/


























































































































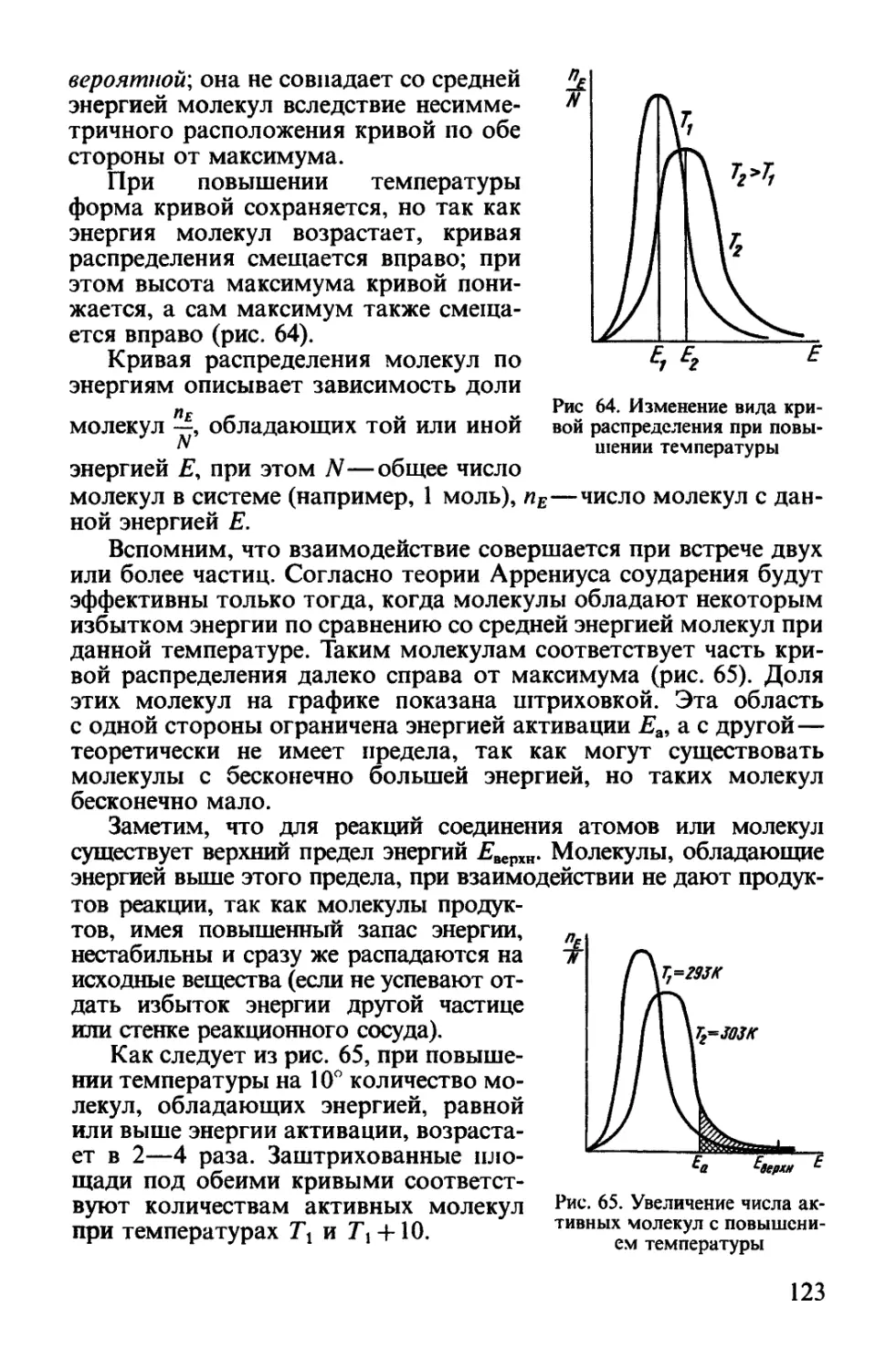


















































































































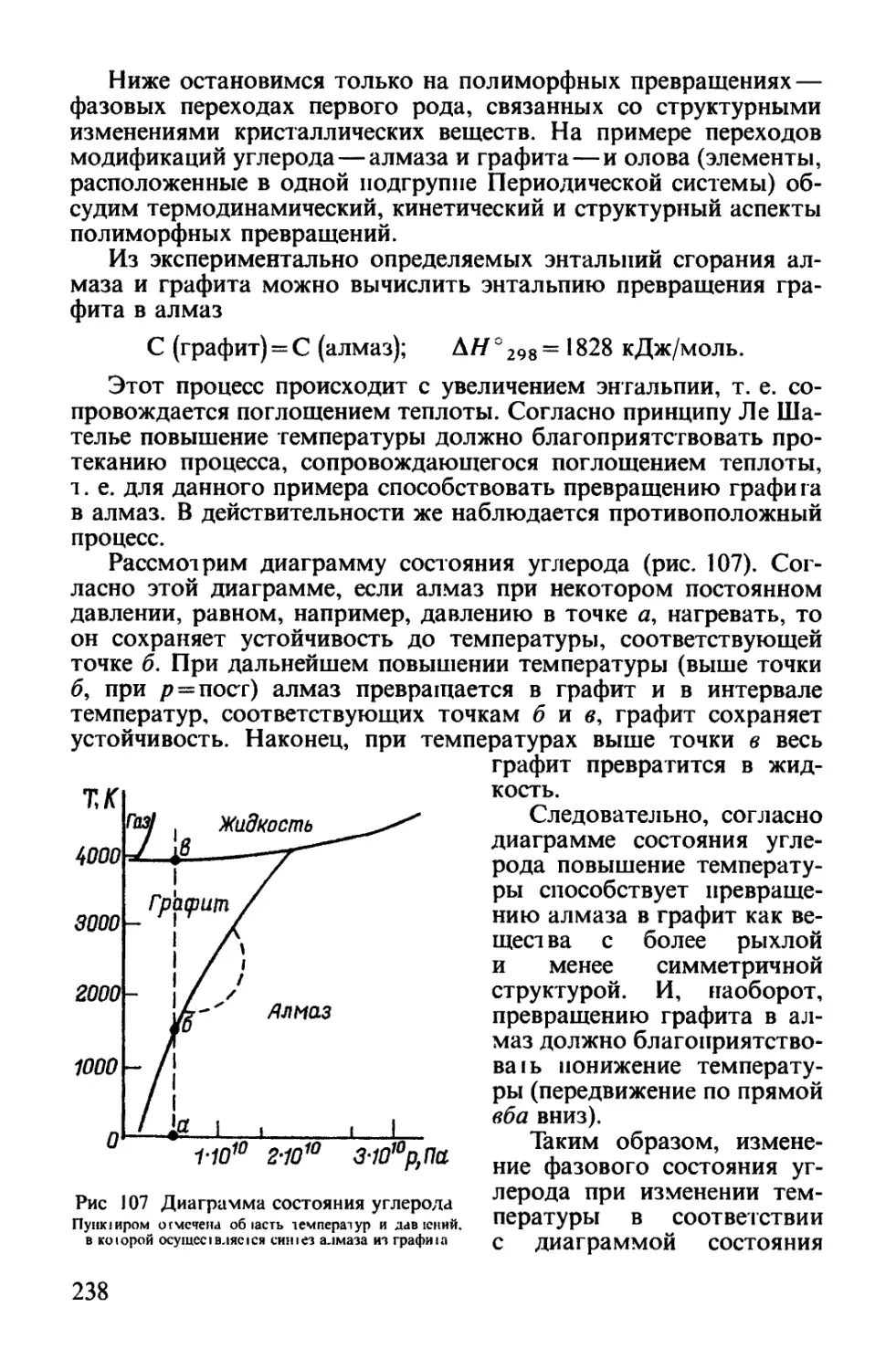













































































































































































Текст
ОХЛайцев
О. С. ЗАЙЦЕВ
ХИМИЯ
Современный краткий курс
Допущено Министерством общего и профессионального образования
Российской Федерации в качестве учебника для студентов,
обучающихся по направлению и специальности «Химия»
«Агар»
Москва
1997
ББК54.317.Я73
УДК 541@75.8)
Зайцев О. С. Химия. Современный краткий курс.
Учебное пособие.—М.: Агар, 1997.—416 с, ил.
В учебнике излагаются основные представления главных учений
химической науки—о направлении химических процессов, об их скорости,
о строении вещества и о периодичности в изменении свойств элементов
и их соединений. Эти самые общие представления позволят будущему
специалисту решать наиболее часто встречающиеся в практической
деятельности химические проблемы.
Учебник имеет широкий профиль использования—для студентов
университетов и студентов высших учебных заведений, изучающих курсы
химии или общей химии. Учебник будет полезным преподавателям химии
лицеев и колледжей, а также учителям химии, работающим по
углубленным программам.
Учебник написан в соответствии с государственным образовательным
стандартом учебной дисциплины «Общая и неорганическая химия».
ISBN 5-89218-017-4 © Издательство «Агар», 1997
ПРЕДИСЛОВИЕ
Современная общая и неорганическая химия стала не
описательной, а количественной наукой со сложным математическим
аппаратом. Это, а также чрезвычайно большой объем
информации, накопленный химической наукой, затрудняет ее изучение
на младших курсах вуза.
Этот учебник общей химии существенно отличается от других
подобных. Прежде всего он рассчитан в значительной мере на
самостоятельное изучение предмета. Большинство важнейших
соотношений и формул в пособии выведено с помощью
логических приемов, без использования высшей математики.
Эта книга будет понятна всем закончившим среднюю школу.
Однако следует помнить, что материал расположен так, что,
пропустив или не поняв какую-то его часть, впоследствии не
удастся разобраться в новом материале. Рекомендуется работать
с карандашом в руке и обязательно проверять выводы всех
формул и самостоятельно проводить все расчеты. В некоторых
случаях использованы простейшие методы высшей математики.
Читатель, еще не знакомый с элементами высшей математики,
может эти места пропустить, но потом к ним возвратиться,
чтобы окончательно понять содержание текста и еще раз
убедиться в огромном значении математики для развития
современной науки.
Основная цель книги—развить у студентов химическое
мышление, чтобы будущий специалист мог не только самостоятельно
решать различные химические проблемы, но и перенести общие
методы научной работы в работу по специальности. Поэтому
желательно не запоминание материала книги, а понимание
основных закономерностей и методов химии. Как показал опыт,
для усвоения материала достаточно один раз внимательно
прочитать (проработать) текст.
Пособие не охватывает всего материала программы. Освоив
с его помощью теоретические основы химии, студенты уже
достаточно подготовлены к самостоятельному освоению ряда частных
пунктов программы.
Все замечания по учебнику и советы по его изменению автор
просит присылать по адресу: 119899, Москва, Воробьевы Горы,
Московский университет, химический факультет, кафедра общей
химии, лаборатория методики обучения химии.
ГЛАВА 1
УЧЕНИЕ О СТРОЕНИИ ВЕЩЕСТВА
§ 1. МАТЕРИЯ—ВЕЩЕСТВО И ПОЛЕ
Весь окружающий нас мир, все бесконечное множество
существующих в мире объектов и систем, форм движения,
отношений и взаимодействий—все это есть материя. Материя без
движения немыслима. Движение—это изменение вообще, всякое
взаимодействие материальных объектов, это свойство и способ
существования материи. Движение происходит в пространстве
и времени, которые являются всеобщими формами
существования материи. Пространство и время не существуют вне материи
и независимо от нее.
Человек может изучать только движущуюся материю, так как только
движущаяся материя дает сигналы и информацию о своих Свойствах.
Это позволяет описывать материю и движение при помощи
пространственных и временных характеристик. Пространственные характеристики вещества:
положение тел относительно друг друга, их размеры, тип симметрии,
межъядерные расстояния, углы между связями и т. п. Временные характеристики:
длительность процессов, продолжительность между моментами совершения событий,
ритмичность процессов, период полупревращения, константа скорости и т. п.
Любой материальный объект обладает пространственно-временными свойствами.
Материя бесконечно многообразна. Это объясняется тем, что
она состоит из различного типа дискретных частиц, находящихся
до взаимодействии. Не исключена возможность, что
последовательность структурных составляющих материи бесконечна.
Частицы, еще недавно считавшиеся простыми, оказывается, имеют
сложное строение (например, протоны и нейтроны).
Многочисленность типов частиц и огромное разнообразие способов их
сочетания обусловливают существование бесконечно большого
числа различных объектов окружающего нас мира.
Для выявления сущности химических объектов и явлений
важно рассмотреть только некоторые дискретные частицы
материи—элементарные частицы, являющиеся специфическими
квантами материи. Можно выделить следующие общие
характеристики известных в настоящее время элементарных частиц:
масса, заряд, спин, время жизни.
Масса—одна из основных характеристик материи,
определяющая ее инерционные и гравитационные свойства. Природа
массы— одна из сложнейших и нерешенных проблем современной
науки. Те виды материи, дискретные частицы которых имеют
конечную массу покоя, называются веществом. Веществом
являются такие элементарные частицы, как протон, нейтрон,
электрон и др. Сочетанием этих элементарных частиц образуются
ядра атомов, атомы, молекулы, ионы, кристаллы, живые
организмы и т. п. Электрон—элементарная частица, обладающая
наименьшей массой: те = 0,91095 • 10 " 27 г. Масса протона
mp=1836 me, масса нейтрона mn = 1840 me. Масса нейтрино, по-
видимому, равна нулю.
Те виды материи, дискретные частицы которых не имеют
массы покоя, называются полями. Таковы электромагнитное
и гравитационное поля. Квант электромагнитного поля
называется фотоном. Масса покоя фотона равна нулю, а скорость равна
скорости света, и невозможно найти систему отсчета, в которой
фотон покоится.
Поле и вещество находятся в неразрывной связи друг с
другом. Так, частицы вещества связаны между собой при помощи
поля, будь то ядерное поле, связывающее протоны и нейтроны
в ядре, электромагнитное поле, притягивающее два магнита, или
же поле тяготения.
Заряд элементарных частиц определяет их электромагнитное
взаимодействие. Имеется два вида электрических зарядов,
которые по из взаимному притягиванию или отталкиванию условно
считают положительными и отрицательными. Электрону
приписывают отрицательный заряд, а заряды других частиц
определяются по их отношению к заряду электрона. Электроны имеют
наименьший электрический заряд е~ = —1,602*10 Кл. Заряд
протона положителен и численно равен заряду электрона.
Элементарная частица и ее античастица имеют заряды
противоположных знаков.
Спин—определяет собственный момент количества движения
элементарной частицы, не связанный с ее перемещением как
целого. Спин измеряется в единицах кванта действия, или
постоянной Планка, и равен некоторому характерному для каждого
сорта частиц целому или полуцелому числу, называемому
спиновым квантовым числом, умноженному на постоянную Планка.
Спин фотона равен единице; спины электрона, нейтрона и
протона равны половине.
Стабильность частицы—ее время жизни (т)—количественно
характеризует ее устойчивость в различных состояниях. Так, все
перечисленные, за исключением нейтрона, частицы являются ус-
тойчивыми. Среднее время жизни электрона в свободном
состоянии т>5*1021 лет, а протона т> 2-10 лет. Нейтрон устойчив
только в составе стабильных атомных ядер» Свободный
находящийся в вакууме нейтрон нестабилен. Среднее время его жизни
t»16 мин; он распадается на протон, электрон и антинейтрино:
n = p+e+v.
Нейтрино и фотон—устойчивые элементарные частицы.
Некоторые другие частицы живут только около 1О™20 с.
Соотношением чисел протонов и нейтронов в ядре
определяется его устойчивость и в конечном счете распространенность
элемента в природе.
Качественно отличные друг от друга формы движения
материи взаимосвязаны, так как они могут превращаться одна в
другую. Количественно движение измеряется энергией.
Энергия—мера движения и взаимодействия всех видов
материи.
Невозможно исчезновение каких-либо форм материи без
возникновения других ее форм, так же как исчезновение одних форм
движения без возникновения других. Как материя не создается
и не исчезает, так не творится и не исчезает движение.
Закон неуничтожимости материи и
движения—фундаментальный закон естествознания. Следствием этого закона
являются закон сохранения массы и энергии и первый закон
термодинамики. Закон Iecca является, в свою очередь, следствием первого
закона термодинамики. Экспериментально доказана
эквивалентность возникающих и исчезающих форм движения материи.
Закон сохранения и превращения энергии формулируется так:
энергия не творится и не исчезает, любая форма энергии способна
Превращаться в эквивалентное количество энергии любой другой
"формы.
[ Закон сохранения материи в применении к химическим
процессам формулируется так: масса веществу вступающих в
химическую реакцию, всегда равна массе веществ, образующихся в
результате реакции.
Закон сохранения массы при его экспериментальной проверке оказался
точным, однако изучение явлений образования и распада атомных ядер показывает
Ьго приближенный характер. Это вызвано тем, что масса и энергия
взаимосвязаны, причем так, что когда энергия вещества увеличивается, его масса возрастает,
«наоборот.
., , Если в химической реакции выделяется теплота (Д#<0), то масса продуктов
реакции должна быть меньше массы исходных веществ. При поглощении
реакцией теплоты (Д#>0), наоборот, масса продуктов реакции должна быть больше
"массы реагентов. При химических реакциях изменения массы столь малы, что не
могут быть определены экспериментально, но они становятся определимы при
ядерных реакциях, сопровождающихся выделением огромных количеств энергии.
А. Эйнштейн показал, что между массой тела и его энергией
существует взаимосвязь, выражаемая формулой
E=rnc2,
где Е—энергия; т—масса; с—коэффициент
пропорциональности, равный скорости света в вакууме. Это уравнение может
быть представлено в виде
что можно сформулировать так: каждое тело, изменяющее
энергию (АЕ)9 одновременно изменяет в эквивалентном количестве
свою массу (Am). Если тело теряет (испускает) энергию, то
одновременно имеет место эквивалентное уменьшение массы
и наоборот.
Ввиду чрезвычайно большого численного значения
с2—квадрата скорости света—даже крайне малые изменения массы
должны привести к огромным изменениям энергии.
При сообщении телу энергии в 1 Дж его масса увеличивается
на 1,11-10~14г (что не обнаруживается современными
измерительными приборами). Если же произошло уменьшение массы
продуктов реакции на 1 г по сравнению с массой исходных
веществ (в ядерных реакциях или в реакциях между
элементарными частицами), то выделилось эквивалентное количество
энергии, равное 9 • 1013 Дж:
1 г = 9,0 1013 Дж = 5,62 1023эВ, 1 Дж = 1,11 • 1(Г14 г.
В спектроскопии и в атомной физике принято энергии выражать в электрон-
вольтах (эВ). Электронвольт—единица энергии, равная энергии, которую
приобретает частица с зарядом, равным заряду электрона, проходя разность
потенциалов 1 В. 1 эВ = 1,60219-109 Дж. 1 эВ на 1 моль соответствует 96,48 кДж.
Масса и энергия—взаимосвязанные свойства материи.
Энергия движущегося тела увеличивается при увеличении скорости
его движения, и одновременно увеличивается масса движущегося
тела. Взаимозависимость массы тела т и его скорости v
выражается соотношением
т =
где с—скорость света в вакууме, а т0—масса покоя. Масса
покоя численно равна массе тела, когда его скорость i; равна
нулю. Масса покоящегося тела минимальна, она безгранично
возрастает при приближении скорости тела к скорости света; при
этом одновременно возрастает и его энергия. При скоростях
движения тела, близких к скорости света, зависимость энергии
тела Е от его скорости и описывается уже не формулой
а соотношением
из которого следует, что при скорости, стремящейся к скорости
света, энергия тела стремится к бесконечности. Если же масса
покоя тела равна нулю, то его скорость может достигнуть
скорости света. Так, фотоны и нейтрино, имеющие нулевые массы
покоя, движутся со скоростью света. Скорость
света—максимальная скорость передачи в пространстве любых
взаимодействий (в том числе и сигналов при передаче информации).
Масса всегда связана с энергией и наоборот. Поэтому, строго
говоря, закон сохранения массы и закон сохранения энергии
являются единым законом неуничтожимости материи и
движения.
Способность частиц к взаимодействию приводит к
образованию агрегатов частиц и материальных объектов, которые всегда
обладают упорядоченностью и системной организацией.
Упорядоченность любых объектов проявляется во взаимодействии их
элементов, что вызывает взаимное изменение исходных тел. Это
мы наблюдаем, в частности, при прохождении химических
реакций. При взаимодействиях образуются системы, в которых связь
между составными частями—элементами—более прочна, чем
связь элементов системы с окружающей средой или с элементами
других систем. Человек может познавать только объекты,
обладающие системной организацией, и изучать процессы,
совершающиеся между системами или между элементами систем.
§ 2. СТРОЕНИЕ ЯДРА АТОМА
Свойства вещества, как вида материи, состоящего из
дискретных частиц, имеющих массу покоя, определяются типом и
количеством объединившихся частиц и описываются
периодическим законом: свойства химических элементов являются
периодической функцией положительного заряда ядра атомов
элементов.
Напомним, что химический элемент—это вид атомов с
одинаковым положительным зарядом ядра. Отсюда вытекает
важнейшее положение химии: свойства элементов определяются
зарядом ядра. Заряды ядер, в свою очередь, связаны с их строением,
так как только определенное сочетание числа протонов и
нейтронов может приводить к существующим устойчивым ядрам с
известной атомной массой.
Атом представляет собой сложную систему находящихся в
движении и взаимодействии элементарных частиц. Экспериментально
установлено, что каждый атом состоит из двух областей, несущих
противоположные заряды. Заряд области, где сосредоточена
почти вся масса атома, условно принято считать положительным. Эта
область названа ядром атома.
На некотором расстоянии от ядра располагаются области
с противоположным зарядом—так называемые электронные ор-
битали. Орбитали — это области определенной вероятности
нахождения электрона. Заряд электронов принято считать
отрицательным. Область положительного заряда
атома—ядро—несмотря на преобладание ее массы очень невелика по размерам.
Носителем положительного заряда ядра является протон.
За исключением ядра атома водорода, ядра состоят не только
из протонов. Согласно протонно-нейтронной теории атомных
ядер они содержат также и нейтроны. Нейтрон и протон—
частицы, почти одинаковые по массе, но нейтрон не обладает
зарядом.
Число протонов в ядре равно заряду ядра, если выражать
заряд в единицах заряда электрона. Сумма числа протонов и
числа нейтронов равна массовому числу А, т. е. массе атома,
выраженной в единицах атомных масс и округленной до целых
единиц. Масса одного атома может быть определена делением
массы 1 моль атомов элемента на постоянную Авогадро. Масса
атома чрезвычайно мала. Обычно ее выражают в атомных
единицах массы (а. е. м.). В качестве атомной единицы массы
принята — массы атома изотопа углерода \?С:
1 1 12
1 а. е. м. = =
1
г=1,66-10~24г =
12 N 6,023 Ю23
= 1,49-100 Дж = 9,ЗЫ08~эВ.
В табл. 1 сопоставлены массы и заряды электрона, протона
и нейтрона в различных системах измерения.
Таблица 1
Част ица
Электрон
Протон
Нейтрон
Обозначения
_?е или е
+ \рили р
in или п
•
9,110-
1,673
1,675-
10
10
10
-28
-24
-24
Масса покоя
а е м
5,49-10
1,007276
1,008665
в единицах
Mdccbi
электрона
1
1836
1839
ю
-1,6-10
1,6 10"
0
Зарял
-19
19
в единицах
заряда
электрона
+ 1
0
Масса электрона почти в 1840 раз меньше масс протона и
нейтрона. Поэтому масса атома практически равна сумме масс
10
протонов и нейтронов в ядре. Отсюда легко подсчитать число
N нейтронов в ядре, зная массовое число А ядра и число
протонов Z, совпадающее с порядковым номером в периодической
системе элементов:
N=A-Z.
Ядра с одинаковым числом протонов Z, но различным числом нейтронов
называются изотопами. Сейчас принято пользоваться следующим способом
обозначения ядер атомов элементов* перед символом вверху пишется ею атомная
масса Л, а внизу заряд ядра или число протонов, т. е. порядковый номер Z.
Например, для изотопа урана запись 2Ц U означает, что этот вид атомов
элемента урана характеризуется ядрами с порядковым номером 92 (или же, что то же,
в его ядрах содержится по 92 протона) и массовым числом 238 (т. е сумма чисел
протонов и нейтронов, составляющих массу ядра, равна 238). Откуда число
нейтронов равно 238—92= 146
Для обозначения элементарных частиц перед символом частицы вверху
указывают ее массу, принимая массу протона или нейтрона за единицу, а массу
электрона условно за нуль, а внизу—условный заряд по отношению к электрону,
заряд которого принят за — 1, например: электрон -°гс, протон jp, нейтрон ои,
нейтрино qV.
Порядковый номер всех изотопов одного и того же элемента один и тот же.
Так, порядковый номер у всех изотопов урана 92: Щи, 212\1Ь 29?U, 292*J.
Изотопы одного и того же элемента занимают одну и ту же клетку в
периодической системе. Большинство элементов, встречающихся в природе,—это
смеси изотопов. Поэтому, а также благодаря связи между частицами в ядре, атомные
массы элементов в периодической системе, определяемые экспериментально,— не
целые, а дробные числа.
Пуст ъаиа2,а5, .., а„ — процентные содержания различных изотопов с
атомными массами АХь А2, Аъ, ., Ап Тогда средняя атомная масса А данного
элемента равна:
100
Например, бор состоит из двух изотопов ^В и П5В. Атомная масса бора
равна 10,811. Вычислим содержание в боре каждого изотопа, приняв процентное
содержание 1%Ъ за вь а И5В за а2 и зная, что я, +а2 —100%.
10,84 =
100
Получаем 18,9% 105В и 81,1% П5В
Протоны и нейтроны в ядре атома связаны между собой
ядерными силами; природа этих сил не выяснена, но
количественно их можно оценить. Энергия разрозненных протонов и
нейтронов больше их энергии в составе атомного ядра. Образование
ядра из этих частиц сопровождается выделением энергии.
В соответствии с соотношением Эйнштейна Е = тс2 масса
ядра должна быть меньше суммы масс составляющих его частиц.
Эта разность называется дефектом массы. Чем больше дефект
массы, т. е. чем больше выделилось энергии при образовании
ядра, тем оно устойчивее. Например, ядро атома лития ]Li
образовано из четырех нейтронов и трех протонов. Сумма масс
11
частиц равна: 4 • 1,008665 + 31,007276 = 7,056488 а. е. м. Масса
ядра атома лития равна 7,01601 а. е. м., откуда дефект массы
0,04048 а. е. м.
В расчете на 1 моль ядер атомов лития это соответствует
выделению энергии:
?=0,04048 • 1,49-10 ~10- 6,02 -1023 = 0,36-1013 Дж/моль.
Энергии, выделяющиеся в результате ядерных превращений,
в миллионы раз больше энергий обычных химических реакций.
Существуют ядерные реакции, сопровождающиеся поглощением
энергии, например, реакция
поглощает 1,16* 10 й Дж/моль.
Образование атомного ядра из нейтронов и протонов
сопровождается выделением энергии. По количеству выделившейся
энергии можно судить об энергии связи между частицами: чем
больше выделяется энергии, тем устойчивее ядра. Энергию связи
вычисляют делением величины энергии, определенной по дефекту
массы, на массовое число.
Энергия связи показывает, какая часть дефекта массы и
соответствующей ему выделившейся энергии приходится на одну
частицу. Часто дефект массы просто делят на массовое число
и получают величину, пропорциональную энергии связи. На
рис. 1 представлены отношения дефекта массы к массовому
числу в зависимости от массового числа. Минимумы на кривой для
гелия, углерода и кислорода указывают на устойчивость их ядер.
Кривая медленно понижается к железу и никелю (область
максимальной устойчивости атомных ядер), а потом снова
повышается.
Эта кривая иллюстрирует два вида методов получения ядерной энергии.
1. Синтез атомных ядер с энергией связи, отвечающей минимуму на кривой
из более легких атомных ядер. При этом выделяется наиболее значительное
О SO WO 150 200 А
Рис. I. Устойчивость атомных ядер в зависимости от массового числа
12
количество энергии на единицу массы (энергия, получаемая при термоядерных
взрывах и в экспериментальных установках).
2. Так как энергия образования тяжелых ядер меньше, чем ядер, относящихся
к области минимума данной кривой, то распад тяжелых атомных ядер на средние
по массе должен сопровождаться также выделением энергии (энергия, получаемая
в атомных реакторах).
Устойчивость атомного ядра связана с соотношением в нем
числа нейтронов и протонов. Массовые числа атомов до 42 почти
вдвое больше заряда ядра: tHe, l|C, ^O, 17CI, 2оСа. Число
нейтронов в них равно числу протонов или меньше его. У более
тяжелых элементов число нейтронов преобладает над числом
протонов. Так, в ядре 2Ц U на 92 протона приходится 146 нейтронов,
т. е. число нейтронов почти в 1,6 раза больше числа протонов.
На рис. 2 показана область ядер, устойчивых по соотношению
числа протонов и нейтронов. Пунктирная линия отвечает
равному числу протонов и нейтронов. Несмотря на то что нейтрон не
имеет заряда, взаимодействие протонов и нейтронов играет
огромную роль при образовании устойчивых ядер.
Распространенность атомов в земной коре связана с устойчивостью
атомных ядер: чем устойчивее ядро, тем больше содержание атомов
в земной коре.
Сопоставление числа протонов и нейтронов в ядре с
содержанием атомов в земной коре позволило обнаружить интересные
закономерности:
Содержание в
земной коре, %
Число протонов
Число нейтронов
Примеры
88
четное
четное
*|О, ft Si
11
нечетное
четное
?ЗА1,
0,98
четное
нечетное
зо Zn,
0,02
нечетное
нечетное
1б|Еи
Пк
S'iPt
Оказалось, что в земной коре
наиболее распространены изотопы с четным
зарядом ядра и четным массовым
числом, т. е. содержащие четные числа
протонов и нейтронов. На втором месте
находятся изотопы с нечетным зарядом
ядра и нечетным массовым числом, т. е.
содержащие четное число нейтронов и
нечетное число протонов. На долю
остальных атомных ядер приходится около
1%. Чрезвычайно ярко подобная
закономерность проявляется в семействе
лантаноидов. Распространенность
лантаноидов сравнивается на рис. 3 с
распространенностью кислорода. У
лантаноидов ясно обнаруживается большая
150
50
I
0 50 100
Число протонов
Рис. 2. Устойчивость
атомных ядер в
зависимости от соотношения числа
нейтронов и протонов
13
La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu
57 58 59 60 61 62 63 6b 65 66 67 68 69 70 71
Рис. З. Распространенность лантаноидов
распространенность элементов с четными порядковыми
номерами.
Это свойство ядер объясняется такой моделью ядерных
оболочек, когда они строятся по аналогии с электронными
оболочками или энергетическими электронными уровнями. Согласно
этой модели в ядре существуют энергетические уровни протонов
и нейтронов, заполняемые парами протонов и нейтронов с
антипараллельными спинами. Поэтому при четном числе протонов
и нейтронов ядро более стабильно.
Если расположить элементы в порядке увеличения числа
протонов в ядре или же, что то же, в соответствии с возрастанием
заряда ядра, то обнаруживается, что химические и физические
свойства являются периодической функцией заряда ядра. Число
протонов у ядер атомов элементов этой последовательности
равномерно увеличивается на единицу; через определенные
промежутки повторяются элементы, проявляющие в своих свойствах
определенное сходство.
Каждый химический элемент в этой последовательности
однозначно характеризуется числом протонов в ядре или зарядом
ядра, так как этим определяется число электронов у электрически
нейтрального атома и тем самым химический характер элемента.
Периодическая система Д. И. Менделеева основана на том, что
химические и физические свойства атомов являются
периодической функцией заряда ядра.
Атомные массы элементов возрастают в том же направлении,
что и заряды ядер, однако свойства элементов не могут считаться
периодической функцией атомной массы. Если расположить
элементы строго по возрастанию атомных масс, то калий и аргон,
иод и теллур, никель и кобальт, протактиний и торий следует
переставить местами, но тогда нарушится периодичность в
изменении свойств.
Порядок следования элементов в периодической системе
обозначается порядковыми номерами^ численно совпадающими с
зарядами ядер и числами протонов в ядрах атомов элементов.
14
Итак, если элементы расположить по возрастанию зарядов их
ядер Z, начиная от водорода с Z=l, то в получившемся ряду
элементов обнаружится периодическое повторение химических
и физических свойств. Элементы с особо ярко выраженным
сходством (не считая водород и гелий) следуют сначала через 8,
а затем через 18 порядковых чисел. Это наиболее отчетливо
видно, если расположить элементы со сходными свойствами
один под другим.
Водород и гелий занимают особое положение в
периодической системе элементов, образуя первый короткий период. Затем
следуют два коротких периода по 8 элементов, а далее два
длинных периода по 18 элементов и еще два длинных периода по
32 элемента.
Сходные элементы располагаются в вертикальных столбцах,
называемых группами или подгруппами в зависимости от
принятой формы периодической системы.
Числа элементов, входящих в периоды различной длины,
относятся друг к другу как 2:8:18:32 или как
B-12):B-22):B*32):B-42). Это наводит на мысль, что
увеличение числа элементов в периодах подчиняется некоторому
простому закону.
Представления о строении атомов объясняют причину разной
и строго определенной длины периодов и, следовательно,
причину периодической повторяемости свойств элементов.
§ 3. ОСНОВНЫЕ ЗАКОНЫ АТОМНО-МОЛЕКУЛЯРНОЙ ТЕОРИИ
Количественное (по массе или объему) изучение многих
реакций и объяснение результатов эксперимента приводит к так
называемым стехиометрическим законам, подтверждающим
атомно-молекулярное учение. Исторически именно эти законы
утвердили в химии атомно-молекулярное учение.
Изучение массовых соотношений, в которых одни вещества
соединяются с другими, привело к закону эквивалентов;
отношения масс, вступающих в химическую реакцию веществ,
равны или кратны мольным массам их эквивалентов.
Эквивалентом называется некоторая реальная или условная
химическая частица, которая может присоединять или выделять
в реакции один атом или ион водорода. Единица количества
эквивалента—моль. Масса 1 моль эквивалента—это мольная
(молярная) масса эквивалента.
Мольной массой эквивалента (или эквивалентной массой
элемента) называют массу элемента, которая присоединяет или
замещает в соединении 1,008 массовых частей водорода или 8,000
массовых частей кислорода (или 12 массовых частей углерода
12 С).
15
Экспериментально найти мольную массу эквивалента
несложно: следует определить массовое количество элемента,
соединяющегося или с 1,008 массовыми частями водорода, или с 8
массовыми частями кислорода.
Например, при окислении 0,253 г металлического магния получено 0,420
оксида магния. Следовательно, 0,253 г Mg прореагировало с 0,420—0,253=0,167 г
кислорода Узнаем количество магния, которое прореагирует с 8 г кислорода,
воспользовавшись пропорцией:
0,253 г магния—0,167 г кислорода
х » » — 8 » »
,12.,.
0,167
Полученное число и есть мольная масса эквивалента магния.
Многие химические элементы образуют друг с другом более
одного соединения. Согласно закону кратных отношений в этом
случае массы одного вещества, приходящиеся на одну и ту же
массу другого вещества, относятся как небольшие целые числа.
В табл. 2 приведен процентный состав оксидов азота и отношения массы
кислорода к массе азота в каждом оксиде. Разделив все отношения на самое
малое число 0,58, получим, что они относятся друг к другу как 1*2:3:4:5.
Таблица 2
Оксид
N2O
NO
N2O3
NO2
N2O5
63,7
46,7
36,8
30,4
25,9
36,7
53,3
63,2
69,6
74,1
о
N
0,58
U4
1,72
2,29
2,86
о
N 0,58
1
2
3
4
5
Этот закон подтверждает дискретность вещества, а также то,
что все атомы одного химического элемента одинаковы и
обладают строго определенной массой.
Закон объемных отношений: объемы вступающих в
реакцию газов относятся друг к другу, а также к объемам
получающихся газообразных продуктов как небольшие целые числа.
Так, один объем водорода без остатка реагирует с одним
объемом хлора, образуя два объема хлороводорода. Объемы
прореагировавших и получающегося газов относятся друг к
другу как 1:1:2.
На основании закона кратных отношений можно
предположить, что одинаковые объемы разных газов (при равных
температурах и давлении) содержат одинаковое число атомов. Это
предположение объясняет, почему один объем водорода реагиру-
16
ет с одним объемом хлора, но совершенно непонятно, почему
образуется не один объем хлороводорода, а два, т. е. почему
в данной реакции объем системы не изменяется. Такое
противоречие возникает для большинства реакций между газами, будь
то реакции между водородом и кислородом или реакции между
азотом и кислородом с образованием различных оксидов азота.
Для объяснения этого противоречия Авогадро предположил,
что простые газы—водород, кислород, азот, хлор и
др.—состоят не из отдельных атомов, а из молекул, в каждую из
которых входят по два одинаковых атома (это гипотеза
Авогадро, ее идея была высказана еще Ломоносовым).
Закон Авогадро: в равных объемах любых газов
(независимо от того, из какого числа атомов состоят молекулы) при
одинаковой температуре и одном и том же давлении содержится
равное число молекул.
Во всех реакциях с водородом, кислородом, азотом, хлором
и другими простыми газами объем продукта никогда не бывает
больше, чем удвоенный объем прореагировавшего газа. Из этого
следует, что молекулы простых газов состоят из двух атомов
каждая.
Основываясь на законе Авогадро, можно определить, во
сколько раз молекула одного газообразного вещества тяжелее
молекулы другого. Предположим, что в сосуде взвешиванием
определена масса qx одного газа, а затем при той же температуре
и том же давлении масса q2 другого газа. Пусть масса одной
молекулы первого газа равна ть тогда масса молекул этого газа,
содержащихся в сосуде, будет
qt=nmu A)
Масса того же числа молекул второго газа, содержащихся в том
же объеме,
q2=nm2. B)
Подставляем из уравнения B) n=q2/m2 в уравнение A):
Чг Я\ ™\
qi——ml или —=—.
т2 Яг *п2
Отношение qi/q2 называется относительной плотностью
первого газа по второму и показывает, во сколько раз одна
молекула первого газа тяжелее молекулы второго.
Для разных веществ массы 1 моль вещества различны, но
число молекул в каждом моле вещества одно и то же и
составляет постоянную Авогадро N=6,02-1023. Отсюда следует,
что для веществ в газообразном состоянии при одинаковых
условиях (Г, р) объемы 1 моль вещества одинаковы. Объем,
занимаемый молем газа при нормальных условиях (Г=273 К,
/>=101 325 Па), равен 22,4 л. Очевидно, масса 22,4 л газа при
17
нормальных условиях численно равна его молекулярной массе.
На этом основан способ определения молекулярных масс
газообразных веществ.
Плотность d одного газа А по другому Б есть отношение
, масса 22,4 л газа А Мол. масса А
масса 22,4 л газа Б Мол. масса Б
Обычно определяют плотность газа по воздуху. Масса 1 л
воздуха равна 1,293 г. Масса 22,4 л воздуха (или средняя
молярная масса воздуха) равна 22,4 • 1,293 = 28,9 г. Поэтому плотность
по воздуху любого газа с молекулярной массой М */=Л//28,9,
откуда M=28,9rf.
Определение молекулярной массы газа заключается в
измерении его плотности по отношению к воздуху.
Закон постоянства состава в его исторически
устаревшей формулировке утверждает количественную определенность
каждого химического соединения: каждое химическое соединение
независимо от способа его получения всегда имеет один и тот же
состав.
Сейчас трудно оценить значение этого закона для развития
химии. Первое время после его открытия этот закон несомненно
способствовал развитию препаративной химии, однако
впоследствии сильно затормозил изучение большого класса веществ, ему
не подчиняющихся. Только в последнее время началось широкое
исследование и практическое использование достижений химии
соединений переменного состава.
Вес газообразные вещества действительно имеют определенный состав
независимо от способа их получения. Например, диоксид уигсрода. полученный
любым способом.
С(к) + О2(г) = СО2(г)
2CO(i) + O2(r) = 2CO2(r)
2СН4A)+4О2<г) = 4Н2О(г) + 2СО2(г)
СаСОз (к) = СаО (к)+СО2 (г)
СаСОз (к) + 2НС1 (р-р)=СаС12 (р-р) + Н2О (ж)+СО2 (г)
СО2(к)=СО2(г)
все1да имеет один и тот же состав и обладает всегда одними и теми же
свойствами. Это имеет большое практическое значение, так как позволяет, не
делая всякий раз химический анализ, индентифицировать химические соединения,
пользуясь строго определенным сочетанием количественных характеристик
свойств, заранее определенных для данного вещества.
С другой стороны, если получить оксид железа FeO—его обычно называют
вюститом—различными методами, то обнаружится, что продукт будет иметь
состав и свойства (например, плотность, электрическую проводимость, цвет,
реакционную способность, температуру плавления и т п.), разные в зависимости
от способа получения. При этом состав оксида железа может изменяться в
интервале FeOi.02 —FeOi.19
18
Оксид гитана TiO как индивидуальное соединение существует не только при
соотношении TiO= 1*1 в соответствии с законом кратных отношений, но и в
широком интервале составов от ТЮ(К7 до TiOi 3
Открытие и исследование соединений переменного состава
потребовало введения ограничивающих условий в закон кратных
отношений и закон постоянства состава. Теперь закон
постоянства состава следует формулировать следующим образом:
химические соединения с молекулярной структурой имеют один и тот
же состав и свойства независимо от способа получения;
химические соединения в кристаллическом и жидком состояниях имеют
один и тот же состав только при строгом соблюдении
постоянными всех условий эксперимента.
Закон кратных отношений следует формулировать
следующим образом: если два элемента образуют друг с другом
несколько соединений с молекулярной структурой, то на одно и то
же массовое количество одного из них приходятся такие
количества другого, которые относятся между собой как целые числа.
Законы эквивалентов, кратных отношений и постоянства
состава несомненно доказывают, что имеются некоторые строго
определенные соотношения, в которых один элемент реагирует
с другим. Атом каждого элемента обладает строго определенной
способностью соединяться с другими. Здесь мы подходим к
понятию валентности.
Это понятие очень широкое и определяется уровнем знаний
о строении вещества. По мере изучения теории строения
вещества понятие валентности непрерывно расширяется и изменяется.
В общем значении валентность есть количественная
характеристика химического поведения атома. Ее можно определить как
способность атома элемента присоединять к себе (или замещать
в молекулах различных соединений) определенное число атомов
других элементов.
Мерой валентности может служить число атомов водорода,
которое атом данного элемента может присоединить или
заменить. Атом одновалентного элемента всегда присоединяет к себе
или замещает один атом другого одновалентного элемента; атом
двухвалентного элемента может присоединить или заместить два
атома одновалентного элемента или один атом другого
двухвалентного элемента, и т. д. Поэтому о валент ности элемента
можно судить по составу его соединений не только с водородом,
но и с кислородом и любым другим элементом, валентность
которого известна.
Валентное ib элемента в данном соединении можно
определить, если известны его атомная масса и мольная масса
эквивалента, из соотношения:
атомная масса
валентность = -
мольная масса эквивалента
19
Численное значение заряда на атоме вообще никогда не
бывает равным валентности. Например, в молекуле N2O заряд
на атоме кислорода равен—0,81 заряда электрона, а в молекуле
С12О7 всего—0,01, хотя в обеих молекулах кислород
двухвалентен.
Роль стехиометрических законов в науке исключительно
велика, так как они исторически явились базой современного учения
о строении вещества. Благодаря им мы имеем представление об
атомах как пределе делимости вещества и о молекуле как
предельном количестве вещества, обладающем теми же химическими
свойствами, что и все вещество. Следует иметь в виду, что
молекуле можно приписать на основании общности состава те же
свойства, что и у всего вещества, но большинство свойств
вещества определяется именно благодаря объединению в нем большого
количества молекул. Мы уже знаем, что нельзя говорить о
температуре одной молекулы. Точно так же нельзя говорить о размерах
одной молекулы, ее плотности, ее теплоемкости, цвете и т. п.
Даже такие главнейшие характеристики молекулы, как
межъядерные расстояния, энергии связей, углы между связями и др.,
существенно изменяются в зависимости от того, изолирована
молекула (в газовой фазе) или находится во взаимодействии (в
жидкости или в кристалле).
Заметим, что термин «атомная масса» неточен. Правильнее
говорить «мольная масса атомов элемента» или, сокращенно,
«мольная масса данного элемента».
§ 4. ПОВЕДЕНИЕ ЭЛЕКТРОНА В АТОМЕ
Стехиометрические законы и вытекающие из них
представления о валентности дают косвенное указание на сложность атома,
однако наиболее убедительным, но также косвенным,
доказательством сложности атома является периодический закон,
открытый Д. И. Менделеевым. Ряд других явлений, а также
результаты прямых опытов окончательно доказывают сложность
атомов. Атом представляет собой систему из ядра и некоторого
числа электронов, равного числу протонов, распределенных
в пространстве вокруг ядра.
Современная теория строения атома основана на законах,
описывающих поведение микрообъектов—микрочастиц,
элементарных частиц и электронов в том числе. Поскольку массы
и размеры микрообъектов чрезвычайно малы, поведение
микрообъектов принципиально—и качественно и
количественно—отличается от закономерностей поведения макрообъектов—тел
нашего окружения, изучаемых классической физикой.
Представление о поведении электронов как микрочастиц
основано на следующих трех главнейших положениях: 1) кван-
20
товый характер энергетических изменений; 2) двойственное кор-
пускулярно-волновое поведение; 3) неопределенность положения
и скорости (если их измерять одновременно).
Остановимся кратко на этих представлениях—только в той
мере, насколько это помогает объяснению химических явлений.
Квантовый характер энергетических изменений. В системе
микрообъектов энергия поглощается и испускается не непрерывно,
а дискретно, отдельными порциями—квантами.
Энергия системы микрообъектов может принимать только
определенные значения, кратные числам квантов, и энергия такого
рода систем может изменяться скачкообразно на величину кванта
или нескольких квантов, или, как говорят, энергия квантуется.
Энергия кванта Е связана с частотой излучения v простым
соотношением (формула Планка):
где h—постоянная Планка, А = 6,626-104 Джс. При этом
частота колебаний v и длина волны X связаны так, что их
произведение равно скорости света с (с = 2,991 •108 м/с):
Xv=с.
Из этих соотношений, в частности, следует, что чем меньше
длина волны или чем больше частота колебаний, тем больше
энергия кванта и наоборот. Поэтому ультрафиолетовые лучи
обладают большей энергией, чем лучи видимого света.
Квантовый характер энергетических изменений особенно ярко
проявляется в спектрах веществ, и в первую очередь—в атомных
спектрах. Спектры получаются при испускании или поглощении
света. Атомные спектры состоят из отдельных спектральных
линий. Каждая спектральная линия характеризуется
определенной частотой колебаний v света и соответствует строго
определенному энергетическому квантовому переходу между
различными уровнями энергии:
Атомные спектры возникают при переходах между
энергетическими уровнями внешних электронов атомов.
В спектроскопии принято характеризовать испускаемый
атомом спектр или длиной волны А,, или величиной, обратной
длине волны, называемой волновым числом, показывающим
число длин волн, укладывающихся в одном метре. В дальнейшем
будем обозначать частоту колебаний буквой v, а волновое число
v0. Все упомянутые величины связаны соотношением
1 V
X с
где с—скорость света.
21
Каждому элементу отвечает свой спектр, т. е. каждому виду
атомов присущ свой характеризующий его прерывистый
(дискретный) ряд значений внутренней энергии. Отсюда следует
невозможность для атома непрерывно изменять свое состояние
и в связи с этим невозможность как поглощать, так и испускать
энергию любыми, в том числе бесконечно малыми, порциями.
Атом элемента, поглотив энергию, через некоторый очень
короткий промежуток времени снова самопроизвольно
возвращается в первоначальное состояние, излучая поглощенную
энергию. Минимальная порция поглощенной или излученной энергии
носит название кванта энергии. Излучение и поглощение энергии
совершается только в количестве одного кванта, т. е. энергии
определенной чаыот или длины волны, определяемых
формулой Планка,
Из этих рассуждений следует, что у атома должно быть
некоторое состояние с наименьшей энергией. Это состояние
называется основным, или нормальным (Еосн). При поглощении
энергии hv атом переходит в состояние с большей энергией—
в так называемое возбужденное состояние (Еъо&).
Возвращаясь в основное сосюяние, атом излучает то же
количество энергии hv:
Изложенные выше свойства атомов—дискретность значений
их внутренней энергии и поглощение и испускание ими света
квантами—были сформулированы Бором A913) в виде двух
постулатов.
Первый постулат Бора. Атомы могут существовать, не
изменяя своей энергии, т. е. не излучая и не поглощая ее, только
в определенных состояниях, которые образуют дискретный ряд
значений энергии Еи Е2, Е3, ..., причем атом, испуская или
поглощая энергию, скачкообразно переходит из одного
состояния в другое.
Второй постулат Бора. При переходе из одного
состояния в другое атом испускает или поглощает один квант энергии,
частота которого определяется из уравнения
AE=hv.
Постулаты Бора были сформулированы для модели вращающегося
электрона, от которой сейчас отказались.
Итак, поглощение или испускание атомом энергии может
быть связано только с изменением энергетического состояния
электрона. В соответствии со вторым постулатом Бора должны
существовать определенные уровни энергии, на которых
находится электрон, и он может занимать только эти уровни, а не
какие-либо промежуточные энергетические состояния.
По-видимому, должен быть один и только один уровень с минимальной
22
энергией, ниже которого электрон
уже не имеет возможности
опуститься. Этот уровень и называется
основным (рис. 4). Из основного
состояния электрон переходит в
возбужденное при поглощении
определенного количества энергии—кванта
энергии /*v. В возбужденном
состоянии атом может находиться очень
короткое время, и в среднем уже че-
1081(Г9
?\
Рис-
р р р
рез 10-8-1(Г9 с электрон испуска-
ет энергию и перескакивает на
нижерасположенный энергетический уровень или же, в конце концов,
на уровень основного состояния. Испускать энергию, как и
поглощать ее, он может только квантами (Av).
Корпускулярно-волновое поведение. Свет обладает рядом
свойств, которые одновременно присущи и волнам и частицам.
Дифракция и интерференция света говорят о его волновом
характере, а явление фотоэффекта — о его поведении как потока
частиц. Подобная корпускулярно-волновая природа свойственна не
только свету, но и многим другим частицам. Как известно,
энергия кванта излучения зависит от частоты (по формуле
Планка). В то же время масса частицы т связана с ее энергией
Е и скоростью с формулой Эйнштейна. Объединим обе формулы
и получим
hv=mc2.
Подставляя v = c/A,, получаем hc/\=mc2 и приходим к
соотношению между скоростью частицы, ее массой и длиной волны:
тс
Одним из общих свойств материи является ее
«двойственность» (дуализм): материя (вещество и поле) обладает
одновременно и корпускулярными и волновыми свойствами.
Соотношение «волна—частица» таково, что с уменьшением массы частицы
ее волновые свойства усиливаются, а корпускулярные
ослабляются, а у излучений с ростом энергии или, что то же, с
увеличением частоты и уменьшением длины волны происходит усиление
корпускулярных свойств.
Чем больше энергия частицы, тем ближе ее поведение к
поведению макротел в классической механике. При увеличении
энергии электрона в атоме водорода квантовые уровни энергии
сближаются и образуют сплошную полосу в спектре, так что линии
невозможно различить.
Неопределенность положения и скорости. Раньше считалось (и
так считал Бор), что электрон вращается вокруг ядра, подобно
23
вращению планет вокруг Солнца. Но если рассчитать скорость
электрона на орбите, то тогда оказывается невозможным
одновременное определение положения атома на данной орбите.
И*наоборот, если определить положение электрона на орбите, то
тогда оказывается невозможным расчет скорости электрона.
Мы в таком случае говорим, что имеет место
неопределенность в определении скорости электрона и неопределенность
в положении электрона. Согласно принципу неопределенности
Гейзенберга произведение неопределенности скорости и
неопределенности положения не может быть меньше чем h/2nm:
—,
2кт
где h — постоянная Планка; т — масса частицы (или любого
другого тела); Ах—неопределенность положения электрона (на
оси х)\ Аих — неопределенность в скорости (составляющей
скорости вдоль оси jc).
Если по эти формуле рассчитать скорость движения
электрона но первой, ближайшей к ядру op6nie, окажется, что
неопределенность в определении скороеi и равна самой скорости!
Принцип неопределенности применим ко всем без исключения
объектам, однако у окружающих нас макротел неопределенности
положения и скорости настолько малы, что не могут быть
обнаружены.
Можно утверждать, что в микромире вообще принципиально
нельзя определить точно положение и скорость частиц, т. е.
микрочастицы вообще не имеют вполне точных положений и
скоростей. Отсюда следует важный вывод: если нельзя точно найти
координаш и скорости электронов, то нельзя ючно описать
размеры и формы их орбиты, а также размер и форму атома. Все, что
можно сказать о положении электрона в атоме,—это только
вероятность его нахождения в какой-либо области пространства
вблизи ядра. Поэтому представление об электроне, вращающемся
по орбите вокруг ядра, не соответствуе1 свойствам микромира.
Вероятность положения электрона обычно выражают
квадратом волновой функции х|/2. Эта вероятность определяет среднюю
плотность положений электрона в атоме и обрисовывает
положение электрона в виде размазанного облака, которое называют ор-
биталыо. Волновая функция входит в уравнение Шредингера,
решения которого позволяют описать поведение электрона в атоме.
§ 5. ЭЛЕКТРОННОЕ СТРОЕНИЕ АТОМА ВОДОРОДА
Квантовый характер энергетических изменений, корпускуляр-
но-волновое поведение микрочастиц, неопределенность
положения и скорости микрочаежцы — это те новые качества микроми-
24
pa, которые оигичают его от мира, движение в котором
подчиняется законам классической механики.
Квантовая механика описывае1 движение электрона в атоме
при помощи так называемой волновой функции v|/. Общий вид
этой функции находится из уравнения Шредингера, которое
связывает волновую функцию vj/ с потенциальной энергией
электрона Епот и его полной энергией Е:
где первые три члена — это сумма вт орых производных волновой
функции по координатам х, у и z; m — масса электрона; И—
постоянная Планка.
Решение уравнения Шредингера связано с большими
математическими трудностями. Оно точно решено для атома водорода
и для одноэлектронных ионов Не + , Li2+ и Н2 . Решая уравнение
Шредингера, находят значения \(/ функций, характеризующие
вероятность нахождения электрона в некотором пространстве
вблизи ядра агома, а также определяют возможные энергешчес-
кие состояния электрона.
Это уравнение описывает вероятность нахождения электрона
в некотором объеме пространства.
Вероятность местонахождения электрона зависит oi его
энергетического состояния. Электрон может находиться в любом
месте пространства, но в облает, где значения ф2 выше, он
пребывает чаще и эти области соответствуют минимальной
энергии электрона. Совокупность Meci пространства, где квадрат
функции \|/2 имеет максимальное значение, называется
электронной орбиталью илич более наглядно,— электронным облаком
атома.
Плотность участков электронного облака пропорциональна
вероятности нахождения в них электрона. Величина \|/2 выражает
верояшость на единицу объема. Верояпюсгь нахождения
электрона в некотором сферическом слое называют плотностью
электронного облака или, короче, электронной плотностью.
Решение уравнения Шредингера даже для атома водорода —
очень сложная задача, гак как эт уравнение имее! бесконечно
большое число решений в связи с тем, что энергия электрона
может принимать бесконечно большое число квантованных
значений. Однако все решения можно разделить на 1ри серии и
ограничиться только теми значениями энергии, которые один
электрон может принимать в поле ядра аюма водорода (протона).
Три серии решений уравнения объединяются значениями
связанных между собой квантовых чисел.
Главное квантовое число п может принимать значение любых
целых чисел от 1 до бесконечное!и:
25
/7= К 2, 3, ..., оо.
Нам понадобятся юлько семь первых значений числа я.
Побочное (орбитальное) квантовое число I может принимать
значения от нуля до п — 1:
/=0, 1,2 л-1.
Необходимость введения побочного квантового числа
подтверждается изучением спектра атома водорода. Спектральные
линии, отвечающие переходам с одного уровня на другой, часто
состоят из нескольких очень близко расположенных отдельных
линий. Причем линия п=\ состоит только из одной линии,
линия п = 2 состоит из двух линий, или, как говорят, расщеплена
на две линии, линия п — Ъ расщеплена на три близко
расположенные линии и т. д. Это указывает на незначительное
различие в энергии электронов одного и того же энергетического
уровня. Поэтому уровни, кроме первого, разделяют на
подуровни, которые обозначают буквами s, /?, rf, / соответственно
значениям /—0, 1, 2, 3.
При и = 2 побочное квантовое число принимает значения 0 и 1;
это означает, что уровень п = 2 состоит из двух подуровней: sup.
При и = 3 / принимает три значения: 0, 1 и 2—третий
энергетический уровень состоит из трех подуровней: s9 p и d. Четвертый
энергетический уровень состоит из четырех подуровней: s,p, dnf.
Число подуровней в каждом данном уровне равно главному
квантовому числу.
В табл. 3 приведены значения главного квантового числа и,
соответствующие им побочные квантовые числа и обозначения
отвечающих им орбиталей.
п
I
2
3
4
0
0,
0,
0,
1
1,
1,
t
2
2,3
Таблица 3
Обозначение
подуровня или
орбитали
Ь
Ъ, 2р
35, Зр, 3d
45, 4р, 4d, 4/
0,
1,
2,
3,
s
Р
d
f
-2,
~3, -2,
m
-1,
-1,
-1,
0
о,
о,
0,
1
1,
1,
Таблица 4
2
2,3
Число
орбиталей
1
3
5
7
Орбитальное квантовое число определяет форму поверхности
максимальной вероятности нахождения электрона и ее
симметрию. Только 5-орбитали имеют вид сферической поверхности,
у других орбиталей форма иная. Их возможное число на каждом
подуровне задается третьим квантовым числом.
Магнитное квантовое число т зависит от побочного
квантового числа / и может принимать значения от — /, проходя через
нуль, до +/:
26
m=-/... 0... +/.
Число орбиталей с данным значением / равно B/+1), что
показано в табл. 4.
Физический смысл магнитного квантового числа заключается
в следующем. В спектрах атомов, помещенных во внешнее магнит-
ное поле, обнаруживается дополнительное расщепление
спектральных линий. Возникновение новых близколежащих линий
свидетельствует о* том, что в магнитном поле энергия электронов
изменяется. Но это возможно только в случае различной взаимной
ориентации электронных облаков. Обнаружилось, что все s-под-
уровни в несильном постоянном магнитном поле не
расщепляются. Это говорит о том, что 5-электронное облако имеет шаровую
симметрию (рис. 5, я). р-Подуровни расщепляются в магнитном
поле на три близкие линии. Это указывает на три различных тица
ориентации /7-облаков в пространстве. Расчеты показывают, что
область максимального значения квадрата волновой функции р-
орбиталей имеет форму, напоминающую гантель (рис. 5, б).
Так как возможны три различных направления р-орбиталей
в пространстве, то удобно их расположить вдоль координатных
осей и обозначить рх, ру и pz.
rf-Подуровень дает в магнитном поле пять близколежащих
линий, что указывает на пять различных способов их ориентации
в пространстве (рис. 5, в). Форма ^/-облаков значительно сложнее
формы /ьоблаков. Четыре d-облака схожи по внешнему виду
между собой—каждое из них как бы составлено из двух
расположенных под прямым углом гантелей.
Принято три такие d-орбитали располагать в трех плоскостях
xz, yz и ху между координатными осями. Их обозначают
Рис 5. Форма электронных орбит алей
27
соответственно dxz, dyz9 dxy. Четвертую tZ-орбиталь, так же
как и орбиталь dxy, располагают в плоскости ху, но направляют
вдоль координатных осей х и у. Эту орбиталь обозначают
dx2_y2. Форма пятой орбитали отличается от остальных: она
напоминает гантель, но между шарами в плоскости ху лежит
кольцевая область повышенной электронной плотности. Эту
rf-орбиталь принято располагать перпендикулярно плоскости
dxi_ 2 и dxy орбиталей, т. е. направляя ее вдоль оси z (как
и ороиталь р2). Ее обозначают dz2.
Несмотря на различную форму rf-орбиталей электронная
плотность для каждой орбитали одинакова, что говорит о
равновероятности нахождения электрона на любой из этих орбиталей
и их энергетической равноценности.
Когда на одном уровне имеется несколько подуровней s, /?,
d и/, то энергия их возрастает в направлении от л1 к /*.
Принято каждую орбиталь символически изображать
квадратом. ^-Подуровень состоит из одного квадрата, р-подуровень—
из трех квадратов, J-подуровень—из пяти и,
наконец,/-подуровень—из семи квадратов.
?
Иногда каждый такой квадрат, или же орбиталь, называют
электронной ячейкой.
Подуровни обозначают буквами s, p, d или /, а цифрой перед
буквой указывают номер уровня или главное квантовое число.
Например, запись Ър означает, что речь идет о р-подуровне
третьего уровня, т. е. п — 3, /== 1. 4/означав г/-подуровень
четвертого энергетического уровня: и=4, 1—3.
В невозбужденном атоме водорода электрон находится на
орбитали Is. При возбуждении, в зависимости от количества
полученной энергии, электрон может занять любой уровень и
любой подуровень. При обратном переходе в первоначальное
основное состояние электрон излучает кванты энергии соотве1ству-
ющей частоты.
На рис. 6, а изображена энергетическая диаграмма уровней
энергии атома водорода, но масштаб для удобства изменен:
сокращено расстояние между вышележащими уровнями. На
каждом уровне квадратами изображены орбитали подуровней.
Энергетические различия между подуровнями также для наглядности
не соответствуют масштабу диаграммы.
Ниже (рис. 6, б) энергетическая диаграмма уровней и
подуровней приведена в общепринятом, сжатом по вертикальной оси
изображении. Следует всегда помнить, пользуясь такой
диаграммой, что для атома водорода энергия незначительно возрастает
с ростом побочного квантового числа /=0, 1, 2, 3..., т. е. на одном
28
I
МММ
1*0
6
5
4
S
2
1
S
g
f
d
P
5
Энергия подуровней
Рис. 6. Энергетическая диаграмма уровней и подуровней
уровне при переходе по подуровням s—p—d—/имеет место рост
энергии.
Для описания поведения одного электрона в атоме водорода
вполне достаточно трех квантовых чисел. Для двухэлектронного
атома и всех остальных многоэлектронных атомов необходимо
введение четвертого квантового числа.
29
§ 6. МНОГОЭЛККТРОННЫК АТОМЫ
Для полной характеристики поведения электрона в атоме
оказалось недостаточно трех рассмотренных выше квантовых
чисел. Тщательное изучение гонкой структуры спектральных
линий показало, что два электрона, имеющие одни и те же значения
и, / и w, т. е. одни и те же энергетические характеристики, ту же
форму орбитали и одинаковую ориентацию, могут различаться
некоторым особым магнитным свойством, которое не поддается
объяснению с точки зрения классической механики и
обусловливает магнитный момент электрона. Эти два электрона
вращаются в различных направлениях (например, по часовой стрелке
и против). Это вращение сообщает электрону магнитный и
механический моменты, что характеризуется величиной, получившей
название спина. Спин электрона может принимать два
противоположных значения, поэтому вводится спиновое квантовое
число, которое мы будем для простоты обозначать символом
S (в литературе часто пользуются обозначением ms). Спиновое
квантовое число принимает только два значения.
Электроны с положительным S— +- или отрицательным
5= — - спинами обозначаются стрелками, направленными
соответственно вверх или вниз и помещаемыми в квадрат,
изображающий орбшаль. Так, символ _^JIH обозначает основное
состояние электрона атома водорода, находящегося на первом
энергетическом уровне я=1 в 5-состоянии (/=0) и имеющего
спин S= +-.
Для атома водорода, т. е. для системы «один протон и один
электрон», спин электрона не влияет на энергетические или
другие характеристики атома, но без представления о спине не
удается объяснить возникновение молекулы Н2 из двух
нейтральных атомов водорода. Благодаря введению спинового
квантового числа подсчитывается число электронов на любом уровне
и подуровне и объясняются магнитные свойства агомов, ионов
и молекул.
У атомов, имеющих больше одного элекфона, в
соответствии с многочисленными теоре1ическими предпосылками,
подтвержденными экспериментальными данными, Fie может быть
двух электронов или более с одинаковыми значениями всех
четырех квантовых чисел Это принцип (или запрет) Паули,
30
Любые два электрона в атоме должны отличаться по крайней
мере значением одного из квантовых чисел. Двум электронам
атома «запрещено» быть во всех отношениях похожими друг на
друга.
Атом водорода в основном состоянии характеризуется
следующим набором квантовых чисел: и= 1, /=0, ди = 0, 5= + !/2-
В этой электронной орбитали нельзя разместить еще один
электрон с тем же набором квантовых чисел, т. е. состояние |Т
недопустимо, но можно поместить еще один электрон,
отличающийся значением спинового квантового числа: п— 1, /=0, w = 0,
5= — V2- что изобразится так:
Принцип Паули запрещает нахождение на той же орбитали
третьего электрона, так как это означало бы, что у двух из них
все четыре квантовых числа одинаковы. На одной орбитали или
в одной ячейке может находиться только два электрона, причем
только с противоположными спинами.
Два электрона, находящиеся на одной орбитали, i.e.
имеющие одинаковые значения квантовых чисел и, / и ди, но
различные значения спинового квантового числа, называются спарен-
ными. Электроны, размещенные по одному на орбиталях одного
и того же подуровня, называются неспаренными. Например, в
состоянии I имеется три неспаренных электрона, в состоянии II
имеется два спаренных электрона и два неспаренных, а в
состоянии III все электроны спарены:
птпл штп
i и ш
Наличие спаренных или неспаренных электронов в атомах,
ионах или молекулах устанавливается экспериментально
изучением магнитных свойств. Вещества с неспаренными электронами
парамагнитны, т. е. эти вещества проводят магнишые силовые
линии лучше, чем вакуум, и магнит ное поле вт ягивает эти
вещества. Это вызвано взаимодействием спинов электронов как
элементарных магнитов с внешним магнитным полем.
Парамагнитны атомы водорода. Парамагнитна молекула NO2, имеющая
один неспаренный электрон.
Вещества, имеющие спаренные электроны, диамагнитны, т. е.
они проводят магнитные силовые линии хуже, чем вакуум, и
магнитное поле их выталкивает. Диамагнитны молекулы Н2, N2O4,
атомы Не, Аг и другие вещества.
Увеличение числа электронов в атоме подчиняется
определенным законам, что приводит к строгому расположению элементов
в периодической системе и периодическому повторению их
31
свойств. Рассмотрим некоторые основные положения
электронного строения атомов, исходя из следующих посылок.
Подобие энергетических уровней атомов уровням энергии атома
водорода. Уравнение Шредингера решено только для одноэлект-
ронных систем. Состояние электронов в многоэлектронных
атомах может быть найдено при помощи различных приближений.
Изучение спектров показало, что в многоэлектронных атомах
относительное энергетическое расположение уровней и
подуровней, форма электронных орбиталей и их направление в основном
повторяют картину одноэлектронного атома водорода.
Учитывая это, применяя принцип Паули и пользуясь набором четырех
квантовых чисел, нетрудно рассчитать число подуровней на
каждом уровне (по числу значений /для данного я), число орбиталей
на каждом подуровне (но числу возможных значений т для
данного значения /) и, зная, что на каждой орбитали не может
быть больше двух электронов E=+-, 5= — ], число
электронов на каждом подуровне (удвоенное число значений т) и
каждом уровне удвоенное число всех орбиталей уровня.
Например, на первом энергетическом уровне могут находиться только два
электрона, так как при п = 1 / может принимать только одно значение /=л—1=0,
возможно только ^-состояние. При /=0 т=0, т. с. имеется одна s-орбиталь, на
которой размещаются два электрона с разными спинами S= + */2, 5= — 1/2
Другой пример. Четвертый уровень, л=4 и / может принимать четыре
значения 0, 1,2, 3, т. е. четвертый уровень разделяется на четыре подуровня.
При /=0 т—0—это j-орбиталь с двумя электронами. При /=1 т— — 1, 0,
+1 — это ^-подуровень с тремя орбиталями, каждая из которых содержит
по два электрона. Всего на /^-подуровне 6 электронов. При 1—2 т =—2, —1,
О, +1, +2, т. е.—это ^-подуровень с пятью орбиталями, содержащий 10
электронов Наконец при /=3 т— — 3, —2, —1, 0, +1, +2, +3, те имеется
еще один подуровень / с семью орбиталями, содержащими 14 электронов
Всего на четвертом уровне содержится 2 + 6+10+14=32 электрона Строение
этого подуровня можно представить схемой.
ti
s
tilti
ti
p
ti Iti Iti
ti tJ
d
- ti |ti ti 1
'i|ti ti
ti
/
Принцип наименьшей энергии. При заполнении электронами
уровней и подуровней последовательность размещения
электронов в атоме должна отвечать как наименьшей энергии электрона,
так и наименьшей энергии атома в целом. Электрон не занимает
вышележащий уровень, если в нижележащем уровне есть места,
располагаясь на которых, он будет обладать меньшей энергией.
Этот принцип выражает общие термодинамические требования
к устойчивости систем: максимуму устойчивости соответствует
минимум энергии. В состоянии максимальной устойчивости
электронной системы в атоме связь электронов с ядром наиболее
прочна.
32
Правило 0'+/), или правило Клечковского. Можно
предположить, что последовательное заполнение электронами
уровней и подуровней должно происходить согласно схеме (рис. 6),
т. е. по мере заполнения одной орбитали заполняется другая,
по мере заполнения одного подуровня заполняется
следующий и по мере заполнения одного уровня начинается в гой
же последовательности заполнение другого уровня. Однако
вследствие взаимодействия электронов между собой в много-
электронных атомах порядок заполнения уровней и
подуровней отличается от ожидаемого в соответствии с
расположением энергетических уровней в агоме водорода. Правило суммы
(п + 1) позволяет предвидеть отличия в расположении
энергетических уровней у многоэлектронных атомов от агома
водорода.
Энергия электрона определяется в основном значениями
главного п и побочного / квантовых чисел, и поэтому сначала
заполняются те подуровни, для которых сумма значений
квантовых чисел п+1 является наименьшей. Например, энергия
подуровня As меньше, чем 3d, так как для 4$«подуровня
и + /=4+0=4, а для 3</-подуровня « + /=3 + 2 = 5, поэтому
электрон должен сперва вступать на подуровень 4s, а не на 3d
(хотя в атоме водорода энергетически более выгоден
подуровень 3d).
Если для двух подуровней суммы значений п и / равны, то
сначала идет заполнение подуровня с меньшим значением и.
Например, на подуровнях 3d, Ар и 5s суммы значений п и /
равны пяти Crf:3 + 2 = 5, 4/?:4+1 =5, 5s: 5+0 = 5). В этом
случае сначала заполняется подуровень с меньшим значением
главного квантового числа и, т. е. в такой последова1елыюсти:
3d— Ар — 5s.
На рис. 7, а показано схематически относительное
расположение энергетических уровней и подуровней многоэлектронных
атомов, найденное при помощи подсчета сумм (п + 1) для
подуровней различных уровней. Только первые два уровня
повторяют структуру атома водорода. После заполнения первых двух
уровней и 3s- и 3/?-подуровней следующего уровня электрон не
вступает, как это ожидалось бы, на 3^/-подуровень (см. рис. 6),
а, минуя его, идет на 4^-подуровень.
Появление электрона на 49-подуровнс. а не на 3^-подуровнс обусловлено
заслонением (экранированием) ядра плотными и симметричными орбиталями
нижележащих уровней В связи с этим сил притяжения ядра не хватает на
удержание электронов на 3^-подуровнс третьего уровня и состояние 45
оказывается энергетически более выюдным по сравнению с 3d Аналотичная каргина
обнаруживается и у всех вышележащих уровней, так 4/-подуровснь заполняется
только после заполнения 6?-подуровня
Несмотря на отличия многоэлектронных атомов от
атома водорода, мы будем пользоваться диаграммой заполне-
2 2452 33
п-6)
(суродняп^)
(с уровня п-5)
~ягт
Рис. 7. Порядок заполнения электронами
энергетических уровней и подуровней
ния уровней и подуровней водородоподобного атома (см.
рис. 1,6) с учетом последовательности заполнения
энергетических уровней и подуровней многоэлектронного атома
(рис. 7, б). ^
Ниже мы будем пользоваться упрощенной энергетической
диаграммой распределения электронов по уровням и
подуровням:
34
7
6
5
4
3
2
1
Р
d
f
Правило Гунда. Порядок заполнения электронами подуровней
формулируется правилом Гунда: в подуровнях р-, d- и /-орбитали
сперва заполняются одиночными, неспаренными электронами
и лишь потом происходит их заполнение вторыми электронами,
т. е. спаривание. Иными словами, при заполнении электронами
подуровня суммарное спиновое число подуровня должно быть
максимальным. Например, состояние (I) имеет суммарное
спиновое число YJ>= "I (--=+-, а состояние (II) того же подуровня
ПТТТЛ
с тем же числом электронов имеет XS= +-+-+-=+-. Второму
состоянию отвечает большее суммарное спиновое число, и это
состояние предпочтительнее первого.
В сложных молекулах и в кристаллах, а также во внешнем
магнитном поле правило Гунда часто не соблюдается (теория
поля лигандов и теория кристаллического поля).
Случаи повышенной устойчивости подуровней. Обнаружено,
что подуровни /?, d и / обладают повышенной устойчивостью,
когда они не заполнены, заполнены наполовину (неспаренными
электронами) и заполнены полностью. Рассмотрим это явление
на примере/-подуровня.
Состояния с одним электроном на подуровне или с одним
электроном свыше наполовину заполненного подуровня
неустойчивы, и часто это приводит к переходу электрона на другой
подуровень с приобретением более устойчивого состояния:
f°
JJ
35
Если до устойчивого состояния недостает одного электрона,
часто на этот подуровень переходит электрон с другого
подуровня:
ititititnni
6
f
§ 7. ПЕРИОДИЧЕСКАЯ СИСТЕМА И ЭЛЕКТРОННАЯ СТРУКТУРА АТОМОВ
Самое низкое значение атомной массы у атома водорода.
Расположим элементы в порядке возрастания атомных
номеров так, чтобы каждый последующий элемент отличался от
предыдущего на один протон в ядре и один электрон в
электронной оболочке, и мы получим так называемый менделеевский ряд
элементов—непрерывную последовательность элементов.
Распределение электронов по уровням и подуровням в атомах
элементов этого ряда подчиняется рассмотренным ранее
принципам и правилам распределения электронов многоэлектронных
атомов.
Водород—первый элемент в этом ряду. Единственный
электрон его атома должен занимать самый низкий энергетический
уровень п—\. Для л = 1 /=0 и т = 0. Его электронная структура
приведена на с. 30. Ее можно представить формулой Н Lv1, где
Н—символ элемента, первая цифра означает номер уровня (/Д
буква—подуровень (s для /=0, р для /= 1, d для /=2,/для /=3),
цифра справа вверху показывает число электронов на данном
подуровне.
На орбита л и 15 может быть размещен согласно
принципу Паули еще один электрон с противоположным спином.
Это имеет место в атоме гелия (№ 2), имеющем структуру
Не Is2:
Не lllTl
Гелием заканчивается заполнение первого электронного уровня
и первый самый короткий период периодической системы.
Электрон атома лития (№ 3) вступает во второй уровень п = 2,
а сам элемент литий открывает новый — второй—период
системы. Электронная структура атома лития Li Is22sl:
36
Обращает на себя
внимание, что и водород, и литий,
имея по одному неспаренному
электрону, проявляют во всех
своих соединениях
одновалентность (например, LiH, LiF,
Н2О), а гелий, обладая двумя
спаренными электронами,
совершенно не склонен к
образованию химических соединений,
он—нульвалентен. Очень
часто, особенно у элементов
главных подгрупп, их валентность совпадает с числом неспаренных
электронов в атоме.
Следующий электрон—атома бериллия (№ 4)—заканчивает
25-подуровень. Его структура Be Is2"
г х
Рис 8. Электронная плотность s-орби-
талей
На рис. 8, а изображено распределение электронной
плотности в атоме, имеющем два заполненных первых s-подуровня,
а на рис. 8, б—разрез вдоль оси Ох, Первый острый максимум
на кривой электронной плотности соответствует Ь-орбитали,
второй максимум, ниже и менее острый,— 2$-орбитали. Между
этими двумя максимумами находится область, где электронная
плотность не равна нулю и где имеется определенная
вероятность нахождения и Is- и 2я-электронов.
Хотя у бериллия нет неспаренных электронов, он проявляет
в своих соединениях валентность, равную двум (например,
в ВеН2, BeF2, ВеО и др.). Это объясняется тем, что подуровни 2s
и 2р энергетически очень близки и незначительной затраты
энергии достаточно для перевода одного 2я-электрона на 2р-подуро-
вень; при этом возникает возбужденное состояние с двумя неспа-
ренными электронами:
37
Процесс возбуждения и распаривания электронов можно
записать в виде уравнения
Be ls22s2^Be*ls22sl2pl.
Именно этим и объясняется двухвалентность бериллия.
Бор (№ 5) в невозбужденном состоянии имеет один неспарен-
ный электрон. Известны его неустойчивые и малочисленные
соединения типа ВН. В большинстве же соединений бор
трехвалентен (В2О3, Н3ВО3, ВН3), что объясняется легкостью
распаривания Zf-элек фонов:
Следующий электрон — у атома углерода (№ 6)—в
соответствии с правилом Гунда вступает на р-орбигаль. Углерод, имея
два неспаренных электрона, проявляет двухвалентность
(например, в СО), но чаще—четырехвалентность (СО2, СН4 и т. д.).
В это состояние атом переходит при возбуждении:
После углерода в ряду элементов кислород—фтор—неон
проходит понижение единственно возможного валентного
состояния:
Неон, как и гелий,—нульвалентен, так как для распаривания
его электронов необходимо их перевести на более высокий
третий уровень, что связано с очень большими энергетическими
затратами.
Наличие неспаренных электронов у нейтральных атомов под-
чверждается магнитными измерениями. В ряду N—О—F—Ne
парамагнетизм уменьшается, неон обладает диамагнитными
свойствами.
Неоном заканчивается заполнение второго энергетического
уровня и соответственно—второй период системы Менделеева.
38
Натрий, имея один электрон на Зд-подуровне, открывает третий
период системы Менделеева. Натрий, магний, алюминий и
кремний повторяют электронные структуры внешних энергетических
уровней лития, бериллия, бора и углерода:
з 1
г U
И
Р
Mill
3
Mg2
1
\\
\\
\\
\\
\\
р
Mill
d
3
А12
1
3
Si 2
1
U
H
llil
s
w
w
w
w
.—
¦
w
w
p
t
w
p
w
1 1 1
*
Они обнаруживают те же валентные состояния, и их соединения
имеют аналогичные формулы (LiOH—NaOH, ВеС12—MgCl2,
В2О3—А12О3, СО2 —SiO2 и т. п.).
Элементы третьего периода отличаются от элементов
второго тем, что у них атомы на третьем уровне имеют свободный
rf-лодуровень. Если в ряду Na—Mg—Al—Si свободный rf-подуро-
вень не влияет на валентные состояния элементов, то
валентности фосфора, серы и хлора отличаются от валентностей азота,
кислорода и фтора. Это объясняется возможностью
распаривания электронов и перевода отдельных электронов на свободный
З^-подуровень:
3
Р 2
1
\\
\\
\\
\
\
\\
Р
t
Mill
и*
и»
¦¦
н
t
р
t
н
1 1 II
Согласно спиновой теории валентности азот может быть только
трехвалентным (например, в соединениях N2O3 или NH3), а фосфор—трехвалентным
в невозбужденном состоянии атома (РН3, РС13, РгО3) и пятивалентным при
возбуждении 3/>-подуровня и переходе одного электрона на З^-подуровень (РС15,
39
P2Os, H,PO4). Точно так же кислород имеет только одно валентное состояние 2,
а сера — три валентных состояния* 2, 4 и 6 Состояния 4 и 6 возникают при
переходе электронов на 3^-подуровснь (SO2. H2SO3, SO3, H2SO4). Хлор в отличие
от фтора, проявлявшего только одновалентность (HF, F2O), имеет валентные
состояния 1. \ 5 и 7 (НС1, НСЮ, НС1О2. НСЮ3, НС1О4). Однако аргон —элемент,
у которою заканчивается заполнение 3/^подуровня,— практически нульвалентен
Эго объясняется исключительной устойчивостью полностью законченного
подуровня
По правилу (п+1) следующий электрон у элемента № 19 калия
вступает не на З^-подуровень, а на 4л\ начиная тем самым
четвертый период системы Менделеева. Очередной электрон кальция
Са \s22s22pe3s23pe4s2 направляется на тот же подуровень 4s:
Са
Ml
з tl
Ml
Ml
ti
tl
ti
tl
p
ti
ti
ZEU-H:
Кальций, как и магний, двухвалентен (СаО, СаС12).
Подуровни Ар и 3d имеют одинаковую сумму п + 1: 4+1=5
и 3 + 2 = 5 соответственно. По правилу (п + 1) энергетически более
выгоден подуровень с меньшим значением главного квантового
числа, т. е. подуровень 3d. Поэтому, начиная с № 21 скандия
и кончая № 30 цинком, десять электронов вступают на второй,
считая от внешнего, подуровень 3d.
Заполнение пяти орбиталей 3</-подуровня проходит в
соответствии с правилом Гунда: сначала непарными электронами,
а затем идет их спаривание:
4
3
Сг
2
1
t
п
tl
tl
tl
tl
tl
tl
p
tl
ti
t
t
t
t
t
1 I 1 1 1 1 1
/
d
Электронная структура хрома Сг \s22s2lpb3s23pe3dsAsx
интересна тем, что один электрон с 4.у-подуровня перешел на 3d-
подуровень, после чего подуровень приобрел более устойчивое
заполненное наполовину состояние. Следующий электрон у
марганца вступает на 4л-нодуровень, и начиная с железа электроны
на 3<У-подуровне начинают спариваться. У меди, как и у хрома,
один электрон с 4л-подуровня переходит на 3^-подуровень,
придавая ему устойчивое состояние полного заполнения Си Ь22.у2
2pb3s23p*3diO4sl:
40
4
3
Си
2
1
t
tl
tl
tl
tl
tl
tl
tl
P
tl
tl
tl
tl
tl
tl
tl
IE
1
d
У цинка 45- и 3*/-подуровни полностью заполнены.
Валентные состояния элементов от Sc до Zn многообразны,
что вызвано энергетической близостью 4s- и 3^-подуровней.
Скандий проявляет только трехвалентность за счет двух 4s-
электронов и одного З^-электрона. Остальные элементы в
отличие от скандия обычно имеют валентное состояние два и,
кроме того, набор других валентностей, причем максимальная
равна сумме числа s-электронов и числа неспаренных J-элек-
тронов.
У элемента галлия, следующего за цинком, очередной
электрон вступает на 4/>-подуровень Ga \s22s22p63s23p63dl04s24pl:
Ga
mi
Ml
' tl
• ti
t
ti
tl
ti
tl
p
tl
tl
tl
tl
tl
tl
d
tl
Mill
Элементы 4-го периода, у которых заполняется Зр-иодуровень
(Ga, Ge, As, Se, Br и Кг), имеют те же валентные состояния, что
и аналогичные элементы 3-го периода, что объясняется
одинаковым характером заполнения ^-подуровней.
У 18 элементов 5-го периода повторяется та же
последовательность заполнения 5^-, 4d- и 5/7-подуровней, что и у элементов
предыдущего периода. Валентные состояния элементов 5-го
периода в основном те же, что и у элементов 4-го.
Энергетически подуровни 5s и 4d очень близки, и часто один
электрон с 55-подуровня переходил на 4</-подуровень. Поэтому
у Nb, Mo, Tc, Ru, Rh и Ag на 55-подуровне находится только один
электрон, а у палладия вообще в невозбуждешюм состоянии
5мюдуровень не заполнен (это единственный элемент
периодической системы, не имеющий s-электронов). Валентные состояния
элементов 5-го периода с заполняющимся ^/-подуровнем
примерно те же самые, что и у элементов 4-го периода, за исключением
того, что рутений в отличие от железа проявляет валентные
состояния 7 и 8.
С цезия (№ 55) начинается заполнение бл-подуровня и
открывается новый, 6-й период системы Менделеева. У бария
41
(№ 56) на 6^-подуровне два электрона. Следующий электрон,
согласно правилу п + /, должен вступить на 4/-подуровень.
Однако вследствие очень близких энергий 4f- и 5^-подуровней и
особой устойчивости незаполненного 4/подуровня у лантана (№ 57)
электрон вступает на 5*/-подуровень. У церия, следующего за
лантаном, распределение электронов подчинено правилу (« + /),
поэтому на 4/-подуровне у него находится 2 электрона — один
новый, а другой — перешедший с 5</-подуровня.
В дальнейшем заполнение 4/подуровня происходит согласно
правилу Гунда: 4/-подуровень заполняется (у европия) неспарен-
ными электронами, а затем образуются пары электронов.
4/-Подуровень, заполненный неспаренными электронами,
обладает повышенной устойчивостью, поэтому у гадолиния,
следующего за европием, на 4/-подуровне сохраняется прежнее
количество электронов G), а новый электрон вступает в 5</-подуровень,
нарушая правило (п + l). У тербия очередной электрон вступает
на 4/-подуровень и происходит переход электрона с 5^-подуров-
ня. Иттербием заканчивается заполнение 4/-подуровня и уровня
cw=4. Новый электрон входит на 5*/-подуровень, создавая
электронную конфигурацию атома лютеция, отличающуюся от
конфигурации атома лантана только полностью заполненным 4/
подуровнем.
Обычно в периодической системе сразу за барием помещают
лантан, а следующие четырнадцать элементов выносят за
пределы таблицы. Если бы правило (п+l) соблюдалось, то лантан
был бы не d-элементом, а первым /-элементом. Заполнение 4/-
подуровня заканчивается у иттербия, и следующий за ним
лютеций
Lu
Ml
5 tl
Ml
3 tl
2 tl
Ml
tl
tl
tl
tl
tl
tl
tl
tl
p
tl
tl
tl
tl
t
tl
tl
tl
tl
tl
tl
d
tl
tl
tl
tl
tl
tl
tl
tl
tl
tl
tl
по существу уже не является /-элементом. Поэтому в таблице
Менделеева под скандием и иприем правильнее расположить
лютеций как первый ^/-элемент, а все 14 элементов перед ним,
включая и лантан, вынести в особую группу лантаноидов за
пределы системы. Это и сделано в прилагаемом варианте
периодической системы (см. таблицу на первом форзаце).
Химические свойства элементов главным образом связаны со
структурой внешних электронных уровней. Изменение числа
42
электронов на третьем снаружи 4/:подуровне слабо отражается
на химических свойствах элементов. Поэтому все /-элементы
очень похожи друг на друга. Все лантаноиды проявляют
валентное состояние 3, которое для них наиболее характерно.
Наиболее устойчиво это валентное состояние у лантана, гадолиния
и лютеция, так как у них в этом состоянии возникаю! устойчивые
конфигурации 4/°, Ajn и 4/14 соответственно. Одновалентное
состояние для лантаноидов неизвестно. Валентности 2 и 4
малохарактерны, но их наличие можно связать с особой устойчивостью
незаполненного 4/°-, наполовину заполненного Ау - и полностью
заполненного ^^подуровня. Валентное состояние 2 имеют
европий и иттербий из-за их электронных конфигураций (сокращен-
то) 4/75</° б*5 и 4/145</° б*2.
Валентное состояние 4 имеют церий и тербий вследствие
строения их атомов 4/25</°6$2 и 4f*Serbs1. У них при
образовании связи участвуют два /-электрона и два распаренных л-элек-
троеа.
После лантаноидов в 6-м периоде идут элементы, у которых
заполняется 5</-подуровень, заканчивающиеся ртутью, а затем—
по радон включительно—элементы с заполняющимся 6/ьпод-
уровнем.
В 7-м периоде имеется два элемента—франций и радий,
имеющие внешние ^-электроны; далее, начиная с актиния,
следуют четырнадцать элементов с заполняющимся 5/-гюдуровнем,
образующие семейство актиноидов. Лоуренсий, как и лютеций,
имеет один 6</-электрон, за ним следуют элементы с
заполняющимся ^/-подуровнем. 7-й период не завершен.
По химическим свойствам актиноиды похожи как друг на
друга, так и на лантаноиды, что объясняется одинаковым в
большинстве случаев строением трех наружных уровней. Наиболее
распространено у актиноидов валентное состояние 3. Актиноиды
проявляют более высокие валентные состояния, чем лантаноиды.
Из-за высоких валентных состояний раньше некоторые из
актиноидов помещали в основные подгруппы. Для тория,
протактиния и урана высшие валентности D, 5 и 6 соответственно)
совпадают с номерами групп, в которых они могут быть
последовательно расположены за актинием.
Для № 104 характерна валентность 4, и он во многом
напоминает гафний, цирконий и титан. Формально элемент № 105
должен быть аналогом тантала, элемент № 106—вольфрама
и т. д., но, поскольку энергии 5/- и 6</-подуровпей очень
близки, можно ожидать проявления более высоких валентных
состояний.
Все рассмотренные примеры показывают, что с ростом
заряда ядра периодически повторяются сходные электронные
структуры и, следовательно, свойства элементов. При
рассмотрении электронных структур атомов элементов становился
43
очевидной связь расположения атомов в периодической системе
с их строением. Здесь можно отметить следующие
закономерности:
1. Все элементы располагаются в порядке их атомных
номеров, т. е. в порядке увеличения числа протонов в ядре. Весьма
вероятно, что периодическая повторяемость внутриядерных
структур, составленных из протонов и нейтронов, отражается на
периодически повторяющихся электронных структурах.
2. Химические элементы по структуре невозбужденных
атомов располагаются в горизонтальные и вертикальные ряды,
носящие название п е р и о д о в и групп.
3. Период—это горизонтальный ряд элементов,
расположенных в порядке непрерывного роста числа протонов в ядре; в
атомах элементов одного периода происходит заполнение
электронами близких по энергии подуровней. Номер периода совпадает
со значением последнего заполняемого или заполненного $-под-
уровня. Энергия ^-подуровня определяется главным квантовым
числом л, поэтому в периоде находятся элементы, в атомах
которых заполняются подуровни, близкие по энергии,
задаваемой квантовым числом и.
Элементы с особо ярко выраженным сходством следуют один
за другим сначала через 8, затем через 18 и через 32 порядковых
номера. Поэтому различаются малые и большие периоды. Длина
периода определяется числом подуровней, заполняющихся при
формировании периода: 1-й период—^-подуровень—2 элемента;
2-й и 3-й периоды—s- и р-подуровни—8 элементов; 4-й и 5-й
периоды—-s-, р- и ^-подуровни—18 элементов; 6-й (и 7-й)
периоды—s-, р-, d- и/-подуровни—32 элемента.
4. Группа — вертикальный ряд элементов. В группе
объединены элементы, атомы которых различаются числом
энергетических уровней, но имеют одинаковую структуру внешних
электронных уровней. Элементы одной группы имеют сходные
валентные состояния и, следовательно, формулы образуемых
соединений. В группе сверху вниз увеличивается число
энергетических уровней и в связи с этим наблюдается закономерное
изменение многих свойств атомов, простых веществ и
однотипных соединений. Элементы групп разделяются на подгруппы.
Элементы, у которых заполняются s- и р-подуровни, образуют
главные подгруппы, а элементы, у которых заполняется rf-под-
уровень,— побочные. Сходство элементов главных и побочных
подгрупп заключается в величине проявляемой максимальной
(спиновой) валентности.
5. По виду заполняемого электронами подуровня элементы
разделяют на s-, р-, d- и/-последовательности,
Каждые два первых элемента всех периодов, у которых
сначала появляются один, а затем второй s-электроны, называются
s-элементами. Последовательность элементов, у которых проис-
44
ходит изменение числа р-электронов от одного до шести,
называется р-элементами. d-Элементы—это элементы, расположенные
между вторым ^-элементом и первым /^-элементом 4-го и
последующих периодов. Аналогично выделяются последовательности
f-элементов, входящих в 6-й и последующие периоды
периодической системы.
6. Форма периодической системы, по существу, повторяет
энергетическую диаграмму заполнения электронами уровней
и подуровней многоэлектронного атома.
Предложено более 400 вариантов формы периодической
системы элементов Д. И. Менделеева. Один из короткопериодных
вариантов системы см. на первом форзаце. Один из длинных
вариантов таблицы приводится на втором форзаце книги.
§ 8. ПЕРИОДИЧНОСТЬ СВОЙСТВ АТОМОВ
Периодичность свойств элементов особенно ярко выражена
в структуре электронных уровней атомов и проявляется в
зависящих от нее свойствах. Так как структура влияет на свойства
молекул, то очень часто в ряду однотипных молекул и
однотипных реакций наблюдается периодичность в изменении ряда
свойств.
Однотипными молекулами и соединениями называются такие
молекулы и соединения, которые имеют аналогичные формулы,
различающиеся лишь одним элементом, причем эти элементы
принадлежат к одной группе (или подгруппе) периодической
системы и находятся в этих молекулах и соединениях в одинаковом
валентном состоянии.
Однотипными химическими реакциями называются такие
реакции, в которых участвуют однотипные молекулы или соединения,
находящиеся в одинаковых агрегатных состояниях, причем стехи-
ометрические коэффициенты при формулах соответствующих
веществ равны.
Закономерное изменение свойств замечается также для более
сложных молекул, жидкостей, кристаллов.
Но поскольку свойства сложных систем слагаются из целого
ряда свойств, которые могут изменяться в противоположных
направлениях, явление периодичности или закономерное
изменение свойств таких систем в периоде или в подгруппе часто не
обнаруживается. В ряду простых однотипных реакций часто
проявляется закономерное изменение характеристики реакции, но для
сложных реакций вследствие суммирования величин, изменения
которых возможны в противоположных направлениях, иногда
закономерность в изменении характеристики реакции нарушается.
Свойства элементов, определяемые электронной структурой
атома, изменяются периодически, как и сами электронные
45
струк!уры. Важнейшей характеристикой атома является его
размер, однако вследствие вероятностного характера нахождения
электронов на энергетических уровнях и подуровнях и волновой
природы их движения размеры атомов не могут быть точно
определены. В качестве радиуса атома принимается эффективный
радиус, г. е. расстояние от ядра атома до области максимальной
плотности внешней электронной орбитали. Его рассчитывают
по межъядерным расстояниям в молекулах и кристаллах.
Атомные (и ионные) радиусы изменяются периодически по
мере увеличения заряда ядра. В периоде слева направо радиусы
уменьшаются, в подгруппе сверху вниз обычно увеличиваются
(табл. 5).
Размеры реагирующих атомов и межъядерные расстояния
в образовавшихся молекулах сильно влияют на
термодинамические и кинетические характеристики процесса и свойства
соединений.
Таблица 5
Агом
н
Li
Na
К
Rb
Cs
F
Cl
Г. HM
0,046
0.155
0,189
0,236
0,248
0,268
0,064
0,121
Лгом
Br
I
Be
В
С
N
О
г им
0,150
0.165
0,113
0,091
0,077
0,071
0,066
Атом
Nc
Mg
Al
Si
P
S
Лг
/ IIM
0,160
0,160
0Л43
0,134
0J30
0,125
0,192
Простейшей химической реакцией, характеризующей свойства
свободных атомов, является процесс отрыва электронов от
невозбужденного атома:
Э = Э++е-, Д//иониз.
Изменение энтальпии в этом процессе численно равно количеству
энергии, необходимой для отрыва электрона от невозбужденного
атома, т. е. равно энергии ионизации.
Обычно, чтобы определить энергию ионизации, исследуют
пределы спектров, т. е. частот, при которых отдельные линии
сливаются в сплошную полосу. Последнее означает, что в этой
области энергий электрон захватывается или испускается без
квантования энергии (кинетическая энергия не квантуется и
поэтому возможно непрерывное поглощение или испускание частот),
что соответствует уходу электрона из поля ядра, т. е. его огрыву
от атома.
Энергия ионизации количественно характеризует способность
атома удерживать электроны, что является важной харак1еристи-
46
500-
5 6 7 8
группа
Рис. 8а. Энергии ионизации
атомов 2-го и 3-го периодов
кой его химической активности. Для
многоэлектронных атомов определены
последовательные энергии ионизации,
отвечающие отрыву первого, второго,
третьего и т. д. электронов 1и /2,
/3 и т. д. Обычно удаление электрона
тем легче, чем больше номер периода,
т. е. чем дальше находится
энергетический уровень от ядра.
В периоде первые энергии
ионизации атомов элементов возрастают, но
не равномерно (рис. 8а). Во 2-м
периоде самая низкая энергия ионизации
у лития, так как он легко теряет один
неспаренный наружный электрон.
Первая энергия ионизации бора ниже
энергии ионизации бериллия, что
объясняется отрывом первого неспаренного р-
электрона. От бора к азоту по мере
заполнения р-подуровня неспаренными электронами энергии
ионизации равномерно возрастают, но у кислорода монотонность
изменения энергии ионизации нарушается. У нейтрального атома
азота все три />-орбитали заполнены по одному электрону.
Наполовину заполненный подуровень обладает повышенной
устойчивостью. Введение одного электрона в такой подуровень
приводит к ослаблению его связи с ядром, и поэтому первая энергия
ионизации кислорода ниже, чем азота. От кислорода к неону
первые энергии ионизации снова равномерно возрастают.
Точно такая же картина в характере изменения первых энергий
ионизации наблюдается и в других периодах, но отличия в
стабильности незаполненного, наполовину заполненного и
полностью заполненного ^-подуровня постепенно сглаживаются при
возрастании номера периода.
Пользуясь значениями энергий ионизации, можно рассчитать
энтальпии реакций обмена электронами и предсказать их
направление.
Например, определим, возможен ли процесс передачи электрона атома лития
положительно заряженному иону натрия:
Li(r)+Na»=Li+(r)+Na(r), АЯр_цаи.
Энтальпии ионизации (энергии ионизации) для лития и натрия практически
равны:
Li = Li++«T, A#!=520 кДж/моль,
Na = Na++<T, Д#2=496 кДж/моль.
Запишем эти уравнения так, чтобы при суммировании можно было получить
искомое уравнение:
Li-Li++e~, АЯ1 = 520 кДж/моль
47
Na*+c = Na, -A//2= -496 кДж/моль
V=Li4 +Na. AHV m = AHx -AH2 = 24 кДж/моль*
Положительное значение АНР ции указывает на невозможность этого процесса
Противоположный процесс
Li+ (r) + Na(r) = Li(r) }-Na+(r). АЯ--24 кДж/моль
сопровождающийся выделением энср! ии и уменьшением энтальпии, возможен.
Рассмотренная реакция является простейшей окислительно-
восстановительной реакцией. Окислительно-восстановительными
реакциями называют такие реакции, которые сопровождаются
передачей одного или нескольких электронов от одних атомов,
ионов или молекул к другим атомам, ионам или молекулам. При
окислительно-восстановительных реакциях протекают два
взаимосвязанных процесса — процесс окисления и процесс
восстановления. Процесс окисления связан с отдачей электронов, а процесс
восстановления—с присоединением электронов.
Те атомы, ионы или молекулы, которые отдают электроны,
называются восстановителями. Те атомы, ионы или молекулы,
которые присоединяют электроны, называются окислителями.
При окислительно-восстановительной реакции окислитель
восстанавливается, а восстановитель окисляется.
Следовательно, реакция передачи атомом натрия электрона иону лития со-
стит из процесса окисления
и процесса восстановления
Li *+<»- = Li
Атомы Na отдают электроны — эго восаановители, ионы Li+ принимают элск-
гроны—'л о окислит ели.
В ?'-подгруппе сверху вниз энергии ионизации понижаются,
следова!ельно, восстановительная способность
агомов—способность отдавать электроны — возрастает и цезий — один из
сильнейших восстановителей. Окислительная способность ионов,
наоборот, возрастает в подгруппе снизу вверх и ион Li+ — наиболее
сильный окислитель. Такое поведение свободных атомов и ионов
не всегда распространяется на их другие состояния — металлы
и водные растворы солей, так как из-за наложения различных
процессов характер изменений свойства может нарушиться.
В периоде у л- и /^-элементов энергии ионизации в общем
увеличиваются. Это означает, что способность к отдаче
электронов уменьшается, т. е. восстановительные свойства атомов
уменьшаются, а окислительные свойства положительно
заряженных ионов возрастают. Так как сверху вниз в подгруппе энергии
ионизации атомов понижаются, то, следовательно, ион F+ —
один из сильнейших окислителей среди простых положительно
однозарядных ионов в свободном состоянии.
48
Определим, возможен ли процесс
F+(r)+Cs(r)^F(iL-Cs+(r), ЛЯР ции
при следующих значениях энтальпий ионизации (энергий ионизации):
F=F+ +*", Af/= 1681 кДж/моль,
Cs = Cs* +e\ Af 1=316 кДж/моль.
Откуда
Д^р цИИ^ -1681 + 376= -1305 кДж/моль
Реакция термодинамически очень вероятна и вследствие большого уменьшения
энтальпии сильно смещена вправо Наоборот, противоположная реакция
получения иона F* невозможна.
Если энергия ионизации характеризует способность атомов
удерживать электроны, то энергетическое изменение в другой
простейшей реакции
характеризует стремление к присоединению атомом электрона
с образованием отрицательного иона. Изменение энтальпии
в этом случае носит название сродства к электрону. Сродство
к электрону—это энергия, выделяющаяся при захвате электрона,
или энергия, которую нужно
затратить для присоединения
электрона к атому (или иону
или молекуле).
Для большинства атомов
присоединение электрона
сопровождается выделением
энергии ((?>0). Наименьшие
численные значения сродства
к электрону у атомов с
заполненными s и ^-подуровнями—
гелия, бериллия, неона, магния
и др. Сродство к электрону
у атома азота и фосфора
меньше, чем у соседних /^-элементов
периода. Это говорит о
повышенной устойчивости
незаполненного, наполовину и
полностью заполненного подуровня
(рис. 9).
Для образования ионов
Ве~, Ne~, Mg~ и других
требуется затрата энергии, поэтому
эти ионы неустойчивы.
Присоединение одного электрона
к атомам углерода, кислорода,
-300^
Рис 9. Сродство к электрону
49
фтора, серы и другим сопровождается выделением энергии. Это
говорит о том, что имеющиеся у нейтральных атомов электроны
не компенсируют полностью сил притяжения противоположно
заряженного ядра. Однако сил ядра уже не хватает на удержание
двух (или более) электронов. Поэтому одноатомные
многозарядные отрицательные ионы (типа О2"", S2~, N3~) в свободном
состоянии неустойчивы:
О+2*Г =О2~, Д#=712 кДж/моль
S + 2e~=S2~, Atf=38 кДж/моль
В подгруппе сверху вниз энергия сродства должна
уменьшаться, однако при переходе от фтора к хлору и от кислорода
к сере неожиданно обнаруживается ее увеличение. Вероятно,
это связано с наличием J-орбиталей у атомов элементов 3-го
периода.
