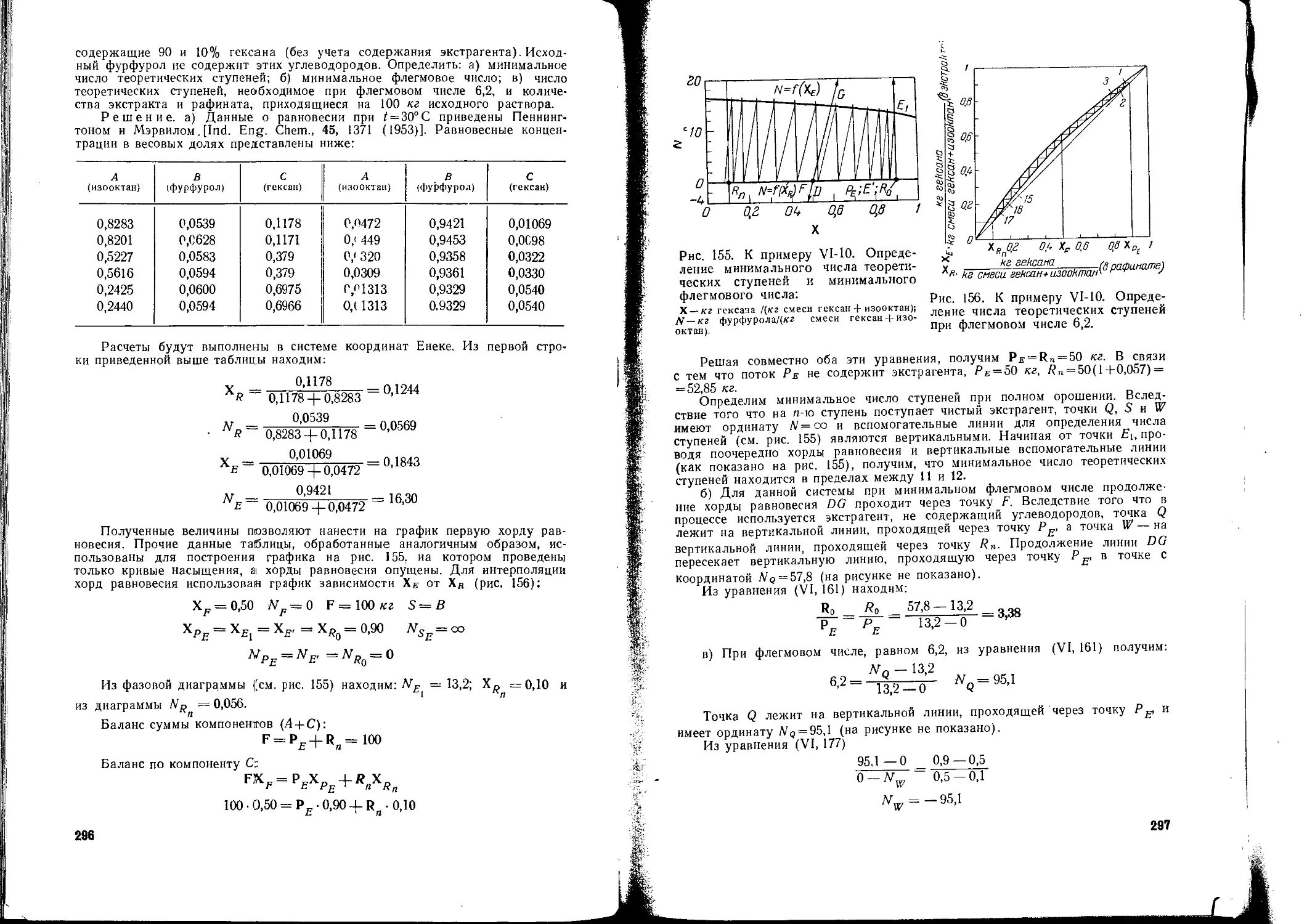
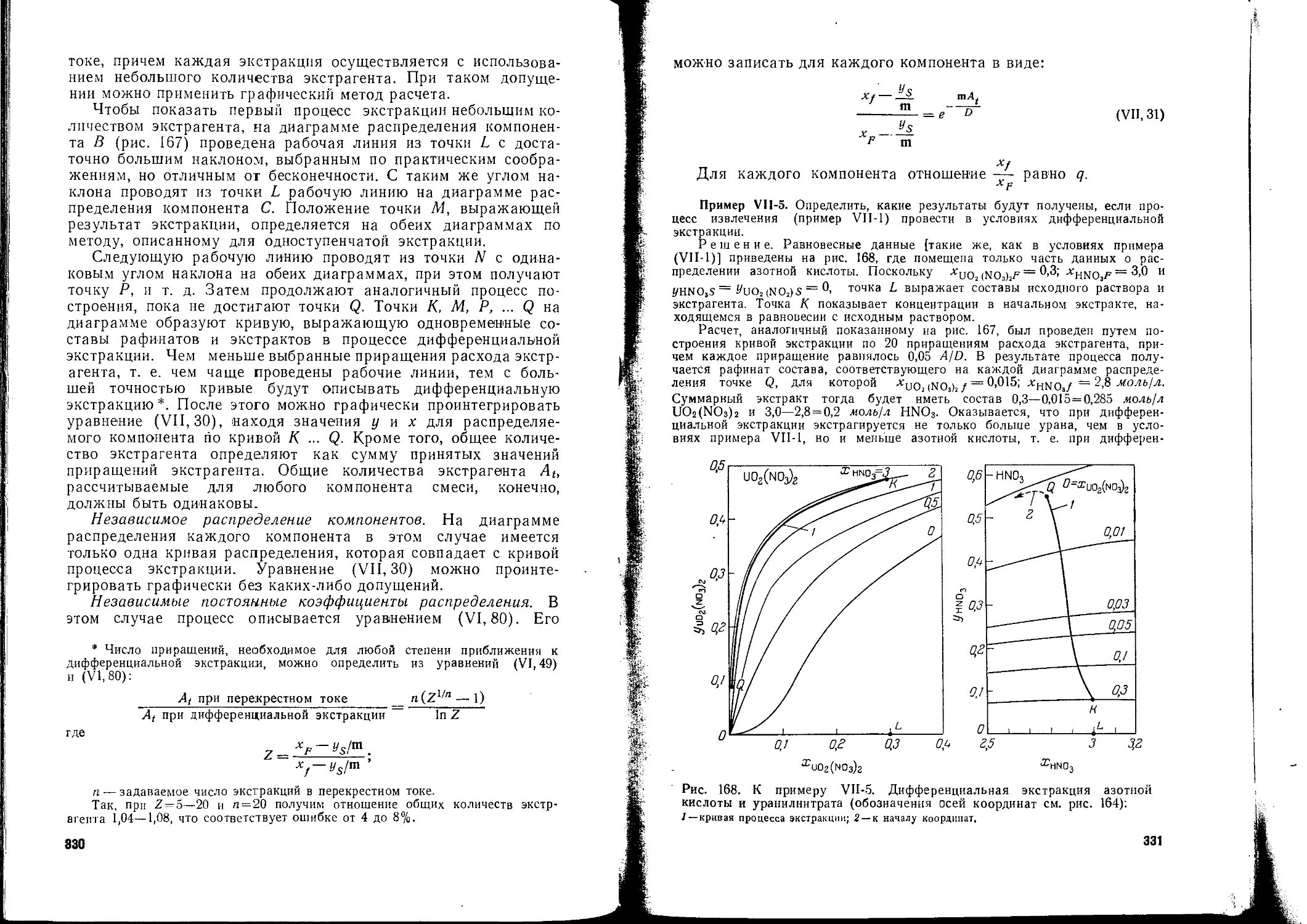
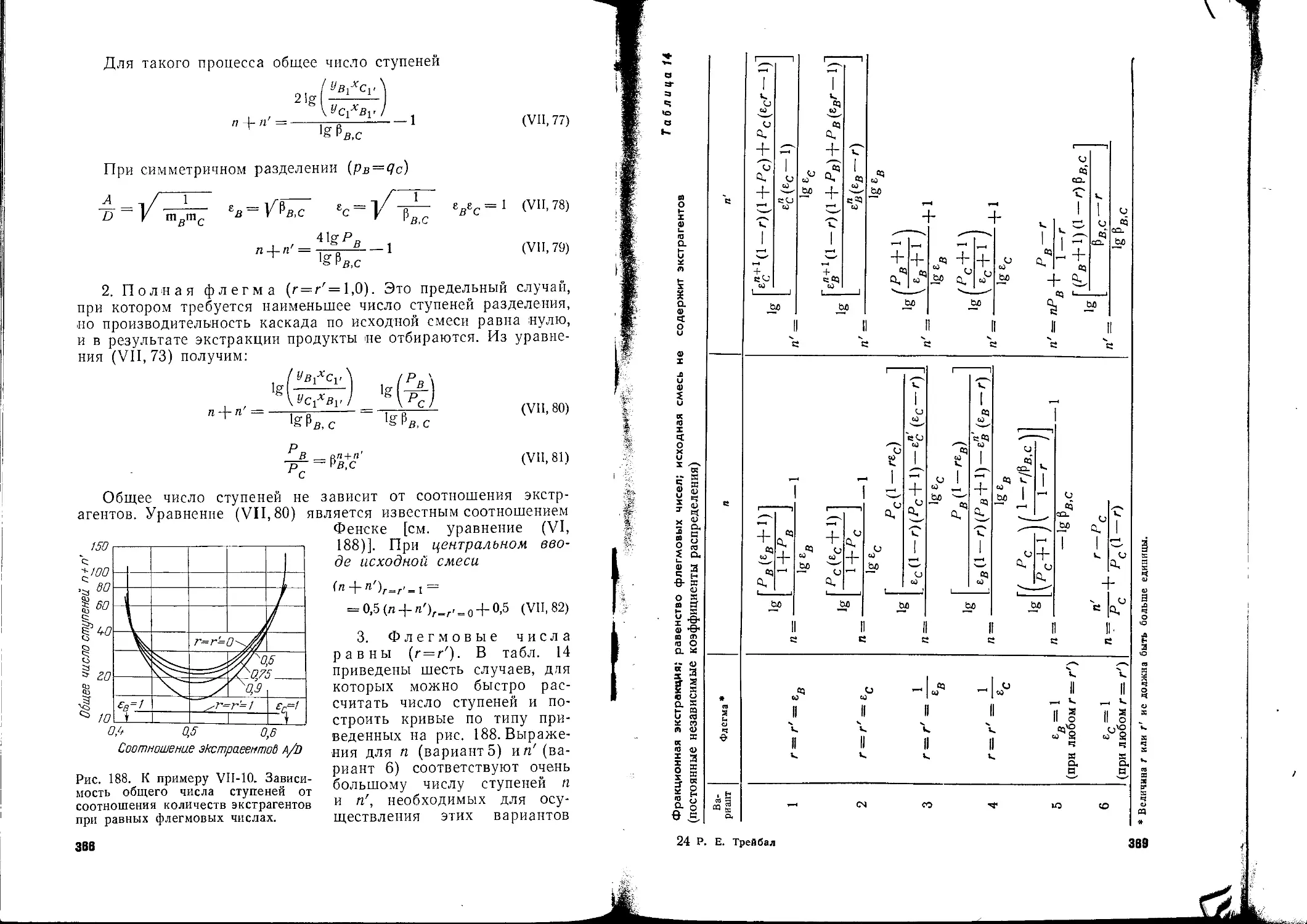
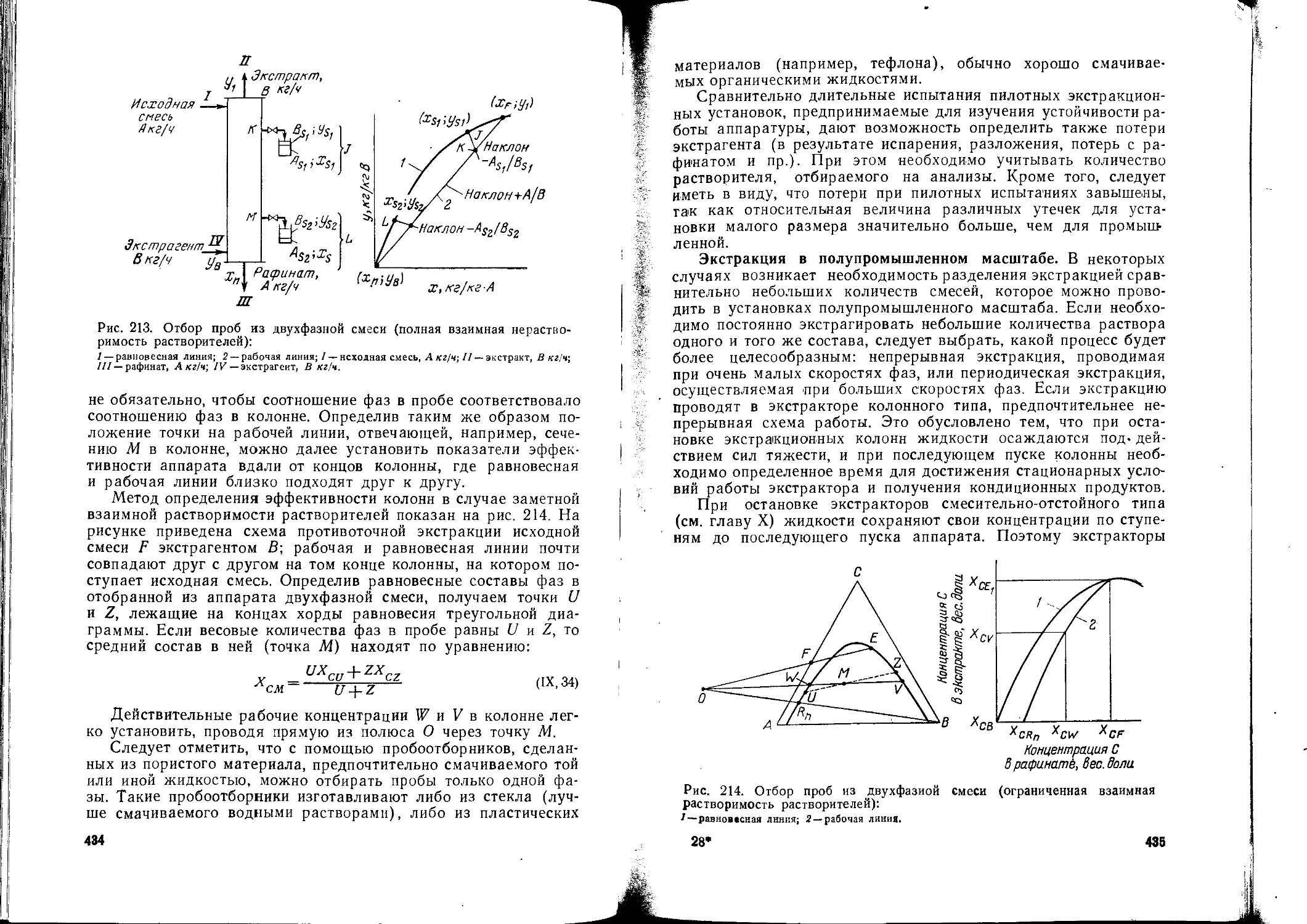
/









































































































































































































































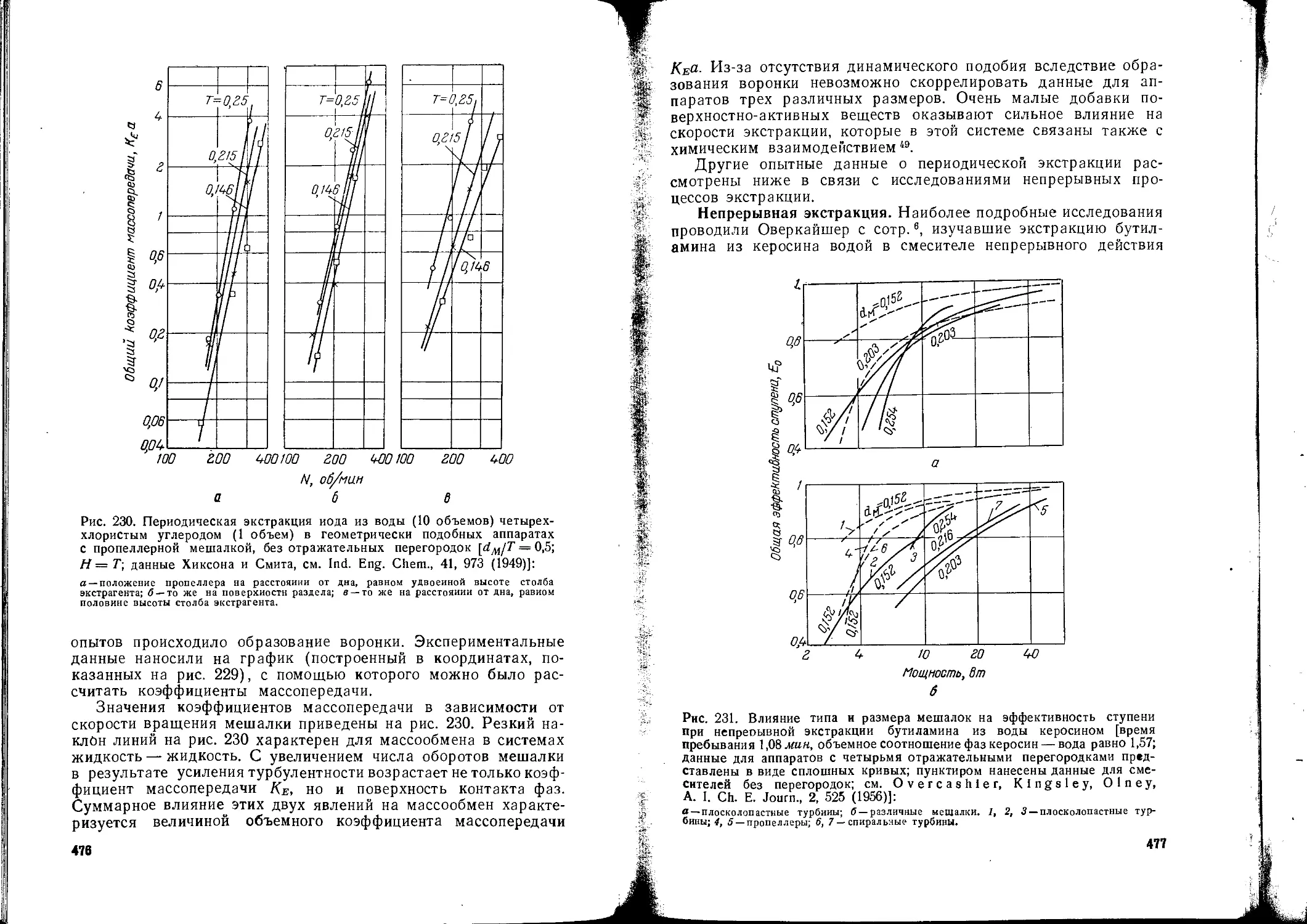














































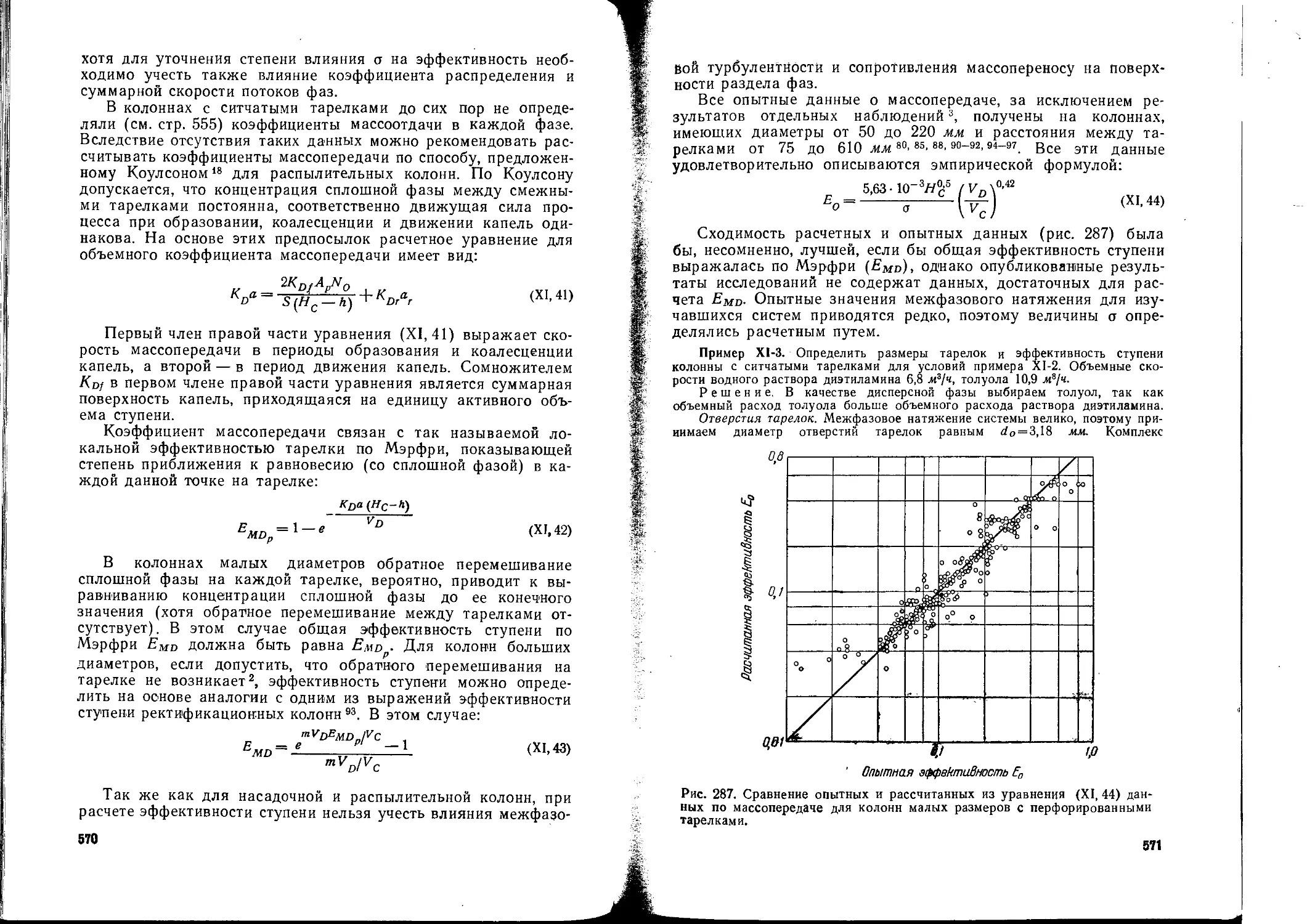

















































































Текст
R.TREVBAL
LIQUID
EXTRACTION
2-ND ED
• •-»•*•..
MC GRAW = HILL BOOK COMPANY INC.
N . V. • I 9 О 3
2GG0 r<
Г
P.T P E Й БАЛ
ЖИДКОСТНАЯ
ЭКСТРАКЦИЯ
Перевод с английского
под редакцией
докт. техн. наук
С.3.КАГАНА
JV3
■с
оснс
♦у из а
CtMiw
основной фонд
Семот»*«льСкого БВ.'лИУ
9/
ИЗДАТЕЛЬСТВО ..ХИМИЯ"
МОСКВА • 1966
УДК ИЛИ .5
Т.66
В книге рассмотрены применение жидкостной
экстракции в различных отраслях химической
промышленности, статика и кинетика процессов
экстракции, выбор экстрагентов и методов их
регенерации. Большое внимание уделено
диффузии и механизму массопередачи в системах
жидкость — жидкость, а также методам расчета
процессов жидкостной экстракции для
различных схем взаимодействия контактируемых
жидкостей (для трех- и многокомпонентных систем).
Специальная глава посвящена методике
изучения процессов экстракции в лабораторных усло~
виях. Подробно рассмотрены конструкции и
характеристики различных типов экстракционного
оборудования, дана технико-экономическая
оценка экстракторов, приведены задачи для
самостоятельной проработки.
Книга является ценным пособием для
широкого круга инженерно-технических и научных
работников химической, нефтехимической и
других отраслей промышленности, в которых
применяется разделение жидких смесей. Книга
окажет также большую помощь преподавателям,
аспирантам и студентам старших курсов высших
учебных заведений.
Перевод с английского
Ю. И. НОВАЛЕВА и В. Г. ТРУХАНОВА
СОДЕРЖАНИЕ
От редактора *
Предисловие — "■
ГЛАВА I
ВВЕДЕНИЕ. ЖИДКОСТНАЯ ЭКСТРАКЦИЯ
В ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
ГЛАВА IV
ВЫБОР ЭКСТРАГЕНТА
И ЕГО РЕГЕНЕРАЦИЯ
15
ГЛАВА II
ЗАКОНОМЕРНОСТИ
РАСПРЕДЕЛЕНИЯ ВЕЩЕСТВА
МЕЖДУ ЖИДКИМИ ФАЗАМИ 20
Двухкомпонентные системы 21
Трехкомпонентные системы 29
Четырехкомпонентные системы 59
Многокомпонентные системы — ~ 65
Условные обозначения 66
Литература 68
ГЛАВА III
РАСЧЕТ РАВНОВЕСИЯ В СИСТЕМАХ
жидкость — жидкость 70
__ _ 71
Идеальные растворы —
Неидеальные бинарные растворы '4
Неидеальные тройные растворы — - — 106
Расчет равновесия по коэффициентам активности 109
Другие методы расчета равновесия — 125
Условные обозначения - 136
Литература 138
139
Выбор экстрагента - — . 139
Регенерация экстрагента - — — 151
Условные обозначения - - 166
Литература - - - 166
ГЛАВА V
ДИФФУЗИЯ И МАССОПЕРЕДАЧА 168
Молекулярная диффузия — 168
Конвективная диффузия _ - 188
Массопередача через межфазовую поверхность 195
Условные обозначения 216
Литература - — 219
ГЛАВА VI
МЕТОДЫ РАСЧЕТА РАЗЛИЧНЫХ ВАРИАНТОВ
ПРОЦЕССА ЭКСТРАКЦИИ
(ТРЕХКОМПОНЕНТНЫЕ СИСТЕМЫ) 222
Ступенчатое взаимодействие исходной смеси с экс-
трагентом - — 222
Одноступенчатая экстракция - 228
Многоступеичатая экстракция при перекрестном
токе - 236
Дифференциальная экстракция 248
Противоточная многоступенчатая экстракция - — 255
Противоточная многоступенчатая экстракция с
флегмой 282
Условные Обозначения - - - 303
Литература 305
ГЛАВА VII
МЕТОДЫ РАСЧЕТА РАЗЛИЧНЫХ ВАРИАНТОВ
ПРОЦЕССА ЭКСТРАКЦИИ
(МНОГОКОМПОНЕНТНЫЕ СИСТЕМЫ) 306
Ступенчатое взаимодействие исходной смеси с
экстр агентом 306
Одноступенчатая экстракция 308
Многоступенчатая экстракция при перекрестном
токе 322
Дифференциальная экстракция 328
Противоточная многоступенчатая экстракция 332
Противоточная многоступенчатая экстракция с
флегмой . _ 343
Фракционная экстракция (многоступенчатая
противоточная экстракция двумя экстрагентами) 346
Упрощенный расчет многокомпонентных систем 379
Условные обозначения з81
Литература 382
глава viii
методы расчета высоты
непрерывных противоточных
экстракторов 384
Единицы переноса массы 385
Коэффициенты массопередачи 394
Общие и частные диффузионные сопротивления — 396
Условные обозначения _ 401
Литература 403
лабораторная и полупромышленная
экстракция 404
Методы лабораторной экстракции 405
Жидкостная экстракция в аналитической химии 423
Пилотные и полупромышленные установки 431
Условные обозначения 438
Литература - 440
ГЛАВА X
СМЕСИТЕЛЬНО-ОТСТОЙНЫЕ ЭКСТРАКТОРЫ
Общие положения
442
442
Аппараты с мешалками — - - 453
Проточные смесители - - — - *"5
Эмульсии и дисперсии — - *Э1
Смесительно-отстойные экстракторы в
промышленности 507
Условные обозначения - - 512
Литература - - 514
ГЛАВА XI
ЭКСТРАКТОРЫ С НЕПРЕРЫВНЫМ
(ДИФФЕРЕНЦИАЛЬНЫМ) КОНТАКТОМ 518
Общие характеристики
Аппараты с орошаемыми стенками (пленочные
аппараты) 530
Распылительные колонны - 531
Колонны с перегородками — - 546
Насадочные колонны - 547
Колонны с перфорированными (ситчатыми)
тарелками 562
Экстракторы с механическим перемешиванием фаз 573
Роторно-кольцевые экстракторы 574
Роторно-дисковые экстракторы (РДЭ) — 576
Экстракторы типа «Mixco» 582
Экстракторы Шайбеля - 584
Пульсационные экстракторы - 587
Центробежные экстракторы - 597
Условные обозначения - 600
Литература 603
ГЛАВА XII
НЕКОТОРЫЕ ВОПРОСЫ ЭКОНОМИКИ
ПРОЦЕССОВ ЭКСТРАКЦИИ 609
ГЛАВА XIII
ЭКСТРАКЦИЯ В ПРОМЫШЛЕННОСТИ
Экстракция в процессах нефтехимического синтеза
Экстракция в технологии жиров, масел н
родственных процессах
Экстракция в коксохимической промышленности
Экстракция в фармацевтической промышленности
Экстракция в технологии органических веществ
Экстракция в технологии неорганических веществ
Экстракция в процессах разделения металлов '/""65!
Избранная литература 659
ГЛАВА XIV
ЗАДАЧИ 662
Дополнительная литература 684
Предметный указатель 712
Именной указатель . 720
ОТ РЕДАКТОРА
экстракция из растворов с помощью
избирательных растворителей как один из
эффективных методов разделения и очистки
жидких смесей получила широкое
распространение лишь в последние 30—35 лет.
Несмотря на столь короткий срок,
достигнуты значительные успехи в области
разработки теории экстракционных процессов
и их аппаратурно-технологического
оформления.
Все возрастающий интерес к жидкостной
экстракции обусловлен в первую очередь
развитием ядерной техники, процессов
органического синтеза в химической
промышленности, а также процессов разделения и
очистки различных продуктов в
нефтеперерабатывающей промышленности.
Экстракционные методы разделения весьма
перспективны для тех производств, где
требуются высокая степень извлечения
чувствительных к повышенным температурам
веществ, регенерация ценных продуктов или
удаление вредных примесей из
разбавленных растворов, а также разделение смесей,
состоящих из компонентов с близкими
физико-химическими свойствами.
В периодической литературе ежегодно
появляются сотни статей, посвященных теории
и практике экстракционных процессов, но
до сих пор весьма ограничено число
трудов, обобщающих многочисленные
опубликованные материалы на современном
научном уровне.
Предлагаемая вниманию читателя книга
Р. Е. Трейбала «Жидкостная экстракция»,
8
выпущенная вторым изданием в Нью-Йорке
в 1963 г. (первое издание было
опубликовано в 1951 г.), представляет собой одну
из лучших монографий по вопросам
жидкостной экстракции, хотя формально книга
написана в качестве учебного пособия для
студентов высших учебных заведений.
Книга Р. Е. Трейбала выгодно отличается от
других монографий по жидкостной
экстракции, переведенных на русский язык '■2, тем,
что в ней наряду с характеристиками
фазовых равновесий и схемами проведения
экстракционных процессов подробно
рассмотрены вопросы массопередачи в
системах жидкость — жидкость и инженерные
расчеты экстракторов. В этом отношении
монография близка к изданной ранее серии
статей Г. Пратта3, но значительно
превосходит последние по широте и степени
обобщения материала. Книга снабжена весьма
полезными примерами и задачами, в
основном прикладного характера (для
самостоятельного решения), которые позволяют
закрепить и глубже освоить материал по
различным научно-теоретическим разделам.
При наличии ряда достоинств книга
Р. Е. Трейбала, если рассматривать ее в
свете требований, предъявляемых к
монографии по жидкостной экстракции, не
лишена определенных недостатков. Так,
например, как отмечалось4, в ней чрезмерно
подробно изложены методы расчета числа
теоретических ступеней для разных схем
1 Л. Аль дер с, Жидкостная экстракция, Издат-
инлит, 1957.
2 3. Зюлковский, Жидкостная экстракция в
химической промышленности, Госхимиздат, 1963.
3 Жидкостная экстракция, Сб. статей под ред.
А. Г. Касаткина, Госхимиздат, 1958.
4 Новые книги за рубежом, серия Б, № 1, 135
(1965).
8
экстракционного процесса и недостаточно
полно освещены некоторые важные
вопросы физико-химической гидродинамики
(дробление диспергируемой фазы на капли,
продольное перемешивание в сплошной и
диспергируемой фазах и т. д.), методы
аналитического расчета с использованием
счетно-решающих машин, вопросы теории
равновесий при взаимодействии электролитов
с неэлектролитами и др. Следует вместе с
тем отметить, что проблемы химии
экстракции в настоящее время настолько
разрослись и приобрели столь большое значение,
что им должны посвящаться специальные
монографии '.
В книге Трейбала совершенно
недостаточно отражены также исследования
советских ученых в области статики и кинетики
экстракционных процессов по созданию
высокоэффективных конструкций и
разработке методов расчета и моделирования
экстракторов2.
Подготавливая перевод к печати, мы
ограничились исправлением замеченных
опечаток и пересчетом большинства величин
в Международную систему единиц (СИ),
не внеся каких-либо дополнений или
изменений в оригинал. При этом учитывалось,
что советский читатель, желающий более
подробно ознакомиться с последними
достижениями в области изучения механизма
массопередачи, кинетики процессов экст-
1 В. В. Фомин, Химия экстракционных
процессов, Атомиздат, 1960.
2 Экстракция (теория, применение, аппаратура),
сб. статей, вып. 1 и 2, Госхимиздат, 1962; Процессы
жидкостной экстракции; Труды научно-технического
совещания, Гостоптехиздат, 1963; Процессы
жидкостной экстракции и хемосорбции, Сборник трудов 1Г
Всесоюзного совещания по жидкостной экстракции
и хемосорбции, изд. «Химия», 1966.
10
ракции и экстракционной аппаратуры,
сумеет воспользоваться отечественными
монографиями, посвященными
физико-химическим основам жидкостной экстракции • и
отдельным типам экстракторов
интенсивного действия 2>3.
Мы сочли также целесообразным
дополнить библиографию перечнем работ по
жидкостной экстракции, опубликованных в
отечественной литературе в последние годы.
Кроме того, приведен краткий указатель
зарубежной литературы по экстракции
жидкость — жидкость за период,
прошедший после выхода второго издания книги
Трейбала (1963—1965 гг.).
Можно надеяться, что книга Р. Е.
Трейбала в русском переводе окажет
существенную пользу широкому кругу инженеров,
научных работников, аспирантов и
студентов, соприкасающихся с теоретическими и
прикладными вопросами процессов
экстракции в системах жидкость — жидкость.
1 Б. И. Броунштейн, А. С. Железняк,
Физико-химические основы жидкостной экстракции,
нзд. «Химия», 1966.
2 И. Д. Шкоропад, И. В. Лысковцев,
Центробежные жидкостные экстракторы, Машгиз,
1962.
3 С. М. К а р п а ч е в а, Е. И. Захаров,
Л. С. Р а г и н с к и й, В. М. Муратов,
Пульсирующие экстракторы, Атомиздат, 1964.
С. 3. Каган
ПРЕДИСЛОВИЕ
Э а годы, прошедшие со времени выхода
в свет первого издания книги, процессы
жидкостной экстракции получили
значительное развитие. Жидкостная экстракция
сформировалась в научную дисциплину,
которую преподают в американских
учебных заведениях в качестве
самостоятельного курса студентам, изучающим
процессы и аппараты химической технологии, в
то время, как прежде даже в учебниках
вопросам жидкостной экстракции не
уделялось достаточного внимания.
В настоящее время жидкостная
экстракция является промышленным способом
разделения смесей, тогда как еще недавно
она применялась лишь в отдельных
производствах. Чем больше знаний будет
накоплено в области проектирования процессов
жидкостной экстракции, тем более
широкое и разнообразное применение получат
эти процессы.
Материал отдельных глав второго
издания книги приведен в соответствие с
современным состоянием исследований в области
жидкостной экстракции. Значительно
расширены главы, в которых рассматриваются
вопросы регенерации растворителя (экстр-
агента), а также проблемы экстракции в
многокомпонентных системах и экстракции
с двумя растворителями. Введены новые
главы, посвященные лабораторным
методам исследования жидкостной экстракции
и вопросам экономики экстракционных
процессов. Полностью обновлены приводимые
в конце книги задачи, причем все они уже
12
предлагались для решения студентам. Для
большинства задач приведены краткие
ответы.
Количество работ по жидкостной
экстракции, опубликованных со времени
выпуска первого издания, превышает количество
работ, выполненных за все предыдущие
годы. Поэтому перечень цитируемой
литературы в значительной степени сокращен.
Не представлялось более возможным
рассмотреть все известные
экспериментальные данные или даже существенную
часть их. При подготовке данного издания
чаще, чем раньше, приходилось решать, что
включать в книгу и что нет. Из-за
недостатка объема пришлось ограничиться лишь
кратким описанием промышленных
экстракционных процессов (глава XIII).
Несмотря на значительное число
опубликованных работ, было бы неправильным
полагать, что выяснены все основные
вопросы, связанные с применением жидкостной
экстракции. Экстракция все более широко
внедряется в промышленности,
возможности ее использования возрастают, и трудно
ожидать уже сейчас полного и
всестороннего разрешения всех проблем жидкостной
экстракции.
В интересной книге Дерри и Вильямса
«Краткая история технологии» описывается
применение жидкостной экстракции в
древнем Риме для отделения золота и серебра
от меди. Этот процесс был разработан в то
время, когда ничего не было известно о
коэффициентах активности, правиле фаз или
коэффициентах массопередачи.
Аналогичная ситуация возможна и в будущем при
внедрении жидкостной экстракции в новые
производства. Обгоняя теорию, практика
13
идет вперед, несмотря на неполноту
теоретических представлений, и, развиваясь,
ставит перед теорией новые задачи.
Поэтому читатель не найдет в книге ответов на
все интересующие его вопросы. Однако
автор надеется, что книга окажется по
крайней мере полезной; при этом его утешает
мысль о том, что аналогичные трудности
испытывали также создатели гораздо более
значительных произведений.
«Тот, кто пишет книгу, идет на очень
большой риск, так как нет ничего более не- .
возможного, чем сочинение труда, который
мог бы получить одобрение каждого
читателя».
Сервантес, «Дон-Кихот»
Автор благодарен многочисленным
фирмам и техническим обществам за
предоставленную возможность воспроизвести
принадлежащие им материалы,
Нью-Йоркскому университету за создание условий,
облегчивших подготовку этого издания, и,
конечно, всем химикам и инженерам, труды
которых составили основу данной книги.
Автор считает себя ответственным за
правильность освещения их работ в том виде,
в каком они представлены в книге.
Р. Трейбал
ВВЕДЕНИЕ.
ЖИДКОСТНАЯ ЭКСТРАКЦИЯ
В ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
уцг идкостная экстракция представляет со-
"■ бой метод разделения компонентов
раствора и является одним из массообменных
процессов химической технологии. Массооб-
менные процессы основаны на
неодинаковом равновесном распределении веществ
между двумя взаимнонерастворимыми
фазами. В случае жидкостной экстракции обе
фазы, между которыми происходит массо-
обмен, являются жидкими.
Таким образом, при разделении
экстракцией компоненты раствора распределяются
между двумя взаимно нерастворимыми
жидкостями. Например, уксусная кислота
может выделяться из водных растворов
экстракцией этилацетатом. Смесь уксусной
кислоты и ацетона можно разделить,
смешивая ее с водой и хлороформом; при этом
кислота будет предпочтительно переходить
в воду, а ацетон — в хлороформ
На практике обычно представляется
возможность выбора того или иного метода
разделения смесей. Существуют
химические методы, основанные на химическом
взаимодействии одного из разделяемых
веществ с соответствующим реагентом,
приводящие к выделению этого вещества из
раствора или его разрушению;
механические методы, например флотация, и
физические методы, применяемые в массообмен-
ных процессах. В последнем случае часто
оказывается возможным выбирать между
различными массообменными методами.
/"Все массообменные процессы можно
разделить на две большие группы. К
первой из них следует отнести процессы, в
которых осуществляется непосредственное
разделение смесей в отсутствие каких-либо
добавочных веществ, например растворителей
15
(дистилляция и ректификация, выпаривание, кристаллизация, а
также зонная плавка). Вторая группа массообменных
процессов, в которых разделение происходит с использованием
добавочного компонента, включает жидкостную экстракцию,
экстрактивную и азеотропную перегонку, абсорбцию, адсорбцию и
ионный обмену Характерной особенностью массообменных
процессов, относящихся к первой группе, является образование двух
фаз (между которыми происходит массообмен) при нагреваниг
или охлаждении исходного раствора. Так, при дистилляции
первоначальная смесь нагревается, и компоненты ее
распределяются между жидкостью и образующимся паром.
ГМассообменные процессы первой группы, в которых
осуществляется непосредственное разделение компонентов
раствора, обладают определенными преимуществами по сравнению с
массообменными процессами второй группы. Присутствие
дополнительного вещества, например избирательного растворителя
(экстрагента) при жидкостной экстракции, приводит к
усложнению процесса^ Растворитель должен быть химически
инертным и не вызывать коррозии аппаратов; это затрудняет выбор
конструкционных материалов для экстракционной аппаратуры.
Иногда приходится считаться с необходимостью иметь в
распоряжении значительные количества растворителя, что связано
с относительно большими затратами; нужно также возмещать
неизбежные потери растворителя в процессе. После экстракции
извлекаемый компонент снова находится в растворе, и для его
выделения необходима та или иная система регенерации
экстрагента. Все это увеличивает стоимость процесса разделения.
Кроме того, при разделении смесей с помощью массообменных
процессов второй группы увеличивается вероятность
загрязнения получаемых продуктов посторонними примесями.
Вместе с тем благодаря присутствию вещества,
отличающегося по химическим свойствам от компонентов исходной смеси,
часто осуществляют весьма тонкое разделение смесей, которое
во многих случаях невозможно выполнить другими способами.
При перегонке химический состав пара и жидкости одинаков,
поэтому в данном процессе нельзя ожидать большой разницы
в коэффициентах распределения компонентов. Действительно,
этим методом обычно вообще невозможно осуществить
достаточно полного разделения смеси.
ГПри жидкостной экстракции, как правило, имеется
возможность подбора из многочисленных экстрагентов растворителя,
обеспечивающего наилучшее разделение. С помощью
жидкостной экстракции можно разделить компоненты смеси по их
химической природе, а не по физическим свойствам (например, по
величинам давлений их паров). Это часто обусловливает
высокую эффективность жидкостной экстракции как метода
разделения смесей.,
16
Жидкостная экстракция применяется в тех случаях, когда
прямые методы разделения смесей непригодны или когда,
несмотря на недостатки экстракционных способов разделения,
затраты на другие методы оказываются большими. Наряду с
экстракцией можно исгтдазовать и другие массообменные
процессы второй гругапы.ГНиже перечислены основные области
возможного применения жидкостной экстракции.
1. Экстракцию можно использовать вместо методов
непосредственного разделения смесей (например, вместо
ректификации), если последние стоят дороже. Сюда относятся:
а) Разделение близкокипящих жидкостей. Типичным
примером служит разделение бутадиена (^Кип. = —4,75° С) и бутиленов
(^кип. от —5 до —6° С), которое трудно осуществить дистилля-
ционными методами; однако разделение этих компонентов мож-
V^ho легко провести экстракцией медно-аммиачным ацетатным
^ раствором.
гсч б) Разделение жидких смесей с малой относительной лету-
^~ честью компонентов. Так, например, в системе вода — уксусная
^кислота, несмотря на сравнительно большую разницу
темперами тур кипения, относительная летучесть невелика. Кроме того,
^^при выделении уксусной кислоты из разбавленных водных
растворов перегонкой необходимо отгонять значительные объемы
воды. Гораздо экономичнее извлекать уксусную кислоту из
разбавленных водных растворов экстракцией этилацетатом или
смесью этилацетата и бензола.
в) Разделение соединений, температуры кипения которых
столь велики, что их обычно разделяют при помощи
высоковакуумной, или молекулярной дистилляции. Например, высшие
жирные кислоты и витамины можно выделять из природных
масел экстракцией жидким пропаном вместо применения для
этой цели глубокого вакуума (молекулярной дистилляции).
г) Удешевление процесса концентрирования растворов
выпариванием. Так, бензойная кислота может быть выделена из
разбавленных водных растворов выпариванием воды (теплота
испарения 2,26-106 дж/кг). Выгоднее, однако, предварительно
экстрагировать бензойную кислоту бензолом. При этом можно
повысить концентрацию кислоты в растворе в 10 раз (теплота
испарения бензола составляет лишь 0,4- 106 дж/кг).
д) Замена фракционной кристаллизации. Так, стоимость
разделения тантала и ниобия фракционной кристаллизацией
двойных фторидов калия и этих металлов очень велика.
Поэтому в настоящее время широкое применение получил метод
разделения тантала и ниобия, основанный на экстракции их ме-
тилизобутилкетоном из солянокислых растворов.
2. Экстракцию можно применять для разделения смесей в
тех случаях, когда методы непосредственного разделения
компонентов оказываются непригодными:
2 Р. Е. Трейбал ,- , .^.,и - й"Т^7^ГгП "
1СН#ЗН.'.Й ФОНД j
Сеаа<дч1*Л1,ского ВЬ.-АИУ^
а) Разделение веществ, чувствительных к воздействию
повышенных температур. Пенициллин и большую часть других
антибиотиков, находящихся, например, в разбавленном
ферментативном растворе, нельзя концентрировать выпариванием из-за
разрушения этих соединений при температурах кипения воды.
Для концентрирования подобных соединений успешно
используют экстракцию органическими растворителями; при этом
антибиотики очищаются также от других продуктов
ферментативного превращения.
б) Разделение веществ, образующих азеотропные смеси. Так,
метилэтилкетон и воду можно разделить экстракцией воды
концентрированным рассолом хлорида кальция или экстракцией
кетона трихлорэтаном. Как известно, азеотропные смеси не
могут быть разделены дистилляцией.
в) Разделение сложных смесей на классы соединений
одинакового химического строения, температуры кипения которых
перекрывают друг друга. Например, ароматические углеводороды
(бензол, толуол, ксилолы) отделяются от предельных
углеводородов, кипящих в том же интервале температур, экстракцией
жидкой двуокисью серы, фурфуролом или диэтиленгликолем.
3. Экстракцию можно применять вместо более дорогих
химических методов разделения. Так, например, уран и ванадий
можно раздельно извлекать из растворов после выщелачивания
этих металлов из руд (и при этом отделять от других
элементов) экстракцией растворами алкилфосфорных кислот в
керосине при различных рН.
4. Методы жидкостной экстракции можно использовать
также в других процессах, где они еще не получили широкого
распространения. Выход продуктов реакции в жидкой фазе
можно в значительной степени повысить непрерывной экстракцией
одного из конечных продуктов. Теплообмен при
непосредственном контакте между двумя несмешивающимися
жидкостями протекает весьма интенсивно, что обусловлено
отсутствием разделяющей теплоносители твердой стенки. Процесс
переноса тепла при наличии стенки замедляется, причем
особенно значительно при отложении загрязнений на ее
поверхностях. Преимущества теплообмена при непосредственном
контакте жидкостей используют в промышленном масштабе в
процессе получения глицерина и жирных кислот гидролизом жидких
жиров при высоких температур_ахЛ
Указанные выше случаи применения экстракции
рассматриваются в книге главным образом в связи со схемами
экстракционных процессов и вопросами проектирования экстракционной
аппаратуры.
Жидкостная экстракция, как и другие массообменные
процессы, находит применение в лабораторном и промышленном
масштабах. Несмотря на то, что основное внимание будет
18
уделено промышленному использованию экстракционных
процессов, лабораторные методы экстракции также представляют
интерес.
Лабораторная экстракция применяется для химического
анализа сложных смесей и является первоначальной стадией
разработки производственных экстракционных процессов. Разделение
многих трудноразделяемых смесей впервые было осуществлено
в лаборатории для аналитических целей и стало представлять
промышленный интерес, лишь когда появилась потребность
в производстве одного из разделяемых веществ. Необходимость
оценивать технические и экономические аспекты применения
результатов лабораторной экстракции в промышленном
масштабе возникает довольно часто. По этим причинам в книге
рассматриваются также методы лабораторной экстракции.
2»
ЗАКОНОМЕРНОСТИ
РАСПРЕДЕЛЕНИЯ ВЕЩЕСТВА
МЕЖДУ ЖИДКИМИ ФАЗАМИ
При исследовании фазовых равновесий
большое значение имеют правило фаз1
и закономерности распределения.
Правило фаз. Для систем жидкость —
жидкость это правило может быть
выражено в простой форме:
F = N— ф + 2 (11,1)
где F — число степеней свободы, или число
независимых переменных (температура,
давление и концентрация), которые должны
быть фиксированы для того, чтобы
равновесная система была полностью определена.
N — число компонентов, или наименьшее
число независимо изменяющихся
компонентов, необходимое для выражения состава
каждой фаЭы,
Ф — число фаз.
Фазой называется гомогенная часть
системы, ограниченная поверхностями
раздела, которая может механически отделяться
от остальной части системы. Вывод
правила фаз и более подробное его
рассмотрение приводятся в специальной
литературе2.
Правило фаз применимо только к
равновесным системам, причем каждое
дополнительное ограничение, накладываемое на
систему, уменьшает величину F на единицу.
Законы распределения. Эти законы в
основном эмпирические и выражают
обобщенные зависимости между
концентрациями компонентов в разных фазах
равновесной системы. К сожалению, полученные
до сих пор обобщения не являются
универсальными.
В дальнейшем при изображении
равновесных соотношений между фазами с
помощью фазовых диаграмм будут иопользо-
20
ваться следующие обозначения. Чистые компоненты будут
обозначены буквами А, В я С, а растворы — другими буквами,
например G, Н и т. д. Эти символы на фазовой диаграмме
указывают состав раствора. Так, А означает А кг вещества,
определенного точкой А фазовой диаграммы, G—G кг раствора,
состав которого выражается точкой G на диаграмме, и т. д.
Концентрации в весовых долях будут обозначены буквой X с
соответствующими индексами. Первая буква индекса относится
к компоненту, концентрация которого указывается, вторая
буква индекса обозначает раствор, состав которого описывается,
или компонент, количественно преобладающий в этом
растворе.
Так, например, приводимые ниже обозначения имеют
следующий смысл:
Xcg — весовая доля С в растворе G;
ХАВ — весовая доля А в фазе компонента В;
Хвв — весовая доля В в фазе компонента В и т. д.
ДВУХКОМПОНЕНТНЫЕ СИСТЕМЫ
Двухкомпонентные жидкие системы можно
классифицировать в зависимости от того, полностью или только частично
смешиваются друг с другом компоненты системы. Для
жидкостной экстракции представляют интерес лишь системы с
ограниченной взаимной растворимостью. Для практических целей
можно иногда считать, что компоненты полностью
нерастворимы, но в действительности все жидкости в той или иной
степени растворимы друг в друге.
Рассмотрим жидкости А и В, частично растворяющиеся друг
в друге. Если сначала добавлять А в небольших количествах к
В, жидкости будут полностью растворяться. В этом случае
N=2 и ф = 2 (жидкость и пар), поэтому F—2 — 2 + 2 = 2. Система
бивариантна и температура, давление и концентрация могут в
определенных пределах независимо изменяться попарно без
изменения числа фаз. Так, например, температура и концентрация
могут независимо изменяться без появления новой фазы, но
давление будет фиксировано до тех пор, пока жидкость и пар
находятся в равновесии, и не сможет произвольно изменяться.
По мере добавления к раствору компонента А в конце
концов будет достигнут предел растворимости А ъ В при данной
температуре; дальнейшее добавление А приведет к
возникновению двух жидких фаз, представляющих сабой насыщенные
растворы А в В и В в А. Появление еще одной жидкой фазы
делает систему моновариантной, и теперь только одна из основных
переменных может произвольно изменяться. Например,
добавление при постоянной температуре еще большего количества А
-вызовет лишь изменение относительных количеств фаз без
21
I
!
ft
ЮОУоА
СостаВ
100%B
Рис. 1. Диаграмма температура —
состав для бинарной системы
с верхней критической
температурой растворения:
/ — одна жидкая фаза; // — две жидкие
фазы.
изменения их состава и давления
паров. При достаточном
добавлении компонента А система
становится, однако, снова
однофазной, когда компонент В
оказывается полностью растворенным в
А. Таким образом, в
определенном интервале составов системы
в целом существуют (приданной
температуре) две жидкие фазы
постоянного состава,
представляющие собой насыщенные
растворы. Изменение состава этих
насыщенных растворов с
изменением температуры удобно
изображать графически.
На рис. 1 изображены
составы насыщенных жидких фаз,
находящихся в равновесии друг с
другом, в зависимости от
температуры для системы, подобной
описанной выше. На этой
диаграмме давление не является постоянным, а равно
равновесному давлению паров; состав пара не показан. Кривая KDM
изображает зависимость состава насыщенных растворов
компонента В в А от температуры, кривая LGM — ту же
зависимость для растворов А в В. Область диаграммы над кривой
соответствует смесям, образующим одну жидкую фазу (I), a
область под кривой представляет собой смеси, которые
образуют два взаимно насыщенных жидких раствора (II).
Рассмотрим смесь, средний состав и температура которой
заданы точкой S. Два насыщенных раствора, образованных этой
смесью, обозначены точками D и G; такие растворы называют
сопряженными, а горизонтальную линию, соединяющую точки
D и G — хордой равновесия или конодой (либо просто нодой).
Смесь, средний состав которой соответствует какой-либо
точке на линии DG между D и G, при равновесии образует одни
и те же насыщенные растворы (D и G); однако относительные
количества этих растворов зависят от среднего состава смеси.
Для смеси S материальный баланс в отсутствие паровой фазы
выражается равенством:
S = D + G (11,2)
где S, D н G — весовые количества растворов.
Если это уравнение решить совместно с уравнением
материального баланса для компонента В
SXBS = DXBD + GXBQ (ИЗ)
22
получим следующее выражение:
D XRr — XRV GS
— = _ва §s_ = ^^ (II 4)
G Xbs-Xbd SD
Из зависимости (II, 4) следует, что относительные весовые
количества насыщенных фаз обратно пропорциональны длинам
отрезков соответствующей хорды равновесия. Это правило (его
обычно называют правилом рычага) дает удобный
графический метод составления материального баланса. Оно применимо
также в том случае, если составы выражены в мольных долях,
а количества смеси и насыщенных растворов — в молях.
Системы с верхней критической температурой растворения.
Примером такой системы может служить система бутанол —
вода (см. рис. 1). Для подобных систем растворимость
компонента А в В и В в А увеличивается с возрастанием температуры.
При некоторой температуре сопряженные растворы становятся
идентичными по составу; соответственно поверхность раздела
между ними исчезает. Эта температура, называемая
критической температурой растворения (К.-Т.Р.), на рис. 1 обозначена
точкой М. Выше данной температуры компоненты А и В
смешиваются друг с другом в любых отношениях. Во многих случаях
верхняя часть кривой растворимости весьма полога3, например,
для системы фенол — вода. Несмотря на это, точка М является
максимумом кривой растворимости, хотя, как правило, она не
соответствует среднему составу смеси, и чаще всего кривая
растворимости несимметрична.
К.Т.Р. соответствует точке, в которой сходятся две ветви
кривой растворимости. Линия постоянной температуры
(изотерма) является касательной к кривой при этой температуре.
Согласно правилу фаз, применительно к указанной точке имеем:
N=2; <p = 3 (две жидкости и пар); F = 2 —3 + 2—1 =0. Число
степеней свободы уменьшается на единицу вследствие ограни*
чения, накладываемого на систему: в данной точке фазы имеют
один и тот же состав. Это ограничение приводит к тому, что
в присутствии паровой фазы система в точке М инвариантна.
Если внешнее давление выше давления пара системы, последняя
находится в жидком состоянии. Величина К.Т.Р. в этом случае
зависит от давления.
Системы с нижней критической температурой растворения.
На рис. 2 изображена типичная зависимость температура —
состав для систем, у которых взаимная растворимость
компонентов возрастает с уменьшением температуры. Примером такой
системы является система триэтиламин — вода. Кривая раство~
римости охватывает область, в которой образуются
сопряженные растворы. Область под кривой соответствует смесям,
образующим один жидкий раствор. Для таких систем (как и для
систем с верхней К-Т.Р.) площадь между ветвями кривой
23
WO%A
Состав
ЮО%В
растворимости можно
представить заполненной
горизонтальными хордами, соединяющими
сопряженные растворы. Самая
нижняя точка кривой
соответствует К- Т. Р. системы.
Применение правила фаз к различным
частям диаграммы приводит к тем
же выводам, что и для систем с
верхней критической
температурой растворения.
Системы с верхней и нижней
критическими температурами
растворения. Для некоторых
частично смешивающихся жидкостей
полная растворимость возможна
как выше верхней К. Т. Р., так и
ниже нижней К. Т. Р. Поэтому
их кривая растворимости имеет
вид, изображенный на рис. 3.
Известно несколько примеров,
когда составы смесей, соответствующих верхней и нижней
критическим точкам, почти одинаковы. Однако это не является
общим правилом.
Системы без критической температуры растворения.
Большое число бинарных жидких смесей образует системы без
критических точек. В таких системах при изменении температуры
(до наступления полной взаимной растворимости компонентов)
либо выпадает твердая фаза, либо достигаются критические
условия, при которых состав и плотность пара становятся
одинаковыми с составом и плотностью одной из жидких фаз. Приме-
Рис . 2. Диаграмма температура —
состав для бинарной системы
с нижней критической
температурой растворения:
/ — одна жидкая фаза; // — две жидкие
фазы.
Таблица 1
Системы с верхней критической температурой растворения
А
Вода
Вода
Вода
Вода
Двуокись серы
Двуокись серы
Компоненты
В
Метилацетат
«-Бутанол
Фурфурол
Фенол
Циклогексан
м-Гексан
К.Т. Р.
"С
10S
125,2
122,7
66,0
13,5
10,1
Состав смесн
в критической
точке
вес. % *
52,5
32,5
51
34,0
55,6
71
• Содержание компонента В.
24
Таблица 2
Системы с нижней критической температурой растворения
Компоненты
А
Вода
Вода
Вода
Вода
в
Диэтиламин
Триэтиламин
1-Метилпипериднн
4-Метилпиперидин
K.T.P.
°с
143,5
18,7
48,3
189,5
Состав смеси
в критической точке*
вес, %
37,5
50
16,7
36,2
• Содержание компонента В.
I
I
ром может служить система эфир — вода, и, по всей
вероятности, к этой категории относятся системы, образованные
практически нерастворимыми жидкостями. В табл. 1—3 приведены
несколько примеров систем описанных выше типов.
Влияние давления. Приведенные выше диаграммы
температура— состав соответствуют давлению системы, т. е.
равновесному давлению паров, которое изменяется с температурой, а при
существовании одной жидкой фазы — также с составом. Однако
опытом установлено, что изменение растворимости
малорастворимых жидкостей, вызванное внешним давлением, очень
невелико и в большинстве случаев им можно пренебречь. Характер
влияния давления можно определить из принципа Ле Шателье:
если растворение компонентов
сопровождается увеличением
объема, повышенное давление будет
способствовать уменьшению
растворимости, и наоборот. В табл.
4 приведены примеры изменения
критической температуры
растворения с изменением давления.
Заметный эффект может вызвать
быстрое центрифугирование4, при
котором создается высокое
гидростатическое давление.
Если диаграмму
температуру—состав для системы с
верхней К.Т.Р. построить при
постоянном давлении, меньшем равно-
/00% А
Состав
100% В
Весного давления паров при Рис. 3. Диаграмма температура—
Л.1.Р., то полная растворимость состав для бинарной системы
* Жидком состоянии не будет до- с верхней и нижней критическими
<**игнута. С повышением темпе- температурами растворения:
1>атуры, когда давление системы ф^на жидкая фаза: "~две жидкне
25
Таблица 3
Системы, имеющие верхнюю и нижнюю критические температуры
растворения
Компоненты
А
Вода
Вода
Вода
Вода
Вода
Вода
в
Моно-н-бутиловый эфир эти-
ленгликоля
Моноизобутиловый эфир эти-
ленгликоля
2-Пропиловый эфир 1,2-про-
пиленгликоля
2,6-Диметилпиридин
Никотин
Метилэтилкетон
К.Т.Р.
°с
49,1
128
24,5
150,4
42,6
162
45,3
164,9
60,8
208
—6
150
Состав смеси
в критической точке *
вес. %
24,8
24,8
24,6
28
' 35
35
27,2
33,8
29
32
31
45
* Содержание компонента В.
Таблица 4
Влияние давления на критическую температуру растворимости *
Компоненты
А | В
Вода
Вода
Вода
Вода
Нитрометан
Пропионитрил
Метилаль
Сукцинонитрил
Температура
103,3
111,0
160,3
52,3
Интервал
давления
am
1—150
5—165
20—64
10—160
At/hP
—Р/Ю8
-0,02
—0,21
—0,003
» International Critical Tables, V.1II, McOrow-Hlll Book Company Inc., New York, 1928.
достигнет значения, при котором построена диаграмма,
начнется процесс испарения, и тогда следует рассматривать
равновесие с учетом паровой фазы. Так, на рис. 4 приведена
диаграмма температура — состав для системы анилин — вода (при
745 мм рт. ст.). Ветви кривой растворимости, сходящиеся в
точке М (167,5° С), прерываются при температуре 99° С,
которой соответствует начало испарения. Такой характер имеют
диаграммы для систем, у которых давление паров равновесных
жидких фаз больше давления паров чистых компонентов.
Когда давление паров в области существования двух жидких фаз
имеет значение, промежуточное между величинами давлений
паров чистых компонентов, для системы пар — жидкость
наблюдается равновесие иного типа, но кривая растворимости и
в этом случае прерывается,
2$
Влияние диспергирования.
Если одна из жидкостей
диспергирована в другой в виде
очень мелких капель,
растворимость из-за большой
кривизны поверхности
увеличивается. Этот эффект
количественно описывается
уравнением
где f—летучесть в
диспергированном состоянии;
f° — летучесть в недиспер-
гированном состоянии;
dp — диаметр капли.
Диспергирование
применяется в большей части
процессов жидкостной
экстракции, однако размер капель
обычно слишком велик для
того, чтобы оказать заметное
влияние на растворимость.
Пример П-1. При каком размере капель растворимость бензола,
диспергированного в воде, увеличится на 5%?
Решение. При 20° С (7=293° К) межфазовое натяжение между
бензолом и водой а=0,035 н/м (для фаз, взаимно насыщенных при обычной
растворимости); мольный объем бензола К=0,0889 м3/кмоль, R=
=8,314 • 103 дж/кмоль°К. При увеличении растворимости на 5% отношение
flf°=l,05. Подстановка в уравнение (11,5) дает dp = 1,05 • 10~7 м, или 0,1 мк.
Считается, что стабильные эмульсии, которые не могут использоваться в
экстракции, имеют размер капель 1—1,5 мк. Отсюда ясно, что значительного
изменения растворимости вследствие диспергирования, практически не
происходит.
Влияние загрязнений на критическую температуру
растворения. Добавление даже небольших количеств третьего
компонента может значительно изменить К.Т.Р. Так, например, при
добавлении 0,2% воды к ледяной уксусной кислоте К.Т.Р. системы
кислота — циклогексан изменяется от 4,2 до ~8,2°С.
На этом явлении основаны некоторые методы анализа.
Такие системы следует, строго говоря, рассматривать как трехком-
понентные. Распределение загрязнений между равновесными
фазами обычно изменяет не только К.Т.Р., но и критический
состав смеси. Так, например, можно найти содержание
ароматических углеводородов в смеси с парафиновыми, определяя
температуру, при которой равные объемы раствора углеводородов
и анилина полностью смешиваются («анилиновая точка»).
Рис. 4. Диаграмма температура -—
состав для бинарной системы при
давлении, меньшем давления паров
при критической температуре
растворения:
/—пар; //—жидкость I+пар; ///—жидкость 1;
IV — жидкость 1 -(-жидкость 2; К-жидкость 2;
VI — жидкость 2 +пар.
27
Из-за изменения критического состава произвольное
использование метода смешения равных объемов жидкостей приводит
к тому, что наблюдаемая температура смеси может отличаться
от истинной К.Т.Р. Однако ввиду того, что кривые
растворимости имеют пологую верхнюю часть, отличие этой температуры
от истинной К.Т.Р. незначительно. Таким же способом можно
легко анализировать состав растворов воды и окиси дейтерия,
наблюдая смешиваемость раствора с 3-пиколином. Этот
растворитель полностью смешивается с водой, но ограниченно
растворяется в окиси дейтерия, образуя замкнутую кривую
растворимости5 (К.Т.Р. равны 38,5 и 117,0° С).
Если загрязнения распределяются приблизительно
равномерно между жидкостями, ограниченно растворимыми друг в
друге, область несмешиваемости в общем случае уменьшается
(верхняя К-Т.Р. понижается). Если же загрязнения
концентрируются в одной из фаз, область ограниченной растворимости
увеличивается6 (верхняя К.Т.Р. повышается). Последнее
явление представляет собой один из видов «высаливания».
Экспериментальное определение кривой растворимости.
Обычно применяют два основных метода определения кривой
растворимости, основанные на проведении эксперимента при
постоянном составе смеси или при постоянной температуре. По
первому способу гетерогенную смесь двух компонентов
известного среднего состава взвешивают в толстостенной пробирке.
Пробирку запаивают, оставляя некоторое свободное
пространство, чтобы жидкость в ней могла расширяться. Запаянную
пробирку встряхивают в бане, температуру в которой медленно
повышают (или понижают) до тех пор, пока два жидких слоя
не превратятся в однофазный раствор. Опыт повторяют при
различных составах смеси, получая в результате каждого опыта
одну точку на кривой растворимости. Для контроля опыт
можно повторить в обратном направлении, отмечая температуру,
при которой однофазный раствор образует гетерогенную смесь.
Температуру обычно можно определить с точностью до 0,1 —
0,01° С.
В случае определения растворимости при постоянной
температуре некоторое количество одного из компонентов «титруют»
в термостате другим компонентом до тех пор, пока при
встряхивании не будет наблюдаться легкое помутнение. Обычно
нахождение кривой растворимости при постоянной температуре
более удобно для определения участков кривой растворимости,
которые удалены от К.Т.Р., а также участков кривой, где она
почти параллельна оси температур. Метод нахождения кривой
растворимости при постоянном составе смеси удобен для
определения растворимости вблизи К.Т.Р., где кривая
растворимости почти параллельна оси составов. Результаты, получаемые
обоими методами, обычно хорошо согласуются, так как влия-
28
'йие давления на растворимость в большей части случаев
нерачительно.
Иногда можно определить растворимость компонентов
химическим анализом. По методу, предложенному Хиллом 7,
приготовляют две гетерогенные смеси разного, но известного состава
и выдерживают их при постоянной температуре до наступления
равновесия. Состав фаз в том и другом случае одинаков,
поэтому по известным объемам и количествам исходных смесей
можно рассчитать составы и плотности равновесных растворов.
Экспериментальные методы определения растворимости при
обычных температурах и давлениях подробно описаны в обзоре
Циммермана8, а при высоких температурах и давлениях — в
работе Кураты и Кона 9.
Опытные данные о растворимости. Сведения о взаимной
растворимости или нерастворимости большого числа органических
жидкостей приведены Друри 10. Аналогичные сведения для
углеводородов вместе с данными о критических температурах
растворения опубликованы Фрэнсисом "• 12.
ТРЕХКОМПОНЕНТНЫЕ СИСТЕМЫ
Влияние давления на равновесие в системах жидкость —
жидкость незначительно. Поэтому можно ограничиться
рассмотрением конденсированных фаз и исследовать зависимость
равновесия только от температуры и состава смеси. Чаще всего
равновесие трехкомпонентных систем представляют графически.
Концентрации изображают в треугольных координатах, а
температуры откладывают по оси, перпендикулярной к плоскости
треугольника составов.
Треугольные координаты. Состав тройных смесей можно
изображать при помощи равностороннего треугольника,
используя следующее его свойство: сумма перпендикуляров,
опущенных из любой точки внутри треугольника на его стороны, равна
высоте треугольника'. Длина высоты треугольника
приравнивается 100%, а длины соответствующих перпендикуляров —
процентному содержанию каждого из трех компонентов в смеси
(рис. 5). Вершины треугольника представляют собой
соответственно чистые компоненты А, В и С. Любая точка на боковой
стороне треугольника изображает бинарную смесь
компонентов. Так, точка G обозначает смесь, содержащую 40%
компонента С и 60% компонента В. Точки внутри треугольника
представляют собой тройные смеси: например, в смеси,
обозначенной точкой М, содержится 20% компонента А, 40% компонента
а и 40 /о компонентов С. При некоторых расчетах, выполняемых
с помощью треугольных диаграмм, вводят понятие о
гипотетических смесях, состав которых изображают точками, лежащими
вне треугольника ABC,
28
Если D кг смеси, представленной точкой D на рис. 5,
смешать с Е кг раствора, обозначенного точкой Е, то полученная
смесь будет иметь состав, характеризующийся точкой F на
прямой DE, причем положение точки F на прямой DE определяется
из соотношения
-£—S- (11,5а)
D EF
Справедливость зависимости (II, 5а) может быть доказана
с помощью следующих рассуждений (рис. 6). Из общего
уравнения материального баланса процесса смешения
D + E = F (11,6)
и материального баланса по компоненту С
DXC.n+EXC.E = FX>
СЕ
CF
исключая F, получаем:
CF
D
JSJL
XCE~XCF
Но XCf = FN, XCd=DM и ХСЕ=ЕК; следовательно
Е ^ Ш — 'DM _ 51 Ш
D ~ ~~ -
EK — FN
ES
EF
(", 7)
(11,8)
(11,9)
Аналогично, если из смеси F тем или иным способом
отделить часть ее, состава Е, остаток будет иметь состав,
соответствующий точке D, лежащей на прямой EF, причем и в этом
случае применима зависимость (II, 5а).
Таким образом, расчет количественных соотношений между
фазами можно выполнять либо при помощи измерения
отрезков на треугольной диаграмме, либо аналитически по
уравнению (11,8).
А,ии аи so со го
Рис. 5. Треугольная диаграмма.
AM N К
Рис. 6. Правило рычага.
Из зависимости (II, 5а) следует, что все точки, лежащие на
одной прямой (например, AG на рис. 5), образуют смеси с
постоянным отношением двух компонентов (в данном случае С
и В) при меняющемся количестве третьего компонента. Чем
ближе точка Н к вершине А, тем богаче компонентом А смесь
Я; если компонент А полностью удалить из этой смеси, оста-
не'тся бинарный раствор, состав которого определяется
точкой G.
Тройные системы, применяемые в процессах жидкостной
экстракции. Если все три компонента смешиваются в любых
отношениях, образуемая ими система не может быть
использована в процессах жидкостной экстракции.
Системы с ограниченной взаимной растворимостью
компонентов, представляющие интерес для жидкостной экстракции,
могут быть классифицированы следующим образом:
Тип I. Системы с одной парой частично смешивающихся
жидкостей.
Тип II. Системы с двумя парами частично смешивающихся
жидкостей.
Тип III. Системы с тремя парами частично смешивающихся
жидкостей.
Тип IV. Системы, образующие твердые фазы.
Во всех системах, за исключением системы типа IV, все
компоненты при данной температуре находятся в жидком
состоянии.
Тип I. Системы с одной парой частично
смешивающихся жидкостей. Системы этого типа
встречаются наиболее часто и имеют вид, представленный изотермой
на рис. 7. В системах такого рода пары жидкостей А—С и
В—С при данной температуре смешиваются во всех
отношениях, а жидкости А и В — частично. Точки D и Е представляют
собой насыщенные растворы в
этой бинарной системе.
Типичным примером является
система бензол
(Л)—вода(£)—этанол (С). Все смеси
компонентов, соответствующие точкам,
лежащим вне площади,
ограниченной кривой DNPLE,
представляют собой гомогенные
однофазные жидкие растворы, а
смеси, отвечающие точкам
внутри^ области, ограниченной
этой кривой и линией DE,
образуют двухфазные растворы.
Кривая DNPLE
представляет собой насыщенные рас-
Рис. 7. Тройная система жидкость —
жидкость (тип I).
31
творы и называется кривой растворимости или бинодальной
кривой. Обычно эта кривая выпукла вверх. Некоторые
описанные в литературе случаи изменения направления выпуклости на
отдельных участках бинодальной кривой объяснялись влиянием
четвертого компонента, примесей или компонента,
образующегося при химическом взаимодействии трех других компонентов 13.
Если смесь состава М образует два несмешивающихся
жидких раствора соответственно составов L и N, точка М лежит
на прямой LN, являющейся хордой равновесия. Все смеси,
состав которых соответствует точкам на линии LN, образуют
равновесные растворы одного и того же состава, а их
относительные количества могут быть определены по правилу рычага.
Область существования гетерогенных растворов можно
представить как бы заполненной бесчисленным множеством хорд
равновесия, из которых лишь несколько показано на рис. 7.
Обычно хорды равновесия непараллельны и медленно изменяют
в одном направлении свой наклон с изменением концентрации.
Случаи изменения направления наклона на противоположное
довольно часты. Примером такой системы служит система
бензол— пиридин — вода; подобные системы называются «солыо-
тропными» 14.
Сольютропия часто исчезает, если концентрации выражать
не в весовых, а в мольных долях15, особенно в тех случаях,
когда одним из компонентов,
составляющих взаимно
нерастворимую пару, является вода. Для
системы, показанной на рис. 7,
компонент С распределяется
неравномерно между фазами; его
Рис. 8. Тройная система без
тройной критической температуры
растворения.
Рис. 9. Изотермы растворимости в
системе без тройной критической
температуры растворения.
32
концентрация в фазе В будет
больше. По мере увеличения
концентрации С взаимная
растворимость А и В возрастает
и в точке Р (критическая
точка) обе ветви кривой
растворимости сходятся. Как
правило, эта точка не является
максимумом на бинодальной
кривой. Хорды равновесия с
увеличением концентрации
компонента С становятся короче и
в точке Р исчезают. В точке Р
существуют две жидкие фазы
одинакового состава и
плотности, поэтому данная точка
представляет собой истинную
критическую точку.
Применение правила
фаз. Согласно правилу фаз,
в трехкомпонентных системах
F=5—ф, а при постоянных
температуре и давлении F=
= 3—ф. Для смесей,
состоящих из одной жидкой фазы, рения.
F=2, т. е., чтобы определить
систему, необходимо задать два состава. При наличии двух
жидких фаз система моновариантна. В критической точке
ввиду ограничения, связанного с идентичностью фаз, система
инвариантна. Если для бинарных систем критическую
температуру растворения определяют при фиксированном давлении,
критическую точку в тройных системах находят только при
одновременно фиксированных температуре и давлении.
Влияние температуры. При постоянном давлении и
переменных температуре и составе тройная система
изображается треугольной призмой. Для систем рассмотренных выше
типов представляют интерес два случая:
.'■ Система без тройной К-Т.Р. (рис. 8). Кривая в плоскости
АВ — АВ выражает зависимость растворимости от температуры
для бинарной смеси А и В- К.Т.Р. этой бинарной смеси
—точка Р5. Точки Pit P2, Р3 и Р4 — критические точки бинодальных
кривых для различных температур; линия, которая соединяет
указанные точки, идет до точки Р5, являющейся одной из би-
иак-Нт"п>КТР- Эта линия не имеет максимума внутри призмы
AR-l-AR достигает наибольшего значения только в плоскости
вя /тпТ' е" В отсУтствие компонента С. Следовательно,
тройная К. 1.Р. в этой системе отсутствует,
3 Р. Е. Трейбал gg
Рис. 10. Тройная система с тройной
критической температурой раство-
'Ч
На рис. 9 показаны проекции бинодальных кривых на
основание призмы. Примером систем такого типа является система
дифенилгексан — докозан — фурфурол 16.
2. Системы, имеющие тройную К.Т.Р. (рис. 10). В этом
случае кривая, проходящая через критические точки Ри Р2, Р3,
Pi и Р5, достигает максимального значения в некоторой точке
Р6. Эта точка служит, следовательно, тройной критической
температурой растворения. Кривая, проведенная через критические
точки, проходит далее через точку Рз до бинарной К.Т.Р.,
соответствующей точке Рт.
Проекции бинодальных кривых для ряда температур на
основание призмы показаны на рис. 11. Для некоторого
интервала температур, лежащего между температурами,
соответствующими точкам Р7 и Р6, например для температуры 4,
изотермы растворимости являются замкнутыми кривыми с двумя
критическими точками Р$ и Р$. Одновременно бинарные
системы всех трех компонентов при этих температурах полностью
смешиваются. Примером таких систем служит система вода —
фенол — ацетон с тройной К.Т.Р. при 92° С и бинарной К-Т.Р.
(вода — фенол) при 66° С 17. Влияние температуры
иллюстрировалось на примере систем типа I с верхней К. Т, Р. Нетрудно
представить себе возможный характер пространственных
диаграмм для систем, имеющих нижнюю или как нижнюю, так и
верхнюю К. Т. Р. В общем случае с изменением температуры
изменяются не только величина области гетерогенных систем,
но и наклоны хорд, т. е. распределение компонента С между
фазами. Последний эффект относительно невелик при
умеренном изменении температур, но в большей части случаев им
нельзя пренебречь.
Рис. 11. Изотермы растворимости Рис. 12. Тройная система жидкость —
в системе с тройной критической жидкость (тип II).
температурой растворения.
' С А А 'it .,£ A t2 в
Рис. 13. Превращение системы типа II в систему типа I при изменении
температуры.
94
3*
Тип II. Системы с двумя парами частично
смешивающихся жидкостей (рис. 12). В этих системах при
данной температуре две пары жидкостей А — В и В — С
растворяются частично, а С и А смешиваются в любых отношениях.
Как и для систем типа I, область диаграммы, ограниченная
кривыми растворимости, соответствует смесям, образующим
две жидкие фазы. Состав равновесных фаз определяется
крайними точками хорд равновесия. Системы этого типа не могут
иметь критической точки. К таким системам относятся,
например, системы н-гептан(А)—анилин(В)—метилциклогексан(С) 18
и хлорбензол(Л)—вода(В)—метилэтилкетон(С) 19.
Системы данного типа часто образуются при изменении
температуры из системы типа I. В качестве примера на рис. 13
приведена система я-гексан(Л)—анилин (В)—метилциклогек-
сан(С)20 для температур tu t2 и t3, равных соответственно 25;
34,5 и 45° С.
Другой пример изменения характера диаграммы равновесия
при изменении температуры показан на рис. 14, на котором
Л 2в°с в А 22ос в
Рис. 15. Система зтиленгликоль (А) — лауриловый спирт (В) — нитро-
метан (С):
I —одна жидкая фаза; /7 —две жидких фазы; III —три жидких фазы; IV — жидкая
фаза + лаурилозый спирт.
33
Рис. 16. Тройная система, образующая твердую фазу:
/—одна жидкая фаза; // — две жидкие фазы; /// — жидкая фаза + твердый компонент С.
изображена система
метанол(Л)—изооктан(В)—нитробензол (С) 21. Температуры tu U, tz, t4, U равны соответственно 25;
15; 14,1; 14 и 10° С. На этом рисунке представляет интерес
точка D — в ней сходятся две критические точки Р' и Р.
Возможны еще более сложные диаграммы равновесия для систем
данного типа. В частности, для некоторых систем, указанных
Фрэнсисом п, возможно образование трех жидких фаз. В
приведенных примерах область расслаивания уменьшалась с
увеличением температуры. Известны, однако, и системы типа II,
состоящие из пар компонентов с нижними критическими
температурами растворения 22.
Тип III. Системы с тремя парами частично
смешивающихся жидкостей. Эти системы встречаются
сравнительно редко. Диаграммы равновесия для них при
изменении температуры могут принимать очень сложный вид.
Поскольку системы такого типа не представляют интереса для
экстракции, ниже будет рассмотрен только один пример
подобной системы.
37
А
Рис. 17. Высаливание с помощью
твердого вещества:
/ — одна жидкая фазаН-твеРДый компонент С;
// — две жидкие фазы; /// — жидкость D +
+ жидкость £+ твердый компонент С.
На рис. 15 показана
система этиленгликоль (А)—ла-
уриловый спирт (В) —нитро-
метан(С) 13. При 29° С эта
система имеет три различные
области расслаивания. При 28° С
области ограниченной
растворимости увеличиваются, и
появляется небольшой участок,
соответствующий
одновременному существованию трех
жидких фаз. Любая тройная смесь
внутри треугольника DEF
образует взаимно
нерастворимые растворы D, Е и F. При
22° С этот участок
увеличивается и, так как эта
температура ниже точки плавления
лаурилового спирта,
появляется область существования твердой фазы. В системах типа III
бинодальные кривые могут встречаться, как было показано на
рис. 14, образуя комбинацию систем типов I и II и.
Тип IV. Системы, образующие твердую фазу.
Ниже будет рассмотрено только несколько относительно
простых примеров подобных систем, представляющих интерес для
жидкостной экстракции.
На рис. 16 изображена наиболее часто встречающаяся
система такого типа. При температуре ti компоненты А и В—
ограниченно растворимые жидкости, а С — твердое вещество.
Растворимость последнего в чистых А и В обозначена
соответственно точками D и Е. Линия DE является кривой
растворимости вещества С в смесях компонентов А и В. Например,
тройная смесь F образует насыщенный раствор G и кристаллы
вещества С. Кривая JPH ограничивает область существования
двух жидких фаз, как в системах типа I. Между двумя
областями существования гетерогенной системы лежит область, где
имеется лишь одна жидкая фаза. При более низкой температуре
4 взаимная растворимость уменьшается, и области
гетерогенности увеличиваются. При еще более низкой температуре г3 би-
нодальная кривая пересекает кривую растворимости твердого
вещества. Любая тройная смесь, лежащая внутри треугольника
CKL, образует три фазы: одну твердую —С и два насыщенных
жидких раствора К и L. Примером служит система
анилин^) —изооктан(В) —нафталин(С).
В некоторых системах, как показано на рис. 17, область
расслаивания двух жидких фаз не достигает оси А—В.
Уменьшение температуры, однако, может вызвать увеличение области
88
кгсо3
^хкгсо3-|-н2о
расслаивания. При
температуре, равной К-Т.Р.
бинарной системы АВ,
критическая точка Р
коснется оси А—В.
При дальнейшем
охлаждении диаграмма
растворимости этой
системы станет подобной
диаграмме
растворимости на рис. 16 для
температуры U-
При образовании ттл
гидратов или других этанол*—*-
сольватных соединений
диаграмма раствори- Рис. 18. Система карбонат калия —вода —
мости может значи- этанол (схематически),
тельно усложниться.
На рис. 18 приведен сравнительно простой случай таких
систем2-23. Насыщенный раствор карбоната калия в воде (точ
ка N) находится в равновесии с гидратированной солью.
Растворимость К2С03 в спирте (точка Р) очень мала. Если к смеси
воды и этанола, обозначенной точкой М, добавлять безводный
карбонат, в точке D система будет состоять из двух жидких
фаз, в точке £ — из растворов F и G и твердого гидрата, а в
точке Н в равновесии будут находиться безводный карбонат
калия, твердый гидрат его и насыщенный раствор /. Перегонка
раствора / дает дистиллят состава К. Так как в
действительности точка / лежит гораздо ближе к точке Р, чем это
показано на диаграмме, К будет представлять собой почти чистый
этиловый спирт. Описанный процесс является одним из видов
высаливания и часто используется для дегидратации
органических веществ.
Значение этого способа дегидратации для практики возрастает, если
систему поддерживать в жидком состоянии. В данном случае можно
использовать непрерывнодействующую аппаратуру для жидкостной экстракции.
Рассмотрим, например, систему, показанную на рис. 19. Область
расслаивания двух жидких фаз здесь больше, чем на рис. 18, и примыкает к оси А
(метилэтилкетон)—В (вода). Точки S (12,6% воды) и Т (77,6% воды)
соответствуют взаимной растворимости воды и метилэтилкетона. Так как
растворимость хлорида кальция в кетоне незначительна, для практических
целей можно считать, что точка G на рис. 19 (отвечающая точке О на
рис. 18) находится иа оси А—В. Можно принять также, что точки,
соответствующие точкам Р и / на рис. 18, лежат на той же оси между точками А
и б на рис. 19. Хорды равновесия, соединяющие кривые растворимости,
аналогичные кривым PJ и JG (см. рис. 18), с хлоридом кальция и его
гидратом, на рис. 19 ие показаны.
Воспользоваться ограниченной растворимостью компонентов для
дегидратации метилэтилкетона перегонкой в данном случае нельзя, поскольку
Метилэтилкетон и вода образуют азеотропную смесь (11% воды), состав
CaCl
2-6H20
AGS
Рис. 19. Система метилэтилкетон —
вода — хлорид кальция при
температуре 25—26° С.
Кристаллогидрат
Рнс. 20. Равновесие между
паровой, жидкой н твердой
фазами.
которой лежит в области существования одной жидкой фазы А.
Максимальная степень дегидратации метилэтилкетона высаливанием с помощью
хлорида кальция, возможная при поддержании системы в жидком состоянии,
соответствует точке G (содержание воды в смеси, отвечающей этой точке,
гораздо меньше, чем в азеотропной смеси).
На рис. 20 изображен замкнутый кольцеобразный сосуд, в который
помещены насыщенные растворы (обозначенные на рис. 19 точками G и F) и
кристаллы гидрата хлорида кальция; кроме того, часть объема сосуда
занята паром V, находящимся в контакте с обеими жидкими фазами. При
равновесии давления паров воды над растворами G и F и кристаллами
должны быть одинаковы. Следовательно, концентрация воды в растворе G
достигнет величины, при которой давление паров воды над этим раствором
будет таким же, что и над насыщенным рассолом F или над кристаллами
гидрата 24. Раствор G практически не
содержит хлорида, а концентрация
кетона в растворе F так мала, что
можно принять давление паров
воды над ним равным давлению ее
паров над насыщенным раствором
хлорида кальция или его гидрата.
Согласно расчетам Мейсснера и
Стокса24, на рис. 21 приведены
кривые парциальных давлений воды над
ее смесями с метилэтилкетоном при
трех температурах. На этих кривых
отмечены также точки, отвечающие
упругостям паров над насыщенными
водными растворами* некоторых
хлоридов. Абсцисса, соответствующая
каждой такой точке, показывает
максимально возможную степень
дегидратации при использовании того
или иного хлорида при данной
температуре. Результаты расчета были
подтверждены экспериментально
Мейсснером и Стоксом. При 40° С с
помощью насыщенного рассола хло-
1
I.
100
во
60
со
го
1 S
■l4
<Ь
Щ
w
в
6
с
ис
/
/
/■
/
1
СаС12
\у
/
1
чщ
^\
•^NaCl
^ А
<• №
Ф
y^i
<t
а
хз
хг
t
о,г ер о,в i г 4 б в ш
Концентрация воды
в метилэтилкетоне, Вес %
Рис. 21. Дегидратация
метилэтилкетона иысаливанием.
40
I" ряда кальция можно получить, например, метилэтилкетон, содержащий 0,6%
•"' воды. Из солей, указанных на рис. 21, наиболее эффективен хлорид лнтня,
• причем эффективность его мало изменяется с температурой.
Прн высокой взаимной растворимости компонентов для анализа
процессов, подобных рассмотренному процессу дегидратации высаливанием, сле-
, дует применять методы, приведенные в главе III.
Экспериментальное определение равновесия в трехкомпо-
нентных системах. Если можно определить химическим
анализом содержание двух из трех компонентов, составляющих смесь,
могут быть определены одновременно хорды равновесия и би-
нодальная кривая. Так, если смесь среднего состава М (рис. 22)
встряхивать некоторое время при постоянной температуре, после
отстаивания образуются два слоя N и О. Анализ каждого из
этих слоев даст составы двух растворов, находящихся в
равновесии. Повторяя встряхивание и отстаивание (их удобнее всего
выполнять в делительной воронке) при разных составах
исходной смеси, можно получить необходимые данные для построения
полной диаграммы растворимости трехкомпонентной системы.
Однако содержание двух компонентов в смеси обычно трудно
найти химическим анализом. В таких случаях хорды
равновесия и бинодальную кривую следует определять отдельно. Бино-
дальную кривую чаще всего определяют методом титрования до
помутнения. Приготавливают определенное количество
гомогенной смеси известного состава, например смесь К (см. рис. 22). К
ней постепенно добавляют при постоянной температуре чистый
компонент В. Когда состав системы окажется на бинодальной
кривой (точка L на рис. 22), смесь помутнеет. Состав смеси в
точке L может быть установлен из материального баланса.
Таким образом, можно определить ветвь бинодальной кривой,
отвечающей смесям, в которых преобладает компонент А. Для
определения остальной части
бинодальной смеси С и В
известного состава необходимо
титровать компонентом А.
Методом титрования
нельзя, однако, определить
положение хорд равновесия на
диаграмме; это может быть
выяснено другим способом. Так,
если несложно определить
химическим анализом
содержание одного из трех
компонентов, то, зная содержание этого
компонента в равновесных
слоях (например, в тех же
слоях А/ и О, полученных из сме- Рис. 22. Определение равновесия
си' М), легко найти полный в тройной системе.
41
состав этих слоев, поскольку Они должны лежать на бинодаль-
ной кривой.
Методы исследования равновесия тройных систем подробно
излагаются в работах Мак-Дональда 25, Богина 26 и Хэнда 27. Для
определения равновесия при высоких температурах и давлениях
необходима .специальная аппаратура9'28-30. Иногда для анализа
составов смесей используют метод меченых атомов 31.
В ряде случаев анализ всех трех компонентов системы
затруднителен. Однако обычно возможно определение вдоль би-
нодальной кривой какого-либо физического свойства системы,
заметно меняющегося с изменением концентрации. Изменение
этого свойства может быть использовано вместо чисто
аналитических методов. Вследствие простоты определения в качестве
такого свойства чаще всего выбирают плотность или
коэффициент преломления. Так, если иметь зависимость плотности от
весовой доли компонента С вдоль бинодальной кривой,
определение плотностей уже упоминавшихся слоев N и О даст
возможность установить положение точек N и О на диаграмме.
Приведенные выше методы определения равновесия тройных
систем описаны выше на примере системы типа I. С
соответствующими изменениями их можно применять и для систем
других типов.
Средний состав М должен лежать на прямой, соединяющей
составы равновесных фаз, поэтому материальный баланс дает
возможность контролировать точность эксперимента. Вместе с
тем материальный баланс совместно с правилом рычага может
быть использован для быстрого, хотя и менее точного
определения состава равновесных фаз. Так, если смесь известного
среднего состава М приготовить в мерном цилиндре, объемы фаз
можно найти непосредственно. Зная плотности равновесных
слоев (удобнее всего для их определения в данном случае
пользоваться весами Вестфаля), можно вычислить относительные
количества слоев. Положение точек N и О на диаграмме может
быть установлено при соблюдении соотношения
Простой метод нахождения точек О и N, согласно уравне^
нию (II, 10), описан Отмером и Тобиасом 32.
Положение критической точки можно определить
экспериментально 33. Для этого находят подбором такую двухфазную
смесь (см. рис. 22), при добавлении к которой компонента С
поверхность раздела между фазами вплоть до наступления
полной растворимости, соответствующей критической точке Р,
будет располагаться около середины сосуда. Если же добавлять С
к смеси другого состава, межфазовая поверхность будет
перемещаться вверх или вниз. Ниже рассмотрен более удобный эм-
42
с w
F J
AD F В D
Рис. 23. Прямоугольные координаты.
лирический способ определения критической точки обработкой
данных, относящихся к хордам равновесия.
Равновесие трехкомпонентных систем в прямоугольных
координатах. Для изображения равновесия в тройных системах
обычно пользуются треугольной диаграммой (в виде
равностороннего треугольника), с помощью которой можно удобно
выполнять различные расчеты по жидкостной экстракции. Однако
обычно треугольные диаграммы издаются только одного
размера; кроме того, для ясности чертежа часто возникает
необходимость в различных масштабах величин, откладываемых на
осях координат. Вследствие этого для
тройных систем применяют также
некоторые диаграммы в прямоугольных
координатах.
Один из видов таких диаграмм —
зависимость Хв отХс — представляет собой
по существу прямоугольный
треугольник. На рис. 23 в этих координатах
изображена система типа I, причем для
сравнения на том же рисунке она
изображена с помощью треугольной
диаграммы. Содержание компонента А в
отданном случае определяется по разности. ^ ;
Другая система координат,
разработанная Енеке34, показана на рис. 24
для систем типов I и II. Преимущества
таких координат описаны ниже, пока же
следует отметить, что масштабы на осях
диаграмм в координатах Енеке в
случае необходимости могут быть неодина-
%С
Рис. 24. Системы типа I (а)
и типа II (б) в координа-
. тах Енеке (К — крити-
ковыми. Для диаграмм в прямоугольных ческая точка).
43
координатах правило рычага [уравнение (II, 9)] также
применимо. При использовании координат Енеке это правило
можно применять, если весовые количества смесей определять без
учета компонента В.
Корреляция равновесных данных
для тройных систем
Интерполяция и экстраполяция хорд равновесия. Для
многих описанных в литературе систем жидкость — жидкость
экспериментально определялось только несколько хорд равновесия.
Прямая интерполяция, и особенно экстраполяция хорд
равновесия на треугольных диаграммах, как правило, приводят к очень
неточным результатам. Для определения положения
неизвестных хорд равновесия по известным предложено несколько
методов, рассматриваемых ниже.
Графическая интерполяция на треугольной диаграмме. Пусть
линия DE на рис. 25 — одна из хорд равновесия, найденных
экспериментально. Проведем прямую DG, параллельную стороне
треугольника СВ, и прямую EF параллельно стороне АС. Эти
линии пересекутся в точке Н.
С Корреляционная кривая РН
проводится через ряд полученных
таким образом точек
пересечения вспомогательных линий,
построенных по известным хордам
равновесия. Прямые,
проведенные из любой точки этой кривой
параллельно АС и ВС, пересекут
бинодальную кривую в точках,
соответствующих составам
равновесных растворов. Линия РН
является кривой (хотя кривизна
«»f" „SL ГраФическая
интерполяция хорд ра&вовесия.
44
/ /в ее обычно не велика), причем
она обязательно проходит через
критическую точку.
Указанный метод дает
достаточно точные результаты при
интерполяции в тех случаях,
когда известны по крайней мере
три или четыре хорды
равновесия. Однако из-за кривизны
корреляционной кривой он
недостаточно точен при экстраполяции
на значительные расстояния.
Точное положение критической
точки можно найти этим спосо-
^
Рис. 26. Графическая интерполяция
хорд равновесия по методу Шервуда.
бом только в том случае, если
известны хорды равновесия,
лежащие близко к ней. Метод
широко применяется в
„International Critical Tables" 35.
На рис. 26 показана
модификация этого метода,
предложенная Шервудом 36. Удобство
построения, приведенного на
рисунке, заключается в том,
что корреляционная кривая
находится на площади
треугольника составов. Оба мето- д
да могут быть использованы в
случае применения
прямоугольных систем координат,
показанных на рис. 23 и 24.
Хэнд27 предложил интересный метод изображения тройных
систем, согласно которому все хорды равновесия оказываются
параллельными основанию треугольника диаграммы. К
сожалению, способ Хэнда не всегда применим, так как лишь немногие
системы жидкость — жидкость поддаются такой обработке. В
некоторых случаях при продолжении хорд за пределы бино-
дальной кривой они пересекаются в одной точке на
продолжении основания треугольника37. Можно математически
показать 38, что это свойство соблюдается только для систем,
следующих правилу Хэнда, и оно, следовательно, также не
является общим. Однако, если хотя бы две хорды равновесия
пересекаются в одной точке на продолжении основания
треугольника, весьма вероятно, что и другие хорды будут проходить
через эту точку. Интерполяцию и экстраполяцию хорд
равновесия производят в данном случае очень просто. В частности,
критическую точку определяют, проводя из точки пересечения
хорд равновесия линию, касательную к бинодальной кривой.
Кривые распределения. Для корреляции опытных данных и
облегчения их интерполяции предложены различные способы
графического изображения зависимости между концентрациями
равновесных растворов. Желательно, чтобы указанные
зависимости были для всех систем линейными. В этом случае
экстраполяция экспериментальных данных облегчается и при наличии
только двух хорд равновесия можно находить положение всех
остальных.
Простейшая кривая распределения представляет собой
зависимость концентрации компонента С в фазе А в функции от
концентрации С в фазе В. На рис. 27 показаны такие кривые
вместе с соответствующими им диаграммами. На рис. 27, а
изображена система типа I. Точка D на кривой распределения
4Б
соответствует харде RE на треугольной диаграмме. Таким
образом, каждая хорда равновесия представлена одной точкой на
кривой распределения. Кривая проходит через максимум и
заканчивается на диагонали осей координат (точка G). Эта точка
соответствует критической точке на треугольной диаграмме.
Кривая распределения может проходить выше или ниже
диагонали в зависимости от того, какой из ограниченно растворимых
Рис. 27. Кривые распределения:
а—система типа I; б —система типа П; в — сольютропная система;
г — распределение твердого вещества.
48
компонентов системы обозначен как компонент А. Отношение
XcbIXca в любой точке кривой называется коэффициентом
распределения. Этот коэффициент для случая, показанного на
рис. 27, а, монотонно уменьшается с увеличением
концентрации Сив критической точке становится равным единице.
Для систем типа II (рис. 27,6) кривая распределения
оканчивается точкой, соответствующей растворимости в бинарной
системе ВС. В сольютропных системах, как показано на
рис. 27, в, кривая распределения пересекает диагональ осей
координат. Точка пересечения представляет собой
горизонтальную хорду RE. На рис. 27, г изображена кривая, характерная
для распределения большей части твердых веществ. Из
диаграммы видно, что коэффициент распределения при всех
концентрациях довольно близок к отношению растворимостей
вещества С в чистых компонентах А и В, предстазленных точками
F, G и Я. Это тем ближе к истине, чем меньше взаимная
растворимость компонентов А и В.
Согласно простейшему закону распределения, коэффициент
распределения тс = ХСв/ХсА должен быть постоянным и не
зависеть от концентрации компонента С. Однако из диаграмм на
рис. 27 следует, что в общем случае этот закон не соблюдается;
лишь при очень низких концентрациях С коэффициенты
распределения почти постоянны. Можно принять, что изменение т, по
крайней мере частично, обусловлено изменением взаимной
растворимости компонентов Л и В с увеличением концентрации С.
На таком допущении основан ряд эмпирических методов
обработки равновесных данных. Поэтому в некоторых методах
учитывается главным образом равновесное распределение
компонентов Л и В, а не С.
Эмпирические методы обработки равновесных данных.
1. Бахман39 на основе данных Бранкера, Хюнтера и Нэша40
установил, что равновесие систем типа I во многих случаях
можно описать сравнительно простым выражением
Хвв = г + Чхвв1хаа) (".ID
где г и Ь — константы.
Из этого уравнения следует, что зависимость Хввот Хвв/ХАА
должна быть линейной.
2. Отмер и Тобиас32 нашли, что для равновесных составов
зависимость величины (1—^аа)1^аа от (1—XBB)jXBB в
логарифмических координатах представляет собой прямую линию,
Удобную для интерполяции и экстраполяции.
3. Методы, описанные в п. 1 и 2, обладают следующим
основным недостатком: при их использовании на осях координат
диаграммы не указывается концентрация распределяемого
компонента С.
47
Хэнд21 предложил метод,
свободный от этого
недостатка. Он показал, что при
равновесии зависимость ХСА/ХАА
от Хсв/Хвв в
логарифмических координатах обычно
линейна. Ранее такой метод
изображения равновесных данных
предложил Бэнкрофт4I. Эта
линейная зависимость может
быть описана уравнением
X.
ев
Ya xaaJ
Определение критической
X
вв
№
(II, 12)
Физический смысл констант
k и г указан в литературе42
и будет рассмотрен в главе III.
На основе метода Хэнда
предложен 43 простой способ
определения критической точки.
На рис. 28 наряду с зависимостью (II, 12) для хорд равновесия
нанесена бинодальная кривая в виде зависимости Хс/Хв от
ХС/ХА (ХА, Хв и Хс — концентрации компонентов на бинодаль-
ной кривой). Для критической точки имеем:
Рис. 28.
точки:
/ — бинодальная кривая (Хс/Хв от XqIXaY
2 — хорды равновесия
(Хсв/ХвВ от ХСА/ХАА).
\Х7в)Р'\х<вАл)Р~\
В/р
х,
СА
X
АА Iр
ХСВ
X
АВ/р
т,
(П, 13)
(II, 14)
Индекс р означает, что концентрации относятся к
критической точке. Данная точка является как бы пределом, к
которому стремятся хорды равновесия, поэтому координаты
(ХСА/ХАА)Р и (Хсв/Хвв) v должны соответствовать
одновременно корреляции для хорд равновесия и для бинодальной кривой.
Следовательно, экстраполируя прямую, выражающую
корреляцию для хорд равновесия, до пересечения с бинодальной
кривой, можно определить положение критической точки Р (см.
рис. 28).
На рис. 29—32 изображены равновесные данные для одних и
тех же систем типа I в различных координатах. Для двух из
приведенных систем (этанол — н-бутанол — вода и 1,4-диоксан—
бензол — вода) имеются экспериментальные данные вплоть до
критических точек. Первая из них характеризуется необычно
высокой взаимной растворимостью компонентов А и В (вода и
48
н-бутанол). Это приводит к тому, что максимальная
концентрация распределяемого компонента составляет лишь 14,9%.
Система изопропиловый спирт — бензол — вода является солью-
тропной. Обращение коэффициента распределения для эгой
системы видно из кривой распределения, показанной на рис. 29.
В координатах Бахмана (рис. 30) данные о распределении
диоксана и изопропилового спирта между водой и бензолом
изображаются кривыми, в то время как в координатах Отмера—•
Тобиаса (рис. 31), а также Хэнда (рис. 32) для всех систем,
кроме сольютропной системы изопропиловый спирт — бензол —
вода, получаются прямые линии (вплоть до критических точек).
Методы Отмера — Тобиаса и Хэнда применялись к большому
числу других систем жидкость — жидкость, причем во всех
случаях были достигнуты хорошие результаты. Анализ данных для
системы этанол — н-бутанол — вода показывает, что хорда
равновесия при самой низкой концентрации этанола,
по-видимому, определена неточно; это четко видно на рис. 32.
Альдерс 44 нашел, что при распределении органических
соединений с нормальной цепью, являющихся членами
гомологического ряда, между одной и той же парой растворителей
логарифм коэффициента
распределения при бесконечном
разбавлении изменяется линейно с
изменением числа атомов углерода ff
к-
i?0,6
|
i
i.
лвв
° 1
х г
д 3
о 4
У.х
"л-
^7
0,2
Of Off OS
Концентрация С 8 фазе а (хса), вес. доя
Рис. 29. Кривые распределения для
систем типа I:
1 — система вода (А) — 1, 1, 2-трихлорэтан {В)~
ацетон (С) при 25° С; 2 — система вода (Л) —
бензол {В) — диоксан (С) при 25° С; 3—
система вода (Л) —бензол (В) — изопропиловый
спирт (С) при 25° С; 4 —система вода (А) — н-
бутанол {В) — этиловый спирт (С) при 20° С;
л—критическая точка.
0,3 0,S Q7 Cjff
Ы
Рис. 30. Координаты Бахмана39:
/ — система вода (А) — 1,1, 2-трихлорэтан(В) —
ацетон (С) при 25° С; 2 —система вода (А) —
бензол В — диоксан (С) —при 25° С; 3
—система вода (Л) —бензол (В) — изопропиловый
спирт (С) при 25° С; 4 — система вода (Л) — к-
бутанол (В) —этиловый спирт (С) при 20° С;
К— критическая точка.
4 Р. Е. Трейбал
49
-2£А
ХАА
Opt' OpSOpu 0/ Q2 0,Ш 1,0
тг
о /.
ори О' цг o,uopt г и в ю
1-х.
*м
Рис. 31. Координаты Отмера — То-
биаса32:
1 — система вода(Л) —1,1,2-трихлорэтан (В)—
ацетои (С) при 25° С; 2 —система вода (Л) —
беизол (В) — диоксаи (С) при 25° С;
3—система вода (Л) —бензол (В) — изопропиловый
спирт (С) при 25° С; 4 —система вода (А) — н-
бутаиол (В) — этиловый спирт (С) при 20° С;
К— критическая точка.
ХСД
ХАА
OPIOPSOPU QJ OtOjfOfil
3
*|Ч
qpu oj цго^ор/ г и в ю
ХСА
ХАА
Рис. 32. Координаты Хэнда 27:
1 — система вода (Л) — 1,1, 2-трихлорэтаи (В)—
ацетои (С) при 25° С; 2 — система вода(Л) —
беизол (В) —диоксаи (С) при 25° С; 3 —
система вода (Л) —бензол (В) —изопропиловый
спирт (С) при 25° С; 4— система вода (Л) —и-
бутанол (В) —этиловый спирт (С) при 20° С;
л—критическая точка.
в молекуле распределяемого вещества. Это наблюдение может
оказаться полезным при систематизации равновесных данных,
а также при расчете коэффициентов распределения (см. также
главу III).
Пример П-2. Вартересьян и Фенске [Ind. Eng. Chem., 28, 928 (1936)]
определяли равновесие в системе бензол (А)—вода (В)—этанол (С).
Данные для двух хорд равновесия (составы в вес.%, температура 25°С)
сведены в таблицу.
Б
ХАА
0,9525
0,8000
еизольный слог
ХВА
0,003
0,0255
ХСА
0,0445
0,1745
ХАВ
0,0102
0,2045
Водный слой
ХВВ
0,6298
0,2740
хсв
0,3600
0,5215
Рассчитать по этим данным полную диаграмму равновесия и сравнить
расчетные величины с опытными.
Решение. Помещенные в таблице равновесные данные изображены
на рис. 33 в координатах Хэнда (см. рис. 28). Проведенные через них
прямые представляют собой в координатах Хэнда бинодальную кривую и
корреляционную прямую для хорд равновесия. Данные, необходимые для по-
50
Хд ХАА
Рис. 33. К примеру Н-2.
строения диаграммы равновесия, определяют с помощью полученных прямых
следующим образом. Расмотрнм точку G, для которой Хс/Хв=9,2 и
ЯСДД = 0,12. Из этих соотношений с учетом того, что ХА+ХВ+ХС = \,
вычисляем: Яаа = 0,883; Ява = 0,012 и ХСа = 0,0\5. Соответствующий точке G
равновесный раствор должен находиться в точке Н, для которой точно
таким же образом получаем Яав=0,072; Авв = 0,422 и Ясв = 0,506.
Аналогично определяют и другие хорды равновесия, данные для которых
представлены в следующей таблице:
ХАА
0,986
0,972
0,833
0,684
0,577
ХВА
0,002
0,003
0,012
0,052
0,083
ХСА
0,012
0,025
0,105
0,264
0,340
ХАВ
0,0005
0,003
0,072
0,393
0,577
Этанол
хвв
0,833
0,733
0,422
0,153
0,083
о /
V,J
хсв
0,167
0,264
0,506
0,454
0,340
Бензол '
bffofc
Рис. 34. Система бензол — вода — этанол:
./ — расчетные точки; 2 —точки, использованные при расчете; 3 — опытная
бинодальиая кривая.
4*
61
0,5 \—
0.5 \
0£
£
^
0,3
о.г
0J
^ _
/ з X
I / •г
Расчетные значения представлены на
рис. 34. Они хорошо согласуются с
экспериментальными данными Вартересьяна и Фен-
ске. Соответствие опытных и расчетных
кривых распределения показано на рнс. 35.
Закон распределения с учетом
диссоциации и ассоциации молекул.
Если даже взаимная
растворимость компонентов А и В при
малых концентрациях
распределяемого вещества С является постоянной,
закон распределения может не
соблюдаться из-за диссоциации или
ассоциации компонента С. Нернст45
предположил, что закон
распределения можно считать во многих
случаях применимым, если исходить
не из общей концентрации
распределяемого компонента, а из
концентрации молекул, находящихся в
одинаковом состоянии, например
ассоциированных молекул.
Получающиеся при таком подходе
простые закономерности, обсуждаемые далее, вряд ли будут
справедливы в случае высоких концентраций распределяемого
компонента.
Ниже будут рассмотрены некоторые частные случаи на основе
применения гипотезы Нернста.
1. Распределяемый компонент не
претерпевает никаких изменений ни в одной из фаз. Для
этого случая закон распределения имеет вид:
0,1 0,8 0,3 O.U
ХСА, вес. Воли
Рнс. 35. Распределение этанола
между бензолом и водой (к
примеру Н-2):
1 — расчетные точки; 2 —точки,
использованные для расчета; 3 —
опытная кривая распределения.
СВ
СА
= тпс = const
(Н, 15)
фазе
ми р
А
а с
(бо-
п р е-
2. Диссоциация компонента С в
гатой компонентом А); между фаза
деляются только недиссоциированные
молекулы С. Равновесие в этом случае можно описать следующим
уравнением:
~ -^ ~ -^. п'С' + п'С"
(фаза В)
Отсюда закон распределения:
п,' [С]В
(фаза А)
= const
(II, 16)
Здесь пг'с относится к распределению только недиссоцииро-
ванных молекул. Квадратные скобки указывают на то, что кон-
52
центрация вещества, стоящего в скобках, должна быть
выражена в молях на литр, поскольку константы равновесия обычно
относят к этим концентрациям. Так, величина [С]А означает
концентрацию (в моль/л) недиссоциированного компонента в
фазе А.
Концентрация С в фазе А, определенная аналитически, как
правило, представляет собой общую концентрацию [Ст]а,
которой учитываются диссоциированные и недиссоциированные
молекулы. Если а — степень диссоциации компонента С в фазе А,
то [С]А = [СгЦ(1—«)• Тогда из уравнения (II, 16) следует:
, [С]д mr
const (11,17)
'"С ТСт-ГдО-») !-«
где т"с = [С]в1[Ст\А коэффициент распределения, отнесенный к
суммарным концентрациям.
Поскольку
[С]ВМС
Хсв ~ 1000рл <"•18)
\СТ\.МГ
X = 1 Г>А—£. m if*
са Ю00рл ( ' Щ
и плотности при умеренных изменениях концентраций остаются
почти постоянными, из уравнения (II, 17) получаем
ев _ m"> _ mc _ const (П щ
ХСА(\-а) "<■■ 1-а
Степень диссоциации обычно меняется с концентрацией
[Ст]а, и, следовательно, коэффициент распределения т или т"
(обычно определяемый экспериментально) не является
постоянным. Если применить к диссоциации закон действия масс,
константу диссоциации /СДИс. можно рассчитать по уравнению
[СГа\С"]а" (n'a[CT]Af (n"a[CT]Af
Kwc-~ WA [Сг]л(1-а) (П'21)
Если величина /СДИс- известна, по уравнению (11,21) можно
определить а и использовать его значение в уравнениях (II, 17)
и (11,20). Кроме того, комбинируя уравнения (11,21) и (II, 17),
находим:
/ , /чл'/ "J1" г Xs, 1 \п'+п"-
тпс (п ) (я ) (а \СТ\А)
(я')"' (я')"" (в[Сг]5'+в*-1 + *д,
(11,22)
Если при диссоциации С получаются два одновалентных
иона, уравнение (11,22) преобразуется к виду:
„ m'ra [C_i.
а1СГ]А+КАИС
53
Рассмотрим, например, случай, когда С является слабой одноосновной
органической кислотой, к которой может быть применен закон действия
масс46. Тогда alCT]A =(H+]A и уравнение (11,23) примет форму:
т'с[Н + ]л т'г
(11,24)
[Н + ]л + /Сд„с. j , К
[Н+]
А
Для слабой кислоты в щелочном растворе [Н+]<^/(ДЯс. Поэтому
уравнение (11,24) упростится:
т'[Н + ]
(Н.25)
Логарифмируя уравнение (11,25), получаем
lg т"с = lg [Н+]л + lgт'с - lg/Сдис. (II,26)
lgm"c = -pH + lgm'c-lgKMc. (11,27)
При высоких рН зависи мость lg mc от рН выражается прямой линией
с наклоном —1. Величина отрезка, отсекаемого этой прямой на оси ординат,
дает возможность определить /(ДИс. и т'. Значения /(ДИс. и тс можно
найти также из зависимости величины 1/тс от 1/[Н+]а, которая в простых
координатах будет представлять собой, согласно уравнению (11,24), прямую
с наклоном KMCJmc. Отрезюк, отсекаемый этой прямой на оси ординат47,
равен 1/тс. С другой стороны, если величина /СДИс. заранее известна, по
приведенным уравнениям можно рассчитать изменение коэффициента
распределения в зависимости от рН.
