хорошо адсорбируется осадком, {адсорбентом) в том случае, если осадок имеет поверхностный заряд, противоположный но знаку заряду адсорбируемого элемента, и адсорбируемое соединение трудно растворимо в данном растворителе.
Осадок обладает положительным или отрицательным поверхностным зарядом в зависимости от тоге, имеется ли в растворе, с которым соприкасается осадок, избыток катионов или анниплв, из которых состоит осадок. Например, гипс, осажденный быстро и, следовательно, с развитой поверхностью, добавлением спирта к раствору, содержащему избыток ионов SO£ (при этом осадок приобретает отрицательный поверхностный заряд), почти полностью захватывает из раствора ТйВ за счет адсорбции. Но если осаждепие пшеа ведут из раствора, содержащего избыточные ионы Са* *, то оп очень слабо захватывает ТйВ. Если же гипсу датв медленно кристаллизоваться, т. е. создать такие условия, при которых вследствие малой поверхности кристаллов адсорбция совершенно подавляется, то осадок совсем не захватывает ТйВ, несмотря на то что ТйВ, как изотон свинца, в этих условиях образовывал бы малорастворимое соединение, если бы он присутствовал в весомых количествах. Сульфат свинца, а следовательно, и сульфат тория В не могут входить в кристаллическую решетку гипса, поэтому в соответствии с законом: со осаждения Гапа при медленной кристаллизации гипса нс должно происходить захватывания ТйВ, тогда как по правилу Фаянса следовало бы ожидать соосаждепия ТйВ даже при медленной кристаллизации, что, одиако, противоречит опытным данным. Аналогично ведет себя в данных условиях и ТйХ, являющийся изотопом радия,
РАДИОАКТИВНЫЙ РЯД УРАНА — РАДИЯ
Вещества, постепенно образующиеся из урана (точнее из изотопа ура-на2зз uj за счет его радиоактивного распада до конечного устойчивого свинца, приведены в табл. 62, Как и в табл. 60, генетические связи элементов обозначены стрелками. Как видно из таблицы, сначала из урана через различные промежуточные продукты образуется радий. Это объясняет, почему в природе радий является постоянным спутником урана.
Однако скорость, с которой уран превращается в радий, чрезвычайно мала. Должно пройти более 4,5 млрд, лет, чтобы данное количество урана распалось наполовину. Поэтому интенсивность радиоактивного излучения урана значительно меньше интенсивности излучения значительно более быстро распадающегося радия (Период полураспада 1590 лет). Относительно небольшой по сравнению с ураном продолжительностью жизни радия объясняется и то, что радий ие может накапливаться в значительных количествах в рудах урана. Как следует из уравнения (13) стр. 562, в радиоактивном равновесии с 1000 кг урана (точнее с содержащимся в нем ураном I) будет находиться только 0,332 г радия.
Один из промежуточных продуктов превращения урана в радий является изотопом исходного элемента ряда, т. о. урана, который, как и последний, имеет очень небольшую скорость распада. Поэтому обычный уран всегда представляет собой смесь обоих этих изотопов. В радиохимии их называют уран I и уран II соответственно. Кроме того, обычный уран содержит еще один изотоп — актиноуран, названный так потому, что оп является исходным элементом радиоактивного ряда актиния (ср. стр. 574).
То, что природный у pan состоит из трех изотопов, впервые было установлено на основании радиологических наблюдений. Например, существование обоих изотопов — урана I и урана II — вытекало из того, что чистый в химическом смысле уран испускает два отчетливо различающихся вида а-частиц, а именно частицы с пробегом
Радиоактивность и изотопия
571
2,6 и с пробегом 3,2 с.ч *. При помощи соотношения Гейгера — Натт опа из этих величин спачала было получено предварительное соотношение для количеств урана Гм урана II, равпое 1000: 1. В дальнейшем масс-спектроскопическим методом удалось доказать присутствие в естественном уране трех изотопов с массовыми числами 238, 235 и 234, т. о. урана I, актиноурана и урана II. Этим путем Нир (Nier, 1939) впервые точпо определил соотношение ко,;гичести атих изотопов и нашел, что па 100 000 атомов 236 U (Cl) приходится 720 атомов -S4J i.Acl.ii и 5 или 6 атомов “3,U (UII). Согласно новейшим определениям констант распада 1’1 и C1I, для атомного соотношения 333 и : 234 U в природном уране получается следующая величина: 1 : 5,14-Ю5. Отсюда состав природного урана выражается в атомных процентах следующим образом: 99,280% 23aU, 0,715% 235Г, 005% гзщ.илив ю совых процентах: 99,289% ами, 0,706% -35U и 0,005% 234U. Из этого состава на основании величин масс изотопов урана, приведенных в табл. 72, для массы природного урана получено значение 238,108 и для атомного леса — 238,04. Химическим путем для атомного веса природного урана получено значение 238,07.
УранП образуется из урана! за счет одного a-распада с последующими двумя 0-распадами. Оба 0-нзлучающие промежуточные продукта получили название уран X п уран Х2. Уран Х15 являющийся изотопом тория, легко может быть химически отделен от урана. Он был открыт уже в 1900 г. Круксом (Crookes). Существование урана Х2 сначала было установлено на основании законов радиоактивного сдвига, которые требуют, если уран I и уран II являются изотопами одного и того же элемента, не одного, а двух 0-распадов после а-распада урана 1. Отделить химическим путем уран Ха от урана X г, из которого оп образуется, удалось в 1913 г. Фаянсу. Поскольку по своим химическим свойствам п в соответствии с законами радиоактивного сдвига уран Х= должен был занять в ту пору еще не занятое в периодической системе место аналога тантала, то он, как новый химический элемент, получил собственное имя. Его назвали «бревием» из-за его очень небольшого периода полураспада (1,14 мин). Но после открытия долгоживущего протактиния для обозначения атомов с порядковым номером 91 обычно применяют это новое название, и в настоящее время необходимость в особом названии для урана Х2 отпала.
При 0-распаде урана X[t кроме урана Х3, может образовываться также другое вещество, а именно уран Z. Этот изотоп был открыт Ганом в 1921 г. Ган выделил его из (практически свободных от протактиния) солей урана точно так же, как протактиний был выделен из остатков от переработки урановой смоляной руды (см. стр. 133). Ган прибавил к раствору солен урана небольшое количество соли тантала и затем высадил тантал обратно. Уран Z отличается от уранаХ2 относительно большим периодом полураспада (6,7 часа), а от протактиния, кроме меньшего периода полураспада, еще и тем, что он распадается с испусканием 0-частиц. Активность урана Z ио сравнению с активностью находящегося в радиоактивном равновесии о ним урана Х4 очень мала: она составляет едва ли более 0,001 последней. Следовательно, уран Z должен быть очепь тщательно отделен от урана Хь чтобы его можно было идентифицировать по собственной кривой распада. Уран Z, как и уран Х2, образуется при 0-распаде урана Xj. 0-Распад урана Х2 приводит к образованию урана II. Следова
* 11 р об сг а-ча сти ц а кти и оу р а па оч ень ма л о от л ича ется от и р об е га а-частиц у р ап а I. Существование этого третьего изотопа впервые было предположено на основании наблюдения, что актиний, как и радий, всегда присутствует в урановых минералах и всегда в постоянном соотношении к содержанию урапа. Первоначально следовало бы рассмотреть возможность двоякого распада урана I или урана II, Который приводил бы к образованию актиноурана. Однако эта возможность отпала, после того как было установлено, что атомный вес протактиния выражается нечетным числом (231). Согласно законам радиоактивного сдвига, в таком случае атомный вес начального члена ряда также Должен быть нечетным, а уран I и урав II оба имеют четный атомный вес (табл, 62}.
572
Глава J J
тельно, p-распад урана Z также должен приводить к урану II, так как уран Z образуется, как и уран Хг, из самого урана Хь а не из его изотопа. Таким образом, уран Х2 и уран Z являются своеобразным примером того, когда две разновидности атомов одного и того же элемента имеют не только одинаковые химические свойства, ио и одинаковые атомные веса (т. е. являются одновременно изотопами и изобарами), а испуская качественно одно и то же излучение (£-излучение), переходят в один и тот же продукт. Это явление называют ядерной изомерией. У искусственно полученных радиоактивных атомов это явление наблюдается довольно часто (см. стр. 621 и сл.).
Ранее считали, что образование урана Х2 и урана Z из урапа X, объясняется Звояии.м рарпайв.н 'последнего. Однако оказалось, что этот процесс протекает иначе . Уран Z образуется но непосредственно из урапа Х1( а из некоторой части первоначально образующегося урана Х2. Основная часть урана Х2 (99,85%), испуская р-частицы, непосредственно переходит в уран П. Меньшая часть урана Х2 (0,15%) претерпевает иное превращение. Опо Протекает с испусканием у-изл учения и называется «изомерным переходом» (ИП) (подробнее об этом см. на стр. 623). При этом «изомерном переходе», т, е. процессе, который сопровождается испусканием не а- или Р-частиц, а только у-квантов, образуется уран Z. У искусственно полученных радиоактивных атомов образование ядерпых изомеров благодаря таким процессам наблюдается довольно часто. Но, кроме изомерных переходов, существуют и некоторые другие процессы, которые могут приводить к образованию ядерных изомеров. Эти процессы будут рассмотрены в гл. 13.
Ионий, продукт распада урапа II и материнское вещество радия, впервые был обнаружен Болтвудом (Boltwood) в 1907 г. в урановых минералах. Так как ионий является изотопом тория и пе может быть отделен от него химическим путем и так как минералы тория обычно содержат некоторое количество урана, а вместе с тем и иония, то ториевые препараты всегда в большей или меньшей степени содержат ионий. Благодаря его большой продолжительности жизни активность ионпя за время наблюдения заметно пе изменяется. Напротив, другие изотопы тория все имеют малые периоды полураспада.
Важнейшим элементом, входящим в радиоактивный ряд урапа —радия, является сам радий. Его химические свойства уже были рассмотрены в т. I. Радий распадается совершенно аналогично торию X. Как и в последнем случае, первоначально образуется некоторый газ, получивший название эманации радия. Затем оп переходит через радий А, радий В и двояко распадающийся радий С в радий D (см. табл. 62), точно так же как эманация тория в торий D. Однако, если последний является устойчивым конечным членом радиоактивного ряда, радий D распадается дальше, переходя через радий Е и радий F (который идентичен выделенному из урановых руд полонию) в радий G — устойчивый конечный член радиоактивного ряда урана — радия.
В радиоактивном ряду урапа — радия двоякий распад встречается не два, как в ряду тория, а четыре раза. Двоякий распад радия Л соответствует такому распаду тория А, а распад радия С — распаду тория С. Кроме того, существуют еще два разветвления ряда, не имеющие аналогов в ряду тория. Как показали новейшие наблюдения, один из двух продуктов двоякого распада радия Л, а именно радиоастатин, в свою очередь распадается двояко, образуя, с одной стороны, радии С, а с Другой— газообразный продукт — эманацию. Она имеет тот же порядковый номер, что и эманация радия, но другой атомный вес и существенно отличный период полураспада, Следовательпо, в радиоактивном ряду радия встречаются в общем две эманации. Только эманация, образую
Радиоактивность и изотопия
573
щаяся при распаде радиоастатина, встречается *в чрезвычайно малых количествах. Поэтому ее образование не поддается наблюдению, если не применять специальных методов. Четвертое разветвление ряда наблюдается в случае радия Е. Основная часть радия Е распадается с испусканием ^-частиц и с образованием радия F (полония). Одпако очень небольшая часть радия Е (0,00005%) распадается, испуская а-частицы. При этом образуется короткоживущий изотоп таллия (2OST1). Этот изотоп распадается с испусканием (3-частпц, тогда как радий F — с испусканием а-частиц. В обоих случаях образуется радий G — конечный продукт радиоактивного ряда урана — радия.
На основании законов радиоактивного сдвига Содди — Фаянса для радия G получается порядковый номер 82 и атомный вес 206. Следовательно, по химическим свойствам он должеп быть идентичен свинцу, отличаясь от пего только споим атомным весом. То же справедливо и для тория D, который по законам радиоактивного сдвига должен иметь порядковый номер 82 и атомный вес 208. Экспериментальное доказательство того, что свинец, содержащийся в урановых рудах (урановый свинец), действительно имеет атомный вес 206, а свинец, присутствующий в рудах тория (ториевый св и псп),— атомный вес 208, тогда как атомный вес обычного свинца равен 207,2, было особенно убедительным доказательством справедливости законов радиоактивного сдвига, а тем самым и всей теории атомного распада, из которой следуют эти законы.
Поскольку урановый свинец образуется из урана (атомный вес 238,07) л общем через 8 a-расиадоп, то в соответствии с теорией атомного распада, или законов радиоактивного сдвига, вес каждого атома урапового свинца должеп быть меньше веса атома урана па восьмикратную массу атома гелия, что дает атомный вес 238,07—(8 х 4,003) = =206,05 *. Соответственно для ториевого свинца получаем атомный вес, равный 232,05 (атомный вес тория) — (6 х 4,003) = 208,03 *. Действительно, Ричардс (Richards) и Хенигшмид (Honlgschmid, 1914 и сл.) независимо показали химическим путем, что свинец из очень чистых урановых минералов имеет атомщдй вес 206, а свинец из возможно более чистых минералов тория имеет атомный вес 208. Хенигшмид нашел, что свинец из чистейшей восточно африканской урановой смоляной руды имеет атомный вес 206,05, из норвежской урановой смоляной руды — атомный вес 206,06, тогда как свинец из норвежского торита, содержащего наряду с 30,1 % тория только 0,45% урана, имел атомный вес 207,90, откуда для атомного веса свинца из совершенно свободного от урана тория получается величина 207,97. Как уже упоминалось, чтобы различать изотопы, отличающиеся по массе, химические символы элементов сопровождают массовым числом, т. е. округленным значением атомного веса. Следовательно, ураповый свинец, т. е. свинец с массовым числом 206, запишется символом 206РЪ, ториевый свинец (свинец с массовым числом 208) —символ ом 20SPb. Обычный, свинец с атомным весом 207,21, как впервые показал Астоп (1927) масс-спектрометрическими измерениями, представляет собой смесь нескольких изотопов. Астон нашел три изотопа 203РЬ, 207РЬ и 208РЬ, количества которых относятся между собой как 100:95: 222. В урановом свипце это отношение, по даппым Астона, равно 100 : 11 ; 4. То, что изотоп aosph не отсутствовал совершенно в урановом свинце, объявляется тем, что этот свинец был получен из уранового минерала, который содержал некоторое количество тория. Удивительным было то, что содержание изотона 5||7РЬ было даже больше содержания изотопа 21)8РЬ, тогда как в обычиом свинце отношение их количеств обратное. Следовательно, присутствие гс,7РЬ нельзя было объяснить примесью обычного свинца, а напрашивается вывод, что ‘О-РЬ, как и 2о6Р1>, образуется в результате радиоактивного распада. Можно было предположить, что этот изотоп образовался из изотопа 2S6U (актиноурана) в радиоактивном ряду актиния. Существование этого изотопа урана и. генетические отношения в радиоактивном ряду актиния.были, как уже упоминалось, позднее достоверно уста по л лены совершенно иным путем. В табл. 60, 62 и 63 жирным шрифтом выделены атомные веса, найденные химическими методами. Набранные обычным шрпф-
* Кроме тог о,~ небольшое уменьшение массы обусловлено громадным выделением энергии, связанной с излучением (см. стр. 609). В этих расчетах потеря массы нс учитывается. Она составляет 0,05 и 0,04 атомной единицы массы соответственно.
574
Глава. П
том значения атомных весов, или массовых чисел, найдены на основании законов радиоактивных сдвигов.
Впоследствии оказалось, что, кроме приведенных выше трех изотопов, в обычном свинце в небольших количествах присутствует и четвертый изотоп, 2U4Pb. По данным Коллинза [Collins, Pliys. Rev., 88, 1275 (1952)], отношение количеств этих изотопов в обычном свинце равно
2п4рь ; мерь ; 2о;рь . й08РЪ=Ю0 : 1845 :1561: 3840.
Из этих данных рассчитан приведенный в табл. 65 состав свинца в атомных процентах. Но отношение! количеств изотонов свинца в свинцовых рудах, свободных от урапа и тория, может н('1:!к,.1лько колебаться в зависимости от их происхождения, как это установил Нир ].Х 1 е г, Phys. Rev., 60, 112 (1941)]. Это, вероятно, объясняется тем, что древние породы: из которых на протяжении истории Земли выделились эти свинцовые руды, содержали уран и торий. Чем позднее произошло выделение руды из древних пород, дем больше «радиогенного* свинца Примешано к первоначальному свинцу. В пользу этого объяснения говорит также то, что в исследованных Ниром рудах изотопы и -и“РЬ всегда обогащены перадиогенным изотопом 204РЬ практически в тоц отношении, которое можпо было бы ожидать, если бы в древней породе отношение Th : Li приблизительно соответствовало среднему значению этого отношения в земной. коре.
РАДИОАКТИВНЫЙ РЯД АКТИНИЯ
Вещества, входящие в радиоактивный ряд актиния, приведены в табл. 63. Начальным членом этого ряда является актиноуран (уран-235). Несмотря на то что обычный уран содержит только 0,71% актиноурана,, скорость превращенЕГЯ урапа в актиний относится к скорости превращения его в радий, как 4,6 к 95,4, так как продолжительность жпзнп актиноурана значительно меньше продолжительности жизни урана L, Актиноуран, испуская а-частицу, превращается в уран Y изотах тория, однако очень короткоживущий. Из урана Y при его 0-pacnars образуется рассмотренный в гл. 4 протактиний. Последний, как долго живущий элемент, содержится в урановых рудах подобно радию в относительно больших количествах (см. стр. 134). Напротив, концентрации актиния в урановых рудах в связи с его относительно небольшой продолжительностью жизни по сравнению с концентрацией радия ничтожен Актиний распадается двояко. Образующийся при его (J-распаде радиоактиний занимает в ряду актиния такое же положение, как радиоторгт. в ряду тория и ионий в ряду урана — радия. Наряду с ним в нобольцшг количествах образуется за счет а-распада актиния аналог цезия — франций.
Франций также распадается разветвление, как показали в 1953 г. Хайд (Hyi и Гиорсо (Ghiorso), Основная часть норе ходит, испуская 0-частицы, в актинов Но вместе с том: испускаются и а-частицы и пря этом образуется иаотоп acmezzz., В общем в ряду актиния встречаются два изотона астатина. В табл. 63 они обозпн^-т как AcAtl и Ac At II. Актгию астатин II образуется при 0-распаде актиния А сн^т: с актинием В, образующимся в значительно больших количествах за счет а-рспхх. актиния А. Он занимает в ряду актипия нодожепие, подобное яоложенпю пззт ~-астатина в радиоактивных рядах тория и урана — радия, и был открыт приблпзптт‘.г..т одновременно с этими изотопами Карликом и Берпертом (Karlik, Rerncrt, 194 "
За исключенном различия, вносимого присутствием франция, : в радиоактивном ряду актиния, начиная с радиоактиния. прт~ совершенно аналогично распаду в ряду тория. Начиная с рал^’-'тг:' радиоактивный ряд актиния совпадает в основных чертшг □ урана — радия. Как и ряды тория и урана — радпя, рш -
чивается изотопом свинца — актинием D (массовое ч~;.' _
Радиоактивность и изотопия
575
Несмотря на то что в урановом свинце наряду с образующимся при распаде ура-па I изотопом свинца 2<ЙРЬ всегда присутствует образующийся из актиноурана изотон £<'"РЬ, последний оказывает на атомный вес свинца очень небольшое влияние, так как его количество очень незначительно по сравнению с изотопом 2()6РЬ. Если рассчитать атомный вес уранового свинца с учетом приведённого на стр. 573 отношения количеств изотопов гоь’РЬп-07РЬ, то полученное значение будет всего на 0,09 атомной единицы веса выше, чем рассчитанное там же без учета содержания изотопа Z07Pb. Если же одновременно учитывать уменьшение массы на 0,05 атомной единицы, связанное с излучением энергии, то теория атомного распада приводит к значению 206,09 для атомного веса уранового свинца. Эта величина хорошо согласуется с найденной эксперимента ль-по Хенигшмидом (206,05 или 206,06);
РАДИОАКТИВНЫЙ РЯД НЕПТУНИЯ
Кроме членов радиоактивных рядов тория, урана — радиям актинии, в природе существуют еще члены четвертого радиоактивного ряда — ряда нептуния. То, что нептуний и продукты его распада существуют в природе, а именно в урановых рудах, доказал в 1952 г. Пеппард. Однако они встречаются в очень малых количествах. Содержание нептуния в урановой смоляной ,руде составляет максимум 1,8 10-:10% от содержания в ней урана. По этой причине присутствие в природе этого элемента и продуктов его распада практически не имеет никакого значения. Исследования радиоактивных свойств членов ряда нептуния проведены поэтому не с природными, а с искусственно полученными атомами. Устойчивым конечным продуктом в ряду нептуния является не изотоп свинца, как в трех других радиоактивных рядах, а изотоп таллия (205Т1). Его предпоследним членом является (чрезвычайно слабо радиоактивный) чистый элемент висмут. Остальные сведения о радиоактивном ряде нептуния можно найти в конце гл. 14. Там же, кроме того, будет показано, что и остальные три радиоактивных ряда имеют некоторое число генетически связанных с ними искусственно полученных членов.
ПОЛОЖЕНИЕ РАДИОАКТИВНЫХ ЭЛЕМЕНТОВ В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ
Несмотря на то что радиоактивные превращения приводят к образованию большого числа различных веществ, отчетливо различающихся между собой своими радиоактивными свойствами (например, периодами полураспада и природой образующихся из них продуктов распада), эти вещества вследствие явления изотопии занимают небольшое число мест в периодической системе. Порядковые номера веществ, входящих в три рассмотренных выше радиоактивных ряда, включая начальные и конечные члены этих рядов лежат между 81 и 92. На каждое из этих 12 мест с порядковыми номерами 81—92 приходится по крайней мере один из входящих в радиоактивные ряды сортов атомов, но в большинстве случаев на каждое место приходится несколько (до семи) изотопов. Порядковые номера 81 и 82, кроме радиоактивных атомов, имеют и устойчивые элементы, а именно таллий и свинец. Все элементы, имеющие порядковые номера выше 82, радиоактивны *.
Но и среди встречающихся в природе элементов с порядковыми номерами меньше 81 имеется несколько, для которых удалось доказать наличие радиоактивности. Уже давно известно, что элементы лютеций (71),
* О ради о активности висмута (порядковый номер 83) см. т. I, стр. 727. Данные Холубея (H.olubei) и Кошуа (Cauehois, 1940) о существовании стабильного изотопа полония (84) следует считать недостаточно надежными.
576
Глава 11
самарий (62), рубидий (37) и калий (19) слабо радиоактивны. Недавно усовершенствованными методами удалось обнаружить радиоактивность рения (75), гафния (72), гадолиния (64), неодима (60), лантана (57) и индия (49). Но все десять названных элементов значительно менее радиоактивны, чем элементы с более высокими порядковыми номерами (за исключением чрезвычайно слабо радиоактивного висмута). Торий, из всех начальных членов радиоактивных рядов имеющий наибольшую продолжительность жизни, испускает (без учета излучений его продуктов распада) на каждый грамм-атом за равный промежуток времени примерно в 70 раз больше частиц, чем самарий, и в 800 раз больше, чем калий.
Радиоактивность рения, гафния, лютеция, гадолиния, самария, неодима, ланта-ца, индия, рубидия и калия обусловлена тем, что эти элементы содержат неустойчивые изотопы. По скорость распада этих неустойчивых изотопов чрезвычайно мала. Она во всех случаях (за единствегпалм исключением 40К, см. ниже) меньше скорости распада тория. содержит до 0,10 ат.% изотопа i‘4Hf. Этот изотоп распадается, испу-
ская в-лучн ’с периодом полураспада 4-1013 лет. Лютеций содержит неустойчивый изотоп Этот изотоп излучает ^-частицы и период полураспада его равен 2,4-101*’ лет. Содержание 17SLu в лютеции достигает 2,5 ат. %. Гадолиний содержит до 0,20 ат. % неустойчивого изотопа162 Gd с периодом полураспада 9-1014 лет, который испускает а-лучи. Содержание неустойчивого изотопа 14"Stu в самарии составляет 14,62%.. Этот изотоп также ct-радноактивен с периодом полураспада 1,4-1011 лет. Неодим содержит 5,63% р-радиоактив но го 150 Nd (период полураспада 5-1010лет). Лантан и индий особенно слабо радиоактивны. Лантан содержит 0,089% изотопа 13SLa, превращающегося за счет электронного захвата (см. стр. 616) с периодом полураспада 2-1011лет, индий на 95,77% состоит из распадающегося с испусканием р-частиц (период полураспада 6- 10Ы лет) изотопа 1161П. Рубидий содержит 27,85% р-излучателя 87Rb, период полураспада которого равен 4,6-101° лет. Изотоп 40К, содержание которого в калии достигает 0,012%, имеет период полураспада 1,18-10* лет, главным образом за счет испускания р-частиц, но наряду с этим происходит и электронный захват. В случае рения, как и в случае индии, доминирующим по сравнению со стабильным изотопом (iesRe) является неустойчивый изотоп (i8"Re) (см. табл. 65). Но скорость распада ы'Ие, так же как и скорость распада п31и, чрезвычайно мала — его период полураспада равен 4- 101а лет.
Химия радиоактивных веществ. Совокупность элементарных веществ отличающихся устойчивостью и атомным весом, которые по порядковому номеру занимают одно место в периодической системе, называют плеядой. Плеяде дают название по тому ее члену, который имеет наибольшую продолжительность жизни. Таким образом, для встречающихся в естественных радиоактивных рядах элементов получаются следующие плеяды: таллия, свипца, висмута, полония, астатина, радона, радия, актиния, тория, протактиния и урапа. Б последней колонке табл. 60 приведены вещества, которые входят в отдельную плеяду наряду с эломепгом, приведенным в первой колонке.
Химические свойства элементов, по которым даны названия плеядам, уже были рассмотрены. Тем самым определены и химические свойства остальных членов плеяды, так как они практически полностью совпадают.. Различия члепов плеяды, проявляющиеся в их радиоактивных свойствах,, но влияют па их химические свойства. Только при химических работах с такими продуктами радиоактивного распада следует учитывать, чга они обычно имеются в невесомых количествах. Следовательно, чтобы осадить их пз раствора необходимо добавить неактивные изотопы илп если таковые не существуют, то другие неактивные вещества, отвечающие закону осаждения Гана, в таких количествах, чтобы получить вадд-мые и фильтруемые осадки.
Радиоактивные вещества обнаруживают по их излучению. обнаружения по радиоактивному излучению является наиболее чутжи™-
Радиоактивность и изотопия 577
тельным методом, какой существует для любого вещества. Для идентификации в большинстве случаев пользуются определением константы распада. Количество вещества определяют по интенсивности излучения.
Несмотря на то что вещества, относящиеся к одной и той же плеяде, в химическом смысле ведут себя практически одинаково и, после того как они уже смешаны, не могут быть разделены при помощи химических реакций, все же легко получить их в чистом виде за счет радиоактин ног о распада чистого исходного продукта. Например, исходя из чистых соединений радия, получают эманацию радии, свободную от эманаций тория и актиния. Аналогично из эманации радия получают ее продукты распада, свободные от продуктов распада веществ, входящих в радиоактивные ряды тория и актиния.
Разделение продуктов распада, не являющихся изотопами одного и того же элемента, можно осуществить химическими методами или использовать различие, папримёр, в летучести при нагревании. Часто получение какого-либо продукта распада в чистом состоянии оказывается возможным благодаря различным скоростям его образования и распада.
Например, радий А получают практически в чистом состоянии, если отрицательно заряженную металлическую проволоку подвергают в течение всего нескольких секунд действию эманации радия. Напротив, если такую проволоку подвергать действию эманации в течение нескольких часов, а затем выждать еще минут 20—30, то радий А, который выделился на проволоке, практически полностью распадается, а активность, которую обнаруживает проволока, обусловлена радием В и радием С Радий С получается чистым, если проволоку недолго нагревать при темно-красном калении. При температурах выше 600° радий В быстро испаряется (улетучивается), тогда как радий С начинает возгоняться только при температуре 1100°. Удобнее всего получать чистый радий В, помещая пад пластинкой, на которой осажден радий А, другую пластипку, заряженную отрицательно. П ри своем распаде радий А образует лучи отдачи, состоящие из радия В. Эти лучи улавливаются отрицательно заряженной пластинкой. Приведенных примеров достаточно, чтобы дать представление о том, как радиоактивные продукты распада, которые можно в некоторых случаях использовать для химических исследований (см. ниже), можно получить в чистом виде. Более подробные сведения о используемых для этого методиках можно найти в специальной литературе.
Применение радио активных методов в химии
Поскольку короткоживущие радиоактивные вещества благодаря их интенсивному излучению можно обнаружить в количествах гораздо меньших, чем это возможно при помощи других реакций, то эти радиоактивные вещества позволяют проводить такие исследования, при которых другие методы обнаружения и определения оказываются непригодными вследствие их очень небольшой чувствительности.
Радиоактивные вещества как индикаторы. Методы радиоактивных индикаторов основаны на явлении изотопии радиоактивных атомов с неактивными. Чтобы исследовать поведение последних, вместо них можно использовать радиоактивные изотопы или прибавить к исследуемому неактивному веществу его активный изотоп. Этот изотоп будет далее сопровождать неактивный изотоп при всех последующих операциях и благодаря своему излучению позволит определить даже невесомые количества неактивного вещества.
Этим методом, например, Папет и Хевеши определили растворимость в воде хромата и сульфида свинца. Опи осаждали из раствора, содержащего наряду с известным количеством хлорида свинца также электроскопически измеренное количество радия D, сульфид пли хромат смеси изотопов, встряхивали эти осадки с водой до насыщения и измеряли радиоактивность сухого остатка после испарения пробы насыщенного
’Л Г. PeiwriT
578
Рллев 11
раствора. Из этих иаиерешгй рассчитывали количества свинца, присутствующего в растворе в виде хромата или сульфида, поскольку его количество в данный момент времени * должно находиться в таком же отношении к количеству радия D, как и в исходном растворе.
В других случаях радиоактивные вещества используют для доказательства существования соединений, которые (пока еще ие известны условия их получения) не могут^быть получеиы в таких количествах, чтобы их можно было обнаружить другими методами. Радиоактивные изотопы можно использовать не только для доказательства существования таких соединений, ио и для нахождения паилучших условий их получения. Используя такие результаты, удалось получить в обнаруживаемых количествах и неактивные соединения. Этим дутом Панет открыл существование висмутистого и свинцовистого водорода (см. т. I,. стр. 733 и 604).
Другим, интересным применением радиоактивных веществ в качестве индикаторов является их использование для исследования реакций замещения в однородных с химической точки зрения веществах. Так, Хевеши (1920) показал, что атомы свпшщ в твердом свинце или ионы свинца в твердых солях свинца не остаются неподвижные на своих местах, а они более или менее часто в зависимости от температуры обмениваются местами. Это следует из того, что радий D, являющийся изотопом свинца, шх-фувдирует с поверхности внутрь свинца и точно так яге радий D в виде иона щтффуя—--рует внутрь хлорида свинца.
Возможности применения радиоактивных веществ как индикаторсд вначале были ограничены тем, что существует лишь несколько устойчивых или практически устойчивых элементов, для которых имеются ралг -активные изотопы, входящие в естественные радиоактивные ряэт? В последнее время благодаря открытию искусственной радиоактивно^-область применения этих методов очень расширилась.
Поведение веществ в минимальных концентрациях. Благодаря легкому обЕгр-жеиию радиоактивные вещества можно использовать также для исследования ш7—г--закономерностей, определяющих поведение веществ в минимальных ко>-щетпра-:.ххх Такно исследования, например, необходимы для выяснения причин aaxcaiua постхр них веществ, часто наблюдающегося при выпадании осадков из растворов. Так лось показать, что такой захват может быть обусловлен, кроме включений маточЁ -раствора, также образованием смешанных кристаллов, адсорбцией на яоверхс:.тт выпадающего осадка и другими явлениями. По данным Гана (1934), в тех саухх:;:;. когда в соответствии с законом, справедливым для больших количеств веществ, eztx. ожидать образования смешанных кристаллов, такой захват может быть вызван сГ_.с зованиен так называемых «аномальных смешанных кристаллов». Кроме того, ест происходить «внутренняя адсорбция», т. о. внедрение посторонних веществ естст кристаллов при их выделении из растворов. Такое внедрение происходит под дяГ-^т-- -сил, родственных силам, вызывающим адсорбцию на поверхности.
Методы эмапирозанпл. Образование поверхности веществ и ее изменена? ~ ?. нагревании, разрыхление кристаллов при нагревании (см. стр. 783) и другие анз=’..“_- -ные процессы можпо количественно изучить при помощи «методов эманирязхст;: Эти методы основаны на том, что в изучаемом веществе гомогенно распределяют р; активное вещество, образующее эманацию. Отношение количества эманации, chzz: щейся из данного вещества, к се количеству, образующемуся за равный прст ; времени, называют панирующей способностью» этого вещества. Эта спгс;:б~ .
тем больше, чем более рыхдо построено это вещество. Из гомогенного кристаллу стенного из ионов, при обычных температурах эманация практически совершешст с.: ляется, тогда как игшество губчатой структуры или кристалл с мноточЕхст". z . .г
* Конечно, при таких определениях следует учитывать уменьшение г:стст .шип
радиоактивного вещества со временем, которое можпо найти из величины ест. в..._ >..даты распада.
Радиоактивность и изотопия
579
пустотами в решетке (см. стр. 780) обладают большой эманирующей способностью При помощи метода эманировапия удалось, папример, показать, что температура, при которой начинается реакция между двумя твердыми веществами, равна температуре, при которой появляется достаточное количество пустот в кристалле одного из них (см. гл. 19). Эта температура разрыхления примерло равна половине абсолютной температуры плавления, что ужо рапсе было установлено Тамманом (1928) другим путем, а именно из наблюдений над реакциями твердых веществ или спеканием порошков. Ган (1934) методом эмалирования исследовал процессы старения препаратов окисв железа(Ш). Так ему удалось показать, что окись железа)III), осажденная из водных растворов, при температуре около 400° претерпевает быстро протекающий процесс спонтанной кристаллизации, затем при температурах выше 800° происходит разрыхление решетки, которое сопровождается полным испарением последних частей воды, еще присутствующей в препарате. Около 1000° происходит окончательное образование упорядоченной чистой рсгпетки гематита (см. также стр.'293 и т. I, стр. 391). В случае компактных гомогенных веществ метод эмалирования позволяет произвести очень точное определение истинной величины поверхности (см. первое примечание к стр. 721).
37*
Глава 12
ИЗОТОПИЯ СТАБИЛЬНЫХ ЭЛЕМЕНТОВ
Так же как и у радиоактивных элементов, явление изотопии паблю-даетсяу стабильных элементов. Но если изотопы радиоактивных элементов отличаются друг от друга, кроме атомного веса, также своими радиоак* тинными свойствами, изотопы стабильных элементов различаются только атомным весом, или массовым числом- Выяснить, состоит ли данный элемент из атомов с различной массой или все его атомы имеют одинаковую массу, можно различными методами. Важнейшими из них являются так называемая масс-спектрография и исследование полосатых спектров.
Маес-спсктрографяя основана па том, что каналовыо лучи отклоняются в магнитном и электрическом полях. В обоих случаях отклонение пропорционально отношенпш е/гп, т. о. отношению заряда к массе электроположительных частиц, ив которых состоят каналовые лучи. Но это отклонение по-разному зависит от скорости частиц к. Тогда как отклонение в магнитном поле обратно пропорционально скорости частиц, отклонение в электрическом поле пропорционально квадрату скорости частицы. В электрическом поле отклонение происходит о направлении поля, в магнитном — перпендикулярно к нему (так же как двигается в магнитном поле проводник, по которому пдёз ток). Предположим, что имеется нападений луч, который состоит только из частнд с одинаковым отношением заряда к массе е/т, но с различной скоростью. Если этс? луч пропустить через гомогенное электрическое поле, направленное перпендикуляре" к нему, например между двумя противоположено заряженными пластинами, то пуч расщепится в направлении силовых липцй поля и на фотопластинке, поставленнгй перпендикулярно направлению луча п чернеющей под его действием, вместо черней точки получится непрерывная вертикальная черпая полоса (если имеется непрерывный ряд значений скорости, что, как показывает опыт, обычпо и наблюдается). Если М& трическое поле направлено горизонтально, то на фотопластинке появится горизонтальная полоса. По если выключить электрическое поле, а вместо него включить, точее; так же направленное магнитное поле, то получим непрерывную черную полосу, которая, однако, будет направлена перпендикулярно первой. Если же одновременно вклеэ-чить оба поля, в результате чего каждая частица попадет на пластипку после тстч. как она подвергнется действию как магнитного, так и электрического полей и испытелл как вертикальное, так и горизонтальное отклонение, то па пластинке получим кривуш подобную приведенной па рис. 71. Эта кривая представляет собой ветвь пара б оды, тле как координаты каждой ее точки определяются соотношениями
el , е 1
-----s ; у=Ъ — . т о3 т v
Здесь а и Ъ — постоянные, определяемые условиями опыта. Комбинируя сСл уравнения, получим
р2 ~ или 1
ат ’
если, ввести новую постоянную величину
Ь-в р= —: . ат
Уравнение (2) представляет собой известное из аналитической геометрии ур"^ -нио параболы.
Если имеется канал оный луч, частицы которого характеризуются рчт—-значениями отношения заряда К массе е/т, то получим соответстпейнэ д
Изотония стабильных элементов
581
чениям р ветви нескольких парабол, а именно стольких, сколько имеется значений отношения е/m для частиц данного луча. Величины постоянных а и b определяются из условий опыта. Впрочем, этот метод впервые был разработан для катодных лучей, которые, так же как и капаловые, отклоняются магнитным и электрическим полями, только вследствие противоположных знаков заряда отклонение происходит в противоположные стороны. Определение величины е/т у катодных лучей и явилось первым определением отношения заряда электрона к его массе. Определение е/m в случае
Риг "1; Отклонение к ап адовых лучей в электрическом и магнитном полях.
капаловых лучей было с па чало предпринято с целью определения величин зарядов, которые наблюдаются у входящих в их состав атомов. При этом оказалось, что заряды атомов в Папановых лучах, как правило, малы п в большинстве случаев равны 1 или 2. При применении достаточно спльпых колем кривые, отвечающие увеличению заряда вдвое, лежат достаточно та .тег, о русс от друга. В 1312 г. Томсон исследовал каиаловые лучи, образующиеся в цаполненной неоном разрядной трубке. Эти каналовые лучи давали кривые почернения, г-п.цветётвуюшие одно- п двукратно ионизированному неону, т. е. существенно различным значениям отношения заряда н массе е/т=г/го и e/m = a/20. По рядом с ветвью параболы, отвечающей однократно ионизированному неону, Томсон обнаружил еще одну кривую, для которой отношение заряда к массе образующих ее частиц равнялось 1/22. Сначала он считал, что эту кривую следует приписать примесям в неопе. Одпако оказалось,что это нс так, и Томсон пришел к выводу, что обычный неон с атомным весом 20 содержит, вероятно, небольшие количества другого инертного газа с атомным весом 22. Впоследствии этот вывод нашел полное подтверждение. Это был первый случай, в котором изотопный состав элемента был определен по отклонениям каналовых лучей.
Позднее метод капаловых лучей был подробно разработан в работах Астона (1919). Для повышения чувствительности метода Астон направил магнитное ноле не параллельно электрическому, а перпендикулярно ему и выбрал условия опыта такими, чтобы для лучей с равным отношением е/т, по с различной скоростью электрическое отклонение полностью компенсировалось магнитным. В этом случае (подробный вывод приводен не будет) каждая ветвь параболы сжимается в точкуДВсе действие лучей концентрируется, таким образом, в нескольких дискретных точках или коротких линиях, если работу ведут со щелевой диафрагмой. Конечно, такие линии проявляются гораздо отчетливее, чем протяженные параболические кривые. Полученные этим методом фотографии очень напоминают по внешнему виду обычные спектры. Смесь изотопов разлагается на свои составные части так ;ке, как пемонохроматический свет разлагается спектрограф ом. Это нашло свое отражение в предложенном Астоном для этого метода названии «масс-спектрографил».
Следует также упомянуть, что Демпстер (1918) разработал метод анализа каналовых лучей, основанный на отклонении частиц, имеющих равные скорости, только магнитным полем. Постоянная скорость частиц достигается тем, что медленные ионы, испускаемые нагретыми солями нт. п., ускоряются сильным электрическим полем. В этом случае отклонение в магнитном поле слу;кит непосредственной мерой величины отношения е/т. Метод Демпстера применяют во многих случаях, в которых метод Астона неприменим, так как в методе Астона исследуемые элементы, или по крайней мере их соединения, должны быть газообразными. в разрядной трубке низкого давления.
В аппарате Демпстера (1935) комбинируется фокусирующее действие радиального электрического поля с разлагающим действием гомогенного магнитного поля.. Этот принцип «двойной фокусировки» применяют почти во всех современных прецизионных масс-спектрографах *.']Тз нпх следует указать па масс-спектрограф Нира (Nier,
* Обзор новых типов прецизионных масс-спектрографов и принципов их действия дан в статье Матта уха lM a t. t и и с h J., Naturw., 39, 557 (1952)].
582
Глава 12
1949—1951). В этом приборе частицы с различными массами'регистрируются не в различных местах фотопластинки, а в качестве меры массы таких частиц используют напряжение, которое должно иметь радиальное электрическое поле, чтобы частицы прошли через установленную в конце аппарата щель.
Определение масс отдельных разновидностей атомов, выполненное на современных приборах, значительно точнее всех других определений атомных весов, проводимых другими применяемыми в химии методами. Но для сравнения С атомным весом смешанных элементов (см. стр. 585), кроме массы отдельного вида атомов, должны быть известны и соотношения количеств различных атомов в естественной смеси изотопов. Определение таких соотношений значительно менее точно, чем определение величины массы отдельной частицы. Поэтому физически установленные средние величины атомных весов (см. стр. 587 и с л.) смешанных элементов в настоящее время далеко не во всех случаях являются более точными, чем химически определенные атомные веса.
Изотопия п полосатые спектры. Энергия многоатомной газовой молекулы складывается из ее поступательной анергии (т. е. кинетической энергии ее поступательного движения), ее вращательной анергии, колебательной энергии ее атомов и энергии связи ее электронов. Благодаря квантовому характеру изменений трех последних составляющих образуется совокупность спектральных линий, которая наблюдается в полосатых спектрах молекул. В области очень длинных волн (инфракрасной области спектра), где вследствие относительно небольшой энергии квантов света еще не происходит изменения орбит электронов, проявляется только «вращательно-колебательный спектр». В этом спектре квантованные изменения колебательной энергии определяют полозкениг полос, а изменения вращательной анергии — расстояние между линиями, из которых состоят отдельные полосы. В более коротковолновой области также проявляются системы полос соответствующие вращатель но-колебательным спектрам. В этом случае они накладываются на каждую линию, обусловленную изменением орбит электронов. Поскольку колебательная и вращательная энергии зависят от массы колеблющихся и вращающихся вокруг общего центра тяжести атомов, то две молекулы, содержащие химически эквивалентные, но различающиеся по массе атомы, должны давись различные полосатые спектры. Поэтому, например, положение полос в спектре 14№5 несколько отличается от их положения в спектре 14N2, и еще больший сдвиг наблюдается при переходе от 14N2 к 15N3. Точно так же молекула 13С16О дает несколько пнюЭ полосатый спектр, чем спектр молекулы I2CisO, а последний в свою очередь отличаетгл от спектра молекулы i^C^O. И несмотря на то что расстояние между линиями внутри данной полосы мало зависит от различия масс изотопов, сдвиг линий, обусловленкдЗ различиями в колебательной энергии, может быть весьма значительным. Сидькзт всего он проявляется у элементов с небольшими атомными весами. Например, дх* молекул 11N2 и I3N3 ои составляет в области360.около 12жр. Поэтому исслепов^т— полосатых ыцуотров позволяет установить изотопный состав легких элементов в том случае, когда содержание отдельных изотопов очень мало. Из наблюдений сюю-сатых спектров в 1929 г. впервые было найдено, что кислород и углерод не сеетт— ив атомов с равными массами, как до этого принимали на основании масс-спектргтр, фических измерений. Впрочем, с тех пор чувствительность масс-спектрографпчэсЕ'г.-метода была значительно повышена. По сравнению с оптическими методами он юр маним в значительно более широкой области. Оба метода позволяют не только уехюг вить существование, но и определить (по интенсивности почернения фотоплаеы__...
соотношение изотопов в смеси.
Из уравнения (13) (т. I, стр. НО) следует, что различие в массах изотопов досюр проявляться также в линейчатом спектре атома. Этот эффект в случае тяжелых г.-ментов настолько мал, что он не имеет никакого значения дня выяснения сущигтю ния изотопов. Однако благодаря этому эффекту впервые было установлено сущегтЕ„_ ние тяжелого водорода SH — дейтерия (см. стр. 591).
Разделение изотопов. В общем случае для разделения изотопов ir::: иен о и ъз о в ат ь только такие процессы, на протекание которых скнгдт существенное влияние масса атома. Поэтому обычно химические ptm л не применяют, так как массы атомов в общем практически не -
влияния па их протекание.
Валентные силы, определяющий течение химических реакций, в сгрзую ""а зависят от числа и типа связи электронов во «внешней оболочке» лтгта... Чртг -тс электронов определяется зарядом ядра атома, а вид связи, или пртчюзгхь юьз , : . ..
Изотопия, стабильных элементов
583
нов, к ait следует из теории строения атома, практически также определяется только зарядом ядра. Однако уравнение (13а) (т. I, стр, 110) показывает, что прочность связи электрона не является совершенно независимой от массы атомного ядра. Но в то же время из этого уравнения видно, что даже в случае водорода, где различие в массах атомов наиболее ярко выражено, влияние массы атома на анергию связи чрезвычайно мало, В общем случае такое влияние практически не имеет значения для химического поведения, а если это справедливо для водорода, то тем более справедливо для более тяжелых элементов.
Впрочем, существенное влияние массы атома на его химическое поведение можно усмотреть в зависимости ат массы вращательной и колебательной энергий молекул, поскольку последние могут оказывать влияние на сродство химических реакций, например на положение химических равновесий (ср. т. I, гл. 5). Однако это влияние настолько мало, что оно практически проявляется только тогда, когда отношение масс изотопов существенно превышает 1. Это отношение более всего отличается от единицы для изотопов водорода. При увеличении атомного веса оно очень быстро уменьшается, так как массы изотопов одного и того же элемента отличаются друг от друга только на несколько единиц. Если для ИН и Ш отношение масс равно 2 : 1, то для изотопов лития ^Li и 6Li оно падает до 1,17 : 1. Поэтому водород является практически единственным элементом, для которого можно говорить о химически различном поведении изотопов (в количественном, но не в качественном отношении) (см. стр. 595 и сл.).
Наиболее непосредственно сказывается различие масс на поведений заряженных частиц в электрическом и магнитном полях. Поэтому при помощи масс-спектраграфов достигается непосредственное полное разложение смеси изогоной на ее составные части. Однако первоначально количества изотопов, которые получали этим способом, были настолько малы, что дальнейшие исследования с этими изотопами проводить было невозможно, за исключением некоторых особых случаев (см. т. I, стр, 184). Однако ввиду необходимости применения чистых изотопов или сильно обогащенных смесей изотопов для решения различных проблем (см. стр. 591 и 596 н с л.) получение чистых изотопов пли обогащение в крупных масштабах представляет значительный интерес (см. об этом стр. 648). Поэтому для этих целей позднее были сконструированы (Smythe, Rum-bough 1934; Walcher, 1938) масс-спектрографы значительно большей интенсивности, сначала для получения чистых изотопов для таких исследований, которые (например, превращения атомов) могли быть проведены с минимальным количеством вещества, а затем и масс-спектрографы для разделения изотопов в больших масштабах *.
Кроме электромагнитного метода, основанного на тех же физических принципах, что и масс-спектрография для разделения изотопов, используют в основном следующие методы:
1. Диффузионные методы. В соответствии с законом Грэма и Бунзена (см. стр. 781) легкие молекулы газа диффундируют через порвстук) перегородку легче, чем тяжелые. Чтобы при этом получить значительный сдвиг соотношения количеств молекул газа, массы которых различаются весьма незначительно, диффузионный процесс следует вести долго и проводить так, чтобы газ последовательно проходил через большое число диффузионных ячеек (по принципу часто применяющихся в технике перегонки «фракционных колонн»). 13первые такой аппарат был сконструирован Герцем (Hertz, 1932). Но так как применение пористой перегородки влечет за собой потери, позднее (1934) Герц заменил аппарат, состоящий из диффузионных ячеек, заполненных глиняными трубками, аппаратом с ртутными диффузионными насосами. Каждый насос отсасывает больше легких, чем тяжелых составных частей разделяемой газовой смеси. При помощи такого аппарата, состоящего из 48 насосов, Барвич (Barwich, 1930) сумел за 5 час количественно разделить изотопы неона 2(,Ne и —Ne. Де Гемптип (Фе Hemptinne) и Капрон (Capron) сумели на аппарате Герца с 51 насосом за 30 час получить 75 %-ное обогащение изотопом 1аС, содержание которого в обычном углероде составляет только 1,1%, Правда, количества отдельных изотопов, которые практически можно получить этим
* В США уже в период второй мировой войны масс-спектрограф особой конструкции применяли для разделения изотопов в большом масштабе (см. стр. 648).
584
Глава 12
методом, в общем довольно малы, так как работы ведут при очень низких давлениях, т. е- с очень разреженными газами. И тем не менее позднее в США диффузионные методы были использованы для разделения изотопов урана в больших масштабах (см. стр, 648).
2, Дистилляционные метод#. Частичное разделение изотопов можно осуществить нри «идеальной дистипднции». Этот метод основан па том, что в каждую единицу времени из жидкости в газовую фазу переходит больше легких атомов или молекул, чем тяжелых. Если создать такие условия, нри которых эти перешедшие в газовую фазу молекулы стали бы конденсироваться, прежде чем они благодаря взаимным столкновениям нерейдут опять в жидкость (что можно достигнуть, помещая вад жидкостью холодную поверхность на расстоянии, не слишком превышающем среднюю длину свободного пробега молекулы), то конденсат обогащается болев легким изотопом, а более тяжелый накапливается в остающейся жидкости. Этот метод прост по выполнению, но не привозит, впрочем, к значительному обогащению и дает небольшие количества обогащенного продукта. Однако для некоторых определенных целей он может быть с успехом использовав (см. т. I, стр. 184),
Больптпе количества обогащенных продуктов можно получить, применяя многократную перегонку на дистилляционных колоннах (ректификационных колоннах}.. Б этих. случаях целесообразнее всего работать вблизи тройной точки (см. т. I, стр. 72),, так как различия в упругости паров изотопов максимальны в твердом состоянии. Но таг цац и в этих условиях различия в упругости паров все же очень малы, следует применять многоступенчатые дистилляционные колонны. Кеесом (Keesoin, 1934) на 85-ступенчатой колонне, исходя из 420 л обычного неона, который содержит 90,5% i0Ke е 9,2% 22Ne (наряду с 0,3% 21Ne), получил 4 л неона, в котором обогащение по наиболее легкому изотопу 30Ne составляло 95,5%, и 5л неона, обогащенного на 83,5% самыы тяжелым изотопом 33Ne.
3. РовЗеленке щюшопое при. полимцк тепловой Оифйбузин. Простотой аппаратуры и большой эффективностью разделения отличается разработанный в 1938 г. Клузиуслп (Clusius) и Динелем (Dickel) метод разделительных трубок. В основе его лежит явление, называемое тепловой диффузией, состоящее в том, что если в газе создать перепел температур, то одновременно создается и перепад концентраций •, что в определенные условиях сопровождается диффузией газа, нри которой более легкие молекулы опятх обгоняют более тяжелые. Сконструированная Клузиусом разделительная трубя:: представляет собой вертикально установленную стеклянную трубку, наполненную разделяемым газом при атмосферном давлении. Длина такой трубки составляет 6— 10 м опа окружена охлаждаемой водяной рубашкой, по оси трубки натянута проволока. Как только по этой проволоке проходит электрический ток, вследствие паденгг температуры от центра к стенкам начинается падение концентрации газа в обратил: направлении, что приводит к диффузии от стенок к проволоке. Поскольку при этгм более легкие молекулы движутся быстрее, пространство около проволоки сбогаищекг ими. Нагревание газа от проволоки одновременно приводило к возникновению дошл— ния газа около проволоки вверх, а около Степок — вниз, что долго мешало достпе--иию равновесия тепловой диффузии. Так разделение благодаря тепловой диффуегг которое само по себе очень незначительно, продолжали до тех пор, пока практичен:., все более легкие молекулы пе собирались в верхней части трубки, а более тяжелый — в нижней. При помощи агрегата, состоящего из нескольких таких трубок, Клус^: достиг обогащения изотопом 3,С1 с 24,6 до 99,4% на одном, а изотопом 35С1 с »й до 99,6% па другом конце «разделительной трубки». При длительной работеустагтшг: ежедневно можно было получать 10—30 мл обогащенных продуктов на обоих
। трубок. Этим же методом он получил по 2,5 л чистых 20Ne и 22Ne. Такой же метод С*.;: им с успехом применен для получения 8,Кг и ®йКг, а позднее (1943) — 1аОа и
4. Методы центрифугирования (газовая центрифуга). Разделение центриф%с. ванием имеет особое значение для разделения смесей изотопов элементов с болыддд атомными весами, так как в этом методе эффективность разделения зависит не от тения масс, как в рассмотренных выше методах, а от разницы в величинах масс.. t. тому она пе зависит от того, ведутся ли работы с чистым элементом или с его-сеедглЕ:- -ем. Элемент пли ого соединение, подлежащие разделению па изотопы, помещают п гг образной или парообразной форме в центрифугу с чрезвычайно большой вращения (ультрацентрифугу, см, стр. 712). Под действием центробежной сгез % : тяжелые изотопы накапливаются па периферии, а более легкие — у (полой) осп i фуги и могут быть легко откачаны. Та, что методы центрифугирования нрипппы”^ ч пригодны для разделения изотопов, впервые было показано Мулликеном (1922. п L.x.1
* Можно наблюдать и обратный эффект: появление разности температур тдг'-:-ствие создания перепада концентраций при диффузии двух газов друг в друге .72 sins, Naturw., 30, 711 (1942)].
Изотопия стабильных элементов
585
сом (Веашз, 1938). Позднее этот метод был значительно улучшен Хартеком (Harteck) и Гротом (Groth) и приметп для обогащения урана изотопом 23iU (см. стр. 648 и сл.).
5. Методы распределения. При распределении какого-либо вещества между двумя растворителями химически однозначные, но различные по массе молекулы ведут себя несколько различно.. При длительном повторении этой операции (на аппаратах типа колонн) можно достигнуть более или менее значительного изменения изотопного состава. Это же справедливо и для распределения между растворителем и газовой фазой. При взаимодействии аммиака с водным раствором сульфата аммония * в 621-ступепчатой ректификационной колонке Юри (Urey, 1937) смог повысить содержание изотопа 15N с 0,38 до 2;35%. В дальнейшем ему удалось даже при последовательном использовании нескольких колонн получать ежедневно по 2,2 г азота с содержанием 70,6% 16N. При обмене лития между амальгамой лития и хлоридом лития, растворенным в этиловом спирте, Льюис (Lewis) сумел достигнуть обогащения изотоном еЫ с 7,9 до 16,3%.
6, Элехтролшаич& кое разделение. В случае легких изотопов разделения лли значительного обогащения можно достигнуть длительным электролизом. По целому ряду причин особенно удобно электролитическое разделение изотопов водорода. Поэтому в этом случае электролиз особенно эффективен (подробнее см. на стр. 592). Для других элементов эффективность электролитического разделения значительно меньше. Тем не менее удалось показать, что электролизом водных растворов солей лития можно существенно сдвинуть соотношение изотопов ®Li и 3Ы. Даже для относительно более тяжелого калия при подходящих условиях эксперимента (последовательная установка нескольких ячеек) электролитическим путем можно достигнуть заметного изменения соотношений количеств изотопов даК и 41К (Harteck, 1353). Точно так же можно провести обогащение изотопом гбМц при противоточном электролизе магния (Martin, 1958). При электролизе наблюдалось также обогащение изотопами 18Q и пО (Johnston, 1935).
Чистые элементы и смешанные элементы. Те элементы, которые состоят только ив атомов с равными массами, называют чистыми элементами в отличие от элементов, атомы которых имеют различные массы. Последние называют смешанными элементами. В табл. 64 приведены те элементы, которые в соответствий с уровнем современных представлений считаются чистыми. Для большинства из них доказано, что они, во всяком случае, пе содержат более 0,2% атомов другого изотопа,
В первой колонке табл. 64 указаны порядковые номера элементов, химические символы которых помещены во второй колонке. В третьей колонке указаны «массы» атомов **. Масса атома, называемая также «физическим атомным весом», показывает, во сколько раз масса атома больше l/w массы атома изотопа кислорода КО. В последних колонках таблицы приведены (химические) атомные веса ***, рассчитанные на основании масс-спектрографических или ядерпо-физических (см. ниже) определений масс атомов, и атомные веса, определенные химическими ^ или физико-химическими методами. Обо последние величины даны по отношению к атомному весу обычного кислорода, принятого за 16,0000. Смысл величин, приведенных в четвертой колонке таблицы (дефекты масс), будет раскрыт позднее (стр. 609 и сл.) Из таблицы видно, что значения атомных весов, полученные масс-спектрографическим и ядерно-физи-ческим методами, прекрасно совпадают со значениями, полученными химическими методами. Кроме того, видно, что величины масс атомов очень мало отличаются от целых чисел. Но отклонения от целочисленных
* Присутствие сульфата аммония улучшает разделение, так как в равновесии I5NHa+(HNHJ'[i5NHJ‘ концентрация ионов [i6NH4]‘ несколько выше концентрации ионов [ 14N IL j ". Поэтому в растворе концентрируется более легко растворимый в воде 15NH3.
** Величины, данные в скобках, рассчитаны по методу, указанному на стр. 610, из массовых чисел и упаковочных множителей.
*** Об отношении «химического атомного веса» к «физическому атомному весу» см. на стр. 590.
586
Глава 12
Чистые элементы
Таблица 64
Z Элемент Масса Дефект массы ТЕМ Химический атомный вей (ио отношению к 0—16)
рассчитано по знача нию массы значение, приводимое в международных таблицах
4 Be 9,01505 62,48 9,01257 9,013
9 F 19,00443 158,79 18,99920 19,00
11 Na 22,9964 201,1 22,9901 22,991
13 Al 26,99011 241,65 26,9827 26,98
15 Р 30,98358 282,45 30,9751 30,975
21 Sc 44,97010 416,72 44,9577 44,96
25 Мп 54,95581 517,53 54,9407 54,94
27: Со 58,9510 556,6 58,9348 58,94
33 Аз 74,943 703 74,922 74,91
39 У (88,940) —1 88,916 88,92
41 Nb (92,942) —1 92,916 92,91
45 Rh 102,949 938 102,921 102,91
53 I 126,944 1153 126,909 126,91
55 Cs (132,947) —1 132,910 132,91
59 Рг 140,951 1266 140,912 140,92
65 Tb (158,978) — 158,934 158,93
67 Но (164,987) — 164,942 164,94
69 Til (168,993) —1 168,947 168,94
73 Та 181,003 1562 180,953 180,95
79 Au 197,039 1664 196,985 197,0
83 Bi 209,047 1762 208,990 209,00
89 Ac 227,098 1867 227,036 227
91 Pa 231,108 1892 231,044 231
значений, как видно из данных табл. 64 (вероятная ошибка последних составляет в большинстве случаев лишь несколько единиц в последнем десятичном разряде), все-таки лежат за пределами ошибок. Величины масс атомов, округленные до целочисленных значений, которые, как будет видно позднее, имеют важное физическое значение, называют массовыми числами атомов.
Замечательно; что, за исключением бериллия (который если и содержит, то не более 0,001% другого изотопа), все чистые элементы имеют нечетные порядковые номера, Это объясняется тем, что элементы с нечетными порядковыми номерами в общем менее устойчивы, чем соседние с ними по периодической системе элементы, имеющие четные порядковые номера (см. стр. 634). Это отражается и на распространенности элементов в природе (ср. рис. 90). Для элементов В1, Ас, Ра известны вообще только неустойчивые (радиоактивные) изотопы. Остальные изотопы этих трех радиоактивных элементов имеют настолько малые периоды полураспада по сравнению с глав-пым представителем плеяды, что в природе эти элементы существуют как чистые.
В табл. 65 приведены изотопические разновидности атомов, из которых состоят устойчивые (т. е. нерадиоактивные) смешанные элементы.
Таблица бб
Состав стабильных смешанных элементов
1 1 Элемент Состав, am. % Cpeb^si ееличииа массы атомньш вес (ио отношению к 0~16) рассчитано значение, при-
по средней величине массы водимое в международных таблицах
7 Н 99,984% iH, 0,016% EH (D) 1,00831 1,00802 1,0080
Не 0,00013% 3He, 99,99987% 4He 4,00388 4,00278 4,003
3 Li 7,30% eLi, 92,70% ’Li 6,9451 6,9432 6,940
5 В 18,83% t0B, 81,17% nB 10,8251 10,8221 10,82
6 С 98,9% 12C, 1,1% 13C 12,0149 12,0116 12,011
N 99,62% 14N, 0,38% ISN 14,0113 14,0075 14,008
3 О 99,7628% ‘«0, 0,0355% ”0, 0,2017% 18O | 16,00440 16,00000 16,0000
10 Ne 90,51 % 20Ne, 0,28% aiNe, 9,21% «Ne 20,1767 20,171 20,183
12 Mg 78,60%24 Mg, 10,ll%25Mg, 11,29% «Mg | 24,3195 24,313 24,32
14 Si 92,18% aaSi, 4,71% «Si, 3,11% MSi 28,095 28,087 28,09
16 S 95,060% S-’S, 0,742% 33S, 4,182% 3iS, 0,016% «8 | 32,0738 32,0650 32,066
п Cl 75,4% 35C1, 24,6% 3'Ci 35,471 35,461 35,457
18 Ar 0,35% «Аг, 0,08% «Аг, 99,57% 40Ar 39,960 39,949 39,944
19 К 93,259% 39К,0,0119%«К(Э-и К-распад) 6,729% 41K [ 39,111 39,100 39,100
20 Ca 96,92% 4®Ca, 0,64% 4iCa, 0,129% «Ca, 2,13% «Ca, 0,003% 48Ca, 0,178% 48Ca | 40,091 40,080 40,08
22 Ti 7,95% 4®Ti, 7,75% 47Ti, 73,45% 48Ti, 5,51% 49Ti, 5,34% “Ti } 47,889 47,876 47,90
23 V 0,25% “V, 99,75% SIV 50,958 50,944 50,95
24 Cr 4,31 % “Cr, 83,76% “Cr, 9,55% “Cr, 2,38% «Cr I 52,014 52,000 52,01
26 Fe 5,81% «Fe, 91,64% fi6Fe, 2,21%S7Fe, 0,34% «Fe | 55,866 55,851 55,85
28 Ni 67,77% ««Ni, 26,16% “Ni, 1,25% «’Ni, 3,66 % “Ni, l,16%«Ni J 58,729 58,713 58,71
29 Cu 68,94% «Он, 31,06% “Cu 63,570 63,553 63,54
30 Zn 48,89% ®4Zn, 27,81% ««Zn, 4,07%87Zn, 18,61% “Zn, 0,62% «Zn | 65,408 65,390 65,38
31 Ga 60,16% “Ga, 39,84% ’4Ga 69;750 69,731 69,72
32 Ge 21,2% 70Ge, 27,3% ’’Ge, 7,9%78Ge, 37,1% 74Ge, 6,5%78Ge 1 72,654 72,634 72,60
34 Se 0,87% 74Se, 9,02% 76Se, 7,58% 77Se, 23,52% 58Se, 49,82% “Se, 9,19% 82Se 1 79.015 78,993 78,96
36 Br 50,53% 7®Br, 49,47% 8IBr 79,933 79,911 79,916
36 Kr 0,342 % 78Kr, 2,228% “Kr, 11,50% 82Kr, 11,48% 84<r, 57,02% 84Kr, 17,43% “Kr . 83,833 83,810 83,80
зт Rb 72,15% ®Rb, 27,85% 8’Rb ^-pasndi') , f 85,487 85,464 85,48
38 Sr 0,55% «Sr, 9,75% 8eSr, 6,96% 87Sr, 82,74% “Sr I 87,648 87,624 87.63
Продолжение табл, 65
fl 1 Элемент Состав, am. % Средняя величина : массы Химический атомный вес (по отношению'к.О^б)
рассчитав по медлен величине массы значение,приводимое в международных та&пщах
40 Zr 51,46% wZr, 11,23% ®‘Zr, 17,11% t£Zr, 17,40% “Zr, 2,80% «Zr 91,257 91,232 91,22
42 Мо .15,84% fi8Mo, 9,04% «Mo, 15,72% 16,53% «Mo, 9,46% ®’Mo, 23,78% «’Mo, 9,63% ‘«Mo 95,92 95,89 95,95
44 Ви 5,68% *Ru, 2,22% MRu, 12,81% MRu,
12,70% l0SRu, 16,98% ‘«‘Bn, 31,34% ‘«Bu, 18,27% 1MBu 101,06 101,03 101,1
46 Pd 0,8% wsPd, 9,3%‘“Pd, 22,6%‘«sPd,
27,l%‘eaPd, 26,7% ‘««Pd, 13,5% “®Pd 106,569 106,540 106,4
47 Ag- 51,92% lwAg, 48,08% 1MAg 107,901 107,871 107,880
48 Cd 1,215% “«Cd, 0,875% ‘««Cd,
12,39% 110Cd, 12,75% nlCd, 24,07% naCd, 12,26% “«Cd, 28,86% n4Cd, 7,58% “«Cd 112,480 112,429 112,41
49 In 4,23% “3In, 95,77% 115In (fi-pacnad) 114,856 114,824 114,82
50 Sn 0,94% “«Sn, 0,65% 114Sn, 0,33% “®Sn,
14,36% 114Sn, 7,51% “TSh, 24,21% llsSn, 8,45% “*Sn, 33,11%««8п, 4,61%iaiSn, 5,83% 1M8n 118,767 118,734 118,70
51 Sb 57,25% 1HSb, 42,75% 1MSb f 121,798 121,765 121,76
52 Те 0,09%‘«Те, 2,43%laaTe, 0,85% 1 «Те,
4,59% 1MTe, 6,98% ‘“«Те, 18,70% 1ИТе, 31,85%‘««Те, 34,51% 136Te 127,674 127,639 127,61
54 Xe 0,094% ‘«Xe, 0,066% lwXe,
1,92% 128Xe, 26,44% 1KXe, 131,349 131,313
4,08% l5aXe, 21,18% “*Xe, 26,89% 133Xe, 10,44% 1MXe, 131,3
8,87%ieeXe
56 Ba 0,102% i«Ba, 0,098% “*Ba,
2,42% 6,59% ,55Ba, 7,81% 11,32% 18TBa, 71,66% »«B» 137,372 137,334 137,36
57 La. 0,089% ia»La (K-pacimA) 99,911% :soLa 138,96 138,92 138,92
58 Ce 0,19% 1ЖСе, 0,25% 1S4>, 88,49% ««Ge, 140,158 140,12 140,13
11,07% 141Ce
60 Nd 27,10% “Wd, 12,14%‘“Nd,
23,84% ‘«Nd, 8,29% ‘«Nd, 17,26%‘«Nd, 5,74%‘«Nd, 5,63% “«Nd (/3-pacnaA) ' 144,295 144,26 144,27
Продолжение табл. 65
ХамичеагиИ
5й* S Cl i Средняя атомный вес (по отношению к 0*16)
Порядка i номе £7ccr?jw3, а.зд. % величина массы рассчитано по средней величине массы значение ^приводимое в международных та&лицах
<ът
62 Sin 2,95% i«Sm, 14,62% 147Sm (tt-pacnab),
10,97% “8Sm, 13,56% «’Sm, 7,27% lbcSm, 27,34% “sSm, 150,473 150,43 150,35
23,29% w«Sm
63 Ей 47,77% 151Eu, 52,23% 1MEu 152,014 151,97 152,0
54 Gd 0,20% 152Gd (a-pamd), 2,15% 1MGd,
14,78% 1KGd, 20,59% 1MGd, 15,71% 1E’Gd, 24,78% laeGd, 21,79% 156,98 156,94 157-26
56 Dy 0,05% “«Dy, 0,05% 16eDy, 0,1% 1K,Dy,
21,l%ieiDy, 26,6% 1MDv, 24,8% ««Dy, 27,3% ie4Dy 162,53 162,49 162,51
68 Er 0,l%le2Er, 1,5% 16iEr, 32,9% 168Er,
24,4% W7Er, 26,9% !C8Er, 14,2% I70Er 167,23 167,18 167,27
70 Yb 0,140% lssYb, 3,04% 170Yb,
14,34% 171 Yb, 21,88% 172¥b, 16,18% 173Yb, 31,77 % 171Yb, 173,09 173,04 173,04
12,65%17«Yb
71 Lu 97,5%17fiLu, 2,5%176Lu (0-pacnad), 175,028 174,98 174,99
72 Hf 0,19% ™B£(a-pacnad) , 5,26% 17«Hf,
18,51% 177Hf, 27,16% 178Hf, 13,79% 178Hf, 35,09% U0Hf 1 178,54 178,49 178,50
74 w 0,16% lsaW, 26,35% le8W, 14,32% 1MW, 30,68% lfilW, 28,49% ,8eW 183,98 183,93 183,86
75 Re 37,07% 18ERe, 62,93% W7Re (fi-patnad), 186,275 186,22 186,22
76 Os 0,018% I840s, 1,582% 18«Os,
: 1,64% 1S70s, 13,27% ««Os. 16,14% 184)s,: 26,38% I8DGs, 190,312 190,26 190,2
40,97% lt30s
77 If 38,5% 1MIr, 61,5% 1S3If 192,27 192,22 192,2
78 Pt 0,006% 18°Pt, 0,784% 182Pt,
30,2% i»Pt, 35,2% 19SPt, 26,6% “«Pt, 7,21% 1MPt 195,185 195,13 195,09
80 Hg 0,15% »»Hg, 10,12% 1B8Hg,
17,04% 23,25% 2»Hg, 13,18% E01Hg, 29,54% 202Hg, 6,72%s<«Hg 200,65 200,60 200,61
81 Tl 29,46% 2WT1, 70,54 % 3MT! 204,468 204,41 204,39
82 Pb '1,36%»«H>, 25,12%2WTb,
21,25%207Pb, 52,27%2oePb | 207,272 207,215 207,21
690
Глава 12
Восемь чрезвычайно слабо радиоактивных элементов, К, Rb, La, Nd, Sm, Gd) Lu, Hf, которые в основном состоят из устойчивых разновидностей атомов, также включены в эту таблицу. Точно так же в нее включены и In и Re. У обоих этих элементов неустойчивым является основной изотоп, но они настолько слабо радиоактивны (их периоды полураспада составляют 6-Ю^и 4-1012 лет соответственно), что практически ведут себя как устойчивые элементы. Остальные радиоактивные элементы не включены в таблицу, так как их изотопы уже были охарактеризованы при рассмотрении радиоактивных рядов. В третьей колонке табл. 65 приведено содержание отдельного изотопа в смешанном элементе. Изотопы различают по индеЕюу массового числа, стоящему вверху слева перед химическим символом элемента. Приведенные в четвертой колонке таблицы «средние значения масс* получены по правилу смешения * из масс отдельных изотопов с учетом содержания последних в смеси. При их вычислении учитывали отклонение масс атомов от целочисленных значений. Все остальные колонки соответствуют аналогичным колонкам табл. 64.
Точно так же, как массы атомов чистых элементов, массы атомов смешанных элементов очень мало отклоняются от целочисленных значений. Это показано на примерах, приведенных в табл. 71 (стр. 611). То, что химические атомные веса большинства элементов значительно отличаются от целых чисел, объясняется тем, что они состоят иа изотопов с различными массами.
Точные значения масс обоих наиболее тяжелых изотопов кислорода равны: 17О = 17,004520 и 1ЙО = 18,004875, если массу более легкого изотопа принять равной 1еО = 16,000000. Поэтому для смешанного элемента кислорода, с учетом его изотопного состава, получается атомный вес, равный 16,00440. Одна шестнадцатая этой величины является единицей химического атомного веса. Поскольку эта единица в 1,000275 раза больше, чем единица, в которой выражены массы, атомов, то последние, если их нужно выразить по шкале химических атомных весов, следует разделить на 1,000275. Другими словами, величины химических атомных весов, основанных на предположении О = 16, получаются делением величин масс атомов, рассчитанных на основании предположения 1еО — 16 («физических атомных весов*), на 1,000275.
Естественные колебания в изотопном составе кислорода, как уже указывалось, очень незначительны. Однако эти колебания существенно отражаются на последних знаках в факторе пересчета. Поэтому было предложено принять величину фактора пересчета в химические атомные веса равной 1,000275. Это должно означать, что основал шкалы химических атомных весов служит такая смесь изотопов кислорода, атомный вес которой в 1,000275 раза больше атомного веса изотопа icO. Однако До сих пор ни дия одного элемента, за исключением серебра, химическим путем ие получено настолько точных значений атомного веса, чтобы па эти величины оказывали влияние незначительные колебания изотопного состава обычного кислорода.
Различие между «физическим» и «химическим» атомными весами и при условном фиксировании величины фактора пересчета все же мало удобно. Это: приводит к тому, что все константы, в которые входит атомный вес (например, газовая постоянная и число Авогадро), имеют два численно различных значения в зависимости от того, какая «физическая» или «химическая» шкала атомных весов положена в их основу. Поэтому в последнее время (Nier, Olandor, Mattauch) появляются предложения основывать как шкалу масс атомов, так и химическую шкалу атомных весов на 12С=12. Это основание для атомных весов имеет то преимущество, что, с одкой стороны, значения масс атомов можно установит/, с той же точностью, что и на основании 1ЙО = 16, а с другой — все до сих пор принятые значения химических атомных весов нуждаются л очень незначительном изменении (см. табл. 1 в приложении).
* По правилу смешения свойство какой-либо смеси (принимая, что оно является аддитивной фушщией свойств составных частей) подучают, складывая произведения процентного содержания каждой составной части на величину, характеризующую интенсивность данного свойства, и деля полученную сумму на 100.
Изотопия стабильных элементов
591
Применение изотопов. Чистые изотопы или обогащенные смеси изотопов часто можно успешно применить для исследования химических реакций.
Так* Полянки (Polanyi, 1934), применяя Н318О, удалось показать, что гидролиз сложных эфиров протекает по схеме А, а не по схеме Б:
------- п = сн3- с/° 4-CsHnOH
ХЮ-С6К11 + Н|-1в0—Н Х18ОН '•
(А)
= СНз—с
CI-L- С< ;
(Б)
Аминовый эфир Уксусная Алсалиныи
дтдгмой кислоты каслота спирт
Амиловый спирт, образующийся при гидролизе, содержит обычный кислород, тогда как образующаяся вместе с ним уксусная кислота содержит столько же изотопа и0, сколько его содержала применявшаяся для гидролиза вода.
На примере этерификации бензойной кислоты содержащим 18 О метиловым спиртом, Робертс (Roberts) и Юри (Urey) в 1938г. показали, что образование эфира карбоновой кислоты и спирта точно так же происходит таким образом, что От спирта отщепляется атом Н, а от кислоты — группа ОН, из которых образуется вода:
7»
С-ЩХ|он + и.г“о-сн.
== CeHs—с
У0
Х18о—сн9
4-нон
Бензойная кислота
Метиловый спирт
Метиловый эфир бензойной кислоты
Соединения, в которых изменено обычное соотношение изотопов, можно также применять для решения аналогичных вопросов в области биологии и медицины.
До сих пор основная часть таких исследований проведена с тяжелым изотопом водорода — дейтерием. Многочисленные исследования проведены и с углеродом, обогащенным изотопом 13С (особенно в области биохимии).
ДЕЙТЕРИИ И ОКИСЬ ДЕЙТЕРИЯ
Тяжелый изотоп водорода 2Н называют «тяжелым водородом» или з дейтерием (то бебтероу — второй). Поэтому вместо символа гН обычно" пользуются символом D. Изотоп водорода 1Н называют «легким водородом» или протием (то лрштоу — первый). Ядро атома протия называют протоном, а ядро атома дейтерия D+—дейтроном. Для окиси дейтерия D2O (2Н3О) также применяют тривиальное название «тяжелая вода».
Третий изотоп водорода 3Н, называемый тритием (то tqitov — третий), получается при искусственном превращении атомов. Тритий (химический символ Т) неустойчив и распадается, испуская р“-частицу. По последним измерениям его период полураспада равен 12,41 лет (Johnes, 1951).
Несмотря на довольно небольшой период полураспада, тритий, хотя и в мини-мальных количествах, встречается в природе. Он образуется в верхних слоях атмосферы благодаря пдериому процессу, вызываемому космическими лучами, и за счет диффузии и конвекции (в виде молекул НТ) постепенно попадает на земную поверхность. Как нашли Хартек и Фальтингс (Fallings, 1950), в 10 воздуха в среднем содержится 1 атом трития, а во всей атмосфере—примерно 1 .ноль. По данным фон Гроссе (V. Grosse), в сильно концентрированной тяжелой воде можно достигнуть миллионнократного обогащения тритием. Лед с высоким содержанием трития лгоминесцирует. Искусственно тритий можно получить п результате ядерных процессов, приведенных в табл. 73. Об использовании трития в атомпых бомбах см. на Стр. 646 и сл.
592 Глава 12
Исторические сведения. После открытия изотопов кислорода (Giauque, 1929) оказалось, кто найденная Астоном масс-споктрографическим методом (1927) величина массы атома водорода, пересчитанная на оснований шкалы химических атомных весов, дает для атомного веса водорода величину, на 0,02% меньшую, чем найдено химическим путем. Поскольку эта разница, несмотря на свою малую величину, выходила за пределы ошибок очень тщательных измерений, Вирге (Birge) в 1931 г. пришел к выводу, Что в обычном водороде должен содержаться в небольших количествах более тяжелый изотоп. В конце 1931 г. Юри установил в атомном спектре водорода, который с целью обогащения тяжелым изотопом был пропущен через дистилляционные колонны, две очень слабые линии, которые соответствовали линиям, получающимся, если в уравнение (13а) (стр. 110, т. I) для М ввести вдвое большую величину, чем величина М в обычной серии Бальмера. Уошберн (Washburn) и Юри вскоре пашли, что. при электролизе воды она значительно обогащается тяжелым изотопом: водорода; и уже, в 1933 г. Льюис и МакДональд (MacDonald) сумели получить длительным электролизом несколько миллилитров «тяжелой воды» D3O практически в чистом виде.
Получение. Получение «тяжелой воды» в технических масштабах производят длительным электролизом воды (содержащей NaOH). При этом раствор все более и более обогащается Г>20 в той же мере, как уменьшается ого объем. Поскольку первые стадии обогащения связаны с относительно очень большим расходом электрической энергии, то исходным материалом обычно служит вода, которая уже нескблько обогащена О3О иными методами (см. ниже).
Водород, выделяющийся при электролизе воды, обогащается изотбпом Ш. Отношение количеств 1Н : 2Н в таком гаэе в зависимости от условий проведения электролиза превышает в 3—20 раз это отношение в поде. Причины очень высокой эффективности электролитического разделения еще не достаточно ясны. В водороде, который находится в равновесии изотопного обмена с водой (см. стр. 596), отношение 1Н : ЙН только в 3 раза выше этого отношения в воде. Поэтому обычно считают, что преимущественное выделение легкого изотопа Щ при электролизе объясняется различной скоростью разрядки ионов Н’ и D’. Это может быть вызвано тем, что высота потенциального барьера, преодолеваемого при разрядке ионов (см, стр, 742 и сл.), различна для обоих изотопов. Кроме того, вероятно, что скорость, с которой атомы объединяются в молекулы, для Н больше, чем для D. Несмотря па значительно большую эффективность разделения изотопов при электролизе воды ио сравнению с другими методами, все же если «фактор разделения» (коэффициент разделения, т. е. отношение величины Щ/2Н в выделяющемся газе к такой же величине в растворе) равен 5, из. 1000 л обычной воды получают только 10 -кл 98%-пой DaO. Электрическая энергия, требующаяся для разложения 1000 л воды, если работать с напряжением на электролитической ячейке 3,6 в, составляет примерно 1000 кет ч. Для получения 99,99%-ной D2O требуется уже в пять раз больше обычной воды и, следовательно, в пять раз больше электрической энергии. Поэтому в действительности исходят не из обычной воды, а из уже обогащенной D20. Известное количество так ой обогащенной в оды можно по лучить из технических электролизеров, например из долго работавших аккумуляторов *. В других случаях для первоначального обогащения , можпо воспользоваться методами химического обмена (см. стр. 596), например обменом между водой и се ров о дор одой,, Исходным материалом может служить водород, обогащенный D2 при помощи других методов разделения изотопов (см. стр. 583 и сл.). Кроме того, можво сжигать водород, выделяющийся из растворов с высоким содержанием D2O, и полученную воду вновь подвергать электролизу. Этими методами можно снизить расход электроэнергии др 100 квт-ч на 1 г DaO.
Газообразный дейтерий можно получить разложением окиси дейтерия натрием, раскаленным железом или вольфрамом или электролизом окиси дейтерия, содержащей безводный карбонат натрия. (Практически чистая окись дейтерия имеется в продаже.) Дейтерий можно получать и непосредственно из газообразного водорода при помощи диффузионных
* Если в электролизерах разлагающаяся вода постоянно заменяется свежен (обычной), то обогащение со D2O происходят до тех пор, пока в выделяющемся из нее водороде отношение содержания 1Н к содержанию D не будет равно их отношению в обычной воде.
Изотопия, стабильных элементов
593
методов, описанных на стр. 583. Однако этот метод значительно более громоздок, чем получение из воды электролизом относительно легко выделяющейся чистой окиси дейтерия.
Свойства. По своим свойствам дейтерий не очень отличается от «легкого водорода» (протия), а тем самым и от обычного водорода. Свойства обычного водорода, который содержит очень небольшие количества дейтерия, существенно не отличаются от свойств чистого протия.
Точное значение химического атомного веса для D равно 2,014190, а для ХН —* 1,007869 *.
к
Обычный водород состоит из молекул *Н2, Р2 а так же HD. Поскольку при обычных температурах реакция Н2 + D2 = 2HD с заметной скоростью пе идет, то в чистом виде можно получить и HD (дейтероводород). В табл. 66 приведены некоторые свойства, которыми обладают все три названных вещества в чистом состоянии.
Дейтерий (но не дейтероводород HD) точно так же, как и легкий водород, существует в двух модификациях: орто- и парадейтерий, (o-D2 и p-D2). При комнатной температуре дейтерий в равновесном состоянии состоит из 2 ч. орто- и 1 ч. пара-дейтерия. При абсолютном нуле устойчив чистый o-D2. Следовательно, относительная устойчивость модификаций в случае дейтерия обратна устойчивости модификаций легкого водорода (см. т. I, стр. 65).
Из величин моментов инерции **, пайдштпых для молекул D3, HD и ’Н3 (см, табл. 66), следует, что расстояния между центрами атомов в этих молекулах отличаются между собой максимум на несколько сотых ангстрема.
Таблица 66
Свойства дейтерия D2, дейтеро в о до рода HD и протия ЧЕС
ИВ 1Н2
Молекулярный вес 4,02838 3,02206 2,01574
Температура кипения, °К 23,6 20,38
Температура плавления, °К 18,65 16,60 13,95
Давление паров при температуре плавления, 121 — 54
мм рт ст
Давление паров в тройной точке, -е.ч рт ст 128,5 95 53,8
Теплота возгонки при 0°К, кал [г 274,0 228 183,4
Нормальный потенциал, в +0,003 — 0,000
Перенапряжение, е 0,93 0,80
Теплота образования молекул из атомов, 104,5 103,5 102,7
ккал [моль
Момент инерции, г-с.м- 9,25-10-« 6,19-10-41 4,67-10-4’
"Теплой ров одно ст ь X при 0° 2.92+0-* — 4,13-10-4
Линии атомного спектра дейтерия, вычисленные по уравнению (13а), стр. НО т. I, приведены в табл. 67, где одновременно указаны положения этих линий, измеренные Юри (Urey, 1932), Райком (Rank, 1932) и Баллардом (Ballard, 1933). Из смещения этих линий по сравнению с лилиями в спектре водорода следует, что потенциал ионизации атомов D на 0,00365 в выше потенциала ионизации атомов Н. Электролитический потен-
* Эти величины получены пересчетом приведенных в табл, 71 масс атомов («физических атомных весов») в химические атомные веса.
** Момент инерции двухатомной молекулы определяется равенством I =
/ т± т^ > и — массы обоих атомов.
38 Г. Реми
594
Глава 12
Таблица 67
Линии в спектре атома дейтерия
Серин Лайлтана («2 = 1) ni «= 2 П| = 3 ni4 пд = 5 тц = 6 ni = 7
^Т>ассч> ^набл> А 1215,33 1215,33 1025,44 : 1025,44 972,27 972,27 949,48 949,48 -937,55 937,53 .. 930,49 ; 930,49
Серия Бальмера (н2=2) пд=3 nL—6 "1 о
^рассч’ А ^-паблт А 6561,00 6560,99 4860,00 4860,00 4339,28 4339.27 4100,62 4100,63
циал растворения дейтерия (в соприкосновении с окисью дейтерия) также примерно на 0,003 в выше* потенциала растворения водорода (в соприкосновении с водой). Следов ат ель по, дейтерий несколько более инертен, чем водород,
Энергия активации (см. стр. 742 и сл.) для Г)2 примерно на 0,85 кнал/молъ выше анергии активации для Н2. Этим объясняется то, что все реакции с участием Da протекают медленнее, чем с участием Н2 (см. стр. 595 и сл.).
Важнейшие свойства «тяжелой воды» (окиси дейтерия) приведены в табл. 68. Как видно из сравнения с данными табл. 11, т. I, стр. 70,
Таблица 6S
Свойства окиси дейтерия
Температура замерзания, °C (при 760 JK41 рт. ст.) 3,82 Поверхностное натяжение при 20°, динасы. 67,8
Температура кипели я, °C 101,42 Вязкость при 20°, санти- : 1,26
(прн 760 .ч.« рт. ст.) пуаз
Плотность при 20° 1,1050 Диэлектрическая про- 77,5
Критическая температу- 371,5 ницаемость при 25°
ра, °C Показатель пре ломл е- : 1,3284
Температура максимальной 11,6 НИЯ Лд При 2(1°
плотности, °C Ионное произведение 1,0.10-15
Удельная теплоемкость 1,02 при 25°
нри 15°, кал Молярное понижение 2,05
Теплота плавления, кал/г 75,86 температуры замер-'
Теплота испарения при 0е, 556,6 зания, °C
кал! г
некоторые ее свойства существенно отличаются от свойств обычной воды. Например, температура замерзания лежит па 3,8° выше: температуры замерзания Н2О, а максимум плотности приходится не на 4°, а на 11,6°.
* Если принять, что температурные коэффициенты потенциалов растворения Ds и Н2 примерно одинаковы, то из приблизительного равенства различий в потенциалах ионизации н потенциалах растворения следует, что теплота гидратации иона D" примерно па 1 ккал больше, чом иона Н‘, поскольку для распада молекулы D, на два D атома (ври О "К) требуется на 0,90 в калЬ'-атом больше, чем для распада молекулы Н2. Такал большая затрата работы должна при нашем предноложенпп компенсироваться большей теплотой гидратации иона D*.
Иготопия стабильных элементов
595
Кристаллическая решетка D20 подобна кристаллической решетке Н20 (см. -г. I, стр. 72). Размеры гексагональной а-тсмсптарной ячейки, по Mero (Megaw, 1034), при 0“ следующие: а = 4,517, с 7,354 Л для D2O и а — 4,514, с = 7,352 А для Н2О.
Свойства -смесей Н2О — D3O примерно аддитивны свойствам обоих составных частей. Особенностью этой смеси является исключительно небольшое изменение объема при их смешиваний. Поэтому плотность воды является примерно линейной функцией содержания в пей D2O.
В эквимолярной смеси Н2О и D2O присутствуют в- основном молекулы HOD, образование которых протекает с малым выделением тепла: НоО-Д-ЬоО -т>- 2HOD-P + 0,036 ккал. Ионное произведение [D*] X [OD'l при 25° примерно в 8 раз меньше ионного пропав ед спи я [Н\] X [ОН'). Электролитическая подвижность ионов в D2O в несколько раз меньше, чем в Н2О. Особенно заметно различие подвижностей ионов D’ и ионов Н‘. Подвижность последних примерно в 1,5 раза больше подвижности ионов D". растворимость солей в D2O большей частью ниже, чем в Н2О( если ее относить к эквимолярным количествам растворителя. Одиако существуют соли, которые растворяются в D2O легче, чем в Н2О.
Определение содержания D3O (или DOH) в воде обычно производят по плотности иля интерферометрически (используя: различней показате^’ лях преломления). Содержание дейтерия в газообразном водороде легче всего определить измерением теплопроводности.
Отношение содержания атомов D : И в обычной воде равно 1 : 6000. Независимо от происхождения воды, существенных изменений этой величины пе обнаруживается. Однако в некоторых очень солспых внутренних озерах содержание D значительно повышено (в Мертвом море на 0.003 ат.%). Па больших глубинах в оксанах вода, по-види-мому, обогащена дейтерием (примерно па 0,0023 ат. % па глубине 3000 ж). В некоторых жидкостях растительного и животного происхождения (молоко, кровь, фруктовые соки) это отношение выше на несколько тысячных атомного процента. Это относится и к минералам.
Поскольку обычный кислород представляет собой смесь трех: изотонов, в обычной воде, кроме молекул Н-Л^О, НВЫО и D2ibo, существуют и соответствующие соединения 1’0 й ISO. Следовательно, в общем существует 9 видов простых молекул. В связи с частым использованием воды в качестве стандартного вещества для определения физических констант очень существенным было установление постоянства относительного количества различного вида молекул воды в обычной воде. При электролизе воды в ней наряду с дейтерием могут накапливаться и тяжелые изотопы кислорода. Благодаря обменной реакции (см. ниже) с безводородным соединением, которое содержит изотоны кислорода в обычном соотношении, например с обычной двуокисью серы, можно устранить избыток тяжелых изотопов кислорода. Физические константы, приведенные в табл. 68, относятся к П2О, содержащей изотопы кислорода в обычном соотношении,
Химическое поведение. Дейтерий и соединения дейтерия по своим химическим свойствам вполне анало гичищ во д о р о дул его с о единениям. Однако существенные различия замечены в коэффициентах равновесия и особенно в скоростях реакции.
Различие в скоростях реакций в общем тем больше, чем ниже температура, при которой протекает реакция. Например, Н2 реагируете С12 в присутствии СО в качестве катализатора при 0s примерно в 13 раз быстрее, при З53 примерно в 10 раз быстрее, чем D2 (Rollefsou, 1934), И, реагирует с О2 в присутствии Никеля при 180° в 2,4 раза быстрее, а с N2O при 160° — р 2,5 раза быстрее, чем D, (Melville, 1934). Превращение р-H., в о-Н2 при комнатной температуре в присутствии 0о в качество катализатора протекает в 16 раз быстрое, чем соответствующее превращение о-1)2 н ь-1)2. Вследствие меньшей реакционной способности соединений дейтерия по сравнению с соединениями водорода реакции в тяжелой воде протекают значительно медленнее, чем в обычной воде, если данной реакции предшествует обмен атомов Н па D. Например, каталитически ускоряемый ионами ОН' распад питрамида по уравнению H2N-I\d3^PI2O + + N2O в тяжелой воде идет значительно медленнее, чем в легкой, так как из этого соединения сначала образуется при замене атомов Н на атомы D дейтеронитрамид D2N- NO2. Следовательно, в тяжелой воде наблюдается уже распад йвйтеронитрамида, идущий примерно в 5 раз медленее, чем распад обычного питрамида. Разнпчня (правда,
38*
596
Глава 12
но столь значительные) в скоростях реакций в обычной и тяжелой воде наблюдаются также в тех случаях, когда вода оказывает влияние па скорость реакции только благодаря своим физическим свойствам (вязкости, диэлектрической проницаемости, способности к сольватации и т. д.: см. гл. 18). И в этих случаях реакции в тяжелой воде идут медленнее, чем в оончкой. Однако существуют и такие реакции, скорость которых в тяжелой воде выше, чем в обычной. В большинстве случаев это реакции, которые катализируются типами Н‘ или D’ и ионами ОН' или OD'. Из теории гомогенного катализа следует, что в этих случаях увеличение скорости реакции вызвано большей прочное тын сцеп.теиия дейтронов по сравнению с протонами (Reitz, 1938).
Н2 и О2 при обычной температуре непосредственно пе реагируют, а только прп повышенных темпера турах или при соприкосновении с накаленной никелевой проволокой.
Такая реакция протекает не до конца, а приводит к равновесию
H2+D3
- 2HD. _[НР1В.
’ [Н2] X [Dj
= Ki.
Прп комнатной температуре равно 3,3, при 400°—3,8.’ Следовательно, при повышении температуры равновесие сдвигается в сторону образования HD.
Равновесие реакций Л2-гUС172; 11D-|-HC1 и ГЬЯ-Р! 7^ HD~[-HI, константы
равновесия которых
[НР]Х{ИСЦ _ „ |НР[Х[Н1] __
(И2] X [DC11 я2, [ТЫ X [D11 3
при 125° равны /7=1.5 и К3 = 2,0, также с повышением температуры сдвигается в сторону образования HD.
Константа равновесия Х4 реакции
и2о+нВ^иоп+нг;
при 20° равна 3,1. Следовательно, газообразный водород содержит в три раза меньше дейтерия, чем вода, с которой он находится в равновесии. В присутствии подходящих катализаторов равновесие устанавливается при комнатной температуре. Условия для установления равновесия выполняются и в некоторых случаях выделения водорода из растворов, так, например, при разложении формиата натрия палладиевой черпаю или ферментом (ИС00лда4- Н2О=Н2 + КаНСО3) или при электролитическом выделении водорода на покрытом платиновой чернью электроде. Фаркащ (Farkas, 1934) смог показать, что в этих условиях отношение Н : D в выделяющемся водороде действительно соответствует равновесному. Впрочем, при обычном электролитическом выделении водорода освобождается значительно больше легкого водорода (стр. 592).
Обменные реакции с окисью дейтерия. Если в тяжелую воду ввести соединения, содержащие ионогенносвязаннъш водород, то при комнатной температуре ои мгновенно заменится на дейтерий, Например, НС1 реагирует с D2O неизмеримо быстро по уравнению НС1 + D3O = DC1 + HOD. Благодаря таким обменным реакциям можно доказать, что в аммиаке все атомы Н способны к обмену. Аналогично аммиаку ведут себя и все его производные, например метиламин CH3-NH2, анилин CSH5-NH2 и ацетанилид GH3-GO-NH-C6H5. В этих и в других аналогичных им соединениях атомы Н, связанные с атомами углерода, не замещаются на атомы дейтерия при комнатной температуре. Точно так же, как атомы И, связанные с галогенами или азотом, на атомы D замещаются и атомы Н, связанные с кислородом шш серой, например оба атома водорода в перекиси водорода, а также атомы Н в органических гидроксильных группах, присутствующих в кислотах, фенолах и спиртах.
Если атомы И связаны с атомами С, по соседству с которыми расположены группы СО илц NO2, при действии D;,0 происходит не мгновенное, а постепенное замещение
Изотопия стабильных элементов
597
атомов Н атомами D. Скорость такого обмена зависит от скорости, с которой кетоформа такого соединения переходит в спольпуго форму [например, СИ3—СО—СН3 в СН3—С(ОН)=СН2] или нитро соединение переходит в свою ацпформу [например, СН3—NO2 в СН2 = NO(OH)].
При повышенных температурах, а такл:е в присутствии катализаторов могут обмениваться и такие атомы Н, которые связаны неионогепно. Примерами таких реакций могут служить упоминавшиеся выше реакции между Н2 и D2> между II2 и DC1 или DI и между Н2 и IIOD. В органических соединениях медленный обмен атомов Н, связанных с атомами С, часто можно вызвать действием на эти соединения «тяжелой серной кислоты»— D^SO,, или HDSO,.,.
Как и в рассмотренном выше обмене с И2, таки при обмене ионогенно св я зап пых атомов Н па атомы D устанавливается некоторое равновесие. Это следует имен, ввиду, если необходимо провести обменные реакции с водой, которая наряду с молекулами D20 содержит значительное количество молекул Н2О.
На основании наблюдений, что при действии тяжелой воды мгновенно обмениваются на атомы дейтерия только ионогенно связанные атомы Н, обменные реакции с тяжелой водой можно использовать для установления ионогенного или неионогенного характера связи атомов Н. Например, оказалось, что кислота Н2РО2 (см. т. I, стр. 678 и сл.) обменивает на дейтерий только один атом Н. Уже из других свойств этого соединения следовало, что оно содержит только один ионогенно связанный атом И. Это было окончательно подтверждено обменной реакцией с тяжелой водой.
Обменные реакции с т я я; едой водой часто можно использовать для выяснения механизма реакции. Кроме того, опи имеют существенную роль в выяснении биологических и физиологически-химических вопросов. Было показано, что дейтерий в организме усваивается так же, как и водород (только поглощается относительно меньше D, чем Н). В высоких концентрациях окись дейтерия вредна, по-видимому, потому, что она нарушает равновесия реакций в организме. На высшие организмы высокие концентрации окиси дейтерия действуют смертельно. В малых концентрациях она безвредна. Поэтому ее мозкно использовать как «индикатор» для исследования обмена веществ. Если дейтерий прочно удерживается в составе данного соединения, то можно точно проследить путь, который это соединение проходит в организме.
Остальные соединения дейтерия. Наряду с окисью дейтерия известно уже много других его соединений. Для их получения используют в основном те реакции, которые служат и для получения соединений обычного водорода. Дейтерированные аналоги таких соединений, содержащие ионогенно связанный водород, проще всего получать обменными реакциями.
Точно так же, как и окись дейтерия, другие соединения дейтерия часто существенно отличаются но своим свойствам от соединений обычного водорода, В большинстве случаев их температуры плавления лежат пиже (по иногда, как, например, для DaO, выше), чем температуры плавления соединений обычного водорода. Давление паров этих соединений при низких температурах значительно меньше, а при высоких зва-чнтельно больше, чем у обычных соединений водорода. Их теплоты плавления и испарения в большинстве случаев заметно отличаются от соответствующих теплот соеди^ нений водорода. И прежде всего такие различия обнаруживаются л температурах, при которых происходит переход одних модификаций в другие. Этому отвечают и существенные различия в теплотах превращений. Например, в твердом состоянии CHS при температуре 20,45 К претерпевает превращение, которое сопровождается выделением 15,7 хал/молъ. Соответствующее превращение CD4 происходит при 26,3° К и при Этом выделяется 58,7 кал/молъ. Интересно, что ND4Br при —104° претерпевает превращение, которое по наблюдается у NH4Br. Обеим температурам превращения NH,Br, лежащим прн+141^:И —38,8°, отвечают температуры превращения ND4Br при ф-1320 и —58,5° (Clusius, 1938),
Гл а в а 13 ИСКУССТВЕННЫЕ ПРЕВРАЩЕНИЯ АТОМОВ (Ядернам химия)
Превращения атомов под действием а-лучей. В 1919 г. Резерфорд установил, что ядра атомов водорода (протоны) образуются в качестве положительных вторичных лучей не только при прохождении а-частиц через соединения, содержащие водород (см, стр. 553), но и при их прохождении через воздух или азот, но не кислород. Действительно, в азоте образовывалось протонов на 25% больше, чем в воздухе. Из этого наблюдения Резерфорд сделал вывод, что частицы Н+ (протоны) выбиваются а-частпцами :из ядер атомов азота. Что в этом случае действительно образуются протоны (ядра атома водорода), можно было установить по их отклонению^ в магнитном поле. Это открытие имело чрезвычайно важное значение, таю как представляло собой первое наблюдение, искусственного атомного превращения. При радиоактивных превращениях атомы распадаются сами по себе, произвольно, но здесь впервые паука столкнулась со случаем, когда превращение атомного ядра, а тем самым и изменение природы химического элемента были осуществлены искусственно бомбардировкой «-частицами. Впоследствии соответствующий превращения атомных ядер под влиянием бомбардировки «-частицами наблюдались для многих элементов.
Превращения, которые происходят с атомными ядрами в результате таких процессов, как показали дальнейшие исследования, заключаются не только в том, что из ядра атома выбивается ядро атома водорода. Более того, как правило, а-частица, выбившая протон, входит в ядро на его место. Атомное ядро, образовавшееся в результате этого процесса, обычно им еет не на единицу меньшие массовое число и заряд, а па 3 единицы большее массовое число и: на 1 единицу больший заряд (соответственно разнице масс и зарядов Неа+ и Н*). А поскольку заряд ядра равен порядковому номеру элемента, то, например, из “ N в результате отщепления протона при бомбардировке а-частиц ами образуется изотоп кислорода *'О, а не изотоп углерода J’C, как сначала можно было полагать.
Нейтроны. В 1930 г. Боте (Bothe) нашел, что при многих превращениях атомов под влиянием а-частиц, и прежде всего при а-облучении бериллия, из атомных ядер выбиваются не протоны^ а лучи нового вида. Вскоре Чедвик (Chadwick) показал, что эти лучи состоят из частиц, масса которых приблизительно равна массе прогона, но которые незаряжёны. Эти частицы назвали нейтронами (символ п). Нейтроны образуются при а-облученнп бериллия в результате процесса “Be + kt = |3С + *п. Так как нейтрон представляет собой атомное ядро, не имеющее заряда, то часто его рассматривают как химический элемент с порядковым номером 0. Вместе с протонами нейтроны являются элементарными составными частями всех атомов (см. стр. 608). Но в свободном состоянии нейтрон
Искусственные превращения атомов
599
неустойчив. Спустя в среднем 20 мин нейтрон самопроизвольно «распадается» на протон и электрон. Как видно из данных, приведенных в табл. 69, этот распад сопровождается заметной потерей массы.
Таблица 69
Заряды н массы элементарных частиц, дейтрона и ядра гелия
Название Символ Заряд2' Масса
Ядро гелия («-частица) 4Но3+ или а 2Л. 4,002782
Дейтрон D+ или d l-f- 2,014196
Протон 1Ht или р l-f- 1,0075973
Нейтрон п 0 : 1,0089899
Электрон Q или е~ 1 — 0,0005486
Позитрон © или е+ 1 + 0,0005486
а Единицей заряда служит элсмептарпый (см. стр. 107, т. I).
квант электричества
Поскольку нейтралы не им итог заряда, они беспрепятственно проходят через алектронпуго оболочку атомов. Слдцовательпо, пейтропы пе выбивают электроны из атомных оболочек (т. е. не обладают ионизирующим действием). Поэтому их проникающая способность велика. При достаточной начальной скорости они проходят даже сквозь сдой свинца толщиной 0,5 м. Нейтроны тормозятся лишь в редких случаях, когда они сталкиваются с атомными ядрами. Наибольшую потерю скорости они испытывают, как следует из законов упругого удара *, при столкновении с ядрами равной массы, и прежде всего ядрами водорода. В соответствии с этим опыт показывает, что' особенно сильно потоки пейтронбв тормозятся соединениями, содержащими водород. Например, слой парафина толщиной 20 с.и полностью задерживаетпейтропы с макси-
мальными начальными скоростями.
Нейтроны, находящиеся в тепловом равновесии с окружающей средой, т, е. такие нейтроны, кинетическая анергия которых равна кинетической энергии молекул газа при заданной температуре, называют тепловыми нейтронами. В соответствии с уравнением (14) гл. 17 средняя кинетическая энергия молекул газа, а следовательно, и теп-
ловых нейтронов
равна А. — Т.
1 2 Na
Коэффициент
называется константой Больцмана.
Он равен 1,3804 х
ХЮ-19 ерг. или 8,610 10_з эв]град. Таким образом, при Т=293° К средняя кинетическая энергия молекул газа будет равна з/,. 293-8,616.10_8 0,038 ев. Для 'нейтронов обычно
приводят по среднюю, а-так называемую вероятную кинетическую эйергию, т. е. кинетическую энергию, которой отвечает наиболее часто встречающаяся при данной температуре скорость. Эта анергия равна кТ н при обычной температуре (Т — 293°) составляет 0,025 эв. Даже нри 3000’К кинетическая энергия тепловых нейтронов (0,258 ев) все еще мала по сравнению с кинетической энергией нейтронов, образующихся при ядерных реакциях. Такие нейтроны часто имеют энергию в несколько миллионов электрон-вольт. Поэтому нейтроны, образующиеся в результате ядериых реакций,
* Если сталкиваются два шара равной массы, то при центральном упругом ударе они обмениваются скоростями. Следовательно, если сначала ударяемый шар находился в состоянии: покоя, то после столкновения скорость ударяющего шара равна пулю. Н ацротив, если ударяющий шар сталкивается с твердой степной, то он отскакивает от п ее с тон же скоростью. В общем случае для центрального упругого удара справедливо с оотнбшенпе (получающееся комбинацией двух уравнений, приведенных в примечании
- ет о т j С< -I- tn.’.C
на стр. о53): щ — 2 -i_n ." "—с1> С1 и —скорости шаров с массами и т2
до удара, i’(— скорость, которую будет иметь шар с массой после удара.
600 Глава 15
называют «быстрыми нейтронами», а те нейтроны, кинетическая энергия которых лежит в области значений кинетической энергии тепловых нейтронов,—Умедленными нейтронами».
Позитроны.При некоторых атомных превращениях (см. стр. 616исл.) испускаются лучи еще одного вида — позитронные лучи. Онн состоят из быстро движущихся частиц, масса которых равна массе электрона, а заряд равен, но противоположен но знаку (следовательно, положительный} заряду электрона. Поэтому позитроны часто называют «положительными электронами» в отличие от обычных — «отрицательных электронов!».
Позитрон был открыт Андерсоном (Anderson) в 1932 г. при исследовании так называемых «космических лучей» *. Вскоре после этого было найдено, что позитроны часто обнаруживаются при ядерных превращениях, вызываемых «-частицами. Продолжительность жизни позитрона очень мала. Он по является неустойчивым в собственном смысле этого слова, а исчезает при столкновениях с электроном; обе частицы исчезают, а их масса превращается в у-излучение (ем. стр. 609).
Мезоны. Под действием космических лучей могут образовываться частицы некоторых других видов, так называемые мезоны (т. е. «средние частицы», поскольку их масса обычно лежит между массой электрона и протона). Существуют различные виды мезонов. Чаще всего наблюдаются такие мезоны, масса покоя Которых в 210 раз больше массы покоя электрона (же). Среднее время жизни таких мезонов (р-мезонев) составляет 2,15-10^ сек. Они могут иметь как положительный, так и отрицательный заряд, абсолютное значение которого равно заряду электрона. Наблюдаются и электро-нейтралъные мезоны, например, такие, масса которых равна 265 те, а среднее время жизни 10'1'1 сек (л°-мезоны). Кроме того, известны (положительные и отрицательные) мезоны с массой 276 те (л-мезоны, среднее время зкиани 2,5- 1O'S сек). Вероятно, существуют и еще значительно более тяжелые мезоны (с массой в 2200 раз тяжелее массы электрона). Но они чрезвычайно быстро распадаются (средиее время жизни 10“1о— 10"э сек} с образованием более легких мезонов. При распаде мезонов с массой 210 те образуются в случае отрицательных мезонов электроны, в случае положительных — позитроны. Кроме того, считают, что наряду с этими частицами образуются но два нейтрино (см. стр. 632).
Элементарные частицы. Электрон, позитрон, мезон, нейтрон и протон называют элементарными частицами», так как они являются наименьшими частицами, которые до сих пор удавалось наблюдать **,
В табл. 69 приведены точные значения масс протона, нейтрона, электрона и позитрона. Поскольку в результате атомных превращений часто из ядер атомов выбиваются дейтропы и ядра гелия, то они также включены в табл. 69.
В настоящее время к элементарным частицам относят также фотоны (т. е. кванты света ftv) и нейтрино (масса покоя которого во всяком случае меньше одной тысячной массы иокоя электрона, а возможно, и вообще равна нулю, как масса покоя фотонов). Природа элементарных частиц во многих отношениях еще загадочна. Они отнюдь пе представляют собой окончательных неизменных единиц. С другой стороны, оказывается невозможным рассматривать их состоящими из еще более простых частиц. В настоящее время склоняются к тому, чтобы рассматривать различные частицы как разные «формы проявления» или «состояния» чего-то, что еще пё вполне четко определено, и что может выступать как в форме материи (т. е. частиц с коночной массой покоя), так и в форме анергии.
Различные виды атомных превращений. Было показано, что превращения атомов могут происходить пе только под действием а-частиц, по
* «Космическими лучами» или «космическим излучением» называют [открытое Гессом (Hess) в 1912 г,] чрезвычайно проникающее излучение, приходящее на Землю из космоса.
ж* Вероятно, существует и «антипротон», т. е. частица, масса которой равна массе протона, а заряд равен, но противоположен по знаку заряду протона. Считается, что существует и «антинейтрон», превращающийся при 0“-распаде не в протон, как обычный нейтрон, а в антипротон. По-видимому, существует и два вида нейтрино-. считается установленным, что нейтрино, образующиеся при 0+-распаде (см. стр. 616), ведут себя иначе, чем образующиеся при р“-распаде «антинейтрино».
Искусственные превращения атомов 601
и под действием протонов, дейтронов или нейтронов. Кроме того, атомные превращения происходят и под действием Y-лучей *.