В такого типа процессах кислород, сера и особенно галогены
играют роль сильных окислителей. В периоде среди s- и /7-элемен-
тов окислительная способность атомов возрастает, а сверху вниз
в подгруппе понижается. Чем труднее атом отдает электрон, тем
сильнее он его притягивает и тем больше склонность атома
к захвату дополнительного электрона. Следовательно,
способность к присоединению электрона усиливается как с ростом
величины сродства к электрону, так и с ростом энергии
ионизации.
Элементы, атомы которых склонны к приему электронов,—окислители —
расположены в правом верхнем углу периодической системы (без ^-элементов).
Элементы, атомы которых склонны к отдаче электронов,— восстановители —
находятся в левом нижнем углу периодической системы
Деление элементов на окислители и восстановители очень условно, так как
окислительно-восстановительная способность одного вещества зависит от
природы другого участника реакции.
Способность атомов легко отдавать электроны свойственна атомам
металлов В сгруктуре металлов как простых веществ атомы удерживаются
переданными ими в общее пользование электронами. Это и обусловливает проявления
особых металлических свойств (блеск, электрическая проводимость и др.). Все
^/-элементы вследствие способности их атомов легко отдавать электроны и
проявлять восстановительные свойства относятся к металлам.
Простые вещества, атомы которых прочно удерживают свои электроны,
являются неметаллами.
§ 9. ХИМИЧЕСКАЯ СВЯЗЬ. МЕТОД ВАЛЕНТНЫХ СВЯЗЕЙ
Учение о строении вещества объясняет причины
многообразия химических соединений и их состав, строение молекул
и структуры веществ в различных агрегатных состояниях. Учения
о направлении процесса, о его скорости и о строении вещества
теснейшим образом связаны и взаимно влияют на развитие друг
друга. Самую общую картину такого влияния можно предста-
50
вить так. Из данных химической термодинамики рассчитывают
энер!ию химических связей, чго дает возможность судить о их
природе. Из сопоставления энергий химических связей большого
числа молекул делают выводы о прочности связей других
молекул, и если обнаруживается несовпадение экспериментальных
и расчетных данных, это становшся толчком для дальнейшей
разработки вопросов leopnn ciроения. Сведения о структуре
молекул позволяю! судить о механизме взаимодействия и на
основании их иногда удае!ся предсказывал» cKopocib реакций.
Если наблюдаемые и 1еорегические данные расходятся, то это
говорит о том, что механизм реакции представлен неправильно
или же мы располагаем несовершенными знаниями о структуре
участвующих молекул.
Современные физические методы позволяют
экспериментально определять структуру молекул. Несоответствие полученных
данных о струкгуре с выводами на основании энергий связи
U механизмов реакции является побудительной причиной
дальнейшей разработки более совершенных физических и химических
методов.
Единственным критерием химического взаимодействия
атомов, ионов или молекул является изменение электронной
плотности. Образовавшаяся новая молекула устойчива только
в том случае, если энергия возникших частиц ниже энергии
исходных, т. е. если имеет место выделение энергии, или. чго то
же, уменьшение эшальпии.
Так как ядра агомов исключительно малы по сравнению
с размерами атомных орбиталей, то химическое взаимодействие
объясняется взаимодействием электронов реагирующих частиц.
Современная теория химической связи основана на
предположении, что при образовании связи двумя частицами в пространстве
между ядрами электронная плотное 1Ь возрастает. Не известно ни
одного экспериментального факта, противоречащего этому
принципу.
Исходя из этого, обратимся к реакции взаимодействия двух
атомов водорода:
Н(г) + Н(г) = Н2(г) АН =-432.1 кДж/моль
Изменение энтальпии в л ой реакции характеризует прочность
связи между атомами в молекуле.
Энергия связи Н2 равна 432,1 кДж/моль (при ОК). Это то
количество энергии, которое следует затражть на разрыв 1 моль
молекул водорода.
Следовательно, система, сосюящая из разделенных aiомов
водорода, неустойчива по сравнению с системой из тех же
атомов, но объединившихся в молекулы (при невысоких
температурах).
51
0fi5 OJQ 0J5 0,20 0%Z5
Межьядерное расстояниеум
Рис. Ш. Образование
химической связи между двумя
атомами водорода
Химическая связь образуется,
когда энергия взаимодействующих
атомов понижается. На рис. 10 приведена
кривая а зависимости потенциальной
энергии двух атомов водорода от
расстояния между их ядрами. Сложный
характер кривой объясняется тем, что
между двумя атомами водорода
действуют силы двух типов: силы
притяжения между ядром одного атома
и электроном другого и силы
отталкивания между ядрами и электронами
разных атомов.
По мере сближения атомов силы
притяжения постепенно ослабевают,
а силы отталкивания, наоборот,
возрастают. В результате сначала имеет
место понижение энергии двух
атомов. Затем, когда силы
отталкивания и притяжения выравниваются,
достигается состояние минимума
потенциальной энергии, отвечающего 432,1 кДж/моль и
межъядерному расстоянию /=0,074 нм. Далее происходит резкое
увеличение энергии вследствие возрастания отталкивания между двумя
ядрами. Именно минимум на кривой энергии отвечает наиболее
устойчивому состоянию системы двух атомов—состоянию
образования химической связи и возникновению молекулы
водорода.
Несмотря на кажущуюся правильность рассуждений,
подобная модель не объясняет тот факт, что половина всех
взаимодействующих атомов водорода не обнаруживает сил притяжения,
и их сближение сопровождается только силами отталкивания,
что представлено кривой б на рис. 10. По-видимому, кроме
упомянутых электростатических сил притяжения и отталкивания
должны существовать другие силы, ответственные за
образование химической связи. Природа этих сил связана с волновым
характером поведения электрона в атоме.
Химическая связь может образоваться только при условии,
что спины электронов взаимодействующих атомов антипарал-
лельны. При этом электронные облака, обладающие волновыми
свойствами, накладываются друг на друга в зоне между ядрами
атомов, и в месте их перекрывания электронная плотность
возрастает. Но электронные облака с параллельными спинами не
способны к перекрыванию: при сближении они отталкиваются
и связь не возникает.
Так как в рассматриваемом случае связь осуществляется
двумя одинаковыми атомами, то область перекрывания электрон-
52
ных облаков притягивается обоими ядрами с равной силой и
поэтому максимум электронной плотности располагается точно
посредине между ядрами. Такая химическая связь называется
неполярной. При частичном смещении области перекрывания к
одному из атомов образуется полярная связь. При полярной связи
вероятность нахождения электрона у ядра одного из атомов
выше, чем у другого.
Теоретически можно подойти к случаю, когда область
перекрывания полностью смещена к одному из атомов. Это
означает, что электрон одного атома перешел в сферу
притяжения другого атома и вероятность нахождения двух
электронов у одного ядра равна 100%. Такая связь называется
ионной. Однако не известно ни одной молекулы с полным
перемещением пары электронов к одному из ядер. Если
известны молекулы с чисто неполярной связью (это двухатомные
молекулы одного элемента), то чисто ионная связь не
встречается.
Способность атома перетягивать на себя область
перекрывания электронных облаков зависит как от способности этого
атома принимать электроны, т. е. от его сродства к электрону,
так и от способности другого атома отдавать электрон, т. е. от
его энергии ионизации.
Сумма энергии ионизации и энергии сродства к электрону
называют электроотрицательностью, ЭО:
АЯиониз+АЯсрод = ЭО.
Электроотрицательность позволяет сравнивать атомы по их
способности оттягивать электронную плотность при
образовании химической связи. Электроотрицательность фтора
ЭОР = Д#„0Н1П+АЯсрод = 1680,6+322,3 % 2000 кДж/моль,
электроотрицательность лития
3OLi = Д#иониз+Д#срод=520,1 + 59,8 « 580 кДж/моль.
Следовательно, способность оттягивать на себя электронную
плотность у фтора больше и молекула LiF сильно полярна.
Поскольку сродство к электрону не определено для всех
элементов, а для других получены неточные величины, при
оценке способности атомов оттягивать электронную плотность
и определении степени полярности химической связи
пользуются относительными электроотрицательностями. Обычно
электроотрицательность фтора, как элемента с
максимальной ЭО, принимают равной 4,0, а лития—1,0. Относительные
электроотрицательности атомов других элементов приведены
в табл. 6.
53
Таблица 6
Шкала электроогрицателъиости
н
2.1
Li
1.0
Na
0.9
К
0 8
Rb
0,8
Cs
0.7
Не
Be
1,5
Mg
1,2
Са
1,0
Sr
1,0
Ва
0.9
Sc
КЗ
Y
1.3
Ti
1 6
Zr
1,6
V
1,6
Nb
1.6
Cr
1 в
Mo
1.6
Mn
1 6
Tc
1,6
Fe
1,6
Ru
1.6
Co
1,6
Rh
1.6
Ni
1.6
Pd
1,6
Cu
1,6
Ag
1,6
Zn
1.6
Cd
1.6
В
2 0
Al
1,5
Ga
1,7
In
1,7
С
2,5
Si
1,8
Ge
1.7
Sn
1.7
N
3 0
P
2,1
As
2,0
Sb
1,8
О
3,5
S
2.5
Se
2,4
Те
2.1
F
4 0
Cl
3,0
Br
2,8
1
2,4
Ne
Ar
Kr
Xe
Наименьшее значение электроотрицательности имеет Cs.
В периоде значения ЭО возрастают, а сверху вниз по подгруппе
падают. Металлы имеют самые низкие значения ЭО,
неметаллы— высокие. У всех ^/-элементов, кроме Sc и Y, ЭО=1,6; у Sc
и?ЭО=1,3.
Определим направление смещения электронного облака в молекуле СО:
ЭОС=2,5, ЭОО=3,5, следовательно, можно ожидать смещения зоны
перекрывания электронных облаков С и О в сторону атома кислорода (Это, однако, еще не
позволяет сделать вывод о дипольном характере молекулы СО.)
Для молекулы BrCl ЭОВт=2,8, ЭОа = 3,0, следовательно, зона перекрывания
несколько смещена в сторону хлора.
Молекула CsF составлена из двух атомов с максимальной разностью элект-
роотрицательностей ЭОС, = 0,7, ЭОР = 4,0. Электрон цезия в сильнейшей степени
смещен в сторону фтора.
Связь, образованная атомами, резко отличающимися по
электроотрицательности, ионная. В действительности все ионные
связи имеют полярный характер. Чем больше различие в электроот-
рицательностях атомов, тем сильнее выражен полярный характер
химической связи. Смещение
электронной плотности происходит
в сторону ядра того из атомов,
который более электроотрицателен
и расположен выше и правее на
шкале электроотрицательности, т. е.
обладает более сильными
окислительными свойствами.
Молекула водорода по строению
Рис. п. Модель молекулы водо- самая простая из огромного много-
рода образия других (рис. 11).
54
Остановимся на
двухатомных молекулах,
образованных водородом
с другими элементами.
Форма электронной
орбитали атома гелия
такая же, как и у водорода.
Два 5-электрона образуют
двухэлектронное облако.
В соответствии с
принципом Паули вхождение
третьего электрона в эту
Рис. 12 Образование молекулы LiH
орбиталь невозможно, перекрывания орбиталей атома гелия
с орбиталями других атомов не происходит, и гелий в
невозбужденном состоянии с атомом водорода, как и с другими а томами,
не образует молекулу.
Форма электронных орбиталей атома лития похожа на форму
орбцталей водорода, но у атома лития имеется два д-подуровня.
В молекуле LiH перекрываются 25-орбиталь лития и Lv-орбиталь
водорода (рис. 12). Внутренние орбитали не оказывают
значительного влияния на химическую связь, и в дальнейшем мы их
изображать не будем.
Молекула ВеН образуется после возбуждения атома бериллия
и перехода одного 2я-олектрона на 2/?-подуровень с последующим
перекрыванием 2/?-орбитали с hv-орбиталью Н, как это
изображено на рис. 13.
Совершенно анало!ичио образуются и другие двухатомные
молекулы гидридов. Например, молекула HF. Атом фтора в
нормальном состоянии имеет один электрон на 2/?-подуровне,
и возбуждения атома не требуется. Молекула HF образуется
Be
u
¦¦
s
J\ f | f
P ' -Щ
s
p
<
f
к
Шбодные
орбитам
Н
Рис. 13 Образование молекулы ВсН
55
Рис. 14. Возбужденный атом бериллия
перекрыванием 2рг -орбитали
атома фтора и Is1 -орбитали
атома водорода. Вследствие
больших энергетических
трудностей перевода хотя бы
одного 2/7-электрона атома неона
на более высокий 3^-подуро-
вень не обнаружено молекул,
образованных атомом неона
с водородом или с другими
атомами.
Среди двухатомных
гидридов элементов второго
периода только молекулы LiH
и HF устойчивы, остальные молекулы ВеН, ВН, СН, NH и ОН
не отвечают валентным возможностям соответствующих
элементов и обнаружены лишь при высоких температурах в газовом
состоянии.
Вдоль периода от Li к F увеличение электроотрицательности
изменяет направление смещения электронной плотности от
водорода к атому другого элемента.
Молекула ВеН известна в газовом состоянии, но она резко
уступает по устойчивости молекуле ВеН2. Структура молекулы
ВеН2 не объясняется рассмотренными ранее представлениями
о простом перекрывании электронных облаков.
Действительно, электронная структура возбужденного атома
бериллия имеет вид, изображенный на рис. 14. Перекрывание
с одним атомом водорода легко объяснимо B/?-орбиталь
бериллия), но неясно, как и в каком направлении происходит
перекрывание с орбиталью второго атома водорода. Это перекрывание
может произойти в нескольких различных местах, например,
указанных стрелками, и, следовательно, угол между связями
Be—Н может быть различным. Перекрывание орбитали атома
водорода с 2/ьорбиталью атома бериллия и его же 2^-орбиталью,
очевидно, энергетически не равноценно, и две связи Be—Н
должны отличаться своими энергиями.
Между тем экспериментально и на основании многих
теоретических предпосылок установлено, что:
1) угол между связями в молекуле ВеН2 равен 180°, т. е. все
три атома расположены на одной прямой Н—Be—Н;
2) обе связи энергетически совершенно равноценны;
3) момент диполя молекулы ВеН2 равен нулю, хотя связи
из-за различия электроотрицательностей элементов полярны.
Ответ на возникшее противоречие дает представление о
гибридизации орбиталей. Гибридизация — это процесс
перераспределения электронных плотностей близких по энергии орбиталей, кото-
рый приводит к их полной равноценности. Гибридные орбитали
56
$-орёиталь и р-ордиталь —»- две гибридные sp-орбиташ
Рис. 15. sp-Гибридизация
имеют одинаковую электронную плотность и, следовательно,
форму. На рис. 15 показан процесс гибридизации орбиталей
s и р с образованием двух равноценных гибридных sp-орби-
талей.
Гибридная орбиталь имеет большую вытянутость по одну
сторону от ядра, чем по другую. В перекрывании с другими
орбиталями участвуют только более вытянутые части гибридных
орбиталей. Основное преимущество гибридной орбитали—ее
высокая степень направленности, что способствует более
глубокому перекрыванию с орбиталью присоединяющегося атома.
Образование более прочных и симметрично расположенных
связей приводит к энергетически более выгодному состоянию.
Молекула ВеН2 (рис. 16) имеет две равноценные связи Be—Н,
расположенные под углом 180°. Несмотря на полярность каждой связи,
вызванную различием электроотрицательностей бериллия и
водорода, молекула в целом неполярна и ее электрический момент
диполя равен нулю. Из-за взаимного перекрывания всех неспа-
ренных электронов молекула диамагнитна.
Совершенно аналогичные рассуждения приводят к выводам
о структуре неустойчивой молекулы ВН3 (рис. 17). Молекула
ВН3 плоская, все три связи В—Н расположены на одной
н
Рис. 16. Молекула ВеН2
Рис 17 Образование молекулы ВН3
57
Рис. 18 Тетраэлрическос
расположение связей в молекуле
метана
плоское 1 и, углы между связями равны
по 120 . Молекула неполярна и
диамагнитна.
Теперь можно сформулировать
новое представление о валентности.
Область перекрывания образуется двумя
электронами с противоположными
спинами, поэтому число областей
перекрывания, равное числу неспаренных
электронов каждого атома, равно
числу связей атома с другими атомами,
или его валентности.
Перейдем к рассмотрению
структуры молекулы метана. Именно для объяснения факта четырех-
валетности углерода в органических соединениях и было
разработано понятие гибридизации. Без гибридизации в
возбужденном состоянии углерод имеет три 2/ьэлектрона и один 2д-элек-
трон. Можно было бы ожидать, что углерод способен образовать
три одинаковые связи перекрыванием 2/;-орбиталей и одну связь
другого типа перекрыванием 2^-орбитали. Тогда углы между
тремя связями первого типа должны были бы равняться 90°
и отличаться от угла с четвертой связью. Однако все углы равны
точно I09°28\ Такой угол получается, если соединить центр
тяжести тетраэдра—правильной пирамиды с треугольным
основанием—с его вершинами (рис. 18).
Углерод в молекуле метана находится в 5р3-гибридном
состоянии (рис. 19). Угол между гибридными sp 3-орбиталями равен
109с28'. Вследствие отсутствия неспаренных электронов молекула
диамагнитна. Молекула неполярна, хотя связи С—Н полярны.
В метане водород несет очень слабый положительный заряд
+ 0,01 е.
В подгруппе сверху вниз у молекул
SiH4, GeH4 и SnH4 сохраняется тетра-
эдрический угол между связями и,
следовательно, одно и то же состояние
гибридизации sp3.
Элементы главной подгруппы
V группы образуют гидриды ЭН 3 двух
разных структур. Три наружные р-ор-
битали атомов этих элементов могут
перекрываться с орбиталями трех
атомов водорода, при этом между
связями сохраняются примерно те же
углы, что и между /7-орбиталями, т. е. 90°
(рис. 20).
Рис 19 Образование молеку- Действительно, в молекулах РН3,
лы метана AsH3h SbH3 углы между связями поч-
58
sp3-гибридизация
Рис. 20. Молекула РН3
Рис 21. Молекула аммиака
ти точно равны 90°, что свидетельствует об отсутствии
гибридизации. Однако в молекуле NH3 угол HNH равен 107°, что очень
близко к величине 109°28' угла между связями при тетраэдриче-
ском их расположении. Именно поэтому следует допустить sp3-
гибридизацию электронов в атоме азота. При этом гибридизует-
ся 2^-орбиталь, заполненная парой электронов, и три 2/?-орбита-
ли, имеющие по одному электрону. В результате получается
четыре 5р3-гибридных орбитали, из которых одна заполнена
парой электронов (рис. 21), Остальные три гибридные орбитали
перекрываются с орбиталями трех атомов водорода. Молекула
диамагнитна, но полярна из-за неполной компенсации моментов
диполя отдельных связей.
Разные по составу молекулы СН4, NH3 и Н2О имеют
одинаковое пространственное расположение связей. Этот
удивительный факт несомненно подтверждает гибридизацию орбиталей.
В атоме О электроны второго уровня претерпевают s/?
^гибридизацию с образованием двух заполненных гибридных орбита-
лей и двух орбиталей, имеющих по
одному электрону (рис. 22).
Вследствие отсутствия неспаренных
электронов молекула Н2О
диамагнитна. Ряд важнейших свойств жидкой
воды как растворителя обусловлен
высокой полярностью ее молекулы
(»4=6913I0-3(fKjiM).
Сверху вниз в подгруппе угол
между связями у гидридов
Н2О—H2S—H2Se—Н2Те
уменьшается скачком, достигая 90°:
sps-гибридизация
Н2О
H2S
104,5°
92°
H2Se
Н2Те
91°
90°
Рис. 22 Молекула воды
59
что говорит об изменении механизма образования молекул.
Молекулы H2S, H2Se и Н2Те образуются перекрыванием негибридных
2/ьорбиталей. По-видимому, это вызвано ослаблением
способности наружных электронных орбиталей гибридизоваться с
ростом числа электронных уровней. Полагают, что даже в молекуле
HF наружные орбитали фтора находятся в л/?3-гибридном
состоянии. Соединения элементов второго периода резко отличаются от
сходных соединений элементов следующих периодов. Гидриды
NH3, H2O и HF чрезвычайно сильно отличаются по свойствам
(например, по способности образовать водородные связи) от
аналогичных гидридов элементов следующих периодов. Это
позволяет сделать предположение о д/?3-гибридизации в атоме фтора.
У всех рассмотренных выше молекул, кроме молекулы
метана, углы между связями отклоняются от тетраэдрического 109°28/
и прямого 90°. Это обусловлено различием сил взаимного
отталкивания орбиталей, заполненных только парой электронов, и
орбиталей, перекрывающихся с атомами водорода. Орбитали с
парой электронов, т. е. не имеющие области перекрывания,
обладают большим отталкивающим эффектом по сравнению
с орбиталями, образующими связь.
В молекуле NH3 имеется одна ,ур3~гибридная орбиталь,
заполненная парой электронов, и она отталкивает в направлении от
себя три остальные орбитали связи N—Н, уменьшая угол до
107,3°. В молекуле Н2О две орбитали с парами электронов. Их
влияние на орбитали связи еще сильнее, и угол НОН
уменьшается до 104,5°. В молекулах H2S, H2Se и Н2Те углы между связями
равны 92, 91 и 90°. Здесь сказывается в большей степени эффект
отталкивания орбиталей связи, который ослабляется при
увеличении длины связи.
Способ образования других молекул принципиально не
отличается от уже рассмотренного. Большой интерес представляет
изучение связей атомов углерода между собой и с атомами
других элементов. Остановимся на строении молекул этана,
этилена и ацетилена. Углы между всеми связями в молекуле этана
почти точно равны между
собой (рис. 23) и не отличаются
от углов С—Н в молекуле
метана. Следовательно, атомы
углерода находятся в
состоянии 5/?3-гибридизации.
Молекула С2Н6 диамагнитна и не
имеет электрического
момента диполя. Энергия связи
С—С в молекуле этана равна
^335 кДж/моль.
В молекуле этилена С2Н4
Рис. 23 Молекула этана углы между связями составля-
60
к>т примерно по 120°. Из
этого можно сделать
вывод о 5р2-гибридизации
наружного электронного
уровня атома углерода.
Энергия связи между
двумя атомами углерода
в молекуле этилена
равна «592 кДж/моль.
Если бы в молекуле
этилена атомы
углерода были связаны такой
связью, как в молекуле
этана, то можно было
Рис 24. Молекула этилена
бы ожидать близких энергий связи в этих молекулах. Однако это
не так. Энергия связи между атомами углерода в этилене чуть ли
не в 2 раза больше, чем в этане.
На рис. 24 изображена структура молекулы С2Н4 в
предположении 5/>2-гибридизации атома углерода. Все связи С—Н
в молекуле этилена лежат в одной плоскости под углами около
120°. У каждого атома углерода имеется по одной негибридной
/м>рбитали, содержащей по одному электрону. «Гантели» этих
орбиталей расположены перпендикулярно плоскости рисунка.
Для объяснения столь большой разницы E92 — 335 = 257 кДж/
моль) в энергиях связи атомов углерода в молекулах этилена
и этана следует принять, что эти негибридные орбитали также
могут взаимодействовать, но боковыми областями своих
гантелеобразных орбиталей. Взаимодействие на рис. 24
изображено волнистой линией. Связь, образованная таким
способом, называется п-связъю.
У связей, с которыми мы имели дело ранее, область
перекрывания орбиталей находится па прямой, соединяющей ядра двух
взаимодействующих атомов. Такая связь, образованная орбиталями,
перекрывающимися в области между ядрами, называется а-связью. Ею
образованы молекулы галогенов, галогеноводородов, СН4, СС14,
СгН6, NH3 и т. п. В молекуле СН4 имеется четыре а-связи (С—Н).
В молекуле С2Н6 шесть а-связей С—Н и одна а-связь С—С.
В молекуле С2Н4 четыре связи С—Н и одна связь С—С —
также а-связи. Но атомы углерода связаны между собой
дополнительно одной я-связькх По-видимому, на долю я-связи
приходится 257 кДж/моль, если предположить, что а-связи С—С в
этане и этилене одинаковы.
Молекула ацетилена С2Н2 линейная, что говорит в пользу
^-гибридизации электронов атомов углерода. Расположение
орбиталей в молекуле С2Н2 схематически изображено на рис. 25.
Энергия связи между атомами углерода в молекуле ацетилена
резко отличается от энергий связи в молекулах этана и этилена.
61
Рис 25. Молекула ацетилена
Энергия связи между атомами углерода в молекуле ацетилена
равна ^811 кДж/моль.
Связи С—Н и С—С каждого атома С образованы
гибридными орбиталями, расположенными на прямой, соединяющей
ядра атомов» Две «гантели» негибридных орбиталей каждого
атома С расположены в двух перпендикулярных плоскостях. Их
взаимодействие боковыми областями приводит к образованию
двух л-связей, в результате чего связь О=Ю в ацетилене обладас!
наибольшей энергией. Однако образование второй тс-связи не
вносит в энергию связи такого же вклада, как образование к-
связи в молекуле этилена Оабл. 7).
В структурной формуле химического соединения одинарная
а-связь обозначается одной черточкой; двойная
связь—комбинация а- и я-связи — обозначайся двумя черточками; тройная
связь—комбинация ст- и двух я-связей—фемя черточками. Из
табл. 7 видно, что энергия двойной связи не равна удвоенной
энергии одинарной связи, а энергия тройной связи не равна ее
утроенной энергии Энергетические вклады каждой новой пары
электронов неэквивалентны.
Та блица 7
Связь
С—С
с=с
с==с
Тип
тибри инации
V
Характер
СВЯЗИ
1ст
1а+2гс
Энергия
спязи,
кДж'мо ib
315
592
811
Разница
кДж'мо н>
257
219
Углерод—совершенно особый элемент. Он способен
образовывать огромное количество органических соединений, так как
обладает особым сочетанием ряда свойств, отличающих его oi
остальных элеменюв. Связь С—С значительно прочнее связей
между двумя aiомами других элементов той же подгруппы.
Ниже сравниваются значения ЛЯ (кДж/моль) для связей между
атомами элементов подгруппы ушерода:
62
д#св. д//ся.
к Д ж'моль кДжмоль
С—С 343 Ge—Ge 188
Si—Si 222 Sn —Sn 154
Кроме того, у атома углерода велика склонность к
гибридизации внешних электронных орбита лей.
Благодаря малым размерам атома углерода все связи с его
участием довольно прочны. Так, я-связи, возникающие при
боковом перекрывании р-орбиталей, у углерода обладают
повышенной прочностью и сильно перекрываются. У других элементов
я-связи намного слабее и даже вообще не образуются. Связь
углерода с водородом также более прочна, чем аналогичные
связи других элементов подгруппы. Интересно, что связи с
атомами галогенов у углерода слабее, чем у кремния: меньшая
прочность связей в CF4 и СС14 по сравнению с SiF4 или SiCl4
вызвана меньшими размерами атома углерода и возникающим
благодаря этому более сильным отталкиванием атомов
галогенов друг от друга.
Один из наиболее сложных вопросов теоретической
органической химии—это природа связей между атомами углерода в
ароматических соединениях, в частности, в молекуле бензола.
Молекула бензола С6Н6 плоская, углы между связями составляют
120°, что позволяет предположить 5/?2-гибридизацию углерода.
Обычно формулу бензола изображают в виде правильного
шестиугольника, в котором одной черточкой изображена а-связь,
а двумя—а- и я-связи:
СН
нс/\н
.1
Казалось бы, в бензоле связь С—С должна быть длиннее связи
С=С как более прочной. Однако изучение кристаллической
структуры бензола показывает, что все расстояния между
атомами С в бензольном кольце одинаковы и формулу бензола следует
писать так:
63
н
Рис. 26.
бензола
Н
молекулы
Рис 27. я-Связи в молекуле
бензола
Это лучше всего объясняется тем, что боковые части
негибридных /ьорбиталей атомов углерода перекрываются с орбиталями
двух соседних атомов углерода. В этом случае все связи между
атомами углерода становятся одинаковыми.
На рис. 26 показан вид молекулы бензола «сверху» и только
гибридные 5/>2-орбитали, образующие а-связи. На рис. 27
молекула С6Н6 показана «сбоку» и видны только негибридные р-
орбитали, которые боковым перекрыванием создают л-связь.
Энергия связи между атомами углерода в молекуле бензола
* 505 кДж/моль.
Энергия связи С—С в этане равна 335 кДж/моль, в этилене
для связи С==С имеем 592 кДж/моль. Средняя величина
C35+ 592)/2=464 кДж/моль довольно близка к энергии связи
между атомами углерода в бензоле. Это показывает, что связи
между атомами углерода в бензоле являются промежуточными
между одинарной и двойной. Поэтому иногда формулу бензола
изображают так:
где связь С—С означает связь, промежуточную между С—С-
и О=С-связями. Таким образом, двойная связь С=С в бензоле
состоит из одной а- и одной половинной л-связи. Электроны на
тс-орбиталях движутся свободно по замкнутому шестиугольнику;
они называются подвижными, или делокализованными. Это
говорит о гом, что молекула бензола может иметь свойства,
сближающие ее с металлическими проводниками тока. Для делокализа-
ции электронов необходимо, чтобы они принадлежали системе
64
атомов, т. е. чтобы атомная орбиталь одного атома
перекрывалась с орбиталями нескольких других.
Все рассмотренные ранее молекулы имели связи,
образованные следующим способом: каждый из атомов дает для
перекрывания по одному электрону на связь. Химическая связь,
образованная этим способом, называется ковалептной.
Принципиально другой, но приводящий к тем же результатам
способ образования связи состоит в том, что один атом
предоставляет одну пару электронов, находящихся на орбитали в
спаренном состоянии, а другой атом — пустую орбиталь. В
результате возникает связь, образованная парой электронов, ставшей
теперь общей для обоих атомов.
Частица (атом, ион, молекула), предоставляющая
электронную пару, называется донором; частица, предоставляющая
пустую орбиталь, т. е. принимающая электронную пару, называется
акцептором.
Так, образование молекулы Н2 можно осуществить
взаимодействием протона Н+ и иона водорода Н~ (гидрид-ион)
О и Ш
s s
При сближении этих ионов двухэлектронная орбиталь иона
Н" притягивается к протону и превращается в общую для обоих
ядер орбиталь с зоной перекрывания, расположенной точно
посредине между ядрами двух атомов. Возникает обычная
неполярная связь со всеми присущими ей свойствами.
Принято говорить, что такая связь образуется по донорно-
акцепторному механизму. Часто эту связь называют донорно-
акцепторной. Образовавшаяся донорно-акцепторная связь уже
ничем не отличается по своей природе от ковалентной.
Образование химической связи по донорно-акцепторному
механизму— явление чрезвычайно распространенное. В реакции
молекула аммиака играет роль донора электронной пары (одна
заполненная гибридная sp -орбиталь), а протон играет роль
акцептора (рис. 28). В образовавшемся ионе NH4 все связи,
несмотря на различные их происхождения, энергетически равноценны,
а.все углы между связями равны 109°28', как в молекуле СН4.
Иногда донорно-акцепторную связь изображают стрелкой,
направленной от донора к акцептору:
(H3N-+H)+
В молекуле NH4CJ атом азота занимает центральное место
и называется комплексообразователем. Вокруг него расположены,
3 2452
65
=+н+
Рис. 28. Образование иона аммония
или, как говорят, координированы, четыре атома водорода,
которые носят название лигандов. Лиганды составляют внутреннюю
координационную сферу. Число лигандов (так называемых моно-
дентантных, т. е. занимающих одно место), окружающих
центральный атом (или ион), называется координационным числом.
Ионы хлора составляют внешнюю координационную сферу.
Границу внутренней сферы при написании формул сложных
комплексных соединений отделяют от внешней квадратными скобками. Так,
формулу NH4C1 мо#жно было бы записать в виде [NH4]C1.
Часто о природе связи судят по ее прочности. Так, энергия
связи в молекуле азота N2 равна %942 кДж/моль. Это самая
прочная связь среди двухатомных молекул. Энергия связи С=С
в молекуле ацетилена ниже (811 кДж/моль), поэтому можно
предположить, что и в молекуле N2 атомы азота связаны
тройной связью (а + 2я). Модель молекулы N2 представлена на
рис. 29.
Рассмотренный способ описания химической связи,
исходящий из положения, что атомы удерживаются общими
электронными парами, т. е. зонами их
перекрывания, называется
методом валентных связей.
Этот метод, используя
представление о негибридных
и гибридных орбиталях,
в очень многих случаях
позволяет правильно объяснять
и предсказывать углы между
связями и структуру молекул
и, наоборот, по известным
углам судить о состоянии орби-
талей, связывающих атомы.
Но, как и в любой научной
теории, есть ряд фактов, ко-
Рис. 29. Связи в молекуле N2 торые не объясняются при по-
66
Рис. 30. Молекула кислорода по
методу валентных связей
мощи метода валентных
связей. Вот некоторые из них.
1. В молекуле кислорода
согласно методу валентных
связей связь между атомами
двойная (а + л), две орбитали
с неспаренными электронами
перекрываются (рис. 30), и
молекула должна быть
диамагнитной. В действительности же
она парамагнитная.
2. С помощью метода
валентных связей невозможно
объяснить крайне малую
энергию связи в молекулах Ве2,
Mg2, Ca2 и других той же
подгруппы (окончательно не решен даже вопрос об их
существовании).
3. При отрыве электрона от нейтральной молекулы, т. е. при
ее ионизации, все связи в образовавшемся ионе сохраняют свою
равноценность. Так, в ионе СН4 все четыре связи равноценны,
хотя, казалось бы, электрон должен был оторваться от одной из
них. Создается впечатление, что электрон оторвался «частями»,
равномерно от каждой связи.
4. У некоторых молекул (например, F2 или О2) при отрыве
электрона связь между атомами становится прочнее. Этот факт
совершенно необъясним, если полагать, что связь образуется
парами электронов.
Эти и другие факты способствовали разработке новой
теории— метода молекулярных орбиталей.
§ 10. ХИМИЧЕСКАЯ СВЯЗЬ. МЕТОД МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
Согласно методу валентных связей химическая связь
образуется при перекрывания электронных орбиталей, содержащих
совместно пару электронов с противоположными спинами. В
области перекрывания электронных облаков (антипараллельные
спины) электронная плотность выше, чем в других областях
пространства вокруг каждого ядра. Область более высокой
электронной плотности соответствует большей вероятности
пребывания электрона.
Так как в области перекрывания облаков вероятность
нахождения электронов двух ядер одинакова, то через область
перекрывания может осуществляться обмен электронами и после
образования связи между атомами водорода имеется одинаковая
вероятность встретить электрон ядра А как у этого ядра, так
67
Рис. 31. Операции суммирования и вычитания электронных плотностей при
образовании химической связи между двумя атомными 5-орбиталями
и у другого ядра В. То же самое и для электрона ядра В.
Следовательно, два электрона двух атомов принадлежат сразу
обоим атомам, т. е. образуется одно общее электронное облако,
как говорят, молекулярная орбиталь.
В основе метода молекулярных орбиталей и лежит
представление об общих молекулярных орбиталях. Электроны в атоме
принадлежат не каждому атому в отдельности, а всей молекуле
в целом, образуя молекулярные орбитали.
Перекрывание атомных орбиталей соответствует
математической операции сложения электронных плотностей, при которой
возникает молекулярная орбиталь. Операция сложения
осуществляется только с атомными орбиталями, имеющими
антипараллельные спины.
Если спины электронов атомных орбиталей параллельны, то
эти электроны отталкиваются, что соответствует операции
вычитания электронных плотностей. Так как в молекулах имеются
орбитали как с антипаралллельными спинами, так и с
параллельными, то необходимо при описании электронного состояния
учитывать и взаимодействие орбиталей, отвечающее операции
вычитания.
На рис. 31, я показана операция суммирования электронных
плотностей двух атомных ^-орбиталей и диаграмма электронной
плотности образовавшейся молекулы. Так как оба атома
одинаковы, образуется неполярная ковалентная связь.
68
На рис. 31,6 показана операция вычитания электронных
плотностей двух атомных s-орбиталей с параллельными
спинами.
Указанный метод расчета химической связи называется
методом линейной комбинации атомных орбиталей в молекулярную
орбиталь (ЛКАО—МО), или же просто методом молекулярных
орбиталей.
При операции суммирования, когда электронная плотность
в области между ядрами возрастает и образуется связь, энергия
системы атомов понижается. Молекулярную орбиталь, которая
возникает при суммировании атомных и которая энергетически
более выгодна, называют связывающей.
По аналогии со связывающими орбиталями вводится
представление о разрыхляющих орбиталях, получаемых операцией
вычитания: когда электронная плотность между ядрами равна
нулю, атомы расталкиваются и энергия системы двух атомов
выше при том же межъядерном расстоянии, что и для
связывающей молекулярной орбитали. На рис. 32 показано образование
связывающей и разрыхляющей молекулярных орбиталей из
атомных 5-орбиталей.
Так как каждые две атомные орбитали дают одну
связывающую и одну разрыхляющую орбитали, то общее число
молекулярных орбиталей равно общему числу атомных. Число
электронов на молекулярных орбиталях равно сумме электронов на
атомных орбиталях.
Заполнение молекулярных орбиталей электронами
подчиняется всем законам заполнения атомных орбиталей.
1. Молекулярные орбитали заполняются в порядке
увеличения их энергии. Заполнение молекулярных уровней начинается
с орбитали самого низкого энергетического уровня.
Энергия
атомы
Рис. 32. Образование связывающих и разрыхляющих молекулярных 5-орби-
талей
69
рйзр Энергетическое состояние молекулы за-
s висит от числа комбинаций операций сум-
/ \ мирования и вычитания, проводимых
/ \ с атомными орбиталями при формирова-
s / \ s нии молекулярных орбиталей.
—( ) 2. В соответствии с принципом Паули
\ / в молекуле не может быть двух электронов
N / со всеми одинаковыми квантовыми числа-
\ I ми (и одновременно находящихся на одной
6$ш молекулярной орбитали).
АО МО АО 3. Заполнение молекулярных орбита-
лей одного энергетического уровня подчи-
Рис 33 Энергетическая няется правилу Гунда. Сперва энергетиче-
диаграмма молекуляр- ски равноценные молекулярные орбитали
ных 5-орбигалей заполняются одиночными электронами,
а затем происходит спаривание новых
электронов с имеющимися.
Схему формирования молекулярных орбиталей (рис. 32)
принято изображать в виде энергетической диаграммы
молекулярных орбиталей, где по вертикали огкладывают значение энергии,
причем уровни атомных орбиталей (АО) располагают по обе
стороны от уровней молекулярных орбиталей (МО)
получающейся молекулы (рис. 33).
При заполнении электронами системы из двух ядер
водорода (Н2+) (табл. 8) первый электрон вступит на
связывающую орбиталь и получим заряженную молекулу HJ, второй
электрон поступит на ту же орбиталь и будет образована
нейтральная молекула водорода Н2. Наличие двух электронов
на а^-орбитали приведет к упрочению системы: энергия связи
в молекуле Н2 несравненно выше, чем в Н2 . Третий электрон
вступает на разрыхляющую а?*зр-орбиталь, поэтому молекула
Щ очень непрочна. Молекулярная система, состоящая из двух
протонов и четырех электронов в невозбуждешюм состоянии
(Н2~), вообще не обнаружена. Вероятно, она невозможна
вследствие равенства числа связывающих и разрыхляющих
электронов.
Двухатомная молекула гелия Не2 должна была бы содержать
также четыре электрона—два связывающих и два
разрыхляющих, поэтому в невозбужденном состоянии она неизвестна.
Однако эта же молекула с двумя связывающими и одним
разрыхляющим электроном (Не2) устойчива и действительно реально
существует.
Данные по энергиям связи подтверждаются межъядерными
расстояниями—чем выше энергия связи, тем меньше расстояние
между ядрами.
При наличии в молекуле только спаренных электронов она
диамагнитна, неспаренные электроны делают ее парамагнитной.
70
Таблица 8
Молекула
орбитали
МО *Г
АО Is
МО <™
Д//СВ,
кДж Аюль
Межъядерно
расстояние.
нм
Магнитные
свойства
молекулы
(Н2+2)
Г-1
/ 1
1 1
1 1
\ /
и-/
е
п
-н' >-
Ч !
260
0,Ю6
парамагнитная
н2
П
432
0,074
диамагнитная
rh
tti' 'ff
V
17
парамагнитная
(Н2)
*
Не?
•f
' 1 1
250
0,108
парамагнитная
(Hc2)
#: to-
У молекул элементов второго периода все молекулярные
Ь-орбитали заполнены, как в воображаемой молекуле Не2,
и происходит заполнение 2s- и молекулярных 2/ьорбиталей. Две
атомные 2я-орбитали дают две молекулярные орбитали:
а!**3 и а5"р, на которых может быть размещено всего четыре
электрона. Из трех атомных 2рх-, 2ру- и 2р2-орбиталей каждого
атома образуются три связывающие и три разрыхляющие
молекулярные орбитали. Перекрывание атомных рл-орбиталей
приведет к образованию двух молекулярных
a^-орбиталей—связывающей и разрыхляющей—в соответствии с операциями сложения
и вычитания электронных плотностей (рис. 34), причем
энергетический уровень <?2В/3 ниже уровня <у^р.
Две другие орбитали рг и ру перекрываются боковыми
поверхностями с образованием я-связей. Перекрывание />2-орбиталей
АО
АО
Рис 34. Молекулярные стрл.-орбитали
71
Перекрыбанив
Рис. 35. Молекулярные я/^-орбитали
двух атомов совершенно аналогично перекрыванию их
/7у-орбиталей. Рассмотрим это на примере /?г-орбиталей.
Операции сложения и вычитания (рис. 35) дают две молекулярные
орбитали я"/3 и я§а^, причем энергия первой ниже. Такая же
картина имеет место2 и с атомными /^-орбиталями, дающими
молекулярные nc™ и я ^-орбитали.
Сё' е
улр ^р
а^-Связываниё' сильнее кр - или л:^-связывания, поэтому
можно ожидать, что энергетический уровень молекулярной
орбита™™ б бй ! ™"
р ур ур р
ли о™™ будет расположен ниже уровня орбиталей я! и п™".
Точно так же расталкивание /^-орбиталей сильнее, чем ру- иЯп
/7г-орбиталей, и уровень молекулярной орбитали а^ должен
лежать выше уровней гс5арзр и л§арР, как это показано на рис. 36, а.
В действительности я?е уровень а|вр^ перемещен несколько
выше уровней nf™ и 7t2Bp3 (рис. 36, б). Такое перемещение уровней
вызвано следующими причинами. Энергетически атомные 2s-
Рис 36 Два типа диаграмм молекулярных орбиталей
72
в
Рис 37 Взаимодействие связывающих орбиталей
и 2р-орбитали близки. При образовании двухатомной молекулы
на линии, соединяющей два ядра, происходит одновременное
перекрывание атомных s- и />х-орбиталей (рис. 37), что приводит
к взаимному отталкиванию областей перекрывания и
повышению энергии молекулярной а ^-орбитали.
На рис. 38 показаны уровни молекулярных орбиталей,
образованных комбинацией Is-, 2s- и 2/ьуровней двух одинаковых
атомов. Так как заполненные молекулярные орбитали
a is113 и а?*зр не участвуют в связывании, при обсуждении связей
в молекулах 2-го периода мы их не будем принимать во
внимание.
Сведения о порядке заполнения электронами молекулярных
орбиталей при
образовании двухатомных молекул
2-го периода и их свойствах
приведены в табл. 9.
Молекула Li2
образована атомами первого
элемента 2-го периода. У нее оба
электрона находятся на
связывающей а2*яз-орбита-
ли. Молекула
диамагнитна. По сравнению с
молекулой Н2 в молекуле Li2
энергия связи ниже,
а межъядерное расстояние
выше.
В молекуле бериллия
числа связывающих и
разрыхляющих электронов
равны, как и в молекуле
гелия. По-видимому,
невозбужденная молекула Ве2 не
существует, хотя по
некоторым данным ее удается
обнаружить. Возможно,
это связано с частичным
перемещением элек-
1
I
A02S
MOffg»
I \
I t
\-
АО IS
\ I
\ I
Рис 38 Молекулярные орбитали
двухатомных молекул второго периода
73
Таблица 9
Молекул**
*?;; *«?
о°гГх
свяч — связ
aSfp
Количество
связывающих
электронов
Количество
разрыхляющих
электронов
Избыток
связывающих
электронов
ЛЯСВ, кДж
Межъядерное
расстояние, нм
Количество
нсспарснных
электронов
Магнитные
свойства
молекул
Li 2
-ft
2
0
2
107
0,2672
0
Диамагнитная
Ве2
-ft
4fc
2
2
0
0 или 54
оо(?)
0(?)
4- 4-
-ft
4
2
2
274
0J590
2
Парамагнитная
с2
6
2
4
603
0,1242
0
Диамагнитная
N2
-* "ft
8
2
6
942
0,10975
0
Диамагнитная
о2
4- 4-
-ft
4t -ft
8
4
4
494
0,1207
2
Парамагнитная
F2
4t 4t
4t
4t -ft
8
6
2
155
0,1418
0
Диамагнитная
(Ne2)
4t
4t -ft
4t
-ft 4t
i
8
8
0
0
00
0
тронной плотное i и с <т$"р-орбигали на более высокие
связывающие л2р-орбитали. Здесь же заметим, что возбужденная
молекула Не2 действительно существует и спектроскопически
доказан переход электрона на энергетически более высокую
молекулярную а2*Я1-орбигаль.
В молекуле В2 два очередных электрона вступают на две
равноценные молекулярные я!- и я2Вр3-°рбитали, а в молекуле
С2 эти электроны спариваются с двумя новыми. Молекула
В2 — парамагнитна, а С2—диамагнитна, что подтверждает
заполнение равноценных молекулярных уровней в соответствии
с правилом Гунда и уже обсужденное смещение уровня
молекулярной а2Вр3-орбитали выше nf™ и nf™. (В противном случае
молекула В21эыла бы диамагнитна, а молекула С2 — парамагнитна.)
В молекуле N2 парами электронов заполнены все
связывающие молекулярные /7-орбитали. Молекула диамагнитна. В ряду
молекул В2—С2—N2 по мере заполнения связывающих
молекулярных орбиталей энергии связи молекул возрастают, а
межъядерные расстояния уменьшаются. Молекула N2 — наиболее
прочная среди двухатомных молекул, составленных из одинаковых
атомов.
По мере заполнения электронами разрыхляющих орбиталей
в ряду N2—О2—F2 энергия связи молекул уменьшается и
межъядерные расстояния возрастают. Невозбужденная молекула Ne2
неизвестна (равенство числа электронов на связывающих и
разрыхляющих орбиталях).
Парамагнетизм кислорода, необъяснимый с помощью
метода валентных связей, легко объясняется с помощью метода
молекулярных орбиталей: каждая молекулярная орбиталь
я§арр и 71§арэр имеет по одному неспаренному электрону.
&4етод Молекулярных орбиталей хорошо объясняет причины
неожиданных изменений энергий связи молекулярных ионов. Так,
ион Ве^, получающийся отрывом разрыхляющего электрона от
гипотетической молекулы Ве2, действительно существует.
, Добавление одного электрона к молекулам N2, O2 или
F2 приводит к ослаблению связи между атомами в молекулярных
ионах, а отрыв электрона от молекул О2, F2 или воображаемой
;Молекулы Ne2 приводит к усилению связи. В первом случае
прибавляются разрыхляющие электроны, ослабляющие связь,
а во втором—отрываются разрыхляющие электроны, что
приводит к ослаблению сил электронного отталкивания и упрочению
связи.
В большинстве случаев удаление электрона со связывающей
орбитали уменьшает энергию связи, а с разрыхляющей —
увеличивает ее, прибавление электрона на связывающую орбиталь
усиливает связь, а удаление—ослабляет. Одним из исключений
из этого правила являются двухатомные молекулы s ^элементов
(щелочных металлов).
75
\ В.атом с большей
\электроотрицп
\электроотрицп -
хтельностью
А0
Рис. 39. Уровни энергии молекулярных
орбиталей молекулы из двух
различных атомов
Для характеристики связи
в молекулах, составленных из
различных атомов,
используется тот же принцип построения
энергетических диаграмм
молекулярных орбиталей, но
учитываются
электроотрицательности атомов.
Электроотрицательность атома определяет
вклад его атомных орбиталей
в молекулярные. Атомные ор-
битали более
электроотрицательного атома принимают
большее участие в формировании связывающих орбиталей
и меньшее—в формировании разрыхляющих, и наоборот.
Поэтому атомные орбитали более электроотрицательного атома
будут энергетически ближе расположены к молекулярным
связывающим орбиталям и дальше от молекулярных
разрыхляющих. У менее электроотрицательного атома, наоборот, его
атомные орбитали энергетически сильнее отличаются от
связывающих молекулярных и приближаются к разрыхляющим (рис. 39).
Рассмотрим некоторые примеры построения энергетических
диаграмм молекулярных орбиталей молекул, состоящих из
разных атомов.
В молекуле метана СН4 углерод находится в состоянии sp3-
гибридизации, что обусловливает энергетическую равноценность
всех четырех связей и углы между связями по 109° 28'. Так как на
4 атома водорода приходится 4 связи, в молекуле метана все
молекулярные орбитали связывающие и не имеется
разрыхляющих орбиталей (рис. 40).
В молекуле аммиака NH3 азот также находится в состоянии
5р3-гибридизации, но одна гибридная орбиталь, заполненная
парой электронов, почти не участвует в связывании (рис. 41). Все
связи N—Н энергетически равноценны, но углы между связями
Рис. 40. Молекулярные орбитали молекулы СН4
76
2s
w ,i t i (
«-*
KH,
-разр
6b5
\ fs fs fs
/ H H H
// H H H
H H H
+p
Л
Рис. 41. Сравнение молекулярных орбиталей NH3 и NH4
меньше, чем в молекуле СН4, что объясняется отталкивающим
действием электронной пары. При образовании иона аммония
NH4 атом азота как донор отдает эту пару протону, и ион NH^
имеет совершенно равноценные четыре связи с углами, как в
молекуле СН4.
Подобным образом
строятся молекулярные орбитали
молекул Н2О и HF. Молекула HF
имеет те же молекулярные
орбитали (рис. 42), что и
молекула LiH, за исключением того,
что уровни атомных орбиталей
фтора расположены ниже
водорода (ЭО F > ЭО н).
Метод молекулярных
орбиталей объясняет
исключительно высокую энергию связи
молекулы СО. В молекуле СО
заполнены только
связывающие молекулярные орбитали, hf
Полученные перекрыванием р- Рис. 42. Молекулярные орбитали моле-
атомных орбиталей, как кулыНИ
77
fs fs fs
н я н \
\ /
'чин*-1'
HfH=H
\ fs fs fs
U-h-
/ H H H
4f>
разр
с вяз
рсзр
0$phti6p-is
Рис 43. Сравнение молекулярных орбиталей молекул этана (а), этилена (б) и
ацетилена (в)
и в молекуле N2. Эти молекулы имеют сходные энергетические
диаграммы молекулярных орбиталей.
Заметим, что совершенно аналогичные энергетические
диаграммы описывают состояния молекулярных ионов CN~ и NO+,
также отличающихся высокими энергиями связи. Молекула CN
имеет меньшую энергию связи по сравнению с ионом CN ~, так
как у нее на один связывающий электрон меньше. Молекула NO,
78
наоборот, менее прочна по сравнению с ионом NO+, так как у нее
один электрон находится на разрыхляющей орбитали.
Остановимся на очень важных для органической химии
диаграммах молекулярных орбиталей молекул, содержащих
одинарные, двойные и тройные связи.
В молекуле этана С2Н6 атомы углерода находятся в
состоянии .ур3-гибридизации. Энергетические диаграммы орбиталей
изображены на рис. 43, а. В молекуле этилена С2Н4, имеющей
атомы углерода в состоянии ,ур2-гибридизации, молекулярные
орбитали размещаются согласно схеме рис. 43, б. Двойная связь
О=С обусловлена одной связывающей молекулярной а*р13—sp2-
орбиталью и одной nf™-. 2p -орбиталью, расположенной ниже.
По методу молекулярных орбиталей в ряде случаев и-свя-
зывание сильнее, чем а-.
В молекуле ацетилена С2Н2 атомы углерода, имеющие sp-
гибридизацию, связаны между собой тройной связью, состоящей
из а- и двух л-связей. Энергетическая диаграмма молекулы
приведена на рис. 43, в.
Все рассмотренные нами диаграммы энергетических уровней
молекулярных орбиталей двухатомных молекул, состоящих из
разных атомов, упрощены и не учитывают некоторые другие
эффекты электронных взаимодействий.
Из приведенных диаграмм видно, что органические вещества,
состоящие только из атомов водорода и углерода, какие бы связи
они ни имели—одинарные, двойные или тройные,—должны
обладать диамагнитными свойствами. Введение или отрыв
электронов от подобных молекул должно приводить к ослаблению
связи между атомами.
Метод молекулярных орбиталей—наиболее широко
используемый в последнее время общий метод описания связей,
пригодный для большинства соединений.
ГЛАВА 2
УЧЕНИЕ О НАПРАВЛЕНИИ ХИМИЧЕСКИХ ПРОЦЕССОВ
§ П. ИЗМЕНЕНИЕ ВНУТРЕННЕЙ ЭНЕРГИИ
В ХИМИЧЕСКОЙ РЕАКЦИИ
Химическая термодинамика и кинетика непосредственно
связаны с практикой (выход продукта реакции и скорость его
образования). Теория строения не обладает этой особенностью, но
ее значение для развития всей химии в целом огромно: она
позволяет предсказывать пути и условия получения тех или иных
веществ, получать вещества с заданными свойствами, проводить
анализ вещества и т. п.
Сейчас мы переходим к изучению простейших представлений
химической термодинамики.
В каждом теле, в каждом веществе заключена энергия. Эта
находящаяся в скрытом виде энергия носит название внутренней
энергии U. Ее количество зависит от количества вещества, его
состава и состояния и характеризует запас энергии системы, ее
энергетическое состояние. Система—это тело или группа тел,
мысленно или при помощи поверхностей раздела обособляемых
от окружающей среды, являющейся также системой.
Содержание энергии в системе определяет ее устойчивость:
чем оно больше, тем менее устойчива система. Системы с
большим содержанием энергии и, следовательно, неустойчивые
стремятся перейти в более устойчивое состояние (или же в самое
устойчивое при данных условиях—равновесное состояние). Они
выделяют избыток энергии в окружающую среду.
Химическая термодинамика изучает условия устойчивости
химических систем и законы, по которым системы переходят из
одного состояния в другое.
Внутренняя энергия системы включает целый ряд различных
видов энергии, в том числе энергию поступательного,
вращательного и колебательного движения молекул, энергию связи между
молекулами, энергию связи атомов в молекулах, энергию, зави-
80
Рис. 44. Определение
теплового эффекта при
постоянном объеме
сящую от распределения электронов по
уровням и подуровням в атомных и
молекулярных орбит алях, энергию связи
ядерных частиц в ядре и другие виды энергии,
еще пока не известные нам. Во внутреннюю
энергию системы не входят кинетическая
энергия всей системы в целом и ее
потенциальная энергия положения в поле
тяготения.
Абсолютную величину внутренней
энергии определить невозможно, и в этом нет
необходимости, так как в практической
деятельности важно знать изменения энергии
при переходе систем из одного
энергетического состояния в другое. Можно судить
лишь об изменении внутренней энергии
системы при ее переходе в другое состояние по
количеству энергии, отданной системой
окружающей среде или принятой от
окружающей среды. При этом очень важно одно
условие: объем системы не должен
изменяться в процессе ее перехода из одного состояния в другое.
Рассмотрим систему из 1 моль цинка и 1 моль серной
кислоты. Поместим эти вещества в цилиндр с поршнем, который
передвигается без трения и снабжен устройством для
отключения его движения. Проведем два опыта при условиях,
когда: 1) поршень не передвигается, т.е. объем постоянный
(K=const), а давление изменяется, 2) поршень может свободно
передвигаться, т. е. объем изменяется, а давление постоянно
(р=const).
Схема первого опыта представлена на рис. 44. Тепловой
эффект легко измерить, если реакцию проводить в калориметре,
представляющем собой сосуд с известным количеством воды.
Реакционную камеру (в нашем случае цилиндр с поршнем и
реагирующими веществами) полностью погрузим в воду. Зная
температуру воды до начала реакции и после ее окончания и
воспользовавшись численным значением теплоемкости воды,
вычисляем количество теплоты, полученное водой, или, что то же,
тепловой эффект химической реакции. В результате
взаимодействия 1 моль цинка с 1 моль серной кислоты выделилось 165,7 кДж
теплоты. Это можно записать гак:
Zn(K)+H2SO4(p-p)-ZnSO4(p-p)+H2(r) +
+165,7 кДж/моль (F= const).
В связи с тем, что в научной литературе до 1980 г. данные приводились
в калориях, напомним, что 1 Дж= 107 эрг = 0,239 кал, 1 кал=4,184 Дж
81
Уравнение реакции, записанное в таком виде, называется
термохимическим, а количество выделенной или поглощенной
теплоты называется тепловым эффектом реакции. Тепловой
эффект, измеренный при постоянном объеме, называется изо-
хорным тепловым эффектом и обозначается Qv. Буквенными
индексами при химических формулах реагентов и продуктов
обозначены их агрегатные состояния: к—кристаллическое
(раньше писали тв—твердое); ж—жидкое; р-р—раствор; г—
газ. Указание агрегатного состояния участвующих в реакции
веществ очень важно, так как величина теплового эффекта
одной и той же реакции меняется в зависимости от того,
в каком агрегатном состоянии они находились. Например, при
взаимодействии водорода и кислорода с образованием жидкой
воды выделяется теплоты больше, чем при образовании
газообразной воды (пар).
Очевидно, что в данной химической реакции выделилось
определенное количество теплоты за счет изменения внутренней
энергии реагирующей системы при переходе ее из начального
состояния в конечное:
Состояние I —> Состояние 2
Обозначим внутреннюю энергию исходных веществ (/ь
а внутреннюю энергию продуктов U2.
В термодинамике принято из величины, характеризующей
конечное состояние 2 системы, вычитать величину,
характеризующую ее начальное состояние 1. Разность обозначается знаком
А (дельта) и называется изменением соответствующего свойства
или характеристики в рассматриваемом процессе. Так,
AU=U2—U1 есть изменение внутренней энергии при переходе
системы из состояния 1 в состояние 2 (или, для данного опыта,
изменение внутренней энергии в процессе растворения цинка
в серной кислоте).
Так как в эксперименте было обнаружено выделение теплоты,
то очевидно, что запас внутренней энергии реагентов был выше,
чем запас внутренней энергии продуктов реакции, т. е. Ut > U29
поэтому разность At/—отрицательная величина: U2 — U\ <0 или
А?/<0. Для наглядности изменения энергии в системах удобно
изображать так называемыми энергетическими диаграммами.
По вертикальной оси (ось ординат) откладывают значения
энергии в начальном и конечном состояниях (и в промежуточных,
если это требуется), а по горизонтальной оси (ось абсцисс)—
значения сложной функции межъядерных расстояний всех
веществ, участвующих в реакции. Именно с этой функцией
приходится иметь дело при изучении скоростей химических реакций.
Условно разделим ось абсцисс на две части—левую для
реагентов, а правую для продуктовой пусть это будет ось хода
химической реакции (рис. 45).
82
-Ли
В реакции взаимодействия цинка с
серной кислотой имело место уменьшение
внутренней энергии системы Af/<0, но
это уменьшение произошло за счет
выделившейся геплогы, т. е. благодаря
положительному изохорному тепловому
эффекту Qv>0- Очевидно, по абсолютной
величине Qv и At/ равны и отличаются ход реакции
только знаками:
Рис. 45. Изменение внут-
— Qv. ренней энергии в реакции
цинка с раствором серной
Изменение внутренней энергии равно кислоты
изохорному тепловому эффекту, но с
обратным знаком.
Термохимический способ написания уравнения реакции в
последнее время заменен термодинамическим. Это такой способ
написания уравнения реакции, когда после уравнения
записывается изменение энергетического состояния в системе:
Zn(K)+H2SO4 (p-p)=ZnSo4(p-p)+H2 (r), Д1/= -165,7 кДж/моль.
Чтобы ответить на вопрос, чем определяется соотношение
энергий у реагентов и продуктов, запишем уравнение процесса
в ионном виде:
Zii(k)+2H+ (p-p)=Zn2+ (р-р)+Н2 (г), А(/= -165,7 кДж/моль.
По-видимому, реакция проходит самопроизвольно и с
уменьшением внутренней энергии благодаря тому, что сумма энергий
связи между атомами цинка в его кристаллической решетке
и между ионами водорода и молекулами воды их гидратных
оболочек больше суммы энергий связи между атомами водорода
в молекуле Н2 и между ионами цинка и молекулами воды их
гидратных оболочек.
В практической деятельности важно не только получить
определенное количество продуктов с заданными свойствами или
количество энергии, но и время, в течение которого они
образуются в ходе реакции. В ряде случаев реакцию необходимо
провести быстрее, в других—замедлить.
Рассмотрение энергетической диаграммы с уменьшением
внутренней энергии системы (/х > 02 ставит важный вопрос:
почему реакция проходит довольно медленно, а очень часто даже
трудно обнаружить ее протекание.
На реакцию влияет ряд условий*
1) с изменением концентрации изменяется не только скоросг* химической
реакции, но и величина изменения внутренней энергии AU. Значение 4(/,
записанное справа от уравнения реакции, соответствует только тому числу молей
реагентов и продуктов, которое равно их стехиометрическим коэффициентам
в уравнении реакции;
83
Ход реакции
Рис. 46. Энергетическая
диаграмма реакции
2) температура сильно влияет на скорость
реакции и на величины внутренней энергии веществ,
но незначительно—на само изменение внутренней
энергии при прохождении процесса,
3) скорость реакции зависит от чистоты
реагентов, поверхности реагирующих веществ, от
присутствия посторонних веществ (например,
присутствие каких-то ионов может ускорить или
замедлить реакцию)
Возникает впечатление, что
существует какое-то препятствие, мешающее
системе быстро перейти из состояния
с большей энергией в состояние с
меньшей энергией. Действительно, реагенты
должны преодолеть энергетический
барьер для превращения в продукты, так
как при сближении атомов (молекул) реагирующих веществ их
электронные оболочки взаимно отталкиваются вследствие
одинаковых по знаку зарядов. Взаимодействие возможно только для
тех частиц, которые обладают некоторым избытком энергии,
достаточным для преодоления сил отталкивания, т. е. для
преодоления энергетического барьера (рис. 46).
Численно величина энергетического барьера выражается
энергией активации ?а. Преодолеть энергетический барьер могут
только атомы или молекулы, обладающие избыточной энергией,
равной или большей энергии активации. Чем ниже барьер, т. е.
чем меньше величина энергии активации, тем большее число
частиц способно преодолеть его и тем выше скорость реакции.
При Ел1>Ел1 реакция с Еа2 протекает медленнее, чем реакция
с?а1.
Некоторые посторонние вещества (катализаторы), не
расходуясь в реакции, способны понижать энергию активации. В ряде
случаев, зная тип и энергию связи реагирующих частиц и
катализатора, можно предсказать не только At/ реакции, но и ее
скорость.
Следует особо подчеркнуть, что значение изменения
внутренней энергия At/ не зависит от скорости реакции. Какова бы ни
была высота энергетического барьера (?а1 или ?а2), разность
U2—Ut сохраняет свое значение постоянным. Это приводит
к чрезвычайно важному выводу (следствие I закона
термодинамики, называемое законом Гее с а): тепловой эффект
химической реакции (и At/) не зависит от пути реакции.
Химическая термодинамика учитывает лишь начальное и
конечное состояния химических систем. Скоростями реакции и
характером промежуточных превращений занимается химическая
кинетика.
Эксперимент с растворением цинка в серной кислоте
проведем теперь при точно тех же условиях, но при изменяющемся
84
объеме системы (поршень свободно—без трения —
перемещается в стенках цилиндра). Водород, образующийся в результате
реакции, поднимает поршень, и давление в системе сохраняется
постоянным:
Zn (k) + H2SO4 (p-p)=ZnSO4 (p-p) + H2(r)+
+163,2 кДж/моль (p — const).
Калориметром измеряется тепловой эффект при постоянном
давлении Qpt называемый изобарным тепловым эффектом.
Для данной реакции изобарный тепловой эффект меньше изохорного Qv>Qp
на величину Qv — (?,,= 165,7—163,2 = 2,5 кДж/моль (при 7=300 К). Для
некоторых реакций Qv = QP, а для других, наоборот,QP>QV> Так,
Н2(г) + С12(г)=2НС1(г) Qp = Qv
2Н2(г) + О2(г) = 2Н2О (г) QP>QV и QV-QP= -2,5 кДж/моль
3H2(r)+N2(r)=:2NH3(r) QP>QV и Qv-Qp= -5,0 кДж/моль
Разница между изохорным и изобарным тепловыми эффектами зависит от
изменения числа молей газообразных веществ при реакции. Если число молей
газообразных веществ не изменяется, то изобарный и изохорный тепловые
эффекты совпадают. Если в реакции имеет место уменьшение объема, то изобарный
эффект больше изохорного, если же наоборот, как в случае растворения цинка,
происходит увеличение числа молей газообразных веществ, то Qv>QP- Причина
этого состоит в том, что при изменении объема система или совершает работу,
или над ней совершается работа.
Предположим, что перед началом реакции поршень
находился на отметке Лх (рис. 47). После растворения цинка выделился
1 моль водорода и для сохранения р=const поршень поднялся до
отметки й2, т. е. переместился на расстояние h2—h1 — Ah (из
конечного значения вычитается начальное!). Перемещение поршня
в цилиндре совершается против силы F внешнего давления.
Работа равна произведению силы на путь:
Пусть р есть внешнее давление, преодолеваемое поршнем (при
условии невесомости поршня и отсутствия трения
давления водорода под поршнем равно внешнему
давлению). Если площадь поперечного сечения
равна S, то F—pS.
Выполненная поршнем работа
Но SAh есть изменение объема ДК Поэтому
Изобарный тепловой эффект меньше
изохорного теплового эффекта для реакции, идущей рис 47. Работа
с увеличением числа молей газообразных веществ, расширения
85
на величину, равную работе расширения, т. е. работе, которую
система совершает над ее окружением:
§ 12. ИЗМЕНЕНИЕ ЭНТАЛЬПИИ В ХИМИЧЕСКОЙ РЕАКЦИИ
Если химическая реакция сопровождается изменением числа
молей газообразных веществ, то уравнение
Менделеева—Клапейрона
можно записать
гдер—давление, Па;
V— объем, л;
А V—изменение объема газообразных веществ при
прохождении реакции, АК= Кпрод- Кисх;
п—число молей газообразных веществ;
An—изменение числа молей газообразных веществ при
прохождении реакции, Дл=ипрод—ппех;
Т—температура, К;
R—газовая постоянная, J? = 8314- =8,314 Дж/(Кмоль).
К * моль
До введения СИ использовалось значение Л—1,98 кал/(К • моль).
Далее получаем
A=pAV=AnRT.
Это соотношение позволяет вычислить работу расширения при
прохождении химической реакции.
Принято считать работу положительной, если она
совершается системой над окружающей средой. Работа,
которая совершается над рассматриваемой системой,
отрицательна.
. Работа расширения при химическом процессе не зависит от
природы реагирующих веществ, а только от температуры (при
р —const). Каждый 1 моль образующегося в реакции газа (Дл= 1)
производит работу
Если реакция проходит при стандартной температуре B98 К,
т. е. чуть выше комнатной), то работа расширения 1 моль
идеального газа
86
,4= [8.314 Дж/(К • моль) ] • 298,15 К = 2479 Дж/моль »
^2,5 кДж/моль.
Вспомнив соотношение
Qp = Qv-pAV=Qv-AnRT
и подставив в него выражение, связывающее изменение
внутренней энергии At/ и изохорный тепловой эффект Qv,
получаем
Qp= -AU-pAV= -AU-AnRT
или
-Qp = AU+pAV=AU+AnRT.
Сумму AU+pAV называют изменением энтальпии и обозначают
АН:
AH=AU+pAV=AU+AnRT.
Таким образом, изменение энтальпии в химической реакции
равно изменению внутренней энергии плюс работа расширения или
же изменение энтальпии в реакции равно изобарному тепловому
эффекту, взятому с противоположным знаком:
Чаще всего химические эксперименты, как и технологические
процессы и химические превращения в окружающей среде,
совершаются при постоянном давлении, поэтому в дальнейшем будем
чаще пользоваться значениями изменений энтальпии, которые
приведены в большинстве термодинамических таблиц и
справочников. Пользуясь этими формулами, по величине АН легко
вычислить AC/, Qp и Qv.
, Изменение энтальпии в химическом процессе всегда
записывают справа от уравнения реакции и относят к тому числу
молей реагентов и продуктов, которое соответствует уравнению
реакции:
Zn (к) + Н 2SO4 (р-р) = ZnSO4 (р-р)+Н 2 (г),
Д//= -163,2 кДж/моль.
Энтальпийная диаграмма этой реакции аналогична диаграмме
для внутренней энергии.
Для данной реакции А//—А(У = — 163,2 — (—165,7) —2,5 кДж/моль.
Действительно, Д#-ДС/=ДлЛГ=1 -8.314 10"' 298 = 2,48 кДж/моль. Для газовых
реакций, протекающих без изменения числа молей газообразных веществ, для реакций
между твердыми веществами и для реакций в растворах, если они проходят без
заметного изменения объема, изменения энтальпии и внутренней энергии, гак же
как Qp и Qv, совпадают. Разность между АН и AU тем больше, чем выше
температура и чем больше Aw
87
§ 13. ЗАКОН ГЕССА
Первый закон термодинамики — это одно из выражений
закона сохранения энергии; энергия не творится из ничего
и не исчезает бесследно, а только превращается из одной формы
в другую в эквивалентных количествах. В 1748 г. М. В.
Ломоносов сформулировал закон сохранения материи и
движения как всеобщий естественный закон: «...все встречающиеся
в природе изменения происходят так, что если к чему-либо нечто
прибавилось, то это отнимется от чего-либо другого. Так,
сколько материи прибавляется какому-либо телу, столько же теряется
у другого, сколько часов я затрачиваю на сон, столько же
отнимаю от бодрствования и т. д. Так как это всеобщий закон
природы, то он распространяется и на правила движения: тело,
которое своим толчком возбуждает другое к движению, столько
же теряет от своего движения, сколько сообщает другому, им
двинутому».
Количественная оценка движения содержится в понятии
энергии. Различные формы движения эквивалентны, поэтому
эквивалентны и различные формы энергии.
Первый закон термодинамики устанавливает связь между
количеством теплоты, полученной или выделенной в процессе,
количеством произведенной или полученной работы и
изменением внутренней энергии. Не обнаружено ни одного исключения из
этого закона. Ни одно из следствий, к которым он приводит, не
находится в противоречии с опытом.
Первый закон термодинамики имеет несколько
формулировок, но все они выражают одну идею: неуничтожимость
и эквивалентность различных видов энергии при их
взаимных переходах. Отсюда следует, что в изолированной системе
сумма всех видов энергии есть величина постоянная.
Работа—это одна из форм перехода энергии, и поэтому
создать вечный двигатель, т. е. машину, которая производила бы
работу из ничего, не затрачивая на это соответствующего
количества энергии, невозможно. Иначе говоря, вечный двигатель
(первого рода) невозможен.
Первый закон термодинамики находит в химических расчетах
свое применение в виде закона Гесса, открытого российским
химиком Г. И. Гессом в 1840 г.: изменение энтальпии (или
тепловой эффект процесса) зависит только от вида и состояния
исходных веществ и продуктов и не зависит от пути перехода.
Если бы тепловые эффекты (или АН или AU) при одинаковых
начальных и конечных состояниях при различных путях
осуществления процесса были неодинаковы, то, совершая реакцию в
прямом направлении но одному пути, а затем в обратном
направлении по другому пути, можно было бы за счет разности энергии
получать энергию из ничего, т. е. построить вечный двигатель.
88
Закон Гесса
устанавливает, что если из данных
исходных веществ (реаген- состояние^
тов) можно различными
способами получить
заданные конечные продукты, то
независимо от способов
получения, от вида
промежуточных реакций, от того
или иного механизма
реакции, от скорости процесса рис 48 превращения исходных ве-
И Т.П. суммарное измене- ществ в продукты реакции
ние внутренней энергии или
энтальпии будет одним и тем же.
Вычисления по закону Iecca можно производить, пользуясь
или изменениями энтальпии, или изменениями внутренней
энергии, так как закон Гесса строго выполняется или только для
изобарных процессов (/?=пост, Qp, ДЯ), или только для изохор-
ных (F=nocT, Qv, AU). Если имеются данные для процессов
обоих видов (ДЯ и А?/), то, пользуясь уже известными
формулами, их необходимо пересчитать для одинаковых условий,
т. е. для постоянного объема или давления.
Рассуждения об энергетической равноценности путей перехода
системы из одного состояния в другое применим к химическим
процессам. Представим себе процесс превращения исходных
веществ (состояние 1) в продукты (состояние 2) несколькими
различными путями (рис. 48): 1) непосредственно реакцией,
изменение энтальпии которой равно AHt; 2) рядом реакций с
изменениями энтальпий Д#2, ДЯ3, ДЯ4, А#5; 3) другим рядом реакций
с ДЯ6 и Д#7.
Согласно закону Iecca суммы изменений энтальпий для
различных путей реакции должны быть равны, т. е. все энтальпии
должны быть связаны между собой равенствами:
Важнейшим практическим применением вышесказанного
является возможность расчета неизвестного изменения энтальпии
какой-либо одной реакции, входящей в цикл, если для остальных
реакций цикла изменения энтальпии известны. Так, если,
например, Д Я6 неизвестно, то его легко определить, воспользовавшись
значениями остальных изменений энтальпии:
Д Я6 = Д#! - АН, = ДЯ2 + ДЯ з + Д #4 + АЯ5 - ЛЯ7
Рассмотрим принципы использования закона Гесса на
примере образования воды из газообразных кислорода и водорода. На
рис. 49 изображена энтальпийная диаграмма образования воды
в двух различных состояниях—жидком и газообразном:
89
= ~285,85 кДж/моль,
= Н2О(г)
= -241,84 кДж/моль.
Уровень энтальпии газообразной воды расположен на
диаграмме выше уровня жидкой воды. Это и понятно, так как для
испарения 1 моль воды затрачивается 44,01 кДж теплоты,
Qvicn^ —44,01 кДж/моль, А//исп=44,01 кДж/моль. В обратном
процессе—конденсации — это количество теплоты выделяется,
бконд=+44ДI кДж/моль, АЯКОНД=—44,01 кДж/моль, что можно
записать так:
Н2О(ж) = Н2О(г) АЯИСП = 44,01 кДж/моль
Н 2О (г) = Н 2О (ж) А#конд = - 44,01 к Дж/мол ь.
Следовательно, существуют два пути, по которым можно из
водорода и кислорода получить жидкую воду: 1) проводить
реакцию так, чтобы сразу получалась жидкая вода, в этом случае
изменение энтальпии составит Д#н,о(ж) = —285,85 кДж/моль,
2) проводить реакцию так, чтобы вначале получалась
газообразная вода, при этом изменение энтальпии составит
184
р
н2о<г) = —241,84 к Дж/мол ь, и далее конденсировать пар в
жидкость. В процессе конденсации изменение энтальпии составит
А#конд= —44,01 кДж/моль. Если теперь сложить A#HiO(r)
и А#конд, то получим изменение энтальпии при образовании
жидкой воды:
АЯ„2о(ж)= -241,84 + (-44,0!)= -285,85 кДж/моль.
Очевидно, изменение энтальпии при образовании жидкой воды
из простых веществ не зависит от способа ее получения.
Из того, что суммарное изменение энтальпии в круговом
процессе равно нулю, вытекают некоторые важные следствия:
1. Энтальпия разложения химического соединения равна, но
противоположна по знаку энтальпии его образования из тех же
продуктов разложения,
находящихся в том же состоянии (и при тех
же условиях):
AH,
обр1
-AH,
разл*
Ход реакции
Рис 49. Энтальпийная диа1рамма
образования волы
2. Разность энтальпий реакций,
идущих от одинаковых начальных
состояний исходных веществ
к двум различным конечным
состояниям продукта реакции, есть
энтальпия перехода продукта
реакции из одного конечного состояния
90
в другое. Пример одной из таких реакций уже рассмотрен: из
энтальпий образования Н2О(г) и Н2О(ж) вычисляется энтальпия
процесса конденсации.
3. Разность энтальпий реакций, идущих от различных
начальных состояний исходного вещества к одинаковому конечному
состоянию продукта, есть энтальпия перехода исходного
вещества из одного состояния в другое.
При расчетах энтальпий особое значение имеет энтальпия
образования. Энтальпией образования называется изменение
энтальпии в реакции образования 1 моль химического соединения из
простых веществ, устойчивых при данных условиях. Например,
энтальпия образования SiO2 равна 908 кДж/моль. Это значит, что
при соединении 1 моль кремния и 1 моль кислорода происходит
уменьшение энтальпии на 908 кДж (выделяется 908 кДж теплоты):
Si (к) + О2 (г) = SiO2 (к) А Я о°бр SiO2(K) = - 908 кДж.
Энтальпии образования относят к 1 моль образующегося
соединения и, как правило, в справочных таблицах приводят для
температуры 25 °С B98 К) и давления 101 325 Па, т. е. для
стандартных условий. Энтальпии образования при стандартных
условиях называют стандартными энтальпиями образования из
простых веществ и обозначают А#^р, 298- Ниже будем их называть
просто энтальпиями образования и обозначать Д#обр-
Температуру в индексе будем указывать только для тех случаев, когда она
отличается от 298 К.
Энтальпии образования простых веществ в их наиболее
устойчивых состояниях при стандартном давлении и стандартной
температуре принимаются равными нулю (например, для Н2(г),
О2(г), Fe(K), Са(к), Si (к)). Углерод при 298 К может
существовать в виде графита и алмаза. Так как графит является
устойчивой при стандартных условиях формой существования углерода,
то А#?>рС(графйТ) = 0, в то время как АЯы>рС(алмаз) = 2,1 кДж/моль.
По энтальпиям образования участников реакции вычисляются
стандартные энтальпии реакций. Например, для вычисления АН
реакции оксидов кальция и кремния с образованием силиката
кальция
СаО (к) + SiO2 (к) = CaSiO3 (к) АН %т
необходимо знать Д#?>рСао<к), A//?6pSiO2(If) и A//S>PcaSio3<K),
значения которых приведены ниже:
Соединение СаО(к) SiO2(K) CaSiO3(ic)
A#S>P,298, кДж/моль -636 -908 -1582
Комбинируем и складываем уравнения реакций образования всех
участников реакции из простых веществ, так чтобы получить
искомое уравнение:
91
= Са(к)+1о2(г)
SiO2(K) =
Са (к) + Si (к)+-О2 (г) = CaSiO3 (к)
A#o6pCaSiO3<K>
СаО (к)+SiO2 (к) = CaSiO3 (к)
Следовательно:
АН р.иии = АН обр CaSiO3(E)"" (А// обр СаО (к) + АН обр SiO2 (к)) ~
= -1582-(-636-908)= -38 кДж/моль.
Из этого расчета, иллюстрируемого диаграммой (рис. 50), видно,
что если через ^AH%^nvojl обозначить сумму энтальпий
образования продуктов реакции, а через ?Д#5бР.„сх.в-в—такую же
сумму для исходных веществ, то стандартная энтальпия реакции
равна:
АЯ р.иии = ? АН обр. прод—2 АН обр. исх. в.в.
Энтальпия реакции равна разности сумм энтальпий
образования продуктов реакции и энтальпий образования исходных
веществ.
В органической химии часто приходится иметь дело с
энтальпиями сгорания. Энтальпией сгорания называется изменение
энтальпии в реакции взаимодействия соединения с кислородом
с образованием соответствующих устойчивых при данных
условиях оксидов Н2О (ж), СО2 (г), SiO2 (к). По энтальпиям сгорания
участников реакции можно рассчитать энтальпию какой-либо
реакции.
Например, так найдена энтальпия превращения алмаза в
графит, хотя непосредственно
ее пока измерить невозможно.
Она была определена по
энтальпиям сгорания алмаза и
графита:
С (графит)+О2 (г) = СО2 (г)
A#x = —393,3 кДж/моль
С (алмаз)+О2 (г)=СО2 (г)
ДЯ2= -395,4 кДж/моль.
Уровни энтальпии реаген-
Рис. 50. Энтальпийная диаграмма ре- ТОВ И продукта реакции пред-
акции образования силиката кальция Ставлены на диаграмме
из оксидов кальция и кремния (рис. 51). Изменение энтальпии
92
при превращении 1 моль углерода
в форме алмаза в 1 моль углерода
в форме графита составляет:
С (алмаз) = С (графит) А Нпрсър =
= А//Сгор С (алмаз) ~~ АНcrop С (графит)=
= АН2 - А#! = - 395,4 - (- 393,3) =
= — 2Л кДж/моль.
1
Ход реакции
Рис. 51 Энтальпийная диаграмма
сгорания алмаза и графита
Энтальпия реакции равна
взятой со знаком минус разности
сумм энтальпий сгорания
продуктов реакции и энтальпий сгорания исходных веществ:
А// р.ции = — (LA//сгор прод — LA/i сгор> исх. внв).
Обратите внимание, что при вычислениях, связанных с
энтальпиями сгорания, в отличие от энтальпий образования, перед
разностью сумм энтальпий стоит знак минус.
Реакция проходит в направлении повышения устойчивости
системы: чем прочнее связи в веществах, тем ниже энергетический
запас системы. Но в ряде случаев энергетических представлений
оказывается недостаточно для суждения о направлении реакции.
Причем замечено, что чем ниже температура, тем большее число
химических процессов идет в направлении уменьшения
энергетического запаса системы и, наоборот, чем выше температура, тем
больше исключений из этого правила.
§ 14. ИЗМЕНЕНИЕ ЭНТРОПИИ В ХИМИЧЕСКОМ ПРОЦЕССЕ
Большинство реакций сопровождается выделением теплоты,
т. е. уменьшением энтальпии. Около 95% всех неорганических
соединений при стандартных условиях и температуре образуется
с выделением теплоты и уменьшением энтальпии. Таким
образом, подавляющее большинство сложных веществ обладает
меньшим запасом внутренней энергии, чем те простые вещества,
из которых они образуются. На основании этого и других
соображений французский химик Бертло и английский химик Томсен
в середине XIX в. сформулировали следующий принцип: любой
химический процесс должен сопровождаться выделением теплоты.
Бертло и Томсен исходили из аналогии в поведении химических и
механических систем. Известно, что тело» находящееся на наклонной плоскости, стремится
скатиться вниз из положения с более высокой потенциальной энергией в
положение с более низкой потенциальной энергией. В обратном направлении оно
переместится только при затрате работы. Бертло и Томсен полагали, что и реакции
будут идти только в направлении уменьшения содержания энергии, т. е. с
выделением энергии в виде теплоты Но химическая форма движения гораздо сложнее
механической. Скоро обнаружились исключения из правила
93
Известно много процессов, в которых теплота поглощается,
например, иногда при растворении. Если растворяемое вешес!во
не реагирует с растворителем с выделением теплоты, то при
растворении наблюдается понижение температуры раствора. Так,
нитрат натрия растворяется в воде с поглощением 1еплогы и
увеличением эшальнии:
NaNO3 + 400H2OM = NaNO3(p-p), А//расгв = 21,3 кДж/моль.
Важный технологический процесс получения смеси оксида
углерода (II) и водорода
С(к)+Н2О(г) = СО(г) + Н2(г)
также сопровождается поглощением теплоты, Д#>0.
Процесс смешения двух газов протекает самопроизвольно
и без изменения энергетического состояния, тогда как обратный
процесс—разделение газовой смеси на сосгавляющие ее
компоненты—самопроизвольно не протекает и требует затраты
работы. Следовательно, движущей силой смешения газов не
является энергия и сведений об энергетических изменениях в системе
недостаточно для предсказания направления процесса.
Вероятно, существуют две движущие силы самопроизвольного процесса,
одна стремится уменьшить энерюсодержание и выделить теплоту, а вторая пока
еще нам неизвестна, но в ряде случаев она превосходит первую Изучение этих
движущих сил следует провести раздельно. По-видимому, следует найти процесс,
в котором максимально преобладает первая сила без заметного воздействия
второй, и другой процесс, в котором, наоборот, преобладает вторая сила без
влияния первой.
Изучить первую движущую силу реакции, связанную с уменьшением энерю-
содержания, можно на примере любой реакции с очень большим тепловым
эффектом. Что касается второй движущей силы, то найти процесс,
удовлетворяющий поставленным условиям, значительно труднее, потому что таких реакций
при комнатных температурах мало и многие из них сопровождаются
энергетическими изменениями.
Процесс смешения двух газов, не реагирующих друг с
другом,— одна из лучших моделей для изучения этого явления.
Больше всего таким условиям отвечают инертные газы,
например, гелий и неон, их атомы имеют полностью заполненные
электронные уровни и малые размеры, благодаря чему
исключено возникновение в этих газах межмолекулярного
взаимодействия типа сил Ван-дер-Ваальса.
Пусть два газа—гелий и неон—находятся по отдельности
при одинаковых температуре и давлении в двух одинаковых
частях сосуда, разделенных перегородкой (рис. 52). Назовем эго
состояние системы состоянием 1. Удалим перегородку, не
изменяя энергетического запаса системы. Хотя давления газов
справа и слева от места расположения перегородки одинаковы,
начнется процесс смешения газов, и через некоторое время
молекулы гелия и неона будут равномерно распределены по всему
94
Не
Ne
объему системы. Новое,
конечное состояние
системы назовем состоянием 2.
Процесс смешения га- состояние 1 Состояние 2
зов, т. е. переход их из со-
СТОЯНИЯ I в состояние 2, Р„с. 52. Процесс смешения газов
проходил без изменения
энтальпии, так как при комнатной температуре какое-либо
взаимодействие между молекулами практически исключено (оно
появляется при низких температурах, близких к кипению) и давления
газов равны.
Газы, разделенные перегородкой (состояние 1), распределены
в системе с большим порядком по сравнению со смесью газов
(состояние 2).
Степень порядка 2 < степени порядка 1.
По-видимому, движущей силой смешения газов является их
стремление перейти в состояние с меньшим порядком. Отсюда
следует вывод: самопроизвольный процесс без изменения
энергетического состояния системы совершается только в направлении,
при котором порядок в системе уменьшается.
В химической термодинамике редко пользуются
выражениями «порядок» или «степень порядка», а применяют обычно
выражение, характеризующее противоположное
свойство—«беспорядок» или «степень беспорядка». Степень порядка и степень
беспорядка находятся в обратной зависимости друг от друга: чем
выше значение одного, тем ниже значение другого. Газы до
смешения, в состоянии 1, имели меньшую степень беспорядка,
чем после смешения, в состоянии 2.
Степень беспорядка 2 > степени беспорядка 1.
Теперь имеем такую формулировку полученного вывода:
самопроизвольный процесс, проходящий без изменения
энергетического состояния системы, совершается только в направлении, при
котором беспорядок в системе возрастает.
Трудно представить, чтобы два газа, не разделенные
перегородкой, не смешались. Такое состояние 1 системы маловероятно.
Состояние 2—равномерное распределение молекул газа по всему
объему—более вероятно. Движущей силой смешения газов
является тенденция перейти в более вероятное состояние.
Вероятность состояния 2 > вероятности состояния 1.
Вывод: самопроизвольный процесс, проходящий без изменения
энергетического состояния системы, совершается только в
направлении, при котором система переходит в более вероятное
состояние.
Вычислить степень порядка или степень беспорядка
невозможно, но вероятность расположения молекул в том или ином
месте системы поддается вычислению. Однако здесь мы сталки-
95
ваемся с одним очень существенным обстоятельством, сильно
усложняющим все дальнейшие рассуждения. Если смешиваются
одинаковые количества нереагирующих газов, то смесь будет
обладать суммой их первоначальных запасов энергии (следствие
первого закона термодинамики). Если объединяются две системы
в одну, то энергетический запас объединенной системы в
простейшем случае равен сумме энергетических запасов исходных
систем. Но аналогичные рассуждения к вероятностям не
применимы. Вероятность существования объединенной системы вовсе
не равна сумме вероятностей существования первоначальных
систем, так как вероятности не складываются, а перемножаются.
В то же время для определения направления процесса
необходимо производить суммирование двух движущих сил.
Это означает, что вероятность следует представить в виде
такой функции, которая вела бы себя как энергия, т. е. при
объединении систем подвергалась бы операции сложения, а не
умножения и измерялась бы в тех же единицах, что и энергия
(вероятность — безразмерная величина). Только при
удовлетворении этих условий можно осуществить суммирование двух
движущих сил реакции для получения некоторой суммарной движущей
силы, ответственной за самопроизвольное протекание реакции
в рассматриваемом направлении.
Представим две системы, имеющие вероятности Wt и W2.
Совместная их вероятность равна не сумме, а произведению:
W— W± W2. Требуется найти функцию от вероятности, чтобы вид
этой функции был таков, что когда вероятности перемножаются,
значения функции складываются. Мы знаем, что логарифм
произведения равен сумме логарифмов перемножаемых чисел:
In (ab) = In a + In А. Следовательно, функция от вероятности может
быть только логарифмической. Эта функция называется
энтропией S. Таким образом, зависимость S—f{W) на один моль
вещества с коэффициентом пропорциональности R (газовая постоянная)
имеет вид:
S=R\nW.
Энтропия — это логарифмическое выражение вероятности
существования системы.
Заметим, что здесь идет речь о 1срмодинамической вероятности, которая
принимает значения больше единицы, в отличие от вероятности математической,
значения которой меньше единицы
Размерность эшропии совпадает с размерностью газовой
постоянной. Энтропия измеряется в Дж/(К - моль). Ранее в качестве
единицы измерения часто пользовались энтропийной единицей
(э. е.). 1 э. е. = 1 кал/(К- моль) = 4,184 ДжДКмоль).
Чтобы значения энтропии веществ были сопоставимы, их, как
и изменения энтальпии, относят к строго определенным, стан-
96
дартным условиям: Г=298 К B5'' С), р — 101 325 Па; для
растворенных веществ или веществ в смеси с другими веществами за
стандартное принимают их состояние при концентрации
(активности), равной единице. Энтропия в этих условиях обозначается
5 §98 и называется стандартной энтропией.
Проверим, удовлетворяет ли выражение логарифмической
зависимости энтропии от верояшости сформулированным
требованиям сложения энтропии при перемножении вероятностей.
Пусть две совершенно не связанные системы обладают
вероятностями Wi и W2 и соответственно энтропиями Sx и 52. Общая
энтропия двух систем
5=5,+52,
а их совместная вероятность
Далее получаем
5=51+52 = Л1п Wt + R\n W2 = R\n(Wl W2) = Rln W.
Таким образом, при сложении энтропии отвечающие им
вероятности перемножаются.
С точки зрения химической термодинамики энтропия — это
логарифмическое выражение вероятности существования
веществ или различных их форм. Зная энтропии веществ —
участников реакции, можно определить суммы энтропии продуктов и
реагентов, т. е. вероятности существования при данных условиях
веществ в форме продуктов и реагентов. Разность этих сумм
показывает, какое состояние более вероятно — продуктов или
реагентов, т. е. позволяет судить о вероятности того или иного
направления химической реакции. Для этого необходимо
определить знак изменения энтропии при самопроизвольном процессе.
Возвратимся к системе двух газов. Пусть в состоянии 1,
когда газы не смешаны, система характеризуется вероятностью
Wx и энтропией 51 = /?1пЖ1. После удаления перегородки и
окончания самопроизвольного процесса смешения газов
система переходит в состояние 2 с вероятностью W2 и энтропией
8 R\W
2 2
Чтобы оценить изменение энтропии при переходе (состояние
1)->(состояние 2), следует, как обычно, из значения свойства,
характеризующего конечное состояние, вычесть значение того же
свойства, характеризующего начальное состояние:
Д5=52-51 = /?1п W2-R\n H^! = it In —.
Распределение молекул по всему объему системы — процесс
самопроизвольный. Обратный процесс—разделение разнородных
4 2452 97
молекул по обе стороны воображаемой
перегородки—невозможен (если, конечно, в системе содержится не слишком малое
количество частиц) или возможен, но с затратой работы. Поэтому
W2> Wu S2>SX и ASSSX)
Энтропия 2 > энтропии 1.
Отсюда следует вывод: процесс, проходящий без изменения
энергетического состояния системы, совершается
самопроизвольно только в направлении, при котором энтропия в системе
возрастает.
Так как вероятность системы и степени ее беспорядка и
порядка связаны между собой, то энтропия также является мерой
порядка и беспорядка системы.
Системы с высокой степенью порядка и соответственно
низкой степенью беспорядка характеризуются низкими энтропиями,
и наоборот. Так, энтропия моля атомов углерода в структуре
алмаза 5298 (алмаз) = 2,439 Дж/(К моль), а для того же
количества атомов, но в форме графита S °98 (графит) = 5,694 Дж/(К * моль).
Из известных веществ алмаз имеет самое низкое значение энтропии при
комнатной температуре, т. е. обладает исключительно высокой степенью порядка
в расположении атомов. Четыре электрона второго уровня атома углерода
в структуре алмаза находятся в 5/;3-гибридном состоянии. Из каждого атома
исходит по четыре совершенно одинаковых гибридных о рб и тал и, которые вес
перекрываются с точно такими же орбиталями четырех соседних атомов углерода
с образованием а-связей. Межъядерное расстояние равно 0,154 нм. Вес углы
между связями равны по 109,5° (тетраэдр). Порядок в расположении атомов
исключительно велик (рис. 53).
В структуре графита три электрона второго уровня атома углерода
находятся в sp -гибридном состоянии. При этом каждый атом связан с тремя
другими благодаря перекрыванию как гибридных орбиталей с возникновением сг-
связей, так и негибридных /?-орбиталей с возникновением тс-связей. Все связи
осуществляются в одной плоскости и направлены под углом 120J. Плоскости
(слои) атомов углерода в структуре графита взаимодействуют между собой
крайне слабо (силы Ван-дер-Ваальса). В плоскости расстояние между атомами
С—С 0,142 нм, а расстояние между атомами двух различных плоскостей
0,335 нм (рис. 54). Неравномерность распределения атомов в структуре вещества
приводит к повышению энтропии. Слабость связи между плоскостями в
структуре графита обусловливает легкость скольжения плоскостей относительно друг
друга и мягкость графита.
В большинстве случаев выполняется правило—чем тверже
вещество, тем ниже его энтропия. Так, углерод в форме алмаза
значительно тверже кремния; энтропия алмаза B,439 Дж/
(К моль)) ниже энтропии кристаллического кремния A8,8 Дж/
(К • моль)), имеющего ту же структуру, тот же тип связи, но
большую ее длину. Чем более компактна структура, тем ниже
энтропия. Сверху вниз в подгруппе системы Менделеева для
простых веществ с одинаковой структурой или для однотипных
соединений энтропия возрастает.
Процессы расширения вещества сопровождаются ростом
энтропии и, наоборот, в процессах уменьшения объема, сжа-
98
Рис. 53. Структура алмаза
Рис 54 Структура графита
тия, когда число частиц в единице объема возрастает, энтропия
падает.
При увеличении давления энтропия уменьшается, при
повышении температуры — возрастает.
В кристаллах расположение молекул или атомов значительно
более упорядочено, чем в жидкостях, а в последних более
упорядочено по сравнению с газовым состоянием того же вещества.
Ниже сравниваются энтропии воды (Дж/(К • моль)) для
различных ее агрегатных состояний:
Кристалл (лсд) 43,9
Жидкость 66,9
1аз (пар) 188,7
Интересно, что среди кристаллов других соединений у льда одно из
наименьших значений энтропии, что указывает на очень высокую упорядоченность и
симметричность расположения молекул
воды в кристалле. Благодаря
.^-гибридному состоянию четырех
электронов второго уровня атома кислорода
из него исходят четыре гибридные ор-
битали, две из которых перекрываются
с я-орбиталями атомов водорода,
образуя сх-связи, а две другие способны
к дополнительному взаимодействию
(водородная связь) с соседними
молекулами воды. Поэтому структура льда
(рис. 55) напоминает структуру алмаза
Несколько выше энтропия жидкой
воды и значительно выше энтропия
газообразной воды.
При 25° С лед существовать
не может, он самопроизвольно
(хотя и с поглощением
теплоты, Д#пл>0) превращается
в жидкую воду—плавится.
Вычислим изменение энтропии рис 55
в процессе плавления льда:
Кристаллическая струю ура
льда
99
Н2О(К) — Н2О(ж)
= 66,9-43,9-23,0 Дж/(К моль).
Переход из менее вероятного состояния (лед) в более вероятное
состояние (жидкость) сопровождается возрастанием энтропии,
А5пл>0.
При 25 С жидкая вода постепенно испаряется. Ее молекулы
самопроизвольно (хотя и с поглощением теплоты, Д//исп>0)
переходят в газовую фазу и равномерно распределяются в
окружающем пространстве. Вычислим изменение энтропии в процессе
испарения воды:
= 188,7-66,9 = 121,8 ДжДКмоль).
Причина большого различия AS „л и AS?Cfl заключается в том, что при
плавлении льда в жидкой воде сохраняются большие группы молекул воды со
структурой льда, тогда как при испарении воды в ее паре они находя! ся в основном
в виде отдельных молекул или объединены в очень небольшие группы (Н2ОJ,
(Н2ОK, которые из-за относительно больших расстояний между молекулами
в газообразном состоянии практически между собой не взаимодействуют.
Энтропии плавления и испарения Н2О сильно отличаются от
энтропии испарения H2S, H2Se и Н2Те (см. рис. 56), у которых
способность образовывать водородные связи значительно
понижена.
Аналогичная закономерность в изменении температур,
энтальпий и энтропии фазовых переходов наблюдается и у
водородных соединений главных подгрупп V и VII групп. Следовательно,
сильно электроотрицательные элементы второго периода F,
О и N проявляют высокую склонность к образованию
водородной связи, что мало характерно для их аналогов из нижележащих
периодов.
110
1
-15
±100
2 3 4 5 п
(Н2О) (H2S) (H2Se) (H2Te)
2 3 k 5n
(Н2О) (H2S) (H2Se) (H2Te)
Рис. 56. Изменение энтропии плавления (а) и испарения (б) в ряду водородных
соединений элементов VI группы
100
На примере процессов перехода
воды из одного агрегатного
состояния в другое можно показать,
что закон Iecca формально
применим к энтропиям и с ними можно
совершать такие же операции, как
и с энтальпиями. Лед может быть
переведен в пар двумя путями.
Первый—через плавление и
последующее испарение жидкости.
В этом случае изменение энтропии
составляет
= 23,0+121,8= 144 Дж/(К моль).
Второй путь — непосредственный Рис 57 Изменение энтальпии
переход молекулы воды из льда и энтропии при фазовых псрсхо-
в пар—сублимация (возгонка). Из- дах
менение энтропии при сублимации равно
А СО __ оО рО
^^ 298,субл — ^ 298,Н2О (г) ~~ ^ 298,Н2О (к) =
= 188,7-43,9 = 144,8 Дж/(К-моль).
Таким образом, изменение энтропии, как и изменение
энтальпии, не зависит от способа перехода, при условии, что исходное
и конечное состояния вещества одинаковы. На рис. 57 показаны
изменения энтропии и энтальпии при переходах воды из одного
агрегатного состояния в другое и ясно видна аналогия с
расчетами по закону Iecca.
Следовательно, закон Гесса формально применим к энтропи-
ям—с ними можно совершать такие же операции, как и с
энтальпиями: изменение энтропии в процессе не зависит от пути
процесса при условии, что исходное и конечное состояния веществ
одинаковы.Изменение энтропии вычисляется как разность суммы
энтропии продуктов реакции и суммы энтропии исходных веществ:
део __
&& р-ции —
о у с-о
прод -~ *« & исх. в-в-
Часто знак изменения энтропии в реакции можно предсказать
в зависимости от состояний участвующих в реакции веществ.
При диссоциации газообразной воды на кислород и водород
объем продуктов реакции больше объема исходных веществ и,
по-видимому, сумма энтропии 1 моль Н2 и 0,5 моль О2 больше
энтропии 1 моль Н2О (г):
(г) = Н2(г)+1о2(г)
= 188,7 Дж/(К моль),
101
S н2 (г) =130,5 Дж/(Кмоль), So2<o = 205,0 Дж/(Кмоль),
Взаимодействие кристаллического оксида кальция и оксида
углерода (IV) с образованием кристаллического карбоната
кальция B98 К) сопровождается уменьшением объема за счет того,
что в реакции поглощается газообразный оксид углерода (IV).
Можно ожидать, что энтропия системы будет уменьшаться:
СаО (к)+СО2 (г) = СаСО3 (к)
S СаСО3 (к) < (S СаО(к) + $ СО2 (г))»
Sgaco3<K) = 88,7 Дж/(К • моль), 58аО(К)-39,7 Дж/(К • моль);
58о2(г)-213,8Дж/(К-моль),
= - 164,8 Дж/(К моль).
Если же реакция проходит без изменения объема, то
предсказать изменение энтропии невозможно. В этом случае следует
обращаться к справочным таблицам и, пользуясь численными
значениями стандартных энтропии всех участников реакции,
вычислять изменение энтропии.
Значения стандартных энтропии веществ определяются по зависимости
теплоемкости вещества от температуры при его нагревании от абсолютного
нуля температуры до 298 К. Принято считать, что при О К энтропия
вещества равна нулю, так как при этой температуре расположение атомов или
молекул в кристаллической структуре характеризуется максимальным порядком.
При 0 К атомы или молекулы в кристалле неподвижны, а при нагревании
приобретают различного рода движение (например, колеба1ельное). Чем выше
температура, тем сильнее колебательное движение атомов и групп атомов,
тем сильнее нарушение порядка в структуре кристалла, тем выше степень
беспорядка и тем выше энтропия. В то же время чем больше различных видов
движения у элементов кристаллической структуры и чем интенсивнее это
движение, тем выше теплоемкость Специальными математическими методами по
зависимое!и теплоемкости вещества от температуры рассчитывается энтропия
вещества.
Представление о равенстве нулю энтропии совершенного
кристалла любого вещества при абсолютном нуле температуры
составляет суть третьего закона термодинамики.
§ 15. ИЗОБАРНЫЙ ПОТЕНЦИАЛ РЕАКЦИИ
Как видно из сказанного выше, для процессов, происходящих
в природе, характерно стремление перейти в состояние с
наименьшей энергией и выделить теплоту, т. е. понизить энтальпию,
или стремление перейти в наиболее вероятное состояние с
максимально допустимой в данных условиях степенью беспорядка,
т. е. повысить энтропию.
102
Если в процессе степень беспорядка не изменяется (А5=0), то
направление процесса определяется изменением энтальпии и
процесс проходит самопроизвольно в направлении уменьшения
энтальпии.
Если в процессе не происходит энергетических изменений
(А#=0), то фактором, определяющим направление реакции,
является энтропия и процесс пойдет самопроизвольно в
направлении, при котором степень беспорядка возрастает, т. е. в сторону
увеличения энтропии.
В химических процессах одновременно изменяются
энергетический запас системы и степень ее беспорядка. Самопроизвольно
процесс проходит в том направлении, в котором общая
суммарная движущая сила реакции будет уменьшаться.
Отсюда следует вывод, который запишем так: общая
движущая сила процесса —{запас энергии в конечном состоянии—запас
энергии в начальном состоянии)—{степень беспорядка в конечном
состоянии—степень беспорядка в начальном состоянии).
Знак минус перед вторыми скобками ставится для того, чтобы
показать, что в самопроизвольном процессе без изменения
энергетического состояния системы изменение общей движущей силы
процесса является отрицательной величиной.
