/































































































































































































Текст
NON-AQUEOUS НЕВОДНЫЕ
SOLVENT SYSTEMS РАСТВОРИТЕЛИ
Edited by Под редакцией
Т. С. WADDINGTON Т. ВАДДИНГТОНА
School of Molecular Sciences, Перевод с английского
University of Warwick, М- Б. Бравермана, Л. В. Васильева,
Coventry, England В. П. Маширева
196 5
ACADEMIC PRESS ИЗДАТЕЛЬСТВО «ХИМИЯ»
LONDON AND NEW YORK МОСКВА I 5Г Г1
УДК 66.062.18
Н 40
Пенодные растворители. Под ред. Ваддипгтопа Т. Перев. с англ.
Изд. «Химия», 1971 г., 376 с.
В книге изложены современные представления о неводных
растворителях. Каждая глава написана крупным специалистом,
имеющим большой опыт работы в данной области.
Первые главы книги посвящены трем наиболее важным рас-
растворителям протонного типа: аммиаку, фтористому водороду
и серной кислоте. В третьей главе описаны галогеноводороды—
кислые растворители с низкой диэлектрической проницаемостью.
Несколько глав посвящено растворителям апротонного типа:
жидкому сернистому ангидриду, галогенидам и оксигалогенидам
элементов V группы периодической системы, интергалогенидам,
координирующим растворителям. В последней главе описаны
свойства расплавов ионного типа.
Книга является ценным пособием для научных сотрудников,
использующих в своих работах жидкие неводные растворители
и занимающихся теорией растворов. Она будет полезной для
аспирантов и студентов старших курсов химических факуль-
факультетов институтов.
В книге содержится: 67 рисунков, 50 таблиц, 1159 библиогра-
библиографических ссылок.
2-5-',
132-69
ИЗ ПРЕДИСЛОВИЯ К АНГЛИЙСКОМУ ИЗДАНИЮ
Круг вопросов, охватываемый понятием «системы неводных рас-
растворителей», настолько обширен, а число различных растворителей,
исследованных хотя бы частично, настолько велико, что лишь в мно-
многотомной работе можно полностью рассмотреть все проблемы, сто-
стоящие перед исследователями в этой области. Для того чтобы дать
в одном томе обзор неводных растворителей, приходится или огра-
ограничиться поверхностным рассмотрением большого числа раствори-
растворителей, многие из которых лишь незначительно отличаются друг
от друга, или достаточно глубоко обсудить свойства ограниченного
числа растворителей. Авторы пошли по второму пути.
Выбор растворителя определялся его практическим значением
и свойствами. Первые главы книги посвящены трем наиболее важ-
важным неводным растворителям протонного типа: жидкому аммиаку,
безводному фтористому водороду и серной кислоте. Эти растворители
нашли широкое применение в препаративной химии и при физиче-
физических измерениях, кроме того, все они, подобно воде, являются
хорошими растворителями для органических и неорганических
веществ. По-видимому, в области исследования именно этих раство-
растворителей происходит наиболее интенсивное накопление количествен-
количественных данных. Однако для неводных растворителей в литературе
содержится все же крайне мало сведений даже о таких простых
количественных термодинамических величинах, как теплоты раство-
растворения галогенпдов щелочных металлов. Поэтому практически невоз-
невозможно сравнить энергии сольватации простых ионов в различных
растворителях, хотя эти сведения были бы весьма интересны.
В книгу включена глава о галогеноводородах — протонных
растворителях с низкой диэлектрической проницаемостью, облада-
обладающих сильными кислотными свойствами. Их удобно использовать
для изучения ионов, образование которых в любых других раствори-
растворителях затруднено, но число количественных физических данных
здесь еще меньше, чем для указанных выше протонных раствори-
растворителей (NH3, HP, H2SO4).
Остальные главы книги посвящены апротонным растворителям.
В качестве типичного представителя этого класса растворителей
рассмотрен жидкий сернистый ангидрид. Это соединение широко при-
применяется в органической и металлоорганической химии в качестве
ПРЕДИСЛОВИЕ
реакционной среды. Однако лишь в последнее время благодаря
работам Норриса и сотр., изучавших реакции изотопного обмена
в этом растворителе, а также исключительно детальным исследова-
исследованиям и точным измерениям Личтина и сотр., изучавших проводи-
проводимость этих растворов, становится ясной та роль, которую играет
сернистый ангидрид в химических реакциях.
В двух главах, посвященных галогенидам и оксйгалогенидам
элементов V группы периодической системы и галогенам и интерга-
интергалогенам как растворителям, рассматриваются системы, в которых
роль растворителя сводится к отдаче и обмену галоген-иона. Сделан
критический обзор данных, помогающий понять протекание этих
процессов.
В пятой главе книги изложены сведения о координирующих
растворителях — соединениях, содержащих хотя бы один атом
(либо О, либо N) с неподеленной электронной парой.
Последняя глава посвящена расплавленным солям как раство-
растворителям. Эти системы сильно ионизированы и обладают высокрц
проводимостью даже в отсутствие растворенных в них соединений.
Методы изучения таких растворителей заметно отличаются от мето-
методов, гшименяемых для изучения большинства растворителей, жидких
нрп комнатной температуре.
Материал в книге изложен на современном уровне знаний, каж-
каждая глава налисана специалистом в данной области.
Т. Вапдингтон,
ГЛАВА ПЕРВАЯ
ЖИДКИЙ АММИАК
В. Л. ДЖОЛЛИ, К. ДЖ. ХОЛЛ АД А
I. Введение 8
II. Физические свойства аммиака 8
III. Физические свойства растворов в жидком аммиаке 10
A. Растворы неэлектролитов 10
Б. Растворы электролитов 11
1. Мольные объемы 11
2. Термохимия 12
3. Электрохимия 13
B. Растворы металлов 15
1. Фазовые диаграммы 15
2. Мольные объемы - • 17
3. Термохимия 18
4. Электропроводность 19
5. Магнитные свойства 20
6. Спектры поглощения 21
7. Модели 21
IV. Реакции в жидком аммиаке 23
A. Сравнение аммиака с другими растворителями • . 23
1. Растворимость 23
2. Диапазон окислительных потенциалов 25
3. Кислотно-основной интервал 27
4. Метод работы с жидким аммиаком 29
Б. Ионная сольватация 29
1. Термодинамика процессов аммонизации н гидратации ... 29
2. Кинетика обменных реакций в аммиаке 30
B. Кислотно-основные реакции 31
1. Природа аммоний- и амид-ионов 31
2. Определение концентрации аммоний- и амид-ионов .... 32
3. Обменные реакции, катализируемые основаниями 35
4. Реакции, катализируемые кислотами 37
Г. Реакции в растворах металлов в аммиаке 37
1. Общий обзор 37
2. Реакция металлов с аммиаком 40
3. Реакции металлов с ионом аммония в аммиаке 41
4. Реакцпя взаимодействия натрия в аммиаке с этиловым спир-
спиртом 42
5. Сравнение с растворами металлов в других растпорителях 44
Литература 44
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
I. ВВЕДЕНИЕ
Исследования в области химии жидкого аммиака проводятся
настолько интенсивно, что для подробного их рассмотрения потребо-
потребовалась бы специальная монография. При написании этой главы мы
решили не пытаться охватить все аспекты химии растворителя,
а рассмотреть лишь те вопросы, которые поддаются количественной
интерпретации, уделив большое внимание термодинамике и кине-
кинетике. Такой подход стал возможен только благодаря многочислен-
многочисленным физико-химическим исследованиям, выполненным в последние
годы. Однако мы считаем, что еще много предстоит сделать для систе-
систематизации результатов исследований.
В различных книгах и обзорных статьях, выпущенных до сих
пор, химия жидкого аммиака рассматривается, скорее, качественно.
Основные принципы систематизации сведений обычно аналогичны
тем, которыми руководствуются в химии водных растворов. Ниже
приведены библиографические ссылки на наиболее важные книги
и статьи, рекомендуемые в качестве источников информации по тем
вопросам, которые не рассмотрены или о которых лишь упоми-
упоминается в данной монографии. По общим вопросам — это ра-
работы 4> 47> 49> 84,i4i,i57. п0 специальным вопросам — аммонолпз 4в,
амиды щелочных металлов 9'10'117, растворы металлов в амми-
аммиаке 92,108,116,144^ реакции в растворах металлов в аммиаке 15> 1в>147,
химическая термодинамика 91.
II. ФИЗИЧЕСКИЕ СВОЙСТВА АММИАКА
Этот раздел является справочным, в нем в краткой форме пред-
представлены наиболее важные физические свойства аммиака *:
А#пл== 1 >352 ккал/моль 128
Д^кип = 5,581 ккал/моль 128
АЩ = —11,04 ккал/моль I3i
}= — 3,976 ккал/моль iai
t,
г
S° = 46,01 калI{град ¦ моль) 134
Ср = 8,523 кал/(град ¦ моль) 134
х (жидкий ГШз) 71 я» 1 .10"И о.ч~1 • см~1
« = 1,325 (при 16° С и ^ = 5899 А) «
Поверхностное натяжение жидкого аммиакаЫ2:
*- °С -75,3 -69.0 -67,0 -58,3 -52,0 -39,4
сг*, эРг/см2 43,45 42,26 41,71 39,65 38,18 35,38
* Ошибка ±0,15 эрг/см2.
с= 23,41 — 0,337И — 0,000943*2 (—75° C<t< — 39° С).
Диэлектрическая проницаемость жидкого аммиака57
—60 ±10
26,7
-33
B3) *
5
18,94
15
17,82
23
16,90
3 5
16,26
Интерполирована.
* Величины АН), AF}, S°, C'p определены при 25° С.
II. ФИЗИЧЕСКИЕ СВОЙСТВА
Таблица 1. Теплоемкость* аммиакате
т, °к
20
30
40
50
60
70
80
90
100
0,368
1,033
1.841
2,663
3,474
4,232
4,954
5,612
6.246
г, °к
110
120
130
140
150
160
170
180
190
ср
6,877
7,497
8,102
8,699
9,272
9,846
10,42
11,03
11,71 (тв.)
г, °к
200
210
220
230
240
17,58 (ж.)
17,75
17,90
18,03
18,12
* 0° С принят равным 273,10° К и температурная шкала заметно отличается от Меж
дуняродной шкалы температур.
Таблица 2. Давление паров * над аммиаком 2
т, ск
/, °с
Над твердым ам
175
180
185
190
195
195,46
—98,16
—93,16
—88,16
—83,16
—78,16
—77,70
Над ж и д к и м а мм
200
210
220
230
239,78
-73,16
-63,16
—53,16
—43,16
—33,38
Р, мм pm. ст.
vi и а к о м **
4,85
8,78
15,42
26,27
43,57
45,58
(тройная точка)
и а к о м ***
64,92
133,21
253,97
454,28
760
(точка кипения)
Т, °К
240
250
260
270
280
290
298,16
300
320
340
360
380
400
405,6
t, "С
-33,16
—23,16
—13,16
—3,16
6.84
16,84
25,00
26,84
46,84
66 84
86,84
106,84
126,84
132,4
Р, Л1Д( pm. cm.
768,43
1 237,5
1 914,8
2 857,1
4 130,0
5804,2
7 520,5
7 956,6
14 028
23 089
35 973
53 597
77 334
85 400
(критическая
точка)
* 0° С равен 273,16° К.
** Вычислено по уравнению Ig Р = 9,98379—
1627,22
Т
*** Вычислено по уравнению lgP = 9,95028 — 147^'17 —0,0038603Т (для Г<2аО°К).
Кристаллическая структура твердого аммиака 145 — кубическая
решетка, четыре формульные единицы^в элементарной ячейке; при
—185° G а0 = 5,2253, /сХ = 5,2358 А. Плотность, рассчитанная
по рентгеноструктурным данным, р = 0,7881 г/см3.
Криоскопическая постоянная
Кк —
1000 АНп
10
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Таблица 3. Вязкость жидкого аммиака
t, °с
-33,5
-26
-10
—4
5
10
1], СПЗ
0,2543
0,230
0,183
0,170
0,1618
0,152
Литература
42
165, 166
165, 166
165, 166
130
165, 166
/, °с
15
20
25
30
50
1), СПЗ
0,1457
0,1479
0,1411
0,1350
0.1345
0,138
0,125
Литература
130, 171,
172
¦171, 172
130, 171,
172
165, 166
165, 166
Эбулиоскопическая постоянная
С _ КИП
Плотность жидкого аммиака 2S:
t, °С —70 —60 —50 —40 —34 —33 —30
р, г/с.чз 0,7253 0,7138 0,7020 0,6900 0,6826 0,6814 0,6776
t, °С —20 —10 0 10 20 25 30
р, г/е.тз 0.6650 0,6520 0,6386 0,6247 0,6103 0,6028 0,5952
III. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
В ЖИДКОМ АММИАКЕ
А. Растворы неэлектролитов
Жидкий аммиак является превосходным растворителем для мно-
многих неэлектролитов, которые в воде практически нерастворимы.
Большая часть исследований направлена на решение практических
задач, например, разделение трудноразделимых смесей методами
экстракции и кристаллизации.
Исследования в области фазовых состояний некоторых систем
углеводород — жидкий аммиак были выполнены Ишида 78"82 и
Фенске с сотр.44 Было показано, что экспериментальные данные
о критическом составе и коэффициентах активности лучше всего
трактовать на основании теории растворимости, предположив, что
молекулы жидкого аммиака ассоциированы в димер.
Имеются данные о теплотах растворения некоторых неэлектро-
неэлектролитов в жидком аммиаке. Шмидтом с сотр.138 было замечено, что
мольные теплоты растворения нормальных спиртов в жидком амми-
аммиаке уменьшаются от 1960 кал для метилового спирта до 100 кал для
нормального бутилового спирта. Ганн и Грин 58 нашли, что мольные
теплоты растворения воды и метиламина не зависят от концентрации.
III. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ В АММИАКЕ
11
Примечательно, что растворы азота 152, водорода1вз, гелия и
аргона 30 в жидком аммиаке подчиняются закону Генри вплоть до
давлений ~ 100 атм. Лефранкуа и Ванискотт 114 изучали влияние
температуры на растворимость смеси N2 + 3H2 в жидком аммиаке
и обнаружили, что растворимость этой смеси находится в кубической
зависимости от температуры, измеренной в градусах Цельсия.
Было проведено сравнение спектров (в диапазоне длин волн от
2500 до 6000 А) растворов некоторых нитрофенолов и нитронафтолов
в жидком аммиаке 160> 161со спектрами тех же соединений в 0,005 М
водном растворе NaOH. Спектры обеих систем весьма похожи,
но в первом случае пики в спектрах растворенных веществ сдвинуты
в сторону больших длин волн, что объяснено образованием кислотно-
основных комплексов.
Б. Растворы электролитов
Почти все виды измерений, проведенные в водных растворах
электролитов, выполнены также и для растворов электролитов
в жидком аммиаке, хотя работать с последним гораздо труднее, чем
с водой. Остановимся на некоторых наиболее важных результатах
и выводах.
/. Мольные объемы
Плотности растворов галогенидов некоторых щелочных металлов
и галогенидов аммония в жидком аммиаке определены при различ-
различных температурах и концентрациях 85> "> 10°. Джонсон и Мартене 85
показали, что плотность раствора изменяется линейно с изменением
температуры, и кажущийся мольный объем V является линейной
функцией с '* (в моль/л). Эта закономерность аналогична полученной
для водных растворов. Позже Ганн и Грин 59 определили кажущийся
мольный объем нескольких электролитов в жидком аммиаке при
0° С и обнаружили, что формы кривых, выражающих зависимость
кажущегося мольного объема V от с , весьма схожи для галогени-
галогенидов некоторых щелочных металлов, галогенидов аммония и нитрата
бария. Правило аддитивности ионов соблюдается даже при концент-
концентрациях, при которых можно было бы ожидать образования значитель-
значительного количества ионных пар. Экстраполяцией, используя вычислен-
вычисленные константы ассоциации, Ганн и Грин получили приведенные
ниже значения мольных объемов некоторых солей в жидком аммиаке
при бесконечном разбавлении Fo (для сравнения приведены мольные
объемы тех же солей в воде)в2:
Соль NaCl Nal KI
в NH3 при 0° С —38 —15 —6
в Н2О при 25° С 16 4 35,1 45,4
12
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Из этих данных можно найти, что
0,Хаг
V0,N-ai (HaO)-F0 NaC1(H2O) = 18J eMs
Таким образом
-VOi и_ (NH3) «* Vo> j_ (H2O)-Ffl] c,_ (H2O)
V
Следовательно
o,
V,
t K+ (NH8)-70| Na+ (NH)8 ~ VOt K+ (HaO)-Vo_ Na+ (ЩО)
Так как в настоящее время нет абсолютно правильного способа
определения Vo для отдельного иона, истинные величины ионных
объемов все еще остаются неизвестными.
Значения ионных объемов, приведенные в работе Джолли 92Т
получены при допущении, что FOj к+ = VOi a--
Воспользовавшись уравнением, подобным уравнению Геплера вв,
можно рассчитать FOi Na+; в жидком аммиаке при 0° С он равен
—41,5 см3. Уравнение Каутри и Лейдлера 27 дает величину —34 см3.
Если взять среднее арифметическое этих значений, то
Аналогично
Fo,cr- =
0, I-"
22
Среднеарифметические значения, по-видимому, более точные
2. Термохимия
Теплоты растворения некоторых электролитов в жидком аммиаке
были измерены при температурах 25 и —33° С (при мольном отноше-
отношении растворителя и растворенного вещества 500 : 1) Ганном и Гри-
Грином 68 (табл. 4). Было замечено, что величины теплот растворения
в аммиаке больше, чем в воде, и при 25° С больше, чем при —33° С.
Для разбавленных растворов KI в жидком аммиаке тангенс
угла наклона кривой зависимости теплоты растворения от с 1г равен
'!!
27 ккал
!г/молъ!г. Это значение почти в двадцать раз превы-
превышает величину, рассчитанную по методу Дебая — Хюккеля, что,
несомненно, указывает на высокую степень ассоциации ионов
III. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ В АММИАКЕ
13
Таблица 4. Теплоты растворения некоторых веществ в воде
и в жидком аммиаке
Вещество
П2О (ж)
CH3NH2 (ж)
HgI-2
NaCl
KI
Csl
NH4C1
NH4I
Ba(NO3J
в Н2О при 2 5° С134
0
-5,0
—
+ 1,02
+4,95
+ 7,9
+3,71
-J-3,3
+ 9,96
ЛНраств' ™ал/моль
в NH3 при 25° С ss
-3,32
+0,50
(—20,8)
—6,75
—9,44
—5,27
—8,23
-16,10
—15 31
в NH, при —33° С 6»
— 2,81
—
(—20,15)
-1,57
—7,89
—
—6,95
—13,37
—
в жидком аммиаке. Поэтому тепловые эффекты, сопровождающие раз-
разбавление, являются главным образом следствием диссоциации ион-
ионных пар и сольватации образующихся ионов.
3. Электрохимия
а. Электропроводность. Были определены электропроводности
растворов нескольких солей и кислот в жидком аммиаке. К сожале-
сожалению, сходимость результатов по эквивалентной электропроводности,
полученных различными исследователями, редко такая же хорошая,
как для водных растворов. Некоторые наиболее достоверные значе-
значения электропроводностей приведены в табл. 6. Экспериментальные
данные не совсем точно согласуются между собой. Так, если исполь-
использовать данные работы п, определенные при — 34° С, получим в то
Л0,К+~Л0, Na+
время как по данным работы 1в4, при —33,5° С
•^0, KNOs~ 0, КаХОз = 39 = ^-0, К+~~^0, Na+
Моносзон и Плесков 1в4 использовали данные Франклина и
Кейди 51 по определению чисел переноса и приписали Я,Ор+ ионам
щелочных металлов, а АОр_ — нитрат-иону. Но приписывать Я9 _
ионам галогенов нет оснований, так как отсутствует сходимость зна-
значений электропроводности нитратных и галогенидных растворов.
Было замечено, что константы ассоциации галогенидов калия
изменяются в соответствии с относительными размерами галоген-
ионов. Весьма странным является то обстоятельство, что расстояние
а
наибольшего сближения а между галоген-ионами и калием, которые
лучше других подчиняются расширенной теории электропроводности
14
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Онзагера — Фуосса, больше в жидком аммиаке, чем в воде или мета-
о
ноле 97. Величина а для водных растворов часто практически равна
сумме радиусов ионов 54, но это, по-видимому, не так для растворов
галогенидов щелочных металлов в жидком аммиаке.
б. Полярография. Полярографические исследования с примене-
применением капельного ртутного электрода, несмотря на трудности в ра-
работе, связанные с тем, что точка замерзания ртути (—38,9° С) и тем-
температура кипения жидкого аммиака (—33,4° С) очень близки, спо-
способствовали получению более ясных представлений о природе
растворов электролитов в жидком аммиаке. Подпрограммы могут
Таблица 5. Эквивалентная электропроводность,
расстояние наибольшего сближения ионов
и константы ассоциации растворов солей в жидком аммиаке
(и в воде при 25° С)
Электролит
КС1
КВг
KI
NaBr
LiNO3
LiNO3
NaNO3
NaNO3
KNO3
KNO-j
RbNO3
CsNOa
KNH2
', °c
-34
-34
—34
—34
—33 5
-40
—33,5
-40
—33 5
—40
-40
—40
—33
Ло в NHj,
см-1 (ом • г-экв)
348,0
346,98
345,1
314,3
299
290
315
300
354
338
344
345
343
Ло в Н20.
см21 (ом-г-же)
149,9
151,9
150,4
128,3
110,1
110,1
121,6
121,6
145,0
145,0
149,2
148,7
—¦
а-\0',
А
6,7
7,8
6,6
5,9
.
,
—
—
—
—
—
ассоц
1060
453
183
263
,
13 700
Литература
71*
71
71
164
164
164
164
164
164
164
164
164
65
* Данные этих авторов были недавно обработаны Кэем97 с использованием теории
электропроводности Онзагера— Фуосса в расширенном варианте •*.
показать, протекает ли восстановление в одну или несколько ступеней
и тем самым помочь понять природу образующихся промежуточных
соединений.
Латинен и Шоумакер m показали, что в процессе полярографи-
полярографических измерений в жидком аммиаке на ртутном аноде образуется
Hg2+-noH и что электрод Hg — Hg2+ в этой среде является обра-
обратимым.
В табл. 6 представлены потенциалы полуволны некоторых про-
процессов восстановления в жидком аммиаке, определенные с электро-
электродом РЬ/0,1 н. Pb(NO3J.
Если катион инертного электролита не восстанавливается, пла-
платиновый и ртутный электроды ведут >еебя в жидком аммиаке как
нерастворимые электроды. Стандартный потенциал такого электрода
III. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ В АММИАКЕ
15
Таблица 6. Потенциалы полуволны в жидком аммиаке при —36° С
Восстанавливаемый катион
Li +
Na +
К+
Rb+
Cs+
Tl+
CU2+-
Cu+ —
NHJ
Pb2+_
Cd2+-
Ni2+ —
Ca2+ —
Sr2+_
Ba2+ —
—*¦ Cu +
> амальгама
—>- амальгама
->Cd
->Ni
->Ca
->-Sr
->-Ba
-1,67
—1 31
-1,24
-1,21
-1,15
+0,15
+0,16
-0,21 „
-1,37 1
—0,01
—0,45
—0,79
—0,89
—1,96
—1,68
—1,59
Обратимость
Обр.
Обр.
Обр.
Обр.
Обр.
Обр.
Обр.
Необр.
Обр.
Обр.
Обр.
Необр.
Необр.
—
—
—¦
Литература
109, 110
109, 110
109, ПО
109, ПО
109, ПО
112
112
112
112
122
122
122
122
124
124
124
относительно стандартного потенциала водородного электрода при
—36° С равен —1,9 в.
Шаап с сотр.137 использовали для полярографических измерений
при комнатной температуре ячейку, работающую при высоком дав-
давлении.
В. Растворы металлов
Несмотря на большое число работ, посвященных растворам метал-
металлов в аммиаке (они были изучены впервые Вейлом 151 в 1864 г.), их
природа еще не совсем ясна. Все растворы металлов в жидком амми-
аммиаке неустойчивы и разлагаются с образованием водорода и амида
металла. Так как эта реакция каталитически ускоряется некоторыми
примесями, работа с такими растворами должна проводиться с боль-
большой осторожностью и с особой тщательностью.
Неустойчивость металл-аммиачных растворов является причиной
многих несоответствий в результатах, полученных различными
исследователями. Обычно принято считать, что при растворении
в жидком аммиаке (по крайней мере в очень разбавленных растворах)
молекула металла диссоциирует на ион и электрон(ы).
1. Фазовые диаграммы
В жидком аммиаке растворяются металлы, обладающие низ-
низкими ионизационными потенциалами и высокими энергиями соль-
сольватации, т. е. щелочные металлы (табл. 7), щелочноземельные ме-
металлы, более тяжелые, чем бериллий, и те редкоземельные элементы,
которые проявляют в окисленном состоянии валентность +2.
16
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Щелочноземельные металлы, а также европий и иттербий обра-
образуют твердые гексааммиакаты, а сами не существуют в равновесии
со своими растворами. Гексааммиакат кальция выделяется при
при концентрации кальция 10 ат. %, при 0° С — когда
концентрация кальция достигает
11 ат. %. Величина мольного отно-
отношения аммиака к гексааммиакату
лежит в пределах от 4 до 5, что
примерно равно мольному отноше-
отношению аммиака к металлу в раство-
растворах щелочных металлов.
Сьенко ue рассмотрел систему
металл — аммиак и предложил элек-
электронную модель, согласно которой
в аммиаке должно растворяться
3,9 ат. % металла, что прекрасно
согласуется с наблюдаемыми величи-
величинами растворимости.
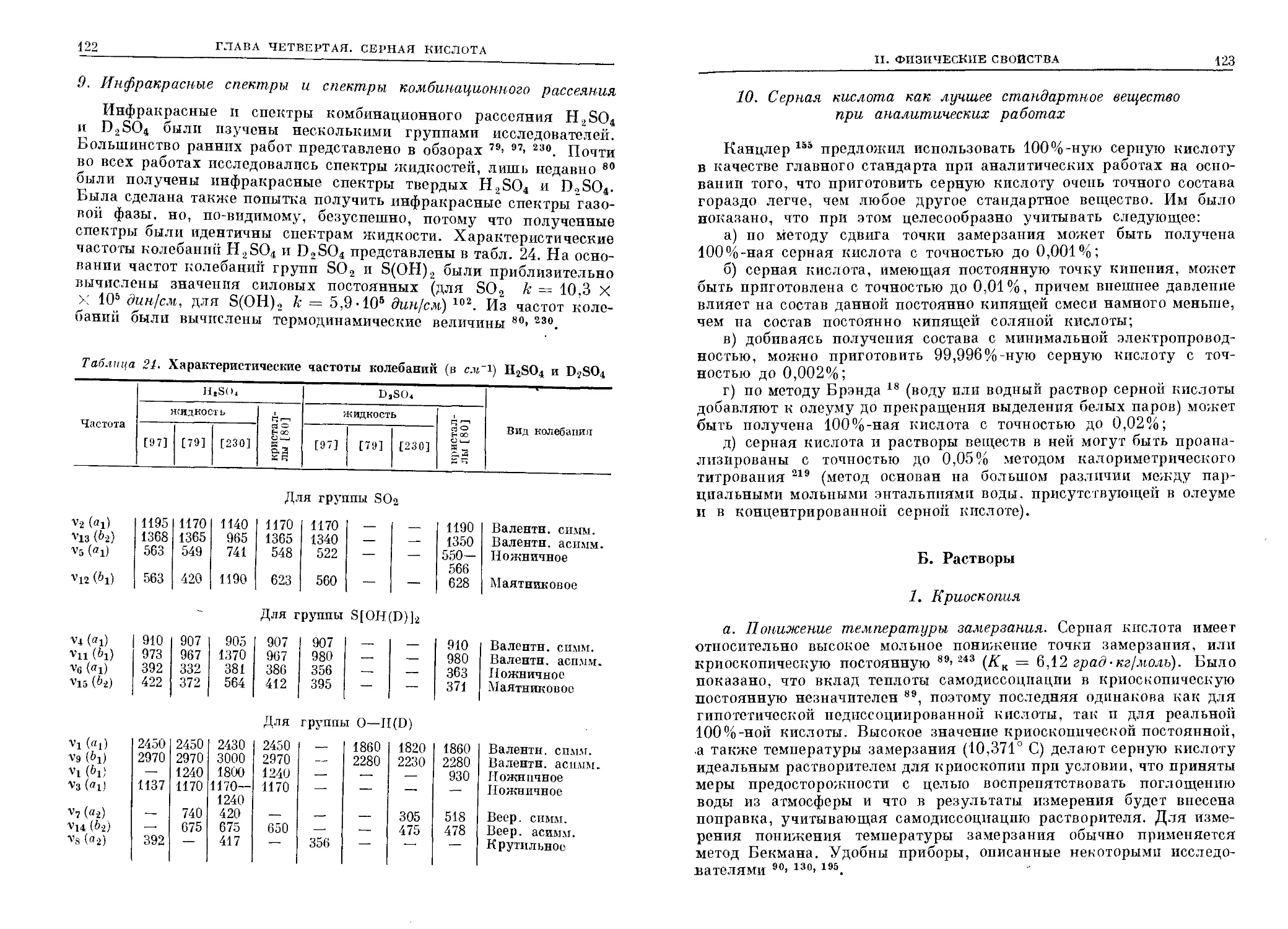
Часть фазовой диаграммы си-
системы натрий — аммиак приведена
на рис. 1. Характерными особен-
особенностями этой диаграммы, как и
III. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ В АММИАКЕ
17
-120
г «,;/ в to lt
Содержание Na , am. °/o
id
Рис. 1.
Фазовая диаграмма92
Na—NH,.
пиитими этой диаграммы, как и
вообще диаграмм системы металл — аммиак, являются: 1) крутизна
кривой растворимости металла (либо гексааммиаката); 2) низкая
температура эвтектической точки; 3) наличие в жидкой фазе области
Таблица 7. Растворимость щелочных металлов в жидком аммиаке
Металл
Li89, юз
, 135
i 104, 135
CS72
t, °С
0
QQ 0
-39,4
—63 5
22
0
—30
—33,8
—33,5
—50
—70
—105
0
—33,2
-50
-100
—33,5
—50
Число грамм-атомов
металла на 10 00 г NH3
16,31
15,66
16.25
15.41
9,56
10,00
10,63
10,72
10,93
10,89
11,29
И 79
12,4
11,86
12 3
12,2
12,05
25,1
Число молей П1,
на грамм-атом металла
3,60
3,75
3,61 '
3,81
6,14
5,87
5,52
5,48
5 37
5,39
5,20
4,98
4,68; 4,74
4,95
5,05; 4,79
4,82
4,87
2,34
несмешиваемости, которая соответствует одновременному существо-
существованию двух различных растворов металла.
В области несмешиваемости более концентрированная фаза
окрашена в золотистый цвет и имеет меньшую плотность, чем фаза
голубого цвета, существующая при значительно меньших концентра-
концентрациях и при болеё"""низких температурах. Аналогичная картина
наблюдается для всех щелочных и щелочноземельных металлов, за
исключением цезия. Однако есть и некоторые различия в диаграм-
диаграммах систем щелочной и щелочноземельный металл — аммиак. В то
время как наибольшая критическая температура растворения в си-
системе натрий — аммиак равна —41,6° С, наибольшая критическая
температура растворения в кальций-аммиачных растворах 94 лежит
выше —50° С. Концентрация, при которой начинает появляться вто-
вторая фаза, для кальциевых растворов гораздо ниже, чем для натрие-
натриевых el, а эвтектическая температура намного ниже у систем щелоч-
щелочной металл — аммиак. Эвтектические точки некоторых систем:
Металл Li К Cs Ca Sr Ba
t, °С —185 —157 —118 —87 —89 —89
Содержание металла, ат. % 22 15 — 12 7 7,7
2. Мольные объемы
При растворении металлов в жидком аммиаке происходит значи-
значительное увеличение суммарного объема. Ниже приведены плотности
насыщенных растворов щелочных металлов при различных темпера-
температурах:
для лития83
t, °С 19 —80
р, г/сиз 0,477 0,495
для натрия88
t, °С —31,6 —33,3 —40,7 —47 —51
р, г/смз 0,576 0,578 0,581 0,585 0,587
для калия88
t, "С —33,3 —39,0 —41,0 —46,4 —49,6
р, г/слC 0,625 0,627 0,629 0,636 0,638
Ганн и Грин 59 обнаружили, что кажущиеся мольные объемы
щелочных металлов при 0° С незначительно изменяются с изменением
концентрации от 0,1 до 1,0 моль/л. Авторы приводят такие зна-
значения парциальных мольных объемов при бесконечном разбавлении:
Fo,Na=57 см3
Fo к=65
2 Заказ 90
18
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Используя величины мольных объемов при бесконечном разба-
разбавлении для KI и Nal, которые были даны ранее, можно вычислить
мольный объем сольватированного электрона при бесконечном раз-
разбавлении:
V0,Na~vo, NaI = 57-(-15) = 72 CM» = VOtt-Vo< x_
Так как Fo, i- = 22 см3, то Vo, e — 94 см3/моль. Этот объем соответ-
соответствует сфере радиусом 3,34 А. Расчеты других авторов 92р75 также
дают для радиуса сферы, занятой электроном, величину —3 А.
3. Термохимия
Щелочные металлы, за исключением лития, имеют очень малые
теплоты растворения; наоборот, щелочноземельные металлы, образу-
образующие твердые гексааммиакаты при растворении в жидком аммиаке,
выделяют большие количества тепла (табл. 8).
III. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ В АММИАКЕ
Таблица &
Металл
Li
Na
Rb
Cs
Ca
. Теплоты растворения металлов в аммиаке
с, моль/л
0,067
0,4
0,07
, 0,13
0,19
0,023
—
АН, ккал/молъ
-9,65
+1,4*
0,0
0,0
0,0
—19,7
—20,7
—19,0
при —33° С
Литература
24
107
107, 139
139
139
156
25, 155
25, 155
* В присутствии Nal теплота растворения Na больше'0.
Значительный интерес представляет величина теплоты аммонн-
зации электрона (т. е. теплоты перехода его из газовой фазы в жид-
жидкий аммиак). По расчетам Коултера 2S, она равна —11 ккал/молъ.
Это значение по абсолютной величине, по-видимому, слишком мало
по сравнению с определенной экспериментально 63 пороговой энер-
энергией фотоэлектрического эффекта, равной 33 ккал/молъ; величина
теплоты аммонизации электрона, равная —40 ккал/молъ 92, предста-
представляется нам более приемлемой. По теоретическим расчетам Джорт-
нера 95, теплота растворения электрона равна —39 ккал/молъ, а ам;
ионизированный электрон находится в сфере радиусом 3.2—3,4 А.
19
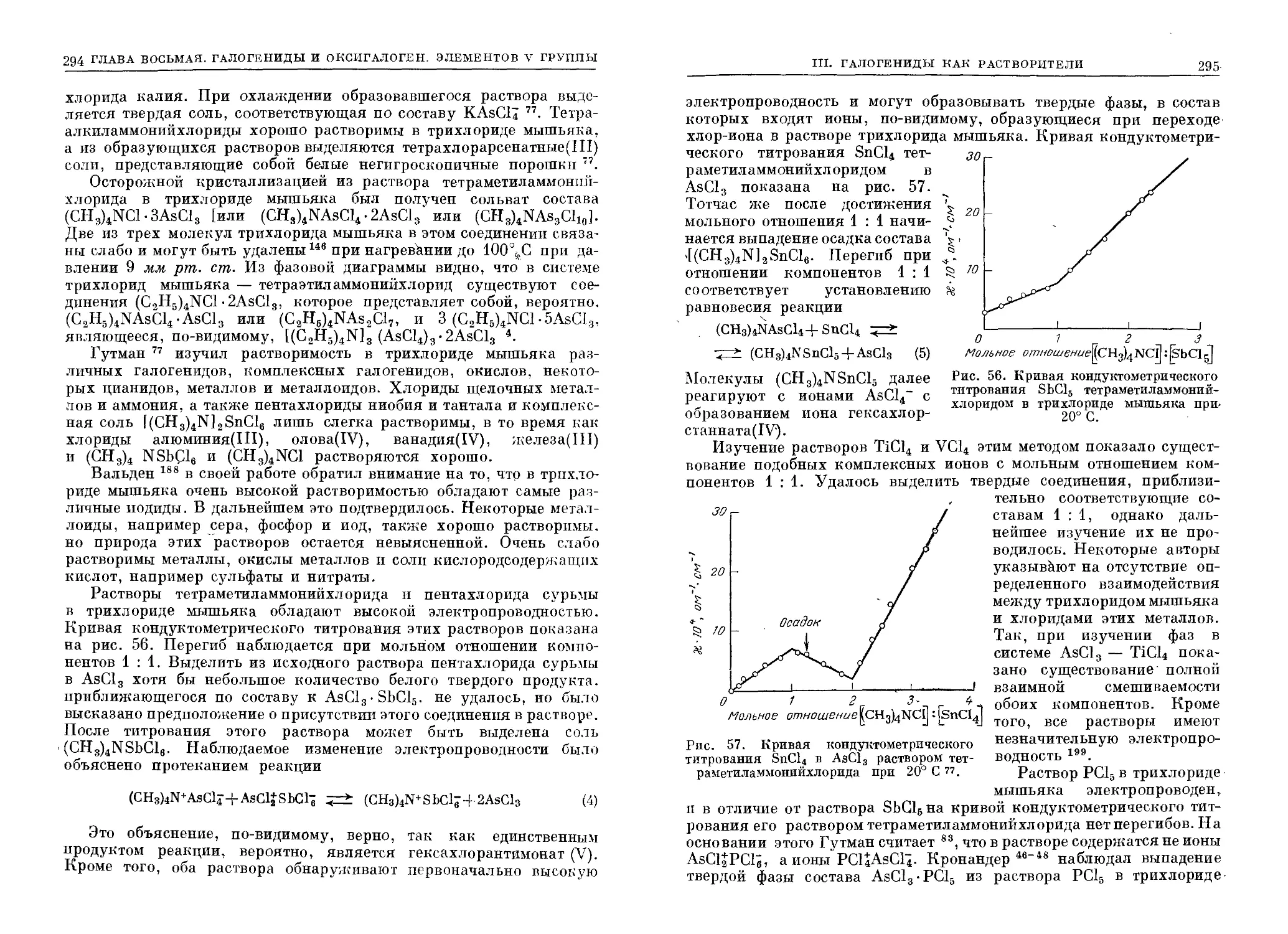
woo
4. Электропроводность
Эквивалентная электропроводность растворов металлов в жидком
аммиаке больше, чем электропроводность любых других электроли-
электролитов в любом из известных растворителей. Удельные электропровод-
электропроводности концентрированных растворов являются величинами того же
порядка, что и электропроводности металлов, а эквивалентные
электропроводности разбавленных растворов достигают величин
«=; 1000 *, т. е. примерно в 5—10 раз больше электропроводностей
растворов солей в воде 104. JZ00
Кривые эквивалентных элек-
электропроводностей растворов ли-
лития, натрия и калия в аммиаке
приведены на рис. 2. Следует
заметить, что при увеличении
концентрации, начиная от бес-
бесконечного разбавления, экви-
эквивалентная электропроводность
уменьшается, достигает ми-
минимума при концентрации
0,04 молъ/л, после чего резко
возрастает.
Изучение чисел переноса t
в растворах натрия показало,
что подвижность электрона
Я_ достигает минимума при
Д
5
800
600
О
5
г з 4
ig v
Рис. 2. Зависимость эквивалентной
электропроводности растворов щелоч-
щелочных металлов в жидком аммиаке при
—33,5° С от разбавления. V — число
литров жидкого аммиака (р =
концентрации 0,04 молъ/л, в то = 0*^674 г/см?), в которых растворен
время как подвижность иона 1 г-атом металла 92.
натрия Х+ уменьшается непре-
непрерывно с увеличением концентрации 41. Отношение tjt_ изме-
изменяется в пределах от 7 (разбавленные растворы) до 280 (кон-
(концентрация натрия около 0,9 молъ/лI02. Данные измерения чисел
переноса показывают, что вид кривых электропроводности опреде-
определяется главным образом подвижностью электрона.
Температурный коэффициент удельной электропроводности для
натрия -г- равен 1,9% на градус для разбавленных
&t X-33,5°C
растворов; он достигает максимума D% на градус) при концентра-
концентрации около 1 молъ/л, после чего с увеличением концентрации металла
уменьшается почти до нуля105> 1Ов. Причиной этого является не
изменение вязкости раствора, а различие механизмов проводимости
в разных концентрационных областях. Обзор различных механизмов,
которые были предложены для областей низкой проводимости, есть
в работе Джолли 92.
* По-видимому, см2/(ом-г-вке). — Прим. ред.
2*
20
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Эффект Вина для растворов щелочных металлов в жидком амми-
аммиаке 115 при напряженности поля 15 000 в/см составляет 4%. Такое же
увеличение проводимости в водных растворах электролитов проис-
происходит при напряженности поля, приблизительно равной 100 000 в/см.
5. Магнитные свойства
Мольная магнитная восприимчивость раствора щелочного ме-
металла при бесконечном разбавлении приближается к величине
N\io/kT. Однако при увеличении концентрации мольная воспри-
восприимчивость быстро уменьшается 52. Сравнение статической восприим-
восприимчивости натрия и калия со значением восприимчивости, полученным
при измерении парамагнитного резонанса, позволяет сделать вывод ",
что радиус сферы, в которой находится электрон, составляет 3,0 А.
Это значение хорошо согласуется с величиной, рассчитанной ра-
ранее из парциальных мольных объемов. Ширина линии парамагнит-
парамагнитного резонанса составляет для калиевых растворов лишь около
0,03 гс, что было интерпретировано как следствие «стесненного»
сверхблизкого взаимодействия между неспаренными электронами п
протонами, ограничивающими сферу, в которой находится элек-
электрон 9в. Ширина линии изменяется прямо пропорционально измене-
изменению вязкости 118.
Время релаксации 7в спин-спинового взаимодействия Т\ и время
релаксации спин-решеточного взаимодействия Т2 в разбавленных
растворах (а для калия и в концентрированных растворах) примерно
равны; в интервале средних концентраций Тх больше Т2. В пределах
ограниченного интервала концентраций Т1 и Т2 приблизительно
равны для растворов лития, натрия и кальция 32. На основании
равенства Т\ и Т2 заключили, что электроны взаимодействуют в ам-
аммиаке с ядрами азота, а не с протонами. Время релаксации в зави-
зависимости от концентрации и температуры изменяется от 1 до
3 мксек 32>1S1.
В настоящее время хорошо известно, что ЯМР-поглощение в рас-
растворах металлов происходит в более сильных полях 101. Мак-Коннел
и Холм 121 измерили химический сдвиг для 14N и 23Na в натрий-
аммиачном растворе. Они нашли, с одной стороны, что средняя кон-
контактная плотность сверхблизкого взаимодействия 23JNa и неспарен-
ного Электрона при отношении [NH3] : [Na] в растворе от 50 до 400
составляет 3 • 10 — 5 • 10, т. е. так же велика, как контактная
плотность в изолированном атоме натрия. При отношениях
[РШ3] : []\a] меньше 50 контактная плотность заметно увеличивается.
С другой стороны, средняя контактная плотность сверхблизкого
взаимодействия 14]\ и неспаренного электрона в растворах любых
концентраций составляет лишь 10% контактной плотности изолиро-
изолированного атома азота. Это подтверждает предположение о том, что
электроны взаимодействуют с ядрами азота 1S1. Температурная зави-
III. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ В АММИАКЕ
21
симость химического сдвига для натриевых растворов в интервале
стократного изменения концентраций была измерена Акривосом и
Питцером х.
6. Спектры поглощения
Спектры растворов щелочных и щелочноземельных металлов,
по крайней мере кальция, в жидком аммиаке, по существу, одина-
одинаковы ъъ в1. Для них характерно наличие широкой полосы поглоще-
поглощения с пиком приблизительно при 15 000 А. Поглощение в коротко-
коротковолновой области обусловливает голубой цвет разбавленных раство-
растворов металлов в аммиаке.
Растворы щелочных металлов подчиняются закону Ламберта —
Бэра вплоть до концентраций 0,2 моль/л. Прио более высоких кон-
концентрациях и в области длин волн выше 20 000 А может наблюдаться
положительное отклонение от закона. Растворы кальция вплоть
до концентраций, при которых происходит разделение фаз, также
подчиняются закону Ламберта — Бэра, но в области очень больших
длин волн. Мольный коэффициент поглощения растворов кальция
(для всех длин волн) почти вдвое больше мольного коэффициента
поглощения растворов щелочных металлов.
Сообщалось, что добавление Nal к растворам натрия вызывает
появление новой полосы поглощения при 8000 А; однако этот пик
не наблюдался другими авторами, проводившими независимые экс-
эксперименты 55. Вполне возможно, что пик при 8000 А связан с образо-
образованием короткоживущих соединений с периодом жизни в несколько
минут.
В интервале длин волн от 1000 до 20 000 А были получены 7
спектры отражения растворов натрия при отношениях [NH3] : [Na]
от 5 до 168. В спектрах разбавленных растворов имеется сильный
пик приблизительно при 1500 А. В концентрированных растворах
наблюдается интенсивное отражение в широком интервале длин
волн.
7. Модели
В заключение будет полезно кратко рассмотреть некоторые микро-
микроскопические модели, которые были предложены для растворов
металлов в аммиаке, в частности в области малых и средних кон-
концентраций.
На основании своих ранних работ по электропроводности рас-
растворов металл — аммиак Краус предложил считать, что в очень
разбавленных растворах металл диссоциирует на аммонизированные
катион и электрон:
М = М+ + е A)
При увеличении концентрации эти ионы ассоциируют, в ре-
результате чего электропроводность уменьшается. С дальнейшим
22
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
увеличением концентрации раствор по своим электрическим свой-
свойствам становится подобен металлам, и эквивалентная электропро-
электропроводность его резко возрастает.
С развитием исследований магнитных свойств растворов металлов
стало очевидно, что модель Крауса, предложенная на основании
данных об электропроводности, неверна, так как в ней не учиты-
учитывается образование спаренных электронов. Магнитные свойства
системы металл — аммиак были объяснены Хастером 74, а также
Фридом и Шугерменом ъ- существованием равновесия между ионами
и диамагнитными днмерами атомов металлов:
Беккер и сотр.6 показали, что данные об электропроводности
и магнитной восприимчивости систем металл — аммиак могут быть
приближенно объяснены с учетом обоих приведенных выше равно-
равновесных уравнений. Однако Арнольд и Паттерсон 3 нашли, что кон-
константы равновесия этих процессов, вычисленные при обработке
электрохимических данных, не согласуются с константами, получен-
полученными из данных по магнитным свойствам систем. Они разрешили
это несоответствие, приняв существование других диамагнитных
ионов (М~) и получили следующие значения констант равновесия
= 9,7-10-4
C)
D)
E)
Подобные модели ионов и частиц продолжают совершенствоваться.
Все данные достаточно хорошо объяснимы, если предположить,
что электрон, находящийся в сферической полости, остается неизме-
неизмененным и в том случае, когда образуются ионы и соединения типа
М, М~ и М2. Соединение М может быть описано как пара ионов,
в которой аммонизированные ион металла и электрон удерживаются
вместе силами электростатического взаимодействия с небольшим
искажением сферы аммонизированного электрона. Ионы и соедине-
соединения М~ и М2 можно интерпретировать как квадрупольные ионные
образования, состоящие из одного или двух (в случае М2) аммонизи-
аммонизированных катионов и двух аммонизированных электронов. Волновые
функции для двух электронов в соединении М2 Достаточно перекры-
перекрываются, так что энергия синглетного состояния меньше энергии
триплетного состояния более чем на кТ.
Растворы с высокой концентрацией металла, в которых катионы
металла аммонизированы, подобны расплавам металлов 55.
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
23
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
А. Сравнение аммиака с другими растворителями
1. Растворимость
Наиболее важным свойством растворителя является его способ-
способность растворять другие вещества. Последняя зависит как от свойств
растворителя, так и от свойств растворяемых веществ, поэтому очень
важно знать, какие именно вещества способен растворять данный
растворитель. Гильдебранд 69 рассмотрел свойства жидкого аммиака,
которые определяют ту или иную растворимость веществ в нем и
которые важны для сравнения растворимостей веществ в жидком
аммиаке и в других растворителях. К этим свойствам относятся
диэлектрическая проницаемость, основность аммиака, способность
к образованию водородных связей и т. д. Ниже будут приведены
данные о растворимости в качестве иллюстрации значения этих
свойств.
Диэлектрическая проницаемость аммиака (е «^ 23 при темпера-
температуре кипения) значительно меньше, чем диэлектрическая проница-
проницаемость воды (& = 78,5), однако она гораздо больше диэлектрической
проницаемости уксусной кислоты (е = 6,4). Поэтому естественно
ожидать, что значения растворимостей ионных солей в аммиаке
находятся, в общем, между величинами растворимостей тех же солей
в воде и в уксусной кислоте *.
Растворимости некоторых солей (в г/100 г растворителя) в этих
трех растворителях при 25° С приведены ниже 84:
Соль LiNC>3
Растворимость
в Н20 . . , 52,2
в NH3^ . . . 243 7
в СНзСООН 10,3 C0° С)
NaNO3 KNO3 NH4OAc
NaCl
91,8
97,6
0,17
37,8
10,4
0,18
1
48 D° С)
253,2
39,3
37
3,02
0.073 C0°
С)
Соли, содержащие двух- и трехвалентные анионы, такие, как
сульфаты, карбонаты и фосфаты, практически нерастворимы в жид-
жидком аммиаке. Энергия сольватации ионов этих солей в аммиаке
недостаточна для того, чтобы компенсировать высокую энергию
кристаллической решетки соли.
Аномально высокая температура кипения аммиака служит дока-
доказательством того, что аммиак проявляет склонность к образованию
водородных связей. Вещества, которые способны к образованию
водородных связей с аммиаком, хорошо в нем растворяются, так же
как и в других растворителях (воде, фтористом водороде),
* Это не относится к солям, анионы которых легко поляризуются, так как
начинают проявляться силы Ван-дер-Ваальса.
24
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
образующих водородные связи. К таким веществам относятся
сахара, сложные эфиры, амины и фенолы.
То, что аммиак является сильным основанием, обусловливает
высокую растворимость в нем карбоновых кислот, спиртов и фено-
фенолов. Во многих случаях при рассмотрении растворимости трудно
отличить явления, вызываемые основностью аммиака, от эффектов,
связанных с его способностью к образованию водородных связей.
Свойством аммиака, близким по характеру к основным, является
склонность его молекул координироваться по отношению к ионам
переходных элементов, таких, как Ш2+, Cu2+, Zn2+, Ag+ и т. д.
Соли таких металлов обычно хорошо растворимы в аммиаке. Это
свойство аммиака убедительно иллюстрируется высокими величи-
величинами растворимости в нем галогенидов серебра (см. ниже).
Молекулы, химически не реагирующие друг с другом, тем не
менее притягиваются под действием сил Лондона или сил Ван-дер-
Ваальса *. У молекул, которые не только имеют высокие дипольные
моменты, но и, обладая достаточно большим количеством электро-
электронов, способны легко поляризоваться, силы Лондона могут превысить
эффект дипольной ориентации. Вклад разных эффектов в полную
ван-дер-ваальсову энергию **:
Вещество Н20 ?Шз
Дисперсионный потенциал, эрг • смв 47 93
Ориентационный потенциал, эрг ¦ смв igo §4
Потенциал взаимодействия диполь —индуциро-
—индуцированный диполь, эрг •смв 10 10
Нетрудно заметить, что потенциал притяжения воды опреде-
определяется главным образом ее дипольным моментом, тогда как у амми-
аммиака он определяется в равной степени дипольным моментом и поляри-
поляризуемостью молекулы. Так, хотя аммиак обычно более слабый раство-
растворитель для солей ионного типа и сильно полярных молекул, чем вода,
он является лучшим растворителем для неполярных молекул,
* Различают три фактора, с помощью которых можно вполне удовлетво-
удовлетворительно подсчитать ван-дер-ваальсовы силы притяжения между молекулами:
ориентационный эффект — взаимная ориентация полярных молекул и
возникновение электростатического притяжения диполей (диполь-дипольное
взаимодействие);
взаимодействие «диполь — индуцированный диполь», т. е. взаимодействие
между полярной молекулой и диполем, индуцированным ею в соседней моле-
молекуле;
дисперсионный эффект — лондоновские силы, возникающие между любыми
молекулами, независимо от их строения, вследствие сихронизации мгновенных
диполей взаимодействующих частиц.
Чем более полярна или поляризуема молекула, тем сильнее "будут прояв-
проявляться первые два эффекта; для симметричных молекул единственно действу-
действующими являются лондоновские силы. — Прим. перев.
** Цитируется по книге Барнард А., Теоретические основы неорганиче-
неорганической химии, Изд. «Мир», 1968.
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
25
в особенности с большим числом электронов. По-видимому, большей
поляризуемостью тиосульфат-иона по сравнению с сульфат-ионом
и объясняется то, что растворимость тиосульфата натрия составляет
0,17 г/100 г жидкого аммиака, в то время как сульфат натрия не обла-
обладает заметной растворимостью в РШ3. На примере галогенидов сереб-
серебра можно проиллюстрировать, как с увеличением ионной поляризу-
поляризуемости при переходе от хлоридов к иодидам повышается раствори-
растворимость в ряду этих солей:
Соль
Растворимость
при 25° С, г 1100 г NH3
AgCl AgBr
0,83 5,92
Agl
206,8
Весьма полезным критерием при рассмотрении растворимости
является «параметр растворимости», равный V&EHCJV (где Л^п —
энергия испарения, а V — объем в см3). В отсутствие сильного
дипольного и кислотно-основного эффектов вещества взаимно раство-
растворимы в наибольшей степени тогда, когда их параметры раствори-
растворимости являются величинами одного порядка (табл. 9).
Таблица 9. Параметры растворимости и растворимость некоторых соединений
в аммиаке''0
Соединение
к-Гексан
Четыреххлористый угле-
род
Бензол
Желтый фосфор
Аммиак
Вода
Ртуть
(ДЕ„сп^I/2
7.3
8,6
9,2
13,1
13,1
23,8
30,7
Растворимость в жидком аммиаке
5,1% при 20° С 7!) (сравн. это значение с
растворимостью в воде при 15,5° С,
равной 0,014%)
Растворим
Умеренно растворим
Растворим
Полная смешиваемость вследствие об-
образования водородных связей
Практически нерастворима
2. Диапазон окислительных потенциалов *
Если рассмотреть с термодинамической точки зрения все реакции
в жидком аммиаке (в отличие от оценки этих реакций с кинетиче-
кинетических позиций), то окажется, что наиболее сильным окислителем,
способным существовать в жидком аммиаке, является азот, а наи-
наиболее сильным восстановителем — водород. Ниже приведены пары
* Таблица окислительных потенциалов в аммиаке и применение ее к химии
жидкого аммиака рассмотрены в работе Джолли 91.
26
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
NH3 —]\„ и Н.2—NH3 и их потенциалы в вольтах при 25° С в кислых
и основных растворах: в кислых растворах A М H
#0=0
F)
= \ N2 + 3NIIt + Зе:
в основных растворах A М РШ2)
Зг 1
2
о = - 0,04
-е;
1
= -?-N2 + 2NHs+3e; ?о=1,55
G)
(8)
(9)
Рис. 3. Зависимость окисли-
окислительных потенциалов в жид-
жидком аммиаке от рН при 25° С.
Совершенно очевидно, что вряд ли
можно найти ионы, термодинамически
стабильные в аммиаке, так как интер-
интервал их окислительных потенциалов не
должен быть более 0,04 в. Однако во-
водород-аммиачная и азот-аммиачная
пары обычно имеют перенапряжение
порядка 1 в. Таким образом, в кислых
растворах диапазон потенциалов для
ионов в растворах в аммиаке факти-
фактически составляет от +1,0 до —1,0 е.
В основных растворах, где водород-ам-
водород-аммиачная и азот-аммиачная пары
1,6 в, диапазон потенциалов составляет
в жидком аммиаке можно
имеют потенциалы по
от 2,6 до 0,6 в. Следовательно,
работать с веществами, которые являются чрезвычайно сильными
восстановителями (например, со щелочными металлами), и с исклю-
исключительно сильными окислителями (такими, как перманганаты, озо-
ниды и пероксиды).
На рис. 3 показана зависимость окислительных потенциалов-
некоторых пар от рН раствора (рН = 0 соответствует 1 М раствору
NHJ, а рН = 27 соответствует 1 М раствору РШ^). Пунктирными
линиями на рис. 3 обозначены приблизительные границы зоны устой-
устойчивости окислительно-восстановительных пар. Таким образом, мы
видим, что аммиачный электрон еам термодинамически неустойчив,
и при всех значениях рН из растворов в NH3 должен выделяться
водород. Однако в щелочных растворах и в отсутствие катализато-
катализаторов скорость разложения очень мала. Вообще, при восстановлении
аммиака твердыми металлами эффект перенапряжения ничтожно
мал. Так, хотя кривая для пары Zn— Zn11 лежит внутри области,
ограниченной пунктирными линиями, при действии металлического
цинка на аммиак водород выделяется как в кислых, так и в щелочных
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
27
растворах 9. Суммарная реакция, протекающая в щелочных раство-
растворах, может быть представлена уравнением
Zn + 2NHJ + 2NH3 = Zn (NH2)J" + H2
A0)
Металлический таллий, безусловно, инертен по отношению к ам-
аммиаку при всех значениях рН; оказалось также, что ни нон Т1+,
ни ион T1(NH2)J не реагирует с аммиаком 9. В кислых растворах
нитрат-ион, по существу, инертен по отношению к аммиаку, но в ще-
щелочных растворах происходит медлен- so
ное выделение азота 8:
и
Циклогептатриен °енэол
Аммиак
U0 - Трифенилметан
(И)
3. Кислотно-основной интервал
Основность аммиака приблизитель-
приблизительно в 10 12 раз выше основности воды,
а кислотность аммиака в 1025 раз ниже
кислотности воды. Следовательно, ки-
кислоты, p^L которых в водных растворах
меньше 12, в жидком аммиаке являются
сильными кислотами, а кислоты, ])К
которых в водных растворах больше,
39, в жидком аммиаке практически не
30
го
1
¦о
О. ГО
-10
Анилин
Ац&тонитрил
-Ацетон
Метиловый Вода
спирт уксус-
уксусная кис
лота
_ Нитро-
метан
Уксусная
п
кислота
- Нон Океания
Соляная
кислота
- Иодистоводород)
тая кислота
Рис. 4. Диапазоны рАГ воз-
возй фф
проявляют кислотных свойств. Кислоты « с^ФыФеР„елНяПнГоторь«
средней силы в жидком аммиаке явля- растворителей iz9.
ются слабыми кислотами и могут быть
дифференцированы в соответствии с их кислотной силой. На рис. 4
показаны пределы pif для различных растворителей, в которых
возможна дифференциация силы кислоты. Большинство значений
рК заимствовано из работы Крэма 20; значения рК для НС1 и HI
взяты из работы Полинга 129. В качестве растворителей рассмотрены
уксусная кислота, вода, аммиак и бензол. На том же рисунке ука-
указаны значения рК некоторых кислот в водных растворах. Следует
отметить, что в связи с меньшей величиной константы ионизации
аммиака по сравнению с другими протонными растворителями в нем
можно изучать кислоты с гораздо более широким диапазоном силы
кислоты. Кроме того, поскольку основность аммиака выше основ-
основности уксусной кислоты и воды, в нем можно дифференцировать
более слабые кислоты, чем в этих растворителях. Бензол — апро-
тонный растворитель и в нем в принципе можно изучать раствор
любой кислоты, однако на практике часто приходится сталкиваться
с рядом трудностей при растворении ионных соединений в бензоле.
Кислоты, p^L которых в водном растворе меньше 12, полностью
реагируют с аммиаком, образуя соответствующие аммонийные соли.
Однако не следует думать, что эти кислоты (или аммонийные соли)
28
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
являются сильными в обычном смысле, т. е. полностью диссоции-
диссоциируют в жидком аммиаке на независимые ионы. Все соли, включая
аммонийные, в аммиаке диссоциированы слабо. В этом случае сила
кислоты определяется константой равновесия реакции
HX+NH3=(NHJ)(X-) A2)
Однако обычно предпочитают обозначать ионы как независимые и не
указывают на образование пар ионов даже тогда, когда эти ионы —
основные продукты реакции. Но надо всегда помнить о том, что
в умеренно концентрированных растворах происходит ионная агре-
агрегация. Так, мы обычно пишем
HOAc + NH3=NHJ + OA(T A3)
даже несмотря на то, что константа диссоциации ионной пары аце-
ацетата аммония 1в2 равна 7,7 • 10~5.
Основания, сопряженные кислоты которых в водной среде имеют
р/? больше 39, в жидком аммиаке практически полностью подверга-
подвергаются аммонолизу с образованием амид-иона. Ниже приведено два
примера таких реакций аммонолиза:
A4)
A5)
KH+NH3 =
Na2O + NH3 = NaNH2 + NaOH
Константы ионизации Н2 и ОН в водных растворах оценены
величинами 108 и <10~зв соответственно113. Таким образом, кри-
критерий протекания реакции для основного аммонолиза в этих слу-
случаях соблюдается, хотя почти на пределе.
Некоторые окислители реагируют с жидким аммиаком, образуя
азотсодержащие продукты, которые можно рассматривать либо как
продукты окислительно-восстановительных реакций, либо как про-
продукты аммонолиза. Рассмотрим, например, реакции аммиака с хло-
хлором, бромом и иодом:
X2+2NHs = NHi + X- + NH2X A6)
(Галогенамин NH2X далее вступает в реакцию, протекающую с го-
гораздо меньшей скоростью, в результате которой образуются такие
соединения, как гидразин и азот.) Начальная реакция A6) может
рассматриваться как реакция самоокисления — восстановления га-
галогена до окислительных состояний +1 и —1, но в равной степени
справедливо -рассматривать ее и как аммонолитический разрыв
связи X—X. Подобным же образом рассмотрим следующую реак-
реакцию
48.
A7)
ЗК+ + 3NH? + NOj = Nj + ЗКОН + NH3
Эта реакция соответствует окислению амида до азида нитрат-ионом,
но она может рассматриваться и как основной аммонолиз нитрат-
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
29
иона (ион O = N<( превращается в ион N=N = N за счет замены
ХО~
- + +/О- + _
группы O=N на N=N и группы N< на N=N).
4. Метод работы с жидким аммиаком
Серьезным недостатком жидкого аммиака как растворителя
является низкая температура кипения (—33,4° С). Однако во мно-
многих случаях при работе с жидким аммиаком нет необходимости охла-
охлаждать систему. Теплота испарения жидкого аммиака велика, так что
его можно держать при температуре кипения в открытых сосудах,
например в химических стаканах, колбах и других, при этом быст-
быстрого испарения не происходит. Скорость испарения аммиака, нахо-
находящегося в сосудах Дыоара (даже не посеребренных, д^ля удобства
наблюдения) практически ничтожна. Давление паров .над жидким
аммиаком при комнатной температуре достаточно низко (8—10 атм),
поэтому с ним можно работать в запаянных.стеклянных ампулах, не
опасаясь взрыва. Методика работы с жидким аммиаком подробно
описана Аудрицем и Клайнбергом *, Франклином49, Джолли 93
п Сандерсоном 13в.
Вязкость аммиака составляет *Д вязкости воды при комнатной
температуре. Небольшая вязкость аммиака дает определенные пре-
преимущества, в частности, при фильтровании растворов и проведении
гетерогенных реакций, в которых огромную роль играют процессы
диффузии растворенных соединений.
Б. Ионная сольватация
1. Термодинамика процессов аммонизации и гидратации
Свободные энергии образования пар противоположно заряжен-
заряженных ионов в аммиаке приблизительно равны соответствующим зна-
значениям свободной энергии образования в воде, если рассматривать
ионы, которые не дают аномально прочных комплексов в амми-
аке
90, 91.
Ионная пара ...
—А^обР ПРИ 25° С,
ккал/молъ
в Н2О ....
в NH3 ....
Na+F"
128,7
124,4
Li+I~
82,6
83
Rb+N(
93,9
90,3
Rb+NOj NHJBr" Na+C10j
43,6
42,6
63,2
61,3
Исходя из этих данных, резонно сделать вывод о том, что сво-
свободная энергия перехода такого индивидуального иона от воды
30
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
к аммиаку близка к нулю *. Свободную энергию перехода иона,
образующего прочные комплексы с аммиаком, назовем «экспери-
«экспериментальной» свободной энергией перехода. Интересно сравнить ее
значения со значениями свободной энергии реакций типа
протекающих в водных растворах. Если в качестве стандартных
растворов использовать 1 м растворы в воде и аммиаке, свободная
энергия (в ккал/молъ) такой реакции выражается уравнением
Д/$ = — 1,36 (lg К + х lg 55) A9)
где К — обычная константа комплексообразования в водных растворах.
Ниже сравниваются рассчитанные и «экспериментальные» сво-
свободные энергии перехода иона от воды к аммиаку при 25° С:
Ион
X
?^., ккал/молъ . .
п, ккал/молъ , ,
4
36
40
Си2+
R
to to,
со со (
#12+
А
см см
2
20
20
Н+
1
15
16
Ag
2
14
17
Нетрудно заметить очень близкое сходство величин соответству-
соответствующих свободных энергий. Следовательно, можно оценить стандарт-
стандартную свободную энергию образования иона в жидком аммиаке по урав-
уравнению
Д/?бр (NHS) = Д^обр (Н2О) + 16Z -1,36 (lg К+х lg 55) B0)
Подобные же уравнения выведены для расчета ионных энтропии
и теплот образования в аммиаке91.
2. Кинетика обменных реакций в аммиаке
Аммонизированный ион водорода, или ион аммония, — простей-
простейший ион, существующий в жидком аммиаке. Скорость, с которой
протоны переходят от ионов аммония к молекулам аммиака (или ско-
скорость, с которой молекулы растворителя — аммиака — замещают
молекулы аммиака в аммонизированных ионах), настолько высока,
* Абсолютное значение изменения свободной энергии перехода иона из
водного раствора в аммиачный раствор (AFn°) — мера склонности иона коорди-
координировать молекулы воды или аммиака. Не существует точного эксперимен-
экспериментального метода оценки этой величины свободной энергии. Для расчета ДР",°
выведено полуэмпирическое уравнение
(NH3) - 16Z -
(Н*О)
где Д^обр(^Нз) и Д/^обр Н.,0 — свободные энергии образования ионов в соот-
соответствующих растворителях, a Z — заряд иона (с учетом его знака)s0. —
Прим. перев.
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
31
что ее невозможно измерить классическими химическими мето-
методами 125. Однако Огг12в показал возможность измерения скорости
зтого обмена методом ЯМР. Спектр протонного магнитного резо-
резонанса чистого жидкого аммиака состоит из трех пиков, появление
которых обусловлено спин-спиновым -взаимодействием протонов
с ядрами14 IN. Наличие следов аммонийной соли меняет картину
спектра: вместо триплетной появляется синглетная линия. Это из-
изменение и приписывается обменной реакции
NHg + NHJ ^ NHJ + NH3 B1)
Грубо оценив концентрацию ионов аммония, вызывающую «сжатие»
триплета. Огг рассчитал константу скорости реакции B1), оказав-
оказавшуюся равной 5-Ю8 л/(молъ-сек). Того же порядка оказались рас-
рассчитанные им константы скоростей реакций
NHg + NHj ^ NHj + NHg B2)
и
NH3-fH2O ^ NHJ+OH- B3)
Висендейнжер и др.153, использовав для экспериментов 15N, по-
показали, что скорость обмена молекулами аммиака между раствори-
растворителем и ионами Ag (NH3)j, Cu (NH3)^+ и Ni (NH3J+ также очень
высока, и ее нельзя замерить классическими методами, но скорость
обмена молекулами аммиака между ионами Сг (NH3);j+ и Со (NH3)jj+
п растворителем сравнительно невелика. Была измерена константа
скорости для бимолекулярной реакции с ионами Cr (NH3)|+. Она
равна 1,3-10"в л/(молъ-сек). Хант и др.73 по уширению линии ЯМР
измерили скорость реакции обмена атомами 14N между ионами
Ni (NH3)|+ и молекулами аммиака-растворителя. На основании
того, что скорость соответствующей обменной реакции в водном ра-
растворе не зависит от концентрации аммиака и практически имеет
такое же значение, как и скорость обмена в безводном аммиаке, авто-
авторы предположили, что реакция мономолекулярна, и сообщили, что
при 25° С константа скорости ее к = 4,7-104 сек'1.
В. Кислотно-основные реакции
1. Природа аммоний- и амид-ионов
Аммоний-ион и амид-ион являются аналогами ионов Н3О+
и ОН~. Так, реакция нейтрализации сильной кислоты сильным осно-
основанием в аммиаке может быть представлена уравнением
NH+4-NH7 —у 2NH3 B4)
что аналогично уравнению
+ + он-
Н,0
B5)
32
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Однако аналогия нарушается, если сравнивать подвижности ионов.
В воде ионная эквивалентная электропроводность Н3О+ и ОН"
в несколько раз больше, чем у обычных одновалентных ионов:
Ион Na+ NHJ H+ NOj NHj ОН"
Яо, сжа/(ол» -г-экв)
в Н2О при 25° С120 . . . . 50,1 73,4 349 8 71,4 — 198,0
в NH3 при —33,5° С "I-"г 158 142 — 177 166 «з —
Эти аномально высокие значения эквивалентной электропровод-
электропроводности объясняются механизмом, впервые описанным Гроттгусом.
Миграция иона Н3О~ происходит путем приближения протона иона
гидроксония к соседней молекуле воды на такое расстояние, которое
необходимо для частичного разрыва его связи с атомом кислорода
и для образования связи с атомом кислорода другой молекулы воды:
Н
I
Н
н-о-н.., о-н
н н
I I
н-о ...н-о-н
Миграция иона ОН" происходит совершенно аналогично:
Н Н н Н
О ... Н-0 —>¦ О-Н ... О
Но подобные процессы не оказывают заметного влияния на под-
подвижность ионов NHj и NHj в аммиаке, так как водородная связь
в этом растворителе гораздо более слабая (чем слабее водородная
связь, тем больше величина энергетического барьера между двумя
равновесными состояниями протона). Из приведенных выше данных
видно, что подвижности аммоний- и амид-ионов в аммиаке близки
к подвижностям обычных одновалентных ионов в этом растворителе.
Константы скоростей реакций B1) и B2), как уже указывалось,
равны — 5-Ю8 л](моль• сек). Константы скоростей соответствующих
им водных реакций119
—> Н2О + Н3О+ B6)
—> Н2О + ОН" B7)
равны 1,1-1010 и 5-Ю9 л/(молъ-сек).
2. Определение концентрации аммоний- и амид-ионов
На электродах, покрытых платиновой чернью, перенапряжение
водорода в жидком аммиаке мало, поэтому эти электроды, находя-
находящиеся в равновесии с водородом, могут быть использованы в ка-
качестве обратимых водородных электродов. Плесков иМоносзон167, 168
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
33
применили водородный электрод в аммиаке при —50° С для опреде-
определения коэффициентов активности NH4NO3 и РШ4С1. Построив
ячейку
Pt, Н210,1 М NH4NO31 насыщенный раствор KNO3 | KNH21 Pt, Н2
они определили константу самоионизации жидкого аммиака при
-50° С:
Воспользовавшись значением ¦.123 теплоты реакции ЛЯ° =
= 26,1 ккал/молъ, получим К = 5,1 -107 при 25° С. Если сравнить
это значение с величиной К — 2,2-108, рассчитанной из свобод-
свободной энергии образования амида натрия26, то видно, что почти во всех
случаях с достаточной точностью можно принять К = 10" . Если
построить шкалу рН, воспользовавшись уравнением рН =
= — lg [NHj ], получим, что рН жидкого аммиака при 25° С
равен 13,5.
Цинтл и Неймайр 169 обнаружили, что воспроизводимые резуль-
результаты получаются при применении хингидронного электрода с кон-
концентрационной ячейкой
Pt | хингидрон (сг), NH4Cl (сг) || хпнгидрон (е2), NH4Cl (e2) | Pt
Измерения проводились при t = 50° С.
Хейн и Берджин в7 пытались использовать стеклянные электроды
для работы с растворами в жидком аммиаке. Для этого они восполь-
воспользовались следующей ячейкой:
РЬ | 0,1 М Pb (NO3J, NH4NO3 (с2) | стеклянная мембрана | 0,Ш РЬ (NO3J,
0,Ш NH4NO3 | РЬ
Авторы обнаружили, что все стекла непригодны для создания сте-
стеклянного электрода в NH3. Оказалось, что ионы аммония в жидком
аммиаке не находятся в равновесии с ионами на поверхности стеклян-
стеклянного электрода. Вероятно, аммиак настолько дегидратирует стекло,
что протоны в стекле теряют подвижность. Интересно было бы про-
проверить возможность использования листовой катионообменной смолы
в аммонийной форме в качестве мембраны, чувствительной к измене-
изменению рН.
В жидком аммиаке изучались свойства ряда кислотно-основных
индикаторов, в том числе некоторых применявшихся в водных рас-
растворах 50' 1в9. В табл. 10 приведены данные об изменении окраски
индикаторов в кислых и основных растворах.
Катрелл и др.31 с помощью спектрофотометрического метода
определили константы ионизации и диссоциации слабых кислот
о- и га-нитроацетанилидов в аммиаке. Эти соединения в кислых рас-
растворах бесцветны, а в щелочных имеют желто-коричневую окраску.
3 Заказ 90
34
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Таблица 10. Кислотно-основные индикаторы в жидком аммиаке
Индикатор
Фенолфталеин
Кармин
Сафранин. Т
«-Иитрианилпн
И-1ТИТРОЗНИЛИ II
о-Нитр''анилин
Цвет раствора
в чистом NH»
Розовый
Темно-красный
Малиновый
Желтый
Желтый
Желтый
в основном растворе
Темно-красный
Синий
Синий
Зеленый
Оранжевый
Красный
в кислом растворе
Бесцветный
К расный
Малиновый
Желтый
Желтый
Желтый
Р1ониза>шя с образованием ионных пар может быть представлена
общим уравнением
а диссоциация — уравнением
[NHj-X-
[HX]
[NHfl[X-]
[NHJX-]
Ниже приведены значения равновесных констант ионизации и дис-
диссоциации изомеров нитроацетанилида при бесконечном разбавлении:
Изомер орто- пара-
КИ 2,2-10 9,3-10~2
А'д„сс 2,2 • 10~4 0,89 • 10~4
В растворах с очень высоким значением рН концентрация амид-
нона может быть определена спектрофотометрически. Максимум
поглощения амид-иона 127 лежит при 3410 А.
Уатт и др.150 провели потенциометрическое титрование растворов
некоторых кислот в жидком аммиаке амидом калия. Была исполь-
использована система разностных индикаторных электродов. Один электрод
погружался в титруемый раствор, второй — в исходный раствор,
соединенный с титруемым раствором капилляром149'158. На основании
положений точек перегиба смесей авторы расположили кислоты
по убыванию их кислотной силы в следующий ряд: NH+ >
> HsNC(NH)NH+> H!NC(S)NH2>H!NC(O)NH2> H,NC(NH)NH2>
> H2NC(S)NH- >H2NC(O)NH->H2NC(NH)NH->NH3. К сожа-
сожалению, этот метод неприемлем для количественного измерения
силы кислоты.
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
35
3. Обменные реакции, катализируемые основаниями
При растворении слабых кислот в жидком ND3 происходит обмен
водорода кислоты на дейтерий. Константы скоростей 1Т0 реакций
обмена дейтерием между ND3 и углеводородом при 120" С (без до-
добавления амида калия в качестве катализатора) сравниваются ниже
со значениями трК, измеренными 29 в воде:
Углеводород
к, сек~* . .
Индеп
4 • 101
— 21
Флуорен
2•10
— 31
Трифенилметан
2-10"'
— 40
Дифенилмстан
7 • КГ9
— 42
Следует отметить, что чем выше кислотность углеводорода, тем
с большей скоростью он обменивается водородом с аммиаком.
По всей видимости, эти константы скорости соответствуют константам
скорости реакций ионизации углеводородов.
Такие чрезвычайно слабые кислоты, как бензол, не вступают
в реакцию обмена с дейтероаммиаком в отсутствие катализатора.
Реакция протекает в присутствии такого сильного основания, как
амид калия. Механизм обмена, вероятно, можно представить сле-
следующими уравнениями:
RH + NDJ > R-+NHDj B8)
R-+ND3 >- RDH-NDJ B9)
Ниже приведены некоторые экспериментально определенные кон-
константы скорости реакций обмена дейтерием между растворами амида
калия в ND3 и бензолом 17°:
Концентрация ами-
амида калия, мольjл 0,010 0,014 0,021 0,039 ** 0,19 0,43
к *, сек"^
при0°С . . . 4- 10"в — 7,3 - 10"в — — 8,9 • 10~5
при 25° С . . . 4,4-10^ 5,7 • 10~5 8,6-10"^ 1,8-КГ* 4,2 • 10~4 —
при 40° С . . . 1,6-10 3,1 • 10~4 — — — —
* Константы рассчитаны из предположения, что скорость не зависит от ьчн^ентрашш
амида калия, « = fe [бензол].
** При— 30° С ft= 4 - Ю-7 сек-1.
Нетрудно заметить, что в разбавленных растворах константа
скорости приблизительно прямо пропорциональна концентрации
амида калия, а при более высоких концентрациях кажущиеся кон-
константы скорости уменьшаются. Такое уменьшение каталитической
активности, вероятно, связано с образованием ионных пар.
Было обнаружено, что скорость обмена водорода, находящегося
в орто-положении замещенного бензола, сильно зависит от электро-
электроотрицательности групп заместителя. Так, константа скорости реакции
обмена для фторбензола более чем в 107 раз превышает константу ско-
скорости реакции с толуолом. Построив график: логарифм относительных
3*
36
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
констант скорости обмена для замещенных бензолов в ND3 —
значения рК в водных растворах соответствующих замещенных
уксусной кислоты, получим прямую линию, показанную на рис. 5.
Это еще одно доказательство существования зависимости между
скоростью ионного обмена и кислотностью углеводорода.
Реакции обмена водородом между органическими соединениями
и аммиаком могут быть использованы для получения дейтерирован-
ных соединений. Таким способом можно полностью дейтерировать
бензол, нафталин, фенантрен, трифенилме-
тан, дифенилметан, дибензил и пиридин 170.
Уилмарт и Дейтон 1и заметили, что про-
процесс конверсии параводорода в ортоводород,
катализируется в жидком аммиаке амидом
калия. Было обнаружено, что обмен газо-
газообразным дейтерием протекает в этом рас-
растворе приблизительно с той же скоростью;
начальным продуктом реакции является HD.
В обоих случаях скорость оказалась про-
пропорциональной концентрации амид-иона и
.-/1—i—1—i—КШ2] водорода (параводорода или дейтерия). Кон-
Константа скорости при —50° С равна
130 л/(моль'сек). Полученные данные были
объяснены с помощью следующего меха-
механизма:
D2 + NHj —у D- + NH2D C0)
D"+NH3 —>- HD+NHj C1)
Изучение реакции обмена дейтерием в ши-
широком интервале концентраций и температур
указывает на то, что эта реакция ката-
катализируется как свободными амид-ионами,
так и недиссоциированными молекулами
амида калия 5. Рассчитанные константы скорости при —61° G
равны: &№1- = 36 л/(молъ-сек) и &KNH =1,7 л/(молъ • сек). Энер-
Энергия активации реакции с амид-ионом оказалась равной 7,5 ккал/молъ.
Из скоростей обмена D2, HD и НТ, а также из скорости конверсии
параводорода были рассчитаны следующие изотопные эффекты:
2*Hd/*d, = 1-28 ±0,03
&HD/fcHT = 1,6^ ±0,07
2AWAD2 = 2>36 ± °>30
Катализируемая амидом реакция обмена водорода с аммиаком
представляет огромный интерес, так как может быть положена в ос-
основу метода концентрирования дейтерия в аммиаке 17> 20> 37> 64'133> 1з3.
Рис. 5. Корреляция меж-
между логарифмом относи-
относительных констант скоро-
скорости обмена замещенных
бензолов в ND3 и зна-
значениями рК соответ-
соответствующих замещенных
уксусной кислоты в воде
(формулы при точках—
заместитель X).
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
37
}4. Реакции, катализируемые кислотами
Многие сложные эфиры в жидком аммиаке подвергаются аммо-
нолизу:
RCO2R + NH3 у RCONH2 + ROH C2)
Процесс аммонолиза катализируется солями аммония аналогично
тому, как процесс гидролиза катализируется водными кислотами.
Но ряд каталитической активности солей аммония обратен ряду
кислотной силы, полученному на основании данных об электропро-
электропроводности растворов. Например, при изучении процесса аммонолиза
этилбензоата было найдено 43, что каталитический эффект умень-
уменьшается в ряду бензоат аммония > РШ4С1 > РШ4Вг > ]\Н4СЮ4.
Позднее было замечено, что натриевые соли также способны катали-.
зировать аммонолиз сложных эфиров, хотя эффективность этих элек-
электролитов как катализаторов меньше, чем эффективность солей ам-
аммония *.
Обмен азотом между амидами карбоновых кислот и жидким
аммиаком изучался88 с использованием аммиака, меченного изото-
изотопом 15N. Процесс обмена катализируется хлоридом аммония. Для
этой реакции предложен следующий механизм:
ОН2
i
C-i
I
NH2
C3)
Г. Реакции в растворах металлов в аммиаке
1. Общий обзор
Известно огромное число реакций синтеза, протекающих в ра-
растворах металлов в аммиаке 15'1в>47>147'148, однако о механизме этих
реакций практически ничего не известно. Тем не менее из рассмотре-
рассмотрения продуктов этих реакций можно сделать некоторые выводы.
Можно считать, что начальной стадией большинства реакций, про-
протекающих в растворах металлов в жидком аммиаке, является один
из следующих процессов:
1) присоединение электрона без разрыва связи
X"
C4}
* Совершенно ясно, что все эти случаи катализированного аммонолиза
сложных эфиров лучше рассматривать как примеры катализа под действием
электролитов *.
38
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
2) присоединение одного электрона с разрывом связи
eaM + X~Y > X.+Y- C3)
3) присоединение двух электронов с разрывом связи
2eaM + X-Y —> X- + Y- C6)
Присоединение электрона без разрыва связи. В результате реак-
реакции электрона с молекулярным кислородом и нитрит-ионом обра-
образуются продукты, кинетически довольно устойчивые в жидком
аммиаке:
+ О O C7)
1 CS)
Скорости этих реакций в жидком аммиаке не определены, хотя
их удалось измерить в воде33'38, даже несмотря на то что в Н30
электрон и ноны Ог и NOjj" могут существовать лишь в течение
крайне малого времени.
Представляют интерес некоторые реакции восстановления ионов
переходных элементов:
N01"
м + MnOj
MnOJ"
C9)
D0)
D1)
D2)
D3)
> Ni(CN)|-
>¦ Pd(CN)f-
—> Pt(NHs)*
2eaM + Pt(en)f+ >- Pt(enJ*
Присоединение одного электрона с разрывом связи. К этой группе
реакций относятся реакции электрона с протонными кислотами.
Ниже в качестве примера будут рассмотрены кинетика и механизм
•следующих реакций:
*aM + NHs > NHJ + 4-Hj D4)
D.3)
' 2
D6)
Реакция электрона с органическими сульфидами может быть
объяснена образованием промежуточных радикалов R ¦:
вам + RaS > RS-+4R2 D?)
В качестве несколько необычного примера можно привести реакцию
eaM+(C2H5KSnBr > (C2H5KSn. + Br- D8)
в которой образуется устойчивый радикал.
* Символ en — этндендпампн. — Прим. ред.
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
39-
Разрыв связи в результате присоединения двух электронов. В
случае в результате реакции образуются либо два аниона,
бианпон:
этом?
либо-
D9)
C6H5NH--|-NHj E0>
2eaM + C6H5-N=O >- CeH5-N-—О" E1)
Однако обычно один из анионов подвергается аммонолизу, как,
например, в следующих реакциях:
2eaM + N=N+-Cr *¦ N=N + 02-
E2).
E3)
E4)
E5).
OH- + NHJ
OH- + NHJ
C2Hj
nh.
RCH-—CH2
I
2NH3
RCH2CH3+2NHi
Было замечено, что многие реакции восстановления в растворах
щелочных металлов в жидком аммиаке протекают только в присут-
присутствии протонной кислоты (например, спирта или водыI5'16. Так,,
бензол практически не реагирует с раствором натрия в аммиаке,,
но в присутствии спирта происходит восстановление до 1,4-дигид-
робензола. По-видимому, спирт или другая протонная кислота иг-
играют в этих реакциях роль буфера. Можно считать, что реакции
восстановления соединений X до Н2Х протекают по следующему
механизму.
х т=±. х- d^± X2-
-е -е
н+ i 1 —н+
! п+
нх —-> нх-
40
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
В большинстве случаев промежуточный ион Х2~ термодинамически
очень неустойчив, поэтому реакция практически не протекает через
образование этого промежуточного комплекса. Отношение между
концентрациями НХ и Х~ зависит от рН раствора. В отсутствие
лротонной кислоты рН вскоре достигает очень высоких значений.
Следовательно, хотя НХ — исключительно слабая кислота, кон-
концентрация НХ будет уменьшаться, и реакция, по существу, пре-
прекратится. В присутствии же протонной кислоты образуется буфер-
буферный раствор, рН которого зависит от силы кислоты. Если добавлена
достаточно сильная кислота, отношение [НХ1 : [Х~] будет высоким,
м реакция восстановления пройдет в заметной степени.
2. Реакция металлов с аммиаком
Все растворы металлов в аммиаке неустойчивы. При длительном
хранении или в присутствии соответствующего катализатора рас-
раствор разлагается с образованием амида металла и выделением во-
водорода:
M + zNH3 —> ~Я2 + М(ПЯг)х E6)
Суммарная реакция для растворов натрия или калия (амиды кото-
которых растворимы) может быть представлена уравнением
NH,+-|
E7)
Если использовать чистые реагенты и чистую аппаратуру, то ско-
скорость разложения холодных растворов может составить0,1% в день 35.
Варшавский146 отмечал, что начало процесса выделения водорода
из растворов металлов в аммиаке в стеклянном сосуде вызывает
реакция между металлом и сильно адсорбированным на поверхности
•стекла слоем воды. Эту воду можно удалить с поверхности стекла
пирекс, выдерживая сосуд в вакууме при 400° С в течение 200 ч.
Реакция взаимодействия калия с жидким аммиаком при комнат-
комнатной температуре изучалась Чоу и др.23 спектрофотометрическим
методом. Было показано, что эта реакция первого порядка по металлу
до степени превращения — 75%. Полупериод реакции U/, для на-
начальных концентраций 2-10—1,13• 10~3 м равен 9—13 ч. По-
Поскольку в различных реакционных сосудах были получены разные
значения скорости реакции, можно предполагать, что реакция ката-
катализируется стенками сосуда. Варшавский (неопубликованные ре-
результаты) заметил, что разложение разбавленных « 10"* м) рас-
растворов натрия в аммиаке при —78° С в присутствии платиновой
•фольги протекает как реакция нулевого порядка по натрию. Он
определил константу скорости к — 1,5-10~9 моль Н 2/(сн2 • ч). Пер-
Первый и нулевой порядок реакции указывают на то, что скорости
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
не зависят от концентрации аммоний- и амид-ионов в изученном диа-
диапазоне концентраций.
Значительно больший интерес, чем описанные выше поверх-
поверхностно-катализируемые реакции, представляет гомогенная реакция
электрона с аммиаком. Вероятно, эта реакция протекает настолько-
медленно, что ее невозможно наблюдать. Если допустить, что скорость
гомогенной реакции меньше, чем скорость гетерогенной реакции
(как считают Чоу и др.), для константы скорости реакции первого-
порядка получим величину /с<1,5-10 сек'1. Сравните эту кон-
константу с константой скорости относительно быстрой реакции взаи-
взаимодействия электрона в водных растворах с молекулой воды (к <С
<4,4-104 сек-1K9:
?водн + Н20 —*• Н + ОН- E8)
Большое различие химических активностей электронов в воде
и в аммиаке не должно удивлять читателя-. Большая энергия акти-
активации реакции в жидком аммиаке
H+NHJ
E9)
по сравнению с реакцией в водной среде соответствует измеренным
теплотам реакции: +33 ккал/молъ для реакции в жидком аммиаке
и —10 ккал/молъ для реакции в водной среде.
Механизм гомогенной реакции неизвестен. Можно считать, что
атомарный водород, образующийся на начальной стадии, ассоции-
ассоциирует
2Н —> Н2 F0)
или что он реагирует с электронами, образуя гидрид-ион
Н+гам > И"
F1)
F2)
3. Реакции металлов с ионом аммония в аммиаке
Ион аммония очень быстро взаимодействует с электроном в ам-
аммиаке:
NH4++eaM —> NH3+jH3 F3)
Полупериод реакции в растворе с концентрацией электронов 10 ж
и ионов аммония 10"* м меньше 1 сек. Отсюда нижний предел для кон-
константы скорости реакции второго порядка равен 104 л!(молъ-сек).
Мы считаем, что константа скорости реакции NH| + еш не должна
превышать константу скорости реакции обмена протонами между
NHJ и NH3, для которой получена величина 5-Ю8 л/(молъ-сек).
Следовательно, константа скорости реакции F3) должна лежать
в пределах 104—5 -108 лЦмолъ-сек).
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
Константа скорости соответствующей реакции взаимодействия
электрона и протона в воде39
i "Г НВОдН
н
F4)
составляет 2,3-1010 л/(молъ-сек). Существует по крайней мере два
способа, с помощью которых можно определить скорость реакции
взаимодействия электрона и иона аммония в жидком аммиаке.
Во-первых, можно использовать методы импульсного радиолиза38
и фотолиза143, которые применялись для изучения реакций с электро-
электроном в воде. Во-вторых, можно изучать реакцию разложения буфер-
буферных растворов металлов, в которых концентрация иона аммония
такова, что реакция протекает со скоростью, удобной для замера.
В следующем разделе мы остановимся на интерпретации данных,
полученных этим методом.
4. Реакция взаимодействия натрия в аммиаке
с этиловым спиртом
Келли и др.98 изучили эту реакцию при —33,4° С, измеряя во вре-
времени количество выделяющегося водорода:
+ -i-H.2 F5)
Результаты опыта представлены ниже (натрий был взят в избытке)98:
t, сек 3 10 30 60 120 240 480 720
Степень превращения
спирта, % 1,2 6,5 14,1 19,8 25,4 31,5 37,7 42,5
t, мин 20 28 40 60 80 120 180 300 480
¦Степень превращения
спирта, % 49,5 56,5 62,0 68,7 76,0 84,0 91,0 97,5 100,0
Примечание. Начальная концентрация натрия 0,96-4, этилового спирта —
0,34 м.
Авторы этой работы отмечали, что до степени превращения спирта,
равной 25%, реакция протекает по первому порядку по спирту
и фактически нулевому порядку по натрию. В течение последующих
25% превращения спирта реакция не подчиняется определенному
кинетическому закону. После 50% превращения спирта реакция
протекает как реакция первого порядка и по спирту, и по натрию,
т. е. суммарный порядок реакции равен 2. По нашему мнению, лучше
всего полученные результаты объясняются следующим механизмом;
C2H5OH + NH3 ^=± NHJ + C2H5O- F6)
NHj + вам —"* NH3 + yH.2 F7)
IV. РЕАКЦИИ В ЖИДКОМ АММИАКЕ
43-
Если допустить, что установившаяся концентрация иона аммония
мала, то кинетическое уравнение может быть представлено в виде
_ h [С2Н5ОН] [еам]
dt
Качественно результаты экспериментов подчиняются этому кинети-
кинетическому уравнению. Для начального момента, когда концентрация
СоН50~ мала, кинетическое уравнение можно упростить:
dt
В этих условиях стадией, опреде-
определяющей скорость реакции, явля-
является ионизация этилового спирта.
В конце реакции, когда (к.2/кя)
[С2Н5СГ
[еа
кинетическое
уравнение упрощается до
d [С2Н5ОН] кг [С2Н5ОН] [еам]
700
80
60
20
!О
ю2 го3
t, сек
1ОЬ
Рис. 6. Зависимость степени превра-
превращения спирта от времени в реакции
взаимодействия натрия с этиловым
спиртом в жидком аммиаке9».
dt кг/к3 [С2Н5О-]
Скорость реакции в этом случае
определяется стадией взаимодей-
взаимодействия иона аммония с электро-
электроном. О точности, с которой экспе-
экспериментальные данные количест-
количественно удовлетворяют суммарному
кинетическому уравнению, можно
судить по кривой Пауэлла,изобра-
Пауэлла,изображенной на рис. б53. График представлен в координатах степень пре-
превращения спирта / — время t. Точки на графике соответствуют экс-
экспериментальным данным, а кривая рассчитана из суммарного ки-
кинетического уравнения; приняты следующие значения констант:
к1 = 0,0303 сек'1 и k2fks = 460. Не совсем понятно расхождение
между экспериментальными точками и расчетной кривой в области
/ = 10 ¦*- 40%. Нет оснований считать, что это расхождение зависит
от образования ионных пар, или от протекания начальной реакции
натрия с аммиаком, или от того, что это реакция первого порядка
по спирту и по натрию.
Величина p/sTa спирта в воде29 равна ~ 17. Воспользовавшись
правилом, что величины рК в аммиаке на 12 единиц меньше, чем
в воде, и приняв константу диссоциации ионных пар NHJOCgHs
в аммиаке равной 10, можно рассчитать суммарную константу
ионизации и диссоциации этилового спирта в аммиаке. Она равна
A-j/Aa = 10"9. Отсюда, зная величины кг и kjk3, можно рассчитать
константу скорости реакции взаимодействия электрона с ионом ам-
аммония А*з ^ 105 л/(моль-сек). Следует отметить, что эта величина
лежит в указанных выше пределах.
44
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
ЛИТЕРАТУРА.
45
5. Сравнение с растворами металлов
в других растворителях
Растворы щелочных металлов в алифатических аминах широко
хшпользуются в органической химии в качестве восстановителей16iie.
Амины как растворители имеют ряд преимуществ перед аммиаком:
более высокие точки кипения, большие растворимости в них орга-
органических соединений, большую восстановительную способность рас-
растворов металлов в них. Однако растворы металлов в аминах зна-
значительно менее устойчивы и быстро разлагаются на амид и водород.
Даун и др.40 показали, что калий и NaK слабо растворяются
'¦(— 10~4 м) в некоторых простых эфирах, образуя нестойкие рас-
растворы синего цвета. Наибольшая растворимость наблюдается в ди-
метиловом эфире этиленгликоля, 1-метоксиметилтетрагидрофуране
и циклическом тетрамере окиси пропилена. Растворимостью калия
в таких эфирах объясняется успешное использование этих раствори-
растворителей при различных реакциях калия, например для реакции с кар-
•бонилами переходных металлов, в результате которой образуются
карбонилаты калия:
2К + Мп2(СОI0 > 2Мп(СО)б+2К+ F8)
Ароматические углеводороды, например нафталин, реагируют
с растворами калия и натрия в диметиловом эфире этиленгликоля
с образованием зеленых растворов соединений. Реакция протекает
по механизму присоединения электронов140:
K-f-C10Hs —> K++C10Hg F9)
Эти сравнительно устойчивые растворы получили широкое распро-
распространение в качестве сильных восстановителей в тех случаях, когда
требуется избежать сольволиза протонными растворителями. Так,
Чатт и Уотсон21 получили нейтральные комплексы переходных
металлов типа М [(СНд^РСНаСНаР^Нз^Ь восстановлением комп-
комплексов более высокой валентности раствором нафталенида натрия
в тетрагидрофуране.
ЛИТЕРАТУРА
1. Acrivos J. V., Pitzer К. S., J. Phys. Chem., 66 1693 A962).
2. Armstrong G. Т., USA National Bureau of Standards, Report 2626,
A Critical Review of the Literature Relating to the Vapour Pressure of Ammo-
Ammonia and Trideuteroammonia, Washington, 1953.
3. Arnold E., Patterson A., in «Solutions Metal-Ammoniac», New
York, 1964.
4. Audrieth L. F., Kleinberg J., Non-Aqueous Solvents, New
York, 1953.
5. В а г - E 1 i К., К 1 in F. S., J. Chem. Soc, 1962, 1378.
6. В е с k e г E., L i n d q u i s t R. H. et al., J. Chem. Phys., 25, 971 A956).
7. Beck man T. A., P i t z e r K. S., J. Phys. Chem., 65, 1527 A961).
8. Bergstrom F. W., J. Am. Chem. Soc, 62, 2381 A940).
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
В e r g s t г о m F. W., F e r n e 1 i u s W. C, Chem. Rev., 12, 43 A933).
В e r g s t г о m F. W., F e г n e 1 i u s W. C, Chem. Rev., 20, 413 A937).
Berthoud A., Helv. chim. acta, 1, 84 A918).
А., С. А., 12, 1849 A918).
, MacDinald D. К. С, Nature, 159, 811 A947).
, M а с D о n a 1 d D. K. C, Trans. Faraday Soc, 44, 735
Berthoud
Birch A. J
Birch A. J
A948).
Birch A. J
Birch A. J
Bourke P.
В о ur k e P.
Brown J.,
Brown J.,
С h a t t J., W a t s о n H. R., J
Y. P., Pri bble M.
С h ou D.
A963).
Clark H.
Coulter
A951).
Coulter
Coulter
A959).
Couture
Quart. Rev. (London), 4, 69 A950).
., Smith H., Quart. Rev (London), 12, 17 A958).
J., Lee J. C, Trans. Inst. Chem. Eng., 39, 280 A961).
J., Lee J. С, С. А., 55, 26615i A961).
Roberts N. W., англ. пат., 867848 A951).
Roberts N. W., С А., 56, Pi 48c A962).
Chem. Soc, 1962, 2545.
J. et al., J. Am. Chem. Soc,
O.
L.
L.
L.
Horslield A.
V., Monchick
et al., J. Chem. Soc, 1959,
L., J. Am. Chem. Soc,
85, 3530
2478.
73, 5867
V., J. Phys. Chem., 57, 553 A953).
V., S i ncl ai r J. R. et al., J. Am. Chem. Soc, 81, .2986
A. M., Laidler K.
J., Can. J. Chem., 34, 1209 A956).
С г a g о е С, Н a r p e r D., Bur. Stds. Sc. Pp., 420, 313 A921).
Cram D. J., Chem. Eng. News, 93, 92 A963).
Cseko G., С rnides I., J. Inorg. a. Nucl. Chem., 14, 139 A960).
Cuthrell R. E., Fohn E. C. et al., Paper 30 presented before the
Division of Inorganic Chemistry, American -Chemical Society Meeting, New
York, 1963, p. 13N—14N.
Cutler D., Powles J. G., Proc Phys. Soc. (London), 80, 130
A962).
Czapski G., Schwarz H. A., J. Phys. Chem., 66, 471 A962).
De Grotthuss С J. Т., Ann. chim., 58, 54 A806).
D e w a 1 d I. F., Lspoutre G., J. Am. Chem. Soc, 76, 3369 A954).
Dirian G., Grandcollot P., Comm. energie at. (France), Rapp.
№ 1981, 1961.
Dirian G., Grande allot P., С A., 55, 26623d A961).
Dorf man L. M., Science, 141, 493 A963).
Dorfman L. M., Taub I. A., J. Am. Chem. Soc, 85, 2370 AЭ63).
Down J. L., L e w i s J. et al., J. Chem. Soc, 1959, 3767.
Dye J. L., S a n k u e r R. F. et al., J. Am. Chem. Soc, 82, 4797 A960).
E 1 s e у H. M., J. Am. Chem. Soc, 42, 2454 A920).
Fellinger
A938).
Fens e M.
A955).
F e n s k e M.
Fernelius
F о w 1 e s G.
A962).
Franklin
Franklin
Franklin
Franklin
L. L., Audrieth
R., McCormick
R., McCormick
W. C,
W. A.,
Bowman
Nicholls
F., J. Am'. Chem. Soc, 60, 579
. H. et al., A. I. Ch. E., J., 1 335
R. H. et al., С. А., 50, 2154b A956).
G. В., Chem. Rev., 26, 3 A940).
D., Quart. Rev. (London), 16, 19
E. C, J. Am. Chem. Soc, 56, 568 A934).
E. C, The Nitrogen System of Compounds, New York, 1935.
E. C, Kraus С A., Am. Chem. J., 23, 227 A900).
E. C, Cady H. P., J. Am. Chem. Soc, 26, 499 A904).
Freed S., S u g a r m a n N., J. Chem. Phys., 11, 354 A943).
Frost A. A., Pearson R. G., Kinetics and Mechanism, 2nd ed.,
New York, 1961.
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
55,
56.
57,
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72
73!
74.
75.
76.
77.
78.
79.
80.
81.
82,
83.
84,
85,
86,
87,
89.
90.
91.
92.
93.
94.
95.
90.
97.
98.
99.
100
101
F u о s s R. M., А с с a s с i n a F., Electrolytic Conductance, New York,
1959.
G ol M., J oily
Gold M., Jolly
W. L., Tnorg. Chem., 1, 818 A962).
W. L. et al., J. Am. Chem. Soc, 84, 2264 A962).
J. Phys. Chem., 64, 1066 A960).
J. Chem. Phys., 36, 363 A962).
J. Chem. Phys., 36, 368 A962).
L., Inorg. Chem., 2, 1076 A963).
Grubb H. M., Chit turn J. F. et al., J. Am. Chem. Soc, 85, 776
A963).
G u n n S. R., G r e e n L. R.,
G u n n S. R., Green L. R.,
G u n n S. R., Green L. R.,
H all ad a C. J., J о 1 1 у W.
Harned H. S., О w e n В. В., The Physical Chemistry of Electrolytic
Solutions, 3rd ed., New York, 1958.
Has ing J., Ann. Phys., 37, 509 A940).
Haul R., I h 1 о Н. et al., Chem. Ing. T,echn., 33, 713 A961).
H awes W. W., J. Am. Chem. Soc, 55, 4422 A933).
He pier L. G., J. Phys. Chem., 61, 1426 A957).
H e у n A. H. А., В e r g i n M J., J. Am. Chem., Soc, 75, 5120 A953).
Heyns К., В г о с к m a n n R. et al., Ann., 614, 97 A958).
J. H., J. Chem. Educ, 25, 74 A948).
R. L., Regular Solutions, New York,
J. H., Scott
Hildebrand
Hildebrand
1962.
H n i z d a V. F., Kraus С A., J. Am. Chem.
H о d g i n s J. W., Can. J. Res., 27, 861 A949).
Hunt J. P., Dodjen '"
Huster E., Ann. Phys.,
Hutchison С A., Jr.,
A961).
Hutchison C. A., Jr.,
A961).
Hutchison С A., Jr.,
A953).
I s h i d a K., Kogyo Kagaku Zasshi, 60,-864 A957).
I s h i d a K., Bull. Chem. Soc. Japan, 31, 143 A958).
I s h i d а К., С. А., 53, 9801h A959).
K., Bull. Chem. Soc Japan, 33, 693 A960).
К., С. А., 54, 236S7f A960).
Soc, 71, 1565 A949).
H. W. et al., Inorg. Chem., 2, 478 A963).
33, 477 A938).
O'Reilly D. E., J. Chem. Phys., 34, 163
O'R e i 1 1 у D. E., J. Chem. Phys., 34, 1279
Pastor R. C, J. Chem. Phys., 21, 1959
I s h i d a
I s h i d a
Jaffe H., Z. Phys., 93, 741 A953).'
J a n d e r G., Die Chemie in Wasserahnlichen Losungsmitteln, Berlin, 1949.
J о h n s о n
J о h n s о n
Johnson
Johnson
A950).
Johnson
Jolly W.
Jolly
Jolly
Jolly
J oily
New York,
J о r t n e r
Kaplan
W. C, M a r t e n s R. I., J. Am. Chem. Soc, 58, 15 A936).
W. C, Meyer A. W., J. Phys. Chem., 33, 1922 A929).
W. C, Meyer A. W., J. Am. Chem. Soc, 54, 3621 A932).
W.C., Meyer A. W. et al., J. Am. Chem., Soc, 72, 1842
W,
W.
W.
W,
W. C, Piskur M. M.. J. Phys. Chem., 37, 93 A933).
L., J. Phys. Chem-., 58, 250 A954).
L., J. Chem. Educ, 33, 512 A956).
L., Progr. Inorg. Chem., 1, 235 A959).
L., Synthetic Inorganic Chemistry, New York, 1960.
L., Hall ad a C. J. et al., Solutions Metal-Ammoniac,
1964.
J., J. Chem. Phys., 30, 839 A959).
J., Kittel C, J. Chem. Phys., 21, 1429 A953).
Kay R. L., J. Am. Chem. Soc, 82, 2099 A960).
Kelly E. J., S e с о r H. Y. et al., J. Am. Chem. Soc, 84, 3611 A962).
К i к u'c hi S., Kudo S., J. Soc. chem. Ind., Japan, 47, 302 A944).
Riku с h i S.. Kudo S., С. А., 42, 6208i A948).
Knight W. D., Phys. Rev., 76, 1259 A949).
ЛИТЕРАТУРА
47
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
117.
118.
119.
120.
121.
122
123!
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
С, J.
W., J.
W., J.
W., J.
C, J. Am. Chem. Soc.
30, 83 A953).
C. J., J. Am. Chem.
Am. Chem. Soc, 47, 725 A925).
Am. Chem. Soc, 43, 2529 A921).
Am. Chem. Soc, 44, 1941 A922).
Am. Chem. Soc, 45, 2551 A923).
56, 2297 A934)
Nyman С J., J. Am. Chem. Soc, 70. 3002 A948).
Am.
Shoemaker С. Е., J.
Shoemaker С. Е., J.
Jr., С. г.,
Solutions
Chem. Soc,
Am. Chem. Soc,
'48).
72,
Kraus С A., J. Am. Chem. Soc.^ 36, 864 A914).
Kraus С A., Johnson W.
Kraus С A., Luc ass e W.
Kraus С A., L u с a s s e W.
Kraus С A., Lucasse W.
Kraus С A., S с h m i d t F.
Kraus С A., J. Chem. Educ,
Laitinen H. A., Nyman C. J., J. Am. Chem. Soc, 70. 2241
A948).
Laitinen H. A.,
Laitinen H. A.,
663 A950).
Laitinen H. A.,
4975 A950).
Latimet W. M., Oxidation Potentials, 2nd ed., New York. 1952. p. 34.
Латимер В. М., Окислительные состояния элементов п их потен-
потенциалы в водных растворах, Издатинлит, 1954.
Lefrancois В., V a n i s с о t t е С, Chal. et Ind., 41, 183 A960).
Lepoutre G., Patterson A.,"
Lepoutre G., Sienko M. J.,
York, 1964.
L e v i n e R.,
Levy R. A.,
72,
240, 1644 A955).
Metal-Ammoniac,
New
F e r n e 1 i u s W. C, Chem. Rev., 54, 449 A954;.
Phys. Rev., 102, 31 A956).
Loewenstein A., Szoke A., J. Am. Chem. Soc, 84, 1151 A962).
Maclnnes D. A.. The Principles of Electrochemistry, New York, 1939.
M с С о n n e 1 1 H. M., Holm С. Н., J. Chem. Phys., 26, 1517 A957).
M с E 1 г о у A. D., Laitinen H. A., J. Phys. Chem., 57, 564 A953).
Mulder H. D., Schmidt F. C, J. Am. Chem. Soc, 73, 5575 A951).
Nyman С J., J. Am. Chem. Soc, 71, 3914 A949).
Nyman С J.,Si Chang Fung, Dodger H. W.,J. Am. Chem.
Soc, 72, 1033 A950).
Ogg R. A., Jr., Disc. Farad. Soc, № 17, 215 A954).
Ogg R. A., Jr., Leighton P. A., Bergstrom F. \V.. J. Am.
Chem. Soc, 55, 1754 A933).
R., Giauque W. F., J. Am. Chem. Soc.
59, 254
3rd ed., New York,
Overstreet
A937).
Pauling L., The Nature of the Chemical Bond,
1960, p. 621.
Plank С J., H u n t H., J. Am. Chem. Soc, 61, 3590 A939).
Pollak V. L., J. Chem. Phys., 34, 864 A961).
Re bora P. L., Energia nucl., 9, 338 A962).
Re bora P. L., С A., 57, 16078 A962).
Rossini F. D., Wag man D. D. et al., USA National Bureau of
Standards, Circular 500, Selected Values of Chemical Thermodynamic Proper-
Properties, Washington, 1952.
Ruff O., Giesel E., Ber., 39, 828 A906).
Sanderson R. Т., Vacuum Manipulation of Volatile Compounds,
New York, 1948.
S с h a a p W. В., С о n 1 e у R. F. et al., Analyt. Chem., 33, 498 A961).
Schmidt F. C, S о t t у s i a k J. et al., J. Am. Chem. Soc. 63, 2669
A941).
Schmidt
A938).
Scott
A936).
Sisler H.
Stairs R.
F. C, S t u d e r F. J. et al., J. Am. Chem. Soc, 60, 2780
N. D., Walker J. F. et al., J. Am. Chem. Soc, 58, 2442
H., Chemistry in Non-Aqueous Solvents, New York, 1961.
A., Sienko M. J., J. Am. Chem. Soc, 78, 920 A956).
48
ГЛАВА ПЕРВАЯ. ЖИДКИЙ АММИАК
ГЛАВА ВТОРАЯ
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
170.
171.
172.
Swenson G. W., Zwicker E. F. et al., Science, 141, 1042 A963).
Symons M. С R., Quart. Rev. (London), 13, 99 A959).
Vegard L., Hillesund S., Avhandl. Norske Videnskaps-Akad.,
Oslo I. Mat.-Naturv. Klasse, № 8, 1942; Wyckoff R. W. G., Crystal
Structures, v. 1, New York, 1948, 1951.
Warshawsky I., J. Inorg. a. Nucl. Chem., 25, 601 A963).
Watt G. W., Chem. Rev., 46, 289, 317 A950).
J. Chem. Educ, 34, 533 A957).
Sowards D. M., J. Electroch. Soc, 102, 46 A955).
So wards D. M. et al., J. Am. Chem. Soc, 77, 5835
G.
G.
G.
W.,
W.,
w.,
Watt
Watt
Watt
A955).
Weyl W., Ann. Phys., 121, 601 A864).
W i e b e R., T r e m e а г п е Т. Н., J. Am. Chem. Soc, 55, 975 A933).
W i e s e n d a n g e r H. U. D., Jones W. H. et al., J. Chem. hys.,
27,668 A957).
W. K., D ay t о n J. C, J. Am. Chem. Soc, 75, 4553
Wilmarth
A953).
Wolsky S.
W olsky S.
6196 A952).
Yost D. M., Russell H., Jr., Systematic
New York, 1944.
Z i n 11 E., G о u b e a u J. et al., Z. phys. Chem., A154, 1 A931).
Z i n 11 E., N e u m а у г S., Ber., 63B, 237 A930).
Дыхно Н. М., Ш ат е н ште йн А. И., ЖФХ, 22, 461
Дыхно Н. М., Ш ат е нште йн А. И., ЖФХ, 25, 670
Гурьянова Е. Н., Плесков В. А., ЖФХ, 8, 345
Ипатьев В. В., Теодорович В. П., ЖОХ 2, 305
Моносзон А. М., Плесков В. A., Z. phys., Chem.
156, 176 A931).
Пиневич Г., Холодильная техника, 20,
Пи не вич Г., С. А., 43, 8813е A948).
Плесков В. А., Моносзон А. М.,
1, 713 A935).
Плесков В. А., Моносзон А. М., Acta Phys.-chim.
615 A935).
Шатенштейн А. И., Acta Phys.-chim.
Шатенштейн А. И., Изотопный обмен и замещение водорода
в органических соединениях. Изд. АН СССР, 1960.
Шатенштейн А. И., Израилевич Е. А., Ладыжни-
к о в а Н. И., ЖФХ, 23, 497 A949).
Шатенштейн А. И., Израилевич Е. А., Ладыжни-
кова Н. И., С. А., 43, 6024с A949).
P., Thesis, Boston University, Massachusetts, 1952.
P., Zdanuk E. J., et al., J. Am. Chem. Soc, 74,
Inorganic Chemistry,
A948).
A951).
A936).
A932).
Abt. A.,
Acta
№ 3, 30 A948).
Phys.-chim. URSS,
URSS, 2,
URSS, iO, 121 A939).
ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
X. X. ХАЙ МЕН, ДЖ. ДЖ. КАЦ
I. Введение 49
А. Развитие исследований в области химии жидкого фтористого
водорода 49
Б. Аппаратура для работы с жидким фтористым водородом .... 50
II. Свойства чистого жидкого фтористого водорода 53
A. Физические свойства 53
Б. Колебательные спектры поглощения 55
B. Структура 58
1. Самоионизация 58
2. Молекулярная ассоциация 59
Г. Кислотные свойства 61
III. Поведение различных веществ при растворении в жидком фтористом
водороде 64
A. Фториды металлов 65
Б. Ионизирующиеся фториды неметаллов (некислые) 66
B. Акцепторы протона 67
Г. Кислоты в роли акцепторов фтор-ионов 70
Д. Соли 73
Е. Вещества, растворяющиеся без ионизации 74
IV. Поведение некоторых биологически важных веществ в жидком
фтористом водороде 75
Литература 78
I. ВВЕДЕНИЕ
А. Развитие исследований в области
химии жидкого фтористого водорода
Жидкий фтористый водород является типичным представителем
класса неводных ионизирующих растворителей. Ионы, образующиеся
при самоионизации этого соединения, являются самыми малыми
по размерам и наиболее подвижными среди всех других положитель-
положительных и отрицательных ионов. До недавнего времени изучение этого
растворителя задерживалось вследствие его разрушительного дей-
действия на стекло и кварц, применявшихся в качестве конструкцион-
конструкционных материалов, и его губительного действия на человеческий орга-
организм. Однако потребность в гексафториде урана, необходимом для
4 Заказ 90
50
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
ВВЕДЕНИЕ
51
разделения изотопов урана, вызвала интерес к фтористому водороду
и к химии фтора вообще. Было создано много типов лабораторных
аппаратов, с помощью которых стало возможным дальнейшее изу-
изучение свойств фтористого водорода. Наиболее существенным дости-
достижением в деле создания аппаратуры для работы с фтористым водо-
водородом явилось производство фторсодержащих пластмасс, стойких
по отношению к фтористому водороду. Из политетрафторэтилена
и полихлортрифторэтилена изготовляются пластины, стержни
и трубы, которые легко обрабатываются, причем изделия из поли-
полихлортрифторэтилена прозрачны.
В настоящее время фтористый водород широко применяется при по-
получении элементарного фтора, при синтезе неорганических и орга-
органических соединений фтора, а также как растворитель. В этой главе
основное внимание уделяется свойствам фтористого водорода как
растворителя. Полные обзоры работ по другим направлениям хи-
химии фтористого водорода сделаны Саймонсом54' 74> 75.
Б. Аппаратура для работы с жидким фтористым водородом
Современная техника дает возможность проводить все физико-
химические исследования растворов веществ в жидком фтористом
водороде, которые раньше осуществлялись лишь в водных растворах.