Протон н дейтрон можпо значительно легче, чем ядра гелия (а-частицы), ввести в атом до о ядро, так как протон и дейтрон вследствие их меньшего по сравнению с ядром, гелия заряда испытывают меньшее отталкивание атомными ядрами. Еще легче ввести' в ядро нейтроны, почему их и используют чаще всего для исскуственного получения радиоактивных изотопов (см. стр. 617 и сл.).
Ядерные превращения можно представить при помощи соответствующих уравнений, как вто обычно делают для простых химических реакций. Поэтом*/ ту область ядерной физики, которая специально занимается ядерными реакциями, называют «.ядерной химией» **.
Для ядерных превращений справедлив следующий закон: при каждом превращении в ядре остаются постоянными как сумма массовых чисел, так и сумма зарядов частиц, принимающих участие в реакции.
Примерами атомных превращений могут служить следующие уравнения:
*?B + £a=’£C + fd
^B+^igC+lp
+ = I
^Al + ^=^A[ + fp ! ГзЛ'+^=^и> i»Al --{p •- jfMg-f-u11 nAi-H"=^Mg+Jp
isA.[ —jaAJ •+ Y
Дя + {Р=4Ве+у
^Br|-Jn^Br-|-2Jra
inH-Y =JP“Tofl
Обычно частицы, используемые для бомбардировки атомных ядер или выбиваемые из них, обозначают малыми буквами: протоны — р, дейтроны — d, ядра гелия — а и нейтроны — п. В приведенных выше уравнениях для иллюстрации сформулированного закона все символы снабжены массовыми числами и зарядами ядер. Н о-обычно ограничиваются только указанием массовых чисел участвующих в реакции атомов (например, 27Al-[-</=25Mg-|-a), так как остальные величины определяются написанными символами. Часто используют краткую запись ядерных реакций, при которой бомбардирующая и выбиваемая частицы отделяются запятой и заключаются в скобки, нерод ними записывают символ исходног о, а после них — образующегося атома. Например, вместо 2’Al-|-tf — S5Mg~pa кратко записывают 2?А1 (d, a) 25Mg. Соответственно реакции, вызываемые облучением дейтронами и сопровождающиеся испусканием a-частиц, кратко называют (а, а)-реакциями, реакции, вызываемые дейтронами и сояровождащиеся испусканием кротонов,— (г/, р)-реакциями и т. д.
Как показывают приведенные примеры, один и тот ;ке атом может претерпеть множество различных превращений. При этом могут образоваться атомы как с большим, так и с меньшим порядковым номером, а также атомы с тем же порядковым номером, но другим: массовым числом (каотовы), а кроме того,— атомы с другим порядковым номером, но с равной массой (изобары). На рис. 72 показано, как при различных реакциях, протекающих под действием протонов, нейтронов, дейтронов, а- и у-частиц, изменяются массовое число А и порядковый номер Z атомного ядра. Стрелки указывают, как и насколько изменяются нрй данной ядерной реакции A a Z. Например,.
* Удалось вызвать атомные превращения и под действием очень быстрых электронов. Вместо a-чаетпц (ядер ОДе) иногда применяют ядра 3Нс, а в последнее время — и ускоренные ядра С и N.
** Основная задача ядерной cfuauKu заключается в выяснении природы ядерных сил. Одним из важнейших вспомогательных средств для этого является изучение превращений веществ при ядерных реакциях п освобождающейся при этих реакциях энергии. Исследования в этой области сначала можно было осуществлять только чисто физическими методами и поэтому их стали рассматривать как часть физики. Только после открытия искусственной радиоактивности (см. стр. 616) оказалось возможным использование в этой области и химических методов, и одновременно химики получили важнейшее новое средство для исследования химических проблем (см. стр. 625 и сл.).
602
.Глава 13
при (а, р)-реакции массойое число увеличивается на 3 единицы, а порядковый номер— на i; при (<7, р)- или (в, у)-реакции массовое число увеличивается па 1 единицу, а порядковый номер остается неизменным; при (л, р)- реакции порядковый номер уменьшается на 1 единицу, а массовое число остается неизменным ит, д.
Для счета частиц, испускаемых при ядерных реакциях, в большинстве случаев используют счетчики. Гейгера—Мюллера. Они представляют собой аксиально натянутую в трубке проволоку, к которой приложено напряжение в несколько тысяч вольт. Ионы, образуемые выбиваемой в процессе ядерйой реакции частицей (см.; стр. .540), настолько ускоряются раэпостыо напряжений между стенкой трубки и проволокой, что
Порядковый номер
Р и <. 72. Изменение порядкового номера и массового числа при искусственных ядерных превращениях.
онив свою очередь ионизируют другие молекулы газа. При этом разряжающее действие ионов настолько усиливается, что прохождение через счетчик каждой отдельной частицы легко обнаруживается как импульс тока. Для точного исследования ядерных реакций важнейшим методом является наблюдение траекторий частиц в камере Вильсона (см. стр. 540). Для наблюдения особенно быстрых частиц используют пузырьковые камеры Глаяера. В этих камерах траектории частиц выглядят как маленькие пузырьки пара, которые образуют частицы па своем пути, проходя через легкошшящую жидкость (например, жидкий водород). Аналогичные траектории образуются и в эмульсионном. слое фотопластинок после их проявления. Такие пластипки используют прежде всего для исследования очень неустойчивых частиц.
Поскольку число частиц, испускаемых при: радиоактивном распаде, а следовательно, и число превращений атомов, происходящих в единицу времени, можно измерить с очень большой точностью, это число нсиольйуют как .меру радиоактивности. Поэтому активность (естественного и искусственного) радиопренарата выражают числом превращений в секунду (преер!оек). Большие активности обычно выражают
Искусственные превращения, атомов
603
в единицах wop и»; при этом кюри определяют соотношением 1 кюри= 3,700 000-IO10 npeepiceu. Раньше кюри определяли как такое количество радиоактивного вещества, в котором в единицу времени происходит такое же число ядериых превращений, как в 1 г радия. Это определение затем было отвергнуто, так как если его принять за основу, то коэффициент пересчета преер/сек в кюри становится зависимым от того, какая величина принята для периста полураспада радия.
В большинстве искусственных ядерных превращений происходят так называемые реакции вытеснения, т. о. реакции, при которых бомбардирующая частица входит в ядро, выбивая из него ppyrvro частицу, Эти ядерные реакции соответствуют реакциям замещения в обычной химии. Но в области ядерной химии существуют и аналоги реакций присоединения, термической и фотохимической диссоциаций обычной химии.
ЯДерные превращения, объединенные в первые две колонки уравнений на стр. 601, относятся к' реакциям. ьт.иещдаия. Две первые реакции третьей колонки являются реакциями присоединения, В результате этих реакций бомбардирующая частица захватывается ядром, которое в свою"очередь не испускает никакой другой частицы. Освобождающаяся при этом энергия испускается В' форме у-излучения. Две следующие реакции третьей колонки представляют собой ядерные реакции диссоциации, которые (как н реакции Термической диссоциации молекул) вызываются кинетической энергией сталкивающихся частиц. Последняя реакция третьей колонки является примером фотохимической (вызываемой действием у-лучей) ядерной диссоциации.
В настоящее время известен уже Целый ряд обратимых ядерных реакций; Примерами таких реакций могут служить
’П-гл D+y; JLi-|-a ив-f-n; ПВ-f-a «N-f-re.
Энергии ядериых реакций. Энергия, освобождающаяся в результате ядерных реакций, огромна но сравнению с энергией, которая выделяется при обычных химических реакциях, Например, при превращении бериллия под действием протонов в изотоп лития 6Li и гелий освобождается энергия в 2,13 млн. ва (Мов), что составляет 49,1 млп. нкал/г-атом Be (см. т. I, стр. 166). В реакции 18О(р,а) i-ipj освобождается энергия в 3,97 Мое (соответственно 91,5- 10в ккал/г-атом], в реакции а) ®гС— 5,02 Маа (115,7-106 ккал1г-атом), в реакции 7Li (р, a) dHe — 17,33 М?в (399,5-10*> ккал/е-атом), а в реакции 6Li(rf, а) 4Не — 22,28 Мэк (513,6-1()8 к'кал/ееатом). Следовательно, энергий, выделяющиеся в результате, ядерных превращений, в миллион раз больше энергий обычных химических реакций. Впрочем,: существуют и эндотермические ядерные реакции, но и в этих случаях поглощаемая энергия имеет такой же порядок величины. Например, в реакции ЦЙ(а, р) 1ТО потребляется 1,20 Мае (27,7-10® ккал/я-атом), в реакций D (у, п) LH — 2,19 Mae (50,5-10® >:ка.г/г-атом).
Использованию громадной энергии, выделяющейся при таких ядерных превращениях, препятствуют чрезвычайно низкие выходы этих реакций. Так как диаметр ядра очень мал по сравнению с диаметром атома (ср. т. I, стр. 106), только незначительная часть частиц, проходящих через атом, сталкивается с ядром. Кроме того, лишь часть таких столкновений приводит к проникновению частицы в столкнувшееся с ним ядро и, следовательно, к ядерной реакции. Например, па 100 000 а-частйц, из которых каждая на своем пути длиной в 1 см проходит примерно через 10 000 молекул Нг< (и, следовательно, ионизирует их), образуется только один протон (т. с. при проход^’ депииа-частицами 1()э молекул только в одном случае происходит столкновение с ядром водорода), При бомбардировке а-частицами молекул N2 число образующихся в тех же условиях протонов еще в десять раз меньше. Следовательно, только 1/10 столкновений с ядрами:азота приводит к ядерным реакциям, или, иными словами, на КДО столкновений а-частиц с молекулами N2 только одна приводит к ядерной реакции. В общем случае выходы (т. е. число атомов, подвергшихся ядерной реакции, отнесенное к числу бомбардирующих частиц), найденные до'настоящего времени для ядерных реакций йод действием а-частиц, дейтронов или протонов, лежат в интервале 10_5_10т11. Гораздо большие выходы характеризуют реакции под действием нейтронов. Впрочем, сами нейтроны, как правило, получаются в результате реакций, проходящих с очень малыми выходами (см. стр. 617).' Однако существует одни вид ядерных реакций, в результате которых можно получить без затраты, энергии любое число нейтронов. Это рассматриваемые на стр. 639 и сл. реакции деления ядер.
Приведенные данные о выходах ядерных реакций, происходящих под действием а-частнц, протонов и дейтронов, относятся к тому случаю, ко г Да: кинетические энергии бомбардирующих частиц достаточны для преодоления значительных сил отталкивания, действующих в силу закона Кулона со стороны атомных ядер на приближающиеся к ним положительно заряженные частицы. Испускаемые радиоактивными веществами а-чаетпцы, под Действием которых были проведены первые ядерные реакции (и которые часто используют Для этих целей и в настоящее время), имеют кинетические энер
fJ04
1’лава 13
гии иорядка нескольких миллионов электроп-в о лът; Однако оказалось, что в случае протонов и дейтронов достаточны кинетические энергии в несколько сотеп тысяч электрон-вольт. Но если удовлетвориться и такой энергией стол к по вопия, то все же получение нучка частиц, обладающих равными скоростями, отнюдь не так просто. Особенно дорогостоящая аппаратура требуется для получения бомбардирующих частиц-с энергией 1 Мае и выше.
Получение потоков бомбардирующих частиц. Старейшим методом получения частиц с высокими скоростями является метод послеускорения Вина (Wien, 1925). Для проведения ядерных реакций оп впервые был использован Кокрофтом (Cockroft, 1932), Этот метод основал иа том, что каналовые лучи, полученные в газообразном гелии, водороде или дейтерии, после выхода из зоны ионизации ускоряются сильным электрическим полом. Этот метод позволяет получить сравнительно высокие концентрации ускоренных частиц (сила тока достигает нескольких сотен микроампер). Но если пользоваться обычпыми способами получения высокого напряжения, то с трудом можпо получить частнцьг с энергией выше 100 000 ов. Для подучения ускоряющего напряжения 1 млн. t и выше необходимо применение специальных приборов: сконструированного вап де-Графом (1932) генератора высокого напряжения,, принцип которого восходит К старым электрическим машинам, иди каскадного умножителя напряжения Спмсяшя, который представляет собой комбинацию повышающего тралс’формагора переменного тока с системой конденсаторов и выпрямителей. Частицы с очень высоким энергиями непосредственно получают при помощи изобретенного в 1932 г. Лоуренсом циклотрона. Это устройство, в котором положительные ионы (в большинстве случаен Н+ или Не®+) ускоряются благодаря тому, что они движутся в сильном магнитном иоле по спиральным траекториям, проходя несколько сотен раз через электрическое поле напряжением ~ 5090 в, прежде чем покинут аппарат. Благодаря синхронизированной со временем полного оборота частиц (которое в магнитном поде по зависит от радлуса орбиты) перемене полюсов электрического поля ионы ускоряются в одну сторону. Следовательно, ускорение иопов при каждом прохождении через электрическое поле суммируется и можпо получить скорости, отвечающие разности напряжений в несколько миллионов вольт.
В общем случае нейтроны не нуждаются в ускорении, чтобы вызвать ядерную реакцию, так как они пе имеют электрического заряда и не подвержены действию кулоновских сил со сторопы ядра. Однако часто «быстрые» пойтроны вызывают совершенно другие ядерцые реакции, чем «медленные». Нейтроны получаются в результате ядерных процессов: или в реакциях типа рассматриваемых на стр, 617, или при делении атомов, проводимом в так называемых ядерных реакторах. Поскольку большие промышленные ядерные реакторы (атомные котлы) в настоящее время служат мощными источниками пейтропов, сейчас используют в основном реакции под действием нейтронов. Но это отнюдь не означает, что получение быстрых положительно заряженных бомбардирующих частиц потеряло свое значение. Продукты ядерных превращений, получаемые с их помощью, часто Отличаются от тех, которые можно получить под действием нейтронов.
(1)
Сечение ядерных реакций. Число нейтронов, проходящих в секунду в определенном направлении через единицу площади (1 сла), называется потоком нейтронов Ф. Если пейтропы проходят через поглощающее их вещество, то число поглощаемых нейтронов тем больше, чем больше нейтронов попадает в это вещество. Кроме того, поглощение зависит от скорости нейтронов и. Если толщина поглощающего слоя настолько мала, что при прохождении его скорость нейтронов практически не изменяется, то уменьшение Ф в направлении х можно представить уравнением
Здесь Л\ означает число атомных ядер поглощающего вещества в 1 смя (число Лошмита), ап — постоянная, которую называют «яРгректиан.ьгм. сечением» атомных ядер данного вещества. Эта постоянная действительно имеет размерность «е.м2», и для наглядности ее можно рассматривать как площадь поперечного сечения атомных ядер, па которую падает поток нейтронов. Если бы каждый нейтрон, центр которого попадает на эту площадь, определяемую диаметром атомного ядра, поглощался, а ни один нейтрон, не попавший па эту площадь, пе поглощался, то эффективное сечение было бы равно действительной площади сечения атомного ядра. Последняя имеет размеры порядка 10-24 сз<-. Однако эффективное сечение а может быть как значительно меньше действительной площади сечепия ядра, так и значительно больше со. По величину 10~-4 с.ч'-используют как единицу измерения п. Ее называют «1 барн», следовательно, 1барн ~ = 10'24 елЛ
Искусственные превращения атомов 605
Эффективное сечение, определяемое уравнением (1), правильнее называть общим эфф>ективным сечением at.’ Ослабление потока нейтронов, которым определяется ц(, складывается из ослабления вследствие отражения и рассеяния нейтронов на атомных ядрах и ослабления за счет захвата нейтронов атомными ядрами. При этих захватах могут протекать различные ядернътс реакции, и для каждой из них можпо определить и измерить особое эффективное сечение атомных ядер.
Точно так же, как и для поглощения нейтронов или различных вызываемых нецтропами ядерных процессов, определяется эффективное сечение для поглощения других частиц —' протонов, дейтронов, а-частиц и даже фотонов (у-лучей), а также для вызываемых этими частицами ядерных реакций. Определение эффективных сечений для отдельных ядерных процессов важно потому, что, как будет показало ниже, с их помощью можно рассчитать выход искусственно получаемых атомных ядер для каждого данного вида излучений.
Эффективное сечение зависит, кроме вида частицы и характеристик атомпого ядра, также от энергии бомбардирующих частиц (т/о ти2 или Av). Вероятность захвата нейтронов ядром в общем случае тем выше, чем больше время их пребывания вблизи ядра. Поэтому обычно вероятность захвата нейтрона уменьшается с увеличением их скорости. Напротив, для положительно заряженных частиц эффективное сечение вызываемых ими ядерных реакций при увеличении кинетической энергии частиц сначала возрастает и начинает постепенно уменьшаться в большинстве случаев только при довольно высоких значениях энергии. Это объясняется том, что для проникновения положительно заряженной частицы в атомное ядро она сначала долж'на обладать энергией, достаточной для преодоления отталкивания со стороны одноимонпо заряженного ядра. Для эндотермических ядерных реакций эффективное сечение реакции при энергиях ниже некоторой определенной величины равно нулю. Поскольку частица по обладает энергией, необходимой для такой реакции, эта реакция вообще не происходит.
Если энергия, освобождающаяся при захвате частицы атомным ядром, достаточна, чтобы перевести такое вновь образовавшееся ядро в «возбужденное» состояние, т. е, в состояние с повышешшй энергией, из которого ядро должно перейти в нормальное состояние за счет испускания у-кванта, то вероятность захвата частицы в этом случае особенно велика. Это выражается в том, что на кривой зависимости эффективного сечения от энергии частиц появляются резкие максимумы. Их называют резонансными пиками, так как с точки зрения волновой механики это явление можно рассматривать как «резонансный эффект». Особо высокие резонансные ники захвата нейтронов часто наблюдаются при энергии частиц пиже 10 ва. В этом интервале энергии эффективное сечение, там, где лежат резонансные пики, в несколько сотен тысяч pas больше действительной площади сечения атомного ядра.
Для поглоиюния очень медленнных, особенно тепловых, нейтропов эффективное сечение зависит от кристаллической структуры облучаемого вещества. Длина волны?,, ' , h характеризующая в соответствии с основным уравнением волновой механики л = — (см, т. I, гл. 3) тепловой нейтрон, ио величине того же порядка, что и расстояния между центрами атомов в кристалле. Поэтому при прохождении таких нейтронов через кристаллы наблюдаются интерференционные явления и, как следствие, обнаруживаст-пя ослабление потека нейтронов в зависимости от кристаллической структуры. В неко-торых случаях это явление можно использовать для технического испытания материалов. там, где непригодно испытание при помощи рентгеновских лучей.
На рис. 73—76 дани примеры зависимости аффективных сечений ядер от скорости, или кинетической энергии, бомбардирующих частиц. Как видно по верхней кривой рис. 73, в случае лития общее эффективное сечение для поглощения нейтронов закономерно уменьшается с увеличением кинетической энергии или с увеличением •скорости нейтронов. Как показывает наклон кривой, в этом случае эффективное сечение является липоцшяг функцией 1.7», т. е. линейпой функцией времени пребывания нейтрона возле атомпого ядра. Такая зависимость встречается часто, например в случае бора. Но бывает я там, что в некоторой широкой области мало зависит от энергии нейтронов. Примером может служить рис, 74. В отдельных местах кривой, приведенной на рис. 74, имеются резонансные пики, расположенные при довольно высоких значениях энергии. На пижней кривой рис. 73, представляющей зависимость эффективного сечения ядерной реакции 6Li -|- п — аИ 4- а от энергии нейтронов, также наблюдается отчетливый резонансный пик я снова при относительно больших энергиях Е. Другие примеры зависимости эффективного сечения от кинетической энергии нейтронов приведены на рис. 75 и 7G. Кривая рис. 76 показывает изменение эффективного сечения деления ядер 23"Np (ср. стр. 644). Как следует ид этой кривой, относится к тем ядрам, деление которых вызывается только быстрыми, а не медленными нейтронами. Данные табл. 70 показывают, что эффективные сечения могут чрезвычайно сильно различаться в зависимости от ядерной реакции.
606
Глава 13
Выходы при ядерных реакциях. Число N бомбардирующих частиц, поглощаемое в секунду тонким споем вещества, сечением q, равно в соответствии с уравнением (1^
V- ——(j(,V£,<Po.rfr.
Число частиц, поглощаемое в секунду 1 с.и3 вещества, составляет в таком случае •= ojNl®- С ДРУгой стороны, оно было бы равно числу Nr ядер
Рис. 73. Зависимость сечения ядер Li от кинетическом энергии нейтронов.
Е —.кинетическая энергия нейтронов (ее или Мэа); аа— сечение (барн) для ядерной реакции: *Ы 4-1п = 3Н4-'|а (для В, мэв); aj—Общее сечение поглощения нейтронов ядрами лития (для В,. аа).
атомов, образующихся ежесекундно в единице объема благодаря определенной ядерной реакции, если бы бомбардирующие частицы вызывали только эту реакцию. Но из всех частиц только — расходуется в даппон ядерной реакции, если — эффективен
Таблица 70
Сечения различных ядерных реакций • л- : . • • •
Реакция Энергия бомбардирующих частиц Сечение, барн Энергия Сомбардит рующих частиц •Сечение, бйрп :
2Н H-n=sH + y 0,025 se 0,0003 — —
2Ц + ^=8Не + р 0,1 Moe 0,023 1 Мае 0,09
SH + y = iH + n 2,6 » 0,00066 6,2 » 0,0012
6Li + «=®H-|-a 0,025 se 900 - 1 » 0,33
’Li + р= 2a 0,1 Mas 0,00008 1 » ' 0,00009
’Li -|-p=z’Be + n 2 о 0,23 — —
WB-ra=’Li—a 0,025 эв 2500 1 » 0,45
^N-f- >i =14 C -|-p 0,025 » 1,2 2,8 » 0,04
23Nan = з’Ха у 0,025 » 0,41 1000 35 3,33
S’»A14»=S8A1-|-V 0,025 » 0,1 0,5 Мяв 0,0018.
107 Ag 4- n=W8Ag + у 0,025 » 3 1 » 0,09
i57Gd:+«=W8Gd + y 0,025 » 270000 0,2 эв 19 200
Искусственные превращения, атомов
607
ное сечение этой реакции, а ст; — общее эффективное сечение. Выход Л'д атомных ядер, образующихся в этой реакции, будет, таким образом, равен
ЛГв==огйДГьФ. (2)
Следовательно, эффективное сечение сгд является непосредственной мерой выхода ядерной реакции дли данного вещества в зависимости от потока бомбардирующих частиц Ф.
Умножая уравнение (2) на продолжительность облучепия, получаем число ядерных превращений, происшедших за все это время в каждом кубическом сантиметре
Рис, 74. Зависимость общего сечения Of ядер бериллия от кинетической энергии £ нейтронов.
Е, ae; <j(, барн.
вещества, при предположении, что число атомных ядер, способных участвовать в этой реакции, за время облучения существенно нё меняется, В противном случае следует учесть изменение этой величины во времени.
Е, Мзв
Р и с. 75. Зависимость обйдего сечения сг* ядер урапа от кинетической энергии нейтронов.
Р и с. 76. Зависимость сёчепця деления Одед ядер 237 Np от кинетической энергии нейтронов.
Практически особо важным является расчет выхода при получении искусственных радиоэлементов цри помощи ядерных реакций- В этом случае из числа атомных ядер, образовавшихся за период времени dt под действием бомбардирующих частиц, падо вычесть число атомных ядер, исчезнувших за это же время в результате радпо-актинпого распада. Следовательно, скорость образования радиоактивных ядер в каж-
608:
Глава 13
дом кубическом сантиметре вещества равна
dN
, (3)
В атом выражении Л — постоянная распада радиоэлемента, образующегося в результате ядерной реакции с эффективным сечением пи. Интегрируя уравнение (3), получаем
<4)
Здесь Лг( означает количество радиоактивных ядер в 1 е.иЗ вещества, накопившееся за время t, Уравнение (6) имеет ту же форму, что и уравнение (14) па стр. 562, которое описывает накопление радиоактивного продукта распада, образующегося из долгоживущего материнского вещества. ^Максимальное количество радиоактивных атомных ядер, которое может образоваться в 1 с.к3 вещества при его облучении, полу-чем, полагая в у равнении (4) t — со,
ШфФ , (5)
Л
Это количество получится в том случае, если время облучения велико по сравнению с периодом полураспада образующегося радиоэлемента. Уравнение (5) показывает, что главным фактором, определяющим максимальную концентрацию, в которой можно получить радио а л ом онт за счет ядерпой реакции, является поток бомбардирующих частиц Ф. В ядерных реакторах, которые служат для получения плутония, И в которых одновременно получаются искусственные радиоэлементы, поступающие в продажу, величины потоков нейтронов в большинстве случаев равны 10Ji — 1012 нейтрон- см2- сек-1. Количества веществ, которые подвергают такому облучению, составляют 1—10 е. В энергетических ядерпых реакторах ноток нейтронов часто значительно больше (10^— 10*5 нейтрон- см~2-сек-1).
Строение атомных ядер. Из наблюдения, что при распаде естественных радиоэлементов испускаются ядра гелия, или электроны, был сначала сделай вывод, что ядра атомов тяжелых элементов состоят из ядер гелия и электронов. Однако при искусственных ядерных реакциях из атомных ядер испускаются частицы значительно меньше ядер гелия — протоны и нейтроны. Поэтому в настоящее время принимают, что составными частями атомных ядер являются протоны и нейтроны. Как следует из ядерно-физически х исследований, внутри ядер кулоновское отталкивание про-топов преодолевается дополнительными силами притяжения, которые, правда, действуют лишь на очень близких расстояниях. Поэтому гипотеза о существовании внутри ядра отрицательно заряженных частиц (электронов), введенная для объяснения устойчивости атомных ядер, оказывается излишней. Напротив, целый ряд фактов свидетельствуют против этой гипотезы, и поэтому сейчас нейтроны и протоны рассматривают как единственные составные части атомных ядер. Обо эти частицы называют общим именем нуклоны.
Частое испускание электронов при радиоактивном распаде находит объяснение в том, что нейтрон может превратиться в протон п электрон: п = Н+-[- е~- Однако из этого нельзя делать вывода о том, что нейтрон состоит из протона и электрона, так как протон в свою очередь может превратиться в нейтрон и позитрон: Н+— п 4- г+. Поэтому в настоящее время склонны считать, что протоп и нейтрон являются двумя (отличающимися зарядами) состояниями одной и той же частицы- — нуклона. Испускание ядер гелия при радиоактивном распаде ядер тяжелых элементов объясняют особенно большой устойчивостью ядер гелия (см. стр. 610). Вероятно, ядра гелия образуются пе в процессе распада, а предварительно внутри ядер тяжелых элементов.
Данные ядерной физики подтвердили — правда, в существенно иной форме'— выдвинутую в 1815 г. врачом Проутом (Prout) гипотезу о том, что все химические элементы в конечном счете построены из водорода
Ис.кусс-тсеиные превращения, атомов
609
как первоосновы. Эта гипотеза была выдвинута в предположении, что атомные веса веек элементов являются целыми кратными атомного веса водорода. Одпако вскоре точные определения атомных весов показали, что это предположение неверно. В настоящее время известие, чем объясняется отклонение атомных весов от целочисленных значений. Частично это объясняется тем, что большинство элементов представляет собой смесь изотопов. Но и отдельные изотопы и чистые элементы отнюдь не имеют целочисленных атомных весов, если их отнести к атомному весу водорода, принятому за единицу. Это объясняется потерей массы, являющейся в соответствии с «принципом эквивалентности'» следствием громадного выделения энергии нри образовании атомных ядер из протонов й нейтропов.
Согласно прпицппу эквивалентности массы и энергии (Hase no hr], 1904, Einstein, 1905), каждое тело, теряющее энергию, одновременно теряет и часть своей массы равную
(6)
где ДМ — измене нпе массы (г); ДЕ — изменение энергии (эрг); с — скорость света (см/сек). Согласно этому соотношению, изменению энергии в 1 Мэв, или
1,60207- 10~G эрг (см, т, I. стр. 137), соответствует потеря массы в
= 1,78254’ 10~27 г. Если эту величину, найденную для одного атома, пересчитать па 1 г-атом (умножая на Л'л — 6,0230(5-1023), то получим значение 1,07363-10-3 г для изменения массы при изменении энергии в 1 Мае на атом. Обычно изменение массы выражают в единицах ТЕМ, т. е. в тысячных долях единицы массы (см. стр. 585). Поскольку 1 ТЕМ равна 1/1000,275 химической единицы атомного веса, на которой основано определение грамм-атома, то, Чтобы выразить полученную выше величину изменения массы в единицах ТЕМ, ее надо умножить на 1000,275. Отсюда следует, что выделение энергии, равной 1 Мэв, соответствует потере массы 1,073925 ТЕМ, и наоборот, потеря массы в 1 ТЕМ соответствует выделению энергии 0,93116 Мае.
Справедливость принципа* эквивалентности массы и энергии в настоящее время надо считать окончательно экспериментально установленной, так как для целого ряда ядерных реакций можно непосредственно показать, что энергии бомбардирующих частиц и энергии выбиваемых частиц (найденные по величинам их пробега) отличаются на величину, которая соответствует, согласно уравнению (6), изменению массы в этой яДернойfреакции. Исключительно наглядно проявляется потеря массы, связанная с выделением энергии, при реакции позитрона с электроном. При этом нейтрализуются заряды и исчезает масса т каждой частйцы и освобождаются два кванта у-излучения, каждый из которых имеет энергию hv = m-c*’. Можно наблюдать и процесс, обратный только что рассмотренному: образование позитрона и электрона из у-излучения с до- !д*‘ ста годно бояыти частотой *.
Дефект массы и упаковочный множитель. Из принципа эквивалентности следует, что при образовании устойчивого атомного ядра из протонов и нейтронов всегда теряется некоторая масса, так как только при этом’ условии будет выделяться энергия. Разница между массой атомного ядра и суммой масс, входящих в его состав свободных протонов и нейтронов, называется дефектом масса соответствующего атомного ядра.
Дейтрон, который, как следует из приведенной на стр. 603 ядерной реакции, состоящий из протона ц нейтрона, имеет дефект массы, равный 1,007597 + 1,008990 — —2,014196 == 0,002301 единицы массы (см. табл. 69), иля 2,391 ТЕМ. Поскольку ядро *Нез+ гелия имеет примерно вчетверо большую массу, но только вдвое больший заряд,
* Частота «излучения аннигиляции», получающаяся при подстановке в приведенное выше уравнение числовых значений h, т и с, равна v — 1,23-10ао сек-1, а его длина волпы 7. = 0,0244 А. При «образовании парт частота должна быть но крайней мере вдвое больше, так как из 1у-квапта образуются две частицы.
39 I1. речи
610
Глава 13
чем протон, можно принять, что оно состоит из 2 протонов и 2 нейтронов. Следовательно, дефект массы равен: 2(1,007597+1,008990) — 4,002782=0,030392 единицы массы, или 30,392 ТЕМ. Ядро изотопа гелия 3Не, как непосредственно следует из реакции его образования, D+(++3He'-+, построено из 2 протонов и 1 нейтрона. Его масса равна 3,01588, откуда для дефекта массы подучаем значение 8,30 ТЕМ. Аналогично можно рассчитать'дефекты массы дкя .ядер остальных элементов, если известны массы их отдельных изотопов, с достаточной точностью найденные масс-спёктрографи-ческим методом, пли, наоборот, можно найти массы изотопов по дефектам массы, вычисленным из наблюдаемого изменения энергии при атомных превращениях *.
Число протонов и нейтронов, принимающих участие в образовании атомного ядра, в настоящее время для большого числа элементов можно непосредственно найти из наблюдаемых ядерных реакций. При этом была обнаружена очень простая закономерность, позволяющая, находить число нейтронов и протонов и для тех случаев, когда его нельзя получить из непосредствен пых наблюдений. Этот закон гласит: общее число составных частей ядра (протонов и нейтронов) равно массовому числу данного атома. Поскольку электроны но принимают участия в построении атомных ядер, то, кроме того, число протонов в данном атомном ядре равно его заряду (порядковому номеру). Отсюда следует:, число нейтронов в данном атомном ядре равно разнице между его массовым числом и порядковым номером.
То, что между массовым числом атома (его округленным атомным весом)’и общим числом входящих в состав его ядра частиц существует столь простое соотношение, объясняется тем счастливым обстоятельством, что в основу величин атомных весов был положен, по предложению Берцелиуса, атомный вес кислорода 16. Если бы основой служил атомный вес водорода 1, то вследствие дефекта массы но было бы никакой простои связи между округленным значением атомного веса и числом нуклонов, из которых состоит данное ядро. Например, в таком случае висмут, ядро которого построено из 209 нуклонов, вместо атомного веса 209,00 имел бы атомный вес 207,31. Для чистых элементов и отдельных изотопов вообще не получалось бы приблизительно целочисленных значений атомных весов.
Отношение разницы между массой атома М и его массовым числом А к его массовому числу, т. е. выражение (М А)/А, называется упаковочным множителем / данного атома. Если в некоторой системе координат представить зависимость между о массовыми числами и упаковочными множителями, вычисленными по разнице между точно определенными массами и массовыми числами, то получим кривую, при помощи которой можно найти приближенные значения упаковочных множителей для тех атомов, точные значения масс которых еще не известны (Н a h ii О., Вёт., 73 А, 22(1940)]. По уравнению М — А (1 + /) для этйх элементов можно также с довольно большой точностью рассчитать массы атомов.
В табл. 71 приведены упаковочные множители и дефекты массы для отдельных изотопов легких элементов. Массы атомов этих элементов были определены с большой точностью, частично масс-спектрографически, частично по величине энергии, выделяющейся в результате атомных превращений. В этой таблице приведены также значения внергии образования ядер ‘атомов из протонов и нейтронов (млн. йкал/г-атом), ятобы еще яснее показать, сколь велика энергия, освобождающаяся при образовании атомного ядра иа протонов и нейтронов. В ядерной физике дефекты массы обычно выражают в мегаэлектрон-вольтах (Мае). Эти величины можно получить, умножая величину дефекта массы (ТЕМ) на 0,93116 (см. стр. 609). Кроме того, и табл. 71 (в последней колонке) приведены отношения дефектов массы ДМ (ТЕМ) к общему числу нуклонов в ядре А. Эти величины, следовательно, показывают, какая часть дефекта массы, а тем самым и энергии связи приходится в среднем на одлн нуклон. Как видпо, эта величина особенно велика для ядер гелия. ОтсюдЯ можно сделать вывод об исключительно большой устойчивости ядер гелия. Правда, для более тяжелых атомов энергия образования, приходящаяся па каждый нуклон, в большинстве случаев еще выше,
* Вместо масс ядер можно вычислить и массы нейтральных атомов (ядро + + электроны оболочки). Поскольку при каждой реакции, протекающей между двумя атомными ядрами или между атомным ядром и нейтроном, сумма зарядов ядер не изменяется, то при нахождении разности массы электронов оболочки взаимно исключаются. Об изменении массы при радиоактивном распаде см, стр, 617.
Таблица 71
Массы, упаковочные множители н дефекты массы изотопов элементов Н — Zn (У элементов, имеющих несколько изотопов, основной выделен жирным шрифтом)
Порядковый номер Элемент Изотоп goHoduinsH a iron/ Масса Упаковочный множитель f-104 Дефект ТЕМ массы млн. ккал/ г-атом Дефект МаССЫ .ТЕМ
Массовое число
1 Водород *Н 0 1,008146 81,46 — — —
Дейтерий *H(D) 1 2,014744 73,72 2,391 51,37 1,196
Тритий 3Н(Т) ямюйми 2 3,01700 56,67 9,13 196,0 3,042
2 Гелий 3Не 1 3,01698 56,60 8,30 178,3 2,767
4Не 2 4,003880 9,70 30,392 652,70 7,598
3 Литий 61д 3 6,01695 28,25 34,46 740,0 5,743
’Li 4 7,01817 25,96 42,23 906,9 6,032
4 Бериллии *Ве i 5 9,01505 16,72 62;48 1341,9 6,943
5 Бор юВ 5 10,01611 16,11 69,57 1494,1 6,957
14В 6 11,01281 11,65 81,86 1758,0 7,442
6 Углерод 13 с 6 12,003823 3,186 98,992 2126,0 8,249
«С 7 13,00751 5,78 104,29 2239,9 8,023
- 7 Дзот 14N 7 14,007515 5,368 112,436 2414,7 8,031
15N 8 15,004902 3,268 124,038 2663,9 8,269
8 Кислород 160 8 16,000000 0 137,086 2944,1 8,568 ’
17О 9 17,004520 2,659 141,556 3040,1 8,327
18Q 10 18,004875 2,708 150,191 3225,6 8,344
9 Фтор Itp 10 19,004427 2,330 158.785 3410,1 8,357
10 Неон “Ne 10 19,998775 - 0,6125 172,583 3706,4 8,629
2INe 11 21,00045 + 0,214 179,90 3863,5 8,567
22Ne 12 21,99836 - 0*745 190,98 4101,5 8,681
11 Натрий 13Na 12 22,9964 - 1,57 201,1 4319 : 8,74
12 Магний 34Mg 12 23,9928 - 3,00 212,8 4571 8.87
“Mg 13 24,9943 - 2,28 220,3 4732 8,81
36Mg 14 25,9899 -3,88 233,7 5019 8,99 У
13 Алюминий 37 Al 14 26,99011 -3,663 241,65 5190 8,950
14 Кремний isSi 14 27,98580 -5,071 254,10 : 5457 9,075
as Si 15 28,98571 — 4,928 263,18 5652 9,075
3°Si 10 29,98331 - 5,563 274,57 5897 9,152
15 Фосфор 3ip 16 30,98358 — 5,297 282,45 6066 9,111
16 Сёра 33S 16 31,98225 - 5,547 291,92 6269 9,123
33S 17 32,98203 -5,445 301,13 6467 9,125
34g 18 33,97873 -6,256 313,42 6731 9,218
17 Хлор 33 Cl 18 34,98005 — 5,700 320,25 6878 9,150
3’C1 20 36,97767 -6,035 340,61 7315 9,206
. 13 Аргон 33 Ar 18 35,97900 — 5,833 329,44 7075 9,151
33 Ar 20 37,97491 -6,603 351,51 7549 9,250
«Ar 22 39,97514 - 6,215 369,26 7930 9,232
39*
Продолжение табл.
datvott гтзохфьгдоу Элемент Изотоп Число нейтронов Масса Упаковочный множитель f-Ю* Дефект массы Дефект массы „ж>
ТЕМ ккал/ гратом Массовое число
19 Калий «К 20 38,97606 — 6,138 35831 7699 9,193
«К 21 39,97654 —5,865 367,01 7882 9,176
«К 22 40,97490 — 6,122 377,65 8111 9,211
20 Кальций *°Са 20 39,97529 —6,177 367,43 7891 9,186
«Са 22 41,97216 —6,629 388,54 8344 9,251
«Са 23 42,97251 — 6,393 397,18 8530 9,237
«Са 24 43,96924 — 6,991 409,44 8793 9,305
«Са 26 (45,9685) (—6,85) (428,2) (9195) (9,308
«Са 28 47,96778 —6,712 446,86 9597 9,309
21 Скандий «$б 24 44,97010 —6,644 416,72 8950 9,260
22 Титан «Ti 24 45,96697 — 7,180 428,00 9192 9,304
«Ti 25 46,96668 — 7,089 437,28 9381 9,304
4вТ1 26 47,96317 — 7,673 449,78 9660 9,370
49Ti 27 48,96358 — 7,433 458.36 9844 9,354
S0Ti 28 49,96077 — 7,846 470,16 10097 9,403
2S Ванадий soy 27 49,9633 —7,34 466,8 10025 9336
siy 28 50,96052 — 7,741 478,55 10278 9,383
24 Хром 50Cr 26 49,96210 — 7,580 467,14 10032 9,343
5aCr 28 51,95707 — 8,256 490,15 10527 9,426
5SCr 29 52,95772 — 7,977 498,49 10706 9,405
S4Cr 30 53,9563 — 8,09 508,9 10929 9,424
2& Марганец 5SMn 30 54,95581 — 8,035 517,53 11115 9,410
26 Железа MFe 28 53,95704 —Л,956 506,47 10877 9,379
5Те 30 55,95272 — 8,443 528,77 11356 9,442 ...
57Fe 31 56,95359 — 8,142 536,89 11530 9,419
5®Fe 32 57,9520 — 8,28 547,5 11758 9,429
27 Кобальт B9Co 32 58,9510 — 8,31 556,6 11954 9,434
28 Никель 5®Ni 30 57,95345 — 8,026 544,33 11690 9,385
60Ni 32 59,94901 — 8,498 566,75 12172 9,446
61Ni 33 60,94907 — 8,349 575,68 12364 9,437
6=Ni 34 61,94681 —8,579 586,93 12605 9,467
64Ni 36 63,94755 — 8,195 604,17 12975 9,440
29 Медь «Си 34 62,94929 — 8,054 592,63 12727 9,407
«Си 36 64,94835 — 7,946 611,52 13133 9,408
30 Цинк 64Zn 34 63,949 55 — 7,883 600,48" 12896 9,383
«Zn 36 65,94722 — 7,997 620,79 13332 9,406
«Zn 37 66,94815 — 7,739 628,85 13505 9,386
e8Zn 38 67,94686 — 7,815 639,13 13726 9,399
’«Zn 40 69,94779 — 7,459 656,18 14092 9,374
Искусственные превращения атомов
613
ио если бы это было не так, то эти ядра вообще не были бы устойчивыми. Например, ядра 12С, 1в0 и 20Ne, количества протонов и нейтронов в которых кратны их количеств у в ядрах гелия, в случае нх образования в каком-либо процессе самопроизвольно распадались бы на ядра гелия, если бы энергия образования, приходящаяся в этих ядрах на каждый нуклон, была бы меньше, чем для ядра гелия. На рис. 77 представлена зависимость величины отношения ДЛ1/Л от массовых чисел атомов А. Как видно, кривая, имеющая резкий максимум для 4Не и еще два других значительно менее резких максимума для 1аС и i -О, постепенно повышается к железу и никелю, а затем медленно падает. То, что элементы, атомные веса которых больше атомных весов железа и никеля,-устойчивы, несмотря на то что приходящийся на один нуклон дефект массы у них все более и более уменьшается, объясняется тем, что атомные ядра этих элементов содержат псе возрастающий избыток нейтронов по сравнению с протонами. У этих элементов уменьшение величины ДлИ/Л меньше, чем выигрыш энергии за счет присоединения избыточных нейтронов. Кальций является последним элементом, у которого существует изотоп (S[JCa), состоящий из равного числа протонов н нейтронов. Если же число протонов в ядре больше 20, то устойчивыми являются только те изотопы, которые имеют избыток нейтронов, причем этот избыток должен становиться тем больше, чем больше число протонов. Это служит причиной того, что величины, половин атомных весов всех элементов, начиная с кальция, возрастают в среднем быстрее их порядковых номеров (см. т. I, стр. 261).
Как видно из рис. 77, у элементов, которые тяжелее углерода, дефект массы, приходящийся на каждый протон и нейтрон, колеблется всего лишь в пределах 8,03—9,47 ТЕМ. В ядре ЮО на каждый нуклон приходится Дефект массы 8,57 ТЕМ, Следовательно, отдельные нуклоны в ядрах элементов, следующих за углеродом (по расчету), не более чем: на j),054% легче иля не более чём на 0,090% тяжелее отдельного нуклона в ядре изотопа кислорода Ы'О (1/161вО). Этим объясняется то, что 'массы отдельных изотопов так же чрезвычайно мало отличаются от целых чисел, т. е. от массовых чисел (ведь если бы протоны и нейтроны во всех ядрах имели бы такую же массу, как в ядре 1ВО, то массы атомов были бы равны массовым чиелд.и). Напротив, по отношению к массам, которые имеют протон и нейтрон в свободном состоянии, их массы в ядрах названных элементов на 0,80—0,95% меньше.
Массы ядер продуктов радиоактивного распада. На основании принципа эквивалентности из измерений энергий распада можно с большой точностью вычислить массы ядер всех членов радиоактивного ряда, если известна масса только одного члена ряда. В табл. 72 указаны полученные таким способом массы ядер для элементов главнейших встречающихся в природе радиоактивных рядов. Различия между приведенными в таблице дефектами массы для различных изотопов * содержат максимальную ошибку в несколько сотых мегаэлектрон-вольта, или ТЕМ. Значения отдельных дефектов массы можно найти с несколько большей ошибкой, так как пределы ошибки зависят от точности, с которой масс-спектрографически определены массы изотопов, приведенных в примечании *. Для членов радиоактивных рядов тория и урана—радия ..ошибка в значениях дефектов массы, приведенных в табл. 72, вероятно, не превышает нескольких десятых ТЕМ, так что значения их масс вычислены с точностью не менее нескольких тысячных процента **. Значения масс М найдены из значений дефектов массы ДМ (в единицах ТЕМ) по уравнению
Jlf=Z. 1,0081459+U—2)1,0089899—-^-, (7)
* Величины различий дефектов масс {Мое) взяты из обзорной работы Пфлюгге и Маттауха (Pfliigge, Mattaucb, Phys. Zejtschr., 44, 181 (1943). Все значения дефектов массы отличаются от приведенных в этой работе на величину’ 13,85 Мэв, которую надо прибавить, чтобы привести в соответствие дефекты массы с новыми результатами определения масс Jre, ]Н, 2||Pb, 2^JTh, -jjiU и (Bainbridge, 1951; Fowler, 1951; Nier, 1951; Stanford, 1951; Dackworlh, 1952; Geiger, 1952; Richards, 1952; Ogata, 1953; Mattauch, 1954),
*’ В пределах этих ошибок они согласуются с величинами, недавно вычисленными Снборгом [J. Inorg. Nucl. Chern., 1, 3 (1955)|.
Рис. 77. Зависимость дефекта массы, приходящегося в среднем на каждый пук л он в ядре, от массового числа*
ч
Таблица 72
Массы, упаковочные множители и дефекты массы естественных радиоактивных изотопов .
§1 й! I Изотоп Массовое ^Число нейтронов Масса Упаковав нъш jvfHoafcU- J Мзв Дефект массы Дефект массы ’ТЕМ Массовое число
МЛН. ккал/ з-атом ТЕМ
81 АсС" 207 126 207,039 1,894 1632,68 37656 1753,38 8,4704
ThC" 208 127 208,045 2,159 1635,71 37725 1756,63 8,4453
RaC" 210 129 210,055 2,600 1643,45 37904 1764,94 8,4045
82 RaG 206 124 206,035 1,704 1627,27 37531 1747,57 8,4833
AcD 207 125 207,037 1,773 1634,15 37690 1754,96 8,4781
ThD 208 J 26 208,039 1,865 1640,64 37 839 1761,93 8,4708
RaD 210 128 210,048 2,286 1648,83 38028 1770,72 8,4320
AcB 21] 129 211,053 2,502 1652,72 38118 1774,90 8,4118
ThB 212 130 212,057 2,693 1657,08 38218 1779,58 8,3943
RaB 214 132 214,066 3,107 1665,05 38402 1788,14 8,3558 . < • .
83 RaE 210 127 210,047 2,238 1648,90 38030 1770,80 8,4324
AcC 211 128 211,050 2,389 1654,13 38150 1776,41 8,4190
ThC 212 129 212,056 2,623 1657,70 38233 1780,25 8,3974
RaC 214 131 214,075 3,486 1656,73 38210 1779,21 8,3141
84 RaP(Po) 210 126 210,045 2,138 1650,07 38057 1772,05 8,4384
AcC' 211 127 211,049 2,318 1654,75 38165 1777,08 8,4222
ThC' 212 128 212,052 2,472 1659,89 38283 1782,60 8,4085
RaC' 214 130 214,060 2,818 1669,£0 38498 - 1792,60 8,3766
AcA 215 131 215,065 3,014 1673,42 38595 1797,13 8,3587
ThA 216 132 216,068 3,171 1678,30 38710 1802,46 8,3447
RaA 218 134 218,077 3,537 1687,12 38911 1811,84 8,3112
86 AcEm 21» 133 219,076 3,479 1694,68 39085 1819,96 8,3103
ThEm 220 .134 220,079 3,605 1700,19 39213 1825,88 8,2994
RaEm(Rn) 222 136 222,087 3,919 1709,74 39433 1836,14 8,2709
87 AcK(Fr) 223 136 223,089 3,978 1715,76 39572 1842,60 8,2628
88 AcX 223 135 223,088 3,928 1716,05 39578 1842,91 8,2642
ThX 224 136 224,090 3,996 1722,60 39730 1849,95 8,2587
Ra 226 138 226,096 4,243 1733,36 39978 1861,50 8,2367
MsThi 228 140 228,103 4,509 1743,68 40216; 1872,59 8,2131
89 Ac 227 138 227,098 4,326 1738,86 40104 1867,41 8,2265
xVsTh2 228 139 228,102 4,474 1743,72 40217 1872,63 8,2133
90 RdAc 227 137 227,097 4,278 1739;09 40110 1867,66 8,2276 ;
RdTh 228 138 228,099 4,360 1745,28 40252 1874,30 8,2206
Io 230 140 230,105 4,570 1756,79 40518. 1886,66 8,2029
UY 231 141 231,109 4,706 1761,76 40633 1892,00 8,1905
Th 232 142 232,111 4,802 1767,59 4076? 1898,26 8,1822
UX! 234 144 234,119 5,068 1777,66 40999 1909,08 8,1584
91 Pa 231 140 231,108* 4,662 1761,92 40636 1892,17 8,1912
UXt 234 143 234,118 5,021 1777,86 41004 1909,29 8,1594
92 UII 234 142 234,114 4,880 1780,20 41058 1911,80 8,1701
AcU 235 143 235,118 5,004 1785,38 41177 1917,37 8,1590
U1 | 238 146 [ 238,127 | 5,340 | 1801,65 | 41553 1934,84 8,1296
616
Глава 13
где Z — заряд ядра; А — его массовое число; 1,0081459— масса атома ТН; 1,0089899 — масса нейтрона. И наоборот, это уравнение можно использовать для нахождения дефектов массы для тех изотопов, массы которых измерены непосредственно масс-сшзктрографичсски. Эти величины являются основой для расчета остальных дефектов массы при помощи разниц в их величинах, находимых из энергий распада.
ИСКУССТВЕННАЯ РАДИОАКТИВНОСТЬ
В 1934 г. Ирэн Кюри и Фредерик Жолио, исследуя позитронное излучение, возникающее при бомбардировке алюминия а-частицами, нашли, что это излучение но прекращается тотчас же после удаления испускающего а-частицы препарата (полония), а исчезает постепенно и притом по закону радиоактивного распада [см. уравнение (4), стр; 550]. Предположение о том, что в этом случае речь идет об искусственно вызванной радиоактивности, было вскоре подтверждено дальнейшими наблюдениями. Оказалось, что активирование алюминия поддействием а-частиц объясняется тем, что сначала из алюминия образуется неустойчивый изотоп фосфора, который затем самопроизвольно с периодом полураспада 3,2 мин превращается, испуская позитроны, в устойчивый изотоп кремния
3,2 мин
^А1-На=!?Р-ЬК ^Р—->^Si + e+ (8>
Сейчас известно уже очень большое число искусственно полученных радиоактивных изотопов. И лишь для нескольких элементов еще не наблюдалось «активирования», т. е. превращения в неустойчивые и поэтому самопроизвольно распадающиеся изотопы,
Виды радиоактивного распада. Большинство искусственно полученных радиоактивных изотопов превращается в устойчивые ядра с испусканием электронов или позитронов. Оба этих типа превращений называют р-распадом.
Те ядра, неустойчивость которых вызвана слишком высоким содержанием нейтронов по сравнению с содержанием протонов, испускают электроны (р “-распад). Те ядра, неустойчивость которых вызвана слишком высоким содержанием протонов по сравнению с Содержанием нейтронов, испускают позитроны ф+-распад). Первые стабилизируются "за счот превращения нейтрона, испускающего электрон, в протон. II наоборот, те ядра, которые неустойчивы вследствие слишком высокого содержания протонов, могут переходить в устойчивые ядра благодаря превращению протона, с испусканием позитрона, в нейтрон. Это может происходить и по-другому, а именно за счет захвата ядром электрона из электронной оболочки атома. Этот электрон реагирует с протоном и образуется нейтрон. Освобождающаяся при этом энергия испускается * в виде нейтрино (см. стр. 557). В этих случаях говорят об электронном захвате, или К-захвате (поскольку обычпо захватываемый ядром электрон относится к ближайшей А'-оболочко (см. т. I, стр. 257).
* При этом иногда возникает вторичное у-излучение, как эта часто наблюдалось и для а- и p-р аспидов (ср. стр. 548). Кроме того, за атомным превращением за счет электронного захвата обычно следует испускание характеристического рентгеновского излучения элемента, образующегося в результате атомного превращения, поскольку А'-о бол очка пополняется электроном из более удаленной оболочки (gm. pzc. 55 нэ стр. 258 т. I).
Искусственные превращения атомов
617
Так же как и испускаемые ядром электроны, испускаемые позитроны могут иметь все возможные скорости. Энергии -распада аналогично энергии ^“-распада определяется максимальной кинетической энергией испускаемых частиц. Следовательно, надо принять, что разница между максимальной энергией и меньшими энергиями позитронов испускается в виде пейтрино.
Радиоактивные превращения за счет испускания позитронов наблюдаются только у искусственно полученных радиоактивных ядер. Превращения за счет электронного захвата также наблюдаются и у встречающихся в природе ядер, но только в исключительных случаях (см. стр. 576). Как правило, радиоактивные превращения атомов природных элементов происходят с испусканием электронов или а-частиц. Превращения с испусканием электронов особенно часто наблюдаются для искусственно полученных радиоактивных ядер. И, напротив, превращения, связанные с испусканием а-частиц, наблюдаются только у тех искусственных ядер, которые идентичны ядрам, входящим в природные радиоактивные ряды или подобно им имеют исключительно большие массы. Это относится прежде всего к трансурановым элементам. Большинство изотопов этих элементов распадается с испусканием а-частиц (см. табл. 86—89).
Если при радиоактивном распаде, сопровождающемся испусканием электрона, порядковый номер элемента увеличивается на 1, то при испускании позитрона или захвате электропа порядковый номер элемента уменьшается па 1. Массовое число во всех этих случаях не изменяется.
Примеры:
й,8 егк 72 Сек
«Не-----> SLi-f-e-; -------> ^О-Н+. (9)
53 д.11
Электронный захват: jBe -f- е~->gLi 4~ v (v — нейтрино) (9а)
Над стрелками указаны значения периодов полураспада расположенных слева неустойчивых ядер. Период полураспада, так же как и в случае естественных неустойчивых ядер, является характеристическим свойством ядра. Следовательно, он не зависит от внешних условии и от процесса, в котором образовалось неустойчивое ядро.
Прн радиоактивных превращениях масса уменьшается на величину, эквивалентную выделяющейся янцргшг Если это изменение не учитывать, то при испускании электрона масса нейтрального атома не изменяется, так как вследствие увеличения заряда ядра электронная оболочка присоединяет еще один электрон. Масса остается неизменной, если не учитывать ее изменения; связанного с испусканием энергии, и при превращениях за счет электронного захвата из электронной оболочки. Напротив, при испускании позитрона масса нейтрального атома уменьшается на две массы электрона, а имеппо на массу позитрона и еще массу электрона, который теряется, электронной оболочкой вследствие уменьшения заряда ядра.
Для получения радиоактивных изотопов используют те же методы, что и дия проведения других искусственных превращений атомов. Но особенно часто для этой цели используют нейтроны. В лаборатории * нейтроны получают обычно при действия излучения радия иа бериллий пли при действии искусственно ускоренных дейтронов на дейтерий (например, в форме льда D20): JBe -f- fa = + Ja; 4~ = ’He 4- Ja.
Так как во многих случаях медленные нейтроны более эффективны, чем быстрые (вследствие большого времени пребывания вблизи ядер), их в большинстве случаев тормо-вят при помощи парафина или воды. Но для многих процессов [например, (л, 2п} и (л, Зл)[ используют особенно сильно ускоренные нейтроны, которые получают, например, бомбардировкой лития быстрыми дейтронами; ’Li(rf,a)sBe.
В табл. 73 приведен ряд искусственно полученных неустойчивых изотопов. В последней колонке указаны устойчивые ядра, из которых можно получить при помощи описанных выше “процессов эти радиоактивные ядра. В таблице приведена только небольшая часть многочисленных
* Техническими источниками нейтронов являются ядерные реакторы. Об этом см. стр. 647.
Таблица 73
Сводная таблица важнейших неустойчивых изотопов
Порядковый номер Элемент Кеустой : чиоый. изотоп Распад Период полураспада Продукт распада.. Способы получения неустойчивых изотопов
1 Водород *Н Р~ 12,46 лет *Не »H(d,p);»He(nfp);
(Тритий) |Li(n,«)
4. бериллии ’Be К 43 дн : ’Li (р, n); *Li(d,nj; ^В(р,а)
6 Углерод j;c Г 20,35 мин 1JB A°B(p,y);\°B(d,n); ’gC (n, 2n); lJN (p,, a) ,
Углерод Г 5,6-103лет “N “B(a, p); l|C(n,y);
•»C.(d,p); “N(n-P); ‘£0(11, a)
7 Дзот ’’К Г 10,13 мин ^B(a,n); »C(p,n);
^(d.n); ,3C(p,7); "*N (n, 2n); '„‘N (y, n)
8 Кислород ”0 Г 2,2 мин “N. *?N (p,y); ИЛ (d,n); "0(n,2n); «О(у,:п)
9 Фтор мр 0+ 1,92 час \3О «0 (p, n) ; “F(n,'2n)i; «F ty.n); “Ne (d,a)
11 Натрий Г 15,1 час “Na (d,p); “Na(n,y);
“Mg(d, a); «Mg (n, p); ^Al(n,a)
т Магний “Mg j3- 9,6 мин ^Mg(d,p); “Mg(B,X}; isAl(n, p)
13 Алюминии 5А1 г 2,30 мин “Si «Mg(a,p):“Al(d,p); ^Al (n,}*); ^Si(n,p);
14 Кремний № j3- 2,62 час sip 15* uSi(d, p); ,“Si(n,y); аДР(п, p); «S(n, a)
15 Фосфор ззр 15* 0- 14,07 дн T.s ”Si(a,p); s’P(d,p); “S(n,pJ; «S(d,a); «Cl(n,a)
16 Сера fi- 87,1 дн nC1 «S(d,p); £01 (n,p); j;ci(d,a)
17 Хлор 0- 38,5 мин ?’Cl(d, p); %С1(П,УГ;
19 Калии 0- 12,4 час ЙСа tJK(d, p) ; «K(n,y); £Са (n, p); JJSc (n,a)
20 Кальций so^a p- 152 дн «Sc P); “Св (n, 7); "Sc (n, p)
Кальций «с» 30 мин 2,5 час я* «Са(n,7); £JCe(d,p)
Цродолжение табл. 73
ПоряЬ-ковый номер Элемент Hetjcnjoib чивый изотоп Распад Период полураспада Продукт распада Способы получения ‘ неустойчивых изотопов
21 Скандии 86 дн £Ti ^Са(а, р); “Sc (d, р); “Sc (п, у)
22 Титан «Ti Г 72 дн 51U 23 т «Ti (d, p); “Ti(n,7) f
23 Ванадий 4Ву аа т л* ЧЗ+ 16,0 дн 16,0 дн «Ti g® «Sb(a,n);«Ti(d, n); «Ti(p, nh «Cr(d,(x) !
Ванадии 52 V 23 ’ г 3,9 мин яСг 24 V1 '•XV(d,p);.5y.(4,y); |^Cr (n, p); “Mn (n, a)
24 Храм 49Пг у 41,9 мин 49V 23 ’ “Ti(a, n); «Cr (n, 2n); “Or (y, n)
25 Марганец «Мп Г 2,59 часа ••Ее oK'r (a, p); «Мп (n, y); |fEe(n,p);«Fe(d,«); f"Co(n,a) •*
26 Железо *9Fe 26Г с р- 46,3 дн ЙСо «Ее (d, p); «Ee(n, y); «Co (n, p)
27 Кобальт “Со 18,0 час «Ее *JEe (p, 7); ^Ee(d,n)
Кобальт «Со р- 6,26 лет «Ni «Со(п,7);«Со^Р); “Ni(n,p); «Ni(d,a)
28 Никель «Ni Г 2,6 час “Ou $Ni(d, p); «Ni(n,y); «Cu(n, p);«Zn(n,i)
29 Медь gCu £+ 10,1 мая «Ni «Co (a, n); °“Ni(p, n); “Си (n, 2й); «Си (p, y)
Медь «Си ук .V 12,88 час 12.88 час 12,88 час «Ni !zn «Ni (p,n); «Ctt(n,y); 2»Cu (d, p); SJOu (n, 2n); •;Cu (y,’n); fjZn(n, p)
30 Цинк г 38,3 мин “Си «Ni(a,n); «Cu(p,n); ’ «Cu(d,2n);£Zn(n,2n); й2п(У«“)
33 Мышьяк г^' \р+ 17,5 дн 17,5 дн wSe 34cw HGa («, n); ”Ga (d, n); ”Ge(p,n); |«As(n, 2n); "As (7. п); (Ф«)"
Мышьяк "Ав ?- 1,187 дн "Ge (p, п); ."Ая (n,/); £fe(^p);£Se(d.«): "Br(n, aj
34 Селен 3^ к 9,5 дн ”Ав ”Ae(d,5n)
Селен £Se 7к 7,1 час 7,1 час 73 Ди. 33“ НА» "Ge (a, n); »Ae (d, 4n)
35 Вром z^' >*/i+ 18,5 мин 18,5 мин ГвКг «Зе 2Se(p,n); "Br(d, p); «Вг(п,у);«Вг(п,2п); HBr (7, n)
Продолжение табл. 73
Порядковый saxtsp- Элемент Неустойчивый изотоп Распад Период полураспада Продукт распада Способы получения неустойчивых изотопов
35 Бром (’Вг So fi- 1,63 дн fJSe(p,n); «JSe(d,2n); «Вг (d, р); «Вг(п, у); £КЬ(п,а)
38 Стронций «Sr fl- 54,5 дн “Sr(d.p); g8r(n,y), 5Y(n,p)
47 Серебро *«Ag Р,К '*fl*‘ 8,2 дн 24,3 мин ^Pd i«Pd «•Rb(a,n);«:Pd(p>n), ‘«Pd(d,n); ™Ag(n,2nh «Ag (y, n); ‘«Cd (n, p)
48 Кадмий ‘«Cd /А~ <*fl- 2,4 дн 43 дн ‘‘<Cd(d, p) ;l}*Cd(n,y), «In (n, p)
50 Олово ’SB" 1,25 дн «)8Ъ ‘£Sn(d,p),«[Sii(n,n '“Sb (d, a)
Олово fl~ 2,05 дн ‘IJSb ^Te(n,aj
51 Сурьма ^Sb fl- 60 дн '"Те ‘«Sb (d, p);*«Sb(n,y), ™Te(d,a); «1(п,а).
52 Теллур i«Te 1 г 9,3 час 90 дн 137 Т 1Я?Т 63 * ^e(d,p);^(n,y), ^J(«.P)
53 Иод шт S31 fl' 13,3 дн *«Хе Y?Sb(a, n), ‘JjTe (d,n);. ^е(р,п)ЛП(п,2н)
Иод Шт 55 1 fl- 24,99 мин № ^(р, И); >”1(И, У)
56 Барии № fl- 1,40 час ‘«Ba(d, p);‘“Ba(n,y) «La (n, p); ‘JJCe (n, a)
57 Лантан *"La fl- 1,67 дн ^Се *“L&(d,p); 7’Ьа(п,у); дач^р)
78 Платина fl 29 мин ^Ап 1SPb(n,-7);1«pb-(d, p), ^Hg(n, a)
79 Золото »«Ап fl- 2,66 дн № *«Pt(d, 2n);^Au(d, pj *"Au(n, y); ‘«Hg (n, p)
80 Ртуть № fl- 43,5 дн “Hg (n,y);«Tl(n, P)
Ртуть № fl- 5,5 мин «SHg (h, y);»JTl (n, p}„ «JPb (n, a)
неустойчивых изотопов, которые получены к настоящему времени. Например, можно получить по крайней мере семь различных неустойчивых изотонов меди (с периодами полураспада от 80 сек до 140 дней) и по крайней мере восемь неустойчивых изотопов цинка (с периодами полураспада от 35 мин до 7 мес). В таблице указаны преимущественно те изотопы, которые вследствие их способов получения и периодов полураспада особенно пригодны для использования в качестве индикаторов при химических исследованиях (см. стр. 626).
Искусственные превращения атамов
621
Как л в естественных радиоактивных рядах, для многих искусственных радиоактивных ядер наблюдается деоякий распад. В табл. 73 приведены три примера: 64Cu % т-i As и 8ОВ г.
Кроме двоякого распада, который с равными периодами полураспада ** приводит к двум различным продуктам распада, в некоторых случаях, например 49Са, 1(wAg; Ш,С<1 и *|JTe(CM. табл. 73), наблюдается образование одного и того оке продукта распада с двумя различными периодами полураспада. Ядра С одинаковыми массовыми числами и одинаковыми порядковыми номерами, которые с различными периодами полураспада переходят в один и тот же продукт распада, например
/
<‘н яиц 2,5 час
-----• и «Са--------> ^Sc.4-e~, - (10)
называют изомерами. Следовательно, в таких случаях говорят о ядерной изомерии.
Если продукт распада неустойчивого ядра также неустойчив и он снова распадается; то говорят о рядах распада, например:
27 мин 2Т5 час
^е’------> ^Вг-Н-; ОБг-------> 5?Кг+е-. (11)
Ряды распада более чем с двумя членами известны для искусственных изотопов только в исключительных случаях. Это прежде всего многочленные ряды распада, начинающиеся с изотопов многих трансурановых элементов (см. г.ч. 14). Это связано с тем, что для этих наиболее тяжелых элементов, кроме p-распада, наблюдается и а-расиад, точно так же как и у естественных радиоактивных элементов урана I, актиноурана и тория. Ряды распада с большим числом членов часто наблюдаются и для элементов средней массы, которые образуются в результате деления наиболее тяжелых ядер под действием нейтронов.
Зеркальные ядра.<Среди излучателей позитронов интересную группу образуют те ядра, число протонов в которых на 1 больше числа нейтронов. При испускании позитрона они превращаются в ядра, число нейтронов в которых па 1 больше числа протонов. Если ядро, содержащее р протонов и п нейтронов, обозначить как (р -}-• я)-ядро, то, например, (6+5)-ядро превращается в (5-|-б)-ядро, (7-}-6)-ядро в (6-)-7)-ядро и т. д. Поэтому эту группу излучателей позитронов называют ^зеркальными ядрами». Известен непрерывный ряд зеркальных ядер с зарядами от 6 до 22. В этом ряду периоды полураспада регулярно уменьшаются. Энергии распада в том я;е направлении возрастают, причем так, что существует примерно линейная связь между логарифмами постоянных распада и логарифмами энергий распада. Следовательно, зеркальные ядра (с известным разбросом) лежат па кривой Саржента, и именно на той, которая является приблизительно прямолинейным продолжением верхней из двух кривых, па которых располагаются естественные радиоэлементы (рис. 78 и 79).
Скалыпапнс ядер. В общем' случае под действием бомбардирующих частиц из атомных ядер выбивается непосредственно лишь одна или две (в редких случаях три) частицы (нейтрон, протон, дейтрон или а-частица). Однако под действием очень быст-
* В случае ”4Сп речь идет о двояком распаде потому, что образуются два различных продукта распада (fl«Ni и e4Zn). То, что один из них образуется двумя различными путями (за счет электронного захвата или благодаря испусканию позитрона), не имеет значения. По другому обстоит дело в случае 1<)eAg, где два превращения, приводящие к одному И тому же продукту распада, имеют различные периоды полураспада. Следовательно, у K^Ag наблюдается случай ядерной изомерии.
** То, что двоякий распад пе может характеризоваться различными периодами полураспада, или постоянными распада, следует непосредственно из определения постоянной^распада А, которая в силу уравнения (11) (стр. 561) характеризует скорость уменьшения количества активного вещества за время dt. Это определение не содержит Оказаний па то, происходит ли такое уменьшение благодаря одному радиоактивному процессу или за счет нескольких таких процессов, протекающих параллельно.
622
Глава 13
рых частиц можно наблюдать такие процессы, при которых из атомного ядра вывевается значительно больше частиц, из которых оно построено, В таких случаях говорят
Р и с. 78, Кривые Саржента для ядер естественных радиоэлементов.
Р и с. 79, Кривая Саржента дне зеркальных ядер.
• зеркальные ядра; о естественные радиоэлементы.
о процессах скалывания ядра- Сейчас известно много примеров таких процессов скалывания. Например, при облучении дейтронами с энергией 200 Mae ’i>As непосредственно переходит в ^Мн, а=^и при облучении а-частицами с энергией 380 Мяв переходит ] непосредственно в 4JW; 1
заАзЦ-|с? —^Мп-|-9}р-}- 12^р, 1
s$U-Ha=J&W-}-201p+35K :
Следовательно, очень быстрая a-частица способна при одном столкновении с яд рек , урана отколоть рт него в общей сложности 55 нуклонов (20 протонов и 35 нейтронов" j В краткой записи эти уравнения выглядят так; 1
«Аа (d, 9р 12н) 5вМп и S3S.U (a, 20р 35n) i8?w; ]
1 Другими примерами реакции скалывания могут служить;
31Р (d, 9р 7n) ”N; 32S (d, 10р 7n) i’N; 76As (d, 10p 16») 51Cr; j
TOGe (d, ,р5гг) «3Ge; 121 Sb (d, 6p 16») WPd; S38U (a, 6p 12n) 224Ra. ,
Часто несколько реакций скалывания идут параллельно. При облучении ^As дейтронами с энергией 190 Мее наблюдается образование не мёнее чем 38 различных ядер. За исключением немногих, образующихся в результате обычных ядерных реакций^ все остальные ядра образуются в результате реакций скалывания.
Изомерия ядер. Впервые изомерия ядер наблюдалась Ханом в 1921 г. на примере входящих в радиоактивный ряд урана—радия урана Х2 п урана Z, Долгое время эти ядра были единственными известными ядрами-изомерами. Только после того как удалось искусственно получить радиоактивные изотопы, были обнаружены другие и притом; многочисленные случаи ядерной изомерии.
Явление ядерной изомерий основано на том, что атомные ядра могут существовать в различных энергетических состояниях,. так что для них, кроме нормальных
Искусственные превращения атомов:
623
состояний, можно наблюдать и «возбужденные» состояния. Обычно атомное ядро переходит из возбужденного состояния в нормальное мгновенно (вероятно, менее чем за 10"iS ceK;;). При этом разница в Энергиях обоих состоянии пспускаетояв виде ^излучения (см. стр. 548); Одпако среди переходов между энергетическими состояниями ядер, так же как и между энергетическими состояниями внешних оболочек атома, существуют так называемые «запрещенные» переходы, т, е, такие переходы, которые очень мало вероятны и поэтому происходят очень редио или вообще не происходят. Возбужденно е^состояние, переход из которого в нормальное (основное) состояние очень мало
а
Неустойчивое состояние
6 .
Неустойчивое состояние
в
Неустойчивое состояние
Материнское ядро
Т, Основное состояние
Материнское ядро
Т
Основное состояние
Основное состояние
(устойчивое)
J' Продукт распада
Продукт распада
Рис. 80. Три вида радиоактивных превращений изомерных атомных ядер. а — независимый распад; 6 — р.чслал после иаоясрлого переход а; в — изомерный переход атомного ядра^из метастабильного состояния в основное состояние для устойчивого элемента.
Г — период полураспада.
вероятен, называют метастабильным состоянием. Изомерные атомные ядра представляют собой ядра с равными массами и порядковыми номерами, одно из которых находится е нормальном, а другое в метастабильном (возбужденном) состоянии ,*/ Если распадается ядро,: находящееся в метастабильном возбужденном состоянии, например испуская р-частицу, то при этом образуется тот ясе продукт, что и при распаде атомного ядра, находящегося в основном состоянии, но;распад метастабильного ядра характеризуется иным периодом полураспада. Такой вид распада приведен на рис. 80а. В этих случаях говорят о ядерной изомерии с независимым распадом, В отличие от другого типа распада (рис. 806). при котором продукт распада образуется не непосредственно из метастабильного возбужденного ядра, а таким образом, что это ядро сначала с периодом полураспада Tt переходит, йену ска я у-кв ант, в основное состояние, < а из последнего с периодом полурасцада Т2 (например, с испусканием р-частиц) образуется продукт распада. Период полураспада Tj тем больше, чем менее вероятен переход материнского ядра из метастабильного возбужденного состояния в основное состояние. Такой переход называют изомерным переходом, а изомерные атомные ядра, находящиеся в возбужденном состоянии, обозначают звездочкой, стоящей справа рядом
Y
с химическим символом ядра. Например, запись 44Sc*---——> 44Sc означает, чтовозбу-
2,44 дн
жденное метастабильное ядро изотопа скандия с массовым, числом 44 переходит с периодом полураспада 2,44 дня в основное состояние. Изотоп скандия 448с неустойчив и в основном состоянии и распадается с периодом полураспада З,92 часа, В результате сгО' |3+
распада испускается позитрон и образуется изотоп кальция: ;)JSc------ад 44Са, Следо-
3,92 час
вательно, 'если распад нормального ядра 44Sc происходит С периодом полураспада 3,92 часа, то для, периода полураспада изомерного ядра м8с* получается значение 2,44 дня, так как в этом случае скоростью, определяющей радиоактивный распад, будет скорость образования нормальных ядер 44 Sc из ядер 44ScВ этом отношении положе
* Известны случаи, когда, кроме нормального, существует два мета стабильных состояния. В этих случаях существует три изомерных ядра.
624
Глава 13
ние совершенно аналогично наблюдаемым скоростям при последовательных химических реакциях, где также скорость всего процесса определяется наиболее медленной стадией (см. стр, 738), Если изомерный переход происходит быстрее, чем распад с испусканием частицы, то скорость будет определяться скоростью последнего процесса. В качестве примера такого распада можно привести радиоактивный изотоп кобальта е'А2о, мета стабильное ядро которого с'>Со* довольно быстро, испуская у-квант, переходит в основное состояние, тогда как образовавшийся нормальный в°Со распадается
с испусканием 0-частицы в изотоп 60Ni только в течение нескольких лет: SSCo* —>
£JCo--------Некоторая определенная часть ядер «°C о * (прнмерно 10%) распа-
5,24 лет
дается непосредственно, испуская ^“-частицы, т, е. по уравнению ’°Со* ——-->2aNi а.
i0,7 мин ,
Следовательно, изомерия ядер в0Со в общих чертах соответствует схеме, приведенной на рис. 8Сш, лишь с тем отличием, что основное состояние ядра вОСо связано
7х=1,14мия
Т,=10,7 мин
iO% 90%
? -«г
fl
60Со* ®Сп
Мзв
Тг~ 5,24 года
60Ыг
Рис, 81. Схема распада обоих изомеров й0Со,
ВД2 (возбужденное яйро я*Ра)
99,85% 0,15%
ттгп г
fl fl fl fl
UZ (осооаяое 'состояние ядра Е»Ра)
[ЯГ
Рис. 82. Схема распада обоих изомерных ядер ILX, и UZ.
Цифры, стоящие у стрелок на рис. 80 и 81, указывают энергии распада или у-иэ л учения ( Мао). К скобках приведены количественные соотношения различных групп р-л учей.
изомерным переходом с возбужденным состоянием. Подробно схема распада обоих изомеров кобальта приведена па рис 81. Как видно из этой схемы, ни при одном из 0-распадов не образуется непосредственно ядро 60Ni в основном состоянии. В обоих случаях оно сначала получается в возбужденном состоянии, из которого затем мгновенно переходит в основное состояние, испуская у-квант.
Совершенно аналогично обстоит дело и в случае изомерных ядер UX2 и CZ (рис. 82). UX2 представляет собой ядро 234Ра, находящееся в метастабидьном возбужденном состоянии, UZ — то же ядро, по в основном состоянии. UX2 на 99,85% переходит непосредственно в ЦП по уравнению аЙ1Ра*---------——Ио 0,15% благо-
. 1,14 мин
а То, что период полураспада для испускания частицы возбужденный ядром равен периоду полураспада для изомерного перехода ядра с испусканием у-излучепия, следует непосредственно из того, что каждый из этих процессов приводит к уменьшению числа возбужденных атомов. Следовательно, в этом отношении здесь дело обстоит совершенно так же, как при двояком распаде (ем, примечание ** к стр. 621),
Искусственные превращения, атомов
625
даря изомерному переходу сначала превращается в нормальные ядра 234 Ра (уран Z), а затем распадается с образованием 234U (уран II): Ра ------->2^U.
6,7 час
Отличие от предыдущего случая заключается только в том, что, как видно из рис, 82, UX3 и. UZ испускают несколько групп р-лучей, тогда как еоСо* и 30Со испускают только одну группу р-пучсй.
Третий вид радиоактивного превращения, обусловленного изомерией ядер, наблюдается дня тех ядер, которые могут существовать в мета стабильном возбужденном состоянии, а в основном состоянии устойчивы, т. е, радиоактивно не распадаются, В этом случае наблюдается только один период полураспада, отвечающий переходу ядра из метастабильпого возбужденного состояния в основное, сопровождающемуся испусканием у-л у чей (рис. 80 в). Несколько примеров трех различных видов радиоактивного распада, обусловленного изомерией ядер, приведено в табл. 74.
Применение искусственно активированных элементов. Искусственно полученные радиоактивные изотопы можно использовать, как и естественные радиоактивные вещества, в качестве индикаторов (см, стр, 577). Искусственное получение радиоактивных изотопов в принципе позволяет распространить метод радиоактивных индикаторов на любые элементы. При этом важным средством исследования обогащается не только химия (и особенно область кинетики реакций), но биология и медицина. Точно так же все больше применяют искусственно полученные радиоактивные изотопы в качестве индикаторов в технике. Кроме применения в качестве индикаторов, или «меченых агпомовь (англ, tracers), искусственно активированные элементы во все возрастающем объеме используют как источники излучений вместо препаратов радия и рентгеновских трубок. Они намного дешевле радия и имеют перед рентгеновскими аппаратами то преимущество, что очень компактны, транспортабельны и легче приспособляются для использования в конкретных целях. Именно этим объясняется, что искусственно активированные элементы начинают вытеснять, например, в области лучевой терапии рентгеновские аппараты и прежде всего препараты радия.
Широкое применение искусственно активированных элементов возможно потому, что большинство из них — а именно те, которые образуются при делении урана или легко получаются при облучении нейтронами,— являются побочными продуктами при работе атомпых реакторов. Эти радиоэлементы, или вещества, в состав которых они входят, появились в продаже наряду с обычными химическими реактивами. Ряд искусственно полученных радиоизотопов приведен в табл. 75 и 76,
В этих таблицах указаны только те радиоизотопы, которые обладают либо чистым Р-излучепием, либо особенно сильным у-излучением. Последнее особенно важио дня. их использования в качестве источников излучения., Кроме изотопов, приведенных в обеих этих таблицах, в продаже имеются многочисленные радиоактивные изотопы других элементов. Наибольшее .применение находят Йод-131, фосфор-32, углерод-14, натрйй-24, золото-198, сера-35, кобальт-60, калий-42, кальций-45, железо-55 и -59, стрбиций-89 и -90, бром-82 и иридий-182.
В промышленности в качестве источника излучения особенно широко используется в0С о, например для испытания материалов. 60Со имеет очень проникающее излучение и относительно большую продолжительность жизни, Эти свойства кобальта-60 лежат в основе ужа сп о го действия «кобальтовой бомбы#. Если атомную бомбу снабдить оболочкой из обычного кобальта (fieCo), то выделяющиеся при взрыве бомбы нейтроны превращают этот кобальт в кобальт-60 по уравнению 69Со (к, у) еоСо. А так как кобальт-60 испаряется в процессе образования, а затем выпадает в виде пыли, то взрыв такой бомбы может привести к радиоактивному заражению: обширных областей на долгие годы.
В терапии радиоизотопы применяют но только дня внешнего облучения, но многие из пйх вводят внутрь. Некоторые элементы накапливаются в определенных органах тела, папример иод в щитовидной железе, фосфор в костях. Следовательно, если в тело ввести медикаменты, содержащие радиойод или радиофосфор (а такие медикаменты уже имеются в продаже в готовом для приема виде), то эти радиоэлементы самостоятельно попадут в то место организма, где желательно действие их излуче-40 Г. Реми
626
Глава 13
Таблица 7{
Радиоактивные превращения изомерных атомных ядер
Независимый распад Распад после изомерного перехода и пепасреоап&рцны'й распад
sc* * 2,5 vac J«Cr ел Эн ,^Ь-73Г*'^П мНв 2.вв8и ’ ”AU 21 мая ‘ ЯТс^7*“Мо ‘«In— 40 13 сек *t:sb-nL^> ворг» - ,ж>вгJ-*’ «ВГ 4,64 рт ” у 3“ _ 1ЭТТв * - 1«ТТл -*137 Т “ 90 Эя ’ й 531 liSe-~ -’’Вг 34 13,6 ЛШ» S5 . г ^Кг
А * вЛ-Ы •>Вг- 18,5Л’“" м Д+
18.5 мид « '^7^7'^ ИИЬ 1J7 1 $2 9,3 гас м t«s 1 ' “ ss ™те-/-—> >g;i ва 25jW4W 61
Узомерпый переход ядра ;ycsiouvuooeo о основном состоянии, из метастобшияого состояния в основное состояние
«Ge* ”Ge. S3Kr*- У -“Кг; •’Sr* У "Sr,
5 Ю"т$ек i.as^c 2,75чдс
'“’Ag* * 6 44.3сек l(,7Ag, K»Ag*. r 39,2 сек *”»Ag, ‘«Cd* У 2,8 деш/ "3Cd
it’In* * ► ITIIn. У '«Те* У
1,74 vac 4,5yac 1,2 10 '«л
>37 Ba*—— ► ls’Ba, •’Au*- У ► ‘’Au, •«Pb* У «‘РЪ,
2,<И muk 7i4ctK 1,10 час
ния. В последнее время начинают исследовать возможность получения радиоактивных излучений при помощи ядерных реакций, проводимых непосредственно в больных тканях. Например, были сделаны попытки накапливать бор в раковых ткапях и превращать его там в литий, облучая нейтронами,. Тогда образующиеся нри ядерной реакции (n, a)J а-частицы будут оказывать концентрированное биологическое действие иа ткань в непосредственной близости от атомов бора. В биологии радиоактивные источники излучений используют, например, для стимулирования мутаций, т. е. существенных изменений в носителях наследственности. Эти опыты открывают новые пути исследования в генетике.
В химии- искусствсппо активированные элементы применяют в основном в качестве индикаторов (меченые атомы).
Например, используя в качестве индикатора iesAu, удалось показать, что золото и платину нельзя количественно разделять при помощи перекиси водорода, несмотря на то что порознь только золото, но пе платила, восстали вливается под действием перекиси водорода (Erbacher, 1935). Используя радиоактивный изотоп серы 35S, удалось показать, что тиосеряая кислота в полном соответствии с приписываемой
Искусственные превращения атомов
627
Таблица 75
Торговые радиоизотопы с чистым р~-из;а учением
Изотоп Период полураспади З-.jучеТК ДГэе Изотон Период полураспада Максимальная анергия 0-лучей, jVse
ЗН 12,46 лсы и.0186 osr 54,5 дц 1,463
МС 5589 э 0,155 soy з₽ 1 2,54 » 2,18
asi 157 мин 1.48 43 Ag 7,5 >> 1,04
ьр 14,07 дп 1,69 *“₽т 13,7 » 0.922
87.1 0.169 ЩРт 2,26 лет 0,223
аоСа 152 ; 0,254 204Т1 3,5 » 0,783
Таблица 76
Торговые радиоизотопы с
особо жестким у-пзлучеппем
Изотоп Период полураспада Распад Максимальная энергия 3”-лучсй, Мэе Энергия у-л у чей, Мэе
^Na 15,10 час ₽- 1,39 2,755; 1,380
12,44 » Р- 3,58; 1,99 1,51
У Ми 2.59 » Р- 2,86; 1,05; 0,75 1,77; 0,822
^Fe 46.3 ,-ш Р~ 0,46; 0,257 1,30; 1,10
а’Со 5,26 гтт Р’ 0,318 1,33; 1,17
«Си 12.88 час р-, Р+Л 0,57 (р+0,66) 1,34
0AS 1,187 ди р- 3,15; 2,56; 1,5; 0,4 1,70; 1,20; 0,55
*?Rb 19.5 i> ₽- 1,80; 0,724 1,08
i^Sb till » г 2,29; 1,69; 0,95; 0,68; 0,50 2,04; 1, 71; 0,730
w?La 1,67 » р- 2,26; 1,67; 1,32 1,63; 0,87
19,1 час р- 2,23; 0,66 1,74; 0,49; 0,424
ЩТа 117 д,и р- 0,53 1,22; 1,13; 0,22; 0,15
ей структурной формулой (см, т, I, стр, 770) имеет два атома сери, которые ведут себя химически различно. Они не вступают друг с Другом в реакцию изотопного обмена (Andersen, 1936). Интересно так же то, что марганец, существующий в форме ионов MnOj не вступает в реакции изотопного обмена с ионами Ми"' или Ми"*, тогда как ионы Ми" и Мп'” тотчас обмениваются зарядами (Polissar, 1936). Кроме того, было показано, что иод в таких органических соединениях, как СгН51, связан неионогенно. Если СгН51 активировать медленными нейтронами, то образуется lasj, который (вследствие испускания у-лучей) отщепляется от материнской молекулы. Под действием воды, в которой растворен какой-либо восстановитель, активированный иод переходит в ионы I' и в этой форме легко может быть отделен от С2Н51 (Szilard, 1934). Если бы иод был связан в С2Н51 ионогенно, то это разделение было бы невозможным. Благодаря такому поведению активированных галогенов (бром и хлор ведут себя аналогично) их можно выделять, даже если опи присутствуют в виде следов (Erhacher, 1936). Это имеет большое значение для изучения поведения веществ, присутствующих в виде следов. Для обнаружения и определении следов элементов в последнее время большое значение приобрел ра&ирактивациопнъгй анализ. Он основан на том, что атомные ядра элементов, присутствующих в виде следов, превращают в неустойчивые атомные ядра (в большинстве случаев облучая нейтронами), которые обнаруживают затем по их характеристическому излучению.
Применение «моченых атомов» для отличия одного атома химического элемента от других, подобных ему (что осуществляют введением в химическое соединение радиоактивного изотопа), особенно часто используют в органической ^имии и биохимии. Например, этим методом удилось показать, что СО2, которая появляется в организме кик продукт разложения уксусной кислоты, образуется исключительно из ее карбоксильной группы, а не из одновременно сгорающей метильной группы. Для доказало»
628
Глава 13
тельства этого была синтезирована уксусная кислота, в которой радиоактивно «мгт»-ли» атом углерода в метильной группе, но не в группе СО ОН(т. е. определенная чгздь. молекул содержала радиоактивный углерод). Как оказалось, двуокись углерода, епрщ-зовавшаяся: в организме из этой уксусной кислоты, не была радиоактивна, сл ед слан тёльпо, она даже частично пе могла образоваться при сгорании метильных групп. Этш пример показывает, что применение изотопов * позволяет сравнительно 'просто изучи»,, тё вопросы; решение которых ипыми путями затруднительно или новее невозможна Тан, раньше нельзя было решить, происходят ли в зеленых растениях на свету охн?'-временно ассимиляция СО2 и дыхание с выделением СО2 или один из этих процесса», происходит на свету, а другой в темноте. Применение СО2, меченной радиоактиннд» углеродом, показали, что оба'процесса идут одновременно.
Другими примерами пспользоьания радиоактивных изотопов в качестве индикаторов при биологических исследованиях могут служить следующие. Применяя активов изотоп фосфора, удалось установить, как фосфор, усвояемый животными в виде ссед®-ненпй, со временем распределяется между различными органами и через какое в реет выделяется организмом. Далее, удалось показать, что сера, которая очень илец усваивается организмом животного, если ее вводить в элементарной форме или в форме неорганических соединений, легко усваивается белком животного, если ее вводите в виде определенных органических соединений, например цистина. Растения лет усваивают серу как в виде сульфатов, так и в виде двуокиси серы. Эти нёорганическЕД соед:гп<п|Ия тотчас же и]) ев решаются в органические. Перейос серы, например, сх листьев к корням иди наоборот происходит таким образом, что сера сначала превращается в сульфат, а затем откладывается в тканях в виде органических соединений. Инее-каторныс методы исследования оказались очень плодотворными и при изучении уевлх-ванпя и использования микроэлементов в организме. Для исследования превращений^ происходящих с соединениями углерода, их часто «метят», кроме радиоактивных игл-топов ИС (короткоживущий) и i*C (долгоживущий)**, также неактивным углеродах, обогащая его изотопом 1SC (см. стр. 591).
Искусственно активированные элементы используют в качество индикатора и в технике. Например, их используют для контроля движения нефти в трубопроводах. Они позволяют Про следить путь, по которому движется определенный элем^л в крупном техническом процессе. Так, используя «меченые атомы» можно установить, куда попадет германий, часто содержащийся в цинковых рудах, на каждых Этане процесса получения цинка, хотя Содержание германия очень невелико (Guesl 1948). Радиоактивные «метки» позволяют исследовать действие трения на разлнчннг детали машин, а также изнашивание подошв обуви, автопокрышек и покрытий дорог.
Реакцип «горячих атомов», Упоминавшееся ранее выдёлепие актив и ров аника нейтронами галогенов из органических соединении представляет собой пример tee называемых ^реакций горячих атомов». Под ними подразумевают такие химические реакции, которые непосредственно вызываются ядерными процессами, причем химпчз-ские связи разрываются под действием энергии, выделяющейся при этом процесса. В некоторых случаях реакции горячих атомов дают возможность разделить ядерник изомеры, В общем случае разделение ядерных изомеров невозможно, так как они еэ только химически эквивалентны, но и имеют равные массы. Но если имеются два^ядер-них изомера, которые связаны между собой изомерным переходом (см. стр. 622 и сл.), го часто у-из л учение, испускаемое при изомерном переходе, можно использовать для разделения этих изомеров. Ня пример, если к раствору теллура тов, в котором присутствуют оба ядерных изомера ките* и 131Те с периодами полураспада 29 час и 25 мне соответственно, добавить небольшое количество пеактивного теллурита щелочного металла, а затем его вновь химически отделить от теллурата, то получается препарат, в котором содержится только тот ядерный изомер, период полураспада которого равез 25 мип. Это объясняется следующим образом: часть у-лучей, которые испускаются при переходе ядра isi.Te* в нормальное ядро ШТе претерпевает «внутреннюю конверсию» (см. стр. 556), выбивая электрон из К- или Z-обблочки образующегося прн изомерном переходе ядра с периодом полураспада 25 мин. Благодаря процессу, который известен как «эффект Оже», энергия, освобождающаяся при заполнении вновь внутренней электропной оболочки, переносится на валентные электроны и вызывает
* Вместо радиоактивных изотопов в качестве «меченых атомов» можно использовать и устойчивые изотопы, изменяя за счет их введения естественнее соотнопЕенкл изотопов (см. стр. 591). Однако применение радиоактивных изотопов значительно удобнее.
** Об использовании относительно долго живущего изотопа 14С см., например, Angew. Chem,, 63,89 (1951). О получении NaCN, меченного 1JC, из торговых меченых карбонатов см. J. Am. Chem, Soc., 72, 4268 (1950).
Искусственные превращения атомов
629
разрыв химической связи между теллуром и кислородом в теллурат-ионе, т, е. вызывает переход.его в теллурит-иоп. Поэтому часть «25-минутного* теллура можно отделить в виде теллурита. Диалогично можно провести разделение и других генетически связанных ядерных изомеров.
Защита от излучений. Ввиду все возрастающего применения радиоактивных веществ в лабораториях и в технике необходимо особо указать на весьма серьезную опасность для здоровья, которая связана с работой с этими веществами, если не используют необходимые средства защиты. Опасность особенно усложняется тем, что человеческий организм не имеет никакого органа чувств, предупреждающего о действии радиоактивных излучений. Прежде чем человек почувствует -какое-либо ухудшение здоровья, вредное действие излучения оказывается уже настолько сильным, что неизбежно приведет к смерти. Поражение радиоактивными лучами протекает скрыто во времени. Могут пройти недели, месяцы и даже годы, прежде чем в полном объеме проявятся последствия такого поражения.
Следует учитывать увеличение поражения при длительном- облучении. Облучение, ‘ которое при кратковременном и одиночном воздействии совершенно безопасно, может привести к серьезным заболеваниям, если оно повторяется в течение длительного времени. Легкое поражение становится заметным по появляющемуся через некоторое время покраснению кожи на об л ученном месте. Прн интенсивном или длительном облучении в конце концов могут образоваться язвы, которые часто переходят в плохо заживающие раны. Е наиболее серьезных случаях может развиться раковая болезнь. Но прежде чем она обнаружится, могут пройти годы. При превышении допустимых доз облучения возможно также развитие тяжелых заболеваний крови (например, лейкемии), особенно если превышение доз допускается систематически.
Чтобы при работе с радиоактивными веществами избегнуть поражения излучением, следует соблюдать допустимые дозы облучения и принимать меры, предупреждающие их превышение. Это не представляет особых трудностей, если (как это часто бывает в случае использования радиоактивных веществ как индикаторов) активность препаратов, с которыми производится работа, не превышает значительно 1 ркюри (или 3,7 • 104 распад!мин, см, стр. 602). В этих случаях достаточно надеть резиновые перчатки и брать сосуды с активными препаратами достаточно длинными щипцами. Простейший способ защиты от излучения слабо радиоактивных веществ — работа на достаточно далеком расстоянии от них. Если работа производится с лучами, которые па их сравнительно коротком пути в воздухе практически не ослабляются за счет поглощения, то (если размеры источника излучения малы по сравнению с расстоянием от пего) интенсивность излучения обратно пропорциональна квадрату расстояния от пего. Еще быстрее уменьшается интенсивность излучения, если оно ослабляется за счет поглощения. Если пробирку, содержащую небольшое количество радиоактивного вещества, взять непосредственно в руку, то расстояние от источника излучения составит, вероятно, около 3 мм. Если же воспользоваться тигельными щипцами обычной длины, то это расстояние уже будет равно примерно 170мм-,. Интенсивность излучения при этом уменьшится по крайней мере в отношении 170а : За = •= 3200 : 1, ш в таком же отношении увеличится время, за которое будет получена допустимая доза облучения.
Под допустимыми дозами понимают дозы излучения, которые не следует превышать, чтобы избегнуть вредного для здоровья действия излучений. Для рентгеновских
630
Глава 13
лучей дозу излучения обычно измеряют в рентгенах всекунду (рентген/сек). При эгок рентген определяется ко,к такое количество рентгеновских или у-лучей, которое в 1 <м*® сухого воздуха приОа и нормальном, давлении образует столько ионов, что их общий заряд равен 1 электростатической единице каждого знака. Опыты показали, что эпергпя, требующаяся для образования пары ионов в воздухе, составляет в среднем 32,5 эа. Поскольку однократно заряженный ион несет заряд 4,80- 1О*10 ал. ст, ед. (см. т, I, стр. 110), 1 ли3 сухого воздуха При нормальных условиях весит 0,001293 г, а 1 за = =1, 601.10-12 эрг, то анергия, рассеиваемая в 1 г воздуха при поглощении 1 рентгена,
Для б иолог и -t ес к. о го действия излучений существенна энергия, которая поглощается тканями, ища. При поглощении 1 рентгена тканями тела они воспринимают энергию, большую чом 84 эре /г. Например, рентгеновское излучение в 1 рентген рассеивает в 1 в воды 93 эрг, а в 1 г костной ткани около 150 эрг, Кромедого, корпускулярное излучение (а-лучи, ^-лучи, нейтроны) оказывает на ткани тела существенно иное воздействие, чем рентгеновские или у-лучи, причем значительно большее. Чтобы производить расчеты, по предло;кеиию Паркера (Parker, 1948) были введены две единицы действия радиоактивных лучей; 1 реп (сокращение от «roentgen equivalent physical»— (ривически.й эквивалент, рентгена) — такая доза лучей, которая передает 1 г облучаемого вещества столько же энергии, сколько передает 1 г сухого воздуха (при 0° и 760 .«.и рт ст) 1 рентген (т. е. 83,8 эрг/г). ,
1 реб (сокращение от «roentgen equivalent biological»-- биологический эквивалент рентгена) — такая доза излучения, которая оказывает такое же биологическое действие, как 1 рентген рентаеновского или у-иэлучения.
В общем молшо сказать, что для р-л у чей 1 реп = 1 реб; ди я а-лучей * 1 реп = = 10—20 реб; для медленных («тепловых») нейтронов 1 реп — 5 реб и для быстрых нейтронов 1 реп = 10 реб.
В последнее время все чаще в качестве единицы Дозы излучения вместо «реп» ^употребляют «рад». 1 рад — такая доза излучения, которая передает облучаемому "веществу энергию в 100 эрг1г. Следовательно, 1 рад соответствует 1,19 реп.
Безвредные дозы облучения (допустимые дозы) составляют в случае рентгеновских, у- или (3-лучей или медленных нейтронов 0,3 реп в неделю, для быстрых нейтронов — 0,03 реп в педелю и для а-лучей — 0,015 реп в неделю. При^ этом принимают, что неделя состоит из 40 рабочих часов. Приведенные Допустимые Дозы относятся к облучению всего тела. Но если облучению подвергается только относительно малая площадь тела, то допустима несколько большая доза. Например, если облучение ограничивается только кистями рук и предплечиями, то допустимая доза примерно в пять раз больше, чем для нсего гола. В медицине, нанример, при облучении раковых опухолей, работают со значительно большими дозами излучений. Одпако в этих случаях облучению подвергают очень небольшой участок тела и, кроме того, оно и применяется как раз пз-за своего биологического действия. В настоящее время еще нельзя удовлетворительно объяснить, почему интенсивное облучение способно как вызывать раковые заболевания, так и излечивать их.
При работе с радиоактивными веществами в количествах, превышающих 1 ркюри, должны соблюдаться особые меры предосторожности, которые связаны не только с охраной здоровья работающего, но и с предотвращением «радиоактивного заражения» лабораторных помещений и сосудов. Особенно относится это к работам с веществами, активность которых составляет 1 мкюри (3,7.10“ распад/сек) и более. Работу с такими веществами производят в специально приспособленных лабораториях, так называемых «горячих» лабораториях, оборудование которых позволяет производить издали манипуляции с находящимися за защитной стеной радио препаратами и сосудами, в которых они содержатся.
Устойчивость ядер. Вопрос о природе"«ядерных сил», т. е. сил, благодаря которым компенсируется кулоновское отталкивание протонов, еще окончательно не выяснен. Ч агто считают, что эти силы являются «резонансными силами» в смысле волновой мо.г.чпцщ! (си. т. I, стр. 156). Во всяком случае, опыт показывает, что ядерные силы только тогда компенсируют кулоновское отталкивание, когда отношение числа протонов в ядре I,- числу пейтропов в нем находится в определенных пределах. Ядра с небольшим числом протонов в общем наиболее устойчивы тогда, когда число нейтронов в них
* Вследствие малой проникающей способности а-лучей, внешние поражения от них едва ли следует учитывать, конечно, если приняты простейшие меры предосторожности. Однако вдыхание а-излучающих веществ в форме пыли может вызвать поражение внутренних органов, точно так же как и несоблюдение допустимых доз прп использовании а-излучающих препаратов для медицинских целей.
Искусственные превращения атомов
631
равно числу протонов ила превышает его на 1. Чем больше число протонов, тем больше сдвигается максимум устойчивости в сторону увеличения избытка нейтронов над протонами, Как видно из рис, 83, на котором приведена зависимость числа нейтронов от
32 31 30 29 28 27
26 ~ 25 -24 -
23-22 -21 -20 -19 -18 -17 -16 ~ 15 -Й -13 -12 ~ 11 ~
10 9 8 7
6 5
4 3
2
-о
HeLi
Be
Me
Na 1
Mg
Si
। А1 к‘5И vT7n SCbArKcaSCTiVGrAn
о о о о • о о •
d о • о • о • о •
- Н
6 с
о
N
о о
о
о F
о о
Р
о
о
о
о
о о
О •
о •
о о
о о о
о о о
о о о
k а I I—I । । t,±,.L | ।—। I I I т । ; I—1_1_|—1_| 1 2 3 4 5 6 7 8 9 1011 12 1314 15 16 17 18 19202! 22232425
Число протонов Z
Рис. 83, Числа прогонов п нейтронов в ядрах атомов элементов с порядковыми числами от 0 до 25, * устойчивые ядра; О неустойчивые ядра. Ядра е равным числом нейтронов и протонов лежат на пунктирной прямой.
числа протонов в ядрах устойчивых и искусственно полученных неустойчивых элементов, ядра оказываются неустойчивыми как в тех случаях, когда отношение числа нейтронов к числу протопов слишком мало, так и и тех случаях, когда оно слишком велико.
Однако ноустойчивое’ядро совсем не обязательно должно мгновенно распадаться, а обладает, если ого энергия не слишком сильно превосходит энергию продуктов распада, только определенной вероятностью распада. Эта вероятность находит свое числовое выражение в во личине постоянной распа да [см. уравнение (4), стр. 550]. Это объясняется, как впервые показал Гамов (Gamow, 1928) на примере радиоактивного а-раснада, следующим: если графически представить зависимость потенциальной энергии электроположительной частицы от ее расстояния от центра ядра, то получим кривую, имеющую вид, приведенный па рис. 84 При этом изменение потенциала вне ядра описывается законом Кулона. В устойчивом ядреэлектроположительные частицы имеют потенциальную энергию мспьше, чем их энергия вне ядра, даже при бесконечном удалении от центра ядра. В неустойчивом ядре потенциальная энергия электроположительных частиц больше, чем опа была бы при бесконечном удалении от центра ядра. Если потенциальная энергия такой частицы меньше, чем энергия, отвечающая вершине потенциального барьера, окружающего ядро, то она не сможет в со от
632 Глава 13
ветствии с законами классической механики покинуть ядро до тех пор, дока внешнж силы не сообщат ей энергию, достаточную для преодоления потенциального барьера. Напротив, согласно волновой механике, существует определенная вероятность твтс, что частица, образно говоря, «просочится» * через потенциальный барьер. Чиепк атомов, в которых происходит этот процесс в единицу времени, прямо пропорционально этой вероятности. Следовательно, числовая величина. постоянной раепаба рветц вероятности, выхода частицы ив ядра. Чем меньше та часть потенциального барьера, под которой должна пройти частица, тем больше вероятность выхода частицы и теи больше одновременно кинетическая энергия, которую будет иметь частица во выход* из ядра благодаря, образно говоря, скатыванию с потенциальной горки. Отсюда ценз-Средственпо следует закон Гейгера — Наттола [см. уравнение (9), стр. 551].
Если при а-расиаде одинаковых атомов все частицы испускаются с одинаковой скоростью, то при р-pa спаде, как уже указывалось (см. стр. 556 и 617), электроны и позитроны выходят из ядер со ско-
ростями, которые непрерывно рас- I предслены в значительном иитервале. I То наблюдение, что ядро теряет энер- 1 гию, отвечающую не средней, а лкг- I ксимальной эцергии р-частиц, застав- | ляет предполо;кить, что при p-рас- 1 паде, кроме позитронов или электро- I нов, атомные ядра испускают еще ! (и притом незаряженные) частицы, на долю которых и приходится та часть энергии, которая отличает энергию “ большинства р-частиц от максимальной энергии. Этими частицами оказываются часто упоминавшиеся нейтрино. ,
& Энергия образования ядер. Точной мерой энергии образования ядро, т. в. энергии, которая выдели-
Р и с. 84. Гамовскпй потенциальный барьер. Е — анергия, освобождающаяся при удалении положительно заряженной частицы на расстояние г = си. Радиус ядра т„ — это такое расстояние от центра ядра, на котором иамепение потенциала начинает отклоняться от предписываемого законом Кулона (пунктирная линия).
ется при образовании атомного ядра из его со ставных" частой (нуклонов), является дефект массы данного атомного ядра. Однако не для всех атомов известны массы с такой точностью, чтобы можпо было найти [дефект массы с той точностью, которая требуется для расчета энергии образования. Поэтому интересно, что энергию образования ядер с удовлетворительным приближением можно рассчитать на основе теории ядерных сил. Эта теория еще не разработала настолько, чтобы с ее помощью можпо было без введения вспомогательных эмпирических данных рассчитывать точные значения энергии образования ядер. Однако все эмпирические константы, используемые при таких расчетах, одинаковы для всех элементов. Поэтому при помощи приведенных ниже формул Можно рассчитать энергию образования и для таких атомов, которые в условиях их получения мгновенно распадаются и массы которых поэтому вообще невозможно определить экспериментально. Эта возможность будет использована позднее (при расчетах, связанных с делением ядер).
Формула для приближенного расчета энергии образования ядер получается, согласно Вайцзекеру (Weizsacker, 1935), следующим образом: исходим из того, что, согласно экспериментальным данным, все атомные ядра имеют практически одинаковую плотность. Поэтому атомные ядра можно сравнить с каплями жидкости, плотность которых также не зависит от их радиуса (^капельная модель» атомного ядра). Кроме того, экспериментально установлено, что ядерныв силы в противоположность кулоновским силам имеют очень малый радиус действия (около 3-10""13 сл). Они действуют только между двумя нуклонами, которые непосредственно соприкасаются. Это приводит к тому, что энергия, освобождающаяся при сближении нуклонов под действием ядерных сил, в первом приближении пропорциональна числу нуклонов, т. е. массовому числу А. Коэффициент пропорциональности (имеющий размерность энергии) обозначают at.
Опыты но изучению взаимного столкновения нейтронов и нейтронов с протонами показали, что силы притяжения, которые действуют между двумя соприкасающимися нейтронами, значительно слабее, чем силы, действующие между нейтроном и про-
* Соответственно возможно просачивание сквозь потенциальный барьер извне внутрь ядра. Этим объясняется способность положительно заряженных частиц проникать внутрь ядра даже в том случае, если их кинетическая энергия меньше, чем энергия, требующаяся, согласно расчету, для преодоления кулоновского отталкивания.
Искусственные превращения атомов
633
тоном. Поэтому энергия образования атомного ядра, имеющего избыток аейтровов над протонами, меньше чем «j/1. Чтобы учесть меньшую связь между нейтронами, из величины а^А надо вычесть член -Дй Здесь а»— новая постоянная, а (Л ч* 2Z)—
избыток нейтронов над протонам.
В атомном ядре протоны вследствие их одноименных зарядов отталкиваются друг от друга. Работа, совершаемая при введении протонов в ядро против этих сил
Z2
отталкивания, в сумме пропорциональна —, где г — радиус ядра. Поскольку г3 про-Г
порционален массе ядра, г пропорционален \‘~А, и, следовательно, из энергии о бра-22
аования ядра надо еще вычесть член, равный а3 (а3 — постоянный коэффи-у Л
циент пропорциональности.)
Далее следует учесть, что нуклоны на поверхности ядра связаны слабее, чем внутри него. Этот поверхностный эффект, совершенно аналогичный поверхностному натяжению в жидкостях, пропорционален г% или Л2'3. При этом энергия образования уменьшается па «4Лг/з,
И, наконец, следует учесть влияние спина. Как и электроны в электронной оболочке атома, так и пуклоиы в ядре обладают некоторым спином. Протоны с противоположными спинами могут образовывать пары, точно так же как и электроны («насыщение спинов»). Это же относится и к нейтронам; однако образования пар между протоном и нейтроном не происходит. В атомном ядре, которое содержит четное число-как протонов, так и нейтронов (четио-четпое ядро), все нуклоны спарены. Этим насыщением спинов объясняют особую устойчивость четно-четных атомных ядер. Если же атомное ядро содержит’ четное число протонов л почетное число нейтронов (четнонечетное ядро) или нечетное число протонов и четкое число нейтронов (нечетно-четноо ядро), то один из его нуклонов не спарсп. Такое ядро, в обп(ем менее устойчиво, чем ядро с полным насыщением спинов. Если же ядро содержит нечетное число как протонов, так и нейтронов (нечетно-нечетное ядро), то в нем оказываются два неспаренкых , нуклона. Такие ядра устойчивы только в редких случаях. При вычислении энергии образования влияние спина можно учесть тем, что в уравнение ввести донолнитель-11г „ ....
нвдй член -j-, в котором ц5 имеет положительный или отрицательный знаки зависимости от того* спарены ли в пем все нуклоны или имеется два неспаренных. Если неспарен, только одни нуклон, то а6 = 0.
В общем формула для расчета энергии образования атомного ядра\Ея (Мае) имеет вид
_ . (Л-22)
Z? Д ™- Я1 — ct 2 — —
А
(12).
где oj = 14,0, а5 = ф-130 для четно-четного ядра,
= 19,3, а5 — —130 для нечетно-нечетного ядра,
а3 — 0,585, а5 — 0 для четно-нечетного ядра, л4 = 13,05, а5 = 0 для нечетно-четного ядра.
Значение константы а3 можно найти из теоретических соображений. Остальные-значёния постоянных определены эмпирически, В табл. 77 для нескольких атомных ядер сопоставлены энергии образования, вычисленные по формуле (12), с найденными из дефектов массы.
Таблица 77
Сравнение энергии образования ядер, рассчитанных теоретически, с найденными из дефектов массы
(Цифры указывают энергию образования в мега электро я-вольтах)
Изотоп 40Аг 18АГ 19К ^Са ||сг ssFe . S^TH
Ея рассч. 347 333 342 452 486 544 1760 1771 1777 1796
ЕЛ из ДМ 344 334 342 456 492 552 1768 1780 1785 1802
4534
Глава 13
Ядерные правила. Как следует из вышеизложенного, количественная теория ядерных сил настолько развита, что позволяет рассчитывать энергия образования атомных ядер с достаточно хорошим приближением к экспериментально установленным величинам. Но она еще неспособна давать общие указания на то, с какими массовыми числами могут быть связаны определенные значения заряда ядра, т. е. какие из ядер с данным порядковым номером будут устойчивы. Все же на основании экспериментального материала было установлено несколько правил, касающихся состава устойчивых ядер. Важнейшие из них следующие:
1. Не существует устойчивых изобаров, заряд которых различался бы на единицу (пр а вил о Маттау ха).