Так, для распределения 2,4-ксиленола между фосфатным буфером (А)
и циклогексаном (В) при рН=6,65 (т. е. в условиях, когда диссоциация
незначительна), величина пгс = тс= 7,06. Константа /(дис. заранее известна
и равна 0,36- Ю"10.
С помощью этих данных по уравнению (11,27) можно найти
коэффициент распределения т'с при рН= 11,08:
lg т"с = — .11,08 + lg 7,06 — lg (0,36 • 10-10)
тс = 1,65
Опытное значение46 тс равно 1,63. Если подобно тому, как в
приведенном примере, степень диссоциации в нейтральном растворе незначительна,
добавление минеральной кислоты к раствору не вызовет заметного
изменения коэффициента распределения. Однако, если величины /(ДИс. и {Н+]
одного порядка, при добавлении сильной кислоты тс увеличится.
Приведенный пример показывает также, как может быть использовано применение
буферного раствора (не растворяющегося в органической фазе) для
поддержания в процессе экстракцши, если это желательно, постоянной величины
рН в водной фазе.
3. Ассоциация в фазе В; распределяются
только н е а ссоци и ро в а н н ы.е молекулы.
Равновесие в этом случае выражается уравнением
С i± С т± - Сп
(фала А) (фаза В)
54
Если а' — степень ассоциации в фазе В и [СТ]В — общая
концентрация компонента С в той же фазе, то [С]в = [СгЬ(1—а')
и [Сп]п = а'[Ст]в/п — соответственно концентрации неассоцииро-
ванного и ассоциированного компонента С. Согласно закону
действия масс, константа ассоциации
(a'\CT]Jn)lln
и распределение неассоциированного компонента С описывается
уравнением:
, [Сг]д(1-сО (а'1стУп)1"1
с [С]А Ка [С]
Следовательно
(«' lCT)B)Va
= const (II, 29)
\Ch
= m'Kanlln = const (II, 30)
Часто бывает, что значение а' приближается к единице.
В данном случае величина [Ст]^п/[С]А (или ХСв11п/Хса)
близка к постоянной. Поэтому зависимость между равновесными
концентрациями в логарифмических координатах должна
изображаться прямой. По наклону этой прямой можно определить
величину п.
4. Диссоциация в фазе Л; ассоциация в
фазе В; распределяются только неассоциирован-
ные и недиссоциированные молекулы.
Уравнение равновесия:
п'С' + п"С" ^± С ^± С ^> (1/п) Сп
В фазе А концентрации ионов равны п'ос[Ст]а и п"о[Ст]а и
концентрации недиссоциирующих молекул — [СГЦ(1—а). В фазе
В концентрации неассоциированных молекул равны [Ст]в(1—а')
и концентрации ассоциированных молекул — ос'[Ст]в1п.
Коэффициент распределения
, _ [Сг]д(1-а')
!Я(1-а')/*с
:тых npeo6f
[ег1д_К*« С1 -«>]""
const (И,31)
или, что то же самое, ХСв{\—<z')IXCa{\—«)• С помощью
уравнения (П,28) после простых преобразований получаем:
(Н,32)
Если значение а' приближается к единице, а а — к нулю, то
зависимость [Ст]в от [Ст]а в логарифмических координатах
66
должна изображаться прямой с наклоном, равным п. Из
уравнения (11,32) также следует:
К"
а' [Суп
|[е7-]д-[е7-]в(1-°'))/я
[Crjj-iB^fCr] (1-a)
Пример 11-3. В таблице помещены данные о распределении бензойной
кислоты (С) между водой (Л) и бензолом (В) при 25°С:
[Cj-Jb, моль/л
0,001491
0,020
0,050
0,1492
0,200
f-rl-A' моль/л
0,000854
0,00373
0,00579
0,0108
0,01276
[СГ]В/[СТ]А
1,75
5,36
8,64
13,83
15,69
Значения коэффициентов распределения для данной системы, как видно
из приведенных величин [Ст]в1[Ст\а, значительно отличаются друг от друга.
В логарифмических координатах указанные величины дают прямую с
наклоном, равным 1,91 (рнс. 36). Можно принять п = 2. Константа диссоциации
бензойной кислоты в воде ЛДИс. = 6,6 • 10~5. Поскольку в данном случае п'—
<=п"=1, из уравнения (11,21) получаем
. + (/С. + 4[Сг]л/Сдис,)0-5
!
4?
0.2
0.1
0,01
-*„■
i
0,001
0,0006^001 0,01 002
[Ci]A, моль/л
Рнс. 36. Распределение
бензойной кислоты между
бензолом (В) и водой (А) (к
примеру И-3).
2[СТ]
При [Ст]а =0,000854 это уравнение дает
<х=0,22 и 1—<х=0,78; при [СТ]А =0,01276
имеем: а=0,0695 и 1—а=0,9305.
Подставляя эти и соответствующие им величины
для [Ст]в в уравнение (11,33) при я = 2,
находим
/С2„ =
0,001491 — т'с (0,000854) (0,78)
2 [т'с (0,000854) (0,78)]2
0,200— т'с (0,01276) (0,9305)
2 [т'с (0,01276) (0.9305)]2
откуда тс —
1,42 и Ка = 17,95.
Те же значения Ка. получаются при
использовании для расчета других данных
из приведенной таблицы. Это указывает на
то, что происходит димеризация бензойной
кислоты в органической фазе.
68
Рассмотренными примерами
не исчерпываются все
возможные типы равновесий.
Существуют более сложные случаи, когда
распределяемый компонент
реагирует химически с одним из
растворителей или с реагентом,
присутствующим в одной из фаз. В
частности, большое практическое
значение имеют такие процессы
при экстракции металлов
органическими растворителями.
Общий подход к изучению
равновесия в подобных системах должен
быть таким же, как и в
рассмотренных выше случаях.
Системы типа II. На рис. 37 изображена кривая
распределения для системы типа II. На осях координат отложены
концентрации распределяемого компонента С в равновесных
фазах, отнесенные к сумме концентраций компонентов А и С без
учета компонента В. Кривые распределения в этих координатах
для систем типа II по виду напоминают кривые равновесия для
многих систем пар—жидкость при постоянном давлении.
Кривая распределения в данном случае может быть описана
уравнением
Рис. 37. Распределение
компонента С в системе типа И.
X
ев
X
СА
= р
X
АВ
X
АА
ИЛИ
ХСВКХСВ+ХАВ)
Р-
хсаКХса + хаа)
1-хсв/(хсв+хав) 1-хсаКхса + Хаа)
(II, 34)
(И, 35)
Величина р является мерой легкости разделения
компонентов Л и С и называется селективностью. Для некоторых
систем р— величина постоянная. Так, в системе н-гептан(Л) —
анилин(5) — метилциклогексан(С) 18 р= 1,90. Эта постоянная
величина представляет собой отношение осмотических давлений
компонентов Л и С, насыщенных В. Однако в большей части
случаев р переменная величина.
Влияние температуры на равновесие. Зависимость
коэффициента распределения от температуры может быть определена
при условии, что изменение взаимной растворимости
компонентов А и В при изменении температуры незначительно. В этом
случае для раствора компонента С в фазе А может быть
использована зависимость48:
dlnyQA = Щ^
(11.36)
67
где уСА—коэффициент активности С в растворе (стандартным
считается здесь состояние чистого компонента С);
Нса — парциальная мольная энтальпия компонента С в
растворе;
Не — мольная энтальпия чистого компонента С.
Напишем аналогичное уравнение для фазы В и вычтем из
первого выражения второе. После интегрирования
(предполагая постоянство разности энтальпий) получим:
В этом уравнении Нса—Пев представляет собой мольную
теплоту переноса распределяемого вещества из одной фазы в
другую при данной температуре. / — постоянная интегрирования.
Если ас — активность компонента С, а хс — его мольная доля,
то Yc=ac/*c- Так как при равновесии аСА = асв, YcVycb=
^XcbIxca- Для разбавленных растворов
ХСВ _, \СТ\ВМВ?А _ тСМВ?А _ ХСВМВ МВ
ХСА [СТ\АМАРВ МА?В ~ХСАМА~тСМА ^ '
и, следовательно
или
Нг. — HrR М„
lgmc = 23RT +7~ ЪЦГ ("-40)
А
Таким образом^ зависимость lgmc от ЦТ будет линейной с
наклоном (Нса—Нев)/2,3 R-
Отмер и Фэкар 49, комбинируя уравнение (11,39),
написанное в дифференциальной форме:
— (нгд — TJrr,)dT
dlnmc= ( CARJ,2 cs (Ц.41)
с уравнением Клапейрона — Клаузиуса
XdT
RT2
dlnP=-pTT- (П.42)
получили
dinmc __ НСВ — НСА
dlnP
(11,43)
Принимая правую часть этого уравнения постоянной и
интегрируя, получаем _
Следовательно, зависимость тс от давления паров Р,
соответствующего данной температуре, выражается_в
логарифмических координатах прямой с наклоном (НСв—#са)А- В
данном случае в качестве шкалы температур использована
зависимость давления паров одного из компонентов от температуры.
При этом предпочтительнее пользоваться данными о давлении
паров одного из растворителей (А или В). В связи с тем, что тс
может изменяться с ХСа, данные о коэффициентах
распределения должны соответствовать одним и тем же концентрациям.
Влияние температуры на равновесие будет рассмотрено
также в главе III. Обзор опытных данных о равновесии тройных
систем, опубликованных до 1952 г., дан Фрэнсисом50. Сведения
о более поздних работах по равновесию в тройных системах
приводятся в ежегодных обзорах51.
ЧЕТЫРЕХКОМПОНЕНТНЫЕ СИСТЕМЫ
Изображение состава системы. Для изображения состава че-
тырехкомпонентных систем, даже при постоянной температуре,
необходима пространственная модель. Для этого предлагалось,
например, использовать равностороннюю треугольную призму,
основание которой является обычным треугольным
изображением одной из тройных систем, а высота выражает состав по
отношению к четвертому компоненту. Однако более ясное
изображение таких систем достигается с помощью правильного
тетраэдра 52'53. Каждая из боковых поверхностей правильного
тетраэдра представляет собой одну из возможных в системе
комбинаций трех компонентов (см. рис. 38). В случае
необходимости использование ортогональных проекций на одну из
боковых сторон тетраэдра позволяет изобразить систему в
обычных треугольных координатах.
Допустим, например, что точке Р
отвечает процентное содержание
компонентов А, В, С и D,
соответственно равное Ха, Хв, Хс и
XD. Если спроектировать эту
точку на плоскость ABC, положение
ее проекции Р' можно
определить только тремя
концентрациями А, В я С: Х'А, Х'в и Х'с.
Можно показать, что
ХА = ХА + "3~ В
у
Х'В = ХВ+^- (11,45) С
X Рис. 38. Изображение четырех-
X' = X -I — компонентных систем с помощью
с с 3 тетраэдра.
59
По другому методу тройные системы изображаются
прямоугольными треугольниками, образующими тетраэдр, у которого три
из четырех сторон взаимно перпендикулярны54.
Из-за трудоемкости экспериментального исследования число
изученных четырехкомлонентных систем незначительно.
Брейкер, Хюнтер и Нэш 62 довольно подробно исследовали систему
хлороформ — ацетон — уксусная кислота — вода при 25° С. Эта
четырехкомпонентная система состоит из следующих тройных
систем:
1. Хлороформ — вода — уксусная кислота (тип I).
2. Хлороформ — вода —ацетон (тип I).
3. Хлороформ — ацетон — уксусная кислота (полностью
смешиваются).
4. Вода — ацетон — уксусная кислота (полностью
смешиваются).
Такая система схематически изображена на рис. 39. Бино-
дальная кривая MJLRK является кривой растворимости для
системы хлороформ — уксусная кислота — вода, прямая JR — одна
из хорд равновесия этой системы. Аналогично кривая MUNVK
представляет собой бинодальную кривую тройной системы
хлороформ— вода — ацетои, UV — одна из ее хорд равновесия. Для
этой системы обнаружена довольно простая связь между
хордами равновесия для тройных систем и хордамичетырехкомпо-
нентной системы: Построим плоскость, проходящую через
какую-либо из хорд для тройной системы (например, UV), и
вершину тетраэдра, соответствующую 100% уксусной кислоты, а
также плоскость, проходящую через хорду JR и вершину,
отвечающую 100% ацетона. Линия пересечения построенных
плоскостей является одной из хорд четырехкомпонентной системы.
Ацетон
Уксусная
' кислота
Хлороформ
М
И~Вода
Рис. 39. Система хлороформ — вода — уксусная
кислота —■ ацетон при 25° С (схематически).
60
Рнс. 40. Четырехкомпонентная
система с двумя несмвшнвающнмнся
парами компонентов.
Этому правилу подчиняются
также некоторые системы,
встречающиеся в практике экстракции
нефтяного сырья смешанными
растворителями53. Однако
правило не носит общего характера
и применимо только в частных
случаях.
Графические расчеты с
помощью диаграмм для
четырехкомлонентных систем, даже в
системах, следующих упомянутому
правилу, очень сложны.
Поэтому Смит55 предложил для двух
тройных систем с ограниченной
взаимной растворимостью,
входящих в четырехкомпонентную
систему, изображать хорды
равновесия по методу Хэнда, т. е. в
виде двух прямых линий (см.
рис. 32). Тогда точки, лежащие между этими линиями, будут
представлять собой хорды равновесия для
четырехкомпонентной системы.
Четырехкомпонентные системы, в которые входят две
тройные системы типа I и одна типа II, также представляют
значительный интерес для жидкостной экстракции. Характерным
примером может служить изображенная на рис. 40 система
вода— бензол — этилизовалериат — этанол56. В ряде
исследований 56>57 изучали проблему получения равновесных данных для
четырехкомлонентных систем, исходя из данных для
составляющих их тройных систем. Однако зависимостей, имеющих общее
значение, получено не было. Описана также
четырехкомпонентная система, образующая три жидкие фазы 58.
Кривые распределения. Если рассматривать В и С как
вещества, распределяемые между несмешивающимися
растворителями А и D, можно ожидать, что равновесные концентрации
компонентов В и С будут связаны зависимостями, подобными
показанным на рис. 41. На рис. 41, а приведено равновесное
распределение компонента В в зависимости от концентрации
компонента С (Xcds Xcd, и т. д.) в фазе D. Аналогично на
рис. 41,6 показано распределение компонента С в функции от
концентрации компонента В в той же фазе D. В некоторых
системах, особенно при малых суммарных концентрациях
распределяемых веществ, компоненты В и С распределяются
независимо друг от друга, и, следовательно, для каждого из них будет
одна кривая распределения. В случае применимости простей-
61
а б
Рис. 41. Кривые распределения для четырехкомпонентных систем.
шего закона распределения эти кривые, очевидно, превратятся
в прямые линии. Если коэффициенты распределения равны:
X
ВА
X
их отношение
BD
X
тс= х
СА
CD
Р =
хва1хса
xbdIxcd
(И, 46)
(Н, 47)
называется селективностью. Селективность является мерой
легкости разделения компонентов В и С. В общем случае она
зависит от их суммарной концентрации и от концентрации
каждого компонента в отдельности.
В тех случаях, когда вещества В и С порознь подчиняются
закону распределения, их влияние на распределение друг друга
может быть рассчитано. Примером служит распределение
диссоциирующих соединений,, образующих общий ион.
Рассмотрим, например, распределение одноосновной кислоты В между
органическим растворителем {.А) и водой (D). При этом допустим что
взаимная растворимость А и D не изменяется при умеренном изменении
концентрации В. Применяя к данному случаю уравнение (11,24), получаем
*»№+],
где
[Д]д __
(И, 48)
Аналогично для одноосновшой кислоты С
[Я
[CT]D [Н+Ь+Кдисс
*с[Н + ]д
(И, 49)
62
аС = *дис, С
Если присутствуют обе кислоты, то
Ионы водорода образуются в данном случае прн диссоциации обеих
кислот:
1н+1в = ав1вт]о + ас1стЬ ("-51>
Следовательно
Подобное выражение получается н для Кдас, с. Если каждая из
величин ав и ас гораздо меньше единицы, можно показать, что для водного
раствора, содержащего обе кислоты
aj~Vjhf \В 1 4-АГ ГС 1 / (4,53)
1^Д„С,В[^Ь+^ДИС.,С[СГЬ/ (П,54)
Если С — более сильная кислота, то [Н+]д = etc [Сг] или рН =
= lg(l/ac[Cr]ri), и из уравнения (11,51) следует, что диссоциация слабой
кислоты подавлена. При постоянном тпв ее концентрация в органической
фазе (фаза А) больше. Добавление к водному слою минеральной кислоты
вызовет в большей степени переход в органическую фазу более слабой
органической кислоты. Если обе кислоты диссоциированы очень незначительно,
эффект будет, конечно, малым.
Прн подщелачиванни водной фазы [™+]д <С^ ^дис. для °бенх кислот.
Тогда из уравнения (И, 50) следует
__в_ = в "дне, с (П> 55)
тС '"С^дис, В
Если оба отношения Кдис, с/Кцвс, в и тв/т'с больше или меньше
единицы, добавление щелочи приведет к увеличению селективности. Брэнкер47
обнаружил, что чем большая доля основания взаимодействует с более
сильной кислотой, тем меньше его расход. Этот принцип был использован Уоке-
ром59 для улучшения разделения т- н р-крезолов. Константы диссоциации
этих соединении в воде равны соответственно 0,98-Ю-10 и 0,67 • 10-10.
Коэффициенты распределения т'в и т'с указанных изомеров между водой н
бензолом одинаковы, однако между раствором NaOH и бензолом они
распределяются с селективностью 6=0,98/0,67=1,46 [согласно уравнению (II, 55)].
Опытная величина, полученная Уокером, равнялась 1,43.
Пример 11-4. В системе эфир (А)—уксусная кислота (В)—вода (D)
концентрации [ВтЬ = 0,02 моль/л прн 18°С соответствует равновесная
концентрация в эфнре [В]а = 0,00966. Следовательно, коэффициент
распределения гПд = 0,00966/0,02 = 0,483. Константа диссоциации уксусной кислоты в
воде Кдис, в=1,75-Ю-5. При распределении между водой и эфиром
муравьиной кислоты (С): [СтЬ=0,02, [С]А = 0,00746, т"с= 0,373, АГдис., с =
= 1,76 • I0-4. Рассчитать равновесные концентрации в эфирном слое, если в
водной фазе присутствуют совместно уксусная и муравьиная кислоты при
концентрации каждой, равной 0,02 моль/л,
63
Решение. Степень диссоциации уксусной кислоты в отсутствие
муравьиной
_ — Кдис, в + (^дис, в + 4 [Вт\р ^дис, в)
- - 1,75• 10-5 +[3,06•10-10 + 4-0,02-1.75- 10"5]°'5 _ nn9Q9
j-^щ и,ад«
Следовательно, коэффициент распределения недиссоциированной кислоты,
принимаемый постоянным, пгв = тд/(1 — ад) = 0,483/(1 — 0,0292) = 0,498.
Подобный расчет для муравьиной кислоты (в отсутствие уксусной) дает
ас =0,0896 и т'с = 0,373 (1 — 0,0896) = 0,409. -
В присутствии обеих кислот с концентрацией rg i = iQ l = 0,02 при
помощи уравнения (11,53) получаем
= , ,; -si 1
а0= 1,75-Ю-0'I-
u
75 • 110" 30,02 + 1,76 -10- q ■ 0,02.
0,5
= 0,00890
Аналогично из уравнения (11,54) ас=0,0894. Таким образом,
диссоциация уксусной кислоты в значительной мере подавлена, в то время как
степень диссоциации муравьиной кислоты почти не изменилась. Из уравнения
(II, 51) следует:
[Н + Ь = 0,00890) (0,02) + 0,0894 (0,02) = 0,001966
Концентрации в эфирном слое можно теперь найти по уравнению (11,48)!
0,498 • 0,001966
0,001966+1,75- Ю-5
: 0,493
[В] = 0,012 • 0,493 = 0,00986 моль/л
(0,409-0,001966 по.с
тс = ■ ■ j- = 0,376
0,0011966 + 1,76-Ю-4
[С]А = 0,02 - 0,376 = 0,00752 моль/л
Рассчитанная по этим концентрациям селективность
тп 0 493
P = ^ = W=1'312
Из коэффициентов распределения муравьиной и уксусной кислот
независимо друг от друга получилось бы значение (5=0,483/0,373=1,295. При
подщелачивании водного слоя |МОжно ожидать, что селективность, согласно
уравнению (11,55), будет равна:
04,98- 1,76- КГ' 2
0,409 ■ 1,75 • Ю-5
Однако при той же концентрации кислот количество каждой из них,
экстрагированное органической фазой, будет меньше из-за образования не-
экстрагируемых солей.
Вследствие изменения взаимной растворимости
растворителей и соответственно величины т' расчеты, подобные
приведенным выше, при высоких концентрациях распределяемых ве-
§4
ществ не будут точны. Эти расчеты не могут также заменить
экспериментального определения равновесия; они полезны лишь
для получения ориентировочных данных.
Высаливание. К четырехкомпонентным относятся системы,
образуемые при добавлении к тройным системам неэкстрагируе-
мых соединений, способствующих увеличению коэффициентов
распределения. Так, равновесная концентрация уксусной
кислоты в эфире может быть увеличена при добавлении к водному
слою ацетата натрия. Обусловлено это подавлением
диссоциации уксусной кислоты сильно диссоциированной солью, что, в
соответствии с изложенным выше, приводит к повышению
коэффициента распределения. Действие высаливающего агента
обычно осуществляется, как в приведенном случае, через общий
ион или в результате гидролиза с образованием, например, иона
водорода. При распределении неэлектролита (С) добавление
электролита часто вызывает изменение коэффициента
распределения в соответствии с правилом Сеченова60432:
*
lgHl£.= kX (И, 56)
тс S
где тс — коэффициент распределения в отсутствие соли;
пгс — коэффициент распределения в присутствии соли с
концентрацией Xs;
k — константа, отражающая влияние концентрации соли.
Известно, что в присутствии некоторых солей растворимость
как органических, так и неорганических веществ в воде
увеличивается. Это явление называется гидротропией. Особенно велик
эффект, оказываемый олеатом и даже бутиратом натрия, суль-
фонатами (например, ксилилсульфонатом натрия). Добавление
их к воде может привести к увеличению растворимости
органических жидкостей в ней в сотни раз63-65. В присутствии таких
солей характер равновесия можно полностью изменить, что
позволяет осуществлять экстракционные процессы, проведение
которых в отсутствие этих добавок оказалось бы невозможным.
Примеры применения подобных методов экстракции приведены
Ламбом и Винзором66.
МНОГОКОМПОНЕНТНЫЕ СИСТЕМЫ
Графическое изображение равновесия в системах с числом
компонентов более четырех весьма затруднительно. Вместе с
тем в ряде многокомпонентных систем жидкость — жидкость,
применяемых в промышленной практике, содержится столь
большое число компонентов, что их даже трудно
идентифицировать. Для большей части таких систем имеется возможность
группировать компоненты по химической природе последних и
рассматривать каждую систему состоящей из групп соединений.
§ P. g. Трейбад
85
150л Пример подхода к иссследова-
нию статики экстракции
сложных промышленных смесей
приведен ниже.
В состав растительных масел
(например, соевого масла)
входят ряд эфиров жирных кислот
и глицерина, как насыщенных,
так и ненасыщенных, различаю-
]2q / 1 / \ щихся по степени насыщения.
^РФУРоп Количество ненасыщенных соеди-
Рис. 42. Применение йодного .числа нений в маслах обычно оцени-
в качестве меры состава расти- вается «йодным числом», т. е.
тельного масла. числом сантиграммов иода,
поглощаемого 1 г масла. Йодное
число является аддитивным свойством: если 100 г масла с
йодным числом 50 смешать с таким же количеством масла,
имеющим йодное число 100, то 200 г полученной смеси будет иметь
йодное число 75. Йодное число смеси двух масел может (если
известно йодное число исходных масел) указывать их
относительные количества в смеси. Следовательно, йодное число
можно использовать (например, при выделении из масла
экстракцией фурфуролом части его с высоким йодным числом)
вместо концентраций на одной из координатных осей
треугольной диаграммы67 (см. рис. 42). Подобно этому, для
характеристики нефтяных масел по содержанию в них парафинов можно
применять значения вязкостно-весовых констант масел (см.
главу XIII).
Полученные таким образом диаграммы не являются
истинными диаграммами равновесия. Равновесные составы для них
обычно определяют однократной экстракцией. Так как
различные компоненты системы могут оказывать влияние на
распределение друг друга, а ком поненты даже одной группы соединений
распределяться по-разнюму, изменения объемного соотношения
экстрагента и экстрагируемого раствора должны влиять на
равновесие (в отличие от истинных трехкомпонентных систем).
Применение таких диаграмм равновесия к многостадийной
экстракции, а также вообще к условиям, отличным от условий,
при которых получены равновесные данные, может привести к
значительным ошибкам., Этот вопрос рассматривается Альдер-
сом68( см. также главы 'VII и IX).
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
А — компонент paiCTBopa; количество компонента А.
а — активность (з:а стандартное состояние выбран чистый
компонент).
В— компонент раствора; количество компонента В,
Ь --константа.
С — компонент раствора; количество компонента С.
dp — диаметр частицы, м.
F— число степеней свободы.
F — раствор, обозначенный точкой F; количество
раствора F.
j_ — летучесть, н/м2.
Н — парциальная энтальпия вещества в растворе,
дж/кмоль.
Н° — энтальпия чистого вещества, дж/кмоль.
Н+ — ион водорода.
/ — константа интегрирования.
Ка — константа равновесия реакции ассоциации,
^дис. — константа равновесия реакции диссоциации.
k — константа.
М — молекулярный вес.
т — коэффициент распределения, отношение равновесных
суммарных концентраций (в вес. долях)
растворенного вещества.
т! — коэффициент распределения, отношение равновесных
концентраций (в моль/л) растворенного вещества,
находящегося в простой молекулярной форме.
т" — коэффициент распределения, отношение равновесных
полных концентраций (в моль/л) распределяемого
вещества.
т!" — коэффициент распределения, отношение равновесных
концентраций (в вес. долях) распределяемого
вещества, 'находящегося в простой молекулярной форме,
N — число компонентов.
п — число молекул мономера в полимере.
п', п" — числа ионов.
Р — давление паров, н/м2.
pH-lg(l/[H1).
Р— универсальная газовая постоянная, дж/кмоль, °К.
г — константа.
S — раствор, обозначенный точкой 5; количество
раствора S.
Т — абсолютная температура, °К.
V — мольный объем, м3/кмоль.
X — концентрация, вес. доли.
х — концентрация, мол. доли.
а — степень диссоциации.
а' — степень ассоциации.
Р — селективность.
Y—коэффициент активности (при выборе в качестве
стандартного состояния чистого компонента).
5»
67
X — теплота испарения, дж/кмоль.
р — плотность, кг/м3.
а — межфазовое натяжение, н/м.
Ф — число фаз, находящихся в равновесии.
[ ] — концентрации, моль/л.
Индексы
А, В, С, D — относятся к компонентам А, В, С, D
(так, ХАв — весовая доля А в фазе В).
р — критическая точка.
Т — полная концентрация распределяемого вещества во
всех молекулярных формах.
ЛИТЕРАТУРА
1. Gibbs, J. Willard, Trans. Conn. Acad. Arts Sci., 3, 152 (1876).
2. J. R i с с i, The Phase Rule a. Heterogeneous Equilibrium, D. Van Nostrand
Company, Inc., Princeton, New York, 1951.
3. O. K. Rice, Chem. Revs., 44, № 1, 69 (1949).
4. J. H. H i 1 d e b r a n d, B. I. Alders, J. W. Beams, H. M. Dixon,
J. Phys. Chem., 58, 577 (1954).
5. J. D. Cox, J. Chem. Soc, 1952, 4601.
6. J. Timmermans, J. Lewis, Disc. Faraday Soc, № 15, 195 (1953)
7. A. E. H i 11, J. Am. Chem. Soc, 45, 1143 (1923).
8. H. K. Zimmerman, Chem. Revs., 51, 25 (1952).
9. F. Kurata, J. P. Kohn,, Petrol. Process., 11, № 12, 57 (1956).
10. J. S. Drury, Ind. Eng. Chem., 44, 2744 (1952); 51, 1491 (1959).
11. A. W. Fr a.n с is, J. Phys. Chem., 58, 1099 (1954).
12. A. W. Francis, Critical Solution Temperatures, Advances in Chemistry
Series, № 31, American Chemical Society, 1961.
13. A. W. F r a n с i s, J. Phys. Chem., 60, 20 (1956).
14. A. S. Smith, Ind. Eng. Chem., 42, 1206 (1950).
15. G. N. Vriens, E. С. М e d с a 1 f, Ind. Eng. Chem., 45, 1098 (1953).
16. S. W. Briggs, E. W. Comings, Ind. Eng. Chem., 35, 411 (1943).
17. F. A. H. Schrein em a:kers, Z. Physik. Chem., 25, 545 (1898).
18. K. A. Varteressian, M. R. F e n s k e, Ind. Eng. Chem., 29, 270 (1937).
19. M. Newman, С. В. Hay worth, R. E. Treybal, Ind. Eng. Chem.,
41, 2039 (1949).
20. D. De B. Darwent, С A. Winkler, J. Phys. Chem., 47, 442 (1943),
21. A. W. Francis, J. Am. Chem. Soc, 76, 393 (1954).
22. A W H i x s о n, J. В. В о с k e 1 m a n n, Trans. Am. Inst. Chem. Engrs.,
38, 891 (1942).
23. G. В Frankforter, F.. С F г а г у, J. Phys. Chem., 17, 402 (1913).
24 H. P. Meissner, С A. Stokes, Ind. Eng. Chem., 36, 816 (1944).
25. H. J. Mc D о n a 1 d, J. Am. Chem. Soc, 62, 3183 (1940).
26. С D. Bogin, Ind. Eng. Chem., 16, 380 (1924).
27. D. B. Hand, J. Phys. Chem., 34, 1961 (1930).
28. G. B. Arnold, С A. Coghlan, Ind. Eng. Chem., 42, 177 (1950).
29. E. D. Butler, С. В. Miles, С. S. Kuhn, Ind. Eng. Chem., 38, 147
(1946).
30. D. A. Drew, A. N. Hi x son, Trans. Am. Inst. Chem. Engrs., 40, 675
(1944)
31. H. E. Hughes, J. O. Ma lone y, Chem. Eng. Progr., 48, 192 (1952).
32. D. F. Othmer, P. E. Tobias, Ind. Eng. Chem., 34, 690, 693 (1942).
68
33. A. E. Hill, in «A Treatise on Physical Chemistry», H. S. Taylor, Ed.,
2nd ed., D. Van Nostrand Company, Inc., Princeton, New York, 1930,
p. 573.
34. E. Jane eke, Z. anorg. Chem., 51, 132 (1906).
35. International Critical Tables, v. Ill, McGraw-Hill Book Company, Inc., New
York, 1928, p. 393ff.
36. Т. К. Sherwood, Absorption and Extraction, 1st ed., McGraw-Hill Book
Company, Inc., New York, 1937, p. 242.
37. Тара сей ков Д. Н., Паульсен И. A., Acta physicochim. URSS, 11,
75 (1939).
38. R. L. P i 11 о t о n, A. S. Т. М., Spec. Tech. Pub., 238, 5 (1958).
39. I. Bachm an, Ind. Eng. Chem., Anal. Ed., 12, 38 (1940).
40. A. V. Brancker, T. G. Hunter, A. W. Nash, Ind. Eng. Chem., Anal.
Ed., 12, 35 (1940).
41. W. D. В a n с г о f t, Phys. Rev., 3, 120 (1895).
42. R. E. Treybal, Ind. Eng. Chem., 36, 875 (1944).
43. R. E. Treybal, L. D. Weber, J. F. Daley, Ind. Eng. Chem., 38,
817 (1946).
44. L. A 1 d e r s, Appl. Sci. Research, A4, 171 (1954).
45. W. Nernst, Z. physic. Chem., 8, 110 (1891).
46. С Golumbic, M. О г с h i n, S. We Her, J. Am. Chem. Soc, 71, 2624
(1949).
47. A. V. В r a n с k e r, J. Soc. Chem. Ind., 66, 453 (1947).
48. О. А. Н о u g e n, К. М. Watson, R. A. R a g a t z, Chemical Process
Principles, pt. 2, John Wiley a. Sons, Ine., New York, 1959.
49. D. F. Othmer, M. S. Thakar, Ind. Eng. Chem., 44, 1654 (1952).
50. A. W. Francis, Solubilities of Inorganic a. Organic Compounds, A.
Seidell a W. F. Linke, Eds., suppl. to 3rd ed., D. Van Nostrand Company,
Inc., Princeton, New York, 1952.
51. R. E. Treybal, Ind. Eng. Chem., 43, 79 (1951); 44, 53 (1952) н т. д.
(ежегодные обзоры).
52. А. V. В г a n k е г, Т. G. Hunter, A. W. Nash, J. Phys. Chem., 44, 683
(1940).
53. A. V. Brancker, T. G. Hunter, A. W. Nash, Ind. Eng. Chem., 33,
880 (1941).
54. J. H. W i e g a n d, Ind. Eng. Chem., Anal. Ed., 15, 380 (1943).
55. I. C. Smith, Ind. Eng. Chem., 36, 68 (1944).
56. Y. Chang, R. W. M о u 11 о n, Ind. Eng. Chem., 45, 2350 (1953).
57. R. G. H. Prince, Chem. Eng. Sci., 3, 175 (1954).
58. G. M. H a r t w i g, G. С Hood, R. L. M а у с о с к, J. Phys. Chem., 59,
52 (1955).
59. С. A. Walker, Ing. Eng. Chem., 42, 1226 (1950).
60. J. Setschenow, Z. Phys. Chem., 4, 117 (1889).
61. L. E. S w abb, E. L. Мог gan, Chem. Eng. Progr. Symp. Ser., 48, № 3,
40 (1954).
62. K. E. Whitehead, С J. G e a n к о р 1 i s, Ind. Eng. Chem., 47, 2114
(1955).
63. H. S. Booth, H. E. Ever son, Ind. Eng. Chem., 40, 1491 (1948); 41,
2627 (1949); 42, 1536 (1950).
64. A. D о b у - D u с 1 e a u x, Chem. Ztg., 76, 805 (1952).
65. H. В. К 1 evens, Chem. Revs., 47, 1 (1950).
66. E. С Lumb, P. A. W i n s о r, Ind. Eng. Chem., 45, 1086 (1953).
67. R. R. R u t h r u f f, D. F. W i 1 с о с к, Trans. Am, Inst. Chem. Engrs., 37.
M 649 (1941).
68. L. A 1 d e r s, J. Inst, Petrol., 42, 228 (1956).
РАСЧЕТ РАВНОВЕСИЯ В СИСТЕМАХ
ЖИДКОСТЬ — ЖИДКОСТЬ
при расчетах процессов экстракции и
"экстракционной аппаратуры необходимы
возможно более точные данные о
равновесии для соответствующих систем
жидкость — жидкость.
Как показано в последующих главах,
обоснованный выбор схемы процесса,
определение скоростей, при которых будет
протекать экстракция, оптимальное объемное
соотношение экстрагента и исходного
раствора, а также выяснение изменения
концентраций фаз при экстракции возможны
лишь на основе анализа фазовых диаграмм
равновесия. В связи с тем, что подробно
исследовано сравнительно немного трех-
компонентных систем и еще меньшее число
четырехкомпонентных, обычно возникает
необходимость в экспериментальном
определении равновесия для новых
экстракционных процессов.
При выяснении возможности
применения жидкостной экстракции как метода
разделения в каждом конкретном случае
нужно располагать равновесными данными
о нескольких системах для сравнительной
оценки различных экстрагентов. При этом
желательно иметь возможность, хотя бы
ориентировочно, определять расчетом
фазовое равновесие для этих систем, не
прибегая к эксперименту. Таким путем могут
быть заранее исключены многие
растворители, первоначально намечаемые для
использования в качестве экстрагентов, и
экспериментальные исследования
равновесия можно ограничить наиболее
перспективными системами. В некоторых случаях
представляет большую ценность уже одно
лишь указание о направлении
распределения вещества между двумя растворите"
лями.
70
ИДЕАЛЬНЫЕ РАСТВОРЫ
Идеальные растворы обладают следующими свойствами:
1. Смешение компонентов раствора не изменяет средней
величины сил межмолекулярного взаимодействия.
2. Объем раствора является аддитивным свойством объемов
компонентов во всем интервале концентраций.
3. Смешение компонентов раствора в любых соотношениях
происходит без поглощения или выделения тепла.
4. Давление паров раствора представляет собой линейную
функцию концентрации (в мол. долях) во всем интервале
составов.
Только очень немногие смеси могут образовывать идеальные
растворы. Для этого необходимо, чтобы молекулы компонентов
раствора были близки по строению, размерам и химической
природе.
Растворы, близкие к идеальным, образуют оптические
изомеры, вещества, являющиеся ближайшими членами
гомологического ряда, и подобные им смеси. Всякий реальный раствор
может лишь в той или иной мере приближаться по свойствам к
идеальному.
Применяемые при экстракции системы с ограниченной
растворимостью значительно отличаются от идеальных, и именно на
этом основана возможность осуществления самого процесса
экстракции. Степень отклонения растворов от идеальности
проявляется в отклонениях их свойств от перечисленных выше
свойств идеальных растворов. Изучение этих отклонений дает
возможность предсказывать поведение соответствующих систем
в процессе экстракции. Для расчета равновесия практически
наиболее важным свойством идеального раствора является
давление его паров, так как о температурах кипения растворов,
давлении паров, составах азеотропных смесей и равновесии
пар — жидкость накоплено значительное, количество данных.
Классификация соединений по признаку влияния
межмолекулярных сил на свойства растворов также может быть весьма
полезной при расчетах. Такие свойства, как изменение объема
раствора при смешении и теплоты растворения, имеют меньшее
значение вследствие ограниченности соответствующих
экспериментальных данных.
Закон Рауля. В простейшей форме закон Рауля может быть
сформулирован следующим образом: для идеального раствора
при условии, что паровая фаза ведет себя подобно идеальному
газу, парциальное давление р любого компонента равно
произведению давления пара р этого компонента в чистом виде на
его мольную долю х в растворе. Следовательно, для бинарного
раствора, состоящего из компонентов А и В, имеем:
Рл = хлрл Ре = хвре = (1~хА>р8 (IIIll)
71
f*const
Концентрация А В жидкости,
мол. Вола
Рис. 43. Закон Рауля для
бинарного раствора.
Давление Р равно сумме
парциальных давлений компонентов,
поэтому
Р = РА + Р
в'
= *дРд + (1-*д)^<Ш'2>
Из уравнения (111,2) видно, что
общее давление является линейной
функцией состава раствора. Состав
пара определяется уравнениями:
-1а = .
Ув=1~уА-
(1
Р
~ха)Рв
(Ш, 3)
Приведенные выше зависимости показаны графически на
рис. 43.
Мольная доля каждого компонента смеси прямо
пропорциональна числу его молекул; каждая молекула в паровой фазе
над раствором может рассматриваться как «испарившаяся» из
жидкости. Поэтому концентрацию любого компонента в паре
можно считать мерой склонности вещества к испарению1.
Соответственно возможна другая формулировка закона Рауля:
склонность молекул к испарению в идеальном растворе прямо
пропорциональна их концентрации в жидкости.
Пример III-1. Смеси «-пентана и к-гексана могут рассматриваться как
растворы, близкие по свойствам к идеальным. Рассчитать состав пара,
находящегося в равновесии с раствором, содержащим 0,3 мол. доли пентана
при 25° С.
Решение. Давления паров чистых пентана (А) и гексана (В) равны
соответственно рд=508 мм рт. ст. и рв = 149 мм рт. ст. при 25°С. Так как
*а = 0,3, согласно уравнению (III, 1), парциальные давления компонентов
составляют:
р = 0,3 ■ 508 = 152,4 мм pm. cm.
рв = 0,7 • 149 = 104,3 мм рт. ст.
Общее давление Р= 152,4 + 104,3=256,7 мм рт. ст. Следовательно, по
уравнению (111,3) мольная доля пентана в паровой фазе уА = 152,4/256,7=
= 0,594, а мольная доля гексана ув = 104,3/256,7=0,406.
Летучесть (чистые вещества). При высоких температурах и
давлениях законы идеальных газов не применимы. Однако
простая форма уравнений, описывающих свойства идеальных газов,
справедлива и для реальных газов при использовании гГонятия
летучести, введенного Льюисом2. Для чистого вещества при
постоянной температуре имеем3:
df>=*vdp (III, 4)
П
Если применимы законы идеальных газов, то
RT
v = — (III, 5)
Р
и, следовательно
dF = #7" dp/p = RT d In p (III, 6)
Летучесть / вещества определяется из следующего уравнения
для изохорно-изотермического потенциала, совпадающего по
форме с уравнением для идеальных газов:
dF=RTdlnf = vdp (III, 7)
или в интегральной форме (при постоянной температуре):
р
F — F° = RT In-L- = Г vdp (III, 8)
Р"
Верхний индекс 0 относится к некоторому произвольно
выбранному стандартному состоянию. Удобно принять f° = p° в
условиях, при которых вещество ведет себя подобно идеальному
газу. Величину р° можно принимать равной произвольному
невысокому давлению (обычно 1 ат). Летучесть можно
рассматривать как некоторое исправленное давление, при помощи
которого свойства реальных газов описываются уравнениями для
идеальных газов. Летучесть рассчитывают с достаточной
точностью из обобщенных зависимостей летучести от приведенных
температуры и давления 3>4. Отношение ///° называется
активностью а.
Летучесть растворов. Закон Рауля выражается через
летучести следующим образом:
где /д — летучесть чистого компонента А в жидком состоянии
• при температуре и давлении раствора;
/д — летучесть компонента А в растворе и в равновесном
с ним паре.
Такое выражение закона Рауля является более строгим,
так как не требует, чтобы паровая фаза была идеальной.
При равновесии летучесть компонента во всех фазах
одинакова3. Активность в растворах определяют так же, как и для
чистых веществ, т. е.
RT\na= RT\n-^ = T — F* (III, 10)
a=-I (111,11)
где /°— летучесть в стандартном состоянии.
Выбор стандартного состояния зависит от природы раствора.
Для растворов неэлектролитов удобнее всего принимать за стан-
73
дартное состояние чистого вещества при температуре раствора.
Давление в стандартном состоянии можно принимать равным
1 ат, давлению паров вещества при данной температуре или
давлению над раствором. ]В условиях, достаточно далеких от
критического состояния, при малых парциальных мольных объемах
влияние давления в стандартном состоянии на величину
активности незначительно. Для растворов электролитов иногда
удобнее выбирать в качестве стандартного состояния бесконечно
разбавленный раствор три соответствующих температуре и
давлении.
При выборе состояния чистого вещества в качестве
стандартного закон Рауля принимает вид:
aA*=f = xA aB = xB (111,12)
IA
Как было указано выше, в равновесных гетерогенных
системах летучесть каждого компонента одинакова во всех фазах.
Поэтому, если выбрало одинаковое стандартное состояние,
активность компонента во всех фазах одинакова. Отношение
а/х называется коэффициентом активности у. Для идеальных
растворов
YA = YB=1,0 (IH.13)
НЕИДЕАЛЬНЫЕ БИНАРНЫЕ РАСТВОРЫ
Отклонение реальных растворов от идеальных проявляется
в том, что парциальное давление, летучесть, активность и
коэффициент активности не подчиняются простым линейным
зависимостям, свойственным идеальным растворам. В качестве меры
отклонения от идеальнюсти обычно используют коэффициент
активности у. Так, для (бинарного раствора компонентов А и В
имеем: _ _
v* = T-77r ^lT = -h (IIU4)
ха- ха1а хв хв<в
Здесь летучести в стандартном состоянии fA и f°B являются
летучестями чистых компонентов при температуре и давлении
раствора (обычно можно пользоваться значением летучести
компонента при давлении, р)авном давлению его паров). Если к
паровой фазе применимы законы идеальных газов, получим:
Y = ^ = _El_==2^. Y_i5L__L__J^l („us)
А ха хаРа хаРа В хв хвРв хвРв
Коэффициент активности, следовательно, равен отношению
действительной летучесгги, парциального давления или
склонности вещества к испарению к величине того же параметра в
74
идеальном растворе при той же концентрации. Если
коэффициент активности больше 1,0, склонность вещества к испарению
больше, чем для идеального раствора, что соответствует
положительному* отклонению от закона Рауля. Если же
коэффициент активности меньше 1,0, склонность вещества к переходу
из одного состояния в другое меньше значения того же
параметра для идеального раствора, т. е. отклонения от закона
Рауля отрицательны *.
Пример II1-2. Прн 760 мм рт. ст. раствор этилового спирта (0,30 мол.
доли) н этилацетата имеет температуру кипения 72,2° С. Равновесная
концентрация этилового спирта в парах уа = 0,356. Рассчитать активности и
коэффициенты активности компонентов, предполагая, что к паровой фазе
применимы законы идеальных газов.
Решение. При 72,2°С давления паров чистых компонентов
соответственно этанола и этилацетата равны: рА =591,6; рв =639,7 мм рт. ст.
Кроме того, имеем:
х А = 0,30 хв = 1,0 — 0,30 = 0,70
уд = 0,356 ув= 1,0 — 0,356 = 0,644
Из уравнения (III, 15) получаем:
_ 0,356 ■ 760 _
Y^~~ 0,30 -591,6 -1'324
аА = уАхА = 1,524 • 0,30 = 0,657
0,644 • 760
УВ~ 0,70-639,7 -1'093
ав = увхв = 1,093 • 0,70 = 0,765
Если отклонениями паров от законов идеальных газов
пренебречь нельзя, следует вводить поправки на неидеальность
паровой фазы. В этом случае, согласно методу, предложенному
Бенедиктом с сотр.5, коэффициент активности может быть
выражен следующим образом:
УАР Ca-p)(va-ba)
YA = 3T7-<? RT (Ш- I6)
А" А
где Уд — мольный объем компонента А в жидком состоянии;
ВА — второй вириальный коэффициент в уравнении
состояния компонента A [vA = RTIP + BA]. Величину ВА
можно получить из данных о сжимаемости ВА =
= (RT/P)(Z—1) или, если необходимо, из
обобщенных зависимостей для коэффициентов
сжимаемости 4>6.
Уравнение Бенедикта применимо, если изменение объема при
смешении незначительно. Другие, более строгие методы учета
* Термины «положительные» и «отрицательные» относятся к знаку
логарифма Y-
75
неидеальности паровой фазы приведены в монографии Рида и
Шервуда6.
Пример 111-3. По данным Отмера с сотр. [Ind. Eng. Chem., 43, 707
(1951)], при давлении Р=5,08 ат и температуре 126,3е С для системы этанол
(А) — вода (В) составы жидкости н пара равны соответственно хА = 0,533 и
У а = 0,640. Рассчитать коэффициент активности этанола.
Решение. Температура Г= 126,3+273,1 =399,4°К. Давление паров
этанола при 126,3°С составляет 5 ат (р=4,91 • 105 «/.и2). Критические
температура и давление этанола равны соответственно 516,2° К и 63,1 ат (РКр.=
=61,9 • Ш5 к/ж2). Отсюда приведенная температура составляет 399,4/516,2=
=0,773 и приведенное давление равно: 5,0/63,1=0,0792. Из обобщенной
зависимости6 имеем: Z=0,930; Я=8,314-103 дж■ кмоль~1 град~1. Следовательно,
ВА = (RT/P) (Z — 1) = 8,314 • 103 • 399,4 (0,930 — 1) (5,08 • 9,81 ■ 104) =
= — 0,452 м3/кмоль
При 126,3° С мольный объем этанола Уа = 66,5-10~3 м31кмоль. Величина
(ра~р)(уа~ваУ^т
г ' =0,9987. Подставляя вычисленные значения в
уравнение (III, 16), получаем:
_ 0,640 • 5,08 • 0,9987 _
УА - 0,533-5,0 ~ 1М
На рис. 44—48 приведены типичные зависимости
парциального давления, активностей и коэффициентов активностей для
ряда систем, причем коэффициенты активности нанесены на
графики в логарифмическом масштабе.
На рис. 44 показаны соответствующие зависимости для
системы с положительными отклонениями от закона Рауля (такие
системы наиболее распространены). Если положительные
отклонения велики, а давления паров чистых компонентов не сильно
отличаются друг от друга, кривая общего давления обычно
проходит через максимум (рис. 45), т. е. система образует азео-
тропную смесь. Для азеотропного состава х=у, поэтому
Р Р
Y,= y» = № 17)
А РА в Рв
На рис. 46 приведены те же зависимости для системы с
ограниченной взаимной растворимостью компонентов. В
двухфазной области между равновесными концентрациями xt и х2
парциальные давления и активности каждого компонента
одинаковы в обеих фазах. Когда общее давление достигает
максимума, кривая общего давления имеет плоский участок. Этот
случай соответствует образованию гетероазеотропной смеси.
Сравнительно редкий пример системы с положительными и
отрицательными отклонениями приведен на рис. 47. На рис. 48
представлена система с большими отрицательными
отклонениями, образующая азеотропную смесь.
Во всех приведенных примерах по мере приближения
концентрации компонента к единице его свойства приближаются
76
250
^ 200
X /50
о
7
6
5
а а з
щг
I
о о,г о,и о,б qe 1
Концентрация ацетона, мол. воли
в
РиС'//п4' Парциальные давления (а),
активности (о) и коэффициенты активности (в) для
системы ацетон — вода при 25° С [данные Бэра,
a«i?inKapa и ФеРг!°соиа, см. J. Phys. Chem.,
», 1310(1930); коэффициенты активности
удовлетворяют уравнениям Ваи-Лаара второго
«орядка при ААВ = Ig6,5 и АВА = lg4 (кри-
ш£-Иа иижием рисунке); пунктиром показаны
твора?ТСТВуКИЦИе Лииии для иДеальи0го Рас-
\°
\°
А цетон
V>
Boda^/t
1^
700
600
% 500
иоо
%300
1
15.200
100
О
1
ъ0,8
А
■<
02
К О
1 /
1 /
1/
р
cs2
ч.
/
/
/
Ацетон
ч.
>
\д
"ч ■?
ч. \
-^
/ /
1/
1
ч ^
/
/
/
X
V
/Ацетон
ч
ч
ч
ч
\ д
ч т
\ \
\
1 6
V
t з
£ г
t >
§0,9С
Ik
Ацетон^
4f%
А л
0,2 ofi 0,6 qe 1
1нцентрщия CSZ, мол. доли
6
Рис. 45. Парциальные давления (а),
активности (б) и коэффициенты активности (в) для
системы сероуглерод —ацетон при 35,17° С
[данные Завидского, см. Z. physlk. chem.,
35, 129 (1900); коэффициенты активности
удовлетворяют уравнениям Ваи-Лаара второго
порядка при А = lg 4,0 и А = lg 6,6
(кривые на иижием рисунке); пунктиром показаны
соответствующие лииии для идеального
раствора].
к соответствующим свойствам в
вые парциального давления и
Две
х, жидкие фазы хг
О
во
во
£30
к го
<§ is
| W
« °
16
•а.
"S- р
|^
#?
(■у
s
\
\
\-Анилин
\
X
м
к
V
\
\
у
/
/
S,
^
Л
*<
—•*!
гу№
& L
Л V<7#Z
'"'б
4? о/, о,б 0,8 1
Две
fHuffkus фазы т
1 2 о
Концентрация анилит,иолдо.ш
В
идеальном растворе. Так, кри-
активности касаются
соответствующих прямых для
идеальных растворов вблизи х=
= 1,0, а коэффициент
активности с увеличением х стремится
к 1,0. Это свойство характерно
для всех систем и может быть
использовано при проверке
экспериментальных данных.
Поскольку величина
коэффициента активности очень
чувствительна даже к небольшим
экспериментальным ошибкам,
а последние возрастают при
очень малых или очень
высоких концентрациях, в области
концентраций, близких к х=0
или х=\, обычно
наблюдается значительный разброс
значений коэффициентов
активностей. Поэтому, если имеется
только одно значение
коэффициента активности при
малых или высоких
концентрациях, оно может оказаться
неточным.
При наличии ассоциации
или диссоциации молекул по
мере приближения мольной
доли компонента к единице
коэффициент активности не
будет стремиться к 1,0, если
для расчета концентраций не
пользоваться истинными
молекулярными весами.
Рис. 46. Парциальные давления (а),
активности (б) и коэффициенты
активности (в) для системы анилин —
вода при 100° С [данные Грисволда,
Андре и Гарланда, см. Ind. Eng.
Chem., 32, 878, (1940); коэффициенты
активности удовлетворяют
уравнениям Ван-Лаара второго порядка при
АВА — lg 71 и АВА = lg 6 (кривые на
нижнем рисунке); пунктиром
показаны соответствующие линии для
идеального раствора].
76
О
В
5
4>
?N ?
§|
§Г| г
э 1
^ 1
\-~Зтанол
Хлороформ
1
0,9
О 0£ 0и 0,6 0,8 I
Концентрация этанола, мол. Воли
3S0
300
5 ZSO
гоо
150
<S| 100
50
д
/
А
S.
V
/ >
/ /
f f
Р
/
ш
й&ч
ч
V
Л
г
\ Ацетон
1
Хлороформ
1
ii
If
/
0,8
0,6
Ofi
03
О 0,2 0,4 0,6 0,8 1
Концентрация ацетона, мол. доли
Ацетон-^ef
Хлороформ]
Рис. 47. Парциальные давления (а),
активности (б) и коэффициенты
активности (в) для системы этанол —
хлороформ при 45° С [данные Скэт-
чарда и Рэимонда, см. J. Am. Chem.
Soc, 60, 1278 (1938); коэффициенты
активности удовлетворяют
уравнениям Маргулеса третьего порядка
при ААВ = lg 5,0 и АВА = lg 1,65
(кривые на -нижнем рисунке); пунктиром
показаны соответствующие линии
Для идеального раствора].
Рис. 48. Парциальные давления (а),
активности (б) и коэффициенты
активности (в) для системы ацетон —
хлороформ при 35,17° С [данные
Завидского, см. Z. physik chem., 35,
129 (1900); коэффициенты активности
удовлетворяют уравнениям
Маргулеса третьего порядка при ААВ =
= lg 0,39 и Лвд = ^0,51 (кривые на
нижнем рисунке); пунктиром
показаны соответствующие линии для
идеального раствора].
Уравнение Гиббса — Дюгема. Для любого экстенсивного
свойства раствора можно написать4:
**(%L+''&L+--°
При постоянной температуре, согласно уравнению (111,10),
находим:
dF = RTd In f (III, 19)
Тогда уравнение (III, 18) принимает вид:
, А +xJ , в +...=0 (111,20)
дХА 1г,Р *\ дХА )т,Р
Для бинарных растворов
ха + хв-1 (ш'21)
и
dxA= — dxB (111,22)
Поэтому
<Э1п7д\ /дЫТ
"[
Это и есть уравнение Гиббса — Дюгема. Его можно
написать также через производные коэффициентов активности:
din у, \ f dlnya\
-*А.,-*'Ы?1.Г <"|'24)
Уравнение (111,24) можно использовать при проверке
термодинамической консистентности данных для коэффициентов-
активности3'7, для чего удобнее написать его в следующей форме:
[(*'пУл)А*л1г,р 'в
[(<»ПУв)1дхА1т,Р~ ХА
Из этого уравнения видно, что отношение наклонов
зависимостей lgv от хА должно равняться отношениям (—хв/хА).
Если коэффициенты активности определяют из данных по
давлениям паров, поддерживать постоянными температуру и
давление одновременно невозможно. Этот вопрос рассмотрен в
работах Ибля и Доджа8, а также Ван-Несса9. Как будет показано,
однако, влияние температуры и давления на коэффициенты
активности часто невелико и уравнение (111,25) можно написать
в виде:
(dlgyA)/dxA хв
(^SyB)/dxA—^ (Ш-26>
Это уравнение можно применять в большей части случаев
либо при постоянной температуре, либо при постоянном
давлении. Данные, не удовлетворяющие уравнению (111,26), следует
80
считать термодинамически
неконсистентными; ими следует
пользоваться с
соответствующими ограничениями. В
интегральной форме уравнение
(111,26) имеет вид:
1ВУЛ
или
IgY,
Г 1м.
J хя
dlgyR (111,27)
'л-1
- J -J*- d Ig yA (HI, 28)
«N?
Рис. 49. Проверка
термодинамической консистентности по Редлиху —
Кистеру.
Выражения (111,27) и (111,28) можно применять для
расчета коэффициентов активности одного из компонентов, если
коэффициенты активности другого компонента известны для всего
интервала составов. Действительно, как видно из
уравнения (111,27), площадь под кривой зависимости хв/ха (ось
ординат) от lgys (ось абсцисс) в пределах от хА = \ (илихв/хА = 0)
до хА должна быть равна \gyA. Аналогично, если известно уА,
можно определить ув [уравнение (111,28)].
Редлих и Кистер 10 показали, что
. = i
, -о
Ig ~4- dx л = 0
Y„ A
(III, 29)
Это условие является необходимым, но не достаточным для
того, чтобы экспериментальные данные удовлетворяли
уравнению Гиббса — Дюгема. В соответствии с этим условием площади
под кривой lg(YA/vs) B функции от хА, расположенные выше и
ниже нулевой ординаты, должны быть равными (рис. 49). Вся
заштрихованная площадь между указанной кривой и ординатой
1&(Уа1ув)=0 должна быть алгебраически равна нулю.
Описанные графические методы полезны для установления
термодинамической консистентности экспериментальных данных
и построения термодинамически консистентных кривых при
наличии данных с большим разбросом.
Интегральные формы уравнения Гиббса — Дюгема.
Уменьшение свободной энергии при растворении пА молей
компонента Л в чистом компоненте В, согласно уравнению (III, 10),
равно: _
»д^-^) = "д«п4 О».30)
1а
6 Р. Е. Трейбал
61
Для идеального раствора ]а1(а=ха и уравнение (111,30)
принимает вид:
пл(Гв-^в)=пАЯТ1пхА (Ш. 31)
или
Па?а = njfiT InxA + nAFA (III, 32)
Соответственно для компонента б в идеальном растворе
имеем: _
nB(FB-FB) = nBRT\nxB (Ш.ЗЗ)
nBFB = nBRT\nxB + nBFB (111,34)
Следовательно, для идеального раствора полная свободная
энергия раствора
F = (пАТА + nBFB)m = nARTIn*д + ПдЛ7-In*в + nAF°A + nBF% (III, 35)
Аналогично в случае неидеального раствора, для которого
f/f° = yx, получим:
("А + «Лвенд. = "д^Г1П ХА + nBRT 1П ХВ +
+ "д^д + "я^ + "дЯПп^'д + "яЯ7'1пув (1«. 36)
' Разность свободных энергий для идеального и неидеального
растворов называется11 избыточной свободной энергией и
обозначается FE. Очевидно
FE=nART\nyA + nBRT\nyB (Ш,37)
F = nART\nxA + nBRTlnxB + nAFA + nBF°B + FE (111,38)
Дифференцируя уравнение (111,38) по пА, находим
^! = ?А = ЛГ1п*д + /^+^=ЛЛп*А + ^ + ЛГ1пуА (Ш. 39)
ПА А
Следовательно
RT\nyA=^- (IH.40)
"А
ИЛИ
д (п .-\-п0) F„
*""*А= *£/ (Ш'41)
А
ЛПпуи =
^ + "*)F£ (111,42)
в <Эгс
в
Вооль 12 показал, что большая часть известных интегральных
форм уравнения Гиббса — Дюгема представляет собой
упрощенные модификации уравнений, связывающих избыточную свобод-
82
ную энергию с энергией взаимодействия различных
молекулярных групп из двух, трех, четырех молекул и более. Число,
молекул в наибольших группах, принимаемых в рассмотрение,
определяет порядок уравнения. Так, если учитываются группы,
состоящие из двух и трех молекул, получают уравнение третьего
порядка. В уравнении третьего порядка для бинарной смеси А
и В величина
FJ2,3 RT— сумме вкладов молекулярных групп А к В; А, А и В,
а также А, В и В (III, 43)
Вклад каждой из этих групп в величину избыточной
свободной энергии раствора принимают равным xAxBkAB, хахаХв^аав
и т. д. Дифференцируя полученную сумму в соответствии с
уравнениями (111,41) и (111,42), находим по существу
полуэмпирические зависимости между у и х. С помощью подобных
зависимостей описываются, например, кривые для коэффициентов
активности, приведенные на рис. 44—48, причем эти
зависимости одновременно удовлетворяют уравнению Гиббса —
Дюгема.
Зависимость избыточной свободной энергии от концентрации
можно выражать также просто эмпирическим степенным рядом.
Чем сложнее молекулярные группы, учитываемые уравнением
(111,43), или чем больше членов взято в степенном ряду, тем
большее число эмпирических констант содержит получаемое
уравнение и соответственно тем более широкий круг различных
систем это уравнение может описывать. Одновременно, чем
меньше констант содержит уравнение, тем меньше
экспериментальных данных необходимо для их вычисления, но число
систем, к которым оно может применяться, более ограниченно.
Далее будет показано, что возможен чисто теоретический
подход к вычислению коэффициентов активности, имеющий,
однако, практическую ценность только в отдельных случаях.
Известны уравнения Гиббса — Дюгема в их интегральной
форме с одной, двумя, тремя и более константами.
Уравнения с одной константой
(симметричные системы). Эти уравнения имеют вид:
>g У а = аав*в «В У в = аваха (Ш, 44)
ААВ = АВА
Здесь
А = lim Ig у при х -> 0 или
конечное значение lgv^ или 'ёУд
Ава = lim lg ув ПРИ хв "*" ° или
конечное значение Ig у„ или lg yB
6*
(III, 45)
(Ш, 46)
(Ш, 47)
83
Уравнения (111,44) называют симметричными, так как
константы ААв и Ава равны и зависимости lg у от ХА представляют
собой зеркальное изображение друг друга. Лишь очень
небольшое число систем может быть описано при помощи уравнений
(111,44). Поэтому последние имеют ограниченное применение и
представляют интерес главным образом как предельные случаи
более сложных уравнений с двумя константами.
Уравнения с двумя константами. Чаще всего
применяют следующие уравнения:
/. Уравнения Ван-Лаара второго порядка 7<13
1ёУл = -
АВ
м C+VVVi,)2
А
ВА
В £+АВАХв1ААВХА?
ИЛИ
ВА~В1"АВГА>
*JgY„\2
^Ы'+Ш)
XAlS'
ха12Уа\2
а^1Ы1+Ш)
(Ш, 48)
(Ш, 49)
(Ш,
№
50)
51)
Константы ААВ и АВА имеют то же значение [уравнения
(111,46) и (III, 47)],-что и в уравнениях (111,44).
Первоначально эти константы связывали с константами уравнения
состояния Ван-дер-Ваальса. Однако в настоящее время принимают,
что они носят чисто эмпирический характер.
Примеры применения уравнений (111,48) — (111,51)
приведены на рис. 44—46.
2. Уравнения Маргулеса третьего порядка7'1*
или
•g Уд = (2АВА - ААВ) хв + 2 (ААВ - АВА) хв
<g У в = (2 Au - АВА) х\ \- 2 (АВА - ААВ) х\
(*B-xA)lgyA , 2IgYB
{
ААВ =
в)1^
~А
2'gY,
ВА
(III, 52)
(III, 53)
(III, 54)
(III, 55)
где ААВ и АВА также определяются уравнениями (111,46) и
(Ш,47).
Соответствующие примеры представлены на рис. 47 и 48.
Уравнения Маргулеса наиболее пригодны для относительно
симметричных систем; они удобны тем, что могут выражать мак-
84
симум или минимум на кривой зависимости lg у от состава*.
Дифференцирование уравнений Маргулеса показывает, что
экстремальные значения lgy возможны, если АвА/ААВ>2,0. Воол,
однако, показал, что в таких сильно несимметричных системах
уравнения Маргулеса не пригодны для количественного
описания экспериментальных данных; более подходящим критерием
появления экстремума является неравенство АВА/ААВ>2 +
+ 2,3 АВА/4. Для сильно несимметричных систем без
экстремумов уравнения Маргулеса не применимы. Если ААВ=АВА, то
уравнения Маргулеса сводятся к уравнениям (111,44).
Уравнения Ван-Лаара могут удовлетворительно описывать
системы с высокими значениями величин ААВ и АВл и более
несимметричные, чем системы, характеризуемые уравнениями
Маргулеса. Однако к системам с очень большими отношениями
АВА/ААв уравнения Ван-Лаара не применимы. В тех случаях,
когда отношение АВА/ААВ близко к двум, уравнения
Ван-Лаара лучше соответствуют опытным данным по сравнению с
уравнениями Маргулеса. Однако уравнения Ван-Лаара более
ограничены в отношении форм кривых, которые можно получить,
так как эти уравнения не могут отражать максимума или
минимума ira кривых зависимости \gy от концентрации, а также
изменения знака \gy.
Если АВА = ААВ, то уравнения Ван-Лаара также сводятся
к уравнениям (111,44). Поэтому для систем, близких к
симметричным, уравнения Маргулеса и Ван-Лаара дают почти
одинаковые результаты.
Сильно несимметричные системы, а также системы, в
которых происходит ассоциация, лучше описывать уравнениями с
числом констант более двух.
Уравнения с тремя и более константами.
Уравнения типа Маргулеса и Ван-Лаара, содержащие три и более
констант, были выведены Воолем12. Блэк15 ввел в уравнения
Ван-Лаара дополнительные члены. Однако более удобными для
практического применения оказались уравнения Редлиха и Ки-
стера10',6, представляющие собой уравнения четвертого
порядка:
J ^yA = xB[BAB-CAB + DAB + (4CAB-8DAB)xA + l2DABxA] (III, 56)
l ^yB^^A[BAB + CAB+DAB-(ACAB + 8DAB)xB+\2DABxB] (111,57)
или
'* 71 = BAB (XB - XA) + CAB (&XAXB - 0 +
'B
+ DAB {хв - xa) (4 - bxAxB + x\) (III, 58)
* Если lg у а имеет максимум, то lg у в при тон же концентрации имеет
минимум.
65
Эти уравнения относятся к уравнениям типа Маргулеса.
Если Dab = 0, то они сводятся к уравнениям (111,52) и (111,53);
при этом ААВ = ВАВ—Cab и Ава = Вав + Сав. Если необходимо,
в уравнения Редлиха и Кистера можно ввести дополнительные
члены. Однако с помощью этих уравнений в их первоначальном
виде также может быть описана большая часть систем.
Влияние температуры. Зависимость коэффициента
активности от температуры выражается следующим уравнением4'7:
/д'пул\ _ НА-Н\
\ дТ )р —
RT2
(III, 59)
где НА — парциальная мольная энтальпия компонента А в
растворе;
НА — мольная энтальпия чистого компонента А при той же
температуре.
Уравнение (III, 59) можно привести к виду:
А - - s (III, 60)
d(\/T) 2.303Я
где АН3— парциальная мольная теплота растворения
компонента А.
Аналогичное уравнение можно написать и для компонента В.
В случае бесконечно разбавленного раствора уравнение (111,60)
принимает вид:
dAAB дЯо
(III, 61)
d(\/T) 2.303Я
где AHrs— теплота растворения А при бесконечном
разбавлении.
Из наклона зависимости lgVA от 1/7" для какой-либо
величины хА можно определить дифференциальную теплоту
растворения, или при хА = 0 — теплоту растворения при бесконечном
разбавлении. Обратным путем можно установить зависимость
от температуры коэффициентов активности; для этого наиболее
удобно пользоваться уравнением (111,61). К сожалению,
данные о теплотах растворения ограничены.
Обычно при образовании растворов с положительными
отклонениями от закона Рауля происходит поглощение тепла.
Следовательно, теплота растворения АН3 положительна и уА будет
уменьшаться с возрастанием температуры. Для систем с
отрицательными отклонениями величина AHS, как правило,
отрицательна. Другими словами, в обоих случаях при увеличении
температуры отклонение системы от закона Рауля уменьшается.
Исключение из этого правила составляют системы с нижней
критической температурой растворения, в которых по крайней
мере вблизи от К-Т.Р.
отклонения от закона Рауля
увеличиваются при повышении
температуры.
Ряд авторов, в том числе
Бенедикт с сотр.5, вводят
температуру в зависимости у от
концентрации в виде произведения
Т lg у. При хА = 0 эта величина
равна ТААВ, откуда следует
простая приближенная зависимость:
-^ = ^2 (Ш,62)
ЛАВ1 ' 1
Это уравнение не имеет
общего значения, но при отсутствии
экспериментальных данных
может служить полезной
аппроксимацией. Как будет показано
ниже, влияние температуры на
величину коэффициента
активности для многих систем можно
определить с помощью табл. 6 и
7 (стр. 100).
4*
I
t
1
1
о го°с
о40Т
*55°С
л 75"С
• 7в,3-Ю0°С
6"
0,85
0.8
~'~о о,г о,и o,6 o,g i
Концентрация этанола, мол. доли
Рис. 50. Влияние температуры на
и„ „ ,„ кп л. коэффициенты активности этило-
На рис. 50 приведены коэф- ВОГо спирта в водных растворах,
фициенты активности этанола в
водном растворе при 20; 40; 55 и 75° С ". Значения
коэффициентов активности, рассчитанные по равновесным данным для пара
и жидкости при постоянном давлении (1 ат) и температурах
от 78,3 до 100° С ,8, также показаны на рисунке. Ясно, что в этой
системе изменение коэффициентов
активности с температурой
невелико. Коэффициенты активности для
большей части других систем
также мало изменяются с
температурой; поэтому температурной
зависимостью коэффициентов
активностей часто можно пренебречь. На
рис. 51 показана зависимость lgyA
при хА = 0 (т. е. величины ААВ) от
температуры для той же системы
этанол — вода, вычисленная по
уравнению (111,61).
Пример II1-4. Исходя из величины АА в
при 40° С и данных по теплотам
растворения, вычислить и сравнить с опытными
данными величину ААВ при 55° С для
системы этанол (А) —вода (В).
*
0,75
0.7
0.65
0,6
•>
гв гв
3.2 3(t
0/т) -/о3, °н
Рис. 51. Зависимость констан-
ты ^ав от температуры для
системы этанол (А) — вода (В)-
87
Решение. При 40°С, согласно рис. 50, величина AAB = \g5,90 = 0,7709.
Интегральные теплоты растворения этанола в воде для 0; 17,33 и 42,05° С
приведены в «International Critical Tables» (том V). Экстраполяция 'этих
данных к нулевой концентрации приводит к следующим величинам теплот
растворения при бесконечном разбавлении:
t,°C
AH'S ■ 10й дж/кмоль
Из уравнения (111,61) имеем:
I/T,
ААВ, = ААВ2 + J
1/Г2
о
-16
' АЯ-
2,303/?
17,33
-12,6
d (1/Г)
42,05
—8,5
Следовательно, при 55° С
0,00305
^ = <^+2,303.8314
J ДЯ^(1/Г)
0,003195
Интегрирование можно выполнить графически, определяя площадь под
кривой зависимости Mis от 1/7" между ординатами, соответствующими 1/7*=
= 0,00305 и 0,003195 (см. рис. 52). Эта площадь соответствует величине
ИЗО дж/кмоль°К. Поэтому при 55° С
А , „ = 0,7709 4- ^»,1130^., = 0,829
А в
2,303 ■ 8314
-S
что отвечает уА (при хА=0), равному 6,75. Согласно опытным данным, ААв
и у а равны соответственно 0,8195 и 6,60.
При умеренных изменениях температур коэффициенты
активности изменяются настолько незначительно, что данные при
постоянном давлении обычно достаточны для применения
различных интегральных форм уравнения Гиббса — Дюгема.
Применение интегральных форм уравнения Гиббса
—Дюгема. Как указывалось, уравнение Гиббса — Дюгема и
следствия из него [уравнение (111,29) и рис. 49] можно применить
для проверки термодинамической
консистентности и уточнения
экспериментальных данных.
Различные интегральные формы
этого уравнения [уравнения
(111,44) —(111,58)] особенно
полезны для расширения
информации, которую можно получить на
основании опытных данных для
бинарных систем (иногда даже на
основе данных одного измерения),
а также для вычисления
коэффициентов активности тройных
систем, исходя из их значений для
0/0-to3, "к
Рис. 52. Теплота растворения эти- ,..,„.. „„„„,,„ ,,~ ,,„ „„
лового спирта в воде при беско- ^1еМ' ИСХ°ДЯ из их значении для
нечном разбавлении. бинарных систем. Для этого
необходимо определить эмпирические константы, входящие в
указанные уравнения.
1. Определение зависимости коэффициентов
активности от концентрации по данным о
равновесии пар — жидкость. Имеется большое количество
опытных данных о равновесии пар — жидкость, которые
приводятся в технической литературе19,20. Из этих данных могут
быть рассчитаны коэффициенты активности; константы,
входящие в соответствующее уравнение, можно затем определить,
пользуясь, например, методом наименьших квадратов.
Более просто и в то же время с достаточной точностью
константы можно найти при помощи графической зависимости,
изображенной на рис. 49. Проведя через опытные точки кривую
и учитывая указанное выше требование о равенстве площадей,
константы можно определить по нескольким точкам на кривой,
применяя данные табл. 5. Например, для уравнений Редлиха—
Кистера САВ характеризуется величиной \ц(ул/ув) при л:А = 0,5;
ВАв определяется из значений lgyA/ув при л:А = 0,1464 и 0,8536
и Dab —из lg ул/ув при ха= 0,2959 и 0,7041. Остальные точки
могут служить для контроля — положение кривой при
необходимости можно уточнить. Подобный расчет произведен в
примере Ш-11 (стр. 112).
Для определения констант в уравнениях Ван-Лаара второго
порядка кроме конечных величин при Ха = 0 и 1,0 имеется
сравнительно мало промежуточных точек (хА = 0,5 и \gyA/\B=®)-
Однако для систем с положительными отклонениями от закона
Рауля применимость уравнений Ван-Лаара можно установить
на основе зависимости (lgYA)0'5 от (lgys)0'5. которая в этом
случае должна быть прямолинейной; кроме того, на оси ординат
и оси абсцисс должны отсекаться отрезки, равные
соответственно А°ав и Aba- В случае симметричных уравнений с одной
константой зависимость на рис. 49 будет изображаться прямой
линией.
2. Определение зависимости коэффициентов
активности от концентрации по одной точке
равновесия пар — жидкость. При наличии только одной
опытной точки по равновесию пар — жидкость (Р, х, у)
коэффициенты активности можно рассчитать, пользуясь уравнением
(111,14) или (111,15). Подставляя значения коэффициентов
активности и концентраций в соответствующее уравнение с
двумя константами, определяют ААв и АВа- После этого
можно вычислить значения коэффициентов активности каждого
компонента раствора во всем интервале концентраций. Наибольшая
точность расчета достигается в том случае, если известная
равновесная точка лежит в интервале концентраций
приблизительно между х = 0,25 и л; = 0,75. В частном случае могут быть
89
41
m I
I
■е-
■е-
m
О
s
х
о
о
ч
а>
а
5
I
а
>•
£
«и
£ °
S fc-
щ о
а) a
и о
и н
сз а
a
Э°о
Е в я
si*
О.
>,
Ч
•ч
Ч т(>
+
5
ч
■ч
ч
■ч
03
ч
■ч
2
о
г^
о"
ч
ч
оэ
ю
о
О
+
ч
•ч
Q
Ч
•ч
03
О)
оо
О
Ч"
о"
•ч
<л
+
ч
•ч
1
ч
•ч
03
082
те
го
Ч
•ч
Q
|
ч
•ч
03
го
t~-
1Л
ч
«j*
ч
•ч
|
ч
■ч
03
t-
о
о"
1
"4
■ч
Q
1
ч
■ч
1
ч
03
1
?
ч
ч
+
ч
ч
оо
О
Ч
•ч
ч
■ч
+
•ч
ч
оо
оо
О)
о"
Ч
•Ч
•Ч
ч
+
ч
•ч
•ч
н
•ч
ч
ч
•ч
•ч
ч
•ч
н
!М-Ч
+
•ч
н
м-Ч
«8
щ
•ч
_ч
5
■ч
_ч
ч
■ч
1Л
о
в.
в
«*
<.о
те
о
го
IN
О
О!
ю
8
о
1Л
о
,_!
Ч"
о
о
t^
00
78
о
СО
ГО
85
о
Р
т-Н
.4? ч
известны состав и температура кипения азеотропной смеси при
данном давлении. Так как для азеотропного состава х = у, то
Y* =
Ра
Р
Рв
(III, 17)
Подробные данные об азеотропных смесях приведены в книге
Хорсли 2I.
Пример 111-5. Азеотропная смесь этилацетата (А) и этанола (В) 2I при
760 мм рт. ст. содержит 30,98 вес.% этанола. Температура кипения смеси
71,8° С. Рассчитать константы уравнения Ван-Лаара и коэффициенты
активности. Сравнить их с коэффициентами активности, вычисленными из
равновесных данных при 1 ат.
Решение. Концентрация 30,98 вес.% этанола соответствует
концентрации 0,462 мол. доли. Следовательно, хв = 0,462 и хА = \ — 0,462 = 0,538. При
71,8°С давление паров этилацетата рл=631 мм рт. ст., а давление паров
этанола рв = 581 мм рт. ст.; Р = 760 мм рт. ст. Из уравнения (111,17) для
азеотропной смеси находим:
760
= -^f = 1,204
Р _
РА ~ "631
Р 760
Р„ 581
1,307
'в
Из уравнений (111,50) и (111,51) получаем:
i 1 on. (л I 0,462 lg 1,307 \2
ААВ = 'g 1-Ш I1 + 0,538 lg 1,204 ) =
= 0,4057
i=lg 1,307 (
В А
АВ
0,538 lg 1,204\2_
+ 0,462 lg 1,307 j ~
= 0,3781
0,4057
ВА
0,3781
= 1,073
Уравнения Ван-Лаара [уравнения
(111,48) и (111,49)], следовательно,
имеют вид:
0,4057
ЫчА-
|г
if
I
I,
0.3
о\
8
IL^X
у^Д
•а
"1
<^Ь
Q2
I 1,073л:. \
Of» 0.6 0.6
lgY« =
В
0,3781
кв
1,073л;,
На рис. 53 сравниваются
коэффициенты активности, рассчитанные по
этим уравнениям, с коэффициентами
активности^ вычисленными из данных
Концентрация этилацетата,
мол. доли
Рис. 53. Коэффициенты
активности в системе этилацетат —
этанол (при 760 мм рт. ст.);
светлые точки — данные Фэнеса и
Лейтона; темные точки — данные
Грисволда и Уинзауэра; кривые
рассчитаны из азеотропного
состава (обозначенного на рисунке
крестом).
91
о равновесии пар—жидкость при 760 мм рт. ст., полученных Фэнесом [Ind.
Eng. Chem., 29, 709 (1937)] и Грисволдом [Ind. Eng. Chem., 41, 2352 (1949)].
3. Определение зависимости коэффициентов
активности от концентрации по температурам
кипения растворов или по давлениям паров.
Часто бывают известны температуры кипения растворов или
общее давление паров в зависимости от концентрации и
отсутствуют данные о составе равновесных паров. Тогда, выбрав
подходящее уравнение, можно определить коэффициенты
активности во всем интервале концентраций по методу,
предложенному Карлсоном и Кольборном7. Из уравнения (III, 15)
следует:
тл =
*ЛР
А^А
р~уаРаха
(III, 63)
(III, 64)
^в^в
Когда хв приближается к 1,0, ув также стремится к
единице; поэтому в первом приближении можно принять, что
р— P*xR
ул(прихл->0) = -
и аналогично
ув(пРихв->0) =
Р —
Рвхв
(III, 65)
(III, 66)
Зная температуры кипения разбавленных растворов (с
малой концентрацией компонента А), можно приближенно
рассчитать уА. Экстраполируя зависимость уА от хА, построенную в
полулогарифмических координатах, можно определить
приближенное значение ул при хА = 0, т. е. Аав- Аналогичным путем
находят АВА. Подстановка полученных констант в одну из
интегральных форм уравнения Гиббса — Дюгема позволяет
рассчитать более точные значения коэффициентов активности в
области разбавленных растворов; таким образом, методом
последовательного приближения можно получить действительные
значения ААв и АВа. Затем можно рассчитать коэффициенты
активности во всем интервале концентраций.
Пример 111-6. В «International Critical Tables» (том III) приведены
давления паров над растворами этанола (А) и толуола (В) при 32,3°С:
ХА
Р,мм pm.cm.
ХА
Р, мм рт. ст.
0
93,0
0,6680
249,2
0,0420
141,2
0,7627
248,2
0,1775
214,8
0,8369
244,4
0,3089
233,1
0,8802
243,0
0,4613
242,1
0,9599
230,9
0,5765
244,2
1,000
219,5
82
Рассчитать по этим данным константы Ван-Лаара.
Решение. При 32,3 С имеем: р^ =219,5 мм рт. ст.; рв = 93,0 мм рт. ст.
По уравнению (111,65) в первом приближении находим:
Р — 93,0 х„
У а (при*д->0)= 219
' л
Рассчитанные по этому уравнению приближенные значения уА сведены
в таблицу:
ХА
0,0420
0,1775
0,3089
хв
0,9580
0,8225
0,6911
93'%
89,0
76,5
64,3
219,5х„
Л
9,22
38,98
67,9
Р — 93,0.г _
52,2
138,3
168,8
Прнбл. у .
5,67
3,55
2,485
Полученные приближенные значения уА откладываем в
полулогарифмических координатах в функции от хА и экстраполяцией находим величину
уА (при хА=0), равную 6,65. Величина A^s = lg 6,65=0,8228.
Аналогично по уравнению (111,66) рассчитывают приближенные
значения ув:
ХА
0,9599
0,8802
0,8369
хв
0,0401
0,1198
0,1631
93,0ж„
3,73
11,14
15,19
219.5жд
Л
211
193,3
183,9
Я-219.5.Г.
Л
19,9
49,7
60,5
ПрибЛ. Yd
D
5,35
4,46
3,99
Тем же путем находим AeA = ig 5,88=0,7694. Проверяем полученные
значения ААВ и АВА, рассчитывая по уравнению Ван-Лаара коэффициенты
активности и по ним — давление Р из зависимости
Р=УаРаха + УвРвхв
Результаты расчетов приведены в таблице:
ХА
0,0420
0,3089
0,5765
0,7627
0,8802
0,9599
УА
5,624
2,382
1,368
1,102
1,025
1,002
Ч
1,003
1,204
1,873
2,884
4,036
5,176
Р
(рассчитанное^
мм рт. ст.
141,3
239,1
246,7
247,8
242,9
230,3
Рассчитанные таким образом величины Р удовлетворительно
согласуются с опытными данными, и полученные приближенные значения Алв и Авл
93
можно принять за окончательные. Более точные величины можно найти, если
повторить расчет по уравнениям (111,65) и (111,66), используя
рассчитанные выше величины у л и ув.
4. Расчеткоэффициентовактивностиизсоста-
вов жидкости и пара при известном давлении
(температура не известна). Метод расчета предложен
Карлсоном и Кольборном 7. Температуру находят в первом
приближении, применяя закон Рауля к компоненту, находящемуся
в большем количестве. Это дает возможность определить
приближенные значения коэффициентов активности компонента,
присутствующего в меньшем количестве. С помощью графика,
построенного в полулогарифмических координатах, находят в
первом приближении величины Аав и Aba- При подстановке
полученных значений ААв и АВА в одно из уравнений (111,44) —
(111,58) находят более точные величины коэффициентов
активности и далее методом последовательных приближений
определяют окончательные значения Аав и Ава-
5. Расчет коэффициентов активности из
данных о взаимной растворимости компонентов
раствора7-22. По мере того как компоненты раствора
химически все более различаются друг от друга, отклонения раствора от
закона Рауля возрастают. Большие положительные отклонения
приводят в конце концов к ограниченной взаимной
растворимости компонентов. Так, в системах спирт — вода метиловый,
этиловый и пропиловый спирты полностью смешиваются с
водой; однако растворы этих спиртов отличаются все
увеличивающимися отклонениями от закона Рауля. Бутиловый спирт, для
растворов которого в воде характерны очень большие
отклонения от закона Рауля, растворяется в ней ограниченно.
Аналогично ведут себя и системы кетон — вода. Ацетон и вода
смешиваются полностью с умеренными отклонениями от идеальности,
тогда как метилэтилкетон и вода ограниченно смешиваются и
обнаруживают большие положительные отклонения от закона
Рауля.
В любых двух находящихся в равновесии фазах, в том
числе и жидких, летучести каждого компонента одинаковы.
Если одно и то же стандартное состояние выбрано для
компонента в каждой фазе, то активности его в равновесных фазах
также одинаковы. Таким образом
(III, 67)
AA АВ
Следовательно
ХАА УАВ
ХАВ УАА
аВА = ЛВВ
ХВА _ УВВ
ХВВ ЧВА
(III, 68)
В интервале концентраций между пределами растворимости
кажущиеся значения коэффициентов активностей изменяются
94
обратно пропорционально концентрациям, отнесенным ко всей
смеси. Совместное решение уравнения (111,68) и
соответствующего уравнения для коэффициентов активности с двумя
константами позволяет из данных о взаимной растворимости
определить Аав и Aba- Так, из уравнения Маргулеса третьего
порядка получаем:
Л4 =—= , бл, (Ш) 69)
Ав
(В А 1/2 2\ iBA wo 3 \
2 ~д 1) \х ва — хвв) — 21 ^д J \х ва — хвв)
а 2 На-*ввУ(*3ва-*вв)] *^\(*аа-*ав)-* (Аа-Лв)] ^~
АВ ВА АД
АВА * Ми-ЪНЛл-Лв)] *Х-~А{*2ва-*2вв)-> (4а-*вв)} ьУ*
AA BA
(III, 70)
Уравнения Ван-Лаара второго порядка дают следующую
зависимость:
(III, 71)
А —
ЛАВ
(
\
ААВ _
АВА
'g
1
j , AabxaaY
АВАХВА1
(ХАА , ХАВ
\ ХВА ХВВ
АА , АВ
ХВА ХВВ
ХАВ
ХАА
1
\ ЛВАХВВ1
(*УВ\
ХАА 2
)\ ,g *ВЛ )
V хвв)
2ХААХВВ^~-
ЛАА
ХВАХВВ^ ХВА
ЛВВ
(Ш, 72)
Концентрации в уравнениях (111,69) — (111,72) равны
концентрациям равновесных насыщенных растворов.
В том и другом случае по второму уравнению можно
рассчитать Aab/Ава, а затем из первого уравнения определить Алв.
На рис. 54 приведено графическое решение уравнений (111,71)
и (III, 72), которыми можно, в частности, пользоваться для опре-
95
Рис. 54. Решение уравнений Ваи-Лаара применительно к взаимной
растворимости компонентов [Colburn, Schoenborn, Trans. Am. Inst.
Chem. Engrs., 41, 421 (1945); пунктиром показана критическая кривая
растворимости].
деления растворимостей, если известны величины ААВ. и АВА
(см. пример III, 14, стр. 120).
Для уравнений с тремя константами (например, уравнений
Редлиха — Кистера) необходимы дополнительные данные для
получения третьей константы. Из уравнений (111,56), (111,57) и
(111,68) следует:
•g —" = Вав (Лв - **ва) + Cab [*вв (**ав - 0 - 4л {^аа - Щ +
АВ
+ DAB \х\в (1 - 8хАВ + \2х\в) - хВА (1 - 8хАА + \2х2АА)} (III, 73)
'g ~" = Вав (Лв - Ла) + Cab [Лв 0 - ^вв) ~ Аа 0 - 4*вл)] +
вв
+ DAB [хАВ (1 - 8хвв + 124в) - х\А (1 - 8хВА + 124л) ("I. 74)
Подстановка в эти уравнения данных о взаимной
растворимости приводит к двум уравнениям с тремя неизвестными. В
качестве третьего уравнения может служить уравнение (111,58),
95
1.3
V
0.9
«=— Область
>^ нерастворимости
>,
-я
^1-
$ь=
£1
=fc=z
Область-
TS
1 х
- растворимости -
" МММ!
1
-s
_
ч
ь
\
о
0.1 а
I
02. <§
0.3
(Ц. Ч"
0.5
$
если имеется хотя бы одно до- >,s
полнительное измерение,
например, по равновесию пар
—жидкость.
Для систем с близкой
взаимной растворимостью
компонентов (т. е. когда хАВ
приблизительно равно хВА) величина ААВ
по значению близка АВА. В ° 0,!
этом случае можно ожидать, что
применимы уравнения как Мар-
гулеса, так и Ван-Лаара. Для
заметно несимметричных систем
следует применять уравнения
Редлиха — Кистера. По мере
уменьшения взаимной
растворимости эти уравнения приводят к
все более и более высоким конечным значениям
коэффициентов активности.
В критической точке уравнения (111,69) — (III,74)
становятся неопределенными, так как хАА = хАВ и хВА — хвв. Хильде-
бранд23 показал, однако, что в критической точке (d\na)/dx и
(d2 In a) /dx2 равны нулю. При этих условиях из уравнений Ван-
Лаара, например, следует:
0,3 Ofi- 05 0,3 0/ 0,8 0,9
АВА
Рис. 55. Графическое решение
уравнений (III, 75) и (III, 76) для
определения растворимости (на
оси ординат справа отложены
составы в критической точке
растворимости; кривая —
критическая кривая растворимости).
ЛАВ
\-х\
ЛВА
2х,
ААВ —
5,862(1— xAf
(2-xAf(l-xA)
(111, 75)
(III, 76)
Эти уравнения могут применяться для расчета констант по
составам в критической точке. Так, например, если система
симметрична, т. е. если в критической точке х = 0,05, то ААВ=АВД =
= 0,868.
Смеси, для которых константы А больше вычисленных по
уравнениям (111,75) и (111,76), должны (если к ним применимы
уравнения Ван-Лаара второго порядка) ограниченно
смешиваться. Для определения растворимости удобно пользоваться
рис. 55, на котором графически изображены зависимости
(111,75) и (111,76). Если значениям констант ААВ и АВА
соответствует точка, лежащая выше кривой, изображенной на этом
рисунке, компоненты ограниченно растворимы друг в друге;
точка под кривой указывает на полную взаимную
растворимость. Концентрации хА на правой оси ординат относятся к
составам в критической точке. С увеличением степени
несимметричности надежность такого способа определения взаимной
7 Р- Е. Трейбал
97
растворимости уменьшается. Эти вопросы будут обсуждены
ниже при рассмотрении «регулярных» растворов.
Пример II1-7. Взаимная растворимость метилэтилкетона (А) и воды (В)
при 40° С равна 18,6 и 90,1 вес.% кетона17. Рассчитать для этой системы
константы Аав v. Aba и сравнить их с величинами, рассчитанными по
данным о равновесии пар—жидкость.
Решение.
Расчет по данным о растворимости. В растворе, содержащем 18,6%
кетона и 81,4% воды, мольные доли кетона и воды равны 0,054 и 0,946; в
растворе, содержащем 90,1% кетона и 9,9% воды, — соответственно 0,695 и
0,305. Следовательно
хлв = 0,054
хАА = 0,695
х = 0,946 (в водной фазе)
ВА
■ 0,305 (в фазе кетона)
Так как растворимости компонентов заметно отличаются друг от друга,
применяем уравнения Ван-Лаара. Согласно уравнению (111,72), имеем:
'0,695 , 0,054 \ lg (0,054/0,695)
АВ
, 0,305 -1" 0,946 ) lg (0,305/0,946)
ВА
0,695 , 0,054 2 (0,695 • 0,054) lg (0,054/0,695)
■=1,860
0,305 п 0,946 (0,305 • 0,946) lg (0,305/0,946)
По уравнению (111,71) находим:
0,054
lg
ААВ~ "
0,69,5
1
1
■ = 1,455
[1 + 1,36 (0,695/0,305)]2 [1 + 1,86 (0,054/0,946)]2
1,455
Ава =
1,860
= 0.781
Полученным значениям ААВ и Aba соответствуют:
уА= 28,51 (прихд = 0)
YB=6,04 (прихд=1)
Расчет по данным о равновесии пар—жидкость. Отмер и Бененати [lnd.
Eng. Chem., 37, 299 (1945)] приводят равновесные данные для этой системы
при нескольких давлениях. Воспользуемся данными при 200 мм рт. ст., так
как температуры кипения при указанном давлении близки к 40е С.
Температуры кипения и составы вместе с рассчитанными по ним [уравнение
(111, 15)] коэффициентами активности приведены ниже:
t, °с
41,5
40,6
40,0
39,9
39,8
41,3
66,4
*А
1,000
0,903
0,813
0,777
0,674
0,038
0
УА
1,000
0,825
0,763
0,750
0,726
0,709
0
Y.4
1,000
1,009
1,065
1,102
1,237
20,0
0
чв
6,31
4,59
4,06
3,07
1,021
1,000
98
Из зависимости ул и ув от *л (в полулогарифмических координатах)
получаем: при хА = 0 величина Ya = 29 и lg ул~ААв = 1,462; при хА = \
величина ув=8,2 и lg ув = Ав а = 0,914. Эти значения хорошо согласуются с
величинами, полученными из данных о взаимной растворимости.
Пример III-8. Для смеси метилэтилкетона (А) и воды (В), содержащей
45 вес.% кетона, критическая температура растворения равна 150°С ".
Рассчитать константы Ван-Лаара при этой температуре.
Решение. Концентрации метилэтилкетона, равной 45 вес.%,
соответствует величина а-д = 0,1696 мол. доли. Из уравнения (111,75) имеем:
Аяа 1 —(0,1696)2
АВ v 4 19е;
АВА ~ 2-0,1696— (0.1696)2 '
По уравнению (111,76) находим:
А 5,862 (1-0.1696)2
лав ~ (2 — 0Д696)2 [1 — (0Д696)2] '
^л = 1д25=0,397
Сравнение этих констант с величинами, полученными п предыдущем
примере, еще раз иллюстрирует влияние температуры на величину
коэффициентов активности.
6. Определение коэффициентов активности
при отсутствии экспериментальных данных. Для
так называемых «регулярных» растворов можно определить
коэффициенты активности чисто теоретическим путем, не прибегая
к экспериментальным данным. Для растворов других типов это
в настоящее время невозможно. Однако предельные значения
коэффициентов активности при бесконечном разбавлении
определяли для многих бинарных систем, и оказалось возможным
связать их величины с молекулярной структурой соединений,
составляющих систему. Эти зависимости наиболее пригодны
для расчета коэффициентов активности соединений, являющихся
членами одного и того же гомологического ряда. В случае
применимости для определения коэффициентов активности
уравнения с двумя константами можно, следовательно, полностью
рассчитать зависимость у—х. При наличии дополнительных
данных для расчета коэффициентов активности можно
использовать и более сложные уравнения.
Большое количество констант для определения
коэффициентов активности вместе с соответствующими корреляционными
уравнениями приведено в табл. 6 и la — 7e2i. С их помощью
можно рассчитать у'А со средним отклонением 8% и Аав с
точностью около 3%- (В оригинальных работах, по которым
составлены таблицы, подробно обсуждаются величины отклонений.)
Следующие примеры иллюстрируют применение этих
корреляций (стр. 106); использование их при расчетах процессов
экстракции показано также в примерах 111-14 и Ш-15.
7*
99
Таблица 6
Константы в эмпирических уравнениях для коэффициентов активности (гомологические ряды растворенных веществ
и растворителей)
Растворенное вещество А
н-Кислоты
н-Первичиые спирты
и-Вторнчные спирты
н-Третичные спирты
Спирты
н-Аллиловые спирты
н-Альдегиды
н-Алкенальдегиды
н-Кетоны
Растворитель В
Вода
Вода
Вода
Вода
Вода
Вода
Вода
Вода
Вода
/, °с
25
50
100
25
60
100
25
60
100
25
60
100
25
60
100
25
60
100
25
60
100
25
60
100
25
60
100
а
—1,00
—0,80 '
—0,620
—0,995
—0,755
—0,420
—1,220
—1,023
—0,870
—1,740
—1,477
—1,291
—0,525
—0,33
—0,15
—1,180
—0,929
—0,650
—0,780
—0,400
—0,03
—0,720
—0,540
—0,298
—1,475
—1,040
—0,621
е
0,622
0,590
0,517
0,622
0,583
0,517
0,622
0,583
0,517
0,622
0,583
0,517
0,622
0,583
0,517
0,622
0,583
0,517
0,622
0,583
0,517
0,622
0,583
0,517
0,622
0,583
0,517
1
0,490
0,290
0,140
0,558
0,460
0,230
0,170
0,252
0,400
0,170
0,252
0,400
0,475
0,390
0,340
0,558
0,460
0,230
0,320
0,210
0
0,320
0,210
0
0,500
0,330
0,200
Т]
—
—
0
0
0
—
0
0
0
—
—
—
0
0
0
/
е
0
0
0
0
0
0
—
—
—
0
0
0
0
0
0
0
0
0
/
Уравнение
(а)
(а)
(а)
(а)
(а)
(а)
(*)
(Ь)
(Ь)
(с)
(с)
(с)
(d)
(d)
(d)
(а)
(а)
(а)
(а)
(а)
(а)
(а)
(а)
(а)
(Ь)
(Ь)
(*)
н-Ацетали
и-Эфиры (простые)
н-Нитрилы
н-Алкеннитрилы
н-Эфиры (сложные)
н-Эфиры муравьиной
кислоты
н-Моноалкилхлориды
н-Парафины
н-Алкилпроизводные
бензола
н-Спирты
н-Кетоны
Вода
Вода
Вода
Кетоны
Вода
Вода
Вода
Вода
Вода
Вода
Вода
Вода
Вода
Парафины
Парафины
н-Спирты
Вторичные
спирты
н-Кетоны
н-Спирты
25
60
100
20
25
60
100
25
60
100
20
20
20
16
25
25
60
100
25
60
100
25
60
100
80
25
60
100
25
60
100
—2,556
—2,184
—1,780
—0,770
—0,587
—0,368
—0,095
—0,520
-0,323
—0,074
—0,930
—0,585
1,265
0,688
3,554
1,960
1,460
1,070
0,0877
0,016
—0,067
0,760
0,680
0,617
1,208
1,857
1,493
1,231
—0,088
—0,035
—0,035
0,622
0,583
0,517
0,640
0,622
0,583
0,517
0,622
0,583
0,517
0,640
0,640
0,640
0,642
0,622
0
0
0
0
0
0
0
0
0
0
0
0
0
0,176
0,138
0,112
0,486
0,451
0,426
0,195
0,760
0,413
0
0,760
0,413
0
0,260
0,260
0,073
0
-0,466
0,475
0,390
0,340
0,757
0,680
0,605
0
0
0
0
0
0
0
0,50
0,33
0,20
—0,00049
—0,00057
—0,00061
—0,00049
—0,00057
—0,00061
—0,00049
—0,00057
—0,00061
—
—
—
—
0
0
0
0
0
0
—
0
0
0
—
(•)
(е)
(О
(Ь)
(а)
(а)
(а)
(а)
(а)
(а)
(Ь)
(а)
(а)
(а)
(Л
—0,630
—0,440
—0,280
—0,690
—1,019
—0,73
—0,557
—0,630
—0,440
—0,280
(d)
(d)
(d)
(b)
(b)
(a)
(a)
(a)
(c)
(c)
(c)
(c)
(g)
(g)
(g)
Продолшение табл. В
Растворенное вещество А
Альдегиды
Эфиры (сложные)
Ацетали
Парафины
Растворитель В
я-Спирты
я-Спирты
я-Спирты
Кетоны
t, °с
25
60
25
60
100
60
25
60
90
а
—0,701
—0,239
0,212
0,055
0
-1,10
—
—
—
е
0,176
0,138
0,176
0,138
0,112
0,138
0,1821
0,1145
0,0746
1
0,320
0,210
0,260
0,240
0,220
0,451
—
—
—
т| '
—0,00049
—0,00057
' -0,00049
—0,00057
—0,00061
—0,00057
-0,00049
—0,00057
—0,00061
е
—0,630
—0,440
—0,630
—0,440
—0,280
—0,440
0,402
0,402
0,402
Уравнение
(h)
(h)
(g)
(g)
(g)
(i)
U)
(i)
U)
Примечания: 1. Корреляционные уравнения для коэффициентов
активности
Ф) [^A = AAB-a + ENA+i (^7_ + ^Г) + Т| (NA~NbY
AB [na na na) [nb nb)
B \NA NA NA )
(e) lg v'
'А = ААВ = а + тА + Ч^- + ^~ + ^А
\NA NA Na)
W» ^-^ = 0 + ^+8^-4)
e
.NA
"A "A
(h) ,g 1'a-AAB = a+ ^ + ^+ Ч ("А-"В)*+ 1^
/ NA / 1 1 2 \ 9
\" в "в I
где NA, Ng — общее число атомов углерода;
Nf, N", Nm — число атомов углерода в соответствующих цепях
разветвленных соединений, имеющих полярные группы (так, для трет-
бутанола N' = N" = N'" = 2).
2. Таблица заимствована из литературы: Plerotti, Deal, Dert,
Ind. Eng. Chem., 51, 95(1955).
7a — 7в. Константы в эмпирических уравнениях для коэффициентов активности (растворы углеводородов — членов
гомологических рядов — в некоторых растворителях) Таблица 7а
Температура
°С
20
50
70
90
Температура
"С
25
70
130
25
70
130
Т|
—0,00049
—0,00055
—0,00058
-0,00061
гептан
0305
0,1668
0,1212
0,1874
0,1478
,0,1051
гептаи
метил-
этилкетон
фурфурол
0
0
0
0
0,0455
0,033
0,025
0,019
0,0937
0,0878
0,0810
0,0686
Растворитель В
феиол
этанол
Величина е
0,0625
0,0590
0,0586
0,0581
0,088
0,073
0,065
0,059
Растворитель В
метилэтил-
кетон
фурфурол
феиол
0,1435
0,1142
0,0875
0,1152
0,0836
0,0531
0,1421
0,1054
0,0734
0,207с
0,175^
0,142'
)
Г
0,217*
0,167..
0,118,
1
0,24(
0,18
0,14!
)6
0
Ю
этанол
Тр
г
Величина 0
0,2125
0,1575
0,1035
(
Величина х
0,2
ОД
од
425
753
169
триэтиленгли-
коль
диэтилен-
гликоль
этилен-
глнколь
0,161
0,134
0,191
0,179
0,173
0,158
0,275
0,249
0,236
0,226
Таблица 76
иэтилен-
ликоль
диэтилен-
гликоль
этилен-
гликоль
),181
3,129
3,0767
0,2022
0,1472
0,0996
0,275
0,2195
0,1492
0,3124
0,2406
0,1569
0,3180
0,2545
0,1919
0,4147
0,3516
0,2772
Таблица 7в
пература
25
50
70
90
25
50
70
90
25
50
70
90
25
50
70
90
25
50
70
90
Растворенное вещество
внение
Парафины
Алкилзамещенные
циклогексана
Алкилзамещенные
бензола
Алкилзамещенные
нафталина
Алкилзамещенные
тетралина
(а)
(а)
(а)
(а)
(а)
Растворитель В
метилэтил-
ФУРФУРол
фенол
триэтилен-
гликоль
диэтилен-
гликоль
о
о
о
о
—0,260
—0,220
—0,195
—0,180
—0,466
—0,390
—0,362
—0,350
—0,10
—0,14
—0,173
—0,204
0,28
0,24
0,21
0,19
0
О
О
О
0,18
0,131
0,09
0,328
0,243
0,225
0,202
0,53
0,53
0,53
0,53
0,244
0,220
0,335
0,332
0,331
0,330
6,70
0,650
0,581
0,480
0,277
0,240
0,239
0,169
0,141
0,215
0,232
0,179
0,217
Величина а
0,916
0,756
0,737
0,771
1,26
1,120
1,020
0,930
0,67
0,55
0,45
0,44
0,870
0,755
0,690
0,620
1,20
1,040
0,935
0,843
0,694
0,580
0,500
0,420
0,46
0,40
0,39
—
0,652
0,528
0,447
0,373
0,595
0,54
0,497
0,445
0,378
0,364
0,371
0,348
0,580
0,570
0,590
0,910
1,06
1,01
0,972
0,925
1,011
0,938
0,900
0,862
1,06
1,03
1,02
0,72
0,68
1,46
1,25
0,800
0,740
0,75
0,83
1,00
0,893
этилен-
гликоль
0,875
0,815
0,725
0,72
1,675
1,61
1,550
1,505
1,08
1,00
0,96
0,935
1,00
1,00
0,991
1,01
1,43
1,38
1,33
1,28 /
1,208
1;154
1,089
—
2,36
2,22
2,08
—
1,595
1,51
1,43
—
1,92
1,82
1,765
—
—
—
—
25
50
70
90
25
70
130
/
Алкилзамещенные (а)
декалина
Незамещенные
ароматические
соединения; нафтены;
соединения нафте-
ноароматического
строения
(6)
(Ь)
—0,43
—0,368
—0,355
—0,320
1,176 2
1,845 3
0,846 2
1,362 3
0,544 2
0,846 з
■ '"■■■!•'"■■•::■■"
—
—
0,356
—
—1,072
—0,886
—0,6305
:: • :' я*!*.;...
0,871
—
0,80
—
—0,7305
—0,625
—0,504
^iiiSTr*.:?'?
1,54
1,367
1,253
1,166
—0,230
—0,080
+0,020
щщщщ
1,411
1,285
1,161
1,078
—0,383
—0,226
—0,197
ЩШРИНР*Р
—
—
—
—
—0,485
—0,212
+0,47
—
1,906
—
1,68
—0,406
—0,186
+0,095
2,46
2,25
2,07
2,06
—0,377
—0,0775
+0,181
—
—
—
—
—0,154
—0,0174
+0,229
Примечания к табл. 7а—7в.
1. Корреляционные уравнения для коэффициентов активности:
^l^'A-AAB = a + eNP+-iN^2)
-t](NA+NBf
где Nд, N3 — обшее число атомов углерода;
I
I
N —число «ароматических» атомов углерода, включая =С —; =СН—; атомы углерода, соединяющие нафтеновые кольца —С—Н
«нафтеновые» атомы углерода, находящиеся в а-положении к ароматическому ядру;
/V_ — число «нафтеновых» атомов углерода, не учтенных в /V ;
г —число колец.
Примеры:
Бутилдекалии: Np = 4; N а = 2; Nл = 8; Л^ =
Бутилтетралин: N„ = 4; Na = 8; N п = 2; Nд =
г = 2
г=2
2. Конденсированные ароматические соединения типа нафталина.
3. Ароматические соединения типа дифенила.
4. Таблица заимствована из литературы: PlerottI, Deal, D е г г, Ind. Eng. Chem., 51, 95 (1959).
Пример 111-9. Определить константы Ван-Лаара для системы этанол-
вода при 100°С.
Решение.
Этанол при бесконечном разбавлении в воде. Этанол — А, вода — В. Из
табл. 6 находим а=—0,420; е = 0,517; £ = 0,230; 9 = 0; NA=2. Следовательно,
по уравнению (а) табл. 6 получаем:
'g Уа = аАВ = - 0Д20 + 0,517 • 2 + ^^- = 0,769
у'А = 5,875
По данным Джонса, Кольборна и Шенборна18, опытная величина y=5,85.
Вода при бесконечном разбавлении в этаноле. Этанол — В, вода — А.
Из табл. 6 имеем: а = 0,617; е=|=0; 0=—0,280; NB = 2. По уравнению (а)
табл. 6 находим:
•g Уа = ААВ = 0,617 - -°^°- = 0,477
У А = 3,0
Джонс, Кольборн и Шенборн приводят опытное значение уА = 2,80 при
78,3°С (1 ат). Если проделать расчет, интерполировав константы из табл. 6
к этой температуре, получим уА = 2,94.
Приняв Аав = 0,769 и ЛВл = 0,477, с помощью уравнений (111,48) и
(111,49) можно рассчитать полностью зависимость у — х.
Пример 111-10. Определить, какой из низших кетонов неограниченно
смешивается с водой.
Решение. Способом, показанным в примере 9, с помощью табл. 6
получены следующие величины констант А при 25°С (кетон — А, вода — В):
Кетон
Диметилкетон (ацетон)
Метилэтилкетон
Диэтилкетон
"А
2
2
3
NA
2
3
3
ААВ
0,891
1,430
1,968
АВА
0,838
1,008
1,178
Пользуясь рис. 55, определяем, что ацетон смешивается с водой
полностью, тогда как метилэтилкетон и высшие кетоны ограниченно
растворяются в воде. Поскольку константы для диэтилкетона больше
соответствующих констант для метилэтилкетона, можно сделать вывод, что
растворимость диэтилкетона в воде меньше. Это, конечно, согласуется с опытом.
Количественное определение растворимости см. пример 111-14.
НЕИДЕАЛЬНЫЕ ТРОЙНЫЕ РАСТВОРЫ
Основные закономерности, установленные для идеальных
растворов, например закон Рауля в его различных формах,
применимы к растворам с любым числом компонентов. Для
тройных смесей, так же как и для бинарных, можно получить
различные зависимости, позволяющие рассчитать коэффициенты
106
активности. Обзор различных методов подхода к решению этой
задачи дан Воолем |2.
Простейшими эмпирическими уравнениями для
коэффициентов активности в тройных системах являются уравнения,
которые могут быть получены только по данным для трех бинарных
систем, составляющих тройную систему. В таких случаях
постулируют, что тройная система может быть полностью рассчитана
без каких-либо непосредственно относящихся к ней данных.
В большей части случаев, однако, гораздо более точное
описание тройных систем достигается при включении в уравнения
дополнительных членов, причем для этого необходимы некоторые
сведения, касающиеся тройной системы. Чем больше таких
дополнительных членов, тем точнее уравнение, но при этом оно
становится менее удобным для расчетов.
Уравнения второго порядка для тройных
систем, состоящих из трех симметричных
бинарных систем. Для таких систем Бенедиктом5 выведены
следующие уравнения:
•g УА = ААВХ\ + ААСХС + ХВХС (ААВ + ААС ~ АВс) (Ш- 77)
•g У В = ААВХА + АВСХС + ХАХС (ААВ + АВС ~ ААс) (Ш> 78)
lg УС = ААСХА + АВСХ1 + ХАХВ (ААС + АВС ~ ААв) ("L 79)
Константы в этих уравнениях равны константам, полученным
на основе предельных значений коэффициентов активности, для
трех бинарных систем, каждая из которых является
симметричной:
ААВ =
ААВ =
ЛАС =
ЛАС =
АВС =
АВС =
= Hmlgv^
lim lg yB
lim lg yA
lim lg yc
lim lg yB
limlgYc
при
при
при
при
при
при
х^0
хв^0
ХА^°
хс^°
хв^°
хс^0
и
и
и
и
и
и
хв^
ХА^
хс^
ХА^
хс^
хв^
Так как в этих уравнениях отсутствуют дополнительные
константы, которые можно было бы определить только по данным
для тройных систем, коэффициенты активности для тройной
системы можно рассчитать, ограничиваясь данными для трех
бинарных систем. Важно отметить, что даже для этого наименее
сложного случая простая интерполяция данных для бинарных
растворов невозможна. Интерполяция величин lgy была бы
допустима, если в уравнениях (111,77) — (111,79) опустить члены
—Авс, —ААС и —Алв- Обычно константы положительны,
поэтому значения lg y будут ниже средних арифметических
велики, 80)
(111,81)
(111,82)
107
чин, полученных на основе данных для соответствующих
бинарных систем. Линейная интерполяция \gyA, например,
возможна только в том случае, если смесь В—С является идеальной.
Уравнения Ван-Лаара второго порядка. Эти
уравнения имеют вид:
КВЛАВ
ВА
^АВ
+ х2сА
СЛАС
А \2
С А
АС
igyA =
+""(£)
ХА + ХВ
№
Gfc)
ААВ + ААС~
мш
Асв\аса)\
2
(III, 83)
Выражение для lgYs можно получить заменой индексов: В
на Л; С на В и Л на С. Так, в уравнении для lg ув величина хв
в уравнении (111,83) заменяется на хс\ ААВ— на АВс', ААС—
на АВа и т. д. Аналогично в уравнении для ус величина хс в
уравнении (111,83) заменяется на хв\ АВА — на ААС и т. д.