Если процесс проводится при постоянном давлении, то общая
движущая сила процесса называется изменением изобарного
{р=пост) потенциала и обозначается AG (Г=пост). Как обычно,
изменение изобарного потенциала в реакции есть разность между
суммой изобарных потенциалов продуктов и исходных веществ:
Запасы энергии продуктов и исходных веществ при
постоянном давлении есть энтальпии, поэтому выражение в первых
скобках можно записать Н2-~Я1=АЯ.
Во вторых скобках записана разность степеней беспорядка
в системе в конечном и начальном состояниях. Запас энергии, или
Д#, выражается в Дж/моль. Так как складывать и вычитать
можно только величины одинаковой размерности, то следует
выразить член во вторых скобках в тех же единицах, что и
энтальпия.
Степень беспорядка и энтропия связаны между собой
пропорциональной зависимостью.
Такой же зависимостью связаны степень беспорядка с
температурой. Действительно, чем выше температура, тем больший
объем занимает газ, тем сильнее испарение жидкости, тем
больше сложных молекул распадается на более простые молекулы
и атомы. Огромное число других явлений указывает на то, что
с повышением температуры возрастает степень беспорядка.
Поскольку значение степени беспорядка вычислить
невозможно, коэффициент пропорциональности можно перенести в са-
103
мо значение степени беспорядка. Поэтому можно записать
выражение:
степень беспорядка = TS.
Вводя выражение степени беспорядка в словесную формулу
общей движущей силы, получаем
где TS2 — величина, связанная со степенью беспорядка в
конечном состоянии (продукты реакции), TSt—то же, для начального
состояния (реагенты). Обратите внимание, что, не имея
возможности вычислять абсолютные значения беспорядка системы в
конечном и начальном состояниях, мы пришли к выражению,
включающему разность степеней беспорядка в двух состояниях,
которая вычисляется. Упрощаем выведенное соотношение:
Таким образом, зная зависимости одних свойств от других и
воспользовавшись приемом логического вывода формулы, мы
получили важнейшее термодинамическое соотношение:
В этой формуле
прод — Yj Д^обр. исх в-в*
Теперь понятно, почему в зависимости энтропии от вероятности
коэффициентом была выбрана газовая постоянная: размерность
энтропии совпадает с размерностью газовой постоянной, и член
TAS имеет ту же размерность, что и энтальпия. Это
действительно позволяет суммировать две движущие силы реакции
AH—TAS и получить суммарную движущую силу AG,
измеряемую в тех же единицах, что и энтальпия.
Изменение изобарного потенциала учитывает одновременно
изменения энергетического запаса системы и степени ее
беспорядка. Стремление системы перейти в состояние с минимальным
изобарным потенциалом теперь уподобляется шару,
двигающемуся в сторону положения с минимальной потенциальной
энергией. В любой самопроизвольной реакции происходит превращение
исходных веществ, имеющих некоторую величину изобарного
потенциала, в продукты, имеющие меньшую его величину.
Поэтому все самопроизвольные реакции характеризуются
отрицательным значением изменения изобарного потенциала:
AG<0.
104
Если в процессе энтропия системы не изменяется (А5=0), то
фактором, определяющим направление реакции, будет изменение
энтальпии. Уменьшение энтальпии (Д//<0) соответствует
уменьшению изобарного потенциала (AG<0). В этом случае
самопроизвольно протекает реакция, идущая с выделением теплоты, что
согласуется с принципом Бертло—Томсена.
Если в процессе энтальпия системы не изменяется (А//=0), то
самопроизвольно система может перейти только в состояние
с большей энтропией (Д5>0); при эюм из-за знака минус перед
членом TAS изменение изобарного потенциала будет
отрицательной величиной (AG<0).
Из уравнения AG=Д#— TAS следует:
1) если Д#<0 и А5>0, то всегда AG<0, т. е. реакция с
выделением тепло i ы и увеличением степени беспорядка возможна при
всех температурах;
2) если А#>0 и А5<0, то всегда AG>0, т. е. реакция с
поглощением теплоты и увеличением степени порядка невозможна ни
при каких условиях;
3) во всех остальных случаях (А//<0, А5<0 и А#>0, А5>0)
знак AG зависит от соотношения членов АН и TAS. Реакция
возможна, только если она сопровождается уменьшением
изобарного потенциала;
4) при комнатной температуре, когда значение Т невелико,
значение произведения TAS также невелико, и обычно изменение
энтальпии превосходит TAS. Поэтому большинство реакций,
протекающих при комнатной температуре,— это реакции с
выделением 1епло1Ы (ДЯ<0). Чем выше температура, тем большее
значение приобретает член TAS и при высоких температурах
даже реакции с поглощением теплоты (А#>0) становятся
самопроизвольными. При абсолютном нуле изменение изобарного
по1енциала реакции равно изменению энтальпии и возможны
только реакции с выделением 1еплоты (А#<0).
Знак изменения изобарного потенциала указывает на
направление реакции. При ДС>0 реакция не проходит в прямом
направлении (т. е. в том направлении, как написано уравнение реакции),
возможен лишь обратный процесс (для него AG будет уже
отрицательной величиной). При AG = 0 все вещества находятся
в химическом равновесии и внешне незаметно никаких изменений
и процессов в системе. При AG<0 реакция протекает в прямом
направлении.
Значения AG зависят от концентраций реагирующих веществ.
Для сравнения движущих сил реакций и для выяснения
влияния температуры на потенциал реакции нужно пользоваться
сопоставимыми концентрациями. Принято для
характеристики химических процессов пользоваться стандартным
состоянием, при котором концентрации или парциальные
давления всех участников химического процесса равны единице
105
A моль/л, 101 325 Па). В этом случае при протекании
химической реакции потенциал изменяется на AG0 (Дж/моль).
Обычно в таблицах приводятся термодинамические величины для
298 К, и тогда говорят, что процесс совершается при
стандартной температуре и стандартных условиях, а величина ДС^в
называется стандартным изменением изобарно-изотермического
потенциала, или просто изобарным потенциалом реакции.
Изобарные потенциалы образования простых веществ равны нулю.
В реакции
Н2(г)+1о2(г) = Н2О(г)
изменение изобарного потенциала при взаимодействии 1 моль
газообразного водорода и г/2 моль газообразного кислорода
с образованием 1 моль газообразной воды при 298 К
р-ции — AU обр Н2О (г) — I AU обр Н2 (г) "Г ~
= —228,6 кДж/моль.
Для характеристики процессов, идущих при постоянном
объеме, используется изохорный потенциал AF (точнее, изохорно-
изотермический, К=пост, 7*= пост) и изменение внутренней
энергии At/:
AF=AU-TAS.
Самопроизвольная реакция возможна только при AF<0. Для AF
рассуждения точно те же, что и для AG.
Изменения энтальпии и внутренней энергии связаны
формулой
поэтому
Следовательно, численно AG и AF отличаются на работу
расширения.
В последнее время функцию G стали называть свободной
энергией Тиббса, а функцию F—свободной энергией Гельм-
гольца.
Заключение о возможности самопроизвольного прохождения
процесса (AG<0, AF<0, А5>0) не означает, что в
действительности процесс будет протекать. Это связано с тем, что
термодинамический подход принимает во внимание только конечное
и начальное состояния, не учитывая, что могут быть промежу-
106
точные состояния, переход к
которым сопровождается
увеличением G или F(AGnp>0), (AFnp>0)
(рис. 58), или же могут
существовать механические преграды или
сопротивления, не дающие
возможность проходить процессу
или резко снижающие его
скорость. Поэтому при исследовании
возможности осуществления
процесса кроме термодинамических
данных необходимо учитывать
Ход реакции
Рис 58. Повышение изобарного
потенциала на промежуточной стадии
самопроизвольной реакции
и кинетические и искать способы ускорения (а иногда замедления)
реакции. Сейчас мы переходим к кинетике химических реакций—
учению о скорости реакций и ее зависимости от различных
факторов.
107
ГЛАВА 3
УЧЕНИЕ О СКОРОСТИ ХИМИЧЕСКИХ ПРОЦЕССОВ
§ 16. СКОРОСТЬ ХИМИЧЕСКОЙ РЕАКЦИИ И ЕЕ ЗАВИСИМОСТЬ
ОТ КОНЦЕНТРАЦИИ
Любой процесс, любой переход системы из одного состояния
в другое протекает во времени. Представление о малой или
высокой скорости процесса условно и связано с нашей обыденной
оценкой величин интервалов времени. Очень медленные
процессы мы не замечаем и поэтому считаем, что они не проходят,
очень быстрыми пока еще не умеем управлять.
Раздел химии, рассматривающий скорости и механизмы
химических процессов, называется химической кинетикой. Понятие
скорости относится одновременно и к процессу и к объекту
и характеризуется изменением какой-либо характеристики
объекта (положение или свойство) в единицу времени. Скорость
химической реакции показывает число химических
взаимодействий, приводящих к образованию продукта реакции, в единице объема
или на единице поверхности за единицу времени. Измеряется
скорость реакции изменением концентрации реагирующего вещества
в единицу времени.
Пусть за промежуток времени от tt до /2 концентрация
некоторого реагирующего вещества уменьшилась с Ct до С2.
Скорость реакции в данный промежуток времени
Поскольку концентрация реагирующего вещества во времени
уменьшается, С2<Сх и (С2 — Сг)<0. Однако, поскольку скорость
не может быть отрицательной величиной, если ее определяют по
расходу реагирующего вещества, перед отношением
в правой части уравнения ставится знак минус; если же ее
определяют по увеличению концентрации продукта
реакции, то она положительна. В общем случае
108
o±±?.
t2~tx At
Отношение (C2-Ct)/(t2-tl)
выражает среднюю скорость
реакции v в интервале времени
/2 —/i. Средняя скорость реакции
тем ближе к истинной, чем
меньше промежуток времени t2 — 'i
(рис. 59). За бесконечно малый
промежуток времени dt
концентрация изменится на бесконечно
малое значение dC и, значит,
отношение dC/dt измеряет
истинную скорость реакции в момент /:
v=±dC/dt.
В любой химической реакции реагенты расходуются и
поэтому скорость реакции уменьшается. Отсюда следует, что скорость
реакции зависит от концентраций реагирующих веществ и ее
следует относить к какому-то определенному моменту времени.
Рассмотрим реакцию
Рис. 59 Средняя и истинная
скорости реакции
проходящую при постоянных температуре и объеме. Уравнение
химической реакции, написанное в таком виде, означает, что
1 моль А, 1 моль В и 2 моль D. реагируя между собой, дают по
1 моль веществ F и L. Реакция „между молекулами реагирующих
веществ осуществляется только при соударении молекул. При
увеличении концентраций реагирующих веществ число
соударений должно возрасти, а скорость увеличиться. Весь вопрос в том,
как возрастает скорость при увеличении концентрации каждого
из реагирующих веществ, скажем, в 2—3 раза. Можно ли
представить зависимость скорости от концентрации в виде уравнения,
позволяющего рассчитывать скорость для любых концентраций?
Можно ли, зная вид сгехиометрического уравнения реакции,
судить о зависимости скорости от концентрации?
Оказывается, что вообще для большинства реакций стехи-
ометрическое уравнение не может дать сведений о зависимости
скорости реакции от концентрации. Даже утверждение, что с
увеличением концентрации какого-либо реагирующего вещества
скорость реакции возрастает, иногда оказывается неверным. Есть
реакции, на скорост ь которых увеличение концентрации
реагирующего вещества не оказывает влияния. Есть реакции, скорость
которых возрастает при увеличении концентрации молекул
веществ, не записываемых в уравнении реакции и, казалось бы, не
участвующих в процессе (например, молекулы инертных газов).
109
При одних условиях зависимость скорости от концентрации
одна, а при других—другая. Для очень большого числа реакций
зависимость скорости от концентраций реагирующих веществ
определяется экспериментально.
Выведенное на основе экспериментальных данных уравнение
математически выражает зависимость скорости от концентраций
реагирующих веществ и показывает, что скорость химической
реакции пропорциональна произведению концентраций реагирующих
веществ в степени некоторых чисел, определяемых опытным
путем. Будем называть уравнения подобного типа,
устанавливающие экспериментально определенную зависимость скорости
от концентраций реагирующих веществ, кинетическими
уравнениями реакции.
В общем случае для реакции
aA+bB+dD+ ... =/F + /L+ ...
экспериментально определенная зависимость скорости от
концентрации выражается кинетическим уравнением:
Коэффициент пропорциональности к называется константой
скорости химической реакции. Ее значение определяется по
известному значению скорости при заданных концентрациях
реагентов А, В и D:
Для вычисления константы скорости реакции следует
определить скорость реакции при равенстве единице всех
концентраций (или парциальных давлений газа) или же так подобрать
концентрации реагентов, чтобы их произведение было равно
единице. В этом случае скорость численно равна константе
скорости реакции.
Константа скорости зависит от тех же факторов что и
скорость данной реакции, но не зависит от концентраций
реагирующих веществ и времени. Для характеристики химических
реакций значения скоростей непригодны, так как скорость реакции
меняется во времени. Константа скорости характеризует реакцию
для определенных условий (температура, давление, количество
катализатора и т. п.). Реакции, проходящие при строго
одинаковых условиях, можно сравнивать по их константам скоростей.
В этом кинетическом уравнении показатели степени а,
Р и 8 могут быть любыми небольшими числами, чаще всего
равными 1 или 2, редко 3. При некоторых особых условиях
проведения реакции показатель степени может равняться нулю.
Известно немало реакций с дробными показателями.
110
Иногда показатель степени концентрации данного вещества
совпадает с его стехиометрическим коэффициентом в уравнении
реакции. Для элементарных реакций, т. е. реакций, проходящих
в одну стадию и именно так, как написано уравнение, показатели
степени в кинетическом уравнении совпадают со стехиометриче-
скими коэффициентами реагентов в уравнении реакции.
Показатель степени концентрации реагирующего вещества
в кинетическом уравнении реакции (а, р, 8) называется порядком
реакции по данному веществу.
Общим порядком химической реакции, или просто порядком
реакции, называется величина, равная сумме показателей степени
концентраций реагентов в кинетическом уравнении реакции.
а+Р+5. Порядок реакции равен сумме порядков по
реагирующим веществам.
В качестве примеров реакций различного порядка можно привести
следующие реакции
Реакции первого порядка:
разложение газообразного N2O5
2N2O5(r)=4NO2(r)+O2(r)
разложение пероксида водорода в водном растворе
2Н2О2(р-р)==2Н2О(ж) + О2(г)
разложение хлорида лиазобензола в водном растворе
C6H5NNCl(p-p) = C6H5Cl(p-pL-N2(r).
Скорость расходования реагента в этих реакциях зависит от его концентрации по
кинетическому уравнению v — kC
Реакции второго порядка:
взаимодействие водорода и паров иода в 1азовой фазе
Н2(г)+12(г) = 2Н1(г), i7=*CH2C,2.
Эта реакция—отдельно по водороду и по иоду — первого порядка, но
суммарный порядок ее равен двум
Димеризация оксида азота в газовой фазе
2NO(r) = N2O2(r), v = kCjo
протекает как реакция второго порядка.
Реакции третьего порядка очень редки. Так, реакция
Cl2(r) + 2NO(r) = 2NOCl(r), v=kCC]2 CN2O
по компонентам является реакцией первого и в юрою порядков, но суммарный
порядок ее равен трем.
Интересно отметить, что реакции с одинаковым видом
химического уравнения часто имеют разные порядки. Иод и бром
находятся в одной подгруппе периодической системы, тем не
менее реакция взаимодействия иода с водородом
r) = 2HI(r), v=kCH2(r)Cl2ir)
111
— реакция второго порядка, а реакция водорода с бромом
вообще не имеет порядка (ее порядок непрерывно изменяется по мере
прохождения реакции).
Вид кинетического уравнения, а следовательно, и порядок
реакции могут зависеть от соотношения реагентов. При
большом избытке одного из реагентов его концентрация практически
не меняется и ее изменение поэтому не влияет на скорость
реакции.
Так, гидролиз сахара с образованием глюкозы и фруктозы
66
сахар глюкоза фрукл оэа
обычно проходит при большом избытке воды, поэтому
концентрация воды в ходе реакции практически не изменяется и не влияет
на скорость реакции. Кинетическое уравнение реакции
1> = ^Сс12н22о11-
Реакция разложения оксида азота N2O5 в газовой фазе
протекает как реакция первого порядка, но если при этом присутствует
твердый оксид азота (V), то расход газообразного оксида не
влияет на его концентрацию благодаря испарению с поверхности
твердого оксида. Испарение—лимитирующая стадия процесса,
имеет нулевой порядок. Поэтому процесс описывается
кинетическим уравнением нулевого порядка и=кС^гО5 = к.
Порядок газовых реакций, проходящих на твердых
катализаторах, часто ниже, чем в отсутствие катализаторов. Например,
горение оксида углерода на поверхности кристаллического
оксида ванадия V2O5 описывается кинетическим уравнением
первого порядка:
2СО (г) + О2 (г) = 2СО2 (г).
(K)
=кС
со
и скорость не зависит от концентрации кислорода.
Реакция взаимодействия водорода с кислородом
2Н2(г) + О2(г) = 2Н2О(г)
на поверхности твердых катализаторов (например, CdO) имеет
первый и дробные порядки в зависимости от парциальных
давлений кислорода и водорода (табл. 10).
Таблица 10
Рн2 Па
130—2600
2600—8000
30600—50600
Рог Па
26600—53 300
20000—53 300
1330—5 300
Вид кинетического
уравнения
Суммарный
порядок
1
0,5
0,7
112
Посмотрим, как изменяется концентрация вещества
(количество исходного вещества или продукта реакции) во времени, т. е.
по мере прохождения реакции.
Если реакция протекает по нулевому порядку, то для нее
v=-dS=kC°=k.
dt
Перепишем выражение в виде
dC=-kdt
и возьмем неопределенный интеграл от обеих частей уравнения
\dC~-\kdU
тогда
C=-kt+By
где В—постоянная интегрирования.
В начальный момент времени /=0, а концентрация С=С0.
Подставляя эти значения, получаем
Теперь, заменяя постоянную интегрирования В на Со,
получаем
C=C0-kt.
Следовательно, в реакциях нулевого порядка концентрация
линейно уменьшается со временем (рис. 60, а).
Константа скорости реакции нулевого порядка вычисляется
по формуле
где Со—начальная концентрация; С—концентрация в момент
времени /. Константа скорости нулевого порядка измеряется
1 1
в моль л -с
г о
I
1 0
Рис 60. Изменение концентрации со временем в реакциях:
а—нулевого порядка, б—первого порядка; в—второго порядка
из
Наряду с константой скорости для характеристики реакций
часто пользуются величиной, называемой временем
полупревращения tm. Время полупревращения—промежуток времени, в
течение которого реагирует половина взятого (исходного)
количества вещества.
Пусть f = /i/2 и С=С0/2. Получаем C0/2 = C0—kti/2, откуда
*i/2 = Со/2*.
Следовательно, для реакции нулевого порядка время
полупревращения пропорционально начальной концентрации исходного
вещества.
Перейдем к реакциям первого порядка. Кинетическое
уравнение скорости реакции первого порядка имеет вид
и —= ~*~ == fCv^.
di
Отделим переменные (время, концентрация) и проинтегрируем:
Находим постоянную интегрирования 5, считая, что в
начальный момент времени t — О концентрация реагента С=С0:
In Со = В.
Поэтому
Следовательно, для реакций первого порядка характерна линейная
зависимость логарифма концентрации от времени (рис. 60, б):
k = - In — или к = 2,
Константа скорости реакции первого порядка измеряется в с.
Время полупревращения (для реакций первого порядка чаще
говорят—период полураспада) находим тем же приемом. Для
t = tii2nC=C0/2
к = 2,303 — lg -^ = 2,303 — lg 2,
Ul2 Q)/2 /i/2
откуда
0,69
Эта формула используется для характеристики процессов
радиоактивного распада, которые являются процессами первого
114
порядка и подчиняются всем уравнениям реакций первого
порядка. Интересно, что для реакций первого порядка период
полупревращения не зависит от начальной концентрации исходного
вещества, а константа скорости реакции первого порядка обратно
пропорциональна периоду полупревращения.
Для реакции первого порядка характерно то, что время и
концентрация связаны зависимостью
*/=-1п—.
Со
Поэтому при изменении обеих концентраций в одинаковое
число раз t не изменяется. Из этого следует, что если для одной
и той же реакции в одном опыте концентрация через некоторый
промежуток времени уменьшится в два раза, то и в других
опытах за гот же промежуток времени она уменьшится также
в два раза. Это означает, что в одинаковые промежутки времени
реагируют одинаковые доли взятого количества исходного
вещества. Так, если за первый час в реакцию вступило 10% взятого
количества вещества, то за следующий час прореагирует 10% от
оставшихся 90% исходного вещества.
Величина, обратная константе скорости реакции первого
порядка,
имеет размерность времени и характеризует среднюю
продолжительность жизни исходных молекул в условиях опыта.
Зависимость константы скорости реакции первого порядка от
отношения концентраций позволяет при расчетах заменять
концентрации пропорциональными им величинами — количес!вами
веществ, давлениями, показателями светопреломления и т. п.
Вывод уравнения зависимости концентрации от времени для
реакции второго порядка рассмотрим только для простейшего
случая, когда концентрации двух реагирующих веществ
одинаковы: 2А = D. Запишем кинетическое уравнение:
vkC\
dt
После разделения неременных, интегрирования и определения
постоянной интегрирования получаем:
при / = U С =С0 и 1/Со^^, откуда
I 1
115
Следовательно, для реакций второго порядка при равенстве
начальных концентраций реагирующих веществ наблюдается
линейная зависимость величины обратной концентрации от времени
(рис. 60, в).
Константа скорости вычисляется по формуле
и измеряется в л • с * • моль *.
Вычислим время полупревращения веществ. При t=ti/2
С = Со/2, откуда
В отличие от реакции первого порядка период
полупревращения реакций второго порядка обратно пропорционален начальной
концентрации.
Реакции третьего порядка очень редки и не представляют
большого практического интереса.
Порядок реакции в целом равен сумме порядков по
компонентам. Чтобы узнать порядок п по какому-либо компоненту,
достаточно экспериментально определить, как зависит скорость
реакции от концентрации данного компонента. ^
Предположим, что при увеличении концентрации вещества
в 2,5 раза скорость реакции возросла в 4,0 раза. Найдем порядок
реакции по этому веществу. Запишем кинетическое уравнение
реакции для данного компонента (предполагая, что
концентрации остальных веществ постоянны) до изменения концентрации
и после:
v=k-cn
Разделим второе уравнение на первое и вычислим п
4,0=2,5"
lg450=«lg2,5
1^0 0^^
lg 2,5 0,40 '
Итак, порядок реакции по этому веществу равен 1,5.
Нетрудно рассчитать, как изменится скорость при заданном
изменении концентрации. Пусть концентрация увеличена в 3
раза. Тогда при п = 1,5 скорость возрастет в 3Ь5 = 5,2 раза.
В общем случае, если экспериментально обнаружено, что
увеличение концентрации реагирующего вещества в т раз приве-
116
ло к возрастанию скорости в / раз, ю порядок реакции по
данному компоненту равен
lg m
Определив экспериментально зависимость скорости реакции
от концентраций реагирующих веществ, можно ее использовать
для вычисления скорости при других концентрациях и
предсказывать изменение скорости при изменении концентраций
реагирующих веществ или изменении некоторых внешних условий.
Например, предскажем, как изменится скорость реакции
при увеличении общего давления в 2 раза, если реакция проходит в газовом
состоянии. При повышении давления в 2 раза концентрация каждого вещества
в газовой фазе возрастает в 2 раза
Пусть Vi —скорость при некотором давлении р, a v2—скорость при давлении
в 2 раза более высоком. Тогда
у2_кBСАУBСвHBСвУ
кааа
Следовательно, скорость возрастает в 8 раз. При разбавлении газовой смеси
инертным газом в 3 раза можно ожидать уменьшения скорости
вЗгЗ°-32 = 27раз.
Реакции первого порядка представляют особый интерес, так
как для них действительны те же законы, что и для процессов
радиоактивного распада.
Так, для изотопа углерода ^С, постоянно образующегося
в верхних слоях атмосферы действием нейтронов космических
лучей на атомы азота
период полураспада /1/2 = 5570 лет. Содержание в атмосфере
углерода 14С несмотря на его распад сохраняется постоянным.
В составе углекислого газа углерод 14С участвует во всех
процессах биологического круговорота и отношение 14С/12С=10~16
остается постоянным в веществе живых организмов или
растений, пока они участвуют в процессах жизнедеятельности. После
смерти живого организма новые порции изотопа 14С не
поступают в ткани и его содержание уменьшается в соответствии с
законом химической кинетики реакции первого порядка: каждые
5570 лет количество 14С в смеси с 12С уменьшается вдвое.
1 грамм углерода, свежевыделенного из живого организма
или растения, испускает 16 р-частиц в минуту, полученный из
животных или растительных остатков, пролежавших в земле
117
JZ
5570 лет, будет показывать
8 распадов в минуту, а еще
через 5570 лет—4 распада и т. д.
|^_4—^^ (Рис- 61>-
Чтобы определить, когда было
срублено дерево, по найденному при
раскопках его куску, древесину
сжигают в токе кислорода, а образовавший-
0 2000 4000 6000 8000 1000012000 ся диоксид углерода поглощают рас-
t, годы твором гидроксида кальция и
исследуют активность карбоната кальция
Рис- 61 Радиоактивный распад изото- Предположим, образец показал 12,5
па 14С распада в минуту в пересчете на 1 г
углерода. По графику определяем
возраст образца — 2000 лет. Те же результаты получаем расчетом:
=^, *вЙйГ1'24 Ю год, Л= 1,24
/ /U
Отношение начальной Со и имеющейся в данный момент времени концентрации
С заменяем отношением пропорциональных им величин—числами отвечающих
им распадов:
1,24-10 = 2,303 - lg —, /=1990 лет.
Эта методика пригодна для определения возраста от 400 до 40000 лет.
Остановимся на одной особенности процессов
радиоактивного распада (это относится и к химическим реакциям). Как уже
отмечалось, величина, обратная константе скорости реакции
первого порядка, имеет размерность времени и называется средним
временем жизни атома (или молекулы). Так, среднее время жизни
атома 14С равно
1 г|/2 5570
Это число означает, что в среднем атом 14С живет 3843 года, но
ничего не говорит о времени жизни данного атома. О периоде
полураспада одного атома бессмысленно говорить, период
полураспада двух атомов также нельзя определить, но с увеличением
числа атомов быстро возрастает вероятность того, что за интервал
времени tXf2 распадется половина их исходного количества и из
неопределенной для малого числа атомов величины мы получаем
точную величину периода полураспада для большого числа атомов.
Как и законы термодинамики, законы химической кинетики
носят вероятностный, или статистический, характер, т. е.
пригодны и точны только для большого числа частиц. Плавная кривая на
рис. 62 представляет скорость распада большого числа частиц.
При малом числе распадов кривая распада превращается в
ломаную линию с тем более сильными отклонениями от плавной
кривой, чем меньше распадов происходит в единицу времени. Для по-
11R
0,5
I
W5
Рис. 62. Вид кривой распада в зависимости от числа частиц
лучения плавной кривой следует или увеличивать количество
распадающегося вещества, или же увеличивать время наблюдения.
Плавная кинетическая кривая—это очень точное приближение
к наиболее вероятной скорости процесса в каждый момент времени.
Хотя реакции первого порядка и процессы радиоактивного
распада описываются одними и теми же законами, природа их
различна. Скорость химических реакций в отличие от процессов
радиоактивного распада зависит от температуры, присутствия
посторонних веществ и других факторов. Период полураспада
радиоактивного вещества не зависит от его исходного количест-
,ва. Это указывает на то, что распад атомного ядра не связан
с присутствием других ядер, но непонятно, почему одно ядро
в данный момент распадается, а другое нет. По той же причине
очень трудно предположить, что в реакциях первого порядка
происходит распад некоторых отдельных молекул. Известно
очень много кристаллических веществ, термодинамически
неустойчивых при данных условиях (для них изменение изобарного
потенциала образования положительная величина AG<0), но
кинетически устойчивых. Часто признаков распада таких
кристаллов обнаружить не удается даже самыми точными методами.
Скорость же радиоактивного распада почти не зависит от
агрегатного состояния вещества и его химического состава.
В заключение этого раздела поговорим о кинетике простых,
или элементарных, реакций.
Необходимым условием химического взаимодействия
молекул является их столкновение. Именно в момент
столкновения электроны какого-либо атома, иона или молекулы попадают
в сферу действия ядер и электронов другой частицы и
совершаются переходы электронов на другие энергетические уровни
119
и подуровни, перекрывание электронных облаков и их смещение
к другим атомам.
Число молекул, одновременным взаимодействием между
которыми в момент столкновения осуществляется акт химического
превращения, называется молекулярностью реакции. Реакции
бывают мономолекулярные (одномолекулярные), бимолекулярные
(двухмолекулярные) и гримолекулярные (трехмолекулярные).
Тримолекулярные реакции очень редки, так как одновременное
столкновение трех молекул очень маловероятно. Молекуляр-
ности реакций выше трех неизвестны.
К мономолекулярным реакциям относят реакции разложения
молекул и внутримолекулярных перегруппировок, например:
12 (г) = 21 (г), и=кС\2(г). Скорость мономолекулярных реакций
описывается кинетическим уравнением реакции первого порядка.
Примеры бимолекулярных реакций:
2NO(r) = N2O2(r), v = kC2NO{r)
О2 (г) + Н(г) = НО (г) + О (г), v = kCO2{r)CHir)
НВг (г)+Н (Г) = Н2 (г) + Вг (г), v = kCHBTir)CHiT)
Скорость бимолекулярной реакции описывается кинетическим
уравнением реакции второго порядка.
Пример тримолекулярной реакции:
С12(г) + 21ЧО (r) = 2NOCI (г), v = кСа а (г) С?о (г).
Скорость тримолекулярных реакций описывается кинетическим
уравнением реакции третьего порядка.
В приведенных примерах реакций молекулярность и порядок
реакции совпадают, но в общем случае они различны. Зная1
молекулярность реакции, можно сразу же сделать вывод о ее
порядке и о влиянии изменения концентраций на скорость
реакции. Если уравнение реакции точно отражает ее ход, т. е. все
атомы (или молекулы) исходных веществ сталкиваются
одновременно и участвуют в элементарном акте химического
превращения, то скорость химической реакции прямо пропорциональна
произведению концентраций реагирующих веществ, возведенных в
степени их стехиометрических коэффициентов. Так формулируется
закон действия масс (первая формулировка; другая относится
к константе равновесия) — основной закон химической
кинетики— в приложении к простым реакциям.
§ 17. ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА СКОРОСТЬ ХИМИЧЕСКИХ РЕАКЦИЙ
При повышении температуры обычно скорость химических
реакций увеличивается в соответствии с правилом Вант-Гоффа,
которое является весьма грубым приближением. Правило Вант-
МО
Гоффа утверждает, что при повышении температуры на каждые'
10° скорость реакции (константа скорости) увеличивается в 2—4
раза.
Значительно более точно зависимость константы скорости or
температуры Г описывается уравнением
Здесь А и В—постоянные.
Чтобы определить значения постоянных А и В, нужно знать минимум два
значения константы скорости при любых произвольно выбранных двух
температурах Пусть при температуре Тг константа скорости реакции равна ки а при
температуре Т2 равна к2 Составляем систему из двух уравнений с двумя
неизвестными А и В-
А
А
Вычитая первое уравнение из второго, получаем
А А
In /г2-1п *, = -— + —
'2 ' t
Далее
. к2 А(Т2~Т,)
in — = ,
откуда
ТХТ2 t к2
1 2~ М «1
Подставляя А в любое из двух уравнений при известных к и Т, вычисляем
постоянную В. Подстановкой А и В в уравнение получаем уравнение зависимости
константы скорости от 1смпературы Ниже увидим, как тесно связана постоянная
А с энергией активации
Почему при повышении температуры скорость химической
реакции возрастает?
При сближении двух реагирующих чаешц наступает момент,
когда электронное облако одной частицы попадает в сферу
действия другой. Вследствие одинакового знака зарядов
электронных оболочек частицы взаимно отталкиваются и, если
они не обладают достаточной кинетической энергией,
разлетаются в разные стороны, что и происходит с огромным
количеством взаимодействующих частиц. Но те частицы,
которые обладают некоторым минимальным избыточным
относительно среднего запасом энергии, сближаются настолько
близко, что происходит перекрывание наружных электронных
121
облаков всех сталкивающихся частиц в одно общее молекулярное
облако.
Энергия активации—это избыточное количество энергии (по
сравнению со средней ее величиной), которым должна обладать
молекула в момент столкновения, чтобы быть способной к
химическому взаимодействию. Те молекулы, которые обладают таким
количеством энергии, называются активными. В основе теории
активных столкновений лежат положения молекулярно-кинетиче-
ской теории газов и предположение об энергетическом барьере,
численно равном энергии активации.
Можно предположить, что повышение температуры
сказывается на числе активных молекул. Посмотрим, как влияет
температура на энергию молекул газа. Молекулы реагирующих
веществ находятся в непрерывном движении. Количество молекул,
обладающих той или иной кинетической энергией (или
скоростью), определяется законом Максвелла—Больцмана
распределения молекул по скоростям и энергиям. Только столкновения
молекул, обладающих энергией, равной или большей энергии
активации, эффективны и ведут к химическому превращению.
Ниже приведено распределение молекул (для 1 моль в %)
некоторого газа по группам, характеризующимся определенным
интервалом энергии (Дж/моль).
0—1300
1300—2500
2500—3800
3800—5000
1
22
43
24
5000—6300
6300—7500
Больше
7500
7
2
1
На рис. 63 эти данные представлены графически. Площади,
получаемые умножением числа молекул на длину интервала энергии,
представляют собой ту часть энергии молекул газа, которая
приходится на данную группу молекул. Чем меньше интервал
энергий, тем уже прямоугольники и тем
точнее описывается распределение
молекул.
В предельном случае, когда интервалы
энергий бесконечно малы,
прямоугольники превращаются в прямые линии,
перпендикулярные оси энергии. Концы этих
прямых опишут кривую, называемую
кривой распределения молекул по энергиям
(распределение Максвелла—Больцмана).
Подобного типа кривые начинаются из
точки начала координат, имеют
максимум, более растянуты в сторону высоких
энергий и не пересекаются нигде с гори-
Рис. 63. Распределение зонтальной осью. Энергия, отвечающая
молекул по энергиям максимуму кривой, называется наиболее
10
122
вероятной*, она не совпадает со средней
энергией молекул вследствие
несимметричного расположения кривой по обе
стороны от максимума.
При повышении температуры
форма кривой сохраняется, но так как
энергия молекул возрастает, кривая
распределения смещается вправо; при
этом высота максимума кривой
понижается, а сам максимум также
смещается вправо (рис. 64).
Кривая распределения молекул по
энергиям описывает зависимость доли
сг
молекул —, обладающих той или иной
N
Рис 64. Изменение вида
кривой распределения при
повышении температуры
энергией Е% при этом N—общее число
молекул в системе (например, 1 моль), пЕ—число молекул с
данной энергией Е.
Вспомним, что взаимодействие совершается при встрече двух
или более частиц. Согласно теории Аррениуса соударения будут
эффективны только тогда, когда молекулы обладают некоторым
избытком энергии по сравнению со средней энергией молекул при
данной температуре. Таким молекулам соответствует часть
кривой распределения далеко справа от максимума (рис. 65). Доля
этих молекул на графике показана штриховкой. Эта область
с одной стороны ограничена энергией активации 2?а, а с другой—
теоретически не имеет предела, так как могут существовать
молекулы с бесконечно большей энергией, но таких молекул
бесконечно мало.
Заметим, что для реакций соединения атомов или молекул
существует верхний предел энергий ?верХн- Молекулы, обладающие
энергией выше этого предела, при взаимодействии не дают
продуктов реакции, так как молекулы
продуктов, имея повышенный запас энергии,
нестабильны и сразу же распадаются на
исходные вещества (если не успевают
отдать избыток энергии другой частице
или стенке реакционного сосуда).
Как следует из рис. 65, при
повышении температуры на 10° количество
молекул, обладающих энергией, равной
или выше энергии активации,
возрастает в 2—4 раза. Заштрихованные
площади под обеими кривыми
соответствуют количествам активных молекул Рис- 65- Увеличение числа ак-
при температурах Тх и Г, +10. ™BHbIXJS^™""
293К
123
Заштрихованная площадь под кривой G\ + 10) примерно
в 3 раза (в 2—4 раза) больше такой же площади под кривой Тх.
Увеличение количества акт ивных молекул в 2—4 раза при
увеличении температуры на 10° хорошо согласуется с
экспериментально наблюдаемым увеличением скоростей химических реакций
в то же число раз.
Отношение числа активных молекул пл к общему числу
молекул N9 т. е. доля активных молекул, равно
Еа—энергия активации; R—газовая постоянная; е = 2,7183—
основание натуральных логарифмов.
Очевидно, чем больше активных молекул в реакционной
системе, тем выше скорость реакции и соответственно константа
скорости.
Следовательно, константа скорости химической реакции
определяется долей активных молекул, т. е. числом эффективных
столкновений. Поэтому
Это—уравнение Аррениуса, показывающее, что при постоянной
температуре константа скорости определяется энергией
активации. Чем выше численное значение энергии активации, тем
меньше в реакционной смеси активных молекул, тем меньше число
эффективных соударений и тем, следовательно, ниже константа
скорости и сама скорость химической реакции.
При комнатной температуре обычно проходят реакции с энергиями актива-
ции 60—105 кДж/моль. Если ?а<60 кДж/моль, реакция совершается
чрезвычайно быстро, а если Ея> 105 кДж/моль, скорость ее настолько мала, что
прохождение реакции нереально
На энергетической (энтальпийной) диаграмме реакции
(рис. 66, а) энергии активации прямой реакции отвечает разность
между максимумом на кривой изменения энергии в ходе реакции
и уровнем энергии исходных веществ. Возникает вопрос—в
каком состоянии находятся реагирующие вещества, когда они
поднялись к вершине кривой.
Переходным состоянием системы, или переходным
активным комплексом, называется состояние системы
реагирующих веществ, соответствующее максимальной энергии на пути
реакции (точка К), Энергия активации—это количество
дополнительной энергии, необходимой для перехода системы из
исходного состояния в состояние активного комплекса. Разность
уровней энергии в исходной точке / и конечной точке 2 есть
изменение энтальпии, которое не зависит от пути реакции,
124
a
Ход реакции
Рис. 66. Реакция взаимодействия иода и водорода
т. е. от энергетического состояния системы в переходном
состоянии.
Предполагают, что реакция между газообразными
водородом и иодом протекает через образование комплекса «H2l2»
(Рис. 66,6):
H2 + I2 = «H2I2» = 2HI.
Реакция совершается через постепенное ослабление связей
у исходных молекул и усиление их у молекул продуктов реакции.
Энергию активации можно рассматривать как изменение
энтальпии при образовании комплекса из исходных веществ.
Энергия, выделяющаяся при переходе системы из состояния
комплекса в конечное, численно равна энергии активации
обратного процесса. Энергия активации обратного процесса больше
энергии активации прямого процесса для реакций,
сопровождающихся выделением теплоты (АЯ<0), на величину изменения
энтальпии:
= Ai.o6p"
-'а. пр*
Переходный комплекс только на первый взгляд представляет
собой молекулу, обладающую обычными термодинамическими
свойствами. Имеется ряд принципиальных отличий переходного
комплекса от обычной молекулы. Главная особенность любой
молекулы состоит в том, что она всегда обладает некоторым
минимумом энергии. Это обусловливает ее стабильность, в то
время как переходный комплекс обладает некоторым
максимумом энергии, характеризуемым энергией активации. Поэтому
переходный комплекс нестабилен. На энергетической кривой
ему соответствует некий максимум—вершина энергетического
125
барьера, после которого неизбежно следует понижение энергии.
В этом отношении он отличается от промежуточных соединений,
которым на энергетической кривой обязательно соответствует
хотя бы очень маленький минимум.
Переходный комплекс отличается or молекулы и в том
отношении, что энергии связей, длины связей и углы между связями
в переходном комплексе искажены и не отвечают средним
значениям тех же величин у молекул.
Величина энергии активации определяет скорость химической
реакции. Она зависит от строения электронных оболочек
взаимодействующих атомов и молекул и образующегося из них
переходного комплекса. Величина теплового эффекта реакции или
изменения энтальпии зависит от соотношения энергий связи
реагентов и продуктов реакции, т. е. также от способности
наружных электронных оболочек к взаимному перекрыванию или
смещению электронной плотности. В конечном счете и скорость
реакции, и количество образующегося продукта определяются
электронным строением всех участвующих в процессе веществ.
В уравнении
= Се
RT
к—константа скорости, определяемая опытным путем (из
скорости прохождения реакции или же времени ее прохождения), Т—
температура, при которой проводится определение константы
скорости, R—газовая постоянная. Таким образом, в этом
уравнении только два неизвестных: коэффициент С и энергия
активации Еп.
Если уравнение содержит два неизвестных, то для их
определения достаточно составить систему минимум из двух уравнений,
т. е. знать по крайней мере два значения константы скорости
кг и к2 при двух температурах, соответственно 7\ и Г2.
Прологарифмируем уравнение:
1пк = \п
(се"-),
т*=1„с--|.
Сравним это уравнение с уравнением:
называемым уравнением Аррениуса.
Видно, что
126
B=In С
и
откуда
Таким образом, энергия активации равна постоянной А в
уравнении Аррениуса, умноженной на газовую постоянную.
Составим систему из двух уравнений
HI х
= \пС-^г.
Л/2
Решив систему, находим:
?а ЛIn.
Переходя к десятичным логарифмам (коэффициент перевода
2,303) и подставляя численное значение газовой постоянной,
получим энергию активации:
?, = 8,314-2,303-?Ц^18?=19,14^18?.
Энергия активации называется истинной энергией активации^
если она относится только к той реакции, которая протекает
в соответствии с написанным уравнением. Чаще всего на
практике, изучая зависимость скорости какого-либо процесса от
температуры, вычисляют эффективную энергию активации, которая
служит только лишь для нахождений скоростей и констант
скоростей при любых интересующих температурах.
Под символом логарифма в формуле находится отношение
констант скоростей процесса при двух температурах, поэтому
константы можно заменить любыми пропорциональными им
количественными выражениями свойств при тех же
температурах, например, вместо константы скорости можно подставить
обратное время прохождения реакции (до некоторого
определенного состояния, предела):
Ea = R——rln--.
Например, измеряя число сокращений мышцы лягушки при двух
температурах, вычисляют энергию ее активации. Измеряя время
127
Рис. 67. Виды
зависимости скорости реакции от
температуры
вытекания одного и того же объема
жидкости или газа через малое отверстие при
различных температурах, находят
энергию активации этого физического
процесса, и т. п.
Известны реакции, скорость которых
зависит от температуры более сложным
образом. На рис. 67 приведены кривые
типичной зависимости скорости от
температуры (/) и зависимости, которая часто
обнаруживается у каталитических
реакций B). До некоторой температуры
Тм скорость возрастает с ростом
температуры по обычным законам, но потом
начинает резко падать. Объясняется это
явление уменьшением активности катализатора при высоких
температурах. Особенно часто это наблюдается для реакций,
проходящих в живых организмах: около температуры Тм
биологический катализатор (фермент) распадается и процесс
прекращается.
Не только энергия активации (высота энергетического
барьера) влияет на скорость реакции. Большую роль играют размеры
и форма молекул, а также место расположения реакционноспо-
собных атомов или групп атомов в молекуле. Далеко не всякое
столкновение молекул с энергией, равной или большей ?а, ведет
к химическому взаимодействию.
Рассмотрим столкновение двух атомов водорода.
Электронная плотность в атоме водорода распределена равномерно по
шаровой поверхности, и поэтому безразлично, какими местами
столкнутся реагирующие атомы. Перекрывание орбиталей
произойдет в любом месте (рис. 68).
Если же сближаются атомы водорода и галогена, то
небезразлично место их соприкосновения. Электронная плотность
валентных орбиталей галогена распределена в пространстве сложным
образом. На рис. 69 зачернены две /7-орбитали атома галогена,
имеющие по 2 спаренных электрона. Если атом водорода
подходит к этим двум орбиталям, перекрывание невозможно и реакция
не происходит. Только одна р-орбиталь, имеющая 1 электрон (не
зачернена), может
перекрываться с шаровой сферой
электронной плотности атома Н,
образуя химическую связь.
Еще сложнее ситуация
при сближении молекул
водорода и галогена.
Требование надлежащей ориентации
делается еще жестче.
Н
Рис. 68. Взаимодействие двух атомов
водорода
128
г нг
Рис. 69 Взаимодействие атомов галогена и водорода
Обозначим вероятность необходимой для взаимодействия
ориентации молекул при столкновении W. Тогда
„,_ Число способов ориентации, приводящих к взаимодействию
Общее число возможных способов ориентации
Натуральный логарифм величины W, умноженный на
постоянную Больцмана, называется энтропией активации:
Умножив 5 а на постоянную Авогадро, получаем энтропию
активации для 1 моль:
S* = S'%N=Nk\n lV=R\n W.
Таким образом, вероятность необходимой ориентации
h
Чем больше вероятность необходимой для реакции
ориентации, т. е. чем больше значение энтропии активации, и чем
меньше значение энергии активации, тем выше скорость реакции
и соответственно константа скорости. Таким образом,
где Z—коэффициент пропорциональности.
Коэффициент Z для бимолекулярных реакций — это число столкновений
частиц в единице объема за единицу времени A с, 1 см3)
Это уравнение называют основным уравнением химической
_
кинетики. Множитель е R называется стерическим множителем,
а также стерическим фактором или фактором вероятности. В
него не входит температура, так как она не влияет на ориентацию
молекул.
5 2452
129
Сгерический фактор учитывав не только определенную
ориентацию реагирующих частиц, но и продолжительность контакта
в момент столкновения. Если время контакта частиц очень мало,
частицы не успевают осуществить перераспределение
электронных плотностей и, отталкиваясь, расходятся без изменения. Сте-
рический фактор может иметь самые различные значения.
Встречаются реакции, у которых величина е ~* больше единицы или же
очень мала A0 ~8). Стерический фактор связан с изменением
энтропии в ходе реакции.
§ 18. КИНЕТИКА СЛОЖНЫХ РЕАКЦИЙ
В большинстве случаев химическое уравнение показывает
лишь количественное соотношение реагирующих веществ. Только
химическое уравнение элементарных процессов говорит
одновременно о механизме процесса и о количественных соотношениях
реагентов и продуктов. Уравнения химической реакции часто
пишут с дробными коэффициентами, хотя участвовать в реакции
могут только целые частицы. Одно и то же уравнение можно
записать с различными стехиометрическими коэффициентами,
что очень удобно для расчетов с использованием закона Iecca.
Имеется огромное число уравнений реакций с суммой стехио-
метрических коэффициентов реагирующих веществ выше трех,
хотя вероятность прохождения тримолекулярной реакции очень
мала. Большинство уравнений не дает указаний на
действительный механизм протекающего процесса. То, что некоторые
реакции записываются с большими стехиометрическими
коэффициентами, по-видимому, говорит о том, что такие реакции протекают
через ряд других, более простых процессов, а само уравнение
процесса представляет собой лишь алгебраическую сумму
уравнений элементарных стадий. Остановимся на трех типах реакций:
последовательных, параллельных и обратимых.
Последовательными реакциями называются реакции
вида А=Б=В==Г, где вещество А является исходным, вещество
Г—продуктом, а вещества Б и В — промежуточными (играют
одновременно роль и продуктов и реагентов для
соответствующих отдельных стадий).
Наиболее важная особенность последовательных реакций
состоит в том, что если одна из стадий обладает значительно
меньшей скоростью, чем остальные, то общая скорость реакции
образования конечного продукта определяется скоростью именно
этой стадии. Следовательно, лимитирующей (ограничивающей)
стадией процесса будет та, у которой более высокая энергия
активации (или малая энтропия активации).
130
АИ
8
ход реакции
а
Ход реакции
5
Рис 70 'Энтальпийная диаграмма посдсдоватс imimx реакций
На рис 7() для реакций, проходящих через две стадии рредешвдены случаи,
Koi;w димитируютей С1адией явдясгся первая (a) (Л\ , > /:u ,,) и вгорая (г;)
{Елj<ha „) стадии. Число С1адий и величины их шергий акшвации в соогветс!-
вии с первым законом термодинамики не влияют на изменение жтальпии при
переходе системы из состояния А в сосюянис Н
Экспериментальное определение механизма химического
процесса и представление его в виде отдельных сшдий имеет
большое практическое значение для подбора ка1ализаторов. Из-за
специфического действия каюлизаюра ею следует подбирать
юлько для самой медленной С1адии, но не для всей реакции
в целом.
Параллельными называются реакции, идущие
одновременно по нескольким направлениям с образованием различных
продуктов.
Если реакции различаются но скорости, то реакцию,
обладающую большей скоростью, называют основной (главной),
а остальные побочными. Основная реакция, являясь наиболее
быстрой, имеет самое низкое при данных условиях значение
энергии активации.
Рассмотрим подробнее наиболее просюй случай двух
параллельных мономолекулярных реакций
характеризующихся соответственно константами скорост ей
kFt и /св, энергиями активации ?а,АБ и ?а.лв- Если энергия
активации реакции А=Б больше энергии активации реакции
А=В, ^а,лг»>^а.лв. то в основном будет проходить реакция
А=В и в смеси продуктов реакции будет преобладать вещество
В. При другом соотношении энергий активации ?га<лв>^а,ль
будет проходить реакция А=Б и продуктом реакции следует
ожидать вещество Б. При равенстве энергий активации обеих
реакций возможно образование смеси Б и В.
131
и (T\^h* •гч Следовательно, от энергии
активации зависит прохождение
реакции по тому или иному пути:
осуществляется стадия, имеющая
более низкое значение энергии ак-
ход реакции тивации.
Если при данных условиях
Рис 71 Энтальпийная диаграмма термодинамически возможны два
параллельных реакций или более наПравлениЙ реакции,
то преобладание того или иного направления и количества
продуктов, получаемых по различным направлениям, определяются
соотношением скоростей (н энергий активаций) этих реакций,
а не соотношениями термодинамической устойчивости
продуктов реакций.
На рис 71 представлена энтальпийная диаграмма двух параллельных
реакций Пусть /:а.лв>?а аб а Д#лв>А#лб< т. е термодинамически более вероятно
образование вещества В, однако из-за меньшею значения энергии активации
образуется вещество Б Если энср!ия активации ?\Б много ниже ЕАВ, в качестве
продукта реакции получас 1ся практически чистое вещество Б, которое, однако,
термодинамически неустойчиво. Если энер1ии активации ?АВ и ?АБ не слишком
отличаются друг от друга, в начале реакции получается смесь продуктов Б и В,
количества которых определяются исключительно соотноиюнием энергий
активации ?АВ и ?АЬ.
Однако вещество Б, обладая большим запасом энергии, неустойчиво и можно
ожидать его превращения в вещество В, устойчивое при данных условиях Если
процесс Б=В характериз>егея высокой энергией активации Яр>в. го количество
вещества Б не будет изменяться во времени Однако, если энергия активации
этого превращения ?'ьв не высока, постепенно вещество Б, по закону реакции
первого порядка, превратится в вещество В и по прошествии некоторого времени
образуется термодинамически устойчивое вещество В
Заметим, что на примере этого процесса был рассмотрен случай двух
параллельных реакций I и И, причем реакция II проходит через две последовательные
с 1 алии.
В большинстве реальных процессов происходит наложение
параллельных и последовательных реакций. На рис. 72
представлен пример такого превращения.
Переход А==В может совершаться непосредственно по пути I,
но энергия активации его слишком велика и практически процесс
проходит не по двум параллельным путям I и И, а по пути II,
который составляется из двух последовательных стадий, каждая
из которых имеет Fie слишком высокие энергии активации.
Введение катализатора в реакционную смесь ускоряет
прямую и обратную реакции одного (или нескольких) из многих
возможных параллельных пулей, причем это совсем
необязательно термодинамически самый выгодный путь. Одно из объяснений
каталитического действия состоит в следующем. Катализатор
К и реагирующее вещество А образуют промежуточное
соединение АК, которое реагирует с другим исходным веществом В (или
просто распадается) с образованием продуктов реакции В и
исходного катализатора К:
132
Ход реакции
Рис 72 Два пути реакции
Ход реакции
Рис. 73. Реакция с участием
катализатора
В + АК-ВАК-АВ + К.
Энергетическая диаграмма такой реакции представлена на
рис. 73. В данном примере катализа!ор К, взаимодействуя с
исходным веществом А и давая промежуточное соединение А К,
изменяет электронную плотность наружных орбиталсй А и
облегчает реакцию с веществом В. Образующийся переходный
комплекс ВАК распадается на исходный катализатор К и
обладающую избыточным запасом энергии молекулу (АВ)*, которые
расположены на гом же энергетическом уровне, что и исходные
вещее 1ва. Далее молекула (АВ)*, выделяя энергию,
количественно равную изменению энтальпии в процессе А+В —АВ,
переходит в устойчивое состояние АВ. Следовательно, в данном
примере возможны два параллельных пути: I — не каталитический,
проходящий через высокий энергетический барьер, и II—со
значительно меньшей энергией активации благодаря воздействию
катализатора. Пуги I и II — параллельные, но путь II состоит из
трех промежуточных С1адий с невысокими энергиями активации.
Несмотря на энергетическую равноценность обоих nyieft реакция
проходит по в 1 орому пути, как более быст рому.
Ускоряющее действие кашлизатора не обязательно должно
состоять в понижении энергетического барьера. Катализатор
может способствовать достижению необходимой для
взаимодействия молекул ориентации, повышая энтропию активации.
§ 19. ОБРАТИМЫЕ РЕАКЦИИ И КОНСТАНТА РАВНОВЕСИЯ
Реакции, которые протекаю! одновременно в двух
противоположных направлениях, называются обратимыми. Обратимые
реакции имеют исключительное значение.
Рассмотрим реакцию
133
В начальный момеш времени, когда концеш рации вещества
Л и В максимальны, скороеи> максимальна С," [счением времени
копнет рации реагенюв уменьшаются и скороеп> падает, но
в любой момен1 времени она определяется уравнением
Скорое I ь взаимодейс i вия исходных вещее i в на швом с ко-
рентью прямой реакции vuv% а ее копсинту — коиегангой
скорости прямой реакции А'||р.
Реакция между А и В приводив к образоваьшо веществ D и F4
молекулы которых при столкновениях могу г вновь naib вещества
А и В. Чем выше концеш рации D и F, тем верояшее их взаимо-
лейс!вие и переход в вещес1ва А и В. Скорость этою процесса
назовем скоростью обратной реакции уобр. а ее
конским)—константой скорости обраiной реакции А'оГ)р. Скороеib обратной
реакции описывается уравнением
В начальный момент времени, когда в реакционной системе
нет веществ D и F, ообр —0; с течением времени vlxp уменьшается,
а 1>обр возрастает.
Изменение скоросчеи прямой и обрашой реакций во времени
представлено на рис. 74. По мере прохождения реакции
наступает такой момент времени, когда скорости прямой и обратной
реакций делаю 1ся равными, кривые 1>пр и уобр сливаются в одну
прямую линию, параллельную оси времени, а скорость
обратимой реакции становшея равной нулю. Такое состояние системы
называется состоянием равновесия. При равновесии
концентрации всех участников реакции постоянны и не изменяются со
временем, хотя одновременно осуществляю 1ся прямая и
обратная реакции. Концентрации (молярные) веществ в состоянии
равновесия принято обозначать формулой вещества в
квадратных скобках. При равновесии
поэтому равны и правые части уравнений:
V
*np[A][B]=*o6p[D][F].
Отношение двух постоянных величин
. ^'нр/^обр есть также величина постоян-
t пая. Ее обозначают символом К и на-
Рис 74. Скорость прямой зывают константой химического рав-
и обра i ной реакций НОвесия'
134
[а] [в] w
Вообще говоря, все химические реакции обратимы. Если
в реакционной смеси анализ не обнаруживает присутствия
продуктов реакции и если есть уверенное ib, чго смесь
равновесная, т. е. скорое ib реакции не заторможена, то говорят,
что реакция не проходит и ее константа равновесия крайне
мала. Наоборот, если после осуществления реакции в
реакционной смеси не удается обнаружить исходных веществ, то
процесс считается необратимым и его константа равновесия
очень велика.
Понятие обратимой реакции не соответствует
термодинамическому понятию «обратимый процесс». Обратимые химические
реакции являются обратимыми в термодинамическом смысле
только в непосредственной близости от состояния равновесия,
когда скорость прямой и обратной реакций отличаются на
бесконечно малую величину.
Напишем уравнение какой-либо реакции в общем виде:
В уравнение G1) концентрации веществ входя! в степенях сгехиометрических
коэффициентов. Поскольку любую сложную химическую реакцию можно
представить в виде ряда последовательных простых реакций, то для каждой из них
можно записать кинетические уравнения прямой и обратной реакций и
соответствующие им выражения для константы равновесия При суммировании
последовательных реакций их константы равновесия перемножаются и поэтому в
суммарное выражение для константы равновесия равновесные концентрации войдут
в степенях их стехиомстрических коэффициентов:
PNFK
[ANB]*
Смысл этого уравнения можно выразить так: для одной и той же
температуры отношение произведений равновесных концентраций
(в степенях их стехиометрических коэффициентов) веществ
в правой и левой частях уравнения химического процесса
представляет постоянную величину.
Это вторая формулировка закона действующих масс.
Постоянство состава системы не означает состояния
равновесия. Смесь Н2 и О2 может при комнатной 1емпературе
сохраняться бесконечно долго, но при этих условиях она
не находится в химическом равновесии. Достаточно
преодолеть активационный барьер (например, внесением
катализатора), как реакция осуществляется с чрезвычайно высокой
скоростью.
Истинное химическое равновесие характеризуется
следующими признаками:
1. При сохранении внешних условий состояние системы не
изменяется во времени.
135
2. При изменении условий (введение дополнительных
количеств реагирующих веществ, изменение давления или
температуры) сжмема приходи! к новому состоянию равновесия.
3. К состоянию равновесия система может подойти с
противоположных сторон.
Так, например, равновесное состояние в системе Н2(г)-Н2(г) = 2Н1(г),
характеризующееся (для одной и той же температуры) константой равновесия
[НЮ)]2
К-
[Н2(г)][Ь(г)]
может быть достигнуто как исходя только из веществ Н2 и 12, стоящих в левой
части уравнения, так и только из чистого HI. В первом случае Н2 и 12 будут
реагировать до момента достижения смесью Н2, 12 и HI равновесного состава, во
втором случае иодоводород будет распада! ься на водород и иод до дост ижсния
того же равновесного состава смеси
Численные значения концентраций Н2, 12 и HI могут быть очень различны
в зависимости от исходных составов, но всегда для любого равновесного
состояния соотношение концентраций Н2, 12 и HI (или любых дру«их веществ) таково,
что значение константы равновесия К постоянно
Если vnp>vo6p или кпр>кобр, то при равновесии концентрации
продуктов реакции выше концентраций исходных веществ и
равновесие, как принято говорить, смещено вправо. При vo6p>vnp
или ко6р>кар равновесие смещено влево, т. е. в сторону исходных
веществ. В первом случае, если анализом не удается обнаружить
вещества А и В, считается, что реакция полностью прошла; во
втором случае, если не удается обнаружить вещества D и F,
считается, что А и В не реагируют друг с другом.
Увеличением или уменьшением концентрации участвующих
в реакции веществ можно изменять скорости прямой и обратной
реакций и смещать равновесие в ту или иную сторону. Например,
в реакционной равновесной системе
A + 2B-2D
увеличим концентрацию В в 3 раза. До ее изменения скорость
прямой реакции ^пР = ^пр[А][В]2; после увеличения концентрации
В в 3 раза ^р = А:пр[А]C[В]г. Отношение
*[А1C[В]J 2
показывает, что скорость прямой реакции возросла в 9 раз; это
должно привести к увеличению концентрации продукта реакции
D. Если увеличить концентрацию D в 2 раза, то скорость
обратной реакции повысится в 4 раза (в степени стехиометрического
коэффициента) и равновесие сместится в сторону образования
веществ А и В. Уменьшение концентрации вещеегва D приводит
к смещению равновесия вправо.
Независимость константы равновесия от концентраций веществ,
участвующих в равновесии, рассмотрим на примере системы, сосчоящей из кислорода
136
и оксидов серы SO2 и SO3. Безразлично, как будет записано уравнение —
продукты или исходные вещества будут помещены справа от знака равенства, так как
в равновесной системе продуктами или исходными веществами их называю!
только условно, по месту в уравнении реакции. Запишем оба варианта уравнения
реакции.
2SO2 ^O2-2SO3 (I) и 2SO3 = 2SO2 + O2 (II)
Выражения для константы равновесия обоих вариантов
[SO3]2 [O2][SO2]2
'~[Oa][SO2J2'*"—W3)~
При увеличении концентрации кислорода для сохранения постоянными
констант АГ, или Ktt концентрация SO2 должна уменьшиться, a SO3 возрасти, т. е
равновесие в любом случае смещается в сторону образования SO3
При увеличении давления в системе в п раз произведение [O2][SO2]2
возрастает в пп2~пъ раз, а произведение [SO3r—только в п2 раз. Для сохранения
постоянными констант К\ и К» произведение [O2][SO2]2 должно уменьшиться,
a [SO3 ]2 возрасти, т. е равновесие сместится в сторону образования SO3.
Влияние изменения внешних условий—концентрации,
давления, температуры и других факторов—определяется принципом
Ле Шателье: если на систему, находящуюся в равновесии,
производится воздействие, равновесие смещается в сторону той
реакции (прямой или обратной), которая ослабляет воздействие.
Увеличение концентрации кислорода в равновесной системе
SO2(r) — O2(r)—SO3(r) приводит к тому, что в системе начинается реакция (I),
связанная с уменьшением его концен грации, и равновесие смещается вправо.
При увеличении давления равновесие сметается в сторону реакции, которая
сопровождается уменьшением числа молей газообразных веществ. Увеличение
давления в рассматриваемой системе приведет к уменьшению объема благодаря
процессу (I), так как число молей газообразных веществ уменьшается и опять
равновесие смещается вправо.
Для реакций, проходящих без изменения объема, например,
между газообразными водородом и хлором, изменение давления
практически не сказывается на смещении равновесия.
Разбавление равновесной реакционной смеси инертным газом действует
на равновесие аналогично уменьшению давления. Для
равновесий между газообразными веществами удобнее пользоваться не
молярными концентрациями (моль/л), а парциальными
давлениями газообразных веществ.
Константа равновесия, выражаемая через концентрации,
обозначается символом Кс, а константа равновесия, выражаемая
через парциальные давления веществ,—символом Ку
Наличие равновесных стадий в последовательных реакциях
часто изменяет вид ожидаемой кинетической зависимости.
Например, скорость распада озона
2О3(г) = ЗО2(г)
зависит не только от концентрации озона, но и от концентрации
кислорода и описывается следующим экспериментально
найденным кинетическим уравнением:
137
Полагают, что распад озона проходит через две стадии:
первая стадия отличается быстрым достижением равновесного
состояния, вторая стадия — медленная:
Оз = О2 + О (I) быстрая, равновесная,
О + О3 = 2О2 (II) медленная.
Скорость второй стадии определяет скорость процесса
в целом:
v = k2CoCOy
Концентрация атомов кислорода зависит от численного
значения константы равновесия первой стадии:
Подставив в уравнение скорости равновесную концентрацию
кислорода
получаем выражение, правильно описывающее результаты
эксперимента:
и-к к С°ъс -к?С°3
V — /С2Л1—- СОз — Л —— .
Это указывает на ю, что предложенный механизм реакции,
вероятно (но не обязательно), правилен.
Ранее было показано, что константа равновесия есть
отношение констант скоростей прямой и обратной реакций. Константа
скорости реакции связана с энергией активации и энтропией
активации выражением k^Ze RreR. Подставим его для констант
скоростей прямой и обратной реакций в формулу константы
равновесия реакции
A+B=C+D
v, [CHDJ *пр Zlipe*e ^
I/*JIDJ лобр ^обр '-'обр
Отношение Znp/ZoQP—величина постоянная. Перенесем ее в
левую часть и будем считать К^ константой равновесия К:
138
А-р
Проведем преобразования:
Rl
Разность Еоър-
По этому
-?пр равна
AS
K=eRe
-АН.
АН
ГА 5-АЯ
"~~RT
где AS—разнос г ь эшропий активации прямой и обра той
реакций. Возьмем натуральный логарифм правой и левой час гей
уравнения #
TAS-AH
1 ъ- I R?" TAS-AH
In A = lru' Л| =
о1 куда
- RT\n K = AH-
Обозначим А //— ТА S через АО':
Функция A//— TAS, как известно, называйся изменением изобар-
но-изотермического потенциала АС. Для стндарпшх условий
При замене натурального логарифма на десяшчпый
(In« = 2,303 Iga) и подстановке значения газовой постоянной
/? = 8,314 Дж/(Кмоль) получаем удобную для использования
формулу
ДС° = А//°- TAS°= - 19,14 7'1g Kr
Проведенный вывод показывает, что Konciania равновесия ие
зависит oi энергии ак!ивации. От нее зависш лишь время досж-
жеиия сосюяния равновесия. Поэтому катализаторы не влияют на
коне lain у равновесия, а снижая энергию активации и повышая
энтропию акшвации, ускоряют лосжжение равновесия. Констан-
ia равновесия зависит от ieMiiepaiypbi, oi изменения эшальиии
(тепловою эффекта) в реакции и oi изменения энтропии.
139
Из полученных уравнений следует: чем больше значение
константы равновесия, т. e. чем больше в равновесной смеси
содержание веществ, сюящих в правой части уравнения реакции
(продукты), тем больше отрицательное значение АС Если константа
равновесия меньше единицы, то AG положительная величина.
Чем меньше К, т. с. чем меньше в равновесной смеси содержание
продуктов реакции и больше содержание веществ, стоящих в
левой части уравнения, тем больше значение АС, т. е. оно связано
со степенью смещения равновесия. Знак АС указывает на
направление смещения равновесия.
При выводе формулы изменения изобарно-изотермического
потенциала предполагалось, что эксперимент проводится при
постоянном давлении, и энергетические уровни исходных веществ
и продуктов обозначались через Н{ и Н2, а их изменение АН
называлось изменением энтальпии. Константа равновесия
выражалась через парциальные давления компонентов. Если
эксперимент проводится при постоянном объеме, энергетические
состояния исходных веществ и продуктов принято обозначать через
U{ и С/2, а изменение AU называется изменением внутренней
энергии. В этом случае изменение потенциала системы
обозначают обычно через AF и называют изменением изохорно-изотер-
мического потенциала:
AF=AU-TAS= -19,14 TigКс(К=пост).
В уравнении
AH-TAS=-RT\nK
константа равновесия К определяется экспериментально при
заданной температуре Т. Таким образом, уравнение имеет два
неизвестных АН и AS, которые можно рассчитать, если известно
минимум два значения константы равновесия К± и К2 при двух
температурах—соответственно Тх и Г2.
Решая систему двух уравнений
АН - T2AS = -RT2\nK2,
находим АН:
Подставляя найденное значение АН в одно из уравнений
системы, рассчитывают AS.
Зная АН и AS реакции, можно рассчитать АС при
интересующей температуре и величину константы равновесия.
140
Вместо отношения констант равновесия можно пользоваться
отношением значений любых свойств, пропорционально
связанных с константами равновесия.
Часто последнее уравнение записываю! в другом виде:
Эта формула интересна тем, что показывает влияние
температуры на равновесие. Очевидно, что для одного и того же
температурного интервала (Т2 — Т{ =гюст) чем больше
численное значение АЯ, тем сильнее сказывается влияние температуры
на равновесие. Если реакция не сопровождается изменением
энтальпии, то температура не должна влиять на состояние
равновесия.
При повышении температуры в реакциях с выделением
теплоты (А//<0) член lnjK2/Ki<0, тогда К2/К1<\ и К2<К^
т. е. константа равновесия К2 при температуре Т2 меньше
константы Кх при температуре Тг. Следовательно, для реакций
с Д//<0 при повышении температуры равновесие смещается
в сторону образования веществ, стоящих в левой части
уравнения.
При Д#>0 \пК2/Кх>0, К2/К{>\ и К2>Кх. Следовательно,
для реакций с А#>0 при повышении температуры равновесие
смещается вправо.
В общем случае можно сформулировать правило: при
повышении температуры в равновесной химической системе равновесие
смещается в сторону реакции, сопровождающейся увеличением
энтальпии, т. е. в сторону процесса, препятствующего внешнему
воздействию.
Это есть применение принципа Ле Шателье к влиянию
темпера 1уры на равновесие. Другими словами, повышение
температуры приводит к уменьшению константы равновесия реакции
с уменьшением энтальпии и к росту константы равновесия
реакции с увеличением энтальпии.
Уравнение AG = A//— TAS является линейным уравнением
зависимости AG0 от температуры при предположении, что
Д#° и Д5° от температуры не зависят. Однако в
действительности Д#° и А5° зависят от температуры (и тем сильнее,
чем больше сумма теплоемкостей продуктов реакции отличается
от суммы теплоемкостей реагентов). В большинстве случаев,
даже несмотря на зависимость А//0 и А5° or температуры,
обнаруживается линейная зависимость АС0 от температуры.
Это объясняется тем, что температурные поправки в членах
Д//° и А5° имеют противоположные знаки. Таким образом,
для большинства случаев эт уравнение можно считать
уравнением прямой.
141
§ 20. ПРОВЫЕНИК ХИМИЧЕСКОЙ РЕАКЦИИ ПРИ СТАНДАРТНЫХ
И 1ШГ\1ШР1НМ\ ХЛОВИЯХ
Для сравнения олнотипных реакций их след>сч рассма грива п>
при сопоставимых условиях. За исковые принято считать
стандартные условии При "них условиях концетраимн всех вещее <п
в системе равны по I моль/л или их парциальные давления
составляю! по 101 325 Па. проичсанио химич^с^и реакции
сопровождается стандартным изменением ичобарною ио1енциала
AG0 или стандартным изменением изохорного потенциала AF0
соо1всгсгвенио Если в дополнение к 'л им условиям процесс
совершается при стандартен leMucpaiype 298 К B5 С), ю
изменения потенциалов обозначаются АО 298 и Д^298-
В связи с введением СИ константы равновесия реакций, сопровождающихся
изменением числа молей газообразных веществ. 6>uvi отличаться от значений,
вычисленных ранее A aiM- 101 325 Па) По>гому. чтобы парциальные давления
комионешов, вычисленные in конекппы равновесия, совпадали в СИ и в прежней
системе, в констаму равновесия Кр подставляю гея значения парциальных
давлений компонентов, выраженные не в Па. а в атм. или. что ю же самое, частные oi
деления парциальных давлений в Па на 101 325
В качестве примера рассмотрим равновесие между диоксидом
азота NO2 и его димером N2O4.
Изучаемая реакция ишерсенл во многих оiношениях Диоксид NO2 — «аз
красно-бурою uBCia Цвет обус ювлен одним неспаренным )iuciponoM аюма
азота Впагодаря эгому хтекфону две молекулы NO2 обьединяются в димер
|ч|:Од—бесцветный газ Связь между аюмами а юта осуществ 1яе!ся электронной
парой Молекула N2O4 — плоская, расстояние между атомами азота довольно
велико AЛ5 10 10 м), однако вращение ipynii NO2 вокруг оси N — N жергетичс-
ски сильно затруднено Потому прешолакшч. что атомы азота связаны тг-
связыо
При димеризаиии и обратном процессе распада N2O4 crpyKiypa NO2 не
искажаемся, >ют О — N—О не изменяемся A3^ ) и скорости процессов дпмериза-
ции и распада ве жки
Энергия iiKiиьашш процесса личеришцим очень тика, но сюрический факгор
равен 0.010. чго неско п>ко ограничивает скорость ре акции Встсдспшс небо н.нтой
энергии сйя$и между атомами азота ~жер1ия активации реакции распада лнмера
также ненстика Поному равновесие очень бысмро смешается в ту или иную
сюрону при изменении температ>ры. давления или концентраций peaiируюпшх
вещест в
О смещении равновесия в iу или ин\ю сторон) можно судить
по изменению UBeia реакционной смеси:
2 240 P
крлсио- бесиисчный /}no,
oypi lit
При повышении 1смперат>ры красно-бурая окраска газовой
смеси NO2 — N?O4 становится все более темной, чю юиорт
о CMeuieiiHH равновесия в стороя> образования NOi и о юм. чго
реакция в соогвеютвии с принципом ^ie Ulaiejue сонровожлае1ся
уменьшением )нтальпии.
142
Подтвердим качественные выводы расчетами. Ниже приведены
термодинамические характеристики оксидов
кДж/Moib Дж/(К моль)
NO, (г) . . 33,47 240,2
N2O4@ . . • 9,62 303,8
Изменение энтальпии в реакции при стандартной температуре А//рЦИи298 =
= Д#?брм2о4-2Д#?брЖ>2=9,62-2-33,47= -57,32 кДж/моль.
Знак изменения энтальпии совпадает с предсказанным по изменению окраски
газовой смеси при повышении температуры. Изменение энтропии А5рЦИи298 =
= SSo4-25go2 = 303,8-2 240,2=-176,6 Дж/(К моль)
Энтропия продуктов реакции меньше энтропии исходных веществ Продукты
реакции характеризуются меньшей степенью беспорядка по сравнению с
исходными веществами Если в качестве движущей силы реакции рассматривать только
изменение степени беспорядка, не учитывая энергетического вклада, то реакция
самопроизвольно должна была бы протекать в сторону образования диоксида
азота как вещества, имеющего большую степень беспорядка. Если же
рассматривать только энергетические изменения как движущую силу реакции, то реакция
должна самопроизвольно протекать в сторону образования веществ,
обладающих меньшим запасом энергии, т е. в сторону образования димера
Зависимость изобарного потенциала от температуры
описывается уравнением
А/^0 ALT ТАС GH 1Л i 1 ПИ ЛТ
При комнатной (стандартной) температуре 25°С B98 К)
изменение изобарного потенциала
ДС?298= -57320+176,6-298= -57320 + 52627= -4693 Дж/моль.
В этих условиях смесь с парциальными давлениями NO2 и N2O4
по 101 325 Па самопроизвольно превращается в равновесную
газовую смесь с уменьшением изобарного потенциала на 4693
Дж/моль.
Рассчитаем константу равновесия при 298 К:
А<?298 = -8,314-298-2,303lg?p= -5709lg#p= -4693 Дж/моль,
откуда
4693 _
= 5709~~
Следовательно, из исходной газовой смеси с pN c^—Pno,— 1 атм
самопроизвольно при 298 К получается смесь cVno4 = 6,64 pxo,
(атм).
При охлаждении (поместим стеклянный сосуд в гающий лед)
окраска смеси NO2 и N2O4 становится бледно-бурой, почти
бесцветной, что говорит о смещении равновесия в сторону
бесцветного N2O4.
143
Учитывая линейную зависимость изменения изобарного
потенциала от температуры, рассчитаем его для 0° С B73 К):
= -57320+176,6-273= -57 320+48212= -9108 Дж/моль.
При понижении температуры изменение изобарного
потенциала становится все более отрицательной величиной. Вероятность
самопроизвольного протекания реакции димеризации
возрастает. Константа равновесия при 273 К равна:
=-8,314-273 -2,303 \gKp= -5230lgKp= -9108 Дж/моль
lgKp = ^ 1,741
Кр=\0и1*1 ==55,14 атм.
Константа равновесия при понижении температуры возросла.
При 273 К из смеси с Pn,o4=Pno2=1 атм самопроизвольно
с уменьшением изобарного потенциала на 9108 Дж/моль
получается смесь с/?No4 = 55,14 p^Oz (атм).
При 0° С выход димера возрастает. Если бы была поставлена
задача получать N2O4 из NO2, то следовало бы пользоваться
наиболее низкими температурами (если только скорость реакции
при понижении температуры не слишком сильно замедляется).
Посмотрим теперь, как поведет себя реакционная система при
повышении температуры. Поместим стеклянный баллон со
смесью в кипящую воду. Окраска усилится, что свидетельствует
о смещении равновесия в сторону образования красно-бурого
NO2. При 100°СC73К)
AG§73 =- 57 320 + 176,6 • 373 =- 57 320 + 65 872 =+ 8552 Дж/моль.
При этой температуре изобарный потенциал реакции
становится уже положительной величиной — член TAS0 превосходит
член А#°. Равновесие смещается в сторону веществ с большей
степенью беспорядка. Положительная величина изменения
изобарного потенциала реакции делает маловероятным протекание
реакции вправо (при стандартных давлениях компонентов).
Константа равновесия при 373 К равна:
3=-8,314• 373-2,303lgtfp=-7145lgtfp=:8552 Дж/моль
Следовательно, при 100 С из исходной газовой смеси
с pNo2 =Лч о4 = 1 атм получается смесь с pNO4 = 6,354 • 10 р?о,. При
этом изобарный потенциал изменяется на 8552 Дж/моль.
По сравнению с ранее рассмотренными температурами в
равновесной газовой смеси парциальное давление N2O4 значительно
144
меньше и равновесие смещено в сторону исходных веществ.
Самопроизвольно протекает реакция распада димера.
При какой температуре состав исходной газовой смеси
с p^o=pNo=\ атм не изменяется и равен составу равновесной
газовой смеси? Этому условию отвечает ДС° —0. Поэтому
°=-57 320+176,6 7=0, откуда
При 5ГС исходная газовая смесь является одновременно равновесной
смесью Скорости прямой и обратной реакций равны Движущая сила реакции равна
нулю, так как при этой температуре энталышйный и энтропийный члены равны
ГЛ5.
При более высокой температуре TAS°>AH° и реакция протекает в сторону
образования NO2 благодаря увеличению беспорядка в системе
N2O4 = 2NO2, Д//°>0.
Этот процесс проходи i с поглощением геплоты и не может быть объяснен
принципом Бертло—Томсена.
При температуре ниже 51° С реакция протекает в сторону уменьшения
энтальпии.
и подчиняется принципу Бертло—Томсена
Если взять чистый диоксид, то он димеризуется при любых условиях, но
полнота прохождения димеризации зависит от температуры* при более низких
температурах большая часть NO2 превратится в димер, при высоких — очень
небольшая.
Чистый димер N2O4 (г) также при любых условиях распадается, но при
низких температурах распадается незначительная его часть, при высоких —
большая часть.
При любой температуре превращение одного оксида в другой
происходит до тех пор, пока не достигается состояние равновесия
с одной строго определенной для данной температуры
константой равновесия. Можно брать в качестве исходных различные
смеси. В зависимости от их состава и температуры процесс идет
в ту или другую сторону до достижения равновесия. В любом
случае, если только заведомо не берутся равновесные смеси,
имеет место процесс, который протекает самопроизвольно.
Договоримся считать термодинамически самопроизвольным
процесс, который протекает в смеси компонентов с их
парциальными давлениями по 1 атм A01325) Па или концентрациями по
1 моль 1л в направлении, при котором изобарный или изохорный
потенциалы системы уменьшаются.
Например, в системе NO2 — N2O4 при температурах ниже
324,6 К самопроизвольным является процесс димеризации; при
температурах выше 324,6 К самопроизволен противоположный
процесс распада димера.
Из этого примера можно сделать выводы: при низких
температурах вероятны реакции, идущие с выделением теплоты или
145
уменьшением энтальпии, при высоких температурах вероятны
реакции, в которых энтропия возрастает (степень беспорядка
возрастает).
Результаты расчетов равновесия NO2 — N2O4 приведены
в табл. 11.
Таблица 11
/, °С
0
25
51
100
г. к
273
298
324
373
Дж/моль
-57320
-57320
-57320
-57320
Д5°,
Дж/(моль К)
-176,6
-176,6
-176,6
-176,6
TAS0,
Дж/моль
-48212
-52627
-57320
-65872
Дж/моль
-9108
-4693
0
+ 8552
АГР, атм *
55,14
6,637
1
0,06354
Из таблицы видно, что для данной реакции повышение
температуры на 100° С приводит к изменению изобарного
потенциала почти на 20 000 Дж/моль и понижению константы равновесия
в 1000 раз.
Часто требуется знать состав равновесной газовой смеси.
Проведем расчет состава для равновесия NO2 — N2O4; принципы
этого расчета могут быть использованы и для других равновесий.
Пусть р—общее давление в смеси. Тогда Pno2+Pn2o4=P и константа
равновесия примет вид
Решим уравнение относительно рмо2'
Рассчитаем парциальные давления компонентов при р— 1 атм. Это—условия не
стандартные (при стандартных условиях р=^о2+^2о4=1-Ы =2 атм), но
константы равновесия, вычисленные при стандартных условиях (табл 11), сохраняют
свои численные значения и при иных концентрациях и давлениях.
При 298 К Кр=6,637 атм
-1±VH-4*6,6371
1 2-6,637 ™
±^/27,548 -1 ±5,2486
13,274 13,274
рНо2=0,32 атм
или
146
С юловаre 1ьно, при екждаршой icvuicpai>pc и общем давлении I01 325 Па
пр" гыгноиосип обратимей смесь с илрчильными швюниямм /ч<»,— 42450 Па
и /ч о ~ 10) 325- *2 4^0 ^68 8X0 Па Pe$v !ыаты раосюв гшршш imimx давлений
kom'uviouioh в равновесных га юных смесях при рамичныч ючпературах
приведешь и мо 1 12
0
2S
51
100
"v :: 1
-9108
- 4693
0
'- 8552
П .~ ]
0.544 10 '
0,654 10 J
0,986 10 *
0,625 10 "
Г ' ^ 1
12 760
62 640
95610
Та б л и на 12
Р\ о,* Па
88 570
68 880
Я8 690
5 720
При повышении температуры на 100 константа равновесия уменьшается
почти в 1000 раз парциальное давление NO2 возраеиет в 7.5 рача. а парциальное
давление N2C>4. уменьшается в 15 раз
В практической научной работе сгандаригые условия
проведения реакций почш никогда не осуществляются. Чаще всего
приходится иметь дело с произвольными количествами компонентов
и определять, возможна или невозможна реакция в данном
конкретном случае, каковы константа равновесия и выход вещества
(зависящий 1акже о г скорости реакции).
При неетандаршых условиях AF и AG реакции
вычисляются но формулам
7
Рн
19.14 Т lg~~
У4 />в
В тгих уравнениях.
AG — изменение изобарного потенциала (ДжЧю.иО при любых
коицешрациях и т парциа м>иых давлениях комнопешов
реагирующей системы.
AG — изменение июоариою iioieiiimaja (Джмо1ь) при стан-
дар п!ых условиях, С | -СВ — Сп = Сн= 1 моль л,
Р » ' Рп = Рп = Ри=\ aiM,
AF и AF° — изменения изочорного iioicHmiaja,
i ¦ С н. ( /;, ( п— iio»>i>u копис». i рицин ivojb, о вешесч* при
нееымдаршыч уоюпняч.
/* <* 14. PihPii— нобые плрмиачьиые лав юияя (ai\i)
н\)н исокшларшых чс:и»ьиях.
147
Приведенные термодинамические соотношения называются
уравнениями изотермы химической реакции.
При нестандаршых условиях реакция проходит при условии,
что AG<0.
Если в уравнение изотермы подставить
AG = -RT\nKp,
то получим соотношения
р\рьъ
PdPh
При помощи этих соотношений по известной константе
равновесия и заданным парциальным давлениям компонентов быстро
определяется знак изобарного потенциала и, следовательно,
возможность прохождения реакции.
Если исходная газовая смесь характеризуется отношением
РоРн
то дробь за знаком логарифма будет меньше единицы, AG<0
и реакция оказывается термодинамически возможной. Это
означает, что в системе имейся избыток исходных веществ или
недостаток продуктов по отношению к их равновесным
парциальным давлениям и система самопроизвольно переходит в
равновесное состояние благодаря химическому процессу.
При
дробь за знаком логарифма равна единице, Д<7 = 0 и взятая
газовая смесь является равновесной.
При
PdPu
AG>0, в системе имеется избыток продуктов по отношению
к равновесному состоянию и прохождение реакции в прямом
направлении невозможно. Реакция проходит в обратном
направлении до тех пор, пока не будет достигнуто равновесное
состояние.
148
Для реакции димеризации диоксида азога при 25 С
ДС= -4693 Дж/моль.
Определим возможность прохождения реакции при общем давлении
101 325 На и при следующих начальных парциальных давлениях веществ
а) /)N?Oj--= 100000 Па. /чс>2=П25 Па,
б) /?N2oa = 68880 Па. /;NO; = 32450 Па:
») /Чо4- П25 Па- /N,o4=W0000 Па
Решение
a /?N,o4^0,9869 лм. /?No2 = 0,OI31 атм,
0,9869
AG- -4693 {• 8,314 298 In— 5=-4693 + 21 459*16 770 Дж;моль
Положительное значение АО' твори г о невозможности прохождения реакции
димеризации Наоборот, при данных условиях имеет месю распад молекул
N2O4. который будем продолжаться до достижения системой состояния
равновесия При и ом изобарный исхенпиал системы изменится на 16770 Дж/моль
б. /7n2o4 = 0,6798 атм. pNO2 =0,3202 атм,
0,6798
АС= -4693 +-8,314-298 In — j= -4693 + 4689*0 Дж/моль.
Исходная «азовая смесь является равновесной
в рк^4 = 0,0131 «им, pso% = 0,9869 атм.
АО =-4693 + 8,314 298 In М!^ -4693-10 681 ^ - 15 370 Дж/моль
0,9869
В данном случае АО<0 и процесс димеризации проходит до тех пор, пока не
будет достигнуто состояние равновесия при этом изобарный потенциал
уменьшится на 15 370 Дж/моль
Анализ большого числа экспериментальных данных
показывает, что если отрицшельное значение стандартного изобарного
потенциала реакции превышает 40 кДж/моль, то реакция
возможна при практически любых нестандартных условиях. Однако
это ничего не говорит о кинетической осуществимое i и реакции.
Подчеркнем еще раз, что для реакций между
кристаллическими вещесл вами или для реакций между жидкими веществами
и в растворах без участия газообразных веществ, а также с
участием газов, но при условии, что не происходит изменение числа
молей газа (Дя = 0), численные значения ЛG и AF совпадают,
149
ГЛАВЛ 4
ФАЗОВЫЕ СОСТОЯНИЯ ВЕЩЕСТВ
И ФАЗОВЫЕ РАВНОВЕСИЯ
§ 21. ФАЗОВЫЕ СОСТОЯНИЯ И ФАЗОВЫЕ ПЕРЕХОДЫ
По ощущениям, которые производят различные вещества на
органы чувств человека, они могут бьпь разделены на три группы:
газообразные, жидкие и твердые (криааллические). Каждая из этих
групп указывает на определенное фазовое состояние вещества.
Под фазой понимаю 1 часть системы, которая имеет во всех
точках один и гот же состав и обладает одинаковыми
термодинамическими свойствами. Таким образом, фаза преде 1авляет
собой термодинамически равновесное состояние веществ—фазовое
состояние. Переход вещества из одного фазового состояния
в другое — фазовый переход—сопровождайся скачкообразным
изменением свойств, например плотности, теплоемкости,
внутренней энер! ии, энтропии и др.
Фазовые переходы разделяю 1ся на два класса. К фазовым
переходам первого рода относятся испарение, возгонка,
плавление, полиморфные переходы и i. д. Эти переходы
сопровождаются выделением или поглощением теплоты и изменением объема
фазы. Фазовые переходы второго рода не обладают этими
качествами. Примерами фазовых переходов второго рода могут
служить 1акие процессы, как переход железа из ферромагнитного
состояния в парамагнитное a-Fe—p-Fe при 769 С без изменения
кристаллической структуры металла'и при сохранении объема
фаз (изменение эшропии в этом переходе равно нулю); переход
металла в сверхпроводящее состояние; переход жидкого гелия
в сверхтекучее состояние.
Вода при атмосферном давлении A01 325 Па) при
температуре ниже 0' С находится в кристаллическом состоянии (лед). При
0 С она переходит в жидкое состояние (при эт их условиях
жидкость и лед сосуществуют в состоянии равновесия), в интервале
температур 0—100 С вода представляет собой жидкость, при
150
100' С она кипит (равновесие жидкость—газ) и переходит в
газообразное состояние.
Приведенные ниже значения плотное!и р. стандартной
теплоемкости Ср,298 и стандартной энтропии S298 Для воды
иллюстрируют зависимость свойств от ее состояния:
р I СМ Ч С ? *ад S 2ЧЯ
Дж'(К мо ib) Дж (К мо ib)
Газ 0,5977-10 A00 С) 33,6 188/7
Жидкость 0.99987@' С) 75,3 66,9
Кристалл 0,9168 @ С) 37.7 43,9
Из этих данных видно, что энтропия наиболее четко
указывает на повышение с i рук i урной организации воды при ее переходе
из газообразного состояния в жидкое и далее в кристаллическое.
Газы не имеют собственной поверхности и собственного
объема, они занимают полностью емкость того сосуда, в который
они помещены. Газы обладают неограниченной способностью
к расширению при повышении температуры и понижении
давления. Расстояние между молекулами в газах во много раз больше
размеров самих молекул, а межмолекулярные взаимодействия
слабы, и молекулы движутся практически независимо друг от
друга. Расположение частиц в газовой системе почти полностью
хаотично, и энтропия газа намного выше энтропии вещества
в других состояниях.
Жидкости сочетают свойства газообразного и
кристаллического состояний. Они имеют поверхность и собственный объем.
Молекулы жидкого вещества связаны между собой более
прочными межмолекулярными силами, и упорядоченность в
расположении частиц жидкой системы намного выше, чем у газов. В
некоторых жидкостях (вода) отдельные очень небольшие ее объемы
имеют упорядоченность, близкую к кристаллической.
Кристаллы, как и все твердые тела, имеют собственную
поверхность и объем, коюрые не изменяются в гравитационном
поле. Расстояния между частицами в кристаллах значительно
меньше, чем в газах, а межмолекулярные и межатомные
взаимодействия намного сильнее, чем в газах и жидкостях. Частицы
в кристаллическом веществе распределяются в некотором
закономерном порядке, образуя крис!аллическую решетку. Частицы,
составляющие кристаллическую решетку, достаточно прочно
закреплены на своих Meciax и совершают колебательные движения
около некоторых положений, называемых углами
кристаллической решетки. Энтропия вещества в кристаллическом состоянии
ниже энтропии жидкое!и и газа. Отличительная особенность
кристалла состоит в том, что его свойства неодинаковы в
различных направлениях (анизотропия). Для обозначения жидкого
и кристаллического состояний вещес!ва приняю также название
«конденсированное сост ояние».
151
Рис 75. Фазовые переходы веществ
при изменении температуры и
давления
Существование вещества
в том или ином фазовом
состоянии зависит от температуры
и давления. Повышение
температуры всегда приводит к
переходу кристалл—>жидкость—>газ
и росту степени беспорядка
в системе. Увеличение давления
оказывает на вещество обратное
влияние. На рис. 75 приведена
схема, показывающая переходы
из одного фазового состояния
в другое в зависимости от
температуры и давления.
Переход газа в жидкое и
кристаллическое состояния
называется конденсацией. Иногда
процесс перехода газа в жидкость
называют сжижением, а в
кристалл— десублимацией. Переход
вещества из жидкого или кристаллического состояния в
газообразное часто называют парообразованием.
Пар—газообразное состояние вещества в условиях
равновесия газовой фазы с жидкой или кристаллической фазой того же
вещества. Например, пары Н 2О могут представлять собой
равновесную систему водяной пар—жидкая вода или водяной пар—
лед («водяной пар над льдом»).
Испарение—процесс парообразования, совершающийся на
поверхности жидкости (или кристалла). Если пар образуется при
испарении жидкости не только с поверхности, но и в ее объеме,
то происходит кипение. При кипении давление пара над
жидкостью равно внешнему давлению (атмосферному).
Переход вещества из кристаллического состояния в
газообразное называют сублимацией, или возгонкой. Переход жидкости
в кристалл—кристаллизация, или отвердевание. Процесс
отвердевания, протекающий при невысоких температурах,—
замерзание. Обратный процесс перехода вещества из кристаллического
состояния в жидкое—плавление.
Фазовые переходы сопровождаются выделением или
поглощением теплоты и значительным изменением энтропии. Если
фазовый переход вещества совершается при повышении
температуры (возгонка, плавление, испарение), то он сопровождается
поглощением теплоты и для него характерно увеличение
энтальпии, Д#>0. Энтропия вещества в результате такого перехода
возрастает, AS>0. Если переход совершается при понижении
температуры (конденсация, сжижение, отвердевание), то он
сопровождается выделением теплоты и для него характерно Д#<0.
152
Энтропия вещества при таком переходе понижается, Д5<0.
Принято при символе изменения энтальпии и энтропии указывать
название (в сокращенном виде) соответствующих фазовых
переходов, например А//исп, ASnjl. Кристаллические состояния одного
и того же вещества Moiyi различаться по свойствам и строению,
и тогда говорят, что данное вещееjво существует в различных
модификациях. Явление сущес!вования нескольких
кристаллических модификаций у данного вещества называется
полиморфизмом, а переход из одной модификации в другую—полиморфным
превращением.
Не следует путать полиморфизм с аллотропией — явлением существования
элемента в виде различных простых веществ независимо oi их фазового
состояния Например, кислород О2 и озон О3 — аллотропные формы кислорода,
существующие в газообразном, жидком и кристаллическом состояниях Графит
и алмаз—аллотропные формы углерода и одновременно его кристаллические
модификации Понятия «аллотропии» и «полиморфизма» совпадают для
кристаллического состояния простого
Направление полиморфною превращения, как и фазового
перехода, определяется соот ношением изобарных по1енциалов
фаз. Эти переходы совершаю 1ся в направлении уменьшения
изобарного потенциала, т. е. если АG<0.
Скорость перехода вещества из одного фазового состояния
в другое зависит от скороеiи подвода и отвода теплоты. Однако
при некоторых условиях фазовый переход может быть кинетически
заторможен, и поэтому получаются перегретые или
переохлажденные жидкости и пересыщенные растворы. Состояния
переохлажденного газа (пара) или перегретого кристалла не наблюдаются.
Скорости полиморфных превращений могут быть самыми
различными. При обычных условиях термодинамически
устойчива кристаллическая модификация углерода—графит. Тем не
менее переход алмаза в графит не наблюдается даже при
сравнительно высоких температурах, что обусловлено высокой энергией
активации перехода.
Термодинамически неусюйчивое при данных условиях
состояние вещества, коюрое может длительное время существовать
из-за кинетической неосуществимости перехода, называется ме-
тастабильным.
§ 22. УСЛОВИЯ ФАЗОВЫХ РАВНОВЕСИЙ
Возможность перехода вещества из одного фазового
состояния в другое (из одной фазы в другую) определяется одним из
общих законов химии и физики — правилом фаз Гиббса. Правило
фаз Гиббса применимо к равновесным системам и является
выражением второго закона термодинамики в приложении к фазовым
равновесиям.
153
Прежде чем перейти к определению условий фазовых
равновесий и рассмо!рению правила фаз Гиббса, введем некоторые
важные термодинамические понятия.
Система—тело или группа тел, находящихся во
взаимодействии и физически или мысленно обособленных or окружающей
среды, которая, в свою очередь, также является системой.
Свойства систем можно разделить на два типа по характеру
их зависимости от количества вещества. Экстенсивные
свойства—это свойства, которые суммируются при соединении тел.
Такими свойствами являкмся длина, объем, масса, внутренняя
энергия, энтальпия, теплоемкость, энтропия и др. Если масса
системы увеличивается в п раз, то во столько же раз возрастают
значения экстенсивных свойств. Экстенсивные свойства
пропорциональны массе системы.
Интенсивные свойства—это свойства, не зависящие от массы
системы. Температура относится к интенсивным свойствам —
температура некоторого тела сохраняется постоянной при
делении его на части. Давление—также интенсивное свойство. То же
касается и плотности, которая является производной от
экстенсивных свойств, а именно массы и объема. Если значение
экстенсивного свойства выражается на единицу массы или количества
вещества, то оно становится интенсивным. Таковы мольные
массы, объем, энтальпия образования вещества, изменение
энтальпии в реакции, теплоемкость, энтропия и г. п. Численные
значения этих свойств не зависят о г массы системы.
В зависимости от наличия или отсутствия в системе
поверхностей раздела между ее отдельными частями системы
классифицируют на гомогенные и гетерогенные.
Гомогенная система не имеет поверхностей раздела, т. е. это
сплошная, однородная система. Для гомогенной системы
интенсивные свойства постоянны.
Гетерогенная система—система, отдельные части которой
различаются по свойствам и разграничены поверхностями
раздела.
Гомогенной системой является, например, жидкая вода (при
этом не следует принимать во внимание поверхности раздела со
льдом, со своим паром или со стенками сосуда, в котором она
находится). Кусок льда, рассматриваемый как сплошное и
однородное тело и мысленно выделенный внутри другого большего
куска льда,— гомогенная система.
Гетерогенные системы составляют жидкая вода и лед, или
жидкая вода и пар, или одновременно жидкая вода, лед и пар.
Вода, лед и пар—это различные фазовые состояния одного
вещества—воды.
Фаза—совокупность гомогенных частей системы,
одинаковых по свойствам и ограниченных от других частей системы
поверхностями раздела. Гомогенные системы состоят только из
154
одной фазы. Гетерогенные системы содержа! больше одной фазы.
По числу фаз системы разделяю! на однофазные, двухфазные,
фехфазные и т. д. (многофазные). Например, смесь газообраз-
пых водорода, азога и аммиака Н2<1)—N2(I)—NH3(rJ, равновесие
в которой описывается уравнением
— гомогенная система, имеющая одну фазу.
Системы С(к)—О2A)— СО2(г) и СаСО3<к) — СаО<к) — СО2(Г),
содержащие кристаллические и газообразные вещества и
равновесия в которых описываются уравнениями
С(к) + О2(г) = СО2A,; СаСО3(к) - CaO(It) + CO2(r,,
— тсрогенные сисмемы. Первая из этих систем—двухфазная,
сосюящая из одной твердой и одной газовой фазы, вторая —
трехфазная, состоящая из двух твердых и одной газовой фазы.
Составные части системы — простые вещества или
химические соединения, входящие в систему, которые могут быть
выделены и могут существовать в изолированном виде. Составными
частями раствора хлорида натрия являются вода и NaCl, но не
ионы С1" и Na + , гак как они не могут бьпь выделены в
химических реакциях и не могут существовать изолированно.
Компоненты системы—это ie составные части системы,
равновесные концентрации которых можно изменять произвольно
(в некоторых пределах), не вызывая изменения числа и вида
фаз з системе. Иногда о такого типа компонентах говорят
как о независимых компонентах. Понятия «составная часть»
и «компонент» совпадают, если в системе нет химического
взаимодействия.
Число независимых компонентов—это наименьшее число
составных частей системы минус наименьшее число равновесий,
характеризующих все химические взаимодействия в
рассматриваемой системе, и минус число уравнений, необходимых для
расчета концентраций всех веществ в системе.
При высоких температурах равновесная система Н2(г)—
С>2<г) — Н2О(Г) имеет три составные части, а именно водород,
кислород и газообразную воду, но число компонентов равно
двум, так как имеет место равновесие:
При низких температурах (комнатных), когда химическое
взаимодействие не проходит (крайне низкая скорость реакции),
число компонентов в этой системе равно трем и совпадает с числом
составных частей системы.
При нагревании кристаллическою хлорида аммония в
системе NH4C1(IC)—НС1A)—NH3(I) устанавливается равновесие
Число составных частей этой системы равно трем, а число
155
независимых компонентов только единице, так как в системе
осуществляется равновесие*
и концентрации газообразных аммиака и хлороводорода связаны
равенством парциальных давлений /?\н3=^нс1- Однако если
нагревание хлорида аммония проводить в атмосфере 1азообразных
хлороводорода или аммиака или же в равновесную систему
ввесш дополнительные количества Э1их соединений, то число
компонентов возрастает и становится равным двум вследствие
неравенства парциальных давлений составных частей.
По числу независимых компонентов системы разделяю! на
однокомпонентные, двухкомттентные, трехкомпоиеитиые и т. д.
(многокомпонентные).
Равновесие любой сиаемы, гомогенной или гетерогенной,
зависит от условий ее существования—температуры, давления,
концентрации компонентов, наличия различных полей и др.
Число условий, которые можно менять (в определенных пределах)
без изменения числа и вида фаз в системе, называется числом
степеней свободы системы. Жидкая вода и водяной пар могут
находиться в равновесии при некоторых различных температурах
и давлениях, но каждой температуре отвечает строго
определенное давление и, наоборот, каждому давлению водяного пара
соответствует строго определенная температура. Следовательно,
равновесная однокомпонентная система жидкость — газ имеет
одну степень свободы.
По числу степеней свободы С системы разделяют на
безвариантные (С=0), одновариантные (С=1), двухвариантиые (С = 2),
трехвариантные (С—Ъ) и г. д. (многовариантные).
Температура и давление—это два важнейших фактора,
влияющие на состояние равновесия в системе. Кроме температуры
и давления на равновесие могут влиять и такие факторы, как
электрическое, магнитное, гравитационное поля.
Число степеней свободы определяет максимальное число
факторов, которые могут изменяться независимо один oi другого
и которые можно произвольно изменять в известных пределах, не
нарушая числа и вида фаз.
Число степеней свободы С равновесной 1ермодинамической
системы определяется по формуле:
где К—число компонентов (независимых);
ф — число фаз;
п—число внешних факторов, влияющих на равновесие
системы.
Это и есть правило фаз Любса A876 г.), которое
формулируется следующим образом:
156
число степеней свободы равновесной термодинамической
системы равно числу независимых компонентов системы минус число
фаз плюс число факторов, влияющих на равновесие.
Правило фаз позволяет по числу степеней свободы
предсказывать поведение системы при изменении одного, двух или более
внешних условий (при сохранении остальных параметров
постоянными) и вычислять максимальное число фаз, которые могут
находиться в равновесии при данных условиях. При помощи
правила фаз можно предсказать термодинамически, возможно
(или невозможно) существование изучаемой системы.
В обычных условиях влияние различных полей на равновесие
настолько незначительно, что его не учитывают и рассматривают
только два фактора: температура и давление, т. е. я = 2. Для
таких систем число степеней свободы равно:
Если в системе температура или давление сохраняются
постоянными, то число параметров состояния снижается на единицу.
Тогда
С=К-Ф+\.
Если же в системе поддерживаются постоянными и температура,
и давление (л = 0), то
Число степеней свободы может быть равно нулю или целому
положительному числу, т. е. С^О.
Так как число степеней свободы не может быть
отрицательным, то число фаз в равновесной термодинамической системе не
может быть больше /f+2 (в общем случае К-\-п).
Число степеней свободы равновесной системы возрастае1
с увеличением числа компонентов и понижается с увеличением
числа сосуществующих фаз.
§ 23. ДИАГРАММА СОСТОЯНИЯ ОДНОКОМПОНЕНТНОЙ СИСТЕМЫ
Для иллюстрации правила фаз Гиббса рассмотрим
простейший вариант термодинамической системы — однокомпо-
нентную систему. В однокомпонентных системах все фазы
содержат одно и то же вещество, существующее в различных
состояниях. Найдем максимальное число фаз, возможное в равновесной
однокомпонентной системе. Число фаз в системе максимально
только тогда, когда система безвариантна, т. е. при С=0 или
#Ф 2 0
Так как для однокомионентной системы /Г=1, то Ф = 3. Из
этого следует, что ни одно индивидуальное вещество не может
157
обра юна! ь сипемм и* б^пес чем «;к ч ;4!ьноь~еньр fpj's * 4iu
cymeciByei 10лько одно сочекпчю »»пч т'й 1емчсра1>ры и *ав-
ючн* »ipn ко юром ip>; фаш еинокомчо» сигмой сипгчч—
криссалл, жидкоеiь и j аз — мог v i начоди гься в равновесии V: ю-
вие равновесия ipex фаз сиаемм харак1еризустся так
называемой тройной точкой.