Это правило, впсрвыев 1934г. установленное эмпирическиМаттаухом, -следует рассматривать, как показало дальнейшее развитие ядерной физики, как строгий закон (Jensen, 1939). Несколько очевидных исключении относится к долгоживущим изотопам, ядра которых изменяются главным образом вследствие электронного захвата.
2. При четно» массовом числе устойчивые изобары всегда имеют четное число протонов и четное число нейтронов (правило Харкинс а). Отсюда следует: у устойчивых ядер никогда не бывает одновременно нечетного числа протонов и нечетного числа нейтронов.
Некоторые легкие ядра (“Н, ®1л, и “N) не подчиняются этому правилу.
3. Для каждого нечетного массового числа существует только одно-единстсеннве устойчивое ядро.
Ядра с четным числом протонов могут иметь как четные, так и нечетные массовые числа. Напротив, ядра с нечетным числом протонов, согласно правилу 2, могут иметь только нечетные массовые числа, и из них исключаются, кроме того, согласно правилу 3, те массовые числа, которые /встречаются у ядер с другим числом протонов. Этим объясняется то, что элементы с нечетными порядковыми номерами большей частью нредстав--лепы только одним устойчивым ядром (см. табл. 64). Если элементы, с нечетными порядковыми числами являются смешанными элементами, они никогда не бывают представлены более чем двумя устойчивыми ядрами (см. табл. 65). Это согласуется со следующим правилом:
4. Каждый химический элемент имеет по крайней мере одно устойчивое ядро с нечетным массовым числом и максимум два ядра с нечетными массовыми числами (правило Астона).
Исключения из этого правила представляют элементы с порядковыми номерами 18 и 58, а также 43 и 61. Первые два (Аг и Се) со стоят до л ько из изотопов с четными, массовыми числами, дня двух Других (Тс и Рт) известны только неустойчивые атомы.
Правило Астоиа не охватывает элементы с особо высокими значениями порядкового номера, а именно элементы с порядковыми номерами более 82. Для этих элементов, как уже говорилось, устойчивые изотопы вообще не известны.
Искусстве иные новые элементы. Сначала искусственно полученными атомами были только неустойчивые изотопы уже известных химических элементов. Но позднее при помощи ядерных реакций удалось получить и такие элементы, которые до того не были найдены в природе п, как оказалось, в некоторых случаях вообще не могут существовать в природе. Искусственное получение этих элементов заполнило существовавшие до тех пор пустые места в периодической системе. Но искусственно были получены и такие элементы, порядковые номера которых больше поряд-
Искусственные превращения атомов
635
нового номера урана, на котором заканчивалась раньте периодическая система. Это так называемый трансурановые элементы, им и будет посвящена следующая глава.
Первым не найденным (или, по крайней мере, достоверно не найденным) в природе элементом, полученным искусственно при помощи ядерной реакции был элемент с порядковым номером 43 (технеций). В 1937 г., когда образование этого элемента было впервые обнаружено при облучении молибдена дейтронами, в периодической системе было четыре незанятых моста. Кроме моста для элемента с порядковым номером 43, вакантными оставались места для элементов с порядковыми номерами 61, 85 и 87. Впоследствии удалось искусственно получить многочисленные неустойчивые изотопы этих элементов (см, табл. 78). Но пи для одного из них пе было достоверно установлено существование хотя би одного устойчивого изотопа. В результате дальнейших исследований изотопов и ядер оказалось очень мало вероятным, чтобы местам с порядковыми номерами 43, 61, 85 и 87 соответствовали, кроме неустойчивых атомов, также и устойчивые.
В табл. 78 приведены известные к настоящему времени изотопы элементов с порядковыми номерами 43, 61, 85 и 87. В этой таблице приведены только те изотопы, массовые числа которых достоверно известны или (эти случаи отмечены знаком вопроса) установлены с большой вероятностью. Для большинства этих элементов известно еще значительное число изотопов. Например, для технеция подучено по крайней мере 20 различных изотопов (если считать и ядерные изомеры).
Их можно различать по периодам полураспада, но не для всех из них достоверно известны значения массовых чисел. Изотопы, являющиеся членами естественных радиоактивных рядов, указаны во второй колонке таблицы жирным шрифтом. Звездочка, стоящая при символе элемента во второй колонке, показывает, что соответствующее атомное ядро при данном способе получения образуется в таком состоянии, из которого оно без испускания или поглощении частицы, а только с выделением энергии переходят с периодом полураспада, указанным в третьей колонке, в менее богатое энергией состояние. Этот вид ядерпото превращения обозначен в четвертой колонке знаком у. В остальных случаях самопроизвольные ядерные превращения указаны в четвертой колонке таблицы, так же как и в табл. 73. Некоторые изотопы, приведенные в табл. 78, образуются как продукты деления урана. Этот тип образования обозначен в таблице знаком «U (я, деление)». Из таблицы видно, что изотопы технеция и прометия испытывают те же превращения, которые вообще характерны для искусственно полученных радиоэлементов легких и средних масс, а изотопы тяжелых элементов — астатина и франция, напротив, подвержены а- или р_-распаду. Это относится как к естественным, так и к искусственным изотопам франция и астатина, и только для некоторых из изотопов астатина наряду с а-распад ом происходит и электронный захват.
Существование устойчивых ядер с порядковыми номерами 43, 61, 85 и 87 противоречило бы правилу Матта у ха, если бы массовые числа этих ядер лежали в некоторой, рассматриваемой как вероятная области. Для устойчивого изотопа элемента 43 в соответствии с его положением в периодической системе следовало бы ожидать массового числа примерно 98. Но все массовые числа от 94 и 102 уже заняты устойчивыми^ изотопами соседних элементов 42 и 44 (Мо и Ни) (см. табл. 65). Поэтому по правилу Маттауха не должно существовать устойчивого ядра с порядковым номером 43, массовое число которого лежало бы менаду 94 и 102. В то же время маловероятно, чтобы существовало устойчивое ядро с порядковым номером 43, массовое число которого было бы меньше 94 или больше 102. Ядра, массовые числа которых сильно отличаются от ожидаемых в первую очередь в силу положения этих ядер в периодической системе, всегда менее устойчивы, чем тс ядра, массовые числа которых лежат близко к ожидаемым величинам. Поскольку в интервале масс 94—102 нельзя ожидать существования устойчивого изотона элемента 43, он пе может существовать и вне пределов этих значений масс. Действительно, наблюдения показали, что оба наиболее долгоживущих изотопа элемента 43 (технеций) имеют массовые числа 98 и 99, а изотопы с массовыми числами больше 102 или меньше 94, если только они были получены, оказываются очень короткоживущими (см. табл. 78). Следовательно, существование устойчивого изотопа технеция оказывается очень мало вероятным.
Аналогично обстоит дело и в случае элемент,а 61. Массовые числа его наиболее устойчивых изотопов должны находиться около 147. Однако все массовые числа от 142 до 150 уже заняты устойчивыми изотопами элементов 60 и 62. Эти примеры указывают па гораздо большую устойчивость ядер с четным числом пр о топов но сравнению с ядрами с нечетным числом протонов. Искусственно (с использованием ядерной реакции) элемент 61 впервые быд получен в 1942 г. Лоу (.Law), Пулом (Pool), Курбатовым и Купля ом (Quill) ва циклотроне. Уже через несколько дот (1946) другой изотоп элемен-
636
Глава 13
та 61 был открыт Корнеллом (Coryell) вместе с Марипским (Marinsky) и Кленденином (Glendenin) среди продуктов деления урана (си. стр. 639 и сл.). Изотоп элемента 61 образуется при 3"-распаде изотопа неодима с периодом полураспада 11,6 дия ив свою очередь превращается в устойчивый ивотоп самария с периодом" полураспада 3,7 года. Теперь элемент 61 назван прометием (символ Рт). Это название должно указывать на то, что выделение этого элемента произошло на пороге века атомной энергии, открытие которой можно сравнить с подвигом Прометея, которому люди обязаны, открытием огня.
Таблица 78
Изотопы элементов 43, 61, 85 и 84
Изотоп Период полураспада Распад Продукт распада Способ получения
4,6 мин Г SJMo ^Mo(d, 2п)
“Тот 2,7 час Г* НМ® “Mo (d, п)
^с* 50 мин V :*Mo(p,n):«Mo(d,2n)
< 50 лгин К «Мо «Тс* у
ГвТс 20 час К SSMo «Мо («, р); «Мо (р, и); «Мо (d, 2n); «Ru - 0+
«Тс 62 де — . h £>V- «Мо «Мо «Мо(а, р); «Мо (а, п); «Мо (d,2n);«Mo (р,п)
0,8% ?
«Тс 4,2 дн К I’MO «Mo (d,n);«Mo(d, 2п); «Мо (р, n);“Nb(a, п)
сч Ёт »sTc* 91 дн V :;тс «МО (d, n); °JMo (р, в);
S X № в «Тс 10s—10е лет г JjMo (d, п); «Мо (d, 2n)
5 с Е о «Тс* 6,6 час V «Тс U Th (п,йеление);
S 2,12-10* лет» Г я»* »ГГе* -* у
^Тс? 1,33 жая /л “®Ru «®Мо (d, 2п)
™Тс 14,3 мин Г «*Вп \«Mo(d, nh ^Ru(y, р); ^Ru (п, пр)
4,5 мин Г ^Ru(n,P)
\^Тс 1,2 мин э- 5«|Ru гЙЕи (n, пр)
^Тс 18 мин г «;ви “*Bu (n, p)
™Ts ~ 15 мт “JRu U (n^w); ^JMo —►
Продолэгсение табл. 78
Изотоп Период полураспада Распад Продукт распада Способ получения
20 лия«-х У ’«Nd (р, 2п)
^«Рт 285 дн К да ’«Рг (а, 2n), ’«Nd (р, п)
\«Рт ~ 300 дн К да ’«Рг (а, п); ’«Nd (р, п)
’«Вт ~ 30 лет К ’JtSrn (ii, у ); ’jJSm К
§ Е ’«Рт ~ 1 год р- да да (р, п)
§ ’«Рт 2,6 лет 0- да U (П,бренча); ’j’Nd j8"
ё »JPm 5,3 дн /в да ’«Nd (а,р); да (d,2n); ’«Nd (р, nh “’Рт (в, у)
Б о § ’«Pm (изомер) 42 дн 2,27 дн F г да да да (р.п) U (п,йыяще); ’floNd —* Д“ ; да (Р,2п)
’68Рт 2,7 час «Зт ’«Nd (р, п)
™At 8,3 час Ь< 8 . ' ' С" HaiJPo 2ЙВ, ™Bi (а, Зв)
к да
1,5 нас "<1%* а оо ^вца.гпьда^эрзгп)
' § ^At 0,25 сек а 2«Bi Р4 да («, п)
<3 ^At ~ 10'‘wit О. да я’?Ег -> а
: I ~ 10"4 сек а дас дал да
S • s£At ~ 1СГ8сйг а дас дал- КН да-а
В О «At 0,018 «л а да да — а
S; ^At 1,5—2,0 сек $0^»- а oj%* даас дап Во дал—
=^At 0,9 лгин а 0" 2да ио даЕт да-а
"Fr 5 • 10’"3 сек а да вмАс -> а
§ “?Fr 0,02 сек •к а да |юАс — а
а* л 1 <“Fr 27,5 сек а да <мАс —>• а
*’Fr 4,9 лшн а «At Q& «»Ас — а
5 с о Е о ^Fr 21 лгин а 4SSj/>- да «»АсХ «’Ас—а
638
Глава, 13
В настоящее время известно большое число изотопов прометия (около 10), Многие из них образуются в весомых количествах в атомных реакторах как продукты деления урана. Одпако их выделение связано со значительными трудностями вследствие чрезвычайно высокой радиоактивности продуктов деления (см. стр. 630 и 648).
Из того что технеций неустойчив, нельзя сразу делать вывод о том, что этот элемент не встречается в природе- Несмотря на то что период полураспада даже его самого долгоживущего изотопа (08Тс) псдостаточен, чтрбй сохранились значительные количества технеция с момента застывания земной коры, все же возможно, что в некоторых молибденовых рудах под действием нейтронов постоянно образуются минимальные количества 0зТс. Благодаря большому периоду полураспада 9STc (Ю5—10° лет) это могло привести к тому, что за время геологических эпох накопились значительные количества технеция. В. и И. Ноддаки считали, что им удалось открыть элемент 43 рентгенографически вместе с рением уже в 1925 г. (см, стр. 250), Недавно Герр (Негг) следующим образом показал большую вероятность присутствия ®8Тс в молибденовых рудах: специально подготовленный препарат, получающийся при переработке молибденовых руд, он нодверг интенсивному облучению нейтронами. Оказалось, что после такого облучения препарат обнаруживал радиоактивное излучение, характерное для ®9Тс* (см. табл. 78). Препарат был предварительно тщательно очищен от молибдена (из которого также мог бы образоваться 99Тс*), и поэтому результат опыта можно объяснить, только предположив, что 99Тс* образуется за счет ядерной реакции Я8Тс(п, у) si'Tc* па присутствовавшего в препарате технеция-98 (наличие которого ввиду слабой активности при этих условиях непосредственно обнаружить не удается).
О получении весовых количеств технеция из продуктов деления урана си. на стр. 252.
Элементы 85 и 87 (астатин At и франций Fr) относятся к той области периодической системы, в которой вообще не существует устойчивых элементов. Следовательно, надо принять, что существуют только неустойчивые изотопы элементов 85 и 87, и притом только короткоживущие, так как элементы с нечетными порядковыми номерами вообще оказываются более короткоживущими, чом соседние элементы с четными порядковыми номерами. Радиоактивные элементы с не очень большими периодами полураспада могут встречаться в природе в значительных количествах только тогда, когда они постоянно образуются в результате распада элементов с большой продолжительностью жизни. Из таких элементов известны только уран (Ш и AcU) и торий. Поэтому можно было бы ожидать, что элементы 85 и 87, если они встречаются в природе, являются членами естественных радиоактивных рядов. И действительно, в 1939 г. Перей (Рогу) установила, что актиний наряду с р -лучами испускает небольшое количество (около 1%) а-лучей, т. е. обнаруживает двоякий распад. Но в соответствии с законом сдвига при радиоактивном распаде актиний, испуская а-частицу, переходит в элемент с порядковым номером 87 (и массовым числом 223) — экацезий, который впоследствии был назван францием, (Fr). Сейчас известно уже 8 изотопов франция. Наиболее долгоживущий изотоп 328Fr — член радиоактивного ряда актиния. Его период полураспада равен 21 мин и основная часть его (см. табл. 63} переходит, испуская р"-частицы в АсХ. Оказалось, что франций легко соосаждается с перхлоратами или хлороплатинами рубидия и цезия и, следовательно, как можно было ожидать по его положению в периодической системе, обнаруживает свойства щелочного металла. Элемент 85’ (экаиод, называемый теперь астатином) впервые был искусственно получен Сегрс (1910) при облучении висмута а-частицамн высокой энергии: 2з3В1(а, 2га) ““At. Его период полураспада равен 7,5 часа, п он обнаруживает двоякий распад: около 59,1% за счет захвата электрона превращается в 211Ро, который, испуская а-частицу, переходит в 2О7РЬ, а 40,9% претерпевает а-распад, превращаясь в 2O'Bi, из которого затем также образуется 20ГРЬ. То, что астатин является членом естественного радиоактивного ряда, было доказано в 1943 г. Карликом (Karlik) и Бер-
Искусствшные превращения атомов
639’
пертом (Bernert). Эти исследователи установили, что RaA, а также ТЬА и АсА. обнаруживают двоякий распад, приводящий к образованию-a*?A.t или соответственно 2^Л1 и 2^At (доля второго пути распада составляет 0,031, 0,014 и 0,0005% соответственно). Эти три изотопа астатина еще более короткоживущие, чем первый, полученный искусственно 2”At. Они очень быстро, испуская а-частпцы (пробег равен 5,53, 6,84 и 8,0 см соответственно), переходят в RaC, ThC или АсС. Членом радиоактивного ряда актиния, как было найдено позднее, является еще один изотоп астатина s)’At (см. стр. 569). В общем к настоящему времени известно не менее 17 изотопов астатина. Наиболее долгоживущий из них 2™At имеет период полураспада 8,3 часа и за счет электронного захвата превращается в полоний. Но некоторая часть ядер этого изотопа (0,17%), испуская а-частицы, превращается в 206В1, который затем (главным образом за счет электронного захвата) переходит в аойРЬ.
Деление ядер. При облучении урана нейтронами вместо образования, неустойчивого ядра с большим массовым числом (за счет внедрения нейтрона в ядро урана) может происходить, как доказал впервые Ган (Hahu, 1939), «деление» атомного ядра с образованием неустойчивых ядер средней массы, которые в дальнейшем в свою очередь радиоактивно распадаются. Оказалось, что нейтрон внедряется только в изотоп урана 23SU, и причем только тогда, когда этот изотоп урана облучают медленными нейтронами. Под действием медленных нейтронов из 23aU сначала образуется неустойчивый изотоп , который, испуская р “-частицу, переходит в элемент 93 (нептуний). Этот процесс будет подробнее рассмотрен в следующей главе. Деление ядер изотопа 338 U происходит только в случае облучения быстрыми нейтронами. Напротив, ядра изотопа-28 “U делятся и при действии медленных нейтронов. Это имеет большое значение, так как в естественном уране всегда присутствует изотоп 235U (актиноуран), правда, в очень небольших количествах (0,706 вес.%).
Среди осколков, на которые делится ядро урана-235, обнаружены следующие-элементы: 35Вг; Кг; 37Kb; 38Sr; 39 Y; 42М0; 4зТс; ^Sb; 5STe; вз1; 54X0; 5$Сз; sgBa; sjLa; ggCe; 59Pr; 6ON<1; ejPm; «sSm; 6gEu Для большинства из них параллельно образуются различные неустойчивые изотопы. Вероятно, лишь некоторые из этих неустойчивых ядер являются непосредственными осколками ядра урана, например;
Э2и + ол=5в^а + звК-г и aaU-l-en^^^r-j-^Xe. (13)
Большинство остальных изотопов образуются при вторичных ядерных реакциях из1 первичных осколков, например:
р- р“ Р“
к4Хе —---> S5Cs —--> 5вВа ——----> етЬа ~~—S8Ce (уст.). (14)
04 15 мин 33 мин 00 300 час °' Зв час 50 « ' v ’
Эти ядерныо превращения, сопровождающиеся испусканием р “-частиц, пе наменяют массового числа осколков. Изменяется лишь их химический характер. В общем при делении урана под действием медленных нейтронов образуется не менее 34 различных элементов. Массовые числа отдельных изотопов, как видно из рис. 85, лежат в области 72—162. На рис. 85 [взятом из работы Зигеля (Siegel, J. Am. Chem. Soc., 68, 2411 (1946)] приведена зависимость экспериментально установленных выходов отдельных продуктов деления от массового числа. Лишь в очень редких случаях ядро урана-235 делится на два приблизительно равные по массе осколка. Массовые числа подавляющего большинства осколков лежат в интервале 95—139. Следовательно, деление, кат; правило, происходит несимметрично — один из осколков имеет относительно мепыпую массу, другой — большую.
Первое время предполагали, что продукты превращения урана всегда имеют-больший заряд ядра, чем уран. Поэтому сначала их принимали за «трансурановые эле-ментт.ы (экарсний, экаосмий, экаиридий и т. д.) (Fermi, 1934, Hahn, Meitner, 1935 и сл.), Однако более подробное химическое исследование показало, что большинство-
640
Глава 13
этих элементов имеет не большие, а меныцие, чем уран, порядковые номера. Например, оказалось, что элемент, принимаемый ранее за «экаплатину», химически ('идентичен иоду. Это полученное химическим путем Доказательство того, что ядра наиболее тяжелых элементов могут распадаться на крупные осколки, сыграло большую роль в развитии ядерной физики. Вскоре стало понятно, что это явление открывает возможность для использования атомной энергий, и эта возможность очень скоро была осуществлена.
Деление тяжелых атомных ядер, и особенно ядра урана-235, имеет фундаментальное значение для использования атомной энергии, так как
в этом процессе освобождаются избыточные нейтроны (что в свою очередь объясняется тем, что в ядрах тяжелых элементов отношение числа нейтронов к числу прогонов больше, чем в ядрах элементов средней массы, которые образуются при делении). Каждый из нейтронов, освобождающихся при делении ядра урана, при подходящих условиях может вызвать деление еще одного ядра урана-235 с одновременным образованием еще одного нейтрона. Если принять меры, предотвращающие присутствие посторонних элементов (которые могут захватывать нейтроны) и использовать достаточно большое количество урана, чтобы не происходило утечки большого числа нейтронов, а напротив, подавляющее большинство из них сталкивалось бы с ядрами урана-235, вызывая их деление, то число
Маконе число
Рис. 85. Кривая распределения но массам выхода продуктов деления урана-235.
нейтронов будет непрерывно возрастать, и при этом (если процесс не тормозить) с большой скоростью (атомная бомба). Если же этот процесс соответствующим образом тормозить, то его можно использовать для получения энергии (атомная энергетическая установка). Этот процесс приводит к взрыву, если, например, оп происходит в чистом уране-235, при условии, что количество последнего достаточно велико, чтобы предотвратить утечку нейтронов. Если же он происходит в естественном уране, который наряду с ураном-238 содержит только 0,7% урана-235, то часть нейтронов расходуется на образование: плутония (этот элемент образуется из у рана-238 через S33U и 233Np; см. стр. 665). В этом случае процесс легко провести так, чтобы число нейтронов це превышало некоторой максимальной величины. При этом условии он будет протекать непрерывно. В этой форме процесс деления урана сначала был технически использован для получения плутония. Позднее перешли к тому, чтобы
его использовать для получения энергии. Количество энергии, которое освобождается при делении урапа-235, составляет около 200 Мэе! атом. следовательно, около 4,6-103 ккал!г-атом. Приблизительно 11% этой
энергии выделяется при радиоактивном распаде продуктов деления. В атомных реакторах (атомных «котиах»), кроме того, энергия выделяется при образовании плутония из урана-238. В настоящее время практичвезн
Искусственные превращения атомов
641
получаемая энергия составляет около 6-10я ккал!кг разделившегося ура-па-235. Эта энергия равна теплоте сгорания примерно 800 т каменного угля. '
Деление плутония (точнее плутония-239) происходит совершенно аналогично делению урана. Одна из атомных бомб, примененных в конце последней войны, содержала уран-235, другая — плутоний. Плутоний проще, чем уран-235, можно получить, чистым в больших количествах, так как разделение изотопов представляет особенно большие трудности и проведение этого процесса в больших технических масштабах требует огромных средств.
Делиться могут также торий и протактиний. Но делятся они, как и уран-238, только под влиянием быстрых нейтронов, причем таких, энергия которых равна по меньшей мере 1 Afae. Для процессов, па которых основано получение плутония и связанное с дим использование атомной энергии, по ряду определенных причин быстрые нейтроны пе пригодны (ем, стр. 665 и сл.). Деление ядер может быть вызвано бомбардирующими частицами с высокими энергиями — протонами, дейтронами или сеча стица ми > у-Л учи высокой энергии также способны вызвать деление ядер. Частицы с особо высокими энергиями могут вызвать деление атомных ядер висмута, свинца, таллия, ртути, золота, платины и таптала. Однакб чем меньше порядковый номер элемента, тем больше должна быть энергия бомбардирующих частиц, чтобы произошло деление ядра. Для деления тантала используют а-частнцы с энергией 400 Мае, а для деления ядер элементов с порядковыми номерами меньше, чем у тантала, даже такой огромной энергии бомбардирующих частиц недостаточно. В дальнейшем будет показано, почему это так. Одновременно будет дано объяснение тому, что существует лишь ограниченное число ядер, которые делятся под действием, пе только быстрых, но и медленных нейтронов.
Расчет энергии деления ядер. То, что при делении тяжелого атомного ядра на два ядра средней массы происходит выделение энергии, непосредственно следует, согласно принципу эквивалентности, из связанных с делением потерь массы. Что масса должна уменьшаться, видно из рис. 77. Размер такого уменьшения зависит, конечно, от того, какие изотопы образуются при делении ядра. Среди продуктов деления урана-235 под действием медленных нейтронов, как уже указывалось, чаще всего встречаются атомы, массовые числа которых равны 95 и 139., Сумма этих массовых чисел равна 234, а не 236, так как на каждый нейтрон, сначала захватываемый ядром 235U, при делении его на два названных осколка испускаются два нейтрона. В данном случае устойчивыми ядрами, образующимися при р “-распаде первичных осколков с массовыми числами 95 и 193, являются 35Мо и ly9La. Если подставить значения масс в уравнение
гз5и+1я==^Мо-Н3!,Ьа-)-21щ то получим
235,118 4-1,009=94,9394-138,955 4-2x1,009 +ДМ,
Отсюда потеря массы составляет 0,215 единицы массы, или 215 ТЕМ. Это соответствует энергии 215 X 0,931 = 200 Мэе (см. стр. 609).
Практически та же величина была получена экспериментально. Была измерена кинетическая энергия образующихся при делении ядра частиц и у-из л учения. При этом получены значения энергии от 174 до 182 Мэв. Эти величины относятся к энергии, освобождающейся непосредственно при делении. Последующий радиоактивный распад осколков даст дополнительную энергию около 21 Мзв. Следовательно, экспериментально найденная суммарная энергия (от 195 до 203 Мэв) хорошо согласуется с расчетной.
При вычислении энергии деления учитывалось только образование 95мо и 1з9Ца, т. е. образование только двух различных продуктов. Однако экспериментально найденные величины относятся к образованию большого числа разнообразных продуктов деления, как это и происходит в действительности. Но это не приводит к существенным 41 Г. Реми
642
Глава 13
различиям в энергии, выделяющейся при делении. Это станет очевидным, если вычислить анергии деления в общей форме, что можно осуществить следующим образцах совершенно отвлечемся от того, какие образуются отдельные продукты деление; а будем исходить только из экспериментального факта, что массовые числа осколкз деления лежат в интервале 72 и 162 (см. рис. 85), а массовые числа более легкого и более тяжелого осколков в сумме равны 234 (или близкой к этому числу величине^. Из рис, 77 можно заключить, что в ядре с массовым числом 236, которое, прежде чес произойдет деление, образуется из ядра урапа-235 при захвате им нейтрона, в среднем на каждый нуклон приходится дефект массы в 8,05 ТЕМ. В той области, к которой относятся массовые числа продуктов деления, т. е. области массовых чисел 72—162, как видно из рис. 77, на каждый нуклон приходится дефект массы в 9,05 ТЕМ. Теперь представим себе, что 2 ядра из этой области образовались из отдельных нуклонов. Если массовые числа этих ядер дополняют друг друга до 234, то для построения таких ядер потребуется 234 нуклона. Каждый из этих нуклонов при образовании из них обоих ядер теряет примерло 9,05 ТЕМ. Следовательно, в сумме образуется дефект массы в 9,05 X 234 = 2118 ТЕМ. При образовании же из отдельных нуклонов ядра гз“К теряется масса в 236 X 8,05 = 1900 ТЕМ. Разница (2118—1900 = 218) представляет собой среднюю потерю массы при- делении урана-235 под действием медленных нейтронов, а освобождающаяся при этом энергия равна 218 X 0,931 = 203 Мэе на атом Следов ат ел ьп о, при вызываемом медленными нейтронами делении ядер урапа-235 выделяется в среднем 200 Мэв на каждый атом урапа-235 независимо от того, какие конкретно продукты деления образуются в каждом отдельном случае. Единственное Допущение заключается в том, что массовые числа обоих продуктов деления всегда дополняют друг Друга до 234, т. е. при каждом делении освобождается в среднем 2 нейтрона. Для каждого последующего нейтрона, испускаемого в процессе деления, потеря массы в этом процессе уменьшается в среднем на 9,05 ТЕМ. Однако отсюда не следует, что энергия, освобождающаяся при делении ядер, для каждого дополнительного испускаемого нейтрона уменьшается примерно па 4%, Если нейтроны имеют возможность соединяться с другими атомными ядрами (а это, как правило, так и бывает), то освобождающаяся при этом энергия более или менее компенсирует недостаток анергии, подучающийся нри образовании продуктов деления. Энергия деления 200 Мав/атам соответствует электрической энергии ~23 ООО квпг-ч./s урана-235. При современном состоянии реакторной техники * в тепловых турбинах для получения электрического тока используют примерно 1/3 этой энергии. *
Теория деления ядер. Из того факта, что нри делении атомных ядер тяжелых элементов на ядра элементов среднем массы происходит потеря массы, а следовательно, выделяется энергия, следует, что ядра тяжелых элементов неустойчивы, или мета-стабильны. Но самопроизвольного деления мгновенно но происходит, поэтому при делении необходимо преодолеть некоторый потенциальный барьер точно так же, как мри радиоактивном распаде (см. стр. 631—632}, однако для деления этот барьер часто значительно выше. Ядро может преодолеть потенциальный барьер, если ему сообщить необходимую для этого «энергию активации» (см. стр, 743). У некоторых атомных ядер для этого достаточна энергия, которая выделяется при внедрении в ядро нейтрона. Эти атомные ядра делятся уже под действием медленных нейтронов. Ио обычно этой энергии недостаточно, так как требуется дополнительная энергия, которая и передается ядру быстрыми нейтронами в форме кинетической энергии.
Силы, которые удерживают нуклоны в ядре, формально можно сравнить с силами, действующими между молекулами жидкости. Точно так Ж6 как капли жидкости в нормальном состоянии имеют форму шара и поверхностное натяжение препятствует изменению этой формы, так и атомные ядра под действием ядерных сил принимают в нормальном состоянии форму шара и сопротивляются ее изменению. Если на каплю жидкости действует сила, растягивающая ее в некотором определенном направлении, то капля спачала принимает форму эллипсоида. Но капля стремится вернуть себе форму шара и начинает в этих условиях совершать деформационные колебания вокруг угол наиболее устойчивей формы. При атом форма капли все больше удаляется от шарообразной, т. е. эллипсоид тем более вытягивается, чем больше сила, вызывающая такие деформационные колебания капли. Однако растяжение эллипсоида не может происходить бесконечно. Как только достигнута известная степень растяжения в середине эллипсоида появляется перемычка, становящаяся все более отчетливой, пока капля пе разорвется посередине. Так образуются две капли, каждая из которых вновь приобретает форму шара. Аналогично ведет себя атомное ядро, которое благодаря поглощенной энергии начинает совершать деформационные колебания. Л. Мейтнер
* О ядерных реакторах см. стр. 647.
Искусственные превращения атомов
643
и Фриту удалось исходя иа этих представлений сделать физически понятным процесс деления ядер непосредственно после того, как он был открыт Ганом и Штрассманном. Вскоре после атого количественная теория была разработана Бором и Уилером.
Различие между разрывом капли жидкости и делением ядра, как правило, заключается в том, что в первом случае процесс происходит с потреблением энергии, тогда как во втором — энергия выделяется *. Но, во всяком случае, оба процесса протекают совершенно аналогично до того момента, когда происходит преодоление потенциального барьера, т. е. начинает образовываться перемычка, В обоих случаях, пока не достигнуто это состояние, для деформации должна совершаться работа. Различие состоит только в том, что при разрыве капли жидкости энергия падает от вершины потенциального барьера до некоторого уровня, лежащего выше исходного энергетического уровня капли, тогда как при делении ядер конечный энергетический уровень, как правило, лежит ниже исходного.
Энергия, требующаяся для преодоления потенциального барьера, а следовательно, и работа, необходимая для такой деформации, при которой образуется перемычка, в случае капли жидкости может быть найдена из величины радиуса капли и поверхностного патяжения. В случае атомных ядер эту энергию можно найти из числа нуклонов в ядре и ядсрш.+х сил. Очевидно, ядро тем легче деформируется, чем слабее силы притяжения, действующие между нуклонами. Между нейтронами и протонами ядра действуют, как уже указывалось, силы притяжения, величина которых возрастает с увеличением числа нуклонов в ядре, т. е. с увеличением массового числа А. Но протоны вследствие их одноименного заряда отталкиваются, и сила этого отталкивания приблизительно пропорциональна Z2, где Z — общее число протонов в ядре, или заряд ядра (см. сто. 633). Из капельной модели ядра следует, что энергия активации для
Z2
деления уменьшается при увеличении отношения — . По вычислениям Бора и Уилера
7А
энергия активации уменьшается до нуля, когда величина —j- достигает 45. Это случится у элемента, заряд ядра которого будет равен примерно 120. Элемент с таким или большим порядковым покером будет не мета ста бил иным, как остальные тяжелые элементы, а абсолютно неустойчивым. Если бы удалось получить такой элемент, то он бы мгновенно (точнее, за время 10"12 сек) делился на ядра средней массы,
Z3
Для ядра 233 U величина —р равна 36,0; энергия активации для процесса деления
Z2
ядра23аС составляет 5,3 Мае. Для ядра 233U = 35,6 и энергия активации равна
Z2
5,9 Мае. Для Я’Р« отношение -j- равно 37,0, для 233и — 36,4, а для Та — около 29,5. Энергии активации для процессов деления 23ИРн н 233U (об их значении для процессов деления ядер еще будет идти речь ниже), следовательно, меньше, чем для 23 3U. Напротив, энергия активации для деления тантала значительно больше, и этим объясняется то, что этот процесс происходит только под действием бомбардирующих частиц с чрезвычайно высокой Энергией (а-частицы с энергией 400 Мэе).
Но одной разницей в 0,6 Мае между энергиями активации 238U и 235U нельзя объяснить деления 23«U медленными нейтронами (например, нейтронами с энергией 0,025 ав), так как для деления 233 U требуются нейтроны с энергией по крайней мере 1,0 Мэе. Необходимо также учитывать разницу в энергиях, выделяющихся при внедрении нейтрона в ядро. При помощи уравнения (12), стр. 633 , можно рассчитать энергию образования ядер -3SU, 233 U,233 U и 239U. Разница в энергиях образования 233U и 286 U составляет 6,6 Мае, а разница энергий образования 23eIJ и 2SBU — 5,3 Мае. Если ядро 233U захватит нейтрон, то выделится, следовательно, энергия, равная 6,6 Мае. Эта энергия значительно превосходит анергию активации, требующуюся для деления ядра a35U (5,3 Мае), и в этом случае нет надобности в том, чтобы захватываемый нейтрон сообщал ядру еще какую-либо дополнительную энергию. Поэтому деление ядер 233и может вызываться медленными нейтронами. Если нейтрон захвачен ядром 233U, то выделяется энергия 5,3 Мае, но в этом случае энергия активации, требующаяся для деления ядра, составляет 5,9 Мае. Следовательно, ядру необходимо сообщить дополнительную энергию, т. е. деление может быть вызвано только быстрыми нейтронами, которые могут передать ядру недостающую энергию за счет своей кинетической энергии.
* Используя очень сильно ускоренные протоны, удалось вызвать деление и относительно легких ядер (например, -j- ip = з«С1 4- 2'А1 4- i«). Эти реакции деления эндотермичны.
41*
644
Глава 13
Разница в энергиях, выделяющихся в процессах 2j|‘U -р и 1 JJ51J и 2^U -J- п -> -*• ”’»U, обусловлена главным образом последним членом в уравнении (42). При превращении четно-нечетного ядра в четно-четное этот член положителен, при превращении четно-четпого ядра в четпо-нечетпое — отрицателен. Точно так же этот член положителен при внедрении нейтрона в нечетно-нечетное ядро (превращающееся в нечетно-четное) и отрицателен при захвате нейтрона нечетно-четным ядром. Этим объясняется то, что аналогично ®3SU медленными нейтронами Делятся два других четно-не четных ядра и 2^Рп, тогда как ядра 2^Ра и 3^Np, оба относящиеся к нечетночетным ядрам, делятся только под действием быстрых нейтронов. При этом энергия активации для ядра 2;,1Ра незначительно больше энергии активации для 2S7Np и даже меньше энергии активации для335U. (Для 23iPa отношение равно 35,9, дця 237Np —
36,6). Ядро 1Sp'h, относящееся к четно-четным ядрам, делится также только под действием быстрых нейтронов. Энергия, выделяющаяся при захвате нейтрона этим ядром, равна примерно 5,3 Иве, тогда как энергия активации для деления 2^Th еще больше, /а
чем для238 U (отношение — в случае 232Th равно 35,0), А
Спонтанное деление ядер. На стр. 632 было указано, что в соответствии с представлениями волновой механики существует определенная вероятность того, что а-частпца может проникнуть через потенциальный барьер, окружающий ядро. То же справедливо и для осколков, образующихся при делении ядер. Только вероятность того, что такая большая частица проникает через потенциальный барьер, значительно меньше, чем для а-частицы. Но поскольку она все же существует, то деление ядер, хотя и чрезвычайно редко, может происходить спонтанно (самопроизвольно). Уже говорилось, что спонтанное деление удалось наблюдать для урана вскоре после открытия искусственного деления под действием нейтронов. Впоследствии был открыт целый ряд других процессов спонтанного деления. Они протекают по тем же законам, что и радиоактивный распад, и можно, как и для последнего, определить ((периоды полураспада», т. е. время, требующееся для деления половины имеющегося количества атомов. В табл. 79 приведены значения периодов полураспада Для деления ряда атомных ядер (см. стр. 679, а также табл. 86 и 88).
Таблица 79
Периоды полураспада при споит айном делении ядер
Ядро 23t)Th гзать 2з1ра 2S2U 233Ц £3l$j 235U ЗЗВЦ
Период полураспада >i;5-10T7 1,4-1013 1,2-1016 3,6-Ю1- >3-10W 2-10W 1,9-iOH 8,!)-101э
Ядро a^Np £38Kp азбрн ззйрц 23Spu 241 Am 24ОС1Л 212Сщ 24 4 Cm
Период полураспада >4-1016 >5.1012-0,5-IO’ 2,45-101° 5,6-1015 1,24018,1,9401 7,2-103 i/(.107
Как установил Сиборг (1952), для четно-четных ядер существует линейное соотношение между периодом полураспада для спонтанного деления и отношением .
7,2
Экстраполяцией полученных чисел до значения —45 рассчитывают период полу-
распада, который приблизительно соответствует среднему времени жизни, теоретически предсказанному Бором п Уилером на основании капельной модели ядра.
Ядра е почетным числом нуклонов менее склонны к спонтанному делению, чем ядра с четным числом нуклонов. Например, период полураспада значительна больше, чем период полураспада 238 U. Следовательно, эакопомсрности спонтанного деления отличаются от закономерностей деления под действием нейтронов. Этого
Искусственные превращения атомов 645
можно было ожидать, так как спонтанное и искусственное деление представляют собой совершенно различные процессы. Например, прн искусственном делении ядра 235U из него вначале при захвате нейтрона образуется ядро 23eU (в сильно возбужденном состоянии), которое и делится, тогда как спонтанное деление 23aU происходит непо~ срвдственна с самим ядром 235 U.
Цепные ядерные реакции. Если при каждом делении ядра под действием 1 нейтрона образуются 2 нейтрона и условия таковы, что каждый из этих нейтронов вновь может вызвать деление ядра, то в системе, где происходит деление ядер, число нейтронов может очень быстро возрастать, так как в этих условиях при каждом делении число нейтронов возрастает вдвое: от 1 до 2, затем до 4, 8,16, 32 и т. п. После n-ного деления оно равно 21*. Так, получается, что после, например, 85-го деления в системе будет примерно 4 Юуа нейтронов. Этого количества достаточно, чтобы вызвать деление примерно 15 кг урана-235. При этом выделится около 3-1011 ккал тепла. Время, за которое произойдет 85 делений, составляет менее одной миллионной доли секунды. Следовательно, реакция протекает со взрывом, причем с громадной силой.
Реакции, которые подобно только что рассмотренной, характеризуются тем, что каждая пз них вызывает одну или несколько других реакций, называются цепными реакциями. Цепные реакции часто встречаются в химических процессах (см. гл. 17). В отличие от химических цепных реакций, цепные реакции, связанные с ядернымп превращениями, называются ядерными цепными реакциями. Среди них различают управляемые и неуправляемые. В приведенном выше примере речь шла о неуправляемой ядерной ценной реакции. Управляемая ядерная цепная реакция протекает тогда, когда условия выбирают так, чтобы из всех нейтронов, которые выделяются при делении ядра под действием 1 нейтрона, на дальнейшие деления использовался бы только один нейтрон. Если это так, то скорость протекания процесса остается постоянной. Как это практически достигается, будет рассмотрено в следующей главе при описании процессов получения плутония, для чего используется управляемая цепная ядерная реакция.
Когда происходит деление ядра 233 U под действием нейтрона, то часто освобождается больше 2 нейтронов. В среднем их число равно 2,5 на каждое деление, если оно вызвано медленными нейтронами. Прн делении 23ePu в среднем освобождается 3 нейтрона. Но пз этих трех нейтронов в среднем только 2 могут вызвать дальнейшее деление, так как в 1f3 случаев захвата нейтронов ядрами 233Ри происходит не деление, а реакция 23ЭРн + п = 240Рн + у. При захвате нейтронов ядрами 233U реакция 233 U -|- п = 23<iU + у происходит в г/0 случаев. Приведенные значения справедливы для деления под действием медленных нейтронов, но в данном случае можно не учитывать различий, наблюдающихся для быстрых нейтронов. Следовательно, практически дед о обстоит так, как будто при каждом акте деления ядер 233 U или 233 Р и освобождается 2 нейтрона и каждый из этих нейтронов способен вызвать дальнейшее деление.
Одиако должны быть выполнены два условия, чтобы в относительно небольшом количестве делящегося вещества действительно протекала цепная реакция: во-первых, в уране-235 или плутонии-239 не должно быть примесей, способных захватывать нейтроны, т. е< процесс должен проводиться с чистым ураном-235 или плутонием-239. Во-вторых, пз системы не должно теряться больше нейтронов, чем их образуется вновь в течение данного времени и может быть использовано для дальнейшего деления ядер. Число нейтронов, которые образуются в определенный момент времени за счет деления ядер, пропорционально количеству делящегося вещества. Для данного вещества опо, следовательно, пропорционально его объему. Число нейтронов, которые за это же время теряет система, пропорционально поверхности этой системы. Представим себе, что уран-235 пли нлутопий-239 изготовлены в форме шара, тогда число вновь образующихся нейтронов будет пропорционально третьей степени, а число теряемых нейтронов — второй степени радиуса такого шара. Следовательно, должно существовать определенное значение радиуса, при котором будут равны количества вновь обра
646
Глава 13
зующихся и теряемых системой нейтронов. Как только это значение будет превзойдено, число нейтронов в системе будет постоянно увеличиваться, причем с такой большой скоростью, что реакция тотчас же приведет к взрыву. Поэтому это значение радиуса называют критическим радиусом системы. В общем случае говорят о критической, величине системы, так как аналогичные соображения справедливы и для неигар о об ранных систем. Если система, в которой могут происходить деления ядер, не достигла своей критической величины, то реакции деления не только не приведут ко взрыву, но, если они не будут поддерживаться введением дополнительных нейтронов извне, могут очень быстро совершенно прекратиться, как только источник нейтронов будет удален (см. стр. 667 и ел.). Напротив, для шара, радиус которого больше критического, Достаточно одного-единственного нейтрона, чтобы вызвать цепную реакцию и тем самым взрыв. На этом основано использование урана-235 и плутония-239 в атомных бомбах.
Количество тепла, выделяющегося при делении 1 кг урана-235, составляет примерно 2-101° ккал (см. стр. 641 и сл.). Примерно столько же тепла выделяется при делении плутония. Если принять, что атомная бомба содержит 10 кг урана-235, то получается, что при полном делении этого урана выделится столько тепла, сколько выделяется при взрыве 200 000 m тринитротолуола. Уже небольшой части этой выделяющейся за очень короткое время энергии достаточно, чтобы произвести опустошительное действие. При взрыве такой бомбы развивается температура примерно в 10 млн. градусов, сравнимая с температурой внутри Солнца. Но разрушающее действие атомной бомбы только частично основано на внезапном сильном расширении газов, вызванном чрезвычайно высокой температурой, и образующейся при этом ударной волне, как это происходит и при обычных взрывах. Существенная часть обусловлена непосредственно чрезвычайно высоким световым давлением. Кроме того, разрушающее действие атомной бомбы связано и с очень сильным тепловым излучением, действующим даже на больших расстояниях от центра взрыва. К тому же атомная бомба обладает поражающим биологическим действием в связи с возникающим при ее взрыве проникающим гамма-излучением, а также вследствие долго сохраняющегося радиоактивного излучения продуктов деления.
Атомная бомба состоит в принципе из двух кусков урана-235 или плутония, каждый из которых не достигает критической величины, но при соединении их критическая величина превышается. Сначала куски отделены друг от друга толстым слоем парафина, который совершенно не пропускает нейтронов. Взрыв происходит при соединении кусков, и притом мгновенно, поскольку нейтроны (одного-едннственно-го из которых достаточно, чтобы вызвать полную цепную реакцию) всегда присутствуют благодаря действию космического излучения. Чтобы взрыв не произошел преждевременно, соединение кусков надо произвести чрезвычайно быстро. Это достигается тем, что один кусок «выстреливается» в другой при помощи своего рода пушки. Но несмотря на это в реакцию вступает только часть «взрывчатого вещества», так как выделение тепла происходит настолько быстро и в таком большом количестве, что металл испаряется и его атомы рассеиваются, прежде чем значительная часть их вступит в реакцию деления. Однако и в том случае, если произойдет деление только одного процента делящегося вещества атомной бомбы, действие ее будет чудовищным по своей разрушительной силе, как это видно из приведенных выше данных. Рассеянно атомов при взрыве можпо несколько замедлить, если окружить бомбу оболочкой из тяжелого металла. Атомы этого металла вследствие своей инерции будут оказывать известное сопротивление расширению окружаемого ими взрывчатого вещества. Кроме того, они отражают нейтроны, попадающие на оболочку. Оба эти эффекта приводят к тому, что доля атомных ядер, подвергающихся делению, возрастает.
Под воздействием очень высоких температур, развивающихся при взрыве урановой или плутониевой атомной бомбы, могут непосредственно произойти ядерные реакции, например реакции типа
зТ-рТ^Не4-2n-f-11,4 Mss (15)
и
зТ-рО=Ч1е-|-п-|-17,6 Мое. (16)
В этих реакциях на каждую единицу веса реагирующего вещества выделяется значительно больше энергии, чем прп делении урана-235 или плутония. Так, энергия, выделяющаяся при реакция (15), превосходит энергию, выделяющуюся прп делении, более чем вдвое, а при реакции (16) — более чем в четыре раза. Эти реакции исноль-зуют в водородной бомбе. Необходимый для этого тритий Т получают облучением нейтронами лития в ядерном реакторе (см. ниже). При этом литий, содержащий 7,3% в Li, реагирует по уравнению 6 Li + п = 4 Не -f- Т. Чтобы наполнить бомбу тритием или смесью трития и дейтерия, эти вещества сжижают при охлаждении. Они должны
Искусственные превращения атомов
647
сохраняться при низкой температуре до момента взрыва. Возникающие при этом трудности преодолевают тем, что вместо Элементарных трития и дейтерия используют эти изотопы в форме гидридов легких Металлов. Поскольку тритий радиоактивно распадается (период полураспада 12,46 года), его можно хранить ограниченное время, «Запаливание» водородпой бомбы осуществляется взрывом плутония. Следовательно, н водородной бомбе в качестве взрывчатых веществ объединены плутоний и водород (последний в форме Т и О).
Нейтроны, выделяющиеся при делении ядер, имеют кинетическую энергию больше 1 Мэв, Следовательно, они способны вызывать деление не только ядер 235U Или езорц, ио и расщеплять ядра 23sU. Тем пе менее в обычном уране цепная реакция описанного выше типа нс происходит по той причине, что большинство быстрых- нейтралов при столкновении с ядрами 238 U пе вызывают реакции деления, а только отражаются или рассеиваются. При этом нейтрон, как правило, передает большую часть своей энергии ядру урана, которое, возбуждаясь, испускает у-л учи, а замедленные нейтроны уже не способны вызвать деление ядер 238 U. И только при соблюдении определенных условий они оказываются способными вызвать деление ядер 238 U. Об этом речь будет идти в следующей главе.
Ядерные реакторы. Для использования атомной энергии в энергетических установках, для получения плутония и в других целях неуправляемые цсппые реакции непригодны. Следует использовать управляемые цепные реакции, т. е. реакции, протекающие с постоянной скоростью. Устройства, в которых протекают такие управляемые цепные реакции, называются ядерными реакторами или атомными котлами.
Каждый ядерпый реактор (атомный котел) для своей работы ну издается в так называемом ядерном горючем, т. е. и веществе, в котором может протекать цепная ядерная реакция. Это вещество одновременно служит источником энергии. Обычно ядерный реактор содержит также питающее вещество, т. е. такое вещество, которое само по себе не способно поддерживать цепную ядеряую реакцию, но может превращаться в реакторе в такое вещество, которое способно вступать в цепную реакцию, т. е. в ядерное горючее. «Горючее» и «питающее» вещества вместе образуют рабочее вещество реактора. Обычный урап содержит 0,7% ядерного горючего (236U), а остальное имеет характер «ядерного питающего вещества»: при работе уранового реактора наряду с расходованием 235U как ядерного горючего часть 233U превращается в плутоний (23эРи), который в свою очередь может служить ядерпым горючим.
Разновидностью атомов, которая, возможно, в будущем получит важное значение как ядерноегорючее, является изотоп урана 233U. Эти ядра, как и ядра 235U и азвРи,- м* под действием медленных нейтронов делятся и могут быть получены из тория (который в природе значительно более распространен, чем уран) по ядерной реакции
, 0* . ₽-
si’2Th-|-Jn=a^Th ----> 233ра ------(17)
40 10 23 мин 41 27,4 дн 42 v '
Уран-233 является а-излучате.чем с периодом полураспада 163 000 лет. В настоящее время его уже получают в технических масштабах, окружая урановый реактор оболочкой из тория. Медленные нейтроны, испускаемые урановым реактором, поглощаются торием, являющимся чистым элементом с массовым числом 232, переводя его в радиоактивный торий-233, из которого через протактинии-233 образуется изотоп урана-233.
В зависимости от целей, для которых они используются, ядерпые реакторы делят па реакторы для получения плутония (так называемые промышленные реакторы), энергетические реакторы и исследовательские реакторы. Промышленные реакторы служат главным образом для получения чистых делящихся веществ, например плутония. Они будут рассмотрены в следующей главе. Энергетические реакторы служат в первую очередь для использования атомной энергии в тепловых машинах, напри
648
Глава 13
мер в паровых турбинах. Их устройство приспособлено для этих целей, но принцип действия совершенно аналогичен принципу действия промышленных реакторов. Исследовательские реакторы, как видно из .самого названия, служат для целей исследования, в первую очередь для изучения свойств нейтронов и их действия. Используя уран, сильно обогащенный изотопом “35С!, или даже чистый плутоний, эти реакторы можно сделать очень небольших размеров. Мощность большинства исследовательских реакторов невелика (часто менее 1 кюп), но, как правило, они сконструированы так, что дают особо интенсивные потоки нейтронов. Поэтому они позволяют получать препараты, сильно обогащенные искусственно актин про и энными атомами. Установки для получения искусственно активированных элементов часто сооружаются и на других ядерных реакторах (см. стр. 670).
Возможность использования продуктов Зеленая (образующихся в больших масштабах прп работе ядерных реакторов) в качестве источников излучения также имеет хозяйственное- значение. В будущем, вероятно, большое значение приобретет использование возникающих при делении ядер лучей- отдачи непосредственно для проведения химических реакций ч технических масштабах. Лучи отдачи, например, настолько сильно активируют молекулы N2 и О-,, что они уже при обычных температурах реагируют с образованием NO. Трудности, связанные с очень небольшим пробегом лучей отдачи (см. стр. 552), как показал Харток (Harteck, 1960), можпо преодолеть, используя ядернЫЙ реактор особой конструкции.
Техническое пол у чеппе урана-235. Для технического получения чистого урана-235 в период последней мировой войны в США были разработаны два промышленных метода; метод калутрона и диффузионный метод. Налутрон представляет собой особый тип масс-спектрографа. Однако принцип его работы отличен от принципа работы обычных масс-спектрографов и подобен принципу работы циклотропа. Первое устройство конструкции Лоуренса (декабрь 1941) давало в час примерно 1 мг чистого 235U. Осенью 1943 г. получение урана-235 на клаутроне началось на большом эаводе (Окридж близ Клинтона, шт. Теннесси). На той же площади (180 к.цЗ) позднее был построен завод, который работал на диффузионном принципе на ик'в (5000 ступеней разделения, 6000 центробежных насосов, несколько гектаров диффузионной поверхности). Постепенно общая мощность обоих заводов достигла 3 кг сзьц в день. В Германии Хартек и Грот (1942) с успехом применяли метод, в котором разделение обоих изотопов урана проводили центрифугированием UFe . Вследствие трудностей военного времени было изготовлено лишь несколько таких центрифуг, да и те работали очень недолго. В настоящее время получение урана-235 для атомных бомб потеряло свое значение, так как для этих целей столь же пригоден и легко получаемый в чистом виде плутоний. Однако методы, разработанные для технического получения, урана-235', интересны потому, что в упрощенном впде их можно использовать для обогащения урана изотопом -ззц. Получение довольно сильно обогащенного урана далеко не так дорого, как получение совершенно чистого урана-235. Благодаря применению в качестве ядерного горючего урана, значительно обогащенного ураном-235, можно значительно уменьшить размеры ядерных реакторов. Это имеет большое значение, так как только при этом условии можпо перечти к использованию атомной энергии не только в стационарных, но и подвижных установках.
Глава 14
ТРАНСУРАНОВЫЕ ЭЛЕМЕНТЫ
Пагяд-Ки 1'ЫЙ Элемент Символ Атомный вес валентность
93 Нептуний Np 237 II, III, IV, V, VI
94 Плутонии Ри 242 II, III, IV, v, VI
95 Америции Am 243 II, III, IV, V, VI
96 Кюрий Cm 247 III
97 Берклии Bk 2-17 III, IV
98 Калифорний Cf 251 III
99 Эйнштейнии Fs 254 III
100 Фермий Ftn 253 III
101 Менделевий Nd 256 III
102 Нобелий Ko 255 III
Приведенные здесь атомные веса каждого элемента представляют собой массовое число наиболее долгоживущего из известных в настоящее время изотопов.
Общие сведения. Все трансурановые элементы, т. е, элементы, порядковые номера которых больше’ порядкового номера урана, получены искусственно — в результате атомных превращений. Два из них, нептуний и плутонии, встречаются, как будет показано позднее, и в природе (в урановых рудах), но в таких небольших количествах, что это совершенно не имеет значения для их получения. Поэтому, как и остальные трансурановые элементы, нептуний и плутоний практически получают только-искусственно при помощи ядерных реакций.
Все трансурановые элементы неустойчивы. Для большинства из них известно значительное число изотонов. В табл. 80 сопоставлены некоторые из них *. В таблицу включены также важнейшие, способы получения и важнейшие радиоактивные свойства отдельных изотопов. Среди них нет ни одного, который бы нс распадался радиоактивно, а также ни одного, продолжительность жизни которого была бы близка к продолжительности жизни последнего из встречающихся в природе в больших количествах элементов — урана.
Важнейшим элементом в ряду трансурановых элементов является плутоний. Его наиболее долгоживущий изотоп (M3Pu) имеет период полураспада около 4-10й лет. Поскольку возраст Земли более чем в тысячу
* Большее число изотопов трапсураноных элементов приведено в табл, 86—89.
Изотопы трапе урановых элементов
Таблица ST)
Элемент Изотоп Период полураспада Вид распада Знерзия излучения, ЛГэв Реакция образовался
Нептуний S3 »»Np iMNp a30Np a36Np* 2EeNp sa,Np M8Np wsNp 53 мин 4,4 дн 410 дн 22 час ~ б-КРлет 2,25* 10е лет 2,10 дн 2,35 дн ‘к / ‘ЗЗ’Л / । 8 к к Х/1 ’ч/* Х/1 а 6,28 у 1,0 3+ 0,8 а 5,06 Z/J" 0,52; 0,48 4,87; 4,79; 4,77; 4,52; р~ 1,25; 0,28 0,25 у 1,1 0,64; 0,43; 0,33; 0,21 «6U (d,6n); 13SU(d,9c 335 U (р, 2и); »“U (d, 3n) идр IMU (d,2n); mU (а, р Зп) идр. »«U (d,n);“sU(d.4n); ISTNp (n, 2п) и др. tMU (d, 4n) и др s3’U-*^-;M1Am—а >38U (d,2n); a3SU (a, p); i3’Np (n, y; J3»U a»U (d,n)
Плутоний 94 mpu 334 Pu >3aPu 387 Рц 138рц ИВрц a«Pu M1Pu MaPu марц wpu a«Pu 36 мин 8,5 час 2,85 лет 45,6 дн 86,4 лет 2,436 • 10е лет 6,58 - IO* лет 13,0 лет 3,73-10s лет 4,98 час 10,1 час 10,85 дн <* *се <'Y X а <К а а а а а 0- 0- 6,58 а 6,19 5,76; 5,72; 5,61; 5,45 <х 5,65; 5,36 5,49; 5,45; 5,35; 4,61 5,15; 5,13; 4,66 5,16; 5,12; 4,85 0- 0,0205 а 4,893; 4,848 4,90; 4,854 0,58; 0,49; 0,37 0,33; 0,15 S36U (a, 7n) S39U (a,3n); ®JSU (a, 5n); >38Cm -* a S36Np-p'; '«Cm-.j; SS8U (a,3n)udp «Ст-ка; MDU (a, 2n); ®MU (a, 5n) и dp. M8Np-48-; MBU (a, 4n); ®3’Pu (n, у) c & 1MNp-*/3"; ®43Cm-» a; i3eU (a, 3n) ~U(«, 2n); Е«Сгп->я MeU (a, n); S4°Pu (n, y} B41Pu (n,y) M1Pu (n,y) B14Pu (n, y) ®«Pu (n, y)
4
Продолжение табл. 80
Элемент Изотоп Период полураспада Вид распада. Энергия излучения, Мэв Реакция образования
Америции аз’Ат 1,3 час /К а 6,01 ”»Pu (d, 4n); MTPu(d,2n)
95 338Ат 1,86 час К 33BPu (d, 2n)
-зэ Am 12 час Ч у 0,285 а 5,77 **эВк-«-а; 339Pu(p,n), e3BPu(d,2n);33’Np(a,2n) и dp
840Am 2,1 дн К У 1,3 M’Pu (d, n); M’Np (a, n)
341 Am 458 лет а 5,54; 5,476; «‘Pu—0', ’“Cin-b/T,
/Г 5,433 84SBt->a
MaAm* 16,01 час 0- 0,667, 0,625 «‘Am (n, y)
MSAm 100 лет 6' 0,585 “’Am (n, y)
348 Am 7,95 103лт а 5,34; 5,27; ’«Am (n.y); «8Pu->3
5,22
Л-
144 Am 26 мин 3' 1.5 “Am (n, y)
246 Am 25 мин 0~ 2,41; 2,29; 2,10; 1,60; 1,35 348Pu->/3"; S4iAm (n, y)
у 1,07
Кюрии ’«Cm 26,8 дн а 6,25; 6,21 «’Pu (a,3n); 4<4Cf-a
96 «‘Cm 35 дн z^ а 5,95; 5,89 MBPu (a, 2n)
a4aCm 162,5 дн а 6,110; 6,066 29aPu (a,n), *4’Am-*/?', *4®Cf-*a
343 Cm 35 лет с а 5,99; 5,78 5,73; 5,58 ’«Cm (п, у), «»Вк—A
a44Cm 19 лет а 5,80; 5,76; «‘Am (a, p); !44Am-*0’,
5,52 848Cf->a
345Cm 1,4 • 104лет а 5,45; 5,36 ’«Bt-KiT; 349Cf-a. B44Cm (n, y)
=4eCm 5 10э лет а 5,37 ’«Am-+^-; i6<lCf->a, S4SCm (n, y)
’“’Cm 4—9 10’ лет а — 83BPu (8n,y20-)
берклий 97 a48Bk 4,5 час ск ча а 6,72; 6,55; 6,20 141 Am (a,2n)
a«Bk 4,95 дн а 6,33; 6,15; M4Cm (d, n); ’«Cm (a, pl
ч 5,90
34’Bk 7 10я лет а — 34’«-►A; 3S1Es->a
3“>Bk 290 дн Z4" Д- 0,10 353Ee-+a
Ч а 5,40; 5,08
»«Bk 3,13 час /Г 1,80 ос 5,54 MSBk (n, y)
Чс у 0,9
Продолжение табл. 80
Элемент Изотоп Период полураспада Вид распада Энергия излечения, Мэе Реакция образования
Калифорнии a«Cf 25 мин а 7,15 !«Сш (а, 2n); mU(12C, 6п).
98 *к а 7,11 «8и (MN, р 7п)
2«Cf 44 мин < *а 6,75; 6.71; MSCm (а. п);244 Ст (а. Зп)
«*Cf 35,7 час а 6,47 «4Ст («, 2n); “’U (1гС, 4п).
6,26 35|Тш^а;
350 дн а 6,19; 5,82; S38U (14N, рЗп); 245Ст(а,1Ц
®‘>Cf 360 лет а 5,57 24 9 В к
250Cf 9,3 лет сс 6,15; 6,02; я»Вк->^, aieCf (п,у>
5,98 25->Ет->-а
asiQf 660 лет а — ssepu (12П.У4/Г);
6,12; 6,07; 23SU (13п, У 6 0")
««Of 2,2 лет а 5,97 MlCf (п, у)
asBQf 19 дн 0 0,27 a53Cf (n, у)
Эйнштейний a46Es 7,35 мин И а 7,35 U8U (14N, 6n)
99 *а
E4SEs 25 мин а 6,87 24SCf (d, 3n)
Чх
249Ee 2 час а 6,76 2«Bk (a, in)
з&оЕе 8 час К 24»Bk(oi,3n);2S,)Ein-*A: udp.
2S3Es 20,03 дн а 6,636; 6,59; 2a3Cf^0- udp.
6,24
я- 0~ 1.04;
254Ез* 38,5 час Ст 0,389;. 253Es (ii, у) и dp.
у 0,680
284Es 280 а 6,42 ”3Es (n, y) u dp: Ifr
2S5Es 40 дн г — a64Es (n, у) и bp.
Фермий !5“Fm 30 мин а 7,43 2ISU'(160,4n);
100 „К 2«Cm(12C, <x2n)
asitfm 7 час < а 6,89 31eCf (a, 2n)
!5!Fm
4,5 дн с *а а 6,94 3S2Cf (<x, 3ii) 239Pu (15n, у 60);
aslFm
3,24 час а 7,20; 7,16, 338U (16n,y 80);
7,06 254Es*->0
!lsFm 21,5 час а 7,03 25iEs->0_ и dp.
Менделевий 101 25-Md. ~ 30 мин к , — 2MEs (a, n)
Нобелий 264No 3 сек а — 24eCm (lflC, 4n);
102 Z55No? ~ 10 мин а — 24eCm (1SC, 5n) 21,1Cm(13C.4n);8,1Pu(leO.2n)
Трансурановые: элементы 653
раз превышает эту величину, то даже если бы плутоний первоначально присутствовал в ней, он пе мог бы сохраниться до настоящего времени. Плутоний, содержащийся в урановых рудах, должен быть вторичного происхождения. При этом речь идет не о наиболее долгоживущем изотопе, а об изотопе 239Ри. Его образование в природе происходит, вероятно; тем Же путем, что и получение плутония (й39Ри) в настоящее время в технике,
Требующиеся для этого нейтроны могут образовываться в результате спонтанного деления урана. Несмотря на то что такое самопроизвольное деление происходит чрезвычайно медлепно, все же оно было открыто вскоре после открытия деления урана под действием нейтронов. Впервые спонтанное деление наблюдали в 1940 г. Флеров и Петржак. Эти исследователи нашли для него период полураспада примерно 1014 лет. По новейшим измерениям, точное значение периода полураспада для спонтанного деления урана составляет 0,80- 10м лет, следовательно, примерно на миллион атомов урана, распадающихся с испусканием а-частиц, приходится один, который делится на две приблизительно равные части. Отсюда в 1 г обычного урана в среднем происходит одно спонтанное деление ядра в 1 мин, что соответствует выделению в среднем двух нейтронов в минуту. Другая возможность появления нейтронов в урановых рудах заключается во взаимодействии а-частиц, испускаемых ураном и продуктами его распада, с легкими элементами, содержащимися в урановых рудах.
Трансурановые элементы представляют собой особую группу не только потому, что все они являются неустойчивыми, получаемыми лишь искусственно элементами, но и в силу их химических свойств. Трансурановые элементы не являются аналогами рения и платиновых металлов. Они не осаждаются, как последние, сероводородом из кислых растворов; нептуний и плутоний пе образуют летучих окислов, характерных для рения и осмия. Особое положение трансурановых элементов в периодической системе вызвано теми же причинами, что и особое положение лантанидов. Оно объясняется строением их атомов. Точно так же как в случае лантанидов происходит заполнение 4f-уровня, в атомах трансурановых элементов происходит заполнение 5 f-уровня. Отсюда попятно, почему трансурановые элементы по мере увеличения их порядкового номера по своему химическому поведению все более напоминают лантаниды.
То, что первые три трансурановых элемента — нептуний, плутоний н америций — в отличие от лаптаппдов могут существовать в высшем со стоянию о кис ления, а имеппо шестивалентном, объясняется менее прочной связью 5/-электронов в атомах трансурановых элементов но сравнению со связью 4/-электронов в атомах лантанидов. Положение совершенно аналогично положению для 3d-, 4d- и 5 d-электро нов в атомах переходных металлов. Уменьшение силы связи d-электронов с увеличением главного квантового числа приводит к тому, что у переходных металлов способность существовать в высшем валентном состоянии, согласно известному правилу, в каждой группе увеличивается в направлении сверху вниз. Этого же следует ожидать и при сравнении трансурановых элементов с лантанидами, но крайней мере для первых членов ряда трансуранов.
Еще окончательно не решен вопрос о том, начинается ли заполнение 5/-уровня уже в следующем за актинием элементе, тории (Seaborg, 1949), или только начиная с нептуния (Dawsi)n, 1952). Но, по-видимому^ можно считать твердо установленным (особенно на основании магнитных измерении), что даже если заполнение 5/-уровня начинается с нептуния, то уже у следующего за ним элемента, плутония, по крайней мере пять электронов относится к 5/-уровпю. Начина яс плутония число электронов, находящихся на 5/-уровне, регулярно возрастает с увеличением порядкового номера элемента.
То, что у нептуния или плутония имеется но пять электронов на 5/-уровпе, тогда как предыдущие элементы не имеют ни одного электрона, относящегося к этому уровню, становится понятным, если предположить, что при увеличении заряда ядра сила связи 5/-электронов возрастает скорее, чем сила связи 6d-электронов. В таком случае кривые, отражающие зависимость силы связи электронов от заряда ядра, должны иметь такой вид, который схематически показан па рис. 86. На рисунке эти кривые 'пересекаются между ураном и неитупиом. Если исходить из этих кривых, то у эле
654
Глава lx
ментов, предшествующих нептунию, бй-уровень лежит пиже, чем 5/-уровень, а у элементов, следующих за нептунием, 6<1-уровень выше, чем 5/-уровень. Если это тан, то в атомах первых -элементов электроны должны располагаться яа fid-уровне, а в атомах последних — па 5/-уровне.
Существенное различие во взглядах Сиборга и Доусона заключается только в истолковании электронного строения атомов элементов, предшествующих трансурановым. В отношении электронного строения атомов трансурановых элементов взгляды обоих исследователей s основном совпадают. Особенно это откосится к. кюрию, для которого оба автора принимают электронное строение 5/!6d7s2, что полностью соответствует электронному строению гадолнния 4/!5<i6s2. Отсюда можно заключить, что кюрий среди трансурановых элементов занимает такое же особое положение, как гадолиний среди лантанидов (см. стр. 514). Это подтверждается поведением предшествующих
Порядковый номер
Рис. 86. Устойчивость 5/- и бй-энергетпческих уровней в атомах элементов, следующих за актинием.
и особенно следующих за кюрием элементов. Следовательно, по расположению каждого элемента из группы трансурановых относительно элемента кюрия можно определить, какому из лантанидов оп наиболее близок:
е1Рш вг£тп и Eu в4<И вз/ГЬ 66Dy в7Но egEr вэТп 70Y‘b ?iLu
аз^Р B4^*u де Cm ajBk agCi ggEs mol in toiMd 102N0 (103).
У кюрия и следующих за ним трансурановых элементов очень отчетливо проявляется родство с соответствующими лантанидами. У нептуния, плутония и америция также наблюдается такое сходство, но оно проявляется только в тех соединениях^ в которых названные трансурановые элементы находятся в тех же валентных состог?-ниях, что и соответствующие лантаниды. В остальном сходство нептуния, плутония и америция с лантанидами несколько стирается вследствие способности этих элементов существовать в высших валентных состояниях. Прежде всего это относятся к нептунию и плутонию, которые по своей способности быть шестивалентными и свойствам соединений, в которых они шестивалентны, обнаруживают близкое родство со своим левым соседом по периодической системе — ураном. Впрочем, их склонность быть шестивалентными выражена не столь ярко, как у урана, а следующий за ними элемент — америций с трудом переходит в шестивалентное состояние. Его наиболее устойчивое состояние — треззвалентное, и в этом отношении он значительно ближе к европию, чем к у рапу.
Насколько близки трансурановые элементы и лантаниды (кроме сходства строения их электронных оболочек) и по валентностям, видно пз табл. 81. Для сравнения в таблице приведены также четыре элемента предшествующие трансурановым (вместе с лантаном и соответствующими лантанидами), а именно актиний, торий, протактиний и уран. То, что нептуний и плутоний, кроме сходства с лантанидами, обнаруживают близкое родство с ураном, отчетливо видно из приведенных в этой таблице данных.
Трансурановые элементы
655
Таблица 81
Строение атомов и валентности элементов ряда лантана и ряда актиния
Ряд лантанидов Ряд актинидов
элемент электронная кояфиздрация валент~ яостъ элемент электронная конфигурация валент-адояю T
по Сиборгу Л по Лоусону
67Ъа 5d С>з- ш 8вАс 6d 7* 6d 7a® IO
адСе 4) 3d б г? ni.IV soTh 5f 6d 7 s® fid® 7 s® n, ш,П
(йРг 4/s 6# III, IV, V ш «Ра 5/® 6d 7s® fid® 7s® V
«Nd 4/< 6а2 eiU Зр 6d 7s® fid4 7s® п,ш, IV, у, VI
ejPm 4f* 1П 5 1 ssNp 5/* Sd 7r 5P 7aig) п, Ш, IV, V, Vf
«Вт fis8 II, ш 1 „Ри 5p 6d 7 s® 5p6d 7s® II, in, IV v. VI
«Ей 4р 6 г II, ш со ь „Ат 5/’ 7 s® 5/6 fid 7 s® П, Ш, IV, V, VI
4f> 3d 6& ш 1 «Ст 5/’ 3d 7s® 5f fid 7 s® Ш
«ть 4Р 5d 6? HI.IV 1 5p 6d 7a® 5/6 fid 7s® Ш, IV
«Ру 4р 3d fis® ш 5P 6d 7a® 5/6 fid 7s® TO
а) В некоторых случаях Cnfiopr, кроме указанных, рассматряьает и другие электронные конфигурации, например вместо 5/-i3cf7.<2T
6) Для Np, по Доусону, возможна и конфигурация 6d^7s3.
Пэ международной номенклатуре элементы от 57 до 71 (от La до Lu) называют элементами ряда лантана», а элементы с 89 (Ас) до 103 образуют «ряд актиния». Элементы, в атомах которых происходит заполнение 5/-уровпя и которые вследствие этого очень похожи на актиний, называют актинидами по аналогии с лантанидами (от Се до Lu), в атомах которых происходит заполнение бру рое ня. Сиборг, который ввел: термин «актиниды», считает, как уже было отмечено, что заполнение 5/-уровня начинается уже с элемента, следующего непосредственно за актинием,— тория, подобно тому как заполнение 4/-уровня начинается непосредственно у следующего за лантаном элемента — церия. Если же исходить из представлений Доусона, основывающихся па наблюдениях магнитного поведения, то названия «актиниды» не будет соответствовать торию, протактинию и урапу. Группа актинидов в таком случае будет начинаться с нептуния. '
Поскольку еще однозначно не установлено, как распределены по уровням 5/-и 6rf-электропы в следующих за актинием элементах, в табл. 81 приведены электрон-' ные конфигурации элементов по Сиборгу [Nucleonics, 5, 16 (1949)) и Доусону [Nucleonics, 10, 39 (1952)]. Из химического поведения элементов нельзя сделать надежных выводов об их электронной конфигурации. Химическое поведение в первую Очередь определяется прочностью связи валентных электронов, а больше ли эта прочность связи для отдельных электронов на уровне 5/ или 6 д', не имеет существенного значения, так как в к а и: дом отдельном случае электроны в нормальном состоянии нейтрального атома находятся на наиболее пизких энергетических уровнях, т. е. в состояниях, где они прочнее всего связаны. Но вопрос о распределении электронов но уровням 5/ и 6d имеет значение для понимания многих свойств соединений трансурановых элементов и прежде всего их магнитных свойств. О них см. ниже.
В табл. 81 не включены полученные только недавно элементы с порядковыми номерами от 99 до 103, так как с ними ле проведено еще никаких исследований, на основании которых можпо было бы сделать выводы о распределении их электронов на уровнях 5/ и 6(7. По своему химическому поведению все эти элементы, вне всякого сомнения, являются аналогами лантанидов—Но, Ег, Ти и Yb.
Все трансурановые элементы могут быть положительно тредвалент-нымн. Это относится и к урану, но соединения урана(Ш) в водных растворах очень неустойчивы. Значительно устойчивее соединения непту-Ш1я(111). При дальнейшем увеличении порядкового номера элемента
656
Глава, 24
устойчивость его соединений, в которых он трехвалентен, быстро возрастает. Уже плутонии образует многочисленные соединения, в которых он трехв а лентой. Для америция, как уже было отмечено, трехвалентное состояние наиболее устойчиво. Кюрий и калифорний, насколько известно, вообще существуют только в трохвалентном состоянии*, так же как и их аналоги в ряду лантанидов — гадолиний и диспрозий. Все тригалогепиды МХ3 элементов ряда актинидов изоморфны друг другу и соответствующим тригалогенидам актиния. Все они изотипны трйгалогенидам редкоземельных металлов.
Как и уран, нептуний, плутоний и америций образуют соединения в четыреотзалснтном состоянии. У нептуния и плутония чётырехвалепт-ное состояние наиболее устойчиво. Соединения ypana(IV) в водном растворе легко переходят в соединения урана(У1), а соединения америция(1У)~— в соединения америция(Ш). Тетрахлориды названных трансурановых элементов, насколько известно, изоморфны между собой и UC14 и ThCI4.
Урану, нептунию, плутонию и америцию свойственно и пятмвалонт-ное состояние. Однако у всех этих элементов соединения, в которых они пятивалентны, по своей устойчивости уступают соединениям других валентных состояний. Наименее устойчивы они в случае америция.
Нептуний, плутоний и америций, как и уран, могут существовать в znecmwB ал ситном состоянии. Но если для урана шестивалентное состояние в растворе является наиболее устойчивым, в случае нептуния и плутония и в еще большей степени америция оно менее устойчиво, чем четырехва-ленТное. Для следующих за америцием трансурановых элементов вообще не известно соединений в шести- или пятивалентном состоянии. Соединения нептупила [ХрО21Х2 и пл утонил а [РпО2]Х2, соответствующие соединениям уранила [UO2JX2, имеют подобную кристаллическую структуру. Точно так же изотипны между собой двойные ацетаты NafUOJRs, Na[NpO2]R3, Na[PuO2]R2 и Ха[АтО21Нз> где R — ацетатный остаток.
В общем по химическим свойствам америций стоит ближе к элементам семейства лантана, чем к урану. Трансурановые элементы, следующие за америцием, полностью аналогичны лантанидам. Каждый из этих трансурановых элементов можпо рассматривать как аналог того элемента группы лантана, который стоит выше его, если кюрий расположить подзадо-лшпы (см. стр. 654). Следовательно, америций будет аналогом европия, берклий — тербия, калифорний — диспрозия. С известными оговорками нептунии и плутоний также можно рассматривать в качестве аналогов лантанидов прометия и самария.
В табл. 82 приведены рассчитанные из плотностей металлов атомные объемы и кажущиеся ионные радиусы отдельных элементов ряда актиния. Приведенные значения ионных радиусов основаны главным образом на данных Захариазена. Радиусы шестивалентных иопов найдены следующим образом: из рентгенометрически установленных расстоянии М «—>-0 вычтена величина 1,33 А (радиус иона кислорода). Данные таблицы показывают, что в ряду актиния с увеличением порядкового номера элемента ионные радиусы уменьшаются так же, как и в ряду лантана (см. стр. 575). Изменение атомных объемов в зависимости от порядкового номера также совершенно аналогично такому изменению для элементов ряда лантана. Так же как и европий (ср. рис. 5S, стр. 522) в ряду лантала, америций в ряду актиния имеет самый большой атомный объем.
Связь между элементами ряда актиния и лантана проявляется и в отношении их магнитных свойств. В табл. 83 указаны значения магнитной восприимчивости ионов обоих рядов. Указанные в этой таблице числа /-электронов даны в предположении, что пз всех внешних электронов наиболее прочно связаны /-электроны. Иными словами, принято, что, например, атом плутония с конфигурацией легче теря-
* См. примечание на стр. 679,— Прим, персе.
Трансурановые элементы.
657
Таблица 82
Плотности, атомпые объемы, атомные радиусы й полные радиусы актинидов
Элементы soTh ЧцРа ми saNp мРи ФбАш
Плотность 11,71 15,37 19,00 20,45 19,74 11,9
Атомный объем 19,8 15,2 12,5 11,6 12,11 20,5
Атомный радиус, А 1,82 1,61 1,48 1,31 1,56 1,82
Радиус иона М3+, А — — 1,04 1,02 1,01 1,00
Радиус попа М4+, А 1,10 1,06 0,89 0,88 0,86 0,85
Радиус иона М«+, А — 0,58 0,57 0,56 —
Таблица 83
Молярные магнитные восприимчивости Хмол ионов лантанидов п актинидов в водном растворе при комнатной температуре
:Йои Се4* — СеЗ+ | _ — РгЗ+ Nd»+ Раз* Sms* Енз* Gd3+
Несло 4/-электронов 0 — 1 — 2 3 4 5 6 7
Хмол Диа-маг-нитен +2,35 + 5,05 + 5,05 + 1,0 +3,60 4-24,6
Ион ТЦ4* Np4* :U1* Nps* p(1e* Nplt РЦ4* Раз* Ащз* СтЗ+
Число 5/-электрон б в 0 0 1 2 2 2 3 А 5 6 7
Хмол' Дна- АТ11Г- 1ШТ0Ц Диа-маг-кятен +2,05 +3,75 4-4,10 -^3,50 +3,95 4-1,65 4-0,25 4-0,70 4-26,5
ет свои два s-электрона и один rf-эпектрон, чем хотя бы один из пяти /-электронов, так что при образования иона Ри3+ конфигурация переходит из 5/ь6Д7»'2 в 5/6. То, что. в атомах всех элементов ряда актиния 7^-электроны наименее прочно связаны, не вызывает никакого сомпепня. Предположение, что иопы Ри3+, Ри4+ и Np:1+ имеют конфигурации 5/> и 5/э соответственно, подтверждается намерениями мх магнитной восприимчивости- Для попов с 1 или 2 внешними электронами измеренные величины магнитной восприимчивости лежат между теми, которые можно ожидать при допущении, что эти электроны относятся к /-уровню, и теми, которые отвечают наличию //-эле кт ровен. Частично они несколько ближе тс значениям1, соответствующим d-электронам. Какой из этих двух возможностей отдать предпочтение на основании проведенных в настоящее время измерений, решить невозможно. Для многих ионов ряда актиния уровни 5/ и (W лежат настолько близко, друг к другу, что можно говорить об их смешении, В общем изменение магнитной восприимчивости ионов в зависимости от числа внешних электронов в ряду актинон совершенно аналогично ее изменению в ряду лантана. Как среди лаптяпидов ион гадолиния отличается особенно сильным парамагнетизмом, так и среди актинидов особенно сильно парамагнитен ион кюрия, т. е аналог гадолиния в ряду актиния. Особо низкому значешню магнитной восприимчивости иона Sin3+ соответствует особо малая магнитная восприимчивость
42 г. Реми
658
Глава 14
иона Рн3+. Соответствие простирается настолько далеко, что минимуму на кривсЁ зависимости магнитной восприимчивости от температуры для соединений самария, кщг установил Доусон (1951), соответствует такой же минимум на кривой х — Т для соединений плутония.
Как и соединения лаптанидов, соединения трансуранов обладают характерными спектрами поглощения. Спектры поглощении соединений трансуранов часто да яг в деталях близки спектрам поглощения соединений соответствующих им лантанидох Интересно, что появление в ряду лантанидов совершенно бесцветного в водном растворе иона повторяется п в ряду трансурановых элементов: подобно иону Gd’’" аналогичный ему ион Ст"' не поглощает в видимой области спектра в отличие от остальных ионов трансуранов(III), которые, как и соответствующие им ионы лантапидов(Ш), окрашены.
Исторические сведения. В 1936 г. Ган и Мейтнер наблюдали, что при облучении урана медленными нейтронами образуется £ “-излучатель с периодом полураспада 23 мин, который невозможно химически отделить от урана. Как позднее выяснилось, это был изотоп урана SSBU. Он образуется при захвате ядром гзв и нейтрона по уравнению: 2^U Jh = 3^U + у. Естественно, что образующийся нри ^“-распаде элемент должен иметь заряд ядра, равный 93. То, что облучение урана медленными нейтронами приводит через изотоп урана в качестве промежуточного продукта к образованию трансуранового элемента, первым из исследователей понял Ган, Но из-за отсутствия в его распоряжении достаточно сильного источника нейтронов он не смог непосредственно идентифицировать Полученный ин новый элемент.. Виервые^это удалось осуществить американским исследователям МакМиллану (McMillan) п Эй бе ас он у (Ahelsoii) в 1940 г. Опи наблюдали, что прн облучении урана медленными нейтронами происходит образование -излучателя с периодом полураспада 2,1 дня, который можно химически отделить от урана, и доказали, что открытый ими новый радиоактивный изотон является образующимся нри распаде 23,1 U элементом с зарядом ядра 93. МакМиллан и Эйбелсон назвали его нептунием, так как он в периодической системе следует за ураном, так же как планета Нептун в Солнечной системе следует за плане-
той Уран.
Химическое поведение нептуния сначала можно было исследовать только метода
ми радиохимии (г. е. с применением «носителей»). Это положение изменилось после открытия в 1942 г. долгоживущего изотопа нептуния (2371Хр, период полураспада которого 2,25'10й лет), Этот изотоп был получен при бомбардировке урана быстрыми
нейтронами по реаиции 2^и(п,2я) —-—>• 2jjJNp. С этого времени стала возможна
- ?
работа с чистыми соединениями нептуния, без соедивении-носителеи, правда, вначале с количествами в несколько микрограммов. Но вскоре было установлено, что аз“Кр образуется в качестве побочного продукта в результате ядерных реакций, происходящих в атомпых котлах. Несколько миллиграммов, полученного таким образом
нептуния впервые было выделено в 1944 г., и уже вскоре после зтого для химических исследований стали доступны количества в несколько дециграммов нептуния.
Спустя несколько месяцев после открытия нептуния был открыт второй элемент из ряда трансуранов — плутоний. В конце 1940 г, Сяббрг, "МакМиллан, Уол и Кенг неди (Seaborg, McMillan, Wahl, Kennedy) наблюдали, что при облучении урана (в виде U3O8) дейтронами с большой энергией образуется очень короткоживущий изотоп нептуния B3eNp, который в свою очередь, испуская р “-частицу, превращается в новый a-и злу чате ль с периодом полураспада ~80 чет. Этому новому элементу с порядковым > номер ом 94 открывшие его исследователи дали имя плутоний в честь следующей за Нептуном планеты Плутон.
Изотон плутония 23йРи, образующийся при р “-распаде 333Np, был идентифицирован в 1941 г. Кеннеди, Си бортом, Сегре и У о лом. Он оказался «-излучателем с периодом полураспада 24 000 лет. Оказалось, что этот изотон плутония подобно урану-235
делится под действием медленных пейтронов и, следовательно, его можно использовать для техже целей, что и урап-235. Поэтому уже вскоре после открытия 239 Р и было начато его получение в крупных масштабах. В настоящее время плутоний-239 непрерывно получают в больших количествах. ,
Элементы с порядковыми номерами 95 и 96 были открыты в 1944 г. Сиборгом, Джеймсом, Морганом и Гиорсо (James, Morgan, Ghiorso). Первый из этих элементов был назван америцием вследствие его химического сходства с европием из ряда лантанидов. Второй элемент был назван кюрием в честь супругов Кюри и одновременно, чтобы подчеркнуть его особое сходство с гадолинием, названным в честь финского химика Гадолйпа. Первым из этих двух трансурановых элементов был открыт кюрий, а пе предшествующий ему но порядковому номеру америций. Впервые кюрий был получен при облучении а-частицами 239Ри на циклотроне: 2^Pu + ;ta = 2^Сш-|- fpi. Когда
Трансурановые элементы
659
была предпринята попытка получить аналогичным образом 241 Рп из ^ззц, то оказалось, что образующийся при этом изотоп плутония, характеризующийся очень слабым Р "-и а лучением, превращается в «-излучатель с периодом полураспада около 500 лет:
$8U-Ha=^iPu+jB; i^Pu-------51-^ g^Am ° > .
13 лет 458 лет
При облучении й44Лт нейтронами вскоре был получен новый изотоп америция 242 Am, который, испуская р"-частицу, превращается в изотоп кюрия 242Сш;
й4Ат + «п = Ат+у; Г62Ат -2-^ gf Ст —. 1в час 162 ли
Получающийся в результате этой реакции кюрий-242 был первым изотопом кюрия, полученным в свободном от носителя состоянии. Первые ультрамикрохимические исследования были выполнены с этим изотопом в 1947 г. Вернером и Перлманом. До тех нор химическое поведение кюрия изучалось только радиохимически (т. е. с использованием носителей).
В конце 1949 г. Томпсон, Гиорсо и Сиборг на циклотроне Калифорнийского университета в Беркли получили еще два трансурановых элемента, названных берклием и калифорнием. Они образовывались при облучении америция или соответственно кюрия а-частица ми с энергией 35 Мэе:
^Am-Ha = ^3Bk+2j«; ^Cm-Ka^Cf
Подобно остальным трансуранам оба этих элемента неустойчивы. Берклий-243 в основном за счет электронного захвата превращается в кюрий. Наряду с этим берклий-243 мощет испускать ii a-частицы, превращаясь в америций. Его период полураспада равен 4,5 час. Калифорний-245 также способен распадаться как за счет электронного захвати, так п испуская а-чагтпчу. Его период полураспада равен 44 мин. Впервые этот элемент был получеп (при его открытии) всего в количестве примерно 5000 атомов. Для его получения бомбардировке a-частицами было подвергнуто всего лишь несколько микрограммов кюрия. Изотоп калифорния с несколько большим периодом полураспада (24«С1, период полураспада 35,7 час) был получен позднее на циклотроне в Беркли при бомбардировке урана ускоренными ионам С4+:
^U-h^C=i^Cf4-44n.
Этот изотоп представляет собой и-излучатель, при распаде которого образуется а4гСш. С другой стороны, он получается также при облучении a-частицами кюрия, а именно 244Ст. Химическое исследование чрезвычайно небольших количеств, в которых были доступны берклий и калифорний, можно было, конечно, провести только с использованием носителей. В качестве последних использовали соединения соответствующих лапта пидов.
То же справедливо и в отношении элементов 99 и 100. Эти элементы были открыты в течение 1952—1953 гг. приблизительно одновременно двумя группами американских исследователей, изучавших продукты превращения урана, который (при термоядерном взрыве) за короткое время подвергся действию чрезвычайно интенсивного потока нейтронов. В честь Эйнштейна и Ферми вновь открытые элементы были названы эйнштейнием и фермием (химические символы соответственно Ез и Fm). Отделить их от химически очень похожих трансурановых элементов Cf, Bk, Cm и Am удалось на специальной катиоиообмепной смоле. Катионы 'Ез и Fm вытесняются в промывающий раствор катионами перечисленных трансурановых элементов, и поэтому элюирование происходит в следующем порядке:
цщГш"’ — даЕз’" —9sCf‘" — втВк"* —geClU"'—В5Аш"‘.
В случае лантанидов элюирование в этих же условиях происходит в последовательности
б8Ег"’—етТ1о"‘—ввОу“‘—вдТЬ*" — e4Gd"‘ —63Еп‘“.
Следов а тел ьп о, на основании их поведения при элюировании можно сразу сделать выводы о порядковых номерах двух новых элементов, открытых по характерным для них периодам полураспада и энергиям распада. Оба эти элемента являются «-излучателями с пер иода мп поя у распада 20 дней и 15 час соответственно и имеют массовые числа 253 и 255. В дальнейшем были открыты и другие изотопы этих элементов. Так, Мсландер с сотрудниками в 1954 г. получил в Стокгольме при облучении урана ионами 1вО0+ с высокой энергией изотон фермии с массовым числом 250 и периодом полураспада 30 мин. Облучая 23SL) понами 4|NS\ разогнанными до высоких скоростей, Гиорсо с сотрудниками в 1953 г, получил короткоживущий изотоп эйнштейния 246Es
42*
660
Глава 14
(период полураспада 7,35 мин), Наиболее долгоживущим из известных к настоягтЕЛу времени1 изотопов эйнштейния является 254Ез — a-и злу чате ль с периодом полураспада 280 дней. Период полураспада наиболее долгоживущего изотопа фермия (25э₽дь равен 4,5 дня.
Элемент 101 был открыт Сибортом, Гиорсо и их сотрудниками при облучена 233 Es ускоренными на циклотроне а-частицами, Эти исследователи получили изотсе элемента 101 с массовым числом 256. Вначале они получали в опыте в среднем тольен два атома этого элемента; ин удалось отделить эти атомы при помощи ионообменной смолы от калифорния, эйнштейния и фермия и идентифицировать их по излучение образующегося при их распаде фермия. Новый элемент был назвав .менделевием в честь Д. И. Менделеева. Оказалось, что этот изотон за счет электронного захвата переходет в 3MFm. Период полураспада равен примерно 30 мин. Изотоп 253 рш затем спонтанна делится с периодом полураспада около 3 час.
В 1957 г. Филдс (Fields) совместно с группой шведских исследователей сообщил, что им удалось открыть элемент 102 в виде а-излучателя с периодом полураспада 10— 12 мн п и массовым числом 253. Эти ядра были пол учены при облучении а<иС1п лона-мп 13С,+ с высокой энергией. Идентифицирование нового элемента проводили хрома-тографпчески. Так как эти опыты проводили в Нобелевском институте в Стокгольма, новый элемент был назван нобелием. Гиорсо, Сиборг и их сотрудники в 1958 г. получили новый изотоп нобелия с массовым числом 254. Эти исследователи облучали 2ИСтл ионами !;ц}4+ иЛИ ЩС*+. И этот изотоп оказался а-пзлучателем с периодом полураспада около 3 сек. Получение нобелия пз ®'HCin американские исследователи повторить пе смогли, И действительно, как впоследствии пашли Филдс н сотрудники, нобелий образуется не из 24JCm, а из 2111 Ст, причем, вероятно, в виде изотопа нобелия 2S8No. Тот же изотоп нобелия был получен Флеровым с сотрудниками (1958) при облучении 241 Pii ионами 160а+ . Идентифицирование очень короткоживущего изотона 384No производили по a-излучению образующегося при его распаде фермия (см. стр. 679).
Расп ростра пение в природе. Как оказалось, трансурановые элементы в минимальных количествах существуют и в, естественных условиях. Уже в 1941—1942 гг. Сиборг и Перлман установили, что урановая смоляная руда и карнотит содержат плутонии, а именно плутоний-239. Впоследствии было показано, что урановые руды всегда содержат плутоний. Содержание плутония в них находится всегда примерно в постоянном отношении к содержанию урана. В среднем содержание плутония равно 10"12 % содержания урана.
Если, как это принято, образование естественного плутония происходит в урановых рудах таким же образом, как и техническое получение плутония, т. е. благодаря последовательным ядерпым процессам
*FU + п =rU------g?Pu, (1)
23 мин 2,3 Дн
то в урановых рудах неизбежно должен присутствовать нёптунпй-239. Но в состоя нии радиоактивного равновесия количество нептуния равно лить 2,6-10~® % от содержания плутония. Поэтому в 1 а урановой смоляной руды должно содержаться менее 6 атомов 2j8Np. Столь малые количества нельзя обнаружить даже при помощи очень чувствительных методов радиоактивного анализа. Это легко понять, если представить себе, что количество нептуния-239, присутствующее при радиоактивном равновесии в 1 кг урановой смоляной руды, испускает Только одну fi"-частицу в минуту. Напротив, в урановых рудах удалось обнаружить другой изотоп нептуния, долгоживущий 237Np. Но и он присутствует в урановых рудах лишь в исключительно малых количествах — около 210"|О% рт содержания урана. Он образуется, вероятно, по реакции a9;U(», 2л) Д >н ^usNp- Этот изотоп нептуния является начальным членом приведенного на стр. 682 радиоактивного ряда нептуния. Испуская а-частицу, он переходит х изотоп протактиния 2 311Ра, а из последнего за счет р"-распада образуется 233U. Содержание233 U в природе оценивается в 0,003 % содержания 238(j, Считают, что 233U образуется в природе (кроме распада 23!Np) также й иным Путем, а именно за счет ядерных реакций
232Th(n, y)233Th ——> 233Ра_______—>аззи. (2)
2 3,0 мин 27,4 дн
Не исключено, что, кроме плутония и нептуния, за счет ядерных реакций в природе образу ютел и другие трансурановые элементы. Однако вследствие их малых периодов полураспада вряд ли можно рассчитывать, что они присутствуют в природе в таких количествах, чтобы их можно было пен ос ре дет веяно обнаружить.
Трансурановые элементы
661
НЕПТУНИЙ (NEPTUNIUM) Np
Нептуний является как раз тем трансурановым элементом, который больше всех остальных напоминает уран. Он существует в тех же валентных состояниях, что и уран. Но в отличие от урана его наиболее устойчивые соединения относятся к чстырехвалентному состоянию.;
Картину относительной устойчивости состояний окисления нептуния дает следующая схема окислительно-восстановительных потенциалов, полученная в 1951 г. на основании измерения Коэна (Cohen) и Хайндмена (Hind шапп):
4-0,679а__________ I + 0,150в_________________________+ 0,7546- т „ +1,18Ев
ХрШ------—> NpIV------> Npv------► NpVI
I._________I____;_____J j
+ 0,452В I_______+ 0,943s_____]
Приведенные в этой схеме данные относятся к потенциалам следующих реакций ненов нептуния (иди 259 и pH 0):
Np- Np”" + r; Np*'"4-2II2O [NpOa]’-p4H'4-c; [NpOJ' [NpO2]'‘-ре.
Ионы Np”" более устойчивы, чом ионы I)””, но уже ионы Fe’” способны их окислить до ионов [Np^O2]L (Huizenga, Magnusson, 1951). С другой стороны, как показал Хайндмен (1951), соединения непт)Нпя(У) в концентрированных растворах хлорной кислоты подвержены окислительно-восстановительной реакции 2Npv NplA + NpVI.
Первые химические исследования со свободным от носителя нептунием были выполнены в 1944 г. Магнуссоном и Л а-П 1а пел л ем (La Chapello). Эти исследования проводили с несколькими микрограммами долгоживущего изотопа, 235 Np, пол ученного облучением урапа быстрыми нейтронами, Чтобы облегчить определение нептуния, к нему добавляли в качестве индикатора небольшие количества сильно радиоактивного 23BNp. Возможность работать со .значительно большими количествами появилась позднее, после получения нептуния-237 в качестве побочного продукта при техническом получении плутония. Выход нептуния при этом, составляет около 0,1 % от образующегося количества плутония.
Нептуний представляет собой серебристый блестящий, довольно устойчивый на воздухе металл (йпикк — 1.9,5, т. пл. 640°). Он существует в нескольких модификациях. Устойчивый при обычных температурах a-Np образует ромбическую решетку, которую можно рассматривать как деформированную кубическую объемноцентрированмую , решетку. Эле-' мептарная ячейка, состоящая из 8 атомов, имеет следующие размеры: а = 4,723 А, b = 4, 887 к, с = 6,663 A (rfpcHTr = 20,45)/ При 278° происходит превращение в тетрагональную модификацию ф-Np). При температурах выше 550s существует третья модификация (y-Np), устойчивая до температуры плавления (Zachariasen, 1952).
Нептуний является довольно сильно электроположительным металлом. В электрохимическом ряду напряжений он стоит рядом с алюминием. Но он несколько более инертен, чем алюминий, и более инертен, чем уран и плутоний (см. стр. 665). Металлический нептуний можно получить нагреванием NpF3 с Ва мри 1200°.
СОЕДИНЕНИЯ НЕПТУНИЯ
Трифторид нептуния NpF3 получают нагреванием NpO2 со смесью I1F и И2 до 500°:
NpO2 4- SIJF-f-1/2На = NpF3 + 2Н8О.
662
Глава 14
Если NpO2 или NpF3 нагревать со смесью HF и 02, то образуется тетрафторид нептуния:
NpOz4- 4HF = NpF4+ 2На0; NpF3 ф- HF+1/4Oa= i/2H2O.
Тетрахлорид нептуния, КрС14 можно получить нагреванием NpO3 в парах СС14. Если тетрахлорид нагревать в токе водорода до 450°, то он восстанавливается до трихлорида:
NpO2 2СС14 == NpGl4-p 2СОС12; NpC14-J- 1/гНг= NpCl3 -|-НС1.
Эти реакции полностью аналогичны тем, которые используются для препаративного получения UC14 и UC13. И по своим свойствам эти соединения нептуния напоминают соответствующие соединения урана. Точно так же гексафторид нептуния NpF0 полностью аналогичен гексафториду урана UF6. Его можпо получить нагреванием NpF3 в токе фтора.
Если на смесь Np02 с избыточным количеством металлического алюминия действовать парами брома, то образуется NpBr3. В чистом состоянии это соединение можно получить, испаряя одновременно образующийся Л1Вг3 при 250°, а затеи возгонял i\pBr3 (при 800°). Точно так же можно получить триодид, Npl3. Если в парах брома нагревать смесь МрО2 с Л Г, не допуская избытка последнего, то получается тетра-бромид Np В г4, возгоняющийся уже при 500°. Рентгенометрические исследования перечисленных галогенидов нептуния показали, что все они до мельчайших подробностей строения аналогичны соответствующим соединениям урана.
С азотом и углеродом нептуний образует кристаллизующиеся щотипу NaCI соединения NpN(a = 4,887 A, Zachariasen, 1949) и NpC(a = 5 004 A; Templeton, 1952). С фосфором и кремнием образуются соединения Мр3Р4 и NpSi2. При нагревании УрО2 со смесью H2S и Сз2 Сефт (Siieft) и Фрид (Fried, 1949) получили сульфид Np2S3, изоморфный U2S3.
Из окйслов нептуния наиболее устойчива двуокись NpO2. Ее можно нагревать на воздухе и при этом ее состав но изменяется. Однако ее можно окислить до двойного окисла Np3O8, но.такое окисление протекает труднее, чем окисление UO2. Это можно осуществить, например, нагреванием NpO2 с NO2 (Katz, 1949). Np3Og изоморфна U3Og. Менее устойчива, по-видимому, соответствующая UO монокись NpO.
Соединения нептуния в состояниях- окисления III, IV, V и VI можно получить и в водных растворах. В водном растворе соединения непту-ния(Ш) можно получить, например, электролитическим восстановлением: но это следует производить без доступа воздуха, так как кислород воз-^. духа окисляет в водном растворе соединения нептуния(П1) до соединений нептуння(ГУ).
Из соединений нептуния(1¥) фторид, иодат и гидроокись (или гидрат окиси) практически нерастворимы в воде. Но их можно перевести в раствор действием серпой кислоты и окислять броматом до соединений нептуния) VI). Соединения нептуния(V) можно получить электролитическим окислением соединений нептупия(ГУ) или окислением последних соединениями железа(Ш), Сернистая кислота восстанавливает соединения и епт уния (V) ипептуния(У1) до соединений нептуния(1У)-
Шестивалентный нептуний образует как нептуниты [пептунаты(А1)1, так и соединения нептунила [соединения нептунила(\Ч)[. Иептунаты соответствуют уранатам, а соединения нептунила соединениям уранила. Как и соли уранила, соли нептунила склонны к образованию двойных и комплексных солей. Примером таких соединений может служить натрий-нептунил ацетат Na [КрО2](С2Н3О2)з, изоморфный натрину ранил ацетату.
Двойные и комплексные соли (ацидо-соли) образуют также соли нептуния(V), в большинстве своем содержащие радикал [NpOa]+ [радикал нептунии а (V).), и соли нептуния(1У), прежде всего галогениды NpX4.
Трансурановые элементы
663
Пря добавлении к растворам солей неЕггупня(У) ионов ОН' выпадает гидроокись нептуния(У). Растворением последней в 1 п. соляной кислоте, добавлением щавелевой кислоты, растворенной в тр ет-бутилов ом спирте, и промыванием бледно-зеленого осадка эфиром в 1952 г. Гибсон (Cibsoa), Грузи (Gruen) и Кац (Katz) получили чистый оксалат нентунила(¥) [Np02irJC204-2H20. Водный раствор этого соединения дает абсорбционные полосы, характерные для соединений нептунпя(У); однако эти полосы несколько искажены, что указывает на образование наряду с ионами простых солей ионов комплексных оксалатов.
Промежуточную окись Np3Og можно получить действием NO2 при 300е на гидро-* окиси нептуш1я(1¥) или нептуния(У) или на нитрат нептуния(1¥), а также при нагревании на воздухе до 275—400° динептуната аммония (NH4)2Np2O7-HaO. Если же прокаливание лроводитыгрп теми ера туре 770е, то образуется NpO,. Нагреванием LWg с NO, до температур выше 250е можно получить UO3. В случае нептуния соответствующим окисел NpO3 получить не удается. При нагревании на воздухе Np30s сначала непрерывно отщепляет кислород, но при этом ее кристаллическая рещетка не изменяется. При 600° происходит резкое выделение кислорода и образуется 1УрО3. Этот окисел не разлагается даже при нагревании в вакууме до 1100°.
При растворении Np30g в 1н. хлорной кислоте образуется смесь перхлоратов рентуния(У) и пептуния(УГ), тогда как, по данным Мидлера (Miller) и Дина (Dean, 1952), при растворении в кислотах П3Оа образуется смесь солей урана(1У), урана(У) и ypana(Vl),
От плутония и других трансурановых элементов, а также от лаптаиа и лантанидов: нептуний можно отделить, добавляя при обычной температуре к сернокислому раствору, содержащему, в се эти элементы, бромат натрия, а затем плавиковую кислоту. Бромат натрия при обычных температурах окисляет только пептупий. При этом образуются иопы Нептунпла(У1) [Np О >], которые не осаждаются ионами фтора. Напротив, все трансурановые элементы, существующие » этих условиях в трёх- или четырехпалоптном состоянии, а также лантан и лантаниды образуют нерастворимые фториды.
ПЛУТОНИЙ (PLUTONIUM) Pu
Плутоний подобно нептунию существует в тех же состояниях, что и уран. Но в шестивалентное состояние перевести его еще труднее, чем нептуний. В общем плутоний существует в основном в четырехъ ал енти ом состоянии. Но по отношению ко многим элементам его трез’валент-пое состояние даже более устойчиво, чем четырехвалентное. Например, в тех же условиях, при которых образуются тетрахлориды и тстрабромиды урана и нептуния, плутоний образует соответствующие тригалогениды. Аналогично в оксисульфидах различного^ состава UviO2S, NplVOS, Pu*nQ2S отчетливо проявляется тенденция плутония существовать в более низком состоянии окисления. ""-У“
Склонность к существованию в высших состояниях окисления уменьшается при переходе от урана к плутонию и далее к кюрию. Это проявляется также в составе окислов, как видно из следующего обзора:
ио NpO PuO AmO
Pu2O3 Cm2O3
Pn4O7
ио2 NpO, Pu02 AmO2
и4о9
1-А
и3о8 Np3Oa
иОз
Как и уран, плутоний образует гидрид PuHj. С азотом и углеродом плутоний образует соединения PuiM и РпС. которые ииотипны соответствующим соединениям урана и пептуиия.
664
Глава 14
Подобно нептунию плутоний может существовать в водных растворах в виде ионов всех состояний окисления, за исключением двухвалентного. В кислых растворах (если не образуются комплексные ионы) плутонии существует в виде следующих ионов:
Pu'“, Ри"”, [РиО2]‘ И [РиО2]".
Относительные устойчивости ионов плутония и урана можно определить из следующих потенциалов:
: , _________—0,01
Г — 0,6йа + 0,550 -р 0,0бв-^
уш------л. ПУ > ini
+ 1,05 в з
1,00968
I + 0,9828 +1,1348„ „ + 0,912В +„
ри1П_!_2ГХри1У >РцУ-----L—j.puVT
+ 1,02 so
0,04 в
+ о,з1в
Приведенные окислительно-восстановительные потенциалы (при 25° и’ pH 0) рассчитаны па основании измерений Кониина (Connick, 1944—1951) и Хайндмена (1944). Иа этих значений потенциалов видно, например, что реакция [РиО2]"-{-1Г + 3/2Н,= = Ри”' -|- 2Н2О протекает с уменьшением свободной энергии (69,8 KKo/a-tHW [Рн.0г1 ') (см. т. I,: гл. 5), тогда как соответствующая реакция урана требует затраты энергии (0,5 ккалla-ион [11О2Г'). Уменьшение свободной энергий при восстановлении иона [NpO2]“ до иона Np"‘ составляет 47,0 ккал/г-иоп. Для окисло'нпя иона Ри"'; до иона Ри'"' требуется затрата энергии, равная 22,65 ккал/а-цон, а для окисления Np‘,“ до Np‘"‘— только 3,5 к кал la-ион, Ион IP", если только он не стабилизирован за счет комплексообразования, в водных растворах вообще неустойчив. Он окисляется до иона U"": уже ионами Н‘, и этот процесс экзотермичен и сопровождается выделением 14,5 ккал/г-ион, если активность ионов Д в растворе равна 1.
Изморены также теплоты восстановления ионов [РпО2]" и Ри"” до иона Pii”' (Evans, 1945; Connick, 1951). Они равны:
(PuOa]"4-H--|-S/aH2=Pii," + 2H2O+77,8 ккал, (3)
Pu""+1/2H2=Pu'";4*H'-)-i3,5 «вал, (4)
В первом случае теплота восстановления больше, а во втором меньше, чем изменение свободной энергии реакции. Следовательно, первая реакция сопровождается, уменьшением, а вторая — увеличением энтропии (Д5——26,6 и -|-Зи,6 кал/ёрад соответственно).
Между ионами Рн‘“ и [РнО2]" может устанавливаться зависящее, от pH раствора равновесие, которому отвечает уравнение '•
2Pu"' + (PuO2]"-|-4H- 7^ ЗРн-- .’; 2Н2О.
ГРЩ”]2 X fPuO*’]
Для константы равновесия, Ki = ~ > Каша (Kasha, 1945) при
ионной силе, равной 1, и 25°, получил значение 0,040 в 1 и, НСЮ4 и 40,2 в 0,1 н, НС1О4. Следовательно, К£ пропорциональна |Н]~3, тогда как по 'уравнению реакции константа равновесия должна была бы быть пропорциональна (Н']“4 * * * В * *, Причина этого расхождения еще
Аналогичное
не выяснена.
равновесие устанавливается и для реакции
(5)
[Ри-]Х[РпОГ]г
Рн— 4-2[РнО2]”4-2Н2О 3[PuO2p-)-4H*.
По данным Конника (1944), константа равновесия К2 =
в 0,5 н. растворе HCI при 2511 имеет величину 1,2 • 104. Поэтому в сильнокислом растворе ионы [PuO2J‘ могут присутствовать лишь в очень небольших концентрациях. Однако при уменьшении концентрации ионов Н' положение равновесия сильно сдвигается в сторону ионов [РиО2)‘. Согласно Каша; при иониои силе, равной 1 в 1 н. ; растворе НС1О4, доля пятивалентного плутония в равновесии с другими состояниями
Трансурановые алементы665
окисления составляет только примерно 0,5%, но в 0,01 н, растворе НСЮ4 — у же более 60%.
Но и в довольно кислых растворах ионы [РцО2]' можно получить в значительных концентрациях, например электролитическим восстановлением иопов [РиО2]”, так как окислительно-восстановительная реакция, которая приводит к указанному выще равновесию; является довольно медленным процессом.
Во всех состояниях окисления, в которых плутоний может существовать В: виде ионов в водных растворах, он способен также образовывать ацидо-ионы. Особенно ярко выражено его стремление к образованию ацидо-ионов с оксалат-, ацетат- и сульфат-и одами. Менее активно он образует ацидо-ионы с хлорид- и цитр ат-ио на ми. С нерхпорат-йонами плутоний ацидо-комплексов не образует.
Металлический плутоний получают нагреванием фторида плутония с парами бария; это металл темно-серого цвета (с?ЯЕКи = 19,74; г/ре11ТГ = - 19,82).
Для плутония известно 6 различных модификаций. 'При обычных температурах плутоний монок,чинен (а-Рп). При 122° он се значительным расширением переходит В другую модификацию неизвестной структуры ф-Рп, д?ПИКн=17,65)., Прн. 206^ происходит переход в ромбическую модификацию (у-Рп). Между 319 и 451е плутоний имеет трапе центрированную кубическую решетку (<5-Ри). При Дальнейшем повышении температуры сначала происходит переход в тетрагональную модификацию и наконец (при 476°) — б модификацию с объемноцентрированнои кубической решеткой.