Константы в этих уравнениях также определяются только по
данным для бинарных систем:
AAB=Vlml£VA ПрИ ХА^° И Х£
\im\gy„ при хв->0 и х
в а'
A = \\m\gy. при
-О и
АСА = \\m\gyc при xQ^>0 и х .-
ABC = \\m\gyB при -*в->0 и хс
ACB = \im\gyc ПРИ хг^>^ и хв~
Система А — В
Система А — С
Система В — С
(III, 84)
Вооль показал, что эти уравнения применимы лишь при
соблюдении условия:
ев
АСАААВ
ВС
ААСАВА
(III, 85)
Как и в предыдущем случае, коэффициенты активности для
тройных систем можно рассчитать по данным для бинарных
систем. Уравнения Ван-Лаара в форме, модифицированной Блэ-
ком, выведены 15 также для тройных систем. Эти уравнения не
содержат тройных констант и подчиняются ограничению
(111,85).
Уравнения с тройными константами. Эти урав-
нения могут быть очень сложными и зависят от тройных
эффектов, учитываемых при составлении молекулярных групп для
уравнения (111,43). Наиболее применимыми оказались
следующие:
108
Уравнения Маргулеса третьего порядка с одной тройной
константой 12:
Iff У А = 4 [Л4В + 2хА (АВА - ААВ)\ + ХС [ААС + 2х А (АСА ~ ААс)] +
+ ХВХС ■ [°>5 (АВА + ААВ + АСА + ААС ~ АВС ~ АСв) +
+ ХА (АВА ~ ААВ + АСА - ААС) + (ХВ ~ ХС) (АВС ~ АСв) ~ 0 ~ 2х А) Щ
(111,86)
Уравнения Редлиха — Кистера четвертого порядка с тремя
тройными константами 10> 16:
£ У А = 0 - ХА) (ХВВАВ + ХСВСА) + ХВ {[2хА (1~ХА + Х в) ~ Х в] С АВ +
+ [4 + »A& (1 - хА) - хАхв (4 - 6хА + Зхв)] DAB) +
+ хс{[хс-2ха(1-ха+хс)\Сса +
+ \х\ + 3хА (1 - ха) - хахс (4 - 6жл + а*с)] DCA} -
-xbxc{Bbc + (xb-xc)\2Cbc + 4xb-xc)dbc]-(1-2xa)g-
- О- - *Ха) (ХВ ~ХС)Н + (ХС - Ха) J\ + XAJ\ <Ш. 87)
В обоих случаях выражения для ув и ус можно получить
циклической перестановкой индексов. Величины Е, G, Я и J
являются тройными константами, при расчете которых
необходимы данные для тройных систем (например, данные о
равновесии пар — жидкость или жидкость — жидкость в тройных систе'
мах). Для определения трех тройных констант в уравнениях
Редлиха — Кистера необходима только одна точка по
равновесию в тройной системе. В частных случаях тройные константы
могут быть равны нулю. Если Я=/ = 0, уравнения Редлиха —
Кистера сводятся к уравнениям (111,86). Если G, Я, / и D
равны нулю, то уравнения (111,87) приводятся к уравнениям
(111,86), в которых £ = 0.
Более сложные уравнения, содержащие дополнительные
тройные константы, описаны Воолем12. Приближенный метод
расчета коэффициентов активности для тройных систем по
данным для бинарных предложен25 Шайбелем и Фридлэндом.
РАСЧЕТ РАВНОВЕСИЯ
ПО КОЭФФИЦИЕНТАМ АКТИВНОСТИ
Как указывалось в начале главы, желательно иметь
возможность рассчитывать распределение растворенного вещества
между частично смешивающимися растворителями с помощью
минимального числа исходных данных. Выше были рассмотрены
пути решения этой проблемы. В последующем изложении под
компонентом В подразумевается растворитель — экстрагент для
выделения распределяемого вещества С из растворов А — С.
109
аСА = йСВ
оме того
ЛСА = ^САХСА
ЛАА = аАВ аВА '
асв ~ Чсвхсв
= авв
и т. д.
Ранее отмечалось, что при выборе одинакового стандартного
состояния активности вещества в каждой фазе при равновесии
равны:
(III, 88)
(III, 89)
Общий метод, которым следует предпочтительно
пользоваться при расчете равновесия, заключается в следующем.
Определяют величины коэффициентов активности в трех бинарных
системах (А—В, А—С и В—С), применяя для получения
большего числа исходных данных интегральные формы уравнения
Гиббса — Дюгема для бинарных систем. При помощи
интегральных форм уравнений Гиббса — Дюгема для тройных
систем рассчитывают значения коэффициентов активности и
активностей в тройных системах. Далее определяют равновесные
составы жидких фаз, исходя из условия, что активности всех
трех компонентов в каждой фазе одинаковы.
Последний этап расчета можно пояснить с помощью рис. 56.
На этом рисунке приведена типичная тройная диаграмма для
систем типа I. На диаграмме показана одна из хорд равновесия
KL, Активности компонентов А, В я С в растворе К должны
быть равны соответственно активностям А, В и С в раствореL.
Кривые VK и LR представляют собой растворы с постоянными
значениями активностей компонента С. Аналогично кривым ТК
и LS соответствуют растворы с постоянной активностью
компонента В, а кривым UK и LW—с постоянной активностью А.
При помощи уравнений для коэффициентов активности в
тройных системах могут быть найдены точки пересечения этих трех
кривых постоянных активностей (в данном примере точки К и
L). Таким образом можно определять как хорды равновесия,
так и кривую растворимости.
Поскольку уравнения для коэффициентов активности
являются неявными относительно х, следует пользоваться
следующим методом. По уравнению для коэффициентов активности
тройной системы рассчитывают коэффициенты активности ус и
активности ас при постоянных величинах хв (для растворов
с преобладающим содержанием компонента А) и для каждого
значения хв определяют составы, соответствующие некоторым
фиксированным значениям ас- Для составов с преобладающим
содержанием компонента В повторяют расчет при постоянных
значениях хА, охватывая таким образом полностью
предполагаемую область ограниченной растворимости.
Координаты точек на треугольной диаграмме для каждой
фиксированной величины ас образуют линии постоянных ас,
как показано на рис. 58 (стр. 114). В связи с тем что
уравнения для коэффициентов активности непрерывны относительно.г,
110
рассчитанные кривые ас также
будут непрерывны, хотя в
действительности они не будуг
существовать в области
несмешиваемости жидкостей. Далее
рассчитывают величины уА и аА,
а также ув и ав вдоль одной из
линий ас. Затем активности аА
и ав изображают в функции от
хв как для растворов с
преобладанием компонента А, так и для
растворов с преобладанием В _
(см. рис. 59, стр. 115). Из гра- ат м N w в
фика на этом рисунке нетрудно п сс .
^ „ Г J ttstha "ИС. 56. АКТИВНОСТИ В ТРОЙНЫХ
найти четырехугольник JK.LM, системах,
соответствующий двум таким
значениям хв, для которых одновременно удовлетворяются
равенства аАА — аАВ и аВА = авв. Так как график построен для
постоянного значения ас, точки на линии ас, определяемые
полученными величинами хв, удовлетворяют условиям (111,88).
Эти точки можно перенести на фазовую диаграмму (точки F и
G на рис. 58); они являются концами хорды равновесия.
Повторение аналогичного расчета для других значений ас
позволяет получить все данные, необходимые при построении
фазовой диаграммы. Концентрации в мольных долях можно
пересчитать в весовые доли, что удобнее для расчетов.
При построении фазовой диаграммы с достаточной
точностью необходимо, как правило, пользоваться уравнениями для
коэффициентов активности в тройных системах, содержащими
константы, которые можно определить только по данным для
тройных систем. Тройное равновесие пар — жидкость для
полностью смешивающихся систем можно достаточно точно
рассчитать, исходя только из данных для бинарных систем. Однако
при ограниченной смешиваемости компонентов этот метод
непригоден для большей части систем, используемых в процессах
жидкостной экстракции.
Имеются, конечно, исключения; расчет равновесия в тройных
системах только по данным для бинарных систем оказывается,
по-видимому, более успешным в тех случаях, когда область
нерастворимости в бинарной системе АВ (для систем типа I или
в обеих ограниченно растворимых парах для систем типа II)
почти симметрична. В качестве исходных данных для тройных
систем (в дополнение к данным для бинарных), которые удобно
применять при расчете тройного равновесия, могут служить
известная хорда равновесия или данные о равновесии пар —
жидкость. Можно пользоваться также данными об азеотропных
составах, исключая те случаи, когда азеотропная смесь образо-
111
08
Цъ
&
вана двумя несмешивающими-
ся жидкостями и неизвестны
составы каждой из жидких
фаз (в этом случае не
требуется знания состава пара).
Если известны две (или более
для сольютропных систем)
хорды равновесия, для
расчета фазовой диаграммы
применим также значительно более
простой метод,
использованный в примере П-2 (стр. 50).
Пример 111-11. Рассчитать
фазовую диаграмму равновесия для
системы типа 1: вода( А)—этилацетат
(В) —этанол (С).
Решение. Чтобы
воспользоваться уравнениями Редлиха —
Кистера (111,87), необходимо
определить константы в уравнениях
(III, 56) —(III, 58) для бинарных
систем.
Вода (А) —этанол (С).
Коэффициенты активности рассчитываем на основе данных Джонса и др.21 по
равновесию пар—жидкость при 760 мм рт. ст. Полученные коэффициенты
активности приведены в виде зависимости lg(\c!yA) от хс на рис. 57.
Определение констант в уравнении (111,58) проводилось следующим
образом (см. табл. 5):
при х~ = 0,5
1&(УсАл) = -°'0900=Ссл/2
-/
г
0,6
ол
0,2
-0.2
-Ofr
' О 0,2 Ofi- 0,5 0,8 1
хв,хс, мол. дола
Рис. 57. Определение констант
Редлиха— Кистера (к примеру III-11):
/ — вода (Л) —этанол (С); 2 —этилацетат (В) —
этанол (С),
СГ. = — 0,1800
СА
При
хс = 0,1464
\
[ё (УсДл) = 0>4113 = °'7твсА - ссл/4
ВГЛ = 0,5180
СА
при
lg(Yc/Y^) = -0,3220
хс == 0,8536
- 0,7071ВСА-ССД/4
В
С А'
: 0,5190
Положение кривой на рис. 57 можно исправить, чтобы величина ВСа
приняла постоянное значение; но, как будет видно из дальнейшего,
достаточно взять среднее значение Вса = 0,5185:
при хс = 0,2959
>£ (Ус/У а) = °'1498 = О-4082 (Вса - 2°сл/3) + Ссл/4
£>„л= 0,0618
СА
при хс = 0,7041
£ (Ус/Уа) = - °'2464 = - а4082 (ВСА ~ 2Dc л/3) + Сса/4
£>сл= 0,0378
112
Среднее значение Оса =0,0498. Зависимость на рис. 57, рассчитанная С
помощью полученных значений констант, хорошо согласуется с
экспериментальной.
Этилацетат (В) — этанол (С). На основе данных Грисволда с сотр. [Ind.
Eng. Chem., 41, 2352 (1949)] по равновесию пар—жидкость при 760 ммрт.ст.
находим аналогичным способом следующие значения констант: 5ВС = 0,3505;
Свс——0,0280; DBc=—0,0303. На рис. 57 приведены экспериментальные
точки и кривая, рассчитанная на основе полученных значений констант. При
этом достигается хорошее совпадение опытных и расчетных данных.
Этилацетат (В) — вода (А). Грисволд с сотр. в указанной выше работе
приводит следующие данные о взаимной растворимости этилацетата и воды
при 70°С: *ав = 0,232; *вв = 0,768; *дД=0,986; *ва=0,014. При подстановке
этих значений в уравнения Редлиха — Кистера (111,73) и (111,74) получаем
уравнения:
0,5896ВЛД — 0,04304СЛД -
- 0,06006 D
АВ
■■ 0,6284
0,9184В +1,0293 С „ + 0.76153Z? = 1,7392
АВ
АВ
Грисволдом с сотр. опубликованы также следующие данные о
равновесии пар—жидкость при 760 мм рт. ст.:
ХА
0,060
0,042
0,024
хв
0,940
0,958
0,976
YЛ
6,91
8,33
8,30
УВ
1,01
0,998
0,995
>8 (¥лЛв)
0,83569
0,92169
0,92117
Значения ув должны быть во всех случаях больше единицы. Однако
при столь больших величинах хв некоторые неточности при определении \в
возможны и обычно бывают значительно больше.
Воспользуемся этими данными для определения константы Dab- При
подстановке данных о равновесии пар—жидкость из первой строки таблицы
в уравнение (111,58) и решении последнего совместно с зависимостями,
полученными на основе данных о взаимной растворимости компонентов,
находим значение £>лВ = 0,3365. Аналогично, используя остальные данные о
равновесии пар—жидкость, получаем Z)ab = 0,3735 и /)ав=0,3350. Среднее
значение £>ав = 0,3483, откуда находим ВАв= 1,1367 и CAB=0,4804.
Тройные константы. Грисволд приводит состав равновесных жидких фаз
при 70°С (одна хорда равновесия):
*4Л = °'917 -*ВЛ = 0'030
хАВ = 0,465 *м = 0,415
хСА = 0,053
vcb
= 0,120
Так как активности компонента А в обеих фазах должны быть
одинаковы, согласно уравнению (111,89) имеем:
'& аА = '2 У АЛ + '2 ХАА = ]S УАВ + ]S *АВ
Подстановка в это уравнение значений хАа и *ав из данных Грисволда
и выражений для lg у, полученных из уравнений Редлиха — Кистера для
тройных систем (111,87), в которые введены найденные ранее значения
бинарных констант, приводит к линейному уравнению с тремя константами G,
8 Р. Е. Трейбал
113
С-этанол
CD
о
Этилацетат
Рис. 58. Система вода — этилацетат — этанол [концентрации в мольных
долях (к примеру III-11)].
Н и J. Такие же уравнения можно написать для ав и ас; в результате
получаем систему трех уравнений с тремя неизвестными. Решение ее дает
0=1.301; #=9,400 и 7=1,708. Следовательно, константы в уравнении (111,87)
равны:
ВАВ= 1,1367
САВ = 0,4804
Олв = 0,3483
ВВС = °'3505
Свс = —0,0280
DBC = — 0,0303
ВСА = °'5185
ССА = — 0,1800
°сл = °'0498
G = 1,301
Я = 9,400
/= 1,708
Тройное равновесие. Точки D и Е на рис. 58 характеризуют взаимную
раствориместь воды и этилацетата. С помощью полученного уравнения
рассчитываем ус и ас для линий хв = 0; 0,03; 0,06; 0,09 и 0,12 и Хд = 0,2; 0,3;
0,4; 0,5; 0,6 и 0,7. Из зависимости ас от хс при постоянных Хд или Хв
определяем координаты линий ac = const (см. рис. 58).
Для ас = 0,2 были рассчитаны величины аА и ав:
ХА
0,360
0,400
0,500
0,894
0,939
0,952
хв
0,515
0,495
0,445
0,090
0,030
0
хс
0,125
0,105
0,055
0,016
0,031
0,048
УА
2,601
2,435
2,108
1,078
1,018
1,006
УВ
1,371
1,443
1,643
16,04
29,32
34,37
«Л
0,936
0,975
1,054
0,965
0,956
0,957
ав
0,707
0,715
0,731
1,444
0,880
0
Результаты расчета изображены графически на рис. 59. Из
прямоугольника, построенного таким образом, чтобы обеспечить равенство активностей
как компонента А, так и компонента В в обеих фазах, находим равновесные
составы на линии ас = 0,2; хВа=0,022 и хвв = 0,502 (точки F и G на рис. 58).
Повторяя расчет, находим положение еще нескольких хорд равновесия.
На рис. 60 показаны полученная расчетом бинодальная кривая и
корреляционная линия для хорд равновесия в сравнении с данными Грисволда с
сотр. Как видно из рис. 60, совпадение расчетных и экспериментальных
данных отличное. На том же графике приведена конода, при помощи кото-
114
рой найдены значения тройных
констант в уравнениях для
коэффициентов активности.
Расчет без тройных
констант G, Н и J приводят к
плохому совпадению с опытными
данными при значениях хс,
больших 0,05; область
взаимной растворимости,
определенная таким образом, выходит
за пределы хс=0,3.
Приведенный в этом
примере расчет очень
трудоемок. Положение би-
нодальной кривой часто
бывает известно, и тогда
расчет диаграммы
равновесия можно
значительно упростить. В
данном случае достаточно
найти только значения ас
на бинодальной кривой,
положение которой
известно.
Пример Ш-12. Рассчитать равновесие в системе типа II: «-гептан {А) —
анилин (5)—циклогексан (С) при 25° С.
Решение.
н-Гептан (А) — анилин (В). Данные о взаимной растворимости
компонентов А и В при 25° С—приведены в работе Хюнтера и Брауна [Ind. Eng.
Chem., 39, 1343 (1947)]:
слой гептана
055
Рис. 59. Определение хорды равновесия
при ас =0,2 (к примеру 111-11).
93,20
6,80
т —П °/
*с и'У\
xfV/\gd-\
A/sf V
Вода
вес.
вес.
%
%
гептана
анилина
С-этанол
v \/ V
А /\ 2 /\
\/—Л?^"~"
/yTV"-7^A—
\/ /f \/ \7
хАА = отт2
хВА = 0,0728
\j з\/ \
7\^vv\
лА-^О-^Лл
Этилацетат
Рис. 60. Система вода — этилацетат — этанол при 70° С [к примеру (Ш-11);
сравнение результатов, полученных на основе уравнений Редлиха —
Кистера, с опытными данными Грисволда, Чу и Уинзауэра, см. Ind. Eng.
Chem., 41, 2352 (1949); концентрации выражены в мол. долях]:
1 — расчетная корреляционная кривая для хорд равновесия; 2 — хорда равновесия, по
которой рассчитывались тройные константы; 3 — расчетная бинодальная кривая; 4 —
опытная корреляционная кривая1 для хорд равновесия; 5 —опытные данные.
8*
115
слой анилина
6,50 вес. % гептана
93,50 вес. % анилина
хАВ = 0,0607
xDD = 0,9393
На
Подставляя эти величины в уравнения Ван-Лаара (III, 71) и (111,72),
получаем:
Ав'
■■ 1,359
^ВА
: 1,290
Для нахождения констант можно воспользоваться также рис. 54.
Анилин (В) — циклогексан (С). Расчет проводим на основе данных,
имеющихся в упомянутой выше работе Хюнтера и Брауна (при 25°С):
слой циклогексана
83,50 вес. % цнклогексана
16,50 вес. % анилина
хсс = 0,8488
хвс~ 0,1512
слой анилина
26,20 вес. % цнклогексана
73,80 вес. % анилина
*св
= 0,2820
хвв = 0,7180
Из уравнений Ван-Лаара (111,71) и (111,72) или из графика на рис. 54
находим:
Авс= 1,120
Асв = 0,867
н-Гептан (А) — циклогексан (С). Для этой системы отсутствуют какие-
либо данные, которые можно использовать для расчета коэффициентов
активности. Можно, однако, считать
указанную систему близкой к
идеальной (см. пример 11,17). Тогда-
/
I
g &
I
4 *'
1\*/
L-/
-
/
&у
,&А
~г
ЛСА
АС
= 0
СА
\с
= 1,0
цг ofi off ojs
Будем вести расчет на основе
уравнения Ван-Лаара для тройных систем
(111,83). После подстановки
значений констант и упрощений
получаем:
_ \М7хв — 0,\69\хАхв
™С ~ /у Л- г -LI 9Q9v \2
(ХС + ХА + 1&2хвГ
концентрация циклоггксана хс,
мол. дали
Из этого уравнения
рассчитываем коэффициенты активности вдоль
г, „. . бинодальной кривой, пользуясь дан-
Рис. 61. Активности цнклогексана Иыми Хюнтера и Брауна (см. выше)
в системе гептан (А) — анилин (В) — „о взаимной растворимости в трой-
циклогексан (С): иой системе при 25° С. Результаты
/ — в фазе анилина; 2 —в фазе гептана. расчета сведены в таблицу;
118
Слой
Углеводородный
Анилиновый
*А
0,9272
0,687
0,667
0,426
0,248
0,1248
0
0,0607
0,0368
0,0219
0
хв
0,0728
0,0850
0,0835
0,1019
0,1210
0,1428
0,1512
0,9393
0,853
0,785
0,718
хс
ХСА
0
0,227
0,249
0,472
0,630
0,731
0,8488
хсв
0
0,1098
0,1938
0,282
VC
ЧСА
0,9920
0,9992
1,0015
1,0165
1,0355
1,058
1,0725
VCB
6,046
4,721
3,681
3,251
ас
аСА
0
0,227
0,250
0,480
0,652
0,774
0,910
^СВ
0
0,518
0,714
0,916
На рис. 61 изображены зависимости аСл от
Хса и аСв от хсв, с помощью которых легко
иайти при условии равенства активностей
равновесные величины Хса и Хсв-
0,280 0,480 0,600 0,705 0,795 0,849
0,05 0,10 0,15 0,20 0,25 0,282
хсв
Результаты расчета в сравнении с опытными
данными Хюнтера и Брауна приведены на
графике (рис. 62). Сходимость опытных н
расчетных данных хуже, чем в предыдущем примере;
однако следует учесть, что расчет производился
только по данным о взаимной растворимости в
двух из трех бинарных систем. Кроме того,
вызывает сомнения точность опытных данных
Хюнтера и Брауна о взаимной растворимости в
системе анилин — гептан, так как их результаты
несколько отличаются от результатов
соответствующих измерений Вертересьяна и Фенске [Ind.
Eng. Chem., 29, 270 (1937)]. Для подобных
расчетов желательно располагать очень точными
исходными данными.
Селективность. Как будет показано
ниже (глава IV), селективность
растворителя является- более важным
показателем при оценке пригодности
растворителя как экстрагента, чем коэффициент
распределения. Селективность
компоненту
з
<§ 06
0,2
о
о/
о/
0,1 0,2 0,3
хсв, мол. доли
Рис. 62. Распределение
цнклогексана (С) между
«-гептаном (А) и
анилином (В) при 25° С
[расчетные данные
нанесены в виде сплошной
кривой; точки —
опытные данные Хюнтера и
Брауна, см. Ind. Eng.
Chem., 39, 1343 (1947)].
117
та В в отношении разделения компонентов С и А определяется
выражением
Рс,л = ^
Св/ХАВ
са/хаа
(111,90)
причем сюда входят равновесные концентрации. В связи с тем
что х—а/у и при равновесии асв = аСА и аАВ = аАА, имеем:
R _ Уса/Уаа
VC,A~
Усв/У.
АВ
УСАХАА
УСВХАВ
(111,91)
Следовательно, селективность можно рассчитать по данным,
используемым при расчете равновесия. Чтобы разделение было
возможно, величина р должна превышать единицу.
Системы с взаимно несмешивающимися растворителями.
Часто из-за недостатка данных о тройном равновесии или для
упрощения расчетов делают допущение, что взаимная
растворимость ограниченно смешивающихся растворителей не влияет
на активность распределяемого вещества. Так, если требуется
рассчитать распределение этанола между бензолом и водой,
можно пренебречь взаимной растворимостью воды и бензола
и ее влиянием на активность этанола. В таких случаях
коэффициенты активности распределяемого вещества С в бинарных
растворах АС и ВС можно определить при помощи
сравнительно малого числа опытных данных, используя уравнения для
коэффициентов активности в бинарных системах. После расчета
активностей равновесные величины хСА и хсв определяют,
исходя из условия равенства23 активностей компонента С.
Применимость такого способа расчета тщательно
анализировалась26 и было показано, что в тех случаях, когда взаимная
растворимость компонентов А и В
очень мала, можно ожидать
сравнительно хороших результатов
при низких концентрациях
распределяемого вещества С. В
случае же заметной взаимной
растворимости растворителей
можно получить только
качественные данные о направлении
распределения.
Пример 111-13. Рассчитать
распределение ацетона (С) между водой (Л) и
хлороформом (В), пренебрегая
взаимной растворимостью воды и
хлороформа.
Решение.
Ацетон — вода. Коэффициенты
активности и активности ацетона при 25° С
приведены на рис. 44 (см. стр. 77).
1
0£
§ 0,2
I
а-с
л О/Пй
ССА^
V
гУ
Лас
в о/па
:св
0,2 0,Ь 0,6 Q8 I
Концентрация ацетона
хс, мол. дола
Рис. 63. Активности ацетона в
смесях с водой (кривая /) и с
хлороформом (кривая 2).
118
Ацетон — хлороформ. Коэффициенты
активности и активности ацетона в этой системе
представлены для температуры 35,2° С на рис. 48 (см.
стр. 79).
Активности ацетона в этих бинарных
системах изображены графически в координатах,
показанных на рис. 63. Пренебрегая взаимной
растворимостью воды и хлороформа, определяем, исходя
из условия равенства активностей ацетона,
равновесные концентрации его в обеих фазах:
х 0 0,01 0,03 0,06 0,08 0,10 0,15 0,2
х* 0 0,137 0,305 0,44 0,495 0,550 0,635 0,680
ев
Рассчитанная кривая распределения вместе с
экспериментальными данными Хэнда27 для
температуры 25° С показана на рис. 64. Как видно
из графика, хорошее соответствие с опытом
достигается при низких концентрациях ацетона, но
по мере увеличения взаимной растворимости
воды и хлороформа с возрастанием концентрации
ацетона расхождение увеличивается. Тем не
менее можно сделать вывод, что распределение
сильно сдвинуто в сторону хлороформа. Даже
такой качественный вывод в ряде случаев может
оказаться весьма полезным.
0,8
0,6
I
I 0£
I
о—
о
0,2
0,2 0,Ь
хСА,мол. доли
Рис. 64. Распределение
ацетона (С) между
водой (А) и
хлороформом (В) при 25° С
[расчетные данные
нанесены в виде сплошной
кривой; точки — опытные
данные Хэнда 27].
аСА — УСАХСА
г
асв —Усвхсв
Коэффициенты распределения при
низких концентрациях. Кривые
активность — концентрация для растворенного
вещества С в тройных смесях имеют общую форму, показан*
ную на рис. 65. Уравнения касательных к этим кривым в
начале координат имеют вид:
(III, 92)
(III, 93)
где коэффициент активности у'с относится к бесконечному
разбавлению, или к нулевой концентрации С. Следовательно, для
равновесных растворов (в случае
равенства активностей) при
нулевой концентрации С имеем:
Усахса = У с 3х с в (И1194)
Поэтому наклон кривой
распределения в начале координат, или
коэффициент распределения при
нулевой концентрации С, равен:
1св
Уса
хс , мол. доли
Рис. 65. Начальный наклон
кривых активность — концентрация.
ссл
Уев
(111,95)
или
lg«' = lgYc4-l&Ycs («1.96)
119
Величины у'с и т' легко определить из уравнений (III, 83) —
(111,87), принимая хс равным нулю и подставляя в них
взаимные растворимости компонентов А и В (определения констант
Е, G, Н и / для тройных смесей при этом не требуется).
Ситуация еще больше упрощается, если А и В очень мало растворимы
друг в друге, т. е. если хАВ и хВА приближаются к нулю. Тогда
значения у'с равны предельным значениям коэффициентов
активности в бинарных системах, одним из компонентов которых
является С. Поэтому
т' = ф=ю(дсл-лЫ (Ш97)
х
слили
^т' = АсА-Асв (ш-98)
Уравнения (111,97) и (111,98) позволяют очень быстро
определить характер распределения. Такая информация часто
бывает достаточной,, для того чтобы исключить тот или иной
растворитель из рассмотрения при подборе экстрагента. Если же
взаимная растворимость А и В мала, с помощью этих
уравнений можно рассчитать равновесное распределение
количественно. В противном случае следует пользоваться уравнением
(111,96).
При низких концентрациях распределяемого вещества можно
также приближенно рассчитать селективность. Комбинируя
уравнения (111,90) и (111,95), получаем:
Рсл = т' ~- (III, 99)
ХАВ-
где $'с,а~ селективность В по отношению к С при нулевой
концентрации С.
Селективность, следовательно, можно найти по известному
коэффициенту распределения и данным о взаимной
растворимости компонентов А и В.
Пример 111-14. Рассчитать коэффициент распределения и селективность
в системе вода (А) — н-бутанол (В) — н-пропиловый спирт (С) при 38° С
для бесконечно разбавленного раствора пропилового спирта
Решение.
а) Константы в уравнениях для бинарных систем можно определить
любым из описанных способов в зависимости от характера исходных
данных. Воспользуемся, например, данными табл. 6 (см. стр. 100). Графической
интерполяцией констант для нормальных спиртов при темпепатуре 38° С
получаем: а= -0,920; е=0,612 и 1=0,534. Следовательно, для раствора
и-пропилового спирта в воде по уравнению (а) табл. 6 находим:
*СА = — 0,920 + 0,612 • 3 + 0,534/3 = 1,086
Система к-пропиловый спирт — и-бутанол должна быть близкой к
идеальной, поэтому Лсв=Лвс=0. Тогда по уравнению (111,97):
т! = 101'086 = 12,2
120 *
б) Более точно величины т' и р можно определить следующим образом:
н-Пропиловый спирт в воде— Л с а = 1,086 (см. выше). Вода в н-пропи-
ловом спирте — для 38° С по уравнению (а) табл. 6 находим:
ААС = 0,730 — 0,560/3 = 0,543
н-Пропиловый спирт — н-бутиловый спирт—принимаем систему
идеальной; тогда
Асв = Авс = ° и Асв/Авс = 1$
н-Бутиловый спирт в воде — по уравнению (а) табл. 6 имеем:
АВА = — 0,920 + 0,614 • 4 + 0,534/4 = 1,602
Вода в н-бутиловом спирте — из уравнения (а) табл. 6 следует;
ААВ = °'730 — °-560/4 = °-590
Определяем с помощью рис. 54 взаимную растворимость «-бутилового
спирта и воды. Полученным значениям Аав и АВа могут соответствовать
растворимости, приведенные в следующей таблице (первые два столбца
определялись из графика иа рис. 54, а по иим рассчитывались остальные):
хва/хав
0,02
0,05
0,10
0,20
ААВ = 0,590
ХАВ
0,362
0,408
0,450
0,522
ХВА
0.С0724
0,0204
0,0450
0,1044
хав/хва
30
20
10
АВА = Х,602
ХВА
0,0230
0,0269
0,0306
ХАВ
0,690
0,538
0,306
Используя приведенные в таблице величины, полученные иа основе
значений ААв и АВа, можно построить зависимости хав от хва. Точка
пересечения линий *ав=0,425 и хва =0,029 определяет взаимную растворимость
воды и «-бутилового спирта.
Подставляя полученные выше значения коистаит А в уравнения Ван-
Лаара для тройных смесей, находим при *с=0:
, 0,272лг^ — 0,2585л: Ахв
IgYc= (0,500л-л + л-в)2
Отсюда имеем:
при хАВ = 0,425 хвв = 0,575 Ig усв = — 0,0225
при хВА = 0,029 л-дд= 0,971 \gyCA = 0,94\
Уравнение (111,96) дает:
Ig m' = 0,941 + 0,0225 = 0,964 m' = 9,3
Согласно уравнению (111,99), имеем:
РС>Л = 9'30425" J
121
Результаты расчета показывают, что распределение пропилового спирта
сильно сдвинуто в пользу бутанола (при выражении концентраций в
мольных долях). Кроме того, бутанол отличается значительно более высокой
селективностью по отношению к пропиловому спирту, чем к воде.
Примечание. Мак-Кенте и др. [Ind. Eng. Chetn., 45, 454 (1953)]
исследовали равновесие для рассматриваемой системы при 38° С. Согласно их
данным, коэффициент распределения для бесконечно разбавленного раствора
(при выражении концентраций в мольных долях) т'=Ю и селективность
Р'=19. Взаимная растворимость бутанола и воды найдена равной: хАВ =
= 0,525; Хва =0,017. Учитывая значительную несимметричность системы и
высокую взаимную растворимость компонентов А и В, следует считать, что
рассчитанные величины очень хорошо согласуются с этими опытными
данными.
Размерности концентраций. Методы расчета распределения,
рассмотренные выше, предусматривали выражение
концентраций в мольных долях. Коэффициенты распределения,
выраженные в весовых долях, отличаются по численному значению от
коэффициентов распределения в мольных долях, но характер
распределения в обоих случаях обычно одинаков. Лишь в
редких случаях, при необычной комбинации молекулярных весов,
он может изменяться. Например, в системе этанол — этилаце-
тат — вода равновесные концентрации выше в водном слое,
если их выражать в весовых долях, и, наоборот, выше в
эфирном слое, если пользоваться для выражения концентраций
мольными долями.
Когда бинодальная кривая известна, переход от одной
системы размерностей для концентраций к другой не представляет
затруднений. В тех случаях, когда распределение рассчитывают
только по данным для бинарных систем, при нулевой
концентрации С имеем:
^св хсв
лСА ХСА
'x'ab/MWa + Xbb/MWb
xaaImWa + X'baImWb\
(III, 100)
где Х'АД, х'вв, Х'АВ yl Х'ВА — взаимные растворимости
компонентов А и В.
Если можно считать А я В практически взаимно
нерастворимыми, так что Х'ВВ = Х'АА = 1 и Х'АВ = Х'ВА = 0, то
приближенно:
Х'св x'CBMWA
к:=^ш; (,IU01)
Величина селективности от способа выражения
концентраций не зависит.
Многокомпонентные системы
В процессе экстракции с двумя растворителями
(фракционная, или дробная экстракция) одновременно распределяются
два вещества между двумя ограниченно смешивающимися
122
растворителями, образуя таким образом четырехкбмпонентнукЭ
систему. Обозначим распределяемые вещества буквами В я С,
а растворители— Л и D. Коэффициенты распределения будут
тогда выражаться следующим образом:
т=?Ш- тг=^+- (111,102)
Селективность в таких системах определяется как
В = ™s = хвахср (Ш 103)
В'° тС XCAXBD
Поскольку активности каждого вещества при равновесии
одинаковы во всех фазах
уса*вр_ (Ш) Ю4)
CD^BA
В' С УгпУ
В системах, состоящих из большого числа компонентов,
распределяемых между двумя растворителями (такие системы
встречаются, например, во многих процессах
нефтепереработки), уравнения (III, 103) и (III, 104) можно применять к любой
паре распределяемых веществ.
Вооль12, Редлих и Кистер 10'16 и др. показали, как
составляются уравнения для коэффициентов активности в системах,
состоящих из четырех компонентов и более. Уравнения,
предложенные Блэком2, обладают тем преимуществом, что
эмпирические константы, входящие в них, можно определить только из
данных для бинарных систем. Эти уравнения существенно
упрощаются, если распределяемые вещества являются членами
одного гомологического ряда. Однако все известные зависимости
для коэффициентов активности в многокомпонентных системах
сложны, и ни одна из них не проверялась на системах,
представляющих интерес для жидкостной экстракции, т. е. на
системах, образующих две жидкие фазы. Поэтому при расчете
равновесия в таких системах лучше всего ограничиться
определением р для систем с малой взаимной растворимостью
растворителей при бесконечном разбавлении распределяемых веществ.
Даже такие ограниченные данные весьма полезны.
Рассмотрим, например, разделение алкилзамещенных бензола и
парафиновых углеводородов дробной экстракцией по методу, предложенному
Пьеротти и др.24. Если экстрагентами являются фурфурол (А) и к-гептан
(D), предельные значения коэффициентов активности при бесконечном
разбавлении в этих растворителях при 70° С можно определить из табл. 7в (см.
стр. 104):
Парафиновые углеводороды (С) в фурфуроле (А):
\g у'СА = 0,737 + 0,0810iVc - 0,00058 (Nc - 5)2
123
Парафиновые углеводороды (С) в «-гептане ф)
lgYco = -0,00058 (Nc-7f
где Nc — число атомов углерода в молекуле углеводорода.
Так как
тс = x'caIxcd = Усо/у'са
и
\gm'c =\gVcD — lgy'cA
подставляя в это уравнение выражение для Yc получаем:
\gm'c =— 0,7509 — 0,0787-/Vc
Алкилпроизводные бензола (В) в фурфуроле (А)
О 469
\g у'ВА = 0,45 + 0№ШрВ - U|JDf - 0,00058 (ЛГ - 5)2
Алкилпроизводные бензола (В) в «-гептане (£>)
Ig Ybd = 0,225 - „U'J0T2 - 0,00058 (NB - 7)2
и
4™'в = — 0,2389 —0,0810ЛГрВ+0,00232ЛГв
Здесь iVpB и #в соответственно число алкильных и общее число атомов
углерода в алкилзамещенном бензоле. Так как NpB = Nb — 6
\gm'B = 0,2471 — 0,0787^в
Зависимости Ig m' от N изображаются параллельными прямыми,
приведенными иа рис. 66, а.
Далее имеем:
Ig Рв, с = te т'в ~ Iff «с = О."8 - 0,0787 (tfB - ЛГС)
Для Nb^Nc коэффициент Рв с = 10; величина Рд с = 1, когда WB—
NC = \2J, или округленно, когда Л?в— iVc = 12.
Предположим, что на разделение поступает смесь парафиновых
углеводородов, для которых Nc колеблется от 5 до 14, и алкилпроизводных
бензола, для которых Nb*=6—13. Пусть разделение экономически выгодно, если
Рв с равно трем или более. Для $в,С~ 3 разность NB — Nc=6,6.
Следовательно, для полного выделения алкилзамещенных бензола было бы
необходимо допустить присутствие в ароматической фракции парафиновых
углеводородов с числом атомов углерода, равным 13 — 6,6=6,4 или с шестью
атомами углерода и менее. Если присутствие парафиновых углеводородов в
ароматической фракции недопустимо, должны будут теряться ароматические
углеводороды, содержащие 5+6,6=11,6 атомов углерода, т. е. округленно —
двенадцать атомов углерода и более. Это иллюстрируется графически на
рис. 66.
Если в качестве экстрагентов использовать этиленгликоль (А) и
«-гептан (D), расчетом, аналогичным предыдущему, можно получить:
\gm'c =—1Д80 — 0,2302iVc
\gm'B =0,105 — 0,2302JVB
$'Bi c = 1,285 — 0,2302 (NB - Nc)
124
О 5 10 15 20 0 5 10 /5 20
Число атомов углерода, N
а 6
Рис. 66. Распределение углеводородов между фурфуролом и гептаном (а)
и этиленгликолем и гептаном (б) при 70° С:
1 — алкилпроизводные бензола; 2 — предельные углеводороды.
Для NB = NC коэффициент Р^; с = 19; величина Р^с —1,0, когда NB —
Nc — 5,6 или NB — Nc=5. Для Рв с = 3 разность NB — ЛГС = 3,5.
Зависимости \gmc от N для этой системы показаны на рис. 66,6.
При разделении описанной выше смеси для полного выделения алкил-
бензолов потребовалось бы допустить присутствие в ароматизированном
продукте парафинов с числом атомов углерода, равным 13 — 3,5=9,5, т. е.
с девятью атомами углерода и менее. Следовательно, гликоль пригоден для
выделения высокооктанового бензина из широкой (по молекулярным весам)
фракции. Если необходимо получить чисто ароматический продукт, следует
учитывать неизбежность потери алкилбензолов с числом атомов углерода,
равным 5+3,5 = 8,5 или округленно с девятью атомами углерода и более.
Вместе с тем гликоль обладает высокой селективностью (Рв> с = 19) при
выделении бензола, толуола и ксилола, например, из парафиновых
углеводородов, содержащих шесть и более атомов углерода.
Несмотря на то что селективности изменяются при переходе к реальным
значениям концентраций, подобные общие закономерности позволяют
получить полезную информацию, тем более, что в промышленных процессах
экстракции обычно применяют большие объемы растворителей. Таким
образом, растворители можно характеризовать, во-первых, величиной Рв с ПРИ
Nb = Nc, которая выражает селективность разделения по типу
углеводородов, и, во-вторых, при §'в с = 1,0 величиной NB — Nc, которая, наоборот,
является мерой способности растворителя к выделению широких (по
молекулярных весам) фракций.
ДРУГИЕ МЕТОДЫ РАСЧЕТА РАВНОВЕСИЯ
При отсутствии данных для расчета активностей во многих
случаях характер распределения можно определить другими
методами. Эти методы обычно позволяют лишь качественно
оценить распределение и менее надежны, чем описанные выше.
125
Критические температуры растворения (К. Т. Р.). На рис. 67,
изображена типичная тройная система, каждая из бинарных пар
которой имеет верхнюю К. Т. Р. Предположим, что каждая из
бинарных кривых растворимости симметрична. Очевидно, в этом
случае [уравнения (111,75) и (111,76)] константы в
уравнениях Ван-Лаара для каждой пары компонентов будут при
соответствующих температурах равны. Так как обычно теплота
растворения уменьшается с возрастанием температуры, каждая
из бинарных систем с увеличением температуры приближается
к идеальной. Следовательно, для произвольной температуры
коэффициенты активности наименьшие в системе В — С, средние
по величине в системе С — Ли самые большие в системе А— В.
Рассмотрим теперь систему типа I при температуре tx. Из
наших рассуждений о сравнительной величине коэффициентов
активностей вытекает, что компонент С обладает большей
избирательностью по отношению к фазе, богатой компонентом В.
Отсюда следует, что из двух взаимно несмешивающихся
растворителей более селективный тот, который обладает более
низкой критической температурой растворения с распределяемым
веществом С (в данном случае растворитель В). Рассмотрим
теперь систему типа II, находящуюся при температуре t2. В этой
д системе компонент А может
служить для разделения
компонентов В и С. Качественный
анализ коэффициентов
активностей показывает, что в
системе А—В склонность к
испарению выше, чем в системе
А—С. Следовательно,
компонент А преимущественно
экстрагирует С. Таким образом,
растворитель опять имеет
большую селективность по
отношению к компоненту, с
которым он обладает более
низкой К.Т.Р. Подобные
рассуждения можно применить и к
системам с нижней К.Т.Р.
Выводы о селективности,
основанные на величине К.Т.Р.,
вообще говоря, обоснованы
для реальных систем, однако
они не всегда оправдываются
из-за влияния других факто-
Рис. 67. Критическая температура Ров- Так> изменение теплот ис-
растворения и селективность. парения с температурой
различно для разных растворов,
126
и кривые растворимости редко бывают симметричными.
Поэтому на основе значений критических температур растворения
можно сделать только грубо приближенное заключение о
направлении распределения, которое не всегда надежно.
Полуколичественные данные можно получить только для
растворов, близких к регулярным, энтропия смешения которых
приближается к энтропии смешения идеальных растворов.
Френсис28 показал полезность такого подхода при
рассмотрении экстракции нежелательных примесей из нефтяных
фракций. В указанной работе Френсиса представлен критический
обзор имеющихся данных о критических температурах
растворения.
Для характеристики распределения в процессах
селективной очистки продуктов нефтепереработки часто пользуются
селективностью анилина как растворителя. Мерой селективности
служат значения анилиновой точки.
Анилиновой точкой называется температура, при которой
смесь равных объемов анилина и углеводорода разделяется на
два слоя. Хотя эта температура не идентична критической
температуре растворения, поскольку кривые растворимости в
большей части случаев несимметричны, она, как показал Френсис,
близка по величине к К. Т. Р. Анилиновые точки для различных
углеводородов, а также данные о К- Т. Р. углеводородов с
другими растворителями приведены в литературе28,29.
Используя в качестве указателя селективности данного
растворителя в процессах разделения различных углеводородов
разность К. Т. Р. этого растворителя с соответствующими
углеводородами, Френсис показал влияние химической структуры
растворителя на селективность. Подобно этому, Дрю и Хик-
сон30 и Хиксон и Бокельман 31 установили связь между К.Т.Р.
пропана с различными жирными кислотами и их эфирами и
селективностью пропана как растворителя для разделения смесей
жирных кислот и их эфиров.
Водородные связи и внутреннее давление. Из изложенного
выше следует, что распределение определяется степенью
отклонения смесей от идеальности. Степень отклонения от
идеальности можно определить на основе способности соединений
образовывать водородные связи, а также на основе величины
внутреннего давления.
Молекулы многих соединений полярны, т. е. имеют диполь-
ный момент, обусловленный неравномерным распределением
электронов в ковалентных связях атомов. Например, в
молекуле спирта электронная пара, образующая связь между
кислородом и водородом, сдвинута к атому кислорода:
R-6: Н
127
Следовательно, водород в молекуле спирта относительно
положительно заряжен, а остальная часть молекулы заряжена
отрицательно. Дипольный момент служит мерой этого явления.
Обычно молекулы имеют несколько групп (образующих
диполи), которые вследствие симметрии компенсируют друг друга,
уменьшая общий дипольный момент. Полярные молекулы имеют
тенденцию к ассоциации с расположением атомов водорода
между отрицательно заряженными частями соседних молекул:
R R R R
I I I I
Н—0---Н—О-.-Н—0---Н—О
Водород способен также образовывать связи с азотом и
фтором; такие атомы называются «донорами». Кроме того, водород
может образовывать связь между каким-либо атомом-донором
и углеродом, если к последнему присоединена достаточно
сильная отрицательная группировка. Некоторые из этих связей
сильны, другие сравнительно слабы. Ниже приведена
классификация жидкостей по водородным связям 32.
Класс I
Жидкости, способные к образованию трехмерных цепей
прочных водородных связей, например: вода, гликоль, глицерин,
аминоспирты, гидроксиламин, оксикислоты, многоатомные
фенолы, амиды и др. Соединения типа нитрометана и ацетони-
трила также образуют трехмерные цепи водородных связей,
однако гораздо более слабых, чем связи соединений, имеющих
группы ОН и NH. Поэтому соединения данных типов отнесены
к классу II.
Класс II
Жидкости, молекулы которых содержат как активные атомы
водорода, так и атомы-доноры (кислород, азот и фтор).
Примеры: спирты, кислоты, фенолы, первичные и вторичные амины,
оксимы, нитросоединения и нитрилы с атомом водорода в сс-по-
ложении, аммиак, гидразин, фтористый водород, цианистый
водород и др.
Класс III
Жидкости, в молекулах которых имеются атомы-доноры, но
нет активных атомов водорода: простые и сложные эфиры, ке-
тоны, альдегиды, третичные амины (включая соединения
пиридинового ряда), нитросоединения и нитрилы без атома водорода
в сс-положении и др.
№
Класс IV
Жидкости, молекулы которых содержат активные атомы
водорода, но не имеют атомов-доноров. К этому классу относятся
соединения, имеющие один или два атома хлора при атоме
углерода, у которого стоит водород, а также один атом хлора у
имеющего водород атома углерода и один или более атомов
хлора у соседнего атома углерода (СНСЦ, СН2С12, СНзСНС12,
СН8С1—СНаС1, СН,С1—СНС1—СН2С1, СН2С1—СНС12 и др.).
Класс V
Все прочие жидкости, т. е. жидкости, не способные к
образованию водородных связей: углеводороды, сероуглерод,
сульфиды, меркаптаны, галоидоуглеводороды, не попавшие в класс IV,
и такие неметаллические соединения, как иод, фосфор и сера.
Растворение друг в друге веществ, ассоциированных
посредством водородных связей, может привести либо к распаду
последних, либо к образованию новых связей. Так, при
растворении спирта в воде водородные связи в воде и спирте могут
разрушиться. При этом возможно образование новых связей
между молекулами воды и спирта:
Н R H H R
I I I I I
Н—О- • Н—О- ■ Н—О- • Н—О- ■ Н—О
Если размер углеводородной цепи в молекуле спирта велик,
слабый отрицательный заряд в этой молекуле не оказывает
заметного воздействия на сильные водородные связи между
молекулами воды. Такие спирты мало растворимы в воде. Влияние
водородной связи и других межмолекулярных сил на
растворимость обсуждали Гильдебранд и Скотт23,
Эвелл, Харрисон и Берг 32 разделили все соединения на пять
групп по их способности к образованию водородных связей (см.
выше). Как отмечают эти авторы, разрушение водородных
связей при смешении жидкостей приводит к положительным
отклонениям от закона Рауля, а образование водородных связей — к
отрицательным отклонениям. Прочность образующихся
водородных связей в общем случае указывает на степень
отклонения от закона Рауля. Данные о характере отклонений от
идеальности по Эвеллу, Харрисону и Бергу приведены в табл. 8.
Следует отметить, что для любых смесей жидкостей,
относящихся к классам III, IV и V, за исключением смесей классов
III и IV, новые водородные связи не образуются. Многие из
таких смесей относятся к категории регулярных растворов,
энтропия смешения которых приближается к энтропии смешения для
идеальных смесей.
9 Р. Е. Трейбал
129
Та б л ища 8
Характер отклонений от идеальности и водородные связи
Классы
I + V
II + V
III + IV
14-IV
II + IV
н
I-
I-
II —1
II-
hl
-II
-III
-II
LIII
III-
III —
III-
IV-
IV-
V-
-III
-V
-V
г-IV
-V
-V
Водородная связь
Водородные связи только
разрушаются
Водородные связи только
образуются
Происходит разрушение
и образование водородных
связей; однако разрушение
связей в жидкостях
классов I и II оказывает более
важное воздействие
Происходят распад и
образование водородных связей
Водородные связи
отсутствуют
Отклонения
Положительные; классы I+V
часто ограниченно взаимно
растворимы
Отрицательные
Положительные; классы
I и IV часто ограниченно
взаимно растворимы
Обычно положительные; при
наличии сложных групп
иногда возможно
образование азеотропных смесей
с максимальной
температурой кипения
Квазиидеальные системы;
обычно положительные
отклонения или
идеальность; при образовании
азеотропных смесей
последние обладают
минимальной температурой
кипения
Гильдебранд и Скотт23 нашли, что для регулярных
растворов отклонение от идеальности можно описать следующими
уравнениями:
V
RT\naA = RT
1п*л + *а(1-тИ-
+ Va*b{M>a,b? О"."»)
RT\naB = RT
la*R + +A[*-
+ УВФ2а(^а,в)2 ("U06)
Здесь ф представляет собой объемную долю компонента в
смеси, причем
VA*A
А VAXA + VBXt
Ф
VBXB
в V х А- V х
V A A^ V ВХВ
(III, 107)
В выражениях (111,107) величина V—мольный объем
компонента.
130
Параметры растворимости некоторых веществ при 25° С
Вещество
Четыреххлористый углерод
Четырехфтористый углерод
(при —128° С)
Четыреххлористый германий
Четыреххлористый кремний
Четыреххлористый титан
Гексафторуран
Двуокись углерода (при —50° С)
Двуокись серы (при —10° С)
Пропан
н-Бутан
Изобутан
н-Пентаи
Изопентан
2,2-Диметилпропан
н-Гексан
н-Гептан
н-Октан
Изооктан
н-Нонан
н-Гексадекан
Циклопентан
Циклогексан
Метилциклогексан
Бензол
Толуол
Этилбензол
о-Ксилол
л-Ксилол
л-Ксилол
Стирол
1-Бутен
цыс-2-Бутен
транс -2-Бутеи
Изобутен
1,3-Б)тадиен
Формула
СС14
CF4
ОеС14
S1CI4
Т1С14
UFe
со2
so2
с3н8
с4н10
с4н,0
с5н12
с5н12
с5н12
с6н14
с7н16
С8Н18
С8Н18
CgH20
С)бНз4
с5н10
С6Н12
С7Н14
с6н6
С7Н8
С8Н10
СвНю
С8Н10
С8Н10
с8н8
С4Н8
С4Н8
С4Н8
С4Н8
С4Н6
6
(аж/л3)0,5хю-3
17,6
15,7
16,6
15,6
18,4
18,2
18,2
12,3
13,7
12,8
14,4
13,8
12,8
14,9
15,2
15,4
14,0
15,6
16,4
16,6
16,8
16,1
18,7
18,2
18,0
18,4
17,9
17,7
19,0
13,7
14,7
14,3
13,7
14,5
V
м3/кмоль
0,097
0,045
0,115
0,115
0,111
0,096
0,038
0,044
0,089
0,101
0,105
0,116
0,117
0,122
0,132
0,147
0,164
0,166
0,180
0,295
0,095
0,109
0,128
0,089
0,107
0,123
0,121
0,123
0,124
0,115
0,095
0,091
0,091
0,094
0,088
9*
131
Продолжений табл. 9
Вещество
•Перфтор-н-бутан
Перфтор-н-пентан
Перфтор-н-гексан
Перфтор-н-гептан
Перфторбензол
Перфтортолуол
Метил енхлорид
Хлороформ
Хлористый этил
Дихлорэтан
Нитрометан
Сероуглерод
Акрилонитрил
Пиридин
Хлорбензол
Нитробензол
Формула
C4F10
C5F,2
CeF14
C?Fie
CeFa
C7F8
CH2CI2
CHCI3
C2H5CI
C2H4CI2
CH3N02
cs2
C8H3N
C5H5N
CeH5CI
CeH5N02
6
10,6
11,2
11,5
11,7
11,6
15,7
19,8
19,0
17,4
20,0
25,8
20,5
21,5
21,8
19,4
20,5
V
M'jKMOAb
0,163
0,183
0,205
0,227
0,115
0,142
0,065
0,081
0,073
0,079
0,054
0,061
0,066
0,081
0,102
0,103
Примечание. Hlldebrand, Scott, Solubility of Nonelectrolytes, Relnhold
Publishing Corporation, New York, 1950.
В случаях, когда мольные объемы компонентов почти
одинаковы, уравнения (III, 105) и (III, 106) преобразуются к виду:
RT In yA = Уаф\ (Дод Bf (III, 108)
RT In YB = V^\ (Дбдв)2 (III, 109)
В этих уравнениях б — параметр растворимости,
являющийся характерным свойством данного вещества, а Аб — разность
параметров растворимости двух веществ. Для неполярных
веществ параметр б равен корню квадратному из величины
изменения внутренней энергии при испарении, отнесенной к единице
объема, и служит мерой внутреннего давления:
Следовательно, значение б можно найти с помощью
величины скрытой теплоты испарения. Таким образом, для
регулярных растворов неполярных веществ коэффициенты активности
132
можно рассчитать, не прибегая к определению эмпирических
констант. Конечно, при этом не следует ожидать очень точных
результатов. Уравнения (III, 108) и (III, 109) часто можно
применять для слабо полярных соединений, если подставлять в эти
уравнения эмпирические величины параметров растворимости.
Однако в данном случае теряется чисто теоретический характер
этих уравнений.
Значения V и б {величины б рассчитаны по уравнению
(111,110)] для некоторых веществ при 25° С приведены в
табл. 9. Влияние температуры23 выражается уравнением
dIn6. = _l,25 (IH.lll)
din V
Таким образом, lg6 при изменении температуры меняется
линейно с lg V. Чем больше разница в величинах б для
компонентов бинарного раствора, тем больше положительные
отклонения от закона Рауля.
Применяя условия Гильдебранда, использованные ранее для
вывода уравнений (111,75) и (111,76), к уравнениям (111,105)
и (III, 106), получаем для критической точки растворимости
ограниченно смешивающихся веществ:
Ф IV \°.5
^\vj (Ш,112)
\0,5
*А
2V.Va№iRf
К.Т.Р.= * *v *•*' (111,113)
R(yf+Vff
Эти уравнения еще раз показывают наличие связи между
критической температурой растворения и отклонениями
растворов от идеальности.
Пример 111-15. Найти растворитель, который может оказаться
пригодным для экстракции ацетона из водных растворов.
Решение. В соответствии с классификацией жидкостей по
способности образовывать водородные связи ацетон относится к классу III, а вода —
к классу I (см. стр. 128). Можно ожидать, что водные растворы ацетона
•будут иметь положительные отклонения от закона Рауля (табл. 8).
Следовательно, для того чтобы коэффициенты распределения были наибольшими,
растворы ацетона в экстрагенте должны иметь отрицательные отклонения от
закона Рауля. Из табл. 8 следует, что жидкости, относящиеся к классу IV,
должны обеспечивать высокие коэффициенты распределения. Галоидоугле-
.водороды (класс V) также можно использовать в качестве экстрагентов, так
как их смеси с ацетоном в худшем случае будут характеризоваться очень
слабыми положительными отклонениями. Величины коэффициентов
распределения для ряда растворителей приведены ниже;
133
Растворитель
Хлороформ
1, 1, 2-трихлорэтан
Хлорбензол
Тетрахлорэтан
Класс
IV
IV
V
V
Коэффициент распределения при низких
концентрациях
ацетон в слое растворителя
ацетон в водном слое
вес. %
1,83 при 25° С
1,47 при 25° С
1,0 при 25—26° С
2,37 при 25—26° С
МОЛ. %
7,85 при 25° С
14,58 при 25° С
5,91 при 25° С
18,2 при 25° С
Однако, как будет показано в главе IV, при выборе растворителя
следует основываться не только на величинах коэффициентов распределения,
но и учитывать ряд других факторов.
Пример 111-16. Оценить селективность перфторгептана (В) как
растворителя (экстрагента) для разделения бензола (А) и к-гептана (С).
Решение. Водородные связи в системе отсутствуют. Из табл. 9
находим:
6Д = 18,7 103; бв=11,7-103; бс = 15,2-103;
Дбл,в = <18'7 — * 1,7>'103 = 7000; Д5в,с = <15,2 — 11,7) • 103 = 3500
Следовательно, растворы перфторгептан — бензол должны проявлять
большие положительные отклонения от закона Рауля, чем растворы
перфторгептан — к-гептан. Поэтому гептан должен селективно экстрагироваться
перфторгептаиом. Тот же вывод может быть сделан при сопоставлении
критических температур растворения:
перфторгептан-к-гептан
К. Т. Р. = 50° С
перфторгептан — бензол
К. Т. Р. = 113,5° С
Большинство растворителей, применяемых для разделения
ароматических и парафиновых углеводородов, имеют б > 18 700 и, следовательно,
селективно экстрагируют ароматические углеводороды. Селективная
экстракция парафиновых углеводородов особенно удобна, когда они присутствуют
в углеводородной смеси в малых количествах. Применение перфторуглеводо-
родов для этой цели явилось предметом нескольких патентов (см., например,
Egan, пат. США, 2582197, 1952).
Пример III-17. Критические температуры растворения в системе
к-гептан (А)—анилин (В)—циклогексан (С) равны: 70°С для пары анилин —
к-гептан; 31° С для пары анилин—циклогексаи; к-гептан и циклогексан
смешиваются.
Рассчитать селективность анилина для разделения к-гептана и цикло-
гексана.
Решение. В данном случае подлежит разделению система типа II. По
значениям критических температур растворения можно заключить, что
циклогексаи будет распределяться между углеводородной и анилиновой
фазами в значительной мере в пользу первой (эта фаза близка к идеальной);
тем не менее анилин будет селективно экстрагировать циклогексан в
сравнении с к-гептаном. Для получения более определенных, количественных
выводов проведем некоторые расчеты,
134
Анилин (В)-н-гептан (А). Для грубой оценки распределения
воспользуемся величинами V и б при 25° С. Так как анилин обладает некоторой
полярностью, его параметр растворимости лучше определить на основе
К Т Р чем из уравнения (111,110). Величина VB=92-10-3. Из табл. 9
находим:' Va = 0,147; бл = 15,2-103; К. Т. Р. = 343° К. По уравнению (111,113)
находим:
Этому будет соответствовать 8В = 15 200+7080=22 280. Полученная
величина более вероятна, чем 15 200 — 7080=8120, так как по уравнению (III, 110)
величина 8В = 20 000.
Согласно уравнению (III, 105)
RT In уА = RT [In ФА - In хА + Фв (1 - VA/VB)] + УАФ2В (Д6Д Bf
При ха = Фа=0 и 0в = 1,О находим:
In уАВ = 2,ЖААВ = iaL*. + i-LL+VA (Л6ДВ)2/ДГ
v в в
При 25° С (298° К) имеем:
о oaq , 1 °>U7 i г 0Д47 0,147 (7080)2
2<306ААВ-[п'оЩ+ 0,092 + 8314-298
°ТКУАнилин! В)- циклогексан (С). Величина VB =0,092. Из табл 9
находим: Vc = 0,109; бс = 16,8-133; К-Т. Р. = 304°К. По уравнению (111,113)
л* rmnf^0 002n5)f 304"8314 У'5-7М"
дбв,с = (0'109 +0-092 ^ 2-0,109-0,192 J
бв= 16 800+ 7140 = 23 940
Значения 8В не совпадают. Это указывает на то, что их следует
рассматривать просто как эмпирические величины. Уравнение (III, 105) дает
при £С = 0С=О; 0В=1,О и Г=298°К:
Vc , Vc Ус(^с,в)2
1пу'св = %ЖАсв = 1п— + 1--у- + д= =
0Д09 0Д09 , 0,109 - (7140)'
ТЩй"1- 0,092 "1" 8314-298
откуда Асв = 0,965. Л
н-Гептан (А) — циклогексан (С). Вследствие близости мольных объемов
можно воспользоваться уравнением (III, 108):
УСФа(^а,с?
1п Уса = RT
При *с = 0с=О; 0а = 1,0; Г=298°К получаем:
, 0,109(16 800-15 200)2
1п Уса = А<л»лсд — 8314 2д8
откуда Аса= 0,045.
Аналогично можно найти А Ас = 0,0605. Эта система является почти
идеальной.
135
Для селективности циклогексаиа по отношению к гептану, комбинируя
уравнения (111,98) и (III, 99), получаем:
ЛАВ
Приближенно
lg(xaaIxab) » ^(уав/Уаа) = 'g Уав = ЛАВ
поскольку Yaa~ 1.0-
'g Рс,л = Лсл - ^св + Аав = 0.045 - 0.965 +1,272 = 0,352
$С,А — 2>25
Коэффициент распределения циклогексаиа (анилин/углеводород) равен:
\g т' = АСА — Асв = 0,045 — 0,965 = — 0,920
откуда т'=0,12.
Если выражать коэффициент распределения как отношение
концентрации в углеводородном слое к концентрации в анилиновом слое, 1/ш'=
= 1/0,12=8,3.
Примечание. Опытная величина селективности равна 1,46, а
коэффициент распределения (углеводород/анилин) составляет, по
опытным данным, 12,6 [Hunter, Brown, Ind. Eng. Chem., 39, 1343 (1947)].
Та же система рассматривалась в примере Ш-12 (см. стр. 115), где
было найдено 1/т'=0,28/0,05=5,6. Можно сравнить константы Л,
полученные в примере 111-12 и в данном слунае. Величины Лев отлично
согласуются; подтверждается близость к идеальности пары С — Л,
однако величины Л ав недостаточно хорошо совпадают друг с другом.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
А — предел lg\ при х—► () в бинарном растворе;
компонент раствора.
а — активность.
В—компонент раствора; в уравнении (III,
16)—второй вириальный коэффициент.
С — компонент раствора; константа в уравнениях для
коэффициентов активности.
D — компонент раствора; константа в уравнениях для
коэффициентов активности.
Е — константа в уравнениях для коэффициентов
активности.
Д£исп. — внутренняя энергия испарения, дж/кмоль.
F— свободная энергия, дж/кмоль.
F—парциальная свободная энергия вещества в
растворе, химический потенциал, дж/кмоль.
F£— полная избыточная свободная энергия
неидеального раствора, дж.
F — полная свободная энергия раствора, дж,
f — летучесть чистого вещества, н/м2.
138
f — парциальная летучесть вещества, н/м2.
О—константа в уравнениях для коэффициентов
активности.
Н — константа в уравнениях для коэффициентов
активности.
Н° — энтальпия чистого вещества, дж/кмоль.
Н — парциальная энтальпия вещества в растворе,
дж/кмоль.
&Jis — парциальная теплота растворения, дж/кмоль.
Д^исп. — энтальпия испарения, дж/кмоль.
J—константа в уравнениях для коэффициентов
активности,
m — коэффициент распределения, мол. доли/мол. доли.
MW—молекулярный вес.
N — число атомов углерода в молекуле.
п — число молей.
р—парциальное давление паров компонента над
раствором, н/м2.
р — общее давление, н/м2.
R — универсальная газовая постоянная (/? = 8314 дж X
X/смоль-1 >град-1).
г — число углеводородных колец в молекуле.
Т — абсолютная температура, °К.
V — удельный объем в жидком состоянии, м3/кмоль.
v — удельный объем в газообразном состоянии,
м3/кмоль.
X — концентрация в жидкой фазе, вес. доли.
х—концентрация в жидкой фазе, мол. доли.
у — концентрация в газовой фазе, мол. доли.
а, е, |, tj, 0, к — эмпирические константы.
Р — селективность.
Y — коэффициент активности.
б — параметр растворимости, (дж/м3)0-5.
ф — концентрация в жидкой фазе, объемн. доли.
Индексы
А, В, С— относятся к компонентам А, В, С.
АВ — относится к компоненту А в фазе с
преобладающим содержанием компонента В.
а — относится к соединениям ароматического ряда.
п — относится к соединениям нафтенового ряда.
р— относится к соединениям парафинового ряда.
Верхние индексы
о — относится к стандартному состоянию.
'— относится к нулевой концентрации; в примечании
к табл. 6 — цепочка углеродных атомов.
137
ЛИТЕРАТУРА
1. G. N. Lewis, M. Randall, Thermodynamics a. the Free Energy of
Chemical Substances, McGraw-Hill Book Company, Inc., New York, 1923.
2. G. N. Le wis, Proc. Am. Acad. Arts. Sci., 37, 49 (1901).
3. B. F. Dodge, Chemical Engineering Thermodynamics, McGraw-Hill Book
Company, Inc., New York, 1944.
4. J. M. Smith, H. С Van Ness, Introduction to Chemical Engineering
Thermodynamics, 2nd ed., McGraw-Hill Book Company, Inc., New York,
1959.
5. M. Benedict, С A. Johnson, E. Solomon, L. С Rubin, Trans.
Am. Inst. Chem. Engrs., 41, 371 (1945).
6. R. С R e i d, Т. К. Sherwood, The Properties of Gases a. Liquids,
McGraw-Hill Book Company, Inc., New York, 1958.
7. H. С Carlson, A. P. С о 1 b u r n, Ind. Eng. Chem., 34, 581 (1942).
8. N. V. I Ы, B. F. D о d g e, Chem. Eng. Sci., 2, 120 (1953).
9. H. С Van Ne ss, Chem. Eng. Sci., 10, 225 (1959).
10. O. Redlich, А. Т. К i s t e r, Ind. Eng. Chem., 40, 341, 345 (1948);
J. Chem. Phys., 15, 849 (1947).
11. G. Scatchard, W. J. Hamer, J. Am. Chem. Soc, 57, 1805 (1935).
12. K. Wohl, Trans. Am. Inst. Chem. Engrs., 42, 215 (1946); Chem Eng
Progr., 49, 218 (1953).
13. J. J. Van La a r, Z. physik. Chem., 72, 723 (1910); 185, 35 (1929).
14. M. Mar gules, Sitzber. Acad. Wiss. Wien. Math.-naturw. KL, 104, 1243
(1895).
15. С Black, Ind. Eng. Chem., 50, 391 (1958); 51, 211 (1959); A. I. Ch.
E. Journ., 5, 249 (1959).
16.0. Redlich, А. Т. К i s t e г, С. Е. Turnquist, Chem. Eng. Progr.
Sympos. Series, № 2, 48, 49 (1952).
17. International Critical Tables, McGraw-Hill Book Company, Inc., New York,
1926.
18. С A. Jones, A. P. С о lb urn, E. M. S с h о e n b о г n, Ind. Eng. Chem,
35, 666 (1943).
19. J. С Chu, S. L. Wang, S. L. Le v y, R. P au 1, Vapor-Liquid Equilibrium
Data, J. W. Edwards, Publishers, Inc., Ann Arbor, Mich., 1956.
20. E. Hal a, J. Pick, V. Fried, O. V i 1 i u m, Vapour-Liquid Equilibrium,
2nd ed. (английский перевод Д. Стандарта), Pergamon Press, London, 1958.
21. L. H. Horsley, Azeotropic Data, American Chemical Society, Washington,
D. S., 1952.
22. A. P. Colburn, E. M. S с h о e n b о г n, Trans. Am. Inst. Chem Engrs.,
41, 421 (1945).
23. J. H. H i 1 d e b r a n d, R. L. Scott, The Solubility of Nonelectrolytes, 3rd
ed., Reinhold Publishing Corporation, New York, 1950.
24. G. J. Pierotti, С A. Deal, E. L. Derr, Ind. Eng. Chem., 51, 95
(1959).
25. E. G. Scheibel, D. Friedland, Ind. Eng. Chem., 39, 1329 (1947),
26. R. E. Treyb a 1, Ind. Eng. Chem., 36, 875 (1944).
27. D. B. H an d, J. Phys. Chem., 34, 1961 (1930).
28. A. W. Francis, Ind. Eng. Chem., 36, 764, 1096 (1944); Cricital
Solution Temperatures, American Chemical Society, Washington, D. C, 1961.
29. H. M. Woo db urn, K. Smith, H. Tetervsky, Ind. Eng. Chem 36,
588 (1944).
30. D. A. Drew, A. W. H i x s о п, Trans. Am. Inst. Chem. Engrs., 40. 675
(1944).
31. A. W. Hixson, J. B. Bockelmann, Trans. Am. Inst. Chem. Engrs.,
38, 891 (1942).
32. R. H. Ewe II, J. M. Harrison, L. Berg, Ind. Eng. Chem., 36, 871
(1944).
33. J. H. Hi Id e brand, Chem. Eng. Progr. Sympos. Series, 48, № 3, 3 (1952).
ВЫБОР ЭКСТРАГЕНТА
И ЕГО РЕГЕНЕРАЦИЯ
ВЫБОР ЭКСТРАГЕНТА
При выборе избирательного растворителя
для проведения данного процесса
жидкостной экстракции можно
руководствоваться различными требованиями.
Последние часто противоречивы, и поэтому
невозможно подобрать такой экстрагент,
который удовлетворял бы одновременно всем
предъявляемым к нему требованиям. В
таких случаях приходится принимать
компромиссное решение, учитывая
относительное значение различных факторов,
влияющих на выбор экстр агента.
Селективность. Основным свойством,
которым должен обладать экстрагент,
является селективность. Указанное свойство
характеризует способность экстрагента
предпочтительно извлекать один из двух
компонентов раствора. С этой точки зрения
наиболее подходит тот экстрагент,
который растворяет максимальное количество
одного компонента и минимальное
количество другого... \
Рассмотрим тройную систему,
представленную на рис. 68. Точка М соответствует
раствору компонентов А я С, который
необходимо разделить с помощью экстраген-
та В. Точка S выражает состав
двухфазной смеси, получаемой при добавлении В
к исходному раствору (точка М). После
интенсивного перемешивания, приводящего
к установлению равновесия в фазах, и
последующего отстаивания жидкостей
получают две фазы Т и R, находящиеся в
равновесии друг с другом. Линия RT является
хордой равновесия. Обычно экстрагент В
регенерируют из обеих фаз. Если В
полностью удаляется из фазы R, образуется
бинарный раствор D; точка Е соответствует
139
Рис. 68. Селективная экстракция С Рис. 69. Селективная экстракция С
из раствора в А экстрагентом В. из раствора в А экстрагентом В'.
фазе Т, полностью освобожденной от экстрагента В- В
результате этого процесса из исходного раствора М получают два
раствора — раствор D, обогащенный компонентом А, и раствор £,
обогащенный компонентом С. В случае полного выделения
экстрагента точки Е и D находятся на максимальном
расстоянии друг от друга.
Однако если для разделения раствора компонентов Л и С
использовать экстрагент В' (рис. 69), то, несмотря на
принципиальную возможность разделения (растворы Е и D имеют
различные составы), эффективность его не будет столь велика, как
в первом случае. Это объясняется различием наклонов хорд
равновесия. Проведем мысленно на рис. 69 хорду равновесия,
которая точно совпадает с прямой В'М. В данном случае
составы полученных растворов после удаления экстрагента В'
равны составу исходной смеси. Таким образом, для разделения
растворов компонентов А и С экстрагент В более селективен,
чем В'. Аналогично для разделения указанной бинарной смеси
можно сравнивать по селективности ряд экстр агентов. Как
следует из рис. 68 и рис. 69,Лбелективность экстрагента для
данной системы зависит от концентрации распределяемого
вещества в исходном растворе.
Количественно селективность можно более наглядно
показать на графике зависимости концентрации распределяемого
вещества С в D от концентрации С в Е, т. е. с помощью кривой
распределения компонента С, построенной в координатах:
концентрация компонента С в свободном от В растворе D и
концентрация компонента С в свободном от В растворе Е. Для
каждой тройной системы можно построить две такие диаграммы
селективности в зависимости от того, какая из частично
растворимых жидкостей тройной системы рассматривается как экстра-
140
гент. Кривые селективности
также, как и кривые распределения,
проходят через начало
координат, обычно, но не всегда имеют
максимум и оканчиваются на
диагонали графика в
критической точке. Экстрагент тем более
селективен, чем больше
расстояние по вертикали между кривой
селективности и диагональю.
Если кривая селективности
совмещена с диагональю,
разделение смеси с помощью данного
экстрагента невозможно. Кривые
селективности во многих
отношениях аналогичны диаграмме
Мак-Кэба — Тиле, используемой
в процессах ректификации для
изображения равновесий между
жидкостью и паром.
На рис. 70 представлены
кривые селективности для тройных
систем типа I (типичная
диаграмма растворимости подобных
смесей показана на рис. 68):
бензол— вода — этанол1 при 25° С
и этилацетат — вода — этанол 2
при 20° С. Распределяемое
вещество— этанол в обоих
случаях предпочтительно растворяется
в водной фазе. Кривые на рис. 70
показывают, что при низких концентрациях вода представляет
собой более селективный растворитель, чем бензол или этил-
ацетат. Если же для разделения смеси этанол — вода
необходимо сделать выбор между бензолом и этилацетатом,
предпочтение следует отдать бензолу как более селективному
растворителю. Однако ни бензол, ни этилацетат по другим
соображениям не являются наиболее подходящими экстрагентами.
Для системы типа II, представленной на рис. 71, с помощью
диаграммы селективности (рис. 72) аналогично может быть
проведено сравнение селективности различных растворителей (экс-
трагентов). Обычно селективность экстрагентов для систем
типа II несколько ниже, чем для систем типа I.
Численное значение селективности (5 можно рассчитать так
же, как величину относительной летучести в процессах
ректификации. Так, селективность экстрагента В по отношению к
компоненту С определяется как частное от деления отношения С
I
I
1
0,6
о.в
ОА
I
о,г
I
iV
'' s^
л
/
/
о
%:
/
4 цг оа од
Содержание этанола в фазе
рафината, вес. доли
Рис. 70. Диаграммы
селективности для систем типа I [две
верхние кривые— система бензол (А)—
вода (В) — этанол (С) при 25° С;
две нижние кривые — система
этилацетат (Л)—вода (В) —
этанол (С) при 20° С; содержание
этанола указано без учета
содержания экстрагента]:
/, 5—экстрагент —вода; 2 — экстрагент —
бензол; 4 — экстрагент — этилацетат.
141
к Л в фазе экстракта на
отношение С к А в фазе рафината:
хсвКхсв + хав) ^
1-1ХСв/(ХСВ + ХАв)1
хса/(Хса + хаа)
= р
с'а1-1хса/(хса + хаа)}
(IV, 1)
R XCBXAA
РСА— Y ЯХ,
(IV, 2)
Рис. 71. Селективная экстракция в
системах типа II.
СА АВ
В выражения для
определения селективности входят
равновесные концентрации, и
численные значения
селективности не зависят от способа выражения концентраций
(мольные или весовые доли). Селективность р для некоторых
систем 3'4 является практически постоянной величиной. В этом
случае селективность может быть использована для
корреляции хорд равновесия [уравнения (11,34) и (11,35)]. Для
большей части систем жидкость — жидкость (см. рис. 73, на
котором приведены данные рис. 70 и 72) величина р сильно
зависит^, от концентрации.
'Величина селективности для процессов экстракции имеет то
же значение, что и относительная летучесть для процессов
ректификации. Для практических ;
целей необходимо, чтобы р была
возможно больше единицы. При | ^ пд
значениях р, мало отличающих- a*
ся от единицы, увеличиваются
габаритные размеры
оборудования, потребное число ступеней
разделения, а также в общем
случае возрастают капитальные
затраты и эксплуатационные
расходы. При р=1 разделение
смеси экстракцией невозможно.
Высокая селективность по
отношению к экстрагируемому
компоненту, который, как правило,
составляет меньшую долю
исходного раствора, обусловливает
более экономичное проведение
процесса в результате снижения
количества циркулирующего экс
^<§ 0,6
■>
ХГ °
С1/
ff1/
0.2
0,U 0,6 0,8 t
Концентрация С В фазе
рафината, вес. доли
Рис. 72. Диаграммы селективности
для систем типа II
(концентрации С указаны без учета
содержания компонента В):
/ — система к-гептан (А) — анилин (В) —
метилциклогексаи (С) при 25° С; 2
—-система рафинированное хлопковое масло
^ - - (Л) —пропан (В) —олеиновая кислота (С)
трагента. Это относится как к ПрИ 98,5° с.
142
системам типа II (см. пример
III-16, стр. 134), так и к
системам типа I.
Рассмотрим зависимость
между растворимостью и
селективностью. Совместное решение
уравнений (11,15) и (IV, 2)
позволяет установить, что
селективность растворителя В по
отношению к компоненту С
связана с коэффициентом
распределения тс зависимостью:
"с X.
$С,А = П<
АА
(IV, 3)
АВ
В связи с тем что ХАА/ХАВ
больше единицы, для систем с
коэффициентом распределения
т>1 всегда сохраняется условие
р>1. Для систем с
коэффициентом распределения т< 1 или для
систем, у которых коэффициент
распределения, изменяясь от
т<1 до т>1, проходит через
значение т—\ (сольютропные
системы), величина р также
может иметь значения,
превышающие единицу, за исключением
Со
I
J
I
«5
шо
2000
1000
600
ш
гоо
то
60
ьо
20
ю
е
г
о
Концентрация С 6 фазе А,
ХСА, вес. доли
_0 0£ Otf 0,8 0,8 1
„О
_~Щ,
,tl
•
) ^Ч,
■ 4
-\х
\1 '
X
Д
о
*"
о /
02
3
и -
с\<
•
0,3 0U 02
0,1 0,2
Концентрация с 6 фазе В,
ХС8г6ес. доли
Рис. 73. Зависимость
селективности от концентрации компонента С
в фазах:
Системы типа I: / — бензол (А) — вода
(В) —этанол (С) при 25° С; 2 —этилацетат
(Л) —вода (В) —этанол (С) при 20° С.
Системы типа II: 3 — гептан (Л) —анилин
(В) —метилциклогексаи (С) при 25° С; 4 —
рафинированное хлопковое масло.
(А) — пропан (В) —олеиновая кислота (С)
при 98,5° С
случаев, когда взаимная раство
римость А и В велика.
Для четырехкомпонентных систем, у которых компоненты В
и С распределяются между двумя несмешивающимися
растворителями А и D, селективность связана с коэффициентами
распределения следующими зависимостями:
X
тв= X
ВА
X
BD
^В,С ~ т
тс= X
СА
CD
XBAXCD
XCAXBD
(IV, 4)
(IV, 5)
где р — селективность пары растворителей по отношению к В.
Как и для тройных систем, успешное проведение процесса
возможно при соблюдении условия р>1.
Коэффициент распределения. Из уравнения (IV, 3) видно,
что коэффициент распределения т имеет важное значение как
фактор, влияющий на селективность. Чем больше величина т,
143
тем выше селективность. Если какой-либо экстрагент обладает
малым коэффициентом распределения, но по другим причинам
его применение желательно, то в некоторых случаях (когда
экстрагируемое вещество диссоциировано) коэффициент
распределения можно увеличить изменением рН раствора (см.
главу II). Изменение рН раствора с целью получения более
высоких коэффициентов распределения, а также введение в систему
буферного раствора (буфера) для поддержания
благоприятного значения рН в ходе процесса экстракции широко
используют на практике (особенно в процессах разделения металлов).
Правильный выбор рН в таких случаях может привести к
резкому возрастанию селективности в результате увеличения тв
и уменьшения тс в уравнении (IV, 5). Другой способ
увеличения величины коэффициента распределения — введение в
систему неэкстрагируемой соли, т. е. применение «высаливания»
(см. главу II).
Емкость экстрагента. Если экстрагент не обладает большой
емкостью, т. е. способностью растворять относительно большое
количество избирательно извлекаемого компонента, то,
несмотря на его высокую селективность, применение экстрагента
может оказаться неэкономичным из-за необходимости иметь в
экстракционной системе большое количество циркулирующего
экстрагента. Поэтому экстрагент должен иметь не только
высокий коэффициент распределения тс, но и возможно большую
величину XCbJ Указанному требованию обычно удовлетворяют
системы типа I (рис. 68), даже если бинодальная кривая не
охватывает большую область фазовой диаграммы.
Высокая емкость экстрагента имеет особенно важное
значение для систем, у которых распределяемым компонентом
является твердое вещество (см. рис. 16). Предположим, например,
что для разделения смеси хлорида кобальта и воды
применяется н-гептан, обладающий высокой селективностью по отношению
к хлориду кобальта (Р = 9) и, следовательно, высокой
разделяющей способностью5. Однако из-за. низкой растворимости СоС12
в н-гептане (0,04%) для экстракции 1 кг соли необходимо
минимально 2500 кг w-гептана; в то же время наименьший расход
н-бутанола, имеющего меньшую, но достаточно высокую
селективность (р = 5), составляет 1,9 кг на 1 кг экстрагируемой соли
(растворимость СоС12 в н-бутаноле 34,6%). По этой причине
следует считать н-бутанол значительно лучшим экстрагентом,
чем н-гептан._)
При разделении систем типа II на практике часто
необходимо выделять компоненты Л и С из почти идеальных
растворов, ^например, в нефтеперерабатывающей промышленности (А
и С — углеводороды). Поэтому трудно найти экстрагент,
который, обладая высокой растворимостью по отношению к
компоненту С, одновременно плохо растворяет компонент А, что
144
\
обусловливает низкую селективность экстрагента. В подобных
процессах обычно повышают селективность, уменьшая емкость
экстрагента (например, при выделении ароматических
углеводородов из парафиновых углеводородов экстракцией не
безводным, а содержащим воду диэтиленгликолем). При этом
повышение селективности обеспечивается в большей мере в
результате увеличения отношения ХАА/ХАВ [см. уравнение (IV, 3)],
чем путем возможного увеличения коэффициента
распределения тс, который в действительности может уменьшаться. В
таких случаях должно быть найдено компромиссное соотношение
между селективностью и емкостью экстрагента.
ГВ условиях фракционной экстракции, как и при экстракции
тройных систем, недостаточно иметь лишь благоприятное
соотношение коэффициентов распределения в уравнении (IV, 5);
емкость экстрагента, или достижимая концентрация
распределяемого вещества в экстрагенте, также должна быть достаточно
высокой для уменьшения количества регенерируемого
экстрагента. Например, в процессе распределения хлоридов никеля и
кобальта между этил ацетатом и водой достигается значение
селективности этилацетата по отношению к хлориду кобальта \
примерно равное 50. Несмотря на прекрасную селективность
этилацетата, его нельзя считать хорошим экстрагентом,
поскольку в этилацетате может раствориться лишь около 1%
хлорида кобальта, что приведет к необходимости иметь в системе
слишком большое количество экстрагента.
Растворимость экстрагента. Для систем типа I желательно,
чтобы экстрагент В и исходный растворитель А обладали
наименьшей взаимной растворимостью. Это приводит к
увеличению селективности в результате возрастания отношения
Хаа/Хав в уравнении (IV, 3), а также, как следует из рис. 74,
расширяет пределы концентраций компонентов бинарных смесей
компонентов Л и С, которые могут извлекаться экстрагентом В.
Рис. 74. Влияние растворимости экстрагента на процесс экстракции.
10 Р. Е. Трейбал 145
Для обеих систем, показанных на рисунке, экстрагент В можно
использовать лишь при разделении смесей А я С, состав
которых отвечает точкам, лежащим на отрезке AM, так как только
в этом интервале концентраций при добавлении В возможно
образование двух жидких фаз.
Из сопоставления рисунков 74, а н 74, б следует, что
использование экстрагента В в системе, представленной на рис. 74, а,
будет более эффективным. Кроме того, тз случае взаимной
нерастворимости А к В упрощается регенерация экстрагента и
отпадает необходимость в дополнительной обработке получаемого
рафината для выделения растворенного в нем экстрагента^ Для
систем типа II 'незначительная взаимная растворимость В в А
и С обычно приводит к увеличению селективности, но
ограничивает, как указывалось выше, емкость экстрагента. В этом
случае должно быть найдено компромиссное соотношение
указанных величин. 'Существенное изменение взаимной растворимости
компонентов иногда может быть достигнуто при умеренном
изменении температуры^,
Регенерируемость. Во всех процессах жидкостной
экстракции необходимо выделять экстрагент из продуктов экстракции,
чтобы предотвратить загрязнение этих продуктов экстрагентом,
а также свести к минимуму его потери. Если экстрагент
выделяется посредством ректификации, важное значение имеют
летучесть и теплота парообразования экстрагента. Для
регенерации экстрагента помимо ректификации применяют также другие
методы. Проблема регенерации экстрагента, имеющая очень
важное значение для экономичного осуществления процесса,
рассматривается ниже.
Плотность. ЧЛри выборе экстрагента существенную роль
играет также разность плотностей фаз, которая должна быть
возможно большей. Разность плотностей фаз определяет не
только скорость расслаивания несмешивающихся жидкостей, но
и производительность экстракционного оборудования.
Недостаточно сравнивать плотности исходного раствора и чистого
экстрагента, так как изменение взаимной растворимости этих
жидкостей при их смешении приводит к изменению плотностей фаз.
В непрерывных экстракторах важно по всей рабочей высоте
(длине) аппарата поддерживать определенную разность
плотностей контактирующих фаз.
На рис. 75 показана зависимость плотностей насыщенных
слоев фаз от концентрации распределяемого вещества для
тройной системы типа I (ацетон — 1, 1, 2-трихлорэтан — вода;
распределяемое вещество — ацетон). Плотности равновесных фаз
соединены хордами6. Из рис. 75 следует, что плотность водной
фазы данной системы всегда меньше плотности фазы трихлор-
этана и что разность плотностей фаз во всем диапазоне
изменения концентраций ацетона достаточно велика. В критической
146
Критическая
точка
' О /0 20 30 U0 50 ВО 70
Концентрация ацетона, Вес. %
Рис. 75. Плотность равновесных
растворов системы вода (А) — 1,1,2-
трихлорэтан (В) — ацетон (С) при
25° С6:
1 — водные растворы; 2 —растворы в три-
хлорэтане.
Концентрация метилэтилкетона,
вес. %
Рис. 76. Плотность равновесных
растворов системы вода (А) — трихлор-
этилен (В) — метилэтилкетон (С)
при 25° С7:
/ — водные растворы; 2 — растворы в трихлор-
этилене.
точке плотности фаз, находящихся в равновесии друг с другом,
одинаковы. На рис. 76 приведена аналогичная зависимость для
тройной системы типа II: вода — трихлорэтилен —
метилэтилкетон.
Для этой системы характерно изменение знака разности
плотностей равновесных фаз. В данном случае применение
аппаратов с непрерывным контактом фаз невозможно^'
Фрэнсис8 назвал хорды, соединяющие растворы равной
плотности, изопикническими. Рассмотрев данные о плотностях около
100 систем, он отметил, что изопикнические хорды встречаются
довольно часто, хотя это важное свойство редко отмечалось в
оригинальных работах. Изопикнические хорды наблюдаются для
систем различных типов и могут исчезать при изменении
температуры. Подобные явления встречаются также в
многокомпонентных системах, разделение которых имеет промышленное
значение, например, в нефтеперерабатывающей
промышленности. Если такую систему предста-вить как псевдотройную (см.,
например, рис. 42), она образует не одну пару растворов,
соответствующих определенной изопикнической хорде, а ряд
растворов, занимающих некоторую область фазовой диаграммы.
Межфазовое натяжение. Для ускорения коалесценции
несмешивающихся жидкостей при их разделении (отстаивании)
необходимо, чтобы межфазовое натяжение было достаточно
высоким. Чрезмерно большое межфазовое натяжение приводит к
увеличению энергии, затрачиваемой на создание дисперсии.
Жидкости с малым межфазовым натяжением образуют
стабильные эмульсии, возникновение которых очень трудно
предотвратить.
10*
147
Для тройных систем проведено ограниченное число
измерений межфазового натяжения, но известно, что межфазовое
натяжение двух частично растворимых жидкостей наиболее велико
в отсутствие'.распределяемого вещества и равно нулю в
критической точке. Межфазовое натяжение взаимно насыщенных
бинарных жидких систем можно связать9 со способностью
жидкостей образовывать водородные связи и их мольными объемами.
Донауэ и Бэртел 10 получили эмпирическую корреляцию для
межфазового натяжения, выразив его в зависимости от
взаимной величины растворимости, выраженной в мольных долях
(хАв и хва) компонента, находящегося в меньшем количестве
в каждой из фаз. Автор нашел, что модифицированные
координаты Донауэ — Бэртела (рис. 77) могут быть использованы для
коррелирования данных по межфазовым натяжениям тройных
систем типа I. Координаты на рис. 77 преобразуются в
координаты Донауэ — Бэртела для бинарных систем (хСа = 0 и хСв —
= 0). Величины межфазовых натяжений для тройных систем
приведены по данным Мэрфи и др. и, при этом были исключены
некоторые системы, у которых данные для пар несмешиваю-
щихся жидкостей не соответствуют величинам, отвечающим
принятой корреляции. Полученная кривая удовлетворительно
описывает также данные для систем метилэтилкетон — вода —
уксусная кислота и циклогексанол — вода — уксусная кислота,
которые для большей ясности графика не нанесены на рис. 77.
Известное правило Антонова, согласно которому межфазовое
<и
3
t
тяте,
ч
8
8
ч
§•
I
t
1
£
50
W
30
^2tf
а
па
10
0,OOOU OflOl OPOU 0,01 0,Ои 0,1 0£ 1
Концентрация в равновесных растворах
х&в+хвй+ £с^Хсв > мал. доли
Рис. 77. Зависимость межфазового иатяжеиия от взаимной
растворимости для бинарных и тройных жидких смесей (критическая точка имеет
ординату 0 и абсциссу 1):
1 — различные бинарные системы; тройные системы: 2—трнхлорэтаи (Л) —вода (В)
—ацетон (С); 3 — гексаи (Л) —вода (В) —ацетон (С); 4 — к-бутанол (Л)—вода (В) —уксусная
кислота (С); 5 — фурфурол (Л) —вода (В) —уксусная кислота (С); б —нитрометан (А) —
вода (В) —уксусная кислота (С); 7 — гексан (Л) —вода (В) —уксусная кислота (С).
148
N.
7N
ц.
X
А
^
а
S»
•
/
о 2
D 3
■ 4
v 5"
А 6
д
/
натяжение двух взаимно насыщенных жидких фаз равно
разности между их поверхностными натяжениями (на границе с
воздухом), недостаточно надежно.
Опытные данные на рис. 77 аналогично другим данным, пуб--
ликуемым в химической литературе, обычно относятся к
химически чистым веществам и поэтому, вероятно, должны
рассматриваться как максимальные значения межфазовых натяжений,
'существенное влияние на величину межфазового натяжения
оказывают (наиболее часто в направлении его снижения) даже
очень небольшие количества примесей. Особенно значительно
влияние тех примесей, которые абсорбируются на поверхности
раздела фаз (поверхностно-активные вещества и им подобные).
Поэтому технические жидкости почти всегда обладают
межфазовым натяжением, меньшим стандартного^ В связи с этим
для оценки экстрагентов измерения межфазового натяжения
следует проводить на реальных (применяемых в
промышленных условиях) жидкостях. Полезные практические сведения
можно получить в результате встряхивания исходного раствора
с намеченным экстрагентом в делительной воронке. Если за
5 или по крайней мере за 10 мин не произойдет достаточно
четкого разделения фаз, осуществление процесса сепарации фаз
после экстракции в промышленном масштабе, по-видимому,
окажется затруднительным.
Реакционная способность и стабильность. Обычно
химическая реакция между экстрагентом и компонентами исходного
раствора, приводящая к образованию новых веществ,
нежелательна, так как она сопровождается снижением выхода
целевого продукта, усложнением стадии регенерации экстрагента и
может привести к увеличению потерь последнего. Вместе с тем
такая химическая реакция обычно увеличивает коэффициент
распределения вещества, распределяемого между фазами, и по
этой причине может быть иногда желательной. Если, однако,
реакция необратима, стоимость возможной регенерации
экстрагента будет, вероятно, очень велика. Вследствие этого
необходимо, чтобы химическая реакция была обратимой. Например,
уран можно извлечь из разбавленных кислых растворов
некоторыми алкилами фосфорной кислоты, химически
взаимодействующими с исходным раствором. При этом экстрагент регенерируют
экстракцией из него урана более концентрированной кислотой.
Продукты реакции, образующиеся в процессе экстракции,
иногда являются целевыми конечными продуктами.
Необходимо, чтобы экстрагент при взаимодействии с
исходным раствором был стабильным; например, экстрагент не
должен изменяться под действием таких окислителей, как
азотная кислота, если она присутствует в исходном растворе.
Экстрагент не должен полимеризоваться или окисляться
кислородом воздуха, а также изменяться при многократном нагревании
149
вплоть до максимальной температуры, достигаемой в процессе
регенерации его ректификацией.
Следует избегать таких экстрагентов как эфиры, которые
образуют взрывоопасные перекиси. Необходимо также
принимать во внимание возможность гидролиза экстрагентов,
который может привести к образованию коррозионноактивных
соединений. Так, например, при использовании хлорированных
растворителей в присутствии воды и пара может
образовываться соляная кислота. В специальных случаях, например при
экстракции радиоактивных веществ, радиация может снизить
экстрагирующую способность растворителя и затруднить
эксплуатацию экстракционной установки. В таких условиях потери
экстрагента составляют иногда существенную часть всех
издержек производства.
Коррозионная активность. Чтобы снизить стоимость
экстракционного оборудования, желательно изготовлять его из обычных
конструкционных материалов. Поэтому выбор экстрагента
следует проводить с учетом его коррозионной активности^уВ
настоящее время расширяются возможности применения новейших
конструкционных материалов относительно низкой стоимости.
'Некоторые современные промышленные экстракционные
установки целиком изготовлены из пластических материалов или
металлов, футерованных пластмассой^ При выборе подобных
конструкционных материалов следует учитывать также
экономичность их использования, связанную со стоимостью получаемых
продуктов.
Вязкость. С уменьшением вязкости экстрагента
уменьшаются затраты энергии на перекачивание и перемешивание,
увеличивается скорость тепло- и массопередачи, а также
скорость осаждения дисперсий. Иногда для уменьшения вязкости
экстрагента его смешивают с инертным растворителем,
например, в процессе экстракции урана трибутилфосфат смешивают
с керосином или другими углеводородами.
Давление пара. Низкое давление пара экстрагента
обеспечивает лучшие условия хранения экстрагента и дает возможность
проводить процесс экстракции при атмосферном или умеренном
избыточном давлении; одновременно уменьшаются потери
экстрагента. Иногда экстракцию проводят при повышенном
давлении, чтобы облегчить регенерацию экстрагента методами, в
которых используется высокая летучесть последнего. Например,
в процессах очистки нефтепродуктов обычно применяют в
качестве селективных растворителей жидкие пропан и двуокись
серы, высокая летучесть которых позволяет эффективно
осуществлять последующий процесс регенерации экстрагента.
Точка замерзания. Экстрагент должен иметь температуру
замерзания достаточно низкую, чтобы обеспечить его хранение
и использование в холодное время года.
150
Воспламеняемость. Это свойство экстрагента обычно
характеризуется его температурой вспышки, которая в целях
пожаробезопасное™ должна быть достаточно высокой. Если в
процессе экстракции используется горячий экстрагент, он должен
не только иметь достаточно высокую температуру вспышки, но
и обладать узким диапазоном пределов взрываемости его
паров в смеси с воздухом.
Токсичность. Использование в промышленности
высокотоксичных экстрагентов сопряжено с трудностями выполнения
требований техники безопасности, и их применения следует по
возможности избегать. Токсичные экстрагенты не должны
применяться в пищевой и парфюмерной промышленности.
Стоимость. Низкая стоимость и доступность экстрагента
(в количествах, необходимых для его промышленного
применения) также являются важными характеристиками, обычно за-w
висящими друг от друга. Несмотря на то что экстрагент реге-ч
нерируют после экстракции, обычно бывает необходимо вводить
в процесс некоторое количество свежего экстрагента для
восполнения необратимых потерь. ; Кроме того, большие потери
экстрагента связаны с увеличением непроизводительных
расходов. Из рассмотренных выше требований, которым должен
удовлетворять экстрагент, наиболее важными являются
селективность, регенерируемость, межфазовое натяжение, плотность
и реакционная способность. Остальные свойства с технической
точки зрения менее важны, но должны приниматься во внимание
при выборе экстрагента и определении стоимости проведения
процесса.
Подробный обзор промышленного применения экстрагентов,
весьма полезный для оценки их свойств (исключая
селективность), опубликован Мелланом12. Сведения об использовании
многих экстрагентов в различных процессах экстракционного
разделения содержатся также в патентной литературе.
РЕГЕНЕРАЦИЯ ЭКСТРАГЕНТА
Удаление экстрагента из продуктов экстракции, т. е. его
регенерация, почти всегда необходимо не только для его
повторного использования, но и для получения свободных от
экстрагента продуктов. Во многих случаях стоимость регенерации
экстрагента составляет большую часть стоимости всего
процесса разделения, поэтому вопросам выбора схемы регенерации
экстрагента следует уделять особое внимание. При
возможности выбора, следует выбирать тот экстрагент, применение
которого обеспечит наименьшую стоимость всего процесса,
включая стадии экстракции и регенерации.
Процессы регенерации экстрагента и экстракции
взаимосвязаны, поэтому процесс регенерации нельзя рассматривать вне
151
связи со схемой процесса экстракции. В процессах экстракции
тройных или псевдотройных систем одним растворителем экс-
трагент В применяют для разделения бинарной (или
псевдобинарной) смеси компонентов Л и С. В результате получают
раствор, обогащенный экстрагентом, — экстракт и раствор,
обедненный экстрагентом, — рафинат. Предположим, что
желательно получить компоненты Л и С в возможно более чистом виде,
в пределах, допускаемых свойствами системы.
В процессе фракционной экстракции, или экстракции двумя
растворителями, невозможно установить различие между
экстрактом и рафинатом. Процесс экстракции обычно проводят в
многоступенчатых аппаратах, обеспечивающих эффект
разделения, достигаемый при многократном контактировании в
одноступенчатых аппаратах. Многоступенчатые аппараты
выполняют различных типов, однако на приводимых ниже схемах они,
как правило, будут условно изображаться в виде аппаратов
колонного типа.
Наиболее распространенным способом регенерации экстра-
гента является ректификация. Однако часто могут быть
применены более дешевые методы регенерации, рассматриваемые
ниже.
Ректификационные методы. Регенерация экстрагента с
помощью ректификации может быть проведена в том случае,
когда относительная летучесть компонентов растворов,
образующихся после экстракции, достаточна для осуществления этого
процесса.
При проведении процесса регенерации экстрагента методом
ректификации важное значение имеет температура кипения
экстрагента, которая может быть больше или меньше температур
кипения других составляющих системы. В связи с тем что
большую долю экстракта составляет экстрагент, количество тепла,
необходимого для ректификации экстракта, а также размеры
ректификационного оборудования окажутся, вероятно,
меньшими, если экстрагент будет высококипящим компонентом
разделяемой смеси. В данном случае вещество, подлежащее
выделению, получается в виде дистиллята и поэтому не должно
содержать высококипящих примесей, таких, как смолы, полимеры
и т. п. Однако при этом высококипящие примеси будут
аккумулироваться в циркулирующем экстрагенте, для очистки
которого может оказаться необходимой дополнительная
периодическая или непрерывная ректификация. В случае применения
экстрагента с очень высокой температурой кипения для
достаточно полного его отделения может потребоваться применение
ректификации в вакууме; вероятность разложения экстрагента
при высокотемпературной ректификации будет возрастать.
Если применяют низкокипящий экстрагент, регенерируемый
в виде дистиллята, для снижения стоимости ректификации же-
152
лательно, чтобы теплота парообразования экстрагента была
невысокой. В этом случае периодической повторной перегонки
экстрагента, как правило, не требуется, но высококипящие
примеси останутся в продукте. Ниже будет показано, что экстра-
генты этого типа часто удаляются из растворов в виде гетеро-
азеотропов; поэтому желательно, чтобы содержание
экстрагента в гетероазеотропной смеси было по возможности высоким
для уменьшения общего количества подлежащей испарению
жидкости.
На приводимых ниже схемах процессов регенерации
экстрагента показана лишь основная аппаратура без соблюдения
масштаба. Вспомогательное оборудование (насосы,
теплообменники, используемые с целью утилизации тепла) для упрощения
схем не изображено. Если схема приводится вместе с фазовой
диаграммой, последняя изображается лишь в принципиальном
виде: конкретные системы могут иметь разные составы. На
фазовой диаграмме составы различных потоков обозначают
аналогично обозначению тех же потоков на схеме. Так, например,
N на технологической схеме соответствует производительности
JV кг/сек потока состава ./V на фазовой диаграмме. Различные
потоки, имеющие один и тот же состав, будут отмечаться теми
же буквами, но с разными индексами (например, N и N').
Изображение компонента в круглых скобках, например (С),
обозначает, что данный компонент может быть получен любой
степени чистоты в зависимости от используемого способа
регенерации. Условно принято, что экстракт имеет меньшую
плотность, чем рафинат.
В дальнейшем будут рассмотрены наиболее часто
встречающиеся случаи. Если необходимые характеристики подлежащей
ректификации системы заранее неизвестны, многие из них могут
быть получены на лабораторной ректификационной установке,
предложенной Эвеллом и Велчем 13.
С л у ч а й 1
Система типа I. Условия: температура кипения компонента С
выше, чем компонента А; температура кипения компонента В
выше, чем гетероазеотропной смеси А и В; компоненты В и С
не образуют азеотропной смеси; тройные азеотропы
отсутствуют. Чтобы оценить возможности регенерации экстрагента,
желательно рассмотреть те характеристики системы, которые
определяют способность его выделения методами дистилляции.
Как видно из рис. 78, наименьшей температурой кипения
обладают двухфазные смеси К и L, которым соответствует пар
состава D. Двухфазным смесям Н и J отвечает равновесный пар
состава М. Каждая точка пунктирной линии GD определяет
состав пара какой-либо двухфазной смеси. Примечательно, что
153
линия состава паров,
находящихся в равновесии с
двухфазными жидкими смесями,
расположена вне области
диаграммы, соответствующей
полной взаимной растворимости
компонентов (гомогенным
смесям). Приведенная диаграмма
подробно рассмотрена в
книге Рикки и.
Кривые в области
диаграммы, соответствующей
гомогенным смесям, показывают
изменение состава остатков,
получаемых в процессе
периодической ректификации, когда
не образуется азеотропной
смеси А—С. Растворы с
составами, отвечающими точкам на диаграмме, которые лежат справа
от кривой PC, т. е. обогащенные компонентом В (Р —
критическая точка), при периодической ректификации дают остатки,
составы которых приближаются к составу, отвечающему
точке С. Растворы, соответствующие точкам, лежащим слева от PC,
т. е. обогащенные компонентом А, также дают остатки составов,
близких к точке С, но получаемые из растворов, обогащенных
Ректификация Ректификация
Экстрактор ^тракта рафшшта
I
F
Рис. 78. Дистилляциоккые
характеристики системы типа I (случай 1):
7 —хорда равновесия для системы жидкость-
жидкость; 2 — хорды равновесия для системы
пар —жидкость.
Рис. 79. Схема регенерации экстрагекта (случай 1):
7 — сборни* экстракта; 2, 3 —отстойники; 4 — сборник рафината; 5 — сборник экстрагент^;
[ытание; II —коночный экстракт; ///— свежий Элстрагент.
154
компонентом А. Если компоненты А и С образуют азеотропную
смесь с максимальной температурой кипения, кривая из точки Р
будет направлена к точке, отвечающей составу этой
азеотропной смеси. Если образуется азеотропная смесь с минимальной
температурой кипения, при ректификации жидкостей,
обогащенных компонентом А, будет получен окончательный остаток
состава, отвечающего точке А.
На рис. 79 показана схема экстракции и регенерации экс-
трагента для случая, когда компоненты А и В заметно взаимно
растворимы. На рис. 80 приведена соответствующая фазовая
диаграмма. В результате ректификации экстракта Е получают
остаток (С) и пар Q, выходящий из ректификационной
колонны сверху. После конденсации последнего образуются две
равновесные жидкие фазы V и К, которые разделяются в
отстойнике. Принято, что температура отстаивания равна температуре
экстракции. В результате отстаивания получают один слой,
содержащий в основном регенерированный экстрагент V и, если
необходимо, флегму V для орошения колонны, а также слой К,
обедненный экстрагентом. Этот слой вместе с рафинатом также
подвергают ректификации, в результате которой получают
конечный рафинат / и пар U. Образующиеся после конденсации пара U
жидкие фазы также разделяются в отстойнике. Если хорды
равновесия, соединяющие точки, которые соответствуют составам
фаз, получаемым в отстойниках, проходят на фазовой диаграмме
F+S=E+R
T=J+M
U=M+N
Рис.
Экстракция
С
Ректификация
рафината
Ректификация экстракта
С
q=v+v'+K
H=V+H
E=(C) +H
.. „ G \ Хорда
ВВевеше раднодесия
cbetkeao экстраеента
V+m=Z
Z+B=S
Хорда раднодесия
Э. Фазовые диаграммы для систем жидкость
■жидкость (случай 1).
155
Очень близко друг к Другу, Для обеих ректификационных колонн
можно использовать общие конденсатор и отстойник.
Регенерируемый экстрагент в смеси со вновь вводимым в
процесс свежим экстрагентом В образует экстрагент S,
направляемый в экстрактор. Следует подчеркнуть, что точка S на
фазовой диаграмме должна лежать ниже продолжения хорды
равновесия RW (см. рис. 80, ректификация рафината),
начинающейся в точке R, соответствующей рафинату R, иначе рафинат
данного состава в экстракторе не может быть получен. Чем
ниже содержание С в S, тем меньше требуется ступеней
экстракции и тем меньше необходимое соотношение экстрагента
и исходного раствора.
Как видно из рис. 80 (ректификация экстракта), если
экстракт имеет состав, соответствующий точке Е на диаграмме,
содержание компонента В в экстракте более чем достаточно для
того, чтобы удалить А из экстракта; при подаче экстракта в
дистилляционную колонну сверху процесс разделения экстракта
можно проводить без флегмы. Если же точка Е на бинодальной
кривой будет расположена несколько левее, чем показано на
рис. 80, содержание В в экстракте может оказаться
недостаточным для проведения дистилляции. Эту трудность можно
преодолеть при использовании большого флегмового числа V /Н.
Подача флегмы в колонну равнозначна разбавлению экстракта
растворителем. Необходимо отдавать предпочтение такому
растворителю, который обеспечивает уменьшение числа ступеней
ректификации ,5. В первом приближении можно применять
правило: составы экстрактов должны соответствовать точкам,
располагающимся на бинодальной кривой справа от точки L,
которая лежит на линии, соединяющей С с точкой G,
соответствующей гетероазеотропному составу пара.
Если А и В практически взаимно нерастворимы, бинодаль-
ная кривая в интересующей нас области почти совпадает с
линиями А—С и В—С, и растворы, соответствующие точкам R и К,
практически не содержат компонента В. В этом случае
ректификацию рафината можно не проводить.
Наличие азеотропной смеси компонентов В и С, обладающей
максимальной температурой кипения, приводит к тому, что
получение чистого С становится невозможным. При этом состав
нижнего продукта, получаемого при ректификации экстракта,
близок к азеотропному. Когда азеотропная смесь В—С имеет
минимальную температуру кипения, выделение компонента С
возможно, если относительная летучесть гетероазеотропной
смеси А—В настолько высока, что она будет преобладать в
дистилляте. При относительно невысокой температуре в
конденсаторе азеотропная смесь образует две жидкие фазы и ее
можно разделить отстаиванием. Разделение азеотропных
смесей А—С не встречает затруднений.
156
Случай 2
Система типа I. Условия: порядок температур кипения
компонентов В>С>А> гетероазеотропной смеси А и В;
компоненты В и С не образуют азеотропной смеси В—С; тройные азео-
тропные смеси отсутствуют. Если В является высококипящим
компонентом, то гетероазеотропная смесь А — В, вероятно,
будет обогащена компонентом А. Нижние продукты, получаемые
при ректификации экстракта, будут иметь составы,
отвечающие точкам, перемещающимся по направлению к вершине В на
треугольной диаграмме.
Рассмотрим рис. 81, на котором показан лишь процесс
ректификации экстракта. В этом случае ректификация экстракта
предназначена, скорее, для полного разделения смеси А я С
(если позволяет относительная летучесть), чем для разделения
В и С. Пар, поднимающийся из колонны для ректификации
экстракта, конденсируется, образуя две жидкие фазы. Весь
обогащенный экстрагентом слой V используют в качестве флегмы,
чтобы иметь возможно более низкое содержание компонента С
в верхнем продукте. Слой К, обогащенный компонентом А,
содержит такое количество С, что его нецелесообразно смешивать
с рафинатом; вместо этого его возвращают в экстрактор в
точке, где содержание компонента С соответствует его
содержанию в слое К-
Нижний продукт /, выходящий из ректификационной колонны
для экстракта, обычно является бинарным раствором
компонентов В—С, который разделяется в другой
ректификационной колонне. В том
случае, если взаимная
растворимость
компонентов А и В существенна,
рафинат обрабатывают
так же, как в случае 1;
поэтому для осуще-
F+S+K=E+R
E=J+H
Ректификация
экстракта
Экстрактор
а о
Рис. 81. Схема регенерации экстрагента {а) и фазовая диаграмма {б) для
случая 2:
1 — сборник экстракта; 2 — отстойник; 3 — сборник экстрагента; / — питание; // — на
ректификацию рафината; III — регенерированный экстрагент (после ректификации рафината);
IV — свежий экстрагент В; К —конечный экстракт.
157
ствления данного процесса обычно требуются три
ректификационные колонны. Если компоненты А я В практически взаимно
нерастворимы, ректификацию рафината можно не проводить.
Наличие азеотропной смеси А — С в этом случае не создает
затруднений, но из азеотропных смесей В — С, обладающих
максимальной или минимальной температурой кипения,
достаточно чистый компонент С и хорошо регенерированный экстр-
агент В обычными способами получить нельзя.
В другом случае для ректификации экстрагента может быть
использована специальная колонна, нижний продукт из которой
будет соответствовать составу (В). Верхний продукт (конечный
экстракт) представляет собой в основном бинарный раствор
компонентов А — С, состав которого будет выражаться точкой,
получаемой при пересечении продолжения линии BE со
стороной А — С на треугольной диаграмме (рис. 81). Если бинодаль-
ная кривая ограничивает небольшую область на диаграмме (при
этом концентрация ХСЕ мала), получаемый продукт С будет
недостаточно чистым. Однако соотношения летучестей в системе
иногда не позволяют применить другой способ. Введением
флегмы в экстрактор (см. главу VI) можно увеличить значение ХСЕ,
но соотношение С/А в конечном экстракте все равно не может
быть больше отношения, определяемого тангенсом касательной,
проведенной из точки В к бинодальной кривой.
Случай 3
Система типа I. Условия: порядок температур кипения ком-
поненов: В>А>гетероазеотропной смеси А и В>С;
компоненты В и С не образуют азеотропной смеси В — С; тройные азео-
тропные смеси отсутствуют. На рис. 82 представлена схема
разделения для случая, когда А я В практически взаимно
нерастворимы 16' 17. Ректификация рафината на этой схеме не
предусмотрена, так как концентрация экстрагента в рафинате
невелика. Распределяемое вещество С имеет минимальную
температуру кипения и при ректификации экстракта (С)
выделяется как дистиллят. Отделяемая от пара жидкая смесь TV
разделяется в отстойнике с образованием двух фаз, составы которых
(при температуре в экстракторе) характеризуются на
диаграмме точками Я и /. Если отстаивание более выгодно проводить
при высокой температуре, смесь TV после остаивания охлаждают.
Раствор Я, содержащий очень небольшое количество
экстрагента, возвращают в экстрактор для извлечения из него
компонента С. Обогащенный экстрагентом слой / вновь направляют
в ректификационную колонну для окончательного выделения
экстрагента, имеющего состав, отвечающий точке Т. Экстра-
гент S, подаваемый в экстрактор, содержит меньше
компонента С, чем фаза W. Если в смеси N содержится очень мало С
158
Ректификации
экстракта
Рис. 82. Схема регенерации экстрагента (а) и фазовая диаграмма (б) для
случая 3 (экстрагент В не растворим в А):
7_сбориик экстракта; 2 —отстойник; 3—сборник экстрагента; / — питание; // — рафииат;
III — свежий экстрагент; IV— конечный экстракт.
(при этом точка / лежит ниже W), устраняется необходимость
в нижней (исчерпывающей) части ректификационной колонны,
и после отстаивания жидкая фаза / будет представлять собой
регенерированный экстрагент, а фаза Я может добавляться к
рафинату.