Эти исследования включают изучение электропроводности, давления
паров, спектрофотометрию в ультрафиолетовой, видимой и инфра-
инфракрасной областях, спектры комбинационного рассеяния, протонный
магнитный резонанс, поляриметрию и рефрактометрию.
Измерение электропроводности удобно проводить в аппаратах
(рис. 7) из полихлортрифторэтилена (Кел-Ф) с герметичными ячей-
ячейками (рис. 8) 62> 63. Очень чистый фтористый водород может быть
приготовлен дистилляцией технического продукта также в аппара-
аппаратуре из полихлортрифторэтилена69. Полученный таким способом,
он обладает более низкой электропроводностью по сравнению с фто-
фтористым водородом, содержащим примеси, и пригоден в качестве ра-
растворителя при измерении электропроводности растворов.
Исследование спектров поглощения растворов в HF в видимой
и ультрафиолетовой областях стало возможным благодаря примене-
применению ячеек, снабженных окнами из синтетического сапфира (А12О3),
которые очень хорошо пропускают свет и в то же время не подвер-
подвержены разрушающему действию фтористого водорода (рис. 9). Для изу-
изучения инфракрасных спектров успешно применяются ячейки с ок-
окнами из хлорида серебра; окна при необходимости можно сделать
сменными (рис. 10)в2. При изучении спектров комбинационного рас-
рассеяния можно применять в качестве ячеек сапфировые трубки; в по-
последнее время часто применяются трубки из полихлортрифторэти-
полихлортрифторэтилена. Трубки меньших размеров, выполненные из полихлорфтор-
этилена, удобно использовать при изучении ядерного магнитного
резонанса.
Простота механической обработки труб и заготовок из полихлор-
полихлортрифторэтилена облегчила создание удобных конструкций вакуумных
систем, предназначенных для работы с жидким фтористым водородом.
Из этого прозрачного полимера методом экструзии можно изгото-
изготовлять краны, тройники, колена, переходники и другие аналогичные
Разборная ячейка
для сильно электро-
электропроводных растворов
Разборная ячейка
дли слабо электро-
электропроводных растворов
Ячейка
Смеситель-
Рис. 7. Прибор для измерения электропроводности жидкого фтористого водо-
водорода:
1 — электроды и проводники из платаны; 2 — платино-родиевый проводник термопары;
з — втулка иэ политетрафторэтилена, центрирующая электроды; 4 — пористый диск из поли-
полихлортрифторэтилена; 5 — коническая пробка из политетрафторэтилена для создания уплот-
уплотнения в местах ввода проводников; 6 — нажимной металлический диск, сдавливающий
конические элементы из политетрафторэтилена для более надежного уплотнения; 7 — под-
поджимная гайка из никеля или никелевого сплава с резьбой 8/4" (съемная нарезная втулка,
на которую навинчивается гайка, может быть выполнена из любого конструкционного ме-
металла); 8 — гибкий трубопровод из полихлортрифторэтилена; 9— изоляционная трубка из
полихлортрифторэтилена; 10— сосуд из полихлортрифторэтилена (может быть изготовлен
путем литья, токарной обработки или экструзии); 11 — узел уплотнения крышки (крышка из
полихлорфторэтилена, болты и фланцы металлические, кольцевое уплотнение из политетра-
политетрафторэтилена); 12 — вентиль, может быть выполнен из полимера или металла.
приспособления. Резьбу можно выполнять непосредственно на пласт-
пластмассе, однако ее обычно предпочитают делать на металле. В связи
с этим многие приспособления содержат полихлортрифторэтилен
в виде прокладок, помещенных между нарезными или зафланцован
ными металлическими деталями, как было показано на рис. 7 и 8.
Металлические кольца, снабженные резьбой, охватывают с обеих
сторон зафланцованные пластмассовые трубы. Полагают, что соеди-
соединения или уплотнения такого типа найдут широкое примене-
применение 1,2,28,30,34_
И. СВОЙСТВА ЖИДКОГО ФТОРИСТОГО ВОДОРОДА
Рис. 8.
я—
Политетрафторэтилен (тефлон) непрозрачен; он мягче, чем по-
лихлортрифторэтилен и способен выдержать более высокую темпера-
температуру. Однако в связи с тем что работы с жидким фтористым водо-
водородом проводят обычно при комнатной и более низкой температуре
лучше использовать легкоплавкие, но легче обрабатываемые пласт-
пластмассы. Для работы с фтористым водородом пригодны и другие фтор-
содержащие полимеры (например, Витон А).
11
1°
Рис. 10. Ячейка с переменной длиной светового пути для изучения спектров
жидкого фтористого водорода:
кольцо из политетрафторэтилена; 11 —
д.-:
II. СВОЙСТВА ЧИСТОГО ЖИДКОГО ФТОРИСТОГО ВОДОРОДА
А. Физические свойства
В этом разделе приведены данные, которые позволяют составить
достаточно ясное представление о свойствах жидкого фтористого
водорода как растворителя.
Термодинамические константы HF26:
«пл=— 83,37° С, или 189,79 ±0 02° К
1^ГЛ3^296
Шм« * КаЛ/?' ИЛИ °'939 ю«/-«о^ь* (при 1Ш)
исп = 89,45 ±0,20 кале, или 1,789 ккал/МОлъ* \>t™)
Snn = 4,942 кал/(град ¦ моль) \-^wn)
исп = 6,117 кал/(град ¦ моль)
Га37^'9 кал^Р"д ¦ *°лъ) (при 292,61° К и 741 мм рт. ст.)
газ (мономер HF) = 41,5 кал/(град ¦ моль) (при 298,16° К)
* Молекулярный вес принят равным 20,006 г.
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
*> °С —50 —25 0 25 50 100 1Г.П
Р, г/см* ... U231 1,0606 1,002 0,9546 29 0,90813 0,796" 0,646"
Плотность в критической точке A88° С) равна13 0,29 ± 0,03 г/см3
Вязкость жидкого HF ":
*> сс —50 —25 о
*Ь спз 0,570 0,350 0,256
*!' сст 0,507 0,330 0,256
Поверхностное натяжение жидкого HF78:
*- Iе -81,8 -23,2 0 182
or, динjсм 17,7 12,0 10,1 8,62
Показатель преломления HF58:
»25° с= 1,15436 + 0,001025X2
п*>° с= 1Д574
Дп^/At = —0,0004
Диэлектрическая проницаемость жидкого HF18:
*• °С -73 -42 -27 0
Е 175 134 111 84
Электропроводность15-69 жидкого HF:
*- °с _ -15 о
у., ом 1-см 1 —1,4-10~5 ~ 10~6
Некоторые из приведенных выше величин были получены по-
видимому, для неочищенного HF, и поэтому должны быть проверены
На основании данных о теплотах образования фторидов, напри-
например SiF4, определенных калориметрически, и о теплоте гидролиза
фторидов, протекающего по реакции SiF4+ 2H,0 ->- SiO2 + 4HF
Федер и др.11 вычислили величину образования" газообразного мо-
мономера HF, ДЯобр = —64,9 ккал/моль при 25° С.
Давление паров жидкого фтористого водорода было измерено
Джерри и Дэвисом 32. Их данные описываются любым из двух урав-
уравнений:
B)
lg P=- 1.91173 —
+ 3,21542% Т
II. СВОЙСТВА ЖИДКОГО ФТОРИСТОГО ВОДОРОДА
55
Ниже приведены давления паров HF при различных температу-
температурах, вычисленные по этим двум уравнениям.
t, °С —80 —50 —25 0 10 20
Р, .iut pm. ст., рассчитанное
по уравнению A) 32 5,55 34,82 123,7 363,8 536,2 773,2
по уравнению B) is 484 33,52 122,5 363,8 536,7 774,1
t, °C 25 50 100 150 188*
Р, мм рт. ст., рассчитанное
по уравнению A) 32 921,4 2069,0 7891,0 — —
по уравнению B) is 922,5 2069,0 7894,0 23 460 48 690
* Критическая температура 188±3 °С; критическое давление 48 700±2 500 ммрт. ст.
Теплота испарения, найденная Джерри и Дэвисом по кривой
зависимости давления паров от температуры, при tKiin равна
1,608 ккал/моль, что не согласуется, однако, с данными других
исследователей 26>54. Различие между обоими значениями Д/7ИСП
не было удовлетворительно объяснено.
Физические свойства фтористого водорода свидетельствуют о том,
что это не совсем обычный растворитель. Высокая точка кипения,
широкий температурный интервал жидкого состояния и высокая
диэлектрическая проницаемость позволяют предположить, что фто-
фтористый водород, как и вода, представляет собой ассоциированную
жидкость; в системе с фтористым водородом должна заметно про-
проявляться способность к образованию водородной связи и передаче
протона, раствор должен быть ионизирован. Значения поверхност-
поверхностного натяжения и вязкости жидкого фтористого водорода значительно
ниже соответствующих величин для воды, что указывает на отсутствие
в структуре жидкого HF трехмерного каркаса, подобного наблю-
наблюдаемому у воды и безводной серной кислоты. Структура жидкого
фтористого водорода заметно отличается от структуры других раство-
растворителей с высокой диэлектрической проницаемостью.
Б. КОЛЕБАТЕЛЬНЫЕ СПЕКТРЫ ПОГЛОЩЕНИЯ
Изучение спектров поглощения фтористого водорода необходимо
при исследовании молекулярных свойств его в различных агрегат-
агрегатных состояниях.
Для получения колебательного и вращательного спектров газо-
газообразного мономера HF измерения необходимо проводить при по-
пониженном давлении и умеренной скорости подъема температуры
(это обеспечивает полный распад молекул полимера). В качестве
материалов для окон измерительных ячеек пригодны фторид каль-
кальция, хлорид серебра и полиэтилен. Характеристическая частота
валентных колебаний мономера HF расположена при 3 мк; враща-
вращательные линии находятся в длинноволновой области инфракрасного
спектра. Весьма точные спектроскопические измерения были
56
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
И. СВОЙСТВА ЖИДКОГО ФТОРИСТОГО ВОДОРОДА
57
выполнены Купером42'43 и Ротшилдом68. Ниже приведены наиболее
важные спектроскопические параметры мономерного газа HF:
Во = 20,56 ye(s>e = 0,533
А»=О,00211 Ве = 20,95
со,, = 4137,25 De = 0,00213
= 88,73 ае = 0,789
Ye =f 0,0087
Примечание. Принятые в спектроскопии обозначения см. Герцберг Г.,
Колебательные и вращательные спектры многоатомных молекул, Издатинлит, 1950.
vo(O<
@.
(О.
(о.
•1) =3 961,64 см-1
-2) = 7 751,26 см-1
¦3) = 11 372,7 см-1
•4) = 14 831,6 см-1
700 е-
10 -
10000 ШО
5000*
Рис. 11. ИК-спектр жидкого фтористого водорода53 (тол-
(толщина слоя жидкости в ячейке 6 мк; звездочки на рисунке
обозначают, что масштаб изменен).
При пониженных температурах и повышенных давлениях про-
происходит ассоциация молекул фтористого водорода.
ИК-спектры поглощения80
помогают выявить природу
полимерных образований в
газообразном HF. Для спект-
спектра полимера HF, так же
как и для спектра мономер-
мономерного газа, характерно нали-
наличие полосы поглощения
в области 3 мк, которая
обусловлена валентными ко-
колебаниями протонов в ассо-
циатах.
Жидкий фтористый водо-
водород дает ряд ИК-полос погло-
поглощения (рис. И и 12), обус-
обусловленных, по-видимому, вы-
высокочастотными колебаниями
в ассоциированных молеку-
молекулах жидкости 28>63. Наличие
О.О1 -
0,00! г
0,000!
1,5 2,0 2,5 3,0 3,5
Л,,МК
Рис. 12. Часть ИК-спектра жидкого фто- широкой и интенсивной поло-
fi
Рис. 13. Ячейка для изучения ИК-спект^ов поглощения рас-
растворов в жидком фтористом водороде:
1 — прокладка; 2 — эластичная диафрагма, позволяющая менять длину
светового пути; 3 — окно из хлорида серебра; 4 — упругая шайба; 5 —
нарезная упругая шайба; 6 — гайка для регулирования толщины слоя.
Таблица 11. Частоты (см~1) в спектрах поглощения
чистых кристаллических HF и DF
р
ристого водорода as.
сы в области 3 мк затрудняет
vDF по '2
2527
2284
855
715
570
405
по «
3581 (пшр.)
3414 (узк.)
3270 (шир.)
3060 (узк.)
1200
971
790
555
515
370
VHF
по 1в
3590 (шир.)
3420 (узк.)
3270 (шир.)
3060 (узк.)
1204
(955-1010)
547
366
58
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
изучение ИК-спектров разбавленных растворов в HF. Изучение рас-
растворов высокой концентрации более эффективно 2.
Считают, что при снятии ИК-спектров жидкого фтористого водо-
водорода лучшим материалом для окон ячеек является хлорид серебра.
Так же как и жидкая вода, фтористый водород интенсивно поглощает
лучи в инфракрасной области спектра, поэтому необходимо, чтобы
путь светового луча был возможно короче. На рис. 13 показана
ячейка, применяемая при исследовании ИК-спектров; она характе-
характеризуется очень коротким световым путем, длину которого можно
изменять.
Были изучены спектры поглощения твердого фтористого водо-
водорода (табл. 11). Интерпретация отдельных линий спектра осно-
основана на некоторых допущениях относительно структуры твердого
вещества, предполагающих ассоциацию молекул.
В. СТРУКТУРА
Физические свойства жидкого фтористого водорода указывают
на то, что его структура сложна. Очевидно, здесь играют роль по край-
крайней мере два типа молекулярного взаимодействия, а именно само-
самоионизация и межмолекулярная ассоциация.
1. Самоионизация
У фтористого водорода, как и у большинства протонных ионизи-
ионизирующих растворителей, ионные частицы, образующиеся в резуль-
результате самоионизации, вероятно, являются частью молекулярной
сетки, построенной за счет водородной связи; индивидуальных мо-
молекул в жидкости нет.
Существование несольватированного протона нереально.
Процесс самоионизации формально можно представить уравне-
уравнением
2HF ^
C)
Важно отметить, что протон присоединяется не к мономеру, а к мо-
молекулам ассоциатов. Ион фтора также сильно сольватирован.
Степень самоионизации в жидком фтористом водороде пока точно
не установлена. Наиболее распространенным критерием для ее
установления является электропроводность, однако истинная ми-
минимальная величина этого параметра еще не определена. Примесь
воды (либо окислов металлов, которые реагируя с HF, образуют
воду) сильно увеличивает электропроводность фтористого водорода.
При очистке HF необходимо удалить не только воду и окислы ме-
металлов, но и примеси никеля, так как поверхность никеля покрыта
окисной пленкой. Фреденхаген и Каденбах15, применив платиновую
аппаратуру, смогли приготовить сухой фтористый водород, электро-
II. СВОЙСТВА ЖИДКОГО ФТОРИСТОГО ВОДОРОДА
59
проводность которого 1,4-10 5 ом г-см~г при 0° С. В настоящее
время известно, что это значение соответствует содержанию воды
менее 0,0002%. Совсем недавно дистилляцией в полихлортрифтор-
этиленовой колонне был получен фтористый водород с электропро-
электропроводностью до 10"в ом~г-см~г. Однако фтористый водород такой сте-
степени чистоты изучен недостаточно.
Если принять, что эквивалентная электропроводность чистого
HF при 0° С равна 700 см2/(ом-г-экв), а удельная электропровод-
электропроводность равна 10~в ом'1 -см'1, то эти значения соответствуют концентра-
концентрации ионов Н+ или ?~ 1,4-10~6 г-ион/л, или константе самоиониза-
самоионизации Khf = 2-103. Действительная величина константы самоио-
самоионизации будет, вероятно, ниже, и в отсутствие примесей может быть
близкой к значению соответствующей константы воды.
И фтор-ион, и протон в жидком фтористом водороде показывают
высокую ионную электропроводность, которая, несомненно, имеет
цепной характер аналогично электропроводности в воде и безводной
серной кислоте. Хотя низкая вязкость HF затушевывает разницу
между явлением цепной электропроводности и обычным перемеще-
перемещением ионов, а для точного измерения электропроводности разбавлен-
разбавленных растворов сильных кислот необходимо использовать растворы
в достаточной мере свободные от примесей, что сопряжено с экспе-
экспериментальными трудностями, цепной характер электропроводности
жидкого фтористого водорода сейчас установлен 27>зв.
2. Молекулярная ассоциация
Непосредственное определение фактора ассоциации или формы
молекулярных ассоциатов в жидкой фазе невозможно. Но экспери-
эксперименты показывают, что при переходе из жидкого состояния в газо-
газообразное среднее число молекулярных образований изменяется не-
незначительно, так как фтористый водород обладает по сравнению с дру-
другими ассоциированными жидкостями низкой теплотой испарения
и достаточно высокой теплотой диссоциации молекулярных ассо-
ассоциатов. В связи с этим ассоциацию HF изучали в газовой фазе.
Измерения плотности насыщенного пара, находящегося в равнове-
равновесии с жидкостью, позволяют считать, что среднее число простых мо-
молекул фтористого водорода в молекулярных образованиях соста-
составляет приблизительно 3,5. Молекулярные образования в жидкости
имеют, по-видимому, такие же размеры, как и газовые ассоциаты.
Зависимость фактора ассоциации от температуры и давления хорошо
изучена (табл. 12).
Исследования инфракрасных спектров поглощения, выполненные
Смитом80, позволили выяснить причину высокого молекулярного
веса частиц газа и подтвердили предположение Саймонса и Гиль-
дебранда78 о том, что при повышенных давлениях в молекулярной
структуре газообразного HF преобладают ассоциаты состава (HF)«.
60
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
0®
Измерения диэлектрической поляризации упрочили гипотезу о коль-
кольцевой структуре молекулы гексамера (рис. 14). При пониженных
давлениях 78 в структуре HF преобладают тетрамер, димер и моно-
мономер78. Однако, по данным Сми-
Смита, концентрация циклического
гексамера всегда выше, чем те-
трамера, даже при очень низких
давлениях, когда содержание
ассоциатов в газовой фазе ста-
становится ничтожно малым. Этот
автор считает маловероятным
существование линейной зигза-
зигзагообразной цепи полимера (Н F),(
(где п колеблется вокруг неко-
некоторого среднего значения, ко-
которое возрастает с увеличением
давленияL'83. Фрэнк и Мейер 12
признают, что преобладают ас-
социаты кольцевого типа, но
константы равновесия, рассчи-
рассчитанные ими, учитывают суще-
Рис. 14. Некоторые нз возможных
структур ассоциатов фтористого водо-
водорода.
Таблица 12. Зависимость фактора ассоциации
газообразного фтористого водорода
от температуры и давления
t, "С
0,0
19,5
25,0
26,0
38,0
50,0
100,0
160,0
200,0
300,0
Р, мм рт. ст.
363,8
244,5
200,2
760,0
921,4
488,5
401,0
342.5
639,0
501,0
407,0
2 069,0
7 891,0
26 500,0
37 600,0
58 800,0
57 5000
173 000,0 .
z *
4,717
3,118
2,597
3,76
3,553 '
1,708
1,422
1,279
1,354
1,184
1,118
3,015
2,453
1,70
1,32
2,05
1,09
1,43
Литература
32
46
46
32
32
46
46
46
46
46
46
32
32
32
13, 82
13, 82
13, 82
13, 82
* п—показатель ассоциации—число молекул НГ в ассо-
циате; Z—фактор ассоциации—усредненное значение п при дан-
данной температуре и давлении.—Прим. иерее.
II. СВОЙСТВА ЖИДКОГО ФТОРИСТОГО ВОДОРОДА
61
ствование как цепного, так и кольцевого полимера. Свойства
жидкого HF, такие, как плотность, кислотность, спектр про-
протонного магнитного резонанса, зависят от фактора ассоциации
и конфигурации молекулярных образований. Последние, в свою
очередь, сильно зависят от присутствия ионизирующих фтористый
водород примесей. Так, плотность водных растворов фтористого
водорода имеет максимум р = 1,25 г/см3 при 0° С и 75%-ном содер-
содержании HF. Эта величина на 25% больше плотности каждого отдель-
отдельного компонента. Изменение плотности можно объяснить переходом
кольцевых ассоциатов в чистом жидком HF в линейные цепочки,
если жидкий фтористый водород содержит примеси, т. е. в условиях
значительной ионизации. Раствор HF— Н2О максимальной плот-
плотности характеризуется высоким значением электропроводности
(около 1 ом'1 -см'1). Рассчитано, что в таких растворах более 2%
атомов фтора присутствует в виде ионов. Смещение равновесия коль-
кольцевых и цепочечных образований можно использовать также для опи-
описания поведения других акцепторов протонов в фтористом водороде.
Изучение структуры твердого HF с помощью дифракции рент-
рентгеновских лучей и ИК-спектров поглощения показывает, что кри-
кристаллы HF состоят из линейных зигзагообразных цепочек 3> 72
(см. рис. 14). В связи с этим обращает на себя внимание плотность
твердого фтористого водорода44 р = 1,653 г/см3 при —93,8° С. Из-
Изменение объема при плавлении весьма значительно.
Различие форм молекулярных образований в твердом и газообраз-
газообразном Н F не нашло удовлетворительного объяснения.
Г. Кислотные свойства
Жидкий фтористый водород с давних пор считается очень сильной
кислотой. В жидком фтористом водороде хорошо растворяются мно-
многие органические соединения с различными функциональными груп-
группами. Обычно такие растворы проявляют электропроводность ион-
ионного типа, хотя диссоциация растворенного вещества на ионы едва
ли здесь происходит. Данное явление проще всего объяснить перехо-
переходом протона молекулы растворителя к молекуле растворенного
вещества 74:
R + HF ?Z± RH + + F" D)
Однако в разбавленном водном растворе HF является очень сла-
слабой кислотой57. На первый взгляд слабокислый характер разбавлен-
разбавленных водных растворов фтористоводородной кислоты представляется
неожиданным, но тем не менее его следует принимать во внимание
при описании кислотно-основного взаимодействия между молекулами.
Устойчивость молекулы галогеноводорода к ионизации в водном рас-
растворе 58, как и в газовой фазе, возрастает в квадратичной зависи-
зависимости от разности электроотрицательностей галогена и водорода.
Следовательно, кислотность водных растворов галогеноводородов
62
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
уменьшается в ряду HI > HBr > HG1 > HF. В самом деле, кон-
константы ионизации кислот приближенно подсчитываются в зависи-
зависимости от различий в электроотрицательности анионов, что и было
выполнено Полингом. Он указывает, что зависимость устойчивости
молекул от электроотрицательности входящих в них анионов спра-
справедлива также для гидридов элементов шестой группы периодической
системы элементов. Так, вода является более слабой кислотой, чем
гидриды серы, селена и теллура. Различия между кислотностями
Н20 и Н2Те, с одной стороны, и HF и HI, — с другой, вполне сопо-
сопоставимы. Эти положения могут быть приняты без строгих доказа-
доказательств, но удивительно, что степень протекания реакции
H2O + HF ^=t
(¦">)
в разбавленном водном растворе определяется величиной константы
ионизации в7
„ [НзО+] [F ] ,„,
*и = [HF] F)
равной* C -5- 7) 10. Удивление вызывает и то, что константа ре-
реакции
In + HA —> InH+ + A" G)
в безводном HF в 1017 раз больше, чем в чистой Н20.
Мы уже говорили, что в жидком фтористом водороде, так же как
и в жидкой Н20, практически все молекулы связаны в ассоциаты.
Механизм перехода протона от матрицы (т. е. группы ассоцииро-
ассоциированных молекул) HF к отдельной молекуле Н20 коренным образом
отличается от механизма перехода протона из отдельной молекулы HF
к матрице Н20. В первом случае фтор-ион, остающийся после перехода
протона, является частью относительно устойчивой зигзагообразной
цепочки (рис. 15). Во втором случае фтор-ионы сольватируются только
молекулами воды. Следовательно, легкость, с которой матрица теряет
протон, увеличивается по мере роста концентрации фтористого водоро-
водорода в системе. Попытка объяснить это как простое присоединение фтор-
иона к молекуле HF с образованием иона HF2 является грубым
* К сожалению, точное значение константы не определено. Обычно иони-
ионизация трактуется как двухстадийный процесс:
v [H8O+][F-]
HF
HF+F" ^ZT HFJ
[HF?}
[HF] [F-j
Значения Klt приведенные в литературе, колеблются54 в пределах B,4—
—7,2)-10-4, а Кг — в пределах от 5 до 25. Экспериментальные данные не под-
подтверждают постоянства величины К.
II. СВОЙСТВА ЖИДКОГО ФТОРИСТОГО ВОДОРОДА
63
приближением. Вот почему не следует удивляться невозможности
найти удовлетворительную константу равновесия.
По-видимому, среди всех имеющихся показателей кислотности
наиболее подходящим для сильных протонных кислот является Но —
кислотная функция Гаммета 21~23. Он предложил оценивать кислот-
кислотность любых растворов по степени превращения индикатора основа-
основания в его ионную форму:
^ . (8)
где pKjn — показатель константы диссоциации индикатора как катионной
кислоты; с1п и е1пН+ — концентрации индикатора основайия и его иона.
hf;
СЮ
(HFLH+
(HF)aF-
(HFLF"
(hpLp-
Рис. 15. Предполагаемая конфигурация сольватированньгх ионов
Н+ и F" в жидком HF.
При выборе индикатора обращают внимание на то, чтобы инди-
индикатор был основанием, не имел заряда и чтобы нейтральная и ион-
ионная формы его имели заметно различающиеся спектры поглощения.
Данный метод был проверен Полем и Лонгом5в. Ими же были найдены
константы диссоциации индикаторов (Кщ) для ряда веществ. Зна-
Значения кислотной функции Но жидкого HF при наличии некоторых
примесей:
Концентрация при-
примеси, моль/л . .
н«о
NaF
#28, 29
<КГ4 —2,5-10-4 <10-з 0,1 1,0 1,0
—10,98 —10,65 —9,72 —10,2 —9,6 —8,86 —8,4
Шкала Но предназначена для изображения непрерывного ряда
значений кислотности. Для безводной H2SO4 Но = —И, т. е. почти
64
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
такая же, как у жидкого HF. Эти значения показывают, что данные
вещества являются исключительно сильными кислотами.
Кислотность безводного фтористого водорода, а также его раз-
разбавленных растворов должна быть связана с равновесием между
кольцевыми и цепочечными образованиями. Хаймен и сотр.29 пред-
предположили, что фтор-ион и сольватированный протон устойчивы в це-
цепочечных образованиях, а нейтральная молекула HF — в кольце-
кольцевых. Форма кривой зависимости кислотной функции от содержания
примесей связана с перестройкой образования из одного в другое.
Для обоснования этой гипотезы необходимо детально исследовать
структуру жидкого фтористого водорода.
III. ПОВЕДЕНИЕ РАЗЛИЧНЫХ ВЕЩЕСТВ
ПРИ РАСТВОРЕНИИ В ЖИДКОМ
ФТОРИСТОМ ВОДОРОДЕ
Как уже упоминалось, жидкий фтористый водород — ионизиру-
ионизирующий растворитель, обладающий малым молекулярным весом, высо-
высокой летучестью, очень большой величиной диэлектрической проницае-
проницаемости и сильной кислотностью. Он, по существу, не проявляет ни оки-
окислительных, ни восстановительных способностей. Если фтористый
водород может быть восстановлен большинством веществ, которые
восстанавливают воду (при этом выделяется водород), то окислению
он подвергается труднее. До элементарного фтора он не может быть
окислен ни одним химическим окислителем. Следовательно, жидкий
фтористый водород можно использовать для приготовления раство-
растворов очень сильных окислителей.
Наиболее подробно изучены растворы простых фторидов в HF.
Как и большинство электроотрицательных элементов, фтор обра-
образует простые фториды со всеми элементами, за исключением гелия,
неона и аргона. Фториды могут быть классифицированы в зависи-
зависимости от вида ионов, которые они образуют при растворении в жид-
жидком фтористом водороде. Фториды элементов I—IV группы периоди-
периодической системы образуют металлсодержащий катион и HFj:
HF + MF* ZZ? MFjU+HFj
(9)
Фториды неметаллов при растворении в жидком HF либо иони-
ионизируются по уравнению (9), либо присоединяют фтор-ион:
HF + 3F*
A0)
Какая из этих реакций преобладает и в какой степени, зависит
от ряда факторов, среди которых очень важным и часто решающим
является структура фторида и образующихся ионов.
Фториды, которые при растворении в HF вызывают увеличение
концентрации иона HFj в системе, считаются основаниями, хотя
Ш. ПОВЕДЕНИЕ ВЕЩЕСТВ ВО ФТОРИСТОМ ВОДОРОДЕ
65
в обычном представлении их растворы могут быть очень кислыми.
Вещества, которые вызывают увеличение концентрации иона H2F+,
' рассматриваются как кислоты.
А. Фториды металлов
Фториды металлов — простейшие вещества, способные раство-
растворяться в жидком фтористом водороде. В процессе их растворения
образуется новый катион (например, Na+) и увеличивается кон-
концентрация иона HF2, т. е. они являются основаниями и соответ-
соответствуют гидроокисям металлов в водных растворах. Данные о раствори-
мостях некоторых фторидов, взятые большей частью из работы 31,
приведены в табл. 13. Растворимость многих труднорастворимых
фторидов в жидком фтористом водороде увеличивается в присутствии
некоторых комплексообразователей, таких, как уксусная кислота,
лимонная кислота, метилцианид, 7,10-фенантролин, 8-оксихино-
лин, дитизон и окись углерода 8.
Таблица 13. Растворимость фторидов металлов во фтористом водороде
Фторид
LiF
NaF
KF
RbF
CsF
(NH4F)
AgF
Hg2F2
T1F
CuF2
AgF2
CaF2
SrF2
BaF2
BeF2
MgF2
ZnF2
CdF2
HgF2
Раствори-
Растворимость,
г/100 г
10,3
30,1
36,5
110.0
199,0
32,6
83,2
0,87
580,0
0,010
0,048
0,817
14 83
5,60
0.015
0,025
0,024
0,201
0,54
t, °c
12
11
8
20
10
17
19
12
12
12
12
12
12
12
11
12
14
14
12
Фторид
PbF2
FeF2
CrF2
MF2
A1F3
CeF3
TIF3
SbF3
BiF3
M11F3
FeF3
C0F3
ZrF4
CeF4
TI1F4
NbF6
TaF3
SbF3
Раствори-
Растворимость,
г/100 г
2,62
0,006
0,036
0 037
<0,002
0,043
0,081
0,536
0,010
0,164
0,008
0,257
0,009
0,10
< 0,006
6,8
15,2
00
t, "С
12
12
14
12
И
12
12
12
12
12
12
12
12
12
18
25
25
25 •
Разбавленные растворы фторидов щелочных металлов в жидком
фтористом водороде ионизированы почти полностью. Кривые за-
зависимости эквивалентной электропроводности растворов фторидов
натрия и калия в жидком HF от концентрации (с1/*) при различных
температурах представлены74'78 на рис. 16. К сожалению, погреш-
погрешность измерения электропроводности в HF больше, чем в водной
5 Заказ 90
66
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
системе, но, по-видимому, эквивалентные электропроводности рас-
растворов фторидов натрия и калия в жидком фтористом водороде-
при бесконечном разбавлении при 0° С должны составлять около-
400 см2/(ом ¦ г-же). Килпатрик и Льюис зв показали, что в этих раство-
растворах число переноса фтор-иона равно приблизительно 70%. Эта ве-
величина меньше, чем число переноса аниона в аналогичной водной
системе (раствор КОН в воде), что объясняется меньшей вязкостыа
жидкого фтористого водорода и цепным механизмом электропро-
электропроводности фтор-иона.
500 г-
^ A-KF
o-NaF
Ш. ПОВЕДЕНИЕ ВЕЩЕСТВ ВО ФТОРИСТОМ ВОДОРОДЕ
67
0.3
моль
1л'
Рис. 16. Изотермы эквивалентной электропроводности рас-
растворов фторидов натрия и калия во фтористом водороде.
Очевидно, как отмечалось выше, фтор-ион во фтористоводород-
фтористоводородной системе сильно сольватирован: по крайней мере образуется ион
HF2, но, по-видимому, возможно образование и более крупных
ионов H.2F3 и даже H4Fs. Инфракрасные спектры поглощения рас-
растворов веществ в HF показывают наличие водородной связи во фто-
ридном сольвате2. Известно, что многие фториды металлов могут
быть выделены из растворов в жидком фтористом водороде в виде
солей с анионом HF?.
Б. Ионизирующиеся фториды неметаллов (некислые)
Фториды неметаллов обычно представляют собой летучие жид-
жидкости. Наиболее характерными представителями этой группы со-
соединений являются галогенофториды. Трифторид хлора и трифторид
брома полностью смешиваются с фтористым водородом и заметно
ионизируются e4~ee:
CIF3 + HF ^ ClFJ + HF»
BrF3 + HF ^
(И)
A2)
Так как эти галогенофториды во фтористоводородной системе являются
основаниями, сам фтористый водород в такой системе проявляет
кислотные свойства.
Растворы галогенофторидов в жидком фтористом водороде могут
быть нейтрализованы двумя способами:
1) кислым раствором пятифтористой сурьмы
—>- BrF2SbFe+ HF| A3)
2) основным раствором трифторида брома
A4)
В процессе нейтрализации можно измерять электропроводность
системы; образовавшуюся соль удается выделить упариванием рас-
растворителя.
Несмотря на ,то что многие химические реакции в системе гало-
генофторид — фтористый водород можно объяснить через взаимо-
взаимодействие ионов, в действительности для протекания реакций тре-
требуется незначительная ионизация. Для большинства систем макси-
максимальная степень ионизации составляет менее 1 %.
В. Акцепторы протона
Изучение растворов фторидов — оснований не вызывает особого
интереса, так как фтористый водород выступает здесь формально
в роли акцептора фтор-иона, что для него менее характерно, нежели
ионизация с отщеплением протона и присоединением Н+ к молекуле
растворенного вещества. Последнее наблюдается при растворении
в HF и органических, и неорганических веществ, причем среди не-
неорганических веществ особый интерес представляет вода. При до-
добавлении ее к безводному фтористому водороду образуются преиму-
преимущественно ионы Н3О+ и HF2
H2O + 2HF
A5)
что подтверждается резким увеличением электропроводности.
Но, по данным Фреденхагена, вода увеличивает электропроводность
фтористого водорода в меньшей степени, нежели такие же концентра-
концентрации фторидов натрия и калия или таких акцепторов протонов, как
метиловый спирт и уксусная кислота. По-видимому, это можно
объяснить объединением молекул Н2О в ассоциаты (Н2О)Я-Н3О+
или (H2O)n-(HF)n-H3O+, имеющие водородные связи и несущие
один протон:
A6)
ROH + 2HF > (ROH)H++HFJ A7)
Следовательно, по основности 1 моль ROH соответствует п моль Н2О.
68
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
Многочисленные органические соединения, растворимые во фто-
фтористом водороде *, ведут себя как основания. У большинства тех
из них, которые содержат атомы кислорода, азота или серы
и являются координационно-ненасыщенными, есть свободная элек-
электронная пара, способная к связыванию протонов. В эту категорию
входит очень большое количество органических соединений: спирты,
кислоты, альдегиды,, кетоны, эфиры и амины 5V4. Все они во фтори-
фтористом водороде сильно ионизируются.
г Уксусная
III. ПОВЕДЕНИЕ ВЕЩЕСТВ ВО ФТОРИСТОМ ВОДОРОДЕ
69
о го 40 60 во юо
Содержание HF, мол. %
Рис. 17. Зависимость удельной
электропроводности растворов ук-
уксусной и трифторуксусной кислот
в жидком HF от концентрации.
о го ьо во во юо
Содержание HF, мал. %
Рис. 18. Зависимость удельной элек-
электропроводности растворов этанола и
трифторэтанола в жидком HF от кон-
концентрации.
По-видимому, многие органические соединения являются акцепто-
акцепторами протонов и могут быть выделены неизменными после отгонки
фтористого водорода. Некоторые вещества, однако, в результате
растворения в HF претерпевают превращения. Бутадиен и другие
непредельные соединения полимеризуются и подвергаются пере-
перестройке. Часто образуются сильно окрашенные растворы. Только
в отдельных случаях удается подробно изучить поведение растворен-
растворенного вещества, причем не очень сложного. Доказано, что присоеди-
присоединение протона обычно происходит в первой стадии реакции.
В жидком фтористом водороде растворим ряд .ароматических
углеводородов. Представляет некоторый интерес поведение аромати-
ароматических ядер как акцепторов протонов, в связи с чем проведен ряд
* Предельные углеводороды сравнительно мало растворимы в HF.
исследований. Например, вопрос об относительной основности ме-
тилбензолов во фтористоводородном растворе был выяснен методом
измерения электропроводности 37, термохимическими методами50 и ис-
исследованиями ядерного магнитного резонанса52.
Простые закономерности, которым подчиняется переход протона
во фтористом водороде, представлены на рис. 17 и 18. Уксусная
и трифторуксусная кислоты, этиловый спирт и трифторэтанол сме-
смешиваются с безводным фтористым
водородом без химического взаи-
взаимодействия. Разбавленный раствор
уксусной кислоты в безводном фто-
фтористом водороде практически пол-
полностью ионизирован. При этом об- ,п-з
разуется катион уксусной кислоты:
| Нитробензол/
10
10
10'
10
10
^ 10~
A8) ^
Трифторуксусная кислота обла- ^ 70'
дает более сильными кислотными SJ
свойствами и соответственно более \п-в
слабыми основными свойствами,
чем уксусная кислота, поэтому
естественно, что раствор трифтор-
трифторуксусной кислоты во фтористом
водороде имеет более низкую элек-
электропроводность, чем раствор ук-
уксусной кислоты. Следует отметить,
что более низкая степень иониза-
ионизации связана с уменьшением основ-
основности CFgCOOH, вызванным замещением водорода метильной группы
на фтор. Таким образом, кислотность молекулы HF не является:
определяющим фактором. Предполагают, что раствор этилового
спирта во фтористом водороде почти полностью ионизирован. Однако
растворы трифторэтанола, как и растворы трифторуксусной кислоты,
ионизированы слабо. Это обстоятельство вызвано уменьшением
основности CF3CH2OH, потому что трифторэтанол едва ли можно-
квалифицировать как сильную кислоту.
Наконец, следует обратить внимание на характер изменения!
электропроводности растворов веществ с разной диэлектрической
проницаемостью во фтористом водороде в зависимости от концентра-
концентрации (рис. 19). Нитробензол, слабое основание с высокой диэлектри-
диэлектрической проницаемостью, и диэтиловый эфир, сильное основание-
с низкой диэлектрической проницаемостью, смешиваются с безвод-
безводным фтористым водородом в любых соотношениях. Концентрирован-
Концентрированные растворы диэтилового эфира во фтористом водороде почти не про-
проводят электрический ток, однако разбавленные растворы почти:
о го ьо 60 so wo
Содержание HF ,мол.%
Рис. 19. Зависимость удельной;
электропроводности растворов ни-
нитробензола и диэтилового эфира.
в жидком HF от концентрации.
70
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВС
)ДОРОД
лолностью ионизированы. Нитробензол в умеренно концентрирован-
концентрированном растворе имеет значительную электропроводность, хотя он слабо
ионизирован даже в очень разбавленном растворе. Во всякой си-
системе кислота — основание переход протона зависит от относитель-
относительной силы обоих акцепторов протона, но ионизация и высокая элек-
электропроводность были обнаружены лишь в растворителях с высокой
диэлектрической проницаемостью.
Поведение нитробензола в жидком фтористом водороде можно
"Сравнить с поведением его з дымящей серной кислоте. В обеих ки-
кислотах нитробензол играет роль кислотно-основного индикатора.
Необходимо отметить, что процесс растворения в таких сильных
кислотах, как жидкий фтористый водород и серная кислота, обычно
сопровождающийся переходом протона и ионизацией, может про-
происходить и без перехода протона, когда растворяемым веществом
является слабое основание. В этих случаях процесс растворения
трактуется как слабое кислотно-основное взаимодействие, хотя
и не влекущее за собой перехода атома и образования связен, ко-
которые можно было бы идентифицировать при изучении спектров
поглощения или другими простыми способами 22>ЗЙ.
Такие термины, как диполь-дипольное взаимодействие, кислотно-
основное взаимодействие и переход протона, часто не совсем точно
употребляются для описания взаимодействия растворителя с раство-
растворенным веществом, но дают более или менее правильную картину
происходящего процесса.
Для изучения ряда растворов различных концентраций при-
применялся метод ИК-спектроскопии2. В качестве растворенных ве-
веществ использовалось большинство упомянутых соединений: диэтп-
ловый эфир, этиловый спирт, трифторэтанол, нитробензол и др.
Было показано, как влияют дипольная ориентация и процессы иони-
ионизации на колебательные спектры растворов в HF, а также указан
лредел концентраций, в котором преобладает каждый фактор.
Г. Кислоты в роли акцепторов фтор-ионов
Согласно кислотной функции Гаммета (Но^ —11), безводный фто-
фтористый водород представляет собой очень сильную кислоту. Точное
значение //„ установить не удается, потому что сам индикатор влияет
на концентрацию анионов и катионов. Значение И0, близкое к —12,
.наблюдаемое у растворов веществ в жидком фтористом водороде,
указывает на присутствие избыточного количества кислотных
ионов 29.
Так как фтористый водород обладает сильными кислотными
^свойствами, лишь немногие вещества, растворенные в нем, будут
вести себя как кислоты. Очень незначительное число соединений
способно увеличивать концентрацию протонизированных молекул
растворителя (H2F+). Хлорная и фторсульфоновая кислоты
III. ПОВЕДЕНИЕ ВЕЩЕСТВ ВО ФТОРИСТОМ ВОДОРОДЕ
71
являются достаточно сильными и стойкими веществами, что позво-
позволяет отнести их к этой категории. Однако наиболее важную и инте-
интересную группу кислот, растворяющихся в жидком фтористом водо-
водороде, составляют акцепторы фтор-иона. Большинство этих соедине-
соединений являются фторидами элементов V группы, из которых в литера-
литературе чаще всего упоминается пентафторид сурьмы.
Прибавление к фтористому водороду акцептора фтор-иона дает
такой же результат, как и введение донора протона, т. е. происходит
увеличение концентрации сольватированных протонов:
SbF5+2HF -
Что пентафториды сурьмы
и мышьяка могут выступать
как акцепторы фтор-иона, было
неоднократно установлено в"9>
27, 29, 30
Электропроводность раство-
растворов HF — SbF5 весьма велика.
Если в эту систему вводить
такое основание, как вода, то
электропроводность уменьшает-
уменьшается, и пентафторид сурьмы мо-
может быть оттитрован таким спо-
способом. При дальнейшем добав-
добавлении воды электропровод-
A9)
0,010
0,005
0 0,01 0,О2 0,03
Прибавлено Н2О, моль/л
Рис. 20. Кривая кондуктометрического
титрования раствора пентафторида
сурьмы во фтористом водороде водой.
Начальная концентрация SbF6 равна
0,02 моль/'л.
ность увеличивается 30 (рис. 20).
Спектры комбинационного
рассеяния растворов пентафто-
ридов сурьмы и мышьяка в жидком фтористом водороде дают наиболее
веские доказательства того, что названные соединения ведут себя
в этом растворителе как кислоты. В спектрах разбавленных раство-
растворов отчетливо видна линия, принадлежащая октаэдрическим ионам
AsFe или SbFe. Электропроводность раствора AsF5 несколько
меньше, чем SbF5, следовательно, пентафторид мышьяка является
более слабой кислотой.
Пентафториды ниобия и тантала ограниченно растворяются
в безводном фтористом водороде, но вполне достаточно для того, чтобы
считать их кислотами, хотя и несколько более слабыми, чем соеди-
соединения мышьяка и сурьмы. Кроме фторидов элементов V группы,
по-видимому, только трифторид бора является акцептором фтор-
ионов, проявляя в растворе HF кислотные свойства. Низкая раство-
растворимость BF3 (а также низкая электропроводность растворов HF —
BF3. означает, что это вещество является довольно слабой кислотой38.
Фториды элементов IV группы, такие, как тетрафторид титана
и тетрафторид кремния, практически нерастворимы в безводном фто-
фтористом водороде и не вызывают увеличения концентрации водородных
72
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
ионов в чистом растворителе. Однако в многофазных системах
тетрафторид титана, по-видимому, увеличивает растворимость не-
некоторых углеводородов, выступая при этом как катализатор в про-,
цессе перестройки структуры углеводородов 47~49. Поведение TiF4
во многом напоминает поведение TaF5 и MoF5 в аналогичных си-
системах; оно связано с кислотными свойствами TiF4 и TaF5. Раство-
Растворимость и кислотные свойства труднорастворимых слабокислых
'фторидов сильно возрастают в присутствии акцептора протона:
R + TaF5-f-HF —>¦ RH+-f-TaFe B0)
R-f-2TiF4-f-HF > RH+-f-Ti2F; B1)
Фториды можно легко классифицировать по их кислотно-основ-
кислотно-основным свойствам. Кислотность фторидов усиливается в направлении
от фторидов щелочных металлов, являющихся основаниями, к пента-
фторидам металлов V группы, которые в большинстве случаев ведут
себя как кислоты. В общих чертах эта закономерность аналогична
поведению окислов, растворенных в воде:
сильные основания
NaF + HF —
Na2O + H2O
слабые кислоты
A1F3+ HF-f-NaF —5
Al2O3-f-3H2O+NaOH -
«сильные кислоты
SbF5+2HF !
S03 + 3Ho0 э
Cl2O7+3H2O —)
> Na+-f~HFj
> 2Na+-f-2OH~
>• Na++AIFj* + HF
—у 2Na-f-2Al(OH)J*
> H2F+ + SbFe
- 2H3O"-f-SOf-
> 2H3O++2C1O7
B2)
B3)
B4)
B5
B6)
B7)
B8)
Различия в поведении окислов и фторидов наблюдаются, по-
видимому, как результат влияния особых геометрических стабили-
стабилизирующих факторов. Различие между переносом единичного электри-
электрического заряда, связанного с фтор-ионом, и переносом электронной
пары, связанной с ионом.кислорода, делает эту аналогию менее точ-
точной. В системе HF устойчив одновалентный гексафторидный анион
MFe, имеющий вид октаэдра. Кислород не образует анионов анало-
аналогичной структуры, и в случае пятиокиси сурьмы кислотные свойства
проявляются только при взаимодействии с сильными основаниями:
Sb2O5 + 2NaOH + 5H2O > 2Na+-f-2Sb (ОН); * B9)
В воде очень сильными являются кислоты с тетраэдрическим одно-
одновалентным анионом (содержащим четыре атома кислорода), образо-
* В действительности анионы, содержащие алюминий или сурьму, имеют
«более сложный состав, который зависит от концентрации раствора.
III. ПОВЕДЕНИЕ ВЕЩЕСТВ ВО ФТОРИСТОМ ВОДОРОДЕ 7*
ванным элементом VII группы, например ClOj, а также кислоты,
производные от элементов VI группы, например H2SO4. Но из этих
кислородсодержащих кислот только хлорная кислота, как имеющая
ClOl-ион, а также, по-видимому, фторсульфоновая являются до-
достаточно сильными для того, чтобы быть устойчивыми в жидком
фтористом водороде.
Растворимость и ионизация хлористого или бромистого водо-
водорода в жидком фтористом водороде пока мало изучены. Фреденха-
ген 1в первоначально предположил, что растворимость этих кислот
в HF очень мала. Однако оказалось, что из фтористоводородного
раствора (при низких температурах вблизи точки плавления HF)
можно осадить нерастворимый хлорид серебра. Вероятно, раствори-
растворимость этих галоидных кислот в жидком фтористом водороде больше,,
чем полагали раньше 14.
Д. Соли
Хотя протонизации во фтористом водороде подвергаются очень
многие соединения, число типичных солей весьма ограничено.
Те соединения, которые в воде ведут себя как соли, в жидком
фтористом водороде подвергаются сольволизу. Например, нитрат
калия превращается 10 в KF, H3O+ F~ и NOJF":
KNO3-f-6HF ^ГГ K+-f-NO+-f H3O+-f-3HFj C0)
Перхлорат калия, по-видимому, является истинной солью:
KC1O4 + HF ^ K+ + C1OJ + HF C1)
Сульфаты щелочных металлов обычно переходят во фторсуль-
фонаты, которые далее ведут себя как типичные соли. Анионы дру-
других кислородсодержащих кислот образуют в процессе растворе-
растворения в HF катионы Н3О+, и окисел или оксифторид. Получающееся
кислородное соединение может реагировать далее до образования
свободного элемента. Так, хлораты выделяют двуокись хлора, а бро-
маты восстанавливаются до свободного .брома.
Двойные соли щелочных металлов и трифторидов бора, а также
фторидов элементов V группы в жидком фтористом водороде ведут
себя как типичные соли. Некоторые из них были идентифицированы
Клиффордом и Моррисом 7. Сообщается, что AgPF6, NaPF6 и
Ba(PFeJ растворяются в жидком фтористом водороде; NaAsFe
и NaSbFe растворяются плохо e, a KBF4 — хорошо.
При упаривании растворов BaF2 или NaF совместно с малолету-
малолетучими кислотами TeFfi и GeF4 образуются твердые вещества, кото-
которым приписывается формула Ba(TeF7K и NaGeF5. Существование
во фтористом водороде иона TeF7 не доказано. В жидком фтористом
водороде удалось идентифицировать лишь небольшое число ани-
анионов; свойства солей, которые они образуют, также мало изучены.
.74
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
IV. ПОВЕДЕНИЕ БИОЛОГИЧЕСКИ ВАЖНЫХ ВЕЩЕСТВ
75.
Е. Вещества, растворяющиеся без ионизации
Растворение в жидком фтористом водороде не обязательно со-
сопровождается ионными процессами, хотя переход протона проис-
происходит в большом числе случаев. Как отмечалось выше, при раство-
растворении слабых оснований * в безводном фтористом водороде наблю-
наблюдается лишь незначительное увеличение электропроводности. Вслед-
Вследствие высокой диэлектрической проницаемости фтористого водорода
водородная связь обычно вызывает переход протона и значитель-
значительную ионизацию в разбавленном растворе. Если этого не происходит,
можно сделать вывод, что тенденция к образованию водородной
связи очень слаба. Кислотно-основное взаимодействие и водородная
связь настолько важны для понимания процесса растворения в кис-
кислых растворителях, подобных HF, что мы не совсем уверены в том,
как трактовать растворение слабых оснований, смешивающихся
с HF в любых отношениях. На практике они могут быть использо-
использованы как разбавители фтористого водорода. В качестве примера
можно привести трифторуксусную кислоту и жидкую двуокись
серы. Они смешиваются с фтористым водородом в любых отношениях
и, по-видимому, без значительного ослабления кислотных свойств
HF.
Существует значительное количество инертных и практически
нерастворимых соединений. К умеренно растворимым и не образу-
образующим ионов веществам относятся многие простые неорганические
соединения, в том числе некоторые простые фториды.
Роль геометрической стабилизации при растворении кислот
уже рассматривалась. Большинство пентафторидов обладает кислот-
кислотными свойствами, потому что эти соединения склонны образовывать
октаэдрические симметричные анионы с шестью атомами фтора.
Предполагают, что устойчивые молекулы гексафторидов растворя-
растворяются в жидком фтористом водороде без перехода фтор-иона. На это
указывают проведенные исследования. В системе гексафторид ура-
урана — фтористый водород имеется область несмешивающихся жидких
растворов, причем растворимость твердого гексафторида урана
во фтористом водороде весьма мала и данных, свидетельствующих
об ионизации или об образовании соединений, не получено 70. Изу-
Изучение методом ядерного магнитного резонанса некоторых гекса-
гексафторидов, содержащих фтористый водород, подтверждает отсутствие
взаимодействия 55. Систематические исследования разбавленных рас-
растворов гексафторидов во фтористом водороде до сих пор не прово-
проводились .
Среди гексафторидов исключение составляет гексафторид ксенона.
Это соединение обнаруживает высокую растворимость в жидком фто-
* К таким основаниям относятся органические соединения, основные
свойства которых ослаблены введением тех или иных группировок, например
нитро- или трифторметильной групп.
рнстом водороде, быстрый обмен фтором и значительную иониза-
ионизацию 25> -7. Это согласуется с данными других исследователей о томг
что симметрия гексафторида ксенона более низкого порядка, чем
октаэдрическая 20> 81.
Октаэдрпческие симметричные соединения, растворяющиеся
в жидком фтористом водороде,— примеры соединений, химически
не взаимодействующих с этим растворителем. Среди других соеди-
соединений, не реагирующих с растворителем, следует отметить тетра-
адрические SiF4 и CF|5 и плоский тетрагональный XeF|'25.
IV. ПОВЕДЕНИЕ НЕКОТОРЫХ БИОЛОГИЧЕСКИ
ВАЖНЫХ ВЕЩЕСТВ
В ЖИДКОМ ФТОРИСТОМ ВОДОРОДЕ
На первый взгляд может показаться, что такое агрессивное ве-
вещество, как безводный фтористый водород, едва ли будет пригодно
в качестве растворителя сложных и неустойчивых органических
соединений. Однако углеводы и белки хорошо растворяются в без-
безводном фтористом водороде, претерпевая лишь незначительные-
изменения. Несмотря на то что безводный фтористый водород яв-
является одним из наиболее гигроскопичных среди всех известных
веществ, растворение в нем органических соединений, способных
терять воду, часто происходит без дегидратации.
В процессе растворения необходимо отводить выделяющееся тепло, иначе-
температура раствора сильно возрастет. Растворы углеводов и белков следует
готовить в хорошо проводящих тепло сосудах при достаточно низкой темпе-
температуре.
Фреденхаген и Каденбах 17, по-видимому, первыми обнаружили,
что целлюлоза легко растворяется в жидком фтористом водороде.
Раствор целлюлозы в HF проводит электрический ток. Вещество,
выделенное из таких растворов,^ Фреденхаген идентифицировал
как глюкозан, который при неглубоком гидролизе образует глю-
глюкозу. Этот процесс был предложен для осахаривания целлюлозы.
Более поздние работы пролили мало света на процессы, происхо-
происходящие при растворении углеводов в жидком фтористом водороде.
Сравнительно недавно Педерсен 59~81 изучил поведение некоторых
эфиров Сахаров в безводном фтористом водороде. Длительная об-
обработка пента-О-ацетил-C-Б-глюкопиранозы безводным фтористым
водородом приводит к образованию производных маннозы и альт-
розы; происходит частичный гидролиз и изомеризация. По-види-
По-видимому, нужны особые предосторожности для того, чтобы растворы
не поглощали воду. Однако и в таких условиях жидкий фтористый
водород медленно реагирует с эфирами Сахаров. Поведение этих
веществ в совершенно свободном от воды жидком фтористом водороде
представляет большой интерес.
76
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
IV. ПОВЕДЕНИЕ БИОЛОГИЧЕСКИ ВАЖНЫХ ВЕЩЕСТВ
77
Безводный фтористый водород является прекрасным раствори-
растворителем для белков. В нем легко растворяются белки, растворимые
в воде, а также многие нерастворимые в воде волокнистые белки,
например шелковое волокно. Хорошо растворимы в жидком HF
рибонуклеазы, инсулин, трипсин, альбумин сыворотки, глобулин
¦сыворотки, эдестин, гемоглобин и коллаген. При этом возможны
химические реакции, но они не нарушают биологических свойств
белковых веществ. Из раствора в жидком фтористом водороде можно
выделить инсулин, почти полностью сохранив его биологические
^свойства 33. Рибонуклеазы и лизоцимы можно растворить в жидком
HF или в смеси HF—S02. Выделенные из раствора путем отгонки
растворителя эти вещества также не теряют своих ферментных
свойств при условии, что температура процесса отгонки достаточно
низкая, а продолжительность небольшая 39. При более высоких
температурах происходит инактивация фермента. Это связано,
по-видимому, с разрывом небольшого числа пептидных связей,
что приводит к увеличению гидродинамического объема вещества,
хотя и не вызывает уменьшения молекулярного веса. Исследования
в жидком HF белков, имеющих дисульфидные связи, убедительно
показывают, что последние не разрываются:
Безводный фтористый водород вызывает набухание коллагена
и дезориентацию его, но, вероятно, не разрушает пептидные связи.
Отмечаемый иногда низкий молекулярный вес желатина, выделен-
выделенного из раствора в жидком фтористом водороде, очевидно, объяс-
няется гидролизом под действием воды, случайно попавшей в рас-
раствор HF.
При изучении растворов белков в Н F обращает на себя внимание
процесс полимеризации- N-карбоксиангидридов, приводящий к об-
образованию поли-а-аминокислот40. Таким образом могут быть
получены полипептиды, состоящие из 30 молекул. Относительно
небольшая длина цепи полимера объясняется, вероятно, побочной
реакцией — выделением окиси углерода из N-карбоксиангид-
рида 41.
Химическое поведение некоторых белков, растворенных во фто-
фтористом водороде, сравнительно недавно было изучено Гессом и
и сотр.45 71> 73. Их исследования показали, что серии- и треонин-
содержащие пептиды при растворении в жидком HF подвергаются
NjO-ацильной перегруппировке. Она происходит в результате
разрыва пептидной связи с аминокислотой, содержащей гидроксиль-
ную группу в боковой алифатической цепи, с последующей мигра-
миграцией ацила и образованием сложноэфирнои группы (азот пептидной
связи превращается в свободную амино-группу). Давно известно,
что О-ацилирование протекает в сильнокислой среде (например,
под действием концентрированной H2SO4) и зависит от величины
рН раствора. Считают, что оксиоксазолидин является промежу-
промежуточным продуктом М,0-ацильной перегруппировки:
он
/С СН2 О
'I I I
HN-CH-C-NHR'
СИ
н+
НО/
он-
HN-
-CH-C-NHR'
н+ ,
¦¦-*-
R
С СН2 О
о^ | I
4rz^ +H;iN СН—C-NHR'
C2)
Так как эфирная группа, образовавшаяся в результате миграции
ацила, в сильной степени подвержена гидролизу, этим способом
можно пользоваться для химического разрыва пептидных цепей
в пептидных связях, примыкающих к серии- и треонинсодержащим
остаткам. Характерный разрыв наблюдается также в С-метиониль-
ных пептидных связях.
Названные работы Гесса содержат наиболее детальный анализ
химических явлений, происходящих при растворении белков в жид-
жидком фтористом водороде. Реакции, протекающие при этом, типичны
только для такой среды и могут быть применены для установления
структуры белка. Зингер 79 недавно изучил влияние неводных рас-
растворителей на строение белков, причем обратил особое внимание
на действие сильных протогенных растворителей.
Было замечено, что спектры поглощения раствора железосодер-
железосодержащих белков цитохрома с и гемоглобина в жидком фтористом во-
водороде и в воде похожи друг на друга. Установление этого факта
способствовало изучению свойств других координационных соеди-
соединений металлов в жидком фтористом водороде. Способность комплек-
комплексов металлов противостоять действию фтористого водорода, по-ви-
по-видимому, характерна для фталоцианинов металлов, аминов кобаль-
та(Ш) и многих других координационных соединений, растворя-
растворяющихся без разрушения комплекса. Особый интерес представляют
биологически важные координационные соединения металлов —
хлорофилл и витамин В12. Спектры поглощения растворов хлоро-
хлорофилла в жидком фтористом водороде аналогичны его спектрам в бо-
более обычных растворителях. Раствор витамина В12 в жидком HF
имеет яркий оливково-зеленый цвет, а цвет самого витамина В12
ярко-красный. Витамин В12, представляющий собой координаци-
координационное соединение кобальта(Ш), не разрушается при растворении
в жидком фтористом водороде, несмотря на сложную структуру
и наличие многочисленных функциональных групп. Он может быть
легко выделен из раствора HF и при этом не теряет своих
свойств.
78
ГЛАВА ВТОРАЯ. ЖИДКИЙ ФТОРИСТЫЙ ВОДОРОД
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28,
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
Adams R.
Adams R.
A t о j i M.,
Briegleb
Brown Т.
1963, p. 263.
Clifford
A957).
Clifford
A957).
Clifford
Clifford
57 A957).
Del Greco
ЛИТЕРАТУРА
M., К a t z J. J., J. Opt. Soc. Am., 46, 895 A956).
M., К at z J. J., J. Mol. Spectrosc, 1, 306 A957).
Lipscomb W. N., Acta cryst., 7, 173 A954).
G., Strohmeier W., Z. Electrochem., 57, 668 A953).
H., Whipple E. В., Noble Gas Compounds, Chicago,
A. F., Kongpricha S., J. Inorg. Nucl. Chem., 5, 7&
A. F., Morris A. G., J. Inorg. a. Nucl. Chem., 5, 71
A. F., Sargent J., J. Am. Chem. Soc, 79, 4041 A957).
A. F., Beachell H. С et al., J. Inorg. a. Nucl. Chem., 5,
F e d о г
A963).
F r an с к Е.
F r an с к Е.
F. P., Gryder J. W., J. Phys. Chem., 65, 922 A961).
H. M., Hubbard W. N. et al., J. Phys. Chem., 67, 1148
Fredenhagen
Fredenhagen
A929).
Fredenhagen
A930).
Fredenhagen
Fredenhagen
Giguere P. A.,
G i 11 e s p i e R.
H a m m e t t
H a m m e t t
H a m m e t t
Herzberg
U., Meyer F., Z.
U., Sp alt hof f
H.
K.
Z. anorg. Chem.,
Cadenbach
Elektrochem., 63, 571 A959).
W., Z. Elektrochem., 61, 348
K., Cadenbach G., Z.
A957).
178, 289
phys. Chem., 146A, 245
242, 23 A939).
G., Z. anorg. Chem.,
L.
L.
L.
G.,
K., Cadenbach G., Angew. Chem., 46, 113 A933).
K., D a h m 1 о s J., Z. anorg. Chem., 178, 272 A929).
, Zengin N., Can. J. Chem., 36, 1013 A958).
J., Noble Gas Compounds, Chicago, 1963, p. 333.
P., Chem. Rev., 16, 67 A935).
P., Physical Organic Chemistry, New York, 1940.
P.,Deyrup A. J., J. Am. Chem. Soc, 54, 2721 A932).
Molecular Spectra and Molecular Structure. I. Spectra
of Diatomic Molecules, 2nd ed., New York, 1950.
Hindman J. C, Svirmickas A. Noble Gas Compounds, Chicago,
1963, p. 251.
Hu J. H., White D. et al., J. Am. Chem. Soc, 75, 1232 A953).
H у m a n H. H., Quarterman L. A., Noble Gas Compounds, Chi-
Chicago, 1963, p. 275
H у m a n H. H.,
3668 A957).
H у m a n H. H.,
1963, p. 63T.
H у m a n H. H.,
A961).
J а с h e A. W., С a d у G. H., J. Phys. Chem., 56, 1106 A952).
Jarry R. L., Davis W. J., J. Phys. Chem., 57, 600 A953).
К a t z J. J., Arch. Bioch. Biophys., 51, 293 A954).
К atz J. J., Ну man H. H., Rev. Sci. Instrum., 24, 1066 A953).
К il patrick M., H у m a n H. H., ~
Kilpatrik M., Lewis J. I., J.
Kilpatrick M., Luborsky F.,
Kilpatrick M., Luborsky F., J. Am. Chem. Soc, 76, 5863 A954).
Koch A. L., L a m о n t W. A. et al., Arch. Bioch. Biophys., 63, 06
A956).
К о p p 1 e K. D., К a t z J. J., J. Am. Chem. Soc, 78, 6199 A956).
Kopple 1. I)., Quarterman L. A. et al., J. Org. Chem., 27,
1062 A962).
Kilpatrick M. et air, J. Am. Chem. Soc, 79,
Lane T. I. et al., 145th Meeting A. C. S. Abstracts,
Quarterman L. A. et al., J. Phys. Chem., 65, 123
J. Am. Chem. Soc, 80, 77 A958).'
Am. Chem. Soc, 78, 5186 A956).
J. Am. Chem. Soc, 75, 577 A953).
ЛИТЕРАТУРА
79
42.
43.
44.
45.
46.
47.
48.
49.
.50.
51.
52.
53.
54.
¦55.
56.
57.
58.
59.
¦60.
61.
62.
63.
64.
€5.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
G. A., J. Mol. Spectrosc. 2, 75 A958).
G. A., S m i t h D. F. et al., J. Chem. Phys., 25, 275 A956).
L., F i s с h e r W. et al., Z. anorg. Chem., 206. 61 A932).
A. V. et al., Biochem. Biophys. Res. Com-
R. W., H i I'd e b r an d J. H. et al., J. Am. Chem. Soc, 65, 182,
К u i p e r s
К u i p e r s
Le Boucher
Lenard J. I., Schally
num., 14, 498 A964).
Long
A943).
McCaulay
McCaulay
McCaulay
3009 A956).
M а с к о г Е.
MacLean
MacLean С,
D.
D.
D.
A.,
A.,
A.,
Lien A. P., J. Am. Chem. Soc, 75, 2013 A951).
Lien A. P., J. Am. Chem. Soc, 79, 2495 A957).
Higley W. S. et al., J. Am. Chem. Soc, 78,
L., H о f s t г a A. et al., Trans. Farad. Soc, 54, 186 A958).
C, Mack or E. L., J. Chem. Phys., 34, 2207 A961).
" Mack or E. L., Disc. Farad. Soc, № 34, 165 A962).
M а у b u г у R. H., G о r d о n S. et al., J. Chem. Phys., 23, 1277 A955).
M e 11 о r's Comprehensive Treatise on Inorganic and Theoretical Chemistry,
Part I, London, Chapter I, Fluojine Section 3, 72—84. The preparation of
hydrofluoric acid. Section 4,85—146. The physical and chemical properties
and uses of hydrogen fluoride and its aqueous solutions.
Muetterties E. L., Phillips W. D., J. Am. Chem. Soc, 81,
1084 A959).
Paul M. A., Long F. A., Chem. Rev., 57, 1 A957).
Pauling L., J. Chem. Educ, 33, № 1, 16 A956).
Perkins A., J. Phys. Chem., 68, 654 A964).
Pedersen C, Acta chem. scand., 16, 1831 A962).
C, Acta chem. scand., 17, 673 A963).
Fletcher H. G., Jr., J. Am. Chem. Soc, 82, 941
C,
Pedersen
Pedersen
A960).
Q uarterm an L. А., Ну man H.H. et al., J. Phys. Chem., 61, 912 A957).
L. А., Ну man H. H., ot al., J. Phys. Chem., 65,
Quarterman
90 A961).
Rogers M. Т.,
A956).
Rogers M. Т.,
Speirs J. L. et al., J. Am. Chem. Soc, 78, 3288
Speirs J. L. et al., J. Phys. Chem., 61, 366 A956).
Rogers M. Т., S p e i r s J. L. et al., J. Am. Chem. Soc, 78, 936 A956).
Roth W. A., Ann. chim. phys., 542, 35 A939).
Rothschild W. G., J. Opt. Soc. Am. 54, 20 A964).
Runner M. E., Balog G. et al., J. Am. Chem. Soc, 5183 A956).
L. et al., J. Phys. Chem., 57, 541 A953).
H. et al.
R.
K.
J. Am. Chem. Soc. 84, 4921
Rutledg G. P., Jarry
Sakakibara S., Shin
A962).
Sastri M. L. N., Ho rn ig D. F., J. Chem. Phys., 39, 3497 A963).
Shin K. H., Sakakibara S. et al., Biochom. Biophys. Res. Com-
H.,
num., 8, 288 A962).
H.
H.
H.
Fluorine Chemistry, v. I, New York, 1950, p. 225.
Fluorine Chemistry, v. V, New York, 1964~ p. 2.
Bouknight J. W., J. Am. Chem. Soc, 54,
Simons
Simons
Simons
A932).
Simons
A944).
Simons
A924).
Singer S. J., Adv. Protein Chem., 17, 1
" D. F., J. Chem. Phys., 28, 1040
129
1070
J. H., Dresdner R. D., J. Am. Chem. Soc, 66,
Hilde brand J. H., J.Am. Chem. Soc, 46, 2183
A962).
A958).
J. H.
Smith
Smith D. F., Noble Gas Compounds, Chicago, 1963, p. 295.
Spalthoff W., Fran с к Е. U., Z. Elektrochem., 61, 993 A957).
Strohmeier W., Briegleb G., Z. Electrochem., 57, 662 A953).
Veis А., К a t z J. J., Biochim. et Biophys. acta, 22, 96 A956).
ГЛАВА ТРЕТЬЯ
II. СВОЙСТВА ЖИДКИХ ГАЛОГЕНОВОДОРОДОВ
81
ГАЛОГЕНОВОДОРОДЫ КАК
ИОНИЗИРУЮЩИЕ РАСТВОРИТЕЛИ
М. Е. ПИЧ, Т. ВАДДИНГТОН
I. Введение 80
II. Свойства жидких галогеноводородов 81
А. Физические свойства 81
Б. Самоионизация 82
III. Свойства растворов в галогеноводородах п экспериментальные методы
исследования 84
A. Электропроводность , 84
Б. Спектроскопия 89
B. Фазовые диаграммы 90
Г. Криоскопия и эбулиоскопия 91
Д. Препаративные методы 91
IV. Реакции в жидких галогеноводородах 92
A. Кислотно-основные реакции 92
1. Основания 92
2. Кислоты gg
Б. Реакции сольволиза 104
B. Окислительно-восстановительные реакции 105
Литература 107
I. ВВЕДЕНИЕ
Галогеноводороды (НС1, HBr, HI) представляют определенный
интерес как ионизирующие растворители, особенно в сравнении
с жидким HF, на который они так похожи по своим свойствам и от
которого в то же время так отличаются. Наибольшая часть материала,
обсуждаемого в данной главе, относится к жидкому хлористому
водороду. Первые эксперименты с этим веществом как растворителем
были проведены более века назад Гором 14, причем наблюдения были
визуальными и носили качественный характер. Из 66 опробованных
им соединений только 10 растворились в НС1, и Гор заключил, что
«жидкий хлористый водород имеет, в общем, ничтожную растворя-
растворяющую силу для твердых веществ».
Несмотря на это заключение, изучение хлористого, бромистого
и йодистого водорода как неводных растворителей продолжалось
примерно в течение десятилетия в самом начале XX века в Канаде.
Основные результаты этих исследований были изложены в обширном
труде, опубликованном в Англии и Германии 50> 51.
В работе приведены физические и термодинамические константы
галогеноводородов, данные о растворимости, электропроводности и
данные эбулиоскопических измерений, числа переноса в НВг, опреде-
определенные по методу Гитторфа, и изложено подробное обсуждение
результатов.
Непосредственное изучение некоторых реакций в жидком НО
было затем возобновлено в конце пятидесятых годов в Кембридже 58,
а свойства жидких НВг и HI были изучены недавно Ваддингтоном
и Уайтом 6°-62, Кленбергом и Колыпуттером 30.
Галогеноводороды, за исключением фтористого, при комнатной
температуре являются газами. Они обычно изучаются при темпе-
температурах —111,6° С (плавление сероуглерода) и —95,0° С (плавле-
(плавление толуола) для НС1, —83,6° С (плавление этилового эфира уксус-
уксусной кислоты) для НВг и —45,2° С (плавление хлорбензола) для HI.
II. СВОЙСТВА ЖИДКИХ ГАЛОГЕНОВОДОРОДОВ
А. Физические свойства
Некоторые физические свойства жидких галогеноводородов и
воды (для сравнения) представлены в табл. 14. Заметно очевидное
сходство физических свойств воды и фтористого водорода, с одной
Таблица 14. Некоторые физические свойства галогеноводородов и
Свойство
Молекулярный вес
Плотность жидкости, г/см3
Температура, °С
плавления
кипения
Температурный интервал жид-
жидкого состояния
Теплота, ккал/молъ
плавления
испарения
Постоянная Трутона
Диэлектрическая проница-
проницаемость жидкости
Удельная электропроводность,
MKOM'l- ¦ СМ~1
Вязкость, спз
Характеристическая частота
связи Н—Г (газ), см~1
Н2О
18,0
1,00
@°С)
0
100
100
144018
972018
26,0
84 2
@°С)
0,05И
A8° С)
1,00
B2° С)
3568 **
HF
20,0
1,2
(—80° С)
—83,0
19,5
102,5
1094
723018
24,7
175
(-73° С)
0.1*4
(—80° С)
0,24
@°С)
396116
НС1
36,5
1,187
(—114°С)
—114,6
—84,1
30,5
476
3860
20,4
9 28
(-95° С)
0,003512
(—85° С)
0,5150
(—95° С)
288616
НВг
80,9
2,603
(-84° С)
—88,5
—67,0
21,5
600
4210
20,4
7,018
(-85° С)
0,001482
(-84° С)
0,8350
(-67° С)
2558"
воды *
HI
127,9
2,85
(-47° С)
—50,9
—35,0
15,9
686
4724
19,8
3,3918
(-50° С)
0,0008549
(—45° С)
1,355о
(—35,4° С)
2230"
лора
* Вое величины без указанной ссылки на литературу заимствованы из книги Мед-
** Величина для связи Н—ОН, цитируется по работе 2S.
6 Заказ 90
82 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
II. СВОЙСТВА ЖИДКИХ ГАЛОГЕНОВОДОРОДОВ
83
стороны, и других галогеноводородов между собой, с другой
стороны, и большое различие между физическими свойствами этих
двух групп. У НС1, НВг и HI относительно узкие области жидкого
состояния, что приводит к некоторым экспериментальным трудно-
трудностям, и низкие значения диэлектрической проницаемости. След-
Следствием последнего является то, что в этих галогеноводородах раство-
растворяются только соли с низкими энергиями кристаллической решетки,
такие, как галогениды тетраалкиламмония, а соли, такие, как хло-
хлористый аммоний, с высокой энергией кристаллической решетки,
нерастворимы. Другим следствием низкой диэлектрической прони-
проницаемости является сложный характер изменения эквивалентной элек-
электропроводности с концентрацией. Постоянные Трутона высоки для
воды и фтористого водорода, что указывает на ассоциацию молекул
за счет водородных связей. В, других галогеноводородах водород-
водородная связь хотя и может возникнуть, но она гораздо слабее.
Б. Самоионизация
Электропроводность чистых жидких галогеноводородов, за исклю-
исключением фтористого водорода, несколько меньше, чем воды, но, чтобы
объяснить эти значения, необходимо предположить определенный
тип самоионизации. По аналогии с ионизацией воды, учитывая соль-
сольватацию ионов в растворе, процесс самоионизации галогеноводоро-
галогеноводородов можно представить в виде
ЗНГ ^zT Н2Г+ + НГ? A)
Единственным доказательством существования иона Н2Г+ в рас-
растворе служит ион H2F+, который был обнаружен вместе с ионом
SbFe в системе HF—SbF5. Его существование доказано на основа-
основании данных об электропроводности, ИК-спектров и спектров ком-
комбинационного рассеяния 27. Ионы Н2С1+, Н2Вг+ и Н21+ ни в рас-
растворе, ни в твердом состоянии еще не обнаружены.
Ион Н2С1+ может существовать в соединениях HG1 • НСЮ^, которое взрыво-
взрывоопасно; в HC1-H2SO4, которое нельзя Выделить в твердом состоянии19, и
в HCl-HBr31. To, что последнее соединение, как утверждается, имеет струк-
структуру Н2С1+Вг~, а не Н2Вг+С1~, недостаточно обоснованно, так как оно построено
на приравнивании иона Н2С1+ иону К+ н иона Н2Вг+ иону Rb+ (для этих ионов
одинаковы заряд ядра и количество электронов) и сравнении энергий кристалли-
кристаллической решетки бромистого калия и хлористого рубидия*.
Существование иона НГг было подтверждено открытием ионов
? и HClJ- Ион HFJ был известен давно, а о существовании HCli
в качестве промежуточного продукта некоторых органических ре-
реакций в нитробензоле 22 сначала предположили; он был получен 55>56
в виде (СН3L М+НС1г и некоторых соединений с ионом карбония 47.
Сообщение о получении Вестом 64 Cs+HCli позднее не подтвердилось;
соединение на самом деле отвечало формуле CsCl-3/4HCb3/4H2O35>53
со структурой 4Cs+Cl~-SHgO+Cl". Соединение Cs+HCl5 было полу-
получено из хлористого водорода и хлорида цезия при —78° С; оно ха-
характеризуется высоким давлением диссоциации D,4 атм при ком-
комнатной температуре 52). ИК-спектры некоторых из этих соединений
подтверждают47'5? существование НС1г и свидетельствуют о линей-
линейной структуре этого иона, т. е. (С1—Н—С1)~. Симметричное положение
протона в нем было подтверждено термодинамическими измерени-
измерениями2. Теплота образования иона HF? в газовой фазе, как было
рассчитано 55, составляет 58 ккал/молъ. Теплота образования для
иона НГг с большими анионами Г, как сообщают, достигает предель-
предельных величин, равных 14,2 ккал/молъ для HClJ, 12,8 ккал/молъ для
НВгг и 12,4 ккал/молъ для иона HIJ 53. Ионы НС1г и НВгг были
выделены в виде солей троповой кислоты С7ЩНГг, которые были по-
получены реакцией раствора тропенилметилового эфира с насыщен-
насыщенным эфирным раствором галогеноводорода 20. Ион HBri был также
получен в виде тетра-н-бутиламмониевой и тетрафениларсониевой
солей 60, ион Н1г — в виде тетра-к-бутиламмониевой соли. Удалось
получить46 соли смешанных ионов, таких, как НС1Вг~ и HC1I". Дан-
Данные ИК-спектроскопии для всех этих ионов приведены в табл. 15.
Ион
HF2
HC1J
НВгг
HIj
НВгСГ
НС1Г
Таблица 15. Частоты *
Соль
KHF2
(CH3LNHG12
(CH3LNHC12
(k-C4H9LNH4C12
Si(acacKHCl2**
(C2H5LNHBr2
(K-C4H9LNHBr2
(k-C4H9LNHI2
(K-C4H9LNHBrCl
(/t-C4H9LNHClI
ионов HFj в
V3, CM~l
1450
1565
1565
1540
1625
1670
1690
1650
1650
2000
ИК-спектрах
V,, CM*'
1223
1180
1160
1150
1150
1170
1170
1165
1100
990
Литература
55, 56
53
53
53
53
53
53
46
46
* v3 — частота ассиметрического уширения; чг—частота деформационного колебания.
** Символ асас—ацетилацетонато—СНзСОСН=ССНз.—Прим. ред.
о-
Если самоионизацию галогеноводородов представить в виде
уравнения A) и рассматривать как кислотно-основное взаимодей-
взаимодействие, то кислоты могут быть определены как доноры протона или
акцепторы галоген-иона, а основания — как акцепторы протона или
доноры галоген-иона (под галоген-ионом .мы подразумеваем соль-
ватированный ион НГг, под протоном — Н2Г+). Следовательно,
хлористый водород обнаруживает свойства такого растворителя,
как AsCl3 — акцептор С1-ионов 17, — и некоторых кислотных раство-
растворителей, таких, как серная кислота
9>ю *
* См. главу четвертую.
84 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
III. СВОЙСТВА РАСТВОРОВ В ГАЛОГЕНОВОДОРОДАХ
И ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
А. Электропроводность
Исследование электропроводности * является основным ме-
методом изучения этих растворителей. Спектральный анализ пока
широко не используется. Электропроводность растворов жидкого
НС1 и НВг изучалась Стилом и сотр.50>51. В последнее время при
изучении этих растворителей широко применяется кондуктометри-
ческое титрование. Электропроводность чистых галогеноводородов
приведена в табл. 14, а электропроводность растворенных веществ
в этих растворителях — в табл. 16.
Таблица 16. Электропроводность некоторых веществ в галогеноводородах
Галогеноводо-
род
НС1
НВг
ш
растворенное
вещество
РС15
' (CH3LNCl
ВС13
BF3
POF3
(CH3LNBr
SnBr4
PhsCHsNI
Пиридин
с, моль/л
0,36
0,32
0,32
1,43
0,25
0,023
0,064
0,10
0,38
я,
мком-1 • см~1
9350
6790
0,13
0,09
3,6
9,8
0,024
243
125
л,
см* /(ом- моль)
25,9
21,5
4,0 • 10
0,6 • 10-4
1,5-10-2
0,432
3,8-10-4
2,55
0,32
Раствор
Основной 58
Основной 58
Кислый 88
Кислый 58
Основной 40
Основной 60
Кислый во
Основной 30
Основной30
Из этих данных видно, что электропроводность растворов осно-
оснований намного выше электропроводности кислот. Это объясняется
сильной кислотностью галогеноводородов, и поэтому очень трудно
найти кислоту более сильную, чем сами растворители. Электропро-
Электропроводность в жидком НС1 больше, чем в НВг и HI. Механизм электро-
электропроводности неизвестен, но можно предположить, что в основных
растворах НС1 она обусловлена переходом иона хлора, а именно
Cl—Н—С1" . . . Н—С1
С1-Н . . . Cl-H-Cl-
Если этот механизм отражает истинную картину проводимости,
то эквивалентные электропроводности при бесконечном разбавле-
разбавлении оснований с одинаковой валентностью катиона должны быть
* Измерение удельной электропроводности — наиболее чувствительный
метод из числа применяющихся для контроля чистоты галогеноводородов;
измерение давления паров дает лишь грубую ориентировку.
III. СВОЙСТВА РАСТВОРОВ В ГАЛОГЕНОВОДОРОДАХ
85
приблизительно одинаковыми. К сожалению, измерить их с доста-
достаточной степенью точности пока невозможно 42.
Числа переноса в жидком НС1 еще не измерены, но это сделано
для некоторых растворов в бромистом водороде 50>51:
Растворенное ве-
вещество . . . .
с, молъ/л . . . .
Среднее значение
t+
(С2Н5JО
1,0 0,5
(G2H5KN
0,62—0,75 1,04
GH3COGII3
1,0 1,82
GH3GOG6Hi3
0,9 1,8
0,39 0,77
0,82 0,18 0,22 0,35 0,38 0,95
Нетрудно заметить, что числа переноса катионов увеличиваются
при возрастании концентрации растворенного вещества. Это ука-
указывает, по-видимому, на возрастание степени комплексообразования
катиона с увеличением концентрации.
Рис. 21. Зависимость
электропроводности от
концентрации для рас-
растворов в жидком хлори-
хлористом водороде:
1 — (C2H5).,NH+Cl-;2—HCN;
3—(С2Н5JО; 4~CH.,CONH2;
5 — CH3CN; 6 —(CH3JS (no
внутренней шкале); 7 —
(СН3)г S (по внешней шка-
шкале); 8 — (CH,KN; 9— (СН.,K
NH+BC1J.
^
0,3
Была изучена
от концентрации
42,50,51,62 зависимость электропроводности Л
с в НС1 (рис. 21), НВг, HI и найдено, что Л
стремится к минимуму при низких значениях ус (с — молярная
концентрация). Экстраполяция этих кривых до бесконечного
86 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
разбавления очень трудна. Была определена42 концентрация смиНТ
которой соответствует минимальное значение Л, для хлористоводо-
хлористоводородных растворов оснований (димётилсульфида и триметиламина),
соли (тетрахлорбората триметиламмония) и слабого электролита
ацетилхлорида. Эти значения представлены в табл. 17.
Таблица 17. Значения* смнн, Лмин и Ло для растворов в хлористом водороде *2
III. СВОЙСТВА РАСТВОРОВ В ГАЛОГЕНОВОДОРОДАХ
87
растворенное
вещество
(CH3JS
(CH3KN -
CI-I3KNHBC14
CH3COCI
(а)
0,01%
0,0064
0,01538
0,0625
смин
(б)
0,0197
0,0069
0,0133
0,114
0,020
0,0072
0,01514
0,0603
К
(а)
0,85
0,85
0,79
0,00485
ин
0,76
0,87
0,80
0,00516
Ло
(а)
3,65
1,90
2,50
0,0151
* Определены по графикам: (а) Д—с It; (б) \сЧ'—с; (в) lg Л—Ige-
Галогеноводороды, за исключением HF, имеют низкие диэлектри-
диэлектрические проницаемости. Фуосс и Краус 6'32 изучили изменение
электропроводности с концентрацией в галогеноводородах с низкой е
в зависимости от величины диэлектрической проницаемости и обна-
обнаружили, что с увеличением е растворителя минимум кривой lg Л —lg с
сдвигается в область более высоких концентраций. В водных раство-
растворах этот минимум находится в области концентраций, слишком
высоких для наблюдений.
Изменение электропроводности с концентрацией в растворах
хлористого водорода очень похоже на наблюдавшееся Фуоссом и
Краусом изменение электропроводности в растворителях с примерно
такой же диэлектрической проницаемостью (при е = 9,0,смин= 0,025,
Лмин = 5,6, Ло = 40). Чтобы объяснить данные, представленные в
табл. 17, было принято, что возможно два вида ионизации для
ионной пары А+ В":
B)
C)
D)
Ионизация по типу B) происходит в очень разбавленных растворах,
а по типу C) и D) — в более концентрированных. Этими тремя
уравнениями может быть описана ионизация сильных электролитов
в растворах галогеноводородов 42. Можно рассчитать и константы
диссоциации К2, К3 и /С4. Изменение электропроводности растворов
слабых электролитов с концентрацией более сложно и не может
быть так легко объяснено.
При дальнейшем обсуждении для удобства представим иониза-
ионизацию всех солей, проходящей по типу B), хотя, по-видимому, имеется
и ионизация по типам C) и D).
кон-
ти-
ти2 3
12 Л
в -
-
4 -
\ >v^ S if
'*--0w_ 6 Л
1 1 1
! \/
\! 8 -
\-o—<^-°—°
]
- 3
-2
Рис. 22. Кривые
дуктометрического
трования в жидком хло-
хлористом водороде:
1 — 0,37MPh2C=CH2 + BCl3;
2 — 0,75 М Ph3N + ВС13;
3 — 0,29 M(CH3JS + ВС13;
4 — 0,26M(CH3JS + -| PF5;
5 — 0,33 М Ph3As +-|. РРБ;
6 — 0.30М Ph3As+ ВС13; 7—
0,22 М Ph3PO + ВС13; 8 —
0,21 М Ph3PO+ -§- PPS.
О
0 7 2 3 0 J 2 3
W: M
За реакциями, протекающими с изменением видов ионов в рас-
растворе, можно проследить кондуктометрически, например, если это
кислотно-основная реакция 58
или окислительно-восстановительная реакция
РС13+С]2 + НС1 —*¦ PClJHGlj F)
На рис. 22 показано несколько графиков * кондуктометрического
титрования оснований в НС1 сольвокислотами трихлорида бора
или пятифтористого фосфора.
Пятифтористый фосфор действует как источник кислоты HPF6;
в результате реакции с основанием образуются только гексафтор-
* Здесь и на последующих рисунках этой главы по оси абсцисс отложено
мольное отношение кислоты А к основанию В. — Прим. ред.
88 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
III. СВОЙСТВА РАСТВОРОВ В ГАЛОГЕНОВОДОРОДАХ
89
фосфаты. Трихлорид бора может реагировать с основаниями, кото-
которые легко отдают ион хлора, как, например, хлорид тетраметил-
аммония.
При кондуктометрическом титровании возможно протекание
реакций четырех различных типов:
1) с образованием соли
Ph3N + BCl3; (CH3JS+PF5; Ph3PO + PF5
2) с образованием аддукта
(CH3JS+BCl3
3) с ионизацией
Ph2C=CH2-f BCl3
4) с смешением указанных выше типов
Ph3PO + BCl3
По форме кривых кондуктометрического титрования часто
можно определить, какая из четырех типов реакций протекает.
При образовании соли и аддукта электропроводность по мере до-
добавления кислоты неуклонно понижается до эквивалентной точки;
она достигает очень низкого значения, когда в растворе образуется
аддукт (см. рис. 22, 3)\ в случае образования соли электропровод-
электропроводность несколько выше (см. рис. 22, 2, 4). Это хорошо иллюстриру-
иллюстрируется значениями начальной мольной электропроводности и электро-
электропроводности в эквивалентной точке Лэкв при различных титрова-
титрованиях, приведенными в табл. 18.
Когда Лэкв меньше 0,1 см1!(ом -моль), можно допустить, что в рас-
растворе образовался аддукт, если, конечно, во время титрования не
Таблица 18. Начальная мольная электропроводность
и электропроводность в эквивалентных точках
при кондуктометрическом титровании растворов в НС1
Основание
Ph3N
Ph3As
(CH3JS
Ph2C=CHo
PhC=CH
(CH3JO
PhCN
Ph2SO4
Кислота
BC13
BC13
PFr,
BC13
PF5
PC13
РС1з
PC13
BC13
BCI3
Лв
12,6
39,5
40,3
10,05
7,8
1,77
0,72
0,43
1,43
6,65
Лэкв
7,8
20,4
27,8
0,033
4,1
23,8
0,68
0,012
0,0052
1,60
Лв/Лэкв
1,62
1,94
1,45
304,6
1,90
0,074
1,06
25,8
27,5
4,15
Продукт
в растворе
Соль 38
Соль 38
Соль 43
Аддукт 38
Соль 43
Соль 39
Соль 39
Аддукт 38
Аддукт 39
Соль «
образовался осадок. Используя этот метод, можно обнаружить
в растворе образование солей, которые неустойчивы при комнатной
температуре. Например, трихлорид бора, несомненно, реагирует
с трифениларсином с образованием соли Ph3AsH+BClI, но при ком-
комнатной температуре удалось выделить только аддукт PhgAsBCl3.
Стоит отметить, что кондуктометрпческая кривая вблизи точки
эквивалентности обычно плавная, если образуется аддукт, но имеет
резкий перелом, если образуется соль.
Если во время титрования происходит ионизация, электропро-
электропроводность возрастает по мере добавления кислоты (см. рис. 22, Т),
затем понижается, вероятно, вследствие того, что соль более устой-
устойчива к ионизации, чем свободное основание. Иногда нельзя отнести
кривую ни к одному из указанных типов, как, например, при реак-
реакции окиси трифенилфосфина с трихлоридом бора (см. рис. 22, 7);
в этой реакции, вероятно, происходит и ионизация, и образование
соли.
Б. Спектроскопия
Спектры растворов галогеноводородов изучены очень мало.
Это объясняется тем, что для проведения эксперимента приходится
применять низкие температуры и весьма высокие давления. Были
изучены ЯМР-спектры некоторых растворов в НС1 при комнатной
температуре с использованием трубки под давлением. Они под-
подтвердили присутствие ионов (PhJC+(CH3) и PhC2+(CH3) в растворах
несимметричного дифенилэтилена и фенилацетилена в жидком хло-
хлористом водороде.
ИК-, УФ- и спектры комбинационного рассеяния растворов
в жидких галогеноводородах не были изучены ни при комнатной,
ни при низких температурах. В некоторых случаях спектры, как,
например, УФ-спектры растворов ароматических олефинов, могли
бы подтвердить предполагаемый тип ионизации 39:
Ph2C=CH2 + 2HCl
Ph2C+(CH3) + HClj
G)
— электропроводность своСодного основания в НС], см2/(ом¦ моль).
Спектрометрические измерения широко применялись при изучении
растворов во фтористом водороде, где можно проводить измерения
почти при комнатной температуре (см. гл. вторую, III, Г).
Спектры комбинационного рассеяния были использованы при
изучении некоторых продуктов присоединения хлористого водо-
водорода. Спектры соединений СН3СОМН2-НС1 и (CH3CONH2J-HC1
показывают, что первый продукт присоединения, возможно, отве-
отвечает структуре СН3СОМЩС1~; спектр гемигидрохлорида трудно объ-
объяснить 29. Спектры соединений диметилового эфира с хлористым во-
водородом (СН3JО-НС1, (СЦзJО-ЗНС1 и (СН3JО-4НС1 показывают,
что последние два соединения являются ионными, тогда как
90 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
моногидрохлорид имеет структуру с водородными связями. Соеди-
Соединения (CH3JO-DCl и (СН3JО-НВг имеют ионную структуру с
протонизированным кислородом 54.
В. Фазовые диаграммы
Изучение фазовых диаграмм, в которых одним из компонентов'
является галогеноводород, не раскрывает полностью природы рас-
растворов, но дает информацию об образовании некоторых возможных
соединений. Фазовые диаграммы воды и хлористого водорода пока-
показали, что образуются три соединения 45: НС1-ЗН2О (tnjl = —24,4° С),
НСЬ2Н2О AПЛ = -17,7° С) и НС1-Н2О (tnn = -15,4° С). Послед-
Последнее соединение нерастворимо и образуется, когда в жидком хлори-
хлористом водороде присутствует небольшое количество воды 58. У него
ионная кристаллическая структура (НаО+СГ), каждый атом водо-
водорода соединен водородными связями с ближайшим атомом хлора в7.
Фазовые диаграммы сольвокислот и сольвооснований могут
дать ценную информацию о природе сольватированных протона и
галоген-ионов. В системе ВС13—НС1 не обнаружено образования
каких-либо соединений 15, из чего можно заключить, что трихлорид
бора должен быть очень слабой сольвокислотой в хлористом водо-
водороде. Фазовая диаграмма НС1—С12 показывает, что образуются со-
соединения 2НС1-С1, (ta]1 = -12ГС) и НС1-С12 (tm = -115° С) в5.
Однако нет доказательств того, что растворы хлора в хлористом водо-
водороде являются кислыми, хотя они могут действовать как окислители
(Дж. А. Зальтхауз, Т. Ваддингтон, неопубликованные результаты).
Были изучены диаграммы соединений, которые являются соль-
вооснованиями в хлористом или бромистом водороде 33. В системе
этиловый эфир — хлористый водород обнаружены соединения
(С2Н5JО-НС1 (/ПЛ = -92°С), (С2Н5JО-2НС1 (*пл = -88° С),
(С2Н5JО-5НС1 (tnn = —89° С) 33. Согласно этим данным, по-види-
по-видимому, разумно предположить, что эфир акцептирует протон, обра-
образуя (С2Н5JОН+, а ион хлора сольватируется, образуя СЦНО)*,
где х = 0, 1 или 4.
Совсем недавно были изучены фазовые диаграммы соединений
с я-связями в хлористом водороде. Было найдено, что некоторые
алифатические олефины и ацетилены образуют с НС1 соединения
составов 1:1;2:1и4:1; все они имеют низкую точку плавления.
Аналогичные результаты были получены для аддуктов 1 : 1 и 2 : 1
(НС1 : Аг =углеводород) 3.
Для ацетонитрила, как системы с я-связями 37, в хлористом
водороде были обнаружены соединения CH3CN -НС1 (tnJI = —63,2° С),
2CH3CN-3HC1 (*ПЛ = -88°С), CH3CN-5HC1 (*пл = -123,6° С) и
CH3CN-7HC1 (tnjI =— 125,0° С), которые можно представить как
CH3CNH+C1(HC1)^. ИК-спектры CH3CN-2HC1 указывают, что
это соединение ионное, т. е. CH3C=NH+HCli28. Соединения
III. СВОЙСТВА РАСТВОРОВ В ГАЛОГЕНОВОДОРОДАХ
91
«-(C4H9LNC1(HC1J, H-(C4H9LNBr(HClJ и н-(С4Н9LМ(НС1).2 были
недавно выделены при низкой температуре. Полные фазовые диа-
л а
граммы их не изучены
46
Г. Криоскопия и эбулиоскопия
Для растворов некоторых веществ в хлористом, бромистом,
и йодистом водороде были проведены криоскопические 1 и эбулио-
скопические 50>5г измерения, однако результаты этих измерений
чрезвычайно трудно объяснить вследствие сложной схемы иониза-
ионизации. Так, Бекман и Уинтиг х заключили, что в растворе образуются
какие-то ассоциаты. Трудностей в интерпретации результатов
для некоторых веществ можно избежать, если сопоставить резуль-
результаты криоскопических и эбулиоскопических измерений с данными
об изменевгии электропроводности при разбавлении.
Д. Препаративные методы
Благодаря низкой температуре кипения и легкости испарения
жидкие галогеноводороды как растворители являются хорошей
средой для приготовления некоторых препаратов. Используя гало-
галогеноводороды и соответствующие тригалогеннды бора, легко при-
приготовить тетрагалогенидбораты 57~61. Особенно легко получаются
тетрахлорбораты (избыток растворителя и ВС13 может быть удален
перегонкой при температуре около —80° С). Гексахлордибораты
могут быть приготовлены из В2С14 таким же образом 24. Этот метод
применим при реакциях между растворителем и веществом, если
избыток одного из них может быть удален с растворителем или от-
отгонкой в высоком вакууме. Если оба реагента нелетучи, нужно
быть особенно осторожным при интерпретации результатов, так как
продукт реакции может быть эквимолярной смесью исходных ре-
реагентов. Большинство соединений, полученных в жидких галогено-
водородах, было выделено в конце кондуктометрических титрований.
Хотя полученные количества могли быть порядка миллпмоля, этого
было достаточно для анализа и исследования необходимых характе-
характеристик. Используя некоторые реакционные ячейки, возможно изучать
образование продуктов реакции по наблюдаемому увеличению веса.
Реакции сольволиза, где один продукт летучий, могут быть изучены
аналогично. Однако если один из продуктов нерастворим, а другой —
растворим, то растворенное вещество может быть удалено декан-
декантацией в обращенную Y-образную ячейку и растворитель затем
удален испарением 38. Реакции сольволиза — не лучший метод
получения хлористых, бромистых и йодистых соединений, так как
чаще они могут быть получены другими более легкими способами.
Этот метод хорош для получения небольших количеств нитрилхло-
рида высокой степени чистоты. В качестве исходных продуктов
берут пятиокись азота и жидкий хлористый водород.
92 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
IV. РЕАКЦИИ В ЖИДКИХ ГАЛОГЕНОВОДОРОДАХ
Гор 14 показал, что многие металлы, неметаллы и простые неорга-
неорганические вещества нерастворимы в жидком хлористом водороде.
Однако он наблюдал некоторые реакции сольволиза, так как, по его
сообщению, отдельные вещества, например сульфид кадмия, стано-
становились белыми, и полученное твердое вещество не содержало серы.
Растворимость большого числа неорганических веществ была изу-
изучена Стилом и др.50'51. Авторы обнаружили, что ионные галогениды,
например галогениды щелочных металлов, нерастворимы в хлори-
хлористом водороде, а хлориды с ковалентной связью, такие, как хлорид
олова и оксихлорид фосфора, растворимы. После того как была уста-
установлена сильная кислотность4 и низкая диэлектрическая проница-
проницаемость галогеноводородов, последующим исследователям стало легче
находить вещества, растворяющиеся в этих растворителях.
А. Кислотно-основные реакции
Галогеноводороды являются растворителями с сильными кис-
кислотными свойствами. Их кислотность возрастает в ряду HF <^ НС1 <^
< HBr < HI. Следовательно, очень трудно найти какие-либо ве-
вещества, которые ведут себя как кислоты, особенно в хлористом,
бромистом и йодистом водороде. Наоборот, существует очень много
веществ, являющихся основаниями в галогеноводородах; некото-
некоторые из них являются сильными электролитами.
1. Основания
Основные растворы в галогеноводородах можно получить двумя
различными способами:
а) из солей, которые легко ионизируются, образуя свободный
галоген-ион, сольватированный в растворе, НГ?; типичными при-
примерами таких соединений являются галогениды тетраалкиламмо-
ния R4№T;
б) из соединений, содержащих атомы с неподеленной парой
электронов или я-связь н легко акцептирующих протон, например
трифениламина в жидком хлористом водороде 38. Электропроводность
растворов оснований в хлористом водороде приведена в табл. 16 и 19.
а. Соли как доноры галоген-ионов. Солей, которые ведут себя как
основания в галогеноводородах, очень немного, так как они должны
быть ионными в твердом состоянии при комнатной температуре,
иметь большой катион и низкую энергию кристаллической решетки
вследствие низких значений диэлектрических проницаемостей гало-
галогеноводородов. Этим требованиям удовлетворяют галогениды эле-
элементов V группы зо,58.бо,б2 типа rjmr+Г-.
IV. РЕАКЦИИ В ГАЛОГЕНОВОДОРОДАХ
93
Таблица 19. Электропроводность
Растворенное
вещество
Ph3CCl
Ph3SiCl
Ph3As
AsH,
H2S
(CH3JO
(CH3JS
Ph2C=CH2
CHgCN
(CH3JCO
СНзСОЫ
Ph2SO
COC12
некоторых
в хлористом водороде 38 40 при —95
с, моль/л
0,25
Насыщенный раствор
0,20
Насыщенный раствор
Насыщенный раствор
0,26
0,35
0,37
0,42
0,41
0,30
Насыщенный раствор
0,39
У.
мком~1 • см~
10 640
1,53
7700
26,3
2,8 -
114,4
3950
650
1220
4300
2,9
1500
0,27
основании
°С
¦
Л, см'/(ом-моль)
42,9
—
39,3
—
—
0,43
11,3
1,77
2,94
10,4
0,0097
—
0,0007
Основные растворы получаются также при растворении пента-
хлорида и пентафторида фосфора в соответствующем раствори-
растворителе:
РС15(тв)+2НС1 z=? PClf + HClj
(8)
после реакций
Такие растворы являются источниками ионов PFJ;
с сольвокислотой образуются соответствующие соли.
Хотя пентахлорид фосфора существует в твердом состоянии как PClJPCle,
невозможно было установить существование иона РС1^ в жидком хлористом
водороде58; действительно, имеется удивительно мало доказательств наличия
иона РС1^ в солях с обычными катионами, несмотря на то, что известны гекса-
хлорфосфаты катионов в С.2Н3СС12- РС1? 68 и (C13P=N— (PC^N)*—PC13)+5.
Галогениды углерода, в которых связь С—Г легко разрушается
с образованием устойчивого карбониевого иона, также ведут- себя
как основания в НГ. Ион Ph3C+ может быть легко получен из три-
фенилметилхлорида в хлористом водороде 38 и, вероятно, в йоди-
йодистом водороде 30. Растворы с этим ионом ярко окрашены, что, по-
видимому, обусловлено именно Ph3C+, а не HFJ, так как большин-
большинство других растворов оснований бесцветны. Попытки получить
ион Ph3M+ (M=Si, Ge, Sn, Pb) растворением хлоридов Ph3MCl в
хлористом водороде не удались 38; в соединениях с оловом и свин-
свинцом наблюдалось частичное замещение фенильной группы галогеном.
б. Химические соединения с элементами IV, V и VI групп как
акцепторы протона. Изучать реакции протонизации этих соединений
в жидком хлористом водороде легче, чем в серной кислоте, так как
нет нежелательных побочных реакций сульфирования 9.
Трифенил- и триметиламины являются сильными основаниями
в жидком хлористом водороде
38,39
Свежеполученный препарат
94 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
трифениламина при растворении в НС1 образует бледно-коричне-
бледно-коричневый раствор, но при использовании препарата, выдержанного в те-
течение шести месяцев, а затем сублимированного в вакууме, раствор
был голубым; это изменение цвета, по-видимому, зависит не от кон-
концентрации, а является следствием образования иона Ph3NH+, так как
некоторые ионы такого типа, как известно, имеют голубой цвет 38.
Возможна протонизация азота в гидразобензоле в хлористом водо-
водороде, но повторное присоединение протона приводит к образованию
бензидина 38. Пиридин действует как превосходное основание во всех
трех растворителях 58'60>61 при добавлении ВГ3 образуются тетра-
галогенбораты пиридина.
Атом фосфора в фосфине и трифенилфосфине при растворении
в НГ также легко прот;онизируется 30>38. При растворении фосфина
в НС1 и НВг образуется ион фосфония, при взаимодействии с гало-
генидами бора получаются соответствующие тетрагалогенбораты
фосфония 58>61. Однако получить таким же образом ионы дифосфо-
ния при растворении дифосфина не удалось из-за неустойчивости
этих ионов 38. Трифениларсин легко присоединяет протон в хлористом
водороде
Ph3As + 2HCl > PhgAsH+HCla (9)
но получить такую же протонизированную форму при комнатной
температуре при реакции с ВС13 в виде тетрахлорбората нельзя из-за
образования аддукта
Ph3AsH+ + ВС13
Ph3AsH +
Ph3As • BGI3
A0)
Попытки использовать арсин AsH3 для получения арсониевых солей
мышьяка были безуспешными, так как арсин реагирует с раствори-
растворителем. Соединения Ph3Sb и Ph3Bi не образуют устойчивых прото-
визированных продуктов в жидком хлористом водороде; они реа-
реагируют с растворителем и частично хлорируются 38. '
Были исследованы 38 основные свойства различных производных
кислорода и серы типа ROR' и RSR' в жидком НС1. Более ранние
исследования показывают, что можно обнаружить протонизирован-
ные формы спиртов и эфиров в НС1, НВг, HI и что некоторые из
этих растворов хорошо проводят ток 63. Вода и сероводород нерас-
нерастворимы в жидком хлористом водороде 38> 58, но растворяются,
когда один или оба атома водорода в них замещены на метильную
или фенильную группу. Диметилсульфид, судя по электропровод-
электропроводности, — одно из самых сильных оснований в галогеноводородах
(см. табл. 19).
Кондуктометрическое титрование этих оснований трихлоридом
бора в НС1 не дает много полезной информации, за исключением
IV. РЕАКЦИИ В ГАЛОГЕНОВОДОРОДАХ
95
случаев титрования диметилового эфира и сульфида, когда в растворе
образуется аддукт (см. рис. 22, 3). После реакции с трихлоридом
бора из этих растворов нельзя выделить какие-либо протонизиро-
ванные по кислороду или сере соединения. Неустойчивый протони-
зированный диметилсульфид может быть выделен 43 после реакции
с PF5 в НС1.
в. Химические соединения с п-связъю как акцепторы протона.
Ароматические олефины, такие, как 1,1-дифенилэтилен, легко про-
тонизируются с образованием иона карбония:
Ph2G=GH2
Такие реакции наблюда-
наблюдаются в HC1hbH2SO4 13>39.
Кондуктометрическое ти-
титрование 1,1-дифенилэти-
лена с ВС13 в жидком хло-
хлористом водороде указы-
указывает на интересное явле-
явление ионизации, происхо-
происходящее по мере присоеди-
присоединения кислоты (рис. 23):
острый пик конца титро-
титрования соответствует моль-
мольному отношению [ВС13] :
:[Ph2 = CH2], равному 1:1.
В подобных опытах со
стиролом и а-метилстиро-
Ph2G (CH3) +
A1)
1,5
[вС1з]:[РЬ2С=СН2] [вС1д]:[РЬС=Сн]
Рис. 23. Кривые кондуктометрического ти-
титрования Ph2G=CH2 и PhC=CH трихлори-
трихлоридом бора в жидком хлористом водороде.
лом ионизация является доказательством образования иона кар-
карбония. Если по кривой кондуктометрического титрования трудно
определить тип реакции, происходящей в растворе, основы-
основываются главным образом на окрашивании растворов. По цвету рас-
раствора можно судить об устойчивости ионов карбония, которые
должны образоваться: (СН3KС+, PhCH3HC+, Ph(CH3J C+<Ph2CH3C+
<Ph3O.
Ион карбония с двойным зарядом [С6(СН3M]С1С2+ был обнаружен
в растворе пентаметилтрихлорметилбензола в серной кислоте 21.
Кондуктометрическое титрование раствора фенилацетилена трихло-
трихлоридом бора в хлористом водороде показало, что образуется двух-
зарядный карбониевый ион PhCH3C2+ 39, причем точка конца титро-
титрования находилась при мольном отношении [ВС13] : [PhC=sCH],
равном 2 :1 (см. рис. 23):
PhC=CH + 4HCl —> PhCH3G2+ + 2HClj A2)
РЬСН3С2+ + 2НС17+2ВС1з —> PhGH3G2++2BClj-+2HGl A3)
,9,3 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
При растворении дифенилацетилена в НС1 образуется очень сла-
слабый основной раствор и нет достаточных доказательств существо-
существования иона PhC2+ -CH2Ph.
Нитрилы содержат тройную связь, которая легко протонизи-
руется, и со всеми галогеноводородами можно приготовить соедине-
соединения типа CH3CN-2Hr. ИК-спектры этих соединений указывают28
на наличие двух различных структур в НС1 и HBr : CHgC=]NH+HCl?
и CH3BrC=NH2Br". Ацето- и бензонитрилы являются умеренно
сильными основаниями 39 в жидком НС1. В реакциях всех этих ос-
основных растворов в НГ с трихлоридом бора не было возможным
выделить тетрахлорбораты и таким образом провести дальнейшее
исследование структуры катиона. Аддукт был выделен лишь с ни-
нитрилами смолистых продуктов. Невозможно сказать, образовался ли
ацетонитриловый аддукт в ходе титрования или при последующем
нагревании раствора до комнатной температуры, так как происхо-
происходило осаждение во время титрования. В реакции ацетонитрила с пен-
тафторидом фосфора не удалось выделить устойчивую соль или
аддукт 43.
Азосоединения также содержат л-связь, которая должна присое-
присоединять протон. После реакций азобензола и ж-азотолуола в НС1
с ВС13 были выделены устойчивые тетрахлорбораты 39. Положение
протона нельзя определить из ИК-спектррв, но измерения 66 $Ка
и УФ-спектры протонизированных азосоединений 8 наводят на мысль,
что протон локализован на связи. Азоксибензол и нитрозобензол
являются умеренно сильными основаниями в жидком хлористом
водороде. Можно выделить аддукт азоксибензола с трихлоридом
бором, структура которого точно не известна, как и место первона-
первоначального присоединения протона. Нитрозобензол в НС1 реагирует
с ВС13 с образованием соединения, которое взрывается уже при тем-
температуре ниже комнатной, но из этого раствора может быть выделен
гидрохлорид неизвестной структуры 39.
г. Присоединение протона соединениями, содержащими кислород
с двойной связью. Карбонил-, нитро-, фосфорил-, сульфо-группы
имеют по крайней мере один атом кислорода, связанный с централь-
центральным атомом двойной связью. Обычно считают, что эта связь поляр-
полярная, особенно в карбонильной группе
;с+ -о"
и кислородный атом должен легко присоединять протон. В жидком
хлористом водороде было изучено довольно большое число органи-
органических соединений, содержащих эти группы. Электропроводность
растворов таких соединений представлена в табл. 20.
Из данных этой таблицы видно, что большинство карбонильных
соединений — сильные основания, за исключением альдегидов.
IV. РЕАКЦИИ В ГАЛОГЕНОВОДОРОДАХ
97
Таблица 20. Электропроводность растворов некоторых соединений,
содержащих кислород с двойной связью,
В ЖИДКОМ ХЛОРИСТОМ ВОДОрОДе 39,40
Соединение
СИзСНО
PhCHO
(СН3JСО
Ph2CO
CH3CONH2
PhCONH2
PhCO2C2H5
CH3NO2
PhNOa
(CH3JSO
Ph2SO
(CH3JSO2
Ph2SO2
PhgPO
с, моль/л
Насыщенный раствор
Насыщенных"! раствор
0,41
0,25
Насыщенный раствор
0,23
0,33
Насыщенный раствор
0,31
Насыщенный раствор
Насыщенный раствор
0,23
0,21
0,22
%, МК0М~1 • СЛГ~ *
1,60
1,81
4300
1810
1640
800
3200
1,3
490
1200
1500
47
110
2200
Л, см'J(ом-моль)
—
10,4
7,15
—
3,48
9,74
—
0,16
—
—
0,20
0,51
9,61
У амидов и эфиров присоединение протона к карбонильному
кислороду может не произойти, например известно 49 , что ацетамид
в хлористом водороде CH3CONH3Cr протонизирован по азоту.
Реакции большинства карбонильных соединений с трихлоридом
бора весьма сложны. Так, образовавшийся аддукт бензальдегида
3PhCHO-2BCl3 подвергается затем пиролизу и превращается в
бензилиденхлорид 7. Соединения, содержащие карбонильные группы,
всегда образуют с ВС13 в жидком НС1 аддукты, а не тетрахлорбо-
тетрахлорбораты 39. Хлористоводородная окись триметиламина (CH3KNOH+C1~
и окись трифенилфосфина действуют как сильные сольвооснования.
Из раствора окиси трифенилфосфина в НС1 может быть выделен
гидрохлорид, а после реакции с пентафторидом фосфора — гекса-
фторфосфат. С трихлоридом бора получается смесь тетрахлорбората
и аддукта. Протонизированная форма окиси триметиламина не со-
сохраняется в реакции с ВС13; в этом случае образуется аддукт. Ана-
Аналогичные реакции наблюдаются для растворов днметил- и дифенил-
сульфоксида в жидком хлористом водороде; раствор получается
умеренно основным, но после реакции с трихлоридом бора образуется
аддукт. С пентафторидом фосфора получается гексафторфосфат
диметилсульфоксида 40> 43.
Основные свойства нитросоединений и сульфонов выражены
значительно слабее, чем основные свойства соединений, содержа-
содержащих только один атом кислорода с двойной связью. Это, вероятно,
нроисходит из-за существования резонансных форм, эффективно
7 заказ 90
ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
уменьшающих заряд на атомах кислорода, например, у окиси три-
метиламина и нитрометана:
СНз—N ->О<
сн/
СНз-N-CT и CH3-N<
/0-
^0
СНз—
yO
Хлорангидриды кислот, например хлористый бензоил, в жидком:
НС1 могут либо присоединять протон к карбонильному кислороду,
либо отдавать ион хлора:-
A5)
PhC0Cl+2HCl ^ir
PhCOCl + HCl ^=Г
Экспериментальные данные наводят на мысль, что соединение иони-
ионизируется по уравнению A5). Если бы происходила ионизация по
уравнению A6), то должен-^был бы произойти сольволиз бензойной
кислоты
РЬСО2Н + ЗНС1=РЬСО+НС1г + Н3О+СГ A7)
какового в действительности не наблюдается 39.
Еще одним доказательством, подтверждающим этот механизм,
являются реакции дифенилфосфорилхлорида Ph2POCl в хлористом
водороде. Если это соединение ионизируется с потерей иона хлора,
как показано в уравнении A6), тогда продукт реакции с трихлори-
дом бора должен иметь структуру Рп2РО+ВСЦ. Но все данные под-
подтверждают формулу PhaPClO-BCl", т. е. первоначально образуется
тетрахлорборат, который при удалении растворителя теряет моле-
молекулу НС1:
РЬ2РСЮН+-НС1а+ВС1з
A8)
РЬ2РС1О-ВС13
Попытки приготовить ион РС130Н+ при взаимодействии сильных
аквакислот с хлорокисью фосфора в хлористом водороде также
оказались неудачными 40.
Большинство таких хлорсодержащих соединений в жидком НС1
являются слабыми основаниями, за исключением дифенилфосфо-
дифенилфосфорилхлорида; с ВС13 они либо образуют аддукты, часто неустойчи-
неустойчивые, либо не образуют соединений 40. Некоторые реакции хлористого
нитрозила в НС1 можно объяснить, предположив, что растворенное
вещество не ионизируется 59.
При рассмотрении мольной электропроводности хлористоводо-
хлористоводородных растворов соединений обнаружена интересная зависимость
между электропроводностью основания и отрицательным индук-
индуктивным влиянием лигандов, присоединенных к атому, который про-
IV. РЕАКЦИИ В ГАЛОГЕНОВОДОРОДАХ
99
тонизируется. Приводим зависимость константы ионизации для за-
замещенных уксусной кислоты ХСООН от лиганда X 40:
X СНз Н Ph ОСН3 I Br Cl F
К-105 1,34 2,1 5,6 33,5 75 138 155 217
Изменение мольной электропроводности растворов в НС1 в зави-
зависимости от лиганда 40:
Соединение СН3ОСН3 CH3OH CH3OPh PhOH PhOPh
А, см*/(ом-моль) . . . 0,43 0,13 0,021 0,019 0,0017
Соединение CH3SCH3 CH3SH PhSH PhSPh
А, см*/(ом-моль) ... 11,8 0,054 0,030 0,15
Соединение X2SO2 X3PO PhCOX
где X СН3 Ph Ph Cl F Ph Cl OC2H5
А, см*I(ом-моль) . . . 0,20 0,51 9,61 0,20 0,015 7,15 0,046 0,74
Если основность, определяемая мольной электропроводностью '
при сравнительном разбавлении, обусловлена отрицательным за-
зарядом на кислороде или сере, то должна быть прямая связь с отри-
отрицательным индуктивным влиянием, измеренным при ионизации
в воде соответствующей замещенной уксусной кислоты. Есть не-
несколько исключений, как показывают приведенные выше данные.