Плутоний является довольно сильно электроположительным металлом. Его положение в электрохимическом ряду напряжений определяется из сравнения следующих нормальных потенциалов, отнесенных к нормальному водородному электроду:
А1/АР" U/U*’* Pu/Pu"’ Np/Np’“ Мп/Мп" — 1^69 в —1Ф6 й — 1,5 ff « 1,4 в .1,1 в
Практическое значение плутония обусловлено способностью его изотопа 23йРп расщепляться как медленными, так и быстрыми нейтронами.. Это свойство наряду с плутонием присуще и урану-235. Нес.мптря на то что уран-235, как составная часть: естественного урана, имеется в природе в довольно больших количествах, а плутоний можно получать только искусственно из изотона урана-238 при помощи ядерных реакций, -j тем не мепее чистый плутоний легче получать в больших, технических масштабах, чем уран-235. Уран-235 можно получить в чистом состоянии только после разделения изотопов, проведение которого в больших масштабах требует огромных затрат вспомогательных средств. Напротив, плутоний, после того как он получен при помощи ядерных реакций, можно отделить от урана чисто химическими методами. К тому же при крупномасштабном техническом получении плутония в качестве ценных побочных продуктов получаются другие радиоактивные элементы. Кроме того, производство плутония в промышленных: масштабах можно провести так, что выделяющаяся при этом огромная энергия будет использована в силовых установках.
Получение плутония. Техническое получение плутония основано па том,: что при действии медленных нейтронов па составляющий основную часть естественного урана изотоп 23SU происходят следующие ядерные реакции:
аз8О4-л —> 239U -_р~ .. 23»Np__> йзврц. (б>
23 мин 2,31 дн
666
Глава 14
Необходимые для этого нейтроны образуются при делении ядер изотопа 235U, также содержащегося в естественном уране.
При делении ядер 335 U на каждый нейтрон, расходующийся на деление, образуется по крайней мере два новых нейтрона. Если условия таковы, что каждый из этих нейтронов может в спою очередь вызвать одно деление, то число нейтронов, как уже говорилось в гл. 13, очень быстро возрастает и реакция протекает со взрывом - чудовищной силы (атомная бомба). В отличие от таких «неуправляемых ядерпых реакций», протекание которых приводит к взрыву, для получения плутония используют «управляемые ядерные реакции».
Если реакции деления происходят не в чистом уране-235, а в естественном уране, который содержит значительно больше 338U, чем то освобождающиеся при делении нейтроны значительно чаще сталкиваются с ядрами чем с ядрами 236U. И несмотря на то что лишь часть столкновении нейтронов с ядрами 238U приводит к захвату нейтрона ядром с образованием новых ядер 239U, все же вследствие поглощения ядрами 338U только часть нейтронов, образующихся при делении, снова встретится с ядрами 235U и вызовет новые деления. Если принять для простоты, что при каждом делении ядер 235U образуется два нейтрона и что один из них реагирует с ядром 93SU, образуя 239Ри (через 239U и 233Np), а другой вызывает новый акт деления ®®®U, то число свободных нейтронов длительное время будет оставаться постоянным и, следовательно, реакция будет проходить с постоянной скоростью. Эти условия приблизительно выполняются при техническом получении плутония.
То, что только половила нейтронов захватывается ядрами 239U, хотя их содержание в естественном уране в 139 раз больше содержания ядер 235U, обусловлено тем, что «сечение захвата» (см. стр. 604) нейтронов ядрами ass и значительно меньше «сечения захвата» нейтронов ядрами 235 U. Это относится к так называемым «тепловым» нейтронам, кинетическая энергия которых при обычных температурах равна в среднем 0,025 se. В еще большей степени это справедливо для нейтронов с большей кинетической энергией. Но между ними (примерно при 5 ев) лежит так называемый «резонансный пик», т. е. узкая область, в которой сече пи е захвата нейтронов для 239 U имеет чрезвычайно большое значение (несколько тысяч барн). Нейтроны, освобождающиеся при делении, сначала имеют очень большую кинетическую энергию (1—2 Когда опи сталкиваются с ядрами a39U, то почти во всех случаях они рассеиваются ими и поглощения пе происходит *. При каждом столкновении скорость нейтронов уменьшается, так как каждый раз часть их кинетической энергии передается тому ядру,
Таблица 84
Тормозящее действие различных атомных ядер на нейтроны «й-
Атомное ядро П D не Be с О
Массовое число 1 2 4 9 12 16
Потеря энергии при каждом столкновении, % 63 52 35 48 14 11
Число столкновений, тро-б у тощее с я д.'.пг т при оженил нейтрона до тепловых скоростей 18 25 42 90 114 150
‘Сечение захвата нейтронов, барн 0,3 0,001 0 0,01 0,005 0,002
* Очень небольшая часть быстрых нейтронов захватывается ядрами 338U, вызывая их деление.
Трансурановые элементы
667
с которым они сталкиваются (см. стр. 553). Так, их кинетическая энергия в конце концов падает до 5 эе. Если нейтрон с такой энергией вновь столкнется с ядром, то он обязательно поглотится, но деления ядра не произойдет. Следовательно, чтобы осталось достаточное число нейтронов для продолжения деления ®s6U, надо позаботиться о том, чтобы торможение нейтронов происходило в веществе, сечение захвата нейтронов которого имеет возможно малую величину. Вещество, которое можно использовать для этого, называют замедлителем, а процесс торможения нейтронов до скоростей тепловых нейтронов — термализацией. От хорошего замедлителя требуется, чтобы не только его сечение захвата нейтронов было возможно меньше, но и число столкновений, нужное для замедления нейтрона, было небольшим, так как даже при малом сечении некоторая доля столкновений все же приводит к захвату нейтронов. Из законов столкновения следует, что наибольшим тормозящим действием будут обладать те атомные ядра, масса которых наименьшая (ср. стр. 559). Данные табл. 84 цокааы-
вают, что из всех веществ, которые практически можно рассматривать как замедлители в процессе получения плутония, наиболее пригодны тяжелая вода (D3O) и графит. Графит проще получить в нужных количествах, чем тяжелую воду, по он должен быть очепь чистым, так как уже небольшие загрязнения (примеси) значительно увеличивают поглощение нейтронов.
На рис. 87 приведена схема процессов, которые в соответствии с изложенным выше играют основную роль при получении плутония. Если процесс протекает по такой схеме, то на каждый целящийся атом*3"!! должен образовываться один атом плутония. Практически это приблизительно так.
Необходимое для спокойного протекания процесса условие, заключающееся в том, что из всех образующихся при делении нейтронов один обязательно должен вызвать повое деление, не обеспечивается только тем, что в процесс вводится замедлитель. Для этого нужно учитывать и ряд других обстоятельств, о которых речь пойдет в следующих разделах.
Коэффициент размножения. Предположим, что в системе, в которой может идти деление ядер под действием нейтронов, число таких делений в данный момент времени равно z. Из образующихся при этом нейтронов /-s могут вновь вызвать деления ядер. В таком случае множитель / называют коэффициентом размножения данной системы. Для процесса, который протекает по схеме, приведенной па рис. 87, f = 1. Если в данный момент 100 пей
Медленный
нейтрон
Р и с. 87. Схема получения плутония.
тронов вызвал и деления ядер и 105 из обра-
зовавшихся в результате этих делений нейтронов вызвали новые деления, то f = 1,05. Число пейтропов, которые вызовут третью серйю делений, будет равнов таком случае 100X1 >05x1,05,ан-наясерия делений будет вызва-
на 100 X 1,05 й-1 нейтронами. Если вначале имелся один-сдннствешшй нейтрон и коэффициент размножения равен 1,05, то в тот момент, когда происходит двухсотая серия делений, число пейтропов будет уже равняться 16 000, к началу пятисотой серии делений — уже 38 млрд., а к началу тысячной — более чем тысяче триллионов. Следовательно, число нейтронов очепь быстро возрастает, если коэффициент размножения существен
668
Глава 14
но больше 1. II, наоборот, оно быстро падает, если f меньше- 1. При коэффициенте размножения 0,95 число нейтронов через, 400 серий делений упадет от 1 млрд, до 1.
Поэтому для спокойного протекания процесса получения плутония величину коэффициента размножения необходимо длительное время поддерживать по возможности ближе к единице. В противном случае реакция или затухнет, или скорость ее начнет так быстро возрастать, что появится опасность взрыва.
...На величину коэффициента размножения влияют многие процессы. Каковы они и насколько учет их позволяет варьировать величину коэффициента размножения, видно из следующего.
1. До сих пор было принято, что па каждый медленный нейтрон, расходующийся в акте деления, образуются 2 быстрых нейтрона. В действительности в среднем образуется их несколько больше. Обозначим это число /рЕсли каждый из z медлен-^ ных нейтронов вызывает один акт деления, то число нейтронов в системе в результате этих делений возрастет от г до z-fj.
2. Нейтроны, образующиеся при делении ядер, имеют кинетическую энергию 1—2 Мае. Поэтому они могут вызвать деление ядер 23SU. Несмотря на то что сечение такого деления очень мало, такие деления все же происходят. Случается также, что быстрый нейтрон, сталкиваясь с ядром 235 U, вызывает ого деление. В результате таких делений число нейтронов снова увеличивается с s-/j до z Множитель / 2 называют коэффициентом деления на быстрых нейтронах. В большинстве случаев он равен 1,03.
3. Часть нейтронов, прежде чем замедлитель затормозит их до тепловых скоростей, сталкивается с ядрами MSU при таких скоростях, при которых они поглощаются этими ядрами, но вызывая их деления. В. связи с этим число нейтронов в системе уменьшается от до г- /г/3-/з- Множитель /3 (всегда меньше 1) учитывает вероятность того, что нейтроны поглотятся ядрами 238О прежде, чем они замедлятся до скоростей тепловых нейтронов. Поскольку такое поглощение обусловлено существованием «резонансного пика» 238U при 5 as, fa называют вероятностью резонансного поглощения.
4. При торможении, нейтронов замедлителем поглощается хотя и малая, ио все же заметная часть нейтронов. Еще одна, и притом очень значительная часть нейтро-нов, после торможения расходуется па превращение 233О в плутоний. Кроме того, часть нейтронов поглощается примесями и иными посторонними, веществами. В результате всех этих процессов число нейтронов в системе уменьшается от до'
z-fa'fa-fa'fa- Чем больше Д, тем больше число тепловых нейтронов, которые благодаря делению ядер 235U вызовут образование новых нейтронов. Поэтому множитедь /4 называют коэффициентом использования тепловых нейтронов.
В системе неограниченных размеров доля нейтронов, образовавшихся в результате z делений, которые после замедления вызовут новые акты Деления, будет Опре-Z * f 4 * /о Г«Э * f к -1. -VI
делиться отношением -—, Это отношение по размерности совпадает с коэф-фициентом размножения Поэтому для таком системы справедливы равенства
и /^./,./3.^ (7)
Из четырех множителей, входящих в состав коэффициента размножения, /3 и /4 (а в известной мере и /,) зависят от условий эксперимента. Легко попять, что на величину/4 основное влияние оказывает природа замедлителя. Кроме того, можно показать, что вероятность резонансного поглощения /3 значительно больше в тех случаях, когда компактные, куски урана укладывают в графит; чем при использовании мелкозернистых, смесей урана и графита. Преимущественное использование металлического урана, а не урана в виде одислов, также вызвано влиянием такой замены на величину множителей (3 и (4.
Критическая величина лдерпого реактора. Формула (7) справедлива для системы неограниченных размеров. Но в случае ядерного реактора имеется система,размеры которой ограничены. В этом случае потеря нейтронов возможна и вследствие выхода их через поверхность системы. В связи с такими потерями коэффициент размножения может значительно уменьшиться. Число нейтронов, выходящих из системы реактора-пропорццбнально его поверхности, а; число образующихся в реакторе нейтронов-пропорционально его объему. Если реактор имеет форму шара, то с увеличением радиуса шара отношение числа выходящих из системы нейтронов к числу образующихся в ней нейтронов уменьшается. Любой ядерный реактор, для которого рассчитанный по уравнению (7) коэффициент размножения больше 1, должен иметь такие размеры,.
Трансу раковые элементы
669
чтобы утечка нейтронов через поверхность компенсировалась приростом их числа за счет делений. В этом случае множитель, учитывающий утечку нейтронов через поверхность, который обозначим через, /е, будет точно равен 1.
Если при 100 делениях образуется столько нейтронов, что при отсутствии утечки нейтронов из реактора 105 из них могли бы вызвать новые акты деления, то при утечке 5 из этих 105 нейтронов останется только 100 нейтронов, способных вызвать вторую серию делений. Следовательно, из-за утечки нейтронов из реактора коэффициент размножения снизится с / == 1,05 до fe — 1,00 й деление ядер в реакторе будет протекать не с все возрастающей скоростью, как это было бы в системе не огр а ничейных размеров, а скорость деления останется постоянной. Размеры ядерного реактора,: при которых увеличение числа нейтронов в нем как раз компенсируется утечкой нейтронов, называются его критической величиной. Критическая величина реактора зависит от состава смеси изотопов, используемой в реакторе,,а кроме того, и бт вида и коли- " чества замедлителя, рода и количества примесей, которые захватывают часть нейтронов. Так, нри применении в качестве замедлителя тяжелой воды критическая величина реактора значительно меньше, чем при использований графита.
Per у пирую nine стержни. Процесс деления может' самостоятельно протекать в реакторе только в том случае, если его величина по меньшей мере равна критической. На практике реактор строят таким, что его величина несколько превышает критическую, а часть нейтронов улавливают регулирующими. стержнями из кадмия или бористой стали. Кадмий и бор имеют особо высокие значения сочеппй захвата мед-ленных нейтронов, в противоположность, например, алюминию, сечение захвата которого очень мало. Опуская регулирующие стержни в реактор на различную глубину, моя;по добиться того, что число нейтронов в реакторе будет оставаться постоянным. Если опо возрастает, то регулирующие стержни опускают ниже, если уменьшается — то поднимают выше.
Запаздывающие нейтроны. То, что скорость деления в ядерной реакторе можно поддерживать постоянной при помощи регулирующих стержней, объясняется тем обстоятельством, что при делении ядер часть нейтронов освобождается не тотчас же, а с некоторым запозданием. Иначе регулирование было бы невозможным из-за очень большой скорости, с которой возрастает или уменьшается число актов деления в реакторе, если по той или иной причине изменяется коэффициент размножения.
Нейтроны, освобождающиеся при делении ядер, испускаются не делящимся ядром, а его осколками. Однако большая часть нейтронов испускается мгновенно вслед за делением ядра (вероятно, в течение Ю-12 сек). Но небольшая часть нейтронов (около 0,75%) освобождается с известным запозданием, в большинстве случаев спустя несколько секунд. Если в данный момент времени ядерпый реактор работает так, что ого коэффициент размножения равен /е —,-1,0055, то число актов деления в каждой серии делений возрастает в отношении 10 055 : 10 000. Но из 16 055 нейтронов, которые вызывают новую последовательность делений, только 9980 освобождаются тотчас же. Следовател ыю, к первым 10 000 делений спустя очень небольшое время добавляются 9980 делений второй серии, а остальные 75 делений второй серии происходят только спустя несколько секунд. Но общее число нейтронов в реакторе возрастет только в том случае, если число актов деления дополнится теми делениями,, которые вызываются «запаздывающими нейтронами», Поэтому такое возрастание происходит настолько медленно, что остается достаточно времени для опускания регулирующих стержней с целью уменьшения коэффициента размножения, прежде чем увеличение -числа нейтронов примет угрожающие размеры.
Атомный котел. Ядерные реакторы, использующиеся для технического получения плутония, обычно называют атомными котлами. Первые самостоятельно работавшие устройства такого рода были выполнены так, что в них последовательно чередовались уложенные друг на друга слои графита и урановых слитков (частично окись урана). Позднее стали использовать большие графитовые блоки с просверленными в них: каналами, в которые вводят стержни или бруски из металлического урана, заключенные в герметичные алюмпппевыс гильзы. Такое устройство обеспечивает непрерывную работу реактора: урановые стержни вводимые с передней стороны, проходят с любой скоростью через котел и могут быть извлечены с задней стороны. При выгрузке пз реактора урановые стержни содержат около 0,1% плутония (включая еще не превратившийся в него нептуний).
670
Глава 14
За один рабочий цикл нельзя провести процесс так, чтобы разделился весь уран-235 и образовалось соответствующее количество плутония, так как при все возрастающем обогащении стержней плутонием он во все большей степени будет подвергаться делению на медленных нейтронах, что в конце концов приведет к тому, что плутоний будет исчезать в результате деления с такой же скоростью, с какой он будет образовываться. Кроме того, накапливающиеся продукты деления урана-235 ведут себя как примеси, поглощающие нейтроны. Следовательно, они уменьшают коэффициент размножения реактора, что может, наконец, привести к тому, что /в станет меньше 1 и реактор прекратит работу.
Рис. 88. Схематический разрез атомного котла.
т — урановые стержни; 2 — алюминиевые капсулы; з — графит — замедлитель; 4 — регулирующий стержень; S — защитная оболочка из бетона; в — держатель из графита для алюминиевых ампул с веществом. активируемым нейтронами.
В первых атомных котлах в качестве замедлителя использовали графит, Впоследствии были построены и такие реакторы, в которых замедлителем служила тяжелая вода. Схематический разрез атомного котла, работающего на графите, приведен на рис. 88.
Кроме трубчатых алюминиевых гильз (в которые заключены урановые стержни) и графитовых блоков, служащих замедлителем, на рисунке показан и один из «регулирующих стержней». Он представляет собой круглый стержень из кадмия или бори-стой стали, перемещающийся вверх и вниз. Большинство атомных котлов (например, в Окридже и Брукхейвене в США, в Чок-Ривер в Канаде, в Харуэлле и Эймерсхеме в Англии, в Шатниьонс во Франции) снабжено специальными приспособлениями, позволяющими использовать образующиеся в реакторе нейтроны для одновременного получения искусственных радиоэлементов, например иода-131, фосфора-32, углерода-14. На рис. 88 показан принцип этого устройства. В реактор через специальное отверстие вдвигают прямоугольный графитовый брусок, имеющий пазы для алюминиевых капсул. Вещества, которые хотят активировать нейтронами реактора, помещают в эти алюминиевые капсулы. Затем графитовый брусок с капсулами вдвигают в реактор и спустя некоторое время выдвигают обратно, чтобы вынуть препараты. Размеры реактора, подобного изображенному на рис. 88, примерно равны размерам пятиэтажного дома. Реактор окружен толстой бетонной оболочкой. Она служит,
Трансурановые элементы
671
с одной стороны, для защиты персонала от вредного действия нейтронов, с Другой— бетонная оболочка отражает обратно большую часть выходяящх из реактора пейтропов, благодаря чему повышается коэффициент размножения реактора. Вследствие выделения в реакторе большого количества тепла необходимо хорошее его охлаждение. Сначала реакторы охлаждали воздухом, позднее перешли к охлаждению водой. Если ядерный реактор доллгеп приводить в действие тепловые иашипьг. то вместо воды целесообразнее приме пять вы сококипящие охлаждающие жидкости, которые непрерывно циркулируют между реактором и теплообменником, в котором они нагревают воду. Горячий водяной пар затем используют, как обычно, в паровых; турбинах.
Первое опытное устройство для проведения само поддерживающейся реакции деления урана было сооружено в-Колумбийском университете в Нью-Йорке и начало работать в июле 1941 г. Оно состояло из больших кусков технически чистой окиси урана общим весом 7000 заложенных в графит. Работа этого устройства еще не была само поддерживающейся, так как примеси поглощали слишком много нейтронов. Первая установка, работавшая в само поддерживающемся режиме, т. е. без подвода нейтронов извне, была пущена в эксплуатацию 2 декабря 1942 г. на территории Чикагского университета. Одна ко она служила только для исследовательских целей. Первый атомный котел, дававший непрерывную продукцию плутония, был построен в Окридже, шт, Теннесси. Он начал работать в ноябре 1943 г. при мощности 800 кет и к середине 1944 г., после многочисленных переделок достиг мощности более 2000 кет, что соответствует ежедневной продукции плутония около 2 а. Первый большой завод для получения плутония был пущен осенью 1944 г. в Хэифорде (на северо-западе США). Он имел три атомных котла и занимал площадь 1600 я.и'2. Общая тепловая мощность этих трех атомных котлов сначала составляла примерно 1 млн. кет (по порядку величины пример [[О рапная общей мощности гидроэлектростанций Германии в 1933 г.). Производство плутония па хэпф орденом заводе должно бы до составлять с начала лета 1945 г. 1 ко/ сутки. В настоящее время я,черные реакторы, кроме США, работают и в Других странах. Опп служат отчасти для получения плутония, отчасти для получения этгоргпп. По насколько известно, заводов, приближающихся по мощности к заводам в Хэнфорде, пе существует.
Получение чистого плутония. Урановые стержни или бруски, содержащие плутоний и нептуний, после выгрузки из реактора падают в большие резервуары с водой. В них они остаются продолжительное время, пока не закончится превращение нептуния в плутоний. При атом одновременно распадаются короткоживущие изотопы продуктов деления. Благодаря этому значительно уменьшается первоначально чрезвычайно высокая интенсивность радиоактивного излучения. И тем не менее интенсивность излучения остается настолько высокой, что -дальнейшая химическая переработка должна производиться в окруженных бетонными стенами и частично расположенных под землей помещениях с дистанционным управлением.
При такой химической переработке содержащий плутоний уран сначала раство- У ряют в кислоте, а затем окисляют до соли урана(УТ) в таких условиях, при которых плутоний пе окисляется. Плутоний(1У) осаждают из раствора добавлением подходящего носителя. После фильтрования осадок вновь растворяют и окисляют плуто-ний(1У) до плутония(У1). Затем снова осаждают носитель. При этом он захватывает часть продуктов деления. Те продукты деления, которые не захватываются носителем, не захватываются н плутонием, если его после удаления носителя вновь восстановить до плутония(IV) п затем осадить в тех же условиях, при которых перед этим осаждался носитель. Чтобы получить совершенно чистый плутоний, эти реакции о кисления, восстановления и осаждения повторяют несколько раз.
В принципе этот метод довольно прост. Но чтобы получить совершенно чистый плутоний, надо провести приблизительно тридцать химических реакций. Поэтому требуются сотни отдельных операций, каждую из которых проводят автоматически или с дистанционным управ,гением, чтобы избежать сильного радиоактивного излучения. Па хэнфордском завале цех Для извлечения чистого плутония из материалов, выгруженных из атомных реакторов, выглядит как голый, лишенный окон, длинный огромный бетонный блок. Во внутри он скрывает множество установок, аппаратов, сосудов п контрольных приборов, которые запутанной системой труб п проводов связаны между собой и с внешним миром. Первые проекты таких цехов были созданы уже тогда, когда еще никто не видел невооруженным глазом самого плутония или его соединений. Видимые количества соединения плутония, свободного от носителя, были впервые получены в августе 1942 г. Окончательный вид процессы получения чистого плутония.
672 Глава .14
использующиеся на хэнфордском заводе, приобрели на основании опытов, при провес ;дении которых в распоряжении исследователей- имелась в ‘ общей сложности только крупинка плутония величиной с булавочную головку (0,5 м-г). Этот плутоний был получен не в ядерном реакторе, а на циклотроне. Большинство этих опытов было про1* ведено с такими количествами вещества, которые исчислялись микрограммами. Нети средственное перенесение лабораторных опытов в крупное техническое производство, т. с. переход от микрограммов к килограммам, означало увеличение масштабов в отношении 1 : 1 000 000 0(10. То, что отважились пойти на такой скачок и что он счастливо завершился, указывает на надежность и точность результатов, которые молено получить современными ультрамиярохимичоскими методами, при которых работают с «пробирками» и «колбами» емкостью 1 /100 000—4/10 .ил.1
Реакторы-размножители. Если плутоний получают по схеме, приведенной на рис. 87, то?из: данного количества урана (после многократных рабочих циклов) можно получить в конце концов столько плутония, сколько первоначально содержалось в уране изотопа 2;!BU, но пе больше. Следовательно, из 1000 кг урана этим способом можно получить в лучшем случае 7 кг плутония. Но для получения энергии, плутоний можно прибавлять к оставшемуся 2380 и использовать его для превращения нового количества 338U в 23аРн. Но при этом расходуется эквивалентное количество плутония. Следовательно, выход плутония не повышается/ Это справедливо при определенных предпосылках, т. е. что из образующихся при каждом делении нейтронов в среднем один расходуется на дальнейшие деления, а один тратится на превращение 238U в плутоний. Однако было показано, что число нейтронов, которые превращают ?®3U в 23аРц, можно несколько увеличить, т, е. на 1 г-атом Рп, подвергающегося делению, получить за счет превращения 238U несколько больше 1 г-атома Ри. Ядёр-ные реакторы, сконструированные таким образом, что в них осуществляется этот эффект, называются реакторами-размножителями. Они открывают принципиальную возможность исходя из данного количества плутония многократным повторением процесса (при одновременном использовании выделяющейся ядерной энергии) получить из 238Ц большее количество плутония, не расходуя при этом азШ. Насколько это важно, станет ясно, если учесть, что мировые запасы 23SU более чем в сто раз превышают мировые запасы 235U.
СОЕДИНЕНИЯ ПЛУТОНИЯ
Из многочисленных соединений плутония важнейшими являются следующие: окислы., галогениды, и соли плутония в различных состояниях окисления.
Соли плутония и их водные растворы имеют характерную окраску и их спектры поглощения напоминают спектры поглощения редкоземельных элементов. Водные растворы солей плутония(Ш) ярко-голубого цвета, солей плутония(ГУ) — бледно-красного, солей плутония(У1) — оранжевого, а при сильном разбавлений — розового. Эти цвета наблюдаются в тех случаях, когда в растворах не происходит образования комплексных соединений, присутствие которых сопровождается изменением цвета.
Соли плутоиия(Ш) в водных растворах окисляются кислородом воздуха до солей плутония(1У). Это следует из значений окислительно-восстановительных потенциалов этих соединений (см. стр. 669 и табл, 112, т. I) и было подтверждено опытом. Перманганат при обычных температурах тотчас же окисляет соли плутония(Ш) до плутония (ГУ). Но при температурах выше 60° при окислении перманганатом: образуются соли плутония^!) Или соли плутонила(У1). Ионы урана(1У) восстанавливают ионы плутония(ГУ) до ионов плутоний (III).
Трансурановые элементы 673
Иовы плутония(Ш) значительно менее склонны к образованию комплексных соединений, чем ионы плутопия(1У), которые обнаруживают сильную тенденцию к комплексообразованию. Особенно часто образуются ацидо-ионы. Поэтому, например, в солянокислом растворе для окислительно-восстановительного потенциала пл утони я( IV) получается иное (примерно на 0,025 в меньше) значение, чем в перхлоратном растворе. G перхлорат-ионами плутоний не образует комплексов ни в одпом окислительном состоянии. С иолами хлора трехвалентный плутоний образует очень слабые, а четырехвалентный — значительно более устойчивые комплексы. В связи с этим уменьшается величина изменения свободной энергий, связанной с превращением Puiv в Риш. И наоборот, в солянокислых растворах изменение свободной энергии при превращении PuVl ->• PuIV больше, чем в перхлоратных растворах, так как сильная склонность PuIV к комплексообразованию способствует этому переходу. Но в этом случае различие окислительно-восстановительных потенциалов в солянокислых п перхлоратных растворах меньше (0,019 в), так как в солянокислых растворах РиУ1 обнаруживает хотя и небольшую, по заметную тенденцию к образованию комплексов с ионами хлора. Образование комплексов плутония отчетливо обнаруживается и вследствие его влияния на спектры поглощения. Иногда оно становится заметным по явному изменению цвета раствора. Например, растворы нитрата плутония(1¥) не имеют характерного бледно-красного цвета гидратированных ионов плутония(1у), а в результате образования нитрато-комилексов окрашены в золеный цвет.
Из растворов солей плутония(ТП) аммиак осаждает гидроокись плу-тонйя(111}, выпадающую в виде грязно-голубого легко окисляющегося на воздухе осадка. Из растворов солей плутония (IV) при до ба в лении аммиака выпадает б.тедпо-зслепый слизистый осадок гидроокиси плупгонияЦУ), который при прокаливании переходит в темно-коричневую, нерастворимую в иоде двуокись плутония РиО2. Двуокись получается также при прокаливании нитрата иди подата плутонпя(ГУ). Из растворов солей плуто-ния(У1) или солей плутонила(У1) щелочи осаждают плутоний в виде плутоната или полиплутоната только в тех случаях, когда концентрация плутония сравнительно высока. Плутонаты.щелочных металлов отличаются от соответствующих соединений урана своей значительно большей растворимостью. Значительно менее растворим, по сравнению с плутонатами щелочных металлов, нлутонат бария. Это соединение егце нс было получено в чистом виде, но, по-видимому, оно представляет собой полиплутонат, например ВаРи3О1О.
Окислы. Известны следующие окислы плутония: PuO, Pu203, PU4O7 и РпО2. Окислы РпО п I’uOq изотипны соответствующим окнелам урана, нептуния и америция. Моноокиси этих металлов имеют структуру каменной соли, а двуокиси подобно двуокиси тория — структуру плавикового шпата. Ри2О3 не изотипен полуторной окиси самария аналогичного состава. Этот окисел кристаллизуется не по тину Sc203, как полуторная окись самария, а имеет ту же кристаллическую решетку, что и гексагональная модификация ЬагО» (см. рис. 15) с а = 3,840 и с = 5,957 А. Рп2О3 образует смешанные кристаллы со всеми полуторными окислами редкоземельных металлов, кристаллизующимися по этому типу (Templeton, 1952).
Перекись плутония. Если к раствору соли плутопия(1¥) добавить перекись водорода, то вследствие образования перекисных комплексных соединений Он приобретает кроваво-красный цвет. При увеличении концентрации перекиси водорода выпадает интенсивно зеленый осадок, состав которого, если не учитывать содержание в нем воды, отвечает формуле В этом соединении плутоний присутствует в четырех-валецтном состоянии, п оно изоморфно соединению тория аналогичного состава (Кash-land, 1945). Из растворов солей плутонила(У1) получается, по-вндимому, тот же осадок, но значительно медленнее (Harvey, 1947).
Сульфиды. Плутоппй образует полуторный. сульфид Pu2S3 п оксисульфид Pu2O2S. Полуторный сульфид получается при нагревании РпС13 в токе сероводорода. Он кристаллизуется в кубической системе и изотипен La,S3, Ce2S3, Ас283 и Am2S3. Оксисульфид, образующийся при нагревании 1’пО2 в токе сероводорода при 1250’, изотипен 43 г. Реми
674 Глава 14
La3O3S и Ce2O2S, Эти соединения кристаллизуются в гексагональной системе, во их структура родственна структуре полуторных сульфидов (Zachariasen, 1949).
Фторнды. Трифторид плутония PuF3 выпадает в виде фиолетового осадка из растворов солей плутония( 111) при добавлении к ним фторида щелочного металла. Если его нагревать в токе кислорода при температуре около 600°, то оп окисляется, образуя Pub'., и РпО3.
Тетрафторид плутония PuF4 выпадает из растворов солей плутония (IV) прн добавлении к ним фторид-ионов. Состав этого осадка телесного цвета отвечает формуле PuF4-21/2H2O. При слабом нагреваний он переходит в безводный зеленый PhF4. Это соединение“разиагается только при нагревании в вакууме до температур выше 900°, образуя по уравнению PuF4 PuF3 1/%&?,, трифторид плутония. Некоторые наблюдения подтверждают предположение, что при этом в качестве пр омея:у точного продукта образуется PuFs (Fried).
Тетрафторид плутония явился первым соединением плутоция, которое было получено свободным от носителя. Оно было выделено Каннингемом (Cunningham, 1942) из нитрата урацила, который долгое время облучали нейтронами. Нейтроны получали при реакции бериллия с ускоренными па циклотроне дейтронами (по уравнению “Вс -|- р/ =''(В -|- jn). Из облученного препарата сначала выделяли основную часть недаиоггнвшегося нитрата уранила, для чего раствор встряхивали с эфиром. Затем после добавлении слабого восстановителя плутоний и нептуний осаждали в виде фторидов вместе с фторидами церия и лантана, использовавшимися в качестве носителей. Нерастворимые впводе фториды выпариванием с концентрированной серной кислотой переводили в растворимые сульфаты. Водный раствор последних обрабатывали броматом при обычной температуре. При этом нептуний окислялся до Йона (NpO2]" и при новом осаждении плавиковой кислотой оставался в растворе. Плутоний, выпавший в осадок вместе с носителями, снова превращали в сульфат ,и к .раствору последнего добавляли аммиак. Выпадавшие при этом гидроокиси растворяли в азотной кислоте. При добавлении персульфата я нитрата серебра плутоний окислялся до ионов [РпО3|" и на Этот раз при осаждении плавиковой Кислотой оставался, в растворе, тогда как носители выпадали в осадок в виде фторидов. Освобожденный от носителей раствор выпаривали с концентрированной серной кислотой. После разбавления водой вновь добавляли плавиковую кислоту и получали осадок,, состоявший из чистого фторида плутония)IV). Описанный метод работы интересе^.тем, что он демонстрирует принцип, при помощи которого плутоний проще всего можно отделить от наиболее близких ему элементов, использовав различие в отношений плутония и родственных _ ему элементов к окислителям и восстановителям.
Тетрафторид плутония в противоположность трифториду скдснёп к образованию двойных; Пли комплексных, солей, например NafPuFji, K(I'uF5| и RbfPuFj] (все эти соли изотиппы K[UFS[). Плутоний, по данным Андерсона (Anderson, 1949), не образует фторидной соли,* аналогичной Си[СеРй]2,а па против, кроме пентафторо-сол ей, Андерсоном были получены соединения типа М^РиоКдЕнопафтородпплутонатыПУ)]. Тетрафторид плутония с LaF3 образует аномальные твердые растворы, поэтому ионы Ри— со осаждаются не только с CeF4, но и с LaF3, Для СеР4п UF4, радиусы катионов которых близки к радиусу Pirt+, образование аномальных твердых растворов c.IiaFg было доказано рентгенометрически (Schlyter, Sillen, 1950).
Гексафторид плутония РиР6 образуется при действии F? на,РпЕ4 при 600? и конденсируется в холодной части прибора в форме бесцветного кристаллического порошка (т. н.ч, 50,7°, т? кйи. 62,3°). Он нзотипен UF0 и NpFe, но в отличие от них легко распадается с отщеплением F2.
Хлориды и бромиды. Хлорид плутония(1У) можно получить только в водном растворе. В твердом состоянии он неустойчив, хотя Ри1П в солянокислом растворе очень легко окислить до PulV. При упаривании солянокислых растворов PulV происходит их разложение вследствие гидролиза ими, если он затруднен, с выделением хлора. При прямом соединении Ри с С13 получается PiiCl3, который не изменяется даже при нагревании и жидком хлоре под давленном. Однако в токе хлора РпС)3 легче возгоняется, чем в отсутствие хлора, откуда можно заключить, что неустойчивый в Твердом состоянии РиСЦ при высоких температурах (600—800°) в газовой фазе может находиться в равновесии с РиС13 и С12,
Трцхяорид плутония I’uCl-, кроме непосредственного соединения элементов, получается также различными другими способами, например нагреванием РнС>2 со смесью Н2 и IIC1 или с СОЦ или S3C12. Проще всего безводное соединение получать упариванием раствора хлорида нлутоння( II1) и обезвоживанием получающегося при этом гидрата нагреванием в токе НС1. ТрихЙбрпд плутония легко растворяется в воде. Растворы его окрашены в пурпурно-красный цвет, безводное соединение — зеле
Трансурановые элементы
675
новато-голубо го циста. С парами воды РиС13 образует гидраты с 1; 3 и 6 молекулами воды. С NH3 и H2S ири 800—1000° трихлорид плутония реагирует с образованием puN и J’u2S,(.
При нагревании РиС13-6Н2О в запаянной ампуле получается оксихлррид; плутония РнОСЦ. Это соединение, как п РиС13, зеленовато-голубого цвета, по в отличие от него нерастворимо в воде, а растворяется только в кислотах.
Трибромид плутония РцВг3 получается так же, как и PuGI,, и очень близок ему . по своим свойствам и химическому поведению. Безводный РиВг5 зеленого цвета. Он при-; тягивает пары воды из воздуха, образуя зеленый ;Ке гексагидрат РиВг3-6П2О, и расплывается в конце концов в пурпурно-красный раствор. РпВг3-6Н2О изоморфен РпС13-6112О и NdCl -i-lMt.
Можно получить и оксибромид РпОВт, соответствующий оксихлориду. Напротив, PuBr4 в твердом состоянии также неустойчив, как и Р«С14. Можно получить водные растворы, бромида п..1утония(1¥), одпако бромид плутония(1У) и в водном растворе восстанавливается НВг до бромида нлутония(ПГ).
РцВг3 кристаллизуется в кубической системе и нзотипен NdBr3> 8шВг3 Lal3, VI3 и РпГ3. Напротив, Рi[С 1у имеет гексагональную решетку (а — 7,380; с — 4,238 А) и нзотипен следующим галогенидам;
ис!з РгС13 иВг3 РгВг3
(1=7,428 7,41 7,926 7,92 А
с=4,312 4,25 4,432 4,38: А
Другие данные, относящиеся к тригалогенпдам плутония, приведены в табл, 85.
Таблица 85
Температуры плавления, теплоты плавления, испарения и возгонки ti оитроппя плавления трига.'югснпдоп плутония
Соединение Температура плавления, :°с Теп.Tin та плавления, ?;»ал/лголё Те и ло-та. испарения, Теплота возгонки, К^ЬД/лЕОЛЬ Энтропия плавления, кал/.нол,е>
РйРз 1170 7,9- 88,7 96,6 5,5
PuClj 760 15,2 57,6 72,8 14,7
РиВгз 680 13; 4 56,5 69,9 14,0
СОЛИ ПЛУТОНИЯ
Соли трех-, четырех- и шестивалентного плутония образуются легко., д* Напротив, соли: пятивалентного плутония вследствие склонности его к диспропорционированию получаются с трудом. Соли нлутония(П) получить нс удастся.
Соли плутоппя(И1); частично гидролитически расщепляются водой, если гидролиз пе подавляется добавлением избытка кислоты. Еще более склонны к гидролизу соли плутонияЦУ). Сол и плутония(У1), если только речь идет ие о прочных комплексных соединениях, содержат плуто-iihh(VI) в форме радикала, плутопила [PuQJ3*,. аналогичного радикалу уранила [иог]',!+. Соединения плутонмла [РиО2]Х2 мало гидролизуются п в большинстве случаев хорошо кристаллизуются.
('очи нлутоппяОП). К солям влутопия(Ш), кроме солей кислородных Дислот, относятся также и уже упоминавшиеся гр и галогениды. Хлорид и бромид имеют отчетливо выраженный характер силон: они легко растворяются в воде и кристаллизуются пз водных растворов в виде гидратов. Трифгорид мал о растворим. С NaF он образует двойную соль NaPuIG. Светло-коричневый иодат Pu(IO3)3 плохо растворяется в воде. Бледно-краспый. трихлороацетат плутоппя(1П) Рп(СС1а- СОО)» мало растворяется в воде, по хорошо — в ацетоне. Очень легко растворяется перхлорат нлутония(Щ). Довольно легко растворяются фиолетовый оксалат и сульфат плутония(Ш), Послед-
43':
«76
Глава 14
ний легко образует двойные соли, из которых многие довольно мало раствор ими как это видно из следующих примеров;
Соединение КРц(5О4)2.5Н2О K5Pu(SO4)4 RbPu(SO4)2.4H2O
Цвет Растворимость, Лавандово-синий Почти бесцветный Светлый лавандевост [Ний
а Рн па 1 л 4,8 0,54 0,50
Соединение CsPu(SO4)2-4H2O (NH4)Ph(3O4)2.4H2O
Цвет Растворимость, ' Светлый лавандово-синий Светло-голубой
г Ри на 1 л 0,34 1,30.
Соли пл утопия (IV). Из солей плутоння(ГУ) особое место занимает нитрат, в связи с его чрезвычайной склонностью к образованию комплексов, хотя вообще нитраты редко образуют комплексные ионы. В водных растворах нитрата пл утони я( IV), по данным Хайндмена (1944), наряду с ионами [Pu(NOs)eI" присутствуют также ионы [Pu(NO3)j"'. Гексанитрато-соль (Nli4)2Pu(NO3)e изоморфна (ХН4)2Се(МО3)в и (NH4)2Tb(NO3)6. Хлорид плутония(1¥) в водных растворах, содержащих достаточное количество ионов хлора, существует в виде комплексных ионов IPuCl6]n. Кроме того, по Хайндмену, в хлорйдных растворах нлутопия(1¥) могут существовать и ионы [PuCljJ’.B сильно разбавленных растворах тенденция хлорида к образованию комплексных соединений выражена слабее, чем у нитрата, Из не слишком разбавленных растворов можно выделить кристаллические светло-желтые гексахлоро-соли, например CSglPuCl^j и [(СН3)4Х] (РнС1в]. Сульфат плутвния(1¥) кристаллизуется из водных растворов! н виде кораллево-красного или красно-коричневого тетрагидрата Рн(8О4)2-4Нг.О. Столько же молекул воды содержат гидраты сульфатов ypana(IV), церия(1У), тория и циркония. Сульфат плутопия( IV) с сульфатами щелочных металлов образует довольно трудно растворимые двойные соли М4 [Рп(8О4)4]-пН2О. Опи зеленого цвета и по составу аналогичны двойным щелочным сульфатам ypana(iv), цория(1У), тория и циркония. При гидролизе нейтрального сульфата образуется светлый с оропато-зеленый Pu2O(SO4)3‘ 8НаО. И в этом отношении плутоний похож на уран, торий и цирконий, которые также образуют аналогичные ок си сульфаты. При добавлении уШу>ат-ионов из растворов солей плутония(IV) выпадает желатинообразпый, почти бесцветный осадок. Он представляет собой смесь фосфатов, состав которых» зависит от" условий осаждения. Смиту YSmith, 1944) удалось получить кристаллическое светло-коричневое соединение Рн3(РО4)4-а:Н2О. Оно оказалось изотйппым соответствующим фосфатам церия(1У) и тория. Как почти все соли плутонияЦУ), фосфаты также способны образовывать в растворе комплексные ионы. Благодаря атому можно значительно увеличить растворимость малорастворимого фосфата плутония.
Соли ив утонил а( VI). Соли 'плутонила( VI), т. е. соединения общей формулы [PuVIO2lX2, значительно менее устойчивы, чем соли уранила. По в остальном их свойства совершенно аналогичны свойствам солей уранила. Легко растворимые соли Ш1утонила(¥1) получаются в водном растворе при окислении солей плутопня( IV) сильными окислителями, например солями церия (IV), бпхроматом, перманганатом прн нагревании или электролитическим окислением. При концентрировании растворов эти соли легко получаются в кристаллическом виде. Трудно растворимые соли йлутонялa(V I) можно получить двойным обменом из лег ко растворимых солей. В качестве примеров солей нлутонила(У1), кроме ацетата [РцО2] [C2HsO2Ia? 2И2О и уже упоминавшейся двойной соли последнего с ацетатом натрия, можно указать легко-растворяющийсл нитрат [РнО2] [ХО3[2-6Н2О и почти бесцветный студенистый фторид [PuO2JF2-^II2O. Фторид плохо растворяется в воде, во за счет комплексообразования растворяется в плавиковой кислоте. Из розового раствора при добавлении фторида щелочного металла можно выделить двойные или комплексные соли также розового цвета. Если к раствору нитрата плутонила(у1) Добавить ионы Г Fe(CN)6j'", то из него выпадет красно-коричневый осадок гексацианоферрата (Ш) плутонила(У1) [PuO2].,[Fe(CN)eJ2- Напротив, ионы [Fc(CN)e]"" восстанавливают ион [РнО2)” с образованием черного осадка.
Трансурановые элементы
677
АМЕРИЦИЙ (AMERICIUM) Am
Америций в соответствии с его положением в ряду актинидов можпо рассматривать кап аналог европия. Как и у европия, его основное состояние окисления 3" *.
Особая устой'швость трехпалентного состояния проявляется прежде всего на примере галогенидов америция. В тех же условиях, при которых нептуний образует тстрагалогеииды, америций дает тригалогениды. Например, при нагревании АтОг со смесью HF и Ог образуется трифторид америция AmF3. Нагревание АтО2 в парах СС14 приводит к образованию трихлорида америция AmCI3. При нагревании смеси АшО3 и А1 с парами брома всегда получается только трибромид америция AmBr3, а пе тетрабромид. Айалогичпо получается и трииодид америция Аш13. Тетрагалоге-нпды америция нельзя получить никаким иным способом. Например, Фрид (Fried, 1951) установил, что при пагревапии AmF3 с элементарным фтором до 500—700° тетрафторид америция пе образуется.
Галогениды америция изотнцны тригалогенидам нептуния и плутония. AmF3 —• светло-красный, ArnCl, и AinBr3 — бесцветные, ^Aml3 — желтоватый, АшС13 возгоняется при 850°, AmBr3 — в интервале 850—900’, Aml3 — при —900°.
Кроме галогенидов, америций и в других солях существует обычно в трехвалент-пом состоянии. Растворы солей амсриция(Ш) имеют характерный красный цвет. Теплота образования иона Am'", т. с. тепловой аффект реакции Am -I- 3HCI + aq = = Ат"г ЗС1' + 1 ’ДП», равен, ио данным Уэсгрума и ййринга (Weslrum, Eyring, 1951), J-160 7о,ля f .5 и. НС1 н 25°). Аналогичные теплоты образовании полу-
чены и для ионов лантанидов (например, для La " 167ты/моль, дляРг"' 166 ккал/моль). Для иона Ат"" рассчитанное значение теплоты образования составляет + 112 ккал 1г- ион.
По отношению к кислороду трехвалентное состояние пе является для америция предпочтительным. Несмотря на то что из растворов солей америция(Ш) при добавлении к ним аммиака или натриевой щелочи выпадает гидроокись америцня(Ш) Ат(0Н)3 (светло-красный студенистый осадок), из безводных окислов америция известны только мопооксид АтО и двуокись АтО2. Монооксид имеет структуру каменной соли (а = 4,95 А) и, следовательно, пзотипеп UO, NpO и РоО. Двуокись получается при нагревании гидроокиси или нитрата америция(III) на воздухе. Опа нс отщепляет кислород, даже если ее нагревать на воздухе до 1000°.
Если па черную двуокись подействовать разбавленной соляной кислотой, то она ц? с выделением газа и с сильным увеличением объема превращается в светло-красный продукт еще неизвестного состава (возможно, AmOCl). Растворение этого продукта происходит лишь постепенно. Раствор имеет характерным для солей америция(Ш) красный цвет.
Ащ(ОН)3 растворяется в концентрированном растворе К2СО3. Если к этому раствору добавить АаОС! и пагреть его, то выпадает темный осадок, представляющий собой гидроокись америция(1У) или америция(V) **. При нагревании на воздухе этот осадок превращается в АшО2.
* При переводе исключены все сведения, относящиеся к америцию(11), так как в настоящее время твердо установлено, что америций не может существовать в водном растворе в виде Катиона Дгп 2+ (аналогичного Енй+). Приводимые в оригинале сведения взяты из ранней работы, посвященной изучению химий америция индикаторными методами. Эти данные впоследствии не были подтверждены при работе с весовыми количествами америция (см. Сиборг и К а ц, Химия актинидных элементов, Атомиздат, Москва, I960.— Прим, перге.)
** В действительности эта двойной карбопат амсриция(У) и калия. См. Г. П, Я к о в л е в и Д. С. Горбенко - Ге риапов, Сб. докладов советских ученых, представленных на I женевскую конференции) по мнрпому использованию атомной энергии. Исследование л области геологии, химии и металлургии, Изд. АН СССР, Москва, Стр. 236, 1955.— Прим, персе.
678
Глава 14
Нагреванием AmO2 со смесью H2S и CS2 до 1400—1500° Фрид получил сульфид Am2S3, изотипный La2S3.
Подобно урану, нептунию и плутонию америции, кроме трех-и четырехъ ал снтпого, может быть также пяти- и шестивалентным. Однако соединения америция(У) и амернция(У1) еще менее устойчивы, чём соответствующие соединения плутония, Поэтому сходство америция с ураном проявляется в значительно меньшей степени, чём сходство с ураном предшествующих америцию трансурановых элементов. Но в тех случаях, когда америций в своих соединениях оказывается пяти- пли шестивалентным, свойства этих соединений, если не считать их гораздо меньшей устойчивости, совершенно аналогичны свойствам соответствующих соединений урана.
Соединения америция(У) можно получить электролитическим окислением соединений амерпция( 111), Они очень мало устойчивы, по не настолько сильно^ как соединения питутония(У), и менсо склонны к дпспронорцпопировапию, ибо п соолпиешгя амерпцпя(У1) неустойчивы. И все же Эспри (Asprey), Стефеиоу (Stephanpu) й Попне-мен (Реппеman, 1951) наблюдали,' что при добавлении к раствору америцпя(у) в 0,3 н. растворе НСЮ4 такого количества H2SO4;. чтобы раствор стал 4 и. по H2SQ4, происходит диспропорционирование но уравнению ЗАт'1 ==. Ат111 + 2Ат^, что можно установить по спектру поглощения раствора. Растворы солей америция(Щ) имеют характерные полосы поглощения при 504 и 811 йц. Растворы солей америция(V) поглощают при 514 и 714 лщ, а растворы солей америция(¥1) имеют сильную узкую полосу поглощения ври 991—992 .ир, а также сильно поглощают в ультрафиолетовой области спектра.
Чистые растворы солей америция(У1) были получены Эспри с сотрудниками при окислении Ат111 в солянокислых плп перхлоратных растворах персульфатом а миопия, Эти растворы имеют чистый желтый цвет и пе дают осадка при добавлении ионов F'. При добавлении ацетата натрия можно получить тетраэдрические кристаллы натрииамерицил(у1)ацетата Na[AmO2(C2Ef3O2)3[. Он кристаллизуется в кубической системе (а == 10,6 А) и изоморфен соответствующим соединениям уранила, нептунила и пл утони л а. Показатели преломления двойных солей составляют: для уранила — 1,501, плутонила — 1,518 и амерпцила — 1,528. Нитрат амери-цила(У1), как и нитрат уранила, можно извлечь из водного раствора встряхппа, нием с эфиром.
Металлический америций впервые был получен Уэструмом и Эпрмн-гом (1951) восстановлением AmF3 металлическим барием при 1100°. Работу производили с количествами америция от 0,04 др 0,2 ,мг; тигельки из . ВеО нагревали в вы соков аку умной мйкропечн. Америций представляет собой с сребрист о-белый, ковкий металл, кристаллизующийся йо типу магния (а — 3,642, с — 11,76А; йпикн —: И»7, , dpeHTP = 11,87). Уже при небольшом нагревании он соединяется с II,, образуя гидрид америция.
От редких земель, которые применяют в: качестве носителей при выделении америция, Ат можно отделить дробной кристаллизацией фторидов, так как AniF3 растворяется несколько лучше, чем фториды редких земель.: Другой путь, который также позволяет осуществить разделение, заключается л применении ионообменной смолы. При этом поступают следующим образом: солянокислый раствор, в котором присутствуют различные ионы, меллеппп пропускают через колонку, наполпеппую подходящей йонообмеппой смолой. При этом ионы америция Ат"' и редких земель удерживаются на “смоле. И если затем эту смолу промыть слабокислым раствором цитрата аммония, то в раствор перейдут в первую очередь те ионы, которые образуют наиболее прочные комплексы с цитрат-ирпами. Первые порции цитратного раствора, выходящие из колонки,, содержат ионы лантана и церия. Затем вымываются ионы америция и только потом ионы остальных редких земель.
Трансурановые элементы
679
КЮРИЙ (CURIUM) Cm, БЕРКЛИЙ (BERKELIUM) Вк. КАЛИФОРНИИ (CALIFORNIUM) Cf
Трансурановые элемеити 9GCm, ^Вк и 9SCf являются аналогами e4Gd, 65Tb и e6Dy и по своему поведению особенно близки перечисленным лантал пидам. ,Так же как ото редкоземельное элементы с трудом поддаются разделению, так и отделение перечисленных трансурановых элементов друг от друга л от редких земель представляет значительные трудности. В таких случаях используют те же методы, которые применяют для разделения редких земель. Работа с к Юрием, берклием и. к ал ифорнйем осложняется еще и тем, что эти элементы особенно сильно радиоактивны. Вода при i растворении соединений этих элементов нагревается под действием их собственного излучения и разлагается с выделением газов.
Кюрий, как и гадолиний, насколько известно в настоящее время, бывает трезвалюпным *. Растворы его солей бесцветны. Они поглощают только в ультрафиолетовой. области спектра, как и растворы солей гадолиния. Отсюда, а также из магнитных свойств этих воединопий мошна заключить, что электронная конфигурация иона июрпя совертлигию аналогична о.тектропнон конфигурации иона тадблнния, т: е. в ноне Сиг’* [(мается 5/'"-оболочка, соответствующая -оболочке в ноне GdB+ (см. С'Г]>. 636 — 657).
Из растворов содой кюрия при добавлении аммиака выпадает гидроокись Gin(0H)3, при добавлении H2F2 — фторид СшК, — практически нерастворимый осадок. Прп прокаливании гидроокиси па воздухе получается полуторная окись СпъО-. Следовательно, при этом не образуется двуокись, как в случае америция. Металлический кюрий был : впервые получен Крейном (Gruне) в 1951 г.
Берклий, как и его аналог тербии, может быть переведен Я четырехвалентное состояние, но, как и тербий, обычно бывает шрейалептиым.
ЯРлмрерный подобью диспрозию известен только в жреявахентном состоянии. *0п, как и кюрий и берклий, проявляет свойства, типичные для редких земель. Ий для одного из этих трех трансурановых элементов не известны такие реакции, в которых их поведение отличалось бы от поведения редкоземельных элементов.
ЭЙНШТЕЙНИЙ (EINSTEINIUM) Es, ФЕРМИЙ (FERMIUM) Fm МЕНДЕЛЕВИЙ (MENDELEVIUM) Md
По химическому поведению элементы MEs, ,wFm и i0IMd являются аналогами лантанидов 67Но, 08Еги еэТи. Отличительной чертой этих элементов с особо высокими порядковыми номерами и атомными весами является большая скорость спонтанного делепйя ядер. Это свойство проявляется уже у тяжелых изотопов калифорния. Период полураспада для спонтанного деления и случае 2i0Cf составляет около 5000, лет (примерно в 400 раз больше, чем для а-распада), для ?’2Cf Он равен 60 годам, для 2«4Fm — только примерно 200 дням, а для 230Fm —даже только 3 час. В связи со все возрастающем тенденцией к спонтанному делению ядер можно предположить, что должен существовать предел практического получения ядер с еще большими порядков ими номерами и атомными весами.
* В последнее время получены соединения CniflV), правда, малоустойчивые, например CmF4 (Asp re у eta!., J. Am, Chem, Soc., 1955—1957).— Прим, персе.
680
Глава 14
НОБЕЛИЙ (NOBELIUM) No
Вследствие очень малого периода полураспада химические свойства нобелия еще не были непосредственно изучены. Однако доказательства того, что оп является элементом с порядковым номером 102 (и, следовательно, аналогом 70Yb), получены химическим путем. Элемент, образующийся при а-распаде нобелия, был химически идентифицирован как фермий. Наибольшее количество Fin, которое имелось в распоряжении исследователей в каждом опыте, составляло около 40 атомов.
РАДИОАКТИВНЫЕ РЯДЫ ТРАНСУРАНОВЫХ ЭЛЕМЕНТОВ
С наиболее тяжелых из встречающихся в природе неустойчивых элементов, предшествующих трансурановым элементам, начинаются, как было сказано в гл. 11, длинные радиоактивные ряды. Все радиоактивные ряды трансурановых элементов включаются в радиоактивные ряды естественных элементов. Это значит, что среди продуктов радиоактивного распада трансурановых элементов через известное число превращений всегда появляется такой изотоп, который является членом естественного радиоактивного ряда, и дальнейший распад происходит так же, как и в естественном радиоактивном ряду.
Так как радио активный распад тяжелых элементов сопровождается или уменьшением массового числа на 4 единицы (а-распад) или массовое число вообще не изменяется (р-распад, А'-захват), то массовые числа всех членов данного радио активного ряда могут быть выражены одпой формулой. Массовое число изотопа тория, с которого начинается радиоактивный ряд тория, равно 232. Это число является целым кратным 4, Поэтому массовые числа всех членов радиоактивного ряда тория являются целыми кратными 4 и, следовательно, могут быть выражены формулой 4п,. где п — целое число. Аналогично массовые числа членов радиоактивного ряда урана — радия передаются формулой 4я + 2, а членов радиоактивного ряда актиния — 4« 3.
Радиоактивный ряд нептуния. Кроме того, имеется и такой радиоактивный ряд, для массовых чисел членов которого справедлива формула 4л + 1. По наиболее дойгоживущему изотопу, входящему в этот ряд, он получил название радиоактивного ряда нептуния. Долгоживущим^ изотопом нептуния, входящим в этот радиоактивный ряд, является 237Np. Так как этот изотоп встречается и в природе (хотя и в очень малых количествах), то радиоактивный ряд нептуния также можно считать естественным радиоактивным рядом,
В радиоактивном ряду нептуния встречаются только те атомы, которые не входят пи в один из других радиоактивных рядов.- Но все члены радиоактивного ряда являются изотопами члепов других радиоактивных рядов. Предпоследний/ членом радиоактивного ряда нептуния является очень слабо радиоактивный висмут. Устойчивый конечный член этого ряда — таллий. 13 этом состоит самое существенное отличие радиоактивного ряда нептуния от трех остальных радиоактивных рядов, где нсегда устойчивым конечным членом является свинец.
. Радиоактивные ряды других трансурановых элементов. Из табл. 86—89 видно, к каким естественным радиоактивным рядам относятся изотопы трансурановых элементов. Распад трансурановых элементов с массовыми числами М = 4л всегда в конце концов приводит к элементам, которые являются членами радиоактивного ряда тория
Таблица 86
102
101
100
99
98
97
96
95
94
95
92
91
90
89
88
87
86
556Ез
Радиоактивные ряды трансурановых элементов, I
Радиоактивный ряд 4п
,?56Fm^<^
sseMd
г!2Гт
Спонтанный 25гр-васпад я&ра
8 час ss:Cf
2,2 лет
а -140
^0
<х
г44Ст 4’7''°5 Л4Ат^С лет о Л,-
ссшчас rcJ^Es
Mai л 125
^Гг 0,8%1ман
а 3^0
а 4,4 0,006% час
жАт 1
34<cf
а
25 мне
жс™
w'Pu
19 лет
1С.
<а*
26,S й дн
6,6-да3 л’
лет
1270' cq
| лет
232Th
„ 1,4-Ю10 лет т
v
^MsTh/w
85
84
83
82
81
«ери.
232у гз2Ра<^ <х
70 л лет к'Йр
™Pd*
’' а 22
^RdTh 2% :
^eMsTh2 ,
7OU
22Л * л года
2,5 час
1,9
гг4ТЬХ
Л
Раоис-акотиеный
гмЕг
И2Ри
а
v-
22ВЦ
36 мин
80%
7 мин
Z2«Th
быстро
2ZQRa
«г йгсда
ряд тория 50 1'
* сек 216 Ет
г,6ТЬАЬ
I
Радиоактивный ряд тория
Очень iuanpo
mThC'
Paho-u активный
ряд тория
Таблица 87
Z
102
.101
100
99
98
97
96
95
94
93
92
91
$0
89
88
87
86
85
84
83
82
Радиоактивные ряды трансурановых элементов,!!
Радиоактивны!! ряд нептуния (4n+1)
“’Fm
t,i>* a 4,5 ’ Ю% дн
20 249р£>'
?" е>-|
2*98k"^S л|4„0£ .<*“5ВЬ'*^
О.ООЩ
wEs
<х 0,1%
6f^cf
44
О,^" 1,4-Ю4 ^АпГ" лет р
241Nd-^C- 13 0003% гз7и:
а.
•_ 2S7Np
<К;
гять-^
гззРа
2час
34% I мин
х 35 1%„дя 458 z-srp.^
лет и
Йр* 456 0г003%
~ 0р01%\жт
J.4a;
*|1Д
WC
J^Ac 1 4°
и дн
221РГ а и мин
1Г
2,7At 0,0',8
47 мин
2,25-10s —-лет
G(
<- -0005% 1тос
1,3 гззри г^^1^20 Р 0,1%*™
35 ггэЛ г; )tg6^'
V10 ас j здС 2%,, гг5ТЪ
« 7-5 , < ма1
22’Ra
1,63-Ю5 лет
0,1%,
г2эти 1 7-Ю3
т
<х риот/w
2,7 Em
а
а 4% .гоар^'
[Отель «Змсдаро
,213Ро
4 7- 1O~S
। Лчас а ^РЪ^’2,5-10<7
лет ,
2°5TL
*
81
Таблица S8
Радиоактивные ряды трансурановых элементов, Ш
/ j Радиоактивный ряд(4пч-2)
102 ®*N°
101
100
99
98
97
95
95
94
93
92
91
90
89
38
87
85
85
84
2S*Cf
25^1
-sfV16 35 ?
Спонтанный распад ядра 56 дн
83
82
31
а З сек
»«Fm
280 дн
JSQBk
Спонтанный распад ядра а'
,.2-104лет
™Ат'ф*
^Fm
30 2«Es
Я лкаг
а 3,24
>ftjac
w>Cf
ивВ!{ w \лет
5-Ю3 лет к
ISO*1’
242pQje<-.
3,7-105 лет
of
258UI |4,5-Юэ • лет а* гэЧ’Х, *
ji2Am
л Mil#
WSQf
35,7 час
а
242Вк
24£Ст :5'?\|?бг15 а\дн
сг 400
0,2% лет f
23SNp г'
к г¥Ри
г’«Ат
9#
? гз^Ст li-V 'г' а
2Д час
j»*
cf
23фц
86,4
лёт k^-nP 1% 1
<г^я ,г
ЛЗ EJO0
o' 8,5
' 1% час
р'
234т г
иЛ2 активный ряд урака-радия
20.8 d дв
E2STh
30,9 мин
222Ra
а33 сек
2,г‘ Ет
I Радио-активный ряд урана-радия
Таблица 89
Радиоактивные ряды трансурановых элементов, &
Радиоактивный ряд(4п+3)
102
101
100
99
98
97
96
95
94
93
92
91
90
89
88
87
86
85
84
83
25SEs
82
31
. 2S5Fm
255мо(?)
а |~Юл1шг
251 Fm
** 1215" 25Гт- ^1*?
! йЬ«с М7час
* '’• а 7 5
0,5
247BkJ
5^.
7-Ю3
,лет г
Агт) 99,7%
8-)0э Л лет >«'
25ICf
а
660 лет
jrtiib
мзВк
М7Ст
4-/0т
лет' б л!
^Ри
а
of
ZJSjj
: « 4.5 0,15% час г4т *
35 лет к
^Ри 0,304%
2,44-Ж4
“ лет it.
s5®*
0,001% дн
2i5AcU
Радиоактивный ряд актиния
12 у ас ”6Np.^ .* 410
23SCm ^0:
26
0,003% мин 231J
К
™Np
53
231 ра
I Радио-активный ряд актиния
,,7 227RdAc
Радио-
1% 1лад
227Ра
58 30% мии ггзАс
активный ряд актиния 2 мин
2(3Fr
10'4 сек
sl5AcAt
I
Радиоактивный ряд актинах
о
Tpuficypakcanc элементы
685
(радиоактивный ряд 4я), Точно так же радиоактивные ряды трансурановых элементов -с массовыми числами М = + 1 и конце концов впадают в радиоактивный ряд неп-
туния. Аналогично распад трансурановых элементов с массовыми числами 4л + 2 приводит к радиоактивному ряду урана — радия, а с массовыми числами 4л 4- 3 — к радиоактивному ряду актипия.
К продуктам распада 232Рн, принадлежащего к радиоактивному ряду 4л, относится инертный газ пт, являющийся изотопом эманации тория. Среди продуктов распада нептуниевого радиоактивного ряда эманации нет, хотя изотоп эманации встречается в боковой линии нептуниевого радиоактивного ряда, а именно средн: продуктов распада Это ядро представляет собой такой изотоп урана, который не встречается л природе и получается толмго в результате ядерных реакций. Среди продуктов распята 234Ри (радиоактивный ряд —4п4-2) также существует эманация р18Егп). В этом месте радиоактивный ряд впадает в радиоактивный ряд урана радия. То, что в этом радиоактивном ряду, кроме эманации радия, встречается еще одна эманация, уже указывалось ранее. Кроме радиоактивных рядов, образованных продуктами распада трансурановых элементов, В радиоактивный ряд урана — радия впадает еще одип радиоактивный ряд, образованный продуктами распада искусственно получеппо: о изотопа протактиния:
91 1 а---------—— .-ас
1,5 мин
----2—> $SFr _-----» sl4At ------------IJ’BaE -ч, .
Быстро Очень быстро Очень быстро
К В a F (полонию}, образующемуся в естественном радиоактивном ряду при распаде ВаЕ, приводит также распад искусственно полученного изотопа астатина;
к
g^At Д|° ВаЕ. Как уже было указано ранее, RaE переходит в ВаС (урановый
-свинец) по только через BaF, по и в очень незначительной степени (0,00005%) через жороткоживупшй изотоп таллия:
»J»RaE —^Т1.—«osRaG.
5 дн 4,2 мин
Глава 15
РАСПРОСТРАНЕНИЕ ЭЛЕМЕНТОВ; ! ГЕОХИМИЯ
Состав земной коры. Очевидно распространенность различных элементов на Земле весьма пе одинакова. Это ясно следует уже из данных, которые были приведены для отдельных элементов. Сопоставление долей, которые приходятся на отдельные элементы в среднем составе земной коры, приведены в табл. 90. Под «земной корой» при этом понимают каменную кору Земли на глубину до 16 к.и от уровня моря, причем к пен причисляют также океанские и внутренние воды, а также воздушную оболочку. Последнюю называют атмосферой, общую массу воды -г гидросферой, а каменную кору литосферой.
На литосферулприходится (при расчете до указанной глубины) 93,06 вес. % земной коры, да гидросферу 6,91% н на атмосферу 0,03%. ,
По данным Кларка, литосфера состоит, на 95,0% из вулканических пород, на 4,0% из сланцев, на 0,75% из песчаных и па 0,35% из известковых пород.
Состав гидросферы, по существу определяется составом морской поды. Согласно Вернадскому (1924) и Свердрупу (Sverdrup, 1942, ср. также Мазон), средний состав последней в весовых процентах выражается примерно следующими цифрами;
О 86,03 Вг 0,0065 Ar 5-10-s Pb. 4,5-10-’ ::
н 10,70 С 0,0053 Rb 2-10-s •’ Sc 4-10-’
GI 1,90 N 0,0017 Li 1-10-5 Sn 3d0"’
Na Mg 1,06 0,127 Sr В 0,0013 0,00046 : P Cu 1-10-5 9-10-5 Cs 2-10 ’ IJ 1 • 10 ’
s 0,088 Si . 0,00035 Ba 5-lQ-e Ga 5-40-8
Са 0,040 Al 0,00019 I 5-10-e Ag 2-10-s ;
К. 0,038 Fe F 0,00015 0,00014 As Zn Mn 2-10-e 1-40-6 ГАО"6 An 6- 10_]й Ra 3-10 11
В незначительных концентрациях в морской воде обнаруживается еще ряд других элементов, из которых Cd, Сг, TI, Sb и Zr содержатся в морских организмах.
Атмосфера вблизи поверхности Земли цри среднем содержа пни водяных паров 0,27% состоит на 75,31% из азота, на 22,95% из кислорода, на 1,43% из инертных газов и иа 0,03—0,04_из двуокиси углерода *. Содержание водяных паров сильно колеблется в заииснмостн от температуры и других условий (при 0° воздух может содержать воду в виде паров максимум 0,43 вес.%, однако при 40е уже до 5,82 вес. %). В остальном же состав атмосферы очень равномерен. Ранее считали, что он быстро изменяется с высотой иод действием силы тяжести, Полагали, что на высоте примерно выше 12 км происходит постоянное изменение в соотношении составных частей атмосферы в пользу более легких. Рассчитали, что на высоте выше 20 км в атмосфере больше нс содержится аргона, Выше 85 к.м из нее должен будто бы исчезнуть такще кислород, а выше 100 жат — азот, после чего атмосфера должна состоять только
* Эти данные также приведены в: весовых процентах.
Распространение эле.чентое; геохимия ; 687
из водорода, возможно в смеси с 1% гелия. Исследовании проб воздуха, которые были взяты с большой высоты при помощи ракет, этого пе подтвердили. До 60 км пе было установлено никаких изменении в соотношении составных частей атмосферы; только еще выше было замечено такое изменение (Paneth, 1950—1951). Следовательно, конвекция, которая препятствует «расслаиванию» атмосферы, проявляется на гораздо большей высоте, чем это считали ранее. Без сомнения, однако, можно считать, что Isa очень больших высотах в атмосфере начинает наконец преобладать водород. Под действием ультрафиолетовых лучей солнца он отчасти распадается на атомы, которые в свою очередь частично ионизируются ультрафиолетовыми лучами. Атомы водорода и в еще большей степени образовавшиеся в результате ионизации цротопы оказываются В состоянии покидать поле тяготения Земли. Для жизни па Земле это имеет исключительное значение, ибо водород появляется в атмосфере главным образом вследствие того, что пары воды в высоких слоях .атмосферы разлагаются ультрафиолетовыми лучами на составные части. Освобождающийся при этом кислород диффундирует постепенно к поверхности Земли; это приводит к тому, что, хотя в точение всей истории Земли кислород постоянно используется в большей степени окислительными процессами, чем отдается земной поверхностью в воздушное пространство, атмосфера все же содержит еще достаточное количество кислорода, чтобы поддерживать жизнь. С возрастанием атомного веса элемента способность покидать поле тяготения Земли быстро снижается. Кислород практически не уходит в мировое пространство.’ Уже в случае дейтерия часть атомов, которые докидают Землю, значите ль по меньше, чем в случае водорода. Поэтому, как показал Хартёк (Harteck, 1949), атмосферный водород заметно обогащается дейтерием. В атмосферном водороде было найдено соотношение D : Н 1 : 4500, в то время как в обычной воде оно составляет 1 : 6<X)t). Так как плотность атмосферы, даже если отвлечься от изменения ее состава, быстро падает с ростом высоты *, то пес части атмосферы, лежащей выше 100 км, совершенно ничтожен в сравнении < общим весом. Одггяйо некоторая часть атмосферы, хотя и в крайне разреженном состоянии, простирается даже выше 200 км. Это подтверждается тем. что на таких высотах наблюдается еще «падение звезд», свечение которых обусловлено трением, испытываемым ими в земной атмосфере.
При рассмотрении табл. 90 становится ясным, что имеется сравнительно небольшое число элементов, которые по весу образуют главную часть земной коры. Почти половина веса земной коры приходится уже только на кислород, более четверти — на кремний. Двенадцать наиболее часто встречаемых элементов составляют вместе уже 99,5% земной коры; следовательно, на все другие, вместе взятые, элементы; приходится только 0,005 общего веса. За вычетом двух первых, десятков на другие элементы остается только 0,0007 общего веса. На 52 элемента, следующих за торием, следовательно, на 56,5% общего числа встречающихся: в природе элементов,. приходится вместе только 0,0001 общего веса земной коры.
Следует отметить далее, что в общем составе земной коры имеется, сравнительно большая часть элементов, которые обычно считаются «редкими»; это прежде всего титан, кроме того, цирконий и ванадий. Даже большинство элементов «редких земель» в общем активнее участвуют в строении земной коры, Чем некоторые обычные тяжелые металлы , например висмут, ртуть, кадмий и сурьма. Титан относится даже к наиболее распространенным элементам: как видно из табл. 90, он принимает почти такое же большое участие в строении земной коры, как 80 элементов, следующие за фосфором, вместе взятые.
Если вместо весовых процентов расчет произвести в атомных процентах (см. табл. 91), то водород перемещается на третье место, в то время как. кислород и кремний сохраняют свои места. Углерод, также передвигается вперед па несколько мест, то же относится к азоту, в то вромл как железо с четвертого места
* Вблизи уровня моря при подъеме па 11 .«• давление падает примерно ла 1 .и.и рт ст. Для большей разницы высот следует учесть, что давление воздуха падает с высотой не равномерно, а по экспоненциальному закону. Еслп пренебречь изменением состава воздуха, для высоты 100 км лавдеппе равно 0,001 мм рт ст.
Таблица 90 Относительная распространенность отдельных элементов (Состав земной коры, включая водную я воздушную оболочки, дан в весовых процентах)
% %
1 / Кислород 49,501 44. Самарий 6,0-10-4,
2. Кремний 25,80 45. Гадолиний 5,9-10-4
3. Алюмипнй 7,57 46. Германий 5,6-10-4
4. Железо 4,70 47. Мышьяк 5,5-10-4
5. Кальций 3,38 48, Бериллий 5,3-10-4
б. Натрий 2,63 QQ £4 49. Празеодим 5,2-10-4
7. Калий 2,41 ' УУ.,01 50. Скандий 5,1-10-4
8. Магний 1,95 51. Гафний 4,2-10-4
9. Водород 0,88 52. Диспрозий 4,2-10-4
10. Титан 0,41 53, АргСш 3,6-10-1
11, Хлор 0,19 54. Уран 2,9-10-4.
12, Фосфор 0,09 55. Иттербии 2,5-10-4
13. Углерод 0,087’,| 56. Эрбий 2,3-10-4
14. Марганец 0,085 57. Гольмий 1,1-10-4
15. Сера 0,048 58. Европий 9,9-10-4
16. Азот 0,030 59. Тербий 8,5-10-s
17. Рубидий 0,029 60. Селен 8-10-s
18. Фтор 0,028 61. Лютеции 7,0-10-5
19. Барий 6,026 U, 42 62. Сурьма : 6,5.10-5
20. Цирконий 0,021 63, Ртуть 4-10-5
21. Хром 0,019 64. Кадмий , 3-10-5
22. Никель 0,015 65. Таллий 3-10-5
23. Стронций 0,014 66, Висмут 2-10-5
.24. Ванадий 0,014 67, Тулий 1,9-10-в
25. Цинк 0,012 1 68. Индий 1-10-5
26. Медь 0,010 69. Серебро — 110“5
27. Вольфрам 0,0064 76. Иод 6-10-в
28. Литий 0,0060 71. Рутений ~2-10-с -тГ
29. Церии 0,0043 72. Осмий - l-10-e
30. Кобальт 0,0037 73. Палладий — 1.10-4,
31. Олово 0,0035 74, Теллур : — 1-10-в
32. Иттрий 0,0026 0 06 75. Золото ~ 5-10-7
33. Неодим 0,0022 76. Платина ’ ~ 5-10-7
34. Ниобий 0,0019 77. Неон. 5-10-’
35. Свинец 0,0018 78. Гелий 4,2-10-7
36. Лантан 0,0017 79. Иридий —1-10-7
37. Бор 0,0016 80. Родий - Н Ю’7
38, Галлий 0,0014 81. Рений ~ 1 10-7
39. Молибден 0,0014 1 8‘Л Лантан 1,9-10-8
40. Торий 0.0011
83. Ксепон 2,4-10-9
41, Тантал 8-10-т
42, Цезий 6.5- 10-4 84. Радин 9,5-10-41
43, Брон 6-10-4 85. Протактиний 9,0-10-и
Распространение элементов; геохимия
689
Продолжение табл. 90
% %
86, Актиний ,6,1-10-14 : 90. Плутоний 2-10-1®
87. Полонии 2,1-10“14 91. Франций 1,3.10-31
88. Радон 6,2-Ю-16 92. Астатин 3• 10“ 24
89. Нептуний 4.10-17
Таблица 9.1
Содержание наиболее распространенных элементов в земной коре в атомных процентах
1. О 55,00 7 . Fe 1,49 13. Р 0,052 19. Сг 0,0064 25; Zn 0,0033
2. Si 16,35 8. Mg 1,42 14. N 0,038 20. Rb 0,0060 26. Sr 0,0029
3. Н 15,52 9. К 1,10 15. Мп 0,028 21. V 0,0049 27. Cu 0,0028
4. AI 4,99 10. Ti 0,152 16. S 0,027 22. Ni 0,0046 28. В 0,0027
5. Na 2,03 И. С 0,129 17. F 0,026 23. Zr 0,0040 29. Co 0,0012
6. Са 1,50 12. С] 0,095 18. Li 0,016 24. Ba 0,0034 30. Be 0,0011
перемещается па седьмое, ниже натрия п кальция. Титан сохраняет свое место в качестве десятого члена ряда, даже при расчете в атомных процентах.
Табл. 91 содержит 30 наиболее распространенных элементов, расположенных в ряд по их распространенности, выраженной в атомных процентах. Эти элементы не только наиболее распространенные, но и находятся также среди наиболее важных Для человечества элементов. В трех первых колонках таблицы помещены важнейшие неметаллы, важнейшие легкие металлы и важнейший тяжелый металл (железо). Среди элементов, стоящих в трех первых колонках, оказываются все так называемые жизненно важные элементы, например С, О, Н, N, К, Са, Mg, Fe, Р и S, без которых растительная и животная жизнь невозможны.
Первые 16 элементов, приведенные в табл. 91, составляют 99,9 ат. % коры Земли, а 30 элементов более чем 99,99 ат.%. На остальные 62 элемента, встречающихся в земной коре, приходится округленно 0,02 ат.%.
Распространение и доступность элементов. Многие элементы, принимающие сравнительно большое участие в строении' земной коры, например титан, цирконий и ванадий, обычно причисляются к «редким». В то же время такие металлы, как свинец, олово, висмут и ртуть (которые в соответствии с современным состоянием наших знаний, вместе взятые, не так распространены, как один ванадий) обычно отнюдь не считаются «редкими». Это связано с тем, что последние металлы накапливаются в значительных количествах в определенных «месторождениях» и поэтому легко доступны, в то время как титан, цирконий и ванадий сравнительно редко встречаются в больших скоплениях. Однако в незначительных количествах они встречаются почти во всех породах *, и это приводит к тому, что они в общей массе превосходят металлы, встречающиеся преимущественно в месторождениях, ограниченных какими-то местами.
* Следует отметить_ значительное обогащение многими «редкими» элементами, такими, как Ge, Ga, Sc, У Мо, золы малозольных каменных углей (Goldschmidt, 1930" Thilo, 1934).
44 г, Реми
690 Глава 15
В связи е тем, Что многие металлы концентрированно встречаются лишь в отдельных местах, в так называемых рудных месторождениях, появляется значительная трудность при оценке их количеств по отношению к тем элементам, которые широко распространены в качестве составных частей породы, хотя обычно лишь л незначительных концентрациях. В то время как о распространенности собственно образующих породу минералов (и, следовательно, о распространенности содержащихся в них элементов, включая редкие) сведения достаточно надежны, число и протяженность рудных месторождений удается оценить только приблизительно. Поэтому данные табл. 90, относящиеся к подобным металлам, обладают не такой надежностью,, как данные для элементов, образующих породу. Особенно относится это к редко встречающимся металлам, таким, как серебро, висмут, ртуть й золото; Для этих металлов в результате появления новых данных могут произойти еще существенные изменения приведенных числовых значений. Напротив, данные, который'в настоящее время приняты для оценки сравнительного распространения основных образующих породу элементов, в большинстве случаев лишь мало отклоняются от тех, которые были, рассчитаны К.иарком уже 40 лет : назад * **.
Несмотря на то что некоторые металлы сконцентрированы в отдельных 'месторождениях, наряду с этим они встречаются также в ничтожных концентрациях, в очень многих породах. В таких случаях нахождение в породах, которое ранее часто не принимали во внимание, может иметь большое значение для оценки среднего распространения металла и земной коре. Так, ранее на основании распростри ценности собственных руд сроднее содержание цинка в земной коре принимали около 0,005%. Однако затем Иоддаки (I- iind W. ffoddack, 1934) нашли, что изверженные породы содержат в среднем по меньшей мере 0,007% цинка. Так как изверженные породы составляют примерно 88,4% от веса земпой коры (включая гидросферу и атмосферу), то опи содержат в общем больше цинка, чем имеется этого металла в форме руд в цинковых месторождениях. Еще больше на распространенность платиновых металлов влпяет их нахождении л минимальных концентрациях в породах, панример в олив и новых породах и во многих распространенных рудах, например в хромистом' железняке и в молибденовом блеске. По данным Нодцака^ если принять во внимание нахождение в породах, содержание элементов в земной коре возрастает для рутения примерно в 15 000 раз, для радия в 6000 раз, для палладия и платины более чем в 100 000 раз, для осмия в 1000 раз и для иридия примерно в 700 раз по сравнению с теми величинами, которые получают из нахождения этих металлов в месторождениях платиповых руд1=¥.
Цифры, получающиеся для распространенности таких элементов, которые встречаются в общем только в незначительных количествах (однако в очень многих породах), зависят от точности, с которой незначительные концентрации этих элементов удаётся определять. Это приводит к тому, что новые дапцыо о распространенности этих элементов часто значительно отклоняются от старых. Во многих случаях для определенной группы элементов взаимпоо количественно о соотношение известно существенно точнее, чем их общее содержание в составе земной коры. Это относится, например, к элементам редких земель (ср, стр. 693). Точно так же, по данным Гольдшмидта (1937), количественные соотношения как наиболее верные можно рассматривать для следующих металлов:
’ -"Г
Rh • Ir : Pd;: Pt: Ag : Ач=1:1 :10 : 5:100: 5,
в то время как данные об их концентрации л земной коре (табл. 90), возможно, правильны только по порядку величины. Количественное соотношение РЬ : Th : U в земной коре в соответствии: с новыми исследованиями равно 10,584 : 0,156.
Даже в случае таких металлов, которые встречаются в рудных месторождениях в очень больших количествах, часть их, которая там находится,
* Еще не вполне ясна сравнительная распространенность Sfh Ба, В противоположность приводимым обычно данным, Нивопбург (Nieuwenbnrg, 1935 и сл.) нашел при анализе изверженных пород, что содержание стронция значительно превышает содержание бария.
** Для сравнения здесь положены в основу данные, Предложенные Бергом в 1929 г.: Ин Rh I-10’12. Pd 6-Ю-11, Os 9-10~i8, Ir 5.10-1®, Pt 1,2-10-1°
(в частях От 1). Ии данных табл. 90, которые, кроме сведений Нодддка, базируются па цифрах Гольдшмидта (1937), получаются отчасти более низкие относительные числа, чем приведенные выше. Однако в том й в другом случаях содержание платиновых металлов оказывается много больше, чем рассчитано lie содержании этих металлов в месторождениях платиновых руд.
Распространение элементов; геохимия 691
исчезающе мала по сравнению с общим содержанием их в слоях земной коры, легко доступных для горной разработки. Это следует из данных табл . 92. Если сравнить мировой запас руд, приведенных в таблице металл лов, в месторождениях, известных к 1930 г;, с годовым мировым потреблением. то станет ясно, что для многих важных металлов старые месторождения были бы уже исчерпаны, если бы не были открыты новые.
Таблица 92‘
Мировой запас некоторых металлов л известных месторождениях [По данным I. и W. Npddack (1936)1
Металл" Мировое потребление в 1030 г., m Известный к 1930 г* мировой запас в месторождениях, гл Общее количество в земной коре ТОЛЩИНОЙ 1 Ы1, m : Отношение содержание / содержа-з месторо- I ниё ждениях Г в земной 7 коре
Fe 0,08-10» 73-109 1,3-101° 5.6-10"°
Мп 2,0-10° 460-10° 2,2-lOH 2,1-10"*
Сг 0,17-100 1,5-10° 9,2-10|:| 1,6-10’°
Ni 0,065-10° 51-10° 5,0.10*3 1,0-10’®
Си 1,35-10° 93-10й 2,8-1013 3,3-10’®
Zn 1,40.10° 26-10° 5,6-101» 4,6-10’’
РЬ 1,63-10° 13-10° 2,3-Ю12 5,7-10-®
Sn 0,18-10° 5,6-10° 1,7-1012 3,3-10’®
Открытия новых месторождений следует, очевидно, ожидать й в будущем еще в значительных количествах. Однако если даже в результате новых открытий мировые запасы возрастут в десять или сто раз, содержание месторождений останется незначительным в сравнении с общим содержанием рассматриваемого металла в земной коре. Об этом свидетельствуют данные, приведенные в последней колонке таблицы. Они полу-. чаются, если числа третьей: колонки разделить на числа четвертой колонки. Часть металла, которая не находится в месторождениях, следовательно, та часть, которая распределена по общей массе земной коры, обозначается как дисперсная часть металла. Эта часть приобретает зна-чение для технического производства постольку, поскольку обильные месторождения теперь истощаются. Уже в настоящее время переработка бедных пород (например, путем флотации) играет большую роль для получения некоторых металлов. Из последней колонки табл. 92 видно, что для различных металлов соотношение между частью, встречающейся в рудных месторождениях, и дисперсной частью не подвергается очень большим колебаниям. В общем, следовательно, металл тем в больших количествах встречается в форме руд, чем больше его вообще в составе земной коры.
В тесной связи со средним содержанием различных элементов в земпой коре находится также их появление в форме отдельных минералов, в которых они присутствуют как существенная составная часть, Те элементы, которые более всего участвуют в строении земной коры, чаще всего выступают и как минера лообразующнв элементы. Число минералов, отличающихся гго своему химическому составу, составляет, по данным Иодцаков, в общем около 1800. Среди них 1097, следовательно, около 61%, в качестве существенной составной части содержат кислород. Затем следуют кремний (21%), железо (20%), сера (19,8%) и алюминий (17%), которые являются 44*
692
Глава 15
существенной составной частью большинства минералов. Болес редкие элементы, такие, как бериллий, хром, цирконий, олово, ниобий, тантал, молибден и вольфрам, образуют значительно меньше минералов — не более двадцати. Особенно редкие элементы, такие, как скандий, германий, рутений и палладий, образуют только одно или два соединения, которые встречаются в минералах. Для некоторых особо редких элементов — галлия, родия, индия и рения — вообще не известны соединения, встречающиеся в виде минералов. Однако соответствие между распространенностью и участием в минералообразовапии не полное. Яркое исключение представляет серебро, для которого известно нс мопее 71 минерала, хотя этот металл в целом в образовании земной коры участвует пе более, чем некоторые элементы, для которых не известно ни одного собственного минерала. Для некоторых элементов существует небольшое чйсло собственных минералов, несмотря на их распространенность. Это связано с тем, что эти элементы выделяются при минералообразовании большей частью в виде изоморфной примеси вместе с другими более ра сп р ост ра не иными элементами, что может полностью препятствовать образованию собственных минералов даже такими элементами, которые не особенно редки. Например, не известно никаких минералов рубидия и гафния, хотя оба элемента встречаются чаще, чем бериллий, для которого известно 20 минералов. Напротив, известно значительное число минералов, содержащих соединения рубидия или гафния в качестве изоморфных примесей.
Закономерности в количественных соотношениях элементов; правило Гаркинса. Как видно из табл. 90, среди сильно распространенных элементов находятся преимущественно элементы первых рядов периодической системы, среди мало распространенных — преимущественно элементы последних рядов. Таким образом, в земной коре в основном преобладают элементы с относительно низкими атомными весами. Далее, неметаллы и легкие металлы преобладают в земной коре по сравнению с тяжелыми металлами. Среди 12 наиболее распространенных элементов (табл. 90) находятся пять неметаллов и шесть легких металлов; среди них имеется только один тяжелый металл. Еще ярче выступают эти закономерности, если вместо весовых процентов взять атомные проценты. Три наиболее часто встречающихся элемента (данные в атомных процентах, см. табл. 91) — неметаллы, три следующих по распространенности элемента — легкие металлы. Из 18 наиболее часто встречающихся элементов, приведенных в табл. 90, половина неметаллов; из остальных 74 найденных в природе элементов примерно только Ve часть относится к неметаллам.
Гаркинсом (Harkins) в 1917 г. было установлено правило, гласящее, что элементы с нечетными порядковыми номерами в общем более редки? чем соседние элементы с четными порядковыми номерами. Рис. 89, на котором порядковые номера нанесены на абсциссу, а десятичные логарифмы распространенности (выраженной в атомных процентах) — па ординату, показывает, в какой степени это правило выполняется для состава земной коры. Острия кривой, направленные вниз, соответствуют элементам с нечетными порядковыми номерами, а острия, направленные вверх,— элементам с четными порядковыми номерами.
Особо четкое подтверждение правила Гаркинса дал Гольдшмидт своими исследованиями относительной распространенности элементов редких земель. На рис. 90 на ординату нанесены числа атомов каждого элемента ряда лаптава, которые приходятся в среднем на 100 атомов иттрия. Ливии, соединяющие точки ординат, дают ярко выраженную зигзагообразную кривую, вершины которой, направленные вверх, соответствуют всем элементам с четными порядковыми номерами.
Имеется, однако, несколько отчетливых исключений из правила Гаркинса. Например," инертные газы (соответствующие ем точки на
Порядковый номер
Рис. 89. Зависимость между распространенностью и порядковыми^ номерами элементов.
<?/
Ld Се Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tu Yb Lu
Рис. 90. Относительная распространенность редкоземельных элементов (по Гольдшмидту).
694
Глава 15
рис. 89 окантованы), хотя н обладают нотными порядковыми номерами, распространены гораздо реже, чем их соседние элементы *.
Незначительное содержание инертных газов в атмосфере объясняется, но мнению Гартека (Наг t е с k Р.,: Angew, Chem., 63, 1 (1951)1, тем, что под влиянием действия атомных ядер, извергаемых протуберанцами Солнца, часть земной атмосферы постоянно «отделяется^. В течение всей земной истории это приводит к тому, что атмосфера обедняется газами, которые постоянно но поставляются в атмосферу в таких количествах, чтобы компенсировать потери (как это происходит в случае водорода, кислорода и азота).
Солнце испускает главным образом протоны, а также ядра гелия. Часть из них достигает Земли. Они входят в земпую атмосферу со скоростью W00—1500 км/сек и плотностью 101—106 частиц/см3. Вследствие огромной скорости протоны способны при стййкновецнц передавать значительному числу частиц земной атмосферы такую энергию движения, что последние становятся в состоянии покинуть поле тяготения Земли. В случае частички, находящейся вблизи земной поверхности, энергия движения, которую она должна иметь, чтобы преодолеть поле тяготопия Земли, соответствует скорости 11,5 км/сек. Если частичка находится в более высоких слоях атмосферы, то скорость, приводящая к ее отрыву, соответственно меньше; однако, как можно рассчитать на основании закона распределения Максвелла (см: гя. 17), ни одна сколько-нибудь значительная по весу газовая частица, из которых состоит земная атмосфера, при обычной температуре не имеет такой скорости. Исключение представляют атомы водорода. Так как в высших слоях атмосферы газы находятся преимущественно в атомарной форме, то даже без подвода энергии со стороны, солнечных протуберанцев водород постоянно отрывается от земной атмосферы. Благодаря подводу энергии часть водорода, которая отрывается из земной атмосферы, увеличивается. Кроме того, благодаря этому даже более тяжелые атомы, которые достигают высоких слоев атмосферы, становятся способными к выходу из поля тяготения. Водород и кислород в свою очередь постоянно поставляются в верхние слои атмосферы в результате фотохимического разложения паров воды (ср. стр. 687), Азот также поставляется вследствие распада азотистых соединений по меньшей мере в тех же количествах, в каких он уходит из атмосферы. Другое положение создается в случае инертных газов, Неон, криптон и ксеноп практически вообще не восполняются, Правда^ предполагают, что небольшие количества криптона и ксенона поставляются в атмосферу как продукты спонтанного расщепления урана. Это расщепление происходит, однако, настолько медленно, что оно не компенсирует их потери в результате отщепления. За геологические эпохи это расщепление может привести по крайней мере лишь к сдвигу изотопного соотношения у этих газов. В точение истории Земли аргона также больше уходит из атмосферы, чем вновь образуется в результате радиоактивного распада 10К, Однако образованием 40 А г при распаде калия объясняется значительное преобладание этого изотона в теперешнем аргоне по сравнению с другим изотопом. Гелий является том элементом, который вслед за Водородом более всего удаляется из атмосферы под влиянием излучения протонов Солнцем/ Поэтому этот инертный газ, особенно распространенный на Солнце и на других звездах, лишь в очень незначительном количестве содержится в земной атмосфере; .По данным Гольдшмидта (1038), его количество составляет около 1% того количества гелия, которое образовалось (в виде а-лучей) путем радиоактивного распада со времени возникновения Земли. Следовательно, под действием идущего от Солнца корпускулярного излучения отрывается не только первоначально содержащийся в земной атмосфере гелий, но также и большая часть вновь образующегося в результате радиоактивного распада гелия, поскольку последний переходит в атмосферу, а не включается в земную кору.
Состав внутренних частей Земли. В то время как каменная кора Земли в среднем обладает наибо.чьптей плотностью 2,7, для средней плотности всего земного шара оказывается более высокое значение, а именно 5,5.
* Другие отклонения происходят, может быть, потому, что в основу кривой, приведенной на рис. 89, положена распространенность элементов в земной коре, а не во всем земном таре, Средняя распространенность элементов, вытекающая из анализа метеоритов, не обнаруживает отклонении От правила Гаркинса, проявляющихся парне. 89 у Элементов Бе (порядковый номер 4), Mg (порядковый помер 12) и S (порядковый номер 16),
Распроспгранение злементов; геохимия 695
Следовательно, внутренняя часть Земли состоит из более тяжелых (по удельному весу) веществ, чем кора Земли, и поэтому ее состав должен значительно отклоняться от состава коры Земли. Только давлением, производимым на внутреннюю часть Земли массой внешней оболочки, отнюдь не удается объяснить значительное различие в их плотностях; сжимаемость силикатов, из которых преимущественно состоит каменная кора, для этого слишком недостаточна, Внутренняя часть Земли Должна состоять в среднем из: элементов, плотность которых существенно выше плотности каменной коры. Предполагают, что ядро Земли состоит в основном из железа. Это предположение, с одной стороны, подтверждается принятым для ядра удельным весом, который примерно соответствует удельному весу железа, и, кроме того, тем, что Земля обладает сильным магнитным полем, преобладанием линяй желоза в солнечном спектре, а также, составом железных метеоритов. Железные метеориты состоят в среднем примерно на 91% из железа и па 8—9% из никеля. Для земного ядра принимают примерно тот же состав, т. е. его рассматривают по существу как сплав железа с никелем, к которому примешаны лишь сравнительно незначительные количества других веществ *.
Кроме Fe и Ni, железные метеориты содержат небольшие количества других; веществ, главным образом Со, Р, S, С, Си, Мп, а часто также отчетливо определяемые количества pt. Однако о си при.меси в «’родием составляют только около 0,5—1% общего веса желейного метеорита. Чаще, мем железные. встречаются каменные метеориты.
Таблица. 93
Средний состав метеоритов
(Элементы расположены в ряд по их распространенности.
Числа соответствуют содержанию элементов в весовых процентах)
1. О 36,3 15.11 0,058 29. Be 6,7-10-* 43. Yb 1,7-10-4 57. Ho 6,0-10^5
2. Fe 24,1 16. Cl 0,047 30. Y 5,5-10-« 44.: Ilf 1,6-10-4 58. Lu 5,4-10-5
3. Si 17,96 17. Cu 0,018 31. Sc 4,9-10-4 45. Cd 1,6-10-4 59. Tb 5,2-10 •'
4.. Mg 13,55 18. Zr 0,008 32. Ga 4,6-10-4 46. В 1,5-10-4 60. Rh 4,7-10-5
5. S 2,01 19. Zn 0,0076 33. Li 4,4.10-4 47, Ru 1,4-10-4 61. Nb 4,1-10-5
6. Ni 1,45 20. Ge 0,0053 34...Mo 3,6-10-4 48. Ag 1,35-16-4 62. It 3,8-10-5
7. Ca 1,43 21, V 0,0048 35, Nd 3,0-10-4 49. I 1,25-10-4 63:, Th 3,1.10~5
.8. A] 1,12 22. F 0,004 36. Ba 2,9-10-4 50, Os 1,2-10-4 64. '.la 2,8-10-5
9. Na 0,75 23. Br 0,0025 37. Ce 2,1-10-4 51. Cs 1,1-10-4 65, Eu 2,7-10-5
10, Cr 0,27 21. Sr 0,0023 38, Dy 2,1-10-4 52, Sm l ,.l-10-* (iti. Au 2,5-16-5
11. Mn 0,24 25, As 0,0018 39. La 1,9-10-4 53. Be 9,3-10-5 (17. In 2,.Q-l()-5
12. P 0,15 26. W 0,0016 40, Pt 1,9-10-4 54, Pd 9,2-10-5 68, Bi 2,0-10-5
13. Co 0,11 27. Sn 0,0014 41. Gd 1,7-10-4 55. Pr 8,8-10-5 69. Ti 1,5.10-5
14, К 0,09 28. Kb 0,0008 42. Er 1,7-10-4 56. Sb 6,4-10-5 70. Tc 1,3-10-5 71.. Re 0,8-10-5
1—14 вместе 99,83 15—28 вместе 0,1631 29—42 вместе 0,0047 : 43—56 вмеСто 0,0017
Для среднего состава метеоритов принимают данные, приведенные в табл, 93 [согласно Urey И. С., Phys, iiev., 88, 248 (1952)], Среднее содержание урана в метеоритах лежит, вероптпо, ниже 10“5%, Однако более важным является количественное соотношение Не : U, так как па него можно определить продолжительность жизни метеорита (ср, стр, 700).
* Существенно другие взгляды защищает Куп [Kuhn, Naturw., 30, 689 (1942)].
696
Глава, 15
Наблюдения за распространением сейсмических воли показали, что во внутренней части Земли в трех местах проявляется прерывистое изменение плотности, сжимаемости и твердости. Первая граница непрерывности лежит сравнительно близко к поверхности Земли — на глубине 120 км, Предполагают, что состав земного шара на этой глубине и глубже, до ——1200 км, где лежит вторая граница непрерывности, в основном тот же, что и состав изверженных пород, наблюдаемых на поверхности Земли. Однако при исключительно высоком: давлении, господствующем на этой глубине, стабильны отчасти другие минералы, чем вблизи поверхности Земли, а именно минералы с большей плотностью (3,6—4). Такие,
D=2,8
пМ \ "~Я- Силикатная оболочка
1 Д—ffi-4 Эклогитовая оболочка
' (силикаты под высоким давлением)
i j—Сульфидно-окисная оболочка
\ 0=5—6 /
1
Металлическое • ядро (Ni- Fe)
\D~8 /
\
I
\/
Рис. 91. Схема разреза Земли (по Гольдшмидту).
устойчивые только при высоком давлении минералы (метастабильные при обычном давлений) встречаются иногда также на поверхности Земли, куда они попадают в результате вулканического извержения и т. п. К ним относится, например, алмаз. Их называют теперь в большинстве случаев эклогитами, а образуемую ими оболочку внутри .Земли в соответствии с этим — эклогитовой оболочкой (рис. 91).
Металлическое ядро с плотностью 8—9, состоящее по существу из никелевого железа, начинается ниже третьей границы непрерывности — на глубине примерно 2900 км *. Несмотря на высокую температуру, металлическое ядро, как и вся внутренняя часть Земли,— твердое вследствие действующего на него высокого давления. Если бы оно или какая-нибудь другая существенная внутренняя часть Земли были жидкие, это было бы заметно при изучении приливов в твердой Земле.
Между второй и третьей границей непрерывности, следовательно, в золе, которая лежит примерно на глубине 1200—2900 км, простирается промежуточный слой, обладающий плотностью 5—6. Гольдшмидт (1922) предположил, что этот слой состоит из смеси сульфидов и окислов тяжелых металлов, главным образом железа. Он сравнил Землю (в то время полагали, что Земля много времени назад была расплавлена) с расплавом доменной печгг, в которой также происходит разделение на металлический основной слон, сульфи дно -окисную промежуточную зону и силикатный поверхностный слой. Аналогичные представления примерно в то же время были развиты Тамманом.
* Радиус Земли у экватора составляет 6377,4
Распространение элементов; геохимия
697
Геохимический закон распределения элементов. Наука, которая занимается изучением химического состава земного шара в целом, называется геохимией. Ее первой задачей является исследование распределения элементов в земном шаре и изучение закономерностей, в соответствии с которыми это происходит. Полагают, что для имеющегося теперь распределения элементов в земном шаре решающее значение на протяжении земной истории имели главным образом следующие три фактора: во-пер-выХ, тяжесть, под действием которой элементы: более высокого удельного веса постепенно должны были накапливаться внутри Земли, в то время как легкие элементы перемещались к поверхности. Во-вторых, различная химическая, способность к образованшо соединений, в результате которой с охлаждением массы Земли элементы соединялись между собой и благодаря которой даже металлы с высоким удельным весом, ио сильным сродством к кислороду, например железо, смогли накапливаться в земной коре. В-третьих, различная кристаллизационная способность и вместе с тем влияния, обусловленные различйем кристаллического строения. Последние приводят к тому, что вещества, накапливающиеся вблизи поверхности Земли, при затвердевании ее коры различным образом распределялись в минералах и породах, так что теперь одни вещества объединены почти всегда вместе, а другие — резко отделены от них.
Определяющее значение для распределения отдельных элементов между тяжелым металлическим ядром, образующим впутреинюю часть Земли, и силикатным слоем, лежащим вследствие своей легкости на поверхности, в период, когда вследствие Жидкого состояли я возможен еще обмен веществ до установлении равновесия, должна иметь прежде всего различная сила электроположительного характера элементов. Наиболее электроположительные металлы вследствие высокого сродства к кислороду Должны скапливаться в силикатном слое, менее электроположительные—в железном ядре. Так как силикатный слой богат железом, то металлы менее благородные, чем Железо, могут находиться в металлическом ядре только в виде следов. Напротив, металлы более благородные, чем железо, могут присутствовать в металлическом ядре в значительно больших концентрациях, чем в силикатном слое, как это можно заключить по составу железных метеоритов (например, никель). Аналогичные соображения можно привести относительно рас предел е^нии различных металлов между силикатным и сульфидпым слоями. Следовательно, по составу земной коры до некоторой степени удается сделать заключение о составе ядра Земли (Тапинаи, 1923).
Если средний состав земной коры, соответственно отклонение его от состава ядра Земли, удается объяснить, как результат действия силы тяжести и химического сродства, то можно предположить, что разлпчпое распределение отдельных веществ в земной коре вследствие затвердевания се происходит так же, как и в лабораторном эксперименте — обособление компонентов прп затвердевании расплавов. Это разделение при затвердевший земной коры происходило тем в большей степени, чем медленнее шло охлаждение в сравнении с лабораторным опытом. Важную роль при этом играла сила тяжести. Из выделившихся вначале минералов наиб о нее тяжелые (такие, как магнитный железняк, хромистый железняк, оливин, пироксен) опускались вниз. Только в сравнительно редких случаях они были позднее вновь вынесены на поверхность Земли, па пример при вулканических извержениях. Легкие силикаты напротив, обычно перемещались вверх, то же относится и к газообразно-выделяю-Щимся продуктам, например летучим соединениям тяжелых металлов, особенно их галогепопроизводным. Этим объясняется то, что определенные количества тяжёлых металлов выносились поверх, вместо того чтобы опускаться. Точно так яге соединения тяжелых металлов, растворенные в воде или в легких расплавленных жидкостях, могли подниматься вверх. Следует, пожалуй, предположить, что большая часть разрабатываемых в настоящее время месторождений тяжелых металлов достигла доступных нам сфер именно таким путем. Прежде всего это относится к месторождениям свинцовых, цинковых, олова иных, молибденовых, вольфрамовых, серебряных и золотых руд. Иными путями разделение веществ происходило и происходит еще в настоящее время внутри и па поверхности затвердевшей коры Земли благодаря воздействию гидросферы и атмосферы, т. е. прежде всего благодаря процессам выветривания и седиментации.
«98
Глава, 15
Подобно тому как для состава кристаллов, выделившихся ив расплава, большое значение имеет способность к образованию твердых растворов, эта же способность имела значение и для состава кристаллов, образовавшихся при затвердевании коры: Земли. Зтим обусловливается совместное существование многих веществ. Давно известно, что химически аналогичные вещества способны совместно кристаллизоваться. Однако, как показал Гольдшмидт, для этой способности, кроме химической близости, большое значение имеют также: атомные и ионные радиусы, В общем только такие вещества способны взаимно заменять друг друга в кристаллах в больших количествах, которые при химической аналогии, соответственно при аналогичном строении внешних оболочек, одновременно обладают также близко совпадающими атомными или ионными радиусами. При этом близость атомных или ионных радиусов часто имеет более важное значение, чем аналогия в химическом поведении. Например, аналог магния бериллий никогда не находят в виде изоморфной примеси в минералах магния, напротив, в них часто находят никель, далёко ^стоящий в периодической система от магния. Ионный радиус бериллия, как видно из рис. 3 на стр; 37 т. I, значительно отличается от радиуса магния, в то время как радиус никеля практически совпадает с радиусом магния. Постоянное совместное нахождение редкоземельных . металлов также обусловлено небольшими различиями между радиусами их ионов, которые несут одинаковый заряд. Напротив, с алюминием, который/обладаёт значительно меньшим ионным радиусом, чем элементы редких земель, последние не встречаются вместо, хотя они и обладают общим свойством — трехвалентностьго. Галлий, ионный радиус которого очень мало отличается от ионного радиуса алюминия, наоборот, постоянно сопутствует алюминию.
Изоморфизм соединений может привести и тому, что два (или даже несколько) элемента при затвердевании земной коры постоянно сопровождают друг друга в выделившихся кристаллах. Если речь идет при этом о двух элементах,.концентрация которых в затвердевающей магме существенно различалась, то последнее (различие концентраций) проявится также и в минералах, образованных в результате затвердевания, Если к тому же подобные элементы обнаруживают по своим химическим свойствам большое сходство, то возможно, что более распространенный элемент совершенно «закроет» более редкий элемент, тате что последний трудно обнаружить И трудно выделить в чистом состоянии. В таких случаях говорят о маскировке. Наиболее типичным примером этого является маскировка гафния цирконием, которая привела к тому, что^ гафний, был открыт сравнительно поздно, хотя он в значительных количествах содержится во всех'минералах циркония ив общем не очень редок (ср, табл. 90), В случае гафния, помимо изоморфизма соединений, обнаружение его затрудняла дакже исключительно полная химическая аналогия. Чаще бывают такие случаи, когда при незначительной химической аналогии маскировка объясняется тем, что одно соединение очень сильно количественно преобладает над другим. Такой случай представляет, например, маскировка галлия алюминием. Галлий считают обычно одним из наиболее редких элементов. Однако галлий встречается в земной коре в больших количествах, чем, например серебро, висмут и ртуть. Его встречают постоянно как спутника алюминия, которому он в количественном отношении сильно уступает (как одному из наиболее распространенных металлов); так, гаЯлий, хотя и встречается во всех минералах алюминия, однако всегда только в чрезвычайно небольших концентрациях. Галлий относится поэтому, хотя он встречается всюду, к таким веществам, которые сравнительно труднодоступны, ив этом смысле их можно считать редкими элементами.
На распределение различных элементов в верхних слоях земной коры влияет также жизнедеятельность организмов. Вследствие селективной способности живых организмов поглощать определенные вещества и накапливать их, жизненные процессы на Земле приводят’ к разделению очень значительных количеств веществ. В качестве примера следует указать на
Распространение алиментов; геохимия 699
мощнще известковые отложения, которые возникли в основном благо-даря известковым выделениям растений и животных (известняки, раковинные известняки, кораллы).
Распространенность радиоактивных элементов и их влияние па тепловой баланс Земли. Средн данных, приведенных в табл. 90, данные о распространенности радиоактивных алиментов в земной коре заслуживают особого обсуждения, Они позволяют судить о том, какое влияние оказывает выделение тепла, связанное с радиоактивным распадомj па тепловой балапс Земли. Тепловая энергия, которую излучает за год 1 г урана (включая находящиеся с ним в равновесии продукты распада), составляет 0,79 нал. 1 г тория излучает за год 0.23 Нал, 1 г калия -4,24-10“4"Нал и 1 г рубидия — 2,38-10“4 кал. Так как на 1 г земной коры в среднем приходится 2,9-10-6 г урана, 1,1-10“6 г тория, 2,4-10“2 г калия и 2,9-10“4 г рубидия, то ежегодное количество тепла, .выделяющееся в результате радиоактивного распада, составляет в среднем примерно 7;8- 10~а' калгг -земпой коры. (Из этого количества 2,3- 10~в Kajt, падают на уран, 2,5-10“° кал на торий и 3,0-10~6 кал на калий; на рубидий приходится мецее чем 0,01-10“* кал. Отсюда видно, что калий, несмотря на незначительную радиоактивность, вследствие своего сильного распространения принимает большое участие в радиоактивном выделении тепла). Однако радиоактивные элементы пе равномерно распределены в земной коре, а накоплены в ее верхних слоях; такими образом здесь ^радиоактивное выделение тепла значительно больше, чем в среднем. Оно составляет, по данным Гольмеса. и Лоусона (Holmes, Lawson, 1926) для гранитной породы в среднем 15,9-10"6 кал,'? ,ежегодно, из которых на уран приходится 7,1 • 10“’’ кал, на торий 4,6-10“е кал и на калий 4,2-10'° кал.
... Выделение тепла содержащимися в земной коре радиоактивными веществами имеет огромное значение для теплового баланса Земли. Более чем три четверти .постоянно излучаемого Землей в мировое пространство собственного тепла поставляется радиоактивными процессами. Воз этого был бы совершенно непонятен опре-деляемыц па основании гоолого-падеонтологических данных н подтверждаемый радиохимическими исследованиями возраст Земли, примерно 2,5—3 млрд, лет.