Если А я В обладают значительной взаимной
растворимостью16-17, применима схема регенерации экстрагента,
показанная на рис. 83. На треугольной диаграмме приведены две би-
нодальные кривые (при температуре экстракции tx и при более
высокой температуре t2). Так же, как и в предыдущем варианте,
после выделения (С) из экстракта в ректификационной колонне
для экстракта образуется двухфазная жидкая смесь N, которая
после расслаивания дает две равновесные фазы D и V (при
температуре U). После охлаждения обогащенного экстрагентом
слоя V до температуры tx из него осаждается фаза К,
обедненная экстрагентом. Последнюю вместе с рафинатом R и
обедненной экстрагентом фазой D из отстойника направляют в
ректификационную колонну для рафината с целью получения
конечного рафината (А). Образующиеся при этом пары состава Р
с небольшим содержанием С подают в ректификационную
колонну для экстракта.
Если же содержание (С) в смеси ./V относительно велико,
жидкие фазы, полученные после отстаивания, обрабатывают по
способу, показанному на рис. 82. При отдельной ректификации
159
Ректификация
экстракт
Рис. 83. Схема регенерации экстрагента (а) и фазовая диаграмма (б) для
случая 3 (экстрагент В значительно растворим в А):
/ — сборник экстракта; 2,—.сборник экстрагента (ti); 3~сборник рафината; 4 — отстойник;
/ — питание; // — свежий экстрагент; /// — конечный экстракт; IV — конечный рафинат.
рафината будут получаться пары Р (см. рис. 83), которые
могут быть направлены в нижнюю часть колонны для
ректификации экстракта (см. рис. 82), и остаток (А) (см. рис. 83).
В обоих случаях можно поддерживать температуру в
кипятильнике куба ректификационной колонны для экстракта
равной температуре кипения (В) и получать Л и С из экстрактов,
так как В является самым высококипящим компонентом.
Состав конечного экстракта, получаемого при этом, будет
соответствовать точке, пересечения продолжения линии BE со
стороной А—С. Упрощение схемы регенерации нежелательно, так
как приводит к ухудшению чистоты конечного экстракта.
С л у ч а й 4
Система типа II. Экстракция с флегмой. Условия:
температуры кипения А, В и С выше температур кипения гетероазео-
тропных смесей А — С и В — С; А — С — бинарная азеотроп-
ная смесь (смесь А — С, обладая минимальной температурой
кипения, кипит при температуре, более высокой, чем гетероазео-
тропные смеси); тройные азеотропные смеси отсутствуют. Схема
регенерации для этого случая представлена на рис. 84, а соот-
160
ветствующая фазовая
диаграмма— на рис. 85.
Рассмотрим сначала
рис. 85, а. Исходную смесь
F обрабатывают экстра-
гентом, по составу
близким к В. После
экстракции образуются экстракт
Е и рафинат R. Чтобы
экстракт Е содержал
возможно большее
количество С, экстрактор (в
соответствии с
принципами, изложенными в главе
VI) орошают флегмой L
с высоким содержанием
С. В таких системах
двухфазные жидкие смеси
(например, смесь,
соответствующая хорде
равновесия MN) обычно
образуют пар равновесного
состава (например, V), который характеризуется точкой,
лежащей на кривой аа'. Кривая аа' соединяет гетероазеотропную
смесь А—В (точка а) с гетероазеотропной смесью В—С
(точка а'). При конденсации такого пара образуются две жидкие
фазы. При ректификации фазы, обогащенной компонентом В,
в качестве окончательного кубового остатка получают
компонент В.
Ректификация жидких смесей, лежащих на ветви бинодаль-
ной кривой, обращенной к стороне А — С треугольной
диаграммы, приводит к получению кубового остатка, представляющего
собой компонент С (при условии, что С имеет более высокую
температуру кипения, чем А). Если в системе возможно
образование азеотропной смеси А — С с минимальной температурой
кипения, в кубовом остатке будут получены либо компонент А,
либо С в зависимости от того, каким из этих компонентов
обогащена поступающая на ректификацию смесь.
Рассмотрим рис. 84 и 85,6. После ректификации
экстракта £ в кубе получают регенерированный экстрагент (В) и на
верху колонны пар К, состав которого соответствует точке,
лежащей вблизи кривой аа'. После отстаивания жидкостей,
получаемых при конденсации паров Z, обогащенный экстрагентом
слой / возвращают в ректификационную колонну в качестве
флегмы, а обедненный экстрагентом слой L разделяется на два
потока: L', который направляют в качестве флегмы в
экстрактор, и L", который дистиллируют для получения обогащенного
Экстрактор
Ректификация Ректификация
экстракта рафината
В Ш
Рис. 84. Схема регенерации экстрагента
(случай 4; экстракция с флегмой):
/ — сборник экстракта; 2 —отстойники; 3 — сборник
рафината; 4 — сборник экстрагента; / — питание;
// — свежий экстрагент; /// — конечный экстракт;
IV — конечный рафинат.
Н Р. Е. Трейбал
161
с
Рис. 85. Фазовые диаграммы (случай 4):
7 —хорда равновесия для системы жидкость —жидкость; 2 —хорда равновесия для системы
пар —жидкость.
компонентом С продукта Н. Образующийся при этом пар Р
можно сконденсировать в том же конденсаторе, что и пар К.
При ректификации рафината образуется пар W, который
после конденсации и последующего отстаивания дает флегму U
и обогащенный экстрагентом слой Q. Если W и, следовательно,
Q будут относительно более богаты компонентом С, слой Q
можно подвергнуть дополнительной ректификации в четвертой
колонне, пары из которой можно направить в конденсатор, где
конденсируются пары W, а кубовый остаток — в хранилище для
экстрагента. Регенерированный экстрагент S будет содержать
С меньше, чем любая смесь, лежащая на продолжении хорды
равновесия, проходящей через точку R. При построении
диаграммы на рис. 85, б сделано допущение, что температуры при
отстаивании равны температуре в экстракторе.
Азеотропные смеси с экстрагентом. Любая азеотропная
смесь экстрагента и экстрагируемого компонента является
нежелательной. Если эта азеотропная смесь обладает минимумом
температуры кипения, ее получают в качестве верхнего
продукта. При этом экстрагент выделяют в достаточно чистом виде,
в то время как экстрагируемый компонент получают нечистым.
Иногда его можно подвергнуть дополнительной обработке
методом, описанным для случая 1 (см. выше),
162
При некоторых условиях можно использовать другие способы
регенерации 18, например по схеме, изображенной на рис. 86. На
этом рисунке показана технологическая схема разделения смеси
3- и 4-пиколинов бензолом (экстрагирующим предпочтительно
3-пиколин) и буферным водным раствором фосфата натрия
(экстрагирующим 4-пиколин). Ректификацией раствора 3-пико-
лина в бензоле в качестве нижнего продукта получают
3-пиколин; присутствие даже незначительного количества воды
улучшает отделение бензола.
При ректификации водного продукта на верху колонны
получают азеотропную смесь воды и 4-пиколина. При добавлении
бензола к парам, поступающим в конденсатор, после
конденсации образуются две жидкие фазы, причем 4-пиколин переходит
из водного слоя в бензольный. После ректификации
бензольного слоя получают бензол (верхний продукт) и 4-пиколин
(нижний продукт).
При таком способе выделения продуктов необходимо иметь
в системе большое количество рециркулирующего бензола, если
коэффициент распределения 4-пиколина в бензоле недостаточно
высок. Поэтому для экстракции 4-пиколина из водного слоя
после ректификации вместо одноступенчатого отстойника
необходимо использовать многоступенчатый экстрактор. Можно
также непосредственно извлечь 4-пиколин бензолом сразу после
основной экстракции, если коэффициент распределения окажется
достаточно благоприятным, причем в этом случае водный
раствор фосфата не
подвергают ректификации. Иногда
имеет преимущества
применение азеотропной
дистилляции с использованием
нерастворимого третьего
компонента. Разделение
бинарных азеотропных смесей с
максимумом температуры
кипения также
затруднительно, если не применить
схемы, аналогичной
описанной выше.
Тройные азеотропные
смеси могут быть
обычными (т. е. образованными из
гомогенного раствора) или
гетероазеотропными
(образованными из смесей двух
жидкостей), причем эти
азеотропные смеси обычно
обладают минимумом тем-
Фракционная
экстракция
Л
£2.
ш
ш
ж
X
^н
и ш
k~z?
ж
Рис. 86. Схема разделения пиколинов
методом фракционной экстракции 18:
J—питание (3- и 4-пиколииы); //— бензол +
+ 3-пиколин; /// — бензол; IV — азеотропная
смесь вода-f 4-пиколни; К —бензол ^4-пиколин;
VI — водный слой; VII — бензольный слон;
VIII — водный раствор фосфата натрия; /X
—бензол; X —водный раствор фосфата иатрня +
4-пиколин; XI — 3-пиколин; XII — 4-пиколии.
11*
163
пературы кипения. В первом случае едва ли можно предложить
экономичную схему регенерации экстрагента.
При разделении тройных гетероазеотропных смесей точка,
соответствующая составу пара М, образовавшегося из
жидкостей Н и J (см. рис. 78), будет лежать на хорде равновесия HJ.
Разделение возможно после конденсации пара и отстаивания
конденсата, причем обогащенный компонентом В слой
направляют в виде флегмы в ректификационную колонну, а слой,
обогащенный компонентом Л, выводят из процесса.
Трудности, которые могут возникнуть при разделении такой
системы, связаны с возможностью образования бинарной азео-
тропной смеси В — С, обладающей минимумом температуры
кипения. В качестве примера разделения подобных систем можно
привести процессы дистилляции, применяемые при
дегидратации спирта бензолом. Следует, однако, отметить, что азеотроп-
ная дистилляция смесей с тройными азеотропами является
более экономичным процессом, чем экстракция. Сведения об азео-
тропных смесях приведены в таблицах Хорсли ,9.
Перегонка с водяным паром. В любой из приведенных схем,
в тех случаях когда кубовый остаток, получаемый при
ректификации, представлят собой почти чистую воду, может быть
использована перегонка с острым паром. Последний подают в
нижнюю часть дистилляционной колонны.
При разделении неводных систем, компоненты которых не
растворяются в воде, экстрагент можно отделить от прочих
компонентов перегонкой с водяным паром. При использовании легко
летучих растворителей такой способ регенерации более
экономичен, чем ректификация. Перегонку с водяным паром
применяют также при обработке высококипящих жидкостей для
снижения содержания экстрагента в продукте вместо ректификации
в вакууме или ректификации при чрезмерно высоких
температурах в кипятильнике. Водяной пар, а также другие вещества
иногда используют в качестве добавочного компонента для
увеличения обычно низкой относительной летучести нерастворимых
в воде жидкостей20.
Выпаривание. Если распределяемый компонент нелетуч,
экстрагент может быть выделен из экстракта выпариванием.
Эту операцию применяют при низких температуре кипения и
теплоте парообразования экстрагента. В некоторых случаях,
если относительная летучесть распределяемого компонента
очень мала, большую часть растворителя можно регенерировать
при однократном испарении. Остаток либо подвергают
ректификации, либо обрабатывают другими способами. Так
регенерируют, например, низкокипящие экстрагенты, подобные
пропану или двуокиси серы, при отделении их, например, от
высококипящих компонентов нефтяных масел.
164
Кристаллизация. При охлаждении экстракта
распределяемое вещество может выкристаллизовываться. Выделение
экстрагента из рафината и в этом случае обычно проводят с
помощью ректификации. г-^
Жидкостная экстракция. 'Если относительная летучесть
экстрагента и распределяемого компонента низка или если в
системе образуются нежелательные азеотропные смеси,
экстрагент можно извлечь из этой смеси с помощью экстракции
новым экстрагентом, который можно в дальнейшем отделить тем
или иным способом.. Такой метод используют, например, при
концентрировании разбавленных растворов соляной кислоты
экстракцией смесью пентанолов; полученный экстракт
(раствор НС1 в смеси пентанолов) нельзя разделить дистилляцион-
ными методами2l. Пентанолы извлекают из водного рафината
и экстракта экстракцией бензолом, который затем отделяют
перегонкой.
Экстракцию применяют также в тех случаях, когда
образуется большой объем рафината и стоимость его нагрева до
температуры ректификации высока даже при использовании
эффективных теплообменников для утилизации тепла [например,
экстракция гексона (метилизобутилкетона) из насыщенного
водного рафината высококипящими углеводородами].
Коэффициент распределения в данном случае очень высок, и поэтому
после экстракции образуется относительно небольшое
количество раствора гексона в масле, из которого затем с помощью
дистилляции регенерируют гексон. Масло практически
совершенно нерастворимо в воде, так что рафинат выводится из
процесса без дальнейшей обработки.
Другим примером является извлечение экстрагента диэти-
ленгликоля из парафинового рафината и даже из экстракта
(после перегонки с водяным паром) водой в «Удекс-процессе»
для разделения парафиновых и ароматических углеводородов22.
Иногда экстрагируют из экстракта распределяемое вещество;
например, пенициллин извлекают водным буферным раствором
при рН = 7. Экстракт образуется в процессе экстракции
пенициллина амилацетатом из ферментативных растворов при
рН = 2. После вторичной экстракции одновременно с
пенициллином получают регенерированный экстрагент.
х- Наконец, еще одним примером является извлечение урановой
соли водой из экстракта; последний образуется при экстракции
урана из его растворов в азотной кислоте раствором трибутил-
фосфата в керосине. С помощью вторичной экстракции
получают чистую урановую соль и одновременно регенерируют
экстрагент.;
Химическая реакция. Регенерация экстрагента с помощью
химической реакции применяется в том случае, когда
образующееся химическое соединение представляет собой целевой
<65ч
продукт. Например, никотин экстрагируют из щелоков
табачного производства керосином. Затем экстракт обрабатывают
серной кислотой и получают водный раствор сульфата никотина
(целевой продукт) и очищенный от никотина керосин,
пригодный для дальнейшего использования. Тантал, экстрагированный
гексоном в виде фтористых соединений, осаждают из экстракта
аммиаком. Образующаяся гидроокись является исходным
материалом для получения чистого металла. В других случаях
химическую реакцию используют, чтобы достичь высокой степени
очистки регенерируемого экстрагента, например легкого масла,
применяемого в процессе извлечения фенолов из водных
аммиачных растворов (побочных продуктов коксохимического
производства). Очистка легкого масла достигается извлечением
фенолов водным раствором каустической соды.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
$с,а—селективность экстрагента по отношению к
компоненту С в растворе А — С.
т — коэффициент распределения, равный отношению
концентраций распределяемого вещества в равновесных
жидких фазах.
X — концентрация, вес. доли.
х— концентрация, мол. доли.
Индексы
А, В и т. д. — компоненты А, В и г. д.
АВ — Л в растворе, обогащенном В.
ЛИТЕРАТУРА
1. Е. R. Washburn, R. Void, J. Am. Chem. Soc, 53, 3237 (1931).
2. D. Q. В е е с h, S. Q 1 a s s t о n e, J. Chem. Soc, 1938, 67.
3. T. F. Brown, Ind. Eng. Chem., 40, 103 (1948).
4. K- A. Varteressi an, M. R. Fenske, Ind. Eng. Chem., 29,270(1937).
5. L. Garwin, A. N. H i x s о n, Ind. Eng. Chem., 41, 2298 (1949).
6. R. E. Treybal, L. D. Weber, J. F. Daley, Ind. Eng. Chem., 38, 817
(1946).
7. M. Newman, С. В. Н а у w о г t h, R. E. Treybal, Ind. Eng. Chem.,
41 2039 (1949).
8. A.'W. Francis, Ind. Eng. Chem., 45, 2789 (1953); 46, 205 (1954).
9. L. A. Girifalco, R. J. Good, J. Phys. Chem., 61, 904 (1957).
10. D. J. Donahue, F. E. В art ell, J. Phys. Chem., 56, 480 (1952).
11. N. F. Murphy, J. E. Lastovica, J. G. F a 11 i s, Ind. Eng. Chem.,
49, 1035 (1957).
12. I. M e 1 1 a n, Source Book of Industrial Solvents; v. I Pure Hydrocarbons;
v. II Halogenated Hydrocarbons; v. Ill Monohydric Alcohols, 1957, 1959;
and later volumes; Reinhold Publishing Corporation, New York.
13. R. H. E well, L. M. Welch, Ind. Eng. Chem., 37, 1224 (1945),
166
14. J. Ricci, The Phase Rule a. Heterogeneous Equilibrium, D. Van Nostrand
Company, Inc., Princeton, New York, 1951.
15 P E ao-lesf ield, В. К. Kelly, J. F. Short, Ind. Chemist, 29, 147,
243 (1953).
16. D. F. Othmer, R. L. R a t с 1 i f f e, Ind. Eng. Chem., 35, 798 (1943).
17. D F. Othmer, E. T г eu ge r, Trans. Am. Inst. Chem. Engrs., 37, 597
(1941).
18. A. E. Karr, E. G. Scheibel, Ind. Eng. Chem., 46, 1583 (1954).
19. L. H. H о г s 1 e y, Azeotropic Data, American Chemical Society, Washington,
D C. 1952.
20 D. F. Othmer, Chem. Eng. Progr., 54, № 7, 48 (1958).
21. E D. Crittendon, A. N. H i x s о n, Ind. Eng. Chem., 46, 265 (1954),
22. H. W. Grote, Chem. Eng. Progr., 54, № 8, 43 (1958).
ДИФФУЗИЯ И МАССОПЕРЕДАЧА
Рассмотрим многофазную систему, не
находящуюся в равновесии. Опыт
показывает, что такая система изменяется
самопроизвольно, стремясь к состоянию
равновесия, при котором температура и
химический потенциал или активность каждого
компонента одинаковы во всех точках
системы.
Изменения, происходящие в любой из
фаз системы (при постоянной
температуре), приводят в конечном счете к
равномерному распределению каждого
компонента в данной фазе. Указанное
самопроизвольное перемещение различных
компонентов, вследствие которого их
концентрации становятся постоянными, называется
диффузией. Если, например, каплю спирта
поместить в лабораторный стакан с водой,
спирт в конце концов распространится во
всем объеме воды; если жидкость в
стакане неподвижна, продолжительность
процесса может быть очень велика. Этот процесс
можно значительно ускорить
перемешиванием жидкости, но конечный результат
будет одинаков: одна жидкость (спирт)
равномерно распределится в другой (воде).
Таким образом, необходимо различать
два явления: молекулярную диффузию,
или самопроизвольное приближение к
равномерному распределению в молекулярном
масштабе внутри неподвижной жидкости,
и явление равномерного распределения,
достигаемое принудительным движением
массы жидкости.
МОЛЕКУЛЯРНАЯ ДИФФУЗИЯ
В приведенном выше примере при
отсутствии принудительного движения
жидкости, когда концентрация спирта
становится одинаковой во всех ее точках, ника-
166
ких дальнейших самопроизвольных изменений системы не
происходит. Однако до тех пор, пока не достигнута равномерность
распределения, система самопроизвольно изменяется, причем
растворенное вещество переходит из области с большей
концентрацией в область с меньшей концентрацией. Скорость, с
которой растворенное вещество в любой точке движется в
произвольном направлении, должна поэтому зависеть от градиента
концентрации в данной точке для заданного направления.
Пусть в растворе компонентов А и В мольная массовая
скорость в заданном направлении, или поток растворенного
вещества Л, составляет JA молей А в единицу времени на единицу
поверхности, нормальной к направлению движения. Тогда
коэффициент диффузии DA компонента А можно определить как
отношение потока Jа к градиенту концентрации dcAjdz:
Это выражение называется первым законом Фика*.
Обычно коэффициент диффузии — величина переменная для
данной системы и зависит от концентрации и температуры.
Истинной «движущей силой» диффузии является, по-видимому,
не концентрация, как следует из уравнения (V, 1), а химический
потенциал или активность2. Выше уже отмечалось, что
самопроизвольное изменение системы прекращается, когда
выравниваются не концентрации, а активности компонентов системы.
Однако для определения коэффициентов диффузии, как
правило, используют концентрации.
Поток / в уравнении (V, 1) представляет собой мольную
массовую скорость, определяемую по отношению к средней мольной
скорости vM в растворе3'4. Рассмотрим некоторый объем
раствора компонентов Л и В с площадью поперечного сечения,
равной единице, в котором происходит молекулярная диффузия.
Для простоты примем, что раствор идеальный; поэтому при
протекании диффузии объем раствора остается постоянным и
движения жидкости не происходит. Тогда, если компонент А
диффундирует слева направо с объемной скоростью, равной
линейной скорости vA, то скорость диффузии vB компонента В в
противоположном направлении vB =—vA (поскольку объем
раствора останется постоянным), или
VA + VB = ° <V'2>
Объемы слева и справа от некоторой неподвижной точки
остаются в этом случае постоянными. Мольная массовая
скорость компонента А относительно этой точки составит NA=*
= vAcA, а мольная массовая скорость компонента В равна
NB=vBcB, где сА и св — мольные концентрации соответственно
компонентов А и В.
169
Результирующая мольная скорость компонентов
относительно неподвижной точки равна NA + NB- Средняя мольная
скорость
cava + cbvb
(V.3)
где с — суммарная мольная концентрация.
Поскольку концентрации компонентов не равны друг другу,
скорость vM не равна нулю. Если бы оказалось возможным
наблюдать процесс диффузии, перемещаясь со скоростью vM,
обнаружить молекулярный поток было бы нельзя. Число молей
раствора слева и справа от гипотетического движущегося
наблюдателя будет в этом случае оставаться постоянным.
Мольная массовая скорость, или поток NA относительно некоторой
фиксированной точки пространства, должен быть больше
потока JА на количество компонента А, соответствующее
объемной скорости vM, или
NA = VMCA + JA (V'4>
откуда
с, дс,
NA = (NA + NB)ir-DAl)f (V'5>
Таким образом, при диффузии в идеальных растворах
существуют молекулярный и массовый потоки. В неидеальных
растворах должен существовать также объемный поток. Для
определения коэффициентов диффузии можно пользоваться
любым из указанных выше потоков. При этом для получения
одинакового результата следует учитывать соотношения между
потоками. В работе Бэрда и др.4 этот вопрос рассмотрен подробно.
Принимая для бинарного раствора
сА + св=с (V.6)
напишем уравнение, аналогичное уравнению (V, 5), для
компонента В. Складывая это уравнение с уравнением (V, 5), получим
дс. дсв
- D л -Н- = D „ S&- (V, 7)
а дг в dz
Отсюда следует, что JA = —JB. Далее в результате
подстановки выражения (V, 6) в уравнение (V, 7) приходим к выводу,
что DA = DB (при данных концентрации и температуре).
Для объема жидкости с площадью поперечного сечения,
равной единице, и длиной dz скорость, с которой компонент А
поступает в сечение z = z, равна NAz, а скорость, с которой он
покидает сечение z + dz, составляет NAz+ (dNA/dz)dz. Скорость
уменьшения количества компонента А в этом объеме равна
разности указанных выше количеств, или
д (с. dz)
170
Если vM = 0 и DA = const, в результате совместного решения
уравнений(V, 1) и (V, 4) с учетом уравнения (V, 8) получают
второй закон Фика:
дсл дЧя
-Ж = °л-ёФ CV.9)
Выводы, сделанные выше, относятся только к одномерной
диффузии. Все приведенные выражения можно написать также
в трех измерениях — для координат да, у и z. Тогда при DA =
= const уравнение (V, 9) принимает вид:
дс, (дЧ, д2сА дЧА
19 А \ dw2
дв л \ dw* ' ду* ' dz"2
(V, 10)
Для системы, движущейся со скоростью v, материальный
баланс выражается уравнением
с. / дс, дс, дс.\ (дЧ. дЧ. дЧ Л
Члены уравнения (V, 11), в которые входит скорость,
описывают конвективный перенос. Размерность D—(длина )2/время.
Обычно применяемые размерности связаны следующей
зависимостью:
3,87 (Д см2/сек) = (Д фуш2/ч) (V, 12)
Экспериментальное определение и численные значения
коэффициентов молекулярной диффузии. Обзоры экспериментальных
методов определения коэффициентов диффузии в жидкостях
приведены в литературе5,6. Самым простым методом, точность
которого достаточна в большей части случаев для практических
целей, является метод, связанный с применением пористых
мембран, наиболее часто изготовляемых из спекшегося стекла7-11.
Растворы, имеющие различную концентрацию, помещают по обе
стороны пористой перегородки, и диффузия происходит в порах
мембраны. Каналы в мембране весьма узки и могут тормозить
конвективные токи; поэтому с помощью пористых мембран
можно измерять истинную молекулярную диффузию. Мембрану
калибруют, используя раствор с известным коэффициентом
диффузии, так как ни длина пор, ни площадь их поперечного
сечения неизвестны.
Установлено, что для систем с обычной вязкостью D имеет
величину порядка Ю-9 м2/сек. Значения D зависят от
концентрации. Различают дифференциальное, или мгновенное, и
интегральное, или среднее, значения D в данном интервале
концентраций. Коэффициенты самодиффузии описывают движение
молекул относительно молекул того же вида в одно- или
многокомпонентных системах; эти коэффициенты нельзя определить
непосредственно. Величины коэффициентов самодиффузии из-
171
меряют с помощью «меченых» атомов — изотопов тех веществ,
для которых определяются указанные коэффициенты.
Наиболее полные данные о коэффициентах диффузии для
неэлектролитов приведены Джонсоном и Бэббом12 и для
электролитов— Харнедом и Оуэном13.
Теория диффузии в жидкостях. При наличии точного
объяснения механизма молекулярной диффузии можно было бы
рассчитывать коэффициенты диффузии, исходя из других свойств
растворов; такой расчет необходим при отсутствии опытных
данных о величинах коэффициентов диффузии. Кинетическая
теория оказалась чрезвычайно полезной для расчета
коэффициентов диффузии и других характеристик переноса в газах.'
Однако теория диффузии в .жидкостях до сих пор разработана
недостаточно, что затрудняет точный расчет коэффициентов
этого процесса.
Данный раздел посвящен определению коэффициентов
диффузии в разбавленных и концентрированных растворах.
Коэффициенты диффузии
неэлектролитов
Разбавленные растворы. Для броуновского движения
коллоидных частиц компонента Л, радиус которых велик пе
сравнению с частицами растворителя В, можно применить закон
Стокса. Исходя из этой предпосылки, Эйнштейн получил 14
уравнение, известное как уравнение Стокса — Эйнштейна:
где DAB — коэффициент диффузии компонента А в
разбавленном растворе его в В;
к — постоянная Больцмана;
гА — радиус частицы;
\хв — вязкость растворителя.
Законом Стокса предполагается, что диффундирующие
частицы движутся через сплошную среду. Это положение не
применимо для диффузии молекул, так как молекулы
растворителя и распределяемого вещества сравнимы по величине.
Поэтому выражение (V, 13) нельзя использовать для расчета
коэффициентов диффузии растворенных веществ с относительно
малым размером молекул.
Сэзерленд !5 нашел, что если частицы диффундирующего
вещества и растворителя одинаковы, уравнение (V, 13) принимает
вид:
172
Для чистой жидкости при расположении молекул в виде
плотно упакованной кубической решетки
(V./n)ll>
ГА--^Г~ <v-15)
где V — мольный объем жидкости;
п — число Авогадро.
Тогда для самодиффузии уравнение (V, 14) преобразуется
к виду:
Это уравнение можно применять для расчета коэффициента
самодиффузии многих жидкостей с точностью примерно 10—
12%.
Теорию абсолютных скоростей Эйринга и др.16-19 можно
успешно использовать для выявления связи между диффузией
и вязкостью. Согласно этой теории, вязкость является мерой
скорости переноса количества движения в жидкости в
результате движения молекул от точки, обладающей большей
скоростью, к точке, движущейся с меньшей скоростью. Диффузия
же характеризует скорость переноса материи за счет
молекулярного движения; поэтому диффузия и вязкость тесно связаны
друг с другом.
По концепции Эйринга, идеальная жидкость имеет
решетчатую структуру, причем молекулы расположены в
гексагональной или кубической решетке. Молекулы упакованы неплотно,
образуя «дырки». Движение одного слоя относительно другого
требует движения молекул из одного равновесного положения
в другое, для чего необходимо наличие «дырок» в жидкости.
Эта концепция приводит к зависимости: *
«*-тетг (V-17>
где Я,, — расстояние между двумя слоями молекул;
Х2 — расстояние между соответствующими молекулами в
направлении движения;
Х3 — среднее расстояние между соответствующими
молекулами в направлении, перпендикулярном к
направлению движения;
К — расстояние между положениями равновесия в
направлении движения;
k' — константа скорости.
Применение теории абсолютных скоростей приводит к
выражению
ц = *vw*r (Vj18)
173
где Евяз, — энергия активации для вязкого течения;
^" — константа, учитывающая молекулярные объемы и
температуру.
В более ранних теориях ^вяз. связывали с энергией
испарения, так как энергия, необходимая для образования «дырки» в
жидкости размером в одну молекулу, равна внутренней энергии
испарения:
l±Ev = liHv — RT (V, 19)
где Д#„ — энтальпия испарения.
Было найдено, что
Здесь п = 3—4 для большей части жидкостей, причем
значение п меньше для ассоциированных жидкостей, подобных
воде; это значит, что размер «дырки» составляет лишь долю
размера молекулы. Позднее Эйринг выдвинул гипотезу о том,
что структура жидкости с «дырками» аналогична
газообразному состоянию, а регулярная структура — твердому состоянию
и что величина Еъпз. связана с энергией сублимации и мольными
объемами вещества в жидком и твердом состояниях. Таким
путем можно, например, очень точно рассчитать вязкость
сжиженных редких газов. Величина k", по-видимому, мало зависит
от температуры, поскольку известно, что построение зависимости
lg (д, от \/Т дает для большинства жидкостей линию, близкую
к прямой. Наклон этой линии позволяет определить Евяз..
Применяя теорию Эйринга к идеальным жидкостям,
получают зависимость
DAB = W (V, 20)
При допущении, что величины % и k' для диффузии и
вязкости одинаковы, находят:
Это выражение аналогично по форме уравнениям Стокса —
Эйнштейна и Сэзерленда. Величина кг/Хгкз связана с
молекулярным объемом, и поэтому произведение D\y должно быть
функцией молекулярного объема. Как известно, температурный
коэффициент при диффузии больших молекул равен
температурному коэффициенту вязкости растворителя; следовательно,
уравнение Стокса — Эйнштейна можно считать предельным случаем
уравнения (V, 21). Это объясняется тем, что при диффузии
больших молекул скорость определяется «скачком» молекулы
растворителя от одного положения равновесия к другому,
приводящим к перемещению диффундирующей молекулы в
освобождающееся пространство.
174
Если использовать теорию абсолютных скоростей для
определения константы k', для расчета коэффициента диффузии
(в сильно разбавленных растворах) можно получить уравнение
DAB = (—)''' — eAS,R ■ e~E^JRT = k'"e~E^-lRT (V, 22)
где VB — мольный объем растворителя;
h — постоянная Планка;
AS — энтропия активации диффузии;
/Гдиф. — энергия активации диффузии.
Вследствие подобия молекулярной диффузии и вязкости их
энергии активации можно считать равными:
Адиф. = ^вяз. (V, 23)
Для самодиффузии в уравнении (V, 21) имеем: Xi = X2=ta =
— Х = 2гА и, как указывалось выше, 2гА= (Уд/п)1'»; уравнение
(V,21) приводится к виду:
Как показывает сравнение уравнения (V, 16) с уравнением
(V, 24), последнее не согласуется с опытом. Позднее теория
Эйринга была усовершенствована, и для простой кубической20
или квазигексагональной21 упаковки молекул было получено
уравнение
Уравнение (V, 25) согласуется с опытными данными по
самодиффузии. Было показано22, что температурная зависимость
коэффициента самодиффузии в широких пределах изменения
температуры более точно описывается первым выражением
правой части уравнения (V, 22) или уравнением (V, 25).
Для бинарных разбавленных растворов уравнение (V,22)
дает абсолютные значения DA, отличающиеся от опытных часто
более чем в 2 раза. Температурная зависимость D в небольшом
интервале температур удовлетворительно описывается
уравнением (V, 22), если константа k'" не зависит от температуры и
зависимость D от \/Т изображается линией, весьма близкой к
прямой.
Применение методов статистической механики к данной
проблеме23'24 пока что не дало практически полезных результатов.
В связи с отсутствием теории, позволяющей надежно
определять D, приходится обращаться к эмпирическим методам.
Несколько эмпирических методов определения величин D в раз'-
бавленных растворах неэлектролитов критически рассмотрены
175
Ридом и Шервудом 25, причем Для расчетов рекомендуется
корреляция Уилки и Чэна26'27. Учитывая, что теоретически
величина DAbIIbIT должна быть функцией мольного объема, Уилки
и Чэн получили корреляцию опытных данных о DAb с точностью
около 10%- Уравнение, предложенное ими, имеет вид"
7,4 • Ю-8 (ч>Мв)0'5 Т
*.
»*» = т^ (V'26)
А
АВ ^у0,6
где DAB — коэффициент диффузии компонента А (для
разбавленного раствора А) в В, см2/сек;
Мв — масса 1 моль растворителя;
Т — температура, °К;
ц'— вязкость раствора, спз;
VA — мольный объем растворенного вещества при нор- *
мальной температуре кипения, см3/моль (для воды I
VA = 75,6). Ш
Величина ф в этом уравнении представляет собой фактор
ассоциации растворителя. Значения ф для разных растворителей
приведены ниже:
Вода ' . 2,6
Метанол 1,9
Этанол 1,5
Неассоциированные растворители
(например, бензол или этиловый
эфир) 1,0
Уравнение (V, 26) не пригодно для комплексообразующих
систем, например для растворов иода в ароматических
углеводородах. Мольный объем растворяемого вещества при
нормальной температуре кипения можно определить с помощью правила
Коппа, согласно которому мольный объем является аддитивной
функцией атомных объемов, составляющих молекулу. Атомные
и мольные объемы приведены в табл. 10.
Было отмечено28, что по уравнению (V, 26) можно
рассчитывать D для воды, растворенной в органических
растворителях, только в том случае, если вводить в расчет учетверенный
мольный объем воды (4X18,9 = 75,6); это подтверждает
ассоциацию ее при растворении в органических растворителях.
Пригодность уравнения (V, 26) для растворителей, отличающихся
высокой вязкостью, равной, например, 100 спз или более,
представляется сомнительной29.
Для растворителей, фактор ассоциации которых неизвестен,
величину ф можно найти подстановкой в уравнение (V, 26)
данных о диффузии других веществ в этих растворителях. Если
* Уравнение (V, 26) верно только при указанных ниже размерностях
входящих в него величин.
176
Атомные и мольные объемы
Таблица 10
Атомный объем
Углерод
Водород
Хлор
Бром
Иод
Сера
Азот
в первичных аминах
во вторичных аминах
Кислород
в метиловых эфирах
(простых)
в метиловых эфирах
(сложных)
в высших эфирах (простых
и сложных)
в кислотах
Бензольное кольцо
Нафталиновое кольцо
14,8
3,7
24,6
27,0
37,0
25,6
15,6
10,5
12,0
7,4
9,9
9,1
11,0
12,0
15,0
30,0
Мольный объем
н2
о2
N2
Воздух
СО
со2
so2
NO
N20
NH3
н2о
H20
COS
С12
Br2
I»
H2S
14,3
25,6
31,2
29,9
30,7
34,0
44,8
23,6
36,4
25,8
18,9*
75,6 **
51,5
48,4
53,2
71,5
32,9
* Растворитель.
** Растворенное вещество.
значение величины ф нельзя определить, то для расчета DAb
применимо эмпирическое уравнение Шайбеля30; оно имеет
вид *:
DAB = 8,2- Ю-8-
1+iV/
v>
(V.27)
Это уравнение является модификацией корреляции Уилки26.
Шайбель отмечает, что уравнение (V, 27) пригодно как для
ассоциированных, так и для неассоциированных растворителей
при условии, что Va>Vb- Им даны следующие рекомендации
для расчета DAb (В— растворитель):
В — вода; при Va<.Vb принимается Va=Vb\
В — метанол; уравнение применимо в случае Va^1,5VV,
В — бензол; при УА<2Ув принимается Va = 2Vb.
Другие испытанные растворители; при Ул<2,5Ув, прини«
мается УА = 2,5УВ.
* В уравнение (V, 27) необходимо подставлять величины с
размерностями, указанными для уравнения (V, 26),
12 Р. Е. Трейбал
177
Концентрированные растворы. Пауэлл, Розевуа и Эйринг19
показали, что коэффициент диффузии в концентрированных рас-
творах является функцией активности растворенного вещества
в растворе:
/ din a ,\ I din v. \
Da, ко„ц. = *>АВ (^) = DAB (l + -^ (V,28)
где уА — коэффициент активности растворенного вещества;
хА — мольная доля растворенного вещества.
Подстановка DАВ из уравнения (V, 21) дает:
К.кТ din а.
Если используются средние значения %i и Я-гЯ-з и они
изменяются линейно в зависимости от мольной доли, величина
d In a /d In х
должна быть также линейной функцией мольной доли
растворенного вещества при постоянной температуре. Таким'образом,
уравнение (V,29) может быть представлено в виде26:
где DAB\\B относится к коэффициенту диффузии А в сильно
разбавленном растворе его в В и Dba^a— к коэффициенту
диффузии В в сильно разбавленном растворе его в А.
Величина dlnajdln хА легко определяется по наклону
линии зависимости логарифма активности от логарифма мольной
доли А. Последнюю зависимость в свою очередь можно
получить из данных о давлении паров или другими способами,
указанными в главе III. Однако область применения уравнения
(V, 30) ограничена, вероятно, растворами, энергии активации
которых для диффузии и вязкости равны18. Для некоторых
систем величины
(P^)kohu.
d In aAfd In x.
имеют максимум. Как видно из рис. 87, этот эффект для
некоторых систем может быть резко выражен, и поэтому уравнение
(V, 30) не всегда надежно.
Пример V-1. Рассчитать коэффициент диффузии бромнафталина в
разбавленном растворе его в этиловом спирте при 20° С (опытное значение
0,76-10"5 см2/сек).
Решение.
Метод Уилки и Чэна. Для бромнафталина величину Vа можно
определить по данным табл. 10:
VА = 10 • 14,8 + 7 • 3,7 + 27,0 — 30 = 170,9 см3/моль
178
л, Ацетон-бензол
Ацетон-вода
Концентрация ацетона, мол. доли
Рис. 87. Зависимость коэффициентов диффузии ацетона от
концентрации для систем ацетон — бензол и ацетон—вода
при 25,15° С [J. Phys. Chem., 62, 404 (1958)].
При 20° С вязкость этанола ц'= 1,192 спз; ф=1,5; Мв = 46,05; 7=293° К.
Из уравнения (V, 26) находим £>АВ=0,70-10-5 см2/сек.
Метод Шайбеля. Величина VB = 2 • 14,8+6 • 3,7+7,4=59,2. Из
уравнения (V, 27) имеем £>АВ = 0,74-10~5 см21сек.
Пример V-2. Определить коэффициент диффузии этанола в воде как
функцию концентрации при 10° С и сравнить с опытными данными.
Решение. Обозначим компоненты раствора: этанол — А, вода — В.
Для определения DAB находим: VA = 59,2 (см. пример V-1); фв = 2,6; Мв =
= 18,02; Г=283°К. Из уравнения (V, 26) получаем ДАВЦв/7=4,36 • 10 8.
Для определения DBA находим: VB=75,6; MA =46,05; ф=1,5. Из
уравнения (V, 26) находим £>ваЦа/7=4,6 • 10~8.
Расчет должен быть проведен с помощью уравнения (V, 30).
Коэффициенты активности этанола в водных растворах взяты из данных рис. 50
(см. гл. III). Эти данные были нанесены на график зависимости
IgyA — lgxA; измерены также наклоны кривой при различных значениях
хА. Вязкости растворов этанол —вода при 10°С взяты из справочника*.
Табличные данные и расчеты приведены ниже:
0
0,02
0,0416
0,0899
0,1439
—0,0875
—0,123
—0,270
—0,442
Р
DBA»A |
°АВ»В
,+
4,355 10
4,350 • 10'
4,338 • 10'
4,325-10
\ Т /к
3,96 • 10
3,82 • 10
3,17-10
2,41 • 10
1,308
1,60
2,16
3,24
4,10
Рассчитанные
значения
D Я-Ю«
А
смЦсек
0,945
0,700
0,500
0,277
0,167
* International Critical Tables, v, V, p, 22.
12*
179
-а;
0,5
V*
к
0 0,05 0,1 0,15
Концентраций, мол. доли
Рнс. 88. Зависимость
коэффициента дирфузии этанола
в водном растворе от
концентрации при 10° С:
1 —рассчитанная кривая; 2
—определено из опыта.
Hi
ш
1.5
%%%0,3
70Т
-/
г
Д
^■^
го"с
/•**.
/
2,9 3 3,1 3,2 3,3 J,4 3,5
Т
L -/О3, °>
'К
Рис. 89. Влияние температуры на
коэффициент диффузии маннита в
в водном растворе: ,
/ — коэффициент диффузии маннита; // —
вязкость воды; 1 — определено из опыта; 2 —
рассчитанная кривая.
В том же справочнике (т. V, стр. 70) помещены значения
коэффициентов диффузии этанола в его водных растворах при 10° С для концентраций,
больших 3,75 моль/л. Эти концентрации были переведены в мольные доли с
помощью данных о плотности растворов *. Расчетные и опытные данные
сравниваются на рис. 88.
Пример V-3. Коэффициент диффузии маннита в воде при 20° С (в
разбавленном растворе).** равен 0,56• 10-5 см2/сек. Определить коэффициенты
диффузии маинита при температурах до 70° С и сравнить с опытными
данными.
Решение. Согласно (V, 23) имеем: £ДЯф.=£вяз.. Вследствие того
что раствор разбавленный, зависимость вязкости воды от температуры
можно использовать для определения температурной зависимости D. В
соответствии с уравнениями (V, 18) и (V, 22) линия полулогарифмической
зависимости ц и D от 1/Г должна иметь наклоны, равные, но противоположные по
знаку. Соответственно на рис. 89 приведена зависимость вязкости воды от
1/Г по данным «Chemical Engeneers' Handbook». На график нанесено также
значение D при 20° С (обозначено на рисунке крестиком) и через эту точку
проведена кривая с наклоном, равным по величине, но противоположным по
знаку наклону кривой вязкости. Полученная зависимость дает значения D
при любой температуре и может быть сравнена с опытными данными. Так,
при 70° С рассчитанное значение Z>= 1,47 • Ю-5 см2/сек, опытное* —
1,56- Ю-5.
Можно использовать также вывод Уилки и Чэна о том, что TjDAB\\,B
сравнительно мало зависит от температуры. Поскольку раствор сильно
разбавленный, цв можно принять равной вязкости воды. При 20° С имеем:
Т __ 273 + 20 _,
DAR»
АВ^В
0,56- Ю-0- 1,005
:520-105
При 70° С для воды ц = 0,4061 спз и
D
273 + 70
520-105-0,4061
= \,62-\0~5 смУсек
* Chemical Engineers' Handbook, McGraw-Hill Book Company, Inc., New
York, 3rd ed., p. 188.
** International Critical Tables, v. V, p. 71.
180
ч
Коэффициенты диффузии электролитов
Разбавленные растворы. Диффузия электролитов
осложняется диссоциацией молекул на ионы. Измерения
электропроводности показали, что разные ионы имеют различную
подвижность, и, следовательно, можно предположить, что они будут
диффундировать с неодинаковой скоростью. Это привело бы,
однако, к высоким локальным концентрациям положительно или
отрицательно заряженных ионов и замедлению движения
быстро движущихся ионов под действием электростатических сил.
В действительности ионы диффундируют с одинаковыми
скоростями, и раствор остается электрически нейтральным.
Для полностью диссоциированных растворов Нернст31
показал, что при бесконечном разбавлении коэффициент диффузии
электролита следующим образом связан с подвижностью
ионов *:
_ R'TU+\J- 1 1 1 \
Дразб.- и+ + и_ (уг+рг) (V.31)
где U+ и U-—соответственно абсолютные подвижности
(скорости) катиона и аниона под действием силы
в 1 дину при бесконечном разбавлении, смХ
Хсек'1 • дин-1;
Z+ и Z~ — соответственно валентности катиона и
аниона;
/?' = 8,314 • 107—газовая постоянная, эрг • моль~1 • град~1;
^разб. — коэффициент диффузии при бесконечном
разбавлении, см2/сек;
Т — температура, °К.
Подвижности ионов можно получить из измерений
электропроводности.
В табл. 11 приведены найденные таким способом
значения подвижностей, пересчитанные из таблиц Партингто-
на32. Значения U, помещенные в таблице, равны величинам X
по Харнеду и Оуэну13, деленным на F2-107, где F = 96 500 —
число Фарадея.
Величины U, определенные при температуре tu для
получения их значений при другой температуре tz пересчитывают по
уравнению
Г2 + а(гг-г,)1
U'> 42-a(r2-r,)J
где а — температурный коэффициент.
Концентрированные растворы. Для концентрированных
растворов необходимо ввести поправку, в которую входит
Уравнение (V, 31) верно только при указанных ниже размерностях
входящих в него величин.
181
_ Т аб л и ца 11
Подвижности ионов (в см ■ сен ■ дии~ ) при бесконечном
разбавлении при 18° С
Катион
Li+
Na+
Ag+
К+
т1+
Rb+
Cs+
Н+
NH+
i/2 Be=+
i/2 Мп2+
V, Со2+
V2 Ni2 +
1/2 Fe*+
Ve pe8+
7s Cr*+
V, Mgs+
Vi zn2+
V, Cu2+
./2 Cd2+
./2 Sr2+
1/2 Ca2+
Vi Ba2 +
V. рь2+
Vi Ra2+
7s Al3+
7з La'+
7з Sm3+
7« Th4+
У+.10"
35,8
46,6
58,0
69,4
70,8
72,5
73,0
337
69,5
30,1
47,2
46,2
47,2
48,3
65,5
48,4
49,3
50,5
49,3
49,9
55,7
55,7
59,5
65,3
62,4
43,0
65,5
57,5
25,3
a-io"
265
244
229
217
215
214
212
154
222
—
—
—
—
—
—
256
254
245
247
247
239
240
239
—
—
—
Аиион
F-
cr
J-
Br"
SCN~
JO3-
C2H3°2~
СЮ3-
ВгОГ
jo^-
cio4-
N03-
OH_
Vi c2o^-
Vi so^-
Vl Cr°4~
1/2 CO2,-
«/« Fe (CN6)4
t/"-10"
50,1
70,4
71,6
72,7
60,9
36,5
37,6
59,2
51,1
51,6
68,8
66,4
187
67,6
73,5
77,4
64,5
102
a-io<
238
216
213
215
221
234
238
215
—
—
—
180
231
227
—
270
коэффициент активности, а также множитель, учитывающий
влияние концентрации на подвижности ионов:
/ mdlnv. \
Око„ц. = Оразб. \\ + dm ±) f (m) (V, 32)
где m — мольность раствора;
у — средний ионный коэффициент активности
электролита (отнесенный к мольности раствора);
Дш) — поправка, учитывающая влияние концентрации на
подвижности ионов.
182
При низких концентрациях член уравнения (V, 32), в
который входит коэффициент активности, имеет доминирующее
значение. Подробные сведения о предложенных методах
теоретического расчета f(m) приведены в работах Харнеда33, а также
Харнеда и Оуэна13. Показано, что рассмотренные
теоретические методы не применимы, особенно для
высококонцентрированных растворов. Гордоном предложено34 эмпирическое
уравнение *
/(т) = —= ё. (v,33)
cbvb »
где Vв — парциальный мольный объем воды в растворе,
см3/моль;
ев — количество молей воды в 1 см3 раствора;
{Ад — вязкость воды;
\i — вязкость раствора.
Это уравнение рекомендуется, в частности, для
электролитов, диссоциирующих на два одновалентных иона; для ионов
большей валентности оно менее пригодно.
Пример V-4. Определить коэффициент диффузии хлорида натрия в воде
в функции от концентрации при 18° С и сравнить с опытными данными.
Решение. Коэффициент диффузии при бесконечном разбавлении
определяется на основе подвижиостеи ионов, рассчитанных по уравнению (V, 31).
При 18° С (табл. 11)
для Na+ U+=46,6-10-17 см-сек'1-дин'1
для СГ U~ = 70,4 • Ю-17 см-сек'1 ■ дин'1.
Т = 273 +18 = 291° К
z+ = z- = i
п 8,314-107- 291 -46,6 -70,4 (КГ?7)2 /1 , 1\ , „„ ,„_5 ,.
Dpa36- " (46,6 + 70,4) -ИГ" (Т + Т) = 1ЛИ-10 СМ'СеК
Комбинируя уравнения (V, 32) и (V, 33), получают следующее
выражение для D с учетом поправки на концентрацию раствора:
\ dm ) cBVB \i
Средние коэффициенты активности \± при различной мольиости
подучены Гласстоном ** для температуры 25° С. Они приводятся к 18" С с
помощью зависимости
lg
Yi8°c LA I 1 1
"y^T = 4-576v 1273+18 ~~ 273 + 25
)
где_у —число ионов (для NaCl величина v=2);
La — относительная парциальная мольная энтальпия NaCl в растворе,
кал/моль.
* Уравнение (V, 33) верно только при указанных ниже размерностях
входящих в него величии.
' ** Thermodynamics for Chemists, D. Van Nostrand Company, Inc.,
Princeton, New York, 1947, p. 402.
1.0
9>
5: If*
О)
<3>
Я
\
V
1
1
^ж^~
г
о
о
о
0,5
'Р
&
Значения La приведены
Льисом и Рендаллом *.
Величина 1 + (тд In Y±/dm)
рассчитывается графически в виде
l + (d\gy±/d\nm) no наклону
кривой lgY± B Функции от
lgm; эту же величину можно
определить
дифференцированием эмпирических выражений
для у± в функции от т.
Значения св для различных
мольных концентраций
определялись из плотностей растворов
по справочным данным **. Те
же данные использовались для
расчета парциальных мольных
объемов в Vв по методу,
описанному Льюисом и Рендаллом.
Относительные вязкости ц/цв для растворов NaCl при 18° С приведены
в «International Critikal Tables» (т. V, стр. 15). Результаты расчетов
представлены ниже.
'Концентрация ( мольность)0'5'
Рис. 90. Зависимость коэффициента
диффузии NaCl в воде от концентрации при
18° С:
1 — рассчитанная кривая; 2 — определено из опыта.
Мольность NaCl
m
0,001
0,005
0,01
0,02
0,05
0,1
0,2
0,5
1,0
2,0
3,0
и
X
а
а
-Н
0,966
0,929
0,904
0,875
0,823
0,778
0,732
0,679
0,656
0,670
0,719
и
СО
= ч
а, о
5 *
—5
—12
—30
-122
—332
—561
—738
и
3
ОО
X
а.
а
~Н
0,966
0,929
0,904
0,875
0,823
0,778
0,732
0,676
0,651
0,662
0,708
■н
¥-
•а
-
d log m
0,979
0,968
0,958
0,943
0,922
0,915
0,915
0,915
0,995
1,080
1,1
74
ем
к
Ч
О
Ч
CJ
0,0555
0,0555
0,0555
0,0555
0,0554
0,0553
0,0552
0,0549
0,0544
0,0533
0,0524
■О
О
18,05
18,05
18,05
18,05
18,05
18,05
18,05
18,03
18,00
17,95
17,91
*l*
1,00
1,00
1,00
0,998
0,995
0,992
0,983
0,962
0,922
0,838
0,753
О)
О
Я
н
О)
D"
и
п)
а.
1,327
1,313
1,300
1,278
1,244
1,230
1,222
1,205
1,265
1,280
1,278
Полученные данные сравниваются с экспериментальными (рис. 90),
приведенными в «International Critical Tables» (v. V, p. 67).
* Thermodynamics and the Free Energy of Chemical Substances, McGraw-
Hill Book Company, Inc., New York, 1923, p. 92.
** Chemical Engenders' Handbook, McGraw-Hill Book Company, Inc., New
York, 1950,
184
На рис. 91 показаны результаты
аналогичных расчетов для растворов
электролитов с концентрацией от 0,01
до 0,2 н. [т. е. для области
концентраций, в которой поправочный член
f(m) практически равен единице] и
температурой от 10 до 18° С. Все
точки на графике относятся к различным
электролитам. При указанных
концентрациях опытные данные хорошо
согласуются с расчетными.
Коэффициент диффузии слабого
электролита, например раствора
уксусной кислоты в воде, представляет
собой среднюю величину,
отражающую влияние индивидуальных ионов
и недиссоциированных молекул 35:
RT I д\пу,\
1
2.5"!
1.5
Q5\
о
'Of 1,5 2 2,5 3
Опытное значение
V:109r мг/сек
Рис. 91. Сравнение опытных
значений коэффициентов
диффузии в водных
растворах сильных электролитов
с рассчитанными.
(1/U),
'ср.
В этом выражении средняя подвижность ионов определяется
по уравнению
(±\ _ a(l/U+ + l/U~) + q~a)R'T/Da
\UJcp. 1+а
(V, 35)
где а — степень диссоциации;
Dtt — коэффициент диффузии недиссоциированного
электролита.
При бесконечном разбавлении, когда величина а, а также
поправка на концентрацию раствора равны единице, уравнение
(V, 34) приводится к уравнению Нернста [уравнение (V,31)].
При высоких концентрациях, когда а стремится к нулю,
уравнение (V, 34) переходит в уравнение (V, 32), сходное с
уравнением для неэлектролитов.
Простейшие случаи
молекулярной диффузии
Уравнения (V, 5), (V, 9) и (V, 10) можно проинтегрировать,
если диффузия протекает как в стационарных, так и в
нестационарных условиях. Для большей части практически важных
случаев методы интегрирования указанных выше уравнений
разработаны довольно подробно4,6. Ниже для иллюстрации
приведено несколько простых примеров.
Установившаяся одномерная диффузия в неподвижной
жидкости. В стационарных условиях величины NA и NB постоянны.
189
Если принять, что величины DA и с также постоянны, то
интегрирование уравнения (V, 5) дает:
Принимая концентрацию на уровне Z\ равной cAl, а на
уровне z2 равной Са2 и обозначая сА/с=хА; с=(р/М)ср_ и гг—Zi = z,
из уравнения (V, 36) получим:
^+"*-—'» NA,(NA + NB)-cAjc <V'37>
или
NA DA(P/M)cp NA/(NA + NB)-Xb2
NA—N-+W; 'г ЫМЖМа + Мв)-Ха- <V'38>
Величины NA и NB рассматриваются как положительные,
если диффузия направлена от Z\ к г2. Зависимость между NA
и Nb определяется свойствами системы. Например, если
разбавленный раствор уксусной кислоты (А) в воде (В)
контактирует с бензолом, то только уксусная кислота переходит в
бензол, и поэтому величина NA положительна в направлении к
поверхности раздела фаз, a NB=0- Возможны и другие
зависимости между NA и. Ns (см. пример V-6).
Подобно тому как было принято для системы
разбавленный водный раствор уксусной кислоты — бензол, величину NB
на практике часто принимают равной нулю. В этом случае
уравнение (V, 38) приводится к виду:
Учитывая, что
ха, + хв, = ха2 + хв, = l>° (V. 40)
уравнение (V, 39) можно записать в виде:
Д,(р/а.> (^,-,„)
ZXBM
где
у — fii~XBl
BM~\axBjxB; (V.42)
иогоПрТтвРорГтолОЩПиРноТ2'Ь10-зТСрТс ЛГ" ЭТЗН0Ла ЧеРез пленкУ в°Д"
нола 0,74-10-9 м^сек. ' )ш КоэФФиВДент диффузии эта-
Решение. При 14,0 вес.% этанола мольная доля его составляет»
14,0/46,05
ХА, ~ 14,0/46,05 + 86,0/18,02 ~ '
При хА =0,060 мольная доля для воды хв =0,94; средний молекулир-
ный вес /И =19,7 и р/М=976/19,7=49,6 кмоль/м3.
Аналогично 9,6 вес.% этанола соответствует хА =0,040; хв =0,960;
/И=19,2 и p/Af=51,l. Отсюда имеем:
0,960-0,940
хвм~ In(0,960/0,940) -и,уои
,„,,,, 51,1+49,6 -п.
(P/^f)cp. = к = 50,4
2
г = 2.10~3ж.
После подстановки этих значений в уравнение (V, 41) получаем:
NA = 3,92 • 10~7 кмоль ■ м~2 ■ сек'1
Пример V-6. Капля метилэтилкетона (МЭК), насыщенная водой
(90,0 вес.% МЭК и 10,0 вес.% воды), диаметром 0,5-Ю-2 м растворяется
в большом объеме воды при 25°С (вода неподвижна). Концентрация МЭК
в воде у поверхности капли равна растворимости МЭК в воде (24,4 вес.%
МЭК; плотность 962 кг/м3). Определить скорость растворения капли.
Решение. Считаем каплю сферической. Диффузия происходит в
радиальном направлении от поверхности капли наружу; величина поверхности,
через которую происходит диффузия, возрастает пропорционально квадрату
расстояния от капли. Если скорости растворения компонентов капли
обозначить Qa и Qb молей в единицу времени и считать их постоянными,
соответствующие потоки на расстоянии г от центра капли составят:
N. = -rA- ЛГ„ = -р^з- (V.43)
А 4яг2 в 4яг2 v
и уравнение (V, 5) примет вид:
Qa _(Qa + Qb)-ca d ±a_
4nr2 4ncr2 A dr
Интегрирование при постоянных DA и с дает:
['А-Т^^\*ЯА+ЩРа°Ае'-*<>°* (V.44)
Обозначая концентрацию на расстоянии Г\ через сА, на расстоянии гг —
Слг; сА1С^хА; с=(р//И)Ср. и подставляя уравнение (V, 43) в уравнение
(V, 44), получаем:
А- I А' ^(Р/Л*У ^ щ "*<№* +Nb,)-xa2
"*+"* rj_ri 1П NAjiNAi+NB)-xAi V'45)
Пусть МЭК — компонент А, вода — компонент В. Коэффициент
диффузии DA, рассчитанный по уравнению (V, 26), равен 0,975-№9 м2/сек.
Мольная доли компонента А в капле составляет:
9°-0/72-0 = 0692
90,0/72,0 +10,0/18,02 и,°
187
Соответственно мольная доля компонента В в капле составляет
1—0,692 = 0,308. Капля насыщена компонентом В. Следовательно, капля не
может терять компонент А без уменьшения в ней содержания компонента В
в отношении А/В = NA/NB = 0,692/0,308 нли NA/(NA+NB) =0,692. Оба
вещества диффундируют в одном направлении.
В воде у поверхности капли г=0,25-10"2 ж; хА, = (24,4/72,0)/(24,4/72,0+
+75,6/18,02) =0,0748, что соответствует среднему молекулярному весу 22,06
н р/М, равному 962/22,06=43,5 кмоль/м*. В объеме воды хАг =0 и р/М=
= 10а/18,02=55,5 кмоль/м3. Следовательно, величина (р/Л1)ср.= (43,5+55,5)/2=
=49,5. Если считать величину г2 значительно превосходящей ги то
г2/Гх _ г2 1
r2 — r, r2-rl rx
и согласно уравнению (V, 45), получаем: *
х, . ., 0,975-10~9.49,5 . 0,692 — 0 „,„, _„
NA, + NB, = ~ . -а 'П — = 2,19-10 6лгжолй-л-2-Сйл:-1
0,25-10 2 0,692 — 0,0748
Так как поверхность капли равна я (0,5 ■ 10"2)2=0 785 • Ю"4 ж2, скорость
растворения капли Qa + Qb = 2,19 • 10'6(0,785 • 10т1) = 1,72 ■ Ю"10 кмоль/сек; из
этого количества 69,2 мол.% приходится на долю МЭК и 30,8 мол.% — на
долю воды. Если учесть плотность капли, это соответствует 0,018% объема
капли в секунду. Поскольку по мере растворения капля становится меньше,
полученный результат выражает лишь мгновенную скорость растворения для
капли диаметром 0,5-Ю-2 ж. Капля легче окружающей среды, и в
действительности она будет подниматься с определенной скоростью. Поэтому
следовало бы учесть соответствующее уменьшение величины г2 — Г\, что можно
сделать при помощи коэффициента массопередачи (см. пример V-7).
В большей части экстракционных процессов вследствие
изменения концентрации распределяемого вещества изменяется
взаимная растворимость компонентов. Поэтому одновременно
происходит диффузия нескольких компонентов. В подобных
случаях коэффициент диффузии каждого компонента связан (по
крайней мере теоретически) сложной зависимостью с
коэффициентами диффузии для бинарных систем и величинами
потоков 4. Однако сведения о значениях коэффициентов диффузии в
жидкостях и о зависимостях этих коэффициентов от
концентраций ограничены, а имеющиеся уравнения для жидкостей
недостаточно проверены. По указанным причинам данный вопрос
в дальнейшем рассматриваться не будет.
В случае разбавленных растворов нескольких
диффундирующих компонентов можно использовать значения коэффициента
диффузии для отдельных бинарных систем, входящих в
многокомпонентную смесь.
КОНВЕКТИВНАЯ ДИФФУЗИЯ
Существуют два вида движения жидкостей —ламинарное и
турбулентное. При ламинарном движении тонкие слои или
пленки жидкости скользят относительно друг друга, причем сосед-
188
ние слои не перемешиваются. В условиях турбулентного потока
на основное движение жидкости накладываются дополнительные
скорости, что приводит к взаимному перемешиванию слоев
движущейся жидкости.
Диффузия в ламинарном потоке. При ламинарном движении,
несмотря на отсутствие макроскопического перемешивания
соседних слоев жидкости, в случае наличия разностей
концентраций, происходит молекулярная диффузия, приводящая к
поперечному молекулярному перемещению компонентов в слоях и
между слоями жидкости. Если уравнение движения жидкости
известно, уравнение (V, 11) можно проинтегрировать, чтобы
получить общую зависимость для скорости массопереноса.
Свойства жидкости (вязкость и плотность) зависят от
концентрации ее компонентов; соответственно дифференциальные
уравнения диффузии и движения являются взаимозависимыми,
что затрудняет их решение. Поэтому всегда допускают, что
свойства жидкости постоянны. Путем решения уравнения
диффузии в ламинарном потоке при таком допущении удалось
получить ценные зависимости для изучения массопереноса в
движущейся капле. Некоторые из этих решений будут
рассмотрены ниже.
Диффузия в турбулентном потоке. При турбулентном
движении вторичные скорости, накладывающиеся на основной
поток, изменяются во времени и в пространстве. Распределение
этих скоростей, характеризующих турбулентность потока,
хаотичное; поэтому для изучения турбулентного движения должны
применяться статистические методы. Наиболее подробно
разработаны теории, основанные на некоторых упрощенных моделях
механизма турбулентного движения. Например, допускается, что
вихри в турбулентно движущейся жидкости перемещаются из
одной точки потока в другую, где они исчезают, смешиваясь с
потоком; возникновение и перемещение вихрей носит
неупорядоченный характер. При этом вихри переносят свойства
жидкости из точки возникновения вихрей в точку, где они
разрушаются. Такая гипотеза дает наглядное представление о том,
каким образом в турбулентном потоке очень быстро
усредняются концентрации растворенного вещества.
Вихрь, движущийся из одной точки турбулентного потока в
другую, может обмениваться растворенным веществом с вихрем,
движущимся в противоположном направлении. Если в разных
точках жидкости концентрация растворенного вещества
различна, обмен вихрями приводит к переносу вещества, скорость
которого пропорциональна разности концентраций и объемной
скорости обмена. Последняя выражается произведением
линейной скорости на единицу площади. Если вихри перемещаются
с определенной скоростью на данное расстояние, поток вещества,
189
переносимого вихрями, можно выразить с помощью градиента
концентраций в форме уравнения (V,l).
где /, —турбулентный диффузионный поток;
е — коэффициент вихревой, или турбулентной, диффузии.
Наряду с турбулентным переносом осуществляется перенос
в результате молекулярной диффузии. При этом полный поток
распределяемого компонента А можно определить по уравнению
■^г = -(^ + в)-ЗГ- <v'47>
где (DA + e)—полный коэффициент диффузии, учитывающий
влияние как молекулярного, так и турбулентного переноса.
В отличие от D, коэффициент турбулентной диффузии е не
зависит от физических свойств жидкости, если последние не
связаны с интенсивностью турбулентного движения.
Пока что имеется мало данных о численных значениях е в
условиях развитого турбулентного движения. Однако известно,
что величина е на несколько порядков больше величины D.
В таких условиях поток распределяемого компонента в
основном не зависит от D. В жидкостях, движущихся с относительно
малой скоростью, можно, наоборот, пренебречь влиянием е;
наконец, в покоящихся жидкостях перенос вещества определяется
только величиной D.
В турбулентном потоке, имеющем фиксированную границу,
например при турбулентном движении жидкости в трубе,
известно, что скорость непосредственно на внутренней поверхности
трубы равна нулю. В слое, прилегающем к стенке трубы,
преобладает ламинарное движение, и лишь в ядре потока движение
турбулентное. Если растворенное вещество диффундирует из
ядра потока к стенке трубы, на перенос на этом пути влияют
в различной мере величины D и е и необходимо учитывать
различное относительное значение последних.
Скорость массопереноса можно рассчитать, если известен
закон распределения скоростей и может быть постулирована
закономерность скорости уменьшения е в радиальном
направлении от центра к стенке трубы. Наиболее плодотворными
оказались те расчеты для турбулентного потока в трубе 36>37, в
которых принималось, что величина е обращается в нуль только
на стенке трубы 38.
Таким образом, существует постепенный переход от области,
в которой преобладающую роль играет турбулентная диффузия,
к области у стенки трубы, где преобладает молекулярный
перенос. В системах жидкость — жидкость, в отличие от систем
твердое тело — жидкость, граница раздела фаз подвижная.
190
Даже в простейшем случае, когда две несмешивающиеся
жидкости движутся в трубопроводе одна над другой, скорость на
границе раздела этих жидкостей обычно не равна нулю.
Обстановка усложняется, если между находящимися в контакте
жидкостями происходит диффузия. Подобная сложная
гидродинамическая обстановка создается при движении капель в
сплошной жидкой среде. Опубликованный обзор работ по
турбулентной диффузии39 позволяет заключить, что пока имеется
мало данных, пригодных для непосредственного использования
в расчетах.
Коэффициенты массоотдачи. В тех случаях, когда
распределение скоростей и влияние турбулентной диффузии неизвестны
и, следовательно, скорость переноса массы нельзя
непосредственно рассчитать, ее определяют опытным путем. Полученные
опытные данные стремятся скоррелировать, обычно используя
для этого коэффициенты массоотдачи k, которые находят из
уравнений
NA = к АсА = kccp ЬхА = к (p/Af )cp. Л*л (V, 48)
Величины АсА или АхА представляют собой разности
концентраций на пути, проходимом диффундирующими частицами.
Коэффициент массоотдачи k учитывает влияние на скорость
переноса режима движения жидкости, а также коэффициентов
молекулярной и турбулентной диффузии. В настоящее время
существуют две основные точки зрения iHa механизм массопере-
дачи: пленочная теория и теория проникновения,
или теория обновления поверхности.
Пленочная теория. Эта наиболее простая теория массопере-
дачи представляет собой развитие идей, ранее выдвинутых
Льюисом40. Пленочная теория допускает, что в движущейся
жидкости имеются ламинарные и турбулентные области;
скорость массопередачи определяется скоростью молекулярной
диффузии, а влияние турбулентности проявляется в увеличении
потока вещества N'а и может быть охарактеризовано величиной
диффузионного пути — «эффективной толщиной пленки».
В уравнении (V,38), полученном для переноса в стационарных
условиях, расстояние z приобретает, таким образом, значение
«эффективной» толщины, меньшей, чем действительное
расстояние, на котором наблюдается падение концентраций, но
оказывающей сопротивление молекулярной диффузии, равное общему
сопротивлению. Обозначив в уравнении (V,38) DA(p/M)cp/z = F,
получим
дг =F( "л \,п^/(^+^)-^
"а г[ма + мвУ*ма/(ка + мв)-Ха ,<v,49)
где F — коэффициент массопереноса по Кольбурну — Дрю41.
181
Сравнивая уравнения (V, 41), (V, 48) и (V, 49), находим:
F NA 1
k = "ФЩГ 'NA + NB- [NaKNa + Nb)-хА]м (V'50)
где [NAl(NA + NB)—хА]м — среднее логарифмическое значение
этих величин для положений 1 и 2.
Из уравнения (V, 49) можно заключить, что коэффициент F
меньше зависит от уровней концентраций, чем k, хотя
вследствие зависимости DA от концентрации значение F также в
некоторой степени связано с величиной концентрации. Расчет с
помощью коэффициента k может привести к затруднениям в
случае некоторых соотношений между потоками N А и NB.
Несмотря на это, в расчетной практике почти всегда применяют
коэффициент k. При высоких скоростях массопередачи, когда
потоки N а и NB оказывают существенное влияние на величину
членов, выражающих скорость в уравнении (V, 11), необходимо
вводить в величину F дополнительные поправки3'4. Однако
небольшая точность опытных данных об экстракции не позволяет
пока надежно уточнять величину F.
Допустим, что поток вещества или коэффициент диффузии
не влияет на «эффективную» толщину г. Тогда можно считать,
что для различных систем при прочих равных условиях
величина k должна линейно зависеть от D, а величина z будет
изменяться со степенью турбулентности, выражаемой, например,
величиной критерия Рейнольдса. Вывод о пропорциональности &
величине D противоречит практическим данным, согласно
которым k зависит от D в меньшей степени- Вместе с тем, следует
отметить, что многие опытные данные получены в
несопоставимых условиях и обработаны в виде эмпирических
зависимостей, затрудняющих выяснение истинной зависимости k от D.
Теория проникновения. Теория проникновения впервые
предложена Хигби42 специально для определения скорости
растворения газового пузыря, поднимающегося в жидкости, но
основной принцип, положенный в ее основу, имеет общее значение.
Если распределяемый компонент А переходит через
поверхность раздела фаз в неподвижную жидкость, в которой
начальная концентрация компонента А равна сАг, проникновение А
в жидкость от границы раздела (на которой концентрация А
постоянна и равна сА,) осуществляется в результате
молекулярной диффузии. При незначительной продолжительности
контакта (экспозиции) глубина проникновения распределяемого
компонента невелика, поэтому перенос вещества можно
рассматривать как неустановившийся процесс. Интегрированием
уравнения (V, 9) можно получить мгновенную скорость
массопередачи:
192
При непрерывном процессе массопередачи описанный выше
механизм взаимодействия фаз повторяется многократно, причем
принимают, что в промежутках между последовательными
контактами происходит полное перемешивание жидкости.
Уравнение (V, 51) применимо также для жидкостей, движущихся ла-
минарно и параллельно границе раздела фаз, при условии
настолько малой глубины проникновения, что влиянием изменения
скорости движения жидкости можно пренебречь. Применяя
уравнение (V, 51) к определенным элементарным процессам,
получают уравнение для осредненного потока вещества NA в
форме, аналогичной уравнению (V, 51), но с коэффициентом
пропорциональности, отличным от я, под знаком радикала.
Например, для случая, рассмотренного Хигби, можно принять время
контакта жидкости и газового пузыря равным времени всплыва-
ния пузыря на расстояние, эквивалентное его диаметру. Тогда
средняя скорость переноса определяется по уравнению
^ = "/^T(<4-4) (V. 52)
Теория обновления поверхности. Описанные выше теории
были развиты Данквертсом43, предложившим теорию
обновления поверхности для описания переноса в турбулентном потоке
жидкости. Данквертс допускает, что вихри непрерывно
переносят элементарные объемы жидкости из ядра потока с
постоянной концентрацией сд к поверхности раздела фаз. Здесь вихри
задерживаются весьма короткий промежуток времени, в
течение которого распределяемое вещество проникает в жидкость
в стационарных условиях вследствие молекулярной диффузии
(размеры вихрей таковы, что глубина проникновения каждого
вихря очень мала). По истечении короткого времени пребывания
(времени экспозиции) данный вихрь уносится в основной объем
жидкости (ядро потока) и замещается новым вихрем,
омывающим поверхность раздела. При этом
NA = V^A1'(CA1-^) (V.53)
где s — средняя скорость обновления поверхности вихрями.
Данквертс показал, что такой механизм совместим с
допущением о преобладании переноса турбулентной диффузией в ядре
потока.
Из сравнения уравнений (V, 48) и (V, 53) следует, что в
данном случае k зависит от D в степени 0,5. В действительности
показатель степени при D изменяется от 0 (см., например,
работу Льюиса 44) до 2/3 и более. Таким образом, ни пленочная
теория, ни теория проникновения не могут претендовать на
13 Р. Е. Трейбал
193
точное описание процесса. Тур и Марчелло 45 предложили
комбинированную теорию, согласно которой в зависимости от
продолжительности контакта показатель степени при D изменяется
от 0,5 (короткое время контакта) до 1,0 (длительное время
контакта). При изменении показателей степени при D в указанных
пределах теория Тура и Марчелло может лучше объяснить
экспериментальные данные, чем пленочная теория и теория
проникновения. М. X. Кишиневский46 разработал вариант
теории обновления поверхности без учета влияния молекулярной
диффузии.
Критерии подобия. В любом случае коэффициент k зависит
от скорости молекулярной диффузии D и гидродинамических
факторов, влияющих на движения жидкости. Обычно в
эмпирических корреляциях и в теоретических исследованиях связь
между k и указанными параметрами выражают в виде
зависимости между безразмерными комплексами (критериями
подобия), в которые входят эти величины. Ниже приводятся
важнейшие критерии подобия, используемые в расчетных
зависимостях по массопередаче.
Критерий Шервуда Sh = kL/D (где L некоторый
определяющий линейный размер) единственный безразмерный комплекс,
в который входит коэффициент массоотдачи k. Этот критерий
можно рассматривать как отношение размера L к
«эффективной толщине пленки»; он является диффузионным аналогом
теплового критерия Нуссельта.
Критерий Рейнольдса Re = Lvp/\x, величина которого
определяет степень турбулентности, выражает отношение
инерционных сил к силам вязкости и имеет большое значение для
описания процессов, протекающих в условиях вынужденной
конвекции.
Критерий Грасгофа Gr = gL3Ap/p(p/[x)2 применяется при
описании процессов, происходящих в условиях естественной
конвекции.
Критерий Фруда, или отношение инерционных и
гравитационных сил Fr = v2/Lg, применяется в условиях, когда возникает
поверхностное волнообразование и т. п.
Критерий Вебера We=pv2L/a выражает отношение сил
инерции к силам поверхностного (межфазового) натяжения;
используется, в частности, при описании процессов, в которых
осуществляется контактирование несмешивающихся фаз.
Критерий Шмидта Sc = n/pD— диффузионный аналог
теплового критерия Прандтля; выражает отношение скоростей
переноса количества движения (ц/р) и массы (D).
Критерий Пекле Pe=Lv/D представляет собой произведение
критериев Рейнольдса и Шмидта.
Для описания процессов используют также некоторые другие
критерии подобия, которые будут рассмотрены в дальнейшем.
194
МАССОПЕРЕДАЧА ЧЕРЕЗ МЕЖФАЗОВУЮ
ПОВЕРХНОСТЬ
При контактировании исходного раствора с несмешиваю-
щимся избирательным растворителем (экстрагентом) для
экстракции распределяемого компонента последний переходит из
исходного раствора в растворитель через границу раздела фаз,
или межфазовую поверхность. Процесс перехода продолжается
до тех пор, пока химический потенциал или активность
распределяемого компонента (отнесенная к некоторому стандартному
состоянию) не станет одинаковым в каждой фазе, т. е. пока не
установится состояние равновесия.
В главе II было отмечено, что при равновесии
экстрагируемый компонент равномерно распределен в пределах данной
фазы, но концентрации его в фазах неодинаковы.
Соответственно растворенное вещество может переходить из фазы с
меньшей концентрацией в фазу с большей концентрацией, если
этот переход осуществляется в направлении падения
химического потенциала или активности. В процессе массопередачи
вследствие разности концентраций между основной массой
исходного раствора (рафината) и границей раздела фаз
распределяемый компонент движется к межфазовой поверхности. Под
действием разности концентраций между межфазовой
поверхностью и фазой растворителя (экстракта) распределяемое
вещество переходит далее от поверхности в экстракт. При этом
на границе раздела фаз нарушается непрерывность изменения
концентрации.
На рис. 92 показано изменение концентраций в процессе
массопередачи (принято, что межфазовая поверхность
перпендикулярна к плоскости графика, на котором она изображается
линией). Жидкие фазы обозначены буквами Е и R; с —
концентрация распределяемого компонента. Фазы обычно движутся
турбулентно, так что в объеме жидкостей в результате
турбулентной диффузии экстрагируемый компонент распределен
равномерно. Если предположить, что вблизи поверхности раздела
фаз поток ламинарен (это нельзя считать обязательным во всех
случаях*), основное изменение концентраций должно
наблюдаться именно в этой области, так как величина D значительно
меньше е.
Обозначим среднюю концентрацию распределяемого
компонента в объеме фазы R, через cR, на поверхности раздела фаз —
через cRi\ в фазе Е— соответственно через сЕ и cEi. По
причинам, указанным выше, нельзя выразить движущую силу
* Поверхность раздела фаз в системе жидкость — жидкость может быть
в некоторых случаях турбулентной; см., например, работу Льюиса44,
который в опытах на аппарате, изображенном на рис. 96 (стр. 200), нашел, что
k не зависит от D.
13*
195
Направление
нассопереда чи
Р
I
^§
§|
I
.
Фаза Я
^~"^\
v;
\Ei
V^ _Се
ФазаЕ
Поверхность
раздела
Расстояние
Рис. 92. Градиенты
концентраций вблизи поверхности раздела
жидких фаз.
процесса массопередачи в виде
разности cR—сЕ. Скорость
массопередачи внутри каждой фазы
может быть выражена в форме
уравнения (V, 48). Если
рассматривать установившийся процесс,
скорость подвода вещества к
поверхности раздела фаз в фазе R
должна быть равна скорости
переноса вещества от поверхности
раздела фаз в фазу Е:
NA = kR (CR - CRi) = kE (СЕ~Се) (V' 54>
где kR и kE— коэффициенты
массопередачи в соответствующих
фазах (коэффициенты массоот-
дачи).
Двухпленочная теория
массопередачи. Вследствие того, что
основное изменение концентраций происходит в пограничных
слоях, имеющих чрезвычайно малую толщину, опытным путем
невозможно определить значение концентраций cEi и сш на
поверхности раздела фаз. Практически имеется возможность
находить лишь значения концентраций сЕ и cR в объеме каждой
фазы. Уитман47,48 сделал предположение, что концентрации
Cm и cEi на поверхности раздела фаз равны равновесным и что
общее сопротивление массопередаче определяется на основе
аддитивности фазовых сопротивлений, которые являются
величинами, обратными коэффициентам массопередачи k в фазах.
Равновесие на поверхности раздела фаз означает равенство
химических потенциалов, вследствие чего поверхность раздела не
оказывает сопротивления переходу вещества из фазы в фазу.
Эти положения лежат в основе так называемой двухпленоч-
ной теории, которой больше соответствует название «теория
аддитивности сопротивлений». Хотя эта концепция
первоначально была предложена применительно к пленочной теории,
она используется в теории проникновения и других, а также
при изучении и анализе коэффициентов k. Графическая
интерпретация этой теории приведена на рис. 93. Здесь показана
кривая равновесия и нанесена точка Р, выражающая составы
жидких фаз; координатами точки Р являются объемные
концентрации распределяемого компонента в фазах; координатами
точки Т на кривой равновесия — равновесные концентрации на
поверхности фаз. Линия ТР имеет наклон, равный
Асс
СЕ1-СЕ
_ R
Дсг
CRi-°R
(V, 55)
196
Общие
коэффициенты массопередачи. Если
известна кривая
равновесия (рис. 93),
то концентрацию с*Е
распределяемого
компонента в фазе Е,
равновесную с
концентрацией сд, можно
использовать для измерения
cR. Тогда скорость
массопередачи из объема
фазы R в объем фазы
Е можно выразить
уравнением
na = Ke{c*e-ce):
Кривая равновесия
kj
гй? Я? Р
I c
■KEhc0E
Cr
Концентрация распределяемого
компонента в фазе R
Рис. 93. Графическая интерпретация теории
аддитивности сопротивлений.
(V.56)
Соответственно cR
можно использовать
как меру сЕ:
na = Kr{cr-Cr) = KRbc0R
(V.57)
В этих уравнениях КЕ и Kr являются общими
коэффициентами массопередачи в двухфазных системах. Концентрации
гсены также на графике рис. 92.
т" — тангенсы углов наклона линий ST и TU на
(V, 58)
cR и
Если т' и
рис. 93, то
Асоя = Дся + /й"Дс/?
N. N. m"N,
А_ А. Л.
КЕ~ kE^
R
1
К,
1
(V.59)
(V.60)
Аналогично
К,
m'k.
(V.61)
Уравнения (V, 60) и (V, 61) выражают принцип
аддитивности сопротивлений Уитмана и показывают, что общие
коэффициенты массопередачи можно получить из частных (фазовых)
коэффициентов, или коэффициентов массоотдачи, если
последние известны,
197
Когда движущую силу Процесса массопередачи выражают
через разность активностей, скорость массопередачи
определяется по уравнению
NA = kRa (*R ~ аш) = kBa (?Bl ~ *в) <V' 62)
где а —активность диффундирующего (распределяемого)
компонента.
При равновесии на поверхности раздела фаз aRi = aEi.
Соответственно скорость массопередачи
NA = KaiaR-aE) (V,63)
и общий коэффициент массопередачи
1 J—Ьтг— (V.64)
К.
Ra
кЕа
Если распределяемый компонент предпочтительно
растворяется в фазе Е, то т' становится очень большим, a Kr
примерно равным kR. В этом случае основное сопротивление массопе-
редаче сосредоточено к фазе R. Графическая интерпретация
данного варианта процесса массопередачи показана на рис. 94,
из которого видно, что Дсок—Асн-
Аналогично, если наклон т" очень мал, то при равновесии
распределяемый компонент предпочтительно растворяется в фазе
/?; общий коэффициент КЕ становится примерно равным kE, и
скорость массопередачи контролируется сопротивлением в
фазе Е (рис. 95). В некоторых случаях линия равновесия на
рис. 93 является прямой и т'=т" = т; при этом соотношение
между общими коэффициентами
массопередачи упрощается:
KR = mKE (V.65)
Обоснованность правила
аддитивности. При выводе уравнений (V, 55) —
(V, 65) было постулировано, что на
^Кривая
равновесия
Концентрация распределяемого
компонента д фазе Я
Рис. 94. Схема массопередачи для
случая, когда основное
сопротивление сосредоточено в фазе R.
4i
о. S> S
III
ill
м
Кривая равновесия
Cri Cr
Концентрация распределяемое/)
/(оппонента S фазе я
Рис. 95. Схема массопередачи для
случая, когда основное сопротивление
сосредоточено в фазе Е.
198
поверхности раздела фаз устанавливается статическое
равновесие и что сопротивления массопередаче в фазах
(выраженные величинами, обратными k) аддитивны. Поэтому весьма
важно установить обоснованность принятых допущений и
пределы их применимости.
Для систем газ — жидкость было показано 49, что массопере-
дача может приводить к образованию вблизи межфазной
поверхности слоев с неравновесными составами. Однако скорость
массопередачи при этом должна быть очень высока, вследствие
чего данным эффектом для систем жидкость — жидкость, в
которых скорость диффузии мала, вероятно, можно пренебречь.
Уорд и Брукс50 оптическим методом измерили концентрацию
низших жирных кислот на поверхности раздела фаз в системе
вода — жирная кислота — толуол (жирная кислота —
распределяемый компонент). Они нашли, что отношение концентраций
распределяемого компонента вблизи поверхности раздела в
обеих фазах очень точно соответствует равновесному. Не было
обнаружено наличия энергетического барьера переходу
вещества через поверхность раздела; если такой барьер и
существует, его величина, по-видимому, настолько мала, что не
поддается измерению.
С другой стороны, Дриккамер косвенно показал, что при
определенных условиях может существовать сопротивление
поверхности раздела переходу вещества из фазы в фазу. Если
предположить, что концентрации на поверхности раздела не
равновесны, коэффициент массопередачи на поверхности kt (и
соответствующее ему сопротивление 1/fej) можно определить из
уравнения
NA = ki(mCRi-CEl) (V.68)
Интегрируя уравнение Фика для двухфазной системы при
этом условии, получим распределение концентраций в жидких
фазах во времени. Распределение концентраций различно в
зависимости от величины kt SI>52, которая стремится к
бесконечности, если сопротивление поверхности отсутствует. Измерением
концентраций в функции от времени в диффузионной ячейке
обнаружено53-54 наличие существенных сопротивлений
поверхности раздела при переходе двуокиси серы через поверхность
раздела в -системе S02 — гептан, серной кислоты — в системе
фенол — вода и серы через различные поверхности раздела.
Высказано предположение, что у полярных веществ
молекулы должны быть определенным образом ориентированы и при
переходе через поверхность раздела могут задерживаться те
молекулы, ориентация которых неблагоприятна для такого
перехода. Возможно, что этим явлением обусловлена способность
образовывать водородные связи. Переход распределяемого
компонента через поверхность раздела может сопровождаться
199
предварительной абсорбцией его на поверхности раздела или
хемосорбцией, если сопротивление на поверхности очень
велико. В этом случае для перехода через поверхность раздела
молекулы должны обладать определенной величиной энергии
активации.
Гордон и Шервуд55 изучали массопередачу в ячейке с
перемешиванием, показанной на рис. 96. Скорость перемешивания
в ячейке оказалась достаточно высокой, чтобы исключить чисто
молекулярный перенос, и недостаточной, чтобы вызвать
возмущение поверхности раздела фаз, величина которой была
известной и постоянной.
Коэффициенты массопередачи для каждой фазы определяли
при постоянной скорости перемешивания по методу,
предложенному Кольбурном и Уэлшем56. По этому методу одна из
жидких фаз (изобутанол) заранее насыщалась другой фазой
(водой) и обе фазы приводились в соприкосновение. Поскольку
разность концентраций существовала в данных условиях
только в одной фазе (водной), измеренное значение скорости
массопередачи было равным скорости массопередачи в этой фазе,
характеризуемой величиной k. Аналогично, применяя чистый
изобутанол и насыщенную им воду, получали коэффициенты
массоотдачи для фазы изобутанола.
При исследовании скорости массопередачи различных
органических кислот и оснований из одной фазы (насыщенной) в
другую было показано, что опытные коэффициенты
массопередачи можно получить расчетом из частных коэффициентов
массопередачи изобутанола в воде и воды в изобутаноле (с учетом
поправки на величину коэффициента диффузии при условии, что
&ocD0'5). Этим подтверждается принцип аддитивности фазовых
противлении и косвенно — принцип равновесия фаз на
поверхности раздела (в опытах коэффициент распределения т
изменялся в 7600 раз). Аддитивность сопротивлений фаз также
подтверждена в опытах по
экстракции из капель 57.
Следовательно, если
отсутствуют некоторые
специфические условия, подобные тем,
которые наблюдались в
опытах Дриккамера, то постулаты
Уитмана о равновесии на
поверхности раздела фаз и
аддитивности фазовых
сопротивлений всегда корректны. Тем не
менее имеются условия, при
которых уравнения (V, 60) и
Рис.96. Схема ячейки с леремеши- (V.61) не применимы вслед-
ванием. ствие того, что измеренное
Поверхность
раздела
Фаз
200
общее сопротивление (1//С) значительно больше или меньше
величины сопротивления, определяемого из коэффициентов
массоотдачи k. Эти аномалии можно рассматривать как следствие
некоторых дополнительных условий, не учтенных при выводе
уравнений (V, 60) и (V, 61), или неправильного определения
коэффициентов и, возможно, других причин. В ряде случаев
указанные аномалии имеют важное значение, и некоторые из
них будут рассмотрены ниже.
1. Изменения температуры на поверхности раздела фаз. Из-
за различных теплот растворения компонента в жидких фазах,
между которыми он распределяется, при массопередаче должно
происходить перемещение или абсорбция тепла и
соответственно изменение температуры поверхности раздела фаз.
Вследствие этого (см. главу II) могут изменяться локальные значения
коэффициента распределения и физические свойства фаз
(плотность, вязкость), влияющие на величину k. Обычно данный
эффект невелик, однако, в некоторых случаях он весьма
значителен. Например, при экстракции уксусной кислоты из
изобутанола водой, содержащей едкий натр, в результате
нейтрализации выделяется тепло.
Установлено, что увеличение температуры может превысить
11°С в зависимости от предполагаемой скорости диссипации
тепла с поверхности раздела58. В этих условиях недопустимо
применять значения k, отнесенные к температуре основного
объема жидкости.
2. Сопротивление поверхности при адсорбции малых
количеств поверхностно-активных веществ. Поверхностно-активные
вещества могут адсорбироваться на поверхности раздела фаз.
Присутствие некоторых из них даже в очень малых
концентрациях приводит к значительному снижению скорости
массопередачи. Например, введение 6-Ю-5 г поверхностно-активного
вещества на 100 мл жидкости снижает скорость массопередачи
более чем на 68% 59. Оказывая такое действие, поверхностно-
активные вещества вносят дополнительное сопротивление
массопередаче, которое должно учитываться третьим слагаемым,
вводимым в правую часть уравнений (V,60) и (V,61). Хотя
механизм массопередачи в присутствии поверхностно-активных
веществ еще недостаточно изучен, их действие может быть
различным.
Блокирование поверхности. Известно, что
некоторые вещества (например, цетиловый спирт), когда они
растекаются в виде мономолекулярной пленки по поверхности воды,
снижают скорость выпаривания в результате уменьшения
поверхности, через которую должны проходить молекулы воды.
Однако это явление весьма специфично; не все
мономолекулярные пленки в равной мере оказывают такое действие. Дрика-
мерБз, а также Гордон и Шервуд55 не обнаружили влияния
201
поверхностно-активных веществ в опытах, описанных выше. Если
бы влияние таких веществ заключалось в уменьшении
поверхности раздела фаз, эффект их действия был бы пропорционален
концентрации на этой поверхности и проявлялся в уменьшении
граничного (межфазового) натяжения. Однако Гарнер и Хэйл60
не обнаружили связи между измеренной поверхностной
концентрацией и сопротивлением поверхности. По-видимому, подобное
объяснение действия поверхностно-активных веществ
недостаточно.
Изменение подвижности поверхности.
Поверхность раздела фаз в системе жидкость — жидкость обычно
обладает подвижностью, причем движение одной жидкости может
легко передаваться через эту поверхность другой и оказывать
влияние на величину k в последней. Известно, что очень малые
количества некоторых веществ, адсорбируясь на поверхности
раздела, увеличивают ее жесткость и препятствуют нормальному
перемещению поверхности. При этом снижаются значения
коэффициентов массоотдачи k.
Льюис44, проводя опыты в диффузионной ячейке, нашел, что
неподвижная протеиновая пленка, адсорбированная на
поверхности раздела фаз, заметно уменьшает скорость процесса массо-
передачи, в то время как подвижные пленки других
поверхностно-активных веществ не оказывают подобного действия.
Движение капель одной жидкости в другой обычно происходит
при скоростях, более высоких, чем движение твердых сфер. Это
объясняется подвижностью поверхности жидкость — жидкость,
в результате которой возникает циркуляция жидкости внутри
движущихся капель. Если присутствие поверхностно-активных
веществ делает поверхность капли менее подвижной, можно
ожидать, что в присутствии этих веществ скорость подъема
(или падения) капель будет уменьшаться, достигая в пределе
скорости движения твердых сфер; скорость экстракции при этом
должна замедляться. Такая гипотеза подтверждена
экспериментально 59-62.
Предложена теория 63, согласно которой при движении капли
поверхностно-активное вещество смывается в ее кормовую часть,
приводя к образованию градиента граничного натяжения вдоль
поверхности капли. Градиент граничного натяжения
препятствует движению поверхности, вследствие чего поведение капли
становится подобным поведению твердой сферы. Если массо-
передача через различные участки поверхности раздела фаз
происходит с различными скоростями (как в случае
движущейся капли), вдоль поверхности раздела могут образовываться
градиенты концентраций и соответственно градиенты
граничного (межфазового) натяжения.
Взаимодействие адсорбированных веществ и
экстрагируемого компонента. Хатчинсон64 высказал
202
гипотезу о том, что экстрагируемый компонент может
адсорбироваться поверхностно-активным веществом, в свою очередь
адсорбированным на поверхности раздела. Существует ряд
доказательств59,60'65 в пользу наличия этого эффекта. Тот факт,
что данное поверхностно-активное вещество не в одинаковой
мере замедляет скорость массопередачи для различных
компонентов, подтверждает эту теорию66.
Некоторые эффекты, вызываемые поверхностно-активными
веществами, очень трудно обнаружить. Уэст67,68 нашел, что
бензол, однократно проходя через трубку из пластмассы «Ту-
gon», растворяет поверхностно-активный пластификатор в
количестве, достаточном, чтобы эта примесь могла сильно влиять
на скорость экстракции. Понижая межфазовое натяжение,
поверхностно-активные вещества могут влиять на форму капли и
тем самым на величину k путем изменения гидродинамических
условий при взаимодействии фаз.
3. Межфазовая турбулентность. Эта турбулентность
представляет собой спонтанную активность поверхности,
возникающую тогда, когда в соприкосновение приводятся две жидкие
неравновесные фазы. В результате появления межфазовой
турбулентности капли, погруженные в другую жидкость,
испытывают воздействие беспорядочных пульсаций, эррупции
(«извержения» с поверхности раздела распределяемого компонента) и
интенсивной внутренней циркуляции69-72.
Это явление сопровождается также спонтанным
эмульгированием, пульсированием и волнообразованием на поверхности
раздела фаз73. Такая активность поверхности приводит к
увеличению скорости массопередачи вследствие возрастания
величины k (по сравнению со значением, коэффициента
массоотдачи k при стабильной межфазовой поверхности) и увеличения
поверхности массопередачи. В некоторых случаях скорость
массопередачи возрастает в десять раз и более 44>66'п-74. Часто
отмечается, что межфазовая турбулентность возникает только при
определенном направлении массопередачи и заметно
подавляется поверхностно-активными веществами. Этот эффект влияет
также на производительность экстрактора 75.
Межфазовая турбулентность вызывается, по-видимому,
градиентом межфазового натяжения, возникающим при наличии
градиента концентрации на поверхности раздела фаз, или так
называемым эффектом Марангони. Такое объяснение
подтверждается некоторыми экспериментальными данными76'77. Исходя
из подобных представлений, Стернлинг и Скривен78 описали
нестабильность поверхности раздела фаз математически,
использовав для этого уравнения движения и диффузии. Они
получили результаты, подтверждающие, что нестабильность
поверхности раздела фаз должна наблюдаться при определенном
направлении массопередачи. Возникновению межфазовой
203
турбулентности могут способствовать: а) переход
распределяемого компонента из фазы с более высокой вязкостью; б)
переход распределяемого компонента из фазы, в которой его
коэффициент диффузии более низок, чем в другой фазе; в) большая
разность кинематических вязкостей фаз и коэффициентов
диффузии в фазах; г) большие концентрационные градиенты
вблизи поверхности раздела;; д) малые вязкости фаз и
коэффициенты диффузии в обеих фазах; е) сильное изменение граничного
(межфазового) натяжемия с концентрацией; ж) отсутствие
поверхностно-активных веществ, которые увеличивают
поверхностную вязкость; з) высокая дисперсность системы и
соответственно большая межфазовая поверхность.
4. Химическая реакция. Если экстрагируемый компонент
подвергается химическому взаимодействию (в дополнение к
тепловому эффекту и межфазо'вой турбулентности) химическая
реакция может также влиять на движущую силу процесса мас-
сопередачи.
Химическая реакция увеличивает или уменьшает скорость
экстракции (в зависимости от скорости самой реакции),
приводит к образованию новых химических веществ и т. п.
Другими словами, она вызыьчает эффекты, которые не учитываются
уравнениями (V, 60) и (V, 61). Некоторые реакции,
протекающие в системах жидкость — жидкость, например процессы
сольватации или ■ димеризации диффундирующего
(распределяемого) компонента не могут быть с достаточной точностью
учтены.
Химическая реакция.. В качестве примера влияния
химической реакции на скорость экстракции рассмотрим случай, когда
экстрагируемый компонент, прежде чем перейти из рафината
в экстракт, образует с :экстрагентом сольваты. Такая реакция
происходит при экстрактии уранилнитрата из воды метилизобу-
тилкетоном и другими растворителями 79:
U02 (N03)2 • 6Н20 -Ц- 2S ;£ U02 (N03)2 • 2S • 4Н20 + 2Н20
где S — молекула растворителя.
Эту реакцию можно рассматривать как гетерогенную,
происходящую на поверхности раздела фаз. В данном случае
концентрации растворителя и воды в соответствующих фазах
настолько велики, что эту [реакцию можно считать реакцией
псевдопервого порядка. Обозначим константы скорости прямой и
обратной реакций соответственно через kx и k2- Тогда, если с —
концентрация распределяемого компонента А, при
стационарном состоянии скорость массопередачи 44>79:
NA = КСМ ~ k2CEl= k\ (CRi ~ if Ce) = ^ ^Rl ~ ^ (V' 67)
204
Здесь c*Ri определяется с помощью коэффициента
равновесного распределения, принятого постоянным:
т = cEi/c*m = ceIcr
В этом случае отношение концентраций равно отношению
констант скоростей реакций и определяется константой
химического равновесия.
Частные и общий коэффициенты массопередачи
определяются обычным способом:
NA = kR (CR - CRi) = kE (CEi - CE) = mkE{CRi ~ Cr) = KR (CR — C*R) (V, 68)
поскольку
CR - CR = (CR - CRi) + (CRi - CRi) + (CRi ~ 4) <V' 69>
Путем комбинирования приведенных уравнений получают:
Если скорость реакции невелика (как в рассматриваемом
примере), появляется дополнительное сопротивление
поверхности раздела фаз, выражаемое членом 1/fei. Если пренебречь
влиянием химической реакции и рассчитывать общий
коэффициент массопередачи Kr, учитывая только kR и kB, это приведет
к результатам, не соответствующим опытным данным. Мурдох
и Пратт79 рассмотрели также случай, когда на поверхности
контакта фаз протекает реакция не первого порядка и т не
является постоянной величиной.
Примером более сложных химических процессов,
сопутствующих жидкостной экстракции, служит экстракция уксусной
кислоты из органического растворителя водой, содержащей
гидроокись натрия. Уксусная кислота реагирует с гидроокисью
натрия моментально и необратимо. Согласно пленочной теории,
можно показать, что кислота диффундирует в воду и реакция
происходит в «пленке» водной фазы, примыкающей к
поверхности раздела фаз.
Результатом реакции является в конечном счете уменьшение
толщины «пленки» (в воде), через которую диффундирует
кислота, и увеличение скорости экстракции по сравнению с
экстракцией в отсутствие гидроокиси натрия. Скорость экстракции
долж:на возрастать с увеличением концентрации щелочи,
которая при этом быстрее подводится к поверхности раздела фаз.
Если концентрация щелочи будет достаточно высока, щелочь
проникнет через поверхность раздела в органический растворитель
и свяжет кислоту до того, как последняя достигнет поверхности
раздела фаз. Положение зоны реакции по отношению к
поверхности раздела зависит также от относительной величины коэф-
205
фициентов диффузии реагентов. Описанные закономерности
протекания экстракции, осложненной химической реакцией,
подтверждены экспериментально58.
Ниже представлен перечень работ, в которых рассмотрены
теория подобных процессов с позиций пленочной теории,
теории проникновения, а также теории обновления поверхности
контакта фаз:
Гомогенные реакции
Литература
Реакция первого порядка
обратимая, быстрая 80
обратимая, медленная .... 81; 82
необратимая, медленная . . . 4; 43; 82—84
Реакция второго порядка
обратимая, быстрая 80; 85
необратимая, быстрая ... 43; 82; 84
необратимая, медленная . . 86
Реакция л-ro порядка
необратимая, медленная ... 4
Полимеризация (ассоциация)
обратимая, быстрая 80; 87
Гетерогенные реакции
(в системах жидкость — жидкость)
обратимая, медленная,
сольватация 44; 79
В некоторых работах приводятся решения уравнений
диффузии совместно с уравнениями химической кинетики и
движения жидкостей. Однако точность этих решений недостаточно
подтверждена экспериментально.
Локальные коэффициенты массопередачи. Все
рассмотренные выше вопросы, связанные со скоростью массопередачи,
относятся к локальным или мгновенным) коэффициентам
массопередачи. В реальном процессе экстракции вследствие
изменения концентрации распределяемого компонента движущая сила
процесса, физические свойства веществ, гидродинамические
условия и другие параметры, от которых зависят величины
локальных частных и общих коэффициентов массопередачи,
изменяются в пространстве (неодинаковы по величине в различных
точках экстракционного аппарата) или во времени. Поэтому на
практике скорость массопередачи должна рассматриваться
совместно с уравнениями материального баланса.
Экстракция единичными каплями. Массопередача при
экстракции обычно происходит между системой капель
(дисперсная фаза) и другой жидкостью (сплошная фаза). При этом
дисперсная фаза движется через сплошную под действием
гравитационных сил. Закономерности массопередачи в таких усло-
208
Зкдидалентный диаметр
сферы, dp
Рис. 97. Типичные зависимости
скоростей осаждения капель в
бесконечной жидкой среде от
эквивалентного диаметра
сферы (обозначения см. стр. 208).
igRe
Рис. 98. Зависимость коэффициента
сопротивления движению капель от
критерия Рейнольдса для капли:
/ — область применения закона Стокса;
2 — кривые для различных систем
жидкость—жидкость; 3 —кривая для твердых
сфер.
виях рассмотрены в главе XI. Ниже приведены сведения,
относящиеся к предельному случаю — экстракции из единичной
капли или внутрь ее. До изложения вопросов массопередачи
рассмотрим закономерности движения капли.
Скорость осаждения единичной капли. Для любой пары
жидкостей скорость осаждения, т. е. постоянная скорость подъема
или падения капли (в зависимости от плотности капли
относительно плотности сплошной фазы) изменяется в функции от
размера капли, следуя одной из зависимостей,
представленных кривыми 'на рис. 97. Форма капель зависит от их размеров
и физических свойств жидкости, поэтому обычно размер капли
выражают через эквивалентный диаметр dv — диаметр сферы,
имеющей тот же объем, что и капля. Однако поведение капель
(рис. 97) отличается от поведения твердых сфер, постоянные
скорости падения или подъема которых систематически
возрастают с увеличением диаметра сферы.
Более наглядно отличие в движении капель и твердых сфер
видно из рис. 98, на котором сравниваются коэффициенты
сопротивления среды при движении в ней капель и твердых сфер.
В большей части случаев капли малого диаметра движутся с
большей скоростью, чем твердые сферы того же размера и
плотности, так как коэффициент сопротивления CD для капли
меньше, чем для твердой сферы. Это является следствием
подвижности поверхности капли, причем поверхность движется в
направлении от передней неподвижной точки к корме капли под
действием срезающих усилий и внутренней циркуляции
жидкости в капле (см. рис. 99) 88>89. Если вязкость сплошной фазы
велика, циркуляция внутри капли может происходить при любом
207
Рис. 99. Схема циркуляции вну
три медленно движущихся жид
ких капель.
значении критерия Рейнольдса для
капли и наблюдалась90 даже при
Re=0,0003.
Очень мелкая капля имеет
почти сферическую форму, но уже при
dp = 2—3 мм ее форма заметно
отличается от сферической, что
приводит к большему отличию
скорости осаждения такой капли от
соответствующей скорости для
сферической капли. Несферичность капли
Направление обусловлена давлением, вызывае-
капли мым ее Движением в сплошной
фазе и приводящим к увеличению ее
фронтальной поверхности. Крупные
капли имеют нестабильную форму,
приближающуюся к сплющенной
эллипсоидальной, особенно тогда, когда сплошная фаза
отличается малой вязкостью.
Капли периодически осциллируют, принимая
последовательно шарообразную и эллипсоидальную форму. Форма очень
крупных капель становится неопределенной; осцилляция их
совершенно беспорядочна. Форма и осцилляция капель в конечном
счете влияют на скорость осаждения, которая либо достигает
максимума, либо проходит через максимум (см. рис. 97).
Соответственно увеличивается и коэффициент трения, который с
некоторого момента начинает превышать коэффициент трения
для твердой частицы. Величина максимума зависит от
физических свойств жидкости. На рис. 97 кривая А относится к очень
чистым жидкостям, кривые В и D — к жидкостям с небольшим
количеством примесей или поверхностно-активных веществ.
Кривая, имеющая форму кривой С, встречается редко91. Примеси
поверхностно-активных веществ способствуют уменьшению
подвижности поверхности капли, различия в поведении мелких
капель и твердых сфер и гасят осцилляцию крупных капель.
При очень малых значениях критерия Рейнольдса для
капли (Re<l), если принять, что капля имеет сферическую форму,
скорость осаждения можно рассчитать, исходя из уравнений
Стокса —Навье88:
vr
АР^4 ( ! + iyi*c
18ц
I 1 + Цр/цс\
(V, 71)
В этом уравнении величина, стоящая перед скобками,
представляет собой скорость осаждения твердой сферической
частицы по закону Стокса. В скобках приведена поправка,
учитывающая влияние внутренней циркуляции на скорость движения
208
капли. Из уравнения (V, 71) следует, что при очень малой
вязкости жидкости в капле последняя будет двигаться со скоростью
осаждения, примерно в 1,5 раза большей, чем скорость
осаждения твердой сферической частицы.
В процессах жидкостной экстракции критерий Re обычно
превышает 50, поэтому уравнения Стокса — Навье неприменимы.
Скорость осаждения капель в этих случаях определяют из
эмпирических уравнений. Наиболее подробные исследования
скорости осаждения капель проведены Кинтнером 91~94. Анализ
размерностей приводит к следующей обобщенной
зависимости 92, 95.
Re = f (CD,We)
(V, 72)
Лучшая корреляция для капель, движущихся в жидкостях
относительно невысокой вязкости (порядка 1 спз), приведена
на рис. 100. График построен «а основе уравнения (V, 72),
причем величина Р на графике выражается следующим образом:
Р =
4Re4
Р2с*3
3CD We3
№с АР
(V, 73)
Из корреляции можно определить значения максимальных
скоростей движения капель, соответствующих максимальной
скорости на кривой А (см. рис. 97). Максимальную скорость
находят при значении ординаты на рис. 100, равном ~70,
причем этой скорости соответствует переходный размер капли
dpi, вычисляемый из уравнения
\0,5
ipf-
:7,25
А рию , <V'74) '000
Значения скоростей осажде- ^ ^
ния капель можно быстро опре- 5- Z
делить также по преобразован- ^ too
ному безразмерному уравнению Ч?- в
Кли и Трейбала95, которое в %■
размерном виде (в системе СГС) ^
имеет следующий вид для
каждой из двух областей
изменения скорости осаждения:
при dp < dpt
%
Vf
0,481 Ap°'58rf°'70
Pc V-c
при dp > dpt
6,25.10-3Др0'28ц°(;10а0-18
vt = -
P°d55
(V, 75)
(V, 76)
г
i
зи s i г
4 в щ г <<■ в too
Re/Pa'5
14 P. E. Трейба
Рис. 100. Корреляция Хью—Кинт-
нера92 для определения
скоростей осаждения капель.
208
Уравнение (V, 76) не отражает уменьшения скорости
осаждения капли с увеличением ее размера; эту зависимость можно
установить по рис. 100. При пользовании корреляционным
графиком (см. рис. 100) и уравнениями средние отклонения от
опытных данных составляют менее чем 10% для области dv<.dvt
и около 15% для области dp>dpt. На практике присутствие
даже незначительных количеств загрязнений может в
несколько раз изменить коэффициент сопротивления движению капли
в данной системе при том же значении критерия Рейнольдса м;
поэтому скорость осаждения капель можно определить лишь
приближенно.
При расчете скорости осаждения капель в сплошной фазе,
вязкость которой значительна, но не превышает ~ 30 спз,
Джонсон и Брайда97 предложили умножать ординату графика,
изображенного на рис. 100, на величину (цН2О/цс)0,14> где цНг0—
вязкость воды. При очень высоких вязкостях сплошной фазы
эта поправка неприменима94, и при Re<10 коэффициенты
сопротивления капли и твердых шарообразных частиц
оказываются примерно равными.
Для капель, движущихся в вертикальной трубе малого
диаметра d, но не меньшего 2dp, скорость осаждения капель91
v = vtyi — [■
d \2~|1,43
(V.77)
При dp/d = 0,l87 поправочный коэффициент равен 0,95.
Отношение dp/d на практике всегда намного меньше указанного
выше, поэтому отмеченный «пристенный эффект», вероятно, не
имеет большого значения. Хармати98 предложил для движения
капель в трубах малых диаметров (при Re>500) корреляцию,
учитывающую форму капли.
Литература по этому вопросу весьма обширна (наиболее
полная библиография приводится в статьях Кинтнера).
Скорость массопередачи в дисперсной фазе. Ниже будут
рассмотрены несколько теоретических решений этой проблемы,
которые можно использовать для сравнения с предельным слу-
чаем — массопередачей для твердых сфер.
Твердые шарообразные частицы. При отсутствии
сопротивления массопередаче в сплошной фазе нестационарное изменение
средней концентрации распределяемого компонента cD со
временем 0 выражается уравнением
_±vJ_
я •*" п
cD — cDi л £-
' — 4D0it
, 4
2л29^
J"
1 — exp 1
— 4D0Jt29y
> 4 JJ
0,5
(V.78)
aw
В этом уравнении c°D—начальная равномерная
концентрация распределяемого компонента; cDi — постоянная
концентрация на поверхности раздела фаз. Таким образом, левая часть
уравнения (V, 78) показывает степень приближения
концентрации распределяемого компонента к равновесной со сплошной
фазой. Первое выражение правой части уравнения—точное99,
второе — эмпирическая аппроксимация100. Коэффициент массо-
отдачи, если принять движущую силу пропорциональной
разности концентраций, можно определить по уравнению
2я2£>„
(V, 79)
*0 = '
Мп
Шарообразные капли с внутренней циркуляцией. Крониг и
Бринк101, использовав линии тока Адамара, подобные
показанным на рис. 99, но с центром циркуляции, совпадающим с
центром большого круга сферы, решили уравнение диффузии для
сферы при отсутствии сопротивления массопередаче в сплошной
фазе. Строго это решение применимо лишь при Re<l, однако
Спеллс89 наблюдал линии тока Адамара и при больших
значениях критерия Рейнольдса. Уравнение Кронига— Бринка имеет
вид:
"о" Т 2j An ехР (!'
CD — CD
= 1-
CD — CDl
л-1
tn64D0e
-{-
exp
17.9D
ь — i
D dn
- 2,25 • 4 • D0it29'
0,5
(V.80)
(V.81)
Второе выражение правой части уравнения (V, 80)
представляет собой эмпирическую аппроксимацию 102.
Сравнение показывает, что каплю можно рассматривать как
твердую сферу, эффективный коэффициент диффузии которой
в 2,25—2,7 раза больше истинного коэффициента молекулярной
диффузии DD для твердой сферы.
Хандлос и Барон 103 проанализировали случай, когда
движение жидкости в капле полностью турбулентно, причем (в
отличие от линий тока, показанных на рис. 99) происходит по
концентрическим окружностям. При циркуляции жидкость в
течение одного оборота полностью перемешивается в радиальном
направлении между соседними линиями тока. Учитывая, что
средняя скорость циркуляции связана со скоростью капли,
Хандлос и Барон получили следующее выражение для определения
14*
211
величины коэффициента турбулентной диффузии:
dPvt(Qr*-8r + 3)
(V,82)
1 + И0/Ис
где r = 4r'/dv — безразмерная координата (г'—радиус линии
тока).
Решение уравнения диффузии совместно с уравнением
неразрывности потока для случая, когда сопротивлением в сплошной
фазе можно пренебречь, позволило вывести для коэффициента
массоотдачи следующее выражение:
, 0,00375^ ,v Яо.
k° = T+^ (V'83)
или
kndn 0,00375dnvt/DD
_£_i=Shn =— p ti D ^ o,00375Ред (V,84)
DD D 1 + И0/Ис
В уравнении (V, 84) величина Ре о—модифицированный
критерий Пекле. Сравнивая полученные соотношения с
уравнением (V, 79), можно сделать вывод, что при наличии полностью
развитого турбулентного движения жидкости в капле
последнюю можно рассматривать как твердую сферу, для которой
эффективный коэффициент диффузии DD в 0,00057Рео раз
больше истинного коэффициента молекулярной диффузии для
твердой сферы. Каплю с полностью развитым турбулентным
движением жидкости в ней нельзя отождествлять с
осциллирующей каплей, однако математическое описание массопередачи
внутри капель согласно модели Хандлоса и Барона наиболее
близко соответствует действительности.
Для сравнения полученных выражений с
экспериментальными данными необходимо, чтобы последние были найдены в
условиях, когда сопротивлением в сплошной фазе можно
пренебречь. Это возможно, например, если капля чистой жидкости
проходит через слой сплошной фазы, насыщенной данной
жидкостью. При таких условиях градиент концентрации имеется
только внутри капли, а концентрация на поверхности капли
постоянна и соответствует растворимости распределяемого
компонента. Кроме того, необходимо устранить влияние «концевых
эффектов», возникающих, например, при образовании капли на
конце сопла.