Малейшее изменение темпера*уры или давления
относительно значений 1(*мисри1уры для даи.юиим. отвечающих iройной
ючке, приводит к исчезновению одной или двух фаз и
превращению их в другие фазы
Однокомноненшые ежмемы. состоящие из двух фаз, voryi
быть трех типов:
кристалл—жидкоеib; крис«алл — нар: жидкость — нар
Число степеней свободы лвухфаз>?ой системы равно-
Это означает, что каждой температуре отвечает
одно-единственное значение давления и, наоборот, любое давление в
двухфазной однокомтюнен»ной системе реализуется только при
строго определенной температуре. Следовательно, нагревание любых
двух сосуществующих фаз должно сопровождаться олновремеп-
но строго определенным изменением давления, т. е давление
и температура двух фаз должны изменяться по с i рог о
определенной зависимости p~f(T), которая может бьпь выражена
уравнением или графически представлена кривой.
Равновесие между жидкой и газовой фазами вещества
магматически описывается уравнением зависимости давления
насыщенного пара над жидкостью от температуры, а графически
изображается кривой давления насыщенного пара над жидкостью
(рис. 76, а, кривая «ж = г»). Равновесие между кристаллом и
паром описывается уравнением зависимости давления насыщен-
Р\
Рис 76 Кривые фаговых равновесий {а) и диаграмма
состояния олнокомпонентной системы {б)
Символами к ж и г ибтнач^на сот во ici ценно криси i (ическая (!вор
дая) жидкая и f ловля (пареная) фл<»
158
ного пара над кристаллом от температуры и изображается
кривой давления пара над кристаллом (кривая «к = г»). Равновесие
между жидкостью и кристаллом выражается зависимостью
между температурой плавления вещества и давлением и
изображается так называемой кривой плавкости (кривая «к = ж»).
Каждая из трех кривых фазовых равновесий показывает те
соотношения между температурой и давлением, при которых
химические потенциалы двух сосуществующих фаз равны.
Очевидно, что точка пересечения этих кривых отвечает
значениям температуры и давления, при которых сосуществуют все
три фазы. Этому условию удовлетворяет только одна точка —
тройная точка. В тройной точке давление пара над жидкостью
и кристаллом одно и то же (рис. 76, б).
Если в состоянии, соответствующем тройной точке,
трехфазная однокомпонентная система безвариантна (С=0), то во
всех остальных точках любой из трех кривых система
обладает одной степенью свободы (С=1). Если система
двухфазная, можно произвольно изменять лишь одну переменную
величину (температуру или давление), другая же переменная
величина (давление или температура) будет изменяться как функция
первой.
Область, заключенная между кривыми, называемая полем,
характеризует совокупность значений температур и давлений,
при которых устойчива одна фаза. Так, между кривыми «к = ж»
и «ж = г» лежит поле жидкости (ж), между кривыми «ж = г»
и «к = г» лежит поле газа (г) и между кривыми «к = ж» и «к = г»—
поле кристаллического состояния (к) (см. рис. 76, б).
При любых значениях температуры и давления внутри поля
система однофазна. Число степеней свободы однофазной
системы равно:
Следовательно, можно изменять произвольно в определенных
пределах (в пределах поля) давление и температуру, не вызывая
при этом изменения числа и вида фаз.
Диаграмма, подобная представленной на рис. 76, б,
выражающая зависимость состояния системы и фазовых равновесий
в ней от внешних условий (и от состава для более чем одноком-
понентных систем), называется фазовой диаграммой, или
диаграммой состояния.
По диаграмме состояния легко определить характер
поведения системы при изменении внешних условий или же
предсказать число сосуществующих фаз при заданных внешних
условиях.
Пусть точка Кх (рис. 77) характеризует состояние
кристаллического вещества при температуре ТК1 и давлениирх. Процесс
нагревания вещества при постоянном давлении рх соответствует
159
h
Рис. 77. Графическое
определение температур
фазовых переходов в одноком-
понентной системе при
повышении температуры
)
перемещению точки Кх по прямой
КХПХИХГХ. На участке КХПХ
подводимая теплота поглощается
кристаллической решеткой вещества. Так как
давление в системе постоянно и изменяется
только один параметр состояния,
а именно температура, то число
степеней свободы следует определять по
формуле С—К—Ф+1. На участке КХПХ
число степеней свободы С=1, поэтому
изменение температуры не должно
приводить к изменению числа фаз, т. е.
система остается однофазной.
В точке Я,, т. е. при температуре
Тии прямая К1П1И1Г1 пересекает
кривую зависимости температуры
плавления от давления, точка Ях соответствует
началу процесса плавления вещества. В этой точке система
двухфазна и безвадэиантна, поэтому температура в системе при
заданном давлении не может изменяться до тех пор, пока все
кристаллы не превратятся в жидкость. Это означает, что подводимая
к системе теплота расходуется на плавление вещества, т. е.
количество подводимой теплоты равно энтальпии процесса плавления
(в расчете на 1 моль вещества):
А(к, = А(ж)-?пл или
А — А . A LJ
гх(к) ^*(ж)> и||пл*
После того как вся кристаллическая фаза превратится в
жидкость, система снова становится однофазной и одновариантной.
Дальнейшее подведение теплоты идет на повышение
кинетической энергии молекул жидкости.
Точке пересечения Их (см. рис. 77) соответствует начало
процесса равновесного испарения жидкости. Жидкость
испаряется при любых температурах, кипение же происходит только
при строго определенной температуре, при которой
давление насыщенного пара достигает значения внешнего давления.
Кривая давления насыщенного пара жидкости выражает не
только зависимость этого давления от температуры, но также
показывает зависимость температуры кипения от давления в
системе.
У всех веществ давление насыщенного пара с повышением
температуры возрастает, и на диаграммах состояния кривая
давления пара отклонена вправо от оси «р» (см. рис. 77). Увеличение
давления приводит к повышению температуры кипения, и
наоборот, при уменьшении давления жидкости кипят при более низких
температурах. Различие процессов испарения и кипения носит
160
скорее кинетический характер, поэтому понятия «энтальпия
кипения» и «энтальпия испарения» совпадаю! и в термодинамике
обычно говорят только об энтальпии испарения, хотя часто
относят ее к температуре кипения.
В точке #i система двухфазна; она состоит из пара и
жидкости. Поэтому при постоянном давлении система безвариантна
и температура в системе не изменяется. Вся подводимая теплота
расходуется на разрыв связей между молекулами жидкости и
перевод их в газовую фазу, т. е. количество теплоты есть энтальпия
испарения:
А(Ж) = А(Г); Л/Уисп.
Когда вся жидкость превратится в пар, система снова
становится однофазной и одновариантной. При дальнейшем
подведении теплоты она расходуется на повышение кинетической
энергии молекул газа.
Выше с помощью диаграммы состояния было рассмотрено
поведение вещества при давлении большем, чем при давлении,
соответствующем тройной точке. Если нагревать
кристаллическое вещество при постоянном давлении ниже тройной точки
(р2)> то до момента достижения давления насыщенного пара
кристалла система будет однофазной и одновариантной. Когда
температура повысится до значения, отвечающего температуре
на кривой давления пара, система скачкообразно превращается
в двухфазную, теряет одну степень свободы и при постоянном
давлении становится безвариантной, т. е. ее температура не
изменяется, пока все кристаллы не перейдут в газообразное
состояние. Это означает, что вся подводимая к системе теплота
расходуется на разрыв связей между частицами в кристаллической
решетке (вещество сублимируется):
А (К) = А (Г) — Q субл или
А(К) = А(Г); А//субл.
Таким образом, воспользовавшись диаграммой состояния,
можно предсказать поведение системы при нагревании.
Рассмотрим, как экспериментально определяются значения
температуры или давления фазовых переходов, г. е. как
осуществляется построение диаграммы состояния.
Пусть при постоянном давлении рх кристалл некоторого
вещества нагревают, начиная от температуры Тк, и через
некоторые определенные промежутки времени C0 с, 1 мин или 10 мин)
измеряют температуру этого вещества. Построением графика
температуры как функции времени Г=/(/) получают кривую
изменения температуры образца во времени—кривую нагревания
(рис. 78).
6 2452 161
Рис. 78 Кривые нагревания при постоянном
давлении рх A) и р2 B); р2>Р\
Участок К1П1
отвечает равномерному
нагреванию кристалла и
соответствует участку Кх #i
на диаграмме состояния
(см. рис. 77).
При некоторой
температуре Тп j начинается
плавление вещества и вся
подводимая к системе
теплота расходуется на
этот процесс. В
результате кристалл превращается
в безвариантную
двухфазную систему, и при постоянном давлении, заданном методикой
эксперимента, температура ее также должна оставаться
постоянной. Процесс плавления отображается на кривой нагревания
горизонтальным отрезком прямой ПХП\. Длина этого отрезка
зависит от скорости нагревания, массы вещества и энтальпии
плавления.
По окончании плавления всего вещества последующее
нагревание снова вызывает повышение температуры системы
(жидкости), чему соответствует участок П\ИХ на кривой нагревания.
Этот участок кривой характеризует однофазную и одновариант-
ную жидкую систему. При нагревании в определенный момент
жидкость приобретает температуру, при которой химические
потенциалы жидкости и пара становятся равными, и образуется
двухфазная система из жидкости и насыщенного пара. На кривой
нагревания отмечается горизонтальный отрезок И±И\Ь
свидетельствующий о постоянстве температуры системы.
После превращения всей жидкости в пар (газ) температура
системы при дальнейшем нагревании постепенно повышается,
чему отвечает участок И\ГХ. При достижении газом высокой
температ уры возможно протекание различных процессов распада
молекул, которые требуют дополнительных количеств теплоты,
что приводит к более или менее значительным изменениям
в форме кривой температура — время.
Проведение одного эксперимента, подобного описанному,
позволяет получить следующие данные: температуру фазового
перехода, равную температуре, которой соответствует
горизонтальный отрезок на кривой нагревания, и равновесное давление
фазового перехода, равное заданному в эксперименте давлению
(это может быть атмосферное давление, при котором проводится
опыт). При температуре кипения заданное давление отвечает
равновесному давлению насыщенного пара жидкости.
Одна кривая нагревания, полученная в рассмотренном выше
эксперименте, позволяет нанести одну точку соответствующей
162
кривой фазового превращения на диаграмме состояния. Для
построения всей кривой не обязательно проводить много опытов,
достаточно по крайней мере двух. Для этого проводят еще один
цикл нагревания кристаллического вещества при более высоком
давлении р2 и строят вторую кривую нагревания 2 (см. рис. 78).
На этой кривой имеются два горизонтальных участка ПгП'г
и И2И'2, отвечающие соответственно плавлению вещества и
кипению вещества при давлении р2.
Ниже показано, как, зная минимум две температуры фазового
перехода при двух давлениях, можно построить часть кривой
диаграммы состояния однокомпонентной системы.
Остановимся сначала на кривой давления насыщенного пара
жидкости. Вспомним, что равновесное давление пара численно
равно константе равновесия процесса его образования, т. е.
Кр=рГ. Константа равновесия зависит от температуры:
Составим систему двух уравнений для двух известных значений
давления пара (давление, задаваемое в опыте) при двух
температурах (температура, соответствующая горизонтальным участкам
на кривых нагревания):
-ДГ,1п/>,=Д#Л-:Г,Д5с\
Решением этой системы уравнений относительно неизвестных
величин Д#с и А5° можно найти значения энтальпии и энтропии
испарения, а также уравнение зависимости давления пара от
температуры:
RT\ AH°TAS~ или
/R;b=-AH /Л.
По выведенному уравнению, вычислив давления пара при
нескольких промежуточных температурах (интерполяцией) и при
нескольких температурах выше и ниже изученных
(экстраполяцией), легко построить искомую кривую нагревания.
При перемещении но кривой давления пара над жидкостью
в область высоких температур и давлений свойства газа и
жидкости все более сближаются и наконец наступает критическое
состояние, при котором различия между жидкостью и газом
исчезают. Достижение критического состояния отображается на
кривой критической точкой, которой отвечают строго
определенные критическое давление и критическая температура.
В критической точке все термодинамические свойства сосущее!-
вующих фаз становятся одинаковыми, поэтому система в
критической точке безвариантна. Выше критической точки ни при
б* 163
каком давлении не происходит разделения вещества на две
фазы — жидкую и газообразную.
Наряду с кривыми нагревания для построения диаграмм
состояния часто используют кривые охлаждения. Для этого
нагревают вещество до достаточно высокой температуры, дают ему
самопроизвольно охладиться и регистрируют температуру через
определенные промежутки времени.
При нагревании и охлаждении веществ возможны явления
перегревания и переохлаждения. Нагреть кристалл выше
температуры плавления не удается, но переохлаждение жидкости ниже
этой температуры—явление очень распространенное. Также
очень легко происходит перегревание жидкости выше
температуры кипения. В результате этих явлений температура,
соответствующая горизонтальному участку на кривой нагревания или
охлаждения, может не отвечать температурам равновесных
фазовых переходов. Чтобы избежать подобного рода неточностей,
строят несколько кривых нагревания и охлаждения при
различных скоростях изменения температуры или принимают
специальные экспериментальные приемы, позволяющие избежать
перенагревания или переохлаждения.
Аналогичные закономерности, характеризующие процесс
испарения жидкости, относят и к процессу возгонки (сублимации).
Однако при изучении этих процессов с участием кристаллической
фазы следует учитывать возможность полиморфных
превращений кристаллического вещесгва и указывать, к какой
модификации относятся полученные данные.
Следует отметить, что способ построения диаграмм
состояния по кривым температура—время применяется в основном
для определения кривой плавкости (плавления) и областей
устойчивости полиморфных модификаций, если превращения
сопровождаются достаточно большими тепловыми эффектами.
Сведения о фазовом состоянии системы можно получить
и другим способом, а именно измерением давления
насыщенного пара над жидкостью или кристаллическим веществом во
времени при изменении внешнего давления при постоянной
температуре. По мере понижения давления пара над веществом
система приходит в состояние, характеризуемое точкой на
кривой испарения или возгонки. При этом однокомпонентная
система становится двухфазной. Пока все вещество (кристаллы,
жидкость) не превратится в пар, равновесное давление в системе
не может быть понижено. На кривой давление насыщенного
пара—время, построенной аналогично кривой нагревания,
обнаруживается горизонтальный участок, соответствующий
процессу возгонки или кипения.
Проведя по крайней мере два эксперимента по изучению
изменения давления насыщенного пара во времени (при двух
значениях температуры) над твердым веществом (возгонка) и над
164
жидкостью (исиарение) и обработав результаты описанным выше
способом, можно построить кривые возгонки и испарения на
диаграмме состояния вещества.
Экспериментально определить положение тройной точки
довольно сложно, так как фудно подобрать такие температуру
и давление, при которых сосуществуют в равновесии три фазы —
кристалл, жидкость и насыщенный пар. Однако температуру
и давление тройной точки, как точки пересечения кривых
давлений пара над жидкостью и кристаллом, легко вычислить по
уравнениям давления пара над жидкостью и над кристаллом,
приняв равными температуры или давления в обоих уравнениях.
Из экспериментально найденных значений температуры
фазового перехода Т и изменения энтальпии АЯ° легко рассчитать
изменение энтропии А5° при фазовом переходе. Изобарные
потенциалы веществ фаз, находящихся в равновесии, равны.
Поэтому при равновесии
Откуда
= Atf /Г.
Данные по изменению энтропии в фазовом переходе позволяют
судить о характере превращения веществ и их строении.
§ 24. ДИАГРАММА СОСТОЯНИЯ ВОДЫ. СВОЙСТВА ВОДЫ
Диаграмма состояния воды в области невысоких давлений
представлена на рис. 79. Только при одном сочетании значений
температуры и давления, а именно 0,0076° С и 610, 381 Па —
тройная точка (точка О на диаграмме),— одновременно
сосуществуют три фазы. Следует отметить, что температура 0° С,
которую считают
температурой плавления льда, отвечает
равновесию между льдом и
водой, насыщенной воздухом при
давлении 1,013 • Ю5 Па.
Кривая ОА показывает
равновесные условия между льдом
и паром и характеризует
зависимость давления
насыщенного пара льда от
температу\В
2.20Ю7 А-
ры. Кривая ОВ отвечает
равновесным условиям между
льдом и жидкостью и
представляет зависимость
температуры замерзания воды от
давления. Кривая ОК изображает
1tQ13'W5
00076100 373t,°C
Рис 79 Диаграмма состояния воды.
Масштаб произвольный
165
равновесные условия между жидкостью и паром и представляет
зависимость давления насыщенного пара жидкой воды от
температуры или зависимость температуры кипения воды от
внешнего давления.
Точка К—критическая точка воды. Выше температуры 373° С
вода не может находиться в жидком состоянии ни при каком
столь угодно большем давлении, чем 219,82-105 Па. Выше
критической температуры ни при каком давлении не происходит
разделения на жидкую и газообразную фазы. В областях,
лежащих за пределами кривой давления насыщенного пара и
критической точки, можно осуществить непрерывный переход жидкости
в пар и пара в жидкость ж«->г (этот переход на диаграмме показан
стрелками).
По сравнению с диаграммами состояния других веществ
диаграмма состояния воды обнаруживает ряд особенностей. В
жидкой воде молекулы расположены очень упорядочение благодаря
сильным взаимодействиям, обусловленным в основном
водородными связями, что сказывается на наклоне кривых плавления
и испарения и на значениях относящихся к ним
термодинамических данных.
В ряду водородных соединений элементов VI группы
Н2О—H2S—H2Se—Н2Те температура плавления почти линейно
увеличивается для трех последних веществ (рис. 80). Если бы
зависимость температуры плавления от номера периода п соб-
3 4 5п
(нго) (h2s) (H2se)
'г 3 4 5л
(ЩО) fcfeS) (HiSe) (Н2Те)
Рис 80 Изменение температур плавления {а) и
кипения (б) в ряду водородных соединений элементов
VI группы
и—номер периода Периодической сиаемы, в ко юром находится
Э1СМСНГ
166
людалась во всей группе, то лед плавился бы примерно при 170 К
(—100° С). Однако температура плавления воды резко нарушает
ожидаемую закономерность (почти на 100° С выше).
По-видимому, это объясняется значительно более прочной кристаллической
решет кой твердой воды по сравнению с кристаллическими
решетками других гидридов.
Эта повышенная прочность обусловлена двумя главными причинами: 1)
взаимодействием молекул через водородные связи и 2) углом между связями в
молекуле воды блаюдаря sp -гибридизации внешних электронных оболочек атома
кислорода близким к 109,5° (тетраэдрическому углу), в то время как в молекулах
остальных гидридов из-за отсутствия гибридизации углы близки к 90 . Благодаря
5/?3-гибридизаиии и тетраэдрическому углу атом кислорода каждой молекулы
Н2О в структуре льда связан двумя связями через заполненные 5/?3-орбитали
с атомом водорода двух соседних молекул воды. Одновременно каждая молекула
воды еще двумя связями своих атомов водорода соединена с двумя другими
молекулами воды В результате координационное число кислорода в структуре
льда равно четырем и каждая молекула воды окружена четырьмя ближайшими
соседями Все водородные связи между молекулами энергетически равноценны,
и кристаллическая структура льда напоминает структуру алмаза, если атом
углерода мысленно заменить на молекулу воды В кристаллических соединениях
H2S, H2Se и Н2Те из-за отсутствия sp -гибридизации центрального атома связи
между молекулами обусловлены в основном силами Ван-дер-Ваальса, которые
в структуре льда играют значительно меньшую роль.
Анализ закономерностей в изменении термодинамических
характеристик фазовых переходов веществ дает возможность
получить представление о внутреннем строении и, в частности, о
наличии межмолекулярных взаимодействий и водородных связей,
исходя из значений легко экспериментально определяемых
температур плавления и кипения, давления пара над фазами,
плотностей фаз и других свойств.
Следует отметить, что фазовые переходы (испарение,
сублимация и плавление)—это не только физические процессы
изменения состояния, но и в значительной степени химические процессы
разрыва и образования связей, сопровождающиеся изменением
энтальпии и энтропии в системе.
Изменение энтальпии при плавлении льда также аномально
по сравнению с H2S, H2Se и Н2Те (рис. 81). Плавление льда
требует значительно больше теплоты, чем это можно ожидать из
сравнения энтальпий плавления других водородных соединений
той же группы элементов.
Плавление льда сопровождается уменьшением объема. Это
имеет огромное значение для обеспечения жизни животных и
растительных организмов при низких температурах: плавающий лед
защищает нижние слои воды от замерзания. Уменьшение объема
составляет ~ 10% и в согласии с принципом Ле Шателье с
повышением давления температура плавления льда понижается. Это
находит отражение в наклоне кривой плавления по отношению к оси
давления. Повышение давления на 130-105 Па приводит к тому,
что температура плавления льда становится равной — 1° С.
167
2 3
(Н2О)
5п г з 4 Sn
(н2те) (н2о) (h2s) (H2se)
Рис 81. Изменение энтальпии плавления (а) и
испарения (б) в ряду водородных соединений элементов
VI группы
Исследование процесса перехода молекул воды из жидкой фазы в
газообразную также доказывает существование водородных связей и высокий порядок
расположения молекул воды в структуре жидкости Температуры кипения
водородных соединений элементов IV группы H2S, H2Se и Н2Те линейно возрастают
с номером периода, в котором расположен элемент (см. рис 80), и, если бы
подобная закономерное! ь сохранялась у воды, она кипела бы примерно при
180 К (% — 90° С). Истинная же температура кипения воды 100г С, т. е. почти на
200° С выше ожидаемой, что объясняется наличием в структуре воды водородных
связей, препятствующих разрушению льдоподобной жидкой структуры.
Изменение энтальпии при испарении жидкости может быть
вычислено, если известны, как минимум, два значения давления
пара при двух температурах. Разрыв водородных связей в
структуре жидкой воды требует дополнительного количества энергии
при испарении, что сказывается на аномально высокой энтальпии
испарения жидкой воды (рис. 81).
Аналогичная аномалия воды наблюдается и при
рассмотрении энтропии плавления. Из сопоставления энтропии плавления
водородных соединений элементов VI группы следует, что при
плавлении льда происходит несравненно большее изменение
в степени порядка. Структура льда характеризуется большей
упорядоченностью в расположении молекул по сравнению со
структурами других гидридов.
168
ГЛАВА5
ГАЗОВОЕ СОСТОЯНИЕ И РЕАКЦИИ В ГАЗОВЫХ ФАЗАХ
§ 25. ТЕПЛОЕМКОСТЬ ГАЗОВ
При высоких температурах и низких концентрациях или
давлениях в газовой системе (расстояния между частицами при этих
условиях велики и намного превосходят их собственные размеры)
частицы могут свободно перемещаться, не взаимодействуя друг
с другом, и состояние вещества соответствует максимальной
степени беспорядка—поведение газовой системы отвечает
поведению идеального газа.
Для повышения температуры различных веществ, и в том
числе газообразных, на одно и то же число градусов
требуются различные количества теплоты. При этом
обнаруживается, что чем сложнее молекула и чем выше молекулярная
масса газообразного вещества, тем больше теплоты следует
затратить. Для того чтобы можно было сравнивать газы по
их способности повышать температуру при подведении
теплоты, пользуются количественно мерой, называемой
теплоемкостью.
Теплоемкость—это отношение количества теплоты,
получаемого веществом при нагревании или отдаваемого при охлаждении,
к соответствующему изменению температуры вещества. Если
теплоемкость относится к единице массы вещества, то она
называется удельной теплоемкостью, к 1 моль вещества—это молярная
теплоемкость.
В зависимости от условий измерения теплоемкости
различают изохорную Су и изобарную Ср теплоемкости. Изохор-
ная теплоемкость определяется при условии, когда при
нагревании (охлаждении) объем системы сохраняется постоянным и вся
сообщаемая (отводимая) теплота расходуется на увеличение
(снижение) внутренней энергии тела. Если измерение теплоем-
169
кости проводится при постоянном давлении, часть теплоты идет
на повышение внутренней энергии вещества, численно равное
изохорной теплоемкое!и, а другая часть — на работу расширения
А против внешнего давления. Определяемая при 1аких условиях
теплоемкость называется изобарной, она равна:
Если для [ вер дых тел и жидкостей изменение объема
при изменении температуры незначительно и CP^CV, то
для газов работа расширения А должна бьпь учтена.
Известно, что при нагревании 1 моль газа при постоянном давлении
на 1 К (или Г С) производится работа, численно равная
универсальной газовой постоянной /1 = 8,314 Дж/(К* моль).
Следовательно,
R = СУ + 8,314 [Дж/(К-моль)].
Кинетическая энергия 1 моль одноатомного газа равна
При изменении температуры одноатомного газа на 1 К при
постоянном объеме изменение энергии составляет
Изменение энергии Д?, очевидно, представляет изохорную
теплоемкость газа
CV = 3/2^ = 3/2 8,314 Дж/(К-моль)* 12,471 Дж/(Кмоль)^
^ 12 Дж/(К моль).
Изобарная теплоемкость одноа томного газа равна
Cp = Cv + R = 20.785 Дж/(Кмоль)*21 Дж/(Кмоль).
Экспериментально определенные изобарные и изохорные
теплоемкости гелия, неона, аргона и других одноа томных газов
совпадают с вычисленными значениями и не зависят от мольной
массы: Cp,298,i5 = 20,786 Дж/(К*моль).
Теплоемкость газов характеризуег среднюю энергию
теплового движения его молекул. Поступательное движение частицы
в пространстве в любом произвольном направлении может
бьпь разложено по трем взаимно перпендикулярным
направлениям или, что ю же самое, но трем координашым осям.
В этом случае говорят, что частица имеет три степени
свободы поступательного движения. Если бы молекула двигалась
только по одной координатной оси, т. е. обладала бы одной
170
Рис 82 Степени свободы движения
двухатомной молекулы
/ 2 3—nocmiare ibiioe движение 4 5—врашл-
гс ibtioe движение
степенью свободы,
кинетическая энергия поступательного
движения одной молекулы
равнялась 11гкъТ{къ — постоянная
Больцмана), а одного моля
F,02 • 1023 молекул)— l/2 RT.
При движении молекулы газа
по плоской поверхности ее
движение описывается изменением
координат по двум осям, и
молекула имеет две степени
свободы; в этом случае
кинетическая энергия 1 моль газа
составляет 2/2 RT= RT, При
движении в пространстве по трем координатным осям молекулы
обладают уже тремя степенями свободы, и кинетическая энергия
1 моль газа равна 3/2 RT.
Таким образом, на каждую степень свободы движения
молекулы приходится энергия, равная г/2къТ, а в пересчете на
1 моль газа—xliRT. Эта величина, как условно принято
считать, не зависит от вида движения молекулы. Если молекула
имеет более трех степеней свободы движения, т. е. кроме
поступательного движения совершает, например, вращательное
движение в одном направлении, то и на вращательное
движение приходится энергия, также равная г/2 RT в расчете на 1 моль
газа.
Двухатомные молекулы имеют пять степеней свободы: три
степени свободы поступательного движения и две степени
свободы вращательного движения (рис. 82). Вращательное движение
двухатомных молекул осуществляется вокруг осей,
перпендикулярных линии, соединяющей атомы. Вращение вокруг линии
(оси), соединяющей атомы, требует очень малого количества
энергии, которое можно не учитывать.
Таким образом, теплоемкости двухатомного газа,
рассчитанные как энергия, приходящаяся на пять степеней свободы,
должны составлять:
Cv = 5 • V2 # = 20,785 Дж/(К моль)*21 Дж/(К • моль),
Cp = Cv + R = 29,099 Дж/(Кмоль)^29 Дж/(Кмоль).
Однако теплоемкости многих двухатомных газов,
полученные опытным путем, отличаются от вычисленных значений и
зависят от температуры. Это объясняется тем, что при
повышении температуры энергия расходуется также на
внутримолекулярные колебания и другие виды движений, не учитываемых
в формулах. Теплоемкости некоторых двухатомных газов
приведены в табл. 13.
\п\
Та б л и ц а 13
Теплоемкость Ср |в Дж/(К моль)| некоюрых газов при 298,15 К
Двухатомные газы
Газ
н?
о2
N?
СО
NO
F?
Cl2
<¦',
28,832
29.351
29,124
29,112
29,874
31,338
33,941
Газ
h
HF
НС1
НВг
HI
с.
36,054
36,890
29,137
29,133
29,142
29,149
Газ
о.,
Н2О
н2о2
со?
so2
Трех- и многоатомные газь
с.
39,246
33,577
43,137
37J12
39,874
Газ
N2O
NO2
NO3
N2O3
N2O4
N2O5
i
38.618
37,489
46,861
65,270
78,659
94.977
По мере увеличения мольной массы газа возрастает
отклонение теплоемкости от расчетной величины. Это объясняется
увеличением вклада энергии вращения молекулы вдоль оси,
соединяющей атомы, а также тем, что часть подводимой энергии
расходуется на разрушение межмолекулярных сил.
У трехатомных молекул появляется третья степень
вращательного движения, поэтому расчетные теплоемкости
составляют:
Cv = 6 • 1/2R = ЗЯ = 24,942 Дж/(К • моль)« 25 Дж//(К • моль),
СР = ЗЛ + /? = 4Л = 33,256 Дж/(К • моль)« 33 Дж//(К • моль).
Теплоемкости газов, молекулы которых состоят из трех и
более атомов, сильно отличаются от расчетных значений и
возрастают по мере увеличения числа атомов в молекуле (см. табл. 13).
Изучение теплоемкостей дает ценные сведения о структуре
вещества, а также о термодинамике и кинетике процессов,
проходящих в нем при изменении температуры. Рассмотрим несколько
примеров.
Сопоставление теплоемкостей SO2 и СО2 при низких
температурах показывает, что молекула SO2 имеет шесть степеней
свободы и ведет себя как трехатомная, а молекула СО2—только
пять степеней свободы, т. е. поведение ее соответствует
двухатомной молекуле. Недостаток одной степени свободы
объясняется тем, что молекула СО2—линейная и вместо трех степеней
свободы вращательного движения она имеет только две.
Линейность молекулы СО2 подтверждается отсутствием у нее диполь-
ного момента. Факт линейности молекулы СО2 позволяет
предположить, что углерод находится в состоянии лр-гибридизации
и каждая связь С—О составлена из одной а-связи и одной
тс-связи. Плоскости двух я-связей расположены относительно
друг друга под углом 90°. Высокий дипольный момент молекулы
SO2, нелинейность молекулы, вытекающая из теплоемкости газа
172
и угол между связями, равный 119,5°, говорят о л/?2-гибридном
состоянии атома серы в молекуле SO2.
При стандартной температуре экспериментальное значение
изобарной теплоемкости диоксида углерода, Ср=37,11 Дж/
(К • моль), заметно превосходит расчетные значения, 33 Дж/
(К • моль), что вызвано различного рода колебательными
движениями атомов в молекуле. Таким образом, сведения о теплоем-
костях газов позволяют выяснить характер внутримолекулярных
движений.
При сравнении теплоемкостей простейших углеводородов
обнаруживается резкое уменьшение теплоемкости при переходе от
этана к этилену и ацетилену:
сн4 с2н6 с2н4 с2н2
C"V 298, Дж/(К- моль) 35,606 52,677 42,928 44,057
Это вызвано появлением у этилена и ацетилена соответственно
одной и двух я-связей. В молекуле этана группы СН3 могут
вращаться вокруг а-связи С—С, что приводит к повышению
теплоемкости. В молекуле этилена вращение групп СН2 вокруг
связи С=С запрещено из-за тс-связи. Молекула ацетилена
линейна, теплоемкость ацетилена хотя и намного ниже, чем этана, но
выше, чем этилена. Отсутствие угловых связей в этой молекуле
(ее линейность) приводит к росту вклада колебательного
движения (вдоль линии Н—С==С—Н) в теплоемкость этого вещества.
На основании данных по изучению теплоемкости
молекулярного водорода было открыто явление вращения протонов в
ядрах атомов и введено представление о спине частиц,
составляющих ядро.
§ 26. РЕАКЦИИ ВЕЩЕСТВ В ГАЗОВОМ СОСТОЯНИИ
В научно-исследовательской и технологической практике
часто пользуются смесями газов. Газовыми смесями являются и все
встречающиеся в природе газовые системы. Если газы в смеси
находятся при низком давлении и взаимодействием между
молекулами можно пренебречь, то можно считать, что компоненты
смеси ведут себя независимо друг от друга. Смеси,
удовлетворяющие этому условию, называются идеальными.
Газовые смеси правильнее считать растворами, т. е.
однофазными системами переменного состава, состоящими из двух или
более компонентов. Смесь идеальных газов, подчиняющихся
уравнению Менделеева—Клапейрона, представляет собой
идеальный расгвор газов.
Каждому газу в смеси отвечает его собственное парциальное
давление. Парциальным давлением компонента газовой смеси
173
называется то давление, которое оказывал бы газ, входящий
в смесь, если бы из нее были удалены все остальные газы
при условии сохранения постоянными температуры и объема.
Общее давление газовой смеси определяется законом
Дальтона A801 г.): общее давление смеси газов, занимающей
определенный объем, равно сумме парциальных давлений, которыми обладал
бы каждый отдельно взятый газ, если бы он занимал объем,
равный объему смеси газов (при Т = пост):
где р—общее давление;
Ль Pi* Ръ, •••> Л—парциальные давления компонентов.
Рассмотрим в качестве примера реакцию водорода с
газообразным иодом.
Система, состоящая из газообразных водорода, иода и иодо-
водорода, реагирующих по уравнению
2
; Кр = ,
имее1 два независимых компонента. Это означает, что, имея
любые два из составляющих систему веществ, можно получить
равновесную систему, состоящую из трех веществ, осуществив
реакцию или в прямом, или в обратном направлениях.
Так как давление в соответствии с принципом Ле Шателье не
влияет на равновесие этой реакции, то я=1, и поэтому число
степеней свободы для системы Н2 —12— HI равно:
С=2—1 + 1=2.
Система Н2 —12 — HI двухвариантна, и равновесие определяется
температурой и составом исходной смеси.
Равновесную систему Н2 —12 — HI можно получить также,
исходя только из иодоводорода:
Следовательно, условие равновесия этой системы можно
выразить также (помимо константы равновесия) через равенство
нарциальных давлений водорода и иода: Pn2—Pi2-
Для этой равновесной системы число независимых
компонентов уменьшается на два: /Г=3 — 2== 1, т. е. в этом случае система
ведет себя как однокомпонентная. Действительно, равновесная
система Н2 —12 — HI может быть получена из одного
иодоводорода. Из этого примера видно, что число независимых
компонентов есть наименьшее число составляющих систему веществ,
достаточное для определения состава любой фазы системы.
Процессу образования иодоводорода по реакции
174
при 410 С отвечает константа скорости прямой реакции
/спр = 0,0659 (условные ед.) и константа скорости обратной
реакции /собр = 0,00137. Напомним, что константа скорости реакции не
зависит от времени ее измерения. Частное от деления констант
скоростей прямой и обратной реакций, определенных в любой
момент времени, даже далекий от состояния равновесия,
представляет собой константу равновесия. Для системы Н2 —12 — HI
константа равновесия составляет:
А-р = /fc=Лпр/А:овр = 0,0659/0,00137=48.
По известному значению константы равновесия при данной
температуре и по исходным концентрациям (парциальным
давлениям) веществ можно вычислить их равновесные концентрации.
Проиллюстрируем такой расчет на следующих примерах.
В сосуде емкостью 1 л смешаны при 410° С по 1 моль
водорода и иода. Если к моменту наступления равновесия
прореагировало х моль водорода и такое же количество иода, то
значит, образовалось 2х моль иодоводорода и осталось по A —
х) моль водорода и иода. Так как объем газовой системы равен
1 л, то равновесные молярные концентрации веществ, входящих
в сие тему, составя г:
[Н2] = [12] = 1 — х моль/л,
[Н1] = 2л: моль/л.
Подставим эти концентрации в выражение константы
равновесия:
[HI]2 _ 4v2
^Тн7]р7Г(^
Решая это уравнение, получаем х=0,776 и находим равновесные
концентрации веществ:
[Н2] = [12] = 1-0,776 = 0,224 моль/л,
[HI] = 2 • 0,776 = 1,552 моль/л.
Пусть исходная газовая смесь состоит из 2 моль водорода
и 1 моль иода. Если к моменту равновесия прореагировало
х моль иода, то должно прореагировать такое же количество
водорода, образоваться 2х моль иодоводорода и остаться B-х)
моль водорода и A — х) моль иода.
Из уравнения
4х
2
находим х=0,932 моль/л и вычисляем равновесные
концентрации компонентов системы:
175
[Н 2] = 2 - 0,932 = 1,068 моль/л,
[12]= 1 -0,932 = 0,068 моль/л,
[HI] = 2 0,932= 1,864 моль/л.
Следовательно, увеличение начальной концентрации
водорода приводит к более полному расходованию иода и увеличению
выхода иодоводорода.
По известным равновесным концентрациям кроме константы
равновесия можно вычислить исходные концентрации
реагирующих веществ. Предположим, в системе Н2 —12 — HI равновесие
установилось при следующих концентрациях веществ:
[Н2] = 0,004 моль/л; [12] = 0,025 моль/л; [HI]=0,08 моль/л.
Согласно уравнению реакции из 1 моль Н2 и 1 моль 12
образуется 2 моль HI. Следовательно, для образования 0,08 моль
HI требуется 0,04 моль Н2 и 0,04 моль 12. Таким образом,
начальные концентрации веществ должны быть равны:
С„2 = 0,04+0,04 = 0,08 моль/л,
Ch=0,025 + 0,04 = 0,065 моль/л.
Пользуясь аналогичными рассуждениями, можно вычислить
равновесные концентрации веществ в реакционной системе, если
известны исходные концентрации и равновесная концентрация
одного из реагентов.
Предполагают, что образование HI из Н2 и 12 совершается
через переходное состояние Н212.
Чтобы предложить механизм реакции, необходимо знать
кинетическое уравнение процесса (порядок по компонентам), состав
реагирующей смеси и учесть, что наиболее предпочтительны
стадии, проходящие через столкновение двух частиц
(бимолекулярные реакции). Экспериментально определяемое кинетическое
уравнение иногда удается объяснить с помощью механизма,
включающего стадию, в которой быстро достигается состояние
равновесия.
В качестве примера рассмотрим реакцию
которая подчиняется кинетическому уравнению скорости
третьего порядка:
v~kC noCh2-
В связи с тем, что в реагирующей смеси спектроскопическим
методом были обнаружены молекулы пероксида водорода,
можно предположить, что процесс состоит из двух стадий:
176
A) 2NO + H2 = N2 + H2O2 (медленная)
B) H2O2 + H2 = 2H2O (быстрая)
Первая стадия, являясь более медленной, обусловливает
наблюдающийся третий порядок процесса. Однако такое
объяснение не может считаться удовлетворительным из-за того, что
вероятность одновременного столкновения двух молекул NO
и молекулы Н2 крайне мала. Поэтому был предложен другой
механизм реакции, включающий фи стадии:
A) 2NO = N2O2 (быстро достигается
равновесие)
B) N2O2 + H2=:N2 + H2O2 (медленная)
C) Н2О2 + Н2 = 2Н2О (быстрая)
Вторая стадия, как самая медленная, лимитирует скорость
всего процесса. Скорость этой стадии выражается уравнением:
v2 —k2C^2o2CU2.
Но концентрация N2O2 определяется равновесными условиями
стадии A):
Откуда находим, что
Подставляя концентрацию N2O2 в кинетическое уравнение
второй стадии, получаем:
v2 — k2KcC noCh2 = ^C noCh2-
Это уравнение совпадает с кинетическим уравнением,
выведенным из экспериментальных данных по зависимости скорости
от концентрации реагирующих веществ.
Среди газовых реакций нередко встречаются реакции,
имеющие дробный порядок, чаще всего 1/2 и 3/2. Обычно это говорит
об участии атомов, образующихся при диссоциации молекул.
§ 27. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Реакции, которые протекают под действием света,
называются фотохимическими. Под светом понимается видимый свет,
инфракрасное и ультрафиолетовое излучения. Эффективность
действия света зависит от его энергии: чем короче длина волны
(т. е. чем больше смещено излучение в ультрафиолетовую
область спектра), тем выше энергия излучаемых фотонов и тем
сильнее воздействие кванта света на облучаемую частицу—атом,
ион или молекулу.
177
С термодинамической точки зрения фотохимические реакции
можно разделить на два класса. Один класс объединяет реакции,
которые в данных условиях термодинамически
(самопроизвольно) проходить не способны, для их протекания необходима
затрата энергии, которая передается в виде световой энергии.
Важнейшим примером такого процесса является фотосинтез,
осуществляемый растениями. Он состоит из серии реакций, которые
в сумме описываются уравнением синтеза углеводов из диоксида
углерода и воды:
6СО2 + 6Н2О=С6Н12О6 + 6О2.
ыюкоза
Эта реакция сопровождается увеличением энтальпии,
уменьшением энтропии и, следовательно, увеличением изобарного
потенциала (AG>0). К этому же классу реакций относятся
фотографические процессы, в основе которых лежит способность галоге-
нидов серебра разлагаться под действием света.
Ко второму классу фотохимических реакций относятся
реакции, которые термодинамически способны при данных условиях
протекать самопроизвольно, но не протекают из-за высокой
энергии активации. Для этих реакций свет играет роль
возбудителя, инициатора реакции.
Остановимся на реакциях второго типа. Механизм действия
света на развитие реакции проявляется по-разному. При
поглощении реагирующей частицей кванта света hv могут протекать
следующие процессы, являющиеся начальными стадиями
фотохимической реакции:
1) переход частицы в возбужденное состояние
Повышенный запас энергии частицы делает ее способной
к реакции с другими частицами;
2) диссоциация частицы на атомы или другие частицы
3) ионизация молекулы или атома при поглощении кванта
света:
Соотношение между количеством поглощенной энергии и
количеством прореагировавшего вещества впервые было
установлено К. А. Тимирязевым, доказавшим, что эти количества строго
подчиняются закону сохранения энергии. Позже А. Эйнштейн
вывел формулу, являющуюся математическим выражением
закона фотохимической эквивалентности, согласно которому каждая
молекула, реагирующая под действием света, поглощает только
один квант световой энергии. Следовательно, в любой элементар-
178
ной реакции может принимать участие только один квант света,
т. е. число прореагировавших молекул должно равняться числу
поглощенных квантов.
Количество энергии, поглощенное 1 моль прореагировавшего
вещества, выражается уравнением:
где NA — постоянная Авогадро, моль \ h— постоянная Планка; v—частота
колебаний света, с"!; X—длина волны света, м; с—скорость света, м -с.
Например, оранжевый свет имеет длину волны Х=6-10 ~7 м,
т. е. его энергия равна:
р_6,02-Ю23-6,626-10~34 3,0 108 _
ь По^
= 1,994 • 105 Дж/моль^ 199 кДж/моль.
Этой энергии достаточно только для того, чтобы разорвать
не слишком прочную химическую связь. В видимом свете
наименьшей энергией обладает красная часть спектра (къ
7,5-10~7 м, ?=158 кДж/моль), наибольшей—фиолетовая часть
(к&4-10~7 м, ?=297 кДж/моль). Невидимые ультрафиолетовые
лучи имеют еще большую энергию, чем и объясняется их высокая
фотохимическая активность.
Под действием ультрафиолетового света с длиной волны
«2-10~7 м, т. е. с энергией «628 кДж/моль, молекулы хлора,
брома и иода распадаются на атомы:
Ультрафиолетовые лучи способны разрушать и более
сложные молекулы:
H2O2 + Av = 2OH,
CH3CHO+/*v =
(CH3JCO+Av =
Существует много реакций, в которых число
прореагировавших молекул не равно числу поглощенных квантов. Поэтому
для характеристики фотохимических реакций введено понятие
квантового выхода у. Квантовым выходом называется отношение
числа прореагировавших (разложившихся или образовавшихся)
молекул к числу поглощенных квантов.
Для истинно фотохимических процессов (элементарных
процессов) квантовый выход всегда равен единице и именно эти
процессы подчиняются закону фотохимической эквивалентности.
179
Такие процессы называются первичными. Первичные процессы
непосредственно вызываются действием света в отличие от
вторичных, не требующих освещения, но протекающих вслед за
первичными с участием продуктов первичных процессов.
В зависимости от квантового выхода фотохимические
процессы можно разделить на три группы.
1. Реакции с квантовым выходом, равным единице (у= 1). Это
элементарные химические реакции и небольшое число других
реакций, например, разложение сероводорода в бензольном
растворе или образование из водорода и кислорода пероксида
водорода [Х, = B,070 — 2,537) • 10 ~7 м, поглощающая квант света
молекула О2 ].
2. Реакция с квантовым выходом меньше единицы (у<1).
Примером такой реакции может служить разложение аммиака
[^ = B,0-2,2) • 10 -^м]:
Квантовый выход этой реакции равен 0,14—0,20 при 20° С
и 0,5 при 500° С. Механизм реакции можно представить
следующим образом:
NH3+Av — NH2 + H; у = 1 (первичный процесс)
NH H NH H
2 2
NH + NH = N2 + H2 (вторичные процессы)
NH H H
Большая часть продуктов первичной реакции вновь образует
молекулы исходного вещества, что и является причиной столь
низкого квантового выхода. Увеличение квантового выхода с
повышением температуры объясняется тем, что вероятность
реакции
с ростом температуры уменьшается (по принципу Ле Шателье
равновесие смещается влево).
Большинство фотохимических газовых реакций, проходящих
при невысоких давлениях, имеет квантовый выход меньше
единицы. Это вызвано тем, что образующиеся в первичной реакции
возбужденные молекулы, не успевая прореагировать, испускают
квант света, переходя в обычное состояние. При повышении
давления вероятность столкновения возбужденной молекулы
с другой молекулой увеличивается и квантовый выход
возрастает. Однако иногда увеличение давления в системе, достигаемое
введением нереагирующих веществ, в частности, благородных
газов, снижает квантовый выход. Это объясняется тем, что при
столкновении с активными молекулами не участвующим в
реакции молекулам передается избыточная энергия. По этой же
причине квантовый выход фотохимических реакций в растворах так-
180
же часто бывает ниже единицы—молекулам растворителя
передается избыточная энергия частиц—продуктов первичной
реакции.
Квантовый выход реакции фотосинтеза составляет ^0,1. Под
действием света молекула хлорофилла X переходит в активное
состояние, активная молекула затем отдает свою энергию
взаимодействующим молекулам СО2 и Н2О:
X+Av = X*
CO2 + H2O+X*=V6C6Hl2O6 + O2 + X.
Следовательно, для синтеза одной молекулы глюкозы и
образования шести молекул кислорода требуется шесть квантов
света.
3. Реакции с квантовым выходом больше единицы (у> 1). Эти
реакции, в свою очередь, можно разделить на две подгруппы.
К первой подгруппе относятся процессы, квантовый выход
которых равен 2 или 3 или близок к этим числам; во вторую
подгруппу входят реакции с квантовым выходом от 10 до 106 и выше.
Для реакции образования бромоводорода из простых веществ
[i=E,00 —5,78)* 10 м] и обратного процесса распада молекул
[?1 = B,07-2,54)-10-7м]
квантовый выход соответственно равен у = 0—2 (в зависимости
от условий реакции) и у = 2.
Фотохимический распад иодоводорода характеризуется
следующими процессами:
HI + /iv=
A)
B)
C)
D)
E)
F)
G)
H+l; y=l
H + HI = H2 +1;
H + I2 = HI + I*
I + H2 = HI + H
I +HI = I2 + H;
И 1 И TJ ,
П "T" П — *~i2»
i +i=i2;
(первичный i
A#=- 137,6
AH=-145,6
[; Atf= +137,6
АЯ= +.145,6
ДЯ= -432,0
АЯ=-148,8
АЯ=-294,4
iponecc)
кДж
кДж
кДж
кДж
кДж
кДж
кДж
Из указанных вторичных процессов A)—G) возможно
протекание не всех реакций. Рассмотрим, какими приемами следует
пользоваться для отбора реакций, имеющих реальные значения
АЯ. Реакции C) и D) проходят с увеличением энтальпии. Их
энергии активации должны быть по крайней мере выше
указанных значений АЯ, поэтому протекание этих реакций
маловероятно. Это означает, что атомы иода расходуются в других
реакциях, а атомы водорода образуются только по первичной реакции.
Из-за малой концентрации атомов водорода столкновение двух
атомов водорода по реакции E) маловероятно, и эта реакция
181
также должна быть исключена из рассмотрения. По той же
причине следует отказаться и от реакции G). Вследствие высокой
концентрации молекул HI практически все атомы водорода
взаимодействуют по реакции A). Увеличение количества атомов
иода в результате протекания реакции A) говорит о возможности
соединения двух атомов иода в молекулу по реакции F).
Образовавшиеся молекулы 12 удаляются из сферы реакции,
исключая тем самым реакцию B).
Следовательно, фотохимический процесс разложения иодово-
дорода сводится к одной первичной и двум вторичным реакциям:
Суммарная реакция выражается уравнением:
Действительно, один квант света превращает две молекулы иодо-
водорода в молекулы водорода и иода.
Под действием ультрафиолетового облучения кислород
переходит в озон. Этим процессом объясняется образование в
верхних слоях атмосферы озонового слоя, поглощающего
ультрафиолетовое солнечное излучение. Благодаря этому коротковолновая
часть солнечной радиации, опасная для живых организмов и
растительности, не достигает земной поверхности.
Реакция образования озона в зависимости от условий и ее
механизмов может проходить с квантовыми выходами, равными
двум или трем.
Предполагают, что в верхних слоях атмосферы (на высоте
25 км от поверхности Земли) под действием солнечной
ультрафиолетовой радиации молекулы кислорода распадаются на
атомы, которые, взаимодействуя с молекулами О2, дают молекулы
озона:
у=1,
2-О3; 7=1.
Суммарно процесс записывается в виде уравнения:
7 = 2.
Обнаружено, что при давлении кислорода — 5 • 106 Па квантовый
выход реакции возрастает до трех. Предполагают, что при этих
условиях первичным процессом является возбуждение молекулы
кислорода, которая из-за высокого давления не успевает
распадаться на атомы и взаимодействует при тройном столкновении
с двумя другими невозбужденными молекулами кислорода:
182
Следовательно, квантовый выход реакции равен трем, так как
один квант света приводи! к участию в реакции трех молекул
кислорода:
3O2+Av-2O3.
Распад молекулы озона в зависимости от внешних условий
может проходить по различным направлениям. При малых
концентрациях озона под действием ультрафиолетовой радиации
разложение совершается в две стадии:
О3+О = 2О2.
Эксперимент показывает, что скорость разложения озона
зависит не только от его концентрации, но и от концентрации
кислорода:
-dCoJdt = kCO32/CO2.
Слой с максимальной концентрацией озона располагается
на высоте около 25 км над поверхностью Земли. В этом слое
реакции образования и распада озона протекают наиболее
интенсивно.
Не меньшее значение для жизни на Земле имеют ионы NO+,
NO2~, О2 + , O2 и другие, образующиеся на высотах 100—400 км
по различным ионно-молекулярным реакциям.
Положительно заряженные молекулярные ионы не
образуются путем прямого отрыва электрона от нейтральной молекулы,
так как при поглощении кван i а света молекула переходит в
возбужденное состояние и энергия успевает распределиться по
молекуле прежде, чем произойдет отрыв от нее электрона.
Учитывая достаточно высокую концентрацию в атмосфере
молекул N2 и О2, а также то, что энергия связи О—О меньше
энергии связи N—N, можно предположить следующий механизм
образования иона NO + :
+ Av = O+O (первичный процесс)
В этом процессе первичной реакцией является
фотохимическая диссоциация молекулы кислорода на атомы. Следующая
стадия представляе! собой фотохимическую ионизацию атома
кислорода. Двойное столкновение иона О+ и молекулы азота
приводит к образованию заряженной частицы NO+.
По аналогичному механизму бимолекулярной реакции
происходит образование заряженной частицы О2 + :
О2 + О+=О2++О.
183
Отрицательно заряженная частица О2 образуется в
результате тримолекулярной реакции:
(электрон—это тоже реагирующая частица).
Вторая молекула кислорода служит для отвода избыточной
энергии.
Атомы кислорода, образовавшиеся по первичной реакции,
могут присоединять электрон:
Молекулы обычно не присоединяют электроны, поэтому
можно предположить, что образование обнаруженного в верхних
слоях атмосферы иона NO2" происходит по реакции:
NO2 + O"=NO2 +0.
Таким образом, процесс формирования сложных
молекулярных ионов включает в себя в качестве первичных реакций
диссоциацию двухатомных молекул и ионизацию атомов, и только
во вторичных процессах заряженные атомы обмениваются
зарядами со сложными молекулами.
Реакции с квантовым выходом много больше единицы—это
цепные реакции. Заметим, что все цепные реакции
термодинамически возможны (AG<0).
Световое излучение является одним из самых сильных
факторов, способствующих прохождению химических процессов. Под
действием света и особенно ультрафиолетового излучения
разрушаются многие неорганические и органические вещества.
Кванты света легко переводят частицы в возбужденное состояние,
а при достаточной энергии их ионизируют, т. е. под действием
кванта света от частицы отрывается электрон—образуется
положительный ион.
Возбужденная частица, образовавшаяся в результате
поглощения кванта света, имеет избыточную энергию, благодаря
которой электроны в ней переходят на более высокие энергетические
уровни и подуровни. Обратный переход из возбужденного
состояния в основное сопровождается испусканием кванта света.
Это явление называется люминесценцией.
Возможны два пути перехода частицы из возбужденного
состояния в основное (рис. 83). Первый путь—это
непосредственный переход в основное состояние, в результате чего развивается
резонансная люминесценция (раньше это явление называли
флюоресценцией).
Второй путь состоит в том, что частица, прежде чем
возвратиться в основное состояние, переходит в метастабильное
(неустойчивое) энергетически более низкое состояние. Этот переход
не сопровождается излучением света. Далее совершается переход
184
•и
Hi
Метастабияьное
состояние
II
ч
В ОСНОВНОе СОСТОЯНИе С ИСПуСКа- Воэбужденное состояние
нием кванта света, но другой
частоты — спонтанная
люминесценция (раньше это явление
называлось фосфоресценцией).
Основное отличие
флюоресценции от фосфоресценции
состоит в том, что
флюоресценция происходит в течение очень
короткого промежутка
времени (fclO~8 с), это быстрозату-
хающая (резонансная)
люминесценция . Фосфоресценция—
длительная (спонтанная)
люминесценция— происходит в течение значительно большего
промежутка времени. Вещество (обычно кристаллы или жидкости)
может фосфоресцировать в течение нескольких секунд и даже
часов после прекращения облучения.
В возбужденном состоянии молекулы проявляют резко
отличные свойства. Например, при возбуждении молекулы ацетилена
она из линейной превращается в угловую с увеличением
расстояния между атомами углерода:
Основное состояние
Рис 83 Схема возникновения
резонансной и спонтанной люминесценции
0,1208 НМ
Z-1800
0Д385НМi /6?O58HM
т/0,1058 НМ
Z =120°
Возбужденная молекула кислорода отличается по свойствам от
молекулы в основном состоянии. При возбуждении два электрона
с я/азр- и пРь разр-молекулярных орбиталей переходят в спаренное
состояние на2 ст2рлгразр-молекулярную орбиталь (рис. 84). Это син-
глетное состояние молекулы кислорода. Синглетная молекула
кислорода диамагнитна. Она способна вызывать реакции окисления,
не идущие под действием нормального кислорода. В таком
состоянии молекула кислорода напоминает молекулу этилена, для
которой характерны реакции присоединения. Действительно, наиболее
типичные реакции синглетного кислорода—образование
циклических соединений с органическими молекулами, имеющими у
крайних углеродных атомов двойные связи:
02сннгл+
сн=
нс-сн2—о
I I
НС—СНг—О
Отметим, что облучение светом смещает равновесие
химических реакций в ту же сторону, что и повышение температуры,
185
рээр разр
*Ру J * Рх
4-+-
4*-
г. е. увеличивается константа
равновесия реакции, идущей
с поглощением теплоты или
возрастанием энтальпии,
и уменьшается константа
равновесия реакции с Q>0 или
А#<0. Заметное смещение
равновесия наблюдается
только у реакций с квантовым
выходом, близким к единице.
Отличительной
особенностью фотохимических реакций
является независимость их
скорости от температуры, т. е.
энергия активации реакции равна нулю. Скорость
фотохимической реакции определяется только вероятностью поглощения
квантов света, которая практически не зависит от температуры.
Рис 84. Распределение электронов по
орбиталям молекулы кислорода в
основном тринлстном и возбужденном
синглетном состояниях
§ 28. ЦЕПНЫЕ РЕАКЦИИ
Ряд химических реакций протекает через стадию образования
активных частиц—чаще всего это свободные атомы,
неустойчивые молекулы или радикалы. Активные частицы вступают
в реакции с исходными веществами, в результате снова возникают
активные частицы. Такая последовательность периодически
повторяющихся химических процессов называется цепной реакцией.
Цепные реакции—это совокупность последовательно
протекающих реакций, в которой реагентами очередного
развивающегося процесса служат продукты предыдущего процесса.
Типичным примером цепной реакции может служить взрыво-
подобный процесс взаимодействия хлора с водородом, для
развития которого реакционную смесь достаточно осветить на
ничтожно малое время. Этот процесс начинается со стадии
химического распада молекулы хлора на атомы при поглощении кванта
света — зарождения цепи:
За этой стадией следует группа непрерывно повторяющихся
реакций с участием активных частиц и образованием новых активных
частиц без воздействия света—продолжение цепи:
Кроме этих реакций одновременно протекают процессы,
которые приводят к исчезновению активных частиц—обрыву цепи.
186
Реакции обрыва цепи могут проходить при столкновении
активных частиц со стенками реакционного сосуда, при тройном
соударении с частицей М, к которой переходит выделившаяся
энергия, при взаимодействии с различными примесями,
например, молекулами кислорода:
Несмотря на постоянно идущий процесс обрыва в цепи
последовательно протекающих реакций их число (длина цепи) может
достигать 10—100 тысяч.
Цепные реакции по ряду признаков отличаются от обычных
реакций, происходящих с перегруппировкой атомов. Так,
скорость цепных реакций зависит от размеров, формы и материала
реакционного сосуда (на его стенках проходит дезактивация
частиц, т. е. обрыв цепи), от добавок посторонних веществ и т. п.
Кроме того, цепные реакции характеризуются очень высоким
квантовым выходом. Например, в процессе взаимодействия
водорода и хлора одному поглощенному кванту света отвечает
около ста тысяч прореагировавших молекул хлора и водорода,
т.е. у=1105. Другая особенность цепных реакций состоит
в том, что в ходе этих реакций одна из стадий, протекающая
самопроизвольно с уменьшением энтальпии (или AG), может
вызвать процессы, сопровождающиеся увеличением энтальпии.
Большая скорость цепных реакций объясняется высокой
реакционной способностью активных частиц, вызывающих развитие
цепной реакции. Эти частицы имеют одиночные неспаренные
электроны или не полностью насыщенные химические связи.
Реакции зарождения цепи могут происходить не только под
действием квантов света. Так, введение в смесь водорода и хлора
ничтожного количества паров натрия или калия приводит к
цепной реакции. В этом случае зарождение цепи обусловлено
реакцией:
Cl2(r) + Na(r) = NaCl<r)+Cl(r).
Реакция прямого взаимодействия молекул водорода и хлора в их
смеси практически не протекает из-за очень высокой энергии
активации. Не обнаружены также реакции соединения атомов
водорода и хлора с образованием хлороводорода. Это
объясняется как низкими концентрациями атомов водорода и хлора, так
и тем, что энергия возникающей молекулы НС1, содержащая
избыточные энергии атомов и энергию связи, очень велика, и эта
молекула сразу же снова распадается на аюмы.
По такой же энергетической причине реакции обрыва цепи
очень редко проходят при соударении активных частиц. Чтобы
предотвратить распад молекулы, избы гок энергии должен быть
187
передан или стенке реакционного сосуда, или какой-либо другой
частице. Именно поэтому скорости цепных реакций сильно
зависят от размера, формы и материала стенок сосуда, от давления
и от присутствия других газообразных веществ.
Цепной механизм взаимодействия галогенов с водородом,
особенно характерный для фтора и хлора, позволяет объяснить
значения энергии активации элементарных реакций между
атомами и молекулами галогенов и водорода (табл. 14).
Таблица 14
Энергия активации отдельных стадий взаимодействия атомов
и молекул водорода и галогенов (Г— F, CI, Вг, 1)
№
1
2
3
4
5
Реакция
Г2 + Н2 = 2НГ
Г2 = 2Г
p_j_ н2 —НГ + Н
Г2-ЬН = НГ+Г
НГ + Н = Н2 + Г
F
>Ю5
155
33
17
151
а
210
250
21
8
17
Е , кДж/мо-и
Вг
172
188
75
5
4
172
142
138
0
4
Протекание реакции образования любого галогеноводорода
через стадию прямого взаимодействия молекулярных галогенов
и водорода (I) затруднено и маловероятно для всех галогенов
из-за высокой энергии активации и из-за необходимости
столкновения двух частиц. Зарождение цепной реакции между
галогенами и водородом начинается со стадии диссоциации B), которая
имеет очень высокую энергию активации, хотя и заметно
снижающуюся при переходе от хлора к иоду. Несмотря на высокую
энергию активации, атомы галогенов, получающиеся при
возбуждении по реакции B) в небольшом количестве,
взаимодействуя с молекулой водорода C), дают молекулу галогеноводорода
и атом водорода. Последний по реакции с молекулой галогена D)
образует молекулу продукта (галогеноводород) и атом галогена,
вновь повторяющий стадию C). Реакция D) благодаря невысокой
энергии активации проходит легко для всех галогенов. По
реакции E) уменьшается концентрация атомов водорода, что
способствует обрыву цепи. Из-за высокой энергии активации стадии
E) с участием фтора обрыв цепи по этой реакции, по-видимому,
не происходит, но этот процесс протекает с другими галогенами.
Таким образом, реакцией, ответственной за развитие цепи,
является стадия C)—взаимодействие между атомом галогена
и молекулой водорода. При переходе вниз по подгруппе
галогенов энергия активации процесса C) возрастает, а энергия
активации обрыва цепи E) понижается, вследствие чего цепной
механизм реакции с участием иода, а также брома ослабляется.
188
Порядок реакции брома с водородом зависит от концентраций
реагентов и непрерывно изменяется в ходе реакции, что говорит
об изменении механизма процесса. Взаимодействие иода с
водородом проходит частично по цепному механизму, но в основном
через образование промежуточного комплекса H2I2.
Механизмы реакций, даже простых, могут быть очень
сложными, и по мере развития экспериментальной техники сведения
о кинетике реакции дополняются новыми деталями и
представления о механизме изменяются. Высокая скорость
реакции—необязательный признак цепной реакции (см. пример с реакцией
образования иодоводорода). Реакцию хлорирования этилена
с образованием дихлорэтана
C2H4+CI2-C2H4CI2
можно описать прямым взаимодействием этилена и хлора через
стадию образования активного комплекса:
СУ'91 а а
нс
:Г —>н-с-снн
н к \
н ы н к
Однако вероятность расположения молекул при их
столкновении, благоприятствующая образованию комплекса, очень мала,
энтропия активации процесса низка и скорость протекания
реакции по этому пути должна быть невысокой. Поэтому
предпочтителен другой механизм, а именно цепной, по которому реакция
проходит несравненно быстрее.
Если в системе образуются атомы хлора, например, при
действии света на реакционную смесь, процесс осуществляется по
цепному механизму. Атом хлора легко присоединяется по
двойной связи С2Н4 с возникновением молекулы C2H4CI, которая
способна оторвать один атом хлора от молекулы СЬ для
образования конечного продукта C2H4CI2. Эти стадии имеют
невысокие энергии активации, и такой путь протекания процесса
обеспечивает более высокую его скорость.
Цепной механизм процесса взаимодействия этилена с хлором
может быть представлен следующими стадиями:
A) СЬ = С1 + С1
B) С2Н4 + СЫС2Н4С1
C)
D)
E)
F) С1 + С1 = С12.
189
Из этих реакций стадии A) и B) являются
последовательными, так как атом С1, образующийся на первой стадии, служит
исходной частицей для второй стадии. Стадии D) и E) протекают
параллельно, так как в обоих процессах в качестве реагента
принимает участие одна и та же частица. Таким образом,
рассматриваемый цепной процесс включает
последовательно-параллельные стадии. Две стадии реакций называются
последовательно-параллельными, если они являются параллельными по
отношению к одной и последовательными по отношению к другой
из участвующих в этих стадиях частиц. Такими являются стадии
B) и D). По отношению к О эти стадии являются
параллельными, а по отношению к C2H4CI—последовательными.
Реакции полимеризации также могут быть отнесены к ценным
процессам, причем число стадий и длина цепи в процессе
определяют длину полимерной молекулы.
Рассмотренные выше процессы относятся к 1ииу неразветв-
ленных цепных реакций.
Если в результате одного первичного процесса возникают две
или больше активные частицы, процесс называется
разветвленным цепным (рис. 85). Из-за роста числа активных частиц
скорость разветвленного процесса в целом в начальный момент
времени будет лавинообразно возраст ать до того момент а, когда
в результате снижения концентраций реагирующих веществ
скорости всех реакций не начнут уменьшаться.
Путь развития цепного процесса в большой степени связан
с условиями, при которых протекает взаимодействие веществ
(давление и темпера тура). Рассмотрим это на примере реакции
водорода с кислородом.
Взаимодействие водорода и кислорода при низких давлениях
и высоких температурах (^900 С) протекает как сильно
разветвленная цепная реакция. Первичный химический акт в системе—
реакция образования атомов. Энергия связи в молекулах
водорода и кислорода соответственно равна 432,0 и 493,6 кДж/моль,
поэтому можно ожидать,
что диссоциация молекулы
водорода, а не кислорода да-
/ ет свободные атомы, кото-
—•—•—¦ рые вызывают развитие
цепного процесса:
A)
^ B) Н+О2 =
C) О
D)
Рис 85. Скема разветвленного цепного . По реакции A) образуют-
процесса (а) и процесса с редкоразветвлен- *-я Две активные частицы, КО-
ной цепью F) торые на стадии B) также
190
дают по две активных частицы. Стадии C)—D) продолжают
цепь. Образование в стадиях зарождения и продолжения цепи
в среднем более чем одной активной частицы (а иногда и трех)
приводит к скачкообразному, самоускоряющемуся,
лавинообразному процессу.
Та же реакция между кислородом и водородом при высоких
давлениях и более низкой температуре E00° С) протекает как
процесс с редкоразветвленной цепью (см. рис. 85, б). При высоких
давлениях, когда появляется возможность передачи избытка
энергии другой частице М, взаимодействие атома водорода с
молекулой кислорода приводит к образованию неустойчивой
молекулы НОг:
Bа) Н + О2 + М = НО2 + М*.
Этот элементарный процесс—тримолекулярная реакция.
Молекула НОг может взаимодействовать с атомом водорода,
образовавшимся по уже известным стадиям цепного процесса:
E) НО2 + Н = 2ОН.
Дальнейшие превращения молекул и атомов те же, что
и в разветвленном цепном процессе.
При высоких температурах и низких давлениях большее
значение приобретает стадия B) по сравнению со стадией Bа),
протекающей в основном при высоких давлениях и низких
температурах. На стадии B) возникают две активные частицы и
далее развивается процесс по схеме с сильно разветвленными
цепями. На стадии Bа) образуется только одна активная частица
и реакция проходит по схеме с редкоразветвленными цепями.
При протекании процесса, включающего стадию Bа)
образования частицы НОг (при высоких давлениях), в реакционной
смеси обнаруживаются молекулы пероксида водорода, которые
могут распадаться или на две молекулы ОН, или на молекулу
воды и атом кислорода:
F) НО2 + Н2 = Н2О2 + Н
G) Н2О2 = 2ОН
(8) Н2О2 = Н2О+О.
Начало цепной реакции между водородом и кислородом
может быть положено не только диссоциацией молекулы водорода
на атомы, но и прямым взаимодействием молекул Ш и О2 (хотя
оно менее вероятно):
(9) Н2 + О2-НО2 + Н,
A0) Н2+О2 = 2ОН.
Образование молекул ОН по реакции A0) можно представить
как процесс с формированием промежуточного комплекса (рис.
191
н—н щ ,н Hf <н н н н н
/ \Г \
н н
о. .о о' *> о^ 4о
Активный
комплекс
Рис. 86 Схема взаимодействия кислорода и водорода с образованием активного
комплекса «Н2О2»
86). Промежуточный активный комплекс Н2О2, как
предполагают, имеет плоское строение в отличие от молекулы пероксида
водорода ШСЬ, в которой связи Н — О расположены под углом
95° к связи О—О, а относительно друг друга—под углом 120°.
Нет никаких экспериментальных данных, подтверждающих
возможность образования молекулы воды из молекул водорода
и кислорода, как это обычно пишут:
В этом процессе вероятность одновременной встречи трех
частиц очень мала, еще более мала вероятность такого
расположения молекул в пространстве, которое привело бы к
химическому взаимодействию (энтропия активации имеет очень
небольшое значение). Молекула воды образуется почти
исключительно по стадии C) взаимодействия ОН иНгив незначительном
количестве по стадии (8) и по реакции
A1) НО2 + Н2 = Н2О + ОН.
Интересно отметить, что в приведенном наборе реакций A) —
F) нет ни одной реакции, по которой образовалась бы только
одна частица, например:
Это объясняется тем, что одна часшца должна нести очень
большую избыточную энергию. Кроме того, исходя из
геометрических представлений о характере связей в молекулах исходных
веществ и продуктов, нельзя расположить в пространстве
молекулу водорода и атом кислорода, чтобы в одну стадию
произошло перераспределение электронов и образование новых связей.
В реакционной смеси водорода и кислорода в очень
незначительных количествах обнаружены и более сложные молекулы:
НОз, НО4, Н2Оз, Н2О4 и др.
Скорость разветвленных цепных реакций сильно зависит от
давления, причем для многих процессов с повышением давления
скорость резко возрастает и реакция протекает как взрыв, но при
дальнейшем повышении давления скорость реакции резко умень-
192
шается. Это объясняется тем, что при низком давлении
столкновения между частицами очень редки и активные частицы, не
прореагировав, отдают свою энергию стенкам сосуда. При
высоком давлении вероятность столкновений активных частиц с
молекулами конечного продукта реакции или с молекулами
примесных веществ М возрастает, что приводит к обрыву цепи. Только
в некотором интервале давлений реакции проходят как сильно
разветвленные и могут сопровождаться взрывом.
Кинетическим закономерностям газовых цепных реакций
подчиняются многие взрывные процессы и, в частности, один из
важнейших процессов—деление атомных ядер под действием
нейтронов. Нейтроны особождаются при делении ядер урана-235.
Это объясняется тем, что в ядрах атомов тяжелых элементов
отношение числа нейтронов к числу протонов больше, чем в
ядрах атомов элементов средней массы, образующихся при
делении. Каждый из нейтронов, выделившихся при делении ядра, при
достаточно высокой плотности вещества может вызвать деление
еще одного ядра с одновременным образованием в среднем более
чем одного нейтрона. В результате число делений и число
нейтронов будет лавинообразно возрастать (принцип атомной
бомбы). Если этот процесс тормозить, уменьшая концентрацию
делящихся атомных ядер в смеси, или вводить другие вещества,
поглощающие нейтроны, то можно регулировать скорость
процесса. Этот прием используется в атомных энергетических
установках.
7 2452
ГЛАВА 6
ЖИДКОЕ СОСТОЯНИЕ И РЕАКЦИИ В ЖИДКИХ ФАЗАХ
§ 29. ЖИДКОЕ СОСТОЯНИЕ ВЕЩЕСТВА
Силы взаимодействия между частицами жидкости
значительно больше по сравнению с силами, действующими в газовых
системах. В результате в жидкостях могут возникать
упорядоченные участки, которые распадаются и снова образуются, поэтому
энтропия жидкости ниже энтропии газа. В то же время малая
прочность связей между частицами в жидкости обусловливает ее
подвижность и текучесть. Сжимаемость жидкостей намного
меньше, а плотность намного больше, чем у газов; количественно
эти свойства близки к свойствам кристаллических тел. Их
зависимость от температуры значительно меньше, чем у газов, но
несколько сильнее, чем у твердых тел.
По многим свойствам жидкость занимает промежуточное
положение между газами и кристаллическими (твердыми)
веществами, и для описания поведения жидкостей используются
закономерности, присущие газовому и кристаллическому состояниям.
Однако внутреннее строение жидкостей значительно сложнее
внутреннего строения газов и кристаллов.
Поведение жидкостей зависит от их химической природы
и внешних условий. Как правило, чем ниже температура и чем
ближе температура жидкости к температуре ее кристаллизации,
тем в большей степени некоторые свойства жидкости
приближаются к свойствам твердых веществ. И наоборот, чем выше
температура и чем ближе она к температуре кипения, тем больше
сходства в поведении жидкостей и газов.
Влияние природы жидкости на ее поведение определяется
силами взаимодействия между молекулами. Чем более полярны
молекулы жидкого вещества, тем сильнее взаимодействие между
ними и тем ближе по строению и поведению жидкости к твердым
телам. Например, такие полярные вещества, как вода, аммиак,
серная кислота, в жидком состоянии по поведению ближе к твер-
194
Рис 87 Схема образования ассоциатов
полярных молекул
дым веществам, а
неполярные жидкости типа
бензола или толуола,
наоборот, по свойствам
ближе к газам.
Чтобы понять
характер взаимодействия
между частицами, следует
остановиться на типах
связи, которая может
возникать между частицами.
Тип связи между
частицами вещества в жидком
состоянии в зависимости от
энергии связи может изменяться в очень широких пределах — от
межмолекулярных сил (сил Ван-дер-Ваальса) до истинно
химической связи.
Напомним, что силы Ван-дер-Ваальса—это силы притяжения
между молекулами вещества в газообразном, жидком и твердом
состояниях. Эти силы могут возникать как между полярными,
так и неполярными молекулами.
Каждая полярная молекула, являясь диполем, ориентирует
окружающие ее молекулы так, чтобы их противоположно
заряженные части располагались ближе друг к другу. При этом
возможно образование не только пар молекул, но и групп—ассоциа-
тов, построенных различно в зависимости от формы молекул.
На рис. 87 представлены различные ассоциаты полярных
молекул; ассоциаты изображены на плоскости, в действительности
же молекулы располагаются в трехмерном пространстве, образуя
упорядоченные микрообъемы.
Возникающее электростатическое притяжение между
полярными молекулами обусловлено ориентационным эффектом.
Изучение структуры жидкостей показало, что они состоят из
упорядоченных групп молекул, которые непрерывно образуются, в
результате теплового движения распадаются и снова образуются из
тех же и других частиц. Число частиц в ассоциате зависит от
полярности молекул и температуры.
Межмолекулярное взаимодействие существует и между
неполярными молекулами. Благодаря непрерывному перемещению
электронов в атоме центры положительного и отрицательного
зарядов в какой-то момент времени могут не совпадать,
возникает мгновенный диполь, который, смещая электронные плотности
в соседних молекулах, превращает их также в диполи.
Образование мгновенных диполей и их вклад в структуру жидкости носит
название дисперсионного эффекта.
Выделяют и третий тип межмолекулярного
взаимодействия— индукционное, приводящее к проявлению индукционного
195
О эффекта. Суть этого эффекта состоит
в том, что электрическое поле одной
молекулы усиливает диполь второй
молекулы, что приводит к росту сил притяжения.
Индукционное взаимодействие особенно
ярко проявляется, если среди полярных
Гтвия8 шорной иТеполя?: молекУл Распределены неполярные
моленной молекул (проявление кулы, тогда электрическое поле полярной
индукционного эффекта) молекулы индуцирует, наводит диполь
в неполярной молекуле (рис. 88).
Дисперсионное, индукционное и ориентационное
взаимодействия проявляются одновременно и способствуют образованию
групп молекул и упорядоченных участков жидкости.
Дисперсионным эффектом объясняется взаимодействие
между атомами благородных газов, приводящее к их сжижению. Чем
больше размеры атомов, тем легче проявляется этот эффект
и тем выше температура кипения жидкостей (и температура
плавления). В основном дисперсионным эффектом обусловлено
взаимодействие практически неполярных молекул СО.
Взаимодействие молекул НС1 также вызвано в первую очередь
дисперсионным эффектом, далее в порядке ослабления следуют
индукционный и ориентационный эффекты. Иной вклад вносят
различные эффекты во взаимодействие молекул воды: наиболее сильно
проявляется ориентационное взаимодействие, далее следуют
дисперсионное и индукционное.
Высокая упорядоченность расположения молекул многих
жидкостей, например, воды, фтороводорода, аммиака и спирта,
не может быть объяснена только действием сил Ван-дер-Ваальса.
Для объяснения структуры жидкости используется представление
о водородной связи. Образование этой связи обусловлено тем, что
электронная орбиталь атома водорода имеет сферическую
симметрию и формирует одну связь в результате перекрывания
с орбиталью другого атома, при этом у атома водорода
остаются возможности для взаимодействия с другими атомами и
образуется водородная связь.
Водородная связь имеет наибольшую прочность в
комбинациях водорода с наиболее электроотрицательными элементами
второго периода Периодической системы: фтором, кислородом
и азотом. Считают, что связь атома водорода с атомами этих
элементов сильно полярна и атом водорода приобретает
положительный заряд, который способствует сближению
электронных облаков другого атома с атомом водорода с образованием
водородной связи. Однако такое объяснение возникновения
водородной связи совершенно недостаточно хотя бы потому, что
электроотрицательность хлора равна электроотрицательности
азота, но хлор почти не участвует в образовании водородной
связи.
196
Предполагают, что в жидком фтороводороде за счет
водородных связей молекулы образуют зигзагообразные цепи (HF)n,
которые также обнаружены в кристаллической и газообразной
фазах:
Электронная s-орбиталь атома водорода, содержащая один
электрон, перекрывается с р-орбиталью атома фтора, имеющей
один электрон, и дополнительно с такой же р-орбиталью атома
фтора другой молекулы HR В результате все молекулы HF
в цепи соединяются друг с другом через атомы водорода.
Наряду с электростатическим взаимодействием в
образовании водородной связи, как предполагают, существенную роль
играет донорно-акцепторное взаимодействие особого типа. Так,
атом фтора, участвующий в водородной связи, может передавать
пару электронов в частичное пользование атому водорода, у
которого единственный электрон сильно оттянут в направлении
к другому атому фтора.
Одна молекула воды может образовать четыре связи с
другими молекулами за счет двух атомов водорода и двух гибридных
несвязывающих орбита л ей атомов кислорода, имеющих по паре
электронов. При этом возникают пространственные полимеры
(Н2О)„. С энергетической точки зрения образование их даже
более предпочтительно, чем димеров или линейных и
плоскостных структур. Лед представляет собой кристалл, в котором
каждый атом кислорода связан с двумя атомами водорода своей
молекулы воды, которые связаны с двумя другими молекулами
воды и с двумя атомами водорода еще двух других молекул. При
плавлении льда эти очень упорядоченные структуры
разрушаются только частично, так что вода представляет собой «осколки»
структур льда, «плавающих» среди более мелких группировок
молекул воды.
В жидкой воде на долю ориентационного взаимодействия,
т. е. взаимодействия диполь-диполь и через водородную связь,
приходится ^75% от всех сил, действующих между молекулами,
на долю индукционного взаимодействия (благодаря наведенным
диполям) %5% и на долю дисперсионного взаимодействия
(мгновенный диполь) «20%. Для сравнения укажем, что в
жидком гелии осуществляется только дисперсионное взаимодействие
A00%).
Жидкий аммиак также имеет очень упорядоченную структуру,
но тенденция к упорядочению у него значительно слабее, чем
у воды. Это объясняется более низкой электроотрицательностью
азота и благодаря этому меньшей способностью образовывать
197
водородную связь, а также наличием одной орбитали с парой
электронов в молекуле NH3. Вследствие этого одна молекула
аммиака может образовать дополнительно связь только с одним
атомом водорода. Позтому молекулы аммиака образуют в
основном плоскостные структуры, а не пространственные,
типичные для жидкой воды.
Сопоставление структур жидких аммиака, воды и фтороводо-
рода показывает, что тип структуры — плоскостная, объемная
или линейная—зависит как от числа атомов водорода в
молекуле, гак и от числа заполненных парами электронов
.^-гибридных орбиталей, способных участвовать в донорно-акцепторном
взаимодействии с атомами водорода других молекул:
Число Чис ю заполненных Число Ciovk
атомов v/?J-op6maneii связей "^ УР
водорода
NH3 3 1 3 плоскостная
Н2О 2 2 4 объемная
HF 1 3 2 линейная
Явление структурообразования было обнаружено даже у
неполярных жидкостей, например, у жидкого бензола, тетрахлорида
углерода, а также у расплавленных металлов, солей и других
соединений. Структуры жидких мет аллов имеют некоторые общие
черты со структурами твердых металлов. Аналогичные явления
характерны и для расплавленных оксидов металлов и их солей.
Один из методов изучения структуры жидкостей основан на
исследовании вязкости. Вязкость отражает способность вещества
оказывать сопротивление перемещению одной его части
относительно другой. Вязкость очень чувствительна к молярной массе,
строению молекул и межмолекулярным взаимодействиям.
Гидродинамические теории течения газов и жидкостей
практически одинаковы, но механизмы течения этих систем, т. е.
механизмы смещения частиц относительно друг друга, различны.
Это подтверждается сравнением влияния температуры и
давления на вязкость газов и жидкостей.
Для веществ в газовом состоянии характерно, что их вязкость
увеличивается с повышением температуры и почти не зависит
от давления. Для жидкостей вязкость увеличивается с ростом
давления и уменьшается (экспоненциально) с повышением
температуры.
Свойства воды обнаруживают исключение из этого правила, ее
вязкость при температурах ниже 25° С с ростом давления
снижается, достигая минимального значения, и затем возрастает. Вязкость
воды при 20° С равна 1,002 мПа • с, что принимается в качестве
эталона вязкости. Вязкость низкомолекулярных однотипных
соединений в жидком состоянии растет с увеличением молярной массы.
Для большинства жидкостей зависимость вязкости г| (или
коэффициента вязкости) от температуры описывается уравнением:
198
Рис 89. Зависимость
количества Q нормальной
(/) и аномальной B)
жидкости, протекающей через
капилляр, от давления
где А — постоянная;
R—универсальная газовая
постоянная;
Г— температура;
?вязк—энергия активации вязкого
течения, т. е. тот энергетический
барьер, который следует
преодолеть для смещения слоя 1 моль
частиц относительно другого
такого же слоя.
Зависимость вязкости от давления
существенно различается у
неструктурированных (нормальных) и
структурированных (аномальных) жидкостей. У
нормальных жидкостей проявляется прямо
пропорциональная зависимость между
количеством жидкости, протекающей в единицу времени через
капилляр, и давлением, действующим на жидкость (рис. 89,
кривая 7). Вязкость структурированных жидкостей не подчиняется
этому правилу. При течении аномальной жидкости работа
затрачивается не только на преодоление сопротивления слоев
жидкости, но и на разрушение ее структуры. Графическая зависимость
количества вытекающей жидкости от давления в этих случаях
имеет вид кривой, выпуклой к оси давления (рис. 89, кривая 2).
Другим структурно зависимым свойством жидкостей,
и в частности воды, является плотность. Вода представляет
собой совокупность мельчайших объемов со структурой льда,
полимерных образований (Н2О)И и отдельных молекул. Упаковка
молекул в жидкости более
плотная, чем в структуре
льда, и плотность воды
выше плотности льда.
Наибольшая плотность
жидкой воды наблюдается
при +4,0° С, ниже и выше
этой температуры
плотность воды меньше
(рис. 90, кривая У).
Можно предположить,
что такая зависимость
плотности воды от
температуры отражает две
различные температурные
зависимости. Первая из
них отвечает тепловому
1,000
О
2 b 6 t,°C
Рис 90 Зависимость плотности жидкой воды
от температуры:
/—суммарная зависимость, 2—составляющая,
обусловленная тепловым расширением жидкости, 3—составляющая,
обусловленная разрушением структуры
199
расширению жидкости. С повышением температуры в результате
теплового расширения плотность должна уменьшаться (прямая
2 на рис. 90). Вторая составляющая (прямая 3) отражает
изменение степени упаковки молекул при повышении температуры.
Из-за разрушения рыхлых льдоподобных структур плотность
возрастает с повышением температуру. При температуре ниже
+ 4° С вторая составляющая (прямая 3) преобладает над первой
составляющей (прямая 2), и плотность возрастает с повышением
температуры. Выше +4° С зависимости изменяются.
Зависимость плотности воды от температуры представляет собой сумму
этих двух составляющих и поэтому имеет максимум при +4° С.
Чистые жидкости практически не встречаются в природе.
Большинство природных жидких систем представляют собой
растворы. Почти всегда человек имеет дело с растворами.
Изучение свойств чистых жидкостей обычно проводят для сравнения
поведения чистой жидкости с поведением раствора и объяснения
и предсказания свойств растворов, используемых в практической
деятельности.
§ 50. ЭЛЕКТРОЛИТИЧЕСКАЯ ДИССОЦИАЦИЯ ВЕЩЕСТВ
В ЖИДКОМ СОСТОЯНИИ
Многие жидкие вещества способны диссоциировать —
распадаться на ионы. Способность диссоциировать характерна и для
жидких веществ в растворах, и для многих твердых веществ
в расплавленном состоянии или растворенных в каком-либо
растворителе, в частности, в воде. Такие вещества называются
электролитами, а процесс распада электролитов на
ионы—электролитической диссоциацией. Теория электролитической диссоциации
веществ в водных растворах разработана шведским ученым Ар-
рениусом; основные закономерности этого процесса,
протекающего в водных растворах, рассмотрены ниже, в §§ 44 и 45.
Чистая вода почти не проводит электрический ток, и ее
раньше рассматривали как вещество, практически не
диссоциирующее на ионы. Однако вода очень слабо, но диссоциирует
с образованием двух противоположно заряженных ионов.
Упрощенно этот процесс можно записать так:
Н2О = Н++ОНГ.
Константа равновесия этого процесса выражается уравнением:
[н+][он-]
[н2о] '
Так как вода — очень слабый электролит и только ничтожная
часть ее молекул находится в состоянии диссоциации, можно
считать, что концентрация воды соответствует числу молей Н2О
в 1 л воды A000 г), т. е. равна
200
[Н2О]= 1000/18,02 = 55,5 моль/л.
Подставим это значение в константу равновесия
или
При постоянной температуре произведение равновесных
молярных концентраций [Н+ ] [ОН" ]—постоянная величина.
Она называется ионным произведением воды и обозначается
символом Кв:
где К—константа диссоциации воды.
Если в выражение изменения изобарного потенциала
диссоциации воды
AG°=-RT\nK
вместо константы диссоциации К подставить ионное
произведение воды ЛГВ, то получим величину AG°, характеризующую
направление и меру диссоциации воды. Обычно процесс
диссоциации воды принято количественно выражать не константой
диссоциации, а именно ионным произведением воды, которое
широко используется для вычисления ряда физико-химических
характеристик водных растворов. Поэтому изменение
изобарного потенциала диссоциации воды относят не к константе
равновесия диссоциации, а к ионному произведению воды. Данные
в табл. 15 показывают изменение ионного произведения воды
и отвечающие им изобарные потенциалы в интервале температур
от 10 до 25° С.
Таблица 15
Ионное произведение воды и изобарный потенциал диссоциации воды
при различных температурах
Температура
С
10
15
20
25
к
283,2
288,2
293,2
298,2
кь ю14
0,2918
0,4505
0,6814
1,008
-14,5349
-14,3463
-14,1666
-13,9965
ДС% кДж/моль
78,8
79,1
79,5
79,9
При дальнейшем повышении температуры ионное произведение
возрастает. При 120° С оно равно 1 -10" , достигает максимума
Я^=6,5-1(Г12 при 220° С и затем снижается до 3,5-10~14 при
критической температуре воды 374е С (температура, при которой (и
выше) газ уже ни при каком давлении не переходит в жидкость).
201
С повышением температуры изменение изобарного потенциала принимает
увеличивающиеся положительные значения Казалось бы, из этой зависимости
можно сделать вывод, что с ростом температуры термодинамическая
вероятность процесса диссоциации воды снижается, хотя константа равновесия
возрастает. Такое противоречие объясняется следующим образом
Изобарный потенциал процесса всегда уменьшается (становится все более
отрицательной величиной) с ростом константы равновесия, но при этом
температура, при которой протекает процесс, должна оставаться постоянной Для
диссоциации воды с повышением температуры константа возрастает, но логарифм
константы, оставаясь отрицательной величиной, уменьшается по абсолютному
значению В произведении 71g К температура возрастает, а логарифм константы
равновесия уменьшается, но поскольку степень роста температуры выше по
сравнению с уменьшением логарифма константы, все произведение в целом
увеличивается с повышением температуры. Следовательно, по температурному
изменению изобарного потенциала не всегда можно судить о характере
изменения константы равновесия и направлении его смещения.
Для получения значений энтальпии и энтропии диссоциации воды составим
систему двух уравнений для AGC с двумя неизвестными Д#° и ASC, например,
для температур 20 и 25° С:
79 501 = Д/Г-293,2 AS°,
79886 = ДЯ:)-298,2Д5С
Решив систему, находим Д//с = 56924 Дж/моль и AS° = -77,0 Дж/(К моль)
для интервала 20—25' С. В этом интервале температур AG 'J зависит от
температуры (в предположении, что А#у и AS~ от нее не зависят) согласно уравнению:
AGc = 56924+77,0r.
Для средней температуры интервала 22,5° С находим:
AG° = 56924+ 77,0 • 295,7=79693 Дж/моль.
Точно так же вычисляются термодинамические характеристики диссоциации воды
при других температурах
В табл 16 приведены значения А5° и Д#с в интервалах температур двух их
соседних значений, взятых из табл 15. с ПОВЫ1Пением температуры
Кв возрастает, что говорит о
согласии с принципом Ле Шателье об
эндотермичности процесса
диссоциации (AH°mcti>0). С ростом
температуры изменение
энтальпии уменьшается чем выше
температура, тем слабее связи в
молекуле Н2О и тем меньше энергии
требуется на их разрыв.
Изменения энтропии для всех
температур отрицательны. При
диссоциации молекулы Н2О
образуются два иона Казалось бы,
беспорядок в системе должен
возрастать, энтропия системы ионов Н+ и ОН' должна быть выше энтропии
молекулярной воды и изменение энтропии должно быть положительной
величиной. Приведенные в табл. 16 отрицательные значения А5^ могут быть
объяснены на основании данных о структуре воды. Упрощенное объяснение состоит
в том, что ионы Н+ и ОН~, образующиеся при диссоциации воды, сильно
взаимодействуют с окружающими молекулами воды
Таблица 16
Изменение энтропии и 'энтальнии диссоциации
воды при различных темпера гурах
Интервал
темпера iyp, *с
10—15
15—20
20—25
AS ,
Дж/(К моль)
-70,0
-73,0
-77,0
АН ,
кДж/моль
58.962
58,097
56,924
202
В чистой воде ион Н + — ядро атома водорода, лишенное
электронных оболочек—практически не существует. Благодаря
очень малому размеру и относительно большому заряду ион Н +
проникает в электронные оболочки молекул Н2О и образует ион
гидроксония (точнее, катион оксония) Н3О+:
Н++Н2О = Н3О+.
Доказано, что концентрация свободных ионов Н+ в чистой воде
составляет не более 1 10~36 моль/л, поэтому процесс
диссоциации воды правильнее записать так:
2Н2О = Н3О++ОН".
Образующиеся ионы Н3О+ и ОН" окружаются молекулами
воды, которые вследствие дипольного характера ориентируются
вокруг ионов частями диполей с противоположным ионам ОН ~
и Н3О+ знаком заряда. Схематически явление диссоциации воды
изображено на рис. 91.
Таким образом, диссоциация воды приводит к образованию
упорядоченных структур, и в результате диссоциации происходит
общее уменьшение энтропии системы. При повышении
температуры эти структуры, конечно, разрушаются, что должно привести
к росту энтропии. Однако диссоциация воды сильнее подвержена
влиянию температуры, чем процесс разрушения структур,
и уменьшение энтропии в системе вследствие образования
упорядоченных структур превышает ее возрастание из-за термического
разрушения. В результате с повышением температуры изменение
энтропии при диссоциации воды уменьшается, А5°<0
(разумеется, до определенного предела, после чего она, по-видимому,
начнет увеличиваться).
Таковы противоречивые особенности термодинамики
процесса диссоциации воды. Они обусловлены самой природой воды,
в частности, дипольным характером ее молекулы, особенностями
гибридизации электронных орбиталей атома О, способностью
образовывать водородные связи, а также тенденцией ионов Н +
п н2о = (V
Рис. 91 Схема диссоциации воды
203
и ОН к гидратации (например, Н+ существует в виде ионов
Н3О+, Н5О2 + , Н7О3+, НлН2О+).
Учитывая природу ионов, образующихся при диссоциации,
этот процесс для воды следовало бы записывать таким образом:
A+/и+«)Н2О=(Н-тН2О)Ч(ОН-«Н2О)~.
Образование противоположно заряженных ионов из
нейтральных молекул (ионизация, самоионизация, автопротолиз) —
явление, свойственное электролитам. В отсутствие воды или
других электролитов диссоциация серной кислоты происходит
согласно уравнению:
В небольшой степени протекает диссоциация безводного фторо-
водорода (электропроводность жидкого HF очень низкая):
Диссоциация жидкого аммиака также ничтожно мала:
Диссоциация жидкой азотной кислоты сопровождается
образованием молекулы воды:
Способность жидких веществ к диссоциации (ионизации)
количественно характеризуется константой равновесия этого
процесса. Однако более принято пользоваться не константами
равновесия, а ионными произведениями, которые, как и ионное
произведение воды, представляют собой произведение константы
равновесия на число молей вещества, содержащихся в 1 л этого
вещества (т. е. на молярную концентрацию вещества). Ионные
произведения некоторых жидкостей приведены в табл. 17.
Таблица 17
Ионное произведение некоторых жидких веществ при 20° С
Вещество
H2SO4
нсоон
СНзСООН
н2о
СН3ОН
с2н5он
NH3
Уравнение диссоциации
2H2SO4 = H3SO4+ + HSO4"
2НСООН-НСООН24 +НСОО"
2СН3СООН = СН3СООСН2+ +СН,СО(Г
2Н2О-Н3О++ОН-
2СН3ОН-СН3ОН2+ +СН3О
2С2Н5ОН = С2Н5ОН2"+С2Н5О
2NH3 = NH4++NH2-
Ионное
произведение
МО
5-Ю
3 103
1 104
2 107
8-КГ20
3 КГ32
(при 50 С)
204
Процессы, связанные с диссоциацией молекул веществ и
взаимодействием образующихся ионов между собой, а также с
молекулами растворителя, используются в теориях кислот и оснований.
Процессы, проходящие в жидкой воде, аммиаке, серной
кислоте и других жидких веществах, осуществляются путем
перехода протона от одной молекулы к другой. Вещества, частицы
которого (молекулы, ионы) отдают протоны, относятся к классу
кислот, а вещества, частицы которого присоединяют протоны,
составляют класс оснований. Такие определения трактуются в
рамках теории кислот и оснований, разработанной Й. Брёнстедом.
Согласно этой теории функции кислот и оснований могут
выполнять как молекулы, так и ионы, причем продукты реакции также
становятся кислотой и основанием:
H2SO4 + Н2О - HSO4" + Н3О\
кислота I основание 1 основание 2 кислота 2
Эта теория ограничена веществами, в состав которых входит
водород, хотя имеется большое число веществ кислотного
характера, не содержащих водорода. Согласно другой теории (Г.
Льюис) кислотой называется вещество, присоединяющее пару
электронов, а основанием—вещество, отдающее пару электронов. Таким
образом, кислота—акцептор пары электронов, а основание—
донор электронной пары. Эта теория включает не только
кислоты и основания в традиционном их понимании, но и электронно
ненасыщенные соединения, например, галогениды бора,
алюминия, олова, оксиды ряда металлов.
В реакции
= H3N — BF3
BF3—кислота, a NH3 — основание. (В полученном соединении
осуществляется донорно-акценторное взаимодействие,
обозначаемое стрелкой.)
В подобного тина кислотно-основных реакциях происходит
обобществление электронной пары основания.
В зависимости от условий некоторые вещества проявляют
либо кислотные, либо основные свойства. Они называются ам-
фотерными соединениями (или амфолитами). Жидкие Н2О, NH3,
HNO3, H2SO4—амфотерные вещества, так как образующиеся
при их самоионизации ионы—и кислоты, и основания. Ион
Н3О+—кислота, он может отдавать протон, а ион
ОН"—основание, так как может принимать протон:
Н3О+ = Н+ + Н2О; ОН" + Н+-Н2О.
кисло Iа основание
Просуммировав оба уравнения, получаем уравнение
диссоциации воды, в котором одна из исходных молекул воды проявляет
себя как основание, а другая—как кислота:
205
2Н2О = Н3О+ + ОН".
кислота основание
Точно так же в жидкой серной кислоте ион H3SO4+ —кислота,
а ион HSO4~—основание:
H2SO4 + H2SO4 = H3SO4+ + HS04"
кислота I основание 1 кислота 2 основание 2
В приведенных примерах процесс ионизации обусловлен
способностью молекул и отщеплять, и присоединять протон.
Возможны процессы ионизации, сопровождающиеся переходом
других частиц, например:
В этих процессах ионы BF2+ и SbCl2 + —кислоты, а ионы
BF4~ и SbQ4~ — основания. Исходные молекулы—амфотерные
электролиты: одна из молекул в приведенных уравнениях
проявляет свойства основания, а другая—кислоты.
Противоположный процесс—соединение ионов, играющих
роль кислоты и основания, с образованием электрически
нейтральных молекул (также кислоты и основания) называется
нейтрализацией:
Н3О+ + ОН" = 2Н2О{Ж),
кислота основание
SbCl2+ + SbCl4- = 2SbCl3<«).
кислота основание *»
Изложенные представления о кислотно-основных равновесиях
лежат в основе современной трактовки кислотно-основных
свойств веществ в растворах. Действительно, чистая вода всегда
будет представлять собой нейтральную среду, так как
содержание ионов Н3О+ и ОН" одинаково. Чистая серная кислота также
имеет нейтральную реакцию из-за равенства концентраций ионов
H2SO4+ и HSO4". Свойства этих жидкостей резко изменяются
при введении в воду даже малых количеств серной кислоты или,
наоборот, при добавлении к серной кислоте малых количеств
воды. Сравнение по ионным произведениям процессов:
2Н2О-Н3О++ОН"; #=11014,
2H2SO4 = H3SO4++HSO4-; #=11(Г5
показывает, что в водном растворе ион H3SO4+ проявляет
большую склонность к отдаче протона иону ОН", чем ион Н3О+,
передающий протон иону HSO4~. В результате протекает
процесс нейтрализации:
H3SO4+ + ОН" =H2SO4 + H2O.
кислота основание
206
Запишем уравнение реакции нейтрализации в виде суммы
двух уравнений:
4 34
2O = H3O++OFT
= H3O++H2SO4+H2
или H2SO4+H2O=:H3O++HSO4-
Таким образом, в рамках теории электролитической
диссоциации понятия «кислота» и «основание» имеют следующие
определения.
Кислота—вещество, которое в растворе диссоциирует с
образованием катиона, представляющего собой продукт
присоединения протона Н+ к молекуле растворителя.
Основание—вещество, которое в растворе диссоциирует с
образованием гидроксид-иона.
ГЛАВА 7
КРИСТАЛЛИЧЕСКОЕ СОСТОЯНИЕ И РЕАКЦИИ
В ТВЕРДЫХ ФАЗАХ
§ 31. СТРОЕНИЕ КРИСТАЛЛА
Наиболее сильное взаимодействие между частицами
проявляется в кристаллическом состоянии вещества. Сила этого
взаимодействия такова, что частицы образуют определенную
пространственную структуру—кристалл, в котором они закономерно
расположены на фиксированном расстоянии друг от друга.
Кристалл ограничен плоскими гранями, которые пересекаются
по прямым линиям—ребрам. Углы между гранями обусловлены
внутренним строением кристалла и зависят от типа химической
связи между частицами, от ее энергии, углов и числа связей
меж,цу частицами. Существование кристаллов является
следствием исключительно высокого порядка в расположении частиц,
составляющих кристалл.
Внешняя форма кристаллов одного и того же вещества может
быть различной, но если все углы между гранями равны, можно
предположить, что кристаллы имеют одинаковый состав.
Различие в формах кристаллов одного и того же вещества
обусловлено тем, что кристаллы формируются в условиях неравных
скоростей роста граней.
На рис. 92 показаны некоторые возможные формы
кристаллов кварца, образованные сочетанием призмы с двумя
шестигранными пирамидами.
Символами /, Я и /77 обозначены грани,
углы между которыми одинаковы.
Все кристаллы кварца можно
сделать совершенно одинаковыми
путем шлифования на определен-
Рис. 92. Некоторые формы кристал- "Ую глубину различных граней по
лов кварца параллельным плоскостям.
208
Наличие у кристаллов граней и постоянство углов между
ними свидетельствуют о том, что структура кристалла
образована частицами, расположенными на строго определенных
расстояниях друг от друга. Пространственная совокупность частиц
в структуре твердого тела образует кристаллическую решетку—
присущее кристаллу периодически повторяющееся в трех
измерениях правильное расположение частиц (атомов, ионов, молекул).
Кристаллическая решетка—это математическое
(геометрическое) понятие; оно может быть определено как группа точек,
получающихся при взаимном трехкратном пересечении в
пространстве плоскостей трех семейств, причем все плоскости
каждого семейства параллельны и равноудалены друг от друга.
Точки пересечения всех трех семейств плоскостей называются
узлами кристаллической решетки, а точки, расположенные между
узлами на равном от них расстоянии,—междоузлиями.
Весь объем кристалла делится тремя семействами
параллельных плоскостей на параллелепипеды, называемые
элементарными ячейками. Именно правильная и периодическая
повторяемость расположения частиц в структуре кристалла позволяет
представить решетку в виде множества элементарных ячеек.
Элементарная ячейка—наименьший объем кристаллической
решетки, с помощью которой можно построить (мысленно) всю
структуру кристалла путем последовательного приложения таких
ячеек друг к другу в трех пространственных направлениях, т. е.
путем параллельного переноса (трансляции) ячейки в трех
направлениях (рис. 93). Для описания кристаллической решетки
достаточно знать расположение частиц в элементарной ячейке,
которое характеризуется ее параметрами. Параметры элементарной
ячейки включают длины ее ребер—периоды идентичности, или
периоды решетки, а, Ь, с и углы между ребрами, а, р, у. Число
частиц, непосредственно окружающих данную частицу в
кристаллической решетке и расположенных на ближайших и одинаковых
расстояниях от центральной частицы,—это ее координационное
число.
Многие свойства кристаллов
(механические, оптические, электрические,
магнитные и др.) зависят от направления их
измерения. Проявление неодинаковых
физических свойств кристалла по его
разным направлениям называется
анизотропией. Анизотропия вызвана тем, что
внешнее воздействие на кристалл
осуществляется чер'ез различное число узлов
кристаллической решетки, которое
определяется направлением воздействия (рис. р^* Jzl Схема построе-
с\л\ ж/* ния структуры кристйл~
94). Количественные характеристики ме- ла из элементарных
ханических, оптических, электрических, ячеек
209
о
о о о
Рис. 94 Схема,
поясняющая причину анизотропии
кристалла.
С i ре 1ки показывают направления
внешнего воздействия на кршпа.и
магнитных свойств анизотропных
веществ зависят от направления измерения
и меняются при повороте тела. У
аморфных веществ, газов и большинства
жидкостей свойства одинаковы во всех
направлениях, т. е. они изотропны.
Анизотропными могут быть
некоторые жидкости (жидкие кристаллы, см.
ниже). Ряд твердых веществ под
действием электрических или магнитных
полей, а также после механических
воздействий (удары, ковка) становятся
анизотропными.
Изучение анизотропии кристаллов
дает ценные сведения об их внутреннем
строении и характере связей.