Ра д 11 ох н ми ч некое оп ределение возраста мин ера лов. Bosрост- о преде ленных минералов и, таким образом, возраст породы, в которой они находятся, можно сравнительно с большой точностью определить из отношения содержания урана или тория в этих минералах к содержанию свинца. «Возрастом» минералов считается время, которое прошло с момента их затвердевания. Из скорости распада урана, следует (см. гл. 11), что из 1г урана ежегодно образуется 1,35.10“10 а свипцак Если разделить количество уранового свинца (20°РЬ), найденное па каждый грамм урана, на 1,35-10-10, то получают возраст минерала. Таким же образом удается определить возраст из отношения ториевый свипец ; торий или также из отношения актиносвинец: актиноуран. Конечно, для этого должен быть известен изотопный состав свинца в рассматриваемом минерале. Из содержания 2114 РЪ путем,умножения на относительные числа, приведонные па стр. 574, получают часть изотонов 20еРЬ, г07РЬиг**РЬ перадиогенного происхождения. Полученное таким образом содержаний изотопов в нормальном (нерадноген-ном) свинце следует вычесть нз найденного содержания с тем, чтобы получить коли чество свинца, образованное путем радиоактивного распада. Если возраст минерала очень велик, то следует также Принять во внимание, что количества урана, со ответа ственно тория, с течением времени уменьшались в результате радиоактивного распада. Расчет довольно вроет при применении уравнения, (4), приведенного на стр. 550, Если имеющееся содержание урана в'мпнерале (точнее урана I) обозначить iVuj, а содержание уранового свинца Л'Ла1-, то первоначальное содержание урана оказывается Лг-щ + так как на образование каждого атома уранового свинца (RaG) затрачивается один атом урана I. Уравнение (4) принимает форму: Лгш= (jVui Нт А'Паб) e~Xf, где X — Константа распада урана I, a t — возраст минерала. Путем преобразования получим
4-l==p?J ИЛИ Inf 1V
A’ui \ Лит J
Следовательно, если атомное соотношение известно, возраст минерала I удается очень просто рассчитать по формуле
2,8026 ]ng (
( = —--------. (1)
700
Глава 15
Подстановкой или вместо и констант распада тория или актино-ЛТЬ ЛАеи ЛШ
урапа в качестве Л получают два соответствующих уравнения, следовательно в общем три уравнения, из которых каждое независимо от других позволяет определять возраст минерала па основании изотопного состава, содержащегося в ном свипца и содержания в нем урана или тория. Например, Ниер (Nier, 1941) нашел для свинца из самарскита известного происхождения изотопное соотношение s°4Pb ; *’ciePb : 2U7Ph ; й08РЬ== = 0,167 : 100 : 7,60 : 21,3. Следовательно, на 100 г-атомов 20sPb в минерале содержится 0,167-18,45 — 3,08 г-атомов 2015 РЬ нерадиогенного происхождения, т. е. 96,92 г-атома радиогенного 2|ырЪ (радия G). Для нерадпогенных ао7РЬ и "“РЬ соответствующие числа: 0,167-15,61 = 2,61 и 0,167-38,40 = 6,41, и, таким образом, AcD и ThD: 7,60—2,61 = 4,99 и 21,3—6,4 = 14,9. Химический анализ показал, что минерал содержит: 6,91% U, 3,05% Th и 0,314% РЬ (пес. %), Для содержащегося в минерале свинца пи изотопного состава получается среднее массовое число 206,39. Массовое число Щ составляет 238,12, На 96,92 g-атома RaG приходится, согласно приведенным выше данным 100 г~атомов 30вРЬ, т. о. 129,07 г-атомов всего свинца.
, RaG 0,314-238,12-96,92 .
Таким образом, атомное соотношение составляет цт д — и, и 394
Ш 2ио,ЗУ 0,91 • 12У,и/
Отсюда в соответствии с уравнением (1) t = 2,52- 10s лет, если подставить Лиг — . ...... т й ThD 0,314-232,11-14,89
= 1,535-10“™ год \ Таким же образом получают ч ле ыю .л- — 0,01336
In JUo.dJ-o,Vo-1ХУ,Ш
и тем самым, если подставить Хть=4,99-10-и год"1, то t = 2,66-iO8 лет хорошо сов-шщает со значением, рассчитанным по содержанию RaG. Третье значение, 1=
... о,. . -,о тт AcD „ AcD
= 2,S0- 10s лет, Ниер получил из соотношения ц—ж. Его применили вместо у—гт , xtciL> ACU
чтобы исключить из расчета не очень точно известную константу распада AcU. Для канадской смоляной обманки Ниер нашел возраст 1000 млн. лет (t = 1,00-10й, 0,95- 10s, 1,03- 10s). Еще существенно более высокий возраст около 2500 млн. лет, был установлен им для канадского монацита.
Определение возраста минералов указанным путем помогло проверить геологические и палеонтологические данные и найти для самых старых седиментационных пород возраст примерно 2500 млн, лет. Так как эти породы выделились из моря, то это число одновременно дало низшую границу образования океана и затвердевания земной поверхности.
Другая возможность установления возраста дается определением отношения в минералах гелий : уран (или гелий : радий). В этом случае при расчете исходят из того, что 1 а урана вместо со своими продуктами распада ежегодно производит 1,1-IO-7 juji гелия. На этом основании удается также определять возраст метеоритов (Paneth, 1928 и сл.). Для самых старых из них получается возраст 2800 млп. лет. Этот возраст совпадает по порядку .величин со временем, которое принимают астрономы как время, прошедшее с возникновения солнечной системы.
Другие методы радиохимического определения возраста минералов основаны на образовании 40Са в результате ^“-распада 40К (калиево-кальциевый метод; Ahrens, 1951) и на образовании ™Аг из 40К путем захвата: электрона (налиево-аргонпый метод; Gentner, 1950), Последний метод считается особенно надежным (Schilliber, Russe], 1954; Wasserburg, Hayden, 1955;’ ср. также Sttassmann, Z. Naturf,, Ila, 277 (1956)]; однако при пользовании этим методом следует учесть, что в течение геологических эпох часть аргона из кристаллов диффундирует наружу. Гентнер (1953) разработал способ количественного учета этого влияния. При определении возраста минералов гелиевым методом следует также принять во внимание влияние диффузии. Согласно Панету, это иначе происходит в метеоритах, которые часто состоят из монокристаллов, полностью удерживающих гелий.
Очень точным, однако ограниченным в применении, является рубидиево-строп-циевый метод определения возраста минераиов (Hahn, Walling, i938; Ahrens, 1949). Изотоп стронция s7Sr, который содержится в обычном стронции в количестве 6,96 ат, % , бо.чце или менее сильно концентрируется в минералах, содержащих рубидий, иногда присутствует там даже в чистом состоянии. Он образуется в результате радиоактивного распада 87ПЬ, содержащегося в рубидии в количестве 27,85 ат.%: s'Rb — ——> s"Sr. Из соотношения стр опций-8 7 : рубидий, так же как из соотношения свинец: уран удается определить возраст минералов и тем самым возраст пород в которых они содержатся. Следует конечно вычесть из найденного содержания ®7Sr содержание, приходящееся па нормальную изотопную смесь. Относительно применимости «обращенного стронциевого метода» для определения возраста минералов, не содерншщнх рубидия^ см. Walling' Е., Z. Naturf., 4а, 153 (1949).
Р ас пр ос траление алементов; геохимия
701
Определение возраста минералов, содержащих рений, можно производить цо редиево-осмиевому методу (Herr, 1955). Последний основан на том, что 18'Re, содержащийся в рении в количестве 62,9%, распадается с образованием 18’0е. Чтобы измерить исключительно малые количества осмия, которые при этом образуются, можно определить общее содержание осмия в минерале спектрофотометрически, а псрадио-гепный осмий определить путем активирования нейтронами. 18'0а нейтронами не активируется, так как 183 О я стабилен. Разница между общим содержанием и содержанием активируемого осмия дает содержание радиогенного осмия. Определение содержания рения также можно производить методом активирования нейтронами.
Метод, позволяющий установить, когда породы попали на поверхность Земли или когда они там образовались, основан на определении содержания радиоактивного ЗСС1, который образуется из МС1 в результате захвата нейтронов под действием космического излучении: ®5С1 (к, у) ®6С1 [F г а и с k е Н. W., Nucleonics, 13, 64 (1955)).
Для определения возраста органических веществ используют радио-угмероднын способ, открытый Либби, который был рассмотрен в т, I. Этот способ удается применить также к некоторым известковым породам (лапример, натечным известнякам).
Распространение элементов вне Земли. О внеземном распространении элементов нам дают сведения, с одной стороны, падающие из мирового пространства метеориты, с другой стороны—спектральные исследования светящихся звезд. Аналитическое исследование многочисленных метеоритов, средние данные о составе которых были уже приведены на стр. 695, показали, что эти небесные тела состоят из тех же веществ, которые встречаются на Земле. По существу тс же данные получены для светящихся звезд при спектральном анализе испускаемого ими света. Однако количественные соотношения различных элементов на звездах отчасти иные, чем на Земле. Например, в атмосфере Солнца, судя по спектру, господствуют кальций и железо, а затем водород и натрий. В общем на самых горячих звездах преобладающими оказываются наиболее легкие элементы, прежде всего водород и гелий, в то время как по мере охлаждения в спектрах вес более преобладают линии тяжелых элементов, например калия, кальция, железа и титана. В итоге можно сказать, что в доступных наблюдению частях вселенной по существу встречаются те же Химические элементы, что и на Земле.
Числовые данные относительно участия отдельных элементов в строении вселенной отличаются еще большой неопределенностью. Все же на основании новых результатов различимо исследователи дают значения, которые в целом сравнительно хорошо согласуются. Об этом свидетельствует табл, 94, в которой приведены числа относи-тельной распространенности элементов, рассчитанные Броуном (Rev. mod. Phys., 21, 625 (1949)1 п К випер ом (Kuiper) из астрономических наблюдений и из состава метеоритов. Элементы в ней расположены в порядке зарядов ядер Z. -Литий, бериллий и бор не приведены, потому что аги элементы совершенно неустойчивы при температурах, которые господствуют внутри звезд. Для остальных не приведенных в таблице элементов можно предположить, что их количества относятся к кремнию примерно так же, как в метеоритах.
В данных табл. 94 не отражена часть материи, которая распределена как космическая пыль пли находится в форме газа в межзвездном пространстве. О составе этой части материи, масса которой оценивается примерно как 50% общей массы космоса, пока очень мало известно. Приведенные в табл. 95 данные о распространенности некоторых элементов в межзвездной материи получены из данных Да нема (Dunham, 1939) и Струве (Struve, 1941) в предположении, что в межзвездной материи соотношение Са : Si такое же, кик и в звездной материи.
Мелсду относительной распространенностью элементов в космосе и свойствами ядер имеется определенная связь, поэтому возможно более точные сведения о распространенности элементов представляют большой иптерес для ядерной физики. Опо имеет также важное значение для объяснения возникновения различных элементов. Кроме того, из соотношения земной распространенности к космической можно сделать заключения, которые позволят глубже прогон,"путь в историю возникновения Земли, в закономерности распределения веществ внутри Земли п на ее поверхности и в общие законы геохимического распределения элементов.
702
Глава 15
Таблица 94
Относительная распространённость элементов во вселенной
(Числа показывают, сколько грамм-атомов данного элемента приходится на 1 г-атом кремния)
Z Элемент Распространенность 2 Элемент Распространенность
по Крауну no Huntiepy no Bpayny по Квиперу
1 Н 35 000 37 000 19 К 0,0069 0,0057
2 Не 3 500 5100 20 Са 0,067 0,072
6 С 8 4,1 21 Sc . . 0,000018 ' —
7" N 16 8,9 22 Ti 0,0026 : 0,0028
8 О 22 ! 15 23 V 0,00025 —
9 F 0,009 0,12 24 Cr 0,0095 0,0081
10 Nl! 0,9—24 17 25 Mn 0,0077 - 6,0082
11 Na 0,046 0,062 26 Fo 1,83 0,62
12 Mg 0,89 1,8 27 Co 0,0099 ; 0,0032
13 Al 0,088 0,083 28 Ni 0,134 0,038
14 Si 1,000 1,000 29 Cu 0,00046 • г
15 P 0,613 0,020 30 7П 0,00016 0,0029
16 ' S 0,35 0,46 36 Kr 0,000087
17 Cl 0,017 0,40 54 : Xe 0,0000015 —
18 Ar 0,013—0,22 2,5 •. -
Таблица 95
Атомная распространенность некоторых элементов' в межзвездной: материи
Элемент
Н G О’ Na К Са. Ti
Распространенность, г-атрм.[\ г-атом Si 200000'—! 000 000 1,5 1000 1 0,2 0,07 o,ooq3
Образованно элементов в космосе. В настоящее время можно определенно сказать, что на. Солнце и внутри других -звезд происходит постоянное превращение водорода в гелий; образованно гелиевых ядер из протонов является единственным процессом, на основании которого можно объяснить, почему сохраняются высокие температуры звезд, несмотря на' сильное излучение тепла в космическое пространство. При температурах между 10 и 20 млн. .градусов, которые, например, господствуют внутри Солнца, превращение водорода в гелий может происходить двумя путями: в результате так называемой «про-тон-пр стопной реакции» и «углерод-азотного цикла».
Протон-протоппая реакция через образование дейтерия ведет к получению 3Не, затем путем столкновения двух ядер чНе—к 4Не, причем вновь освобождаются два протопа:
1Ц(р, e7)3D; 2D(p, у)3Ие; 3Не(3Не, SjaJ^IIe-
Распространение элементов; геохимия
70S
Углерод-азотный цикл протекает следующим образом:
:->i2C(p, y)13N; 13Q; 13С(р, y)*«N; MN(j>, y)l50; 160i5N; i5N(p, a)lsO—j
При этом также четырьмя протонами, которые требуются для реакции, смотря по обстоятельствам, образуется ! ядро 4Не (а-частица). Энергия, которая освобождается при образовании 1 г 4Не из протонов, составляет примерно 150 млн. ккал.
Относительно условий, при которых могли образовываться элементы с более ... высокими порядковыми номерами и атомными весами, чем гелий, а возможно, даже ещо И: образуются, можно строить лишь обширные догадки. Можно, правда, показать, Что прп температурах примерно 200 млн. градусов может происходить образование 12С путем соединения ядер 4Не между собой. Благодаря соединению с новыми ядрами 4Г1о может, вероятно, происходить также образование более тяжелых элементов — вплоть до 4(:’Са; однако захвату еще новых ядер 4Не препятствует растущее электростатическое отталкивайт:е. Более тяжелые элементы могут быть построены, если в распо-ряжепии имеются свободные нейтроны. Доказательством того, что при определенных условиях это происходит, было бы подтверждение наблюдений Мсрилла (Meri jl, 1952), который будто бы спектрально-аналитически обнаружил в некоторых звездах образование технеция; таг; как все изотопы технеция нестабильны и период полураспада самого долгоживущего из пих (9®Тс равен 105—10н лет) незначителен в сравнении с возрастом этих звезд, технеций ire может быть их первоначальной составной частью. : Если он фактически существует па определенных звездах, то он должен там возникать. Это может ироисх1.>;нгтв только при захвате нейтронов. Действительно, известны ядерные регтыггып. дающие нейтроны, которые осуществимы в условиях, господствующих па некоторых звездах, паирпмор;
2°.Ne(p, yfiNa; 2iNa Д 2iNe; 2INe(a, ra)«Mg. :
Имеются даже признаки того [ср. St. Tomes vary, NatttrwiB., 44, 321 (1957)], что при определенных процессах в космосе (о которых имеются сведения благодаря неожиданной вспышке новой звезды) поток нейтронов так сплел, 'что там могут возникнут!, элементы с особо высокими порядковыми номерами (например, калифорний 98С1). Если все это подтвердится, то происхождение более тяжелых элементов можно, вероятно, будет объяснить предположением^ что общая космическая материя но крайней мере уже один раз участвовала в таких звездных взрывах, которые проис- ; ходят при всцыгпке новых звезд.
Гл а в а 16
КОЛЛОИДНАЯ ХИМИЯ.
ХИМИЯ ПОВЕРХНОСТНЫХ ЯВЛЕНИЙ
Общие сведения. При исследовании диффузии растворенных веществ Грэм (I860) обнаружил, что различные вещества ведут себя по-разному при пропускании их раствора через какую-нибудь Мембрану, например из пергаментной бумаги. Вещества, которые при обычных условиях аморфны (такие, как клей и гуммиарабик), полностью задерживаются такой мембраной, в то время как хорошо кристаллизующиеся вещества проходят беспрепятственно при условии, что по обе стороны мембраны находится один и тот жо растворитель. Грэм нашел, что те и другие вещества отличаются и еще целым рядом свойств. «Клееподобные» вещества он называл коллоидами (хоХ.Аа —- клей), остальные кристаллоидами, а растворы коллоидов в отличие от «истинных растворов», т. е. растворов кристаллических веществ, золями.
Впоследствии было обнаружено, что способность образовывать коллоидные растворы не является свойством лишь некоторых веществ. В настоящее время известно, что в принципе все вещества при определенных условиях могут образовывать коллоидные растворы.. Хотя для таких веществ, как клей, белок, двуокись Кремния, окись алюминия, это свойство особенно характерно, удается также приготовить коллоидные растворы даже таких ярко выраженных кристаллоидов, как хлорид натрия (см. т. I, стр. 215). Вследствие этого в настоящее время различают ужо не коллоидные и кристаллоидные вещества, а говорит лишь о коллоидном и кристаллоидном состоянии веществ.
Учение о коллоидах в настоящее время развилось в отдельную области науки. Явления, связанные с особенностями поведения веществ в коллоидном состоянии, встречаются почти во всех областях химии. Коллоидные химические процессы часто играют весьма значительную роль и в технике. Исключительно важное значение имеет изучение природы коллоидного состояния и открытие его закономерностей для развития биологической химии.
Помимо чрезвычайно небольшой диффузионной способности, вещества в коллоидном состоянии обладают другими характерными свойствами: очень низким осмотическим давлением их растворов, ярко выраженной способностью рассеивать свет (эффект Тиндаля, ср. стр. 710), а также способностью выпадать в осадок («коагулировать») прн небольших изменениях внешних условий (добавлении электролита, повышении температуры).
Перечисленные выше свойства обусловлены тем, что величина частиц веществ, находящихся в коллоидном состоянии, больше, чем размеры атомов и обычных молекул. С другой стороны, коллоидные растворы, так inc как и истинные растворы, могут оставаться совершенно прозрач-
Коллоидная химия. Химия поверхностных явлений 705
ними. При исследовании под микроскопом в проходящем свете в коллоидных растворах не заметно никакой оптической неоднородности. Они также полностью проходят через обычную фильтровальную бумагу. Так как разрешающая способность микроскопа измеряется половиной длины волны света, т. е. не превышает 200 игр, а размеры молекул, как правило, не более 1 игр (i0~7 см), то интервал 200—1 игр мощно считать приблизительно верхней и нижней границами диаметра коллоидных частиц. Более точное измерение (см. стр. 710) показало, что в этом интервале величина коллоидных частиц может быть любой. Следовательно, все растворы, у которых размеры частиц лежат в указанной области, обладают свойствами, характерными для коллоидных растворов. Таким образом, коллоидным состоянием веществ называется такое состояние, при котором величина его частиц лежит внутри определенных границ {от 1 до 200 mu) *.
Имеются молекулы, размеры которых во всех трех измерениях превышают 1 тр («молекулы-великаны»). Например, диаметр молекул яичного альбумина достигает 4,34 тр, а спирали гемоцианина даже 24 mp (Svedberg). Среди неорганических веществ гигантские молекулы встречаются у гетерополикислот и их солей. Такие вещества (ъэуноллоидыь, по Во. Оствальду) в растворе ведут себя как типичные коллоиды.
В интервале 100—500 тц устойчивость коллоидных растворов быстро падает, они переходят в так называемые суспензии. Суспензиями называют взвесь более крупных частичек, различимых под микроскопом и задерживаемых фильтровальной бумагой. В отличие от коллоидных растворов суспензии не могут существовать долгое время; под действием силы тяжести суспендированные частицы постепенно оседают па дно (седиментация).
Частицы во взвешенном состоянии, видимые под микроскопом, называют микронами. Частицы, невидимые под обычным микроскопом, но различаемые под ультрамикроскопом (см. стр. 710), — су бмикро нам и (пределы размеров 2-10”5—Ь10'5), а невидимые и под ультрамикроскопом — амикронами.
Классификация дисперсных систем. По степени измельченности {степень дисперсности) ** различают грубодисперсные, коллоидно-дисперсные и молекулярно-дисперсные вещества. Вещество, находящееся в более или менее топко раздробленном состоянии, называют диспергированным веществом, а гомогенное вещество, в котором оно распределено, дисперсионной средой; оба вещества вместе — дисперсной системой. В зависимости от агрегатного состояния диспергированного вещества и дисперсионной среды различают, по Во. Оствальду, восемь групп коллоидных систем, указанных в табл. 96.
Наиболее характерные свойства коллоидного состояния рассмотрены ниже, в главе о коллоидных растворах, поскольку последние являются важнейшими и наиболее изученными среди коллоидных систем. Краткая характеристика важнейших свойств других коллоидных систем приведена на стр. 731.
Исторические сведения. Основоположником ученияо коллоидах является Грэм. Им введопы основные понятия в этой области, хотя отдельные исследования были проведены раньше. Широкое изучение химии коллоидов было начато только в начале
* Указанные размеры не следует рассматривать как строго определенные границы существования коллоидных частиц. Иа границах области коллоидного состояния веществ постоянно наблюдаются свойства, присущие соседним областям. Помимо этого играет роль форма частиц и другие их свойства, а также природа растворителя. Одпако очень существенного влияния па границы коллоидного состояния веществ опи но оказывают.
** Степень дисперсности D, но данным Во. Оствальда, определяется отношен нем общей поверхности диспергированных частиц О к их объему V. Следовательно, D ~ O/V. 45 г. Реми
706
Глава 16
Таблица S6
Классификация коллоидных систем
Дисперсионная среда Дисперсная фаза Название Пример
Твердое пе- Г Твердое вещество Твердые води (стеклозо Jriij кристалле зол и и др-) Рубиновое стекло (см. стр, 444)
щество Жидкость я
Газ Твердые коллоидные пены Пемза (частично грубодисперсная)
Твердое вещество Коллоидные растворы пли золи (гидрозоли, органозоли и т. д.) Коллоидный* раствор золота (см. стр. 451), аквадаг, коллаг, ойлдаг (см, т. I, стр. 462)
Жидкость Жидкость Коллоидные эмульсии (эмульсоиды) t Молоко (частично грубо-дпсперспое), раствор желатины
Газ Коллоидные пены Мыльная пена (частично грубодисперсная)
Газ - Твердое вещество Коллоидная пыль (аэрозоли) Дым боевых веществ
Жидкость Туман Атмосферный тумаи; туман серпой кислоты (см. т. I, Стр. 759).
текущего столетия. Изобретение ультрамикроскопа (Siedcstopf, Zsigmondy, 190 Я) позволило впервые непосредственно наблюдать коллоидные частицы и приблизительно в то же время удалось установить основные закономерности их поведения (Sutherland, 1905; v. Smoluchowski, 1906). Изучение пределов существовании коллоидных.частиц и систематизация их в отдельную область зпаппя принадлежит Во, Оствальду. Позднее большой вклад в технику эксперимента и теорию внесли Бредит, фон Вей-марн, Перрен, Дюкло, Сведберг, Лангмюр и Фрейндлих (Bredig, v. WeiiHani, Perrin. Duciaux, Svedberg, Langmuir, Freundlich). В настоящее время, особенно благодаря связи учопия о коллоидах с явлениями, происходящими на поверхности раздела фаз (капиллярные явления и адсорбция), значительно углубились знания о поведении веществ в коллоидном состоянии.
КОЛЛОИДНЫЕ РАСТВОРЫ
По.Грэму, в зависимости от того, какое вещество является дисперсионной средой — вода, спирт, эфир или иное органическое вещество, — коллоидные растворы называют гидрозолями, алкозолями, этерозолями и вообще органозолями. Соответственно среди твердых коллоидно-диспер
Коллоидная химия. Химия поверхностных явлений
707
сных систем различают кристалл оз о ли, стек лоз оли (yilreus — стеклянный) ит. д. Объемистые осадки (гели), выделяющиеся из золей (при коагуляции), называют гидрогелями, алкогелями и т. д. Если гель легко переходит снова в золь, говорят об обратимых, а в другом случае — о необратимых коллоидах.
Системы, дисперсные частицы которых одинаковой величины, называют изодисперсными, а такие системы, в которых частицы различаются по величине,— поли дисперсными. В общем коллоидные растворы в болен или менее значительной степени являются полидиснерсными. Если требуется получить однородные золи с одинаковыми по размеру частицами, то следует подбирать особые условия.
Получение. Коллоидные растворы получают либо диспергированием, исходя из грубодисперсных веществ, либо конденсацией молекулярно-дисперсных, т. е. находящихся в виде истинных растворов веществ.
Среди дисперсионных методов наиболее важным является электрическое распыление, впервые примененное Бредигом (Brodig, 1898). Этот метод особенно удобеп для приготовления золей металлов. Он заключается в том, что между двумя охлаждаемыми водой металлическими проволоками создают электрическую дугу. Для получения органозолей удобнее осуществлять распыление по Сведооргу — при помощи высокочастотной искры, подобной разряду ленденеких банок.
В некоторых случаях вещество при простом соприкосновении с какой-либо жидкостью сразу а:е переходит в коллоидный раствор. Это явленно может служить помехой Ири промывании осадное. Например, некоторые сульфиды (CuS, HgS, FeS, ZnS) и гидроокиси при промывании чистой водой особенно склонны к образованию гидрозолей, что приводит к потере вещества в процессе фильтрования. Во избежание потерь осадки промывают водой, содержащей электролит. Электролиты могут вызвать и обратное явление — переход вещества в раствор в виде гидрогеля *. Например, р-оловяппую кислоту можно перевести в коллоидный раствор при помощи разбавленного раствора едкого кали. Или можно сначала обработать ее концентрированной соляной кислотой, а затем водой (ср. т. I, стр, 578). В таких случаях говорят о пептизации, по аналогия с процессом растворения белка под действием пищевого фермента пепсина, содержащегося в желудочном соке. Вещество, вызывающее процесс пептизации, называют пептигатором.
Пептизацию веществ, находящихся в форме геля, часто можпо вызвать введением электролита. Иногда могут пептизироваться и кристаллические вещества. Так, получают гидрозоли Ti, Zr, Мо и W, попеременно обрабатывая тонко из мельченный металл кислотой и щелочью, а в промежутке промывая его водой (ср. стр. 182).
Перевести вещество в коллоидный раствор можно чисто механически путем очень тонкого распыления. При таком распылении в качестве разбавителя удобно добавлять индифферентное твердое вещество, легко растворимое в данной дисперсионной среде. Например, гидрозоли серы, селепа, теллура и различных металлов были получены фон Веймарном (v. Weimara, 1923) с использованием тростникового сахара в качестве нейтрального разбавителя.
Весьма распространенными методами приготовления золей являются методы конденсации. Большинство из них основано на проведении химических реакций образования труднорастворимого осадка в условиях, неблагоприятных для быстрого роста первоначально образовавшихся частиц (например, охлаждение, сильное разбавление) и исключающих по возможности влияние факторов, вызывающих коагуляцию коллоидно-растворенных веществ (см. стр. 715).
аг
* Объяснение различного действия электролитов см. па стр. 712 и сл.
45*
708
Глава. 16
Помимо химических реакций, для получения золей конденсацией используют и другие процессы. Например, золи фосфора и серы можно приготовить вливанием спиртовых растворов этих веществ в воду (v. Weimarn, 1911). Жальников (1927) получал гидрозоли металлов путем совместной конденсации их пересыщенных паров с дисперсионной средой на стенке стакана, охлаждаемого жидким воздухом.
Особенно легко водные коллоидные растворы образуют следующие вещества:
1) сульфиды мышьяка, сурьмы, олова, висмута, меди, ртути, никеля, цинка м кадмия;
2) окислы или гидроокиси железа и алюминия;
3) двуокиси кремния, германия, олова, титана и циркония, пятиокиси сурьмы п ванадия, а также трехокиси молибдепа и вольфрама;
4) благо род игге я полублагородные металлы: золото, платина, палладий, серебро, ртуть, медь п висмут;
5) многие органические вещества, например белок и крахмал (ср. стр. 716).
Вещества, перечисленные в пунктах 1—3, настолько склонны к образованию золей, что При осаждении их из растворов необходимо создавать особые условия (нагревание, присутствие электролита: ср, стр. 715), иначе они частично или полностью останутся в растворе. Это особенно важно учитывать при осаждении этих веществ из очень разбавленных или очень концентрированных растворов; именно в обоих этих случаях, как показал фон Веймарн (1908), создаются наиболее благоприятные условия для образования коллоидных частиц в момент осаждения веществ.
Гидрозоли металлов, указанных в пункте 4, помимо электрического распыления, можпо легко получить восстановлением их соединений в растворе. Гидрозоли металлов используют в аналитической химии, для катализа и для медицинских целей,
При помощи химических реакций, а также й другими способами необходимый золь нельзя приготовить сразу в чистом состоянии; он всегда в той или иной мере содержит примеси электролита или других присутствующих в растворе молекулярно-дисперсных веществ. Для устранения примесей, от которых часто в значительной степени зависит устойчивость золя, применяют диализ.
Диализ, Под диализом (SixAvaig — разделение) понимают разделение веществ на основе их различной способности диффундировать через мембраны. Диализатор Грэма состоит из, стеклянного стакана, дном которого служит пергаментная бумага. В него наливают раствор, подлежащий диализу, и укрепляют во втором, более широком стакане, заполненном чистым растворителем, например водой. В настоящее время в лаборатории используют в большинстве случаев мешочки из пергаментной бумаги, коллодия или других подобных веществ, которые помещают в цилиндр или стакан с чистым растворителем.
Такне мешочки можно легко наготовить по методу Зигмонди. Тщательно вымытую и высушенную пробирку ополаскивают раствором коллодия, дают пленке коллодия немного подсохнуть, после чего заполняют пробирку водой. Образовавшийся мешочек спустя некоторое время легко можно отделить от стенок пробирки. Для диализа больших количеств коллоида имеются производительные и быстро действующие аппараты.
Диализ, помимо отделения коллоидных растворов от примесей, часто применяют для доказательства коллоидного характера находящихся в растворе веществ. В технике диализ широко применяют для очггстки природных и искусственно получаемых коллоидов.
Скорость диалпза зависит от размера поверхности мембраны, количества жидкости, подлежащей диализу, и от разности концентрации вещества, находящегося по обо стороны мембраны. Поэтому растворитель по внешнем стакане следует чаще обновлять (лучше всего вести диализ с проточным растворителем). Иногда для ускорения диализа работают прц нагревании (Schmidt, 1930).
Коллоидная химия. Химия поверхностных явлений
70S
Особенно сильно ускоряется процесс длализа под влиянием электрического' тока. Электродиализ позволяет достигнуть такой степени очистки от электролита, которая при обычном диализе вообще недостижима-. Благодаря этому его в первую очередь применяют для очистки так называемых био коллоидов (протеидов, полисахаридов, пектиновых веществ, ферментов, гормонов и т. и,). Очень простой способ электродиализа, удобный для лабораторной практики, описан Рейнером (Reiner 1926) (ср. также Ch. Dker б, Kolloid-Z., 41, 243, 315 (1927)].
Ультрафильтрование. Ввиду непроницаемости коллодиевых мембран для коллоидных веществ их можно использовать для отделения от молекулярно-дисперсных веществ, присутствующих в растворе, а также и от растворителя, так как он способен проникать через такие мембраны. Однако, чтобы растворитель уходил из раствора, требуется создать некоторое давление, и чтобы ограничиться атмосферным давлением, с обратной стороны мембраны следует создавать небольшой вакуум (при посредстве водоструйного насоса). На этом основан процесс улътрафилътрова-ния.
Как показал Во. Оствальд (1918), ультрафильтр можно легко изготовить, покрыв фильтровальную бумагу тонким слоем коллодия. Приготовленный таким способом ультрафильтр позволяет фильтровать коллоидные растворы, без отсаеывательной склянки («спонтан-ультрафильтр»). Имеются так называемые «мембранные фильтры» (Zsigmondy Bachmann, 1918) с различной пористостью. В отлично от обычных коллодиевых фильтров их можно сушить, не нарушая пропускающей способности, Часто применяют также ультрафильтр Беххольд-Кёнига (1924).
УльтрафиЛ ьтровапцо находит раз постороннее применение. Помимо коллоидной химии, его удобно использовать в аналитической и препаративной химки для быстрого отделения студенистых или очень мелко кристаллических осадков (Jander, 1919). В настоящее время ультрафиль-трованио широко применяют в биохимии, физиологической химии, биологии, бактериологии и медицине.
Оптические свойства золя. Свет, падающий на золь, частично отражается коллоидными частицами, частично рассеивается, частично поглощается (превращается в другие виды энергии, преимущественно тепло) и частично проходит сквозь раствор.
Изменение интенсивности света за счет поглощения при прохождении через золь, подчиняется, как и в случае истинных растворов, закону Ламберта. В соответствии с а тян законом экстинкция пропорциональна толщине поглощающего слоя. Экстинкцией называют выражение log (Zo/Z) , где Zo — интенсивность падающего, а I — интенсшчгость проходящего света. Следовательно,
iog-^-=a.x, .(О
где х — толщина слоя; a — коэффициент пропорциональности, называемый ковффи'-циентом зкетипнции. С другой стороны, имеет силу закон Бера, в соответствии с которым свото по гл отдающая способность вещества не зависит от толщины слоя при равном сечении. Из этого следует, что экстинкция пропорциональна концентрации:
Iog^?- = E-c.z. (2)
Если концентрация с выражена в молях на 1 л (моль/л)1 то в называют молярным ковр-акстинкции или молярным коэффициентом погашения. Так как закон Бера выполняется только при условии, когда изменение концентрации не влияет на состояние частиц, поглощающих свет (например, изменение степени дисперсности), измеряя поглощение света золем и истинным раствором при их различной концентрации, можно сделать вывод о влиянии растворителя на рассеивающее вещество. Для одного итого же вещества при переходе от молекулярно-дисперсного состояния поглощение света по мере укрупнения частиц сначала возрастает, а затем падает. Максимальное поглощение, как правило, приходится на коллоидное состояние. Селективно поглощающие вещества (окрашенные), следовательно, наиболее интенсивную окраску
710 ’ Глава 16
имени в коллоидном состоянии. Это имеет значение, например, при производстве малярных красок.
Как правило, тон окраски по мере укрупнения частиц изменяется от красного к синему. Еще Фарадей наблюдал это явление на золе золота. При размере частиц менее 80 тр в диаметре они имеют интенсивно красный цвет. Если добавить вещество, вызывающее укрупнение частиц за счес частичной коагуляции, окраска изменяется до темно-синей через фиолетовую. Помимо размера частиц, на окраску влияют еще и другие факторы (например. форма частиц). Поэтому только по окраске нельзя судить о размере частиц.
Основным характерным свойством существующих в коллоидном состоянии частиц является их способность сильно рассеивать падающий свет, Рассеянный свет оказывается всегда частично поляризованным. Проходящий через коллоидный раствор луч света вследствие рассеяния становится видлмым в лем. Такое явление, наблюдаемое в тонкодисперспых суспензиях и взвесях веществ в газах, было названо эффектом Тиндаля по имени ученого, впервые наблюдавшего его (Tyndall, 1869). Вещества в молекулярно-дисперсном состоянии аффекта Тиндаля не обнаруживают- *.
Ультра ми про скопил. Благодаря рассеянию света коллоидные частицы в проходящем свете выглядят светящимися точками и поэтому становятся видимыми под микроскопом, несмотря на небольшие размеры, если наблюдать их не в направлении световых лучей, а перпендикулярно к ним. Это свойство лежит в основе ультрамикроскопа, изобретенного Зиденто-пфом и Зигмонди **, который при достаточно сильном освещении позволяет наблюдать частицы диаметром в несколько миллимикронов.
Число, размеры и форма коллоидных частиц в золях. При помощи, ультрамикроскопа можно подсчитывать число коллоидных частиц в определенном объеме. Если, известно количество вещества, образующего аоль, и число частиц, то мощно определить размеры отдельной частицы. Для частиц кубической формы диаметр или длину ребра I вычисляют по формуле:
- 1=V^^T’ (3>
у n-d
где п — число коллоидных частиц в 1 сл*3; d — плотность вещества, образующего коллоидный раствор; с — концентрация (г/смя). Чтобы определить диаметр частиц шаровидной формы, подкоренное выражение следует умножить па 6/л. Формула (3) применима только для изодпспе.рсного золя. Полидлсперсный золь предварительно следует разделить ультра фильтрованием через мембранные фильтры Зигмонди с различной пористостью (ср. стр. 709) на приблизительно вводисперсные. Однако точную оценку размеров частиц в таких случаях получают при помощи ультрацёнгрифугиро-вания (см. стр. 712). Определение размера частиц в золях, суспензиях и других дисперсных системах, включающее еще целый ряд методов, объединяют под названием «дисперсионного анализа».
Частицы, имеющие примерно одинаковые размеры во всех трех измерениях, принято называть «шаровидными» в отличие от частиц пластинчатой и палочкообразной формы. В системах с любой не газообразной дисперсионной средой отклонения от шарообразной формы большей частью лишь в том случае влияют на свойства, если они очень веники. Зоди, состоящие из частиц пластинчатой или палочкообразной формы, в потоке являются двояко преломляющими (анизотропия текущей среды). При рассмотрении протекающего золя между скрещенными, призмами Николя наблюдается посветиение поли зрения. Характер двойного преломления в потоке у золей пластинчатой и палочкообразной форм различен (отрицательный у первых и положительный у вторых.) Анизотропию текущего золи с частицами, сильно отличающимися от шаровидной формы, можно наблюдать часто и невооруженным глазом в виде слабо мерцающего света или образования полосы при перемешивании. В золях с очень вытянутым н частицами, на пример в постаревшем золе V2O5, при прикосновении появляется
* Молекулы газов и жидкостей хотя и рассеивают свет («релеевское рассеянно»), однако вызываемое имя рассеяние ничтожно мало но сравнению с рассеянием коллоидными частицами; ср. т. 1, стр. 343.
По существу, на таком же принципе основаны так называемые «ультралевдеп-саторы», которые можно применять в соединении с обычным микроскопом.
Коллоидная химия. Химия поверхностных явлений
711
великолепный шелковистый блеск. Под ультрамикроскопом у частиц, сильно отличающихся от шаровидной"формы и с интенсивным броуновским движением (см. ниже), часто наблюдается характерное искрение.
Число частиц в золях, как и в истинных растворах, может изменяться в широком интервале. В золях обычной концентрации, как правило, оно колеблется от 10® до 1014 в 1 с.ч3. Чем меньше частицы, тем, естественно, их больше при одной и той же аналитической концентрации. Однако даже в наиболее тонко дисперсных золях, не говоря уже о золях эуколлолдов и родственных им лиофильных веществ, число частиц редко превышает 101в в 1 с.к3, без того чтобы золь не коагулировал. Такая верхняя граница приблизительно соответствует ионной концентрации в 1 с.и3 0,00001 н. раствора NaCl. В среднем число частиц вг золях лиофобных коллоидов составляет менее миллионной части числа частиц кристаллоидов в растворах с обычными концентрациями.
Осмотические свойства коллоидных растворов. Вещества, находящиеся в коллоидном растворе, вследствие небольшого по сравнению с истинными растворами числа частиц обнаруживают очень небольшое осмотическое давление. Следовательно, и вызываемые им понижение давления пара и температуры замерзания, а также повышение температуры кипения едва заметны. Измеренное у золей гидрофобных коллоидов (ср. 716) понижение температуры замерзания па одну тысячную г рад уса, по-видимому, обусловлено главным образом постоянно присутствующим в таких золях электролитом. Электролит оказывает влияние также и на непосредственное измерение осмотического давления, которое для коллоидов легко осуществимо, так как не составляет труда изготовить непроницаемую мембрану для коллоидных частиц и полностью проницаемую Для всех остальных.
Осмотическое давление типичных коллоидов, хотя оно и чрезвычайно мало по сравнению с кристаллоидами, одпако является еще, как это можно вычислить из числа частиц, измеримой величиной. Например, вычислено, что для золя, содержащего 50 a As.jSi3 в 1 лт при радиусе частиц 2,5 та осмотическое давление составляет около 15 с.и вод. ст. При этом предполагали, что содержащийся в золо электролит не влияет па осмотическое давление. Одпако, в случае если электролит полностью проходит через мембрану, такое влияние существует, как теоретически впервые показал Допнан (Doniian, 1911). Таким образом, цри определении числа коллоидных частиц или их размеров в каком-нибудь золе необходимо учитывать влияние электролита.
Броуновское движение. При помощи ультрамикроскопа было обнаружено, что взвешенные в жидкости или газе коллоидные частицы находятся в непрерывном интенсивном беспорядочном движении. Такое явление можно наблюдать но только в случае коллоидных частиц, но и для частиц, видимых под обычным микроскопом, и чем меньше частицы, тем более интенсивно их движение. Это явление было открыто английским ботаником Броуном в 1827 г. (первоначально для цветочной пыльцы.) Пак удалось доказать, броуновское движенце обусловлено соударениями молекул с малыми частицами. Броуновское движение тем сильнее, чем меньше вязкость дисперсионной среды. Следовательно, оно наиболее интенсивно у веществ, взвешенных в газах.
Благодаря броуновскому движению вещество, находящееся во взвешенном состоянии, прп условий, что оно достаточно тонко диспергировано, не оседает под действием силы тяжести, а распределяется в дисперсионной среде.
Подобно тому как в газах (например, в воздухе) плотность с увеличением высоты закономерно уменьшается, такое же явление экспериментально наблюдал Перрен (Perrin) и для взвешенных частиц. Из кинетической теории газов следует, что высота слоя, в котором плотность газа убывает на определенную величину, на пример наполовину, обратно пропорциональна: молекулярному весу газа. Соответственно, высота слоя, в котором концентрация взвешенных частиц (незаряженных) уменьшается в определенном соотношении, обратно пропорциональна весу частиц *. Перрен нашел,
* Если концентрацию частиц на высоте и обозначить q и с2, массу частицы т, плотность ее d, а плотность дисперсионной среды do, то
где ХЛ — число Авогадро; Л — газовая постоянная; Т — абсолютная температура; у — ускорение свободного падения.
712
Глава. 16
что в случае частиц мастикса с диаметром 1,2 р в воде в качестве дисперсионной среды высота слоя, в котором плотность убывает наполовину, равна 6 р; для частиц того же вещества, но диаметром 0,12 р эта высота была бы уже 6 мм, а диаметром 0,612 р—6 м.
Ультрацентрифуга. Если силу тяжести, под влиянием которой происходит уменьшение концентрации частиц по мере увеличения высоты, заменить центробежной силой, то высота слоя уменьшается в том же отношении, в каком центробежная сила Превосходит силу тяжести. На этом основано действие ультрацентрифуги, сконструированной Сведбергом. Она позволяет развить центробежную силу,- превосходящую силу тяжести в 10в раз. Как наблюдал Перрен на микроскопических, но еще видимых частицах, иод действием такой большой силы устанавливается равновесие в седиментации не только в случае коллоидов, но и веществ, ня ход я пти тс я в молекулярно-дисперсном состоянии (с молекулярным весом менео 200). При помощи ультрацентрифуги можно определять массу или размеры частиц в тонкодисперсных золях, а также молекулярные веса растворенных веществ, я особенно веществ с высокими молекулярными весами. Если в растворе имеются вещества с различным молекулярным весом, то под действием силового поля ультрацентрифуги опи оседают с различной скоростью.
В золях, которые содержат менее тонко диспергированные частицы, размеры частиц можно определить и при помощи обычных лабораторных центрифуг.
Электрические свойства коллоидов; коагуляция и пептизация. Коллоидные частицы, суспендированные в жидкости, передвигаются в электрическом заряд. Коллоиды
поле. Из этого следует, что они несут электрический металлов и сульфидов металлов, как правило, заряжены отрицательно, коллоиды окислов и гидроокисей металлов, напротив, обычпо заряжены положительно.
Рис. 92. золя окиси в электрическом поле.
Движение коллоидных, а также грубодиснерсных частиц в электрическом поле называют катафорезом или электрофорезом. Его можно наблюдать на простом опыте (рис. 92). Если в U-образную трубку поместить коллоидный раствор гидроокиси железа(Щ), сверху в каждое колено трубки осторожно долить чистой воды и погрузить в нее электроды, несущие достаточно высокое напряжение, (например, 220 в), то можно наблюдать, как постепенно обе границы коричневого коллоидного раствора переместятся в сторону отрицательного электрода. Следовательно, частицы гидроокиси железа заряжены положительно. Если вести опыт: до тех нор, пока коллоидные частицы не достигнут отрицательного электрода, то они там разрядятся и выделятся в виде хлопьев. Скорость перемещения коллоидных частиц при катафорезе в общем того же порядка, что и скорость движения ионов при электролизе. Обычно она лежит в пределах 3- 10-i—5- 10~4 ем] сек при градиенте напряжения 1 е!см. Для ионов Na‘ и К' con тв етств у тоито величины 4,4-10*4 и 6,7- 1б“4 см/сек (при 18°).
Электрический заряд коллоидных частиц обусловлен адсорбированными на его поверхности ионами.
Например, находящаяся в коллоидном растворе окись железа(ТП) обычно адсорбирует на своей поверхности ионы Н‘. Если к коллоидному раствору добавить сильно разбавленное едкое кали, то заряды коллоидных частиц частично или полностью нейтрализуются, так как иопы ОН' соединяются с адсорбированными ранее ионами Н'. Это подтверждается уменьшением скорости движения коллоидных частиц в электрическом поле. По мере уменьшения скорости устойчивость золя падает, и когда, наконец, скорость становится равпой нулю, золь коагулирует *. Таким
Движение
железа
* Обычно коагуляция наступает раньше, чем заряд снизится до нуля (ср. стр, 713).
Коллоидная химия. Химия. поверхностных явлений 713
же образом ведут себя золи очень многих, особенно неорганических, веществ. Следовательно, чтобы золи подобных веществ были устойчивыми, коллоидные частицы должны нести заряд.
Электрический заряд требуется для того, чтобы, сталкиваясь в результате броуновского движения (или приближаясь друг к другу достаточно близко), частицы не слипались, а вновь расходились. Если же^ заряд исчезает, то частицы под влиянием criitf поверхностного натяжения (см. стр. 720) стремятся уменьшить свою поверхность; в результате они слипаются, образуя грубо дисперсные агрегаты, которые выпадают в осадок, т. е. происходит коагуляция,
Из теории коагуляции следует, что свертывание отрицательно заряженных коллоидных частиц зависит от катионов, а положительнозаряженных—от анионов прибавляемого электролита. Коагулирующее действие ионов возрастает по мере увеличения их заряда (правило коагуляции Шульца — Гарди). Молярные концентрации одно-, двух- и щретвалентных ионов неорганических веществ, отвечающих одному и тому же коагулирующему действию, относятся как 500 ; 10 ; 1. Неорганические ионы с одинаковым заря дом ведут себя почти одинаково. Только ионы ОН', и особенно ионы Н", проявляют гораздо более сильное коагулирующее Действие, чем другие однозарядные ионы.
Ьез особых доказательств понятно п „ ___й
что коагулирующее действие копов уве- Г п °. 93. Зависимость адсорбции от доплачивается с ростом заряда. Одвако то, центрации.
что коагулирующая Способность возра-
стает чрезвычайно сильно, требует особого объяснения, Как показал Фрейндлих (Freundlich, 1907), причина Итого в зависимости адсорбируемости ионов от их концентрации. Из рис. 93 следует, что зависимость количества адсорбированных ионов от концентрации имеет вид круто поднимающейся кривой (ср. стр, 722 и сл.). Если обозначить количество одновалентных ионов, адсорбируемых из раствора с концентрацией ео через af, то для нейтрализации такого же заряда коллоида йву-гвалентпых ионов потребуется вдвое меньше. Но ординате <ц/2 соответствует абсцисса сг, которая, как видно-из рисунка, составляет 1/1о от щ, При этом предполагается, что адсорбция ионов-пе зависит от их валентности. Для большинства неорганических ионов это с тем или, иным приблия!опием выполняется (ср. стр, 725). А ионы Н’ И ОН' адсорбируются значительно сильнее, чем остальные неорганические ионы. Поэтому коагулирующее действие их значительно больше по сравнению с другими однозарядными неорганическими ионами. Адсорбционная способность органических ионов носит индивидуальный характер,В связи с этим дня пих не существует простой зависимости между коагулирующей способностью и зарядом. В общем органические ионы адсорбируются сильнее, чем неорганические, и соответственно значительно превосходят их по коагулирующей способ пости. Если сравнить коагулирующую способность, отнесенную по к общему количеству электролита в растворе, а К тому, которое адсорбируется коллоидом, и выразить это количество в эквивалентах, то оказывается, что в пределах ошибки опыта для одного и того же коллоида получаются одинаковые Числа рля любого электролита. Следовательно, для коагуляции коллоида требуется такое количество любого электролита, которое уменьшает заряд коллоида настолько, что прекращается электростатическое отталкивание частиц. Минимальное значение разности потенциалов между коллоидными частицами л раствором, обусловливающее коагуляцию, называется «критическим потенциалом» данного золя. В табл. 97 приведена коагулирующая способность некоторых электролитов по отношению к золю AsgS3. Ввиду того
714
Глава 16
что коллоидные частицы в таком золе заряжены отрицательно (вследствие адсорбции ионов S"), решающее значение для коагуляции имеют катионы добавляемого электролита. Как видно из приведенных данных, количества электролита, необходимые для коагуляции, довольно малы. Кроме того, эти данные свидетельствуют о некоторой .зависимости коагулирующей способности от заряда аниона. Объясняется это тем, что вместо активностей, от которых непосредственно зависит адсорбируемость, в таблице приведены аналитические концентрации ионов. А активность, естественно, зависит также от апиопа.
I
В некоторых случаях нри добавлении электролита может произойти перезарядка коллоидных частиц. Например,1 частицы золя золота, заряженные обычно отрицательно, под влиянием раствора соли алюминия
Таблица 97
Коагулирующая способность электролитов
(Привод о иные в таблице данные соответствуют концентрациям электролита (ммоль/л), обеспечивающим одинаковую во времени коагуляцию золя, содержащего 1,85 г As3S3 в.1 л (по данным Фрейндлиха')] -
Электролиты с однозарядными катионами Электролиты с диухзаряднымя катионами Электролиты е трехэа ряд дыми катионами Электролиты с органическими катионами
I.iGl 58 : Na.Cl 51 К (11 50 К NO., ВО 1/2KgS04 65 НС1 31 I/2H3SO4 30 MgClg 0,72 MgSO4 0,81 СаС13 0,65 StG13 0,64 BaClg 0,69 ’ZnClg 0,69 UO2(KO3)2 0,64 А1С13 0,093 A1(NO3)3 0,095 i/2Ala(SO4)3 0,096 Ce(NO3)3 0.080 Анилин солянокислый 2,5 Морфнп солянокислый 0,42 Кристаллвио.чет 0,16 Новый фуксин 0,11 Бензидин азотнокпе- 0,087 лый Хнпин сернокислый 0,24
могут стать положительно заряженными. Если ввести очень небольшое количество этого электролита, частицы золота разряжаются и коагулируют; если добавить его много, то коагуляция не наступает и в электрическом поле частицы движутся в противоположном направлении в соответствии с переменой их заряда.
Добавляя очень небольшие количества ионов с одноименным зарядом, молено в некоторых случаях усилить заряд коллоидных частиц и благодаря этому стибнлиэи-роеатъ золь. Например, золи металлов стабилизируются сильно разбавленными растворами гидроокисей щелочных металлов вследствие того, что ионы ОН' хорошо адсорбируются даже из очень слабо щелочных растворов. Если увеличить копцеятра-цито щелочи, то адсорбируется и катион, и наступает коагуляция. У многоосповных гидроокисей, таких, как Вз(0Н)3, преобладающим является действие катиона.
Приобретением коллоидными частицами заряда (благодаря адсорбции ионов), объясняется явление пептизации. Если на гель, образовавшийся в результате разрядки, или на иное вещество, достаточно пористое, чтобы в него мог проникнуть электролит, подействовать электролитом, ионы которого хорошо адсорбируются, то составляющие такое вещество частицы, заряжаясь, отталкиваются друг от друга, и вещество переходит в коллоидный раствор. Так как ионы Н' и ОН' адсорбируются особенно хорошо, они, как правило, и вызывают пептизацию.
В процессе пептизации может наступить состояние равновесия между золем и пептизирующимся веществом, подобно тому как в истинных растворах существует
Коллоидная, химия. Химия поверхностных явлений
715
^равновесие между раствором и осадком. Однако между этими равновесиями существует заметная разница; е отличие от. истинных растворов для данного количества .жидкости и пептизирующего вещества концентрация золя, зависит от количества осадка', при этом пептизированная часть осадка достигает максимума при некотором среднем количестве осадка {правило Оствальда). Это объясняется следующим образом: -заряд частиц, обусловленный адсорбцией ионов, достигает максимума при определенной концентрации электролита (см. стр. 730). Поэтому при определенной концентрации нентизатора достигается оптимальное пептизирующее действие. Чем больше количество твердой фазы, тем больше адсорбируется на ней пептизирующего вещества, поэтому количество пептизирующего вещества, оставшегося в растворе, зависит Ют количества твердой фазы и, следовательно, при одинаковом исходном количестве пептизирующего вещества может быть разпица между конечной его концентрацией я той, которая соответствует оптимальному пептизирующему действию.
Число соударений между коллоидными частицами и, следовательно, скорость коагуляции возрастают ио мере увеличения температуры и концентрации золя. Помимо действия электрического тока, к основным факторам, вызывающим коагуляцию коллоидов, принадлежат следующие:
1) добавление электролита,
2) кипячение золя,
3) упаривание.
Коагуляция па ступает также под действием золя с противоположно заряженными частицами. Вследствие этого не всякие коллоидные растворы можпо перемешивать.
Таким образом, в аналитической и препаративной практике для осаждения .веществ, оставшихся в коллоидной форме в растворе, как правило, поступают таким образом: добавляют к раствору подходящий электролит (чаще всего ЦС1, 11СгН3О2 или NH,-H3O) и некоторое время кипятят. Если это не приводит к достижению цели, «оагуляцию осуществляют упариванием досуха.
Те вещества, которые при обработке водой (в общем растворителем,
из которого коагулировал гель) вновь превращаются из теля в золь,
называют обратимыми коллоидами в отличие от необратимых, которые
без пептизирующего средства неспособны переходить ляющее большинство неорганических веществ принадлежит к числу необратимых коллоидов. Даже те из них, которые, будучи в рыхлом и водусодер-жащем состоянии, обратимы, по мере увеличения частиц теряют свою обратимость. Подобное увеличение частиц происходит, например, при многократном упаривании двуокиси кремния сконцентрированной СОЛЯНОЙ кислотой. Этот процесс используют в аналитической практике для количественного отделения двуокиси кремния.
Мицеллы. Поскольку коллоидные частицы в любом -золе заряжены, а раствор в целом электропейтрален, вокруг
в раствор. Подав-
Н'Н‘
Н‘Н-
Рис. 94. Схема ми-
целлы.
коллоидных частиц должны группироваться кристаллоидные
частицы с противоположным зарядом, т. е. электролитические ионы. Вследствие электростатического притяжения они достаточно плотно окружают каждую
коллоидную частицу, однако непосредственно с нею, как это происходит при адсорбции, пе соединяются *. Весь комплекс, т. е. коллоидную частицу с принадлежащими ей нейтрализующими ионами (так называемыми <т ретиво ионами»), называют, согласно Мальфптано (Malfitano, 1904) и Дюкло (Duclaux, 1907), мицеллой. Жидкость, в которой «плавают» мицеллы, называют меж мицеллярной жидкостью. На ряс. 94 схематически покапана мицелла :и:т ASgSj, полученного действием HgS па раствор соли As(Hl).
* В том, что соеднпеппя пе происходит, повинны те же силы, которые отталкивают иопы в обычном растворе электролита.
716
Глава 16
Межмнцеллярную жидкость можно отделить от мицелл ультрафильтрованием. Если при этом не происходит коагуляции, т. е. мицеллы сохраняются, то коллоид можно снова перевести в золь, прибавляя нейтральный диспергатор, например чистую воду. Однако, если мицеллы уже разрушены коагуляцией, перевести коллоидные частицы (если речь идет о лиофобном коллоиде, ср. ниже) снова в раствор возможно только путем новой зарядки, при которой мицеллы образуются вновь.
а
Лиофильные и лиофобные коллоиды. Как было отмечено ранее, не у всех золей устойчивость связана с зарядом частиц. Имеются вещества, которые удерживаются в коллоидном растворе, по существу, теми же силами, что и кристаллоиды, т. е. силами, которые обусловливают сольватацию. Такие вещества называют лиофильными (qiiAeiv — любить) коллоидами в отличие от всех остальных, называемых лиофобными ((pofteiv — бояться). Соответственно в зависимости от отношения к воде различают гидрофильные и гидрофобные коллоиды.
Типичными гидрофильными коллоидами являются клей, желатина, агар, крахмал, декстрин и многие другие органические вещества (прежде всего большинство белков). Из неорганических веществ к ним принадлежат гидроокись кремния, гидраты двуокиси олова и пятиокиси сурьмы, а также тонко дисперсные формы молибденовой и вольфрамовой кислот и молибденовой и вольфрамовой сини. Одвако эти вещества уже довольно близки: к гидрофобным коллоидам. Резкой границы между гидрофильными и гидрофобными коллоидами пе существует.
Типичные лиофильные или гидрофильные коллои^ды можно разряжать без коагуляции иоиами Н* или ОН'. В растворе их частицы сильно-сольватированы или гидратированы. Вследствие этого они по оптическим свойствам мало отличаются от дисперсионной среды и поэтому неясно-различимы под ультрамикроскопом, а часто вообще ие видны. По той же причине их золи обнаруживают весьма слабый эффект Тиндаля. Лиофильные коллоиды сильно повышают вязкость дисперсионной среды, в то время как лиофобные почти пе изменяют ее. В отличие от лиофобных лиофильные коллоиды сильно влияют и на поверхностное цатяжение дисперсионной среды.
Особо высокая вязкость золой лиофильных веществ обусловлена, по-видимому, сильной сольватацией содержащихся в таких золях коллоидных частиц (по крайней мере в подавляющем большинстве случаев). При прочих равных условиях вязкость в области молекулярной и коллоидной дисперсности тем больше, чем крупнее частицы. Следовательно, если радиус частиц возрастает за счет сольватации, это влечет за собой, увеличение вязкости. Вообще лиофильное вещество образует с дисперсионной средой, в которой оно плохо растворимо, более вязкий золь, чем с растворителем, обладающим лучшими растворяющими свойствами. Это находится в соответствии с предположением о влиянии сольватации. Все же часто оказывается, что хуже растворяющая дисперсионная среда вызывает гораздо меньшее распределение диспергируемого вещества, чем лучше растворяющая. В качестве примера лиофильных золей, обладающих высокой вязкостью, следует привести следующие:
Чистая вода Трисульфид Желатина Каучук Нитроцеллю- Целлюлоза
(щш 20°) мышьяка в в ваде в бензоле лоза в реактиве
вйде ( 1%-ИЫЙ) (1%-НЫЙ) в бутиловом Швейцера
(3%-пый, при спирте (1%-ный)
20°) {(%-ный)
0,01005 0,01043 3 35 — 65 180 пула
Таким образом, 1%-ный раствор желатины повышает вязкость воды почти в 10 000 раз сильнее, чем 3%-пый раствор гидрофобного трисульфида мышьяка. Однако вязкость золей лиофильных коллоидов может сильно колебаться у одного и того же вещества и при одних и тех же концентрациях и температуре. В значительной степени это зависит от способа приготовления золя и часто ог продолжительности выдерживания. Что касается последнего, то при выстаивании мицеллы в такого рода золях претерпевают
Коллоидная химия. Химия поверхностных явлений
717
изменения — степень дисперсности * либо уменьшается, либо увеличивается. Те в дру-гие процессы, происходящие с мицеллами (например, изменение степени сольватации), вызывают значительное изменение вязкости золя. Таким образом, измерение вязкости является важнейшим способом изучения золей лиофильных коллоидов. В технике оно служит мерой оценки такого рода веществ.
Оказалось, что лиофильные и лиофобные коллоиды в растворе несут электрический заряд**. Во многих случаях этот заряд можно устранить добавлением ионов Н' или ОН' (в зависимости от знака заряда), пе изменяя при этом устойчивости золя. В результате коллоид перестает передвигаться в электрическом поле. pH раствора, при котором это достигается, называют изоэлектрической точкой соответствующего коллоида (Michaelis) ***. При дальнейшем изменении pH коллоид снова начинает перемещаться, следовательно, оп снова приобретает заряд. Для лиофильных коллоидов очень характерна легкость, с которой они меняют заряд при введении в раствор ионов Н' или ОН'.
Так как устойчивость золя почти не зависят от заряда частиц, лиофильные коллоиды в общем мало чувствительны к действию электролитов. Лишь при сравнительно высокой концентрации электролитов происходит коагуляция. Однако этот процесс в соответствии с правилом Шульца — Гарди зависит нс только от заряда ионов, вызывающих^ коагуляцию, по и от других их свойств, влияющих также и на растворимость кристаллоидно-растворенных веществ, но-видимому- в первую очередь от гидратации (или сольватации) этих ионов.
Геди И студии
Желатинизация и яабз'хание. Наиболее характерным свойством лиофильных коллоидов является способность их золей, если они по слишком разбавлены, застывать в виде однородной компактной мягкой, но довольдо эластичной массы. Ее называют студнем, а процесс ее образования — студнеобразованием или желатинизацией. Студень может содержать весьма значительное количество дисперсионной среды (при известных обстоятельствах более 99%). Если концентрация коллоидного вещества в студне не очень высока, то он ведет себя, не учитывая механических свойств, почти так же, как жидкий золь. Например, скорость диффузии кристаллоидов в студпо практически равна таковой в золе, электрические свойства не изменяются и химические реакции в студне протекают так же, как и в обычном растворе ****. Одпако с увеличением концентрации коллоидных частиц в студне скорость диффузии растворенных кристал
лоидов уменьшается.
* Под «агрегацией» понимают понижение степени дисперсности вследствие слипания частиц, первоначально еще на приводящее к коагуляции.
У лиофильных коллоидов заряд обусловлен иногда пе адсорбцией ионов, а электролитической диссоциацией, подобно веществам, находящимся в молскулярно-дисперсном состоянии. В таком случае устранение заряда есть не что иное, как подавление диссоциации.
*** Такнм же образом определяют изоэлектрическую точку для амфотерных кристаллоидов.
**** В студне отсутствуют помехи, обусловленные конвекцией. Вследствие этого в процессе диффузии одного вещества в студепь, который содержит Другое вещество, реагирующее с первым с образованием осадка, наблюдается образование так называемых слоев Лпзеганга (например, при диффузии нитрата серебра в студень, содержащий бихромат). [Обтякнение образования сдоев см. Wi. Ostwald, Z. physi-kaJ. Chain., 23, 365 (1897) н Wo. О .? t w a 1 d, Kolloid-Z„ 36, Erg.-Bd. (Z s i g ти о n-dy-Fest.scftr., 1925), 380; 40, 144 (1926). J Подобные слои иожпо получить 11 другими способами, если предотвратить мешающую их образованию конвекцию, например проводя реакцию в капиллярах.
718
Глава 16
Студии, образующиеся при однородном застывании золя, отличаются от прсту®-] тов, образующихся в результате коагуляции коллоидов, не только по способу ocj£>w-1 вания, но п по свойствам. Такие студни называют целями в узком смысле. О^нгк»1 название «гель» употребляют и в более широком смысле для обозначения как npcjprat- ! тов коагуляции (гели в узком смысле), так и студней. Предлагали гели, получв^ш^» в результате коагуляции называть «коагелямц», однако такое название до сих с:» не привилось. Студии иногда называют также «желе» или «желатина».
При длительном «высыхании» студни могут превратиться в сове--шснно твердую массу. В большинстве случаев при этом образуются пр; дукты, напоминающие роговое вещество (например, у клея и желатины : последние при добавлении дисперсионной среды способны снова пре.вра щаться в студни. Такой процесс превращения студня называют на£-_ ханием *.
Другие студни (например, студепь кремневой кислоты) при высушивании уыеп^ шаются в объеме лишь до определенного предела. При дальнейшем высушивании czzr внезапно теряют прозрачность, а пространство, из которого уходит вода, заполняется воздухом («точка превращения», си. стр. 729, а также т. 1, стр. 537). При этом объис либо пе изменяется вовсе, либо только очень незначительно. Вода способна внось. заполнить пустое пространство. Однако студент, кремневой кислоты после высушивания и последующей обработки водой уже не набухает; другими словами, он уже не возвращается в первоначальное Студнеобразное состояние и, более того, приобретает свойства, роднящие его с гелем кремневой кислоты, полученным в результате коагуляции. Такого рода студни, которые при высыхании утрачивают способность к набуханию и, следовательпо, свойства студня, навыкают ненабухающими в отличие от упомянутых выше — набухающих.
Некоторые студни при нагревании разжижаются. Если их охладить, до первоначальной температуры, то через некоторое время они вновь застывают. Общеизвестным является процесс застывания желатины. Известны также студни и гели, которые разжижаются в результате встряхивания при* постоянной температуре (еще лучше под действием ультразвука). Это явление носит название тиксотропии ((h'£tg — соприкосновение,. тролЬ; — изменение).
Синерезис. Студни могут сжиматься не только при высушивании, но и при стоянии на влажном воздухе (или на воздухе, насыщенном парами дисперсионной среды). При атом из них самопроизвольно выделяется разбавленный коллоидный раствор, (и одновременно присутствующие в растворе кристаллоиды). Это явление впервые-, было описайо Грэмом и названо синерезисом (ouvaigelv — прессовать).
Структура студней. Микроскопическое и ультрампкросконическое исследование-студией весьма осложняется тем обстоятельством, что студпе образующие вещества в набухшем состоянии очень мало отличаются от дисперсионной среды по способности преломлять свет. Устранить эту трудность впервые удалось Бютптлн (Biitschli, 1892) путем замены воды в гидросгуднях другой дисперсионной средой, например спиртоьг или бензолом. Под микроскопом ему удалось установить, что студни имеют частично сетчатую, частично сотовую структуру. Последнюю он объяснил таким образом, что-коллоидное вещество как бы образует связующую пенообразную массу, пустоты в которой заполнены жидкой дисперсионной средой. Более поздние ультрамикросконп-ческце изучения студней показали (Zsigmondy, Bachmann, 1911), что «соты» образуются благодаря прослойкам жидкости, т. е. за с,чет той можмпцеллярной жидкости, которая разделяет мицеллы, сближающиеся в процессе студнеобрааовання. Следовательно, пенистая структура принадлежит дисперсионной среде или межмицелл ярпой ашдкости, заполняющей пространство между мицеллами коллоида. Однако вполне-вероятно, что при более тесном сближении мицеллы частично слипаются между собой,
* Термин «набухание» употребляют также в болею широком смысле для обозначения явления сильного увеличения объема по верх постно активно го вещества т происходящего при добавлении воды независимо от того, образуется при этом студень или нет.
Коллоидная химия. Химия поверхностных явлений. 719“
Если предположить, что этот процесс происходит достаточно полно, то становится попятным возникновение только ячеистой или губчатой структуры, что для некоторых студней наблюдал еще Вютшли. На основании многих наблюдений фон Веймарп (v. Wei-шагп, 1908) и Пул (Poole, 1925) пришли в основном к тому же выводу относительно структуры студней. Более поздние представления об их структуре основаны на предположении, что дисперсионная среда связанно распределена цо всему студню и неотделима от него в виде капелек, проходящих через мембрану. Подтверждением этому служит почти беспрепятственная диффузия в разбавленных студнях. С этой точки зрения нопятны и другие явления, наблюдаемые у студней, например при высушивании студня кремневой кислоты (см. т. I, стр. 537).
Пластические массы. По своей структуре студни родственны так называемым пластическим. массам, таким, как глиняная масса, употребляемая для изготовления керамических изделий, смолам и другим пластмассам, продуктам из целлюлозы (например1, целлулоид, лак), а также химически однородным веществам (каучуку и гуттаперче). Для понимания свойств всех этих веществ учение о коллоидах имеет огромное значение.
Оксигидраты. Применение методов коллоидной химии (в совокуи-пости с рентгенометрическими) позволило также донять природу так называемых оксигидратов, т. е. желатинообразных осадков, образующихся при осаждении из водных растворов многих веществ в виде окислов или гидроокисей. Типичпыми представителями оксигидрагов являются гели SiOE, GeOs, SnO2, TiO2, ZrO2, а также A12O3, Cr2O3 и FcEO3. Эти вещества образуют гели, которые в свежеприготовленной форме настолько тонко> раздроблены, что при рентгенометрическом исследовании оказываются' аморфными. Вследствие очень сильно развитой поверхности они удерживают воду за счет сил адсорбции, а частично, по-видимому, за счет капиллярных явлений настолько прочно, что и теперь еще только в отдельных случаях удалось доказать (ср. т. I, стр. 393 и 579), насколько вода связана таким образом и насколько химически.
Если гели выдерживать в присутствии осадителя, то большинство ns пих с течением времени превращаются в осадки с кристаллическими свойствами, обнаруживаемыми рентгенометрически (-«старение» осадков) (Fricke, Huttig, Biltz W., Bohm, Simon,. Ko J Usch utter), При этом в некоторых случаях (например, SiO2, GeO2, SnO3, TiO2^ ZrO2) атомы располагаются в решетках окислов, в других (папример, ALOy, ZnOr CdO) — в решетках гидроокисей. Ответить на вопрос, в каких случаях аморфный осадок представляет собой окись или гидроокись, невозможно без дополнительных исследований. В таких случаях аморфный осадок называют «окенгидратом» (в отличие от гидроокиси). я»
Рентгенометрическими измерениями установлено, что при старении окси гидрате в появляются структуры, характерные для окислов, но не для гидроокисей. Это-свойственно, как видно из приведенных выше примеров, преимущественно веществам, образованным элементами IV группы периодической системы. По обе стороны от этой группы в главных подгруппах очень быстро возрастает способность к образованию Хорошо выраженных гидроокисей. А для большинства веществ побочных подгрупп также наблюдается образование окислов при старении осадков.
Рентгенометрическими исследованиями такого рода гелей можно не только установить, являются ди образующие их вещества аморфными или кристаллическими (в последнем случае при сравнении с рентгенограммами известных соединений можно определить, в форме какого соединения находятся вещества), но и определить размеры частиц. Как было указано в гл. 7 т. I, интерференция рентгеновских лучей, получаемая по методу Дебая — Шерера, тем более размыта, чем мельче кристаллики. Если выполнены определенные условия, размер частиц можно определять по ширине интерференционных полос. Проводи сравнение с другими способами определения размера частиц (ср. стр. 710—712), следует учесть, что так называемая «рентгенометрическая величина частниц указывает па область пространства, которую занимает решетка одного однородного кристалла. А поскольку коллоидные частицы или мицеллы, как правило, не монокристаллы, а кристаллические агрегаты, то исходя из рентгенометри-вдек» пай денной величины частиц в общем пользя сделать вывод о размере мицелл.
720
Глава 16
Защитные коллоиды. Лиофильные коллоиды способны сообщать свою низкую чувствительность к электролитам лиофобным коллоидам, с частицами которых они соединяются. Это свойство можно успешно использовать для стабилизации лиофобных коллоидов. Применяемые для подобных целей лиофильные коллоиды называют защитными коллоидами.
Например, если к Ю мл 0,005 %-йог о красного гидрозоля золота прилить 1 .м.г 10%-ного раствора NaCl, то окраска золя быстро переходит в фиолетовую, что вскоре влечет за собой полную коагуляцию частиц золота. Одпако если предварительно к раствору золота добавить гидрофильный коллоид, например желатину, то изменение окраски, обусловленное укрупнением частиц и последующая коагуляция не происходят. Количество защитного коллоида (мг), достаточное при определенных ускокиях для предотвращения изменения окраски гидрозоля золота и последующей коагуляции, называют, по предложению Зигмонди, золотым числом. По аналогии в зависимости от того, какой гидрофобный коллоид подлежит защите, говорят о серебряном числе, серном числе, число берлинской лазури и т. п., вообще о защитном числе.
В условиях вышеприведенного опыта золотое число желатины и клея равно 0,005, альбумина 0,2, картофельного крахмала 25. Таким образом, количества коллоида, обеспечивающие одно и то же защитное действие, для разных защитных коллоидов весьма различны. Защитное действие зависит также от состояния, в котором находится защитный коллоид. Например, свежеприготовленный гидрат двуокиси олова действует как защитный коллоид Для золота (кассиев золотой пурпур; ср. стр, 451); напротив, «постаревший? гидрат двуокиси олова защитных свойств не проявляет. Гель, приготовленный упарнвапием воля, обладающего защитным свойством, можно снова растворять, не опасаясь, что защитные свойства его исчезнут. Это свойство находит применение при получении растворимых гелей лиофобных веществ, главным образом металлов (например, протаргол; см. стр. 425). Количества защитного коллоида, необходимые для стабилизации золя, часто весьма невелики. Как видно из приведенных выше чисел, достаточно 0,005 мг желатины, чтобы защитить в 100 раз большее количество золота от действия 0,2 и. раствора NaCI.
Если количество взятого лиофильного коллоида значительно меньше защитного числа, то у многих золей наблюдается повышенная чувствительность по отношению К электролитам («сенсибилизация»),
Поверхностная активность коллоидных веществ. По сравнению с грубодиспорсными веществами коллоидные частицы обладают чрезвычайно сильно развитой поверхностью. Из 10,5 а серебра, в компактном состоянии занимающего объем куба с длиной ребра 1 сл* и общей поверхностью 6 сл*2, при измельчении до размеров кубика с длиной ребра 100 шр получают 1016 таких кубиков с общей поверхностью 600 000 сл*2. Если измельчение довести до частиц диаметром 1 шр, то число кубиков возра-^. стет до 10s1, а суммарная поверхность до 60 млн. сл*2. Такое чрезвычайно сильное увеличение поверхности прн переходе от грубодиснерсното к коллоидному состоянию влечет за собой сильное возрастание так называемых поверхностных сил. Ввиду этого вещества, находящиеся в кодлоидном состоянии, обладают сильно выраженной «поверхностной активностью». Это проявляется, например, в их высокой адсорбционной способности. Этим объясняется широкое применение коллоидных веществ в качестве адсорбентов. Зная адсорбционную способность вещества, можно определить величину поверхности и размеры частиц коллоидных веществ. С поверх- ' постной активностью связано также значительное повышение каталитического действия некоторых веществ при переводе их в коллоидное состояние (ср. гл. 17).
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ
Возникновение поверхностных сил объясняется неполным валентным насыщением атомов или ионов, расположенных на поверхности. Например, у хлорида натрия каждый из ионов, лежащих внутри кристалла,
Коллоидная, химия.. Химия поверхностных явлений 721
окружен шестью противоположно заряженными ионами, а ион, лежащий на грани куба,— только пятью (ср. т. Г, рис. 44). Следовательно, ионы, расположенные на поверхности, являются координационно ненасыщенными. У веществ, кристаллизующихся в объемноцентриров энных кубических решетках, таких, как Mo, W, a-Fe, каждый атом, расположенных! на гранях куба, имеет 4 незанятых координационных места. Это характерно также и для металлов с гране центрированной ..решеткой, таких, как Си, Ag, Ап и у-Fe. Сказанное относится соответственно и к веществам, кристаллизующимся в любых других решетках. У атомов или ионов, расположенных па ребрах или в углах, число координационно ненасыщенных мест еще больше, например у NaCl для ионов, лежащих на ребрах куба, оно равно 2, а в углах — 3. Энергию, которая может выделиться в результате насыщения этих координационно свободных валентностей на поверхности вещества (включая ребра й углы), называют поверхностной энергией этого вещества. Как следует из сказанного выше, поверхностная энергия данного вещества зависит от формы его существования. Эта энергия очень сильно возрастает по мере степени измельчения соответствующего вещества. Причем следует отметить, что измельчение сопровождается не только увеличением собственно поверхности, но и в гораздо большей степени увеличением общей длины ребер и числа углов *.
Поверхностные силы имеются к у кристаллов, образованных молекулами. В атом случае они обусловлены тем, что у молекул, расположенных на поверхности, имеются ненасыщенные ванДерваальсовы силы (ср. т. I, гл, 9). То же справедливо и для жидкостей.
Уменьшение поверхностной энергии, т. с. насыщение валентностей, выходящих на поверхность, может происходить либо в результате поглощения поверхностью другого вещества (адсорбция), либо за счет сокращения самой поверхности. Последнее возможно при изменении способа измельчения или уменьшении степени измельчения. Жидкие вещества приобретают форму шара, если на них не действуют посторонние силы, так как при данном объеме шар имеет наименьшую поверхность. Со временем мелкие капельки укрупняются (агломерация). Кристаллики твердых веществ также стремятся к увеличению,. Этот процесс может Происходить при непосредственном срастании (рекристаллизация, ср. гл. 19), а также в результате роста более крупных кристаллов за счет более мелких (при этом последние либо испаряются, либо переходят в раствор).
Увеличение диплопия пара и растворимости по мере измельчения вещества непосредственно связано с возрастанием энергия, зависящей от измельчения. Однако явно подобные изменения давления пара и растворимости становятся заметными лишь при очень тонком измельчении (близком к коллоидно-дисперсной области). У веществ, обладающих сравнительно малой растворимостью (например, гипс), увеличение растворимости при уменьшении зернистости хорошо заметно. Для очень трудно растворимых веществ растворимость, как правило, определяется не с такой точностью, которая позволяла бы проследить за влиянием степени зернистости па величину растворимости. Однако уменьшение поверхности вследствие укруп-гтепия зерен большей частью наглядно видно благодари снижению адсорбционной
* Если куб объемом 1 см3 измельчить до кубиков с длиной ребра 100 тц, то общая поверхность возрастает в птвошеппи 10й : 1, общая длина ребер — в отношении 1010 ; 1, а общее число углов--н отпошщтгш П)1г> . д Следует, одпако, otmisthti,, что пи одно TBtjwoe вещество пе обладает совершенно гладкой поверхностью, так что реальный куо всегда имеет большую поверхность и большее число ребер и углов, чем получается при вычислении из его идеальной конфигурации.
46 г Реми
722 Глава 16
способности осадков по мере «старения» У легко растворимых веществ укрупнение верен происходит настолько быстро, что практически они в очень тонко раздробленном состоянии никогда не успевают контактировать с растворителем.
АДСОРБЦИЯ И АБСОРБЦИЯ
Под адсорбцией понимают поглощение какого-нибудь газообразного или растворенного вещества поверхностью твердого или жидкого вещества. Если при этом поглощаемое вещество распространяется по всему объему (например, при поглощении водорода палладием или при растворении газов в жидкостях), то процесс называют абсорбцией. Пористые вещества, обладающие капиллярной структурой, способны поглощать газы или пары за счет капиллярных сил (ср. стр. 728). Такой процесс называют капиллярной конденсацией. Если де известно, к какому из перечисленных способов сводится поглощение какого-нибудь вещества, или если речь идет о процессах, в которых эти: явления перекрываются, то употребляют общее выражение сорбция.
Тело, на поверхности которого происходит адсорбция, называют адсорбирующим, материалом, или адсорбентом, вещество, которое адсорбируется,— адсорбируемым веществом. В процессе сорбции различают соответственно сорбент и сорбируемое вещество. Удаление сорбированного вещества с поверхности или из объема сорбента называется десорбцией.
В процессе адсорбции выделяется тепло, так как с ней связано уменьшение поверхностной энергии {тепло адсорбции}. Следовательно, по принципу Ле-Шателье, с увеличением температуры адсорбция уменьшается. То же справедливо и для сорбции (см. рис. 96, стр. 724).
При постоянной температуре адсорбция пропорциональна количеству (точнее, поверхности) адсорбента/0на1 зависит также от концентрации адсорбируемого вещества в газовой фазе или в растворе, из которого происходит адсорбция. Зависимость адсорбции от концентрации существенно отличается от зависимости, наблюдаемой при поглощении газообразного или растворенного вещества растворителем. В то время как в последнем случае поглощение прямо пропорционально плотности газа или концентрации, в процессе адсорбции, как правило, вначале при очень небольших концентрациях адсорбируемого вещества адсорбция сильно возрастает с увеличением концентрации, а при более высоком содержании адсорбируемого вещества возрастает лишь незначительно. Это следует из рис. 95, где кривая I соответствует зависимости абсорбции газа жидкостью от его давления в соответствии с законом Генри; кривые II— V соответствуют адсорбции газов твердыми веществами также в зависимости от Давления газа.
По закону Генри, растворимость газа в жидкости (а также в твердом веществе) пропорциональна его давлению. Таким образом,
coast; (4)
где с —- концентрация газа в жидкости и р — давление (или'парциальное давление) в газовой фазе. Закон Гепри справедлив в том случае, когда растворенное вещество не вступает в соединение с растворителем и молекулы при переходе в раствор не расщепляются. При соблюдении этих условий уравнение (4) применимо также и для абсорбции газов твердыми веществами. Если, как в случае растворения водорода в палладии, происходит расщепление молекулы на два атома, то растворимость цро-
* При «старении» осадков, помимо укрупнения зерен, иногда происходят ещб и другие превращения^ например изменение модификации.
Коллоидная химия. Химия поверхностных явлений.
723
порциона льна корню квадратному из величины давления недиссоциированного газа с = 1//7- const
Закон распределения вещества между растворителем и газовой фазой аналогичен закону распределения растворенного вещества, между двумя растворителями
— = COHSt, (5);
с2 i
где всг —концентрации растворенного вещества в растворителях 1 и 2. Другими словами; вещества распределяется между двумя растворителями в постоянном соотношении при условии, что в обеих фазах оно имеет одинаковый молекулярный вес.
0,1 0,2 0,3 Ofi 0,5 0,6 0,7 0,8 Q5 1,0 1,1 1,2 Концентрация с .
Рис. 95. Изотерма абсорбции (/) и изотермы адсорбции [1I V}.
Если в одном растворителе вещество существует в мономолекул яр ной форме А, а в др у- 1 гои — в более высокомолекулярной форме ЛЛ, то при распределении имеет место равновесие «Л 7’ А„, т. е. , 4
С| - ' .
—™ const, (закон распределения Нернста). . (6)
Подобное же соотношение эмпирически найдено для зависимости/ адсорбции от давления газа иди концентрации адсорбируемого вещества;
a — k~cl^n (изотерма адсорбции Оствальда}, (7)
В этом уравнении а — количество адсорбированного вещества, приходящееся на одну весовую часть * адсорбента; с — концентрация адсорбируемого вещества в газовой фазе или в растворе, из которых осуществляется адсорбция; к и ,п — константы, зависящие от природы адсорбируемого вещества и адсорбента и от температуры. Однако аналогия между уравнениями (6) и (7) чисто формальная. Опа не имеет никакой внутренней связи. Уравпепие (7) в отличие от уравнения (6) нельзя вывести теоретически и можно показать, что оно применимо только в ограниченной области, концентраций. На его ограниченную применимость указывал еще Оствальд, впервые предложивший эту формулу (1905). Одпако на практике ее применяют часто. Значение п, как правило, лежит В ; интервале 1,5—5. Логарифмируя уравнение (7), получают
log a— log k -j- 1/n-Iog е, т. е. уравнение прямой,
* Поскольку реальная величина поверхности у твердых веществ в большинстве случаев не известна (ср. сноску * на стр. 721), количество адсорбированного вещества относят обычпо к единице веса, предполагая, что для одного и того же адсорбента поверхность пропорциональна весовому количеству.
46*
724
Глава 16
Теоретически изотермы адсорбции были обоснованы Лангмюром (1918) и другими. Исходя из предположения, что процесс адсорбции происходит главным образом в мономолекулярном слое, Лангмюр вывел формулу
в которой а — ноютчество адсорбированного вещества на единицу веса адсорбента; р — его давление (пли концентрация) в газовой фазе; а и р — Константы, зависящие от условий опыта. При условиях опыта, соответствующих тем, при которых эта формула была выведена, уравнение хорошо согласуется с экспериментальными данными. Однако применимость его ограничена, так как в большинстве случаев лежащие в его основе предположения невыполнимы. Общепринятой является изотерма адсорбции, найденная Брунауором, Эмметтом и Теллером (Brunauer, Emmett, Teller, 1937—1938):
ер-Ь-р
(Po-P)
(9)
где а — количество адсорбированного газа; р —давление адсорбированного газа; Ро — давление при насыщении; «0 и b — константы. Эта формула справедлива также для адсорбции с образованием более толстых слоев. Если на адсорбенте образуется
насыщенный мономолок ул яр ный слой; то о=.л0. Если выбрать подходящий газ, то это соотношение выполняется довольно точно. Таким образом, можно определить действ шпелыпро величину поверхности адсорбента (способ БЭТ).
Для особо сильно сорбирующих веществ, па пример активированного угля, которые сорбируют главным образом за счет капиллярной конденсации (ср. пиже), справедливо соотношение Реми ’* ~
или,
и Крона (Remy, Krohn, 1937):
<« + а)2=*(р + (5),
в л огарифмической форме, 2 log (n + a)=logff+Iog (р+Р), и р имеют те же значения, что и в
(10) у рав непричем
где а ниях (8) и (9): К, а и [} — константы, часто' К = а2/р.
Сорбция газов. Сорбцию газов поверхностно активными веществами в настоящее время широко используют, например, для извлечения паров легколетучих растворителей. Для этой цели в первую очередь применяют пористые сорбционные материалы с сильно развитой внутренней поверхностью, такие, как специально обработанный древесный уголь (активированный уголь) и силикагель. У таких веществ на процесс адсорбции газов иногда накладывается явление капиллярной конденсации (см. стр. 728), которая часто играет даже решающую роль. Характер зависимости сорбции газов активированным углем от природы газа, давления и температуры показан па рис. 96 (ср. также т. I, стр. 464).
Скорость адсорбции газов в большинстве случаев очепь велика. Для веществ с открытой поверхностью адсорбционное равновесие устанавливается, как правило, в течение нескольких секунд. У веществ типа угля и силикагеля, для проникновения во внутреннюю поверхность которых адсорбируемое вещество должно продиффундировать через узкие и довольно длинные поры, равновесие устанавливается соответственно медленнее.
Р и с. 96. Сорбция различных газов активированным углем в зависимо сти о т темп ер ат у ры (Remy, Непс, 1932).
Ордината — температура, €С; абсцисса — количество сорбированного газа (мл), приведенное: к ja" и 1 атм.
Коллоидная химия. Химия поверхностных явлений
725
Активированная адсорбции. В некоторых случаях уменьшение количества адсорбированного газа с увеличением температуры происходит лишь до определенного значения ее, и при дальнейшем повышении температуры адсорбция вдруг сильно возрастает. Так как тепловой аффект адсорбции положителен (см. стр. 722), это явление может быть обусловлено только наложением па процесс адсорбции других процессов, поглощающих тепло. В некоторых случаях удалось показать, что таким процессом является расщепление адсорбирующихся молекул. на атомы й свободные радикалы. В других на процесс адсорбции, сопровождающийся выделением тепла, накладывается растворение адсорбируемого вещества в адсорбенте. Оба процесса (расщепление и растворение) могут идти одновременно. Например, Нг адсорбируется № или Си только до температуры 100° К. Выше этой температуры наряду с адсорбцией происходит раещеп.тепие иа атомы Н и одновременно растворение атомов Н в металле (Benton, 1931). Описанные явления могут происходить также в том случае, когда образуется химическое соединение с сорбентом (^хемосорбция»), поскольку образованию химического соединения, как правило, предшествует расщепление реагирующих молекул (см. гл. 17). В каждом случае возрастание адсорбции с увеличением температуры обусловлено тем, что возникает процесс, требующий затраты определенной энергии активации (см. стр. 740). В связи с этим процесс такой адсорбции называют активированной адсорбцией (Taylor, 1931).
Адсорбция по растворов. Между адсорбцией газов и адсорбцией веществ из растворов не существует в общем простой взаимосвязи. Адсорбенты, весьма активные по отношению к газам, отнюдь не всегда являются хорошими поглотителями дня растворенных веществ, и наоборот. Одним из решающих факторов при адсорбции растворенных веществ яь.т.тетея растворитель. Растворитель, который сам хорошо адсорбируется, понижает адсорбируемость растворенного в пем вещества.
Адсорбция электролитов. Из растворов электролитов, как правило, адсорбируются иолы какого-нибудь одного вида — катионы h.'jii анионы — в зависимости от природы адсорбента. Часто адсорбция электролитов сопровождается изменением значения pH растворов. Например, если внести гидрат двуокиси марганца в раствор хлорида калия, то преимущественно будут адсорбироваться ионы К’, а раствор при этом становится кислым. Объясняется это тем, что тидрат двуокиси марганца уводит из раствора ионы К.’ и отдает в раствор иопы Н‘. Электролиты, как правило, адсорбируются в относительно небольших количествах. Адсорбируемость простых ионов с одинаковым знаком в большинстве случаев не сильно различается; в общем ноны тем сильнее адсорбируются, чем меньше они гидратированы и чем выше их заряд.
Селективная адсорбция. В результате адсорбции какого-нибудь легко адсорбируемого вещества снижается поглотительная способность адсорбента по отношению к другому, труднее адсорбируемому веществу. Вследствие этого из смеси веществ с существенно различающейся адсорбируемостью тфактически извлекаются только наиболее легко адсорби- и» руемые {селективная, адсорбция).
Селективная адсорбция из раствора лежит в основе так называемых хроматографических енпсейае разделения веществ *. Если раствор, содержащий вещества А, В, С и т. д. с существенно различной адсорбируемостью, пропустить через стек ляпнуто колонку, заполненную подходящим адсорбентом (см. рис. 97), то в верхней се части концентрируется практически только наиболее хорошо адсорбируемое вещество А, Только после того как оно полностью уйдет из раствора, в следующем, более глубоко лежащем слое, поверхность которого еще свободна, адсорбируется вещество В. Когда и это вещество полностью извлечено из раствора, в следующем слое адсорбируется вещество С и т. д. Если селективной адсорбции подвергаются окрашенные вещества, то очень четко виден ряд окрашенных слоёв (отсюда название «хроматография»). Путем последующего промывания чистым растворителем (селективная десорбция) можпо еще более увеличить эффект разделения.
* Метод хроматов рафия покой адсорбции (хроматография) был открыт русским ботаником М. С. Цветом в 191 ”>б г. Ему удалось разделить красящее вещество листьев, использовав порошок СаС(),> в качестве адсорбента. Позднее этот способ был развит главным образом Куном, Цехмейстерпм и Карцером, Его с успехом применяют в основном для разделения органических природных веществ.
726
Глава 16
Эффективность некоторых веществ, применяемых для хроматографического разделения, определяется пе Столько адсорбирующей способностью их, сколько способностью образовывать твердые растворы с другими веществами. В таком случае растворенные вещества распределяются между твердой и жидкой фазами по закону распределения Нернста (см. 723): при этом, как правило, п == 1. В отличие от адсорбционной хроматографии последнюю называют распределительной хроматографией.
В настоящее время хроматографию используют не только в органическом химии и биохимии, но также й в неорганической химии. Часто ее применяют теперь в форме хроматографии на бумаге. Для этого вместо _ зернистого вещества, помещаемого в колонку, например И поверхностноактивной окиси алюминии., селективную адсор-™ опию осуществляют при помбщи полосок фильтровальной
.и] бумаги. Такой способ позволяет работать-с весьма нёболь-И шими количествами веществ,
Ц Возможность применения хроматографии для разде-
В ления ионов неорганических веществ впервые была пока-=4 зана Швабом и Джокером (Schwab, lockers, 1937) на очень Н наглядном примере. В препаративной неорганической химии она имеет особенно большое значение для получения чистёгх редких элементов и для разделения трансурановых эле-/ \ ментов. С’ этой Целью, как правило, используют ионообмен-
/ \ ныв синтетические смолы, (см. т. Д стр. 81). Их действие
/ \ основано, правда, не на селективной адсорбций, а на химй-
ческих превращениях самой смолы (ионный обмен). Но по-скольку этот процесс также носит селективный характер, Рис. 97. методика работы с Ионообменными смолами по существу Хроматогра- такая же, как и при адсорбционной хроматографии. Для фпческая некоторых аналитических целей хроматография оказывается колонка. более: предпочтительной. При помощи бумажной хроматогра-фии в некоторых случаях можно особенно прюсто и быстро анализировать вещества при высоком их разведении, например определять ионы щелочных и щелочноземельных металлов в водопроводной воде и в минеральных водах.
Селективной адсорбцией из водных растворов объясняется известное в геологии и почвоведении явление высокого обогащения некоторыми веществами слоёв земной коры, пропитанных коллоидными соединениями (см. т. I, стр. 185).
Техническое извлечение летучих растворителей из их паров, сильно разбавленных воздухом, при посредстве активированного угля или силикагеля также основано па селективной адсорбции, а частично — на селективной капиллярной конденсации. Хроматографию часто используют для разделения газовых смесей (газовая хроматография).
Мопомолекулярные поверхностные слон. Явлению адсорбции родственно явление растекания масел и т. п. па поверхности воды. Если поверхность воды достаточно велика но сравнению с количеством масла, то можно считать, что на воде образуется его мономолекулярный слой. Если вещество носит полярный характер (например, Жирные кислоты), то при образовании м о помол скул ирного слоя, как показал Лангмюр (1917), нее молекулы располагаются перпендикулярно к поверхности воды так, что\ их полярный копец (в случае жирных кислот группа СООН) обращен к-воде. Несомненно, что при адсорбции на поверхности твердых веществ имеет место аналогичное явление. Другими словами, и здесь наблюдается тенденция к образованию мономолокулярных поверхностных слоев с направленной адсорбцией полярных молекул.
Коллоидная химия. Химия поверхностных явлений 727
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ И КАПИЛЛЯРНЫЕ СИЛЫ
Поверхностное натяжение. Свободная поверхностная энергия, отнесенная к единице поверхности, называется поверхностным натяжением. У жидкостей поверхностное натяжение можно непосредственно измерить благодаря их способности изменять свою поверхность в зависимости от формы сосуда.
Приращение свободной энергии ДЕ цри увеличении поверхности жидкости на некоторую величину АО соответствует работе, которую необходимо затратить на ото увеличение'.
Ы?=а-&0, (11)
где о — поверхностное натяжение. Отсюда поверхностное натяжение соответствует размерности работы на единицу поверхности или силы на единицу длины и измеряется в динах на 1 с.« (дин/см.). -
Поверхностное патяжение зависит от природы обоих веществ, соприкасающихся на границе раздела фаз. Например,; поверхностное натяжение воды по отношению к воздуху равно 72,7 дин)см, а относительно ртути 413 дин^см (при 20°). С увеличением температуры поверхностное натяжение уменьшается. Вследствие этого при увеличении поверхности поглощается тепло. Общая поверхностная энергия складывается из затраченной теплоты и произведенной работы. Следовательно, последняя соответствует лишь свободной поверхностной энергия.
Закон адсорбция Гиббса. Непосредственная зависимость между поверхностным натяжением жидкости и адсорбцией была термодинамически установлена Гиббсом, Кратко эту зависимость можно сформулировать так: если гав или находящееся с растворе вещество понижает поверхностное натяжение, то оно концентрируется на поверхности жидкости (положительная адсорбция), если еаз или находящееся е растворе вещество повышает поверхностное натяжение, то концентрация его на поверхности меньше, чем в объеме (отрицательная адсорбция).
Поскольку при растворении неорганических солей в воде ее поверхностное натяжение относительно воздуха увеличивается, концентрация соли непосредственно на поверхности раствора меньше, чем внутри объема; известно, что в растворах некоторых содей поверхностный слой образован чистой водойо
Каппл.тяршде трубки. Поверхностным натяжением объясняется также явление повышения уровня смачивающих жидкостей в узких трубках (капиллярах). Если капилляр опустить в какую-нибудь смачивающую жидкость, то его стенки покрываются. плойкой жидкости. Поверхностное натяжение в данном случае стремится уменьшить поверхность этих пленок, что способствует всасыванию жидкости в капилляр. Работа, которая затрачивается для подъема столба жидкости весом r2nfeg$ па высоту д (где г — радиус трубки, g ускорение свободного падения й з — плотность жидкости), на отрезке dh трубки равна r^nhgs-dh. Уменьшение поверхности, вызываемое подъемом жидкости на отрезке dh в трубке, составляет 2ти. dh. Умно- с'й' жая это выражение на о, получают в соответствии с уравнением (И) уменьшение свободной энергии, обусловленное сокращением поверхности. Для равновесного состоявши оно должно быть равно работе, затрачиваемой на перемещение столба жидкости, т. е. r-ahes. dh~a-2rii-dh, откуда следует
(12)
На основании уравпепия (12). поверхностное натяжение жидкости можно измерять, зная высоту подъема жидкости в капилляре h и его радиус г.
Если жидкость не смачивает стенки трубки (например, ртуть, стекло), мениск жидкости в трубке находится ниже уровня жидкости снаружи трубки. Смещение мениска вниз называют7 капиллярной депрессией. В1 подобном сл у чаеносма кивающее стенки вещество стремится уменьшить свою поверхность. В соответствии с приведенным выше выводом его поверхностное натяжение можно рассчитать по формуле (12), в которой h — капиллярная депрессия.
Кривизна поверхности и давление пара. Известно, что мениски жидкости в трубках вогнуты, если жидкость смачивает стенки трубки, и выпуклы, если не смачивает. Вследствие того что смачивающая жидкость в трубке находится на более высоком уровне, чем вне ее, столб иарв над вогнутым мениском меньше, чем пад плоской поверхностью жидкости вне трубки. Л так как в состоянии равновесия давление пара
728
Глава 16
над поверхностью жидкости должно быть одним и тем же и в трубке и впе ее и равным собственно давлению пара данной жидкости, напрашивается вывод, что одно и то же вещество, если его поверхность вогнута, имеет меньшее давление пара, чем в случае плоской поверхности. Наоборот, над выпуклой поверхностью давление пара более высокое, чем над плоской. Этот вывод справедлив для любой поверхности жидкости, например для поверхности капель. Давление пара над выпуклой поверхностью возрастает по мере увеличения кривизны. Томсон (Thomson, 1871) термодинамически установил следующее соотношение:
Й_М 2о Л1 1 Л
ln р, ~ RT ' s С r3 rt) ’ (13)
гдо pj и Р2 — давление пара капель с радиусами и г3; о — поверхностное натяжение; а — плотность жидкости; М — молекулярный вес ее пара. Увеличение давленая пара
Поглощенное количество жидкости , лгл
Р и с. 98. Увеличение и уменьшение давления пара над гелем кремневой кислоты (по Андерсону).
над выпуклой поверхностью достигает заметной величины лишь в области коллоидного размельчения веществ. Давление пара капель воды радиусом 10~4 ем всего на 0,1% превышает давление пара над плоской поверхностью воды. Однако, если радкус уменьшить до 10“в с.к, давление пара возрастает па 10%, а при г™10~7 см — почти на 100%.
Капиллярная конденсация. Понижение давления пара над вогнутой поверхностью может привести к тому, что в очень тонких капиллярах, например в высушенном геле и в других пористых веществах (древесный уголь и др.), наступает конденсация ненасыщенного пара. Это явление называют имбибицией или капиллярной конденсацией. Ее можно наблюдать, например, при поглощении паров воды высушенным студнем кремневой кислоты при постоянной температуре и возрастающем давлений пара (см. рис. 98).
Вначале водяные пары адсорбируются высушенным гелем (поверхностью его капилляров). При этом давление пара довольно быстро повышается. Гель остается
Коллоидная химия. Химия поверхностных явлений
729
о. поглощение и выделение наблюдается значительный
Рис. 99. Электроосмос.
прозрачным. Однако, начиная с точки Ог, гель мутнеет, а в точке О2 снова становится прозрачным. Обе точки носят название «точек превращения», В области между этими точками с увеличением поглощения воды давление пара возрастает лишь незначительно. Если в геле содержится воздух, процесс поглощения и отдачи паров воды не строго обратим. А именно поглощению и выделению пара воды, как показано стрелками на рис. 98, соответствуют различные кривые давления пара («гистерезис»). Выше точки <?2 обе кривые сливаются снова в одну, т. паров воды опять происходит обратимо. Одновременно рост давления пара с увеличением содержания воды, пока в точке О.. пе будет достигнуто давление насыщенного пара при данной температуре. Естественно, начиная с этой точки пар воды может в любом количестве конденсироваться на голе при постоянном давлении.
За исключением петель гистерезиса, кривая по своему виду напоминает кривые изотермического разложения иля синтеза химического соединения (ср. т. I, стр. 99, рис. 15). Однако в случае, приведенном на рис. 98, почти горизонтальный участок кривой между точками и <?2 обусловлен не появлением химического соединения, а тем обстоятельством, что в этой области происходит капиллярная конденсация пара воды. Если бы все капилляры в геле имели одинаковое сечение, то конденсация паров воды в них происходила бы при постоянном давлении пара. Но поскольку размеры их различны, вначале заполняются наиболее тонкое капилляры, а по мере увеличения давления — следующие, мениск в которых менее выпуклый и, следовательно, менее понижено давление. Помутнение геля в области между точками превращения объясняется наличием пузырьков воздуха в капиллярах при неполном заполнении их ;кидкостыо. Той работой, которую следует затратить на удаление воздуха из капил-
ляров, и обусловлено возникновение «гистерезиса». Если перед опытом полностью удалить воздух, гистерезис исчезает (Patrjk, 1920), Если вместо паров воды на гель действуют пары другого вещества, например пары бензола, точка <р2 достигается в тот момент, когда образовался равный объем жидкости, т. е. когда заполнены пустоты (см. рис. 91з).
Зная давление пара, при котором происходит капиллярная конденсация, можно вычислить диаметр капилляров. У геля, для которого получены кривые рис. 98, в точке С?! диаметр капилляра получился 2,6 тр, в точке О2 —- 5,6 тр. Эти величины практически не зависят от того, давление пара какого вещества использовали при расчете в точках О| п О2 (воды, спирта или бензола). Найденные величины показывают, что капиллярная конденсация имеет существенное значение только для очень тонко пористых веществ и только в том случае, если они попадают в область давления пара, близкую к давлению насыщения адсорбируемого вещества.
Электро капиллярные явления. Если к обоим концам капиллярной трубки, заполненной жидкостью, приложить разность электрического потенциала, то жидкость в трубке начинает смещаться, что является признаком ее заряженное™ относительно стенок трубки. В каком направлении она переместится, следовательно, положительно или отрицательно она заряжена относительно стенки, зависит от природы жидкости истенки. Наиболее просто такое явление можпо продемонстрировать, заменив один капилляр системой капилляров, например глиняным цилиндром. Если такой цилиндр, предварительно заполненный жидкостью, например водой, закрыть пробкой, снабженной вертикальной трубкой, то при определенном направлении тока жидкость в трубке поднимется (см. рис. 99). Такое явление называют электроосмосом.
Приобретение жидкостью заряда относительно стенки или непосредственно прилегающего к ней неподвижного слоя жидкости возможно благодаря тому, что аз нее в жидкость, находящуюся внутри трубки, или обратно перемещаются поны. Следока-
730
Глава 16
тёльно, рассматриваемое явление основано на приобретении заряда В: результате адсорбции. Изучение этого способствует дальнейшему пониманию природы адсорбционных процессов. Описанное выше явление катафореза основано на тех же принципах, что и электроосмос. Различие между ними только в том, что в первом случае передвигаются твердые частицы относительно жидкости, а во втором — жидкость относительно твердой стенки.
В соответствии с законом о действии и противодействии можно полагать,'что если приложение разности, потенциалов вызывает движение жидкости в капиллярной трубке, то и обратно, движение жидкости в капил-ляре должно приводить к возникновению разности потенциалов между его концами. Это действительно и наблюдается при протекании жидкости через капилляр. Такой потенциал называют потенциалом потока.
Измерением потенциала потока можно непосредственно определить разность потенциалов между жидкостью и веществом, с ней соприкасающимся, или, что то же самое, скачок потенциала между жидкостью й твердый веществом. Этот скачок потенциала, по Гельмгольцу (1879), равен
1=^.
где г; — вязкость; й — удельная электропроводность текущей жидкости; D — ее диэлектрическая проницаемость; Р — давление, под которым она проходит через капилляр; Е — измеряемый потенциал потока.
Подобная формула справедлива для электроосмоса. .Объем о жидкости, проходящей в единицу времени через капилляр (или диафрагму) диаметром q под действием разности потенциалов G. равен
4гсту
Скорость и, с которой жидкость движется в капилляре, равна и=^2-. 4лт|
Уравнение (16) справедливо и для скорости движения при катафорезе частиц, суспендированных в жидкости; только вместо множителя 4 (для частнц цилиндрическом формы) появляется множитель 6 для частиц шарообразной формы. D, j. и1] в уравнениях (15) и (16) те же, что и в уравнении (14).
Разность потенциалов £ является непосредственном мерой заряда твердого вещества относительно жидкости, возникающего вследствие адсорбции ионов. Таким образом, измерением этой величины можно непосредственно судить о процессах адсорбции и заряжения, имеющих определяющее значение для коагуляции и пептизации коллоидных веществ. Так, можно показать, что из электролита, добавленного к жидкости, преимущественно адсорбируется один вид ионов (катион или аййоп). Вместе с тем оказывается, что одновременно адсорбируется и небольшое количество Других ионов. С увеличением концентрации электролита наблюдается уменьшение величины т. е. заряд твердого вещества относительно жидкости падает. Это явление становится попятным при рассмотрении типичной кривой адсорбции (рис. 95, стр. 723). Когда йонцентрацпя 'наиболее адсорбируемого иона в растворе достигает определенной величины, адсорбированное количество этого йода при дальнейшем увеличении концентрации возрастает очень незначительно, в то время как максимум на кривой адсорбции менее адсорбируемого иона еще не достигнут. Следовательно, по мере повышения концентрации электролита после некоторой определенной величины происходит адсорбция менее адсорбируемого ио на, а адсорбция более адсорбируемого иопа остается практически постоянной. Количественное соотношение обоих ионов в поверхностном сдое изменяется при этом в пользу менее адсорбируемого иона и, следовательно, заряд, обусловленный наиболее адсорбируемым ионом, должен понижаться.
Заряд пузырьков газа в жидкости. Если пузырьки газа пропускать через жидкость, то они, как впервые установлено Куном (Coehn, 1914), приобретают электрический заряд. Величина и знак заряда зависят рт концентрации раствора (ср. Remy, Koch, 1924). В кислых растворах, как правило, пузырьки газа заряжаются положит тельпо, в щелочных т-отрицательно. При пропускании газа в щелочной раствор через пористую перегородку (например, фильтр из глины) образуются настолько.
Коллоидная химия. Химия поверхностных явлений , 731.
мелкие пузырьки, лто жидкость выглядит мутной, как молоко. Если через такой же фильтр пропускать газ в кислый раствор, образуются крупные, прилипающие к стенкам пузырьки. Это объясняется тем, что большинство пористых веществ заряжено отрицательно относительно и кислых и щелочных растворов. Если пузырьки га за-заряжены тоже отрицательно, то, выходя из капилляров, они сразу отталкиваются от стенки. В противном случае опи прилипают к стенке и-могут достигнуть Значительной; величины (Kautsky, 1926). Это обстоятельство следует учитывать, когда дело касается непосредственного контакта газа или аэрозоля с адсорбирующими жидкостями.
Эмульсин и эмульсоиды
Жидкость, в которой суспендированы капельки другой жидкости, называют эмульсией. Коллоидные эмульсии по предложению Во. Оствальда (19,07) называют эмульсоидами * в отличие от коллоидных растворов твердых веществ, называемых суспенсоидами. Примером эмульсии служит молоко, в котором диспергированы панельки жира.
Наиболее охотно образуют эмульсии между собой жидкости, мало различающиеся по плотности й величине поверхностного натяЖеиия. Поверхностное натяжение является причиной агломерации Капелек жидкости. Вследствие этого эмульсин устойчивы в большинстве-случаев только при условии, если они содержат так называемые «эмульгаторы», которые адсорбируются на поверхности капелек и изменяют их свойства.
Эмульсии имеют важное значение в 'технике. Большая роль принадлежит им в производстве масел, жиров, каучука, лаков и красок, Клея и смол. Эмульсии применяют также в целлюлозной, в бумажной промышленности, при изготовлении текстильного подокна ц выделке кож. Водные эмульсии асфальта и других подобных ому веществ применяют для обеспыливания улиц.
Разрушение эмульсий является важной проблемой в процессах-очистки сточных вод. Для очистки от масла конденсаторов паровых машин, в которых- образуются чрезвычайно устойчивые эмульсий, в возрастающей мере применяют электролитическое обезжиривание. Оно основано на том; что капельки масла, заряженные отрицательно относительно воды, катаферетически поступают на анод, а с него адсорбируются образующимся при электролизе осадком (например, основным карбонатом железа при использовании железных электродов),
К коллоидным эмульсиям и эмульсоидам принадлежит, по-видимому, большое число золей лиофильных коллоидов органических веществ. Например, желатину в жидком состоянии рассматривают как гидрозоль, В качестве примера эмульсоида гидрофобного коллоида можно привести золь ртути. ..*?
Дэродисперсные системы
Пепы. Пенами называют дисперсные системы, в которых жидкая дисперсионная среда в виде тонких сферических оболочек окружает газообразное диспергированное вещество. Диспергированное вещество в таком случае может быть в форме видимых простым глазом, или микроскопических, или ультрамикроскбпических газовых пузырьков, В последнем случае речь идет о коллоидных пенах.
"Для устойчивости пен поверхностное натяжение имеет еще большее значение, чем для эмульсий. Так же как п в случае эмульсий, пены в общем тем более устойчивы, чем меньше поверхностное натяжение жидкости но сравнению с газом. Имеет значение й вязкость жидкости.