Типичные данные о массопередаче в каплях воды, падающих
в слое сплошной фазы (этилацетат, н-бутанол, циклогексанол),
приведены на рис. 101 104. Из рисунка следует, что при Re<50
применима модель Кронига — Бринка. Согласно модели
Хандлоса и Барона, отношение Do /DD для н-бутанола должно быть
порядка 100, так что при более высоких значениях критерия
Реганольдса движение внутри капли приближается к турбулент-
212
ному, принятому в указанной
модели.
Уравнения (V, 78) —(V, 84)
применимы также к процессам
теплопередачи, протекающим в
тех же условиях, для которых
были получены эти уравнения
[в уравнения (V, 78) —(V, 84)
при этом вместо концентрации
и коэффициента диффузии
должны подставляться соответственно
температура и коэффициент
теплопроводности]. Кальдербанк и
Корчинский 102, анализируя
работы по тепло- и массопередаче в
каплях, нашли, что отношение
D'D/DD равно 1,8—3,3 даже при
очень больших величинах Re для
неосциллируюших капель и что
это отношение значительно
больше, если капли осциллируют.
Хандлос и Барон103 отметили
80
60
UO
Н
го
too
ьо
у/ 1
г (
*-о
О °
600
го
/о
о
ио
/о
°
Г
\°°
э
-
600
л
60
во
2
Q
ш
10
го
Критерий Re для капли
Рис. 101. Сравнение различных
мотелей массопередачи в
дисперсной фазе |03:
/ — этилацетат; // — н-бутанол; /// —
циклогексанол; / — модель Хандлоса и Ба-
хорошее соответствие уравнения
(V, 84) опытным данным для
быстро движущихся капель. В их
ОПЫТаХ, ОДНаКО, ИЗМерЯЛИСЬ Об- рона; 2—модель Кронига и Бринка.
щие коэффициенты
массопередачи, и можно было лишь делать допущения о механизме
диффузионного сопротивления в сплошной фазе.
Скорость массопередачи в сплошной фазе. Эту скорость
определяли для твердых сфер и капель с внутренней циркуляцией.
Твердые сферы. Массопередача для твердых сфер
представляет собой предельный случай, соответствующий наименьшей
скорости массопередачи в сплошной фазе. Штайибергер и Трей-
бал105 предложили следующую корреляцию для многочислен*
ных данных о тепло- и массопередаче в газовой и жидкой
сплошной фазах:
Shc-
Sh'+ 0,347 (Re Sc£5)0'62
(V, 85)
где Sh' определяет скорость массопередачи в результате
естественной конвекции и молекулярной диффузии в неподвижной
жидкости.
Величину Sh' находят из уравнений:
при Qrc Scc < 108 Sh' = 2 + 0,569 (Grc Scc)0,250 (V, 86)
при Grc Scc > 108 Sh' = 2 + 0,0254 (Grc Scc),/s ScW44 (y> 87)
213
В критерии Грасгофа (Gr) величина Др является разностью
плотностей сплошной фазы на поверхности твердой сферы и в
объеме сплошной фазы.
-Капли с внутренней циркуляцией. Для потенциального
потока сплошной фазы (цс = 0) и больших значений критерия Ре
имеем 106:
Sh с = изРе0/ = 1,13 (4г^)°'5 <v> 88>
Это уравнение можно получить также из уравнения (V, 52)
теории проникновения Хигби при условии, что Q = dp/vt.
Указанное уравнение успешно используют для интерпретации многих
данных о массопередаче в сплошной фазе. Однако если
подвижность поверхности капли уменьшается вследствие
адсорбции на ней некоторых веществ, то эффективное значение 0
сильно увеличивается67.
Гриффите107 получил несколько соотношений, аналогичных
по форме уравнению (V, 85), без учета естественной конвекции.
В этих соотношениях учитывается, однако, степень подвижности
поверхности капли (но не межфазовая турбулентность),
обусловленная градиентом граничного натяжения. Однако такие
уравнения практически трудно применять из-за недостаточности
данных о градиентах граничного натяжения.
Бауман и др.108 рассчитали скорость массопередачи для
капель при Re<l с использованием линий тока Адамара, но без
учета естественной конвекции. Элцинга и Банчеро109
предложили эмпирическую корреляцию по тепло- и массопередаче для
капли с внутренней циркуляцией (при 3600< Ре<22 500):
Эти же авторы нашли, что осцилляция увеличивает значение
Shc на 45%. Были предложены66 также другие уравнения для
коэффициентов массопередачи в сплошной фазе, аналогичные
по форме уравнению (V, 85).
Таким образом, в настоящее время можно рассчитать только
предельные значения коэффициентов массопередачи либо для
неосциллирующих капель, которые ведут себя подобно твердым
сферам, либо для неосциллирующих капель с полной внутренней
циркуляцией. Осцилляция капель и межфазовая турбулентность
значительно увеличивают коэффициенты массопередачи, причем
степень увеличения последних пока что не поддается расчету.
Пример V-7. Определить скорость растворения капли метилэтилкетона в
условиях примера V-6, предполагая, что капля свободно движется через
сплошную фазу (воду). Физические свойства дисперсной фазы (капли):
pD=837 кг/м2 (0,837 г/см3); \ю = 6-1Ст4 кг ■ м~' ■ сек-'(6 • 10-3 пз). Физические
214
свойства сплошной фазы: рс=962 кг/м2 (0,962 г/см3); цс = 1,45-10-3 кг-лг'Х
Хсек ' (1,45- 10"2 пз). Межфазовое натяжение З-Ю"4 кг/сек2 (0,3 дин/см).
Решение.
Скорость подъема капли. Эту скорость определим для
взаимонасыщенных фаз, так как в условии дано равновесное значение межфазового
натяжения. Подстановкой этой величины в уравнение (V, 73) получаем Р =
=4710. Из уравнения (V, 74) вычисляем dvt = 1,93 • 10~3 м. Поскольку dp >
> dPt, из уравнения (V, 76) находим постоянную скорость движения капли
0*=5,27-10"2 м/сек.
Ордината на рис. 100 равна 485. Ей соответствует абсцисса, равная 48.
Отсюда определяем и( = 5,15 • 10~2 м/сек. Экспериментально определено95
значение и( = 5,5-10~2 м/сек. В дальнейшем будем использовать
экспериментальное значение vt.
Скорость массопередачи. Если капля не осциллирует и на ее
поверхности ие адсорбированы поверхностно-активные вещества, величина
коэффициента массоотдачи, вероятно, находится в пределах значений, получаемых
по уравнениям (V, 88) и (V, 89). Критерий Пекле равен
ре= 5,5 .ИГ*-5-10-3 =
9,75- КГ10
Подстановка его в уравнение (V, 89) дает Shc = 0,518 • Ре°'5=276; по
уравнению (V, 88) получаем Shc=600. Если на поверхности капли
адсорбированы примеси, из уравнения (V, 85) при Сгс=70 200, She = 1545 и Re=183
получим She =94,5. По условию капли не содержат примесей поверхностно-
активных веществ и имеют размер, более чем в два раза превышающий
переходный диаметр. Поэтому капли, по-видимому, осциллируют, и наиболее
реальным из всех сравниваемых значений критерия Шервуда является
наибольшее She =600. Отсюда
600О„
kc= с =\,\7.\0~*м-сек 1
ар
Из предыдущего примера (р/М)ср.=0,0485 и АхА =0,0748. По
уравнению (V, 48) величина N А =4,25 • 10~4 кмоль ■ мт2 • сек-'. Поэтому *
4 9^ 1П~4
NA, + nb< = о 692— = 6>2'10_4 кмоль -м~2- сек~Х
что в 300 раз превышает скорость, соответствующую переносу только в
результате молекулярной диффузии (см. пример V-6).
В примере V-7 вместо более обоснованного уравнения
(V, 49) используется уравнение (V, 48), так как в имеющиеся
корреляции обычно входит коэффициент массоотдачи k. При
расчете этого коэффициента следует учитывать условия, для
которых получена данная эмпирическая корреляция. Эти
трудности не возникают при расчете коэффициентов массопере-
носа F. В качестве примера рассмотрим случай, когда (см.
пример V-7) чистый метилэтилкетон (фаза 1) контактирует с
чистой водой (фаза 2). При этом кетон переходит из фазы 1 в
фазу 2, вода — в противоположном направлении. Для этого
случая (при установившемся массообмене), согласно уравнению
(V, 48), имеем:
NA = kl (p/Af), (1 - хАП)= k2 (p,M)2 (xAn - 0)
215
Из этого уравнения
*! (Р/М)2ХА12
k^~ (9IM)l(\-xAn)
Если на поверхности раздела фаз устанавливается
равновесие, то хАц и хАц представляют собой постоянные концентрации
кетона в равновесных насыщенных растворах при данной
температуре. Отсюда следует, что отношение kjk2 также является
постоянной величиной; при этом, однако, известно, что
коэффициенты k могут изменяться совершенно взаимно независимо.
С другой стороны, можно сделать заключение, согласно
которому на поверхности раздела между фазами не достигается
равновесие, что противоречит опыту.
Эта дилемма не возникает при использовании уравнения
(V.49):
Л _ ■ "ANA + NB)-° 1"ЖЫА + ЫВ)-ХАП
F2 NaI(Na + Nb)-xai2I Na/(Na + Nb)-1
Уравнение (V, 49) допускает постоянство хАц и хАа-
Изменение отношения Fi/F2 компенсируется изменением AfA/(AfA + AfB).
Отметим, что при переходе такого распределяемого
вещества, как, например, уксусная кислота, из одной насыщенной
фазы в другую равновесные концентрации на межфазовой
поверхности не остаются постоянными. При этом изменения
отношения kR/kE в уравнении (V,55) связаны с изменением
положения точки Т на рис. 93.
По цричинам, отмеченным выше, для обработки результатов
опытов, особенно при массопередаче между двумя чистыми
жидкостями, более оправданно пользоваться уравнением (V, 49),
чем уравнением (V,48).
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
Ап — корень характеристического уравнения.
а — активность.
CD='4ApdpgJ(3pcvf) — коэффициент сопротивления
(безразмерный).
с — концентрация, кмоль/м3.
D — коэффициент молекулярной диффузии,
м2/сек.
D'—эффективный коэффициент диффузии,
м2/сек.
d — диаметр цилиндра, м.
dp — диаметр сферы, имеющий объем,
равный объему капли, м.
£дИф. — энергия активации диффузии, дж/кмоль.
216
^вяз. — энергия активации вязкого течения,
дж/кмоль.
&EV — внутренняя энергия испарения,
дж/кмоль.
F — коэффициент массопереноса,
кмоль
м2 ■ сек • (кмоль/м3) '
g — ускорение свободного падения, м/сек2.
А//р — энтальпия испарения, дж/кмоль.
J—диффузионный поток, отнесенный к
мольной скорости, кмоль • м~2 • сек~1.
К — общий коэффициент массопередачи,
КМОЛЬ
м2 ■ сек ■ (кмоль/м3)
k — коэффицент массоотдачи,
кмоль
мг • сек • (кмоль/м3)
k=l,38- 10" дж/°К — постоянная Больцмана.
к', k", k'", kx, k2 — константы скорости.
L — расстояние, м.
М — масса 1 моль, кг/кмоль.
т, т', т" — коэффициенты равновесного
распределения (безразмерные).
m — концентрация, мольность.
./V—массовый поток относительно
неподвижной точки, кмоль • м~2 • сек~[.
Qr = gd3pAp • pc/|ic — критерий Грасгофа для капли
(безразмерный).
Pe = ReSc — критерий Пекле (безразмерный).
Re = dpvtpc/\ic — критерий Рейнольдса для капли
(безразмерный).
Sc = n/pD — критерий Шмидта (безразмерный).
Sh = kdp/D — критерий Шервуда для капли
(безразмерный).
We = pcv2dp/o— критерий Вебера для капли
(безразмерный).
п = 6,06-1026 молекул/кжоль — число Аво-
гадро.
Р — безразмерная величина, определяемая
из уравнения (V,73).
Q — скорость массопередачи, кмоль/сек.
^ = 8314 дж• кмоль'1 -град'1 —
универсальная газовая постоянная.
г — радиус, м.
S — средняя скорость обновления
поверхности раздела фаз.
217
Т — абсолютная температура, ° К.
U — абсолютная подвижность иона при
бесконечном разбавлении (м • сек~х X
Хк"1).
V — мольный объем, м3/кмоль.
v —линейная скорость, м/сек.
vt - постоянная скорость движения капли
(скорость осаждения), м/сек.
х — концентрация, мол. доля.
Z — валентность иона.
z — расстояние, ж.
а — фракционная степень диссоциации в
уравнении (V, 35); температурный
коэффициент (в табл. 11).
у — коэффициент активности.
Y± — средний коэффициент активности
ионов в электролите, отнесенный к мо-
ляльности.
А — разность.
е — коэффициент турбулентной диффузии,
м2/сек.
6 — время, сек.
X, Хи Я2, Я3— межмолекулярные расстояния в
уравнении (V, 17).
\л — вязкость, н • сек/м2.
я= 3,1416.
р— плотность, кг/м3.
а — межфазовое натяжение, н/м.
Ф — фактор ассоциации в уравнении
(V.26).
ф„—корень характеристического уравне-
ния.
Нижние индексы
А — компонент А, диффундирующее вещество,
а — активность.
ср. — среднее значение.
В— компонент В, экстрагент.
С — сплошная фаза,
конц. — концентрированный.
D — дисперсная фаза,
разб. — разбавленный, при бесконечном разбавлении,
Е — экстракт.
/ — значение на поверхности.
М — средняя мольная; средняя.
О — общий.
218
Й. — рафинат.
Т— общий.
t— турбулентный [уравнение (V, 46)]; постоянная скорость
(осаждения) [уравнение (V, 71)]; переходный
[уравнение (V, 74)].
и — недиссоциированный.
w — направление w.
у—направление у.
z — направление г.
1,2— положения 1,2.
Верхние индексы
* — при равновесии.
0—начальное значение.
ЛИТЕРАТУРА
1. A. F i с k, Ann. Physik, 94, 59 (1855).
2. L. О n s a g e r, R. M. F u о s s, J. Phys. Chem., 36, 2759 (1932).
3. С. Н. Bedingfield, Т. В. Drew, Ind. Eng. Chem., 42, 1164 (1950).
4. R. B. Bird, W. E. Stewart, E. N. L i g h t f о о t, Transport
Phenomena, John Wiley a. Sons, Inc., New York, 1960.
5. A. L. G e d d e s, R. B. Pontius, Physical Methods of Organic Chemistry,
A. Weissberger (ed.), pt. 1, 3rd ed., Interscience Publishers, Inc., New
York, 1959.
6. W. J о s t, Diffusion in Solids, Liquids and Gases, Academic Press, Inc.,
New York, 1952.
7. F. A. L. D u 11 i e n, L. W. S h e m i 11, Nature, 187, 767 (1960).
8. E. R. G i 11 i 1 a n d, R. F. В a d d о u r, D. J. Goldstein, Can. J. Chem.
Eng., 35, 10 (1957).
9. A. R. Gord on, Ann. N. Y. Acad. Sci., 46, 285 (1945).
10. G. S. Hartley, D. F. R u n n i с 1 e s, Proc. Roy. Soc. (London), A168,
401 (1938).
11. J. W. Mc Bain, Т. Н. Liu, J. Am. Chem. Soc, 53, 59 (1931).
12. P. A. J о h n s о n, A. L. В a b b, Chem. Revs., 56, 387 (1956).
13. H. S. Harned, В. В. Owen, The Physical Chemistry of Electrolyte
Solutions, 3rd ed., Reinhold Publishing Corporation, New York, 1958.
14. A. Einstein, Ann. Physik, 17, 549 (1905); 19, 371 (1906).
15. G. В. В. М. Sutherland, Phil. Mag., 9, 781 (1905).
16. H. Eyring, J. Chem. Phys., 4, 283 (1936).
17. S. G 1 a s s t о n e, K. J. L a i d 1 e r, H. Eyring, The Theory of Rate
Processes, McGraw-Hill Book Company, Inc., New York, 1941.
18. J. F. К i n с a i d, H. Eyring, A. E. S t e a r n, Chem. Revs., 28, 301
(1944).
19. R. E. Powell, W. E. R о s e v e a r e, H. Eyring, Ind. Eng. Chem.,
33, 430 (1941).
20. J. С. М. Li P. Chang, J. Chem. Phys., 23, 518 (1955).
21. H. Eyring, T. Ree, D. M. Grant, R. С Hurt, Z. Elektrochem., 64,
146 (1960).
22. K. K. Innes, I. F. Albright, Ind. Eng. Chem., 49, 1793 (1957).
23. R. J. В ear man, J. С. К i г к w о о d, J. Chem. Phys., 28, 136 (1958),
24. S. A. Rice, J. G. К i г к w о о d, J. Chem. Phys., 31, 901 (1959).
25. R. C. R e i d, Т. К. Sherwood, The Properties of Gases and Liquids,
McGraw-Hill Book Company, Inc., New York, 1958.
219
26. С. R. Wi Ike, Chem. Eng. Progr., 45, 218 (1949).
27. С R. Wi Ike, P. Chang, A. I. Ch. E. Journ., 1, 264 (1955).
28. D. R. О 1 a n d e r, A. I. Ch. E. Journ., 7, 175 (1961).
29. H. H. Reamer, С H. Duffy, В. Н. Sage, Ind. Eng. Chem., 48, 285
(1956).
30. E. Q. S с h e i b e 1, Ind. Eng. Chem., 46, 2007 (1954).
31. W. Nernst, Z. physik. Chem,. 2, 613 (1888).
32. J. R. Partington, Treatise on Physical Chemistry, H. S. Tayler (ed.),
2nd ed., D. Van Nostrand Company, Inc., Princeton, New York, 1930,
p. 673.
33. H. S. Harned, Chem. Revs., 40, 461 (1947); Disc. Faraday Soc, 7,
№ 24 (1957).
34. A. R. Gordon, J. Chem. Phys., 5, 522 (1937).
35. V. Vitagliano, P. A. Lyons, J. Am. Chem. Soc, 78, 4538 (1956).
36. R. Q. Deissler, Natl. Advisory Comm. Aeronaut. Repts., 1955, 1210.
37. С S. Lin, R. W. M о u 11 о n, Q. L. Putnam, Ind. Eng. Chem., 45,
636 (1953).
38. E. V. Murphree, Ind. Eng. Chem., 24, 726 (1932).
39. J. В. О p f e 11, B. W. Sag e, Advances in Chemical Engineering,
T. B. Drew and J. H. Hoopes (ed.), v. I, Academic Press, Inc., New York,
1956.
40. W. K. Lewis, Ind. Eng. Chem., 8, 825 (1916).
41. A. P. Co lb urn, Т. В. Drew, Trans. Am. Inst. Chem. Engrs., 33, 197
(1937).
42. R. Higbie, Trans. Am. Inst. Chem. Engrs., 31, 365 (1935).
43. P. V. Danckwerts, Ind. Eng. Chem., 43, 1460 (1951); A. I. Ch.
E. Journ., 1, 456 (1955).
44. J. B. Lewis, Chem. Eng. Sci., 3, 248, 260 (1954); 8, 295 (1958).
45. H. L. To or, J. M. Marchello, A. I. Ch. E. Journ., 4, 97 (1958).
46. M. X. Кишиневский, А. В. Памфилов, ЖПХ, 22, 1173 (1949);
24, 542 (1951); 27, 382 (1954).
47. W. K. Lewis, W. Q. Whitman, Ind. Eng. Chem'., 16, 1215 (1924).
48. W. Q. W h i t m a n, Chem. Met. Eng., 29, 147 (1923).
49. R. W. Schrage, A. Theoretical S.tudy of Interphase Mass Transfer,
Columbia University Press, New York, 1953.
50. A. F. H. Ward, L. H. Brooks, Trans. Farad. Soc, 48, 1124 (1952)
51. P. L. Auer, E. W. Murbach, J. Chem. Phys, 22, 1054 (1954).
52. E. J. Scott, L. H. Tung, H. Q. D r i с k a m e r, J. Chem. Phys, 19,
1075 (1951).
53. J. H. Sinfelt, H. Q. D r i с k a m e r, J. Chem. Phys, 23, 1095 (1955).
54. L. H. Tung, H. Q. Drickamer, J. Chem. Phys, 20, 10 (1952).
55. K. F. Cordon, T. K. Sherwood, Chem. Eng. Progr. Symp. Ser, 50,
№ 10, 15 (1954); A. I. Ch. E. Journ, 1, 129 (1955).
56. A. P. С о 1 b u r n, D.Q.Welsh, Trans. Am. Inst. Chem. Engrs, 38,
179 (1942).
67. K. Fujinawa, Y. Nakaiki, Т. К u г с h a r a, Chem. Eng. (Japan),
22, 420, 540 (1958).
58. R. S e a r 1 e, K. F. Q о r d о n, A. I. Ch. E. Journ, 3, 490 (1957).
59. K. P. Lindland, S. Q. Terjersen, Chem. Eng. Sci, 5, 1 (1956).
60. F. H. Garner, A. R. Hale, Chem. Eng. Sci, 2, 157 (1953).
61. F. H. Garner, A. H. P. Skelland, Ind. Eng. Chem, 48, 51 (1956).
62. S. G. Terjersen, Dechema Monograph, 32, 190 (1959).
63. А. Ф р у м к и н, В. Л е в и ч, ЖФХ, 21, 1183 (1947).
64. Е. Hutchinson, J. Phys. Colloid Chem, 52, 897 (1948).
65. G. В о у е - С h r i s t e n s e n, S. G. Terjersen, Chem. Eng. Sci, 9,
225 (1959).
66. F. H. G a r n e r, A. R. H a 1 e, J. Appl. Chem, 5, 653 (1953).
67. F. B. West, J. A. Hermann, A. I. Chong, L. E. K- Thomas, Ind.
Eng. Chem, 44, 625 (1952).
220
4
68. F. В West, P. A. Robinson, А. С. М о r g e n t h a 1 e r, J. R. Beck,
D. K. McGregor, Ind. Eng. Chem, 43, 234 (1951).
69. H. Kroepelin, H.-J. Neumann, Naturwiss, 43, 347 (1956); 45, 540
(1958).
70. H. Kroepelin, E. P r 6 11, Naturwiss, 45, 333 (1958); Erdol u. Kohle,
12 344 (1959)
71. J.'b. Lewis.H. R. С Pratt, Nature, 171, 1155 (1953).
72. K. Si g wart, H. N a s s e n s t e i n, Naturwiss, 42, 458 (1955).
73. T. K. Sherwood, J. С Wei, Ind. Eng. Chem, 49, 1030 (1957).
74. A. E. Karr, E. G. Scheibel, Chem. Eng. Progr. Symp. Ser, 50, № 10,
73 (1954).
75. H. R. С Pratt et al. Trans. Inst. Chem. Engrs. (London), 29, 89, 110,
126 (1951); 31, 57, 69 (1953).
76. D. A. H а у d о n, Nature, 176, 839 (1955).
77. D. A. Hay don, T. V. D a v i e s, Proc Roy. Soc. (London), A243, 483,
492 (1958).
78. С V. Stern ling, L. E. S с r i v e n, A. I. Ch. E. Journ, 5, 514 (1959).
79. R. Murdoch, H. R. С Pratt, Trans. Inst. Chem. Engrs. (London), 31,
307 (1953).
80. D. R. Olande r, A. 1. Ch. E. Journ, 6, 233 (1960).
81. P. V. Danckwerts, A. M. Kennedy, Trans. Inst. Chem. Engrs.
(London), 32, S49 (1954).
82. T. K. Sherwood, R. L. P i g f о г d, Absorption and Extraction, 2nd ed,
McGraw-Hill Book Company, Inc., New York, 1952.
83. P. V. Dankwerts, Trans. Farad. Soc, 46, 300, 701 (1950).
84. S. Hatta, Tehn. Repts. Tohoku Imp. Univ., 8, 1 (1928—1929); 10, 119
(1932).
85. H. С W e b e r, K. N i 1 s s о n, Ind. Eng. Chem, 18, 1070 (1926).
86. D. W. Van Krevelin, P. J. Hoftijzer, Rev. trav. chim, 67, ^32,
563 (1948).
87. J. D. Ra a 1, A. I. Johnson, Can. J. Chem. Eng, 37, 218 (1959).
88. J. S. Hadamard, Compt. rend. 152, 1735 (1911); 154, 109 (1912).
89. K. E. Spells, Proc. Roy. Soc. (London), B65, 541 (1952).
90. F. H. Garner, К. В. М a t h u r, V. G. Jensen, Nature, 180, 331
(1957).
91. J. R. Strom, R. С. К i n t n e r, A. I. Ch. E. Journ, 4, 153 (1958).
92. S. Hu, R. С Kintner, A. I. Ch. Journ, 1, 42 (1955).
93. M. V. Mhatre, R. С Kintner, Ind. Eng. Chem, 51, 865 (1959).
94. M. W a r s h а у, Е. В о g u s z, M. Johnson, R. С Kintner, Can. J.
Chem. Eng, 37, 29 (1959).
95. A. J. Klee, R. E. T г е у b a 1, A. I. Ch. E. Journ, 2, 444 (1956).
96. F. H. Garner, A. H. P. Skelland, Chem. Eng. Sci, 4, 149 (1955).
97. A. I. Johnson, L. В r a i d a, Can. J. Chem. Eng, 35, 165 (1957).
98. T. Z H a r m a t h y, A. I. Ch. E. Journ, 6, 281 (1960).
99. А. В Newman, Trans. Am. Inst. Chem. Engrs, 27, 310 (1931).
100. T. Vermeulen, Ind. Eng. Chem, 45, 1664 (1953).
101. R. Kronig, J. С Brink, Appl. Sci. Res., A2, 142 (1950).
102. P. H. Calderbank, I. J. В. К о г с h i n s k i, Chem. Eng. Sci, 6, 65
(1956).
103. A. E. Handlos, T. Baron, A. I. Ch. E. Journ, 3, 127 (1957).
104. A. I. Johnson, A. E. Hamielec, A. I. Ch. E. Journ, 6, 145 (1960).
105. R. L. S t e i n b e r g e r, R. E. T г е у b a 1, A. I. Ch. E. Journ, 6, 227
(1960).
106. J. В о u s s i n e s q, J. math, pures appl, 11, 285 (1905).
107. R. M. Griffith, Chem. Eng. Sci, 12, 198 (1960).
108. С W. Bowman, D. M. Ward, A. I. Johnson, O. Trass, Can. J.
Chem. Eng, 39, 9 (1961).
109. E. R. E 1 z i n g a, J. Т. В a n с h e r o, Chem. Eng. Progr. Symp. Ser, 55,
№ 29, 149 (1959),
МЕТОДЫ РАСЧЕТА
РАЗЛИЧНЫХ ВАРИАНТОВ
ПРОЦЕССА ЭКСТРАКЦИИ
(ТРЕХКОМПОНЕНТНЫЕ СИСТЕМЫ)
СТУПЕНЧАТОЕ ВЗАИМОДЕЙСТВИЕ
ИСХОДНОЙ СМЕСИ С ЭКСТРАГЕНТОМ
разделение компонентов раствора экс-
■ тракцией можно проводить различными
путями в зависимости от свойств системы
жидкость — жидкость и схемы
расположения аппаратуры. Обычно применяют
следующую классификацию процессов
экстракции по этим признакам:
/. Системы с одним экстрагентом,
включая состоящие из трех компонентов — двух
компонентов, подлежащих разделению, и
экстрагента, а также системы, которые
можно упрощенно рассматривать как
эквивалентные трехкомпонентным системам.
Ступенчатое взаимодействие.
К этой категории можно отнести такие
схемы расположения аппаратов, при которых
экстрагент и смесь, подлежащая
разделению, приводятся в тесный контакт друг с
другом до достижения равновесия, а затем
отделяются. Процесс можно многократно
повторять и проводить при различных
схемах взаимного движения потоков.
Непрерывное взаимодействие.
В этом случае экстрагент и смесь,
подлежащая разделению, непрерывно
взаимодействуют друг с другом в соответствующей
аппаратуре. Равновесие обычно не
достигается.
2. Системы со смесью экстрагентов.
К этой категории процессов относится
экстракция раствором, состоящим по меньшей
мере из двух компонентов, причем
соотношение растворимостей в них таково, что
данную систему нельзя рассматривать как
222
эквивалентную тройной системе. Здесь возможны ступенчатое
и непрерывное взаимодействие.
3. Система с двумя экстрагентами (дробная, или
фракционная экстракция). При такой схеме экстракции смесь,
подлежащая разделению, распределяется между двумя несмешиваю-
щимися экстрагентами. Система состоит по меньшей мере из
четырех компонентов. Здесь также возможны ступенчатое и
непрерывное взаимодействие.
В этой главе рассматривается только ступенчатое
взаимодействие с одним экстрагентом.
Аналогия с дистилляцией. При изучении процессов
экстракции часто бывает полезным проводить параллель между ними
и процессами дистилляции, которые обычно лучше изучены.
^"Ащ^югию этих двух методов разделения отмечали несколько
авторов1-2. В процессах дистилляции смесь двух веществ
разделяется вследствие образования двух фаз, жидкой и паровой,
при подводе тепла. Разделение возможно благодаря тому, что
относительные концентрации компонентов в обеих фазах
различны. Последующая конденсация паров сопровождается
отводом тепла.
При экстракции две жидкие фазы образуются в результате
введения экстрагента, не смешивающегося с исходной смесью.
Введение экстрагента при экстракции аналогично вводу тепла в
Таблица 12
Аналогия между процессами экстракции и дистилляции
Экстракция
Введение экстрагента
Смешение экстраигеита с исходным
раствором
Удаление экстрагента
Отделение (регенерация) экстрагента
Фаза экстракта, насыщенная экстрагентом
Фаза экстракта, содержащая экстрагент
в количестве большем, чем при
насыщении
Фаза рафииата, содержащая экстрагент
в количестве меньшем, чем при
насыщении
Фаза рафииата, насыщенная экстрагентом
Двухфазная жидкая смесь
Селективность
Изменение температуры
Дистилляция
Введение тепла
Кипячение
Отвод тепла
Конденсация паров
Пар в точке кипения
Перегретый пар
Жидкость ниже точки кипения
Жидкость в точке кипения
Смесь жидкости и пара
Относительная летучесть
Изменение давления
процессах дистилляции. Относительные
концентрации компонентов, подлежащих
разделению, в обеих фазах различны, поэтому
при отстаивании фаз достигается
определенная степень их разделения. Регенерация
п 1n„ ~ экстрагента из экстракта аналогична кон-
Рис. 102. Схема иде- у v *
альной (теоретиче- денсации паров при дистилляции. Анало-
ской) ступени: гия между процессами экстракции и ди-
S-экстрагент; /?-исход- СТИЛЛЯЦИИ НЭГЛЯДНО ПОКЭЗаНа В Табл. 12.
£-ГстрВа0к? ^7c?pt™aTB Определения. Приведем несколько опре-
рафинат находятся в рав- делений, необходимых для дальнейшего
новесии). /-.
изложения. Ступенью называется
некоторый объем аппарата, в котором раствор, подлежащий
разделению, смешивается с нерастворимым в нем экстрагентом (при
этом система стремится к равновесию), и затем получаемая
смесь разделяется на две несмешиваемые жидкости, которые
выводятся из процесса. Одна из фаз, покидающих ступень,
богатая экстрагентом, называется экстрактом, а другая фаза,
бедная экстрагентом, — рафинатом.
Теоретической, или идеальной, ступенью является ступень,
на которой продолжительность взаимодействия между фазами
достаточна для достижения равновесия между ними. Таким
образом, экстракт и рафинат, покидающие теоретическую
ступень, представляют собой растворы, находящиеся в равновесии
друг с другом. Существует много модификаций и комбинаций
смесителей и отстойников, применяемых для проведения
процессов экстракции. Идеальная ступень схематично будет
обозначаться в дальнейшем кружком, как показано на рис. 102.
Расчетные диаграммы. Соотношения между количествами
равновесных фаз, число ступеней, концентрации и весовые
количества потоков в экстракционном процессе наиболее просто
и наглядно определяют графически с помощью диаграмм,
построенных для равновесных условий. В расчетной практике
используют диаграммы различных типов, в большей или меньшей
мере удобные для расчетов. Выбор диаграммы зависит от1
характера равновесия системы и способа проведения процесса.
Ниже будут рассмотрены три типа подобных диаграмм.
Треугольные координаты. Обычно данные о равновесии для
тройных систем изображают, применяя треугольные диаграммы.
С помощью этих диаграмм можно получить все данные о
концентрациях в таких системах, поэтому треугольные диаграммы
используют чаще других. Общие свойства треугольных
координат описаны в главе II.
В данной главе приняты следующие обозначения на
диаграммах, построенных в треугольных координатах: А, В и С —
чистые компоненты тройной системы; А и С — основные
компоненты исходного раствора, подлежащего разделению; В —
224
хА+хс
а б
Рис. 103. Системы координат для расчета процесса экстракции:
а —треугольная; б —Енеке.
чистый экстрагент. В тройных системах типа I компоненты А
и В частично смешиваются друг с другом; в системах типа II
частично смешиваются пары компонентов А — В и В — С.
Символы А, В и С обозначают также весовые количества
соответствующих компонентов. Так, например, А обозначает А кг
компонента А; В соответствует В кг компонента В и т. д.
Символы F, Е и R обозначают соответственно составы и весовые
количества питания, экстракта и рафината. Например, буква F
соответствует F кг питания и составу смеси, определяемому
положением точки F на треугольной диаграмме. Прочие растворы
и смеси обозначены другими символами, но в каждом случае
соответствующие символы выражают не только весовое
количество, но и состав, показываемый на фазовой диаграмме.
Весовые доли обозначены буквой X с соответствующими
индексами, как указано в главе II. Так, ХАВ выражает весовую
долю компонента А в растворе, обогащенном В; ХСЕ — весовая
доля компонента С в экстракте; XBF — весовая доля компонента
В в питании. Вместо весовых количеств и долей можно
использовать соответственно числа молей и мольные доли.
Номера ступеней обозначены цифрами; цифра в индексе
означает, что дайн а я величина относится к соответствующей
ступени. Например, ХСНг — весовая доля компонента С в рафи-
нате ступени 2; Rn — весовое количество рафината, получаемого
на п-й ступени, и т. д.
Длины линий, например, отрезка прямой между точками О
и Р на фазовой диаграмме (рис. 103, а) будем обозначать ОР.
Если к смеси состава Р добавить смесь состава О, получим
новую смесь, состав которой определяется положением точки К на
прямой линии ОР, при этом
О ~КР
(VI, 1)
р m
15 Р. 5- Трейбал
226
Диалогично, выделяя из смеси К раствор состава Р, получим
смесь О на продолжении прямой РК. Хорды равновесия
показаны на диаграммах линиями более тонкими, чем другие.
Диаграмма Енеке. В системе координат Енеке, описанной в
главе II, строят зависимость между значениями X (весовая
доля компонента С в растворе, отнесенная к суммарному
количеству в нем компонентов Л и С без компонента В) на оси
абсцисс и N (весовая доля компонента В в растворе,
отнесенная к суммарному количеству в нем компонентов А и С) на оси
ординат, как показано на рис. 103,6. Таким образом:
Х= „ *с„ N= X» (VI.2)
ХА + ХС ХА + ХС
Как и в треугольных диаграммах, символы Е, A, R и т. д.
выражают весовые количества и состав жидкостей Е, А и R.
Символы, набранные прямым жирным шрифтом, обозначают
весовое количество рассматриваемого потока жидкости без
учета содержания в нем компонента В. Так, R2 обозначает
суммарное весовое количество компонентов А и С в рафинате
ступени 2.
Общее весовое количество потока Р и его весовое количество
без учета компонента В связаны между собой следующим
образом:
P = P(l + iVp) (VI, 3)
Весовое количество компонента В в растворе Р определяется
зависимостью:
Bp=PNp (VI, 4)
Длина линии между точками О и Р обозначена ОР. Так же,
как и на треугольной диаграмме, вместо весовых количеств
и весовых долей на диаграмме Енеке могут быть нанесены
числа молей и мольные доли.
Если смесь состава О смешать со смесью состава^ Р,
образующаяся смесь будет иметь состав, соответствующий точке К
на прямой ОР, причем и в этом случае применимо соотношение
0 КР (VI, 5)
Аналогично, выделяя Р из К, получим точку О на
продолжении прямой РК- Хорды равновесия и бинодальные кривые
(линии, соединяющие концы хорд равновесия) обозначены
соответственно тонкими и жирными линиями.
Простая диаграмма распределения. Такая диаграмма
строится в координатах ХСа (ось абсцисс) и ХСв (ось ординат), как
показано на рис. 27. В случае необходимости будет
использоваться модификация подобной диаграммы, Применяемая, когда
компоненты А и В практически взаимно нерастворимы.
Обычно расчеты можно проводить с помощью любой из
этих диаграмм, однако для некоторых систем при определенных
условиях та или иная диаграмма может оказаться
непригодной для графических расчетов. Прочие свойства диаграмм,
которые здесь не рассматриваются, соотношения между различными
диаграммами, а также другие применимые системы координат
подробно разобраны в серии статей Рэнделла и Лонгтина '.
Способы организации процесса экстракции. Ступенчатую
экстракцию с одним экстрагентом можно проводить различными
способами. В зависимости от взаимного расположения ступеней
получают различные результаты разделения. Ниже приведены
следующие варианты организации процесса:
V 1. Одноступенчатая экстракция. В этом процессе используют
одну ступень, где раствор, подлежащий разделению, и экстр-
агент взаимодействуют однократно, после чего экстракт и ра-
финат разделяются. Процесс можно проводить периодически
или непрерывно. Аналогичным процессом в дистилляции
является однократное испарение или однократная дистилляция
(перегонка в равновесных условиях).
2. Дифференциальная экстракция. Этот процесс является
периодическим. Определенное количество исходного раствора
взаимодействует с весьма малыми количествами экстрагента,
при этом образуются соответствующие количества экстракта.
Процесс подобен дифференциальной дистилляции и может
быть назван, по Вартерисиану и Фенске3, «бесконечной
ступенью с параллельным током жидкостей».
3. Многоступенчатая экстракция с перекрестным током.
Такой процесс осуществляется периодически или непрерывно и
представляет собой многократно повторяемую одноступенчатую
экстракцию, причем рафинат после каждой ступени вновь
взаимодействует со свежим экстрагентом. В пределе (при
бесконечном числе ступеней) этот процесс становится аналогичным
дифференциальной экстракции.
4. Многоступенчатая противоточная экстракция. Процесс
проводят в каскаде ступеней. Экстрагент и подлежащий
разделению раствор поступают с противоположных концов каскада,
причем экстракты и рафинаты каждой ступени движутся
противотоком друг к другу. Этот процесс скорее аналогичен
абсорбции, чем какому-либо виду дистилляции. Процесс
осуществляется непрерывно, но может быть имитирован путем
проведения ряда периодических процессов (например, в
лабораторных условиях часто применяют многоступенчатую псевдопроти-
воточную экстракцию).
5. Многоступенчатая противоточная экстракция с флегмой.
Непрерывный процесс, аналогичный фракционной дистилля-
15*
227
ции. Исходный раствор, подлежащий разделению, обычно
подают в среднюю часть каскада ступеней, а экстрагент — с одного
из концов каскада. Фазы экстракта и рафината движутся
противотоком, флегма поступает со стороны выхода экстракта из
каскада.
ОДНОСТУПЕНЧАТАЯ ЭКСТРАКЦИЯ
Треугольные диаграммы. Схема движения жидкостей с
указанием весов различных фаз показана3 на рис. 102. Обычно
раствор, подлежащий разделению, состоит только из
компонентов А и С, а экстрагент представляет собой чистый компоненте.
Однако в общем случае все три компонента могут
присутствовать в этих фазах, как показано на рис. 104 (точки F и S).
Кривая RDEG на этом рисунке является типичной бинодальной
кривой, или кривой растворимости, для систем типа /,
ограничивающей область сосуществования двух жидких фаз.
Материальный баланс процесса имеет вид:
F-\-S=R-\-E=M (VI, 6)
где М — смесь исходного раствора и экстрагента.
Точка, соответствующая составу смеси М, как показано на
диаграмме, расположена в двухфазной области. Положение
точки М может быть определено графически на линии FS
диаграммы с помощью соотношения:
1- = Ш- (VI, 7)
5 FM
или аналитически расчетом состава этой смеси с помощью
материальных балансов. Так, баланс по компоненту С имеет вид:
™Cf + Sxcs = MXcm <V1'8>
Рис. 104. Экстракция при однократном взаимодействии.
228
откуда
_FXcp + SXcs_FXCF + SXcs
см м F + S' (V1,9)
Аналогично из баланса по компоненту В получаем:
FX +SX„. FXRp + SXR.
X = —BF 2i-= BF ' bs_ /vi im
вм м F-\-S (W ' '
Уравнения (VI, 9) и (VI, 10) определяют положение точки М.
Чтобы рассчитать количество экстрагента, необходимое для
получения смеси состава М при данном количестве исходного
раствора, уравнение (VI, 10) решают относительно S:
с— f(xbm~xbf) ,,n m
BS ВМ
В идеальной ступени достигается равновесие; из двухфазной
смеси М получают экстракт Е и рафинат R, составы которых
выражаются на диаграмме точками, лежащими на
противоположных концах хорды равновесия, проходящей через точку М.
Составы фаз Е и R определяют по диаграмме (иногда
применяя интерполяцию хорд равновесия методом последовательного
приближения). Весовые количества этих фаз можно рассчитать
или определить графически, используя соотношение
* Ш ОП. 12)
Е RM
Весовые количества экстракта и рафината можно также
рассчитать из уравнений материальных балансов. Так, баланс по
компоненту С имеет вид:
EXCE + RXCR=MXCM (V'113)
Решая это уравнение совместно с уравнением (VI,6),
получают:
Е = ХСМ_Х CR (VI, 14)
Определив Е, с помощью уравнения (VI, 6) рассчитывают R.
Если точка М находится вне двухфазной области диаграммы,
разделение компонентов исходного раствора методом
экстракции невозможно. Соответственно этому можно определить
минимальное количество экстрагента, при введении которого в
процесс точка М расположится в точке D диаграммы
равновесия (на бинодальной кривой). Это количество экстрагента
выражается зависимостью:
XBS~XBD
-—"(ж)8
229
При проведении процесса
с минимальным количеством
экстрагента Smto получают
максимальное количество ра-
фината состава D; экстракт
при этом не образуется.
Можно определить также
максимальное количество
экстрагента, при введении
которого в процесс точка М
совпадает с точкой G:
•->тах — *" I '
(aF\ F(xB0-xBF)
Рис. 105. Процесс экстракции в тео- \GS J X — X _
ретической ступени.
В последнем случае образуется максимальное количество
экстракта и не образуется рафинат.
Эффективность ступени. В теоретической (идеальной)
ступени образуются две фазы, экстракт и рафинат, которые
находятся в равновесии. Однако если в ступени фазы недостаточно
интенсивно перемешиваются друг с другом или в ступени не
обеспечивается необходимое время контакта между экстрагентом и
исходным раствором, жидкости, выходящие из ступени, не
будут находиться в равновесии, то есть эффективность ступени
будет меньше 100%.
Рассмотрим рис. 105. При взаимодействии экстрагента S
с исходным раствором F одна из жидких фаз диспергируется
в другой, при этом (как обычно принимают) на поверхности
раздела фаз очень быстро достигается состояние равновесия,
соответствующее концентрациям насыщения (точки D и G).
В основной массе жидкостей (точки F и S) должны быть в
конечном счете также достигнуты равновесные концентрации,
отвечающие точкам R и Е соответственно.
Изменение концентраций в фазах, показанное на рис. 105,
зависит от относительных скоростей диффузии компонентов.
Если на ступени в результате взаимодействия фаз достигаются
составы R и Е, такая ступень является идеальной. Методы
определения эффективности ступени, или степени приближения
к идеальности, рассматриваются в главе X.
Регенерация экстрагента. Проблемы регенерации
экстрагента, а также составы регенерированного экстрагента и
получаемых конечных продуктов связаны с методами регенерации и
степенью разделяемости фаз. Эти вопросы освещаются в
главе IV.
Диаграмма Енеке. Соответствующие расчеты для схемы
процесса, показанной на рис. 102, могут быть проведены по диа-
230
грамме Енеке. Весовые количества различных потоков без
компонента В обозначены F, S, E, R и М. Координатами точки,
соответствующей составу питания, являются
/ кг В \ I кг С \
Р\кгА + кгС ) Р\кгА + кгС )
и т. п. На рис. 106, а показана диаграмма Енеке для систем
типа I (диаграмма для систем типа II идентична, но кривая
растворимости для таких систем соответствует приведенной на
рис. 24,6). Точки F и S выражают составы питания и
экстрагента и наносятся на диаграмму первыми. Положение
точки М на диаграмме определяется или графически:
MS
FM
(VI, 17)
или аналитически из уравнений материального баланса. Так,
из баланса по сумме компонентов Л и С получим:
F+S=E+R=M (VI, 18)
Из баланса по компоненту В
FNF+SNS = MNM (VI, 19)
Следовательно,
N
FNp + SN FN +SN
м~
М F + S
(VI, 20)
Из баланса по компоненту С
FX. + SX.
FXf + SXs
MX
FX. + SX,
(VI, 21)
чи
м
F + S
(VI, 22)
Уравнения (VI, 20) и (VI, 22)
выражают координаты точки М.
При определении количества
экстрагента, необходимого для
получения смеси состава М,
уравнение (VI, 20) решают
относительно S:
S = -
ns~nm
ft/
a)
<!
V
xe
R
J
A
a—ii—■
tS
m]
\d
'At
Py
""Л
/
X
/P
(VI, 23)
Xr Am
Рис. 106. Экстракция при одно-
/^„„^ , кратном взаимодействии, изобра-
Составы экстракта и рафина- ж*иная в координатах Енеке (а),
та находят графически по хорде и диаграмма распределения (б).
231
равновесия, проходящей через точку М, как показано на
рисунке. Весовые количества этих жидкостей можно определить
либо графически с помощью соотношения
R ЕМ (VI, 24)
Е MR
либо из материальных балансов.
Баланс по компоненту В
ENB + WR=MNM (VI, 25)
Решая это уравнение совместно с уравнением (VI, 18),
получают:
Е = М м_ R (VI, 26)
Е R
Далее из уравнения (VI, 18) определяют R.
Минимальное количество экстрагента соответствует
перемещению точки М в точку D (см. рис. 106, а) и соответствует
F(Nn — NF)
Sn,in= lD_NF) (VI, 27)
Максимальное количество экстрагента потребуется при
перемещении точки М в точку G:
Sn,ax= N _NF (VI.28)
;vs о
В реальных условиях количество экстрагента должно быть
промежуточным между этими предельными количествами.
В случае, когда экстрагентом является чистый компонент В
и, следовательно, Ns = oot точка S на рис. 106, а имеет
бесконечную ординату и линия, соответствующая вводу экстрагента,
на диаграмме вертикальна. Поэтому точка М лежит
непосредственно «ад точкой F. Ранее полученные уравнения
упрощаются, так как S = 0; SNS = В; Xs = 0 и F = М. Вместо
уравнений (VI, 23), (VI, 27) и (VI, 28) получают систему
уравнений для расчета В:
b = f(nm-Nf) (vi-29)
Bmla = F(ND-NF) (VI, 30)
Bm!LX^F(NQ-NF) (VI, 31)
Диаграмма распределения. Один из видов диаграммы
распределения показан на рис. 106, б, на котором приведена
зависимость концентрации распределяемого компонента в экстракте,
рассчитанной без учета содержания экстрагента (ось ординат).
от концентрации того же компонента в рафинате, также
рассчитанной без учета содержания экстрагента (ось абсцисс).
Хорду равновесия RE на рис. 106, а можно спроектировать, как
показано на рисунке, с тем чтобы получить точку / на кривой
равновесия. Проекции других хорд равновесия, не показанные
на рисунке, дадут кривую распределения LJP,
заканчивающуюся в критической точке Р.
Процесс одноступенчатой экстракции изображается в этих
координатах «рабочей линией» JM, наклон которой в
соответствии с уравнением (VI, 24) равен — R/E. Когда компоненты
А и С содержатся в экстракте S в том же отношении, что и
в питании, или когда в процессе используется экстрагент,
представляющий собой чистый компонент В (при этом линия FM
на рис. 106, а вертикальна), величина Хм = Xjr. В то же время
диаграмма, приведенная и а рис. 106, б, становится сходной
с диаграммой процесса мгновенного испарения при
одноступенчатой дистилляции, аналогом которой является процесс
одноступенчатой экстракции.
Типичные задачи по расчету процесса одноступенчатой
экстракции. Основными величинами в экстракционном процессе
являются F, ХР, Е, ХЕ, R, XR, S и Xs. Из этих величин обычно
бывают известны F, XF и Xs. Из других величин только одна
выбирается произвольно, после чего остальные могут быть
определены с помощью диаграмм равновесия.
Пример VI-1. Сто кг раствора, содержащего 25 вес.% ацетона (С) и
75 вес.% воды (А), экстрагируется метилизобутилкетоном, или МИК (В),
при 25° С. Рассчитать: а) минимальное количество экстрагента; б)
максимальное количество экстрагента; в) весовые количества экстракта и рафи-
ната (без учета содержания в них экстрагента), приходящиеся на 100 кг
экстрагента, и количество (в процентах) экстрагированного ацетона; г)
максимально возможную чистоту компонента С в конечном экстракте; д)
максимально возможную чистоту компонента А в рафинате.
Решение. Используем данные по равновесию, полученные Отмером,
Уайтом и Тройгером [Ind. Eng. Chem., 33, 1240 (1941)]. Расчеты будут
проведены по треугольной диаграмме и диаграмме Енеке.
а) Определяем положение точки F, соответствующей составу исходного
раствора, по треугольной диаграмме (рис. 107). В качестве экстрагента
используем чистый компонент В. Минимальное количество экстрагента
образует с исходным раствором смесь, соответствующую точке D. Тогда F= 100;
Xbd = 0,036.
Из уравнения (VI, 15)
F(X —X ) 100(0,036 — 0)
Лшш - -«mm - хх - j _ 0 „36 - *" Кг
Применим диаграмму Енеке (рис. 108):
x,=-Sr=°'25
W = О F = 100 кг Nn = 0,04
Г D
233
Из уравнения (VI, 30)
Bmin = F (ND — Np) = 100 (0,04 — 0) = 4 кг
Расхождения в ответах, полученные при решении задачи с помощью
различных диаграмм, объясняются ошибками, неизбежными при графическом
методе расчета — построении диаграмм в различных системах координат.
б) При максимальном количестве экстрагента последний образует смесь
с исходным раствором, соответствующую точке О. В треугольных
координатах XBG = 0,97.
Из уравнения (VI, 16)
F(XRn — XBB) 100(0,97 — 0)
а _ ^ ВО BF) v ' ' Ч01П vi
~ xbs~xbo ~ 1-°'97
По диаграмме Енеке iVG=32.
Из уравнения (VI, 31)
Bmax = F (NQ — Np) = 100 (32 — 0) = 3200 кг
в) В треугольных координатах S=B=100 кг.
Из уравнения (VI, 6)
F + S = M-E-\-R 100 -f 100 = Af = 200 кг
По уравнению (VI, 10)
FX +SXR. 100.0+100-1
X = MF BS— nv»
вм м - 200 ~~V,M
Определим положение точки М на диаграмме. Величина ХСм=0,125.
С помощью корреляционной кривой хорд равновесия и соответствующих
С-ацетон
Вода мин
Рис. 107. К примеру VI-1. Система ацетон — вода — метилизобутнлкетон
при 25° С [Ii,d.* Eng. Chem., 33, 1240 (1941)].
234
данных проведем через точку М хорду
равновесия и определим положение точек
Е и R. Из графика найдем ХСе = 0,15;
Хве = 0,818; Хсн = 0,09; Хвя=0,023.
По уравнению (VI, 14)
£ =
m(xcm-Xcr)
X.
се'
CR) __
х.
CR
200(0,125-
-0,09)
= 116,8 кг
~ 0,15 — 0,09
Из уравнения (VI, 6)
R = M — £ = 200— 116,8 = 83,2 кг
Линию BE продолжим до точки £',
которая соответствует составу экстракта
(без учета содержания экстрагента). Из
графика найдем Хс£' = 0,829; А'в£'=0.Ко-
личество экстракта (без учета веса
экстрагента) равно: £ '=£(1— А"ВЕ) = 116,8Х
Х(1 — 0,818) =21,3 кг. Количество рафина-
та (без учета веса экстрагента)
составляет: R'=F — £'=100— 21,3=78,7 кг.
Количество экстрагируемого ацетона:
Е'Х,
СЕ'
FX.
100 =
21,3 • 0,829
CF
100 • 0,25
100=70,5?
Рис. 108. К примеру VI-1 (дна-
грамма Енеке).
По диаграмме Енеке имеем: £=100 кг; S = 0; SNs=B;
■■ 100 кг.
Из уравнения (VI, 20)
Xs = 0; M=F=
N
FN +SN 100 • 0-f 100
s _
м
Из уравнения (VI, 22)
X
м
FXF+SXS
100
1,0
м
м
= хс
;0,25
После определения положения точки М с помощью хорды равновесия,
проходящей через эту точку, получим координаты точек £ и R: #Е=4,55;
NR = 0,0235.
По уравнению (VI, 26)
М (NM — NR) _ 100(1—0,0235)
N„—Nr
4,55 — 0,0235
= 21,5 кг = Е'
'Е R
Из уравнения (VI, 18)
R = М — Е = 100—21,5 = 78,5 кг = R'
Из уравнения (VI, 3)
£ = Е (1 +N£) = 21,5 (1 -f 4,55) = 119,3 кг
Я = R (1 -Н N ) = 78,5 (1 + 0,0235) = 80,3 кг
235
г) Если конечный экстракт
получают ректификацией
[нормальные температуры кипения
компонентов ^=100° С и tB =
= 116° С; гетероазеотропная смесь
А я В (24,3% А) кипит при
87,9° С], способ регенерации
аналогичен описанному в главе IV,
раздел «Ректификационные
методы», случай 3 и рис. 82. Ацетон
можно получить любой степени
чистоты.
д) Рассмотрим рис. 82. Воду
можно отделить ректификацией.
Все расчеты можно
проводить с помощью треугольной
диаграммы, показанной на рис. 103.
Расчет, приведенный в
пункте «в» примера (VI-1),
был выполнен для
различных количеств экстрагента,
приходящихся на заданное
количество исходного
раствора. В результате удалось получить графические
зависимости, показанные на рис. 109. Степень экстракции ацетона
быстро возрастает с увеличением количества экстрагента и
достигает примерно 90% при 300 кг экстрагента на 100 кг исходного
раствора. При дальнейшем увеличении количества экстрагента
степень экстракции повышается незначительно и это приводит
ко все возрастающему разбавлению экстракта.
Максимальная концентрация ацетона в экстракте (без учета
содержания экстрагента) соответствует точке Р. Ее получают
как точку пересечения стороны АС треугольника и касательной,
проведенной из вершины В к бинодальной кривой (см.
рис. 107). Кривая 3 (см. рис. 109) показывает селективность
экстрагента при разделении ацетона и воды. Такие кривые
типичны для систем данного типа, хотя следует иметь в виду,
что различия в равновесных данных могут оказывать
существенное влияние на ход кривых.
МНОГОСТУПЕНЧАТАЯ ЭКСТРАКЦИЯ
ПРИ ПЕРЕКРЕСТНОМ ТОКЕ*
В этом процессе рафинат после первой ступени экстракции
взаимодействует в следующей ступени со свежей порцией
экстрагента того же состава, что и на предыдущей ступени (см.
рис. ПО, а). Таким путем достигается более низкая концентра-
* Процесс экстракции в перекрестном токе автор часто называет
многоступенчатой прямоточной экстракцией. — Прим. ред.
Количество экстрагента кг/ЮОке
исходного раствора
Рис. 109. Экстракция уксусной кислоты
из 25%-ного водного раствора метил-
изобутилкетоном при 25° С:
/ — минимальный расход экстрагента; 2~содер-
жание ацетона в экстракте (без учета
содержания экстрагента) в %; 3—степень извлечения
ацетона в %; 4 — количество экстракта (без
учета содержания экстрагента) в кг на 100 кг
исходного раствора; 5 —максимальный расход
экстрагента.
236
Рис. ПО. Многоступенчатая экстракция при
перекрестном токе.
ция компонента С в рафинате. На различных ступенях можно
использовать различные количества экстрагента.
Метод расчета многоступенчатой экстракции при
перекрестном токе с помощью треугольной диаграммы показам на
рис. 110,6. Рафинат после каждой предыдущей ступени
является исходным раствором на каждой последующей ступени.
Соответственно этому для расчета процесса на первой ступени
многоступенчатой прямоточной экстракции применимы
уравнения (VI, 6) — (VI, 16) для одноступенчатой экстракции при
замене S, Е, М и R соответственно на Si, E\, М\ и R\. Аналогично
для любой (например, m-й) ступени применимы уравнения
(VI, 6) — (VI, 14) при замене F на Rm-\ и R, Е, М и S
соответственно на Rm, Em, Мт и Sm. Для всех ступеней экстракции,
кроме первой, невозможно найти минимальное количество
экстрагента, поскольку исходные растворы на этих ступенях
являются насыщенными. Максимальное количество экстрагента,
так же как и для процесса одноступенчатой экстракции,
должно быть таким, чтобы при смешении исходного раствора с экстр-
агентом образовывались две жидкие фазы. Схема
графического расчета для систем типа II показана на рис. 111.
_ Регенерация экстрагента. Рафинат, выходящий с последней,
я-и ступени, подвергают обработке для регенерации
экстрагента. Экстракты различных ступеней обычно объединяют и из
237
со
Ч
F
4L
R,l
гг^
Щм
Rnf^
Ц?
^^^
^cj\
л^
=~^г=
\ Е'
^кг
[V5* $\
X,
в
Еп
Рис. 111. Многоступенчатая
экстракция при перекрестном токе для
систем типа II.
Рнс. 112. Многоступенчатая
экстракция при перекрестном токе
(диаграмма Енеке).
получаемой смеси регенерируют экстрагент, хотя в принципе
возможно регенерировать экстрагент из экстракта каждой
ступени в отдельности. Экстракт Е, получаемый смешением
экстрактов отдельных ступеней, имеет следующие
характеристики:
E = Et + Et + Et + ... +ЕП
X,
Е1ХСЕ{ + Е^СЕг + Е3ХСЕ3 + ■ ■
+ ЕпХСЕ„
СЕ'
ElXBEl+E2XBE2 + ESXBEs + •-• +EnXBEn
X „„= —в ' "
'BE
(VI, 32)
(VI, 33)
(VI, 34)
В связи с тем что кривая растворимости обычно вопнута
вниз, смесь Е должна иметь состав, соответствующий
двухфазной области диаграммы.
Чистота продуктов. Наиболее чистый компонент А в
конечном рафинате (что отвечает максимальной степени экстракции
компонента С) получают тогда, когда л-я хорда равновесия,
соответствующая последней ступени, при продолжении
проходит через точку S. Это условие выполнимо при бесконечно
большом числе ступеней.
Диаграмма Енеке. Схема расчета в координатах Енеке
показана на рис. 112. Метод расчета также несложен и сводится
к многократному повторению схемы расчета для одной
ступени. Поэтому для любой m-й ступени применимы уравнения
(VI, 17) — (VI, 26), если в каждом из них заменить S, E, R, М
288
и F на Sm, Em, Rm, Mm и Rm_i. Экстракт, получаемый
смешением экстрактов отдельных ступеней, имеет следующие
характеристики:
Е = Е, + Е2 + Е3+ ... +ЕП (VI, 35)
ENE = E,WBi + E2NEi + E3NE> + ... + Е^ (VI, 36)
и
v Е1Х£1 + Е2?Ч + ЕзХ£з+ ••• +ЕпЕЕп (VI, 37)
Если в качестве экстрагента используют чистый компонент
В, линии построения, которые обычно радиально расходятся из
точки S, получают вертикальное направление.
Изменение температуры. Если, согласно данным о
равновесии и по другим соображениям, желательно проведение
процесса экстракции в отдельных ступенях при различных
температурах, при расчете в любой системе координат используют
соответствующую данной ступени кривую растворимости и
строят хорды равновесия, как показано на рис. 113. На этом
рисунке приведена схема двухступенчатой экстракции при
различных температурах ti и t2 на каждой ступени. Все уравнения,
указанные выше, применимы и в этом случае.
Типичные задачи по расчету процесса многоступенчатой
прямоточной экстракции. Концентрации экстрагента, питания
(исходного раствора) и количество последнего обычно известны.
Основными дополнительными переменными являются общее
количество экстрагента, распределение экстрагента по ступеням
и составы экстракта и рафината. В дополнение к
фиксированным величинам, указанным выше, следует определить: а) число
ступеней и количество экстрагента для каждой ступени; б)
составы экстракта и рафината на каждой ступени; в) состав
Рис. 113. Двухступенчатая экстракция при различных температурах на
каждой ступени:
—треугольная диаграмма; б —диаграмма Енеке.
239
1
5: « //
§1
6 °э
«со
8
Весовое отношение компонентов
С и А в рафинатах, х
Рис. 114. Многоступенчатая экстракция
при перекрестном токе в случае несме-
шивающихся растворителей:
1 — равновесная кривая; 2—наклон (— А1В •
3 —наклон (— A/Bs); 4 —наклон (— А/В2у, 5
—наклон (—А/В,).
конечного рафината и общее
количество экстрагента с
распределением его по
ступеням; г) состав конечного
рафината, число ступеней и
распределение общего
количества экстрагента по
ступеням. Определить
величины, указанные в двух
последних пунктах, можно
лишь методом
последовательных приближений. При
фиксированном количестве
экстрагента и числе
ступеней, равном бесконечности,
получают тот же результат,
что и для
дифференциальной экстракции.
Несмешивающиеся
растворители. Если принять, что
жидкости А я В
совершенно не смешиваются друг с другом или по крайней мере их
взаимная растворимость в условиях проведения экстракции не
изменяется при изменении концентрации распределяемого
компонента С в фазах, расчеты могут быть упрощены4.
Предположим, что в исходном растворе и рафинате после каждой
ступени содержится А кг компонента А и что экстракт и экстр-
агент после каждой ступени содержат одинаковое количество
компонента В. Тогда в системе координат у = ХСв/Хвв (для
растворов экстракта) и х=ХСА/ХАА (для растворов рафината)
можно провести кривую равновесного распределения,
соответствующую температуре процесса экстракции, как показано на рис. 114.
Пусть экстракт с пг-й ступени имеет концентрацию ут,
а рафинат —концентрацию хт. Концентрации компонента С
будут у8 (в экстрагенте) и xF (в исходном растворе). Тогда
общие весовые количества различных потоков определятся из
уравнений:
F*=A(l+xp) (VI, 38)
sm = Bm(l+ys) <VI'39>
Rm = A(l+xm) (VI, 40)
Ет*=Вт(\ + Ут) (VI, 41)
Для любой tn-Pi ступени можно составить уравнение баланса
по компоненту С:
Ах , + В у„ = В у + Ахт (VI, 42)
или „ _„ л
(VI, 43)
хт-\
в„
240
Уравнение (VI,43) представляет собой уравнение прямой
линии (в координатах рис. 114) с углом наклона —А/Вт,
проходящей через точки с координатами (хт, ут) и (xm-i, ys)-
Потоки, выходящие из теоретической ступени, находятся в
равновесии друг с другом, поэтому точка с координатами (хт, ут)
будет находиться на кривой равновесного распределения.
Соответственно для первой ступени линия с наклоном —А\ВХ
выйдет из точки V (xF, y8) и пересечет равновесную кривую в точке
Т (xi} yx). Построение для второй ступени начинают из точки U
и продолжают расчет до тех пор, пока конечный экстракт и
рафинат не достигнут составов, соответствующих точке W. Ту
же самую концентрацию компонента С в рафинате можно
было бы получить и в одной ступени (линия WV) при
соответственно большем расходе экстрагента.
Аналогичная диаграмма и метод расчета могут быть
использованы и при выражении концентраций в других единицах.
Так, если раствор является разбавленным (по компоненту С)
и в процессе экстракции не происходит заметного изменения
плотности, величины хну можно выразить как отношения
весовых количеств компонента С к единице объема раствора.
В этом случае F, S, Е и R измеряются в объемных единицах.
Можно выразить также х как отношение весового количества
компонента С в фазе к объему компонента А, а у — как
отношение весового количества компонента С к объему
компонента В. В этом случае А я В измеряются также в объемных
единицах.
Закон распределения. В частном случае, когда экстрагент и
исходный раствор полностью взаимно нерастворимы, применим
закон распределения
Ут = тхт (VI, 44)
где m — постоянный коэффициент распределения.
Индекс т указывает, что закон распределения применим
к любой (m-й) ступени. Пусть В обозначает количество
компонента В в экстрагенте на каждой ступени, одинаковое для всех
ступеней. Составим баланс для (т+1)-й ступени по
компоненту С:
В(Уш+1-Уз) = А(хт-хт+1) (VI. 45)
Решая это уравнение совместно с уравнением равновесия
на (т+1)-й ступени и исключая величину ym+i (принимая
в—тВ/А), получим:
В
ys-A
""'-<-' 6+1 ' е+1
16 Р. Е. Трейбал
(VI, 46)
241
0,000/
3 k 5 6 7 в
Число теоретических ступеней, п
Рис. 115. Многоступенчатая экстракция при перекрестном токе в случае
несмешивающихся растворителей (экстрагент распределен по ступеням
равномерно; е—фактор экстракции для каждой ступени).
Уравнение (VI, 46) является линейным разностным
уравнением первого порядка, решение которого имеет вид
, _/, _Es.)(_L.y+ys.
хп-\хр тУ U+l/ m
(VI, 47)
Это уравнение можно преобразовать к следующей более
удобной форме:
lg
к,-Ч)/(«-Ч)
lg(e+l)
_(xF-ys!m\lln
\ха-У3/т I
(VI, 48)
(VI, 49)
242
Удобное для расчетов графическое решение уравнений
(VI, 47) — (VI, 49) приведено на рис. 115.
Общее количество экстрагента Bt = пВ, и концентрация
экстракта
А(х„ — х\
Следует подчеркнуть значение величины фактора
экстракции е, который, как станет ясным из последующего изложения,
играет важную роль в проведении процессов экстракции. В
соответствии с определением m величина е имеет следующий
физический смысл:
тВ количество распределяемого вещества в экстракте ... _.
А количество распределяемого вещества в рафинате '
Из уравнения (VI, 47) следует, что для заданного общего
количества экстрагента, распределяющегося по ступеням
экстракции, увеличение числа ступеней приведет к увеличению
общего количества экстрагированного вещества или
уменьшению величины хп.
Однако, как будет показано ниже (см. раздел
«Дифференциальная экстракция»), величина хп не достигает нуля даже
при бесконечном числе ступеней и применении чистого (без
распределяемого вещества) экстрагента.
Рассмотрим вопрос о распределении общего количества экстрагента по
ступеням экстракции, необходимом для получения заданной степени
экстракции при наименьшем расходе экстрагента.
Пусть процесс осуществляется в установке из двух ступеней. Для
первой ступени
В. х„— х.
—г = — — (VI, 52)
Для второй ступени
В2 __ х1—х2 (VI, 53)
A mx2-ys
Тогда, если обозначить через Bt общее количество экстрагента для
двух ступеней, то
В, В.А-В- х„ — хЛ хЛ—х~
-J-= l\ 2 =—Л- L_l_J ?_ (VI, 54)
A A mxx — ys mx2 — ys
Для определения минимального количества Bt обеспечим условие
d(Bt/A)/dxl=0, откуда
m
16* 243
Подстановка в уравнения (VI, 51) и (VI, 53) дает:
A A m
Из уравнения (VI, 56)- следует, что наилучшим является распределение
экстрагента поровну между ступенями.
При трех ступенях экстракции уравнение (VI, 52) применимо для первой
ступени. Раствор, поступающий на экстракцию в последующие две ступени,
имеет концентрацию х\. Чтобы на этих ступенях расход экстрагента был
наименьшим, количество его должно распределяться поровну в соответствии
с уравнением (VI, 56), в которое вместо величин xF и хг нужно подставить
величины х\ и xs. Следовательно
— -^—) —1 (VI, 57)
Снова определив d(Bt/A),ldx] = Ot найдем:
Следовательно, минимальный общий расход экстрагента при трех
ступенях прямоточной экстракции также достигается в случае равномерного
распределения экстрагента по ступеням. Этот вывод можно распространить на
любое число ступеней.
Арис и др.6 показали, что равномерное распределение экстрагента по
ступеням экстракции наиболее экономично тогда, когда принимается в расчет
стоимость экстрагента при фиксированной концентрации ys. При определении
оптимальных условий проведения процесса экстракции необходимо, однако,
принимать во внимание стоимость ступеней, взаимозависимость стоимости
регенерации экстрагента и t/s, а также стоимость экстрагируемого компонента
(см. главу XII). Равномерное распределение экстрагента не является
наиболее экономичным при нелинейной равновесной зависимости; при сложных
равновесных зависимостях расчет целесообразно проводить на электронных
машинах6. Если коэффицкент распределения m изменяется незначительно
с изменением концентрации, в приведенные выше уравнения можно с
достаточной точностью подставлять среднюю геометрическую величину из значений
коэффициентов распределения (гг^гпг) •
Разработаны7 методы расчета составов рафината и
экстракта, изменяющихся во времени в случае внезапного изменения
концентрации экстрагируемого компонента в исходном
растворе, поступающем на разделение.
Пример VI-2. Обрабатывают 1,1,2-трихлорэтаном (В) при перекрестном
токе 100 кг 50%-ного раствора ацетона (С) в воде {А), в результате чего
концентрация ацетона в исходном растворе снижается до 10%. На каждой
ступени экстракции используют 25 кг экстрагента. Рассчитать число
ступеней и концентрации экстрактов. Экстракция проводится при
температуре 25° С.
Решение. Равновесные данные для этой системы опубликованы в
литературе [Ind. Eng. Chem., 38, 817 (1946)]. Рассмотрим рис. 116. Имеем:
F=100 кг; ХСу = 0,50; XBF = 0; S = 25 кг (для каждой ступени); Хса = 0;
Xlls=l,0.
>-ys/]
m V/s
■yjm
(VI, 56)
£.
mx,
244
С-ацетоп
Вода.
Трихлорэтан
Рис. 116. Многоступенчатая экстракция ацетона из воды трихлорэтаном
при перекрестном токе.
Для первой ступени, согласно уравнению (VI, 9)
FXcp + SlXcs _ 100-0,50 + 0
X,
СМ,
Из уравнения (VI, 10)
F + St
100+125
= 0,40
FXaB + S.Xat! 0 + 25-1,0
у BF~ 1 BS | л 20
лвм,~ F + Si 100 + 25
С помощью полученных данных можно определить положение точки М{:
Afi=F+Si = 100+25= 125 кг. Через точку Mi проводим хорду равновесия,
используя корреляцию для хорд равновесия. Из диаграммы, приведенной на
рис. 116, находим координаты точки 7?i: хса, =0,35; -^д/?, = 0,64; ^^=0,02.
Координаты точки £,: XCEi = 0,475; XAEi = 0,04; XBEi = 0,485.
По уравнению (VI, 14)
£,=
Ml(XCMi — XCRi) 125 (0,40—0,35)
Х СЕ, — XCR,
Из уравнения (VI, 6)
Rl=Ml — El--
0,475—0,35
125 — 50 = 75 кг
= 50 кг
245
I
Аналогичный расчет ведется для последующих ступеней; его результаты
представлены ниже:
Показатели
м2
R*
Ег
м3
Rs
Ез
М<
R*
Е<
*с
0,262
0,223
0,325
0,159
0,134
0,204
0,0925
0,075
0,120
ха
0/80
0,769
0,013
0,547
0,860
0,007
0,5945
0,920
0,005
*в
0,258
0,008
0,662
0,294
0,006
0,789
0,313
0,005
0,875
Количество
кг
100
61,8
38,2
86,8
55,8
31,0
80,8
50,3
30,5
Так как Хср =0,134 и XCRt = 0,075, для достижения заданной
концентрации в конечном рафинате .Х"^ =0,1 трех ступеней недостаточно, а
четырех — слишком много. Таким образом, необходимо около 3,5 ступеней.
Для получения рафниата заданной концентрации возможны три варианта
проведения процесса: а) использовать три ступени и несколько увеличить
подачу экстрагента на каждую ступень, определяя его количество нз опыта,
пока не будет обеспечено условие Хср =0,1; б) применить четыре ступени
и несколько уменьшить подачу экстрагента на каждую ступень, так же
определяя его количество методом последовательных приближений; в) принять
концентрацию ацетона в рафинате 7,5%- Следует отметить, что если общая
эффективность ступени составит I -4— I • 100 = 87,5%, будет достаточно
четырех реальных ступеней.
Принимая вариант «в», получим: количество конечного рафниата 50,3 кг,
который содержит 0,075-50,3=3,77 кг ацетона; 0,005-50,3=0,25 кг
экстрагента и 0,92-50,3=46,3 кг воды. Количество суммарного экстракта £=50+
+38,2+31,0+30,5=149,7 кг. Конечный экстракт содержит 50 — 3,77=46,2 кг
ацетона. Концентрации экстрактов:
46 2
*„„ =-т^=г =0,309
СЕ'
149,7
X
чпп /"общее количество \ ппс
100 кг —0,25 кг
\ экстрагента /
/количество экстрагента\
\ в рафинате /
BE
149,7
= 0,666
По полученным данным можно определить на диаграмме положение
точки Е.
Пример VI-3. В системе вода (А) — трнхлорэтан (В)—ацетон (С) вода
и трнхлорэтан практически взаимно нерастворимы до концентрации ацетона
в воде, не превышающей 27%. Коэффициент распределения m при этом
остается практически постоянным и равным 1,65. Количество исходного
раствора (для экстракции в перекрестном токе), содержащего 25% ацетона и
75% воды, составляет 1000 кг. Используется экстрагент, получаемый после
его регенерации, содержащий 1% ацетона и 99% трихлорэтана.
246
Определить: а) общее количество экстрагента, необходимого для
достижения концентрации ацетона в рафинате, равной 1% на пяти ступенях
экстракции (при равномерном распределении экстрагента по ступеням) и
концентрацию ацетона в суммарном экстракте; б) провести расчет по пункту
а) для одной ступени; в) число ступеней, если 2830 кг экстрагента
равномерно распределены по ступеням экстракции; г) до какой степени должен
быть очищен от ацетона регенерированный экстракт, чтобы уменьшить
содержание ацетона в конечном рафинате до 0,5%, если прн пяти ступенях
экстракции использовать 3000 кг трихлорэтана, поступающего с
регенерированным экстрагентом.
Решение
а) Согласно исходным данным, имеем:
хр = 0,25/0,75 = 0,333 кг ацетона/лгг воды
А = 0,75 • 1000 = 750 кг воды
ys = 0,01/0,99 = 0,0101 кг ацетона/лгг трихлорэтана
п = 5 хп = 0,01/0,99 = 0,0101 кг ацетона/лгг трихлорэтана
Далее
0 0101 °'0101
хп~ У*/т • Гб5~
" S ' =0,01217
V-V» 0,333—»-
1,65
При л=5 нз рнс. 115 определяем е= \,4=тВ/А = 1,655/750; 5=637 кг
трихлорэтана на ступень илн 637/0,99 = 644 кг регенерированного
экстрагента на ступень. Общее количество экстрагента 5-644=3220 кг.
Из уравнения (VI, 50)
750(0,333 — 0,0101)
Уц—~~ 3220 (-0,0101=0,0854 кг ацетона/кг трихлорэтана
0,0854-100 _._
—j~0gO— = '»87% ацетона в суммарном экстракте
б) Величина я=1. По уравнению (VI, 49)
rafi
А
( 0,01217 )~1=81'2
или
81.2Л 81,2-750 „RQm
а — ——— = —j-gc— = 36 900 кг трихлорэтана
36900 ...
0 qq =ol 3UU кг регенерированного экстрагента
в) Величина 5,=2830 • 0,99=2800 кг трихлорэтана. Расчет проводим
методом последовательного приближения. При л=7 величина В = 2870/7 =
— wu кг на ступень; величина е = тВ/А = 1,65 • 400/750 = 0,88.
Окончательно находим:
Уз
Хп m
- = 0,01217
к»к л прежде.
ys
F га
247
При п = 7 из рис. 115 определяем е = 0,88 (проверка). Следовательно,
п = 7 ступеней.
г) Имеем: Bt — 3000, я = 5; В = 600 кг на ступень; *„ = 0,005/0,995 =
= 0,00502, тВ/А = 1,65 • 600/750 = 1,32.
Из рнс. 115 получаем:
m
0,00502 -
хр
m
: 0.0145 =
1,65
0,333-
1,65
0,00035 кг ацетона/к-г тркхлорэтана
0,00035 ■ 100
1,00035
= 0,034% ацетона
ДИФФЕРЕНЦИАЛЬНАЯ ЭКСТРАКЦИЯ
Дифференциальная экстракция, как отмечалось ранее, во
многих отношениях подобна дифференциальной дистилляции.
Ола не применяется в промышленности и представляет интерес
в основном в лабораторных условиях вследствие того, что этот
процесс соответствует случаю предельного увеличения числа
ступеней при прямоточной экстракции. Подобно аналогичному
процессу дистилляции, реальный процесс может лишь в той
или иной степени приближаться к идеальному процессу,
описанному ниже.
Пусть сосуд, показанный на рис. 117, заполняют исходным
раствором F, после чего экстрагент S в небольших
количествах вводят снизу сосуда (если его плотность меньше, чем
плотность исходного раствора). Если исходный раствор не
насыщен, т. е. точка, соответствующая его составу, не
расположена на бинодальной кривой фазовой диаграммы, первые
порции экстрагента будут растворяться, пока исходный раствор не
- Мешалка
Слой_ з нстрак/па
Экстракт
_ Слой
рафишата
Экстрагент
Рнс. 117. Принципиальная схема Рнс. 118. Процесс дифференциальной
дифференциальной экстракции. экстракции в треугольной диаграмме.
248
станет насыщенным. Дальнейший ввод экстрагента приводит
к образованию слоя экстрагента, который собирается сверху и
тотчас же удаляется из сосуда. Сосуд снабжен мешалкой, и
при перемешивании экстрагент в нем задерживается; поэтому
экстракт всегда находится в равновесии с остающимся в сосуде
рафинатом. Процесс проводится периодически.
Расчеты будут выполнены с помощью треугольной
диаграммы (рис. 118). Количество экстрагента, которое нужно
ввести в процесс для насыщения исходного раствора и которое
соответствует перемещению точки F на бинодальную кривую
в точку R0, можно определить из баланса по компоненту В:
^^"Ш^^х**0^ (VI'59)
4*V>/ abs~abr„
На следующей стадии процесса, после того как начинает
образовываться слой экстракта, состав рафилата
характеризуется положением точки R. При добавлении очень малого
количества экстрагента dS количество рафината уменьшается
на величину dR, что приводит к увеличению слоя экстракта Е
(на противоположном конце хорды равновесия) на величину
dE. Новый рафилат, количество которого R—dR, имеет состав:
%ar — dXAR; XBR — dXBR; XCr — dXCR. Общий материальный
баланс процесса:
/? + rfS = (/?—dR) + dE (VI, 60)
dS = dE—dR (VI, 61)
Баланс по компоненту С:
*XCR + XCSdS=W-dRKXCR-dXCR)+XCEdE (VI>62>
XcsdS = XCEdE — RdXCR — XCRdR (VI, 63)
Баланс по компоненту А:
RXAR + XAS dS = (* - dR) (XAR - dXAR) + XAE dE <VI> 64>
XASdS = XAEdE-*dXAR-XARd* (VI. 65)
Исключая dS и dE из уравнений (VI, 61), (VI, 63) и (VI, 65),
получаем:
dR
R
dXCR
_ XCS~XCE
XCS~XCR
XCS~XCE
dxtn
AR
XAS — XAE
XAS — XAR
XAS — XAE
(VI, 66)
249
Интегрирование в соответствующих пределах дает:
dXr
Ы1Ь-) ~R-= J
CR
xCRf (xcs~xce)
XARt
XCS — XCR
XAS~XAR
XCS~XCE
XAS~XAEi
с
XARf{XAS-
-xae)
dXAR
XCS — XCR
. XCS — XCE
XAS~
XAS~
~XAR
-xae\
(VI, 67)
Это уравнение аналогично закону Релея для дистилляции и
может быть представлено графически. Первый интеграл — это
площадь под кривой, абсциссой которой является XCR, а
ординатой — выражение
1
(*,
CS
■X
СЕ.
XCS XCR
XAS~XAR
X.
CS
■X
СЕ
X
AS
■X
AEJ
Концентра ции Х-сеу Хае и Xqr, X-ar определяют на
противоположных концах хорд равновесия. Второй интеграл находят
графически подобным образом.
Величину Ef — количество конечного экстракта (не
находящегося в равновесии с Rf) можно найти исключением dS из
уравнений (VI, 61) и (VI, 63):
dE =
dR{Xcs-XCR)-RdXCR
X
(VI, 68)
cs
СЕ
Величина XCs постоянна, соответственно
(*cs-xcr0) r„
.-/*£_ J
d[(XCS-XCR)R]
X,
(XCS~XCRf)Rf
CS
■X
(VI, 69)
CE
Этот интеграл можно определить по площади под
кривой, построенной в координатах 1/(Хса—ХСе)—ордината
(Хсз—XCr)R — абсцисса, с использованием данных,
полученных при решении уравнения (VI, 67). Количество расходуемого
экстрагента находят из соотношения
S/ = Ef + Rr
(VI, 70)
К этой величине следует добавить количество экстрагента,
необходимое для насыщения исходного раствора и определяе-
290
мое уравнением (VI, 59). Состав конечного экстракта можно
получить из уравнений материальных балансов:
л V^ + *'*^.-*/***/ (VI.72)
ХВЕ,= ^-ХСЕГХАЕ, (V^73)
Для случая3'8, когда экстрагент состоит из чистого
компонента В и ХС8=ХАВ=0, уравнение (VI, 67) приводится к виду:
„,_§>_ Г **с* Г dX*« (VI. 74)
Rf J .. Хли ХсА .,■> „ l*AR ACR\
Rf vJ X (Xa* Х°Л xL X (^AR-.^cr
C*l св\*7в Хсе) Хдл/М
Из уравнения (VI, 69):
XAE XCE ,
Ef-
RoXcr,,
d (XCRR)
1 ^f1 (VI.75)
Rf
Для систем типа II:
*f*CRt CE
(J = 4^ • 4^ = const (VI, 76)
C'A XAE XCR
где Pc.a — селективность (см. главы II и IV). В этом случае,
если экстрагент состоит лишь из компонента В, подстановка 9
в уравнение (VI, 74) дает:
,п ■#■ = Т-^-Г (Ы Тр— рС л 1п £*") О". 77)
Rf Рсд-1 V Cff/ ARfl
Это уравнение аналогично интегралу закона Релея при
постоянной относительной летучести.
Пример VI-4. Обрабатывают по методу дифференциальной экстракции
4.1,2-трихлорэтаном при 25° С 100 кг 50%-ного раствора ацетона (С) в
воде (А). Концентрация ацетона в воде после экстракции снижается до 10%.
Рассчитать требуемое количество экстрагента, а также концентрации н
количества экстракта и рафнната.
Решение. Равновесные данные [Ind. Eng. Chera., 38, 817 (1946)]
приведены на рис. 119. Точка F, соответствующая исходному раствору, лежит-
ва линии FB, которая определяет также положение точки Ro- Имеем: FB =
-100 /C3;S=£; xj=0; XBS=XBB = 1,0; ХВЯ> = 0,045; XCRa = 0,478; XARt =
"*0,477. Точка Rf расположена на кривой растворимости и соответствует
Ю% ацетона. Находим: XCR =0,100; XAR =0,895.
251
По уравнению (VI, 59)
й _ f(.xbr0 — xbf)
100(0,045 — 0)
xbs — xbr,
1,0 — 0,045
4,71 кг
Следовательно, R0= 100 + 4,71 = 104,7 кг.
Согласно уравнению (VI, 74)
ХСо-0,478
/?, 104,7 C%f rf*
/?
'СЯ
А"
AS.
Л".
А"
АЕ
X
CR
СЕ J
ХЛ%=0,477
\
XAR
dX
X
\х
AR
АЕ
AR
X.
CR
X
АЕ
X.
СЕ.
Хорды равновесия на рис. 119 проведены на основании расчетов, часть
которых представлена ниже:
XCR
1
0,478
0,409
0,3088
0,2763
0,2600
0,1704
0,1000
Xar
2
0,477
0,5700
0,6795
0,7133
0,7300
0,8223
0,8950
*СЕ
3
0,592
0,5395
0,4297
0,3939
0,3706
0,2514
0,150
ХАЕ
4
0,107
. 0,0605
0,0311
0,0240
0,0209
0,0110
0,0060
~
х 1х
о:
t
X
о
X
5
0,463
0,2135
0,1100
0,0878
0,0788
0,0536
0,0470
-
0:1 ft]
ОС
X X
а:11ц
ч; ч;
X |х
X*
6
3,44
1,905
1,52
1,437
1,395
1,227
1,123
104,7
1П —
7
0,0000
0,2379
0,4274
0,4807
0,5057
0,6328
0,7217
R
8
104,7
82,6
68,3
64,8
63,2
55,5
51,0
xcrR
9
50,0
33,8
21,1
17,91
16,43
9,46
5,10
1
ХСЕ
10
1,690
1,853
2,325
2,54
2,795
3,98
6,67
Данные вертикальных столбцов 1—4 получены графически н
соответствуют концам хорд равновесия. Графическое интегрирование кривой,
построенной по данным столбцов 1 н 5, дает значение первого интеграла
уравнения, а ннтегрнрованне кривой по данным столбцов 2 н 6 — второго
интеграла. Значения разности интегралов в соответствии с выражением для
In Ro/R приведены в столбце 7. В столбце 8 даны значения R,
соответствующие определенному значению Xqr- Значение R. прн
равно 51,0 кг. По уравнению (VI, 75)
Ro*cr0
diXCRR)
104,7-0,478
XCR — XCR
7
= 0,100
c/
- J
RjXCRf
4XcR4)
X.
CE
51,0-0,100
X
CE
252
С-ацетон
Вода Трихлорэтан
Рнс. 119. Дифференциальная экстракция в системе ацетон — вода —
трихлорэтан [равновесие по данным, приведенным в Ind. Eng. Chem., 38,
817 (1946)].
Этот интеграл определяется графически прн построении кривой по
данным столбцов 9 н 10. Площадь под кривой соответствует £/=112,2 кг.
Согласно уравнению (VI, 70)
Sf = Bf == Ef -f Rf — R0 = 112,2 -4- 51,0 — 104,7 = 58,5 кг
Общее количество экстрагента:
Bf + Внач. = 58,5 -4- 4,71 = 63,2 кг
Из уравнений (VI, 71) и (VI, 73) рассчитаем концентрации Ef:
ХСЕ = 0,400; ХАЕ = 0,0383; ХВЕ == 0,562. Точка, соответствующая этому
составу экстракта, находится в двухфазной области диаграммы. Общий
экстракт будет образовывать два жидких слоя.
Несмешивающиеся растворители. Пусть исходный раствор
содержит А кг компонента А и имеет концентрацию
распределяемого компонента С, равную xF. Примем концентрацию
распределяемого компонента С в экстрагенте В равной ys. В
момент, когда концентрация рафината равна х, добавление
экстрагента в количестве dB изменяет концентрацию в экстракте
До у, а концентрацию в рафинате — на величину dx.
Баланс по компоненту С:
— Adx = (y — ys)dB (VI, 78)
253
Общее количество экстрагента:
Xf—ш -
= е
ys
х „ -
f га
тВ{
А
Bf xp
Bt= [ dB = A[ dX (VI, 79)
J J У — У*
О х^ л
Этот интеграл можно определить с помощью графика
зависимости \/(у — у в) от х. Значения хну берут на кривой
равновесного распределения.
Закон распределения. Если коэффициент распределения в
уравнении (VI,44) постоянен, подстановка его в уравнение
(VI, 79) дает:
(VI, 80)
Этот результат можно получить также из уравнения
(VI, 48), принимая, что количество экстрагента, приходящееся
на каждую ступень, равно Bt/n, и определяя значение предела
при условии, что величина п стремится к бесконечности. Из
уравнения (VI, 80) следует10, что даже при чистом экстрагенте
и бесконечном числе ступеней разделения в перекрестном токе
величина Xf (при конечном количестве экстрагента) не может
быть равна нулю *.
Степень экстракции распределяемого компонента всегда
возрастает при увеличении количества экстрагента для любого
числа ступеней, работающих в перекрестном токе. Однако
степень извлечения распределяемого компонента на п ступенях
разделения относительно степени извлечения при бесконечном
числе ступеней разделения (и том же количестве экстрагента)
всегда проходит через минимум при увеличении количества
экстрагента 5>10.
Минимум обычно находится вблизи значений тВ/А = 1,6—1,8
в зависимости от числа ступеней. При п = Ъ степень экстракции
составляет не менее 94% от степени экстракции при
бесконечном числе ступеней разделения.
* При равновесном распределении вида у=тхг, где г< 1,0, н чистом
экстрагенте полностью извлечь распределяемый компонент из исходного раствора
можно при
АХр
"' ш(1-г)
При г!> 1,0 необходимое количество экстрагента равно бесконечности.
254
ПРОТИВОТОЧНАЯ МНОГОСТУПЕНЧАТАЯ
ЭКСТРАКЦИЯ
При этом способе экстракции используют каскад ступеней,
в который исходный раствор и экстрагент вводят с
противоположных концов. Рафинат и экстракт движутся противотоком,
как схематично показано на рис. 120. Процесс проводится
непрерывно.
Треугольная диаграмма. Допустим, что на диаграмме4'11
(рис. 121) положение точек F, Eu Rn и S известно.
Материальный баланс для всей установки:
F + 5 = £, + /?л = Л1 (VI, 81)
или
/=• — £, = /?„ —5 = 0 (VI, 82)
Следовательно, если продолжить линии EiF и SRn, точка О
(рабочая точка, или полюс) будет находиться на их
пересечении. Материальный баланс:
для 1-й ступени
F + Em+i =£, + /?„, (VI, 83)
или
/=•-£, = Rm — Em+1 = О (У, 84)
для m-й ступени
Rm-i+Em+> = Rm + Em (VI, 85)
Ят-l— £т = /?т-£т + 1=0 (VI, 86)
Из уравнений (VI, 84) и (VI, 86) следует, что экстракт
(т + 1)-й ступени Em+t можно определить по рафинату т-и
ступени Rm, продолжив линию ORm до пересечения с ветвью
бинодальной кривой, соответствующей фазе, обогащенной
компонентом В. Так как рассматриваются теоретические ступени,
экстракт Ет и рафинат Rm будут находиться в равновесии друг
с другом (на противоположных концах хорд равновесия).
Следовательно, Ri можно определить на противоположном конце
хорды равновесия, проходящей через точку Ей Е2 — на
продолжении линии ORu R2 — на хорде равновесия, проходящей через
Е2, и т. д. Рабочая точка О может быть расположена со стороны
исходного раствора или со стороны экстрагента в зависимости
от относительных количеств исходного раствора и экстрагента
и наклона хорд равновесия.
Е, Ег . Е3 ti, сф cm^i с л &
Рнс. 120. Принципиальная схема многоступенчатой противоточной
экстракции.
с
Рис. 121. Многоступенчатая противоточная экстракция.
Положение точки М и состав соответствующей ей смеси
исходного раствора и экстрагента рассчитывают по уравнениям
(VI, 7), (VI, 9) и (VI, 10). Количества £\ и R„ можно
определить совместным решением уравнения (VI, 81) с уравнением
материального баланса по компоненту С:
£,=
м(хсм-хс*а)
XCEi — XCRn
(VI, 87)
Для любой т-й ступени общий баланс [уравнение (VI, 85)]
можно представить в виде:
Em+l = Rm + Em-Rrn_1 (VI, 88)
Материальный баланс по компоненту А для т-й ступени
т АЕ„
R ,Хлп + Е ,ХЛВ = R Хяо +Е X
т-\ ARm_l ■ m + l АЕт + 1 m ARm ' m
Решаем совместно эти два уравнения:
Rn
*„,_,(■
X
AR„
— Хь*
-1 АЕт +
,)+ЕЛ}
АЕ,
m + l
■X
АЕ„
X
AR„
-X
(VI, 89)
(VI. 90)
АЕ„
С помощью уравнения (VI, 90) рассчитываем величину Rt
(принимая т — 1 =F и т= 1); затем по уравнению (VI, 84) —
величину Е2 (при т=\)\ по уравнению (VI, 90) —величину
R2 (при т = 2); Е3 находим из уравнения (VI, 84) при т=2
286
и т. д. Таким путем (после определения соответствующих
концентраций) из треугольной диаграммы можно рассчитать
количества всех экстрактов и рафинатов. Способ расчета в
прямоугольных координатах идентичен.
Предельные количества экстрагента. Из треугольной
диаграммы (рис. 121) видно, что если при продолжении хорды
равновесия она пройдет через рабочую точку О,
последовательное определение числа ступеней методом расчета от ступени к
ступени окажется невозможным. Даже для того, чтобы
достичь в процессе экстракции извлечения, соответствующего
данной хорде, потребовалось бы бесконечное число ступеней
разделения, что отвечает ректификации с бесконечно малым флегмо-
вым числом. Поэтому в реальных условиях линия RnS не
должна совпадать с хордой равновесия, иначе на конце
установки, соответствующем входу экстрагента, движущая сила
процесса может оказаться равной нулю. Чем дальше отстоит
точка О от точки Rn, тем большее количество экстрагента
необходимо для проведения процесса. Соответственно для определения
минимального количества экстрагента можно применить
следующий графический способ (см. рис. 122).
Продолжим линию RnS и все нанесенные да диаграмму
хорды равновесия проведем до пересечения с продолжением
линии RnS. Точка пересечения, наиболее удаленная от Rn (на
рисунке — хорда равновесия HJ и точка пересечения О'),
соответствует минимальному расходу экстрагента. Действительное
положение рабочей точки О, отвечающее количеству
экстрагента, большему минимального, должно выбираться для каждого
конкретного процесса. В большей части случаев положение
рабочей точки при минимальном расходе экстрагента
определяет хорда GD, проходящая через точку F. Если хорды
равновесия наклонены книзу в направлении к вершине В, точки
Рис. 122. К определению минимального расхода экстрагента в процессе
многоступенчатой протнвоточной экстракции.
17 Р. Е. Трейбал
257
с
Рнс. 123. Двухступенчатая протнвоточная экстракция прн разных
температурах на ступенях.
пересечения их с линией RnS будут располагаться со стороны
вершины треугольника, соответствующей экстрагенту В. В этом
случае чем ближе точка пересечения расположена к точке S,
тем больше необходимый расход экстрагента.
Для сольютропных систем, у которых хорды равновесия с
возрастанием концентрации компонента С меняют направление
на противоположное, хорды равновесия при продолжении могут
пересекать линию RnS на противоположных сторонах
диаграммы- Пересечения со стороны вершины треугольника, отвечающей
экстрагенту, соответствуют большим соотношениям экстрагента
и исходного раствора, что должно быть учтено при расчете
минимального расхода экстрагента- Максимальное количество
экстрагента может достигать величины, при которой полностью
растворяется исходный
раствор, как в случае
одноступенчатой экстракции.
Изменение температуры.
Процесс экстракции при
разных температурах на
различных ступенях экстракции
легко представить на
треугольной диаграмме п,
используя для этого хорды
равновесия и кривые
растворимости,
соответствующие температуре, поддер-
В живаемой на данной ступе-
1 ХА и ни. Все уравнения, приве-
Рнс. 124. Графический расчет в случае Денные ранее, применимы и
значительного удаления рабочей точки В ЭТОМ случае. На рис. 123
от треугольной диаграммы:. представлена типичная диа-
258
грамма двухступенчатой экстракции при температурах на
ступенях, равных ti и t2.
Графоаналитический расчет. Иногда рабочая точка О
располагается на значительном расстоянии от фазовой диаграммы,
что затрудняет проведение расчета. В этих случаях возможно
применение следующего приема12 (см. рис. 124): после
определения положения точек F, Rn, S и Et (Et — из материального
баланса) продолжают линию FEt до пересечения со стороной
ВС диаграммы в точке /, а линию RnS — до пересечения с ВС
в точке L.
Уравнение линии FJ:
& XC=\XCFXA*CJ)XA + XCJ (VI-91>
Уравнение линии R„L:
J XC-[XC*X *CL)XA + XCL ^1,92)
3" \ ARn I
Уравнения (VI, 91) и (VI, 92) позволяют получить значения
_ координат точки О:
хао = Пс xCJ~xCL х (VI-93>
XAF XAR„
Координату XCo можно определить подстановкой величины
ХА0, найденной по уравнению (VI, 93), в- уравнение (VI, 92).
Положение точки К, лежащей на продолжении линии ORmEm+\,
можно найти из уравнения
хск = [хС° -х"т)Хао + Хсо (VI.94)
\ ARm ЛА01
Поэтому, определив положение точки R{ на конце хорды
равновесия, проходящей через Е{, рассчитывают величину Хск
и наносят точку К- Проводя линию RtK, получают точку Ег на
бинодальной кривой и т. д. Эти уравнения и способ расчета
применимы также для равносторонней треугольной диаграммы
и в тех случаях, когда точка О лежит на диаграмме со стороны,
отвечающей экстрагенту. Они могут быть использованы и при
расчете по диаграмме Енеке (см. ниже).
Диаграмма распределения. Если число ступеней велико,
расчет часто более удобно проводить с помощью простой
диаграммы распределения (с координатами ХСА и Хсв) в
сочетании с треугольной диаграммой3. Баланс по компоненту С для
ступеней от п-й до (т+1)-й:
SXCS + KmXCRm = RnXCRn + Em + lXCEm+1 (VI. 95)
it!" I?»
258
СНп-1 ЛСКг
Концентрация С врафинате ХСА, Sec доли,
а 6
Рнс. 125. Построение процесса многоступенчатой противоточнон
экстракции в прямоугольных координатах:
/ — равновесная кривая Х„„ =f(XCD \, // — раб
— рабочая линия X
СЕ,
-f(*.
(Xc«J
Это уравнение можно преобразовать:
Е
X
cr„
. л. у
т+\
X
SX
CS
СЕ,
/71+1
Л»
(VI, 96)
Уравнение (VI,96) является уравнением рабочей кривой,
связывающей в координатах ХСА и ХСв величины Хсе , и
Xcr . так как величины S, XCs, Rn и Xqr постоянны.
Рассмотрим рис. 125,6, на который нанесена кривая
равновесного распределения Хсе —f{XcRm), построенная по рав-
XCR,XCFXCA
Рнс. 126. Пересечение рабочей н равновесной линий:
I — равновесная линия; 2 — рабочая линия.
260
новесным данным рис. 125, а.
Из рабочей точки О
произвольно проведем линии,
подобные линии OEm+i.
Пересечения этих линий с бинодальной
кривой дают координаты Rm
и Ет+\. Эти линии не
совпадают с линиями, нанесенными
ранее для определения числа
ступеней. Соответствующие
координаты переносят на
диаграмму распределения и
получают рабочую линию.
Проводя ступенчатую линию
между рабочей и равновесной
кривыми, находят число
ступеней и концентрации экстракта
и рафината. При минимальном
расходе экстрагента (число
ступеней равно бесконечности)
равновесная и рабочая линии
касаются друг друга, причем
в точке их касания движущая
сила процесса равна нулю.
Необходимо, однако,
отметить, что если концентрация
компонента С в критической
точке меньше, чем XCf, рабо-
Рис. 127. Протнвоточная экстракция:
,. а —в координатах Енеке; б — в
прямоугольная И равновесная ЛИНИИ бу- Ных координатах.
дут пересекаться, как
показано для двухступенчатого процесса на рис. 126. Возможность
подобной ситуации отмечалась Рэндэлом и Лонгтином ' для
некоторых процессов абсорбции. Важно также отметить, что при
определении числа теоретических ступеней уравнение (VI,95)
имеет значение только при целых значениях т, а кривые
рабочие линии (рис. 125,6 и 126,6) в действительности не
являются непрерывными, а лишь условно проведены по точкам
(-^ся,, Xcf), (Xce2, Xcr) и т. д. Растворы, составы которых
определяются координатами рабочей линии между указанными
выше точками, реально в процессе не существуют.
Расчеты от ступени к ступени. Можно также определить
число ступеней аналитически путем расчета от ступени к ступени.
Этот метод нецелесообразно применять для трехкомпонантных
систем; в дам-ном случае результаты можно получить более
быстро с помощью графического расчета. Метод расчета от
ступени к ступени иногда бывает необходимо использовать при
расчетах экстракции в многокомпонентных системах, для кото-
261
рых его обычно и применяют. Использование этого метода
иллюстрируется в приведенном ниже примере VI-5.
Диаграмма Енеке. Схема расчета для систем типа II
показана на рис. 127, а. Зная положение точек F, S, Ех и Rn,
положение точки М можно определить графически или с помощью
уравнений (VI, 17) —(VI, 22). Для всей установки:
F-fS = E, + R„=M (VI, 97)
F-E! = R„-S (VI, 98)
Для ступеней от 1-й до т-й:
F + Em+I = E, + Rm (VI, 99)
F-E1 = Rm-Em+1 (VI, 100)
Следовательно
F-E, = Rm-Em+1=R„-S = 0 (VI. 101)
причем точка О является общей для всех ступеней. Она^опреде-
ляется графически на пересечении продолжений линий EtF и
' SRn или по ее координатам.
Баланс экстрагента:
™Р-Е1"ВГ0"° = **"*П-*"3 (VU02)
°ТКУДа F^-EA* R^ -SNS
Баланс по компоненту С:
откуда
™F - E,X£l = ОХо = К»хЛя - SXs <VI> Ш)
FX„-E.X_ R„X -SX
х0= ;_Е;£- —4t=s- (VU05)
Число ступеней определяют попеременным проведением
линий из точки О и хорд равновесия, так же как в треугольной
диаграмме. Количество М можно рассчитать из уравнения
(VI, 97). После этого находят
Е1=мЫМг (VU06)
И R„=M-E, (VI, 107)
Для любой ступени
Rm+1 + Em+1=Rm + Em (VI, 108)
R м J-E N' =R Nn + Е N- (VI, 109)
откуда
^_«-(^,-^)+'.Р'д„.-"0 (VU10)
Дт £т+1
262
Уравнения (VI, 110) и (VI, 100), применяемые совместно,
позволяют рассчитать весовые количества различных
экстрактов и рафинатов. Если в качестве экстрагента используется
чистый компонент В, то Хо = Хл и линии SF и SRn будут
вертикальными, так как точка S удалена в бесконечность. Кроме
того, число ступеней можно рассчитать, пользуясь диаграммой,
показанной на рис. 127,6.
Типичные задачи по расчету процесса многоступенчатой про-
тивоточнсй экстракции. Обычно F, XF и Х8 для данного
процесса экстракции известны. Прочие переменные (количество
экстрагента S, число ступеней п и составы экстракта и рафи-
ната Хе, и Xr ) можно определить попарно следующим
образом: a) S и п — определить графически методом
последовательных приближений положение точки Е\, затем — Rn на
продолжении линии EiM и построить на диаграмме необходимое число
ступеней так, чтобы Rn и Еп лежали на противоположных
концах хорды равновесия; б) л и Хе1 или Xr/ — определить
положение точки S методом последовательных приближений, пока
число ступеней не будет построено так же, как при заданных S
и п [см. пункт «а»]; в) S и XEl или Хцп — определить М и
Xr или Хе1 . О и затем найти прямым расчетом величину п;
г) Xr и Хе{ —определить положение Rm, Eu M, О и
рассчитать значения S и п.
Пример VI-5. Экстрагируют 1,1,2-грнхлорэтаном 100 кг/ч. 50%-ного
раствора ацетона (С) в воде (А). Расход экстрагента 30 кг/ч. Процесс
экстракции— многоступенчатый протнвоточный; температура 25° С. Концентрация
ацетона в исходном растворе после экстракции снижается до 10%.
Определить число ступеней, а также концентрации и весовые количества различных
потоков.
Решение. Равновесные данные опубликованы в литературе [Ind. Eng.
Chem., 38, 817 (1946)]. Воспользуемся рис. 128, на котором отметим точки,
соответствующие F, S (чистый В) и Rn- Расчет ведем на 1 ч. Получим:
f = iook2 b = s = 30kz xbf = o xbs = xbb = i,o
По уравнению (VI, 10)
FXRR + SXR. 100-0 + 30-1,0
у Br ' Bi> I п oqi
лвм~ f+s ~ 100+30 -и-АИ
^ = /=• + 5= 100 + 30 = 130 кг
Точка М лежит на линии FB; продолжая линию RnM, определяют
положение точки Е\. Продолжая линии EtF и BRn до пересечения, находят
точку О. Точка Ri расположена на хорде равновесия, проходящей через Е\\
точку £2 находят, продолжая линию ORi, точку /?2—на хорде равновесия,
выходящей из £г, и т. д. Из диаграммы получаем следующие значения
концентраций:
Номер ступени 12 3 4 5 6
Экстракт, ХСЕ. . 0,557 0,489 0,403 0,300 0,180 0,038
Рафннат, XQR . . 0,438 0,363 0,287 0,206 0,120 0,025
263
&
о
я
о
S
Я'
я
о.
о
а
S
н
о
а,
с
с
о
я
S
а
л
Из диаграммы видно, что для
получения заданной концентрации
XCg =0,10 достаточно около 5,2
теоретических ступени. Чтобы число
теоретических ступеней было целым и
равнялось пяти или шести, надо
было бы найти подбором
необходимое количество экстрагента.
Принимаем (не изменяя количества экстра- *<\->t
гента) 5,2 теоретических ступени.
По уравнению (VI, 87)
СО0
too
10
Ujl Cfc
£, =
м(хсм-хсяп)
0,1
X
С£, XCRn
130(0,384 — 0,10)
= 80,8 кг
~~ 0,557 — 0,10
По уравнению (VI, 81)
/?„ = М — £, = 130—80,8 =
= 49,2 кг (1096-нын раствор ацетона)
Количество неэкстрагированного
ацетона:
49,2-0,10 = 4,92 кг
0,01
ороь
о
*-^
.
-
.
-
"в
«с
Ка
р^
02
0Л
OjB
Концентрация ацетона
брафинате (xcR= Xc^, Sec. доли
Рнс. 129. Коэффициенты
равновесного распределения для системы
ацетон — вода — трихлорэтан при
25° С (Р — критическая точка).
Количество экстрагированного ацетона:
50 — 4,92=45,1 кг, или 90,2% от содержания ацетона в исходном растворе.
Для первой ступени по уравнению (VI, 90)
F (XAF — ХАЕ2) + Е\ (ХАЕ2 — ХАЕ,) _
/?!'
X
ARX~
■X
АЕ-,
100 (0,50 — 0,044) -f 80,8 (0,044 — 0,073)
/?2 =
0,531 — 0,044
Согласно уравнению (VI, 84)
Е2= R1—F-\-Ei =89,0—100-)-80,8=69,8 кг
Для второй ступени в соответствии с уравнением (VI, 90)
#1 (XAR, — ХАЕг) + Е2 (ХАЕ, ~ ХАЕ2) __
= 89,0 кг
XAR2 — ХАЕ2
■■ 73,5 кг
= 89,0 (0,531 — 0,027) -4- 69,8 (0,027 — 0,044)
— 0,622 — 0,027
По уравнению (VI, 34)
Е3 = /?2 — F -f £, = 73,5 — 100 4- 80,8 = 54,3 кг
Аналогично можно рассчитать количества прочих экстрактов н рафн-
натов.
Расчеты от ступени к ступени. Решим теперь приведенный выше
пример аналитическим способом, применимым для любого числа компонентов.
265
Однако следует отметить, что для случая разделения трехкомпоиеитиой
системы рассмотренный выше графический метод расчета является более
простым и быстрым.
В случае расчета по методу от ступени к ступени необходимы график
зависимости коэффициентов равновесного распределения К,п = ХЕ /XR
т m
для каждого компонента от концентрации одного из компонентов при
равновесии, а также график равновесных данных, подобный приведенному на
рис. 128. Первый из этих графиков показан иа рис. 129, где дана
зависимость К для ацетона, воды и 1, 1,2-трихлорэтаиа от ХСа— концентрации
ацетона в водном растворе.
Расчет ведем иа 1 ч. По условию количество исходного раствора 100 кг
(50 кг ацетона и 50 кг воды); XCf=0,50; XAF — Q,bQ. Количество экстраген-
та S=30 кг; XBS=l,0; XAS = XCs = 0. Рафинат представляет собой
насыщенный раствор, XCR = 0.10. На кривой растворимости получим XBR =0,00613
и XAR = 0,8939 (это аналогично расчету температуры кипения в процессе
дистилляции). Экстрагеит после первой ступени Е\ также должен быть
насыщен, это позволяет рассчитать состав Е\. Такой расчет лучше провести на
фазовой диаграмме по данным предыдущего расчета (X AF = 0,073;
ХВЕ< = 0,370; XCEi = 0,557)..
Общий материальный баланс:
F + S = Et + Rn
100 + 30= Ei+Rn
Баланс по компоненту С:
50 + 0 = 0,557£14-0,10/?п
Решая эти уравнения совместно, получим £i=80,8 кг; Rn=49,2 кг.
Исходя из состава конечного рафината, составим следующую таблицу:
Компонент
А
В
С
Сумма
XR
0,8939
0,00613
0,100
1,000
кг
43,98
0,30
4,92
49,20
Кп
0,0067
137,5
1,50
—
**» = *»**„
0,0060
0,844
0,1500
1,000
Значения К взяты из графика (рис 129). Сумма ХЕ должна быть
равна единице, если только равновесные данные правильно нанесены на график
и правильно используются в расчете. Чтобы перейти к следующей ступени
(за последней), составим общий баланс для гс-й и (п—1)-й ступеней:
Яп-1 = Rn + Е„ — S
Балансы для каждого компонента в форме уравнений (VI, 96)
перепишем в соответствии с принятыми индексами для каждого компонента.
Расчет проводим методом последовательных приближений до тех пор, пока
состав рафината с (п—1)-й ступени не будет соответствовать составу иа
бинодальной кривой. Так, например, примем: £„ = 40 кг:
Rn-i = 49,2 -4- 40 — 30 = 59,2 кг
266
[49,2 • 0,00613 + 40 • 0,844 — 30-1] = 0,0685
Уравнение (VI, 96) для В дает;
х L
*BRn-l~ 59,2
Для С имеем:
хго = -4о- [49,2 • 0,1 -4- 40 • 0,15 — 30 • 0] = 0,222
Эти концентрации ие лежат иа бинодальной кривой диаграммы. Если
принять £„ = 35,7 кг, то Д„_, = 54,9 кг; XAR _ =0,8053; XBR _ =0,0075;
: 0,1872. Таким же способом проводим расчет для прочих сту-
X
С#„
пеней и результаты расчетов сводим в таблицу:
Ступень
п
Сумма . . .
и—1
Сумма . . .
и—2
Сумма. . .
и—3
Сумма . . .
и —4
Сумма . . .
и—5
Компонент
А
В
С
А
В
С
А
В
С
А
В
С
А
В
С
А
В
С
XR
0,89.39
0,0061
0,1000
1,000
0,8053
0,0075
0,1872
1,000
0,722
0,010
0,268
1,000
0,640
0,0142
0,346
1,000
0,554
0,025
0,421
1,000
0,453
0,055
0,492
кг
43,98
0,30
4,92
49,2
44,21
0,41
10,28
54,90
44,54
0,62
16,54
61,70
45,24
1,00
24,46
70,70
46,64
2,11
35,45
84,20
*т
0,0067
137,5
1,5
0,0157
95,00
1,46
0,032
59,6
1,42
0,064
34,5
1,36
0,120
15,7
1,29
ЧГ^Чп
0,006
0,844
0,150
1,000 •
0,0127
0,714
0,273
1,000
0,023
0,596
0,381
1,000
0,041
0,489
0,470
1,000
0,066
0,391
0,543
1,000
Ет
кг
0,21
30,13
5,36
35,70
0,54
30,36
11,60
42,50
1,18
30,70
19,62
51,50
2,67
31,78
30,55
65,00
6,04
35,78
49,78
91,50
Отношение XCRIXAR для (п — 4)-й ступени равно 0,761, а для (п — 5)-й
ступени составляет 1,088. Так как отношение этих компонентов в исходном
растворе равно 1,00, точка ввода исходной смеси находится между (п — 4)-й
и (п—5)-й ступенями. Следовательно, эта точка лежит между 5-й и 6-й
ступенями каскада.
Концентрации и весовые количества жидкостей, рассчитанные по методу
от ступени к ступени, не совпадают с теми же величинами, определенными
графическим способом, потому что для проведения процесса требуется ие
Целое число ступеней. Кроме того, расчет от ступени к ступени начинают с
и-й ступени, в то время как графический расчет — с 1-й ступени. При целом
числе ступеней оба метода должны дать одинаковый результат.