Проявление веществом анизотропии доказывает, что его структура
представлена правильным пространственным расположением частиц.
Типичным примером вещества с ярко выраженной
анизотропией является графит. Кристаллическая структура графита
представлена параллельными слоями атомов углерода. Все углы
между связями равны 120° С (д/?2-гибридизация орбиталей
атомов углерода). Энергия связи между атомами в слое «168
Дж/моль; слои связаны силами Ван-дер-Ваальса с энергией связи
в десять раз более слабой («17Дж/моль), Это и является
причиной особых механических свойств графита—легкости
скольжения слоев относительно друг друга и смазочных (мажущих) его
качеств.
При нагревании графита до «430° С происходит сжатие
кристалла по направлению, параллельному слоям, а при более
высоких температурах — расширение. При нагревании кристалл
расширяется по направлению, перпендикулярному слоям, причем
в 20 раз сильнее, чем в направлении слоев.
Теплопроводность вдоль слоев в пять раз выше по сравнению
с теплопроводностью в перпендикулярном направлении.
Электропроводность в направлении слоя, т. е. вдоль плоскостей
перекрывания я-орбиталей, близка к металлической и в сотни раз
больше электропроводности в перпендикулярном направлении.
Магнитная восприимчивость диамагнитного графита в
направлении вдоль слоев примерно в 50 раз выше, чем в
перпендикулярном направлении.
По природе входящих в состав кристалла частиц и по типу
химической связи кристаллические решетки подразделяются на
молекулярные, ионные, атомные (ковалентные) и металлические.
В узлах молекулярных решеток располагаются молекулы
вещества. Вещества, имеющие молекулярные решетки, обычно имеют
низкие температуры плавления и кипения, высокое давление на-
210
Рис. 95. Молекулярная решетка кристаллов аргона (а) и иода (б)
сыщенного пара. К такого типа веществам относятся, например,
твердые Н2, О2, N2, галогены, СО2, все благородные газы (хотя
они одноатомны) и многие органические вещества.
Кристаллические Аг и 12 имеют одинаковые решетки (рис. 95).
Координационное число для атома аргона равно 12. Связь между частицами
в решетке осуществляется силами Ван-дер-Ваальса.
В узлах ионных решеток находятся ионы противоположных
зарядов. Связь между частицами осуществляется за счет
электростатических сил притяжения, хотя может налагаться и сила
взаимодействия перекрывающихся орбиталей. Ионные кристаллы
отличаются высокой температурой плавления. Такую решетку
имеют кристаллы NaCl с координационным числом натрия
6 и CsCl с координационным числом цезия 8 (рис. 96). Как
правило, для хлоридов щелочных металлов при переходе вниз по
подгруппе элементов их координационные числа возрастают.
В узлах кристаллической решетки могут находиться не только
простые ионы, но и сложные, например, как в кристаллах
NH4NO3 и (NH4JSO4.
В узлах атомной решетки располагаются атомы, связанные
друг с другом ковалентной связью. Вещества с такого типа
решетками имеют высокую температуру плавления и низкое
давление насыщенного пара. Характерным примером является
алмаз, в кристаллической решетке которого все межъядерные
расстояния и все углы между связями равны. Это обусловливает
его очень низкую энтропию и как следствие—высокую
твердость.
Рис 96 Ионная решетка кристалла хлорида натрия (я),
элементарная ячейка его (б) и кристалла хлорида цезия (в)
211
Металлическая решетка является
разновидностью атомной и отличается тем, что в ее
узлах находятся атомы и положительно
заряженные ионы (катионы). В пространстве между
узлами перемещаются электроны,
обеспечивающие электронейтральность вещества. Эти
подвижные электроны придают металлам ха-
Рис 97 Атомная Рактерные свойства: металлический блеск, вы-
решетка кристалла сокие электропроводность и теплопровод-
натрия ность, пластичность и др. На рис. 97
изображена элементарная ячейка металлического
натрия, координационное число атома натрия равно 8.
Классификация кристаллов по типу связи основана на
изучении электропроводности, теплопроводности, температуры
плавления, теплоемкости, состава продуктов разложения кристалла
и продуктов реакций с другими веществами.
Существует классификация кристаллических структур,
критерием которой служит характер межъядерных расстояний между
частицами. По этой классификации выделяют следующие
структуры: координационные, цепные, ленточные, слоистые,
островные и смешанные.
Координационными называются кристаллические решетки,
в которых каждая частица окружена частицами, находящимися
от нее на равных расстояниях и связанными одинаковой по типу
и прочности химической связью. К координационным
структурам относятся структуры NaCl, CsCl, металлов, алмаза и др.
Цепные структуры образуются, если действие прочной
химической связи распространяется между атомами или группами
атомов вдоль некоторого направления. Таким образом, каждая
частица связана наиболее прочно только с двумя другими.
Таковы зигзагообразные молекулярные структуры селена и теллура,
некоторые силикаты. Между собой цепи атомов связаны
слабыми силами Ван-дер-Ваальса.
Цепи, соединенные поперечными связями, образуют ленты.
Известны ленточные структуры среди силикатов.
Дальнейшее соединение в плоскости ленточных структур
приводит к формированию слоистых структур. Эти структуры также
характерны для силикатов. Ярко выраженной слоистой
структурой обладает графит, слои атомов углерода в котором также
связаны силами Ван-дер-Ваальса. Межъядерные расстояния
в слое значительно меньше межъядерного расстояния между
слоями.
Островные кристаллические структуры содержат
«островки»—группы из ограниченного числа частиц. Внутри
«островка» химические связи прочнее и короче, чем между
«островками». Островные структуры имеют силикаты. К этому типу
структур относят также кристаллы, в узлах кристаллической
212
решетки которых находятся молекулы, сложные и комплексные
ионы. Например, в кристаллах K4[Fe(CNN] роль «островков»
играют ионы [Fe(CNN]4~. К островной можно причислить
структуру кристаллического иода.
В смешанных структурах обнаруживаются две или более выше
рассмотренных структур.
В зависимости от внешних условий (температура, давление)
некоторые вещества способны существовать в нескольких
состояниях с различной кристаллической структурой, называемых
полиморфными модификациями. Так, графит и алмаз—полиморфные
модификации углерода, серое и белое олово—модификации
металлического олова, арагонит и кальцит—полиморфные
модификации карбоната кальция СаСО3.
Полиморфные модификации обычно обозначают буквами
греческого алфавита а, Р, у, 5 и т. д. в порядке повышения
температурного интервала существования модификации.
Например, серое олово, устойчивое ниже 286,4 К A3,2° С), обозначают
как a-Sn, а белое олово, устойчивое выше этой температуры,—
p-Sn.
Структура решетки низкотемпературного серого олова (a-Sn)
такая же, как и у алмаза, но со значительно большим
межъядерным расстоянием, а = 0,646 нм (у алмаза я = 0,154нм). Белое
олово (P-Sn) имеет кристаллическую решетку, которая
получается от предыдущей сжатием вдоль одного направления.
Часто обнаруживается, тю два вещества образуют кристаллы
одинаковой формы и структуры. Такое явление называют
изоморфизмом. Например, кристаллы NaNO3 и СаСО3 близки по форме
и межъядерным расстояниям, но различаются по другим
свойствам. Обычно понятие изоморфизма используется для описания
явления образования кристаллических фаз переменного состава
вследствие взаимной замещаемости частиц (атомов, молекул,
групп) в узлах кристаллической решетки. Это явление правильнее
называть замещением или растворением. Например, в
кристаллической структуре КС1 хлорид-ионы могут быть замещены на
бромид-ионы, а в структуре КВг бромид-ионы—на
хлорид-ионы, при этом могут образоваться кристаллы состава КОВ
01
Многие изоморфные вещества обладают способностью
кристаллизоваться совместно с образованием общей
кристаллической решетки, узлы которой заняты различными частицами.
Такого типа кристаллы переменного состава называют
смешанными кристаллами (или твердыми растворами замещения).
Например, карбонаты марганца и магния МпСО3 и MgCO3
изоморфны и при совместной кристаллизации образуют кристаллы
MM^O
Явление изоморфизма проявляется, если однотипен
молекулярный (формульный) состав веществ, элементарные ячейки
213
имеют аналогичное строение и близкие размеры, а радиусы
ионов отличаются не более чем на 10—15% и их заряды
одинаковы. Последнее требование иногда необязательно.
Осадок BaS04, выпадающий из раствора, содержащего
КМпО4, имеет красную окраску, которая не исчезает при
промывании осадка водой, т. е. КМпО4 распределен не на поверхности
кристаллов BaS04, а внутри их. Следовательно, BaSO4 и КМпО4
образуют смешанные кристаллы. Действительно, КМпО4
изоморфен BaSO4, хотя по химическим свойствам обе соли имеют мало
общих признаков и заряды катионов и анионов в обеих солях
различны. Однако ионные радиусы довольно близки:
ЛВа2+ =0,143. /?к+ =0.133, Rsol~ =0,230, ЛМпо4 = 0,240 нм. Но
основная причина проявления изоморфизма состоит в том, что
расстояния катион—анион в кристаллах обеих солей одинаковы:
Ва2 + — SO42~ d=0,143+0,230 = 0,373 нм,
К+—МпО4~ d= ОД33+0,240-0,373 нм.
В свою очередь, BaSO4 изоморфен K[BF4], в результате чего
возможно образование изоморфных составов в системе
KMnO4—BaSO4—K[BF4].
Близость значений радиусов ионов приводит к тому, что
часто изоморфными оказываются кристаллы с катионами
элементов, расположенных по диагонали в Периодической системе,
например Li+— Mg2 + , Na+—Ca2 + , Be2*— Al3 + . Еще более
интересно явление, когда два одинаковых иона замещаются
одновременно на два разных иона, но так, что сумма радиусов
замещающих ионов примерно совпадает с удвоенным радиусом
замещаемо о иона, а суммарный заряд ионов сохраняется:
2Са ' A,04 HM) = Na+ @,098 нм) + Ьа3+ @,104 нм),
2Ti44 @,064 »M)-Fe3+ @,067 HM)+Nb5+ @,066 нм).
В некоторых минералах, содержащих Na, Ca, A1 и Si,
возможно одновременное замещение кальция и алюминия на натрий
и кремний с сохранением суммарного заряда ионов, но с
изменением сумм их радиусов:
Са2+ @,104 нм) + А13+ @,057 HM) = Na+ @,098 нм) +
+ Si4+ @,039 нм).
Такое явление объясняется тем, что замещение алюминия
и кремния происходит в группах А1О45~ и SiO44~, которые
близки по размерам.
В истории определения атомных масс элементов немалую роль сыграл
изоморфизм, позволивший предсказать валентность изучаемого элемента и далее
по значению его эквивалентной массы (молекулярной массы эквивалента)
вычислить атомную массу. Так, оказалось, что соли гольмия и тулия кристаллизуются
214
совместно с солями некоторых трехвалентных элементов. Отсюда был сделан
вывод о трехвалентности гольмия и тулия и установлены их атомные массы.
Изоморфное замещение—это процесс образования твердых
растворов замещения. Твердые растворы другого
типа—растворы внедрения—образуются, если радиусы внедряющихся атомов
или ионов сильно различаются, а размеры пустот между узлами
решетки близки к размерам внедряющегося атома. Это значит,
что размер внедряющихся атомов должен быть меньше размеров
атомов основной кристаллической решетки (обычно не более 0,6
от размеров атома растворяемого вещества).
Некоторые вещества способны существовать в состоянии,
промежуточном между жидким и кристаллическим, они образуют так
называемые жидкие кристаллы. Жидкие кристаллы обладают
одновременно рядом свойств жидкости, например, текучестью,
и кристалла, например анизотропией. Жидкокристаллическое
состояние обнаруживается в определенном температурном
интервале, ниже которого вещества ведут себя как кристаллы, а выше—
как жидкости. Жидкие кристаллы образуют органические
вещества, молекулы которых имеют форму палочек, вытянутых
пластинок и дисков. Например, и-азоксианизол
J-0-СНз
проявляет жидкокристаллическое состояние между 114 и 135° С.
Повышенный порядок в расположении молекул у жидких
кристаллов обусловлен особым строением молекул, способных
взаимодействовать боковыми частями, что приводит к взаимной
ориентации молекул. В зависимости от типа ориентации молекул
жидкие кристаллы разделяются на несколько групп.
Обсудим строение жидких кристаллов, построенных из
палочкообразных молекул (рис. 98). У нематических жидких
4
'/*////////.
Рис. 98. Строение жидких кристаллов:
о—иематические, б—холестерические. в—смектические
215
кристаллов оси молекул ориентированы с небольшими
отклонениями вдоль некоторого направления (рис. 98, а). У хо-
лестерических жидких кристаллов оси молекул ориентированы
не во всем объеме кристалла, как у нематических кристаллов,
а только в плоскостях (рис. 98, б). Направление ориентации
молекул в двух соседних плоскостях различается на небольшой
угол @,5°), поэтому при переходе от одного слоя молекул
к другому молекулы поворачиваются подобно винту. В слое
молекулы перемещаются, сохраняя взаимную ориентацию, т. е.
в плоскости жидкий кристалл ведет себя как жидкость. Хо-
лестерическое состояние является по степени упорядочения
промежуточным между нематическим и наиболее
упорядоченным—смектическим состоянием. У смектических жидких
кристаллов (рис. 98, в) оси молекул ориентированы одинаково
во всем объеме, но в дополнение к этому молекулы расположены
слоями. Молекулы этого типа кристаллов в середине имеют
циклическую группу атомов, и взаимодействие групп приводит
к параллельной укладке молекул и образованию слоев.
Некоторые жидкие кристаллы могут находиться и в смектиче-
ском, и в нематическом состояниях. Фазовые превращения таких
веществ из крист аллического состояния в жидкое при повышении
температуры проходят по схеме: кристалл-»смектическая
фаза ->нематическая фаза -»жидкость. Все эти
превращения—фазовые переходы первого рода, сопровождающиеся изменением
внутренней энергии, плотности и энтропии системы. Энтальпия
перехода жидкого кристалла в жидкость в десятки раз меньше
энтальпии плавления, а энтальпия перехода смектической фазы
в нематическую еще меньше.
Вследствие низкой энергии связи между ориентированными
молекулами жидкие кристаллы легко изменяют свою структуру
под действием электрических и магнитных полей, света, давления,
температуры. Время отклика на внешнее воздействие может
составлять 1 • 10" с. Благодаря таким свойствам жидкие кристаллы
нашли широкое применение в индикаторных устройствах
электронных часов и микрокалькуляторов, дисплеях и телевизионных
экранах. Обнаружено каталитическое действие
жидкокристаллической среды — ориентация молекул в структуре жидкого
кристалла способствует взаимной ориентации реагирующих молекул.
Делаются попытки объяснить некоторые процессы,
протекающие в живых организмах, с помощью теории жидких
кристаллов. Протоплазма живой клетки по ряду свойств близка
к жидкокристаллическому состоянию. Распространение импульса
возбуждения но нерву может быть описано переходами
различных жидкокристаллических состояний. Возникновение жизни
на уровне самосборки надмолекулярных структур, возможно,
по своему механизму близко к образованию
жидкокристаллических структур.
216
§ 32. ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА КРИСТАЛЛОВ
По способности проводить электрический ток вещества
делятся на проводники, полупроводники и изоляторы (диэлектрики).
Такое деление довольно условно. Нет веществ, абсолютно не
способных проводить электрический ток, и иногда трудно
отнести вещество к тому или иному классу. Электропроводимость
зависит от температуры, давления, чистоты вещества
(содержание примесей), кристаллической структуры (ср., например, алмаз
и графит, белое и серое олово), характера химических связей
и других факторов.
Проводники электрического тока по типу
электропроводимости делятся на электронные, или проводники первого рода, и
ионные, или проводники второго рода. Электронные проводники —
это металлы в кристаллическом и жидком состояниях,
проводимость в них осуществляется перемещением электронов. Для
электронной проводимости характерно то, что прохождение тока не
сопровождается химическим превращением вещества.
Ионные проводники—это растворы и расплавы
электролитов, проводимость в них осуществляется перемещением
положительных и отрицательных ионов. Характерной особенностью их
является то, что в месте подвода тока посредством
металлического контакта (проводника с электронной проводимостью)
меняется механизм передачи тока, ионы разряжаются, а
нейтральные частицы приобретают заряд и гаким образом происходят
различные химические превращения.
Известно немало веществ со смешанным характером
проводимости. К таким проводникам относятся оксиды и сульфиды при
высоких температурах, газы в состоянии плазмы и др.
Как указано выше, электропроводимость вещества зависит от
кристаллической структуры. Типичный пример тому—алмаз
и графит, представляющие собой аллотропные модификации
углерода, из которых алмаз практически не проводит
электрический ток, а графит проявляет высокую
электропроводимость.
Неодинаковая электропроводимость алмаза и графита
объясняется различием электронного строения атомов, типа
химической связи и кристаллической структуры.
В структуре алмаза внешние электронные орбитали атома
углерода находятся в состоянии л/?3-гибридизации и все четыре
гибридные $р3-орбитали перекрываются с шкими же орбиталями
соседних атомов углерода. Области перекрывания орбиталей
располагаются точно посредине между двумя атомами углерода,
и пара перекрывающихся электронов принадлежит только этим
двум атомам. Из-за малых размеров атомных орбиталей каждая
область перекрывания не соприкасается с соседней, что
исключает переход электронов из одной области перекрывания в другую,
217
т. е. перемещение электронов по кристаллической решетке,
поэтому электропроводность алмаза ничтожно мала.
В структуре графита внешние электронные орбитали атома
углерода находятся в состоянии ,у/?2-гибридизации. Углы между
связями С—С равны 120° и атомы углерода располагаются в
одной плоскости. В отличие от алмаза графит хорошо проводит
электрический ток. Это явление можно объяснить, допустив, что
все негибридные /ьорбитали, расположенные перпендикулярно
плоскости 5/?2-гибридных орбиталей, перекрываются между
собой боковыми частями, при этом возникает к-связь. В результате
образуется одно «размазанное» электронное /?-облако,
расположенное выше и ниже плоскости 5/?2-гибридных орбиталей. Такие
«размазанные» электроны не локализованы и ответственны за
электропроводимость графита.
Если на монокристалле графита укрепить электроды
(проводники первого рода) перпендикулярно атомным слоям углерода,
то под действием электрического поля электроны будут
смещаться вдоль л-связей перекрывающихся негибридных орбиталей, что
и обусловливает высокую электропроводность графита. Если же
электроды укрепить параллельно слоям, то ток через графит не
проходит. По методу молекулярных орбиталей проводимость
графита вдоль плоскостей атомов можно объяснить
образованием единой для всех атомов молекулярной я-орбитали,
простирающейся на всю плоскость.
Для объяснения высокой электропроводимости металлов
была предложена модель, согласно которой в кристаллической
решетке металла имеются свободно движущиеся электроны,
проявляющие себя в междоузлиях кристаллической решетки подобно
молекулам газа. Если это действительно так, то составляющая
теплоемкости металла, обусловленная кинетической энергией
электронов, должна (R/2) • 3 «12 Дж/(К' * моль), и тогда общая
теплоемкость металла, определяемая суммой электронной и
решеточной [(/?/2) -6«24 Дж/(К моль)] составляющих, будет
равна 37—38 Дж/(К • моль). Однако теплоемкость металла
приблизительно равна (Л/2) -6^25 Дж/(К • моль) (правило Дюлонга
и Пти). Таким образом, теория электронного газа не может
объяснить причин проявления металлом ряда свойств.
В металлах кристаллическая решетка состоит частично из
атомов и положительно заряженных ионов и некоторого
количества электронов, которые перемещаются в пространстве между
узлами решетки. Правильнее было бы говорить, что эти нелока-
лизованные электроны способны передвигаться по
молекулярным орбиталям, связывающим атомы в структуру кристалла.
Метод молекулярных орбиталей, распространенный на
ансамбль частиц, связанных в кристалле, называется зонной
теорией кристаллов. Согласно методу молекулярных орбиталей
взаимодействие атомов приводит к образованию связывающих, раз-
218
рыхляющих и несвязывающих молекулярных орбиталей. Это
также означает, что при взаимодействии атомов происходит
расщепление атомных энергетических состояний электронов на
молекулярные.
Обсудим это явление на примере рассмотрения химического
связывания атомов лития. При взаимодейс!вии двух атомов
лития (Lil.y22iVl) образуется одна связывающая и одна
«разрыхляющая молекулярные орбитали, т. е. каждые два атомных
энергетических состояния переходят в два молекулярных состояния;
в системе из четырех атомов лития возникает четыре
энергетических состояния по числу молекулярных орбиталей (две
связывающие и две разрыхляющие). В системе, состоящей из 1 моль Li,
каждое атомное энергетическое состояние расщепляется на
NA молекулярных состояний (NA = 6,02 • 1023), так как образуется
NA молекулярных орбиталей. Поскольку число NA очень велико,
соседние молекулярные орбитали энергетически настолько
близки друг к другу (различие составляет всего 10~22 эВ), что
изменение энергии электронов в jVa молекулярных орбиталях можно
представить как непрерывную полосу энергетических уровней.
Такое энергетическое состояние NA атомов носит название
энергетической зоны.
На рис. 99 представлены энергетическая диаграмма атомной
и молекулярных орбиталей и энергетическая зона
кристаллического лития.
В зависимости от типа атомных орбшалей ($-, /?-, я?-, /-
подуровни) энергетические зоны кристалла разделяются на s-, p-,
rf-, и /-зоны. Орбитали энергетической зоны заполняются как
обычные молекулярные орбитали парами электронов в порядке
возрастания энергии и в соответствии с принципом Паули.
Поэтому максимальное число электронов равно 2NA в д-зоне, 6NA
в р-зоне, 107VA в af-зоне и 14NA
в/-зоне. Зона, которую занимают
электроны, участвующие в
образовании химических связей между
атомами кристалла, называется
валентной. Зона, расположенная
энергетически выше валентной,
называется зоной проводимости.
Расположение зон (ближе или
дальше друг от друга) и их
заполненность электронами
обусловливают свойства кристалла как
диэлектрика (изолятора),
полупроводника и проводника. При
условии перекрывания валентной
зоны и зоны проводимости
вещество ведет себя как проводник.
М
М0разр
3 Е?
АО
MOf«P
мо™
I 1
4V-
Var^ атомов
Рис 99 Схема образования
энергетической зоны кристалла лития
219
Рис 100 Энергетические зоны диэлектрика (а)ч полупроводника F) и
металла (в)
Если зоны не перекрываются, достаточно далеко удалены друг о i
друга и валентная зона полностью заполнена электронами,
вещество проявляет свойства диэлектрика. Энергетический разрыв
между зоной проводимости и валентной зоной называется
запрещенной зоной. Количественно способность веществ проводить
электрический ток оценивается по ширине запрещенной зоны АЕ.
У диэлектриков ширина запрещенной зоны выше 3 эВ, у
полупроводников от 3 до 0,1 эВ и у проводников (металлов)
запрещенная зона отсутствует, АЕ=0 (рис. 100).
Способность хорошо проводить электрический ток при
подведении невысокой разности потенциалов проявляется только при
условии, что перемещение электронов не требует преодоления
высоких энергетических барьеров. Это возможно, если в зоне
проводимости имеются незанятые уровни, т. е. перемещение
электронов можно представить как их движение по
незаполненным полностью и энергетически близким молекулярным орбита-
лям кристалла. Если в кристалле имеются энергетически далекие
друг от друга заполненные и свободные зоны, то переход
электронов из нижележащей заполненной в вышележащую
свободную зону возможен только при очень высокой разности
потенциалов. Скорость такого процесса описывается кинетическим
уравнением цепной реакции; этот процесс называется пробоем
диэлектрика. Он появляется в резком, скачкообразном
увеличении электрической проводимости материала.
Обсудим электрические свойства алмаза, кремния, графита
и металлов группы I Периодической системы элементов.
У алмаза электроны атомов углерода заполняют валентную
зону. Перевод электронов в зону проводимости требует высоких
энергий—ширина запрещенной зоны составляет Д?=5,7 эВ,
поэтому алмаз—диэлектрик (хотя по ряду других свойств его
относят к полупроводникам). Кремний имеет структуру алмаза,
и у него также заполнена валентная зона, но вследствие
энергетической близости зоны проводимости и валентной зоны
220
(Д2?=1,1 эВ) кремний проявляет свойства полупроводника.
У графита валентная зона, содержащая 2р-негибридные
электроны, и зона проводимости перекрываются, и эта модификация
углерода, не являясь металлом, хорошо проводит электрический
ток.
Элементы главной и побочной подгрупп I группы
Периодической системы (Li, Na, К, Rb, Cs и Си, Ag, Аи) на внешнем
энергетическом уровне имеют по одному s-электрону. В
кристаллах этих металлов валентная зона s-орбиталей заполнена
наполовину и энергетическое состояние электронов легко изменяется
в пределах зоны, поэтому кристаллы электропроводны.
Мерой связанности электрона в решетке металла служит
определяемая экспериментально работа выхода электрона.
Работой выхода электрона называется количество энергии, которое
необходимо затратить для удаления электрона из металла. Эта
величина может быть определена измерением наименьшей
частоты (энергии) волн света, обеспечивающей выделение электронов
из кристалла {фотоэлектрический эффект), или измерением
минимальной температуры, при которой происходит выход
электронов из металла (термоэлектронная эмиссия). Работа выхода
электрона обычно на 2—5 эВ меньше энергии ионизации
свободных атомов, так как включает кинетическую энергию электронов
в кристалле.
На рис. 101 представлено распределение электронов по
уровням и подуровням невозбужденного атома меди и энергетические
уровни электронов в изолированном атоме Си и в кристалле,
иллюстрирующие образование зоны проводимости.
Подуровни Зр и 3s и остальные нижележащие подуровни
в изолированном атоме и атоме в кристаллической решетке
совершенно одинаковы, подуровни 3d и 4s у атомов в
кристаллической решетке расширены в зоны, вследствие чего энергия,
необходимая для удаления электрона из металла, т. е. работа
выхода электрона Евых, меньше, чем энергия для его удаления из
свободного атома, т. е. энергия ионизации /ион.
Атом меди имеет только один валентный электрон, и
валентная 45-зона заполнена наполовину. В результате
перекрывания 45-зоны с З^-зоной 45-зона может принимать с З^-зоны
дополнительные электроны. Если к металлу приложить разность
потенциалов, энергетическое состояние электронов изменяется
в пределах этих двух перекрывающихся зон. Перекрывание зон
равноценно образованию молекулярных орбиталей,
распределяющихся по всему объему кристалла, что позволяет электронам
перемещаться в электрическом поле.
Как видно из рис. 101, З^-подуровень, даже заполненный
полностью, нельзя рассматривать как внутренний энергетический
подуровень, не участвующий в образовании химических связей.
Атом калия также имеет один 4я-электрон, однако его внутренние
221
4
3
г
1
t
¦¦
¦¦
liil
$
f
p
¦¦
i
d
¦¦
МММ!
f
Рис. 101. Электронная
конфигурация невозбужденного
атома меди (а) и
энергетические уровни электронов в
атоме и кристалле Си (б).
400
4000
40000
3s
2o
2р
2s
Атом Си
Кристалл Си
шесть 2/?-электронов не перекрываются и в формировании связи
не участвуют. Это приводит к значительно большей энергии
связи между атомами в кристалле меди, чем в кристалле калия:
на перевод I моль кристаллической меди в газообразное
состояние изолированных атомов требуется 339 кДж/моль (АН атоми-
зации), а для калия—всего 92 кДж/моль.
Ряд свойств переходных металлов—высокие температуры
плавления и кипения, большая энтальпия атомизации,
сравнительно малые межъядерные расстояния, высокая твердость—
свидетельствуют о том, что большая часть валентных электронов
участвует в формировании направленных связей между атомами,
222
а число свободных электронов ненамного превышает единицу на
атом. Например, в кристалле циркония на один атом приходится
1,4 свободных электрона, а в кристалле ниобия—1,2 электрона;
остальные электроны образуют прочные ковалентные связи
между атомами.
В полупроводниках ширина запрещенной зоны невелика; это
означает, что для перевода электронов из вален гной зоны в зону
проводимости требуется невысокая энергия, для обеспечения
которой достаточно нагревания или освещения вещества. Так, при
действии одного кванта света один электрон в решетке кремния
переходит в зону проводимости, а вместо него в валентной зоне
образуется одна положительно заряженная (относительно
электронов) дырка.
Если к полупроводнику приложить невысокую разность
потенциалов, то это вызовет движение электронов в зоне
проводимости (дырочная проводимость я-типа) и одновременное
перемещение дырок (дырочная проводимость р-типа). Движение дырки
происходит по следующему механизму: электрон, находящийся
рядом с дыркой, занимает ее положение, при этом на его месте
снова возникает положительно заряженная дырка. Соседний
электрон осуществляет подобный переход, и т. д. Таким образом,
в валентной зоне дырки будут перемещаться в сторону
отрицательного электрода, а в зоне проводимости электроны будут
двигаться в сторону положительного электрода (рис. 102).
Проводимость такого типа называется собственной.
Следует иметь в виду, что движение дырок — это не движение
носителя положительного заряда в электрическом поле, а
следствие перескоков электронов. Перемещение носителя
положительного заряда означало бы перемещение положительного иона и,
следовательно, перенос вещества в электрическом поле.
Известны также ионные полупроводники, в кот орых
электрический ток обусловлен движением ионов. В электронных
приборах ионные полупроводники не используются, гак как перенос
вещества приводит к изменению состава и струк туры материала,
вследствие чего изменяются его свойства. В электронных
полупроводниках ток переносится
электронами и дырками, и
приборы на основе таких
полупроводников очень стабильны.
В кристаллической решетке
полупроводников с собственной
проводимостью число
электронов равно числу дырок (п=р).
Типичными полупроводниками
среди простых веществ являются Рис 102 Схема характеризующая мс_
Кремнии, германии, селен, тел- ханизм собственной проводимости по-
лур. Некоторые другие простые лупроводника
223
Зона проводимости
Валеитиая зона
риоды
1
2
3
4
5
6
ГРУППЫ
г
н
Li
Na
К
Rb
Cs
11
Не
Be
Mg
Ca
Sr
Ba
III
В
Al
Ga
In
Tl
IV
С
Si
Ge
Sn
Pb
V
N
P
As
Sb
Bi
VI
0
5
Se
Те
Po
VII
F
Cl
Bf
1
At
VIII
Ne
Ar
Kf
Xe
Rn
С - аямаз;
Sn- серое олово
Рис 103 Положение полупроводников
(простые вещества) в Периодической
системе элементов
вещества в кристаллическом
состоянии также проявляют
полупроводниковые свойства
в зависимости от условий. Эти
полупроводники
располагаются в Периодической системе
между металлами и
неметаллами (рис. 103).
Электрические свойства
большинства
полупроводящих веществ объясняются
тем, что число электронов
в кристаллической решетке не
равно числу дырок. Такое
состояние достигается
введением примесей (легированием).
Примесь, атомы которой
в кристаллической решетке
основного вещества отдают
электроны, называется донорной. У полупроводников с донорны-
ми примесями и»/?, и они относятся к полупроводникам л-типа,
т. е. с электронной проводимостью. Примесь, атомы которой
захватывают электроны от атомов основного вещества,
называется акцепторной. У полупроводников с акцепторными
примесями />»и, и они являются полупроводниками /кгипа с дырочной
проводимостью. Характер проводимости, достигаемый
легированием, можно предсказать сравнением обычных валентных
состояний атомов примеси и основного вещества. Если валентность
атомов примеси выше валентности основного вещества, то
атомы примеси отдают электроны, й»ри примесь донорная. Если
же валентность атомов примеси ниже, то ее атомы захватывают
электроны, /?»п и примесь акцепторная. Легирование всегда
повышает проводимость полупроводника.
Фосфор, мышьяк или сурьма (имеющие электронное строение
внешнего энергетического уровня s2p3 и проявляющие
валентность 5), будучи введенными в кристаллические решетки
германия или олова (электронное строение внешнего уровня s2p2,
валентность 4), ведут себя как донорные примеси, т. е. отдают
электроны и создают проводимость «-типа. Если же в германий
или кремний ввести бор, алюминий, галлий или индий
(электронное строение внешнего уровня s2p1, валентность 3), то атомы
примеси захватывают четвертый электрон и полупроводник
обнаруживает проводимость /кгипа.
В рассмотренных примерах атомы примеси замещают в
кристаллической решетке атомы простого вещества (образуются
твердые растворы замещения). Если же в германий или кремний
ввести литий Bs1), то его атомы, имеющие малые размеры,
224
будут внедряться в междоузлия (образуются твердые растворы
внедрения) и отдавать свои электроны, т. е. создавать
проводимость я-типа.
Полупроводниковые свойства проявляют многие химические
соединения. Такие свойства имеют соединения р-элементов
III группы с /^-элементами V группы, например GaP, InP, InSb
(среднее число валентных электронов, приходящихся на один
атом в соединении, равно 4). Соединения р-элементов II
и VI групп, как, например, ZnS, ZnTe, ZnSe, CdS, CdSe, CdTe,—
также полупроводники (на один атом соединения приходится
четыре валентных электрона). Особенно много
полупроводниковых соединений образуют /7-элементы VI группы, например, PbS,
PbTe, SnTe, GeTe, Cu2O (используется в выпрямителях), Bi?O3
(используется в термоэлементах) и т. п. Известно немало
проводников и более сложного состава, например, GaAs^P, _х,
InxGa1_xSb, ZnSnP2, CdGeAs2.
Обнаружены полупроводники и среди кристаллов
органических веществ. К ним относятся многие органические красители (в
том числе хлорофилл), нафталин и др. Для органических
полупроводников важно наличие сопряженных связей в молекуле.
Электропроводимость металлов выше 104 Ом~1 *см~\
диэлектриков— ниже 10 ~10 Ом -см (при 298 К), проводимость
полупроводников лежит между этими значениями. Однако
главное отличие полупроводников от металлов состоит не в
количественной оценке электропроводности, а в характере зависимости
проводимости от температуры (рис. 104). Температурная
зависимость проводимости металлов определяется временем
свободного пробега электронов. С повышением температуры тепловые
колебания атомов в узлах кристаллической решетки
усиливаются, что приводит к увеличению взаимодействия их с электронами
и к понижению проводимости.
В полупроводниках число электронов, переходящих через
запрещенную зону в зону проводимости,
и число дырок, образующихся в
валентной зоне, возрастает с повышением
температуры: электропроводность
полупроводников с повышением температуры
увеличивается (до определенного
предела, пока полупроводник не начинает вести
себя как металл). Таким образом,
разделение веществ на металлы и
полупроводники отражает не только меньшую
электропроводность полупроводников, но
и отличие в знаке температурного
коэффициента электропроводности.
Представления о валентных зонах
и зонах проводимости могут быть ис-
1/т
Рис. 104 Зависимость
электропроводимости
металла (/) и
полупроводника B) от температуры
8 2452
225
пользованы для объяснения ряда процессов, протекающих в
кристаллических веществах, например, для объяснения
люминесценции—испускания электромагнитного излучения после
возбуждения вещества видимым или ультрафиолетовым светом,
облучением частицами (электронами, а-частицами) и т. п.
Механизм люминесценции можно описать тремя стадиями:
1) поглощение энергии веществом с переходом электронов в
возбужденное состояние, 2) сохранение энергии в возбужденном
состоянии и 3) излучение энергии в результате возвращения из
возбужденного состояния в обычное. Поглощение энергии
кристаллом происходит при возбуждении электрона из валентной
зоны в зону проводимости с одновременным образованием
дырки в валентной зоне. При этом энергия сохраняется, пока
электрон находится в зоне проводимости. Излучение возникает при
возвращении электрона из зоны проводимости в валентную зону
и взаимодейс! вии его с дыркой.
Продолжительность люминесценции связана с тем, что
электрону, находящемуся в зоне проводимости, требуется некоторая
дополнительная энергия (энергия активации) для перехода на
основной энергетический уровень и взаимодействие электрона
с дыркой в валентной зоне также протекает не мгновенно. По
длительности свечения различают резонансную люминесценцию,
или флюоресценцию, и спонтанную люминесценцию, или
фосфоресценцию.
Вещества, способные люминесцировать,—люминофоры —
применяются в лампах дневного света, в экранах телевизоров,
в устройствах для преобразования различных видов энергии
в световую. В люминесцентных лампах используют кристаллы
MgWO4, (ZnBeJSiO4 и др. Для покрытия экранов цветных
телевизоров используют люминофоры: ZnS (синий), ZnSe (зеленый)
и Zn3(PO4J (красный).
§ 33. МАГНИТНЫЕ СВОЙСТВА КРИСТАЛЛОВ
По характеру поведения в магнитном поле вещества
разделяют на диамагнитные, парамагнитные и ферромагнитные,
различающиеся по магнитной восприимчивости. Магнитная
восприимчивость определяет способность вещества
взаимодействовать с магнитным полем. Диамагнитные вещества (диамагнети-
ки) обладают отрицательной магнитной восприимчивостью —
они приобретают магнитный момент с направлением,
противоположным направлению внешнего магнитного поля.
Парамагнитные вещества (парамагнетики), наоборот, обладают
положительной магнитной восприимчивостью и во внешнем магнитном
поле приобретают магнитный момент с направлением внешнего
магнитного поля.
226
Диамагнитные вещества ослабляют
магнитное поле (по сравнению с
напряженностью магнит ного поля в вакууме) и,
как следствие, выталкиваются из
пространства между полюсами магнита
(рис. 105). Парамагнитные вещества
усиливают магнитное поле и втягиваются
в него.
Магнитные свойства веществ
обусловлены наличием в атомах электронов
и нуклонов и количественно
определяются орбитальным и спиновым
магнитными моментами этих частиц,
возникающими в результате их внутреннего
движения в атоме. Наибольший вклад вносят
магнитные моменты электронных
оболочек.
Орбитальный магнитный момент
электрона зависит от побочного
(орбитального) квантового числа /:
а
N
Рис. 105. Распределение
магнитных силовых
линий в вакууме (а) и при
внесении в магнитное
поле диамагнитного (б)
и парамагнитного (в)
вещества
где е и т—соответственно заряд и масса электрона;
h—постоянная Планка.
Величина eh/Dnm) является единицей магнитного момента
и называется магнетоном Бора. Магнетон Бора численно равен
магнитному орбитальному моменту /7-электрона. Магнетон Бора
для одного электрона имеет значение 9,27* 10~24 А-м2
(9,26-101 гаусс см*).
Орбитальным магнитным моментом обладают все
электроны, кроме тех, у которых /=0, т. е. \it имеют /ь, d- и/-электроны.
При обычных условиях у большинства веществ магнитные
моменты всей системы электронов (и ядерных частиц) почти
полностью скомпенсированы. При внесении вещества во внешнее
магнитное поле происходит определенная ориентация
собственных магнитных моментов электронов. Таким образом,
проявляется диамагнетизм.
Диамагнетизм свойственен всем веществам. Упрощенно его
возникновение можно представить следующим образом. При
внесении в магнитное поле замкнутого электрического контура
в нем возникает ток, что приводит к появлению вокруг контура
магнитного поля, противоположного по направлению внешнему
полю. Электронную орбиталь атома можно представить как
конгур при внесении вещества в магнитное поле, электронные
орбитали всей системы электронов индуцируют магнитное поле,
8*
227
противоположное по направлению внешнему полю. Так
возникает орбитальный магнетизм.
Диамагнитными являются все атомы и ионы, имеющие
заполненные s-9 />-. d- или /-подуровни. Соединения,
состоящие из диамагнитных ионов, в жидком и кристаллическом
состояниях также диамагнитны. В результате взаимодействия
атомов и молекул веществ в жидком и кристаллическом
состояниях наблюдаемый диамагнетизм веществ в этих
состояниях, как правило, ниже, чем у веществ в газообразном
состоянии.
Парамагнетизмом характеризуются вещества, у которых ядра
атомов, атомы, ионы, молекулы имеют собственный
магнитный момент, но в отсутствие внешнего магнитного поля
магнитные моменты всех частиц ориентированы беспорядочно и
намагниченность парамагнетиков равна нулю. Под действием
внешнего поля магнитные моменты ориентируются по направлению
поля.
Парамагнетизм может быть обусловлен следующими
основными причинами: движением электронов в оболочке атома
(орбитальный парамагнетизм), наличием спинового
магнитного момента неспаренных электронов (спиновый
парамагнетизм) и магнитного момента ядер атомов (ядерный
парамагнетизм). Последняя составляющая парамагнетизма
примерно в 1000 раз меньше двух первых, и при изучении
магнитных свойств химических соединений ее не
учитывают.
Наибольший вклад в парамагнитный эффект вносит спиновый
магнитный момент электронов ц5, который обусловлен
движением свободного электрона вокруг собственной оси. Величина
Для атома или иона, имеющего п неспаренных электронов со
спином s=1/2, суммарное спиновое квантовое число равно s = n/2
и спиновый магнитный момент составляет:
или же в магнетонах Бора:
Откуда магнитный спиновый момент одного электрона (неспа-
ренного) составляет:
^ = ^/3= 1,73 магнетона Бора.
228
Ниже приведены суммарные спиновые магнитные моменты (в
магнетонах Бора) для систем, содержащих до семи неспаренных
электронов (максимальное число возможных неспаренных
электронов в атоме):
л 0 1 234567
ц, 0 1,73 2,83 3,87 4,90 5,92 6,94 7,95
Следует обратить внимание на то, что магнитный момент
не изменяется прямо пропорционально числу неспаренных
электронов. Так, увеличение числа неспаренных электронов в два
раза не приводит к двухкратному росту магнитного момента
системы.
Соединения, содержащие парамагнитные ионы,
обнаруживают парамагнитные свойства, причем с ростом ионности связей
в кристаллической решетке парамагнетизм усиливается. При
образовании химических связей посредством перекрывания
неспаренных электронов парамагнетизм атомов ослабляется и может
вообще настолько уменьшиться, чго вещество начнет проявлять
диамагнитные свойства. Например, газообразные атомы NaC^1)
и С1C523/?5) парамагнитны, так как имеют по одному неспарен-
ному электрону, а газообразные молекулы Na2 и С12
диамагнитны, поскольку при образовании неполярной связи происходит
спаривание электронов. Диамагнетизм присущ жидким и
кристаллическим хлору и натрию. В то же время ионы Na+C^°)
и С1~ Cs23p6) диамагнитны из-за отсутствия несиаренных
электронов, и диамагнетизм сохраняется у NaCl в растворе и
кристалле.
Диамагнетизм, вызванный орбитальным движением
электронов, присущ всем веществам, имеющим и спаренные, и неспарен-
ные электроны, а парамагнетизм—только веществам,
обладающим неснаренными электронами. При экспериментальном
изучении магнитных свойств веществ определяют суммарный
эффект, вызванный диа- и парамагнетизмом. Так как
диамагнетизм веществ выражен намного слабее парамагнетизма, при
оценке магнитных свойств парамагнетиков диамагнетизм не
учитывают.
В табл. 18 сопоставлены вычисленные и экспериментально
определенные магнитные моменты ионов ^/-элементов
четвертого периода в кристаллах хлоридов, нитратов, сульфатов и
других солей, а также в их водных растворах. Обнаруживается
хорошее соответствие экспериментальных и вычисленных
значений. Вместе с тем, если ^-подуровень заполнен более чем
наполовину, то экспериментальные моменты оказываются всегда
выше вычисленных значений спиновых моментов. Это означает,
что орбитальный магнитный момент скомпенсирован не
полностью.
229
Таблица 18
Вычисленные н экспериментально определенные магнитные моменты ионов
{/-элементов четвертою периода Периодической системы
Ион
Sc3+
Ti3<
Ti2*
v2+
Cr2 +
Mn2t
Fe2+
Co2 +
Ni2 +
Cu2f
Zn2'
Число «/-электронов
всего
0
1
2
3
4
5
6
7
8
9
10
неспаренных
0
1
2
3
4
5
4
3
2
1
0
ц, магнетоны Бора
расчет
0
1,73
2,83
3,87
4,90
5,92
4,90
3,87
2,83
1,73
0
жеперимен!
0
1,75
2,76
3,86
4,80
5,96
-5,3
-4,7
-3,1
-2,0
0
Из группы парамагнитных веществ выделяют
ферромагнитные вещества, которые делят на собственно ферромагнитные
и антиферромагнитные.
Ферромагнетизм—такое магнитное состояние
кристаллического вещества, при котором магнитные моменты всей
совокупности электронов имеют параллельную ориентацию независимо
от наличия внешнего магнитного поля. Если в отсутствие
внешнего магнитного поля намагниченность парамагнитного
вещества равна нулю, то у ферромагнетиков (и антиферромагнетиков)
она имеет высокое положительное значение. Постоянная
намагниченность ферромагнетиков вызвана сильным взаимодействием
атомов или ионов кристаллической решетки, приводящим к
образованию областей, так называемых доменов, с параллельно
ориентированными магнитными моментами.
К простым веществам, проявляющим ферромагнитные
свойства при комнатной температуре, относятся rf-элементы VIII группы
4 периода—железо, кобальт и никель. Ферромагнитны и многие
сплавы на их основе, а также некоторые оксиды, нитриды и другие
соединения, например Fe3O4, CrO2, Mn4N, CrTe.
Неспаренные электроны атома металла или катиона в
кристаллической решетке обладают параллельными спинами, если:
а) имеется большое число (более 8) ближайших соседних атомов
(ионов) с неспаренными электронами, б) неспаренный электрон
находится достаточно далеко от ядра атома (d- или/-электроны),
в) строго соблюдается определенное межъядерное расстояние,
которое должно быть больше, чем это требуется для образования
химической связи посредством перекрывания электронных ор-
биталей, но в то же время оно не может быть слишком велико
(югда электроны перестают взаимодействовать).
230
Первым двум требованиям удовлетворяют большинство d-
и/-элементов, а третьему—только Fe, Co и Ni (Gd и Dy—при
низких температурах).
Антиферромагнетизм вызван таким взаимодействием ионов
и электронов, при котором спины электронов располагаются
навстречу друг другу (антипараллельно). Чаще всего это
обнаруживается у кристаллов, в которых между ионами,
обладающими магнитными моментами, находятся ионы, не имеющие
магнитных моментов. Так, в кристалле МпО магнитный момент
одного иона Мп2+ имеет направление, противоположное
направлению (антипараллелен) магнитных моментов ближайших двух
ионов Мп2 + , между которыми находится ион кислорода. В
кристаллах типа МпО магнитные моменты электронов
компенсируются.
Диамагнитные, парамагнитные и ферромагнитные вещества
различаются не только по их отношению к внешнему
магнитному полю, но и к температуре.
На свойства диамагнитных веществ напряженность
магнитного поля и температура не оказывают влияния. На
парамагнитные вещества внешнее магнитное поле не влияет, но магнитная
восприимчивость их зависит обратно пропорционально
абсолютной температуре. Это объясняется следующим образом. Каждая
частица парамагнитного вещества обладает постоянным
магнитным моментом, определяемым числом неспаренных электронов.
В отсутствие внешнего магнитного поля суммарный магнитный
спиновый момент равен нулю вследствие хаотического
направления спинов, а при наложении магнитного поля происходит
ориентация спинов, атомов и молекул в магнитном поле.
Повышение температуры ослабляет ориентацию во внешнем магнитном
поле, и парамагнетизм уменьшается в соответствии с формулой
{закон Кюри):
где X—магнитная восприимчивость; С—константа (константа
Кюри); 9—некоторая постоянная (температура).
Магнитные свойства ферромагнитных веществ зависят и от
напряженности магнитного поля, и от температуры.
Повышение температуры приводит к понижению магнитной
восприимчивости, и при некоторой температуре, называемой
температурой Кюри, она резко снижается. Выше температуры Кюри
ферромагнетик ведет себя как парамагнитное вещество. Для
ферромагнитных веществ постоянная 0 равна температуре
Кюри.
Таким образом, признаки сходства и различия диамагнитных,
парамагнитных и ферромагнитных веществ обусловливаются
зависимостью их магнитной восприимчивости от рассмотренных
двух факторов:
231
Диамагнетики
Парамагнетики
Ферромагнетики
noe no 1С
не влияет
»с влияет
влияет
Темпера ivpa
нс влияет
влияет
влияет
Как показано выше, магнитные свойства веществ
определяются электронным строением атомов, ионов и молекул и типом
химической связи, поэтому на основании магнитных свойств
вещества можно сделать заключение о его строении и указать на
валентные состояния (степени окисления) атомов и тип
химической связи в молекуле. Проиллюстрируем это следующим
примером. При окислении фосфора во влажном воздухе наряду с
фосфорной Н3РО4 и фосфористой Н3РО3 кислотами образуется еще
одна кислота, которая при добавлении NaOH осаждается в виде
соли состава Na:H:P:O = 1:1:1:3. Этому составу отвечае! формула
NaHPO3—фосфорноватокислый натрий (соль фосфорноватой
кислоты Н2РО3). Изучение магнитных свойств соединения
NaHPO3 показало, что оно диамагнитно, как и сама
кристаллическая кислота Н2РО3. Но в этом случае приведенные выше
формулы кислоты и соли неверны, так как в соо1ветствии с
такой записью молекулы содержат нечетное число электронов,
что указывает на парамагнетизм соединений. Диамагнетизму
должна отвечать не содержащая неспаренных элек фонов
молекула, записанная удвоенной химической формулой:
НО ОН
-.и
Hi
ОН
Правильность этой формулы по;иверждается 1акже данными по
определению молекулярной массы соединения.
По результатам магнитных измерений иногда можно сделать
вывод о валентном состоянии (степени окисления) элементов. Все
соединения двух- и четырехвалентной платины, например, PtBr2
или PtBr4, диамагнитны, что указывает на отсутствие в молекуле
неспаренных электронов. Тогда соединения с трехвалентной
платиной, например, PtBr3, должны быть парамагнитны. Однако
в действительности они диамагнитны, и эт позволяет
предположить, что рассматриваемые вещества—двойные соли или
двойные (молекулярные) соединения. Поэтому соль состава PtBr3
правильнее представить в виде Pt2Br6 или PtBr2 • PtBr4, т. е. как
соединение, содержащее двух- и четырехвалентную платину.
Большинство солей серебра содержат ион Ag+, который
имеет заполненные подуровни, поэтому соли серебра диамагнитны.
Но в то же время встречаются парамагнитные комплексные соли
серебра, что говорит о существовании двухвалентного серебра,
232
причем магнитные восприимчивости этих солей такие же, как
и у обычных солей двухвалентной меди.
Парамагнетизм кислорода в кристаллическом, жидком и
газообразном состояниях явился в свое время отправной точкой
для разработки метода молекулярных орбиталей. Магнитные
свойства молекулярных ионов кислорода (О2~, О2+) и
кристаллических соединений, ими образованных, подтверждают
предложенные распределения электронов по молекулярным орбита-
лям и хорошо согласуются с энергетическими свойствами ионов
и межъядерными расстояниями.
По результатам магнитных измерений можно подтвердить
или опровергнуть приписываемую веществу структуру. Так, для
двухвалентного никеля характерно координационное число
четыре, проявляемое в тетраэдрической и плоско-квадратной
кристаллической структурах. При координационном числе четыре вокруг
комплексообразовагеля расположены четыре лиганда, которые,
будучи донорами, предоставляют четыре пары электронов. При
образовании тетраэдрической структуры эти электроны должны
находиться в состоянии ^-гибридизации, а при образовании
плоской квадратной—
в состоянии dsp
^гибридизации. На рис. 106, а
представлено
распределение электронов по
уровням и подуровням
в изолированном атоме
никеля. При
образовании иона Ni2+ атом
теряет два 4$-электрона
(рис. 106, б): ион Ni2"
имеет четыре незанятых
орбитали на 4s- и 4р-
подуровнях, которые
могут принимать пары
электронов при
связывании по донорно-акцеп-
торному типу. Так как
энергии подуровней 3d,
4у и 4р близки, то на рис.
106, в эти подуровни
изображены рядом друг
с другом (так принято
для наглядности при
обсуждении строения
комплексных ионов).
К2 [NiF4] парамагни-
4
3
2
1
н
п
14
llil
я
t4
J4
44
t4
р
Н
П
U
14
14
А
t
II II II 1
f
¦4
14
¦4
14
t4
U
¦4
Р
t4
14
Н
н
а
\
¦
1 1 1 1 1 1 1
f
Тетраэдрический
комплекс
Рис' }°ч6- Распределение электронов в атоме ни-
келя (о), в изолированном ионе Nr (б, в) и в ио-
нах Ni , входящих в тетраэдрический (г) и пло-
ско-квадратный (д) комплексы
нах
233
тны, и экспериментально измеренный магнитный момент
совпадает с вычисленным при условии, что в комплексе имеются два
неспаренных электрона: 2,8 магнетона Бора. Из этого следует,
что электронные пары ионов фтора входят на As и 4р-подуровни
иона никеля. Так как все четыре связи равноценны, то,
по-видимому, имеет место лр3-гибридизация (рис. 106, г), откуда
вытекает, что четыре связи иона никеля (комплексообразователя)
направлены к вершинам тетраэдра, т. е. комплекс К2 [NiF4] имеет
тетраэдрическую структуру.
Кристаллы другой соли К2 [Ni(CNL] диамагнитны.
Диамагнетизм может проявляться только в том случае, если химическая
связь формируется путем спаривания двух 3^-электронов и
последующего распределения четырех донорных электронных пар на
освободившейся одной З^-орбитали и имеющихся одной 4s-
и двух 4р-орбиталях (рис. 106, д). Для обеспечения энергетической
равноценности четырех связей необходима Л/?2-гибридизация,
а такой тип гибридизации приводит к расположению четырех
связей в одной плоскости, т. е. плоско-квадратному строению
комплекса.
§ 34. ЭНЕРГЕТИЧЕСКИЕ СВОЙСТВА КРИСТАЛЛОВ
Для количественной характеристики прочности
кристаллической решетки используется представление об энергии
кристаллической решетки. Это то количество энергии, которое
необходимо для перевода 1 моль атомов или молекул
кристаллического вещества в газообразное состояние. Таким образом,
энергия кристаллической решетки—это га энергия, которую
необходимо затратить, чтобы разделить и удалить на бесконечно
большое расстояние частицы, образующие кристаллическую
решетку. При этом обязательно указывается состав частиц газовой
фазы, а ими могут быть атомы и молекулы, составляющие
кристаллическую решетку, продукты их ионизации, диссоциации
и ассоциации.
Рассмотрим в качестве примера определение энергетических
характеристик алмаза и графита.
Алмаз—термодинамически неустойчивая модификация
углерода при давлении 101325 Па и любых температурах, и он
должен самопроизвольно переходить в
графит—термодинамически устойчивую модификацию углерода. Существование
алмаза при комнатной температуре объясняется исключительно
высокой энергией активации перехода в графит.
При переводе 1 моль атомов углерода из структуры алмаза
в состояние невозбужденных изолированных газообразных
атомов углерода разрываются все связи, удерживающие атомы
в кристаллической решетке. Энтальпия сублимации (атомизации)
234
алмаза характеризует прочность его кристаллической решетки.
Определить экспериментально энтальпию сублимации алмаза
невозможно—при низких температурах у алмаза крайне низкое
давление пара, при высоких же температурах, когда становится
реальным измерение давления его пара, он превращается
в графи г.
Энтальпия сублимации графита может быть найдена по
данным измерения давления пара при высоких температурах,
поскольку при этих условиях алмаз превращается в графит и
становится реальным измерение давления пара углерода. Ниже
приведены значения давления пара одноатомных молекул углерода:
Г, К 1700 2000 2400 3000
р, Па 1,31 Ю~9 2,72-ИГ6 3,76 10 4,68
Для процесса перехода графита в газообразное состояние
константа равновесия равна давлению пара углерода, т. е.
С (графит) = С (газ); К=рС{газ)
По известной зависимости давления пара углерода от
температуры можно найти термодинамические характеристики этого
процесса: изменение энтальпии АН и изменение энтропии AS. Для
этого необходимо составить и решить систему из двух (как
минимум) уравнений:
AH-TiAS=-RTl\npu
AH-T2AS=-RT2\np2.
Установлено, что для процесса сублимации (атомизации)
графита Д#° = 715,0 кДж/моль и AS° = 155,2 Дж/(К моль).
Чтобы вычислить энтальпию сублимации алмаза, нужно
знать энтальпию перехода алмаза в графит, которую можно
определить экспериментально по разности энтальпий сгорания
алмаза и графита, АН°= — 1,9 кДж/моль.
Из соотношений
С (алмаз) = С (графит); А Я° = - 1,9 кДж
С (графит) = С (газ); А /Г = 715,0 кДж
получаем
С (алмаз) = С (газ); А /Г = 713,1 кДж.
Таким образом, на переход атомов углерода из структуры
алмаза в состояние одноатомного газа закачивается количество
теплоты, равное 713,1 кДж/моль.
Валентные орбитали атома углерода в алмазе находятся в
состоянии 5/?3-гибридизации и каждый атом углерода связан а-
связями с четырьмя другими атомами. Поэтому на каждый атом
235
С в решетке алмаза приходятся две связи С—С. Тогда энергия
связи С—С в алмазе должна быть равна половине энергии,
требующейся на разрыв всех связей при переходе атомов
углерода из структуры алмаза в газовое состояние, т.е. 713,1:2^
356,6 кДж/моль.
В графите атомы углерода находятся в состоянии яр
^гибридизации, при этом каждый атом углерода связан с тремя другими
вследствие перекрывания гибридных орбиталей. При переводе
1 моль атомов графита в состояние газообразных атомов
разрывается 1,5 связи (в расчете на каждый атом углерода).
Поскольку энтальпия сублимации графита равна 715,0 кДж/моль,
энергия связи С—С в графите составляет 715,0:1,5=476,7 кДж/
моль. Из приведенных данных видно, что связь атомов углерода
в графите значительно прочнее связи в алмазе. В решетке алмаза
межъядерное расстояние составляет 0,154 нм, в структуре
графита расстояние С—С в плоскости перекрывания 5р2-орбиталей
равно 0,142 нм. Как известно, графит по твердости не может
сравниться с алмазом и намного ему уступает. Алмаз—самый
твердый из встречающихся в природе веществ. Это объясняется
тем, что, хотя в плоскости перекрывания л/?2-орбиталей атомы
углерода связаны значительно прочнее, чем в алмазе, сами
плоскости атомов взаимодействуют друг с другом крайне слабо (силы
Ван-дер-Ваальса) и расстояние между ними составляет 0,335 нм.
Слабость связи между атомными плоскостями приводит к
легкому скольжению плоскостей 5р2-гибридных связей относительно
друг друга, что и обусловливает мягкость графита.
Следовательно, энергетические характеристики кристаллов
и их физические свойства (твердость и другие) можно
сопоставлять только при условии однотипности кристаллических
структур и связей.
Энергетическое состояние ионного кристалла обычно
сравнивают не с энергетическим состоянием молекул или атомов в
газовой фазе, а с состоянием ионов, образующих решетку.
Энергией кристаллической решетки ионного соединения, А//кр,
называют то количество энергии, которое требуется для удаления
1 моль ионов из решетки на расстояние, при котором силы
взаимодействия между ионами бесконечно малы. Например, для
кристалла NaCl стандартная энергия кристаллической решетки равна:
NaCl{K) = Na+(r) + Cl"(r); АН°кр, = 753 кДж/моль.
Энергия решетки характеризует силу взаимодействия между
ионами, которая удерживает их в кристаллической решетке. Чем
выше А//кр, тем сильнее связаны ионы между собой. Большим
значениям АЯкр обычно соответствуют высокие температуры
и энтальпии плавления, так как чем прочнее кристаллическая
решетка, тем больше теплоты нужно затратить на ее разрушение
при плавлении. Так, для LiF и Nal (прочность связей в кристалле
236
фторида лития выше) стандартная энергия решетки А//°кр
соответственно равна 1029 и 694 кДж/моль, а температура плавления
составляет 870 и 651° С.
Для однотипных соединений с одинаковой структурой
наблюдается связь между А#°кр и их механическими свойствами.
Обычно чем выше АЯ°кр, тем прочнее решетка и тем тверже ионный
кристалл.
Энергия кристаллической решетки зависит от радиуса и
заряда ионов. С увеличением ионного радиуса Д#%р уменьшается.
Например, для NaCl и NaBr (RC\- <Rbt~) А#скр соответственно
равна 753 и 736 кДж/моль. С увеличением заряда иона энергия
решетки возрастает. Так, А#'кр для кристаллов Na2O и СаО
равна 2520 и 3520 кДж/моль.
§ 35. ФАЗОВЫЕ ПЕРЕХОДЫ В КРИСТАЛЛИЧЕСКИХ ВЕЩЕСТВАХ
Как было показано в § 22, переход вещества из одной фазы
в другую совершается при изменении внешних
условий—температуры, давления, напряженности электромагнитного поля и т. п.
Фазовый переход обнаруживается по скачкообразному
изменению свойств вещества при непрерывном изменении какого-либо
внешнего условия.
Все многообразие фазовых переходов классифицируется на
фазовые переходы первого и второго родов. При фазовом
переходе первого рода выделяется или поглощается определенное
количество теплоты, изменяются объем и плотность вещества, его
энтропия, теплоемкость и т. п. Фазовые переходы первого рода —
плавление, испарение, возгонка, полиморфное превращение и
другие—характеризуются равенством изобарных потенциалов двух
сосуществующих в равновесии фаз. В отличие от фазовых
переходов первого рода для фазовых переходов второго рода
свойственно не только равенство изобарных потенциалов, но и равенство
энтропии, объемов и плотностей фаз. К фазовым переходам
второго рода относятся магнитные превращения при температуре
Кюри, переход вещества в сверхпроводящее сое гояние, появление
сверхтекучести у гелия, переход из парамагнишого состояния
в ферромагнитное и др. Одно из объяснений фазовых переходов
второго рода состоит в изменении симметрии частиц системы,
например, переход системы част иц с беспорядочно
направленными спинами в систему частиц с преимущественной ориентацией
спинов или переход из неупорядоченного распределения атомов А
и В по узлам кристаллической решетки в упорядоченное.
Не всегда можно точно отнести переход к тому или иному
роду, так как возможны фазовые переходы первого рода,
сопровождающиеся незначительным изменением плотности,
теплоемкости и малой теплотой переходами т. п.
237
Ниже остановимся только на полиморфных превращениях —
фазовых переходах первого рода, связанных со структурными
изменениями кристаллических веществ. На примере переходов
модификаций углерода — алмаза и графита—и олова (элементы,
расположенные в одной подгруппе Периодической системы)
обсудим термодинамический, кинетический и структурный аспекты
полиморфных превращений.
Из экспериментально определяемых энтальпий сгорания
алмаза и графита можно вычислить энтальпию превращения
графита в алмаз
С (графит) = С (алмаз); АН ° 19% = 1828 кДж/моль.
Этот процесс происходит с увеличением энтальпии, т. е.
сопровождается поглощением теплоты. Согласно принципу Ле Ша-
телье повышение температуры должно благоприятствовать
протеканию процесса, сопровождающегося поглощением теплоты,
1. е. для данного примера способствовать превращению графи га
в алмаз. В действительности же наблюдается противоположный
процесс.
Рассмотрим диаграмму состояния углерода (рис. 107).
Согласно этой диаграмме, если алмаз при некотором постоянном
давлении, равном, например, давлению в точке а, нагревать, то
он сохраняет устойчивость до температуры, соответствующей
точке 6. При дальнейшем повышении температуры (выше точки
б, при р = пост) алмаз превращается в графит и в интервале
температур, соответствующих точкам бив, графит сохраняет
устойчивость. Наконец, при температурах выше точки в весь
графит превратится в
жидкость.
Следовательно, согласно
диаграмме состояния
углерода повышение
температуры способствует
превращению алмаза в графит как ве-
щества с более рыхлой
и менее симметричной
структурой. И, наоборот,
превращению графита в
алмаз должно благонриятство-
вагь понижение
температуры (передвижение по прямой
вба вниз).
Таким образом,
изменение фазового состояния
углерода при изменении
температуры в соответствии
с диаграммой состояния
4000
3000
2000
1000
1-ю10 2-м10 з-ююр,па
Рис 107 Диаграмма состояния углерода
Пушииром отмечена об «асть температур и дав 1емий,
в ко юрой осуществляемся сишез алмаза m графит
238
противоположно выводу, получаемому с использованием
принципа Ле Шателье. Такое противоречие можно объяснить
следующим образом.
Точно определить направление процесса можно на основе
второго закона термодинамики. Для этого рассчитаем изменение
энтропии и изобарного потенциала при переходе графита в
алмаз. Изменение энтропии составит:
AS °298 = S °алмаз - S °графит=2,368 - 5,740 = - 3,372 Дж/(К ¦ моль).
Зная АЯ°298 и ASro2989 можно рассчитать уравнение
температурной зависимости изобарного потенциала перехода графита
в алмаз:
Д<7° = Д#°-:ГД5О = 1828 + 3,372 Г.
При стандартной температуре и стандартном давлении
A(j°298 Для рассматриваемого процесса равно:
АС°298= 1828 + 3,372-298,15 = 2833 Дж/моль.
При комнатной и любой другой температуре процесс
перехода графита в алмаз характеризуется положительным значением
изменения изобарного потенциала, и при стандартном давлении
101 325 Па переход графита в алмаз невозможен (рис. 108). Чем
выше температура, тем более положительным становится А G°
фазового перехода графит-*алмаз и тем термодинамически
менее вероятен процесс, хотя по изменению энтальпии повышение
температуры должно благоприятствовать этому процессу.
Таким образом, рассмотренный пример показывает, что для
некоторых процессов принцип Ле Шателье не способен
предсказывать влияние температуры на его направление. Это относится
к реакциям, идущим с поглощением теплоты (АЯ>0) и
одновременно сопровождающимся уменьшением энтропии (А5<0,
увеличение степени порядка).
Превращению графита в
алмаз должны способствовать
низкие температуры и высокие
давления. Тем не менее при низких
температурах ни прямой, ни
обратный процессы не
наблюдаются. Термодинамически
неустойчивый при стандартной
температуре и давлении, алмаз
в течение длительного времени
не изменяется и в нем не
обнаруживается ни малейших
примесей графита. Энергия
активации фазовых переходов
модификаций углерода очень велика
500 1000 1500Т,К
Рис. 108. Температурная
зависимость изменения энтальпии Д#с
и изобарного потенциала AG "
перехода графит->алмаз
239
и обусловлена необходимостью коренной перестройки
кристаллических структур.
На диаграмме состояния (см. рис. 107) пунктиром отмечена
область температур и давлений, наиболее благоприятная с точки
зрения термодинамических и кинетических представлений для
превращения графита в алмаз и используемая в технологии
синтеза промышленных алмазов. Из диаграммы видно, что
превращение графита в алмаз становится возможным при высоких
температурах, порядка 2000—4000 К, и высоких давлениях,
1 • 109—1 • 1010 Па. Процесс ускоряется в присутствии жидких
металлов VIII группы, служащих катализаторами.
Противоположный переход алмаза в графит термодинамически возможен
при всех температурах. Термодинамически нестабильное
вещество— алмаз — оказывается тем не менее кинетически
чрезвычайно стабильным, и переход алмаза в графит наблюдается только
при высоких температурах. Процесс уже заметно протекает при
1000—1500° С (без доступа кислорода). Кристалл алмаза
постепенно начинает темнеть с вершин и ребер, затем он весь
покрывается черной коркой графита. При 2000—3000° С (без доступа
кислорода) фазовое превращение протекает чрезвычайно быстро.
Рассмотренные полиморфные превращения — это простейшие
твердофазные реакции. Обычно при повышении температуры
скорость твердофазных реакций возрастает (переход белого олова
в серое—одно из исключений). Это обусловлено тем, что с
повышением температуры колебания атомов или ионов в узлах
кристаллической решетки около своего равновесного положения
усиливаются. При достижении значения энергии этих колебаний,
достаточного для преодоления сил связи между частицами,
начинается обмен частиц в кристаллической решетке, приводящий
к образованию нового вещества.
Предложено много объяснений механизмов твердофазной
реакции. Один из них заключается в следующем. Твердофазная
реакция обычно протекает сравнительно медленно и развивается
не сразу во всем объеме кристалла, а в отдельных его областях,
называемых зародышами. Эти области должны иметь
повышенную энергию, благодаря чему они проявляют более высокую
реакционную способность. Такими участками в кристалле могут
быть вакансии, посторонние атомы, расположенные в узлах или
междоузлиях кристаллической решетки, атомы основного
вещества, смещенные из своих нормальных положений, и другие
дефекты кристаллической решетки. Так как наибольшее число
дефектов находится на поверхности кристалла, твердофазные
реакции обычно начинаются на поверхности и распространяются
в глубь кристалла.
Зародыш представляет собой небольшую группу атомов
(молекул или других структурных единиц) с правильным и
характерным для стабильного при новых условиях кристалла расположе-
240
Зародыша ядер
" ^
.поверхность т<жш
Рис. 109 Схема развития
твердофазной реакции
нием. Образование зародышей
можно рассматривать как воз-
никновение дисперсной
системы, изучаемой коллоидной
химией. По
последовательности расположения частиц
в зародыше происходит
дальнейшая укладка слоев частиц
и рост ядер.
Если зародыш находится
внутри кристалла, ядро
принимает сферическую форму, если
зародыш расположен на
поверхности кристалла, ядро
приобретает частично
сферическую форму. На рис. 109 представлена схема развития реакции на
поверхности кристалла и в глубине его. По мере протекания
реакции возрастает поверхность ядер, т. е. поверхность раздела
фаз исходного вещества и продукта. Это приводит к росту
скорости реакции. В момент начала объединения ядер поверхность
раздела и соответственно скорость достигают максимального
значения, затем поверхность раздела начинает уменьшаться
и скорость реакции снижается.
Для реакций с участием твердых фаз обычное определение
понятия «скорость реакции» непригодно, так как процесс
протекает не в объеме (газа или жидкости), а в основном на
поверхности раздела фаз исходного вещества и продукта. Под
скоростью твердофазной реакции понимают изменение во времени
степени превращения вещества:
где а—степень превращения вещества.
Степень превращения вещества а представляет собой
относительное изменение количества исходного реагирующего вещества
к моменту времени /:
* = {N0-Nt)/N09
где iV0—начальное количество исходного вещества;
Nt—количество вещества в момент времени /.
На рис. ПО, а показана кривая зависимости степени
превращения от времени. Участок / соответствует периоду индукции,
в котором реакция проходит очень медленно и с очень низким
ускорением. Затем следует участок ускорения 2,
продолжающийся до точки перегиба кривой (обозначенной, крестиком). Участок
3 отвечает замедлению реакции. Скорость реакции в данный
момент времени определяется по углу наклона касательной
в точке кинетической кривой, соответствующей этому времени.
241
Рис. 110. Кинетическая кривая (а) и зависимость скорости процесса
от степени превращения вещества F) для твердофазных реакций
Так, в момент времени i' степень превращения равна а ', а тангенс
угла наклона ср касательной равен скорости процесса в этот
момент времени:
tg <p = da/dt.
Скорость реакции максимальна в точке перегиба кинетической
кривой. Пересечение касательной, проведенной в точке перегиба,
с осью абсцисс дает отрезок времени, соответствующий
индукционному периоду *инд. На рис. ПО, б те же данные представлены
в виде зависимости скорости реакции от степени превращения
вещества. Максимум на кривой отвечает такому состоянию
процесса, при котором поверхность раздела превращающихся фаз
и скорость реакции максимальны.
Кинетические кривые типа кривых превращения модификаций
олова имею! S-образную форму. Полиморфное превращение —
наиболее простой тип гетерогенного процесса, и если кинетика
излучаемого гетерогенного процесса описывается S-образной
кривой, то считают, что промежуточные продукты не
получаются (при образовании промежуточных соединений кривая имеет
другой вид). 5-образную форму имеют кинетические кривые
многих других гетерогенных процессов, например, дегидратации
медного купороса CuSO4 * 5H2O, восстановления водородом
оксидов никеля, меди, германия.
В явлениях, описываемых такой кривой, процесс начинается
с образования продуктами реакции зародышей и ядер. Иногда
начальные стадии проходят настолько быстро, что они не
наблюдаются. Стадии могут иметь другие соотношения по скоростям,
поэтому экспериментальные кинетические кривые твердофазных
реакций сильно различаются и их математически обрабатывают
по различным уравнениям. Предложено более десяти
кинетических уравнений твердофазных реакций.
Наиболее удовлетворительно описывает кинетику
твердофазных реакций уравнение Ерофеева—Колмогорова:
242
где а—степень превращения вещества;
t—время;
п и к — постоянные;
п зависит от природы веществ и механизма процессов, она
может иметь смысл порядка реакции; константа к при л=1
совпадает с константой скорости реакции.
Постоянные кип определяют графически по
логарифмической зависимое!и а от /. Для перевода уравнения в
логарифмическую форму, удобную для построения графической
зависимости, дважды прологарифмируем это уравнение:
In In = In к + n In t.
1 —a
В десяшчных логарифмах это выражение имее! вид:
1 —ос
Таким образом, двойной логарифм от 1/A—а) представляет
собой линейную функцию логарифма времени. Для нахождения
кип строят график зависимое! и lg lg [1/A — a)] or lg/. Тангенс
угла наклона полученной прямой к оси абсцисс дает постоянную
п. Для определения к прямую
продолжают до пересечения при lg r=0, которое
соответствует значению lglg [1/A—а)], и,
иодставив эти два условия в уравнение,
вычисляют к.
Постоянные пик можно определить
и другим способом. При условии
прямолинейной зависимости а от t составляют
систему из двух уравнений для двух
известных степеней превращения вещества
а при двух значениях / и решением этой
системы находят постоянные пик.
Обычно экспериментальные результаты
по изучению кинетики 1вердофазных
реакций не дают логарифмическую зависимость
точно в виде прямой, и ее представляют как рис 111 Представление
СОСТОЯЩУЮ ИЗ Двух ИЛИ трех Отрезков кинетической кривой «сте-
(рис. 111). Тогда для каждого участка кри- пень превращения вешесг-
ВОЙ НахОДЯ! ЗНачеНИЯ ПОСЮЯННЫХ к И П И ПО с^ь^иТТине^нь^зави-
ним судят о механизме реакции. симосгей
243
ГЛАВА 8
ПРОЦЕССЫ НА ГРАНИЦЕ РАЗДЕЛА ФАЗ
§ 36. РЕАКЦИИ НА ГРАНИЦЕ КРИСТАЛЛ—ГАЗ
Гетерогенные процессы, проходящие на поверхности раздела
кристалл—газ, чрезвычайно многообразны; такие реакции
широко используются в химической технологии. Из множества этих
реакций ниже рассмотрены четыре группы процессов: горение
твердого топлива, восстановление оксидов металлов, процессы,
лежащие в основе газовой коррозии, и гетерогенный катализ.
К процессам горения относят
окислительно-восстановительные реакции, сопровождающиеся выделением теплоты и
светового излучения. Таковы, например, реакции металлов с
галогенами, с монооксидом углерода, реакция между углеродом
и кислородом.
Ранее предполагали, что при горении углерода сначала
получается монооксид углерода, который сгорает в газовой фазе
с образованием диоксида углерода:
, Д#=-123,1 кДж/моль,
СО(Г)+ 1/2О2(г) = СО2{г), Д#= -285,4 кДж/моль.
Согласно современным представлениям при горении углерода
образуются одновременно оба оксида
С(К) + О2(Г) = СО2(Г), Д#= -408,6 кДж/моль,
С(к>+ l/2O2(r) = CO(r), АЯ= -123,1 кДж/моль.
т. е. процесс проходит не через две последовательные стадии
(хотя этот путь и возможен для некоторой части сгорающего
вещества), а через параллельные стадии.
Процесс усложняется взаимодействием выделяющегося
диоксида углерода с углеродом
С(К)+СО2(Г) = 2СО(Г); Д#= 162,3 кДж/моль.
244
В равновесной газовой смеси, полученной при сгорании
углерода, содержатся СО2, СО и О2 в количествах, зависящих от
температуры и давления (в соответствии с принципом Ле Шате-
лье). Эти количества можно вычислить, исходя из значений
констант равновесия.
Несмотря на то что углерод по отношению к кислороду
воздуха термодинамически неустойчив, самовозгорания
материалов, содержащих углерод, при комнатной температуре не
происходит.
По современным представлениям сгорание углерода в общем
случае можно представить в виде пяти стадий:
а) диффузия кислорода к поверхности углерода,
б) адсорбция кислорода на поверхности углерода,
в) взаимодействие кислорода с углеродом на поверхности,
г) десорбция продуктов реакции (СО и СО2),
д) диффузия продуктов реакции от поверхности.
Как известно, наиболее медленная стадия определяет скорость
всего процесса. Первая и последняя из перечисленных стадий—
это диффузионные процессы. Для снижения их влияния на
скорость горения реакционную смесь перемешивают и усиливают
поток воздуха. Чем выше температура, тем меньше количество
адсорбированных и десорбированных веществ на поверхности
угля, т. е. тем выше скорость адсорбции и десорбции, поэтому при
температуре выше 600° С процессы адсорбции и десорбции
перестают оказывать влияние на общую скорость реакции.
В экспериментах, в которых удавалось лимитирующей
стадией сделать сам процесс сгорания углерода (стадия в), было
установлено, что до 1300° С реакция протекает по первому
порядку с энергией активации »100 кДж/моль; при более высоких
температурах порядок реакции понижается до нуля и энергия
активации возрастает до ?50 кДж/моль.
При горении углерода продукты реакции удаляются с
реакционной поверхности. Описание процессов, в которых образуются
нелетучие кристаллические продукты, значительно сложнее.
Таковы многие реакции, лежащие в основе металлургических
процессов, например:
4FeS2{K)+llO2(r)==2Fe2O3(K)+8SO2(r)
3Fe2O3(K)+CO(r) = 2Fe3O4<K)+CO2(r)
Подобного типа процессы состоят, по крайней мере, из
следующих стадий;
а) диффузия газообразного исходного вещества к
поверхности исходного кристаллического вещества,
б) адсорбция газообразного исходного вещества на
поверхности исходного кристаллического вещества,
245
в) реакция адсорбированного газа с исходными кристаллами,
г) адсорбция газообразного исходного вещества на
поверхности кристаллического продукта,
д) диффузия газообразного исходного вещества через слой
кристаллического продукта к границе раздела кристаллических
фаз,
е) взаимодействие исходных веществ на границе раздела
продукта реакции и исходного вещества,
ж) диффузия газообразного продукта от границы раздела
кристаллических фаз через слой кристаллического продукта
реакции к поверхносш,
з) десорбция газообразного продукта с поверхности
кристаллического продукта,
и) диффузия газообразного продукта от поверхности
кристаллического продукта в газовую фазу.
Стадии бив проходят только в начале процесса, далее
последовательно протекают семь стадий: а, г—и. Для
достижения максимального и быстрого выхода продукта реакции важно,
чтобы каждая стадия проходила быстро и не лимитировала
протекание других процессов и чтобы положение равновесия
стадии е было максимально смещено в сторону продуктов
реакции. В подобных гетерогенных процессах очень часто
кинетический фактор протекания реакции преобладает над
термодинамическим.
В выражение константы равновесия гетерогенной реакции
концентрации кристаллических фаз не входят (если не
образуются твердые растворы). Это объясняется тем, что какое бы
количество (очень много, очень мало) кристаллической фазы ни
находилось в равновесной системе, концентрация ее (выраженная
в моль/л) в егда одна и та же.
Другую руппу практически важных гетерогенных процессов,
протекающих на границе раздела кристалл—газ, составляют
реакции восстановления оксидов металлов.
Рассмотрим процесс восстановления оксида железа
водородом:
Fc5O4(K)+4H2(r) = 3Fe(K)+4H2O<r); K=p\o/p\2.
Одновременное существование двух кристаллических фаз Fe3O4
и Fe возможно юлько при строго определенных значениях
отношений р4н,о//;4нн соответствующих равновесию системы.
Если это oiношение меньше значения константы равновесия,
процесс протекает в направлении образования продуктов Fe и Н2О
(оксид железа восстанавливается), если же р*н2о1р*н2 больше
константы равновесия, процесс проходит в противоположном
направлении — окисления железа. В любых условиях процесс
совершается до момента достижения равновесного отношения
PArw2olP4r\\1 — К и> если рассматриваемое отношение не достигает
246
значения константы равновесия, процесс проходит в том или
ином направлении до полного исчезновения одной из
кристаллических фаз.
Ниже приведены термодинамические характеристики веществ
рассматриваемого процесса:
А" 248 обр ^ 2Ч8«
к Дж, моль Дж' (К мол ь)
FeK 0 27,15
Н2ОГ -241,82 188,72
Н2| 0 130,52
Fe3O4l? -1117,13 146,19
Рассчитаем изменение изобарного потенциала реакции:
Д//°= -4-241,82~(-1117,13)= 149,85 кДж/моль
А5° = 3 -27,15+4-188,72-4 130,52-146,19= +168,06 Дж/(К моль).
Следоват ельно,
AG= 149850- 168,06 Г[Дж/моль]
АС п298,, 5 = 149 850 - 168.06 • 298,15 « 99 740 Дж/моль.
Положительное значение АС 298 свидетельствует о
невозможности протекания процесса при стандартных условиях G=298 К,
А*2=/>н2о=Ю1325Па).
Найдем температуру, при которой процесс может протекать
самопроизвольно при стандаршых условиях (АС =0):
Т= 149850/168% 892 К E94° С).
Таким образом, газовая смесь водорода и паров воды
сРн2=Аьо восстанавливает Fe3O4 при температуре выше 594° С.
При этой температуре окислительные и восстановительные
способности участвующих в процессе веществ равны. При /<594° С
железо проявляет более сильные восстановительные свойства,
чем водород, при />594 С, наоборот, восстановительная
способность водорода выше, чем железа (только для
рассматриваемой системы!).
Процесс восстановления Fe3O4 водородом при стандартной
температуре невозможен, так как ДСС298 = 99,7 кДж/моль (т.е.
АС°298>0), но термодинамически вероятен противоположный
процесс:
3Fe(K)+4H2O(r) = Fe3O4(K)+4H2(r); AG°298= -99,7 кДж/моль.
Эта реакция лежит в основе одного из многочисленных
процессов газовой коррозии—разрушения металлов и сплавов при
различных температурных и концентрационных условиях.
Коррозия развивается при взаимодействии металлов с внешней
средой; в результате коррозии резко ухудшаются эксплуатационные
247
основной причиной коррозии следует считать
термодинамическую неустойчивость системы металл — вещество окружающей
среды. Чем более отрицательно значение изобарного потенциала
для какой-либо реакции, протекающей в этой системе, тем ниже
коррозионная стойкость металла. Однако для железа и других
металлов и их сплавов коррозионная стойкость определяется
кинетическими факторами. Действительно, изобарные
потенциалы образования оксидов хрома и алюминия (—1059 кДж/моль
для Сг2О3, —1582 кДж/моль для А12О3) намного ниже, чем у
оксида железа ( — 740 кДж/моль для Fe2O3), однако коррозионная
стойкость железа часто оказывается значительно ниже, чем
хрома или алюминия.
Одна из причин коррозионной стойкости металлов состоит
в образовании на их поверхности пленки из продуктов
взаимодействия с веществами внешней среды. Такая пленка должна
быть механически прочной и непроницаемой для веществ
коррозионной среды, и она защищает металл от дальнейшего
разрушения.
Кроме паров Н2О коррозионно агрессивным по отношению
ко многим металлам является другой компонент окружающей
среды, а именно кислород. Окисление металлов кислородом—
также один из распространенных процессов коррозии. При
окислении железа чистым кислородом или кислородом воздуха
образуется окалина (ржавчина), в которой слои оксидов железа
располагаются в последовательности понижения содержания
в них кислорода по мере приближения к поверхности металла:
О2—Fe2O3—Fe3O4—FeO—Fe. Окисление железа проходит не
только на поверхности железа, но и в слое окалины, так как
оксиды железа проницаемы как для кислорода, так и для железа,
и в слое оксида происходит встречное движение атомов и ионов
кислорода и железа.
Плотности оксидов железа и самого железа различны,
поэтому слой оксидов легко отслаивается от поверхности металла, что
обеспечивает доступ окислителю и ускоряет процесс коррозии.
Пленка оксида алюминия не пропускает ни кислород, ни
алюминий, поэтому алюминий на воздухе (даже при сильном
нагревании) вполне устойчив.
Железо способно окисляться и кислородом, и водой, а их
совместное воздействие резко увеличивает скорость коррозии.
Процесс коррозии (ржавления) железа при этих условиях
выражается следующим суммарным уравнением:
4Fe+2H2O + 3O2-2(Fe2O3H2O).
Состав Fe2O3H2O—это по существу кислота простейшей
формулы HFeO2 (или FeOOH).
Скорость коррозии оценивают обычно двумя методами: по
убыли металла с единицы площади его поверхности и по скоро-
248
сти уменьшения слоя металла. Если скорость коррозии
составляет менее 0,001 мм/год, то металл или сплав считаются
совершенно стойкими, при скорости коррозии более 10 мм/год,
наоборот,— нестойкими.
Обеспечение снижения скорости коррозии — важнейшая
теоретическая и практическая проблема. Решение этой проблемы
носит комплексный, системный характер, поскольку коррозия —
это не один химический процесс, а множество процессов, каждый
из которых подвержен влиянию разных по своей природе
факторов, причем процессы взаимно влияют друг на друга. Поэтому
при разработке методов защиты от коррозии необходимо
учитывать, что одни факторы, замедляющие какую-либо стадию
коррозионного процесса, могут ускорять другие стадии и приводить
к противоположному влиянию на весь процесс в целом.
Известно, что литий крайне нестоек но отношению к
кислороду, но введение лития (^0,6% ат.) в никель приводит к
замедлению окисления никеля. Это объясняется тем, что в образующейся
окалине оксид лития Li2O уменьшает концентрацию катионных
вакансий, что понижает их подвижность в слое окалины и тем
самым снижае i скорость окисления в целом. В то же время добавка
металла, образующего в оксиде никеля более высокозарядные
ионы, приводит к увеличению концентрации катионных вакансий.
Так, добавка хрома (до 5% ат.) в никель приводит к росту скорости
окисления никеля и снижению его коррозионной стойкости.
В связи с тем, что перенос вещества при образовании окалины
в ходе высокотемпературной газовой коррозии происходит путем
переноса вещества по вакансиям кристаллической решетки
оксида, скорость окисления металла связана с проводимостью
окалины и она тем ниже, чем меньше эта проводимость. Поэтому
вещества, снижающие проводимость (ионную и электронную)
оксидов окалины, снижают и скорость коррозии.
Особую группу гетерогенных процессов составляют реакции,
протекающие в газовой (или жидкой) фазе с участием твердого
катализатора; такие реакции рассмотрены в следующем
параграфе.
§ 37. ГЕТЕРОГЕННЫЙ КАТАЛИЗ
При гетерогенном катализе скорость химической реакции
ускоряется кристаллическим веществом — катализатором, на
поверхности которого происходит промежуточное химическое
взаимодействие его с реагирующими веществами. Гетерогенный
катализ— яркий пример объекта системного научного
исследования, объединяющего основные учения химии—термодинамики,
кинетики, теории строения вещества и периодичности свойств
элементов.
249
Гетерогенно-каталитическая реакция совершается через ряд
стадий, из которых важнейшими являются следующие:
а) диффузия реагирующих веществ к поверхности
катализатора,
б) адсорбция реагентов на поверхности катализатора,
в) реакция на поверхности катализатора,
г) десорбция продуктов с поверхности катализатора,
д) диффузия продуктов от поверхности катализатора.
Катализатор увеличивает скорость химического процесса
и изменяет путь его прохождения. Скорость химического
процесса возрастает в результате понижения энергии активации и
повышения энтропии активации под влиянием катализатора. В общем
виде скорость реакции, протекающей на поверхности
кристаллического катализатора, можно описать уравнением:
v = vJl = Zexp [-?акт/(Я7)]ехр [SUJR],
где v—скорость реакции;
vn—скорость реакции, отнесенная к единице площади
поверхности;
П — площадь поверхности катализатора;
Еакт—энергия активации;
5акт—энтропия активации;
Z—функция, характеризующая влияние концентрации
реагентов на скорость реакции.
На рис. 112 изображена энергетическая диаграмма реакции,
проходящей с участием гетерогенного катализатора A) и без него
B). От аналогичной энергетической диаграммы с участием
гомогенного катализатора данная диаграмма отличается теплотами
адсорбции и десорбции. Адсорбция молекул реагирующих
веществ на поверхности
катализатора обычно сопровождается
выделением теплоты (А#адс<0).
Чем прочнее связи исходных
молекул с атомами на поверхности
катализатора, тем больше
теплота адсорбции. Очень сильное
адсорбционное взаимодействие
может препятствовать
прохождению основного химического
процесса.
Молекулы продуктов,
образующиеся в акте химического
взаимодействия на поверхности
катализатора, должны быть
оторваны от поверхности, на
что затрачивается теплота
десорбции (Д#дес>0). Слишком вы-
ход реакции
Рис. 112. Энергетическая диаграмма
реакции с катализатором A) и без
него B)
250
сокая теплота десорбции также может препятствовать
прохождению процесса.
Таким образом, только некоторое оптимальное сочетание
значений ?"акт, А#адс и АЯдес способствует каталитическому
ускорению процесса. Если энтальпии адсорбции или десорбции близки
к энергии активации, то стадия адсорбции или десорбции
становится лимитирующей и для ускорения реакции необходимо
обеспечивать активацию этих стадий. Обычно повышение
температуры ускоряет процессы адсорбции и десорбции и сокращает время
установления адсорбционного и десорбционного равновесия.
В гетерогенном катализе большую роль играет адсорбция,
так как каталитическая реакция протекает в мономолекулярном
адсорбционном слое. Однако сама по себе адсорбция не приводит
к ускорению реакции, хотя при этом и происходит локальное
увеличение концентрации реагирующих веществ и ослабление
некоторых химических связей в молекулах. Для эффективности
химического взаимодействия адсорбированные молекулы
должны быть определенным образом расположены на поверхности по
отношению не только друг к другу, но и к атомам и группам
атомов на поверхности катализатора. При этом происходит
понижение энергии активации реакции за счет перераспределения
энергий связи и повышение энтропии активации.
Для объяснения механизма гетерогенного катализа
предложено несколько теорий, причем общим является представление об
активных центрах—активных участках (точках) на поверхности
катализатора. Ниже рассмотрена теория А. А. Баландина,
основанная на структурных, энергетических и кинетических
представлениях, связанных с положением катализирующего элемента
в Периодической системе. Согласно теории А. А. Баландина
реагирующие молекулы располагаются на поверхности ка гализато-
ра мономолекулярным слоем, при этом наиболее активные
участки поверхности ориентируют молекулы строго
определенными связями, в результате происходит перераспределение
электронных плотностей в реагирующих молекулах и образование
новых связей.
Активные центры катализатора, на которых совершается
акт химического превращения, называются мультиплетами, а
сама теория, использующая представления о мулыиплетах,—
мулътиплетной. Активный центр, состоящий из двух точек
(атомов, групп), носит название дублета, из трех — триплета,
из шести—секстета. Все известные гетерогенно-каталитические
реакции, объясняемые мультиплетной теорией, могут быть
описаны дублетным, триплетным и секстетным механизмами и их
сочетанием.
Как следует из схемы, приведенной на рис. 113, реакция
AB+CD=AC+BD
251
к» »к
АВ
CD
+AC+BD
Рис 113. Схема реакции AB+CD = AC + BD, протекающей на
поверхности катализатора
К—активный цешр катализа юра
с участием катализатора протекает через стадию образования
мультиплетного комплекса с последующим его разложением.
Два атома К на поверхности катализатора формируют
простейший мультиплет—дублет. Например, образование диэтило-
вого эфира
= (С2Н5JО + Н2О
может быть объяснено дублетным механизмом:
CH3-CH2-f~O-H
!
!к
СН3—CHa-
Однако эта реакция может проходить и без гетерогенного
катализатора. Вместе с тем ряд реакций осуществляется только
на поверхности катализатора по мультиплетному механизму.
Рассмотрим одну из наиболее интересных каталитических
реакций — взаимодействие циклогексена С6Н10 с образованием
бензола С6Н6 и циклогексана С6Н12:
В газовой фазе эта реакция невозможна, поскольку
эффективное столкновение трех молекул циклогексена, приводящее к
химическому взаимодействию, маловероятно. Однако реакция
проходит на платине (и палладии).
Кристаллическая решетка платины принадлежит к кубической
системе. Молекула циклогексена имеет форму правильного
шестиугольника. В рассматриваемой реакционной системе атомная
структура катализатора и реагирующие молекулы обладают
одним общим качеством—элементами симметрии третьего поряд-
252
ка. В кристалле платины такой
порядок расположения атомов
присущ только октаэдрической
грани. Поверхность этой грани
может быть представлена
тремя семействами параллельных
прямых, пересекающихся под
углом 60°. В узлах
расположены атомы платины. Таким
образом, поверхность грани
кристалла платины—это мно- и .., п
v Рис. 114 Схема реакции
жеСТВО равносторонних треу- ЗС6Н10 = СбН6 + 2С6Н12, протекающей
ГОЛЬНИКОВ С атомами Платины на поверхности октаэдрической грани
В вершинах (рИС. 114). кристалла платины
При прохождении гетеро-
генно-каталитической реакции шестиугольные молекулы
циклогексена налагаются на треугольные структуры атомов
платины. Каждая молекула циклогексена удерживается на поверхности
катализатора тремя атомами платины, из которых только два
каталитически активны (на рис. 114 они выделены большими
черными кружками). Три молекулы циклогексена располагаются
так, что две связи С—С одной молекулы оказываются вблизи
связей С=С двух других молекул, которые и оттягивают на себя
атомы водорода (изображено стрелками). В данной схеме
процесса на три молекулы циклогексена приходится шесть
каталитически активных атомов платины, объединенных в два триплета
(они обведены пунктиром).
Если бы рассматриваемая реакция осуществлялась в
результате тройного столкновения частиц, то ее скорость подчинялась
бы уравнению третьего порядка:
где к—константа скорости реакции;
С—концентрация реагирующих веществ.
В действительности эта реакция имеет порядок, равный 3/2:
v=kCZ12.
Такой порядок реакции подтверждает предположение о том, что
на три молекулы циклогексена приходятся два активных
центра—два триплета.
То, что в продуктах реакции циклогексана получается в два
раза больше бензола и не обнаруживается других веществ,
является прекрасным доказательством мультиплетной теории.
В подобного типа гетерогенно-каталитических реакциях
участвуют не молекулы целиком, а группы их атомов, которые
должны находиться на расстояниях, обеспечивающих
возможность формирования новых связей. В то же время на поверхности
253
кристалла на молекулы или молекулу воздействует не один атом,
а несколько (мулыиплет). Взаимодействующие молекулы и
соответствующие мулыиплеты образуюi определенную химическую
систему—мулыпиплетный комплекс (подобный активному
комплексу), неустойчивый по отношению к продуктам реакции.
Образование мультиплетного комплекса возможно при строгом
структурном и энергетическом соответствии всех элементов
системы.
Требование структурного соответствия элементов
реакционной системы выражается принципом геометрического
соответствия. Согласно принципу геометрического соответствия для
осуществления каталитического процесса чрезвычайно важен способ
наложения реагирующих молекул на поверхность катализатора,
определяемый размерами атомов и их расположением на
поверхности катализатора. По мультиплетной теории каталитически
активными металлами в реакциях, подобных рассмотренной,
могут быть только те, которые обладают гранецентрированной
кубической и гексагональной решетками с межъядерными
расстояниями от 0,249 до 0,2775 нм, и твердые растворы этих
металлов. Этим условиям удовлетворяют все ^-элементы
VIII группы Периодической системы (за исключением железа):
Со, Ni, Ru, Rn, Pd, Os, Ir, Pt, а также Тс, Re и Zn. Межъядерные
расстояния в кристаллических структурах железа, хрома,
ванадия, молибдена, вольфрама укладываются в пределы
межъядерных расстояний, требуемых теорией, однако эти металлы не
могут катализировать реакции по мультиплетному механизму
из-за их кубических объемно центрированных решеток (в других
реакциях эти металлы могут быть катализаторами. Это еще раз
показывает, что катализ—системное явление, требующее для
объяснения привлечения многих теорий). Сплав 75% Со и 25% Fe
имеет кубическую гранецентрированную решетку и
каталитически активен в реакции гидрирования бензола, а сплав 50% Со
и 50% Fe в этой реакции не активен—у этого сплава кубическая
объемно центрированная решетка.
Для осуществления гетерогенно-каталитической реакции
недостаточно одного геометрического соответствия между
реакционными центрами реагирующих молекул и активными центрами
катализатора. Должно соблюдаться определенное соответствие
между энергиями связи в реагирующих молекулах, энергиями
связи между атомами молекул и активными центрами
катализатора и, наконец, между энергиями связи в молекулах
образующихся веществ. Все эти связи не должны быть ни слишком
прочными, ни слишком слабыми. В противном случае или не
происходит необходимого ослабления в связях исходных
молекул, или же исходные или образующиеся молекулы не смогут
оторваться от поверхности катализатора. Если подобными
молекулами являются молекулы веществ, не участвующие в основной
254
реакции, но присутствующие в зоне реакции, то происходит
отравление каталитической поверхности.
Явление отравления катализатора объясняется не только
сильной адсорбцией посторонних веществ, но и очень прочной
связью молекул, участвующих в реакции, с активными центрами
поверхности. В то же время некоторые вещества—промоторы —
усиливают активность катализатора. Теория объясняет это
явление тем, что посторонние атомы на поверхности катализатора
создают более благоприятные геометрические и энергетические
условия для прохождения реакции.
На основе принципа энергетического соответствия
разработаны способы вычисления энергии активации по энергиям связей
между атомами реагирующих молекул и атомами катализатора.
Руководствуясь мультиплетной теорией, А. А. Баландин
предсказал каталитическую активность в реакциях
гидрогенизации родия для олефинов, никеля для ацетилена, рутения для
соединений с карбонильной связью. С помощью этой теории
предсказана каталитическая активность рения в реакции
дегидрогенизации циклогексана: рений по параметрам
кристаллической решетки и структуре соответствует секстегному механизму
катализа.
§ 38. ОБРАЗОВАНИЕ КРИСТАЛЛОВ ИЗ ЖИДКОЙ ФАЗЫ
Наиболее высокий уровень химической организации вещества
достигается в его кристаллическом состоянии. Расположение
атомов и молекул в кристаллической структуре максимально
упорядочено. В кристаллическом состоянии порядок
расположения атомов и молекул наиболее высок по сравнению с порядком
в жидком и газообразном состояниях, а энтропия вещества в
кристаллическом состоянии минимальна. Так же, как жидкость имеет
некоторые свойства, характерные для газообразного состояния,
кристаллические вещества обладают рядом свойств жидкостей,
а жидкости—свойствами кристаллических веществ. При
переходе от одного уровня организации вещества к другому одни
свойства исчезают, другие видоизменяются и появляются новые.
Взаимосвязь уровня организации веществ и их свойств
хорошо прослеживается при обсуждении процессов образования
кристаллов из жидкой (а также из газообразной) фазы.
Вещества кристаллизуются из раствора, если при данной
температуре раствор пересыщен. Для приготовления пересыщенного
раствора необходимо знать растворимость вещества в данном
растворителе при соответствующей температуре и зависимость
(хотя бы качественную) растворимости от температуры. Если
растворимость вещества с повышением температуры возрастает,
что характерно для многих кристаллических веществ, то для
255
приготовления пересыщенного раствора достаточно ввести
в определенное количество растворителя избыточное количество
растворяемого вещества, нагреть раствор до полного
растворения вещества и затем его медленно охладить. Из растворов
солей, растворимость которых в воде понижается с повышением
температуры (Li2SO4 • Н2О, КС1 и др.), кристаллы можно
выделить нагреванием насыщенного раствора. Если растворимость
вещества почти не зависит от температуры, как, например, у
хлорида натрия, то кристаллы нельзя получить при изменении
температуры насыщенного раствора (кристаллы таких солей можно
выделить при испарении растворителя).
В любом случае пересыщения раствора и образования
кристаллов можно достичь удалением растворителя путем его
испарения или введением в раствор посторонних веществ,
связывающих растворитель или уменьшающих растворимость
растворенного вещества.
Кристаллизация — самопроизвольный фазовый переход
вещества из состояния пересыщенной или переохлажденной среды
в кристаллическое состояние. При таком переходе изобарный
потенциал системы уменьшается, т. е. АС? < 0, и процесс
кристаллизации происходит до тех пор, пока изменение изобарного
потенциала станет равным нулю, ДG = 0, т. е. не образуется насыщенный
при данных условиях раствор. Если А<7 > 0, то раствор не насыщен,
самопроизвольная кристаллизация невозможна и протекает
противоположный процесс—растворение (или плавление).
Превращение жидкой фазы в кристаллическую совершается
с разнообразными скоростями и происходит не сразу во всем
объеме, а сначала процесс развивается в некоторых участках —
центрах кристаллизации, или зародышах.
Процесс кристаллизации складывается из двух стадий:
образование зародышей и их рост.
Переохлажденная или пересыщенная среда может долго
сохраняться, не изменяясь и не кристаллизуясь. Однако при
достижении некоторого предельного при данных условиях
пересыщения возникает крайне быстро множество агрегатов молекул,
мелких кристаллов—зародышей, которые растут до достижения
системой равновесного состояния (AG = 0). Способность системы
долго находиться в пересыщенном метастабильном состоянии
свидетельствует об очень высокой энергии активации
образования кристаллических зародышей.
Один из возможных механизмов образования кристаллов
в растворе соли КА состоит в следующем. В растворах сильных
электролитов возможно образование ионных пар:
Чем выше концентрации растворов, тем выше вероятность
образования ионных пар и более сложных ассоциатов:
256
=(Л'К'А
И I. Д.
Можно предположить, что, например, в расiворс NaCl ионы
натрия координируют вокруг себя шесть ионов С1~, а хлорид-
ионы— шесть ионов Na":
[NaCU [C1Na6].
Каждый из ионов в этом кристаллическом зародыше служит
центром координации других иро1ивоположио заряженных
ионов.
Далее процесс криааллизации можхм развиваться в двух
направлениях в зависимости от cooj ношения скоростей
образования зародышей и их роста.
Скорость образования зародышей i>, измеряется числом
зародышей л, образующихся в единицу времени
Uj =cIn,'(lL
Скорое Jb роста зародышей (скорость роста кристалла)
v2 определяется числом молекул, атомов, ионных пар /и,
переходящих из раствора на поверхносп> кристаллического зародыша
в единицу времени:
v2— —clm/clt.
При образовании крис1аллической фазы оба процесса
накладываются друг на друга. При и, >v2 скорость образования
зародышей велика и растворенное вещество переходит в состояние
зародышей, в результате получается высокодисперсная
кристаллическая фаза. При v2>vx формируются грубодисперсные фазы
(крупные частицы, большие кристаллы).
К процессам образования зародышей, их роста или
растворения применимы все те положения, о которых пойдет речь при
рассмотрении термодинамических, кинетических и структурных
факторов стабильности коллоидных систем. Коллоидные
частицы могут служить центрами кристаллизации. Иногда достаточно
ввести в пересыщенный раствор моль кристаллизующегося
вещества, как начинайся кристаллизация. Если рост ядер-зародышей
приостанавливается вследствие возникновения на них
адсорбционного слоя ионов, то образуйся высокодисперсная
мелкокристаллическая фаза, обладающая юй или иной степенью агрега-
тивной устойчивое!и.
Таким образом, представления химии коллоидных сиеiем
в полной мере могут быть использованы для обсуждения
9 24S2 257
^ процессов
кристаллизации малорастворимых
веществ. Следует огме-
ц гить, чго переход веще-
r l 7 ства в коллоидное
состояние при охлажде-
цент рированных раст-
1>ис 115 Зависимость скоросги образования за- ВОРОВ' ПРИ ИХ У^РИВа-
ролышсй Vi и скоросги роста кристалла нии и ПРИ введении дру-
v2 в переохлажденной среде от температуры ГИХ веществ,
уменьшала г.,-г юндах растворимое! ь,
не обнаруживается.