* «Эмульсоидами» часто называют также золи лиофильных, а «су сне Неоплат» золи лиофобных коллоидов независимо от агрегатного состояния дисперсной фазы. Однако употребление обоих наименований нежелательно, так как это может повлечь за собой ошибки.
732
Глава 16
В попах диспергированное вещество также заряжено относительно дисперсионной среды. Чей выше заряд, тем более устойчива пена. Растворенные в дисперсионной среде вещества изменяют поверхностное натяжение и заряд и поэтому способны весьма значительно влиять на устойчивость пен. Чистая вода вследствие высокого поверхностного натяжения относительно воздуха по может образовывать устойчивых пен. А добавленном, например, мыла ее можно превратить в хороший пенообразователь.
Чрезвычайно сильно влияют па устойчивость пен тонко распределенные в них. твердые вещества. В частности, л иофобные вещества повышают устойчивость пеп. Лиофильные вещества либо совсем не влияют, либо влияют очень слабо. Зависимость пенообразования от твердых веществ и других добавок имеет большое значение при флотации- руд (см. стр. 181).
Некоторые производные глюкозы, так называемые сапонины, обладают свойством очень сильно понижать поверхностное натяжение воды. Благодаря этому они служат прекрасными пенообразователями. Добавка 10 мг сапонина на 1л воды Делает се уже весьма пепообразующей. То, что подобные вещества действуют уже в таких небольших количествах, объясняется тем, что они в соответствии с законом адсорбции Гиббса почти полностью концентрируются в поверхностном слое (см. стр. 727).
Аэрозоли, Аэрозолями (ocrtQ — воздух) называют системы, образованные воздухом или другими газами и взвешенными в них твердыми или жидкими частицами. Если взвешено твердое вещество, их называют* пылями, если жидкое — туманами. Очень тонкодисперсные аэрозоли часто образуются при горении твердых или жидких веществ. Аэрозоли, содержащие продукты горения, называют дымом *.
К аэрозолям относятся, например, атмосферный туман и облака^ В очень тонких слоях тонкодисперсные аэрозоли выглядят лишь слабо мутными. Аэрозоли встречаются очень часто: так, обычный воздух, строго говоря, не чистый газ, а аэрозоль. В проходящем свете видны содержащиеся в нем пылевидные частицы («солнечные пылинки»; ср. стр. 710). Более плотные аэрозоли часто образуются при химических и физических процессах, в которых участвуют газы и жидкости (или их пары), а также при взаимодействии газов с твердыми веществами в условиях высоких температур.
Устойчивостъ аэрозолей существенно зависит часто от интенсивности броуновского движения взвешенных частиц. Если оно мало, аэрозоль оказывается устойчивым, когда оседанию взвешенных частиц препятствует конвекция несущего их газа. Содержащиеся в подобных аэрозолях взвешенные вещества устойчивы к химическому воздействию. Так. туманы, содержащие в качестве взвешенных частиц капельки концентрированной серной кислоты, почти беспрепятственно проходят через воду; еще меньше они поглощаются концентрированной калиевой щелочью (Remy, 1924, 1927). Объясняется это тем, что взвешенные частицы вслед- ? ствиё малого броуновского ;движепия большей частью совершенно не соприкасаются с адсорбентом. Это обстоятельство следует принимать во внимание в аналитической практике при испытании газов на чистоту в том случае, когда примеси находятся во взвешенном состоянии.
Прежде считали, что существенную роль в устойчивости туманов играет равновесное давление пара над поверхностью капелек. Как показано на стр, 727, давление пара над выпуклой поверхностью болыпе, чем над плоской. Вследствие этого раствор в виде достаточно малых капелек может иметь такое же давление, как чистый растворитель с плоской или слабо искривленной поверхностью. Следовательно, давление пара не влияет па устойчивость тумана. Однако, если капельки соединяются с растворит где и пли между собой, кривизна всякий раз сокращается и давление падает, что сопровождается выделением энергии.
* Иногда «дымом» называют также коллоидные пыли. Ввиду особого практического значения аэрозолей, образующихся в процессах горения (к которым принадлежит обжиг и т. п.), Они нуждаются в особом обозначении. Рекомендуется называть их просто дымом. В дыме, образующемся при горении, часто сосуществуют твердые и жидкие пылевидные вещества.
Коллоидная химия. Химия поверхностных явлений 733
Очень тонко диспергированные взвешенные в газах частицы, если они не заряжены и аэрозоль пе сильно разбавлен, довольно быстро слипаются между собой. При этом частицы тумана стремятся достигнуть определенной величины (около 2,5 •10~4 сл»). Если эта величина достигнута, то даже в относительно концентрированных аэрозолях агломерация практически прекращается (Remy, 1924).
В отличие от атмосферных туманов в аэроволях, образующихся в результате химических процессов, взвешенные частицы в общем не заряжены (Remy, 1924). Лишь с течением времени они приобретают заряд благодаря адсорбируемым из воздуха ионам газа. При этом образуется примерно равное число положительных и отрицательных частиц (lander, 19331. В случае очень тонко дисперсных аэрозолей с сильным броуновским движением заряд, вероятно, существенно влияет на устойчивость. При меньшей дисперсности, однако, это не имеет места. Влияние поверхностного натяжения иа устойчивость аэрозолей до сих пор не установлено.
Для устранения взвешенных частиц из газов в технике используют главным образом два способа. Относительно грубодиспергиронэнные взвешенные частицы удаляют седиментацией (в пылеосадительных каморах). Тонкодиспергиров энные удобнее осаждать электрическим путем при помощи тока высокого напряжения [способ Котрелла — Мёллера (Cottrell — Moller)]. Серия разветвленных электродов заряжает взвешенные частицы, которые оседают затем на противоположно заряженных (или заземленных) плоских электродах.
Для обеспыливания газов часто применяют суконные фильтры, а также центробежные осадители. В последнем случае поток газа при помощи турбины приводится во вращательное движение; взвешенные частицы увлекаются центробежной силой к стенкам и оседают на них. Действие суконных фильтров на тонкодиспергировэнные взвептеппыо частицы основано, вероятно, также па центрифугировании, которое возникает при прохождении частиц через многократно изотпутые узкие каналы фильтра. Е о л ее грубые частицы улавливаются, естественно, благодаря отсеивающему действию суконпых фильтров.
Центрифугирующую стюсобность. проявляющуюся при прохождении частиц через многократно изогнутые тонкие каналы, используют также в специальных противогазах для защиты от вредных веществ на промышленных предприятиях и от химических отравляющих веществ. Обычные противогазы, защитный слой которых состоит из простых или химических адсорбентов, задерживают только газообразные ядовитые вещества. Л так называемые высокоэффективные фильтры снабжены специальным слоем, улавливающим па основе центрифугирующей способности твердые иди жидкие взвешенные вещества из атмосферы.
ДИФФОРМНЫЕ СИСТЕМЫ*
Помимо измельчения, значительного увеличения поверхности вещества можно добиться unMi. Ht-Hне.и его формы. Если кубик раскатать в тонкую пластинку, его поверхность увеличится, побудет на 2/3 меньше по сравнению с общей поверхностью отдельных кубиков (с длиной ребра, равной толщине пластинки), вырезанных из этой пластинки. Еслп из кубика вытянуть проволоку с сечением, равным сечению отдельного Кубика, то поверхность будет па г/$ меньше по сравнению с суммарной поверхностью отдельных кубиков. Иэнр'цмер, поверхность кубика объемом 1 ем9 возрастает при делении на кубики с длиной ребра 10 тпц от 6 см- До 600 л*в, при вытягивании в проволоку диаметром 10 — до 100 ,иа, а при измельчении на пластинки толщиной 10.ир —
до 200 л»2. Так же как в коллоидных системах, в системах, образующихся при резком изменении формы. для которых «Оствальд предложил наименования чдщфформные системы», большую роль играют силы поверхностного натяжения. Примером характерной дифформпой системы служат упомянутые на стр. 726 мономолекул я рные поверхностные слоп-
* Системы с ани To меч’рпчпыми (удлиненными) частицами.— Прим. род.
Гл а в а 17
КАТАЛИЗ И КИНЕТИКА РЕАКЦИИ’
Катализ. Под катализом понимают явление ускорения реакций в присутствии веществ, которые не расходуются в реакции, а .как бы влияют на нее одним своим присутствием. Такие вещества называют катализаторами. Их действие особенно ярко проявляется в реакциях, скорость которых в отсутствие катализаторов практически равна нулю. Так, перекись водорода в водном растворе в отсутствие посторонних веществ, по-видимому, совершенно устойчива, т. е. в течение определенного времени не наблюдается никаких признаков ее разложения. Однако при добавлении некоторых веществ, например мелкораздробленной платины,происходит ее бурное разложение (ср. т, I, стр. 77). Совершенно аналогично при пропусканий гремучего газа над губчатой платиной при обычпой температуре происходит его вспышка. В таких случаях речь идет о «возбуждении» реакций посторонним веществом, называемым «катализатор» (хптаХиеьт — возбуждать). Если, как в приведенном примере, катализатор представляет собой самостоятельную фазу, говорят о гетерогенном катализе. В таком случае катализатор называют также иногда контактом. Если же катализатор образует с реагирующим веществом однофазную (гомогенную) систему, как, например, при'ускорении разложения перекиси водорода в водных растворах в присутствии ионов OB', речь идет о гомогенном катализе. Вещество или смесь веществ, превращения кртот рых ускоряются катализатором, называют субстратом катализа.
Нередко в такой системе могут происходить различные реакции. Например, смесь СО и Н2 может реагировать е образованием СН3ОН, СН4 и Н20 или жидких углеводородов (см. т. I, стр, 471). В зависимости от вида катализатора одна из указанных реакций может ускориться настолько, что практически будет иметь место лишь она одна. В таких случаях говорят о кагграелекн<|.«. действии ката лизатеров. , '
Поскольку катализатор, возбуждающий или ускоряющий реакцию, сам не расходуется в ней, в принципе бля превращения сколь угодно больших количеств веществ требуется минимальное количество катализатора. Практически действие катализатора часто ограничивается тем, что он постепенно расходуется в другой реакции, которая'может случайно идти параллельно главной — каталцтйчёскрн (ср. т. I, стр. 760). Кроме того, посторонние вещества, случайно попавшие в: реакционную смесь, могут изменять свойства катализатора, вследствие чего он теряет каталитическую активность. В таких случаях говорят об «отравлении» катализатора, а обусловливающие его вещества называют «каталитическими ядами». Отравление катализаторов имеет большею значение в первую очередь в гетерогенном катализе; в гомогенном катализе такую отрицательную роль играют побочные реакции.
*) В редактировании перевода гл, 17 принимал участие В. С. Гурман.
Катализ и кинетика реакций " 735
Ввиду того что катализатор должвв прийти е еоприкоскзеение с мопекулаъш реагирующих веществ, ускорение реакции зависит, естественна, и от количества ката-лизатора. Чей оно больше, тем большее число молекул субстрата в единицу времени могут в результате диффузии прийти в соприкосновение' с катализатором. В гомогенном катализе число молекул субстрата, соприкасающихся с катализатором в единицу . времени, зависит от концентрации катализатора, а в гетерогенном — от его поверхности. Поэтому действие данного катализатора тем значительнее, чем больше степень его измельчения. Отсюда становится ясной особая каталитическая активность веществ в коллоидном состоянии. Однако еще более значительным может быть каталитические действие растворенных веществ (в состоянии молекулярной дисперсности), если они По своей химической природе способны значительно увеличить скорость реакции. Коллоидная платина, ио данным Бредига, приводит к разложению перекиси водорода в количествах, во много миллионов раз превышающих содержание катализатора. Каталитическое ускорение самоокисления сернистой кислоты ионами меди наблюдается при разбавлении 1; 1 млрд,, а каталитическое действие ионов серебра на гемолиз (растворение красных кровяных телец) наблюдается даже при его сб.отиошепии е реагирующим веществом 1 ; 1000 биллионов (так называемое «олигодипамическое» действие серебра), j
Каталитические процессы имеют огромное значение в химической промышленности. Кроме процессов основного неорганического синтеза, описанных в т. I, существует еще бесконечное множество реакций органического и неорганического синтеза, в которых используются Катализаторы.
Биологическое значение катализа. Исключительно велико значение катализа в процессах жизпедеятельности организмов. Органические вещества, являющиеся катализаторами, называют ферментами пли энзимами. Раньше предполагали, что ферменты отличаются от псоргапических катализаторов в основном специфичност ыо своего действия, т. о. ускоряют лишь строго определенные реакции совершенно ,о пре -деленных веществ. Однако в дальнейшем было показано, что ,и ферменты нй всегда действуют специфично, а с другой стороны, неорганическим катализаторам при соответствующем составе удается придать в большинстве случаев специфичность действия.
Исторические сведения. Отдельные примеры каталитических процессов.... были известны ужо во второй половине XVIII в. Так, в 1782 г. Шееле ускорял реакцию этерификации минеральными кислотами (ионами Н-). Особое внимание исследователей привлекло наблюдение Доберейнера (1823) о воспламенении водорода на воздухе при соприкосновении его с губчатой платиной при обычной температуре («огниво» Доберейнера). Определение понятия «катализ» было дано впервые в 1835 г, Берцелиусом, называвшим катализаторами вещества, «одно лишь присутствие которых вызывает химическую деятельность, не наблюдающуюся без этих веществ». Введение в определение катализаторов понятия времени, т. е, рассмотрение их как веществ, обладающих свойством ускорять реакцию, принадлежит Вильгельму Оствальду (1888). В этом -определении впервые дан путь к объяснению сущности каталитических процессов.' Однако прошло еще десятилетие, пока стало возможным на основе количественного изучения кинетики химических процессов и механизма реакций более глубоко проникнуть В природу каталитических процессов. Исследования смешанных катализаторов (состоящих из многих веществ) получили развитие в основном благодаря использованию их в промышленном синтезе аммиака и проводились начиная с 1909 г. в первую очередь Митташем [М j t t a s с h А., Вег., 59, 13 (1926); Z. Eick troche m., 36, 569 .(1930)].
Теория катализа. Еще во времена Берцелиуса многие исследователи высказывали предположение о том, что роль катализатора: заключается в образовании особо реакционноспособных промежуточных продуктов с теми веществами, которые они «возбуждают». Дальнейшие превращения этих продуктов приводят к выделению катализатора в свободном состоянии; Образование в камерном процессе таких промежуточных продуктов, и прежде всего питрозилглдросульфата * (раньше называвшегося «нитрозилсернон кислотой»), было установлено Дэви еще в 1812 г.
* Ср. г. I, стр. 760.
736 Глава 77
Противники теории катализа ссылались па реакции, ускоряемые платиной и аналогичными металлами. Казалось совершенно невероятным, чтобы такие инертные металлы, как платина, могли давать промежуточные соединения с водородом, кислородом или перекисью водорода. Некоторые исследователи, например Либих, пошли еще дальше и действительно каталитическими считали лишь те реакции, при которых удавалось обнаружить появление промежуточных соединений субстрата с веществами, возбуждающими реакцию. Однако для объяснения гомогенного каталина теория промежуточных реакций применялась с успехом, й в результате было доказано существование большого числа промежуточных соединений катализатора с субстратом. Действие контактных веществ в гетерогенном катализа еще долгое время рассматривали лишь как способность к уплотнению субстрата на поверхности катализатора за счет адсорбции.
В настоящей время образование промежуточных продуктов предполагается пе только в гомогенном, но и в гетерогенном катализе; тем не менее, как правило, речь идет не о соединениях в обычном смысле слова, а о продуктах разнообразной природы, образовавшихся за счет «побочных валентностей». Часто эти продукты не имеют определенного стехиометрического состава, но далее и при наличии такового продолжительность их существования так мала, что изолировать весомые количества комплексов оказывается практически невозможным. Образование таких исключительно короткоживущих промежуточных продуктов можно обнаружить лишь при помощи методов, получивших свое развитие при изучении механизма химических реакций,
В общем случае было показано, что химическая реакция,— в присутствии и в отсутствие катализатора—протекает, как правило, более сложно, чем это отряжают обычные химические уравнения, содержащие лишь начальные и конечные продукты. Даже если в химических уравнениях отражено образование промежуточных Соединений, обычпо принимают во внимание только продукты, образующиеся в весомых количествах. Между тем, исследование механизмов реакций доказало, что химические процессы очень часто идут через стадию саобобкьж радикалов и других промежуточных продуктов, которые ио могут быть изолированы вследствие их исключительной нестойкости. И важнейшая функция катализаторов заключается как раз в том, что они способствуют образованию таких промежуточных продуктов или тому, что их дальнейшие превращения протекают в совершенно определенном направлении.
Вследствие важнейшего значения, которое имеет исследование скорости реакций и их механизмов для объяснения сущности катализа, описанию современной теории каталитических процессов предпосылается, краткое изложение важнейших снедений о кинетике реакций, т. е. учение о скорости и механизмах реакций.
КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИИ
Скорость реакции. Скорость реакции определяется изменением концентрации с реагирующего вещества со временем t, т. е. dc'idt, где de изменение концентрации за бесконечно малым промежуток времени*.
* Изменение концентрации при определении скорости реакции может относиться к бее ко ималому промежутку времени, так как концентрация зависит от времени. Изменою».' концентрации в течение конечного промежутка времени, устанавливаемое экспериментально, получается при интегрировании дифференциальных Уравнений. Если вещества, вступающие в реакцию, существуют в одинаковых концентрациях, при интегрировании уравнений (1) — (4) получаем: Ju с0/<ц = k-1 (la): i/ci — l/c0 = i (2a); */2 (l/c| — l/co) = A'1 (3a); c0 — ct — k-1 (is). В mix c0 --начальная концентрация исходных веществ, a "t — концентрация пх л момепт времени t.
Катализ w кинетика реакций 737
Уменьшение концентрации (—de) веществ, вступающих в реакцию, эквивалентно увеличению концентрации (-f-cic) ее продуктов. Скорость реакции зависит от температуры и концентраций веществ, вступающих в ре акцию. Характер зависимости скорости от концентрации определяет так называемый порядок реакции.
Порядок реакций. Для реакции, в.которой изменяется концентрация лишь одного из исходных веществ, справедливо уравнение
где к — константа, зависящая от природы исходного вещества и других условий опыта; Это значит, что скорость реакции пропорциональна концентрации исходного вещества. В таком случае речь идет о реакции первого порядка.
Если два вещества реагируют по уравнению * А -j- В • == С, то для реакций в гоио-генных системах (гомогенных реакций) обычно
.. (2) о$ -
где сд и св — концентрации веществ А и В. Такие реакции называют реакциями второго порядка.
Если три вещества реагируют между собой по уравнению A-|-B-]-C=D, то скорость реакции пропорциональна концентрациям веществ сд, св и cq:
—(3)
В .данном случае речь идет о реакции третьего порядка.
Реакции, имеющие, порядок выше третьего, почти не встречаются. Зато среди реакций, протекающих главным образом на поверхности фаз (гетерогенных), часты случаи, когда скорость практически не зависит от концентраций веществ, участвую? щих в реакции:
В таких случаях говорят о реакциях нулевого порядка.
При одинаковых концентрациях исходных веществ в правой части уравнений (2) и (3) получаются выражения: к -с® и к-с3. Соответствующие выражения в уравнениях (1) и (4) имеют вид к-с1 и й-с°. Следовательно, порядок реакции по веществу определяется показателем степени с. Известны также реакции, для которых этот показатель имеет нецелочисленное значение (ср. стр. 739).
Порядок реакции с точки зрения молекулярно-кинетической теорий* При реакции, протекающей в общем случае по уравнению А + В =С, например
Нг4-1г=2Н1, (5)
скорость, очевидно- определяется частотой столкновений молекул обоих реагирующих веществ. Число столкновений между двумя одинаковыми молекулами в соответствии с кинетической теорией газов пропорционально квадрату их количества в данном объеме, т. е. квадрату их молярной концентрации. Число столкновений между двумя различными молекулами пропорционально произведению молярных концентраций обоих видов молекул. Если скорость реакции подчиняется уравнению (2), это служит указанием на то, что превращение осуществляется при столкновении двух различных молекул. Это справедливо, например, для реакции водорода **
**При этом несущественно, образуется ли один или несколько продуктов реакции, пока но начинает сказываться вторичное образование исходных веществ при обратной реакции. Чем ближе реакция к положению равновесия, тем большим становится влияние обратной реакции. Тогда экспериментально определяемая скорость реакции равна разнице в скоростях прямой и обратной реакций.
47 г. Реми
738'
Глава Т?
с йодом в газовой фазе. Уравнение (5) отражает, следовательно, не только количественные соотношения между исходными и конечными продуктами реакции, но и механизм реакции. Как уже было отмечено, в большинстве случаев обычные химические уравнения не дают представления о механизме реакции.
Чтобы произошла реакция распада Ш на Н2 и 13, тоже должны столкнуться две молекулы (2Н1), Поэтому и для указанной реакции справедливо уравнение (2), где ca=<?b = chi; скорость распада, следовательно, пропорциональна квадрату концентрации HI. Другим примером гомогенных реакций второго порядка может служить термическое разложение закиси азота ио уравнению 2М3О = 2N24-O2 и окиси хлора: 2С12О = 2С12-|-О3.
Если в реакции, протекающей по общей схеме А. + В = С, вещество В содержится в столь высокой концентрации по сравнению с А, что изменение его концентрации уловить невозможно, то концентрация с% в уравнении (2) входит в значение константы к. Тогда уравнение (2) превращается в -уравнение (1). Этот случай часто встречается в гомогенных реакциях, происходящих в растворах, когда растворитель принимает участие в реакции.
В качестве примера можно привести инверсию сахарозы, протекающую в соответствии с уравненном Cj2H22Ofj-|-HaO = 2GsHI3Oe. Обычно исследования проводят при таких концентрациях, что уменьшение количества воды в процессе реакции исчезающе мало в сравнении с общим ее содержанием. Поэтому рассматриваемое превращение оказывается реакцией первого порядка.
Часто реакции в действительности имеют более низкий порядок по сравнению с тем, который можно было бы ожидать в соответствии с обычным уравнением. Так, распад пятиокиси азота-2N3O5 = 4NO3 + О3 как в газовой фазе, так и в среде индифферентных растворителей (например, СС14) протекает как реакция первого порядка, а не второго, как мощно было ожидать, если бы приведенное уравнение отражало механизм реакции. Можно показать, что в таких случаях превращение проходит через промежуточные стадии. Чтобы отразить механизм реакции, следует суммарное уравнение представить в виде нескольких составляющих. Тогда понятно, что экспериментально найденная скорость реакции определяется наиболее медленно протекающей ее стадией.
Иногда при измерении скорости реакции обнаруживается более высокий порядок, чем тот, который имеют промежуточные стадии. Например, реакция, происходящая Ио уравнению
2NO + C12=2NOCI, (б)
судя по измерениям скорости, имеет третий порядок. По данным Траутца (Trautz, 1924), она идет в две стадии
NO-J-C^NOCI* (ба)
NOC124-NO=2NOC1. (66)
Если предположить, что реакция (66) протекает медленнее, чем (6а), последняя может привести к равновесию, для которого по закону действия масс [NOj* [Gl3] =kt [NOC12]. Скорость реакции (66) выражается уравнением
- ll^l=fc2.[NOCl2]-[KO].
Если подставить в пего концентрацию NOClj в положении равновесия (6а) j получим
-®=^-[NOp.(Gl3J, at
т. е. ту же зависимость скорости от концентрации, которая получается при столкновении трех частиц в нредположении непосредственной реакции по уравнению (6).
.ашййЙЬь*
Каталиа и кинетика реакций _________.739
Одновременные столкновения трех молекул происходят значительно реже, чем обычные столкновения (двух частиц). В газе при атмосферном давлении и обычной температуре отношение числа тройных соударений к количеству двойных составляет около 1 : 1000* **. Поэтому, вообще говоря, значительно более вероятно, что реакция между тремя молекулами происходит путем двух двойных соударений, вместо одного тройного.
Молекулярность реакций. Реакции, в элементарном акте которых претерпевает превращение лишь одна молекула, называются мономоле-кулярными' если происходит взаимодействие двух молекул, реакция бимо-лекулярна, при соударении трех молекул — тримолекулярна. Вследствие исключительно небольшой вероятности одновременного соударения четырех молекул реакции выше тримолекулярных представляются совершенно нереальными. Даже тримолекулярные реакции осуществляются крайне редко.
Кроме простых, известны сложные реакции, в частности процессы, протекающие через промежуточные стадии. Отдельные стадии такой сложной реакции называются последовательными, (консекутивнъгми реакциями). Например,
Сложная реакция: А Щ В С =D,
Последовательные Г Л -р В = Е, реакции: X Е + С = D.
Реакции, отражающие действительные процессы взаимодействия молекул, т. е. не только результат превращения, по и его механизм, называются элементарными реакциями.
Если одна из последовательных реакций протекает значительно медленнее, чем остальные, то, как уже было указано, практически только она определяет скорость всего процесса. Следовательно, порядок суммарной реакции, определяется молекулярностью наиболее медленно протекающей из се стадий. Если жо скорости последовательных реакций лишь мало отличаются друг от друга, то порядок реакции зависит от каждой из ппх. Поэтому нор един случаи отклонения порядка реакций от целочисленных значении.
Многие процессы, обычно рассматриваемые как гомогенные, при более детальном исследовании оказываются либо’протекающими на стенках реакционного сосуда, либо происходящими каким-либо иным путем, чем это выражает обычное уравнение; Например, реакция 2СО-|-О2 = 2СО2 обычно протекает на стенке сосуда, хотя варывиой процесс происходит в газовой фазе; он связан с присутствием паров воды, благодаря которым становятся возможными последовательные реакции:
СО + Н2О= со2+нк; нг+о2=нгог; 2Нг02=2Н20+ О2.
Приведенный пример одновременно иллюстрирует каталитическое ускорение газофазной реакции парами воды, что наблюдается довольно часто *41.
Если сравнить число столкновений между газообразными молекулами, вычисленное по кинетической теории газов, с количеством прореагировавших молекул, найденным по скорости реакции, то оказывается, что в случае газофазных реакций, протекающих при атмосферном давле
* Это справедливо для молекул, построенных из малого числа атомов. Вероятность Того, что молекула А встретится с молекулой. Б как раз во время столкновения с другой молекулой В, относится к вероятности столкновения с одной молекулой В, как диаметр молекул В к средней длине их свободного пробега.
** Иногда предполагают каталитическое действие следов водяного пара и в тех случаях, когда реакция останавливается про интенсивном высушивании компонентов. Однако в действительности торможение реакций связано с «отравлением» системы посторонними веществами [см. В о cl е n a t о i и, Z. phys. Chem., В20, 451 (1933); Rodebush, J. Am. Chem. Soc., 55, 1742 (1933)].
47*
740
Глаеа 17
нии (скорость которых легко определить экспериментально), только одно из 1010—10й столкновений приводит к химическому взаимодействию. Следовательно, только очень малая часть Сталкивающихся молекул способна к химической реакции; отсюда :следует предположение, что реакционноспособные молекулы должны чем-то отличаться от остальных. Назовем их пока «алтпивнььии» молекулами. В чем состоит сущность «активирования», будет показано ниже.
Скорость реакции и температура. Аррениус (1889) эмпирически установил,. что зависимость скорости реакции от температуры в широком интервале выражается уравнением
d In к __ А
dT НТ1 ’
где к — константа скорости реакции; Т абсолютная температура; R — газовая постоянная; А — величина, имеющая размерность энергии * **. Аррениус считал эту энергию «теплотой активации», т. е. количеством тепла, которое нужно сообщить молекулам, чтобы перевести их из обычного состояния, в котором они неспособны вступать в реакцию, в «активное». Уравнение Аррениуса справедливо как для гомогенных, так и для гетерогенных реакций. С его помощью можно вычислить энергию активации по данным скорости реакции иди, наоборот, если известна энергия активации, найти скорость реакции**.
Необходимость «активирования» газовых молекул объясняет тот факт, что для проведения газофазной реакции очень часто требуется сообщать энергию реагирующим молекулам, даже в случае сильно экзотермического процесса. Если энергия сообщается в форме тепла, то говорят о возбуждении реакции воспламенением:
Фотохимические реакции. Фотохимическими, называются реакции, возбуждаемые светом. Прирост энергии е2 ~ сообщаемый одной молекуле при поглощении света, так же как ц в случае атома (ср. т. I, стр. 109),: определяется частотой колебания v поглощаемого света по уравнению
е3—et = /i.v. (8)
Поэтому при исследовании фотохимических реакций можно непосредственно определить энергию, необходимую для активирования одной моле^ кулы. Кроме того, из общей энергии облучения с частотой v можно определить число активных молекул. Коэффициент E/hv, (где Ё энергия облучения) определяет число Световых квантов и равен числу активированных молекул (так как каждой отдельной молекуле для активирования требуется один квант Лv). Если энергия, необходимая для определенного изменения состояния молекулы (например, перехода одного электрона на более высокий уровень или диссоциации молекул на атомы), известна из других исследований, то волновое число v излучения, способного вызвать определенную реакцию, позволяет определить и природу изменения, состояния, которое должна претерпеть молекула, чтобы она могла реагировать с рассматриваемым веществом.
* Уравнение (7) не просто аналогии ио уравнению изохоры реакции, предло-женаому В авт-Гоффам (т. I, стр, 58),— его можно вывести из этого уравнения. Указанное соотношение находится в строгом соответствии с экспериментом лишь в том случае, когда содержит еще один дополнительный член; однако, как показывает опыт, в большинстве случаев его можно пе принимать во внимание (ср. примечание к стр. 748).
** Для вычислений скорости реакции необходимо зпать не только энергию активации, но также и пр ед экспоненциальный фактор,— Прим. ред.
Катализ и кинетика реакций . 741
Сказанное можно пояснить на примере фотохимической реакции Я2 и С/j. Взаимодействие водорода с хлором Н2+С 1а==2НС1 инициируется светом; длина волны которого равна или меньше 4785 А. Отсюда следует, во-первых, что для возбуждения реакции необходимо активировать только хлор, но не водород, тал как водород не поглощает света в указанной области. Квант света, соответствующий данной длине волны* составляет Л с/Х= 4,10-10-12 spa (ср. т. I, стр. 108 и НО). Энергия, необходимая для расщепления молекулы С12 на два атома С1, равна 3,93-10_1? эре. Это значение подучено при делении величины, приведенной в табл. 113 (стр. 826 т. I), на число Авогадро (с учетом того, что 1 зрг соответствует 2,39-10-3хй.г). Итак, Квант света сообщает энергию, необходимую для расщепления молекул CI2 на атомы *. Атомьгхлора в противоположность молекулам СИ при обычной температуре могут реагировать с молекулами Н2. Таким образом объясняется инициирование светом реакции между хлором и водородом при обычной температуре.
Цепные реакции. При сравнении количества хлористого водорода, образующегося при фотохимической реакции между хлором и водородом, с общим количеством световой энергии облучения оказывается, что образуется несравненно больше НС1, чем возникает атомов С1 при облучении молекул С12. Каждый световой квант может расщепить на атомы только одну молекулу С13. Тем пе менее Бодентптёйп (Bodenstein, 1913 г. и сл.) обнаружил, что часто более 100 000, а при известных условиях й более 3 000 000 молекул С13 взаимодействуют с водородом под действием одного кванта света. Это явление впервые в 1918 г. Нернст объяснил следующим образом: образованно первых атомов хлора по реакции
Cb-|-iv=2Cl (реакция «зарождения цепи») (9)
завершается бесконечным рядом повторяющихся процессов:
(10а) (Юб) (реакция «продолжения цепи») (10в)
Обе реакции (10а) й (106) непрерывно чередуются, пока не произойдет случайно побочная реакция, например:
НЦ-С1=ИС1 («обрыв цепи»)’, (11)
поглощающая атомы Н и CI. Было показано, что «цепные реакции» такого типа исключительно часты. Вместо свободных атомов в качестве промежуточных продуктов могут образовываться и свободные радикалы **.
Чем позже произойдет обрыв реакционной цепи, тем большие количества веществ будут участвовать в определенном числе реакций продолжения цепи. Так как в приведенном примере при элементарных реакциях, из которых построена цепь, освобождающиеся атомы II или С1 всегда существуют наряду с молекулами С12 или Н2,-с Которыми они могут вступать в дальнейшее взаимодействие, то реакция (11)-обрыв
цепи — происходит исключительно редко. Значительно менее благоприятной становится перспектива дальнейшего развития цепи при наличии в газовой смеси nocni о ранних веществ, которые могут перехватывать промежуточные продукты ценных реакций—. свободные атомы и радикалы. Уже минимальные количества таких примесей (например, кислорода при реакции хлора с водородом) значительно укорачивают длину реакционной цени. Поданным Боденштейна, обрыв цепи в случае реакции прак
H+ci2=Hci+ci
С1 + На=НС1+Н
* Отдельные молекулы СБ поглощают несколько большую световую энергию, чем это необходимо для расщепления на атомы. Это приводит к;тому, что наряду с обычным образуется «возбужденный» атом С1, т. е. атом, в котором один электрон находится на более высоком уровне энергии, чем в нормальном состоянии.
:f;f В дальнейшем большой вклад» в развитие учения о цепных реакциях был внесен академиком Н. Ц. Семеновым и его школой (см., например, Н. Н. Семенов, Цепные реакции, Госхимиздат, 1934; Н. И. Семенов, О некоторых проблемах химической кинетики и реакционной способности, Изд. ЛИ СССР, 1958.)— Прим. ред.
742
Глава 17
тически вообще никогда не наступает в результате реакции (11)*, а всегда из-за присутствия в реакционной смеси следов примесей. Вообще особая чувствительность к присутствию в реакционной смеси примесей является характерным свойством цепных реакций. К цепным реакциям отнюдь не относятся все фотохимические процессы. Известны и такие реакции, когда при действии одного светового кванта происходит превращение только одной молекулы. С другой стороны, цепные реакции,не ограничиваются фотохимическими процессами. Кроме света, для инициирования цепных реакций (возбуждения молекул) с успехом можно применять и другие виды энергии (тепловую или
Рис. 100. Переход через потенциальный барьер при фотохимическом активировании.
Пример: схема реакции хлора с водородом. .
а-излучение). Приведенная в качестве примера цепная реакция может возникнуть и вследствие попадания атомарного водорода в реакционную смесь, Кроме того, в каче-стве^нсрвичной можно использовать реакцию Na4-CI2=NaCl-|-CI.
Важную роль играют цепные реакции в процессах горения, происходящих с образованием пламени. Нарастание их приводит к переходу горения во взрыв. Его часто удается предотвратить добавлением постороппих веществ, приводящих уже на первых стадиях к возникновению реакции обрыва цепи или к подавлению реакции зарождения цепи. На этом основано действие антидетонаторов в двигателях внутреннего сгорания (ср. т. I, стр. 598).
Энергия активации и потенциальный барьер. На рис. 100 (схематически) показано изменение химического потенциала (т. е. способности
* Вероятность рекомбинации л газовой фазе свободных атомов значительно меньше, чем свободных радикалов. В отличие от радикалов свободные атомы могут рекомбинироваться только при тройном ссуда рении, В нем должен участвовать (любом) третий партнер, на который может распределиться часть энергии соударения, В противном случае произойдет совершенно упругий удар, в результате которого атомы должны разлететься с относительными скоростями, равными тем, с которыми они сближались.
Катал из й кинетика реакций 743
производить работу за счет химической реакции) при реакции хлора с водородом, инициируемой фотохимическим путём. При этом для простоты будем рассматривать только взаимодействие двух молекул С1а с двумя молекулами Н2 и предположим, что реакционная цепь обрывается уже на третьем члене благодаря процессу И 4- Cl = НС1.
В этом случае весь процесс можно представить уравнениями (9), (40а) — (10в) и (11). При атом образуются четыре молекулы HCI. Что касается энергии их образования 7—i=lG,01-10~ао «ал\ то химическая энергия системы перед началом
\ u,U2o 10 у
реакции выше, чем в конечном состоянии. Эта. разность энергии между начальным и конечным состояниями на рис. 100 обозначена Et.
Несмотря на относительно большой выигрыш энергии при образовании ИС1 (22,75 ккал на 1 моль НС1), система не может перейти из начального состояния (2С1а + 2Н3) в конечное (4НС1). Опыт показывает, что если предотвратить реакции на стенке сосуда, смесь хлора с водородом в темноте может оставаться сколько угодно долго без признаков взаимодействия. Если игнорировать возможность инициирования реакции каталитическим путем, то для начала реакции необходимо перевести систему на более высокий энергетический уровень. Для наглядности применяют такое выражение: начальное и конечное состояния отделены друг от друга тотенциалъным барьером». Система должна быть «поднята» на его высоту, прежде чем начнется процесс, связанный с выделением энергии, приводящий и конечному состоянию (см. рис. 100).
В данном случае имеют место те же соотношения, что и для радиоактивных атомов, распаду которых тоже препятствует существование потенциального барьера. Существует однако и различие, заключающееся в том, что потенциал ядерной реакции (рис. 84, стр. 632) зависит от расстояния между центрами ядер, в то время как химический потенциал системы на рис. 100 зависит от времени.
В приведенной реакции существование потенциального барьера обусловлено тем, что молекулы С1Й не способны к реакции с молекулами На; такой способностью обладают лишь атомы хлора. Следовательно, прежде чем начнется реакция, должна быть затрачена работа на расщепление одной из молекул хлора на атомы. Работа, идущая на ато расщепление, представляет собой ^энергию активации» рассматриваемой системы. Для указанной реакции она составляет, как уже упоминалось, 3,93-1Сг13 эрг, или 9,41 1О-ао кал*. Итак, эта величина определяет высоту потенциальпого барьера, препятствующего непосредственной реакции уг хлора с водородом.
Достаточно сообщить одной молекуле CJS энергию 9,41 -IO-20 кал, чтобы реакция началась. Если молекула расщепляется под действием света, то по законам квантовой механики один из атомов С1 оказывается в «возбужденном состоянию, поэтому работа расщепления возрастает до 9,81-10~20 нал (Д2 на рис. 100). Можно предполагать, что этот «возбужденный» атом хлора прореагирует первым с молекулой Н2 в соответствии с уравнением (10а). Тогда выделится энергия Е?. =0,41 • 10-20 кал **. Следующая за ней реакция (106) сопровождается выделением энергии £?,= 7,50- 1О-20 кал. Между тем и второй атом хлора, возникающий в результате первичной реакции, вступает в реакцию с И2. Выделяющаяся при реакции энергия (£'5 = 0,01-10'20 кал) оказывается меньше Es па величину «энергии возбуждения» ***. В соответствии с первоначальным предположением процесс в данном случае ограничивается реакцией Н-|-С1=НС1.
* Так как энергия активации обычно измеряется в тепловых единицах (кал пли ккал), ее часто называют таюзе «теплотой активации».
** Значения энергии IE,, Еь и Е$ можно легко вычислить из величин, приведенных в табл. 7, 113 и 114 т. I, тс.ч же путем, что н Ev
*** Возбужденное состояние атома в приведенном случае, по-вицимому, не оказывает влияния на реакционную способность.
1!А
Глава, 17
Несмотря на значительную экзотермичность (Ее=16,90- 10’20 кал), вероятность ее крайне мала (ср. стр, 741); Суммарная энергия, выделяющаяся я результате реакция как видно из рис. 100, равна сумме первоначальной энергии системы Ei и энергии излучения 7?2 = hv. Следовательно, световая энергия поглощается при реакции. В Ыпол» йаключаетеа существенное отличие ^ототимиясских вроце&ое от каталитически^:.
Если в первичном фотохимическом процессе образуется цепь из 200 000 звеньев, то в реакцию вступает 100 000 молекул С12 и Н2 — количество, кратное числу молекул в предыдущем случае. Тогда Ег (несмотря на то что она бесконечно мала по сравнению с Еt) относится к химической энергии всех ыолокуи, участвующих в реакции; том не менее остается справедливым положение о том, что энергия, выделяющаяся при реакции, превышает энергию, выделившуюся в случае, если бы процесс шел без облучения, на всличипу Ау.
Термическое активирование. Как указывалось выше, для активирования молекул, кроме световой, можно использовать и другие виды энергии, например тепловую. На основании кинетической теории газов можно
вычислить, какая доля от о(ь щего числа молекул при дайнойтемпературе обладает тепловой энергией, превышающей энергию активации..
Теплосодержание Q идеального одноатомного газа в соответствии с ки нетической теорией газов равно сумме кинетической энергии его молекул. Если обозначить их число через А, получим
Q^V-Vbwh2, (12)
где т масса отдельных моле-
Р ис. 101. Распределение газовых молекул по скоростям в соответствии с законом Максвелла.
Кул, аи2- так называемая передняя квадратичная скорость» их поступательного движения. Конечно, не все молекулы обладают равными скоростями — скорости .отдельных молекул распределнкГСея по закону Максвелла (закон распределения скоростей), как показано на рис. 101. На нем по оси абсцисс отложены скорости отдельных молекул газа, причем за единицу принята наиболее часто встречающаяся скорость — «наиболее вероятная скорость» Иц,. По оси ординат отложат 1
жена значения ir • т- , т. е. доля общего числа молекул, скорости которых ложатг N du' „ г ...
в определенной области du, деленная на ширину этой области. Например, число молекул, скорости которых составляют 0,8—1,2 наиболее вероятного значения, отмечено на рис. 101 заштрихованной областью; оно равно 0,8.0,4, т. е: составляет около трети общего числа молекул. В том виде, как это представлено па рис. 101, форма кривой распределения не зависит от температуры. Среднее из всех значений скоростей и,п несколько больше, чем «наиболее вероятная скорость» иш. В формулу (12) входит, однако, не квадрат этого среднего значения (т._е. це в^)., а средняя величина квадратов отдельных скоростей *. Ее обозначают как в3,- а корень из нее в.
Для газа, находящегося под давлением р, кинетическая теория дает
1 - 1 JY -
р=2-. nmu2=±.-L^mlli, (13)
jV -
где п — ——-число молекул в единице объема. Для одного моля это уравпепие перейдет в
1 V л _
р-= А . —•— юи3 (где Л\—число Авогадро). (13а)
3 в
* Значения и,в, ит и в2 связаны между собой- следующим_образом: ит — — 2щцД/зГ =- 1,128вш; «3 = 3/3 uw. Следовательно, в = Т/й2 = 1,225 • вш=
= 1,085 ит.
Катализ и кинетика реакций. 745
Подставляя в уравнение (13а) значение р из уравнения газового состояния для 1 моля р- ?> — НТ, получим
1 — 1 3 Й
у;Уллшг=Й71 или — - Г. (14)
i? Z Л. 2V. д
Следовательно; средняя кинетическая анергия молекул (постунательная) пропорциональна абсолютной температуре. В соответствии с ЭТИМ «наиболее вероятная скоростью «и, растет (гак же как и и) пропорционально ^/Т. Итак, если рассматривать множество скоростей не в зависимости от и/И(Д, как это делается на рис. 101, а непосредственно от скорости и, то с повышением температуры максимум на кривой сдвигается в сторону более высоких скоростей.
В го время как уравнения (13) н (14) справедливы для идеальных газов вообще, уравнение (12) относится лишь к одноатомному идеальному газу. Теплосодержание многоатомных газов обусловлено пе только энергией поступательного движения, но м вращательной энергией молекул, а также колебательной энергией атомов, составляющих эти молекулы. Это выражается, как известно, в соотношении удельных теплоёмкостей при постоянном давлении и постоянном объеме <р/с0 — различном для одно- и многоатомных газов *. Следующие соображения, в которых учитывается только поступательная энергия, в основном справедливы й для двух других видов энергии.
Из закона распределения Максвелла следует, что доля молекул газа (а), У которых компонента поступательной энергии в направлении линии, соединяющей центры молекул, в момент столкновения оказывается большей, чем А, составляет
a=~RT_
Это выражение одновременно указывает па вероятность столкновения двух молекул, при котором сумма поступательной энергии компонентов, расположенных на линии, соединяющей их центры (A t + А 2), превышает величину А. Отсюда следует: а в уравнении (15) означает отношение числа столкновений, при которых At ~р А2 2: А, к общему числу соударений между молекулами. Если1 рассматривать А как энергию активации химической реакции, то а означает ту часть общего количества молекул, которая может реагировать между собой при соударениях. Это обусловлено тем, что в момент столкновения энергией, необходимой для осуществления химической реакции, обладают только молекулы, для которых суммарная
Таблица 98
Зависимость зпачеппйа (=e~“4',Jsl) от энергии активации А {кал/молъ) и температуры Т (“К)
(Только значения а, обведенные рамкой, приводят к реакциям, протекающим с измеримыми скоростями при обычном давлении)
A 1000 10 000 20 000 40 000 50 000 100 000.
Т=300° 1,87-10-1 .. 5,09-10-8 2,59-10-15 6,73’10'30 3,43-10-3'? l,18.10-?3
600° 4,32-lG-i 2,26-10-* 5,09.10-8 2,59-10'15 5,86’10-1» 3,43-10-37
1000° 6,04-10-1 6,49-Ю-з 4,21-IO"5 1,77-10'9 1,15’10-й 1,32-10-22
1500° 7,15-10-1 3,48-10-2 1,21-10-3 1,53-10'3 5,69-10-8 2,59-10-15
2000° 7,77-10-1 8,05-10-2 6,49-10“3 4,21’10'5 ' 3,18-10-e 1,15-10-n
3000° 8,45-10-1 1,87-Ю-i 3,48-10-2 1,21’10'3 2,26’10-* 5,09-10-s
* Относительно зависимости вращательной и колебательной энергии от тем ратуры см., например, Е иск е и -Wicke, GmadriB der Physical. Chemie, 10 A (1959), стр. 159 и сл.
746
Глава 17
энергия при столкновении достигает или даже превосходит энергию активации. Следовательно, скорость реакции пропорциональна величине о. В табл. 98 приведены значения о. для различных температур и анергий активации, вычисленные по уравнению (15). Из этих данных следует, что величина а очепь сильно зависит и от температуры, и от энергии активации.
На основании значений а можпо вычислить скорость, например для гомогенной бимолекулярной газофазной реакции в предположении, что взаимодействие имеет место лишь при столкновениях, энергия которых больше, чем энергия активации *.
Для простоты рассмотрим реакцию между двумя Молекулами одного вида (например, распад йодистого водородапо уравнению 2Ш = Нг + 1Д-Скорость реакции в этом случае оказывается вдвое больше, чем число столкновений, приводящих к реакции, так как при каждом из таких соударений реагируют между собой две молекулы. Итак, скорость выражается следующим образом:
- Щ~=2а‘-2-’ <16)
где п — общее число молекул реагирующего вещества в 1 см3 и Z— число столкновений (см. далее) одной молекулы **. Тогда, следовательно, в случае если а больше чем 10”7, превращение происходит моментально, а если а меньше 101а, реакция вообще не наблюдается. Эти значения справедливы для реакций, происходящих при обычном давлении (порядок величии нс изменяется, если вместо реакций между одинаковыми молекулами рассматривать взаимодействие различных молекул). С изменением давления эти предельные значения несколько смещаются. Аналогичным образом в случае цепных реакций этот сдвиг происходит в соответствии со средней длиной цепи, так, для цепи из 100 000 звеньев смещение составляет примерно пять порядков.
Указанные выше границы области измеримых скоростей *** можно определить следующим образом. Назовем реакциями, «происходящими моментально», те, в которых положение равновесия достигается так быстро, Что через 1 сек в реакционной массе уже содержится максимум 0,1% исходного вещества:
nt £ 0,001 "о (ДЛЯ t = l сек). (17а)
Реакциями, которые «вообще не наблюдаются», будем считать такие, в которые но истечении одного дня вступает максимум 0,1% исходного вещества:
по—nt £ 0,001дй (для г=8.64'104 сек) (17б)
В уравнениях (17а) и (176) л0 означает число молекул в 1 сл3 исходного "вещества в момент времени 0 и nt — соответствующую величину в момент времени t. Значения а, для которых справедливы эти условия, могут быть вычислены по уравнению (16). В этом уравнении Z означает число столкиотеиий ойной лме л » el сев. Кинетическая теория
* Неточность такого предположения (как показывает сравнение вычисленных значений скорости с измеренными экспериментально) при реакциях между простыми молекулами практически компенсируется ошибкой за счет того, что не принимается во внимание пи вращательная, ни колебательная энергия.
** Если одна молекула в 1 сек сталкивается с 2 другими молекулами, то л молекул в 1 сек претерпевают сыдкиопсния с п'2 молекулами. Если п — общее число молекул в 1 елг3, то число соударении равно 1/г 'Л, так как в каждом столкновении участвуют две молекулы.
*** Отсюда но следует, что скорости реакций, лежащие в этой области, действительно всегда удается точно измерить. Область, в которой это возможно, зависит от природы реакции и точности эксперимента. В общем случае она бывает значительно меньше, чем это задается уравнениями (17а) и (17б).
Катализ и кинетика реакций
747
газов дает для этой величины следующее выражение:
2=-|/2-л-а2-й-Л(, ' (18)
где ц — число молекул газа в 1 с.п3; и — корень из среднего значения квадрата их скорости; с — диаметр молекул. Для иллюстрации нарядна величии «числа столкновений» в табл. 99 приведены некоторые из пих *.
Таблица 99
Число соударений (Z) между молекулами газов при 0° и 1 атм
О, С1а NO ИгО И2О
2=4,14-10» 6,21-10» 4,86-10» 5,91-10» 8,85-10» сек"2
С03 CS2 NII3 СН4
Z—5,76-10» 8,67-10» 8,36-10» 7,69-10» сек'2-
При подстановке значения Z из уравнения (18) в (16) получаем:
—=а-1/2-л (19)
at
откуда интегрированием находим:
11 —
— ~~—а-1/2-к^2-и-1. (19а)
HQ г
Выразив отсюда щ через пр и подставив его в (17а), получим;
1000—1 сца —-------=— •
Д/2 л. о- и < пр
Аналогичным путем неравенство (176) приводит к выражению
0,001
а2 —.—_--------------—
8,64-104-'|/2-и-ог-«-П(
в котором произведение "|/2 этоаинд в соответствии с уравнением (18) означает число соударений и момент времени 0 и "]/2 ян2иц( — в момент £. Для вычисления приме рнт.гх значений а в обоих случаях подставим округленное среднее значение Z из 999 1-10-3
табл. 99, т. е. число 7-10». Тогда ctj а ^-^=1,43-10'’иаг £ 8 64-10*.7-10» = 1,65-Ю~13. Итак, значения 10“’ и 10“18 дают примерные границы изменения величины а в реакциях, протекающих с измеримой скоростью. Значении с:, каки числа соударений, необходимые для их расчета, относятся к температуре 6е С и давлению 1 атм. Однако н области температур, указанной в табл, 98, порядок величины а не изменяется с температурой. Так как и возрастает пропорционально ~\/Т, Z увеличивается примерно лишь л 3,3 раза при возрастании Т от 273 до 3000е **. В соответствии с уравнением (18) Z пр о нор ди опально давлению, так как, п находится от него в прямой зависимости.
Если известна энергия активации, то, как следует из сказанного-.? выше, скорость реакции можно вычислить непосредственно по уравнению молекулярно-кинетической теории ***. С учетом уравнения (15) уравнение (19) может быть записано в форме
<20>
* Числа соударений, приведенные в табл. 99, можно вычислить из величины впу-
„ и 0,30967-й.й2
треннего трения т| газа по формуле кинетической теории газов Z=-p — —----------,
£ Ц
где I — длина свободного пробега молекулы газа и d — плотность газа.
** В некоторых расчетах, кроме зависимости и от температуры, следует учитывать и аналогичную зависимость ст.
*** В действительности правая часть уравнения (20) содержит еще стерический фактор р, учнтывашгцнй необходимость ориентации взаимодействующих молекул, благоприятной для протекания реакции, р 1 и может достигать значений 10-4— 10-8.— Прим. ред.
748
Глава. 17
где с — концентрация (молъ/л) и NA — число Авогадро. Так как и мало изменяется в зависимости от Т, выражение jggjj- Уа'ПО3 и можно приближенно заменить константой С. Тогда уравнение (20) запишется так: А
= ЛГ.<Л (21)
__А
Произведение С-е RT соответствует коэффициенту к в уравнении (2) на стр. 737, если его отнести к концентрации, выраженной "в молях на 1 литр.
Уравнение (20) справедливо для бимолекулярных! реакций между молекулами одного вида (например, 2HI = 1г + Н2). Если речь идётЦ бимолекулярных реакциях, происходящих между молекулами различного вида (например,, ,1г + Hj = 2HI), то в уравнении (20) й па до заменить па вместо о подставить среднее значение
О1 “Д Й2 **
-Г-~—, а вместо — произведение ct-cz.
Следует учесть, что уравнение (20) выведено в предположении, что энергия, необходимая для активации при соударениях, без существенной ошибки может быть принята равной сумме поступательной анергии обеих молекул. Для сложных молекул, колебательная энергия которых в общем значительно больше поступательной, - А опа также должна быть принята во внимание. Тогда в уравнении (20) 4лен е it'!’ заменяется значительно более сложным: выражением.
Уравнение
А
k=C-e 1<Т (22)
при логарифмировании даст
Ini=lnC—А.. (23)
В результате дифференцирования получаем
» (in
dT RT% ' ' '
Уравнение (24) идентично эмпирическому уравнению (7) на стр. 740, на оспове которого еще Аррениус рассматривал величину А как тепловую «энергию активации».
Зависимость энергии активации от механизма реакции. По уравнению (23) можно вычислить энергию активации реакции, если известны константы скорости & для различных температур *. На графике зависимость In k от 1 /У выражается прямой, наклон которой соответствует значению AIR. Энергия’активации реакции разложения Йодистого водорода по уравнению 2HI = 12 + Н2, вычисленная указанным методом по данным табл. 100, составляет 44 ккал на 1 моль HI. Энергия диссоциации молекул HI на атомы равна 70,8 ккал/молъ. В противоположность реакции хлора с водородом, инициируемой фотохимическим путем, термическая диссоциация подпетого водорода не проходит через стадию свободных атомов. Часто достаточно одного энергичного столкновения двух молекул HI,
* Если учитывать температурную зависимость А (что обычно не требуется), то вместо уравнения (23) получим:
In й=1п С~^ + -11пГ.
Катализ и кинетика реакций
749
чтобы произошло превращение
Если бы для осуществления реакции требовалось расщепление молекул HI на атомы, олоргия (теплота) активации составила бы 71 ккал/моль вместо 44 ккал/моль. Этот пример показывает, что в противоположность тепловому аффекту реакции', который не зависит от ее отдельных стадий,
Таблица, 100
Константы скорости реакций образования (fct) и диссоциации (7г3) III, по данным Бодецштёйна
[Л-j иди А-2 = —с* ... относятся к равным начальным концентрациям исходных cn,cfi
веществ (с0 и ct [.ноль/л], t [сек]). Т — абсолютная температура),
г 556* 575° 62Э° С47° 666° 683“ 700° 716°’ 781’
.1,52-10’" 1,22-10-* 3,02-iO-5 8,59-10-5 2,20-10-4 5,12-10-4 1.16-Ю-з 2.50-Ю-з 3,95-10-2
Тс2 4.44-10-5 1,32-10-* 2.52-Ю-з 5,23-1О-з 1.41-10-2 2,46-10-й 6.42 -10"2 1,40-10-1 1.34-10°
теплота активации обусловливается механизмом реакции. Это положение имеет принципиальное значение для объяснения природы катализа.
Реакция 12 и Н2 с образованием HI происходит также в результате непосредственного присоединения атомов при столкновении молекул. В данном случае энергия активации (40 ккал /моль 12) оказывается Достаточной для диссоциации 12 па 21 (энергия диссоциации 35,5 ккал/моль). Одиако процесс
14-Н2 = Н1 + Н — 32 ккал (25)
сильно эндотермичен. Он будет происходить лишь в том случае, если при столкновении атома с молекулой Н2 их суммарная тепловая энергия по крайней мере достигнет этой величины, Вероятность такого столкновения, вычисленная по уравнению (15), даЖеТпри 1000° К составляет примерно -лишь 1-10-3. Из значений степени диссоциации |пр и 100О’ К (табл. 113, стр. 826, т, 1) следует, что вероятность соударения двух атомов I составляет 5,4-10“ 2 общего числа столкновений, Если предположить, что,, в среднем в одном из 1000 столкновений между атомами 1 участвует третья молекула, ' т. е. может происходить рекомбинация * атомов I, то на 500 атомов I, превращающихся в 12, приходится максимум один, реагирующий с Н2 с образованием HI. по уравнению (25). Непосредственный расчет коэффициентов скорости по уравнению (20) показывает, что при каждом соударении с энергией -40 ккал/молъ происходит одно превращение. Итак, несмотря на относительно сильную термическую диссоциацию иода, в реакции участвуют не свободные атомы I, а непосредственно молсъулм 12 и Н^. В этом заключается и причина того, что энергии активации обеих реакций — прямой я обратной — мало различаются. Если бы в обоих процессах участвовали свободные атомы, энергия активации реакции разложения HI должна бы была, быть значительно больше, чем для реакции образования HI.
Квантовомсхагт ячее кий расчет энергии активации. Теория химической связи (ср. т. I, гл. 5) в простых случаях позволяет представить энергию системы как функцию расстояния между ядрами атомов, а также типом связи электронов. На этом основании иногда удается вычислить энергию активации, необходимую для перехода систем из одного ^состояния в другое, Однако следует показать, например, что в дей-
* Ср. примечание на стр. 741, Если третьей частицей, участвующей в столкновении, является молекула И2, то становится возможным процесс 21 4- Н2 = 2HL Имеет ли он значение в действительности, пока пе ясно.
750
Глава, 17
ствнтельностк непрерывный переход системы из состояния 12 4" Н., в состояние 2HI возможен без промежуточной стадии — диссоциации на атомы. Подробнее этот момент здесь пе будет рассмотрен. Краткое представление о принципах квантово-механических расчетов энергии активации приведено в работах Шумахера и Фран-кенбургера. S с h цin a с h е г; Chemische Casreaktionen, S. 487, Dresden, 1938; W. Frankenbiirger, Katalytische Umsetzungcn in homogenen «nd enzymatischen Systemen, S. 444, Leipzig, 1937).
КИНЕТИЧЕСКОЕ РАССМОТРЕНИЕ КАТАЛИЗА
Приведенные выше соображения касались в первую очередь газовых систем. Однако основные положения можно распространить па жидкие и многофазные * системы. Из них следует, что скорость реакции в системе при данной температуре можно однозначно установить по энергии активации, если извне энергия не поступает пи в каких иных формах, кроме тепловой **.
Так как, по определению, катализатор можно выделить из реагирующей системы в неизмененном состоянии, он не может сообщать системе энергию ***, Следовательно, он влияет только на факторы, определяющие скорость реакции. К таким факторам относятся о. и Z в уравнении (16). Сюда же относится и фактор, учитывающий вероятность того, что столкновения, в которых молекулы обладают энергией, необходимой для химического взаимодействия, действительно приводят к реакции. В уравнении (16) эта «(вероятность реакциям предполагается равной 1. Для молекул с более сложным строением она значительно меньше. Катализатор может повысить вероятность (пример см. па стр. 753); однако реакция под влиянием этого фактора ускоряется не очень сильно. Число столкновений Z может быть увеличено в присутствии постороннего вещества в первую очередь за счет уплотнения реагирующих веществ путем адсорбции на его поверхности или конденсации в капиллярах. Однако этот эффект даже в случае катализаторов, обладающих значительной поверхностной активностью, имеет лишь подчиненное значение по сравнению с влиянием катализатора па фактор а, т. е. в соответствия с уравнением (15) па энергию активации А (ср, стр. 753). В большинстве случаев действие катализатора заключается только в понижении энергии активации. Учитывая, что энергия активации зависит от механизма реакции, это можно объяснить тем, что в присутствии катализатора реакция, идет по иному механизму, чем в отсутствие его. Каким окажется этот новый механизм, зависит от природы реагирующей системы и вида катализатора. Однако всегда катализатор должен каким-либо способом участвовать в определенной стадии реакции. Как правило, энергия активации снижается за счет образования промежуточного продукта, которое в отсутствие катализатора пе имеет места. Для реакции образования этого продукта необходима мешаная энергия активации, чем в случае промежуточного продукта, определяющего скорость реакции и возникающего в отсутствие катали
* В многофазных системах с кор octi, реакции определяется скоростью диффузии к поверхности раздела и от ясс. Если скорость диффузии мала по сравнению со скоростью химической реакции, го она является решающей дня протекания реак-_. Цин (ср. стр, 788 и си.).
** См. прим, иереи. к стр. 747,
*** В мкой-mo промежуток времени катализатор может сообщать энергию системе, в которой он инициирует реакцию; однако в дальнейшем ходе реакции эта энергия полностью возвращается к нему (если отвлечься от случайных обстоятельств, не имеющих отношения к существу катализа).
Катализ и. кинетика реакций
751
затора. При этом оказывается несущественным, является ли такой «промежуточный продукт» химическим соединением в обычном смысле пли речь идет о твердом растворе, образующемся на поверхности катализатора, или о любой другой системе, возникающей при действии сил, исходящих от катализатора. Таким образом, промежуточные продукты могут быть самыми разнообразными. И в принципе для каталитического Действия пе имеет значения, изменяется ли в присутствии катализатора механизм всех пли только некоторых стадий реакции.
В общем каждая каталитическая реакция, происходящая цо схеме
А + В=АВ, (26)
состоит минимум из двух последовательных стадий, которые можно представить следующим образом (Катализатор обозначен через К):
А + К=АК, (26а)
АК4-В=АВ4-К. (266)
В данном случае АК — любой продукт, образуемый катализатором; \ при этом речь идет не о химическом соединении в обычном смысле. Конечно, существенно, что при реакции, представленной уравнением (266), катгдлгг-затор снова выделяется в исходной форме.
Для ускорения реакций за счет образования «промежуточных соединений» между катализатором и субстратом или составной частью его важно как раз, чтобы промежуточные продукты оказывались неустойчивыми. Если образуется относительно устойчивое соединение, то, как правило, ото приводит к торможению реакции. В то же время при образовании очень малоустойчивого продукта реакция легко развивается дальше. Поэтому вещества, образующие вообще очень прочные соединения (например, щелочные металлы), мало активны в каталитическом отношении. Торможение каталитического действия вследствие образования устойчивых промежуточных соединений можно проиллюстрировать также на упомянутом на стр. 313 влиянии кобальта на СН4 и СО. Этот металл каталитически ускоряет разложение метана или окиси углерода лишь выше температур, при которых стабилен карбид кобальта.
Процесс, происходящий с участием катализатора, не обязательно должен быть первой стадией реакции. Ему могут предшествовать стадии, происходящие с заметь ной скоростью и в отсутствие катализатора. Однако вследствие высокого потенциального барьера какой-нибудь из стадий образование конечных продуктов происходит чрезвычайно медленно.
В некоторых случаях скорость реакции повышается благодаря тому, что реакция, инициируемая катализатором, в отличие от нормальной реакции является цепной. Но даже и в этом случае сущность действия катализатора заключается в снижении энергии активации одной реакции — в данном случае той, с которой связано зарождение реакционной цепи *. Одпако по величине ускорения реакции, обусловленной действием катализатора, нельзя сразу сделать заключение о возникновении цепной реакции. Следует иметь в виду, что значительное снижение энергии активации, как следует из данных табл. 98 (стр. 745), может само по себе привести к огромному возрастанию скорости реакции.
В дальнейшем характер действия катализаторов будет продемонстрирован па некоторых типичных примерах. Однако необходимо отме
* Иногда реакция, инициируемая каталитически (цеппая реакция), даже в присутствии катализатора имеет более высокую энергию активации, чем нормальная реакция (происходящая без образования реакционной цепи). Тогда большая скорость каталитическом реакции по сравнению с нормальной связана с ее цепным механизмом, а то, что опа вообще имеет место, обусловлено снижением иод действием катализатора энергии активации реакции зарождения или продолжения цепи.
752
Глава 17
тить, что, поскольку сущность каталитического действия в настоящее время следует рассматривать и, объяснять при помощи кинетических представлений, определенные выводы о механизме катализа в конкретных реакциях можно сделать лишь в ограниченном числе случаев.
Гомогенный катализ. Настоящие гомогенные каталитические реакции в газовой фазе — происходящие под действием газообразных катализаторов внутри реакционного объема, а не на поверхности (например, на стенках сосуда) — довольно редки. Примером их может служить реакция окиси азота с хлором, которая ускоряется в присутствия брома (v. KiB А., 1923 и СЛ;). В отсутствие брома реакция идет по уравнению (с образованием NOC12 в качестве промежуточного продукта, ср. стр. 738):
2NO4-C12=2NOC1. (27)
В . присутствии брома
2NO J- Br2=2NOBr, (28)
2NOBr-f-Cl3=2NOGl;-|-Br2. * (29)
Энергия активации реакций (28) и (29) меньше, чем в случае реакции (27), Поэтому образование NOC1 ускоряется вследствие промежуточного образования NOBr, который при взаимодействии с С12 опять дает Вг2.
Термическая диссоциация диметилового эфира (СНЭ)2О, каталитически ускоряемая водородом (Hinshelwood, 1927), могла бы служить примером гомогенных каталитических газовых реакций, при которых не появляется промежуточный; продукт, в образовании которого участвовал бы катализатор; действие катализатора заключается, лишь в том, что при соударениях он переносит енергию нВ те молекулы, которые следует активировать (при дальнейших столкновениях он опять приобретает эту энергию). Этот распад происходит следующим образом:
СН3-О—СН3=СН44-СО-ЬН2 (30)
и является реакцией первого порядка. Скорость распада определяется числом молекул (СН3)3О, вращательная и колебательная энергия которых достаточна для разрыва связей С—О; Экспериментально было показано, что энергия, необходимая для этого, может передаваться посредством столкновений молекул ,(СН3)2О и помимо этого, особенно легко при соударениях молекул (СН3)2О с молекулами Н2, но не с какими-либо другими молекулами, например Не, N2 или СО2. Так как Водород при дальнейших столкновениях вновь приобретает утраченную энергию, приведенную реакцию следует рассматривать как настоящую каталитическую.
Значительно чаще, чем гомогенные каталитические реакции, в газовой фазе встречаются гомогенные жидкофазные каталитические реакции. Особое значение среди реакций в жидкой фазе имеет ускорение процессов (в первую очередь окисления и восстановления) солями тяжелых металлов, а также кислотами и основаниями (ср. гл. 18, стр, 762).
Каталитическое действие солей тяжелых металлов часто основано па том, что их катионы в результате присоединения или отщепления электронов способствуют превращению молекул субстрата в свободные радикалы или неустойчивые ионы. Часто это приводит к цепным реакциям (ср. т. I, стр. 823 и сл.). Одпако с этими процессами связало такое значительное снижение энергии активации, что и без цепной реакции происходит очопь резкое возрастание скорости.
В случае ускорения реакции в присутствии кислот речь идет главным образом о промежуточном образовании продуктов присоединения протонов к молекулам субстрата, отличающихся особой реакционной способностью (низкой энергией активации). Ввиду того что, как правило, к субстрату присоединяются не ионы гидррксо-. ния IH30j+, а протоны, не редко в слабо диссоциированных или индифферентных растворителях кислоты обладают более сильным каталитическим действием, чём в водном растворе (ср, стр. 762 и сл.). По той же причине каталитическое действие, связанное с отщеплением протонов, может иметь место не только при действии кислот (в обычном смысле), но и других веществ, например иОнов NHJ. Каталитическое дей
Катализ и кинетика реакций
753
ствие оснований часто определяется тем, что их гидроксильные ионы отрывают протоны от молекул субстрата. В таких случаях катализаторами являются основания (в обычном смысле) и одновременно другие вещества, которые имеют тенденцию к присоединению протонов, например ноны слабых кислот.
Гетерогенный катализ. Существенное отличие гетерогенных каталитических реакций от гомогенных заключается лишь в том, что в первом случае реакция происходит на границе раздела двух фаз (катализатора и субстрата). И в случае гетерогенных реакций причиной их ускорения является, как правило, снижение энергии активации под действием катализатора. Переход молекул субстрата в реакционную, т. е. более легко активируемую форму, происходит под действием поверхностных сил (на поверхности раздела фаз) или имеющихся там свободных валентностей (ср. стр. 720 и сл.). Действие этих сил может привести к расщеплению абсорбированных молекул на атомы, правда, в большинстве случаев происходит только ослабление связи между атомами в свободных молекулах. Но даже такая «деформация», как следует из данных квантовой механики, приводит к значительному возрастанию реакционной способности.
Так как в твердых веществах особо большое число свободных валентностей расположено на ребрах и углах, именно эти места поверхности вболыняпстве случаев обладают особой каталитической активностью («активные центры» — по Лангмюру и Тейлору). Топкое измельчение катализаторов (применение их в коллоидном состоянии) или придание их поверхности шероховатости может поэтому привести к возрастанию каталитической активности не только за счет увеличения поверхности, но и вследствие связанного с ним увеличения числа активных центров.
Кроме снижения энергии активации за счет деформации (поверхности), существует еще одпа возможность влияния поверхности раздела фаз на скор о стр реакции. Однако это сказывается только в исключительных случаях. Так, в процессе рекомбинации свободных атомов твердая стенка при соударении может играть роль третьего партнера, необходимого для передачи энергии (ср. примечание па стр. 741). Далее, благодаря ориентации молекул:, связанной с адсорбцией, возрастает (или уменьшается) вероятность того, что при столкновении молеь-улы соприкоснутся своими реакционными центрами (ускорение реакции за счет повышения ее «вероятности», ср. стр. 750). Даже простое уплотнение субстрата на поверхности катализатора, т. е. увеличение его концентрации вследствие адсорбции, приводит (путем повышения числа соударений) к ускорению реакции. Практически, однако, этот эффект редко бывает существенным. Обычпо не удается ограничиться им для объяснения реально наблюдаемых ускорений реакций. Например, газы Н2 и О2 при адсорбции на фарфоре уплотняются в мопомо л окулярном слое цри 900° К примерно в шестнадцать раз (что следует из данных по теплотам адсорбции). Если бы только уплотнение было причиной каталитического ускорения реакции, скорость взаимодействия в гремучем газе должна была быть цронорциональной увеличению числа столкновений. Так как речь идет о три-молекулярной реакции, опа была бы пропорциональной третьей степени от степени уплотнения, т, о. в среднем отношение скоростей составляло бы 4000 : 1. В действительности, однако, как показал Боденщтепп (Bodenstein, 1899), реакция гремучего газа ускоряется в присутствии фарфора при данных условиях примерно в 20 000 000 раз. Снизанное с адсорбцией уплотнение субстрата на поверхности катализатора в большинстве случаев практически очепь мало влияет на каталитическое ускорение (по сравнению с «деформирующим действием»). В соответствии с этим поло-жепиом, как правило, не удается обнаружить связи между адсорбционной способностью различных веществ и их каталитической активностью.
Наибольшее значение имеют также гетерогенные каталитические реакции, в которых катализатор находится в твердом состоянии, а субстрат — и газообразном, жидком или в растворе. Ускорение газофазных реакций катализаторами, существующими в жидком или растворенном 48 г. геми
754 Глава 17
состоянии, применяют практически также часто. Относительно редки случаи, когда катализатор существует в менее плотном агрегатном состоянии, чем субстрат. В качестве примера можно упомянуть ускорение некоторых реакций между твердыми веществами в присутствии паров воды.
Очень часты случаи, когда химические реакции или превращения физического характера (полиморфные превращения) твердая веществ каталитически ускоряются в присутствии другого твердого вещества. Это может происходить по различным причинам.; Например, вследствие того что при внесении постороннего вещества увеличивается число едырок» в кристаллической решетке реагирующего вещества (ср. стр. 783). Кроме того, причиной ускорения реакции может быть понижение температуры плавления исходных веществ или одного из пих вследствие промежуточного образования твердых растворов. И, наконец, катализатор может явиться зародышем при кристаллизации продукта реакции. Последнее обстоятельство является причиной очень часто наблюдаемого аут окатал ат ическо&о ускорения реакций твердых веществ (с твердыми, жидкими или газообразными), в результате которых образуются твердые продукты. “ •
Аутокатализ, Аутокатализом называется ускорение реакции за счет каталитического действия одного из еепродуктов. Это явление проявляется в том, что скорость реакции сначала возрастает (в соответствий с количеством катализатора, образовавшегося в результате реакции) и только через некоторое.время начинает Падать в связи с резким снижением концентраций исходных веществ, перекрывающим каталитическое действие. Часто скорость аутокаталитической реакции становится заметной лишь через длительное время после ее начала. Но с этого момента происходит дальнейшее, иногда очень резкое, возрастание скорости, .вплоть до бурного протекания реакции. Если в реакционную смесь вначале ввести продукт реакции, ответственный за аутокатализ, реакция мгновенно начинается и происходит с большой скоростью.
Явление аутокатализа встречается довольно часто. Наряду с; названной выше группой реакций можно упомянуть ускорение окислеиия азотной кислотой при введении окйслов: азота или окисления перманганатом калия при добавлении солей марганца(П).
Направляющее действие катализаторов. Если постепенно повышать температуру вещества или смеси веществ, способных реагировать различным образом, в первую очередь осуществляется превращение, имеющее наименьшую энергию активации. Этот вывод непосредственно следует из сказанного в предыдущем разделе относительно кинетики реакций. То же, конечно, справедливо и для реакций, происходящих в присутствии катализатора. Однако катализатор нужен в данном случае совсем пе для снижения энергии активаций той реакции, для которой в отсутствие катализатора энергия активации оказывается наименьшей. Рассмотрим случаи, когда в отсутствие катализатора реакция может происходить двумя путями: один из них — нормальный — с меньшей энергией активации, другой — с более высокой энергией активации. Если у второго процесса прп помощи катализатора снизить энергию активации настолько, что она станет ниже соответствующей величины нормального процесса, то в этом случае образуются совершенно иные конечные продукты, чем при некаталитической реакции. Разные катализаторы могут снижать энергию активации различных реакций. От природы катализатора зависит й то, какая из реакций будет идти. Так объясняется направляющее действие катализатора.
"'г
Катализ и кинетика реакций
755
Например, термический распад мстил этилового эфира CHS-Q..C2HB в отсутствие катализатора протекает следующим образом:?
н-----г
, I । t
HjC-i-C—O-j-GH3 = СН4 + СО •+ СН4. (31)
Г I !
—-н
А в присутствии подходящих катализаторов, например иода, разложение является реакцией первого порядка *:
нэс-с-о4сн3= Н3С-С=О + СН4. (32}
I j. 1
н - н • :
Соответственно ведут себя и другие эфиры (Closing, Hinshelwood, 1930), например диэтиловый эфир: ’ 7 ’
Без кагпаяизатара: Н3С^С— О-гСН"гСН3 = СН4 4- СО 4- 1/2С2Н4 4? СН4, ! I ; I ; ♦
L-- Н : Н---------J :
(33)
Н ..-I
। : *
С катализа шаром1. Н3С—С — О-рСН —С Н3 = Н3С~ С= О + Н3С“СН3. /34J
I i I I
НН н
Если системе сообщить энергию активации [47 ккал для С11:, ()-С2Н;;, 53 ккал для (СгН5)2О] в отсутствие катализатора, разрыв различных связей в молекуле происходит в равной степени. В присутствии же катализатора, одна из связей ослабляется настолько, что для ее разрыва оказывается достаточной небольшая энергия активации (38 и 34 ккал соответственно). Вследствие этого под действием катализатора распад происходит при значительно более низкой температуре и совершенно иным путем. Упомянутый па стр. 752 распад диметилового эфира (энергия активации 65 ккал)-не ускоряется в присутствии иода; В соответствии с этим в данном случае введение, иода в сферу реакции по изменит характера распада.
Отравление катализаторов. Вещества, снижающие или вообще подавляющие активность катализатора, называются каталитическими ядами. Их действие основано на образований с катализатором малоактивных дли вообще не реакционноспособных соединений. В случае каталитически, инициируемых цепных реакций «яды», реагируя с промежуточными продуктами, вызывают преждевременный обрыв цени. В обоих случаях пе обязательно должно происходить образование химических/ соединений с каталитическими ядами. Например, при гетерогенном катализе для этого достаточно адсорбции яда на поверхности катализатора. Твёрдые катализаторы особенно чувствительны к ядам, ввиду того что их поверх-, ность резко уменьшается при адсорбции очень небольших количеств посторонних веществ. "К роме того, ад с о р бция пр он сх о д ит, п р еим у ществ ей -нб на «активных центрах», т. щ как раз в местах, отличающихся особо высокой каталитической активностью.
В зависимости от того, может ли быть устранено отравление при удалении яда ив реакционной смеси. говорят о чвремеипом» или «постоянном// отравлении. Причиной временного отравления может быть, например, слишком высокая концентрация
* Ацетальдегид, образующийся в результате реакции первого порядна, распадается по реакции второго порядка СН5-СПО = СН4 + СО.
756
Глава. 17
продукта реакции па поверхности катализатора. В таком случае его можно в значительной степени устранить увеличением скорости потока или перемешиванием.
Каталитические яды используют иногда для устранения нежелательных реакций («защитное отравлением).
Отрицательный катализ. В соответствии с определением явления катализа, отрицательный катализ заключается в том, что реакция, протекающая без катализатора, в присутствии некоторых веществ получает отрицательное ускорение, т. е. замедляется. Это может происходить, например, вследствие обрыва нормальной цепи в присутствии такого катализатора. На атом, как уже упоминалось, основано действие антидетонаторов.
Несмотря на то что в общем отрицательный катализатор следует отличать от каталитического яда. часто бывает трудно решить, о каком из этих явлений идет речь. Иногда реакция, протекающая внешне как кек атлантическая, в действительности пе инициирует с; я следами (позитивного) катализатора вследствие того, что его действие тормозится присутствием другого вещества (отрицательного катализатора).
Смешанные катализаторы. Каталитическое действие смеси двух или большего числа веществ обычно не равно сумме каталитических активностей компонентов. Опо может быть меньше, но может быть и значительно больше.
Так, Реми (1924) обнаружил, что относительная каталитическая активность Rh и Pt в реакции гремучего газа составляет 22 и 34, а сплава этих металлов — 100. При сплавлении Rli с Ru, совершенно неактивным по отношению к разбавленной гремучей смеси, каталитическая активность Rh при комнатной температуре возрастает от 22 до 34, а при иных условиях даже от 56 до 137. Как видно из этого примера, повы-гпепие активности катализатора достигается даже прн получении смеси его с веществом, которое само по себе вообще не обладает каталитическим действием в рассматриваемой реакции.
Часто достаточно добавки даже очень небольших количеств вещества для значительного повышения каталитической активности. Такие вещества носят названия «активаторов». Помимо этого, активности катализаторов, работающих в твердом состоянии, можно значительно увеличить ц осаждением их на веществах с сильно развитой поверхностью, например платины на асбесте. Вещества, действие которых заключается в увеличении поверхности катализатора, называются «носителями». Носитель? может одновременно действовать и как активатор. Это следует из того, что различные носители, обладающие практически равной поверхностью, могут оказывать совершенно различное влияние на осажденные на них катализаторы.
Смешанные катализаторы имеют исключительно большое значение в промышленных гетерогенных каталитических реакциях. Каталитические процессы в технике лишь в редких случаях проводят на простых катализаторах. В настоящее время почти всегда используют смешанные катализатор!.|, ввиду того что активность веществ, обладающих разносторонними каталитическими свойствами (например, железа), возрастает в совершенно определенном направлении при введении соответствующих активаторов н носителей. Так, железо оказывается особенно активным в реакции синтеза аммиака в присутствии добавок окиси алюминия. Добавление окиси висмута повышает активность железа при каталитическом окислении аммиака до окиси азота, Введение окиси хрома повышает его активноств при взаимодействии окиси углерода с водяным паром с образованием двуокиси углерода и водорода. Чувствительность катали
Катализ и кинетика реакций 757
заторов к действию ядов также можно значительно снизить, а иногда вообще практически устранить добавками соответствующих веществ.
В качестве примера действия смешанных катализаторов в гомогенных' реакциях можно привести наблюдение Прайса (Price, 1898). Он показал, что при одновременном присутствен солей медн и железа в реакции окисления водных растворов йодистого водорода введение пер оксосульфатов ускоряет взаимодействие более чем вдвое пр сравнению с тем, что имело бы место при простом суммировании активностей обоих веществ. Несмотря на то что случаи повышения активности мри добавлении к катализатору других веществ были известны и раньше, Прайс первым обратил внимание па неаддитивность действия смешанных катализаторов. Брод (Brode, 1901), продолжая опыты Прайса, впервые установил, что на активность катализатора могут оказывать влияние и такие вещества, которые сами по собе не обладают или обладают слабым каталитическим действием.
В последующие годы, как уже упоминалось, смешанные катализаторы подробно исследовались в работах Митташа. Причины активирующего действия могут быть различными. Если речь идет об ускорении взаимодействия двух веществ, то может быть, что катализатор А будет активировать исключительно или почти исключительно молекулы одного из веществ, а катализатор В — молекулы другого компонента. Если же смесь А и В активирует одновременно молекулы, обоих видов, в результате возникает особенно значительное ускорение реакции. В случае твердых катализаторов действие активаторов часто основано на том, что они оказывают влияние на величину зерна катализатора или, внося искажения в решетку, увеличивают число «активных центров» *. Кроме того, в сплаве двух веществ поверхностные силы, могут быть совершенно иными, чем у компонентов, причем сферы действия их атомов взаимно перекрываются и изменяются.
Катализаторы Ренея. Реней (Ranay, 1925—1927) предложил метод приготовления особо активных форм некоторых металлических катализаторов. Он основан па том, что каталитически активный металл сплавляют с неактивным, сплав измельчают и неактивный металл извлекают из сплава. Из металлов Репея чаще всего применяют никель. Его получают обычно сплавлением никеля с алюминием. Грубо или тонкоразмо-лотый сплав обрабатывают раствором щелочи, тщательно промывают остающийся после выщелачивания черный или черно-серый порошок. Последний чувствителен к действию воздуха, часто даже иирофорен, поэтому его нужно хранить под слоем: ‘воды, спирта, бензина и т. ,д.
Никелг, Ренея по структуре не отличается от обычного никеля, однако существует в более мелкокристаллической форме (диаметр кристаллов 40—80 А). Он содержит еще некоторое количество алюминия (до нескольких процентов), который но-види-мому, влияет на каталитическую активность никеля, и, кроме того, значительные количества водорода (доЗЗат.%), а по своим электрохимическим свойствам аналогичен платиновой черни, насЬгщснной водородом. Никель Ренея амальгамируется ртутью (пякель, не содержащий водорода, не дает амальгам). Нитриты и цитраты восстанавливаются пиколем Ренея до аммиака, перманганат — до двуокиси марганца. Хлораты, броматы, йодаты, молибдаты и вольфраматы также могут восстанавливаться ям. С водой никель Репея постепенно реагирует, выделяя водород и образуй гидроокись пикеля(Г1)- Однако в присутствии легко окисляющихся веществ, которые могут быть акцепторами кислорода, никель Ренея не изменяется и является только катализатором окисления. Например, гипо фосфиты окисляются водой в его присутствии до фосфитов по уравнению РН20. Щ П,О ^ PTIO" + Н‘ +ГГ3. Реакция протекает количественно,
* С такими искажениями решетки и аналогичными изменениями связаны особые каталитические свойства вшцестп и «активньгх переходных состояниях», часто встречающихся в реакциях между твердыми веществами (ср. стр. 786).
758 Глаза 17
и ев можно использовать для ацидиметрического определения гипофосфитов. Никель Ренея ускоряет и окисление кислородом воздуха. В технике его используют в основном как катализатор гидрирования. Вследствие особенно высокой активности этот катализатор значительно ускоряет реакцию. Кроме того, он позволяет проводить при низких температурах гидрирование таких соединений, у которых при более высоких температурах оно осложняется побочными реакциями. '
Металлы Ренея применяют и в качестве катализаторов дегидрирования. Кобальт Ренея — отличный катализатор Hpit синтезе бензина из СО и Н3. При этом он является не только катализатором гидрирования, но одновременно и катализатором полимеризации, особенно если он получен не из сплава. Со-Al, а выщелачиванием сплава Co-Si. По данным Гер гл отца н Лисснера (Herglotz, L issuer, 1949), кобальт Ренея можно с успехом применять для обессеривания органических соединений при каталитическом превращении серу органических соединений в сероводород. Кобальт Ренея представляет собой гексагональную модификацию Со,втовремякак кобальтовые катализаторы, полученные обычным путем (например, нагреванием СоО в токё Н2), содержат кубическую модификацию Сю. Обе модификации кобальта каталитически активны, в то время как никель обладает каталитическими свойствами лишь в кубической форме.
Мера каталитической активности. Активность катализатора определяется отношением констант скоростей к катализированной и неката-лизированной реакций. Константы скорости относятся, естественно, к одной температуре. Кроме того, в гомогенном катализе активность относится к определенной концентрации, а в гетерогенномк определенной поверхности катализатора. У особо важных катализаторов — твердых веществ — действительная величина поверхности в лучшем случае известна лишь приблизительно. К тому же при равных поверхностях число «активных центров», определяющее каталитическую активность, может быть совершенно различным. В таких случаях каталитическую активность можно определить значительно более точно и выразить снижением энергии активации под действием катализатора. Это, конечно, справедливо в предположении, что в наблюдаемых случаях на ускорение реакции вообще (или. по крайней мере существенно) не влияют другие факторы, которые могут привести к снижению энергии активации.
Г]Так как с уменьшением энергии активации снижается и температура, при которой реакция гладко протекает, с заметной /скоростью; ронмкение температуры реакции также может .служить дня приближенной оценки каталитической активности. Если реакции протекают без катализатора иным путем, чем вГего присутствии,, или в тех случаях, когда констапты скорости или энергия активации некаталитической реакции не известны, можно сравнивать лишь относительные активности различных катализаторов. Практически пользуются имёппо ими.
Катализ и химическое равновесие. Как уже упоминалось, пз определений понятия катализатор следует, что он ускоряет реакцию, не отдавая и не принимая энергию реагирующей системы. Отсюда следует далее, что равновесия сисщемы катализатор не сдвигает [поскольку сдвиг равновесия связан с изменением энергии системы (ср. т. I, стр. 168)). С кинетической точки зрения химическое равновесие определяется тем, что в этом состоянии реакции протекают с равными скоростями в обоих направлениях, Следовательно, образование соединения происходит с той же скоростью, что н его распад; катализатор ускоряет протекание реакции в обоих направлениях в одинаковой степени. Катализатор, ускоряющий образование одного соединения, ускоряет:, следовательно, в той же мере, и его распад. Этим часто пользуются при исследовании активности катализаторов. Например, для определения активности различных катализаторов в синтез о аммиака, вместо ускорения образования аммиака
Катализ и кинетика реакций
759
можно изучать реакцию его диссоциации. Однако положение о. том, что каталитическая активность не зависит от направления реакции', строго выполняется лишь для систем, которые: находятся вблизи положения равновесия.
ИНДУЦИРОВАННЫЕ РЕАКЦИИ
Индуцированными называются реакции, которые инициируются или ускоряются добавками веществ, расходуемых в реакции. Их действие основано именно на этом и не связано со случайными побочными процессами. Ввиду того что катализатор не сообщает энергии системе, реакции которой он ускоряет, в его присутствии могут осуществляйся лишь такие процессы, в результате которых выделяется энергия. В то же время индуцированные реакции могут быть связаны и с поглощением энергии. Энергия, которая должна подводиться к системе, чтобы реакция могла происходить, выделяется при превращении вещества, инициирующего: реакцию. Следовательно, этот процесс всегда должен быть экзотермическим. .
В качестве примера индуцированной реакции можно привести окисление мышьяковистой кислоты бромнов ат ой кислотой в водном растворе;
НВгО3 + ЗН3АёО3=ЗН3ЛзО4-|-11Вг. '
Несмотря на то что эта реакция экзотермична, самопроизвольно она протекает крайне медленно, Она значительно ускоряется при добавлений сернистой кислоты, которая при этом окисляется;
HBrO3+3H2SO3==3H^SO4+HBr, "
н этим процесс коренным Образом отличается от каталитического,
, Вели реакция,, инициируемая введением индуктора *, самопроизвольно не может идти, между максимальным количеством реагирующего вещества (актора) и соответствующим количеством индуктора всегда существует простое стехиометрическое соотношение. В этом случае реакции называются сопряженными. Примером ; их могут служить реакции сопряженного окислении, сопровождающие процесс образования перекиси водорода, в частности, окисление соединений: титана(1П) в щелочном растворе (си. стр. 73), где на каждый окисляющийся атом Ti(III) образует-ся максимум 1А молекулы Н3О2 (индуцированной в данном случае является реакция окисления Н2О до НаО2). Вели речь идет о такой индуцированной реакции, которая может происходить и самопроизвольно; тогда «фактор индукции», т. е. соотношение количеств актора и индуктора, колеблется в широких пределах и может принимать относительно высокие значения. Например, при окислении в воддом растворе иодид- ?’ ионов, индуцируемом ванадиевой кислотой, фактор индукции ц зависимости от. условий опыта изменяется от 1/1ММ до 12 (Вгау, 1933),
* В русской литературе для компонентов сопряженных реакций
A-j-B- --1М, .
Л«С->А
приняты следующие обозначения: А — актор; В -- акцептор;, С — вещество, инициирующее первую реакцию,-г- индуктор1. В приведенном примере сопря?кенпого окисления Ti(III) по схеме
О3 I-Ц2О - > ПпОо,
t>2 -Ti(III) —> Ti(l V),
О2 — актор; Н2О — акцептор; Т1(Ш) — ип,дуктор. (См., например Н. А. Шилов, О сопряжеппых реакциях окисления, Москва, 1905). —Прим, персе.
Глава 18
РЕАКЦИИ В НЕВОДНЫХ РАСТВОРАХ
Общие сведения. Несмотря на то что вода в качестве растворителя имеет в неорганической химии в общем основное значение, в настоящее время все больше начинают применять другие растворители. Неводные растворители часто используют, например, в препаративных целях и для определения молекулярных весов. Это делают главным образом тогда, когда работают с веществами, которые в воде нерастворимы или ею разлагаются. Общее изучение поведения веществ в неводных растворителях и сравнение его с поведением в воде также очень важно для неорганической химии. Это особенно важно для углубления представлений о сущности процесса растворения и о природе протекающих в растворах реакций. При этом оказывается, что водные растворы отнюдь не представляют собой прототип любых растворов, а лишь составляют их особый случай (правда, практически очень важный).
Растворители (без учета расплавленных металлов) можпо подразделить на две группы: иа такие, которые в состоянии вызывать электролитическую диссоциацию растворенных в них веществ, и на такие, которые не могут способствовать электролитической диссоциации (пли вызывают только крайне слабую диссоциацию). Последние называют индифферентными растворителями. Однако резкой границы между этими двумя группами но удается провести.
С растворением, кроме того, может быть связано разложение переходящего в раствор вещества, сопровождающееся присоединением составных частей растворителя к составным частям растворенного вещества по схеме двойного обмена:
AB-J-CD AC+BD.
В таком случае говорят о сольволизе. Часто сольволиз приводит к установлению равновесия, так же как гидролиз (ср. т. I, стр. 877 и сл.), который является частным случаем сольволиза.
Индифферентные растворители. Растворители, которые не вызывают вообще или вызывают только крайне незначительную диссоциацию, как известно, имеют основное значение в органической химии. Ъ неорганической химии их применяют ограниченно. 'Это связано с тем, что «индифферентные растворители» могут растворять в заметных количествах чаще всего только такие вещества, которые образуют в кристаллическом состоянии молекулярную решетку или но меньшей мере такую решетку, которая приближается к молекулярном. Это справедливо для небольшого числа неорганических веществ (ср. т. I, гл. 9). Поэтому большинство неорганических веществ нерастворимо в индифферентных растворителях, и прежде всего в таких, которые применяют при обычной или при несколько повышенной температуре. Наиболее употребимыми индифферентными растворителями для неорганических веществ являются четыреххлористый угле
Реакции, в неводных растворах 761
род, хлороформ и сероуглерод, К ним можно также причислить эфир, который вызывает лишь крайне незначительную диссоциацию. Аналогично эфиру ведет себя в этом отношении дноксан (1,4-диэтилендиоксид) >СНг — СН2.
Оф фО, который в последнее время часто с успехом используют
^GHj-CHZ
в качестве растворителя неорганических веществ.
Как уже ранее было отмечено (т. I, стр. 101), переход веществ в раствор сопровождается их сил мата ц ией. Вероятно, он даже большей частью обусловлен сольватацией, ибо оказалось, что в общем растворимость веществ В; индифферентных растворителях тем больше, чем больше теплота их сольватации. То обстоятельство, что для растворенных веществ справедливы те же законы, что и Для веществ в газообразном состоянии (прежде всего имея-в виду формальную аналогию между осмотическим и газовым давлением), привело к тому, что одно время мало внимания уделяли взаимодействию между растворенными веществами и растворителем. Растворенные вещества рассматривали как бы находящимися в состоянии, которое почтя полностью соответствует газовому состоянию. Несостоятельность этого представления убедительно-показан Фроденхагсп (Fredenhagen, 1932 и сл.).
То, что сольватация весьма существенно влияет па напряжение выделения металлов ия растворов, на примере водных растворов было показано уже в т. I (стр. 181 и сл.). Сделанные там выводы имеют соответствующее значение также и для неводных растворов. Конечно, это влияние тем меньше, чем меньше теплоты сольватации: (в рассматриваемом случае—ионов).
Растворитель оказывает очень сильное влияние на ход химической реакции. Издавна известно, что скорость химической реакции существенно зависит от природы растворителя, причем даже тогда, когда растворители совершенно определенно не Оказывают диссоциирующего действия.
Большое практическое значение имеет то обстоятельство, что часто даже направление, в котором протекает химическая реакция, зависит от растворителя. Так, реакция1
< 2HI + S H2S + J2
в газовой фазе протекает слева направо, а в водном растворе справа налево. Последнее обстоятельство обусловлено теплотой растворения HI в воде. В сухом четыреххлори-етом углероде H2S и 12 не реагируют, в эфирном растворе в отсутствие воды они также не реагируют или реагируют только в той степени, которая определяется постепенным использованием HI для образования этилиодида.
Очень часто влияние растворителя на направление реакции обусловлено тем, что при течении реакции в одном или другом направлении образуется вещество, нерастворимое в соответствующем растворителе. Согласно принципу Л е-Ш ателье, это-должно привести к сдвигу равновесия в сторону образования вещества , выделяющегося из раствора (жндкой фазы). Как в случае водного раствора, так н неводных растворов, это используют часто для препаративных целей.
Существует обширный материал, касающийся растворимости неорганических и органических веществ в индифферентных и прочих растворителях. Одпако систематических исследований взаимодействия между индифферентными растворителями и растворенными в них веществами проведено сравнительно немного (кроме установления состава сольватов, полученных из таких растворов в кристаллическом состоянии). Широкие-исследования электропроводности неводных растворов были проведены прежде всего Вальденом (Walden). В более поздних работах большое внимание уделено выяснению поведения в неводных растворителях кислот (а также других веществ, которые ведут1 себя в водном растворе подобно кислотам).
ПОВЕДЕНИЕ КИСЛОТ В НЕВОДНЫХ РАСТВОРИТЕЛЯХ
Давно известно, что кислоты в не иодных растворах (причем даже-и таких, в которых они диссоциированы крайне незначительно) часто оказывают более сильное каталитическое действие, чем в водном растворе.
'762- ...... Глава 18
Так как в водном растворе каталитическое действие кислот (например, ускорение ими расщепления диазоуксусногр эфира) пропорционально концентрации ионов водорода (ср. т. I, стр. 102), то этот факт казался необъяснимым, поскольку взаимодействием растворенного вещества с растворителем пренебрегали. Однако, если учесть, что?в водном растворе свободные ядра Н+ (протоны) вследствие сильного сродства к ним молекул воды (образования попов [НзО]’) могут находиться лишь в крайне небольшом количестве (стр. т. I, стр. 100), в то время как в неводных растворах протоны часто связаны существенно слабее, то сразу Становится понятным, что па реакции, которые каталитически ускоряются протонами (как это имеет место при расщеплении диазоуксуспого эфира)*, в неводных растворах кислоты могут оказывать большее влияние, чем в водном, растворе
То, что в водном растворе каталитическое действие кислот в общей оказывается пропорциональным концентрации, ионов [Н3О|*, связано с тем, что концентрация Протонов в водном растворе пропорциональна концентрации ионов (Н-,ОГ. В связи с тем, что вода связывает кротоны с образованием ионов [Н30)', действие протонов ослабляется', в общем .считают, что активность водородного соединения в растворителе, обусловленная отщеплением протонов^ проявляется тем сильнее, нем слабее растворитель связывает протоны. Отсюда следует, что свойства водородных соединении, обусловленные их способностью отщеплять протоны, зависят главным образом от протонного обмена с растворителем. Л
Теория кислот и оснований (теория протонного обмена) Бренстедщ Согласно Бренстеду, вещества отдающие цротоны в растворе, называются кислотами, а вещества, присоединяющие протоны,— основаниями ***, Благодаря тому что кислота D отщепляет протон по уравнению D = Е ;+ 4- Н+, она становится соответствующим основанием Е. Если основание Е принимает протон, оно становится соответствующей тсислбтой D.
Например, ион С1” является соответствующим основанием кислоты НС1; ион ;[NH4]+ — соответствующей кислотой основания NH3; ион ОН” является соответствующим основанием кислоты Н,О, которая в свою очередь является соответствующим основанием кислоты |11;Ор.
Как следует из последнего примера, одно и то же вещество, может фигурировать как в качестве кислоты, так и в качестве основания в зависимости от рассматриваемой реакции. Такие вещества называют амфотерными. В соответствии с; определением соответствующие кислоты и основания имеют всегда различный 'заряд. Это также следует из приведенных примеров. :-pf
Так как при химических реакциях протоны никогда не бывают в свободномсостоянии в больших количествах, то, чтобы заметные количества кислоты D, смогли превратиться в соответствующее основание E j,:должно присутствовать другое основание Е2, которое присоединяло бы отщепляющиеся протоны, переходя в соответствующую кислоту D2:
I)i + E2 D2 j 14. (I)
* Почти в любом случае каталитического ускорения реакции «ионами водорода» речь идет о действии протонов. Очень редко специфическим ускоряющим действием5 ; обладают ионы [Т13О]*.
** То же относится ко всем определениям «силы» кислот, которые основаны непосредственно на измерении иротоипой активности и лишь косвенно дают меру активности ионов [Н3Ор, например к определению «концентрация попов водорода» кислых растворов потенциометрическим, путем. а ~
*** В оригинале кислоты, но теории Бренстеда, называют диспротидами, а основания □.иирстиЗа.ча. Эти термины не нашли широкого распространения в русской литературе. В переводе терминам кислота и основание чаще всего придается смысл, используемый в теории Бренстеда.— Прим, персе,
Реакции в неводных растворах
763
Примерами реакции протонного обмена такого типа являются:1
Кис дота 1 Ос нона нис 2 Кислота 2 Основание 1
НС1 + NH3 <== [NHJT |- : С1-
ИС1 |- 11,0 = [H3OJ+ CI-
H,0 + NH3 = [NI-Id+ -J- ОН"..
Благодаря тому что подобные реакции рассматривают со времени Бренстеда {1923 и сл.) сточки зрения протвкноеа обмена, оказалось, во-первых, что ряд процессов, между которыми раньше не предполагали никакой связи, относится в основном к процессу одном и того жетипа. Такими процессами являются насыщение кислоты анги-дрооснованпем (первый пример); электролитическая диссоциация кислоты (второй пример) а образование основания из ангпдрооснованйяиводы (третий пример). Во-вторых, сразу появляется возможность: рассматривать поведение киелот в различных растворителях с единой точки зрения. Если определить кислотный характер как способность отщеплять протоны, то не будет в основном никакого различия между поведением кислоты в отношении воды ц в отношении аммиака пли любого другого основания *. В-третьих, важен тот факт, что способность про дал ять свойства кислот отнюдь во ограничивается веществами, которые могут функционировать в водном растворе как кислоты. Кроме последних, кислотным характером, обладают не только такие ионы, как (NH4]+, но и, смотря по выбору растворителя или партнера по реак-. ции. даже такие вещества, которые ле способны отдавать протоны непосредственно ^молекулам ЩО. Например^ Бонгоффер (Bonlioeffer, 1936) показал, что даже элементарный водород может функционировать в качестве кислоты в отношении иОна гидро, ксила. Он нашел, что если действовать водородом при 100° на тяжелую воду (D^O) и прежде всего на воду, содержащую щелочь, то в заметной степени происходит протон, пый обмен по уравнению
. j DQ-+ Н-Ц Н-j-D-i-OD^ DOH + HD-F OD"
L------
Из теории протонного обмена следует важный вывод: если при дер лсйваться традиционного определения понятий кислот и оснований (с уточнением последних) **, то говорлть о проявлении кислого и основного характера имеет смысл только в водных растворах: в неводных растворах путем отщепления протонов не могут образоваться попы гидроксония, а соединения, функционирующие в водных растворах как основания, ничем по отличаются в неводных растворах от солей. Напротив, в водном растворе кислоты и основаниями традиционном смысле) отличаются от солей главным образом тем, что при электролитической диссоциации они образуют собепгвенные ионы растворителя 1Н30]', соответственно ОН'. Б качестве меры силы кислот и оснований используют тогда величину, характеризующую количество образующихся ионов [И3О] или ОН', т. е. степень диссоциации в водном растворе. Сущность нейтрализации заключается в соединении ионов [Н30]* и 0(1' с образованием воды. .
Если взять за основу это определение, то, согласно Я ндер у.,, в невод н ых растворах^ можно говорить только о «кислотогаоЗобвй.ч» я «ослованиелойобноаг» поведении, а не о «кислотном» и «основном» поведении. Если, напротив, определить .«кислоты» как соединения, способные: к отщеплению протонов (не обращая внимания на растворитель), то сущность нейтрализации может быть отражена в пйлно.ч протекании реакции протонного обмена в соответствии с уравнением (1), стр. 762. «Основаниями» с этой точки зрения являются соединения, способные нейтрализовать кислоты, т. е. вещества, присоединяющие протоны, G этой же точки зрения, соединения, которые отщепляют ионы гидроксила, даже в водном растворе не отличаются от солей,~С другой стороны,
* .Не только все вещества, которые могут образовывать с кислотами продукты присоединения солео бра злого характера (цацример, эфир, альдегиды, кетоны; ср. т. I, стр. 748), до и очень многие другие вещества, в которых растворимы кислоты, ведут себя по отношению к ним как основания (правда, в различной степени). Нельзя не отметить, что имеются также практически важные свойства кислот, которые связаны с особой, .природой воды, а именно те свойства, которые обусловлены образованием ионов [Н30р (лапример, высокая электропроводность кислот в водных растворах). (Ср. также примечание * на стр. 762).
** Кислотами называют такие соединения, которые в водном растворе путем отщепления протона образуют ионы гидроксония [Н30[+, а основаниями такие, которые отщепляют в водном растворе ионы гидроксила £>Н~.
764
Глава 18
в смысле второго определения к «кислотам» причисляют даже ионы солей, если они способны к отщеплению протона, следовательно, например, ионы [Л’Н4]'. Таким образом, имеется альтернатива: либо ограничить употребление понятий «кислота» и «основание» водными растворами, либо отказаться от традиционного определения «кислот» и «оснований», назвав вещества, отдающие протоны, «кислотами», вещества, присоединяющие протоны, «основаниями» и причислив соединения, которые в водном растворе отщепляют,ионы гидроксила, к солям. Последнее сделал Брепстед, который назвал в соответствии с этим разработанную им (1923) теорию протонного обмена «теорией кислотно-основной функции». Однако вследствие исключительного практического значения воды в качестве растворителя пе следует отказываться от названий обоих классов веществ, занимающих ио отношению к вобе особое положение в связи с тем, что они растворяются в ней с образованием собственных ионов атого растворителя- [H3Of или ОН'. Практически едва ли возможно ввести для этих двух классов веществ вместо обычных названий «кислоты» и «основания» новые названия. Одпако, если названия «Кислота» и «основание» применяют также одновременно к вновь введенным Бренстедом понятиям — отщепляющим протоны веществам и присоединяющим протоны веществам, то легко могут возникнуть недоразумения *.
Индифферентными растворителями, по мнению Бренстеда, являются такие растворители, которые пе обладают характером ни кислот, ни оснований. Их можно назвать также апротидиыми растворителями. При растворении в апротидном растворителе вещества, способного к отщеплению-или присоединению протонов, не могут, конечно, образовываться электролитические ионы, независимо от того, имеет растворитель высокую или низкую диэлектрическую проницаемость.
Если растворить чистую пикриновую кислоту в индифферентном растворителе, например бензоле, то образуется бесцветный раствор, так как недиссоциированная пикриновая кислота бесцветна. Если, однако, добавить к раствору основание, например' анилин или метанол, то в соответствии с общей схемой протонного обмена (уравнение (1), стр, 762( образуются пикрат-ионы и раствор становится желтым:
С6И2(1ТО2)3.0Н + CsH5.NH2 = C6HS.NH+ + СбН,(ИОг)з-О- (2>
Щ (Пикриновая Es (Лан лиц) Щ (А ни линий- Ei (Пикрат-ион) кислота) ион)
CeH2(NO2)3-OH + CH3OU = CHs-Ollf + C6H£(NO2)3.O- (3>
Dj (Пикриновая Ес (Метанол) Щ (Метилон- Ei (Пикрат-ион) кислота) , сони й-ион
Те же соображения можно в общем применить к равновесию диссоциации цветных индикаторов, к которым можно отнести и пикриновую кислоту. Зависимость-равновесия диссоциация (и вместе с тем окраски) этих индикаторов от концентрации ионов (Н30]' в водном растворе представляет собой только частный случай равновесия ** протонного обмена по общей схеме уравнения (1), стр. 762:
C6U2(NO2)3.OH + Н2О [Н3О]+ + C6H2(NO2)3O“ (4)
Кислота 1 Основание 2 Кислота 2 Основание 1
Идет ли речь в случае кислоты о попах [НЭО]+ или о любых других кислотных ионах, имеет значение лишь для определения положения равновесия, но в остальном в общем безразлично. Поэтому в индифферентных растворителях кислоты можно титровать (ангндрооснованиямн или другими основаниями) также с применением цветных индикаторов, как и в водпом растворе. Очень слабые кислоты, пли «протогенные» вещества, не обладающие кислотным характером, следует титровать только в неводном растворе. Цветные ипднкаторы можпо применять как для колориметрического определения концентрации ионов [Н3О[‘ в водных растворах, так И для колориметрического определения способности кислот отдавать протоны в неводных растворителях, в частности в индифферентных растворителях. Пикриновая кислота является примером кислотного индикатора (в поднес оц пиро ванном состоянии). Об основных индикаторах можно сказать то же, с той лишь разницей, что в этом случае роль кислоты 1
* По терминологии Бренстеда, например, ионы [NH4)‘ и (Н3О|‘ являются
кислотами, ионы [SO4] Д" и С1' — основаниями, а соединения КОН и Са(ОН)2—-солями. Бренстед применял иногда также названия «протогенпые» л «протофильные» вещества.
Реакции в неводных растворах
765
в общем уравнении протонного обмена Играют не недиссоцинрованные молекулы, а ионы, образованные в результате присоединения протопов.
Протонное сродство. Под протонным сродством молекулы или иола понимают максимальную анергию, которая может быть получена при присоединении ими протона. Непосредственное измерение протонного сродства встречает еще затруднения, однако легко удается сравнить величины протонного сродства различных веществ. Это удается, например, путем определении положения равновесия протонного обмена по уравнению (1), стр. 762. Если сравнить две кислоты п D2 в одном и том же растворителе *, то равновесие, определяемое уравнением (1), тем сильнее оказывается сдвинутым слева направо, чем больше протонное сродство основания Е2 в сравнении со сродством основания Ер чем легче, следовательно, кислота Dj отщепляет протоны и чем труднее это делает кислота D2. Кислоты удается расположить в ряд па основании их различной способности к отщеплению протонов, например (по Бренстеду):
1. Хлористый водород
2. Трпхлоруксусная кислота
3. Дихлор уксусная кислота
4. Пикриновая кислота
5. Монохлоруксусная кислота
6. Салициловая кислота
7, о~Х лор бензой па я кислота
8. .и-Хлорбытзойиая кислота
9. Ион бепзиламмония
10. Муравьиная кислота
11. Фопилуксусная кислота
12. Бензойная кислота
13. Уксусная кислота
14. Иоп изо амил аммония
15. Ион цинеридиния.
В этом ряду, который построен Для растворов в индифферентном растворителе (бензоле) склонность к Отщеплению протонов падает от хлористого водорода к иону пиперидиния. Эта последовательность пе слишком отличается от последовательности тех же веществ в водном растворе, в котором склонность к отщеплению протонов обычно измеряется величиной, характеризующей образование иопов [Н,О|+. Однако в случае сильных кислот наблюдается существенное различие в том отношении, что исследования в. индифферентных растворителях действительно показывают различную склонность их к отщеплению протонов, в то время как в водном растворе это различие не проявляется или проявляется очень неотчетливо, таи как взаимодействие с Лг0 по уравнению (1) у них у всех практически протекает полностью. На тот факт, что вода оказывает такое «нивелирующее» действие на силу кислот, ранее указывал уже Гаити (Hantzsch).
Кац показали Шварценбах (Schwarzenbach, 1930) и Вйберг (Wiberg, 1939), склонность к отщеплению протопов удается определить потенциометрически, так как разность потенциалов между водородным электродом и (водным или неводпым) раствором зависит <)Т концентрации протопов (точнее, от протонной активности). Можно построить «ряд напряжений» кислот, в котором бин расположатся по своей склонности к отщеплению протонов, так же как металлы и прочие восстановители располагаются в ряду напряжений на основании потенциометрически определенной способности к отдаче электронов (ср. т. -I, стр. 50 и сл. и стр. 820).
I ..