Это этилбензоат, где может также произойти протонизация кисло-
кислорода этокси-группы, и дифенилсульф,ид, сульфоны и дифенилфосфо-
рилхлорид, где возможна также протонизация ароматического ядра.
Протонизация ароматического ядра приводит к возникновению
сильно окрашенных соединений; такие растворы в жидком НС1
были получены с анизолом (бледно-розовый) и дифенилсульфидом
(пурпурный) 38, окисью трифенилфосфина (бледно-желтый) и дифенил-
фосфорилхлоридом (желтый) 40. Мольная электропроводность фос-
форилхлорида в ряду соединений Х3РО лежит между электропровод-
электропроводностью окиси трифенилфосфина и фосфорилфторида. Таким образом
подтверждается, что ионизация, вероятно, протекает через, присое-
присоединение протона, а не потерю иона хлора.
2. Кислоты
Имеются два типа кислот в растворах галогеноводородов, как
это следует из двух различных, но дополняющих друг друга опреде-
определений кислот. Это или акцепторы галоген-ионов, например галогенид-
ные соединения бора, или доноры протонов, как, например, сильные
ак'вакислоты. Данные об электропроводности растворов кислот
в НГ приведены в табл. 16 и 21.
а. Химические соединения с элементами III группы как акцепторы
галоген-ионов. Соединения бора хорошо известны как- акцепторы
100 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
Таблица 21. Электропроводность растворов кислот в
Раствори-
Растворитель
HCI
НВг
Растворенное
вещество
CH3SO3H
CF3SO3H
CISO3H
SiF4
PF5
SO2
Р4ОЮ
BCI3
BF3
Al2Bre
SnBr4
с, моль}л
Насыщенный раствор
Насыщенный раствор
0,38
0,13
0,25
Насыщенный раствор
Насыщенный раствор
0,32
1,43
Насыщенный раствор
0,064
мком~1 -см~1
9,0
2,0
2,7
0,98
1,9
0,15
0,82
0,13
0,09
0,073
0,024
галогеноводородах
Л,
см* / (ом • моль)
—
7,0-10
7,7-Ю
7,5 • 10
—
4,0 • 10
0,6 • 10
—
3,8 • 10" 4
Лите-
Литература
40
40
58
43
43
40
40
58
58
60
60
- 7 т-
галоген-ионов; тригалогениды бора являются сольвокислотами в со-
соответствующих галогеноводородах:
ВГ3 + 2НГ ;=Г Н2Г + ВГГ A9)
Степень ионизации незначи-
незначительна; фазовая диаграмма
раствора ВС13 в НС1 показы-
показывает, что в этой системе не об-
образуется 15 никаких соединений.
Тем не менее очень легко
приготовить тетрагалогенбо-
раты из основания и тригало-
генида бора в соответствующем
растворителе 57> 58> eo> ei. При
интерпретации реакции трихло-
рида бора в качестве сольво-
кислоты с основанием, образу-'
ющимся в результате протони-
зации в растворителе, могут
встретиться некоторые трудно-
трудности вследствие образования аддукта. Например, с диметилсульф-
оксидом 40 протекает реакция
Рис. 24. Кривые кондуктометрического
титрования галогедидов бора (А) хло-
хлоридом тетраметиламмония (В) в жидком
хлористом водороде:
1 — (CH3LNC1 + BCh; 2 — (CH3)<NC1 + BF3.
(CH3JSOH+HClj-fBCl3 > (СНзJ SOH+BCli"
I
(CH3JSO-BC13
B0)
Однако во многих случаях на кривых кондуктометрического титро-
титрования обнаруживаются резкие перегибы в конечных точках (рис 24).
IV. РЕАКЦИИ В ГАЛОГЕНОВОДОРОДАХ
101
Так, при использовании тетрахлордиборана в качестве сольво-
кислоты в НС1 образуются соли гексахлордиборной кислоты Н2В2С1е4:
2(CH3LN+HClj + B.2Cl4 —у [(CH3LN+]2B2C1|-+2HC1 B1)
Перегиб наблюдается при мольном отношении основания к B2Cl4r
равном 2:1. Акцепторные свойства других соединений бора в жид-
Рис. 25. Кривые кондук-
кондуктометрического титрова-
титрования в жидком броми-
бромистом водороде:
1 — SnBr. + РуНВг; 2 —
SnBr, + РОВг3; 3 — ВВГ„ +
+ (CH3LNBr; 4—ВВГ3 +
+ РуНВг.
0 05 1,0 1,5 2,0 О 0,5 1,0 1,5 2,0
И: Н
ком хлористом водороде оказываются до некоторой степени ограни-
ограниченными. При растворении трифторида бора в хлористом водороде
получаются соли трифторхлорборной ки-
кислоты HBF3C1; некоторые из солей выде-
выделены и уже изучены 57> 58. Этот метод
является единственным известным мето-
методом приготовления таких солей. Кондук-
тометрические кривые растворов BF3 так-
также имеют резко выраженные точки
конца титрования при мольном отношении
компонентов 1 : 1 (см. рис. 24). Однако
диборан, триэтилбор, трифенилбор и ди-
метилборхлорид не обладают кислотными
свойствами в хлористом водороде; очень
слабой сольвокислотой является метил-
И : И
д
водороде (В):
i — Gecv, 2 — Snci4.
Рис. 26. Кривые кондукто-
кондуктометрического титрования
бордихлорид (М. Е. Пич, Т. Ваддингтон, хлоридов IV груяпы (А)
неопубликованные результаты). В жидком
бромистом водороде соединения типа
BR3Br~ не получены: BF3 но проявлял
кислотных свойств, а ВС13 полностью пере-
перешел в трибромид60'82. Но ВВг3 является кислотой в НВг, и на кривой
кондуктометрического титрования с донорами иона брома наблю-
наблюдается резкий перегиб при мольном отношении 1 : 1 (рис. 25).
б. Химические соединения с элементами IV группы как акцепторы
галоген-ионов. Тетрахлорид германия не проявляет кислотных свойств
102 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
в хлористом водороде 57 (рис. 26), а галогениды олова являются
кислотами в хлористом и бромистом водороде 58> в0> в2. Их можно
обнаружить и выделить при титровании соли гексахлор- п гексабром-
оловянной кислоты.
Были предприняты попытки получить ионы Si F4C1|~ и GeF4Clf~ ,
применяя соответствующие фториды в качестве кислот в хлористом
водороде. Однако тетрафторид кремния не проявлял никакой тенден-
тенденции к акцептированию хлор-иона, а тетрафторид германия хотя
и является кислотой в НС1, но не удалось выделить определенный
продукт вследствие сольволиза 43.
в. Химические соединения с элементами V группы как акцепторы
галоген-ионов. Трихлорид фосфора нейтрален в жидком хлористом
водороде 58: он не проявляет кислотных свойств и не подвергается
¦сольволизу; трифторид мышьяка, наоборот, нацело сольволпзован
в НС1, поэтому его кислотные свойства не могли быть исследованы 43.
Кислотные свойства других трехвалентных хлоридов V группы
не изучены.
Пентахлорид и пентабромид фосфора, как уже указывалось,
являются сольвооснованиями в соответствующих галогеноводо-
родах, и нет доказательств образования иона РГд в растворе 58> 60>ег.
Пентахлорид сурьмы является очень сильным акцептором электро-
электронов и должен быть в состоянии образовать ион SbCl^ с основаниями
в НС1 однако о таких реакциях ничего не сообщалось.
Свойства пентафторидов элементов V группы в хлористом водо-
водороде более полно исследованы. Пентафторид фосфора реагирует
с сильными основаниями и образует гексафторфосфаты:
2(CH3LNCl+3PF5 > 2(CH3LNPF6 + PF3Cl2 B2)
Кривые кондуктометрического титрования имеют резкий перегиб
при мольном отношении основания к PF5, равном 2 : 3 (см. рис. 22);
к сожалению, в этих экспериментах не удалось выделить какую-либо
ковалентную форму дихлортрифторида фосфора (tm = —125° С,
^кип — 7,1° С), так как его содержание в растворе составляло 0,5%
от веса растворителя.
При использовании пентахлорида фосфора в качестве основания
в жидком НС1 были получены ионная и ковалентная формы ди-
дихлортрифторида фосфора:
2PC15+3PF5 > PC1JPFJ + PF8C12 B3)
Нет данных, указывающих, что происходит внутренняя конверсия
этих двух форм PF'3C12-
Пентафторид фосфора был применен в качестве сольвокислоты
в реакциях с основаниями, которые образуют аддукты с ВС13 в гало-
геноводородах, например с диметилсульфоксидом; во всех случаях
в растворе образуется гексафторфосфат, но иногда при нагревании
до комнатной температуры он разлагается. Пентафторид фосфора
IV. РЕАКЦИИ В ГАЛОГЕНОВОДОРОДАХ
103-
является, таким образом, самой сильной кислотой из до сих пор най-
найденных в жидком хлористом водороде, которая не образует аддуктов
с протонизированными основаниями 43.
Пентафторид мышьяка сольволизуется в НС1 до гексафторарсе-
ната тетрахлормышьяка:
2AsFs+4HCl > AsCl4AsFe+4HF B4>
Подобный сольволиз, или частичный сольволиз, наблюдался и с пен-
тафторидом сурьмы, но не представлялась возможность выделить
определенный продукт 43; в реакции нитрозилхлорида и пентафто-
рида сурьмы в НС1 удалось выделить хлорпентафторантимонат
нитрозила, но роль растворителя в этих реакциях неясна 59.
Механизм внешне похожих реакций пентафторида фосфора и
мышьяка в жидком хлористом водороде неизвестен. Различие между
ними состоит в том, что диспропорционирование пентафторида
фосфора происходит только в присутствии сильных оснований и при
этом не образуется свободный фтористый водород:
2MC1 + 3PF5 >- 2MPFe + PF3Cl2 B5>-
Ни одна реакция не приводит к образованию устойчивого иона
MF5C1~, хотя он и может быть промежуточным продуктом, как пока-
показано в схеме:
PF5Cl-+PF5
PF^l+HClj
PF5CI- + HCI
> PFe+PF4Cl
B6),
PFe+PF3Cl2
Аналогичная схема объясняет и реакцию пентафторида мышьяка.-
В ходе реакции образуются свободные ионы фтора, которые тут же
реагируют с растворителем, образуя фтористоводородную кислоту:.
AsF6Cr+HCl
AsF6cr+2HCl
B7).
2AsF3Cl2
AsCIMsFe
Соединения PF4C1, AsF4Cl, AsF3Cl2 неизвестны, но PF3Cl2r
lJPFe и AsClJAsFj известны и являются относительно устой-
устойчивыми 25> 2в. В связи с этим интересно заметить: соединение
SbCl4SbFg известно 26, но нет указаний на то, что оно образуется
в растворе хлористого водорода 43.
г. Химические соединения с элементами VII группы как акцеп-
акцепторы галоген-ионов. Реакции окисления этих соединений будут
104 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
рассматриваться в последующем разделе, а здесь мы обсудим их
свойства как кислот Льюиса в жидких галогеноводородах. Ни хлор
в жидком НС1, ни бром в жидком НВг не выступают в качестве ак-
акцепторов галоген-ионов. Иод слабо растворим в хлористом водороде
и тоже не является акцептором, и нет доказательств существования
таких ионов, как 12С1~ или I3. Однако монохлорид брома и иода
являются акцепторами галоген-ионов в хлористом водороде, и на
кондуктометрических кривых титрования хлоридов тетраалкил-
аммония были получены точки конца титрования при мольном от-
отношении 1:1; были выделены соединения R4NBr2Cl и R4NIC12
(Дж. А. Зальтхауз и Т. Ваддингтон, неопубликованные результаты).
д. Сильные аквакислоты и ангидриды. Оксикислоты, например
HNO3 и Н3РО4, не оказываются достаточно сильными для того,
чтобы титровать сольвокислоты в жидких галогеноводородах *°.
Трифторметан, метан и хлорсерная кислота также не показали
кислотных свойств в жидком хлористом водороде 40' 58. Из раствора
хлорсерной кислоты можно выделить несколько новых хлористых
сульфонилов, но реакция могла произойти и после удаления рас-
растворителя.
Бромистый водород и йодистый водород, являющиеся очень
сильными кислотами в водных растворах, были исследованы в жид-
жидком НС1. При титровании с галогенидами тетраметиламмония на-
наблюдается постоянное уменьшение электропроводности, перегиба
на кривой нет (Дж. А. Зальтхауз и Т. Ваддингтон, неопубликован-
неопубликованные результаты). После удаления растворителя остаются соответ-
соответственно бромид или иодид тетраметиламмония. С галогенидами,
содержащими большой катион, такой, как тетра-к-бутиламмоний,
образуются смешанные гидродигалогениды (K-C4H9LINHBrCl и
(h-C4H9LNHC1I 4б (см. табл. 21). Таким образом, оказывается, что
и бромистый, и йодистый водород могут быть кислотами в жидком
хлористом водороде, т. е. протекают реакции
НС1;
НС1Вг- + НС1
HC1I- + HC1
B8)
B9)
Подобно некоторым кислотам в воде они так слабы, что конец тит-
титрования с ними нельзя определить кондуктометрически. Ангидриды
кислот, такие, как четырехокись азота, пятиокись фосфора и дву-
двуокись серы, не обнаруживают кислотных свойств в жидком хлористом
водороде 40.
Б. Реакции сольволиза
Можно сказать, что происходит реакция сольволиза, когда ли-
ганд замещается атомом галогена растворителя:
МХ + НГ
МГ + НХ
C0)
IV. РЕАКЦИИ В ГАЛОГЕНОВОДОРОДАХ
105
Этот тип реакции наблюдается в. хлористоводородных растворах
соединений, содержащих фенил, гидроксил или фтор. Единственным
гидроксильным соединением, которое подвергается сольволизу, яв-
является трифенилкарбинол 38:
РЬзСОН+ЗНС!
Ph3C+HClj+H3O+Cr
C1)
Протеканию этой реакции способствует нерастворимость хлорида
гидроксония или чрезвычайная устойчивость иона трифенилметил-
карбония, или, что более вероятно, и то и другое. Бензойная кислота
и ее этиловый эфир не подвергаются 39 сольволизу в НС1, что не-
несколько неожиданно, так как этот эфир быстро сольволизуется
в серной кислоте 9.
Хлорид трифенилолова сольволизуется в хлористом водороде
с количественным выходом до дифенилдихлорида олова и бензо-
бензола 38:
Ph3SnCl + HCl —> Ph2SnCl2-j-PhH C2)
Механизм этой реакции неясен. Сходный тип сольволиза наблюдался
также в растворах трифенилхлоридов металлов IV группы Ph3MCl
и трифенилпроизводных элементов V группы Ph3M в хлористом во-
водороде 38.
Наиболее часто наблюдаемыми реакциями сольволиза являются
реакции замещения атома легкого галогена более тяжелым. Сильные
фторирующие агенты, такие, как трифторид сурьмы, реагируют
в растворе НС1, образуя хлорид и фтористый водород 43:
SbCl3 + 3HF
C3)
Трифторид бора не сольволизуется в хлористом или бромистом всЗ
дороде 68> 80" 62. Трихлорид бора реагирует с бромистым водородом,
образуя трибромид 60> б2. Трихлорид и трибромид сольволизуются
в йодистом водороде
30.
ВС1з + ЗШ
В13 + ЗНС1
C4)
Это различие в поведении галогенидов бора указывает на большую
устойчивость связи В — F. Трифенилметилхлорид и тетрахлорид
германия сольволизуются в HI; в обоих случаях получается иодид30.
В. Окислительно-восстановительные реакции
Реакции окисления и восстановления в растворах галогено-
водородов можно представить следующим образом:
Г"
1
Н2
Евос
C5)
C6)
где Е — стандартный окислительно-восстановнтольный потенциал.
106 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
Если, как в воде, Евос„ > ?окисл, вещества с Е < Явосст будут
восстанавливать растворитель, а с й)> Еокисл — окислять раство-
растворитель. Термодинамически наиболее устойчивым окисляющим аген-
агентом в жидком хлористом водороде должен быть хлор, который не
проявляет никаких кислотных свойств.
Хлор, бром и монохлорид иода легко растворимы в НС1, поэтому
можно исследовать реакции их окисления кондуктометрически.
Иод почти нерастворим в жидком НС1
и в хлористоводородных растворах га-
логенидов. Во всех реакциях, которые
были изучены до настоящего времени
он, по-видимому, не является окис-
окислителем.
Хлор окисляет иод-ион в две ста-
стадии: сначала до Юг, а затем до IC1J;
обе стадии можно обнаружить кондук-
кондуктометрически (рис. 27). Бром-ион окис-
окисляется хлором до BrClg, конец реакции
также можно обнаружить кондуктоме-
кондуктометрически (рис. 27). Бром окисляет
иод-ион до IBrg, конечная точка нахо-
находится при мольном отношении брома
к иод-иону, равном 1 : 1 (Дж. А. Заль-
тхауз и Т. Ваддингтон, неопубликован-
неопубликованные результаты).
Реакции монохлорида иода проте-
Рис. 27. Кривые кондуктоме- кают несколько сложнее. На кривой
титрования IC1 иод-ионом имеются две
эквивалентные точки при отношении
[IC1] : [I—], равном 1 : 1 и 2 : 1.
Вначале образуется элементарный
иод
Г + 1С1 + НС1 —v 12,1.+НС1? (il)
а затем ион IClj
ICl + HCli —*• HCl + ICl; C8)
Вторая стадия является реакцией кислоты по Льюису в растворителе,
а не реакцией окисления — восстановления.
Реакции галоген-ионов с окисляющими агентами в растворителе
дают возможность объяснить более сложные реакции окисления
в растворителе, как, например, окисление трихлорида фосфора.
Это соединение — плохой проводник тока в жидком НС1, но оно
окисляется хлором в пентахлорид, являющийся хорошим провод-
проводником, поэтому за реакцией можно проследить кондуктометрически.
Конец титрования отвечает отношению компонентов 1:1, что соот-
соответствует реакции
РС1з + С12+НС1 —> PClJ + HCla C9)
Лгрического титрования хлора
(А) в жидком хлористом водо-
водороде галогенидами тетраал-
киламмония (В):
1 — С12 + (CH3LNI; 2 — С1, +
+ (CH3LNBr; з — С1, 4- (CHSLNC1
ЛИТЕРАТУРА
107
фосфора бромом ведет к образованию иона
РС13 + Вг2 + НС1 >- РС13Вг+ + НС1Вг~ D0)
Окисление трихлорида
РС13Вг+:
После удаления растворителя ион распадается, но если прибавить
трихлорид бора, то можно выделить соль РС13Вг+ ВС1~ (Дж. А. Зальт-
хауз, Т. Ваддингтон, неопубликованные результаты).
3.
4.
5
6,
7.
8,
9.
10,
И.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
J. A., Quart.
Robinson-E. A., Adv. inorg
A962).
(London), 8, 40
Chem. Radio-
Radioed., London,
ЛИТЕРАТУРА
В е с k m a n n E., W a e n t i g P., Z. anorg. Chem., 67, 17 A910).
Chang S., West rum E. F., Jr., J. Chem. Phys., 36, 2571 A962).
Cook D., L u p i e n Y. et al., Can. J. Chem., 34, 957, 964 A956).
Del Fresno C, Z. anorg. Chem., 170, 222 A928).
Eluek E., Z. anorg. Chem., 315, 191 A962).
Fuoss R. M., Krauss С A., J. Am. Chem. Soc, 55, 2387 A933).
F газе г M. J., Gerrard W. et al., J. Chem. Soc, 1957, 639.
Gerson F., Heilbronner E., Helv. chim. acta, 45, 51
Gillespie R. J., Leisten J. A., Quart. Rev.
A954).
Gillespie R. J.,
chem., 1, 385 A959).
Glasstone S., Textbook of Physical Chemistry. 2nd.
p. 637, 647, 891.
Gloekler G., Peck R. E., J. Chem. Phys., 4, 658 A936).
Gold V., Туе F. L., J. chem. Soc, 1952, 2172.
Gore G., Phil. Mag., D) 29, 541 A865).
Graff W., С. г., 197, 754 A933).
Grange P., L a s с о m b e J. et al., Spectrochim. acta, 16, 981 A960).
Gutmann V., Quart. Rev., 10, 451 A956).
Handbook of Chemistry and Physics, 39th ed. Chemical Rubber Publishing;
Co., 1957-1958.
Hantzseh A., Ber., 63B, 1789 A930).
Harmon K. M., Davis S., J. Am. Chem. Soc, 84, 4359 A962).
Hart H., F i s h R. W., J. Am. Chem. Soc, 80, 5894 A958).
Herbrandson H. F., Dickerson R. T. et al., J. Am. Chem.
Soc, 76, 4046 A954).
Herzberg G., Infrared and Raman Spectra of Polyatomic Molecules,
New York, 1945, p. 280.
H о 1 1 i d а у А. К., Р е а с h M. E. et al., Proc. Chem. Soc. 1961, 220.
" " , J. Chem. Educ, 40, 125 A963).
, Gallagher W. P., Inorg. Chem., 2, 433 A963).
Quarterman L. A. et al., J. Phys. Chem., 65, 125
R.
, R.
H.,
Holmes R
Holmes R
H у m a n H.
A961).
Janz G. J., Danyluk S. S. et al., J. Am. Chem. Soc, 81, 3846,
3850, 3854 A959).
Kahovec L., Knollmiiller K., Z.
F., Kohlschiitter H.
phys. Chem., B51, 49 A941).
W., Z. Naturforsch., 16b,
Klanberg
69 A961).
К 1 e m e n с А., К о h 1 О., Z. anorg. Chem., 168, 163 A927).
Krauss С A., Fuoss R. M., J. Am. Chem. Soc, 55", 21 A933).
M a a s О., М с I n t о s h D., J. Am. Chem. Soc, 35, 535 A913).
Mclntosh D., J. Am. Chem. Soc, 30, 1097 A908).
Maki A. G., West R'., Inorg. Chem., 2, 657 A963).
Mel lor J. W., Comprehensive Treatise on Inorganic and Theoretical
Chemistry, Supplement 2, Part 1, London, 1961, p. 415.
108 ГЛАВА ТРЕТЬЯ. ГАЛОГЕНОВОДОРОДЫ КАК ИОНИЗИР. РАСТВОРИТЕЛИ
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
Murray F. E., S chnei der W. G., Can. J. Ghem., 33, 797 A955).
Peach
Peach
Peach
Peach
Peach
Peach
M.
M.
M.
M.
M.
M. E.
E.
E.
E.
E.
E.
W a d d i n g t о n T. C, J. Chem. Soc, 1961, 1238.
Waddington Т. С, J
W a d d i n g t о п Т. С, J
Waddington Т. С, J
Waddington Т. С, J.
Waddington Т. С, J.
Chem. Soc, 1962, 600.
Chem. Soc, 1962, 2680.
Chem. Soc, 1962, 3450.
Chem. Soc, 1963, 69.
Chem. Soc, 1963, 799.
E., Waddington Т. С, J. Chem. Soc, 1963, 799.
Runner M. E., В a 1 о g G. et al., J. Am. Chem. Soc, 78, 5183 A956).
Rupert E. F., J. Chem. Soc, 31, 851 A909).
Salt house J. A., Waddington Т. С, J. Chem. Soc, 1965,
6291.
A., J. Chem. Soc, 1958, 2558.
W.
ij IX Cl 1_ L/ J ' ¦ IT» 1Ц. U* V>HV 1111 Ъ_/ \J^ Л Л Л. 4J 4J\mf у Ш* ^J C_
Sharp D. W. A., Adv. Fluorine Chem., 1, 105 A960).
Smyth С P., H i t с h с о с к С. S., J. Am. Chem. Soc, 55, 1830 A933).
Mclntosh D., et al., Phil. Trans., """ "" """"
S t e e 1 е В. D.,
S t e e 1 е В. D.,
Vallee R. Е.
A962).
Vallee R. Е.,
Vi d al e G. L.,
Waddington
Waddington
Waddington
Waddington
2332.
Waddington
A960).
Waddington
Waddington
W a d d i n s t
Mclntosh D. et al., Z. phys. Chem., 55, 129 A906).
McDaniel D. H., J. Am. Chem. Soc, 84, 3412
H., Inorg. Chem., 2, 997 A963).
_., J. Am. Chem. Soc, 78, 294 A956).
Trans. Farad. Soc, 54, 25 A958).
J. Chem. Soc, 1958, 1708.
F., Naturwis. 46, 578 A959).
McDaniel D.
Taylor R. С
Т. С, ""
т с
Т. С.' К 1 an Ь о г g
Т. С, К 1 а п Ь е г g
Т. С, К 1 а п Ь е г g
F., J. Chem. Soc', 1960, 2329\
F., Z. anorg. Chem., 304, 185
Proc. Chem. Soc, I960, 85.
Proc chem. Soc, 1960, 315.
J. Chem. Soc, 1963, 2701.
Т. С, White J. A.,
Т. С, White J. A.,
Waddington T. C, White J. A., J. Chem. Soc, 1963, 2701.
Walker J. W., Mclntosh D. et al., J. Chem. Soc, 85, 1098 A904).
West R., J. Am. Chem. Soc, 79, 4568 A957).
Wheat J. A., Browne A. W., J. Am. Chem. Soc, 62, 1577 A940).
Y e h S. J., J a f f ё e H. H., J. Am. Chem. Soc, 81, 3279 A959).
Yoon Y. K., Carpenter G. В., Acta cryst., 12, 17 A959).
Кирсанов О. В. и Федорова Г. К., ДАН УРСР, 1086 A960).
ГЛАВА ЧЕТВЕРТАЯ
СЕРНАЯ КИСЛОТА
Р. Дж. Джиллеспи, Е. А. Робинсон
I. Введение 110
A. Основания 111
1. Соли неорганических кислот 111
2. Простые органические основания 112
3. Сложные основания 113
Б. Кислоты 115
B. Неэлектролиты 115
II. Физические свойства серной кислоты п ее растворов п эксперимен-
экспериментальные методы их исследования 116
А. Серная кислота 116
1. Структура 116
2. Самодиссоциация 116
3. Температура замерзания 119
4. Плотность 119
5. Вязкость 119
6. Диэлектрическая проницаемость 120
7. Электропроводность 120
8. Теплоемкость и теплота плавления 121
9. Инфракрасные спектры и спектры комбинацпонного рассея-
рассеяния 122
10. Серная кислота как лучшее стандартное вещество при ана-
аналитических работах 123
Б. Растворы 123
1. Криоскопия 123
2. Плотность и кажущийся мольный объем 131
3. Вязкость 132
4. Электропроводность 133
5. Термодинамические свойства 143
6. Спектроскопия растворов серной кислоты 144
7. Определение кислотной функции Гаммета 146
III. Растворы неорганических веществ в серной кислоте 150
A. Вода и трехокись серы 150
Б. Бор 152
B. Кремний , . 155
Г. Олово и свинец 158
Д. Азот и фосфор 161
Е. Мышьяк, сурьма, висмут 163
Ж. Ванадий, хром, марганец 166
3. Сера, селен, теллур 168
И. Галогены 169
К. Комплексы переходных металлов , 173
Л. Образование хелатов 175
но
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
IV. Растворы органических веществ в серной кислоте 177"
A. Простые основания 177
Б. Многоосновные соединения 177
B. Амиды и мочевины 1791
Г. Сложные эфиры 181
Д. Ангидриды карбоновых кислот 182
Е. Простыв эфиры 184
Ж. Нитрилы 185
3. Карбоновые кислоты 185
И. Ионы карбония 187
1. Сопряженные кислоты ароматических углеводородов и поло-
положительные ионы 187
2. Ионы триарилкарбония 188
3. Ионы диарилкарбония 189
4. Моноарилкарбониевые ионы 190
5. Положительные двухзарядные ионы карбония 191
6. Алифатические ионы карбонпя 192
7. Ионы полиенов 193
К. Сульфирование ароматических соединений 194
Литература 195
I. ВВЕДЕНИЕ
Благодаря высокой диэлектрической проницаемости, сильной
полярности молекул и их способности к образованию прочных
водородных связей серная кислота служит хорошим растворителем
для соединений, являющихся электролитами. Последние в зависи-
зависимости от поведения в растворителе разделяются на кислоты и осно-
основания. Кислоты образуют положительно заряженный ион H3SO*
НА + H2SO4=H3SOJ+A- A)
а основания — отрицательно заряженный ион HSOJ
I. ВВЕДЕНИЕ
111
. B)
Вследствие высокой кислотности серной кислоты большинство элек-
электролитов ведет себя в ней как основание. Серная кислота оказывает
такое же нивелирующее действие на силу оснований, как вода на
силу кислот. Веществ, являющихся в серной кислоте кислотами,
насчитывается очень мало. Некоторые вещества не проявляют себя
в серной кислоте как электролиты, хотя являются таковыми в вод-
водных растворах; многие соединения, которые не обладают в воде
основными свойствами, в том числе вещества, ведущие себя в водном
растворе как кислоты, в серной кислоте легко протонизируются.
Существует много электролитов, устойчивых в воде, однако они
неустойчивы в серной кислоте.
Серная кислота оказалась, в частности, ценным растворителем
для таких слабых оснований, как кетоны и нитросоединения, а также
оказалась пригодной для приготовления устойчивых растворов
таких реакционноспособных ионов, как ион карбония, NO2 и 1?.
Последние неустойчивы в более основных растворителях, например
ъ воде.
Как растворитель серная кислота во многих отношениях напоми-
напоминает воду, несмотря на разницу в их кислотности. Сравнение элек-
электропроводности, вязкости, плотности и активности растворов элек-
электролитов в серной кислоте и в воде помогает понять общие свойства
электролитов.
Уже имеется несколько обзоров свойств серной кислоты как
растворителя 8в> 91> 9в. Материалы настоящей главы дают общее
представление о системе, в которой растворителем является серная
кислота, и охватывают новейшие исследования свойств веществ
в этом растворителе.
А. Основания
1. Соли неорганических кислот
Соли многих неорганических кислот не растворяются в серной
кислоте, как, например, AgCl, CuBr2, A1C13 и А1РО4, или подвер-
подвергаются полному сольволизу:
NH4C1O4 + H2SO4 = NHJ + HSOJ + HC1O4
C)
E)
Такие соли образуют сильноосновные растворы. Сольволиз происхо-
происходит вследствие относительно высокой концентрации иона H3SOJ, по-
появляющегося в результате самодиссоциации серной кислоты, а также
потому, что кислоты, от которых эти соли происходят, в H2SO4
либо очень слабы, либо совсем не проявляют кислотных свойств.
Так, в сернокислом растворе хлорная кислота является очень слабой
кислотой, а фосфорная и азотная кислоты ведут себя как основания.
Поэтому работы исследователей ограничились главным образом
изучением нормальных и кислых сульфатов. Однако из этого нельзя
делать вывод о том, что химия растворов в серной кислоте не пред-
представляет сколько-нибудь значительного интереса, поскольку в H2SO4
известно много катионов, которые не существуют в водных растворах.
Нормальные и кислые сульфаты в H2SO4 подобны окислам
и гидроокисям в водном растворе, т. е. являются сильными основа-
основаниями. Кислые сульфаты щелочных н некоторых других металлов
в сернокислом растворе ведут себя как полностью ионизированные
электролиты:
= к++он-
112
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
I. ВВЕДЕНИЕ
113
Нормальные сульфаты превращаются в кислые, что формально соот-
соответствует поведению окислов металлов в воде:
K2S О4 + H2S O4 = 2К+ + 2 HS О J
G)
При растворении окислов и гидроокисей в серной кислоте происхо-
происходит превращение их в сульфат или кислый сульфат, причем степень
превращения для различных элементов различна. Так, окислы
и гидроокиси более электроположительных металлов полностью
превращаются в соответствующие кислые сульфаты, в то время как
фосфорная кислота лишь незначительно протонизируется и не обра-
образует сульфатов. В общем, может образовываться большое число
соединений, состав которых изменяется от продуктов растЁорения
окислов и гидроокисей в воде до продуктов растворения нормальных
и кислых сульфатов в серной кислоте.
2. Простые органические основания
Очень многие вещества ведут себя как основания: они присоеди-
присоединяют протон и образуют кислоты, сопряженные основаниям. Напри-
Например, органические вещества, за исключением углеводородов алифа-
алифатического ряда, некоторых ароматических углеводородов и их гало-
галоидных производных, растворимы в серной кислоте. Это происходит,
очевидно, за счет атомов О, N и Р, имеющих неподеленную пару
электронов, которая способна акцептировать протон молекулы серной
кислоты. Процесс растворения можно представить следующим
образом:
кетонов
карбоновых кислот
сложных эфиров
аминов
RCO2H + H2SO4 = RCO2H % + HSOj
RCO2R' + H2SO4= RCO2R'H+ + IISOJ
RNH2 + H2SO4 = RNHJ + HSOJ
амидов
RCONH2 + H2SO4=RC(OH)NH2 + HSOI
и, наконец, фосфинов
R3P + H2SO4=R3PH++HSOJ-
О)
A0)
A1)
A2)
A3)
Даже трифениламин и трифенилфосфин, которые обычно считаются
очень слабыми основаниями, в серной кислоте ведут себя как силь-
сильные основания. Некоторые вещества, такие, как нитросоединения,
диалкилсульфоны и нитрилы, обладающие очень слабыми основ-
основными свойствами в воде, в серной кислоте протонизированы непол-
неполностью.
Молекулы, содержащие более одной основной группы, часто
образуют многозарядные катионы. Например, о-фенилендиамин
присоединяет два протона, некоторые аминокислоты присоединяют
протоны и к NH2, и к СО2Н, а у гексаметилентетрамина протонизи-
руются четыре атома азота.
При комнатной температуре сернокислые растворы очень многих
органических соединений вполне устойчивы, причем если раствор
вылить в мелкораздробленный лед, то можно выделить растворенное
органическое вещество в неизмененном виде. Протонизирование
способствует снятию химической активности соединения и, таким
образом, предотвращает последующее протекание реакции, например
сульфирования ароматических углеводородов.
Примерами неорганических веществ, которые ведут себя как
сильные основания, являются вода и фосфорная кислота:
A4)
A5)
h2o+h2so4 —>-
h3po4 + h2so4 —>- h4poj+hso4
Двуокись селена является слабым основанием:
SeO2 + H2SO4 ^=± HSeOJ+HSO«
3. Сложные основания
A6)
Многие окислы и гидраты окислов ведут себя в H2SO4 как осно-
основания. К простейшим веществам такого типа относятся соединения
с общей формулой ХОН, которые под действием серной кислоты пре-
превращаются в кислые сульфаты XSO4H:
XOH+2H2SO4 =
++HSO-
A7)
Основанием является, например, этиловый спирт, который превра-
превращается в кислый этилсульфат:
C2H5OH+2H2SO4=C2H5SO4H + H3O+ + HSO; A8)
Аналогичным образом йодноватая кислота превращается в кислый
иодилсульфат
HIO3+H2SO4=IO2HSO4-|-H3O+-|-HSO7 A9)
который далее может реагировать как основание или образует
кислоту, сопряженную основанию
IO2SO4H-f H2SO4=IO2SO4HJ-fHSO7 B0)
либо ионизируется до 10, и HSO,.
Азотная кислота, трифенилкарбинол и мезитиленкарбоновая
кислота образуют кислые сульфаты типа XSO4H, которые полностью
8 заказ 90
114
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
I. ВВЕДЕНИЕ
115
ионизируются до Х+ и HSO4. В итоге ионизация этих соединений
может быть представлена следующими уравнениями:
B1)
B2)
B3)
Недавно было показано, что в сернокислом растворе могут быть
получены ионы, содержащие две симметричные группы, в центре
каждой из которых расположен ион карбония:
(С6Н5JС-
ОН
—C(CeH6J+4H2SO4 =
ОН
B4)
Многие из этих катионов являются очень сильными электрофиль-
ными агентами и могут существовать только в очень слабоосновных
растворах, какие получаются на основе серной кислоты или в неко-
некоторых апротонных растворителях. Их образование в H2SO4 обуслов-
обусловлено не только высокой кислотностью серной кислоты, но и очень
низкой химической активностью воды в разбавленном растворе
H2SO4. Последнее обстоятельство можно объяснить интенсивным
связыванием воды в ионы Н3О+. Преимущественное использование
серной кислоты по сравнению с использованием апротонных раство-
растворителей для получения таких ионов объясняется тем, что она является
хорошим растворителем для электролитов. В серной кислоте могут
быть получены относительно концентрированные растворы этих
реакционноспособных ионов.
Некоторые ангидриды также реагируют с серной кислотой,
образуя кислый сульфат и воду:
Гексаметилсилоксан и окись мышьяка (III) реагируют аналогично:
B6)
B7)
Кислый сульфат триметилсилана не является электролитом; AsOSO4H
частично ионизирован. Азотный ангидрид образует полностью
ионизированный кислый сульфат нитрония:
B8)
Б. Кислоты
Большинство веществ, являющихся в водном растворе кислотами,
в серной кислоте не проявляет кислотных свойств и ведет себя как
основания:
B9)
C0)
C1)
H3BO3+6HaSO4= B(HS04)i- + 3H3O + 2HS04- C2)
C3)
' C4)
Даже хлорная кислота, которую считают самой сильной из мине-
минеральных кислот, в серной кислоте лишь слегка ионизирована, а пер-
перхлораты металлов подвергаются полному сольволизу по уравне-
уравнению E). Кислотами в серной кислоте являются пиросерная кислота
H2S2O7, трисерная кислота H2S3O10 и другие полисерные кислоты,
обнаруженные в олеуме, но все они — слабые кислоты. Очень сла-
слабой кислотой является также и фторсульфоновая кислота HSO3F.
Доказано, что в серной кислоте может существовать сильная
кислота —¦ комплексная тетрагидросульфато-борная кислота
HB(HSO4L- Раствор этой кислоты может быть получен путем раство-
растворения борной кислоты в олеуме:
C5)
Таким же образом удалось получить некоторые другие комплексные
сульфатокислоты, например гексагидросульфато-оловянную кислоту
H2Sn(HSO4)e и гексагидросульфато-свинцовую кислоту H2Pb(HSO4N.
В. Неэлектролиты
Соединений, которые при растворении в серной кислоте ведут
себя как неэлектролиты, насчитывается очень мало. Вещество будет
неэлектролитом в H2SO4, если его молекула является достаточно
основной, чтобы образовать прочные водородные связи с серной
кислотой, но не настолько, чтобы протонизироваться. К неэлектро-
неэлектролитам, известным в настоящее время, относятся алкилсульфофториды
и алкилсульфохлориды, сульфурилхлориды, пикриновая кислота
и некоторые другие полинитроароматические соединения и, вероятно,
дифенилсульфон и большинство сульфоновых кислот. Трихлоруксус-
ная кислота и хлорсульфоновая кислота также могут являться
неэлектролитами, хотя это еще не подтверждено 27> 90> 93> 108>127.
116
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
II. ФИЗИЧЕСКИЕ СВОЙСТВА СЕРНОЙ КИСЛОТЫ
И ЕЕ РАСТВОРОВ
И ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
ИХ ИССЛЕДОВАНИЯ
А. Серная кислота
1. Структура
Высокие вязкость, температура кипения и поверхностное натяже-
натяжение серной кислоты указывают на то, что она является сильно ассо-
ассоциированной жидкостью. Несомненно, что это связано с наличием
прочной водородной связи между молекулами. Было показано, что
в твердом состоянии серная кислота имеет слоистую структуру 207,
в которой каждая молекула связана водородной связью с четырьмя
другими. Определить межатомные расстояния с достаточной точ-
точностью при использовании рентгеновских лучей, к сожалению, не
удалось. Структура серной кислоты в жидком и твердом состояниях
очень похожа; это напоминает сходство между структурами жидкой
воды и льда.
Простейшее представление о структуре молекулы серной кислоты
дает формула I, согласно которой й^-^-ти с72*-орбитали атома серы
используются в двойной связи с атомами кислорода:
S
но/ ^о
Это было подтверждено изучением колебательных спектров H2SO4.
2. Самодиссоциация
Свойства 100%-ной серной кислоты обусловлены в основном ее
значительной самодиссоциацией, и поэтому, чтобы понять поведение
веществ, растворенных в серной кислоте, необходимо знать природу
и степень протекания этого процесса.
Несмотря на высокую кислотность, серная кислота может про-
проявлять и основные свойства. Она относится к главной группе рас-
растворителей, в которую входит вода, жидкий аммиак и жидкий фто-
фтористый водород — амфотерные или амфипротные растворители,
в значительной степени подверженные процессу автопротолиза *
C6)
* Термин «автопротолиз» равнозначен термину «самоионизация»,
бляемому в других главах. — Прим. ред.
употре-
II. ФИЗИЧЕСКИЕ СВОЙСТВА
117
при котором одна из двух молекул ведет себя как кислота, а дру-
другая — как основание. Степень автопротолиза зависит от силы кислоты
и основания. Для серной кислоты эта величина выше, чем для
большинства протонных растворителей. Константы автопротолиза
для некоторых растворителей при 25° С:
Растворитель NH3
—lgKan 29,8
Растворитель Н2О2
-lgXan 9,7
Помимо автопротолиза
С2Н5ОН D2O H2O СЩСООН
' 18,9 14,8 14,0 g 12,6-
HF HCOOH D2SO4 H2SO4 Н3РО4
9,7 6,2 4,3 3,6 ~2
C7)
Вода ионизируется по уравнению
H2O + H2SO4=H3O*-|-HSOj
SO3 образует пиросерную кислоту
SO3 + H2SO4=H2S2O7
которая частично ионизирована как кислота
происходит самодиссоциация молекул серной кислоты, которая
является следствием первичной диссоциации на воду и трехокись
серы
H2SO4=H2O + SO3 C8)
C9)
D0)
а так как ионы H3SO^ и HSOJ находятся в равновесии в результате
реакции автопротолиза, то и ионы Н3О+ и HS2O7 также должны
находиться в равновесии:
2H2SO4=H3O+ + HS2O7 D2)
Процесс D2) был назван реакцией ионной самодегидратации,
константа равновесия которой получила обозначение Кл. Уравне-
Уравнения C7), C9), D1) и D2) удобны для описания процесса полной
самодиссоциации серной кислоты. Значения констант равновесия
реакций самодиссоциации серной кислоты представлены ниже:
ti°C ; 10 25 40
tfan = [H3S04"][HS0S-] 1,7-Ю-* 2,7-10'* 4,0 • 10"*
tfa=[H3O+][HS2O7] 3,5-10-5 5,1 • 10'5 8,1 • КГ*
2O7] ..... 1,4-10-2 1,4-10-2 1,4-10-2
118
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Значения при 10° С были получены определением температуры
замерзания сернокислых растворов кислых сульфатов металлов,
воды и пиросерной кислоты, каждый из которых различным путем
подавляет самодиссоциацию серной кислоты 8. Значения Кап и Ка
при 25 и 40° С были получены из значений этих констант при 10° С,
теплот автопротолиза и ионной самодегидратации, которые были
определены Виаттом с сотр. (АЯап = 5,0 ккал/молъ, Д#д =
= 6,0 ккал/молъ) 42- 101- 102- 149.
Ниже приведены значения констант самодиссоциации дидейтеро-
серной кислоты D2SO4:
t, °C 14 25 40
^ = [D3SOJJ[DSO7] 2,9 -10 4,6 • 10 5,2 • 10~5
i^=[D3O+][DS2O7] 5,0-10-5 5,5 • 10 6,0 • 10"»
2,8 • 10'3 2,8 • to 2,8 • 10
0,2 0,2 0,2
Они были получены таким же путем, как и для серной кислоты, но
с гораздо меньшей точностью.
Следует отметить, что значения константы автодейтролиза меньше
константы автопротолиза. Частоты колебаний группы SO2 в моле-
молекуле D2SO4 несколько меньше, чем в молекуле H2SO4. Это означает,
что D2SO4 является более сильным основанием 97' 102, чем H2SO4.
Следовательно, как кислота D2SO4 должна быть слабее H2SO4.
Интересно отметить, что и D2S2O7 в растворе D3SO4 является,
по-видимому, более слабой кислотой, чемН2Э2О7 в растворе H2SO4 93.
В табл. 22 приведены моляльные концентрации продуктов само-
самодиссоциации кислот H2SO4 и D2SO4 при температуре замерзания
соответствующих кислот.
Таблица 22. Моляльные концентрации частиц, т,
образующихся при самодиссоциации H2SO4 и D2SO4
Частица
HSOr
Н,О+
HS2Of
H2S2O7
Н2О
Итого ....
т
0,0150
0,0113
0,0080
0,0044
0,0036
0,0001
0,0424
Частица
DSOJ-
D3SOt
DS2O7
D2S-2O7
D2O
Итого ....
m
0,0112
0,0041
0,0112
0,0049
0,0071
0,0006
0,0391
II. ФИЗИЧЕСКИЕ СВОЙСТВА
119
3. Температура замерзания
Температура замерзания 100%-ной серной кислоты при нормаль-
нормальном атмосферном давлении равна 8>168 10,371° С. 100%-ную кислоту
можно приготовить, добавляя низкопроцентный олеум к концентри-
концентрированной серной кислоте до тех пор, пока не будет достигнута топка
замерзания 10,371° С. Точка замерзания гипотетически полностью
недиссоциированной кислоты, вычисленная из концентраций частиц,
образующихся при самодиссоциации, и криоскопнческой постоян-
постоянной8, равна 10,625° С. На рис. 28
показана часть кривой зависи-
зависимости температуры замерзания
от концентрации SO3 в системе
Н2О—SO3 вблизи состава, от-
отвечающего H2SO4. Теоретиче-
Теоретическая кривая была рассчитана
на основании констант равно-
равновесия, приведенных в разделе
II, А, 2. Небольшое различие
между экспериментальной и
расчетной кривыми может быть
объяснено тем, что растворы
неидеальны. Точка замерзания69
D2SO4 равна 14,34° С.
4. Плотность
Плотности H2SO4 и D2SO4
при 25° С равны соответственно
1,8269 г/см3 и 1,8573 г/см3. Зависимость плотности от температуры
может быть выражена приведенными уравнениями:
для H2SO4 p| = 1,8516-1,000 -10-зг D3)
для D2SO4 pj= 1,8816 —0,980-10-3* D4)
Мольный объем H2SO4 при 25° С равен 53,69 см3, а объем D2SO4
равен 53,89 см3. Предполагают, что увеличение мольного объема
при замещении водорода на дейтерий происходит вследствие большей
длины дейтериевой связи по сравнению с водородной связью 126.
5. Вязкость
При 25° С вязкость H2SO4 равна 24,54 стгз, a D2SO4"— 24,88 спз.
Зависимость вязкости от температуры описывается следующим
уравнением 125:
H2S2O7
Рис. 28. Зависимость температуры за-
замерзания системы Н2О—SO3 от кон-
концентрации SO3:
1 — теоретическая кривая; 2 — эксперимен-
экспериментальная кривая.
) = тHехр(С /RT)
D5)
Величина 2С7]/Г представляет собой энергию активации Ev Она
уменьшается от 6,78 ккал/молъ при 10° С до 5,76 ккал/молъ при 60° С
для H2SO4 и от 6,99 ккал/молъ при 10° С до 5,62 ккал/молъ при 60° С
120
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
для D2SO4. Уменьшение энергии активации объясняется, вероятно,
ослаблением водородных и дейтериевых связей с повышением темпе-
температуры.
6. Диэлектрическая проницаемость
Диэлектрическую проницаемость серной кислоты трудно изме-
измерить в основном вследствие высокой электропроводности кислоты.
В 1953 г. Брэнд и др. 23 применили для измерения диэлектрической
проницаемости волноводный метод, использовав при этом диапазон
частот 100—3000 мггц. Они обнаружили, что на этих частотах про-
происходит дисперсия диэлектри-
диэлектрической проницаемости; стати-
статическую диэлектрическую про-
120\- ""Nv,. ницаемость они получили экс-
экстраполяцией.
Были сделаны две попытки
непосредственно определить ве-
величину статической диэлектри-
диэлектрической проницаемости. Джил-
леспи и Коль 89 определяли
_j i диэлектрическую проницае-
го 25 мость по величине емкости
II. ФИЗИЧЕСКИЕ СВОЙСТВА
121
ч>
ПО
то
10
15
6. "С
Рис. 29. Зависимость диэлектрической
проницаемости серной кислоты от тем-
температуры (числа при точках — литера-
литературный источник данных).
конденсатора, содержащего сер-
серную кислоту в качестве диэлек-
диэлектрика. Измерения проводились
обычным мостовым методом.
Расстояние между обкладками
конденсатора можно было ме-
менять. Это позволило устранить влияние поляризационной емкости
обкладок конденсатора, обусловленной высокой электропроводностью
кислоты. Была сделана попытка 112 применить механический метод,
впервые предложенный Фюртом, согласно которому измерялся вра-
вращающий момент подвешенного в жидкости платинового эллипсоида,
создаваемый электрическим полем, однако высокая электропровод-
электропроводность жидкости вызывала значительные экспериментальные труд-
трудности. Полученные результаты представлены на рис. 29. Они
показывают, что зависимость диэлектрической проницаемости от
температуры для изученного интервала температур приближенно
выражается прямой линией. Значения е, равные 120 при 10° С
и 100 при 25° С, получены интерполяцией.
7. Электропроводность
Высокая электропроводность 100% -ной серной кислоты обус-
обусловлена ионами, образующимися при самодиссоциации. Удельные
электропроводности H2SO4 и D2SO4 при различных температу-
температурах "9- 89. «в приведены в табл. 23.
Таблица 23. Удельная электропроводность H2SO4 и D2SO4
при 100%-ном составе и минимальная (ом i.Cjh)
Кислота
H2SO4
D2SO4
t, "С
9,66
25,00
40,00
10,00
25,00
40,00
х- Ю2 при 100%-ном
составе
0,570
1,0439
1,711
0,133
0,2568
0,446
"мин'102
0,5685
1,0432
1,710
0,2540
Состав, отвечающий * „„,
' МИН
моль Н2О (D2S2O?)/Ke рас-
раствора
0,0023
0,0019
0,0015
0,0045
Минимумы электропроводности в системах Н2О—SO3 и D2O—SO3
хотя и существуют, но выражены нерезко. Они примерно соответ-
соответствуют составам H2SO4 и D2SO4. В системе Н2О—SO3 при темпера-
температурах 10—40° С минимум расположен несколько левее точки с соста-
составом H2SO4 и сдвигается в сторону H2SO4 при возрастании темпера-
температуры. В системе D2O — SO3 при температуре 25° С минимум находится
правее состава D2SO4. Положение минимума зависит от концен-
концентраций и подвижностей всех присутствующих ионов. Последнее
обстоятельство было использовано для определения концентрации
ионов.
Основываясь на данных об электропроводности (см. табл. 23), 100%-ную
серную кислоту можно получить следующим образом. Сначала готовят серно-
сернокислый раствор с минимальной электропроводностью, затем приливают к нему
низкопроцентный олеум до тех пор, пока электропроводность смеси не станет
равной электропроводности 100%-ной кислоты.
Энергия активации для удельной электропроводности серной
кислоты почти такая же, что и для вязкости, однако, как отмечают
Гринвуд и Томпсон 12S, это совпадение случайно и не связано с меха-
механизмом электропроводности серной кислоты.
Электропроводность кристаллов серной кислоты в несколько сот
раз меньше электропроводности жидкости""
125
8. Теплоемкость и теплота плавления
Удельная теплоемкость 149 серной кислоты составляет
0,3373 кал/(град-г). Кривая зависимости удельной теплоемкости от
состава системы Н2О—SO3 имеет небольшой, но четкий максимум
в точке с составом H2SO4. Это вызвано тем, что теплоемкость 100% -ной
серной кислоты дополнительно возрастает эа счет температурного
коэффициента теплоты самодиссоциации157.
Теплота плавления 158 составляет 2560 кал/моль.
122
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
0. Инфракрасные спектры и спектры комбинационного рассеяния
Инфракрасные и спектры комбинационного рассеяния H2SO4
и D2SO4 были изучены несколькими группами исследователей.
Большинство ранних работ представлено в обзорах 79> 97> 230. Почти
во всех работах исследовались спектры жидкостей, лишь недавно 80
были получены инфракрасные спектры твердых H2SO4 и D2SO4.
Была сделана также попытка получить инфракрасные спектры газо-
газовой фазы, но, по-видимому, безуспешно, потому что полученные
спектры были идентичны спектрам жидкости. Характеристические
частоты колебаний H2SO4 и D2SC>4 представлены в табл. 24. На осно-
основании частот колебаний групп SO2 и S(OHJ были приблизительно
вычислены значения силовых постоянных (для SO2 k = 10,3 X
X 105 дин/см, для S(OHJ к = 5,9 -105 дин/см) 102. Из частот коле-
колебаний были вычислены термодинамические величины 80> 230.
II. ФИЗИЧЕСКИЕ СВОЙСТВА
123
Таблица
Частота
24. Характеристические
h.soi
жидкость
[97]
[79]
[230]
кристал-
кристаллы [80]
частоты колебаний
(в см
DaSO4
жидкость
[97]
[79]
[230]
gs
н ос
Olj
i) HaSO4 и D2SO4
Вид колебания
Для группы SO2
з (h)
(«l)
V12
VU Fj)
V6 («l)
Vl5 (Ы
V9
v7(a2)
Vl4 F-2)
V8 («2)
1195
1368
563
563
1170
1365
549
420
1140
965
741
1190
1170
1365
548
623
1170
1340
522
560
—
—
1190
1350
550-
566
628
| 910
973
392
422
907
967
332
372
905
1370
381
564
Для группы
907
967
386
412
907
980
356
395
S[OH(D)]2
—
—
910
980
363
371
2450
2970
—
1137
—
—
392
2450
2970
1240
1170
740
675
2430
3000
1800
1170-
1240
420
675
417
Для
2450
2970
1240
1170
—
650
—
группы О—II(D)
—
—.
356
1860
2280
. .
—
1820
2230
305
475
1860
2280
930
518
478
Валентн. симм.
Валентн. асимм.
Ножничное
Маятниковое
Валентн. симм.
Валентн. асшш.
Ножничное
Маятниковое
Валентн. спмлг.
Валентн. асимм.
Ножничное
Ножничное
Веер. симм.
Веер, асимм.
Крутильное
10. Серная кислота как лучшее стандартное вещество
при аналитических работах
Канцлер 1SS предложил использовать 100%-ную серную кислоту
в качестве главного стандарта при аналитических работах на осно-
основании того, что приготовить серную кислоту очень точного состава
гораздо легче, чем любое другое стандартное вещество. Им было
показано, что при этом целесообразно учитывать следующее:
а) по методу сдвига точки замерзания может быть получена
100%-ная серная кислота с точностью до 0,001%;
б) серная кислота, имеющая постоянную точку кипения, может
быть приготовлена с точностью до 0,01%, причем внешнее давление
влияет на состав данной постоянно кипящей смеси намного меньше,
чем на состав постоянно кипящей соляной кислоты;
в) добиваясь получения состава с минимальной электропровод-
электропроводностью, можно приготовить 99,996 %-ную серную кислоту с точ-
точностью до 0,002%;
г) по методу Брэнда 18 (воду или водный раствор серной кислоты
добавляют к олеуму до прекращения выделения белых паров) может
быть получена 100%-ная кислота с точностью до 0,02%;
д) серная кислота и растворы веществ в ней могут быть проана-
проанализированы с точностью до 0,05% методом калориметрического
титрования 219 (метод основан на большом различии между пар-
парциальными мольными энтальпиями воды, присутствующей в олеуме
и в концентрированной серной кислоте).
Б. Растворы
1. Криоскопия
а. Понижение температуры замерзания. Серная кислота имеет
относительно высокое мольное понижение точки замерзания, или
криоскопическую постоянную 89> 243 (Кк = 6,12 град -кг /моль). Было
показано, что вклад теплоты самодиссоцпации в криоскопическую
постоянную незначителен 89, поэтому последняя одинакова как для
гипотетической недиссоциированной кислоты, так и для реальной
100%-ной кислоты. Высокое значение криоскопической постоянной,
а также температуры замерзания A0,371° С) делают серную кислоту
идеальным растворителем для криоскопии при условии, что приняты
меры предосторожности с целью воспрепятствовать поглощению
воды из атмосферы и что в результаты измерения будет внесена
поправка, учитывающая самодиссоциацшо растворителя. Для изме-
измерения понижения температуры замерзания обычно применяется
метод Бекмана. Удобны приборы, описанные некоторыми исследо-
исследователями 90> 130>196.
124
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Криоскопические измерения могут быть использованы для полу-
получения величины v, т. е. числа молей (молекул и ионов) в растворе,
приходящихся на один моль растворенного вещества, и, следова-
следовательно, изучения процесса ионизации этого вещества.
В общем, можно записать:
9 A +0,002 6) = ККФ ^ ггц
D6)
где Э — понижение температуры замерзания, относительно t3aM гипотетически
недиссоцяированной серной кислоты, равной 10,625° С; Кк — криоскопическая
постоянная; 2mf ~ моляльная концентрация всех частиц растворенного веще-
вещества (включая ионы, образующиеся при самодиссоциации растворителя);
Ф — моляльный осмотический коэффициент 8.
Вообще Ф неизвестен, но можно принять его равным единице.
Далее, если md — общая концентрация частиц растворителя, обра-
образующихся при самодиссоциации, т — моляльная концентрация
растворенного вещества, а для реакции с одним молем этого вещества
требуется s молей растворителя, т. е.
Л+sH2SO4 =/>+<? +Д D7)
то мы получим
в A+0,0028) = *к (_gJ_ + IB<r) D8)
или после упрощения
6A+ 0,002 8 — 0,098 ms)
V~ 6,12 m
m
D9)
Концентрацию частиц, образующихся в результате самодиссоциации
md, можно вычислить для любой концентрации электролита, зная
константы Кап, Ка, Кц^о, и Кн,0, а также уравнения, отражающие,
состав и стехиометрию растворов. Были составлены таблицы вели-
величин md 8 для сильного основания В, сильной кислоты НА, воды,
пиросерной кислоты и комплексного основания ХОН, ионизиру-
ионизирующегося по уравнению
XOH + 2H2SO4 = X++H3O+ + HSO4- E0)
Подобные расчеты могут быть выполнены для более сложных элек-
электролитов. Было показано, что в случае электролита Е, ионизиру-
ионизирующегося по общему уравнению
концентрация ионов и молекул, образовавшихся при самодиссо-
самодиссоциации, для данной стехиометрической концентрации вводимого
основания может быть подсчитана с достаточной точностью, если
принять, что автопротолиз не зависит от других процессов само-
самодиссоциации и что концентрация ионов тл, образовавшихся при
автопротолизе, определяется только концентрацией HSOj, в то
II. ФИЗИЧЕСКИЕ СВОЙСТВА
125
время как концентрация ионов и молекул, образовавшихся при дру-
других процессах самодиссоциации, т^, определяется8 только концен-
концентрацией H3OHSO4. Значения та могут быть получены из таблич-
табличных данных для сильного основания; значения т^ — из табличных
данных для воды. Эти значения приведены в табл. 25. Кроме того,
известно, что md = тх + т~.
тнэоГ
0,00
0,01
0,02
0,03
0,04
0,06
0,08
0,10
0,0263
0,0191
0,0142
0,0110
0,0088
0,0063
0,0045
0,0036
Таблица 25. Значения тх
тНзО+
0,00
0,01
0,02
0,03
0,04
0,06
0,08
0,10
0,0160
0,0081
0,0041
0,0030
0,0020
0,0014
0,0009
0,0009
mHSOl
0,12
0,14
0,16
0,18
0,20
0,24
0,28
И Шр
та
0,0028
0,0023
0,0017
0,0012
0,0010
0,0003
0,0001
тНаО+
0,12
0,14
0,16
0,18
0,20
0,24
0,28
0,0006
0,0006
0,0004
0,0004
0,0003
0,0002
0,0000
Среди первых исследователей возникли существенные разногласия,
вызванные непониманием ими процесса самодиссоциации серной
кислоты. Значение процесса самодиссоциации растворителя было
впервые выяснено Гамметом и Дейрупом 130. Они изучали в качестве
растворителя серную кислоту, содержащую такое количество воды,
которое вызывало понижение температуры замерзания растворителя
до 9,8—10° С. При таком составе в заметной мере удалось подавить
самодиссоциацию растворителя. Тогда выражение для 0, равного
9,8—10° С, принимает вид
E2)
Для понижения точки замерзания кислоты можно использовать
также небольшие количества других оснований, например KHSO4.
Для приблизительной оценки величины v в равенстве E2) многие
исследователи используют значение i, называемого г-фактором
Вант-Гоффа.
Однако описанный прием не позволяет подавить самодиссоциа-
самодиссоциацию полностью. Кроме того, присутствие третьего электролита,
например H3OHSO4 или KHSO4, усложняет происходящие про-
процессы 27' 94. Трудности возникают и тогда, когда растворенное
вещество способно отнимать воду от серной кислоты, как, например,
ангидриды карбоновых кислот. Так, ангидрид уксусной кислоты,
растворенный в серной кислоте, содержащей небольшое количество
воды, вызывает понижение точки замерзания, соответствующее
126
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
v = 2. Из этого факта можно ошибочно заключить, что в данном
случае происходит протонизация:
(СН3СОJО + H2SO4 = (СН3СОJОН+ + HSOJ E3)
Однако понижению точки замерзания 100%-ной H2SO4 соответствует
v = 4. Это обстоятельство и то, что измерениями электропровод-
электропроводности подтверждено образование кислого сернокислого иона, пока-
показывают: ангидрид уксусной кислоты дегидратирует серную кислоту,
образуя RCO2H2-hoh по уравнению
(RCOJO + 3H2 SO4 = 2RCO2HJ + HSOj + HS2Oj E1)
Лайстен 163' 1М отмечал, что различие в криоскопическом пове-
поведении вещества, растворенного в различных смесях серной кислоты,
содержащей небольшие количества воды или других электролитов,
позволяет получить более подробные сведения об ионизации раство-
растворенного вещества, нежели это удается сделать при изучении поведе-
поведения его в каждом из растворителей в отдельности. Лайстен предло-
предложил использовать следующие криоскопические смеси:
а) серную кислоту, содержащую воду, другими словами, содер-
содержащую ионы Н3О+ и HSOJ;
б) серную кислоту, содержащую H2S2O7 и HS2O7;
в) серную кислоту, содержащую HS2O7 и HSO4;
г) серную кислоту, содержащую HgSOJ и Н3О+;
д) серную кислоту, содержащую HgSOJ и H2S2O7.
В общем виде реакция серной килоты с некоторым веществом S
может быть представлена уравнением
S + 7?H2SO4 >¦ aSx + uHSOl + cHsSOJ + rfHaO+efl^SaOj E5)
Увеличение или уменьшение общего числа молей частиц v в растворе
в результате введения одного моля Н2О, H2S2O7, HSO4 или HgSOJ
локазано ниже:
Вводимое вещество Н2О HSOj H2S2O7 II3SOJ
Изменение v в криоскопической смеси
а) 2 1 -2 -1
б) —1 0 1 0
в) 0 1 0 —1
г) 0 —1 0 1
д) —1 —1 1 1
Данное растворенное вещество будет вести себя неодинаково
по меньшей мере в трех из названных криоскопических смесях, и так
как коэффициенты Ъ или с и либо d: или е должны равняться нулю, то
оценка криоскопическпх свойств вещества в трех подходящих
растворителях (криоскопических смесях) дает возможность опреде-
определить коэффициент а и либо Ъ или с, и либо d или е. Таким образом,
однозначно определяется способ ионизации вещества.
б. Осмотические коэффициенты. Трефферс и Гаммет 207 первыми
предположили, что растворы электролитов в H,SO4 ведут себя как
И. ФИЗИЧЕСКИЕ СВОЙСТВА
127
1.08
I.t
•S,
0,96
0,92
Ph2COH
идеальные и что силы взаимодействия между ионами в серной кислоте
весьма малы. Джиллеспи 90 предположил, что это может быть обу-
обусловлено высоким значением диэлектрической проницаемости H2SO4
и показал, что отклонения от идеальности, которые могут наблю-
наблюдаться, можно объяснить сольватацией ионов. При этом он приписал
ионам произвольные числа сольватации. Однако измерения е серной
кислоты (см. П,А,6) показали: диэлектрическая проницаемость
велика, но не настолько, чтобы
можно было игнорировать меж-
межионные силы. Тщательные
измерения температур замерза-
замерзания полностью ионизированных
растворов некоторых кислых
сульфатов позволили обнару-
обнаружить межионные силы и выяс-
выяснить в общих чертах причины
отклонений свойств реальных
растворов от идеальных. Это
было выполнено Бэссом и
Na
0,2
О Л
0,6
Рис. 30. Зависимость осмотического
коэффициента Ф от ионной силы рас-
раствора /:
1 — теоретические кривые с указанными а,
рассчитанные по формуле 1 + Фэл; ПЛ —
предельная линия рассчитанная из уравне-
уравнения Дебая — Хюккеля; 2 — эксперименталь-
экспериментальные кривые.
Джиллеспи 8, применившими
метод криоскопического равно-
равновесия. На основании получен-
полученных результатов по уравнению
D6) были рассчитаны моляль-
ные осмотические коэффи-
коэффициенты. Рис. 30 показывает
зависимость осмотического ко-
коэффициента от ионной силы
раствора в степени 1/2, т. е.
/1/г. Интерпретация получен-
полученных кривых сложна, потому
что они относятся не к раство-
растворам только электролитов, а к смеси электролита с молекулами
и ионами, образующимися при самодиссоциации растворителя.
Концентрация ионов, образующихся в процессе самодиссоциации,
ничтожно мала при относительно высокой концентрации электролита,
однако роль этих ионов увеличивается с уменьшением концентрации
электролита, и при бесконечном разбавлении раствор будет состоять
только из них. Ионная сила раствора не может быть меньше ионной
силы 100%-ной серной кислоты, поэтому при f1* = 0,0189 и Ф = 0,98,
т. е. в случае 100%-ной серной кислоты, все кривые сходятся в одной
точке. Зависимость осмотического коэффициента от концентрации
может быть выражена следующим уравнением:
E6)
128
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
где Фэл — вклад электростатических межионных сил в осмотический коэффи-
коэффициент, который может быть вычислен по теории Дебая — Хюккеля? b — про-
произвольный коэффициент; b^jrii — вклад ионной сольватации в осмотический
коэффициент.
Доля электростатических межионных сил была подсчитана при
диэлектрической проницаемости, равной 120, и для различных зна-
значений наибольшего сближения катиона и аниона (т. е. параметра а).
Значения аи Ъ, которые лучше всего соответствуют эксперименталь-
экспериментальной кривой зависимости осмотического коэффициента от величины
f1'-! приведены в табл. 26.
о
Таблица 26. Параметры а и в и числа сольватации s, рассчитанные
по криоскопическим данным
Катион
Ва2+
Na+
Li+
К+
NH?
Ag+
Н3О+
Ag* (Kb = 1)
Н3О+(Кв = 1)
(CH3JCOH+
CH3PhCOH+
Ph2COH+
(n-CH3CeH4JCOH+
<п-С1С6Н4JСОН+
PhNH3+
Ph2NH2+
Ph3NH+
0
a
10
10
10
10
10
2
2
10
10
—
—
—
—
—
e
0,32
0,14
0,08
0,07
0,015
0
0
0,07
0,06
0,025
0,14
0,31
0,34
0,15
0,025
0,14
0,31
s*
11,5
3,8
2,6
2,4
1,2
_
2,4
2,1
1,5
3,8
7,2
7,8
4,0
1,5
3,8
7,2
6,5
3,0
2,3
2,1
1,2
2,1
1,8
1,0
1,4
13
1,1
0.5
0,8
0,6
0,6
3,0
2,0
3,4
0,6
0,2
4,0
4,0
. .
* Данные первой графы рассчитаны по уравнению E7), второй [графы—по уравнению
E8); в третьей графе помещены значения для водных растворов118.
Из данных этой таблицы нетрудно заметить, что параметр а для всех
растворов в серной кислоте, за исключением H3OHSO4 и AgHSO4,
равен 10 А, что значительно больше соответствующих значений для
электролитов в водных растворах. Эта величина, однако, не будет
казаться очень большой, если допустить, как это сделали Вик и
Айген 237, что для неассоциированных электролитов между двумя
сталкивающимися противоположно заряженными ионами всегда
находится по крайней мере одна молекула растворителя. Действи-
Действительно, основываясь на этом допущении, из кристаллических радиу-
радиусов ионов и значений радиусов молекулы H2SO4 и кислого сульфат-
иона можно получить 8 значение а, приблизительно равное 10 А.
II. ФИЗИЧЕСКИЕ СВОЙСТВА
129
Теоретическая кривая зависимости осмотического коэффициента от
величины I1* при а = 10 и Ъ = 0 представлена на рис. 30.
Для всех доступных концентраций растворов веществ в серной
кислоте вклад электростатических межионных сил в осмотический
коэффициент хотя и не мал, но почти постоянен (см. расчетную кри-
кривую при а = 10 А). Это следствие, скорее всего, высокой диэлектри-
диэлектрической проницаемости и относительно больших размеров молекул
серной кислоты и кислого сульфат-иона. Различия в поведении
электролитов обусловлены различным характером взаимодействия
растворителя с растворенным веществом.
Причина низких величин осмотических коэффициентов раство-
растворов AgHSO4 и H3OHSO4 еще не вполне ясна. По-видимому, разбав-
разбавленные растворы AgHSO4 и H3OHSO4 в серной кислоте диссоцииро-
диссоциированы не полностью. Для AgHSO4 это может быть объяснено отчасти
ковалентным характером связи,- а для H3OHSO4 — сильной водо-
водородной связью между Н3О+ и HSOJ, что обнаружено при изучении
колебательных спектров растворов воды в серной кислоте (Е. А. Ро-
Робинсон, неопубликованные результаты). В самом деле, если принять,
что степень диссоциации для этих двух электролитов равна единице,
то их кривые зависимости Ф от 11г очень похожи на кривые для дру-
других электролитов.
в. Ионная сольватация. Робинсон и Стоке 214 привели метод
количественного расчета чисел сольватации по коэффициентам актив-
активности водных растворов электролитов, предполагая, что сольвати-
рованные ионы и молекулы растворителя образуют идеальные рас-
растворы (т. е. подчиняются закону Рауля). Применив их метод к рас-
растворам веществ в серной кислоте, Бэсс и др.8 показали, что параметр
Ъ связан с числом сольватации s электролита выражением
b=Bs-v)/40,8 E7)
Величины s, вычисленные по этому уравнению, приведены в табл. 26.
Глюкауф 118 отметил, что такой расчет не учитывает энтропию
смешения молекул растворителя и сольватированных ионов, которые
могут иметь очень различные размеры. Он показал, каким образом
можно учесть этот дополнительный эффект. Полученные им числа
сольватации для водных растворов электролитов имели меньшие
значения и изменялись с изменением размеров ионов более законо-
закономерно, чем величины, найденные Робинсоном и Стоксом. Согласно
Глюкауфу, параметр Ъ для сернокислых растворов может быть
вычислен из следующего выражения:
ь=-
ГУ
40,8
E8)
где г — доля мольного объема несольватированного электролита в мольном
объеме растворителя.
9 Заказ 90
130
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Величины чисел сольватации, вычисленные по уравнению E8),
также представлены в табл. 26.
Так как все изученные вещества имели в качестве аниона кислый
сульфат-ион, числа сольватации должны выражать относительные-
числа сольватации катионов. Если вследствие большого размера
кислого сульфат-иона допустить, что он не сольватируется, то эти
величины могли бы рассматриваться как числа сольватации ка-
катионов.
Уравнение E8) позволяет получить более достоверные значения
чисел сольватации для сильно сольватированных и поэтому крупных
ионов бария, а также органических катионов, чем уравнение E7).
Числа сольватации, определенные по уравнению Глюкауфа, при-
приближенно соответствуют ожидаемому изменению s с размером иона.
Поведение лития аномально, у него число сольватации меньше, чем
у натрия. Это объясняется тем, что координационное число иона
лития для молекул серной кислоты равно только трем, в то время как
Для иона натрия оно равно четырем 8' 93. По-видимому, числа соль-
сольватации в серной кислоте больше, чем в воде (см. табл. 26), что
согласуется с большей полярностью молекул серной кислоты по*
сравнению с молекулами воды.
г. Неэлектролиты. 2,4,6-Тринитротолуол дает несколько боль-
большее понижение точки замерзания, чем это следовало бы ожидать
от раствора неэлектролита, поэтому первоначально заключили, что
он является слабым основанием 83. Однако Брэнд и др. 21' 22, исполь-
используя сцектрофотометрические методы исследования, не обнаружили
ионизации этого соединения. Брэйфорд и Виат 27 показали, что*
пикриновая кислота, тринитробензол и тринитротолуол вызы-
вызывают в серной кислоте, содержащей немного воды, более значитель-
значительное, а в H2SO4 с небольшими количествами KHSO4 или H2S2O7 —
меньшее значительное понижение температуры замерзания, чем это
следует ожидать от раствора неэлектролита. Авторы 27 полагают,
что данные вещества действительно являются неэлектролитами, но
вода высаливает их из серной кислоты, a KHSO4 и H2S2O7 оказы-
оказывают обратное действие, т. е. растворимость неэлектролита должна
быть больше в присутствии KHSO4 или H2S2O7 и меньше — в при-
присутствии воды. Это было подтверждено экспериментально 160, хотя
увеличение растворимости неэлектролита в присутствии KHSO4
очень мало.
Джиллеспи исследовал раствор нитробензола в H2SO4 с неболь-
небольшой примесью воды и обнаружил, что он дает аномально высокое
понижение температуры замерзания. На основании этого автор
предположил, что нитробензол ведет себя как слабое основание.
Величина основности нитробензола, полученная из криоскопических
измерений его раствора в серной кислоте с примесью воды 83, однако,
была выше величины основности, полученной измерениями электро-
электропроводности 108. Но если использовать для криоскопических измере-
II. ФИЗИЧЕСКИЕ СВОЙСТВА
131
ний 100%-ную кислоту, то криоскопические данные удовлетвори-
удовлетворительно согласуются с результатами измерений электропровод-
электропроводности
94
2. Плотность и кажущийся мольный объем
Плотности растворов 71> 110 большинства веществ были измерены
при 25° С. Растворы NaHSO4 и KHSO4 были изучены также при 10
и 40° С. По величине кажущегося мольного объема электролита V,
который может быть вычислен из плотности раствора, можно полу-
получить дополнительную информацию о взаимодействии между ионами
электролита и молекулами растворителя. Если принять, что моль-
мольный объем HSO4 = 54 см3 (т. е. такой же, как у H2SO4), и это зна-
значение вычесть из кажущегося мольного объема полностью ионизи-
ионизированного электролита, то получится кажущийся мольный объем
катиона Vm+- Если сравнить затем это значение с объемом катиона,
рассчитанным из его кристаллического радиуса Vm+, разница 8V
покажет сжатие катиона в растворителе. Логично предположить,
что это сжатие пропорционально силе взаимодействия «ион — раство-
растворитель», или сольватации. Тогда, зная число сольватации для ка-
какого-либо иона, например Na+, полученное из криоскопических
Измерений, МОЖНО ВЫЧИСЛИТЬ ИЗ ПрОСТОЙ Пропорции 6FNa+/sNa+ =
=¦ SFm+/sm+числа сольватации s для других катионов. Числа соль-
сольватации, вычисленные таким путем, поразительно совпадают с чис-
числами сольватации, полученными экспериментально из криоскопи-
криоскопических измерений; вероятно, они являются мерой первичной соль-
сольватации.
Кажущиеся мольные объемы катионов в сернокислом растворе
можно сравнить с их парциальными объемами в водном растворе 40,
которые представлены в табл. 27.
Таблица 27¦ Кажущиеся мольные объемы (в см?) и числа сольватации
катионов в сернокислых растворах их кислых сульфатов
Катион
Li
Na
К
Rb
Cs
Ag
Tl
NH4
H3O
Ca
Sr
Ba
V
47
46
53
59
68
53
62
59
61
84
87
96
¦—7
-8
—1
5
14
—1
8
5
7
-24
-21
—12
VM+ (H,O)
-7,0
-7,5
1,7
7,7
15,1
-7,0
7,7
—
12,0
-29,7
—30,2
-24,3
r, A
0,78
0,98
1,33
1,49
165
1,13
1,49
1,48
1,48
1,06
1,27
1,43
v'm+
1,2
2,4
5,9
8,3
¦ 11,3
3,6
8,3
8,2
8,2
3,0
5,8
7,4
ay
-8
-10
—7
-3
3
—5
0
о
—1
—27
27
—19
8расч
2
3
2
Г
0
1,5
0
1
0.3
8
8
о
ъксп
2,3
3,0
2,1
—
—
2,1
—
1,2
1,8
—
—
6,5
132
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Заметно сходство этих величин в двух растворителях. Кутюр
и Лейдлер 40 показали, что парциальный мольный объем ионов
в водном растворе пропорционален заряду z и кубу ионного ра-
радиуса г3:
^(HO) 3-26z E9)
I8z F0)
II. ФИЗИЧЕСКИЕ СВОЙСТВА
13»
Аналогичное соотношение
/,2 1,6
' MHSO4
Рис. 31. Зависимость вязкости сер-
сернокислых растворов некоторых элек-
электролитов от концентрации при
25° С:
1 — NH4S04; 2 — KHS04; 3 — NaHSO<;
4 — LiHSO4; S — Ba(HSO4J; 6—Sr(HSO4J.
по-видимому, справедливо для
кажущегося мольного объема ка^
тиона в сернокислом растворе.
Физический смысл этих эмпири-
эмпирических соотношений еще не вы-
выяснен.
3. Вязкость
График зависимости вязкости
растворов электролитов в серной
кислоте от их концентрации по-
показан на рис. 31. Хотя сама сер-
серная кислота обладает очень вы-
высокой вязкостью, последняя уве-
увеличивается при введении электро-
электролитов в H2SO4, причем влияние
электролитов на вязкость серной
кислоты гораздо больше, чем на
вязкость воды. Это снова указы-
указывает на значительное взаимодей-
взаимодействие между ионами электролита
и молекулами растворителя в
сернокислых растворах. Относи-
^суиипполыл. растворах, относи-
относительная вязкость растворов электролитов может быть выражена
уравнением 145
?1 + Ае''+Вс + Сс* F1)
Для сернокислых растворов значение Ас1/2 невелико, поэтому
вязкость является почти линейной функцией моляльной концентра-
концентрации приблизительно до концентрации 0,4 м для одно-одновалентных
электролитов и 0,2 м для одно-двухвалентных электролитов. Вели-
Величина параметра В для растворов различных электролитов в H2SO4
и Н2О приведена ниже:
Катион Н3О+ NHf K+ Na+ Li+ Ba2+
Величина В
в H2SO4 —0,4 —0,1 0,2 0,8 1,0 4,4
в Н2О 0,07 —0,01 —0,007 0,09 0,15 0,22
Если предположить, что кислый сульфат-ион оказывает незначи-
незначительное влияние на вязкость, то величина вязкости будет опреде-
определяться катионами. Полагают126, что член уравнения F1), содержа-
содержащий В, является мерой взаимодействия иона с растворителем.
Интересно отметить, что ион аммония, как видно по величинам пара-
параметра В, несколько уменьшает вязкость серной кислоты и воды 126.
Ион гидроксония заметно уменьшает вязкость серной кислоты, в то
время как в воде наблюдается обратный эффект. Уменьшение вяз-
вязкости в присутствии иона Н3О+ можно считать результатом того, что-
он разрушает структуру серной кислоты. Очевидно, в этой среде он
не так сильно сольватирован, как в воде.
4. Электропроводность
а. Протонная электропроводность H3SO4 и HSO4. Впервые-
работы по измерению удельной электропроводности сернокислых
растворов выполнили Оддо и Скандола 2о°, Ханч 131, Бержу13,
Кендолл и др. 147, однако полученные ими результаты и выводы
сильно различались. Это объясняется отчасти незнанием ими про-
процесса самодиссоциации и ее влияния на электропроводность раство-
растворов. Только сравнительно недавно были проведены детальные и точ-
точные измерения электропроводности растворов электролитов в серной
кислоте. Удельные электропроводности растворов кислых сульфатов
щелочных металлов и кислых сульфатов некоторых других катио-
катионов 8 приведены в табл. 28. При низких концентрациях электролитов
электропроводность растворов этих веществ почти одинакова.
Очевидно, в этом случае ток переносят главным образом ионы
HSOJ. Это было подтверждено109 измерениями кажущихся чисел пере-
переноса катионов металлов в сернокислых растворах их кислых:
сульфатов при 25е С:
Ион Ag+ K> Na+ Li
mMHSG. 0,2490 0,31 0,62 1,23 0,79 0,56
t+ /.' 0,026 0,022 0,030 0.025 0,021 0,013-
Ион Ba2+ Sr2+
mMHS0< 0,17 0,23 0,31 0,80 0,21 0,82
t+ . * 0,009 0,010 0,008 0,004 0,007 0,003
Результаты этих измерений показывают, что в растворе кислого*
сульфата металла только небольшая часть электрического тока
переносится катионом.
То обстоятельство, что ион H3SO4 обладает аномально высокой
подвижностью, подтверждается результатами кондуктометрического
кислотно-основного титрования в серной кислоте 72> 127: прибавле-
прибавление KHSO4 к раствору кислоты, например к HB(HSO4L, силвно;
о
о
I1
.'I
о а
а, а.
о н
а в
1
с?
а
о
с
В
о
1
ft
оо
«# см со
ОО тч ю
«#" L-o" ю"
I I
со
«а*
I I I I I I I
СО ОЭ СО
U0 |>. -^ч
«а*
«а*
ю"
оо"
см
ГО
ГО 1>- ОО sf О
СМ. 1>-^ |>-^ 00 О
*# «#" *#" *#" ю"
гч
о
uo"
00
го"
СО
«а*
со
ГО
«#"
ГО
ГО
см"
с^
^ч
:м"
ГО
см_
к}<"
,99
го
-^ч
о
со
«*"
|>-
«*~
,32
см
«*"
NI1"
СО
см
«#
СО
СО
го"
1
1
СО
СО
го"
ГО_ *#
го" "
СО СО СО СО СО СО" ГО"
8 I
г го
о
00
го"
"О со со о
1> О •* 41
го го" го" го"
со см
СО OS
оо" см"
го го го го го" го"
со
-^ч Oi
го го го го* см" см"
го
см
см"
ГО О
ГО vt<
см" оо"
со
см
оо"
СО
ГО
см"
О5
го
см"
1,520
1,252
,145
1,068
S?
о_
со
го
UO
1,258
1,068
1,051
1,558
,274
СО
LO
1,073
1,052
со
OS
СМ
|>-
1,08
1,052
со
,29
1,08
1,052
1,58
,26
со
1,075
го
LO
о
со
,29
,08
1053
,590
,278
161
,075
го
LO
о
,530
,249
130
8
1,046
,281
OS
со
,068
о
,61
29
го
со
,073
1,051
3?
270
ГО
UO
.073
1,052
,56
270
го
LO
,073
1,052
со
LO
ГО
LO
073
1,052
,55
270
LO
073
1,052
со
LO
270
го
073
1,052
О
X X
о "
О О
S Р 9 о
ч. О О
о S й §
~\ —м
Zi zl w ii
5 I м
Я X
о <
т* f-ч О
^- W4 НН НН s -
<Н <^Н Ь^ |_j ^^
'Й U ,^J
CJ
а и
II. ФИЗИЧЕСКИЕ СВОЙСТВА
135
уменьшает электропроводность, так как очень подвижный ион H3SOt
заменяется относительно мало подвижным ионом К+. Подвижности
НОЧ ,О
Рис. 32. Механизм
протонной элек-
электропроводности
иона H3SO? в сер-
серной кислоте.
но/ хон
HOv°
HV°
VOH
оч * он
V
но/Лон
оЛш
VOH
о' хон
оч он
не
s
но/ хон
3
ионов в серноп кислоте довольно малы вследствие высокой вязкости
H2SO4. Но ионы H3SOJ и HSOJ, однако, являются наиболее подвиж-
подвижными из всех ионов в H2SO4 и из большинства ионов в воде; их под-
подвижности сравнимы с подвиж-
подвижное тямп сверхпроводящих ионов
Н3О+ и ОН". Высокая вязкость
растворителя не влияет на по-
подвижности ионов H3SO^ и
HSO^, так как их электропро-
электропроводность обусловлена перехо-
переходом протона (как и Н3О+ , и ОН"
в воде). Механизм электропро-
электропроводности для ионов H3SO1[ очень
наглядно изображен на рис. 32.
Последовательное перемещение
протонов вдоль водородных
связей приводит к эффективному
движению ионов H3SO4 через
раствор без необходимости дей-
действительного движения отдель-
отдельных ионов. Детали этого меха-
механизма, в частности природа
стадии, определяющей скорость,
в настоящее время дискутиру-
дискутируются 7в> 123. В общем, для того
чтобы образовались водородные
связи, способные к передаче
протона в нужном направлении,
требуется определенное количество молекулярного движения и
ориентация. Образование таких водородных связей, по-видимому,,
и является стадией, определяющей скорость процесса.
б. Влияние взаимодействия ион — растворитель на подвижность-
HSOj. При низких концентрациях простых оснований в серной
0,7 0,2 0,3
с, моль/кг раствора
Рис. 33. Зависимость удельной элек-
электропроводности растворов некоторых
веществ от концентрации при 25° С:
1 — ацетон; 2 — ди-п-толилкетон A и 2 сме-
смещены по оси ординат на 1,0); 3—CsHSO4;.
4 — KHSO4; S — NaHSOi; 6 — HB(HSO4)«;
7 — H2S2O7 F и 7 смещены по оси абсцисс на
0,05 молъ/кг).
136
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
II. ФИЗИЧЕСКИЕ СВОЙСТВА
137
кислоте разница между величинами их электропроводностей незна-
незначительна, однако при увеличении концентраций эта разница воз-
возрастает. Графики зависимости удельных электропроводностей кислых
сульфатов некоторых одновалентных металлов от концентрации
представлены на рис. 33. Форма кривых, в общем, одинакова.
При низких концентрациях кривые к оси абсцисс обращены выпук-
выпуклостями, что объясняется подавлением самодиссоциации раствори-
растворителя, а затем, при относительно высоких концентрациях, ¦— вогну-
вогнутостями. Это показывает, что подвижность HSO4, очевидно, умень-
уменьшается с увеличением концентрации. Более того, это уменьшение
подвижности для растворов кислых сульфатов различных металлов
различно. Разница между электропроводностями растворов различ-
различных электролитов слишком велика, чтобы ее можно было объяснить
•столь малыми различиями в подвижностях катионов, В связи с этим
был сделан вывод о том, что катионы влияют на подвижность
HSOl-иона 76. т.
Сделав допущение, что вклад ионов металлов в электропровод-
электропроводность невелик, и зная удельные электропроводности растворов
кислых сульфатов, можно вычислить эквивалентные электропровод-
электропроводности, или подвижности кислого сульфат-иона (К_) (табл. 29).
Таблица 29. Подвижность иона HSO4"
в сернокислых
растворах кислых
сульфатов различной моляльной концентрации
Катион элек-
электролита
Li+
Na+
К +
Rb+
Cs+
JNTH4+
0,000
151,2
151,2
151,2
151,2
151,6
151,2
e.oio
148,5
149,0
149,0
—
—
149,0
1— при концентрации
0,020
143,0 "
143,6
144,0
—
—
144,8
0,040
129,9
130,2
131,7
135.5
135;5
132,8
0,060
118,3
119,4
121,5
125,3
125,5
122,3
0,120
98,5
99,8
102,7
106,9
109,2
104,6
0,180
86,2
87,7
91,4
96,1
98,9
93,7
0,24 0
76,7
79,3
83,2
89,0
91,5
86,1
Из этих данных видно, что электропроводность HSOJ для любых
¦концентраций прямо пропорциональна радиусу катиона, т. е. обратно
пропорциональна степени сольватации его. Исходя из этого, ло-
логично предположить, что те молекулы растворителя, которые соль-
ватируют катион, не могут принять участие в процессе передачи
протона так легко, как это делают «свободные» молекулы раство-
растворителя. Таким образом, подвижность кислого сульфат-иона умень-
уменьшается с увеличением концентрации кислого сульфата щелочного
металла пропорционально силе взаимодействия катионов с раствори-
растворителем.
По-видимому, ион лития сильнее взаимодействует с растворите-
растворителем, чем ион натрия, хотя криоскопические измерения дают для Li+
число сольватации меньше, чем для Na+. Можно предположить, что-
взаимодействие ион — растворитель, оцениваемое по величине элек-
электропроводности, не ограничивается одним сольватированным слоем,
а распространяется на несколько других слоев, окружающих ион,
т. е. учитывается вторичная сольватация, в то время как криоско-
криоскопические измерения дают информацию только о первичной сольва-
сольватации.
Следует отметить, что электропроводности растворов H3OHSO4
и AgHSO4 имеют нормальные значения и не свидетельствуют о не-
неполной диссоциации, как это вытекает из криоскопических измере-
измерений. Удовлетворительного объяснения этому пока не найдено.
Возможно, что молекулы воды полностью ионизированы в виде
H3O+HSOJ, но при этом ионные пары диссоциированы непол-
неполностью, а всякая неполная диссоциация не влияет на электропровод-
электропроводность раствора, если продолжительность жизни ионной пары короче,
чем среднее время между последовательными переходами протона.
в. Вклад асимметричного автопротолиза в электропроводность.
Виат 245 отметил, что эквивалентная электропроводность кислого
сульфат-иона очень сильно зависит от концентрации, особенно для
концентраций меньше 0,1 м, в то время как эквивалентная электро-
электропроводность водного раствора НС1 в этом диапазоне концентраций
изменяется всего лишь на 2%. Он предположил, что в величину
электропроводности может вносить вклад асимметричная диссоциа-
диссоциация растворителя под действием внешнего электрического поля, т. е.
в растворе имеется небольшая тенденция к движению протонов от
одних нейтральных молекул к соседним (т. е. к диссоциации) в на-
направлении поля, и это сказывается на электропроводности. Если а —
расстояние, на которое должны сблизиться при диффузии ионы
автопротолиза под действием поля для того, чтобы смогла произойти
нейтрализация комплекса, образованного с помощью водородных
связей, то этот процесс можно рассматривать как процесс удаления
зарядов на расстояние а за время автопротолиза. Последний вносит
специфический вклад в электропроводность. Виат вывел следующее
выражение для вклада асимметричного автопротолиза в электропро-
электропроводность растворителя типа НА:
ха = 3.733 • 10-и кр [Н2А+] [А-] Dр1
F2)
где кр — константа скорости рекомбинации ионов автопротолиза, h — эффек-
эффективная напряженность внутреннего поля прн напряженности внешнего-
поля 1 в/см.
Ясно, что вклад определяется константой автопротолиза, которая
для серной кислоты относительно велика. Виат показал, что хотя
напряженность внутреннего поля h и кр точно не были известны,
вклад асимметричного автопротолиза растворителя в электропровод-
электропроводность 100%-ной кислоты составляет около 30%. Вот почему гораздо
-138
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
II. ФИЗИЧЕСКИЕ СВОЙСТВА
139
меньшая величина константы автопротолиза воды незначительно
увеличивает электропроводность. Если считать, что удельная элек-
электропроводность раствора кислого сульфата металла "&' обусловлена
только ионами H3SO4 и HSOJ, тогда
где т+ и т. — моляльности H3SOj и HSOj;^ р — плотность раствора; X. —
подвижность иона HSO4; |х — отношение подвпжностей ионов H3SOJ и HSOj;
ха — вклад асимметричного автопротолиза.
Если Х_и [I постоянные, то зависимость 103 к' от цт+ + т_ должна
выражаться прямой линией, отсекающей от оси ординат отрезок,
15 -
70
0,08
0,12
Рис. 34. График зависимости удель- Рис. 35. Определение 7 по электропро-
ной электропроводности от jxm++m_ водности:
для раствора KHSO4 в серной ки- i — сильный электролит, у > 1; г — стан-
слоте. График дает ВОЗМОЖНОСТЬ дартный электролит, у = 1 (например,
определить вклад асимметричной KHso4); 3 - слабый электролит, v < 1.
диссоциации в электропроводность
серной кислоты.
равный ха. Исходя из положения минимума электропроводности
системы Н2О — SO3, находящегося вблизи точки с концентрацией
100% H2SO4, можно заключить103, что ц = 1,5. Зная ц, т+ и т_,
вычисленные из константы самодиссоциации растворителя, удалось
подтвердить, что для кислых сульфатов уравнение F3) линейно.
От оси ординат отсекается отрезок ха = 0,38-10; наклон прямой
соответствует Х_ и равен 166 при 25° С (рис.34). Таким образом,
при 25° С вклад асимметричной диссоциации в электропроводность
кислоты составляет 36%, а величина Х_ остается постоянной почти
до концентрации 0,1 м.
г. Определение у. Так как в сернокислом растворе подвижности
ионов H3SO4 и HSOJ гораздо выше, чем других ионов, электро-
электропроводности растворов кислот и оснований в серной кислоте почти
полностью определяются концентрациями H3SO| и HSOJ. Следова-
Следовательно, у, т. е. число молей HSOJ или H3SO4, образованных одним
молем электролита, может быть найдено, исходя из электропровод-
электропроводности раствора какой-либо кислоты или основания. Следует ожидать,
что при данной концентрации электролита электропроводность
дигидросульфата, например Ba(HSO4J, будет приблизительно вдвое
выше электропроводности моногидросульфата, например NaHSO4,
но при низких концентрациях этого не происходит, что объясняется
различной степенью подавления автопротолиза растворителя каждым
из электролитов. Однако если сравнить концентрацию стандартного^
раствора моногидросульфа-
моногидросульфата, например KHSO4, необ-
необходимую для достижения
определенной электропровод-
электропроводности, с концентрацией лю-
любого другого электролита,
имеющего такую же электро-
электропроводность, то можно непо-
непосредственно рассчитать вели-
величину у, так как степени
подавления автопротолиза
для обоих сравниваемых
растворов одинаковы. Рис. 35
иллюстрирует применение
этого метода для сильных и
слабых электролитов. Для
сильного электролита у =
для слабого
„V 0,9 В
'$ 0,92
\ 0,88
& ОМ
О
0,2 0,4 0,6
с, моль /кг раствора
Рис. 36. Зависимость удельной электро-
электропроводности сернокислых растворов не-
неэлектролитов от концентрации при 25° С:
1 — CH3SO2F; 2 — CH3SO2C1; 3 — SO2C12; 4 —
C2H6SO2C1; 5 — Ph2SO2; 6 — о-динитробензол;
7 — 1,3,5-тринитробензол.
= ОВ/ОА, а
у = ОВ/ОС.
Лайлер 16Э попыталась сделать поправку на эффект электропро-
электропроводности ионизированной части слабого электролита, так как боль-
большинство неэлектролитов уменьшает электропроводность серной
кислоты. Ей удалось вычислить константы ионизации основных
нитросоединений, которые гораздо лучше согласуются с константами,
полученными из криоскопических измерений, нежели данные Джил-
леспи и Соломонса 108.
д. Неэлектролиты. Электропроводность растворов неэлектроли-
неэлектролитов ниже, чем электропроводность чистого растворителя (рис. 36).
Это может быть связано с уменьшением подвижностей ионов H3SO4
и HSO4 в присутствии неэлектролита н(или) уменьшением концентра-
концентрации ионов автопротолиза.
Уменьшение подвижностей ионов растворителя может быть
вызвано образованием сильных водородных связей между молеку-
молекулами растворенного вещества и растворителя, которое мешает ориен-
ориентации последних и, следовательно, снижает их эффективность в про-
процессе проводимости, обусловленной передачей протона. Уменьшение
концентрации ионов автопротолиза является результатом уменьшения
140
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
II. ФИЗИЧЕСКИЕ СВОЙСТВА
концентрации «свободных» молекул серной кислоты вследствие
перехода молекул растворителя в сольватную оболочку молекул
растворенного вещества.
Считают, что меньшая электропроводность растворов аромати-
ароматических полинитросоединений объясняется снижением концентра-
концентрации ионов автопротолиза, на основании чего можно рассчитать
соответствующие числа сольватации. Было найдено, что каждая
нитро-группа сольватирована одной молекулой серной кислоты 108.
Последняя работа Лайлер 169 по определению электропроводности
раствора KHSO4 в смеси H2SO4 — SO2C12 показала, что сульфурил-
хлорид влияет на электропроводность 100% -ной кислоты гораздо
больше, чем KHSO4. Из этого следует, что сульфурилхлорид,
являющийся неэлектролитом, больше влияет на концентрации ионов
автопротолиза, нежели на их подвижности. Последнее обстоятель-
обстоятельство подтверждает допущение, используемое при интерпретации
результатов исследования электропроводности полинитросоединений.
Однако такой упрощенный подход не полностью подтверждается
последними исследованиями электропроводности растворов некото-
некоторых алкилсульфохлоридов и алкилсульфофторидов. Графики зави-
зависимости электропроводности растворов некоторых сульфосоединении
от концентрации представлены на рис. 36. Несмотря на то что
CH3SO2F более слабое основание, чем CH3SO2C1, в небольших кон-
концентрациях оно меньше понижает электропроводность серной кис-
кислоты, нежели другие сульфурилгалогениды127. В то же время
относительно высокие концентрации этого соединения увеличивают
электропроводность, которая начинает значительно превосходить
электропроводность чистого растворителя. Этот факт пока не полу-
получил удовлетворительного объяснения.
е. Кондуктометрическое кислотно-основное титрование. Простые
основания. Реакции кислотно-основной нейтрализации проходят
в серной кислоте так же, как и в любом другом амфотерном раство-
растворителе. Реакция нейтрализации является обратной автопротолизу
растворителя, т. е.
H3SOJ + HSOi = 2H2SO4 F4)
Так как подвижности ионов H3SOJ и HSOJ гораздо выше подвиж-
ностей любых других ионов, удобно проводить реакции нейтрализа-
нейтрализации, измеряя при этом электропроводность раствора. По мере до-
добавления основания к кислоте электропроводность систем сначала
уменьшается. При этом сильно электропроводный ион H3SOt свя-
связывается с ионом HSOj. Далее электропроводность проходит через
минимум, после чего снова возрастает, так как в растворе увеличи-
увеличивается концентрация ионов HSO7 (рис. 37). Положение минимума
электропроводности зависит от констант диссоциации кислоты КА
и основания Кь; если известно значение одной константы, можно
вычислить значение другой, исходя из экспериментально определен-
0 0,5 1,0 1,5 2,0 О 0,5 1,0 1,5 2,0 2,5 3,0 3,5 '
Г
Рис. 37. Кривые кондуктометрического титрования слабых
оснований кислотами H2S2O7 и НЕЦНЭО^,, (г — мольное
отношение основания к кислоте):
1 — нитробензол A' — 0,102 м, 1" — 0,153 м по SOs в H2S2O7); 2 —
о-нитротолуол; 3 — м-нитротолуол; 4 — п-нитротолуол.
0,8
Г
1,2 О 0,2 О,Ч О,в 0,8 1,0
г-
Рис. 38. Изменение концентраций частиц (молекул и ионов)
в процессе титрования при 25Q С:
о — реакция сильной кислоты НА с сильным основанием; б — реакция
пиросерной кислоты с сильным основанием.
ного положения минимума электропроводности. Изменение электро-
электропроводности в течение кислотно-основной реакции можно выразить
математически. С этой целью было изучено поведение некоторых
оснований 74. Математическое выражение не учитывает асимметрич-
асимметричный автопротолиз растворителя, но это может сказаться только на
142
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
величине, но не на положении минимума электропроводности при
кислотно-основном титровании. Положение минимума электропро-
электропроводности может быть описано отношением г = Пв/пА, (пъ — число*
молей прибавленного основания, п&— исходное число молей кислоты).
Рис. 38 показывает, как изменяются концентрации всех ионов,
участвующих в реакции сильной кислоты НА и кислоты H2S2O7 при
концентрации кислот таНА или тнгд8о7 = 0,1 м.
Значения гмин могут быть вычислены из следующих выражений:
для реакции основание — НА kl
m
II. ФИЗИЧЕСКИЕ СВОЙСТВА
143
для реакции основание — H2S2O7
Гмин = 0,56 fl+-^) F6>
Расчетные гчин для различных величин Кх и К-в приведены ниже:
Сильная кислота (Ка = °°)
А'в , со 1,0 10 10~2 ю-з
/•„„„ 1,01 1.02 1,19 2,82 19,1
Сильное основание (Къ = со)
КА со 1,0 10"! 10~2 ю-з
гт,н 1,01 0.98 0,89 0,44 0,08
Пиросериая кислота
Къ со 1,0 10-1 ю~2 ю-з
гмяя 0,56 0,57 0,66 1,58 11,3
Вода
КА со 1,0 0,3 10-1 Ю
гыи„" 1.00 1,00 1,00 0,93 0,42
Сложные основания. Минимум электропроводности 0,1 м раствора
H2S2O7 при добавлении к нему простого основания, например
KHSO4. соответствует г = 0,56; однако в случае сложного основа-
основания, например Ph3COH, введенного в 0,1 м раствор H2S2O7, мини-
минимум будет наблюдаться при г ~ .'„' =0,36, потому что при иони-
зации каждая молекула Ph3COH кроме иона трифенилкарбония
образует одну молекулу воды, реагирующую с молекулой H2S2O7.
Следует ожидать, что азотная и мезитиленкарбоновая кислоты
ведут себя аналогично. В случае N2O5 каждая молекула, ионизи-
ионизируясь, образует два иона нитрония и молекулу воды. Минимум
Г) 2Я
электропроводности в этом случае соответствует г = ' =0,22.
Ионизация трихлорметилмезитилена в H2SO4 приводит к образо-
образованию иона карбония и хлорсульфоновоп кислоты (Р. Дж. Джиллеспн
и Е. А. Робинсон, неопубликованные результаты), согласно уравне-
уравнению
(CH3KC6H2CCl3 + 3H2SO4=(CH3KC6H2CClt + HClSO3 + H3O++2HSO; F7)
Было показано, что при кислотно-основном титровании рас-
раствора трихлорметилмезитилена в разбавленном олеуме минимум
электропроводности соответствует г = 0,37. В этом случае
пиросерная кислота служит как для нейтрализации основания
<СН3KС6Н2СС1$ HSO7, так и для образования HC1SO3:
(CH3KC6H2CC13+2H2S2O7=
F8)
Итак, при исходной концентрации H2S2O7, равной 0,1 м, теоретиче-
0 56
ский минимум электропроводности будет соответствовать г = , ' =
p,
= 0,36, что близко к экспериментальной величине 0,37. Мезитилхло-
рид ведет себя аналогично. Таким образом, кондуктометрическое
кислотно-основное титрование H2S2O7 сложным электролитом дает
информацию, дополняющую результаты криоскопии и измерении
электропроводности.
5. Термодинамические свойства
Киркбрайд и Виат 14Э определили парциальные мольные энталь-
энтальпии сернокислых растворов кислых сульфатов калия, аммония,
бария, а также уксусной кислоты и воды при 25° С. Результаты,
полученные ими для воды, хорошо согласуются с данными ранних
и менее подробных исследований Канцлера 156 и были подтверждены
новейшими работами Маунтфорда и Виата190. Было найдено, что
парциальные мольные энтальпии растворов Н2О заметно зависят
от концентрации, так как при низких концентрациях происходит
подавление самодиссоциации растворителя. Авторы обнаружили
также, что при добавлении воды к раствору, содержащему сульфат
металла, парциальная мольная энтальпия воды все же заметно
зависит от концентрации, подтвердив таким образом положение:
одна из стадий самодиссоциации, а именно ионная самодегидрата-
самодегидратация, не полностью подавляется кислыми сульфатами. Ими были
получены величины:
Хап = 3,4-10~4
д
ДЯап = 4,8 ккал/молъ
ДЯД = 6,2 ккал/молъ.
144
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Дакр и Виат 42 непосредственно измерили теплоту реакции
HB(HSO4L+KHSO4 = KB(HSO4L+H2SO4 F9)
Приняв, что HB(HSO4L является сильной кислотой, и учитывая
сольволиз иона H3SOJ, они получили для Д#ап значение 5,0 ±
± 0,2 ккал/молъ, что хорошо согласуется с результата^-косвенного
определения этой величины, представленными вышеЛ/По нашему
мнению, криоскопические данные при 10° С являются самыми точ-
точными характеристиками самодиссоциации, поэтому они были исполь-
использованы наряду со значениями А#ап и А#д для расчета констант
само диссоциации при 25 и 40° С, представленными в разделе Н,А,2.
Но эти данные 103 плохо согласуются с приведенными выше резуль-
результатами.
Теплота бесконечного разбавления насыщенного раствора 2,4,6-
тринитротолуола составляет менее 100 кал/молъ, а теплота растворе-
растворения самого вещества равна 4,6 ккал/молъ, что близко к теплоте
плавления 150. Полученные результаты свидетельствуют о том, что,
по всей вероятности, неэлектролиты находятся в растворе в виде
простых молекул, а не в сольватированной форме и что отклонение
свойств растворов от идеальных происходит вследствие превосход-
превосходства энтропии над тепловыми эффектами 150. Возможно, однако, что
теплота, необходимая для разрыва водородных связей между моле-
молекулами растворителя в процессе внедрения молекулы растворенного
вещества, приблизительно равна теплоте, выделяющейся при обра-
образовании водородных связей между нитро-группами тринитротолуола
и растворителем и что совпадение теплот растворения и плавления
случайно.
6. Спектроскопия растворов серной кислоты
а. Инфракрасные спектры и спектры комбинационного рассеяния.
Спектры комбинационного рассеяния сернокислых растворов, осо-
особенно системы Н2О—SO3, давно изучены исследователями, но
ИК-спектры начали изучаться лишь сравнительно недавно. Это
объясняется тем, что снятие ИК-спектров в H2SO4 невозможно
в обычно применяемых кюветах с окнами из хлорида натрия, в то
время как изучение спектров комбинационного рассеяния можно
успешно проводить в стеклянных кюветах. Недавно было проведено
изучение инфракрасных спектров в тонком слое H2SO4, заключен-
заключенном между пластинами из хлоридов серебра или натрия, покрытых
тонкой полиэтиленовой пленкой.
Сейчас исследованы спектры комбинационного рассеяния и
ИК-спектры 79 олеумов и колебательные спектры растворов воды
в серной кислоте. Полученные результаты будут обсуждаться в раз-
разделе Ш,А. Растворы других веществ в серной кислоте этими мето-
методами изучены сравнительно мало. Так, на основании спектров ком-
II. ФИЗИЧЕСКИЕ СВОЙСТВА
145
бинационного рассеяния было доказано образование линейного
иона NOJ в сернокислых растворах азотного ангидрида и азотной
кислоты 143, причем иону нитрония приписывается частота 1400 см~1,
а иону HSOI — частота 1050 см'1.
б. УФ-спектроскопия. Спектры оснований и их сопряженных
кислот в видимой и ультрафиолетовой областях различны, что исполь-
используется для определения степени ионизации этих соединений, раство-
растворенных в смесях серной кислоты с водой, в частности при измерении
кислотной функции (см. раздел II,Б,7). Таким образом были изу-
изучены растворы нитросоединений и SO|22 в олеуме. Кроме того,
на основании этого метода
было доказано существова-
существование ацил-ионов (см. раз-
раздел IV,Г) и ионов карбония
(см. раздел IV,И) в серной
кислоте.
в. ЯMF'-спектроскопия.
Можно ожидать, что вслед-
вследствие быстрого обмена
протоном между ионом
HSOJ растворителя и этим
же ионом кислых сульфа-
сульфатов металлов, протонный
химический сдвиг будет
линейно зависеть от кон-
концентрации, выраженной в
виде доли протонов р, со-
согласно уравнению
0,2
i
,~ OA
0,5
ОМ
0,02
0,0 в
0,10
p
Рис. 39. Протонный химический сдвиг для
сернокислых растворов MHSO4.
sh,so4)
G0).
Рис. 39 показывает, что кривые зависимости б от р для растворов
кислых сульфатов различных металлов различны 15> 113, хотя обычно
линейны. Следовательно, причина протонного химического сдвига —
результат взаимодействия ионов металлов с растворителем. Наиболь-
Наибольшие сдвиги в область низких полей вызваны теми ионами, которые,
как это показано другими методами, наименее сольватированы.
Проще всего это объяснить тем, что ион HSO4 вызывает большой
сдвиг в область низких полей, а в результате взаимодействия катиона
с растворителем происходит сдвиг в область высоких полей. Порядок
сдвига соответствует степени сольватации, определяемой другими
методами. Так как известно, что протон, осуществляющий водород-
водородную связь, вызывает сдвиг резонанса в область низких полей 203,
сдвиг резонанса в область высоких полей, вызванный, вероятно,
сольватированными ионами, происходит за счет более слабых водо-
водородных связей между сольватирующим слоем и основной массой
10 Заказ 90
146
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
II. ФИЗИЧЕСКИЕ СВОЙСТВА
147
растворителя, а не между самими молекулами растворителя. Дру-
Другими словами, между сольватирующим слоем и основной массой
растворителя имеется область неупорядоченной структуры с отнот
сительно слабыми водородными связями.
Сильный сдвиг в области низких полей, вызванный HOHOvfoHSOj,
представляет большой интерес. Плотность электронного $блака,
•окружающего протон отдельного иона HSOj, должна быть выше
плотности электронного облака, окружающего протон в молекуле
серной кислоты, и, следовательно, отдельный ион HSOj должен
иметь резонанс в более высокой области, чем резонанс H2SO4- Сдвиг
в область низких полей происходит вследствие сильного взаимодей-
взаимодействия с растворителем. Отрицательно заряженные атомы кислорода,
входящие в состав иона HSOJ, вероятно, поляризуют связь О—Н
•окружающих молекул серной кислоты и образуют водородную связь
с ними. Это обстоятельство рассеивает сомнение в том, что ион
HSOJ не сольватирован.
Протонный химический сдвиг помог установить степень иони-
ионизации раствора воды в серной кислоте U3' uo, и хотя полученные
данные хорошо согласуются с результатами исследования спектров
комбинационного рассеяния 247, допущения, на которых основы-
основывается этот метод, трудно подтвердить, поэтому такое совпадение
может быть случайным.
7. Определение кислотной функции Гаммета
Очень удобной количественной характеристикой кислотности
является кислотная функция Щ2":
[ВН+
[В]
G1)
где [ВН+]/[В] — индикаторное отношение (отношение концентраций прото-
низированного и нейтрального индикаторов основания).
Были определены Но для смеси воды с серной кислотой 129 и Н9
для олеумов 19~22. Итоги этой работы подвели Поль и Лонг208.
Индикаторное отношение [ВН+]/1В] в данной среде опреде-
определяется экспериментально спёктрофотометрическим методом по на-
наблюдаемым коэффициентам экстинкции | очень разбавленных рас-
растворов оснований в данной среде и сравнением их с коэффициентом
экстинкции полностью ионизированного основания ?иоц и неионизи-
при одинаковой длине волны. Таким обра-
обрарованного основания
зом
[ВН+]
—Ев
[В] ?„ои-1 '
Если непосредственное определение величины рЛГВн+ невозможно,
то ее можно установить сравнением индикаторного отношения для
основного индикатора В в растворах с непрерывно увеличивающейся
кислотностью и индикаторного отношения для другого основного
индикатора С, для которого величина рЛГСн+ известна, так как при
некоторой кислотности
откуда
[В]
s [С]
[CH+]
1С]
Допускаем, что
'вн+А:
л
G3)
G4>
G5)
где /в и /с — коэффициенты активности оснований В и С, а /дн+ и /сн+ —
коэффициенты активности соответствующих сопряженных кислот.
Экспериментально доказано, что разность ig[BH+]/[B] —
— lg[CH+]/[C] при изменении кислотности остается постоянной.
При этом условии можно построить шкалу И0, независимую от при-
применяемых индикаторов. Однако Йетс недавно показал, что кислот-
кислотность водных растворов серной кислоты E—80% H2SO4) по шкале
Но при использовании 'некоторых замещенных ароматических ами-
амидов в качестве индикаторов отличается от кислотности, полученной
с замещенными ароматическими анилинами 246. Указанное различив-
объясняется разными степенями сольватации сопряженной кислоты
ВН+.
В 1963 г. Йергенсон и Хартер пересмотрели шкалу кислотной
функции Гаммета для водных растворов серной кислоты, содержащих
более 60% H2SO4- Они показали 129, что некоторые ранее определен-
определенные значения ytK неправильны и что индикаторы, использовав-
использовавшиеся предыдущими исследователями, не образуют набор, приемле-
приемлемый для ступенчатого сравнения. При использовании универсальной^
серии индикаторов, состоящей исключительно из первичных анили-
анилинов, была создана новая шкала Но для растворов серной кислоты,
которая с возрастанием концентрации H2SO4 по абсолютной вели-
величине возрастает больше, чем шкала Поля и Лонга. Наиболее слабым
основанием, применявшимся Иергенсоном и Хартером, был пикр-
амид (рК = —10,10); такому значению Но соответствует концентра-
концентрация H2SO4, приблизительно равная 99%.
Недавно Р. Дж. Джиллеспи и Е. А. Робинсон (неопубликован-
(неопубликованные результаты) пересмотрели ряд индикаторных отношений в кон-
концентрированной серной кислоте и в олеуме. Оказалось, что резуль-
результаты выполненных ими измерений с пикрамидом хорошо согласуются
с данными Йергенсона и Хартера. Были использованы в качестве
индикаторов также замещенные нитробензолы; шкала, полученная
10*
148
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
с ними, аналогична шкале с первичными анилинами. Было найдено,
что некоторые из ранее опубликованных Брэндом и др. 21> 22 значе-
значений ? для полностью ионизированных ароматических нитросоеди-
нений ошибочны. Ошибки могли возникнуть из-за трудности опре-
определения коэффициента экстинкции в среде, где индикатор подвер-
подвергается слабому сульфированию, и в случае применения в качестве
индикатора очень слабых оснований, например 2,4-динитротолуола,
из-за неполной протонизации ос-
основания даже в наиболее кислом
из применявшихся растворителей.
Используя сильнокислые среды,
12
10
о,г
о,в
Рис. 40. Зависимость кислотной Рис. 41. Зависимость кислотной функ-
функции Гаммета Яо от состава ции Но сернокислых растворов некото-
системы Н2О—SO3: рых электролитов от концентрации
1 — теоретическая кривая; 2 — экспери- электролита:
ментальная кривая. , _ Hj0+HS0-. g _ CfH,COOHiHSO-;
3 — Na+HSO^; 4 — K+HSOJ.
такие, как фторсульфоновая кислота, растворы пентафторида сурьмы
во фторсульфоновои кислоте и растворы сильной кислоты HB(HSO4L
в серной кислоте, удалось получить более надежные величины
§ион Для некоторых наиболее слабых оснований. В результате была
создана новая шкала функции Яо для концентрированных растворов
серной кислоты и олеумов (рис. 40). Значения Яо для системы
Н2О — SO3 при составах, близких к H2SO4, приведены ниже:
H2SO4, % 96,0 97,0 98,0 99,0 99,5 100 0 100 5
—#о 10,03 10,21 10,40 10,72 11,0 12,08 12,58
H2SO4, % 101,0 101,5 102,0 103,0 104,0 105,0 106 0
—Но 12,74 12,83 12,92 13,06 13,20 13,34 13,50
Делались попытки теоретически установить значения Но для
концентрированных растворов серной кислоты. Такие работы не-
недавно были выполнены Йергенсоном и Хартером 146. Авторы пришли
к выводу, что упрощенные представления Дено и Тафта 50, основан-
II. ФИЗИЧЕСКИЕ СВОЙСТВА
149
ные на допущении об идеальности растворов серной кислоты при
концентрациях ниже 95%, неудовлетворительны. Даже при 100%
H2SO4 кислотные функции Но растворов таких оснований, как
бензойная кислота, NaHSO4, KHS04 и вода, неодинаковы. Из рис. 41
видно, что нельзя игнорировать влияние активности электролита.