Скорость образования зародышей связана со степенью
переохлаждения раствора, т.е. с тем, насколько раствор
перенасыщен.
С увеличением переохлаждения скорость образования
зародышей (кривая vt на рис. 115) вначале возрастает, затем
достигает максимума, после чего снижается; при сильном
переохлаждении она становится очень малой.
Скорость роста кристалла v2 (скорость отложения вещества
на зародышах) с понижением температуры вначале растет, затем
достигает некоторо1 о постоянного максимального значения,
после чего уменьшается.
Скорость процесса кристаллизации в целом определяется
соотношением обеих скоростей. Если максимум скорости
образования зародышей попадает в область высоких скоростей роста
кристалла, то вещество кристаллизус1ся легко и получить
переохлажденную жидкость трудно. Если же скорость образования
зародышей попадает в область температур, при которых
скорость отложения вещее 1ва на зародышах мала, то жидкость
легко переохлаждается и можно получить вещество в
стеклообразном состоянии. Скорость образования зародышей и
последующего pocia кристаллов тем больше, чем дальше от
равновесия находится система.
Наличие плоских граней у формирующихся кристаллов
говорит о том, что дальнейший рост зародышей и мелких кристаллов
происходит путем отложения параллельных слоев вещества на
зародыше или уже образовавшемся кристалле.
Зародившиеся кристаллы продолжают свой рост по
закономерностям, которые обусловливают строго периодическое
повторение в расположении частиц и строго определенную
геометрическую форму кристалла. При росте кристалла всегда соблюдается
следующая закономерность: размер грани кристалла тем
больше, чем меньше скорость ее роста. Именно поэтому изменение
«емпературы и концентрации раствора, т. е. степени его
пересыщения, приводит к образованию кристаллов самого различного
258
внешнего вида для одного и того же соединения. По мере
приближения системы к состоянию равновесия форма растущего
кристалла приближайся к равновесной, т. е. такой, которая
отвечает минимуму суммарной поверхностной энергии граней.
Скорость роста кристаллов зависит от многих факторов.
Рассмотрим важнейшие из них.
Теплота, выделяющаяся при pocie кристалла, должна быть
отведена от его граней, где происходит осаждение вещества
и формирование химических связей. Поэтому на скорость роста
кристалла влияет теплопроводность как самого кристалла, так
и окружающей его жидкости.
Скорость роста кржналла сложным образом зависит от
температуры. Чем ниже температура расплава или раствора, тем
выше степень его переохлаждения и пересыщения и, казалось бы,
больше скорость образования зародышей и роста кристаллов.
Однако в действительности эта скорость возраст ает до
некоторого максимума, после чего понижается вследствие увеличения
вязкости среды и уменьшения скорости движения молекул.
Другой фактор, определяющий скорость роста кристалла,—
скорость диффузии частиц вещества из жидкой фазы к
поверхности растущей грани кристалла. Следовательно, протекают два
одновременно противоположно направленных процесса — отвод
теплоты и доставка к зародышам новых порций частиц.
На скорость образования зародышей в большой степени
влияет наличие в растворе посторонних частиц (пылинки и т. д.),
которые играюi роль центров крисгаллизации. Скорость pocia
кристаллов зависит не только ot температуры раствора или
расплава, но и скорости ее понижения. При быстром понижении
температуры системы образуется мелкокристаллический осадок.
Если быстро охлаждать жидкость, то стадию зародышеобразова-
ния можно «проскочить», и в этом случае происходит застекло-
вывание вещества. Стекло по существу представляет собой очень
вязкую жидкое! ь. Многие органические вещества способны
образовывать стекла, из неорганических веществ к стеклообразова-
нию склонны силикаты, 6opaibi и фосфаты.
Рассмотрим, как отражается переохлаждение на ходе кривых
охлаждения. При охлаждении расплава однокомпонентной
системы (рис. 116, а) в идеальном случае в момен г появления первых
кристаллов (Г11Л) система становится нонвариантной (/? = пост):
Температура в системе сохраняется постоянной до исчезновения
последних капель жидкости, что отражается на кривой
охлаждения горизонтальным участком.
Подобное описание кристаллизации относится к
гипотетическому идеальному процессу. При переохлаждении расплава его
температура становится ниже равновесной температуры начала
9* 259
Рис. 116 Влияние переохлаждения на кривые охлаждения однокомпонентного
расплава (а) и двухкомпонентног о раствора (б):
/—«нормальный» xo;j кривых, 2—кривые для переохлажденных сиеiсм
кристаллизации (плавления). В момент начала кристаллизации
переохлажденного расплава (кривая 2 на рис. 116) под действием
выделяющейся теплоты температура повышается, и тем сильнее,
чем больше скорость кристаллизации, энтальпия плавления,
степень переохлаждения и слабее отвод теплоты кристаллизации.
При большой скорости кристаллизации температура в системе
может приблизиться к температуре плавления вещества.
Поэтому температура начала кристаллизации может оказаться
значительно ниже температуры плавления (которую следует
определить путем повышения температуры кристаллов; перегреть
кристалл выше температуры плавления не удается). Пунктир на рис.
116, а показывает ход изменения температуры при образовании
стеклоподобной фазы.
При кристаллизации из двухкомпонентного раствора эта
система в отличие от чистой жидкости отвердевает не при
постоянной температуре, и процесс кристаллизации проходит как
моновариантный:
На кривой охлаждения при температуре кристаллизации
обнаруживается излом (рис. 116, б, кривая /). У переохлажденного
раствора 1емпература замерзания (начала кристаллизации) ниже,
а в результате выделения теплоты кристаллизации
переохлажденного раствора форма кривой охлаждения искажается (кривая 2 на
рис. 116, б).
Способность веществ переохлаждаться и образовывать
пересыщенные растворы обусловлена высокой энергией активации,
низкой энтропией активации процесса образования центров
кристаллизации и малой скоростью роста зародышей.
Установлено, что скорость образования зародышей
происходит почти по цепному механизму и образовавшиеся кристаллы
катализируют размножение зародышей. Были предложены
различные объяснения этого явления. Возможно, что при быстром
260
росте зародышей на их поверхности появляются игольчатые или
усоподобиые частицы, которые обламываются и служат
центрами крисмаллизации. Более правдоподобно другое объяснение:
броуновское движение зародышей вызывает их столкновения,
и если кинетическая 'энергия зародышей велика, они
раскалываются на несколько частей, коюрые становятся центрами
кристаллизации
Рост криааллического зародыша осуществляйся путем
перемещения грани параллельно самой себе в направлении,
перпендикулярном грани.
Возможны два основных механизма роста граней и всего
крисчалла в целом. Первый механизм состоит в том, что грани
растут послойно. На плоскую поверхность грани (рис. 117, а) на
некотором ее участке осаждается из жидкой фазы частица.
Вероятность того, ч!о она удержится на краю грани или на ее угле,
Рис 117 Схема роста кристаъюв по двум механизмам
а—с юями, о—но спирали
261
намною ниже, чем на некотором серединном участке грани. Это
объясняется тем, что на такую отдельную частицу кроме сил
прямого контакта с поверхностью действуют силы соседних
частиц, что ослабляет способность частицы к перемещению по
поверхности грани. Присоединение к осевшей частице другой
частицы будет более вероятно, чем осаждение этой в юрой частицы
в другом месте грани. Осевшая на поверхности грани частица
играет роль зародыша развития новой грани, поэтому частицу
называют двухмерным (плоским) зародышем. Для роста грани
по этому механизму необходимо сильное пересыщение раствора
(до 40—50%) у поверхности грани, так как вероятность
удержания о 1 дельной частицы на грани очень мала и не превышает
вероятность образования центра кристаллизации.
В то же время известно, что кристаллы растут и при крайне
малых (доли процента) пересыщениях. Это объясняют тем, что
поверхность грани растущего кристалла не идеально плоская,
а имеет различного рода впадины, микротрещины, изломы и т. д.
Другой механизм роста кристалла заключается в том, что
из-за некоторого «винтового» нарушения (винтовой дислокации)
на поверхности кристалла его дальнейший рост происходит как
бы по винтовой лестнице, или по спирали (рис. 117, б). Для роста
кристалла путем присоединения частиц к постепенно
продвигающейся спирали не требуется образования двухмерного
зародыша, поэтому рост возможен при минимальных пересыщениях
раствора.
Как указано выше, форма кристалла определяется скоростью
роста той или иной грани. Существование различных
кристаллических форм свидетельствует о неодинаковых скоростях роста
граней и их кристаллографической неидешичности.
Следует обратить внимание на следующее кажущееся
парадоксальным явление: быстро растущие грани имеют наименьшие
размеры, а медленно растущие—наибольшие. По мере роста
кристалла быстро растущие грани постепенно сокращаются,
исчезают и заменяются медленно растущими.
Наличие в растворе посторонних веществ может вызвать
изменение внешней формы растущего крисшлла. Так, хлорид
натрия в водном растворе кристаллизуется в виде простых кубов
(рис. 118, я), если же раствор содержит немного мочевины
CO(NH2h, то кристаллы приобретают форму кубов со
срезанными вершинами. При еще большем содержании мочевины
в растворе размер граней, срезающих вершины куба,
увеличивается (рис. 118, б, в), а при достаточно высокой концентрации
мочевины именно эти грани формируют кристалл и вместо куба
получается октаэдр (рис. 118, г). По составу и структуре октаэд-
рические кристаллы хлорида натрия ничем не отличаются от
кубических и практически не содержат мочевины. Это явление
можно объяснить по-разному: молекулы мочевины адсорбиру-
262
Рис 118. Формы кристаллов NaCt, выращенных из чистого
раствора (а) и с добавкой мочевины в увеличивающейся
концентрации {б, в, г)
ются или на гранях куба, способствуя их быстрому росту, или же,
что более вероятно,— на гранях октаэдра, замедляя их рост
(скорость самопроизвольно растущих граней кристалла в услови-
Ах, близких к равновесным, должна быть минимальной). В
данном случае проявляется каталитическое влияние постороннего
вещества (мочевины) на скорость роста отдельных граней
кристалла (хлорида натрия).
§ 39. РАСТВОРЕНИЕ, РАСТВОРИМОСТЬ, ПРОИЗВЕДЕНИЕ РАСТВОРИМОСТИ
При соприкосновении двух (или более) различных веществ
в месте контакта начинается процесс проникновения одного
вещества в другое. Взаимное проникновение соприкасающихся
веществ называется диффузией. Она проходит из-за теплового
движения частиц и теоретически не совершается только при О К.
Диффузия присуща веществам в любых агрегатных состояниях
и проходит в направлении понижения концентрации
диффундирующего компонента. Диффузия прекращается при достижении
равномерного распределения частиц вещества в
рассматриваемой системе (или при достижении некоторого равновесного
состояния, насыщения одного компонента системы другим, при
котором дальнейшая диффузия при данных условиях уже не
возможна).
Если диффузия проходит без изменения энергетического
состояния системы (теплота не выделяется и не поглощается), т. е.
АН процесса =0, то диффузия совершается за счет увеличения
энтропии системы, А5>0. Такая диффузия называется
физической, или идеальной. При физической диффузии образуются
идеальные растворы (Д#обр = 0, AS>0). В идеальных растворах
частицы участвующих в диффузии веществ не взаимодействую» ни
между собой, ни с частицами вещества, в которое они
диффундируют. Этим идеальные растворы отличаются от реальных.
В большинстве случаев при диффузии одновременно
совершаются многочисленные процессы взаимодействия частиц от
слабых межмолекулярных ван-дер-ваальсовых до сильных типа
химических реакций. Подобные процессы мы будем называв
263
растворением, а продуюы — растворами. Расiворы бываю!
жидкими и твердыми (кристаллическими), редко гозорят о газовых
растворах.
В этом разделе мы будем обсуждать paciBopcimc крисчал-
лических веществ в жидкости с образованием раствора.
Раствор—это гомогенная (однофазная) система неременного
состава (в зависимости от температ>ры, давления и других
условий), в которой одно или несколько веществ равномерно
распределены в среде другого вещества. Поэюму расiворы могут
состоять из двух и более компонентов, коюрые распределяются
в виде атомов, молекул, ионов или в виде ipynn небольшого
числа этих частиц. Это оi носится к любому агрегатному
состоянию веществ. При распределении компонентов энтропия системы
в большинстве случаев возрастает, но когда при образовании
раствора проходит реакция или образуются большие
упорядоченные группы частиц, энтропия может понижаться.
То вещество, которое растворяе! другое, называемся
растворителем. Раствори i ель в чистом виде существую в том же
агрегатном состоянии, что и образовавшийся расiвор. Если до
растворения оба вещества находились в одном и том же агрегатном
состоянии, то растворителем считают вещество, присутствующее
в растворе в большем количестве. В зависимости oi природы
растворителя растворы бывают водными и неводными.
Иногда говорят о рас1ворении металлов, например, натрия
в воде или цинка в серной кислоте. Не будем такие процессы
называть растворением (это относится также и к процессам
перехода металла в расiвор при электролизе). Будем называть
растворением только такой процесс, при котором растворенное
вещество можно выделить из рас г вора в исходном его сосюянии
такими простыми операциями, как перекристаллизация,
выпаривание и т. п.
Важнейшей характеристикой раствора являе1ся его cociaB,
который выражается концентрациями компонентов Обычно
говорят о концерн рации того компонент, которого в растворе
меньше (т. е. концентрацию растворителя обычно не указывают).
Чисто качественно в зависимости от концентрации жидкого
раствора говорят о разбавленных и концентрированных расi ворах.
Концентрацию раствора выражают различными способами.
Массовом концентрация определяйся как оiношение массы
растворенного вещее iBa g к объему раствора К:
С = ^(кг/м3, г/ли т.п.).
Титр раствора — та же самая массовая концен! рация, но
выраженная числом граммов растворенного вещества в 1 мл
раствора, г/мл.
264
Массовая доли С, некоторою компонента / в растворе,
содержащем А' компонентов, равна отношению массы данного
компонента gj в растворе к сумме масс всех компонентов, включая
и растворитель ( )
где g—массы coo i ветс гвующих компонентов, выраженные в г,
кг или других единицах массы
Сумма массовых долей всех компонентов раствора равна 1.
Если сумма g\+g2 + — +gt + — +gk равна 1 г, то массовая доля
компонента gt численно равна числу граммов этого компонента,
содержащихся в I г раствора.
Наиболее paciipoci ранен способ выражения состава раствора
через массовые проценты («процентная концентрация»). Так, 20%-
ный раствор — это раствор, в 100 i которого содержиich 20 г
этого вещее !ва. Массовый процент (или «процеш по массе»)
равен массовой доле компонент, умноженной на 100. Сумма
массовых процентов всех компонентов равна 100.
Мольная (молярная) концентрация — это число молей
растворенного вещества в I л раствора. Мольная концеи!рация равна
отношению количества н растворенного вещее 1ва в молях к
объему V раствора в литрах:
С = - (моль/л, моль/дм3 и т. п.).
Мольная (молярная) доля Nf компонента / равна отношению
числа молей п( эюго компонента к сумме чисел молей
n1 + n2-\-... + ni + ... + пк всех к компонентов paciBopa:
Сумма мольных долей всех компонентов раствора равна
единице.
При выражении концентрации через количество
растворенного вещества, приходящегося на единицу объема раствора
(мольная концешрация), следует учитывать, что вследствие
зависимости объема рас!ворителя от температуры концентрации
растворенного вещества будет изменяться с температурой. Поэтому
при проведении точных экспериментов растворитель следует
отмерять не по объему, а по массе. Концен! рация, выраженная
числом молей растворенного вещес!ва на 1000 г растворителя,
называется моляльной. Молялышя концентрация — это
отношение количества рассоренного вещее[ва т в молях к массе
растворителя g (кг, г и т. п.):
265
(моль/1000 г растворителя).
т
лрасг
Пользоваться ранее широко применявшейся нормальной
концентрацией сейчас не рекомендуется. Нормальная концентрация
есть мольная концентрация эквивалента (моль/л). Численное
значение нормальной концентрации зависит от реакции, в которой
принимает участие вещество. Для таких веществ как НС1, КОН
или NaOH нормальная и мольная концентрации совпадают. Если
серная кислота реагирует как одноосновная, то для нее также
мольная и нормальная концентрация равны, но, если серная
кислота реа1ирует как двухосновная кислота, численное значение
нормальной концентрации будет в 2 раза меньше мольной. То же
самое можно сказать и о фосфорной кислоте.
Иногда мольная и нормальная концентрации обозначаются
не моль/л, а буквами Мин соответственно, например:
2 М—двумольный 2 н—двунормальный
(двумолярный) раствор
i М — одномольный 1 и—однонормальиый
0,1 М—децимольный 0,1 н—децинормальный
0,01 М—сантимольный 0,01 н—сантинормальный
В лабораторной практике эти обозначения используются для
растворов, точное значение концентрации которых не известно
или не требуется для работы. В остальных случаях рекомендуется
использовать общепринятые обозначения, например,
0,0936 моль/л.
В зависимости от того, образуются в растворе ионы или нет,
растворы подразделяют на два класса—растворы электролитов
и растворы неэлектролитов.
При растворении в воде хорошо растворимого сильного элек-
фолита концентрации ионов определяются в соответствии
с уравнением диссоциации, например, в растворе Na2SO4 с
концентрацией 0,1 моль/л концентрации ионов будут равны:
С 2С С
CNa+ =0,2 моль/л, CSo42+ =0,1 моль/л.
Если растворяется хорошо растворимый слабый электролит,
например, уксусная кислота СН3СООН, концентрации ионов
СН3СОО" иН4 рассчитываются с использованием константы
равновесия диссоциации кислоты.
Абсолютно нерастворимых веществ в природе не существует.
В ю же время существуют вещества с неограниченной
растворимостью друг в друге (например, вода и серная кислота) и
ограниченно растворимые. При ограниченной растворимости
способность вещества образовывать с другим веществом однородную
266
термодинамически устойчивую систему характеризуется
растворимостью. Растворимость выражается концентрацией
насыщенного раствора.
Насыщенный рас i вор получается, когда дальнейшее
растворение данного компонента в растворе прекращается. В
насыщенном растворе концентрация этого компонента максимальна при
данных условиях. Насыщенный раствор всегда должен
находиться в равновесии с кристаллическим компонентом (осадком).
Концентрация этого компонента в растворе называется его
растворимостью. Раствор при концентрациях этого компонента ниже
растворимости называется ненасыщенным.
Раствор, в котором концентрация растворенного вещества
выше его растворимости, называется перенасыщенным.
Перенасыщенный раствор не может находиться в контакте с
кристаллами растворенного компонента. Перенасыщенные растворы
обычно получаются при переохлаждении раствора.
Перенасыщенный рас i вор нестабилен (метастабилеп) и при внесении
затравки или при перемешивании выделяет в осадок избыток
компонента и переходит в насыщенный раствор. Перенасыщения
водного раствора соли можно также достичь добавлением в
раствор веществ, связывающих воду, например, спирта
(растворимость многих солей в водноспиртовых растворах меньше, чем
в чистой воде).
Рас1воримость удобно выражап> в моль/л, однако часто ее
выражают в процентах по массе а, т. е. числом граммов
растворенного вещества в 100 г раствора или числом граммов Ь
растворенного вещества в 100 г растворителя, при этом
\тъ
а —
100 + Л
Зависимость растворимости от температуры определяют по
справочным таблицам или по кривым растворимости (рис. 119).
Если приготовить насыщенный раствор при более высокой
температуре гъ содержащий b2 г вещества на 100 г воды, а затем
охладить до температуры /,, при которой растворимость
составляет Ьи то выпадет осадок (при условии, чго не произошло
переохлаждения и перенасыщения раствора) в количестве Ь2—Ь1 г.
На различной растворимости веществ основан один из
способов очистки веществ—перекристаллизация. Очистка
перекристаллизацией сводится к растворению загрязненного вещества
в подходящем растворителе при повышенной температуре и
последующему выделению кристаллов очищаемого вещества из
перенасыщенного раствора при более низкой температуре.
Очистка перекристаллизацией возможна, если растворимость
вещества заметно зависит от температуры (сравните, например,
КС1 и KNO3).
267
Р, г//00гН2О
20 40 SO SO WO t}°C
Рис 119 Рас i воримос гь солей (г/
100 i волы) в зависимости от
температуры
в воде, то его растворимость
через концентрации ионов в
растворимость NaCl в воде
иСГ:
Насыщенный раствор соли,
который оаается после
отделения выпавших кристаллов,
называется маточным. Некоторое
количество примесей может
увлекаться осадком. При повторной
нерекристаллизации чистота
вещества повышается.
Растворимость Р вещества, но
существу, есть констата
равновесия reieporenHoro процесса
перехода вещества из
кристаллической фазы в рас i вор.
Если вещество не электролит,
то его рас i воримость равна
количеству вещест ва, перешедшему
в раствор:
А(к) = А(р_р) Л>РЛ, моль/л.
Если вещество являе1ся
электролитом и хорошо растворимо
может быть выражена (определена)
насыщенном растворе. Например,
равна концешрациям ионов Na"
/>=с,
NaCl <p р)
= CNa - =Ca- моль/л.
В случае насыщенного раствора солей типа Na3PO4
соотношение растворимости и концентраций ионов зависит от зарядов
ионов. Например, в случае Na3PO4:
— /
)> MOJIb/л.
Точно так же выражают и растворимость малорастворимых
электролитов, но для них чаще пользуются констанюй
равновесия, называемой произведением растворимости.
В насыщенном при данной температуре растворе
малорастворимого электролига KxAy между кристаллами и раствором
устанавливается динамическое равновесие:
Произведение растворимости — это константа iетерогенного
равновесия между кристаллами малорастворимого электролита
и его раствором:
268
В выражение константы равновесия концентрация
кристаллической фазы не входит, так как она постоянна. Следовательно,
в насыщенном растворе малорастворимого электролита
произведение концентраций его ионов в степени их стехиометрических
коэффициентов есть величина постоянная.
Д;,я насыщенного раствора малорастворимой соли Са3(РО4J
в отсутствие сильных элект ролитов произведение растворимости
записывается как произведение равновесных концентраций ионов
в растворе:
Са3(РО4J(к) = ЗСа2 + (р-р)+2РО43~ (Р-р)
ПР-[Са2+]3[РО43-]2.
Рассматривая произведение растворимости как константу
равновесия процесса перехода электролита из кристаллического
состояния в раствор, найдем изменение изобарного потенциала
растворения при любых концентрациях ионов в системе:
(Р-Р)
где Ску+ и Сл* —любые произвольные неравновесные концет-
рации ионов Ку+ иАх~ в растворе.
Это выражение преобразуем так:
Самопроизвольный процесс растворения возможен, если
AG<0, что достигается при
пр <im
Отсюда
CV+CV <ПР.
Следовательно, если произведение концентраций ионов в
степени их сгехиометрических коэффициентов меньше произведения
растворимости для того же малорастворимого электролита, то
кристаллизация из данного раствора невозможна, раствор
ненасыщен и происходит процесс растворения вещества.
При ГУСУ
ПР
ПР = С\у+С\х- =[К
и AG-0.
При равенстве произведения растворимости и произведения
концентраций ионов в степени их стехиометрических
коэффициентов система находится в равновесии и раствор относится к
насыщенным.
cv?V
и ДС>0.
Эю означаем что если произведение концентрации ионов
в растворе больше произведения растворимости, процесс
растворения малорастворимого электролита термодинамически
невозможен, раствор пересыщен и протекает противоположный
процесс — кристаллизация.
Из определенной опытным путем растворимости электролита
можно рассчитать произведение растворимости и, наоборот, зная
произведение растворимое!и электролита, можно рассчитать его
растворимость в воде.
Ниже приведен пример расчета произведения растворимости
гидроксида цинка, если известно, что при комнатной 1емиературе
в 100мл его насыщенного раствора содержится 1,49'10 г
Zn(OHJ. Мольная масса гидроксида цинка 99,39 г/моль. Для
расчета ПР необходимо найги растворимость Р гидроксида
цинка:
п 1,49 !<Г5 1000 , . 1Л_6 ,
Р= = 1,5-10 6 моль/л.
99,39 100 '
В растворе гидроксида цинка
Zn(OHJ(K) = Zn2 + (p-p)-f 2ОН- (р-р)
на каждый ион цинка приходится два гидроксид-иона. Поэтому
[Zn]2+ = 1,5 -10 моль/л и [ОН"]-2 [Zn^+ ] = 3,0-10 моль/л.
Отсюда произведение растворимое!и составит:
Вычисление растворимости по известному значению ПР
проводится в обратной последовательности. Найдем растворимость
гидроксида цинка, для которого ПР= 1,35* 10~17. Примем
растворимость Zn(OHJ за х, тогда концентрации ионов
электролита в растворе составят:
Zn(OHJ (p-p) = Zn2+ (р-р)+2ОН" (р-р).
270
х х 2х
Отсюда
IlP-[Zn2+] [OH-]2=y{2yJ-!,35-1(T17
P=x=.*/\j5. \0Trj4= 1,5-10"ft моль/л.
Таким образом, растворимость Zn(OHJ равна
концешрации ионов Zn2 + в расгворе* [Zn?+]~1.5-10 моль/л;
концентрация гидроксид-ионов в два раза больше: [ОН~ ] = 2[Zn24 ]=^
= 3,0-10~й моль/л.
Следует обратить особое внимание на операцию, которую
часто забывают проводить в аналитических расчетах. Как видно
из приведенного примера, в квадгш возводится удвоенное
значение растворимости: BхJ = BРУ =4Р2, т. е. растворимость
умножается на стехиометрический коэффициент и полученное
произведение возводится в степень, равную стехиометрическому
коэффициенту.
Равновесие кристаллизации и растворе™ я электролиз
подчиняется всем правилам смещения равновесий. При увеличении в
насыщенном растворе концентраций одного из ионов равновесие
смещается влево, т. е. в направлении процесса образования
кристаллической фазы, и, наоборот, при уменьшении его концентрации
равновесие смещается в направлении процесса растворения осадка.
Выясним, как изменится растворимост ь гидроксида цинка при
добавлении в раствор хлорида цинка. Для этого рассчитаем
растворимость Zn(QHJ в 0,001 М растворе ZnCl2. Эта задача
равносильна определению изменения рас!воримости Zn(OHJ
при увеличении концентрации ионов Zn2^ в расгворе до
0,001 моль/л.
В соответствии с уравнением равновесия в системе запишем
выражение для ПР гидроксида цинка при условии
[Zn2T] = 0,001 моль/л:
Zn(OHJ (p-p)-Zn2 + (p-p) + 2OH- (р-р),
х 0,001 2х
ПР-1,35* 107 = [Zn2 + ] [ОН "]? = 0,001 • B vJ-0,004л-2.
Откуда х= 2/Ь^!° .-_ = 5,8 • 10"8.
V 0.004
Следовательно, в 0,001 М растворе ZnCl2 растворимость
Zn(OHJ составляет 5,8 -10~8 моль/л (ср. с 1.5 10 моль/л в
чистой воде, т. е. растворимоеib уменьшилась почш в 250 раз
Рассчитаем рас1воримость Zn(OHJ в 0,001 М растворе КОН*
Zn(OHJ (p-p) = Zn2+ (p-p) + 2OH- (р-р).
д: х 0,001
[Zn2f] [OH-]2=v@.001J.
271
(Следует обратить внимание, чго концентрация гидроксид-ионов
не удваивается при возведении ее в квадрат!)
Откуда
1,35 10-
А~~1
! 3510
1 ИГ6 ' '
Таким образом, в 0,001 М растворе КОН растворимость
Zn(OHJ стала еще меньше. Растворимость малорастворимого
электролита понижается в наибольшей степени при введении
в раствор того иона, стехиометрический коэффициент которого
в формуле электролита выше.
Предсказывать характер изменения растворимости при
введении того или иного вещества в раствор следует с большой
осторожностью. Так, прибавление в насыщенный раствор
Zn (ОНJ большого количества щелочи КОН приводит к
растворению осадка, что объясняется амфогерностью Zn(OHJ:
Zn(OHJ(K) + 2OH- (p-p) = [Zn(OHL]2- (p-p).
При введении в насыщенный раствор Zn (OHJ такого слабого
электролита, как NH4OH, осадок Zn(OHJ растворяется в
результате образования комплексного иона [Zn(NH3L]2 + :
Zn(OHJ(K) + 4NH4OH (p-p) = [Zn(NH3L]2+ (p-p) +
+ 2ОН" (р-р)+4Н2Ож.
Растворение Zn(OHJ объясняется тем, что при введении
в раствор молекул аммиака образуется комплексный ион,
концентрация ионов цинка понижается и произведение
концентраций [Zn2+][OH~]2 становится меньше произведения
растворимости.
Введение в насыщенный раствор малорастворимого
электролита другого, хорошо растворимого, сильного электролита, не
содержащего с ним одноименных ионов, приводит к повышению
растворимости (солевой эффект). Например, при добавлении к
насыщенному раствору Zn(OHJ раствора хлорида натрия
растворимость гидроксида цинка повышается и в растворе
увеличиваются концентрации ионов Zn2+ и ОН". Повышение ионной силы
раствора приводит к увеличению растворимости
малорастворимых электролитов. Так, в морской воде растворимость многих
веществ, мало растворимых в речной воде, заметно возрастает.
В приведенном выше расчете растворимости гидроксида
цинка в растворе хлорида цинка ионная сила раствора, создаваемая
ионами Ъх\}А и С1~, не учитывалась. Если в расчете вместо
концентраций ионов пользоваться активностями, будут получены
более высокие значения растворимости гидроксида цинка.
К произведениям растворимости приложимы те же
термодинамические зависимости, что и к обычным константам равнове-
272
сия. Зная, по крайней мере, два значения произведения
растворимости (или растворимости) какого-либо элеюролита при двух
температурах, можно рассчигам> термодинамические
характеристики процессов растворения и кристаллизации
Пусть при температурах 7\ и Т2 произведение растворимое»и
соответственно равно ПР, и ПР2. Составляем сиаему двух
уравнений:
раетв J = - #^1 In ПР,=АЯ pM1H-TxAS раств.
раств.2 = - RT2 In ПР2 = А// раС1В-7ч2А5 раств
и находим А//"раств и А5 раств или равные им, но прошвополож-
ньге по знаку энтальпию и энтропию кристаллизации:
чр; AS раств=-А5 кр.
Например, произведение расчворимости хлорида серебра при
10° С равно 3,70-10"и, а при 25 С равно 1,56-10" 10; составляем
систему уравнений
-8,314-283,2-2,303 lg 3,70-10 ll=AH рас1В-283,2 AS1 paCTB
-8,314-298,2-2,303 Ig 1,56 10 W = AH расгв-298,2 AS раств,
откуда находим термодинамические харак1еристики рас!ворения
АН '„ас™ = 67,35 кДж/моль; AS раств = 38,12 Дж/(К • моль).
Полученные данные показываю!, что расгворение хлорида
серебра в воде сопровождайся поглощением теплоты, и в
соответствии с принципом Ле Шателье при повышении температуры
растворимость и ПР для AgCl возрастают. Положи гельное
значение изменения энтропии свидетельствует об уменьшении
степени порядка в системе при переходе хлорида серебра из
кристаллического состояния в сосюяние ионов в водном растворе.
Рассчитаем изменение изобарного потенциала растворения
AgCl при 298 К:
AC °pdcl в = АНс - TASс - 67,35 - 0,03812-298 = 55,98 кДж/моль.
При внесении кристаллов AgCl в воду хлорид серебра
растворяется. Для этого процесса изобарный потенциал должен
уменьшаться, расчет же дает положительное значение AG. Такое
противоречие объясняйся тем, что расчетное значение
AG° — 55,98 кДж/моль ошосшея к аандартным условиям
(система, состоящая из ионов серебра и хлорид-ионов
с [Ag'HICr ]=I моль/л).
Такая система может получаться при смешивании, например,
0,5 л 2 М раствора ионов Ag+ и 0,5 л 2 М раствора ионов С1~.
Однако шкой раствор существовать не может. В нем
самопроизвольно проходит процесс кристаллизации, и система переходит
273
в равновесную, содержащую осадок хлорида серебра; этот
процесс сопровождается уменьшением изобарного потенциала:
Ag4 (р-р)+С1" (р-р) = AgCI(K); ДС кр- -55,98 кДж/моль.
Процесс кристаллизации проiекает до тех пор, пока
концентрации ионов в растворе Fie станут равными:
[Ag + ] = [Cr] = yfTP = >//'^6:^'""*I'2e|0 моль/л
(при 25° С).
Выпадение в осадок малорастворимого электролита проходит
быстро, и часто наблюдается образование золя (коллоидного
раствора) с последующей его коагуляцией и формированием
мелкокрис галлического осадка.
Произведение растворимости может быть рассчитано из
термодинамических характеристик кристаллического электролита
и его ионов в водном расiворе. Для расчета ПРЛ?С, используем
справочные значения стандартных энтальпий образования и
энтропии хлорида серебра, ионов серебра и хлорид-ионов:
кДж vo >ь Дж/(К мо п.)
AgCl{K> -I27J 96,11
Ag4 (p-p) 105.-S 72,62
CI" (р-р) -167.1 56.53
Рассчшаем изменение эшальпии и энтропии в процессе paciBo-
репия хлорида серебра:
ДЯ раств- 105,5- 167,1 + 127,1-65,5 кДж/моль,
AS на». = 72,62 + 56,53 - 96.11 = 33,04 Дж/(К моль).
По этим результатам находим температурную зависимость
AG расгв и значение изменения изобарного потенциала (при 298 К):
At? раств = 65 500-33.04 Г.
А<7 расгв, -9« ="-65 500-33,04-298,15 -55600 Дж/моль.
Так как AG = - RT\n ПР. то выражение для ПР при стандарт -
ной температуре запишется в виде:
55600 о 74
2.303 8,314 298,15"
О гкуда
Некоторое расхождение найденного значения ПР с приведенным
выше объясняе1ся гем. что для расчетов были использованы
справочные данные, полученные в резулыате экспериментов
с различной ючнос'1ыо измерения.
274
ГЛАВА9
РАСТВОРЫ
§ 40. ОБЩИЕ СВЕДЕНИЯ О РАСТВОРАХ.
ДАВЛЕНИЕ НАСЫЩЕННОГО ПАРА НАД РАСТВОРОМ
Движущими силами образования растворов являются энталь-
пийный и энтропийный факторы. Энтропийным фактором
объясняется самопроизвольное смешивание двух инертных,
практически не взаимодействующих газов гелия и неона. Чем слабее
взаимодействие молекул растворителя и растворенного
вещества, тем больше роль энтропийного фактора в образовании
раствора. Знак изменения энтропии зависит от степени изменения
порядка в системе до и после процесса растворения. При
растворении газов в жидкости энтропия всегда уменьшается, а при
растворении кристаллов возрастает. Знак изменения энтальпии
растворения определяется знаком суммы всех тепловых эффектов
процессов, сопровождающих растворение, из которых основной
вклад вносят разрушение кристаллической решетки и
взаимодействие образовавшихся ионов с молекулами растворителя
(сольватация).
И растворитель, и растворенное вещество оказывают
значительное влияние друг на друга и взаимно изменяют свои
свойства. Степень этого влияния зависит от природы того и другого
вещества, и оно наиболее существенно проявляется в
диссоциации молекул растворенного вещества или их ассоциации.
Преимущественное прохождение того или иного процесса
определяется концентрацией вещества в растворе, температурой и
соотношением полярностей растворителя и растворенного вещества.
Ассоциация молекул в растворе может осуществляться двумя
различными путями—соединением однородных молекул
(собственно ассоциация) или соединением разнородных молекул
(сольватация). Так, в неполярных растворителях (СС14, С6Н6)
бензойная кислота дает димеры (С6Н5СООНJ, а в полярных
растворителях, с молекулами которых возможна водородная
275
связь (ацетон. фенол), образует сольваты, например,
С,Н5СООН-ОС(СН3J.
Между частицами растворителя и растворенного вещества
возможны самые разнообразные типы взаимодействий:
важнейшие из них следующие:
1) различные по силе взаимодействия между молекулами
растворителя и растворенного вещества, начиная от сил Ван-дер-
Ваальса и кончая образованием бинарного соединения;
2) взаимодействие между молекулами растворителя под
влиянием растворенного вещества и диссоциация молекул
растворителя;
3) ассоциация молекул растворенного вещества иод
действием растворителя;
4) диссоциация молекул растворенного вещества на более
простые молекулы;
5) диссоциация молекул растворенного вещества на ионы;
6) взаимодействие ионов растворенного вещест ва с
молекулами растворителя;
7) взаимодействие молекул двух (или более) растворенных
веществ под влиянием растворителя.
Взаимодействия, проявляющиеся в растворе,
обнаруживаются при сопоставлении свойств чисюго растворителя и растворов
с различной концентрацией растворенного вещества. Связь
между составом и свойствами растворов очень сложна и еще более
усложняется с увеличением концентрации вещества в растворе.
Изменение свойства раствора в зависимости от его состава
может служить в той или иной степени для изучения
взаимодействий между частицами в рас i ворах.
Одно из важнейших термодинамических свойств чистых
жидкостей и растворов—давление насыщенного пара.
Предположим, что происходит испарение чистой жидкости (чистого
растворителя) А (рис. 120.а). Константа равновесия процесса
перехода молекул из жидкой фазы в газообразную численно равна
парциальному давлению насыщенного пара при данной
температуре. Например, для воды:
а а
А А
А
А А
А
А А
А А А
А А
АААААААААА
АААААААААА
АААААААААА
ААААААА А А А
б АЛ
А А А
А А
АЛА
АА В А в А В А 8 В
ВААВАВАВВА
А В А А В А В А А А
ВАВААБАВАВ
Рис 120 Схема испарения чистого растворителя Л (а)
и растворителя из раствора, содержащего вещество
В F)
276
н2о(ж)—
В выражение Konciaiiibi равновесия концентрация жидкой
воды не входит, так как она постоянна.
Тог же нодход используем для описания раствора, состоящего
из растворителя А и растворенного вещества В. При растворении
вещества В в веществе А число молекул А в единице объема
уменьшается, а значит, снижается и их число на единице
поверхности испаряющейся жидкосш (рис. 120,6). Тем самым
уменьшается скорость испарения и снижается парциальное давление
насыщенного пара растворителя над раствором.
Обозначим мольную долю растворителя через NA и мольную
долю растворенного вещества Л'в. Давление пара компонента
А буде*, очевидно, тем выше, чем больше этого компонента
в раст воре:
где к — коне 1 ант а или коэффициент пропорциональности между
парциальным давлением растворителя над раствором и его
мольной долей NA в растворе.
Коэффициент к определяйся подстановкой в это выражение
какого-либо значения рА при данном значении УУЛ. Если мольная
доля растворителя равна единице (/VA = 1), знании имеется чистый
растворитель, давление насыщенного пара которого при данной
температуре составляет /?°Л. Следовательно, для чистого
растворителя коэффициент к ecib /?°Л, т.е. к~р°А. Подставляя это
равенство в последнее уравнение,
получаем:
4?в
Это соотношение является одним из
выражений чакона Рауля A887 г.).
Оно показываем что парциальное
давление насыщенного пара
растворителя над раствором прямо
пропорционально его мольной доле (рис. 121).
Изменение содержания
растворителя, например, в два раза приводи i
к изменению его парциального
давления 1акже в два раза.
Используя аналогичные
рассуждения о i носит ельно растворенного
вещества В, получаем:
Состав, мол. доля
Рис 121 Зависимость
парциальных давлений пара компонентов
Ра и рп раствора и обша о дав
гения насыщенного пара ро6ш нал
раствором от его сооава
277
Это уравнение представляет собой другое выражение закона
Рауля (применительно ко второму компоненту раствора):
парциальное давление насыщенного пара растворенного вещества
прямо пропорционально его мольной доле.
Общее давление пара растворителя и растворенного вещества
равно сумме их парциальных давлений
или />общ = NAp
Учитывая, что ;VA + iVB = 1, получаем:
ИЛИ
Уравнение показывает, что общее давление пара над
раствором изменяется пропорционально мольной доле одного из
компонентов и графически представляется прямой (см. рис. 121).
Действительно, при NA = 0 Р<лъ=Р°ъ ПРИ #в = 0 рОбт=Р°\-
-Р°в+Р%=Р°а.
Зависимость «робщ» на рис. 121 представляет собой диаграмму
состояния бинарный раствор—пар (газ). Ось А В изображает
состав бинарной системы в мольных долях, если его длина равна
единице, или мольных процентах, если его длина принята за
100%. От точки А диаграммы откладываются значения мольной
доли (или мольного процента) компонента В. В точке В мольная
доля компонента В равна единице. Промежуточные составы
характеризуются точками на отрезке АВ, при этом NA + NB=\ (или
100%). На осях ординат наносятся значения давления пара
чистых веществ р°А и р°ъ. Верхняя прямая, соединяющая р°А и /?°в,
описывает давление общего пара над раствором в зависимости
от его состава.
При выводе приведенных выше соотношений предполагалось,
что к насыщенному пару применимы законы идеальных газов,
а сам раствор ведет себя как идеальный. Идеальный раствор—
эт о раствор, в котором силы межмолекулярных взаимодействий
молекул растворителя и растворенных веществ одинаковы. На
свойства такого раствора не влияет, находится ли молекула
некоторого компонента в окружении собственных молекул или
в окружении молекул других компонентов (растворителя или
растворенных веществ). При образовании идеального раствора
энтальпия системы не изменяется (Д#р=0, Qp = 0). По этой же
причине объемы смешиваемых компонентов суммируются.
Каждый компонент в составе идеального раствора ведет себя
независимо от других компонентов.
Давление насыщенного пара растворителя над идеальным
раствором всегда меньше, чем над чистым растворителем, и
понижение давления пара тем больше, чем выше концентрация (или
278
мольная доля) растворенного вещества. Следовательно, для
идеальных растворов понижение давления пара зависи! только от
концентрации растворенных частиц (но не от их природы).
Другие свойства идеальных растворов, такие как 1емиература
замерзания, темпера!ура кипения, осмотическое давление, также
зависят только от концентрации часшц.
Полученная зависимость сохраняв 1ся и тогда, ко!да
растворенное вещество нелетуче, г. е. давление его паров пренебрежимо
мало по сравнению с парциальным давлением раиворителя.
Если /?°в^0, то
или, что то же самое
Это уравнение показывает, чго парциальное давление пара
растворителя (в данном случае общее давление пара над
раствором) пропорционально его мольной доле в идеальном растворе.
Преобразуем полученное уравнение, подставив в него
\N
/>л = A-ЛЪ)/>°а или NB = (p°A
Разность ро.\—р\ называется понижением давления насыщенного
пара, а отношение (р°\—Р\Iр°\—относительным понижением
давления насыщенного пара. Полученное соотношение можно
выразить следующим образом: относительное понижение давления
насыщенного пара растворителя над идеальным раствором равно
мольной доле растворенного вещества. Это другая формулировка
закона Рауля.
Закон Рауля применим только к растворам, близким к
идеальным, когда оба компонента — растворитель и растворенное
вещество—близки по строению и свойствам и силы взаимного
притяжения молекул обоих компонентов примерно одинаковы.
Строго говоря, свойствами идеального раствора не обладает
ни один реальный раствор, за исключением растворов
различного изотопного состава (Н2О—D2O) и смесей оптически
активных изомеров. Однако многие растворы практически ведут
себя как идеальные. Большинство реальных растворов следуют
закону Рауля только при малых значениях мольных долей
растворенного вещества. Чем меньше содержание растворенного
вещества в рас i воре, тем ближе его свойства к свойствам
идеального раствора. Бесконечно разбавленный раствор можно
рассматривать как идеальный При этом условии молекулы
растворенного вещества, распределенные в среде молекул
растворителя, настолько удалены друг от друга, что практически не
взаимодействуют между собой и не влияют на молекулы
растворителя. Поэтому можно принять, чго свойс!ва бесконечно
279
\
СН2(ОСН3J
CS2 CH3COCH3
- в
CHCI3 C5H5N
НоО
Рис 122 Зависимость парциальных и общего давления пара над раствором от
состава для систем, не подчиняющихся закону Рауля.
а—сис1сма лимстксимсгап — ссроуыерот с но южше (ьными oik к>нсииями >мв.1ения пара о—сисмема
аисюн — х.юрофорч с огрицатс 1ьиыми ojk юпениями «—cncievia иирилии — вода с положиicibiibiM и oi-
рицаю 1Ы1ЫМ oik юненинми л 1Я о 1>юю компопома
разбавленного раствора (УУВ<О,ОО1) будут аналогичны свойствам
идеального раствора.
По мере увеличения мольной доли растворенного вещества
проявляются все более заметные о«клонения о г закона Рауля,
действительные значения давления пара перестают отвечать
свойствам идеального paciBopa. Отклонения зависимости
давления пара от состава раствора отиоси!ельно линейной
зависимости в область больших значений давления принято называть
положительными, а отклонения в область меньших значений
давления — отрицательными. Обычно знаки отклонений как
общего давления пара над раствором, гак и парциальных давлений
компонентов одинаковы и сохраняю 1ся при всех составах
рассматриваемой системы.
На рис. 122, я преде 1авлены общие и парциальные давления
пара компоненте в смесях диметоксиметана СН2(ОСН3J и
сероуглерода CS2 (при 35 С). Эга система характеризуется
положительными отклонениями. Пунк! ирными прямыми показано,
как изменялись бы давления пара компонентов и раствора, если
бы раствор был идеальным. Сплошные кривые изображают
действительные зависимости давления о г состава. Только при
небольших содержаниях одного вещества в растворе с другим
зависимость давления пара совпадает с линейной зависимостью,
рассчитанной по закону Рауля.
Отклонения давления пара вызваны различной силой
взаимодействия между однотипными и разнотипными молекулами.
Взаимодействие между молекулами диметоксиметана, так же как
и между молекулами сероуглерода, сильнее, чем между
молекулами диметоксиметана и сероуглерода. Эю приводит к тому, что
молекулы диметоксиме!ана как бы выталкивают из своей среды
молекулы сероуглерода, а молекулы сероуглерода выталкиваю!
280
молекулы диметоксимсмаиа. Взаимное вьпалкивание молекул
равносильно повышению парциального давления компонентов
и общего давления пара над раствором. Таким образом,
положительные отклонения от закона Рауля наблюдаются, когда
молекулы одного компонента раствора взаимодейавуют сильнее, чем
молекулы различных компонентов, а эго облегчает переход их из
жидкой фазы в газовую.
Образование растворов с положительными отклонениями
давления пара сопровождается поглощением теплоты, т. е.
увеличением этальпии ((?<0, Д//>0). Давление пара над
раствором связано с тепловым эффектом его образования:
эндотермический эффект при растворении влечет за собой уменьшение
количества теплоты, необходимой для перехода жидкости в пар,
и это приводит к тому, что процесс испарения 1ермодинамически
протекает легче и давление пара раствора и составляющих его
компонешов повышается. Обычно сисмемы с положительными
отклонениями образуются с некоторым увеличением объема
Причины отклонений давления пара могут быть весьма
различными, по наибольший вклад вносят процессы,
сопровождающиеся изменением размеров чааиц в растворе по сравнению
с размерами частиц чисшх компонентов. При образовании
раствора с положительным отклонением давления пара размеры
частиц уменьшаются вследствие диссоциации (частичной или
полной) ассоциатов частиц, существовавших в структуре чистой
жидкости одною из компонентов. Диссоциация ассоциатов
вызывает поглощение теплоты при растворении, что приводит
к увеличению внутренней энергии раствора, повышению
кинетической энергии молекул жидкости, большей легкости их
испарения и в конечном C4eie к более высокому давлению пара.
Растворы с отрицательными отклонениями (рис. 122, б)
образуются обычно с выделением теплоты (Д//<0), поэтому leiuio-
та испарения компонентов из раствора больше, чем теплота
испарения чистых компонентов, и давление пара рас i вора ниже,
чем ожидалось бы у идеального расiвора. Обычно при
образовании таких растворов имеет место уменьшение объема. Наиболее
важной причиной отрицательных отклонений является
возникновение ассоциатов и соединений между молекулами
компонентов. Обычно комплексы, получающиеся из разнотипных молекул,
имеют переменный cociae и не отвечают простым стехиомет-
рическим соотношениям. Типичным примером раствора с
отрицательными отклонениями может служить система ацеюн —
хлороформ: растворение сопровождается выделением теплоты
и понижением давления пара.
Для многих систем образование и диссоциация ассоциатов
и соединений происходят одновременно, и действие этих
противоположных по характеру процессов на поведение раствора
частично компенсируется. В результате по типу отклонения удается
281
судить о процессе, оказавшем большее влияние на поведение
раствора. Наблюдаемые эксперимешально отклонения
являются результатом наложения противоположных но знаку
отклонений.
Одновременное проявление противоположных отклонений
особенно ярко выражайся у растворов, для которых знак
отклонений давления пара изменяется с изменением мольной доли
компонента. Например, в системе пиридин—вода (рис. 122, в)
в интервале мольных долей пиридина от 1 до 0,6 наблюдаются
отрицательные отклонения, а в интервале от 0,6 до
0—положительные отклонения давления пара пиридина, однако давление
пара раствора во всем интервале концентраций описывается
зависимостью с положительными отклонениями.
Изучение диаграмм «давление пара—cociae» показывает, то
компоненты раствора ведут себя не так, как эт следовало бы
ожидав на основании теоретических предпосылок. Для
идеального раствора парциальное давление пара любого /-го
компонента Pi пропорционально его мольной доле Nh причем коэффициент
пропорциональности численно равен давлению пара над чистым
компонентом pt° при данной температуре:
Мольная доля компонента N( указывает на число частиц
в растворе. Естественно предположить, что наблюдаемые
отклонения от идеальности связаны с отклонениями в числе частиц
по сравнению с тем их числом, которое должно быть в
идеальном растворе при его поведении в соответствии с законом Рауля.
С учетом процессов диссоциации и ассоциации для реальных
растворов мольная доля N( должна быть заменена на некоюрую
другую величину, зависящую от состава системы. Обозначим эту
величину символом ah тогда можно записать:
Если для идеальных растворов отношение pjpi0 равно мольной
доле Ni компонента в растворе, то для реальных растворов это
отношение соответствует величине ah т. е.
Термодинамическая функция а( называется активностью
компонента в растворе. Активность данного компонента раствора —
это величина, которая связана с парциальным давлением пара
этого компонента и другими его термодинамическими
свойствами так же, как в идеальных растворах с давлением
насыщенного пара связана мольная доля этого компонента (его
концентрация). Активность представляет собой расчетную
термодинамическую функцию, характ еризующую меру взаимодействия
молекул компонента, она позволяет судить об отклонении
282
свойств данного компонента в реальном растворе от его cboucib
в идеальном растворе при той же концентрации.
Наряду с активностью компонента сц используют
коэффициент активности/, равный отношению активности к мольной доле
компонента:
Коэффициент активности данного компонента (так же, как
и его активность) характеризует степень отклонения свойств
компонента в расiворе от его свойств в cooiветствующем
идеальном растворе.
Уравнение можно записать не через мольные доли, а через
концентрации:
Концентрация вещества может быть выражена различными
способами, но обычно применяют молярную концентрацию.
Таким образом, активность характеризует ту реальную
концентрацию вещества в рас i воре, которая отвечает поведению
данного вещества в системе. Например, если для 0,01 М раствора
р./р.° = 0,009, то из этого следует, что компонент i ведет себя так,
как будто количество его меньше, чем это следует из молярной
концентрации. Коэффициент акжвности вещества будет равен:
0,009=/0,01; /-0,009/0,01 =0,9.
Чем меньше коэффициент активности, тем заметнее
отличается поведение вещества от поведения, отвечающего его
концентрации. Коэффициенты активности могут быть больше единицы,
но не могут быть отрицательными величинами.
При помощи активности оказалось возможным объяснить
некоторые особенности поведения вещее i в в разных фазах.
Например, многие мономолекулярные реакции в газовой фазе
и в растворе протекают с одинаковыми скоростями, а
бимолекулярные реакции в растворах обычно протекают значительно
быстрее, чем в газовой фазе, и их скорость зависит oi природы
растворителя.
§ 41. МЕТОДЫ ИЗУЧЕНИЯ ЖИДКИХ РАСТВОРОВ
Свойства растворов зависят от природы их компонентов, от
состава и условий существования расiвора и в значительной
степени определяются теми взаимодейс1виями, которые
происходят между частицами растворителя и растворенного вещества.
Есть вещества, коюрые при растворении практически не
изменяют своих свойств. Однако многие вещества при pacieo-
реиии не только сами претерпевают изменения, но и оказывают
283
влияние на свойства растворителя. Причина эгих изменений
связана с химической природой исходных веществ, а также
с межмолекулярными и межионными взаимодействиями,
изменяющими концентрацию растворенного вещества и растворителя
(по сравнению с той, которая устанавливается в отсутствие
этих взаимодействий).
К практически важным свойствам расi воров, изучение
которых позволяет получить информацию о некоторых физико-
химических параметрах веществ, относятся осмотическое
давление, понижение давления насыщенного пара растворителя над
раствором, понижение температуры замерзания и повышение
температуры кипения раствора по сравнению с этими
параметрами для чисюго рас1ворителя. На измерении
концентрационной зависимости этих свойств растворов основаны методы
определения молекулярной массы вещее i в, степени диссоциации
электролитов и др.
Осмотический метод. Растворенное вещество по своему
поведению во многих отношениях напоминает газ. Так, рассоренное
вещество, как и газ, стремится равномерно распределился по
всему объему раствора. Если растворитель привести в
соприкосновение с раствором (другой окраски для удобства наблюдения),
то происходит диффузия—переход молекул растворенного
вещества через поверхность раздела в раствори iель и одновременно
молекул растворителя в расгвор. Такая встречная диффузия
рассоренного вещества и растворителя продолжается до iex пор.
пока система не придет в состояние равновесия.
Одностороннюю диффузию чистого растворителя в раствор
можно количественно оценить с помощью специального
прибора— осмометра, в котором раствор и растворитель отделены
друг от друга перегородкой (мембраной), проницаемой для
молекул растворителя и непроницаемой для молекул растворенного
вещества. Если сосуд / (рис. 123, а), закрытый внизу
полупроницаемой перегородкой 2 и наполненный водным раствором
какого-либо вещества, поместить в сосуд 3 с водой, то вода будет
проходить из сосуда 3 в 7.
Явление самопроизвольного перехода растворителя через
полупроницаемую перегородку в расi вор называется осмосом.
Через некоторое время объем раствора в сосуде / увеличится
и его уровень поднимется выше уровня растворителя в сосуде
3 на высоту А.
В результате увеличения объема раствора в сосуде У
возникает дополнительное гидростатическое давление, называемое
осмотическим. Осмотическое давление можно определить по Bbicoie
h подъема жидкости (см. рис. 123, а). Можно воспользоваться
осмометром специальной консфукпии с поршнем, в который
вмонтированы полупроницаемые перегородки (рис. 123, б).
В этом осмометре растворитель, проникая через полупроница-
284
а
_— —;
i
_
r _.
Рис 123 Схема осмометра простейшей
конструкции {а) и с поршнем (б)
Дма I—еос>д с рас тором 2—мембрана,*—coc>jcpuc
iBopmeieM (водой) дчя 6 I—омегснис ддя раствора. 2—
поршень с по ^проницаемыми перегородками.
S—обеление для paciBopnte 1я. 4—уравновешивающий гр\з
емую перегородку, увели-
чивает объем раствора,
и возникающее осмошче-
ское давление действует
на поршень. Подбором
груза 4 достигают
уравновешивания силы,
обусловленной осмо i ическим
давлением (и поршень
сохраняет свое
первоначальное положение). Этот г руз
и служит мерой
осмотического давления.
Процесс перехода
растворителя в расгвор
самопроизволен, но
обратный процесс выделения
растворителя из раствора
самопроизвольно осу-
щес i вляться не может,
и для разделения
раствора на растворитель и
растворенное вещество
следует затра!ить работу.
Если давление на поршень (см. рис. 123, б) меньше осмотического,
то раствори!ель самопроизвольно проникает в расiвор и
поднимает поршень до тех пор, пока не установится равновесие и
осмотическое давление раствора не сравняется с силой тяжести груза,
действующей на поршень. Если же на поршень дейавует сила,
превышающая осмотическое давление, ю поршень будет
опускаться, при этом растворитель выделяется из раствора.
Обратный осмос может быть использован для опреснения морской воды.
Осмотическое давление зависит от концентрации
растворенного вещества и температуры. Так, при увеличении концентрации
С раствора сахарозы в воде в два раза осмотическое давление
я возрастает также примерно в два раза, при увеличении С в три
раза к возрастав в три раза и т. д.
При повышении температуры на 1 С осмотическое давление
возрастает на V273 часть первоначального значения, при
повышении температуры на 10 С — на !0/27з первоначального значения
и т. д. Экстраполяцией можно найти, что при повышении
температуры Г (в К) в два раза осмотическое давление я возрастает
в два раза.
Объединяя две зависимости (концентрационную и
температурную) и вводя коэффициент пропорциональности /?, получим:
k = CRT.
285
Для нахождения численного значения R подставим значения
я, Си 7*для одного из опытов. Так, при концентрации сахарозы
0,01 моль/л и температуре 0°С осмотическое давление составило
22 700 Па. Огкуда
r = JL =JiZ?L = 8300 Па-л/(К-моль).
Полученный результат интересен тем, что численное
значение коэффициента пропорциональности R в выражении
осмотического давления совпадает со значением универсальной
газовой постоянной. Из этого следует, что осмотическое
давление раствора, содержащего 1 моль сахарозы, равно 2 270000 Па
B2,4 атм), а осмотическое давление раствора, в котором на
22,4 л приходится 1 моль сахарозы, составит 101 325 Па A атм).
Следовательно, при Г—273 К и 71=101325 Па (нормальные
условия) раствор, содержащий 1 моль сахарозы, должен
занимать объем 22,4 л. Этот пример иллюстрирует аналогию
поведения растворенного вещества с поведением его в газовом
состоянии.
Впервые эта аналогия была обнаружена голландским ученым
Вант-ТЪффом. Уравнение осмотического давления носит его имя
и формулируется как закон Вант-Гоффа: осмотическое давление
раствора равно тому давлению, которое оказывало бы
растворенное вещество, если бы, находясь в газообразном состоянии при той
же температуре, оно занимало тот же объем, который занимает
раствор.
Уравнение Вант-Гоффа применимо тлько для разбавленных
растворов.
Осмос и осмо!ическос давтение имеют огромное значение в биологических
явлениях, так как оболочки клеток биологических тканей являются
полупроницаемыми перегородками. Осмотическое давление клеточного сока растений
изменяется от 2,0 105 Па у болотных растений до 4,5 I06 Па у степных. Вследствие
осмоса вода и питательные расi воры поднимаются на значительную высоту по
стволу растений Тканевые жидкости млекопитающих имеют осмотическое
давление F,7—8,1)-10s Па
Осмотическое давление внутри живых клеток обусловливает прочность и
упругость тканей и блаюдаря ему осуществляется солевой обмен живой ткани
с окружающей средой
Пользуясь уравнением Вант-Гоффа, несложно определить
мольную массу вещеава (недиссоциирующего) в растворенном
состоянии. Мольная масса равна числу грамм растворенного
вещества, содержащихся в 22,4 л раствора при 0°С, когда его
осмотическое давление равно 101 325 Па.
Раствор, осмотическое давление которого можно описать
уравнением Вант-Гоффа, т. е. уравнением состояния идеального
газа, называется идеальным раствором. Чем выше концентрация
вещества в растворе, тем в большей степени отличается
поведение реального раствора от поведения идеального расiвора.
286
Рис. 124. Зависимость давления
насыщенною пара чистого paciBopn-
теля (/) и над раствором B. 3) о г
температуры
Концентрация вещества в растворе 3 бо ibiiic
чем в растворе 2
Согласно уравнению Вант-
1оффа осмотическое давление
разбавленного раствора (С<0.01
моль/л) прямо пропорционально
молярной концентрации
растворенного вещества, т. е.
пропорционально числу частиц,
находящихся в данном объеме раствора,
и, как показывают опыты, в ряде
случаев не зависит от природы
растворенного вещества.
Свойства растворов, зависящие от числа
частиц, называются коллигатив-
ными. К коллигативным
свойствам относятся также
относительное понижение давления пара
растворителя, понижение
температуры затвердевания и
повышение температуры кипения раствора. Все эти свойства
пропорциональны числу растворенных частиц.
Эбулиоскопический метод. Понижение давления насыщенного
пара расгворителя над раствором приводит к повышению
температуры кипения раствора по сравнению с температурой кипения
чистого растворителя.
Жидкость кипит, когда давление насыщенного пара равно
внешнему давлению. Если растворенное вещество имеет
пренебрежимо малое давление насыщенного пара (например, над
раствором кристаллических веществ в воде), ю пар над раствором
такого вещества содержит практически чистый растворитель,
и в соответствии с законом Рауля давление насыщенного пара
над раствором всегда меньше давления насыщенного пара чис го-
го растворителя.
На рис. 124 кривая / изображает зависимость давления
насыщенного пара чистого растворителя от температуры. В
точке Т° эта кривая пересекает прямую, отвечающую
постоянному внешнему давлению /7=1,01325* 105 Па, точка абсциссы
Т^кип указывает на температуру кипения жидкости. В
результате растворения в жидкости вещества, обладающего при этой
температуре пренебрежимо малым давлением насыщенного
пара, давление пара растворителя (давление пара над раствором)
уменьшается. Кривая 2, характеризующая температурную
зависимость давления пара над раствором, располагается ниже
кривой /, она пересекает прямую постоянною давления (изобару)
в точке Т\ Эю означает, что раствор кипит при более высокой
температуре Т кип. Раствор с более высокой концентрацией
растворенного вещества кипит при более высокой температуре
fit
287
Следует обратить внимание на важное отличие процессов
кипения чисюго вещества и расi вора. Применяя правило фаз
к равновесию чистая жидкость — пар при постоянной
температуре и постоянном давлении, получим:
т. е. однокомпонентная система не имеет степеней свободы.
Температура кипения раствора (К=2) даже при постоянном
давлении не постоянна, так как система имеет одну степень свободы:
Действительно, темпера тура кипения рас i вора отвечает
появлению первого пузырька пара, т. е. началу процесса кипения.
Если растворенное вещееiво нелетучее, уход из раствора
растворителя в виде пара приводит к увеличению концентрации
вещества в растворе, что, в свою очередь, вызывает непрерывное
повышение температуры кипения.
Обычно при изучении зависимости температуры кипения
раствора от концентрации растворенного вещества используют не
мольные доли, а моляльность раствора т.
Повышение температуры кипения раствора неэлектролита
прямо пропорционально концентрации раствора:
где—т — моляльная концен! рация неэлектролита в растворе;
?—коэффициент пропорциональности, называемый эбули-
оскопической постоянной.
Для волы ?"=0,52.
Рас i воры одной и той же моляльности @,01) глюкозы, 1лице-
рина, мочевины и ряда других не диссоциирующих в воде ве-
meciB кипят при одинаковой TeMiiepaiype A00,0052°С) и
обнаруживают равное повышение температуры кипения раствора
по сравнению с температурой кипения растворителя @,0052° С).
Казалось бы, эбулиоскопическую константу можно
определить как повышение 1емпературы кипения одномоляльного
раствора. Действительно, для одномоляльного раствора ?=ДГКИП.
Однако экснеримешально ну юм измерения 1емпературы кипения
одномоляльного раствора величину ? определять нельзя, так как
одномоляльные растворы — это не разбавленные растворы,
а концентрированные (одномоляльный раствор сахарозы
содержит 342 г его в 1000 г волы).
Для определения эбулиоскопической постоянной
экспериментально измеряют зависимость А Тшп от моляльности для
разбавленных растворов (ш^0,1) и экстраполяцией этой зависимости
к т= 1 находя 1 значение постоянной ?.
По результа«ам измерения повышения 1емпературы кипения
(эбулиоскоиический меюд) можно сулшь о некоторых процессах,
288
протекающих в расiворах, и определять мольную массу
растворенного вещества В основе 1акого определения лежит положение
о том, что мольная масса растворенного вещее!ва соответствую
числу его граммов в 1000 г рас!ворителя, которое вызывает
повышение температуры кипения раствора, численно равное эбу-
лиоскопической посюянной.
Криоскопический метол. Уменьшение давления насыщенного
пара растворителя над раствором нелетучего вещества является
причиной понижения 1емперагуры замерзания раствора (по
сравнению с температурой кристаллизации чистого растворителя).
На диаграмме состояния воды кривая давления пара рас «вора
располагайся ниже кривой давления пара чистой воды.
Следовательно, появление первых кристаллов из рас i вора при
охлаждении происходи! при более низкой 1емперагуре, чем [емпература
плавления чисюго вещества.
Важное отличие процессов плавления чистого раствори 1еля
и раствора состоит в следующем. В соответствии с правилом фаз
чистое вещество плавится при постоянной leMiicparype, если
давление сохраняе1ся постоянным и при плавлении одноком-
понентная система не имеет степеней свободы
с=а:-ф+1 = 1 -2 + 1-0.
Как плавление, так и обратный процесс — крисшллизация жид-
кос iи — совершаю 1ся при одной и той же температуре (если не
происходит переохлаждения жидкое ж).
За (смиературу замерзания раствора принимают температуру
начала его отвердевания, i. e. ieMiieparypy выделения из
раствора первого кристалла твердой фазы.
Для чистого растворителя (чисюго вещества) температура
плавления и температура замерзания совпадают.
Выпадение из раствора даже ничтожно малого количества
рас1ворителя приводит к увеличению концешрации вещества
в рас i воре, понижению давления насыщенного пара рас!вори-
теля над раствором и сооизекмвенно к понижению температуры
появления следующих порций кристаллов рааворителя.
Следовательно, отвердевание раствора происходит в некоюром
интервале leMiicpaiyp в октичие от кристаллизации чистого вещества,
происходящей при посюянной температуре.
Эюг вывод согласуйся с правилом фаз в применении к
раствору. При носюянном давлении двухкомпонентная двухфазная
система раствор — крис!аллы растворителя имеет одну степень
свободы:
С=*:-Ф+1=2-2+ 1 = 1.
Понижение температуры замерзания разбавленных растворов
пропорционально концентрации paciворсиною вещества:
, = Km.
зам '
Ю 2452 289
Коэффициенi пропорциональности К в полученном
уравнении называется криоскопической постоянной, а меюд изучения
рас i воров по температурам отвердевания—криоскопией. Чтобы
найти численное значение криоскопической постоянной,
достаточно хотя бы для одного раствора извесшой моляльности
определи п> температуру начала замерзания раствора. Так, 0,01 мо-
ляльные водные растворы сахара, глюкозы, мочевины и других
(недиссоциирующих) веществ начинают отвердевать при
температуре — 0,0186" С. Следовательно, для воды
#=0,0186/0,01 = 1,86.
Формальный смысл криоскопической постоянной, как и эбу-
лиоскопической постоянной, заключается в том, что они
означают понижение температуры замерзания или повышение
температуры кипения одпомоляльною раствора. Однако для таких
концентрированных растворов использование метода криоскопии
недопустимо, поэтому постоянную К, как и Е* определяют
экстраполяцией.
Так как понижение температуры замерзания и повышение
температуры кипения изменяются пропорционально моляльной
концентрации, а один моль любою вещества содержит
одинаковое число молекул, то значш, А7",ам и АГКИП зависят только от
числа частиц растворенного вещества. Это утверждение
формулируется как следствие из закона Рауля: понижение температуры
замерзания раствора и повышение температуры кипения раствора
пропорциональны числу частиц растворенного вещества.
Криоскопия, как и эбулиоскопия, применяется для
определения молыюй массы и других параметров растворенных веществ.
Для повышения iочноеги измерения А7\ам следуем пользоваться
рас1ворителями с высокими значениями криоскопической
постоянной, например, камфорой (К = 39,8).
Криоскопический метод наиболее час? о используется при
изучении водных растворов, так как он экспериментально намного
проще методов, основанных на измерении понижения давления
пара, осмотическою давления растворов и эбулиоскопии.
Криоскопия—один из наиболее точных методов определения
активности растворителя в реальном растворе.
§ 42. ПРОЦЕССЫ КООРДИНАЦИИ В РАСТВОРАХ
Взаимодействия между частицами как одного, так и разных
видов очень разнообразны Чрезвычайно широк и интервал
энергий взаимодействий — от слабых сил Ван-дер-Ваальса до
высоких энергий химических связей.
В системах, состоящих из частиц одного вида, возможны
следующие взаимодейст вия.
290
1. Взаимодежмвие между частицами, приводящее к
образованию полимерных молекул и структур. Так, в результате действия
сил Ван-дер-Ваальса между атомами благородных газов или
между молекулами О2, N2, С12 и другими эти вещества могут
существовать в жидком состоянии. Энергия этих взаимодействий
может достигать 20 кДж/моль.
Способноеib веществ, содержащих агомы водорода и одного
из следующих элементов—азота, кислорода и
фтора,—образовывать водородные связи между молекулами приводит к
формированию айсоциатов молекул и жидких структур.
2. Взаимодействие двух чаепщ, приводящее к образованию
двух противоположно заряженных ионов, что обусловливает
электропроводность чистых жидкостей, например:
2Н2О-Н3О++ОНГ
или
+ NO3".
Такого типа процессы диссоциации (ионизации) молекул
жидкостей приводят к упорядочению систем в результате
сольватации образующихся ионов:
и т. д.
Действительно, в растворе HF кроме иона HF2~ обнаружены
ионы H2F3" и менее устойчивый FH2 + или [H(HF)] + .
При понижении температуры склонность молекул к
ассоциации возрастает, происходит образование все более крупных
агрегатов и в конечном итоге—кристаллических зародышей и
кристаллов.
Межмолекулярные взаимодействия еще более усложняются
при внесении в жидкость (растворении) молекул другого сорта:
введенное вещество оказывает влияние на взаимодействие между
молекулами растворителя и, кроме того, появляются новые
взаимодействия между частицами разного сорта, существенно
зависящие от их концентраций. Можно выделить следующие виды
взаимодействий между молекулами (или частицами) разной
природы.
3. Взаимодействие молекул растворителя и молекул
растворенного вещества с образованием нового соединения.
Энергетический интервал эгих взаимодействий очень велик—от слабых
взаимодействий, проявляющихся в отклонении от состояния
ю* 291
идеального раствора (например, при образовании раствора гелия
и неона) до химических взаимодействий (химических реакций).
Например, расшорснис триоксида серы в воде приводи! к
образованию серной кисло 1ы:
И 2О<Ж) + SO 3(к) = Н 25О4(Ж)-
Эта хорошо известная реакция, как и многие другие, итересна
тем, что реагирую 1 две молекулы, аюмы которых находятся
в наивысших степенях окисления. Несмотря на кажущуюся
простоту реакции и хорошо изученную cipyiciypy молекулы серной
кислоты
но/No
объяснение механизма процесса и свойств образовавшейся
молекулы вызывает определенные затруднения: непонятна природа
сил взаимодействия и характер связей в молекуле серной
кислоты. Имеются доказательства, что все атомы кислорода в
молекуле H2SO4 равноценные, а два атома водорода соединены не
с двумя a i омами кислорода, а с группой SO42 . Тогда формула
серной кисло1ы запишется *ак:
H2[SO4].
Такой записью подчеркивается, что атом серы координирует
вокруг себя чешре равноценных атома кислорода, проявляя
координационное число, равное четырем.
Координационное число часто зависит от концентрации
растворенного вещества. Например, в зависимоеi и от cooi ношения
триоксида серы и воды могут быть получены вещества состава:
SO3 • 9Н.О, SO3 • 7Н2О, SO3 5Н2О, SO3 • ЗН2О, SO3 • Н,О,
2SO3H2Oh4SO3H2O.
Объяснение причин взаимодействия нейтральных и валеншо-
насыщенных частиц, не имеющих исспаренных электронов. 1акже
представляет затруднения.
4. Взаимодействие молекул растворителя с молекулами
растворенного вещества, приводящее к диссоциации их на ионы:
НС1 = Н + +СГ или НС1 + Н2О = Н3О++СГ .
Диссоциация веществ в водных растворах может сопровождайся
увеличением общего порядка в системе (Д5<0). Это объясняйся
процессами гидратации образовавшихся ионов:
СН3СООН + (л-Н)Н2О-ЬГ vH2O + CH3COO" • vH2O.
5. Взаимодейс1ние молекул растворителя с ионами
растворенною вещества. Эти взаимодействия шкже чрезвычайно сильно
292
различаются по энергиям. Частицы растворенного вещества,
будучи в меньшем количестве по сравнению с частицами рас1вори-
теля, разумейся, окружены последними. Число окружающих
частиц зависит от соотношения размеров частиц растворителя
и растворенного вещества: чем меньше молекула растворителя
и чем больше ионы растворенного вещества, тем больше молекул
располагается вокруг ионов. В отсутствие взаимодействия время
пребывания молекул растворителя около частицы растворенного
вещества должно быть таким же, как время ее нахождения в
любой другой точке раствора. Однако в связи с тем, что
напряженность электрического поля вокруг иона (или любой растворенной
частицы) иная, чем вокруг частицы растворителя, это условие не
соблюдается. Около иона молекулы рас1ворителя удерживаются
за счет собственных или наводненных диполей. Число молекул
растворителя, составляющих сольватный слой вокруг частицы
растворенного вещества, называется координационным числом
сольватации. При слабых взаимодействиях число сольватации
изменяется во времени; оно зависит от способа его определения,
и обычно указывают среднее его значение, выраженное дробным
числом.
В водном растворе ионы щелочных металлов гидрат ируются,
т. е. их окружают молекулы воды с образованием сравнительно
прочных комплексов М+ пН2О. Ниже приведены относительные
числа Z гидратации ионов щелочных металлов (число
гидратации иона цезия принято за единицу):
Na1
Kb*
Cs+
Приведенные числа гидратации — не целые, хотя, разумеется,
ион может быть окружен только целым числом молекул воды.
Несмотря на го что радиусы ионов возрастают от лития к
цезию, числа гидратации в том же направлении уменьшаются,
хотя, казалось бы, больший ион должен окружаться большим
числом молекул воды. Ион лития, имеющий наименьший радиус,
удерживает вокруг себя наибольшее число молекул, т. е. связь
иона лития с окружающими его молекулами воды наиболее
прочная.
Измерение электропроводности в paciворах однотипных
солей Li, Na, К, Rb и Cs показываем что электропроводность от Li
к Cs возрастает, хотя в этом направлении радиусы катионов
также возрастают. Такая зависимость подтверждает больший
размер гидрагвровашшго иона лития, нем гидратированного
иона цезия (рис. 125).
293
«>,068
0,0<>8
0 i33
0,149
0,165
5.5
3,3
1.8
КЗ
1,0
Рис 125 Соотношение размеров тдрлшронлнных ионов
v-xicvcinoB 1 группы Периодической системы
П>нк1иром отмечена гр.шшы со :ьиамюю с юя
Существование иона гидроксония Н3О + доказано результата-
ми изучения электролиза смеси НВг и Н2О в жидком оксиде серы
SO2 как растворителе. Эксперименты показываю!, что вода
перемещается к кагоду, и это свидетельствует о наличии
положительного заряда на молекуле воды, т. е. свидетельствует об
образовании иона Н3О+ и соединения [Н3О]Вг. Ион гидроксония
обнаружен рентгенографически в кристаллах гидратов HNO3 * Н2О
и НСЮ4Н2О, или [H3O]NO3 и [Н3О]СЮ4.
Образовавшийся ион гидроксония гидратируе гея другими
молекулами воды с возникновением ионов Н5О2 + , H9O4f и,
возможно, других. Эти ионы можно записать как комплексные
[Н(Н2ОJ] и [Н(Н2ОL] + . Следовательно, можно
предположить, что протон Н+ проявляет координационные числа 1,2 и 4.
Ион Н9О4+ окружен другой менее плотной оболочкой
молекул воды, которые связаны значительно слабее с центральным
ионом, чем молекулы воды, входящие в первую оболочку.
Гидроксид-ион в водном растворе также сильно гидратирован.
Наиболее прочно он связан с тремя молекулами воды
(координационное число равно 3) с образованием комплексного иона
Н7О4 ~.
В отличие от катионов д-элеменюв I группы Периодической
системы (Li, Na, К, Rb и Cs) катионы переходных металлов
имеют целые координационные числа гидра 1ации. Для
большинства катионов они равны 4 или 6. Например, в водном растворе
ионов Fe3+ нет, а есть ионы Fe(H2ON . Гидратированные ионы
относят к комплексным ионам (см. ниже), если вода связана
прочно и число молекул связанной воды о!вечает
координационному числу катиона. В то же время комплексный ион окружен
значительно менее прочной гидратной оболочкой с переменным
числом молекул воды.
Выше было рассмотрено взаимодешмвие между
нейтральными молекулами растворителя и ионами в растворе.
Взаимодействия между ионами в расiворе можно классифицировать
следующим образом.
6. Взаимодействие двух ионов; прос1ейтим примером такого
взаимодействия является любое равновесие в растворах слабых
электролитов типа
Н + +СН3СОО -СН3СООН.
294
Степень взаимодействия можег быть увеличена, i. e.
положение равновесия може1 смещаться вправо в соответствии с
принципом Ле Ша гелье.
В растворах сильных электролитов ирожвоположно
заряженные ионы, особенно в концентрированных расjворах, образуют
ионные пары, iройники и другие более сложные образования,
например:
Na^ ч Cl -(Na'CP)
(Na+Cl") + Na+ = (Na + C!~Na4)
(№+СГ)-|-СГ-(С1 Na + CP).
Важнейшим взаимодействием между ионами следует счшагь
донорно-акцепторное, например:
Fe3++6CN~ = [Fe(CNN]3".
Так как в растворе содержатся гидратированные ионы железа, то
процесс правильнее описывать уравнением:
[Fe(H2ON]3++6CN- - [Fe(CNN]3" +6H2O.
Вторая запись отражает процесс вытеснения одного лшанда
другим, что возможно, поскольку ион Fe3 + имеет большую
склонность координировать ион CN , чехМ молекулу воды, т. е.
ион CN" проявляет большую способность быть лигандом.
Следует отметить, что не всегда можно провести че!кую
границу между ионными ассоциатами и комплексными ионами.
7. Взаимодействие между молекулами раствори «еля и
ионными ассоциатами. В связи с тем, что ионы в растворе соль-
ватированы, должны быть сольватированы ионные пары и
другие ионные ассоциаты. Число тех или иных ионных пар в ассо-
циате зависит от концентрации ионов и их относительной
способности взаимодействовать между собой и с молекулами
растворителя.
Продуктами этого типа взаимодейс!вий можно считать
и комплексные ионы с различными лигандами, которые
образуются, например, из исходного комплекса [Fe(CNN]3~ путем
замещения части лигандов, несущих отрицательный заряд, на
молекулы воды:
[Fe(CNMH2O]2", [Fe(CNL(H2OJj\ [Fe(CNK(H2OK],
[Fe(CNJ(H2OLr, [FeCi\(H2OM]2i\
При образовании таких ионов координационное число комплек-
сообразова геля не изменяется.
8. Взаимодействие между ионами и нейтральными
молекулами. При растворении аммиака в ночном расiворе ионов железа
образуются ионы [Fe(NH3N]3" по реакции
295
Fe3f+6NH3=[Kc(NH3)c,]3t
или
N]3 + +6NH3=[Fc(NH3N]3++6H2O.
Эта реакция показывает, что молекулы аммиака обладают
большей склонностью к координации ионом Fe3+ по сравнению
с молекулами воды.
9. Взаимодейавия между несколькими растворенными
частицами и молекулами растворителя. Эта группа объединяет
разнообразные взаимодействия, в том числе процессы образования
различного типа смешанных сольватов, ионных ассоциатов
и смешанных комплексов типа [Fe(NH3JCl2(H2OJ]f,
[Co(NH3K(NO2JH2O]+ и т. п.
Приведенный краткий обзор взаимодействий между
частицами растворенного вещества и молекулами (и ионами) рааво-
рителя показывает исключительную сложноеib полного
описания всей системы частиц в растворе, поэтому обычно выбирают
один или несколько наиболее важных процессов, определяющих
поведение системы, пренебрегая многообразием остальных.
Координацию можно рассматривать как одно из
фундаментальных свойств вещества, заключающееся в способности частиц
взаимодействовать с чааицами того же состава или другими
с образованием более или менее устойчивых агрегатов.
Координация осуществляется и при взаимодействиях, обусловленных
слабыми силами, типа сил Ван-дер-Ваальса, и энергетически
средними силами, как водородная связь, и при сильных
взаимодействиях— формирование ионного, полярного, ковалентного
и в их числе донорио-акцепюрного типов связи. Способность
частиц окружать себя другими частицами — координировать
их — присуща любым веществам. Координация как стремление
одной частицы окружить себя подобными чааицами или
частицами любого другого сорта приводит к отклонению поведения
газов от идеального состояния, образованию сфуктур жидкостей
и кристаллов, простых и сложных молекул, слабосвязанных ассо-
циа i ов ионов и молекул, цепей молекул и т. п.
Любая координация частиц способствует увеличению степени
порядка в системе, поэтому изменение энтропии в результате
этого процесса должно быть отрицательным. Чтобы изобарный
потенциал уменьшался при протекании самопроизвольных
процессов (AG<0), энтальпия в процессах координации также
должна уменьшаться (Д//<0). Следовательно, процессы
взаимодействия частиц сопровождаются выделением теплоты.
В равновесных системах концентрации всех видов части
постоянны и энталышйный член АН равен энтропийному TAS, т. е.
имеет место равенство двух прошводействуюших факторов
упорядочения и разупорядочения системы частиц.
296
Координацией (и другими эффектами) объясняйся
каталитическое действие среды и, в частое г и, рас1вори1еля. Оно
обусловлено различной силой взаимодежмвия молекул рашворшеля
с молекулами исходных веществ, акжвным комплексом и
молекулами продуктов реакции.
§ 43. КОЛЛОИДНОК СОСТОЯНИЕ ВКЩКСТВЧ
Свойства вещества в раздробленном, дисперсном, состоянии
значительно отличаются от свойств того же вещества,
находящегося в недисиерсном состоянии, i. е. в виде куска т вердого тела
или некоторого объема жидкое i и. Так, давление пара воды при
20 С равно 2333 Па, но если измерять давление пара над
каплями воды радиусом 1 мм, го оно окажется выше на 0,003 Па,
а над каплями радиусом 0,01 мм давление паров выше на 0,3 Па
по сравнению с давлением над плоской поверхпос1ью воды.
Кристаллический гидрат оксида алюминия А12О3-ЗН2О (или
А1(ОНK) начинает терять воду при +200 С, а в очень
мелкораздробленном состоянии — при 100 С. Золото в хлороводородной
кислоте не растворяется, однако в высокодисперсном сосюянии
легко переходит в раствор. Рас1воримое!ь CaSO4 в воде
составляет 4.9-10 моль/л, если же CaSO4 находится в виде частиц
размером 2-К) см, то раемворимость повышается до
15-Ю моль/л.
Системы, в которых одно вещество, дисперсная фаза,
распределено в другом, в дисперсионной среде, называю i ся дисперсными.
Существует несколько различных классификаций дисперсных
систем: по размеру частиц, по атрегатному состоянию дисперсной
фазы и дисперсионной среды, по xapaKiepy взаимодействуя
частиц дисперсной фазы с молекулами дисперсионной среды, по
термодинамической и кинетической усюйчивосж дисперсных
систем и 1. и.
В зависимости от размеров частиц дисперсной фазы
дисперсные системы условно можно разделить па две группы:
высокодисперсные системы, или собственно коллоидные
системы, и низкодисперсные системы. Размер частиц
высокодисперсных систем лежит в интервале 10 —10 5 см, что, но
крайней мере, на порядок выше размера чааиц в истинных
растворах (^10 8 см). Размер частиц ни*кодисперсных систем
10 ~4 см и выше.
В реальных дисперсных системах частицы имеют различные
размеры. Распределение частиц по размерам описывается
кривыми, аналогичными кривым распределения молекул газа по
энергиям или скороеi ям.
Классификация дисперсных сие i ем по агрегашому состоянию
дисперсной фазы и дисперсионной среды приведена в 1абл. 19.
297
Та б f и ц а 19
Классификация дисперсных систем по ai pei атному состоянию
iHLiiepcmta
фа и
Пи
Жидкость
Твердое
I0JIO
Ди'.исрсмои
нам среда
Пи
Жидкость
Твердое
гсдо
Газ
Жидкость
Твердое
гело
Газ
Жидкость
Твердое
?СЛО
Название -исмемы
Дисперсная
Пена
Твердая пена
А >розоль
Эмульсия
Твердая эмульсия
Аэрозоль,
порошок
Суспензия и золь
(коллоидный
раствор)
Твердый золь
Примеры
система не образуется
Пена газированной воды,
пузырьки газа в жидкости
Пенопласт, микропористые
резины, пемза
Туман, облака, струя из
аэрозольного баллона
Молоко, сливочное масло,
крем, мазь
Жемчуг, опал
Пыль, дым
Глина, пас га, ил, жидкие
смазочные масла с добавкой
графита или MoSx
Сплавы, цветные стекла,
минералы
Важнейшей отличительной особенностью дисперсных систем,
или коллоидного состояния вещества, является большая площадь
поверхности раздела фаз. В иоверхносшом слое вещееiво
обладает сущес!венно иными свойствами, не похожими на свойства
вещества в большом его объеме и в атомном или молекулярном
состяниях. Большая поверх нос п> раздела фаз вызывает сильное
взаимодействие частиц дисперсной фазы с дисперсионной средой,
которое приводит к тому, что частицы дисперсной фазы
окружало юя молекулами и ионами дисперсионной среды (растворителя)
или же приобретают довольно значительный заряд (возможно
одновременное проявление обоих явлений).
По интенсивности взаимодействия дисперсной фазы с
дисперсионной средой коллоидные системы разделяют на шофобные
и лиофильные (по-гречески лиос—жидкость, фобо — ненавидеть,
бояться, фило — любить). Эти названия указывают на то, что
в лмофильяых коллоидных системах взаимодействие частиц с
молекулами дисперсионной среды сильнее, чем в лиофобных.
Из дисперсныч систем в данном разделе обсуждаются
системы с жидкой дисперсионной средой — золи и эмульсии.
В ЗО1ЧХ, или командных растворах* дисперсной фазой
являйся твердое тело. Следует отметить, что термин «коллоидный
раствор» не совсем правильный, гак как исшнные растворы —
:>тс гомогенные системы с молекулярной степенью
раздробленное ги вещества, а коллоидные растворы — гетерогенные сипс-
мы, обладающие межфазной поверхностью
298
По размерам частиц и по ряду свойств золи занимают
промежуточное положение между истинными растворами и грубодис-
персными системами—суспензиями. Можно предположить, что
в реакциях, протекающих с получением и растворением осадков,
одной из промежуточных с!алий являемся образование и
разрушение коллоидных частиц.
Золи — типичные коллоидные системы, которые наиболее
ярко проявляют свойства, присущие веществу в коллоидном
состоянии.
Рассмотрим процесс образования гидрозоля (дисперсионная
среда — вода) и строение его дисперсных частиц на примере
гидрозоля хлорида серебра. Этот гидрозоль образуется при
медленном приливании водного раствора хлорида натрия NaCl
к водному раствору нитрата серебра AgNO3, взятому в избытке.
Если ввести сразу большую порцию раствора NaCl, то
мгновенно выпадет осадок хлорида серебра и золь не получится. При
постепенном введении раствора NaCl в раствор AgNO3 в системе
в первый момент образуются кристаллические агрегаты (AgCl)n
из ионов Ag4" и СГ, расположенных в том же порядке, что
и в решетке кржмалла AgCl. Агрегаты (AgCl),, адсорбируют на
своей поверхности те ионы, которые составляют
кристаллическую решетку и находятся в растворе в избытке. Этим условиям
отвечают ионы серебра. В результате адсорбции ионов Ag+
кристаллические arpeiaTbi хлорида серебра приобретают
положительный заряд.
Ионы, адсорбирующиеся на поверхности кристаллического
arpeiaTa, называются потенциалопределяющими. Наличие
одноименных зарядов на агрегатах препятствует их объединению
и pociy кристллов AgCl. Таким образом, агрегаты AgCl с
адсорбированными на них ионами приобретают состояние агрегатив-
ной устойчивости.
Кристаллический агрегат (AgCl)n вместе с
потенциалопределяющими ионами Ag4" составляет ядро, К заряженному ядру
притягиваются ионы противоположного заряда — противоионы.
Для рассматриваемой системы водный раствор
AgNO3—кристалл AgCl противоионами будут нитрат-ионы
NO3~. Противоионы, непосредственно примыкающие к ядру,
образуют адсорбционный слой противоионов. За этим слоем
следует диффузный слой тех же противоионов. Концентрация
противоионов диффузного слоя постепенно понижается по мере
удаления от ядра. Между противоионами адсорбционного и
диффузионного слоев устанавливается подвижное равновесие.
Противоионы диффузного слоя ориентируют полярные
молекулы растворителя, создавая дополнительную сольватную
оболочку.
Ядро вместе с противоионами адсорбционного слоя
составляет коллоидную частицу, или гранулу. Коллоидная частица
299
Рис 126 Строение мицеллы золя хлорида
серебра
совместно с прошвоионами
диффузного слоя называс!ся
лшщч.юй. Следует иметь
в виду, что коллоидная
частица всегда заряжена, знак
заряда соответствует знаку
заряда погеиниалопределя-
ющих ионов; мицелла в or-
личие oi гранулы электро-
нейфальна.
С i роение мицеллы
гидрозоля хлорида серебра
преде гавлено на рис. 126.
Формула мицеллы
записывается так:
{(AgCl),, wAg + ,
(/h-.y)NO3~}%\vNO3~
(в фи! урные скобки
заключена гранула). Ионы,
указанные за фигурными
скобками, составляют внешнюю
чаегь мицеллы. Эти ионы
под действием
электрического поля офываются от
мицеллы, и к отрицательно
заряженному элекфоду будет
передвигаться гранула,
заряд которой определяется
ионами серебра, входящими
в ядро.
Строение мицеллы и
заряд гранулы зависят от
способа получения коллоидного
раст вора. Если прилива i ь
раствор нитрата серебра
к рас 1 вору хлорида натрия,
взятому в избытке, то на
поверхности агрегата (AgCl)n
будут адсорбироваться
хлорид-ионы, имеющиеся в
избытке в растворе, а в качестве противоиоиов адсорбционного
и диффузионного слоев буду! выступать ионы натрия. Состав
мицеллы полученного гидрозоля записывается формулой:
{(AgCl),t, wCl \ (w--v)Na
300
Во внешнем элекфическом ноле отрицательно заряженная
гранула, образовавшаяся в эшх условиях, пере.мстаеюя к
положительно заряженному хгсктроду.
В зависимое!и or условий получения золей коэффициенты и,
т и л\ определяющие число чаешц, составляющих мицеллу,
Moryi принимать различные значения. Jleiкость получения
коллоидных часчиц и их усюйчивос1Ь возрас!ают в ряду
AgCl—AgBr— Agl.
Ионы обладают различной способностью адсорбироваться на
поверхности aiperara и ядра коллоидной частицы. При этом
обычно соблюдаются общие правила адсорбции. Согласно
одному из них на поверхности кристалла, как и на поверхности
агрегата коллоидной частицы, преимущественно адсорбируются
ионы, имеющие общий с поверхноомо сосгав или одинаковую
с ней атомную группу.
Способноеп> ионов одинакового заряда адсорбироваться
понижается с уменьшением их размеров (и их массы).
Li4 <NaJU<K* <Rbf <Cs +
Cl" <Вг"<Г.
Ионы с большим зарядом адсорбирую!ся лучше:
Na" <Mg2r<AI3\
Знак заряда коллоидной чаежцы опрелеляеюя зарядом
ионов, адсорбированных агрегатом. На границе раздела
коллоидной частицы и диффузного слоя противоионов возникает двойной
электрический слои.
Если каким-либо nyicM заряд коллоидных частиц уменьшить
или полностью уничтожшь, ю диффузный слой разрушается
тогда частицы получают возможность слипания и укрупнения,
что приведет к понижению устойчивости коллоидной системы.
Процесс укрупнения (слипания) коллоидных частиц,
приводящий к образованию осадка, называют коагуляцией. Явление
осаждения частиц дисперсной фазы под действием силы гяжесш
называю! седиментацией. Подбором соответствующего
электролита осевшие чаепшы можно снова заряди п> и перевеем и в
коллоидный раствор. Переход осадка в золь называется пепти-
зацией
Явления коагуляции и пепгизацин связаны с разрушением
и образованием двойного электрического сюя (и с гидратацией
коллоидных частиц). Двойной электрический слой возникает на
поверхности раздела любых фаз. в частости, дисперсная
частица— раствор, и наиболее четко он обнаруживайся при условии
ионной (или металлической) ефукчуры вещества дисперсной
фазы и тлекфолитиой природы дисперсионной среды. Этот слой
сосюи! из ио1снциалопределяюпшх ионов, фиксированных на
301
поверхности твердой фазы (дисперсной частицы), и
противоположно заряженных ионов — противоионов, находящихся в жидкой
фазе. Вследствие наличия двойною электрического слоя между
твердой и жидкой фазами возникаем разность потенциалов—
поверхно( пшый потенций i ср.
Слой противоионов создает размытую, диффузную ионную
атмосферу. Концентрация iipoi ивоиоиов максимальна вблизи
поверхности частиц, покрытой слоем потенпиалобразующих
ионов, и убывает по мере удаления см границы раздела фаз в глубь
раствора.
При относительном перемещении фаз происходит разрыв
двойного электрического слоя, обычно по диффузному слою,
и дисперсная фаза и дисперсионная среда становятся
противоположно заряженными. В результате на границе разрыва возникает
разность потенциалов, называемая элекгрокинетическим
потенциалом, или ? (дзета)-потенциалом.
Потенциалы <р и ?—важнейшие характеристики устойчивости
дисперсных систем.
При движении мицеллы под действием электрического ноля
она распадается на две части: ядро (кристаллический агрегат
с потенциалопределяющими ионами) перемещается к одному
электроду, а иротивоионы диффузного слоя—к другому. Очень
важно, что вместе с гранулами передвигаются молекулы
жидкости, составляющие их сольватные оболочки (рис. 127). Так как
к одному электроду перемещаются только ионы, а к другому—
заряженные коллоидные частицы, то количества вещества,
выделяющиеся на обоих электродах, сильно различаются.
Явление перемещения во внешнем электрическом поле
коллоидной частицы называется электрофорезом, а перенос жидкой
дисперсионной среды—электроосмосом. Электрофорез и
электроосмос используются не только для исследования дисперсных
систем, но и для обезвоживания
_ различных материалов (глины,
почвы), для очистки воды,
отделения белков, витаминов,
аминокислот и т. п.
Значение ^-потенциала
определяется толщиной диффузного
слоя, которая зависит от
концентрации в растворе не только
ионов, входящих в диффузный
слой, но и общей концентрации
ионов в растворе, т. е. от ионной
силы раствора. Увеличение кон-
7 чеи.ращм электролит, в рас.во-
(а) и ее перемещение в элеприческом Р^ приводит К сжатию диффуз-
поле {6} ного слоя и уменьшению ?-по-
302
тенциала. При некотором значении ионной силы раствора элек-
трокинежческий поднимал равен нулю. Сосюяние системы, при
котором электрокинегический потенциал становится равным
нулю, называется изоэлектрически\л. В изоэлектрическом или близ-
ком к нему состоянии коллоидные системы наименее устойчивы
и коагулируют с наибольшей скоростью. Изоэлектрическое
состояние определяется по отсу!С1Вию передвижения коллоидных
частиц в электрическом поле, что свидетельствует об
электронейтральности частицы.
Под устойчивостью дисперсных систем понимают
способность дисперсной фазы сохранять состояние равномерного
распределения в дисперсионной среде. Различают два вида
устойчивости— агрегаттную и седиментациоиную (иногда ее называют
кинетической, по скорости осаждения частиц).
Агрегативная усюйчивоегь золя понижается с уменьшением
электрокинетического потенциала Термодинамически все
гидрофобные золи неустойчивы. Эта неустойчивость обусловлена
огромной поверхностью дисперсной фазы и ее высокой
поверхностной энергией. Состояние вещества с высокоразвитой
поверхностью всегда менее устойчиво, чем состояние, в котором его
поверхность меньше. Процесс укрупнения частиц при коагуляции
связан с образованием новых связей между ними и
сопровождается выделением теплоты Q>0 или уменьшением энтальпии
А//<0. Укрупнение частиц приводит к увеличению степени
порядка в системе и уменьшению энтропии. При
самопроизвольном процессе коагуляции изобарный потенциал уменьшается,
ДG<0, и этот процесс протекает до установления состояния, при
котором энтальпийный и энтропийный факторы становятся
равными AH—TAS, и тогда AG = 0. Для процессов образования
и разрушения коллоидных систем эти простые положения
усложняются рядом побочных факторов.
Коагуляция—самопроизвольный процесс, возникающий из-за стремления ежмемы
перейти в состояние с более низкой поверхностной энергией и более
низким значением изобарного потенциала.
Понижение поверхностной энергия, к коюрому
самопроизвольно стремятся сисгемы, возможно при объединении частиц
в агрегаты, однако этот процесс может быть очень медленным.
Агрегативная устойчивость, таким образом, включает в себя
термодинамический и кинетический факторы.
Процесс пептизации скоагулированного вещества также
протекает самопроизвольно. Основными движущими силами
пептизации являются образование двойного электрического слоя,
адсорбция ионов на частицах и сольватация частиц. Вещества,
способствующие переходу осадка в золь—пептызаторы —
изменяют структуру двойного слоя, увеличивают толщину
диффузного слоя, повышают ^-потенциал, увеличивают степень
сольватации частиц и защищают их от слипания. Пепютация протекаем
303
с выделением энер1ии (Д//<0) и связана как с увеличением
порядка в еисмеме в результате сольватации, *ак и с ei о
уменьшением вследс!вие раздробления вещества.
Коагуляция может быть обусловлена различными причинами,
наиболее эффективно действие элекiролитов. Минимальная
концентрация электролита в paciBope, вызывающая коагуляцию,
называемся порогом коагуляции. Порог коа!уляции зависш or
природы дисперсной сис!емы, концешрации дисперсной фазы,
а также or скорое»и прибавления электролша, интенсивности
перемешивания, присутсгвия в ежмемс других электролитов и
неэлектролитов.
Коалирующее действие оказывает не весь электролит, а его
ион, заряд которого по знаку прошвоноложен заряду
коллоидной частицы. По отношению к офицагсльно заряженным
частицам золей коагулирующая способность катионов (при любом
одинаковом анионе) возрастет с увеличением массы кантона:
Li+<Na + <K4 <Rbh<Cs+.
Коагулирующая способность анионов в зависимости от их
массы по отношению к положительно заряженным частицам
золей изменяется в противоположном направлении:
CI >Вг">1 .
Чем выше заряд иона, тем сильнее его коагулирующее
действие и тем ниже порог коагуляции.
В зависимости от условий коагуляция может протекать
медленно и быстро. Мед пенная коагуляция продолжается часами
и сутками, а быстрая завершается за секунды и доли секунды.
Скорость быстрой коагуляции не зависит от природы и
концешрации электролига, так как она проходиi при ? = 0.
Медленная коагуляция проходит при некотором значении
^-потенциала и определяется долей эффективных взаимодействий
сталкивающихся мицелл. Медленная коагуляция возможна, если
кинетическая энергия броуновского движения частиц достаточна
для преодоления энергетическою барьера расталкивания двух
чаешц, окруженных ионными атмосферами одинакового заряда
(аналогия с энергией активации)
Скорость медленной коагуляции зависи! от концентрации
электролита, его природы, ионной силы раствора (т. е. является
функцией ц-шненциала), а также oi температуры и вязкости
среды (т. е. от скорости движения частиц).
В отличие от медленной коагуляции при быарой коагуляции
каждое столкновение частиц является эффективным, так как
вследствие ? = 0 силы расталкивания частиц отсутствую!.
Скорость быстрой коагуляции описывается уравнением скорости
реакции второго порядка:
304
где п—число часшц в данный момент времени;
к — константа скорости коагуляции.
Оишчие процесса быстрой коагуляции oi бимолекулярной
реакции второго порядка состоит в том, что в химической
реакции продукты не влияют на дальнейший ход процесса; при
коагуляции слипшиеся частицы продолжают участвовать в
процессе, образуя все более крупные и тяжелые часгицы. Повышение
массы частиц приводит к тому, что они перестают участвовать
в броуновском движении и осаждаются на дно сосуда.
Коагуляция коллоидных систем наступает не юлько иод
действием электролитов, но и при смешении двух золей с
различными знаками зарядов частиц. Это явление называется взаимной
коагуляцией и вызывае1ся нейфализацией зарядов ионов
диффузионных слоев двух видов разноименно заряженных частиц.
Если агрегативная устойчивость коллоидных систем отвечает
их способности сохранять постоянными размеры часшц, то седи-
ментациоиная устойчивость характеризую противодействие
осаждению в поле тяготения. Коллоидные частицы участвуют в
тепловом (броуновском) движении, что обусловливает их
равномерное распределение в достаточно узком слое жидкости.
Способность частиц удерживаться во взвешенном состоянии
зависит от их размеров, массы, вязкости раствора, различия
плотностей дисперсной фазы и дисперсионной среды. Повышение
темпера туры увеличивает скорость броуновского движения
частиц, однако при слишком высоких температурах сталкивающиеся
частицы разрушают свои защитные оболочки из ионов и молекул
растворителя, частицы слипаются, и начинается их седиментация.
Следует отметить, что при одинаковых плотностях
дисперсной фазы и дисперсионной среды система сохраняет седимен-
тационную устойчивость, несмотря на большие размеры
агрегатов слипшихся частиц.
Устойчивость гидрофобных золей сильно повышается при
введении в раствор даже незначительных количеств
высокомолекулярных соединений, растворимых в дисперсионной среде (т. е.
гидрофильных золей). Например, коагуляцию многих золей *а-
медляют или предотвращают желатин, яичный белок, крахмал
и даже сахар (перечислено в порядке уменьшения защитного
действия). Эю явление, называемое колмтдчой защитой,
объясняется адсорбцией этих веществ на поверхности часшц золя. При
этом в результат определенной ориентации групп —ОН,
— СООН, —NH2 макромолекул образуются дополнительные
устойчивые гидратные оболочки, препятствующие слипанию
частиц. Кроме юго, возможность электролишческой диссоциации
по этим группам изменяет (повышаеО электрокинетический
потенциал, что также способствует защите золя oi коатуляции.
Осадок, полученный коагуляцией, обычно сильно загрязнен
примесями. Для промывания его ис следует применяв чистую
305
воду, гак как с удалением посторонних ионов может произойти
перезарядка частиц, пет изация и потеря части осажденжм о
вещее 1ва. Поэтому осадки следует промывать расiворами -шекфо-
литов. Наиболее эффективен раствор хлорида аммония, так как
при прокаливании осадка хлорид аммония улетучивается.
Рассмотрим методы получения гидрофобных золей. Такие
методы перевода веществ в мелкокристаллическое состояние, как
охлаждение пересыщенного раствора, выпаривание
растворителя, введение в раствор веществ, уменьшающих растворимость
вещества, непригодны для синтеза коллоидных систем, 1ак как
при этих процессах кристаллические зародыши (агрегаты)
образуются без формирования двойного электрического слоя.
Любой синтез золя должен включать стадию возникновения
электрокинетического потенциала на достаточно малых
кристаллических агрегатах.
Наиболее часто для получения золей применяют методы
конденсации (укрупнения частиц) и методы диспергирования
(измельчения). В химических методах конденсации могут быть
использованы следующие реакции.
1 Окислительно-восстановительные реакции, например, для
получения гидрозолей металлического золота и серы:
2H2S+O2 =
Образующийся в первом процессе электролит НС1 стабилизирует
частицы золота.
Золь серы без применения стабилизаторов неустойчив.
Последняя реакция используется для изучения влияния
концентрации реагирующих веществ на скорость химической реакции;
о скорости судят но времени появления золя серы определенной
плотности.