«ВОДОПОДОБНЫЕ» РАСТВОРИТЕЛИ
Для познания сущности превращений, происходящих в водных растворах, очень полезно изучение превращений, протекающих в растворителях, которые имеют с водой ряд важных общих свойств: высокая растворяющая способность, незначительная собственная электропроводность
* Положение равновесия зависит от природы растворителя. Если принять что основания X- имеет то же протонное сродство, что и NH3, то но уравнению НХ + + NH3 [NII4]+ + X- в равновесии находятся одинаковые количества НХ, NH3, [NH,4]+ и X - нрц условии, что растворитель пе оказывает никакого влияния иа положение равновесия. Однако если в качестве растворителя используют, например, воду, то по реакции 14Н3 -Н П20 7_ NH3-H2O часть основания NH3 используется па образование йН3-Н2О (соответственно гидратированных молекул NH3). Это, согласно закону действия масс, приводит к сдвигу равновесия НХ -(- NH3 X Д |AII/J+ + X- справа палено. Точно так же в общем случае сдвигается положение равновесия при сольватации (ср. стр. 7(H). Таким же образом приводит к сдвигу равновесия протонного обмена ассоциация растворенных молекул, наблюдаемая в слабя сольватирующем растворителе.
766
Глава 18
и ярко выраженная электропроводность растворов, обусловленная способностью этих растворителей вызывать электролитическую диссоциацию растворенных веществ. Подобные растворители называют, по предложению Яндора (Jander), вводоподобнылш». Такими водоподобными растворителями являются, например, жидкий аммиак, жидкий фтористый водород: и жидкая двуокись серы. Сюда же можно причислить в известном смысле и жидкий сероводород, хотя этот растворитель по своим свойствам во многих отношениях близок к индифферентным растворителям (ср. стр. 772 и сл.). Таким образом, свойства «водоподобных растворителей» болев или менее-отчетливо проявляются у водородных соединений трех элементов, расположенных рядом с кислородом в периодической системе. Однако эти свойства отнюдь не ограничиваются водородными соединениями, а, как показывает пример двуокиси серы, могут проявляться у соединений, которые производятся от воды путем замены водорода или кислорода на другой элемент. Жидкая двуокись серы ведет себя даже более «водоподобно», Чем жидкий сероводород. Условием для проявления растворителем; «водоподобных» свойств является то, что диссоциация (наблюдаемая для чистого растворителя только в слабой мере) осуществляется по схеме, аналогичной диссоциации воды:
2Иг0
2NHs [NHJ+-f-NHj-,
2HF [H2F]++F-,
|П30]++01Г, 2H2S [H3S]+-f- SH-, )
2SO3 SOM- i-|SO3|“- J
(5)
(6).
Общим для этих процессов диссоциации является то, что одна молекула растворителя отдает другой молекуле элементарный ион и благодаря этому возникает пара ионов, несущих противоположные заряды.
Рассмотрение этих процессов диссоциации выходит за пределы реакций протонного обмена. В соответствии с этим аналогии между реакциями, происходящими в различных «водоподобных» растворителях, охватывают значительно более широкупт-область, чем: реакции протонного обмена. Они охватывают; в частности, все процессы нейтрализации * (включая процессы, «аналогичные нейтрализации» в «в одо по дойных» растворителях); далекие сольволиза; затем реакции, аналогичные поведению-амфотврных гидроокисей; реакции, аналогичные образованию оснований из ангидро-оснований и кислот из ангидрокислот, л вообще всю область образования соединений присоединения и внедрения при реакциях раствороппого вещества с растворителем.--^
Способность растворителя вызывать электролитическую диссоциацию растворенных веществ, как правило, тем сильнее, чем больше его диэлектрическая проницаемость **. Правда, это справедливо только тогда, когда процесс диссоциации протекает одинаковым образом в сравниваемых растворителях, что по всегда имеет место (ср. стр. 772 исл.). В табл. 161 сопоставлены диэлектрические проницаемости и некоторые другие свойства перечисленных выше водоподббных растворителей. Из веществ,, приведенных в этой таблице, только фтористый водород имеет почти такую же высокую диэлектрическую проницаемость, как вода. Сероводород и двуокись серы отличаются от воды, помимо своей значительно меньшей диэлектрической проницаемости, также существенно большим молекулярным объемом, С этим, по-видимому, связано-то, что опи, как правило, значительно слабее действуют на растворенное вещество, чем фтористый водород, вода и аммиак. Насколько известно, эти растворители, если, по счптать экстремаль пых разбавлений, никогда не вызывают полной, а лишь частичку ю диссоциацию, Вещества, которые полностью гидролитически расщепляются водой,
* К реакциям протошюго обмена причисляют только те процессы нейтрализации (в обычном смысле), которые происходят при взаимодействии кислот и ангидрооснований. (
** Какая существует зависимость между способностью вызывать электролитическую диссоциацию и диэлектрической проницаемостью, выяснено еще не окончательно.
Реакции- е неводных растворах 767
Таблица ]01
Сопоставление некоторых свойств чводоподобпых» растворителей со свойствами воды
Вода Аммиак Фтористый водород Сероводород Двуокись ’серы
Молекулярный вес £8,016 17,032 £0,01 34,08 64,06
Температура плавления, “с 0,00 —77;" -83,1 --85,60 -72 „5
Температура кипения, °C 100,00 —33,35 +19,54 —60,7 5 -10,0
Удельный вес при температуре кипения 0,958 0,683 0,991 0,093 1,46
Молекулярный объем при 18,81 24,94 . 20,19 34,32 43,9
температуре кипения 83,6
Диэлектрическая нроницае- 87,8 22,0 6 (при 0° 13,8
мость (при 0°) (При—34°) (прп 0°) в жидком состоянии) (при +1.4, 50, В 1КИДКОМ СОСТОЯНИИ)"
Электропроводность я, 4-10-8 4-10-1» 5-10-1 4-10-11 11):
Q-UcAt-i (при 18°) (прп —15°) (при —15°) (при -78°) : (при 0°) -
Вяекость, с.н-1 - г. сек-1 0,002Э (прп 100°) 0,0026 (при -34е) — —' 9,0039 (при 0°)
Молярное повышение тем- 0,513 0 т 34 1,90 0,63 1,45
пера туры кипения в 1000 г растворителя, °C
Продукты диссоциации [ЩО]++ +СОП]- [№,]++ +[NH2J- 1Н2Г]++ +F- а [HsS]++[SHJ- [SO]2++ +ISO3]2-
а Ср. со сноской на стр. 773. • ' '
” ...' ' Г J ' ' '
в этих растворителях обычно лишь частично претерпевают превращения, аналогичные ГИДрс..;? ,-у.
Далее, в этих растворителях наряду с диссоциацией часто проявляется сильная ассоциация молекул растворенного вещества, В то время как способность присоединять молекулы растворителя, напротив, здесь выражена менее отчетливо, чем в случае воды и аммиака. Однако даже в жидком сероводороде и в жидкой, двуокиси серы происходит присоединение молекул растворителя к: молекулам растворенного вещества. (сольватация). Этот вывод был сделан на основании того, что из этих растворов нередко могут получаться продукты присоединения, хотя последние большей частью разла- - д? гаются уже прп комнатной или немного более высокой температуре.
Аналогии между превращениями, происходящими в «водрподобных» растворителях и в водных растворах, проявляются особенно отчетливо «в аммоносистеме», т. е. в жидком аммиаке и в «сульфитосистеме», т. е. в жидкой двуокиси серы. Здесь будут кратко рассмотрены основные виды подобных превращений.
Реакции, аналогичные нейтрализации. Подобно тому как в водном растворе для реакции нейтрализации характерно протекание процесса, выражаемого уравнением (5) на стр. 766 практически полностью справа налево, реакции, «аналогичные нейтрализации», характеризуются тем, что в них процессы, представляемые уравнением (6), также протекают справа налево. Так как вещества, которые в водном растворе образуют [Н3О1*-ионы (путем отщепления протонов), называются кислотами, а вещества, отщепляющие ОН'-ионы — основаниями, то вещества, которые прп диссоциации в «водоподобных» растворителях образуют соот-
768
Глава 18
-встствующие иопы, типичные для рассматриваемого растворителя, целесообразно называть (по предложению Лидера) кислотоподобншш и осно~ ^аниеподобными веществами.
Следовательно, «кислотоподобными» веществами в жидком аммиаке являются те вещества, которые образуют за счет протонного обмена с растворителем NHj-ионы, в ;стдком фтористом водороде — те вещества , которые образуют J H*>F)+-иоиы, в жидком сероводороде — те, которые образуют [H3S]+-ixohh, а в жидкой двуокиси серы — те, которые отщепляют [8О|'-+-ноны. «Основаниеподобнымн» веществами в рассматриваемых системах даляются те вещества, которые образуют NH;-, F--, SH"-, [803]-_-иопы соответственно. Ветцества, аналогичные кислотам и основаниям:, реагируют между собой;в обсуждаемых системах с образованием NH3, HF, HaS и SCU в согласии с уравнением (6) на Стр. 766. При этом точно так же , кар п в водном растворе, образуются соли.
В воде; НС14-К{ОИ]=Ц2О-{-К.С1,
[1<3ОИСЮ4] + К[ОН]==2НгО + К[С1О4].
В аммиаке: [ЫН^С1-ГЩА’ТЫ~2МНз-|-КС1.
Б двуокиси серы: [SOjCI2-|-.K2[SO3]=2SO2q-2.KCI,
[SOl[SCNh+K2[SO3l=2SO2+2K{SGN], [80]Вгг+[(СИ3)4М]г[50з]=230г+2[(СНз)4Б’]Вг.
Сольволиз. Гидролизу в водной системе («аквосистемеа) соответствует аммонолиз в «аммоносистеме», и, вообще, сольволиз в водоподобпых системах. г
Гидролиз: SiCi4+4H2O=4HCl+Si(OH)4 —> S1O2+2H2O.
Аммонолиз: SiCl4-y8NH3—4[NIl4]Cl-ySi(NH2)4’Si(NH)a-|-2NH3.
Гидролиз: GeCl4 + 2H2O —4HCl + GeO2.
Аммонолиз: GeCl4 + 6NH3= 4[NH4]C1 + Go] NH]2.
Ансольвокислоты и ансольвооспопання. Аксо.гъвокислотами, соответственно ансолъвооснованиями, называют по аналогии с ангидрокислотами и ангидро основаниями такие соединения, из которых кислого подобные, соответственно основапиеподобные, соединения образуются только при присоединении молекул растворителя:
Ангидрооснование —> основание; NH3-|-П[ОН] =[NH4]+4-[OH]_.
(В иодаой системе)
А нсольвоо снование —> о снование подобное соединение:
2NH3 + [SO][SO3|=[OS(NH3)2]®+ + {SO3P-.
(В сульфите-системе)
Апсель весе кование —> основан неподобное соединение:
, H2O4-HF=[H3O]+ + F-
(В жидком фтористом, водороде).
Очень многие вещества в отношении жидкого фтористого водорода функционируют как ансольвоо снования (ср. стр. 772 и сл.).
Поведение амфотерных гидроокисей. Поведению амфотерных гидроокисей относительно сильных оснований в водном растворе аналогично поведение соответствующих веществ в «в о до подобных» растворителях. Подобно тому как из водного раствора хлорида алюминия при действии небольшого количества едкого кали сначала выпадает гидрат окиси алюминия, который затем растворяется в избытке едкого кали с образованием гядроксоалюмпяата калия, хлористый алюминий в жидкой двуокиси серы взаимодействует с сульфитом тетраметил аммония, являющимся в этой системе «основание-подобным» веществом, сначала с образованием труднорастворимого сульфита алюминия (также «основапиенодобного» соединения), который затем при добавлении избытка
Реакции, в неводных растворах
сульфита тетраметил аммония снова переходит в раствор в виде сульфит о алюминат а:
А1С1з+ЗК(ОИ) = А1(ОН)з+ЗКС1; А1(ОН)з + ЗК(ОН) = К3[Л1(ОН)в].
А1С13Ч-3/2[(СН3)4Х)2[ЗО3]^173А1е[ЗО3]3 + 3[(СН3)^]С1,
1/2Al2[SO3]3 + 3/2[(CII3)4N]2tSO3] = [(CH3)4NI3[Al(SO3)3].
В рассматриваемом примере аналогия особенно глубокая: в обоих случаях образуются желеобразные осадки, отличающиеся высокой адсорбционной способностью. Подобно гидрату окиси алюминия, выпавшему из водного раствора, сульфит алюмипия, выпавший из жидкой двуокиси серы, при соприкосновении с раствором постепенно переходит в труднорастворимую форму.
Примером аналогичных превращений в жидком аммиаке может служить образование тетраамидоцинката калия:
ZnI2-HKNH2=K2[Zn(Ali2)4]+2KL
Двойные обмены солей. В качестве солей в «водо подобных» растворителях выступают такие электролиты, которые е растворе не имеют общих ионов с растворителем. В соответствии с этим соединения аммония в «аммоносистеме» являются не солями, а аналогами гидратов кислот в «аквосистеме». Соответственно при рассмотрении превращений в жидком фтористом водороде такие фториды, как NaF, считаются не солями, а веществами «аналогичными основаниям» и т. д. Определяющим свойством для двойных обменов солей в «водоподобных» растворителях, так же как в водном растворе, в основном является их различная, растворимость. Так как отношения растворимости в таких растворителях часто существенно отличаются от отношений растворимости в водном растворе, подобные превращения часто с пользой применяют для препаративных целей. С препаративной точки зрения иногда бывает полезно также то обстоятельство, что при превращениях в этих растворителях соли значительно меньше разрушаются за счет сольволиза, чем в воде (ср; стр, .766) .
Рассмотрение некоторых «водоподобных» растворителей
В дальнейшем будут подробно рассмотрены важнейшие свойства названных выше «водоподобных» растворителей и некоторые особенно характерные для них превращения.
Жидкий аммиак является хорошим растворителем для многочисленных неорганических й органических веществ. Он занимает особое место среди «водоподобных» растворителей, так как легко доступен, относительно дешев и имеет разностороннее применение; кроме того, проведение эксперимента в жидком аммиаке не представляет существенных трудностей.
Исследование химических превращений, протекающих в жидком аммиаке, началось с работы Вейлса (WeyJs, 1864). Установлено, что щелочные металлы растворимы в аммиаке. Широкое исследование растворяющей способности жидкого аммиака было проведено Горе (Gore, 1872—1873) я Дайверсом (Divers, 1873); Последний исполь-зовал в Своих опытах не чистый сжиженный аммиак, а раствор нитрата аммония в аммиаке, который образуется непосредственно при действии газообразного аммиака па нитрат аммония ори обычной температуре (лучше при 0°), подобно тому как нри действии водяного пара, соответственно влажного воздуха, па хлористый кальций можно получить раствор хлористого кальция. Систематическое иаучеппе поведения жидкого аммиака в качестве растворителя и превращений, происходящих в нем, проводили начиная с 1898 г. Франклин (Franklin) и Краус (Kraus), а позднее и другие 49 г. Ремя
770
Глава. 18
(преимущественно американские) исследователи. Сравнение с процессами, протекающими в водных растворах, привело Франклина (1912) к обнаружению «а мм оно системы» соединений, которые при растворении в сжиженном аммиаке ведут себя по отношению Друг к другу как кислоты, основания и соли в воде («аквосистемы»),
«Основаниеподобные» вещества в аммонос истеме, по мпениго Франклина, могут быть кратко названы аммонооснованиями, а «кислотоподобные вещества» — аммонокислотами. В качестве аммонооснований функционируют те NH3-соединения, которые в жидком аммиаке отщепляют NHo-попы. Это в первую очередь амиды металлов. В качестве аммонокислот функционируют те соединения, которые образуют в жидком аммиаке ионы за счет отщепления протона. В основном это те же соединения, которые функционируют и в водных растворах как кислоты. Однако следует учесть, что аммонийные соединения [NHJX, относящиеся в «аквосистсме» к солям, в «аммоносистеме1> играют ту же роль, что и гидраты кислот в «ак в о системе». В соответствии с этим соли аммония функционируют в жидком цмлшяке небо б но кисло ига.ч в водном растворе.
Типичным примером кислого подобного поведения солей аммония, растворенных в жидком аммиаке, является способность последних участвовать в реакциях следующего типа:
Mg2Si -Ь 4NH4Br = 2MgBr2 + 4NH3 + SiH4 (Johnson, 1935).
Амиды органических кислот и большая часть амидов неметаллов также имеют характер аммонокислот, подобно тому как соответствующие гидр оксосое дийопия в водных растворах функционируют как кислоты. Органические амины л аммоносистемах соответствуют спиртам в аквоснстемах. Имиды и имины находятся в таких же отношениях к амидам и аминам в аммоносистемах, как окиси к гидроокисям в акво-снстемах, с той разницей, что они способны далее отщеплять аммиак и превращаться в нитриды, которые можно рассматривать в качестве аналогов окйслов.
Подобно воде аммиак обладает ярко выраженной способностью присоединять другие вещества с образованием продуктов присоединения (образование аммиакатов). Со времени применения жидкого аммиака (с которым часто образуются более высокие аммиакаты, чем с газообразным аммиаком) знания об этом классе соединении существенно расширились.
Высокое сродство жидкого аммиака к вебе часто позволяет путем обработки жидким аммиаком удалять воду из таких веществ, из которых опа не может быть удалена непосредственно без глубокого нх разложения («Методы аммиачной экстракции», ср. т. I, стр. 394 и 537).
Кроме того, аммиак обладает способностью вызывать аммонолиз соединений (ср. стр. 768). Этим путем можно получись амиды и имиды, а отщепляя от них аммиак—нитриды. Образование нитридов таким образом полностью соответствует образованию окйслов из гидроокисей.
Образованию основных солей в водных системах при гидролизе соответствует следующее превращение (сольволиз):
2Pbl3+6NH3=Pb(NH2)2-Pb(NH2)I-i-3NH4I.
Элвкюрсыптичсскся Succoциация типичных сален в жидком аммиаке в общем слабее, чем в водном растпоре. С другой сторопЫ, имеются соединения, которые электролитически диссоциируют при растворении в жидком аммиаке, в то время как в водных растворах их диссоциация в заметной мере не происходит. Например, ацетилен, растворенный в жидком аммпатсе, обладает отчетливой электропроводностью. Еще лучше проводят ток бензальдегид CBlTsCIIO и пи тр о метан CH3NO2, Растворы других нитро со единений в жидком аммиаке также имеют значительную электропроводность.
Следует отметить высокую электропроводность растворов щелочных и щелочноземельных металлов в жидком аммиаке. Эти растворы большей частью окрашены в глубоко-синий цвет. Щелочноземельные металлы находятся в растворе в виде аммиакатов, например Ca(NH;J)e. В какой форме присутствуют в растворе щелочные металлы.
Реакции в неводных растворах ' 1 771
пока еще окончательно не выяснено. Раствор щелочного металла в аммиаке более или мепсе быстро (раствор цезия менее чем за минуту, раствор натрия — только в течение месяца) превращается в бесцветный раствор амида щелочного металла М1 NH2.
В общем растворы металлов в аммиаке очень реакционноспособны. Например, при действии О9 на щелочные металлы, растворенные в жидком аммиаке, образуются соответствующие перекиси и наднерекиси (ср. т. I, стр. 202), при действии NO — гипонитриты M.jN2O2, при действии РНЯ — фосфиниды М1РН2. Разнообразные превращения протекают и с органическими соединениями, например с веществами кислотного характера происходят реакции следующего вида:
(С6Н6)3СН+Na = (CBH6)3CNa + ЧгНг.
ЗСгНг + 2Na = 2NaHCa + С2Н4.
С органическими галоидными соединениями (RX) раствор натрия в жидком аммиаке реагирует с образованием галогенидов металла: RX -j- Na = NaX Ц- R. Освобождающийся прн этом органический радикал R взаимодействует или с растворителем, образуя амин (RNII2), имин (R2NH) или нитрид (R3N) и углеводород (RII), или же два радикала R могут соединяться, образуя высший углеводород R — R. Однако часто свободные радикалы сохраняются в растворе. Возможность получения органических и неорганических радикалов при обработке галоидных соединений натрием, растворенным в жидком аммиаке, часто используют в настоящее время. При помощи аналогичных реакций удалось получить неорганические соединения с относительно длинными цепями, например
Sn(CH3)3.Sn(CH3)2.Sn(CH3)2.Sn(CH3)2.Sn(CH3)3.
Так как в кислого подобных органических соединениях, таких, как амиды кислот, имиды, фенолы, тиофенолы, спирты, гидразип и т. д., атомы И могут непосредственно замещаться металлом, растворенным в жидком аммиаке *, а металл замещенное соединение легко взаимодействует в том же растворителе с алкил галогенидами, жидкий аммиак является очень удобным растворителем для алкилирования. Кроме того, жидкий аммиак является очень подходящим растворителем для соединений азота и проведения реакций с ними (Audrieth, 1930). Этот факт объясняется тем, что аммиак находится в таких же отношениях с этими соединениями, как вода с кислородными соединениями, Для которых, как известно, она обладает специфической растворяющей способностью. Нередко в жидком аммиаке можно наблюдать электролитическую диссоциацию таких соединений, для которых в воде диссоциация невозможна вследствие того, что они полностью разрушаются водой, как это, например, имеет место в случае амида натрия (NaNH2) и мотюацотилида патрия NaMC2.
Некоторые интерметаллические соединения также растворяются в жидком аммиаке, претерпевая при этом электролитическую диссоциацию, Например, Цинтль (Zintl, 1931) нашел, что соединение NaPb3 растворяется в жидком аммиаке с образованием [Na(NH3)3|4 [РЪ0] **. Апион (РЬЙ]4“ этого солеобразного соединения заслуживает особого внимания как вследствие его аналогии с полисульфяд- и полииодид-нонами, так и потому, что его существование свидетельствует о том, что свинец может быть отрицательно заряженным, что и следует ожидать на основании его поло линия в периодической системе. В растворе ионы (РЬЭ]4“ агрегируются в частицы, различимые под ультрамикроскопом. Соответствующим образом Цинтль сумел получить соединения fNa(NH3)gJ/1|Sa,,| п lNa(Nl-Is)el3[Sbi]. Если удалить нэ mix аммиак, то останутся несо-леобразно построенные сплавы.
Растворением металлов или солей металлов в жидком аммиаке можно также получать интерметаллические соединения непосредственно из растворов. Например:
9Na+4Zn(CN)2=NaZn4+8NaCN.
Для осуществления двойного обмена в жидком аммиаке (так иге как в водном растворе) решающее значение имеет соотношение растворимостей веществ, участвующих н превращении. Естественно, здесь, так же как и в водных системах, имеется большое число реакций, протекающих
* Вместо Щелочного металла можно также использовать ого амид, растворенный в жидком аммиаке, Взапмолепсттше последнего с кислотоподобными соединениями в жидком аммиаке соответствует взаимодействию оснований и кислот в воде.
** Если соединение содержит больше 9 атомов РЬ па 4 атома Na (ср. т. I, стр. 614), то часть свинца остается перастворенной.
49*
772
Глава 18
в гомогенной фазе и обусловленных другими причинами, например соле-образование из кислото- и основанной од обпых веществ (соответственно взаимодействие аммонийных солей и амидов металлов), обусловленное стремлением NI-I* и NH'-ионов объединиться с образованием 2NH3.
Приведенные ниже уравнения представляют собой примеры двойного обмена в жидком аммиаке. Так как протекание реакции до конца обусловлено нерастворимостью одного из продуктов реакции, соединения, нерастворимые в Ягидком аммиаке, выделены жирным шрифтом.
Ba(NO3), + 2AgCl = Bad 2 J- 2AgNO3, <
Ba(NO3),-|- 2КОСЙН5 = Ва(ОС2Н5)з 4.2KNO3,
AgNH24HN=C(NH2)3=AgN=C(NH2)2-i-NH3,
AgNO3+KNH2=AgNHa4.KNO3,
Pb(NO3)24 2KNH2 = PbNH 4.NH3 + 2KNO3,
3rig(NO3)a+6KNH2 = Hg3N2 + 4NH34 6KNO3.
Трудпорастворимые соединения можно перевести в легко растворимые за счет комплексообразования, как это происходит и в водных системах. Например;
Zii(NH3)2 + 2KNH2+ K2[Zn(NH2)4J.
Растворимость неорганических соединений в жидком аммиаке зависит от теплоты сольватации, подобно растворимости в воде. Примерами для сопоставления растворимости в жидком аммиаке и в воде могут служить данные, приведенные в табл. 102.
Таблица 102
Растворимость солей щелочных металлов в жидком аммиаке и воде, [в молях соли на 1000 г растворителя (Linliard, Stephan, 1932)]
Растворитель NaCl NaBr Nal NaNOs j KCI KBr KI KNO3
Жидкий аммиак 2,20 6,21 8,80 15,00 0,0177 2,26 11,09 1,04
Вода 6,10 . 7,71 10,72 8,62 3,76 4,19 7,72 1,30
Жидкий аммиак был с успехом использован и в качестве растворителя для электролитических целей. Некоторые металлы, электролитическое осуждение которых из водных растворов солей невозможно, осаждают из растворов их солей в жидком аммиаке. Примером может служить бериллий (Booth, 1930—1931). Из растворов в жидком аммиаке электролитически можно осадить и свободные радикалы, такие, как HgCH3 (Kraus, 1913) и Cr(CgH3)4 (Hein, 1926). Если электролизу подвергнуть раствор четвертичной соли аммония [NRJX, растворенной в жидком аммиаке, то у катода появляется глубоко-Сипее окрашивание, характерное для растворов щелочного металла в жидком аммиаке. Из этого факта был сделай вывод о том, что в подобном растворе радикалы NR4 могут находиться преимущественно в свободном состоянии (Schlu-bach, 1920).
*
Жидкий фтористый водород. Свойства жидкого фтористого водорода в основном изучал Фреденхаген (Fredenhagen, 1928 и сл.). Несмотря йа то что диэлектрическая проницаемость жидкого фтористого водорода (ср, табл. 101, стр. 767) не достигает диэлектрической проницаемости воды, он значительно превосходит воду по своей растворяющей способности и влиянию на электролитическую диссоциацию. HF не только растворяет многочисленные неорганические вещества, но и является превосходным растворителем для большого числа органических соединен ни. Относительно веществ, способных к электролитической диссоциации, он часто ведет
Реакции в неводных растворах
773
себя существенно иначе, чем вода. Очень многие вещества, которые в водном растворе обладают ярко выраженным кислотным характером, взаимодействуют с жидким фтористым водородом, подобно тому как аигидрроснования реагируют с водой. Поведение жидкого фтористого водорода относительно солей также в основном обусловлено его особен-' иостями.
Полагая, что собственная диссоциация жидкого фтористого водорода осуществляется по уравнению * 2HF [HZF]+ + F", фториды, способные к отщеплению ионов F-, с точки зрения превращений, происходящих с ними в жидком фтористом водороде, следует рассматривать в качестве основание подобных» веществ. Соединений, которые функционируют в жидком фтористом водороде, как «кислотоподобдыс» вещества, известно очень немного.
То, что жидкий фтористый водород своим поведением относительно многих веществ сильно отличается от воды, основано па его существенно болёе сильном в сравнении с водой кислотном характере. Многим веществам, у которых вода вырывает протоны, фтористый водород отдает протоны. Так, со спиртами н фенолами он реагирует следующим образом: **
ROH + HF [ROiy+J-F-.
Фенолы, функционирующие в водном растворе подобно кислотам, по отношению к жидкому фтористому водороду обнаруживают поведение, аналогичное поведению ангидро о снова нпй (пан ример, NEIa) относительно воды. Подобно тому цак последние под влиянием воды переходят и настоящие основания, фенолы при взаимодействии с IIP превращаются в «основапиенодобпые» вещества (ROII2]F. Соответствующим образом реагирует жидкий фтористый водород с водой, муравьиной кислотой, уксусной кислотой, а также с ацетатами и даже нитратами;
H20 + HF = [H30]++F- II CH3.CO.OK+2HF=K+4.[CH3 C(OH)3]+4-2F-
HCOOII-f-HF=(HC(QII)2l+4.F-
CH3-COOH4-HF = [CH3-C(OH)21++F-|| KNO34.2HF = K+4.[N0(OH)2l+4-2F-
Реакции с названными выше солями соответствуют реакции воды с амидами металлов: KNH2 + 2Н0Н - К++ [NH4]+ 4- 20I-I",
с той только разницей, что при этой реакции наряду с сильным основанием КОН образуется слабое основание NH3-H2O, в то время как в жидком фтористом водороде оба «основаниеподобных» вещества — KF и [CH3CO(OII)2jF или [NO(OH)21F — силь- „ но диссоциированы. '1
Использование жидкого фтористого водорода в качестве растворителя влечет за собой существенно большие экспериментальные трудности, чем те, которые появляются при работе с жидким аммиаком. Однако особый характер воздействия фтористого водорода на растворенные
Поведение фтористого водорода в водном растворе (ср. т. I стр. 839) делает более вероятной гипотезу, в соответствии с которой собственная диссоциация жидкого фтористого, водорода осуществляется преимущественно по уравнению ЗН F 2 [H2F]+ 4* 4- [HF2]“, Если это справедливо, то в качестве «основапненодобных» веществ в системе жидкого фтористого водорода следует рассматривать не нормальные фториды MIF, а гиДрофторпды Mi [HF3J. Нормальные фториды в этой системе тогда будут соответствовать основным ангидридам (окислам) в водной системе. Все же образование [НР2]_-нонов при диссоциации жидкого фтористого водорода пока еще не доказано. До сих пор превращения, протекающие в пем, интерпретируют с точки зрения существования f^-HOTTOB.
** Наряду с этим иногда частично протекают и другие реакции [ROHS]+ 4-4- F~ RF 4- Н2О; Ц20 4- HF [Н3О]+ 4- F”, которые ведут к дальнейшему повышению Электропроводности соответствующих растворов.
774
Глава IS
в нем вещества, обусловленный его сильной кислотной природой, свидетельствует о том, что его применение (особенно для изучения органических соединений) даст новые важные результаты jcp. Fred ев hagen, Z. Elektrochem., 37, 684 (1931)1.
Жидкий сероводород. В соответствии с относительно большим „молекулярным объемом и низкой диэлектрической проницаемостью (см. табл. 101, стр, 767) жидкий сероводород не обладает такими высокими растворяющей и ионизирующей способностями1; как рассмотренные выше растворители. В нем растворимо лишь очень огра-ничоппоо число неорганических соединений. Несколько лучше в жидком сероводороде растворяются органические соединения. Из неорганических соединений растворимы, например, некоторые галогениды, такие, как РС15, РС13, Р13, РВг3 и BiCl3. Их растворы обладают очень слабой электропроводностью. Существенно лучше проводят ток растворы ярко выраженных оснований, например органические амипы. Характерной особенностью этих растворов является понижение молярной электропроводности с разбавлением, В некоторых случаях происходит разрушение соединений, растворённых в жидком сероводороде, за счет взаимодействия с растворителем. Последнее идет, однако, чрезвычайно медленно, даже при комнатной температуре (в занаяпной ампуле), иногда только в течение нескольких педель.
Едва ли можно ожидать, что жидкий сероводород со своими мало подходящими для растворителя свойствами, к которым добавляются экспериментальные трудности, обусловленные низкой температурой его кипепйя, пайдет практическое применение в какой-либо области в качестве растворителя. Поэтому ему посвящены лишь немногие исследования. В частности, пе изучен вопрос о превращениях растворенных в пем веществ. Растворами в жидком сероводороде занимался главным образом Вилкинсон (Wilkinson, 1925 и сл.)
Жидкая двуокись серы. Жидкая двуокись серы, как установил еще Вальдеп (Walden, 1920 и сл.), обладает хорошей растворяющей*способностью для многих неорганических и органических веществ. Из неорганических соединений в ней достаточно легко растворимы главным образом иодиды, бромиды, хлориды, роданиды и ацетаты некоторых легких и тяжелых металлов, а также большинство солей замещенного аммония. Легкорастворимыми органическими соединениями являются прежде всего вещества, содержащие кислород и азот, из углеводородов легко растворяются лишь циклические и ненасыщенные. Различную растворяющую способность жидкой двуокиси серы по отношению к разным группам углеводородов используют для очистки нефти по методу Эделеану.
Превращения веществ, растворенных в жидкой двуокиси серы, систематически исследовал главным образом Яндер (Jander, 1936 и сл.). При этом оказалось, что они протекают вполне аналогично превращениям в водных растворах, если учесть, что в этом растворителе соединения тиопила SOX2 функционируют как «кислотоподобные», а сульфиты Mi[SO3] как «основаниеподобные» вещества (ср. стр. 768). То, что они подобно кислотам и основаниям в водном растворе взаимно «нейтрализуют» Друг друга, Яндер сумел доказать, в частности, кондуктометрическим титрованием. Степень диссоциации «кислотоподобных» и «основание-подобпых» веществ в жидкой двуокиси серы соответствует степени диссоциации слабых кислот и оснований в воде.
Вода ведет себя в жидкой двуокиси серы так, как аммиак в водном растворе, т. е. она функционирует как ангидрон слова пне: 2Н2О + 2SO2 Д [(H20)oS0]a+ 4- [SO3]3-. Образующееся при этом «основапиеподобное» соединение [(H20)2SO) [SO3j дает при взаимодействии с «i;n Слотоподобными» веществами соли:
[(H2O)2SO][SO3]+SOBr2 = [(H2O)zSO)Br2 + 2SO2
за этим превращением можно следить кондуктометрически). Подобно тому как нерастворимые в воде соли, присоединяя аммиак, превращаются в растворимые комплексные соли, в жидкой двуокиси серы тот же процесс осуществляется при присоединении воды: Co(SCN)2 + 2Н2О == [Co(OH2)2](SCN)2.
Реакции в неводных растворах 775
В качестве примера салъволшга в жидкой двуокиси серы следует привести пре-вращение диэтил цинка: Zn(C3H5)3 + S03 ~ ZnO + О8(С2Н^2- Однако жидкая двуокись серы сольватирует растворенные в ней вещества много слабее, чем вода. Например, очень склонные к гидролитическому расщеплению вещества, такие, как SiCl4, SnC]4 и SbCl-, растворяются в ней, не подвергаясь в заметной степени сольволизу.
Другие «водоподобиые» растворители. Свойства других растворителей, ведущих себя в жидком состоянии как более или менее отчетливо «водоподобные», приведены в табл. 103. Растворители расположены в ней в порядив убывания диэлектрических проницаемостей. Особенно высокой диэлектрической проницаемостью отличается синильная кислота. Наиболее употребительным и хорошо изученным в отношении взаимодействия с растворенными веществами растворителем является безводная уксусная кислота (ледяная уксусная кислота).
Таблица 103
Свойства некоторых растворителей, способствующих диссоциации
Назнание Формула Д калек греческая проницае- 1 мость при 25* | Собственная влектро-про КОЛКОСТЬ, Я-1-(мг~1 при 25° Вязкость, Т.Н—1' в сет.~1 при 25’ Температура плавления, °C Температура кипения, °C
Циап истый водород HCN 152 (при О’) 4,5-10—7 0,00242 (При 04 — 15 £6.Ь
Формамид НСОХНг 2- 10-7 9,9077 (при 105’) 2 105 (при 11лен рт. ст.)
Аэотиэя ивслота HNO, >81 (при О’) 1,3в’ 1W (111)11 0е) 0,6227 (при О’) -41,1 84
Муравьиная в и- нсоон 58 6,3-10-ь 0,9163 8,43 100,6
слота
Гидрааин nsh4 51,7 2,5-10-е 0,90005 i.i 113,5
Метанол СП зон 30,3 6-10-8 0,00546 -97,8 64,7
Оти^ювый CUTlpT С3НВОН 23 9-10-» 0,01084 -114,1 7813
Адеток снгсосн3 21 5,5-10-8 9,00316 -94,3 56,3
Пир иди Н CeHsN 12,5 4,5-10-8 0,00882 -50 115,4
X пористый водород НС! 10,2 (при —108°) 2.10-1 (при -ЮО’) 0,00504 (при -100°) -114,8 -84,9
Уксусная кисло- CHsCOOH 7 610-е 0,0116 16,7 118,1
та
Фосгся С0С12 i,3 — — —126 8,2
Теория кислотно-основной функции Льюиса. Для многих целей удобно рассматривать реакции кислот и оснований, а также родственные процессы независимо от растворителя. Это облегчено теорией кислотно-ос нов ной функции, предложенной Льюисом. В соответствии с этой теорией кислый характер вещества обусловлен способностью одного из его атомов объединяться с другим веществом за счет захвата электронной пары этого вещества, необходимой для образования гомеополярной связи. Основной характер, но Льюису, связан со способностью предоставлять пару электронов для образования гомеополяр ной связи. В соответствии со сказанным амфотерными веществами являются те, молекулы которых могут как захватывать, так и предоставлять пару электронов для образования гомеополярной связи.
Следовательно, реакция кислоты с основанием в водном растворе, соответственно реакция иопа гпдроксония с ионом гидроксила, основана, по Льюису, на следующем процессе;
+ =Н:О--Н*-О:Н -----> 2HsO.
L Н 1 ’’ н
При рассмотрении процесса нейтрализации Льюис обращает внимание не на'образование конечного продукта (Н2О), а на образование промежуточного продукта реакшз
776
Глава 18
(гипотетического строения). Если рассматривать реакцию кислоты и основания в код-ком растворе с этой позиции, то очень легко обнаруживается полная аналогия между ней и солеобразоваиием, протекающим без участия растворителя, например образование NH4C1 из HCI и NH3 (взаимодействие кислоты с ангидрооснованием) или образование СаСО3 из СО2 и GaO (взаимодействие ангидрида кислоты с ангидридом основания):
= sCUHsjfcH ----» [СЛГСКВД*;
Н ‘ н
(8)
(9).
О::С
Совершенно аналогично трактуются, по теорий Льюиса,. реакции кислото подобных и о снов анион одр бпых веществ, например:
’:O:S16:T” .Q: sSiOtSsOs'
":О:" —.......:О:“
--->2SO2.
(10)
Аналогичными схемами можно представить многие виды комплексообразования. Следовательно, их можно рассматривать но теории Льюиса, как особый случай кислотно-основной функции; так, образование (ВГь]_-йона п образование продукта присоединения В F3 it NH3 можно представить следующим образом:
Л?: :К:
Ё:;В 4- [:£:]“ = , 01)
Ф •?= J
Н :F= Н
& :В + :N;H= :Е:В : N = H
:К: Н ”.F: И
Аналогии, выявляемые при помощи теории Льюиса в реакциях, представленных уравнениями (7-^12), позволяют понять многие процессы, которые играют существенную роль, особенно it органической химии. Так, действие хлористого алюминия в качестве катализатора определенных реакций (например, синтеза Фриделя — Крафтса) становится понятным; если учесть, что А1С13 роднит с [ Н3Ор-прпом, являющимся, как известно, превосходным катализатором, способность присоединять пару электронов с образованием гомеополярной связи. ,
Взаимодействие кислот и оснований и аналогичные процессы отличаются от окислите льно-восстановительных реакций: так, восстановитель отдает электроны, которые присоединяет окислитель, в то время как при реакции веществ основного характера с веществами кислотного характера, несмотря па то что одно на них предоставляет: другому пару электронов для образования связи, в. образующемся благодаря этому соединении эта электронная пара является общей.
Кислоты реагируют с основаниями моментально, так что зависимость процесса от температуры Не имеет значения. Однако имеются случаи, когда для осуществления реакции необходима энергия активации и вследствие этого превращение протекает со скоростью, зависящем от температуры. Льюис (1939) предположил, что в таких случаях существуют две электромерные формы, не находящиеся в резонансе друг с другом (как это обычно имеет место для электромерных форм), из которых только одна функционирует как кислота или основание.
Реакции в расплавах
К растворителям в широком смысле прина длежат также расплавленные вещества. Поскольку при этом речь идет об индифферентных веществах, которые применяют иногда для определения молекулярного веса (например, камфора), то для них справедливо все то, что сказано для индифферентных растворителей в обычном смысле, от которых эти вещества отличаются лишь более ^высокими температурами затвердевания.
Реакции в неводных растворах,
777
По-другому обстоит дело с расплавленными солями и подобными им веществами,, р Дсп лавы которых обнаруживают заметную алектропроводность. Оли занимают особое положение не только относительно индифферентных растворителей, но и относительно «во до подобных» растворителей, собственная электропроводность которых в общем очень мала но сравнению с электропроводностью растворенных в них электролитов. Из веществ, жидких при обычной температуре, к «солевым расплавам», вероятно, следует причислить безводную азотную кислоту ([ON(OH)2]+ [N0s]“, ср. т, I, стр. 646 и сл.),
В сияли с большим техническим аначеиисм расплавов металлов для электролитического получения металлов (особенно легких) условия осаждения их из расплавов многократно изучались. Напряжение разложения чистого вещества в расплавленном состоянии в общем пе равно напряжению разложения его, если оно растворено-в расплаве другого вещества, так как и здесь справедлив закон, по которому напряжение осаждения зависит от растворителя. Влияние, которое оказывает состав-расплава на напряжение разложения растворенных в нем веществ^ как правило, невелико. однако при точных измерениях пренебрегать им не следует.
Изучение взаимодействия расплавленных металлов с расплавленными солями или окис лают, находящимися-с ними в соприкосновений («равновесие вытеснения»), также имеет техническое значение в связи с использованием обменного взаимодействия металлов и шпаков в металлургических процессах. В общем к равновесиям такого-вида можно сразу применять химический закон действия масс и закон распределения Мер иста, без учета отклонений от идеальных законов, которые могут быть вызваны ассоциацией молекул в расплаве. Однако во многих случаях заметные отклонения от идеальных законов все-таки проявляются. Нередко в подобных расплавах ионы металлов и неметаллов присутствуют в ассоциированном состоянии. Так, на основании наблюдений за скоростями вытеснения Еллииек (Jellinek, 1933 и сл.) сделал вывод о существовании в расплавах соней ионов Li.?+, СН_, С1|_, Вг|~ и
Глава 19
РЕАКЦИИ ТВЕРДЫХ ВЕЩЕСТВ
Общие сведения. Реакции твердых веществ, если к ним отнести все реакции, в которых участвуют (расходуются в ходе реакции) твердые вещества, делятся на две группы: реакции, которые происходят только на поверхности твердого вещества, и реакции, протекающие внутри твердого вещества. Из этих двух групп реакций целесообразно выделить вторую *, назвав ее «реакции в твердых веществах». А такие реакции, в которых участвуют исключительно твердые вещества (и, следовательно, пе участвуют ни жидкости, ни газы), называют «реакциями между твердыми веществами».
Примерами реакций, которые происходят па поверхности твердых веществ (с затратой последних) являются: сгорание С в кислороде воздуха с образованием СО2, растворение Zn в соляной кислоте, ржавление железа. Как показывают эти примеры, в реакциях этой группы могут образовываться газообразные, жидкие (соответственно находящиеся в растворе) и твердые продукты реакций. Одвако при образовании твердого продукта реакции превращение может дальше развиваться на поверхности твердого вещества, вступающего в реакцию, если продукт реакции достаточно порист, чтобы допустить дальнейший приток газов или жидкостей к поверхности.
Непосредственное взаимодействие двух твердых веществ без участия газообразных, жидких или растворенных веществ, возможно только тогда, когда превращение может продолжаться внутри твердых веществ. То же справедливо и в том случае, если при взаимодействии твердого вещества с газом, жидкостью или растворенным веществом образуется продукт реакции, покрывающий поверхность компактным слоем.
Реакции, которые происходят на поверхности твердых веществ, давно известны. Между тем на тот факт, что реакции могут разыгрываться также в твердых веществах, ранее мало обращали внимания, хотя к таким -У реакциям принадлежит термическое разложение твердых веществ, протекающее с выделением газов; многие из этих реакций, такие, как разложение СаСО3 на СаО и СО2 при нагревании, с древнейших времен применяют на практике. Реакции же происходящие только, между твердыми веществами, в которых, следовательно, в качестве исходных, промежуточных и конечных продуктов выступают исключительно твердые вещества, часто считали ранее даже невозможными. Несмотря на то что в отдельных случаях подобные реакции также уже давно нашли практическое применение — например, при цементации железа и при его обезуглероживании (ср. стр. 280) — все же до недавнего времени твердые вещества считали, как правило, малорсакциопноспособными в соответствии со старой поговоркой: «Corpora non agunt nisi fluida» **.
* Для реакции первой группы до сих пор отсутствует краткое паз ванне. Под термином «реакции на твердых веществах» понимают такие, которые разыгрываются на твердых: веществах, без использования последних в ходе реакции, т. е., следовательно, реакции, протекающие па катализаторах.
** Тело не деятельно, если опо пе жидкое (лат.) — Прим, перев.
Реакции, твердых веществ
779
Хотя и в настоящее время существует представление о том, что в общем газы и жидкости гораздо более реакционноспособны, чем твердые вещества, одпако известны многочисленные случаи, когда реакции, между твердыми веществами протекают очень быстро и с сильным выделением тепла. И несмотря на то что это обычно происходит при высокой температуре, однако все же значительно пиже температуры плавления. Систе-матическое изучение подобных реакций и связанных с ними явлений « не только существенно углубит общие знания о природе кристаллического состояния и о механизмах химических реакций, но также позволит решить важные технические проблемы.
К реакциям, протекающим в твердом состоянии, относится также электролитическое разложение твердых солей и других подобных соединений. Как установили Бруни (Bruni, 1913) и Тубандт (Tubandt, 1920 и сл.), многие соли проявляют повышенную электропроводность значительно ниже температуры плавления, например Ag! уже при l iti°. Скорость перемещения иона серебра в твердом соединении Ае! при этой температуре примерно равна скорости перемещения иона серебра в водном растворе нри 25°. При этом следует отметить, что, как правило, в переносе электричества участвует только один из двух видов ионов, иэ которых построены кристаллы соединения, в то время как ионы другого вида остаются неподвижными на своих Местах.
Исторические сведения. Процессы, которые плгеют значение в реакциях, происходящих на поверхности твердых веществ, были установлены еще в период становления физической химии. Превращения же, протекающие в твердых веществах, и другие нрсврапцпгЕп, прежде всего химические реакции, разыгрывающиеся только между твердыми веществами, систематически стали исследовать в более позднее Время. Характерные для этих реакций процессы были изучены впервые Тамма ном (Татктапп, 1905 п СИ.) на металлах. Доказательство диффузии твердых металлов ДРУГ в друга было получено Бруни (Bruni) только в 1911 г. на основании снижения электропроводности, вызванного образованием твердых растворов (см. стр. 30). Широкое исследование реакций между твердыми веществами на смесях, образованных неметаллическими веществами (окиси, соли), первым предпринял Хедвелл (Hedvall, 1910 и сл.). Течение и механизм этих реакций исследовали затем, помимо Хедвелла, главным образом Тамман (1925 и сл.), Лидер (Jander,' 1927 и сл.) и Фишбек (Fischbeck, 1927 и сл.). Изучение при помощи рентгеновского анализа строения решеток кристаллических веществ и данные о силах, действующих между элементами решетки, дали точную физическую основу для толкования явлений, наблюдаемых в реакциях твердых веществ (v. Hevesy, 1922;Snjekal, 1925 и сл.; Jost, 1926 исл,; Wagner, 1930 и сл.). Одновременно они установили, что превращения и взаимодействия в твердом состоянии проходят через переходное состояние, отличающееся особой активностью, так,что некоторые вещества можно получить в особо богатой энергией и ре акционно снос об ной форме путем синтеза в твердом состоянии (Hedvall, 1922 и сл.; Huttig, 1931 и сл.). В настоящее время можно точно описать также «процессы потускнения» и другие подобные явления. Эта теория была развита главным образом трудами Вагнерй (Wagner, 193.3).
Диффузия. Как для реакций, протекающих на поверхности, так и для реакций в твердых веществах основное значение имеют процессы диффузии. Под термином диффузия понимают постепенное смешивание различных находящихся н соприкосновении веществ, происходящее без воздействия внешних сил и обусловленное движением самих молекул.
Для диффузии в растворах справедлив следующий закон: количество растворенного вещества dn, диффундирующее за время dt в направлении от более высоких концентраций раствора к более низким через сечение д, пропорционально падению концентрации в направлении диффузии — dc/dx и сечению д-dn ,, Сс ~л~ — — (1
dt dx
D называют коэффициентом дифф>узии растворенного вещества. Оп показывает количество ветцества в граммах, которое проходит в единицу
780
Глава 19
времени через сечение в 1 елг, если падение концентрации равно 1. Концентрация при этом выражается в граммах на кубический сантиметр (г/с.и3).
Коэффициент диффузии зависит от температуры, природы растворенного вещества и растворителя. Так как растворенные вещества изменяют природу растворителя^ на коэффициент диффузии данного вещества влияют другие вещества, содержащиеся г в том же растворе. В соответствии с этим D также не совершенно независим от концентрации диффундирующего вещества. При равных температурах в одном и том же растворителе коэффициенты диффузии различных, но химически подобных, одинаково построенных и нс сильно сольватированных в растворе веществ обратно пропорциональны корням квадратным из их молекулярных весов:
(2>
если ГБ и D2 — коэффициенты диффузии, а Мх и М2 — молекулярные веса веществ 1 и 2».
Для диффузии иопов в растворах электролитов, которые содержат разные количества посторонние электролитов, справедливо выражение
Ji -г), _
Вг-Пг Уд?! ’
(2а)-
где ц, и ц2 — различные вязкости, обусловленные разным содержанием посторонних электролитов.
Основной закон диффузии, выражаемый уравнением (1), был установлен в 1855 г. Фиком. Он справедлив также й для взаимной диффузии смешивающихся жидкостей, например воды и спирта, а также для взаимной диффузии газов.
При применении закона; диффузии Фика к случаю взаимной диффузии газов целесообразно заменить с в уравнении (1) иа плотность газа, диффундирующего через определенное сечение. Для диффузии второго газа, осуществляемой в противоположном направлении, справедлив тот же коэффициент диффузии, что и для диффузии первого; одпако падение плотности для обоих газов различно. Поэтому через одно и то же сечение в противоположных направлениях переносятся хотя и равные объемы обоих газов, но разные весовые их количества. Коэффициенты диффузии газов зави-сяТ, помимо температуры, от давления. С изменением состава смеси они изменяются незначительно.
С явлением диффуэии газов друг в друга не следует путать прохождение газов через мелкопористую перегородку (например, из неглазуровэнного фарфора), которое таюке называют «диффузией». Для диффузии газов через пористую перегородку справедлив закон Грэма и Бунзена, по которому скорость диффузии газа обратно пропорциональна корню квадратному из его плотности. Аналогичный закон справедлив для эффузии газов. Под этим термином понимают истечение газа из очень малых отверстий в тонкой стенке. Так как по закону Авогардо плотности газов относятся, как их молекулярные веса, то на основании закона Грэма и Бунзена справедлив следующий вывод; скорости эффузии и диффузии газов через пористую перегородку обратно пропорциональны, корням квадратным из не молекулярные весов:
(3)
где и ь-2 — скорости эффузии или диффузии с о ответственно, а М, и М2 — молекулярные веса газов 1 и 2.
Для скорости взаимной диффузии газов без промежуточной диафрагмы этопростое соотношение уже неверно. Одпако, как ноказывает кинетическая теория газов, скорости взаимной диффузии газов находятся в достаточно простой связи с длиной свободного пробега и скоростью отдельных молекул газа. Так как последние обратно пропорциональны корню квадратному из молекулярного веса, то в общем газы диффундируют друг в друга тем быстрее, чем меньше их молекулярный вес.
Реакции твердых веществ
781
Для диффузии в твердых веществах также формально справедлив закон диффузии Фика [уравнение (1) стр. 779], хотя в этом случае механизм диффузионного процесса существенно отличается по своей природе от диффузионного процесса в газах, жидкостях и растворах.
В табл. 104 сопоставлены коэффициенты диффузии в газах, растворах й твердых веществах. Из приведенных данных видно, что диффузия в газах осуществляется много скорее, чем в жидкостях. В твердых веществах скорость диффузии, в общем, даже при повышенных температурах значительно меньше, чем в жидкостях, Однако
Таблица 104
Коэффициенты диффузии различных веществ [с.к2/сек]
Газы в воздухе при 0° и 760 лмг рт ст Вещества, растворенные в воде при 18° Ионы солей в кристаллах Металлы в твердых металлах Неметаллы в мота л.чая
яг 0,685 КС1 1,67-10-5 Св+ вAgl 3,9-10-ь при 480“ Ли йРЬ При 6.5 .10-5 320“ Н в Pd 0,5.10-5 Яри 25“
н2о NHs 9, 103 0,108 ЫС1 Mgs О 4 1,07-10-5 0,56-10-» Ag+ в СВгЗ 3,1- 10-е при 223“ Ag в РЬ Кри 9,1-10-8 285° N в Fe 1,4 -10-7 при 1000“
СО2 0,139 сгн5оп 1,10-10-5 Ag Ь в Си2Те 3,0-10-1 при 330“ Zn в РЬ □pit 1,0-10-10 285“ С й Fe 1,1 fG~a при 1000°
CS2 0,088 CjHe 0,075 Цензом ССЦКНз)^ CaHiaOe Глдосоаа 1,04.10-» 0,57-10-5 012+ в ZnWOi 1,0-10-10 ярл 050“ Си в Ati яря 9,4-10-П 500°
Sr2+ в СаМоОд 1,7-10-’* при 350“ Au B.PL при 1,4-10-11 900“
МоОЗ* в BaWCU 9,6 fQ-11 При 550° Ли в Ag при 5-10-1’ 490“ !
имеются отдельные случаи, когда коэффициенты диффузии в твердых веществах уже при немного более высоких температурах имеют значения, близкие значениям коэффициентов диффузия в жидкостях и растворах при обычных температурах и даже превышающие их. В общем коэффициенты диффузии в твердых веществах колеблются: в очень широких пределах, В то время как коэффициент диффузии Ав и РЬ уже при 250° достигает значения 3,5-10-7 с.«2- с<?л-“х, коэффициент диффузии Мо в W при 2000° составляет лишь 1.O1O-10 <Хкг сек-1. Наименьшие измеренные значения D лежат около 10-гт &иг-сек-1. Однако измерение коэффициентов диффузии ниже 10_12 ел-2-стл'-1 уже представляет существенные трудности.
Температурную зависимость коэффициентов диффузии твердых веществ можно выразить формулой
-10g»=-^ + &, (4)
где Т — абсолютная температура; а и Ь — константы *, зависящие от природы даппого вещества. В качестве примера хода кривой такой температурной зависимости на рис. 102 приведена зависимость отрицательных логарифмов коэффициента диффузии Мо в W от абсолютной температуры **. В то время как в интервале температур
* Константы а и Ъ можно не только определить эмпирически, но также и рассчитать теоретически (ср. стр. 78-i), [см., папример, van Licmpt, Rec. 1га v. chim. Pays-Bas, 51, 114 (1932) |.
** Если построить зависимость log В от 1/Т, то, как показывает уравнение (4), получится прямая.
782
Глава it)
2600—2300° К коэффициент диффузии молибдена уменьшается мопее чем на. 10%, в интервале мопеду 1000 и 700° К оп уменьшается более чем на 70%. Подобное чрезвычайно быстрое уменьшение коэффициента диффузии ниже оиредедеппой температурной границы наблюдается и для других веществ; этим и объясняется тот факт, что даже у тех твердых веществ, которые уже при нескольких сотнях градусов Цельсия обладают относительно хорошей диффузионной способностью, эта способность большей частью падает до неизмеримо малых величин при комнатной температуре.
Взаимодействия между твердыми веществами. Если нагреть смесь двух порошкообразных твердых веществ, то часто можно наблюдать, что при определенной температуре в смеси происходит сильное выделение тепла. Опо выражается внезапным часто очень сильным подъемом кривой
Ряс. 102. Температурная зависимость коэффициента диффузии (D) молибдена в вольфрам.
Р ис. 103. Кривая нагревания ВаО -ф--j-CuSO4 (по Хедвеллу).
нагревания, которая представляет собой зависимость температуры реакционной смеси от продолжительности нагревания (см. рис. 103). Это выделение тепла, происходящее при температуре, которая может лежать значительно ниже температуры плавления и разложения составных частей смеси, следует трактовать, как теплоту реакции, протекающей между двумя твердыми веществами. В примере, приведенном на рис. 103, это превращение следующее: ВаО + CuSO4 = BaSO4 + СиО. То, что такое превращение действительно имеет место, можно доказать аналитическим исследованием продуктов нагревания.
Аналитическим исследованием смесей, нагретых до различных температур, можно определить температуру, при которой произошла реакция, даже и в тех случаях, когда она не может быть установлена по кривой нагревания вследствие слишком небольшой величины теплоты реакции или слишком малой скорости реакции. Удалось показать (Нейvail, 1924; Tammann, 1927), что в подобных случаях превращения не связапы с присутствием низкоплавких загрязнений и не должны включай, газообразные продукты разложения, образовавшиеся в результате незначительной термической диссоциации, и что, следовательно, речь действительно идет о превращениях в твердом состоянии.
Аналогично приведенной выше реакции протекают реакции между основными и кислотными окислами, которые реагируют между собой с образованием солей или двойных окислов, а также реакции труднолотучих кислотных окислов с карбонатами, замещение металла в солях, окисла х и сульфидах па менее благородный металл, двойной обмен солей, например ВаСО3 и CaSO4 с образованием ВайО4 и СаСО3, а также-двойной обмен между окислами металлов и сульфидами, фосфидами, карбидами и силицидами соответственно.
Реакции твердых веществ
788
Для объяснения этих реакций принимают, что с повышением темпе-ратуры атомы или соответственно ионы в кристаллах все сильнее колеблются около положения равновесия. Когда эти колебания достигают определенного значения, на пинается более или менее частый обмел местами в решетке.
Подобный обмеп местами происходит особенно легко в «разрыхленных» кристаллах, т. е. в таких, которые прорезаны мелкими (обычно не обнаруживаемыми под микроскопом) трещинами, копалами и т. д., так как в атом случае свободные поверхности (внутри кристалла) предоставляют атомам пространство для любых сдвигов. Однако обмел местами для атомов возможен и в полностью гомогенных кристаллах благодаря дефектам структур и пустым местам, которые всегда имеются в большем или меньшем числе (ср, со споской на стр. 36). В тех местах, где соприкасаются различные кристаллы, атомы или ионы могут: переходить из одпого кристалла в другой также путем обмена местами. Если речь идет о кристаллическом порошке однородного вещества, то подобный обмен может привести к спеканию зерен порошка, что используют, например, в «металлокерамике» (ср. стр. 21), В связи с этим происходит частичная переориентация плоскостей кристаллических решеток и новое огранепие кристаллитов, т. е. «рекристаллизация» (ср. т. I, стр. 616). В порошкообразной смеси одновременно с обменом местами при соприкосновении кристаллов различных веществ осуществляется химическое изменение, которое путем обме в а местами распространяется внутрь кристалла. Поэтому превращение веществ зависит от скорости, с которой атомы или попы могут перемещаться в кристалле путем обмена местами, и, следовательно, от скорости, их диффузии в кристалле *.
Удалось показать, что скорости, с которыми распространяются реакции в твердых веществах, и температурная зависимость скорости этих реакций подчиняются законам, справедливым для процесса диффузии. Особенно хорошо выполняется «закон корня квадратного» (см. стр-794). Температурная зависимость скорости реакции выражается подобно температурной зависимости скорости диффузии в твердых веществах уравнением (4) стр. 781. Так как По теории перемещения условия для «рекристаллизации» и химического взаимодействия в твердых веществах должны быть одинаковы, гипотеза, по которой процесс обмена местами является решающим для протекания химического взаимодействия, получила дальнейшее подтверждение в работах Таммана, установившего, что температура, при которой происходит взаимодействие между двумя твердыми веществами, почти всегда совпадает с температурой рекристаллизации одного из них.
Как показал Хедвелл, «спекание», рассматриваемое ранее как явление, сопровождающее частичное плавление кристаллических порошков, может происходить уже при тех температурах, когда плавление еше не наступает. В таких случае начинается спрессовывание зерен порошка путем обмена местами, возникающего при соприкосновении одного кристалла с другим, и связанная с ним «рекристаллизация». Последняя может одновременно привести к значительному сокращению объема порошка; это явление известно в технике под названием «усадка». В смесях, составные части которых могут взаимодействовать между собой, этот процесс часто существенно усиливается, так как обмену местами в точках соприкосновения благоприятствует химическое сродство. Выяснение этих явлений очень важно практически, например ДЛЯ трактовки явлений, происходящих при обжиге цемента, а также отчасти и керамики.
Во всех тех случаях, когда не образуется непрерывный ряд твердых растворов, реакции между твердыми веществами,' если их доводят до конца, протекают вплоть до полного исчезновения одного из компонентов.
* Сказанное справедливо для реакций, которые происходят внутри или между твердыми веществами. Взрывные процессы протекают иным образом.
784 Глава 19
Справедливость этого положения следует аз правила фаз. Если, например, имеется смесь ВаО и CuS04, в которой может идти реакция ВаО + CuSO4 = BaSO4 + 4- СиО, то в пей имеется три компоиёнта. (ВаО, СиО и SO3}. Количественные соотношения исходных веществ произвольны, но опи фиксируются как только образуется определенная смесь. Остается еще две степени свободы (давление и температура). Число фаз, которые могут находиться в равновесии между собой, равно тогда в соответствии с правилом фаз 3. Следовательно в равновесии cEaSCC и СиО может находиться еще овна твердая фаза, ВаО или CuSO4, в зависимости от того, какое из этих двух веществ было в избытке в первоначальной смеси.
Если два вещества могут образовать несколько соединений, то вопрос -о том, какое соединение образуется (если взаимодействие происходит в твердом состоянии), Зависит не только от состава смеси, но и от скоростей Диффузии ионов, которые должны обменяться местами в различных
Продолжительность нагревания-, час
Р ис. 104. Зависимость взаимодействия СаО с SiO2 в твердом состоянии от продолжительности нагревания (цо Лидеру).
чфазах. Например, СаО и SiO2 способны образовывать при кристаллизации из расплавов четыре следующих соединения: 3CaO-SiO2;' 2CaO-SiO2; ВСаО -2SiO2 и СаО -Si02. Если же оба вещества взаимодействуют в твердом состоянии, то, как нашел Яндер, независимо от соотношения компонентов в смеси в качестве первичного продукта всегда появляется ортосиликат 2CaO-SiO2. Если имеется избыток SiO2, то ортосиликат превращается -затем в силикат, соответствующий составу смеси. В смеси с молярным отношением 1-1 из ортосиликата сначала возникает только силикат 3CaO-2SiO2, а затем из пего метасилнкат CaO-SiO2 (см. рис. 104).
По мне пню Яндера, реакция протекает следующим образом: в месте соприкосновения кристалла СаО с зерном SiO2 сначала образуется путем обмена местами тонкая оболочка ортосиликата (см. рис. 105, й)< Чтобы реакция могла идти дальше, СаО, который, как показал ряд наблюдений, более подвижен, чем SiO2, должен диффундировать через 2СаО SjO2 (на рисунке через заштрихованную область). Пока слон последнего не слишком толст, эта диффузия Идет так быстро, что на границе фаз ортосиликата и SiOE может происходить дальнейшее образование 2СаО • SiO2. На границе между 2CaO-SiOa и СаО может образовываться 3CaO.Si02, однако образование этого соединения при 1200° протекает так медленно, что ого появление не
Реакции твердых веществ
785
было обнаружено * в опытах , результаты которыхДгрнведены на рис. 104. С увеличением толщины слоя ортосиликата СаО больше не достигает границы'*между фазами 2CaO-Si02 и SiO2 и там путем обмена местами образуется соединение ЗСаО-2SiOz. Благодаря этому возникает сна тала очень тонкий слой этого соединения, для дальнейшего роста которого справедливо все сказанное выше для ортоси лик ада. Последний растёт, далее, па своей границе с фазой 3CaO-2SiO2 B ’ой мере, в которой требуемый для этого СаО доставляется туда путем диффузии. Эта стадия приведена на рис. 105, 6. Если СаО не достигает больше границы фаз 3CaO-2SiO-> H SiO2 в количестве, требуемом дня образования 3CaO-2SiO21 то там образуется ~ метасиликат CaO:SiO2 (рис. 105, в). Когда, наконец, фаза СаО совсем исчезнет, слой ортосилйката будет все
Рис. 105. Схематическое изображение течения реакции между СаО и SiO2 в твердом состоянии.
более и. более уменьшаться за счет диффузии СаО из ортосиликата в направлении фазы SiCL через фазы 3CaO-2SiOa и CaO-SiO2. То же произойдет затем/ и с 3CaO-2SiQ2, если еще будет свободный SiOa (рис. 105, г). Если исходить из смеси с молярным соотношением 1 : 1. то в конце концов останется только метасиликат.
Если бы скорости образования силиката 3CaO-2SiO2 и CaO-SiO2 были исчезающе малы в сравнении со скоростью диффузии СаО через быстр о образующийся ортосиликат, ,то можно было бы получить последний в чистом состоянии (наряду с исходным компонентом, имеющимся в избытке) исходя из смесей СаО — SiO2 в любых соотношениях. Подобные отношения существуют, по-вндийому, для многих веществ. Например, Дикерхофф (Dykerhoff, 1925) нашел, что при нагревании смеси твердого СаО и А12ОЭ из четырех соединений, которые могут образовать эти вещества (ЗСаО-А12Оу; 5СаО- ЗА12О3, СаО-А12О3 и ЗСаО-5А12О3), постоянно выделяется лишь СаО-А12О3 независимо от состава смесей, с которыми работают.
На скорость реакции, в твердом веществе вследствие её зависимости от скорости диффузии атомов (соответственно ионов в кристаллах) сильно влияет размер зерен порошка. Чем мельче зерна порошка, тем скорее доходят реакции до конца. Далее, реакция протекает тем. быстрее, нем сильнее «.искажения решетки», так как . опи облегчают атомам обмен местами.
То, что искажение решетки благоприятствует обмену местами, может быть не только установлено теоретически, но и доказано экспериментально. Например, у твердых солеобразных соединений значительно повышается электропроводность (являющаяся непосредственной мерой способности ионов к обмену местами) в том случае, если искажения решетки усилить внедрением небольших количеств чуждых решетке компонентов. Одновременно повышается и реакционная способность рассматриваемого соединения относительно других твердых веществ.
* Несколько ниже 1200е 3CaO-SiO2 распадается на СаО и 2CaO-Sip2. 50. г, Реми
786
Глава 19
Особо реакционноспособными являются, как правило, так называемые аморфные вещества, у которых вообще нет упорядоченного распределения атомов и которые часто разрыхлены трещинами, каналами и другими внутренними полостями.
Вещества в таком разрыхлённом состоянии часто получают высушиванием гелей или осторожным нагреванием соединений, способных к термической диссоциации, Например, полученная слабым прокаливанием силикагеля аморфная двуокись кремния значительно болоо реакционноспособна, чем мелкойзмельчепный кварц. Очень высокой реакционной способностью в твердом состоянии отличается также окись железа(Ш), полученная термическим разложением сульфата железа(Ш), если его не сильно нагревали. Несмотря на то что полученная таким образом окись желе-за(Ш) по рентгенографическим данным кристаллична, кристаллы ее, однако, сильно «разрыхлены», что следует, между прочим, из значительного уменьшения объема, наблюдаемого при сильном ее нагревании. При этом вследствие рекристаллизации исчезают пустоты и одновременно снижается реакционная способность.
Активные переходные состояния. В тесной связи с только что рассмотренными явлениями стоит появление «активных переходных состояний»
Р и с. 106. Каталитическая активность смеси ZaO^-— Fe2O3 п₽п 250® в зависимости от температуры, до которой смесь была предварительно нагрета (по Хюттигу).
в реакциях, происходящих менаду твердыми веществами. Если постепенно нагревать два твердых вещества (например, ZnO и Fe2O3), которые могут образовать химическое соединение (в данном случае Fe2[ZnO4j), то часто можпо наблюдать возникновение переходных состояний, при которых появляются свойства, сильно отличающиеся как от свойств исходной смеси, так и от свойств конечных продуктов. Так, смесь ZnO и Fea03 после продолжительного нагревания при 400° значительно сильнее каталитически ускоряет реакцию 2СО + О2 = 2СО2, чем та же смесь, не подвергавшаяся подобной предварительной обработке (см. рис. 106). Однако если смесь нагревают нс до 400°, а до 500°, то опа обладает по сравнению с необработанным образцом не только пе повышенной, но даже меньшей каталитической активностью. Нагреванием примерно до 650° вновь достигается усиление каталитической активности. При этой температуре начинает образовываться соединение Fe2[ZnO4]. Однако после того как окончится его образование, продукт полностью теряет Свою каталитическую активность. :
Реакции твердых веществ
787
Аналогично от предварительной обработки зависит гигроскопичность рассмотренной смеси; можно наблюдать также и изменения других свойств в «переходном состоянии». Для практики особо важно, например, при использовании подобных смесей в качество катализаторов повышение каталитической активности в определенных переходных состояниях, следовательно, появление активных переходных состояний.
Появление «активных переходных состояний» может обусловливаться, по Хют-тигу, двумя различными причинами в зависимости от того, образуется ли подобное активное состояние при такой температуре, когда еще пе заметно образования никаких соединений, или только при более высокой температуре, при которой ужо осуществляется диффузия составных частей одного вещества в другое и благодаря этому происходит образование соединений. В первом случае атомы, соответственно по вы, лежащие в местах тесного соприкосновения различных веществ, могут перейти в реакционноспособное состояние благодаря обменному взаимодействию их силовых нолей, или же ионы одного кристалла могут достигать поверхности другого кристалла путем диффузии вдоль внешней поверхности и отщепления. Оба процесса вызывают изменение тех свойств, которые зависят от природы поверхности. Такими свойствами, помимо каталитической активности, являются адсорбционная способность относительно газов и паров и иногда окраска. При дальнейшем повышении температуры атомы соответственно ионы, укрепившиеся па поверхности за счет активированной адсорбции, будут проникать сквозь поверхность и благодаря этому терять свою активность. Так объясняется быстрое падение каталитической активности при температуре выше 400® ъ приведенном примере (рис. 106). При дальнейшем повышении температу-. ры диффузия атомов, соответственно ионов, одного вещества внутрь другого происходят в еще большей мере. Однако образование решетки нового соединения отстает более или мсисе сильно от этой диффузии. Поскольку этот процесс происходит вблизи поверхности, то опа вновь может изменить свои свойства, а именно, находясь как бы в аморфном состоянии, она может вновь развивать повышенную каталитическую активность. Но теперь уже эта активность обусловлена совершенно другими причинами по сравнению с первым случаем, и в соответствии с этим параллельно со вторым активированием происходит изменение других свойств. Так, в смеси ZhO Fe3O3 почти одновременно со вторым увеличением активности происходит возрастание парамагнетизма. Вблизи второго максимума активности парамагнетизм переходят в ферромагнетизм. Одновременно на рентгенограммах становятся видимыми линий, характерные дня соединения Fe2[ZnO4]. В той же мере, в какой усиливаются эти линии, т. е. в какой степени поверхность и соседние с ней слон переходят из состояния неупорядоченного распределения ионов в состояние упорядоченного распределения, активность снова падает.
Появление двух активных промежуточный состояний, разделенных' областью минимальной активности, можно наблюдать, помимо системы ZnO ~ Fe3O3, также и для Других систем, например СнО — Fe2Og, MgO — Ст3О3, СнО — CrsO3. Однако часто наблюдается лишь один максимум активности, вследствие того что либо один из максимумов отсутствует, либо оба максимума объединяются в результате того, что промежуточное состояние активированной адсорбции переходит в состояние активации за счет внутренней диффузии непосредственно, без периода дезактивации, Это справедливо, например, дня системы MgO — FeaOg.
Температуры, при которых происходит разрыхление (активация) поверхности и разрыхление (активация) кристаллической решетки для некоторых классов веществ, в частности для металлов, можно выразить однозначно (для различных веществ) в дробных долях температуры плавления (по абсолютной шкале). Температура разрыхления поверхности составляет около 0,29, температура разрыхления кристаллической решетки 0,42 температуры плавления [Н u t t i g, Kolloid-Z., 98, 263 (1942)]; ср. Lichteneckor L., Z. Elektrochem., 48, 669 (1942).
Условия для появления активного переходного состояния вследствие неупорядоченного распределения атомов на поверхности й вблизи нее создаются также при термическом разложении соединения. В процессе изменения модификации вещество также находится в состоянии повышенной активности.
Поэтому, как показал Хедвелл, AgNO3 реагирует с окислами СаО, SrO и ВаО не только при тех температурах, когда обычно начинаются реакции этих трех окислов , 50*
788' Глава IS
с нитратами и которые для них различны, по й при температуре, примерно равной для всех трех окислов и лежащей чуть выше температуры превращения AgNO3 (160°).
В общем справедливо положение, в соответствии с которым твердое вещество, испытывая какое-либо превращение, сопровождающееся изменением расположения атомов, проходит через промежуточное состояние, в'котором оно много более реакционноспособно, чем в исходном и конечном состояниях (принцип Хедвелла).
Особая реакционная способность смешанных осадков. Особо реакционноспособные смеси твердых веществ часто можно получить при их совместном осаждении в стехиометрических соотношениях из раствора. Так, Фрикке и Дюрр (Fricke, Durr, 1939) нашли, что образование шпинели, наблюдаемое в простой смёси окислов ZnO и Ге,О3 только при температурах 600°, при совместном осаждении обоих/-окислов может быть обнаружено при 60°. Нагреванием до 1200° смеси Ве(ОН)й и у-АЮ(ОН), полученной совместным осаждением, Фрикке и Шредеру (Schroder, 1948) удалось синтезировать хризоберилл. Измерением эманационным методом (ср. стр. 578) наряду с рентгенометрическими исследованиями им удалось установить, что АЮ(ОН), выпадающий вместе с Ве(ОН)2, действует на последний как защитный коллоид и препятствует его кристаллизации ири нагревании. Это защитное действие сохраняется и при температурах, при которых названные вещества, отщепляя воду, превращаются в окислы. Вплоть до пачапа образования хризоберилла (950°) в рентгенограмме: появляются лишь липни у-А12О3.и отсутствуют линии ВеО. При 1200° линии у-А13О3 исчезают и остается ясная добайеграмма хризоберилла, '
Реакции твердых веществ с жидкими. Для реакций твердых веществ с жидкими, в случае если продукт реакции переходит в раствор, справед-< ливы, в сущности, те же законы, что и для растворения твердого вещества п растворителе в обычном смысле.
Как показал Нойс (Noyes, 1897), при равномерном распределении частиц растворяемого вещества в растворителе путем перемешивания скорость растворения пропорциональна величине поверхности твердого вещества, соответственно поверхности соприкосновения его с раствором, а также разнице между имеющейся концентрацией раствора и его концентрацией насыщения: б? (7 •
& -0-(сааСЫщ—с), (5)
где о — поверхность твердого вещества; сяасыщ — концентрация насыщенного раствора- с — концентрацияраствора ко времени t; de— увеличение концентрации за Время dt\ кпостоянная. При сопоставлении этого уравнения с уравнением (1) на стр. 78Q' видно, что определяющим фактором дня скорости растворения является скорость диффузии, растворенного вещества в растворителе, если, как это обычно имеет место, равновесие растворения на поверхности вещества устанавливается так быстро, что слои растворителя, находящиеся в непосредственном соприкосновении с ним, постоянно насыщен ы растворяемым веществом. Как показал Бруннер (Вгипнег, 1904), к = ,
если D — коэффициент диффузии переходящего в раствор вещества; 6 — толщина слоя жидкости, обволакивающей его кристаллы (т. е. слои, который не отделяется от кристаллов при перемешивании); и — объем раствора.
Если при реакции твердого вещества с жидкостью продукт реакции Не переходит в раствор и реакция перемещается внутрь твердого вещества, то ее скорость определяется скоростью, с которой молекулы жидкости или атомы, соответственно ионы твердого вещества, способны диффундировать через твердый продукт реакции. Но при обычной температуре скорость диффузии в твердых веществах, как правило, так мала, что реакции с жидкостями, которые идут таким путём, практически редко имеют значение. По-иному обстоит дело, если нерастворимый продукт реакции очень порист или не крепко пристает к поверхности, В этом слу
Реакции твердые веществ- 789
чае жидкость снова может проникать по каналам й трещинам к вновь освобождающимся частям поверхности. Благодаря этому реакция часто может протекать достаточно быстро даже вплоть до полного разрушения исходного вещества. Это происходит, например, в еду чае действия воды па металлический магний.
Реакции твердых веществ с растворенными. Для реакций твердого вещества с веществами, находящимися: в растворе, если продукт реакции переходит в раствор и, следовательно, превращение идет только на внешней поверхности твердого вещества, справедливо уравнение, найденное Перистом (1904):
7 <6>
где D — коэффициент диффузии растворенного вещества, -реагирующего с твердым веществом; О — внешняя поверхность последнего; 6 - - толщина слоя жидкости, обволакивающей эту поверхность; с—концентра-: ция реагентов: в растворе (кроме обволакивающего слоя); dn, — .увели-., чение количества продукта реакции за время dt (которое эквивалентно уменьшению количества реагирующих веществ за то же время). Уравнение справедливо для того случая, когда продукт реакции равномерно распределяется в жидкости при перемешивании, так чГо в/растворе вплоть до границы слоя жидкости, обволакивающей твердое вещество, существует однородная концентрация с. Толщина 6.обволакивающего слоя зависит от скорости перемешивания. В . общем она измеряется величиной порядка Ю'3 см. Далее, при выводе уравнения Нернста предполагалось, что скорость реакции бесконечно велика в сравнении со скоростью диффузии реагентов, так что концентрация последних непосредственно на поверхности твердого вещества практически может быть ' приравнена к нулю. Эксперимент показал, что это предположение большей частью выполняется.
Уравнение (6) свидетельствует о том, что при сделанных предположениях скорость, с которой твердое вещество разрушается за счет реакции щ растворенным веществом (например, МдО соляной кислотой); определяется исключительно скоростью, с которой молекулы или ионы, реагирующие с твердым веществом, достигают его поверхности в процессе, диффузии. Она определяется па основании' закона диффузии . Фика [уравнение (1), стр. 779 J; если вместо g подставить поверхность твердого веще-„ rfc
сгва О, а вместо — — .падение концентрации вступающих в реакцию растворенных
веществ в обволакивающем слое жидкости (с — 0)/б .= с/6. Если проинтегрировать уравнение (1), допустив, что поверхность твердого вещества остается постоянной во время растворениями принимая во внимание, что справедливо равенство с/ =—— $ тде па — количество реагента, имеющегося в объеме v раствора к началу опыта, а щ — количество реагента, истраченного к моменту времени г, то подучим выражение
In f (7)
ла — п; J C.-V
Это уравнение позволяет рассчитать скорость растворения твердого вещества под влиянием любого растворенного реагента, если экспериментально определить велп-. - . „ „ , £> п
чину о при выбранной скорости перемешивания. Если учесть, что и с = —, то пз сравнения уравнений (S) и (5) следует: скорость растворения твердого вещества в чистой воде и скорость реакции насыщенного водного раствора того же вещества
790
Глава 19
с другим твердым веществом. равны друг другу (ВгипПОГ, 1940). Если реакция па поверхности протекает не с бесконечно большой скоростью, то уравнение Нернста и, естественно, также предшествующее выражение будут^несправедливы,
Для реакций, которые происходят в твердой фазе между твердыми веществами и веществами, переходящими на твердые из раствора, справедливо то же, что и для реакций твердых веществ с жидкостями.
Растворение металлов в кислотах. Особое место среди реакций твердых веществ с растворенными веществами занимает растворение металлов в кислотах, так как для этого существенную роль играет образование локальных элементов. Их значение обсуждалось уже на стр. 462 и сл. на примере цинка; в этом случае их действие проявляется особенно отчетливо.
Как показал Палмаэр (Palmaor, 1929), для скорости растворения металлов В кислотах решающим является активность локальных элементов. Так как их число и Вид постоянно изменяются в процессе растворения в зависимости от высвобождаю-, щихся при этом загрязнений, скорость растворения нельзя обычно выразить формулой, справедливой дли длительного промежутка времени. Для данного состояния внешней поверхности количество металла, переходящего в раствор за время dt, согласно Палмаэру, выражается уравнением
dx Е-И'О .
где Е — электродвижущая сила локального элемента (па величину которой значительное влияние оказывает перенапряжение водорода); и — удельная электропроводность раствора электролита; О — величина поверхности; С — коэффициент, зависящий от других условий опыта. Для величины С среди прочего существенное значение имеет среднее расстояние между электродами локального элемента. Следовательно, данное количество загрязнения, внедрившегося между кристаллитами металла, ускоряет его растворение тем сильнее, чем мельче отдельные кристаллиты.
Ускорение растворения одного металла под влиянием постороннего металла может прекратиться из-за появляющегося па них перенапряжения водорода. Например, растворение цинка в кислотах не облегчается при сплавлении его с небольшими количествами свинца или кадмия, а также при амальгамировании ртутью. Напряжение, которое необходимо приложить для выделения водорода на этих металлах, так велико, что оно не достигается за счет потенциала растворения цинка. Сплавление цинка с более благородными'металлами, которые образуют с ним твердые растворы или соединения, также пе повышает его растворимости в кислотах, если сплавы гомогенны, так как локальные элементы могут образовываться только между веществами;; находящимися в разных фазах
Пассивность. Состояние пассивности, появляющееся у определенных металлов, отличается тем, что металлы в этом состоянии ведут себя подобно благородным металлам. Они устойчивы относительно реагентов, с которыми взаимодействуют в нормальном состоянии, а также обнаруживают существенно более высокий потенциал растворения, чем в нормальном состоянии. Нормальное состояние, чтобы отличить его от пассивного, называют активным состоянием. Переведение металла из активного в пассивное состояние называют «пассивированием». Состояние пассивности можпо вызнать анодной поляризаций или химическими средствами, а именно обработкой известными окислителями, например концентрированной азотной кислотой.
Эле к Тро литической’(«гальванической») поляризацией называют появление между электродами при электролизе или в гальваническом элементе вследствие прохождения тока электродвижущей силы, направленной противоположно электродвижущей силе, вызывающей прохождение тока. Эта противоположная электродвижущая сила обусловлена изменениями либо поверхности электродов, либо обволакивающих их слоев
Реакции твердых веществ 791
раствора электролита. Вызванное прохождением тока при электролизе изменение анода называют анодной, а катода — катодной поляризацией. Простым примером электролитической поляризации является возникновение разности потенциалов между двумя платиновыми электродами, погруженными в серную кислоту после того, как через раствор пропустили электрический ток. Вследствие выделения водорода на катоде, а кислорода на аноде оба электрода приобретают различные потенциалы относительно раствора. Если после выключения первичного тока электроды соединить проволокой, то по ней, до тех пор пока не израсходуются газы, покрывающие электроды, будет проходить ток, противоположный по^направлению первичному току. Возможность получения элементов, дающих электрический ток за счет электролитической поляризации, была практически реализована в аккумуляторах (ср. т, I, стр. 594 и сл.).
Пассивирование при помощи поляризации наступает только у металла, использованного в качестве анода. При применении в качестве катода металл не только не пассивируется, но может быть даже переведен в активное состояние. Пассивирование, вызванное находящимися в растворе химическими агентами, можно отнести на. основании теории локальных элементов к поляризации за счет локальных токов. Однако еще нельзя уверенно сказать, чем вызывается в случае пассивирования эта,поляризация. Некоторые металлы, прежде всего хром и железо, частично пассивируются уже при храпении на воздухе («самопроизвольное пассивирование»). Можно получить сплавы, которые самопроизвольно очень сильно пассивируются (сталь V2A).
Активирование пассивированных металлов легко происходит главным образом иод действием протонов. Поэтому особенно сильным активатором металлов является электролитически полученный водород (катодная поляризация). Активирование происходит также при нагревании в атмосфере водорода, при воздействии кислот, при обработке восстановителями, и, в первую очередь, водородом в момент выделения. Кроме того, активирование достигается и под действием водных растворов солей, прежде всего хлоридов, затем также (в соприкосновении с растворами) механическим путем (при скоблении, ударе, сотрясении), а, по данным Шмида (Schmid Q., 1937), также под влиянием ультразвука. Активирование пассивного металла путем соприкосновения с активным сводится к катодной поляризации.
Явление пассивности было открыто дня железа в конце 'ХVIИв. Фарадей (1836) пытался объяснить его гипотезой об окисном или окисноноДобпом поверхностном слое. Шенбайн (Schonbein) предположил, что пассивность должна о бу ел окликаться физическим изменением металла (например, превращением в аллотропную ^модификацию). Позднее фон Белк (v. Be lek, 1887) развил теорию, по которой пассивность вызывается топким споем кислорода, обволакивающим металл. Эти три различные точки зрения в более или менее сильно измененной форме отстаиваются еще и в настоящее время. Наряду с ними выдвигался ряд других объяснений, которые все же большей частью были недостаточно обоснованы экспериментально; например, раз личные] исследователи развивали взгляд, согласно которому у металлов пассивное, нормальное и активное состояния обусловлены водородом, растворенным в металле. Несмотря на то что: многочисленные исследователи, особенно с начала этого столетия, занимались изучением пассивности, причины этого явления еще и сегодня изучены не до конца.
В известной мере как доработку воззрений Шелбайна в соответствии с современными представлениями о состоянии вещества можно рассматривать высказанную Смитсом (Smits, 1923) и независимо от пего Расселом (Russel, 1926) точку- зрения, по которой различие активного и пассивного состояний обусловлено разной электронной конфигурации й атомов пли но поп, лежащих на поверхности металла. Смите показал, что состояние пассивности можно легко объяснить, если допустить, что в таких металлах, как, например, железо, в равновесии имеются атомы или ионы с разной потенциальной энергией (обусловленной различным квантовым состоянием валентных электронов). Установление этого равновесия происходит в зависимости от обстоя
792 ; Глава 19
тельств со значительным замедлением *, как это, -например, имеет место для равновесия а-и f}-SO3 (ср. т. I, стр. -758). Металл, в котором такое равновесие устанавливается не сразу, должен вести себя подобно твердоед раствору двух металлов. Следовательно, если анодно растворять такой металл, то сначала в раствор перейдут атомы одного вида и при этом потенциал растворения будет сдвигаться в сторону более благородных компонентов до тех пор, пока-только они и останутся на поверхности. Если описанное явление действительно имеет место, то металл достигает высшего состояния пассивности. Если вследствие каких-либо причин, например каталитического воздействия протонов, вновь установится равновесие между двумя состояниями атомов металла, то вновь возникает активное состояние, Теория Смитса, между прочим, хорошо объясняет тот факт, что металлы в пассивном состоянии часто переходят в раствор с иной валентностью, чем активные (ср. стр, 145).
Многие поддерживают представление о .том, что пассивирование основано на образовании окисной оболочки под влиянием окислителей; это, однако, очень спорно. Мюллер видоизменил и обобщил это представление на основании своих наблюдений. Он считает, что пассивность во многих случаях объясняется образованием на металле оболочки, пронизан пой не очень многочисленными мелкими порами. Такая оболочка не обязательно должна состоять из онпсла металла. Она может быть образована и солью металла. Связанное с пассивированием сильное повышение анодного потенциала при постоянной плотности тока следует объяснить, по Мюллеру, .тем, что после покрытия анода оболочкой ток модют проходить только через ее поры. Эта теория пассивности, «теория покрытия», хорошо согласуется с рядом явлений, наблюдаемых при пассивировании металлов в результате анодной поляризации. Однако она не объясняет того факта, что пассивированный металл имеет более высокий потенциал растворения, чем тот же металл в активном состоянии даже при измерениях, проведенных в отсутствие тока (ср, т, I, стр, 166). Можно установить термодинамически, а также показать экспериментально, что измеренный без тока потенциал металла («Потенциал покоя») не изменится, если металл покрыть пористым слоем (например, благородного металла). Тем не мепее этот более высокий «нотепциал покоя» как раз отличает пассивное состояние, Для объяснения более высоких Потенциалов покоя Мюллер допускает, что окисное и солевое покрытия вызывают сдвиг в расположении электронов в атомах поверхности металлов. В этом его взгляды совпадают со взглядами Смитса и Рассела. Однако существенное отличие заключается н том. что, по мнению Мюллера, образование окисной или солевой оболочки должно быть не просто более или менее случайным явлением, сопровождающим пассивирование, а необходнмой предпосылкой: но крайней мере в большинстве случаев для появления пассивности. Одно из главных возражений, выдвинутых против «теории окисных оболочек», достоит в- Том, что для. большинства металлов образование слоя окисла не, приводит к пассивированию. Еще в 1899 г. Гитторф показал, что даже такие металлы, как хром, особенно склонные переходить в пассивное состояние, не обнаруживают пассивности, если их нагреванием па воздухе покрыть отчетливой .пленкой окисла. Наоборот, хром, покрытый пленкой окисла, образованной при нагревании на воздухе, сохраняет активность лучше, чем Хром с блестящей поверхностью.
Гипотеза, в соответствии с которой пассивность обусловлена покрытием поверх-ности металла (или; почти всегда имеющегося на ном в .результате соприкосновения-' с воздухом чрезвычайно тонкого слоя окисла, ср. стр, 795) оболочкой кислорода, удовлетворяет возражениям, выдвинутым против «теории окисных оболочек»! Кроме того, существование газовой оболочки подтверждается рядом наблюдений, а . также общих теоретических соображений. Пассивирующее действие' такой- газовой оболочки усматривают в настоящее время главным образом в, вызываемом еЮ ^насыщении свободных валентностей поверхности. Для этого достаточно мономолекулярпого слоя таза. Возможно также, что речь при этом идет об адсорбированных на поверхности ионах кислорода; которые не выхватывают атомы металла из их решеток, как это происходит при образовании окйслов. Поверхность, покрытая подобной оболочкой, ведет Себя, естественно, иначе,чем по л ерхнреть, покрыта я тольк о сл оем о кисла. Так как, вероятно, все металлы (поскольку они соприкасаются с воздухом) тотчас покрываются тонким слоем окисла, следует допустить (если считать, что пассивность обусловлена слоем окисла), что сдой окисла, образованный при пассивировании, имеет другую природу, чем слой окисла, образующийся при обычпом окислении металла на воздухе. Напротив, теория газовой оболочки для объяснения различия в поведении металла, окисленного обычным образом на воздухе и пассивированного, использует только известное
'* По мнению Смитса, достаточно принять, что установление равновесия между атомами металла, его ионами (с различными аар ядами) и электронами заторможено. Тогда в металле не должны присутствовать атомы или ноны с различной электронной конфигурацией в нейтральном состоянии, соответственно с одинаковым зарядом.
Реакци.и.твер$их веществ
793-
явление адсорбции! Она хорошо согласуется также с тем фантом, что более или менее далеко идущее пассивирование часто наступает уже при простом соприкосновении в воздухом, в то время как дальнейшее окисление на воздухе не усиливает пассивцро- ? сания, а вновь снижает его.
Со свойством некоторых металлов переходить в активное состояние, различное по силе, связана другая особенностьj а именно периодическое изменение скорости рас-творения. Для наглядности этого явления (впервые установленного Ей. Оствальдом для хрома, полученного алюмотермическим способом) Па рис. 107 по абсциссе отложено время, а по ординате скорость растворения хрома в? соляной кислоте, измеренная по количеству водорода, вьщеляемого в единицу времени (данные заимствованы из опытов Брауэра, 1901). Б этих опытах обнаружилось, что колебания скорости растворения
Время
Р п с. 107. Периодическое уменьшение скорости растворения хрома в 2 я. соляной кислоте.
идут; абсолютно параллельно с колебаниями потенциала растворения; величины их были, правда, значительно меньше, чем разница потенциалов между совершенно активным и совершенно пассивным хромом. Уже Оствальд предположил, что это объясняется присутствием в хроме загрязнении. Еонее поздние эксперименты подтвердили такое предположение. Это явление, которое наблюдается также у определенных: сортов железа, вызвано, вероятно, чередующимся пассивированном металла (вследствие анодной поляризации) локальными токами, которые вызываются внедрившимися между его кристаллитами Примесями и последующим повторным активированием огр за счет выделяющегося водорода (вероятно, вследствие диффузии протонов в металл). Ретерт (Rathert, 1914) сумел вызвать для железного электрода (которым ведет себя в разбавленной хромовой кислоте как равномерно пассивированный) периодическое активирование под влиянием водорода, диффундирующего в металл.
Реакции твердых веществ с газами. Так как скорость диффузии н газах очень велика по сравнению со скоростью диффузии в растворах, то де л я ющеив е л ичиной для скора сти ре акций тв е рды х веществ и с г азами (если они протекают с образованием летучих продуктов) гораздо чаще, чем при реакциях с растворенными веществами, является скорость, с которой протекают собственно химические превращения. Это. прежде всего справедливо для реакций твердых веществ с газами прн умеренно-высоких температурах.
При высоких температурах скорость потока газов часто играет значительно бблыпую роль, чем их диффузия, как, на пример,, при сжигании угля. Широко: Известно, что при повышении скорости подачи воздуха сильно ускоряется сгорание угля. Однако подвод воздуха можно усиливать не в любой степени, так как иначе с потоком воздуха будет уноситься слишком много тепла. Поэтому целесообразно повышать скорость сгорания увеличением поверхности угля (сжигание угольной пыли). В общем моЖно утверждать, что идущее при высоких температурах с образованием летучих продуктов взаимодействие твердого вещества: с потоком газа идет совершенно аналогично растворению твердого вещества в потоке жидкости. Скорость, с которой происходит собственно химическое превращение на поверхности, играет в этих реакциях ту же роль, какую при растворении играет скорость, с которой происходит длффузия через обволакивающий слой (ср. стр. 788 и сл.). Так же как последняя зависит от скорости течения жидкости вне этого слоя, скорость химических превращений в газах зависит от скорости движения газа вблизи поверхности твердого тела. Если наглядно изобразить линии течений вокруг кубика угля, сгорающего на воздухе, Й вокруг кубика соли, растворяющегося в воде, то получится почти идентичная картина (Rosin, 1931). В обоих случаях кубик будет подвергаться воздействию главным образом с того конца, который
794
Глава 19
повернут в сторону от направления потока газа или жидкости, так как там образуются наиболее сильные завихрения. у
Исследование механизма подобных реакций имеет технический интерес прежде всего для изучения процессов горения при производстве генераторного газа, при обжиге руды и для доменного процесса.
Если образуется жидкий продукт реакции, то продолжение реакции зависит от скорости, с которой происходит длффузия газов через сдой жидкости. Этот случай имеет практически второстепенное значение, так как образованию слоев жидкости обычно препятствует испарение за счет теплоты реакции.
Практически очень важны реакции твердых веществ с газами, которые протекают с образованием твердого продукта реакции. Если последний образует на поверхности, твердого вещества компактную пленку, то реакция может развиваться только за счет диффузии через твердый слой продукта реакции. Подобными процессами объясняется, например, потускнение многих металлов при нагревании на воздухе или в другом газе, с которым они дают соединения, образующие нерастворимые и компактные пленки. Потускневшие слои при обычной температуре, когда скорость диффузии в них практически равна нулю, защищают металл от дальнейшего воздействия воздуха. Если бы этого не происходило, то большая часть используемых металлов иа воздухе очепь быстро превращалась бы в окислы. На поверхности кислород воздуха реагирует с металлами чрезвычайно быстро уже при обычной температуре.
Время, за которое на металле образуется моно молекул ярпый слой окисла, обычно •составляет доли секунды. Между тем, как: можпо рассчитать из температурной зависимости скорости потускнения, для меди должно было, бы пройти около 1 млрд., а для никеля — даже около 50 триллионов лет, до тех пор пока слой окислов на них в сухом воздухе при комнатной температуре увеличился бы настолько, чтобы появились первые отчетливые следы окрашивания,— желтый в первую очередь*. '
Как уже упоминалось, диффузия в твердых веществах и, следовательно, рост толщины слоев потускнения подчиняются тому же закону, что и диффузия в растворах. Если применить уравнение (1) (стр. 780) к реакции, развитие которой зависит от скорости диффузии одного из компонентов через ком паханый твердый слой, то следует обратить внимание на то, что в этом случае толщина варя х, через который происходит диффузия, не постоянна в противоположность отношениям, обсужденным ла стр, 788 и 789. Здесь она пропорциональна количеству реагирующего вещества («); .т = а-п. Между тем разница в концентрациях ct и с2, которую диффундирующее вещество имеет па обеих границах слоя, постоянна; далее можно принять, что падение концентрации в слое линейно. Тогда для каждой точки внутри слоя справедливо выра-жеиие — ' Вместо q — сечения диффузионного слоя —- вновь подставим ^
О — величину поверхности твердого вещества, которую примем за постоянную. При .этих условиях интегрированием уравнения (1) получим, если ко времени t s== 0 количество прореагировавшего вещества равно О,
а или
„ = -р/2Д'°(с2 —(9)
Этот «закоп корня квадратного? справедлив, как следует из предыдущего, не только дли химических реакция между твердыми веществами и газами, но и для реакций между твердыми и растворенными веществами в тех случаях, когда продукт реакции образует компактный слой, а также для реакций твердых веществ между собой. При приложении его к реакциям между твердыми веществами принимают С£ — 1 и с2 = О,
* Эта интерференционная окраска в среде с показателем преломления 1 соответствует слою толщиной 0,15 ц. Если слой имеет показатель преломления п, то его •толщина будет в п раз меньше.
Реакции твердях веществ
796
Общая форма уравнения (9) не изменится, если допустить, что не только первый но также и второй компонент реакции диффундирует через слой; но тогда в него войдет два коэффициента диффузии. Случай диффузии только одного компонента является, по-видимому, обычным, по крайней мере для солеобразных соединений.
Компактные слои окислов образуются при действий сухого воздуха на металлы, если окисел .металла имеет больший объем, чем эквивалентное .количество металла (Pilling, 1923). В других случаях образуются-пористые слои окислов, через которые кислород может достигать непосредственно поверхности металла. Тогда окисление развивается с постоянной
Рис, 108, Прирост веса меди, цинка и железа прп их окислении на влажном воздухе (по В ер поп у).
скоростью и делается заметным уже при обычной температуре. Это наблюдается для следующих металлов: Li, Na; К, lib, Cs, Mg, Ca, Sr и Ba.
Правило Пиллинга не справедливо для поведения металла на влажном воздухе; в этом случае-на металлах довольно часто образуются пористые слоя, которые не тормозят окисления. Если образуются пористые слои, то скорость окисления может даже расти со временем. Это происходит в тех случаях, когда продукты окисления каталитически ускоряют .окисление, например при ржавлении железа.
"Три только что рассмотренных различных тина реакций твердого вещества -(в частности, металлов с газами) с образованием твердых продуктов реакции отращены кривыми на рис. 108, которые соответствуют увеличению веса Си, Zn и Fe при окислении их на влажном воздухе в зависимости о? времени. Окисление медн протекает, как .свидетельствует параболическая форма кривой, по закону, выражаемому уравнением (9), следовательно, с образованием компактной плепки на металле. Цинк окисляется с постоянной скоростью, следовательпо, с образованием пористой пленки. У железа не только образуется пористая пдепка, но, кроме того, реакция аутокаталитически ускоряется, В соответствии с этим скорость окисления здесь растет со временем.
Аутокатализ про ржавлении железа обычно объясняют образованием локальных элементов (ср. ниже). Для неметаллических веществ аутокаталитическое ускорение реакций, протекающих с образованием твердого продукта реакции, происходит или за счет того, что зародыши кристаллов продукта реакции увеличивают пересыщение границы фаз или за счет тою, что химическая реакционная способность возрастает
796 Глава 19
вследствие соприкосновения одного твердого вещества с другим, следовательно, также-с зародышами кристаллов продукта реакции.
Может случиться, нто скорость окисления металла на воздухе достигнет нормальной зависимости от времени, когда толщина окисного слоя будет определенной величины. Например, окисление никеля при 400е протекает как наблюдал Хауффе (Hauffe, 1954), по «закону корня квадратного» (см. стр. 794)Долько тог да, когда толщина окисного слоя превысит 100 тр; до этого оно протекает существенно медленнее. Хауффе объясняет это действием пространственного заряда в слое окисла, которое осуществляется за счет адсорбции кислорода на поверхности никеля и становится заметным при очень небольшой толщине слоя.
Для дегазации твердых веществ (т, е. для удаления обратимо удерживаемых примесей) Хюттиг (Н u t t i g, Z. anorg, Chem., 249, 134 (1942)] нашел закон, по которому удаление газов происходит с максимальной скоростью при обеих указанных па стр. 787 температурах разрыхлении.
Коррозия я защита от коррозии
Среди разделов науки, для которых очень важно выяснение природы реакций, происходящих с твердыми веществами, основное место занимает исследование коррозии. Народнохозяйственное впадение этой области исследования вытекает из размера тех ценностей, которые ежегодно уничтожаютсякоррозией. Подсчитано, что за период с 1890 по 1923 г. мировое производство железа составляло 1766 мпп. т, а потери железа, вызванные ржавлением, 718 млн. т.
Коррозией (от corrodere — грызть) называют разрушение твердого тела, которое начинается на поверхности вследствие неконтролируемых химических и электрохимических воздействий. Среди веществ, которые; погибают вследствие коррозии, в первую очередь следует назвать металлы, а среди них — в основном железо.
Ржавление железа сводится, как ужо указывалось, к действию локальных элементов. Так как возникающая при этом окись железа образует новый локальный элемент (в котором оНа играет роль катода), то ржавление является аутокаталитически ускоряемым процессом. Зависимостью силы тока в локальных элементах от удельной электропроводности раствора, в соприкосновении с которым находится металл [см. уравнение (8) стр. 790], объясняется большее корродирующее действие морской воды в сравнении о обычной водой. Покрытие из более благородных металлов, таких, как олово я медь, защищает от ржавления, если опо не повреждено и, напротив, ускоряет ржавление, как только повреждается, так как Эти металлы образуют с железом : локальные элементы, в которых последнее функционирует в качестве анода. Между, ,.г_ тем цинковое покрытие защищает железо даже и тогда,; когда оно неплотно, так как в этом случае атакуется менее благородный металл —цинк. Последний разрушается медленно, так как аутокаталитическое ускорение, происходящее в случае образования ржавчины, отпадает. Одпако все же вследствие образования окиси цинка защитное действие может слабеть.
С тем, что компактное железо пе подвергается заметному воздействию пи сухого-воздуха (вследствие образования компактного окиспого слоя, ср. стр, 795), ни чистой, свободной от воздуха воды, а только одновременному воздействию воды и воздуха, на впервый взгляд трудно увязывается тот факт, что железо, ржавеющее под действием влаги, сильнее всего разрушается как раз в тех местах, к которым доступ воздуха наиболее труден. Сложенные вместе железные пластины более всего: разрушаются там, где они соприкасаются. Случается, что железные тросы внутри корродируют, а внешне еще кажутся совершенно прочными. Можно легко показать экспериментально, что железо, «насыщенное» кислородом, имеет более высокий потенциал, чем «ненасыщенное». Следовательно, согласно теории локальных элементов, железо должно пере- ходить в раствор преимущественно в местах, «ненасыщенных» кислородом; таким образом теория полностью объясняет наблюдаемые явления.
Наряду С теорией локальных элементов и общими положениями металлографии; для изучения коррозии металлов имеют прежде всего значение исследования природы поверхностных слоев, возникающих на металлах при различных условиях, например при «процессах потускнения». Для изучения коррозии важно также расширение-знаний р причинах.^ сопутствующих явлениях пассивности и, кроме того, важно пони
J
Реакции, твёрдых веществ 797
мание явлений, протекающих на поверхностях (границах фаз), чему Способствует изучение процессов адсорбции и механизма действия катализаторов при гетерогенном катализе.
В качестве средств предотвращения коррозии металлов в первую очередь следует назвать:
1) применение самостоятельно пассивирующихся сплавов (например, сталь V2A);
2) применение сплавов, на которых: образуются самостоятельно защищающие поверхностные слои (например, гидронал it другие корро-з ионно устойчивые сплавы алюминия);
3) искусственное создание, соответственно укрепление, поверхностных слоев (например, воронение стали, анодирование алюминия);
4) нанесение коррозионноустойчйвых: металлических покрытий. Это производят чаще всего либо электролитическим («гальваническим») путем (например, электролитическое хромирование, никелирование, цинкование), либо напылением. Наряду с этим для некоторых металлов применяют и другие способы (огненная оцинковка, паровая оцинковка);
5) эмалирование (ср. т. I, стр. 551);
6) покрытие лаками или красками.
Железо можно защитить от воздействия кислорода воздуха при высоких температурах (до 1200°), заставляя диффундировать в его поверхность алюминий. Образующийся при прокаливании па воздухе тонкий слой окиси алюминия в значительной •стенепн замедляет дальнейший процесс окисления. Так можно, например, цри 4000° достигнуть 6—8-кратпого увеличения продолжительности службы железа, которое применяют, например, для, изготовления колосников топок.
В железных водопроводных трубах большой частью образуется тонкий сдой карбоната кальция, защищающий их от коррозии. Однако образование такого: защитного слоя происходит только тогда, когда вода содержит достаточно гидрокарбоната .кальция и пе содержит избытка угольной кислоты и когда, кроме того, в ней растворен .кислород. Роль, которую играет при этом кислород, еще полностью не выяснена.
Топохимические реакции А '
Топохимическими реакциями называют такие реакции, при которых •свойства твердых продуктов реакции существенно определяются том, что взаимодействие происходит на твердом веществе, т. е. оно локализовано (голод — место). При топохимических реакциях получаются иногда г "такие вещества,: которые вообще не удается; получить при реакциях в газах или в растворах. В других случаях, несмотря на то что при реакциях в газах или в растворах и получаются вещества того же состава, но по свойствам они явно отличаются от веществ, полученных при реакциях, идущих на поверхности или внутри твердого вещества. Это можно •объяснить тем, чТо твердое исходное вещество благодаря .химической природе или свойствам (папример, Ьтепени дисперсности) определяет течение реакции, или тем, что структура этого вещества, его степень .дисперсности оказывают влияние на свойства продукта реакции.
В то время как реакции обычно подразделяются по характеру их протекания, топохимические реакции разграничивают па основании других классификационных принципов; а именно в зависимости от места, на котором происходит реакция и которое определяет взаимосвязь свойств и природы начального и конечного продуктов. Сообразно с этим топохимические реакции представляют прежде всего значительный препора-тивный интерес, Одпако в настоящее время все в большей степени их изучение1 сводится к расширению представления о механизмах реакции, взаимосвязи структур .и отношений между структурой и химическими свойствами.
798 Глава 19
Понятие «топохимических реакций» было установлено в 1919 г. В. Колыпутте-ром. (V, Kohlschutter) Тогда были известны лишь немногие процессы, которые подходили под это понятие. В дальнейшем Кольшуттер, а позднее и другие исследователи-(Feitknecht, Fricke, U. Hofmann, H. Kohlschiitter) на большом числе веществ смогли показать- взаимосвязь между условиями их образования и их природой. Ранее были приведены уже многие такие примеры. В дальнейшем будут рассмотрены отдельные типичные примеры топохимических реакций, которые полезны для уяснения их сущности.
В зависимости от характера влияния исходных веществ на свойства твердых продуктов реакции топохимические реакции можно подразделить на три типа:
1) реакции, у которых от характера твердых исходных веществ зависят лишь степень дисперсности и развитие поверхности продуктов-реакций, а также число и вид имеющихся в твердых продуктах реакции искажений решеток и дефектов структур;
2) реакции, ори которых твердое исходное вещество влияет на кристаллическую структуру продукта реакции;
3) реакции, при которых твердое исходное вещество определяет также химический состав продукта реакции. Последние: приводят к соединениям, которые другим путем либо вообще не получаются, либо по меньшей мере не получаются в растворе или газовой фазе путем аналогичных взаимодействий.
Влияние исходного вещества на степень дисперсности и развитие поверхности твердого продукта реакции наблюдают часто при получении веществ термическим разложением твердых соединений.
Фейткпехт нашел, например, что если различные соединения кальция нагревать до одной и той же температуры (900°), то получается порошкообразная окись кальция с различными свойствами. В структурном отношении продукты идентичны; однако степень дисперсности очень различна. Объемы одинаковых весовых количеств СаО из СаС3О4, Ca(OH)2j СаСО3, Ca(NO3)2 относятся как 100 : 78 ; 59 : 34,5 соответственно. Скорость, с которой различным образом полученные порошки поглощают при обычной температуре нары воды, надает с уменьшением их объема. Объемы гидроокисей, образующихся путем поглощения паров воды, относятся к исходным веществам и между собой как 164 : 127 ч 102 : 74,5.
Приглашении водой из порошков СаО, приготовленных из СаС2Н4, Са(ОН)2 и СаСО3 удается получить пластичные гели Са(ОН)2 («известковую кашу»). СаО, приготовленная'из Ca(NO3)2, не дает такого геля; также почти не удается получить «известковую кашу» при последующей обработке жидкой водой порошка, Са(ОИ)2, полученного поглощением паров воды СаО. г
Адсорбционные способности и каталитическое действие соединений, полученных термическим разложением или путем реакций в растворе, могут существенно различаться. На это указывалось уже ранее. Кроме того, как показывает приведенный выше пример, у соединений, полученных термическим разложением, эти свойства могут быть выражены различно в зависимости от природы исходного вещества. Правда, на практике при этом значительную роль играет часто то обстоятельство, что для разложения различных исходных веществ используют различные температуры, В соответствии со сказанным ранее это существенно влияет па развитие поверхности и реакционную способность продукта. Однако это влияние, обусловленное ростом гомогенности и кристаллизации с возрастанием температуры, только тогда относится к области топохимии, когда для течения этого процесса существенное значение имеет расположение структурных частичек, определяемое исходным веществом,
В качестве технически важного топохимического явления следует упомянуть о зависимости окраски многих веществ от способа их приготовления. Этим уже издавна пользуются при изготовлении минеральных красок. Например, давно известно, что путем осторожного нагревания сульфата железа(Ш) или аналогичных соединений удается получить окись железа(Ш) с красивой красной окраской; издавна ее использовали в качестве краски. Эта окись железа, которая в отличие от обычной состоит не из ромбоэдрических кристалликов, а из топких чешуек, по данным Хедвелла (1922), обладает такой же кристаллической структурой, как и обычная; она отличается только-особой формой образования. Можно предположит^ что у этой группы топохимических
Реакции. твердых веществ 799’?