Тем не менее в системе Н2О—H2SO4— SO3 в области 100% H2SO4
значения Н 0 довольно хорошо соответствуют изменениям концентра-
концентрации ионов H3SO4 (см. рис. 40), так как для равновесия между про-
протонами и сольватированными
протонами
К = а,
UH2SO,
H3SO4
G6)
G7)
откуда
Яо = - lg[HsSOJ]
'вн+
X /.
К
H3SO4 av
X
G8)
13 -
12 -
где а — активности; / — коэффици-
коэффициенты активности.
с, мол
Рис. 42. Кислотная функция Гаммета
#0 для растворов некоторых кислот
в серной кислоте.
Для разбавленных сернокислых растворов веществ можно допу-
допустить, что величина aH2so4 постоянна, и если пренебречь значе-
значениями коэффициентов активности, то
где к — постоянная.
G9)
Концентрация HgSOJ в 100%-ной H2SO4 при 25° С|[равна
0,135 м, поэтому изменение концентрации H3SOt в зависимости от
состава серной кислоты удобнее всего связать с наблюдаемой вели-
величиной Но для 100%-ной серной кислоты, равной 12,08, тогда
-13,95
(80)
Рассчитанные значения Но для водных растворов несколько ниже,
а для олеума несколько выше экспериментальных, хотя формы
обеих кривых одинаковы (см. рис. 40). Было показано, что аромати-
ароматические нитросоединения вызывают более сильное понижение точки
замерзания водных растворов, чем 100%-ной серной кислоты94.
Аналогично было установлено, что полинитросоединения, которые
ведут себя как неэлектролиты, при растворении в низкопро-
низкопроцентном олеуме вызывают меньшее понижение точки замерзания,
чем это можно было бы ожидать 27. Электролит H3O+HSO4 в вод-
водном растворе серной кислоты оказывает высаливающее действие,
150
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
а в олеуме — наоборот. Этим объясняется различие между расчетной
и экспериментальной величинами Но.
Изменение Но с изменением состава для некоторых систем пред-
представлено 6 на рис. 42. Значения Но для системы H2SO4 — HSO3C1
были определены Пальмом 249.
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
В СЕРНОЙ КИСЛОТЕ
А. Вода и трехокись серы
Существование в системе Н2О — SO3 ряда химических соеди-
соединений признано всеми исследователями. В данной системе имеются
соединения, плавящиеся конгруэнтно: H2SO4 • 4Н2О (—28,366° С),
H2SO4 • 2Н2О (-39,51° С), H2SO4 • Н.О (8,56° С), H2SO4 A0,37° С),
и инконгруэнтно — H2S2O7 C5,15° С), H2SO4 • 6Н2О и H2SO4 • ЗН2О;
последние два при —53,73 и —36,56° С (соответственно) переходят
в тетрагидратную форму 77> 78>15е.
Описание системы Н2О — SO3 будет ограничено знакомством
со свойствами растворов в очень концентрированной серной кислоте.
Криоскопическим методом 8>82 показано, что раствор воды в серной
кислоте, по-видимому, ионизирован не полностью и что эксперимен-
экспериментально определенная константа диссоциации Кцго = 1 моль/кг
растворителя. Однако результаты измерений электропроводности
не свидетельствуют о неполной диссоциации растворов воды в сер-
серной кислоте 11в. Это несоответствие легко объяснить, если предполо-
предположить, что кажущаяся неполнота ионизации Н2О обусловлена обра-
образованием ионной пары Н3О+ • HSO4, продолжительность жизни
которой меньше средней продолжительности перехода протона
между ионами HSOj, обусловливающими электропроводность. По-
Последние исследования Е. А. Робинсона (неопубликованные резуль-
результаты), подтверждают образование ионных пар Н3О+ • HSO4, а час-
частоты 1140 и 1300 см'1 можно приписать валентным колебаниям SO-
групп этих пар.
Недавно были изучены инфракрасные спектры растворов воды
в серной кислоте 79' 230. Много работ посвящено изучению спектров
комбинационного рассеяния «водных растворов серной кислоты.
Янг
248
утверждает, что растворы, составы которых находятся между
H2SO4 и H2SO4 • Н2О, помимо молекул H2SO4, ионов НSO4 и Н3О+
содержат ионы H5SO5. Вероятно, ион H6SOJ представляет собой
сольватированный ион Н3О+, т. е. Н3О+ • H2SO4. Криоскопические
исследования показывают 8, что ион Н3О* сольватирован, по-види-
по-видимому, двумя молекулами H2SO4. Однако Виат 244 утверждает, что
для расчета парциальной мольной энтальпии раствора воды в сер-
серной кислоте необходимо сделать допущение о наличии соединения
Н3О+ ¦ H2SO4.
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
151
Леман 159 на основании имеющихся данных о фазовом составе
системы H2SO4 — SO3 утверждал, что существует H2S2O7, H2S3O10,
H2S4O13 и H2S8O25. Криоскопические исследования показали, что
в разбавленных растворах SO3 в серной кислоте присутствует слабая
кислота 83, вероятно H2S2O7. Была определена константа диссоциа-
диссоциации Kntsao, = 0,014 моль/кг растворителя 8. Значения электро-
электропроводности этих растворов при 10 и 25° С согласуются со степенью
ионизации H2S2O7, вычисленной из этой константы 11в, а положение
минимума электропроводности при кондуктометрическом титровании
низкопроцентного олеума сильным основанием лакже соответствует
КнАО, = 0,014 моль/кг растворителя. Понижение точки замерза-
замерзания, наблюдающееся при добавлении к низкопроцентному олеуму
небольших количеств сульфата аммония, можно объяснить присут-
присутствием в системе помимо H2S2O7, H2S2O7 и S.2O7~ высших поли-
полисерных кислот и их ионов. В частности, было показано, что даже
при такой низкой концентрации, как 0,15 м, в системе присутствует
H2S3O10; уменьшение понижения точки замерзания 0,4 м раствора
при добавлении (NH4JSO4 можно объяснить 83 только присутствием
H2S4O13.
Состав растворов олеума был изучен с помощью спектров комби-
комбинационного рассеяния 98' 188> 231 и инфракрасных спектров 79. Все
исследователи согласны с тем, что спектры доказывают образование
H2S2O7. Полученные спектры они сравнивали 98 со спектром соеди-
соединения S2O5F, которое является изоэлектронным с H2S2O7. Разногла-
Разногласия между исследователями касаются спектральных доказательств
образования H2S3O10. В 1960 г. Вальрафен и Янг заявили, что
можно объяснить спектры, не принимая во внимание высшие поли-
полисерные кислоты, за исключением H2S2O7. Однако при сравнении
спектров олеумов со спектрами S2O5F2 и S3O8F2 было доказано, что
олеумы, концентрация SO3 в которых составляет более 12%, наряду
с H2S2O7 содержат 98 H2S3Oi0.
Было показано, что в колебательном спектре трисульфурильного
соединения наблюдаются две полосы с частотами, близкими -к
700 см'1, между тем как дисульфурильное соединение в этой части
спектра имеет только одну полосу. Колебательные спектры олеумов,
содержащих относительно малое количество SO3, имеют единствен-
единственную полосу с частотой, близкой к 730 см'1 (для S2O5F2 при 733 см'1),
в то время как более концентрированные олеумы имеют две полосы
с частотами вблизи 690 и 735 см'1 (для S3O8F2 при 699 и 724 см'1).
Присутствие H2S3O10 в олеумах подтверждается и другими
характеристиками спектра. Сравнительно недавно Вальрафен 229,
изучая интенсивности некоторых спектральных линий олеумов,
Доказал присутствие H2S3O10. К сожалению, в отличие от спектра
S3O8F2 спектр 84О1ХР2 не имеет четких диагностических признаков,
которые позволили бы убедительно доказать наличие H2S4O13
в олеуме путем сравнения спектра последней со спектром S4OX1F2.
152
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Однако частота симметричных колебаний группы S02, равная
в растворе H2SO4 1195 см'1, а в олеумах состава H2S2O7 и H2S3O1(>
равная 1224 и 1230 см'1 соответственно, при увеличении концентра-
концентрации SO3 сдвигается в сторону более высоких значений. Например,
при концентрации SO3 в олеуме с 92,2% SO3 частота колебаний
равна приблизительно 1260 см'1.
Сдвиг линий, отнесенных к симметричному растягиванию группы
SO2, указывает на возрастание концентраций полисерных кислот,
таких, как H2S4O13, и даже более высоких поликислот в очень
концентрированных олеумах 98. Частота симметричных валентных
колебаний группы SO2 в ациклическом полимере SO3, который
является, по-видимому, полисерной кислотой с очень высоким моле-
молекулярным весом, так как его образование из жидкого примера
катализируется следами воды, также составляет 1260 см'1. В уме-
умеренно концентрированных олеумах SO3 должен находиться в виде
мономера, а в очень концентрированных — как в виде мономера,
так и в виде тримера. Спектры комбинационного рассеяния раство-
растворов SO3 в D2SO4 могут быть исследованы и интерпретированы 98
так же, как спектры системы H2SO4— SO3.
Б. Бор
Борная кислота хорошо растворима в серной кислоте. Известны
различные соединения борной кислоты и окиси бора с серной кисло-
кислотой. Одно из них имеет состав Н3ВО3 • 3SO344 и может рассматри-
рассматриваться как кислый сульфат бора B(HSO4K. На основании понижения
точки замерзания предположили, что это соединение образуется
при растворении окиси бора в серной кислоте 131. Более поздние
измерения точки замерзания 69 показали, что как для растворов
Н3ВО3, так и для В2О3 v = 6, в то время как при образовании
В(HSO4K величина v должна равняться 7 и 8 соответственно, со-
согласно уравнениям
Н3ВОз+ 6H2SO4 = В (HSO4K + ЗН3О+ + 3HSO; (81)
B2O3 + 9H2SO4 = 2B(HSO4K+3H3O+ + 3HSOJ. (82)
Измерения электропроводности показывают, что для борной
кислоты у = 2; а для окиси бора у = 1; это также не согласуется
с уравнениями (81) и (82), которые в обоих случаях дают у = 3.
Однако если кислый сульфат бора соединяется с кислым сульфат-
ионом с образованием кислого сульфат-иона бора
B(HSO4K + HSO4 = B(HSO4)J
по аналогии с реакциями
(83)
(84)
(85)
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
153
то уравнения (81) и (82) можно переписать следующим образом:
1 = B(HSO4)J+3H3O+-|r2HSOj (86)
(87)
В этом случае значения у = G и у = 2 для борной кислоты, v = 6
и у = 1 для окиси бора согласуются с экспериментальными данными.
То обстоятельство, что в растворах образуется B(HSO4L, пред-
представляет большой интерес, ибо этот ион, по-видимому, не подвер-
подвергается сольволизу; соответствующая ему кислота HB(HSO4L в серно-
сернокислом растворе должна быть относительно сильной кислотой.
Растворы кислоты HB(HSO4L могут быть приготовлены путем рас-
растворения борной кислоты или окиси бора в олеуме. При этом удается
связать ион Н3О+ по уравнению
(88)
Взаимодействие может быть представлено результирующими урав-
уравнениями
(89)
(90)
Криоскопические измерения и измерения электропроводности 69-
подтверждают такое протекание реакции и показывают, что кислота
HB(HSO4L сильно ионизирована, Къ = 0,4 молъ/кг. Раствор
HB(HSO4L может быть кондуктометрически оттитрован 72 таким
сильным основанием, как KHSO4:
KHSO4 + HB (HSO4L = KB(HSO4L + H2SO4
основание кислота соль растворитель
Минимум электропроводности наблюдается при
(91)
nKHSO4
raHB(HSO4L
- == 0,98
где nKHS04 — число молей основания, прибавленное к раствору кислоты,
содержащей вначале п*яъ (hso.). моль тетрагидросульфато-борной кислоты.
Точка замерзания раствора мало изменяется, потому что KHSO4
прибавляется до соотношения
nKHS04
i
WHB(HSO4L
= 1,0
т. е. ионы H3SOa" заменяются таким же количеством ионов К+,
а концентрация ионов B(HSO4L остается постоянной. Следовательно,
общее число частиц растворенного вещества сохраняется неизмен-
неизменным. По мере возрастания электропроводности после ее минимума
154
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
происходит понижение точки замерзания вследствие введения из-
избытка KHSO4.
Концентрированные растворы HB(HSO4L были изучены с по-
помощью спектров комбинационного рассеяния ". Спектры этих
растворов довольно сложны. Наряду с характеристическими часто-
частотами H3SO4, H2SO4 и B(HSO4)J в них имеются линии, принадле-
принадлежащие H2S2O7 и H2S3O10. Названные полисерные кислоты получа-
получаются, вероятно, в процессе конденсации ионов B(HSO4O, которые
образуют полимерные ионы, содержащие связи В—О—В:
2B(HSO4)j =
HO4S4
ho4s/
,SO4H
+ H2S2O7+H2SO4
(92)
II
Томпсон и Гринвуд 22в сообщили, что им удалось получить кислоту
HB(HSO4L в результате реакции ВС13 с серной кислотой:
BC]3 + 4H2SO4 = HB(HSO4L + 3HC1 (93)
Исследование спектров комбинационного рассеяния концентри-
концентрированных растворов этой кислоты показало ", что в отсутствие
H2S2O7 образуются связи В—О—В; принимая во внимание это
обстоятельство, а также сложные структуры солей, образование
осадка HB(HSO4L следует считать маловероятным. Вероятнее всего,
продукт реакции представляет собой смесь полисульфато-борных
кислот с серной и пиросерной кислотами с составом, отвечающим
формуле HB(HS<\L.
Соли состава MB(SO4J, М11 [B(SO4J]2 и Mm[B2O(SO4K] были
получены по реакции между сульфатами соответствующих металлов,
борной кислотой и трехокисью серы 218, например:
(NH4JSO4 + 2В(ОНK + 6SO3 = 2NH4[B(SO4J] + 3H2SO4 . (94)
Нагревая кальциевую, стронциевую или бариевую соль и удаляя
тем самым SO3, можно получить соединение состава Мп [B2O(SO4K1.
Соль NH4 [B(SO4J] была получена по реакции нитрида бора с серной
кислотой
BN + 2H2SO4 = NH4[B(SO4J] (95)
Все эти соединения сильно гигроскопичны и подвержены гидро-
гидролизу; считают, что они содержат высокополимерные анионы.
Попытки получить такие соли, как KB(HSO4L, нейтрализацией
растворов HB(HSO4L в серной кислоте не увенчались успехом.
Для того чтобы получить кристаллический продукт, необходимо
приготовить значительно более концентрированные растворы тетра-
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
155
гидросульфато-борной кислоты, чем те, которые применяются для
изучения физических свойств. При соблюдении такого условия полу-
полученные в твердом состоянии соли натрия, калия, аммония и строн-
стронция содержали100 сульфат-ион и бор в соотношении меньшем, чем
4:1. Это аналогично поведению борат-иона в водном растворе.
В разбавленном водном растворе 60 существует В(ОНO; из концен-
концентрированных выделены бораты, содержащие комплексные ионы,
такие, как В4О|~ и В6О*7- Эти ионы образуются в результате поли-
полимеризации В(ОНL и В(ОНK, происходящей с выделением воды.
Составы солей, а также результаты криоскопических измерений и
измерений электропроводности растворов солей можно объяснить 10°,
если допустить, что анионы солей образованы шестичленными
кольцами III, как это показано для соединений IV и V:
хв к
/\ |\
о о
III
,SO4H HO4S /Ox
/ XB B7
o/l Г
О О \ /00
H04Sx
чв в
ho4s/| i о
IV
/Оч
HO4SX/OX/SO4H HO«Sx/"\/i
SO4H
о о
40
I |\SO4H
о о
0
^
0
s
В Ъ'
/\ |\o
о о
0^%
V
В. Кремний
В соответствии с экспериментально определенными значениями
•v и у гексаметилсилоксан74'195'210 ионизируется по уравнению B6).
Прайс210 предположил, что триметилсилангидросульфат ионизи-
ионизируется так же, образуя ион триметилсилана:
(CH3KSiSO4H = (CH3KSi+ + HSOJ
(96)
156
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Однако экспериментально не доказано, что это происходит в зна-
значительной степени74.
В сернокислых растворах (СН3)зЗЮС2НБ, (С2НБKЗЮС2Н5 и
(CH3KSi0H 195 соединения реагируют с растворителем, образуя
соответствующие триалкилсилангидросульфаты:
(97)
(98)
R3S1OH + 2H2SO4= R3SiSO4H + Н3О+ + HSO4
Триметилсилангидросульфат был получен по реакции между олеу-
мом и гексаметилсилоксаном "и.
Недавно Ривилл 211 высказал мнение о непригодности тетраме-
тилсилана в качестве эталонного вещества при изучении ЯМР-спек-
тров сернокислых растворов. Он показал, что (CH3LSi не смешивается
с серной кислотой, однако если эти вещества, помещенные в один
сосуд, взбалтывать при комнатной температуре, то образуется метан
и триметилсилангидросульфат.
Экспериментальные данные исследования раствора диметилди-
этоксисилана 74 соответствуют реакции образования кислых суль-
сульфатов
(99)
а результаты исследования раствора (CH3JSiO4 —¦ реакции 210
A00)
Метилтриэтоксисилан отличается от упомянутых выше соедине-
соединений тем, что для его растворов величины v и у в некоторой степени
зависят от концентрации: в интервале концентраций 0,01—0,06 м v
уменьшается от 8,4 до 7,7, а у от 2,8 до 2,2 74. Вероятно, такие низ-
низкие значения v и у, а также их зависимость от концентрации объяс-
объясняются полимеризацией метилсилантригидросульфата. Общее урав-
уравнение образования линейного полимера может быть записано в виде
CH3Si(OC2H5K+F + 3/«) H2SO4 = -
СНз /СН3
HO4S—Si—О—I Si—0
so4ri
A01)
Очевидно, правильнее было бы представить строение этих поли-
полимеров в виде VI, т. е. состоящим из шестичленных колец, связан-
связанных сульфатными группами, которые образовались в результате
отрыва серной кислоты от линейного полимера:
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
157
СЫз
1/°s
HO4S—Si
1
0
CH3
/Ox
4s(
1
(
СНз
/Ox
Si
i
(
CH3
\/°
X /
Si
i
1
0
CH3
\Л
X /
Si
1
1
0
СНз
Si—SO4H
I
(
1
VI
Эта структура аналогична предполагаемой структуре сложных сульфатных
соединений бора и мышьяка (формулы III и XV).
Экспериментальные данные могут быть удовлетворительно
объяснены образованием смеси следующих полимеров:
» v=7+l- 7=2 + 4"
Димер 2 8,5 2,5
Тример 3 8,0 2,3
Тетрамер 4 7,75 2,25
Результаты криоскопических исследований растворов тетрафе-
нплсилана, тетрабензилсилана и гексафенилдисилана 22Б можно
объяснить, лишь предположив полное расщепление всех связей
Si—С, сопровождающееся образованием соответствующих арил-
сульфоновых кислот; эти кислоты удалось выделить из растворов.
Известно, что расщепление связи Si—С вызывается различными
кислыми и основными реагентами. Была определена скорость реак-
реакции расщепления для фенилтриметилсилана в серной кислоте48.
Однако образование Si(HSO4L, а также анионов Si(HSO4)J или
Si(HSO4)|~ по реакциям
A02)
Ph4Si + 12H2SO4=Si(HSO4)J+4PhSO3H + 4H3O+ + 3HSOl A03)
Ph4Si + 12H2SO4 = Si(HSO4)|' +4PhSO3H + 4H3O+ +2HSOJ A04)
не подтверждается данными криоскопических измерений. Было
обнаружено, что в интервале концентраций 0,005—0,03 м v умень-
уменьшается от 10,8 до 10,3, а у от 2,7 до 2,5. Вполне вероятно, что в дан-
данном случае происходит полимеризация. Результаты криоскопических
измерений и измерений электропроводности раствора Ph4Si 74,
криоскопических измерений растворов тетрабензилсилана, гекса-
158
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
фенилдисилана 225 и [(CH3KSi0]4Si 210 свидетельствуют об образо-
образовании полимера пространственной структуры [Si2O3(HSO4J]n:
SiPh4 -S*^* 4PhSO3H+ 1 n-
A05)
Si[OSi(OCH3Kk
4(CH3KSiSO4H +1' n [Si2O3(HSO4J] „ +
+ 2,5H3O++2,5HSOi" A06)
Соединения Si(OC2H5L, Ph3SiOH, Ph2Si(OHJ, Si(OAcL и
{( || | I SiOH, по-видимому, реагируют с серной кислотой, обра-
зуя нерастворимые полимеры.
Г. Олово и свинец
Оловянная кислота и сульфат олова практически нерастворимы
в серной кислоте, однако ряд солей кислоты H2Sn(HSO4)e был при-
приготовлен упариванием смеси окиси олова, сульфата металла и серной
кислоты59-232,-это Rb2Sn(SO4K, K2Sn(SO4K, Ag2Sn(SO4K-3H2O,
,CaSn(SO4K • 3H2O, PbSn(SO4K • 3H2O. Все такие гидратные соли
содержат три молекулы воды. Их можно представить как соли
тригидросульфато-оловянной кислоты H2Sn(OHK(HSO4K, например,
Ag2Sn(OHK(HSO4K. Присутствие гексагидросульфато-оловянной
кислоты H2Sn(HSO4N в растворе серной кислоты было доказано
на основании измерений температур замерзания и электропровод-
электропроводности растворов Ph4Sn, Ph3SnOH и Sn(OAcL в серной кислоте 104.
Результаты исследований свидетельствуют о возможности следу-
следующих способов ионизации:
Ph4Sn + 14H2SO4=H2Sn(HSO4N+4PhSO3H + 4H3O+ + 4HSOl A07)
Ph3SnOH + 13H2SO4= H2Sn(HSO4N+3PhSO3H + 4H3O++4HSOj A08)
Sn(OAcL + 10H2SO4 = H2Sn(HSO4N+4AC0HJ + 4HSO;- A09)
Значения v и -у уменьшаются с возрастанием концентрации и
соответствуют равновесию между свободной кислотой и ее анионами:
(НО)
A11)
4)? + 24 A11)
Брубейкер 29 изучил растворы Sn(II) в водных растворах серной
кислоты различной концентрации с помощью дзпектрофотометрии,
измерений чисел переноса, а также" исследуя равновесие между
гидроокисью олова и водной H2SO4. Он пришел к заключению, что
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
159
в разбавленном растворе гидроокись олова находится в равновесии
со Sn(SO4J+, а в концентрированных растворах — со Sn(SO4J+,
Sn(SO4J и H2Sn(SO4K.
Тетраметилолово хорошо растворяется в серной кислоте, реакция
протекает по уравнению
(CH3LSn+H2SO4= (CH3KSnHSO4 + CH4T A12)
так же, как при взаимодействии тетраметилолова и концентриро-
концентрированных растворов галогеноводородов 39. Криоскопические измере-
измерения и измерения электропроводности показывают, что получаю-
получающийся триметилоловогидросульфат ионизируется, образуя катион
триметилолова:
(CH3KSnHSO4 = (CH3KSn++HSOj A13)
Эта же реакция может быть записана в виде
(CH3KSnHSO4 + H2SO4 = (CH3KSnSO4H? + HSO;
A14)
На основании криоскопических измерений и измерений электро-
электропроводности невозможно установить, какая из двух реакций про-
протекает в действительности. Были изучены инфракрасные спектры
концентрированных растворов (Е. А. Робинсон, неопубликованные
результаты), которые оказались гораздо сложнее, чем это следовало
бы ожидать для катиона триметилолова (CH3KSn+. Новые исследо-
исследования в области инфракрасных спектров твердых солей триметил-
триметилолова, таких, как перхлорат, нитрат, карбонат и тетрафторборат,
показали, что эти соединения не содержат катиона (CH3KSn+,
анионы ковалентно связаны с оловом, образуя тригональную бипи-
рамиду с координационным числом, равным 5 37' 138> 205. Рентгено-
структурные исследования свидетельствуют о том, что молекула
(CH3KSnF представляет собой тригональную бипирамиду, в центре
которой находится ион олова, связь в структуре осуществляется
через фтор-ионы 37. В водном растворе катион триалкилолова,
вероятно, находится в сольватированной форме 39, т. е. R3Sn0H2.
По-видимому, в сернокислом растворе этот катион превращается
в [(CH3KSnSO4H2]+ или [(CH3KSn(SO4H2J]+. Значения v и у для
раствора сульфата триметилолова равны 4 и 2, что соответствует
уравнению ионизации
[ (CH3KSn]2SO4 + 4H2SO4 = 2(CH3KSnSO4H J
A15)
Сульфат триметилолова, таким образом, является сильным основа-
основанием, между тем как соответствующая ему гидроокись в водном
растворе ведет себя как слабое основание.
Диацетат ди-м-бутилолова реагирует с серной кислотой, образуя
три кислых сульфат-иона на одну молекулу растворенного вещества
(у = 3) 105. Для реакции
(»-C4H9JSn(OAcJ + 4H2SO4 = (»-C4H9JSn(HSO4J+2AcOHJ+2HSOl A16)
160
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
протекания которой следует ожидать по аналогии с поведением
соединений диалкилкремния, у = 2, но наблюдаемое значение у
соответствует образованию однозарядного катиона олова по реакции
(n-C4H9JSn(OAcJ + 5H2SO4=(«-C4H9JSn(HSO4JH+ + 2AcOHJ + 3HSO; A17)
Дигидросульфат ди-м-бутилолова, по-видимому, ведет себя как
сильное основание.
Метилоловянная кислота плохо растворяется в серной кислоте,
что объясняется, вероятно, ее полимерной структурой. Эксперимен-
Экспериментальные значения v и у с ростом концентрации изменяются от 6 до 5
и от 3 до 2 соответственно, согласно реакции
CH3SnOOH + 5H2SO4 = CH3Sn(HSO4K + 2H3O++2HSOj A18)
с последующей ионизацией CH3Sn(HSO4K как слабого основания
CH3Sn(HSO4K + H2SO4 = CH3Sn(HSO4KH+ + HSOj A19)
В отличие от соединений кремния все изученные соединения
олова полностью переходят в соответствующие сульфаты, что объяс-
объясняется меньшей электроотрицательностью олова. По мере увеличе-
увеличения числа HSOl-rpynn, связанных с атомом олова, наблюдается
уменьшение основности кислых сульфатов олова. Поэтому R3SnHSO4
и R2Sn(HSO4J — сильные основания, RSn(HSO4K — слабое осно-
основание, a Sn(HSO4L — кислота.
Так как из растворов в H2SO4 можно осадить комплексные соли
калия и аммондя K2Pb(SO4K и (NH4JPb(SO4K, по аналогии пред-
предположили, что растворы тетраацетата свинца в H2SO4, в которых
образуется Pb(SO4J, а также зеленовато-желтые растворы, получа-
получающиеся при электролизе H2SO4 со свинцовыми электродами, содер-
содержат кислоту H2Pb(SO4K 57'58'63- Нерастворимые соли (NH4JPb(SO4K,
K2Pb(SO4K, Rb2Pb(SO4K и Cs2Pb(SO4K были приготовлены дейст-
действием кислого раствора Pb(SO4J на раствор сульфата металла в сер-
серной кислоте 61.
Криоскопические измерения и измерения электропроводности
показали, что тетраацетат свинца, как и тетраацетат олова, пол-
полностью переходит в тетрагидросульфат 94>105. Так как это соединение
ведет себя как кислота, его можно рассматривать как гексагидро-
сульфато-свинцовую кислоту:
Pb(OAcL+10H2SO4=H2Pb(HSO4)e+4AcOHJ+4HSOj
A20)
Зная зависимость величин v и у от концентрации (в интервале
концентраций0,005—0,55м\ изменяется от 8,2 до 7,5, а у от 3,4 до
2,5), можно рассчитать значения двух констант диссоциации этой
кислоты при 25° С:
A21)
= i,2' 10 моль/кг растворителя
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
161
HPb(HSO4)i + H2SO4 = H3SOJ + Pb(HSO4)=-
#2 = 1,8- Ю~3 моль/кг растворителя
A22)
Медленно нагревая желтый раствор тетраацетата свинца до
100° С, можно получить ярко-желтый осадок сульфата свинца
Pb(SO4J, который при дальнейшем нагревании разлагается на
кислород и PbSCV Сульфат свинца может быть также приготовлен
из PbF2 и концентрированной серной кислоты 26. Как и Sn(SO4J,
это соединение нерастворимо в серной кислоте. Логично предполо-
предположить, что нерастворимость Pb(SO4J, а также солей M2Pb(SO4K
объясняется их полимеризацией, при которой сульфатные группы
играют роль мостикообразователей. Структура образующихся поли-
полимеров не установлена.
Д. Азот и фосфор
Растворы азотной кислоты, нитратов металлов и азотного ангид-
ангидрида в серной кислоте известны как эффективные реагенты при нит-
нитровании ароматических соединений 92. Некоторые исследователи
высказали предположение, что при определенных условиях нитру-
нитрующим агентом могут быть ионы нитрония. Однако существование
NOij было доказано лишь после того, как Ингольд с сотр.90- 119> 143
с помощью криоскопии и спектров комбинационного рассеяния изу-
изучили растворы азотной кислоты и некоторых окислов азота в серной
кислоте и других кислотах, а также приготовили устойчивые соли
NOJ. Количественное образование NOg в растворах азотной кислоты
в серной кислоте было подтверждено измерениями электропровод-
электропроводности ш. Исследования криоскопических свойств этих растворов
показывают, что величина v близка к 4, а измерения электропровод-
электропроводности дают у = 2, что удовлетворяет уравнению
HNO3 + 2H2SO4 = NO+ + H3O+ + 2HSO; A23)
В спектре комбинационного рассеяния есть две интенсивные линии—
при 1050 см'1 и 1400 см'1, которые не могут принадлежать ни моле-
молекулам растворителя, ни молекулам растворенного вещества. Первая
из них принадлежит иону HSO4, а вторая, поляризованная, несом-
несомненно, вызвана линейным ионом NO2, в спектре комбинационного
рассеяния которого и следует ожидать только одну поляризованную
линию. Это было установлено путем исследования спектров комбина-
комбинационного рассеяния смесей азотной кислоты с другими сильными
кислотами, а также солей NO2CIO4 и NO2HS2O7 187. Спектр ком-
комбинационного рассеяния раствора HNO3 в олеуме 33> 34> 186 вклю-
включает спектр олеума и две линии, причем одна (с частотой 1400 см'1)
является частотой симметричных колебаний иона NO2, а другая
(с частотой 1075—1095 см'1) принадлежит иону HS2C>7- Криоскопи-
Криоскопические измерения показывают, что в избытке низкопроцентного
11 Заказ 90
162
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА •
олеума азотная кислота ионизируется 90, образуя ион NOJ по урав-
уравнению
HNO3 + 2H2S2O7 = N0+ + HS2Of + 2H2SO4 A24>
Огг и Рэй 204 изучили ЯМР-спектр14 N в растворах азотной кис-
кислоты в олеуме. Они наблюдали сигнал от азота иона N02 в виде
четко выраженного широкого пика, ширина которого свидетельствует
о большом квадрупольном взаимодействии, обусловленным несим-
несимметричным окружением. Сравнение этого спектра с ЯМР-спектром
линейного азид-иона еще раз доказывает, что ион нитрония также'
линеен. В инфракрасных спектрах растворов азотной кислоты в сер-
серной кислоте есть интенсивная линия с частотой 2360 см, обусло-
обусловленная 175 асимметричным колебанием NOJ.
Аналогично было установлено образование иона нитрония в серно-
сернокислых растворах нитратов металлов -и азотного ангидрида, протека-
an 1 07
ющее в соответствии с уравнениями ""•10'
KNO3+3H2SO4 = NO+ + K+ + H3O+ + 3HSOj A25)
N2O5 + 3H2SO4 = 2NO+ + Н3О+ + 3HS0J A26)
Азотистый ангидрид и нитриты металлов реагируют с серной
кислотой, образуя полностью ионизированные кислые сульфаты
нитрозила
N2O3 + 3H2SO4 = 2NO+ + Н3О+ + 3HS0j A27)
A28)
a N,0, ведет себя аналогично нитрозилнитрату, образуя смесь кис-
лых сульфатов нитрония и нитрозила ""•±о<
A29)
Трифенилфосфин, как и трифениламин, является сильным осно-
основанием, которое при растворении в H2SO4 полностью протонизи-
руется:
Ph3P + H2SO4 = Ph3PH+ + HSOj A30)
Кроме того, это соединение с трудом подвергается сульфированию
и окислению.
Фосфорная кислота в сернокислом растворе является сильным
основанием, что можно видеть, например, по результатам криоско-
пических измерений и измерений электропроводности растворов
КН2РО4104, которые дают значения v = 4e^ = 2, в соответствии
с уравнением
KH2PO4+2H2SO4 = K> + H4POJ + 2HSOj A31)
Для раствора пирофосфата калия v = 12 и у = 5; это указывает
на то, что пирофосфорная кислота дегидратирует серную кислоту,
образуя 104 ион Р(ОН)?
К4Р2О7 + 7H2SO4 = 4К+ + 2Р(ОН) J + HS2O? + 5HSO; A32)
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
163
Криоскопическими измерениями и измерениями электропровод-
электропроводности было установлено, что тример фосфонитрилхлорида (PNC12K
в серной кислоте присоединяет 1,2 протона, а тетрамер протонизи-
руется сильнее, дгрисоединяя приблизительно 2,0 протона 206.
Е. Мышьяк, сурьма, висмут
При комнатной температуре мышьяковый ангидрид нерастворим
в серной кислоте, но несколько растворим при температурах, близ-
близких к точке кипения кислоты. Мышьяковистый ангидрид раство-
растворяется лучше. Криоскопические измерения и измерения электро-
электропроводности 101 показали, что он ионизируется аналогично N2O,,
образуя AsOHSO4:
As2O3 + 3H2SO4 = 2AsOHSO4 + H3O+ + HSOj A33)
Но образующийся NOHSO4 ионизируется полностью, a AsOHSO4
в 0,05 м растворе As2O3 ионизирован лишь на 50%. Если As2O3
ведет себя аналогично N2O3, то следует считать, что значения v = 6
и у = 3 при низких концентрациях свидетельствуют об образовании
полностью ионизированного AsOHSO4. По мере увеличения концен-
концентрации As2O3 от 0,01 до 0,07 м v уменьшается от 6,0 до 4,8, а у при-
принимает значения от 2,6 до 2,1, что объясняется неполнотой диссо-
диссоциации
AsOHSO4 5=± AsO+ + HSOj A34)
Можно предполагать, что при увеличении концентрации As2O3
значения v и у уменьшатся до 4 и 1 соответственно. Эксперименталь-
Экспериментальные данные не противоречат этим представлениям о ходе реакции,
поэтому можно рассчитать приближенное значение константы дис-
диссоциации AsOHSO4 (К = 12 молъ/кг растворителя). Однако v умень-
уменьшается быстрее, чем у. Это легко объяснить, если допустить, что
AsOHSOj при повышенных концентрациях полимеризуется. Напри-
Например, образование димера можно представить в виде уравнения
As2O3 + 3H2SO4 = (AsOHSO4J + Н3О+ + HSOj
A35)
Быстрое образование белого осадка, имеющего состав As2O3 • SO3,
даже из очень разбавленных растворов также свидетельствует об
образовании полимеров с предполагаемым строением VII и VIII:
• —As—0—As—0—As—0— • • •
I
SO4H
L
SO4H
• —As—О—As—О—As—О—As-
I I I I
0 0 0 0
O^"\>
VII
11*
0
s
0
vili
164
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
165
Для завершения этой цепи необходима еще одна молекула серной
кислоты, которая на один конец цепи отдает протон, а на другой —
группу HSOl:
НО—As-!-O— As-i-O—As—О— • • • —As—О—As—SO4H
I ' I [ I I I
SO4H SO4H SO4H SO4H SO4H
IK
Первые два члена структуры IX представляют собой не что иное, как
сольватированные формы AsOHSO4 и (AsOHSO4J, т. е. HOAs(HS04J
и (НО) (SO4H)AsOAs(SO4HJ.
Группы, обозначенные выше как AsO+, правильнее было бы пред-
представить в виде одной из сольватированных форм X или XI
HO-As-
X
-SO4H
Н2О—As—SO4H
1
SO4H
XI
которые могут быть получены из HOAs(HSO4J путем отрыва группы
HSOj или присоединением протона; возможно также образование
димерных ионов XII и XIII
НО—As—О—As— SO4H
I
SO4H
XII
Н2О—As—О—As—SO4H
SO4H SO4H
хш
К сожалению, методами криоскопии и электропроводности невоз-
невозможно отличить AsO+ от сольватированных форм X и XI. Если
для доказательства присутствия несольватированных ионов NO+
и NOJ в сернокислых растворах были успешно применены спектры
комбинационного рассеяния 143, то в данном случае это невозможно,
потому что для получения четких спектров комбинационного рас-
рассеяния необходимы достаточно концентрированные растворы.
В ранних работах сообщается о получении некоторых сульфат-
сульфатных соединений трехвалентного мышьяка, например х> 182> 219
As2O3 • SO3, As2O3 • 2SO3, As2O3 • 3SO3, которые по составу
близки к соединениям, присутствующим в 100% серной кислоте и
в олеуме. Установлено, что в кислородных соединениях трехвалент-
трехвалентного мышьяка атомы мышьяка соединены друг с другом через кис-
кислород, и кольцо XIV является важной структурной единицей в суль-
сульфатных соединениях трехвалентного мышьяка; оно аналогично
кольцу Ш сульфатных соединений бора. Поэтому структуру
As2O3 • SO3 можно представить в виде XV, a As2O3 • 2SO3 — в виде
XVI:
\
As
О
As
О
// \ч
о о
XIV
I
о
As
I
О
As
I
О
As
I
О
//
О
О
О О
XV
О
• —O-S—О—As
II I
О О
о
As—О—S—О—As
I
О
о
о
As
I
О
А
XVI
Трехокись сурьмы плохо растворяется в 100%-ной серной кис-
кислоте даже при температуре кипения. При охлаждении раствора из
него осаждаются белые призматические кристаллы 182 Sb2O3 • 3SO3,
т. е. Sb2(SO4K. Из растворов водной серной кислоты получаются
Sb2O3 • 2SO3 и Sb2O3 • SO3. Было показано, что эти соединения
не полностью гидролизуются водой, в результате чего могут образо-
образоваться SbO • SO4H и (SbOJSO4. Несомненно, что поведение Sb2O3
при растворении в серной кислоте аналогично поведению As2O3,
однако сульфирование сурьмы происходит при более мягких усло-
условиях. Это объясняется более низкой электроотрицательностью Sbm
по сравнению с As111. Были получены 182 некоторые соли кислоты
HSb(SO4J. При комнатной температуре все они нерастворимы
в серной кислоте. Предполагали, что эти соли образованы отдель-
отдельными анионами Sb(SO4J, замыкающимися в кольцо через атом
сурьмы. Однако учитывая нерастворимость их в серной кислоте,
более вероятно предположить, что они содержат полимерные анионы,
в которых сульфатная группа связывает атомы сурьмы.
Сульфат трехвалентного висмута получается при упаривании
Ш2О3 или В1A\О3)з с серной кислотой. В горячей воде он медленно
гидролизуется до соединения висмутила (BiOJSO4. При комнатной
температуре окись и сульфат висмута плохо растворяются в серной
166
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
167
кислоте. Соли щелочных металлов MBi(SO4J образуются при доба-
добавлении сульфатов этих металлов к кипящему раствору Bi2O3 в кон-
концентрированной серной кислоте. Эти соли также трудно растворимы
в серной кислоте. По своей природе они аналогичны соответству-
соответствующим соединениям сурьмы 183.
Ж. Ванадий, хром, марганец
На основании измерений электропроводности предположили 189,
что V2O5 растворяется в 100%-ной H2SO4, образуя равновесную
смесь VO(HSO4K и HOVO(HSO4J. Более детальные исследования
криоскопии и электропроводности показали, что в очень разбавлен-
разбавленном растворе V2O5 и NH4VO3 происходит ионизация и образуется
только VO(HSO4K
A36)
A37)
а с увеличением концентрации образуется полимер [VO(HSO4J]2O104.
Были изучены 189 растворы К2СгО4 в серной кислоте и в разба-
разбавленном олеуме и установлено, что эти растворы относительно
устойчивы, величина у, определенная по электропроводности, при-
приблизительно равна 3. Это значение соответствует реакции
О
О
K2CrO4+4H2SO4==HO-Cr-O-S-OH+2K++H3O+
О О
A38)
Кислота H2CrSO7 описана Джильбертом и сотр.81.
При растворении перманганата калия в серной кислоте, содер-
содержащей небольшое количество воды, образуется раствор зеленого
цвета. Было замечено, что растворы с концентрацией перманганата
менее 0,01 м в отсутствие восстановителей сохраняются в течение
24—36 ч, после чего из них осаждается МпО2216. Более концентриро-
концентрированные растворы быстро мутнеют, и из них выделяется зеленый оса-
осадок, который разлагается до МпО2. Если в качестве растворителя
брали безводную H2SO4 или олеум, то очень быстро разлагались
даже растворы, содержащие менее 0,01 м КМпО4.
Ройер обнаружил, что. криоскопически определенная величина
v зависит от концентрации перманганата калия и составляет при-
примерно 6 при концентрации 0,00358 м и 4,2 при 0,0145 м. Криоскопи-
ческая величина v уменьшается со временем, но через 3—12 ч ста-
становится постоянной, равной приблизительно 4. Замечено, что вели-
величина у, определенная по электропроводности при 25° С, уменьшается
'От 2,6 до 2,0 в течение 20 мин. Экстраполяцией было определено, что
в начальный момент реакции у « 3. Ройер интерпретировал эти
результаты, предположив образование перманганил-иона:
4= К>+Н3О++MnOJ+3HS0J
A39)
Уменьшение величин v и у со временем и с увеличением начальной
концентрации КМпО4 он объясняет выпадением осадка нераствори-
нерастворимого перманганилгидросульфата:
KMnO4+3H2SO4 = К+-{-Н3О+-{-2HSO4-{-MnO3HSO4
A40>
Было замечено 189, что КМпО4 медленно разлагается в 100%-ной
серной кислоте и быстро — в низкопроцентном олеуме; при этом
выделяется кислород и двуокись марганца или другое соединение
четырехвалентного марганца. Была определена величина у, равная
2,0—2,2, но не была исследована зависимость у от времени. Авто-
Авторы 189 пренебрегли зависимостью у от времени и предпочли взять
среднее значение из данных, полученных Ройером. С их точки зрения,
реакция может протекать следующим образом:
Эти же авторы провели кондуктометрическое титрование, при
котором к низкопроцентному олеуму добавлялся КМпО4. Минимум
электропроводности они обнаружили при величине г, равной при-
приблизительно половине ее значения при титровании KHSO4(r — отно-
отношение числа молей растворенного вещества к числу молей H2S2O7).
Таким образом, если при титровании KHSO4 минимум электропро-
электропроводности соответствует г = 0,56, то при титровании КМпО4 он
отвечает г = 0,28. Они предположили, что полученные результаты
объясняются реакцией
KMnO4+2H2S2O7 «= M11O3HSO4 + К> + HS2Oy + H2SO4
A42>
Однако для этой реакции расчетный минимум электропроводности
должен наблюдаться при г = 0,36. Если при разложении раствора
образуется кислород и МпО2, то, по-видимому, происходит не кис-
кислотно-основное взаимодействие, а сульфирование КМпО4 и
MnO2(SO4HK по уравнению
A43).
Минимум электропроводности при этом должен соответствовать
г =0,26, что близко к экспериментальному значению.
К сожалению, приведенные выше данные недостаточны для удо-
удовлетворительного объяснения поведения КМпО4 в серной кислоте.
168
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
3. Сера, селен, теллур
Двуокись серы умеренно растворяется в серной кислоте 185,
образуя раствор, электропроводность которого немного выше элек-
электропроводности растворителя (Р. Дж. Джиллеспи и Е. А. Робин-
Робинсон, неопубликованная работа). По-видимому, происходит очень
слабая ионизация:
SO2 + H2SO4 = HSOJ+HSOl A44)
Голд и Тай 121 обнаружили лишь незначительное отличие спектра
поглощения раствора двуокиси серы в олеуме от спектров индиви-
индивидуальных компонентов, на основании чего сделали вывод о том,
что эти вещества до некоторой степени взаимодействуют между
собой, но без значительной протонизации. Растворимость двуокиси
серы в олеуме выше, чем в 100%-ной H2SO4, и быстро возрастает
с увеличением концентрации SO3, т. е. с ростом кислотности раство-
растворителя 185. Это означает, что основность двуокиси серы, по существу,
обусловливает ее растворимость в серной кислоте и олеуме, вероятно
потому, что переходу протона, т. е. ионизации, предшествует образо-
образование комплекса с водородной связью:
H2SO4+SO2 —> HSO3OH...O=S=O —v HSOr + +HO=S=O A45)
Давно известно, что двуокись селена растворяется в серной
кислоте, образуя ярко-желтый раствор 184. Впоследствии было
показано 70, что в разбавленных растворах двуокись селена ведет
себя как слабое основание:
SeO2 + H2SO4
SeO2H
A46)
Возможно, что ион SeO2H+ присутствует в растворе в виде кислого
сульфата (SeO2H)HSO4. Реакцию двуокиси селена с серной кислотой
правильнее было бы написать следующим образом:
SeO2 + H2SO4
(SeO2H)HSO4 —*¦
A47)
В разбавленном растворе SeO2 в серной кислоте присутствует
также небольшое количество ионов HSe2O4 (XVII) и ионизирован-
ионизированного Se2O3(OH) (HSO4) (XVIII):
о
II
Se
О
ООО
II
Se
НО
О
XVII
i !i Ч ?
Se Se S
НО О О ОН
XVIII
С увеличением концентрации SeO2 образуются относительно боль-
большие количества соединения XVIII и, по-видимому, более высоко-
высокополимерных веществ.
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
169
При добавлении двуокиси селена 70 к олеуму электропровод-
электропроводность системы падает и при мольном отношении [SeO2] : [H2S2O7] =
= 1:1 проходит через минимум. Это проще всего объяснить образо-
образованием почти неионизированного комплекса SeO2 • H2S2O7, кото-
который можно представить и в виде SeO(HSO4J- Существование таких
неионизированных частиц было подтверждено криоскопическими
измерениями, которые показали, что состав смеси в точке минимума
электропроводности соответствует v = 1,3 при исходной концентра-
концентрации SeO2 в H2S2O7, равной 0,1л. Имеются доказательства существо-
существования аналогичной диселенистой кислоты Se2O3(HSO4J.
Кажется странным, что двуокись теллура нерастворима в серной
кислоте 183, так как известно соединение25 2ТеО2 • S03, которое
растворяется в HNO3, НСЮ4 и HSO3F. Из таких растворов удалось
выделить 2ТеО2 • HNOJ61 и 2ТеО2 • НСЮ f. Эти соединения можно
рассматривать как производные дителлуристой кислоты, т. е.
Te2O3(OH)(NO3), Te2O3(SO4), Те2О3(ОН)(СЮ4). Вполне возможно,
что при обработке ТеО2 серной кислотой образуется нерастворимый
высокополимерный сульфат.
И. Галогены
Недавно было показано (Р. Дж. Джиллеспи и Е. К. Робинсон,
неопубликованные результаты), что хлористый водород при низких
концентрациях реагирует с серной кислотой, образуя хлорсульфоно-
вую кислоту с количественным выходом:
A48)
Аналогично реагируют многие хлориды:
r A49)
Хлорная кислота и хлорсульфоновая кислота ведут себя в серной
кислоте как очень слабые кислоты 5.
Растворы йодноватой кислоты в серной кислоте были изучены
методами криоскопии и электропроводности Аротским и сотр.3,
Джиллеспи и Сеньором 1Ов' 107. Более обширные исследования после-
последующих авторов показали, что йодноватая кислота не только про-
тонизирована 3
HIO3 + H2SO4=H2IO?+HSO4- A50)
но и образует иодилгидросульфат IO2HSO4
НЮз+ 2H2SO4= IO2HSO4+ H3O+ -fHSO; A51)
который в растворе находится, вероятно, в сольватированной и поли-
полимерной формах, таких, как XIX и XX. Белый осадок, выделя-
выделяющийся из раствора при стоянии, представляет собой, по-видимому,
длинноцепной (XXI) или пространственный (XXII) полимер. Нет
170
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
доказательств того, что ионы Ю2 или Н2Юг присутствуют в серно-
сернокислых растворах в значительных количествах.
SO4H SO4H SO4H SO4H
l\)H
SO4H
XIX
XX
HO4S—I
0
SO4H SO4H SO4H SO4H SO4H
1
XXI
0
-O-I-O-I-O-I-
so4 so4 so4
I I I
-O—I—O—I—O—I—
II II II
0 0 0
I
I—OH
0 0
II II
0—I-O-I-O
I I
so4 so4
I I
0—I—0-1—0
II II
0 0
XXII
Реакцией иода с раствором йодноватой кислоты в серной кислоте
был получен желтый иодистокислый сульфат (IOJSO4, «сульфат
Кретьена»35' 36. ИК-Спектры показали, что это соединение и анало-
аналогичные ему (IOJSeO4 и ЮЮ3 относятся, вероятно, к ионному типу
и содержат полимерный иодозил-ион (Ю+)?5. Иодозилсульфат
в H2SO4 реагирует с нитробензолом, образуя ж-иодозонитробензол;
это свидетельствует о том, что в растворе присутствует простой
иодозил-катион. Более активные вещества, такие, как бензол и хлор-
хлорбензол, образуют с количественным выходом сульфат диарилиодо-
ния. Массон и Хэнби178 предположили, что последнее вещество
получается из ионов I 3+, присутствующих в небольших количествах
и находящихся в равновесии с Ю+:
1О++2Н+=1»+ + Н2О A52)
Образование иона диарилиодония протекает следующим образом:
A53)
A54)
Равновесие реакции A52) при удалении воды должно смещаться
вправо. Действительно, в олеуме желтый иодозилсульфат переходит
в белое соединение 177; ему соответствует формула I2(SO4K • H2SO4;
его можно рассматривать как частично десольватированную форму
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
17f
I(HSO4K. Криоскопические исследования и исследования электро-
электропроводности растворов иодозилсульфата в серной кислоте пока-
показали 107, что это соединение не неэлектролит, как считают Аротский
и сотр.3, а превращается в кислый сульфат, который в разбавленном
растворе ионизирован на 50%:
(IOJSO4+H2SO4
I0++2HS0r
A55)
В низкопроцентном олеуме IOHSO4, образует IO(HSO4L, а в из-
избытке олеума — полный гидросульфат трехвалентного иода I(HSO4K.
Белый осадок, который получается при действии олеума на иодо-
эилсульфат, представляет собой частично десольватированную
форму I(SO4)(HSO4) со структурой линейного нолимера XXIII или
аналогичного соединения ионного типа [I(SO4)+]n(HSO4")n, тогда
как нормальный сульфат I2(SO4K является, несомненно, простран-
пространственным полимером XXIV:
SO4H SO4H SO4H SO4H SO4H
I I I I I
I I I I I
S04 SO4 S04 S04 S04
ххш
-I-SO4— I-SO4-I-SO4-
S04
S04
3O4
—I-SO4—I—SO4-I-SO4-
XXIV
Следует ожидать, что двуокись иода (Ю2)„ будет вести себя как
соединение I111 и Iv и растворяться в серной кислоте с образованием
IO2HSO4 и IOHSO4107.
Массон 176 показал, что иод растворяется в растворах иодозил-
сульфата в концентрированной серной кислоте, образуя коричневые
растворы, которые реагируют с хлорбензолом, в результате чего
получается смесь хлортрииодбензолов и осадок элементарного иода.
Быстрота протекания реакции и образование вещества коричневого
цвета, устойчивого только в сильнокислых средах, дают основание
полагать, что иод находится в катионной форме. Исходя из стехио-
стехиометрии реакции растворов, содержащих иод и иодозилсульфат в раз-
различных соотношениях с хлорбензолом, Массон пришел к выводу
о существовании ионов I+ , IJ и 1?.
Были определены точки замерзания и электропроводность серно-
сернокислых растворов иода и йодноватой кислоты. Экспериментальные
значения v и у при отношении [12] : [НЮ3] = 7,0 моль соответ-
соответствуют образованию IJ:
A56)
172
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
III. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
173
Замечено, что в таких растворах может раствориться больше
иода, чем это соответствует отношению [12] : [НЮ31 = 7,0 моль,
но температура замерзания и электропроводность остаются постоян-
постоянными и не зависят от количества добавляемого иода. Это можно
объяснить только образованием Ц:
H A57)
При отношении [I] : [НЮ3] = 2,0 моль образуется I+:
A58)
где v=16, v = 8 и v — y = 8.
Если IHSO4 неэлектролит, то эту же реакцию можно написать
следующим "образом:
HIO3 + 2I2 + 8H2SO4 = 5IHSO4 + 3H3O+-(-3HSO4' A59)
для которой v = 11,y = 3hv — 7 = 8. Однако было найдено, что
v с увеличением концентрации уменьшается от 8,5 до 7,7, у — от
35 до 3,3, a v — у — от 5,0 до 4,4, т. е. полученные данные не соот-
соответствуют образованию 1+ или IHSO4. Экспериментальные значения
v и у скорее соответствуют уравнению
4HIO3 + 8I2-(-17H2SO4 = 5l3 + 7H3O+ + 5lOHSO4-(-12HSOi A60)
для которого v = 8,5 ч- 7,25, у = 4,25 ч- 3, v — у = 4,25.
Так как IOHSO4 частично ионизирован, значения v и у, при-
приведенные выше, являются предельными значениями, отвечающими
степеням диссоциации 0 и 100% соответственно. Экспериментальные
значения v и у для очень разбавленных растворов несколько выше,
чем следовало бы ожидать. Очевидно, это означает, что при таких
концентрациях в растворе присутствуют небольшие количества 1+
или IHSO4. Диспропорционирование 1+ может быть представлено
уравнением
4I+ -bH2O + 2HSOj= ГО+ + IJ + 2H2SO4 A61)
из которого ясно, что с возрастанием концентрации равновесие
сдвигается вправо.
Сообщается, что темно-синие растворы, образующиеся при рас-
растворении иода в олеуме, содержат ионы 1+, которым приписываются
полосы поглощения 224 при 640, 500 и 410 ммк. Спектры коричневых
растворов, содержащих 13, имеют полосы поглощения при 290
и 460 ммк. Растворы с мольным отношением [12] : [НЮ31 = 2,0,
что отвечает стехиометрии образования 1+, имеют четкую полосу
поглощения при 640 ммк, характерную для 1+, а также четкую по-
полосу при 460 ммк, частично накладывающуюся на полосы 500 и
410 ммк. Относительные интенсивности поглощения показывают,
что в некоторых из исследованных растворов примерно одна треть
иода присутствует в виде 1+, остальная часть подвергается диспро-
порционированию 224 по уравнению A56). Если вспомнить, что для
спектроскопии применяются менее концентрированные растворы,
чем при криоскопических исследованиях, то меньшая степень диспро-
порционирования первых становится вполне понятной.
Добавление IC1 или 1Вг к сернокислому раствору, содержащему
иод и йодноватую кислоту в мольном отношении [12] : [НЮ3] =
= 2,0, приводит к результатам почти аналогичным тем, которые
получаются при добавлении иода [см. ур. A60)]. На основании
этого заключили, что образуются соответственно 12С1+ и 12Вг+.
К. Комплексы переходных металлов
Появление в области высоких полей ЯМР-спектра характеристи-
характеристических линий, обусловленных водородной связью переходного
металла, указывает на то, что металл-карбонильные и циклопента-
диенильные комплексы протонизированы по металлу 46. Например,
в спектрах комплексов PPh3Fe(COL, AsPh3Fe(COL, (PPh3JFe(CO3)
и (AsPh3JFe(COK, образующих в 98%-ной серной кислоте устойчи-
устойчивые желтые растворы, наблюдается сигнал (т ^ 18), принадлежа-
принадлежащий связи Fe—Н. Относительные интенсивности сигналов от Fe—Н
и фенильных групп свидетельствуют о том, что комплексы играют
роль однокислотных оснований, т. е.
Fe(COLPPh3+ H2SO4= HFe(COLPPhJ
A62)
Ряд трикарбониларенов хрома легко растворяется в серной кис-
кислоте с образованием желтых растворов, которые могут содержать
соответствующие сопряженные кислоты. Эти растворы быстро раз-
разлагаются.
Двухъядерные соединения [С2Н5Мо(СО3)]2 и [C2H5W(COK],
имеющие багрово-красный цвет, в серной кислоте образуют красно-
коричневые растворы. Последние медленно разлагаются в отсут-
отсутствие воздуха и содержат протонизированные комплексы. Химические
сдвиги в ПМР-спектрах, обусловленные протоном, связанным
с металлом, исключительно велики (т = 30 -f- 40). Подтверждением
того, что водород непосредственно связан с атомом металла, является
наличие дополнительных пиков в спектрах соединений вольфрама,
которые могут быть вызваны спариванием спинов протона с изото-
изотопом вольфрама 183W. Дополнительные линии показывают, что протон
некоторым образом связан с обоими атомами металла. Тщательное
изучение спектра позволило установить, что имеется интенсивный
внутримолекулярный обмен протоном между двумя атомами воль-
вольфрама.
Двухъядерный комплекс железа [C5H5Fe(COJ]2 растворяется
в серной кислоте. В ЯМР-спектре его есть сигнал в области высоких
полей, площадь которого составляет 1/10 площади сигнала С5Н5,
174
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
что указывает на одноосновность [C5H5Fe(COJ]2. Криоскопические
исследования дали несколько завышенные значения v, равные
2,3 и 2,4. Причина этого неизвестна. К сожалению, криоскопиче-
криоскопические исследования других соединений такого типа не проводились.
ИК-спектр индивидуального соединения содержит полосу 1785 см'1,
которая приписывается мостиковой карбонильной группе. В спектре
раствора этого соединения в H2SO4 такая полоса отсутствует. Она
отсутствует и в спектре гексафторфосфатной соли, которая полу-
получается путем обработки раствора комплекса в безводном HF пента-
хлоридом фосфора. Таким образом, протонизированные молекулы
состоят, по-видимому, из двух частей —C5H5Fe(COJ, между кото-
которыми существует металлическая связь, а протон, как и в комплексах
вольфрама и молибдена, совершает быстрый внутримолекулярный
переход.
Карбонил-олефин-металлические комплексы в сернокислом рас-
растворе протонизируются неодинаково 47. Водородная связь осуще-
осуществляется не непосредственно с ионом переходного металла; вместо
этого протон присоединяется к олефину лиганда, образуя «ион кар-
бония», связанный с металлкарбонильной частью комплекса и ста-
стабилизируемый ею. Трикарбонилциклооктатетраенжелезо легко рас-
растворяется в серной кислоте, образуя растворы красного цвета.
Спектр протонного резонанса не имеет какой-либо линии в области
высоких полей, характерной для металл-водородной связи; он сви-
свидетельствует о протонизации кольца, т. е. образовании катиона
трикарбонилбицикло[5,1,0]октадиенилжелеза:
--)—Fe(coK
Н+
;V-Fe(coK
A63)
Однако удивительно, что этот спектр существенно отличается от
спектра свободного катиона бицикло[5,1,0]октадиена (см. IV, И, 1).
Ион трикарбонилжелезоциклогептатриена в сернокислом рас-
растворе протонизирован до катиона C7H9Fe(CO3)+:
Те(со)г-СЛ + н+
Д__ге(со)г
A64)
Были исследованы кислые растворы ряда цианистых комплексов
и показано, что они присоединяют протон к CN-группе, а не к атомам
Ш. РАСТВОРЫ НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
175
металлов 217. Например, Н [Fe(phen)(CNL] образует в концентриро-
концентрированной серной кислоте ярко-желтый раствор *. Изменение окраски,
происходящее при разбавлении раствора водой, по-видимому,
результат образования иона [Fe(phen)(CNH)J3+. В отличие от пре-
предыдущих сообщений было показано, что трис-A,10-фенантролин)-
-железо(И) и аналогичный ему комплекс 2,2-дипиридила(Ыру) не
обладают заметным сродством к кислотам. Постепенное изменение
цвета растворов этих комплексов в концентрированной серной кис-
кислоте объясняется их окислением.
Л. Образование хелатов
Мы считаем, что структурные формулы различных соединений,
содержащих сульфатные группы, имеют скорее вид XXV нежели
XXVI, так как нет доказательств того, что SO4-rpynna ведет себя
как бидентатная, но известны некоторые соединения, где она служит
мостикообразователем.
О О
О
\
/
\
V
О
XXV
XXVI
Так как ионные радиусы бора и углерода почти одинаковы, можно
предположить, что их сульфатные соединения должны обладать
аналогичной структурой.
В связи с этим интересно сравнить предполагаемые структуры
сульфатборных соединений со структурой метиленсульфата. Послед-
Последнее соединение, как было показано *, в бензоле имеет димерную
структуру и, следовательно, для него более вероятна структура
XXVII, а не XXVIII:
о о—сн2—о о
V V
О-СНг-О О
KXVII
О О
V Хсн2
/\/
О О
XXVIII
* Символ phen — 1,10-фенантролин. — Прим. ред.
176
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Вполне уместно сравнить их также с триметиленсульфатом, который
в бензоле мономерен 4 и имеет структуру
СЩ-0 О
н2с
Н2-
XXIX
По-видимому, хелатообразование с сульфатной группой воз-
возможно у многих соединений, например у H3Fe(SO4)(C2O4J,
H3Fe(SO4J • С2О4, M2Pb(SO4K, K3Ce(SO4K • Н2О, Sb2(SO4K и т. д.
Однако для всех этих соединений нет прямых доказательств образо-
образования такой связи, и возможно, что в действительности их струк-
структуры сложнее, чем описывает простая молекулярная формула,
и содержат мостиковую сульфатную группу.
Существует несколько соединений, для которых присутствие
мостиковых сульфатных групп можно считать, по-видимому, дока-
доказанным, например диэтилсульфат германия в растворе ацетона, име-
имеющий по данным криоскопических исследований 2 димерную струк-
структуру XXX. Для солей, получающихся по реакции диаминов с бис-
-(тетраэтилдизолотосульфатомN4, предложена структура типа XXXI.
Ge
ххх
{[(C2H6JAu bipyl+}2
2-
Au
т./
Аи
х \с2н5_!
XXXI
Известно несколько случаев, когда сульфатная группа связы-
связывает два атома металла, которые соединены еще и другими мостико-
образователями, например NH2, ОН и Н2О. Накамото и сотр. 191
изучили ИК-спектры соединения XXXII и пришли к заключению,
что сульфатная группа имеет симметрию C2tl, и, следовательно,
является мостикообразователем. Однако Барраклу и Тоб 7 заявили,
что отличить методом ИК-спектроскопии мостиковую сульфатную
группу от хелатной сульфатной группы очень трудно.
(NH3LCo
Co(NH3L
4
KXXII
(NO3K
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
177
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
В СЕРНОЙ КИСЛОТЕ
А. Простые основания
Многие кетоны, альдегиды, карбоновые кислоты, ангидриды,
эфиры, амины, амиды, нитрилы, нитросоединения, сульфоксиды,
сульфоны, а также некоторые этиленовые и ароматические угле-
углеводороды легко превращаются в сопряженные им кислоты 91. В тех
случаях, когда происходит неполная ионизация, относительная
основность определялась методами криоскопии, электропроводности
и спектроскопии. Полученные результаты представлены в табл. 30.
Таблица 30. Константы диссоциации слабых органических оснований,
определенные разными методами
Основание
Нитрометап
Нитробензол
в-Нитротолуол
о-Нитротолуол
л-Нитротолуол
п-Бутшгаитробензол
гс-Хлорнитробензол
о-Хлорнитробензол
м- X лорнитробензо л
2,4-Дшштротолуол
1,2-, 1,3- и 1.4-Динитро-
бензолы
Ацетонитрил
Бензонитрил
Димстилсульфон
Диэтилсульфон
Ди-и-пропилсульфон
Ди-»-бутилсульфон
Тетраметиленсульфон
¦^дисс
по электропро-
электропроводности
0,00251
0,0101 0,013'
0,0951 0,105'
0,0671 0,0737
0,0231 0,0287
—
0,0041 0,00577
0,00247
0,00137
0,0005?
0,00037
0,162
0,072
0,01483
0.01193
0,00513
0.00393
0.00393
по криоско-
криоскопии
0,004 **
0,011*
0,094-1
0,0624
0,0204
0,10*
0,0031
—.
—
—
—
—
—
0,01503
0.01203
0.00483
0,00413
0.00363
по спектро-
спектроскопии
_
0.0075 0,0096 8
0.0805 0,0908
—
0,021 & 0,0258
0,108
0,0055 0,00428
—
0,00118
0.00065
—
—
—
—
—
—
—
—
по титро-
титрованию*
_
0,013в
—.
—.
0,0246
—
—
—
—
—
.—
—
.—.
0,0145*
0,0123»
0,0052»
0.00403
0.0036»
Примечание. Верхний индекс у цифр указывает на литературный источник данных:
i — 10S; 2—171; 3 —127; k—94, 96; Ь — ЮЗ; Ъ — 71, 72, 76; 7 — 169; Ь^21, 22.
*) Титрование основанжя раствором H2Sj07 в HjSOn.
**) Измерена в концентрированной (слабоводной) H2SO4.
Б. Многоосновные соединения
Место присоединения протона и степень протонизации много-
многоосновного соединения зависят от основности каждой группы, поло-
положения ее в пространстве и мезомерного взаимодействия между
группами.
12 заказ 90
178
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Основность амино-группы такова, что обычно в полиаминах все
амино-группы протонизированы независимо от их положения в моле-
молекуле. Например, о-фенилендиамин понижает точку замерзания в три
раза, что указывает на двойную протонизацию 1в4>165. В молекуле
гексаметилентетрамина, как показали измерения электропровод-
электропроводности, протонизированы все четыре атома азота ш. Гуанидин, кото-
который в водном растворе является сильным основанием, интересен тем,
что присоединяет только 1,3 протона 241. Эта неполнота прохождения
второй стадии протонизации иллюстрирует важность резонансного
взаимодействия между основными группами. При резонансе одно-
протонизированный ион прочно стабилизирован; в результате при-
присоединение второго протона будет происходить потеря значительного
количества энергии резонанса:
NH
2\
c=NH2
NH2;
NH2
NH,
^C-NH2
l/
;c-nh2
Определение основности растворов ароматических кетонов и аро-
ароматических карбоновых кислот в смесях серной кислоты с водой
методом УФ-спектроскопии показало, что эти соединения являются
более слабыми основаниями, чем анилин, приблизительно в 10 10
раз вв,в?,222>22з_ Такая же разница в основности наблюдается для
растворов в серной кислоте полиаминов, с одной стороны, и дикето-
нов, дикарбоновых кислот и' кетокарбоновых кислот — с другой.
При этом если две основные группы достаточно близки друг к другу,
то дикетоны, дикарбоновые и кетокарбоновые кислоты в растворе
серной кислоты присоединяют только один протон. С увеличением
расстояния между двумя основными группами непрерывно увеличи-
увеличивается и степень протонизации. Это подтверждается значениями v,
полученными из криоскопических данных (табл. 31I30>193>239'240.
Максимальное значение v = 3,0 в растворах дикарбоновых кислот
не достигается; это еще не получило объяснения. Можно ожидать,
что амино-группа в аминокислотах и аминокетонах будет протонизи-
ровайа полностью, а основность карбонильной или карбоксильной
группы Зудет увеличиваться по мере увеличения расстояния между
ней и амино-группой (см. табл. 31).
Алифатические простые эфиры в H2SO4, по-видимому, полностью
протонизированы по метокси-группе. Эфиры, имеющие ароматиче-
ароматический радикал с СО-группой, протонизируются по кето-группе, но
метокси-группа сильно ослабляет основность последней. Например,
1-метокси-, 2-метокси- и 1,2-диметоксиантрахинон, как и сам антра-
хинон, протонизированы один раз
239
Однако в 1,4-диметокси-,
4,5-диметокси- и 1,4,5,8-тетраметоксиантрахиноне вторая карбониль-
карбонильная группа активирована метоксильной группой и присоединяет
протон, т. е. эти соединения протонизированы дважды 239. Анизол
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
179
Таблица 31.
Значения v для сернокислых растворов
некоторых органических соединений
Соединение
Дибензонл
Дибензонлметан
Дибензонлпропан
Дибензоилпентан
Дибензоилгексан
Дибензонлгептан
Антрахинон
Щавелевая кислота
Малоновая кислота
Янтарная кислота
Глутаровая кислота
Пробковая кислота
Себациновая кислота
ж-Бензоилбензойная
кислота
и-Бензоилбензойная
кислота
Глицин
Р-Алании
у-Амино-«-масляная
кислота
7-Амино-и-валериановая
кислота
п; и'-Диметиламинобен-
зофенон
Формула
PhCOCOPh
PhCOCHjCOPh
PhCO(CH2KCOPh
PhCO(CH2MCOPh
PhCO(CH2NCOPh
PhCO(CH2OCOPh
CeH4(COJCeH4
HO2CCO2H
HO2CCH2CO2H
HO2C(CH2JCO2H
HO2C(CH2 3CO2H
HO2C(CH2NCO2H
HO2C(CH2)8CO2H
J№-HO2CC6H4COPh
n-HO2CC6H4COPli
HO2CCH2NH2
HO2C(CH2JNH2
HO2C(CII2KNH2
HO2C(CH2LNH2
(C Hs)»N-<f~4-C-<^4-N(CHs),
0
. v
1,8
2,1
3,0
2,9
3,0
3,0
2,0—2,2
1,3
2,0
2,5
2,6
2,6
2,6
2,5
2,5
2,2
2,7
2,9
3,0
3,5
и многие его производные легко сульфируются серной кислотой,
однако во фторсульфоновой кислоте анизол при —80° С присоеди-
присоединяет протон и к кольцевому атому углерода, находящемуся в пара-
положении, а не к метоксильному кислороду 15. Хотя анилин при-
присоединяет протон к атому азота, в растворах бис-(диметиламинофе-
нил)-фенилкарбинола и трис-(диметиламинофенил)-карбинола, не-
несмотря на протекающее до конца образование иона карбония, полной
протонизации амино-групп, как показывают значения v, не проис-
происходит 194. По-видимому, основность амино-групп ослабляется сопря-
сопряжением с центральным ионом карбония, в результате чего в 100%-
ной серной кислоте она протонизирована не полностью.
В. Амиды и мочевины
Известно, что в серной кислоте амиды ведут себя как сильные
основания 201, однако место присоединения протона долгое время
оставалось неизвестным. ЯМР-спектры сернокислых растворов
12*
180
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
амидов показали, что амиды присоединяют протон, вероятнее всего,
к кислороду, а не к азоту. Например, было показано 30, что площади
линий протонного резонанса NH2-rpynnbi и метильной группы ацет-
амида, растворенного в серной кислоте, находятся в отношении 2 : 3.
Если бы протон присоединялся к азоту, то наблюдалось бы два пика
равной площади. Было показано, что спин-спиновое взаимодействие
между протонами, связанными с азотом, и протонами метильной
группы N-метилацетамида в 100%-ной H2SO4 вызывает появление
дублетной линии для метильной группы, тогда как протонизация
азота давала бы триплетную линию 12. В ЯМР-спектре сернокислого
раствора при комнатной температуре невозможно наблюдать от-
отдельный сигнал протонизированной карбонильной группы, так как
происходит быстрый обмен протонами с растворителем. Однако
в растворе фторсульфоновой кислоты при пониженных температурах
обмен протонами с растворителем так замедлен, что сигнал от группы
С=ОН+ наблюдается в области очень низких полей14'88.
Лайстен1в2 изучал скорость сольволиза некоторых амидов в
100%-ной H2SO4:
R'C(OH+)NHR + HSOI+2H2SO4 >¦ R'CO2HJ + RNHJ + HSOJ+HSjO^ A65)
Он нашел, что по мере изменения состава растворителя от водного
раствора серной кислоты до 100%-ной H2SO4 реакция сначала про-
протекает по обычной схеме механизма А2, включающей бимолекуляр-
бимолекулярное взаимодействие молекулы воды с кислотой, сопряженной амиду,
а затем сменяется механизмом А1, при котором стадией, определя-
определяющей скорость реакции, является мономолекулярный гетеролиз
N-протонизированного амида:
а)]
б)
в)
г)
R'
;0Н
4NHR
быстро
R'
R'CONH2R+
RNH2
R'CO
медленно
R'CO
A66)
'¦' быстро
быстро
R'CO2H5
Для того чтобы объяснить увеличение скорости реакции при введе-
введении кислот H2S2O7 или HSOgCl и уменьшение скорости при приба-
прибавлении такого основания, как Н3О, Лайстен предположил, что
реакция подвержена кислотному катализу вследствие того, что ста-
стадии (а) и (в) протекают с одинаковой скоростью.
Методом криоскопии было показано, что мочевина, уретан и
N-метилуретан в сернокислом растворе присоединяют один про-
протон 139. Для раствора мочевины это было подтверждено и измере-
измерениями электропроводности. Тетраметилмочевина в 0,05 м растворе
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
181
полностью монопротонизирована, а приблизительно 10% молекул
лротонизированы двукратно (Т. Бирчелл, Дж. Голди и Р. Д. Джпл-
леспи, неопубликованные результаты).
Г. Сложные эфиры
Как правило, сложные эфиры карбоновых кислот в сернокислом
растворе подвергаются сольволизу. Однако некоторые из них, такие,
как метил- и этилбензоат, а также этилацетат реагируют с серной
кислотой при комнатной температуре очень медленно; криоскопиче-
ские исследования показали, что первоначально эфиры просто
протонизируются. Если такой сернокислый раствор вылить на лед,
то почти весь эфир регенерируется 131Д52.203. Принято считать,
хотя это и не доказано, что протон присоединяется к карбониль-
карбонильному кислороду.
Грэхем 124 показал, что реакция сольволиза метплбензоата в 98—
100%-ной серной кислоте имеет нулевой порядок по воде. Автор
пришел к выводу, что эта реакция мономолекулярна. Лайстен 161
криоскопически измерил скорость реакции в водном растворе H2SO4,
в 100%-ной H2SO4 и в низкопроцентном олеуме. Он показал, что
реакция имеет первый порядок, причем константа скорости не зави-
зависит от исходной концентрации эфира и, таким образом, от концен-
концентрации кислого сульфат-иона, так как метилбензоат ионизирован
как сильное основание. Отсюда ясно, что устраняется определяющая
скорость реакции стадия, — взаимодействие кпслогр сульфат-иона
с протонизированным эфиром. Скорости реакции в очень концентри-
концентрированной и в 100%-ной серной кислоте одинаковы, а в олеуме не-
несколько выше; следовательно, вода почти не влияет на реакцию.
Мономолекулярный гетеролиз может происходить разрывом либо
связи ацил — кислород (ААс1), либо связи алкил — кислород
(АД
*
О
R'-C
\
V
Аде ' Лд,к
Электронодонорные заместители в положении R' должны уско-
ускорять гетеролиз по механизму ААо1, а в положении R тормозить его
и соответственно оказывать противоположное действие на гетеролиз
Ааш1- Электроноакцепторные заместители в положении R' должны
тормозить гетеролиз по механизму Аас1 , а в положении R ускорять
его и соответственно оказывать противоположное действие на гете-
гетеролиз AAitl. Грэхем 124 нашел, что скорость сольволиза метилового
эфира га-толуиловой кислоты в четыре раза выше скорости
182
ГЛАВА ЧЕТВЕРТАЯ. 'СЕРНАЯ КИСЛОТА
сольволиза метилбензоата, и сделал вывод о том, что реакция проходит
но типу AacI- Качественное сравнение скоростей гидролиза для
большого числа эфиров было сделано Куном и Корвином 163, а также
Брэдли и Хиллом17. Полученные ими результаты подтверждают
механизм ААс1-
Скорости сольволиза алкилбензоатов изменяются следующим
образом: СН3 > С2Н6 <С мзо-С3Н7 < торете-С4Н9, что свидетельст-
свидетельствует об изменении механизма реакции в этом ряду соединений, начи-
начиная с изопропилбензоата. Лайстен показал, что введение нитро-
группы в гаяра-положение этнлбензоата приводит к уменьшению
скорости сольволиза в 60 раз, в изопропилбензоат — в 200 раз.
Итак, механизм сольволиза этих эфиров изменяется от AacI для
метил- и этилбензоата до A\il для изопропил- и трет-бутилбензоата.
Для этих эфиров начальное значение v = 2,0 по мере протекания
реакции сольволиза возрастает до 3,0. Окончательное уравнение
сольволиза может быть записано в виде
PhCO2CH3 + 2H2SO1=PliCO2HJ + HSOj + CH3HSO4 A67)
Вводя в R' электроноакцепторные заместители, можно вызвать
даже для этиловых эфиров сольволиз с разрывом связи алкил — кис-
кислород. Было найдено, что скорость сольволиза этиловых эфиров
возрастает в ряду гс-нитро- < 4-хлор-З-нитро- <; 3,5-динитробензой-
ная кислота 148.
Величина v для метилового эфира мезитиленкарбоновой кис-
кислоты 227 постоянна и равна 5, т. е. это соединение подвергается
быстрому и полному сольволизу, вероятно, с разрывом связи
ацил — кислород и образованием устойчивого ацил-иона:
СеН2(СНз)зСО2СНз + 2Нг8О4=СеНг(СНз)зСО+ + НзО + + СНзН8О4 + 2Н8Ог A68)
Интересно отметить, что эфиры мезитиленкарбоновой кислоты в вод-
водных растворах плохо гидролизуются. Легче они подвергаются соль-
сольволизу в серной кислоте. Выливая полученный раствор на лед,
можно выделить свободную кислоту. Обратный процесс, т. е. этери-
фикация мезитиленкарбоновой и родственных ей кислот, может быть
проведен путем приливания их сернокислых растворов к соответст-
соответствующему спирту "~
192
Д. Ангидриды карбоновых кислот
Уксусный и бензойный ангидриды понижают точку замерзания
100%-ной серной кислоты в 4 раза 84, а очень концентрированной
кислоты 160 — в 2 раза. Измерение электропроводности этих раство-
растворов показывает, что на каждую молекулу ангидрида образуется один
кислый сульфат-ион (т. е. у = IO5. Полученные результаты,свиде-
результаты,свидетельствуют о следующем механизме ионизации:
A69)
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
183
в 100%-ной H2SO4 и
(RCOJO + H2SO4 + H3O+=2RCO2HJ + HSOi- A70)
в водной концентрированной серной кислоте.
Фталевый ангидрид вызывает понижение точки замерзания
100%-ной серной кислоты только на 10—15% больше, чем неэлектро-
неэлектролиты 13Мв2,198) а измерения электропроводности показывают,68 что
•у == 1,14 -^ 1,18. Вполне вероятно, что это происходит вследствие
неполной протонизацтш. В концентрированной серной кислоте
понижение точки замерзания еще меньше, так как ангидрид пре-
превращается в протонизированную фталевую кислоту 1вз:
A71)
•со
ЧСО2Н
Полное превращение ангидрида в протонизированную фталевую
кислоту соответствовало бы величине v = 0. Но фталевая кислота
вследствие образования этой сопряженной кислоты
СО2Н
+ H2SO4
С02Щ
-f-HSOj
A72)
СО2Н
вызывает несколько больше чем двукратное понижение температуры
замерзания концентрированной серной кислоты, по-видимому, из-за
последующей слабой протонизации второй карбоксильной группы.
Однако v для раствора фталевой кислоты в серной кислоте, содержа-
содержащей HSOJ и HS2O,, только несколько больше единицы 1вз вследствие
того, что HS2O7 дегидратирует кислоту:
| ||
СО2Н
-f-HS2O7=
A73)
сек
Для этой реакции v = 1, если не происходит дальнейшая прото-
низация. Ясно, что кислота и ангидрид вблизи состава, отвеча-
отвечающего 100% H2SO4, находятся в равновесии, незначительный из-
избыток воды или трехокиси серы сдвигает равновесие соответственно
в сторону образования кислоты или ангидрида.
Малеиновая кислота и ее ангидрид ведут себя аналогичным обра-
образом. То же самое можно сказать о янтарной кислоте и ее ангидриде
с той разницей, что протонизация второй карбоксильной группы
кислоты протекает примерно на 50% и что ангидрид также почти
на 50% переходит163 в протонизированное состояние в серной ки-
кислоте, содержащей небольшие количества HSOJ.
184
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Е. Простые эфиры
Алифатические эфиры в H2SO4 ведут себя как сильные или уме-
умеренно сильные основания, v sg 2 в начальный момент 91,144>203.
Такие растворы, однако, неустойчивы, причем точка замерзания
растворов со временем понижается. Оддо и Скандола 203 выделили
через некоторое время после растворения из сернокислых растворов
диэтилового и некоторых других алифатических эфиров небольшие
количества соответствующих алкилгидросульфатов. Это обстоятель-
обстоятельство и то, что значение v в конце концов достигает предельного
значения, равного —4, показывают, что протекает полный сольво-
лиз, согласно следующему уравнению:
Скорости сольволиза многих эфиров могут быть рассчитаны,
исходя из скорости изменения v со временем 144. Полученные данные
свидетельствуют о таком механизме реакции, при котором стадией,
определяющей скорость, является мономолекулярный распад со-
сопряженной кислоты или какого-нибудь другого комплекса эфира
с растворителем, в результате чего образуется более устойчивый
из двух возможных ионов карбония. Несколько удивляет, что ско-
скорость сольволиза уменьшается при добавлении к эфирному раствору
кислого сульфата и еще больше — при добавлении воды. Если бы
комплексами, подвергающимися мономолекулярному распаду, были
протонизированные эфиры, то прибавление кислого сульфата и воды
должно было бы оказывать одинаковый эффект. Исходя из этого
принимается следующее уравнение, описывающее действие раство-
растворителя на растворенное вещество 144:
RR'OH+ + SO3 ^z? RR'SO4H+ A75)
за которым следует
R'SO4H
A76)
SO3H +
Ион карбония, образовавшийся в результате гетеролиза, быстро
соединяется с кислым сульфат-ионом. Наблюдаемый эффект различ-
различного уменьшения скорости при добавлении Н3О и HSOJ объясняется
действием растворителя, самоднссоциация которого
+H3O+ A77)
подавляется более сильно водой, чем кислым сульфат-ионом.
Предварительные опыты с арилалкилэфирами показали, что
в некоторых случаях механизм реакции отличается от ранее описан-
описанного 144 тем, что гетеролизу подвергается протонизированный эфир.
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
185
Однако принимая во внимание то, что в растворах фторсульфоновой
кислоты при низких температурах анизол присоединяет протон
в основном к кольцевому атому углерода, а не к атому кислорода,
необходимо провести дальнейшие исследования для выяснения при-
природы и механизма взаимодействия ароматических эфиров с серной
кислотой.
Ж. Нитрилы
Методом криоскопии было найдено, что в сернокислом растворе
нитрилы полностью ионизированы 132. Недавно это было подтвер-
подтверждено измерениями электропроводности и определением констант
диссоциации ацетонитрила и бензонитрила. Константы диссоциации
равны соответственно 0,16 и 0,07 171. Однако растворы этих нитрилов
неустойчивы и подвергаются сольволизу до соответствующих амидов:
RCN + 2H2SO4 =
H2S2O7
A78)
Скорости реакций определялись по изменению электропроводности
растворов 171. Было найдено, что для ацетонитрпла и бензонитрила
реакция сольволиза имеет второй порядок, а стадией, определяющей
скорость реакции, является взаимодействие ионов HSOJ с протонн-
зированным нитрилом:
+ HSOJ=RCONH2 + S03 A79)
3. Карбоновые кислоты
Большинство алифатических и множество ароматических карбоно-
вых кислот ведут себя как полностью ионизированные, по уравне-
уравнению (9), простые основания 91i131!199. Электроноакцепторные заме-
заместители могут уменьшать основность, в результате чего ионизация
будет неполной. Таким образом, в сернокислом растворе уксусная
кислота является сильным основанием, дихлоруксусная кислота —
слабым основанием, а трихлоруксусная кислота — неэлектролитом111.
Некоторые замещенные бензойной кислоты, например мезитилен-
карбоновая кислота, подвергаются сложной ионизации с выделением
воды и образованием устойчивого ацил-пона 227:
A80)
Для сернокислого раствора мезитиленкарбоновой кислоты 227
{Р. Дж. Джиллеспи и Е. А. Робинсон, неопубликованные результаты)
v = 4 и у = 2; при разбавлении этого раствора водой образуется
кислота, при разбавлении метанолом — метиловый эфир. Другие
кислоты, например 2,3,5,6-тетраметилбензойная и пентаметилбен-
зойная, ведут себя аналогично 193. Для растворов 2,6-диметилбензой-
ной и 3,5-дибром-2,4,6-триметилбензойной кислот значения v равны
соответственно 3,5 и 2,4. Считают, что кислоты ионизируются
186
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
частично по описанному выше «сложному» способу и частично как про-
простые основания. Довольно сходный тип ионизации наблюдается
у некоторых ароматических кетокислот. Так, для раствора о-бензоил-
бензойной кислоты v = 4, что указывает на следующее протекание
ионизации
196.
О
СО2Н
О—
например, муравьиная кислота
+ H3O+ + 2HSOj A81)
Так же ведут себя о-мезитоилбензойная, нафталевая и фенан-
трон-4,5-дикарбоновая кислоты 196.
Многие карбоновые кислоты неустойчивы в серной кислоте и
разлагаются с образованием окиси углерода по реакции
A82)
A83)
A84)
A85)
A86)
щавелевая кислота
(СО2НJ + H2SO4 = CO +СО2 + Н3О + + HSOj
сс-кетокислоты
PhCOCO2
а-оксикислоты
СН2СО2Н
= PhCO2HJ
СН2СО2Н
+ 2H2SO4 = [ +H3O++CO+2HSO7
СН(ОН)СО2Н СН(ОН)+
СН2СО2Н
сно
а также трифенилуксусная, дифенилуксусная и бис-(/г-хлорфенил)-
уксусная кислоты 91i235. Дцциклогексилуксусная и 2,4,6-триметил-
циклогексанкарбоновая кислоты также разлагаются с выделением
окиси углерода (остальные продукты реакции не были идентифици-
идентифицированы) 233.
Была изучена кинетика разложения муравьиной 65, щавеле-
щавелевой «8,170,23^ лимонной 238, бензоилмуравьиной 62, трифенилуксус-
ной 66 и яблочной 236 кислот. Во всех случаях реакции имели первый
порядок по органическому соединению, причем скорость реакции
уменьшалась с увеличением содержания воды в растворителе. .
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
187
Лайлер 17° изучила разложение щавелевой киелоты методом изме-
измерения электропроводности. Была определена степень ионизации
щавелевой кислоты. Полученные результаты хорошо согласуются
с результатами ранее проведенных криоскопических измерений 199>239,
которые показали, что щавелевая кислота протонизирована только
на 30%. Скорости разложения, найденные Лайлер, близки по зна-
значениям к скоростям, которые были определены ранее путем титрова-
титрования раствора перманганатом 168. Было обнаружено, что скорость раз-
разложения больше зависит от концентрации дважды протонизирован-
ной, чем монопротонизированной щавелевой кислоты, и увели-
увеличивается пропорционально концентрации. По-видимому, реакция
происходит путем мономолекулярного разложения дипротонизиро-
ванных частиц, присутствующих в растворе при очень низких кон-
концентрациях щавелевой кислоты 17°. К этому заключению еще раньше
пришел Гаммет 128, основываясь на результатах своих наблюдений,
согласно которым тангенс угла наклона графика зависимости лога-
логарифма константы скорости от Но приблизительно равен 2.
И. Ионы карбония
1. Сопряженные кислоты ароматических углеводородов
и положительные ионы
Ионы карбония образуются прп протонизации и окислении
ароматических углеводородов в серной кислоте, хотя при этом
часто протекают и другие реакции, например сульфирование и
деалкилирование. Так, мезитилен подвергается сульфированию 51,
а гексаметилбензол 166 — одновременно сульфированию и деметили-
рованию; оба они образуют один и тот же продукт — мезитилен-
трисульфоновую кислоту. Однако антрацен и 3,4-бензпирен образуют
устойчивые сопряженные кислоты XXXIV и XXXV 122;
XXXV
Наиболее глубокие исследования протонпзации ароматических углеводо-
углеводородов были выполнены с другими кислыми растворителями, такими, как HF
и HSOgF, т. е. в условиях, когда можно получить растворы более устойчивые,
чем сернокислые 15,43,173,174,179,212.
Циклооктатетраен растворяется в 98%-ной H.2SO4 с образованием
бицикло[5, 1,0]октадиенил-катиона 228. По данным ЯМР-спектро-
188
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
скошга, этот катион имеет структуру XXXV, так как сигналы от
Н,_6 наблюдаются в области спектра, характерной для водорода
ароматического ядра, сигналы от Нх и Н7 получаются в аномально
низком поле, а сигнал Н8а сильно ослаблен действием кругового
тока; очевидно, данный атом расположен вне кольца.
Исследования электронного парамагнитного резонанса показали,
что катион-радикалы образуются в разбавленных сернокислых
растворах при окислении таких углеводородов, как антрацен,
нафтацен и перилен 32>233. Персульфат в присутствии серной кислоты
окисляет 9-метнл- и 9,10-диметилантрацен до соответствующих катио-
катионов. В этих же условиях ксилол окисляется до 2,5-диметилбензо-
семихинона 16-28. Последний радикал может быть получен также путем
восстановления сернокислого раствора хинона дитионатом натрия 16.
Несовпадение результатов этих данных с данными спектроскопии,
которые указывают на то, что антрацен только протонизируется, не
совсем понятно. Возможно, что основным продуктом реакции является
сопряженная кислота, и только незначительное количество исходного
вещества превращается в катион-радикал. Действительно, удалось
показать, что способность углеводорода к образованию в сернокислом
растворе положительного иона зависит от легкости протонизации 151.
Было сделано предположение, что при этом протекают следующие
реакции:
A -f- H2SO4 = AII+ + HSO j
A87)
AH +2H2SO4 = А+ +2Н2О +S02 + IISOj"
Однако такой механизм реагирования пока не подтвержден.
2. Ионы триарилкарбония
Известно, что при растворении трифенилметанола в серной
кислоте происходит ионизация:
РЬзСОН + 2H2SO4 = Ph3C+ + H3O
A88)
При этом образуется устойчивый раствор желтого цвета. Такой
характер ионизации подтверждает спектр поглощения раствора, ко-
который очень похож на спектр проводящего ток раствора трифенил-
метилхлорида в жидкой двуокиси серы 1го>1зз^ величина v = 4 132i203
и реакция сернокислого раствора трифенилметанола со спиртами,
в результате чего образуются простые эфиры:
Было показано, что ионизация три-/г-фенил-, три-/г-хлор- и три-/г-
нитропроцзводных трифенилметанола протекает таким же обра-
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ|ВЕЩЕСТВ
189
зом 194. По-видимому, в этих случаях строение иона карбония лучше
всего представить хиноноидной структурой XXXVI:
(СН3J HN—
С=<
XXXVI
=N (CH3J
3. Ионы диарилкарбония
Устойчивые диарилкарбониевые ионы могут быть получены рас-
растворением соответствующего олефина или спирта в серной кислоте.
Например:
Ph2C=CH2 + H2S O4 = Ph2CCH3+H S OJ A90)
A91)
Ph2c( +H2SO4=Ph2CCH3 + H3O+-f2HSO4
ХСН3
Так, 1,1-дифенилэтилен дает в начальный момент двукратное,
а дифенилметилкарбинол — четырехкратное понижение точки
замерзания серной кислоты 122.
Эти катионы не совсем устойчивы, и точки замерзания растворов
со временем понижаются. УФ-спектры сернокислых растворов-
1,1-дифенилэтилена, трифенилэтилена и антрацена очень похожи
и имеют пики при 2050—3150 и 4250—4350 А. Это сходство легко
понять, предположив, что при протонизации всех трех названных
веществ образуются следующие устойчивые ионы:
С—СН3
C-CH2Ph
сн2 сн
=/
Для растворов ди-/г-хлорфенилкарбинола и ди-/г-хлорфенил-
метилкарбинола, а также димезитилкарбинола v = 4, следовательно,
образуются соответствующие карбониевые ионы. Раствор димезитил-
димезитилкарбинола частично разлагается, вероятно, происходит сульфиро-
сульфирование 194. Для раствора дифенилкарбинола в начальный момент v
несколько больше 4, что также свидетельствует об образовании иона
карбония. Однако дифенилкарбинол быстро реагирует с растворите-
растворителем, который, вероятно, сульфирует его 234. Несколько более устой-
устойчивые растворы могут быть приготовлены путем растворения бенз-
гидрола в четыреххлористом углероде с последующей экстракцией
190
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
серной кислотой. Полученные растворы при выливании в воду,
метанол и ледяную уксусную кислоту дают соответственно дибенз-
гидриловый эфир, метилбензгидриловый эфир и бензгидрил-
ацетат
234
4. Моноарилкарбониевые ионы
Бензиловый спирт, как это впервые показал Канниццаро 31,
в серной кислоте образует нерастворимый полимер розового цвета;
аналогично га-хлорбензиловый спирт образует полимер красного
цвета. У растворов гептаметилбензилового и а,а,2,4,6-пентаметил-
бензилового спиртов значения v быстро изменяются со временем;
экстраполяцией удалось установить, что в начальный момент времени
v = 4 194. Разбавление водой сернокислого раствора а,а,2,4,6-пента-
метилбензилового спирта приводит к образованию 2-мезитилпропи-
лена, количество которого в растворе со временем уменьшается. По-
видимому, происходит сульфирование, в результате которого обра-
образуются растворимые в воде продукты. Так можно получить ион
мезитилдиметилкарбония, который, кроме того, образуется в резуль-
результате растворения а,2,4,6-тетраметилстирола в серной кислоте. Зна-
Значение v в начальный момент реакции, полученное экстраполяцией,
приблизительно равно 2.
Криоскопические исследования показывают, что все моноарил-
моноарилкарбониевые ионы и большинство диарилкарбониевых ионов в серно-
сернокислом растворе неустойчивы, так как подвергаются сульфированию
или полимеризации. Однако некоторые исследователи сообщают,
что можно приготовить растворы большого числа диарил- и моно-
арилкарбониевых ионов, но для этого необходимо поддерживать
минимальные концентрации всех других ионов, с которыми ион
карбония может реагировать. По-видимому, лучший способ для
этого — разбавить уксуснокислый раствор соответствующего спирта
избытком серной кислоты или же большим избытком серной кислоты
экстрагировать раствор циклогексана 123>242. К сожалению, получен-
полученные таким путем растворы настолько разбавлены, что их невозможно
использовать для криоскопических измерений и измерений электро-
электропроводности. Единственным доказательством существования ионов
.карбония в этих растворах является наличие в видимом участке
УФ-спектра полосы поглощения с длиной волны около 400 ммк.
Грейс и Саймоне 123 получили довольно высокие значения коэффи-
коэффициентов экстинкции порядка 104. Вильяме 242 нашел, что коэффи-
коэффициенты экстинкцпи зависят от концентрации и определил их значения,
которые гораздо меньше соответствующих величин, приведенных
Грейсом и Саймонсом для большинства ионов карбония.
Дено и сотр. 61>62 пытались получить некоторые моноарилкарбо-
моноарилкарбониевые ионы в водной серной кислоте. Они наблюдали характери-
характеристическую полосу поглощения при 400 ммк, однако все растворы
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
191
оказались неустойчивыми, и полоса поглощения быстро исчезала.
Было обнаружено, что пентаметилбензиловый спирт разлагается на
гексаметилбензол и пентаметилбензальдегид.
5. Положительные двухзарядные ионы карбония
Криоскопические исследования сернокислых растворов тетра-
фенил-/г-ксилолгликоля и тетрафенилфталеина показывают, что при
ионизации этих соединений образуются положительные двухзаряд-
двухзарядные ионы карбония 137:
Ph Ph Ph Ph
\/ \/
,/C-OH C+
I I +4H2SO4=| I!
/\ / \
Ph Ph Ph Ph
Ph Ph Ph Ph
+ 3H2SO4=|
A92)
A93)
Ph Ph
При разбавлении этих сернокислых растворов водой из них выделяет-
выделяется исходное соединение. Спектры поглощения растворов тетрафенил-о-
ксилолгликоля и соответствующего лета-соединения очень похожи
на спектры пара-соединения. При разбавлении раствора мета-
соединения образуется гликоль, а opmo-соединения — тетрафенил-
фталеин. Отсюда следует сделать вывод, что эти соединения также
образуют положительные двухзарядные ионы карбония.
Харт и Фиш 13i~13a сообщают, что несколько необычный положи-
положительный двухзарядный ион карбония получается при растворении
трихлорметилполиметилбензолов в серной кислоте. Например, для
раствора трихлорметилмезитилена было получено значение v = 5,
которое наряду с другими признаками позволило авторам предста-
представить ионизацию соединения следующим уравнением:
(СН3)зСвНгСС1з+ 2H2SO4=H3C—
Однако при такой интерпретации не учитывается, что НС1 реагирует
с серной кислотой, образуя хлорсульфоновую кислоту по уравне-
уравнению A48).
192
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
Повторные исследования 103>104 показали, что в действительности
образуются положительные однозарядные, а не двухзарядные ионы:
.СНз
A95)
ХСН3
+ НС1- SO3+H3O++2HSOi-
6. Алифатические ионы карбония
Аллильный катион XXXVII образуется при введении смеси
циклодиенов XXXVIIIa и XXXVIII6 в концентрированную серную
кислоту.
XXXVIIIa
ХХХУШб
Об зтом свидетельствуют двукратное понижение точки за-
замерзания полученного раствора, а также ЯМР-спектр, который
имеет четыре линии с химическим сдвигом протонных сигналов отно-
относительно сигнала тетраметилсилана: 1,98; 6,67; 6,93 и 8,79 м. д., —
площади которых относятся как 1 : 4 : 6 : 6 63.
Для сернокислого раствора трициклопропилкарбинола значение v
в начальный момент равно 4; при прибавлении льда в раствор реге-
регенерируется спирт с 63%-ным выходом 64. Предположили, что полу-
полученные результаты свидетельствуют об образовании иона трицикло-
пропилкарбония, но тогда непонятно, почему ЯМР-спектр состоит
только из одной линии. Максимальное поглощение ультрафиолетовых
лучей раствором наблюдается 64 при длине волны 270 ммк.
Бутен-1, бутен-2, пентен-1 и пентен-2 растворяются в концентри-
концентрированной О 75%-ной) серной кислоте. При разбавлении раствора
водой получаются вторичные кислые сульфаты 197. Изобутилен,
триметилэтилен я 2-метилбутен-1 легко растворяются даже в 60%-ной
кислоте, причем разбавление раствора водой приводит к образованию
третичных спиртов. Последние могли получиться вследствие быстрого
гидролиза третичного кислого сульфата, который образуется в на-
начальный момент. Вероятно, при растворении олефинов в 100%-ной
кислоте в начальный момент образуются алкилгидросульфаты.
Во всех этих реакциях, за исключением реакции с этиленом,
в результате которой образуется устойчивый этилгидросульфат,
происходит процесс окисления и полимеризация. Полимеризация,
IV. РАСТВОРЫ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
193
а также алкилирование парафинов и ароматических углеводородов
олефинами в присутствии серной кислоты наводит на мысль, что
яри ионизации алкилгидросульфатов образуется алкилкарбониевый
ион, однако степень ионизации, по-видимому, невелика.
Для сернокислого раствора этанола v = 3 и не изменяется со
временем, ау = 1, что указывает на образование этилгидросуль-
<?ата 83,iii,202_ К-Пропиловый и другие первичные спирты с нераз-
ветвленной цепью образуют растворы, имеющие в начальный момент
бледно-желтую окраску и показывающие трехкратное понижение
точки замерзания, что также свидетельствует об образовании алкил-
гидросульфата 132. Однако затем v заметно увеличивается, цвет
раствора становится темно-красным или коричневым, выделяется
сернистый газ и отделяется слой бесцветного углеводорода. Для
растворов некоторых спиртов с разветвленной цепью в начальный
момент значение v = 2-f-3,Hoco временем оно быстро увеличивается.
Очевидно, при этом протекают те же реакции, что и у большинства
спиртов с неразветвленной цепью. Третичные спирты дают в началь-
начальный момент значения v *=» 2 132,195,202 раньше эт0 объясняли образо-
образованием иона оксония. Однако последняя работа Лайстена 1в4 пока-
показала, что в системе H2SO4 — HS2O7 — HSO4 начальное понижение
точки замерзания очень мало, и подтвердила предположение, появив-
появившееся после изучения растворов в метилсульфоновой кислоте41,
о том, что трет-бутаяол немедленно дегидратируется и полимери-
зуется:
Розенбом и Саймоне 216 сообщают, что устойчивые растворы ионов
третичного алкилкарбония можно получить, приготовив очень
разбавленные растворы олефинов или спиртов по методу, который
раньше применялся для арилолефинов и спиртов. Существование
ионов алкилкарбонила доказывают тем, что в спектре растворов
спиртов и алкилгалогенидов в H2SO4 имеется полоса поглощения
при 293 ммк, интенсивность которой нарастает в течение 7 дней;
такая же усиливающаяся полоса имеется в спектре растворов боль-
большинства олефинов. Авторы приписывают ее иону алкилкарбония,
так как не удалось заметить образования сколько-нибудь заметных
количеств сернистого газа или других продуктов окисления. Дено
и др. 51 сообщили о получении алкенил-катионов.
7. Ионы поливное
Высшие полиены растворяются в серной кислоте, образуя окра-
окрашенные растворы, причем интенсивность окраски возрастает по мере
увеличения длины цепи164. Недавно сообщалось, что 1,6-дифенил-
/ексатриен немедленно окисляется 100%-ной серной кислотой-до
13 Заказ 90
194
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛ)ТV
соответствующего положительного двухзарядного иона 49. Однако
криоскопические исследования Лайстена и Уолтона1вв показали,,
что на каждый моль гексатриена образуются два моля воды, а серни-
сернистый газ не является главным продуктом реакции. Авторы пришли
к выводу, что образовавшийся продукт представляет собой протони-
зированную гексатриендисульфоновую кислоту:
C6H5CH=CH-CH=CH-CH=CHCeH5 + H2SO4
A97>
Место протонизации еще не установлено.
ЛИТЕРАТУРА
195
«редах. Наиболее вероятно, что реакция сульфирования протекает
зю следующему механизму:
/Н
ArH+SO3
Аг'
Аг
.Н
A98)
К. Сульфирование ароматических соединений
Ингольд из составил обзор работ по сульфированию ароматиче-
ароматических соединений, опубликованных до 1953 г. Он пришел к выводу,
что кинетика реакции в слабоводной серной кислоте в большей мере
свидетельствует об активной роли молекул S03, чем ионов H3SO4,
HSO3, так как скорость сульфирования пропорциональна концен-
концентрации ароматического соединения и обратно пропорциональна
квадрату концентрации воды. Брэнд и сотр. 19-22>24 изучили сульфи-
сульфирование ароматических соединений серной кислотой в широком интер-
интервале составов растворителя от водного раствора кислоты до олеума.
Они показали, что для ряда веществ константа скорости реакции
первого порядка почти линейно зависит от состава растворителя,
выраженного в виде —Н0 -f- lg aSOi-
Вероятность того, что сульфирующими агентами являются ионы
HSOj, мала, поскольку скорость сульфирования в дейтерированном
растворителе ниже, а содержание DSO3 в D2SO4, по данным спектро'-
фотометрических исследований, выше, чем HSOJ в H2SO4. Однако
на основании измерений электропроводности было установлено,
что степень ионизации ж-нитротолуола в D2SO4 меньше, чем
bH2SO473, и недавно установлено, что D2SO4, вероятно, является
более слабой кислотой и более слабым основанием, чем H2SO4 127.
Все это означает, что аргумент Брэнда и сотр. несостоятелен.
Тем не менее их заключение о том, что ион HSO3 не
является сульфирующим агентом, по-видимому, правильно, так
как S03 — очень слабое основание. Например, раствор S03
во фторсульфоновой кислоте, которая является значительно более
сильной кислотой, чем серная, не проводит ток в. Таким образом,
концентрация протонизированных ионов S03 в слабоводной серной
кислоте или в низкопроцентном олеуме чрезвычайно мала, и вряд
ли возможно, чтобы этот ион был способен сульфировать в таких
10.
11.
12.
13.
14.
15.
16.
17.
: 18.
19.
20.
21.
22.
3.
24.
25.
26.
27.
28.
29.
30.
' 1
31.
32.
33.
34.
35.
36.
37.
ЛИТЕРАТУРА
Adie R. H., J. Chem. Soc, 57, 450 A890).
Anderson H. H., J. Am. Chem. Soc, 72, 194 A950).
А г о t s k у J., M i s h r a H. C. et al., J. Chem. Soc, 1962, 2582.
Baker W., F i e 1 d F.
Barr J., Gillespie
Barr J., Gillespie
Barraclough C. G.,
Bass S. J., Gillespie
Gillespie
Gillespie
Flowers et
В., J. Chem. Soc, 1932, 86.
R. J. et al., Can. J. Chem., 39, 1266 A961).
R. J. et al., Inorg. Chem., 3, 1142 A964).
To be M. L., J. Chem. Soc, 1961, 1993.
R. J., J. Chem. Soc, I960, 814.
R. J. et al., J. Chem. Soc. 1960, 837.
R. J. et al., J. Chem. Soc, 1960, 821.
J. Chem. Soc, 1960, 4315.
al.
Bass S. J.,
Bass S. J.,
В a ss S. J.,
Berger A.,Lowenstein A. et al., J. Am. Chem. Soc, 81, 62 A959).
Bergius F., Z. phys. Chem., 72, 338 A910).
В ire hall Т., Gillespie R. J., Can. J. Chem., 41, 2642 A963).
Gillespie R. J., Can. J. Chem., 42, 502 A964).
Carrington A., Proc. Chem. Soc, 1961, 385.
., J. Am. Chem. Soc, 77, 1575 A955).
Soc, 1946, 585.
J. Chem. Soc, 1950, 997.
J. Chem. Soc, 1950, 1004.
Horning W. C, J. Chem. Soc, 1952, 3922.
Horning W. С et al., J. Chem. Soc, 1952, 1374.
James J. С et al., J. Chem. Soc, 1953, 2447.
J a r v i e A. W. P. et al., J. Chem. Soc, 1959, 3844.
Chem. Soc, 55, 382 A889).
J.
J.
J.
J.
J.
J.
Т.,
. R.,
A., Hill M. E
С D., J. Chem.
С D.
С D.,
С D.
С D..
С D..
С D.,
В., J.
Birch all
В olt о n J
Bradley
Brand J
Brand
Brand
Brand
Brand
Brand
Brand
В r a u n e г
В г a u n e r
Brayford J. R., W у a 11 P. A. H., J.Chem. Soc. 1955, 2453.
Brivati J. A., Hulme R. et al., Proc. Chem. Soc, 1961, 384.
В ru baker С. Н., J. Am. Chem. Soc, 77, 3265 A955).
В u n t о n C. A., F i g g i s B. N. et al., Advances in Molecular Spectros-
copy», v. I Oxford, 1962, p. 209.
Cannizzaro S., Ann., 92, 113 A854).
Carrington A., Dravieko F. et al., J. Chem. Soc, 1959, 947.
Chedin J., С. г., 292, 220 A936).
С he din J., Ann. chim., 8, 243 A937).
Chretien P., Ann. chim. phys., 15, 367 A895).
Chretien P., С r. 123, 814 A896).
Clark H. C, O'Brien R. J. Inorg. Chem., 2, 1020 A963).
13*
BL, Z. anorg. Chemu 7, 11 A894)._
196
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
ЛИТЕРАТУРА
197
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85:
86.
Clark Н. С, O'Brien R. J., Tr oiler J., Proc. Chem. Soc,
1963, 85.
Coates G. E., Organometallic Compounds New York, 1960, p. 184.
Couture A. M., Laidler K. J., Can. J. Chem., 34, 1209 A956).
Craig R. A., G a r r e t t A. B. et al., J. Am. Chem. Soc, 72, 163 A950).
D а с г е В., W у a t t P. A. H., J. Chem. Soc, 1961, 568.
Dallinga G., Mack о г E. L. et al., Mol. Phys., 1, 123 A958).
d'Arcy R. H., J. Chem. Soc, 55, 159 A889).
D a s e n t W. E., W a d d i n g t о п Т. С, J. Chem. Soc, 1960, 3350.
Davison A., Mcfarlane W., et al., J. Chem. Soc, 1962, 3653.
D a v i s о n A., M с F al a n e W. et al., J. Chem. Soc, 1962, 4821.
Deans F. В., E a b о r n C, J. Chem. Soc, 1959, 2299, 2303.
de Boer
D e n о N.
D о п о N.
D e n о N.
D о и о N.
D e n о N. С.
de Right
, v a n
, T af t
, G г о v e s
, G г о v e s
, R i с h e у
, R i с h e g
der Meij P. H., J. Chem. Soc, 1962, 139.
R. W., J. Am. Chem. Soc, 76, 244 A954).
P. T. et al., J. Am. Chem. Soc, 81, 5790 A959).
Am. Chem. Soc, 82, 4719 (I960)..
P. T. et al., J
H. G. et al., J
H. G. et. al, J
Am. Chem. Soc,
Am. Chem. Soc,
55, 4761 A933).
1498 A962).
84, 2016 A962).
R. E., J. Am. Chem. Soc, __, -.-- x-_
Dittmar H. R, J. Phys. Chem., 33, 533 A929).
D о 1 e z a 1 e к Г., Finkh, Z. anorg. Chem., 50, 82 A960).
Dolezalek F., Finkh K., Z. anorg. Chem., 51, 320 A960).
D г и с e J. G. F., Chem. Eng. News., 128, 33; С. А. 18, 976 A924).
Edwards J. О., М о r r i s о n G. С et al., J. Am. Chem. Soc
166 A955).
E 1 b s K., Fischer F., Z. Elektrochem., 7, 343 A901).
Elliott W.W.,Hammick D. L * ~"
E s с h W., Chem. Ztg., 27, 297 A903).
R. V. G., Gibson С S., J.
F., Sclimid M., Z. anorg
L. A., Hammett
77,
Evans
F i с h t e r
F 1 e x s e r
A938).
F 1 e x s e г
2103 A935).
Flowers
Flowers
Flowers
J. Chem. Soc, 1951, 3402.
Chem. Soc, 1941, 109.
Chem., 98, 141 A916).
L. A., Hammett
L. P., J. Am. Chem. Soc, 60, 885,
L. P. et al., J. Am. Chem. Soc, 57,
9, 155 A959).
Flowers
Flowers
A960).
Flowers
Flowers
A963).
Flowers
Flowers
Gable С
G i a u q u e
62 A952).
G i g u e r e
G i g u e r e
Gilbert _
G i 11 e s p i e
G i 11 e s p i e
Gillespie
G i 1 1 e s p i e
Gillespie
R.
R.
R.
. et al., J. Chem. Soc, 1956, 607.
et al., J. Chem. Soc, 1956, 1925.
et al., J. Inorg. a. Nucl. Chem..,.
et al., J. Chem. Soc, 1960, 845.
, et al., Can. J. Chem., 58 , 1363
et al., J. Chem. Soc, 1958, 667.
. et al., Can. J. Chem., 41, 2464
et al., J. Chem. Soc, 1956, 607.
, . et al., J. Chem. Soc, 1960, 4327.
M., В e t z H. F. et al., J. Am. Chem. Soc, 72, 1445 A950).
W. F., Horning E. E. et al., J. Am. Chem. "Soc, 74,
H.
H.
H.
H.
H.
H.
H.
H.
H,
, Gillespie
, G i 11 e s p i e
, Gillespie
, Gillespie
Gillespie
,Gillespie
, Gillespie
Gillespie
Gillespie
R.
R.
R.
R.
R.
R.
R.
R.
R.
P. A., S a v о i e R., Can. J. Chem., 38, 2467 A960).
P. A., Savoie R., J. Am. Chem. Soc, 85, 287 A963).
L. F., Buckley H. et al., J. Chem. Soc, 1922, 1934.
R. J., J. Chem. Soc, 1950, 2493.
R. J., J. Chem. Soc, 1950, 2516.
R. J., J. Chem. Soc, 1950, 2542.
R. J., J. Chem. Soc, 1950, 2997.
R. J., Rev. Pure a. Appl. Chem. (Australia), 9, 1 A959).
Graham J.
L e i s t e n J.
R. J., Mi 11 en D. J., Quart. Rev. (London), 2, 277
87. G i 11 e s p i e R. J.,
88. G i 11 e s p i e R. J.,
89. G i 11 e s p i e R. J.,
90. G i 1 1 e s p i e R. J.,
91. G i 11 e s p i e R. J.,
A954). -
92. Gillespie
A948).
93. G i 11 e s p i e R. J.,
94. G i 1 1 e s p i e R. J.,
95. Gillespie R. J.,
96. Gillespie R. J.,
System, in «Advances in
New York, 1959, p. 385.
Gillespie R. J., Robinson
Gillespie R. J.
Gillespie R. J.
Gillespie R. J
R. J
R. J
J. Chem. Soc, 1960, 2516.
В ire hall Т., Can. J. Chem.,
Cole R. H., Trans Farad. Soc
41, 148 A963).
52, 1325 A956).
J. Chem. Soc, 1950, 2532.
A., Quart. Rev. (London), 8, 40
Oubridge
Robinson
Robinson
Robinson
J. V., J. Chem. Soc, 1956, 80.
E. A., J. Chem. Soc, 1957, 4233.
E. A., Proc. Chem. Soc, 1957, 145.
E. A., The Sulfuric Acid Solvent
Inorganic Chemistry and Radiochemistry», v. I
97.
98.
99.
100.
101.
102.
103.
104.
105.
Gillespie
Gillespie
A963).
Gillespie
A964).
Gillespie
2428 A965).
Gillespie
Robinson
Robinson
.Robinson
.Robinson
E. A.,
E. A.,
E. A.,
E. A.
Can. J,
Can. J.
Can. J
Can. J.
E. A., Can. J
Robinson E. A., Can
R. J., Robinson E. A., Can. J
R. J., Robinson E. A., J. Am.
Chem., 40, 644 A962).
Chem., 40, 658 A962).
Chem., 40, 784 A962).
Chem., 40, 1009A962).
Chem., 41, 450 A963).
J. Chem., 41, 2074
Chem., 44, 1197 A966).
R. J., К а р о о г R.
E. A., J.
, Robinson
Chem., 42, 2496
Chem. Soc, 87,
E. A., Can. J.,
106
107
108
109
110
111
R.
R.
R.
R.
R.
R.
R.
3,
3,
Senior J. В., Inorg. Chem.,
Senior J. В., Inorg. Chem.,
Solomons C, J. Chem. Soc,
Was if S., J. Chem. Soc, 1953, 209.
J. Chem. Soc, 1953, 215.
J. Chem. Soc, 1953, 221.
F. M., Trans Farad. Soc.
440 A964).
972 A964).
1957, 1796.
W as if S.,
>V as if S.,
White R
Gillespie
Gillespie
Gillespie
Gillespie
Gillespie
Gj 1 1 a q n i a
112. Gillespie R. J., White R. F. M., Trans Farad. Soc, 54, 1846
A958).
Gillespie R. J.
G ill es pie R. J.,
Gillespie R. J., Oubridge
1804.
Gillespie R. J., R о b i n s о n E. A. et al.
4320.
Gillespie R. J., Graham J. et al., J. Chem.
Gluekauf E., Trans. Farad. Soc, 51, 1235 A955).
Goddard D. R., Hughes E. D. et al., J. Chem. Soc, 1950, 2559.
Gold V., H awes B. M. W., J. Chem. Soc, 1951, 2102.
Gold V., T у e F. L., J. Chem. Soc. 1950, 2932.
Gold V., Туе F. L., J. Chem. Soc, 1952, 2172.
Grace J. A., Symons M. С R., J. Chem. Soc, 1959, 958.