267
^ 70
Й: Со
!* и*7
ig-g §
sis зо
44~k
щ_ S. ю
°>аоо
/
^
'
у'
1
г.
1 1
I
!
и А;
1
з
200
100
1
\
\
,з
'1
~
Теперь представляется
возможным сравнить три
типичных способа
проведения процесса экстракции:
дифференциальную
экстракцию, многоступенчатую
прямоточную и
многоступенчатую противоточную
экстракцию. Расчеты, приведенные
в примерах VI-2 и VI-5,
были дополнены расчетами
числа ступеней и составов
экстракта для количеств
экстрагента, отличных от
принятых ранее.
Однако для всех вариан-
Jq Jz So тов расчета принимали по-
Число теоретических ступеней, п стоянными концентрации ис-
г , ° ходного раствора, конечного
рафината и экстрагента.
Результаты расчетов
приведены на рис. 130. Оказалось,
что при данном числе
теоретических ступеней
меньшее количество
экстрагента требуется при
многоступенчатой противоточной экстракции. При этом получают
меньшее количество экстракта и, следовательно, большую
концентрацию распределяемого компонента в экстракте.
Указанные результаты применимы для любого числа
ступеней, кроме одной. На одной ступени для обоих
рассматриваемых способов экстракции (см. рис. 130) получают идентичные
результаты. Увеличение числа ступеней сверх пяти приводит к
незначительным изменениям. Предельные условия экстракции
(число ступеней равно бесконечности) для противоточной
экстракции определяли по методам, описанным при нахождении
минимального расхода экстрагента, для прямоточной
экстракции— по данным примера VI-4 для дифференциальной
экстракции. Ход кривых может существенно отличаться от
приведенных на рис. 130 и,зависит от равновесных данных и составов
экстракта и рафината (см., например, работу Вартериссиана и
Фенске 3).
Рис. 130. Экстракция ацетона из воды
трихлорэтаном:
а — концентрация распределяемого компонента
в экстракте; б —требуемое количество
экстрагента; 7 —противоток; 2—перекрестный ток;
3— перекрестный ток (общее количество
экстрагента).
Пример VI-6. Экстрагируют по многоступенчатой противоточной схеме
чистым бензолом (В) при 25° С 100 кг/ч раствора, содержащего 30% изо-
пропанола (С) и 70% воды (А). Насыщенный конечный рафииат должен
содержать 2% изопропаиола. Определить минимальное количество
экстрагента, которое можно использовать в процессе.
266
Решение. Применяем равновесные данные Олсена и Уошбурна [J. Am.
Chem. Soc, 57, 303 (1935)], приведенные на рис. 131, где показаны некоторые
хорды равновесия. Расчет ведем на 1 ч. Имеем: F=100 кг; Xcf = 0,30; XAf =
= 0,70; XBs = XBB = \fl; S = B; Хся = 0,02.
На диаграмме показано положение точек F и Rn и проведена линия
BRn- Хорды равновесия для этой системы наклонены к вершине А (примерно
до концентрации изопропаиола, равной 24%), а затем изменяют наклон и
ниже указанной концентрации наклонены в сторону вершины В. Хорда
равновесия HJ параллельна линии BRn.
Продолжение хорды равновесия DG, проходящей через точку F,
пересекает линию BRn в точке О'. Поэтому рабочая точка не может находиться
между точками О' и Rn. Любая другая точка, лежащая слева от О', не
удовлетворяет условию, так как она является точкой пересечения линии BRn
с некоторой хордой равновесия, лежащей между DG и HJ. Хорды
равновесия, расположенные ниже HJ, пересекают линию BRn справа от В, и чем
ближе рабочая точка к точке пересечения продолжений линий HJ и BRn,
тем большему количеству экстрагента оиа соответствует. При продолжении
хорд равновесия LN и PQ они пересекают RnB в точке О", ближе всего
расположенной к точке В. Хорды равновесия, проходящие ниже PQ и между
хордами LN и HJ, ПврвСбКЭЮТ Дп В значительно правее точки О". Поэтому
точка О" соответствует наименьшему количеству экстрагента. Проводим
линию 0"F1 пересекающую кривую растворимости в точке Е\. Затем проводим
линии BF и RnE\, которые пересекаются в точке М\ Хвм = 0,69.
Следовательно, из уравнения (VI, 11)
S=B:
F(X
вм Xbf">
100(0,69 — 0)
Xbs~ хвм
1,0 — 0,69
=222 кг
Полученный результат соответствует минимальному расходу экстрагента
в час. Положение действительной рабочей точки должно быть выбрано
между точками О" и В.
Экстракция из нескольких растворов. Иногда возникает
задача провести в одном каскаде ступеней экстракцию из двух и
более растворов различных составов с получением одного ра-
Изопропанол
В Ж
Бензол
рис. 131. К определению минимального количества экстрагента (система
вода — изопропанол — бензоле
269
Рис. 132. Принципиальная схема многоступенчатой противоточной
экстракции из двух растворов.
фината и одного экстракта. Рассмотрим, например, схему
экстракции из двух исходных растворов F и F\ показанную на
рис. 132. Концентрация компонента С в исходном растворе
F' является промежуточной между концентрациями его в
растворах F и Rn- Исходный раствор F' можно ввести в ту точку
каскада, где концентрация С в рафинате точно соответствует
концентрации С в фазе F'-
На рис. 133 показано построение процесса в треугольной
диаграмме. Материальный баланс всей установки:
/? + /?, + S = £, + fl/I = Af (VI, И1)
Принимая F + F' = L и проведя преобразования, получают:
F + F'—El = L — El=Rn — S = 0' (VI.112)
Следовательно, положение рабочей точки О' можно
определить так же, как при экстракции из одного суммарного рас-
Рис. 133. Многоступенчатая противоточная экстракция из двух
растворов.
270
а
*с Рис. 134. Графический расчет числа теоретических ступеней при экстракции
I из двух растворов.
к
твора L. Материальный баланс для ступеней от т-й до п-и:
F' + Rm-i + S = Ra + Em (VI, ИЗ)
F' + Rm-i-Em^Rn-S=(y (VI, 114)
или
Следовательно, положение рабочей точки для ступеней,
лежащих между пг-и и п-и ступенями, определяется так же, как
для экстракции из одного исходного раствора. Для удобства
мы можем рассматривать исходный раствор, поступающий на
m-ю ступень, состоящим из растворов Rm-\ и F'\ при этом
Rm-i+F'=T, и расчет можно провести для рассматриваемой
части каскада таким же способом, как прежде. Материальный
баланс для ступеней от 1-й до (т—1)-й:
F + Em = Rm_l+El (VI, 115)
ИЛИ
F -£,=Лт_,-£и = О (VI, 116)
Следовательно, точка О является рабочей точкой для
части каскада, содержащей ступени от 1-й до (т—1)-й. Комби-
«ируя уравнения (VI, 112) и (VI, 116), найдем:
С-
0 + F' = Rn — S=0'
(VI, 117)
271
Из уравнения (VI, 117) следует, что точка О' лежит на
линии F'O. Полная схема расчета, включающая определение
числа ступеней, приведена на рис. 134. Рабочая точка О
используется для ступеней от 1-й до {т—1)-й. На следующей пг-й
ступени концентрация С в рафинате равна Xcf' и находится
в пределах между Хсцт и Xqr _ • Поэтому для ступеней от
m-й до я-й при расчете используют рабочую точку О'.
Схема расчета в координатах Енеке приведена на рис. 135
и с применением кривой распределения — на рис. 136. Рабочая
линия на последней диаграмме имеет перегиб в точке ХСР'\
верхняя часть рабочей линии получается при проведении
произвольных лучей из точки О (см. рис. 134); нижняя часть —
при проведении подобных лучей из точки О'.
Пример VI-7. Два исходных раствора (F, состоящий из 50% воды и
50% ацетона, и F', содержащий 25% ацетона в воде) экстрагируются в про-
тивоточной установке 1, 1, 2-трихлорэтаном при 25° С. Производительность
установки по каждому раствору 100 кг/ч; расход экстрагента 50 кг/ч. По-
Рис. 135. Расчет многоступенчатой прогивоточной экстракции из двух
растворов в координатах Енеке.
272
"n-i CRm*1 '
СЛт-г
Рис. 136. Расчет многоступенчатой противоточной экстракции из двух
растворов в прямоугольных координатах.
лучаемый рафинат должен содержать 10% ацетона. Рассчитать число
ступеней и определить ступень, на которую следует подавать раствор F'.
Решение. Равновесные данные [Ind. Eng. Chem., 38, 817 (1946)]
приведены на рис. 137. Расчет ведем на 1 ч. Имеем: F=100 кг; Хс/Г=0,50;
^^ = 0,50; Г =100 кг; XCF, = Q,2b; XAF, =0,75; L = F + F' = 200 кг. Отсюда
X,
FXCF + F'XCF, 100 • 0,50 + 100 • 0,25
CL
F + F'
100+100
= 0,375
С-ацетом
Рис. 137. К примеру VI-7.
18 Р. Е. Трейбал
Трихлорэтан
273
70
СО 60
По
«а
§|
||^
^
I 10
О
\
/ (
(*п
•)
ад
з)
(F.E.)
Далее находим: S = B = 50 кг;
X S=X в=1,0; ^„ —
насыщенный раствор; XCR = 0,10.
Определяем положение точек В, F, F',
L и Rn и проводим линию LB.
Координаты точки М:
X
LX..+BX
вм~
BS
200-0 + 50. 1,0
= 0,200
10
£0x^30
200 + 50
М = L + S = 250 кг
Определяем положение точки
М на линии LB. Находим
положение точки Е\ на пересечении
4-0 50 ВО продолжения линии RnM и
кривой насыщения. Определяем
положение рабочей точки О' на
пересечении продолжений линий E\L
и BRn. Продолжаем линии O'F' и
FE\ до пересечения в точке О.
Число ступеней находим в
прямоугольных координатах
(рис. 138). Верхнюю часть
рабочей кривой определяем по точкам Хср и X„D полученным при пересечении
лучей, произвольно проведенных из точки О, с кривой насыщения. Нижнюю
часть рабочей кривой строим аналогично, проводя произвольно лучи из
точки О'. Строя ломаную линию, получаем число теоретических ступеней.
Общее число теоретических ступеней равно 8,7; исходный раствор F' следует
вводить между четвертой и пятой ступенями от точки ввода раствора F
в каскад.
По уравнению (VI, 87)
Концентрация ацетона
В Водных растворах XCR, дес.%
Рис. 138. Определение числа
теоретических ступеней к примеру VI-7.
Е, = -
X
СЕ,
■х
CR„
250(0,30 — 0,10)
0,523 — 0,10
= 118,1 кг
По уравнению (VI, 112)
Л„ = 1 —£,+S = 200—118,1+50 = 131,9 кг
Несмешивающиеся растворители. Так же, как в случае
прямоточной экстракции, к существенным упрощениям расчета
приводит допущение, что жидкости А и В практически
нерастворимы друг в друге в пределах изменения значений
концентраций С, происходящих при экстракции4^13. В настоящем разделе
будут использованы обозначения, принятые ранее: у = Хсв/Хвв,
х = ХСА/ХАА и т. д.; при этом количества компонента А в
исходном растворе и всех рафияатах и компонента В в экстрагенте
и всех экстрактах остаются постоянными. Баланс по
компоненту С для ступеней от 1-й до т-н:
Ах^ + ВУ,п+\-=Ах,п + вУ1 (VI,,118)
274
Это уравнение можно преобразовать к виду:
Ут+1 — "д" хт + У\ '
-BXF
(VI, 119)
Последнее уравнение представляет собой уравнение прямой
линии ym+\ = f(xm), наклон которой равен отношению А/В, так
как у\ и xF являются постоянными для данной установки.
Поскольку т — номер любой ступени, уравнение линии («рабочей
линии») применимо ко всей установке, и эта линия может быть
проведена между двумя точками, координаты которых (xF, t/i)
и (хп, ys). На рис. 139 рабочей является линия GK. Можно
принять, что каждая теоретическая ступень каскада работает по
схеме прямотока, причем на ней достигается состояние
равновесия. Например, для п-й ступени из уравнения (VI, 43)
получим следующее уравнение рабочей линии ступени:
А
(VI, 120)
-ys
' хп-\
в
Это уравнение соответствует линии MN на рис. 139.
Прямоугольник GMPN представляет собой я-ю ступень, а подобные
прямоугольники (показанные на рисунке) — прочие ступени. На
практике проводят лишь линию GK и для определения числа
ступеней строят ступеньки, представляющие собой две стороны
каждого из прямоугольников.
Обычной задачей является определение числа ступеней при
заданных величинах A, xF, xn и у8. Тогда рабочую линию строят
из точки G (см. рис. 139), заканчивая ее в точке, абсцисса
которой xF, а ордината зависит от соотношения потоков
растворителей. При больших расхо-
У
У,
Уг
дах используемого экстраген-
та рабочая линия имеет
небольшой наклон, и для
проведения экстракции необходимо
небольшое число
теоретических ступеней. При малых
расходах экстрагента рабочая ли- у
ния идет более круто и число
ступеней возрастает. При
уменьшении количества
экстрагента точка К на рисунке
будет приближаться к точке D,
для которой число ступеней
равно бесконечности, что
соответствует минимальному
расходу экстрагента. Как
отмечалось выше, если рабочая
линия касается равновесной
ys
Рис. 139. Многоступенчатая противо-
точная экстракция с несмешиваю-
щимися растворителями:
/ — равновесная кривая хт = /(Ут)' // —
рабочая линия xm — f(ym + \) (наклон Л/В);
/// — рабочие лнннн ступеней (наклон — А/В).
18*
27S
кривой в какой-либо точке между хп и xF, наклон этой рабочей
линии соответствует минимальному расходу экстрагента.
Закон распределения. Если при взаимной нерастворимости
исходного раствора и экстрагента применим также закон
распределения [уравнение (VI, 44)], возможны дальнейшие
упрощения и можно не прибегать к графическому методу расчета.
Материальный баланс для ступеней от (т+1)-й до п-и:
Ахт + ВУ5 = Ахп + ВУт+1 (VU21)
Подстановка выражения ym+i — mxm+i [по уравнению (VI,44)]
при допущении, что е = тВ/А, дает:
-г+if—г+*«•+• <VI-122>
Это уравнение представляет собой линейное разностное
уравнение первого порядка, решения которого5 имеют вид:
при е ф 1 Хя = ( V- 51-1/Е ) (т) + 1-У <VI' 123>
при е=1 хт = хр — т(хп — ys/m) (VI, 124)
Решения уравнения (VI, 122) применимы для расчета
концентраций промежуточных рафинатов аналитическим путем.
При т = п можно получить следующие широко применяемые в
расчетах соотношения:
■",""М -фф'^г^х (VI. 125)
*[(¥Н£)(-т)+т]
п = ±±—2 ^у^ ± (VI, 126)
ПРИ&=1 rCfL-^ГТ (VU27)
— ys/m n+l
хт= — ■*„
F л
ysi
(VI, 128)
Уравнения (VI, 125) — (VI, 128) аналогичны известным
уравнениям Кремсера — Брауна — Саудерса 14'15, выведенным ими
для процессов абсорбции *. Графическое решение этих
уравнений приведено на рис. 140. Указанные выражения, хотя в
строгом смысле применимы лишь при полной взаимной
нерастворимости компонентов Л и В и прямой линии распределения, дают
* Эти уравнения еще ранее применялись для процессов выщелачивания
и промывки твердых частиц Тэрнером (Ind. Eng. Chem., 21, 140 (1929)].
276
хорошее соответствие с результатами расчета графическим
методом, при умеренных отклонениях от этих условий. При этом
используют среднее геометрическое значение фактора
экстракции е= (eien),/2.
Фактор экстракции, в дополнение к его прежнему
выражению [уравнение (VI,51)], можно определить также как
отношение наклонов линии равновесия и рабочей линии. При е=1 обе
эти линии параллельны. Рассмотрим рис. 141, а. Если е<1,
рабочая и равновесная линии могут пересечься в точке,
соответствующей точке ввода исходного раствора в каскад. В этом
случае степень экстракции, даже при числе ступеней, равном
бесконечности, измеренная при наиболее низком возможном
/ 2 3U5B7BB10 Щ 20
Число теоретических ступеней, п.
Рис. 140. Многоступенчатая протнвоточная экстракция с иесмешиван>
Щимися растворителями при постоянном коэффициенте распределения.
277
Рис. 141. Противоточная экстракция; предельные случаи (бесконечное
число ступеней):
/ — равновесная линия; 2 —рабочая линия.
значении хп, будет ограничена. Если я=оо, правая часть
уравнения (VI, 125) становится равной е, а ордината на рис. 140 —
равной 1—s.
При е>1 (рис. 141,6) пересечение равновесной и рабочей
линии происходит на конце, соответствующем выходу рафината,
и величина хп ограничивается только значением ys. При я = оо
величина xn — ys/m. При экстрагировании особо ценных
веществ для достижения наибольшей степени извлечения
желательно, чтобы величина е была больше единицы, а величина
ys — возможно меньше.
Пример VI-8. Тысяча кг/ч 25%-ного раствора ацетона в воде
экстрагируется трихлорэтаном при противотоке фаз. Регенерированный экстрагент,
используемый в процессе экстракции, содержит 1% ацетона. Определить:
а) наинизшую концентрацию рафината, достижимую при бесконечном числе
ступеней; б) минимальный расход экстрагента, необходимый для получения
концентрации ацетона в рафинате, равной 1%; в) расход экстрагента,
необходимый для того, чтобы иа пяти ступенях получить концентрацию ацетона
в рафинате 1%, и достигаемую при этом концентрацию ацетона в
экстракте; г) число необходимых ступеней, если при расходе 600 кг/ч экстрагента
надо получить в рафинате 1% ацетона; д) до какой степени должен быть
очищен экстрагент для тога, чтобы получить в конечном рафинате 0,5%
ацетона, если для экстракции в пяти ступенях используется 1200 кг/ч трихлор-
этана, находящегося в регенерированном экстрагенте.
Решение. Обратимся к примеру VI-3. а) Имеем: *F = 0,333 кг
ацетона/кг воды; Л = 750 кг воды/ч; t/s = 0,0101 кг ацетона/кг трихлорэтана; т =
*=1,65. Наименьшую концентрацию рафината найдем как концентрацию
воды, равновесную с концентрацией исходного экстрагента:
ys o.oioi
1,65
= 0,00612 кг ацетона/кг воды
278
б) Находим хп = -^щ = 0,0101 кг ацетона/кг воды. Это случай,
подобный приведенному на рис. 141, а. Далее получаем:
xP-ysim л
0,333 — 0,0101 лп0. 1,65ВШ1П
: 0,987 = ■
0,333 — 0,0101/1,65 ' 750
SmIn = 449 кг трихлорэтана/ч
в) При п=Ъ
yS
Хп 5Г
Уз
F m
0,0101
0,0101
1,65
0,333-
0,0101
1,65
: 0,01217
Из рис. 140 е=2,13=т£/Л = 1,656/750; В=968 кг трихлорэтана/ч, или
968/0,99=978 кг/ч регенерированного экстрагента.
А 750 у,— у, у,— 0,0101
В — 968 хс—х 0,333 — 0,0101
F п
t/i = 0,260 кг ацетона/кг трихлорэтана
г) Имеем: 6 = 600-0,99 = 594 кг/ч; е=т£/Л= (1,65 • 594)/750= 1,285.
Далее находим:
ys
т =0.01217
ys
F т
Из рис. 140 гс=12,1 теоретической ступени,
д) При гс=5 имеем:
0.О05 л г™™ , тВ 1,65-1200
1-0,005 -0-00502 г—Г= 750 ~2'66
Из рис. 140
х„ — ^2- 0,00502
х„ — —
«п _0,005=- ЬЁЁ_
0,333 -
V- т 1,65
ys = 0,00557 кг ацетона/кг трихлорэтана, или ' ^ 0Q557—=0,553% ацетона.
Пример VI-9. В разбавленном растворе, получаемом после
выщелачивания урана (из руды) смесью серной и соляной кислот, уран находится
в виде UO|+. В таком виде он непрерывно отделяется от примесей,
содержащихся в исходном растворе, и концентрируется. Исходный раствор
содержит. 1,5 кг/.и3 урана и непрерывным методом экстрагируется децимолярным
раствором алкилфосфата в керосине. Для регенерации 99,8% урана
необходимы три теоретические ступени. При кислотности исходного раствора,
соответствующей рН=1,0, экстракция примесей незначительна, а коэффициент
279
/ ш
Экстракция F
Реэкстракция
т
z=0
Рис. 142. Принципиальная схема процесса экстракции (к примеру VI-9):
/ — исходный раствор (Л) —раствор, получаемый после выщелачивания урана (из руды);
Я —рафинат; /// — концентрированная НС1 в воде (D); IV — очищенный урановый
концентрат; V — керосин + алкилфосфат (В).
распределения при концентрации урана в органической фазе свыше 6,5 кг/л3
равен
к:г/м3 органического раствора
кг/м3 водного раствора
30
Реэкстракцией ураиа концентрированной соляной кислотой
концентрируют очищенный уран в кислоте и одновременно регенерируют
органический растворитель, вновь используемый в процессе экстракции. Для ре-
экстракции урана и повышения его концентрации в кислоте до 100 кг/м3
необходимы пять теоретических ступеней. Если реэкстракция протекает в
очень кислой среде, ураи предпочтительно переходит в водную фазу и
коэффициент распределения равен
0,017
ikz/m3 органического раствора
кг/м3 водного раствора
Схема процесса представлена на рис. 142. Взаимная растворимость
водных и органических жидкостей невелика, изменение объемов, вызванное
переносом урана (при указанных пределах изменения концентрации ураиа),
также незначительно. Определить скорость циркуляции органической
жидкости (в м3 раствора алкилфосфата на м3 исходного раствора).
Решение. Вследствие взаимной нерастворимости жидкостей и
постоянства их объемов при изменении концентраций величины х и у
выразим в кг ураиа иа 1 м3, а расходы А и В — в м3 раствора в единицу
времени.
Расчет ведем иа 1 м3 исходного раствора. Обозначим: В — количество
органического экстрагента в м3, D — количество концентрированной НС1 в м3.
Содержание урана в исходном растворе 1,5 кг. Остаток урана в рафинате:
1,5(1 — 0,998) =0,003 кг. Содержание урана в жидкости, идущей иа реэкстрак-
цию: 1,5 — 0,003=1,497 кг. Объем кислоты, идущей иа реэкстракцию:
0=1,497/100=0,01497 м3.
Экстракция, х = 1,5; х = 0,003; А = 1; m = 30; е = ЗОВ; л = 3.
Из уравнения (VI, 125))
1„5 — 0,003 (30В)4-
115 ^- (3°S)4
"•5~ 30
-зов
Далее находим:
У\-
1,497
В
260
По этим данным можно определить следующие величины:
В 0,250 0,260 0,267 0,300 0,333 0,400
у .... О 0,0069 0,0150 0,036 0,0498 0,0666
у, 5,988 5,758 5,621 5,026 4,541 3,809
Реэкстракция. При реэкстракции органическая жидкость является
исходным раствором (и рафинатом), а водная фаза — экстрагентом (и
экстрактом). Применим уравнение (VI, 126) с учетом того, что вместо xF в данное
уравнение следует подставлять соответствующие значения у\, вместо хп —
значение ys=0 (концентрированная кислота не содержит урана). Тогда
1 со - концентрация урана в водной фазе
m . — 00,0 " "—
0,017 ' концентрация урана в органической фазе
58,8 • 0,01497 0,88
Е = ■
В В
При 6=0,300 число ступеней для реэкстракции составит:
5,026 / 0,300 \ . 0,300]
' 0,88 ) + 0,88 '
i Г5'026 Л
'ПобзбТ
0,88
g 0,300
4,13
Поскольку на реэкстракции применяется пять ступеней, величина В Ф
=^=0,300. Подстановка других значений приводит к конечному результату
В=0,264 м3 органической жидкости на 1 м3 исходного раствора (для пяти
ступеней реэкстракции).
В случае схемы экстракции, показанной на рис. 142 и
являющейся типичной, комбинацией уравнений Крейсера для обоих
каскадов можно показать, что
хп (4й-0(^-0+(^-0^(4*-О
*, (4*_i)(Bj_i)+(B"*+>_i)Bs(B?-i)
(VI, 129)
m В
где еЕ = —|-
ее— —*-
bs — —A~'
пЕ— число ступеней экстракции;
ns — число ступеней реэкстракции;
у
тЕ — — при равновесии;
ms= — при равновесии;
z— концентрация распределяемого вещества в реэкстраги-
рующей жидкости.
Периодическая экстракция. Противоточная экстракция
всегда проводится по непрерывной схеме. Суммарный эффект про-
тивоточной экстракции можно получить в лаборатории с
помощью периодической экстракции (см. главу IX).
*61
ПРОТИВОТОЧНАЯ МНОГОСТУПЕНЧАТАЯ
ЭКСТРАКЦИЯ С ФЛЕГМОЙ
Противоточная многоступенчатая экстракция с флегмой
представляет собой вариант простой противоточной экстракции
и является по существу аналогом процесса ректификации. В то
время как в схемах экстракции, описанных ниже, наиболее
обогащенный экстракт, выходящий из установки, имеет
концентрацию, близкую к равновесной с исходным раствором,
путем применения флегмы можно получать экстракт,
концентрация которого может быть значительно больше концентрации,
равновесной с питанием (исходным раствором) 2> 1б- 17.
Схема экстракции с флегмой представлена на рис. 143.
Исходный раствор F разделяется на конечный экстракт РЕ и
конечный рафинат Rn, составы которых существенно
отличаются от состава исходного раствора. На ступенях от 1-й до
(f—1)-й происходит обогащение экстракта компонентом С, при
этом получают первичный экстракт Е^ После отделения всего
или большей части экстрагента получают раствор Е'', часть
которого отбирается в виде конечного экстракта РЕ, а другая
часть возвращается на экстракцию в виде флегмы R0;
последняя движется в аппарате противотоком к обогащенному
экстракту.
Установка для отделения экстрагента (аналог конденсатора
в ректификации-) представляет собой обычно дистилляционный
аппарат, в котором экстрагент отгоняется в таком количестве,
чтобы составы £4 и Е' соответствовали точкам, находящимся
по крайней мере на противоположных сторонах бинодальной
кривой треугольной диаграммы. Растворы Е', R0 и РЕ имеют
одинаковый состав и могут быть насыщенными или
ненасыщенными растворами (т. е. точки, отвечающие их составам, могут
находиться на бинодальной кривой треугольной диаграммы или
вне этой кривой).
На ступенях от (f-f-l)-fl до я-й происходит очистка
(исчерпывание) рафината—экстракция компонента С из рафината. В
этой секции каскада, как и в секции каскада, описанной выше,
экстракция производится при противотоке фаз. Расчет
процесса в этой секции проводят аналогично расчету для схемы с
двумя исходными растворами F и R0, принимая во внимание
► Sf
—■ f i s—^^г fe^-^e+i Ef~-^Ef+t ES^ES+1 En^3kcmpaeeHmS
p 4-—<p о ^—'a ' 'n v •ol иГч—/71 CTX^^iC!
_ я • Ro , R1 Re-i Re Rf-ijRf Rs-tRs ^пчРафйнат Rn
Отделение \ Конечный. - \ Исходный,
экстрагента экстракт РЕ ' раствор F
Рис. 143. Принципиальная схема противоточной экстракции с флегмой.
282
Соотношения между количествами экстрактов £4 и Ё',
конечного экстракта РЕ и флегмы R0. Процесс в секции обогащения
экстракта аналогичен процессу, происходящему в
укрепляющей части ректификационной колонны.
Обогащение экстракта. Расчет проводим первоначально с
помощью треугольной диаграммы9, приведенной на рис. 144.
Материальный баланс дистилляциовного аппарата для отделения
перегонкой экстрагента:
£, = S£ + £' (VI, 130)
Тот же баланс с учетом разделения Е' на конечный экстракт
и флегму:
El = SE + PE + R0 (VI'131>
Обозначим:
SE + PE=Q (Vl, 132)
Тогда
£, = Q + /?o (Vl, 133)
Следовательно, Q лежит на линии SEE', между точками Et
и SE. Материальный баланс всей секции обогащения экстракта
с любым числом е ступеней:
Ee+l = SE + PE + *e <VI- 134>
Соответственно
SE + PE=Q = Ee+1-Re (VI, 135)
Рис 144. Расчет секции обогащения экстракта при противоточной
экстракции с флегмой.
263
Таким образом, Q представляет собой общий поток,
направляющийся к концу секции обогащения экстракта, для всех
ступеней установки от 1-й до (f—1)-й. Линия, проведенная из
точки Q внутрь треугольника (см. рис.144), будет пересекать ветвь
кривой растворимости (соответствующую растворам,
обогащенным компонентом В) в точке, концентрация которой отвечает
концентрации экстракта, поступающего на данную ступень;
ветвь, соответствующую растворам, обогащенным компонентом
А— в точке, отвечающей концентрации рафината, выходящего
с той же ступени. Для любой ступени экстракт Ее и рафинат
Re находятся в равновесии и располагаются «а
противоположных концах хорды равновесия. Последовательным построением
хорд равновесия и линий, исходящих из точки Q, можно, как
показано на рис. 144, определить все концентрации по
ступеням и число ступеней от 1-й до (f— 1)-й.
В соответствии с уравнением (VI, 135)
Ee+\Q
Q ReEe+1
Поскольку
(VI, 136)
ReEe+i + Ee+xQ = ReQ (VI, 137)
комбинируя уравнения (VI, 136) и (VI, 137), получим:
AQ
R E j_,Q хаг —X
—— = _ = е+1 ^ (VI, 138)
Е , RQ X.D —X.n
e+l ew ARe AQ
Уравнение (VI, 138) применимо для любой ступени в секции
обогащения экстракта. По аналогии с ректификацией это
отношение можно назвать внутренним флегмовым числом, которое
будет изменяться от ступени к ступени. На конце секции:
(VI, 139)
(VI, 140)
P~^-\-QSE = P~^-E (VI, 141)
Комбинируя уравнения (VI, 139) —(VI, 141) и учитывая, что
составы R0 и РЕ соответствуют одной точке треугольной
диаграммы, получим
Tr-lwll-sH-) (VI, 142)
Е
#0
SE
РЕ
EiQ
RoQ
p~eQ
QSE
284
Рис. 145. Расчет секции исчерпывания рафината при противоточной
экстракции с флегмой.
Уравнение (VI, 142) определяет внешнее флегмовое число на
конце секции обогащения экстракта. Окончательно
R0 {XaEi-xaq){Xape-xase)
Ре {xape-xaex){xaq-Xase)
(VI, 143)
Поэтому точка Q располагается таким образом на линии
SePe, чтобы обеспечить желаемое флегмовое число.
Исчерпывание рафината. Обратимся к рис. 143 и 144.
Материальный баланс для ступеней от 5-й до я-й:
s + RS-i = Es + *n <VI>144)
(VI, 145)
R=EC
*s-i=W
Следовательно, W представляет собой постоянную разность
между экстрактом и рафинатом, движущимися между
соседними ступенями этой секции. На рис. 145 точка,
соответствующая W, лежит на продолжении линии, проходящей через точки
RnHS.
Материальный баланс ступени питания:
Ef-Rf_l = Ef+1-Rf + F (VI, 146)
Поэтому
Q=W+F (VI, 147)
Таким образом, точка W лежит на продолжении линии FQ
и соответствует пересечению последней с продолжением линии
28S
'£л /
.
^--sf/jw
"
пп
S
R'n
р*
Rn$- Любой луч, проведенный в
радиальном направлении из точки W, будет
пересекать ветвь кривой растворимости
(отвечающую растворам, обогащенным
компонентом А) в точке, соответствую-
Рис. 146. Схема примете- щей концентрации экстракта, поступаю-
ния флегмы рафината. щего на любую СТуПень (s) секции
исчерпывания рафината. Учитывая, что
рафинат и экстракт любой ступени располагаются на
противоположных концах хорды равновесия, последовательным
построением, показанным на рисунке, можно определить число
ступеней для секции исчерпывания рафината и все
промежуточные концентрации. Для любой ступени 5 из уравнения
(VI, 145) получим:
Чг'-тг-ё- (VU48)
Комбинируя уравнения (VI, 145) и (VI, 148), получим
внутреннее флегмовое число для секции исчерпывания рафината:
RS-l
Es
ESW
RS_,W
XAES — XAW
X У
(VI, 149)
Внутреннее флегмовое число изменяется от ступени к
ступени. Фиктивную величину XAW можно рассчитать из
уравнения (VI,145) при использовании уравнения материального
баланса по компоненту А.
Далее производится графический расчет этих ступеней.
Начиная со ступени 1 проводят построение, применяя в качестве
рабочей точку Q до тех пор, пока хорда равновесия
(проходящая через точку £/) не пересечет линию FQ. Для расчета
ступеней от f-й до n-й в качестве рабочей используют точку W.
Способ графического расчета в прямоугольных координатах
идентичен указанному выше.
Необходимое количество экстрагента определяется из
материального баланса для всей установки.
S = SE + PE + Rn-F (VI, ISO)
Из этого уравнения видно, что чем больше флегмовое число
в секции обогащения экстракта, тем больше будет необходимый
расход экстрагента.
Применение флегмы рафината. В некоторых процессах
экстракции раньше использовали также флегму рафината, как
показано на схеме рис. 146. Поток рафината Rn распределяется
при этом на две части: флегму R'n и непрерывно удаляемый из
286
установки рафинат PR. Однако следует отметить
нецелесообразность применения подобной схемы процесса ,8. Для данного
исходного раствора и конечного экстракта состав и количество
PR определяются материальным балансом независимо от
величины Rn- Следовательно, W имеет фиксированное значение при
определенных S—PR в случае любых величин Rn, даже когда
#л=0. Смешение R„ и S приводит к тому, что Em+i
располагается на линии RnS (см. рис. 145) в некоторой точке,
находящейся между К и S, в зависимости от величины R„.
Следовательно, число ступеней в каскаде остается
неизменным независимо от того, используется Rn или нет, а флегма
в количестве Rn многократно рециркулирует через n-ю ступень.
Не дает преимуществ применение экстрагента, более близкого
к насыщению (точка К на рис. 145), перед его поступлением в
n-ю ступень.
Эти выводы указывают на некоторое отличие экстракции от процесса
дистилляции, в котором пар нз кипятильника возвращается, подобно «флегме
рафината», в дистилляционную колонну. В процессах дистилляции роль тепла
аналогична роли экстрагента в экстракции и введение тепла в колонну («ди-
стилляционный каскад») соответствует вводу экстрагента в экстракционную
установку. Однако при дистилляции переносчиком тепла является насыщенный
пар, для получения которого часть жидкости (рафината) испаряется. В
процессах же экстракции экстрагент может быть введен в каскад без носителя,
получаемого из рафината.
Минимальное флегмовое число. Как было описано выше,
определение числа ступеней состоит в последовательном
построении хорд равновесия и радиальных линий, выходящих из
точек Q или W. Отсюда следует, что если продолжение какой-
либо хорды равновесия проходит через точку Q в секции
обогащения экстракта или через точку W в секции исчерпывания
рафината, для достижения состава, соответствующего этой
хорде равновесия потребуется бесконечное число ступеней.
Уравнение (VI, 142) показывает, что чем ближе лежит
точка Q к точке SE, тем больше флегмовое число. Поэтому для
секции обогащения экстракта в случае минимального флегмо-
вого числа, при котором необходимо бесконечное число
ступеней, точка Q будет находиться в точке пересечения
продолжения самой ближней к SE хорды равновесия этой секции с
линией E'SE (например, линия DG на рис. 147).
Для секции исчерпывания рафината, как следует из
уравнения (VI, 145), чем ближе точка W расположена к S, тем
больше требуется экстрагента.
Следовательно, при минимальном флегмовом числе в этой
секции точка W должна находиться в точке пересечения
продолжения самой близкой к S хорды равновесия с линией RnS
(линия HJ на рис. 147). Поскольку при этом должно соблю-
267
Рис. 147. Определение минимального флегмового числа.
даться равенство (VI,147), одну из точек (Q или W)
располагают на одном из пересечений, указанных выше, а положение
другой точки выбирают таким образом, чтобы точка F, Q и W
находились на одной прямой линии. При таких условиях по
уравнениям (VI, 142), (VI, 147) и (VI, 150) можно определить
минимальное флегмовое число и наименьший расход экстр-
агента.
Во многих случаях минимальное флегмовое число
определяют9 одновременно для обеих секций каскада по хорде
равновесия LK, которая при продолжении ее проходит через точку F;
однако такой способ не применим к процессу, представленному
на рис. 147. В реальных условиях точка W должна находиться
ближе к точке S, а точка Q — ближе к SE, чем при
определении их положения таким! способом.
Полное орошение. По мере увеличения флегмового числа
возрастает S, а число ступеней в обеих секциях каскада
уменьшается. В пределе количества РЕ и Rn и исходного раствора F
становятся бесконечно малыми по отношению к величинам R0
и S, что соответствует бесконечно большому флегмовому
числу, или так называемому полному орошению; одновременно
число ступеней становится наименьшим. При этом S = SE и
точки Q и W совпадают с точкой S на фазовой диаграмме. Схема
построения процесса показана на рис. 148. В реальном каскаде
работа с полным орошением соответствует уменьшению РЕ и F
до нуля и возвращению всего количества Rn на п-ю ступень.
288
Оптимальное флегмовое число. Для любого процесса число
ступеней может изменяться в пределах от минимального при
полном орошении до бесконечно большого при минимальном
флегмовом числе в зависимости от равновесных данных для
тройной системы жидкость — жидкость, составов исходного
раствора, экстрагента и продуктов разделения. Стоимость
установки должна, таким образом, быстро уменьшаться при увеличении
флегмового числа, но вместе с тем должна возрастать ее
пропускная способность. Следовательно, стоимость установки
должна проходить через минимум в зависимости от величины
флегмового числа.
При увеличении флегмового числа возрастают количества
перерабатываемых жидкостей, стоимость экстрагента, а также
стоимость регенерации экстрагента на единицу исходного
раствора; в связи с этим увеличивается стоимость эксплуатации
установки. Общая стоимость переработки единицы исходного
раствора, которая определяется суммой капитальных и
эксплуатационных затрат, будет проходить через минимум, по
которому определяют оптимальное флегмовое число.
Применение в качестве экстрагента чистого компонента Д.
К числу простых, часто применяемых процессов экстракции
относится процесс, в котором в качестве экстрагента используют
чистый компонент В. Исходный раствор не содержит этого
компонента, а экстракт, отбираемый на конце секции обогащения
последнего (т. е. после регенерации), также является чистым
Рис. 148. Схема расчета при бесконечно большом флегмовом числе
и минимальном числе теоретических ступеней.
19 Р- Е. Трейбал 289
эд
xCRn ХСЯПЧ XCRf+\XDRf-.1XCRf-ZXCR,XCE' XCA
ХСН'„ Х*
хСРа
X,
UCR0
'СР£
Рис. 149. Расчет противоточной экстракции с флегмой при помощи линии
равновесного распределения:
1 — линия равновесного распределения; 2 —рабочая линия.
компонентом В. В этом случае применимы приведенные выше
уравнения; на треугольной диаграмме точка F располагается
на оси А — С, а точка S находится на вершине В. Указанное
изменение не отражается на последующей схеме расчета. При
других возможных модификациях процесса, связанных,
например, с изменением температуры на различных ступенях
экстракции, схема расчета изменяется аналогично тому, как она
изменялась в связи с изменением температуры на ступенях при
других методах экстракции.
Равновесная ланий
Равновесная линия /
Рис. 150. Минимальное флегмовое Рис. 151. Экстракция при полном
число при противоточной многосту- орошении (минимальное число теоре-
пенчатой экстракции. тических ступеней).
290
Расчет с помощью
кривой равновесия. В тех
случаях, когда число ступеней
разделения велико, расчет
обычно проводят в
координатах ХСА — Хсв- Основные
принципы расчета те же,
что и для противоточной
экстракции без флегмы.
Рабочую линию (рис. 149)
строят по концентрациям,
соответствующим точкам
пересечения бинодальной
кривой на треугольной
диаграмме и произвольно
проведенных лучей из рабочих точек
Q и W. Разрыв на
кривой отвечает переходу
построения от точки Q к
точке W. Построением ступеней
между кривой равновесия и
криволинейной рабочей
линией определяют число
теоретических ступеней
разделения.
Условие проведения
процесса при минимальном
флегмовом числе показано
на рис. 150, где точка
касания рабочей и равновесной
линий находится на участке
графика, соответствующего
секции обогащения экстракта. Точка касания может, конечно,
находиться в точке ввода исходного раствора или на участке,
отвечающем секции исчерпывания рафината. Изображение
процесса при наличии полного орошения, соответствующего
минимальному числу теоретических ступеней разделения, приведено
на рис. 151.
Координаты Енеке19. Если расчет в треугольных
координатах оказывается затруднительным (вследствие затемнения
чертежа, связанного с необходимостью проведения большого
количества линий), предпочитают проводить расчет с
использованием координат Енеке. Это обусловлено тем, что применение
координат Енеке позволяет пользоваться различными
масштабами на осях координат.
Рассмотрим рис. 143 и рис. 152. На конце установки,
соответствующем выводу экстракта, материальный баланс для суммы
!8' 291
X/?, Х/»£ */?
Рис. 152. Противоточная
многоступенчатая экстракция с флегмой:
а —в координатах Енеке; б —в прямоугольных
координатах.
компонентов (А + С) имеет вид:
E! = SB + E' (VI, 151)
а для компонента В
E^^S^ + Е'Л^, (VI, 152)
Аналогично, включая орошение флегмой (экстрактом):
Е^в^ + Р^ + Н,, (VI, 153)
ЕЛг, = SeNse + PeNpe + Wr0 (V. 154>
Принимая во внимание, что
Q = S£ + Ря (VI, 155)
получим
E, = Q + R0 (VI, 156)
Уравнение (VI, 156) показывает, что точка Q лежит на
линии SePe между SE и Е'. Для всей секции обогащения
экстракта, включая ступень е:
Ee+l = SE + PE + *e <VI-157>
или
S£ + Pi: = Q = E* + l-Re (VI. 158)
Следовательно, Q является рабочей точкой для расчета
числа ступеней в секции обогащения экстракта. Хорды
равновесия, как обычно, связывают составы рафината и экстракта на
одной ступени. Так же как и в треугольных координатах,
внутреннее флегмовое число составляет:
CT NQ-">
■е+1
Е „ RQ Лг — N.
(VI, 159)
Внешнее флегмовое число:
h.=(Щ (£И£\ = ("q-W**-"»*) (VI i60)
В случае, когда экстрагент, удаляемый с конца секции
обогащения экстракта, после отделения (отгонки) экстрагента
является чистым компонентом В, т. е. SE=S, линия £tQ
вертикальна и
R„ E.Q Nq — Nei
— = =£=-=-х^ хг1- (VI, 161)
Р£ PEEl N^-NPE
Координаты точки Q можно установить из определяющего
эту точку уравнения. Баланс по компоненту В;
^Nq=SeNSe + PeNpe (VU62)
292
откуда
N
SeNse + peNpe
Q
sf + p,
Из баланса по компоненту С
X
SEXSE + РЕХРЕ
Q
SE + PE
(VI, 163)
(VI, 164)
Если на установке для отделения экстрагента получают
чистый компонент В, то S£ = 0; Afs =oo; SeNs' —Be и
N
Q
Р ^ Ре
Е с
XQ~XPE
(VI, 165)
На конце секции исчерпывания рафината материальный
баланс по сумме компонентов (А + С) для ступеней от s до я
имеет вид:
Р,_, + 8=Е, + Р„ (VI, 166)
S — R„ = W = E5 — R5_, (VI, 167)
Следовательно, W является рабочей точкой, из которой
проводят линии построения для определения числа ступеней в
секции исчерпывания рафината (см. рис. 152). Хорды равновесия
соединяют точки, соответствующие составам экстракта и
рафината каждой теоретической ступени. Внутреннее флегмовое
число составляет:
\г-1
Е W
S
Nr
-N.
w
Ks-iW
N
P-s-1
■N.
(VI, 168)
w
Координаты точки W, так же как и точки Q, определяют из
материальных балансов. Баланс по компоненту В:
WW,„ = SAf„ — R JV„ (VI, 169)
Следовательно
Аналогично
w
Nw-
xw~
SNS-RnNRn
S^-R^„
S-R„
S-R„
(VI, 170)
(VI, 171)
Если вводимый в установку экстрагент является чистым
компонентом В, то S = 0; Ns = oo; SNS=BR, откуда
N.
w
■ N
Rn
w
(VI, 172)
293
Материальный баланс для всей установки по сумме
компонентов А и С:
F4-S=RB + PB + SB (VI, 173)
Подстановкой уравнений (VI, 155) и (VI, 167) и некоторых
преобразований получим::
F + W = Q (VI, 174)
Для компонента В
FJ<p + V/Nw=QNQ (VI, 175)
Для компонента С
FXF+WXW,= QXQ (VI. 176)
Решая систему уравмений (VI, 174) — (VI, 176), получим:
"ц-Nf XQ-Xf
Nf: Nw Xp Xw
(VI, 177)
Уравнение (VI,177) пригодно для аналитического
определения координат точек Q или W. Все построение можно
спроектировать на нижний гра(фик (см. рис. 152,6); для этого
проводят произвольные линии из точек Q и W с целью определения
положения рабочей линии.
Минимальное флегмавое число. Основные принципы
построения остаются теми же, «то и при расчете в треугольных
координатах. Если продолжение какой-либо хорды равновесия в
секции обогащения экстракта
проходит через точку Q, для
достижения этой хорды
необходимо бесконечно большое
число ступеней разделения.
Подобно этому, если
продолжение какой-либо хорды
равновесия в секции
исчерпывания рафината проходит через
точку W, этот случай
соответствует бесконечному числу
ступеней разделения. Чем ближе
точка Q лежит к точке SE,
тем больше флегма экстракта
[уравнение (VI, 160)]; чем
дальше W от Rn, тем больше
расход экстрагента
[уравнение (VI, 167)]. Поэтому
минимальное флегмовое число опре-
Рис. 153. Определение минимального Деляют следующим образом
флегмового числа. (рис. 153).
294
Рис. 154. Определение минимального
числа теоретических ступеней при
полном орошении.
Хорды равновесия
секции обогащения экстракта
продолжают до пересечения
с линией E'SE; пусть D —
точка пересечения, ближе
других расположенная к SE.
Хорды равновесия для
секции исчерпывания рафината
продолжают до пересечения
с линией SPR, причем
точка G наиболее удалена от S.
Одну из точек, например G,
принимают за рабочую
точку W, a D — за Q,
учитывая, что F, W и Q
должны лежать на одной
прямой линии, причем Q не
может быть расположена
ниже D, так же как W не
может находиться выше Q.
Из уравнений (VI, 160) и (VI, 161) находят минимальное
флегмовое число. Наиболее часто (но не в случае, показанном на
рисунке) положение точек W и Q при минимальной флегме
определяется пересечением хорды равновесия HJ, которая при
ее продолжении проходит через точку F.
Полное орошение. При наименьшем числе ступеней
производительность установки по исходному раствору равна нулю и
продукты с установки не отбираются. Точки S, SE, Q и W
располагаются в одной точке, как показано «а рис. 154. Если на
последнюю n-ю ступень подается чистый растворитель В, точка
S находится в бесконечности и радиальные линии,
необходимые для построения процесса, становятся вертикальными.
Системы типа I. Все указанные расчеты относятся к
диаграммам для систем типа II. Последовательность графических
расчетов и все приведенные уравнения полностью применимы
также для систем типа I. Однако в то время как для систем
типа II применение флегмы и чистого компонента В в качестве
экстрагента обеспечивает возможность получения рафината и
экстракта, содержащих компоненты А и С, практически любой
чистоты, для систем типа I возможно достижение любой
степени обогащения только рафината; при этом нет
необходимости пользоваться флегмой. Вопрос о том, является ли
использование флегмы желательным для достижения предельной
степени обогащения экстракта у систем типа I, можно решить
только в каждом конкретном случае.
Пример VI-10. Раствор, состоящий из 50% изооктана (Л) и 50% гекса-
на (С), экстрагируется фурфуролом (В); при этом получают продукты,
295
содержащие 90 и 10% гексана (без учета содержания экстрагента).
Исходный фурфурол не содержит этих углеводородов. Определить: а) минимальное
число теоретических ступеней; б) минимальное флегмовое число; в) число
теоретических ступеней, необходимое при флегмовом числе 6,2, и
количества экстракта и рафината, приходящиеся на 100 кг исходного раствора.
Решение, а) Данные о равновесии при ^=30° С приведены Пеннинг-
тоном и Мэрвилом. [Ind. Eng. Chem., 45, 1371 (1953)]. Равновесные
концентрации в весовых долях представлены ниже:
А
(изооктан)
0,8283
0,8201
0,5227
0,5616
0,2425
0,2440
в
(фурфурол)
0,0539
0.С628
0,0583
0,0594
0,0600
0,0594
с
(гексан)
0,1178
0,1171
0,379
0,379
0,6975
0,6966
А
(изооктан)
0,0472
0,| 449
0,' 320
0,0309
0,01313
0,( 1313
в
(фурфурол)
0,9421
0,9453
0,9358
0,9361
0,9329
0.9329
с
(гексан)
0,01069
0.0С98
0,0322
0,0330
0,0540
0,0540
Расчеты будут выполнены в системе координат Енеке. Из первой
строки приведенной выше таблицы находим:
0,1178
R~ 0,1178 + 0,8283
= 0,1244
N
Лг =
0,0539
R~ 0,8283 + 0,1178
0,01069
Е~ 0,01069 + 0,0472
0,9421
= 0,0569
= 0,1843
16,30
Е ~ 0,01069 + 0,0472
Полученные величины пюзволяют нанести на график первую хорду
равновесия. Прочие данные таблицы, обработанные аналогичным образом,
использованы для построения графика на рис. 155, иа котором проведены
только кривые насыщения, ai хорды равновесия опущены. Для интерполяции
хорд равновесия использован график зависимости Хе от Хд (рис. 156):
XF = 0,50 Np-.
0 F=100/C2 S=B
*РЕ = Хя, = Хя, = XRq = 0,90 NSe = оо
^ = ^,=^ = 0
Из фазовой диаграммы ([см. рис. 155) находим: NE = 13,2; Х„ =0,10 и
I п
из диаграммы Nn = 0,056.
"я
Баланс суммы компонентов (А + С):
р = Ря + К„=100
Баланс по компоненту С:.
FXp=PEXpE + RXRn
100 • 0,50 = РЕ ■ 0,90 + Rn • 0,10
296
1
0.8
0.6\
Ofr
о.г
-
\'7
I i i
/
' '—i—
ХЛ ф? 0,1* Хс 0,6 0вХрс I
Рис. 156. К примеру VI-10.
Определение числа теоретических ступеней
при флегмовом числе 6,2.
Рис. 155. К примеру VI-10.
Определение минимального числа
теоретических ступеней и минимального
флегмового числа:
Х — кг гексана /{кг смесн гексан + изооктан);
N — кг фурфурола/(кг смеси гексан +
изооктан).
Решая совместно оба эти уравнения, получим PE = Rn = 50 кг. В связи
с тем что поток РЕ не содержит экстрагента, РЕ = 50 кг, /?„ = 50(1+0,057) =
=52,85 кг.
Определим минимальное число ступеней при полном орошении.
Вследствие того что на n-ю ступень поступает чистый экстрагент, точки Q, S и W
имеют ординату N=co и вспомогательные линии для определения числа
ступеней (см. рис. 155) являются вертикальными. Начиная от точки Ей
проводя поочередно хорды равновесия и вертикальные вспомогательные линии
(как показано на рис. 155), получим, что минимальное число теоретических
ступеней находится в пределах между 11 и 12.
б) Для данной системы при минимальном флегмовом числе
продолжение хорды равновесия DG проходит через точку F. Вследствие того что в
процессе используется экстрагент, не содержащий углеводородов, точка Q
лежит на вертикальной линии, проходящей через точку Р Е, а точка W — на
вертикальной линии, проходящей через точку Rn. Продолжение линии DG
пересекает вертикальную линию, проходящую через точку Р „ в точке с
координатой N(3 = 57,8 (на рисунке не показано).
Из уравнения (VI, 161) находим:
R„ Rn 57,8 — 13,2
13,2 — 0
= 3,38
в) При флегмовом числе, равном 6,2, из уравнения (VI, 161) получим:
6,2:
N,
■13,2
13,2 — 0
NQ = 95,l
Точка Q лежит на вертикальной линии, проходящей через точку Р„ и
имеет ординату NQ = 95,\ (на рисунке не показано).
Из уравнения (VI, 177)
95,1—0 0,9 — 0,5
0 — N.
w
N
W
0,5 — 0,1
-95,1
297
Точка W лежит на вертикальной линии, проходящей через точку Rn
(не показано). Из точек Q и W проведем произвольные лучи и координаты
точек пересечения этих лучей с кривой насыщения на рис. 155 (ХЕ и Хп)
перенесем на график рис. 156, при этом получим рабочую кривую.
Для проведения процесса прн подаче исходного раствора на девятую
ступень от конца секции обогащения экстракта необходимо 17—18
теоретических ступеней разделения.
Из уравнения (VI, 165) S£ = ВЕ = Р£ (NQ— Np \ =50(95,1 — 0) =
-4755 кг. С установки для отделения экстрагента получают 4755 кг
последнего. Из уравнения (VI, 172) BR = S = (NR — Nw\Rn = (0,056+95,1) • 50=
= 4758 кг. На последнюю ступень поступает 4758 кг экстрагента.
Баланс по экстрагенту:
^+в« = рЛг + «Л„тВг
50 ■ 0 + 4758 = 50 ■ 0 -4- 50 • 0,056 + 4755
4758 = 4758
R. = 6,2P =6,2-50 = 310л;г
RQ = R (1 _|_ N j = 310 (1 + 0) = 310 кг
Е'= Р£ + RQ = 50 + 310 = 360 кг
£' = E'(l+W£,) = 360(l-|-0)=360 кг
Et = Е' = 360 кг
Ех = Ej П + NE\ = 360(1 + 13,2) = 5110 кг
Из уравнения (VI, 159)
*! NQ~NE2 95,1-13,4
Е„ Nn — ND 95,1 — 0,0630
:0,86
Из уравнения (VI, 157)
E2 = SiJ+Pi:+Rl = 0 + 50 + Rl
Решая одновременно оба эти уравнения, получим:
Е2 = 357 R! = 307
Е2 = Е2 (1 + NE^ = 357 (1 -4- 13,3) = 5110 кг
У?!= R, (1 -J- W ) = 307 (1 + 0,063) = 326 кг
Количества экстракта и рафината для остальных ступеней в секции
обогащения экстракта можно рассчитать аналогичным способом.
Из уравнения (VI, 167)
W = S — R„ = 0 — 50 = — 50 кг
W = -50 = E„ — R„_,
Из уравнения (VI, 168):
R„-i ^ Ne„-nw 16,1+95,1 _
Еп " NRn_rNw 0,057 + 95,1 -1'1'
298
Решая одновременно оба этн уравнения, находим:
Е„ = 294 кг R„_, = 344 кг
Еп = Е(\ + NE \ = 294 (1 + 16,1) = 5020 кг
*„_! = R„_i (I + NRn_1) = 344 (1 + 0,057) = 364 кг
Аналогично можно определить количество экстракта и рафината в
каждой последующей ступени этой части каскада. Количество экстрагента,
которое может быть удалено из конечного рафината, составляет Rn — Rn =
= 2,85 кг.
Расчеты от ступени к ступени. Эти расчеты проводятся
подобно тому, как было сделано в примере VI-5 (процесс без
использования флегмы). В данном случае расчет можно,
по-видимому, упростить, если вести его от я-й ступени (на выходе
рафината) к ступени питания, как делалось выше, и от 1-й
ступени (конец секции обогащения экстракта) к ступени питания.
В тройных системах ступень, на которую вводится исходный
раствор, определяется, как и ранее, по соотношению значений
Хс/Ха в рафинате и исходной смеси. Такой метод расчета
представляет интерес только для многокомпонентных систем,
поскольку для тройных систем графические методы позволяют
быстрее проводить расчет.
Постоянная селективность3'9. Для некоторых систем типа II
селективность, определяемая уравнением (IV, 2), является
постоянной. Учитывая, что растворы, обогащенные компонентом
А, представляют собой рафинаты, а растворы, обогащенные
компонентом В, — экстракты, и что селективность определяется
с помощью равновесных концентраций, для любой т-и
ступени уравнение (IV, 2) можно преобразовать к виду:
X X
~Х 'с,л ~х (VI'178)
АЕт ARm
При помощи уравнения (VI, 178) расчеты процессов
экстракции с полным орошением и с минимальным флегмовым числом
можно упростить.
Рассмотрим процесс, в котором экстрагентом является
чистый компонент В, а исходный раствор и конечные продукты
насыщены экстрагентом (их составы находятся на ветви бино-
Дальной кривой, соответствующей растворам, обогащенным
компонентом А). В этом случае материальные балансы для п-й
ступени при наличии полного орошения (см. рис. 143) имеют
вид:
Rn-i = En + B (VI, 179)
Rn-lXCRa_1 = E^CE„ <VI'180>
*п-1Х AR^WaE,, <Vf'181>
299
Комбинируя уравнения (VI, 178), (VI, 180) и (VI, 181),
получим:
CRn-
ARn-l
СЕп
X,
CRn
АЕ„
VC,A X
ARn
Аналогично для ступени (п— 1)-й
X
CRn-
X
X.
■ = Р,
СА X
CRn-: „2
=Рс,л
X
CRn
х,
'ARn-2 "" "ARn-i \"ARn
Для всей секции исчерпывания рафияата
X
XCF
X
AF
3С,Л
X
CRn
X
ARn
(VI, 182)
(VI, 183)
(VI, 184)
где «mm — общее число ступеней в секции исчерпывания рафиг
ната при полном орошении.
Аналогично для секции обогащения экстракта
X
срР
X
АРт.
— Remin / С/Л
(VI, 185)
где emln — число ступеней в секции обогащения экстракта при
полном орошении.
Поскольку
X,
cpf
X
APF
Dnmin
Зс,л
X
CRn
X
или
lg
X
срР
X
^mln —■
APf
ARn
X
ARn
X
CRn
"igP,
C,A
(VI, 186)
(VI, 187)
(VI, 188)
Уравнение (VI, 188) выражает известное соотношение,
полученное Фенске для процессов дистилляции.
Для таких систем при минимальном флегмовом числе
касание рабочей и равновесной линий наблюдается на ступени
питания при условии, что исходный раствор насыщен или
содержание экстрагента в рафинатах невелико. Если допустить,
что на конце секции обогащения экстракта в результате
отделения экстрагента получают чистый компонент В,
материальный баланс (без учета компонента В) для (/— 1)-й ступени
выразится уравнением:
- - ■ - (VI, 189)
(VI, 190)
'я + V.
Баланс по компоненту С:
Е/хЯ/=рЯхВ/ + к
*/-»"*/-!
300
Исключая из этих уравнений Е/, получим:
V-i
хя£-х*
(VI, 191)
с/
■7-1
Поскольку на ступени питания рабочая линия касается
равновесной
и
$C,AXRf{l-XEf)
Подстановка значения Х£ в уравнение (VI,191) дает:
V-i
/mln
ГС,А
Рсл
*Рв
1-Х,
(VI, 192)
Тогда с помощью материальных балансов получим:
\PE/mIn V E/mln V PE /mln
N„ —Nr
7
N
Ei
-N
Pe
N
E\
■N,
c/
N„ —N
Если концентрации экстрагента в экстрактах
постоянны, то
VE/mhi \ ГЕ /min
(VI, 193)
и рафинатах
(VI, 194)
Расход экстрагента можно найти из уравнения
материального баланса. Уравнения (VI, 192) и (VI, 193) точны лишь при
определенных условиях, когда селективность рс.л вычисляется
1Ю концентрации
исходного раствора. Если $с,а
существенно изменяется,
то минимальное флегмо-
вое число лучше
определять графически, так как
точка пересечения
рабочей и равновесной линий
может не совпадать со
ступенью, на которую
поступает исходный
раствор.
В том случае, когда
0,6
- о,и
+
с;
0,2
О
0,01 0,02
селективность $,с,а п0"
стоянна, а концентрации
экстрагента в экстрактах
и рафинатах близки к
постоянной величине, для
Ори 0,06 0,1
ЫРе)~ЫРе)п
К/Ре) + '
0,2 0,U Ор 1
Win
Рис. 157. Соотношение между числом
теоретических ступеней и флегмовым
числом при противоточной экстракции
(селективность постоянна).
301
расчета применимо эмпирическое соотношение Джиллилэнда20,
связывающее числа ступеней и флегмовые числа. Это уравнение,
полученное для процессов дистилляции, во многих случаях
справедливо также для процессов экстракции. Модификация
уравнения Джиллилэнда гарименительно к экстракции показана
на рис. 157.
Пример VI-11. Определить более коротким способом число
теоретических ступеней, необходимых по условию примера VI-10.
Решение. Обратимся к таблице равновесных данных в примере VI-10.
Согласно первой строке табл;ицы:
XirvX.D 0,01069-0,8283
рс, а Х,лгЛго 0,0472-0,1178
АЕ CR '
Из прочих строк таблицы аналогично получим следующие значения
Рг .: 1,53; 1,39; 1,59; 1,43 и 1„44. Можно в первом приближении принять, что в
данном случае селективность постоянна.
По уравнению (VI, 188)
Г 0,90 0,90]
gL0,10'0,10j 11n
"т,п:= ЕТ49 '
Пользуясь уравнением (W1, 192), получим:
/R/_i\ 1 Г0-90 /°>Ч1
-р— . --ист -щ-1-40 о? Нд
V Е /mm L '
где 1,49 — среднее значение селективности (3.
Поскольку содержание эжстрагента в экстрактах и рафинатах примерно
постоянно (см. рис. 155), порученное значение (R/-i/PE)min можно принять
равным значению (Ro/fK)min. Абсцисса на рис. 157 составляет
(6,2—3,1)/(6,2+1) =0,43, а ордината равна 0,26. Следовательно, я=16. Этот
результат хорошо согласуется с точным значением п, полученным в
примере VI-10.
Получение флегмы охлаждением или с помощью «анти-
экстрагентов». В большей части случаев флегму для
экстракции получают ректификацией, как показано на рис. 84. В
некоторых процессах экстракции смазочных масел 2| флегму иногда
получают охлаждением (рис. 158). Хотя в процессах
экстракции, используемых в негфтяной промышленности, участвуют
более чем три компонента», протекание этих процессов
изображают в треугольных диаграммах.
На схеме четырехступенчатой установки (см. рис. 158)
ступени 2, 3 и 4 работают при более высокой температуре t2.
Экстракт Е2 охлаждают до температуры tt, после чего взаимная
растворимость 'компонентов уменьшается, и декантацией
отделяют фазы РЕ и Rt. Обедненная экстрагентом фаза Ri
возвращается в процесс в вид№ флегмы, а экстракт РЕ выводится из
установки. Если отношение С/А в фазе РЕ не отличается
существенно от того же отношения в фазе Ео, проводить процесс
по такой схеме не име(ет смысла. Иногда вместо охлаждения
302
Рис. 158. Процесс экстракции с получением флегмы охлаждением.
для получения аналогичного изменения растворимости
компонентов в экстракте в последний вводят дополнительный
компонент. Так, например, добавление воды в экстракт при
экстракции смазочных масел фенолом способствует выделению
углеводородов, которые затем в виде флегмы возвращаются в цикл.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
Символ, обозначающий раствор, указывает не только
состав, но и весовое количество раствора. Моли и мольные доли
во всех случаях можно заменить весовыми количествами и
весовыми долями.
А — компонент А.
В — компонент В, основной компонент экстрагента.
С — компонент С.
Е—раствор экстракта.
Е — раствор экстракта (без учета содержания
компонента В).
е — ступень каскада, находящаяся в секции
обогащения экстракта,
303
F — исходный раствор.
F — исходный раствор (без учета содержания
компонента В).
К—коэффициент распределения, равный для любого
компонента отношению XE/XR при равновесии.
т—любая ступень каскада.
m — коэффициент распределения, равный отношению у/х
при равновесии.
N—весовая доля В в растворе (без учета содержания
в нем компонента В), равная ХВ/(ХА + ХС).
п — общее число ступеней в каскаде; последняя ступень.
РЕ— конечный экстракт.
РЕ — конечный экстракт (без учета содержания в нем
компонента В).
PR — конечный рафинат.
R — раствор рафината.
R — раствор рафината (без учета содержания в нем
компонента В).
S — избирательный растворитель (экстрагент).
S — экстрагент (без учета содержания в нем
компонента В).
SE— экстрагент, выделяемый из экстракта.
SE — экстрагент, выделяемый из экстракта (без учета
содержания компонента В),
s — ступень каскада в секции исчерпывания рафината.
X — весовая доля.
X — весовая доля компонента С в растворе (без учета
содержания компонента В), равная Хс/(ХА-\-Хс).
г_ хса
хлл'
У ХЕВ
$с,а — селективность экстрагента В (по отношению к
компоненту С сравнительно с А).
г = —д фактор экстракции.
Индексы
А, В, С— компоненты А, В, С.
Е — экстракт.
е — ступень е.
F — исходный раствор.
f — ступень, на которую поступает исходный раствор,
max — максимум.
min — минимум.
304
R — рафинат.
5—растворитель (экстрагент).
s — ступень 5.
t — общий.
1, 2 и т. д. — обозначение ступеней.
ЛИТЕРАТУРА
1,. М. Randall, В. Longtin, Ind. Eng. Chem., 30, 1063, 1188, 1311 (1938);
31, 908, 1295 (1939); 32, 125 (1940).
2. R. N. J. S a a 1, W. J. D. Van D i j с k, World Petrol. Congr., pt. 2,
London, 1933, Proc. 2, p. 352.
3. K. A. Varteressian, M. R. Fenske, Ind. Eng. Chem., 28, 1353
(1936).
4. T. W. Evans, Ind. Eng. Chem., 26, 860 (1934).
5. F. M. T i 11 e r, Chem. Eng. Progr., 45, 391 (1949).
6. R. Aris, D. F. Rudd, N. R. Amundsen, Chem. Eng. Sci., 12, 88
(1960).
7. L. Lapidus, N. R. Amundsen, Ind. Eng. Chem., 42, 1071 (1950).
8. T. W. Evans, J. Chem. Educ, 13, 536 (1936); 14, 408 (1937).
9. K. A. Varteressian, M. R. Fenske, Ind. Eng. Chem., 29, 270 (1937).
10. T. W. E va n s, Ind. Eng. Chem., 26, 439 (1934).
11. T. G. Hunter, A. W. Nash, J. Soc. Chem. Ind., 53, 95T (1934).
12. R. J. Hob son, T. Robbins, Birmingham Univ. Chem. Engr., 9, № 2,
42 (1958).
13. T. G. Hunter, A. W. Nash, J. Soc. Chem. Ind., 51, 285T (1932).
14. A. Kremser, Nat. Petrol. News, 22, № 21, 42 (May 21, 1930).
15. M. Souders, G. G. Brown, Ind. Eng. Chem., 24, 519 (1932).
16. T. G. Hunter, A. W. Nash, Ind. Eng. Chem., 27, 836 (1935).
17. E. W. Th iele, Ind. Eng. Chem., 27, 392 (1935).
18. J. F. Wehner, A. I. Ch. E. Journ., 5, 406 (1959).
19. J. O. Maloney, A. E. Schubert, Trans. Am. Inst. Chem. Engrs., 36,
741 (1940).
20. E. R. Gi lliland, Ind. Eng. Chem., 32, 1220 (1940).
21. L. Alders, Liquid — Liquid Extraction, 2nd ed., Elsevier Publishing
Company, Amsterdam, 1959.
20 P. E. Трейбал
МЕТОДЫ РАСЧЕТА
РАЗЛИЧНЫХ ВАРИАНТОВ
ПРОЦЕССА ЭКСТРАКЦИИ
(МНОГОКОМПОНЕНТНЫЕ СИСТЕМЫ)
СТУПЕНЧАТОЕ ВЗАИМОДЕЙСТВИЕ
ИСХОДНОЙ СМЕСИ С ЭКСТРАГЕНТОМ
Экстракция в многокомпонентных
системах проводится тогда, когда исходный
раствор, подлежащий разделению, или
экстрагент содержат более двух
компонентов, или же и исходный раствор и
экстрагент представляют собой смеси
нескольких компонентов.
Если для разделения
многокомпонентного исходного раствора используется один
экстрагент, можно применить любую из
принципиальных схем процесса экстракции,
описанных в главе VI,
Когда требуется полное разделение
исходного раствора на составляющие с
получением достаточно чистых продуктов,
экстрагент должен обладать селективностью
по отношению к какому-либо одному
компоненту смеси; при этом обычно следует
применять многоступенчатый процесс
экстракции. Так же как в процессах
ректификации многокомпонентных смесей, число
необходимых многоступенчатых каскадов
будет на единицу меньше числа
компонентов исходного раствора.
Экстрагент, представляющий собой рас-
тво;р нескольких веществ, будем называть
смешанным. Такие растворы применяют для
улучшения некоторых экстракционных
свойств экстрагента, например
селективности или емкости по растворяемому веще-
ств'у, а также для улучшения некоторых
физических свойств экстрагента, таких, как
точ1ка замерзания или вязкость (см.,
например), влияние буферных растворов на
распределение диссоциированного
растворенного вещества (стр. 54).
ЗОВ
В некоторых случаях активный экстрагент может быть
разбавлен инертным растворителем, причем последний не
участвует в процессе; примером может служить разбавление ал-
килфосфата (применяемого для экстракции урана) керосином
для снижения вязкости экстрагента (см. пример VI-9, стр. 279).
Инертный растворитель не распределяется между фазами,
поэтому расчеты можно проводить по методам, описанным для
трехкомпонентных систем. Однако в общем случае такое
упрощение расчета невозможно. Все принципиальные схемы
процессов экстракции, приведенные в главе VI, применимы также для
процессов со смешанным экстрагентом и с двух- или
многокомпонентным исходным раствором.
Исходный раствор, состоящий из двух или более
компонентов, может быть разделен путем распределения последних
между двумя несмешивающимися экстрагентами. Такой процесс
называется экстракцией двумя растворителями
(экстрагентами). Проводимый по многоступенчатой схеме, он носит
название фракционированной, или фракционной экстракции и
отличается наибольшей разделяющей способностью по сравнению
с другими известными схемами процессов экстракции. Для
достаточно полного разделения компонентов исходного
раствора необходимое число каскадов должно быть на единицу
меньше числа компонентов исходного раствора.
Если любая двухфазная многокомпонентная система
достигает состояния равновесия, в общем случае можно считать,
что все компоненты этой системы находятся в каждой фазе
в равновесных количествах. На практике, когда число
компонентов в системе превышает четыре, изображение равновесных
данных с помощью диаграмм (даже при постоянных
температуре и давлении) оказывается весьма затруднительным. Эта
проблема представляет, вероятно, лишь теоретический интерес,
так как имеется очень мало равновесных данных для четырех-
компонентных систем и еще меньше — данных о равновесии в
системах с большим числом компонентов.
Можно достаточно ясно изображать графически процессы
экстракции в четырехкомпонентных системах. К счастью,
многие из систем с большим числом компонентов можно (для
практических целей) рассматривать состоящими из двух
взаимно нерастворимых, не участвующих в массообмене
растворителей, между которыми распределяются остальные
компоненты смеси. При этом условии данные о равновесии в
подобных системах также можно довольно легко представить
графически. Если же указанное условие не соблюдается, то,
по-видимому, наиболее целесообразно имитировать
экстракционный процесс в лабораторных условиях, как описано в
главе IX.
20*
307
В данной главе рассматриваются типовые принципиальные
схемы процессов экстракции применительно к четырехкомпо-
нентным системам и наиболее простым системам, содержащим
большее число компонентов. Принято, что все ступени являются
идеальными (теоретическими), т. е. фазы, выходящие из любой
ступени, имеют равновесные составы. Процессы
одноступенчатой и многоступенчатой экстракции при перекрестном токе
могут быть организованы по непрерывной (в условиях
установившегося процесса) или периодической схеме;
дифференциальная экстракция является периодическим процессом, а все
многоступенчатые противоточные процессы экстракции
непрерывны.
В то время как для четырехкомпонентных систем имеется
возможность в той или иной мере интуитивно определить число
независимых переменных, которые можно произвольно выбрать
при проектировании процесса, для более сложных систем
данный вопрос усложняется. Эта проблема будет кратко
рассматриваться ниже в соответствии с общим подходом к ее
решению, развитым KeayKoiM'. Применительно к процессам
одноступенчатой экстракции и экстракции смесью экстрагентов
определенные идеи высказывали ряд авторов2~13, однако
приведенные литературные ссылки нельзя рассматривать как
исчерпывающую .библиографию по данным вопросам. Прочие
ссылки будут указаны в тексте, особенно для процесса
фракционной экстракции.
ОДНОСТУПЕНЧАТАЯ ЭКСТРАКЦИЯ
Одноступенчатая экстракция не имеет большого
промышленного значения, так: как достигаемая степень разделения
обычно невелика. Однако изучение этого процесса полезно для
анализа других, практически важных процессов экстракции.
Степени свободы. Если исходный раствор и экстрагент
взаимодействуют на идеальной (теоретической) ступени, имеется
возможность произвольно выбрать некоторые величины,
характеризующие процесс, такие, как расходы фаз, температура и
концентрация одного из участвующих в процессе компонентов;
тогда этими условиями будут автоматически фиксироваться
значения других величин. Для точного анализа процесса важно
знать, сколькими степенями свободы обладает система, с тем
чтобы избежать задания невыполнимых условий.
В общее число Nv гаеременных на ступени входят: а)
интенсивные переменные (температура, давление, концентрации),
число которых для каждого входящего в ступень и выходящего
из нее потока определяется правилом фаз [уравнение (II, 1)]:
с + 2— ф, где с — число компонентов, а ф— число фаз;
б) скорости потоков, одна степень свободы для каждой фазы
308
в каждом из потоков; в) степень обмена энергией, например
обмена теплом между ступенью и окружающей средой.
Однако имеется ряд условий, число которых Nc
ограничивает число произвольно устанавливаемых переменных.
Например, для каждого компонента должен быть соблюден
материальный баланс. Далее, первый закон термодинамики требует,
чтобы соблюдался также баланс энергии. У потоков фаз,
рассматриваемых здесь, кинетическая и потенциальная энергия,
а также совершаемая ими работа невелики, поэтому последний
баланс упрощается и сводится к балансу энтальпии или тепла.
При других обстоятельствах могут возникать иные
ограничительные условия, например идентичность двух потоков. В
любом случае число независимых переменных N{, которые можно
выбрать произвольно, равно разности Nv — Nc. Часто многие из
них будут выявляться при анализе конкретных процессов.
Применяя эти положения к анализу одноступенчатой
экстракции, получим:
Поступающий исходный
раствор (одна фаза, одна
скорость потока) с + 2—l-f-l = c-f-2
Поступающий экстрагент (одна
фаза, одна скорость потока) с —|— 2 — 1 —(— 1 == с —(— 2
Уходящий экстракт н рафннат
(две фазы при равновесии,
две скорости потоков). . . с-\-2—2-(-2 = c-f-2
Теплообмен с окружающей
средой 1 = 1
Всего ... Nv = 3c + 7
Материальный баланс .... с
Баланс энтальпнн 1
Всего ... Nc = c-\-l
Таким образом, число независимых переменных:
N. = N —N = 2с-1-6
I V С <
Из них обычно фиксируются:
Состав, состояние и скорость потока
исходного раствора (нли
количество, если процесс периодический) с -4- 2
Состав и состояние экстрагента ..." c-f-1
Давление в ступени 1
Потерн тепла 1
Всего . . .
2с + 5
Число Ni, выбираемое произвольно, равно единице. Выбор
производится из числа следующих переменных:
"t
Концентрация какого-либо компонента в любой
из выходящих фаз 1
Отношение концентраций двух компонентов
в любой из выходящих фаз 1
Количество любого компонента исходного
раствора, находящееся в выходящей фазе ... 1
Отношение процентного содержания двух
компонентов исходного раствора в выходящей
фазе 1
Расход
экстрагента (или его количество, если
процесс периодический) 1
любой из фаз (или ее количество, если
процесс периодический) 1
Так, например, выбор концентрации только одного из
компонентов в рафинате автоматически фиксирует концентрации
всех остальных компонентов в экстракте и рафинате, а также
расходы экстракта, рафината и экстрагента. Эти переменные
не могут быть выбраны произвольно, при проектировании
установки их можно определить простым расчетом. Далее, нельзя
выбирать последнюю независимую переменную (jV, = 1)
совершенно произвольно. Например, нельзя принимать концентрацию
распределяемого компонента в рафинате такой, чтобы она была
меньше равновесной с экстрагентом данного состава.
Четырехкомпонентные системы. Данные об изотермическом
равновесии в таких системах можно представить графически
в правильном тетраэдре, сторонами которого являются
равносторонние треугольники (рис. 159). Три из четырех сторон
четырехгранника могут быть прямоугольными треугольниками.
Для диаграмм такого типа применимы все геометрические
соотношения, используемые для треугольных диаграмм. Так, если
раствор F смешивается с раствором S, то точка М, отвечающая
составу смеси, располагается на линии FS в соответствии с
отношением
Координаты точки М можно рассчитать при совместном
решении уравнений материальных балансов:
F + S=M (VII, 2)
FXp + SXs=MXM (VII. 3)
В уравнении (VII, 3) концентрация X относится
последовательно к компонентам А, В, С и D.
310
Рис. 159. Изображение изотермического равновесия
четырехкомпонентных систем типа I.
Графические расчеты в объеме четырехгранника проводить
невозможно. Для расчетов можно пользоваться ортогональными
проекциями различных точек и кривых на грани
многогранника. Большими прописными буквами будем обозначать
весовое количество (в случае периодической экстракции) или
весовой расход (в случае непрерывной экстракции) фаз, а также
положение этих фаз в пространственной диаграмме.
Соответствующими малыми прописными буквами будем обозначать
проекции точек на грани. Так, т обозначает проекцию точки М
и т. д. Если точка Р в четырехграннике имеет координаты ХАР,
Хвр, ХСР и XDP, где X — весовая доля, то координаты
проекции р на плоскость ACD будут обозначены ХАр, ХСр и XDp.
Эти координаты могут быть рассчитаны из следующих
соотношений:
Xz
ХАР=ХАР-
хсР = хср ■
ВР
X
ВР
X
Dp'
X
ВР
DP
(VII, 4)
~. На рис. 159 показана наиболее простая из четырехкомпо-
^ нентных систем, имеющая лишь одну пару частично смешиваю-
/ Щихся компонентов А и D. Следовательно, тройные системы
&, 311
^
Рис. 160. Корреляция хорд равновесия чегырехкомпонентных систем.
Рис. 161. Одноступенчатая экстракция трехкомпонентного исходного
раствора однокомпоненткшм экстрагенгом.
на гранях ABD и ACD являются системами первого типа
(см. гл. II). Кривая МР'Т образована сечением тетраэдра
плоскостью, соответствующей постоянной величине Хв, а кривая
tnp't является ее проекцией на основании ACD. Кривая РР'Р"
представляет собой геометрическое место критических точек;
NT является хордой равновесия для тройной системы ABD,
a tit—ее проекцией. Линия KL представляет собой хорду
равновесия четырехком'понентной системы, a kl—ее проекцию. Все
хорды равновесия, начинающиеся на горизонтальной кривой
TLP' между точками Т и Р', заканчиваются на пунктирной
кривой NKP', проекция которой nkp'.
На рис. 160, а показаны соотношения между проекциями.
Необходимо учесть, что точка m лежит на проекции кривой
UPTV (рис. 159), которая на рис. 160 не показана.
Корреляционная кривая р'Ю для хорд равновесия может быть
проведена по методу, показанному на рис. 27. Путем построения
таких корреляционных кривых для других сечений
четырехгранника, отвечающих различным значениям Хв, можно
получить ряд корреляционных кривых, как показано на рис. 160,6.
Аналогичные графики можно построить для более сложных
четырехкомпонентных систем. В дальнейшем при изложении
методики расчета будем допускать, что соответствующих
данных всегда достаточно, чтобы точно определить положение всех
необходимых точек и кривых (хотя в действительности во
многих случаях мы не располагаем полностью всеми данными).
На пространственной диаграмме и соответствующей
проекции на грань ACD (рис. 161, а) дана схема расчета
одноступенчатой экстракции трехкомпонентного исходного раствора/7,
состоящего из компонентов Л, В и С, однокомпонентным экстр-
агентом D. При смешении D и F получается смесь М, которая
образует два равновесных раствора R и Е. Последние должны
лежать на поверхности растворимости. Точки F, D и М
спроектированы на грани ACD и BCD (рис. 161, в и 161, г), на
которых проведены линии fD.
Если фиксированы независимые переменные Nit
перечисленные выше (см. стр. 309), то можно выбрать, например, величину
Xar (в известных пределах). На рис. 161,6 плоскость GHJ,
проведенная при постоянном значении XAR, пересекает поверхность
растворимости по кривой NP, проходящей через точку R. Надо
расположить точку R на этой кривой таким образом, чтобы
линия RE пересекала линию FD в точке М.
Проведем проекцию кривой NP на грани ACD. На рис. 161, в
выберем какую-нибудь точку г на линии Рп и отметим ее
координаты. С помощью корреляции хорд равновесия определим
положение точки е на плоскости ACD и также отметим ее
координаты. На плоскость BCD (рис. 161, г) нанесем точки е
И г. Если линия re на обеих проекциях проходит через точку т,
313
то линии RE и FD
пересекаются и положение точки R
выбрано правильно. Соотношение
Е _ Жя
R ~ 7Ш
(VII, 5)
Рис. 162. Выделение экстрагента D
из продуктов экстракции.
можно рассчитать по длинам
проекций этих отрезков или
по координатам точек. В
случае необходимости расчет
повторяется в той же
последовательности, но при этом
выбирается новое положение точки
г. Количества Е, R, F и D
должны удовлетворять
уравнениям материальных балансов.
Если не удается получить
пересечение в точке М, это означает, что первоначально
выбранная величина XAR превышает ее предельное значение и
требуемая степень извлечения в одной ступени недостижима.
Рассмотрим рис. 1 62, на котором точки R и Е определены
так, как было описано выше. Если относительная летучесть
экстрагента D (по отношению к другим компонентам)
позволяет выделять его полностью, то свободные от экстрагента
продукты будут иметь составы R' и Е', которые находятся на
стороне ABC тетраэдра ((/?' — продукт, обогащенный компонентом
А, а Е' — продукт, обедненный компонентом А).
На рис. 163, а приведен пример использования смешанного
экстрагента S, содержащего компоненты С и D, для экстракции
из двухкоматнентного исходного раствора, включающего
компоненты А и В. Полнюе отделение компонентов С и D от
продуктов экстракции приводит к получению продуктов R' и Е'.
Смесь F можно разделить с помощью экстрагента,
содержащего лишь один компонент D; преимущества использования
смешанного экстрагентга заключаются главным образом в
некотором улучшении селективности иди других свойств
экстрагента. На рис. 163,6 схематично показан процесс разделения
двухкомпонентного исходного раствора F, содержащего
компоненты В и С, двухфазным экстрагентом S, состоящим из
компонентов А и D. Полное извлечение А и D из рафината и
экстракта приводит к получению конечных продуктов R" и Е".
Фактически различия между некоторыми из описанных выше
процессов разделения до некоторой степени условны. Так,
процесс разделения трехкомпонентного исходного раствора одним
экстрагентом, приведенный на рис. 162, можно рассматривать
как процесс экстракцши исходного раствора F' (не
содержащего компонента А) иесмешивающимся экстрагентом S, содер-
314
жащим Компоненты А и D, взятых в соотношении,
соответствующем положению точки М. Метод расчета для этих
процессов один и тот же.
Численный расчет может быть проведен так же, как для
каждой из ступеней в примере VI-5 (см. стр. 263). Этот расчет
требует построения графика зависимости коэффициента
равновесного распределения для каждого компонента от
концентрации двух других компонентов.
Взаимно нерастворимые экстрагенты. Пусть
нерастворимыми друг в друге экстрагентами являются компоненты А и D.
Эти экстрагенты представляют собой либо практические
нерастворимые друг в друге чистые жидкости, либо взаимно
насыщенные растворы частично растворимых жидкостей, при этом
их взаимная растворимость, или распределение между фазами
не изменяется при распределении между ними третьего
компонента.
В общем случае распределение какого-либо вещества
зависит от присутствия другого. Типичным примером является
совместное распределение уранилнитрата и азотной кислоты
между водой и органической фазой, состоящей из керосина и
трибутилфосфата (ТБФ), показанное на рис. 164. Как видно
из рисунка, распределение любого компонента зависит от
концентрации другого. Эту систему можно рассматривать как че-
тырехкомпонентную, поскольку вода, керосин и ТБф не
участвуют в распределении.
Из правила фаз следует, что при равновесии (при
фиксированных значениях давления и температуры) можно задаваться
лишь двумя концентрациями. Так, на диаграмме
распределения и02(МОз)2 точке М соответствуют равновесные
концентрации 0,10 и 0,36 моль/л U02(N03)2 водной и равновесной фаз.
При этих условиях равновесные концентрации HN03 в тех же
Рис. 163. Одноступенчатая экстракция смешанным экстрагентом (а) и двумя
экстрагентами (б).
315
US
0A
^0,2
OJ
■U02(N03)2
yy
IK
^Ms
^C^^^^'
/0,5 /S
Makmn=-I
i \^
0,6
Q5
Oh
§0,3
0,2
s'uo2(no3)2
OA
0,2
OJ
0
-HN03 _
Наклон=-1
/
1/ /St
У 1
z(t>°^lP/
A 0,01/
'Ib^TOJ
^P\- i_
M 1 0,3
V
X
HN03
Phc. 164. Одновременное распределение азотной кислоты н уранилни-
трата между водой н керосином, содержащим 30 объемн. % трнбутил-
фосфата, при 25° С [Ind. Emg. Chera., 59, 145 (1958)]:
■*UO (NO ) ~ концентРаиия U0>2(NO3)2 в водной фазе, моль/л; jr —
концентрация HN03 в водной фазе, моль!'л; y^Q —концентрация UOjtNOjh' в
органической фазе, моль/л; VHN0 — ковщентрация HN03 в органической фазе, моль/л.
фазах составят соответственно 2,0 и 0,13 моль/л (это следует
из положения точки М. на диаграмме распределения HN03).
Если в этом случае происходит распределение третьего
компонента, то концентрацию его можно майти, однако для этого
необходима третья диаграмма распределения, причем на
каждой диаграмме должны быгь указаны два параметра.
Для некоторых систем, например, при распределении
диссоциирующих с образованием общего иона веществ или
веществ, реагирующих с оэдним из компонентов экстрагента,
кривые распределения могут быть рассчитаны на основе данных
о распределении компонентов распределяемого вещества.
Примеры приведены в главе II.
Обычно применяют следующие обозначения:
кг распределяемого вещества У в фазе D
хг D в фазе D
кг распределяемого вешества У в фазе А
\кг А в фазе А
JD
DD
X
JA
X
АА
Если объемы взаимодействующих растворов существенно не
изменяются, то может оказаться удобным выражать
концентрации в единицах массы в;а единицу объема, а количества
жидкостей заменять соответешующими объемами.
31 в
Хвг
Рассмотрим теперь равновесную
ступень, показанную на рис. 165. В и ^
С — два распределяемых
компонента — растворены в растворителе и
составляют исходный раствор. Экстр-
агентом является компонент А.
Для каждого распределяемого
компонента рис. 165. Схема односту-
Dx„ + Ay„ = Dx,+Ay, (VII, 6) пенчатой экстракции (экстр-
f ь 1 ' агенты А н D взаимно не-
D У, — (/„ растворимы).
-А=Т^Гр <VI1"7>
Эти зависимости являются уравнениями прямой на графике
с координатами х ц у, а именно — уравнениями рабочей линии,
проходящей через точки с координатами (xF, ys) и (xlt yt) и
имеющей наклон —D/A. Потоки, покидающие ступень,
находятся в равновесии, так что точка (хи у4) лежит на кривой
равновесного распределения. С помощью кривых равновесия
(например, рис. 164) и рабочей линии можно решить любую
задачу при расчетах жидкостной экстракции. Если свободная
от экстрагентов смесь распределяемых компонентов (питание)
обрабатывается двумя растворителями (экстракция двумя
экстрагентами), расчет проводят при допущении, что исходная
смесь распределяемых компонентов первоначально
взаимодействует только с одним из экстрагентов. Схема расчета
сохраняется в основном неизменной для любого числа
распределяемых компонентов.
Пример VII-1. Водный раствор, содержащий 0,3 моль/л иО2(Ы0з)2 и
3,0 моль1л HN03, экстрагируется в одной ступени равным объемом 30%-ного
ТБФ в керосине прн 25° С. Определить степень экстракции каждого из
распределяемых компонентов.
Решение. Выразим х н у в моль/л, А — в л органической фазы и
D — в л водной фазы; xVO2{NOahF^0,3; xm0iF = 3,0; yVOa (No3)2s=^HN03s =
= 0; — D/A = —1,0.
На рис. 164 точки L соответствуют составам исходных водных
растворов и органического экстрагента. Рабочая лнння проведена на обеих
диаграммах нз точки L с наклоном, равным —1,0. Поскольку количества
экстрагента и исходного раствора установлены, в системе не остается
независимых переменных; конечные растворы также фиксированы и их
концентрации можно определить. Предположим, что на диаграмме распределения
и02(ЫОз)г составы равновесных растворов соответствуют точке N, для
которой *hnos=1.° и ^WVNO,)^0-06-
На диаграмме распределения HN03 этим концентрациям соответствует
точка N, которая не находится на рабочей лнннн и, следовательно, не
отвечает концентрациям выходящих из идеальной (теоретической) ступени
потоков. Аналогично точки Р и Q с диаграммы U02 (N03)2 можно перенести
на диаграмму HN03.
На последней диаграмме точки N, Р и Q образуют пунктирную
линию, пересекающую рабочую линию в точке Т, которая соответствует
равновесным концентрациям выходящих нз теоретической ступени потоков.
317
б точке Т водная фаза имеет следующие концентрации: *hno i = V молЬ/я;
-*иО rNO ) i = 0,025 моль/л, а органическая фаза—#hno i ==0,295 моль/л;
^uo2(N03)21 = 0,274 моль/л. Степень экстракции U02(N03)2 составляет
[0,3—0,025)/0,3]-100=99,2%, а азотной кислоты (HN03) [(3,0—2,7)/3,0] • 100=
= 10%.
Пример VII-2. Водный ра!Ствоэ, содержащий 3,0 моль/л HN03 и не
содержащий U02(N03)2, взаимодействует при 25° С с равным объемом 30%-
ного раствора ТБФ в керросиге. В последнем растворе содержится
0,4 моль/л U02(N03)2 и отсутствует HN03. Определить концентрации
экстракта и рафината.
Решение. Обозначим бугквой А органическую фазу, а буквой D —
водную фазу; Xm2[tiOahp=0; ' #U02(N0,)2S = °'4; -^HNOsf=3.°. #HNO,S =
= 0 моль/л. На рис. 164 по эгим концентрациям нанесены точки / [на
диаграмме U02(N03)2] и L (на диаграмме HN03). Согласно графику —D/A =
——1,0. Следовательно, рабочие линии проходят через точки L и / с этим
наклоном. По аналогии с примером VII-1 точки R на обеих диаграммах
гоответствуют равновесным концентрациям: -*uo2(NO ) l = 0,056; -*hno 1 =
=2,8 моль/л в водной фазе: и г/и0 (мсп 1= 0,348; </HNq i = 0,22 моль/л
в водной фазе. В этом случае распределяемые компоненты перемещаются в
противоположных направлениям (уран вымывается из органической фазы, а
кислота переходит в органическую фазу).
Независимое распределение компонентов. Если
распределение каждого компонента не зависит от концентрации прочих
(что часто «аблюдается для разбавленных растворов, особенно
если распределяемыми компонентами являются недиссоцииро-
ванные или не реагируювдие ни с одним из компонентов экстр-
агента вещества), то диаграммы распределения (аналогичные
рис. 164) для каждого компонента содержат только одну
кривую. Это значительно ущрощает расчеты, которые могут
проводиться для каждого компонента независимо от другого.
Независимые постоянные коэффициенты распределения. При
независимых постоянных коэффициентах распределения (что
часто наблюдается в раззбавленных системах) расчеты
упрощаются еще в большей степени. Допустим, что коэффициенты
распределения m для двух распределяемых компонентов В и С
находят с помощью соотношений:
хв хс
Предположим, что mB;>mc, так что экстрагент А селективно
экстрагирует В, а экстраагент D селективно экстрагирует С.
Селективность пары экстрагентов, с помощью которых
разделяются компоненты В и С„ составляет
Р*.с = -^ <VII'9>
Как было показано в главе IV, для разделения
компонентов В и С необходимо, чтобы селективность была больше
единицы, причем чем больше селективность будет превышать
единицу, тем лучше осуществится разделение. Вместе с тем произ^
318
ведение mBmc не должно сильно отличаться от единицы; в
противном случае при низких концентрациях распределяемого
вещества в одном из экстрагентов может потребоваться
чрезмерно большой расход этого эКСтрагента, что приведет к
значительному возрастанию объемов аппаратуры и усложнению
проблемы регенерации экстрагентов.
Обозначим через рв долю компонента В (от его количества,
поступающего на ступень), уходящего со ступени в фазе,
обогащенной компонентом А, а через ав — долю В, уходящего
с фазой, обогащенной компонентом D u. Тогда
^ = _3^=^ = i^L=8fi (VII, 10)
где е — фактор экстракции.
Следовательно
Рв = -г£г = 1-Чв <™.»>
в
По аналогии обозначим через рс и qc доли компонента С
(от поступающего на ступень количества), уходящего в фазах,
обогащенных соответственно компонентами А и D. При этом
Атпс
где е = —д—
Рс=н% = 1-^с • <VII'12>
Комбинируя уравнения (VII, 11) и (VII, 12), получим:
1в. с _ с (VII, 13)
1 + -L 1- D
1 в
tB ' "Г Атв
Уравнение (VII, 13) показывает, что при mB>mc
увеличение отношения D/A приводит к увеличению Рв/Рс в пределе
стремящемуся к mB/mc. После некоторых преобразований из
уравнения (VII, 13) получим выражение для определения
отношения экстрагентов, необходимого для достижения заданного
значения рв/рс-
1 1в^
А _тс рств
D J^L_i
Рс
(VII, 14)
В атомной технике отношение Рв/Рс известно под названием
«степени разделения».
Для симметричного разделения, т. е. для такого разделения,
при котором доля компонента В в фазе А, выходящей со
319
ступени, равна доле компонента С в фазе D, покидающей ту же
ступень (при этом концентрации могут быть не равны)
J>B = qc=l — pc (VII, 15)
откуда
eB=-J- (VII, 16)
fcc
Из последнего соотношения получим:
D У
- (VII, 17)
- ' "'В"1С
или
е5=У'^7 ec = l/"l^ (V". 18)
Если степень разделешия компонентов В и С
характеризовать величиной (рв—Рс), то
Рв-Рс-ТТГв-ТТ7-с <vll'19>
Производная d(pB—pc)/d(A/D), полученная из уравнения
(VII, 19), при приравнивании ее нулю приводит к выражению,
идентичному уравнению (VII, 17), т. е. максимальная степень
разделения достигается при соотношении экстрагентов,
определенном по уравнению (VI I, 17).
Когда число распределяемых компонентов больше двух,
разделение какой-либо пар>ы компонентов возможно в
соответствии, например, с уравнением (VII, 15), при этом
распределение других компонентов iфиксируется и осуществляется для
каждого в соответствии с (собственным фактором экстракции по
уравнению (VII, 11).
Пример VII-3. Шайбель 15 приводит данные об одновременном
распределении пара(5)- и орто(С)-клоронитробензолов между двумя экстраген-
тами: А — «Skellysolve» (смеесь гептанов) и D — метанол, содержащий
15 объемн. % воды. При распределении 50 г компонентов между 250 мл
каждого растворителя тв = 1.-33 и тс = 0,82; при этом коэффициенты
распределения остаются постоянными при изменении соотношений количеств
распределяемых компонентов, i Определить составы фаз (без учета
содержания экстрагентов) после экстргакции 100 кг смеси, содержащей 33,5% пара-
и 66,5% орто-изомеров, в одкой ступени: а) при симметричном разделении;
б) при получении конечных продуктов одинаковой степени чистоты.
Определить также расходы экстргагентов (экстрагенты взаимно нерастворимы).
Решение, а) Для симметричного разделения используем уравнение
(VII, 17):
А
D
( 1 Г' -if l У'5 0 959 л3гептана
{твтс J 4,1,33-0,82/ ' ж3 метанола
eB=0,S59- 1,33=1,276
ес = 0,959 • 0,82 = 0,786
320
Из уравнения (VII, 11)
е„ 1,276
Рв-ТТ^-Т+Шб^0'560
рс= 1—0,560 = 0,440
Дальнейший расчет проводим на 100 кг исходного раствора. В этом
количестве исходного раствора содержится 33,5 кг пара- и 66,5 кг
орто-изомеров. Слой, обогащенный компонентом А, будет содержать:
0,560-33,5=18,78 кг (39,1%) пара-изомера
0,440-66,5 = 29,3 кг (60,9%) орто-изомера
Всего. . . 48,1 кг
В слое, обогащенном компонентом В:
33,5—18,78 = 14,76 кг (28,4%) пара-изомера
66,5—29,3 =37,2 кг (71,6%) орто-изомера
Всего. . . 52,0 кг
Чтобы можно было использовать данные о распределении компонентов,
концентрации компонентов должны примерно соответствовать
экспериментальным — 50 г на 500 мл общего количества экстрагентов. Следовательно,
для экстракции необходимо общее количество экстрагентов, равное
500 • 10s • 100/50 • 10е = 1 ж5, в котором должно содержаться 0,959/1,959-1 =
= 0,49 м3 гептана.
б) Требование, чтобы продукты имели одинаковую степень чистоты,
налагает условие:
Ув,_ xc,
Ус, ~^И[
Поскольку ув =твхв и ус = тсхс , это условие приводится к виду:
' т„ \о,5
*с, \тв1
Для расчета примем, что все исходное количество распределяемых
компонентов сосредоточено в фазе А, поступающей на экстракцию.
Концентрации этих компонентов в фазе А равны Ув и ус . Тогда хв =-хс = 0.
Из уравнения (VII, 7) получим:
D _ Уд, — У в, ^ Ус, — Ус,
А хв хс
Исключая из полученного уравнения ув и ус (пользуясь
соотношениями равновесного распределения), а также величину хв , на основе
21 Р- Е. Трейбал
321
приведенного выше условия получим:
Поскольку Ус,/Ув0 = 66,5/33,5 =1,985 и (тв/тс)0'5 = 1,274, величина лгС[
отрицательна. Таким образом, требование равной чистоты конечных
продуктов не может быть удовлетворено при экстракции исходного раствора
данного состава на одной ступени при любом соотношении растворителей. Хотя
отношение хв /хс выбирают при проектировании процесса, эта величина
имеет предел, который в данном случае превышен. Если бы исходное
отношение (тв/тс)0'5 было несколько меньшим, чем 1,274, то требуемое условие
могло бы быть удовлетворено.
МНОГОСТУПЕНЧАТАЯ ЭКСТРАКЦИЯ
ПРИ ПЕРЕКРЕСТНОМ ТОКЕ
Принципиальная схема процесса была рассмотрена выше и
показана на рис. ПО (см. стр. 237).
Степени свободы. Общее число независимых переменных для
п ступеней будет в п раз больше, чем для одной ступени, и
равно я(2с + 6), причем свобода выбора п добавляет еще
одну степень свободы. Однако если две ступени связаны между
собой таким образом, что рафинат с одной ступени поступает
в качестве исходного раствора (питания) «а другую, то эти
два потока совершенно идентичны. Таким образом, имеются
с + 2 условия, налагаемые на каждый поток, протекающий
между ступенями, причем в случае п ступеней существует п—1
таких потоков.
С учетом изложенного выше для я-ступенчатого каскада,
работающего при перекрестном токе, число независимых
переменных
Nt = п (2с + 6) + 1 — (п — I) (с + 2) = сп -4- с -4- 4и -4- 3
Фиксированными обычно являются следующие независимые
переменные:
Ni
Состав исходного раствора, состояние н
расход с -\-2
Состав экстрагента и состояние на каждой
ступени п(с-\-\)
Потери тепла и давления на каждой ступени In
Всего . . . cn-\-c-{-3nJr2
322
Для произвольного выбора остается независимых перемен-
Щ ных Ni = n+l. Их можно выбрать из числа следующих
переменных:
Л7
Число ступеней i
Концентрация одного нз компонентов в одном из потоков,
выходящих с какой-либо ступени; каждая концентрация I
Отношение концентраций двух компонентов в одном нз
потоков, выходящих с какой-либо ступени; каждое
отношение 1
Относительное содержание одного из компонентов
исходного раствора в одном из потоков, выходящих с какой-
либо ступени; каждое относительное содержание .... 1
Отношение относительных содержаний двух компонентов
исходного раствора в одном из потоков, выходящих с
какой-нибудь ступени; каждое отношение 1
Общий расход (или количество, если процесс
периодический) экстрагента I
Расход (или количество) экстрагента на какой-нибудь одной
ступени; каждый расход 1
Доля от общего количества экстрагента, приходящаяся на
какую-нибудь одну ступень (количество таких долей
п— I), каждая I
Таким образом, если принять, например, л = 5 (Ni—1) и распределить
I' общее количество экстрагента (Af,-=I) поровну между ступенями (./Vj = 4), то
число произвольно выбранных и общее число независимых переменных
составит ^,= 6. Это означает, что в данных условиях невозможно произвольно
устанавливать какие-либо концентрации в конечных продуктах.
Расчеты в пространственной диаграмме для четырехкомпо-
I*- нентных систем и на диаграммах распределения типа,
показанной на рис. 164, являются простым повторением расчетов
одноступенчатой экстракции, подробно описанных для тройных
систем в главе VI. Наиболее полезные обобщения можно
сделать в случае постоянных независимых коэффициентов
распределения. Эти обобщения могут быть полезны также для
качественного анализа процессов экстракции в более общем случае.
JK Независимые постоянные коэффициенты распределения. Все
■j* соотношения, полученные в главе VI, уравнения (VI, 44) —
,•$. (VI, 57) и рис. 115 применимы к каждому распределяемому
*&• компоненту в отдельности. Чтобы использовать эти соотшоше-
pi ния, считают, что распределяемые компоненты растворены
-*} первоначально в растворителе D, имеющем концентрации xF,
--*. и экстрагируются с помощью экстрагента А, причем начальная
•].( концентрация каждого компонента в экстрагенте равна ys.
JA] Фазы А являются экстрактами, а фаза D — рафинатом, кото-
I: рый переходит от ступени к ступени и многократно экстраги-
• " Руется. Так же как в случае экстракции одного компонента,
,, Равномерное распределение экстрагента по ступеням обеспе-
& Чивает наибольшую степень экстракции каждого компонента (и,
' % 21* 323
следовательно, всех компонентов). Чем больше число ступеней
при данном количестве экстрагента, тем выше степень
экстракции каждого компонещта и соответственно всех компонентов.
При экстракции в перекрестном токе может быть
достигнуто определенное разделение компонентов исходной смеси.
Однако более высокий выход при данной степени разделения
с меньшим количеством экстрагента на меньшем числе
ступеней можно получить Б1 условиях противоточной
многоступенчатой экстракции.
Чистый экстрагент. Допустим, что экстр агент А не содержит
разделяемых компонештов при входе на каждую ступень и что
на каждую ступень подается одинаковое количество
экстрагента. Для первой ступени доля любого компонента исходной
смеси, перешедшего в экстракт, в соответствии с уравнением
(VII, 11) составляет
р. = 7ЧТ (VH, 20)
а доля этого компонента, оставшегося в рафинате
Для второй ступен
?i = l-/>i = 7TT (VH,21)
Ри
7TT)(jh) = ¥TW (VII'22)
и
H(ttt)(ttt)=(FW (VU,23)
где р% и <7г представлянот собой доли компонентов, перешедших
в экстракт и оставшийся в рафинате второй ступени, по
отношению к содержанию их в исходной смеси.
Аналогично после ш-н ступени получим:
Рп = -(ф[уГ (VH, 24)
где рп — доля компонента исходной смеси, перешедшего в
экстракт на /г-й ступени.
Соответственно
?«=(4ж (VII,25)
где qn — доля компонента исходной смеси, оставшегося в
рафинате п-и ступени.
Тогда для всего экстракта:
п
5>» = 1 - *я = (e+|)"1)71- (VII. 26)
л-1
324
Во всех приведенных выше уравнениях экстракционный
фактор s рассчитывается относительно AID для каждой
ступени. Уравнение (VII, 25) аналогично уравнению (VI, 48) при
у3 = 0.
Степень разделения двух любых компонентов В и С
исходной смеси в суммарном экстракте (или в конечном рафинате)
после п ступеней определяется так же, как и для одной
ступени:
п п
^Рвп-^Рсп-Ясп-Чвп (VII, 27)
В_1 я-1
Можно найти максимальное значение этой величины в
зависимости от числа ступеней или от отношения растворителей.
При условии д(дс -\-qB\jdn — Q получим:
ig
п = —
'8(«д + Ъ'
>g(ec+1).
<m
(VII, 28)
где п — номер ступени, иа которой достигается наибольшая
степень разделения компонентов исходной смеси при данном
значении отношения AID, приходящегося на одну ступень.
Аналогично, принимая условие [<Э(<7сл—?в„)]/[д(А^)] = 0.
получим:
ед = —g— = |д'сид-с l J (VII, 29)
Рв,с — Рв,с
Это соотношение позволяет определить отношение AID,
которое приходится на одну ступень и при котором достигается
наибольшая степень разделения для п ступеней. Решая
уравнения (VII, 28) и (VII, 29) совместно, получим я = оо и A/D = 0,
что соответствует условиям проведения дифференциальной
экстракции. Это означает, что наилучшее разделение достигается
в условиях дифференциальной экстракции. Применяя
указанные уравнения раздельно, можно определить ступень, на
которой достигается наивысшая степень разделения любой пары
компонентов при конечном числе ступеней. Можно показать,
что в любом случае для максимального разделения необходимо
равномерное распределение общего количества экстрагента по
ступеням экстракции; вместе с тем в отличие от
одноступенчатой экстракции максимальное разделение не является
симметричным.
325
Пример VI1-4. Раствор, содержащий по 1 кг орто-, мета- и пара-нитро-
анилинов в воде, многократно обрабатывают бензолом, причем на каждой
ступени используют одинаковое количество экстрагента. Коэффициенты
распределения равны соответственно: то = 64,0; тт = 25,0 и mj,=9,3.
Коэффициенты распределения выражают отношение концентраций упомянутых
изомеров в бензоле и в воде. Принято, что коэффициенты распределения в
процессе не изменяются, а вода и бензол взаимно нерастворимы. Определить:
а) какая степень разделения будет достигнута, если использовать
соотношение бензола (А) и воды (D), при котором е0 = 1 для каждой ступени;
б) какое отношение растворителей нужно применить, чтобы получить
максимальное разделение орто- и мета-изомеров на четвертой ступени.
Решение, а) При бо = П1оЛ/£) = 64,0 отношение AID для каждой
ступени экстракции
А ! пшси кг бензола
и 64 кг воды
Такое малое количество бензола обусловлено очень большим значением
коэффициента распределения. Следовательно, em=mmA/D=25,0 • 0,01563=0,391;
8Р = 9,3 • 0,01563 = 0,1453.
Номер ступени, на которой будет достигнута наибольшая степень
разделения орто- и мета-изомеров в рафинате или в суммарном экстракте,
определится из уравнения (VII, 28):
, М£!!±1)_1
g jig (0,391 + 1)J=203
. I 1+1 \ '
lgf l + l )
g \ 0,391 +1 )
т. е. при целом числе ступеней — на второй ступени. На этой ступени
[уравнение (VII, 25)]:
(1 + 1)2
(0,391 +1)2
1_
qP'~~ (0,1453 + l)2
Чт2 in 5ni i i\2 0,519
: 0,760
Следовательно, будет экстрагировано (1—0,25) • 100=75% орто-изоме-
ра, 48,1% мета-изомера и 24,0% пара-изомера. Экстракт второй ступени
будет иметь состав [уравнение (VII, 24)]:
Ро>= (1 + 1)* =°'25
- 0.391 _
Рт' (0.391 + 1)2 — '
°Д453 =0,1105
(0,1453 + 1)2
328
Рафинат, суммарный экстракт и экстракт после второй ступени будут
содержать следующие количества компонентов:
Изомер
Всего ...
Рафинат
0,250 «2=16,4%
0,519 кг = 33,9%
0,760 «2 = 49,7%
1,529 кг
Суммарный экстракт
0,750 кг = 51,0%
0,481 «г = 32,7%
0,240 кг = 16,3%
1,471 кг
Экстракт второй
ступени
0,250 «2 = 44,5%
0,201 «2=35,8%
0,1105 «2= 19,7%
0,5615 кг
Аналогично определяем максимальную степень разделения орто- и пара-
изомеров:
Г lgd + 1) 1
LlR (0,1453+1) J
* Llg (0Д 453+1) J _ono
n= , г i + i 1
Ig 1.0,1453+1 J
0,6
C>4
Of*
О
0,6
Ofr
V
V4
V
\
4
v^
,J\
^
^
—
^1
Следовательно, наибольшая степень разделения достигается на третьей
ступени. Таким же образом можно показать, что максимальная степень
разделения между мета- и
пара-изомерами достигается на пятой ступени
(я=4,55). Количество и концентрации
компонентов на этих ступенях можно
рассчитать аналогично. Подобные
расчеты были выполнены для десяти
ступеней, и их результаты приведены на
рис. 166.
В ряде случаев чистоту или выход
данного компонента можно увеличить
отбором части рафината или
экстракта или смешением экстрактов. Так,
например, если выделить экстракт с
первой ступени, то в нем мы получим
50% орто-изомера с чистотой 55,2%.
Чистоту можно увеличить в результате
некоторого уменьшения выхода при
проведении обратной экстракции —
промывки этого экстракта водой.
Дальнейшее развитие такого приема
приводит в конечном счете к схеме
фракционной экстракции, которая будет
рассмотрена ниже (см. пример VII-11).
Рафинат после седьмой ступени
содержит 38,7% пара-изомера с
чистотой 78,5%, а рафннат восьмой
ступени практически не содержит орто-
изомера. Смешивая некоторые
промежуточные экстракты, можно добиться
лучших результатов и при
получении мета-изомера, хотя при
использовании такой простои схемы его нельзя
получить с высокой степенью чистоты.
С>.
02
О
z5
А
■46
в /О
Рис. 166. К примеру VII-4.
Разделение нитроанилинов при
перекрестном токе:
/ — гс-нитроаннлин; 2 — .«-нптроанилин;
3 — о-нитроанилин; 4—смесь о- й п-нитро-
анилннов; 5— смесь м- и
«-нитроанилинов; 6 — смесь о и .«-нитроанилинов;
qn~Остаточное содержание компонентов
в рафинате после экстракции; ^ —
степень разделения компонентов в рафинате.
327
б) Для пары орто- и мета-изомеров (30, т~64,0/25,0=2,56. Для
получения максимальной степени их разделения при я = 4 необходимо [уравнение
(VII, 29)]:
е0^б4^-=2-5б(2'56'/5-1) =0,392
D 2,56—2,56''
Следовательно, A/D = 0,392/64,0 = 0,00612 кг/кг на каждую ступень
экстракции. Расчеты, проведенные аналогично пункту а), показывают, что
суммарный экстракт после четвертой ступени будет содержать 73,5% орто-
изомера с концентрацией 54,2%.
ДИФФЕРЕНЦИАЛЬНАЯ ЭКСТРАКЦИЯ
Дифференциальная экстракция представляет собой
предельный случай экстракции в перекрестном токе, когда число
ступеней равно бесконечности и на каждую ступень подают
бесконечно малое количество экстрагента. Дифференциальная
экстракция является периодическим процессом (см. рис. 117).
Степени свободы. Так же как для экстракции в перекрестном
токе, при заданных составе, агрегатном состоянии и количестве
исходной смеси, составе и агрегатном состоянии экстрагента,
потерях давления и тепла, для дифференциальной
экстракции число произвольно выбираемых независимых
переменных Ni = n+l. Однако если учесть, что п должно быть равно
бесконечности (одна переменная) и что экстрагент на каждую
ступень подается в бесконечно малых количествах (п — 1
переменных), то Ni = 1. Эту переменную выбирают из следующих
возможных независимых переменных:
Ni
Общее количество
экстрагента 1
конечного рафината 1
суммарного экстракта 1
Концентрация какого-либо компонента в конечном рафинате
или в суммарном экстракте 1
Отношение концентраций двух компонентов в конечном
рафинате или в суммарном экстракте 1
Относительное содержание какого-либо компонента
исходной смеси в конечном рафинате или в суммарном
экстракте 1
Отношение относительных содержаний двух компонентов
исходной смеси в конечном рафинате или в суммарном
экстракте 1
В дальнейшем будет рассмотрен только процесс с
применением взаимно нерастворимых растворителей. Расчет в
пространственной диаграмме производится в принципе аналогично
показанному на рис. 159, однако такой расчет является слишком
медленным и поэтому не пригоден для повседневного
пользования на практике.