реакций зависимость свойств конечного продукта от исходного вещества часто в основном обусловлена тем влиянием, какое оказывает структура (и также степень размельчения) исходного вещества на образование зародышей, на развитие зародышей и процессы диффузии. Кроме того, следует также учесть влияние исходного вещества па число и вид искажений решеток и дефектов структур во вновь возникающей твердой фазе.
Поскольку расположение атомов, соответствующее, вновь возникающей фазе, практически ионностью не реализуется, можно предположить, что имеется (хотя и очень мало) некоторое число атомов или ионов, занимающих свои первоначальные-положепия или удаленные от них нс слишком далеко. Этим объясняется, почему твердые продукты реакции часто легче превращаются в свои исходные вещества, чем в новые соединения. Например, хлорид стронция, полученный оаторо'маьш обезвоживанием StC12-61]3О, быстрее поглощает воду, чем аммиак, в то время как хлорид стронция, приготовленный из SrCl2-8NHs, быстрее абсорбирует газообразный аммиак, чем. пары воды. Далее, окись алюминия, приготовленная термическим разложением нитрата алюминия при не слишком высоких температурах, быстрее растворяется в разбавленной азотной кислоте, чем в уксусной кислоте, в то время как окись алюминия, полученная термическим разложением основного ацетата алюминия, быстрее растворяется в уксусной кислоте. В этой связи следует также упомянуть часто наблюдаемое явление —• активное твердое вещество в том случае проявляет оптимальное каталитическое действие на газообразную систему, если его образование происходило в атмосфере етой газообразной системы.
В качестве примера влияния исходного вещества на кристаллическую структуру продукта реакции следует указать наблюдение Феиткнехта — метастабильная а-гидроокись цинка (ср. стр. 468) образуется постоянна тогда, когда вьпреличиваются водой определенные основные сопи цинка, например основной иодид ципка Zd12 4Zti(ОН)2. Напротив, гидролизом солей цинка в водном растворе обычно получают совершенно аморфные-осадки, которые могут перейти при старении как в окись цинка, так и в гидроокись цинка (как правило, в £-Zn(0H)2].
Основные соли цинка имеют слоистую структуру решетки. <x-Zn(OH)2 также образует слоистую решетцу (ср. стр. 465 и 468). После сказанного понятно, что наблюдающееся у исходных соединений расположение иопов благоприятствует образованию' решетки a-ZufOri).;. Однако предпосылкой для взаимодействия без существенного искажения первоначального порядка ионов является достаточное расстояние между слоями, так чтобы ионы гидроксила могли туда внедряться. В случае основного иодида цинка это обеспечивается внедрением объемистых ионов иода между слоями.
Соединения графита. Как с точки зрения валентного состояния и химической структуры, так и в препаративном отношении особенно интересны топохимические реакции, ведущие к соединениям, которые нельзя получить другим путем. Наиболее старым примером реакций этого типа является образование так называемой графитовой кислоты путем окислений графита хлоратом калия и азотной кислотой (ср. т. I, стр. 461).
Структура графитовой кислоты выяснена рентгенометрическими исследованиями Гофмана (1930 и сл.). Опа производится из структуры графита следующим образом. Между плоскостями слоев (ср, т. I, стр. 463, рис, 84, б) расположены атомы 0, причем, вероятно, так, что с каждым из Двух соседних атомов С одного слоя, поочередно сверху и снизу плоскости слоя, связан один атом О. Таким образом, за каждым кислородным слоем следует углеродный слой (на расстоянии 1,4 А) и затем на том же расстоянии — снова кислородный слой. За последним на существенно большем расстоянии следует снова кислородный слой, который относится к следующему углородному слою (ср. рис. 49,6 т. I, стр. 245, на котором представлено точно такое же расположение слоев).
Межатомное расстояние С. ч—* О составляет i,57 Л. Порядок атомов С в каждом слое тот же, что и в графите. Расстояние С-—’С равпо 1,46 Л (в графите 1,45 А). Напротив, расстояние между углеродными слоями благодаря внедрению кислородных слоев почти в два раза больше расстояний в графите. Слои могут легко еще более раздай-
800 Глава 19
путься, если заставить молекулы воды: цродиф фундировать Внутрь («набухание» графитовой кислоты пе следует смешивать со «вспучиванием», происходящим при сухом нагревании вследствие выделения газа). Полностью безводные препараты графитовой кислоты, по-вйдимому, не удается приготовить, Это связано, вероятно, с тем, что между углеродными слоями располагаются не только атомы О, по и ОН-группы. Кроме того что увлажненная графитовая кислота окрашивает лакмусовую бумагу в красный цвет, в Пользу зтого представления говорит также и то обстоятельство, что она способна обменивать протоны на другие катионы (Thiele, 1937), Правда, отчасти этп кислотные свойства могут быть обусловлены тем, что края углеродных слоев (па границах кристалла) содержат карбоксильные группы. Поэтому Гофман рассматривает графитовую кислоту как окись графита, бол ее “'и ли менее сильно загрязненную кислыми «краевыми соединениями». Однако расположение ОН-грунп между углеродными слоями следует из того факта (Hofmann, 1939), что метилирование ОН-групп вызывает существенное увеличение расстояния между плоскостями слоев.
Обработкой окиси графита сероводородом, т. е, в результате замены атомов О на атомы S, Гофман смог получить //.сульфид графитах.
Ионы HS0,7 также могут располагаться между углеродными слоями графита. Так получают гидросульфит, графита (Hofmann, 1934 й сл.). Он ббразуется при ’Обработке графита концентрированной серной кислотой в присутствии незначительных количеств азотной кислоты или другого окислителя: hHsSO4 -|- графпт -t- 'y/j.O = = [графит. SO4Hj„ + "/аЩО. Образование гидро сульфата графита может, происходить только в присутствии окислителя, так как для связывания ионов HSOy углеродные слои должны зарядиться положительно, отдав электроны. Гидросульфат графита может вновь разложиться до графита. Это разложение происходит ступенчато благодаря тому, что ионы HSOj удаляются слоями: Гидросульфат графита в проходящем свете обладает темпо-синей окраской. Так же как и окись графита, он всегда загрязнен «краевыми соединениями». При внедрении HSOj-rpyno графит расширяется в па правлении осп с. Это наряду с параллельно идущим окислением краев углеродных Слоев с образованием карбоксильных групп ведет к тому, что при более длительном действии серной кислоты вместе с окислением происходит далеко идущее измельчение с образованием коллоидного графита. Наконец, это же является причиной того, что графитовые аноды сильно разрушаются в серной кислоте, в то время как в растворах галогеноводо-родиых кислот они устойчивы.
Гвдррсульфат графита устойчив только под концентрированной серной1 кислотой. Водой он разлагается с выделением СО2. Если между всеми углеродными слоями расположены иопы HSOj, гидросульфат графита имеет состав C|4HS0j- 2H2S04. При обработке другими сильными кислотами молекулы’ серной кислоты и анионы могут быть обратимо заменены с образованием нитрата графита, перхлората графита, :селе-ната графита и т. д. * > '
Структура и состав фторида графита (монофторуглерода) CF (ср. т. I стр. 463), полученного Руффом действием фтора па графит, также определяются тем, что внутри характерных для графита слоев атомы .углерода связаны значительно прочнее, чем в направлении, перпендикулярном слоям.
По даппым Руффа, в монофторуглероде ионы фтора расположены между слоями углерода таким образом, что на каждый углеродный слой С обеих сторон накладываются три слоя фтора. Углеродные-слои благодаря этому раздвигаются па 8,17 А (по сравнению с 3,25 Ав графите). Однако впутриэтих слоев порядок атомов С остается неизменным.
При действии паров щелочных металлов на графит также получаются соединения, структура которых определяется исходным веществом и которые другим путем нельзя получить, а именно КС8, ВЬС8 и CsC8 (Fredenhagerj, 1926).
Как установил рентгенометрически Шлееде (Schleede, 1932), этп Соединения производятся от графита внедрением одного слоя атомов щелочного металла между двумя углеродными слоями решетки графита (см. рис. 109, стр. 801). Благодаря этому внедрению расстояние между углеродными слоями изменяется от 3,35 до 5,34; 5,68
Реакции твердых веществ
801
и 5,94 А ее ответственно. При разложении путем нагревания соединения М*С8 скачко-
образно переходят в соединения МТС^В. Последние отличаются тем, что в них на каждые два углеродных слоя следует только один слой атомов металла.
Фторид графита и щелочные графиты удается получить также из черного углерода (активированного угля или сажи). Это подтверждает данные, что в чёрном угле-
роде имеются те же углеродные слои, что и в графите (ср. т. 1, стр. 465).
Приведенные примеры покавывают, чем обусловливается возможность получения посредством топохимических реакций соединений, которые другим путем но образуются. В исходных веществах должен существовать при этом особый порядок атомов, присущий только твердому состоянию, который настолько устойчив, что при мягких условиях протекающего взаимодействия он сохраняется. Наряду с особо прочными связями в исходном веществе должны быть одновременно другие сравнительно рыхлые связи, которые могут расщепляться под влиянием других веществ при одновременном их присоединении. Такие связи скорее всего существуют у веществ со слоистой стр ip ктурой решетки, так как у них силы связи в одном направлении иные, чем в-
• К
Рис. 109. Элементарная ячейка КС8. а = 4,94 А, с = 21,34 А.
Масштаб рисунка в два раза меньше, чем масштаб решетки графита на рис. 84,
двух других. У веществ с цепочечной,
ленточной и трубчатой структурой эти условия людаться.
также могут часто соб-
Примером топохимического образования т' г
соединений, не получающихся иным путем, ИЗ исходного продукта с трубчатой структурой служит получение сульфидных и полисульфидных пермутитов (ср. т. I, стр. 560). Обменные реакции пермутитов и цеолитов также следует рассматривать как топохимические процессы. Монтмориллонит (ср. т. I, стр. 545 и сл.), который также способен к основному обмену, обладает особо ярко выраженной слоистой структурой (Hofmann, 1933—1937). Распространенным примером топохимических реакций, которые приводят к соединениям, не образующимся другим путем, являются превращения силоксена, описанные в т. I, на стр.518.
51 г. Ремп
ПРИЛОЖЕНИЕ
51*
805
Таблица I
Атомные веса, отнесенные к 0=16 и 12С = 12а
Элемент Символ АТОМНЫЙ" вес по 0=16 Атомный вес по 12С=12
Азот N 14,008 14,0067
Актиний Ас 227 [227]
Алюминий А1 26,98 26,9815
Америций Ат [243] [243]
Apron Аг 39,944 39,948
Астатин At [210] [210]
Барий Ва 137,36 137,34
Бериллии Be 9,013 9,0122
Берклий Bk [247] [247]
Бор В 10,82 10,811
Бром Вт 79,916 79,909
Ванадий V 50,95, 50,942
Висмут В1 209,00 208,980
Водород II 1,0080 1,00797
Вольфрам W 183,86 183,85
Гадолиний Gd 157,26 157,25
Галлий Ga 69,72 6'3,72
Гафний Gf 178,50 178,49
Гелий Не 4,003 4,0026
Германий Ge 72,60 72,59
Гольмий Но 164,94 164,930
Диспрозий By 162,51 162,50
Европий Ей 152,0 151,96
Железо Fe 55,85 55,847
Золото Ан 197,0 196,967
Индий In 114,82 114,82
Иод I 126,91 126,9044
Иридий Ir 192,2 192,2
Иттербий Yb 173,04 173,04
Иттрий Y 88,92 88,905
Кадмий Cd 112,41 112,40
Калий К 39,100 39,102
Калифорний Cf [251] [249]
Кальций Ca 40,08 40,08
Кислород 0 16,0000 15,9994
Кобальт Co 58,94 58,9332
Кремний Si 28,09 28,086
Криптон Kr 83,80 83,80
Ксенон Xe 131,30 131,30
Кюрий Cm [247] [247]
Лантан La 138,92 138,91
Литий Li 6,940 6,939
Лютеций Lu 174,99 174,97
Магний Mg 24,32 24,312 j
Марганец Mn 54,94 54,9381 ’ II
Элемент Символ Атомный вес по 0=16 Атомный вес но 1SG«12
Медь Си 63,54 63,54
Менделевий Md [256] [256]
Молибден Мо 95,95 95,94
Мышьяк As 74,91 74,9216
Натрий Na 22,991 22,9898
Неодим Nd 144,27 144,24
Неон Ne 20,183 20,183
Нептуний. Np [237] [237]
Никель Ni 58,71 58,71
Ниобий Nb 92,91 92,906
Нобелий No [255] [255]
Олово Sn 118,70 118,69
Осмий Os 190,2 190,2
Палладий Pd 106,4 106,4
Платина Pt 195,09 195,09
Плутоний Pu [242] [242]
Полоний Po 210 [210]
Празеодим Pr 140,92 140,907
Прометий Pm [145] [147]
Протактиний Pa 231 [231]
Радий Ha 226,05 [226]
Радон Bn 222 [222]
Рений Re 186,22 186,2
Родий Rh 102,91 102,905
Ртуть Hg 200,61 200,59 <
Рубидий Rb 85,48 85,47
Рутений Ru 101,1 101,07
Самарий Sm 150,35 150,35
Свинец Pb 207,21 207,19
Селен Se 78,96 78,96
Сера S 32,066 32,064
Серебро Ag 107,880 107,870
Скандий Sc 44,96 44,956
Стронций Sr 87,63 87,62
Сурьма Sb 121,76 121,75
Таллий TI 204,39 204,37
Тантал Ta 180,95 180,948
Теллур Те 127,61 127,60
Тербий Tb 158,93 158,924
Технеций Tc [99] [97]
Титап Ti 47,90 47,90
Торий Th 232,05 232,038
Тулий Tu 168,94 168,934
Углерод C 12,011 12,01115
Уран U 238,07 238,03
806
П родолагсение табл. I
Элемент Символ Атомный вес по 0=1 в Атомпый вес по 13С=12 : Элемент Символ Атомный вес по 0=16 Атомный вес по 13С=12
Фермий Fm [253] [253] Цезий Са : 132,91 132,905
Фосфор Р 30,975 30,9738 Церий Се 140,13 140,12
Франций Fr [223] [223] Ципк Zn «65,38 65,37
Фтор F 19,00 19,9984 Цирконий Zr 91,22 91,22
Хлор Cl 35,457 35,453 Эйнштейний Es [254] [254]
Хром Сг 52,01 51,996 Эрбий : Er 167,27 • 467,26
® В таблице атомные веса по углероду, 13С=12, приведены принятыми Международной комиссией по атомным весам на 1962
в соответствии с данными, г. — Прим, рев.
Таблица II
Энергии ионизации в электрон-вольтах
I]—энергия ионизации первой ступени (М=М+-|-0
II—энергия ионизации второй ступени (М+=Ма+ -f-e) и т. д.
Жирным шрифтом указаны энергии, требующиеся для отрыва; одного электрона из оболочки инертного газа. В скобки заключены предполагаемые значения,
Элементы главных подгрупп периодической системы
Л’ uNa: Д&К a?Rb ssC3 4Be nMg asSr sfi-Ba авНа
I 5,40 5,138 4,339 4,176 3,893 9,32 7,64 6,11 ’ 5,69 5,21 5,28
II 75,62 47,29 81,81 27,56 25,1 18,21 15,03 11,87 11,03 10,03 10,14
III 122,42 71,68 45,7 39,68 34,6 153,85 80,15 51,21 48,6 37 (84)
IV 98,88 60,61 52,6 (46) 217,66 109,34 67,3 57,0’4 (49) (46)
SB l»Al 3iGa 81'1’1 .0 n8i SpSli ззЕЬ
I 8,30 5,934 5,98 5,785 6,106 11,26 8,15 7,88 7,332 . 7,42
II 25,15 18,82 20,51 18,87 20,42 24,38 16,34 15,94 14,63 15,03
П1 37,92 28,44 30.70 27,9 29,82 47,86 33,46 34,22 30,66 31,93
IV 259,30 119,96 64,2 57,9 ~50. 64,48 45,13 45,71 39,4 42,11
V 340,13 153,8 (90) (77) (64) 391,99 166,43 93,47 80,9 69,40
VI 190,42 (118) (98) (81) 489,84 204,14 (123) (103) (84)
7N: isP за As 6lSb eo: | : I»® 34 Se : аДе 84Ро
I 14,54 10,’55 9,81 8,64 7,287 13,61 10,36 ; 9,74 9,01 8,2
II 29,60 19,66 18,7 16,7 19,3 35,15 23,41 21,5 18,8 19,4
III 47.43 30,16 28,32 24,87 25,6 54,93 35,06 , 32,0 30,64 27,3
IV 77,45 51,35; 50,14 44,16 45,3 77,39 47,29 42,91 37,83
V 97,86 65,01 62,64 55,67. 56,0 113,87 72,5 68,3 60,29
VI 551,93 220,41 127,57 107,65 94,4 138,08 88,06 81,71 72,35 —
Главная подгруппа седьмой группы Главная подгруппа восьмой, гриппы
iH «01 s;Br sy) ,Не I0Ne is Аг saK-r 54Хе geRn
I 13,595 17,418 13,01 11,84 10,44 24,580 21,56 15,755 14,00 12,127 10,75
П 34,98 23,80 21,6 19,02 54,400 41,07 27,49 24,56 21,21 21,4
Ш 62,65 39,92 35,64 33 63,88 40,90 36,8 32,13 29,4
IV 87,23 53,47 50,21 (42) 97,16 59,79 52,5 (45) (44)
V 114,21 67,83 59,7 (52) 126,4 75,0 64,7 (57) (55)
VI 157,12 96,6 88,6 (77) 157,9 91,0 78,5 (68) (67)
VH 185,14 114,31 103,0 (90) 207,2 124,0 111,0 (96) (97)
VIH 958,8 348,5 193 169,93 239,1 143,4 (127) (110) (Ill)
807:
Продолжение табл. II
Элементы, побочных подгрупп периодической системы
i(Cu | „Ан 30?Й i^Cd soHg 21?C | 3.Y 6?La BsCe | esEu tiLu
I и ш IV 7,72 20,29 36,83 (59) 7,57 21,48 36,10 (52) 9,22 20,5 (30) (44) 9,39 17.96 39,70 (62) 8,99 16,90 44,5 (55) 1 10,43 18,75 34,2 (46) 6,56 12.89 24,75 73,9 6,38 12,23 20,5 61,8 5,61 11,43 19,17 (52) 6,.54 12,3 19,5 36,7 5,67 11,24 .6,15 14,7 (10)
33Ti «Zr T2Hf -ooTh =3v | 4iNb 7зТа- siCr mW j i.U
I п ш IV V VI 6,84 13,57 28,14 43,24 99,8 119 6,84 12,92 24,11 33,98 82,3 98,9 5,5 14,8 (21) (31) 11,5 20,0 28,7 (65) (80) 6,74 14,2 1 29,7 48,0 65,2 128,9 6,88 13,90 28,1 38,3 ~50 110,4 7,7 16,2 (22) (33) (45) 1 II 1 If 6,76 16,49 31 (51) 73 90,6 7,13 15,72 29,6 45,04 61,2 ~67 7,98 17,7 (24) (35) (48) (61) I I I Г । 1 |
1аМп «Тс ,sRe гДе 27C0 2в^ 44Bu : «Rh 7вОя 77Ir
I II ш IV V VI Vii VIII ь 7,43 15,64 33,69 (53) 76 (101) 119,3 196,4 7,23 14,87 31,9 (43) (59) (76) (94) (162) 7,87 16,6 (26) (38) (51) (65) (IS) 7,90 16,18 30,64 (56) (79) 103 (133) 151,23 7,86 17,05 33,49 (53) (82) (109) 133 165 7,63 18,15 36,16 (56) (79) (113) (143) 170 7,36 16,60 30,3 (47) (63) (81) (100) (119) 7Д6 15,92 32,8 (46) (67) (85) (Ю5) (126) 8,33 19,42 (33) (49) (66) (90) (Ш) (132) 8,7 17 (25) (40) (54) (68) (83) (99) 9,2 17,0 (27) (39) (57) (12) ; (88) ( (104) 8,96 18,54 (29) (41) (55) (75) (02) (109)
Таблица III
Распределение электронов по отдельным энергетическим уровням свободных т атомов в нормальном состоянии
Для каждого элемента указано число электронов, которые находятся в нормальном состоянии на уровнях с квантовыми числами, приведенными в головке таблицы.
Конфигурации инертных газов даны жирным шрифтом.
Объяснение символов термов, указанных в последней колонке , таблицы, см. т. I, стр. 283. 1
Порядковый:номер Рентгеновские уровни К L м •W О Р Q Основные термы :
Главное квантовое ЧИСЛО я Побочное квантовое число 1 Символы анергетй-:чесйих уровней 1 0 S 2 2 0 1 к р 3 3 3 0 1 2 • р d 4 4 4 4 0 12 3: : s JJ d / 5 5 5 5 012 3 s р d 1 0 6 0 0 1 2 е р 4. 7 6 , S
1 2 Водород Гелий 1 2
808
Продолжение табл. Ill
0* 0 Рентгеновские К L м N 0 P Q
я о уровни * s - 1 < > *
«а tn Главное квантовое 1 2 2 3 9 3 4 4 4 4 9 5 5 5 в в в 7 Основные
ЧИСЛО п lepmi
Побочное квантовое 0 0 I 0 1 2 0 12 3 0 12 3 0 1 2 0
число I
А Символы внергети- в а р в р d s р d f » p d j >pd a
ё ческих уровней
3 Литий 2 1
4 Бериллий 2 2 l^o
5 Бор 2 2 1 2Л/
6 Углерод 2 2 2 3Po
7 Азот 2 2 3
8 Кислород 2 2 4 3P2
9 Фтор 2 2 5
10 Неон 2 2 6 ^0
И Натрий 2 2 6 1
12 Магний 2 2 6 2 ^0
13 Алюминий 2 2 6 21
14 Кремний 2 2 6 22
15 Фосфор 2 2 6 23 4^/a
16 Сера 2 2 6 24 3^2
17 Хлор 2 2 6 25 S/4
18 Аргон 2 2 6 26 ^0
19 Калий 2 2 6 26 1
20 Кальций 2 2 6 28 2 ^0
21 Скандий 2 2 6 26 1 2 £^3/s
22 Титан 2 2 8 26 2 2 3f2
23 Ванадий 2 2 6 26 3 2 ^3/T
24 Хром 2 2 6 26 5 i ! 7$3
25 Марганец 2 2 6 26 5 2 i
26 Железо 2 2 6 26 6 2 > w4
27 Кобальт 2 2 6 26 7 2 i J
28 Никель 2 2 6 26 8 2 i 3f4
I
29 Медь 2 2 6 2610 1 2%
80»
Порядковый номер 1 Рентгеновские уровни К L м О р Q 7 4 а Основные термы
Главное квантовое число п Побочное квантовое число I Символы энергетических уровней 1 0 S 2 2 0 1 « р 3 3 3 0 1 2 s р d 4 4 0 1 а Р 4 4 2 3 d f 5 5 0 1 s р 5 5 2 3 d 1 в в 0 1 S р в 2 d
30 Цинк 2 2 6 2610 2 ^0
31 Галлий 2 2 6 2610 21 ал/1
32 Германий 2 2 6 2 610 22 3Л)
33 Мышьяк 2 2 6 2610 23
34 Селей 2 2 6 2 610 24
35 Бром 2 2 6 2 610 25
36 Криптон 2 2 6 2610 26 ^0
37 Рубидий 2 2 6 2610 26 1
38 Стронций 2 2 6 2610 26 2 1*?о
39 Иттрий 2 2 6 2610 26 1 2
40 Цирконий 2 2 6 2610 26 2 2 3Л2
41 Ниобий 2 2 6 2610 26 4 1 в2)1/з
42 Молибден 2 2 6 2610 26 5 1 7^3
43 Технеций 2 2 6 2 610 26 (5) (2)
44 Рутений 2 2 6 2610 26 7 1
45 Родий 2 2 6 2610 26 8 1 4^/>
46 Палладий 2 2 6 2 610 26 10 ^0
47 Серебро 2 2 6 2610 26 10 1
48 Кадмий 2 2 6 2 610 26 10 2 1|^0
49 Индий 2 2 6 2 610 26 10 2 1
50 Олово 2 2 6 2 610 26 10 2 i 3^о
51 Сурьма 2 2 6 2 610 26 10 2 1
52 Теллур 2 2 6 2610 26 10 2 4 ЗР2
53 Иод 2 2 6 2 610 26 10 2 аЛ/
54 Ксенон 2 2 6 2 6 10 26 10 2 6 ^0
55 Цезий 2 2 6 2 610 26 10 2 1 а^/»
56 Барий 2 2 6 2610 26 10 2 6 2 ^0
57 Лантан 2 2 6 2610 26 10 2 6 1 2 az)3/s
58 Церий 2 2 6 2 610 26 10(1) 2 6(1) (2) №)
59 Празеодим 2 2 6 2 610 26 10 (3) 2 6 (2)
60 Неодим 2 2 6 2 610 26 10 4 2 6 2 вЛ
61 Прометий 2 2 6 2 610 26 10 (5) 2 ( (2)
62 Самарий 2 2 6 2610 26 10 6 2 ( 2 1
63 Европий 2 2 6 2 6 10 26 10 7 2 f 2
64 Гадолиний 2 2 6 2610 26 10 7 2 6 1 2 j »2)
65 Тербий 2 2 6 2610 26 10 8 2 С 1 2 I
810
Продолжение табл. 111
a ф S Рентгеновские К L лг JV 0 1 3
о и УРОВНИ V
Главное квантовое 1 2 2 3 3 3 4 4 4 4- 5 5 5 6 6 6 1 Основные
g число n Побочное квантовое 0 0 1 о 1 г 0 1 2 3 0 12 3 0 1 ' 2; " c / термы
ЧИСЛО 1
Ci Символы внергети- S 5 р »Р d s р d / P d 1 S P :Д " 3
& ческих уровней
i 66 Диспрозий 2 2 6 2 610 2 610 (9) 2 6 (1), (2)
67 Гольмий 2 2 0 2 610 2 610 (10) 2 6 (1) (2)
68 Эрбий 2 2 6 2 6 10 2610 (11) 2 6 (1) (2)
69 Тулий 2 2 6 2 610 2 610 13 26 . 2 *
70 Иттербий 2 2 6 2 610 2610 14 26 2
71 Лютеций 2 2 6 2 6 10 2 6 10 14 2 6 1 2 2D^
72 Гафний 2 2 6 2 610 2 610 14 2 6. 2 2-
73 Тантал 2 2 6 2 610 2 6 10 14 26 3 2
74 Вольфрам 2 2 6 2 610 26 Ю.14 2 6 4 '2 ' V - 6Z>0
75 Рений 2 2 6 2 610 2 610 14 26 5 ' 2:
76 Осмий 2 2 6 2 610 2 6 10 14 2 6 : 6 2
77 Иридий 2 2 6 2 610 2 6 10 14 2 6 7 2
78 Платина 2 2 6 2 610 2 610 14 2 6 9 /2
79 Золото 2 2 6 2 610 26 10 14 2 6 10 ; i
80 'Ртуть 2 2 6 2 610 2 610 14 26 10 2. ^0
81 Таллий 2 2 6 2 6 10 2 6 10 14 2 6 10 , 21 i/s
82 Свинец 2 2 6. 26 10 2610 14 2 6 10 .... 22 ... •H’o
83 Висмут 2 2 6 2 610 2 6 10 14 2 6 10 2 3
84 Полоний 2 2 6 2 6 10 2 610 14 26 10 2 4 SP2
85 Астатин 2 2 6 2 6 10 2 610 14 2 6 10 2 5 : X 3/2
86 Радон 2 2. 6 26 10 2 610 14 2 6 10 26 Vo
87 Франций 2 2 6 2610 2G1O 14 26 10 _26 : 1
88 Радий 2 2 6 2 610 2610 14 26 10 2 6 ; 2
89 Актиний 2 2 6 2 610 2 6 10 14 26 10 26 1 2
90 Торий 2 2 6 2 610 2 6 40 14: 2 6 10 :26(2) ; (2) .:
91 Протактиний 2 2 6 2 6 10 2 6 10 14 26 10 : 26(3) X2) < c^3/2)
92 Уран 2 2 6 2 6 10 2 6 10 14 26 10 .'2.6 (4) (2) < (SHo)
93 Нептуний 2 2 6 2 6 10 2 640 14 26Ю (5) 2 6 (2) •. —
94 Плутоний 2 2 6 2 6 10: 2 6 10 14 2 610 (5) 26(1) (2 ) —
95 Америций 2 2 6 2 610 2 610 14 2 610 (6) 26(1) (2) —
96 Кюрий 2 2 6 2610 2 6 10 14 2 610 (7) 26(1) (2 ) —
97 Берклий 2 2 6 2 610 2610 14 2 6 10 (8) 26(1) (2 ) —
98 Калифорний 2 2 6 2 6 10 2 610 U 2610 (9) 26(1) (2 )
99 Эйнштейний 2 2 6, 2 610 26 10: 14 2610(10) 26(1) : (2) —"
100 Фермий 2 2 6 ,2610 2 6 10 14 2 6 10 (11) 26(1) (2) r—•
101 Менделевий 2 2 : 6 2 610 2610 14 2 6 10 (12) 26(1) (2) ' • -
102 Нобелий 2 2 6 2610 26 10 14 2 6 10 (13) 26(1) (2 )
Пернодичееная таблица химических элементов
Таблица IV
Слева внизу у каждого элемента указан порядковый помер, вверху — атомный вес. Элементы, у которых все виды атомов нестабильны (радиоактивны), отмечены звеадочками. Элементы, названии которых набраны курсивом, получены искусственно^ но существование их в природе не доказано.
Значение символов электронных конфигурации см. т. I, стр. 145
Группы
I 1 ш IV V VI VI. VIII
Периоды и подгруппы
Ряды электронная конфигурация побочная главная □обочная главная побочная главная побочная главная побочная главная побочная главная побочная главная побочная главная водгруппа (нулевая группа)
1 1,00797 !Водород 1s 4,0026 гГелим 1s*
2 Первый короткий период Оболочка Не-}- 6,939 зЛмтий 2s 9,0122 ^Бериллий 2s2 10,811 ьБор 2»22р 12,01115 вУглерод 2s42p2 14.0067 7А30Т 2s*2p® 15,9994 аКислород 2s22₽« 18,9984 ,Фтор 21«2р» 20,183 ]фНеон 2$*2р*
3 Второй короткий период Оболочка Ne-| 22,9898 и Натрий За 24,312 12Магммй 3s* 26,9815 рАлюмнмни 3s23p 28,086 ^Кремний 3*23р2 30,9738 „Фосфор 3*23р® 32,064 1НСера 3№3р< 35,453 17Хлор 3s83p6 39,948 „Аргон 3$*3рв
4 Первый длик-иый период Оболочка Ат + 39,102 jgKa.'lllH 4 s 40,08 гоКальцпй 4s* 44,956 2|СККНДМП 344s4 47,90 ггТмтки 3424$* 50,942 гзВкнадим 3^4*2 51,996 2<Хром 3464$ 54,9381 ^Марганец 34*4ss 55,847 ^Железо 34*4$* 58,9332 ^Кобальт 3474s* 58,71 ^Никель 34*4s*
5 Оболочка Аг 4-3<ро-Ь 63,54 геМедь 4s 65,37 зоЦинк 4s* 69,72 31 Галлин 4s24p 72,59 згГермамим 4s24p2 74,9216 ззМышьмк 4$24р3 78,96 здСелем 4$*4р« 79,909 з^Бром 4$*4р* 83,80 2оКрННТОИ 4$*4р*
6 Второй ДЛИННЫЙ период Оболочка Кг-]- 85,47 37Рубндин 5s 87,62 з^Стронцин 5s« 88,905 звИттрин 445s4 91,22 40ЦмрКОШ1й 44*5$* 92,906 цНнобий 4445$ 95,94 /^Молибден 44*5$ 97 4дТео;*<ецый» 4465s* 101,07 102,905 ^Рутений „Родий 44’5s 44*5$ 106,4 ^Палладии 4410
7 Оболочка Кг 107,870 47Серсбро 5s 112.40 шКадмнп 5*2 114,82 4!>11||ДМН 5s25p 118,69 ^Олово 5*‘25р2 121,75 ь(Сурьма 5s25p3 127,60 52Теллур 5s25p< 126,9044 53Н0Д 5s25p5 131,30 54 Ксенон 5$85р*
8 Треткй длинный период Оболочка Хе-}- 132,905 ьзЦеаий 6s 137,34 ^Бармн 6*2 138,91 &7Лантаи 5(76*2 Лантаниды 58-71 178,49 72Гафмий 4/И5446$* 180,948 73 Тантал 4/145da6*2 183,85 74Вольфрам 4/Н5446*2 186,2 7&Ремнй 4/1*5466$* 190,2 192,2 195,09 76Осыий 77Ирндвй 78Платниа 4/м54*6$* 4/1*54*6$* 4/1*54’6$
9 Оболочка Хе + 4/н +5rfio 4- 196,967 79ЗОЛОТО 6s 200,59 «Ртуть 6,2 204,37 81Таллнй 6*’26р 207,19 вгСвинсц 6*26р2 208,980 g3 Висмут» 6*26р’ 210 ^Полоний» 6s*6p< 210 кАстатмп» 6s*6p6 222 деРадои» 6$*6р®
10 Четвертый длинный период Оболочка Rd 4- 223 в7Фраицин* Is 226 деРадпй* 7s2 ^Актиний* 647s2 232,038 УоТормй* 64*7s4 231 ^Протактиний» 6d37s2 238,03 «Уран» 6447$* Трансурановые элементы (см. ниже)
140,12 140,907 144,24 147 150,35 151,96 157,25 158,924 162,50 164,930 167,26 168,934 173,04 174,97
Лантал иды: ьвЦерий ьоПраэеоднм „Неодии ^Прометий* 02Самармй взЕвропмй деГадолммий в,,Тербий иДиспрозвП ^Гольмий «Эрбий тТу.|ВЙ ТоИттербмй 7|Лютеций
Оболочка Хе4- 4/546s2 4/36s* 4/46s* 4/»6s® 4/’6s2 4/’546s* 4/05465* 4/»5d6s* 4/'»5<76s* 4/i‘5<76s2 4/l«6s8 4/«6s« 4/14546s*
237 242 243 247 247 249 254 253 256 255 257
Трнисураноиые: взНентувий» гцПлутомий» ^Америций* ^Кюрий* ^Берклий* «я Кал uffiopttuit* эо-Тй Htametf кий • \&Фермий* миМемЭелееий» 102» 103е
Оболочка Rn-]- 5/»7s* 5f*fid7s* 5f«ed7s* bfbdls* 5/»647s» 5/*647*-2 5/i*647s«? 5/“647s*7 5/l*647s2? 5/«647s2? 5/l4647s*?
а В табл. IV указаны атомные аеса по данным Международной комиссии по атомным весам на 1962 г,- Прим. ред.
6 В J961 г. был искусственно синтезирован изотоп элемента 103, названного лоуренсием. Его массовое число предполагается равным 257.— Прим. ред.
в В 1966 г. в СССР в Объединенном институте ядерных исследовании был обнаружен элемент 104.— Прим. реО.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбция 722— 726
Адский камень 433
Адсорбция 722—727 _
активированная 725
закон 72/
из растворов 725
селективная 725
тепло 722
электролитов 725
Азурит 395
Аккумулятор Элисона 335
Активаторы 756
Активирование 791
Активные осадки 558
Активные переходные состояния 786
Актиниды 14
Актшшп 47—51, 59—61, 538, 569, 574
; брОМИД 61
валентность 47, 48
галогениды 61
ионы 47
искусственный 538
кристаллическая решетка 50
нейтральные атомы 47
общие сведения 47—51
окись 48, 61
окспфторид 61
оксихлорид 61
положение в периодической системе 6'9
радиоактивность ВО
радиоактивный ряд 569, 574
распространение в природе 59 соединения 47, 49, 50, 60, 61 — /гомсополярные 50 — содеобразные 47, 49
\ — фосфат безводный 60
— халькогениды 61
— хлорид 61
— энергия ионизации 49
Актиний D 574
Актиноуран 60, 569, 570
Америций 649-660, 676-678, 680
валентцость 655
галогениды 677
изотопы 651
исторические сведения 658
металлический 678
обтцйе сведения 649
окислы 677
распространение в природе 660
Амикроны 705
Аммиакаты 409, 770
Аммины 409
Аммонолиз 770
Аморфные вещества 786
Амфотерные вещества 762, 768
гидроокиси 768
Анатаз 77
Ан сольвокислоты 768
Ансол ьв оценивания 768
Антинейтрон 6'00
Антипротон 600
Апатитовой группы соединения 119
Атакамит 395
Астатин 638
Астона правило 634
Атмосфера 686
Атом 585, 586, 598—648
искусственные превращения 598—648
масса 585
массовые числа 586
меченый 626
превращения под действием а-лучей 598
строение ядра 608
Атомная бомба 640
Атомная энергетическая установка 640
Атомный котел 669
Аутокатализ 754
Аутупит 207
Аэродисперспые системы 731
Аэрозоли 732
Бадделеит 87
Белопозит 180
Белый никелевый колчедан 331, 337
Берклий 649—660, 673—680
валентность 655
изотопы 651
исторические сведения 658
общие сведения 649
распространение в природе 060
Берлинская лазурь 307
Берцелианит 399
Бессемеровский процесс 396
Бильтца и Вайбке правило 455
Бихроматы 146, 148, 171—175
Ионы 171
калия 174, 175
натрия 174
серебра 172
Блеклые руды 395, 487
Болотная руда 271
Больцмапа константа: 599
Бомбардирующие частицы 604
Бора—Зоммерфельда теория 15
812
Предметный указатель
Бордосская жидкость 404
Борпит 395
Браунит 232
Брейтгауптит 331
Брепстеда теория кислот и оснований 762
Броггорит 207
Бронзит 300
Бронзы 198, 402
Броуновское движение 711
Брохантит 414
Бупзепит 334
Бурый железняк 271, 292
Валентность 12
Валентные силы 582
электроны 39
Ванадаты 116, 118, 119
Ванадий 105—122
аналитические сведения 121
гидрид 106
исторические сведения 109
кристаллическая решетка 106 общие сведения 105—107 окислы 107, 110
— теплоты образования НО получение 109 применение 110
пятиокиси теплота образования 107 распространение и природе 107 свойства 109
соединения 108, 110—121
сплавы, 107
Ванадий(П), 111, 112
ацидовападаты 111
гексациапованадат калия 112
гидроокись 112
окись 111
соединения 111, 112
сульфат 112
сульфатованадаты 112
хлорид 112
Ванадий(Ш) 111—114 ацидовападаты ill бромид 113 гексацианованадат калия 113 гидроокись 112 иодид 113 окись 112
оксаЛатованадаты 113
рода нова надаты 114
соединения 111—114
сульфат ИЗ
сульфит 114
фторид 113
фторованадаты 113
хлорид 112
Банадий(1У) ill, 114—116
гипованадаты 111
окись 114
оксалатованадаты 116
о ксосульфатова надаты 115
роданованадаты 116
соединения Ilf, 114—116
сульфид 116
сульфитов анадаты 116
тетрафторид 115 тетрахлорид 114 тиованадаты 116 фторованадаты 115 Ванадий(У) ill, 116—122 ацидовападаты 111 ванадиевые кислоты 118, 119 галогениды 121 галогенова надаты 121 гексафторованадаты 121 окситрихлорид 121 пентасульфид 120 пероксованадаты 111 пер оксосоединения 120 пятиокись 111, 116—118 — кристаллическая решетка 117 соединения 111, 116—122 тиованадаты 111, 120
Ванадил 114, 115 дихлорид 114 сульфат 115 Ванадинит 107, 119 Вестона нормальный элемент 491 Вивианит 300
Видемана и Франца правило 23
Видна 313
Виллемит 458, 474
«Водоподобные» растворители 765, 769’ Вольтаит 305
Вольфрам 34, 136—142, 191—206, 381 аналитические сведения 206 атомный радиус 138 ацидо-соедипепия 205 валентность 136, 194 галогениды 194, 195 гексавольфрамовая кислота 197 гексакарбонил 381 дивольфрамат 197 ионные радиусы 138 исторические сведения 191 карбид 193 коллоидный 192 кремневольфрамовая кислота 200 кристаллическая решетка 137 метавольфрамовая кислота 197, 202,. 203 общие сведения 136—139 i
Окислы 139, 195, 196 •
— молярные теплоты образования 139 оксалато-соли 205 оксогалогениды 205 оксртиовольфраматы 195 пассивируемоеть 192 пер оксо вольфраматы 199 пероксовольфрамовая кислота 199 поливольфраматосиликаты 200 поливольфраматы 198 получение 192 применение 192, 193 расплавленный 192 распространение в природе 191 свойства 192, 205, 206 соединения 194—206 — обзор 142, 194 — с металлами 142
Предметный указатель
813
соединения с неметаллами 142 сплавы 134, 139—142
сульфиды 195
температура кипепия 137
тиовольфраматы 195
Вольфраматоарсепаты 200
Вольфраматобораты 200
Вольфрамата кремневая кислота 201
Больфраматофосфаты 200
Вольфраматы 136, 193, 196—205 кальция 198
мета(вольфраматы) 196
натрия 193, 198
нормальный 196
нар а (вольфраматы) 196
Вольфрамит 191
Вольфрамовая кислота 196—205
охра 191
сиш, 199
Вольфрамовые бронзы 198
Вторичные лучи 552, 553 отрицательные 552 положительные 553
Вюртцит 407, 469
Вюстит 290
Гадолиний 51, 511—530, 534, 535
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515
ионы 547
исторические сведения 523
кристаллическая решетка 520
общие сведения 511
окись 517
получение 529
применение 529
распространение в природе 528
свойства 520
соли 517
Галмей 458, 473, 474
Галогенокарбопилы металлов, обзор 376—381
Гальванический элемент 461
Гальваническое серебрение 435
Гапа законы 568
теория двойного распада тория С 559
Гаркипса правило 692
Гарниерит 331
Гаусманпит 231
Гафпнй 62-68, 95-98, 576
валентность 64
галогениды 64, 98
изотоп 576
исторические сведения 95
кристаллическая решетка 63, 95
молекулярные объемы простых соединений 96
нитрид 68
общие сведения 62—65
окислы 63, 96
отделение от циркония 97
отношение к водороду 63
работа ионизации 64
соединения 96, 97
удельная электропроводность 68
Геденбергит 300
Гейгера—Мюллера счетчики 602
Гейслера сплавы 229
Гели 707, 717—720, 729
точки превращения 729
Генри закон 722 :
Герсдорфит 331 I
Гетер оно л икис лоты 119, 137, 200, 201,
203
кристаллическая решетка 201
Гетит 292, 304
Гиббса закон адсорбции 727
Гидратная изомерия 74
Гидрозоли 706
Гидросфера 686
Гиперстен 300
Гистерезис 729
Глазера пузырьковые камеры 602
Гольмий 511—530, 535
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515 ’
исторические сведения 523
криетал дичее кая решетка 520
оощие сведения 511
окись 517
получение 529
применение 529
распространение в природе 528
свойства 520
содн 517
Графит 799, 800
коллоидный 800
соединения 799
фторид 800
Гримма правило 31
Гринокит 476, 481
Гроссмана реактив 343
Групповой реагент на анионы кислот 433
Грэма и Бунзепа закон 780
Гюбнерит 191
Гюгпетова зелень 154
Дагерротипия 431
Деварда енлав 403
Дейтерий 591—597
исторические сведения 592
модификации 593
окись 591—597
— свойства 594
— кристаллическая решетка 595
— обменные реакции 596
получение 592
свойства 593
скорости реакций 595
соединения 597
спектр атома 594
химическое поведение 595
Дейтроп 591
Демпстера аппарат 581
Дефект массы 609
Дефекты структуры 36, 37
814 Предметный указатель
Диализ 708
Диоптаз 395
Дискроэит 420
Дисперсионный анализ 710
Дисперсные системы 705
Диспрозий 511—530, 535
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515
исторические сведения 523
кристаллическая решетка 520
общие сведения 511
окись 517
получение 529
применение 529
распространение в природе 528
свойства 520
соли 517
Дифформиыё системы 733
Диффузия 779, 780
закон диффузии в растворах 779
коэффициент диффузии 780
Доменный процесс 273
Дюрра на соль 325
Европий 511—530, 534, 535: аналитические сведения 535 атомный объем 522 валентность 512, 527 гидроокись 515 исторические сведения 523 кристаллическая решетка 520 общие сведения 511 окись 517 получение 529 применение '529 распространение в природе 528 свойства 520 соли 517
Желатинизация 717
Железа группа 259, 260, 263
Железистосинеродистая кислота 301
Железная шпинель 294
Железный колчедан 271
купорос 289, 299
шпат 271, 299
Железо 45, 226, 259—311, 691, 795, 796
абсорбция водорода 268
аналитические сведения 311
атомарное 286
борид 45
валентность 260, 263
гидраты окислов 290
гидроксотио-соли 297
гидроокись 268
двойные сульфиды 297
дисульфид 268, 296
иододипитрозилжелёзо 310
иопный радиус 263
исторические сведения 272
карбопилы 309
ковкое 287
кристаллическая решетка 262 литое 276, 277, 279, 287 модификации 280, 281, 283 моносульфид 268 общие сведения 259—263 окислы 268, 290, 294 пассивирование 282 получение 272—278 распространение в природе 271, 691 растворимость неметаллов в железе 286
ржавлеяиё 279, 795, 796 сварочное 276, 287 силицид 45 система Fe—Сг 226, 284 — Fe — Ми 226, 286 — Fe — Ni 284 — Fe — Si 285 соединения 269, 289—311 — комплексные 269 i: — нитроаил-соединеция 290, 310 — нитропруссидные 290 — пруссидиые 290:-’ — сернистые 295 сплавы 226, 263, 282—284
— с углеродом 282—284 тетранитрозил 310 техническое 272, 282, 287 электронная конфигурация 270 Железо(П) 289—291, 295, 297—301 аммиакаты 289 ацидо-соли 289 бромид 298 галогениды 297 гпдрокарбонат 300 гидроксо-соли 289 гидроокись 291 дисульфатоферраты 299 дитирнат 297 иодид 298 карбонат 299 нитрит 298 окись 290 оксалат 300 оксалатоферраты 300 т?
перхлорат 298 роданид 298 силикаты 300 соединения 284 соли 297—301 сульфат 299 сульфид 295 сульфит 297 : тиосульфат 297 фосфаты 300 фторид 297 фтороферраты 297 хлорид 268, 290 298 хлороферраты 298 цианид 300 цианоферрат калия 301 цианрферраты 300, 301
— попы 301
Железо(Ш) 289—294, 302-307 аммиакаты 289 ацидо-соли 289
Предметный указатель 815
бромид 304
галогениды 302 — 304
гидроксо-соли 289, 293
гидроокись 292, 293
— амфотерность 293
патрат 305 s
окись 291—293
— модификации 292, 293
— применение 292
оксалат 306
оксалатоферраты 306
оксихлорид 304:
перекись 290
перхлорат 304
— гидролиз 304
роданид 306
род ано ферраты 306
силикаты 306
соединения 289
соли 302, 306
сульфаты 290, 305
— двойные 305
: — основные 305
ферраты 293, 294
фосфат 3Q6
фторид 302
фтор о ферраты 302
хлорид 289, 302
хлороферраты 302
цианоферраты 307
Железо(ГУ) 289, 309
ферраты 289, 309
Железо(УТ) 289, 309
Желтая кровяная соль 300
свинцовая руда 180
Желтый никелевый колчедан 330, 336
Жидкая двуокись серы 774 (см. т. I)
Жидкий аммиак 769
сероводород 774
фтористый водород 772
Замедлитель 666
Земельные кислоты 105
Земная кора, состав 686
Зеркальная изомерия 329
Золотая кислота 446, 448' : ..
соль 449
Золото 39, 40, 386—394, 439—451
алюминат 40
аналитические сведения 451
атомный радиус 389 валентность 39, 386, 445 гидрид 392, 441
— твердый 392
гидрозоли 443
— получение 443
гремучее 445
ионный радиус 389
ископаемое 439
исторические сведения 393
коллоидное 443, 444
комплексообразование 387, 445 кристаллическая решетка 389, 441
намывное 439
нормальный потенциал 442
общие сведения 386—392
получение 439, 440
— амальгамированием 439
— гидравлической разработкой 439
— квартацией 440
— промыванием 439
—хлорированием 440
— цианидным выщелачиванием 440
— электролитическим отделением 440
поляризующее действие 390
применение 444
проба 444
распределение электронов в атоме 388
распространение в природе 439
растворимость 390, 393, 394
свойства 440 ;
соединения 390, 393, 394, 445—451
! соли 387
.... спектр 387
свлавы 39, 392 :
твердый раствор 389
физические константы 389
Золото(1) 43, 392, 446—448
бромид 447
галогениды 392, 446, 447
иодид 447
карбонилхлорид 447
окись 392, 446 :
— растворимость 392
соединения 446—448
сульфид 392, 448
— растворимость 392
сульфитоаураты 448
тиосульфатааураты 448
хлорид 392, 446 :
— растворимость 392
цианид 447
цианоаураты 447
Золото(Ш) 392, 441 448—451
ацетатоаураты 451 ,
бромид 449
гидроокись 441, 448
иодид 450 ' ।
окись 392, 448 ’’
— растворимость 392
роданоаураты 450
соединения 448—451
соли 450
сульфатов ураты 451
сульфид 392, 451
— растворимость 392
тетрапитратозолото, кислота 450
тетрахлороаурат натрия 449
хлорид 392, 449
— растворимость 392
цианид 450 •
цианоаураты 450
Золотохлористоводородпая кислота 444, 449
Золя стабилизация 714
Изобары 568, 601
Изодисперсные системы 707
816 ПреЗметний указатель
Изоляторы 26
Изомерия Ш6
Изомерный переход 572
Изоморфизм соединений 698
Изо поликислоты 203
Изотопия 566, 580—597, 601
— стабильных элементов 580—597
Изотопы элементов 43, 61, 84, 85, 566, 582, 591, 617—620, 638
— неустойчивые 618—620
— применение 591
— радиоактивность 617
— разделение 582
Ильменит 79
кристаллическая решетка 79
Индий 47
Индифферентные раствернтелл 760, 764
Интерметаллические соединения 40, 41, 43, 55
— правила образования 40
— структура 41
Интерметаллические фазы 28, 33—36, 37—39
Ионизация газов 540
Ионий 572
Ионообменные синтетические смолы 726
Ионы газов 540
Ирвдий 259—263, 267, 343—346, 354, 358—361, 374
аналитические сведения 374
валентность,260
галогениды 360
гидрат двуокиси 359
ионный радиус 263
исторические сведения 344, 358
общие сведения 259—263, 343, 354
окислы 359
получение 344
применение 345
распространение в природе 344, 358
соединения 267, 359—361
— карбонильные 361
— образование 267
соли 360
сплавы 263
сульфиды 360
физичос/тйе свойства 353
химические свойства 358
Искусственно активированные элементы 625
Искусственные полые элементы 634
Иттербиевые земли 535
Иттербий 511—530, 535
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515
исторические сведения 523
кристаллическая рещетка 520
общие сведения 511
окислы 517
получение 529
применение 529
распространение в природе 528
свойства 520
соли 517
Иттриевые земли 534
Иттрий 47—59
аналитические сведения 59
ацетат 54, 58
ацетилид 50
валентность 47, 48
галогениды 56
гидроокись 56
гомеополярпые соединения 50
ионы 47, 54
исторические сведения 51
карбид 50
карбонат 54, 58
кристаллическая решетка 50
нейтральные атомы 47
нитрат 54, 57
общие сведения 47—51
окислы 48, 54, 55
оксалат 54, 58
получение 52, 53
распространение в природе 51
свойства 52
соединения 47, 49, 54—59
— интерметаллические 55
~ перекисные 56
— солеобразные 47, 49
— теплота образования 49
соли комплексные 54
сплавы 50
сульфат 54, 58
сульфид 54
фосфат 54
фторид 54, 56
фторокись 57
хлорид 54, 57
— безводный 57
— Двойной 57
Энергия ионизации 49
Кадмий 39, 41—43, 452—458, 476—486
алкилы 474
амид 481
аналитические сведения 486
атомный радиус 452
аутокомнлексы 479
бромид 482
валентность 39, 452
галогениды 483
• — аутокомплексы 483
— двойные 483;
гидрид 475 _
гидр ок со к адм ийты 481
гидроокись 481
иодид 482
ионпый радиус 452
исторические сведения 476
карбонат 485
кристаллическая решетка 452
нитраты 484
— основные 484
нитрит 484
— двойной 484
нормальный потенциал 477
общие сведения 452
окись 480
Црейметный указатель .. . 817
оксалат 488
поя учение 476
поляризующее действие 455
применение 477
распределение электронов в атомах 454
распростри ненке в природе 476
роданид 484
двойной 484
свойства 476
соединения 41,, 454—-456, 479—486
— алкильные 455
— с водородом 454
- интерметаллические 41
соли 479, 480
— комплексообразование 479
— смешанные основные 480
сплавы 455
сульфат 485
сульфид 4S1
твердые растворы 456
физические константы 453
фторид 482
хлориды 480, 482
— безводный 482
— основной 480
— — кристаллическая решетка 480
цианид 481, 484
— двойной 481
Калаверит 439
Калифорнии 649—660, 678—680
валентность 655
изотопы 652
исторические сведения 658
общие сведения 649
распространение в природе 660
Каломель 489, 4S7
Каломельный электрод 489
Каналовые лучи 539, 581
Капиллярная депрессия 727
конденсация 722, 728
Капиллярные силы 727
трубки 727
Карбо нил гидр иды металлов 376—380, 383
Карбо пил нитр о аил ы мета ллов 384
Карбонилы металлов 375—385
— исторические сведения 375
— обзор 376—380
— свойства 380
— синтез 385
— смешанные 384
— строение 380
— структура 382
Карнотит 107
Касселева зеленая 246
Кассиев золотой пурпур 451
Катализ 734—736, 752/ 756, 758
биологическое значение 735,
гетерогенный 736, 753
гомогенный 736 , 752
исторические сведения 735
отрицательный 756
теория 735
Катализатор 734, 750,'754—757
действие 750, 754
J/4 52 г, Реми
отравление 734, 755:
Ренея 757 f смешанные 756 Каталитической активности мера 758 Катафорез 712, 730 Катодные лучи 539, 581 Квасцы 289, 305, 324 железные 289 желёзо-аммоаийпые 305 желез о-калиевые 305 кал иево-коб альтовые 324 Кленетедтит 305' Киноварь 487, 496 .
Кислотоподобные вещества„768 Кислоты 763 определение 763 Кларка элемент 491 Клевеит 207 Клокманипт 399 Коагуляция 712 Кобальт 43, 45, 259—270, 311—330 абсорбция водорода. 268 аналитические сведения 330 арсениды 317 борид 45 валентность 260, 263, 313 гидроокись 268, 314 дисульфид 268 ионный радиус 263 исторические сведения 311 кристаллическая решетка 262, 312 мопокись 315 моносульфид 268 НитроЗилгаяогеннды 317 « общие сведения 259—263 окислы: 268, 312, 314, 315 получение 312 применение 313 распространение в природе 311 свойства 312 соединения 264—267, 269, 313—330 — карбонильные 317 — комплексные 269 — нитроайльные 317 — образование 264—267 сплавы 43, 263 сульфиды 316;
гное у ль фа тный комплекс 314 хлориды 268, 315 электронная конфигурация 270 Кобальт(П) 313; 318—324 аммиакаты солей 323 ацетат 313 323 /•
бромид 319 :: бромиды основные 321 галогениды; основные 319 иодид, 319 карбонат 323 ./ карбоматокобалЬтаты 323 нитрат 313, 322 ширит 322 нитриты двойные 322 оксалат 323 оксалатокобальтиты 323 роданид 321 родадокрбальтаты 321
818
Предметный указатель
сояв 318—324
сульфат 313, 322
сульфат 322
сульф игокобадьтатьг 322
фторид 319
хлорид 313, 318
— зеленый основной 320
— — двухслойная решетка 320
— розовый основной 320
— — решетка 320
хлорокобальтаты 319
цианид 321
цианокобальтаты 321
Кобальт(Ш) 313, 314, 324—329
аквокомплексы 327
акдоанминкобальт 326
аммины мпогоядерные 329
аммпиные комплексы 326, 327
ацидо-соедипеяия 314, 327
гексанитроцобальтаты 325
гекса нитрокобальтат аммония 325
— калия 325
— натрия 325
— рубидия 325
— цезия 325
гексацианокобальтаты 325
геясациапокобальтовая кислота 325
двухслойные комплексы 328
координационное число 314
ексалатокобальтаты 324
соли 313, 324
— простые 313
сульфат 324
фторид 324
фторокобальтат калня 324
хлорид 324
— аквопептаамминкобальта 326
— гексааммипкобальта 326
— яитропентааммингсобальта 329
— хаоронептаамминкобальта 326
Кобальтовая бомба 625
Кобальтовый блеск 311, 317
колчедан 317
шпейс 311, 317
Ковел лип 395
Кокимбит 305
Колларгол 425
Коллоидная химия 704—733
Коллоидное состояние 704, 705
— общие сведения 704
Коллоидные растворы 706—720
— осмотические свойства 711
— получение 707
частицы 710
— размеры 710
— форма 710
Коллоиды 705, 712, 716, 717, 720
защитные 720
изоэлектрическая точка 717
, исторические сведения 705
лиофильные 716
лиофобные 716
необратимые 715
обратимые 715
поверхностная активность 720
электрические свойства 712
Колорадоит 487
Колошниковые газы 275
Колумбит 250. !
Комплексные ионы 153, 30(
двухслойные 301
Комплексы внедрения 162, 269
Комптона эффект 556
Конарит 341
Конвертор: 272, 277
продувка 272 i
Конвертирование 272, 277
методы 277
Константа распада 549, 550, 577
— определение: 550
Коррозия 796, 797
аатцита 797
Космические лучи 600
Коссияит 487
Коэффициент экстинкции 709
Красная кровяная соль 307
цинковая руда 458, 466
Красный железняк .271, 291
никелевый колчедан 331, 337
К ремнец пиковая руда 458, 474
Кривизна поверхности и давление вара 727
Кристаллическая решетка 36, 37, 42, 44, 45, 407
— вакансия 37
— гексагональная 45
— дислокации 36
— искаженная 36
— линии свсрхструктуры 42
— междоузлия 37
— правильная 44
— типы 42, 407
Ксантокон 420
Куприт 395, 405—407
кристаллическая решетка 406, 407
Кюрий 649—660, 678—680
валентность 655
изотопы 651
исторические сведения 658 7*
общие сведения 649
распространение в природе 660
Лавеса фазы 44
Лангмюра формула 724 '
Лантан 42, 47—59, 524, 526
аналитические сведения 54
ацетат 54, 58
ацетилид 50
валентность 47, 48
галогениды 56
гидрид 49, 53
гидроокись 5В
ионы 47, 54
исторические евдеяпя 51
карбид 50
карбопаты 54, 53
кристаллическая решетка 50
нейтральные атомы 47
нитраты 54, 57
Предметный указатель f
819
нитрид 53
общие сведения 47—-51
окисли 48, 54—59
оксалат 54, 58
получение 52, 53
распространение в природе 51
свойства 52, 53
силицид 55
соединения 47, 49, 54—59, 526
— гомеопблярные .50
— интерметаллические 55
— перекисные 56
— со ле образные 47, 49
— — теплота образования 49
соли комплексные 54
сплавы 50
сульфаты 54, 58
сульфид 54
фосфат 54
фторид 54, 56
хлорид 48, 54
— безводный 57
— двойной 57
чистота 53
энергия ионизации 49
Лантанидное сжатие 17, 516
Лантаниды 14—16, 48, 51
Латупъ 39, 41—43, 463
желтая 463
красная 463
решетка* 43
Лаурит 347
Лимонит 292
Литопон 463. 469
Литосфера 686
Локальные элементы 790
Лучи отдачи 552
Льюиса теория кислотно-основной функции 775
Люминесценция 541
Лютеций '511—530, 535
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515
исторические сведения 523
кристаллическая решетка 520
общие сведения 511,
окись 517
получение 529
применение 529
распространение в природе 528
свойства 520
соли 517
Магнетит 295
Магнитный железняк 271
колчедан 295, 331
Магнуса соль 373
Максвелла закон 744
Малахит 395, 416
Мапганатометрические методы 247
Майга инн 230
Маи гаи нт 233
Манганозит 232
Марганец 42, 43 224—249, 691 аналитические сведения .248 атомный радиус 226 валентность 224, 225, 230 гидроокиси 231 ионный радиус 226 исторические сведения 228 , карбонил 229
' красная окись 231, 232
кристаллическая решетка 42, 43 Модификации 42, 43 нитрид 229 нормальный потенциал 229 общие сведения 224—226 окислы 23 i получение 228 применение 230 распространение в природе 226, 691 растворение в кислотах 229 свойства 229 система Мп — Fe 226 соединения 225, 230—248 — теплоты образования 225 — применение 230 сплавы 226 сульфид 249
Маргапец(П) 229—232, 234, 236—241 аммониймаРганецфосфат 239 арсенат 239 ацетат 239 бораты 240 бромид 237 гидроокись 232 диарее, щтомарганЦовая кислота 239 дисульфид 240 дихлорид 229 иодид 237 ион 236 карбопат 238 — основной 238 нитрат 239 опись 230—232
— модификация 231 оксалат 240 роданид 241 родапоманганаты 241 селеиид 240 силикаты 241 соли 234,: 236—241
— двойные 236 сульфат 230, 237, 238 — двойные соли 238 сульфид 239 сульфит 241 фосфат 239 фторид 237 хлорид 230, 237
— основные соли 237 цианид 241 цпаномапганагы 241
Марганец(Ш) 232, 233, 241—243 ацетат 243 гидрат окиси 233 квасцы 242 окись 232
52*
820 Предметный указатель
оке ал ат ома ига наты 243 соли 241—243
! сульфат 242 сульфатомангапаты 242 триарсепатомаргапцоаая кислота 243 фосфат 243 фторид 242 хлорид: 242 хлороманганаты 242 циансмангаяаты 243
Маргапец(1У) 230, 233-235, 243—247 ацидомангапачы 243 гексахлоромапганат 244 глицерилманганиты 244 двуокись 230, 233—235, 244, 245,247 — амфотерность 235 — гидрат 234, 245 — применение 234 манганаты 244-^246 марганцовая кислота 244 пербкеомангапаты 245 соединения 243—245 сульфат 244 хлорид 244 хлрроманганаты 244
Марганец(у) 230, 245, 246 манганаты 245, 246
MapraneitfVI) 230, 234, 245, 246, 249 манганаты 230, 234 245, 249
Марганец(УП) 234, 235, 246—248 перманганат калия 246, 247 — кальция 246 перманганаты 234, 246 — ноны 235, 247:, 248 семиокись 235
Марганцовая кислота 235, 246
Марганцовые краски 230, 233, 243, 245 — коричневая 233 — синяя 245 — фиолетовая 243 —• черная 233 Марганцовый блеск 240 купорос 238
Марказит 296
Массовое число 568, 573 Масс-спектрография 580 Маттауха правило 634 Медная лазурь 403, 416 Медноборатная жидкость 404 Медно фосфатная жидкость .404 Медный блеск 395, 409 колчедан 296, 395 купорос 404, 414 — техническое приготовление 414 штейн 396
Медь 24, 30, 34, 35, 39—43, 45, 46, 386— 420, 691 аналитические сведения 419 атомный радиус 389 валентность 39, 386 гидрид 392, 399 ионный радиус 389 исторические сведения 393 каталитические свойства 403 _
комплексообразование 387, 404
краски 403 ’
кристаллическая решетка 389 нормальный потенциал 401 общие сведения 386—392 получение 395
— обжиг395 *
— — восстановительный 396 ]
— — с дутьем 396 — плавка па штейн 395 применение 401 и
распределение электронов в атоме 388 распространение д природе 394, 691 : растворение в разбавленной азотной
кислоте 401 У
растворимость 390, 393, 394 ,
рафинирование 398 1
свойства 398 ,
силицид 40 соединения 390, 393, 394, 402—419 —каталитические свойства 403 ;
— образование 390, 393, 394 — применение 402 >
соли 387 спектр 387 сплавы.30, 34, 35, 39, 43, 45,.46, 392, 399 j твердые гидриды 392“ физические константы 389 цементная 401 энергетические уровни 24 Медь(1) 392, 405 —411 ацетат 408 бромид 408 "••
галогениды 408 гидрид 410 нитрид 409 окись 392, 405 ’ ••••••,
— растворимость 392 оксалат 408 роданид 408 соединения 405—411 сульфид 392, 409 растворимость 392 сульфит 408 фторид 408 ...
хлорид 392, 406 ’ <
цианид 408
Медь(П), 392, 411—418
ацетат 415: бромид 413 гексоловыа соли 417 гидроксокупраты 412 ГИДРООКИСЬ 411 ::
иодид 413 .
карбонат 416 нитрат 415 нитрит 415 окись 392, 411
— растворимость. 392 оксалат 416 : :
роданид 416 соединения 411—418 сульфат 414 j
сульфид 392, 418 ч
— растворимость 392 фторцд 412
Предметный указатель
821
хлорид 392, 412
— растворимость 392
хлорокунраты 413
цианид 416
1едь(Ш), 418, 419
окись 418
оксокупраты 418
соединения 418
тетрагидро к со купраты 418
Лежмицеллярпая жидкость 715
Сезоны 600
йезоторий 562
Йенделевий 649—660, 679, 680.
валентность 655
изотоны 652
исторические сведения 658
общие сведения 649
распространение в природе 660
Металлическое состояние, теория 22—26
Метаплоподобпые соединения 20
Металлы 19—46, 48. 790
второго рода 39, 40
общие сведения 19
первого рода ,49, 40
получение 20, 22
растворение в кпслотах 790
структура 41
химическое восстановление соединений 2i>
чистые1 4 [
электропроводность 28
аффективное алектропное число 25
Метеориты 271. 695
Средний состав 695
Микроны 705
Ми.члопа основание 507
Мицеллы 715
Молибден 136—142, 180—191, 381
аналитические сведения 191
атомный радиус 138
ацидо-соли 190
валентность 136, 183
гексакарбонид 381
двуокись 186
дисульфид 185
дпх,дорид 184
— гидрат 184
долекамолибдатофосфорная кислота 137
иоиные радиусы 138
исторические сведения 180
карбид 182
карбонил 139, 381
коллоидный 182
кристаллическая решетка 137
нитрид 182
— теплота образования 182 общие сведения 136—139 окислы 139. 186
— молярные теплоты образования 139
оксифториды |84
пассивирование 182
пентахлорид 183
пероксомолпбденовая кислота 189 получение 180—182
53 г Реми
применение 183
промежуточные окислы 186
пятиокись 186
распространение в природе 180 ~ свойства 182, 183
соединения 142, 183—190
— с кислотами 190
— с металлами 142
— с неметаллами 142
сплавы 139—142
сульфиды 184, 18а
температура кипения 137
— плавления 137
тетрахлорид 184
тиомолибдаты 185
трехокись 186
трисульфид 185
трихлорид 184
фториды 184
хлориды 183, 184
Молибдаты 136, 187—190, 197
аммония 188
димолибдаты 197
оксосульфатомолибдаты 190
оксофторомолибдаты 190
оксохлоромолибдаты 190
пер оксомолибдаты 189
полимолибдаты 187 Молибденовая синь 188, 189
кислота 187—190
— ангидрид 186
Молибденовый блеск 180, 185, 250
Монацит 98
Монетные сплавы 403
Набухание 717
Нагиагит 439
Наращивания метод 83
Негатив 432
Нейтрино 557, 600
Нейтроны 598—600, 610, 617, 668, 669
быстрые 600
медленные 600
теиновые 599, 668
— коэффициент использования 668
Неметаллы 19, 44
соединения 44
Неодим 511—531
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515
исторические сведения 523
кристаллическая решетка 520
общие сведения 511
окись 517
получение 529
применение 529
распространение и природе 528
свойства 520
соли 517
Лепту паты 662
Нептуний 575, 649—663
валентность 655
822
Предметный указатель
'1—F..
изотопы 650
исторические сведения 658
общие сведения 649
радиоактивный ряд 575, 680 „
распространение в природе 660
соединении 661
Нернста закон распределения 723
Нивенит 207
Никелевый купорос 340
Никель 259—271, 330—343, 691 :
абсорбция водорода 268
„ аммиакаты 351
аналитические сведения 343
антимониды 336
арсениды: 336
ацетат 340
борид 45
бромид 338
валентность 260, 263, 333
галогениды основные 338
гидроокись 268, 334, 335
дисульфид 268
йодид 338
ионный радиус 263
исторические сведения 331
карбонат 340
карбонаты двойные 340
карбонил 337
карбонилгидрид 384
комплекс внедрения 271 :
кристаллическая решетка 262, 332
:: мбйбсульфид 268
. никельдв метил глиоксим 342
нитрат 339
нитраты основные 339
нитрит 339
нитриты двойные 339
общие св едения. 259—263
окислы 268, 334—336
— высшие 335
— г’ двойные 336
оксалат 340
получение 331
— карбонильный метод 331, 332
• оксфордский метод 331
— электролитическое рафинирование 332
применение 333
распространение в природе 330, 691
роданид 339
роданиды двойные 339
свойства 332
силикаты 341
соединения 264—267, 269, 333—343
— комплексные 269
соли 338—342
— инутрикомплексные 341, 342
— двойные 340
сплавы 34, 44, 263, 333
сульфат 339
сульфиды 336
— двойные 336
фрсфаты 340
хлорид 268, 338 А.;
хлоропикелаты 338
цианид 338
цианиды двойные 338
электронная дсопфигурация 270
Ниобий 105—107, 122—127
аналитические сведения 127
валентность. 123
гидрид 106
двуокись 123
исторические сведения 122
кристаллическая решетка 106
общие сведения 105--107
получение 122 :
дятиокись 107, 124, 125
;— теплота образования 107
распространение в природе 122
свойства 123
соединения 108, 123—127 ;
сплавы 107
твердые растворы 108. ‘
тетр а хлор ид 123
трихлорид 124
ниобаты(У) 124
ниобиевая кислота 124, 125, 126
НиобиЙ(У) 124—127
ацидониобаты 425
бро.мокись 127
нитрид 127
окситрихлорид 127 -
пентабромид 127:
пентаиодид 127
.пента фтор ид 127
пентахлорид 126
пероксониобаты 126
пероксониобйевая: кислота 126
Е соединения 124 — 127
фторониобат 127
хлорокись 127 “
Ниобит 122, 128
„Нитрозилы металлов 384
Нитропруссиды 290
Нобелий 649—660, 679, 680
валентность 655
изотопы 652
исторические „сведения 658
общие сведения‘ 649
распространение в природе 66СЩ
j Нуклоны 608 i
Онислите.ги.ный плав 249
Оксигидраты 719
Оливин 300
Органозоли 706
Оса)идение из гомогенного раствора 104
Осматы 354
Осмий 259—263, 267, 343—347, 349г 351-354, 374
анал итиче ские св еден ия 374
валентность 260
галогениды 353
двуокись 352
ионный радиус 263
исторические сведения 344
кристаллическая рещетка 262
общие сведения 259—263, 343, 346
окислы 346, 352
открытие 351
Предметный указатель
823
получение 344
применение 346
распространение в природе 344
селенид 349
соединения 267, 352—354
— координационные 353
сплавы 263
сульфиды 349, 353
теллуриды 349
физические свойства 351
химические свойства 351
четырехокись 352
Осмиридий 344, 347...
Оствальда изотерма адсорбции 723
Палладий 259—267, 343—346, 361—366, 374
аммиакаты 365
аналитические сведения 374
валентность: 260, 361
ионный радиус 263
исторические сведения 344, 362
коллоидное состояние 363 ’ кристаллическая решетка 262 общие сведения 259—263, 343, 361 окислы 364
получение 344
применение 345 ,
распространение в природе 344, 362 соединения 264—267, 364—366
— образование 264—267
соли 365
сплавы 263
сульфат 365
сульфиды 365
физические свойства 362
химические свойства 363
хлорид 365
Папета пранило адсорбции 568
Паркера метод 281
Пассивирование 145, 146, 182
Пассивность 790
Патер а ит 180
Патронит 107
Паяльная соль 471
Пейроне соль 372
Пентландит 336
Пептизатор 707
Пептизация 707, 714
Период полураспада 549
Перманганат калия 230
Перовскит 79
Пиллинга правило 795
Пирагирит 420
Пирит 296
Пиритная плавка 397
Пиролюзит 230, 233, 244
серый 233
Пиррхроит 232
Пластические массы 719
Платипа 34, 259—267, 343—346, 361, 366—375
аналитические сведения 374
валентность 260, 361
гидроокиси 369
ионный радиус 263
исторические сведения 344
коллоидная 367
кристаллическая: решетка 262
общие сведения 343, 361..
окислы 369
получение 344 -
применение 345 ;
распространения в йрироде 344, 366
селенид 370
соединения 264—267, 369—374
— комплексные 371
— образование 264—267
: сплавы 34, 263
сульфиды 369
теллурид 370
физические свойства 366
химические свойства 367
хлориды 370
хлороплатинаты 373 ; s
хлороплатиновая кислота 373
цианоплатинаты 374
цианоплатиповая кислота 374:
Платиновая аппаратура 368
чернь 367
Платиновые металлы 259 , 260, 343 , 344, 690
— распространенность 690
руды 344
Платиновый нашатырь 345, 373
Плутоний 641, 649—660, 663—676, 680
бромиды 674
валентность 655
деление 641 '
изотоны 650
ионы 672
исторические сведения 658
коэффициент размножения 667
общие сведения 649 "
окислы 673
перекись 673
получение 667, 671 !
применение 665
распространение в природе 660
соединения 672 ,
соли 675 ' :
сульфиды 673
фториды 673
хлориды 674
Плутоний(Ш) 675
соли 675
Плутоний(1 V) 676
соли 676 L
Плутонил(¥1) 676
соли 676
Побочные подгруппы 11
Поверхностная Энергия вещества 721
Поверхностное натяжение. 727
Поверхностные явления 720
Позитив 732
Позитроны 600
Полианит 233 ,.
Полибазит 420
Полидисперсные системы 707
Поликислоты 118, 136'
Полоний 538
53*
824
Предметный указатель
Полосатые спектры 582
Полуметаллы 19
Полупроводника 26
Постоянная распада 631
Потенциальный барьер 742
Правила устойчивости ядер 634
Празеодим 42, 511—531, 535
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515
исторические сведения 523
кристаллическая решетка 520
общие сведения .511
окислы 517
получение 529
применение 529
распространение в природе 528
свойства 520
соединения 524
соли 517
Преципитат белый плавкий 506 неплавкий 506
Принцип экви вален гп ости массы и энергии 609
Прометий 511, 518, 521, 531, 535
валентность 512, 527
исторические сведения 523, 526
соединения 531 .
Протактиний 105—107, 133—135, 574, 685
галогениды 135
гидрид 106
двуокись 135
изотопы 134, 685
кристаллическая решетка 106
металлический 134
монокись 135
общие сведения 105
окислы 134, 135
открытие 133
получение 134
пятиокись 134, 135
свойства 134
соединения 134, 135
сплавы 107
Протаргол 425
Противогаз 733
Протон 591, 610
Протонное сродство 765
Пруссидные соединения 290, 308
Прустнт 420
Псиломелан 233
Пудлингование 276
Радий 538
Радиоактивное излучение 537—548, 554—
556, 566, 616, 617, 629, 630
— виды 539—541, 543—548,
554—556, 566, 616, 617, 630
— доза 630
— защита 629
— общая характеристика 538
— окрашивающее действие 542
— пропикающая способность 541
— физиологическое действие 543
— химическое действие 542
Радиоактивное равновесие 552, 561
Радиоактивного смещения законы 565, 566
Радиоактивность 537—579 602 616—
648
единица 602
индуцированная 558 искусственная 616—648 определение 537 открытие 537
Радиоактивные вещества 537, 539, 577 — действие 539 — индикаторы 577
Радиоактивные ряды 549
Радиоактивные элементы 575, 576, 6S9 — влияние ла тепловой балакс земли 699
— положение в периодической системе 575
— распространение 699
Радиоактивный распад 561, 616
— виды 616
— закон 561
Радиоактивный ряд урана — радия 567, 570-574
Радио астат ик 572
Радиоизотопы 627
Радиоторий 562
Радиохимическое определение возраста минералов 699
Раммельсбергит 337
Распит 191
Редкие земли 48, 51, 526
— иттриевые 51
— классификация 526
— цериевые 51
Редкие элементы 687
Редкоземельные металлы 698
Редкоземельные элементы 51, 530—535 — соединения 530—535
Режущие сплавы 313
Резодансное поглощение 668
Рейзе хлорид 372
Рейнит 191
Ренея катализатор 757
Ренат(1У) 255, 250
Ренат(У) 255
Рёнат(У1) 255
Репиевая кислота 253, 255
Рений 224—226, 249—258
аналитические сведения 258
атомный радиус 226
бромид 256
валентность 253
галогенокарбонилы 258
гексафтороренат калия 256
гептасульфид 256
Гидрат двуокиси 254
Двуокись 254
диамидореяиевая кислота 25S дисульфид 256 иодид 256 исторические сведения 250 карбонилы 258
кристаллическая решетка 2S общие сведения 224—226 окислы 254
Предметней указатель
825.
оксихлориды 257 оксопентахлороренаты 257 оксо-соли 255 перренаты 255 полуторная окись 254 получение 252 применение 253 распространение в природе 249 рениепая кислота 253, 255 свойства 253 семноипсь 254 соединения 225, 253—258
— теплоты образования 225 спектр 251 сплавы 225 сульфиды 251, 254 тиоперренаты 256 трехокись 254 трпхлорпд 257 фториды 254, 256 хлориды 254, 256, 257 хлоропентакарбонил 258 хлорорецат калия 257 хлорореиат цезия 257 хдороренаты 257 хлорореняевая кислота 257
Рентген 630
Рентгеновские лучи 539
Ринмана зелень 475
Роговое серебро 428
Родий 259—263, 343—346, 354-358, 374 аналитические сведения 374 бромид 357 валентность 260, 356 иодид 357 ионный радиус 263 исторические сведения 344. 354 кристаллическая решетка 262 общие сведения 259—263, 343, 354 окислы 356 получение 344 применение 346 распространение в природе 344, 354 соединения 356—358
— интерметаллические 355
— координационные 357 соли 356
— простые 356 сплавы 283 сульфиды 356 физические свойства 355 химические свойства 355 хлорид 356
Родозохромовые соли 170
Родонит 241
Родохромовые соли 169
Ртутная роговая руда 487 497 чернь 496
Ртутный электрод 180
— вращающийся 180
Ртуть 39, 41, 452-458, 475. 487—510 амальгамы 490 аналитические сведения 509 атомный радиус 452 валентность 39, 452 гидрид 475
иопный радиус 452
исторические сведения 4S7 нормальный потенциал 4S3 общие сведения 452 получение 487 поляризующее действие применение 490
распределение электрСЕ~з з атомах 454
распространение в ирхрдте 487 свойства 487
соединения 41, 454—458.. ^22—509
— алкильные 455, 50S
— интерметаллическЕ5 2Л
— с азотом 506
— с водородом соли 490
— каталитическое -ретине 490 сплавы 455 физические константы
Ртуть(1) 491, 492, 497—4ЙД, £04—506 бромид 499 галогениды 497—499
— кристалличеепгх нелегка. 499
иодид 499
карбопат 506
нитрат 504 нитрит 505 роданид 504 соединения 491, 492 — получение 492 — свойства 492 — строение 491
сульфат 505
фторид 499
хлорид 497, 498
— кристаллдчесная зеигтка 498
Ртуть(Н) 493, 495, 496, 493—£2£ бромид 502
— кристаллическая оепхльа 502 галогениды 499—503 йодаты двойные 503 иодид 502 карбонат 506 нитрат 504
нитрит 505 *
окись 495 оксалат 506
роданид 504
соединения 493
— получение 493
— свойства 493
сульфат 505
сульфид 496, 506
сульфит 506
сульфитомеркураты 506 тио-соли 49& фторид 503 хлорид 498, 499, 501
— двойные соединения 5£И
— кристаллическая рещягш 498, 499
цианид 503
Рубиновая слюда 292
Рубиновое стекло 444
Руссена соли 290
826 - Предметный указатель
Рутенаты 350
Рутениевая красная 350
Рутений '259—263, 266, 343—351, 374
амминные комплексы 350
аммины 350
аналитические сведения 374
валентность 260
гидроокись 348
двуокись 348
ионный радиус 263
исторические сведения 344
карбонилы 351
кристаллическая решетка 262
нитрозил 351 л
общие сведения 259—263, 343, 346
окислы 346, 348
получение 344
применение 346
распространение в природе 344
селепид 349
соединения 266, 267, 34S—351
— координационные 349
— образование 266
сплавы 263
сульфид 348
теллурид 349
тиоксо-соединения 350
физические свойства 347
фториды 349
химические свойства 347
хлориды 349
Рутил 67
кристаллическая решетка 77
Самарий 511 —531, 535 .
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокиси 515
исторические сведения 523
кристаллическая решетка 520
общие сведения 511
окислы 517
получение 529
применение 529
распространение в природе 528
свойства 520
соли 517
Сверхпроводимость 27
Сверхструктуры 31
Сверхструктурные соединения 32
Седиментация 705
Семейство лантанидов 261
Серебрение 424
Серебро 34, 38—40, .386—394, 420—439
алюминат 38, 40
— кристаллическая решетка 40
аналитические сведения 438
атомный радиус 389
валентность 39, 386
гидрид 392
ионный радиус 389
исторические сведения 393
комплексообразование 387
кристаллическая решетка 389
молекулярное 429, 437
общие сведения 386—392
очистка 421, 422
— аффинирование 422
— электролитическое рафинирование 422
получение 420, 421
— способ амальгамирования 421
— — Паркеса 420
— — Патти неон а 420 поляризующее действие 390 применение 424 распределение электронов в атомах 388 Г
распространение в природе 420 растворимость 390, 393, 394 свойства 422 соединения 390, 393,,394, 425—438
— образование-390, 393, 394
соли 387 ,/ .. -
спектр 387 - •• .'
сплавы 34. 39, 392 физические константы 389 фторид 427 7.
Сереброф 43, 392, 425—437
ацетат 435
бромид 429 . •
галогениды 427—432
— применение в фотографии 431 иодид 430 / карбонат 434. нитрат 432 f окись 392, 426,’ 427
— кристаллическая решетка 427
— растворимость 392
— теплота образования 427
оксалат 435
роданид 436 ’• соединения'425—437 сульфат 434 сульфид 392,: 436
— кристаллическая решетка 436
— распространение в природе 436 ;
— растворимость 392:
сульфит 434
тиосульфат 434
фосфат 436
фторид 427
хлорид 392, 428
— растворимость 392
цианид 435
Серебро(ГТ) 437
дифторид 437
окись 437
соединения 437
— кош лек си ые 437
Серёбро(Ш) 438
соединения 438
Серебряный блеск 420, 436
С и дотов а обманка 469
Сиккативы 236
Сильванит 439
Сименса — Мартена процесс 279, 280, 294-
печь 280
Предметный указатель 827
Синерезис 728
Скандий 47—59
алкил производные 50
аналитические сведения 59
ар ил производное 50
ацетат 54, 58
ацетилацетопат 50
ацетилиды 50
валентность 47, 48
галогениды 56
гидроокись 56 ионы 47—54 исторические сведения 51 карбид 50
; карбонаты 54, 58
кристаллическая решетка. 50
нейтральные атомы 47
нитрат 54, 57
общие сведения 47—51
окислы 48, 54, 55
оксалат 54—58
получение 52, 54
распространение в природе 51
свойства 52
соединения 47, 49, 50, 54—59
::— гомеополярные 50 :
— интерметаллические 55
— еолеобразпые 47, 49
— теплота образования 49
соли комплексные 54
сплавы 50
сульфат 54, 57
сульфид 54
фосфат 54
фторид 54, 56
—.двойной 56
фтороскайдиаты 56
хлорид 54, 57
безводный 57
энергия ионизации 49
Смальта 313
Смарагд 143
Смешанные осадки 788
элементы 585
Солевая изомерия 329
Сольватация 716, 761
Сольволиз 760, 768
Сорбент 722
Сорбируемое, вещество 722
, Сорбция газов 724
Состав внутренних частей Земли 694
Спекание 783
Сперрилит 366
Спинтарископ 541-
Сплавы 45, 46
облагораживание 45, 46
термическая обработка 45
Средняя кинетическая энергия молекул 745
Сталь 193, 276, 287—289, 296
быстрорежущая 193
ковкая 287
сернистая 296
специальная 288
твердение 288
Старение осадков 719
Студни 717720 структура 718 /
Субмикропы 705
Сулема 499, 500
Суспензии 705
Суспенсоиды1 731
Сцинтилляция 541
Сфен 79
Тантал 105—108, 128—133
аналитические сведения 133 ...
гидрид 106
исторические сведения 128
карбиды 133
кристаллическая решетка 106
нитрид 133
общие сведения 105—107
получение : 128
применение 129 пятиокись 107 .
— теплота образования 107
распространение в природе 128
свойства 128
соединения 108, 129—133
— гептафторотанталаты 132
— с болев: низкими степенями окисления 132
сплавы 107, 108
длоряд.ы 132
Танта.ЩV) 129—132
метатаптплаты 130
пептафторид 131
пентахлорид 131
пероксотапталаты 130
пероксотанта л оная кислота 130
пятиокись 129
соединения 129—132
танталовая кислота- 130
фторотанталат калия 132
— натрия 132
фторотантала гы 131, 132
Тапталаты 130
Танталиты 130
Твердые растворы 28—33, 5G :
— аномальные 56
— внедрения 29;
— замещения 29
— неограниченные 29
— ограниченные 29
Тенорит 395
Теплота активации 740
Тербиевые земли 534
Тербий 511—530, 534i 535
аналитические сведения 535
атомный объем 522
валентность 512, 527
гидроокись 515
исторические сведения 523
кристаллическая решетка 520
общие сведения 511
окись 517
получение 529
применение 529
, распространение в природе 528
свойства 520
соли, 517
828
Предметный указатель
Термическое активирование 744
разложение соединений 21
Термолизация 666
Террар 85
Тефроит 241
Техпециевая кислота 252
Технеций 224—226, 249—252
атомный радиус 226
изотоп 251
исторические сведения 250
кристаллическая решетка 252
металлический 252
общие сведения 224—226
пертехнеат аммония 252
— светопоглощение 252
получение 250, 252
применение 252
распространение в природе 249
свойства 251
семиокись 252
сплавы 226
сульфид 251
техиеат аммония 251
Тигельная плавка 276
Тиеманнит 487
Тиндаля эффект 710
Титан 62—82
борид 81
бромиды 72, 76
валентность 64, 71, 72
галогениды 64, 72, 74, 76
иодиды 72, 76
исторические сведения 68
карбид 72, 81
квасцы 64
кристаллическая решетка 63—65
молярная теплоемкость 70
нитрид 68, 72, 75
нормальный потенциал 73
общие сведения 62—65
окислительным потенциал 72, 73
окислы 63, 72
отношение к водороду 63, 70
получение 69
применение 70
работа ионивации 64
распространение в природе 68,
свойства 68
соединения 63, 66, 67, 71—82
— простейшие 72
— теплота образования 63
соли 71 ;
сплавы 65—68
строение атома 64
сульфаты 74, 76
сульфиды 72, 80
удельная электропроводность 68
фосфиды 72
фториды 72, 76
хлориды 72, 74, 75
чистый 70
Титан(П) 73
окислительный потенциал 73
Титан(Ш) 73—75
ацидотитанаты 74
гидроокись 75
двойные соли 75
квасцы 75
комплексные соли 74, 75
нитрид 75
окислительный потенциал 73
окись 75
сульфат 74
— двойной 74
хлорид 74
Титан(1¥) 75—85
алкоголяты 76
аналитические сведения 82
ацидотитанаты 77
р-титановая кислота 78
борид 81
двуокись 77—79
дисульфид 81
карбид 81
— получение 81
ортотитановая кислота 78
пероксотитапаты 80
пероксотитановая кислота 80
соединения 81
— с азотом 81
— с углеродом 81
сульфат 76
сульфатотитанаты 76
сульфид 81
тетра бромид 76
четраиодид 76
тетрахлорид 75
титановая кислота 78
фторид 76
фторотитанаты 76
хлоротитанаты 76
Титанаты 79
Титанил 72
Титановая белая 71
Томасова мука 278
Томпак 403
Топохимические реакции 797, 798
— типы 798
Торианит 207
Ториевый свинец 563
Торий 62—68, 98—104, 558, 564, 565
аналитические сведения 104
ацетат 103
бромид 102
валентность 64, 99
галогениды 64
гидроокись 100
двойной распад 565
двуокись 100, 101 !
— взаимодействие с перекисью водорода 101
иодид 102
исторические сведения 98
карбид 103
карбонатодораты 103
кристаллическая решетка 63, 65
нитрат 100, 102
нитратотораты 102
нитрид 68, 104
общие сведения 62—65
окислы 63, 88
оксалат 103
Предметный указатель 829
оксалатотораты 103 отношение к водороду 63 получение 98 применение 99 работа ионизации 64 радиоактивней ряд 564 распространение в природе 98 свойства 98 силикат 103 силицид 103
соединения 63, 66, 67, 99—104
— теплота образования 63 сплавы 65—68 сульфат 103 сульфатотораты 103 фосфат 103 фторид 102 фторотораты 102 хлорид 101
Торий А 560
Торий В 559
Торий С 554, 559
Торий С' 559
Торий D 560
Торий X 560
Торит 207
Тортвейтит 51
тра ас-Соединение 166
Трансурановые элементы 649—685 радиоактивные ряды 680—-685 спектры поглощения 658
Тритий 591
Троилит 295
Троостит 458, 474
Тулий 511—530, 535
аналитические сведения 535 атомный объем 522 валентность 512, 527 гидроокись 515 исторические сведения 523 кристаллическая решетка 520 общие сведения 511 окись 517 получение 529 nil имен еп ио 529 распространение в природе 528 свойства 520 соли 517
Турнбуллева Синь 307
Ульманит 331
Ультрамикроскопия 710
Ультра фильтров ап ио 709
У дыр ацентр ифу га 712
Умангит 399
Умбра 230
Упаковочный множитель 609
Урая 136—142, 206—223, 640 аналитические сведения 222, 223 арсенид 213 атомные радиусы 138 бориды 213 бромиды 214—216 налегтпость 136, 209 галогениды 211, 214—216, 222 гидраты трехокиси 215
гидрид 211, 214; 215
двуокись 217, 218
деление ядра 640
изотопы 215
иодиды 211, 214—216 ?
ионпые радиусы 138
1 исторические сведения 207
карбиды 213, 215 *
карбонилы 139
кристаллическая решетка 137
модификация 211
монокйст 217
нитриды 213, 215, 217
общие сведения 136—139 окислы 139, 214, 215, 217—220 — молярные теплоты образования?
139
перекиси гидрат 219
пербксоурапаты 211, 219
получение 207, 208
применение 209
радиоактивность 209
распространение в природе 207
свойства 208
силициды 213
соединения 209—233
— бинарные 210—213
— интерметаллические 211, 21&
— классификация 211
— органические 210
— с кислотами 220—222
— с металлами 142
— с неметаллами 142
— триураноктоксид 217, 218
сульфиды 217
„ температура кипения 137
— плавления 137
трехокиси 217
фосфиды 213, 214
фториды 214—216
хлориды 214—216
Уран(П) 571
Урап(Ш) 211
иодид 211:
соли 211
Урап(!У) 137, 211, 222
галогениды 222
гидроокись 137
оксалат 222
соли 211, 222
сульфат 222
Уран-235 648
Урап-а 211
Уран-у 215
Уран X, 571
Уран Хг 571
Уран У 60
Урап Z 571, 572
Уранаты 210, 219—22Г
Урапил 137, 210, 211, 220—222
ионы 210
радикал 220
соли 137, 211, 220—222
— ацетат 220, 221
— карбонат 221
— нитрат 220
830 Предметный указатель
соли сульфат 221
— сульфид1 221
— фторид 220 .
— хлорид 137, 220
Уранинит 207
Урановая желтая 209, 219
смоляная руда 206
Ураноцерит 207
Фаматинит 395
Фаянса правило осаждения 568 !
Фелингова жидкость 405
Ферберит 191
Фермий 649—660, 679, 680
валентность 655
изотоны 652
исторические сведения 658
общие сведения 649
распространение в природе 660
Ферровольфрам 192
Ферромарганец 228, 230
Ферромолибден 183
Ферросилиций 289
Ферротитан 69
Феррохром 143
Физический атомный вес 590
Фика закон 780
Флотационный процесс 181
Фольгарда метод 247
Фотоны 600
Фотохимические реакции 740
Франклинит 458
Франции 638
Фришевание 275
Халькаптит 414
Хальколит 207
Харкинса правило 634
Хедвелла принцип 788
Химические реакции 203, 628, 736—801
— аналогичные нейтрализации : 767
— в ^неводных растворах 760—
— воспламенения 740
— горячих атомов 628
। — индуцированные 759
— кинетика "36—759
— конденсаций 203
— механизм 738
— молекулярность 739
— порядок 737
— скорость 736, 740
— сопряженные 759
— твердых веществ 778—801
— — —в расплавах 776
— — — исторические сведения 779
— — — общие сведения 778
— — — с газами 793
— — — с жидкими веществами 783
Химический атомным вес 590
Химическое соединение 39
Хлориты 142
Хризокол 395
Хром 50, 136—180,"226, 304, 381, 691 активное состояние 145 алкилпроизводные 50 аналитические сведения 179, 180 а рил производные 50; атомные радиусы 138 валентность 136 гексакарбонил 139, 381 , ионные радиусы 138 исторические е веде пня 143 - коллоидное состояние 146 Кристаллическая решетка 137: общие сведения 136—139 окислы 139, 148 ;; перекись 177—179 получение 143, 144 применение 146, 147 распространение в природе 142,. 691 свойства 14'1141: соединения 142, 147—179
— металлрргапиче ск ие 149 — с металлами 142 т — с неметаллами 142
соли 304
— гидродиз 304 сплавы 139—143, 226 физические свойства 137 электролитическое осаждение 143 Хром(1) 149 Хрбм(П) 146, 149—151 ацетат 151 бромид 151
. двойной сульфат 151 иодид 151 соединения 149—151 — комплексные 151 соли 146, 149, 150 сульфат 151 сульфид 150 ~ фторид 151 хлорид 150
Хром(Ш) 137, 146—148, 151—170 акво^соедйпения 164 :
« аммиппые комплексы 137, 161—.170 ацетат,158 ,
ацидо-соединенйя 165, 167, 168 бромид,157 гекса аммивхрома соединения 162 гидр оксохрома ты 148, 153 ; гидроокись 137, 152 иодид 157 нитрат 159 окись 148 151—153 оксалат 158 оксалатохроматы 158 роданид 157 роданохроматы 157 соединения 151—170 соли 146, 147, 149, 158, 159 — ацетотрихрома 158 — сульфатохрома 159 сульфат 159, 160 , — кислый 160
сульфид 154
Предметный указатель
831
тиохроматы 154
фторид 157
хлорид 154—157
комплексы 154—157
цианид 157
цианохроматы 157
XpOM(IV) 148
Хром(У) 148, 178, 179
пятиокись 178, 179
соединения 148
XpoM(Vi) 148, 170, 171 ..
дихромовая кислота 148
окись 148, 170, 171
соединения 170
тетрахромовая кислота 148
трихромовая кислота 148
Хроматографические способы разделения веществ 725
Хроматография 726
адсорбционная 726
' бумажная 726
Хроматы 136, 146, 148, 149, 153, 171 — 179
аммония 175
бария 173
гало read замещенные 176, 177
ионы 171
калия 174, 175
натрия 174
пероксохроматы 177—179
Хромил хлористый 137, 176, 177
Хромирование 144
Хромистым железняк 142, 143, 153, 173
Хромовые квйсцы 147, 160, 161
краски 147
стали 146
Хромпер о кс и дна я реакция 178, 179
Царская водка 442
Цветные индикаторы 764
Цейнирит 207
. Цементация 280
Цепные реакции 74I
Цериевые земли 530
Церий 511 —534
аналитические сведения 535
атомный объем 522
валентность 512, 527
исторические сведения 523
карбонат 533
кристаллическая решетка 520
нитраты 532
общие сведения 511
окислы 515, 517, 532
— гидраты 515, 532
оксалат 533
получение 529
применение 529
распространение в природе 528
свойства 520
соединения 524, 531—534
соли 517
сульфаты 533
сульфиды 533
твердые .растворы 524
хлориды 532
Церит 51
Цианоаргептаты 435
Цианокарбонилы металлов 377—381
Цинк 34, 39, 41—44, 452—476, 691, 799
алкилы 474
амальгамирование 461
амид 468
аммиакаты 465
амфотерность 464
аналитические сведения 475
атомный радиус 452
бромид 465, 471
валентность 39, 452, 455, 464
гидрид 474
тидрбокйсь 467, 468, 799
' — кристаллические модкфпка-пии 468
: йодид 465, 471
ионный радиус 452
исторические сведения 458
карбонат 473
кристаллическая решетка 452
нитрат 471 :
— основной 471
нитрид 468
нитрит 472’
нормальный потенциал 46.1
общие сведении 452
окись 465, 466
— каталитическое действие 466
— теплота растворения 466
оксалат 474
основные соли 799
получение 459
поляризующее действие 455
применение 463
расплавленное состояние 463
распределение электронов в атоме 454
распространение в природе. 458, 691
растворений в воде 462
— в кислотах 461
роданид 473
свойства 460
силикат 474
соединения 41, 454—456, 464—475
— алкильные 455
— интерметаллические 41
— плотность 465
— с водородом 454
— температура кипения 465
— — плавления, .465
— теплота образования 465
соли 465
— комплексные 465
— основные 465
~ простые 466
сплавы 34, 39, 42—44, 455
сульфат. 472
сульфид 465, 468
сульфит 473
тиосульфат 473
физические константы 453
аосфид 469
тррпд 465, 471
хлорид 465, 470, 471
832
Предметный указатель
хлорид двойные соли 470
—- основные 471
цианид 473 Цинкат-ион 464 Цинкаты 464 Цинкование 463 Цинковая обманка 458, 468, 469, 487 мыль 459, 463 серая 463 шпинель 45S
Цинковые белила 463, 466
Цинковый купорос 472
Циркон 93
Циркониевая кислота 89 /
Цирконаты 88
Цирконий 62—68, 82—97 аналитические сведения 94 ацетат 92 борид 94 бромид 90 валентность 64, 68 галогениды 64, 89, 90 гексафтороцирконат аммония 91 — калия 91 двуокись 85, 87, 88, 96 — гидрат 88 днхлорид 86 иодид 90 ионы 86 исторические сведения 83 карбид 94 кристаллическая решетка 63—65 нитраты 92 литрид 68, 94 общие сведения 62—65 окислы 63 оксалатоцир коп аты 92 оксихлорид 89 отделенно от гафния 97 отношение к водороду 63, 88 — к другим неметаллам 85 цероксоцирконаты 89 пероксоциркониевая кислота 89 пирофосфат 93 получепиё 83 применение 85 работа ионизации 64 распространение в природе 82 свойства 84 Силикат 93 соединения 63, 66, 67, 86—97 — дициркоиила 86
— теплоты образования 63 сплавы 65—68 строение атома 64 сульфат 91 сульфатоцйрконаты 91 сульфатоциркониевая кислота 91 удельная электропроводность 68 фосфат 92 фосфатоцирконаты 92 фторид 90 фгороциркопаг 90 хлорид 89
Цирконила ^оксалат 92
соединения 86
Цирконйлсерная кислота 91
цис-Соединения 166
Чистые элементы 585
Чугун 228, 230, 270—278, 280, 287
белый 280
зеркальный 228, 230
Швейнфуртская зелень 417
Швейцера реактив 412
Шее ле зелень 417
Шеелит 191
Шпинель 294
Штайнирит 315
Штольцит 191
Шульца — Гарди правило 713
Эдисона аккумулятор 335
Эйнштейний 649—660, 679
валентность 655
изотопы 652
исторические сведения 658
общие сведения 649
распространение в природе 660
Экамарганец 250
Электрокапилллрные явления 729
Электролитическая поляризация 790
Электролиз расплавов 21
растворов 21
Электролиты 714
коагулирующая способность 714
Электронный захват 616
Электроосмос 729
Электропечи 273
Элементы 16, 18, 585, 625, 634, 687, 689,
697, 698, 701, 702
геохимический аакон распределения
697
искусственные 634
образование в космосе 702
переходные 16, 18
побочной подгруппы 16
распространение вне Земли 701
распространенность 689, 698
редкие 687
смешанные 585
чистые 625
Элементарные частицы 600
Эманация 558, 572
радия 572
Эманирования методы 578
Эмульсии 731
Эмульсоиды 731
Энаргит 395
Энергия активации 742, 748, 749
— зависимость от механизма реакции 748
— кваптовомеханический расчет 749
Энергия распада 553 —558
Эрбиевые земли 535
Эрбий 511—530, 535
аналитические сведения 535
Ире'Омет'пый указатель
833
атомный объем 522
валентность 512, 527
гидроокись 515
исторические сведения 523
кристаллическая рещетка 520
общие сведения 511
окислы 517
иолученне 529
применение 529
распространение в природе 528
свойства 520
соли 517
Эрдмана соль 328
Эритро-соли 169
Эритрохромовые соли 169
Этерозоли 706
Эффузия газов 780
Юм-Роз ер и правило 39, 40
Ядерпая изомерия 572, 621
полимерия 330
химия 601
превращения 601
— закон 601
реакции 603, 604
— сечение 604
— типы 603
— энергия- 603
Ядерпый реактор 647, 668, 669, 672
— критическая величина 668
— размножитель 672
— регулирующие стержни 669 Ядро 621—623, 630, 632 , 639 —648 виды радиоактивных превращений 623
деление 639—648
— расчет анергии 641
— спонтанное 644
— теория 642
— цепные реакции 645
зеркальные 621
изомерия 622, 623
метастабя.таное состояние 623
скалывание ядер 622
устойчивость 630
энергия образования 632
Ялпаит 420
ОГЛАВЛЕНИЕ
Предисловие к одиннадцатому изданию........................ 5
Предисловие к девятому и десятому изданиям ................ 5
Важнейшие общие константы ............................. 7
Введение .............................................. 11
Глава 1. Металлы и интерметаллические фазы ......................... ; . 19
Твердые растворы и интерметаллические фазы 28
Твердые растворы...................... . .............. 29
Интерметаллические /фазы............................. . 38
Искажения решеток (дислокации) и дефекты структуры ... 36
Глава 2. Третья группа периодической системы (Побочная подгруппа) 47
Скандий, иттрий, лантан и актиний . .......................... 47
Скандий, иттрий, лантан ................ . , , . ..... 51
Соединения скандия, нттрйя и лантала . . . , . . . . . . 54
Актннпп .................................................. .. . . 59
Соединения актиния........................................ 60
Глава 3, Четвертая группа периодической системы (Побочная подгруппа) 62
Титан, цирконий, гафний и торим ................ . 62.
Титап . ..................................... . 68
Соединения титана ..... 71
Цирконий ................................................. 82
Соедппсяия циркония..................................... 86
Гафний................................. ....... ; .. 95
Торий ....... .................... ............. . .4 98
Соединения тория ....................................... 99
Глава 4. Пятая группа периодической системы (Побочная подгруппа) 105
Группа земельных кислот............ . . . ............... 105
Ванадий ..... , . .......... . , ........................ 107
Соединения ванадия .... . ... , . . . . , ........ 110
Ниобий . . . , , . . . . . . . . . ........ . 122
Соединения ниобия . • • . . • . ................. . 123
Тантал . . .................................. . . . . . 128
Соединения тантала ................:................ .129
Протактиний...................... . ....... ... . .. . 133
Соединения протактиния ,........... . . . . , . ... , , 434
Глава 5, Шестая группа периодической системы (Побочная подгруппа) 136
Хром, молибден, вольфрам и уран ........................ 136
Хром . . . .; / ..................................' , . . 142
Соединения хрома..................................... 147
Амминные комплексы хрома(Ш) . . 161
Молибден ..... . .................... .:. . , ,, . . 180
Соединения молибдена................................. 183
Вольфрам ............................. ; . . . 191
Соединения вольфрама............................... 194
: Уран.................................................... 206
Соединения урана ................................... 209
Классификация соединений урапа...................... 21.1
Соединения урана с кислотами ...................... . . . 220
Оглавление
835-
Глава 6. Седьмая группа периодической системы (Побочная подгруппа) 224
Марганец, технеций, рений................................ , 224
Марганец ..................................................- , 226
Соединения марганца........................................ 230
Технеций и рений . ...................................... . 249
Свойства и поведение технеция......................... . 251
Получение и свойства рения............................... ; 252
Соединения рения ......................................... 253
Глава
7. Восьмая группа периодической системы
259
Металлы группы железа и платиновые металлы............
А. Металлы группы железа .........................
Железо................................................
Получение железа................. ................
Свойства чистого и технического железа............
Соединения железа.................................
Карбопилы и питрознльпые соединения железа........
Кобальт ..........................". ..............
Соединения кобальта.............. , ..............
Никель ...............................................
Соединения никеля ................................
Б. Платиновые металлы............
Диада I; Рутений и осмий......... ................
Рутений...............................................
Соединения рутения................ , ........
Осмий . . ................................... ........
Соединения осмия..................
Диада II: Родий и иридий..........................
РОДИЙ ........................
Соединения родия..................................
Иридий .................................... .........
Соединения иридия . . ............... . . ........
Диада III: Палладий и платина......... :...........
Палладий ................................... . . . . .
Соединения палладия . . . ........................
Платина ............................... ..............
Соединения платины . , . ................
Карбонилы металлов................................. . ,
259
263
271
4.0??
. 309
, 311
. 313
. 330
. 333
. 343
. 346
.. : 347
. 348
. 351
. 352
. 354
, 354
. 356
, 358
. 359
. 361
< 362
. 364
366
. 369:
... 375
Глава 8. Побочная подгруппа первой группы периодической системы 386
Медь, серебро, золото..................................... •; 386
Медь....................................................... 394
Соединения меди............................ ./............ 404
Серебро....................................... , ............ 420
Соединения серебра................................... 425
Галогениды........................................... ; 427
Другие соли серебра........................................ 432
ч Золото ........................................................ 439
Соединения золота..................................... 445
Гл а в а 9. Вторая группа периодической системы (Побочная подгруппа) 452
Цинк, Кадмий, ртуть..................................... . 452
Цинк ........................... • ....................... 458
Соединения цинка .................... 464
Кадмий ..................................................... 476
Соединения кадмия 479
Ртуть......................................................... 487
•у Соединения ртути . , .......................................... 491
Глава 10. Семейство лантанидов.......................................... 511
Важнейшие соединения редкоземельных металлов ...... 530
Соединения церия......................... 531
836
Оглавление
Глава 11. Радиоактивность и изотопия.................................. 537
Важнейшие действия радиоактивных веществ ................ 539
Свойства и природа трех видов радиоактивных излучений . . 543
Объяснение явления радиоактивности на основе теории атомного распада .............................................. 549
Определение энергии распада............................. 553
Эманации и активные осадки. Радиоактивный ряд торин , . , 558
Радиоактивный ряд урана —радия.................... ... . 570
Радиоактивный ряд актиния............................... 574
Радиоактивный ряд нептуния......................... , 575
' Положение радиоактивных элементов в периодической системе 575
Глава 12, Изотопия стабильных элементов................ . , ,. ... 580
Дейтерий и окись дейтерия.............. , ...... . 591
Глава 13. Искусственные превращения атомов (ядерная химия)......... 598
Искусственная радиоактивность.............................. 616
Глава 14, Трансурановые элементы............................... 649
Нептуний.................................................... 661
Соединения нептуния .................................... 661
Плутоний ....................... . ....................... 663
Соединения плутония ..................................... 672
Соли плутония ..................................... .. 675
Америций.................................................... 677
Кгорий, берклий, калифорний .................. . ......... 679
Эйнштейний, фермий, менделевий .............................. 679
Нобелий ...................." . .......................... 680
Радиоактивные ряды трансураловых элементов............... 680
Глава 15, Распространение элементов; геохимия ....................... . 686
Глава 16. Коллоидная химия. Химия поверхностных явлений............ 704
Коллоидные растворы...................................... 706
Поверхностные явления.................................. 720
Адсорбция и абсорбция................................... 722
Поверхностное натяжение и капиллярные силы . ............ 727
Дифформные системы ..................................... 733
Глава 17. Катализ и кинетика реакций'............................ 734
Кинетика химических реакций.................................. 736
Кинетическое рассмотрение катализа .......................... 750
Индуцированные реакции...................................... 759
Глава 18, Реакции в неводных растворах............................ 760
Поведение кислот в неводпых растворителях................ 761
«Водоподобные» растворители ............................. 765
Глава 19. Реакции твердых веществ.................................. 778
Приложение ........................................................ 803
Предметный указатель ............................................ 811
ЗАМЕЧЕННЫЕ ОПЕЧАТКИ том I
Страница Строка Напечатано Следует читать
35 1 сверху удельный вес удельный объем
276 табл. 50 Твердость (по Ритцу) Склерометрическая твердость
614 1 снизу мтектики перетекгзднш
794 24—25 снизу трубки Лота паяльной трубки
794 27 снизу проба Гепара проба на серную печень
795 2 сверху пробы Гепара пробы на серную печень
808 22 сверху Поль Пьер
840 табл. 114 А1 ДР
(19 сверху)
ТОМ II
Страница Строка Напечатано Следует читать
32 32 снизу веществ в гомогенных веществ, в гомогенных сплавах
сплавах
84 20 снизу Zn Zr
112 11 снизу редкими редкоземельными
164 20 сверху переходят переводятся
268 табл. 33 и других элементов и других соединений элементов
282 34 снизу компонентов составляющих
764 2 снизу [ЗО*]Д' [SOJ'
828 23 снизу (ле- 69 68
вый столбец)
833 4 снизу (пра- 622 621
вый столбец)
8и. Л»