Graham J., Ph. D. Thesis, London, 1943.
Greenwood N. N.. Thompson A., J. Chem. Soc, 1959, 3474.
Gurney R. W., Ionic Processes in Solution, London, 1953.
Robinson E. A., Can. J. Chem., 42, 1113 A964).
L. P., Physical Organic Chemistry New York, 1940, p. 248.
L. P., D e у r u p A. J., J. Am. Chem. Soc, 54, 2721 A932).
L. P., Deyrup A. J., J. Am. Chem. Soc, 55, 1900
113.
114.
115.
116.
117.
118.
119.
120.
121.
122.
123.
124,
125.
126.
127.
128.
129.
130.
White R. F. M., Can. J. Chem., 38, 1371 A960).
Hughes E. D. et al., J. Chem. Soc, 1950, 2473.
" " J. V. et al., J. Chem. Soc, 1956,
J. Chem. Soc, 1960,
Soc, 1950, 2504.
131.
Hall S^ K.,
Hammett
Hammett
Hammett
A933).
Hantzsch
A., Z. phys. Chem., 61, 2 7 A908).
198
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
ЛИТЕРАТУРА
199
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
169.
170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
Hantzsch A., Z. phys. Chem.., 65, 41 A909).
Hantzsch A., Ber., 54B, 2573 A921).
Hart H., F i s h R. W., J. Am. Chem. Soc, 80, 5894 A958).
Hart H., Fish R. W., J. Am. Chem. Soc, 82, 5419 A960).
Hart H., Fish R. W., J. Am. Chem. Soc, 83, 4460 A961).
Hart H., Sulzberg J. et al., J. Am. Chem. Soc, 85, 1800 A963).
Hathaway B. J., Webster D. E., Proc Chem. Soc, 1963, 14.
Holstead C, L a m Ь е г t о n A. H. etal., J. Chem. Soc, 1953, 3341.
R e i 11 у С. A., J. Chem. Phys., 27, 1126 A957).
K., Structure and Mechanism in Organic Chemistry Lon-
H о о d G. С
I n g о 1 d С
don, 1953.
I n g о 1 d
I n g о 1 d
J a q u e s
Jones G., Dole M., J. Am. Chem. Soc, 51, 2590 A929).
J о r g e n s о n M. J., H a r r t e r D. R., J. Am. Chem. Soc, 85, 878
A963).
J., A dl e r J. H. et al., J. Am. Chem. Soc, 43, 979 A921).
N., Leisten J. A., Proc. Chem. Soc, 1960, 84.
B. J., W att P. A. H., Trans. Farad. Soc, 54, 483
С. К., Hughes E. D., et
С. К., Millen D. J. et al., J
C, Leisten J. A., J. Chem.
al., Nature, 171, 301 A953).
Chem. Soc. 1950, 2576.
Soc, 1961, 4963.
Kendall
Kershaw D
Kirkbride
A958).
Kirkbride
Soc, 1958, 2100.
A958).
B. J., W у a t t P. A. H., J. Chem.
К on H., Bio is M. S., J. Chem. Phys., 28, 743
Kothner, von P., Ann., 319, 1 A901)'.
Kuhn L. P., Corwin A. H., J. Am. Chem. Soc, 70, 3370 A948).
Winters tein A., Helv. chim. acta, 11, 87 A928).
E., Analyt., Chem. 25, 93 A953).
Giauque W. F., J. Am. Chem.
E.,
Kuhn L. E.
Kunzler J
Kunzler J
A952).
Kunzler J. E.,
Kunzler J. E.,
A952).
Lehmann H. A.,
Leisten J. A., J
Leisten J. A.
Leisten J. A.
Leisten J. A.
Leisten J. A.
Leisten J. A.
Leisten J. A.
Leisten J. A.
Soc, 74, 804
Giauque W. F., J. Am. Chem. Soc, 74, 3472 A952).
Giauque W. F., J. Am. Chem. Soc, 74, 5271
Ber., 21, 17 A953).
Chem. Soc, 1955, 298.
J. Chem. Soc, 1956, 1572.
J. Chem. Soc, 1960, 545.
J. Chem. Soc, 1961, 2191.
J. Chem. Educ, 41, 28 A964).
J. Chem. Soc, 1964, 3173.
Walton P. R., Proc. Chem. Soc. 1963, 60.
Walton R. R., J. Chem. Soc, 1964, 3180.
Lichty D. M., J. Phys. Chem., 11, 225 A907).
Liler M., J. Chem. Soc, 1962, 4272.
Liler M., J. Chem. Soc, 1963, 3106.
Liler M., Kosanovic Dj., J. Chem. Soc, 1958, 1084.
M а с к о г Е. L., Н о f s t r a A. et al., Trans. Farad. Soc, 54, 66 A958).
Mack or E. L., Mol. Phys., 4, 241 A961).
E. L., Disc Farad. Soc, 34, 165 A962).
J. M., J. Chem. Phys., 26, 1665 A957).
1938, 1708.
Argument C, J. Chem. SoC, 1938, 1702.
E., J. Chem. Soc, 1938, 1699.
A. P., J. Am. Chem. Soc, 73, 2013 A951).
Mellor J. \V., A Comprehensive Treatise on Inorganic and Theoretical
Chemistry, v. 9, London, 1922, p. 332.
Mellor J. W., A Comprehensive Treatise on Inorganic and Theoretical
Chemistry, v. 9, London, 1922, p. 580.
MacClean
MacClean
Marcus R.
M a s s о n I.,
M a s s о n I.,
M a s s о n I., H аЪ b у W.
M с С a u 1 e у D. A., tier
C,
C,
A.,
J.
M а с к о г
Fresco
Chem. Soc.
182.
{83.
184.
485.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
200.
201.
202.
203.
204.
205.
206.
207.
208.
209.
210.
211.
212.
213.
214.
215.
216.
217.
218.
219.
220.
221.
222.
223.
224.
225.
226.
227.
228.
Mellor J. W., A Comprehensive Treatise on Inorganic and Theoretical
Chemistry, v. 9, London, 1922, p. 698.
Mellor J. W., A Comprehensive Treatise on Inorganic and Theoretical
Chemistry, v. 11 London, 1922, p. 117.
Meyer J., L a n g e r M., Ber. 60, 285 A927).
Miles F. D., Carson Т., J. Chem. Soc, 1946, 786.
Soc, 1950, 2589.
Soc, 1950, 2600.
Soc, 1950, 2606.
S. R., J.
P. A. H.,
F u j i t a J. et al., J.
Millen D. J., J. Chem.
Millen D. J., J. Chem.
Millen D. J., J. Chem.
Mishra H. C, Symons M.
Mountford G. A., Wyatt
K.
Nakomoto
A957).
Chem. Soc, 1962, 4411.
J. Chem. Soc, 1964, 518.
Am. Chem. Soc, 79, 4904
Newman
Newman
Newman
Newman
Newman
A945).
N о r r i s J
О d d о G.
О ddo
О d d о
О dd о
О d d о
О ddo
О
О
M.
м.
м.
м.
м.
J. Am. Chem. Soc, 63, 2431 A940).
Deno N. С, J. Am. Chem. Soc,
D eno N. C, J
Craig et al.,
К u i v i 1 a H.
73, 3651 A951).
Am. Chem. SocM 73, 3644 A951).
Am. Chem. Soc, 71, 869 A949).
al., J. Am. Chem. Soc, 67, 704
G.
G.
G.
G.
G.
R. A
a w a r a
. F., J о u b e r J. M.,
Casalino A., Gazz.
Casalino A., Gazz.
Scandola
Scandola
Scandola
Scandola
Gazz.
Gazz.
Gazz.
J. Am. Chem. Soc,
, 47 (ii), 232 A917).
, 47 (ii), 200 A917).
, 38, 603 A908).
39 (i), 569 A909).
39 (ii), 1 A909).
49, 873 A927).
E.,
E.,
E.,
E., Gazz., 40 (ii), 163 A910).
R а у J. D., J. Chem. Phys., 25, 1285 A956).
R., Hathaway В. J. et al., Proc. Chem. Soc, 1963, 13.
Paddock N., Quart. Rev. (London), 18, 168 A964).
Pascard R., С r. 240, 2162 A955).
Paul M. A., Long F. A., Chem. Rev., 58, 935 A957).
Pople J. A., Schnei d er W. G. et al., High Resolution Nuclear
Magnetic Resonance New York, 1959.
Price F. P., J. Am. Chem. Soc, 70, 871 A948).
Reavill R. E., J. Chem. Soc, 1964, 519.
Reid C, J. Am. Chem. Soc, 76, 3264 A954).
Robinson E. A.,J. Am. Chem. Soc, 86, 5676 A964).
Robinson R. A., Stokes R. H., Electrolyte Solutions, London,
1948, p. 223.
Rosenbaum J., Symons M. С R., Mol. Phys., 3, 205 A960).
R oyer J. L., J. Inorg. a. Nucl. Chem., 17, 159" A961).
Sch'ilt A. A., J. Am. Chem. Soc, 85, 904 A963).
Schott G., Kibbel H. U., Z. anorg. Chem., 314, 104 A962).
Somiya Т., Proc. Imp. Acad. (Japan), 3, 76 A927).
S о m m e r L. H., Petruska E. W. et al., J. Am. Chem. Soc, 68,
156 A946).
Stavenhagen A., Z. anorg. Chem., 6, 284 A893).
Stewart R.,Yates K., J. Am. Chem. Soc, 80, 6355 A958).
Stewart R., Yates K., J. Am. Chem. Soc, 82, 4059 A960).
Symons M. С R., J. Chem. Soc, 1957, 387.
S z m a n t H. H., Devlin О. М. et al., J. Am. Chem. Soc, 73, 3059
A951).
Thompson A. Greenwood N. N.,
Treffers H. P., Hammett L. P., J
A937)
vonRosenberg J. L., Mahler J. E. et al., J. Am. Chem. Soc,
84, 2842 A962).
J. Chem. Soc, 1959,
Am. Chem. Soc, 59,
736.
1708
200
ГЛАВА ЧЕТВЕРТАЯ. СЕРНАЯ КИСЛОТА
ГЛАВА ПЯТАЯ
229.
230.
231.
232.
233.
234.
235.
236.
237.
238.
239.
240.
241.
242.
243.
244.
245.
246.
247.
248.
249.
Walrafen
Walrafen
Walrafen
Weinland
Weissman
A956).
Welch СМ.,
A950).
Welch СМ.,
Whitford E
W i с к е Von
G. E., J. Chem. Phys., 40, 2326 A964).
G. E., D о d d D. M., Trans. Farad. Soc, 57, 1286 A961).
G. E., Y о u n g T. F., Trans. Farad. Soc, 56, 1419 A960).
R. F., Kuhl H., Z. anorg. Chem., 54, 244 A907).
S. I., de Boer E. et al., J. Chem. Phys., 26, 963
Smith H. A. et al., J. Am. Chem. Soc, 72, 4748
Sm th H. A., J. Am. Chem. Soc, 75, 1412 A953).
L., J. Am. Chem. Soc, 47, 953 A925).
E., Eigen M., Z. Elektrochem., 57, 319 A953).
W i i g E. O., J. Am. Chem. Soc, 52, 4729, 4737, 4742 A930).
Wiles L A., J. Chem. Soc, 1953, 996.
Wiles L. А., В a u г h a n E. C, et al., J. Chem. Soc, 1953, 933.
Williams G., Hardy M. L., J. Chem. Soc, 1953, 2560.
\V i 1 1 i a m s J. F. A., Tetrahedron, 18, 1487 A962).
Wyatt P. A. H., J. Chem. Soc, 1953, 1175.
V у at t P. A. H., Trans. Farad. Soc, 56, 490 A960).
Wyatt P. A. H., Trans. Farad. Soc, 57, 773 A961).
Yates K., Stov ns J. В., Katritzky A. R. et. al., Can.
J. Chem., 42, 1957 A964).
Young T. F., Record of chem. Progress, 12, 81 A951).
Young T. F., The Structure of Electrolytic Solutions, Cha ter 4, New
York, 1959.
Пальм В. А., ДАН СССР, Ю8, 270 A956).
КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ *
Р. С. ДРАГО И К. Ф. ПУРСЕЛЛ
I. Введение 201.
II. Методы обнаружения координации 205
A. УФ-Спектроскопия 205
Б. ИК-Спектроскопия 206
B. ЯМР-Спектроскопия 208
Г. Криоскопия 209
Д. Электропроводность 209
III. Энергетика процесса координации 210
А. Постановка проблемы 210
Б. Оценка энергий стадий 212
IV. Поведение растворенного вещества в некоторых растворителях . . . 217
A. N.N-Диметилацетамид 217
Б. N-Метилацетамид 220
B. Диметилсульфоксид 221
Г. Нитрометан и нитробензол 223
Д. Ацетон 225
Е. Метанол и этанол 227
Ж. Пиридин 228
3. Ацетонитрил 230
И. Тетраметиленсульфон 232
V. Заключение 233
Литература 235
I. ВВЕДЕНИЕ
Образование координационных связей между молекулами рас-
растворенного вещества и растворителя играет важную роль в химии
неводных растворителей. Растворение неорганического вещества
в растворителе почти всегда сопровождается координацией, выра-
выражающейся в специфическом кислотно-основном взаимодействии по
Льюису между растворителем и растворенным веществом. При отсут-
отсутствии значительных взаимодействий, т. е. в некоординирующих
растворителях, таких, как насыщенные углеводороды, четыреххло-
ристый углерод, фторуглеводороды и т. д., растворяется очень мало
* Применяя термин «co-ordinating solvents», авторы данной главы подразу-
подразумевают, что растворение протекает с образованием координационной связи
между молекулами растворяемого вещества и растворителя. При переводе на
русский язык этого термина переводчик предлагает, как наиболее оптимальный
вариант, термин «координирующие растворители». — Прим. перев.
202
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
I. ВВЕДЕНИЕ
203
неорганических веществ. Таким образом, большинство исследований
в неорганической химии было выполнено в координирующих раство-
растворителях, и мы считаем, что соображения, изложенные в этой главе,
уместны при описании химии именно таких неводных растворителей.
В прошлом при объяснении поведения растворенного вещества
во многих растворителях координация растворителя игнорировалась,
а делался упор на его самоионизацию. Например, поведение кислот
и оснований Льюиса в растворителе РОС13 рассматривалось с уче-
учетом самоионизации этого растворителя:
Р0С13 4=Г РОС1*,+ С1~ A)
Под кислотой подразумевают вещество, способствующее увеличению
концентрации катиона POCIJ, а под основанием — растворенное
вещество, способствующее увеличению концентрации аниона С1~.
Прибавление FeCl3 к РОС13 приводит к образованию FeCl^ и, воз-
возможно, POCIJ; следовательно, FeCl3 является кислотой. Эта модель
поведения неводного растворителя основана на теории сольво-
систем 39. Для того чтобы проиллюстрировать применение этой тео-
теории и для того чтобы утвердить альтернативную модель взаимодей-
взаимодействия «растворенное вещество — растворитель», основанную на пред-
предпосылке координации растворителя, мы рассмотрим системы РОС13 —
FeCl3 и РО(ОС2Н5K - FeCl3.
УФ-Спектры поглощения разбавленных растворов FeCl3 (~10~4.м)
в РОС13 показывают, что ион тетрахлорида железа(Ш) FeClJ является
одним из основных поглощающих ионов в числе частиц, присутству-
присутствующих в растворе 4; убедительных доказательств присутствия иона
POCIJ в этом растворе нет. При более высоких концентрациях FeCl3
(около 0,1 ж), т. е. при отсутствии достаточного количества С1~-иона
из растворителя, достаточного для превращения всего количества
FeCl3 в FeCl^, предполагается существование аддукта красного
цвета— FeCl^POCIg)^. По теории сольво-систем здесь имеет место
равновесие
FeCl3+POCl3 4П^Г Cl3FeClPOCl2 ^=±
красный
B)
желтый
Изменение цвета раствора является полностью обратимым и зависит
от концентрации железа. Прибавление донора хлор-иона к красным
растворам FeCl3 изменяет их окраску в характерный желтый цвет,
обусловленный ионом FeClJ.
На кривых кондуктометрического титрования растворов FeCl3
в РОС13 растворимыми хлоридами имеются резкие перегибы при моль-
мольном отношении хлорида к FeCI3, равном 1:1. Согласно теории
сольво-систем, сокращенное ионное уравнение, описывающее процесс
титрования, можно записать следующим образом:
Полное уравнение имеет вид
D)
В противоположность теории срльво-систем, предполагающей
координацию хлора в продуктах присоединения (уравнение 2),
рентгенографическое исследование структур и изучение спектров
комбинационного рассеяния и ИК-спектров показывают, что коор-
координированным атомом в некоторых аддуктах РОС13является кислород.
Рентгенографическое исследование мо-
монокристаллов и>63 SbCl5- ОРС13 и
(TiCl4 • ОРС13J показывает, что ме-
¦ таллы октаэдрально расположены и
связаны с атомом кислорода молекулы
РОС13. Спектры комбинационного рас-
рассеяния соединений А1С13 • ОРС13 и
GaCl3 • OPCI3 также указывает на то, что
координация осуществляется через атом
кислорода 37. Об этом же свидетельст-
свидетельствуют ИК-спектры продуктов соедине-
соединения 82 РОГ3 и TiCl4, SnCl4 и TiBr4.
Сторонники теории сольво-систем 3>62
утверждают, что, хотя координация
кислорода существует в твердых аддук-
аддуктах или в условиях спектральных ис-
исследований, в растворах РОС13 наблю-
наблюдается два конкурирующих типа рав-
равновесия: координация кислорода при
высокой концентрации растворенного
вещества и ионизация растворителя, в
BOO
Рис. 43. Спектры поглощения
растворов FeCl3 в РОС13 и в
РО(ОС2Н6K при [FeCl3] =
= 10-4 м:
1 — РОС13; г — РО(ОС2НВK.
pocit+cr ^rr рось
C)
результате которой образуется хлор-ион, при низкой концентра-
концентрации растворенного вещества. Таким образом, вопрос о том, какие
типы ионов и молекул присутствуют в растворах FeCl3 и РОС13,
остается неразрешенным.
Чтобы показать, что поведение FeCI3 в РОС13 можно объяснить
без учета ионизации растворителя, т. е. без привлечения хлор-иона
растворителя, было исследовано поведение FeCl3 в растворителе,
не содержащем хлора 68. В качестве такого растворителя был выбран
триэтилфосфат ввиду его структурного сходства с РОС13. Полагают,
что триэтилфосфат несколько лучший донор, чем РОС13, но не на-
настолько, чтобы ожидать большую разницу в координирующих свой-
свойствах обоих растворителей. Сходство спектров поглощения разбав-
разбавленных растворов FeCl3 в РОС13 и в РО(ОС2Н5K видно из рис. 43.
Полосы поглощения, характерные для FeCIJ, наблюдаются в обоих
растворителях.
Применение триэтилфосфата в качестве растворителя исключает
возможность образования иона FeClJ с помощью иона хлора,
204
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
II. МЕТОДЫ ОБНАРУЖЕНИЯ КООРДИНАЦИИ
получающегося в результате самоионизации растворителя, как это
предполагалось при использовании РОС13. Образование FeCIJ
в обоих растворителях можно объяснить следующим равновесием:
205
(FeCl3OPY3) ^
E)
Эта интерпретация находится в согласии с экспериментальными дан-
данными, которые указывают на координацию кислорода в продуктах
присоединения с РОС13. ИК-Спектры РО(ОС2Н5K, координирован-
координированного соединениями железа, также показывают, что координация
молекулы растворителя осуществляется через кислородный атом
фосфорила 68. Сравнение уравнений B) и E) показывает существен-
существенную разницу между теорией сольво-систем и предложенным нами
объяснением, которое будет в дальнейшем называться «координа-
«координационная модель». В соответствии с этими двумя моделями в равновесии
участвуют различные ионы и молекулы.
Кривые кондуктометрического титрования 68 в РО(ОС2Н5J и
РОС13 еще раз указывают на сходство этих растворителей. Резкие
перегибы на кривых электропроводности были получены при моль-
мольном отношении хлор-иона к FeCI3 и SbCI3, равном 1:1. Это соответ-
соответствует образованию ионов FeCI4 и SbClJ. Согласно нашей схеме,
уравнения титрования свидетельствуют о превращении всех соеди-
соединений железа(Ш) в ион
/1FeCl2(OPY3)J-i-2/1Cr
/2FcCl(OPY3)l+ + 3/2Cr
/:!Fe(OPY3)|++4/3Cl~
/4FeCl3(OPY3)+/4Cr
> /1
> /2FeCll
/sFeClJ
/4 FeClJ
F)
где /; — мольная доля t-того иона железа от всего железа, присутствующего
в растворе.
Поскольку отношение железа к хлору в растворе FeCI3 в
РО(ОС2Н5K равно 1 : 3, для превращения всего железа в FeCIJ
должен потребоваться один грамм-эквивалент хлор-иона на моль
FeCI3, что и подтверждается спектрофотометрическими данными.
Сравните эти уравнения с уравнениями C) и D), составленными
в соответствии с теорией сольво-систем.
Таким образом, все свойства растворов хлорного железа в РОС13,
которые были использованы для подтверждения теории сольво-си-
сольво-систем, были исследованы для растворов FeCl3 в триэтилфосфате. На
основании этого показано, что ионизация растворителя не является
обязательной для того, чтобы объяснить равновесия и химические
реакции, которые происходят при растворении кислот Льюиса
в РОС13. Координационная модель, используемая при сравнении
равновесий в различных растворителях, сосредоточивает внимание
на донорно-акцепторных свойствах растворителя и на его сольвати-
рующей способности. Следовательно, поведение кислот в донорных
органических растворителях сходно с поведением этих веществ
во многих галогенкислород- и кислородсодержащих растворителях.
В этой главе будут рассматриваться главным образом органиче-
органические растворители. Однако, как показано на примере РОС13
и РО(ОС2Н5K, понятия, развитые здесь, могут быть с успехом при-
применены и для неорганических растворителей.
Основное внимание в этой главе будет уделено разработке общих
положений координационной модели и применению их для объясне-
объяснения поведения неводных растворителей. В первую очередь мы оста-
остановимся на методах установления координации и определения типов
ионов и молекул, присутствующих в растворе, и затем рассмотрим
энергетику взаимодействия между растворителем и растворенным
веществом и каким образом устанавливается состав раствора в раз-
различных растворителях. В заключение рассмотрим поведение раство-
растворенного вещества в нескольких типичных координирующих раствори-
растворителях для определения качественной связи между типами ионов
и молекул в растворе (составом раствора) и главными свойствами
растворителя. Насколько возможно, будут представлены данные,
позволяющие оценить существенные параметры растворителя.
И. МЕТОДЫ ОБНАРУЖЕНИЯ КООРДИНАЦИИ
Прежде чем обсуждать координирующее действие растворителя
необходимо найти методы, которые ее устанавливают. Эти методы
должны неизменно опираться на различие в физических свойствах
свободного донора или акцептора и координированных частиц —
продуктов их взаимодействия. Так как при координации типа ки-
кислота — основание по Льюису следует ожидать изменений в элек-
электронной структуре, можно применять различные спектральные
методы. С успехом применяются криоскопические и кондуктометри-
ческие методы.
А. УФ-Спектроскопия
Одной из самых ранних работ с применением спектроскопии
явилось изучение иода в различных растворителях. Длина волны,
соответствующая переходу я* -у а* иода, меньше в спектрах иода
в координирующих растворителях. Она равна 520 ммк в спектре
газообразного иода, 517 ммк в спектре 12 в четыреххлористом угле-
углероде, 520 ммк для иода в гептане и 500 ммк в бензоле (в бензоле
происходит координация) и еще меньше в более основных раствори-
растворителях8. В УФ-спектрах иода в донорных растворителях появляется
также полоса, характерная для переноса заряда г.
Электронные спектры ионов переходных металлов ценны не
только для обнаружения координации, но и для определения коорди-
координационного числа и структуры этих ионов в растворителях. Особенно
206
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
легко это сделать, если комплекс, существующий в растворе, можно
выделить в твердом состоянии и определить анализом его состав.
На основании электронного спектра комплекса часто можно судить-
о структуре раствора. В работе 29 рассмотрены спектры ионов пере-
переходных металлов 4 периода периодической системы элементов и раз-
различные структуры этих ионов.
Однако на примере растворов FeCI3 в некоторых растворителях
видно, что ионы и молекулы, присутствующие в растворе, и частицы,,
из которых состоят выделяемые из раствора твердые соединенияг
неодинаковы (Р. С. Драго, Р. Л. Карлсон; Р. С. Драго, К. Ф. Пур-
селл). В системе FeCl3 — ДМА (ДМА — диметилацетамид) спектр
различных хлорсодержащих ионов и молекул приходилось опре-
определять, изучая изменения спектра раствора Ре(ДМАN(С1О4Kг
происходящие при увеличении концентрации иона хлора. Типы
присутствующих в растворе частиц были определены на основании
существования изобестических точек и из спектральных изменений
в разных диапазонах концентраций.
Система СоС12 — пиридин в растворе ацетона исследовалась
Кациным и Гебертом 50~52 методом спектроскопии в видимой области.
Для получения стехиометрических комплексов СоС12— пиридин
применялся метод Джоба 43. Уолдбай 90, однако, указал, что приме-
применение этого метода требует особой тщательности, иначе он может
быть ненадежным. В рассматриваемом ниже случае удалось получить
по этому методу комплексы в ацетоне с отношением пиридина к ко-
кобальту 1:1 и 2:1. Исследование влияния пиридина на спектр
СоС1|~ в ацетоне 43 также указало на существование комплекса
1:1, отвечающего формуле CoCIJ • Ру (Ру — пиридин). Подобным
же образом Файн 33 недавно изучил образование СоГ+, СоГ2, CoFj
и СоГ|" в ацетоне (Г — CI, В г, I). Незаполненные координационные
места были заняты молекулами ацетона, например CoFjAc (Ac —
ацетон).
Другим примером приложения спектрофотометрии для изучения
координации является исследование системы [Со(асасJ]„ — Ру
в бензоле в качестве «инертной» среды (асас — ацетонилацетон) 32.
Были получены константы образования следующих соединений
[Со(асасJ]2-Ру; [Со(асасJ] • Ру и [Со(асасJ] • 2Ру.
Обсуждение исследований по применимости спектрофотометрии
для изучения образования комплексов и математическая обработка
данных приведены в работе Ньюмана и Хьюма 72.
Б. ИК-Спектроскопия
Поскольку координация влияет на распределение электронов
в молекуле, при образовании комплекса происходят изменения
в силовых постоянных различных связей и, следовательно, изменения
их колебательных частот. Таким образом, ИК-спектр растворенного
II. МЕТОДЫ ОБНАРУЖЕНИЯ КООРДИНАЦИИ
207
вещества и (или) растворителя можно использовать в качестве
критерия для обнаружения координации, но надо учесть, что отсут-
отсутствие сдвига частот не всегда свидетельствует об отсутствии коорди-
координации. Весьма важно, чтобы такие исследования проводились с рас-
растворами, ибо хорошо известно, что изменение агрегатного состояния
вещества влечет за собой изменение спектра. Например, спектр моле-
молекулы растворителя в твердом состоянии может заметно отличаться
от спектра его в жидком или газообразном состоянии. Это и не
позволяет при исследовании спектра твердого аддукта установить
наличие координации в растворе.
Кроме доказательства наличия координации, изменения в ПК-
-спектре донорной молекулы частот могут указать на то, какой атом
донора участвует в координационной связи. Например, показано,
что CH3CON(CH3J координируется через кислород с кислотами по
Льюису 12, С6Н5ОН *6.*«.™ и некоторыми ионами металлов 13>28.
Многие ионы переходных металлов, как было показано, координи-
координируют диметилсульфоксид, причем донором является атом кислоро-
кислорода 17,18,25,70. сера является донорным атомом в комплексе
Pd[(CH3JSO24+F. В CH3CON(CH3J и в (CH3JSO координация
через кислород уменьшает электронную плотность на кислороде,
понижая порядок связи и уменьшая силовую постоянную и частоту
валентных колебаний.
Следует упомянуть об осложнениях, которые иногда встречаются
при обнаружении координации методом ИК-спектроскопии. Коорди-
Координация, вместо того чтобы уменьшить, в действительности увеличивает
частоту колебаний исследуемой связи вследствие появления другого
.эффекта. Он называется кинематическим эффектом и возникает в ре-
результате образования новой связи (т. е. координационной связи).
Например, когда ацетонитрил координируется к акцептору, то ча-
частота валентного колебания С—N изменяется вследствие изменения
длины связи акцептор — азот. Этот эффект притяжения — отталки-
отталкивания действует так, что увеличивает частоту колебания С—N.
Второй эффект, увеличивающий силовую постоянную связи С—N,
возникает в результате гибридизации связей азота при комплексо-
образовании. Координация азота усиливает s-характер связи С—N
(N-гибридная орбиталь) 9. Поскольку ослабление а-электронной
плотности группы С—N во многих комплексах мало, в результате
координации к азоту нитрилов часто происходит увеличение частоты
колебаний С—N-связи. Надо подчеркнуть, что рассмотренные
выше эффекты и ослабление электронной плотности л- и а-связи
Действуют одновременно, и направление сдвига частоты определяется
суммарным эффектом. Ш
Еще одним эффектом, который, по-видимому, имеет большое зна-
значение для лигандов с низколежащими я*-орбиталями (незаполнен-
(незаполненными), является эффект обратного связывания. Если кислота имеет
электроны на я-орбиталях той же симметрии и почти той же энергии,
208
ГЛАВА ПЯТАЯ, КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
II. МЕТОДЫ ОБНАРУЖЕНИЯ КООРДИНАЦИИ
209
что и незаполненные я*-орбитали лиганда, может произойти связы-
связывание jt-орбиталей 74; это вызывает уменьшение силовой постоянной
донорной кратной связи.
В. ЯМР-Спектроскопия
Этот метод предоставляет огромные возможности для доказатель-
доказательства наличия координации. Например, если происходит образование
водородной связи, то наблюдается химический сдвиг сигнала про-
протона, вовлеченного во взаимодействие, в область низких полей.
Этот сдвиг является доказательством «координации» протона ы,83.
В очень интересном эксперименте Шнайдера и Ривса " было
показано, что в ЯМР-спектре бензольного раствора хлороформа на-
наблюдается резкий сдвиг сигнала протона СНС13 в область высоких
полей, достигающий значения 1,40 м. д. при бесконечном разбавлении.
Этот сдвиг приписали существованию взаимодействия между протоном
хлороформа и бензолом как донором. Сдвиг в область высоких полей
наводит на мысль, что протон хлороформа ориентирован приблизи-
приблизительно под прямым углом к плоскости ароматического кольца вблизи
оси вращения шестого порядка. При этом положении протонный
сдвиг в область высоких полей обусловлен магнитной анизотропией
кольцевого тока в бензоле.
Найдено, что величина постоянной связи таллий — протон
в соединении (СН3JТ1СЮ4 очень чувствительна к растворителю
(К. Д. Шир., Р. С. Драго). В тех растворителях, в которых таллий
проявляет координационное число 6, а группа СН3Т1СН3 линейна,
величины постоянный связи лежат в интервале 410—475 гц, что со-
соответствует донорным свойствам растворителя.
Обнаружено также 58-73, что координация донорных молекул
многими соединениями бора вызывает резонансный сдвиг сигнала
ПВ в спектре аддукта в область более высоких полей. Например, б
сигнала ВС13 приблизительно на 35 м. д. меньше, чем BC1J,
ВС13О(С2Н5J и BC]3C5H5N. Этот сдвиг резонансных частот ПВ был
использован для установления наличия координации в слабой кис-
кислоте Льюиса В(ОС2Н5K.
Недавно описан ЯМР-метод установления координационных
чисел ряда растворенных веществ 42. Рассмотрим принципы метода
на примере одной из изученных систем. В спектрах ядерного магнит-
магнитного резонанса 17О водных растворов (Н217О) трехвалентного алю-
алюминия имеется одиночный сигнал 17О как от координированной
воды, так и от воды, оставшейся вне координационной среды. По
мере добавления иона Со2+ наблюдается два сигнала: один — от
воды, связанной с А13+, и другой от растворителя, быстро обмени-
обменивающегося с водой, образующей координационную сферу иона
Со2+. Парамагнитный ион Со2+ вызывает большой контактный сдвиг
сигнала, относящегося ко всем молекулам воды, слабо связанным
е А13+. В результате этого появляются два отдельных пика, относя-
относящиеся к несвязанной воде и к воде, координированной с А13+. Такой
же результат был получен, когда вместо А13+ были введены Ве2+
и Ga3+, но с Mg2+, Sn2+, Ba2+, Hg2+ или Bi3+ этого не наблюдалось.
Чувствительность экспериментов была ограничена доступной сте-
степенью обогащения воды изотопом 17О, поэтому координационные
числа нельзя было точно установить.
Конник и Файет 1в, используя Н2О, обогащенную до 12% 17О,
и более чувствительную измерительную аппаратуру, повторили,
по существу, те же самые эксперименты и определили координацион-
координационные числа для А13+ и Ве2+, которые оказались равными 6 и 4 соот-
соответственно. Этот метод и аппаратура могут оказаться очень перспек-
перспективными для работы с неводными растворителями.
Свайнхарт и Таубе 87, чтобы установить координационное число
Mg2+, использовали протонный ЯМР. Применяя различные растворы
СН3ОН—Н2О (мольное отношение равно — 10 : 1) с Mg(C104J
и охлаждая их до —75° С, авторы наблюдали отдельные сигналы
для СН3ОН и Н2О, как связанных, так и свободных. Вычисления
дают число сольватации для СН3ОН, равное 5,0, и для Н2О — 0,7;
таким образом, общее координационное число для магния(П) равно
5,7. Отклонение от числа 6,0 следует отнести за счет координации
перхлората.
Г. Криоскопия
Хотя криоскопия и имеет ограниченное применение при исследо-
исследовании сложных систем, этим методом достаточно убедительно может
быть установлено наличие простых ассоциаций (типа 1:1). Это
было продемонстрировано исследованием 91 системы РОС13—МГ„
в нитробензоле при непрерывном изменении концентраций. На гра-
графике изменения 8t (отклонение точки замерзания раствора CeH5NO2
от аддитивных значений) в зависимости от содержания МГ„ (в мол. %)
был обнаружен для FeCl3 и А1С13 максимум при отношении
РОС13 : МГ„, равном 1:1. (Электропроводность растворов при
составах 1 : 1 незначительна.)
Д. Электропроводность
Измерения электропроводности были использованы де Мейном
и Коубеком в5 для установления ионизации FeCl3 при растворении
этой соли в CH3NO2 или в растворах CH3NO2 в СС14. Электропро-
Электропроводность раствора удовлетворяет уравнению Онзагера при концен-
концентрациях FeCl3 от 2,5 • 10 до 8,5 -10~2 м. При спектрофотометриче-
ском (в видимой области) исследовании растворов FeCl3—CH3NO2
авторы нашли также, что эффективный молярный коэффициент
экстинкции (оптическая плотность пропорциональна общей
14 Заказ 90
210
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
концентрации железа) является линейной функцией концентрации
железа. Линейный характер изменения электропроводности с кон-
концентрацией и спектрофотометрические соотношения соблюдаются
только тогда, когда в растворе устанавливаются равновесия, опи-
описываемые уравнениями
к,
2FeCl3 (раств) ч—- Fe2Cle (раств)
Fe2Cle (раств) ^=t FeClJ (раств)+FeClj (раств)
или
Ш. ЭНЕРГЕТИКА ПРОЦЕССА КООРДИНАЦИИ
211
2FeCl3 (раств) ч—- Fe2Cle (раств)
Fe2Cle (раств) ^=± Fe2Cl+(раств)+СГ (раств)
(8)
Необходимо, чтобы Кг была очень мала, а К2 (или К3) очень велика.
Растворенное FeCl3, очевидно, связано в CH3NO2 • FeCI3, и уравне-
уравнения G), по-видимому, являются наиболее вероятными.
III. ЭНЕРГЕТИКА ПРОЦЕССА КООРДИНАЦИИ
Первой стадией процесса координации является образование
начального аддукта между кислотой Льюиса (акцептор электронной
плотности) и основанием Льюиса (донор электронной плотности).
Вторая стадия — последующие реакции, которым может подвер-
подвергаться этот аддукт, например очень часто происходит ионизация.
Во многих случаях последующие реакции проходят до такой сте-
степени, что аддукт при равновесии присутствует в незначительных
количествах.
А. Постановка проблемы
Существенные свойства растворителя, которые определяют сте-
степень ионизации растворенного вещества, можно простейшим обра-
образом описать, рассматривая растворение гипотетического вещества
MX. Допуская, что все молекулы MX имеют по крайней мере р
присоединенных молекул растворителя S, можно записать:
MX • Sp (раств) + S (раств) ^zT М ¦ S++i (раств) + Х~ (раств) (9)
Это уравнение показывает, что координационное число элемента М
равно р + 1.
Термохимический цикл, изображенный на рис. 44, / описывает
вклады тепловых эффектов в равновесную энтальпию. Эти эффекты
определяются разницей между теплотами сольватации MX • Sp
и М • SJ+i, сольватацией X", теплотой парообразования S и раз-
разностью донорных сил Х~ и растворителя, измеряемых различием
в теплотах образования газообразных MX • Sp + S и М ¦ S +
+ х
В донорном растворителе происходит «специфическое» взаимодей-
взаимодействие, т. е. координация, с растворенным веществом (стадия в), и
«неспецифические» взаимодействия * (например, стадии а, г и д).
Как будет показано, при некоторых обстоятельствах теплоты
стадий а, г и д могут включать в себя и теплоты специфиче-
специфических взаимодействий типа образования водородной связи между
растворителем и анионом или полностью координированным ком-
комплексом.
S(r)-
MX-S (растб) + S(pacT&) *-MS* ,(раст6) + Х"(раст5)
(растб) S(pacT) MS
I
MXIr)
Х"(г)
(p
MX-S (г) +¦ S(r)
>- MS^+1(r)+ Х"(г)
Рис. 44. Циклы Борна — Габера для процесса растворения.
Иногда; более удобно разложить стадию в на стадии, изображен-
изображенные на рис. 44, //. Тепловой эффект стадии е — теплота диссоциа-
диссоциации MX • Sp на р молекул растворителя S и на газообразное со-
соединение MX; стадии ж — теплота диссоциации MX; з — потенциал
ионизации и электронное сродство М и X; и — теплота координации
М+ с (р + 1) молекулами S. Этот цикл эквивалентен стадии в (на
рис. 44, /), но предлагает другой путь определения энергии:
MX- Sp (r) + S (г) ^LT М-Sp-+1 + X~ (г) - (io)
Кроме всех перечисленных выше эффектов необходимо учиты-
учитывать три важных дополнительных положения.
1. Для того чтобы положение равновесия соответствовало сум-
суммарному изменению энтальпии всех стадий в цикле для различных
растворителей и растворенных веществ, суммарное изменение
* Термин «неспецифическая сольватация» будет применяться для описания
биполярного взаимодействия, при котором не может произойти координация,
например, биполярного взаимодействия растворителя вне первой координацион-
координационной сферы с некоторыми комплексами металлов.
14*
212
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
III. ЭНЕРГЕТИКА ПРОЦЕССА КООРДИНАЦИИ
213
(И)
энтропии в этих растворителях должно быть постоянным или должно
линейно изменяться с изменением энтальпии.
2. Когда происходит интенсивное образование ионных пар *,
в цикле должны быть учтены и новые виды образующихся ионов,
и энтальпия их образования. Степень образования ионных пар можно
оценить из диэлектрической проницаемости растворителя или,
более точно, из измерений электропроводности некислых ионных
веществ в растворителе. Энергия этого типа взаимодействия очень
значительна в растворителях с низкой диэлектрической проница-
проницаемостью и является важным фактором в объяснении того, почему
в этих растворителях происходит диссоциация растворенных веществ
на ионы.
3. При сравнении поведения различных растворенных веществ
в данном растворителе следует учитывать, что большое значение
имеет различие их координационных чисел. Энергия стадий будет
сильно зависеть от тепловых эффектов и гибридизационных изме-
,нений.
Для более сложного вещества, чем MX, например FeCl3, должно
быть рассмотрено большее число разновидностей ионов:
FeCl3-S+Cr ^I± FeClj+S
FeCl3-S+3S ^rr
FeCl2Si + S ^
FeClSf++S ^ГГ
Каждое равновесие может быть представлено циклом, сходным
с циклом для MX; виды образующихся ионов будут определяться
тем, в какую сторону сдвинуто равновесие каждой стадии. Если в сис-
системе присутствуют твердые фазы, проблема еще более усложняется
вследствие необходимости учета энергий решетки твердых фаз.
Если бы были известны величины энтальпий всех стадий цикла,
то учитывая энтропию и степень образования ионных пар, можно
было бы составить представление о факторах, действующих на поло-
положение равновесия для различных веществ, растворенных в разных
растворителях. К сожалению, для большинства систем невозможно
прямое измерение энтальпий всех стадий процесса и часто пользу-
пользуются косвенным методом определения относительных величин этих
эффектов.
Б. Оценка энергий стадий
Энергия стадии в на рис. 44 может быть качественно оценена по
способности донорного растворителя координировать с растворен-
растворенным веществом. Для этого необходимо исследовать донорные свой-
* Термин «ионные пары» применяется, чтобы обозначить катионно-анпон-
ную ассоциацию, при которой анион не находится в первой координационной
сфере катиона.
ства растворителя по отношению к ряду различных кислот и поста-
постараться установить критерии, действительно характеризующие силу
донора *. Константы устойчивости комплексов и данные рКв не
дают этих сведений, ибо константы описывают общее изменение
свободной энергии комплекса, которое может быть изображено
циклом, сходным с циклом для MX.
Надежные данные о донорных свойствах растворителя можно
получить при изучении донорно-акцепторного взаимодействия в га-
газовой фазе или в некоординирующем растворителе, например в че-
тыреххлористом углероде, гексане и других, для которых разница
в энтальпии сольватации донора плюс акцептор и аддукта невелика.
Для того чтобы понять донорные свойства растворителя, следует
также определить энтальпии для растворов различных кислот в дан-
данном растворителе. Это необходимо, чтобы вычислить энергии донор-
донорно-акцепторного взаимодействия в системах, которые нельзя изме-
измерить непосредственно.
Свойства донора значительно зависят от свойств акцептора.
Например, найдено (Р. И. Нидзиельски, Р. С. Драго и др.), что
(С2Н5JО является более сильным донором по отношению к фенолу
(энтальпия образования аддукта АД"обр = —5 ккал/молъ), чем
(C2H5JS (Д#обр = —4,6 ккал/молъ), но (C2H6JS является лучшим
донором по отношению к иоду (AHoqp = —7,8 ккал/молъ), чем
(С2Н5JО (АЯобр = —4,2 ккал/молъ). Было установлено, что анало-
аналогичное изменение донорной силы по отношению к этим двум кисло-
кислотам проявляется у растворителей CH3CON(CH3J и CH3CSN(CH3J
(Р. И. Нидзиельски и др.). По-видимому, в этих системах деформи-
деформируемый** (C2H5JS более сильно взаимодействует с деформируемым
иодом, чем полярный (С2Н5JО, а последний более сильно взаимодей-
взаимодействует с полярным фенолом. Это правило должно быть общим.
Было сделано много попыток определить величину энтальпии
донорно-акцепторного взаимодействия по смещению колебательных
частот донора или акцептора в спектре вследствие координации.
Показано, что разница частот валентных колебаний гидроксильной
группы фенола в свободном феноле и в аддукте, образованном с по-
помощью водородной связи, находится в линейной зависимости от
энтальпии образования аддукта 45>4в. Имеется и ряд других статей,
в которых отмечается, что величина смещения частоты указывает
на силу донорно-акцепторного взаимодействия. Например, есть
* Термин «сила донора» будет применяться для обозначения относительной
координационной способности донора, измеренной при отсутствии сольватации
и определяемой энтальпией образования аддукта. Термин «основность» будет
употребляться для обозначения относительного положения донорно-акцептор-
донорно-акцепторного равновесия в растворителе.
** Термин «деформируеМб"сть» обозначает легкость изменения электронной
плотности на месте связи в акцепторе и доноре до электронной плотности
в аддукте.
214
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
сообщение об уменьшении частоты валентного колебания карбо-
карбонильной группы ацетофенона при образовании комплекса со следу-
следующими кислотами85: HgCl2A8)*, ZnCl2D7), BF,A07), TiGl4A18),
A1C13A2O), FeCl3A30), AlBr3A30). Только для фенола установлено
соотношение между частотным смещением и энтальпией; в других
случаях наблюдается более сложная зависимость. Действительно,
было показано в9, что, хотя (CH3JSO и (CH2CH2JSO взаимодей-
взаимодействуют в одинаковой степени с кобальтом в Co(R2SO)e+CoCir (это
доказано идентичным положением их в спектрохимических рядах
по отношению к Ni2+), смещения частоты колебаний кислородных
связей равны 51 см'1 и 88 см'1 соответственно, т. е. из ИК-спектров
получился бы неверный вывод о силе взаимодействия. Усложнение
общей картины в этом примере (а часто и в других) возникает вслед-
вследствие наложения колебаний лигандов.
Вероятно, для некоторых систем можно определить донорную
силу растворителя по отношению к некоторым катионным кислотам
по Льюису из спектрохимических рядов. Например, были сообще-
сообщены 28 следующие значения Dq для комплексов Ni2+ с координацион-
координационным числом, равным шести: NH3 A080 см'1), Н2О (860 см'1),
(CH3JSOG73 слГ1), CH3CON(CH3JG69 см'1). Этот ряд соответ-
соответствует ожидаемому порядку изменения донорной силы.
При определении донорной силы необходимо учитывать природу
кислоты — акцептора вследствие того, что значения энергетических
вкладов деформируемости и электростатических взаимодействий
у разных кислот различны, а также ввиду наличия стерических
эффектов. Так, было найдено (М. Д. Джостен и Р. С. Драго), что
донорная сила амидов общей формулы RCONR'R" (где R' и R" —
СН3 или Н) по отношению к иоду и фенолу увеличивается с увели-
увеличением числа метальных групп. Однако значения Dq для [NiSe]2+
убывают в ряду HCON(CH3J > HCON(CH3)H > CH3CONH2 >
> CH3CON(CH3J > CH3CON(CH3)H. Это можно объяснить следу-
следующим образом. Если в амидах R' и R" — алкильные радикалы,
то между координированными лигандами возникает отталкивание
(рис. 45). Для простоты на рис. 45 указаны только два лиганда.
Когда R — водород, наблюдается уменьшение эффекта отталкива-
отталкивания, в результате чего происходит более сильное взаимодействие
иона металла с лигандом, и Dq становится больше, чем в случае,
когда R, R' и R" являются метальными группами. Этот стерический
эффект меньше также в ацетамиде, где и R' и R" — атомы водорода
и только R — метильная группа. Заметный стерический эффект
отталкивания наблюдается, если R и R" — метилы или все R —
алкилы. Если R и R' — метильные группы, то устойчива конфигу-
конфигурация с игракс-положением этих групп. Это подтверждается появле-
* В скобках указана величина смещения максимума поглощения в см~
III. ЭНЕРГЕТИКА ПРОЦЕССА КООРДИНАЦИИ
215
нием слабых индуктивных эффектов. Подобный стерический эффект
проявляется в комплексах хрома(Ш) с координационным числом 6.
Если координация осуществляется в значительной мере за счет
я-связи лиганда, то Dq не может служить показателем донорной
силы лиганда по отношению к иону металла. Например, в комплексе
[Ni(CH3CN)e]2+ Dq гораздо больше, чем можно ожидать при коорди-
координации без участия я-связи.
Таким образом, для интерпретации поведения растворенных
веществ в различных растворителях следует оценивать донорные
свойства растворителя лишь после проверки всех имеющихся коли-
количественных данных. Наибольшее зна- „
чение следует придавать результатам,
которые получены с кислотами, по
своим свойствам более всего похо-
похожими на рассматриваемое раство-
растворенное вещество.
Проблема оценки энергий стадий
а, г и д (см. рис. 44) является еще
более трудной. При отсутствии спе-
специфических взаимодействий будет
преобладать сольватация типа за-
заряд — диполь. Если диэлектричес-
диэлектрическая проницаемость растворителя об-
обусловлена его дипольным моментом
R
Рис. 45. Иллюстрация стериче-
стерических взаимодействий между ли-
энергия неспецифическои сольвата- гандами амидов в комплексах
ции должна определяться главным Ш(амидJ+.
образом величиной диэлектрической
проницаемости. Полярные молекулы растворителя будут вступать
легче всего во взаимодействие с наиболее высокозарядными ио-
ионами и смещать равновесие в сторону образования этих видов
ионов.
Известно, что степень образования ионных пар также будет
зависеть от диэлектрической проницаемости растворителя. Сила F
притяжения или отталкивания между двумя заряженными части-
частицами равна
р ~
91*72
ЕГ2
A2)
где qx и д2 — заряды частиц, разделенных расстоянием г в гомогенной среде
с диэлектрической проницаемостью е.
Если средой является растворитель, то взаимодействие раство-
растворителя с ионом приведет в результате к частичному распределению
заряда в слое растворителя, окружающем ион. Таким образом,
заряд диспергируется, электрические поля ионов быстро ослабляются
и степень образования ионных пар уменьшается. Растворитель
с высокой диэлектрической проницаемостью ослабляет электрическое
216
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
поле иона сильнее, чем растворитель с низкой диэлектрической
проницаемостью.
В отсутствие специфических взаимодействий Кяссоц галогенида
тетраалкиламмония, вычисленная из данных об электропроводности,
и энергия сольватации должны находиться в соответствии с диэлек-
диэлектрической проницаемостью растворителя. Поскольку с галогени-
дамп тетраалкиламмония координация не происходит, донорные
свойства растворителя не должны влиять на это взаимодействие.
Диэлектрическая проницаемость является слабым критерием
сольватации, когда возможны специфические взаимодействия типа
основание — кислота по Льюису между растворенным веществом
и растворителем, ибо эти процессы обычно энергетически более суще-
существенны, чем. неспецифические диэлектрические эффекты. Примеры
этих специфических взаимодействий — образование водородной связи
между растворителем и анионом (в частности с С1~) и между рас-
растворителем и координированными протонными лигандами, такими,
как [Co(NH3N]3+.
Вследствие отсутствия лучших критериев оценки энергий соль-
сольватации применяется константа ассоциации алкиламмониевых со-
солей Кассоп и Z-величина Косовера 557*.
Поскольку основное состояние ионной пары является ионным,
ее энергия понижается сильно сольватирующим растворителем,
тогда как энергия нейтральной молекулы в возбужденном состоянии
повышается, ибо ее диполь ориентирован в другом направлении,
отличном от направления ионной пары. Следовательно, Z-величина
будет больше для полярного или деформируемого растворителя.
Растворители, образующие водородные связи, вступают в специфи-
специфическое взаимодействие с анионом и, таким образом, приводят к ощу-
ощутимой стабилизации основного состояния ионной пары. В результате
Z-величина достигает больших значений. Применительно к циклу
для MX это равносильно тому, что Z-величина будет содержать
вклад от энергии взаимодействия, такого как в стадии g (см. рис. 44).
Так, хотя диэлектрические проницаемости метанола и формамида
равны 32,6 и 109,5, соответствующие Z-величины равны 83,6
и 83,3 ккал/молъ вследствие описанного выше эффекта.
Значения ^ассоц Для алкиламмонийных галогенидов в различных
растворителях можно также применять для доказательства сольвата-
сольватации. На Кассоп влияют и диэлектрическая проницаемость, и анион-
анионная сольватация. Мы считаем, что Кассоц является одним из лучших
критериев для оценки сольватирующих свойств растворителей-
* Z-Величина — эмпирическая величина, характеризующая полярность
растворителя. Она определяется из спектра 1-этил-4-карбометоксипиридинио-
дида в данном растворителе и равна длине волны максимума полосы переноса
заряда (в ккал/молъ).
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
217
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
В НЕКОТОРЫХ РАСТВОРИТЕЛЯХ
Пока очень мало исследований поведения растворенного вещества
в неводных растворителях проведено с достаточной тщательностью,
т. е. так, чтобы они дали исчерпывающие сведения относительно
структуры молекул и ионов, присутствующих в растворе. Следова-
Следовательно, воздействие растворителя на многие растворенные вещества
неизвестно, и нет полной ясности в понимании относительного зна-
значения существенных свойств растворителя. Вследствие недостатка
сведений в настоящее время можно только поверхностно осветить
этот важный вопрос.
Нашим намерением не является исчерпывающий обзор данных,
которые доступны; будет рассмотрено лишь несколько характерных
растворителей, чтобы показать последовательность суждений, кото-
которая предлагается координационной моделью. После рассмотрения
свойств и поведения некоторых растворенных веществ в нескольких
растворителях будет приведено краткое общее обсуждение, в кото-
котором сделана попытка установить связь между экспериментальными
данными и основными свойствами различных растворителей. Вслед-
Вследствие ограниченности сведений некоторые объяснения в последнем
разделе следует рассматривать как предварительные. Мы надеемся,
что это обсуждение укажет подход к унификации поведения раство-
растворенного вещества в неводных растворителях и будет стимулировать
дальнейшую научно-исследовательскую работу в этой области.
А. ^И-Диметилацетамид (ДМА)
Ниже приведены физические свойства днметплацетамида:
tin, °C 20
*Ьп. °С 165,0
о (при 25° С) 60, г/см» 0,9366
е (при 25° С) во 37,8
т] (при 25° С) «о, спз 0,919
хво, ол-1.сле-1 @,8-2,0) • КГ?
Z-ве личина*, ккал/молъ 66,9
Z)g28 по отношению к Шп, аП 769
* Д. Гарт, частное сообщение.
27.
Параметры образования 12-аддукта
—АН, кдж/моль (ккал/молъ) 16,7 D,0)
К, молъ-1 6,9
Параметры образования фепольного аддукта45'46:
—АН, кдж/молъ (ккал/молъ) 26,1 F,4)
К, молъ~1 134
218
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
Предельная электропроводность Ло и константы ассоциации
растворов некоторых одно-одновалентных электролитов в ДМА при
25° С приведены ниже в0:
Электролит KNO3 NaNO3 NaBr NaSO3Ph
Ло, ему (дм ¦ г-экв) 71,6 71,2 69,0 56,8
Кассоц 25 50 17 33
Электролит I (C2H5)NBr (C3H7)NBr CH3NPhSO3Ph
Ло, ему (дм ¦ г-экв) J 75,9 69,4 59,3
Кассоц 20 20 25
1.0 г-
0.9
0,8
I"
0,6
280
зоо зго
А, ммк
зио
Рис. 46. Изменение УФ-спектра
Fe(C104K в 1Ч,1Ч-диметилацетамиде в
зависимости от концентрации при-
прибавленного хлор-иона при
[Fe(C104)sI = 1,28-10-4 м:
1 — 0; 2 — 0,36 . Ю-4 м; 3 — 0,73 • Ю-4 м;
4 — 1,09 ¦ Ю-4 м; s — 1,46 • 10-* м; 6 —
2,19 ¦ 10~4 лс;
г Л
I
I
0,8
as -
2во зго збо
А, ммк
400
Рис. 47. Изменение УФ-спектра
Fe(C104K в N,N-flHMeTnna4eTaMHfle в
зависимости от концентрации прибав-
прибавленного хлор-иона при lFe(C104Kl =
= 2,0-10-4 м:
1 — 4,54 • 10~4 ж; 2 — 5,66 • Ю-4 ж; 3 —
6,79 • Ю-4 м; 4—9,05 ¦ 10-* м.
Соли лития полностью диссоциированы в1 вплоть до концентра-
концентрации 0,02 м; 10,ы+ < VNa+-
По изменению УФ-спектров поглощения раствора Ре(ДМАN(СЮ4K
в зависимости от концентрации добавленного С1~-иона (из FeCl3)
были установлены типы железохлорсодержащих частиц, присутству-
присутствующих в разбавленных растворах хлорного железа в ДМА (рис. 46
и 47). По мере увеличения концентрации хлор-иона вплоть до отно-
отношения [СП : [Fe3+] = 1,8 : 1 наблюдается исчезновение максимума
поглощения при 287 ммк, относящегося к нону [Fe(flMA)e]3+T
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
219
но увеличивается поглощение в области 320 ммк, достигая макси-
максимума при 323 ммк для [СГ] : [Fe3+] = 1,8 : 1. Этот пик приписы-
приписывается иону FeClJ- В области длин волн больше 340 ммк (см.
рис. 46) поглощение вначале увеличивается (кривая 3, [СГ] : [Fe3+l =
.== 0,6 : 1), а затем уменьшается. Одновременно появляется одна
изобестическая точка при отношении [СГ] : [Fe3+] m 0,9 : 1 (кри-
(кривая 4). Поглощение при длине волны г»290 ммк уменьшается
(вплоть до отношения 0,9 : 1), затем увеличивается и" снова умень-
уменьшается при отношении [СГ] : [Fe3+] :з= 2 : 1. В последнем случае
(см. рис. 47) спектр свидетельствует о появлении иона FeClJ,
в спектре появляются две изобестические точки и максимумы погло-
поглощения при 314 и 364 ммк. Спектры на рис. 46 свидетельствуют
об образовании ионов FeCl2+ и FeClJ, причем последний начинает
образовываться при отношении 0,9 : 1.
Присутствие трех типов ионов при соотношениях [СГ] : [Fe3+l =
= @,9 -г- 1,8) : 1 явствует из исчезновения изобестической точки
при 316 ммк. Рис. 47 иллюстрирует образование ионов FeCli из
FeCl(flMA)!+ [FeCl3 (раств) не обнаруживается] и показывает, что
при растворении хлорного железа в ДМА (кривая 3) основными
типами ионов, присутствующих в разбавленных растворах, являются
FeCl2(flMAL и FeClJ. Точные значения констант образования Кп
ионов FeClJ5~" не установлены, но следующий ниже ряд описывает
состояние этой системы:
Из уравнений A3) видно, что поведение FeCl3 в ДМА исчерпы-
исчерпывающе описывается координационной моделью:
flMA + FeCl3 T"* FeCl3-flMA (первоначальный аддукт)
FeCls-ДМА+ЗДМА -^± РеС12(ДМА)? + СГ A3)
cr+FeCls-ДМА ^=± FeClj + ДМА
Поскольку структура исходного аддукта неизвестна, следующая
система уравнений также может описать поведение FeCl3 в ДМА:
Fe2Cle-flMA
A4)
Ре2С16-ДМА+ЗДМА
Последняя реакция указывает на простой механизм образования
FeClJ действием ДМА на Fe.2Cl6 • ДМА:
ДМА
С1— Fe.
/
С1
Cl
С1
ДМА
220
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
Буффани и Данн 12 провели тщательное исследование системы
СоС12 — ДМА. Спектры в видимой области могут быть объяснены
существованием следующих типов ионов: СоСГГ, Со(ДМА)|+,
СоС13(ДМА)~ и СоС1(ДМА)?. Очевидно, нейтральные молекулы
СаС12 • 2ДМА или СоС12 • 4ДМА существуют при очень низких
концентрациях, как обнаружено для комплекса FeCl3-flMA.
Таким образом, поведение системы СоС12 — ДМА также можно
объяснить с помощью координационной модели уравнениями, анало-
аналогичными уравнениям, приведенным выше для FeCl3.
Б. N-Метилацетамид (NMA)
Физические свойства N-метилацетамида:
*ё°л, °С 29,5
'Й„' °С 206
р (при 40° С) 22, г/см3 0 9420
е (при 40° С) 22 165,5
т) (при 403 С) 22, спз 3.019
х22 (при 40° С), ом' 1 ¦ см'1 ..../.. A—3) • 10~7
Z-величина*, ккал/моль 77,9
Dq№ по отношению к Ni11, c.u"i .... 752
* Д. Гарт, частное сообщение.
Энтальпия образования фенольного аддукта45'46:
—АН кдж/молъ (ккал/моль) 19,7 ±13 D,7±3)
Таблица 32. Предельная эквивалентная электропроводность
растворов электролитов в N-метилацетамиде
Электролит
КС1
NaCl
КВг
NaBr
KI
Nal
CsBr
(C2H5LNBr
Ло
17,9
17,8
19,0
18,9
20,7
20,1
20,1
22,1
Электролит
(C2H3LNPi *
НС1
i HPi **
1 NH4CIO4
! NH4NO3
1 NH4NO3
КСЮ4
! Л0
21,3
20,7
20,9
26,5
26,5
24,2
25,2
Электролит
KSCN
KNO3
KPi
NaClO4
NaSCN
NaNO-t
NaPi
Ло
24,5
22,9
20,2
25,0
24,2
22.7
20,2
Примечание. С КС! по (CtHt)«NPi— по данным работы'4, остальные — по дан-
данным работы !2.
* Pi — пикрат.
** ^ассоц, начиная с HPi, равны нулю.
Подвижности С1 , В г и I указывают на интересное направле-
направление в анионной сольватации. Следует ожидать, что в растворителях
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО" ВЕЩЕСТВА
221
с кислотным протоном, таких, как NMA, способных к образованию
водородной связи, анионная сольватация будет высокой. Энергия
сольватации состоит главным образом из энергии специфического
взаимодействия:
| X | • ¦ • Н ¦ ¦ • N-СОСНз
СНз
Предельная анионная электропроводность должна быть обратно
пропорциональна склонности к образованию водородных связей,
что наблюдается для NMA, т. е. Ко увеличивается в такой последо-
последовательности: СГ << Вг~ <1~.
Исследования Драго и др. также подтверждают наличие интен-
интенсивной сольватации. УФ-Спектр FeCl3 в NMA указывает на присут-
присутствие только FeSjj+ и С1~. Это демонстрирует важность учета анион-
анионной сольватации и диэлектрической проницаемости (или Z-величины)
для данного растворителя, поскольку донорная сила NMA почти
такая же, как у ДМА.
В. Диметилсульфоксид (ДМСО)
Физические свойства диметилсульфоксида 81:
/пл, рС 18,4
«кип, рС 189,0
р (при 25° С), г/смз 1,096
е (при 25° С) 46,6
т) (при 25° С), спз 1,96
¦л, om-1-cm'I 3-10"»
Z-величина55, ккал/моль 71,1
Dqn по отношению к Ni11, елП 773
Параметры образования 12-аддукта 26:
—АН, кдж/молъ (ккал/молъ) ........ 18,4D,4)
К, МОАЬ'У 11,6
Параметры образования фенольного аддукта26:
— АН, кдж/молъ (ккал/молъ) 27,2 F,5)
К, моль'1 182
Предельные электропроводности некоторых одно-одновалентных
электролитов в диметилсульфоксиде 81:
Электролит KI Nal KBr NaBr KSCN
Ло, см*/(ом ¦ г-акв) . . . 38,2 37,6 38,4 38,0 43,5
NaSCN KNO3
43,0 41,5
Электролит . . . NaNO3 КСЮ4 NaClO4 KPi NaPi (С4Н9LШ (CH3KNPhI
Ло, см*/(ом ¦ г-экв) 40,8 39,1 38,3 31,7 31,1 35,0 37,8
Эти соли полностью диссоциированы в диметилсульфоксиде
вплоть до концентраций 10" 3 м; ДМСО, по-видимому, является зна-
значительно лучшим сольватирующим растворителем, чем ДМА.
222
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
0,8
0,6
0,2
О
Предварительные исследования Драго показывают, что
ион FeClJ неустойчив в диметилсульфоксиде и ионизируется, обра-
образуя катионы железа(Ш), вероятнее всего Fe(flMCO)i+ и катионные
частицы железа с хлором.
Поведение СоС12 в диметилсульфоксиде и в растворах С1~ в ДМСО
было исследовано П. Ван Дер Воорном (неопубликованные резуль-
результаты) и Буффани и Данном 12. Спектры этих растворов приведены
на рис. 48. Сравнивая эти спект-
спектры с кривыми молярного погаше-
погашения 33 СоС12, СоС13 и СоС14~, можно
сделать качественные предполо-
предположения относительно того, какие
частицы присутствуют в растворе
в условиях этих экспериментов.
Раствор СоС12, вероятнее всего,
содержит тетраэдрические моле-
молекулы СоС12(ДМСОJ и ионы
СоС13(ДМСО)~; образование анион-
анионного комплекса сопровождается
образованием октаэдрических ио-
ионов Со2+; поглощение, приписыва-
приписываемое этим частицам, наблюдается
ниже 550 ммк. По мере увеличения
концентрации СГ (из Ph3NCH3Cl),
как явствует из спектров погло-
поглощения, увеличивается образование
СоС13(ДМСО)- и СоСЦ-. Было
сообщено 12, что раствор CoClf"
в ДМСО содержит приблизительно
20% тетраэдрических ионов [СоС1|~
и СоС13(ДМСО)~]; остальная часть
ионов Со2+ присутствует в виде
разнообразных октаэдрических
форм.
Интересный эффект наблюдается 67 в растворах NiCl2 в диметил-
диметилсульфоксиде. При комнатной температуре такие растворы окрашены
в светло-зеленый цвет, обусловленный комплексами Ni" с коорди-
координационным числом 6 (спектры в видимой области указывают на окта-
эдрическую конфигурацию Ni"), по-видимому, ионами Ш(ДМСО)!+
и ШСЦДМСО)!. При нагревании приблизительно до 50° С перво-
первоначальный зеленый цвет раствора переходит в интенсивный синий,
характерный для тетраэдрического никеля. Это изменение цвета
является полностью обратимым; при нагревании раствора до 50° С
в течение 6 ч осаждаются крупные синие кристаллы. Данные ана-
анализа кристаллов свидетельствуют о том, что это кристаллы смешан-
смешанного комплекса Ш(ДМСОI+№С1|-.
550
600
650
700
Рис. 48. Изменение спектра СоС12
в диметилсульфоксиде в зависимости
от концентрации прибавленного
хлор-иона:
1 — [СоС12] = 0,002 м; г — Ссо2+] =
= 0,003 м; [С1-] = 0,012 м; 3 — [Сог+] =
= 0,003 м; [С1-] = 0,024 м; 4 — [Со*+] =
= 0,003 м; [С1 ] — большой избыток.
IV- ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
223
Этот же комплекс образуется в растворах NiCl2 в ДМСО, если
добавлять бензол при комнатной температуре. Последний случай
являет.ся примером системы, в которой выделенное твердое вещество
и частицы, находящиеся в растворе при комнатной температуре,
совершенно различны.
Г. Нитрометан и нитробензол (НМ, НБ)
Физические свойства нитрометана и нитробензола 89:
Соединение CH3NO2 C6H5NO2
trm, С —28,5 5,8
«кип. °С '. 101,3 210,8
р (при 25° С), г/слз 1,1312 1,1930
е (при 30° С) 35,9 34,8
т) (при 30° С) спз 0,595 1,634
х (при 25° С), o.w-1-слП 6,56-10"' 9,1 • 10~7
Параметры образования фенольного аддукта45'46:
—АН*, кдж/моль (ккал/молъ) 7,9A,9) 8,56B,04)
* Рассчитано из Д»он фенола.
Очень сложно объяснить результаты, полученные для нитро-
нитрометана как растворителя вследствие трудности получения чистого
растворителя. Буффани и Данн 12 отмечают два метода очистки.
Очистка перегонкой не всегда надежна. Можно с уверенностью
сказать, что если в статье специально не упомянуто об очистке, то
в работе применялся неочищенный нитрометан. Электропроводности
растворов некоторых электролитов приведены в табл. 33.
Таблица 33. Мольная электропроводность растворов
солей в нитрометане
Соль
Fe(HMPA)e(ClO4K
Сг(НМРАN(С1О4)з
А1(НМРА)в(С1О4K
Fe(HMPAL(C104J
Mg(HMPAL(ClO4J
Co(HMPAL(ClO4h
Ш(НМРАL(СЮ4J
Zn(HMPAL(C104J
(Ph3AsCH3JNiCl4
(Ph3AsCH3JCoI4
[(C2H5LN]2MnCl4
(Ph3AsCH3)I
[(CjHWiNJI
Co(R2AsAsR2J(ClO4J
Co(R2AsAsR2K(ClO4K
A,
см2/ (ом- моль)
236
253
241
185
153
178
184
180
173
151
200
85
97
177
262
С, МОЛЬ/А
0,0005
0,005
0,0005
0,002
0,001
0,0105
0,005
0,002
0,0005
0,0005
0,0005
0,0005
0,0005
0,0005
0,0005
Литера-
Литература
24
24
24
и
24
23
23
23
38
38
38
38
38
38
38
224
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
Нитрометан и нитробензол достаточно хорошие сольватирующие
растворители для многих электролитов. В них часто наблюдается
диссоциация молекулярных форм на ионы. Константа ассоциации
для (C4H10LNBr в нитробензоле при 25° С равна 46 84. Как коорди-
координирующие растворители они являются слабыми, но могут действо-
действовать как основания по Льюису.
Донорная сила CH3NO2 и CeH5NO2 неизвестна. Сообщают 26,
что сдвиг частоты ОН-групны фенола нельзя использовать в качестве
критерия донорной силы сульфолана (CH2CH2JSO2, так как фенол
взаимодействует с обоими атомами кислорода. Подобный же эффект
следует ожидать у нитросоединений. Так, рассчитанные значения
энтальпии образования фенольного аддукта, но-видимому, занижены,
и действительные значения нужно измерять непосредственно.
Величина параметра кристаллического ноля Dq неизвестна, но
имеются указания, что нитрометан замещает некоторые лиганды
в координационной сфере ионов металлов. Это может происходить
за счет образования водородной связи между донорным лигандом
и CH3NO2 и не обязательно означает координацию самого нитро-
метана. Однако имеется и прямое доказательство координации
нитрометана.
Наиболее интересное исследование было проведено де Мейном
и Коубеком в5 в системе FeCl3—CHgNO2 и в трехкомпонентной
системе FeCl3—CH3NO2—CC14. Растворы FeCl3 в нитрометане не
подчиняются закону Бэра, но для данного состава раствора суще-
существует линейная зависимость в области всех длин волн между отно-
отношением оптической плотности к общей концентрации FeCl3 и общей
концентрацией FeCl3. Измерения электропроводности этих растворов
показали, что она удовлетворяет уравнению Онзагера, Л =
= Ло — (а +|ЗЛ0I/Т, где с — концентрация FeCl3- Следовало бы
сослаться на оригинальную работу, в которой приведены подроб-
подробности, но рассмотренные эффекты согласуются лишь с одним из сле-
следующих равновесий:
(раств) + FeGl3 (раств)
[комплекс]
^±. FeClj (раств)+FeClJ (раств)
(раств)+FeCl3 (раств) т~*" [комплекс] т""*" Fe
A5)
(раств)
Концентрация комплекса в этих равновесиях (вероятно, Fe2Cl6)
должна быть чрезвычайно малой, чтобы можно было объяснить линей-
линейную зависимость, упомянутую выше. По аналогии с другими раство-
растворителями, описанными в этой работе, первое равновесие предпочти-
предпочтительно.
Буффани и^Данн 12 провели спектрофотометрическое исследова-
исследование растворов СоС12 в нитрометане. Спектроскопические свойства
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
225
этих растворов как-функция концентрации хлор-иона удовлетвори-
удовлетворительно объясняются следующими равновесиями:
СоС12 + 2НМ = СоС12 ¦ 2НМ
СоС12 • 2НМ + С1- = СоС1з(НМГ + НМ A6)
СоС13(НМ)- + СГ = СоС11" 4-НМ
Существование первого равновесия A6) подтверждается данными
об электропроводности раствора СоС12 в CH3NO2. Установить суще-
существование ионов в растворе этим методом не удалось; тщательное
изучение спектров свидетельствует об отсутствии октаэдрических
комплексов.
Ганьо и др. 36 указали, что инфракрасные спектры систем
rc-RC6H4NO2—МХ„ и CH3NO2—МХ„ свидетельствуют о координа-
координации нитросоединений; в спектре наблюдается понижение частоты
асимметричного валентного колебания нитро-группы по сравнению
с ожидаемым. Однако к результатам этой работы надо относиться
с осторожностью, так как изучались спектры твердых веществ.
Очевидно, необходимо продолжить исследования систем с этими
растворителями.
Д. Ацетон (Ац)
Физические свойства ацетона 89:
гпл, °С -95,4
*кип, °С 56,2
р (при 25° С), г/с.цЗ 0,7851
е (при 25° С) 20,7
11 (при 30° С), спз 0,2954
х, 0ЛГ1.МГ1 5,8 ¦ 10~8
Z-величина55, ккал/моль 65,7
Параметры образования 12-аддукта 28:
— ДЯ, кдж/моль (кпал/моль) 13,8C,3)
К, лимь-i 13,5
Предельная эквивалентная электропроводность растворов
электролитов в ацетоне 79:
Электролит .... (C4H9LNI KI Nal KBr NaBr KSCN NaSCN
\0, сму(ом-г-экв) 180,2 «1 193,0 191,0 196,0 194,0 208,0 200,0
202.281
Электролит .... KNO3 NaNO3 KC1O4 NaClO4 KPi NaPi
Ло, сму(ом-г-экв) 202,0 200,0 196,0 194,0 166,0 159,0
#ассоц (C4H9LNI и KSCN равны "соответственно 164 и 294.
Недавно Файн 33 изучил образование комплексов СоГ?,~"
(Г—С1~, Вг~, 1~) в ацетоне. В растворе были найдены СоГ2, СоГ3
и СоГ4". Спектры показывают, что все они тетраэдрической конфи-
конфигурации. Автор допустил, что ацетон занимает координационные
15 Заказ 90
226
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
места, не занятые Г . Используя понятие среднего поля ли-
ганда 18< 19- 4' (очевидно, справедливого для лигандов, близких
друг к другу в снектрохимических' рядах), он вычислил значение Dq'
для ацетона (где Dq' является параметром расщепления поля лиган-
лигандов для тетраэдрического комплекса) из известных значений Dq
для галогенов. При умножении значения D,3±0,4I03 см'1 на % Для
получения lODq — параметра расщепления октаздрического поля
молекул ацетона у Со11 получается удивительно большая величина,
9700 см'1. Файн вычислил также константы образования ком-
комплексов СоГ*~":
[СоГ2]
Частное
К для комплексов
с хлором . .
с бромом . .
с иодом . . .
[Со2+] [Г
3-10»
2-Ю9
>109
[СоГП
[СоГ21 [Г"] [СоГд-] [Г"]
2,2 ¦ 10*
5.4 • 102
42
16
Изучение электропроводности 20 FeCl3 в ацетоне показывает, что
растворенное вещество не полностью диссоциировано. Ионы, очевидно,
одновалентны. Эта система, по-видимому, похожа на систему
FeCl3—HM, рассмотренную выше. Необходимо продолжить исследо-
исследование этой системы.
Фридман и Плэйн 35 опубликовали результаты интересной ра-
работы, посвященной исследованию сольватации Си2+ в водно-ацетоно-
водно-ацетоновых смесях. Спектры поглощения этих растворов в зависимости от
концентрации (в мол. %) Н2О удовлетворительно объясняются
равновесиями
|+ = Си(Н2ОMАц2++Н2О
( '
Авторы заключили, что сила поля лигандов для воды, ацетона
и этанола убывает в ряду вода ^ ацетон ;э=этанол.
Поведение некоторых солей Со11 в ацетоне 50~52 сходно с поведе-
поведением солей Си11. Соли Co(SCNJ, СоС12 и Co(NO3J с увеличением
концентрации X" образуют анионные комплексы типа СоХз и СоХ|~.
К сожалению, в этой работе использовались не безводные соли,
поэтому комплексы, весьма вероятно, содержат координационную
воду. На существование аквакомплексов указывает диаграмма трех-
компонентной системы, опубликованная Кацином и др 49. Из диаг-
диаграммы ясно, что образуются твердые комплексы Co(NO3J-6Н2О,
Co(N03)o-4H20, Co(NO3K-3H2O, Co(NO3J-2H2O и, вероятно,
Со(Ш3),-2Ац.
Кох 53 предположил существование необычных частиц в ацето-
ацетоновых растворах KI и Agl. Соль Agl нерастворима в ацетоне, но
в присутствии 1 моль KI в ацетоне можно растворить до 3 моль Agl;
электропроводность ацетоновых растворов KI в присутствии Agl
гораздо ниже. Это можно объяснить, по-видимому, образованием
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
227
I(AgI)i- Аналогично ведет себя Hgl2. Было бы интересно продолжить
изучение этой системы.
Исследовано образование аддукта 1 : 1 между различными кис-
кислотами по Льюису и альдегидами, а также ароматическими или
алифатическими кетонами 36> 85'86. Единственным выводом, кото-
который можно сделать, основываясь на результатах этих работ, является
то, что координация происходит.
Е. Метанол и этанол
Физические свойства 89 этих растворителей при 25° С:
Растворитель СН3ОН С2Н5ОН
*пл, ?С —97,5 —114,5
гкип, °С 64,51 78,3
р, г 1см? 0,7868 0,7851
е 32,6 24,3
¦л, спз 0,5445 1,078
х, ом.'1-CM-i 1,50-10"» 1,35-10"»
Z-величина55, ккал/молъ . . . 83,6 79,6
Dq * по отношению к Ni11, см"*- 850 —
* В. Имхоф, неопубликованные результаты.
Параметры образования 12-аддукта 88:
— АН, кдж/моль (ккал/моль) 7,97A,90) 8,80B,10)
К, молъ-1 0,47 0,45
При спектрофотометрическом исследовании разбавленных рас-
растворов хлорного железа в метаноле (Р. С. Драго и др.) были найдены
только катионные частицы, содержащие железо и хлор; ионы FeClJ
не были обнаружены. Это можно было предвидеть, если считать, что
метанол является сильным основанием по отношению к катионам
переходных металлов 4 периода (Dq = 850 см'1) и является хорошим
сольватирующим раствором, что доказано Z-величиной и КасС01Х для
(C4H9LNBr. Величина диэлектрической проницаемости метанола
не соответствует его ионизирующей способности. Образование водо-
водородной связи (специфическое взаимодействие) с анионами, по-види-
по-видимому, объясняет большую ^-величину и диссоциацию анионов,
содержащих железо и хлор.
Ниже приведены данные об электропроводности и Кзссоц раство-
растворов электролитов в спиртах:
в метаноле
Электролит LiBrSO AgNO35 KSCN'8 (C4H8LNBr84
Ло, см*/{ом ¦ г-экв) 89,1 — 115,7 96,7
Яассоц — 73,8 11,0 26,0
15*
228
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
в этаноле а
Электролит MgCl2 MgBr2 Mgl2
Ло, см^/(ом-г-экв) 41,8 44,6 47,2
Кассоп 000,0 215,0 180,0
Можно поставить под сомнение Касст Для MgX+. Если сольвата-
сольватация галоген-иона велика, то следует ожидать увеличения Kaccoii
в ряду Ка- <С АГвг- <С Ki-- Однако результаты, приведенные выше,
можно объяснить, если считать, что донорная сила аниона по отно-
отношению к Mg2+ уменьшается от хлора к иоду, а роль этанола сводится
к замещению галогена в комплексном катионе металла. Величины
^ассоц убедительно показывают разницу влияния двухвалентного
и одновалентного катионов на ионную ассоциацию.
Кацин 48 опубликовал спектрофотометрические данные, которые
еще раз подтверждают описанные выше свойства спиртов как раство-
растворителей. Спектры растворов Ni(SCNJ в мзо-С3Н7ОН указывают
на присутствие в значительных количествах только Ni(SCN)+
и Ni(SCNJ вплоть до отношения [SCN~] : [Ni2+] = 4 : 1. В раство-
растворах СоС12 даже при десятикратном избытке иона хлора по отноше-
отношению к кобальту обнаруживаются только СоС1+ и СоС12. Чтобы обра-
образовался СоОз требуются намного большие концентрации С1~. Такой
же снектр получается, если добавлять в изопропиловый спирт CoCll".
Те же закономерности наблюдаются для бромида кобальта(Н).
В растворах Co(SCNJ в изо-С3Н7ОН и С4Н9ОН при большем чем
четырехкратный избытке SCN" по отношению к кобальту существуют
Co(SCNJ и Co(SCNK, но ионов Co(SCN)r не обнаружено.
Были исследованы"спирто-водные смеси, но часто весьма трудно
интерпретировать результаты, полученные для смешанных раство-
растворителей. В работе Маккора 64 исследовалась устойчивость ком-
комплекса Agl2 в смешанном ацетоно-водном и метаноло-водном рас-
растворителях. Устойчивость комплекса возрастает при переходе от
чистой воды в качестве растворителя к смеси ацетона и воды, но
этого не наблюдается при переходе от воды к смеси метанола и воды.
Кроме того, константа образования AglJ одинакова в смеси мета-
метанол — ацетон и в ацетоно-водных смесях. Это указывает на сходство
между спиртами (особенно метанолом) и водой, что объясняется
склонностью к образованию водородных связей.
Ж. Пиридин (Ру)
Физические свойства пиридина 89:
<пл. °С —41,8 т) (при 30° С), спз 0,829
«кип. рС 115,6 х (при 25° С), о.ад-1-сип . . . 4,0 • 10"»
р (при 30° С), г/смз . . . 0,97281 2-величина55, ккал/молъ . . . 64,0
е (при 25° С; 12,3 Dq по отношению к Ni", cm~i ~1000*
* М. Розенталь, Р. С. Драго.
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
229
Параметры образования 12-аддукта 88:
— АН, кдж/молъ (ккал/молъ) 32,7G,8)
К, моль'* 269
Параметры образования фенольного аддукта
— АН, кдж/молъ (ккал/.чолъ) ....
К, моль~±
33,88 (8,07)
Электропроводность и константы ассоциации растворов некото-
некоторых электролитов в пиридине 2:
Электролит . . . NH4Pi KPi NaPi LiPi AgClO4 AgNO3 Py • HN03
Ao, см*/(ом- г-экв) 80,5 65,7 60,5 58,6 81,9 86,9 102,2
' 3580 10 000 23 200 12 000 524 1070 19600
Электролит . . . (C4H9LNOAc (C4H9LNNO3 (GiH9LNI (C4H9LNBr (C4H9LNPi
An, см*/(о.и-г-экв) 76,0 76,6 73,1 75,3 57,7
К
ассоц
5880
2700
2440
4000
780
Электролит
А0*, 0B/@.11
¦ • • >
• г-экв)
KI
80,2
Nal
75.2
КВг
82,9
NaBr
77,9
KNO3
84,7
NaNOa
79,6
* По данным работы тз.
Предварительные эксперименты (Д. Харт, частное сообщение)
по исследованию растворов FeCl3 в пиридине показали, что FeClJ
из FeCl3 не образуется. В растворе присутствуют, вероятно, Fe2Cle
и комплекс Ру —у FeCl3. Однако FeCl4 обладает некоторой устой-
устойчивостью в пиридине. Добавление иона С1" в пиридиновый раствор,
содержащий FeCl4, приводит к увеличению поглощения при 316
и 365 ммк, что свидетельствует о некоторой диссоциации FeClJ.
Структура других растворенных частиц не установлена.
Исследование аддуктов пиридина и замещенных пиридина с SbCl5
и фосфорными кислотами типа PXnF5_n проведено Холмсом и др. 41.
Величины молекулярных весов и электропроводности в нитробен-
зольном растворе показывают, что ионизации не происходит, а обра-
образуются только аддукты 1:1. Авторы приводят энтальпии образова-
образования аддуктов, которые были определены калориметрически в нитро-
нитробензоле, но не были исправлены с учетом различий в неспецифической
сольватации основания и аддукта.
Как упоминалось в разделе II, Кацин 49 исследовал образование
пиридиновых комплексов СоС12 в различных растворителях. Уста-
Установлено существование комплексов Ру-СоС12 1 : 1 и 2 : 1 в ацетоне.
Ион PyCoClj можно получить при добавлении в эту систему избытка
230
С1~-иона. Авторы выделили Со(РуNС12, однако Розенталю и Драго
это не удалось.
Исследование Факлера, упомянутое в разделе II, хорошо иллю-
иллюстрирует влияние координирующего растворителя на типы образу-
образующихся ионов или молекул. Аналогичное исследование
•системы [Ni(acacK]3—С6Н6—Ру было также выполнено Факле-
ром 31. Зависимость спектров поглощения бензольных растворов
[Ni(acacJ]3 от концентрации пиридина указывает на протекание
следующих реакций:
2[Ni(acacJ]3 + 3Py
[Ni(acacJ]2-Py-|-3Py
3[Ni(acacJ]2 ¦ Ру
2Ni(acacJ- 2Py
A8)
Эти исследования интересны тем, что они хорошо иллюстрируют
процесс агрегации растворенного вещества в слабо координирующих
растворителях и влияние координирующего растворителя на эти
агрегаты.
Данные об электропроводности показывают, что электролиты
сильно ассоциированы в пиридине. Наиболее вероятно, что это свя-
связано с низкой диэлектрической проницаемостью пиридина. Соль-
Сольватация аниона через специфическое взаимодействие в пиридине
не происходит, но катион Li+ координирует и сольватирован больше,
чем Na+ и К+. Об этом можно судить по величинам Ло и Кассоп для
обычных солей этих катионов. Очевидно, Na+ и К+ имеют худшие
сольводещамические характеристики.
3. Ацетонитрил
Ацетонитрил — наиболее распространенный из нитрильных
растворителей. Его. физические свойства при 25° С представлены
ниже:
* on
Of
*кип> °С
р.
е
т) (при 30° С), спз
х, ом'1 ¦ см'1 5,9-
2-величина55, ккал/моль 7<
"-"¦ по отношению к Ni11, см'1
-45,7
81,6
0,7768
36,2
0,325
Параметры образования 12-аддукта 26:
— &.Н, кдж/молъ (ккал/моль) 9,6B,3)
К, моль'1 0,40
Параметры образования фенольного а-ддукта26:
— &.Н, кдж/молъ (ккал/моль) 13,8C,3)
К, моль'1 _ 5,0
IV. ПОВЕДЕНИЕ РАСТВОРЕННОГО ВЕЩЕСТВА
231
Электропроводность и константы ассоциации растворов электро-
электролитов в ацетонитриле:
Электролиты * . .
До, см^/(ом • г-вкв)
KI LiC104 NaClO4 КСЮ4 RbC104 CsC104
186,7 183,4 192,3 208,2 203,0 207,6
- 68,4 70,9 97,7 103,7 144,6
данные о KI заимствованы из работы 86, остальные — из работы ".
Электролит * . . .
, Ло, см*/(ом ¦ г-же)
(CH3LNNO3
200,5
23,0
168,2
7,0
193,1
77,5
(CH3LNBr (CH3LNI
192,7 195,3
41,4 27,5
* Данные о (CH3)iNNOj и (CiH.^NNOs взяты из работы10, остальные — из работы'•.
Данные об электропроводности растворов перхлоратов щелочных
металлов в ацетонитриле соответствуют ожидаемому увеличению
сольватации: Cs < Rb < К < Na <[ Li. Значения Kaccoli изме-
изменяются в обратном порядке. Из данных для тетраметиламмониевых
солей видно, что анионы 1~ и NO3 ведут себя в CH3CN одинаково.
Значения Кассоц для галоидных соединений подтверждают ожида-
ожидаемую последовательность увеличения иЬнной сольватации
(Г <ВГ <СГ).
Марсинковский 66 исследовал спектрофотометрически и кондукто-
метрически растворы СоС12 в CH3CN и нашел, что свойства растворов
объясняются следующими равновесиями:
2CoCl2 + 6CH3CN ^=± Co(CH3CN)|+CoCll-
CoCll-+2CH3CN ^± CoCl4(CH3CN)l-
A9)
Хлорное железо диссоциирует в CH3CN, образуя' различные
катионы и анионы (Р. С. Драго и Р. Л. Карлсон):
FeQlg
FeCl2(CH3CN)J
B0)
t FeCl3-CH3CN ^z±
Cl- + FeCl3-CH3CN 5=± FeClJ + CH3CN
Барнес и Хьюм 5 исследовали комплексы, образованные Си11
и Вг~-ионом в ацетонитриле. Они нашли, что СиВгд (тетраэдри-
ческой конфигурации) устойчив в этом растворителе. Комплекс
CuBr^ имеет конфигурацию тригональной бипирамиды с координа-
координационным числом пять:
S
232
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
Л'. ЗАКЛЮЧЕНИЕ
233
Битти и Вебстер « исследовали растворы РС13 и SbCl5 в CH3CN
методом ИК-спектроскопии и выяснили, что РС15 ионизируется
по уравнению
2РС15 5=± PCtf + PCl; B1)
тогда как SbCl5 диссоциирует и координирует CH3CN
2SbCl5 + 2CH3CN ^±-SbCl4(CH3CN)J + SbCle B2)
Эллендт и Круз 30 сообщили данные об электропроводности рас-
растворов HgX2 и указали на образование комплексов HgX3, HgXf
и Hg2X6. Приведены значения свободных энергий (в ккал/молъ)
образования комплексов Hg2Xj и HgXf для следующих реакций
в воде и ацетонитриле:
2HgX2 + R4NX ^=± R4NHg2X5
R4NHgX3 + R4NX ^± (R4NJHgX4 B3)
И. Тетраметиленсульфон (сульфолан, ТМС)
Сульфолан — очень интересный неводныи растворитель; физи-
физические свойства его приведены ниже 14:
'"»• °9Г 28,86
*киП1 с 283
р (при 30° С), г/см? 1 2615
е (при 30° С) '44
т) (при 30° С), спз 9 87
х (при 25° С), ом-1-c.u-i 2-10"8
Z-величина, ккал/молъ 77 5 *
* Д. Гарт, частное сообщение.
Параметры образования 12-аддукта 26:
— АН, кдж/молъ (ккал/моль) 9,2B,2)
К, моль~1 0 73
Параметры образования фенольного аддукта 26:
— АН, кдж/молъ {ккал/молъ) 20,9E,0)
К, моль'1 17 1
Электропроводность и АГасСоц растворов солей в сульфолане 14:
Соль NaSCN CH3N ОГ LL\O3 Ph4AsCl CH3NPh3I
Ao, см*/(о.п-г-экв) —13,5 —12,5 —10,5 —10,5 -
Яассоц 48 77 91 0 0
При работе с сульфоланом встречаются некоторые трудности
вследствие того, что температура плавления его выше комнатной.
Растворы сульфолана более удобны в работе благодаря большой
величине криоскопической постоянной F6,2). Температура плавле-
плавления 3-метилсульфолана несколько ниже.
Найдено, что многие неорганические вещества смешиваются
с сульфоланом и растворяются в нем. Имеющиеся ограниченные
данные о растворимостях указывают на сходство этого растворителя
с SO2. Представляют большой интерес донорные свойства сульфо-
сульфолана. По отношению к иоду его донорная сила сравнима с таковой
для бензола. По отношению к фенолу наблюдается большая донорная
сила, чем ожидалась; по-видимому, оба
атома кислорода сульфолана координи-
координированы к водороду. Изучены также
донорные свойства других сульфонов,
сульфоксидов и сульфитов 26. 2
Спектр FeCl3 в тетраметиленсуль- §
фоне (рис. 49) указывает на устойчи- | ¦
вость FeCl4 в этом растворителе.
о,в
Лангфорд и др. 59 исследовали рас-
С С(С1О) ф
гао
зго
А, мм к
360
Рис. 49. Спектр поглощения
FeCl3 в тетраметиленсульфоне;
IFeCl3] = 2,0-10-4 м.
творы СоС12 иСо(С1О4Jв сульфолане.
Данные об электропроводности раство-
растворов СоСЛ2 указывают на отсутствие за-
заметной ионизации; из растворов синего
цвета можно выделить синий твердый
осадок, который по стехиометрическому
составу отвечает СоС12-ТМС. Судя по форме полос поглощения
и их интенсивности, полагают, что эти комплексы тетраэдрические.
При растворении Со(СЮ4J в ТМС появляется красное окрашива-
окрашивание раствора и образуется комплекс Со(ТМСK(СЮ4J, который можно
выделить. Спектр поглощения этого комплекса подобен спектру
Со(Н2ОI+. Эквивалентная электропроводность при концентрации
1,1 • 10 м равна 18,4 см2/ом (сравните со значениями для одно-
одновалентных электролитов, приведенными выше). Оба эти ком-
комплекса легко подвергаются гидролизу.
V. ЗАКЛЮЧЕНИЕ
Поведение хлорного железа было исследовано во всех рассмотрен-
рассмотренных выше растворителях; это дает возможность сделать некоторые
интересные сравнения свойств растворителей. Следует подчеркнуть
с самого начала, что растворенные вещества, для которых порядок
изменения донорнои силы растворителей отличается от такового для
хлорного железа, ведут себя по-разному. Ацетонитрил, тетрамети-
ленсульфон, нитрометан и нитробензол представляют группу соеди-
соединений, диэлектрическая проницаемость и сольватирующая способ-
способность которых достаточно велики (АГассоц для R4NBr в CH3CN
и C6H5NO2 равны 41 и 46 соответственно; е находятся в интервале
от 36 до 40, ^-величины равны 71,3 и 67 ккал/молъ), а донорные
234
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
силы по отношению к катионному железу(Ш) — слабы. Донорные
свойства триэтилфосфата и ацетона, вероятно, сравнимы со свой-
свойствами приведенных выше растворителей, но триэтилфосфат и ацетон
являются более слабыми сольватирующими растворителями. В этих
шести растворителях FeClJ устойчив, и основными частицами, обра-
образующимися при прибавлении FeCl3 в малых количествах, являются
FeQ2S4 и FeCli, т. е. процессы протекают согласно двум первым
стадиям уравнения A1).
Диметилацетамид — гораздо лучший донор по отношению
к железу(Ш), чем указанные выше растворители, и лучший соль-
ватирующий растворитель, что подтверждено величиной Кассо!1
для R4NBr, но основными частицами при растворении FeCl3 в ди-
метилацетамиде являются все те же FeCl2S4H FeCli; FeClJ в растворе
диссоциирован незначительно. В ДМА, как и в большинстве на-
названных выше растворителей, присутствует, вероятно, некоторое
количество FeCl3S, но оно не было определено^
Донорные свойства N-метилацетамида несколько слабее, чем
ДМА, но сольватирующая способность больше вследствие образова-
образования водородной связи с хлоридом и высокой диэлектрической про-
проницаемости. Ион FeCli неустойчив в этом растворителе, и основными
частицами в разбавленных растворах FeCl3 при равновесии являются
комплексы катионного железа и СГ. В N-метилацетамиде равновесие,
описываемое уравнением A1), смещено в сторону диссоциации хло-
хлорида больше, чем в ДМА, вследствие более сильной сольватирующей
способности N-метилацетамида.
Представляет интерес сравнение ДМА и ДМСО. В последнем
растворителе основными частицами в растворе являются комплексы
катионного железа и хлор-иона; FeCli диссоциирует в этом раство-
растворителе. Донорные свойства ДМА и ДМСО сравнимы, хотя диметил-
-сульфоксид, вероятно, как донор несколько лучше. Константа ассо-
ассоциации для R4NBr, ^-величина и диэлектрическая проницаемость
показывают, что ДМСО является и лучшим сольватирующим рас-
растворителем, чем ДМА. Эти два свойства (особенно сольватирующая
способность) объясняют поведение FeCl3 и FeCli в ДМСО по сравне-
;нию с поведением в ДМА.
Метанол — хороший сольватирующий растворитель, если
возможны специфические взаимодействия, и лишь удовлетворитель-
удовлетворительный, когда эти взаимодействия незначительны. Он является сильным
донором по отношению к некоторым кислотам, например Ni2+, но
слабым донором по отношению к деформируемым кислотам, напри-
например к 12, (С2Н5KРЬС1 и т. д. Следовательно, его поведение как рас-
растворителя будет переменным. В растворах хлорного железа наблю-
наблюдается сильная координация к катионному желез у (III) и сольватация
хлор-иона, вследствие чего FeCli неустойчив. Катнонные частицы
железа и хлор-ион являются основными ионами в разбавленных
растворах хлорного железа в метаноле.
ЛИТЕРАТУРА
233-
Пиридин обладает сильными донорными свойствами, особенно-
по отношению к деформируемым кислотам, но является слабым соль-
сольватирующим растворителем. Основными • частицами, присутству-
присутствующими в растворе FeCli в пиридине являются FeCl3-Py и FeClJr
в растворе FeCl3 в пиридине — FeQ3-Py.
Результаты экспериментов с растворенным (Ph3AsCH3JCoCl4 мо-
могут быть объяснены аналогично. Основными ионами в CH3NO2,
как сообщают, является CoCl3S~. Структура CoCl3S~ неизвестна,,
предположительно считают ее тетраэдрической. В результате доба-
добавления СоС12 в CH3NO2 образуется неэлектропроводный раствор-
CoCl2-2CH3NO2.
Оба амида и диметилсульфоксид являются достаточно хорошими
основаниями и имеют сравнительно высокие диэлектрические про-
проницаемости для того, чтобы вытеснить С1~ из координационной сферы
металла. В результате образуются катионы CoS|+ и CoClSJ в до-
добавление к некоторому количеству CoQ3S~, СоС1|+ и С1". Устой-
Устойчивость CoClf" в этих растворителях уменьшается в следующем
ряду: СН2С12 > CH3NO2 > ДМА > ДМСО. Устойчивость FeCl4
уменьшается в той жа последовательности растворителей, что и для
CoClJ".
Очевидно, что с помощью координационной модели мы смогли
объяснить свойства большого числа неводных растворителей. Учи-
Учитывая сложную природу обсуждаемых процессов, можно утвер-
утверждать, что совпадение между предполагавшимися и наблюдаемыми
свойствами замечательно. Так, несомненно, получилось потому, что-
сравнивались сходные системы с близкой энтропией. Применение
термина «сольватирующий растворитель» основывалось на пред-
предпосылке, что уменьшение степени образования ионных пар связано-
с увеличением степени сольватации; спектроскопические методы,
применявшиеся для исследования диссоциации изучаемых систем,
нечувствительны к оценке степени образования ионных пар и поз-
позволяют установить главным образом количество и тип групп в коор-
координационной сфере металла.
Возможность описать наблюдаемые химические явления, рас-
рассмотренные выше, с позиций координационной модели является
сильнейшим аргументом в поддержку этой модели. Надеемся, что
дальнейшее расширение экспериментальных работ позволит принять
ее в качестве общего подхода.
ЛИТЕРАТУРА
1. Andrews L, .Т., Koefer R. M., Advances in Inorganic Chemistry
and Radiocbemistry, v. 3, New York, 1961, p. 91.
2. Audrieth L. F., Kleinberg J., Non-Aqueous Solvents, New
York, 1953, p. 125.
3. В а а г M., G u t m a n n V., Monatsh., 90, 426 A959).
4. Baaz M., Gut man n V., H u b n e r L., Mohatsh., 91, 537 A960).
236
ГЛАВА ПЯТАЯ. КООРДИНИРУЮЩИЕ РАСТВОРИТЕЛИ
ЛИТЕРАТУРА
237
9.
10.
И.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
Barnes J. С, Hume D. N., Inorg. Chem., 2, 444 A963).
В е a t t i e L. R., Webster M., J. Chem. Soc, 1963, 38.
Beilstein, Bielsteins Handbuch der organischen Chemie, Vierte Auflage,
Vierter Band, Erstes Teil, 1962, p. 124.
Benesi H. A., H i 1 d e b r a n d J. H., J. Am. Chem. Soc, 71, 2703
A949).
Bent H. A., Chem. Rev., 61, 275 A961).
Berns D. S., Fuoss R. M., J. Am. Chem. Soc, 83, 1321 A961).
Branden C, Lindquist
В u f f ag n i S., D u n n Т. М.
Bull W. E., M a dan S. K.,
A963).
Burwell R. L., Langford
A959).
Busby R.
I., Acta chem. scand., 14,
J. Chem. Soc, 1961, 5105.
Willis J. E., Inorg. Chem.,
726 A960).
2, 303
С. Н., J. Am. Chem. Soc, 81, 3799
Griffiths U. S., J. Chem. Soc, 1963, 902.
E., F i a t D. N., J. Chem. Phys., 39, 1349 A963).
~ J. Am. Chem. Soc, 82, 2986 A960).
J. Inorg. a. Nucl. Chem., 17, 62 A961).
D. M. L., Go о dg a me M., J. Am.
E.
Connick R.
Cotton F. A., Francis R
Cotton F. A., Fr an cis R.
Cotton F. A., Goodgame
Chem. Soc, 83, 4690 A961).
Dawson K. R., Belcher
С. А., 45, 10005g A951).
Dawson L. R., Golben M.,
Dawson L.R.,Wilhoit E.
J. Am. Chem. Soc, 79, 3004 A957).
Donoghue J. Т., Drago R.
Donoghue J. Т., Drago R.
Drago R.S.,Meek D. W., J.
R. L., Trans. Ky. Acad. Sci., 13, 129;
Am. Chem.
Holmes
Soc, 74, 4134 A952).
R. R., S e ars P. G.,
Inorg. Chem., 1, 866 A962).
Inorg. Chem., 2, 1158 A963).
Phys. Chem., 65, 1446 A961).
Drago R. S., Way land В., Carlson R. L., J. Am. Chem. Soc,
85, 3125 A963a).
Drago R. S., Carlson R. L., R о s e N. J., W e n z D. A., J. Am.
Chem. Soc, 83, 3572 A961).
Drago R. S., M e e к D. W. et al., Inorg. Chem., 2, 124 A963).
Dunn Т. М., Modern Coordination Chemistry, New York, 1960, p. 229.
G., С r u s e K., Z. phys. Chem., 201, 130 A952).
. P., Jr., J. Am. Chem. Soc, 84, 24 A962).
. P., Jr., Inorg. Chem., 2, 266 A963).
J. Am. Chem. Soc, 84, 1139 A962).
M., Glover К. Н., Trans. Farad. Soc, 51, 1418,
E 1 1 e n d t
Fackler J
Fackler J
Fine D. A.,
French С
A955).
Friedman
G a g n о u x
1322 A958).
G e r d i n g
Spectrochim.
Gill N. S.
G u t m a n n
G u t m a n n
N. J., Plane R. A., Inorg. Chem., 2, 11 A963).
P., Janjic D., Susz B. P., Helv. chim. acta,
H., Konigstein J. A., van der Worm E
acta, 16, 1881 A960).
N у h о 1 m R. S., J. Chem. Soc, 1959, 3997.
V., J. Phys. Chem., 63, 378 A959).
V., Baaz M., Monatsh., 90, 729 A959).
Holmes R. R., Gallagher W. P., Carter R.
Chem., 2, 437 A963).
H., Lemons J. F., Taube H., J.
1427
41,
R.,
Jackson J.
32, 553 A960).
Job P., Ann.
Joesten M.
Joesten M.
Joesten M.
J orgensen
P., Jr.,
Chem.
Inorg.
Phys.,
chim., [10], 9, 113 A928).
D., Thesis, University of
Illinois, 1962.
D., Drago R. S., J. Am. Chem. Soc, 84, 2037 A962).
D., Drago R. S., J. Am. Chem. Soc, 84, 3817 A962).
С. К., Acta chem. scand., 10, 887 A956).
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
К a t z i n L. I., J. Chem. Phys
Katzin L. I., Ferraro J
К a t z i n L. I., G e b e r t E.
Katzin L. I., G e b e r t E.
Katzin L. I., G e b e r t E.
Koch F. K. V., J. Chem. Soc,
Korinek G. J.,Schneider
E. M., J. Am. Chem. Soc.
E. M., J. Am. Chem. Soc.
M., J. Am. Chem. Soc.
H.
Williams R.
К о s о w e r
К о s о w e r
Kosower E.
Landesman
A961).
L a n g f о r d C. H., Langford P. O.,
Lester G. " ~ "
1076 A956).
Lester G. R., Vaughan
Sci., 19, 28 A958).
Lindquist I., Acta chem. scand.,
Lindquist L., Branden
20, 1165 A952).
R., J. Am. Chem. Soc, 72, .3451A950).
J. Am. Chem. Soc, 72, 5455 A950).
J. Am. Chem. Soc, 72, 5464 A950).
J. Am. Chem. Soc, 72, 5659 A950).
1930, 2385.
W. G., Can. J. Chem., 35, 1157 A957).
80, 3253 A958).
80, 3261 A958).
80, 3267 A958).
В., J. Am. Chem. Soc, 83, 2663
R., G о v e r T. A., Sears
J. W., Sears
Inorg. Chem., 1,
P. G., J. Phys.
184 A962).
Chem., 60,
P. G., Trans. Ky. Acad.
12, 135 A958).
С I., Acta cryst., 12, 692 A959).
Mack or E. L., Rec. trav. chim., 7~0, 457 A951).
de Maine P. A. D., Koubek E., J. Inorg. a. Nucl. Chem., 11,
329 A959).
¦ ¦ A. E., Diss. Abs., 22, 97 A961).
Thesis, University of Illinois, 1962, p. 60.
Drago R. S., J. Am. Chem. Soc, 83, 4322 A961).
D r a g о R. S., P i p e r T. S., Inorg. Chem., 1, 285 A962).
Straub D. K., Drago R. S., J. Am. Chem. Soc,
L. W., Can. J. Chem., 35, 251 A957).
Dawson L. R., Trans. Electro-
Marcinkowski
Meek D. W.,
Meek D. W.,
Meek D. W.,
Meek D. W.,
82, 6013 A960).
Mine S., Werblan L., Electrochim. acta, 7, 256 A962).
Newman L., H u m e D. N., J. Am. Chem. Soc, 79, 4571 A957).
О n а к Т. P., L a n d e s m a n H., et al., J. Phys. Chem., 67, 1533 A959).
Orgel L. В., An Introduction to Transition Metal Chemistry. London,
1960, p. 133.
Popov A. I., S к ell у N. E., J. Am. Chem. Soc, 76, 5309 A954).
Schmulbach С D., Drago R. S., J. Am. Chem. Soc, 82, 4484
A960).
Schneider W. G., Reeves
Sears P. G., Holmes R. R
chem. Soc, 102, 145 A955).
Sears P. G., Lester C. R.,
60, 1433 A956).
Sears P. G., McNeer
chem. Soc, 102, 269 A955).
Sears P. G., Wit ho it
60, 169 A956).
Shel don J. C, Tyree S. Y.
Shoolery J. N., Pi m e n t e1
Phys., 23, 1244 A955).
S о d e к H., F u о s s R. M., J. Am. Chem. Soc, 72, 301 A950).
Susz В. Р., С h al an don P., Helv. chim. acta, 61, 1332 A958).
Susz В., С. г., 248, 2569 A959).
Swine hart J. H., Taube H., J. Chem. Phys., 37, 1579 A962).
Tsubomura H., Lang R., J. Am. Chem. Soc, 83, 2085 A961).
Weissberger A., Proskauer E. S., Techniques of Organic
Chemistry, v. VII, New York, 1955.
W о 1 d b у е F., Acta chem. scand., 9, 299 A955).
Войтович В. А., Барабапова А. С, HfflX, 6, 2098 A961).
Dawson L. R., J
R. L., Dawson
E. D., Dawson
. Phys. Chem.,
Trans. Electro-
L. R.,
L. R., J. Phys. Chem.,
J. Am. Chem. Soc, 80, 4775
G. С, Н и g g i n s С. М., J.
A958).
Chem.
ГЛАВА ШЕСТАЯ
II. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
239
ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
т.вАДдингтоа
I. Основные физические свойства жидкого сернистого ангидрида . . . 238
II. Физические свойства растворов в жидком сернистом ангидриде . . . 239
A. Растворимость 239
Б. Сольватация 241
B. Электропроводность и ионизация 243.
Г. Электролиз 247
Д. Электродные потенциалы 248
III. Химические реакции в жидком сернистом ангидриде 249
A. Сольволиз 249
Б. Обменные реакции 250
B. «Амфотерные» реакции * . . 252
Г. Окислительно-восстановительные реакции 253
Д. Комплексообразование 255
IV. Реакции изотопного обмена в жидком сернистом ангидриде 256
V. Заключение 263
Литература 265
I. ОСНОВНЫЕ ФИЗИЧЕСКИЕ СВОЙСТВА ЖИДКОГО
СЕРНИСТОГО АНГИДРИДА
Свойства жидкого сернистого ангидрида как ионизирующего
растворителя начали подробно изучать в девятисотых годах, в числе
первых исследований были работы Вальдена 1(>5-110. Большая часть
исследований, посвященных этому растворителю и выполненных
до 1945 г., изложена в работах Яндера м и Аудрица и Клайнберга *.
Не так давно появился краткий обзор Элвинга и Марковица зъ,
а совсем недавно Личтин 70 сделал обзор работ по физической химии
растворов неорганических и органических соединений в сернистом
ангидриде. Нам представляется нецелесообразным подробно оста-
останавливаться на всех вопросах, поднятых в указанных обзорах,
мы коснемся их лишь бегло, за исключением тех случаев, когда
прежняя интерпретация вызывает сомнения. Но мы остановимся
более подробно на работах по изотопному обмену в сернистом анги-
ангидриде, столь существенных для понимания природы воздействия
этого растворителя на растворенное вещество, которые рассмотрены
более чем кратко.
Основные физические свойства жидкого сернистого ангидрида
приведены ниже:
Гпл = 197,64° К « р = 1,46 г/см* B63,Г К)'1
Гкип = 263,08° К «* и=C—4)-10~8 ом-1.
Д#пл= 1,9691 ккал/моль (при ГПлL4 ЛГк = 0,0393 град/моль
А#кип = 5,96 ккал/моль (при ТКИП) 44 ЛГЭ = 1>48 град/моль
Давление паров сернистого ангидрида 44:
243 253 273 283
284,8 530,6 1159,6 1714
Т, °К .....
Р, мм pm. ст. *
)
-1 «¦
293
2456
* Рассчитано по уравнению
lg/>=_ i8^?. _ о,О15865Г + 0,000015574Г 2 +13,07540
Вязкость жидкого сернистого ангидрида 73>115 в мпз п = 4,03—
'6,0363 t.
Диэлектрическая проницаемость жидкости 73> 89> 103 вычис-
вычисляется по уравнению е = 95,12 ехр (—6,676 • 103 Т), при 273,4° К
е = 15,4. Дипольный момент по Дебаю 69 при 256,9° К ц=1,62. Длина
•связи S—О равна 1,43 А66, угол66 О—S—О составляет 119,5°.
Жидкий сернистый ангидрид широко применяется как раствори-
растворитель, так как обладает большим температурным интервалом жидкого
•состояния. Давление паров его при 0° С достаточно низко, что делает
¦безопасной работу с ним в запаянных стеклянных ампулах; это
весьма важно, поскольку такая методика позволяет исключить
протекание побочных реакций вследствие загрязнений водой и кисло-
кислородом, усложняющих процесс.
II. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
В ЖИДКОМ СЕРНИСТОМ АНГИДРИДЕ
А. Растворимость
Соединения ковалентной природы растворяются в жидком сер-
сернистом ангидриде значительно лучше, чем соединения ионного типа.
Ниже приведены растворимости (в ммолъ/1000 г SO2) в жидком
SO2 при 0° С солей одновалентных неорганических катионов 45> ы:
sor
sor
F"
cr
Br"
r
SCN"
CN"
ClOj
ch3coo-
* При 50°
Li+|
1.55
23,0
2,82
6,0
1490.0
—
3,48
G.
Na+
1,37
н. p.
6,9
н. p.
1,36
1000
80,5
3,67
8,90
K+
1,58
н. p.
3,1
5,5
40,0
2490,0
502,0
2,62
0,61
1,27
—
—
27,2
P-
P-
—
NHJ
2,67
5,07
<27*
1,67
6,0
580,0
6160,0
—
2,14
141,0
T1+
4,96
0,417
н. p.
0,292
0,60
1,81
0,915
0,522
0,43
285,0
Ag
н. p
и. p.
н. p.
20,07
0,159
0,68
0,845
1,42
1,02
240
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
Нетрудно заметить, что лишь растворимости иодидов щелочных
металлов превышают значения 1 моль/1000 г SO2, а растворимости
большинства других солей составляют несколько миллимоль. Все
тетраметиламмонийгалогениды легко растворяются в жидком сер-
сернистом ангидриде, по-видимому, вследствие низкой энергии их
кристаллической решетки.
Растворимости (в ммоль/ЮОО г SO2) некоторых солей двух- и трех-
трехвалентных катионов в жидком SO3 при 0° С 45> 54:
П. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
241
Mg2 +
Ba2+
Zn2+
Cu2+
pjof2-t-
Pb2+
C02 +
Ni2+
A13 +
Sb3+
Bi3+
ci-
1,47
н. p.
11,75
н. p.
3,8
0,69
1,00
н. p.
x. p.
575,0
0,60
ВГ-
1,3
н.р.
¦—
2,06
0,328
—
—
0,60
21,8
3,44
I-
0,50
18,15
3,45
1,17
0.265
0,195
12,2
.—
5,64
0,26
SCN"
.
н. p.
40,4
__
0,632
0,371
н. p.
н. p.
Растворимость ВеС12 — 5,8; HgSO4 — 0,338; Hg(GNJ — 0,556;
Hg(GH3G00J - 2,98; PbS04 - н. p.; PbF3 - 2,16; Pb(GNJ -
0,386; Pb(CH3GOOJ - 2,46; Ni(GH3GOOJ - 0,08 ммоль/ЮОО г S0.2.
Можно видеть, что, за исключением хлоридов алюминия и сурьмы,
ковалентных по своей природе, растворимости большинства солей
не превышают нескольких ммолъ/1000 г SO2. Очень высокая раство-
растворимость в сернистом ангидриде ковалентных соединений хорошо
иллюстрируется на примере таких соединений, как бром, монохлорид
иода, тионилхлорид, тионилбромид, трихлорид бора, сероуглерод,
трихлорид фосфора, трихлорид мышьяка и оксихлорид фосфора,
смешивающихся с жидким SO3 в любых отношениях. Четыреххло-
ристый углерод, тетрахлорид кремния и другие тетрагалогениды
элементов четвертой группы полностью смешиваются с жидким
сернистым ангидридом выше критической температуры растворения,
которая для разных соединений различна 13~16.
В первых работах Вальдена и Центнерзвера 106-110 было показано,
что, за редким исключением, жидкий сернистый ангидрид является
прекрасным растворителем для органических соединений. Амины,
простые эфиры, сложные эфиры, спирты, сульфиды, меркаптаны
и кислоты как алифатические, так и ароматические хорошо раство-
растворяются в жидком сернистом ангидриде. Легко растворяются в SO2
ароматические углеводороды и олефины, в то время как парафины
обладают ограниченной растворимостью.
Хорошо растворяются галоидароматнческие и нитроаромати-
ческие соединения. На селективной растворимости ароматических
углеводородов в жидком сернистом ангидриде основан процесс
рафинирования керосина, разработанный Эделяну. Вода смеши-
смешивается с жидким сернистым ангидридом не полностыр; пределы рас-
растворимости ее при различных температурах точно никогда ие опре-
определялись. Виккерт П1>112 сообщил о существовании соединения
^О2-Н2О, которое остается в виде устойчивого остатка после испа-
испарения раствора воды в сернистом ангидриде при 0° С. Он указал
также, что растворимость воды в жидком сернистом ангидриде при
22° С составляет 2,3 г/100 г SO2. Измерения электропроводности
растворов воды указали на слабую растворимость ее при температу-
температурах ниже 0° С.
Б. Сольватация
Сернистый ангидрид образует устойчивые сольваты со многими
галогенидами щелочных металлов и другими веществами, подобные
кристаллическим гидратам и аммиакатам. Эти сольваты были по-
подробно изучены многими исследователями зв-4о,57,б8< g0 многих
случаях по давлению паров была определена теплота образования
аддуктов. Мольное отношение двуокиси серы к растворенному соеди-
соединению в аддукте обычно колеблется от 1 до 4, но иногда определения
дают значительно большие величины этого отношения. Основные
сведения о сольватах солей щелочных металлов приведены
в табл. 34.