328
Ув
ties
•в
5г
К
s 1Уа
/уЛ
N \
■—хсг
-хс,
—xcf
L
LBf
хв, XBF
ХС, XOf
Рис. 167. Дифференциальная экстракция (взаимно нерастворимые
экстрагенты).
Взаимно нерастворимые экстрагенты. Способ расчета будет
описан для случая распределения двух компонентов В и С,
первоначально растворенных в растворителе D и имеющих
концентрации xBf и Хср. Эти компоненты составляют фазу
исходного раствора. Компоненты из исходного раствора
экстрагируются избирательным растворителем (экстрагентом) А,
начальные концентрации компонентов в котором равны yBS и yes-
Уравнение (VI, 79), описывающее процесс дифференциальной
экстракции, в принятых здесь обозначениях имеет вид:
^\ -.
xBf
dx,
KCF
yBS
J
KCf
dxr
(VII, 30)
*cs
где xBf и Xcf — концентрации компонентов В и С в конечном
рафинате;
At — общее количество экстрагента А.
На рис. 167 показано взаимозависимое распределение
компонентов В и С. Начало экстракции на обеих диаграммах
соответствует положению точки L. При взаимодействии исходного
раствора с первым, бесконечно малым количеством экстрагента
образуется бесконечно малое количество экстракта, который
находится в равновесии с раствором и имеет концентрации,
соответствующие ординатам точки К-
Необходимо определить кривую, которая связывает
одновременно существующие концентрации х и у в фазах для
каждого распределяемого компонента. Обычно для этого процесс
дифференциальной экстракции считают состоящим из ряда
последовательно проводимых процессов экстракции в перекрестном
329
токе, причем каждая экстракция осуществляется с
использованием небольшого количества экстрагента. При таком
допущении можно применить графический метод расчета.
Чтобы показать первый процесс экстракции небольшим
количеством экстрагента, на диаграмме распределения
компонента В (рис. 167) проведена рабочая линия из точки L с
достаточно большим наклоном, выбранным по практическим
соображениям, но отличным от бесконечности. С таким же углом
наклона проводят из точки L рабочую линию на диаграмме
распределения компонента С. Положение точки М, выражающей
результат экстракции, определяется на обеих диаграммах по
методу, описанному для одноступенчатой экстракции.
Следующую рабочую линию проводят из точки N с
одинаковым углом наклона на обеих диаграммах, при этом получают
точку Р, и т. д. Затем продолжают аналогичный процесс
построения, пока не достигают точки Q. Точки К, М, Р, ... Q на
диаграмме образуют кривую, выражающую одновременные
составы рафинатов и экстрактов в процессе дифференциальной
экстракции. Чем меньше выбранные приращения расхода
экстрагента, т. е. чем чаще проведены рабочие линии, тем с
большей точностью кривые будут описывать дифференциальную
экстракцию *. После этого можно графически проинтегрировать
уравнение (VII,30), находя значения у и х для
распределяемого компонента по кривой К ... Q. Кроме того, общее
количество экстрагента определяют как сумму принятых значений
приращений экстрагента. Общие количества экстрагента At,
рассчитываемые для любого компонента смеси, конечно,
должны быть одинаковы.
Независимое распределение компонентов. На диаграмме
распределения каждого компонента в этом случае имеется
только одна кривая распределения, которая совпадает с кривой
процесса экстракции. Уравнение (VII, 30) можно
проинтегрировать графически без каких-либо допущений.
Независимые постоянные коэффициенты распределения. В
этом случае процесс описывается уравнением (VI,80). Его
* Число приращений, необходимое для любой степени приближения к
дифференциальной экстракции, можно определить из уравнений (VI, 49)
и (VI, 80):
At при перекрестном токе __ n(Z1/n — 1)
At при дифференциальной экстракции In Z
где
Zt, xF-yS/m.
xf-ys/m '
п — задаваемое число экстракций в перекрестном токе.
Так, при Z = 5—20 и я = 20 получим отношение общих количеств
экстрагента 1,04—1,08, что соответствует ошибке от 4 до 8%.
330
можно записать для каждого компонента в виде:
хг
m
~у7
m
тА,
(VII, 31)
xf
Для каждого компонента отношение — равно q.
xF
Пример VII-5. Определить, какие результаты будут получены, если
процесс извлечения (пример VII-1) провести в условиях дифференциальной
экстракции.
Решение. Равновесные данные [такие же, как в условиях примера
(VII-1)] приведены на рис. 168, где помещена только часть данных о
распределении азотной кислоты. Поскольку ^ио2 (Ы03)2/г ~ ^'3> -^HNO /=• — 3,0 и
i/HNO s == ^uo2(NOi)S = 0, точка L выражает составы исходного раствора и
экстрагента. Точка К показывает концентрации в начальном экстракте,
находящемся в равновесии с исходным раствором.
Расчет, аналогичный показанному на рис. 167, был проведен путем
построения кривой экстракции по 20 приращениям расхода экстрагента,
причем каждое приращение равнялось 0,05 A/D. В результате процесса
получается рафинат состава, соответствующего на каждой диаграмме
распределения точке Q, для которой xv02,no3)2/= 0,015; xHN0^ =2,8 моль/л.
Суммарный экстракт тогда будет иметь состав 0,3—0,015=0,285 моль/л
иОг(ЫОз)г и 3,0—2,8 = 0,2 моль/л HN03. Оказывается, что при
дифференциальной экстракции экстрагируется не только больше урана, чем в
условиях примера VII-1, но и меньше азотной кислоты, т. е. при дифферен-
0,1 0,2
хио2(мо3)г
0,3
о,в
0,5
/7,4
0,3
0,2
0J
0
-HN03
-
i
Г
г
i
'О.
1
0=хи0г(иа^
0,01
\ 0,03
\ 0,1
\ °'3
н
. iL I
OU
25
Д7Н
&
Рис. 168. К примеру VII-5. Дифференциальная экстракция азотной
кислоты и уранилнитрата (обозначения осей координат см. рис. 164):
1 — кривая процесса экстракции; 2—к началу координат.
331
циальной экстракции достигается более высокая селективность и лучшее
разделение. При дальнейшем вводе экстрагента начинает быстрее
экстрагироваться азотная кислота. Это можно заключить из графика (рис. 168), если
продолжить линию экстракции за точку Q.
ПРОТИВОТОЧНАЯ МНОГОСТУПЕНЧАТАЯ
ЭКСТРАКЦИЯ
Принципиальная схема процесса показана на рис. 120 (см.
стр. 255). Процесс является непрерывным.
Степени свободы. Число независимых переменных
соответствует п ступеням каскада с учетом свободы выбора п — всего
п(2с+.6) + 1. Однако имеются межступенчатые потоки,
являющиеся общими для соседних пар ступеней (всего п— 1),
которые налагают 2(п—1) (с + 2) условий. Следовательно, число
независимых переменных для каскада составит: Л^ = п(2с + 6) +
+ 1 — 2(п — 1) (с + 2)=2с+2гс+5.
Фиксированы следующие независимые переменные:
Ni
Состав питания, состояние и
расход с-)-2
Состав экстрагента и состояние . . <-"4-1
Потери давления и тепла для
каждой ступени In
Всего. . 2с + 2я + 3
Число произвольно выбираемых переменных остается
равным 2. Их можно выбрать из числа следующих переменных:
Число ступеней 1
Расход
экстрагента .
рафината Rn 1
экстракта Е1 ,
Концентрация какого-либо компонента в фазе Rn
или в фазе Et; каждая концентрация .... ,
Отношение концентраций двух компонентов
в фазе Rn или в фазе £,; каждое отношение . 1
Относительное содержание какого-либо
компонента в исходном растворе по отношению
к его содержанию в фа5е Rn или £,; каждое
относительное содержание i
Отношение относительных содержаний двух
компонентов в исходном растворе по
отношению к содержанию в фазе Rn или £,; каждое
относительное содержание л
Четырехкомпонентные системы. Процесс для четырехкомпо-
нентной системы показав на рис. 169, согласно которому
исходный раствор F экстрагируется экстрагентом S. Допустим, что
расход экстрагента и концентрация рафината ^ar„ опреде-
332
лены и тем самым исчерпано число степеней свободы Nt. Общий
материальный баланс для всего каскада
/=•_(_ s = M = R„ + El (VII, 32)
Баланс для каждого компонента
X
м
М
(VII, 33)
Следовательно, можно определить состав и положение
точки М на диаграмме и нанести проекции точек F, S и М на
любые два основания пирамиды (основания ACD и BCD на
Рис. 169. Противоточная многоступенчатая экстракция
(четырехкомпонентные системы).
рис. 169). Так же как и на рис. 161, точка, соответствующая
/?„, должна лежать на кривой пересечения плоскости XAR
поверхностью растворимости; эту кривую можно провести в обеих
проекциях (на рис. 169 не показано). Положение точки Rn на
кривой выбирают произвольно (с последующей проверкой) и
наносят ее на обеих проекциях. На проекции ACD продолжают
линию гпт и выбирают на ней положение точки еь
На проекциях контурных кривых на грани ACD (на рис. 169
не показаны) выбирают значение XBEi. Определяют положение
точки е\ на проекции BCD и проводят линию гпех. Если данная
линия проходит через точку т, значит положение точки £,
выбрано правильно.
Обычно, прежде чем будет удовлетворено указанное
требование, приходится несколько раз выбирать положение точки
в[ на продолжении линии гпт.
Аналогично трехкомпонентным системам [см. уравнения
(IV, 81) —(VI, 86)] из материальных балансов получим
F~E1 = Rn — S = Rm — Em+!=0 (VII, 34)
Таким образом, положение точки О, соответствующее
постоянной разности расходов рафината и экстракта между
соседними ступенями, «аходят пересечением линий EtF и SR„ на
обеих проекциях. Из корреляции хорд равновесия (см. рис. 160)
определяют положение точки ги равновесной с е,, на проекции
ACD.
Путем интерполяции контурных линий на ACD получают
полный состав фазы, соответствующей точке /?,, и проектируют
ее на BCD.
На проекции ACD продолжают линию ог{ и на ней
выбирают положение точки е2. Затем определяют координаты
точки Е2. На проекцию BCD малосят е2, и если линия е2о
проходит через ги то положение точки Е2 выбрано правильно. Если
это требование не выполняется, находят новое положение
точки Е2. Точки, соответствующие составам последующих
экстрактов и рафинатов, определяют таким же способом. Если Еп и R„
будут находиться в равновесии, начальное положение точки Rn
на пересечении плоскости XARn с поверхностью растворимости
выбрано правильно. Если Еп и Rn не находятся в равновесии,
то положение точки Rn изменяют и весь расчет проводят
сначала, пока не будет соблюдено указанное выше условие. Число
ступеней отвечает числу равновесных хорд, использованных в
процессе построения. При задании переменных, отличных от
принятых здесь (расход экстрагента и XARn), схема расчета
должна соответственно измениться.
Для трехкомпонентных систем допускалась возможность
получения расчетом дробного числа ступеней в тех случаях, когда
при целом числе ступеней составы Rn и Еп не находились в
334
равновесии друг с другом. Для тройных систем на действующей
установке небольшим изменением расхода экстрагента можно
получить необходимую концентрацию XARn, а состав
рафината— соответствующим заданному. Однако, несмотря на
достигаемое равновесие составов экстракта и рафината Еп и Rn,
нельзя быть уверенным в том, что для многокомпонентных
систем окончательный состав рафината Rn будет получен даже
при соответствующем изменении соотношения экстрагента и
исходного раствора.
Так же как и для трехкомпонентных систем, существует
минимальное отношение расходов экстрагента и исходного
раствора. Точка О на рис. 169, определенная как наиболее
удаленная точка пересечения продолжений хорд равновесия и линии
SRn, соответствует минимальному расходу экстрагента и
бесконечно большому числу ступеней. Эту точку можно определить и на
проекциях (если точка пересечения определена' на проекциях
правильно, отношения длин линии srn и отрезка от точки гп до
точки пересечения на обеих проекциях должны быть равны).
Для конечного числа ступеней точка О должна быть
расположена на линии SRn еще дальше от тетраэдра; чем больше
это расстояние, тем больше расход экстрагента и меньше
требуется ступеней. По аналогии с трехкомпонентными системами
возможно, что точка О окажется расположенной со стороны S
фазовой диаграммы. Это зависит от наклона хорд равновесия.
В данном случае точка О должна лежать ближе к S, чем
любая точка пересечения RnS с продолжением хорд равновесия.
Расчет от ступени к ступени. Все расчеты можно выполнить
аналитически, от ступени к ступени, точно таким же способом,
как было сделано для трехкомпонентных систем в примере
VI-5 (стр. 263). Для этого необходимо располагать
подробными данными о равновесии, подобными приведенным на
рис. 129. Для четырехкомпонентных систем распределение,
например, компонента А выражается зависимостью Ka = !{Xar,
XBR)\ соответственно значения К для каждого компонента
изображаются семейством кривых.
Для систем с числом компонентов, большим, чем четыре,
положение осложняется; однако возможны некоторые
упрощения, если основные растворители (в четырехкомпонентной
системе А и D) взаимно нерастворимы. В любом случае расчет
проводят методом последовательных приближений. Составом
конечного рафината Rn задаются в начале расчета, а затем в
конце расчета его проверяют. Смит и Бринклей и разработали
способ, сводящий к минимуму число необходимых приближений,
и дали пример численного расчета для системы, аналогичной
по типу системе, показанной на рис. 169. Оландер 16 и Даффин 17
использовали для решения этой задачи высокоскоростные
вычислительные машины.
335
Ув,
Ув,п-1
УВп
Увб
В
/*
/хс,"-/ Ъ
if d
/,fcn
Ун
п/
п-1/
'j Пак
хв, п-1
<L
ПОИ
1 /
В
'А
хв,
м
Ус, п-1
г%
яв. п-г
Xi
■SF
Рис. 170. Противоточная многоступенчатая экстракция (несмешивающиеся
растворители).
Несмешивающиеся растворители. Несмешивающимися
растворителями являются компоненты А и D. Расчет будет
проведен для двух распределяемых компонентов В и С, находящихся
в растворителе D, причем их начальные концентрации
составляют xBf и xCf- Компоненты В, С и D образуют исходный
раствор. Компоненты В и С экстрагируются избирательным
растворителем (экстрагентом) А, причем концентрации
распределяемых компонентов в нем соответственно равны ува и yCs-
Рассмотрим рис. 170, на котором приведены диаграммы
взаимозависимого распределения компонентов. Материальный
баланс для каждого компонента
4 = У1~У* ~ У1~Ут (VII, 35)
Л X г, — X л „ — X .
F n F /п-1
где т соответствует любой ступени каскада.
Это уравнение является уравнением рабочей линии,
проходящей через точки (xF, t/4), (хп, Уз) и (хт-ь Ут) и имеющей
наклон D/A.
Предположим, что величины D/A и хв заданы, тогда число
степеней свободы системы исчерпано. Следовательно,
положение рабочей линии GM на диаграмме распределения
компонента В фиксировано. Значение хСп выбирают произвольно (с
последующей проверкой). Это позволяет нанести рабочую линию
на диаграмму распределения компонента С. Теперь можно
провести построение ступеней на обеих диаграммах, начиная от
точки G:
Диаграмма компонента В. Концентрация ув
находится в равновесии с хв и хс . Это определяет положение
точки Я и после построения ступени — положение точки /.
Диаграмма компонента С. Концентрация ус
находится в равновесии с хс и хв . Это определяет положение
точки Я и после построения ступени — положение точки /.
Диаграмма компонента В. Концентрация ув _
находится в равновесии с хв и хс . Это определяет положение
точки К и, следовательно, положение точки L.
Диаграмма компонента С. Концентрация ус _
находится в равновесии с хв и хс . Это определяет положение
точки К и, следовательно, точки L.
Таким же способом строят все ступени. Положения точек,
соответствующих равновесным растворам, например Я и К,
должны на обеих диаграммах совпадать. Исключение
составляют точки рабочей линии, например G, L, J и М, которые яе
относятся к равновесным растворам. Если после достижения
точки (xF, г/i) на каждой диаграмме получим равное число
ступеней, это доказывает, что значение хс выбрано правильно.
В противном случае задаются новым значением хс и расчет
проводят сначала, пока не будет соблюдено указанное выше
условие.
Вполне возможно, что один из компонентов экстрагируется,
в то время как другой переходит из растворителя А в D. В этом
случае ступенчатая линия на диаграмме реэкстрагируемого
компонента проходит ниже рабочей линии. Как было показано,
один из компонентов может экстрагироваться в одной части
каскада и реэкстрагироваться в другой.
Независимое распределение компонентов. Если компоненты
раствора распределяются независимо друг от друга, для
каждого компонента используется лишь одна кривая распределения.
Равновесные растворы на каждой ступени могут на диаграммах
не совпадать; следовательно, для определения
окончательного состава рафината необходимо провести расчет методом
последовательных приближений.
Пример VII-6. Водный раствор, содержащий 0,3 моль/л U02(N03)2 и
2,0 моль/л HN03, экстрагируется при противотоке фаз экстр агентом —
керосином, содержащим 30 объеми. % трибутилфосфата и 0,048 моль/л
U02(N03)2. Объемное отношение экстрагента и исходного раствора равно
22 Р. Е. Трейбал 337
0,7. Концентрация U02(N03)2 в водном растворе снижается до 0,02 моль/л
Определить число требуемых ступеней и концентрацию HN03 в рафинате
Решение. Равновесные данные (те же, что и в примере VII-1)
приведены на рис. 171, на котором показано увеличенное изображение части
диаграммы распределения HN03. Растворители можно считать несмешиваю-
щимися, концентрации хну выражаются в моль/л, расходы компонентов
А к D -объемные; xVOAN03hp~ 0,3; xVOl(liOAn = 0№ Уио,(КОЛ5 = 0.048;
-^HNOs — 2,0; #hno3S — 0; Ц/Л — 1/0,7 = 1,43. По этим данным определяем
положение точки G на диаграмме U02(N03)2 и с наклоном 1,43 проводим
рабочую линию до точки М, для которой xF=0,3. После нескольких
приближений конечное значение хШОаП принято равным 1,94 моль/л, что позволяет
определить на диаграмме HN03 положение точки G, из которой проведена
рабочая линия с наклоном, равным 1,43.
На диаграмме U02(NO8)2, начиная построение от точки G, определяем
положение точки Н при a:HNOj=1,94, соответствующей продуктам,
выходящим с последней ступени, и аналогично на диаграмме HN03 — положение
точки Н при -^uOjfNOjb п== 0,02. В обоих случаях точка / находится на
рабочей линии и имеет ординату, равную ув. На диаграмме U02(N03)2 точка К
соответствует *HNOi/ = 2,14. На диаграмме HN03 точка К должна быть
определена при -*ruo2(N03)2 A"= ^l1^ это означает, 4toHN03 начинает
вымываться из органической фазы. Строя ступени до точки М, получим, что для
извлечения каждого компонента необходимы три теоретические ступени
(хШО,п= 1>94). Это значение *ныо3п было принято в начале расчета.
В данном случае кислота экстрагируется на ступени, концентрация
урана на которой невелика, и вымывается из органического растворителя, когда
происходит предпочтительная экстракция ураиа при его более высоких
концентрациях 18. Поэтому концентрация кислоты достигает максимального
значения прихНЫОа/= 2,14, что способствует экстракции урана. При низких
концентрациях U02(N03)2 диаграмма подобна приведенной на рис. 170.
иог( моз,)г •Z'hno,
Рис. 171. К примеру VH-6. ПротивоТОЧная экстракция уранилнитрао
и азотной кислоты (обозначения осей координат см. рис. 164).
338
Независимые постоянные коэффициенты, распределения.
В связи с тем, что уравнения (VI, 123) — (VI, 128) и рис. 140
применимы непосредственно для каждого из распределяемых
компонентов независимо от других, окончательный состав
продуктов обычно можно рассчитать 'непосредственно, не прибегая
к методу последовательных приближений. В зависимости от
содержания компонента в исходном экстрагенте один или более
распределяемых компонентов могут «е экстрагироваться или
даже переходить из экстракта в рафина-т. Так, если для какого-
либо компонента ys = tnxF, то этот компонент не будет
переходить из фазы в фазу и для него xn=xF. Если ys>tnxF, то
компонент будет переходить из экстрагента в рафинат. В этом
случае роли компонентов Л и D меняются, и уравнение (VI, 125)
лучше переписать в виде:
шч+1_±
ys~yi \е) е 1-е"
(Г-
1-е"
(VII, 36)
Аналогично в случае процесса обратной экстракции
ординату на рис. 140 заменяют значением (t/i— mxF)/(ys — mxF),
а в качестве параметра используют величину 1/ё.
Как будет показано в следующем примере, если
селективность экстрагента не очень велика, возможная степень
разделения компонентов ограничена аналогично экстракции в
перекрестном токе. Обычно применяют схему процесса,
предназначенную, скорее, для выделения компонентов из исходной смеси,
чем для разделения компонентов друг от друга.
Преимуществами противоточной схемы перед схемой экстракции в
перекрестном токе является уменьшение расхода экстрагента и
числа ступеней.
Обычно лучшее разделение двух компонентов достигается в
том случае, когда отношение A/D выбирают так, чтобы фактор
экстракции ё для одного из компонентов был больше, а для
другого меньше единицы. Это приводит к касанию рабочей
и равновесной линий для двух компонентов на
противоположных концах каскада (рис. 141). Когда требуются высокие
степень разделения и выход обоих компонентов, применяют
центральный ввод исходной смеси в каскад, подобно тому как
показано для процесса фракционной экстракции (см. стр. 348).
Пример VII-7. Смесь, состоящая из 33,5% пара-хлорнитробензола (В)
и 66,5% мета-хлорнитробензола (С), в количестве 100 кг/ч растворяется в
500 кг/ч водного раствора метанола (D) и экстрагируется в противотоке
смесью «Skellysolve» (А) (см. пример VII-3). Экстрагент содержит
компоненты В и С соответственно в количествах 0,005 и 0,005 кг на 1 кг Л, а
постоянные независимые коэффициенты распределения имеют следующие
значения: тв = 1,33 и тс=0,82.
22*
J
Определить: а) максимально возможную степень экстракции
компонента^ из исходного раствора; б) минимальный расход экстрагента при 90%-
ной степени экстракции компонента В из исходного раствора и составы
образующихся при этом продуктов; в) число ступеней и составы
получающихся продуктов при 90%-ной степени экстракции компонента В экстраген-
том, расход которого в 1,5 раза превышает его минимально потребный
расход.
Решение. Имеем:
xbf = W = 0'067kzB/kzZ)
хср = -Ц5. = 0,133 кг С/кг D
yBS = ycs = 0,0°5 кг1кг А
а) Наименьшее значение х , соответствующее максимальной степени
экстракции, достигается, когда концентрация х является равновесной с
экстрой
агентом. Получаем: хд =!/в5/тв=0,005/1,33 = 0,00376 кгВ/кгй. Степень
экстракции компонента В составляет \(хвр — хв )/хвр\ • 100= 94,5%. Это
соответствует числу ступеней, равному бесконечности, или касанию
равновесной и рабочей линий для компонента В на конце каскада, отвечающему
входу экстрагента.
б) При 90%-ной степени экстракции компонента В концентрация
х = 0,01 ^,067) = 0,0067. При минимальном расходе экстрагента (я= со;
см. главу VI):
е = га-
У* D
Ilia
Для компонента В:
0,067 — 0,0067 1,33-Л
.-__ 0,005 — 500
0,067 ~ тж
Л=359 кг/ч (минимальный расход)
Для компонента С:
га „А 0,82 • 359
ec = -zr = ^oo— = 0-588
0,133 — хс
" =0,588
(U33-0'005
0,82
хг = 0,585 кг/кг D
Рафинат (раствор D):
В = 0,0067 • 500 = 3,35 кг/ч = 10,28%
С = 0,0585 • 500 = 29,25 кг/ч = 89,72 %
32,60 кг/ч
340
Экстракт (раствор А):
В = 33,5 — 3,35 -+- 0,005 • 359 = 31,95 кг/ч = 45%
С = 66,5 — 29,25 + 0.005 • 359 = 39,05 кг/ч = 55%
71,00 кг/ч
•При степени экстракции компонента В, равной 90%, невозможно
получить экстракт с содержанием В выше 45% или рафинат с содержанием С
выше 89,72% (при обычной противоточной экстракции данным экстрагентом).
в) Имеем:
xR = 0,0067 А = 1,5 • 359 = 538 кг/ч
1,33-538
500
0,82 • 538
С 500
Ч-М- 0,0067-
1,430
= 0,882
0,005
ТзГ
= 0,0465
xDB-^- 0,067
0,005
bf m0 ' 1,31
Согласно рис. 140, я=5,5 ступеней. Из этого же графика
«/„<. 0,005
—£- = 0,22 = -
0,82
х
!££. 0,133-
0,005
cf mc ' 0,82
хг = 0,0340 кг/кг D
Кроме того, число ступеней я можир найти из уравнения (VI, 126), а
концентрацию х — из уравнения (VI, 125).
Рафинат (раствор D):
В = 0,0067 • 500 = 3,35 кг/ч = 16,48%
С = 0,0340 • 500 = 17,00 кг/ч = 83,52 %
20,35 кг/ч
Экстракт (раствор А):
В = 33,5 — 3,35 -4- 0,005 • 538 = 32,84 кг/ч = 38,6%
С = 66,5 — 17,00 + 0,005 • 538 = 52,19 кг/ч = 61,3%
85,03 кг/ч
Пример VI1-8. Рассчитать процесс экстракции смеси равных количеств
изомеров нитроанилина, вводимой в противоточныи каскад в виде раствора
в воде (D), чистым бензолом {А). Коэффициенты распределения, равные
то=64,0, тт = 25,0 и мр*=9,3, считать постоянными и независимыми.
Решение. Можно разделить орто- и мета-изомеры или мета- и пара-
изомеры. Для выделения орто- и мета-изомеров факторы экстракции можно
принять равными е„=1,5; ет = 25 • е„/64=0,586, ер=9,3 • eo/64=0,218.
341
Рассмотрим рис. 140. При большом числе ступеней разделения,
например я =10, имеем:
хп
х „
of
= 0,006, т. е. экстрагируется 99,4% орто-изомера
-i£!HL= 1 —em= 1—0,586 = 0,414, т. е. экстагируется 58,6% мета-изомера
XmF
—£f- = I —e„= 1—0,218 = 0,782, т. е. экстрагируется 21,8% пара-изомера
XPF
Величины xF для каждого из компонентов равны; соответственно
концентрация (чистота) орто-изомера в экстракте составит:
0,994л: _ 0,994-100
0,994^ + 0,586*^ + 0,218 хрр lW~ 1,798
Принимая число ступеней разделения равным пяти, получим выход
орто-изомера 95,1% при его концентрации 54,2%- Максимальная
концентрация орто-изомера достигается при е0=<1 и очень большом числе ступеней
экстракции. Тогда, учитывая, что при данных условиях доля
экстрагированного вещества равна приблизительно 8, найдем концентрацию орто-изомера
в экстракте (доли xF для каждого компонента равны) по уравнению
е х „-100 m -100
;-°-2f : = г-2 : = 65%
е х „ + е х _ + е х „ га +in +in
о OF ' т mF ' р pF о ' т < р
Эта концентрация достигается при фракционном выходе, равном е0, и
соответствует равновесию с исходной смесью. По мере увеличения е0 до
значений, превышающих единицу, концентрация орто-изомера в экстракте
уменьшается, а выход его увеличивается.
При разделении мета- и пара-изомеров получают рафинат, который
содержит относительно чистый пара-изомер. Полагая е0=3,84, найдем:
8т=25-8„/64=1,5 и 8Р=9,3-80/64=0,558; при большом числе ступеней,
например л=10, из уравнения (VI, 125) для орто-изомера получим:
*оп , е" + '-ео е«-1 3,84-1 _,„-в
■А-—-. - = ^+7 =—тт, г = Ю"
*ор С-1 С-1 з.84'1-1
Из рис. 140 для мета- и пара-изомеров находим:
Хтп 0,006, ^1=1-0,558 = 0,442
XmF XPF
Пара-изомер получим в рафинате с выходом 44,2% и концентрацией,
или чистотой (х „для каждого изомера одинакова), равной
0,442л- р- 100
! PL = 98,6%
Ю-6* в + 0,006л: „ +0,442л: „
OF ' ' mF ' pF
С уменьшением величины г увеличивается выход пара-изомера, но
снижается его чистота.
Если процесс экстракции проводится при этих условиях, бензольный
экстракт можно отмыть водой и получить раствор орто-изомера (высокой
степени чистоты) в бензоле. Для получения же трех относительно чистых
компонентов требуется установка двух каскадов с центральным вводом
питания.
342
ПРОТИВОТОЧНАЯ МНОГОСТУПЕНЧАТАЯ
ЭКСТРАКЦИЯ С ФЛЕГМОЙ
Принципиальная схема этого процесса, проводимого с
возвратом части экстракта, показала на рис. 172. Каскад ступеней
от 1-й до (f—1)-й добавляется к обычному противоточному
каскаду ступеней от f-и до п-й для получения экстракта, более
обогащенного распределяемым компонентом, чем экстракт,
равновесный с исходным раствором.
Степени свободы. Для определения числа степеней свободы
можно рассматривать всю установку состоящей из двух проти-
воточных каскадов, ступени питания и секции получения
флегмы. Таким образом (см. раздел о многоступенчатой противо-
точной экстракции), для двух каскадов:
Для ступеней от (/ + 1)-й до п-а Ni = 2с + 2 (я — /) + 5
Для ступеней от 1-й до (/ + 1)-й N( = 1с + 2 (/ — 1) + 5
Всего Ас + 2л + 8
Рассмотрим теперь точку, в которой рафинат /?/_i и
исходный раствор F смешиваются друг с другом и образуют
поток К:
Поток Nv
Rf-x с + 2
F с + 2
К с + 2
Теплообмен с окружающей средой 1
Всего ... Nv = 3c-\-7
Материальные балансы с
Баланс энтальпии 1
Всего ... с+1
W,. = 3c + 7— (с+1) = 2с + 6
кSF 'Отделение
[ .экстраеента
Рис. 172. Многоступенчатая противоточная экстракция с флегмой
(возвратом части экстракта).
343
Для самой ступени питания Ni = 2c+6; однако это число уменьшается
на с+2, если ступень питания совпадает с точкой ввода исходной смеси, так
как поток К является внутренним. Тогда при подаче исходного раствора на
ступень питания
Nt = (2с + 6) + (2с +6) — (с + 2) = Зс +10
Присоединяя ступень ввода исходной смеси к обоим каскадам, получим
(при четырех внутренних потоках Rs-\\ Rf, Ef и £/+i):
Ni = (Зс 4- 10) + (4c 4- 2л + 8) — 4 (с + 2) = Зс + 2л + 10
Аналогично можно показать, что применительно к установке,
предназначенной для отделения экстрагента (т. е. для получения флегмы), Ni = c+5,
и если это условие наложить на каскад (при внутренних потоках Et и R0),
то для всей установки Л/,-=2с+2я+ 11.
Обычно фиксируются следующие независимые переменные:
Nt
Состав питания, его состояние и расход . с + 2
Состав экстрагента и состояние с-}~1
Потери тепла и давления на каждой ступени 2л
Потери тепла на входе исходного раствора
(обычно равны нулю) 1
Потери тепла и давления на установке
отделения экстрагента 2
Отношение количеств экстрагента и
неэкстрагента в потоке S 1
Организация ввода исходной смеси таким
образом, чтобы число ступеней было
наименьшим 1
Всего . . . 2с4-2л + 8
Число Nt, выбираемое произвольно, равно 3. Это могут быть те же
независимые переменные, которые принимались для противоточной
многоступенчатой экстракции с добавлением в качестве переменной флегмового числа.
Четырехкомпонентные системы. Для таких систем применимы
уравнения (VI, 130) — (VI, 150); построение процесса в
треугольной диаграмме приведено на рис. 173. Для этого можно
допустить, что кроме обычно задаваемых величин (см. выше)
фиксированы XAR , X АЕ{ и флегмовое число. Эти три величины
соответствуют трем имеющимся степеням свободы системы.
Тогда F и S можно изобразить на диаграмме в двух проекциях.
Так же как и на рис. 161, точка Rn должна лежать на кривой
пересечения плоскости XAR с поверхностью растворимости;
положение точки на этой кривой выбирают произвольно (с
последующей проверкой) и затем определяют ее проекции.
Аналогично точка Ei должна лежать на кривой пересечения плоскости
ХАЕ[ и поверхности растворимости (не показана). Ее
положение выбирают на обеих проекциях. По дистилляционным
характеристикам экстракта Ец находят составы регенерированно-
344
го экстрагента SB и конечного продукта РЕ и соответствующие
им точки наносят на обе проекции. При R0+F=L уравнение
материального баланса всей установки имеет вид:
L + S = E{ + Rn = M (VII, 37)
На обеих проекциях проводят линии si и е^гп, которые
пересекаются в точке т. Если отношения отрезков линий sml и
e\.tnrn равны для обеих проекций, то положения точки
пересечения М на фазовой диаграмме и точки £4 выбраны правильно.
Положение этих точек корректируют до тех пор, пока не будет
соблюдено указанное выше условие.
в
ъ;я0;£'
Рис. 173. Противоточная многоступенчатая экстракция с флегмой
(возвратом части экстракта); четырехкомпонентные системы.
345
Положение точки Q (и ее проекции q), являющейся рабочей
точкой для секции обогащения экстракта, определяют по
флегмовому числу; продолжения линий fq и rns на обеих
проекциях пересекаются в точке w. Как и в случае тройных систем,
вспомогательные линии (при определении числа ступеней) для
обеих секций каскада проводят радиально из точек q и w,
изменяя взаимное положение этих точек, если хорда равновесия
пересекает линию qf (что уменьшает п при выбранном флегмо-
вом числе).
Положение точек, соответствующих экстрактам и рафина-
там, находящимся в равновесии, определяют по методу,
описанному в разделе «Многоступенчатая противоточная экстракция».
Если при этом будет найдено, что фазы Еп и Rn имеют
равновесные составы, то .положение точки Rn первоначально было
выбрано правильно. Если это условие не удовлетворяется,
положение точки Rn изменяют и проводят расчет сначала. Число
необходимых теоретических ступеней для каждой секции
каскада соответствует числу хорд равновесия для этих секций.
При минимальном флегмовом числе радиальные линии,
выходящие из точек Q или W, совпадают с хордой равновесия.
В действительности точка Q должна располагаться ближе к
SE, a W — ближе к S, чем при минимальном флегмовом числе.
Иногда для большей ясности построений лучше использовать
проекции на грань ABD (которые заменяют проекции на ACD).
Несмешивающиеся растворители. В этом случае при
регенерации экстрагента на установке для его отделения и получения
потока Ro остаток состоит только из компонентов, которые
смешиваются с экстрагентом D. Следовательно, на конец каскада,
соответствующий выходу экстракта, нужно подавать экстрагент
D. Такая схема лежит в основе процесса фракционной
экстракции, рассматриваемого ниже.
\
ФРАКЦИОННАЯ ЭКСТРАКЦИЯ
(МНОГОСТУПЕНЧАТАЯ ПРОТИВОТОЧНАЯ
ЭКСТРАКЦИЯ ДВУМЯ ЭКСТРАГЕНТАМИ)
Этот способ проведения процесса экстракции (рис. 174)
является наиболее эффективным сравнительно с другими
экстракционными методами разделения. Исходный раствор F вводят в
центральную часть каскада, состоящего из п + п' ступеней,
через которые противотоком движутся два экстрагента А и D;
последние могут быть и не чистыми веществами. На одном или
на обоих концах каскада можно организовать возврат флегмы
(см. рис. 174).
Степени свободы. Рассмотрим наиболее общий случай, когда
флегму используют на обоих концах каскада. Вся установка
состоит из двух противоточных каскадов и ступени питания.
348
Число независимых переменных Nt для двух каскадов и ступени
питания (см. предыдущий раздел) равно Зс+2{п+п') +10. Примем, что флегму
получают на ступени Г. Для установки отделения экстрагента и делителя
флегмы Ni = c+5 (см. предыдущий раздел). Смешение флегмы и
экстрагента А дает ЛГ(=2с+6 (см. предыдущий раздел). Комбинируя делитель
флегмы и смеситель (при одном внутреннем потоке), найдем:
ЛГ, = (с + 5) + (2с + 6)-(с+2) = 2с + 9
То же получим и для ступени 1. Отбирая флегму в двух точках каскада (при
четырех внутренних потоках), определим число степеней свободы для всей
установки:
^ = Зс + 2(л + л') + Ю + 2(2с + 9) —4(с +2) = Зс+2(п + л')+20
Обычно фиксированными являются следующие независимые переменные:
Nt
Состав исходного раствора, состояние и расход . . с —(— 2
Состав обоих экстрагентов и их состояние 2 (с —[— 1)
Потери тепла и давления
на каждой ступени 2 (п -\- п')
в каждом сепараторе экстрагента 4
Потери тепла при смешении флегмы с экстраген-
тами (обычно равные нулю) 2
Потери тепла при вводе исходного раствора
(обычно равные нулю) 1
Относительное содержание экстрагента в каждом
нз регенерированных экстрагентов 2
Выбор места ввода исходного раствора, при
котором сумма п-\-п' наименьшая 1
Всего Зс + 2 (п + п') -f 14
Для произвольного выбора остается Ni=6. Эти величины могут быть
выбраны из числа приведенных ниже:
Число ступеней п-\-п' 1
Отношение п'/п 1
Расход каждого экстрагента 1
Отношение расходов экстрагентов 1
Флегмовое число на каждом конце каскада
(включая число, равное нулю) 1
Концентрация какого-либо компонента в каком-
либо продукте, каждая концентрация .... 1
Отношение концентраций двух компонентов
в каком-либо продукте, каждое отношение . . 1
Отношение относительного содержания какого-
либо компонента в исходном растворе к
содержанию его в каком-либо продукте, каждое
отношение 1
* Отношение относительных содержаний двух
компонентов исходного раствора к их
содержаниям в каком-либо продукте, каждое
отношение 1
347
Таким образом, например, можно выбрать произвольно отношение
концентраций в продуктах, расходы обоих экстрагентов и два флегмовых числа.
Каждое флегмовое число можно выбирать независимо в пределах значений
от нуля до бесконечности (в этом случае обычное понятие о минимальном
флегмовом числе не применимо). Кроме того, существует широкий интервал
отношений количеств экстрагентов и их расходов, в котором можно
провести процесс разделения.
Четырехкомпонентные системы. Для упрощения диаграмм
рассмотрим случай экстракции, когда исходный раствор состоит
только из компонентов В к С, а экстрагенты' являются чистыми
веществами А и D.
На практике возможны отклонения от этих условий. Кроме
того, примем, что установка работает без флегмы на обоих
концах каскада и что фиксированы количества экстрагентов и
отношения компонентов В к С в каждом продукте. Как следует
из рис. 174, для схемы экстракции двумя экстрагентами нет
различия в понятиях «экстракт» и «рафинат»; однако для
удобства потоки между ступенями будут обозначены по-прежнему
буквами Е и R.
На рис. 175 точка F отвечает составу исходного раствора,
а точки К и L — составам продуктов, свободных от
экстрагентов. Плоскость KAD, соответствующая постоянному отношению
В/С, пересекает поверхность растворимости по кривой TEiN;
на этой кривой должна лежать точка, отвечающая продукту Еи
богатому компонентом А (положение этой точки уже
определено). Аналогично плоскость LAD (не показана) пересекает
поверхность по кривой TfoN, на которой должна находиться
Экстрагент А
Е
"Жж.
4
Продукт
Отделение
экстраеента
п(+а)
Подача флегмы на Выходе
экстравента D
Экстрагент D
Отделение
экстраеента
Продукт
Экстрагент D
Подача флегмы на выходе
экстраеента А
Рис. 174. Фракционная экстракция.
348
Рис. 175. Определение состава продуктов при фракционной экстракции
(четырехкомпонентные системы).
точка, соответствующая продукту Rv, богатому
компонентом D, хотя положение точки Ri< еще не известно. Эти кривые
показаны также частично на проекциях ACD и ABD.
Общий материальный баланс имеет вид:
F + A + D = El-\-Rv -4- М (VII, 38)
Для компонента В
FXBF = ElXBEl + RVXBRV = МХВМ <V"- 39>
Для компонента С
FXCP = E,XCEi + RvXCRv = МХСМ (VII, 40)
349
"9 обеих проекциях определяют положение точек f и A+D;
последняя представляет собой состав суммы двух экстр агентов,
взятых в правильном соотношении.
Проекции точки М на линии, соединяющей точки F и A + D,
можно определить по отношению отрезков этой прямой
(отношение для самой прямой и ее проекций одинаково) или
нахождением координат точки М из уравнений материальных
балансов компонентов.
На кривой, проведенной при постоянном отношении В/С,
вибирают положение точки е^ и проводят линию вугп, затем на
второй кривой получают точку г{,. Если отношение отрезков
eimlmrv на обеих проекциях одинаково, то положение точек е,
и гу Определено правильно. После этого можно найти составы
пР0ДУктов экстракции. Например, отношение AID в экстракте
Е, можно определить, продолжив линию С^ до пересечения с
ребро^ AD на проекции грани ACD; из положения точки е, на
проекции ABD находят содержание компонента В, а по
отношению В/С — содержание компонента С. При помощи уравнений
(VII, 38) — (VII, 40) можио рассчитать расходы Е\ и R\.
Материальный балаис для ступеней от Г-й до т'-и
(т'—номер любой ступени, находящейся слева от ступени питания):
/?ffl.+i + A = Em. + Rv (VII, 41)
Следовательно
^•-A = Rm.+i-Em, = W (VII, 42)
Величина W выражает, таким образом, постоянную разность
расходов Е и R между соседними ступенями, имеющими
нумерацию со штрихами.
Аналогично для ступеней от т-\\ до 1-й (т — номер любой
ступени справа от ступени питания):
Em+l + D = Rm + El (VII, 43)
£,-D = £m+1-#m = Q (VII, 44)
Величина Q — постоянная разность расходов Е и R между
соседними ступенями, имеющими нумерацию без штрихов.
Решая совместно уравнения (VII, 38), (VII, 42) и (VII, 44),
получим;
F=Q-\-W (VII, 45)
На рис. 176 показаны составы конечных продуктов £\ и Ri-.
Из уравнения (VII, 44) следует, что точка Q лежит на продол-
350
жении линии DEi и ее положение (а также положение
екцнй) можно определить по величине длин отрезков:
Q EXD exD
Аналогично находят проекцию точки W, лежащей
должении линии AR[. Если положения точек Q и W
лены правильно, то линия qw должна проходить через
На пространственной диаграмме фаза Еу находится в
ве'сии с фазойRy, линия EVW в месте пересечения ее с
ее про-
(VII, 46)
на про-
опреде-
точку /.
равно-
поверх-
Рнс. 176. Расчет числа ступеней при фракционной экстракции
(четырехкомпонентные системы).
351
ностью растворимости определяет положение точки R2- и т. д.
Как показано на диаграмме, растворы Еу, Е2- ... и R\>, R2> ...
образуют на поверхности растворимости кривые. Аналогично
Ri находится в равновесии с Ей Е2 определяют как точку
пересечения линии QRi с поверхностью растворимости и т. д.
Растворы Еи Е2 . . . и Ri, R2 . ■ ■ образуют кривые линии на
поверхности растворимости.
Кривые растворов Е и R для обеих секций каскада должны
каждая в отдельности пересекаться соответственно в точках
£,л+1 = £л. и Rn+\=Rn'- Эти точки пересечения должны в свою
очередь лежать на противоположных концах хорды равновесия,
так как они показывают составы равновесных растворов,
выходящих со ступени питания. На проекциях ступени
определяются так же, как было описано в предыдущих разделах главы.
Если ЕП' и Rnf лежат на противоположных концах хорды
равновесия, то составы конечных продуктов и расходы экстраген-
тов отвечают целому числу ступеней, которое для каждой
секции каскада соответствует числу хорд равновесия. Если кривые
растворов Е или R не пересекаются, — значит соотношение
экстрагентов выбрано вне допустимых пределов и необходимо
проводить расчет, задаваясь другим соотношением экстр-
агентов.
Отметим, что концентрации компонентов В я С
увеличиваются по мере приближения от концов каскада к ступени
питания. Эта закономерность характерна для фракционной
экстракции, проводимой как без флегмы, так и при отборе
флегмы (для большей части значений флегмовых чисел).
Применение флегмы. Если флегма подается на ступень 1, то
при том же отношении AID содержание суммы компонентов
(В + С) в Ei должно возрасти. При этом точка Ei на
пространственной диаграмме перемещается ближе к ребру ВС (при
постоянном отношении В/С), а точка Q — к точке F. Аналогично
использование флегмы на ступени /' перемещает точку W
ближе к F. В результате этого расстояние по кривой растворимости
от точки Ei до точки Еп> и от Ry до Rn+i уменьшается, что
может в ряде случаев (в зависимости от наклона хорд
равновесия или от характера изменения селективности пары
растворителей с концентрацией распределяемого компонента) привести
к уменьшению числа ступеней. Для процессов с флегмой
нельзя сделать каких-либо обобщений, за исключением процессов
со взаимно нерастворимыми экстрагентами и с независимым
распределением компонентов.
Взаимно нерастворимые экстрагенты. В общем случае, когда
распределение компонентов взаимозависимо, поступают так же,
как при 'независимом распределении компонентов. Однако при
этом нужно, чтобы составы на каждой ступени соответствовали
равновесным (аналогично обычной противоточной экстракции).
352
Пример расчета приведен в работе Асселина и Камингса39.
Основные принципы расчета станут более ясными при
рассмотрении случая независимого распределения компонентов.
Взаимно нерастворимые экстрагенты; независимое
распределение компонентов 15,19,20,39^ Принципиальная схема этого
процесса (без применения флегмы) показана на рис. 177.
Распределяемыми компонентами являются вещества В и С; в качестве
экстрагентов используются компоненты А и D. Из схемы видно,
что поступающие на экстракцию экстрагенты содержат
некоторое количество распределяемых компонентов. Это возможно
вследствие использования регенерированных экстрагентов или,
как будет показано ниже, в результате применения флегмы.
Часто случается, что компоненты, подлежащие разделению,
уже растворены в одном из экстрагентов. Если же они
являются твердыми, то их можно предварительно растворить в одном
из экстрагентов, что облегчает их введение в каскад. По этой
причине на схеме показано, что исходный раствор содержит
некоторые количества AF и DF (в исходном растворе может
содержаться один из экстрагентов или оба экстрагента могут
в нем отсутствовать; оба экстрагента в исходном растворе
встречаются очень редко).
Например, уран и ванадий, растворенные в водном растворе
кислоты (раствор после выщелачивания из руд), можно
разделить экстракцией керосином, содержащим алкилфосфат,
который селективно экстрагирует уран. Если воду рассматривать
как экстрагент D, а органическую фазу — как экстрагент А, то
содержание воды в исходном растворе равно DF. В левой
секции каскада, называемой секцией экстракции, уран
экстрагируется органической фазой; водный раствор, выходящий со
ступени Г, имеет очень низкую концентрацию урана и содержит
большое количество ванадия.
Органический растворитель (экстрагент) экстрагирует
некоторое количество ванадия, поскольку его селективность не рав-
Ув0' Увг Увт--1 Увт> Увп--1 Увп+i Увт+, Увт Увг Ув,
У Со Ус; Уст:, Уст- Ус„:, Усп+, Уст+, Уст Усг Ус,
хВ,г хвг. Хвт- хВт,+1 хВп,
ХС ®сг- хСт, хСт,+, хс№
х8п хВт х8т-, ХВ, ХВо
Ami 'XCm ХСт~' ХС< Х°°
растВор
F=B+C+AF+nF
Рис. 177. Фракционная экстракция (взаимно нерастворимые
экстрагенты).
23 Р. Е. Трейбал
353
на бесконечности. В правой секции каскада, называемой
секцией отмывки, ванадий будет вымываться из органической
фазы и возвращаться в секцию экстракции в виде водной фазы.
Это приводит к тому, что органическая фаза, вытекающая со
ступени 1, почти не содержит ванадия и обогащена ураном.
Если отношение количеств экстрагентов для каждой секции
каскада постоянно, то для ступени ввода исходного раствора
D' = D+D
(VII, 47)
А = А'+Ар
Материальный баланс какого-либо одного компонента для
ступеней l'-й — /п'-й:
Л'у0. + D'xn.+1 = A'ym, + Dx[, (VII, 48)
Z- = Ут"~Уо' (VII, 49)
A xm'+l XV
Последнее выражение является уравнением рабочей линии —
прямой в координатах х, у. Прямая имеет наклон, равный
D'lA', и проходит через точку (xv, y0,), соответствующую концу
каскада, и точку (xm,+v УтХ Последняя отвечает составам
растворов, проходящим между двумя соседними ступенями
слева от ступени
питания. Этому уравнению
соответствуют линия
MN для компонента В,
нанесенная на рис. 178
вместе с равновесной
кривой для того же
компонента, и линия
RS на рис. 179 для
компонента С.
Аналогично для
ступеней от т-k до 1-й
материальный баланс
каждого компонента:
= Аух + Dxm (VII, 50)
D
Ут+\ — У\
(VII, 51)
Рис. 178. Построение числа ступеней по
компоненту В:
1 — линия флегмы; 2—рабочая линия (наклон D!A);
3—кривая равновесия; 4 —рабочая линия (наклон D'lA').
Последнее
уравнение является
уравнением рабочей линии —
прямой с наклоном
D/A, проходящей через
точки с координатами
364
хСа'Ус,
Рис. 179. Построение числа ступеней по компоненту С:
1 — линия флегмы; 2 — рабочая лнння (наклон D/A); 3 — кривая равновесия; ^ — рабочая
линия (наклон D'lA').
(*о, #i) и (хт, ym+i). Этому уравнению соответствуют рабочая
линия PQ на рис. 178 для компонента В и рабочая линия TU
на рис. 179 для компонента С («линии флегмы» на этих
рисунках будут рассмотрены ниже).
Если экстрагенты, поступающие с обоих концов каскада, не
содержат распределяемых компонентов, то рабочие линии
начинаются соответственно на осях л; и у. Когда исходный раствор
не содержит экстрагентов, все четыре
рабочие линии параллельны друг _ /V
другу. т'г'
Графическое определение числа
теоретических ступеней проводят
отдельно для каждого каскада и
каждого компонента, причем построения
начинают от точек, соответствующих
концам каскадов (точки М, Р, R и Т), пн
и продолжают в области более высо- m
ких концентраций. Числа ступеней и
концентрации для обоих компонентов
должны совпадать на ступени
питания. Для проверки этого условия
строят график, выражающий
зависимость концентраций компонентов В и хвпн хспд
С в экстрагенте (не содержащемся в Рис ш к 0 ению
исходной смеси) на каждой ступени числа ступеней при фрак-
от номера последней (рис. 180). Число ционной экстракции.
23*
355
ступеней п слева от ступени питания и число ступеней п справа
от нее должны быть равны для каждого из распределяемых
компонентов. Концентрации компонентов В п С в потоке на
ступени питания (при условии, что этот поток не разбавляется
каким-либо экстрагентом, вводимым с исходным раствором на
ступень питания) должны быть одинаковы, независимо от того,
с какого конца каскада велся расчет. Условия,
удовлетворяющие этим требованиям, определяются положением малого
прямоугольника на диаграмме рис. 180.
Экстрагент, вводимый с питанием. При данном общем рас-
го уменьшается21, если принять во внимание
е условия (рис. 181). Это облегчает процесс
личии в системе нескольких распределяемых
PQ и TU — увеличиться.
Пересечение рабочих линий. Как видно из рис. 178 и 179,
рабочие линии при их продолжении пересекаются (исключение
составляет случай, когда исходный раствор не содержит экстр-
агентов). Определим положение точки пересечения рабочих
линий. Пусть точка пересечения имеет координаты хг и уг. Для
компонента / из уравнений (VII, 48) и (VII, 50) получим:
A'vi T А'Уо- + D'xi — D'xv (vn. 52)
и
Ayt = Ayl + Dxt — DxQ (VII, 53)
В этих уравнениях концентрации х я у относятся к
компоненту /. Вычитая одно уравнение из другого и решая их
относительно уи находим:
ID-D'\ Аух-А'уу-Рхй + РГху
Ь = [-А-=Ж) Xi + А=А' (VH'54)
Общий материальный баланс по компоненту /:
JF=Ay1 + A'y0. — Dx0 + D'xv (VII, 55)
где JF — количество компонента в исходном растворе.
Подставляя уравнение (VI, 55) и уравнение (VII, 47) в
уравнение (VII, 54), получим:
DF JF
У1 = -1ГХ1 + 1Г (VII'56)
F F
Это уравнение определяет геометрическое место точек
пересечения рабочих линий и соответствует прямой линии с углом
наклона, равным —DFJAF. Линия отсекает на оси ординат
отрезок IF/AF, на оси абсцисс — отрезок JF/DF и пересекает
кривую равновесия в точке с координатами (xF, yF),
соответствующей концентрациям равновесного раствора на ступени питания.
356
Возможны три частных случая:
1. AF = 0 и Л=Л', откуда xx = xF (исходная концентрация
' распределяемого компонента в DF).
* 2. DF=0 и D = D', откуда yt = yF (исходная концентрация
распределяемого компонента в AF).
3. AF = DF = 0, откуда yi = x{ = oo (рабочие линии парал-
^ лельны).
i Пересечение рабочих линий в общем случае и в частных
Г случаях 1 и 2 показаны на рис. 181.
f Приведенные соотношения удобно применять на практике,
Пересечение рабочих линий. Как видно из рис. 17*8 и Г?9,
рабочие линии при их продолжении пересекаются (исключение
составляет случай, когда исходный раствор не содержит экстр-
циенты распределения не постоянны). Число таких
приближений значительно уменьшается21, если принять во внимание
ограничительные условия (рис. 181). Это облегчает процесс
расчета при наличии в системе нескольких распределяемых
компонентов, даже если первоначально число ступеней не
известно. Число ступеней, рассчитываемых по известным
концентрациям продуктов для двух компонентов, становится
«известным» для остальных компонентов, конечные концентрации
которых должны быть затем определены.
Взаимозависимые коэффициенты распределения. Если
распределение компонентов является взаимозависимым
(равновесные данные изображаются в виде семейства кривых), то
нахождение числа ступеней для каждого каскада необходимо
провести в соответствии с рис. 170; при этом на каждой ступени
должны быть удовлетворены равновесные соотношения для
каждого компонента. В противном случае процесс расчета
аналогичен показанному на рис. 177—180.
Рис. 181. Пересечения рабочих линии:
в —общий случай; 6 — А=0; в —D =0; / — кривая равновесия; 2 — рабочие линии,
357
В некоторых процессах
разделения металлов рН водных
растворов изменяется на
ступени питания из-за ввода
исходного раствора. Так как
распределение металлов обычно
зависит от рН среды, для
экстракционной и отмывной
секций каскадов равновесные
кривые различны, даже если
металлы распределяются
независимо один от другого.
Пример VI1-9. Используем
результаты одного из опытов Шай-
беля 15 по разделению пара- и орто-
изомеров нитрохлорбензола
(соответственно компоненты В и С) двумя
растворителями — гептаном (А) и
86,7%-ным раствором метанола в
воде (D) — по противоточной схеме
(опыт 5). Исходная смесь,
состоящая из 37,5% пара- и 62,5% орто-
изомера, в количестве 2,42 кг/ч.
разбавляется 2,28- Ю~3 м3/ч гептана и
подается на экстракцию. Свежий гептан и свежий метанол в количествах
8,66 - 10~2 и 8,06 • 10~2 м3/ч поступают на крайние ступени каскада. В
обогащенном гептаном продукте содержится 0,89 кг/ч. распределяемых
компонентов, в том числе 81,7% пара- и 18,3% орто-изомера; продукт,
обогащенный метанолом, содержит 1,53 кг/ч распределяемых компонентов, в том
числе 12,0% пара- и 88% орто-изомера. Коэффициенты распределения
рассчитаны по концентрациям, выраженным в кг/м3, и равны тв = 1,10 и
тс =0,679. Взаимной растворимостью растворителей можно пренебречь.
Определить число теоретических ступеней, достигнутое в экстракционной
установке.
Решение. Имеем;
Рис. 182. К примеру VII-9.
Построение числа ступеней по компоненту В:
1 — рабочая лнння (наклон 0,907); 2 — линия
равновесия (наклон 1,1); 3 — рабочая линия
(наклон 0,930); х —-количество л-изомера
а
(в кг) на 1 м' метанола; у„ —количество
я-изомера (в кг) на 1 мъ гептана.
А' = 0,0866 м3/ч.
D
D'
т
Ар = 2,28 -Ю-3 м3/ч А = 0,08888 м3/ч
F
0,0806
0,0866
= 0,930
D'= D = 0,0806 м3/ч
D 0,0806
0,08888
= 0,907
Выразим концентрации х и у в кг компонента/ж3 растворителя. Так как
экстрагенты, вводимые с концов каскада, не содержат распределяемых
компонентов, хщ = xCq = УВо,= Ус0, = 0.
Для продукта, обогащенного гептаном:
„ _ 0,89-0,817
Ч _ 0,08888 = 8'17 кг В/м А
0,89-0,183
Ч = 0,08888 = 1>83 кг С/м А
366
Для продукта, обогащенного
метанолом:
BV
'с,.
0,0806
= 2,28 кг В/м3 D
= 16,7 кг C/m3D
Рабочие линии и кривые
равновесия нанесены на графики (рис. 182
и 183), на которых проведено
также построение числа теоретических
ступеней. На рис. 184 показаны
зависимости концентрации х от
номера ступени. Приводя в
соответствие число ступеней и концентрации,
получим: я'=7,5; «+1=7,1.
Следовательно, общее число ступеней:
л'+л = 7,5 + 6,1 = 13,6
Рис. 183. К примеру VII-9.
Построение числа ступеней по компоненту С:
1 — рабочая линия (наклон 0,907); 2 — линия
равновесия (наклон 0,679); 3—рабочая линия
(наклон 0,930); х — количество о-изомера
(в кг) на 1 м3 метанола; у —количество
о-нзомера (в кг) на 1 м1 гептана.
Применение флегмы. Рассмотрим рис. 185, на котором
показаны лишь концы каскада. Пусть г — доля фазы, обогащенной
компонентом А, возвращаемая на ступень 1 в виде флегмы.
Флегмовое число равно соответственно г/(1—г) кг
возвращаемых компонентов на 1 кг продукта. Для каждого компонента
x0D — rAyx
Соответственно
(VII, 57)
(VII, 58)
Рис. 184. Зависимость
концентраций компонентов от номера
ступени [х — количество компонента
(в кг) на 1 м3 метанола].
Это выражение является
уравнением линий флегмы,
изображенных пунктиром на рис. 178 и
179. Эти линии проходят через
начало координат и точку (*о.
t/i) с наклоном, равным D/rA.
Если г' — доля фазы
(обогащенной D), которая возвращается
в виде флегмы в каскад на
ступень Г при флегмовом числе
г'1(1 — г') кг возвращаемых
компонентов на 1 кг продукта, то
для каждого компонента:
y0,A' = r'xvD' (VII, 59)
00' = *!• I-^) (VII'60)
369
Экстразент
А'
Ув0.
А
Флегма
Г'
Продукт
С+в
0-г) / в
Отделение
экстраеента
У в,' Увг
Ус,- Уог
А' А
Ув,
Ус,
А
п л' ■-»
Хг.
вг'
х,
{С,
с0
Отделение
'экстраеента
Продукт В+С
'Флеема г
Во
Зкстравент
Л
Рнс. 185. Фракционная экстракция с флегмой (взаимно нерастворимые
экстрагенты).
Последнее выражение является уравнением линий флегмы
на рис. 178 и 179, проходящих через начало координат и точку
(xv, г/0,) с наклоном, равным r'D'jA'. При работе с флегмой
рабочие линии берут начало на соответствующих линиях
флегмы.
Наклоны линий флегмы могут быть больше или меньше
наклона кривых равновесия.
При соответствующем подборе флегмовых чисел на обоих
концах каскада любая из двух секций каскада может быть
отмывочной или экстракционной по отношению к каждому из
компонентов В и С.
Так, для ступеней от 1-й до я-й имеем:
1. Если лнння флегмы находится выше кривой равновесия, то
концентрация распределяемого компонента в экстрагенте А на выходе нз ступени /
будет меньше, чем на ступени питания.
2. Если линия флегмы лежит ниже кривой равновесия, то концентрация
распределяемого компонента в экстрагенте А на выходе из ступени 1 будет
больше, чем на ступени питания.
3. Если линия флегмы и кривая равновесия имеют одинаковый наклон ш,
концентрация распределяемого компонента на этих ступенях остается
постоянной.
Для ступеней от Г-й до л'-й имеем:
1. Если лнння флегмы лежит выше кривой равновесия, то
концентрация распределяемого компонента в экстрагенте D на выходе из ступени Г
будет больше, чем на ступени пнтання.
2. Если линия флегмы лежит ниже кривой равновесия, то концентрация
распределяемого компонента в экстрагенте D на выходе из ступени 1' будет
меньше, чем на ступени питания.
3. Если линия флегмы и кривая равновесия имеют одинаковый наклон т,
концентрация распределяемого компонента на этих ступенях остается
постоянной.
Таким образом, подбором флегмового числа можно
организовать процесс так, что концентрация одного из компонентов
будет оставаться постоянной в одной или в обеих секциях
каскада, либо постепенно увеличиваться от одного конца каскада
к другому, либо проходить через максимум (или минимум) на
ступени питания. Так, например, на рис. 186 линии флегмы
лежат между равновесными кривыми, поэтому в данном случае
концентрация компонента В постепенно увеличивается от
ступени Г до ступени 1, в то время как концентрация компонента
С в этом направлении уменьшается.
При флегмовом числе, равном бесконечности, линии флегмы
и рабочие линии совпадают; продолжение рабочих линий
проходит через начало координат, и производительность каскада
по исходному раствору падает до нуля (или количество экстр-
агента, требуемое на единицу количества исходного раствора,
становится равным бесконечности).
Число ступеней каскада, необходимое для дайной степени
извлечения, обычно имеет минимум при некотором соотношении
количеств экстрагентов и, как правило (но не всегда),
уменьшается при использовании флегмы. Отношение количеств
экстрагентов, при котором число ступеней каскада минимально
(если коэффициенты распределения не постоянны), обычно
изменяется в зависимости от флегмового числа, причем в общем
случае получаются очень сложные соотношения. В связи с этим
указанные зависимости лучше рассмотреть для более простого
случая — процесса экстракции при постоянных коэффициентах
распределения.
Если исходная смесь распределяемых компонентов поступает
на экстракцию в виде раствора в одном из экстрагентов, то (при
постоянных соотношениях экстрагентов) в каждой секции
каскада общий расход экстрагентов фиксировав для данной
скорости подачи исходной смеси. При фиксированных расходах
экстрагентов использование флегмы приводит к уменьшению
числа ступеней, и если регенерация экстрагентов осуществляется
с помощью выпаривания, эффект от применения флегмы
достигается без значительных затрат. Если в исходной смеси
ХВ,> Х8а ХС0 ХС,-
Рис. 186. Расположение линий флегмы между кривыми равновесия.
361
экстрагенты отсутствуют, то при фиксированном соотношении
экстрагентов в каскаде их расходы должны быть больше
необходимых для образования двухфазных потоков или
достаточны для растворения компонентов исходной смеси. В этом
случае при возрастании флегмового числа расходы
экстрагентов увеличиваются, но обычно это увеличение мало, так что
применение флегмы часто экономически оправдано.
Независимые постоянные коэффициенты распределения.
В этом случае расчеты можно выполнять аналитически 22~28.
Если в уравнениях, представленных ниже, не приведены
индексы, которыми отмечают распределяемые компоненты, то это
означает, что указанные уравнения могут быть применены к
каждому из компонентов смеси.
Обратимся к рис. 167 и 185. Для ступеней от l'-й до
(п'— 1)-й можно применить уравнение (VI, 125):
vv е'"' —е'
Х„, — Хл
Уо- , D'
(VII, 61)
■ХЛ,Г
где е =
" m " v тА'
тА'
D'
Преобразовывая это уравнение, получим:
Xl,[e'"'_i_r'(E"!'_1-l)]
(VII, 62)
» е' — 1
Для ступеней от 1-й до я-й применим уравнение (VII, 36):
Уп+\—У\ _ Уп+\ — У1 _ 1-е"
Уп+i — тх0 тА i_6"+1
Уп+\ — У\Г •—-р-
(VII, 63)
Или
тА
где е = —д-
,„,_ ».'■-.»■-;.(-.■)] (™,«>
Для ступени питания:
Уп + 1 = тхп- (VH> 65>
В уравнение (VII, 65) могут быть подставлены уравнения
(VII, 62) и (VII, 64). Полученное уравнение можно написать для
каждого из двух разделяемых компонентов и решить
относительно п и п'. Такое аналитическое решение связано с
определенными трудностями, поэтому уравнения (VII, 62) и (VII, 64)
используют для построения кривых типа приведенных на рис. 180,
задаваясь различными значениями величин я и п'. Число
ступеней при соблюдении указанного выше условия сходимости на
ступени питания (см. рис. 180) определяют графически.
362
Экстракция без флегмы (чистые экстрагенты). Подстановка
уравнений (VII, 62) и (VII, 64) в уравнение (VII, 65) приводит
к соотношению:
д, 'g ZB = Щ zc
При AF = 0 имеем:
(VII, 66)
Я(е'— 1)(1— е"+1)
■ 1
е'е" (1 — е)
Ау, _ е'(е'"' —l)e"(l
■Е)
1-р
При DF = 0 имеем:
D'xv
(E'_l)(l_En + 1)
1)0
„л+1
£П + 1(1-
■е)
Р =
\-р
Ау, _ (е'"' — 1)е"+1(1 — е)
(£' — 1)(1— £Л + 1)
Dxv
(VII, 67)
(VII, 68)
(VII, 69)
(VII, 70)
Из уравнений (VII, 66) и (VII, 67) или (VII, 69) получают
две зависимости (для двух распределяемых компонентов),
которые можно решить методом подбора относительно я и я'.
В этих выражениях р — доля компонента исходной смеси,
которая непрерывно выводится в виде продукта с правой
стороны каскада вместе с экстрагентом А. Величину Р можно
назвать «коэффициентом отвода». Соответственно обратную
величину
можно рассматривать как «коэффициент задержки»; в этом
выражении q—доля компонента исходной смеси, которая
непрерывно отводится с левой стороны каскада с экстрагентом D.
Очень часто исходная смесь до подачи на экстракцию
растворяется в одном из экстрагентов, например в DF; тогда AF=0.
В этом случае можно получить простые выражения для
предельных соотношений экстрагентов.
Рассмотрим рис. 181,6. Если линия равновесия — прямая, то
можно показать, что в случае пересечения рабочих линий в
точке пересечения равновесной линии и прямой х = xF число
ступеней равно бесконечности. Используя уравнение (VII, 56) для
этого случая, получим:
V Л /mIn
Рвтс-
Рст
С'"В
fm.
m,
(т) =!
\ ■" /max
1а(1-Л:)-п,с(1-/'в)
~~ f-1
■(тН—с"-»
/-1
(VII, 71) ■
(VII, 72)
степень разделения.
363 .'
Для реального процесса, проводимого при конечном числе
ступеней, D/A должно превосходить минимальное значение,
a D'/A должно быть меньше максимального значения.
Исходная смесь не содержит экстрагентов. Соответствующие
зависимости упрощаются в том случае, когда исходная смесь не
содержит экстрагентов (AF=DF=0). Допустим, что е = е'; тогда
из уравнения (VII, 65) получим:
р Лу,(1-г) J«"'+'-е-/(«"'-«)]«"(!-г) ml73.
\-р D^.(l-r') [E»+i_l_r(eB+1-e)](l-r') ' }
Уравнение (VII, 73) для любого случая можно решить
относительно п и п' в виде:
"'=тЬ7=тЬг (V"'74)
где
z=4^[E(P+1)+w^f] <vn'75>
Отсюда получают два выражения (для двух компонентов),
которые можно решить относительно п и п'. Так как решение
этих уравнений несколько затруднительно, более целесообразно
рассмотреть ряд частных случаев, для которых получение
характеристик каскада упрощается.
1. Процесс без флегмы (г = г' = 0). Уравнение
(VII, 73) приводится к виду:
Р=Л! 111 (VII, 76)
Из этого уравнения следует, что если для данного компонента
8=1, то для того же компонента Р=п'/(п+1). Следовательно,
для каскада с центральным вводом исходной смеси при е=1
компонент будет распределен поровну между получаемыми
продуктами. Для увеличения Р (при этом большая часть
компонента будет выходить со ступени 1, растворенной в экстрагенте
А) необходимо, чтобы величина s для этого компонента была
больше единицы. Аналогично, если коэффициент задержки 1/Р
должен быть больше (при этом большая часть компонента
будет выходить со ступени Г, растворенной в экстрагенте D),
необходимо, чтобы величина s для того же компонента была
меньше единицы.
Для данной степени разделения общее число ступеней и
относительное положение ступени питания сильно зависят от
применяемого соотношения экстрагентов. Типичный случай,
рассмотренный ниже, представлен на рис. 187, из которого видно,
что число ступеней имеет минимум.
На практике в большей части случаев, когда разделяются
два компонента В и С (даже при наличии дополнительных
364
компонентов), значение Рв
выбирают большим единицы, а
значение Рс — меньшим
единицы. Проблема в конечном
счете состоит в том, чтобы
найти наиболее экономичное
число ступеней (которое может не
соответствовать
минимальному), определенное
соотношение расходов экстрагентов и
положение ступени питания в
каскаде. Для этого особенно
часто используют кривые типа
показанных на рис. 187.
Чтобы облегчить построение этих
кривых, следует применить
соотношения 24>25, приведенные в
табл. 13.
во
60
со
I
^ 10
ьтУ
У^П
^П+Г) "^
/Г—.
S'"
ол
05
0.6
Соотношение зкстрагентод a/d
Рис. 187. К примеру VII-10.
Определение числа ступеней при
экстракции без флегмы.
В этой таблице помещены такие шесть значений Л/D, для
которых расчет величин п и п' относительно нетруден. После
подстановки этих значений в уравнение (VII, 76) получают
выражения для определения величин п и я', которые приведены
в таблице.
При объемных соотношениях экстрагентов, определяемых по
уравнениям, приведенным в табл. 13 (варианты 3 и 4), получают
одинаковое общее число ступеней. Соотношение расходов экстр-
агентов, соответствующих минимальному числу ступеней, имеет
величину, промежуточную между этими значениями.
Эмпирическое уравнение Шайбеля 27 для нахождения
минимального числа ступеней (и соответствующее соотношение
расходов экстрагентов, приведенное в таблице) дает результаты,
очень близкие к истинным, хотя это выражение и является
приближенным. Если соотношение расходов экстрагентов выходит
за пределы, соответствующие вариантам 3 и 4, то число
ступеней, необходимое для данной степени разделения,
увеличивается очень быстро при изменении объемного соотношения
экстрагентов. Следовательно, на установке с фиксированным
числом ступеней чистота продуктов сильно зависит даже от
небольшого изменения соотношения экстрагентов, если
установка работает при соотношении, находящемся вне интервала
значений этого параметра, отвечающих вариантам 3 и 4
(табл. 13). Если соотношение экстрагентов находится внутри
указанного интервала, то чистота получаемых продуктов
относительно мало чувствительна к изменению этого соотношения.
В некоторых случаях представляет интерес вариант с
центральным вводом исходной смеси (п' = п + 1), т. е. вариант 3
(см. табл. 13).
365
Таблица 13
Фракционная экстракция без флегмы, исходная смесь не содержит
/ ^.^. \ 24, 25
экстрагентов (постоянные независимые коэффициенты распределения)
Вариант
Соотношение экстрагентов, Л/О
Выражение для л
Выражение для л'
A/D-
V
Ес
= 1/гав
= 1
= Р5,с
А_
D
lg
Yl + iPB-\
2га
л+1
lg
lg
л+1
D
^4_
Р'/'
в, с
га.
У:
гавгас
Рс =
р;,(с+1)№("-1)-1]
РЯ(с+1)-1
л'=/> (л + 1)
lg
log|
1о8Рв,С
У1 + 4Яс-1
IgP
в,с
п = п'
21g
^0 + Рс)
рс(1+рв)
IgP
в. с
п' = 2(п + 1)
lg
21g
1 + Рв
l + pc
'gP^.c
Pc^K.c
Примечания. 1. Приближенные зависимости »:
2[l-0.04 1g(PBPc)lg^jj
Минимум (л + л')= j—=
'g РВ, С
[принимается только положительное значение
логарифма lg (PBPC) ]
соответствующее значение AID:
>е Рв, с
i+
У 4/
ig i/^c
r-lgm.
2. Определение положения ступени питания:
£
В
"в > гс-т+г- у -ф-л
л + 1 _-i/" lgP£
' л/ |/ lgl/Pc
Ig(^B^c)
1 + —i
/'с
Ig^B
рв<Рс-
1+
*Ы>
*Vc
я,*(^
Для такого процесса общее число ступеней
2lg
УвгХСу
УС\ХВу
Igp
в.с
При симметричном разделении (рв=9с)
твтс
п-\-п' =
в, с
(VII, 77)
евес=1 (VII, 78)
^Рв
(VII, 79)
2. Полная флегма (г=г'= 1,0). Это предельный случай,
при котором требуется наименьшее число ступеней разделения,
но производительность каскада по исходной смеси равна нулю,
и в результате экстракции продукты не отбираются. Из
уравнения (VII, 73) получим:
' VBixCy
Ig
п + п' =
Ус1Хвг
Ig
%)
IgPs.c
Igp
в, с
ft
Pr
В ап + п'
~5 Pf
\>В,С
(VII, 80)
(VII, 81)
Общее число ступеней не
агентов. Уравнение (VII, 80) я
150
У оо
%60
|*7
1
I
го
ю
£Р=!
1
r*=r=[l~~~~~d{>f\
■%У^0,5
7^
~"/Г=Г=1
1
С(
,=/
\
ОЛ 0,5 0,6
Соотношение экстраеептоЬ А/В
Рис. 188. К примеру VII-10.
Зависимость общего числа ступеней от
соотношения количеств эксграгентов
при равных флегмовых числах.
зависит от соотношения экстр-
вляется известным соотношением
Фенске [см. уравнение (VI,
188)]. При центральном,
вводе исходной смеси
= 0,5 (л+ «'),_,' = о+ °>5 (VII, 82)
3. Флегм овые числа
равны \г=г'). В табл. 14
приведены шесть случаев, для
которых можно быстро
рассчитать число ступеней и
построить кривые по типу
приведенных на рис. 188.
Выражения для п (вариант 5) и
«'(вариант 6) соответствуют очень
большому числу ступеней п
и п', необходимых для
осуществления этих вариантов
368
\
ч
«о
а
!
I
2 _■
? 3
3 8.
- S
га
о
S
1 я
■8- и
щ
О К
а EJ
h s
i «•
о -в-
ш <п
10 О
Q. К
.. о
5 я
* к
Q. К
h в
и га
а га
п <ц
я
в
|— 1
1—1
1
1
1>
о
й.
+
о
й.
+
1
1
1—1
^
+
с и
^
1
о
со
to
_ад
1 1
1—1
1
1
05
to
05
а.
+
,, °5
и° о.
JM _|_
1
1
1—1
^
+
«05
^
1
1
05
«•«ч
.- ,
ьр
05
(О
ад
+
+
05
J" +
J5P О,
ад
ад
1
О
05
со.
1
Ч
^
1+
05
h 5ь-
■ 1
1
1
о
05
СО.
V *
о
05
■ со.
ад
^
»—•
+
"5
05
й.
1
05
о.
+
*—'
_ад
_^^
1—1
+
о
СО
о
а.
1
о
о.
+
*—•
ад
i— i
о
^
1
о
а,
о
ад
1
1
(0
■^-^
еО
,—i
+
О
а.
^
1
s_*-
О
со
t
ад
1
i i
05
^
О 1
(0 _
ад -—'
~ 05
о.
1
1
05
(0
"~—^
с05
1
+
о.
^~ч
1
1
n_»-
аз
со
t
ад
аз
со
'.—-,'
tj
05
со.
1»
1
^
1
*-*
^—-.
о
о.
1—1
+
о
о.
ьр
о
05
со.
ад
а.
—• о
II
II
II
,-1 V.
II Я
вч<§
ч
S
О.
С
_ V.
II Я
^
ч
S
Q.
С
24 Р. Е. Трейбал
389
s2m
процесса. Наиболее широко применимы варианты 1—4, из
которых по крайней мере два всегда можно использовать для
расчета п и п' при определенных соотношениях количеств экстр-
агентов и различных флегмовых числах. Построение графика
зависимости числа ступеней от г при постоянном отношении
A/D позволяет получить достаточно полное представление о
характеристиках процесса.
В случае центрального ввода питания (п' — п + 1) при е=1
величина Р=1 или р = 0,5 (компонент распределяется поровну
между продуктами процесса); симметричное разделение
(pB = qc) достигается при соотношении экстрагентов,
определяемом по уравнению (VII, 78).
Если флегмовые числа не равны, следует использовать
уравнения (VII, 74) и (VII, 75). Кроме того, можно проводить
расчет от ступени к ступени с графической корректировкой расчета
по ступени питания, т. е. в соответствии со способом,
приведенным на рис. 180. В любом случае соблюдаются следующие
условия:
г = Egi хв и у постоянны, ступени от 1-ой до я -f- 1-ой,
г' = £в\ хв и у _ постоянны, ступени от Г-й до п'-ой,
г = ес; хс и ус постоянны, ступени от 1 -й до п -f- 1-ой,
г' = &с; хс и ус постоянны, ступени от Г-й до п'.
В случае одинаковых флегмовых чисел и постоянных
коэффициентов распределения число ступеней проходит через
минимум при том же соотношении A/D, что и при флегмовом числе,
равном нулю. Если коэффициенты распределения не постоянны,
то отношение экстрагентов, при котором достигается
минимальное число ступеней, зависит от флегмового числа.
Наличие подробных данных о зависимости между числом
ступеней, отношением экстрагентов и флегмой позволяет выбрать
наиболее экономически выгодные условия проведения процесса
экстракции. Эти условия могут «е соответствовать наименьшему
числу ступеней, так как стоимость процесса зависит не только
от числа ступеней, но н от расходов жидкостей и способа
регенерации экстрагентов.
Концентрации компонентов. Во все зависимости,
приведенные выше, входит соотношение количества экстрагентов, но
поскольку исходная смесь не содержит экстрагентов, в этих
зависимостях не учитываются абсолютные количества последних.
Однако абсолютные количества экстрагентов определяют
концентрации распределяемого компонента в различных точках
каскада. В частности, когда расчеты проводят при допущении,
что коэффициенты распределения постоянны и независимы,
важно выбрать такое отношение расходов экстрагентов, при
котором концентрации компонентов не превышали бы тех, для
370
которых применимы упрощенные зависимости по
распределению.
Для каждого компонента / его количество, выходящее из
процесса с продуктом, обогащенным Л, равно JFpj, где JF —
количество компонента, поступающего с исходной смесью
(питанием). Количество компонента /, выходящего со ступени 1 в
экстрагенте А, равно, следовательно, JfPj/(\—г), а его
концентрации на этом конце каскада составляют:
„ ?yJ х, = . ILL—. (VII 83)
yJ'~ (I— r)A J° (\—r)D (vn.oo,
Применяя уравнение (VII, 64) для любой ступени m правой
части каскада, получим общее количество компонента в фазе А:
j pU—tJ — rtAl—tJ-1)]
л,, — f jL J JK - ii (VII 84)
Abm- (1_r)(e«-i_ey) (VIlB4)
Расход компонента в любом потоке фазы D можно найти из
равновесного соотношения:
п I Ay \ Ау
Dx, =-£ \—^-) = —^ (VII,85)
Jm A \ in, j г}
Аналогично для другого конца каскада
х jfO-Pj) , =JfO-P;)r' (VH86)
Общее количество компонента в фазе D для ступени ш' из
уравнения (VII, 62) равно:
'»• (i-r)(e;-i)
m A
где6/=^-.
Количество компонента в потоке А' составляет:
A'yJm, - mjD'xJm, (-£) = t'jD'Xja. (VII, 88)
С помощью этих выражений можно, выбрав необходимые
'расходы экстрагентов, установить определенные концентрации
фаз в любой точке каскада. Если исходную смесь подают на
экстракцию в растворе с одним из экстрагентов, то, выбрав
соотношения экстрагентов в каждой секции каскада,
автоматически фиксируют расходы экстрагентов на единицу исходной смеси.
Расходы экстрагентов, необходимые для поддержания
концентрации распределяемого компонента на данном уровне, в
большей части случаев изменяются относительно слабо при
увеличении флегмового числа; вместе с тем соотношение
экстрагентов сильно влияет на уменьшение требуемого числа ступеней (см.
пример VII-10). Поэтому часто оптимальное (наиболее
экономичное) флегмовое число может оказаться достаточно большим.
24* 371
Пример VII-10. Пара- (В) и орто- (С) изомеры метоксифенола
разделяют экстракцией двумя экстрагентами: 60%-ным водным раствором
этанола {А) и смесью углеводородов (D), содержащей 50% бензина и 50%
бензола. При смешении 20 г компонентов со 100 мл каждого из экстраген-
тов были получены следующие коэффициенты распределения (отношения
концентраций в кг/мг): тв = 2,4 и тс = 1,6 [Ван Дийк и Шаафсма, пат. США
2245945 (1941)].
Необходимо разделить 100 кг/ч смеси, состоящей из 60% пара- и 40%
орто-изомера. Желательно получить пара-изомер чистотой 95% (без учета
содержания экстрагента) при выходе его 90%. Определить характеристики
каскада при допущении, что коэффициенты распределения постоянны и
независимы.
Решение. Расчет проводим на 100 кг исходной смеси, состоящей из
60 кг пара- и 40 кг орто-изомера. Продукт в экстрагенте А содержит
56,84 кг смеси В + С: 95% компонента В (0,90-60=54 кг) и 5% компонента
с(54-оЖ = 2-84-)-
Продукт в экстрагенте D содержит 43,16 кг смеси В + С; 13,9%
компонента В (60—54=6,0 кг) и 86,1% компонента С (40—2,84 = 37,6 кг). Далее
имеем:
44
/>с=^- = °.071
= 9
40
Рс=Т
0,071
- 0,071
= 0,0765
Р
в,с
т 2,4 е
:—£ = Т1Г = 1,50=-£
тс 1,6 ес
Исходная смесь не содержит экстрагентов (AF = DF = 0); экстрагенты
ие содержат компонентов исходного раствора.
а) Процесс без флегмы. Обратимся к табл. 13 (вариант 1). При
/4/Z)= l/mB = 0,417 число ступеней п определяется из уравнения
0,0765
_: 1,5-С+1>[1,5-9С+1>-1]
i^-O+D — 1
Так как величина 1,5-9("+1) мала по сравнению с 1, то
.„+1 1,0765
Следовательно, п'=Рв(п + \) =9 • 6,52=58,7. Аналогично были получены
данные и для остальных вариантов табл. 13:
Вариант
1
2
3
4
5
6
А
D
0,417
0,467
0,498
0,511
0,546
0,625
ев
1,00
1,121
1,196
1,225
1,310
1,500
гс
0,667
0,747
0,798
0,817
0,874
1,00
п
5,5
8,7
10,8
11,5
15,9
73,2
п'
58,7
17,4
11,8
11,0
8,5
5,7
п + п'
64,2
26,1
. 22,6
22,5
24,4
78,9
372
Данные этой таблицы нанесены на график (см. рис. 187). В данном
случае вследствие относительно небольшого значения р интервал величин
отношений количеств экстрагентов, при которых обеспечивается умеренное число
ступеней, невелик. Из графика получаем минимальное число ступеней 22,5
при отношении количеств растворителей Л/£> = 0,504.
Применяя эмпирические зависимости Шайбеля из табл. 13, минимальное
число ступеней получим равным 22,4, а отношение количеств экстрагентов
A/D = 0,506. Совпадение с точным результатом очень хорошее.
б) Процесс с полной флегмой. По уравнению (VII, 80)
находим:
1 9
. , g 0,0765
пЛ-п ' = —;—z—— = 11,8
7 lg 1,5
в) Процесс с равными флегмовыми числами. Так как г
и г' не могут быть больше единицы, воспользуемся только вариантами 2, 3,
5 и 6 табл. 14. Например, примем г=г'=0,75. Тогда для варианта 2 имеем:
тс/1/£> = 0,75 или Л/£> = 0,468; для варианта 3 находим: £>/Лтв = 0,75 или
A/D — 0,555. Прн других флегмовых числах можно использовать также
другие соотношения экстрагентов. Для вариантов 5 и 6 отношение A/D равно
соответственно 0,417 и 0,625. Применяя зависимости, приведенные в табл. 14,
получим следующие результаты:
ЧИСЛО
1
3
9
Т = Т1
0,50
0,75
0,90
Вариант
5
*
**
*
5
2
**
3
6
5
3
##
2
6
А
D
0,417
0,480
0,500
0,540
0,417
0,468
0,500
0,555
0,625
0,417
0,463
0,500
0,563
0,625
п
4,80
8,3
10,0
14,3
3,8
6,3
8,6
16,2
86,8
3,1
3,9
5,9
18,0
137,3
п'
60,2
13,9
11,3
8,1
67,2
15,1
9,7
6,1
3,96
108,9
15,8
8,3
3,7
2,26
п + п'
65,0
22,2
21,3
22,4
71,0
21,4
18,3
22,3
90,8
112,0
19,7
14,2
21,7
139,6
* Рассчитано из уравнений (VII, 74) и (VII, 75).
** Величины лип' получены нз графиков зависимости этих величин от отношения —
На графике (см. рис. 188) нанесены лишь общие числа ступеней п + п'.
Отметим несколько особенностей этого расчета:
1. Для всех найденных комбинаций чисел ступеней, соотношений
расходов экстрагентов и флегмовых чисел получена постоянная степень разделения.
Другие значения степени разделения можно получить в случае применения
неодинаковых флегмовых чисел.
2. Пределы отношений AID для умеренных чисел ступеней разделения
в данном примере невелики вследствие низкого значения р.
373
3. Так как величина п+п' значительно изменяется вие этого узкого
интервала значений A/D, для этого процесса не было необходимости исследовать
влияние флегмового числа при значениях AID, существенно отличных от 0,5.
Такой анализ был проведен для того, чтобы показать более подробно
характеристики каскада.
4. По мере приближения значений ев и ес к 1,0 число ступеней,
необходимых для проведения процесса с флегмой, становится больше числа
ступеней в процессе без флегмы. Это объясняется тем, что при данных
соотношениях количеств экстрагеитов по мере приближения г к 1,0 числа
ступеней лил' стремятся к бесконечности.
г) Расходы экстрагеитов. Расходы экстрагеитов определены
только при Л/£)=0,5 и для т—г'. При г=г'=0 имеем: Л/£)=0,5; ев = 1,2; ес = 0,8.
Уравнения (VII, 84) и (VII, 85), записанные для компонента В, имеют
вид:
bfPb(i-4) Dx __л»вт
Аналогичные уравнения применимы для компонента С. Для 5-й ступени:
60 0,90 (1 — 1,25)
Ау
вь
1,2«
■1.2»
; 194 кг/ч В в А
. 40-0,071(1—0,85)
Аусб = 0,8 W 0,8' = 23>3 кг/Ч С в А
Общее количество компонентов в экстрагеите А составляет: 194,0+23,3 =
= 217,3 кг/ч. Далее иахоДим:
Dx
в5
194
-^ = 161,5 кг/ч В в D
Dxr
23,3
0,8
= 29,2 кг/ч С в D
Общее количество компонентов в экстрагеите Z)= 161,5+29,2=190,7 кг/ч.
Аиалогичио были получены данные для построения графиков (рис. 189 и
190), на которых приведены только общие количества компонентов в
каждом экстрагеите. Наличие Двух минимумов на кривых, соответствующих
600
?
Лгоо
^
г=гЩ9
^^
^*v
Ступень N
/питания
1
о
п'-12 п'-В п'-Ь
п-3 п-7 п-10
п+1
'Номер ступени
Рис. 189. К примеру VII-10. Распределение общего количества
компонентов в фазе А по ступеням каскада.
374
,и
/
^
?гу
,
г=г=
^^^-^75
0^
%$0,5
03
п-3 п-7 п-11
600
$200
О
п-11 п'~в п'-Ь п+1г
Номер ступени
Рис. 190. К примеру VII-10. Распределение общего количества
компонентов в фазе D по ступеням каскада.
г=г'= 0,9, объясняется тем, что при этом флегмовом числе количество
компонента В в экстрагеите увеличивается от ступени 1' до ступени 1, в то
время как количество компонента С уменьшается. При меньших флегмовых
числах количество каждого компонента постоянно возрастает от концов
каскада к ступени питания.
Чтобы поддерживать концентрации иа том же уровне, что и при
определении коэффициента распределения (100 г иа 1 л), их величины не
должны превышать 100 кг компонента/^3 экстрагеита. При флегмовом
числе, равном нулю, максимальное количество компонентов в экстрагеите А на
ступени питания составляет 437 кг/ч (см. рис. 189). Это соответствует
расходу экстрагеита А
Согласно рис. 190, максимальное количество компонентов в экстрагеите
D составляет 396 кг/ч, что соответствует расходу D, равному 396/100=
=3,96 м3/ч. Учитывая, что A/D должно быть равно 0,5, находим расход
экстрагеита D: 4,37-2 = 8,74 м3/ч. Таким же образом для других флегмовых
чисел получим:
Т = Т'
0
0,5
0,75
0,9
1,0
Максимальное количество
компонентов в экстрагентах
кг/ч
А
437
442
445
568
со
D
396
420
430
563
со
Расход А
мЧч
4,37
4,42
4,45
5,68
со
Расход D
8,74
8,84
8,90
11,36
Из полученных результатов видно, что по крайней мере для флегмовых
Чисел, величина которых больше трех (/■=/■'=0,75), расходы экстрагеитов не
375
A
D,
±
Каскад I
■D,
A,-2
Наскад Л
4
/ Сильно отличаются от их расходов при флег-
мовом числе, равном нулю; однако число
необходимых ступеней снижается с 22,5 (без
флегмы) до 18,3 при г=г'=0,75. Это
показывает, что оптимальное флегмовое число может
быть достаточно велико. Возможно также,
ввиду различия в стоимости регенерации
экстрагентов и асимметрии разделения, что
оптимальные условия будут соответствовать
различным флегмовым числам для каждой из
двух секций каскада.
Пример VH-11. Разработать схему для
разделения исходной смеси равных количеств
орто-, мета- и пара-изомеров нитроанилина
экстракцией бензолом (А) и водой (D).
Коэффициенты распределения равны: т„ = 64,0;
тт = 25,0 и тр = 9,3. Необходимо получить все
изомеры с выходом 90%, причем орто- и пара-
изомеры— чистотой 90%.
Решение. Задачу нельзя решить
однозначно, и принятую технологическую схему
нужно рассматривать лишь как отправную
точку для дальнейших расчетов. Схемой
предусматриваются два каскада фракционной экстракции, так как все три
компонента должны быть получены с высокими выходами. Расчеты будут
проведены для процесса без применения флегмы на 1 кг/ч каждого изомера.
В каскаде I (рис. 191) разделяют мета- и пара-изомеры, а продукт Аи
содержащий в основном орго- и мета-изомеры, направляют в каскад II, где
происходит разделение орто- и мета-изомеров. (В каскаде I можно также
провести разделение орто- и мета-изомеров, а поток Di направить в
каскад II.)
Каскад I. Целесообразно принять соотношение экстрагентов по варианту
4 табл. 13 (см. стр. 366):
Рис. 191. К примеру VII-11.
Разделение трехкомпонент-
ной смеси:
/ — исходная смесь: 1 кг о-нзо-
мера + 1 кг лс-нзомера-Н кг
л-нзомера; 2—1,9 кг о-изомера;
1,9 кг лс-нзомера и 0,1 кг л-изо-
мера; 3 — 0,9 кг о-нзомера, 0,1кг
л£-нзомера н 0,1 кг л-нзомера;
4 —о-и л-изомеры н 0,9 кг
.«-изомера; 5-0,1 кг о-изомера, 0,1 кг
лс-изомера и 0,9 кг л-нзомера.
Dl V тттр У
1
1
25,0 • 9,3 15,25
Для пара-изомера находим:
Рр
Рп=-
од
= 0,1111
Р- \-р„ 1-0,1
Селективность при получении мета- и пара-изомеров равна
ри,р = 25,0/9.3 = 2,69
Число ступеней определяем по уравнениям, приведенным в варианте 4
табл. 13:
o,.r/>m(i+(Um)l
г lgL 0,1111 (\+рт) J
(l+Pm)J !
lg 2,69
'"(тЙЙтг)
lg 2,69
Для орто-изомера имеем:
Л|Шо д_§4!0__420
D,
15,25
376
По уравнению (VII, 76) находим:
(4,20"' —1).4,20л+1
Далее имеем:
4,20n+1 —1
р л-р =1,9 = ^-1-^
'о i гт ' 14-/
1+Р0 "Г \+Р„
Эти четыре уравнения можно легко решить подбором; при этом
получим следующие результаты: Рт=9,1; рт=0,9009; Ро = 600,4; ро=0,99ЯЗ;
л=3,443; л'=4,458. Найденное число ступеней можно скорректировать до
целого значения, например до 4 и 5, с внесением соответствующей поправки
на величину соотношения расходов экстрагента. Продукты, выходящие из
каскада I, имеют следующие составы:
D,
Орто-изомер
Мета-изомер .
Пара-изомер .
0,9983 кг
0,9009 кг
0,1000 кг
Орто-изомер ,
Мета-изомер .
Пара-изомер .
0,0017 кг (0,1796)
0,0991 кг (9,83%)
0,9000 кг (90,0%)
Всего . . . 1,0008 кг
Каскад II. Соотношения количеств экстрагентов в каскаде различны в
обеих секциях каскада; эти соотношения лучше определить методом
подбора. Примем v?j = 1,0 и Аи = 1,0, тогда Лц=2,0. Так как разделение
близко к симметричному, соотношение экстрагентов, вероятно, должно быть
порядка A/D = (1/64 • 25)0'5 = 1/40. Приняв Dn = 60, получим Л,',/Оп = 1/60
и A../D.. = 2/60 = 1/30; это значение близко к 1/40.
Следовательно
= 1,067
= =г = 0,416
Ро--
_64
60
_25
60
9,3
60
0,9
= 0,155
0,9983
0,9009-
-0,9
0,9009
б£
30
= 2,13
§ = 0,833
9,3
30
= 0,310
Ро
-Ро
Рт =
1-
= 9,16
= 0,001
Применим уравнения (VII, 66), (VII, 69) и (VII, 70):
9,16(1,067— l)(l —2,13n+1) ,
2,13n+1 (1 — 2,13)
lg 1,067
0,001 (0,416 — 1) (1 — 0,833n+1)
0,833"+1 (1 — 0,833)
lg 0,833
377
Отсюда л=28 и_п'=6,7. Кроме того, находим:
Р - (°Л556-7 — 1) • 0,31029 (1 — 0,310) _
р (0,155 — 1) (1 — 0.31029)
Продукты, выходящие из каскада II, содержат:
Лц Du
Орто-изомер . . 0,900 кг (99,9%) Орто-изомер . . 0,0983 кг (8,95%)
Мета-изомер . . 0,001 кг (0,1%) Мета-изомер . . 0,900 кг (81,95%)
Пара-изомер . . 0 (0%) Пара-изомер . . 0,100 кг (9,10%)
Всего. . . 0,901кг (100%) Всего. . . 1,0983 кг (100%)
Необходимая чистота орто-изомера выше той, которая достигается при
данном отношении экстрагентов. Путем лучшего подбора Ац и А1 можно
снизить число необходимых ступеней и получить заданную чистоту орто-
изомера.
Диссоциативная экстракция. Этот процесс представляет
собой разновидность фракционной экстракции; он применяется
для разделения слабых кислот или оснований29-30. В главе II
было показано, что селективность системы двух растворителей
(водного и органического) для разделения двух слабых
органических кислот возрастает при введении в воду щелочи.
Рассмотрим рис. 192. Органические кислоты НВ (более
сильная) и НС (более слабая) вводят в многоступенчатый
каскад в виде собственно кислот или их натриевых солей. Сверху
каскада подают водный раствор NaOH, который
предпочтительно нейтрализует более сильную кислоту, образуя соль NaB;
вместе с органическим растворителем сверху колонны из
каскада выводится слабая кислота НС. В нижней секции каскада
введением более сильной кислоты переводят слабую кислоту в
органический растворитель (глава II); соль более сильной
кислоты удаляется с водой. В известной степени введение в
процесс щелочи и кислоты аналогично вводу флегмы на
соответствующих концах каскада. Степень фракционирования можно
регулировать изменением количества вводимых щелочи и
кислоты. В каскаде необходимо поддерживать градиент рН, что
достаточно трудно; поэтому процесс проводят в
многоступенчатых периодических экстракторах (см. главу IX).
Следует отметить, что в данном случае возможно
применение периодической дифференциальной экстракции31, хотя эта
схема не имеет преимуществ по достигаемой степени
разделения перед 'каскадом с большим числом противоточных ступеней.
При экстракции данным методом кислоты могут быть
растворены в органическом растворителе. В процесс постепенно
вводят водный раствор NaOH, который предпочтительно
нейтрализует часть более сильной кислоты, а водный экстракт, содержа-
376
-—17
^ Ш
ш
щий соль, удаляют. Непрерывно вводя в процесс ш\ [Ш
и отводя малые количества этих жидкостей, f-A
окончательно разделяют смесь. Разделение
будет тем лучше, чем меньше будут количества
вводимых и отводимых продуктов. И—*■ „
Расчеты от ступени к ступени. В качестве
исходных величин для расчета таким методом
должны быть известны составы конечных
продуктов. Распределение компонентов между про- /■ ТТ
дуктами, получаемыми на ступенях каскада,
неизвестно; это распределение нужно найти и Рис. 192. Диссо-
впоследствии проверить. Обычно степень разде- циативная экст-
ления устанавливают по двум компонентам Ракиия-
, J \ тт о I — органический
(«ключевые» компоненты). Число ступеней ка- растворитель (экст-
скада неизвестно, и разделение прочих компо- ^нв'! нс~в°(£ш
нентов будет зависеть от числа ступеней, необ- NaBfNiC);
J J III — органический
ХОДИМЫХ ДЛЯ разделения «КЛЮЧеВЫХ» КОМПОНеН- растворитель + НС;
тов. Затем определяют составы обоих продуктов ££-^то^кгоп-,
(рафината и экстракта) по всем компонентам; к/-мног0ступен-'
о -. чатый каскад;
расчет ступеней проводят с обоих концов каска- v/z-водный рас-
да к его центру, как описано в примере VI-5 HZ+i?So^B.
(см. стр. 263).
Для определения числа ступеней проверяют совпадение
расчета по «ключевым» компонентам, пользуясь способом,
приведенным на рис. 180. Возможно, что при этом предполагаемое
распределение «неключевых» компонентов в продуктах не
будет соблюдаться и не удовлетворит проверке для ступени
питания. Тогда распределение этих компонентов необходимо
изменять до тех пор, пока не будут найдены числа ступеней,
удовлетворяющие сходимости составов на ступени питания.
Процедура расчета аналогична расчету разделения
многокомпонентных систем в процессе ректификации и может быть
выполнена по методу, описанному Тиле и Геддесом32.
Подробный пример такого расчета приводит Шайбель33. Для расчета
можно использовать высокоскоростные вычислительные
машины 1б.
УПРОЩЕННЫЙ РАСЧЕТ
МНОГОКОМПОНЕНТНЫХ СИСТЕМ
Чтобы упростить расчет некоторых очень сложных
процессов и облегчить получение соответствующих данных, часто
сводят многокомпонентную систему к «эквивалентной» тройной
системе. Например, при разделении ароматических и парафиновых
углеводородов, когда общее число компонентов может быть
очень велико, объединяют различные углеводороды в две группы
(ароматические и парафиновые), выражая содержание арома-
379
Рис. 193. Упрощенный расчет четырех-
компонентной системы.
тических соединении в смеси,
с помощью эмпирически
определяемой «вязкостно-весовой
константы» (см. главу II);
затем этот показатель
используют для расчета данной си-
хтемы в треугольных
координатах.
Равновесные данные
обычно получают экстракцией в
делительных воронках
(встряхиванием последних),
применяя различные соотношения
данной смеси и экстрагента.
Если подобные диаграммы
используют для расчетов про-
тивоточной ступенчатой
экстракции, это почти всегда приводит к ошибочным заключениям
вследствие того, что разновесные данные, полученные для одного
соотношения индивидуальных ароматических и парафиновых
углеводородов, не применимы для других соотношений тех же
веществ.
Чтобы проиллюстрировать сущность подобных затруднений,
рассмотрим четырехкомшонентную систему, показанную на
рис. 159. Представим себе, что компонент D — экстрагент, А—
парафиновый углеводород, а В и С—ароматические
углеводороды. Если спроектировать рис. 159 на плоскость ACD, то
кривые растворимости спроектируются на эту плоскость в виде
кривых UpV и Up"V, приведенных на рис. 193. Концы всех
четырехкомпонентных хорд равновесия при их проектировании
на плоскость ACD ложатся на площадь, ограниченную
проекциями кривых растворимости.
Предположим, что исходная смесь F содержит одинаковое
количество компонентов А, В и С. Ее проекция f показана на
рис. 193. Если эта исходная смесь смешивается с экстр агентом D
в одной ступени в таких соотношениях, что получаемая смесь
имеет состав, отвечающий точке ти то на проекции
образующиеся продукты экстракции (находящиеся в равновесии друг
с другом) будут соответствовать точкам е^ и Гь При других
соотношениях F и D будут получены продукты е2 и г2 и т. д.;
в результате получают кривую f/rir2r3e3^2^i У-
Как было показано выше, для такой смеси отношение
количеств компонентов В и С в различных потоках на
многоступенчатой экстракционной установке не равно их соотношению при
одноступенчатой экстракции. Из этого следует, что кривая
Urt ... eiV, строго говоря, соответствует только тем условиям,
для которых она построена (на практике кривые, изображен-
380
ные на рис. 193, искажаются, так как компоненты В и С
наносят на диаграмму как один компонент, находящийся в
вершине С, а точка / располагается на стороне АС диаграммы).
Эти вопросы изучал Альдерс34, опытные данные приводятся
в работе Скоуджена и Роджерса35; в литературе имеется
также много других указаний по расчетам для многокомпонентных
систем. Сложность расчета оказывается тем большей, чем
больше изменяется взаимная растворимость компонентов А и D
при добавлении компонентов исходной смеси и чем больше
различаются компоненты В и С по своей природе.
Если взаимная растворимость экстрагентов незначительна,
то распределение различных компонентов, вероятно, независимо.
На основании этого для углеводородных смесей иногда
оказываются достаточно пригодными графические расчеты36 и
расчеты от ступени к ступени 37.
Эти способы применимы также в других случаях, например,
при распределении редкоземельных металлов, которое для
выбора наилучших условий фракционной экстракции можно
рассматривать как распределение группы металлов между двумя
жидкостями38. Однако такие методы не являются общими.
Когда данные о равновесии получить сложно и его
подробное изучение слишком дорого, рекомендуется применять
лабораторный способ определения равновесий (см. главу IX),
который имитирует непрерывный многоступенчатый процесс и
может быть использован для определения характеристик
каскада.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
А, В, С, D — количества компонентов смеси, кг (для
периодического процесса), кг/ч (для непрерывного
процесса).
с — число компонентов,
F — количество исходной смеси, кг (для
периодического процесса), кг/ч (для непрерывного
процесса).
f — степень очистки.
К—отношение XF/XR при равновесии.
m — коэффициент равновесного распределения.
Nc— число степеней свободы.
N-t — число независимых переменных.
Nv — число переменных.
п, п' — число ступеней.
Р = р/(1-р).
р — доля компонента в фазе А или в продукте.
q — доля компонента в фазе D или в продукте.
г, г'—доля компонента, возвращаемого в каскад в
виде флегмы.
381
X — весовая доля.
х — количество компонента, кг/кг экстрагента D.
у—количество компонента, кг/кг экстрагента А.
e = Am/D — фактор экстракции.
Ру, к ~ mj/~mK — селективность растворителей (экстрагентов) для
разделения компонентов J и К-
f— число фаз.
Индексы
А, В, С, D и т. д. — компоненты А, В, С, D и т. д.
F — исходная смесь.
f — конечное значение.
п — на выходе с n-й ступени.
S— экстрагент.
t — общее значение.
ЛИТЕРАТУРА
1. М. К w a u k, A. I. Ch. E. Journ., 2, 240 (1956).
2. А. V. Brancker, Ind. Chem., 27, 243 (1951); Petroleum (London), 14,
123 (1951).
3. A. V. Brancker, T. G. Hunter, A. W. Nash, Ind. Eng. Chem., 33,
880 (1941).
4. A. J. B. Cruickshank, N. Haertsch, T. G. Hunter, Ind. Eng.
Chem., 42, 2154 (1950).
5. J. D. Gunn, Chem. Eng. Sci. (1964).
6. T.G. Hunter, Ind. Eng. Chem., 34, 963 (1942).
7. P. D. A. Mole, Chem. Eng. Sci., 8, 236 (1958).
8. J. L. Otero de la Gandara, Anales real soc. espafi. fis. у quim.
(Madrid), 48B, 179 (1952).
9. J. E. P о w e r s, Chem. Eng. Progr., 50, 291 (1954).
10. A. R i u s, M. A. Crespi, Anales real soc. espafi, fis. у quim. (Madrid),
48B, 131 (1952).
11. B. D. Smith, W. К- В r i n к 1 e y, A. I. Ch. E. Journ., 6, 446, 451 (1960)
12. J. С Smith, Ind. Eng. Chem., 36, 68 (1944).
13. J. H Wiegand, Ind. Eng. Chem., Anal. Ed., 15, 380 (1943).
14. M. T. Bush, P. M. Den sen, Anal. Chem., 20, 121 (1948).
15. E. G. Sche ibel, Chem. Eng. Progr., 44, 681, 777 (1948).
16. D. R. О I a n d e r, Ind. Eng. Chem., 53, 1 (1961).
17. J. H. Duff in, U. S. Atomic Energy Comm. UCRL-8787 (1959).
18. J. W. Codding, W. D. Haas, F. К- Н e u m a n n, Ind. Eng. Chem., 50,
145 (1958).
19. E. L. Compere, A. R у land, Ind. Eng. Chem., 43, 239 (1951).
20. A. J. F. Martin, R. L. M. S у п g e, Biochem. J., 35, 91 (1941).
21. M. E. Whatley, U. S. Atomic Energy Comm. ORNL-2477 (1958).
22. L. Alders, Liquid-Liquid Extraction, 2nd ed., Elsevier Publishing
Company, Amsterdam, 1959.
23 С. Е. В artel s, G. Kl eiman, Chem. Eng. Progr., 45, 589 (1949).
24 A. Klinkenberg, Chem. Eng. Sci., 1, 86 (1951); Ind. Eng. Chem., 45,
653 (1953).
25. A. Klinkenberg, H. A. Lauwerier, G. H. Reman, Chem. Eng.
Sci., 1, 93 (1951).
26. R. Rometsch, Helv. Chim. Acta, 33, 184 (1950).
382
27. E. G. Sche ibel, Ind. Eng. Chem., 46, 16 (1954).
28. S. Stene, Arkiv Kemi. Mineral. Geol., 18A, № 18, 86 (1944).
29. D. W. M i n t y, R. McNeil, M. R о s s," E. A. S w i n t о n, D E Weiss,
Austr. J. Appl. Sci. 4, 530 (1953).
30. G. H. Twigg, Nature, 163, 1006 (1949).
31. A. V. Brancker, J. Soc. Chem, Ind., 66, 453 (1947).
32. E. W. Thiele, R. L. G e d d e s, Ind. Eng. Chem., 25, 289 (1933).
33. E. G. S с h e i b e I, Petroleum Refiner, 38, № 9, 227 (1959).
34. L. A 1 d e r s, J. Inst. Petrol., 42, 228 (1956).
35. V. G. Skogan, M. С Rogers, Oil Gas J., 45, № 13, 70 (1947).
36. F. H. Poettmann, M. R. Dean, Chem. Eng. Progr., 45, 636 (1949)
37. H. F. Ho pp, R. B. Smith, A. I. Ch. E. Journ., 2, 34 (1956).
38. J. Bochinski, M. S m u t z, F. H. S p e d d i n g, Ind. Eng. Chem., 50,
157 (1958).
39. G. F. Asselin, E. W. Comings, Ind. Eng. Chem., 42, 1198 (1950)
МЕТОДЫ РАСЧЕТА ВЫСОТЫ
НЕПРЕРЫВНЫХ ПРОТИВОТОЧНЫХ
ЭКСТРАКТОРОВ
U сходный раствор и экстрагент можно
привести во взаимный контакт не
только в смесительно-отстойных экстракторах,
состоящих из отдельных ступеней с
промежуточным отстаиванием (рассмотренных в
двух предыдущих главах), но и в
вертикальных противоточпых колоннах, в
которых движение жидкостей происходит под
действием разности плотностей фаз. Так,
например, исходный раствор, имеющий
большую, чем экстрагент, плотность, может
заполнять весь объем аппарата и в
качестве сплошной фазы двигаться по колонне
сверху вниз; в то же время экстрагент,
диспергируемый с помощью распылителей в
нижней части колонны, будет подниматься
вверх в виде капель, распределенных в
объеме сплошной фазы. В общем случае
диспергировать можно любую фазу.
Использование различных насадок,
аналогичных применяемым в процессах
абсорбции и ректификации, перегородок и
перфорированных тарелок, устанавливаемых
внутри колонны, позволяет увеличивать
турбулентность потоков и воздействовать
на другие их характеристики. Процесс
взаимодействия между фазами
осуществляется непрерывно по всей высоте
колонны. Сечение аппаратов такого типа
зависит от расходов жидкостей, проходящих
через колонну, а высота — от необходимой
степени экстракции.
Высота, эквивалентная теоретической
ступени. Наиболее простым методом
расчета высоты колонных экстракторов
является метод, аналогичный обычно
применяемому для абсорбционных и
ректификационных колонн. По этому методу определяют
число идеальных, или теоретических, сту-
384
пеней разделения га, необходимое для данной степени
извлечения, способами, описанными ранее. Высоту аппарата Я
рассчитывают как произведение га на величину ВЭТС — высоту,
эквивалентную теоретической ступени:
Я=л(ВЭТС) (VIII, 1)
Значение ВЭТС для данной системы определяют из опыта.
Как показала опытная проверка для процессов абсорбции и
ректификации, этот метод недостаточно обоснован, поскольку
он базируется на представлении о ступенчатом изменении
концентраций, в то время как в действительности концентрация
распределяемого компонента по высоте колонного аппарата
изменяется непрерывно. В результате установлено, что ВЭТС
значительно изменяется в зависимости от ряда факторов,
определяющих протекание процесса: от свойств системы, скорости
потоков и концентраций, а также от типа применяемого
аппарата. Поэтому почти для каждого конкретного случая
необходимо располагать данными о зависимости ВЭТС от указанных
факторов, с тем чтобы правильно определить высоту колонны.
Способы расчета высоты аппаратов, описанные ниже, в
значительной степени устраняют (по крайней мере теоретически)
этот недостаток ценой некоторого усложнения методики
расчета.
ЕДИНИЦЫ ПЕРЕНОСА МАССЫ
Частные значения единиц переноса. Рассмотрим
экстракционную колонну (рис. 194), в которой фазы рафината и экстракта
движутся противотоком. Фаза рафината, или исходный раствор,
имеет на входе в колонну фиктивную массовую скорость,
рассчитанную на все сечение колонны, Ri кмоль ■ мг2 • ч-1 и
концентрацию распределяемого вещества xR мол. доли; на выходе из
колонны — соответственно Ri w xR. Скорость Rz меньше Ri на
величину, зависящую от количества экстрагированного
вещества. Аналогично экстрагент имеет на входе в колонну
массовую фиктивную скорость Ег кмоль • м~2 • ч'1 и концентрацию хЕ\
на выходе — скорость £\ и концентрацию хЕ,
Концентрация в каждой фазе может быть выражена также
в весовых долях X или в кмоль/м3 раствора с. Примем, что
поверхность контакта фаз на 1 м2 сечения колонны составляет
А м2, а на 1 мъ объема колонны — а м2/м3. В любом сечении
колонны, для которого скорости рафината и экстракта равны
R и Е, бесконечно малое изменение концентрации этих потоков
будет происходить на высоте dH за счет диффузии
распределяемого вещества из фазы R в фазу Е вследствие наличия
градиента концентраций (см. главу V). Если общая скорость мас-
Сопередачи N моль • м~2 • ч~\ то скорость массопередачи на бес-
25 Р. Е. Трейбал
386
Экстракт конечно малой высоте dH можно выразить '■2
х,
±
\k
_jM
п
с помощью уравнения (V,48):
dN = d (RxR) = kR dAcRM (xR - xR.)