2. Образование малорастворимых в воде соединений для
получения золей сульфата бария и хлорида серебра:
или Ba2 + SOr = BaSO4,
или Ag^ +СГ -AgCl.
Стабилизация обеспечивается электролитом КС! или
находящимся в избытке одним из исходных веществ (большой избыток
хлорида калия может перевести AgCl в раствор за счет комплек-
сообразования).
3. Гидролиз: например, этим методом можно получить золь
гидроксида железа:
306
или
Fe3' +3H2O = Fc(OHK f 311
К* метолу конденсации оiмоется мегод замены расиюршеля.
п котором данное вещееiво менее растворимо
Диспергирование вещее 1ва до коллоидною сосюяния
осуществляют химическими и механическими методами К чимическим
методам диспергирования относится петшация. Например,
свежеприготовленный осадок гидрокепда железа переходи! в золь
при обработке большим количеством чисюй воды, при
добавлении раствора хлорида железа определенной коипен фации,
достаточной для образования двойного слоя, или при добавлении в
раствор поверхностно-активного вещества, например, олеата натрия.
Механическое диспергирование твердых вещее i в проводят
дроблением в коллоидных мельницах. Эффективен метод с
использованием электрической дуги: под действием высокой
температуры электрической дуги металл элек!родов испаряется, пары,
попадая в холодную дисперсионную среду, конденсируются
и в тонкодисперсном виде распределяю 1ся в ней. Fxjih на
холодную поверхность нанести слой двух взаимно не растворяющихся
веществ, а затем температуру повысить до расплавления
образовавшегося слоя, то кристаллическая колиоидная сиспема (к-
к) переходит в жидкую (к-ж или ж-ж)
Очень сложно очистить золи oi находящихся в них обычно
в больших количествах электролитов. Избыток электролита
понижает устойчивость золя, но и не содержащий электролит
коллоидный раствор также неустойчив. Для очистки золей обычно
проводят диализ, который основан на прохождении чаешц
(молекул, ионов) низкомолекулярных вещее\в через
полупроницаемую мембрану (пленки из целлофана или коллодия, пленки
животного происхождения, пергаментная бумага). Принцип
метода очистки золя от электролитов путем диализа состоит в
следующем (рис. 128, а). Если широкую трубку, нижний конец
которой закрыт полупроницаемой мембраной, наполнить золем
и трубку погрузиib в другой сосуд с чистой, жепательно
проточной водой, то частицы электролита из золя будут переходить
через мембрану в сосуд с водой. На этом основан принцип
действия диализаторов.
Рис 128 Принцшша 1ьния схем.) диа imniopu {а) и ысктролиали затора (о)
307
Следуе! имс1Ь в виду, чго слишком продолжи[ельный и
полный лиалт можс1 привести к удалению ионов диффузного слоя,
что ириведс! к понижению устойчивое!и золя.
Me юл элсктродыа шза обеспечивает более полное и быстрое
удаление электролита. Метод состоит в том, что золь помещаюi
в сосуд с двумя полупроницаемыми мембранами
(перегородками), рядом с коюрыми в чистой воле расположены элекфолы
(рис. 128, б). Под действием электрического поля ионы
электролита проходят через мембраны к соответствующим электродам
и уносятся про 1 очной водой. По такому принципу рабоииот
элект родиализа торы.
Для очистки золей от электролитов применяют также филм-
рацию. Для эюго золь промывают водой, а затем фильтруюi
через специальные фильтры с очень малым размером пор. Для
ускорения фильтрации процесс проводя! или при повышенном
давлении над филы ром, или при разрежении за филы ром.
Отделение золя или осадка от расiвора электролита часто
проводят центрифугированием, т. е. разделением центробежной
силой фаз, имеющих различные плошосш. После осаждения
вещества раствор сливаю!, осадок взбалтывают в чистой воле
и снова подвергают центрифугированию. Такую операцию
повторяю! многокршно.
Методы исследования золей (определение размера, формы
и заряда коллоидных частиц) основаны на изучении их особых
свойств, в часmociи ошических, обусловленных
гетерогенностью и дисперсностью. Из явлений, возникающих при денегвии
света на золь, наиболее характерно рассеяние CBeia. Это явление
проявляется в виде опагесцепцш. при боковом рассмотрении
золя, через который проходиi свегоаой луч, впуфи коллоидной
системы наблюдается светящийся конус (явление Типлаля).
Рассеяние снега возможно, если размер коллоидных часищ
мепыпс длины волны проходящего све*а и чоказатели
преломления дисперсной фазы и дисперсионной среды различны.
Интенсивное! ь свеюрассеяния резко увеличивается с уменьшением
длины световой волны. В рассеянном свете коллоидные растворы
имею! синева!ый оттенок, а в проходящем—красно-оранжевый
Па явлении рассеяния cBeia золями основаны методы
определения их дисперсного состава.
Знак заряда коллоидной частицы может быть определен
методом электрофореза и jjieicгроосмоса или же очень простым
способом по харлкчер) взаимодействия коллоидных частице
целлюлозой
В во июй орчМС клич г^гуы чел »ю ним *ар>ша» >iся офшше и,Нч>, л нахоля-
пляог в них bo.w—иолож^счьчо Слол1'йа!сльно. плоль полоски филыровл ib-
noii 6\MirH vuc Liiojoia) no jc клпи,пяоы ,1 %*ог>т nope (внгльел jo.jsko частицы,
имеющее i'^» Ac j;.pM,»s ч:о и иелполоза, ) с oipiuiiMJ into «аряжепные TIojo-
жиимьнс, »t(>^<KCriUbiu час»ииы исажшююн на це июлою в самом канате пути
308
Пели в ко1лоилный раствор поместить конец полоски фильтровальной бума!и
и раствор будет перемещаться вдоль нес, то значит, коллоидные частицы
заряжены отрицательно При исследовании бесцветного коллоидного раствора
пропитанную раствором oyMaiy следует высушить и обработать реактивом,
окрашивающим коллоидные частицы.
Подобный капиллярный анализ представляет собой вариант хроматографи-
ческого анализа на бумаге
Как уже отмечалось, многие важнейшие свойства коллоидных
систем в значительной сюпени зависят oi характера
взаимодействия дисперсной фазы и дисперсионной среды.
Рассмотрим основные признаки различия лиофильных и ли-
офобных коллоидных систем. Лиофильные системы
самопроизвольно образуются в жидкое 1ях без участия электролитов или
поверхностно-активных веществ. Так, гидрофильные системы
образуют желатин и крахмал, которые сначала набухают в воде
и затем переходят в раствор; натуральный каучук легко
растворим в бензине, а яичный белок — в воде. К лиофильным
коллоидным сиаемам относятся растворы мыла в воде.
Лиофобные золи с концентрацией дисперсной фазы выше 1%
получить не удается; лиофильные коллоидные системы Moryi
быть очень концентрированными.
Явление мицеллообразования относится только к лиофобным
коллоидам.
Заряд частиц лиофильных коллоидов значительно ниже или
вообще отсутствует. Заряд на час!ице лиофильного коллоида
изменяется очень легко при прибавлении небольших количеств
электроли I ов. Изменение концентрации ионов водорода в
растворе приводит к легкой перезарядке коллоидного раствора.
Лиофильные коллоиды заряжаются отрицательно, если
концентрация водородных ионов меньше, чем в изоэлектрической точке,
и наоборот. В изоэлектрическом состоянии лиофильные системы
в отличие oi лиофобных усюйчивы (кроме некоторых белков).
В электрическом поле лиофильные коллоиды или не
перемещаются, или перемещаются в любом направлении.
Для осаждения лиофильных систем требуются очень большие
количества электролитов. Коагуляцию, наступающую при
добавлении больших количеств электролитов или дегидратирующих
веществ в гидрофильную систему, называют высаливанием. При
высаливании, а также при испарении растворителя или
увеличении концентрации лиофильной системы большинство из них
превращайся в студнеобразные массы—гели. Влияние
температуры на i елеобразование может быть различным: в некоторых
случаях с понижением темпера 1уры образус!ся гель, в других
случаях гель разрушается. При выпаривании или охлаждении
лиофильных золей нолучаеюя мелкокристаллическое вещество (в
отличие от гелей).
Поверхностное натяжение и вязкость лиофобных коллоидных
систем близки к тем же характерис!икам дисперсионной среды.
309
Лиофильные же системы имекн более низкое поверхносшое
натяжение и более высокую вязкость, чем дисперсионная среда.
Электрофорез и элекгроосмос осуществимы для лиофильных
систем только в гом случае, если их частицы заряжены.
Лиофобные коллоидные системы легко обнаруживаются по
способности к светорассеянию; у лиофильных систем эта
способность проявляется слабо
Принципиальное различие лиофилытых и лиофобных
коллоидных систем сосюит в их термодинамических свойствах.
Лиофобные золи — гетерогенные микроге i ерогенные) системы,
и в этом о i ношении их нельзя от носи п> к истинным расiворам.
Лиофильные золи — однофазные системы, обладающие многими
свойствами истинных рас i воров. Вследствие высокой
поверхностной энергии лиофобные дисперсные системы
термодинамически (и кинешчески) неустойчивы. Лиофильные коллоиды
термодинамически устойчивы. От истинных растворов они
отличаются размером часмиц и формой (длинные нитеподобные
и свернутые в клубок молекулярные струк1уры).
К лиофильиым коллоидным системам относят растворы
высокомолекулярных соединений, которые одновременно
проявляло i и некоторые свойства истинных растворов.
Высокомолекулярные соединения принадлежат к другому уровню организации
вещества — уровню макромолекул. Таким образом, между ли-
офильными и лиофобными коллоидными системами имекмея не
менее принципиальные различия и с точки зрения теории
строения вещест ва.
Четкую границу между лиофильностью или лиофобностью
коллоидных систем не всегда можно установить. Так, золь
кремниевой кисло!ы усюйчив в изоэлектрическом состоянии.
Гидрозоли кремниевых кислот, гидроксидов железа или алюминия
при коагуляции удерживаю! большое количество воды и
образуют студнеобразные системы. В то же время студнеобразный
крахмал в водной среде при нагревании переходит в золь,
обладающий многими свойствами шдрофобных систем. В
подобных случаях часто невозможно провести границу между
гетерогенной и юмогенной системами, и правило фаз Гиббса
оказывается неприменимым. Поэтому для лиофильных коллоидных
систем понятия «дисперсной фазы», «дисперсионной среды»,
«золя» и других условны, в той же мере, как понятие «раствор» для
лиофобиых систем.
Важным типом коллоидных систем являются эмульсии —
высокодисперсные системы, в которых дисперсная фаза и
дисперсионная среда являются жидкостями. Образование таких систем
возможно при нерастворимости или очень ограниченной
растворимости одной жидкости в другой. В зависимости от условий
каждая из фаз может бьпь либо дисперсионной средой, либо
дисперсной фазой Например, из масла и воды могут быть полу-
310
чены эмульсии двух типов: «масло в воде» и «вода в масле».
Агрегативная устойчивость эмульсий повышаемся введением
специальных веществ—эмульгаторов, адсорбирующихся на
поверхности капель и препятствующих их слиянию — коа концепции.
Эмульсии, как и золи, разделяются на лиофииьные,
термодинамически устойчивые, и лиофобнме, термодинамически
неустойчивые системы, для стабилизации которых необходимы
эмульгаторы. Седиментационная устойчивость эмульсий определяется
размером капель и различием в плотностях двух жидких фаз.
Область химической науки, занимающаяся изучением
поверхностных явлений и дисперсных систем, называется коллоидной
химией. Она выделена как самостоятельная научная дисциплина
и являеюя примером науки, объединяющей химические и
физические методы и подходы для объяснения сущности коллоидных
явлений.
В дисперсном состоянии может быть любое вещество
неорганического и органического происхождения. Поэтому
коллоидная химия представляет собой как бы общую связующую основу
многих областей науки.
ГЛАВА 10
ПРОЦЕССЫ В ВОДНЫХ РАСТВОРАХ
§ 44. ДИССОЦИАЦИЯ СИЛЬНЫХ И СЛАБЫХ ЭЛЕКТРОЛИТОВ
Имеется ряд простых, но несомненных доказательств того,
что некоторые вещества в водных растворах распадаются на
частицы. Так, водные растворы кислот H2SO4, HNO3, HC1O4,
НС1, СН3СООН и других имеют кислый вкус. В формулах кислот
общей частицей является атом водорода, и можно предположить,
что он (в виде иона) является причиной одинакового вкуса всех
этих веществ. Точно так же LiOH, NaOH, КОН, Ва(ОНJ,
СА(ОН2) и другие основания имеют одинаковый неприятный
горько-мыльный вкус и вызывают на коже ощущение
скольжения. По-видимому, за это свойство ответственны ионы ОН",
входящие в состав соединений.
Процесс смешивания растворов хлорида калия и нитрага
натрия не сопровождается заметным тепловым эффектом, хотя
после выпаривания из смеси могут быть выделены четыре
кристаллических вещества (KG, NaCl, KNO3, NaNO3), поэтому
можно предположить, что в растворе соли полностью
распадаются на ионы, которые в процессе выпаривания образуют
кристаллические вещества.
Существование некоторых вещее i в в водных растворах в виде
частиц, образующихся при распаде молекул растворяемого
вещества, доказывался результа!ами исследования водных растворов
методами измерения давления насыщенного пара над раствором
и осмотического давления и методами эбулиоскопии и
криоскопии. Экспериментально установлено, что осмотическое давление
при 0 СС 0,01 М водных растворов сахарозы, глицерина, этанола
и других органических веществ (растворы которых не проводят
электрический ток) составляе! ^0,23 • 105 Па.
Изучение осмотического давления при тех же условиях 0,01
М водных растворов неорганических солей NaCl, СаС12, А1С13
и других показывает, чго осмотическое давление растворов не
312
постоянно и, по-видимому, зависит or природы растворенного
вещее 1ва, причем неорганические соединения можно разделить
на группы, коюрые в растворах обнаруживают повышение
осмотического давления примерно в целое число раз B, 3, 4, ...) по
сравнению с осмотическим давлением растворов сахарозы,
спирта или глицерина при той же концентрации. При сравнении
свойств рас 1 воров хлоридов выявляется явная зависимость
повышения осмотического давления от числа атомов в молекуле
(формуле) вещества: растворы хлоридов МС1, формульная
единица которых состоит из двух атомов, имеют осмотическое
давление приблизительно в два раза больше, чем у раствора сахарозы,
для хлоридов МС12 с тремя aiомами — в три раза больше и г. д.
Эти числа говорят о том, чю в расiворах NaCl, СаС12 и А1С13
соответственно в два, три и в четыре раза больше частиц по
сравнению с тем числом чаепщ, которое отвечало бы
концентрации растворенного вещества.
Диссоциацию веществ на ионы в рас [воре подтверждают
также результаты криосконических и эбулиоскоиических
исследований. Растворы электролиiов в отличие от растворов
неэлектролитов обнаруживают понижение температуры замерзания
и повышение температуры кипения значительно более высокое,
чем ожидаемое в предположении огсутс!вия диссоциации. Так,
у растворов соли типа NaCl Д7'ым и АТкнп примерно в два раза
выше, СаС12 — в три раза выше.
Сопоставляя эти данные с высокой элекфопроводностью
растворов солей, С. Аррениус A887 г.) выдвинул гипотезу
электролитической диссоциации, согласно которой «молекулы» солей,
кислот и оснований при растворении их в воде диссоциируют на
ионы.
Изучение продуктов электролиза позволяет приписав ионам
положительные или О1рица1ельпые заряды. Очевидно, если
кислота, например, хлорная НСЮ4, диссоциирует, предположим,
на два иона и при электролизе водного раствора на катоде
выделяется водород, то уравнение диссоциации следует записать
так:
= Н4+С1О4~.
В то же время по результатам измерения осмотического давления
перхлорат меди в растворе диссоциирует на три частицы, а при
его электролизе на катоде выделяе!ся медь; следовательно,
диссоциация этой соли протекает по уравнению:
Процесс диссоциации количес[венно характеризуется степенью
диссоциации. Степень диссоциации — это отношение числа
распавшихся на ионы молекул к общему числу молекул
растворенного вещества, обозначается буквой а. Например, если из каждых
313
100 молекул расгворенного вещества 82 распадаются на ионы, то
cienenb диссоциации соединения буде1 равна:
а = 82/100-0,82, или 82%.
Как следуе! из приведенного выше определения, для расчета
степени диссоциации вещества необходимо знать число частиц,
получающихся при диссоциации ето в расiворе. Это число может
быть определено измерением понижения давления пара над
раствором Ар и других свойств, зависящих oi числа частиц в
растворе (так называемых коллшативных свойств).
Экспериментально найденные количественные
характеристики коллигативных свойств раствора больше, чем вычисленные
для юго же раствора по его молярной концентрации в
предположении отсутствия диссоциации. Число, показывающее, во
сколько раз экенеримен i ально измеренное свойство больше
вычисленного, называется изотопическим коэффициентом; его
обозначаю г буквой /. В частноеiи, изотонический коэффициент
можно преде 1авить как отношение:
Экспериментально определяемый изотонический коэффициент
позволяет усыновить общее число частиц в растворе и,
следовательно, рассчитать аепень диссоциации.
Увеличение числа частиц в растворе вызвано диссоциацией
растворенного вещества на ионы, поэтому изотонический коэф-
фициеш равняется отношению числа находящихся в растворе
частиц (ионов и недиссоциироваиных молекул) к общему
исходному числу молекул растворенного вещества.
Обозначим число молей растворенного электролита через С,
а число ионов, образующихся при диссоциации, через п. Если
степень диссоциации равна а, то произведение аС показывает
число распавшихся молей электролита. В результате
диссоциации в растворе образуется п<хС моль ионов. Общее число частиц
в растворе составит:
С-аС+яосС
Изо! онический коэффициент равен:
Откуда получаем соотношение, связывающее три величины
/, аил:
Это cooiношение позволяет вычислить степень диссоциации при
известной формуле соединения по экспериментально
определяемому изотоническому коэффициенту.
По способности к диссоциации электролиты условно
разделяют на сильные, средние и слабые. Электролиты, которые в рас-
314
гворе существуют юлько в виде ионов, принят называй? си ib-
пыми. Те элекчролиты, коюрые в раемворепном состоянии
находятся частично в виде молекул и часшчно в виде ионов,
называют слабыми и средними.
По С1епени диссоциации к сильным опюся1 электролиты,
имеющие в данном растворителе а>30%, к слабым—а<3%
и к средним — 3% < ot ^ 30% Различие сильных и слабых
электролит ов состоит не только в значении cienenn диссоциации, а в
основном в поведении их в растворе, в cienenn оишчия от
идеальных рас i воров»
В водных растворах к сильным электролитам (а>30%)
относя 1ся следующие соединения
1. Многие неорганические кислоты: HNO3, НО, HBr, HI,
НСЮ4, НМ11О4, H2SO4 (в разбавленных paciворах). Сила
некислородных кислот возрастает в ряду одножнных соединений при
переходе вниз по подгруппе кислотообразующего элемента:
HCI—HBr— HI.
Ич бескис юродных г ало1снсодержащих кислот фтороволородная кисло га —
кислота средней силы, чт ооьясниосм высокой энергией связи Н—F,
способностью молекчл HF к ассоциации благодаря водородным связям, взаимодействием
ионов 1~ с мо и%к)лими HF с образованием комплексных ионов HF2~, Н,Ь3
и других более сложных чаешц. В рсз>льтатс концентрация ионов водорода
в водном растворе фюро водород пой кис Ю1Ы оказывается сильно пониженной
Сила однотипных кислородных кислот изменяется в
противоположном направлении, например, кислота НЮ4 слабее
хлорной кислоты НС1О4—самой сильной из известных кислот.
Если элемент образую несколько кислородных кислот, ю
наибольшей силой обладает кислена, в котрой кислотообразующий
элсмеш находится в максимальной степени окисления. Так, в
ряду кислоi НСЮ— НСЮ2—НСЮ3—НС1О4 последняя наиболее
сильная.
2. Гидроксиды элементов главных подгрупп I и II группы
Периодической системы: LiOH, КОН, Са(ОНJ и г. д.,
исключение сос!авляе1 Ве(ОНJ При переходе вниз но подгруппе по мере
усиления металлических свожлв элемент сила 1идроксидов
возрастает. Гидроксиды лантаноидов шпа La(OHK— также сильные
основания.
3. Почти все соли, например NaCI, Na2SO4, Na3PO4.
Как слабые элекфолиты (а<3%), в водных растворах ведут
себя следующие соединения.
1. Органические кислоты, а также неорганические кислоты
H2S, H2SO3, HCN. H2CO3, H2SiO3. H3BO3 и др.
2. 1идроксиды ^/-элементов, например. Си(ОНJ, Сг(ОНK,
Fe(OHJ. Fe(OHK. Основные свойемва пирокеидон одного и \ою
же злемеша в разных степенях окисления усиливаются с
уменьшением степени окисления элемент. Гак. основные свойства
Fe(OHJ выражены сильнее, чем у Fc(OHK (это у1верждсние
315
равносильно iому, что кислошые свойс!ва Fe(OH)^ проявляю 1ся
сильнее, чем у Fe(OHJ).
3. Некоторые соли: ZnCI2, Znl2, CdCb. Cdb. Fe(CNSh,
Mg(CNJ, HgCl2, Hg(CNJ и др.
Между сильными и слабыми электролитами
располагается немногочисленная группа электролитов средней силы
C0%>а^З%). К средним электролитам относятся некоторые
неорганические кислоты, например, фосфорная Н3РО4.
У сильных электролитов степень диссоциации в разбавленных
растворах велика и мало зависит от концетрации paciBopa.
У слабых электролитов, наоборот, степень диссоциации даже
в сильно разбавленных растворах очень мала и уменьшается
с увеличением концентрации раствора.
При диссоциации слабых электролитов в рас i воре
обнаруживаются как продукты диссоциации — ионы, так и исходные
молекулы. Концентрация этих чааиц в расiворе определяется ciene-
нью диссоциации. В растворе слабого бинарного электролита КА
устанавливается равновесие, характеризуемое констанюй
равновесия—константой диссоциации:
КА(р-р)=К+(р-р)+А- ' '
Если в 1 л раствора растворено С моль электролит и cieneHb
диссоциации равна а, то, значит, продиссоциировало аС моль
и образовалось каждого иона пС моль. В состоянии молекул
остается С—а С моль К A. Toi да константа равновесия составит:
-, яС осС я2Г а2
К = —
С-ъС I-а (\-у)У
где V—разбавление раствора.
Разбавление—величина, обратная концентрации (V= 1/C)
и равная объему раствора (в литрах), в котором содержится
1 моль электролита типа КА.
Так как константа равновесия не зависш от концентрации, го
выведенное соотношение выражает зависимость степени
диссоциации от концентрации электролита. Это соотношение называйся
уравнением Оствальда. С помощью этого уравнения можно
рассчитать константы диссоциации слабых элект ролитов типа
СН3СООН и NH4OH. Ниже приведены ос и К для растворов
уксусной кислоты.
С. моль а V. i мо ib of. % К
7,52 10 13,3 1 57 1.85 10 5
9,37 10 3 106Л 0,44 1,85 10
1,47 Ю 6803 30.1 1,87 10 5
316
Таким образом, уменьшение копценграции слабою элекфо-
лига в расiворе приводит к росту степени диссоциации (teopein-
чески в бесконечно разбавленном растворе аспень диссоциации
равна 100%), а константа диссоциации слабого электролиз не
зависит от его концентрации (в разбавленных растворах и при
от су I с 1 вии в рас i воре других злект рол итов).
Применительно к очень слабым электролитам (ос 2:0)
уравнение Оствальда можно записать следующим образом (считая, то
1)
Откуда следуем что степень диссоциации слабых электролитов
изменяется обратно пропорционально корню квадраиюму oi их
концентрации. Если концентрацию уменьши ib в 100 раз (т.е.
разбавление увеличить в 100 раз), то степень диссоциации
возрастает в 10 раз
Поведение сильных элект роли job существенно отличается oi
поведения слабых электролитов.
Для объяснения того факта, чю сильный электролит,
находящийся даже в кристаллическом состоянии в виде ионов,
в растворе имеет степень диссоциации меньше 100%, следует
предположить наличие сильного взаимодействия между ионами
в рас i воре.
В растворе распределение ионов — промежуточное между
беспорядочным распределением молекул жидкости и их
упорядоченным распределением в кристаллической решетке. Согласно
теории сильных электролитов степень диссоциации а = 82%
означает, что, несмотря на то что все «молекулы» диссоциированы,
ионы находятся в свободном состоянии лишь на 82% (остальные
18% приходятся на межионные взаимодействия).
В приведенном выше примере определения степени
диссоциации такого сильного электролита, как NaOH, было получено
значение а, равное 82%. Вычисленное значение степени
диссоциации сильного электролита может показаться неожиданным. ?ак
как согласно современным представлениям сильные электролиты
в водных растворах полностью диссоциированы на ионы. Toi
факт, что по экспериментальным данным счепень диссоциации
сильных элекгролиiов всегда оказывается меньше 100%,
объясняется электростатическим взаимодейсiвием между ионами.
Вследс1вие этого акшвность ионов уменьшается, и все свойства
растворов, зависящие от концентрации ионов, проявляются так.
как если бы она была меньше, чем их образуется при полной
диссоциации элект роли i а.
Поэтому определяемая обычными методами степень
диссоциации сильных элекфолитов является лишь кажущейся. При
разбавлении раствора сильного электролита водой кажущаяся
317
сiснень диссоциации BO3paciaei не noiому, чю происходи! уве-
чичение сгепени диссоциации молекул (условно записываемых
в виде формчльимх единиц, например, NaOH), а вследсмвие
уменьшения среднего расстояния между ионами благодаря
электростатическому и другим взаимодействиям между
противоположно заряженными ионами.
Учитывая полную диссоциацию сильных электролитов в
растворах любых копией фаций, можно было бы ожидать, что их
элекфопроводноаь (молярная) при любых разбавлениях
должна быть равна электропроводности при бесконечном
разбавлении paeiвора. Однако этого не наблюдайся.
Электропроводное гь уменьшае!ся с ростом концентрации электролита, что
может означать усиление взаимодействия между ионами
Вследствие того, чю число ионов в объеме раствора, содержащем
I моль электролша, постоянно, концентрационная зависимость
электропроводное 1 и сильных электролитов може! бьнь
объяснена изменением скорости движения ионов между электродами.
Чем сильнее ионы взаимодежмвуют друг с другом и с
молекулами рас 1воригеля, тем меньше скорость их перемещения.
Это по-разному сказывается на скоростях прямой и обратной
реакций элек}роли!ической диссоциации, и в результате
константа равновесия изменяется в зависимости от концентрации
ионов диссоциирующего электролита и приеуплвия посторонних
ионов.
Значения степени диссоциации одного и юго же сильного
электролита могут различаться при разных меюдах ее
определения, поэтому консташы диссоциации, вычисленные из степени
диссоциации, зависят как от концентрации электролита, так и от
метода измерения а. Таким образом, константы диссоциации
силы-*мх электролитов не являются постоянными. В то же время
нет никаких экспериментальных указаний на наличие в растворе
сильного электролита молекул типа NaCl, Na2SO4 и т. п.. хотя
по значениям степени диссоциации концентрация должна быть
вполне дос ia'i очной для их обнаружения. Все эти проблемы
разрешает теория сильных электролитов.
§ 45. РАВНОВЕСИЯ В ВОДНЫХ РАСТВОРАХ СИЛЬНЫХ ЭЛЕКТРОЛИТОВ.
ИОННАЯ СИЛА И АКТИВНОСТЬ
В отличие от слабых электролиюв сильные электролиты
в растворах обнаруживают особенности в поведении, не соот-
вс!ствующие их полюй диссоциации на ионы. Так, их расчетная
концентрация в растворах оюпывасюя значительно меньше
аналитической концеш рации, определяемой при приготовлении
раствора. Кажущаяся сюпень диссоциации по данным
экспериментальных измерений меньше 100% даже в разбавленных раство-
318
pax; например, в растворах НС1 обнаруживается такая степень
диссоциации:
С моль/л 1 0,1 0,01 0.001
а, % 79 92 97 99
Электропроводность растворов сильных электролитов ниже
той, какую следует ожидать при полной диссоциации молекул
электролита.
Эти особенности свойств растворов сильных электролитов
нельзя обосновать, исходя из теории электролитической
диссоциации, и для их обьяснения были привлечены следующие
теоретические представления.
Выводы о содержании частиц в растворе, основанные на
расчетах свойств растворенных веществ из данных по давлению
насыщенно! о пара над раствором, повышению температуры
кипения и понижению температуры замерзания раствора,
предполагают, что раствор идеален. Приложение теории идеальных
растворов к реальным приводит к различного рода отклонениям,
которые в конечном счете сказываются на значении расчетных
констант равновесия, не являющихся действительными
величинами. Однако отклонения от идеального поведения не могут
объяснить столь сильного кажущегося уменьшения содержания
электролита в растворе.
Другое объяснение исходит из положения о взаимодействии
ионов с молекулами растворителя. В резулыате образования
вокруг иона любого заряда оболочки из молекул растворителя
(сольватация иона) перемещение иона в электрическом поле
замедляется, что уменьшает электропроводность раствора и
приводит к снижению степени электролитической диссоциации.
Различная степень сольватации веществ, участвующих в реакции, по-
разному изменяет скорости прямой и обратной реакций
электролитической диссоциации, что также может привести к
зависимости константы равновесия от общего содержания ионов в
растворе. Однако и эю взаимодействие не может в значительной ciene-
ни сказаться на наблюдаемом поведении сильного электролита
в растворе.
Наконец, особые свойства растворов сильных электролитов
могут быть объяснены, исходя из представления об межионном
взаимодействии, в резулыате которого вокруг каждою
отдельного иона в растворе образуется ионное облако из
противоположно заряженных ионов — ионная атмосфера. Это представление
лежи! в основе 1еории сильных электролитов
Возможность образования ионной атмосферы вытекает из
статистической теории электролитов. Распределение ионов в
растворе следует рассматривать как промежу1 очное между
беспорядочным распределением молекул в жидкости и упорядоченным
319
Рис 129 Ионная
оболочка вокруг катиона
(а) и состояние кагио-
на в электрическом
поле (б)
а
распределением чааиц в кристаплическои
peiiie i ко Ионы в рас i воре в каждый данный
момеш времени распределены не
хаотически, а в некоторой сюпени упорядоченно бла-
юдаря кулоновскому притяжению зарядов
противоположного знака В каждый момент
времени вокруг любого из ионов формиру-
егся оболочка из ионов противоположного
заряда — ионная атмосфера (рис. 129, а).
Под действием внешнего электрического
поля (при измерении электропроводности
раствора) ион частично выходит из
окружающего его ионного облака (рис. 129, б). Это
облако снова формируется в новом
положении иона (явление релаксации), по не
мгновенно (через 10~7—10~9 с). Поэтому при
движении иона в элек i рическом поле число
ионов противоположного заряда за этим ионом больше, чем
перед ним. Такая асимметрия ионной атмосферы вызывает
торможение движения иона, повышает электросопротивление рас-
1 вора и понижает его элект ропроводность. В результате по
данным электропроводности получают заниженное содержание
ионов сильного электролита в растворе и значение степени
диссоциации меньше 100%
Кроме того, асимметрия ионной атмосферы обусловливается
тем, что ионы атмосферы под действием электрического поля
стремятся переместиться к противоположно заряженному
электроду, что [акже снижает электропроводность раствора.
Влияние ионной атмосферы сказывается не только на
электропроводности, но и на скоростях реакций с участием ионов, что
приводит к непостоянству констант равновесия.
Вследствие образования ионной атмосферы к процессу
диссоциации сильных электролитов нельзя применять закон действия
масс, так как в выражение константы равновесия входят
концентрации свободных ионов, которых в реальных растворах нет.
В концентрированных растворах сильных электролитов
расстояния между ионами могут быть настолько малы, что
возможно непосредственное сближение двух ионов про i ивоположного
заряда и образование ионной пары, если кинетическая энергия
теплового движения недостаточна для преодоления сил
взаимного притяжения ионов. Ионная пара в некоторых отношениях
подобна недиссоциирующей молекуле. Содержание ионных пар
изменяется в зависимости от концепт рации электролита от
десятых долей процента до нескольких процентов в очень
концентрированных растворах.
Два иона противоположного и равного заряда, связавшись
в нейтральную ионную пару
320
не участвую! в переносе элекфичества. Ионные пары,
образующиеся из ионов разного по величине заряда
хотя и заряжены, но вносят в электропроводимость значительно
меньший вклад.
Одновременное сближение трех ионов маловероятно, и трои-
пики в основном образуются при столкновении ионной пары
с ионом; тройники могут быть и заряженными, и электрически
нейтральными:
()
К + А"- + К^=(К + А" К4)
Объединение двух ионных пар приводит к образованию
системы заряженных частиц, называемой квадруполем:
Процесс образования и диссоциации ионных групп — ассоциа-
тов — может быть описан при помощи константы равновесия.
Степень ассоциации ионов возрастает с увеличением зарядов
ионов и уменьшением их радиусов. Ассоциация зависит также от
природы растворителя: чем меньше диэлектрическая
проницаемость растворителя, тем больше степень ассоциации ионов. Так,
при добавлении в водный раствор электролита вещества с
меньшей диэлектрической проницаемостью (например, диоксана)
содержание ионных ассоциатов возрастает по мере уменьшения
диэлектрической проницаемости.
Точно установлено образование таких ассоциатов, как ВаС1 + ,
AgCl^", LiCl^T. В некоторых случаях на кулоновские силы
накладываются слабые Ван-дер-Ваальсовы силы и силы типа
водородных и донорно-акцеп горных взаимодейс1вий. При развитии до-
норно-акцепторного взаимодействия трудно провести резкую
границу между ионным ассоциатом и комплексным ионом.
Математическая модель поведения иона в растворе сильного
электролита чрезвычайно сложна. Такая модель должна
характеризовался некоторой функцией от концентрации
электролита, которая учитывает различные электростатические
взаимодействия между ионами. Эту функцию называют ионной
11 24S2 321
силой растворах се приняю обозначать буквой / (или \i).
Ионная сила расiвора определяется как полусумма произведений
концентраций С всех ионов в растворе на квадрат их заряда z:
Если в растворе содержатся тлько однозарядные ионы
бинарного электролита, ионная сила численно равна мольной
концентрации раствора (поэтому в формулу входит множитель 1/2). Так,
ионная сила 1 М раствора хлорида натрия равна:
Ионная сила 1 М раствора La2(SO4K значительно выше:
La2(SO4K = 2La
Ионная сила 0,01 М раствора La2(SO4K равна:
Таким образом, изменение концентрации электролита в п раз
приводит к изменению ионной силы раствора во столько же раз.
Если раствор содержит несколько электролитов в разных
концентрациях, то при вычислении ионной силы учитывается
вклад всех ионов в ионную силу раствора.
Слабые электролиты вносят очень незначительный вклад
в ионную силу раствора, поэтому, если они содержатся в
растворе, их обычно не учитывают при расчете ионной силы. Так,
в водном растворе хлороводородной и уксусной кислот ионная
сила раствора определяется концентрацией хлороводородной
кислоты.
Для учета межионных и межмолекулярных взаимодействий,
как бы снижающих концентрацию электролита в растворе,
введена величина, называемая активностью. Она определяется как
величина, подстановка которой вместо концентрации в
термодинамические соотношения позволяет применять их для описания
рассматриваемых систем. Активность характеризует активную
концентрацию электролита в растворе, она отражает суммарно
все эффекты взаимодействия ионов между собой и с молекулами
растворителя.
Активность — это мера реального поведения вещества в
растворе; степень отклонения поведения реальною раствора от
идеального выражается коэффициентом активности. Следует
отметить, что общепринятого способа выражения единиц
активности и коэффициента активности нет. Иногда активности
приписывают те же единицы измерения, что и концентрации,
например, моль/л.
322
Ак1ивнос1ь а иона связана с его концентрацией с через
коэффициент активности /"простой формулой:
Для водных растворов при 25° С коэффициент активности
/ иона с зарядом г зависит от ионной силы / раствора
соотношением:
Это соотношение является приближенным и дает
удовлетворительные результаты при расчете коэффициента активности
ионов в растворах с ионной силой не выше 0,01.
Рассчитаем активность ионов натрия в 0,01 М растворе NaCl:
/=i/2@,01-l2+0,01-l2)=0,01,
lg/= —0,51 • 12 • 7^0?= -0,051; /= Ю'051 -0,89,
tfNa+=CNa+ \/Na+=0,01 0,89 = 0,0089.
Таким образом, несмофя на то что аналитическая
концентрация ионов Na+ в 0,01 М растворе хлорида натрия равна
0,01 моль/л, состояние ионов в этом растворе отвечает их
активной концентрации, равной 0,0089 моль/л.
Покажем, как скажется введение в раствор другого сильного
электролита на поведении имеющихся в растворе ионов.
Рассчитаем активность ионов натрия в растворе, содержащем в 1 л
0,01 моль NaCl и 0,01 моль K2SO4:
( 4 4 )
= V2@,01 +0,01 +0,02+0,04)-0,04,
+ - -0,51 VOJ04= ~0,102;/Na + = 10-°'l02=0,79,
aNa +=0,01 0,79 = 0,0079.
Следовательно, при введении в 0,01 М раствор хлорида
натрия 0,01 моль/л K2SO4 аналитическая концентрация ионов
натрия остается прежней—0,01 моль/л, однако они ведут себя
в растворе так, будто их содержание отвечает всего 0,0079 моль/л.
Константа равновесия реакции, вычисленная по
концентрациям вещее IB, участвующих в равновесии, называется
аналитической константой равновесия; константа равновесия, вычисленная
по активности веществ, называется термодинамической
константой равновесия.
Аналитическая и термодинамическая константы равновесия
связаны между собой коэффициентами активности, входящими
в выражение закона действия масс. Действительно, для процесса
электролитической диссоциации аналитическая Кан и
термодинамическая КТ константы равновесия составят:
п* 323
[КЛ]
T aKA fKA[KA] [KA] /-KA Лан fKA '
Аналитическая константа равновесия, выраженная через
концентрации ионов, в отличие от термодинамической константы
зависит не только от природы частиц, но и ионной силы
раствора. По мере уменьшения ионной силы раствора и приближения
коэффициента активности иона к единице значения
термодинамической и аналитической констант равновесия сближаются, и для
бесконечно разбавленных растворов обе константы равны
В достаточно концентрированных растворах сильных
электролитов концентрация ионных пар становится заметной, и для
такого равновесия, как
можно записать термодинамическую константу равновесия
лт .
"Na<l
Здесь иод символом NaCl подразумевается ионная пара или
любой другой ассоциат ионов. Выражение аналитической
константы равновесия подобным образом записать нельзя, так как
в растворе нет молекул NaCl.
Ионная сила влияет на многие процессы, проходящие в
растворах сильных электролитов. В частности, ионная сила
сказывается на равновесии в системе малорастворимый электролит—
раствор, т. е. изменение величины / приводит к изменению
растворимости электролита.
Если малорастворимый электролит находится в растворе,
содержащем другие ионы с высокой их концентрацией, или же
кристаллическое вещество значительно растворимо, то вместо
концентраций следует пользоваться активностями ионов, тогда
выражение произведения раст воримости запишется в виде:
Таким образом, в насыщенном растворе электролита
произведение активностей его ионов в степени их стехиометрических
коэффициенте есть величина постоянная.
В качестве примера рассчитаем растворимость Zn(OHJ
в 0,001 М растворе СаС12. Хлорид кальция—сильный электро-
лш, он создает в рас i воре ионную силу, равную
324
Коэффициент активности ионов Zn2f и ОН" равны:
lg//ni =-~0,51-22ч/°Ьз=-0,112. //п. =0,77.
lg/OH =-0,51-1^/0003=-0,027; /Он -0,94,
Активное!и ионов Zn2 + и ОН" равны:
Подставим эти значения активностей в выражение
произведения рааворимости:
nP = tf/n,..iOH =//n> [Zn2l]/2OH [ОН"]2.
Так как растворимость Zn(OHJ численно равна
концентрации ионов Zn2+ в расi воре, то, приняв ее за .х, получаем:
/п2' / "ОН
От куда
Л' = 3/ = J/ -= ,73* 0 b моль/л.
V4//n-/'2O»l V4 °-77 °*94
Сравнение эюго значения растворимости с растворимостью
в чистой воде (х= 1,50- \0'ь моль/л) показывает, что с
повышением ионной силы раствора растворимость малорастворимого
электролита в его насыщенном растворе несколько возрастает
(солевой эффект). При повышении ионной силы раствора
коэффициенты ак[ивнос1и ионов малорас!воримого электролита
уменьшаются. Так как произведение активное г ей ионов
(термодинамическая консгаша равновесия) есть величина постоянная,
то при уменьшении коэффициентов активностей должны
увеличиваться равновесные концентрации ионов малорастворимого
электролита, т. е. растворимость его возрастет.
Ионная сила раствора изменяет не юлько термодинамические
свойства веществ и реакций в водных растворах, но и скорости
реакций, особенно органических.
§ 46. РЕАКЦИИ НЕЙТРАЛИЗАЦИИ. ВОДОРОДНЫЙ ПОКАЗАТЕЛЬ
В реакциях между сильными кислотами и основаниями
(щелочами), протекающими в растворах, всегда выделяется одно и то
же количество теплоты в расчет на I моль образующейся воды,
а именно 58 кДж/моль. В качеемве примера таких реакций
укажем следующие процессы:
325
= H2O + LiNO3
Так как во всех приведенных реакциях образуется одно общее
вещество — вода, то можно предположить, что тепловой эффект
таких реакций в растворе обусловлен образованием воды из
ионов водорода и гидроксид-ионов, находящихся в растворах
реагирующих веществ в одинаковом состоянии. Этот факт очень
важен для доказательства ионного поведения ряда кислот,
оснований и солей в водных растворах.
Все перечисленные реакции—это реакции нейтрализации
сильной кислоты сильным основанием. С учетом равенства
тепловых эффектов уравнение каждой из реакций можно записать
молекулярно-иоиным способом^ в котором сильные
электролиты указывают в виде ионов, а слабые—в виде молекул,
например:
+Li++OH"=H2O + Li++Cl; Д#°=-58 кДж.
Очевидно, одни и те же ионы, входящие в правую и левую части
уравнения реакции, можно не записывать, тогда все
перечисленные шесть реакций могут бьпь выражены одним уравнением:
Н++ОН- = Н2О; A#r=-58 кДж.
Такой способ написания реакций носит название сокращенного
молекулярно-ионного. По этому способу в химическом уравнении
в виде молекул записываются слабые электролиты, газообразные
и нерастворимые вещества и неэлектролиты. Последнее
указанное молекул ярно-ионное уравнение—это выраженное в общем
виде уравнение нейтрализации сильной кислоты сильным
основанием. Уравнение, записанное в обратном направлении,
характеризует процесс диссоциации воды на ионы:
Н2О = Н++ОН"; Д#> = 58 кДж.
Ниже приведены уравнения реакций нейтрализации слабой
кислоты сильным основанием, сильной кислоты слабым
основанием и слабой кислоты слабым основанием, записанные
молекулярным и сокращенным молекулярно-ионным способами:
1) CH3COOH + NaOH = CH3COONa + H2O
СН3СООН + ОН" =СН3СОО" +Н2О.
326
2)
3) CH3COOH + NH4OH = NH4CH3COO + H2O
CH3COOH + NH4OH = NH4H+CH3COO-+H2O.
Реакции нейтрализации широко применяют в аналитической
химии для определения содержания в растворах кислот и
оснований, а также солей.
В аналитической практике растворы электролитов часто
выражают не молекулярными, а эквивалентными концентрациями.
Понятие эквивалента можно распространить и на сложные
соединения — кислоты, основания и соли. Эквивалентом
называется некоторая реальная или условная химическая частица, которая
может присоединять или выделять один ион водорода в кислотно-
основных реакциях (или один электрон в
окислительно-восстановительных реакциях). Единица количества эквивалента — моль.
Масса 1 моль эквивалентов—это молярная масса эквивалента.
При использовании термина «эквивалент» следует указывать
реакцию, к коюрой он относится. Например, молярная масса
эквивалента H2SO4 в реакции
сос!авляет 98 г, а в реакции
H2SO4 + 2NaOH=Na2SO4
равна массе {/2 моль H2SO4, т. е. 98/2 = 49 г.
Отношение эквивалентного количества растворенного
вещества к объему раствора ecib эквивалентная концентрация; она
измеряется в моль/л. Величину, численно равную эквивалентной
концентрации, принято называть нормальностью раствора. В
расчетных формулах нормальность обычно обозначают символом
N, а значение ее указывают числом, равным эквивалентной
концентрации (в моль/л), с последующей буквой «н.» или словом
«нормальный». Так, раствор серной кислоты, содержащий 0,49 г
H2SO4 в 1 л, буде1 децинормальным, или нормальность его
равна 0,1 н., а раствор, содержащий 0,049 г,— сантинормальным
или с нормальностью 0,01 н. (только для реакции, в которой
реагируют два иона водорода!). Нормальностью рекомендуется
выражать состав растворов, если отношение молярноеги
раствора к нормальности меньше единицы; в дру1их случаях cociaB
раствора выражают через молярную концентрацию.
Растворы одинаковой нормальности реагируют в равных
объемах, так как одинаковые объемы содержат равное число
эквивалентов растворенного вещества. Из этого положения
следует , что, если для нейтрализации раствора основания объемом
Vx требуется раствор кислоты объемом К2, то в момент
нейтрализации справедливо соо пюшение:
327
где Nx и Л'2— нормальное и» соогвсгс1всино рас i воров кислоты
и основания.
Записав выражение в виде
приходим к выводу, чю в момент нейтрализации объемы
растворов прореагировавших веществ обратно пропорциональны их
нормалыюстям. Это соотношение лежи! в основе
количественных расчетов по результатам титриметрического анализа.
Для проведения титримегрического анализа используются
различные реакции. Широкое применение в аналитической
практике получили методы кислотно-основного титрования,
основанные на реакциях нейтрализации.
Сущность титриметрического анализа состоит в следующем.
Допустим, чю имеется раствор, содержащий неизвестное количество
гидроксида натрия. Для определения его применяют прием,
называемый титрованием* коюрый заключается в том. чю к точно
измеренному объему исследуемого титруемого раствора (в данном
примере—основания) приливают малыми порциями раствор реактива
(кислоты) точно известной концентрации—титрант—и измеряют
объем этого раствора, пошедший на реакцию с определяемым
веществом (основанием). Затем вычисляют искомую концентрацию.
Момент, когда анализируемое вещество прореагировало со
строго эквивалентным количеством титранта, называется точкой
эквивалентности. При титровании растворов щелочи кислотой
в точке эквивалентности щелочная реакция раствора переходит
в нейтральную или даже кислую (от прибавления одной
последней капли кислоты). Этот переход можно установить
различными способами, в частности, с помощью
индикатора—вещества, изменяющего окраску при изменении реакции раствора.
Момент наблюдаемого изменения окраски раствора называется
конечной точкой титрования', эта точка обычно не совсем точно
совпадает с точкой эквивалентности.
При титровании сильной кисло 1ы сильным основанием в
точке эквивалентности реакция раствора нейтральная. Нейтраль-
ность раствора не означает полного отсутствия ионов водорода
и гидроксид-ионов в результате образования молекул воды, а
отражает равенс!во концентраций этих ионов Си* = Соп . Если
концентрация ионов водорода в растворе превышает
концентрацию гидроксид-ионов, Сн * > Сон , то говоря i, что раствор
имеет кислую реакцию, и наоборот, если концентрация
гидроксид-ионов выше концентрации ионов водорода, Сон >СН«, то
раствор проявляет щелочную (основную) реакцию.
В водных растворах концентрации ионов Н * и ОН"
взаимосвязаны выражением конеiанты диссоциации воды
328
Н2О-Н++ОН
Так как [Н2О]= 1000/18,02 = 55,49 моль/л, то произведение ионов
Hf и ОН"—постоянная для данной температуры величина,
которая называется ионным произведением воды Кп:
Кв = К[Н2О] = 1,8 -КГ16- 55,49- МО4.
Следовательно,
Это произведение, согласно закону действия масс являясь
константой, не зависит от концентраций ионов водорода и гидрок-
сид-ионов и постоянно при данной температуре.
В чистой воде или нейтральном растворе
[Н + ] = [ОН~] или [Н + ]2=1 • 104.
Откуда
[Н + ]=1 -КГ7 моль/л; [ОН"]=1 -КГ7 моль/л.
В кислой среде
[Н + ]> 1 • 10~7 моль/л и соответственно [ОН~]< 1 • 10 ~7 моль/л.
В щелочной среде
[Н + ]< 1 • 10 моль/л и соответственно [ОН~]> 1 • 10~7 моль/л.
Для удобства обычно пользуются не концентрациями [Н+ ]
и [ОН ~ ], а отрицательным логарифмом концентрации,
обозначаемым символами рН и рОН:
pH=-lg[H + ]; pOH=-lg[OH"]
Величина, обозначаемая рН, называется водородным
показателем.
Так как [Н+ ] [ОН" ]= 1 • 10~14 и Ig [H+ ] + lg [ОН" ]= -14, го
для водных растворов выполняется соотношение
Для нейтральных растворов рН = 7, для кислых растворов
рН < 7 и для щелочных растворов рН > 7. Изменение рН на
единицу соответствует изменению концентрации водородных
ионов в десять раз.
Приведем несколько примеров вычисления рН по
концентрации ионов водорода и, наоборот, расчета концентрации ионов
водорода по известному значению рН.
В растворах сильных кислот и оснований типа НС1, HNO3,
КОН, NaOH молярная концентрация ионов водорода и гидро-
ксид-ионов совпадает с молярной концентрацией кислоты или
щелочи. Поэтому в 0,01 М растворе НС1 концентрация ионов
водорода составляет [Н+ ] = 0,01 моль/л и рН = -lg0,01 = 2.
329
В 0,01 М растворе КОН концентрация гидроксид-ионов равна
[ОН~ ]=0,01 моль/л,значит, рОН = -Ig0,01 =2 и рН = 14-рОН = 12.
Если рН = 12,4, то раствор имеет щелочную реакцию.
Концентрации ионов водорода и гидроксид-иоиов в эюм растворе
(-lg [Н+ ]= 12,4) будут равны:
[Hf]-102'4-10°-6-103 = 3,98 403 моль/л,
[ОН~] == 10" |4+12'4= 10"Ь6 = 0,025 моль/л.
В аналижческой практике редко приходится работать с
распорами, рН которых выходит из интервала 0—14. Так, если
[Н ]=1 моль/л, то рН=0; мри [Н + ]= 10 моль/л рН достигает
значения—1. В щелочном растворе с [ОН"]=1 моль/л рОН = 0
и рН = 14; при [ОН" ]= 10 моль/л рОН= -1 и рН = 14-рОН = 15.
При обсуждении величины рН раствора использовалось выражение «ион
водорода» с изображением ci о символом IP. Однако 1аких ионов в растворе нет в связи с их
гидратацией В водном растворе протон прочно гидратирусгся молекулой воды
с образованием иона гидроксония Н3О+ Ионы гидроксония и гидроксид-ионы
гидра тированы несколькими молекулами воды, но связь их с центральными ионами
Н3О+ или ОН менее прочная, поэюму обычно ионы Н3О+ и ОН" не записывают
в гидратированной форме Правильнее в ионных реакциях писать ионы водорода
в виде Н3О* Для просто гы здесь эта форма не используется, к тому же ото
совершенно не сказывается на результатах расчетов концентраций ионов и рН растворов
При расчетах рН использование аналитических концентраций
допустимо только для очень разбавленных растворов, в которых
активное!и практически равны концентрациям ионов. Покажем,
как влияет на расчетное значение рН переход к активности ионов
водорода. В 0,01 М растворе НС1 [Н ]=0,01 моль/л и рН = 2.
Ионная сила лого раствора равна 0,01; коэффициент активности
ионов водорода определяется из соотношения:
-1g /= -0,51 • lVfr0I = -0,051; /=0,89
откуда ян+ =0,01 -0,89 = 0,0089 и рН= -lg 0,0089 = 2,05.
Таким образом, расчет по активноеiям приводит к несколько
большим значениям рН, т. е. реальный раствор оказывается менее
кислотным, чем это отвечает аналитической концентрации ионов
водорода. Добавление различных солей приводит к увеличению
ионной силы раствора, уменьшению коэффициента активности и
активности ионов водорода и большему значению рН среды раствора.
Существуют различные методы определения концентрации
(точнее — активное!и) ионов водорода (и coo\ветсiветшо
концентрации гидроксид-ионов). Один из простейших методов основан
на использовании кислошо-основных индикаторов. В качестве
таких индикаторов могут служить многие органические кислоты
и основания, которые резко изменяют свою окраску в
определенном узком интервале рН. Так, фенолфталеин представляет собой
слабую кислоту, которая в молекулярной форме бесцветна, а ее
анионы принимают красно-фиолетовую окраску.
330
При уменьшении концентрации ионов Н+ и увеличении
концентрации ионов ОН" положение равновесия смещается вправо
и в растворе образуются красно-фиолетовые анионы. При
увеличении копцен!рации ионов Н^ положение равновесия смещается
влево, в область существования молекулярной формы
индикатора, не имеющей окраски.
Кислотной формой индикатора называют форму, которая
преобладает в кислотных растворах, а основной формой—ту,
которая существует в щелочных расiворах. В некотором промежутке
значений рН в растворе может одновременно находи 1ься в
равновесии некоторое количество обеих форм индикатора,
вследствие чего возникает переходная окраска индикатора. Область
значений рН, в которой совершается переход одной формы
индикатора в другую и отмечается изменение окраски
индикатора,— это интервал рН перехода окраски индикатора, или, как
принято называть в аналитической химии, интервал перехода
индикатора. Обычно стараются подобрал ь индикатор по
возможности с узким интервалом (не более двух единиц рН).
В табл. 20 показаны интервалы перехода окраски важнейших
кислотно-основных индикаторов.
Таблица 20
Важнейшие кислотно-основные индикаторы
Индикатор
Метиловый фиолетовый
Тимоловым синий
2,6 Дикитрофенол
2,4-Динитрофеноя
Метиловый оранжевый
Бромфеноловы* синий
Ализариновым красный
Метииовый красный
Лакмус
п-Нитрофенол
Феноловый красный
Фенолфталеин
Тимояфталеии
Ализариновый жеятын
Индиго карминовый
Тринитробеиэол
-ioi;
It/)
желтая 1?
I
|
красная 11,2-2,8
рН
5 4 5 6 7 8 9 Ю 11 12 13 14 15
1 1 1 1 1 1 1 1 1 1 1 1
фиолетовая
желтая 18,0-9 61 синяя
бесцветная 2,4-4,о| «м™
бесцветная [2,8-4,4 желтая
красная
желтая
3,0-4,4 желто-оранжевая
3,0-4,61 синяя
жеята* 4 0-5 8 фиолетовая
красная 14,3-6,2 | желтая
красная | 5,0-8 0 [ синяя
бесцветная 5 6-7 5 I желтая
желтая 16,8-8,4 красная
бесцветная | 8,1-9,8 | красная
бесцветная 9,4-10,5 синяя
бледно-желтая |i 0,0-12,01 желто кормневая
синяя | 11,6-14,0
бесцветная
12 2-14,0
желтая
ораиже
вал
331
Метод определения рН растворов с помощью индикаторов состоит в юм.
что последовательно фиксируют изменение окраски нескольких индикаторов
в отдельных пробах раствора. рН которого требуется установить Испытание
следует начинать с индикатора, имеющего интервал рН перехода окраски в
нейтральной среде. Предположим, л-нитрофенол при введении нескольких ею капель
к 2—3 мл исследуемого раствора показал желтую окраску Это означает, что
рН5*7,5 (см табл 20) Загсм проводят испытание с помощью индикатора,
имеющего интервал рН перехода окраски в щелочной среде Если фенолфталеин
принимает в пробе раствора красную окраску, значит, рН^9,8 Далее переходят
к индикатору, имеющему интервал рН перехода в еще более сильной щелочной
среде В качестве гакою индикатора можно взягь ализариновый жс;имй, если он
принимает бледно-желгую окраску в пробе, и из этою следует, что рН^Ю
Сопоставляя зто показание с результатом предыдущего испытания раствора.
заключаем, что рН^Ю. Точность определения рН этим методом невысока и
составляет примерно ±0,3 единицы рН.
Установление значения рН, при ко юром наступает
нейтрализация кислоты и основания, используется для определения их
концентраций. Рассмотрим в качестве примера определение
содержания в растворе хлороводородной кислоты. Такое
определение проводят методом кислотно-основного титрования (см.
выше). Анализ обычно проводят, приливая по каплям из бюретки
раствор гидроксида калия известной концентрации (гитрант)
к пробе анализируемого раствора, взятой с точно отмеренным
объемом, до насмупления точки эквивалентности.
В ходе титрования по мере прибавления раствора щелочи рН
титруемого раствора непрерывно изменяется. Если известны
количества (объем) прибавленного раствора в отдельные моменты
титрования и соответствующие значения рН титруемого
раствора, то можно установить зависимость рН от объема раствора,
расходуемого на нейтрализацию. Графическое изображение такой
зависимости называется кривой титрования, она показывае! ход
изменения рН при титровании.
На рис. 130 приведены кривые титрования раствора НС1
раствором КОН. Проанализируем сначала ход изменения рН при
титровании 0,1 М раствора НС1 0,1 М раствором КОН
(сплошная линия на рис. 130).
До начала титрования значение рН 0,1 М раствора НС1 равно
1. В начальные моменты титрования, когда в анализируемом
растворе содержится еще много кисло 1Ы, добавляемая щелочь
почти не изменяет рН раствора. По мере введения в титруемый
раствор все большего количества щелочи рН изменяется более
резко.
После прибавления 9 мл раствора КОН концентрация
кислоты уменьшится в 10 раз и станет равной 0,01 моль/л, рН
полученного раствора составит 2. Чтобы уменьшить
концентрацию кислоты в рас 1 воре еще в 10 раз, т.е. чтобы рН = 3,
требуется добавить только 0,9 мл щелочи. После добавления
еще 0,09 мл раствора щелочи рН станет равным 4 и т. д.
Когда будет введено в раствор кислоты точно 10 мл раствора ще-
332
рН
/2
W
&
6
Ч
2
п
-
. Точка нейтральности*
и эквивалентности
1
- .у
- _у
\ 1 1 1
^ "Красная форма Ю,0
/ Интервал перехода
' фенолфталеина ^ ?
Бесцветная форма
Желтая форма 4,4
Интервал перехода
метилового оранжевого
Розовая форма J,/
1 1 1 1 1
8
П 16 Кон,мл
лочи, будет достигнув точка
эквивалентности, рН = 7. При
дальнейшем добавлении щелочи в рас-
твор рН резко возрастает и
раствор показывает основную
реакцию.
Рассмотренный ход изменения
рН на кривой титрования
отражается фемя участками: начальным
почт горизонтальным участком,
поч 1 и вертикальным отрезком
{скачок рН, или скачок
титрования) и вторым почти
горизонтальным участком.
Как видно из рис. 130, кривая
титрования 0,1 М раствора НС1
0,1 М раствором КОН
поднимается вертикально в интервале
рН от 3 до 11. Поэтому
титрование можно проводить с
любым индикаюром, изменяющим
окраску в этом интервале рН.
Наиболее пригодны для титрования сильной кислоты
сильным основанием (и наоборот) фенолфталеин и метиловый
оранжевый.
Чем разбавленнее растворы кислоты и основания, тем меньше
скачок рН. Так, на кривой титрования 0,001 М раствора участок
вертикального подъема кривой только частично попадает
в интервал рН перехода окраски метилового оранжевого и
фенолфталеина. Если титровать 0,0001 М растворы, то скачок
титрования приходится на интервал рН 6—8, он не буде г
фиксироваться указанными индикаторами. Таким образом, для титрования
следует подбирать такой индикатор, для которого интервал рН
перехода окраски совпадает со скачком титрования (его
устанавливают расчетом).
Рис 130 Кривые титрования 0,1
М раствора НС1 0.1 М раствором
КОН (сплошная -пиния) и 0,001
М раствора НС1 0.001 М раствором
КОМ (пунктирная линия)
§ 47. ИОННЫЕ РАВНОВЕСИЯ В РАСТВОРАХ СЛАБЫХ КИСЛОТ
И СЛАБЫХ ОСНОВАНИЙ
Метод расчета концентрации ионов водорода и гидроксид-
ионов в растворах слабых кислот и слабых оснований несколько
сложнее, чем метод расчета для сильных электролитов, и
проводится с использованием констант их диссоциации. В табл. 21
приведены константы диссоциации некоторых слабых
электролитов.
333
Таблица 21
Коне ran гы диссоциации слабых кислот и слабых оснований
О iCKipo.mi
Азслиаая кислота HNO2
Борная кислота Н3ВО3
Вода Н2О
Тидроксид аммония NH4OH
Кремниевая кислота H2SiO3
Муравьиная кислота НСООН
Орюфосфорная кисло i а Н3РО4
Пероксид водорода Н2О2
Сернистая кислота H2SO3
Угольная кислота Н2СО3
Уксусная кислота СН3СООН
Фтороводородная кислота HF
Циановодородная кислота HCN
Щавелевая кисло г а Н2С2О4
С 1>меш>
лиссоки.щии
—
—
—
I
II
—
1
II
III
—
I
И
I
11
—
—
—
I
II
к.
4 10
5,7 100
1,8 10 16
(Кв=1 10 14)
1,79 10
3,2 10 10
1,6-10 12
1.77 10 *
7,51-10 3
6,23 10~8
2,20 10 13
2,4 102
1,30-10
1,20 10" м
4,31 10 7
5,61 10"м
1,86 10 *
7,40 10 4
7,20-100
5,90 10
6,40-10"s
Применим закон действия масс к диссоциации слабой
кислоты, например, уксусной, и запишем выражение константы ее
диссоциации Ккпсл:
сн3соон=сн3соо +ЬГ; л:кисл = [ "^ еоон
Учитывая, что [СН3СОО~] = [Н+] и что уксусная кислота —
слабая кислота, поэтому в состоянии диссоциации находится
малая часть ее молекул, и концентрацию молекул СН3СООН
можно считать равной общей концентрации кислоты Скисл,
получим:
Ккисл = [Н+]2/^
Например, концентрация ионов водорода в 0,1 М и 0,01 М
растворах уксусной кисло 1Ы и его рН равны:
для 0,1 М раствора
'] = N/l,86-100,l = l,36-10~3 моль/л
pH = -lg(l,36-10-3) = 2.87.
334
или для 0,01 М раствора
86-1-0,01 =4,31 * Ю 4 моль/л
pH--lgD,3110~4)-3,36.
Подобным образом рассчитывается рН растворов слабых
оснований, например, гидроксида аммония NH4OH.
(Существование в водном рас 1 воре молекул NH4OH ставится под сомнение,
и считается, что аммиак в водном растворе находится в виде
гидрата NH3*H2O. Однако для простоты будем пользоваться
формулой NH4OH.) В соответствии с законом действия масс
выражение константы диссоциации гидроксида аммония Косп
записывается так:
; C-.
В растворе слабого основания типа NH4OH [NH4+ ] — [ОН" ],
и концентрацию непродиссоциировавших молекул NH4OH
можно считать равной концентрации основания, i. e.
[NH4OH] = COCH. Поэтому
^och^Loh ] /сосн.
Откуда
и далее
рН = 14-рОН
Например, рН 0,01 М раствора гидроксида аммония
составляет 8,25.
Если в растворе сильной кислоты или сильного основания
увеличить концентрацию одноименного иона введением
соответствующей соли, например, в раствор НС1 или NaOH добавить
хлорид натрия, то концентрация ионов водорода и рН
изменяются только в результате изменения активности ионов водорода
при возрастании ионной силы раствора. Если такую же операцию
провести с раствором слабой кислоты или слабого основания, то
наблюдается резкое изменение рН раст вора
Проиллюстрируем эти зависимости на следующих примерах.
В первом примере покажем, как измени 1ся рН 0,01 М раствора
НС1 при внесении в него 0,01 моль/л КО. Для этого сначала
рассчитаем рН 0,01 М раствора НС1 с учетом ионной силы.
Ионная сила этого раствора составляв i:
Коэффициент активности и активность однозарядного иона Н4
в растворе равны:
315
lg /;,. = -0,5 • 12 ,//= -0,05; /,,. = 10" °-05 =0,89
aH+=fH+CH -0,89-0,01=0,0089.
Следова гелыю, значение рН 0.01 M раствора НС1 (с учетом
ионной силы) буде! равно:
Таким образом, расчет рН раствора с учетом ионной силы
приводит к несколько большему значению рН (без учета ионной
силы для 0,01 М раствора НС1 рН = 2).
Теперь рассчитаем рН 0,01 М раствора НО, содержащего
0,01 моль/л КС1. Ионная сила раствора составит
/= j2(Сц -z н* +? а 'z а +f'K' '- к* +Cci 'z а ) =
= 11г @,01 +0,01 +0,01 +0.01) = 0,0.2
Тогда коэффициент активности и активность ионов Н+ в
растворе составят:
lg/-H. =-0,5-12V/O^2=-0,071; /н~ = Ш °-O71=0,85;
а„*= 0,01-0,85 = 0,0085.
ОI сюда значение рН 0,01 М раствора НС1 и КС1 (с учетом
ионной силы) будет равно:
рН = - lg ан > = - lg 0,0085 = 2,07.
Таким образом, введение 0,01 моль/л КС1 в 0,01 М раствор
НС1 приводи! к увеличению рН на 0,02 единицы, т. е. введение
сильного электролита снижае1 активную концентрацию ионов
водорода и тем самым смещает реакцию раствора в щелочную
область.
Во втором примере рассмотрим, как изменится рН раствора
слабой кислоты, например, уксусной СН3СООН, при внесении
в нее ацетата натрия (введение одноименных ионов СН3СОО~).
Согласно принципу Ле Шателье положение равновесия
сн ,соо
СН3СООН=СН3СОО~+Н +
сместится влево в результате увеличения концентрации ионов
СН3СОО~, образующихся при полной диссоциации ацетата
натрия как сильного электролита. Такое смещение равновесия
диссоциации уксусной кислоты означает уменьшение концентрации
ионов водорода, г. е. увеличение рН раствора.
Рассчитаем рН 0,01 М раствора уксусной кислоты,
содержащего 0,01 моль/л ацетата натрия. В выражении константы
равновесия
336
_[СН,СЧЮ-]ЦГ]_ 5
™м [сн.сооп] ' ° и
концентрация ацетат-ионов определяется в основном
концентрацией хорошо диссоциирующей соли NaCH3COO (Cc). Поэюму
можно записать
_с\Гн<]
Л кисл ^
Из этого соотношения находим концентрацию ионов водорода:
[Н+] ^—Г—К86 '°'
= 1,86- IP"s моль/л.
Отсюда рН=4,73.
Таким образом, в резулыате введения в 1 л 0,01 М раствора
уксусной кислоты 0,01 моль NaCH3COO концентрация ионов
водорода уменьшилась в 4,32* 10 4/A,86-10 ~5) = 23 раза, а
значение рН возросло на D,73 —3,36)== 1,37 единицы
D,32* 10"* моль/л и 3,36—соответственно концентрация ионов
водорода в 0,01 М растворе СН3СООН и ею рН, см. выше).
Аналогично при введении в paciBop слабого основания
NH4OH раствора хлорида аммония положение равновесия
диссоциации NH4OH смещается в менее основную область и среда
становится более кислой:
Nll4*
NH4OH = NH4++OH .
Следовательно, одноименный ион (за исключением ионов Н +
и ОН"), введенный в расiвор слабой кислоты или слабого
основания, изменяет рН раствора таким образом, что среда
приближается к нейтральной. Одноименные ионы в 1акого типа
системах ведут себя как нейтрализующие агенты: анион нешра-
лизует слабую кислоту, выполняя роль основания, а катион
нейтрализует слабое основание, выполняя роль кислоты.
При смешивании равных объемов растворов сильных кислот
и оснований одинаковой нормальности образующийся раствор
будет нейтральным. Если же к раствору слабой уксусной кислоты
прилить эквивалентное количество раствора NaOH, т. е. ионов
ОН", то раствор станет щелочным. Коне i ант а равновесия этой
реакции ней грализации имеет вид:
СНзСООН + ОН-=СН3СОО-+Н2О; /Г
Учитывая, что концентрация воды в водном растворе пракшче-
ски остается постоянной, перенесем ее в левую часть выражения
и получим новую консташу равновесия процесса нейтрализации:
337
[СНзСОО-]
[СН3СООН][О1Г]'
Умножим числитель и знаменатель на [Н+ ]:
[сн3соо-][н*]
[сн3соон][он ][
Поскольку [Н+] [ОН"]—это ионное произведение воды Къ,
получаем новое выражение константы равновесия нейтрализации
слабой кислоты сильным основанием:
'10~5-=1,69-109.
Таким образом, константа равновесия нейтрализации слабой
кислоты сильным основанием определяется только отношением
константы диссоциации слабой кислоты к ионному
произведению воды. Для реакции полученное столь высокое значение
константы равновесия нейтрализации уксусной кислоты говорит
о том, что раствор после нейтрализации содержит соль
NaCH3COO—сильный электролит, находящийся в состоянии
ионов СН3СОО~ и Na+; мольная концентрация ионов СН3СОО~
равна исходной концентрации уксусной кислоты.
При смешивании эквивалентных количеств СН3СООН
и NaOH в растворе остаются равные количества непрореагирова-
вших кислоты и щелочи [СН3СООН]= [ОНГ ], поскольку
СН3СООН—слабая кислота и ненродиссоциированная ее часть
не вступает в реакцию с ионами ОН~.
Рассмотренные особенности ионного равновесия в растворах
слабых кислот и оснований определяют специфику их
титрования.
Титрование слабых кислот и оснований отличается от
титрования сильных кислот и оснований. При эквивалентном
количестве слабой кислоты и щелочи раствор будет щелочным,
нейтральная реакция титруемого раствора слабой кислоты достигается
при меньшем (по сравнению с эквивалентным) количестве
добавленной щелочи. При титровании сильных кислот и оснований
точки нейтральности и эквивалентности раствора совпадают.
При титровании же слабых кислот и оснований они различаются,
и следует фиксировать не точку нейтральности раствора, а точку
его эквивалентности (когда завершается взаимодействие обоих
веществ в эквивалентных количествах).
При титровании уксусной кислоты раствором щелочи следует
пользоваться индикатором, изменяющим окраску при изменении
реакции раствора из слабощелочной в сильнощелочную (около
точки эквивалентности последняя капля раствора щелочи
изменяет реакцию раствора не из кислой через нейтральную в щелоч-
338
Красна* форма
Точка ^Интервал перехода
эквивалентностиt фенолфталеина
в.з
I Бесцветная форма
J Точка нейтральности
Желтая форма Ц-у
Интервал перехода
метилового оранжевого
/ Розовая форма
ную, как при титровании кислоты
НС1, а из слабощелочной в более
щелочную). Таким индикатором
при титровании уксусной кислоты
раствором щелочи может
служить фенолфталеин, а при гифо-
вании раствора аммиака
хлороводородной кислотой — метиловый
оранжевый.
На рис. 131 приведена кривая
титрования слабой кислоты
СН3СООН сильным основанием.
Ход изменения рН согласно этой
кривой иной, чем по кривой
титрования сильной кислоты
сильным основанием.
Опитровать слабую кислоту
слабым основанием обычным
прямым методом невозможно,
так как кривая тифовапия не
обнаруживает резкого скачка рН
и плавно проходит точку
эквивалентности при значении рН, зависящем от соотношения констант
диссоциации кисло 1Ы и основания.
3,1
Ч- в 12 1БУ,ю,ш
Рис. 131. Кривая титрования 0,1
М раствора СН3СООН 0,1 М
раствором КОН (сплошная линия).
Для сравнения пунктиром показана начальная
ВСМВЬ КрИВОЙ ТИфОВаПИЯ СИ 1ЫГОЙ КИСЛОТЫ
сильным основанием в тех же концентрациях
§ 48, ГИДРОЛИЗ. РЕАКЦИИ АМФОТЕРНЫХ СОЕДИНЕНИЙ
Водные растворы солей в зависимости от природы
образующих их кислот и оснований могут бьпь кислыми, щелочными
и нейтральными. Так, раствор хлорида аммония имеет кислую
реакцию, раствор ацетата натрия — щелочную, а растворы
ацетата аммония и хлорида натрия почти нейтральны.
Кислотносгь или основность водных растворов солей
объясняется протеканием в них реакции гидролиза. В широком
понимании гидролиз—это любое взаимодействие веществ с
водой; более конкретно гидролиз можно определить как
реакцию соли с водой, приводящую к образованию кислоты и
основания. Таким образом, гидролиз—это процесс, обратный
нейтрализации (реакции между кислотой и основанием с
выделением воды), а константа гидролиза записывается
выражением, обратным выражению константы равновесия
нейтрализации.
Так как большинство солей — сильные электролиты и
находятся в водном растворе в виде ионов, m уравнения реакции
гидролиза можно записать сокращенным ионным способом как
реакцию между ионами и молекулами воды.
339
Характер протекания гидролиза, г. е. природа продуктов
реакции и реакция получающегося расiвора, зависти от сочекшия
силы кислоты и силы основания, образующих соль, поэтому
возможны четыре варианта гидролиза солей.
1. Соль образована слабой кислотой и сильным основанием,
например, NaCH3COO, Na2CO3, NaCN, NaNO2, Na2SO3.
Рассмотрим гидролиз таких солей на примере ацетата натрия. В
воде эта соль находится в состоянии полной диссоциации
NaCH3COOH = Na++CH3COCr.
С водой возможна реакция лишь того иона, который с одним из
ионов воды, Н+ или ОН", дает слабый электролит. Поэтму
взаимодействие иона Na4 с водой неосуществимо (так как
образовавшийся бы при этом NaOH являлся сильным
электролитом), а протекает реакция между ацетаг-ионами и водой с
выделением слабой уксусной кислоты:
СН3СОО +Н2О = СН3СООН + ОН".
Образующиеся гидроксид-ионы обусловливают щелочную
среду раствора, рН > 7.
Данный процесс можно рассматривать как реакцию,
протекающую в направлении, противоположном реакции нейтрализации
слабой уксусной кислоты:
СН3СООН+ОН=СН3СОО +Н2О.
Применяя закон действия масс к данному гидролитическому
равновесию с учетом, что концентрация воды в достаточно
разбавленном растворе практически не изменяется, запишем
выражение константы гидролиза ацетата натрия (ацетат-иона):
к _[снзсоон][он-]
г " [СНСОО-] '
Умножив числитель и знаменатель на [Н+ ], получим:
_Гсн3соон1[он-][нЧ_ кв _ 1 ш^ =5,о.ш-ю
[СН3СОО-][!Г] ККИС1 K86I0-5" ' '
где Ккксл— константа диссоциации слабой кислоты.
Следовательно, чем меньше константа диссоциации кислоты
^кисл> гем больше константа гидролиза Кг, тем сильнее
положение равновесия реакции смещено вправо и тем выше щелочность
раствора, т. е. больше значение рН.
Рассмотрим метод расчета концентрации гидроксид-ионов
и рН водных растворов солей типа ацетата натрия. Записанное
выше выражение константы гидролиза можно упростиib,
полагая, что мольная концентрация ацетат-ионов в растворе равна
исходной мольной концентрации соли [СН3СОО~ ] — Сс
340
(NaCH3COO полнойьк> расиадае!ся на ионы, концентрация
которых практически равна концентрации соли из-за малой
константы гидролиза). Мольная концентрация образовавшейся
уксусной кислоты равна мольной концентрации гидроксид-ионов
[СН*СООН]= [ОН"] в соответствии с уравнением реакции
гидролиза. Поэтому можно записать:
/гв _ [он ]2
Л. j- — ~" .
Откуда получаем:
[он ¦]=
Например, рН 0,01 М раствора ацетата натрия равен 8,37.
Следует обратить внимание на то, что ранее для расчета
концентрации гидроксид-ионов в точке эквивалентности при
титровании уксусной кислоты тцелочью была выведена та же самая
формула, связывающая константу диссоциации слабой кислоты,
концентрацию ее аниона или соли, ионное произведение воды
с концентрацией гидроксид-ионов. Оказывается, что рН 0,01 М
раствора NaCH3COO и рН раствора, полученного прибавлением
к 0.01 М СН3СООН эквивалентного количества щелочи,
одинаковы.
2. Соль образована сильной кислотой и слабым основанием,
например, NH4C1, (NH4JSO4, NH4NO3. Гидролиз таких солей
разберем на примере хлорида аммония. Эта соль в воде
находится в состоянии полной диссоциации:
Гидролиз иона С1 невозможен, так как он привел бы к
образованию сильной кислоты НС1. При гидролизе иона NH4+
получается слабый электролит —гидроксид аммония:
NH44 +H2O = NH4OH + H\
Ионы водорода обусловливают кислотную реакцию раствора.
Найдем зависимость, связывающую константу гидролиза
с константой диссоциации слабого основания и с концентрацией
ионов водорода
Применяя закон действия масс к рассматриваемому
гидролитическому равновесию и учитывая, что концентрация воды
постоянна, запишем выражение константы гидролиза хлорида
аммония:
_[NIUOH][HJ
[NIVT '
341
Умножив числитель и знаменатель на [ОН ], получим
Кг= [NHTJprl = *«/
где Косн—константа диссоциации слабого основания.
Очевидно, что чем меньше константа диссоциации слабого
основания, образующегося в результате гидролиза, тем выше
константа гидролиза и равновесие гидролиза сильнее смещено
вправо, тем больше кислотность раствора, т, е. меньше рН.
Найдем соотношение между константой диссоциации слабого
основания, копцен грацией соли и [Н+] или рН. Принимая, что
[NH4OH]^ rJP7 ч ооотве!ствии с уравнением гидролиза
и [NH4+] —Сс (гидролизу подвергается малая часть
присутствующих в растворе катионов благодаря невысокой константе
диссоциации основания), приведенное выше выражение константы
гидролиза можно записать в виде:
г % г*
14 оси *- с
Откуда находим:
Эта формула пригодна также для вычисления концентрации
ионов водорода и рН раствора в точке эквивалентности при
титровании слабого основания сильной кислотой (вместо Сс
следует подставить нормальность растворов кислоты и основания).
При титровании раствора слабого основания сильной кислотой
реакция раствора в точке эквивалентности будет не нейтральной,
а кислотной, что объясняется гидролизом катиона слабого
основания, например, рН 0,01 М раствора NH4C1 составит 5,63.
3. Соль образована слабым основанием и слабой кислотой,
например, NH4CH3COO, NH4HCO3, NH4HSO3, NH4NO2.
Гидролиз такой соли рассмотрим на примере ацетата аммония. Эта
соль в воде диссоциирует по уравнению:
= NH4++CH3COO".
Оба иона NH4+ и СН3СОО~ с водой образуют слабые
электролиты— гидроксид аммония и уксусную кислоту:
NH4++H2O =
СН3СОО"+Н2О =
или в суммарном виде
342
В связи с !ем, что консташм диссоциации СН3СООН и NH4OH
примерно одинаковы, копией фации ионов ОН" и Н+ гакже
равны, поэтому раствор аце!ата аммония почти ней[ральный.
В общем случае при гидролизе соли, образованной слабым
основанием и слабой кислоiой, могут получаться нейiральный,
кислый или щелочной растворы в зависимости or того, гидролиз
какого солеобразующе! о иона (кагиона или аниона) преобладав!.
Применяя закон действия масс к рассматриваемому процессу
гидролиза, получим выражение константы гидролиза ацетат а
аммония:
к _[NH4OH][CH3COOH]
' "[NH4-][CH3C(X) ] '
Умножив числитель и знаменатель на произведение [Н+ ] [ОН ],
приходим к выражению:
_[NH4OHJLCH,COOH][H*1[OI! ]_ Кв
'" [NI! + ][CIbC00 HH^OH1] ~~KK;
где Л^ист — константа диссоциации слабой кислоты СН3СООН,
^к„сл = ([СН3СОО [НЧ/[СН3СООН]);
^осн — константа диссоциации слабого основания NH40H,
Как указано выше, реакция раствора при гидролизе солей,
образованных слабыми кислотой и основанием, может бьпь
и кислой, и щелочной (хо!я ионы Н+ и ОН" в суммарное
уравнение реакции гидролиза не входят). Эю зависит от
относительной силы образующихся слабых кислоты и основания.
Так как количества их одинаковы, то, если кислота сильнее
основания (Ккисл> Коси), среда будет слабокислотной, если
основание сильнее кислош (Kocli> Ккксл), среда будет
слабощелочной.
Для вывода формулы расчета рН растворов солей,
образованных слабыми кислотами и основаниями, т. с. растворов,
содержащих эквимолярные количес!ва слабых кислоты и основания,
используем следующий прием. Так как количества ионов
водорода и гидроксид-ионов определяются константами диссоциации
кислоты и основания, m отношение концешраций ионов соот-
ветс1вует отношению констант диссоциации:
[ОН ]
Откуда получаем:
— 5КИС' • ГОН " 1 _^КИС| [Р
~ k" ^- -I к ' гГГч ~ к- ~ТСГ
ло,п лоен \\\ \ Косн [Н
343
Используя это соотношение, находим выражение для [Н+ ]
ирН:
В отличие от двух предыдущих вариантов гидролиза при
гидролизе соли, образованной слабым основанием и слабой
кислотой, рН раствора не зависит от концентрации соли.
Например, рН раствора ацетата аммония равен 6,99.
Реакция раствора ацетата аммония слегка слабокислотная,
так как константа диссоциации уксусной кислоты (К= 1,86 • 10~5)
немного превосходит константу диссоциации гидроксида
аммония (К= 1,79 • 10).
4. Соль образована сильной кислотой и сильным основанием.
К таким солям относятся NaCl, K2SO4, NaNO3 и многие другие.
Растворы этих солей практически нейтральные, рН » 7, так как ни
один из входящих в состав соли ионов не подвергается в водном
растворе гидролизу (рН немного отличается от 7 из-за влияния
ионной силы раствора на диссоциацию воды).
Тот же принцип используется при изучении характера
гидролиза солей многоосновных кислот и оснований. При этом
следует учест ь, что ионы таких солей гидролизуются по ступеням
в соответствии со ступенчатой диссоциацией кислот или
оснований. Так, гидролиз карбоната натрия Na2CO3 протекает в
соответствии со следующими уравнениями:
Первая ступень (I) СО32~ + Н2О = НСО3~+ОН~.
Вторая ступень (II) НСО3" +Н2О = Н2СО3 + ОН~.
Гидролиз этой соли характеризуется двумя константами:
^нсо3- ^н2со3
Поскольку константа диссоциации иона НСО3~
(К2 = 5,6 • 10"! 1) меньше константы диссоциации Н2СО3
(Я:1=4,3* 10~7) примерно на четыре порядка, то во столько же
раз константа гидролиза первой ступени больше константы
гидролиза второй ступени. Это означает, что щелочная реакция
раствора Na2CO3 обусловлена главным образом первой
ступенью гидролиза.
Так как в водном растворе гидролиз совершается в основном
только по первой ступени, рН раствора следует рассчитывать,
1акже пользуясь константой гидролиза первой ступени. Гидролиз
344
иона СО32 " практически не проходит по в юрой аунсни. так как
гидроксид-ионы, образующиеся на первой ыуиени. смещают
в соответствии с принципом Ле Ш ателье положение равновесия
гидролиза по второй ступени влево:
он
НСО3~+Н2О==Н2СО3
Чтобы гидролиз прошел по второй ступени, равновесие
следует сместить вправо добавлением в расiвор кислоты для
связывания гидроксид-ионов в молекулы воды
И'
или ОН~+Ы=Н2О; НСО3~ + Н +=Н2СО3.
Смещение равновесия гидролиза Na2CO3 вправо при прили-
вании к раствору соли кислоты подтверждается выделением
диоксида углерода:
Na2CO3 + 2НС1 = 2NaCl + Н2СО3
СО2(рфL- 2Н f (р р) = Н2СОз(р.Р) - СО2(Г) + Н2О{Ж).
Непрерывное смещение равновесия гидролиза карбона i-иона
вправо при добавлении кислоты coo i ветствует реакции
нейтрализации, в которой роль основания играю i ионы СО32~ и НСО3 .
Именно поэтому возможно шфование Na2CO3 кислотой (или
обратное титрование угольной кислокы раствором щелочи).
При гидролизе Na2CO^ в зависимости oi cieneHH
прохождения реакции образуйся то или иное количество кислотной
соли— гидрокарбоната натрия. В молекулярной форме процесс
гидролиза записывается следующими уравнениями:
Na2CO3 + Н2О = NaHCO3 + NaOH,
Запись уравнения в молекулярной форме используется только
для того, чтобы определить, какие соли и в каком количестве
можно получить при выделении их из водного раем вора
(например, выпариванием воды).
Выражение константы гидролиза любого типа включает
ионное произведение воды и кона анты диссоциации образуюгцихся
слабых электролитов. Так как при повышении температуры
константа диссоциации воды значительно в большей степени
возрастает по сравнению с константами диссоциации слабых кислот
и оснований, то при повышении 1емнера1уры консташа
гидролиза в целом должна увеличиваться и равновесие гидролиза
345
смещается вправо (гидролиз сопровождается поглощением
теплоты, Д//г>0). Действительно, нагреванием удается сместить
равновесие гидролиза вправо, но чаще эю вызвано удалением
одного из продуктов реакции.
Рассмотрим гидролиз сульфида аммония (NH4JS—соли,
образованной слабой двухосновной кислотой и слабым
основанием. Гидролиз этой соли протекав по стадиям:
(NH4JS = 2NH4++S2- (диссоциация)
Первая ступень NH4++S2" +H2O = NH4OH + HS".
Вторая ступень NH4+ + HS+H2O-NH4OH + H2S.
При прибавлении к рас i вору сульфида аммония раствора
гидроксида аммония или сероводорода равновесие гидролиза
смещается влево. Такой гидролиз принято называть обратимым
(все ранее рассмотренные реакции гидролиза были примерами
гидролиза этого типа).
В лабораторной практике обычно процесс гидролиза
проводят в открытых системах. При нагревании раствора сульфида
аммония растворимость аммиака и сероводорода в воде
уменьшается, они уходят из сферы реакции, и равновесие процесса
резко смещается вправо Гидролиз, сопровождающийся уходом
продуктов реакции из зоны реакции или образованием осадка,
часто условно называют необратимым гидролизом (не в 1ермо-
динамическом смысле!). Примерами необратимого гидролиза
могут служить реакции:
А14С3(КL-12Н2О(Ж) =
Рассмотренный выше сокращенный молекулярно-ионный
способ написания уравнений гидролиза, например,
удобен для вычисления консганты гидролиза, рН раствора и
концентраций ионов. С учетом тго, то в водном растворе
практически все катионы гидратированы, уравнение следует записать
в виде:
[А1(Н2ОN]3+
или [А1(Н2ОN]3+=[А1(Н2ОMОН]2+
346
Вторая запись отражает уже не реакцию гидролиза, а
диссоциацию комплексного иона как слабой кислоты. Однако в связи
с тем, чю справочных данных но константам диссоциации
слабых кислот и оснований намного больше, чем по константам
диссоциации комплексных акваионов, для вычисления
концентрации ионов водорода в водных растворах солей приходится
пользоваться уравнениями, записанными как реакция гидролиза
с участием негидратированных ионов.
К рассмотренным реакциям гидролиза примыкают процессы
перехода иона одного состава в другой без изменения степени
окисления (валентного состояния) атомов при изменении рН
раствора. Так, хромат-ион СЮ42~ устойчив в щелочной среде,
а дихромат-ион Сг2О72~ существует в кислой среде, и эти два
иона взаимно превращаются при изменении рН раствора:
2СгО42+2Н+-Сг2О72+Н2О,
Сг2О72-+2ОН~=2СгО42-+Н2О.
Хромат-ион при действии кислоты переходит в дихромат-ион;
дихромат-ион превращается в хромат-ион при действии щелочи.
При составлении уравнения реакций перехода подобного типа
необходимо знать среду (кислотную или щелочную), в которой
осуществляется такой переход. Рассмотрим на примере
превращения СгО42~ и Сг2О72~ рекомендуемые правила составления
уравнения реакций перехода (без изменения степени окисления
атомов):
1) Запишем схему процесса:
СгО42" ^Сг2О72".
2) Уравняем число a i омов металла (Сг) в обеих частях схемы:
2СгО42"—Сг2О72~.
3) В правую часть схемы запишем молекулу воды как продукта
данного превращения:
2СгО42" — Сг2О72+Н2О.
Вода как слабый электролит обусловливает протекание
процесса.
4) Подсчитаем число атомов кислорода в обеих частях схемы.
Если число атомов кислорода в правой части больше, чем
в левой, записываем гидроксид-ион в левой части. Если число
атомов кислорода справа и слева одинаково, в левой части
указываем ион водорода:
2СгО42~ + Н+ — Сг2О72+Н2О.
5) Уравняем числа атомов водорода в правой и левой частях
и записываем уравнение процесса, поставив знак равенства:
2СгО42+2Н+=Сг2О72+Н2О.
347
6) Для окончательной проверки правильности составленного
уравнения устанавливаем равенство числа агомов кислорода
(еще раз) и зарядов ионов в правой и левой чааях уравнения.
Если окажется, что равенство зарядов не соблюдается, то,
значит, рассматриваемый процесс относится к окисли \ елыю-
восстановительным реакциям.
Для составления уравнения обратного перехода Сг2О72"
в СгО42 нельзя записать полученное выше уравнение реакции
в противоположном направлении
Cr2O72 + H2O = 2CrO42" +2H f,
поскольку это будет реакция гидролиза, которая проходит
в очень незначительной степени.
Уравнение перехода иона Сг2О72~ в СгО42~ сос1авляется
в соответс!вии с приведенными выше лапами:
1) Сг2О72 — СгО42".
2) Сг2О72" — 2СгО42".
3) Сг2О72 — 2СгО42" + Н2О.
4) Сг2О72-+ОН" — 2СгО42 + Н2О.
5) Подсчет числа гидроксид-ионов облегчается, если исиользо-
Baib правило: число ОН" должно в два раза превышать
разницу аюмов кислорода в формулах анионов (Сг2О72~
и СгО42 ), кислород воды не учитывается. При этм число
молекул образующей воды будет в два раза меньше, чем
число исходных i идроксид-ионов (в более сложных
уравнениях). Таким образом, составляем равенст во:
Сг2О72" +2ОН -2СгО42" +Н2О
6) Проверяем равенство чисел всех а юмов и зарядов в правой
и левой частях полученного уравнения.
Ион Сг2О72 —пример иона, входящею в состав изополйсоединений Ишпо-
шсоедипепыч—кислоты и их соли со сложным анионом, коюрый образован
двумя или более оксидами одного и юго же кислоюобразующего тлемеша
Существуют также гетеропотсоедииенич — кислоты и их соли со сложным
анионом, который образован двумя или более оксидами различных
кислотообразующих элементов К соединениям такого типа оiносится, например, фосфор-
номолибденовая кислота соеiава Н3РМо]2О40, формулу которой обычно
записывают гак Н3РО4 12МоО3 Гетерополиион PMoJ20403~ получается в кислотной
среде, при добавлении щелочи он разрушас1ся
РО43 +12МоО42 +24*-Г-РМо12О403 + 12Н,О,
РМо,2О403Ч-24ОН =PO4J~4 12MoO42"-f I2H2O
К рассмотренным химическим процессам изменения состава
вещества при изменении рН раствора относятся также реакции,
характеризующие вещество как амфо!ерное.
Амфотерные соединения (амфо1ерные элеюролитм, амфоли-
1ы) в зависимости от условий способны проявлень либо кисло-
348
тные, либо основные свойства. Амфолиты— слабые
электролиты, с сильной кислоiой они обнаруживаю! основные свойства,
а с сильным основанием — кислотные свойства. Амфолитом
является вода (Н2О = Н++ОН ).
Гидроксид алюминия А1(ОНK—характерное амфотерное
соединение, поэтому его осадок может рас1воряться как в
растворах кислот, гак и щелочей:
3 + ОН-=А1О2-+2Н2О.
При изменении среды на противоположную реакции проходят
в обратном направлении:
А13++ЗОН~=А1(ОНK,
При этом, если щелочь или кислота добавлены в избытке,
малорастворимый гидроксид алюминия не осаждается, и реакции
протекают по уравнениям:
Al3+ +4OH" - А!О2- +2Н2О,
Простейшие формулы катиона и аниона А13+ и А1О2" используются в
уравнениях реакции для простоты и облегчения расчетов концентраций частиц в
растворах. В действительности такие ионы не существуют в водном растворе. Ион
А13 f в кислой среде !идратирован шестью молекулами воды [А!(Н,О). ]3 + , в его
состав могут входить и имеющиеся в растворе анионы, например, [А1С1(н2ОM ]2 +
В щелочных растворах алюминий находится в виде иона [А1(ОН).]~ (линейный
ион А1О2 обнаружен только в сильнощелочных растворах при рН > 13)
Понятие амфотерности может быть распросгранено и на
простые вещества: алюминий растворяется в растворах кислот и
щелочей:
К амфотерным электролитам относятся также вещества, у
которых кислотные и основные свойства обусловлены совместным
присутствием кисло i ной и основной групп. Таковы вещества типа
аминокислот NH2RCOOH, содержащие основную группу — NH2
и кислотную группу — СООН.
В водном растворе аминокислоты NH2RCOOH находятся
в состоянии внутренних солей NH3 + RCOO~, т. е. представляют
собой ионы, несущие одновременно отрицательный и
положительный заряды. Основные свойства этих солей, т. е.
взаимодействие с ионами водорода, обусловлены способностью группы
СОО" присоединять протон:
349
NH3 *"RCOO" + H + =NH3RCOOH +
или
NH2RCOOH + H+-NH3RCOOH + .
Взаимодействие с гидроксид-ионами, т. е. проявление
кислотных свойств, осуществляется благодаря способности группы
NH3+ отдавать протон:
или
NH2RCOOH + OH-=NH2RCOO-+H2O.
Получены твердые полимерные материалы, поверхность
которых в зависимости от рН контактирующего раствора способна
проявлять и кислотные, и основные свойства, при этом
осуществляется обмен ионами с раствором. Такие полимеры используются
для разделения сильных и слабых электролитов, для выделения
металлов из растворов и других целей.
§ 49. РАВНОВЕСИЯ В БУФЕРНЫХ РАСТВОРАХ
Горизонтальные участки на кривой титрования сильной
кислоты сильным основанием (см. рис. 130) свидетельствуют о малом
изменении рН раствора в начальный и конечный моменты
титрования. Незначительное изменение рН раствора в начале титрования
объясняется тем, что в растворе кислота находится еще в большем
избытке по отношению к количеству прибавленной щелочи.
Способность раствора поддерживать определенное значение рН
называется буферным действием. Буферное действие раствора
измеряется буферной емкостью, т. е. тем количеством щелочи или кислоты,
которое требуется прибавить к раствору, чтобы значение его рН
изменилось на единицу. На кривой титрования буферная емкое t ь
раствора выражается наклоном кривой к горизонтальной оси, т. е.
производной количества прибавляемого раствора по рН.
При титровании сильной кислоты сильным основанием
буферное действие растворов проявляется только в области очень
низких или очень высоких значений рН. Наименьшую буферную
емкость имеет раствор в точке эквивалентности. К раствору
с рН = 7 достаточно добавить одну каплю раствора кислоты или
щелочи, и рН раствора скачкообразно изменяется.
В химической практике часто возникает необходимость иметь
растворы с устойчивым значением рН в облас1ях, близких к
нейтральности.
Растворы, обладающие способностью поддерживать
определенное значение рН при разбавлении и при введении в раствор
350
некоторых количес1в кислоты или основания, называются
буферными. Согласно этому определению растворы сильных кислот
и оснований, хотя и обладают некоторым буферным действием,
не могут считаться буферными, так как при введении в раствор
сильной кислоты дополнительного количества сильной кислоты
или в раствор щелочи дополнительного количества щелочи рН
растворов изменяется так же, как изменяется рН при разбавлении
растворов.
Наиболее эффективные буферные растворы готовят из смесей
слабой кислоты и ее соли или слабого основания и ее соли,
например CH3COOH + NaCH3COO или NH4OH + NH4C1.
Буферное действие подобных смесей основано на следующих
процессах. Если к ацетатному буферному раствору
CH3COOH + NaCH3COO прибавлять (в пределах буферной
емкости раствора) щелочь, то будет проходить нейтрализация
щелочи слабой кислотой:
сн3соон+он ==сн3соо+н2о.
При добавлении к ацетатному буферному раствору сильной
кислоты ионы водорода связываются анионами слабой кислоты,
образующимися при диссоциации соли:
н+ +сн3сосг =сн3соон.
Таким образом, в результате связывания гидроксид-ионов
и ионов водорода (о г добавляемых сильного основания и
сильной кислоты соответственно) рН буферного раствора
практически не изменится. Приведенные ниже данные иллюстрируют
поведение 0,1 М ацетатного буферного раствора (в сравнении
с водой) при введении в 1 л раствора (воды) 0,01 моль НС1
и 0,01 моль NaOH:
Вода,рН
0,01 Маш на
0
0,01 моль NdOH
2
7
12
4,56
4,65
4,73
Из этих данных видно, что при добавлении кислоты или щелочи
рН водного раствора изменяется на 5 единиц, а ацетатного
буферного раствора—юлько на 0,1 единицы.
Аналогично действие аммиачно-аммонийного буферного
раствора NH4OH-bNH4Cl на введение кислоты или щелочи. Оно
обусловлено протеканием следующих процессов:
NH4++OH=NH4OH.
Буферные растворы могут быть приготовлены также на
основе кислоты и ее кислой соли, на основе двух кислых солей
351
с разными кона антами диссоциации кислотных анионов, а также
на основе соли, подвергающейся 1идролизу, и другой кислой
соли. К таким буферным растворам относятся смеси
H3PO4 + NaH2PO4 (рН<7), NaH2PO4+Na2HPO4 (pH«7),
Na2HPO4-f Na3PO4 (pH>7).
Буферные растворы Na2CO3 + NaHCO3 и Na2HPO4 +
+ NaH2PO4 имеют большое значение для обеспечения
жизнедеятельности организмов, так как они поддерживают постоянство
рН физиологических жидкостей.
Расчет рН ацетатного буферного раствора проводится
аналогично вычислению рН раствора уксусной кислоты, содержащего
ацетат натрия. Подобным же образом рассчитывается рН ам-
миачно-аммонийного буферного раствора как слабого
основания, содержащего сильный электролит NH4C1.
Если буферный раствор приготовлен из эквивалентных
количеств кислоты и ее соли, го CKncJCc~ ' и рН раствора равен
[Н + ] = Ккисл; рН = рКкисл.
Если буферный раствор приготовлен из эквивалентных
количеств основания и ее соли, то рН такого раствора вычисляется из
соотношения:
При разбавлении буферного раствора водой рН изменяется
очень незначительно, гак как, хотя концентрация слабой кислоты
или слабого основания уменьшается, концентрация образуемых
ими ионов водорода или гидроксид-ионов увеличивается за счет
повышения степени диссоциации электролита. Эти два процесса
взаимно компенсируются, и в конечном результате концентрация
ионов водорода или гидроксид-ионов сохраняется постоянной.
В протонной теории водные буферные растворы
рассматривают как системы, сосюящие из слабой кислоты и сопряженного
с ней основания или слабого основания и сопряженной с ним
кислоты. Так, действие ацетатного буферного раствора
обусловлено парой СН3СООН и СН3СОО~. При добавлении в буферный
раствор сильной кислоты ее протоны связываются основанием
СН3СОО", а при введении сильного основания уксусная кислота
отдает ему свои протоны и 1аким образом рН раствора
поддерживается примерно постоянным.
Буферные растворы широко используются в
научно-исследовательской практике и технологии. Они служат для поддержания
постоянного значения рН среды при проведении процессов,
направление и скорость коюрых зависят от рН. Регулирование рН
растворов позволяет направлять различного рода процессы в
желаемую сторону и син1езировать вещества с заданными
свойствами.
352
§ 50. КОМПЛЕКСООБРАЮВЛПИЕ
Если сливать растворы хлорида железа (III) FeCl3 и цианида
калия KCN различных концентраций и определять в смеси ионы
Fe3+ и CN~, то можно установить, что в зависимости от
начальных концентраций солей получаются системы, в которых
аналитически не обнаруживаются свободные ионы Fe3 + или CN~. Это
означает, что один из этих ионов, содержащийся в недостатке,
полностью реагирует с другим. Можно подобрать такие условия
(концентрацию и объем растворов), при которых в полученной
смеси не будут обнаруживаться одновременно ни ионы Fe3 + , ни
ионы CN". Например, при сливании равных объемов точно 0,01
М раствора FeCl3 и 0,06 М раствора KCN (или же 1 объема 0,01
М раствора FeCl3 и 6 объемов 0,01 М раствора KCN) образуется
система, не содержащая ни ионов CN~, ни ионов Fe3 + , что
указывает на протекание реакции:
Fe3 + + 6CN " = [Fe(CNN ]3 ~.
После испарения воды из этого раствора выпадают кристаллы
(бесцветные и красные) двух различных веществ. Бесцветные
кристаллы менее растворимы в воде и мало растворимы в
этаноле, красные кристаллы не растворимы в спирте, но растворяются
в ацетоне.
Качественный химический анализ показывает, что раствор
бесцветных кристаллов содержит ионы калия (обнаруживаются
по окраске пламени горелки) и хлорид-ионы (при действии
нитрата серебра осаждается белый хлорид серебра). Изучение
электропроводности раствора показывает, что одна молекула
вещества при растворении в воде распадается на два иона. Таким
образом, это позволяет установить, что бесцветные кристаллы
представляют собой хлорид калия.
Количественный элементный анализ кристаллов красного
цвета показывает, что вещество содержит 35,63% (масс.) К,
16,96% Fe, 21,89% С и 25,52% N. Соотношение чисел атомов
в этом соединении составляет:
K:Fe:C:N = 35,63/39,10:16,96/55,85:21,89/12,01:
:25,52/14,01 =0,911:0,304:1,823:1,822=0,911/0,304:
:0,304/0,304:1,823/0,304:1,822/0,304 = 3:1:6:6.
Следовательно, простейшая формула вещества—K3FeC6N6.
Раствор красных кристаллов в воде содержит также ионы
калия, но обнаружить в растворе ион CN " и Fe3 + качественными
реакциями не удается. Изучение электропроводности раствора
доказывает, что одна молекула вещества распадается на четыре
•*иона. На основании этих данных можно предположить, что
в растворе наряду с ионами калия содержатся ионы состава
12 2452 353
FeC6N63~. Ион CN очень прочный, и есть все основания
считать, что при взаимодействии ионов Fe3+ и CN" он сохраняет
свое строение и сое i ав.
Следовательно, ион состава FeC6N63~ может быть
представлен формулой [Fe(CNN]3~.
По результатам проведенного анализа можно заключить, что
красные кристаллы представляют собой соединение
K3[Fe(CNN], называемое гексацианоферратом (III) калия.
Исследование подобным же образом системы, получающейся
при сливании растворов сульфата меди CuSO4 и аммиака,
указывает на образование соединения сульфата тетраамминмеди
[Cu(NH3L]SO4. Приведенные примеры иллюстрируют реакции
образования комплексных соединений. В частности,
рассмотренные соединения K3[Fe(CN6)] и [Cu(NH3L]SO4 относятся к ком-
плексным солям* в их состав входят комплексные ионы
[Fe(CN6)]3+ и [Cu(NH3L]2".
При действии на K3[Fe(CNN] концентрированной
хлороводородной кислоты получаегся комплексная кислота H3[Fe(CNN],
а при действии на [Cu(NH3L]SO4 щелочи образуется
комплексное основание [Cu(NH3L](OHJ- Известны также комплексные
неэлектролиты.