Таблица 34. Температура и
Соль
Li I
Nal
NaNCS
NaCH3COO
KF
KBr
Kl
KNCS
KCH3COO
RbF
Rbl
теплота разложения
солей щелочных металлов с сернистым
п*
2
2
4
2
1
^96.97
4
4
0,5
1
2
1
196,97
3
4
'разл'
__,
15
5
—
^>8038>40
__
—1
6
-49
12,5
—
>8036
—
15,3
15,5
о. в
9,487
10,01s'
9,6337
10,538.40
—
—
8,3867.68
9,6737
11338,40
9^67.88
9,75*8.40
—
—
10.567.68
10.93'
Соль
RbNCS
RbCHsCOO
CsNCS
CsCH3COO
Csl
(CH3LNF
(CH3LNC1
(CH3LNBr
(CH3LNI
[(CH3LN]2SO4
еольват on
ангидридом
n
0,5
1
0,5
1
3
4
1
1
2
1
2
1
3
6
'разл.
31,5
^>5№6
19
>806
—
17
15096,97
88
35
41
16
2096,97
28
-2,6
D
10,6437
—
10,1437
10 2538.*
10,8937
—
11 167,68
10 657.68
8 9967.88
10,357'5S
—
11 767,68
8*5367.58
Примечание. Верхний индекс у цифр таблицы указывает литературный источ-
источник.
* Число молей SOs в сольвате на моль соли.
** При атмосферном давлении.
16 Заказ 90
242
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
Первыми исследователями фактически не были получены полные
фазовые диаграммы систем MX—SO2, и вполне возможно, что в этих
системах имеется большое число еще не определенных сольватных
форм. Моноаддукты сернистого ангидрида с тетраметиламмоний-
галогенидами, по-видимому, лучше рассматривать как галогенсуль-
/0-
финаты, структуру ионов которых можно записать как X—S
хЧо
(сравните со структурой галогенсульфонатов). Стойкость их умень-
уменьшается в ряду F^>Cl>Br> I. He выяснено, каким образом
координированы молекулы S02 в полисольватах галогенидов щелоч-
щелочных металлов: к катиону, аниону, либо к обоим ионам одновременно.
Но очевидное возрастание устойчивости аддуктов иодидов щелочных
металлов по сравнению с устойчивостью соответствующих аддуктов
бромидов, а также отсутствие сольватов хлоридов щелочных метал-
металлов (ряд устойчивости в зтом случае обратный по отношению к ряду
для моносольватов с тетраметиламмонийгалогенидами) заставляет
предположить, что связь в этих сольватах возникает за счет переноса
заряда от аниона соли к молекулам сернистого ангидрида. Эта точка
зрения подтверждена спектроскопическим анализом соединений
KI -4SO2 и KNCS-4SO2, Для которых смещение частот колебаний S02
объясняется переносом заряда от отрицательного иона 80.
С ковалентными соединениями сернистый ангидрид образует
многочисленные сольваты, в большинстве случаев являясь акцепто-
акцептором электронов (через атом серы) и лишь в нескольких случаях —
донором электронов (через атом кислорода). Так, фазовая диаграмма
BF3—S02 указывает17 на образование аддукта SO2-BF3. Любо-
Любопытно, что в системе ВС13—S02 в отличие от системы BF3—S03
при низкой температуре образуются два несмешивающихся раствора,
которые, однако, при комнатной температуре становятся полностью
смешивающимися 81>116.
Имеется сообщение б об аддукте S02 с пентахлоридом сурьмы
SO2-SbCl5. Бромид олова и хлорид титана образуют с S02 аддукты,
которым приписывают14 формулы SnBr4-SO2 и (TiCl4J • S02. Тетра-
хлорид циркония образует 16 с S02 аддукт 1:1.
Имеется очень давнее сообщение о существовании аддуктов
(A1C13J-SO2 и A1C13-SO210, но последующие исследования22'101
подтвердили существование лишь A1C13-SO2; по-видимому, это
соединение димерно.
Большинство органических моноаминов образует с сернистым
ангидридом устойчивые моноаддукты. Эти соединения обычно
интенсивно окрашены и хорошо растворимы в жидком сер-
сернистом ангидриде1'9-19-21> 26-40> 49> 50'54> 67> 84~86. Координация
в них, по-видимому, осуществляется через атом азота к атому
серы. Диамины могут присоединять две молекулы S02;
так, ге-фенилендиамин образует re-C6H4(NH2J • 2SO2 50' 84,
II. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
243
а ]Ч,1Ч,]Ч',1Ч'-тетраметил-?г-фенилендиамин образует соединение
W-C6H4[N(CH3J]2-2SO2 26. Утверждение Яндера и Виккерта 62> пз
о том, что растворение триэтиламина в сернистом ангидриде проис-
происходит с образованием следующих соединений:
2(C2H5KN + 2SO2 >¦ 2(C2H5KN • S02 >
I красный
—> [(C2H6KN.SO212 —> [(C2H5KN]2SO2+SO!- A)
II белый
подверглось резкой критике со стороны Бейтмена и др. 9. Эти
авторы нашли, что выделенное Яндером и Виккертом соединение II
в действительности является кислым триэтиламмонийсульфитом
(C,H6KNH+HSO3, образующимся под Действием влаги на красное
соединение I, и что температура плавления кислого сульфита G4—
75° С) хорошо согласуется с температурой плавления, сообщенной
Яндером для соединения II.
Выводы Бейтмена 9 были подтверждены работами других иссле-
исследователей 21> 49> 50. По сообщению Яндера 56' 61, аммиак подобным же
образом образует в жидком сернистом ангидриде соединение
(H3NJSO2+SO3~, но вполне возможно, что и в этом случае наблюда-
наблюдалось либо образование NH4HSO3 из-за присутствия влаги в S02,
либо образование кислоты HSO2NH2.
Окись триэтиламина образует с жидким S02 устойчивый
аддукт (C2H6KNO- S02 68. Структура его может быть представлена
следующим образом:
С2Н6
I Л
С2Н5 —N = O~>S<f
I ^0
с2н5
Несмотря на то что из величин растворимости спиртов и фенолов
в жидком сернистом ангидриде можно сделать заключение о наличии
взаимодействия между этими органическими соединениями и сер-
сернистым ангидридом, фазовые диаграммы этих систем не изучены.
Имеется лишь отдельйое сообщение об образовании неустойчивого
аддукта между фенолом и сернистым ангидридом114. Изучение
УФ-спектров показало, что при взаимодействии сернистого анги-
ангидрида с простыми эфирами и спиртами 33 образуются комплексы
с переносом заряда. Такие комплексы образуются также между
сернистым ангидридом и различными ароматическими моле-
молекулами 2> 3' 78.
В. Электропроводность и ионизация
Содержание большинства работ, посвященных растворам элек-
электролитов в жидком сернистом ангидриде, изложено в обзо-
обзорах *' 35, 54, 70. почти Весь рассмотренный экспериментальный
материал относится к растворам одно-одновалентных электролитов.
16*
244
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
II. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
Жидкий сернистый ангидрид — растворитель с низкой диэлектри-
диэлектрической проницаемостью, поэтому он относится к первому типу рас-
растворителей, описанному Краусом и Фуоссом 43, и на кривой зависи-
зависимости эквивалентной электропроводности раствора от концентра-
концентрации растворенного вещества имеется минимум. В области концентра-
концентраций выше 10 м электропроводность раствора обусловлена главным
образом ионными тройниками. При концентрации приблизительно
10 м наблюдается минимум электропроводности, а ниже этой
концентрации электропроводность возрастает с уменьшением кон-
концентрации, подчиняясь закону разбавления Оствальда. Электро-
Электропроводность в области концентраций менее 10~2 м лишь в незначи-
незначительной степени осложняется образованием ионных тройников,
поэтому в этой области представляется возможным оценить кон-
константы диссоциации ионных пар.
Личтин 70 применил уравнение Шедловского 42 для расчета сте-
степени диссоциации ионных пар в жидком сернистом ангидриде. По-
Подробно с методом расчета можно ознакомиться в главе «Ионизация
и равновесная диссоциация в растворах жидкого сернистого анги-
ангидрида» в книге Личтина70. Суть метода заключается в том, что данные
об электропроводности, нанесенные на график в соответствующих
координатах, представляют собой преобразованное уравнение
Шедловского — прямую линию 32. При этом отрезок, отсекаемый на
оси ординат, дает значение 1/Л0, а наклон прямой равен 1/КА1
(где К — константа диссоциации ионной пары, а Ло — эквивалент-
эквивалентная электропроводность при бесконечном разбавлении). В книге
Личтина, откуда заимствована табл. 35, использована теория ионной
ассоциации Бьеррума п> 12 (для расчета расстояний наибольшего
сближения а в ионных парах). Эти расстояния сравниваются
в табл. 35 с суммой кристаллических радиусов ионов г+ -\- г_ .
Таблица 35. Константы для растворов солей,
состоящих из сферических и тетраэдрических ионов,
в жидком сернистом ангидриде при 0° С
Ионная пара
LiBr
NaBr
КС1
КВг
KI
(CHgkNCl
(CH3LNBr
(CH3LNI
(CHSLNC1O4
(CH3LNBF4
(C2H5LNBr
(C2H5LNI
Ao.
CJK2 / («Ж- МОЛЬ)
189
265
243
249
244
243
236
234
218
215
215
197
0,27
0,48
0,74
1,43
¦ 3,0
10,3
11,8
13,9
8,4
7,9
21
39
о, к
2,70
2,87
2,96
3,28
3,58
4,96
5,25
5,54
4,63
4,56
6,8
10,0
Г+ + Г-.А
2,55
2,91
3,14
3,28
3,50
5,11
5,25
5,47
6,3
6,1
6,6
8,1
Литера-
Литература
75
72
73
73
73
70
73
74
74
74
74
74
Значения предельных электропроводностей растворов солей ука-
указывают на то, что в сернистОхМ ангидриде, как в воде и многих других
растворителях, должен осуществляться значительный гидродинами-
гидродинамический перенос растворителя ионом лития. Представляет, однако,
о
интерес, что в этом растворе, как это видно из значения расстояния а,
по-видимому, происходит лишь очень незначительная, а возможно
и совершенно отсутствует, диссоциация ионных пар.
Личтин указывал, что лишь для одного случая, в частности,
для раствора бромида калия 73> 74> 7б имеется достаточно данных
для расчета константы диссоциации ионной пары во всем интервале
температур. Из табл. 36, также взятой из книги Личтина, видно,
что а меняется очень незначительно с изменением температуры
от —24 до +6° С и хорошо совпадает с суммой ионных радиусов
C,28 А). Кривая зависимости lg Kmcc от 1/Г представляет собой
прямую линию. Отсюда находят Д#дисс — 5,25 ккал/молъ и Д5дисс =
= —36,8 ккал/(моль-град) при 0° С. Уменьшение К с ростом темпе-
температуры объясняется зависимостью диэлектрической проницаемости
растворителя от температуры. Большое отрицательное значение ДЗ'дисс
указывает на гораздо большее взаимодействие растворителя со сво-
свободными ионами, чем с ионной парой. Увеличение предельной экви-
эквивалентной электропроводности с повышением температуры, по-
видимому, связано с уменьшением вязкости растворителя.
Таблица 36. Зависимость К, Ло п а для растворов КВг
в сернистом ангидриде от температуры 70
t, "С
-24,99
-20,58
—15,56
-10,71
л
о
4!
188
202
212
224
О
3,62
2,88
2,51
2,11
а, А
3,41 ±0,0374
3,34+0,0274
3,33 ±0,0374
3,31+0,0374
¦ *, °с
—8,93
—5,25
+0,12
+6,23
о
Si
3
< 3
228
233
249
274
О
п.
1,99
1,80
1,43
1,04
а, А
3,28"з
3,31±0,0271
3,2873
3,23 + 0,02'6
Данные об электропроводности растворов, полученные исследо-
исследователями до Личтина, и их интерпретация авторами, конечно", не
столь надежны, как данные Личтина и сотр. Однако Дутуа и Жир 34
в 1909 г., воспользовавшись простым законом разбавления Ост-
Оствальда, получили значения КДИСС и предельных электропроводностей
одного порядка со значениями, полученными Личтиным с помощью
246
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
более сложного уравнения Шедловского. Некоторые из этих резуль-
результатов мы приводим ниже (при 15° С):
Соль . . . RbBr KBr NH4Br (CH3LNBr Rbl KI NH4I (CH3LNI
A'-103 . . 0,34 0,35 0,16 1,37 0,50 0,57 0,47 1,6S
Предельные
температуре:
I-
Вг"
SCN"
эквнвалентные электропроводности при той же
(CHs).N+
199
194
215
211
NHi
208
208
К+
207
203
-174
Эти данные относятся к диссоциации в жидком сернистом анги-
ангидриде ионных пар соединений, которые в твердом состоянии являются
ионными. Однако имеется и большое количество веществ (например,
трифенилметилхлорид105), ковалентных в твердом состоянии и во
многих обычных растворителях, но образующих электропроводные
растворы в жидком сернистом ангидриде.
Эбулиоскопическим методом был измерен коэффициент Вант-
Гоффа i для растворов ряда соединений в жидком сернистом анги-
ангидриде 54' 59. Коэффициент Вант-Гоффа вычислялся как частное от
деления теоретического молекулярного веса на наблюдаемый моле-
молекулярный вес при условии, что мольная збулиоскопическая постоян-
постоянная равна 1,45 град/моль. Для этих опытов необходимо было иметь
довольно концентрированные растворы, в которых для электролитов
следует ожидать значительного образования ионных тройников.
Зависимость коэффициентов Вант-Гоффа для некоторых соедине-
соединений от концентрации приведена ниже 54> б9:
Разбавление, л/моль 2
Коэффициент Вант-Гоффа для соединений
толуол 1,07
нафталин 0,99
ацетанилид 0,99
со-хлорацетофенон 1,05
тетраэтилмочевина 1,08
ацетилхлорпд 0,83
SbCl3 —
SbCl5 0,91
SnCU —
Н2О 0,62
KSCN 0,50
KBr 0,55
KI 0,63
KSbCle 1,23
(CH3LNC1 1,14
(CH3LNC1O4 1,04
[(CH3LN]2SO4 1,03
PhCCl 0,99
16
32
¦—
—
—
—
—
.—
—
—
0,61
0,67
0,77
1,25
1,05
1,04
102
1,14
1,03
1,00
1,03
1,01
0,75
1,09
0,88
1,00
0,64
0,70
0,85
0,95
1,28
1,03
1,10
1,05
1,26
—
—
—
—
—
—
0,78
0,95
1,08
1,36
1,06
1,26
1,27
1,30
1,01
1,03
0,89
0,90
—
1,11
.
.—
—.
1,01
1,17
1,42
1,22
1,33
1,47
—
И. ФИЗИЧЕСКИЕ СВОЙСТВА РАСТВОРОВ
247
Для всех неэлектролитов, за исключением воды, i в пределах
ошибки эксперимента равен единице. Для бинарных электролитов
значения коэффициентов i находятся в пределах от 0,4 (в очень
концентрированных растворах) до несколько больше 1 (в более
разбавленных растворах), что указывает на большое число различных
ассоциированных частиц.
Г. Электролиз
В ряде работ, результаты которых часто весьма противоречивы,
рассмотрена природа продуктов, образующихся при электролизе
растворов солей в жидком сернистом ангидриде. Впервые продукты
электролиза в жидком сернистом ангидриде изучил Стил 102. Он
сообщил, что в процессе электролиза растворов иодида калия или
натрия в сернистом ангидриде с платиновыми электродами на катоде
выделяется сера, а сила тока, которая вначале очень велика, быстро
падает. Исследования Багстера и Стила 7> 8 показали, что при элек-
электролизе многих иодидов на катоде выделяется не только элементар-
элементарная сера, но и сульфит, а на платиновом аноде — свободный иод.
Металлические аноды, например цинковые и железные, переходят
в раствор в виде иодидов.
Центнерзвер и Друкер 29 получили (при изучении электролиза
иодидов в сернистом ангидриде с платиновыми электродами) резуль-
результаты, отличающиеся от результатов работы Багстера и Стила. Полу-
Полученный ими на катоде осадок имел сильные восстановительные свой-
свойства. Тщательный химический анализ показал, что осадок пред-
представлял собой смесь тиосульфитов и пиросульфитов. Элементарная
сера обнаружена не была.
Этот вывод подтвердился в работе Кейди и Тофта 28, изучавших
электролиз растворов иодида тиоцианата, иодата, хлората п ферри-
цианида калия в жидком сернистом ангидриде. В числе обнаружен-
обнаруженных ими катодных продуктов тиосульфиты, пиросульфиты, соли
тионовой кислоты и др. Наличие среди катодных продуктов, най-
найденных Багстером и Стилом, серы Кепдп и Тофт объяснили при-
примесью воды в сернистом ангидриде.
При электролизе раствора трифеншшетилбромида в жидком
сернистом ангидриде 92 на катоде осаждается трифенилметан. По-
Поскольку в жидком сернистом ангидриде Ph3GBr диссоциирует на
Ph3G+ и Вг", трифенилметан образуется, по-видимому, в результате
обычной нейтрализации катиона Ph3G+ на катоде.
Багстер и Кулинг 6, проводя электролиз смеси воды п броми-
бромистого водорода с сернистым ангидридом, доказали существование
иона гидроксония. Несмотря на то что ни вода, ни бромистый водород
не проводят электрический ток, в сернистом ангидриде смесь их
образует электропроводный раствор. Катодными продуктами элек- ,
тролиза были вода и водород, а анодным продуктом — бром. Кроме
248
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
того, количество выделившейся на катоде воды соответствовало
рассчитанному по закону Фарадея.
При электролизе растворов брома, монобромида иода и трихло-
рида иода в жидком сернистом ангидриде с серебряными катодами 20
на аноде выделяются соответственно бромид, иодид и хлорид се-
серебра. Катодные продукты идентифицированы не были. ,
Д. Электродные потенциалы
Впервые электродные потенциалы в жидком сернистом ангидриде
измерили Багстер и Стил 7> 8. Они получили воспроизводимый потен-
потенциал для таких ячеек, как Zn| ZnBr2 (насыщ.), SO2, Hg2Cl2|Hg.
Присутствие солей, состоящих из обычных ионов, оказывало
такое же влияние на эти потенциалы, как и на соответствующие
потенциалы ячеек с водной средой. Потенциалы замерялись ква-
квадрантным электрометром, поскольку было найдено, что электроды
чрезвычайно легко поляризуются.
Авторы измерили также потенциалы трех металлов — цинка,
кадмия и свинца — в насыщенных растворах их солей относительно
Hg/Hg2Cl2-электрода. Виккерт 1П>112 измерил потенциалы водород-
водородного (Е = 0,3 в) и кислородного (Е = 0,2 в) электродов относительно
Н^/Н^2С12-электрода, использовав в качестве электролита раствор
хлористого водорода произвольной концентрации в жидком сер-
сернистом ангидриде. Эти значения потенциалов одного порядка с полу-
полученными позднее Крузе 31. Последний провел безусловно более
тщательные измерения электродных потенциалов в жидком сернистом
ангидриде. Он определил э. д. с. следующих ячеек:
РЬ, РЬС12 | СГ(в SO2) | Hg2Cl2, Hg
Ag, AgCl | СР(в SO2) I HgaCh, Hg
Ag, AgBr | Br"(B SO2) | Hg2Br2, Hg
В этих ячейках протекают реакции
а) РЬ (тв) + Hg2Cl2 (тв) = РЬС12 (тв) + 2Hg (ж)
б) 2Ag (тв) + Hg2Cl2 (тв) = 2AgCl (тв) +2Hg (ж)
в) 2Ag (тв) + Hg2Br2 (тв) = 2AgBr (тв) +2H^)
III. ХИМИЧЕСКИЕ РЕАКЦИИ В СЕРДИСТОМ АНГИДРИДЕ
249
Эти реакции не зависят, конечно, от концентрации галоген-иона
в сернистом ангидриде и от природы растворителя; для расчета
э. д. с. требуется лишь знание термодинамических данных. Крузе
обнаружил, что в действительности рассчитанные им величины
э. д. с. не очень хорошо совпадают с измеренными, хотя порядок
величин сохраняется:
для ячейки аЕази = 0,36 в, Е°рлсч = 0,55 в;
для ячейки б в начальный момент реакции Е0ЛЗк = 0,04 в; Е%жч =
= 0,049 в;
для ячейки в в начальный момент Е»т = 0,06 в; 2??асч = 0,048 в.
Э. Д. с. в ячейках бив постоянно изменялась во времени,
а в ячейке а она оставалась относительно постоянной. Крузе по-
построил также ячейки
Ag, AgBr | НВг (в SO2) | Н2, Pt
Ag, AgCl | HC1 (в SO2) | H2, Pt
и измерил их э. д. с. при различных концентрациях галогеноводо-
рода. Как и следовало ожидать, получилась линейная зависимость
между э. д. с. и концентрацией галогеноводорода. Однако абсолют-
абсолютные значения э. д. с. расходились со значениями, рассчитанными
термодинамически, и превышали их на 0,2 в.
III. ХИМИЧЕСКИЕ РЕАКЦИИ В ЖИДКОМ]
СЕРНИСТОМ АНГИДРИДЕ
А. Сольволиз
Дпэтилцинк реагирует с жидким сернистым ангидридом уже
при —78° С с образованием диэтилсульфоксида и окиси цинка 1П.112:
B)
Яндер ы сообщил, что при длительном хранении в запаянной
трубке нри комнатной температуре бромиды и иодиды щелочных
металлов медленно реагируют с жидким сернистым ангидридом.
Анализ осадка, выпавшего из раствора иодида калия, показал, что
осадок представляет собой эквимолярную смесь сульфата калия
и серы; в растворе наблюдалось образование иода. В растворе бро-
бромида калия в этих условиях образуется бром и монобромид серы,
в осадке — только сульфат калия. Яндер предложил следующий
механизм сольволиза бромида:
C)
8
4 SOBr2=2SO2
4
= 2K2SO4 + S2Br2-j-Br2
По-видимому, в растворе иодида протекают аналогичные реак-
реакции, за исключением того, что S2I2 нестоек и разлагается на серу
и иод. Таким образом, суммарная реакция сольволиза иодида может
быть записана как
4KI+4SO2=2K2SO4; +2Sj +2I2 D)
Личтин 7в подверг сомнению результаты, полученные Яндером,
и сообщил, что разбавленные растворы бромидов и иодидов вполне
устойчивы в жидком SO2, предварительно дегазованном (с целью
удаления кислорода), а неустойчивость, по крайней мере для рас-
растворов иодидов, объясняется присутствием кислорода. По сообщению
250
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
Яндера bi растворы ацетатов щелочных металлов в жидком сернистом
ангидриде подвергаются сольволизу даже при —50° С. В результате
реакции, по-видимому, образуются сульфит металла и уксусный
ангидрид, которые Яндер рассматривал как продукты разложения
промежуточного тионилацетата.
Летучпе галогениды в жидком сернистом ангидриде ведут себя
по-разному. Галогениды элементов IV группы периодической си-
системы элементов с SO2 не реагируют. Пентахлорид фосфора легко
сольволизуется даже при —50° С, образуя оксихлорид фосфора
и тионилхлорид:
— ¦ "- E)
Аналогично реагирует с SO2 пентабромид фосфора:
F)
Далее реакция сольволпза не протекает. Ни трихлорид сурьмы,
ни пентахлорид сурьмы не подвергаются сольволизу под действием
SO2- Яндер отмечал, что, тогда как реакция E) экзотермична, реак-
реакции дальнейшего сольволиза РОС13
2PC15
и сольволпза SbCl.
>=(P2O5)+5SOCl2
= (SbOCl)-bSOCl2
G)
(8)
должны быть эндотермичны.
Укажем еще на ряд реакций сольволиза, протекающих в рас-
растворах в SO2:
= NbOCl8+SOCl2
12| +2SOC12
(9)
A0)
A1)
Б. Обменные реакции
Большое число реакций в жидком сернистом ангидриде, назван-
названных в некоторых случаях в книге Яндера 54 «реакциями нейтрали-
нейтрализации», но которые, по-видимому, правильнее называть обменными
реакциями, было исследовано Яндером и сотр. 57> 58> 61> 62. Ими
было обнаружено, что сульфиты реагируют с тионилгалогенидами
в SO2, образуя галогениды и сернистый ангидрид:
Cs2 S O3 + S OC12 = 2CsCl + 2 S O2
A2)
A3)
111. ХИМИЧЕСКИЕ РЕАКЦИИ В СЕРНИСТОМ АНГИДРИДЕ
251
Так же реагируют ацетаты:
-f SO(CH3CO2J
j + SO(CH;iCO2J
A4)
A5)
Выделить SO(GH3GO2J не удалось. По-видимому, он разлагается
на уксусный ангидрид и двуокись серы. Но Яндер выделил и про-
проанализировал соединения (С6Н5СН2СО2JЗО и (C1CH2CO2JSO.
О 0,5 Г,0 1,5
Мольное отношение
[S2O|]:[SO(SCNJ]
Рис. 50. Кривая кондуктометричес-
кондуктометрического титрования раствора SO(SCNJ
в сернистом ангидриде сульфитом
калия (ионами S3O62") *.
О 0,5 1,0 1,5 2,0
Мольное отношение
[(CH3LN]2SO2]:[SOCI2j
Рис. 51. Кривая кондуктометрического
титрования раствора SOC12 в серни-
сернистом ангидриде тетраметиламмоний-
сульфитом 62.
Известны и другие обменные реакции:
2NH4SCN+SOC12 = 2NH4C1
4KI+2SOC12 = 4KC1 | +2(SOI2)
2I2-bS \ +SO2
A6)
A7)
Соединение SO(SCNJ устойчиво в разбавленных растворах
в жидком сернистом ангидриде при —15° С, из концентрированных
растворов осаждается аморфное полицпаннстое соединение. Однако
Яндер использовал растворы SO(SCNJ для осуществления таких
реакций, как
A8)
и в процессе кондуктометрического титрования получил конечную
точку при отношении компонентов 1 : 1 (рис. 50) *. Таким же обра-
образом с помощью кондуктометрического метода можно изучить реакцию
сульфита и тионилхлорида (рис. 51).
* На этом и всех последующих рисунках этой главы электропроводность Л
выражена в произвольных единицах. — Прим. ред.
252
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
В. «Амфотерные» реакции Б5-58,в3
Если к раствору хлорида алюминия в жидком сернистом анги-
ангидриде приливать раствор сульфита тетраметиламмония, то образуется
объемистый белый осадок сульфита алюминия. Вероятно, в этом
случае протекает обменная реакция
2AlCl3-|-3[(CHsLNl2SOs=Al2(SOsM + 6(CH3LNCl
[(CH3LN]3A1 (SO3K в растворе
A9)
О 1,5 3,0 Ь,5
Мольное отношение
[к)*]:[А1С1з]
Рис. 52. Кривая кондуктометра-
ческого титрования раствора А1С13
в сернистом ангидриде тетраметил-
аммонийсульфитом ео.
Рис. 53. Кривая кондуктометрического
титрования смеси [(CH3LN]2SO3 и
[(CH3LN]3A1(SO3K в сернистом ангидриде
тионилхлорпдом во. По осп абсцисс отло-
отложено число молей SOC12, добавленных к
смеси 1 моль [(CHgLN]2SO3 + 2 моль
[(CH3LN]3A1(SO3K.
При дальнейшем добавлении сульфита тетраметиламмония осадок
растворяется:
Ali(SOs)8+3t(CH8LN]iSO8=2[(CHeLN]8Al(SO8)8 B0)
По-видимому, в растворе образуется комплексный аннон. При доба-
добавлении к этому раствору тпонилхлорида вновь выпадает осадок
сульфита алюминия:
2[(CHsLN]3Al(SO3)s + 3SOCl2 = Al2(SO3)sJ. + 6(CH3LNCl+3SO2 B1)
Яндер изучал эти реакции методом кондуктометрического титрования
(рис. 52 и 53).
Аналогично ведет себя в сернистом ангидриде треххлорпстып
галлий. При добавлении к его раствору сульфита тетраметиламмония
образуется осадок
B2)
•!• старение
Ga2O3-zSO2 I
III. ХИМИЧЕСКИЕ РЕАКЦИИ В СЕРНИСТОМ АНГИДРИДЕ
253
который растворяется при избытке сульфита тетраметиламмония
в растворе. Так же реагирует в сернистом ангидриде и хлорид олова.
Сначала из раствора осаждается сольват двуокиси олова SnO2-a;SO2
или сульфит олова, который растворяется в избытке суль-
сульфита тетраметиламмония, образуя раствор ортосульфитстанната
[(CH3LN]4Sn(SO3L.
.По сообщению Яндера 64, при добавлении к раствору РС13
в жидком сернистом ангидриде, который сам по себе стоек и бес-
бесцветен, сульфита тетраметиламмония при
—40° С выпадает хлопьевидный осадок
трехокиси фосфора:
P2O3j + 3SO2 +
+ 6(CH3LNC1 B3)
Дальнейшее добавление сульфита вы-
лвает растворение осадка, а из полу-
полученного раствора было выделено соеди-
соединение [(CH3LN]PO2SO2:
(Появление
желтой
окраски
2 4 в в
Мольное отношение
B4)
На кривой кондуктометрического титро-
титрования наблюдаются две конечные точки:
Рис. 54. Кривая кондукто-
кондуктометрического титрования
» раствора РС1» в сернистом
первая при мольном отношении сульфита ангидриде тетраметиламмо-
к РС13, равном 3 : 2, а вторая при отно- нийсульфитом.'
шении 4 : 2 (рис. 54).
Сольваты трехокиси и пентаокиси сурьмы в жидком сернистом
ангидриде также проявляют амфотерные свойства. Яндер и Гехт ъь
пытались показать, что аналогично ведут себя металлы в SOa. Они
обнаружили, что металлические бериллий, алюминий, галлий,
сурьма и свинец не реагируют с сульфитом тетраметиламмония
в жидком сернистом ангидриде, но олово вступает в реакцию. Если
олово и сульфит тетраметиламмония присутствуют в растворе в экви-
молярном соотношении, то выпадает осадок сольватированной окиси
олова:
[(CH3LN]2SO3+(z + l)SO2+Sn=SnO2-a:SCM +[(CH3LN]2S2O3 B5)
В избытке [(СН3L N]2SO3 протекает реакция
Sn-b2[(CH3LN]2SO3+3SO2=[(CH3LN]2Sn(SO3K + [(CH3LN]2S2O3 B6)
Г. Окислительно-восстановительные реакции
Исследован ряд окислительно-восстановительных реакций в жид-
жидком сернистом ангидриде. Сернистый ангидрид при этом обычно
играет роль инертной среды. Сульфит тетраметиламмония
254
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
в сернистом ангидриде быстро окисляется иодом до сульфата б6. Рас-
Раствор хлорного железа количественно окисляет иодид калия до иода:
2FeCl2 + 2KI = 2FeCl2+2KCl + I2 B7)
Точно так же ведет себя по отношению к иодиду калия пента-
хлорид сурьмы:
-------- =3l2-f6KCl+3SbCl3 B8)
Хлорид сурьмы, образующийся по реак-
реакции B8), реагирует далее с КС1 и полу-
получается осадок K3SbCl6:
6KCl+2SbCl3=2K3SbCl6| B9)
Однако добавление избытка пента-
хлорида сурьмы вызывает растворение
осадка с разложением комплексного иона
6 и образованием гексахлорантимоната:
2K3SbCle + 6SbCl5=6KSbCl6+2SbCl3 C0)
На рис. 55 показана кривая кондук-
тометрического титрования раствора
иодида калия в сернистом ангидриде
пентахлоридом сурьмы. На кривой видны
перегибы при мольных отношениях
[SbCl5] : [KI], равных 1 : 2, что соответствует уравнению B8),
и 3 : 2, что соответствует уравнению
г j <¦ s
Мольное отношение
[SbCI5] : [2KI]
Рис. 55. Кривая кондукто-
метрического титрования
раствора KI в сернистом
ангидриде пентахлоридом
сурьмы в0.
2KI+3SbCl6=I2+2KSbCl6 + SbCl3
C1)
являющемуся суммарным уравнением реакций B8), B9) и C0).
Недавно были изучены 98)" реакции нитрозил-соединений с иоди-
дами калия и тетраметиламмония в жидком сернистом ангидриде.
Все нитрозил-соединения, начиная от нитрозилхлорида и кончая
нитрозилфторборатом, реагируют согласно уравнению
C2)
Реакция с этплнитритом (нитрозилэтплатом) протекает по уравнению
C3)
С азидами нитрозил-соединения реагируют следующим образом:
и
C4)
C5)
III. ХИМИЧЕСКИЕ РЕАКЦИИ В СЕРНИСТОМ АНГИДРИДЕ
255
Д. Комплексообразование
В нредыдущем разделе упоминалось об образовании комплексов
K3SbCl6 и KSbCl6. Описаны способ получения и свойства ряда гекса-
хлорантимонатных комплексов в жидком сернистом ангидриде 93~96.
При титровании NOG1, СН3СОС1 и PhCOCl раствором SbCl6 в жид-
жидком SO2 получены кондук"тометрические кривые с резким перегибом
яри мольном отношении компонентов 1 : 1. Из растворов были вы-
выделены соединения NO+SbCle, GH3GO+SbGli и PhGO+SbGle- Ниже
приведены 94>95 значения мольных электропроводностей некоторых
комплексов с SbCl6 в сернистом ангидриде при разбавлении 50 л/моль:
Комплексное
соединение . . KSbCl6 (CH3CO)SbCl6 (RhCO)SbCl6 (NO)SbCl6 SOCl2+2SbCl5
Л, см^/(ом-моль)
при 0°С . . 92,8 80,5 71,5 67,5 0,4
при —70° С 49,0 45,3 37,8 50,0 0,7
Данные об электропроводности раствора SOG12 и SbCl6 в SO2
т,ребуют некоторых комментариев. Сил, как и Яндер 66, считает,
что в этом растворе существует комплекс SO2+(SbCleJ. Однако
данные об электропроводности 79 свидетельствуют о том, что един-
единственным аддуктом тионилхлорида с пентахлоридом сурьмы
является SOGl2-SbCl5. Нет никаких сведений о структуре этого
соединения, к его формуле SOCl+SbCle следует относиться с большой
осторожностью в связи с приведенными выше данными об электро-
электропроводности, а также в связи с тем, что рентгенографические иссле-
исследования 48 структуры аналогичного соединения селена SeOGl2-SbCl
указывают на ковалентную координацию атомов селена по отноше-
отношению к атомам сурьмы через кислород (Gl2SeO -»- SbCl6).
Сил и др. 10° получили комплекс NO2SbGli в жидком сернистом
ангидриде по реакции взаимодействия нитрилхлорида и пентахло-
рида сурьмы. Электропроводность растворов этого соединения
в жидком сернистом ангидриде того же порядка, что и у аналогичных
растворов NOSbCl6 и KSbCl6. При взаимодействии хлорида алюми-
алюминия с бензоилхлоридом 96 и трифторида бора с ацетилфтори-
дом 93>94 в жидком сернистом ангидриде были получены ком-
комплексы PhCO+AlCli и GH3GO+BFJ. На кривой кондуктометриче-
ского титрования трихлорида бора хлоридом калия конечная точка
наблюдается при мольном отношении 1:1, что указывает на обра-
образование в растворе в сернистом ангидриде тетрахлорбората
калия 25.
Еще в 1902 г. Вальден и Центнерзвер 107> 108> 110 заметили, что
добавление иода к растворам иодидов калия и рубидия в жидком
сернистом ангидриде повышает электропроводность раствора; рас-
растворимость иода в присутствии этих электролитов резко возрастает.
Поскольку максимум электропроводности и растворимости наблю-
наблюдался при мольном отношении иодида к иоду, равном 1:1, авторы
256
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
объяснили это образованием в растворах трииодидов металлов М13.
Они получили также доказательства образования комплексных
иодидов кадмия и ртути, показав, что растворимость иадидов кад-
мня и ртути в растворах иодидов калия и рубидия в жидком SO2
повышается 109.
IV. РЕАКЦИИ ИЗОТОПНОГО ОБМЕНА
В ЖИДКОМ СЕРНИСТОМ АНГИДРИДЕ
По-видимому, наиболее сильными аргументами против предло-
предложенного Яндером 64 механизма ионизации жидкого сернистого анги-
ангидрида
3SO2 4=2" SO2++S2O|- C6)
и его объяснения действия SOC12 как кислоты в сернистом ангидриде
SOC12 -^ SO2++2C1" C7)
являются результаты работ по изучению изотопного обмена в сер-
сернистом ангидриде и в подобных системах, изложенные в статье
Норриса 90. Результаты этих работ приведены в табл. 37.
Можно предположить, что если тионилгалогениды и ионизи-
ионизируются, то только по реакции
SOr2 ^ SOr+ + T- C8)
Этот механизм подтверждается: (А) — быстрым обменом радиоактив-
радиоактивным изотопом хлора между тионилхлоридом и ионами СГ в жидком
сернистом ангидриде и (Б) — обменом радиоактивным изотопом
серы между тионилхлоридом и тионилбромидом в жидком сернистом
ангидриде, который протекает быстро и полностью даже при —50° С.
Аргумент (А) наводит на мысль о том, что либо диссоциация идет
по уравнению C8), либо обмен проходит по механизму присоеди-
присоединения: N
SOC12 + C1- ^
C9)
Аргумент (Б) указывает на то, что диссоциация протекает по уравне-
уравнению C8) или происходит прямой переход галоген-иона по урав-
уравнению
SOCl2 + SOBr2 ^Z? SOCl++SOBr2Cr
или через образование переходного комплекса
Оч /СК, /О
S S
D0)
диссоциирующего на молекулы SOClBr. Обмен 18О между серным
ангидридом и растворенной трехокисью серы указывает на возмож-
IV. РЕАКЦИИ ИЗОТОПНОГО ОБМЕНА
257
Таблица 37. Результаты работ по изучению реакций обмена
Растворенное
вещество
SO2
so3
SOC12
SOBr2
SC12
(CH3LNC1
Реакции обмена
в SO2
Прямые доказательства 77 су-
существования обмена атома-
Быстрый обмен атомами *S
' с растворителем 65
Быстрый обмен 52, 88 атомами
180
Обмена атомами *S не про-
происходит 61
Обмена атомами *S не про-
происходит 65' 82
Обмена атомами 18О не про-
происходит 46
Протекает обмен атомамп *S
в присутствии иопов С1~
и молекул SbCl5
и А1С13 23- 24' 47' 6б
Обмена атомами *S не про-
происходит 66'82
Обмена атомами1 «О не про-
происходит *6
Протекает обмен *'> 66 атома-
атомами *S в присутствии ионов
СГ и Вг"
Обмена, атомами *S с SO2 не
происходит 87
в S0C1.
SO-2. растворенный в из-
избытке SOClj. не обмени-
обменивается65'87 атомами *S
Протекает обмен атомамп
*S между SOC12 и SOBr2,
растворенными в SO^ 64
То же в смеси SOClj
и SOBr26*
Степень обмена атомамп
*S с S0C1-2 составляет
45% за 2 ч при 60° С «
Быстрый обмен атомамп
*С1 между SOC12 и СГ
В SO2 83
Примечание. *S — радиоактивный изотоп серы, чаще всего гь S—Л2>ид1. перев.
вость перехода О2~-ионов к сильному акцептору 43> 52> 8S, а отсут-
отсутствие 51 обмена *S подтверждает мысль о том, что обменная реакция
должна проходить либо по уравнению
SO2+SO3 ^ГГ S02++S0r D1)
либо через образование переходных комплексов, например
o-s,
17 Заказ
90
258
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
в которых атомы серы неравноценны. Но следует отметить, что-
электропроводности растворов трехокиси серы в сернистом ангидриде
так же малы, как п электропроводности растворов SOC12. Так,
эквивалентная электропроводность О,\М раствора 54—10"х см2/(ом X
X моль).
Проводились попытки измерить скорость реакции самообмена
180 в жидком сернистом ангидриде, однако при этом возникли серь-
серьезные трудности экспериментального характера, связанные с про-
протеканием в газовой фазе гетерогенно катализированной реакции 52.
Недавно Лпчтин и др. 77, исследуя смесь S16O2 и S18O2 методом
ИК-спентроскопий, показали наличие гомогенного обмена кислоро-
кислородом в газовой фазе и в растворах сернистого ангидрида в четырех-
хлористом углероде и циклогексане. Равновесие устанавливается
в течение одного спектрального измерения, т. е. в течение 10 мин.
Они предположили, что такой обмен протекает через образование
промежуточных циклических соединений, например O = S S = O,
\о/
и отметили, что это предположение подтверждается существованием
некоторой ассоциации сернистого ангидрида как в жидком, так
в твердом состоянии 30' 43.
Скорость обмена *S между тионилгалогенидами и сернистым
ангидридом ничтожно мала. Но было обнаружено, что добавление
галоген-иона сильно катализирует реакцию 47> 82. Обмен *S между
тпонилбромидом и сернистым ангидридом, которые добавляли в рас-
раствор бромида тетраметиламмония, был подробно изучен Гербером
и др. 47. Оказалось, что скорость реакции обмена не зависит от кон-
концентрации тпонилбромида и подчиняется кинетическому уравнению
D2)
где А-= 5,17-106-ехр ( —13200/ДГ) сев.
Был предложен следующий механизм реакции обмена:
SOBr2 ^z? SOBr+-fBr~ Быстрая реакция
к.
*SO2 + Br- ^Ь *SO2Br~ Быстрая реакция
D3)
D4)
к,
SOBr+ + SO§+ Медленная реакция
что приводит к кинетическому уравнению вида
<*(*So6m1 = _|з_
(it /c4
D5)
D6)
IV. РЕАКЦИИ ИЗОТОПНОГО ОБМЕНА
259
Реакция обмена *S между тионилхлоридом и сернистым ангидридом
как растворителем, катализированная добавкой СГ-иона 82, подчи-
подчиняется иному кинетическому уравнению
¦ = к [(CHINCH
dt
[SO2]
Эта реакция изучалась в широком диапазоне составов растворов:
от раствора тионилхлорида в избытке сернистого ангидрида до рас-
раствора сернистого ангидрида в избытке тионилхлорида. Оказалось,
что скорость реакции описывается одним и тем же кинетическим
уравнением, хотя наблюдается трехкратное увеличение константы
скорости при переходе от раствора с избытком сернистого ангидрида
к раствору с избытком тионилхлорида. В том Случае, если в качестве
катализатора реакции обмена в растворах сернистого ангидрида
использовали хлорид тетраметиламмония, к = i,<)8-107 exp
(—14700/RT) л^/(моль2 -сек). Предложенный механизм вписывается
реакциями
*SO2 + C1~ ^Z? *SO2Cr Быстрая реакция
D8)
Медленная реакция
49)
что приводит к кинетическому уравнению D7), где к = к3к1/к„.
В приведенных выше рассуждениях не делалось различия между
концентрацией «свободных» галоген-ионов и полной концентрацией
галогенида, т. е. между свободными галоген-ионами и ионными
парами. Данные об электропроводности, безусловно, указывают
на то, что в растворах галогенидов в жидком сернистом ангидриде
преобладают ионные пары 70.
Было найдено также, что при добавлении хлорида тетраметил-
тетраметиламмония в качестве катализатора скорость изотопного обмена между
тионилхлоридом и сернистым ангидридом почти вдвое больше, чем
при добавлении сравнимой концентрации хлорида рубидия. Подобное
влияние на реакцию обмена S между тионилбромидом и сернистым
ангидридом оказывает добавление бромида тетраметиламмония и бро-
бромида рубидия. Скорость реакции обмена между сернистым ангидри-
ангидридом и тионилхлоридом почти в 7 раз превышает скорость реакции
с тионилбромидом при одинаковых концентрациях, несмотря на
очевидно большую энергию активации A4,7 кшл/молъ для реакции
в хлоридных растворах и 13,2 ккал/молъ для реакции в бромидных
растворах). Вполне возможно, что в случае тионилхлорида равно-
равновесие реакции
SO2 + X- .^r SO2X- E0)
17*
260
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
IV. РЕАКЦИИ ИЗОТОПНОГО ОБМЕНА
261
сдвинуто вправо больше, чем в случае тионилбромида. Это согла-
согласуется с гораздо большей устойчивостью иона SO2F~, но не объясняет-
изменения механизма реакции обмена.
Реакция обмена между SOC12 и SO2, по-видимому, катализи-
катализируется неионными основаниями. Недавно была опубликована ра-
работа, посвященная изучению влияния добавок триэтиламина и аце-
ацетона на скорость реакции обмена 91. Было показано, что для
триэтиламина скорость реакции определяется уравнением
= fe[SO2nSOCl2][(C2H6KN]
dt
в очень широком диапазоне концентраций: от раствора тионил-
тионилхлорида в жидком сернистом ангидриде до раствора сернистого
ангидрида в жидком тионилхлориде. Однако при переходе-
от растворов с высокой концентрацией сернистого ангидрида к рас-
растворам с высокой концентрацией тионилхлорида константа
скорости значительно возрастает. Найденное значение константы
скорости в растворе с избытком тионилхлорида составило 3,42 X
X 10е-ехр (—13 S00/RТ) л2/(молъ2-сек). Для того чтобы описать-
процесс при всех возможных условиях, кинетическое уравнение было»
изменено следующим образом:
dt
- = fc[SOCl2][SO2.
' ISO2] [SOC12 ¦ (C2H5KN] E2>
Соединения, входящие в первый член этого уравнения, домини-
доминируют в разбавленных растворах сернистого ангидрида в тионил-
тионилхлориде. Ацетон оказывает лишь слабое каталитическое воздействие
на реакцию обмена. В рассмотренном диапазоне концентраций реак-
реакция с ним описывается следующим кинетическим уравнением:
[*So6M]
dt
= k [SO2] [SOClal [(СНзJСО]2
E3)
С изменением концентрации реагента не наблюдалось заметных
изменений величины к, для которой получено выражение к = 5,3 X
X 10и-ехр (—24 000/Я71) л3/(моль3-сек).
Реакция обмена серой между тионилхлоридом и сернистым ан-
ангидридом катализируется также пентахлоридом сурьмы и трихлори-
дом алюминия 23) 24> 82. При сравнимых концентрациях скорость
реакции при добавлении этих реагентов составляла 0,01 и 0,001
скорости реакции с добавкой хлорида тетраметиламмония. Реакция
описывается кинетическим уравнением
*So6m]
dt
= fe[SbCl6-SOCl2][SO2]
E4)
что соответствует, механизму
*SOCl2 + SbCl3 4^Ь *SOC12- SbCl6 Быстрая реакция E5)
*SOC12 • SbCl6+ SO2 ^z? *SO2 + SOC12 • SbCl5 Медленная реакция E6)
Во всем интервале составов от избытка сернистого ангидрида до из-
избытка тионилхлорида константы скорости, вероятно, остаются,
по существу, постоянными. Равновесная константа реакции обмена
между S0C1, и SO2 при 0° С К = 0,8, а энтальпия обра-
образования аддукта ДЯ° = 3,6 ккал/молъ, к3 = 0,875-10^-ехр
(_Ю 400/ЯТ) л2!(молъ--сек). Кажущаяся энтропия активации
составляет —51,6 калЦград-молъ). Ниже сравниваются кинетические
уравнения, энергии активации ?акт, предэкспоненциальные множи-
множители к у и энтропии активации 5акт реакций обмена *S между SOC12
и SO2 A) и SOBr2 + SO2 B) в жидком сернистом ангидриде, ката-
катализируемые разными веществами:
реакция A), катализируемая
el- d[*Sd°6"] =fc[cr][soci2][so2i;
?акт = 14,7 ккал/молъ; к1 = 1,08 • 107; S3
29 кал/(град ¦ моль)
at
?аКТ = Ю,4 ккал/молъ; fci = 0,875 ¦ 102; SaKT=-51,6 кал/ (град . моль)
(C2H3KN d^°g»] =fc[SOtl [SOC1»1 [(C2H6)»N1;
?акт=14 6 '(в избытке SO2) и 13,8 ккал/молъ (в избытке SOC12);
fc! = 3,42-10e (в избытке SO2) и 2,06 ¦ 106 (в избытке SOC12); 5aKT =
= _30,7 кал/ (град ¦ моль) (в избытке SO2)
(CHj)jCO
?акт = 24,0 ккал/молъ; kt = 5,3 • 10"; SSKT= — 5,1 кал/(град ¦ моль)
реакция B), катализируемая
Br-
(it
?-акт= 13,2 ккал/молъ; h\ = 4,17 • 10е; 5акт= —30,4 кал/(град ¦ моль)
* Доказано только, что скорость пропорциональна [Вг-].
Бердж п Норрис 23' 2i показали, что если использовать в качестве-
катализатора для реакции изотопного обмена в SO2 смеси SbCl
262
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
•с (CH3LNC1 и (CH3LNC1 с А1С13, то при мольном отношении компо-
компонентов смеси 1 : 1 каталитический эффект минимален.
Изучалось также каталитическое влияние смесей триэтил-
-амина с SbCl5 91. И в этом случае минимальный каталитический эффект
наблюдался при мольном отношении 1:1. причем он был очень
слабым.
Для раскрытия детального механизма реакции обмена весьма
важно знать структуру аддукта SOCl2-SbCl5, который может быть
выделен в твердом состоянии. Как было показано, структура аддукта
SeOCl2- SbCl5 может быть записана в виде Cl.2SeO -> SbCl5. Учиты-
Учитывая низкую электропроводность SOCl2-SbCl5 в жидком SO2, резонно
предположить, что структура этого соединения аналогичная,
т. е. C12SO -> SbCl5.
По-видимому, наиболее интересным выводом из работ по изотоп-
изотопному обмену является то, что в этих реакциях предполагается воз-
возможность образования переходных ионов. Это позволяет пред-
предположить различные механизмы реакции. В реакции между тионил-
'бромидом и сернистым ангидридом, катализированной ионами Вг~,
переходным ионом может быть SO2Br~ • SO., со структурой
V. ЗАКЛЮЧЕНИЕ
263
О
О
В реакции с тионилхлоридом, катализированной ионами С1", пере-
переходными ионами могут быть ионы SO2C1~ • SOC12 со структурой
О О С1-
\ / \ /
S S
/\/\
С1 С1 О
которая предполагает наличие двух мостнковых связей, причем
связи ионов О2~ и С1~ с S различны. В случае обменной реакции
между тионилхлоридом и сернистым ангидридом, катализирован-
катализированной (C2H5KN, в качестве переходного соединения образуется
I(C,H5KN-SO2bSOCl,, структуру которого можно записать следу-
следующим образом:
(C2Ii6KN О С1
V V
/ \ / \
О С1 О
Указанное выше соединение образуется в растворах с высоким
содержанием сернистого ангидрида. В растворах же с высоким содер-
содержанием тионилхлорида образуется [(C2H5KN- SOC12] • SO2 со
структурой
(C2H5KN О О
\ / \ /
S S
/ \ /
С1 С1
О
Каталитическое влияние кислоты по Льюису на скорость реакции
обмена гораздо слабее; предполагаемый механизм реакции обмена,
катализированной SbCl5. включает образование переходных соеди-
соединений (SOC12 • SbCl5) • SO.,. Энтропия активации этой обменной реак-
реакции вдвое превышает энтропию активации любой другой реакции
(—52 кал/(град • моль), что указывает на образование более сложных
переходных соединений.
V. ЗАКЛЮЧЕНИЕ
Пытаясь систематизировать результаты своих обширных иссле-
исследований, Яндер и Виккерт 62, исходя из позиций Кейди и Элси 27,
предложили «сульфитную» теорию реакций в жидком сернистом
ангидриде. Эту теорию Яндер использовал как основной принцип;
в своей монографии при интерпретации изучавшихся им реакций
в жидком сернистом ангидриде. По его мнению, жидкий сернистый
ангидрид подвергается самоионизации путем перехода иона О2~
от одной молекулы SOa к другой:
2SO2 ;p± SO2+ + SO|" E7)
Яндер считал, что ионы SO2+ и SO|" реально существуют подобно
ионам Н3О+ и ОН", образующимся при самоионизации воды. Реак-
Реакция между тионилхлоридом и сульфитом рассматривалась Яндером
как реакция нейтрализации:
Cs2SO3 + SOCl2
2SO2 + 2CsCl
E8)
Многие из реакций, рассмотренных в предыдущих разделах, из-
изучались и интерпретировались Яндером с позиций «сульфитной»
теории. Но, безусловно, нет необходимости постулировать образова-
образование промежуточных ионов или привлекать растворитель для объяс-
объяснения обменной реакции, протекающей в жидком сернистом
ангидриде.
Реакция
[(CH3LN]2SO3-bSOCl2 ?=± 2(CH3LNC1 + 2SO2 E9)
монует, по-видимому, протекать в любом инертном растворителе
и не требует для своего протекания наличия промежуточных ионов
SO2+ или даже SOC1+. Эта реакция может протекать и, вероятно,
264
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
протекает через нуклеофильную атаку SO3' или S20|" на SOC13
с выделением двух С1~-ионов:
,SO|-+SOC12
(SO3-SO)+2C1-
(SO3-SOCl-) + cr —
¦Скорость реакции велика и равновесие
SO|-+SOC12 ^г± 2SO2 + 2C1-
-значительно сдвинуто вправо.
F0)
F1)
титрования при мольном отношении SO*"
На кривой кондуктометрического
_г „л SO|" к SOC12, равном 1 : 1,
наблюдается перегиб. Таким образом, в постулировании самоиони-
зации жидкого сернистого ангидрида нет необходимости. Работами
по изучению изотопного обмена в сернистом ангидриде показано,
что в действительности самоионизация SO2, очевидно, не происходит.
Остается объяснить удивительную растворяющую и ионизиру-
ионизирующую способность этого растворителя. Личтин 70, принимая во вни-
внимание невысокую диэлектрическую проницаемость жидкого серного
ангидрида (е = 15,36 при 0° С), разработал теорию, объясняющую
-свойства растворов ионных солей в нем. Но многие ионные соедине-
соединения с довольно высокой энергией решетки гораздо лучше растворимы
в жидком сернистом ангидриде, чем это можно было бы ожидать,
если учитывать его сравнительно небольшую диэлектрическую
проницаемость. Жидкий сернистый ангидрид обладает также не-
необычной ионизирующей способностью. Так, трифенилметилхлорид,
соединение ковалентное в кристаллическом состоянии и во многих
обычных растворителях, в жидком сернистом ангидриде предста-
представляет собой сильно диссоциированный электролит. Особый интерес,
как указывал Личтин 70, представляет сравнение ионизации трп-
¦фешглметилхлорида в нитробензоле (е = 24,5) и в жидком сернистом
ангидриде (е = 15,4). Трифенилметилхлорид, растворенный в нитро-
нитробензоле, настолько слабый электролит, что константу диссоциации
•его невозможно определить, в то время как в жидком сернистом
ангидриде этот электролит диссоциирован так же сильно, как хлорид
или бромид калия. Такая же картина, как в SO2, наблюдается для
трифенилметилхлорида в жидком хлористом водороде (е = 9,3) 104,
в котором электролит по своей силе сравним с хлоридом тетраметил-
аммония. Это означает, что и в жидком сернистом ангидриде, и в жид-
жидком хлористом водороде должно происходить взаимодействие между
растворителем и С1~-ионами, которое носит совершенно особый,
¦неэлектростатический характер, так как, если судить по величинам
диэлектрической проницаемости и дипольного момента, нитробензол
(и. = 4,24, е = 34,5) должен быть значительно лучшим' ионизиру-
ионизирующим растворителем, чем сернистый ангидрид (j.i = 1,62, е = 15,4)
и хлористый водород (ц = 1,03, е = 9,3). Это особое взаимодействие
>в жидком хлористом водороде объясняется образованием водородных
ЛИТЕРАТУРА
265-
связей. Имеются довольно серьезные основания предполагать, что
в случае сернистого ангидрида это взаимодействие связано если и не
с образованием ковалентных связей, то по крайней мере с переходом
заряда 70'80 (см. II, Б).
Спектроскопическое исследование 70 комплексов сернистого-
ангидрида с С1~-, Вг~- и 1~-ионами показало, что все галогены уси-
усиливают электронный спектр сернистого ангидрида и вызывают бато-
хромные сдвиги. Липпинкотт и Уэлч 80 идентифицировали комплексы
1~-иона с сернистым ангидридом как комплексы с переходом заряда,
и вполне резонно допустить, что такие комплексы с сернистым ан-
ангидридом могут образовывать все галоген-ионы.
В предыдущих разделах рассмотрены многочисленные комплексы
сернистого ангидрида. Интересно отметить, что большинство ком-
комплексов образуется с потенциальными донорами электронов. Ком-
Комплексы сернистого ангидрида с акцепторами электронов сравни-
сравнительно нестойки. Личтин 70 рассчитал из величин степеней ионизации"
замещенных триметилхлоридов, что образование комплексов^
СГ-иона с сернистым ангидридом более чем на 10 ккал/моль умень-
уменьшает свободную энергию ионизации. Подобные же эффекты должны
наблюдаться в случае комплексообразования с Вг~- и 1"-ионами
и с другими одновалентными ионами, например с тиоцианатом. На
свободную энергию растворения ионных галогенидов и тиоцианатов-
комплексообразование, вероятно, должно оказывать такое же вли-
влияние. Этот эффект может приблизительно в 108 раз повысить рас-
растворимость по сравнению с той, которую следует ожидать в «инерт-
«инертном» растворителе с такой же диэлектрической проницаемостью..
ЛИТЕРАТУРА
10.
И.
12.
13.
14.
15.
16.
17.
18.
Andre S., С. г., 130, 174 A900).
Andrews L. J., Chem. Rev., 54, 713 A954).
Andrews L. J.,Keefer R. M., J. Am. Chem. Soc, 73, 4169 A951).
Audrieth L. F., KleinbergJ., Non-Aqueous Solvents, New-
York, 1953.
А у n s 1 e у
Kleinberg J.,
Peacock R. D., Robinson P. L., Chem.
J. Chem.
T7 T7
a. "Ind., 1951, 1117.
Bagster L. S., Cooling
Bagster L.
Bagster L.
Bateman L.
1944, 243.
, Ann. chim. phys., 1, 8 A904).
N., Ergebn. exakt. Naturwiss., 6, 125 A926).
N., K. danske vidensk. Selsk. Math.-fys. Medd
,
В аи d e E
Bjerrum
В jerrum
A926).
Bond P. A
Bond P. A
Bond P. A
Bond P. A
Booth H.
Soc, 117, 693 A920)>
S., Steel e B. 0., Chem. News, 105, 157 A912).
S., Steel e B. 0., Trans. Farad. Soc, 8, 51 A912).
C, Hughes E. D., I ngo 1 d С. К., J. Chem. Soc.
1, № 9, 1
Beach H. Т., J. Am. Chem. Soc, 48, 348 A926).
В el ton W. E., J. Am. Chem. Soc, 67, 1691 A945).
Crone E. В., J. Am. Chem. Soc, 56, 2028 A934).
Stephens W. R., J. Am. Chem. Soc, 51, 2910 A929).
S., Martin D. R., J. Am. Chem. Soc, 64, 2198 A942).
Bright J. R., Fernelius W. S., J. Am. Chem. Soc, 65, 637 A943).
266
ГЛАВА ШЕСТАЯ. ЖИДКИЙ СЕРНИСТЫЙ АНГИДРИД
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
A941).
Bright J. R., J a s p e r J. J., J. Am. Chem. Soc, 63, 3486
Bruner L., Bekier E., Z. phys. Chem., 84, 570 A913).
Bur г А. В.. J. Am. Chem. Soc, 65, 1629 A943).
В i с к e r t о n J. H., J. Am. Chem. Soc, 67, 2261 A945).
, N orris 1. H., J. Am. Chem. Soc, 81, 2324 A959).
, Norris Т.Н., J. Am. Chem. Soc, 81, 2329 A959).
, Freund H., Norris Т. Н., J. Phys. Chem., 63,
Burg А. В.,
Burg А. В.,
В u r g e D. E
В u r g e D. E
В u r g e D. E
1969 A959).
By r d W. E.,
С a dy H. P.,
Cady H. P.,
Inorg. Chem., 1, 762 A962).
Elsey H. M., J. Chem. Educ, 5, 1425 A928).
Toft R., J. Phys. Chem., 29, 1075 A925).
Centnerszwer M., D r u с к е г J., Z. Elektrochem., 29, 210 A923).
Clusius K., Schleich K., Bernstein R. В., Helv. chim.
acta, 40, 252 A962).
Cruse K., Z. Electrochem., 46, 571 A940).
Daggett H. M., J. Am. Chem. Soc, 83, 4977 A951).
de Maine P. A. D-, J. Chem. Phys., 26, 1036 A957).
Gyr E., J. chim. phys., 7, 189 A909).
J., Markowitz J. M., J. Chem. Educ, 37, 75 A960).
Aellig C. Helv. chim. acta, 6, 37 A923).
Kornblum J., Ber.,
Fleischer .Т.,
Fleischer J.,
Fleischer J.,
C, J. Phys. Chem
J.
J.
J.
Am.
Am.
Am
49, 2007 A916)
Chem. Soc, 53,
Chem. Soc, 54,
Chem. Soc, 56
1752 A931).
3902 A932).
870 A934).
48.
49.
50.
1.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
D u t о i t P.
Elving P.
Б p h r a i m F.
E p h r a i m F.,
F о о t e H. W.,
F о о t e H. \V.,
F о о t e H. W.,
Franklin E.
F u о s s R. M.,
F u о s s R. M.,
G i a u q u e W.
A938).
G m e 1 i n's Handbuch der Anorganische Chemie, System № 9, Weinheim,
1953, Bd. B-l, S. 208.
Grigg E. C, Lauder I., Trans. Farad. Soc, 46, 1039 A950).
Her ber R. H., Norris T. H., Huston J. L., J. Am. Chem.
Soc, 76, 2015 A954).
Hermodsson Y., Private Communication quoted by Lindquist I.,
in «Inorganic Adduct Molecules of Oro-compounds», Berlin, 1963.
Hill A. E., J. Am. Chem. Soc, 53, 2598 A931).
A. E., Fitzgerald Т. В., J. Am. Chem. Soc, 57, 250 A935).
' L., J. Am. Chem. Soc, 73, 3049 A951),-
._„_. _ , 15, 675 A911).
Shedlovsky Т., J. Am. Chem. Soc, 71, 1497 A949).
Kraus С A., J. Am. Chem. Soc, 55, 2387 A933).
F., S t e p h e n s о n С. С, J. Am. Chem. Soc, 60, 1389
Hill
Hill
Huston
Huston
Jander
J a n d e r
Jander
Jander
Jander
J a n d e r
Jander
J a n d e r
A937).
Jander
J a n d e r
Jander '
Johnson
J.
J. Am. Chem. ^.„^., .«, и^з
J. L., J. Phys. Chem., 63, 389 A959).
G., Naturwiss, 26, 779 A938).
Die Chemie in Wasseranlichen Losungmitteln, Berlin, 1949.
H e с h t H., Z. anorg. Chem., 250, 287 A943).
I m m i g H., Z. anorg. Chem., 233, 295 A937).
" Z. phys. Chem., A 183, 121 A938).
Z. phys. Chem., A 183, 137 A938).
Z. phys. Chem., A 183, 277 A939).
Immig H., Z. anorg. Chem., 232, 229
M e s e с h H.
Mesech H.,
M e s e с h H.
Knoll H.,
D., Z.
anorg. Chem., 230, 405 A937).
Ullman , , ^„„ vil/^v.
W i с к e r t K., Z. phys! Chem., A 178, 57 A936).
Wendt H., Hecht H., Ber., 77, 698 A944).
F. Jr., Norris Т. Н., J. Am. Chem. Soc, 79, 1584 A957).
V... N n r v ; о т. H., Huston J. L., J. Am. Chem.
Johnson R. E., Norris
Soc, 73, 3052 A951).
К i v e 1 s о n H. D., J. Chem. Phys., 22, 904 A954).
Korezynski A., Glebocka M., Gazz. 50, 1,
ЛИТЕРАТУРА
267
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100,
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
113.
114.
115.
116.
Lecher H. Z., Hardy W.
Le Fevre R. J. W., Dipole
Li с Ji t i n N
York, 1963.
L i с h t i n N
Lichtin N.
Lichtin N
L i с h t
L i с h t
L i с h t
L i с h t
537 A964).
Lichtin
Chem. Soc,
В., J. Am. Chem. Soc, 70, 3789 A948).
Moments, London, 1953.
N., Progress in Physical Organic Chemistry, v. 1, New
N.
N.
N.
N.
N., Glazer H., J. Am. Chem. Soc, 73, 5537 A951).
N., Kliman H., J. Chem. a. Eng.Data, 8, №2, 178 A963).
N., Leftin H. P., J. Phys. Chem., 60, 160 A956).
N., P a p p a s P., Trans. N. Y. Acad. Sci., 20, 143 A957).
N., Rao K. N., J. Phys. Chem., 64, 945 A960).
N., Rao K. N., J. Am. Chem. Soc, 83, 2417 A961).
N., Laubicht I., Pincher S., Inorg. Chem., 3r
N. N., Ullstern R. E., Jr., White J. D., J. Am.
74, 4715 A952).
Lindquist I., Einarsson P., Acta chem. scand., 13, 420 A959).
L i p p i n с о t t E. R., W e 1 с h F. E., Spectrochim. acta, 17, 123 A961).
Martin D. R., J. Am. Chem. Soc, 67, 1088 A945).
Masters В
Masters B.
4252 A956).
Mesech H.,
Michaelis
M о e d e J. A
M u x a r t R.,
J.,
J.
N о r r i s Т. Н., J. Am. Chem. Soc.
Potter N. D., et al., J. Am.
77, 1346 A955).
Chem. Soc, 78,
1938.
, Greifswald,
, 745 A891).
C, J. Am. Chem.
1489 A950).
Dissertation
A., Ber., 24
С u r r a n
С. г., 231,
N а к a t a S., J. Chem. Soc. Japan, 64, 635 A943)
Nickerson J. D., Mclntosh R., Can. J. Res.
Norris Т. Н., J. Phys. Chem., 63, 383 A959).
Potter B. D., Diss. Abs., 23, 1919 A962).
Schlenk W., W e i с к e 1 Т., Herzenstein A., Ann., 372, 1 A910).
Soc, 71, 852 A949).
35, 1325 A957)..
See
See
See
See
See
See
See
Seel
Z. anorg. Chem., 250, 331 A943).
Z. anorg. Chem., 252, 24 A943).
Bauer H., Z. Naturforsch., 26, 397 A947).
Riehl L., Z. anorg. Chem., 282, 293 A955).
Riehl L., Angew. Chem., 67, 32 A953).
В о e z A. K., N о g r a d i J., Z. anorg. Chem., 264, 298 A951).
В о e z A. K., N о g r a d i J., Z. anorg. Chem., 264, 311 A951).
N о g r a d i J., R о s s e R., Z. anorg. Chem., 269, 197 A952).
Silberrad O., J. Chem. Soc, 121, 1015 A922).
Steele В. О., Chem. News, 96, 224 A907).
Vierk A. L., Z. anorg. Chem., 261, 279 A950).
Waddington Т. С, К 1 a n b e r g F., J. Chem. Soc, i960, 2332.
W a 1 d e n P., Ber. deut. keram. Ges., 35, 2018 A902).
W a 1 d e n
W a 1 d e n
Walden
W a 1 d e n
Walden
W i с к е г t
W i с к е г t
W i с к е г t
P., Centnerszwer
P., Centnerszwer
P.,Centnerszwer
P., Centnerszwer
P., Centnerszwer
K., Z. Electrochem., 44,
K., Naturwis., 26, 500
K., Jander G., Ber.
Ber., 32, 2862 A899).
M
M.,Z. phys. Chem./39, 513 A902).
M..Z. anorg. Chem., 30, 145 A902).
"" ~ - 30, 179 A902).
42, 432 A903).
M.
Z. anorg. Chem
M., Z. phys. Chem.
110 A938).
A938).
70 B, 251 A937).
21,
Каштанов Л. И., Соколова Л. Н., ЖОХ,
Лучпнский Г. Л., ЖФХ, 12, 280 A938).
Ш а т е н ш т е й н А. И., Викторов М. М., Acta
URSS, 1, 883 A937).
1484 A951).
phys.-chim.
378 A920).
ГЛАВА СЕДЬМАЯ
II. ГАЛОГЕНЫ
269
ГАЛОГЕНЫ И ИНТЕРГАЛОГЕНЫ
КАК РАСТВОРИТЕЛИ
А. Г. ШАРП
I. Введение 268
II. Галогены 269
A. Хлор . . . .- 269
Б. Бром 270
B. Иод 271
Ш. Интергалогены 273
A. Соединения типа АВ 273
Б. Соединения типа АВ3 275
B. Соединения типа АВ5 278
Г. Соединения типа АВ7 279
Литература 280
I. ВВЕДЕНИЕ
В связи с исключительно высокой реакционной способностью
галогенов н интергалогенов лишь очень небольшое число соедине-
соединений при растворении в них не подвергается химическим превраще-
превращениям, не считая образования ионов и сольватации. Большинство
работ, посвященных галогенам и интергалогенам как раствори-
растворителям, носит чисто качественный характер, а интерпретация полу-
полученных в некоторых работах количественных данных часто весьма
спорна, т. е. эта область неорганической химии — одна из наименее
изученных.
В препаративной неорганической химии в качестве растворителя
некоторое применение получил лишь трифторид брома. С водой
н большинством органических материалов он реагирует со взрывом;
асбест при реакции с ним сильно раскаляется. Работать с трифтори-
дом брома можно только в кварцевой аппаратуре. Хлориды иода,
хотя и менее реакционноспособны, но обладают свойствами, дела-
делающими их непригодными для электрохимических работ, в частности,
в них легко растворяются золото и платина.
Единственный метод, широко применяющийся для исследования
растворителей этого класса, — измерение электропроводности. Он
позволяет судить 6 количестве присутствующих ионов и их подвиж-
подвижности, но мало дает для раскрытия природы ионов. Идентификация
продуктов, выделяющихся на электродах, может пролить некоторый
•свет на эту проблему, однако слишком часто 'взаимосвязь между
шереносчиками тока и продуктами, выделяющимися на электродах,
весьма слаба. Даже если (что бывает очень редко) известны структуры
•твердых аддуктов, содержащих элементы, входящие в состав раство-
растворителя, при растворении могли произойти очень значительные из-
изменения; процессы обмена галоген-ионов часто протекают с большой
•скоростью. Измерения чисел переноса и э. д. с, столь обогатившие
наши знания о природе водных растворов, не принесли заметной
пользы при изучении галогенов и интергалогенов как растворителей.
'Следовательно, зависимости, приведенные в изложенном ниже об-
обзоре, не должны представляться предельно ясными; необходимы .
дальнейшие работы и для уяснения природы растворов в галогенах
и интергалогенах.
Имеется обширная литература о взаимодействии брома, иода,
монохлорида иода и бромида иода с молекулами донорного типа
(в особенности, с органическими молекулами), но в большинстве
этих "работ рассматриваются условия, при которых донорные моле-
молекулы присутствуют в большом избытке. Эти исследования подробно
•освещены в недавней работе Андрюса и Кифера 1.
II. ГАЛОГЕНЫ
Работ, посвященных растворам в жидком фторе, очень немного;
известно, что растворимость хлора в нем незначительна "-. Ниже
приведены в качестве справочного материала некоторые физические
свойства галогенов:
Галоген
А#исп ПРИ 'кип.
ккал/молъ . .
?исп> кал/(мольх
Хград) ....
Т], мпз
—220 —101
-188 —34
1.51 (-190° С) 1,57 (—35° С)
1.52 (—190° С) 2,0 (—35° С)
— 7 • 10~8
Вг|2
-7
59
3,12 B0° С)
3,1 B0° С)
J2g
114
183
3,92 A33° С)
11,1 A18° С)
1,1 ¦ 10~» B5° С) 5,2 • Ю'5 A14° С)
1,56
18,5
4,87
20,4
4,9 (—34° С)
7,25
22,0
10,3 A6° С)
10,6
23,1
19,8 A16° С)
27
Укажем, что удельная электропроводность хлора 27 заметно
выше, чем удельная электропроводность брома. Однако эта вели-
величина получена много лет назад и, по-видимому, должна рассматри-
рассматриваться как верхний предел.
А. Хлор
Известно 9, что хлориды натрия, калия, меди(П), кадмия, алю-
алюминия, свинца(П), церия(Ш) и циркония(ГУ) нерастворимы в жид-
жидком хлоре, а гексахлорид вольфрама лишь слегка растворим.
270 ГЛАВА СЕДЬМАЯ. ГАЛОГЕНЫ И ИНТЕРГАЛОГЕНЫ КАК РАСТВОРИТЕЛИ
Хорошо растворимы в хлоре тетрахлорид углерода, тетрахлорид крем-
кремния, тетрахлорид титана, трихлорид мышьяка, тетрахлорид свинца,
оксихлорид фосфора и хлористая сера. Бильтц и Майнеке 9 не об-
обнаружили признаков образования соединений в растворах первых
четырех хлоридов в жидком хлоре, но Уит и Браун 57 показали,
что существуют соединения СС14-?гС12 (где п = 0,5; 1; 2; 3 и 4),
а также соединения хлора с хлороформом, метилёнхлоридом, метил-
хлоридом и хлористым водородом 55>56.
Хлороформ и метиленхлорид присоединяют максимально три
и две молекулы хлора соответственно, это указывает на то, что атомы
хлора в органическом соединении являются донорами. Но если это
так, то удивительно, почему Уит и Браун не смогли обнаружить
признаков комплексообразования в системе СС14 — Вг2.
Тейлор и Гильдебранд 52 привели следующие данные о понижении
температуры замерзания хлора (измерения были проведены Уэнтигом
и Мак-Интошем 54) при растворении в нем 0,01 мольной доли ука-
указанных ниже соединений:
Соединенно . . (СН3JСО СН3СООС2Н5 (С2Н5JО SnCl4 CC14 С6Н6СН3 СНС13
— At,град . . 0,190 0,190 0,204 0,208 0,210 0,215 0,222
Однако теоретическое значение — At, рассчитанное из скрытой
теплоты плавления, равно 0,27° С, т. е., несомненно, происходит
довольно сильное взаимодействие между хлором и названными
соединениями. Дальнейшие исследования этих систем, а также
сравнение их, например, с системой хлор — гексан, в которой хи-
химическое взаимодействие минимально, следует лишь приветствовать.
Поскольку доказано существование иона C1J в водной среде, следо-
следовало бы изучить также действие жидкого хлора на хлориды круп-
крупных органических катионов. Учитывая' энергию решетки, наиболее
вероятным в этих случаях следует считать образование устойчивого
полихлорида. Необходимо также изучить возможность комплексо-
комплексообразования с кислотами Льюиса, в особенности с пентахлоридом
сурьмы.
Б. Бром
Многие галогениды неметаллов растворимы в броме. Из них надо
отметить тетрахлорид и тетрабромид углерода, трибромнд бора,
тетрабромид кремния, тетрабромид титана, тетрабромид олова,
трибромид мышьяка и трибромид сурьмы 8> 55. Образование в этих
растворах соединений пока не доказано. Трихлорид фосфора раство-
растворяется в броме с образованием хлорбромидов.
Бром образует аддукты со многими органическими основаниями,
такими, как пиридин, хинолин, ацетамид и бензамид. Растворы
этих соединений в броме проводят ток, по-видимому, за счет при-
присутствия ионов (Основание),, Вг+ и Br]j. Но электропроводность
II. ГАЛОГЕНЫ
271
этих растворов изменяется во времени, как предполагают,
вследствие того, что происходит бромированиё органической моле-
молекулы. Интерпретация полученных данных весьма спорна 14> 63.
Растворимость бромидов щелочных металлов в броме ничтожна,
во бромиды, имеющие в своем составе крупный органический ка-
катион, растворяются в броме в заметной степени. Например, экви-
эквивалентная электропроводность 2,01 м раствора бромида тетрабутил-
аммония при 25° С составляет 15,7 см2/(ом-г-экв).
Большая вязкость раствора объясняется, по-видимому, высокой
степенью ассоциации, вероятно, вследствие образования полибро-
мидных анионов 28. Растворы пентахлорида фосфора также прово-
проводят ток и, очевидно, содержат хлор в анионной форме. В растворах
наблюдаются изменения мольной электропроводности с изменением
концентрации. Подобные же растворы образует пентабромид
фосфора 62.
В. Иод
По сравнению с другими галогенами жидкий под как растворитель
более изучен 25, хотя за последнее десятилетие новых работ по хи-
химии этого соединения не появлялось.
Во многих случаях иод ведет себя аномально. Расстояние наиболь-
наибольшего сближения, при котором атомы в твердом теле еще рассматри-
рассматриваются как несвязанные, равно 3,56 А. В молекуле иода это рас-
расстояние равно 2,68 А. Это заставляет предположить наличие доста-
достаточно сильного межмолекулярного взаимодействия, а довольно вы-
высокая энтропия испарения в точке кипения указывает на то, что
и в жидкой фазе такое взаимодействие в значительной степени со-
сохраняется. G ростом температуры диэлектрическая проницаемость
жидкости возрастает с 11,1 при 118° G до 13,0 при 168° С. Электро-
Электропроводность жидкого иода выше, чем электропроводность других
галогенов, но в отличие от электропроводности твердого иода она
уменьшается с ростом температуры. Эти факты обычно объясняют
тем, что проводимость жидкого иода по своей природе частично
электронная, а частично ионная, и ионы — носители тока, вероятно,
термически нестойки.
Сера, селен и теллур растворяются в жидком иоде без химического
взаимодействия. В жидком иоде растворимы также тетраиодид
олова, иодид цинка, трииодид сурьмы, трииодидм ышьяка, иодид
ртути и йодоформ. Как показали криоскопические измерения,
процесс растворения этих соединений также не сопровождается
химическими изменениями. Иодиды кальция, стронция и бария нера-
нерастворимы, а растворимость иодидов щелочных металлов высока.
Как показали криоскопические измерения, в растворах иодидов
щелочных металлов присутствуют ассоциаты, причем наибольшая
степень ассоциации наблюдается в растворах лития, а наименьшая —
в растворах цезия.
272 ГЛАВА СЕДЬМАЯ. ГАЛОГЕНЫ И ИНТЕРГАЛОГЕНЫ КАК РАСТВОРИТЕЛИ
Были измерены электропроводности растворов галогенидов лития,
натрия, калия и рубидия. Удельная электропроводность растворов
иодида натрия ниже, чем удельные электропроводности растворов
других соединений, и с изменением концентрации изменяется не-
незначительно. Удельные электропроводности растворов трех осталь-
остальных иодидов резко возрастают с повышением концентрации, дости-
достигая максимума приблизительно при 10 мол. % соли. Как сооб-
сообщается, удельная электропроводность растворов иодида натрия
уменьшается, а растворов иодидов лития, калия и рубидия, увели-
увеличивается с ростом температуры. Ясно, что в таких растворах при-
присутствуют ассоциаты — тройники и более сложные ионы, а также
ионы М+ и Ig. По-видимому, нецелесообразно пытаться более глу-
глубоко судить о природе растворов иодидов в иоде до тех пор, пока
не подтверждены аномальные свойства растворов иодида натрия
и не проведено более детальное изучение этих растворов.
Самоионизация жидкого иода может быть представлена уравне-
уравнением
212 ;«=± 1+ + 1? A)
На основании этого иодиды щелочных металлов, легко образующие
трииодиды, рассматриваются как основания, а моногалогениды
пода — как кислоты. На кривой кондуктометрического титрования
раствора KI в жидком иоде иодидом брома имеется резкий перегиб
при мольном отношении 1:1, что соответствует реакции
М13 + 1Г 5=? МГ+212 ' B)
За протеканием этого процесса можно проследить с помощью
потенциометрического метода, хотя вследствие того что растворы
моногалогенидов иода в иоде — слабые электролиты, симметрич-
'Ную кривую получить не удается. По грубой оценке, ионное про-
произведение [1+] [1~] равно 10~42. Недавно, однако, ионы 1+ были
идентифицированы в синих растворах иода в олеуме и иода в пента-
фториде иода. Достоверность определения сомнений не вызывает.
В обоих случаях было установлено, что ионы 1+ парамагнитны и мо-
момент их равен 1,7—2*0 цв 3> 5. Сообщения об измерениях магнит-
магнитной восприимчивости растворов моногалогенидов иода в жидком
иоде отсутствуют, но все же весьма сомнительно, чтобы в растворе
могли присутствовать свободные ионы 1+. Можно отметить лишь,
что простейший механизм самоионизации иода, по-видимому, за-
заключается в образовании ионов IJ и IJ, более сложный может вклю-
включать образование и более сложных ионов. Описаны сольволитиче-
ские реакции в жидком иоде, например
KCN + Lj ^=± KI+ICN C)
и амфотерные свойства некоторых солей, растворенных в 12, напри-
например
HgI2+2KI г=± K9,HgI4 D)
III. ИНТЕРГАЛОГЕНЫ
273
III. ИНТЕРГАЛОГЕНЫ
А. Соединения типа АВ
Из соединений, отвечающих общей формуле АВ, как раствори-
растворители изучены лишь монохлорид и монобромид иода. Физические свой-
свойства их приведены ниже 17> 18> ао' а1:
Соединение I'd IBr
twx, °C 27,2 (а), 13,9 (Р) 41
«кип. СС 100 —116
р, г/смз 3,13 D5° С) 3,76 D2° С)
х, om-1-cm'I 4,6 • 10-з C5° С) 3.0 • 10-4D0° С)
Д-^исп ПРП ^кигм ккал/молъ . . 10,0 —
5ИСП, кал/(моль ¦ град) 26,7 —
Ц, мпз 41,9 B8° С) —
При интерпретации данных о свойствах жидких моногалогенидов
иода приходится сталкиваться с трудностью особого рода: эти со-
соединения в заметной степени диссоциируют на свободные галогены.
Для монохлорида иода степень диссоциации при 25° С равна 0,4%,
а при 100° С 1,1%. Соответствующие значения для монобромида
пода17 составляют 8,8 и 13,4%.
Электропроводность, жидкого монохлорида иода значительно
возрастает при растворении в нем хлоридов калия или аммония.
Эквивалентные электропроводности этих растворов при бесконеч-
бесконечном разбавлении (при 35° С) равны соответственно 32 и 26 см2/(ом X
X г-же). Из числа других соединений, имеющих заметную раствори-
растворимость и образующих электропроводные растворы, следует отметить
RbCl, CsCl,KBr, KI, A1C13, AlBr3, PCI5, пиридин, ацетамид и бенз-
амид. Слабо растворимы LiCl, NaCl, AgCl и ВаС12. Тетрахлорид
кремния, тетрахлорид титана и пентахлорид ниобия растворимы
хорошо, но незначительно увеличивают электропроводность раствора.
Добавление SbCl5 повышает электропроводность раствора в гораздо
меньшей степени, чем добавление РС15. Тионилхлорид, смешива-
смешивающийся с монохлоридом иода в любых отношениях, действует как
разбавитель; электропроводность раствора уменьшается пропор-
пропорционально количеству добавляемого тионилхлорида.
Данные об электропроводности трудно интерпретировать из-за
того, что разбавление сопровождается изменениями плотности, вяз-
вязкости и степени диссоциации. Дополнительные сведения в ряде слу-
случаев удалось получить при изучении систем, где в качестве раство-
растворителя использовался раствор монохлорида иода в нитробензоле,
но добавление третьего компонента может вызвать дальнейшие
осложнения.
Самоионизация монохлорида иода, обычно представляемая урав-
уравнением
2IC1 ^z± I+ + IClj E)
18 Заказ 90
274 ГЛАВА СЕДЬМАЯ. ГАЛОГЕНЫ И ИНТЕРГАЛОГЕНЫ КАК РАСТВОРИТЕЛИ
напоминает диссоциацию иода, следует лишь повторить, что весьма
сомнительно присутствие в растворе катиона I + в отличие от таких
ионов, как 12С1 +. Присутствие иона IClJ в твердых солях щелочных
металлов и в соединении PClJIClg установлено с помощью рент-
рентгенографических методов 61, но реальных доказательств существо-
существования несольватированного катиона нет. Изучение фазовых диа-
диаграмм показало, что в системах А1С13 — IC1 и SbCl6 — IC1 нет аддук-
тов, отвечающих составу 1:1, а имеются лишь аддукты с составом
1 : 2 для первой и 1 : 3 для второй системы. Как предполагают,
в этих соединениях имеется ион 12С1 +, аналогичный иону 1С1?
в соединениях 1С13- А1С13 и IC^-SbC^, которые лучше представлять
как IC1J А1С1; и 1С1? SbCle. '
В растворе с избытком монохлорида иода во многих случаях
нет смысла делать различие между ионом I + и его сольватами.
При кондуктометрическом титровании раствора RbCl в IC1 пентахло-
ридом сурьмы или раствора КС1 пентахлоридом ниобия перегиб
кривой наступает при мольном отношении 1 : 1, а при титровании
NH4C1 тетрахлоридом олова —• при мольном отношении 2:1. Это
указывает на образование иона SnCl|~. Соединение PG15 - IC1,
имеющее в твердом состоянии ионную структуру PCIJ ICL J, ведет
себя как кислота по отношению к К1С12 и как основание — по от-
отношению к SbCl5.
Органические основания (пиридин, бензамид, ацетамид) образуют
с IC1 соединения состава 1 : 1 или 1 : 2. По всей видимости, эти сое-
соединения являются соответственно N-донорными комплексами и со-
солями типа (Основание)! + IClJ.
В препаративной химии реакции в жидком монохлориде пода
имеют очень ограниченное применение, так как лишь в редких слу-
случаях удается выделить из растворов чистые продукты. Однако реа-
реагент может оказаться весьма полезным для стабилизации хлор-
комплексов элементов в высоких окислительных состояниях.
Расплавленный бромид иода как растворитель по свойствам очень
похож на монохлорид иода, но стойкие кислоты в IBr пока не выде-
выделены. Бромиды щелочных металлов образуют полигалогениды типа
М1Вг2, а пентабромид фосфора образует соединение 1РВг„. которое
по аналогии с хлор-соединением, по-видимому, лучше представлять
как PBrJ IB гг. В бромиде иода это соединение ведет себя только
как основание. Как ангидрид кислоты действует бромид олова и, как
можно показать кондуктометрическим титрованием, подвергается
нейтрализации, например, по реакции
III. ИНТЕРГАЛОГЕНЫ
275
2RbIBr2+SnBr4
Rb2SnBr6 + 2IBr
F)
Б. Соединения типа АВ3
К этой группе соединений относятся трифторид хлора, три-
фторид брома и трихлорид иода, а также открытый недавно три-
трифторид иода, свойства которого пока мало изучены 41. Физические
свойства жидких интергалогенов типа АВ3 7'17' 30) 81) 34> 37' 38:
Соединение ClFs BrF3 ICI3
(пл, °С -83 9 . 101
(кип, °С 12 126 -
р, г/см* 1,84 A2° С) 2,80 B5° С) -
е 4,6 A2° С) — —
х,ом-1-см-1 6,5 • Ю-» @° С) 8,0 • 10-з B5° С) 8,5 • 10~з (Ю19 С)
Д#испнри гкип, ккал/молъ 6,6 10,2 —
?исп, кал/(град -моль) . . 23.1 25,6 —
ч, мпз . 4,8 A2° С) 22,2 B5° С) -
Свойства трихлорида иода как растворителя в расплавленном
состоянии, когда в заметной степени происходит диссоциация
на монохлорид иода и хлор, не изучены. Но поскольку точно из-
известны структуры твердого трихлорида иода и аддуктов его с КС1,
А1С13 и SbCl5, уместно высказать некоторые замечания о структуре
этого соединения в расплавленном состоянии. Структура твердого
трихлорида иода, состоящую из плоских димерных молекул. В первом
CI-JJJ--CI CI
I ' '3,56 I 94°) '3,61
2,39/ Ч681 / V I
cl_JtfL_>CI С1
приближении можно описать как ионную с резонансным переходом
между двумя формами
-ci
\
/
/
4I
"Cl
\
/
/
СГ
<
в
"Cl
\
/
A
СГ
<
CL
- cr
Cl_
В плоском ионе IC1J, присутствующем в К1С14-Н2О, все углы равны
90 ± 1°, а расстояния I —GI указаны ниже:
С1
С1-
2,42
2,53
-I-
$2,47
2,60
С1
-С1
18*
276 ГЛАВА СЕДЬМАЯ. ГАЛОГЕНЫ И ИНТЕРГАЛОГЕНЫ КАК РАСТВОРИТЕЛЕ
В соединениях 1А1С14 и ISbCl6 атом иода окружен атомами хлора53,
два из которых расположены на расстоянии 2,25—2,33 А, а два дру-
других, дополняющих искаженный квадрат, — на расстоянии 2,85—
3,00 А:
\
/
AI
\
А
\
А!
а
2,8б\
2,25
CI
CI
.2,26
CI
\1/
Sb
CI
3,oos
CI
ex
.2.29
CI
Sb
/IN
В первом приближении такая структура соединений отвечает
формулам Ida А1СЦ и 1СЦ SbCle, но небольшая длина связей
I—С1 (где атомы С1 связаны с атомами Sb или А1) указывает на зна-
значительное катион-анионное взаимодействие. Тем не менее доказа-
доказательство существования ионов IG1J и 1СЦ имеет большое значение
для объяснения химических свойств аддуктов других интергалоге-
интергалогенов, сведения о структуре которых отсутствуют.
Электропроводность трифторида хлора очень мала и нет каких-
либо сообщений о ионных реакциях, протекающих в нем. Однако
заслуживает внимания появившееся недавно сообщение о получении
соединений, содержащих, по-видимому, ионы C1FJ и C1FJ, способ-
способные реагировать по уравнению
C1F*+C1F4 ^=± 2C1F3 G)
При действии фтора на хлориды, бромиды и иодиды щелочных
металлов при температурах 15—250° С образуются соединения
MC1F4, безусловно содержащие ион ClFj (где М — К, Rb или Cs).
Растворимость этих соединений в жидком C1F3 не исследована 4.
Трифторид хлора43»44 образует комплекс 1:1 с SbF8 и BF3.
Оба этих комплексных раствора хорошо проводят ток. На основании
изучения ИК-спектров второму соединению приписана структура
C1FJBFI.
Как указывают измерения ЯМР-спектров, трифторид хлора,
состоящий из Т-образных молекул в твердом и газообразном состоя-
состояниях, в жидком виде должен находиться в равновесии с присутству-
присутствующими в небольших концентрациях димерными молекулами 29.
Из интергалогенов только трифторид брома получил широкое
применение в лабораторной практике, главным образом для полу-
получения комплексных фторидов. Имеется ряд работ, посвященных
изучению этих реакций 19> 26< 64. В данном обзоре мы остановимся
на тех примерах, где BrF3 выступает в роли не только фторирующего
агента, но и растворителя. Трифторид брома фторирует все те co-
coin. ИНТЕРГАЛОГЕНЫ
277
единения, которые в нем растворяются-. Поэтому мы ограничимся
лишь рассмотрением поведения в нем растворимых неорганических
фторидов. Их можно разделить на две группы. К первой группе от-
относятся фториды щелочных металлов, фторид серебраA) и фторид
бария, ко второй группе — фториды золота(Ш), бора, титанаAУ),
кремния, германия(ГУ), ванадия(У), ниобия(У), тантала(У),
фосфора(У), мышьяка(У), сурьмы(У), платины(ГУ), рутения(У)
и ряда других металлов. Представители обеих групп образуют в три-
фториде брома электропроводные растворы, а многие дают с раство-
растворителем стойкие аддукты, например AgBrF4, SbBrF8. Все фториды
¦ первой группы реагируют со фторидами второй группы в BrF3 с об-
образованием комплексных галогенидов. Это легко объяснить тем,
что получаются производные, содержащие ионы BrF4 и BrF2
(аналогично производным 1С13) и взаимодействующие согласно урав-
уравнению ~ '
BrF+SbFj-f Ag+BrFj- ^± AgSbF6 + 2BrF3 (8)
В некоторых случаях для растворов с различным мольным от-
отношением компонентов (в частности, для приведенного выше примера
AgBrF4 + SbBrF8) удалось показать, что электропроводность ми-
минимальна при мольном отношении 1:1.
Тетрафторид олова образует комплекс 1:2с BrF3; реакция этого
аддукта с KBrF4 может быть представлена уравнением
(9)
(BrF$JSnFe-+2KBrF4
K2SnF6
Из огромного числа реакций получения фтор-комплексов с помощью
BrF3 достаточно привести следующие 12'23'45~47'58'59:
Ag + Au
BrF3 +
VF5 + LiF
Ru + KCl
AgBrF4 + BrF2AuF4
у (NOJSnF6
—> NO2SbF6
> LiVFe
—> KRuF6
AgAuF4
A0)
A1)
A2)
A3)
A4)
Трифторид брома не всегда удается полностью удалить из про-
продуктов реакции. Это объясняется сольволизом продуктов реакции
или тем, что взаимодействие кислоты и основания по реакции ней-
нейтрализации
BrFJ + BrFj z=* 2BrF3 A5)
не проходит до конца.
Иногда в трифториде брома происходит стабилизация высоких
окислительных состояний. Так, до сих пор не удалось выделить
тетрафторид палладия, но соли типа K2PdF6 легко выделяются из ра-
растворов хлорпалладитов щелочных металлов в трифториде брома 46.
Из растворов в этом растворителе были получены 47 комплексы
хрома(У) — KCrOF4.
278 ГЛАВА СЕДЬМАЯ. ГАЛОГЕНЫ И ИНТЕРГАЛОГЕНЫ КАК РАСТВОРИТЕЛИ
Следует указать, что структуры кислотного и основного ионов
в системе трифторида брома до сих пор не изучены в должной мере.
В различных работах сообщалось, что анион соединения KBrF4
имеет тетраэдрическую 48>49 или, что представляется более вероят-
вероятным, плоскую 60 конфигурацию; ион BrF| окончательно пока
не идентифицирован. Поскольку BrF3 и SbF8 образуют между собой
ряд соединений 15, вполне возможно, что в этих соединениях имеются
различные катионы. Тем не менее аналогия с аддуктами остается
достаточно близкой. Принимая во внимание большое значение этого
реагента для препаративных целей, можно заключить, что, хотя
механизм процесса самоионизации не выяснен до конца, тем не ме-
менее известная модель служит полезной основой для дальнейших
исследований.
В. Соединения типа АВ3
В настоящее время известны BrF5, IF3 и C1F5. Пентафторид
хлора (?пл .= —100° С) получен недавно при действии фтора на три-
фторид хлора при 350° С и давлении 250 атм. Изучены физические
свойства BrF6 и IF5:
Соединение BrF5 IFc,
tm, pC -61 9,4
«кип. рС 40,5 100,5
р, г/смз 2,46 B5D С) 3,19 B5° С)
8 8 B5° С) 36,2 C5° С)
х, ojk-i ¦ см-1 9-10-8 B5° С) 5,4-Ю"8 B5° С)
Д#исп при tKan, ккал/моль ... 6,7 9,8
5ИСП, ккал/(моль- град) 23,3 26,5
¦Л, мпз 6,2 B4° С) 21,9 B5"? С)
Однако о свойствах C1F6 и BrF6 как растворителей практически
ничего не известно, за исключением того, что трифторид брома
и фтористый водород смешиваются с пентафторидом брома в любых
соотношениях. Возможность комплексообразования пентафторидов
брома и хлора с фторидами щелочных металлов или оловаAУ)
и сурьмы(У) не изучена. (Сообщение о существовании соединения
K2C1F7, или KC1F6-KF, было подвергнуто резкой критике 13 и, по-
видимому, не имеет достаточных оснований).
Свойства пентафторида иода изучены более подробно 13> 32> 29> 34~
38,58, 59_ Многочисленные исследования указывают на то, что пента-
пентафторид иода — ассоциированная жидкость. Изучая зависимость
числа, ширины и положений резонансных линий 19F от температуры,
авторы работы 29 установили, что обмен фтора протекает через об-
образование димерных промежуточных молекул. Небольшая удель-
удельная электропроводность жидкости сильно возрастает при растворе-
растворении в ней иодата калия или пентафторида сурьмы. При электролизе
раствора иодата калия в IF6 на катоде выделяется иод, а эквивалент-
эквивалентная электропроводность его при бесконечном разбавлении равна
III. ИНТЕРГАЛОГЕНЫ
279
35,5 см2/(ом-г-экв). Природа присутствующих в растворе ионов
не установлена.
Пентафторид сурьмы и фторид калия могут реагировать с пен-
пентафторидом иода с образованием соединений ISbF10 и KIFe. Оба
эти соединения хорошо растворимы в горячем и слабо — в холодном
пентафториде иода. Калиевая соль — хорошо измученное соединение,
может быть получено также действием пентафторида иода на нитрат
калия в или иодид калия 22. Кристаллическая структура этого со-
соединения рентгеноструктурным методом не изучена, но, по-видимому,
не приходится сомневаться в правильности формулы K+IFe-
Большие сомнения вызывает природа аддукта пентафторида сурьмы
с IF5. На основании данных об электропроводности раствора и того,
что при взаимодействии KIFe и ISbF10 в IF6 образуется гексафтор-
антимонат калия, этому аддукту приписана формула IF^SbFe.
Процесс-нейтрализации, таким образом, может быть представлен
уравнением
K+IFj + IFJSbFj ^=? KSbFe + 2IF5 A6)
При добавлении BF3 к пентафториду иода повышается его элек-
электропроводность; при пропускании газообразного BF3 через раствор
KF в IF6 образуется калийфторборат. Хорошо растворяется в пен-
пентафториде иода трехокись серы; она также повышает электропро-
электропроводность IF6. Дистилляцией раствора SO3 в IF6 можно получить
азеотропную смесь состава IF6 -1,17 SO3. Вполне возможно, что
в действительности образуется фторсульфонат; такие соединения
получены были другими методами 33.
Аддукты с пентафторидом иода дают6 также N2O5, WO3, MoO3
и-КЮ4. Подробно была изучена система HF—IF5, но каких-либо
соединений в ней обнаружено не было.
В работе по изучению природы растворов синего цвета, получа-
получающихся при растворении иода в IF5, было показано, что они содер-
содержат несольватированный парамагнитный ион I +, образующийся
по реакции
IF5 + 2I2 +± 5I++5IF? A7)
При добавлении фторида калия ион I + соединяется с интергалоге-
интергалогеном; равновесие реакции A7) сдвигается влево, и растворы окра-
окрашиваются в коричневый цвет6. Подобные синие растворы были
получены 6 и с 12О4 в IF5.
В холодном растворе KI в пентафториде иода устойчив также
иод(Ш), существующий в виде соединения KIF4 (в результате ре-
реакции выделяется элементарный иод) 22.
Г. Соединения типа АВ7
Единственным представителем этого класса соединений, извест-
известным в настоящее время, является гептафторид иода, имеющий очень
узкий интервал жидкого состояния. Он плавится при 5—6° С при
280 ГЛАВА СЕДЬМАЯ. ГАЛОГЕНЫ И ИНТЕРГАЛОГЕНЫ КАК РАСТВОРИТЕЛИ
давлении 2 am; давление паров твердого продукта достигает 760 мм
при 4,5° С. Эта же температура является точкой кипения слегка
переохлажденного жидкого" гептафторида иода. Как отмечал Грин-
вуд 17, единственное известное числовое значение SHcn (приписыва-
(приписывается ассоциированной жидкости), равное 26.4 кал/(град-молъ),
неверно; оно рассчитано из теплоты сублимации твердого продукта,
а не из теплоты испарения жидкости. Истинное значение, безусловно,
должно быть значительно ниже.
Сведения о химических свойствах гептафторида иода очень огра-
ограничены. Фториды натрия, калия и рубидия не растворяются и не вза-
взаимодействуют с ним 42. При взаимодействии фторида цезия и гепта-
гептафторида иода при высоких давлениях и умеренных температурах,
по-видимому, может образоваться соединение, содержащее hohIFs; в
скором времени будут опубликованы результаты изучения реакции
2IF7
A8)
протекающей в гептафториде иода.
Имеется краткое сообщение, в котором описываются аддукты
IF7-AsF5 и IF7-3SbF5. Для первого соединения, на основании того
что при взаимодействии с фторидом калия оно разлагается на геп-
тафторид и калийгексафторарсенат 43, предложена структура
IFJ AsFe.
ЛИТЕРА ТУРА
L. J., Keefer R. M., Adv. inorg. Chem. Radiochem.,
К a n d a E., Bull. Chem. Soc. Japan, 12, 455 A937).
Symons M. С R., Quart. Rev. (London), 16,^282
J. L., S il ver t ho r n M, E., I. Am.
N. N., Wh ar m by D. H. W.,
P. L., J. Chem. Soc,
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
Andrews
3, 91 A961).
А о у a m a S.,
Arotsky J.,
A962).
Asprey L. В., Margrave
Chem. Soc, 83, 2955 A961):
Aynsley E. E., Greenwood
J. Chem. Soc, 1963, 5369.
Aynsley E. E., Nichols R., Robinson
1953, 623.
Banks A. A., D a v i e s A., R u d g e A. J., J. Chem. Soc, 1953, 732.
Biltz W., Jeep K., Z. anorg. Chem., 162, 32 A927).
В i 1 t z W., M e i n e с k e E., Z. anorg. Chem., 131, 4 A923).
В о s w i j k K. H., W i e b e n g s E. H., Acta cryst., 7, 417 A954).
El ma R. J., de Boer J. L., Vos A., Acta cryst., 16, 243 A963).
Emeleus H. J., Gutmann V., J. Chem. Soc, 1949, 2979.
E m e 1 ё u s H. J., S h a r p e A. G.
Finkelstein W., Z. pliys. Chem.
Fischer J., L i i m a t a i n e n R.
77, 5848 A955).
G i a u q u e W. F., Powell Т. М., J. Am, Chem. Soc, 61, 1970 A939).
Greenwood N. N., Rev. Pure Appl. Chem., i, 84 A951).
Greenwood N. N., In Mellor's «Comprehensive Treatise on Inorganic
and Theoretical Chemistry, Suppl. II, Part 1, 1956.
Gutmautn V., Angew. Chem., 62, 312 A950).
Gutmann V., Z. anorg. Chem., 264, 151 A951).
Gutmann V., Monatsh., 82, 156 A951).
J. Chem. Soc, 1949, 2206.
121, 46 A926).
Bingle J., J. Am. Chem. Soc,
литература
281
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
.35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
Hargreaves G. В., Р е а с о С k R. D., J. Chem. Soc, 1960,2373.
Hepworth M. A., Peacock R. D., R о b i n s о n P. L., J. Chem.
Soc. 1954, 1197.
Hu J. H., White D., Johnston H. L., J. Am. Chem. Soc, 75,
5642 A953).
J a n d e r G., Die Chemie in Wasserahnlichen Losungsmitteln, Berlin, 1949.
Leech H. R., In Mellor's «Comprehensive Treatise on Inorganic and Theo-
Theoretical Chemistry», Suppl. II, Part I, 1956.
M с I n t о s h D., Proc. Roy. Soc, 16, III, 302 A922).
Moessen G. W., Kraus C. A., Proc Nat. Acad.
1023 A953).
" " Phillips W. D., J.
E. L.,
J. W., J. Am. Chem.
man H. H., К a t z
Muetterties
322 A957).
Oliver G. D., Grisard
Quarterman L. A.,Hy
61, 912 A957).
Rabinowitsch M., Z. phys. Chem., 119, 81 A926).
Rob-erts J. E., Cad у G. H., J. Am. Chem. Soc, 82,
Rogers M. Т., Gar ver E. E.
Rogers M. Т., К at z J. J., J.
" -' - " " R
Sci., USA, 38,
Am. Chem. Soc, 79,
Soc, 74, 2705 A952).
J. J., J. Phys. Chem.,
D.
352 A960).
J. Phys. Chem., 62, 952 A958).
Am. Chem. Soc, 74, 1375 A952).
, Speirs J. L., J. Am. Chem.
Panish M. В., J. Phys. Chem.,
H. В., Speirs J. L., J. Am. Chem.
Rogers M. Т., Pruett
Soc, 77, 5280 A955).
Rogers M. Т., Speirs J. L.
61, 366 A957).
Rogers M. T., Thompson
Soc, 76, 4841 A954).
Rogers M. T. et al., J. Am. Chem. Soc, 76, 4843 A954).
Rogers M. T. et al., J. Am. Chem. Soc, 78, 936 A956).
Schmeisser M., Scharf E., Angew. Chem., 72, 324 A960).
Schumb W. C, Lynch M. A., Ind. Eng. Chem., 42, 1383 A950).
Seel F.,Detmar O., Angew. Chem., 70, 163, 470 A958).
Selig H., Shamir J., Inorg. Chem., 3, 294 A964).
Sharpe A. G., J. Chem. Soc, 1950, 3444.
Sharpe A. G., J. Chem. Soc, 1953, 197.
Sharpe A. G., Woolf A. A., J. Chem. Soc, 1951, 798.
Si eg el S., Acta cryst., 9, 493 A956).
Si eg el S., Acta cryst., 10, 380 A957).
Sly W. G., Marsh R. E., Acta cryst., 10, 378 A957).
Smith D. F., Science, 141, 1039 A963).
Taylor N. W., H il de brand J. H., J. Am. Chem. Soc, 45, 685
A923).
V о n k С G., W i e b e n g a E.
Waentig G., Mclntosh
Wheat J. A., Browne A.
Wheat J. A., Browne A. W.,
A936).
Wheat J. A., Browne A. W.,
Woolf A. A.,
Woolf A. A.,
Woolf A. A.,
Zelezny W. F., В a e n z i g e r N. C, J.
A952).
Плотников В.
Плотников В.
АН УССР, 7,
Рысс И. Г.,
издат, 1956.
Н., Acta cryst., 12, 859 A959).
D., Proc. Roy. Soc. Can., 9, 207 A915).
W., J. Am. Chem. Soc, 58, 2410 A936).
J. Am. Chem. Soc, 60, 1575, 1577
Browne A. W., J. Am. Chem. Soc, 60, 371 A938).
J. Chem. Soc, 1950, 1053.
J. Chem. Soc, 1950, 3678.
Emeleus H. J., J. Chem.
Soc, 1949,
Am. Chem.
2865.
Soc,
74, 6151
А., Я к у б с о н С, Z. phys. Chem., 138, 235 A928).
А., М и х а й л о в с к а я В. И., Зап. инст. химии
V» 1, 85 A940).
Химия фтора и его неорганических соединений, Госхим-
ГЛАВА ВОСЬМАЯ
ГАЛОГЕНИДЫ И ОКСИГАЛОГЕНИДЫ
ЭЛЕМЕНТОВ V ГРУППЫ
Д. С. ПЭЙН
I. Введение 282
II. Экспериментальные методы исследования растворов в галогенидах
и оксигалогенидах элементов V группы 287
A. Рассмотрение качественных данных о растворимости 288
Б. Изучение твердой фазы ' 288
B. Соединения и ионы, образующиеся в результате перехода элек-
г ттРона 288
i. Измерение электропроводности 289
Д. Измерение чисел переноса 290
Е. Потенциометрическое титрование 290
Ж. Титрование с индикатором 291
3. Спектрофотометрические измерения 291
И. Измерение вязкости 291
К. Термохимические измерения ' 292
Л. Реакции изотопного обмена ° 292
М. Эбулиоскопические и криоскопические измерения 292
III. Галогениды как растворители \ 292
A. Трихлорид мышьяка - . . \ \ 293
Б. Трифторид мышьяка .'..'....' 299
B. Трибромид мышьяка ' зОО
Г. Трихлорид сурьмы '..'..'. 303
Д. Триоромид сурьмы 308
Е. Трииодид сурьмы '..'.'. 309
Ж. Пентахлорид сурьмы '..'.'. 309
3. Трихлорид висмута . . . 309
IV. Оксигалогениды как растворители . . . 309
A. Нитрозилхлорид ' зю
Б. Нитрозилфторид .... ^^
B. НитрОНИлфторИД • ¦ • ^gjg
Г. Фосфорилхлорид . . 314
V. Заключение '. . '. 328
Литература q^q
I. ВВЕДЕНИЕ
После того как Кейди 40 установил сходство между процессами
растворения неорганических солей в жидком аммиаке и в воде, вни-
внимание исследователей было обращено на поиски других жидкостей,
обладающих подобными же свойствами. Брюль 37 предположил, что
такими растворителями могут служить галогениды некоторых'эле-
некоторых'элементов, например галогениды фосфора или мышьяка. Но первая
I. ВВЕДЕНИЕ
283
экспериментальная работа была проведена с трпхлоридом сурьмы
в качестве такого растворителя 181.
Классические работы Вальдена 188-190 по неводным растворите-
растворителям посвящены изучению свойств галогенидов и оксигалогенпдов
фосфора и галогенидов мышьяка и сурьмы. Некоторые из этих со-
соединений являются хорошими растворителями для неорганических
солей. Образующиеся растворы обычно обладают высокой электро-
электропроводностью. Процесс растворения интерпретировали как процесс
ионизации и растворителя, и растворенного вещества. До последнего
времени галогениды и оксигалогениды не привлекали к себе присталь-
пристального внимания как растворители. Лишь последние работы, в част-
частности работы австрийских ученых — В. Гутмана и сотр., вызвали
живой интерес к этой группе соединений. В настоящее время в данной
области работает уже большое число ученых, а техника работы с этими
легко гидролизующимися растворителями освоена и проверена доста-
достаточно надежно. В ряде случаев удалось установить природу ионов,
присутствующих в растворе. Основное внимание исследователи об-
обращают на детальное изучение физико-химических свойств этих
систем. Сравнительная простота реакций взаимодействия между рас-
растворителем и ионами, присутствующими в растворе, естественно,
привлекает внимание электрохимиков к этим растворителям, хотя
экспериментальная работа с ними сопряжена с серьезными труд-
трудностями 1в5.
Элементы V группы, за исключением азота, образуют два основ-
основных типа галогенидов — три- и пентагалогениды. Известны и другие
галогениды, например тетраиодидфосфин P2Ij и монохлорид вис-
висмута BiCl. Однако обсуждать возможность использования их в ка-
качестве растворителей, на наш взгляд, нецелесообразно, так как эти
соединения удалось получить в слишком малых количествах.
Оксигалогениды этой группы охватывают очень широкий диапа-
диапазон структур; от простых молекулярных соединений, таких, как
нитрозил- или фосфорилгалогениды, до твердых оксигалогенидов
висмута, имеющих слоистую структуру. Характерной особенностью
химии галогенидов и оксигалогенидов азота, фосфора, мышьяка,
сурьмы и висмута является то, что эти соединения легко взаимодей-
взаимодействуют с нуклеофильными реагентами. Все они также легко гидро-
лизуются, поэтому если результатам эксперимента придается боль-
большое значение, то работу необходимо проводить в абсолютно обез-
обезвоженной системе. Такие условия можно создать, в частности, в ва-
куумированной системе. Надо обратить внимание на то, что все га-
галогениды и оксигалогениды, как правило, реагируют с углеводород-
углеводородными смазками, и необходимо создавать специальные приспособле-
приспособления.
Для стереохимии соединений мышьяка, сурьмы и висмута ха-
характерна тетраэдрическая (четверная) или октаэдрическая (шестер-
(шестерная) координации. В химии фосфора, в особенности его пентагало-
284 ГЛАВА ВОСЬМАЯ. ГАЛОГВНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
генида, тетраэдрическая координация является определяющей. Так,
в твердом пентахлориде фосфора имеется ион РСЦ- Для остальных
элементов V группы, по-видимому, более характерной является окта-
эдрическая координация. В частности, именно этим объясняется
образование комплексных анионов SbClg и AsFg. Стабильность
ионов, особенно октаэдрических, во многом зависит от размера и ве-
величины электроотрицательности присоединенных атомов. Лишь очень
сильно электроотрицательные ионы фтора, хлора и, вероятно, брома
образуют комплексные ионы при участии электронов d-орбиталей,
что весьма существенно для образования соединений с координацион-
координационным числом больше четырех 45. Кроме того, лишь в случае ионов
фтора и хлора соотношения между размерами ионов благоприят-
благоприятствуют образованию соединений с высокими координационными чис-
числами. Значения электроотрицательностей фосфора, мышьяка, сурьмы
и висмута очень близки друг к другу (от 2,06 до 1,67), а соответству-т
ющие электроотрицательности галогенов значительно выше (от 4,0
до 2,2). Благодаря полярному характеру большинства связей между
галогенами и элементами V группы молекулы этих соединений могут
взаимодействовать с ионами и другими полярными молекулами, что
во многих отношениях определяет поведение растворителя. В окси-
галогенидах сильно полярны кислородные связи.
Характерная черта растворителя —его способность переводить
в одну, как правило, жидкую фазу (раствор), одно или несколько
других соединений, обычно твердых веществ. Механизм этого явле-
явления не сводится только к разъединению и сольватации ионов, при-
присутствующих в. кристаллической решетке, а включает также взаи-
взаимодействие между молекулами растворителя и растворенного ве-
вещества, которое часто приводит к образованию совершенно новых
соединений. О наличии или отсутствии подобного взаимодействия
в системе можно судить по алгебраической сумме величин свобод-
свободных энергий отдельных стадий процесса. Числовые значения этих
величин могут сильно меняться для различных систем. Однако для
определенных типов растворителей и растворенных веществ высокое
суммарное значение энергии обычно соответствует высокой раствори-
растворимости. К таким типам соединений относятся тригалогениды, в част-
частности тригалогениды мышьяка и сурьмы, и оксигалогениды, в част-
частности нитрозил- и фосфорилхлориды.
Растворы некоторых соединений в галогенидах и оксигалогени-
дах неэлектропроводны. Эта группа соединений очень немногочис-
немногочисленна и подробно рассматриваться здесь не будет; основное внимание
будет уделено веществам, которые являются электролитами в гало-
галогенидах и оксигалогенидах. Последние играют роль ионизирующих
растворителей. Поскольку свойства водных растворов в значительной
мере определяются высоким значением диэлектрической проницае-
проницаемости воды, по аналогии при рассмотрении неводных растворителей
также обращают внимание на величины е. Из данных табл. 38
Таблица 38. Физические свойства галогенидов
и оксигалогенидов элементов V группы
Соедине-
Соединение
PF3
РС1з
РВг3
Pis
AsF3
AsClJ
AsBrJ
Asl3
SbF3
SLC1S
SbBrJ
Sbl3
BiF3
BiClJ
BiBr3
Bil3
PF5
PC15
PBr3
AsF3
SbFr,
SbCl5
BiF5
FNO
C1NO*
BrNO
FNO2
CINO2
,FNO3
CINO3
POF3
POCIJ
POBr
-151
-112
-40
61
-6
—13
35
- 146
292
73
97
167
725
232
218
408
-94
—
< 100
-80
7
5
< 160
—132,5
-61.5
—55,5
-166
-141
-181
— 107 (в
вакууме
—39
1
56
w °c
—101
74
173
разлаг.
>200
63
130
220
403
субл. 319
221
280
401
—
447
460
>500
-84
субл. 167
разлаг.
> 106
-53
150
— 140
230
—60
-6
~0
—72
-14
-46
18
-40
108
192
S
_
3,5 A7° С)
4,7 B2° С)
3,9 B0° С)
4,1(~65° С)
5,7 «-6° С)
12,6 A7° С)
8,8 C5° С)
7,0 (-150° С)
—
33,0 G5° С)
20,9 (~ 100° С)
13,9 (~ 175° С)
—
—
—
—
—
2,7 A65° С)
2,85 (ж. при
160° С)
—
—
—
3,2B1° С)
—
—
22,5 (—27,5° С)
19,7 (-10,0° С)
13,4 A5,2° С)
—
—
—
—
13,9 B2° С)
—
ц, спз
—
—
—
—
1,225 B0° С)
5,41 C5° С)
4,44 D0° С)
—
—
3,3 (95° С)
6,81 A00° С)
—
—
32,0 B60° С)
—
—
—
—
—
—
—
—
2,16 B5° С)
—
—
0,586 (-27° С)
0,547 (-20° С)
—
—
—
—
—
—
1,15 B5° С)
—
р, г/см3
1,56 B1° С)
2,85 A5° С)
—
2,67 @° С)
2,16 B0° С)
3,33 E0° С)
4,69 B5°ЬС)
4,38 B1° С)
2,44 A78° С)
4,15 B3° С)
4,85
5,32 B0° С)
4,75 B5° С)
5,72 B5° С)
5,88 A7,5° С)
—"
1,60A60° С)
—
3,40 (-73°ЬС)
2,99 B3° С)
2,35 B0° С)
5,40 B5° С)
1,33 (-60°?С)
1,59 (-6°.С)
—
1,49 (-73° С)
1,41 (-15° С)
1,51 (-46° С)
—
—
1,71 @° С)
2,82 D5° С)
* Соединения, обладающие хорошей растворяющей способностью.
286 ГЛАВА ВОСЬМАЯ ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
видно, что диэлектрические проницаемости галогенидов и оксига-
логенидов элементов V группы выше, чем у многих органических
растворителей, но малы по сравнению с диэлектрической проницае-
проницаемостью таких растворителей, как вода (е = 81,1 при 18° С), безвод-
безводный фтористый водород (е = 83,6 при 0° С) или пентафторид пода
(е = 36,2 при 25° С). Однако при взаимодействии растворителя и рас-
растворенного вещества происходит не только ослабление кулоновскпх
сил; галогенид и оксигалогенид по отношению к растворенному ве-
веществу часто выступают как акцептор или донор галоидных ионов.
При этом в процессе растворения образуются новые ионы. Они и вы-
вызывают самый живой интерес исследователей, работающих с невод-
неводными растворителями.
С целью единого подхода к ионизирующим растворителям
при рассмотрении ионов, встречающихся в различных растворителях,
условились применять термины «кислота» и «основание». Опреде-
Определение «кислоты» и «основания», данное Бренстедом и Лоури, оказа-
оказалось совершенно неприменимым к галогенидным и оксигалогенид-
ным растворителям. Более приемлема теория Льюиса, согласно ко-
которой кислота рассматривается исключительно как акцептор электро-
электронов, но и здесь имеются некоторые неясности. Гораздо лучше соот-
соответствует галогенидным растворителям определение кислоты и осно-
основания по теории сольво- систем, развитой Гутманом и Линдквпс-
том 10°, где внимание фокусируется на процессах перехода ионов.
Изучение любой сольво-системы сводится главным образом к вы-
выяснению природы присутствующих ионов. Для интерпретации про-
процесса растворения в свете теории сольво-систем соединений необ-
необходимо разделить суммарный процесс ионообразования на отдель-
отдельные стадии, соответствующие переходу ионов, например, «кислота» -}-
+ галоген-ион = «основание» (сравн. кислота = основание -f- про-
протон в водных системах) или«основание» + растворитель = «кислота»+
+ галоген-ион (сравн. кислота + Н2О = основание + протон). Все
галогенидные растворители анионотропны, что содействует переходу
фтор-, хлор- и иод-иона. При этом «кислоты» являются акцепторами
анионов, а «основания» — донорами анионов. В галогенидном рас-
растворителе МГ„ существует равновесие
г-
мг„ + мг„ 5=
кислота I основание II
: МГй+1 + мг+_х
основание I кислота II
A)
Такой растворитель амфотерен и может реагировать и как кислота,
и как основание. Добавление кислоты к такому растворителю при-
приводит к уменьшению концентрации галоген-иона в растворителе.
Например, добавление акцептора анионов АГ4 приводит к образо-
образованию ионов АГ§~; при этом концентрация ионов Г ~, связанных
с растворителем, уменьшается. Добавление основания вызывает
II. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
287
соответствующее увеличение концентрации галоген-иона за счет
непосредственного высвобождения его из молекулы основания, как,
например, в случае добавления галогенида тетраметиламмония.
Применимость этой теории как единого подхода к так называемым
кислотно-основным реакциям ограничена серьезными трудностями,
связанными с надежной идентификацией ионов, присутствующих
в растворе. Наилучшим образом применима эта теория к простым
галогенидам. Например, в случае трехфтористого мышьяка пере-
переход фтор-иона может быть описан следующим образом:
AsF3
кислота I
f AsF3 ^=Г AsFJ
основание II кислота II
+ AsFj
основание I
B)
В этом^примере основания II и I — доноры фтор-ионов, а кислоты
I и II — акцепторы этих ионов. В растворе в трифториде мышьяка
фторид калия является основанием, так как служит донором фтор-
иона, в растворе при этом образуется анион AsFj, в то время как
пентафторид сурьмы выполняет функции кислоты, присоединяя фтор-
ион с образованием аниона SbFe и катиона AsF?.
Применение теории сольво-систем соединений к растворам в фос-
форилхлориде встретило определенные трудности, связанные с ин-
интерпретацией различных ионов, присутствующих в растворе. Под-
Подробнее эта частная проблема будет рассмотрена позднее. Здесь ука-
укажем лишь, что более простым является подход к ней с позиций льюи-
совских взглядов на кислотно-основное взаимодействие 148. Вообще,
распространять понятия «кислоты» и «основания» на иные классы
растворителей помимо протонных, по-видимому, не имеет смысла,
за исключением разве тех случаев, когда такая интерпретация дает
очевидные преимущества. Толкование реакции катиона с анионом
как «нейтрализации» «кислоты» и «основания» практически не дает
ничего нового для понимания системы. Напротив, применение этих
терминов часто требует дополнительных объяснений, хотя общая
картина от этого яснее не становится.
II. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
РАСТВОРОВ В ГА.ЛОГЕНИДАХ И ОКСИГАЛОГЕНИДАХ
ЭЛЕМЕНТОВ V ГРУППЫ
Выяснение природы проводящих ток растворов всегда составляло
одну из основных задач электрохимии. Однако теории и экспери-
экспериментальные методы электрохимии разрабатывались главным образом,
применительно к водным растворам, и поэтому не могут быть непо-
непосредственно перенесены на системы галогенидов и оксигалогенидов,
хотя в последние годы такие попытки предпринимались.
288 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
А. Рассмотрение качественных данных о рдстворимости
Изучение растворимости самых различных соединений, пусть
даже и не количественное, позволяет в какой-то степени раскрыть
сущность процессов, происходящих при растворении, а также ука-
указывает на общие особенности растворителя. Начиная исследование,
прежде всего интересно установить, растворимы ли в данном раство-
растворителе типичные соли, такие, как галогениды щелочных металлов,
четвертичные аммониевые галогениды, а также отдельные соединения,
например хлориды трехвалентного железа, четырехвалентных олова
и титана и пятивалентной сурьмы. В некоторых случаях изменение
окраски раствора, сопровождающее процесс растворения, может
указывать на наличие комплексообразования. Получив ответ на зти
вопросы, можно не только представить общую картину поведения
различных соединений в данном растворителе, но и выбрать те
из них, на которые в дальнейших исследованиях обращается наиболь-
наибольшее внимание.
Б. Изучение твердой фазы
Природа твердых фаз, выпадающих из растворов при изменении
их концентрации или температуры, имеет особое значение для изу-
изучения растворителя. Эти фазы часто описывают как сольваты, на-
например из раствора пентахлорида сурьмы в фосфорилхлориде вы-
выпадает соединение SbCl5 • РОС13. Иногда образующуюся твердую фазу
рассматривают как комплексное соединение растворителя с рас-
растворенным веществом, например (CHg^N+AsCl^ — комплекс, вы-
выпадающий из раствора хлорида тетраметиламмония в трихлорпде
мышьяка. Лишь в некоторых случаях эти фазы были подвергнуты
полному рентгеноструктурному анализу, но большинство исследо-
исследователей ограничивалось установлением химической формулы осадка,
т. е. отношения числа молекул растворителя к числу молекул рас-
растворенного соединения в осадке. Иногда, сравнивая эти отношения
для ряда растворенных соединений, можно сделать вывод о хими-
химической природе сольвата. Однако трудно с определенностью судить
о взаимосвязи между присутствующими в растворе ионами и выпав-
выпавшей твердой фазой. Дело в том, что лишь при очень низких кон-
концентрациях присутствующие ионы могут находиться в равновесии
с твердой фазой. Если установлено, что осадок — комплексное со-
соединение, резонно считать (если не доказано противоположное), что
в растворе имеются значительные концентрации комплексного иона.
В. Соединения и ионы, образующиеся
в результате перехода электрона
В некоторых случаях, добавляя в раствор соответствующие
металлы, можно изучить эффект перехода электронов к катиону рас-
растворителя. Это особенно полезно, когда образующиеся продукты
II. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
.289
легко могут быть выделены и идентифицированы. Например, обра-
образование окиси азота при добавлении цинка к нитрозилхлориду мо-
может служить доказательством присутствия нитрозил-иона 2. Однако
_этот метод имеет ограниченную применимость в связи с трудностью
изучения нелетучих и растворимых продуктов.
Еще одним методом может явиться электролиз раствора и изу-
изучение электродных продуктов, но результаты могут быть положитель-
положительными, только если продукты легко выделить.
При изучении самоионизации чистого растворителя методом элек-
электролиза встречаются значительные трудности в связи с очень низкой
электропроводностью растворителя; тем не менее в одном случае,
а именно при изучении фосфорилхлорида, этот метод был применен
весьма успешно 179.
Г. Измерение электропроводности
При работе с галогенидами и оксигалогенидами, выступающими
в качестве ионизирующих растворителей, очень большое значение
имеет измерение электропроводности. Самоионизация растворителя
представляет особый интерес и может быть непосредственно опреде-
определена путем измерения электропроводности чистой жидкости. Вполне
понятно, что, кроме того, подобные измерения могут служить кри-
критерием чистоты продукта, так как электропроводность чистых жид-
жидкостей обычно очень низка, а примеси, присутствующие в раствори-
растворителе, повышают электропроводность. Данные об электропроводности
чистых галогенидных растворителей указывают, что электропровод-
электропроводность, по-видимому, соответствует изменению устойчивости анионов
и катионов в ряду РС13 < AsCl3 < SbCl3 < BiCl3.
Хотя измерение электропроводности чистых растворителей пред-
представляет интерес, так как на основании этих данных можно выяснить,
применима ли к неводным растворам современная теория электро-
электролитов, имеющиеся к настоящему времени данные об электропровод-
электропроводности, которые могли бы быть проверены по теории Дебая — Хюк-
келя, весьма скудны. Как правило, исследователи ограничиваются
изучением изменения электропроводности при добавлении раствора
одного растворенного соединения к раствору другого соединения.
Возникающие при этом изменения зависят от природы самой про-
протекающей реакции.
Кольтгофф и Латинен 136 описали формы кривых кондуктометри-
ческого титрования для ряда водных кислот и оснований различной
силы. Эти кривые имеют две характерные особенности. Во-первых,
если в результате реакции образуются ионы, обладающие различной
подвижностью, или суммарное количество ионов уменьшается (или
увеличивается), на кривой зависимости электропроводности от моль-
мольного отношения компонентов в эквивалентной точке наблюдается
перегиб. Во-вторых, изменение электропроводности до и после экви-
19 Заказ 90
290 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
валентной точки обычно характеризуется плавной кривой, часта
прямой линией, и поэтому эквивалентная точка может быть полу-
получена пересечением двух кривых. Форма кривых очень сильно зависит
от самых различных факторов, но все же можно утверждать, чта
наличие такого пересечения двух отрезков кривых свидетельствует
о существовании определенного химического иона при данном моль-
мольном отношении. Этот метод хорошо применим к галогенидным и окси-
галогенидным растворителям вследствие сильной склонности этих
растворителей к образованию комплексных галогенидных ионов
с ионами электролитов и с неэлектролитами.
Д. Измерение чисел переноса
Этот метод является основным при изучении ионов, присутству-
присутствующих в растворе. Однако применение его для систем с галогенидом
или оксигалогенидом как растворителем связано со значительными
экспериментальными трудностями, так как приходится измерять
очень небольшие изменения состава анолита и католита 97.
Е. Потенциометрическое титрование
Галогенидные и оксигалогенидные растворители могут служить
электродами * для определения активности галоген-иона точно так
же, как водородный электрод служит для измерения изменений ак-
активности ионов водорода в водных растворах. Подвижность галоген-
иона играет важную роль в процессе проводимости. Активность ионов
удобно представлять в форме рГ = lg [Г~], предполагая, что коэф-
коэффициент активности равен единице. До сих пор применялись лишь
индикаторы хлор-ионов и их использование ограничивалось потен-
циометрическим титрованием. Хлорсеребряный индикаторный элек-
электрод может быть применен в концентрационной ячейке типа
Ag, AgCl|Cl-(Cl)||Cl-(C2)|AgCl, Ag
в трихлориде мышьяка 5; электрод сравнения, состоящий из раствора
тетраметиламмонийхлорида известной концентрации, отделен от ти-
тровальной ячейки притертой стеклянной пробкой. Однако хлорсе-
хлорсеребряный электрод нельзя применять для изучения растворов в фос-
форилхлориде. В качестве электродных металлов изучались медь,
серебро, золото, магний, цинк, ртуть, алюминий, свинец, молибден
и платина 101. Из них для создания чувствительного индикаторного'
электрода оказался подходящим лишь молибден. Работа этот
электрода обусловлена, по-видимому, установлением равновесия
МоС12 +± Мо2+ + 2СГ. Для более точного измерения э. д. с. в сильно
* Под понятием «электрод» авторы подразумевают металл в электропровод-
электропроводном контакте с электролитом. — Прим. ред.
II. ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ
291
разбавленных растворах успешно использовались каломельный
и хлорный электроды
90
Ж. Титрование с индикатором
Сульфофталеины, хорошо известные в качестве индикаторов во-
водородных ионов в водных системах, могут выполнять также роль
индикаторов хлор-ионов в растворах в фосфорилхлориде ". Обрати-
Обратимые цветовые изменения в РОС13 обычно отличаются от изменений
окраски, возникающих в водных системах. Титрование с исполь-
использованием этих индикаторов может проводиться точно так же, как
и- в водных растворах. Этим методом удается установить стехиоме-
трические соотношения в различных реакциях. Эти индикаторы
можно использовать также для сравнения относительной силы до-
доноров хлЬр-ионав.
3. Спектрофотометрические измерения
С помощью спектроскопических методов исследования успешно
решается проблема идентификации и оценки относительных коли-
количеств различных ионов, присутствующих в растворе. К сожалению,
лишь в сравнительно немногих случаях с помощью спектроскопи-
спектроскопических методов можно однозначно идентифицировать ионы, образу-
образующиеся в растворе галогенида или оксигалогенида. В некоторых слу-
случаях, например при наличии в растворе иона тетрахлорферрата(Ш),
этот метод оказался наиболее эффективным13. Методы инфра-
инфракрасной спектроскопии могут дать ценные сведения о природе соль-
ватов. Так, смещение полосы Р = О в серии фосфорилгалогенидных
комплексов указывает на существование донорной кислородной
связи 177.
Изучение спектров комбинационного рассеяния до сих пор про-
проводилось крайне редко. Об этом приходится только сожалеть, по-
поскольку этот метод исследования особенно хорош для изучения
ионов, обладающих простой симметрией, присутствующих в гало-
галогенидных и оксигалогенидных растворителях. При изучении спек-
спектров комбинационного рассеяния в4 было установлено существова-
существование иона NO+. Однако столь же надежных доказательств сущест-
существования других катионов в галогенидах и оксигалогенидах эле-
элементов V группы получить не удалось.
И. Измерение вязкости
Изучение вязкости водных растворов во многом способствует
пониманию их природы, но такие данные о неводных системах очень
скудны. Единственные известные работы, касающиеся галогенидов
и оксигалогенидов, посвящены измерению вязкости расплавов
GaCl3-POCl3 n и GaBr3POBr3. В этих работах было показано
19*
292 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
существование комплексов в расплаве. Имеется еще лишь одна ра-
работа, посвященная измерению вязкости SbBr3 и ацетона, но дока-
доказательств комплексаобразования в этой работе нет.
К. Термохимические измерения
К сожалению, в литературе отсутствуют данные об изменении
энергии в процессе растворения в галогенидных и оксигалогенид-
ных растворителях. Очень мало данных имеется и о термохимии
многочисленных сольватов. Остается надеяться, что это очевидное
упущение вскоре будет ликвидировано, и, таким образом, системы
в этих растворителях можно будет рассматривать на должном уровне.
Л. Реакции изотопного обмена
Для раскрытия природы ионов, присутствующих в растворах,
особую ценность представляют данные о скоростях изотопного об-
обмена с использованием меченых атомов, так как в этом случае пред-
представляется возможным определить степень взаимного перехода иона
галогена между растворителем и растворенным веществом и интер-
интерпретировать полученные данные с кинетических позиций. К насто-
настоящему времени выполнено очень незначительное количество подоб-
подобных экспериментов, но полученные результаты имеют огромное зна-
значение для понимания природы этих растворов 106>142.
М. Эбулиоскопичеекие и криоскопические измерения
Первые работы, а также недавно выполненные работы в этой
области были посвящены изучению некоторых комплексных галоге-
нидов 102 и природы растворов хлоридов титанаAУ), железа(Ш)
и алюминия(Ш) в фосфорилхлориде 103. При измерениях исполь-
использовался эбулиометр Светославского, снабженный термисторами
для измерения разности температур.
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
Для того чтобы галогенид можно было использовать в качестве
растворителя, он должен отвечать определенным требованиям,
а именно быть доступным в необходимых количествах, точка плавле-
плавления его должна быть ниже комнатной температуры или находиться
близко к ней, а точка кипения — гораздо выше этой температуры;
соединение должно быть устойчивым (не распадаться на компоненты),
иметь подходящие диэлектрическую проницаемость, коэффициент
вязкости и скрытую теплоту испарения. Галогениды, вполне отве-
отвечающие требованиям, предъявляемым к растворителям, приведены
в табл. 38. Общая химия этих соединений рассмотрена в двух не-
недавно появившихся работах вз>т. Мы не будем останавливаться
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
293
на галогенидах азота и иодидах, поскольку эти соединения крайне
нестойки. Как будет показано ниже, из галогенидов гораздо более
подробно исследованы хлориды мышьяка(Ш) и сурьмы(Ш). От-
Отчасти это объясняется их доступностью, отчасти исключительными
свойствами их как растворителей и легкостью работы с ними.
Галогениды элементов V группы могут быть рассмотрены с точки
зрения оценки их донорно-акцепторных свойств. В реакциях с А1С13
и ВС13 только трихлорид и трибромид фосфора проявляют заметные,
хотя и слабые донорные свойства. Трихлориды и трибромиды мышь-
мышьяка и сурьмы являются очень слабыми донорами (видимо, их вообще
нельзя считать донорами). Трихлорид и трибромид фосфора при вза-
взаимодействии с триметиламином показывают слабые акцепторные
свойства 183. Подобные же акцепторные свойства характерны для три-
хлоридов^мышьяка и сурьмы при взаимодействии с триметилфосфином
и триэтиламином 109> m. В связи с наличием у трихлорида фосфора
таких слабых акцепторных и донорных свойств, а также вследствие
небольшой диэлектрической проницаемости растворяющая способ-
способность этого растворителя очень ограничена. Даже такие хорошо
растворимые в большинстве галогенидов соединения, как трихлорид
железа и тетраметиламмонийиодид, в этом растворителе лишь.не-
лишь.незначительно растворяются.
А. Трихлорид мышьяка
Молекула трихлорида мышьяка сильно полярна, имеет форму
пирамиды 51. Диэлектрическая проницаемость жидкого трихлорида
мышьяка довольно высока (е = 12,8 при комнатной температуре).
Температурный интервал жидкого состояния —18 — 130° С при ат-
атмосферном давлении. Благодаря этим свойствам и способности три-
трихлорида мышьяка выступать- в качестве акцептора и донора хлор-
иона при комплексообразовании он является очень хорошим раство-
растворителем-.
Как видно из небольшого значения удельной электропровод-
электропроводности 5 чистого трихлорида мышьяка A,4-10 ом ~х-см ~1), сте-
степень перехода хлор-иона в нем крайне мала. Самоионизация раство-
растворителя протекает по реакции
2AsCl3 —-Z AsClJ+AsGlr C>
Одна молекула AsCl3 играет роль кислоты, присоединяя хлор-
ион с образованием иона AsClJ, а другая — роль основания,
отдавая хлор-ион и образуя: АэСЦ-
Из водных систем удалось получить комплексы трихлорида
мышьяка с хлоридами щелочных металлов, например Rb3 AsaClg.
и Cs3As2Cl9 192> 193, и с аминами, например (C2H5NH3JAsCl5;
(CH3NH3KAs2Cl9, (CH3JNH2AsCl4 и [(CH3KNH]2As3Clli4. При на-
нагревании в AsCl3 можно растворить очень незначительные количества
294 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ Л7 ГРУППЫ
хлорида калия. При охлаждении образовавшегося раствора выде-
выделяется твердая соль, соответствующая по составу KAsClJ 77. Тетра-
алкиламмонийхлориды хорошо растворимы в трихлориде мышьяка,
а из образующихся растворов выделяются тетрахлорарсенатные(Ш)
соли, представляющие собой белые негигроскопичные порошки "'.
Осторожной кристаллизацией из раствора тетраметиламмоний-
тетраметиламмонийхлорида в трихлориде мышьяка был получен сольват состава
(CH3LNCb3AsCl3 [или (CH3LNAsCl4-2AsCl3 или (CH3LNAs3Cli0].
Две из трех молекул трихлорида мышьяка в этом соединении связа-
связаны слабо и могут быть удалены14в при нагревании до 100\С при да-
давлении 9 мм рт. ст. Из фазовой диаграммы видно, что в системе
трихлорид мышьяка — тетраэтиламмонийхлорид существуют сое-
соединения (CoH5LNCl -2AsCl3, которое представляет собой, вероятно,
(C2H5LNAsCl4-AsCl3 или (C2H6LNAs2Cl7, и 3 (CaHBLNCl-5AsCls,
являющееся, по-видимому, [(C2H5LN]3 (AsCl4K-2AsCl3 i.
Гутман 77 изучил растворимость в трихлориде мышьяка раз-
различных галогенидов, комплексных галогенидов, окислов, некото-
некоторых цианидов, металлов и металлоидов. Хлориды щелочных метал-
металлов и аммония, а также пентахлориды ниобия и тантала и комплекс-
комплексная соль f(CH3LN]2SnCl6 лишь слегка растворимы, в то время как
хлориды алюминия(Ш), олова(ГУ), ванадия(ГУ), железа(Ш)
и (СН3L NSbCl6 и (CH3LNC1 растворяются хорошо.
Вальден 188 в своей работе обратил внимание на то, что в трихло-
трихлориде мышьяка очень высокой растворимостью обладают самые раз-
различные иодиды. В дальнейшем это подтвердилось. Некоторые метал-
металлоиды, например сера, фосфор и иод, также хорошо растворимы,
но природа этих растворов остается невыясненной. Очень слабо
растворимы металлы, окислы металлов и соли кислородсодержащих
кислот, например сульфаты и нитраты.
Растворы тетраметиламмонийхлорида и пентахлорида сурьмы
в трихлориде мышьяка обладают высокой электропроводностью.
Кривая кондуктометрического титрования этих растворов показана
на рис. 56. Перегиб наблюдается при мольном отношении компо-
компонентов 1:1. Выделить из исходного раствора пентахлорида сурьмы
в AsCl3 хотя бы небольшое количество белого твердого продукта,
приближающегося по составу к AsCl3-SbCl5, не удалось, но было
высказано предположение о присутствии этого соединения в растворе.
После титрования этого раствора может быть выделена соль
1 (CH3LNSbCle. Наблюдаемое изменение электропроводности было
объяснено протеканием реакции
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
295
(CH3LN+AsCl4-(-AsClJSbCh
(CH3LN+ S ЬС]? -f 2AsCl3
D)
Это объяснение, по-видимому, верно, так как единственным
продуктом реакции, вероятно, является гексахлорантимонат (V).
Кроме того, оба раствора обнаруживают первоначально высокую
электропроводность и могут образовывать твердые фазы, в состав
которых входят ионы, по-видимому, образующиеся при переходе
хлор-иона в растворе трихлорида мышьяка. Кривая кондуктометри-
кондуктометрического титрования SnCl4 тет- ЗОг
раметиламмонийхлоридом в
AsCl3 показана на рис. 57.
Тотчас же после достижения V
мольного отношения 1 : 1 начи- _V
нается выпадение осадка состава ^ i
>[(CH3LN]2SnCl6. Перегиб при J3.
отношении компонентов 1:1 5
соответствует установлению ^
равновесия реакции
(CH3LNAsCl4+ SnCl4 ^=*
^± (CH3LNSnCl5 + AsCl3 E)
Молекулы (CH3LNSnCl5 далее
реагируют с ионами AsQ4~ с
образованием иона гексахлор-
т -
О 12 3
Мольное отнешение^СН3L1ЫС]\ :ЁЬС1 Л
Рис. 56. Кривая кондуктометрического
титрования SbCl5 тетраметиламмоний-
хлоридом в трихлориде мышьяка при-
20° С.
станнатаAУ).
Изучение растворов TiCl4 и VC14 этим методом показало сущест-
существование подобных комплексных ионов с мольным отношением ком-
компонентов 1:1. Удалось выделить твердые соединения, приблизи-
приблизительно соответствующие со-
составам 1:1, однако даль-
дальнейшее изучение их не про-
проводилось. Некоторые авторы
указывают на отсутствие оп-
определенного взаимодействия
между трихлоридом мышьяка
и хлоридами этих металлов.
Так, при изучении фаз в
системе AsCl3 — TiCl4 пока-
показано существование полной
I взаимной смешиваемости
30
20
Ю
к
Осадо,
0
3т
Мольное
Рис. 57. Кривая кондуктометрического
титрования SnCl4 в AsCl3 раствором тет-
тетраметиламмонийхлорида при 20° С ".
обоих компонентов. Кроме
того, все растворы имеют
незначительную электропро-
электропроводность 199.
Раствор РС15 в трихлориде
мышьяка электропроводен,
и в отличие от раствора SbClg на кривой кондуктометрического тит-
титрования его раствором тетраметиламмонийхлорида нетперегибов. На
основании этого Гутман считает 83, что в растворе содержатся не ионы
AsClJPCle, а ионы PCl4AsCl4. Кронандер 46~48 наблюдал выпадение
твердой фазы состава AsCl3-PCl5 из раствора РС15 в трихлориде-
296 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
мышьяка. Однако Кольдитц131, повторивший эту работу, по-
показал, что в действительности из насыщенного раствора выделяются
крупные призматические кристаллы состава 2PCl5-5AsCl3 (tnn =
= 40° С), формула которых PCl4PClJ-5AsCl3- Раствор этого
соединения в AsCl3 обладает заметной электропроводностью (и =
= 6,9 • 10~4 ом~1-см~1 при 25° С и концентрации 0,202 м). Данные
об электропроводности этого соединения в ацетонитриле подтвер-
подтверждают его формулу как гексахлорфосфата(У), однако криоскопи-
ческие измерения показали, что в формульной единице содержится
2,6 частицы. Вероятно, оно может существовать и в виде тетрахлорар-
сената(Ш) PCllAsCl4-l,5AsCls.
По-видимому, в растворе часть этого соединения (до 30%) при-
присутствует в виде тетрахлорарсената(Ш), а остальная — в виде
гексахлорфосфата(У).
В трихлориде мышьяка удобно получать соли тетрахлорфосфо-
ния(У).
Так, соединение РС14РС15Вг~ является продуктом реакции
взаимодействия 134 РС13 с бромом в AsCl3. Кольдитц 130, использовав
трихлорид мышьяка в качестве растворителя, получил из гексафтор-
фосфата(У) тетрахлорфосфония(У), соединения ионного типа, смесь
молекулярного и ионного фторида тетрахлорфосфония(У).
Тетрахлорид теллура растворяется в AsCl3 с образованием элек-
электропроводного раствора. При кондуктометрическом титровании его
раствором тетраметиламмонийхлорида наблюдаются перегибы при
мольных отношениях 1 : 1 и 1 : 2. При отношении 1 :1 может быть
выделено твердое вещество состава (CH3LNCbTeCl4-AsCl3, формулу
которого правильнее записывать в виде (CH3LN+AsCl2TeC]|~,
-а при отношении 1 : 2 образуется соединение [(CH3LN]2TeCl(i.
Соединение с мольным отношением компонентов 1 : 1 можно рас-
рассматривать 80 как «кислую» соль в системе AsCl3.
С пентахлоридом фосфора 8в была получена более сложная кри-
кривая кондуктометрического титрования с перегибами при мольных
отношениях ТеС14 к РС15, равных 2 : 1, 1 : 1 и 1 : 2. При этом вы-
выделялись твердые фазы — сольваты. Исходя из представлений
о сольвокислоте как основании, Гутман предположил, что первый
из этих перегибов, весьма вероятно, соответствует присутствию
в растворе соединения РСЦ (AsCl|K(TeCl?"J, образовавшегося по
реакции 2 моль сольвокислоты (AsCl?JTeClff с 1 моль сольво-
основания PClJAsCll. Твердая фаза, представляющая собой сое-
соединение РС15 ^TeC^-SAsClg, очень легко теряет молекулы раство-
растворителя160, образуя известное соединение 2ТеС14-РС15. Соединению,
образующемуся при мольном отношении 1:1, приписана формула
PClJAsClgTeClf", при потере молекулы растворителя получается
соединение73 РС15-ТеС14.
При кондуктометрическом титровании раствором SnCl4 полу-
'чается иная кривая титрования, при реакции выделяются твердые
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
297
соединения TeCl4-2SnCl4, TeGL, • SnCl4 и 2ТеС14 • SnCl4. В данном
случае наблюдаемые процессы можно объяснить вступлением в ре-
реакцию тетрахлорида теллура в качестве сольвооснования и тетра-
хлорида олова в качестве сольвокислоты:
(AsClJHSnCl-j-4-TeClJAsCll ^=± TeClJAsCl?SnCl§-4-2AsCl3 F)
При титровании пентахлорида сурьмы тетрахлоридом теллура
перегибы на кривой наблюдаются при отношениях 1 : 1 и 1 : 2, что
соответствует выделению из раствора двух твердых фаз. И в этом
случае тетрахлорид теллура действует как сольвооснование, образуя
TeCl4-SbCl5, или TeClJSbCl;, и 2ТеС14- SbCl5 -AsCl3, или
(TeCr8JAsCl;SbCle.
Еще одна группа соединений — алкоксихлорантимонаты
(НО)„С15_-и Sb — растворяются в AsCl3 с образованием проводящих
ток растворов 133. Для SbCl4OC2H5 равновесие сначала устанавли-
устанавливается между ионами SbCl3 (ОС2Н5)+ и SbCl5(OC2H5)~. Однако
дальнейшее протекание медленной реакции с растворителем приво-
приводит к образованию пентахлорида сурьмы и As(OC2H5K, что сопро-
сопровождается изменением электропроводности раствора. Подобным
же образом ведут себя другие представители этой группы соединений.
При растворении С1,^в AsCl3 трихлорид мышьяка способствует
образованию хлор-анионов и хлор-катионов. Это означает, что он
сам при этом должен превращаться либо в пентахлорид мышьяка,
либо в гексахлорарсенат(У) тетрахлорарсония(У). Однако попытки
получить эти соединения успеха не имели вз>1в1. Но при взаимодей-
взаимодействии растворов РС15 или SbCl5 в трихлориде мышьяка с хлором
образуются соединения PCl5-AsCl5 и SbCl5-AsCl5. Оба они, по-ви-
по-видимому, содержат г7>78 катион AsCl4.
Таким образом был получен135 ряд тетрахлорарсониевых(У)
комплексов с различными металлами, например AsC14A1C14.
Поведение пиридина, растворенного в AsCl3, изучалось в не-
нескольких работах. Первой из них явилась работа Вальдена 190, ко-
который обнаружил, что раствор этот электропроводен. Были выделены
сольваты составов: Py2-AsCl3151>178 и Ру AsCl3 50> 67. Пиридин
является сильным электролитом 87 в AsCl3:
C5HBN + 2AsCl3 ^± C5H5NAsClJ + AsClj G)
После удаления растворителя (при комнатной температуре) можно
получить твердое вещество, состав которого колеблется от
Ру • l,6AsCl3 до Ру-l^AsClg. При повышении температуры до 50° С
наблюдается потеря трихлорида мышьяка и образуется Py-AsCl3.
При кондуктометрическом титровании растворов пиридина хлори-
хлоридами олова(ГУ) и ванадия(ГУ) перегибы на кривых наблюдаются
при мольном отношении 1 : 2, при этом выпадают твердые соедине-
соединения, содержащие катион C5H5NAsCl2 и анион гексахлорстанната(ГУ)
или гексахлорванадата(ТУ).
2Q8 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
Изучение физико-химических свойств трихлорида мышьяка как
растворителя до сих пор ограничивалось лишь подтверждением того,
что мольная электропроводность растворов тетраметиламмонийио-
дида в нем вплоть до концентрации 10 м подчиняется уравнению
Дебая — Хюккеля — Онзагера. Проведено было потенциометри-
ческое титрование в AsCl3 с использованием хлорсеребряного элек-
электрода 5. Кривая титрования тетраметиламмонийхлорида пентахло-
ридом сурьмы показана на рис. 58.
Из рисунка видно, что электрод ве-
ведет себя как pCl-электрод в реак-
реакции SbCl5 +Cl-->SbCl6. Приняв,
что величиной потенциала связи
между молекулами жидкости можно
пренебречь и что растворы хлори-
хлоридов тетраметиламмония и сурьмы(У)
в трихлориде мышьяка являются
сильными электролитами, нетрудно
показать справедливость равенства
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
299
500
UOOY-
зооу-
«о
с? гоо\-
юо \-
О 1 2 3b I
Содержание SbCl5,<w
Рис. 58. Кривая потенциометри-
ческого титрования SbCl5 тетра-
метиламмонийхлоридом5 в AsCl3
(конечная точка титрования —
3,01 мл; точка, соответствующая
мольному отношению компонен-
компонентов 1:1, — 2,96 мл).
званных тем, что, во-первых,
где К — ионное произведение.
Отсюда находим, что piST>15,
а ионное произведение [AsClJ] X
X [AsClj] должно быть меньше 106.
Получение более точных потенцио-
метрических данных сопряжено с
рядом серьезных трудностей, вы-
остаются неизвестными потенциалы
связи жидкости и, во-вторых, не определены коэффициенты
активности в растворителях с малой диэлектрической проницае-
проницаемостью. "Кроме того, данные о степени диссоциации растворов элек-
электролитов в трихлориде мышьяка весьма скудны.
До сих пор изучение растворов в трихлориде мышьяка ограничи-
ограничивалось кондуктометриед или носил» препаративный характер. Су-
Существование в растворе иона тетрахлорарсената(Ш) было доказано
исключительно косвенным методом. Из солеи, выделенных из раство-
растворов AsCl3, растворитель довольно легко удаляется. Даже наиболее
стойкая тетраметиламмониевая соль теряет трихлорид мышьяка
при 160° С. Исходя из того, что пять электронных пар распределяются
вокруг центрального атома мышьяка, нон AsClJ должен предста-
представлять собой тригональную бипирамиду, одна из вершин которой
занята неподеленной электронной парой. При этом имеет
место нарушение правильности структуры. Для объяснения высокого
числа переноса хлор-иона 89 в растворе тетраметиламмонийхлорида
в трихлориде мышьяка @,88—0,97) необходимо допустить возмож-
возможность легкого перехода хлор-иона от AsClJ к AsCls. Это свидетель-
свидетельствует о нестабильности иона тетрахлорарсената(Ш) в указанных
растворах 145. Доказательства, подтверждающие существования в рас-
растворах в AsCl3 иона AsCla, основываются исключительно на из-
изменении электропроводности в процессе титрования при определен-
определенных мольных отношениях компонентов. В литературе не встречается
каких-либо иных объяснений этого явления, в связи с чем любые
данные, в какой-то степени характеризующие такие растворы, могут
оказаться весьма полезными.
Б. Трифторид мышьяка
Из-за ~-своих физических свойств
= —6° С, iKIffl = 63° С)
ф (^ Kffl )
и высокой реакционной способности трифторид мышьяка менее при-
пригоден для использования в качестве растворителя, чем трихлорид
мышьяка. Удельная электропроводность его, х = 2,4-Ю омГ1 •см~1
при 25° С, одного порядка с удельной электропроводностью BrF3,
IF5 и HF195.
Вульф и Гринвуд 195 указывали, что способность к комплексо-
образованию между фторидом неметалла и фторидом щелочного ме-
металла в подходящем растворителе определяется хорошей подвиж-
подвижностью фторида неметалла, являющегося в растворе анионообразо-
вателем. Например, электропроводность AsF3 заметно выше, чем
электропроводность SbF5(x = 1,2-10"8 ом~1-см~1), и он легко
взаимодействует с фторидом калия, образуя KAsF4, в то время как
пентафторид сурьмы с этим соединением не реагирует 195. Электро-
Электропроводность трифторида мышьяка возрастает при растворении в нем
фторида калия или пентафторида сурьмы. Из последнего раствора
можно выделить твердое соединение AsF3-SbF5, тогда как при тит-
титровании растворов фторида калия фторидом сурьмы образуется твер-
твердое соединение KSbF6. Предполагается, что при пропускании BF3
через раствор калийтетрахлорарсената(Ш) в AsF3 образуется 195
соединение AsF3-BF3.
На результаты ранних экспериментов по изучению взаимодей-
взаимодействия трифторида мышьяка с галогенами, по-видимому, нельзя
полагаться. В 1955 г. Кольдитц 129 показал, что при барботировании
хлора через AsF3 при 0° С образуется белое кристаллическое соеди-
соединение. Путем измерения электропроводности, а также с помощью
других методов это соединение было идентифицировано как
AsClJAsFe. Оно несколько растворимо в AsGl3 и очень хорошо
растворимо в AsF3. Дальнейшее изучение этой реакции показало,
что в совершенно безводных условиях она не идет, но в присутствии
следов воды протекает гладко 57. Подобные ограничения характерны
для реакции с бромом и иодом, но соединений, содержащих ионы
J или AsIJ, не удалось выделить 56.
300 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
301
Трифторид мышьяка частично благодаря своим свойствам как
растворителя и низкой точке кипения является хорошим фториру-
фторирующим агентом для хлоридов неметаллов. Например 132:
3SbCl5+AsF3 5=± 3SbClJF-+AsCl3 (8)
Однако реакции при этом редко проходят до конца.
Существование твердых тетрафторарсенатных(Ш) солей К+,
Rb+ и Cs+, а также выделение соединения AsF3-SbF5 .наводит
на мысль 92 о существовании самоионизации трифторида мышьяка,
аналогичной самоионизации AsCl3. Катион AsFJ в растворе может
стабилизироваться за счет образования мостиковой структуры,
включающей молекулы растворителя. Анион тетрафторарсената(Ш)
изоэлектронен с тетрафторидом селена, молекула которого имеет
искаженную тригональную бипирамидальную структуру.
Изучение этих растворов с помощью метода ядерного магнитного
резонанса 153 показало, что, хотя в твердых солях и присутствует
ион тетрафторарсената(Ш), он, по-видимому, не является основ-
основным компонентом растворов фторидов в трифториде мышьяка, так
как фторид тотчас же вступает в быстро протекающие обменные
реакции. Смеси трифторида бора и трифторида мышьяка, хотя и по-
показывают несколько повышенную электропроводность, но не имеют
резонансных пиков в ЯМР-спектрах, кроме пиков, соответствующих
исходным соединениям. Эти данные ставят под сомнение возможность
существования нестабильного соединения AsFaBFj, о которой упоми-
упоминалось ранее.
В спектре 19F смеси трифторида мышьяка и пентафторида сурьмы
имеется только один резонансный пик, положение которого зависит
от концентрации. Этот пик расположен между резонансными пи-
пиками чистых соединений. Можно ожидать, что любое соединение
этих фторидов будет иметь неэквивалентные атомы фтора и даст
по меньшей мере два, если не более, пика в ЯМР-спектре. А здесь,
вероятно, происходит быстрый обмен между мостиковыми структу-
структурами:
F2As SbF4
SbF4
''
Подобные мостиковые структуры были предложены в связи с обме-
обменом иона фтора в галогенфторидах 152
В. Трибромид мышьяка
Свойства расплавленного трибромида мышьяка как растворителя
впервые были изучены Вальденом 189. Недавно эти свойства вновь
изучались в работах Яндера и сотр. 113~115. Бромиды щелочных,
щелочноземельных и переходных металлов(П), соли кислородсо-
кислородсодержащих кислот и окислы обладают незначительной растворимостью
в AsBr3. Некоторые бромиды, как, например, бромиды ртути(И), ин-
дия(Ш), теллураAУ) и висмута(Ш), а также'окись и сульфид мышь-
яка(Ш), хлориды ртути(П) и железа(Ш) имеют заметную раствори-
растворимость 167> ш. Очень хорошо растворимы в трибромиде мышьяка бро-
бромиды тетраалкиламмония, бора(Ш), алюминия(Ш) 121> 167> 197, гал-
лия(Ш), оловаAУ), титанаAУ), фосфора(Ш) и (V), сурьмы(Ш) 166
и селенаAУ). Высокой растворимостью в трибромиде мышьяка обла-
обладают и многочисленные органические соединения, в том числе
углеводороды, спирты, кетоны, сложные эфиры и амины.
При взаимодействии AsBr3 с четвертичными аммонийзамещенными
бромидами и органическими основаниями В, образуются сольвйты,
имеющиё^общую формулу R4NBr-AsBr3 и В -HBr-AsBr3. Измерения
электропроводности позволяют предположить, что эти сольваты пра-
правильнее рассматривать как соли с тетрабромарсенатным(Ш) анио-
анионом. Криоскопические измерения показали, что при образовании
аддуктов имеет место равновесие
2R3N+2AsBr3
2R3N-AsBr+
0)
Однако полученные результаты нельзя полностью объяснить, если
не допустить возможности существования ассоциированных молекул
типа (R3N • AsBr3J. Как и для всех слабых электролитов, электроли-
электролитическая диссоциация возрастает с разбавлением.
Добавление перхлората серебра к трибромиду мышьяка при 80° С
вызывает разложение его с выделением брома. Но при 50° С реакция
протекает следующим образом:
AsBr3+AgC104
AsBrJClOjJ-AgBr
(Ю)
Соединение AsBrgClOj не удалось выделить, но раствор обладает вы-
высокой электропроводностью и может быть изучен методом кондукто-
метрического титрования.
По аналогии с другими галогенидными сольво-системами про-
процесс самоионизации AsBr3 выражается следующим образом:
2AsBr3
AsBrf+AsBrj
(И)
Удельная электропроводность чистого растворителя при 35° С
равна 1,6-10~7 ом~1-см~х. Большинство растворов в AsBr3 и реак-
реакций его с различными соединениями может быть интерпретировано
с помощью ионов AsBrJ и AsBrJ. Бромиды алюминия(Ш), гал-
лия(Ш), индия(Ш), бора(Ш), оловаAУ), ртути(П), висмута(Ш),
теллураAУ) в связи со способностью образовывать в AsBr3 комплекс-
комплексные бром-анионы выступают как электролиты, обладающие кислот-
кислотными свойствами. Эту способность подтверждает высокая электро-
электропроводность растворов (см., например, кривые электропроводности
302 ГЛАВА ВОСЬМАЯ.|ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
растворов трибромида Bi и тетрабромида теллура в AsBr3, по-
показанные на рис. 59). Кривые кондуктометрического титрования
раствора тетраэтиламмонийбромида в AsBr3 различными анионобра-
зующими бромидами металлов(Ш) приведены на рис. 60. Для А1Вг3
реакция описывается уравнением
(C2H5LN+
-f- AsBr t + AlBrj
A2)
0,70
: (C2H5LN+-f-AlBr2-f-AsBr3
За протеканием этой реакции можно проследить по потенциометри-
ческим измерениям, использовав золотой индикаторный электрод:
до мольного отношения компонен-
компонентов 1 : 1 наблюдается медленное
изменение потенциала, после чего
он остается постоянным. В
, Предельная
I раствори-
растворимость
0,1
с, моль/л
Рис. 59. Удельная электропроводность113
растворов ТеВг4 и BiBr3 в AsBr3 при
/ г з
Мольное отношение
[MBr3]:[(c2H5LNBr]
Рис. 60. Кривые кондуктометричес-
кондуктометрического титрования-114 тетраэтиламмо-
тетраэтиламмонийбромида растворами бромидов
индия(Ш), алюминия(Ш) и гал-
лия(Ш) в AsBr3 при 93° С.
результате подобных реакций «нейтрализации» могут быть выделены
различные соли, такие, как КА1Вг4, СиА1Вг4, Т1А1Вг4, и др. Такие
же результаты были получены и с другими указанными выше бро-
бромидами, однако в ряде случаев, например у РЬВга, наблюдались
амфотерные свойства.
Применение трибромида мышьяка как растворителя позволяет
получить самые различные комплексные бромиды, которые чрезвы-
чрезвычайно трудно получаются другими методами. Растворяя в нем про-
простые окислы металлов, можно получить безводные бромиды. Окись,
сульфид и селенид мышьяка(Ш) при растворении в AsBr3 образуют
окси-, тио- и селенобромиды:
AsBr3-f-As203 Z=± 3AsOBr A3)
Для достоверной интерпретации кривых кондуктометрического тит-
титрования в системе AsBr3, как и в соответствующих хлоридной и фто-
ридной системах, необходимо провести более детальные исследования.
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
303
Г. Трихлорид сурьмы
Трихлорид сурьмы («Пл = 73° С, tKm ~ 219—223° С) был иссле-
исследован первым в группе галогенидных растворителей. За первой.ра-
первой.работой Толлочко по изучению криоскопических свойств растворов
в SbCl3 60' 61> 182 последовали классические исследования Валь-
дена 188, Бекмана м и Клеменсевича 123-127 Описание общих свойств
трихлорида сурьмы как растворителя неорганических и органиче-
органических соединений приводится в работах многих исследователей 68.
Существенным отличием SbCl3 от других уже указанных гало-
галогенидных растворителей является хорошая растворимость в нем,
по крайней мере после кратковременного нагревания, хлоридов
калия, рубидия, цезия, аммония и таллияA) и хлоридов тетраал-
киламмония. Хорошо растворимы в этом растворителе хлорид, бро-
бромид и иодид ртути(П), фторид и бромид калия. Хлориды лития,
натрия, олова(П), висмута(Ш) и железа(Ш) обладают незначи-
незначительной растворимостью.
Были изучены растворимости окислов и солей различных кисло-
кислородсодержащих кислот. За исключением сульфата и перхлората тет-
раметиламмония, все они либо нерастворимы, либо растворяются
«с разложением 116.
Известно большое число аддуктов, образующихся между SbCl3
и неорганическими соединениями; они представляют собой двойные
и комплексные соли с общими формулами M1SbCl4, M^SbCle, MISb2Cl7
и MjSb2Cl9(MT = Li, Na, К, NH4, Rb, Cs), M"(SbCl4J и M"SbClB
(Mu — Be, Mg, Ca, Sr, Ba). Для многих других неорганических
аддуктов, однако, весьма трудно записать формулу через определен-
определенные ионы, поэтому в дальнейшем об этих соединениях мы не будем
упоминать.
Известны аддукты SbCl3 с углеводородами, а также с органиче-
органическими галоген-, кислород- и серасодержащими соединениями 68
с соотношением компонентов главным образом 1:1. Хорошая рас-
растворимость в трихлориде сурьмы белков и компонентов нуклеино-
нуклеиновых кислот оказывается весьма полезной при изучении ИК-спектров
поглощения групп NH и ОН 137.
Электропроводность трихлорида сурьмы (и = 0,85-10"в ом'1-см'1
при 95° С) указывает на наличие процесса самоионизацин:
2SbCl3 ^±
A4)
Добавление к трихлориду сурьмы хлоридов щелочных металлов
и четвертичных аммониихлоридов вызывает значительное увеличение
электропроводности. На работах, посвященных физико-химии этих
электролитных растворов, мы остановимся позднее.
Кондуктометрическое титрование растворов в SbCl3 приводит
к таким же результатам, как в других галогенидных системах.
Например, перхлорат серебра реагирует с трихлоридом сурьмы
gO4 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
с образованием осадка хлорида серебра, а раствор, по-видимому,
содержит SbCl^ClOj. Подобным образом добавление к SbC]3 хло-
хлоридов алюминия(Ш), сурьмы(У) и теллураAУ) вызывает повыше-
повышение электропроводности растворов, вероятно, за счет образования-
соответствующих хлор-анионов:
AlCl3+SbCl3 ?=*: SbClJ + AlClJ A5)
Криоскопические измерения подтвердили, что трифенилметил-
хлорид является сильным электролитом и диссоциирует в SbCl3
на два иона. По величине коэффициента Вант-Гоффа выяснено также,
что хлориды щелочных металлов и четвертичные аммонийхлориды —
сильные электролиты, хотя в этих случаях наблюдается заметная
зависимость результатов от концентрации.
Для растворов тетрахлоридов теллура и селена коэффициент
Вант-Гоффа лежит в пределах 0,5 —1,0. Следует отметить, что эта
величина не соответствует процессу простого перехода хлор-иона.
Если бы процессы перехода хлор-иона а диссоциации проходили
до конца, коэффициент Вант-Гоффа должен был бы равняться 3.
Для объяснения экспериментальных результатов необходимо до-
допустить возможность значительной ассоциации или протекания
других сложных реакций с участием трихлорида сурьмы, приводя-
приводящих к образованию ионов или молекул, содержащих более одного
атома селена или теллура.
Так же, как и в более очевидной реакции «нейтрализации» тетра-
метиламмонийхлорида [в качестве аналога основания может рас-
рассматриваться (CH^N+SbCli] с SbCljjClOj (аналог кислоты), может
быть проведено титрование 117 сульфата сурьмы(Ш). Получающа-
Получающаяся при этом кривая кондуктометрического титрования имеет пе-
перегибы при отношении 6:1, что, по-видимому, соответствует ре-
реакции
6(CH3LN+SbCli+Sb2(S04K 5=± 3[(CH3LN+]aSO|- + 8SbCl3 A6)
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
305
и при отношении 6 : 4, что соответствует следующей стадии ее
3[(CH3LN+]3SOJ--f-3Sb2(SO4K +=t 6(CH3LN+Sb(SO4)i A7)
Суммарная реакция может быть записана в виде
4Sb2(SO4K+6(CH3LN+SbClJ 5=± 6(CH3LN+Sb(SO4)j + 8SbCl3 A8)
О существовании соединений, содержащих анион Sb(SO4J,
указывалось также в работе Метцла 149. Вполне резонно было бы
ожидать, что подобные комплексные ионы образует перхлорат-ион,
однако на кривой титрования с тетраметиламмонийхлоридом есть
только один перегиб, соответствующий формальной кислотно-основ-
кислотно-основной реакции. Методами кондуктометрического и потенциометриче-
ского титрования (с использованием золотого индикаторного
электрода) установлено полное сходство между этой системой
И системой в AsCl3, как систем, где образуются хлор-анионы. Пока-
Показана возможность протекания некоторых сольволитических реак-
реакций ш. Например, бромид и иодид калия легко растворяются в SbCl3.
0 обоих случаях продуктом реакции является 2КС1 • SbQ3\ Однако
при растворении фторида калия образуется KF-2SbCl3. При взаимо-
взаимодействии с трихлоридом сурьмы окислов, сульфидов, карбонатов
и ацетатов во всех случаях образуются галогениды соответствующих
металлов:
3MnO+2SbCl3 —> 3M«Cl2+Sb303 A9)
Кольдитц 131 получил из раствора хлороформа в присутствии
избытка трихлорида сурьмы аддукт 2РС15'48ЬС13, весьма похожий
Рис. 61. Зависимости экви-
эквивалентной электропровод-
электропроводности 165 растворов гало-
генидов в SbCl3 от ус при
99° С ( эксперимен-
экспериментальная кривая; ——— тео-
теоретическая кривая, наклон
которой соответствует урав-
уравнению Дебая — Хюк-
келя — Онзагера).
Vc -10 , моль/j,
на рассмотренный ранее аддукт трихлорида мышьяка и пентахло-
рида фосфора. Криоскопические измерения в SbCl3 и измерения
электропроводности в метилцианиде показали, что это соединение
является PClJPCl;-4SbCl3.
В появившейся недавно работе Баугана было показано, что
ионизирующие растворители, такие, как галогениды сурьмы(ГП),
диэлектрическая проницаемость которых меньше, чем у воды,
идеально подходят для изучения межионных эффектов в растворите-
растворителях 27> 52> 165. Криоскопическая постоянная, определенная при
использовании высокочистого растворителя и флуорена, антрацена,
бензофенона и дибензила в качестве растворенных соединений,
равна 15,6 ± 0,2 град -кг/моль. Зависимость эквивалентной электро-
электропроводности растворов Т1С1 A), Т1В_г B), КС1 C), КВг D), RbCl E),
NH4C1 F) и NH4Br G) в SbCl3 от j/7линейна (рис. 61). Для теорети-
теоретических прямых е = 30,4, т] = 1,84 спч при 99° С. Близость величин
20 Заказ 90
306 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
предельных электропроводностеи многих изученных бромидов и хло-
хлоридов проиллюстрирована ниже:
Галогеняд ...... Т1С1 TlBr KC1 KBr ? NH4C1 NH4Br RbCl
ло 149 148 158 156 ^ 152 152 155
Она заставляет предположить — хлор- и бром-ионы имеют аномально
высокие подвижности; это подтверждается экспериментальным зна-
значением числа переноса хлор-иона 60' 61, равным —0,9. Такое высокое
число переноса, наблюдаемое также и в трихлориде мышьяка,
обеспечивает достаточную скорость реакции перехода хлор-нона:
SbCls+SbClJ —> SbClj+SbCls B0)
Галогениды щелочных металлов гораздо лучше растворяются
в трихлориде сурьмы, чем в трихлориде мышьяка. Коэффициенты
Вант-Гоффа для некоторых одно-одновалентных электролитов, опре-
определенные из криоскопических данных, при бесконечном разбавлении
стремятся к предельному значению, равному двум.
О неидеальности растворов электролитов можно судить по моль-
мольному осмотическому коэффициенту Ф, определенному из криоско-
криоскопических данных *:
Ф = 1 — l,86m'/2(j @,768eV^) + Ът
где т — моляльная концентрация растворенного вещества; а — расстояние
наибольшего сближения ионов, вА; 6 — константа, учитывающая сольвата-
сольватацию нона.
Из экспериментальных данных, полученных для SbCl3 как раство-
о
рителя, можно рассчитать значения а и сравнить их со значениями,
рассчитанными из межатомных расстояний **, предполагая, что
хлор-ион сольватирован в растворе с образованием SbClJ:
Электролит КС1 CsGl' (CH3LNC1
Значение а, А
теоретическое, r+-fr_ 5,5 5,9 7,7
экспериментальное 2,1 4,5 13,0
Экспериментальные данные для хлоридов калия и цезия указы-
указывают на наличие ассоциации. Параметр Бьеррума для одно-одно-
* В выражении для осмотического коэффициента д = — A-|-г) —
] Где х=В°ат1\ а Д = A^у )''' 10"в (Ж-число
Авогадро). — Прим. перев.
** Теоретическое значение а рассчитывалось как гкатиона + rgbcl_; радиусы
катионов К, Cs, (CH3LN равны соответственно 1,3; 1,7 и 3,5 А,*а г8Ш_ =
= 4,2 А (длина связи Sb— Cl+rcl_ = 2,4 + l,8 = 4,2 к). —Прим. перев.
III. ГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
307
валентного электролита при температуре плавления трихлорида
сурьмы равен 7,3 А, что также свидетельствует об образовании пар
ионов. Для тетраметиламмонийхлорида, являющегося сильным
электролитом, наблюдаемое расстояние наибольшего сближения
катиона и аниона превышает критическое расстояние Бьеррума,
а также и теоретические значения, рассчитанные из предположения
о существовании вокруг катиона (CH3LN+ по крайней мере одной
полной сольватной оболочки.
Значения электропроводности растворов различных органиче-
органических хлоридов в трихлориде сурьмы, измеренные при 75° С, пока-
показали 52, что зти соединения ионизируются главным образом с обра-
образованием иона R 2С1+:
2RC1
B1)
В более разбавленных растворах процесс ионизации протекает
обычно так:
RC1 =± R+ + C1" B2)
Растворы ароматических углеводородов в SbCl3 сильно окрашены
и дают ЭПР-спектры высокого разрешения 27. Сравнение с соответ-
соответствующими спектрами растворов перилена в 98%-ной серной кислоте,
содержащих ион карбония, указывает на наличие в этих растворах
.положительно заряженйых ионов. Но положительные ионы, при-
присутствующие в растворах нафталина в SbCl3 и в H2SO4, различны.
Этим и объясняется появление дополнительных линий в ЭПР-спектре
растворов нафталина. Таким образом, в растворах в трихлориде
сурьмы удобно осуществлять окисление углеводородов. В указанной
выше работе отмечается, что этот метод может оказаться исключи-
исключительно удачным общим способом получения углеводородных ка-
катионов.
Благодаря тому что в растворах трифенилметилхлорида и тетра-
тетраметиламмонийхлорида проведены точные измерения электропровод-
электропроводности, эти системы описаны более надежно по сравнению с осталь-
остальными. Бауган указывал, что для ряда растворителей апротонного
типа произведение Вальдена Яо т] для N(CH3)J составляет около 0,3.
Это должно соответствовать подвижности иона N(CH3)J в трихлориде
сурьмы, равной ~13, что для числа переноса хлор-иона дает вели-
величину 0,87. Кондуктометрическое титрование центахлоридом сурьмы
растворов КС1 или трифенилметилхлорида в SbCl3 дало кривую
титрования, типичную для процесса титрования сильной кислоты
сильным основанием в воде. При этом протекает следующая реакция:
SbClj+SbCl5 +=± SbCls+SbCl^a B3)
и ион SbClJ обладает в отличие от иона SbCl^ .аномально высокой
подвижностью 116~118.
Действительно, если подвижности ионов гексахлорантимоната(У)
и калия или трифенилметил-иона приблизительно равны, то из
20*
308 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
IV- ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
309
формы этой кривой можно сделать вывод, о том, что число переноса
хлор-иона велико @,86). Таким образом, следует считать, что дока-
доказательства высокой подвижности хлор-иона весьма убедительны,
но механизм этой высокой электропроводности недостаточно изучен.
Все же можно предположить возможность существования, хотя,
вероятно, кратковременного, тетрахлорантимонатного(Ш) иона.
Дэвис и Бауган 5а сделали попытку скорректировать данные об
электропроводности, учтя вклад электропроводности самого раство-
растворителя. Они отмечали, что наблюдавшаяся ими удельная электро-
электропроводность чистого растворителя D—6-Ю ом~1-см~1 при 75° С)
была, по-видимому, сильно, завышена. Протекание процесса само-
самоионизации обусловлено, вероятно, наличием примесей (летучего
трихлорида мышьяка или аммонийной соли). Самоионизация быстро
идет при 99° С, но медленнее при 75° С, причем в последнем случае
она вызывается стеклянными стенками сосуда. Тем не менее резуль-
результаты, по крайней мере частично, согласуются со следующей схемой
процесса самоионизации растворителя:
2SbCl3
SbClJ+SbClj
B4)
Итак, трихлорид сурьмы — растворитель, в котором могут быть
получены и изучены простые ионы карбония. Этот растворитель
также весьма удобен для физико-химического изучения межионных
взаимодействий. Однако надо отметить недостаточное количество
прямых доказательств того, какие ионы присутствуют в самом
растворителе в результате самоионизации, а также того, какие ком-
комплексные хлор-анионы образуются в растворе. Укажем также, что
SbCl3 применяют в качестве растворителя при наблюдении основных
колебаний групп N—Н, О—Н и С—Н аминокислот и белков 137> 138.
Д. Трибромид сурьмы
По способности растворять неорганические и органические
соединения трибромид сурьмы сравним с трихлоридом 119. Раствори-
Растворимость бромидов заметно выше, и из растворов получены
MJSb2Br9(MJ-K, NH4, Rb) и M1SbBr4(MI-Tl, (CH3LN). Удельная
электропроводность чистого растворителя равна120 @,9 -ь 1,0) X
X Ю~5 омГ1 • см при 100° С. Как и для SbCl3, при изучении трибро-
мида сурьмы применяются методы измерения электропроводности
и кондуктометрического титрования. Было показано, что при мольном
отношении 1 : 1 в растворе А1Вг3 в SbBr3 наблюдаются максималь-
максимальные вязкость, электропроводность и плотность. Этот расплав отве-
отвечает составу196 SbBr3-AlBrs B
Е. Трииодид сурьмы
Для изучбния низших иодидов сурьмы использовались концен-
концентрационные ячейки с триодидом сурьмы (tm = 166,5 ± 0,5° С)
с сурьмяными электродами при 250° С. Процессы, происходящие
в образующейся окислительно-восстановительной системе, могут
быть интерпретированы как отщепление от трииодида сурьмы двух
электронов. Ток в ячейке возникает в результате переноса электри-
электричества ионами, образовавшимися в самом растворителе, по-види-
по-видимому, ионами SblJ и SblJ. Электропроводность расплавленного
иодида значительно возрастает с добавлением иодида калия, что
объясняется увеличением количества тетраиодантимонатных(Ш)
ионов 44.
Ж. Пентахлорид сурьмы
Большинство неорганических галогенидов плохо растворяется
в пентахлориде сурьмы. Например, растворимость дихлорида ртути
составляет лишь 0,8% при 120° С. Однако трихлорид сурьмы
образует при 40° С 20%-ный раствор 58. При растворении трихлорида
иода в пентахлориде сурьмы образуется 185 соединение ISbCl8.
3. Трихлорид висмута
Изучение расплавленного трихлорида висмута показало, что
наиболее интересным свойством его как растворителя является
способность растворять металлический висмут 35> 36>.42> 43> 122 с обра-
образованием иона Bi|+. Растворы металлического висмута в трихлориде
висмута подчиняются закону Бэра лишь в области малых концентра-
концентраций 30. В области высоких концентраций наблюдаются отклонения 31.
Общая картина процессов, происходящих при растворении, может
быть нредставлена несколькими уравнениями равновесия в раство-
растворах, простейшее из которых
4Bi+
а одно из более сложных
32
Big1
л+
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ *
Вследствие полярности связей в молекуле и стабильности окси-
анионов оксигалогениды обладают хорошей растворяющей способ-
способностью. Они отличаются от галогенидов тем, что могут быть доно-
донорами галоген-ионов и донорами электронов через кислород. Больше
того, они фактически являются лучшими донорами галоген-ионов,
чем галогениды. Хотя число оксигалогенидов азота, фосфора,
мышьяка, сурьмы и висмута очень велико, лишь немногие из них
310 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
имеют достаточно большой температурный интервал пребывания
d жидком состоянии для того, чтобы их можно было использовать
в качестве растворителей. Смешанные галогениды, такие, как POFC12
и POF2C1, малодоступны, чем объясняется их недостаточная изучен-
изученность. Кроме того, в них могут протекать процессы внутренней
перегруппировки173, что приводит к исключительным трудностям
при экспериментальных работах с этими оксигалогенидами.
Общая химия оксигал,огенидов азота изучена достаточно по-
подробно 63. Однако до последнего времени в качестве растворителя
рассматривался лишь нитрозилхлорид. Несомненно, это связано
главным образом с его доступностью, поскольку нет оснований
считать, что подобными свойствами не обладают другие NOF и NO2r.
Оксигалогениды мышьяка, сурьмы и висмута представляют собой
твердые вещества со слоистой структурой. Например, оксихлорид
сурьмы SbOCl состоит из бесконечных слоев состава (Sb6O6Cl4J+,
связанных между собой через хлор-ионы.
А. Нитрозилхлорид
Нитрозилхлорид NOC1, а также фосфорилхлорид РОС13 особенно
пригодны в качестве растворителей в связи с их доступностью и удач-
удачными физико-химическими свойствами (см. табл. 38). Последние
у NOC1 определяются полярностью молекулы, имеющей в своем
составе два сильно электроотрицательных атома 28. Длина связи
N—C1 A,95 А) аномально велика65. В жидком нитрозилхлориде
(е = 19,7 при —10 и 22,5 при —27° С) в заметной степени раство-
растворяются соли нитрозила 38> 39 вследствие сольватации иона NO+.
Сольватация происходит благодаря образованию стабильных аддук-
тов с хлорными мостиковыми связями (О—N—С1—NO)+, стойкость
которых связана с наличием мезомерии между структурами
[:O=N-C1-N = O:]+, [:O = N-C1:1 : N=0: и :O=N: [:Cl-N=O:j.
Вполне понятно, что простые катионы, в частности ионы щелочных
металлов, не сольватируются по указанному выше механизму,
и растворимость галогенидов щелочных металлов поэтому гораздо
ниже, чем растворимость галогенидных солей с нитрозил-катионом.
Такие соединения, как NOA1C14, NOFeCl4 и NOSbCl6, хорошо раст-
растворимы в NOCI и являются сильными электролитами, но есть соли,
в частности (NOJSnCl8, (NOJTiCl6 и NO+HSO4, которые практически
нерастворимы.
Выделено большое число твердых соединений, в состав которых
входят молекулы нитрозилхлорида и молекулы хлорида металла
или металлоида, обычно легко присоединяющие хлор-ион. Соедине-
Соединения эти могут быть получены растворением в нитрозилхлориде
хлорида, а в некоторых случаях даже металла. Иногда для проте-
протекания этой реакции необходимо ввести определенный растворитель;
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
311
для этих целей опробовано множество растворителей, в том числе
безводные галогеноводороды 186. Ниже приведены примеры получен-
полученных соединений этого типа. Соединения состава 1:1 — MClra-NOCl,
где МС1„ - ВС13108' 156~158, BF3 156' ш, А1С18 38' 64, GaCl3157' 158,
1пС18 157« 158, TiCl3157> 158, AsCl3143> 186, SbCl3186, SbCl6 38>170-174- 175,
SbF5 1в6, BiCl3 170- 180> 184, UOgCll, MnCl2 6- 157> 158, FeCl3 39- 17°- 187,
CuCl 6' 39> 18°, АиС13 157- 158- 18°, ZnCl2 6- 157- 158- 18°, HgCl2 157- 158- 184.
Соединения собтава 1 : 2 — MCln-2NOCl, гдеМС1„ — А1С13 39, FeCl3 39,
ZrCl4 98-1вз, ThCl4 163, PdCl, 57> 158, PtCla 6- 157> 158, SnCl4 6' 38- 157- 158,
РЬС14 в- 17°.
В большинстве случаев эти аддукты можно рассматривать как
соли, имеющие в своем составе хлор-анион и нитрозил-катион,—
NO+MC1^+^ и (NO+JMCl^+2. Наличие нитрозил-иона в таких солях,
как NO+C1O4 и NO+HSO4, установлено с достаточной надежностью,
и не вызывает сомнения, что аддукты нитрозилхлорида с различными
хлоридами — соединения ионного типа. В спектре комбинационного
рассеяния аддукта NOC1-A1C13 имеются линии, соответствующие
ионам NO+ и АЮЦ. Однако величина силовой постоянной группы
N0 заставляет предположить, что связь в молекуле по характеру
является не полностью ионной м, т. е хлор, по-видимому, не совсем
переходит к алюминию, и это соединение лучше рассматривать как
А1С13 - • -С1. N0, а не NO+AlClJ. Но можно считать также, что это
соединение имеет следующую структуру с кислородным мостиком:
С1
С1— А1—О—N— C1
С1
Подобным же образом для объяснения данных об электропровод-
электропроводности раствора SbCl5-NOCl в жидком S02 приходится принять
существование в растворе не простых ионов, а более сложных 174> 175.
Для того чтобы с определенностью установить структуру образу-
образующихся аддуктов, необходимо дополнительное изучение этих и Дру-
Других комплексов.
В состав аддуктов хлоридов алюминия и железа(Ш) с N0C1
типа 1 : 2 входит одна слабо связанная молекула нитрозилхлорида.
С такой структурой согласуются величины давления диссоциации 39
этих соединений при 0°-С, равные соответственно 180н22Аммрт. ст.
Электропроводность растворов солей нитрозила в нитрозилхло-
нитрозилхлориде высока. Ниже приведены типичные данные, полученные
при —20° С:
Соль NO+AlClr NO+FeClf NO+SbCl6
х, ом'* ¦ см-1 1,17 • 10 134-10 2,35 • 10"»
с, молъ/л 0,098 0,0099 0,140
312 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ II ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
Мольная электропроводность раствора NO+FeClJ в нитрозил-
хлориде при —10° С удовлетворяет уравнению Шедловского, из
которого получено числовое значение Ао, равное 401,2. Из этих же
данных может быть рассчитана степень диссоциации NOC1 • FeCl3:
с, моль/л 0,328 0,481 0,704 1,53 2,19 4,65 6,13
Степень диссоциации . . . 0,937 0,916 0,885 0,820 0,792 0,717 0,693
При интерпретации этих результатов была использована теория
Дебая — Хюккеля — Онзагера, которая оказалась хорошо приме-
применимой к нитрозилхлоридным сольво-системам. Высокое значение Ао
объясняется большим числом пере-
переноса @,88) иона NO+ в системе.
Большая подвижность иона NO+
(которая требует еще своего под-
подтверждения) предполагает наличие
цепного механизма проводимости,
аналогичного процессу переноса
иона Н+ в водной системе 39. Боль-
Большие подвижности наблюдались также
у хлор-иона в хлоридах сурьмы и
мышьяка(Ш) 176, механизм прово-
проводимости в которых также, по-види-
по-видимому, имеет цепной характер.
Исходя из кислотно-основной кон-
концепции Гутмана и Линдквиста, про-
процесс самоионизации растворителя
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
313
300
200
100
О 0,8 1,6 2,<+
Мольное отношение
jjCH3LNCi]:[NOFeCI4]
Рис. 62. Кривая кондуктометрн-
ческого титрования39 нитрозо- - - ,
тетрахлорферрата(Ш) раствором можно записать следующим образом:
тетраметиламмонийхлорида в ,,i»T^ _^ ... .
NOC1 при —10° С. C1NO -^ NO+(conbBawipoBaH)-f-
+ С1~(сольватирован) B5)
Следовательно, соли нитрозила будут действовать как кислоты,
а хлориды их — как основания. На рис. 62 показаны результаты
кондуктометрического титрования NO+ FeClJ тетраметиламмоний-
хлоридом в NOC1, подтверждающие указанный вывод. В ходе титро-
титрования электропроводность уменьшается (сопротивление увеличи-
увеличивается), что объясняется заменой высокоподвижного нитрозил-иона
менее подвижным тетраметиламмониевым ионом. Титрование соеди-
соединений NO+BFj и NO+ClOj тетраметиламмонийхлоридом показало
уменьшение количества присутствующего нелетучего хлорида и соот-
соответствующее изменение электропроводности, но реакции не проте-
протекали до конца вследствие сольволиза образующихся солей.
Путем растворения фосфатов серебра в нитрозилхлориде могут
быть получены соли нитрозила, содержащие в своем составе поли-
полифосфатные анионы:
4Ag3PO4 + 12ClNO *¦ (NOJP4On+5N2O3-|-12AgCl B6)
3Ag4P2O7-H2ClNO > (NOLP6Ol7-f-4N3O3 + 12AgCl B7)
Ag3P3O9-f-3ClNO
Ag4P4O12+4ClNO
(NOJP4OU + N2O3 -HAgCl
B8)
B9)
В этих реакциях происходит замещение катиона Ag+ нитрозил-ионом,
кроме того, образуются 176 дополнительные связи Р—О—Р.
Пониманию химии растворов в NOC1 во многом способствовало
изучение процессов обмена с использованием меченых атомов.
Быстрый обмен 36С1 между нитрозилхлоридом и хлоридами алюми-
алюминия, галлия, индия(Ш), таллия(Ш), железа(Ш) и сурьмы(У)
подтверждает мысль о том, что эти соединения существуют в растворе
в виде комплексов, в которых атомы хлора равнозначны и быстро
обмениваются 142~144. С помощью метода меченых атомов был изучен
процесс обмена между малорастворимыми хлоридами металлов
и нитрозилхлоридом 141.
Высокая скорость обмена наблюдалась между хлоридами
цинка, кадмия и ртути(П) и нитрозилхлоридом. При этом, вероятно,
образовывались комплексы 1:1. Последнее заключение было сде-
сделано на основании более медленной скорости гетерогенного обмена
между комплексами и растворителем. Обмен не наблюдался для
NaCl и КС1, которые не образуют комплексов, и для хлоридов,
образующих очень стойкие комплексы, например (NO+J(SnCl6J~.
Было обнаружено, что обмен между хлоридом серебра и жидким
нитрозилхлоридом происходит только на свету; вероятно, он про-
протекает через фотохимическое разложение.
Б. Нитрозилфторид
Изучены реакции нитрозилфторида только с различными метал-
металлами, в результате которых часто образуются соли нитрозила
с фтор-анионом 2. Например, при растворении в нитрозилфториде
металлического олова 198 образуется (NOJSnFjj~. Свойства нитро-
нитрозилфторида как растворителя до последнего времени не изучались.
В. Нитронилфторид
Температуры кипения нитронилфторида NO2F и нитронилхло-
рида слишком низкие, поэтому при изучении свойств этих соедине-
соединений как растворителей возникает множество трудностей. Однако
число комплексов, имеющих в составе ион нитрония NO?, очень
велико 107, особенно с фтор-анионами. Из табл. 38 видно, что вряд
ли в этой системе столь энергично проходят процессы, характерные
для сольво-систем, как в нитрозилхлоридной системе.
314 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
. Г. Фосфорилхлорид
Несомненно, что из числа галогенидов и оксигалогенидов элемен-
элементов V группы как растворителей наиболее обширные исследования
проведены с фосфорилхлоридом. Это объясняется главным образом
его доступностью, достаточно широким температурным интерва-
интервалом жидкого состояния, легкостью очистки и высокой растворя-
растворяющей способностью. Во всех работах по изучению фосфорилхлорида
поведение растворенных соединений в нем рассматривалось с самых
различных точек зрения. Таким образом удалось раскрыть общие
свойства системы, однако, для того чтобы окончательно и надежно
определить природу ионного равновесия, существующего в раствори-
растворителе, необходимо еще изучить многие вопросы.
Прежде всего необходимо рассмотреть вопрос о чистоте раствори-
растворителя, так как он имеет первостепенное значение при оценке резуль-
результатов работы. Критерием чистоты растворителя, применявшимся
во многих работах, является его удельная электропроводность.
В первой работе Вальдена 190 использовался растворитель с х ^=
= 1,7 -10~6 ом~1-см~1 при 25° С, а в недавно выполненной работе
Гутмана 80 фосфорилхлорид после повторной перегонки в стеклян-
стеклянном приборе имел при 20° С у, = 1,55 -10 ом~1-см~1. Удельная
электропроводность чистого фосфорилхлорида, по последним дан-
данным 93, равна 2• 10~8 ом~1-см~1 при 20° С. Основными примесями
в этом растворителе являются полифосфорилхлориды и хлористый
водород, образующиеся при гидролизе РОС13. Их удаляют тщатель-
тщательной и многократной фракционной перегонкой. Изучение свойств
этого растворителя должно проводиться в специальной аппаратуре,
обычно изготовляемой полностью из стекла. При этом особое внима-
внимание уделяется необходимости избежать или свести к минимуму
контакт с влагой. Работа с такой аппаратурой сложна.
Для объяснения электропроводности очищенного фосфорилхло-
фосфорилхлорида необходимо допустить наличие в нем процессов самоионизации.
Вальден предложил оригинальный механизм, заключающийся в обра-
образовании серии катионов за счет последовательных отщеплений
хлор-ионов. Однако получить доказательства существования в рас-
растворителе таких ионов не удалось, и принято считать, что в нем
присутствуют только однозарядные ионы. По аналогии с процессами
самоионизации, предложенными и в какой-то степени установлен-
установленными для галогенидных растворителей, можно полагать, что в фос-
форилхлориде протекает следующая реакция:
C0
Гутман 90 предложил более обобщенное уравнение
C1)
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
315
Здесь катион и анион рассматриваются как сольватированные.
Так как удельная электропроводность фосфорилхлорида очень мала
B-Ю-8 ом'1-
см *¦), ясно, что мала и степень его самоионизации;
ионное произведение 93 не превышает величину 9 • 10~14. Потенцио-
метрические измерения 90 дали для ионного произведения величину"
5 • 10~14 при 20° С. Прямой электролиз чистого растворителя осущест-
осуществить невозможно, но при электролизе 0,14 м раствора триэтиламмо-
нийхлорида 179 на аноде выделяется хлор, а на катоде — твердый
полимерный продукт состава РО:
РОС13 .^ P0C1J
^ 1
анод С1~
C2)
катод ЗРОС1? + Зе >• РО+2РОС13
Отношение хлора к моноокиси фосфора составляло 3:1, что
соответствует приведенным выше уравнениям реакций, протекающих
на электродах. Из этого, казалось бы, можно сделать вывод о том,
что в электродных реакциях принимают участие только зти ионы
и тем самым иметь серьезное доказательство существования катиона
РОС12. Однако можно дать альтернативное объяснение, заключа-
заключающееся в том, что ион триэтиламмония нейтрализуется на катоде
и реагирует с растворителем:
(C2H5KNH+ + e —>¦ (C2H6KN + [H] C3)
3[Н] + РОС13 —> РО+ЗНС1
Суммарное уравнение этой реакции может быть записано следующим
образом:
3(C2H5KNH+ + 3e+POCl3 —>- PO + 3(C2H5KNH+ + 3Cr C4)
Число переноса хлор-иона в растворе тетраметиламмонийхлорида
в фосфорилхлориде 90 равно 0,8, что близко к соответствующим
значениям, полученным в хлоридах мышьяка(Ш) и сурьмы(Ш).
Напомним, что столь высокие значения объяснялись протеканием
в растворах процессов переноса, для фосфорилхлорида это согла-
согласуется с рассмотренным выше процессом самоионизации.
Общие свойства фосфорилхлорида как растворителя 81 весьма
похожи на свойства других галогенидных и оксигалогенидных
растворителей. Например, такие соединения, как SiCl4, SiBr4,
SnBr4, легко растворяются в нем без диссоциации или ассоциации 155.
Соединения (CH3LNC1, (C2H5LNC1, PC15, PBr5, AsCl3, BiCl,, BiBr3,
Bil3, ICI3, SCI4, PtCl4154 также хорошо растворимы, но криоскопи-
ческие измерения показали, что эти соединения диссоциируют
в растворе. Соединения А1С13, BBr3155, SbCl5128, SnCl441, ТеС14 140,
TiCl4 172, ВС13 76 растворяются подобно FeCl3, 'но растворимости их
малы. Лишь слегка растворимы хлориды щелочных металлов и аммо-
316 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
ния. Многие иодиды и бромиды растворяются в фосфорилхлориде,
но изменение цвета при этом указывает на протекание реакций.
Растворение в фосфорилхлориде солей различных кислородсо-
кислородсодержащих кислот, например бихромата и перманганата калия,
также сопровождается химическим взаимодействием. Ниже приве-
приведены данные о растворимости, удельной электропроводности насы-
насыщенных растворов некоторых солей щелочных металлов в РОС13
при 20° С и эквивалентной электропроводности при разбавлении
V = 1000 л/моль:
Соль LiCl NaCl KC1 NH4C1 RbCl CsCl
Растворимость, г/л 0,05 0,31 —0,60 0,46 0,87 1,26
x, om-1-cm.-i . . . 6,6-10"e 3,0 -10 3,4-Ю 3,6-Ю 8,3 • 10"8 1,1 • 10
Ло, см2/(ом ¦ г-экв) 4,0 6,4 6,7 6,9 14,6 16,0
Соль KF KBr KI KGN KCNO KSCN
Растворимость, г/л —0,40 0,51 —1,71 —0,73 —0,80 —0,76
х, ол-1 • см-1 ... 2,6 • 10~5 4,3 • 1<Г5 1,2 • 10 3,3 • 10"в 3.1 • 10"в 2,9 • 10~5
Ло, смъ/(ом ¦ г-экв) 6,4 14,5 23,1 7,2 9,0 6,6
Вообще растворимость этих
солей недостаточна для того, чтобы
применять их в работах с фосфо-
рилхлоридом. Удобнее использо-
использовать четвертичные аммониевые
хлориды. Очень мало растворимы
и хлориды щелочноземельных ме-
металлов, серебраA), ртутиA) и тал-
лияA).
Растворимости четвертичных
аммониевых солей высоки 8, и
эквивалентные электропроводно-
электропроводности их близки к значениям, пред-
предсказываемым теорией электроли-
электролитов. Например, зависимость экви-
эквивалентной электропроводности ра-
раствора тетраэтиламмонийхлорида
от ус (рис. 63) во всем измерен-
измеренном интервале концентраций от
1,4-10 до 7,3-Ю-5 М представ-
представляет собой прямую линию. Экспе-
Экспериментальные, определенные из
графика Л — Ус, и вычисленные
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
317
°-(C2H5LNCI
(
a-(c4h9LNCi
¦- (C2H5LNBr
°- (С2н5Lш
- (C2H5LNC1O4
зо V-
0 12 3
Vc-lO2
Рис. 63. Зависимость эквивалентной
электропроводности 8 растворов чет-
четвертичных аммониевых солей в фос-
фосфорилхлориде от Vс (с — молярная
концентрация) при 20° С.
_„ „ л. й9 ±рсщшпа л — у с, и вычисленные
по методу Фуосса 62 значения предельной эквивалентной электро-
электропроводности ряда солей в РОС13 при 20° С приведены ниже:
(C2H5LNC1
53,0
53,0
(C3H7LNG1
47,6
46,3
(C4H9LNC1
44,4
43,0
Соль (C2H5LNBr (G2H5LNI (C2H5LNC1O4
Лэксп 50,0 48,5 54,4
Лтеор 49,0 48,3 53,4
Эти данные хорошо удовлетворяют уравнению Дебая — Хюккеля —
Онзагера 9.
Для определения подвижностей ионов (C2H5LN+, (C3H7LN+,
Gl~, Вг~, I~ и C1O4 можно воспользоваться правилом Вальдена;
ч полученные значения равны соответственно 25,6; 18,9; 15,6; 27,4;
23,4; 22,7 и 27,8. Эти величины сравнимы с подвижностями тех же
ионов в других растворителях (в частности, в воде). Рассчитанные
из данных о подвижностях ионов радиусы Стокса подтверждают
наличие сольватации хлор-, бром- и иод-ионов, причем число моле-
молекул растворителя, приходящихся на один ион, колеблется от 0,5
до 2. Числовые значения радиусов Стокса, полученные из данных
об электропроводности, подтверждаются сравнением их с параме-
параметрами Бьеррума.
Триэтиламин в фосфорилхлориде, по-видимому, электролит,
так как раствор его электропроводен (Ло = 48,5 ± 1). Его поведе-
поведение в РОС13 можно представить уравнением
(C2H5KN + POCl3 ^ [(C2H5KN-POC1JC1-] 4=Г (C3H5KN • POCIJ+GI" C5)
Зная подвижность хлор-иона, можно рассчитать подвижность
катиона, а отсюда и радиус Стокса. Сравнение полученного значе-
значения с теоретическими значениями радиусов ионов [(C2H5KN]3POC12+,
[(C2H5KN3PO3+ и [(C2H5KN]POCl?, свидетельствует о существо-
существовании в растворе катиона [(C2H5KN]POC1J. Он имеет тетраэдриче-
скую конфигурацию, атом фосфора окружен двумя атомами хлора,
одним атомом кислорода и одним атомом азота. Константа равновесия
этой реакции соизмерима с соответствующей константой водной
системы:
(C2H5KN-fH2O ^z^ [(C2H5KNH+OH-] ^ (C2H5KNH++OH- C6)
Однако катион РОС12 в растворе в фосфорилхлориде обладает боль-
большим сродством к триэтиламину, чем сродство протона в воде к нему,
поэтому равновесие заметно смещено вправо. В растворе триэтил-
амингидрохлорида в фосфорилхлориде существует тройное равно-
равновесие:
(C2H5KNH++POC13 Z=? (C2H5KNPOC1J + HC1
-ci-|J+ci- -ci-jj+ci- C7)
(C2H5KN-HC1 (C2H5KN + POC13
Изучая электропроводность этой системы, можно показать, что
сродство протона к триэтиламину в фосфорилхлориде не выше, чем
сродство катиона РОС12. В разбавленных растворах (C2H5KN-HCl
образует примерно в равных количествах ионы (C2H5KNH+
318 ГЛАВА ВОСЬМАЯ. ГАЛОГВНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
, и [(C2H5KN]POC1?. При пропускании азота через раствор элек-
электропроводность его падает, достигая в конечном итоге значения,
соответствующего электропроводности самого триэтиламина9. На кри-
кривой кондуктометрического титрования растворов пиридина в фос-
форилхлориде хлоридами ванадия(Г7), фосфора(У) и тантала(У) пере-
перегибы наступают при мольном отношении компонентов, равном 1 : 2,
что соответствует образованию иона (Ру • POClJ^VCljj" и при отно-
отношении 1:1, что соответствует 88 образованию ионов Pv-POCl?PCl«
и PyPOClJTaCl;.
При растворении в фосфорилхлориде некоторых акцепторов
хлор-ионов, например SbCl5 и ВС13, образуются растворы, облада-
обладающие высокой электропро-
электропроводностью. Особый инте-
интерес представляет раствор
пентахлорида сурьмы, так
как известна структура
твердой фазы, образую-
образующейся в растворе (рис. 64).
Атом сурьмы в молекуле
этого соединения непосред-
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
319
С1
С1
Рис. 64. Структура34 SbCl5-OPCl3.
ственно связан с кислоро-
кислородом 34> 147. Данные об элек-
электропроводности растворов
SbCl5 в РОС13 хорошо ин-
тепретируются равнове-
равновесием 10
Cl5Sb0PCl3
SbClj + POClJ
C8)
Рассчитанная для этого уравнения константа равновесия оказалась
равной 4-10~6. Константа равновесия реакции
C9)
не превышает величину 10"9. Полученные из данных об "электро-
"электропроводности значения подвижностей соответствуют несольватирован-
ным ионам P0G1J и SbClJ. Число переноса иона POCIJ, определенное
Гутманом и Химмлом 98, равно 0,95, но в дальнейшем это значение
не подтвердилось (В. Гутман, частное сообщение). При последующем
изучении этого раствора выяснилось, что при стоянии в концентри-
концентрированных растворах образуются коллоидные агрегаты неопре-
неопределенного состава, в состав которых, по-видимому, входят коор-
координированные атомы кислорода 10. Эти изменения сопровождаются
падением электропроводности раствора. Линдквист 34 отмечал, что,
судя по энергии кристаллической решетки, структура твердого
аддукта пентахлорида сурьмы с фосфорилхлоридом, установленная
ранее Гутманом 9, не должна быть устойчивой. В этой системе воз-
возможны другие процессы ионизации, в частности
Cl3POSbCl5 _
Cl3POSbClJ
Cl2P+OSbCl5
D0)
Однако результаты кондуктометрического титрования SbCl5 в РОС13
тетраметиламмонийхлоридом, показавшие наличие (C2H5LN+ SbCle,
подтвердили существование анионов, содержащих сурьму. При изу-
изучении фазовой диаграммы 139 системы РОС13 — SbCl5 был установлен
состав твердой фазы, которой была приписана формула
РОС13 • 2SbCl5(POCl3 • SbClJSbCle).
В растворе трихлорида бора в фосфорилхлориде существует
равновесие и
ВС13-РОС13 ^ POCIJ+BCI;
D1)
Этот вывод основан на данных кондуктометрического и потенцио-
метрического титрования.
Изучение строения твердого аддукта ВС13-РОС13 с помощью
ИК-спектроскопии привело к двум противоположным точкам зре-
зрения. Согласно одной из них, твердой фазе была приписана струк-
структура 59> 66 POClJBCli, согласно другой, — структура с донорным
атомом кислорода 162> 187. Поскольку неизвестны твердые вещества,
в которых фосфорилхлорид выполняет функцию донора хлора,
по-видимому, следует отдать предпочтение структуре с координиро-
координированными атомами кислорода. При 0° С обмен хлором между фосфо-
фосфорилхлоридом и трихлоридом бора в растворах с высокими концен-
концентрациями фосфорилхлорида происходит с большой скоростью;
в растворах с большим избытком трихлорида бора обмен не проте-
протекает 83. Отсутствие обмена ясно указывает на то, что в растворе
практически нет ионов POCIJ и BCrj. Таким образом, оказывается,
что даже в растворе фосфорилхлорид, скорее, выполняет функции
донора электронной пары кислорода, а не донора хлора. Наблюда-
Наблюдающийся при высоких концентрациях фосфорилхлорида процесс
обмена, по-видимому, обусловливается высокой диэлектрической
проницаемостью растворителя, способствующей ионному обмену:
РОС13 4П1Г POCIJ + СГ
СГ + *С13В-ОРС1з ^ (С1*С13ВОРС1з) ^=± *СГ+ C1SB • OPC1S D2)
Трудно сказать, почему образующийся на этой 'стадии сольвати-
рованный тетрахлорборат(Ш)-ион не образуется в количествах,
по крайней мере достаточных для того, чтобы вызвать быстрый обмен.
Для объяснения электропроводности растворов ВС13 и результатов
кондуктометрического и потенциометрического титрования необхо-
необходимо допустить существование борсодержащего аниона.
320 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
Возможен иной альтернативный механизм ионизации комплекса,
выражающийся уравнением
С13В-ОРС1з -^ ClsB-OPClJ + Cl-j D3)
но этот механизм несовместим с наличием перегиба на кривой кон-
кондуктометрического титрования раствора трихлорида бора в РОС13
тетраэтиламмонийхлоридом.
Спектрофотометрическое изучение системы FeCl3—ВС13—РОС13
также подтвердило, что на ранних стадиях реакции в системе при-
присутствует тетрахлорборат(Ш)-ион, в то время как в системе с тетра-
тетраэтиламмонийхлоридом появляется тетрахлорферратный ион. Даль-
Дальнейшие эксперименты по изучению электропроводности разбавлен-
разбавленных растворов трихлорида бора в фосфорилхлориде (и наоборот)
должны представлять большой интерес 106.
Необходимо опытным путем выяснить роль трихлорида бора
как акцептора хлор-иона. До сих пор известно лишь, что место ВС13
в ряду уменьшающегося сродства к хлор-иону находится между
FeCl3 и TiCl4.
Ниже приведены растворимости галогенидов алюминия в фосфо-
фосфорилхлориде и удельные электропроводности соответствующих рас-
растворов при 20° С:
Соль х A1F3 Ald3 AlBr3 A1I3
Раств«римость,г/1000гРОС13 0,5 60,0 153 170
Цвет раствора бесцветный светло- желто-ко- темно-
желтый ричневый красный
х, ом~1- см'1
при 0,05 м 4,4 • 10'6 3,6 • 10'4 4,5 ¦ 10"* 4,0 • 10"*
при 0,1 м — 7,7 • 10~4 9,1 • 10~4 8,1 • 10"*
при 0,2 м — 1,52-10 1,82-10~s 1,62 • 10~3
Электропроводность раствора А1С13 достигает максимума при
40° С и затем быстро уменьшается с повышением температуры.
Сольват, отвечающий формуле А1С13-РОС13, образуется лишь при
длительном стоянии. В растворе А1Вг3 образуется аналогичное
соединение А1Вг3 • РОС13) а в растворе АП3—соединение АП3 ¦ 2РОС1383.
Изучение фазовой диаграммы А1С13 — РОС13 показало существо-
существование еще двух соединений — А1С13-2РОС13 и А1С13-6РОСЦ4'75,
причем первое из них было выделено 168. Аналогичным образом
ведет себя трихлорид галлия, образующий сольваты GaCl3-POCl3
и GaCl3-2POCl3 69~71. Известны подобные сольваты трибромнда
галлия GaBr3-POCl3 и GaBr3-POBr3, но пока структуры этих соль-
ватов изучены лишь спектроскопически.
Спектр комбинационного рассеяния расплавленного соединения
подтверждает структуру POClJGaClJ, так как положение и поляри-
поляризация четырех из десяти линий спектра соответствуют таковым
в известном спектре иона GaClJ (тетраэдрическая конфигурация,
симметрия Td), а остальные шесть линий могут быть приписаны
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
321
иону РОСЦ (плоская конфигурация, симметрия C2v) 69> 194. На осно-
основании изучения спектра комбинационного рассеяния и ИК-спектров
аддуктов хлоридов алюминия и галлия с фосфорилхлоридом была
предложена также структура с кислородными мостиками 65.
Изменение энергии образования комплекса трихлорида галлия
GaCl3(r) + POCl3(r) 4=^" GaCl3-POCl3(r) D4)
равно —22,6 ккал. Все имеющиеся данные указывают на то, что эти.
твердые аддукты являются в основном комплексами с координи-
координированным атомом кислорода и относительно слабыми взаимодей-
взаимодействиями в кристаллической решетке.
Природу раствора А1С13 в фосфорилхлориде нельзя считать
окончательно выясненной. На кривых кондуктометрического титро-
титрования этого раствора хлоридами цинка(П), олова(Г7) и сурьмы(У)
имеются перегибы при отношениях 2 : 1 (для SnCl4) и 1 : 1 (для
SbCl5 и ZnCl2). При этом происходит осаждение соединений 85
2AlCl3-SnCl4-2POCl3 и AlCl3-SbCl5-3POCl3.
При титровании растворов А1С13 тетраэтиламмонийхлоридом
имеется перегиб при отношении 1 : 1, а вид кривой характерен
для образования растворимого ионного соединения, являющегося
очень слабым акцептором хлора. Более детальное изучение кривой
титрования SbCl5 с А1С13 в РОС13 показало, что в действительности
она сложнее, чем предполагалось в первых экспериментах. В зависи-
зависимости от направления титрования перегибы получаются при раз-
различных составах. Например, при титровании раствора хлорида
алюминия пентахлоридом сурьмы наблюдается незначительный пере-
перегиб при отношении компонентов 1 : 1, но заметный при отношении
1 : 2 вследствие образования комплекса AlCl3-2SbCl5. В то же время
при титровании раствора пентахлорида сурьмы трихлоридом алюми-
алюминия перегибы на кривой наблюдаются при составах 1:1, 1:2
B AlCl3-SbCl5) и, по-видимому, 1:3 C AlCl3-.SbClB). Некоторые
из особенностей кривой кондуктометрического титрования раствора
пентахлорида сурьмы хлоридом алюминия можно объяснить обра-
образованием коллоидных мицелл. Гораздо труднее интерпретировать
их, представив, что это результат медленного установления равно-
равновесия между твердой фазой и раствором.
Рассматривая суммарную картину, полученную из кондукто-
метрических и потенциометрических исследований и препаративных
экспериментов, можно отметить: в растворе существует несколько
типов ионов, что приводит к образованию сольватов указанных
выше составов. Связь фосфорилхлорида с алюминием в этих сольва-
тах вероятнее всего осуществляется через кислородный мостик.
По-видимому, ион AICI4 является единственным анионом в растворе.
Существование ионов А1С1|~ и А1С1|~ не доказано. В растворе при-
присутствуют также ионы А1С1(ОРС13)|+ и А1(ОРС13)в+> являющиеся
продуктами сольватации ионов А1С12+ и А13+- Сольватированный
21 Заказ 90
322 ГЛАВА ВОСЬМАЯ- ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ"
ион AICIJ, по-видимому, не существует. Система усложняется обра-
образованием полимеров типа
~С13РО ОРС13
VI
/1\
_ -С1 С1 С1 _
и даже более сложных двухмерных полимеров 26. Присутствие-
полимерных образований, со временем разрушающихся, подтвер-
подтверждается эбулиоскопическими измерениями. Истинное равновесие
при этом достигается очень медленно 103.
Число переноса тетрахлоралюминатного иона в 0,3 м растворе-
оказалось очень мало и составило лишь 0,04. Это можно объяснить
изменением направления движения ионов 97. Аналогичными экспе-
экспериментами с растворами хлорида алюминия в ацилгалогенидах было^
показано, что алюминий переносится одновременно и к аноду, и
к катоду191. Опыты по определению чисел переноса не могут в зна-
значительной мере подтвердить возможность образования сольватирован-
ного POCIJ и иона A1C1J, так как нетрудно предложить альтерна-
альтернативные более сложные системы, в которых также наблюдаются очень
небольшие числа переноса.
Тетрахлорид титана заметно растворяется в фосфорилхлориде'
с образованием электропроводного желтого раствора, из которого
может быть выделен сольват TiGl4 • 2 PQC1|4. Может быть получен
и сольват TiCl4-POCl3, имеющий димерную структуру. Два окта-
эдрически расположенных атома титана в ней связаны через мостик
из двух атомов хлора. На противоположных концах диагонали
молекулы размещены молекулы фосфорилхлорида 33. Известны три
кристаллические модификации этого соединения. На существование-
кислородных мостиков в структуре твердых сольватов указывают
данные ИК-спектров 177, а также кристаллическая структура трой-
тройного соединения TiCl3(OPCl3I;SbCl;:.
Кривая кондуктометрического титрования растворов тетрахло-
рида титана в фосфорилхлориде тетраэтиламмонийхлоридом 5 имеет
максимум при отношении компонентов 1 : 1 и при отношении 1 : 2.
Однако Гутманом 85 получена кривая, на которой хотя и имеются
перегибы при отношении компонентов 1:1 и 1 : 2, но отсутствует
максимум. Ясно, что интерпретация форм кривых кондуктометри-
кондуктометрического титрования должна проводиться с большой осторожностью.
В процессе титрования осаждается тетраэтиламмонийгексахлортита-
нат(Г7), а, как уже отмечалось ранее, присутствие осадка в системе'
вызывает серьезные трудности при интерпретации формы кривых
кондуктометрического титрования.
Потенциометрическое титрование раствора TiCl4 тетраэтиламмо-
тетраэтиламмонийхлоридом и хлоридами сурьмы(У) и железа(Ш) показало сущест-
существование в растворе ионов TiClg~ и сольватированных ионов
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
323
и TiClJ- Поведение тетрахлорида титана зависит от относительной
донорной или акцепторной силы других хлоридов. Так, в растворе
с SbCl8 преобладают ионы С13Т1(ОРС13)з, а в растворе с FeCl3 —
значительно более слабым акцептором хлор-иона, как показали
фотометрические измерения, существует равновесие
TiCl|(conbB.)FeClJ СГ? ТЮ14(сольв.) + РеС13(сольв.) D5)
Соотношение между процессами отщепления и присоединения
хлор-иона зависит главным образом от концентрации раствора.
В очень разбавленных растворах тетрахлорид титана почти всегда
донор, в концентрированных растворах он часто выступает как
акцептор 21.
Результаты эбулиоскопических измерений, проведенных с экви-
молярными смесями хлорида калия и тетрахлорида титана, показали
присутствие ионов TiCl5POCl3 и TiGl|~. Была выделена 103 твердая
калиевая соль KTiCl5-POCl3. Таким образом, в растворах в изучен-
изученном концентрационном интервале наблюдается равновесие
D6)
Гексахлорид вольфрама легко растворяется в фосфорилхлориде
с образованием раствора красного цвета. При добавлении тетра-
этиламмонийхлорида и хлоридов калия и цезия образуется желтый
раствор, содержащий сольватированные ионы WCl?, которые,
однако, не могут быть выделены в виде соли. Переход хлор-иона
от хлоридов других металлов к гексахлориду вольфрама с образо-
образованием ионов WCI7, как было определено потенциометрическими
и спектрофотометрическими методами, происходит в соответствии
со следующим рядом убывания донорнбй силы хлорида:
С15ТЮРСГ5+СГ 4=?" TiClJT
Cl5TiOPClJ + POCl3 ?=? Cl4Ti(OPCl3J
Cl4Ti(OPCl3J + POCl3 ^ГГ Cl3Ti(OPCl3)J
(C2H5LNC1 ~ KC1 — CsCl > ZnCl2
S11CI4
Данные, свидетельствующие о том, что гексахлорид вольфрама
сам может вести себя как донор хлор-иона, отсутствуют 23.
Трихлорид железа растворяется в фосфорилхлориде с образова-
образованием красно-коричневого раствора при концентрации 0,01 м, но
при разбавлении цвет раствора изменяется и при концентрации
10~4 м раствор окрашен в желтый цвет 90. Из раствора получены
твердые сольваты состава 2FeCl3 -3 POCl^9, FeCl3-POCl^9- 95 и
2FeCl3- POCl3n. УФ-Спектры этих растворов ясно показывают:
в концентрированном растворе отсутствуют тетрахлорферрат(Ш)-
ионы, но разбавление сопровождается изменением окраски раствора,
что соответствует появлению этого иона. В результате упаривания
красного раствора остается коричневый аморфный твердый про-
продукт; при осторожном охлаждении раствора или при добавлении
21*
324 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ' И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
четыреххлористого углерода выделяется красный кристаллический
продукт состава 2FeCl3-3POCl3.
Выдерживая в вакууме коричневый и красный продукты, можно
получить желтый кристаллический FeCl3-POCls .и Коричневый
осадок представляет собой сильно полимеризованный продукт с вы-
высоким содержанием фосфорилхлорида. При добавлении к красно-
коричневому раствору различных хлоридов в нем появляются
тетрахлорферрат(Ш)-ионы, о чем можно судить по спектрам погло-
поглощения раствора и изменению электропроводности. За процессом
образования тетрахлорферрат(Ш)-иона удобно проследить с по-
помощью потенциометрического 21.95, спектрофотометрического 12~15>94,
кондуктометрического 21> 26> и или эбулиоскопического методов 102.
Степень отдачи хлор-иона для разных хлоридов различна. Так,
HgCl2, BC13, TiCl4, SnCl4 и РС15 реагируют с FeCl3, растворенным
в РОС13, образуя сольватированный однозарядный положительный
катион, a ZnCl2 и А1С13, — отдавая два или даже три (А1С13) хлор-
иона. Донорная сила хлоридов по отношению к FeCl3 уменьшается
в ряду16
(C2H5LNC1 ~ КС1 ~ ZnCl2
SbCl5 ~ HgCl2 > ВС18 ~ SnCl4.
TiCl4 > РС15
Г »
Только в некоторых случаях можно считать,' что реакция идет до
конца, например в случае реакции с тетраэтиламмонийхлоридом
и хлоридами алюминия и цинка, что доказано с помощью спектро-
фотометрического метода 15; в спектрах же растворов FeCl3 в Р0С13
с добавками тетрахлорида титана и пентахлорида фосфора всегда
присутствуют линии не входящего в комплекс сольватированного
трихлорида железа 16.
Степень диссоциации, получаемая из эбулиоскопических измере-
измерений тетрахлорферрата(Ш) калия, 102 сравнима с полученной при
измерении электропроводности 94. По-видимому, в растворе происхо-
происходит простое образование ионных пар. Константа диссоциации
KFeClJ03 равняется 3-10, что соизмеримо с константами дис-
диссоциации KSbCle (9-Ю-4) п K(ClBTi • ОРС1.) C-Ю-*) а также
[Al(OPCl3N](FeCl4K(9.10-5).
Фотометрическое изучение 0,01 М растворов FeCl3 в фосфорил-
хлориде в присутствии основания по Льюису В показало, что в этих
растворах происходит переход хлор-иона. Так, пиридин и триэтил-
амин реагируют по реакции
^ C5H5N-
+ FeCl;
D7)
* Донор двух хлор-ионов.
** Донор трех хлор-ионов.
IV. ОКСИГАЛОГЕНИДЫ КАК РАСТВОРИТЕЛИ
325
Основание В отталкивает хлор-ион связи Р—С1, способствуя образо-
образованию катиона В ¦ POClJ-j
С тетраэтиламмонийперманганатом (и хлоратом) протекает реак-
реакция
MnOj + 4FeCl3 —>• Mn3+(conbB.) + 4FeClJ + 2Cl2 D8)
а при взаимодействии с тетраэтиламмонийбромидом образуется
бромтрихлорферрат(Ш)-ион 13.
Появление тетрахлорферрат(Ш)-иона обусловливает электро-
электропроводность растворов FeCl3 в РОС13. Этот ион можно получить
несколькими путями:
а)ч FeCl3 + POCl3 4=Г POClJ + FeClj
б)
в)
г) FeCl3 + B + POCl3 ^=± IT-
Возможность протекания реакции а подтверждается лишь тем,
что тетрахлорферрат(Ш)-ион появляется при разбавлении раство-
растворов трихлорида железа. Однако до сих пор еще не получено ни одного
соединения, в составе которого присутствовал бы ион POCIJ. Хоро-
Хорошим примером реакции типа в может служить реакция взаимодей-
взаимодействия хлорида алюминия с трихлоридом железа. Механизм реакции
г также убедительно доказан. Реакция б для интерпретации свойств
растворов трихлорида железа впервые была предложена Миком
и Драго 148. Растворы трихлорида железа в триэтилфосфате совер-
совершенно аналогичны растворам FeCl3 в фосфорилхлориде. При кон-
концентрации 0,01 м растворы окрашены в красный цвет, добавление
растворителя вызывает изменение окраски, в то время как добавле-
добавление хлорида лития практически не изменяет цвета образующегося
желтого раствора. Таким образом, железо в растворе, вероятно,
находится в форме тетрахлорферрат(Ш)-иона, а образования хлор-
ионов из РОС13 не происходит.
Точно так же можно показать, что кривые кондуктометрического
титрования в триэтилфосфате и в РОС13 аналогичны, откуда можно
сделать вывод, что в растворах в фосфорилхлориде существует
равновесие, описываемое реакцией б. Степень протекания реакции
должна зависеть от ряда факторов, в частности от кислотности
раствора, диэлектрической проницаемости и сольватирующей спо-
способности растворителя. По всей видимости, существенной особен-
особенностью фосфорилхлорида является не способность его подвергаться
самоионизации (этот процесс протекает очень легко), а способность-
его быть донором с помощью кислорода в Одних условиях и донором
хлор-иона — в других 91.
Замещение одного из атомов хлора в фосфорилхлориде на фе-
нильную группу не оказывает существенного влияния на свойства.
326 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
растворителя. Для сравнения приводим свойства обоих раство-
растворителей при 25° С:
Растворитель РОС1з РЬРОСЬ
Р, г/сжЗ 1,648 1,197
*пл> ?С ' 1 3
«кип- °С 108 258
Ц, спз 1,15 4,44
е 13,9 26,0
V, см3 6,7 6,3
х, ом.-1-cM-i 2-Ю"8 9-10"8
Ло для раствора (CzHeJiNCl, см,2/(ом-г-экв) 53 ?у ? 14,5
Данные об электропроводности растворов тетраэтиламмоний-
хлорида в фенилфосфорилхлориде находятся в согласии с теорией
электролитической диссоциации. Зависимость электропроводности
раствора FeCl3 в PhPOCl2 от концентрации не линейна. Отметим,
что подобными же свойствами обладает раствор FeCl3 в РОС13.
На кривых кондуктометрического титрования тетраэтиламмонийхло-
ридом растворов различных хлоридов в PhPOCl2, в частности ВС13,
А1С13, FeCl3, РС15 и SbCI5, наблюдаются перегибы, соответствующие
присоединению одного хлор-иона. Хлориды цинка(И), титанаAУ)
и олова(ГУ) в зависимости от условий присоединяют один или два
хлор-иона. Хлориды бора(Ш), титана(ГУ) и фосфора(У) отдают
один хлор-ион хлоридам железа(Ш) и сурьмы(У), образуя соответ-
соответствующие тетрахлорферратные(Ш) и гексахлорантимонатные(У)
соли 23. С помощью потенциометрических методов удалось подтвер-
подтвердить, что HgCl2, BClj, AlClj, FeCl3, SbCl3, PC15 и SbCl5 отнимают
у тетраэтиламмонийхлорида лишь один хлор-ион, a ZnCl2, TiCl4
и SnCl4. (что видно и из данных об электропроводности) отнимают
один или два хлор-иона, в зависимости от стехиометрического соот-
соотношения компонентов в растворе. Трихлорид мышьяка отдает два
хлор-иона FeCl3 и SbCl5, образуя двухзарядный сольватированный
ион 24.
Спектрофотометрические исследования показали: трихлорид же-
железа растворяется в фенилфосфорилхлориде с образованием красного
сольватированного комплекса, который при добавлении доноров
хлор-ионов, в частности хлоридов щелочных металлов или тетра-
тетраэтиламмонийхлорида, превращается в желтый тетрахлорферрат(Ш)
ион. Донорная сила хлоридов по отношению к FeCl3 в PhPOCL,
уменьшается в ряду
KCl~ (C2H5LNC1 > A1C1J ^TiCl4 > РС15 > ZnClJ >
> BCI3— SnCl4~ A1C1J* > HgCl2 > SbCl3
* Донор одного хлор-иона.
** Донор двух, хлор-ионов.
IV. ОКСИГАЛОГБНИДЫ КАК РАСТВОРИТЕЛИ
327
Этот ряд был установлен фотометрическим методом 15. По-видимому,
существенных различий между трихлоридом железа и пентахлоридом
сурьмы как акцепторами в этом ряду наблюдаться не будет. С по-
помощью потенциометрического метода установлен ряд уменьшения
акцепторных свойств хлоридов
FeCls >SbCl5 » S11CI4 > BCI3 ^HgCl2~ PCls > TiCU > AlCl3 > ZnCb
Этим методом удалось установить также, что способность хлор-иона
к отщеплению в растворе уменьшается в ряду
(C2HbLNC1 > AICI3 > TiCU, BC13 > ZnCl2 > PCl5>SbCl5 > FeCb
Трифенилметилхлорид реагирует в растворах РОС13 или PhPOCl2
с такими акцепторами, «ак хлориды цинка(П), алюминия(Ш),
. железа(Ш), бора(Ш), титана(ГУ), оловаAУ) и сурьмы(У), с обра-
образованием трифенильных солей карбония Рп3С+МС1й+1. В этом случае
акцепторная сила солей по отношению к хлор-иону уменьшается
в ряду
FeCl3 > SbCl5 > SnCU > ВС13 > ZnCl2~TiCl4 > AlCl3~ HgCl2 >
> SbCl3 > PCI5
для фенилфосфорилхлорида как растворителя 19 и в ряду
FeCl3 > SbCl5— ВС13~ SnCl4 > TiCU > А1С13 > ZnCla > HgCl2 > SbCl3 >
>PC15
для фосфорилхлорида 20. В деталях два последних ряда отличаются
от наблюдаемых с другими донорами хлор-ионов в РОС13 и PhPOCl2,
но существенные особенности сохраняются прежними, в частности,
к наиболее сильным акцепторам относятся FeCl3 и SbCl5, а к наибо-
наиболее слабым — SbCl3 и РС15. Приведенные ниже константы образо-
образования комплекса по реакции
SbCl5
РЬ3С+СГ + МС1„ 4Г2- РЬ3С+ + МС1Я+1 E0)
были получены с помощью спектрофотометрических методов:
А1С13 FeCl3 TiCl4 SnCl4
Растворенное вещество ZnCb BCI3
Константа образова-
образования комплекса
Ph3C+MCl;r+i
в РОС13 12 100
в РЬРОСЬ ••• 5,3 10,8
14
0,19
290
130
16
5,3
85
15,5
110
39,4
Следовательно, несмотря на то что диэлектрическая проницае-
проницаемость фосфорилхлорида меньше е фенилфосфорилхлорида, РОС13
в большей степени, чем PhPOCl2, способствует переходу хлор-иона.
Это различие определяется способностью фосфорильной группы
сольватировать анионы и, по-видимому, также и катионы.
328 ГЛАВА-ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
Такое поведение хлоридов как акцепторов или доноров хлор-иона
наблюдается не только в галогенидных и оксигалогенидных раствори-
растворителях. В опубликованных нрдавно работах Гутмана и его сотр. 17> 18>
25, 96, Ю5, иг показано, что хлориды ведут себя подобнцм образом
в очень отличающихся друг от друга растворителях, в частности
в метилцианиде, бензоилхлориде и других. Вполне понятно, что
ряды уменьшения донорнои и акцепторной силы для разных раство-
растворителей различны.
Способность сульфофталеинов служить индикаторами хлор-иона
может оказаться полезной при изучении процессов передачи хлор-
иона. Изменения окраски, происходящие при этом, похожи на те,
которые наблюдаются в водных растворах:
(Красный цвет) А
доноры хлор-иона
В (желтый цвет)
акцепторы хлор-иона
Изменение окраски связано с существованием ионизационного
равновесия "/аналогичного тому, которое встречается в протонных
растворителях. В некоторых случаях, как было показано, добавле-
добавление POClo вызывает появление одной из окрашенных форм ".
V. ЗАКЛЮЧЕНИЕ
До сих пор галогениды и оксигалогениды элементов V группы
как растворители находили весьма незначительное применение,
разве лишь для приготовления хлор-комплексов, которые трудно
получить иными методами. Для получения хлор- или фтор-комплек-
фтор-комплексов лучше всего использовать трихлорид мышьяка, а для получения
бром-комплексов — трибромид мышьяка. Фосфорилхлорид удобен
.для препаративных целей, так как он совершенно безводен, и для
осуществления непосредственного хлорирования- Он обладает высо-
высокой летучестью, поэтому избыток растворителя легко удаляется.
О применении галогенидов как растворителей для электрохимиче-
электрохимических целей уже упоминалось в настоящей главе. В последнее время
появились сообщения о возможности использования фенилфосфорил-
хлорида для аналитических целей, в частности в качестве раствори-
растворителя в электрохимических методах анализа, например в полярогра-
полярографии 53~55>104-
В фосфорилхлориде и трихлориде мышьяка удобно титровать
тетрахлориды титана и олова различными азотистыми основаниями
с использованием бензантрона в качестве индикатора. Наблюдается
значительный разброс результатов, но этот метод все же может
найти применение для аналитических целей 159. Аналогичная картина
наблюдалась и при титровании с использованием в качестве внутрен-
внутреннего индикатора кристаллического фиолетового 160.
В заключение нам хочется подчеркнуть, что поведение галогени-
,дов и оксигалогенидов как растворителей не является необычным.
литература
329-
Водородные связи, в основном определяющие свойства воды, в этих
растворителях отсутствуют, но растворители этой группы проявляют
способность к ассоциации и сольватации других галогенидов, раство-
растворенных в них. Этот процесс хотя и выражен гораздо слабее, чем
процесс образования водородных связей, но тождествен ему по своей
природе. Например, акцепторный характер фосфора делает возмож-
возможным образование фосфорильных мостиков Р=О- • -Р, которые можно
сравнить с водородными связями в воде; свойства фосфорилхлорида
как растворителя нужно рассматривать именно с этих позиций.
Оксигалогениды являются хорошими донорами электронов через
атом кислорода и образуют многочисленные сольватированные
комплексные катионы, а в ряде случаев и анионы. Самоионизация
растворителей несущественна для рассмотрения их свойств, но,
по-видимому, она происходит, хотя и в ограниченных пределах,
так как эти растворители по своей природе являются донорами;
и акцепторами и имеют высокую диэлектрическую проницаемость.
Важность процесса самоионизации в свете теории кислот и основа-
оснований, вероятно, слишком преувеличивалась в прошлом. Отметим
здесь, что подобным образом ведут себя и другие растворители, даже
не содержащие хлор-ионов.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
ЛИТЕРАТУРА
Addison С. С, Hodge N., J. Chem. Soc, 1961, 240.
Addis on С. С, Lewis J., Quart. Rev. (London), 9, 115 A955).
Adolfsson G., В г у n t s e R., Lindquist I., Acta chem. scand.,
14, 949 A960).
A g e r m a n M., Anderson L. H., Acta chem. scand., 12, 477 A958).
Andersson L. H., Lindquist I., Acta chem. scand., 9, 79 A955).
Asmussen R. W., Z. anorg. Chem., 243, 127 A939).
V., Monatsh., 90, 276 A959).
V., Monatsh., 90, 256 A959).
Monatsh.,
В a a z
В a a z
В a a z
В a a z
В a a z
В a a z
В a a z
В a a z
18, 276
В a a z
В a a z
В a a z
В a a z
В a a z
A961).
В a a z
A961).
В a a z
A960).
В a a z
A961).
В a a z
M.
M.
M.
M.
M.
M.
M.
M.
M.
M.
M.
M.
M.
G u t m a n n
G u t m a n n
G u t m a n n
G u t m a n n
G u t m a n n
G u t m a n n
G u t m a n n
G u t m a n n
V.,
90, 744 A959).
V., Monatsh., 90, 426 A959).
A961).
Gutmann
G u t m a n n
G u t m a n n
G u t m a n n
G u t m a n n
V.,
V.,
v.,
v.,
v.,
v.,
v.,
v.,
V.,
H ii b n e r
H u b n e r
H u b n e r
H ii b n e r
H ii b n e r L.