В молекулу комплексного соединения входит центральный
атом (ион)—комплексообразователь. Частицы (ионы или
молекулы), располагающиеся вокруг комплексообразователей,
называются лигандами. Комплексообразователь и лиганды образуют
внутреннюю сферу комплексного соединения. Ионы,
нейтрализующие суммарный заряд ионов внутренней сферы и
располагающиеся более отдаленно от комплексообразователя, образуют
внешнюю сферу (рис. 132). При написании формул комплексных
соединений комплексообразователь с лигандами, т. е.
внутреннюю сферу, заключают в квадратные скобки. Этим
подчеркивается, ч го в растворах комплексный ион почти не диссоциирует на
составляющие его ионы или молекулы. Известно много
комплексных соединений без внешней сферы, например, Fe(COM,
lPt(NH3JCI2].
Если лигандами являются толь-
Лигаиды Компяексообразовагель KQ нейТраЛЬНЫе МОЛеКуЛЫ, ТО За-
4- ряд комплексного иона равен заря-
к*
\ сг /си-
CN-
Fe2+
CN" CN-
Вкешилл
**¦ сфера
\ ду комилексообразователя; если во
Заряд
внутреннюю сферу входят ионы
и молекулы, го заряд комплексно-
_ го иона равен алгебраической сум-
внутренняя ~" ме зарядов комилексообразователя
(комплексны». И ЛИГаНДОВ.
"ои} В большинстве соединений
Рис. 132 Строение гексациано- связь между комплексообразовате-
феррата (И) калия K4[Fc(CNN] лем и лигандом имеет донорно-ак-
354
цепторную природу. Число связей между лигандами и комплек-
сообразователем определяет координационное число последнего.
Если один лиганд соединен с комплексообразователем одной
связью, то координационное число совпадает с числом лигандов,
и такие лиганды называются монодентатными.
Координационные числа центральных ионов в [Fe(CNN]3~ и [Cu(NH3L]2 +
равны 6 и 4, а лиганды CN" и NH3—монодентатные.
Лиганд, образующий с комплексообразователем две связи,
называется бидентатным. Например, ион С2О42~ —
бидентатный и образует с Си2+ соединение К2 [Си(С2О4J ]•
Гидразин H2N—NH2 , этилендиамин H2NCH2CH2NH2—также би-
дентатные лиганды. Такие лиганды, как SO42~ или СО32~, в
зависимости от условий могут быть моно- и бидетатными. Заряд
иона-лиганда не определяет его дентатность. Так, в соединении
К2[СеAМО3)б] координационное число церия равно 12, поскольку
каждый ион NO3~ соединен с ионом церия через два атома
кислорода.
Наиболее часто встречаются комплексообразователи с
координационным числом 6 и 4, менее часто—2 и 8 и еще реже—с
другими. Координационное число иногда зависит от
концентрации ионов-лигандов в растворе. Отмечена также зависимость
координационного числа от температуры.
Для построения названия комплексного соединения используются
следующие правила Если соединение построено из комплексного иона и одного
или нескольких ионов внешней сферы, т. е является комплексной солью, то
первым—в именительном падеже—называется анион, а затем — в родительном
падеже—катион. При названии комплексного иона сначала указываются
лиганды, затем комплексообразователь. Лиганды в комплексном ионе
перечисляются в следующем порядке. 1) гидроксид-ион (если он имеется), простые
анионы и затем в алфавитном порядке органические анионы, 2) нейтральные
молекулы NH3, H2O и др.
Нейтральные лиганды указывают названиями соответствующих молекул,
кроме воды и аммиака, для обозначения их в качестве лиганда применяют
термины «аква» и «аммин». К анионным лигандам прибавляют окончание «о»
(хлоро, сульфато). Число лшандов, если их больше одного, указывают
греческими числительными, ди-, три-, тетра-, пента-, гекса-.
Если комплексный ион—катион, то для названия комплексообразоватсля
используют русское название элемента и указывают его степень окисления (в
скобках римской цифрой). Если комплексный ион — анион, то используется
латинское название элемента-комплексообразователя, к коюрому прибавляется
окончание «ат» и указывается римской цифрой в скобках заряд иона У
нейтральных комплексов (без внешней сферы) центральный атом называется в
именительном падеже, а его степень окисления не указывается.
Примеры названий комплексных соединений, которые помоет усвоить
правила их номенклатуры:
К4 [Fe(CNN ] — гексацианоферрат (II) калия
К3 [Fe(CNN ] — гексацианоферрат (III) калия
К2 [PtCl6 ] —гексахлороплатинат (IV) калия
K[PtNH3Cl5] —пентахлороамминплатинат (IV) калия
[Pt(NH3JCl4 ] —тетрахлородиамминплатина
[Pt(NH3KCl3 ]С1 —хлорид трихлоротриамминплатины (IV)
12* 355
[Pt(NH3MCl]Cl3 —хлорид хлоропснтаамминплатины (IV)
[Pl(NH3N]Cl4 —хлорид гексаамминплатины (IV)
[PtfNI^bNC^CllSO*— сульфат хлоронитротриамминплатины (П)
[Co(NH3K(NO2K ] —тринитротриамминкобальт
Со[СоС14] —тетрахлорокобальт (II) кобальта (II)
Следует обратить внимание, что записанная формула читается в обратной
последовательности ее написания. При написании формулы по ее названию
в квадратных скобках записывают комплексообразователь, затем—лиганды
в обратном порядке их перечисления. Сложные молекулы в качестве лигандов
записывают сокращенно: тиомочевина—thio, пиридин—ру, этилендиамин—еп,
этилендиаминтетрауксусная кислота—edta Перед сложными л иг ан дам и,
имеющими в названии числительное, число лигандов обозначается приставками бис-,
трис- и т д. Сложные лиганды при написании названия иногда заключают
в скобки, например. [Cuen2]Cl2—хлорид бис(этиаендиамин) меди (И).
Растворяясь в воде, комплексные соединения, имеющие
внешнюю сферу, диссоциируют как сильные электролиты на
комплексный ион, состоящий из частиц внутренней сферы, и на ионы
внешней сферы:
K4[Fe(CNN]=4K+ + [Fe(CNN]4",
K3[Fe(CNNl = 3K+ + [Fe(CNN]3",
[Cu(NH3L](OHJ= [Cu(NH3L]2+ +2OH".
Механизм образования комплексных соединений и их
свойства наиболее полно объясняются в рамках теорий валентных
связей и молекулярных орбиталей. В соответствии с методом
валентных связей связь между комплексообразователем и лиган-
дом осуществляется электронной парой, которая поставляется
лигандом, при этом электронная плотность лиганда
перемещается к незаполненным орбиталям комплексообразователя, и в
результате происходит как бы перекрывание свободных орбиталей
комплексообразовагепя (акцептор) с заполненными орбиталями
лиганда (донора).
Большинство лигандов поставляет одну электронную пару,
и число координированных лигандов, т. е. координационное
число комплексообразователя, зависит от числа свободных
электронных орбиталей. ч
Покажем это на примере рассмотрения электронного
строения комплексов железа. На рис. 133, а изображено распределение
электронов в невозбужденном атоме железа. Подуровни 3 s и Ър
полностью заполнены парами электронов, в образовании связи
они не принимают участия, на рис. 133, б даны подуровни с
электронами, принимающими участие в химических процессах, а на
рис. 133, в это электронное строение представлено одной
строкой: 3d64sz4p°4d°. При образовании иона Fe2+ два электрона
с 45-подуровня отрываются и образуется конфигурация
3d64s°4p64d° (рис. 133, г). Ион Fe2+ взаимодействует с лиган-
дами, причем от силы ноля лигандов зависят электронное
строение комплексообразователя и его магнитные свойства.
356
и
3
2
1
«
И
М
ш
м
¦¦
м
f
¦
d
t
I I I I I I I
f
Fe
Fe 1s22s22p63s23p63dHs*
6
Щ
зП
•s1/»
U.
Aim
в
Fe |**|*И1ф| 0 DID I I 1 I 1 I
3d 4s 4p kd
s
Fe2+
3d
п rm 111111
Us h"D 4>d
t ttt ft"—;
О О О О О О
« М W N ^1 с*
X XXX XX
H2O
sp3-au6pu-
дизация
s - &
II
:::схп
tf-гибридизация
по методу
валентРис. 133. Схема образования комплексного иона [
ных связей:
а б в—распрелс 1ение злектронов в атоме Fe, /—распределение электронов в ионе Fe2\ д—элек!ронное
строение молеку ili H^O, v ж—электронное сiроение Fe24 в комплексном ионе
Рассмотрим комплекс железа с лигандами, обладающими
слабым электрическим полем. К таким лигандам относятся,
например, молекулы воды. Шесть молекул воды, отдавая по паре
^/?3-гибридных электронов (рис. 133, д\ заполняют одну 4s, три
Ар и две Ad свободные орбитали иона Fe2+ (рис. 133, е). В
образовавшемся ионе [Fe(H2ON ]2+ все шесть молекул воды
энергетически равноценны, что свидетельствует о я/?3</2-гибридизации
(рис. 133, ж) (следует обратить внимание, что при написании
формулы гибридизации в верхнем индексе указывается не число
электронов, как в формулах электронных конфигураций, а число
гибридных электронных орбиталей, и перечисление гибридных
357
подуровней дается oi нижнего энергетического уровня или
подуровня к более высоколежащим уровням).
В пространстве шесть равноценных орбиталей могут
располагаться одним-единственным образом — в направлении вершин
октаэдра. Октаэдрическое расположение частиц-лигандов вокруг
центральной частицы (комплексообразователя)—самое
выгодное по плотности упаковки и энергии системы. Это и определяет
координационное число Fe2 + , равное шести, хотя еще остается
три свободных, незаполненных орбитали (см. рис. 133, е).
Рентгенографическое изучение комплексов железа
подтверждает эту модель. Подобным образом построен комплекс [FeF6]4~.
При образовании гексаакважелезоA1)-иона и гексафтороже-
лезо(Н)-иона в формировании связи с лигандами Н2О и F
участвуют внешние орбитали четвертого уровня иона железа: As, Ар
и 4*/и не участвуют внутренние орбитали подуровня 3d с неспа-
ренными электронами (см. рис. 133, г, е, ж). Поэтому такого
типа комплексы носят название внешнеорбитальных. Лиганды
типа Н2О или F", использующие для формирования связи
орбитали внешних уровней комплексообразователя,—эю слабые
лиганды. Под действием слабых лигандов одиночные электроны
нижележащего уровня не спариваются, и поэтому ион железа
находится в высокоспиновом состоянии. Данные магнитных
измерений показывают, что ионы [Fe(H2ON]2+ и [FeF6]4"
парамагнитны и их магнитный момент определяется числом неспа-
ренных электронов (четыре 3^-электрона).
Лиганд CN~ образует с ионом Fe2+ комплекс [Fe(CNN]4~.
Несмотря на то же координационное число и ту же окта-
эдрическую структуру, что и у рассмотренных выше
комплексов, магнитные
а свойства этого компле-
П MM jiiiii кса отличаются от
uifLJLLJ У "ЛН ' ' 'J ' ' свойств [Fe(H2ON]2 +
и [FeF6]4 . Лиганд
Ш ? Mil I1II11 CN—сильный лиганд.
3d *s up иа Он стремится войти
a a a i а а в донорно-акцепторное
II I 111 взаимодействие с внут-
ас * г * г ss ренними орбиталями
о о о о а о комплексообразовате-
^^.^.^.^ ля и благодаря своему
1 III iJ сильному полю вытес-
няет одиночные элек-
дизацияг троны на соседние ор-
Рис. 134. Схема образования комплексного иона битали, ЧТО приводит
[Fe(CNN]4 ~- к образованию общих
а—распределение электронов в ионе Fe21": б—донорно-акцеи- ЭЛеКТрОННЫХ Пар
торное взаимодействие комплексообразователя с лигандами, в— ^ ii/i ^:\
распределение электронов в комплексном ионе (рИС. 134, а, О).
358
Fe3*iHmtm I
I I 1 1 i I
Слабые мганды
за
D ггп i i i I i i
us up ua,
t iff tt
3d
:::nm
Чги6
дизация
Сильные лиганды
пинт i i ? i i i i МММ
3d bs bp U(t
tt t ttt
LL L LLL
JIM
d2sp3-гибридизация
Рис 135 Распределение электронов иона Fe3+ в окружении слабых и сильных
лигандов
Вытеснив из орбиталей З^подуровня иона Fe2+ максимально
возможное число неспаренных электронов и образовав из них
электронные пары, сильные лиганды поставляют на
освободившиеся орбитали 3 rf-подуровня, а также на свободные орбитали 4 s-
и 4/7-подуровней иона Fe2+ свои пары электронов. У иона Fe2 +
две освободившиеся Зй?-орбитали, одна 4s- и три 4р-орбитали
способны принять шесть пар электронов от доноров—лигандов
CN~. Все образовавшиеся шесть связей равноценны, что
указывает на й?25р^-гибридизацию (рис. 134, в). Структура
комплексного иона [Fe(CNN ]4~ —октаэдрическая. Комплексы, в которых
комплексообразователь отдает для связи внутренние орбитали,
называются внутриорбитальными. В этих комплексах ион Fe2 +
не имеет неопаренных электронов и находится в низкоспиновом
состоянии.
Совершенно аналогичен подход к рассмотрению механизма
образования химической связи в комплексных соединениях Fe3 + .
Слабые лиганды типа Н2О и F" отдают шесть пар электронов на
свободные 4s-, Ар- и две 4</-орбитали иона Fe3+ (рис. 135). Окта-
эдрическое строение образующихся внешнеорбитальных
комплексов позволяет приписать иону Fe3 + sp3^-гибридизацию
участвующих в образовании связей орбиталей. Благодаря
высокоспиновому состоянию иона Fe3+ комплексные ионы [Fe(H2ON]3 +
или [FeF6]3~ парамагнитны.
Сильные лиганды типа CN " образуют с ионом Fe3+ внутри-
орбитальные комплексы октаэдрического строения, что
указывает на </25/?3-гибридизацию орбиталей иона Fe3 + . Один неспарен-
ный электрон в комплексе [Fe(CNN]3" ответствен за невысокий
парамагнетизм этих ионов.
Часто удается установить конфигурацию комплекса,
сравнивая число известных изомеров этого комплекса с
рассчитанным для гой или иной предполагаемой конфигурации. Среди
359
цис-изомер транс -изомер
Рис 136. Геометрические изомеры квадратного
комплекса [Pt(NH3JCl2]
комплексных соединений известно много соединений одного
состава, различающихся свойствами и, следовательно,
строением. Например, соединению PtCl2 *2NH3 отвечают два изомера,
которые различаются окраской и растворимостью, но растворы
этих изомеров одинаково плохо проводят электрический ток
и хлорид-ионы осаждаются из них медленно. Изучение других
свойств этого вещества показывает, что оно не может быть
сильным электролитом. Поэтому соединению состава
PtG2-2NH3 следует приписать одну и ту же координационную
формулу [Pt(NH3JCl2], предположив, что изомерия вызвана
различным взаимным расположением лигандов.
Если бы этот комплекс имел тетраэдрическую конфигурацию,
то вследствие полной равноценности всех четырех вершин
тетраэдра соединение не могло бы иметь изомеров. Только при
квадратном строении комплекса [Pt(NH3JCl2 ] возможно
существование двух изомеров: i/мг-дихлородиамминплатина (II) и транс-
дихлородиамминилатина (II) (рис. 136).
Рассмотренные структуры являются примером
геометрической изомерии, или цис-транс-изомерии. Метод валентных связей
позволяет предсказывать строение комплексных соединений,
число ожидаемых изомеров и их некоторые свойства.
Кроме геометрической изомерии известны другие виды,
в частности, ионизационная, сольватная, солевая, оптическая
изомерии.
Ионизационная изомерия заключается в различном
распределении ионов между внешней и внутренней сферами.
Кроме различия в свойствах ионизационные изомеры проявляют
неодинаковый характер диссоциации на ионы. Примером таких
изомеров являются [Pt(NH3LBr2 ]С12 и [Pt(NH3LCl2 ]Br2.
Первый из них имеет более интенсивную оранжево-жел гую окраску.
При действии нитрата серебра из раствора первого изомера
выпадает осадок AgCl, из раствора другого—AgBr.
К ионизационной близка сольватная изомерия*
заключающаяся в различном распределении молекул растворителя, в
частности воды {гидратная изомерия), между внутренней и внешней
координационными сферами. Например, составу
Co(NO3K(NH3M *Н2О отвечают два изомера:
[Co(NH3MNO3](NO3JH2Oh [Co(NH3MH2O](NO3K.
360
Рис. 137. Зеркальные изомеры тетраэд-
рического комплекса KL?LnLmLIV
Комилсксообрачова 1 ел ь К располагается в центре
1сгра">лра
Солевая изомерия
проявляется у соединений, содержащих
во внутренней сфере анионы,
которые имени два или более
атома, способных быть
донорами электронных пар, и
поэтому эти анионы могу г
образовывать связь с комплексообра-
зователем различными
способами. Например, ион
NO2~ с кобальтом образует
связи либо через кислород
Со—ONO (нитри i окомплекс), либо через азот Со—NO2 (нит-
рокомплекс). Комплексная соль [Co(NH3)sONO ]C12 имеет
красную окраску, a [Co(NH3MNO2 ]С12 — коричнево-желтую.
Некоторые комплексные соединения способны существовать
в виде двух изомеров, один из которых по своей конфигурации
является зеркальным изображением другого, причем эти
конфигурации не могут быть совмещены в пространстве. Такие
изомеры называются оптическими^ или зеркальными. На рис. 137
показано расположение относительно центрального атома К
четырех различных лигандов в тетраэдрическом комплексе
[KL'U'L111!^]. Оптическая изомерия возможна у тетраэдриче-
ских и ок гаэдрических структур, но невозможна у квадратных.
По числу оптических изомеров можно судить о структуре
комплекса.
Оптические изомеры проявляют одинаковую
электропроводность в растворах при равной концентрации вещества.
Магнитные свойства их также идентичны. Однако свойства, в которых
проявляется асимметричность, оказываются различными. Так,
у оптических изомеров равны, но противоположны по знаку углы
вращения плоскости поляризации. Скорости взаимодействия
оптических изомеров с молекулами или атомами оптически
активного заместителя также различны.
§ 51. РЕАКЦИИ С УЧАСТИЕМ КОМПЛЕКСНЫХ ИОНОВ
Комплексные соли, растворяясь в воде, диссоциируют как
сильные электролиты на комплексной ион и ионы, входящие во
внешнюю сферу. Такое ионное равновесие устанавливается
практически мгновенно. Например, дихлорид хлоропеш аамминхро-
ма (III) и гексацианоферраг (II) калия диссоциируют согласно
уравнениям:
K4[Fe(CNN]-4K++[Fe(CNN]
4"
361
В paci ворах комплексные соли ведут себя как простые соли, и для
их растворов характерны все свойства, присущие растворам элек-
фолитов: повышение температуры кипения, понижение
температуры замерзания, понижение давления пара растворителя над
раствором, наличие осмотического давления,
электропроводимость и др. На основе результатов изучения свойств водных
растворов комплексных соединений можно установить характер
их ионного равновесия, т. е. соотношение числа катионов к числу
анионов в молекуле соединения, и тем самым по составу
определить их строение (координационную формулу).
Образующиеся при растворении комплексных солей
комплексные ионы подвергаются дальнейшей ступенчатой
диссоциации как слабые электролиты. Так, ион [Fe(CNN]4~—далее
диссоциирует по стадиям:
[Fe(CNN]4-=[Fe(CNM]3-+CN-,
[Fe(CNM]3--[Fe(CNL]2-+CN-HT.^
Подобная запись уравнений диссоциации комплексных ионов
в водных растворах не отражает одно из условий протекания
процессов в водных растворах—координационное число ком-
илексообразователя должно сохраняться постоянным. Поэтому
при отрыве от комплексного иона лиганда его место заполняется
молекулой воды:
[Fe(CNMH2O]3+H2O-[Fe(CNL(H2OJJ2+CN-.
Конечным результатом этого процесса является образование
иона [Fe(H2ON]3 + . Таким образом, диссоциация комплексного
иона—это замещение лиганда на молекулу растворителя.
Прочность комплексного иона характеризуется константой
диссоциации, называемой константой нестойкости
комплексного иона. Так, ион [Fe(CNN]4~—диссоциирует по ступеням,
каждая из которых описывается определенным значением
константы диссоциации:
*- +CN"; Kt J
[Fe(CNMl3-=[Fe(CNL]-+CN-; ^2 = LF
[Fe(CNL]-=[Fe(CNK]-+CN-; ^-^
Константу нестойкости комплексов в растворах с ионной силой
выше 0,01 следует выражать через активности ионов (а не
концентрации).
362
В связи с тем, что определение ступенчатых констант
нестойкости (относящихся к каждой последовательной ступени
диссоциации) представляет большие экспериментальные и расчетные
трудности, для некоторых комплексных ионов даются общие
константы нестойкости, т. е. отвечающие полной диссоциации на
все имеющиеся во внутренней сфере лиганды:
[Fe(CNN]-=Fe^+6CN-; K^E
Общая константа нестойкости равна произведению ступенчатых
констант нестойкости
... К
п.
Иногда вместо констант нестойкости пользуются обратными им
величинами—константами устойчивости, относящимися к
процессу образования комплексного иона:
= * / **уст •
Зная концентрацию комплексной соли в растворе и константу
нестойкости комплексного иона, можно рассчитать состав
раствора. Предположим, что имеется 0,01 М раствор
[Cu(NH3L]Cb. Равновесные концентрации ионов СГ
и [Cu(NH3L] в растворе вследствие диссоциации соли как
сильного электролита
равны [Cu(NH3L2+] = 0,01 моль/ли [СГ ] = 0,02 моль/л.
По известной общей константе нестойкости можно оценить
концентрации иона-комплексообразователя и лигандов:
[Cu(NH3L]-=Cu-+4NH3; ^=
Если образовалось х моль Си2*, то аммиака должно
получиться Ах моль. При концентрации иона [Cu(NH3L]24", равной
0,01 моль/л, получим:
?=^j?=2,l • 10~13, откуда х = 0,38 • 10~3 моль/л.
Следовательно, [Си2 + ] % 0,4 • 10 ~ 3 моль/л и [NH3 ]«
«1,6* 10 моль/л.
По известным ступенчатым константам нестойкости можно
вычислять концентрации ионов, образующихся на
соответствующих ступенях диссоциации. Так, для первой диссоциации иона
тетраамминмеди (II) в 0,01 М растворе концентрация
образующихся ионов составит (расчет аналогичен приведенному выше):
363
[Cu(NH3L]2+=[Cu(NH3K]2++NH3;
0.01 v v
*' [Cu(NH3L-] U Ш
^=?^=7,2-10"\ откуда х = 8,5-10 моль/л.
Следовательно, [NH3] и [Cu(NH3K2+ ] = 8,5* 10~3 моль/л
в 0,01 M растворе комплексной соли [Cu(NH3L]Cl2.
Подставляя вычисленную концентрацию иона [Cu(NH3K]2 +
в константу нестойкости по второй ступени, можно получить
концентрацию иона [Cu(NH3J] , и т. д. Подобного типа
расчеты очень приблизительны, так как не учитывают изменения
концентраций ионов при их диссоциации (предполагается, что
исходные концентрации ионов не изменяются из-за очень малого
значения константы нестойкости).
Равновесия диссоциации комплексных ионов подчиняются
всем правилам смещения ионных равновесий. Так, при
добавлении в раствор [Cu(NH3L]Cl2 аммиака концентрация ионов
[Cu(NH3K]2 + уменьшается. Если [Cu(NH3L2+ ] = 0,01 моль/л
и [NH3 ] = 0,01 моль/л, то по закону действия масс получим:
[Cu(NH3L]2+=[Cu(NH3K]2++NH3,
0,01 г 0.01
^ 72.10
-з
к
Лнест~ [Cu(NH3L2 + ] " 0,0!
Откуда л*=7,2* 10~3 моль. Следовательно, [Cu(NH3K2+ ] =
= 7,2* 10" моль/л. Таким образом, введение в раствор
0,01 моль/л аммиака вызывает понижение концентрации иона
[Cu(NH3K ]2+ в 8,5 • 10'3/G,2 • 1<Г3)= 1,2 раза.
§ 51 ЭЛЕКТРОДНЫЕ ПРОЦЕССЫ
Растворимость одного вещества в другом—свойство,
присущее всем веществам. Раст воримость может быть неограниченной
и крайне малой, что зависит от термодинамических свойств
растворяемого вещества и растворителя. Даже при чрезвычайно
малой растворимости одного вещества в другом всегда имеет
место переход веществ через поверхность их контакта. Любой
металл растворяется в воде, однако растворимость в ряду
металлов изменяется в очень широких пределах. Например, щелочные
металлы бурно взаимодействуют с водой, при этом выделяется
из воды водород и в растворе образуются гидроксиды металлов.
Серебро практически не реагирует с водой, тем не менее процесс
364
перехода частиц серебра в воду происходит, и получается так
называемая «серебряная вода». Таким образом, одни металлы
активно растворяются в воде, другие—крайне мало. Ответ на
вопрос, чем обусловлена различная растворимость металлов
в воде, дает отрасль химической науки—электрохимия.
Ниже рассмотрена особая группа гетерогенных процессов—
электрохимические реакции, протекающие на границе раздела
фаз, в частности металл — вода (или раствор соли металла). Эти
реакции характеризуются переносом заряда и вещества через
границу раздела фаз твердое вещество—жидкость.
Если металлическую пластинку, например, медную,
погрузить в воду или раствор соли меди, то из слоя металла,
находящегося на границе с водой, положительно заряженные ионы Си2 +
начнут переходить в воду. При этом в кристаллической решетке
металла оказывается избыток электронов и пластина
приобретает отрицательный заряд. Между отрицательно заряженной
пластиной и перешедшими в раствор положительными ионами
возникает электростатическое притяжение, что препятствует
дальнейшему переходу ионов меди в раствор, т. е. процесс
растворения металла прекращается. Одновременно развивается
противоположный процесс: ионы меди из раствора, подойдя
к поверхности пластины, принимают от нее электроны и
переходят в нейтральное состояние. Через какой-то промежуток
времени устанавливается состояние динамического равновесия, при
котором скорость перехода ионов из металла в раствор равна
скорости разряжения ионов из раствора на металле.
Схематически описанное явление представлено на рис. 138 (ионы металла
для простоты изображены негидратированными).
Равновесие между ионами в растворе и металлом для
рассматриваемого примера выражается уравнением:
В уравнении равновесия электрохимической реакции
электроны принято записывать в
левой части, т. е. записывать
процесс восстановления. Соблюдение
этого способа написания
химических уравнений крайне важно, и,
как будет показано ниже,
пользуясь такими уравнениями, можно
предсказывать направление
окислительно-восстановительных
реакций.
Как видно из приведенного вы-
ше примера, при контакте металла
С раствором его СОЛИ ЭТИ две сопри-
касающиеся фазы приобретают
Рис K8 Схсма а.
ющего „а границе раздела металл
(Си)—вода
365
противоположные заряды, в резулы ai e на поверхности раздела фаз
образуется двойной электрический слой и между металлом и
раствором возникает разность электрических потенциалов.
Образование двойного электрического слоя и возникновение разности
потенциалов на границе металл — вода (раствор) происходит
в соответствии с теми же закономерностями, которые обсуждались
при изучении коллоидного состояния вещества.
К типу электрохимических реакций относятся электродные
процессы, в которых осуществляется перенос носителей заряда
через границу раздела металлический проводник—раствор.
Система, состоящая из электрического проводника и раствора (или
расплава) электролита, в который погружен проводник,
называется электродом. Так, медная пластина, опущенная в водный
раствор CuSO4—типичный электрод.
Состояние равновесия электродного процесса определяется
электродным потенциалом Е, представляющим собой разность
потенциалов на границе металл—электролит. Непосредственно
измерить абсолютное значение электродного потенциала нельзя,
но его можно определить сравнением с известным потенциалом
другого электрода при стандартных условиях — электрода
сравнения. В качестве электрода сравнения применяют водородный
электрод.
Водородный электрод представляет собой платиновую
пластину, опущенную в раствор кислоты (обычно НС1 или H2SO4),
через который пропускается газообразный водород (рис. 139).
Действие этого электрода состоит в следующем.
Газообразный водород не проводит электрического тока, но,
адсорбируясь в водном растворе на поверхности платины, ведет
себя как электрод, аналогичный металлическому. Для увеличения
адсорбирующей способности платину покрывают слоем
губчатой платины (платиновой чернью). Платиновую пластину
опускают в раствор кислоты (обычно НС1 или H2SO4) с
концентрацией (активностью) ионов водорода,
равной единице, и через раствор
пропускают водород так, чтобы происходило
непрерывное соприкосновение
поверхности платины с раствором и
водородом. В результате платина насыщается
водородом. Молекула водорода в
адсорбированном состоянии распадается
на атомы, которые ионизируются
(Н—е" >Н+), и ионы Н+ переходят
в раствор подобно ионам металла.
Одновременно ионы водорода из
раствора, находящиеся вблизи
поверхности платины, принимают электроны
Рис 139. Принципиальная
схема водородного
электрода
(Н++е
-Н). Между этими процес-
366
Рис 140. Схема i альваничсского
элемента, состоящего из медного и
водородного электродов
сами устанавливается
равновесие, которое в упрощенной
форме можно передач ь
уравнением:
Заряд платиновой пластины
зависит от парциального
давления водорода, концентрации
ионов водорода в растворе
и температуры. Потенциал
водородного электрода при
концентрации (активности) в
растворе ионов Н +, равной 1 моль/л, давлении газообразного
водорода в 101 325 Па и при температуре 25° С (стандартные условия)
принят равным нулю.
Электрод, потенциал которого сравнивается с потенциалом
водородного электрода, должен находиться при тех же условиях.
Потенциал электрода, измеренный при стандартных условиях,
т. е. при температуре 25° С, давлении 101325 Па и активности
ионов в растворе, равной единице, называется стандартным
электродным потенциалом (обозначение Е°).
При измерении потенциала изучаемого электрода, например,
медного, медную пластину (с отходящим от нее проводником)
опускают в раствор, содержащий ионы Си2т с концентрацией
(активностью) 1 моль/л, и эту систему соединяют
электролитическим мостиком со стандартным водородным электродом
(рис. 140). Электролитический мостик — это П-образная
стеклянная трубка, заполненная проводящим электрический ток
раствором—обычно насыщенным раствором КС!. Полученное
устройство называется гальванической цепьюь или гальваническим
элементом.
Если цепь разомкнута, на каждом электроде устанавливается
равновесие с отвечающим ему электродным потенциалом:
Cu2 +
H+
=Cu
w;
Си Си
2 +
(Р-Р)"
В замкнутой гальванической цепи электроны с платинового
электрода переходят на медный. Это означает, что равновесие
нарушено и на платиновом электроде совершается реакция, в
результате которой водород превращается в ионы:
!/2Н2(Г)""^ =Н+(
-?'
и? н
Хотя, разумеется, заряд электрода и отвечающий ему знак
потенциала не зависят от способа написания электродного про-
367
цесса, при изменении направления реакции знак потенциала
изменяется на противоположный. Это делается для того, чтобы с
потенциалами можно было проводить такие же операции, как с AG,
т. е. формально использовать закон Гесса.
На медном электроде электроны, перешедшие в платинового
электрода, взаимодействуют с ионами меди, в результате на
электроде осаждается металлическая медь, т. е. проходит
реакция восстановления:
Си2+(р.
Учитывая, что число отданных электронов при окислении и
принятых при восстановлении должно быть одинаковым, запишем
уравнение суммарного процесса:
xz /2**2(т)~~е "~n (р-р)»
=Cu(K); Е°Си/Си
+(р.р) — Н (р.р) + Сик Е—Е°Си1Си +—
Разность электродных потенциалов Е—это электродвижущая
сила (эдс) гальванического элемента. Так как водородный
электрод служит электродом сравнения, для которого ?°н2/н+ =0, то
измеряемая эдс рассматриваемого элемента—это потенциал
медного электрода по отношению к водородному.
Таким образом, потенциалы металлов можно сравнивать по
эдс гальванической цепи с водородным электродом. Поскольку
в гальваническом элементе в качестве электрода сравнения
используется стандартный водородный электрод, то искомый
электродный потенциал будет равен измеренной эдс. Например, если
измеренная эдс гальванической цепи из стандартных
водородного и медного электродов составляет +0,34 В, то, значит,
стандартный потенциал меди равен
Cu2+ +2e~ =Cu; ?°Cu/cu2+ = +0,34 В.
Положительное значение стандартного потенциала меди
говорит о возможности самопроизвольного процесса осаждения
меди и о невозможности противоположного процесса —
растворения меди в кислых растворах с концентрацией ионов водорода
1 моль/л. В гальванической цепи, составленной из цинкового
и водородного электродов, равновесное значение эдс при
стандартных условиях составит —0,76 В:
Zn2++2e"=Zn; ?о2„/2„2+ = -0,76 В.
Отрицательное значение стандартного потенциала цинка
свидетельствует о невозможности проч екания реакции по этому урав-
368
нению; самопроизвольно осуществляется процесс в
противоположном направлении:
Zn'Zn
= +0,76 В,
т. е. металлический цинк растворяется в растворах кислот с
концентрацией ионов водорода 1 моль/л.
Если расположить стандартные электродные потенциалы
металлов в порядке уменьшения их отрицательного значения и
повышения положительного, т. е. в порядке возрастания
электродных потенциалов (табл. 22), то получится ряд стандартных
электродных потенциалов (ранее используемое название—ряд
напряжений металлов).
LV
Na
Zn
Fc
H +
Ряд
Равновесная
электродная реакция
h+e-=Li
+ +<?"=Na
2 + +2е~ =Zn
2t+2e"=Fe
A0 M)+e~ = Ч2Н
стандартных электродных потенциалов
2
Е\ В
-3,05
-2,71
-0,76
-0,44
-0,41
Таблица 22
Равновесная электродная реакция
Sn2++2<T=Sn
H++e" = V2H2
Си2*+2?" = Си
Ag++^"=Ag
Аи3++Зе"=Аи
Е\ В
-0,14
0,00
+0,34
+0,80
+ 1,50
Чем более отрицателен электродный потенциал, тем выше
способность металла посылать ионы в раствор и тем сильнее
проявляет себя металл как восстановитель.
Все металлы, расположенные выше водорода, т. е. имеющие
отрицательное значение электродного потенциала, растворяются
в кислотах с концентрацией (активностью) ионов водорода
1 моль/л.
Если электродный потенциал металла имеет положительный
знак, то металл является окислителем по отношению к водороду
и не вытесняет его из растворов, содержащих по 1 моль/л ионов
водорода и катионов металла, а, наоборот, водород вытесняет
металл из раствора соли.
Рассмотрим гальваническую цепь из стандартных медного
и цинкового электродов (концентрация ионов металлов в
растворах по 1 моль/л). Эдс этого элемента составляет 1,10 В. Это
значение есть разность электродных потенциалов меди и цинка:
Цинк, электродный потенциал которого имеет отрицательное
значение ( — 0,76 В), посылает в раствор большее число катионов,
чем медь, поэтому отрицательный заряд цинкового электрода
будет выше и электроны с цинковой пластины переходят на
369
медную и, соединяясь с катионами меди из раствора вблизи
медного электрода, приводят к осаждению металлической меди
на электроде. Таким образом, на цинковом элекфоде
самопроизвольно проходит реакция окисления цинка, а на
медном—восстановление ионов Си2 + :
Zn-2*T=Zn2 + ; -?°=+0,76B.
Си2+ +2е~ -Си; ?°= +0,34 В.
Суммарная реакция, протекающая в этом гальваническом
элементе, записывается уравнением:
Cu2++Zn = Cu+Zn2f; ?° = 0,76+0,34=1,Ю В.
Для установления направления электродных процессов,
расчета эдс и правильного написания уравнения самопроизвольно
протекающей в гальваническом элементе реакции следует
поступать следующим образом. Пользуясь таблицей стандартных
электродных потенциалов, записывают уравнения реакций для
каждого электрода с указанием значения электродного
потенциала. Электродную реакцию с большим отрицательным или
меньшим положительным значением потенциала переписывают в
обратном направлении (при этом знак потенциала следует
изменить на противоположный). Под этим уравнением записывают
уравнение второй электродной реакции в том виде, в котором
она дана в справочной таблице. Умножают коэффициенты при
формулах веществ на такие числа, чтобы числа принятых и
отданных электронов были равны (следует обратить внимание, что
потенциалы на эти числа не умножаются!). Суммируют оба
уравнения и их потенциалы. Таким путем получают уравнение
самопроизвольно протекающей электродной реакции.
Ниже приведен пример использования данного способа для
определения, будет ли олово растворяться в растворе кислоты
сСн+ = 1 моль/л.
Из табл. 19 выписываем уравнения реакций и значения
потенциалов для олова и водорода:
Sn2+ +2e~ -Sn; ?°= -0,14 В.
В прямом направлении самопроизвольно протекает реакция,
характеризующаяся большим (алгебраическим) значением
потенциала. Так как
2 @,00> -0,14),
то такой реакцией является восстановление ионов водорода.
Уравнение другой реакции, как источника электронов, перепишем
в обратном направлении
Sn = Sn2++2e~; E°= +0,14 В.
370
Общее уравнение реакции, проходящей в гальваническом
элементе, получается суммированием обоих уравнений:
2H"+Sn = Sn2+ + H2; ?° = 0,14 В.
Таким образом, олово растворяется в растворе кислоты
с Сн+ = 1 моль/л.
Определим, буде! ли олово растворяться в воде. Из таблицы
стандартных элекч родных потенциалов находим, что потенциал
?н*\'Н2 лля воды (Сн+ = 10"~7 моль/л) не равен нулю, как
это имело место для растворов с Сн+ = 1 моль/л, а равен
-0,41 В, т. е.
H + +e- = V2H2; ?=-0,41 В при СН+ = Ю-7 моль/л.
Для олова:
Sn2+ +2e~ = Sn; ?° = -0,14 В.
Так как ( — 0,4) >( — 0,41), то в прямом направлении протекает
реакция восстановления ионов олова (как она записана
в табл. 22). Реакция, характеризующаяся меньшим потенциалом,
будет протекать в обратном направлении, т. е.
Следовательно, реакция, протекающая самопроизвольно,
выражается уравнением:
Таким образом, в воде (точнее, в растворе соли с CSn2 + ^ 1 моль/
л) олово не растворяется, а если через раствор соли олова
пропускать водород, то будет осаждаться металлическое олово.
Аналогично решается вопрос, взаимодействует ли натрий
с водой при стандартных условиях. По данным табл. 22
записываем уравнения равновесных электродных реакций:
Na++<?" = Na; ?°=-2,71 В.
Н+ (вода)+^- = 1/2Н2; ?--0,41 В.
Первый процесс протекать не может (алгебраическое значение
электродного потенциала больше для второго процесса).
Следовательно, самопроизвольно идут такие процессы
Na-e"=Na+; ?°=+2,71 В.
Н+ (вода)+е~ = 72Н2; е= -0,41 В.
Суммарное уравнение реакции имеет вид:
Na + H+ (вода) = №} +72Н2; ?=2,71-0,41=2,30 В
или
+ Vr); ?=2,30 В.
371
Таким образом, натрий самопроизвольно реагирует с водой.
Электродные потенциалы металлов зависят от концентрации
(точнее, активности) ионов металла в растворе и от температуры.
Как известно, изменение изобарного потенциала в системе
численно равно работе, совершаемой в результате химической
реакции:
AG=-A.
Работа электрического тока равна произведению числа молей
перенесенных электронов и, постоянной Фарадея F=96484 Кл/моль
и напряжения в электрической цепи. Так как электродный
потенциал— это эдс гальванической цепи с водородным электродом,
то работу электродной реакции можно рассчитать относительно
работы реакции стандартного водородного электрода:
A=nE°F.
Поскольку для водородного электрода принято ?° = 0, то и
работа его реакции также равна нулю и, следовательно, AG°, АН°
и AS° для реакции стандартного водородного электрода также
равны нулю:
1 =0; AS =0.
Подставляя выражение работы в равенство AG = — Л, получаем
или AG — —пЕ F (для стандартных условий).
Изменения изобарного потенциала при нестандартных и
стандартных условиях связаны соотношением
псп
AG = АС ° + RTln ——,
ПСИСХ
где ПСИСХ и ПСпр—соответственно произведение концентраций
(в степени их стехиометрических коэффициентов) продуктов
реакции и исходных веществ.
Объединяя выражения, получаем:
ПС
-nEF= -nE°F+RT\n np
ПСИ
Л/ t НС „о
nF ПГИСХ
Эта формула—уравнение Нернста, позволяющее вычислить
электродные потенциалы при нестандартных условиях. Для
электродного процесса
372
уравнение при 298,15 К приобретает вид:
„ го 2303 8.314 298,15.
Е Е
g
м
0,0592 1
Пользуясь уравнением Нернста, можно рассчитать значение элек-
тродно1 о потенциала при любой концентрации ионов раствора (при
известном значение ?° для данного металла). Например, потенциал
цинкового 'электрода в 0,001 М растворе его соли составит:
Следовательно, при уменьшении концентрации ионов цинка
в растворе потенциал металла становится более отрицательным
(по отношению к стандартному водородному электроду).
Ниже приведены вычисленные по уравнению Нернста
значения электродных потенциалов Е (в В) цинка и меди в растворах
различной концентрации:
Zn Си
0,001 моль/л -0.85 +0,25
0,01 моль/л -0,82 +0,28
0,1 моль/л -0.79 +0,31
1 моль/л -0,76 {?-) +0,34 (Е°)
При увеличении концентрации ионов металла в растворе
электродный потенциал возраставi, что согласуется с
теоретическими закономерностями о смещении положения
химического равновесия.
Рассчитаем эдс медь-цинкового гальванического элемента,
состоящего из цинкового электрода в 0,001 М растворе Zn2+ и
медного электрода в 0,01 М растворе Си2 + . По формуле Нернста
находим электродные потенциалы для данных концентраций
ионов в рас i воре:
°^2-l7= -0,85 В
~А 0,0592, 1 , л-и> о
,34—_-lg_=+0,28B.
Цинковый электрод заряжен отрицательно относительно меди,
и он передает электроны на медь. Следовательно, в
гальваническом элементе протекают процессы:
Zn - 2г ~ = Zn2 ч; - ?Zn/Zn,. = + 0,85 В,
= Cu; ?CuCu2.= +0,28 В.
373
Суммарное уравнение запишется так:
Znw+Cu2+(p.p)=Zn2(p.P)+Cu(K); ?=0,85+0,28 = 1,13 В.
То же значение получается при расчете по формуле
Объединяя выражение работы с зависимостью AG° от Г, А#°,
А5° и коне 1 анты равновесия электрохимической реакции,
получаем
AGo=-nE°F=AH°-TASo=-RT\nK.
Это соотношение позволяет вычислить термодинамические
характеристики AG\ Д#°, AS° и константу равновесия
электрохимического процесса, если известны, по крайней мере, два
значения Е при двух температурах. Для этого достаточно составить
систему из двух уравнений с Е°тл и Е°тл при Гх и Т2:
-nEoT,2F=AH°-T2AS°=-RT2lnKTa.
Для процессов, проходящих в растворах при стандартной
(комнатной) температуре, изменение энтропии невелико, энталь-
пийный фактор АН° намного превосходит энтропийный фактор
TAS°, и поэтому можно принять AG°«A#°. Тогда
AH°*-nE°F.
Пользуясь этой формулой, по измеренному значению
энтальпии (тепловому эффекту) окислительно-восстановительной
реакции можно оценить значение ее эде и, наоборот, вычислив по
табличным значениям электродных потенциалов эде реакции,
сделать вывод об ее энтальпии. Так, для реакции
Zn(K) + Cu%_p) = Zn2VP)+Cu(K); ?°=l,10 В,
AG°=-2 1,10-96500=-212300 Дж/моль,
и Д#°» -212 кДж/моль.
Значения AG0 и АН° можно получить, воспользовавшись
термодинамическими свойствами ионов:
Zn ,. Си ,.
A# обр 298> кДж/моль -153,64 66,94
ДС~обР.298> кДж/моль -147,16 65,56
Отсюда изменение энтальпии и изобарного потенциала в
рассматриваемой реакции окисления цинка ионами Си2+ составит:
374
АНс = - 153,64 - 66,94 = - 220,1 кДж/моль,
ДС = - 147,16-65,56= -212,72 кДж/моль.
Найдем выражение для константы равновесия
электрохимической реакции.
-RT In К=-пЕТч
откуда \пК=пЕ F/(RT).
При 298,15 К константа равновесия равна:
Для процесса, протекающего в медно-цинковом гальваническом
элементе, п = 2 и Е ' = 1,10 В. поэтому
К=?^=1016<90 2 1ЛО=1037'2=1,6-1037.
Столь большая константа равновесия говорит о практически
полном смещении положения равновесия в область
существования продуктов реакции.
Остановимся еще на одном электродном явлении —
возникновении эдс на г ранице контакта растворов с различной
концентрацией ионов. Поскольку электродный потенциал металла
зависит от концентрации раствора, то можно составить
гальваническую цепь из одинаковых металлических электродов,
погруженных в растворы с различной концентрацией ионов этого
металла.
Рассмотрим гальваническую цепь, собранную из двух
цинковых электродов, один из которых погружен в раствор соли цинка
с CZn2-=0,l моль/л, а другой — в раствор с CZni+ =0,01 моль/л.
Потенциалы этих электродов соответственно равны:
Ех = ~0,76~^9~!g~:= -0,79 В,
Электродвижущая сила этого гальванического элемента
составит:
?-=?г~?2=: -0,79-(-0,82)-0.03 В.
Гальванический элемеш, состоящий из такого рода
электродов, называется концентрационным. Чтобы получить
положительное значение эдс, следует из потенциала, возникающего в
более концентрированном растворе, вычесть иО1енциал,
возникающий в менее концентрированном растворе. В общем случае эдс
375
концентрационного элемента вычисляется по формуле (при
298,15 К):
С\>С2.
Если цепь собрана из металла и раствора его соли,
содержащей однозарядный катион, то при различии концентрации соли
в 10 раз эдс составит 0,058 В, при концентрациях, отличающихся
в 100 раз, эдс равна 0,116 В. При тех же условиях цепи с
электролитом, содержащим двухзарядные катионы, показывают эдс
в два раза, а трехзарядные—в три раза более низкие.
Работа концентрационного элемента состоит в том, что
металл, погруженный в более разбавленный раствор, посылает свои
ионы в раствор и, таким образом, растворяется; на электроде,
опущенном в более концентрированный раствор, ионы из
раствора разряжаются. Таким образом, происходит выравнивание
концентрации ионов в обоих электролитах, и элемент работает до
тех пор, пока концентрация ионов не станет одинаковой у обоих
электродов. С точки зрения термодинамических представлений
электрический ток вырабатывается концентрационным
элементом за счет стремления сисгемы перейти в состояние с
равномерным распределением молекул и ионов во всех ее частях, т. е.
в состояние с максимальной энтропией.
Концентрационные элементы широко используют в
химической исследовательской практике для определения многих
важных констант: произведения растворимости, константы
нестойкости комплексного иона, ионного произведения воды,
констант диссоциации кисло i и оснований, для нахождения
концентрации ионов водорода в водных растворах и рН растворов
и т. п.
Ниже дан пример определения произведения растворимости
бромида серебра. Для этого используется гальваническая цепь из
двух электродов (рис. 141), одним из которых служит серебряная
проволока, погруженная в
насыщенный раствор бромида серебра, другой
электрод—серебряная н ровол ока
в 0,01 М растворе нитрата серебра.
В этой цепи возникает эдс, равная
0,245 В, что обусловлено только
различием концентраций ионов серебра на
двух электродах. Вблизи первого
электрода концентрация ионов серебра
определяется произведением
растворимости:
Рис. 141. Схема
концентрированного элемента для
измерения произведения
растворимости бромида серебра
AgBr(K) = A
= [Ag+][Br-];
376
Концентрация ионов серебра у второго электрода задается
условием опыта: [Ag+] = 0,01 моль/л. По известным значениям
эдс и концентрации можно вычислить ПР:
,, п,к 0,0592, 0,01
? = 0,245 = lg
lg ПР= -12,277, откуда ПР = 5,28 • 103.
Измерив эдс и определив произведение растворимости, но
крайней мере, для двух температур, легко вычислить изменение
энтальпии, а также изобарного потенциала и энтропии при
растворении бромида серебра:
Одним из методов определения константы нестойкости
комплексного иона может быть метод, основанный на измерении эдс
концентрационного гальванического элемента. Рассмотрим
возможность применения этого метода для определения константы
нестойкости хлорида диамминсеребра [Ag(NH3J]Cl. В растворе
эта соль диссоциирует как сильный электролит:
[Ag(NH3J]Cl=[Ag(NH3J]++CL-
Недиссоциированные молекулы соли [Ag(NH3J]Cl
практически отсутствуют, и если соль взята в концентрации С моль/л,
то концентрация ионов [Ag(NH3J]+ также составит С моль/л.
Ионы [Ag(NH3J]+ диссоциируют как слабый электролит
[Ag(NH3Jr =Ag+ +2NH3: ^
Для такого усюйчивого иона, как [Ag(NH3J] + , константа
нестойкости—довольно малая величина, поэтому концентрацию
этого иона в растворе можно принять равной концентрации
соли С.
При диссоциации комплексного иона [Ag(NH3J]+ на каждый
образующийся ион серебра приходятся две молекулы аммиака,
поэтому констан i у нестойкости можно записать в виде
_[Ag*] B[
Лнсст У, Т*
откуда [Ag + J^
Для определения констан\ы нестойкосги иона [Ag(NH3J] +
составляют гальванический элемеш, в котором один электрод
представляет собой серебряную проволоку, погруженную в
раствор комплексной соли с концентрацией С моль/л, а другой
177
электрод— также серебряная проволока, но опущенная в раствор
нитрата серебра с концентрацией ионов серебра С". Измеряю i
эдс такого концентрационного элемента и рассчитывают
константу нестойкости комплексной соли, используя формулу:
Определив константу нестойкости, по крайней мере, для двух
температур, легко вычислить термодинамические характеристики
процесса диссоциации и образования комплексного иона.
Наиболее точным методом определения рН является потен-
циометрический метод, основанный на измерении зависимости
потенциала электрода от активности ионов водорода в
исследуемом растворе. Этот метод практически осуществляется с
помощью концентрационной гальванической цепи, составленной из
стандартного водородного электрода и водородного электрода
с неизвестной концентрацией ионов водорода. Предположим, что
эдс гальванической цепи, состоящей из стандартного
водородного электрода (Сг = 1 моль/л, /?н=101 325 Па) и водородного
электрода (Ри2= ' 325 Па) в раств2оре с неизвестным значением
рН, равна 0,4R В B5° С). Концентрацию ионов водорода можно
рассчитать, используя формулу:
?=^,g_L=0,414.
Откуда
-6,992s-7; (V = 1 1(Г7 моль/л.
Следовательно, рН = 7.
Кроме водородного электрода в практике потенциометриче-
ских измерений широко используется стеклянный электрод.
Действие этого электрода основано на свойстве стекла приобретать
отрицательный заряд при погружении его в водный раствор,
и потенциал этого электрода зависи! от концентрации ионов
водорода.
Концентрационные элементы являются источниками
электрических потенциалов и токов между участками с различной
концентрацией ионов в живых организмах, растениях, в земной коре,
океанических водах и т. п.
§ 53. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ РЕАКЦИИ
Ионные реакции, сопровождающиеся изменением степени
окисления атомов в молекулах реагирующих веществ,
называются окислительно-восстановительными реакциями. Типичный про-
378
цесс окисления-восстановления наглядно можно наблюдать
в следующем эксперименте.
Если слить подкисленные эквимолярные растворы перман-
ганата калия, имеющего красно-фиолетовую окраску, и нитрита
натрия, то через некоторое время реакционная смесь
обесцвечивается. Качественный анализ образовавшейся смеси показывает,
что в ней содержится ничтожно мало ионов NO2~ и МпО4
и значительное количество ионов NO3~ и Мп2 . Очевидно,
произошло превращение
NO2", MnO4" — МО3~, Мп2 + .
В отдельности растворенные перманганат калия и нитрит
натрия могут храниться долгое время без изменения,
следовательно, наблюдаемая реакция обусловлена двумя
взаимосвязанными переходами, в результате которых изменяется степень
окисления (валентное состояние) азота и марганца:
NO2~ — N(V; MnO4" —> Мп2 + .
Первый переход представляет собой реакцию окисления,
второй—реакцию восстановления.
Для составления уравнения окислительно-восстановительной
реакции, протекающей в водном растворе, удобно использовать
метод электронно-ионного баланса. Этот метод рассмотрен ниже
на примере составления уравнения реакции между пермангана-
том калия и нитритом калия в водном растворе.
Сначала составляют уравнение реакций окисления и
восстановления. В схеме процесса окисления число атомов азота,
входящих в ионы нитрита и нитрата, одно и то же, число атомов
кислорода разное. Для уравнивания числа атомов кислорода
в левую часть схемы этого перехода записывают молекулу воды
(или ионы ОН ~ для щелочной среды):
Число атомов N и О в обеих частях этой схемы одинаково,
а атомы водорода указаны только в левой части. Для
уравнивания числа атомов водорода справа в схему приписывают
недостающее их число в виде ионов водорода:
В данной записи не выполняется равенство суммарных зарядов:
сумма зарядов слева (—1) не равна сумме зарядов справа (+1).
С учетом правила сохранения зарядов ионное уравнение этого
процесса должно быть записано так:
Аналогичные операции проводят при составлении ионного
уравнения перехода перманганат-иона в двухвалентный ион
марганца:
379
MnO4" — Mn2++4H2O,
+8H+ —Mn2++4H2O,
MnO4" +8H+ +5e" =Mn2+ +4H2O.
Разумеется, при составлении уравнений реакции их не следует
переписывать несколько раз, а нужно последовательно дополнять
недостающими ионами и молекулами и расставлять стехиомет-
рические коэффициенты.
Таким образом получают уравнения двух процессов,
одновременно протекающих в растворе:
В первой реакции происходит присоединение электронов, это
реакция восстановления, а ион МпО4~—окислитель. Во второй
реакции происходит отдача электронов, это реакция окисления^
а ион NO2~—восстановитель.
В общем уравнении окислительно-восстановительного
процесса число принятых окислителем электронов должно равняться
числу электронов, отданных восстановителем, поэтому
умножают стехиометрические коэффициенты первого уравнения на 2,
а второго—на 5 (чтобы получить наименьшее одинаковое число
принятых и отданных электронов) и суммируют оба выражения:
5NCV +5Н2О~ 10*Г = 5NO3~
2МпО4- + 16Н+ + 10е~ =2Мп2+ +8Н2О
5NOr+2MnO4~+6H+=5NO3~+2Mn2+
В левой части уравнения находятся ионы водорода, что
говорит о необходимости проведения реакции в кислой среде. Если
растворы были подкислены серной кислотой, то процесс
описывается следующим молекулярным уравнением:
При составлении молекулярного уравнения требуется
уравнять число катионов, входящих в состав солей.
Метод электронно-ионного баланса используется в основном
для подбора стехиометрических коэффициентов в уравнениях
окислительно-восстановительных реакций, протекающих в
водных растворах. Этот метод имеет то преимущество, что при его
применении необязательно знать степени окисления атомов,
участвующих в реакции ионов.
Коэффициенты в уравнениях реакций, проходящих в газовом
или кристаллическом состоянии, а также в водных растворах,
можно подбирать методом электронного баланса. Разберем этот
метод на примере составления уравнения реакции разложения
хлората калия. Этот процесс протекает по схеме:
380
— КС1 + КС1О4.
В результате этого процесса хлор в состоянии С15+ (в КС1О3)
переходит в С1~ (в КС1) и С1 (в КС1О4). Для нахождения
стехиометрических коэффициентов подбирают множители,
уравнивающие число отданных и принятых электронов:
4С15+=СГ+ЗС1
7 +
Следовательно, молекулярное уравнение
окислительно-восстановительного процесса разложения хлората калия следует
записать так:
4КСЮ3-
Окислительно-восстановительные реакции разделяют на три
группы,
1. Межатомное или межмолекулярное
окисление-восстановление. Это наиболее обширная группа
окислительно-восстановительных процессов. В этих реакциях обмен электронами
происходит между различными по составу частицами (атомами,
ионами, молекулами), например:
Fe(K) + CuSO4 (p-p) = Cuw + FeSO4(p-p),
5Н2О2 + 2KMnO4 + 3H2SO4 - 5O2 + 2MnSO4+K2SO4+8H2O.
2. Самоокисление-самовосстановление, или реакции
диспропорционирования. Эту группу составляют реакции, в которых
восстановителем и окислителем являются атомы одного элемента,
имеющие степень окисления, промежуточную между некоторыми
более высоким и более низким значениями. К этому типу
относится рассмотренная выше реакция разложения КС1О3. В
реакциях диспропорционирования молекулы или атомы одного и
того же вещества реагируют друг с другом как восстановитель
и окислитель вследствие того, что имеющийся в них атом с
промежуточной степенью окисления способен отдавать и принимать
электроны, переходя один—в состояние более высокой степени
окисления, другой—в состояние более низкой степени окисления.
Примеры:
2NO2 + Н2О = Н + + NO3 " + HNO2
3NaOCl = NaClO3 + 2NaCl
2H2MnO4 = 2HMnO4 + MnO2 + 2H2O
+ H2SO4 = KNO3 + 2NO+K2SO4
2K2[Sn(OHL]-K2[Sn(OHN
381
3. Внутримо. 1ску. шрпое окис icnue-eoc станов, iemu\ К ) \ о и
группе относятся превращения молекул, в которых два разных
атома в молекуле вещества могут бьпь но оiношению друг
к другу окислителем и восстановителем. Например:
Как и в процессах других типов, например, при окисли гельпо-
восстановительных взаимодействиях, в
окислительно-восстановительных реакциях окислитель и восстановитель реагируют друг
с другом в эквивалентных количествах. Эквивалентом окислителя
или восстановителя называется некоторая реальная или условная
частица, которая может присоединять или отдавать один электрон
в окислительно-восстановительной реакции. Молярная масса
эквивалента окислителя или восстановителя (масса моля
эквивалента) относится к конкретной реакции. Например, в реакции
+ ЗН2О
молярные массы эквивалента окислителя КМпО4 и
восстановителя КЛМО2 равны соответственно:
ЭКмпо4= 158,0/5 = 31,60 г/моль; 3KNo2 = 85,1/2 = 42,05 г/моль.
В нейтральных и щелочных средах нерманганат-ион
восстанавливается до МпО2 и манганат-иона МпО42 , и молярные массы
эквивалента КМпО4 соответственно равны:
ЭКм„о4 = 158,0/3 = 52,67 г/моль;
42";
МпО4~+<Г =МпО42"; Экмпо^ 158,0/1 = 158,00 г/моль.
На законе эквивалентов основано количественное
определение окислителей и восстановителей методом титрования.
Из титриметрических методов, основанных на окислительно-
восстановительных реакциях, в аналитической практике широкое
применение имеет перманганатометрия, использующая в
качестве титранта раствор перманганата калия. При добавлении в
раствор какого-либо восстановителя темно-фиолетовая окраска
раствора перманганата калия исчезает. При титровании следует
установить момент, когда одна капля раствора перманганата
окрасит весь титруемый рас i вор в неисчезающий в течение 1 —
2 мин бледно-розовый цвет. Этот момент соответствует точке
эквивалентности. По объему раствора перманганата калия,
пошедшему на титрование исследуемого вещества, и известной
концентрации (нормальности) титранта находится концентрация
изучаемого восстановителя в растворе.
382
Кроме перманганатометрии к
окислительно-восстановительным титриметрическим методам анализа относятся иодометрия
(титрант 12), хроматометрия (К2О207), броматометрия (КВЮ3),
цериметрия [Ce(SO4J], ванадатометрия (NaVO3) и др. Точка
эквивалентности при титровании этими методами
устанавливается с помощью окислительно-восстановительных индикаторов.
Каждая окислительно-восстановительная реакция
складывается из двух процессов (полуреакций) окисления и
восстановления, протекающих одновременно с переходом электронов от
восстановителя к окислителю. Эти процессы могут проходить
пространственно раздельно (но не независимо друг от друга),
например, в замкнутой гальванической цепи. Так,
рассмотренную выше реакцию окисления нитрита калия перманганатом
калия можно осуществить следующим образом. Если в один
сосуд налить водный раствор KNO2, а в
другой—подкисленный раствор КМпО4, соединить оба сосуда
электролитическим мостиком, опустить в сосуды пластины из платины
(инертный материал) и соединить их проводником, то в такой
замкнутой гальванической цепи электроизмерительный прибор
покажет наличие тока. Электродвижущая сила полученного
гальванического элемента при стандартных условиях B98,15 К,
СШоА- = Смп2+ = Сн+ = CNo2- = CNo3- =*1 моль/л) равна 0,57 В.
Направление тока в этой гальванической цепи указывает на
переход электронов от платины в растворе KNO2 к платине
в растворе КМпО4. Таким образом, в первом и во втором
сосудах раздельно протекают полуреакции:
NO2" + H2O-2e" =NO3" +2H+,
Рассмотренный гальванический элемент состоит из двух
окислительно-восстановительных электродов Mn2+/MnO4 ~ и
NO2~/NO3~. Электродный потенциал
(окислительно-восстановительный потенциал) таких электродов количественно
характеризует окислительную или восстановительную способность
веществ.
Как было указано выше, электродный потенциал измерить
невозможно, его определяют сравнением с потенциалом
стандартного электрода, в частности водородного, потенциал
которого принят равным нулю. Для этого собирают гальваническую
цепь из окислительно-восстановительного электрода, например,
Мп2+/МпО4~ (платиновая пластина, опущенная в подкисленный
раствор КМпО4), и стандартного водородного электрода и
компенсационным способом измеряют эдс этой цепи.
При работе этого элемента через некоторое время
обнаруживается уменьшение концентрации ионов МпО4~ и
увеличение концентрации ионов Мп2+ в одном сосуде и увеличение
383
концентрации Н+ в другом сосуде. Очевидно, что в системе
проходят процессы:
10
2МпО4~+6Н+ + 5Н2 = 2Мп2++8Н2О
При стандартных условиях, т. е. при 298,15 К, рН2 = 1,013 • 105 Па
и СМпо4_ = СМп2+ = Сн+ = 1 моль/л, электродвижущая сила (эдс)
этого элемента равна 1,51 В. Значит, стандартный электродный
потенциал Мп /МпО4~ составляет 1,51 В. Аналогично
определяют стандартный электродный потенциал системы NO2~/NO3~;
он равен +0,94 В.
Значения стандартных электродных потенциалов,
приводимые в справочных таблицах, относятся к полуреакциям
восстановления (т. е. к процессам, в уравнениях которых прибавляемые
электроны записываются перед знаком равенства).
По значениям стандартных электродных потенциалов можно
определить направление (возможность)
окислительно-восстановительной реакции, протекающей в водном растворе.
Окислительно-восстановительная реакция самопроизвольно
протекает в растворе, если стандартный потенциал системы,
включающей окислитель этой реакции, больше стандартного
потенциала системы, включающей восстановитель реакции. Это
означает также, что окислительно-восстановительная реакция
протекает в водном растворе, если разность стандартных
потенциалов окислительно-восстановительных пар будет
положительной величиной, т. е. электродвижущая сила реакции ?°>0. Так,
?°мп-/Мпо4->?ож>2^оз- (U51 В>0,94В).
Другими словами, направление
окислительно-восстановительной реакции определяется направлением перехода
электронов с электрода (системы веществ), имеющего более высокий
отрицательный или меньший положительный потенциал. На
этом электроде происходит реакция отдачи электронов.
Уравнение этой реакции записывается уравнением, противоположном
тому, которые приводятся в справочных таблицах, а знак
потенциала также изменяется:
NO2 ~+H2O-2e-=NO3-+2H + ; ?c=-0,94B.
Реакция на другом электроде протекает в том направлении,
в котором она записана в таблице потенциалов, так как этот
электрод принимает электроны:
; ?°=1,51 В.
Суммируя одну самопроизвольно идущую электродную
реакцию с другой, протекающей под действием первой, получаем
384
общее уравнение реакции, самопроизвольно протекающей в
гальваническом элемеше. Суммируя электродные потенциалы,
находим эдс элемента: Е = — 0,94+ 1,51 = +0,57 В. Положительное
значение эдс подтверждает возможность протекания реакции
в целом:
; E J=-0,94B
1Qg-=2Mn2++8H2O; ?°=1,51 В
5NO2~+2MnO4-+6H+=5NO3"+2Mn24+3H2O; ?° = 0,57 В
Значительно проще определяется возможность протекания
окислительно-восстановительной реакции, если пользоваться
следующими рекомендациями. Выясним, например,
осуществляется ли реакция в водном растворе между иодидом натрия Nal
и свободным хлором. Для этого следует выписать из справочной
таблицы стандартные электродные потенциалы
соответствующих реакций:
12 + 2*Г=2Г; Е ' = 0,54 В.
?'=1,36 В.
Одно из уравнений переписывают в противоположном
направлении, изменив знак потенциала также на противоположный,
и полученное уравнение суммируют с другим. Суммирование
потенциалов дает значение электродвижущей силы. Если оно
окажется отрицательным, процесс, отражаемый этим
уравнением, в стандартных условиях невозможен. Самопроизвольно будет
протекать противоположный процесс, для которого ?с>0.
Если вторую из указанных реакций записав в обратном
направлении, получим:
=С12; ?С=-1,36В
=2Г; ?=0,54 В
; ?'=-0,82 В
Полученное отрицательное значение эдс показывает, что иод
не способен реагировать с хлорид-ионами. Протекание
противоположного процесса возможно:
2Г+С12 = 12 + 2СГ; ? =+0,82 В.
Окислительные свойства молекул или ионов тем сильнее, чем
больше положительное значение и ниже отрицательное значение
потенциалов.
Самый сильный окислитель—фтор (Ес\ г, = 2,87 В).
Самый сильный восстановитель—литий (Е °Li. ,Li=— 3,03 В). При
переходе от фтора к иоду окислительная способность галогенов
снижается:
13 2452 385
Реакция L , В
F2r2<?"=2F +2.85
Cl2 + 2tf~ = 2C1 +1,36
Br2 + 2*~=2Br~ +1,09
12+2р"=2Г +0,54
Поэюму галоген в состоянии простого вещества, имеющий
более высокое положительное значение стандартного
электродного потенциала, вытесняет другой галоген из раствора,
содержащего простые анионы этого галогена. Так, если через
раствор KI пропускать газообразный хлор, то будет
выделяться иод, а в растворе образуются хлорид-ионы (см, пример
выше).
Правило, что свободный галоген Г2, стоящий левее в ряду
F —C1 —Вг— I, вытесняет из растворов галогенидов следующие за
ним галогены (С12 + 2Вг~=2СГ +Вг2), относится только к
водным растворам солей галогеноводородных кислот. В то же время
свободный иод вытесняет хлор из растворов солей его
кислородсодержащих кислот:
Действительно, две окислительно-восстановительные пары
(полуреакции) в этом процессе имеют следующие значения
стандартных электродных потенциалов:
О; ?° = 1,47В
Поскольку ?'оС|2/СЮз->?1О|2/ю3 » го первая полуреакция
самопроизвольно протекает в том направлении, как она записана.
Вторая полуреакция может проходить как процесс отдачи
электронов, т. е. в противоположном направлении:
12 + 6Н2О-10е=2Ю3 +12Н + ; ?°=~1,19В.
Следовательно, суммарная реакция складывается из процессов:
; ?°=-1,19 В
?° = 1,47» 1,19 = 0,28 В
Окислительно-восстановительные потенциалы и эде окисли-
хельно-восстановительных реакций, протекающих в водном рас-
гворе, зависят от концентрации (точнее, активностей)
участвующих в процессе ионов. Эта зависимость выражается формулой
Нернста
386
где ПСпрод и ПСИСХ — произведение концсш раций (в cieueHH их
стехиометрических коэффициентов) продуктов реакции и
исходных веществ;
п — число молей участвующих в процессе электронов;
Е°—стандартный элекфодный шненциал полуреакции или
стандартная ЭДС окислигелыю-восстановительной реакции;
Е—электродный потенциал полуреакции или ЭДС
окислительно-восстановительной реакции при нестандартных, любых
заданных концентрациях веществ, входящих в произведения ПСпрод
иПСиех.
Подставив в формулу значение констат и скшдартиой
температуры, получаем
"96484" Ь ПСМСЖ " п ё ПССЖ '
Обрати (с внимание на ю.чю дробь под знаком логарифма —
это не константа равновесия, но по внешнему виду похожа на нее.
Концентрации кристаллических фаз и воды в расчетах не
участвуют! Формула Нернста позволяет находить как потенциалы
электродных реакций (полуреакций), 1ак и ЭДС окисли! ел ьно-воесга-
новительной реакции в целом.
Например, для реакции
MnO4 + 8H + + 5t> =Mn2f+4H2O; E =1,51 В.
Уменьшение концентрации нермангана1-ионов до 0,01 моль/л
приводит к снижению по1енциала (при C^ni- = C\r = 1 моль/л):
^ _о 0.0592, Смп- . С1 v,,^.z. , . . лп
§4 — /* I q — I л I IО — I а\4 t\
S ёГ Г8 ~~1'J1 1 ёПП1 j — 1^^0-
э к мпо4 *- и* - и'и| *
На значении окислительно-восстановительного потенциала
сильно сказывается изменение рН раствора. В растворе с рН = 7
(нейтральная среда) при СМпО, =Смп- = • моль/л:
с . С1 0,0592 , 1 Л ос о
?=1,51 lg ---=0,85 В.
Варьируя концентрации вещее i в, учас1вующих в окислитель-
но-восстановигельном процессе, и регулируя рН раствора,
можно изменяв не только эде реакции, но и ее направление (если
потенциалы двух полуреакций не слишком сильно различаются).
По известному значению эде реакции можно вычислить
изменение изобарного по1енциала и коисинту равновесия.
Воспользовавшись известными формулами
и* 387
"=-nE F
AG°= -RT\n K= -2,303RT\gK
рассчитаем AG° и константу равновесия (при 298,15 К) реакции:
5NO2-+2MnO4-+6H+=5NO3"+2Mn2++3H2O;?c-0,57 В
AG°= -w?loF=-10 0,57 -96500= -550050 Дж/моль«
« 550 кДж/моль
lgK=\ 6,90 л? ° = 16,90 • 10 0,57 = 96,33
К= 1096'33 = 100'33 • 1096 = 2,14 • 1096.
Столь высокое значение константы равновесия говорит о
практически полном смещении равновесия в направлении
образования Мп2+ и NO3".
Изменение изобарного потенциала, эдс и константу
равновесия реакции с участием ионов можно рассчитать,
воспользовавшись табличными значениями Atfc и S° участвующих в реакции
веществ.
§ 54. ЭЛЕКТРОХИМИЯ—КОРРОЗИЯ, ЭЛЕКТРОЛИЗ, АККУМУЛЯТОРЫ
Коррозия
Все металлы и изделия из них подвергаются разрушению
в результате химического взаимодействия с окружающими их
веществами. В самом общем случае эти процессы называются
коррозионными, или просто коррозией (механическое разрушение
называется эрозией). Суммарный химический процесс коррозии
в системе «вещество—окружение» всегда самопроизволен,
АС<0, хотя отдельные реакции могут иметь AG>0 и проходить
под воздействием других реакций.
Ниже пойдет речь о коррозии металлов, процессы которой
принято разделять на два типа — химические и
электрохимические.
Химическая, или газовая, коррозия—это коррозия, связанная
со взаимодействием металлических материалов с газовой фазой,
чаще всего с кислородом и во влажной среде.
Электрохимическая, или эмкШрбЛигпная^ коррозия — это
коррозия, обусловленная взаимодействием металлических
материалов с растворами электролитов, чаще всего связанная с
образованием гальванических элементов при контакте различных
металлов или с их нахождением в растворах с различными
388
концентрациями электролита. Любая металлическая
конструкция в обычных условиях представляет собой гальванический
элемент.
В ряде случаев коррозионные процессы обоих типов
протекают одновременно и создают сложнейшую систему
взаимосвязанных реакций, имеющих различные значения изобарных
потенциалов (ЭДС) и констант скорости. Эта система настолько сложна,
что часто для новых материалов или сред коррозия оказывается
непредсказуемой.
Изучение коррозии всегда преследует цель ее предотвращения
или замедления. Коррозия—самопроизвольный (A G < 0) при
данных условиях процесс, и защита от нее основывается на
термодинамических и кинетических принципах. Некоторые
важнейшие способы защиты от коррозии состоят в следующем.
Электрохимическая (электролитная) коррозия
предотвращается контактом разрушающегося от коррозии металла с
металлом, имеющим более отрицательный электродный потенциал,
например, железо в контакте с цинком или покрытое им
(оцинкованное) не подвергается коррозии в связи с тем, что в
образующемся гальваническом элементе растворяется цинк, а на железе
выделяется водород.
Цинк по сравнению с железом обладает большей
способностью к передаче ионов в раствор, поэтому приобретает
отрицательный заряд, переходящий на железо. Возникающая высокая
концентрация электронов на железе препятствует переходу
железа в виде ионов в раствор, что уже защищает железо от
растворения (коррозии). Так как переход ионов железа в раствор
ограничен, поверхность железа не закрыта слоем ионов железа (двойной
электрический слой) и электроны на железе беспрепятственно
участвуют в реакции с водой в нейтральной и щелочной средах
или с ионами водорода в кислой среде. В результате на железе
выделяется водород. Слой водорода (атомарного или
молекулярного) препятствует не только подходу к поверхности железа
коррозионно-агрессивных веществ, но и окислению его
поверхности (водород—восстановитель!).
Электрохимическая коррозия главным образом вызывается
примесями и различного рода неоднородностями, выходящими
на поверхность металла. При соприкосновении металла с
электролитом, которым может быть просто влага, адсорбируемая
или конденсирующаяся на поверхности, возникают
гальванические элементы. В них основной металл играет роль анода, а
загрязнения и примеси других металлов, имеющие более
положительный потенциал, становятся катодами. На катоде обычно
выделяется водород, а анод—основной металл—корродирует.
Процесс усложняется другим процессом, связанным с раство-
peimeM в электролите кислорода, который на катоде может
участвовать в реакции (кислородная деполяризация катода)
389
Эта реакция становшея особенно важной в основной (щелочной)
среде. Поэюму водород образуется в кислоiной среде, а в
нейтральной и основной средах водород обычно не выделяется.
Заметим, что в обычных условиях коррозии водород в свободном
виде также не выделяется, так как окисляется атмосферным
кислородом и превращается в воду уже в момент образования.
Микрогальваническими элементами может быть обьяснена
характерная особенность кинетики взаимодействия металлов
с кисло тми — в течение довольно длительного начального
периода скорость растворения металла и выделения водорода
возрастает. Эю связано с постепенным накоплением на реакционной
поверхности iex включений, которые в начале реакции
находились не на поверхности.
На практике конiакт менее активною, но разрушающеюся
коррозией металла с более активным осуществляется двумя
путями: покрытием поверхности менее активною металла слоем
более активного (оцинкованное железо) или размещением куска
более активного металла в той же среде поблизости от
защищаемого металлическою изделия и соединением обоих металлов
проводником (так называемая протекторная защита).
Защита металла oi электролитной коррозии nyieM
повышения его отрица1слыюго заряда может бьнь осуществлена не
только кон i актом с более активным металлом, но и
подсоединением его к офицательному полюсу внешнею источника
постоянного тока. Защищаемое изделие играет роль каюда (катодная
защита).
Перечисленные способы защиты oi коррозии имеют
термодинамическую основу: повышением отрицательного заряда искус-
С1венно изменяв!ся электродный потенциал металла—ЭДС
гальванического элемеша,— и !ем самым изменяется знак
изобарного потенциала реакции, ответеiвенной за коррозию
данного металла.
Целый ряд способов защиты от коррозии основан на
кинетических принципах: в коррозионную среду вводя 1ся интибиторы
коррозии, поверхнос1ь корродирующего изделия покрывается
слоем какого-либо вещества, препятс!вующего доступу
агрессивных агентов, и др.
Довольно часто железо покрывают слоем олова («лужение»),
которое устойчиво к действию воды обычного солевого состава
в присутствии кислорода воздуха. Поведение луженого железа
в условиях эксплуатации изделий принципиально
противоположно поведению оцинкованного железа. Повреждение слоя цинка на
железе приводиi к разъеданию цинка, чт предохраняет железо
от ржавения. Но повреждение слоя олова приводит к ржавлению
железа при неизменяемости покрытия. При этом также возникает
390
гальванический элемент, но направление перехода элскфонов
в нем иное, чем в случае с оцинкованным железом. Железо
в соответствии со значениями стандартных элек1 родных
потенциалов обладает большей способностью посылать ионы
в раствор и приобретав отрицательный заряд. В cooiBeicr-
вии с этим электроны с железа переходят на олово, на
поверхности которого они, взаимодействуя в кислотной среде
с ионами Н+ или молекулами воды в нейтральной и щелочной
средах, приводят к образованию водорода. Железо же в этих
условиях непрерывно переходит в раствор в виде ионов Fe2 + .
Дальнейшая судьба ионов Fe2+ не зависит от тина покрытия
железа.
Переходящие в раствор с железа ионы Fe2^ начинают
участвовать в многочисленных реакциях, которые схематически
можно представить следующим образом. Они взаимодействую г с
ионами ОН", образующимися в реакции выделения водорода из
воды:
Fe2++2OH"=Fe(OHJ.
Ионы Fe2+ подвергаются гидролизу:
Они взаимодействуют с растворенным в воде углекислым газом
или карбонат-ионами:
Fe2++HCO3~ = FeHCO3~,
Все частицы с двухвалентным железом в окисли тельных условиях
неустойчивы и окисляются до частиц, имеющих железо в
трехвалентном состоянии. Например:
4Fe(OHJ + O2 + 2H2O = 4Fe(OHK.
Основным продуктом ржавления железа ечтают гидроксид
Fe(OHK. При удалении влаги (высушивании) i идроксид
частично отщепляет воду:
и получается вещество, отвечающее составу бурой ржавчины.
Гальванические микроэлементы образуются не только при
контакте двух различных металлов, но и при наличии примесей,
неоднородности в составе металла (сплава), а также при
наличии в металлическом изделии любых участков, оишчающихся
391
друг от друга какими-либо парамефами: температурой,
давлением, плотностью, состоянием поверхности и т. п. Даже
предыстория обработки играет роль в возникновении
коррозии. Наличие деформированного и недеформированного
участков приводит к возникновению разности потенциалов, и
деформированный участок корродирует сильнее
недеформированного.
Коррозия—сложнейшая химическая система, обладающая
огромной и ярко выраженной многоаспектностью способов
воздействия на нее и последствий от ее прохождения. Коррозия
зависит от химической природы вещества, его окружения и
термодинамических и кинетических характерисшк отдельных
реакций и всего процесса в целом, а также от многих других часто не
поддающихся учету факторов (примеси, состояние поверхности,
предыстория получения и обработки изделия, климат,
географические и геологические условия эксплуатации и т. п.). Именно
поэтому коррозия часто непредсказуема, а результаты
лабораторного эксперимента не подтверждаются в условиях реального
использования материала.
Важнейшим показателем коррозии является ее скорость. Она
выражается различными единицами измерения. Часто скорость
коррозии оценивают в изменении (потере) массы на единице
поверхности за некоторый период времени, например г/м2год
или моль/см2 • год. Принято также выражать коррозию
уменьшением толщины изучаемого образца или толщиной
образовавшегося слоя продукта. Скорость электрохимической коррозии
может быть выражена плотностью тока, необходимой для
данного изменения массы или толщины образца в единицу времени.
При действии на металл кислоты скорость растворения может
быть определена объемом выделившегося газа.
По стехиометрическим коэффициентам уравнения реакции
коррозии довольно просты, но по механизму и
элементарным стадиям коррозия относится к числу наиболее сложных
гетерогенных реакций. Скорость коррозии определяется
скоростью наиболее медленной в данных условиях стадии, а она
может быть как химической (переход электрона, окисление
металла и т. п.), так и физической (диффузия электролита или газа)
природы.
Катализатор ускоряет, а ингибитор замедляет обычно лишь
одну из стадий химического процесса. Из-за многостадийное™
процесса коррозии и очень высокой чувствительности скорости
отдельных стадий даже к незначительному изменению внешних
условий поиск ингибитора для какого-либо реального процесса
коррозии теоретически очень затруднен, и часто положительные
результаты обнаруживаются случайно. Любой предполагаемый
ингибитор должен сделать одну из стадий самой медленной
и тем самым лимитирующей весь процесс.
392
Электролиз
При прохождении электрического тока через металлы
(проводники 1-го рода) химические реакции не проходят и металлы
остаются неизменными. Если же электрический ток проходит
через расплав или раствор электролита (проводники 2-го рода),
на границе электролит—металлический проводник (электрод)
происходят различные химические реакции (электролиз) и
образуются новые соединения.
При электролизе катионы перемещаются к отрицательному
электроду (катоду), а анионы — к положительному электроду
(аноду). При этом, однако, не всегда катионы и анионы
электролита разряжаются, принимая или отдавая электроны. Часто в
реакциях электролиза принимает участие растворитель-электролит,
например, вода.
Принципиальное различие между реакциями в
гальваническом элементе и электролизере заключается только в их
направлении и самопроизвольности. В замкнутой цепи гальванического
элемента электрохимическая реакция протекает
самопроизвольно, а в электролизере—только под воздействием электрического
тока внешнего источника.
Следует обратить внимание на название электродов:
в гальваническом элементе отрицательный электрод—анод,
а положительный—катод;
в электролизере, наоборот, отрицательный электрод—катод,
а положительный—анод.
При этом следует помнить, что термины «отрицательный»
и «положительный» всегда относятся к полюсам источника тока,
именно так они и обозначают электроды электролизера. Общее
в этих процессах состой г в том, что как в гальваническом
элементе, так и в электролизере на отрицательном электроде создается
избыток электронов, а на положительном — их недостаток. На
катоде ионы или молекулы восстанавливаются под действием
электронов, на аноде частицы окисляются, отдавая свои
электроны электроду.
Запомните: в электролизере катионы (Mw + ) перемещаются
к катоду ( —), а анионы (Ая~)— к аноду ( + ).
Напряжением разложения электролита при электролизе
называется минимальное напряжение (внешняя ЭДС), которое нужно
приложит ь к электродам. Например, для раствора хлорида цинка
при стандартных условиях
Zn24+2* = Zn E = -76 В,
С12 + 2* = 2СГ ? = + 1,36 В.
и напряжение разложения равно сумме стандартных электродных
потенциалов обоих электродов: 0,76+1,36 = 2,12 В, т. е. напряжение
393
разложения не может бы гь ниже ЭДС соответствующего
гальванического элемента.
Напряжение разложения составляется из потенциалов двух
электродов—потенциалов разряжения ионов.
Потенциал разряжения катиона иногда называют
потенциалом осаждения металла. Это тог минимальный потенциал,
который должен быть приложен к электроду для того, чтобы катион
потерял заряд и произошло осаждение металла. Для некоторых
ионов (Fe , Cu2 + , Ag+, Cd2+) потенциал осаждения близок
к электродному потенциалу, для других же ионов (Fe2 + , Co2 + ,
Ni2 + ) потенциалы осаждения значительно превышают
электродные потенциалы металлов—для электролиза необходимо
определенное перенапряжение.
При электролизе водных растворов электролитов часто
вместо металла на катоде выделяе1ся не металл, а водород. В
кислотных средах водород образуется по реакции
В нейтральных и щелочных средах водород образуется по
реакции с участием молекул воды:
Такие катионы, как Na+ или К+, в водном растворе вообще
не разряжаются, а выделяется водород.
Катионы могут быть сгруппированы по способности разря-
жа г ься в ряд от неразряжающихся до легко разряжающихся. При
этом изменяются и продукты электролиза. Обратите внимание,
что для некоторых катионов возможно одновременное
образование металла и водорода.
Ниже даны катионы в порядке понижения трудности их
разряжения и продукты электролиза:
К a i и ot i ы П ро дукт ы
мек i ролиза
Li4, К\ Na+,Mg*. AP\ Н+ (перенаир ) Н2
Mn2\Zn2\Cr^, Fe2*, Н1 (рН 7) М + Н2
Co2t, Ni24, Sr2 + , Pb2\ H' (рНО) М+Н2
Cu2f. Ag+, Au3+ M
Различное положение водорода в этом ряду объясняется
следующими причинами. Положение водорода между свинцом
и медью соответствует численным значениям стандаршых
электродных потенциалов при См„* =Сн+ = 1 моль/л, г. е. при рН 0.
Положение водорода между железом и кобалыом соответствует
электродному потенциалу водорода в воде при рН
7 (?УН, н I- — —0,414 В). При этих условиях из растворов
могут быть осаждены все металлы, значение Е° которых больше,
чем —0,414 В. Однако па практике кроме кобальта, никеля, оло-
394
ва и свинца удается из водных растворов осадить также цинк,
хром и железо. Это объясняется 1ем, что выделение на катоде
газообразного водорода затрудняется перенапряжением
водорода.
Таким образом, в ряду катионов от Li+ до А13+ металл не
образуется, а при электролизе выделяется водород за счет
восстановления воды. В ряду катионов от Мп2х до РЬ2+ при
электролизе образуются одновременно металл и водород, и, наконец,
в ряду Си2 + — Аи3 образуется только металл.
Следовательно, чем левее стоит металл в ряду стандартных
электродных потенциалов (ряд напряжений), тем труднее
выделить эгот металл электролизом водного раствора.
Если к раствору, содержащему несколько катионов,
приложить постепенно возрастающее напряжение, то электролиз
начинается тогда, когда достигается потенциал осаждения катиона
с самым высоким электродным потенциалом (наиболее
положительным). При электролизе раствора, содержащего ионы цинка
(?° = —0,76 В) и меди (?° = +0,34 В), на катоде вначале
выделяется медь, и лишь после того, как почти все ионы Си2+
разрядятся, начнет выделяться цинк. Таким образом, если в растворе
одновременно содержатся различные катионы, то при
электролизе их можно выделить последовательно в соответствии со
значениями их электродных тменциалов. При этом предполат ает-
ся, что перенапряжение выделения металлов для них примерно
одинаково (и невелико).
Что касайся потенциалов разряжения анионов, то здесь
картина намного сложнее из-за способности воды
участвовать в процессе электролиза. В общем случае можно сказа гь,
что на аноде сначала разряжаются анионы с самым низким
потенциалом (наиболее отрицательные). Если раствор содержит
ионы СГ (?6=1,36 В), Вг" (?°-1,09 В) и I" (?Ъ = 0,54 В),
ю сначала будет образовываться йод, затем бром и, наконец,
хлор. Фгорид-ионы в водном растворе вообще разряжаться
не могут (?° = 2,87 В).
Большинство кислородсодержащих анионов (кроме ацетат-
иона) в водном растворе не разряжаются, вместо них в
кислотных и нейтральных растворах происходит разложение воды:
а в щелочных растворах — разрядка гидроксид-ионов:
2ОН"-2е>=1/2О2
Анионы по их способности разряжаться при электролизе
водных растворов располагаются в следующем ряду от неразряжа-
ющихся в водном раст воре анионов кислородсодержащих кислот
типа SO42~, NO3~ до легкоразряжающихся:
395
Анионы SO42", NO3" И Т. П . ОН О2
F" О2
СГ, Вг", I С12(СЮ-. СЮ3"), Вг2, Ц + О,)
S2" S, SO2( + O2)
Например, при электролизе серной кислоты (графиювые
электроды) происходят следующие процессы:
на катоде 2Н + + 2е = Н2,
на аноде 2Н2О-4е = О2 + Н + .
Суммарное уравнение:
2Н2О = 2Н2+О2,
т. е. при электролизе раствора серной кислоты водород и
кислород выделяются за счет разложения молекул воды. Продукты
электролиза: водород и кислород.
Электролиз раствора сульфата меди:
на катоде Си2 + + 2е = Си,
на аноде 2Н2О-4е = О2
Суммарное уравнение:
или
2CuSO4+2H2O =
Продукты электролиза: медь, кислород, серная кислота
Возможность разряжения аниона зависит от его
концентрации. Так, продукты электролиза концентрированного и
разбавленного растворов NaCl — хлор и кислород соответственно.
Электролиз разбавленного раствора хлорида натрия
проходит без разряжения ионов С1~ (и соответственно ионов Na + ),
т. е. происходит разложение воды. По мере повышения
концентрации соли на аноде вместе с кислородом начинается выделение
хлора, и в концентрированных растворах образуется хлор (с
примесью кислорода):
на катоде 2Н2О + 2е = Н2 + 2ОН ",
на аноде 2С\~ ~2е = С\2.
Суммарное уравнение:
или
Продукты электролиза: водород, хлор и гидроксид натрия.
396
В случае выделения хлора при электролизе растворов
хлоридов на основной процесс образования хлора накладываются
реакции взаимодействия хлора с водой (гидролиз) и последующих
превращений образующихся веществ. Пщролиз хлора проходит
с образованием слабой хлорноватистой кислоты и хлорид-ионов
(соляная кислота):
= Н + +СГ+НСЮ.
Хлорноватистая кислота с образующейся при электролизе
щелочью (точнее, Na+ 4-ОН ~) дает в качестве продукта гипохлорит
натрия NaClO. В щелочной среде суммарное уравнение реакции
имеет вид
С12 + 2NaOH = NaCl + NaClO + Н2О.
При повышенных температурах (кипение воды) гидролиз
хлора проходит с образованием хлорат-иона. Возможные уравнения
реакций:
= С1Оз~+5СГ+6Н\
= СЮ3"+2СГ+ЗН + ,
ЗС1О-=СЮ3"+2СГ.
В щелочной среде суммарное уравнение имеет вид
ЗС12 + 6NaOH - NaClO3 + 5NaCl + ЗН2О.
Электролиз с диафрагмой. При электролизе разбавленного
раствора хлорида натрия к катоду перемещаются ионы Na+, но
выделяется водород:
и концентрируется раствор гидроксида натрия.
К аноду перемещаются хлорид-ионы, но из-за их низкой
концентрации в основном образуется не хлор, а кислород:
и концентрируется раствор соляной кислоты.
Если электролиз проводится в химическом стакане или
другом подобном сосуде, растворы щелочи и кислоты смешиваются
и электролиз сводится к образованию водорода и кислорода за
счет разложения воды. Если же анодное и катодное пространства
разделить перегородкой (диафрагмой), пропускающей
ионы-переносчики тока, но препятствующей смешению нриэлектродных
растворов, то можно в качестве продуктов электролиза получить
растворы кислоты и щелочи.
При электролизе раствора хлорида натрия гидроксид-ионы,
образовавшиеся на катоде по реакции
397
сразу же начинают участвовать в переносе электричества и вместе
с ионами С1~ перемещаются к аноду, где оба иона разряжаются
и образуется смесь кислорода и хлора. Поэтому выход хлора
падает. Если анод изготовлен из угля (графита), то он окисляется
кислородом и образуются оксиды углерода СО и СО2,
загрязняющие хлор. Далее хлор, образующийся на аноде,
взаимодействует с гидроксид-ионами:
Образование гипохлорит-ионов—также нежелательный
процесс (если получение раствора гипохлорита натрия не является
целью). Всех этих нежелательных последствий удается избежать,
если пользоваться диафрагмой, разделяющей катодное и анодное
пространства и задерживающей ионы ОН", но пропускающей
ионы С1~. Наконец, диафрагма препятствует диффузии газов
и позволяет получить более чистый водород.
Если в растворе содержится несколько анионов,
предсказать последовательность их разряжения на аноде сложнее, чем
катионов, но, вообще говоря, соблюдается правило, что в
первую очередь разряжается анион, характеризующийся самым
низким значением потенциала (или самым высоким отрицательным
значением электродного потенциала реакции, проходящей на
аноде).
Все предыдущие рассуждения касались электролиза с
индифферентными нерастворимыми анодами. Ознакомимся с
электролизом, сопровождающимся растворением анода:
Электролиз с растворимым анодом возможен тогда, когда
металл легче отдает электроны, чем ионы С1~, ОН" или
молекулы воды. Например, на медном аноде в растворе хлорида или
сульфата меди хлор или кислород не выделяются, а происходит
переход в раствор ионов Си2 + . Одновременно на катоде те же
ионы разряжаются и осаждается металлическая медь. Таким
образом, электролиз с растворимым анодом сводится к переносу
меди с анода на катод.
Реакция на аноде в большинстве случаев усложняется
многочисленными побочными и часто нежелательными процессами.
Например, образующиеся ионы могу! образовывать оксиды, ги-
дроксиды и их пленки:
На аноде возможно также выделение кислорода:
398
который может участвовать в самых различных реакциях
электролитической системы.
Электролиз с растворимым анодом применяется для очистки
металлов (электрорафшшровапые). При электрорафинировании
меди в электролизер помещают в качестве анода пластины из
очищаемой меди (катод—пласшны из электролитически ранее
очищенной меди). На аноде и катоде проходят процессы
соответственно:
Си (загрязненная) — 2е~ Си2 + ,
Си2*1 +^ = Си (чистая).
При электрорафинировании меди загрязнения из более
благородных металлов типа Ag или Аи в раствор не переходят и
собираются на дне электролизера. Загрязнения из менее благородных
металлов типа Pb, Fe, Zn, как и сама медь, переходят в раствор,
но на катоде не осаждаются и поэтому не загрязняют
осаждающуюся на нем медь. В качестве растворимых анодов могут
быть кроме меди никель, кадмий, алюминий и дру1 ие металлы.
Электролиз с растворимым анодом используется в
гальванотехнике для покрытий одних металлов тонкими слоями других.
При этом покрываемые мегаллом изделия являются при
электролизе катодом, а в качестве анода используется металл
покрытия. Технологически эго очень удобно, так как концентрации
ионов (солей) в электролизном растворе не изменяются.
При образовании газообразных продуктов, особенно
кислорода, в большинстве случаев потенциалы разложения не
соответствуют электродным потенциалам из-за высоких значений
перенапряжения. Перенапряжением называют разность между
реальным напряжением разложения и теоретически
рассчитанным из электродных потенциалов ЭДС соответствующей
реакции. Особенно сильно влияют на величину перенапряжения
природа выделяющегося вещества (заметим, что для хлора, брома
и йода перенапряжение очень незначительно) и материал
электрода. Ниже приведены данные по перенапряжению при
выделении водорода и кислорода на различных катодах и анодах.
Электрод
Pt черненая
Pt блестящая
Fe
Ni
Си
Pb
Перенапряжение. В
Волорол
0,00
0J
0,1—0,2
0.1—0,2
0,2
0.4 -0.6
Кисюрол
0,2—0.3
0,4—0.5
0,2—0.1
0,1—0.3
0,2— 0,3
0 2—0.3
Перенапряжение зависит также от формы электродов, состо-
ния их поверхности, плотности юка, 1емпературы раствора,
интенсивности перемешивания раствора и других факторов.
399
Перенапряжение водорода на железе равно ~0,1 В, а
кислорода на том же материале ^0,3 В. Следовательно,
перенапряжение при электролизе на железных электродах составит
0,1 +0,3 = 0,4 В. Сумма этого значения и теоретически
вычисленного составит минимальное значение напряжения разряжения
соответствующего электролита.
Отношение к перенапряжению—двойственное. С одной
стороны, перенапряжение приводит к повышенному расходу
электроэнергии, с другой стороны, благодаря перенапряжению
удается осаждать из водных расiворов многие металлы, которые по
значениям их С1андар!ных электродных потенциалов осаждаться
не должны. Это Fe, Pb, Sn, Ni, Co, Zn, Cr. Именно благодаря
перенапряжению, а также влиянию концентрации раствора на
электродный потенциал возможны электролитическое
хромирование и никелирование железных изделий, а на ртутном
электроде удается получить из водного раствора даже натрий.
Разряжение в водном растворе ионов О ~, а не ОН ~ в
растворах с высокой концентрацией электролита также объясняется
перенапряжением кислорода. Однако этого перенапряжения
оказывается недостаточно, чтобы произошло разряжение ионов F~
и выделение свободного фтора.
Многие другие кинетические факторы влияют на величину
перенапряжения—скорости переноса частиц к электродам и
отвода продуктов электролиза, скорость процесса разрушения гид-
ратных и других оболочек разряжающихся ионов, скорость
соединения атомов в двухатомные газовые молекулы и т. п.
Аккумуляторы—химические источники тока многократного
действия, по принципу работы не отличающиеся от
гальванических элементов, но накапливающие электрическую энергию при
своем заряде и отдающие ее при разряде.
Наибольшее распространение имеет свинцовый аккумулятор,
состоящий из свинцового катода и анода, представляющего
собой свинец, покрытый слоем диоксида свинца РЬО2. Оба
электрода погружены в общий раствор серной кислоты.
Химические реакции, протекающие на электродах при заряде
и разряде аккумулятора, сложны и окончательно не выяснены.
Но схематически могут быть представлены уравнениями:
заряд , с*г\ 2 -
на катоде PbSO4,K + 2e i=±PbK + SO4 ,
разрял
па аноде РЬ2+ +2Н2О-2ет=±РЬО2.1С+4Н + .
разрял
Суммарная реакция
2PbSO4t
разряд
400
В разряженном состоянии аккумуляторный раствор содержит
12—24% (мае), а в заряженном—28—40%. Обычно степень
заряженное™ контролируют по плотности раствора, которая
отвечает плотности раствора серной кислоты и растворенного
сульфата свинца. При 20° С рекомендуется поддерживать
плотность в заряженном состоянии р= 1,27 г/см3. При плотности
ниже 1,19 аккумулятор считается разряженным на 50% и при
р= 1,11 —разряженным полностью.
ЗАКЛЮЧЕНИЕ
Итак, в современной химии определились четыре основных
учения: о строении вещества, о направлении химических реакций
(химическая термодинамика), о скорости химических реакций
(химическая кинетика) и о периодическом изменении свойств
элементов и их соединений.
Все эти учения тесно связаны между собой. В самом деле,
без знания теории строения вещества нельзя обсуждать
вопросы о направлении и скорости химических реакций; выводы
о строении вещества немыслимы без сведений об энергетических
характеристиках связи; энергии химических связей и
межмолекулярных взаимодействий веществ обусловливают не только
тепловой эффект реакции, но и ее скорость и тот или иной механизм
реакции; классификация элементов основана на распределении
электронов по энергетическим уровням и подуровням a i омов:
периодичность в изменении электронных структур атомов
обусловливает периодичность в изменении свойств атомов, простых
веществ, однотипных соединений и характеристик химических
реакций.
Курс общей химии отличается от всех остальных курсов
химии (физической, органической, коллоидной и др.) тем, что все
изучаемое им сопоставляется и сравнивается на основе
периодического закона и периодической системы элементов Д. И.
Менделеева.
В практическом применении химических знаний нас
интересуют три основных вопроса, связанные со всеми ее теоретическими
положениями:
1. Получение максимального количества желаемого вещества
с заданными свойствами с минимальным использованием
исходных веществ и с минимальными затратами энергии на
осуществление процесса.
2. Получение максимального количества энергии, если в этом
состоит цель процесса (например, теплоты или электрической
энергии), для последующего ее использования.
402
3. Осуществление процесса с требуемой скоростью.
Ответы на эти вопросы можно получить только при
совместном изучении реакции методами химической
термодинамики, химической кинетики и на основе знания теории строения
вещества.
В химической науке тесно связаны между собой химическая
термодинамика, кинетика, теория химической связи и строение
атома. Для описания, объяснения и предсказания любого
химического явления необходимо одновременное использование всех
имеющихся знаний.
ЛИТЕРАТУРА
Обязательная для успешного усвоения курса
1. Зайцев О. С. Исследовательский практикум по общей химии. М.:
Изд-во МГУ, 1994. 480 с.
2. Зайцев О. С. Задачи, упражнения и вопросы по химии. М.: Химия,
1996. 432 с.
Дополнительная
3. Глинка Н, Л. Общая химия. Л.: Химия, 1983 и все последующие
издания.
4. Некрасов Б, В. Основы общей химии. М.: Химия, 1973 и все
последующие издания.
5. Суворов А. В., Никольский А. Б. Общая химия. СПб.: Химия, 1994.
624 с.
6. Лидии Р. А., Аликберова Л, Ю., Логинова Г. Л. Неорганическая
химия в вопросах. М.: Химия, 1991. 256 с.
7. Зайцев О. С. Общая химия. Направление и скорость химических
процессов. Строение вещества. М.: Высшая школа, 1983. 248 с.
8. Зайцев О. С. Общая химия. Состояние веществ и химические
реакции. М: Химия, 1990. 352 с.
ОГЛАВЛЕНИЕ
Предисловие 3
Глава /. Учение о строении вещества 5
§ I. Материя — вещество и поле 5
§ 2. Строение ядра атома 9
§ 3. Основные законы атомно-молекулярной теории 15
§ 4. Поведение электрона в атоме 20
§ 5. Электронное строение атома водорода 24
§ 6 Многоэлектронные атомы 30
§ 7. Периодическая система и электронная структура
атомов 36
§ 8. Периодичность свойств атомов 45
§ 9. Химическая связь. Меюд валентных связей 50
§ 10. Химическая связь. Метод молекулярных орбиталей 67
Глава 2. Учение о направлении химических процессов 80
§ 11 Изменение внуфенней энергии в химической реакции .. 80
§ 12. Изменение энтальпии в химической реакции 86
§ 13. Закон Гесса 88
§ 14. Изменение энтропии в химическом процессе 93
§ 15. Изобарный потенциал реакции 102
Глава 3, Учение о скорости химических процессов 108
§ 16. Скорость химической реакции и ее зависимость от
концентрации 108
§ 17. Влияние температуры на скорость химической реакции 120
§ 18. Кинетика сложных реакций 130
§ 19 Обратимые химические реакции и константа
равновесия 133
§ 20 Проведение химической реакции при стандартных и
нестандартных условиях 142
Глава 4. Фазовые состояния веществ и фазовые равновесия 150
§ 21. Фазовые состояния и фазовые переходы 150
§ 22. Условия фазовых равновесий 153
405
§ 23. Диаграмма состояния однокомпонентной системы 157
§ 24. Диаграмма состояния воды. Свойства воды 165
Глава 5. Газовое состояние и реакции в газовых фазах 169
§ 25. Теплоемкость газов 169
§ 26. Реакции вешеств в 1азовом состоянии 173
§ 27. Фотохимические реакции 177
§ 28. Цепные реакции 186
Глава 6. Жидкое состояние и реакции в жидких фазах 194
§ 29. Жидкое состояние вещества 194
§ 30. Электролитическая диссоциация веществ в жидком
состоянии 200
Глава 7. Кристаллическое состояние и реакции в твердых фазах 208
§ 31. Строение кристалла 208
§ 32. Электрические свойства кристаллов 217
§ 33. Магнитные свойства кристаллов 226
§ 34. Энер1 етические свойства кристаллов 234
§ 35. Фазовые переходы в кристаллических веществах 237
Глава 8. Процессы на границе раздела фаз 244
§ 36. Реакции на границе кристалл—газ 244
§ 37. Гетерогенный катализ 249
§ 38. Образование кристаллов из жидкой фазы 255
§ 39. Растворение, растворимость, произведение
растворимости 263
Глава 9. Растворы 275
§ 40. Общие сведения о растворах. Давление насыщенного
пара над раствором 275
§ 41. Методы изучения жидких растворов 283
§ 42. Процессы координации в растворах 290
§ 43 Коллоидное состояние вещества 297
Глава 10. Процессы в водных растворах 312
§ 44. Диссоциация сильных и слабых электролитов 312
§ 45. Равновесия в водных растворах сильных электролитов.
Ионная сила и активность 318
§ 46. Реакции нейтрализации. Водородный показатель 325
§ 47. Ионные равновесия в растворах слабых кислот и
слабых оснований 333
§48. Гидролиз. Реакции амфотерных соединений 339
§ 49. Равновесия в буферных растворах 350
§ 50. Комплексообразование 353
§ 51 Реакции с участием комплексных ионов 361
406
§ 52. Электродные процессы 364
§ 53. Окислительно-восстановительные реакции 378
§ 54. Электрохимия — коррозия, электролиз,
аккумуляторы 388
Заключение 402
Ли гература 404