H ii b n e r L.
К u n z e O.,
К u n z e O.,
Masaguer
Monatsh.,
Monatsh.,
Monatsh.,
J. Inorg.
91,
91,
92,
694
537
707
a Nucl.
A960).
A960).
A961).
Chem.,
Monatsh., 92, 135 1961).
Monatsh., 92, 272 1961).
Monatsh., 93, 1142 1962).
Monatsh., 93, 1162 1962).
J. R., Monatsh., 92, 582
M., Gutmann V., Masaguer J. R., Monatsh., 92, 590
M., Gutmann V., Talaat M. Y. A., Monatsh., 91, 548
M., Gutmann V., Talaat M. Y. A., Monatsh., 92, 714
M., Gutmann V., West T. S., Monatsh., 92, 164 A961).
330 ГЛАВА ВОСЬМАЯ. ГАЛОГЕНИДЫ И ОКСИГАЛОГЕН. ЭЛЕМЕНТОВ V ГРУППЫ
24
25
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
В a a z M., Gutmann
В a a z M., Gutmann
Baaz M., Gutmann V. etal.,
В a ugh an Е. С, Jones Т. P.
Soc, 1963, 274.
Beckham L. J., Fessler W.
319 A951).
Beckmann E
Boston С R.,
Boston С R.,
Boston С R.,
V. et al., Monatsh., 92, 150 A961).
V. et al., Monatsh, 93, 1416 A962).
Z. anorg. Chem., 310, 302 A961).
S t о о d 1 e у L. G., Proc. Chem.
A., Rise M. A., Chem. Rev., 48,
62, 409
66, 1178
A958).
A962).
, Z. anorg. Chem., 51, 111 A906).
Smith G. P., J. Phys. Chem.,
Smith G. P., J. Phys. Chem., ... .... ,.
_. -., Smith G. P., Ho wick L. C, J. Phvs. Chem.,
67, 1849 A063).
Branden C. I., Lindquist I., Acta chem. scand.,
Branden C. I., Lindquist I., Acta chem. scand.,
В r e d i g M. A., J. Phys. Chem., 63, 978 A959).
Bredig M. A., Levy H. A. et al., J. Phys. Chem., 64, 191 A960).
Briihl J. W., Z. phys. Chem., 27, 319 A898).
Burg А. В., С a m p b e 11 G. W., J. Am. Chem. Soc,
Burg А. В., М с К e n z i e D. E., J. Am. Chem. Soc,
С a d у H. P., J. Phys. Chem., 1, 707 A897).
W., Ann., 98, 213 A856).
, J. Phys. Chem., 62, 1149 A958).
, J. Am. Chem. Soc, 80, 4757 A958).
Albers F. C, .1. Am. Chem. Soc, 82, 533 A960).
14, 726 A960).
17, 353 A963).
70, 1964 A948).
74, 3143 A952).
Casselmann
Corbett J.D
Corbett J.D
Corbett J. D., .. _., , „, „_„ ч.„
Craig D. P., Mac coll A. et al., J. Chem. Soc, 1954, 332, 354.
W., Bull. Soc. chim. France, [2], 19, 499'A873).
W., Ber., 5, 1466 A873).
Cronander A,
С г о n a n d e r A. , , , ч ,.
Cronander A. W., Uppsala Univ. Arsskr., 1 A873).
D ad ape V. V., R а о M. R. A., J. Am. Chem. Soc, 77, 6192 A955).
Dafert O., Melinski Z. A., Ber. 59,788A926).
Daure P., С. г., 188, 1605 A929).
D a v i e s A. G., В a u g h a n E. C, J. Chem. Soc, 1961, 1711.
Dehn H., Gutmann V., Schober G., Monatsh., 93, 1357 A962).
Dehn H., Gutmann V., Schober G., Monatsh., 94, 312 A963).
Dehn H., Schober G., Monatsh., 94, 316 A963).
Dess H. M., Parry R. W., J. Am. Chem. Soc, 78, 5735 A956).
Dess H. M., Parry R. W., Vidale G. L., J. Am. Chem. Soc,
78, 5730 A956).
Ehrlich P., Diet z G., Z. anorg. Chem., 305, 158 A960).
Fraser M. J., Gerrard W., Patel J. K., J. Chem. Soc, 1960,
726.
F г у с z К., Т о 1 1 о с z к о S., Festschrift Univ. Lwow, 1, 1 A912).
Frycz К., Tolloczko S., Zbl.. 1, 91 A913).
Fuoss R. M., J. Am. Chem. Soc, 57, 488 A935).
George J. W., Progr. inorg. Chem., 2, 33 (I960).
Gerding H., Houtgraaf H., Rec. trav. chim.,
Gerding H., Koningstein J. A., Van der
Spectrochim. acta, 16, 881 A960).
Gerrard W., Mo one у E. F., W i 11 i s H. A., J. Chem. Soc, 1961,
4255.
Gibson S., Johnson J. В., Vining D. C,
1930, 1710.
G m e 1 i n, Handbuch der anorganischen Chemie, Part 18B, 1949.
Greenwood N. N., J. Inorg. a. Nucl. Chem., 8, 234 A958).
Greenwood N. N., Perkins P. G., J. Inorg. a. Nucl. Chem., 4,
291 A957).
Greenwood N. N.,Wade K., J. Chem. Soc, 1957, 1516.
72, 21 A953).
Worm E. R:,
J. Chem. Soc,
ЛИТЕРАТУРА
331
72. Greenwood N. N., W о r r a 11 I. J., J. Inorg. a. Nucl. Chem., 6,
34 A958).
73. Gr о ene v el d W. L., Rec. trav. chim. 72, 617 A953).
74. G г о e n e v e 1 d W. L., Z u u r A. P., Rec. trav. chim., 76, 1005 A957).
W. L., Zuur A. P., J. Inorg. a. Nucl. Chem., 8, 241
75.
76.
77.
78.
79.
80.
81.
82.
83.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
95.
96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
Groeneveld
A958).
G u s t a v s о n G., Z. Chem., B), 7, 417 A871).
Gutmann V., Z. anorg. Chem., 266, 331 A951).
Z. anorg
Monatsh.
Monatsh.
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
Gutmann
V.
V.,
V.,
V.,
V-,
v.,
v.,
v.,
v.,
V.,
v.,
v.,
v.,
v.,
V.,
v.,
V.,
V.,
V.,
v.,
V.,
V.,
v.
v.
v.
v.
V.
v.
Chem., 264, 151 A951).
82, 473 A951).
83, 159 A952).
Monatsh., 83, 279 A952).
Monatsh., 83, 164 A952).
Monatsh., 83, 583 A952).
Z. anorg. Chem., 269, 279 A952).
Z. anorg. Chem., 270, 179 A952).
Monatsh., 84, 1191 A953).
Monatsh., 85, 491 A954).
Monatsh., 85, 1077 A954).
Svenck. Kern. Tidskr., 68, 1 A956).
J. Phys. Chem., 63, 378 A959).
Baaz
Baaz
Baaz
Baaz
Baaz
M.
M.
M.
M.
M.
Angew. Chem
Monatsh., 90,
71,
239
H a m p e 1
H i m ml R., Z
H i m ml R., Z.
H u b а с е к Н.,
Lindquist
M a i г i n g e r
Mairinger
M a i r i n g e r
Schober G.,
H a m p e 1 G.,
Monatsh., 90, 271
Monatsh., 90,
G., Monatsh.,
Z. anorg. Chem'., 298, 121 A959).
"' 57 A959).
A959).
A959).
729 A959).
94, 830 A963).
Phys. Chem., 4, 157 A955).
anorg. Chem., 287, 199 A956).
, Monatsh., 94, 1019, 1098 A963).
I., Z. phys. Chem., 203, 250 A954).
F., Z. anorg. Chem., 289, 279 A957).
F., Monatsh., 92, 529 A961).
F., Monatsh., 92, 720 A961).
, Monatsh., 93, 1353 A962).
Masaguer J. R., Monatsh., 94,
822 A963).
106. H e r b e г R. H., J. Am. Chem. Soc, 82, 792 A960).
107. H e t h er i n g t о n G., Robinson P. L., Chem. Soc. Special
Publication, № 11, 23 A957).
lBSTHewitt F., H oil id ay A. K., J. Chem. Soc, 1953, 530.
R., J. Am. Chem. Soc, 82, 5285 A960).
R., J. Inorg. a. Nucl. Chem., 12, 266 A960).
R., В e r t a u t E. F., J. Am. Chem. Soc, 80, 2980, 2983
109.
110.
111.
Holmes
Holmes '.
Holmes :
A958).
112. H u b асе к
A963).
113. J an der G.
114. J a n d e r G.
J a n d e r G.
J a n d e r G.
J a n d e r G.
J a n d e r G.
J a n d e r G.
lander G.
H., Stancie В., Gutmann V., Monatsh., 94, 1118
115.
116.
117.
118.
119.
120.
121.
G ii n t h e r
G u n t h e r
G u n t h e r
Swart K.
Swart
Swart
Weiss
Weiss
K.
K.
J.,
J.
K., Z. anorg. Chem., 297,81 A958).
K., Z. anorg. Chem., 298, 241 A959).
K., Z. anorg. Chem., 302, 155 A959).
H., Z. anorg. Chem., 299, 252 A959).
H., Z. anorg. Chem., 301, 54 A959).
H., Z. anorg. Chem., 301,- 80 A959).
Z. Elektrochem., 61, 1275 A957).
Z. Elektrochem., 63, 1037 A959).
Kendall J.,Crittenden E. D., Miller H. K., J. Am. Chem.
Soc, 45, 963 A923).
.332 ГЛАВА ВОСЬМАЯ. ГАЛОГВНИДЫ И ОКСИГАЛОГВН. ЭЛЕМЕНТОВ V ГРУППЫ
ЛИТЕРАТУРА
333
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
158.
159.
160.
161.
162.
163.
164.
165.
166.
167.
168.
Klemensiewjcz
Klemensiewicz
Klemensiewicz
Keneshea F. J., Cubicciotti D., J. Phys. Chem., 63 1472
A959).
Klemensiewicz Z., Bull. int. Acad. Cracovie, 6, 418 A908)
Z., Z. Phys. Chem., 113, 28 A924).
Z., В a 1 о w n a Z., Roczn. Chem., 10, 481 A930).
~*.iciUvU?.ic.Yi.'a Z., В a 1 о w n a Z., Roczn. Chem., 11, 683 A931).
Klemensiewicz Z., Zebrowska A., Roczn. Chem., 14, 14 1934).
Kohler H., Ber., 13, 875 A880).
Z. anorg. Chem:, 280, 313 A955).
Chem., 286, 307 A956).
Chem., 289, 118 A957).
Chem., 289, 128 A957).
_ o S., Z. anorg. Chem., 302, 88 A959).
F e 1 t z A., Z. anorg. Chem., 293, 286 A957).
Schmidt W., Z. anorg. Chem., 296,188 A958).
M., L a i t i n e n H. A., pH and Electro Titrations,
New York.
R. et al., Science, 110, 300 A949).
R., et al., J. Phys. Chem., 58, 206 A954).
.., Tridot G., С. г., 248, 3439 A959).
Lenher V.,J. Am. Chem. Soc, 30, 740 A908).
Lewis J., Sower by D. В., J. Chem. Soc, 1956, 150.
D. В., J. Chem. Soc, 1957, 336.
D. В., J. Chem. Soc, 1957, 1617.
D. В., J. Chem. Soc, Special Publication,
К о 1 d i t z
К old itz
К о 1 d i t z
К о 1 d i t z
К о 1 d i t z
К old i t z
К о 1 d i t z
Kolhoff
2nd ed., 1941
L а с h e r J
L а с h e r J
L e m a n G.
Z. anorg.
Z. anorg.
Z. anorg.
E n g e 1 s
Lewis J., Sowerby
Lewis J., Sowerby
Lewis J., Sowerby
№ 10, 123 A957).
Lindqujst I.
Lindquist I.
A954).
Lindquist
Meek D. W..
M e t z 1 S., Z.
Metzner R
Acta chem. scand., 9, 73 A955).
Andersson L. H., Acta chem. scand., 8, 128
I., В r a n d e n C. I., Acta chem. scand., 12, 642 A959).
Drag о R. S., J. Am. Chem. Soc, 83, 4322 A961).
anorg.
.„„,6. Chem., 48, 146 A906).
Ann. chim. phys., [7], 15, 254 A898).
гавиивг п., лпп. сшш. puys., i/|, id, <»4 (loao).
M о n t i g n i e E., Bull. Soc. chim. France, [5], 2, 1365 A935).
Muetterties E. L., P h i 11 i p s W. D., J. Am. Chem. Soc, 79,
322 A957).
Mue.tterties
3686 A957).
О d do G., R. С
E) 10, 452 A905).
О d d о G., T e a 1 d i M., Gazz. 33, II, 427 A903).
Olah G. A., Tolg у esi W. S., J. Org. Chem., 26, 2319 A961).
E. L., Phillips W. D., J. Am. Chem. Soc, 79,
Atti, Accad. Line, Rend. Classe Sci. fis. mat. nat.,
Chem. Soc, 1948,
Chem. Soc, 1949,
Analyt. Chem., 31,
1952.
3135.
1495
Partington J. R",- W h у n e s A. L.~ J
Partington J. R., Whynes A. L., J.
Paul R. C, Singh J., Sandhu S. S.,
A959).
Paul R. C, Singh J., Sandhu S. S., J. Indian Chem. Soc,
36, 305 A959).
Payne D. S., Quart. Rev. (London), 15, 173A961).
Peach M. E., Wadding ton Т. С, J. Chem. Soc, 1962, 3450.
Per rot R., Devin С, С. г., 246, 772 A958).
P e t z о 1 d W., Z. anorg. Chem., 214, 355 A933).
Porter G. В., В a u g h a n E. C, J. Chem. Soc, 1958, 744.
P u s i n N. A., L 6 w у S., Z. anorg. Chem., 150, 167 A926).
P u s i n N. A., M а к u с J., Z. anorg. Chem., 237, 177 A938).
Raman K. N. V., M u г t h у A. R. V., Proc Ind. Acad. Sci., 51A,
270 A960).
169.
'170.
171.
172.
173.
174.
175.
176.
177.
178.
179.
180.
181.
182.
183.
184.
185.
186.
187.
188.
189.
190.
191.
192.
193.
194.
195.
196.
197.
198.
199.
Rettgers J. W., Z. Phys. Chem., 11,328 A893).
Rheinbolt H., Wasserfuhr R., Ber., 60,732A927).
Ruff O., Ber., deut. keram. Ges., 37, 4513 A904).
Ruff O., Ipsen R., Ber., 36, 1783 A903).
E., van Wazer J. R., J. Am. Chem.
Schwarzmann
81, 6366 A959).
Seel F., Z. anorg
Seel F., Bauer
Soc,
Chem., 252, 24 A943).
H., Z. Naturforsch., 126, 397 A947).
Seel J. C, Schmutzler R.,Wasem K., Angew. Chem., 71,
340 A959).
Sheldon J. C, Tyree S. Y., J. Am. Chem. Soc, 81, 2290 A959).
Shire у W. В., J. Am. Chem. Soc, 52, 1720 A930).
Spandau H., Beyer A., Preugschat F., Z. anorg. Chem.,
306, 13 A960).
S u d Ьчо г о u g h J. J., J. Chem. Soc, 59, 655 A891).
Tolloczko S., Z. phys. Chem., 30, 705 A899).
Tolloczko S., Bull. int. Acad. Cracovie, 1, 1A901).
Troost W. R., Can. J. Chem., 32, 356 A954).
Van H et eren W. J., Z. anorg. Chem., 22, 277 A899).
Von к С. G., W i e b e n g a E. H., Rec trav. chim. 78, 913 A959)_.
Waddington Т. С, Klanberg Т., Z. anorg.
A960).
Waddington T. C, Klanberg F., J. Chem.
Wai den P., Z. anorg. Chem., 25, 209 A900).
Wai den P., Z. anorg. Chem., 29, 371 A902).
W a 1 d e n P., Z. phys. Chem., 43, 445 A903).
Wertyporoch E., Firla Т., Z. phys. Chem.,
Wheeler H. L., Am. J. Sci., 13], 46, 90A893).
Wheeler H. L., Z. anorg. Chem., 4, 452 A893).
Woodward R. A., Garton G., Roberts
Soc, 1956, 3723.
Woolf A. A., Gre en woo d N. N.. J. Chem. Soc, 1950, 2200.
Горенбейн Е. И., Ж0Х, 15, 729 A945). ,,„„,,„
Избеков В., Плотников В., Z. anorg. Chem., 71, 332 A911).
Сокольский Г. А., Кнунянц И. Л., Изв. АН СССР, ОХН,
1960, 779.
Эйнгорн Л. Н., Укр. хим. ж., 16, № 4, 404 A950).
Chem., 304, 185
Soc, 1960, 2339.
А162, 398 A932).
Н. L., J. Chem.
ГЛАВА ДЕВЯТАЯ
РАСПЛАВЛЕННЫЕ СОЛИ
КАК РАСТВОРИТЕЛИ
Г. БЛЮМ, Дж. В. ХАСТИ
I. Введение 335
II. Природа расплавленных солей 336
A. Типы структурных элементов в расплавах . 336
Б. Дырки и свободный объем в расплавах солей ионного типа . . . 338
B. Функции распределения 339
Г. Природа сил (связей), действующих между ионами в расплавах 339
III. Растворы соль — вода 339
IV. Растворы соль — органическое соединение 340
А. Растворы солей в органическом растворителе 340
1. Растворы солей в ацетоне 340
2. Растворы солей в триазоле и расплавах других органических
веществ 342
3. Растворы хлоридов переходных металлов в метаноле .... 342
Б. Растворы органических соединений в расплавленных солях . . 342
V. Растворы неметаллов в расплавленных солях 343
А. Растворы серы 343
Б. Растворы иода 343
VI. Растворы газов в расплавленных солях 343
А. Системы без химического взаимодействия 344
1. Растворы двуокиси углорода в галогенидах щелочных металлов 344
2. Растворы благородных газов в расплавленных галогенидах 344
Б. Растворы газов, образующих в расплавленных солях комплексные
соединения 345
VII. Растворы металлов в расплавленных солях 346
A. Системы без существенного химического взаимодействия («ме-
(«металлические» растворы) 347
1. Растворы щелочных металлов в расплавах галогенидов ще-
щелочных металлов 347
2. Растворы щелочноземельных металлов в расплавленных гало-
галогенидах щелочноземельных металлов 349
3. Растворы лантаноидов в расплавленных галогенидах ланта-
лантаноидов 350
Б. Системы с сильным взаимодействием между растворителем и рас-
растворенным соединением («неметаллические» растворы) 351
1. Системы с переходными металлами 351
2. Системы с металлами концов больших периодов 353
B. Растворы металлов в солях других металлов (реакции замещения) 354
VIII. Растворы солей в расплавленных солях 356
A. Системы расплавленных солей с областью расслоения 356
1. Система АХ + BY 357
2. Сютема АХ + ВХ 358
3. Система АХ + AY 358
Б. Системы расплавленных солей с полной взаимной растворимостью 358
B. Структура растворов расплавленных солей 359
Г. Сольватация и образование комплексных ионов в расплавленных
солевых смесях 363
I. ВВЕДЕНИЕ
335
Д. Комплексные ионы и кинетика реакций 63
IX. Применение расплавленных солей в качестве растворителей .... 364
A. Реакции в расплавленных солях с участием органических ве-
веществ или летучих неорганических жидкостей 365
1. Реакции фторирования 365
2. Обменные реакции 366
3. Реакции азидов с органосиланами 366
4. Реакции дегидратации спиртов 366
5. Реакции дегидрохлорирования 366
6. Другие препаративные методы, основанные на реакциях
в расплавленных солях 367
7. Получение силана 367
Б. Получение металлов в расплавленных солях 367
1. Промышленный метод получения алюминия 367
2. Получение актиноидов 368
3. -Разделение металлов с близкими свойствами 368
4. Извлечение магния из силикатных минералов 368
5. Извлечение свинца из галенита 368
B. Хроматографическое разделение расплавленных солей 368
Г. Экстракция расплавленными солями 369
Д. Основные направления дальнейших исследований 369
Литература 369
I. ВВЕДЕНИЕ
Расплавленные соли являются хорошими растворителями для
очень многих веществ: от паров неорганических и органических
соединений до тугоплавких металлов (включая платину) и окислов.
Однако о растворимости имеется лишь ограниченное число данных.
Большое многообразие солей и солевых смесей позволило создать
класс растворителей с температурным интервалом жидкого состоя-
состояния, превышающим тысячу градусов. Например, криолит Na3AlFe
плавится при 1003° С, в то время как эвтектическая смесь А1С13 +
+ NaCl + KC1 плавится уже при 89° С. Наиболее широко исполь-
используемой группой солевых расплавов являются галогениды в связи
с высокой устойчивостью их в широком температурном интервале,
доступностью и способностью растворять самые различные вещества.
Расплавы галогенидов щелочных и щелочноземельных металлов
могут быть использованы в качестве растворителей благодаря тому,
что существует много окислителей и восстановителей, по отноше-
отношению к которым эти системы остаются инертными. Так, разность равно-
равновесных потенциалов окислительных и восстановительных процессов,
в которых уже участвует эвтектическая смесь LiCl + КС1, при
700° С составляет примерно 3,5 в, a CsCl + MgCl2 приблизительно
2,6 в
108
в то время как для водных растворов при комнатной темпе-
температуре эта величина равна лишь 1,2 в114.
336 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
И. ПРИРОДА РАСПЛАВЛЕННЫХ СОЛЕЙ
337
II. ПРИРОДА РАСПЛАВЛЕННЫХ СОЛЕЙ
Блюм и Бокрис 8 показали, что для определения структуры рас-
расплавленных солей необходимо знать:
а) тип присутствующих в расплаве структурных элементов —
ионы, молекулы, комплексные ионы и т.д.;
б) природу «дырок» или вакансий в расплаве;
в) функцию распределения относительных позиций структурных
элементов и дырок;
г) природу связей и сил, действующих между различными струк-
структурными элементами в расплаве. %
Этих сведений вполне достаточно для определения структуры
как простых солей, так и солевых смесей, и с этих позиций лучше
всего рассматривать природу расплавленных солей. Например,
распределение дырок и других форм свободного объема имеет реша-
решающее значение для определения транспортных свойств жидкости,
поскольку без наличия свободных объемов в ней процессы переноса
маловероятны. Кроме того, если известны функции распределения,
представляется возможность рассчитать термодинамические функции
расплава.
А. Типы структурных элементов в расплавах
Основным свойством большинства солей является резкое воз-
возрастание электропроводности при плавлении. Это, а также примени-
применимость к солям законов электролиза Фарадея указывает на ионную
природу расплавленных солей. Широкий интервал устойчивости
жидкостей при обычном давлении, составляющий, например, для
NaCl 612 град, указывает на присутствие в расплавленных солях
сильно взаимодействующих ионов. Несмотря на то что расплавы
носят, по существу, ионный характер, у различных солей имеется
разная степень ассоциации; это определяется в основном положением
элемента — катиона в таблице Менделеева. Например, температур-
температурный интервал жидкого состояния для хлорной ртути составляет
только 26 град, и эквивалентная электропроводность ее сравнительно
мала. Это указывает на то, что расплав состоит главным образом из
незаряженных частиц. Бильтц и Клемм 4, проанализировав электро-
электропроводность различных хлоридов, пришли к однозначному выводу
о том, что степень ковалентности хлоридов возрастает по мере сме-
смещения катиона в периодической системе элементов слева направо
и сверху вниз.
Исследования, проведенные с помощью рентгеноструктурного
и нейтронографического методов, позволили установить, что при
плавлении твердых веществ, ионных по своей природе, происходят
сравнительно небольшие структурные изменения, затрагивающие
первую и вторую координационные оболочки. Структурные исследо-
исследования, проведенные Харрисом и др. 55, Даниловыми Красницким 107,
Зарзикки 102>103 и Леви и др. 73, показали, что при плавлении хло-
хлоридов щелочных металлов координационное число ближайших
соседей (ионов другого типа) обычно уменьшается от 6 до 4. Среднее
расстояние между катионом и анионом также несколько уменьшается;
Напротив, расстояния между катионами и между анионами слегка
увеличиваются. Таким образом, структура ионного расплава пред-
представляется как частично нарушенная модель структуры соответству-
соответствующего твердого вещества. Основное различие между расплавом
и ионной кристаллической структурой заключается в том, что при
плавлении полностью исчезает дальний порядок решетки и одно-
одновременно увеличивается мольный объем (для хлоридов щелочных
металлов это увеличение составляет —25%) п. Ближний порядок
кристаллической решетки сохраняется. Следовательно, можно при-
принять, что при плавлении вследствие присутствия ионов другого типа
структура становится более разреженной, и в ней появляются
«дырки>> и вакантные места.
Приняв, что в основном природа расплавленных солей ионная,
можно высказать некоторые соображения о типах присутствующих
ионов. В расплавах галогенидов щелочных металлов величины
молекулярной рефракции соизмеримы с соответствующими величи-
величинами для водных растворов этих солей при бесконечном разбавле-
разбавлении 9. Это позволяет с уверенностью утверждать, что е обоих слу-
случаях состояния ионизации одинаковы, т. е. такие расплавы состоят
только из простых катионов и анионов.
Криоскопическпе измерения растворов таких солей, как NaCl,
в расплавленном NaNO3 93 указывают на то, что растворы идеальны.
Это вероятно лишь в том случае, когда растворение сопровождается
образованием только простых ионов. Однако величина понижения
температуры затвердевания при растворении в NaNO3 двухвалент-
двухвалентных галогенидов тяжелых металлов указывает на отклонения раство-
растворов от идеальности. Отсюда вытекает, что такие галогениды, по-ви-
по-видимому, в растворе частично ассоциированы.
Результаты криоскопических измерений могут быть объяснены,
если допустить, что степень ассоциации галогенидов в растворе
убывает в следующем ряду: CdBr2 > ZnCl2 > CdCl2 > CuCl2 >
j> РЬС12. Значения коэффициентов активности некоторых из этих
солей в расплавленных галогенидных смесях подтверждают указан-
указанный ряд уменьшения степени ассоциации (Г. Блюм и Д. В. Хасти,
неопубликованные результаты). Вполне возможно, что в этих систе-
системах помимо простых ионов существуют комплексные ионы, напри-
например CdCl+, CdCl;, CdClf или даже CdCl4e-
Были определены13 константы ассоциации в расплавленных
нитратах для комплексных ионов CdCl+ и РЬС1+. Во всех раствори-
растворителях эти константы для CdCl+ оказались выше, чем для РЬС1+.
Браунштейн и Линдгрен 14 определили константы для CdBr+ и Cdl+.
Примечательно, что степень ассоциации убывает в ряду 1^> Вг ]>С1.
22 Заказ 90
338
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
В этой же последовательности изменяется поляризуемость
аниона.
Измерения магнитной восприимчивости расплавов дигалогенидов
тяжелых металлов 42 показали невозможность существования в них
однозарядных ионов, например Cd+. При сопоставлении потенциалов
ионизации в этих расплавах становится ясным, что энергетически
более вероятно образование таких ионов, как CdCl+ и CdCl^,
а не ионов Cd+ и С1~. С позиций стереохимии образование иона
CdCl+ вероятнее образования иона CdCl;. В расплаве CdCrv, (в области
температур чуть выше точки плавления) стереохимически возможно
образование еще более крупного иона GdGl|~.
Относительно небольшая скрытая теплота плавления CdCl2 по
сравнению, например, с таковой для NaCl подтверждает, что при
плавлении зтого вещества образуются не ионы Cd2+ и С1~, а лишь
ассоциированные ионы. Бюэ 23 отметил, что частоты спектра ком-
комбинационного рассеяния для расплава аналогичны соответствующим
частотам в спектре твердого вещества. Это также указывает на
равнозначность структурных единиц твердого вещества и расплава.
Электронные и колебательные спектры таких солей, как нитраты,
в структуре которых в твердом состоянии имеются сложные анионы,
указывают на существование тех же анионов в расплавах (например,
в нитратных расплавах существует ион NOj). Спектроскопическим
методом было установлено и существование ионов GaFj в расплавах
Gar3 (Г—а, Вг) 95.
Б. Дырки и свободный объем в расплавах солей
ионного типа
Увеличение объема при плавлении кристаллов ионного типа,
например галогенидов щелочных металлов, составляет 71 в отдель-
отдельных случаях более 25%. Опыты по определению сжимаемости рас-
расплава показали, однако, что свободный объем, в пределах которого
происходят колебания ионов, составляет лишь 2% от мольного
объема 10. Это означает, что большая часть увеличения объема при-
приходится на образующиеся в расплаве дырки. Эти дырки могут обус-
обусловливать перенос ионов через расплавленную среду. Образование
дырок подтверждается также упомянутыми выше опытами по
дифракции рентгеновских лучей.
Опыты Кэмпбелла 26 по измерению электропроводности, по-види-
по-видимому, также указывают на присутствие в расплавленных солях
дырок. Например, электропроводность LiC103 (растворитель) быстро
падает при добавлении 0—0,4 вес. % нитробензола; добавление
О—1,25% метанола вызывает лишь слабое уменьшение электро-
электропроводности. При добавлении 0—6% воды электропроводность
расплава значительно возрастает, что может быть объяснено увели-
увеличением степени ионизации LiC103 сильно полярными молекулами
III. РАСТВОРЫ СОЛЬ — ВОДА
339
воды. Заметное влияние нитробензола на электропроводность вероят-
вероятнее всего объясняется уменьшением числа дырок в расплаве, что
затрудняет движение ионов, определяющее проводимость. Если
стать на эту точку зрения, то понятно, что менее крупные молекулы
метанола в меньшей степени снижают электропроводность LiC103.
В. Функции распределения
По функциям распределения, описывающим порядок в расплав-
расплавленных солях, можно рассчитать термодинамические свойства.
Сравнивая рассчитанные термодинамические свойства с полученными
экспериментальным путем, можно внести поправки в выведенные
функции распределения.
Г. Природа сил (связей), действующих между ионами
в расплавах
В расплавленных галогенидах щелочных металлов силы, действу-
действующие между ионами, являются в основном кулоновскими, т. е.
каждый ион стремится создать себе окружение из противоположно
заряженных ионов. Скрытая теплота плавления этих солей состав-
составляет лишь незначительную долю от величины энергии решетки,
следовательно, при разрушении твердого вещества и образовании
расплава не происходит абсолютно беспорядочного распределения
катионов и анионов. Напротив, каждый ион стремится окружить
себя оболочкой из противоположно заряженных ионов. Как уже
указывалось, в расплавах галогенидов тяжелых металлов (двух-
(двухвалентных) образуются ковалентные связи и комплексные ионы,
такие, как CdCl+.
III. РАСТВОРЫ СОЛЬ — ВОДА
Обычно вода рассматривается как сольватирующий агент, и это
в физическом отношении верно, поскольку сольватируются соли
или ионные компоненты солей, окружая себя сферой, состоящей из
молекул воды. Однако формально можно рассматривать и соли как
сольватирующие агенты.
К настоящему времени большая часть сведений о природе раство-
растворов соль — вода получена для растворов, в которых вода присутство-
присутствовала в избытке. Такие растворы рассматриваются в многочисленных
монографиях, посвященных растворам электролитов.
Эвтектическая смесь LiCl + KC1 способна растворять пары
воды 2Б, причем растворимость последней подчиняется закону Генри
до давления водяных паров 18 ммрт. ст. при 480° С и до 14 мм рт. ст.
при 390° С. При давлениях водяных паров выше 18 мм рт. ст.
при 480° С наблюдается резкий перелом на кривой растворимости
22*
340
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
вследствие протекания гидролиза LiCl (при 390° С таких явлений
не наблюдается).
Вода исключительно прочно удерживается расплавом, что можно
объяснить, по-видимому, высокой плотностью заряда иона Li+.
Действительно, если расплав LiCl + КС], находящийся в равнове-
равновесии с водяным паром, вакуумировать в течение часа, то в нем оста-
останется 4 мм водяного пара на 1 моль LiCl. С другой стороны, если
в течение 15 мин откачивать при 700° С находящуюся в равновесии
с водяным паром расплавленную смесь, состоящую из 62 мол. %
NaCl и 38 мол. % КС1, то в расплаве не обнаруживаются заметные
остаточные количества водяных паров.
Вода может растворяться также в расплавленных нитратах щелоч-
щелочных металлов (Na, К, Cs), по-видимому, за счет внедрения в струк-
ТУРУ расплава.
IV. РАСТВОРЫ СОЛЬ - ОРГАНИЧЕСКОЕ СОЕДИНЕНИЕ
Такие растворы являются единственными в своем роде в связи
с тем, что компоненты растворов резко отличаются друг от друга
природой химических связей. Тем не менее число растворов указан-
указанного типа удивительно велико. Существует два вида таких раство-
растворов: растворы, образующиеся прк добавлении соли к органическому
соединению, и растворы, образующиеся при добавлении органиче-
органического соединения к соли. Растворы первого вида исследованы го-
гораздо полнее.
А. Растворы солей в органическом растворителе
Существование, как показывают фазовые диаграммы, конгруэнтно
плавящихся соединений, образующихся между растворителем и рас-
растворенным веществом, говорит о наличии химического взаимодей-
взаимодействия в растворе. Растворимость для этих систем приведена в табл. 39.
Системы, в которых не происходит образования соединений при
затвердевании или в насыщенном растворе, можно рассматривать как
«физические растворы» без сколько-нибудь сильного химического
взаимодействия растворенного вещества и растворителя. Раствори-
Растворимость для таких систем показана в табл. 40.
1. Растворы солей в ацетоне а
Растворение солей в ацетоне всегда связано с наличием сильного
взаимодействия между растворителем и растворенным веществом;
из таких растворов могут быть выделены твердые соединения.
IV. РАСТВОРЫ СОЛЬ — ОРГАНИЧЕСКОЕ СОЕДИНЕНИЕ
341
Таблица 39- Растворимость (в г на г растворителя) солей
в органических растворителях
(системы с химическим взаимодействием)
Соль
А1С13
А1С13
S11CI4
Cal2
Ca(NO3^
CaBr2
LiBr
ZnBr2
SrBr2
CoCla
PbCb
Растворитель
Анилин
1, 3, 5-Ксилидии
Нитроанизол
Ацетон
Ацетон
Ацетон
Ацетон
Ацетон
Ацетон
Ацетон
Триазол
Состав кон-
конгруэнтно
плавящихся
соединений
1:3
1:3
1:1
1:3
1:1
1:2
1:2
2:1
1:1/2:3
1:1
2:1
Раствори-
Растворимость
—
—
1,129
0,184
0,0292
0,346
3,81
0,00274
0,0725
7,6
(, °С
—
—
50*
50
40
50
50
50
50
200**
Лите-
Литература
115
115
115
2
2
2
2
2
2
2
* Из насыщенного раствора выпадают твердые соединения.
** Образуется красный расплав, температура плавления которого намного выше, чем
t „ чистого триазола (Дж. В. Хасти, неопубликованные результаты).
Таблица 40. Растворимость (в г на г растворителя) еолей
в органических растворителях
(системы без химического взаимодействия)
Соль
LiCl
№Вг.2
Nal
СаС12
ВаВг2
А12Вг6
А12С1в
А1216
РЬС12
NaCl
CsCl
Раствори-
Растворитель
Ацетон
Ацетон
Ацетон
Ацетон
Ацетон
Бензол
Бензол
Бензол
Триазол
Триазол
Триазол
Раство-
Растворимость
0,0061
0,309
0,000213
0,000246
80,11*
4,88
59,57
11,6
2,9
>2,9
t, °с
50
50
50
50
51,5
108,6
110,7
140
132
Примечание
С повышением температуры рас-
растворимость уменьшается2
С повышением температуры раст-
растворимость увеличивается **•
Расплавы обычно бесцветны или
окрашены в бледно-желтый
цвет, в зависимости от кон-
концентрации раствора
* Начиная с системы А12вг«—бензол данные о растворимости выражены в вес. %.
342 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
2. Растворы солей в триазоле и расплавах других
органических веществ
Расплавленный триазол (tm = 121° С) является хорошим раство-
растворителем для большого числа солей (А. Истел и Г. Рутвен, неопубли-
неопубликованные результаты). В ряде случаев, особенно в растворах солей
переходных металлов, наблюдается сильное взаимодействие между
растворителем и растворенным веществом; при охлаждении из таких
растворов выделяются твердые комплексные соединения. При темпе-
температурах, не намного превышающих температуру плавления триазола
(т. е. при температурах порядка 140° С), такие соли, как РЬС13
и хлориды щелочных металлов, образуют бесцветные или бледно-
желтые расплавы. При затвердевании расплава образования новых
соединений не происходит. Однако при более высоких температурах
растворы РЬС12 в триазоле приобретают красную окраску; раствори-
растворимость соли возрастает более чем в сто раз.
Опубликовано еще несколько работ, в которых изучались рас-
растворы солей в органических расплавах. Так, растворы NiCl2 в рас-
расплавленном пиридингидрохлориде 82 были использованы для изу-
изучения спектра поглощения Nin. Подобная же работа проведена
по изучению спектра поглощения UIV в расплавленном пиридиний-
хлориде (*„, = 144° С) 53.
3. Растворы хлоридов переходных металлов в метаноле 37
Определены электропроводности растворов СаС12, СаС12-2Н2О,
СгС13-6Н2О, МпС12-4Н2О, FeCl2-2H2O, CuCl2, СоС12-2Н2О и ZnCl2
в метаноле при 20 и 45° С.
Б. Растворы органических соединений
в расплавленных солях
При высоких температурах, обычных для расплавов, большинство
органических соединений разлагается. Поэтому вполне понятно,
отчего о растворах органических соединений в расплавленных солях
имеется очень мало сведений. Известно, что метанол растворяется
в расплавленном LiC103, не оказывая значительного влияния на
свойства растворителя, и, по-видимому, устойчив в таком расплаве.
Устойчив в нем, вероятно, и нитробензол 26.
Сандермейер с сотр. 86> 91 провели большое число реакций с орга-
органическими веществами, пропуская пары органических соединений
через расплав растворителя. Растворимости этих соединений не-
неизвестны, но ясно, что органические молекулы должны находиться
значительное время в расплаве для того, чтобы смогла осуществиться
реакция либо между самими этими молекулами, либо между орга-
органическими молекулами и ионами растворителя. Высокий выход
V. РАСТВОРЫ НЕМЕТАЛЛОВ
343
продуктов реакции при достаточно большой скорости потока ука-
указывает на значительную растворимость органических компонентов
в расплаве. В качестве растворителей авторы использовали главным
образом хлориды и псевдогалогениды щелочных и щелочноземельных
металлов с рабочей температурой от 89 до 600° С, в зависимости от
выбранного растворителя. В качестве растворенных веществ были
опробованы следующие соединения: силиконы, четыреххлористый
углерод, этан, ацетилен, этилгалогениды, спирты, н-бутилхлорид
и т. д. Реакции, происходящие между указанными соединениями
и растворителем, приведены в конце главы при обсуждении примени-
применимости расплавленных солей в качестве растворителей.
V. РАСТВОРЫ НЕМЕТАЛЛОВ В РАСПЛАВЛЕННЫХ СОЛЯХ
А. Растворы серы
Состояние растворенной серы в различных растворителях изуча-
изучалось лишь немногими исследователями 45' 66> ". Гринберг и др. 47> 48
исследовали спектры поглощения и парамагнитную восприимчи-
восприимчивость растворов серы в расплавленных солях.
При введении серы в эвтектические смеси LiCl, KC1 и LiBr +
+ КВг образуются растворы, окрашенные в синий цвет. Сходство
спектров этих растворов между собой указывает на отсутствие взаи-
взаимодействия между серой и галоген-ионом растворителя. Измерения
парамагнитной восприимчивости указывают на то, что в растворе
содержатся парамагнитные частицы. Вполне возможно, что этими
частицами являются двухатомные молекулы серы, поскольку высшие
полимеры серы имеют красную окраску
45
Б. Растворы иода
Определены 47> 48 спектры поглощения твердого иода в расплаве
смеси KI + Lil и КС1 + LiCl при 400° С. Полученные результаты
указывают на присутствие иона Ig в первой и иона 12С1" во второй
смесях. Значительная растворимость иода A0~3—10~4 м) при 400° С,
по-видимому, объясняется способностью ионов Ig и 12С1" легко
внедряться в квазирешетку солевой смеси.
VI. РАСТВОРЫ ГАЗОВ В РАСПЛАВЛЕННЫХ СОЛЯХ
Большинство работ, посвященных этим растворам, выполнено
применительно к технологии атомных реакторов. Для того чтобы
определить растворимость нереакционноспособных газов в расплав-
расплавленных солях, расплав обычно выдерживают в равновесных условиях
с газом при известном давлении. Растворимость затем находят либо
344
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
анализируя расплав, либо по изменению массы расплава или давле-
давления газа в резервуаре. Необходимо отметить, что при затвердевании
систем, в которых не образуется твердых соединений между компо-
компонентами расплава и газом, растворенные в них газы выделяются.
В зависимости от того, происходит ли химическое взаимодействие
между растворителем и растворенным веществом или оно отсутствует,
растворы газов в расплавленных солях можно разбить на две группы.
А, Системы без химического взаимодействия
1. Растворы двуокиси углерода в галогенидах
щелочных металлов 12> 61
Растворимости газов при различных температурах приблизи-
приблизительно определяются свободным объемом в расплаве. Следовательно,
вполне возможно, что процесс растворения не соцровождается хими-
химической реакцией. Растворимость газов в расплавленном хлориде
калия выше, чем в расплавленном хлориде натрия при той же темпе-
температуре. Это согласуется с тем, что хлористый калий имеет больший
свободный объем по сравнению с хлористым натрием 10, Например,
растворимости, выраженные в молях СО2 на 1 мл растворителя
(при давлении 1 атм), в NaCl при 850 и 950° С равны 4,6-10
и 4,0-Ю, а в КС1 при тех же температурах соответственно 6,0-Ю
и 7-Ю.
2. Растворы благородных газов в расплавленных галогенидах 49
Растворимости Не, Ne, Ar и Хе в смесях расплавов
NaF E3 мол. %) + ZrF4 D7 мол. %) и NaF E0 мол. %) + ZrF4
D6 мол. %)+ UF4 D мол. %) линейно возрастают с повышением
давления газа, уменьшаются с увеличением атомного веса растворен-
растворенного газа и увеличиваются с ростом температуры. В обеих указанных
смесях растворимости и теплоты растворения Не и Хе очень близки
по величине. Следовательно, при растворении благородных газов
расплавленные соли ведут себя подобно обычным жидкостям. Такой же
растворяющей способностью обладают расплавленные эвтектические
смеси LiF D6,5"мол. %) + NaF A1,5 мол. %) + KF D2 мол. %) 3.
Бландером и др. 3 разработана модель, согласно которой раствор
рассматривается как некая жидкость, имеющая то же поверхностное
натяжение, что и растворитель. Свободная энергия образования
различных дырок в ней и размеры различных молекул растворенного
газа одинаковы. Расчеты дают хорошую сходимость с эксперимен-
экспериментальными данными.
VI. РАСТВОРЫ ГАЗОВ
345-
Б. Растворы газов, образующих в расплавленных
солях комплексные соединения
Если газ химически взаимодействует с расплавленным раствори-
растворителем, то он обычно хорошо растворяется. Например, расплав СиС12
B5 мол. %) -f KC1 G5 мол. %) способен поглотить 0,75 моль фтора
на 1 моль расплава 65. Это объясняется протеканием химической
реакции
CuCl2 + 3KC1 + 3F2 = K3[CuF6] + 2 ~ Cl
A)
Примером растворов этого типа могут служить также растворы
паров TiCl4xB эквимолярной расплавленной смеси NaCl -f KC1.
Растворимость TiCl4 в таких растворителях была определена не-
несколькими исследователями 67' 68, но наиболее достоверными пред-
представляются результаты Фленга 43, поскольку его метод заключался
ис. 65. Изотермы реак-
реакций 4з между газообраз-
газообразным TiCl4 и КС1 или
смесью КС1 + NaCl
(взятых в мольном отно-
отношении 1 : 1).
30
в непосредственном наблюдении за увеличением массы при контак-
контактировании солевого раствора с парами тетрахлорида титана и, сле-
следовательно, в нем исключались ошибки, связанные с приготовлением
и отбором проб. Скорость поглощения паров TiCl4 определялась
и в расплаве, и в тонкоизмельченных смесях твердых солей. Из резуль-
результатов опытов (рис. 65) видно, что расплавленная солевая смесь
может растворять до 14 мол. % TiCl4, в то время как твердая соле-
солевая смесь поглощает 33 мол. % TiCl4. Рентгенографическое изучение
твердой фазы показало, что образуется соединение, по-видимому,
согласно реакции
TiCl4 + 2KCl
K2TiCl6
B)
Из данных о растворимости можно заключить, что оптимальный
температурный интервал образования этого соединения при давле-
давлении паров TiCl4, равном 1 атм, лежит в пределах 350—400° С
При температурах выше 800° С в связи с протеканием реакции между
HU и NaCl, вероятно, образуется Na2TiCle.
346
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛЯ
VII. РАСТВОРЫ МЕТАЛЛОВ
Еще одним примером 82 растворов, в которых происходит хими-
химическое взаимодействие между растворителем и растворенным веще-
веществом, являются растворы фтористого водорода в расплаве NaF +
+ ZrF4. В отличие от поведения благородных газов в подобных
растворителях растворимость фтористого водорода с повышением
температуры уменьшается. Гораздо резче проявляется в данном
случае и зависимость растворимости фтористого водорода от природы
растворителя. По-видимому, это связано с относительно высокой
устойчивостью соединений, образующихся между NaF и HF. Раство-
Растворимость фтористого водорода в расплавленных фторидах щелочных
металлов возрастает с увеличением атомного номера металла. Вполне
возможно, что такая зависимость растворимости от природы раство-
растворителя связана с относительной устойчивостью некоторых кислых
фторидов щелочных металлов, наблюдающейся при более низких
температурах96. Высокая растворимость HF во фториде цезия49
была использована для электролитического получения фтора при
комнатной температуре 7е. Доказано, что в этом растворе происходит
значительное химическое взаимодействие с образованием следующих
соединений: CsF-HF, CsF-2HF, CsF-3HF и CsF-6HF.
Известно, что растворимость кислорода и азота в расплавленных
нитратах щелочных металлов незначительна.
VII. РАСТВОРЫ МЕТАЛЛОВ В РАСПЛАВЛЕННЫХ СОЛЯХ
Многие расплавленные соли могут служить растворителями
металлов; в частности, расплавленные галогениды обычно растворяют
металлы, катион которых входит в состав данной соли. Способность
расплавленных галогенидов растворять материнский металл впер-
впервые была замечена по окрашиванию расплава при электролизе
расплавленных хлоридов рубидия и цезия 24. Лорентц и Эйтель 74,
проведя оптическое и рентгеноструктурное изучение твердых фаз,
сделали вывод, что растворы металлов в расплавленных солях
являются коллоидными. Однако Хеймен и Фридландер Б7 показали,
что это предположение неверно, поскольку свойства образующихся
растворов отличаются от свойств обычных коллоидных растворов.
В настоящее время считают, что растворы металлов в расплавленных
солях можно разбить на две группы:
1) растворы, в которых не происходит заметного химического
взаимодействия между растворителем (солью) и растворенным веще-
веществом (металлом); в этом случае металл придает раствору металли-
металлический характер;
2) растворы, в которых наблюдается значительное взаимодей-
взаимодействие между растворителем и растворенным веществом.
Имеются также растворы, не попадающие ни под одну из указан-
указанных групп и обладающие промежуточными свойствами. Различные
теории о природе растворов металлов в расплавленных солях рас-
рассмотрены в монографиях Делимарского и Маркова 109 и Бредига 1е.
347
А. Системы без существенного химического взаимодействия
(«металлические» растворы)
Растворы этого типа образуются в основном при растворении
металлов I и II групп периодической системы элементов, а также
лантаноидов в расплавленных галогенидах этих металлов.
1. Растворы щелочных металлов в расплавах
галогенидов щелочных металлов
Физические свойства этих систем приведены в табл. 41. Критиче-
Критические температуры растворения во фторидах и хлоридах, т. е. темпе-
температуры, при которых достигается неограниченная смешиваемость
соли и металла, уменьшаются в ряду Li^>Na> K^>Rb. Для фто-
фторидов этот температурный интервал лежит в пределах 1325—800° С.
Растворимость металлов в их расплавленных галогенидах возрастает
в обратном ряду: Li<Na<K<Rb<Cs.
Таблица 41. Системы щелочной металл—галогенид щелочного металла
Система
Li — LiF
Li-LiCl
Na—NaF
Na —NaCl
K-KF
K-KC1
Rb-RbF
Rb-RbCl
Gs —CsF
Cs —CsCl
i
ё
452
452
370
370
337
337
312
312
302
302
ы
соли, °:
с
1121
883
1268
1073
1131
1043
1068
995
976
918
Монотекти-
ческое прев-
превращение (для
жидкого
расплава
с низким
содержанием
металла)
о
Е-Г
1120
882
1263
1068
1122
1024
1046
969
ржание
шла,
115
§33
~-1
—0,5
~3
2,1
4,9
10,5
—9
18
Область
расслоения
отсутствует
—
—
а
й
Is?
я"§
is
ffl 0)
CDS
—
10~9
—
3 • 10
10"9
—
—
10'3
10-8
Критическое
состояние
W
о
Е-Г
1603
1453
1353
1177
1063
1063
979
—
—
„
И
мета
я
h
is
40
—
28
50
20
39
21
37
—
—
Раствори-
Растворимость
°
Е-Г
—
—
1273
—
1084
1273
1221
—
1073
—
—
965
—
899
R
метг
и
и
ТО
1§
OS
—
—
7,6
—
2,8
33,0
23,3
—
7,6
—
—
6,0
—
9,0
ература
g
К
40
40
40,61
19
61
19
19
40,60
61
104
40 61
61
40
17
64
* Значения получены экстраполяцией.
348
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
Во всех системах с цезием не наблюдается расслоения фаз. Со-
Совершенно очевидно, что наиболее существенно на растворимость
в системах, рассматриваемых в этом разделе, влияет размер (или,
что более точно, плотность заряда) катиона щелочного металла.
Концентрация металла в растворе при критической температуре
растворения возрастает от фторида к иодиду для натриевых, калие-
калиевых и рубидиевых систем.
На отсутствие сильного химического взаимодействия между
щелочным металлом и галогенидом щелочного металла указывают 16
значения коэффициентов активности солей (у). Эти коэффициенты
свидетельствуют об увеличении положительных отклонений от закона
Рауля с возрастанием концентрации металла, причем обычно эти
отклонения наблюдаются в тех системах, в которых имеются области
расслоения, но отсутствует сильное химическое взаимодействие
между растворителем и растворенным веществом. Весьма приме-
примечательно также, что кривые зависимости величины RTlnyCOJlb от
Металл (где Л металл—мольная доля металла) для систем КВг, RbBr
и CsBr лишь слегка отклоняются от прямолинейных. С достаточным
приближением эти кривые можно считать линейными, и такая линей-
линейность хорошо согласуется со свойствами регулярных растворов.
Такие растворы характеризуются идеальной энтропией
(в случае, если они соответствуют закону Рауля) и лишь слабым
взаимодействием между растворителем и растворенным веще-
веществом.
Было также выдвинуто предположение 79, что избыточная сво-
свободная энергия смешения (т. e.RTlnyCOJlb) вызвана переходом связи
металлического типа в связь ионного типа. Как предсказывает
теория, такое превращение предполагает значительный положитель-
положительный избыток энтропии смешения.
Удельная электропроводность этих расплавов заметно возрастает
при добавлении в расплавленную соль металла и продолжает увели-
увеличиваться с добавлением новых порций металла. Отсюда Бронштейн
и Бредиг 20> 21 сделали вывод, что значительная доля электропровод-
электропроводности расплава приходится на электронную. В 42 мол. °6-ном рас-
растворе калия в иодиде калия доля электронной электропроводности
составляет более 99,5% от суммарной электропроводности раствора.
Для объяснения значений эквивалентной электропроводности этих
систем Бронштейн и Бредиг предположили:
а) в этих растворах происходит перекрывание электронных
орбиталей при сравнительно высоких концентрациях металла;
б) в растворах натрия с высокими концентрациями металла
происходит «обобществление» электронов различных атомов и обра-
образуются, например, двухатомные молекулы Na2 (еще более ярко вы-
выражено спаривание электронов в литиевых системах);
в) доля удельной электропроводности, приходящаяся на метал-
металлический компонент раствора, значительно возрастает при переходе
VII. РАСТВОРЫ МЕТАЛЛОВ
349
от фторидных систем к иодидным, т. е. с увеличением атомного
номера и размера галоген-иона;
г) доля удельной электропроводности, приходящаяся на метал-
металлический компонент раствора, уменьшается от натрия к калию,
т. е. с увеличением размера иона металла.
2. Растворы щелочноземельных металлов в расплавленных
галогенидах щелочноземельных металлов
Лишь недавно удалось получить точные фазовые диаграммы
этих систем. Определена фазовая диаграмма системы Ва—ВаС12,
которая оказалась подобной ранее полученным фазовым диаграммам
систем щелочноземельный металл — галогенид щелочноземельного
металла. Известны также равновесные фазовые диаграммы систем
Са—CaF2°, Са—СаСЦ8, Са—СаВг2 и Са—Calf. Растворимость
металлов в этих системах приведена в табл. 42. Вообще эти системы
Таблица 42. Системы щелочноземельный
Система
Mg —MgCl2
Са —CaF2
Са — СаС12
Са —СаВг2
Са—Са12
Sr— SrF2
Sr-SrCl2
Sr— SrBr2
Sr — Srl2
Ba—BaF2
Ba —BaCl2
Ba—BaCl2
Ba — BaBr2
Ba—Bal2
W
метал;
с
923
1110
1110
1110
1110
1044
1044
1044
1044
1002
1002
1002
1002
1002
W
сопи,
с;
с
Е-.
987
1691
1045
1015
1053
1145
—
—
—
1235
—
—
—
щелочноземельного
Монотекти-
ческое прев-
превращение
(для жидкого
расплава
с низким со-
содержанием
металла)
е-Г
—987
1563
1093
1100
1104
1112
—
—
1163
—
—
—
&5 .
соде
мет;
мол.
0,2
25,5
2,7
2,3
3,8
5,5
—
—
—
15,5
—
—
—
металл -
металла
Эвтектика
м
е
е-Г
—923
1094
1033
1000
1033
—
—
—
—
985
—
—
—
R
R
ета
я
о
S
и
I*
а
соде
мол.
100,0
98,6
2,0
3,0
2,0
—
—
—
—
99,0
—
—
—
— галогенид
Критическое
состояние
М
Е-Г
1595+5
1610 ±5
1610 ±5
1650+5
—
—
—
—
1290
—
—
—
R
[ета
«
1
а .
соде
мол
45
62
64
74
—
—
—
—
50
—
—
—
Раствори-
Растворимость
М
Е-Г
1073
1323
—
—
1273
—
—
1273
1273
—
1273
1273
1273
1323
—
1323
1323
1323
СО
п
I
о
1
3.°
соде
мол.
1,08
1,57
—
—
5,4
—
—
9,66
19,9
—
24,6
36,1
39,3
21,9
—
30,6
36,7
39,4
ер ату р
Лит
16
112
112
80
16
54
16
16
54
54
16
54
54
54
54
16
54
54
54
350
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
подобны системам галогенидов щелочных металлов. В них также
наблюдается быстрое увеличение растворимости с возрастанием
атомного номера металла. Например, максимальная растворимость
вблизи температуры плавления солей в системе Mg—MgCl2 равна
0,5 мол. %, в системе Са—СаС12 — 2,7 мол. %, в системе
Sr— SrCl2 — 5,5 мол. %, в системе Ва—ВаС12 — 20 мол. %.
Для объяснения свойств растворов были предложены различные
уравнения. Например, с учетом данных измерения коэффициентов
активности в систем* Mg—MgCl2 был предложен следующий меха-
механизм процессов растворения: Mg+Mg2+ = (Mg2J+.
Зависимость электропроводности от концентрации также похожа
на аналогичную зависимость в системах щелочной металл — гало-
генид щелочного металла. Различие заключается лишь в том, что
скорость изменения удельной электропроводности с повышением кон-
концентрации металла в системах щелочноземельных металлов не очень
высока.
3. Растворы лантаноидов в расплавленных
галогенидах лантаноидов
Эти растворы имеют менее выраженный металлический характер
по сравнению с растворами, рассмотренными выше. Растворимости
металлов в расплавленных тригалогенидах приведены в табл. 43.
Характерной особенностью этих систем является существование
конгруэнтно и инконгруэнтно плавящихся соединений. В системах
Таблица 4с
Система
La-LaCls
La — LaBr3
La—Lal3
Се —СеС13
Се — СеВг3
Се—Cel3
Рг —РгС13
Рг —РгВг3
Рг —Рг13
Nd — NdCl3
Nd-NdBr3
Nd-Ndl3
Gd —Gdl3
Y-YIs
;. Системы лантаноид
етаг
E
с;
*?#
1193
1193
1193
1068
1068
1068
1208
1208
1208
1297
955
955
1585
1782
W
о
a"
§
1131
1061
1052
1090
1005
1034
1059
966
1011
1032
955
1060
1204
1270
—галогенид лантаноида
Эвтектика
T, °K
1099
1001
1007
1050
960
988
919
852
939
913
—
764
1098
1221
содержа-
содержание
металла,
мол. %
9,0
14,5
8,2
9,0
12,0
8,8
17,0
16,0
11,9
14,0
—
26,5
14,0
12,0
Растворимость
Г, "К
1273
1173
1173
1171
1123
1173
1173
1073
1023
1073
1173
—
1073
1173
1423
содержа-
содержание
металла,
мол. %
12,0
15,5
33
9
33
14
32
19
18
29
31
—
37
14
15
Литера-
Литература
33
33, 81
33, 38
33
32
33, 81
33
33, 38
33, 81
33
38, 39
16
38, 39
33
16
VII. РАСТВОРЫ МЕТАЛЛОВ
351
Nd—Ndl3, Рг—Рг13, Се—Се13 и La—Lal3 найдены устойчивые
твердые соединения, соответствующие составу МГ2. Существуют,
по-видимому, и нестехиометрические соединения, такие, как NdCl2 3,
NdCl2,2, PrI2,5, CeI2,4, Lal2, 4.
Удельная электропроводность растворов металла в соли возра-
возрастает в ряду Nd<Pr<Ce<La<K.
Б. Системы с сильным взаимодействием между растворителем
и растворенным соединением
(«неметаллические» растворы)
«Неметаллическими» растворами металлов в расплавленных солях
этих же металлов называются растворы, в которых наблюдается лишь
очень слабое изменение электропроводности расплава с увеличением
концентрации металла. Это указывает на то, что электроны металла
в расплаве каким-то образом связаны и не принимают участия
в процессе проводимости. При определении, например, типа растворов
висмута в хлориде висмута этот критерий является очень существен-
существенным
36
м --.
Вторым критерием, позволяющим отличать эти системы от «метал-
«металлических» систем, является образование стехиометрических твердых
галогенидов металла более низких валентностей.
1. Системы с переходными металлами
Число работ, посвященных растворам металлов этого типа, не-
невелико. Система Ni—NiCl2 была изучена Джонсоном и др. 62, причем
растворимость металла при эвтектической температуре 980° С соста-
составила 9 мол. %. Авторы пришли к заключению, что процесс растворе-
растворения происходит с образованием ионов Ni+. Однако для интерпрета-
интерпретации понижения температуры замерзания раствора и данных о теплоте
плавления возможны и альтернативные объяснения. Данные о рас-
растворимости приведены в табл. 44.
а. Металлы I группы. Растворимость серебра в расплавленном
хлориде серебра составляет при 700° С 0,06 мол. %.
б. Металлы II группы. Хорошо известны субгалогениды ртути
Hg2F2. Хлорид устойчив до 525° С. При этой температуре он раз-
разлагается на расплав с высоким содержанием соли, имеющий почти
тот же самый состав, и небольшое количество раствора HgCl3 в жид-
жидкой ртути 98 с концентрацией 6,8 мол. %. Гевеши и Левенштейн 5в
определили растворимость Hg, оказавшуюся равной при 350° С
50 мол. % в HgCl2 и 33,6 мол. % в Hgl2.
Очень много внимания уделено изучению систем
Cd—Cdiy.34> 35' 42'5в' '5. Растворимость Cd в CdCl2 при 600° С равна
15,2 мол. %. Диамагнитная природа раствора указывает на то, что
либо растворение металла происходит через образование ионов
352 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
Система
Ni-NiCl2
Ag-AgCl
Zn — ZnCh
Zn —Znl2
Cd —CdCl2
Cd —CdBr2
Cd —Cdl2
Hg-HgCl2
Hg-Hgla
Таблица 44.
'пл
металла, °С
960,8
419,47
419,47
40,0
(вакуум)
40,0
40,0
-38,87
—38,87
Системы с переходными
*пл соли-
°С
455
262
446
568
567
388
276
259
металлами
Растворимость
t, °С
977
490
700
500
500
500
550
600
800
1000
550
600
700
900
400
600
700
950
280
350
400
500
230
280
350
содержание
металла,
мол. %
Q 4
о ,L
0,03
0,06
0,18
8,9 • Ю
0,28
14 0
15,2
21,0
30,0
14,0
13,9
20,0
28,0
2,5
6,07
15,0
25,0
7,0
50,0
18,0
40,0
25,0
35,0
33,6
Литература
со
06
30
30
30
56
31
1, 56
1, 56
1 56
1, 56
56
92
92
92
92
92
92
92
56
98
98
98
56
56
56
Cd2+, либо получается истинный раствор молекул или атомов.
Измерения понижения температуры замерзания указывают на воз-
возможность образования субхлорида Cd2Cl250- При затвердевании это
соединение диспропорционирует, в чем можно убедиться по наличию
в затвердевшем растворе кристаллов металлического кадмия. Данные
измерений коэффициентов активности можно интерпретировать 15
как указывающие на присутствие ионов (Cd2J+, а также на образо-
образование комплексных анионов, таких, как (CdCl4J~.
Все данные свидетельствуют о том, что Cd, вероятнее всего, рас-
растворяется в CdCl2, образуя ионы (Cd2J+. Поэтому количество кадмия,
переходящего в раствор, зависит от концентрации уже присутству-
присутствующих в растворе ионов Cd2+. Следовательно, зная растворимость
металлического кадмия в расплавленных смесях CdCl2-|-NaCl
и CdCl2+KCl, можно дать полуколичественную оценку содержания
свободных ионов Cd2+ в этих растворах. Действительно, присут-
присутствие хлорида калия сильно подавляет растворимость Cd в распла-
расплавленном CdCl2 (в широком концентрационном интервале), вероятно.
VII. РАСТВОРЫ МЕТАЛЛОВ
353
за счет снижения концентрации ионов Cd2+ в этих растворах вслед-
вследствие образования комплексных ионов, таких, как CdCl|~.
Растворимость металла в системах Zn—Znr2 31 составляет
лишь—1 мол. %. По аналогии с кадмиевыми системами столь
низкую растворимость можно объяснить отсутствием свободных
ионов Zn2+ в расплавленном ZnCl2. Вероятно, цинк в растворе
присутствует в форме комплексных ионов. Меньшая растворимость
цинка в этой системе по сравнению с растворимостью кадмия в CdCl2
согласуется с повышенной способностью меньшего по размерам иона
Zn2+ образовывать стабильные комплексные ионы.
800 г-
2. Сщтемы с металлами концов больших периодов *
Корбетт 28> 30' 31 предположил, что растворимость металлов в рас-
расплавленных галогенидах этих металлов зависит от способности
образовывать катионы низших валентных состояний. Как указано
в табл. 45, способность эта для
каждой из групп галогенов возра-
возрастает с увеличением атомного веса
металла. Растворимости увеличи-
увеличиваются в ряду 1<^Вг<^С1.
а. Металлы III и IV групп.
Растворимости металлов этих групп
периодической системы элементов
в расплавленных галогенидах
этих металлов гораздо меньше, чем
соответствующие растворимости
переходных металлов 30'31.
б. Металлы V группы 28. Для
As, Sb и Bi характерно устойчивое
трехвалентное состояние. Раство-
Растворимости металлов в их солях
быстро увеличиваются с возраста-
возрастанием атомного веса металла.
В системе с Bi образуются субга-
логенидыВ1Х. Фазовая диаграмма
системы Bi—BiCl3 показана на
рис. 66.
Следует отметить, что сами соли могут растворяться в металле,
но обычно меньше, чем металл растворяется в соли. Например,
растворимость РЬС12 в РЬ при 1000° С составляет97—1 мол. %.
у BiCl3+(BiCl)
О 20 ^ О 60 80
Недержание Bi , мол. %
1ОО
Рис. 66. Фазовая диаграмма9 систе-
системы Bi—BiCl3:
I — визуальные методы; 2 — декантация;
3 — термический анализ.
* А также с алюминием.
23 Заказ 90
354 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
Таблица
Система
А1-АПз
Ga— GaCl2
Ga—Ga2Cl4
Ga—Ga2Br4
Tl —TlCl
Sn—SnCl2
Sn— SnBr2
Pb-PbCl2
Pb-Pbl2
Sb-SbClg
Sb— Sbl3
Bi —BiCl3
Bi —BiBr3
Bi —Bil3
45. Системы
<пл металла,
°C
659,7
29,78
29,78
29,78
302,0
231,9
231,9
327,4
327,4
630,5
630,5
271,3
271,3
271.3
с металлами
<пл соли-
°С
191,0
170,5
430,0
246,0
215,5
501,0
402,0
73,4
167,0
230,2
218,0
439,0
концов больших периодов
Растворимость
t, "С
423
180
180
180
550
650
500
500
600
700
700
800
440
600
700
270
200
300
400
202
320
450
550
780
205
294
440
538
336
458
содержание
металла,
мол. %
0,3
1 92
3,7
14,0
0,09
0,09
0,0032
0,068
0,02
0,052
0,055
0,123
0,024
0,15
0,41
0,18
1,69
3,5
5,8
28,0
46,0
47,5
28,0
100,0
21,0
57,0
45,0
100,0
48,0
100,0
Литература
30
SO
30
29 31
30
30
30
30
30
30
из
30
16
16
16
31
22, 31
22
22
99
99
33
72
99
100
100
100
100
100
100
В. Растворы металлов в солях других металлов
(реакции замещения)
В этих системах устанавливается равновесие
М, + МПГ ^гГ Mjr + Мц C)
Можно рассчитать константу равновесия К, определив э. д. с.
соответствующей ячейки Якоби — Даниэля. При этом пренебрегают
потенциалом связи на границе двух жидких расплавленных солей.
Например, в ячейке Cd | CdCl2 |РЬС12| Pb происходит реакция
Cd + Pbci2 4^Г CdCl2 + Pb D)
Константа равновесия К определяет растворимость металла в солях
других металлов (табл. 46).
VII. РАСТВОРЫ МЕТАЛЛОВ
355
Таблица 46. Константы равновесия реакций замещения 109
Реакция
Na + LiCl^NaCl + Li
K + NaCl^tKCl + Na
K + Nal^KI + Na
3Na + AlCl3<±3NaCl + Al
Mg + ZnCl2^MgCl2 + Zn
Cd + PbCl2?±CdCl2 + Pb ,
t, °c
900
900
900
825
600
к
0,45
0,87
0,017
~ 5,0 • 10-w
1,6 • 10-5
~ 3,0 • 10-2
В своей монографии Делимарский и Марков 109 привели значения
К для большого числа реакций металлов с расплавленными гало-
генидами. Следует отметить, что для многих металлов равновесие
реакции М+хРЬС12^МС12л:+а;РЬ почти полностью смещено вправо,
а поскольку расплавленный металлический свинец легко может быть
отделен, эта реакция представляет собой удобный метод получения
безводных хлоридов тяжелых металлов. Инман и др. 59 использо-
использовали эту реакцию для получения TJC13 при 650° С:
E)
Для промышленного электролиза очень важно знать электрохи-
электрохимический ряд металлов в нужном растворителе. Делимарский и Мар-
Марков 109 составили таблицу электродных потенциалов металлов
в различных расплавленных электролитах, а также определили
качественно электрохимические ряды металлов в расплавленных
солях (табл. 47), установив что для разных расплавленных солей-
растворителей эти ряды не обязательно будут одинаковыми.
Таблица 47. Электрохимический ряд металлов в смесях
расплавленных солей 1°9
Растворитель
NaCl + AlCl3
NaCl + AlCl3
NaBr + AlBr3
Nal + Allg
t, °c
500
700
700
700
Na, Be,
Cui, i
Na, Al,
Sb"i,
Na, Al,
Sb"l,
Na, Al,
Al
[g11,
Mn
Ni,
Zn,
Bi
Cd,
Электрохимический
, Mn, Tl, Zn
Sb, Bi, Ni
Tl1, Zn, Gd,
Bi
Cd, Pb, Sn",
Ag, Sn", Pb,
, Cd
Pb,
Cui,
Си»,
ряд
, Sn", Pb, Go, Ag,
Ag, Cub Co, Hg",
Ag, Co, Hg11, Ni,
B^Hgi1, Co.Ni.Sb
С повышением температуры происходит изменение электродного
потенциала: в ячейке с реакцией Cd+PbCl2 <± CdCl2+Pb ниже
650° С кадмий вытесняет свинец, а при более высоких температурах
свинец вытесняет кадмий.
23*
356
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
VIII. РАСТВОРЫ СОЛЕЙ В РАСПЛАВЛЕННЫХ СОЛЯХ
В общем, расплавленные соли лучшие растворители солей по
сравнению со всеми остальными классами растворителей. Большин-
Большинство расплавленных солевых систем неограниченно смешивается
друг с другом. Отдельные системы, в которых наблюдается расслое-
расслоение, т. е. образование двух жидких фаз, вызывают повышенный
интерес, так как они могут быть применены для экстракции некото-
некоторых неорганических соединений из многофазных систем.
Расплавленные галогениды обычно полностью смешиваются друг
с другом, особенно если системы истинно бинарные. Однако в систе-
системах расплавленные галогениды — соли с другими анионами может
наблюдаться расслоение. Примером может служить система борат
натрия—галогенид натрия и.
Вопросам растворимости солей в расплавленных солях в прошлом
уделялось мало внимания. Как правило, наиболее четко свойства
систем фиксируются на фазовых диаграммах. Для детального
рассмотрения фазовых диаграмм и их связи с растворимостью солей
читателю следует обратиться к специальной литературе. Фазовые
диаграммы систематизированы в справочнике Ландольта — Берн-
штейна 70.
А. Системы расплавленных солей с областью расслоения
В табл. 48 приведены системы расплавленных солей, в которых
имеется область расслоения. Взаимное расслоение наблюдается
во многих бинарных системах, обычно состоящих из соли алюминия
и соли ионного типа. Примерами могут служить системы 64 А1Вг3
с NaBr, КВг, NH4Bx, СаВг,,Т1Вг и А1С13 с NaCl, KCI, NH4C1, AgCl,
SnCl2, SnCl4.
Области расслоения наблюдаются также в следующих распла-
расплавленных обратимых системах 106:
AgCl или AgBr-LiNO3, NaNO3, KNO3, T1NO3, Ca(NO3J, LuWO4,
LiVO3, NaVO3, KVO3, Na2Cr04, Na2MO4, Na2WO4;
Agl-NaCl, LiCl, NaNO3, KNO3, T1NO3;
TIBr — нитраты;
T1C1 — нитраты;
CdCl2 — некоторые соли кислородсодержащих кислот;
CdBr2 — некоторые соли кислородсодержащих кислот;
LiF-CsCl, CsBr, PbO;
PbO-NaF, KCI, NaCl. RbCl, NaBr, KF, KI;
SiO2—MgF2, CaF2, SrF2, BaF2,
а также в системах Li2SO4 —PbCl2; PbCl2—Ag2S; B2O3—NaCl
и B2O3—KCI.
Многие ученые e4> 105> 106 пытались связать такие факторы, как
положение элементов в периодической системе элементов, темпера-
VIII. РАСТВОРЫ СОЛКИ
357
Таблица 48. Бинарные системы расплавленных солей с областью расслоения
Л5 пп.
Система
Область расслоения,
мол. % второго компонента
Состав конгруэнтно
плавящихся
соединений
10
11
12
13
14
15
16
AlCls-
А1С13-
A1G13-
Hg2Br2-
SnClj-
PbBr2-
AICI3-
PClr,
¦NaCl
¦KCI
-AlBr3
-SbCl3
-AlBr3
-NH4CI
AlBr3 —NaBr
AlBr3 —KBr
AlBr3 — NH4Br
AlBr3 — CaBr2
AgCl-AlCl3
AgBr—AlBr3
T1C1 —AICI3
TIBr—AlBr3
z — AlBr3
74—78,9
2 фазы вблизи 100% A1C13
2 фазы вблизи 100% A1C13
84,6-99.1
8,8-98,6
83,8—99,2
0,2—20,5
2,6-16,3
0,4—22,1
0,5—20,8
0,8—14,0
82,4—99,3
83,0—97,8
85,3—98,8
77,2-99,4
85,8—98,2
1: 1
1: 1
1: 1
1 : 2
x: у
1: 2
x: у
1: 1
x : у
7: 2
2:1
1: 1
x: у
2: 1
1 : 1
x: у
3: 1
2: 1
1: 1
x: у
2: 1
x : у
1: 1
2 соединения
3 соединения
* С 1—7 по данным работы '", остальные по данным работы <
тура, разности внутренних давлений, мольные и атомные объемы
и другие, с наблюдаемой ограниченной смешиваемостью определен-
определенных расплавленных солей. На основании имеющихся сведений раз-
различают несколько типов систем с областью расслоения: АХ—BY;
АХ—ВХ; АХ—AY. Первая из них обратимая, а две остальные —
бинарные системы.
1. Система АХ + BY
Для того чтобы произошло расслоение в системах этого типа
(табл. 49), один из компонентов должен иметь сравнительно неболь-
небольшую полярность, т. е. быть ковалентным соединением, — галогени-
дом, окислом или сульфидом переходного металла или элементов
концов больших периодов таблицы Менделеева, а второй компонент
должен быть типичной солью (обычно — это соль щелочного ме-
металла). Известны исключения из этого правила, так, расслоение
наблюдается в расплавах LiF — CsCl и LiF — CsBr.
358 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
Таблица 49. Солевые системы с областью расслоения *
Система
Область расслоения
мол. %
второго компонента
Система
Область расслоения,
мол. %
второго компонента
FeO— NaF
AgCl~Li2SO4
CdCl2 —Li2SO4
AgNO3-NaCl
HgBr2-AlCl3
TlBr~KNO3
TlI — KNOs
Tl2SO4-AgI
ТШО3—Agl
3,0—94,0
1,5-98,0
6,0—80,0
29,0—76,0
50.0—99,0
14,0—92,5
0,0—97,0
11,0—86,0
1,5—93,0
AlBr3-NaI
AlBr3-KI
Na2B4O7—NaCl
BaCl2—5fa2CO3
Ag2SO4-LiCl
AgNO3-KCl
T1NO3-KI
Til —AgNO3
2,0—13,0
2.0—13,0
66,0-99,8
39,0—62,0
41,0—87,0
32,0^-64,0
24,0—72,0
45,0-53,0
* К таким системам относятся также: AgCl— LiNOs, TINO3—KBr, C.dSO^ —Licl,
AgNOs — LiCl.
2. Система AX -{- BX
Один из компонентов таких систем должен иметь малую поляр-
полярность и содержать многовалентный катион (например, А13+, Bi3+,
Sb3+ и т. д.), а второй компонент должен быть типичной солью по-
полярного типа. Эти условия благоприятны для образования комплекс-
комплексных соединений, хотя образующиеся соединения, как правило,
нестойки.
3. Система АХ + AY
В системах этого типа анионы компонентов должны сильно
различаться размером и плотностью заряда. Примерами таких
систем могут служить смеси силикатов, титанатов и боратов с гало-
генидами щелочных металлов.
Б. Системы расплавленных солей с полной
взаимной растворимостью
Солевые системы, имеющие полную взаимную растворимость
в жидком состоянии, слишком многочисленны, чтобы их все пере-
перечислять. Для более детального ознакомления с равновесиями,
устанавливающимися в этих системах, читатель может обратиться
к справочникам, в которых приводятся фазовые диаграммы.
Определенный интерес представляет растворимость окислов ме-
металлов в расплавленных солях в связи с проблемой окисления метал-
металлов в этих средах. Так, Стерн 85 изучил растворимость в расплавлен-
расплавленном NaGl при 900° С окислов переходных металлов первого большого
периода от Ti до Си. Из данных о растворимости были установлены
следующие закономерности.
1. В заметной степени взаимодействуют с NaCl лишь окислы
металлов, имеющих нечетный атомный номер, т. е. нечетное число
Зй'-электронов. Из окислов остальных элементов исключение со-
VIII. РАСТВОРЫ СОЛЕЙ
359
ставляет окись хрома Сг2О3, хорошо растворяющаяся только в при-
присутствии кислорода.
2. За исключением V2O5, окисляющего ионы С1~, в числе раство-
растворимых окислов есть несколько, устойчивых выше 900° С. Все не-
нерастворимые окислы (к ним следует отнести NiO и Fe2O3, поскольку
растворимости их очень малы) имеют только один окисел, устойчивый
при этих условиях.
3. Для растворимых окислов отношение металла к окиси в рас-
расплаве намного превышает отношение, которое может быть рассчитано,
только исходя из растворимости. Кроме того, стехиометрическое
соотношение металла и кислорода в расплаве не соответствует ни
одному из известных окислов, устойчивых в твердом состоянии.
4. Несмотря на значительный разброс значений растворимостей,
все растворимые окислы, за исключением СиО, растворяются в рас-
расплаве до концентраций 0,001—0,01 мольных долей.
В. Структура растворов расплавленных солей
Структуре смесей расплавленных солей посвящено много ра-
работ Б> 6. Физическая и химическая природа смесей расплавленных
солей изучены лучше, чем природа индивидуальных расплавленных
солей. Для выяснения структуры использовалось множество методов
физической химии, но, к сожалению, лишь немногие системы изучены
достаточно полно. Наибольшее внимание исследователями было
уделено системам дигалогенид тяжелого металла — галогенид ще-
щелочного металла. Эти системы и будут рассмотрены нами в качестве
примеров.
Термодинамические свойства расплавленных систем дигало-
генидов тяжелых металлов с галогенидами щелочных метал-
металлов изменяются в широких пределах, что позволяет установить
природу взаимодействия между растворенным веществом и раство-
растворителем. Большую пользу в этом аспекте приносит введение понятия
термодинамической активности. Термодинамические системы могут
быть описаны как «идеальные», «регулярные» (энтропия растворов
первых двух типов определяется концентрацией) и «комплексные».
В идеальных смесях, по определению, нет заметного взаимодействия
между растворенным веществом и растворителем; распределение
частиц в них может быть полностью неупорядоченным (если пред-
предполагается соответствие закону Рауля) или может создавать квази-
квазирешетку с катионной и анионной полурешетками, расположенными
одна в другой (если предполагается идеальность раствора по Тем-
кину) 116. Ионная природа расплавов подтверждает последний кри-
критерий идеальности. Следует отметить, что для бинарных систем
типа МХ2—АХ экспериментально определенные активности соответ-
соответствуют идеальной модели.
360
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
Системы, содержащие LiCl (например, LiCl—CdCla и LiCl—PbCl2),
по-видимому, очень близки к идеальным. В системах с NaX наблю-
наблюдаются слабые отрицательные отклонения от идеального раствора
по Темкину. Данные о парциальных мольных объемах, электро-
электропроводности, поверхностном натяжении и данные фазовых диаграмм
свидетельствуют о том, что в растворе нет или мало ассоциированных
частиц, т. е. в этих системах должны преобладать условия идеаль-
идеальной энтропии 6. Значение коэффициентов активностей, измеренные
в этих растворах, можно объяснить, допустив, что теплота смешения
растворителя и растворенного вещества не равна нулю, поскольку
имеется кулоновское взаимодействие между ионами раствора.
Это фактически является нулевым приближением к обычной модели
регулярного раствора. Высокие температуры систем расплавленных
солей, по-видимому, благоприятствуют нулевому, а не большему
приближению.
Термодинамические свойства систем МХ2+КХ слишком отли-
отличаются от термодинамических свойств идеальных и регулярных
растворов. Небольшие числовые значения коэффициентов активно-
активностей компонентов растворов указывают на то, что эти компоненты
частично связаны в растворе в виде комплексных ионов. С этим
согласуются данные, полученные другими методами, например
данные об электропроводности, криоскопии, мольных объемах,
спектрах комбинационного рассеяния. Термодинамические свойства
систем МХ2—RbX и МХ2—CsX еще больше отличаются от свойств
идеальных растворов. Это можно объяснить повышением устойчи-
устойчивости комплексных ионов с уменьшением плотности заряда, или
ионного потенциала, катиона щелочного металла.
Блюм ' рассмотрел факторы, определяющие образование ком-
комплексных ионов в расплавленных солевых смесях. Склонностью
некоторых ионов, например Cd2+, Zn2+ и Pb2+, к образованию ком-
комплексных ионов объясняется появление минимумов на изотермах
зависимости эквивалентной электропроводности от состава смеси,
а также возникновение положительных отклонений на изотермах
зависимости мольных объемов от состава. Было показано 52> 9Б,
что УФ- и ИК-спектры и спектры комбинационного рассеяния таких
расплавленных солевых растворов свидетельствуют об образовании
новых ионов при смешении компонентов расплава. Этими новыми
ионами являются комплексные ионы. Например, в системе
KG—CdCla в результате протекания реакций
КС1 +CdCl2 = KCdCl3
2KC] + CdCl2 = K2CdCl4 F)
4КС1 + CdCl2 = K4CdCl6
образуются ионы CdClJ, CdClt" и CdClJ".
Для подавления процесса комплексообразования в расплаве
достаточно ввести в расплав такие сильно поляризующие ионы,
VIII. РАСТВОРЫ СОЛЕЙ
361
100 т-
t
как Li+. По-видимому, ионы Li+ сильно конкурируют с ионами
Cd2+ за ион С1~. Тенденция к подавлению способности таких ионов,
как ионы Cda+, к комплексообразованию ослабевает с возрастанием
ионного радиуса введенного катиона щелочного металла. Вывод
подтверждается последовательным увеличением минимального зна-
значения электропроводности в этих системах в ряду Li->Cs.
Из рис. 67 видно, что если в системе РЬС12—МС1 комплексные
ионы и образуются, то, вероятнее всего, наиболее устойчивыми они
будут в присутствии ионов Cs+, а наименее устойчивыми — в при-
присутствии ионов Li. Подобныеже вы-
выводы можно сделать относительно
влияния добавок хлоридов Li и Na
на комплексообразование в рас-
расплавленном РЬС12. Введение в си-
систему ионов К+, Rb+ и Cs+ приво-
приводит к неизмеримо меньшему „
эффекту ослабления комплексе- ^
образующих свойств ионов Cd2+ *°
и Pb2+. ^
В табл. 50 приведены изучен-
изученные солевые системы и указаны
образующиеся в этих системах
комплексные ионы.
Анализ результатов выполнен-
выполненных работ позволяет отметить сле-
следующие факторы, способствующие
образованию комплексных ионов
в системах, в которых наблю-
наблюдается сильное взаимодействие
между растворителем и раство-
растворенным веществом.
1. Если при изучении фазовых равновесий окажется, что в си-
системе образуются твердые комплексные соединения, то в расплаве
комплексообразование возможно. Этот вывод вполне обоснован,
поскольку структуру расплава можно рассматривать как неупоря-
неупорядоченную структуру твердого тела, и в расплаве, вероятно, суще-
существуют ионы, подобные тем, которые имеются в твердом веществе.
2. Устойчивость комплексных ионов в расплаве повышается
по мере усиления ковалентной природы связи в комплексах, напри-
например с увеличением поляризующей способности галогенидных лиган-
дов при переходе от хлоридов к бромидам.
3. Поскольку образуются комплексы типа МХ„, любые факторы,
усиливающие способность комплексообразующего катиона притяги-
притягивать анионы, будут способствовать процессу комплексообразования.
Неудивительно поэтому, что способность катионов М образовывать
комплексные анионы возрастает в ряду Pb2+<Cd2+<Zn2+<Mg2+.
so -
о го 1+о ~"бо во
Содержание МС1 , мол. %
Рис. 67. Изотермы эквивалентной
электропроводности ' при 720° С:
1 _ pbCl2 + NaCl; 2 — РЬС12 + KG1;
3 — PbCl2 + RbGl; 4 — РЬС12 + CsCl.
362 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
Таблица 50. Солевые системы, в которых образуются комплексные ионы 7
VIII. РАСТВОРЫ СОЛЕЙ
363
Система
KCl-CdCl2
КС1-
КС1-
КС1-
КС1-
KF-
KI-
RbCl-
CsCl-
CsCl-
CsCl-
CsCl-
CsCl-
-ZnCl2
-PbCl2
¦MgCl2
¦CuCl
¦ZrF4
-Cdl2
-PbGl2
¦CoCb
-NiCl2
-CuCl2
-NpCl4
-PbCla
NaF —AIF3
Методы исследования;
изученные свойства системы
Электропроводность, мольные объемы,
спектры комбинационного рассеяния,
э. д. с, давление пара, криоскопия
Электропроводность, спектры комбинаци-
комбинационного рассеяния, э. д. с.
Электропроводность, мольные объемы,
числа переноса, понерхностное натяже-
натяжение, э.д. с, давление пара
Электропроводность, мольные объемы
Электропроводность
Давление пара
Электропроводность, мольные объемы
Э. д. с, поверхностное натяжение, элек-
электропроводность, мольные объемы
УФ-спектры поглощения
УФ-спектры поглощения
УФ-спектры поглощения
УФ-спектры поглощения
Электропроводность, мольные объемы
Электропроводность, поверхностное натя-
натяжение, криоскопия
Постулируе мый
комплексный ион
CdClJ, CdClf-,
CdClf
PbClj,
PbCl|"
MgCls
CuCl|-
ZrFj
Cdir
PbClJ, РЪ
PbCl|-
N1CI4
cucir
s,
PbCH-
AlFf", AlFj
В этой же последовательности возрастает и плотность,
заряда.
Следует отметить, что из фазовых диаграмм не всегда можно
сделать правильный вывод о существовании твердых соединений,
так как многие работы не отличаются достаточной точностью. Это
хорошо видно на примере системы FeCl3—NaCl. Первоначально счи-
считали 27, что на фазовой диаграмме системы имеется лишь одна эвтек-
эвтектика и что комплексные анионы в расплавах образовываться не
должны. Однако измерения коэффициентов активности показали
обратное. Фазовая диаграмма системы показала существование
твердого соединения NaFeCl4 и двух эвтектик вблизи точки плавле-
плавления этого соединения. Следовательно, вполне возможно, что в рас-
расплаве существуют комплексные ионы. Отрицательное влияние
иона Na+ на процесс комплексообразования, по-видимому, менее
сильно, чем противоположное влияние относительно небольшого'
по размерам, но имеющего высокий заряд комплексообразующего
иона Fe3+.
Г. Сольватация и образование комплексных ионов
в расплавленных солевых смесях
Сольватационные эффекты в ионных расплавах могут изменяться
по величине: от незначительной склонности к агрегатообразованию
(что проявляется в виде неидеальной энтропии смешения) до обра-
образования ковалентных комплексных ионов (что проявляется в откло-
отклонениях от идеальной теплоты растворения). Следовательно, образо-
образование комплексных ионов — особый случай сольватации. Комплекс-
Комплексные ионы, такие, как РЬХ(,"-2>~ и CdX(,"~2)~ (где га > 2), имеют ча-
частично ионный характер, и в растворе, конечно, будут происходить
обменные реакции между лигандами комплексов.
В общем,^компоненты смеси, где возможно образование комплекс-
комплексных ионов за счет взаимодействия растворителя и растворенного ве-
вещества, должны смешиваться в любых соотношениях, хотя имеются
исключения из этого правила. Склонность двух компонентов смеси
к расслаиванию проявляется в виде положительных отклонений тер-
термодинамических активностей от идеальных значений. Это хорошо
видно в системе Bi—Cd. Но если и существует взаимодействие между
растворителем и растворенным веществом даже при условии, что энт-
энтропия сохраняет идеальное значение, термодинамические актив-
активности соответствуют значениям, рассчитанным для регулярных ра-
растворов. Эти значения имеют обычно отрицательные отклонения
¦от идеальных, как в расплавленной смеси AgBr — КВг 58. Если
взаимодействие растворителя и растворенного соединения настолько
сильно, что образуется химическая связь, в частности, при появле-
появлении комплексных ионов, то отклонения активности от идеальных
значений должны быть более отрицательными, как, например, в си-
системе CdCl2 — CsCl.
Д. Комплексные ионы и кинетика реакции
Способность катионов к комплексообразованию может быть ис-
использована для контроля за концентрацией реагирующих ионов
в окислительно-восстановительных равновесных реакциях 94. Это
особенно важно при высоких температурах,' когда вполне возможна
коррозия металлических стенок контейнера расплавленными солями.
Так, если коррозия протекает по реакции
2МЗ+ + А=2М2+ + Л2+ G)
то она может быть уменьшена путем введения в систему ионов Х~
с последующим образованием комплексных ионов, например MXj.
Комплексные ионы имеют большие размеры и, следовательно, малую
подвижность, т. е. скорость реакции с ними будет ниже, чем с мень-
меньшими по размерам, и потому более подвижными ионами.
364 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
IX. ПРИМЕНЕНИЕ РАСПЛАВЛЕННЫХ СОЛЕЙ В КАЧЕСТВЕ
РАСТВОРИТЕЛЕЙ
Расплавленные соли являются более удобными растворителями
для некоторых соединений, чем обычные растворители, так как кон-
концентрации реагирующих компонентов в них могут быть очень боль-
большими. Хотя солевые расплавы как растворители по своей природе
отличаются от обычных растворителей, применяющихся при комнат-
комнатной температуре, при рассмотрении реакций в расплавленных солях
сохраняют силу такие понятия, как кислотно-основная диссоциация
и рН.
Можно считать, что в расплавленных нитратах щелочных метал-
металлов происходит кислотно-основная диссоциация
IX. ПРИМЕНЕНИЕ РАСПЛАВЛЕННЫХ [СОЛЕЙ
36S
z? NO+ + O2-
(8)
аналогично диссоциации
Н20 4Г? Н+ + ОН" (9)
Константа диссоциации реакции (8) гораздо меньше, чем константа
диссоциации реакции (9). При пропускании NO2 через ячейку с пла-
платиновым электродом может протекать реакция
NO2 ^=Г NOJ + e A0)
аналогичная реакция на водородном электроде
уН2 ^ Н++е (И)
В расплавленных солях может протекать также известная ки-
кислотно-основная реакция превращения бихромат-иона в хромат-
ион Б. Бихромат калия растворяется в расплавленном NaNOg, при-
придавая ему окраску, характерную для бихромат-иона. При добавле-
добавлении к этому раствору небольшого количества твердого карбоната
натрия в нем проходит кислотно-основная реакция с выделением
из раствора СОа, причем раствор приобретает окраску, характер-
характерную для хромат-иона:
Cr2O|-+CO§- ^ 2СгОГ + СО2 A2)
Часто могут протекать и окислительные реакции, например:
BrOj+5Br- ^ зВг2 + ЗО2- A3)
При добавлении бромата натрия к расплаву нитрата натрия, содер-
содержащему бром-ион, согласно приведенному выше уравнению выде-
выделяется бром.
Расплавленные соли использовали как реакционную среду при
синтезе многих неорганических и органических соединений. Особенно
большое внимание было уделено применению расплавленных солей
для электролиза, в частности при электролитическом получении
металлов. В последние годы расплавленные соли нашли применение-
в процессах разделения и экстракции.
На практике предпочитают использовать эвтектические смеси,
главным образом в связи с возможностью работать с ними при срав-
сравнительно низких температурах. Особенно широко применяется эвтек-
эвтектическая смесь КС1 + LiCl, как наиболее легкоплавкая из смесей
- хлоридов щелочных металлов, поэтому ниже мы кратко опишем
методику ее приготовления.
Методика приготовления эвтектической смеси ЫС1-\-КС1. Гигроскопич-
Гигроскопичность LiCl заставляет применять специальные меры предосторожности при:
получении этого растворителя. Инман и др.Бв получали расплав смешива-
смешиванием расплавленного LiCl (ч.) и расплавленного КС1 (ч. д. а.) в стеклян-
стеклянной колбе с последующей фильтрацией смеси через стеклянную вату в стакан
из стекла пирекс. Далее эвтектическую смесь помещали в электролитическую
ячейку, где ее расплавляли и подвергали вакуумированию до 10~4 мм рт. ст.
Затем через расплав пропускали смесь безводного газообразного хлористого
водорода с очищенным аргоном. Окончательную очистку от следов примесей,
в том числе от растворенного НС1. проводили электролизом расплава в ва-
вакууме с вольфрамовым или молибденовым катодами и угольными анодами
при напряжении ниже потенциала разложения расплава. За степенью очист-
очистки следили либо по падению остаточного тока при постоянном напряжении,
либо по повышению напряжения при постоянном токе.
А. Реакции в расплавленных солях с участием
органических веществ или летучих неорганических жидкостей
Многие органические соединения при барботировании через рас-
расплавленную соль вступают в обменные реакции. Например, при кон-
контактировании хлорсодержащих соединений с расплавленными фто-
фторидами может происходить их фторирование.
1. Реакции фторирования 87> 91
Расплав KF D5%) + NaF D5%) + LiF A0%) оказался хоро-
хорошей средой для фторирования некоторых органических соединений
в интервале температур 500—600° С. Если необходимы более низкие
температуры, можно использовать расплавы KF + ZnCl2 + KC1
или CaF2 + ZnCl2+ KC1. В последнем случае температура расплава
составляет приблизительно 250° С.
Реакции фторирования протекают довольно легко, для этого
часто достаточно пропускать соединение через расплав, энергично
перемешивая его; выход продуктов реакции достаточно высок.
Примеры таких реакций:
(CH3KSiCl+NaF=(CH3KSi
SOCl2 + 2NaF= SOF2+2NaCl
A4)-
366
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
IX. ПРИМЕНЕНИЕ РАСПЛАВЛЕННЫХ СОЛЕЙ
367
2. Обменные реакции
Обменные реакции таких солей, как цианиды, цианаты и тиоциа-
наты, с галогенидами возможны благодаря хорошей растворимости
указанных солей щелочных металлов в расплавах смесей галогени-
дов щелочных и щелочноземельных металлов. Например, в расплаве
LiCl + KC1, взятом в качестве растворителя, реакция
81С14 + цианаты —*¦ Si(NCOL A6)
может протекать с 55%-ным выходом.
3. Реакции азидов с органосиланами 88
Используя в качестве растворителя эвтектическую смесь
ZnCl3E4%) + KC1 D6%) с *M = 228°C при температуре 230 —
250° С, можно получить органоазидосиланы. В эвтектическую смесь
суспендируют и частично растворяют от 10 до 20% NaN3; затем
пропускают органосилан, при этом протекают следующие реакции:
(CH3KSiCl + NaN3 = (CH3KSiN3 + NaCl D3%)
(CH3JSiCl2 + 2NaN3= (CH3JSi(N3J + 2NaClF9,5%)
СНз S iCl3 + 3NaN3 = CH3S i(N3K + 3NaCl
Образуются также CH3SiN3Cl2, CH3Si(N3JCl и (CH3JSiN3Cl.
A7)
4. Реакция дегидратации спиртов 4e
При барботировании спиртовых паров через расплав происходит
дегидратация спиртов при 340° С В качестве газа-носителя обычно
применяют азот. Таким методом получены олефины, содержащие не-
некоторое количество недегидратированного спирта.
5. Реакции дегидрохлорирования 4в
Дегидрохлорирование алкилгалогенидов может быть осуще-
осуществлено в расплаве ZnCl2(85%) + CuCl A5%). При 400° С, опти-
оптимальной температуре дегидрохлорирования «-бутилхлорида, обра-
образуется бутен (95%-ный) и в реакцию вступает 77,5% исходного про-
продукта; при 240° С реагирует лишь 24% исходного к-бутилхлорида.
На этом примере показано преимущество высокотемпературного
солевого расплава как реакционной среды. Но если температура
реакции слишком высока, то часть продуктов может разлагаться
на стеклянных стенках реакционного сосуда. Эквимолярная смесь
А1С13 + NaCl непригодна для проведения этой реакции, так как
происходит частичная полимеризация бутена.
В расплавах, в состав которых входит А1С13, можно провести
^обратную реакцию. Например, при пропускании через расплав
А1С13 + NaCl + CuCl2 при 400° С смеси этана и хлора образуется
сэтилхлорид с выходом 45%. Пропуская этилен и хлористый водо-
водород в эвтектическую смесь А1С13 + NaCl + КС1 Цш= 89° С), можно
также получить этилхлорид с выходом 82%. Остальные 18% этилена
полимеризуются в расплаве либо расщепляются, причем количество
расщепляющегося этилена возрастает с повышением температуры,
т. е. в некоторых случаях требуются солевые смеси с низкими тем-
температурами плавления. По-видимому, наиболее пригодны для исполь-
использования в качестве низкотемпературных растворителей тройные-
эвтектические смеси.
6. Другие препаративные методы, основанные
на реакциях в расплавленных солях
Сандермейер и др. 91 получили винилхлорид с 97%-ным выходом,
пропуская пары 1,1-дихлорэтана через расплавы ZnCl2 + KC1
или ZnCl2 + CuCl при 330° С. Для этой цели могут быть использо-
использованы эквимолярная смесь ацетилена и хлора и расплав, содержащий
соль ртути. В расплаве NaCl + A1C13 при 175° С хорошо протекает-
реакция Фриделя — Крафтса 46.
7. Получение силана 89
Реакции с использованием расплавленных солей в качестве рас-
растворителей нашли промышленное применение для получения силана.
Пары SiCL, пропускают через раствор LiH в эвтектическом расплаве
LiCl + KC1 при 400° С Выход моносилана достигает почти 100%.
Гидрид лития регенерируется электролизом с последующим пропу-
пропусканием через расплав водорода.
Б. Получение металлов в расплавленных солях
1. Промышленный метод получения алюминия
Промышленный метод получения А1 из бокситовых руд основан
на электролизе окиси алюминия в расплавленном криолите Na3AlF6,
используемом в качестве растворителя. Диссоциация расплава про-
происходит в соответствии с уравнением:
Na3AlFe = 3Na+
Ион A1FJ" также частично диссоциирует44:
368 ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
2. Получение актиноидов
Груен и др. в опубликовали результаты лабораторных эксперимен-
экспериментов с целью получения Zr, Th, U и Np из растворов тетрахлоридов
этих элементов в эвтектическом расплаве LiCl -f- KC1 восстановле-
восстановлением металлическим магнием. Металлический уран может быть по-
получен также восстановлением двойной соли Cs2UCl6 магнием в рас-
расплавленной эквимолярной смеси CsCl + MgCl2 при 550° С:
Cs2UCl6-b2Mg=U + 2MgCl2-b2CsCl B0)
Осажденный уран отделяется от расплава фильтрацией и затем про-
промывается. Спектральный анализ показал, что металл не содержит
цезия и магния.
3. Разделение металлов с близкими свойствами
Соли металлов, имеющих похожие свойства, растворенные в со-
солевых расплавах, легко могут быть разделены электролизом. Опи-
Описан метод прямого извлечения молибдена и вольфрама из шеелитовой
руды с применением солевого расплава 101. Затем для раздельного
извлечения металлов из расплава при электролизе применяют раз-
различные плотности тока.
4. Извлечение магния из силикатных минералов
Магний может быть извлечен при электролизе из силикатных ми-
минералов, таких, как алинин и серпентин 69.
5. Извлечение свинца из галенита
Свинец может быть извлечен электролизом галенита в смеси
хлоридов натрия и калия при 700° С. При прохождении тока через
расплав на катоде выделяется свинец, а на аноде — хлор и элемен-
элементарная сера 1U.
Рафинирование свинца может быть проведено электролизом
в расплаве КС1 + LiCl. Хлорид свинца растворяется в растворителе
с образованием эвтектических смесей 109. В последние годы изуча-
изучались методы противоточной многостадийной экстракции расплавлен-
расплавленных металлов расплавами солей 63.
В. Хроматографическое разделение расплавленных солей в2
Приблизительно 0,01 м (концентрация ионов переходных метал-
металлов) растворы хлоридов Fe111, Со", Мп, Си11 и UO|X в расплавлен-
расплавленной эвтектической смеси LiNO3 + KNO3 могут быть разделены в хро-
матографической колонке, заполненной у-А12О3. Сорбированные
ЛИТЕРАТУРА
369
ионы далее селективно элюируются из колонки расплавленной эвтек-
эвтектической смесью нитратов, содержащей хлорид аммония (концент-
(концентрации 3 м). Результат процесса элюирования зависит в основном
от концентрации хлорида в элюенте.
Г. Экстракция расплавленными солями
62
В водных системах особенно эффективными высаливающими аген-
агентами являются нитраты. Неудивительно поэтому, что эффек-
эффективность расплавленных нитратов также очень высока. Ионы пере-
переходных металлов, таких, как Fe111, Co11, Ni11, UVI, Pr111 и Ndni,
растворенные^ в эвтектике LiNO3 + KNO3, количественно экстраги-
экстрагируются трибутилфосфатом при 150° С.
С этой целью можно использовать также неорганические экстра-
генты. Например, расплавленная окись бора В2О3, образующая не-
смешивающуюся фазу с расплавленной эвтектической смесью
LiCl + KC1, способна экстрагировать ионы переходных металлов
из хлоридного растворителя.
Д. Основные направления дальнейших исследований
Применение расплавленных солей в качестве растворителей будет
возрастать и в дальнейшем, так как исследования открывают все но-
новые области возможного использования растворителей этого типа.
По-видимому,наиболее важные разработки будут проведены в области
топливных элементов, извлечения и очистки металлов, химических
реакций с участием органических и неорганических соединений и в
области ядерной энергетики.
ЛИТЕРАТУРА
1. A ten A. H. W., Z. phys. Chem., 73, 578 A910).
2. Bell W. В. G., Rowlands СВ., et al. J. Chem. Soc, 1927, 31
A930).
3. Blander M., G г i m e s W. R., et al. J. Phys. Chem., 63, 1164 A959).
4. В i 1 t z W., К 1 e m m W., Z. anorg. Chem., 152, 267 A926).
5. Blomgren G. E., Van Artsdalen E. R., Ann. Rev. Phys.
Chem., 11, 273 A960).
6. В 1 о о m H., Rev. Pure Appl. Chem., 9, 139 A959).
7. Bloo m H., Pure Appl. Chem., 7, 389 A963).
8. Bloom H., Bockris J., Modern Aspects of Electrochemistry, v. II,
London, 1959, p. 160—261.
9. В 1 о о m H., R h о d e s D. C, J. Phys. Chem., 60, 791 A956).
10. В о с k r i s J. O'M, Richards N. E., Proc. Roy. Soc, A241, 44 A957).
11. Boleslaw L. D., S с h e i d t R. C, XVIII Con. Pure and Appl. Chem.,
1961, 170.
12. В г a 11 a n d D., G г j о t h e i m К., К г о h n С, XIX Con. Pure and
Appl. Chem., 1963.
13. Braunstein J., Blander M., Lindgren R. M., J. Am. Chem.
Soc, 84, 1529 A962).
24 Заказ 90
370
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
ЛИТЕРАТУРА
371
J о h n s о п
Johnson
R., В г е d i g
С о г b e t t J.
14. Braunstein
' A962).
15. В г е d i g M. A.,
16. В re dig M. A.,
UC-4-Chem., 1963.
17. Bre dig M. A.,
A955).
18. Bre dig M. A.,
19. В г е d i g M. A.,
307 A955).
20. Bronstein H
A958).
21. Bronstein H
22. Br uner B. L.,
A961).
23. В ues W., Z
24. В u n s e n N.
25. В u г к h a r d
A957).
26. Campbell
Can. J.
27. Co ok С. М.,
- 110 A961).
28. Corbet t J.
29. Corbet t J.
30. Cor be t t J.
31. С о r b e t t J.
Soc, 79, 3020
32. Cubicciotti
33. Cubicciotti
34. Cubicciotti
35. Cubicciotti
36. Cubicciotti
37. D e Maine P.
18, 286 A961).
38. Druding L. F.,Corbett
39. D г u d i n g L. F., С о г b e t t
40. ~ ' "
J., Lindgren R. M., J. Am. Chem. Soc, 84, 15
J. Chem. Phys., 37, 451 A962).
Mixtures of Metals with Molten Salts, ORNL-33
Bronstein H. R., J. Am. Chem. Soc, 77, 14
W., J. Phys. Chem., 64, 1899 (I960,.
.Smith W., J. Am. Chem. Soc, 77,
R., Bredig M. A., J. Am. Chem. Soc, 80,2077
M. A., J. Phys. Chem., 65, 1220 A961).
D., J. Inorg. a. Nucl. Chem., 20, 62
anorg. Chem., 279, 104 A955).
, Kirchoff R., Pogg. Ann., 113, 345 A861).
W. J., Corbett J. D., J. Am. Chem. Soc, 79, 6361"
A. N., Kartzmark E. M., Williams D. F.,
Chem., 40, 890 A962).
Jr., Dunn W. E., Jr., XVIII Con. Pure and Appl. Chem.,
D.
D.
D.
D.,
A957).
D,
D,
D.
D.
D,
A.
A 1 b e г s F.
Hershaft
W i n b u s h
W i n b u s h
C, J. Am. Chem. Soc,
A., J. Am. Chem. Soc,
S. V., J. Am. Chem. Soc.
S. V., Al b ers F. C,
82, 533 A960).
80, 1530 A958).
77, 3964 A955).
J. Am. Chem.
J. Am. Chem. Soc, 71, 4119 A949).
Clear у G., J. Am. Chem. Soc, 74, 557 A952).
J. Metals, (New York), 5 A953).
Trans. Am. Inst. mech. Engrs., 197, 1106 A953).
J. Chem. Educ, 37, 540 A960).
D., McAlonie G. E., J. Inorg. a. Nucl. Chem.,
J. D., J. Am. Chem. Soc, 81, 5512 A959).
J. D., J. Am. Chem. Soc, 83, 2462 A961).
H'. R.
Phys.
Bredig ML A., J.
Soc, 47, 1287 A951).
Trans. Farad. Soc, 31, 1004
79, 853 A960).
79, 919.A960).
J. Am. Chem. Soc, 78, 3279
Dworkjn A. S., Bronstein
Chem., 66, 572 A962).
41. E 1 e у D. D., King P. J., Trans. Farad
42. Farquharson J., Heymann E.,
A935).
43. Fie ng as S. N., Ann N. Y. Acad. Sci.
44. F о s t e r L. M., Ann. N. Y., Acad. Sci.,
45. Gardner D. M., F r a e n к e 1 G. K.,
A956).
46. Glemser O., Kleine-Weischede K., Ann., 659, 17 A962).
47. G r e e n b e r g J., S u n d h e i m B. R., J. Chem. Phys., 29, 1029 A958).
48. G r e e n b e r g J., S u n d h e i m B. R., G r u e n D. M., J. Chem.
Phys., 29, 461 A958).
49. G r i m e s W. R., Smith N. V., W a t s о n G. M., J. Phys. Chem.,
62, 862 A958).
50. Grjotheim K.,Gronvold F.,Krogh-Moe J.,J. Am. Chem.
Soc, 77, 5824 A955).
51. G r j о t h e i m K., et al., Acta chem. scand., 16, 689 A962).
52. G r u e n D. M., Fried S., et al. Chemistry of Fused Salts. A/CONF,
15/P/940 USA 1958, p. 26. '
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
71.
72.
73.
74.
75.
76.
77.
78.
79.
80.
81.
82.
82.
84.
85.
86.
87.
88.
89.
90.
91.
92.
93.
94.
G r u e n D. M., M с В e t h R. L., J. Inorg. a. Nucl. Chem., 9, 290 A959).
G u n t z А., В e n о i t F., Bull. Soc. chim. France, 35, 709 A924).
Harris R. L, Wood R. E., Ritter H. L., J. Am. Chem. SocT
73, 3151 A951).
Hevesy G., Lowenstein E., Z. anorg. Chem.,
Heymann E., Friedlander E.,_Z. phys. Chem., 148, 177 A930)
Hildebrand
187, 266 A930)
Am. Chem. Soc, 54
J. H., S als t ro m E. J., J.
4257 A932).
I n m а'Ъ D., Hills G. J., Young L., Bockris J., Ann. N. Y.
Acad. Sci., 79, 803 A960).
Johnson J. W., Bredig M. A., J. Phys. Chem., 62, 604 A958).
Johnson J. W., Bredig M. A., J. Phys. Chem., 64, 64 A960).
Johnson J. W., Cubicciotti D., К ell e у СМ., J. Phys.
Chem., 62, 1107 A958).
Josephs on P, R., Jr., Diss. Abs., 22, 2322 A962).
Kendall J., С r i t t e n d e n E. D., M i 11 e r H. K., J. Am. Chem.
Soc, 45, 963 A923). .
К 1 e m m W., H u s s E., Z. anorg. Chem., 258, 221 A949).
Krebs H., Z. Naturiorsch, 12b, 795 A957).
Kreye W. C, Kellogg H. H., Trans. Electrochem. Soc, 104, 504
A957).
К roll W. J., Metallurgy. 9, 366 A955).
Labounski, пат. США 3093558 A963).
Landolt-Bornstein Zahlenwerte und Funktionen, v. II, Part 3. Berlin, 1956.
Landon G. J., Ubbelohde A. R., Trans. Farad. Soc, 52, 647
A956).
Levy H.
Levy H.
762.
Lorentz R., Eitel E., Z. anorg. Chem., 91, 46 A915).
Lorentz R., Eitel W., Pyrosole, Leipzig, 1926.
P. Т., J. Electrochem. Soc, 66, 245 A934).
elli A., Gazz., 38, 137 A908).
A., Bredig M. A., et al., J. Phys. Chem., 64, 1959 A960).
A., Agron P. A., et al., Ann. N. Y. Acad. Sci., 79, Art. 11,
R., Eitel E.,
R., Eitel W.,
F. C, S t г о u
E., M u z z u с 1
Mathers
P a t e r n о
Peterson D., Hinkebein J. A., J. Phys. Chem., 63, 1360 A959).
P i t z e r K., J. Am. Chem. Soc, 84, 2025 A962).
Rogers P. S., Tomlinson J. W., Richardson F- D-, Met
Soc. Conf., 8, 909 A961).
S a 11 а с h R. А., С о r b e t t J. D., Inorg. Chem., 2, 457 A962).
S с h a f f e r J. H., G r i m e s W. R., W a t s о n G. M., J. Phys. Chem.,
63, 1999 A959).
Smith G. P., Electronic Absorption Spectra of Molten Salts, in «Molten
Salt Chemistry», New York, в печати.
Staffanson L. I., The Physical Chemistry of Metals in their Molten
Halides, Ph. D. Thesis, London, 1959.
Stern К. Н., XVIII Con. Pure Appl. Chem., 1961, 177.
Sundermeyer W., Z. anorg. Chem., 313, 290 A962).
W., Z. anorg. Chem., 314, 100 A962).
W., Chem. Ber., 96, 1293 A963).
Glemser O., Angew. Chem., 70,
№ 20, 625
Sundermeyer
Sundermeyer
Snndereyer W.,
A958).
Sundermeyer W., Glemser O., Kleine-Weischede K.,
Chem. Ber., 95, 1829 A962).
Sundermeyer W., Meise W., %. anorg. Chem., 317, 334 A962).
Topol L. E., Laudis A. L., J. Am. Chem. Soc, 82, 6291 A960).
Van Artsdalen E. R., J. Phys. Chem., 60, 172 A956).
Van Artsdalen E. R., The Structure of Electrolytic Solytions,
New York, 1959, p. 411—421.
24*
¦372
ГЛАВА ДЕВЯТАЯ. РАСПЛАВЛЕННЫЕ СОЛИ КАК РАСТВОРИТЕЛИ
95.
'96.
97.
98.
99.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
110.
111.
112.
ИЗ.
114.
115.
116.
Wait S.
Winsor
Y о s i m S.
A960).
Y о s i m S. J.
Y о s i m S. J.
Yosim S. J
C, J anz G. J., Quart. Rev. (London), 17, 225 A963).
V. R., С ad у G. H., J. Am. Chem. Soc, 70, 1500 A948).
, L u с h s i n g e г Е. В., Ann. N. Y. Acad. Sci., 79, 1079
Mayer S. W., J. Phys. Chem., 64, 909 A960).
D arnell A. J., et al. J. Phys. Chem., 63, 230 A959).
.Ransom L. D., et al. J. Phys. Chem., 66, 28 A962).
Zadra, Gomes, пат. США 3075900204 A963).
Z a r z у с к i G., С. г., 224, 758 A957).
Zarzycki G., J. phys. et rad., 18, 65A A957).
Белозерский Н. А., Избранные работы по электрохимии распла-
института прикл. химии № 35, 50 A940).
ЖНХ, 3, 2805 A958).
Усп. хим., 29, 899 A960).
, Красницкий С. Я., ДАН СССР, 101, 661
28
Б.
вленных солей, Сб.
Беляев И. Н.,
Беляев И. Н.,
Данилов В. И.
A955).
Делимарский Ю. К., ЖФХ, 29,
Делимарский Ю. К., Марков
вленных солей, Металлургиздат, 1960.
Делимарский Ю. К., Марков
Гутман Е. В., Колотий А. А.,
New York, 1961, p. 96.
Гульдин И. Т., Бушинская
VIII, New York, 1961, p. 100.
Журин А. И., Металлургия, 10, №4,
Карпачев С. В., Стромберг ~
43 A944).
Латимер В. М., Окислительные состояния
циалы в водных растворах, Издатинлит 1954.
Пушин М., ЖОХ, 18, 1599 A958).
Темкин М., Acta phys.-chem., 20, 411 A945).
A955).
Ф., Электрохимия распла-
Б. Ф., Панченко И. Д.,
Soviet Electrochemistry, VIII,
В. В., Soviet Electrochemistry,
87 A935).
Л. Г., Иордан
Е., ЖПХ 18,
элементов и их потен-
ОГЛАВЛЕНИЕ
Предисловие 5
Глава первая. Жидкий аммиак. В. Л. Джолли, Е. Дж. Холлада 7
Глава вторая. Жидкий фтористый водород. X. X. X аи мен,
Дж. Дж. Кац 49
Глава третья. Галогеноводороды как ионизирующие раствори-
растворители. М. Е. Пич, Т. Ваддингтон 80
Глава четвертая. Серная кислота. Р. Дж. Джиллеспи, Е. А. Ро-
Робинсон Ю9
Глава пятая. Координирующие растворители. Р. С. Драго,
К. Ф. Пурселл 201
Глава шестая. Жидкий сернистый ангидрид. Т. Ваддингтон . . 238
Глава седьмая. Галогены и интергалогены как растворители.
А. Г. Шарп 268
Глава восьмая. Галогениды и оксигалогеннды элементов V
группы. Д. С. Пайн 282
Глава девятая. Расплавленные соли как растворители. Г. Блюм,
Дж. В. Хасти 334
НЕВОДНЫЕ РАСТВОРИТЕЛИ
Издательство «Химия», М., 1971 г.
376 с. УДК 66.062.18
Редактор Г. И. Белан
Технический редактор В. М. Скитина
Художник К. М. Егоров
Корректор Н. И. Попова
Подписано к печати 17/IV 1971 г.
Формат бумаги 60 X ЭО'/ц. Печ. л. 23,5.
Уч.-изд. л. 23,97. Типогр. бум.'М 2.
Тираж 4900 экз. Цена 2 р. '40 к.
Тем. план 1969 г. № 132. Зак. 90.
Ленинградская типография №14
«Красный Печатник» Главполиграфпрома
Комитета по печати при Совете Министров СССР.
Московский пр., 91.