(VIII, 2)
Pcupufiam
Рис. 194. Схема
непрерывной
противоточной
экстракции.
где cRM — среднее из значении cR и cRi.
Это уравнение применимо для разбавленных
растворов или в тех случаях, когда исходный
растворитель и экстрагент совершенно взаимно
нерастворимы, так как исходное уравнение
(V, 48) было получено для диффузии только
распределяемого вещества, а не растворителей.
В других случаях коэффициенты массоотдачи
должны быть скорректированы в соответствии
с уравнением (V, 49).
Массовая скорость R фазы рафината
изменяется по высоте колонны, но количество
исходного растворителя ^(1—xR) остается
постоянным. Следовательно
RdxD
т (VIII, 3)
rf (***) = * 0-*я)*(тгУ =
Коэффициент массоотдачи kR [уравнение (V,50)] зависит от
величины (1 — xR)iM, которая изменяется по высоте колонны.
Произведение же kR(\—xR)iM практически является
величиной постоянной. Кроме того, dA=adH. С учетом этого
уравнение (VIII, 2) можно привести к виду:
0~*fhR)dXR=kKa(l-x«)iMcRM(xR-*m)dH <™М)
(l-xR)tMdxR
(l-XR)(XR-XRd
k„a(\ — хЛ. „с п „ dH
^(1
)IM RM
R
(VIII, 5)
Величины (1—xR)iM и (1— xR) не отличаются значительно
от единицы, поэтому левая часть уравнения (VIII, 5) выражает,
по существу, изменение концентрации dxR, приходящееся на
единицу разности концентраций (xR — xRi), т. е. -на единицу
движущей силы, и представляет собой меру трудности
экстрагирования. Эта величина называется числом единиц переноса и
обозначается Nt. Высоту экстракционной колонны находят как
произведение числа единиц переноса Nt на высоту единицы
переноса Ни определяемую опытным путем. Таким образом
N
tR
Г _Л=
J (1-х
0-XR)lMdXtt
ЛД2
il-XR)(XR~XRl)
? dH H
J ~Н^ ~ HfR
(VIII, 6)
386
Из уравнений (VIII, 5) и (VIII, 6) получают соотношение
между высотой единицы переноса и коэффициентом
массоотдачи:
Н,р = -т—Г.——. (VIII, 7)
и* kRa(l-xR).McRcp
Если движущую силу процесса выразить через разность
концентраций в фазе экстракта, то аналогично мож-но определить
число единиц переноса по фазе экстракта:
£1 Н
г 0—хЛ.ыйхв ,• dH H
tE= f п-хих -х\= ТГ- = ТГ~ (VIII'8)
J (}—xE){xEl — xE) J HfE HtE
N
in
ЛЕ2
""°vc-wElt. ,vl"'9>
Общее число единиц переноса. Как было показано в главе V,
трудности, возникающие при определении равновесных
концентраций на поверхности раздела фаз хЕ% и xRi, привели
к необходимости включить в уравнения общие коэффициенты
массопередачи Ке и Kr, которые выражают скорость
диффузии в функции от общих градиентов концентраций (xR — x*R^
или (хЕ — хЕ) [уравнения (V, 56) и (V, 57)].
Применение общих коэффициентов массопередачи возможно
в тех случаях, когда коэффициент распределения
Хр: Хр Хр
/я = — = — = -#• (VIII, 10)
является постоянным в пределах изменения концентраций, при
которых протекает процесс в аппарате. Это требование служит
серьезным ограничением. Однако на практике уравнения,
полученные на основе общих градиентов концентраций, используют
и в тех случаях, когда коэффициент распределения не является
величиной постоянной. Применяя уравнения (V,56) и (V, 57),
получают:
хR\ xR\
25* 387
N
ЮЕ
ЛЕ\
-J;
£2
0—хв)омах.
(l—xE)(x*E-xE)
л£1
^-= f ■
й?лгг
Я
XE2 {1-Хе)Ы
(X~xe) Я
(i-4)
ЮЕ
НЮЕ =
KEa(1-XE)oMCEl
(VIII, 13)
(VIII, 14)
ср.
Полученные уравнения используют для расчета высоты
аппаратов. Когда основное диффузионное сопротивление
сосредоточено в фазе R (т велико), применяют уравнение (VIII, 11);
когда основное сопротивление сосредоточено в фазе Е (т
мало) —уравнение (VIII, 13).
Величины Nwr или NtoE обычно определяют методом
графического интегрирования соответствующих выражений, для чего
необходимо иметь данные, получаемые из диаграмм типа
описанных в главе VI. На диаграмме процесса непрерывной про-
тивоточной экстракции (рис. 195) нанесены кривая
равновесия и рабочая линия. Методы построения рабочей линии были
рассмотрены ранее. Вертикальное расстояние от любой точки Р
на рабочей линии до кривой равновесия представляет собой
общую движущую силу процесса, выраженную через разность
(хЕ— х£), расстояние по горизонтали — общую движущую
силу, выраженную через разность (xR— x*Ry Для нескольких
точек рабочей линии рассчитываются значения величин
( xr)om
Кривая
раонодесия'
(I — x£){xR — xRj
или
1
£rz %r xrixr
Концентрация Врафинатв
Рис. 195. Рабочая диаграмма
процесса непрерывной противоточной
экстракции.
(i-^)in[(i-4)/(i-^)]
Число точек на рабочей
линии выбирают таким, чтобы
можно было построить плавную
кривую зависимости
соответствующих величин от xR. Площадь под
кривой между конечными
значениями xR1 и хш выражает в
определенном масштабе значение
величины NtoR- Аналогично
получают общее число единиц
переноса по фазе экстракта Ыюе-
Таким же методом можно
получить численные решения
уравнений (VIII, 6) и (VIII, 8), но для
этого необходимо знать положение на кривой равновесия
точки Q, соответствующей равновесным концентрациям на
поверхности раздела фаз.
Упрощенный метод графического интегрирования. Если
значения (1—х*Л и (1—Хд) отличаются друг от друга не более
чем в 2 раза, то использование средней арифметической из
значений (1—хп)0м вместо средней логарифмической
величины приводит3 к ошибке, не превышающей 1,5%. Таким
образом, если
(I— Х*р) + (1 — Хп)
О - хя)ом = * ^V (VI11' 15>
то подстановка в уравнение (VIII, 11) дает:
Г ахо 1 1 — лоо
R1
Аналогично
ЮЕ
Хт
dx^, 1 1 — х
1-х
^пр= I -=-^ h —In- SL (VIII, 17)
£2
В этом случае графически определяют интеграл зависимости
1/(хЛ xR) от xr (для Nt0Ry Часто концентрации компонентов
выражают в весовых долях, так как эти единицы используются
обычно в треугольных диаграммах равновесия, по даиным
которых строят рабочие диаграммы.
Если г — отношение молекулярных весов растворителя и
растворенного вещества, то
X=rX+l-X (VIII'I8>
При подстановке в уравнения (VIII, 11) и (VIII, 13)
получают соотношения, неудобные для непосредственных
практических расчетов. Соответствующие выражения по фазам рафи-
ната и экстракта4, в которых в качестве средней разности
концентраций принято среднее арифметическое значение (1—Х)ом,
приведены ниже:
Г_ 1 1 — Х-. 1 X-.tr—144-1
(VIII, 19)
(VIII, 20)
Y dXR
N — R
Г £
N(0E~J *e-Xe
XE2 C C
, l , l~XR2
f — In sf- -
2 1-*Л1
1 l~xP,
In m -f
2 1 — Xgo
lInv-l)+l
2 ^iC--I) + l
IIn^£l('-I)+I
2 Хю{г-\) + 1
389
В ряде случаев концентрации удобно выражать в весовых
отношениях w = X/(l—X), так как при этом рабочая линия
на диаграмме процесса часто является прямой. Подстановка
в уравнения (VIII, 11) и (VIII, 13) дает:
**i „ . , "я
юл
_LJI—SJom ^_= л__)—ln_J л? (VIIIi2i)
J (l+rwR)(wR-wR) J wR-wR 2 l+rwRi
"R2 R2
WE\ WE\
f (1+rWB)0MdWE = Г fWE _, l lnl+rWEl
J a-\-rwE)(wE — wE) J wE — wE 2 l+rwE2
£2 £2
Эти уравнения получены также с учетом допущения,
аналогичного зависимости (VIII, 15). Подстановка значений
концентраций, выраженных мольными отношениями, в уравнения
(VIII, 11) и (VIII, 13) приводит к уравнениям вида (VIII,21)
и (VIII, 22), в которых отсутствует величина г.
С помощью дальнейших упрощений интегралы в уравнениях
(VIII, 16) — (VIII, 22) можно вычислить аналитически.
Точность выражений, получаемых в результате интегрирования,
зависит от допустимости принимаемых упрощений. Ниже
рассматриваются различные условия интегрирования и
получаемые при этом результаты.
1. Разбавленные растворы (т = const) 12'4. Для
разбавленных растворов величины (1—xR) и (1—хЕ) близки к единице
и потоки R и Е практически постоянны. Тогда материальный
баланс для нижней части колонны (рис. 194) приближенно можно
записать в виде:
4XR-*R2) = 4XE-XEl) <VIII'23)
Подставим в это уравнение xE = mxR и решим его
относительно x*R:
х«==-щё(х*-хю)+^ (Vin,24)
Обозначим фактор экстракции R/rnE через е и введем
значение x*R из предыдущего уравнения в уравнение (VIII, 16).
Интегрирование последнего при этих условиях дает:
In
ЛГ_„ = -
'(XRl-XB7lm\L М | Г
'tOR— 1—1
е
(VIII, 25)
390
Аналогично получим следующее выражение:
In
N,OE —
£1
"да
£2
R2
(1-е) + е
1-е
(VIII, 26)
В связи с тем, что на практике величины т, R и Е при
экстракции изменяются незначительно, для получения более
точных результатов в расчет целесообразнее вводить значения
этих величин, взятых на конце системы, соответствующем
разбавленному раствору (т2, ^2 и Е2), так как этому концу обычно
i
ч41
/
ч°
06
Ц+
Up
qz
uUu
Щич-
Ции
OflB
U,UI
оров
OflOd
UJJU4-
0,003
0,002
пот
Щ.
ЩШ%~
^^
\4^
"■-^
fe
^
^
X
\
\
V
\
\Е(или1/е)\
X
\
\
i5
п
,
1
С или. i/cj цо
-
ч
д
Г]
XI
д
ч
V
\
т
\
L
vv
\.
\
Л
\
^
\
\
\
л
~^^
\
^
\ f
№
\\
\
\\
у
1
0,6
0,6
I
07
1
SL.
1 ■*•—■
/ -
"^ V
/у
¥•
№\
<Г\
V
> \
V
N
\v
в
I Z 3 & 5 6
Ht0RTum7)t0E)
Рис. 196. График для определения числа единиц переноса.
10 12
16
391
соответствует наибольшее число необходимых единиц переноса.
Если в уравнениях (VIII, 25) и (VIII, 26) заменить х на X,
выразить т в виде отношения концентраций X, а через R и Е
выразить весовые расходы соответствующих фаз, то эти
уравнения можно считать решением уравнений (VIII, 19) и (VIII, 20).
Эти же уравнения можно рассматривать также, как решения
уравнений (VIII, 21) и (VIII, 22), если заменить х на до,
выразить т в виде отношения концентраций до, а через R и Е
обозначить весовые расходы свободных от распределяемого
компонента фаз (инертных носителей). Графическое решение
уравнений (VIII, 25) и (VIII, 26) дано на рис. 196.
Сопоставляя уравнения (VIII, 25) и (VIII, 26) с уравнением
Кремсера (см. главу VI), получают следующие используемые
в расчетах соотношения:
"«ж = -гУт7Г Nu>b = тёт NtoR=*NtoE (VIII, 27)
"tOE-^tOR <VIII'28>
ВЭТС = ■ /of?1, ■ = t0E , (VIII, 29)
1 — 1/е е — 1 \ • /
г, ыю*-ыюе ] Ч"Ю«-У"ЮЕ _
~ 1П (NtO*lNtOE) ВЭТС ~ 1П [(VHtO*W/HtOE)] ( ' }
При е=1
ВЭТС = HtOR = НЮЕ П = NtOR = NtOE (VIII, 31)
2. Концентрированные растворы (/и линейно зависит от.*#).
Решение для данного случая было получено Шайбелем и Отме-
ром5. Приводимые ниже результаты, полученные этими
авторами, применимы в широком диапазоне изменения переменных:
хм
г ах г, хп,—хп. u-+-s
#_ = _#! Е1 \п—!— (если s2 положительно) (VIII, 32а)
J xR — xR s u — s
xm _ _ i .
г =fm.—xRllnfm—m'i*E\ -\-v (еслиий;51Х2П0Л0ЖИТельН0) (vill, 326)
т'лХр, — v
*R2
XE1
lR2 "1\ЛЕ2
2 (x x \ s'
y m ,—BV-tg-i — (если s2 отрицательно) (VIII, 32в)
dxE XE1-XE2,„U' + S"
Г __^g_= ле\ *m In u/'tsi) (если (s")2 положительно) (VIII,33a)
J xE — xE s и — s
-V£2
392
v£2 in ""rm "m.
In—^-^ ^i——r [если u'sss" и (s")2 положительно]
j о m^x^—x^ — v
*E2 (VIII, 336)
f = JT tg-'4p- [если (s")2 отрицательно] (VIII, ЗЗв)
s'" " "'
В этих уравнениях
s = V"(*t?i — *#2 + Ч*£2 — m2^£i)2 + 4 (xE1xR2 — *£2-*#i) {m'i — m2)
S' = У 4 (Jr£2-*7?1 — *£1*#2) (mi — m2) — (*Д1 — XR2 + m'\xE2 — m2^£l)2
s" = У (jc^ - *£2 + mxxm- m2xmf + 4 {xRlxm - x^xEl) (tnx - m2)
a'" = Y4 (^2*£1 - Jf^jc^,) (m, - m2) - (*£1 - *£2 + m,jc^ - т^да)3
M — -^tfl + XR2 — m2xE\ — mlxE2
u' = m, jc^ + m2xRl - xm - xm
_ (-у£1-у^2 — -^гг*;?!) (wi — mi)
XR1 XR2 m2xEl "Г mlxE2
/П2ЛГД1 ~ miX^2 — ^£1 + -*£2
Принято:
* * * *
XE\ XE1 I XR\ I XR2
m, = ffl, = ГПл — ffl, =
XR\ XR2 £1 £2
В тех случаях, когда равновесная кривая и рабочая линия
на конце колонны, соответствующем разбавленному раствору,
подходят очень близко друг к другу, коэффициент
распределения гпч должен отвечать наклону линии равновесия при
значении xR2, a m2 — величине, обратной наклону линии равновесия
при значении хЕ2. Прочие случаи, когда кривая равновесия и
рабочая линия являются кривыми более высоких порядков,
рассмотрены Шайбелем и Отмером5. В работах тех же авторов5-6
приводится графическое решение уравнений (VIII, 32) и
(VIII, 33). Эти уравнения можно использовать также в качестве
решений уравнений (VIII, 19—VIII, 22) в случае подстановки
в последние соответствующих величин, указанных выше1.
Аналитический метод интегрирования наиболее удобно
применять для определения NWr при противоточнои непрерывной
экстракции без флегмы. Если аппарат работает с возвратом
флегмы, для исчерпывающей части каскада следует
рассчитывать NtoR, а для укрепляющей — NtOE-
393
КОЭФФИЦИЕНТЫ МАССОПЕРЕДАЧИ
Коэффициенты массопередачи можно непосредственно
использовать для расчета высоты аппарата путем интегрирования
уравиении, из которых они определяются; при этом отпадает
необходимость в расчете высоты единицы переноса Ню.
Согласно уравнению (V, 57), имеем:
dN = KRa (cR - cR) dH = KpacRM (xR ~ xR) (VIII, 34)
В этом уравнении Kru— общий объемный коэффициент
массопередачи. Величину удельной поверхности контакта фаз а в
ряде случаев отдельно определить нельзя. Далее имеем:
.„ dN dN
= ——т *Т = / ST
KRaKCR — CR) KRacRtA(xR~xR)
Однако поскольку
(VIII, 35)
Rdxn
dN = d(RxR) = j—f- (VIII, 3)
/ RM pcp \
dN = dl—т8-8-) (VIII, 36)
то
(VIII, 37)
н-\лн = Г \р* .{
/?2
XR1
Г RdxD
H= — S- 5- (VIII, 38)
■I КгЛС„,.(\ ХЛ1Х Х.Л
хт
I<RaCRM(l—xR)(xR-xR)
Таким образом, высоту экстракционного аппарата Н можно
определить с помощью графического интегрирования функции
1 **v*
от —
Кй*(с*-с/г) р/?
или функции
R
от хг
к^/М1-**)(**-4) R
В обоих случаях необходимые для графического
интегрирования данные получают из рабочих диаграмм процесса,
построенных в соответствующих координатах (концентрациях).
3S4
Если раствор достаточно разбавлен, интеграл можно упростить:
RMD г dcp R л dx
Н = ~ I У ( *\= I У ( *\ (VIIU9)
PR / *<Ra{cR-cR) CR J ^Ra\x-XR)
В уравнении (VIII,39) используются средние значения R,
Mr и рн- Аналогично, выражая движущую силу в
концентрациях фазы Е, получим:
Н = У d(Ey^\ = ^k 'V *в (VIII, 40)
■I КЕа(сЕ—сЕ) рЕ cJ KEa(cE—cE)
Е2
Edx„ E 7" dxc
ХЕ1
г пах,, с. с ал
Н= Г Z-i-ъ г- = - I v , *Е г (VIII, 41)
J КЕасЕМ(\—хЕ)(хЕ-хЕ) с Е J KEa(xE-xE)
*т Е2
Подобные уравнения можно записать также в виде
зависимости высоты аппарата Н от коэффициентов массоотдачи kRa
и- kEa. Однако в качестве движущей силы процесса экстракции
в этом случае следует применять разность концентраций на
поверхности раздела фаз и в объеме соответствующей фазы.
Для сильно разбавленных растворов, когда коэффициент
распределения системы и коэффициент массопередачи можно
рассматривать как постоянные величины, уравнения можно
проинтегрировать аналитически по методам, аналогичным тем,
которые использовались при определении высоты аппарата с
помощью единиц переноса. В этом случае имеем:
R У1 dx
Н
Ср К,
#,.„ "R
"R2
"ср. " хг
г ахп
- Г V (VIII, 42)
a J х—xD
Комбинируя уравнение (VIII, 42) с уравнением (VIII, 24),
интегрируя и решая полученный интеграл совместно с
уравнением материального баланса, получим:
n = V"4P. (** - * *)ср. (vni-43)
где
0*-4)ср - (Х*-*«д-<>Х*-Х'«д (VIII, 44)
XR2 — XR2
395
Подробно эта методика расчета рассмотрена в работах7'8.
Аналогично получим:
N = КЕаНсЕ^ (4 - хЕ)^ = КЕаН (сЕ - сЕ)^ = KRaH(cR - c*R)^
(VIII, 45)
ОБЩИЕ И ЧАСТНЫЕ
ДИФФУЗИОННЫЕ СОПРОТИВЛЕНИЯ
Целесообразно установить связь между общим
диффузионным сопротивлением, обычно использующимся в расчетах, и
частными (фазовыми) сопротивлениями. Зависимость между
этими величинами уже рассматривалась в главе V. Если
коэффициент распределения т представить отношением
концентраций, выраженных в мольных долях, то уравнение (V, 61)
примет вид:
■- - - J- cp- (VIII, 46)
KRa kRa mk£acE
ср.
Умножив каждый член последнего равенства на
Rl(l— xR)omCrCp, получим2:
Htn„ = HiR Л *^ + Ь#Г "if. 1- , ч (VIII>47)
Если основное диффузионное сопротивление
сосредоточено в фазе R, то (1 — xR)iM= (1 — xr)0m и (\ — хе)ш =
= (1 — хЕ). Тогда
^=^+Ш^(Т^7 (VIII'48)
При этом для разбавленных растворов:
Аналогично для разбавленных растворов при основном
сопротивлении в фазе Е получим:
нЮЕ = нт+{п?)Н* (VIII'50)
Эти уравнения часто используются в экспериментальных
работах и показывают определяющее значение величины фактора
экстракции mE/R для выявления, в какой из фаз сосредоточено
основное диффузионное сопротивление. Если предположить,
что HtE и #(Л, зависящие от скорости потоков, — величины
одного и того же порядка, то при увеличении фактора экстракции
основное диффузионное сопротивление будет перемещаться в
фазу R, а при уменьшении этого фактора — в фазу Е.
396
Фактор экстракции, представляющий собой отношение
наклонов линии равновесия к рабочей линии, позволяет также
учитывать экономические показатели при проектировании экстрак-
ционной установки. При значениях фактора экстракции, меньших
единицы (см. главу VI), степень извлечения в случае
непрерывной противоточной экстракции ограничена даже при
бесконечном числе теоретических ступеней разделения или единиц
переноса.
Чтобы содержание распределяемого компонента в конечном
рафинате было низким, фактор экстракции должен быть больше
единицы. Из рис. 196 видно, что чем больше значение mE/R,
тем меньшее число единиц переноса требуется для достижения
заданной степени извлечения и, следовательно, тем меньше
стоимость аппарата. При больших значениях mE/R
увеличивается стоимость регенерации экстрагента вследствие того, что
получается менее концентрированный экстракт. Поэтому важно
определить оптимальное значение mE/R, учитывая при этом
ценность извлекаемого компонента и прочие факторы, влияющие
на стоимость процесса экстракции.
Оптимальное значение фактора mE/R, как показано в
главе XII, приблизительно равно 1,2—2,0'. При значениях фактора
экстракции, лежащих в этих пределах, сопротивление каждой
фазы имеет существенное значение.
В данном случае применение методики расчета высоты
аппарата, основанной на концепциях общей высоты единицы
переноса или общего коэффициента массопередачи без
соблюдения условия постоянства коэффициента распределения т или
фазовых сопротивлений,
менее обоснованно.
Пример VIII-1. Определить число
единиц переноса Nton для
экстракции в условиях примера VI-5.
Решение. Предварительные
расчеты выполнены в примере VI-5.
В соответствии с принятыми
обозначениями в данном случае R\ —
= 100 кг/ч; £2=30 кг/ч; XR1 = 0,50;
•Хл2=0,10; Хе2 = 0; XEi = 0,557 вес.
долей ацетона. Положение рабочей
точки О показано на треугольной
диаграмме (рис. 128). Произвольные
прямые, проведенные из точки О,
пересекают кривую растворимости в
точках, соответствующих
концентрациям л8 на части кривой,
примыкающей к вершине А, и ХЕ —на части
кривой, примыкающей к вершине В
треугольной диаграммы. По этим
данным на рис. 197 построена
рабочая линия. Кривая равновесия
проведена по точкам, соотввтствую-
& °-В
|«8 0,5
¥
%щ,о,з
и*
J о
ft
/-
*?
"У*
&
V
^
~г
0,1 0,2 0,3 0/+ 0,5 0,6
Концентрация ацетона
В Водном растВоре xR, бес. доли
Рис. 197. Рабочая диаграмма (к
примеру VHI-1):
/ — кривая равновесия) 3 — рабочая линия.
397
щим равновесным концентрациям на концах хорд равновесия треугольной
диаграммы. Из графика рис. 197 находим
X
№'■
О
Хт = 0,435
1-^ = 0,9
1 — ^=1.0
1 _ х*т = 0,565 1 — XR1 = 0,5
Поскольку отношение разностей (1 — Хр) и (1 — XR) на концах
аппарата меньше двух и концентрации выражены в весовых долях, для
определения NtoR воспользуемся уравнением (VIII, 19).
По условию примера VI-5 отношение молекулярных весов воды и
ацетона составляет г= 18,02/58,05=0,31.
Из данных рис. 128 следует, что при концентрации, равной конечной
концентрации экстрагируемого компонента в экстракте, взаимная
растворимость воды и экстрагента существенна. Поэтому аналитическое решение
интеграла в уравнении (VIII, 19) неприменимо. На рис. 198 на основе данных
рис. 197 построен график зависимости \j{XR — XRj от XR. Площадь под
кривой, ограниченная значениями XRi = 0,5 и Хщ—0,1, равна 4,98.
Подставляя значения соответствующих величин в уравнение (VIII, 19),
получим:
т
N
dXn
tOR
Хп — ХЕ
1 1
— In =-
■X,
R2
i , хт(г—1)4-1
■**i ' 2
In-
**i(r-l)+l
лпо , 1. (1—0,10) . 1. o,iO(0,3io—i)-f-i ...
Пример VIH-2. Уксусная кислота экстрагируется из 10%-ного водного
раствора метилизобутилкетоном в насадочной колонне с насадкой из
угольных колец Рашига размером 12 мм. Экстрагент является дисперсной
фазой. Концентрация уксусной кислоты после экстракции: в рафинате 0,1%,
в экстракте 6,5%. Скорость водного раствора в колонне 9,15 м3 • м'2 • ч~'.
Скорость массопередачи и равновесные данные для системы водный раствор
уксусной кислоты — метилизобутилкетон приведены в работе Шервуда,
Эванса и Лонгкора [Ind. Eng. Chem., 31, 1144 (1939)]. Рассчитать
необходимую высоту колонны.
Решение. При заданных концентрациях экстрагент и раствор
уксусной кислоты не обладают заметной взаимной растворимостью. Рабочую
диаграмму строим в концентрациях,
выраженных в кг/кг растворителя, и
определяем число единиц переноса
по уравнению (VIII, 21). При этом
расходы фаз R и Е находим в кг
воды или в кг экстрагента
(соответственно) на 1 м2 сечения колонны
в час. Отношение молекулярных
весов кетона и кислоты равно:
16
10
1
\*в
а
?'
мол. вес кетона 100,2
0,1 0.Z
0.3
ГГ.г,
0А 0,5 03
мол. вес кислоты
60,1
: 1,669
Рис. 198. Графическое
интегрирование (к примеру VIII-I).
Плотность исходного раствора
кислоты в воде составляет 1005 кг/ж3.
Через 1 м2 сечения аппарата за 1 ч
398
Проходит 9,15x1005=9200 кг раствора кислоты, который содержит 920 кг
уксусной кислоты и 8280 кг воды. Следовательно
280 К£ВОДЫ_
1/2 . и
а; =
0,10
Я1 1—0,10
= 0,111
кг кислоты
кг воды
0,001 „ ... кг кислоты
wm — "1— nnni = О'001
Я2 1 — 0,001 кг воды
w ==
0,065
& 1 — 0,065
= 0,0695
кг кислоты
кг кетона
„ кг кислоты
wF0 = 0
hi кг кетона
Из баланса по уксусной кислоте имеем:
8280 (0,111 — 0,001)
£=•
0,0695 — 0
= 13100 *г к,етона (16,15-^-
м2 ■ н \ м2-ч
Рабочая диаграмма, равновесная линия на которой построена по дан*
ным Шервуда, Эванса и Лонгкора (см. ссылку выше), представлена на
рис. 199. В связи с тем что растворы достаточно разбавлены и рабочая
линия является прямой, нет необходимости в графическом интегрировании.
Однако ввиду того что коэффициент распределения системы непостоянен,
уравнение (VIII, 25) не дает достаточно точного решения. Поэтому
воспользуемся уравнением (VIII, 32), куда вместо х подставим w, а коэффициент
распределения т определим через W.
1#1
VE1
0,0959
0,0695
= 1,380
Величина т2, обратная наклону кривой равновесия при wR=0,
составляет 1,543. В данном случае величина s2 отрицательна. Поэтому
S = lA (™E2WR1 — WE1WR2) (К — т'г) — (WR1 — WR2 + т'\ WE2 + т2 w E\f =
= V"4(0—0,0695-0,001) (1,380—1,543)—(0,111—0,001+0—1,543-0,0695)2==0,00615
и = wR1 + wR2 — m'2wE1 — m[wE2 = 0,111 -f- 0,001 — 1,543 • 0,0695 — 0 = 0,0047
Из уравнения (VII, 32в|:
#i
dw
Г awR ^
R2
0,08
I
2 (a
V?i
WR2) -i S
s и
__ 2 (0,111— 0,001) -i 0,00615
0,00615 g 0,0047
'*V
"
/
ve
г
/Л
'u/t
/' WE,
o,ou
0,08
а/г
^35,5
, кг кислоты
ыя> кг t/ооы
Рис. 199. Рабочая диаграмма (к при-
При графическом интегриро- меру VII1-2):
вании получаем величину 37,0. / — кривая равновесия; 2 — рабочая линия.
399
Из уравнения (VIII, 21):
м о.с , 1 ,„ 1 + 1.669-0,001 _qt;o
"ton = 35-5 + 2"1п 1 + 1,669-0,111 _ ^
По данным Шервуда, Эванса и Лонгкора (см. ссылку _выше), при
скоростях потоков водного раствора кислоты 9,15 ж3 • ж 2 ■ ч ' и экстрагента
16,15 дз-ж-з-ч-1 коэффициент массопередачи КЕа=48 кмоль ■ м 3-ч ](Ac= 1) '.
Если коэффициент распределения от определен через концентрации,
выраженные в мольных долях, то:
1 1 mcF
КЕа kEa^ kRacpcp
Решая это уравнение совместно с уравнением (VIII, 46), получим:
KRacR^=mKEa%p.
Подстановка в уравнение (VIII, 12) дает:
R
Н,
tOR тКЕасЕср(1-хр)ом
где R выражено в кмоль • м 2 • ч '.
Далее находим:
920 , 8280 ._с „ кмоль
^==-60- + Т8- = 475,3-^тт
8,28 . 8280 .СЛ1 кмоль
*2=Тю-+-Т8- = 460ЛТ^Г
_ .___ кмоль
Среднее значение Н=- 467,7 . _—
м'- ч
При wm =0,111: хт = 0,0322 мол. доли, л:^ = 0,0280 мол. доли; кроме
того
(1-0,0280)-(1-0,0322)
С1 — хт)ом п 1—0,0280 '
2'3« 1—0,0322
При а^2 = 0,001 находим: (1 — xR2)QM = 1,00; среднее значение
Кривая равновесия и рабочая линия процесса расположены близко,
поэтому сЕ^ сЕ. Тогда
794
°ЕМ* = "Щ2 = 7'93 кмоль/м3
Плотность экстракта равна 812 кг/ж3. Отсюда
812-0,065 , 812-0,935 . „. . ,
сем, = 60 1 100,2 = 8'39 КМ0ЛЬ1М
^ЕМ \~ ^ЕМ
с_ = —^ — = 8,16 кмоль/м3
ССр. £
400
Средний наклон кривой равновесия в принятых пределах концентраций
от=3,0. Тогда
HtOR = 3,0-48-8,16-0,985 = °'405 М
По уравнению (VIII, 11) высота колонны составляет:
Н = 35,2 • 0,405 = 14,25 м
Полученный результат должен быть скорректирован с учётом влияния
продольного перемешивания.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
а —удельная (на 1 м3 объема аппарата) поверхность
контакта фаз, м2/м3.
с —концентрация, кмоль/м3.
__ м,~\-см2 . с обозначает общую концентрацию всех
компонентов.
'Ср
см- 2
с + с*
(сЕ c£Jcp —
(CR-cr)c
м
с обозначает общую концентрацию
всех компонентов.
L£i
)-(<
£2"
^Е2)
In
""ЯГ
ЬЕ\
LE2~
LE2
\CRl ~CRV~ \CR2 ~ CR'i)
ln-
c обозначает концентрацию только
распределяемого компонента.
Е — скорость потока фазы экстракта, кмоль • м~2 • ч'1.
И — высота колонны, м.
ВЭТС — высота, эквивалентная теоретической ступени, м.
Ht — высота единицы переноса, м.
К — общий коэффициент массопередачи, кмоль • м~2 • ч_1Х
Х(Ас=1)-'
Ка — общий объемный коэффициент массопередачи,
кмоль • мг3 ■ ч-1 • (Ас = 1) ~1
k — коэффициент массоотдачи, кмоль • м~2 • ч-1 • (Ас=1)~1
ka — объемный коэффициент массоотдачи, кмоль • м~3Х
Хч-1- (Дс=1)-'
М — молекулярный вес.
т = XEtlxRi — наклон кривой равновесия, за исключением
уравнений (VIII, 32) и (VIII, 33).
./V — скорость экстракции, кмоль-м~2-ч-1.
Nt — число единиц переноса.
26 Р. Е. Трейбал . 401
И — число теоретических ступеней.
R — скорость потока фазы рафината, кмоль • м~2 • ч~у.
г — отношение молекулярных весов растворителя и
растворенного компонента.
w — концентрация, кг растворенного вещества на 1 кг
растворителя; w=Xj{\ —X).
X — концентрация растворенного вещества, вес. доли.
х — концентрация растворенного вещества, мол. доли.
xr> x*e — определяются из уравнения (VIII, 10).
/ * \ _ \ХЕ1 — ХЕ\) \ХЕ1 ХЕ2)
\ХЕ ХЕ)ср. — * __
In —
ХЕ2 ХЕ2
(v __*\ _ \XR\ ~XR\)~\Х№~~ Хт)
\XR xR)c$. —
In
#1
XR2 XR2
n r . Q--*e)-{x-xei)
(1 — xe)im- y^Tz
E
1 — xEi
,, „ . 0-xri)-0-xr)
(1 xR)iM - \-Xm
\-XR
a x^ (i-^£)-(i-4)
К xE)OM — J x
In E-
\ — xE
(1-xr)-0-xr)
0 xr)om ~
1
In
\-XR
z=mE/R— фактор экстракции,
p— плотность, кг/ж3.
Индексы
1 — относится к концу колонны, на котором раствор имеет
большую концентрацию.
2 — относится к концу колонны, на котором раствор имеет
меньшую концентрацию.
ср.— среднее значение.
Е— экстракт.
/— на поверхности раздела.
М — среднее значение.
О — общий.
R— рафинат.
402
ЛИТЕРАТУРА
1. А. Р. Colburn, Trans. Am. Inst. Chem. Engrs., 35, 211 (1939).
2. J. C. Elgin, Chemical Engineers' Handbook, J. H. Perry (ed.) 3rd ed.,
McGraw-Hill Book Company, Inc., New York, 1950.
3 J. H. Wiegand, Trans. Am. Inst. Chem. Engrs., 36, 679 (1940).
4. A P. С о 1 b u r n, Ind. Eng. Chem., 33, 459 (1941).
5 E. G. S с h e i b e 1, D. F. О t h m e r, Trans. Am. Inst. Chem. Engrs., 38, 339
(1942).
6. E. G. Scheibel, D. F. Othmer, Ind. Eng. Chem., 34, 1200 (1942).
7. T. G. Hunter, A. W. Nash, J. Soc. Chem. Ind., 51, 285T (1932).
8. T. K. Sherwood, R. L. P i g f о г d, Absorption and Extraction, 2nd ed.,
McGraw-Hill Book Company Inc., New York; 1952.
26*
ЛАБОРАТОРНАЯ
И ПОЛУПРОМЫШЛЕННАЯ ЭКСТРАКЦИЯ
11/идкостная экстракция является одним
^^ из распространенных лабораторных
методов, который применяется в органическом
синтезе, аналитической химии, а также для
получения равновесных данных в системах
жидкость — жидкость (см. главу II).
Однако в данной главе основное внимание
уделено применению лабораторной экстракции
для изучения и разработки экстракционных
процессов. Промышленные смеси,
разделяемые с помощью экстракции, обычно имеют
сложный состав, и методы расчета,
описанные в предыдущих главах, в большей части
нельзя непосредственно применить к ним.
Не всегда можно подобрать растворитель
(экстрагент) на основе литературных
данных или с помощью расчетных методов,
рассмотренных в главе III; если даже это и
возможно, лабораторная проверка экстр-
агента все равно необходима.
В лабораторных условиях (обычно в
стекле) подбирают экстр агенты для
разделения сложных смесей, а также определяют
условия отстаивания промышленных систем
перед испытаниями на укрупненных
опытных установках (пилотные испытания).
Жидкостная экстракция стала одним из
стандартных методов, применяемых во
многих операциях аналитической химии,
причем специальные экстракционные методы и
аппаратура, разработанные для анализа
сложных природных и синтетических
продуктов, также представляют интерес для
инженера-технолога. На основе
полученных этими методами лабораторных
результатов в дальнейшем можно
разработать промышленные экстракционные
процессы.
Пилотные испытания проводят в
аппаратуре более крупных размеров, геометри-
404
чески подобной той, которая будет использоваться в
промышленном масштабе, чаще всего в непрерывном или
полунепрерывном процессе.
МЕТОДЫ ЛАБОРАТОРНОЙ ЭКСТРАКЦИИ
Разрабатывая новые промышленные экстракционные
процессы, не всегда можно полагаться на равновесные данные,
полученные при применении тщательно очищенных веществ, так
как влияние небольших количеств примесей, обычно
присутствующих в технических продуктах, часто нельзя предвидеть.
Смесь, предназначенная для разделения, во многих случаях не
является раствором двух или трех химически чистых
компонентов, а содержит по крайней мере небольшие количества веществ,
поведение которых при экстракции не всегда можно
предусмотреть. Более того, о наличии следов некоторых веществ в
растворе иногда можно судить, лишь сконцентрировав их в одной
из фаз путем многоступенчатой экстракции. Разделяемая смесь
может иметь настолько сложный состав, что попытка
получить детальные равновесные данные, особенно если
компоненты смеси влияют на распределение друг друга, оказалась бы
безуспешной или слишком дорогостоящей. Методы расчета,
описанные в предыдущих главах, к таким системам не применимы.
Иногда, при очень малых концентрациях, кривая равновесного
распределения может принимать неожиданную,
неблагоприятную для экстракции, форму. Это приобретает важное значение,
если необходимо довести остаточную концентрацию
распределяемого вещества в рафинате до нескольких миллионных долей.
Во всех указанных случаях представляется необходимым
осуществление многоступенчатой экстракции в лабораторных
условиях. При проведении лабораторной экстракции основные
параметры (объемное соотношение фаз, число ступеней и др.)
должны соответствовать тем же параметрам в промышленном
процессе, так как определить их расчетом для сложных систем
невозможно. Только при таком условии можно по
лабораторным данным выяснить, какие результаты может дать
экстракция в крупном масштабе. Это значит, в частности, что в лабо- <
ратории обычно необходимо исследовать проце_сс_дративоточной
многоступенчатой экстракции тгоскШТьку в промышленности для .
повышения эффективности процессы экстракции обычно
осуществляют по противоточной схеме. Истинно противоточный
процесс должен быть непрерывным. Однако на стадии
лабораторной разработки обычно имеется недостаточное количество
исходных веществ, чтобы проводить непрерывную экстракцию.
Поэтому используют один из следующих методов:
а) имитацию процесса непрерывной многоступенчатой
экстракции;
405
б) непрерывную экстракцию в миниатюрном экстракторе с
известным числом ступеней.
На практике применяют оба метода. Однако, учитывая, что
при исследовании по второму методу число ступеней неизменно,
а также ввиду затруднительности непрерывной подачи малых
количеств жидкостей чаще используют первый метод.
Имитация непрерывного процесса. Имитацию непрерывной
экстракции осуществляют с помощью ряда следующих друг за
другом по определенной схеме операций периодической
экстракции, выполняемых чаще всего в обычных делительных воронках.
Ряд схем имитации процессов непрерывной экстракции был
предложен много лет назад Янтзеном '.
При использовании различных схем имитации непрерывной
экстракции предполагается, что каждая операция периодической
экстракции эквивалентна одной идеальной (теоретической)
ступени, поэтому жидкости должны быть достаточно хорошо
перемешаны. Для смешения можно применять лабораторные
«трясучки», однако и при простом встряхивании руками равновесие
достигается очень быстро. Было показано2, например, что
50 «переворачиваний» пробирки с экстрагируемой смесью в
течение 1,5 мин при невысокой вязкости жидкостей достаточно для
достижения равновесия между фазами. Равновесие достигается
быстрее, если энергично перемешиваемой смеси давать
возможность несколько раз отстаиваться. Это обусловливается
перемешиванием при коалесценции капель. В некоторых системах
чересчур энергичное перемешивание может привести к
установлению ложного равновесия вследствие адсорбции распределяемого
вещества на поверхности раздела фаз3. Иногда, например при
разделении металлов, может протекать медленная химическая
реакция между экстрагентом и распределяемым веществом.
В таких системах нужно предварительно исследовать скорость
приближения к равновесию.
При лабораторной имитации противоточного процесса
необходимо очень тщательно переносить жидкости из одной
делительной воронки в другую. Чтобы потери жидкостей не были
слишком заметными по отношению к их объемам, нельзя
проводить процесс с очень малыми количествами жидкостей. При
высокой вязкости жидкостей количества их для лабораторного
исследования следует брать большими. Отстаивание эмульсий
должно быть полным. После отбора из делительной воронки
какой-либо фазы необходимо дать жидкостям постоять
некоторое время и отделить после этого вновь отстоявшееся
дополнительное количество жидкости.
При имитации непрерывной экстракции исходную смесь и
экстрагент вводят в процесс в ряде операций однократной
экстракции. При достаточном числе подобных операций должны
быть получены те же результаты, что и в стационарном непре-
405
Рис. 200. Стойка для воронок 4.
рывном процессе экстракции; приближение к условиям
непрерывного процесса является асимптотическим. Соотношение фаз
в операциях периодической экстракции должно быть таким же,
как и в соответствующем непрерывном процессе. Очень важно
не допускать никаких изменений в схеме имитации
непрерывного процесса; в случае нарушения последовательности
выполнения операций необходимо повторить весь процесс сначала.
С этой точки зрения, для проведения лабораторной экстракции
удобно пользоваться различными автоматизированными
устройствами. На рис. 200 показана стойка для делительных
воронок4, в которой оба ряда воронок могут перемещаться как в
верхнее, так и в нижнее положение. С помощью такого
приспособления количество ручных операций сводится к минимуму.
Противоточная экстракция. На рис. 201 показана схема
повторяющихся операций периодической экстракции, которой
необходимо следовать для имитации процесса непрерывной про-
тивоточной экстракции, требующего пяти теоретических
ступеней разделения (см. нижнюю часть рисунка)5-6. Каждый
кружок на рисунке обозначает отдельную операцию экстракции в
делительной воронке. В операции а определенное количество
исходного раствора F экстрагируется селективным
растворителем (экстрагентом) S; экстракт Fa выбрасывается, а рафинат
Ra экстрагируется свежим экстрагентом в воронке Ь. После
этого экстракт подается в воронку /, где взаимодействует с
исходным раствором F, а рафинат экстрагируется в воронке с
и т. д. Экстракты движутся справа налево, рафинаты — в
обратном направлении. Соотношение F n S должно быть равно
соответствующему соотношению в противоточном процессе (см.
рисунок), а количества F и S не должны изменяться в
продолжении всего исследования.
Теоретически результаты, полученные таким методом,
никогда не будут точно совпадать с результатами имитируемого
процесса непрерывной экстракции. Однако они асимптотически
приближаются к последним, и с увеличением числа операций
(горизонтальных рядов на схеме) практически можно получить
сколь угодно близкое совпадение. Если, например, для четырех-
пяти рафинатов, выводимых из схемы процесса справа, не
обнаруживается заметного изменения состава, можно считать, что
достигнуты условия, практически соответствующие
стационарному состоянию (приближение к этому состоянию происходит
медленно, и поэтому необходимо тщательно проверять,
действительно ли достигнуты стационарные условия процесса).
При достижении стационарного состояния жидкости,
находящиеся в воронках 1—5, должны быть во всех отношениях
(объемы, веса, концентрации и все физические свойства)
аналогичны жидкостям, находящимся на соответствующих
ступенях непрерывного процесса. С помощью полученных таким об-
5 S
-q5[h'tpqi-l3(pqfj,t'3
i5[/-*pq+-~+iZ1(p4)M],
t=t c
ej-Ps Ег Ез E« Es
Рис. 201. Схема имитации процесса непрерывной противоточной
экстракции из пяти ступеней в лабораторных условиях.
40В
разом данных можно построить в концентрациях одного из
компонентов равновесную и рабочую линии. Путем построения
зависимостей концентраций в экстракте, выходящем с данной
ступени, от соответствующих концентраций в рафинате, например
Ег от R2 и т. д., получают равновесную линию. Построение же
зависимости между составами экстракта, покидающего данную
ступень, и рафината, поступающего на нее с предыдущей
ступени, например Е2 от Ri и т. д., дает возможность получить
рабочую линию процесса. Подобные построения позволяют,
например, предвидеть случаи сближения рабочей и равновесной
линий. При отборе проб на анализ (например, из Ri и Е3) на
последующую операцию 2 следует подавать одинаковые доли от
количества каждого из указанных растворов (например,
половину их количеств), чтобы поддержать объемное соотношение
фаз неизменным.
Рассмотренный метод позволяет имитировать любое число
ступеней экстракции. Для п ступеней минимально необходимо
ге/2 или («+1)/2 делительных воронок (в зависимости от того,
является ли п числом четным или нечетным) 7.
Несмотря на то что описанный метод исследования
экстракции применяют, когда характер равновесия и другие важные
для экстракции свойства системы не достаточно выяснены,
результаты такого исследования можно интерпретировать . (по
крайней мере в относительно простых случаях), как обычно, с
помощью коэффициентов распределения, величин фактора
экстракции и т. п.
Верхний ряд операций на рис. 201 (от а до е) представляет
собой пять ступеней экстракции в перекрестном токе (исходная
смесь пять раз экстрагируется одинаковым количеством экстр-
агента). Соответственно после указанных операций
концентрация растворенного вещества в рафинате Re будет значительно
меньше концентрации конечного рафината, получаемого в
имитируемом непрерывном процессе. С увеличением числа операций
конечная концентрация в рафинате будет возрастать, все более
приближаясь к соответствующей величине для непрерывной
противоточной экстракции. Соответственно экстракт Еа получается
в результате экстракции чистым экстрагентом, а не экстраген-
том, использовавшимся в четырех предыдущих ступенях; его
состав также не отвечает составу конечного экстракта в
противоточной процессе. Так как в первых операциях рассмотренной
выше схемы распределяемый компонент экстрагируется более
интенсивно, общее его количество, находящееся в процессе,
постепенно увеличивается и концентрации на последующих
ступенях возрастают.
Рассмотрим подробнее частный случай, характеризующийся
взаимной несмешиваемостью экстрагента и растворителя
исходного раствора, а также постоянством коэффициента распределе-
409
ния извлекаемого компонента. Если компонент В — экстрагент,
компонент А — растворитель исходного раствора и m —
коэффициент распределения, то фактор экстракции е = тВ/А. Пусть
доля общего количества поступающего в каждую ступень
экстрагируемого компонента, переходящая после установления
равновесия в экстракт, равна р. Долю общего количества
экстрагируемого компонента, оставшуюся в рафинате, обозначим q.
Величины р и q будут постоянны и равны (см. главы VI и VII):
" = 7+Т * = Т+^ ^
На рис. 201 показаны количества экстрагируемого
компонента в соответствующих растворах, выраженные через р и q.
При этом количество экстрагируемого компонента в исходном
растворе принято равным единице. Примем далее, что число
циклов t равно числу выводимых (отбрасываемых) из процесса
рафинатов. Например, при t=2 из процесса выводятся два ра-
фината и четыре экстракта (четвертый — после операции g).
Число выводимых экстрактов больше соответствующего числа
рафинатов на величину, зависящую от общего числа ступеней
в имитируемом процессе.
Как видно из рисунка, доля извлекаемого компонента в
рафинате выражается степенным рядом, порядок которого зависит
от t. Для каждого числа ступеней получается ряд следующего
вида8:
It = 1" fYi +ЬР9 + Y3 {РЯ? + У a (Pi)" + ■ ■ ■ + \t {РЧУ-'] (IX, 2)
Здесь п — число ступеней в имитируемом противоточном
каскаде (на рис. 201 n = 5), a t — число циклов; qt равно доле
экстрагируемого компонента, остающейся в рафинате после /
циклов. Коэффициенты этого ряда можно вычислить с помощью
следующего уравнения *:
-if
t-\ t-i
п + 1 n + l
V М\ V М\
Ni\(M — Nl)l jU (Ni—iy.iM — Ni+iy.
-S
t-1 (-3
n + l n+l
Ml , V M\
Nj\(M — Nj)l ^ 4U (Nj— l)\(M — Nj +1)1
(IX, 3)
где
M=n + 2/ —3 (IX, 4)
Nt = t—l-t(n + l) (IX, 5)
Nj = t — 2 — j(n+l) (IX, 6)
* Уравнение (IX, 3) можно получить из уравнения (IX, 15), положив п
равным нулю и решив его относительно у (г.
410
Числа i и / могут принимать целые значения of нуля ДО
верхнего предела суммирования, но не должны его превышать;
если верхний предел отрицателен, суммирование не
производится. Аналогично доля растворенного вещества в экстрактах
в общем виде определяется следующим выражением9:
= р 1
Pt = P
\pq
n + l
л/2
2
4 = 1
sin-4
2£я
n + l.
1
Upq <
kit
1Г+Т
1 — Apq cos2
kn
n+l
(IX, 7)
Величина pt представляет собой долю растворенного
вещества в экстракте после t циклов для схемы процесса, показанной
на рис. 201; k принимает все целые значения от 1 до я/2, но не
превышает я/2. В предельном случае при t=oo величины pt и
qt становятся равными соответствующим величинам в
стационарном процессе. Если свежий экстрагент не содержит
экстрагируемого вещества, предельные значения р и q, как следует из
уравнения (VI, 125), можно определить с помощью зависимости
рИ + 1 „
р =1 — q = -^— (IX, 8)
Составы экстрактов и рафинатов после каждого цикла
приближаются к своим предельным значениям не в одинаковой
степени. Для оценки степени приближения к стационарному
состоянию можно брать
отношение pt к ps; qt к qs или (pt +
+ qt) к (ps + qs). На рис. 202
изображены полученные с
помощью уравнений (IX, 7) и
(IX, 8) кривые для степени
приближения экстракта к
стационарному состоянию 6 = 0,90
и 6 = 0,99, причем 6
определялось отношением
ио
30
го
6 = Pjpa
(IX, 9)
й я
-I B
% в
Те же зависимости показа-
* ны на рис. 203 в виде, удобном
Для интерполяции по
величине е. Процессы экстракции
часто осуществляются при
величине фактора экстракции,
близкой к единице; именно при
таких значениях е скорость
приближения к стационарному
состоянию является наименьшей.
8=0,90
1/
Ч/
1'
S
/
/
/
S
/
/
S
S
'
*г
С,
i ?
05,
, *i Л
£-^
^
"*
/
г
г1Г'
е=ца, <+ ~
£=0£;'г__
£=0,1; 10
131,567 89 Ю
Число ступеней,/!
Рис. 202. Степень приближения
экстракта к стационарному
состоянию при имитации противоточной
экстракции в лабораторных условиях.
411
SO
UO
30
20
I
з
о
5
5
/
/ -
' *
|
/ \п=Юступеней
/
у
•
У И
?7
It /
■
"8\
,в\\
10 \
■?'Skl^^A
> Я-.\ х.
' \\ ^
;:к^
4 \\
6=0,99
— <г=о,до
i::!^4^
1 ч<
, \
3 \ ^
0,1 0,2 0,3 0,5 07 1 2 3
Фактор экстракции, с
5 7 W
Рис. 203. Степень приближения экстракта к стационарному состоянию
при имитации непрерывной экстракции в лабораторных условиях.
Чтобы ускорить приближение рафинатов к стационарному
состоянию, Шайбель5 предложил использовать более
концентрированный исходный раствор в первых циклах процесса и
постепенно разбавлять его по мере приближения к стационарному
состоянию. Если величина г равна номеру операции, в которой
вводится исходный раствор, то для г—\ или г=2:
- = !■
(ея+1_1)(е_|_1)'--
а для г>2
ел —е2
>»+' — l)(e + l)L (е + 1)
г 2е Г2
L(e + l)2J
(IX, 10)
(IX, 11)
где хр — концентрация растворенного вещества в исходном
г
растворе;
хр — та же концентрация в случае достижения стационар-
■S
ного состояния.
При таких исходных концентрациях, для того чтобы
получить «стационарные» составы экстрактов и степень приближения
рафинатов к стационарному состоянию, равную 90%,
достаточно проводить число циклов, лишь несколько превышающее
число ступеней в имитируемом процессе.
412
Приведенными выше уравнениями можно пользоваться
только в том случае, если известны коэффициенты распределения
всех распределяемых компонентов. Вместе с тем, если известны
все коэффициенты распределения, отпадает необходимость в
периодической имитации непрерывного процесса. Несмотря на это,
приведенные уравнения оказываются полезными в следующих
случаях:
1. Имитацией непрерывного процесса предварительно
установлена приблизительная величина коэффициента
распределения, и необходимо повторить процесс при различных объемных
соотношениях экстрагента и исходного раствора или при
различном числе ступеней.
2. Известен коэффициент распределения одного или
нескольких компонентов исходного раствора, и необходимо исследовать
поведение при экстракции других компонентов.
В тех случаях, когда взаимная растворимость экстрагента и
растворителя исходного раствора или коэффициенты
распределения изменяются в процессе экстракции, уравнения (IX, 10) и
(IX, 11) могут давать только грубо приближенные результаты.
Противоточная экстракция с флегмой. На рис. 204
изображена схема имитации процесса экстракции с флегмой. Каждый
кружок на схеме означает операцию перемешивания в
делительной воронке, а прямоугольник — операцию отделения
экстрагента для получения флегмы. Начиная осуществление
указанной схемы, необходимо иметь раствор, соответствующий по
составу флегме /?о; такой раствор отсутствует, и поэтому его
нужно получить в самом процессе имитации непрерывного
процесса. Метод получения раствора состава R0 предложен Шайбе-
лем 4; при использовании этого метода довольно быстро
достигается стационарное состояние. Согласно данному методу, в
первую воронку подается количество исходной смеси Fa,
превышающее расход исходной смеси Fs в стационарном состоянии:
Fa = Fs + (n + \-f)K (IX, 12)
В этом уравнении К — количество исходного раствора,
растворяющееся в объеме S экстрагента, подаваемом на
экстракцию; п — число ступеней экстракционного каскада и /—номер
ступени питания (для схемы, изображенной на рис.204, имеем:
n+l-f = 6+l-3 = 4).
Количество экстрагента в операции а также должно быть
больше S: Sa = fS.
Экстракт Еа из первой воронки разделяют на / частей, одна
из которых поступает на операцию с для отделения экстрагента,
а остальное количество (/— 1 частей) идет на экстракцию в
воронку Ь. Экстракт Еь разделяется на /— 1 частей, одна из
которых освобождается от экстрагента (операция d), а оставшаяся
часть поступает в воронку е и т. д. При этом экстракт, из кото-
413
Рис. 204. Схема имитации непрерывной экстракции с флегмой
в лабораторных условиях.
рого выделен экстрагент, возвращается на экстракцию (см.
схему). После того как при осуществлении схемы будет
воспроизведено полное число укрепляющих ступеней экстракции, весь
экстракт (левый край схемы) после отделения экстрагента
разделяется на флегму и конечный экстракт (продукт процесса).
Таким образом можно имитировать противоточную
экстракцию с флегмой. При этой схеме имитации процесса противоточ-
ной экстракции в операциях первых циклов может вообще не
получаться рафинатного слоя4. В данном случае процесс
имитации осуществляют строго по схеме, не отделяя рафинат при
проведении операций до тех пор, пока не образуются
нормальные количества жидкостей. Число циклов, достаточное для
достижения стационарного состояния, определяется измерением
какого-либо свойства экстракта или рафината, как и при
имитации обычной противоточной экстракции.
414
Схема процесса значительно упрощается, если получать
флегму в результате понижения температуры экстракта7 или
с помощью антиэкстрагента (см. главу VI).
Фракционная экстракция. На рис. 205 приведена схема
имитации шестиступенчатого процесса непрерывной экстракции с
двумя растворителями (экстрагентами). Исходная смесь F (в
которой может содержаться некоторое количество одного из
растворителей) разделяется экстракцией с помощью растворителей AaD.
Если число циклов достаточно для достижения стационарного
состояния, соответствующего условиям работы непрерывнодей-
ствующего каскада, то, согласно данной схеме, продукты,
получаемые при проведении последних операций, будут по всем
свойствам аналогичны продуктам на соответствующих ступенях
процесса непрерывной экстракции. Имитация непрерывного проти-
воточного процесса в лабораторных условиях необходима
главным образом в тех случаях, когда компоненты системы влияют
на распределение друг друга. Схему, изображенную на рис. 205,
широко применяли, в частности, при исследовании процессов
разделения металлов10-12. В этих процессах равновесные кривые
для различных секций каскада часто отличаются друг от друга
из-за различия в рН.
д3[нЗр^9(рд^гд(рфЩаЛ
t=5
t—o
п
'^J^=^,
-D
Рис. 205. Схема имитации процесса экстракции с двумя растворителями
В лабораторных условиях.
415
В простейшем случае, когда растворители А и D взаимно
нерастворимы, коэффициенты распределения постоянны и
исходная смесь состоит только из разделяемых компонентов,
результаты имитации процесса фракционной экстракции легко
определить расчетом. Для этого случая на рис. 205 приведены
выражения, определяющие содержание разделяемых
компонентов в обеих фазах для нескольких циклов процесса.
Количества компонентов в фазах выражены в долях от их
содержания в исходном растворе. Значения величин р и q те
же, что и на рис. 201, a pt и qt, представляют собой доли
разделяемых компонентов, находящихся в экстрагентах А и D, для
соответствующих циклов процесса. Счет циклов для каждой
фазы в данном случае отвечает числу получаемых (выводимых из
процесса) конечных продуктов. Так как процесс экстракции с
двумя растворителями, показанный на рис. 205, несимметричен
(питание подается не в среднюю ступень), числа циклов для
каждой фазы не совпадают друг с другом. Фактор экстракции
принят равным Am/D, где m равно отношению равновесной
концентрации распределяемого вещества в фазе экстрагента А к
его равновесной концентрации в экстрагенте D.
Величины pt и qt' выражаются в виде степенных рядов от
произведения pq, причем для каждой комбинации числа
ступеней п' и п+\ получаются различные ряды, имеющие следующий
вид8:
р, = fn+lh+У2Рд+Уз(рд)2+У4(р^3+ ■ ■■+yiШ1'1] (ix,is)
4t' = ч"' [у[ + Y2 РЧ + Ys (Plf +
+ Y4(V)+.--+Y/''(/>?)''-1] (IX-14)
Выражения для расчета коэффициентов у и у' в общем
случае приведены в ряде работ9' 13' и. Удобнее всего пользоваться
формулами, предложенными Шейбелем 14:
/-1 t-2
У
п' + n + l п' +я+1
М\ V Ml
VI Ml у*
2j N,\(M — Ni)1 ~ 2d
I ZJ Nt\(M — Ni)) jU (Ni—l)\(M-Ni+\y.
t-n'-l t-n'-l
где
n'+n + l n' + n + l
у mi v м±
2u Nj\(M — Nj)\~*~ ^ (Nj—l)\(M — N,+ \)\ <IX'15)
;"-0 ;=0
M = n + 2t — 2 (IX, 16)
Ni = t — l—i(n'+n + l) (IX,17)
Nj = t — n'—l—J(n'+n + l) (IX, 18)
Индексы / и / принимают целые значения от нуля до
верхнего предела суммирования (но не превышают его); при отри-
416
цательном верхнем пределе суммирование не производится.
Чтобы получить выражение для у(,, следует подставить в уравнения
(IX, 15) —(IX, 18) величину п+\ вместо п', п' вместо п+\ и t
заменить на f. Числа ступеней п' и м+1 употребляются здесь
в том же значении, что и в главе VII.
Пример IX-1. Рассчитать коэффициент при члене (pq)1 в выражении для
Ра С=8) при лабораторной экстракции по схеме, изображенной на рис. 205.
Решение. Для данного случая t = 8; я = 3; я' = 3 и я'+я + 1 = 7.
t—\
i —1
n' + n+l
t—1
n' + n + l
t—n' — l
7
J — 3— 1
n' + n + l
t — n' — 2
7
-3 — 2
= 1 (i — 1 для первой суммы)
= 0,86 (/ = 0 для второй суммы)
= 0,57 (] = 0 для третьей суммы)
= 0,43 (у = 0 для четзертой суммы)
п' + п + 1 7
M = n+2t — 2 = 3+2-8 — 2= 17
N( = t — 1 — / (п' + п + 1) = 8 — 1 — 0 = 7 (« = 0)
Nt = t— 1 —; (л'-|-я-|-1) = 8— 1 — 1-7 = 0 (/ = 1)
• я' — 1— j(n' +п + 1) = 8 — 3— 1— 0 = 4
17! 17! , 17!
Nj = t-
Ye
17!
17!
7! (10!) "г" 0! (17!) 61(11!) 4! (13!) "Г" 3! (14!)
= 5373
С увеличением числа циклов уравнения (IX, 13) и (IX,14)
приближаются к геометрическим прогрессиям и коэффициенты
при соседних членах ряда изменяются в постоянном
отношении. Если рассчитать столько членов ряда, что отношение
Уг/yt-i при данной точности расчета станет постоянным, то
легко написать выражение для всего ряда при любом числе
циклов. Степень приближения к стационарному состоянию
можно найти далее, сравнивая pt или qt' с их предельными
значениями, определяемыми из уравнения (VII, 76):
(гп' — 1)ел + 1
/>=!-?,=
вл + 1
(IX, 19)
При имитации асимметричного процесса фракционной
экстракции приближение фаз к стационарному состоянию
происходит с разной скоростью (см. рис. 205). Кроме того,
отдельные компоненты в каждом экстрагенте в разной степени
приближаются к стационарному состоянию в зависимости от
соответствующих им значений фактора экстракции е; скорость
достижения стационарного состояния тем меньше, чем ближе
величина е к единице.
27 Р. Е. Трейбал
417
Фракционная экстракция с центральной подачей исходной
смеси. При большом числе циклов и ступеней расчеты по
приведенным выше уравнениям становятся весьма громоздкими.
Если питание подается в центральную ступень, то степень
приближения к стационарному состоянию проще рассчитать по
следующему приближенному эмпирическому выражению 15:
lg a = lg (1 — 6) = [(0,100л'2 — 0,55л' -f 0,80) е"0'90 — t] X
1 lg[4e/(e + l)2]
X
(IX, 20)
0,940 (л')2—0,040л' — 0,40 1 -f 0,03 е
Это уравнение было проверено для широкого интервала
изменения переменных; величина а определяется по уравнению
а=1—6=-
(IX, 21)
где б — степень приближения к стационарному состоянию.
При имитации фракционной экстракции с центральной
подачей питания обе фазы приближаются к стационарному
состоянию в одинаковой степени для данного компонента, но в
разной степени по отношению к компонентам с различными
факторами экстракции (кроме процессов с симметричным
разделением). Точное число циклов, необходимое для достижения
степени приближения к стационарному состоянию, равной 90%,
можно определить из графика, приведенного на рис. 20615.
Уравнение (IX, 20) совпадает с зависимостями, изображенными
на рисунке, с точностью до долей цикла.
Приближение к стационарному состоянию можно ускорить,
увеличив количество (или концентрацию) исходной смеси в
первых операциях; конечная величина общего количества
разделяемых компонентов, находящихся в процессе, достигается,
таким образом, быстрее. Если г—номер операций, в которых
участвует исходный раствор (для первой операции процесса г=1),
то для нахождения расхода исходной смеси можно пользоваться
следующими зависимостями 16:
при г= 1,2
"л+1 4)
ВА
в.
1
2(гЪ
при г>2
В,
BF \\
(еГЧ0(^-1)(^ + 1Г1
2е,
Г-1
(IX, 22)
(IX, 23)
(4+1 +i)(4-i) L(e+i)2
Величины Bps и Врт представляют собой соответственно
количество компонента В в питании в стационарном состоянии и
количество того же компонента в питании, подаваемом на
операцию г. Аналогичные выражения применимы также для любого
418
компонента. При таком
проведении процесса, чтобы
достигнуть степени приближения к
стационарному состоянию,
равдой 90%, достаточно число
циклов t, лишь незначительно
превышающее число ступеней.
Фракционная экстракция с
центральной подачей исходной
смеси (симметричное
разделение). Под симметричным
разделением компонентов В и С
подразумевается случай, когда
Рв = Яс- При симметричном
разделении (если содержание
обоих растворителей в
исходной смеси равно нулю)
должны соблюдаться следующие
условия (см. главу VII):
4-/
тят
вшс
(IX, 24)
(IX, 25)
Ш
200
ъ
1-J
3
ё
ЧО
«С?
С1
>Ч
-*
ло ци
С1
S
too
so
en
ио
го
10
в
в
<+
^-■^
1 .'
t*
^ <-•
^Ш
~п?Ч\Ь
г=ор,
1
L—
s*o.i;'L
;
1 U 8 12 IB 20 2U 28 32 36
Число ступеней,п+1 или п'
Рис. 206. Степень приближения к
стационарному состоянию при имитации
экстракции с двумя растворителями
в лабораторных условиях для случая
подачи исходной смеси в среднюю
ступень (6 = 0,90)^.
В этом случае оба компонента приближаются к
стационарному состоянию в обеих фазах с одинаковой скоростью и
соотношение компонентов на любой стадии процесса равно их
соотношению в стационарном состоянии. В фазе экстрагента А при
всех значениях t имеем:
Рв, Рв
t_ s_
Рс. ~ Рс„
Рв\п
Те)
И
= е
п + 1
(IX, 26)
Соответственно при изменении числа циклов изменяется
только общее количество компонентов, находящихся в процессе.
Таким образом, при симметричном разделении нет
необходимости доводить процесс имитации экстракции с двумя
растворителями до высокой степени приближения к стационарному
процессу. Несколько полных циклов достаточно для получения
необходимых данных. Так как для фазы экстрагента D при
любом числе циклов t=t' отношение концентраций
распределяемого компонента
Яв \п+1
(IX, 27)
Ч*е Ч
«с, <Ч
Яс )
„п + 1
27*
419
отношение концентраций, полученных после некоторого цикла t,
может служить для оценки селективности (Р):
PBt/Pct /вА,сАк /^
Количество компонента В в растворителях А и D в этом
уравнении выражается величинами ВА и BD, а компонента С —
соответственно величинами СА и CD. При таком методе
нахождения селективности отношение концентраций компонентов в
каждой фазе получается большим, чем при простой однократной
экстракции, и величина р определяется точнее.
Чтобы определить соотношение растворителей, необходимое
для симметричного разделения, или установить расчетом
степень приближения к стационарному процессу при лабораторной
имитации фракционной экстракции, необходимо
предварительно располагать данными о величинах коэффициентов
распределения. Если эти данные отсутствуют, следует пользоваться
методом последовательного приближения, повторяя процесс
лабораторной имитации непрерывной экстракции несколько раз и
используя данные, полученные в первых опытах, для
дальнейшего изучения процесса.
Фракционная экстракция с флегмой. Процесс лабораторной
имитации непрерывной фракционной экстракции с флегмой
можно осуществлять на основе схемы, изображенной на рис. 205,
видоизменив ее в соответствии со схемой для обычной
экстракции с флегмой (см. рис. 204). При имитации процесса с
флегмой определенное количество компонента, используемого в
качестве флегмы, в каждом цикле отделяют от экстрагента и
смешивают со вторым экстрагентом перед тем как последний
вводят в процесс.
Лабораторные экстракторы малых размеров. В
лабораторных многоступенчатых экстракторах непрерывного действия
можно проводить как обычную противоточную экстракцию, так
и экстракцию с двумя экстрагентами. В этих аппаратах
непосредственно осуществляются схемы процессов, показанные в
нижней части рис. 201, 204 и 205. Чтобы применять при работе
аппаратов более крупного масштаба результаты, полученные в
лабораторном экстракторе, необходимо знать достигаемое в
последнем число теоретических ступеней. По этой причине для
лабораторной разработки экстракционных процессов обычно
используют аппараты ступенчатого типа с эффективностью
ступени, близкой к 100%.
За последние несколько лет в литературе описано очень
большое число лабораторных экстракторов малого размера.
Ниже будут кратко рассмотрены лишь некоторые аппараты этого
типа, обеспечивающие, по литературным данным, эффектив-
420
ность ступени, равную 100%. Их размеры, число ступеней и
производительность указаны в оригинальных работах. Эти
аппараты могут быть выполнены с любым числом ступеней, а
изменения их производительности легко добиться изменением
размеров аппарата.
Данные о предельной производительности подобных
аппаратов, приводимые в литературе, следует рассматривать, как
ориентировочные, так как производительность аппарата зависит
от физических свойств разделяемой системы жидкость —
жидкость. При очень малЬ|х расходах Фаз (I мл/мин или менее)
поддержание стационарной подачи исходной смеси и
экстрагента является основной трудностью при проведении эксперимента.
Большая часть описанных в литературе конструкции
миниатюрных лабораторных экстракторов относится к экстракторам
смесительно-отстойного типа (см. главу X).
1. Ящичные экстракторы. В качестве малого лабораторного экстрактора
может служить смесительно-отстойный экстрактор типа «Памп-Микс» (см.
рис. 256). Модель такого экстрактора, описанная впервые Копланом,
Дэвидсоном и Зеброски ", состояла из 16 ступеней и имела следующие размеры:
длину 1220 мм, ширину 280 мм и высоту 255 мм. Объем ступени составлял
2,6-10"3 м3 (2,6 л). Производительность аппарата находилась в пределах
(0,75—1,9) • 10~3 м3/мин. Этот экстрактор был исследован Шафером и
Холмсом 18. Позднее были описаны аппараты типа «Памп-Микс» как больших,
так и меньших размеров. Шестнадцатиступенчатый экстрактор размером
355x203x75 мм с объемом ступени 8 мл имел производительность 0,05—
4 мл/мин 13. Производительность другой модели, еще меньших размеров,
составляла 0,2—1 мл/мин (объем ступени 4 мл) 20. Экстракторы таких
размеров очень удобны для работы с радиоактивными материалами, так как
они требуют минимальной защиты и могут иметь дистанционное
управление.
Описан также ящичный экстрактор другого типа с производительностью
30 мл/мин (20 ступеней, размеры 1120X100X65 мм, объем ступени 0,3 л)21.
Для сильно радиоактивных систем можно рекомендовать пульсационные
экстракторы. Известна модель такого аппарата22, имеющая 82 ступени и
производительность 5 мл/мин.
2. В литературе описаны также малые лабораторные экстракторы
других типов:
а) Экстрактор из полиэтилена, имеющий 56 ступеней и
производительность 130 мл/мин, с перемешиванием плосколопастной мешалкой23.
Приблизительные размеры каждой ступени 255X90X255 мм.
б) Экстрактор, выполненный из стекла, производительностью 20 мл/мин,
с перемешиванием мешалкой, имеющей возвратно-поступательное
движение24. Объем ступени 4—10 мл.
в) Двадцатиступенчатый экстрактор размером 915X150X305 мм,
применимый при давлениях до 2,1 кг/см2 и температурах до 66е С, в котором
перемешивание осуществляется в результате возвратно-поступательного
движения перфорированных тарелок25. Объем ступени 600 мл, производительность
2,3 л/мин.
г) Модификация аппарата Крэга (см. рис. 207), работающая по схеме
непрерывного процесса, осуществляемого в результате прерывистой подачи
исходного раствора и обоих экстрагентов. Экстрактор, состоящий из 17
ступеней, выполнен из стекла и пластмасс; для перемешивания жидкостей
аппарату сообщается качательное движение. Суммарный расход жидкостей
составляет около 500 мл за цикл при 10—12 циклах в час26. Существуют и
421
Другие варианты аппарата Крэга 27>28. Описание Практического применения
лабораторных экстракционных аппаратов подобного типа приведено в
литературе 23> 30.
Для выхода любого указанного выше экстрактора
непрерывного действия на стационарный режим требуется некоторое
время. Конечные продукты, получаемые в непрерывном
экстракторе, приближаются по составу к получаемым в условиях
стационарного процесса, подобно экстрактам и рафинатам
отдельных циклов при лабораторной имитации процесса непрерывной
экстракции. На основе этой аналогии можно рассчитать время,
необходимое, чтобы достигнуть данной степени приближения к
стационарному процессу, и соответственно количества жидкостей,
требующихся для проведения исследования. Суммарный расход
жидкостей для каждого полного цикла равен объемной
производительности одной ступени. Если объем ступени равен v, то,
чтобы получить степень приближения к стационарному
состоянию, соответствующую t циклам, нужно ввести в процесс общий
объем жидкостей, равный tv. Для экстрактора с числом
ступеней, равным п, расход жидкостей составит:
tv t
= — полных объемов аппарата (IX, 29)
пи п г \ < i
Эти рассуждения справедливы также для крупных
(промышленных) экстракторов и могут применяться для оценки
длительности пускового периода. Если экстрактор заполняется перед
началом работы исходной смесью, подвергаемой разделению, то
рафинат, вплоть до достижения высокой степени приближения
к стационарному процессу, следует считать некондиционным и
возвращать в процесс. По этой причине как в лабораторных,
так и в промышленных условиях желательно иметь возможно
меньший объем жидкостей, приходящийся на одну ступень
экстрактора.
Пример IX-2. В лабораторном экстракторе ступенчатого типа, имеющем
10 ступеней (объем каждой 0,5 л), производится противоточное извлечение
из некоторой смеси двух компонентов. Объемное соотношение экстрагента
и исходного раствора равно трем. Коэффициенты распределения
компонентов равны соответственно 0,5 и 1. Суммарный объемный расход жидкостей
составляет 50 мл/мин. Определить время, необходимое, чтобы достигнуть
степени приближения к стационарным условиям, равной 99%.
Решение. Скорость приближения к стационарному состоянию
различна для каждого компонента, а также отличается для экстракта и рафината.
Концентрация компонента, для которого фактор экстракции ближе к
единице, будет медленнее приближаться к значению концентрации при
стационарных условиях. В данном случае факторы экстракции равны
соответственно 3-0,5=1,5 и 3-1,0 = 3,0. При я=10 и s =1,5 для получения 8 = 0,99
необходимо число циклов t=2l (см. график на рис. 203). Следовательно,
£/гс=21/10=2,1 объема экстрактора, что соответствует 2,1-0,5-10=10,5 л
суммарного расхода жидкостей (2,625 л исходного раствора и 7,875 л
экстрагента). Время, необходимое для достижения заданной степени приближения
к стационарным условиям, будет равно 10,5/0,05=210 мин. Для первона-
422
чального заполнения экстрактора необходимо еще 0,5-10=5 л жидкости.
Для компонента с фактором экстракции s=3,0, согласно рис. 203, чтобы
достигнуть степени приближения к стационарным условиям, равной 99%,
потребуется 5,5 циклов, или 5,5-0,5/0,05=55 мин после начала получения
рафината.
В ходе решения предполагалось, что соотношение объемов экстракта и
рафината в каждой ступени равно соотношению расходов экстрагента н
исходного раствора.
ЖИДКОСТНАЯ ЭКСТРАКЦИЯ
В АНАЛИТИЧЕСКОЙ ХИМИИ
Жидкостная экстракция все более широко используется как
один из стандартных методов химического анализа. Наибольшее
применение жидкостная экстракция получила для анализа
металлов. Соединения, способные образовывать хелатные
комплексы, обеспечивают чрезвычайно высокие коэффициенты
распределения и позволяют количественно разделять компоненты
одной-двумя операциями равновесной экстракции. Методы
анализа с помощью таких агентов подробно описаны Моррисоном
и Фрейзером31.
Ниже, однако, основное внимание будет уделено
аналитическим методам, применяемым для разделения на компоненты
сложных природных и синтетических смесей. Данные,
полученные такими методами, часто служат основой для разработки
промышленных процессов.
Методы экстракционного анализа сложных систем весьма
многообразны. Они подробно рассматриваются в работах Хе-
кера32-33. До сих пор наибольшее значение имеют методы
лабораторной экстракции, предложенные Крэгом. Для их
практического применения он разработал ряд остроумных устройств.
Крэгу удалось осуществить некоторые эффективные процессы
разделения на предложенной им аппаратуре, и методы
разделения по Крэгу получили широкое распространение. Литература,
посвященная описанию техники эксперимента при
использовании этих методов и способам интерпретации получаемых с их
помощью данных, очень обширна. В нашу задачу не входит ее
подробное рассмотрение; для более детального ознакомления
с методами Крэга можно рекомендовать работы Крэга34,35, Хе-
кера32 и Вайнерона36.
Наибольший интерес представляет разработанный Крэгом
способ «противоточного распределения». Этот метод применяют
Для разделения определенного количества какой-либо смеси при
помощи многократных операций экстракции, проводимых
обычно в экстракторе Крэга.
В верхней половине рис. 207 показаны конструкции
стеклянных трубок, являющихся основной частью этого аппарата. В
положении а НИЖНЯЯ часть трубки заполняется более тяжелой
423
Рис. 207. Трубки Крэга.
жидкостью до уровня бокового отростка а, над которым
располагается легкая жидкость. Жидкости перемешивают,
переворачивая трубку несколько раз из положения а в положение б и
обратно; при этом компоненты системы распределяются между
фазами и устанавливается равновесие. Затем трубку снова
устанавливают в положение а и дают возможность смеси отстояться;
при отстаивании граница раздела фаз образуется на уровне а.
Далее трубку переворачивают в положение б: в результате
легкая фаза перетекает в трубку с, а тяжелая остается в нижней
части d основной трубки. Если теперь снова перевести основную
трубку в положение а, легкая жидкость будет перетекать в
соседнюю трубку через отросток е и патрубок /.
Экстрактор Крэга состит из большого числа таких трубок
(часто число их составляет несколько сот или более); их
взаимное расположение показано на рисунке 207, г. Каждая трубка
соединяется с соседними с помощью патрубка f и отростка е
(рис. 207,е). Повторение описанного выше цикла операций
приводит к перемещению более легкой жидкости из одной трубки
в другую, в то время как тяжелая жидкость всякий раз остается
в одной и той же трубке. Набор трубок укрепляют на одной
станине, иногда в несколько рядов, один над другим.
Последовательность операций перемешивания, отстаивания и
перемещения жидкости из трубки в трубку можно полностью
автоматизировать с помощью соответствующей механической и
электронной аппаратуры36. Отросток g (на рис. 207 показан закрытым)
служит для заполнения и опорожнения трубок, а также для
соединения одного ряда трубок с другим.
424
п
С помощью аппарата Крэга,
который может работать автоматически и
весьма длительно без всякого контроля,
разделяемую смесь можно
экстрагировать десятки или даже сотни тысяч раз
в зависимости от числа трубок в
аппарате. Трубки Крэга изготавливают
разных размеров с полезным объемом
от нескольких миллилитров до одного
литра.
Схема экстракции по методу Крэга
может быть пояснена с помощью рис.
208. В верхней части рисунка (а)
схематически показан ряд из четырех трубок
Крэга. Каждая из них заполнена до
уровня переточного патрубка тяжелым
экстрагентом D. В первую трубку, кроме
того, налит более легкий экстрагент А.
Жидкости предварительно насыщаются
друг другом, чтобы при экстракции не
изменялись объемы фаз. В первую
трубку помещают также небольшое
количество разделяемой смеси (присутствие
распределяемого вещества показано на
схеме косой штриховкой). Количество
смеси должно быть достаточно малым,
чтобы не вызывать заметного изменения
объемов. После этого систему трубок
несколько раз поворачивают, и смесь,
находящаяся в первой трубке, приходит в
равновесие. Этот ряд операций на рисунке схематически
обозначен кружком под первой трубкой (буква F обозначает
распределяемое вещество).
На рис. 208, б трубки находятся в положении, в котором
происходит перетекание жидкостей: экстрагент А с растворенным
в нем веществом попадает во вторую трубку, а в первую
поступает чистый экстрагент А. Эти операции на рисунке также
показаны схематически с помощью кружков под
соответствующими трубками. В положении, показанном на рис. 208, в,
жидкости в обеих трубках перемешиваются, и цикл операций
повторяется (рис. 208,г и 208,5).
На рис. 208 представлена основная схема проведения
экстракции по методу Крэга. Ее отличительные особенности —
однократное добавление распределяемого компонента (в начале
процесса) и стационарное положение тяжелой фазы в каждой
трубке. Смесь разделяемых веществ движется по ряду трубок,
причем комшоненты смеси с большей величиной фактора
Рис. 208. К описанию
основного метода
экстракции по Крэгу.
425
А.
F
D
PF
pgs
i+pSgi
^\P yO
pq/^\p2 D
^yyy>
.D
4*
D
Sp'
2n3)
kp3ql
t=5
,n
\pq
5pq5\
\5ргЧц
tOptq1*
(Р+Ч)г
i3„3
•,IOp3q
Wp3q3
\ЮрЦг
5pbqs
(Р+Ч)з
yp*q
_^_r P
7p+q)s (p+q)s
Рис. 209. Схема основного метода экстракции по Крэгу (шесть циклов).
ол
экстракции (или коэффициента распределения) продвигаются
быстрее. При достаточном числе трубок компоненты смеси
можно разделить практически полностью. На рис. 209 показана
основная схема процесса экстракции по Крэгу, состоящего из 6
циклов. Первая трубка каждого цикла
°-S{ ' ' ! ' находится на схеме слева. После
проведения всех шести циклов
определяется состав обеих фаз во всех
трубках. Результаты разделения в
процессе Крэга 2 г смеси,
состоящей из равных частей компонентов
В и С, показаны на рис. 210.
Соотношение фаз было выбрано
таким, чтобы разделение являлось
симметричным. Кривые В и С
выражают количества
соответствующих компонентов в обеих фазах
для разных трубок; верхняя
кривая относится к суммарному
содержанию компонентов. Хотя опытные
/ 2 3 и 5 5 точки могут относиться только к
Номер трубки,т трубке определенного номера, как
_.„ Л „ будет показано ниже, целесообраз-
фракции Jo™ (симГ *° «роить по экспериментальным
тричное разделение в шести Данным сплошные кривые (поло-
циклах), жение максимума на кривой для
!
§ о г
I
^ 01
о,
\
-
в-1-е
1в
426
3 5 7 9 11 13 15
Номер трубка, т
компонента С, например,
определяется из кривых В и В + С;
а впадина на линии В + С —
из кривых В и С).
Как видно из рис. 210, в
трубках / и 6 соответственно
содержатся почти чистые
(98,5%-ные) компоненты С и
В. Однако выход их невелик
(16,8%). В остальных трубках
оба компонента находятся в
смеси друг с другом.
Результаты разделения той
же омеси при проведении
пятнадцати циклов показаны на
рис. 211. В этом случае
достигается значительно более вы- Рис. 211. Основной метрд экстрак-
сокая степень разделения. Так, Дни по Крэгу (симметричное разде-
в первых пяти трубках сум- ление в пя™адцати циклах),
марно содержится 58,5% от
первоначального количества компонента С, имеющего чистоту,
равную 99,7%. С увеличением числа циклов и трубок
достигается все более полное разделение компонентов. Схему
экстракции по методу Крэга, показанную на рис. 209 (с шестью
или даже пятнадцатью циклами), можно практически
осуществить с помощью обычных делительных воронок. Однако, если
необходимо провести 100, 1000 или более циклов, возникает
необходимость в аппарате, подобном экстрактору Крэга. Следует
указать, что для хорошего разделения веществ (например, для
получения практически чистых компонентов с выходом, равным
90%) при селективности 0=1,5 требуется 447 циклов, а при
Р= 1,1 —13 540 циклов [см. уравнение (IX, 33)].
Обратимся снова к рис. 209. Количество разделяемых
компонентов (в долях от первоначального количества) в трубках
последнего цикла можно рассчитать, приняв коэффициенты
распределения постоянными. Если величинам р и q придать тот же
смысл, что и ранее [уравнение (IX, 1)], то количество исходной
смеси (в долях от первоначального) в каждой фазе можно
получить, разлагая в ряд величину (p + q)1. Доля общего
количества исходной смеси в обеих фазах каждой трубки выражается
Уравнением (см. рис. 209)
(р + Ч)п
(*-1)!
(т — \)\{t— /и)! \e-fl
1
(IX, 30)
В этом уравнении т — номер трубки и / — число циклов
(m=l для трубки, в которую вводится экстрагент Л, а счет
427
циклов начинается с трубки, в которую вводят исходную
смесь) *. Максимум величины (p + q)m будет достигаться в
трубке, номер которой определяется уравнением
"*тах= (*е^11)е+1~Р('-Ц + 1 (IX, 31)
Из этого уравнения легко определить величину фактора
экстракции и, следовательно, коэффициент распределения.
Уравнение применимо также, если mmax не равно целому числу**.
При очень большом числе трубок (равном 40 или более)
кривые распределения компонентов по трубкам (рис. 211)
становятся симметричными относительно ттах и описываются
функцией нормального распределения:
J_
[2я(* —1)е/(е + 1)»]°'ь ""' 2(*-1)е/(е + 1)«
(Р + 1)г= т_.± „_„. , 1Ч.105«РП„ „_„. , 1., <1Х'32>
где г отсчитывается от mmax (в обе стороны) независимо от того,
является ли ттах целым числом или нет. В случае
многокомпонентных систем приведенные уравнения описывают поведение
каждого компонента. В результате получается семейство
кривых, подобных кривым, изображенным на рис. 211.
При разделении двух компонентов наилучшие результаты
получаются при объемном соотношении фаз, соответствующем
симметричному разделению [уравнения (IX, 24) и (1Л, 25)]. Для
определения этого соотношения фаз, однако, необходимы
предварительные данные о величине коэффициентов распределения.
При разделении слабых кислот или оснований можно
достигнуть симметричного разделения, подбирая рН среды или
буферный раствор (см. главу II), в то время как объемное
соотношение фаз для данного аппарата Крэга можно менять в
сравнительно узких пределах. При симметричном разделении число
трубок (и соответственно циклов), достаточное для получения
данного выхода практически чистых компонентов из бинарной
* В большей части литературных источников счет трубок и циклов
начинают с нуля, а не с единицы. Поэтому получающиеся уравнения несколько
видоизменяются. Чтобы сохранить однообразие обозначений, принятых в
первых разделах этой главы, отсчет mat начинают с единицы.
** Более точное выражение находят дифференцированием уравнения
(IX, 30) по от и приравниванием производной нулю:
L «max—1 J"1" ^— Wmax «шах—1
Если величины {t—тт&х) и mmax велики по сравнению с 0,5 и I
соответственно или если mmai= (f+l)/2, то последние два члена сокращаются и
получается уравнение (IX, 31). Из уравнения (IX, 31а) следует:
„_ mn,ax-l.exp 0,5(f+l)-«n,.x (X>316)
■ Я» max ' («шах — 1) (* — «max)
428
смеси В + С, определяется
следующим уравнением (при
е=0,2—5,0 и t>26) 38:
/== L__ + 1 (IX, 33)
Значения величины Т
приведены в табл. 15.
При этом компонент С
будет находиться в трубках от
1-й до /л-й, определяемой по
уравнению:
+ (V7 - 3) ^(ГГту^/(i +£с)
Таблица 15
Величина Т в уравнении (IX, ЗЗ)38
Выход чистого вещества
%
50
60
70
80
90
95
97,5
99,0
99,73
9,0000
10,5820
12,4221
14,7571
18,3312
21,5742
24,6016
31,0806
36,0000
Соответственно компонент В будет находиться в трубках от
m = mm3T.B — (yT — $)V(t — \)e,Bl(\-\-RB) до m = t. Некоторые
другие полезные зависимости для процесса экстракции по Крэ-
гу выведены Никольсом 39.
При исследовании неизвестной системы методом экстракции
ПО Крэгу можно получить ряд важных сведений. Без проведения
химического анализа фаз в трубках по виду зависимости
суммарного количества исходного вещества от номера трубки
можно судить о том, является ли исследуемое вещество смесью или
чистым соединением (по наличию или отсутствию максимумов
на кривой). Как будет показано ниже, иногда наличие даже
одного пика на кривой распределения свидетельствует о том, что
исследуемое вещество является смесью. Искаженный вид
кривых разделения говорит о взаимном влиянии компонентов на
распределение друг друга или о непостоянстве коэффициентов
распределения. Более подробные сведения об интерпретации
результатов экстракции по методу Крэга можно найти в
литературе 40.
Пример IX-3. Два грамма неизвестной смеси были подвергнуты
экстракции по методу Крэга в пятнадцати циклах. Объемы тяжелого и легкого
растворителей в каждой операции поддерживались одинаковыми. После
окончания процесса из содержимого каждой трубки (обеих фаз вместе) были
отогнаны растворители. Полученные остатки взвешивали (но не
анализировали) . Весовые количества смеси, найденные в каждой трубке, приведены на
рисунке 211 (верхняя кривая; кривые для отдельных компонентов,
показанные иа этом рисунке, в данном случае были неизвестны). Какие заключения
можно сделать на основе этих данных?
Решение. Наличие двух максимумов на кривой В + С, проведенной
через опытные точки (рис. 211), свидетельствует о том, что исследуемая
смесь - состоит по крайней мере из двух (возможно, и из большего числа)
компонентов. Предполагая число компонентов равным двум, можно считать,
что первый максимум находится где-то между т = Ъ и /и = 5,4 (эта
неточность обусловлена тем, что кривые нанесены несколько произвольно, причем
429
точность их проведения увеличивается с возрастанием числа трубок).
Принимая mmax = 5,2 и предполагая, что максимум на кривой распределения по
трубкам первого компонента находится в той же точке, можно найти фактор
экстракции для первого компонента смеси (С).
В данном случае по уравнению (IX, 31) имеем:
(15—1)ег
откуда ес = 0,4285 [уравнение (IX,316)] дает ес=0,448.
Следует отметить, что максимум на общей кривой распределения тем
ближе к соответствующему максимуму на кривой распределения одного из
компонентов, чем полнее происходит разделение смеси. По уравнению (IX, 30)
определяем, какое количество компонента С (в долях от его общего
количества) находится в третьей трубке:
(|5~!)! „.. (^Ц\15-\0А285?-1 = 0,11338
^ + ^-(з-11).(15-3)!(7жГ "(0'4285)
Общее количество смеси в третьей трубке было найдено равным 0,1134 г.
По форме кривой распределения можно полагать, что в третьей трубке
находится лишь очень незначительное количество второго компонента; поэтому
полное количество компонента С в исходной смеси должно быть равно
0,1134/0,11338» 1,000 г. Полученный результат подтверждается, если
рассчитать (p+q)2 и (p+q)i и найти аналогичным образом количество
компонента С в смеси. Определив общее количество компонента С из уравнения
(IX, 30), можно построить кривую распределения его по трубкам.
Повторяя расчет для компонента В, получаем ев = 2,333; количество
компонента В в смеси равно также 1 г. Складывая количества компонентов, рас-
. считанные для каждой трубки, находим точки, соответствующие кривой (В+С).
Полученное совпадение подтверждает сделанное ранее предположение о том,
что смесь состоит из двух компонентов. Так как экстракцию проводили
равными объемами растворителей, найденные значения факторов экстракции
совпадают с величинами коэффициентов распределения. Разумеется, не
исключено присутствие в исследуемой смеси других веществ с коэффициентами
распределения, очень близкими или совпадающими с полученными.
Компоненты смеси с очень близкими коэффициентами распределения можно разделить,
увеличив число проводимых циклов. Присутствие веществ, имеющих
одинаковые коэффициенты распределения для данной пары растворителей, можно
обнаружить с помощью других растворителей (экстрагентов).
Вывод о том, что исследуемое вещество не является чистым соединением,
можно сделать и на основании результатов, получаемых лишь при шести
циклах распределения по Крэгу. В качестве примера рассмотрим рис. 212, на
котором приведены данные о распределении исходной смеси по трубкам,
взятые из рис. 210. Опытные точки соединены на рисунке сплошной линией
(прогиб на рис. 210 по этим данным определить нельзя). Максимум на кривой
получен при mmax=3,5. Если предположить, что исследуемое вещество
состоит только из одного компонента, то по уравнению (IX, 316) получаем
(так как число трубок мало, лучше пользоваться этим уравнением):
_ 3,5 — 1 0,5(6 + 1) —3,5 _ 1Л
6 ~ 6 — 3,5 ехр (3,5 — 1) (6 — 3,5) - 1,и
Для т=3 из уравнения (IX, 30) находим:
^^ Ш6-1 „ «чЭ-1-.03125
</> + ?>»- (зЛ^-^Ш'"'^
Поскольку общее количество исследуемого вещества равно 2 г, в третьей
трубке должно быть 2-0,3125=0,625 г. Рассчитывая таким образом количе-
430
ствй вещества в других трубках, получаем
нанесенную на рисунок пунктирную линию.
Расхождение между опытной и расчетной
кривыми свидетельствует о том, что
исследуемое вещество не является чистым
соединением.
Для анализа сложных смесей
используют также другие методы,
иногда более эффективные, чем
описанный выше метод противоточ-
ного распределения Крэга.
Применяют, в частности, метод
экстракции по схеме, подобной
приведенной на рис. 205, но с однократным
вводом испытуемой смеси в начале
процесса (метод «поочередного
вывода фаз» из процесса). Метод
«двойного вывода фаз» проводят по
аналогичной схеме, но при этом
справа и слева от трубки, в
которую вводится исходный раствор, располагается одинаковое
количество трубок. Можно также использовать преимущества,
связанные с применением флегмы. Разработан аппарат для
распределения веществ между тремя жидкими фазами41. Все эти
методы дают возможность разделять очень сложные природные
и синтетические смеси. Такими методами удается выделить
различные гормоны, антибиотики, стерины и алкалоиды, т. е. такие
соединения, для выделения которых трудно применить какие-
либо другие способы.
Номер трубка, т
Рис. 212. Анализ смеси
экстракцией по Крэгу (к примеру
ПИЛОТНЫЕ И ПОЛУПРОМЫШЛЕННЫЕ УСТАНОВКИ
Исследования экстракционных процессов в более крупном
масштабе проводятся на пилотных установках и имеют целью:
а) определение технологических показателей работы
аппаратов, б) изучение процесса в течение сравнительно длительного
периода работы аппаратуры, в частности выяснение вопросов
стабильности экстрагента, влияния накапливающихся в нем
примесей, а также коррозии конструкционных материалов; в)
получение достаточного количества конечных продуктов для
определения расходов исходных материалов на их производство и для
выяснения перспектив сбыта продукции.
Пилотные испытания должны проводиться на экстракторе,
геометрически подобном тому, который будет использоваться в
промышленном масштабе, особенно если необходимо
определить такие характеристики аппарата, как эффективность
ступени, высота, эквивалентная теоретической тарелке, или высота
единицы переноса, а также предельные производительности.
431
Размер аппарата (любого Типа из описанных в главах X и
XI) обычно выбирают, по экономическим соображениям,
наименьшим из тех, которые могут обеспечить получение
необходимых данных. Однако размер экстрактора не должен быть
слишком мал, так как иначе трудно перейти к аппарату промышленных
размеров, ввиду того что вопросы моделирования экстракторов
еще недостаточно разработаны. Проще других моделируются
смесительно-отстойные и различные колонные экстракторы с
механическим перемешиванием. Поэтому испытывать
экстракторы данного типа можно, используя аппараты относительно
малых размеров. При испытании насадочных колонн тип и
размер насадки в пилотной и промышленной установках должны
быть одинаковыми. Вопрос о минимальном размере насадки и
об отношении размера насадки к диаметру колонны рассмотрен
в главе XI.
Материал насадки, тарелок, мешалок и т. п. в пилотной
установке должен быть тем же, что и в будущем промышленном
аппарате. Это небоходимо как для изучения коррозионной
стойкости материалов, так и для получения надежных данных,
касающихся рабочих характеристик аппарата. Последнее
обусловлено тем, что степень смачиваемости материала аппарата той
или иной фазой влияет на ряд факторов (размер капель,
удерживающую способность и др.), оказывающих в свою очередь
значительное влияние на эффективность процесса экстракции.
Гидравлику экстракторов исследуют обычно в аппаратах со
/стеклянными стенками. Однако следует учитывать, что в
аппаратах малых размеров пристенная область имеет относительно
' большие размеры, в связи с этим смачиваемость стенок
аппарата может существенно влиять на движение дисперсной фазы.
При пуске экстрактора рекомендуется сначала заполнить
аппарат сплошной фазой и затем установить необходимую
скорость ее подачи. После этого в случае механических
экстракторов включают мешалку и лишь тогда начинают медленно
подавать дисперсную фазу, постепенно (чтобы избежать
преждевременного захлебывания) увеличивая ее расход до необходимой
величины. При исследовании эффективности экстрактора, т. е.
при определении к. п. д. ступени, высоты единицы переноса и
т. п., пробы для анализа следует отбирать только после того,
как установлен стационарный режим работы аппарата. Способ
приблизительной оценки времени, необходимого для достижения
стационарных условий, дан в примере IX-2.
Для колонных экстракторов, в которых значительные
объемы жидкостей находятся в верхней и нижней отстойных зонах,
стационарный режим устанавливается значительно дольше, чем
в экстракторах других типов. О достижении стационарных
условий работы аппарата свидетельствует постоянство во времени
концентраций получаемых конечных продуктов. Материальный
432
1
\
\
баланс процесса должен сходиться в пределах принятой или
допустимой степени точности. Приближение к стационарным
условиям более надежно определяют сопоставлением степени
извлечения экстрагируемого компонента из рафината со скоростью
возрастания его концентрации в экстракте. Однако для
малых экстракторов такой баланс трудно составить точно. Опыты,
в которых баланс по экстрагируемому компоненту расходится
более чем на 5%, учитывать не следует.
При исследовании эффективности экстракторов равновесная
и рабочая линии процесса иногда сходятся. Это возможно в
экстракторах с большим числом ступеней (или эквивалентных
большому числу ступеней), если фактор экстракции сильно
отличается от единицы. В таком случае фаза, выходящая с
одного из концов экстрактора, практически находится в
равновесии с жидкостью, поступающей в аппарат. К такому проведению
процесса часто стремятся для того, чтобы увеличить степень
извлечения рафината. Однако при этом становится практически
невозможным определение (с помощью рабочей линии,
построенной по концентрациям конечных продуктов экстракции) числа
теоретических ступеней, полученных в процессе.
Для определения числа теоретических ступеней в этом
случае следует отбирать пробы фаз по высоте аппарата, находить
на основе их анализов соответствующие им точки на рабочей
линии процесса и рассчитывать число теоретических ступеней
иа высоте аппарата, заключенной между этими точками.
Пробы, отбираемые из аппарата, должны представлять собой
средний состав жидкости в данном сечении экстрактора. Лучше
отбирать несколько проб из нескольких точек поперечного
сечения аппарата.
Обычно пробы отбирают простым открыванием крана
пробоотборника; при этом отбирается некоторое количество обеих фаз
экстрагируемой системы. В период отбора проб и до начала
анализа процесс массообмена между фазами продолжается.
Поэтому возникает задача определения действительных концентраций
в колонне по составам проб, концентрации в которых не точно
совпадают с соответствующими концентрациями в аппарате.
Проиллюстрируем один из возможных способов решения
этой задачи42 с помощью рис. 213, на котором показана схема
обычной противоточной экстракции; буквами хну обозначены
концентрации экстрагируемого компонента соответственно в ра-
финате и экстракте. Конечные концентрации (xF, r/4) и (хп, ув)
характеризуют рабочую линию процесса. Если отобранную в
сечении К двухфазную смесь привести в состояние равновесия и
определить состав равновесных фаз (xsi и ysi, точка J на
рис. 213), то, зная объемное соотношение фаз в образце, можно
найти точку на рабочей линии процесса, соответствующую
рабочим концентрациям в сечении К колонны. При этом, очевидно,
28 Р. Е, Трейбал
433
Исходная ■
смесь
Акг/ч
U к Экстракт,
y'J в кг/ч
Экстрагент **
Вкг/ч ув
К
и
/ls1yxSi
■"МъМг
ЦУЛ/Г'5) ^V~^
(xF;y,)
Наклон
As,/Bs,
Наклон+А/В
Наклон-ASz/8Sz
Ш
кг/кг-А
Рис. 213. Отбор проб из двухфазной смеси (полная взаимная
нерастворимость растворителей):
/ — равновесная линия; 2 —рабочая линия;/ —исходная смесь, А кг/ч; // — экстракт, В кг/ч;
/// —рафинат, А кг/ч; IV — экстрагеит, В кг/ч.
не обязательно, чтобы соотношение фаз в пробе соответствовало
соотношению фаз в колонне. Определив таким же образом
положение точки на рабочей линии, отвечающей, например,
сечению М в колонне, можно далее установить показатели
эффективности аппарата вдали от концов колонны, где равновесная
и рабочая линии близко подходят друг к другу.
Метод определения эффективности колонн в случае заметной
взаимной растворимости растворителей показан на рис. 214. На
рисунке приведена схема противоточной экстракции исходной
смеси F экстрагентом В; рабочая и равновесная линии почти
совпадают друг с другом на том конце колонны, на котором
поступает исходная смесь. Определив равновесные составы фаз в
отобранной из аппарата двухфазной смеси, получаем точки U
и Z, лежащие на концах хорды равновесия треугольной
диаграммы. Если весовые количества фаз в пробе равны U и Z, то
средний состав в ней (точка М) находят по уравнению:
X
uxclJ+zxi
CZ
см'
U + Z
(IX, 34)
Действительные рабочие концентрации W и V в колонне
легко установить, проводя прямую из полюса О через точку М.
Следует отметить, что с помощью пробоотборников,
сделанных из пористого материала, предпочтительно смачиваемого той
или иной жидкостью, можно отбирать пробы только одной
фазы. Такие пробоотборники изготавливают либо из стекла
(лучше смачиваемого водными растворами), либо из пластических
434
материалов (например, тефлона), обычно хорошо
смачиваемых органическими жидкостями.
Сравнительно длительные испытания пилотных
экстракционных установок, предпринимаемые для изучения устойчивости
работы аппаратуры, дают возможность определить также потери
экстрагента (в результате испарения, разложения, потерь с ра-
финатом и пр.). При этом необходимо учитывать количество
растворителя, отбираемого на анализы. Кроме того, следует
иметь в виду, что потери при пилотных испытаниях завышены,
гак как относительная величина различных утечек для
установки малого размера значительно больше, чем для
промышленной.
Экстракция в полупромышленном масштабе. В некоторых
случаях возникает необходимость разделения экстракцией
сравнительно небольших количеств смесей, которое можно
проводить в установках полупромышленного масштаба. Если
необходимо постоянно экстрагировать небольшие количества раствора
одного и того же состава, следует выбрать, какой процесс будет
более целесообразным: непрерывная экстракция, проводимая
при очень малых скоростях фаз, или периодическая экстракция,
осуществляемая при больших скоростях фаз. Если экстракцию
проводят в экстракторе колонного типа, предпочтительнее
непрерывная схема работы. Это обусловлено тем, что при
остановке экстракционных колонн жидкости осаждаются под.
действием сил тяжести, и при последующем пуске колонны
необходимо определенное время для достижения стационарных
условий работы экстрактора и получения кондиционных продуктов.
При остановке экстракторов смесительно-отстойного типа
(см. главу X) жидкости сохраняют свои концентрации по
ступеням до последующего пуска аппарата. Поэтому экстракторы
Концентрация С
В рафинат ё, вес. доли
Рис. 214. Отбор проб из двухфазной смеси (ограниченная взаимная
растворимость растворителей):
1—равновкная линия; 2 — рабочая линия.
28* 435
- /
s^
в
Рис. 215. Схема периодической
многоступенчатой противоточной
экстракции:
1 — экстрактор; 2 — стадия регенерации
растворителя; 3 —сборник конечного
продукта процесса (рафината).
данного типа можно
эксплуатировать только одну или две
смены в сутки или останавливать на
выходные дни. Окончательный
выбор того или иного режима
работы экстракционного
оборудования должен производиться
на основе экономических
соображений. Непрерывная экстракция
в аппаратуре относительно
малых размеров требует меньших
капитальных затрат, но больших
эксплуатационных расходов, чем
периодическая экстракция в
аппаратах больших размеров.
В некоторых производствах
небольшого масштаба может
оказаться целесообразным
применять многоступенчатую проти-
воточную периодическую
экстракцию. Состав исходной смеси в
таком процессе непрерывно изменяется, как при периодической
ректификации (получившей более широкое применение, чем
соответствующий процесс экстракции). Для эффективного
разделения компонентов исходной смеси методом противоточной
периодической экстракции необходимо, чтобы объем фазы
рафината, находящейся в экстракторе, был малым по сравнению с
общим объемом исходной смеси. Поэтому в таком процессе
лучше всего использовать механические экстракционные колонны
или центробежные экстракторы, так как их удерживающая
способность невелика (см. главу XI).
Некоторые схемы противоточной периодической экстракции
описаны Праттом43. На рис. 215, а изображена схема
периодической экстракции, которую целесообразно использовать в тех
случаях, когда концентрация экстрагируемого соединения (или
соединений) сравнительно мала и экстрактор должен работать,
по существу, как укрепляющая секция экстракционной колонны
с флегмой. Экстрагент, являющийся легкой фазой, смешивается
с исходным раствором в нижней части колонны и поднимается
вверх.
Выходящий сверху колонны экстракт Е отделяется от
растворителя (экстрагента) дистилляцией или отпаркой, после чего
экстрагент возвращается в колонну. Отделенный от
растворителя экстракт Е поступает в сборник, из которого периодически
подается в экстрактор, где играет роль тяжелой фазы (рафи-
нат). Если необходимо, экстракт можно непрерывно выводить
из процесса, при этом количество флегмы должно меняться.
436
Если в исходной смеси содержится
несколько экстрагируемых компонентов, они
могут отбираться в виде фракций
экстракта Е. Первые (во времени)
фракции экстракта будут обогащены
наиболее легко экстрагируемым компонентом.
Схема процесса принципиально не
изменится, если экстрагентом будет служить
более тяжелая фаза. Экстракт в этом
случае выходит из нижней части
колонны, а исходный раствор и экстрагент
поступают сверху. Весь процесс
аналогичен процессу периодической
ректификации.
Если исходный раствор обогащен
экстрагируемым веществом, то процесс
периодической противоточной
экстракции можно проводить по схеме, в
которой экстрактор будет являться как бы
исчерпывающей секцией колонны (рис.
215,6). Схема такого процесса ясна из
рисунка. Если извлекается смесь
веществ, то рафинат можно отбирать по
фракциям. В этом случае первые
фракции рафината будут обогащены
наиболее трудно экстрагируемым
компонентом. На рисунке показана схема процесса
для случая, когда экстрагент является легкой фазой. Если
экстрагентом служит более тяжелая жидкость, то направление
движения фаз меняется на обратное. Этот процесс аналогичен
процессу так называемой обращенной периодической ректификации.
На рис. 216 показана схема противоточной экстракции с
двумя экстрагентами. Приведенная схема является периодическим
вариантом процесса диссоциативной экстракции, описанной в
главе VII. Этот процесс применяют для разделения смеси
органических кислот с разными константами диссоциации,
растворенных в органическом растворителе. Исходный раствор
смешивают с небольшим количеством водного раствора щелочи, после
чего производят разделение (декантацию) смеси. Декантацию
можно осуществлять также не в отдельном аппарате, а в
верхней части колонны. Кислоты после нейтрализации экстрагируют
водой в виде солей. Для лучшей очистки получаемого продукта
часть кислот, не подвергшаяся нейтрализации, вымывается из
водной фазы в многоступенчатом экстракторе поднимающимся
по колонне экстрагентом.
В экстрактор, показанный в нижней части рис. 216, подают
экстрагент и - минеральную кислоту. Этот экстрактор служит
вода
Рис. 216. Схема
периодического процесса
диссоциативной экстракции:
Л 2 — экстракторы; 3 — стадия
регенерации экстрагеита;
4 — смеситель; 5 — отстойник;
/ — водный раствор щелочи;
// — смесь органических
кислот; /// — водный раствор
натриевых солей н кислот;
IV — фракции органических
кислот в растворителе;
V — водный раствор H2SO,;
VI — водный раствор Na2SO(;
VII — экстракт.
437
для выделения из водного экстракта органических кислот,
которые переходят в экстрагент и далее частично отбираются в
качестве конечного продукта. Продукты процесса можно
отбирать фракциями, каждая из которых будет обогащена одной из
кислот, содержащихся в исходной смеси. Вейс26 показал
возможность применения такого процесса для разделения
органических оснований.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
А—исходный раствор (противоточная экстракция);
экстрагент (фракционная экстракция, или экстракция с двумя
растворителями); расход исходного раствора, кг/сек;
количество исходного раствора, кг (периодическая
экстракция).
В — экстрагент (противоточная экстракция);
экстрагируемый компонент (экстракция с двумя растворителями);
расход экстрагента В, кг/сек; количество экстрагента
В, кг (периодическая экстракция).
С — экстрагируемый компонент, расход компонента С,
кг/сек (непрерывная экстракция); количество
компонента С, кг (периодическая экстракция).
D — экстрагент (экстракция с двумя растворителями);
расход экстрагента D, кг/сек (непрерывная экстракция);
количество экстрагента D, кг (периодическая
экстракция).
Е— экстракт, расход экстракта Е кг/сек (непрерывная
экстракция); количество экстракта Е, кг (периодическая
экстракция).
F — исходный раствор; расход исходного раствора F, кг/сек
(непрерывная экстракция); количество исходного
раствора F, кг (периодическая экстракция).
f — число ступеней питания.
К — количество раствора F, растворяющееся в 5 кг
экстрагента.
М, N — величины, используемые для определения у или у'
[уравнения (IX, 3) — (1Х,6) и (IX.15) — (IX, 18)].
т — номер трубки в экстракторе Крэга (счет трубок
начинается с /п=1 для трубки, в которую вводится
экстрагент) .
mmax—номер трубки в экстракторе Крэга, отвечающей
максимуму на кривой распределения концентрации по
трубкам.
m — коэффициент распределения: отношение концентрации
в экстракте к концентрации в рафинате при
равновесии или отношение концентрации в экстрагенте А к кон-
центрации в экстрагенте D при равновесии.
П. — число ступеней противоточной экстракции; число
ступеней в исчерпывающей секции колонны (не считая
ступени питания) при фракционной экстракции.
п' — число ступеней в укрепляющей секции колонны (считая
ступень питания) при фракционной экстракции.
р — доля экстрагируемого компонента, переходящая при
равновесии в экстракт (противоточная экстракция) или
в экстрагент А (экстракция с двумя растворителями).
ps — доля экстрагируемого компонента (от его
первоначального количества), находящаяся при достижении
стационарного состояния в экстракте (противоточная
экстракция) или в экстрагенте А (экстракция с двумя
растворителями) .
pt — доля экстрагируемого компонента в экстракте
(противоточная экстракция) или в растворителе А
(экстракция с двумя растворителями) после t циклов.
q—доля экстрагируемого компонента, .переходящая при
равновесии в рафинат (противоточная экстракция) или
в экстрагент D (экстракция с двумя
растворителями) .
qs — доля экстрагируемого компонента (в долях от его
первоначального количества), находящаяся при
достижении стационарного состояния в рафинате
(противоточная экстракция) или в экстрагенте D (экстракция с
двумя растворителями).
qt — доля экстрагируемого 'компонента в рафинате
(противоточная экстракция) или в экстрагенте D
(экстракция с двумя растворителями) после I циклов.
R — рафинат, расход рафината, кг/сек (непрерывная экс~
тракция); количество рафината, кг (периодическая
экстракция) .
г — число ступеней, в которые подается исходный раствор
(в схемах лабораторной имитации непрерывной
экстракции); номер трубки, считая от /nmax (экстрактор
Крэга).
t, t' — число циклов в схемах имитации непрерывной
экстракции; число циклов в экстракции по методу Крэга
(начиная от t= 1).
v — объем ступени.
X — концентрация в рафинате, вес. доли.
х—концентрация в рафинате или исходном растворе, кг/кг.
у — концентрация в экстракте, кг/кг.
8— фактор экстракции, тВ/А (противоточная экстракция)
или mA/D (экстракция с двумя растворителями или
экстракция по методу Крэга).
$в, с — селективность растворителя в отношении разделения
компонентов В и С. -
439
Y, y'— коэффициенты в уравнениях (IX, 2), (IX, 13) и (IX, 14),
6 - степень приближения к стационарному состоянию.
(1=1—6.
ЛИТЕРАТУРА
1. Е. J а п t z e n, Das fractionierte Distillieren und das fractionierte Verteilen,
Dechema Monographien V № 48. Verlag Chemie, Berlin, 1932.
2. G. T. Barry, Y. Sato, L. С Craig, J. Biol. Chem., 174, 209 (1948).
3. K. A. Allen, W. J. McDowell, J. Phys. Chem., 64, 877 (1960).
4. E. G. Scheibel, Ind. Eng. Chem., 49, 1679 (1957).
5. E. G. Scheibel, Ind. Eng. Chem., 46, 16 (1954).
6. S. Watanabe, K. Morikawa, J. Coc. Chem. Ind. Japan, 36, 585B
(1933).
7. L. Alders, Liquid — Liquid Extraction, 2nd ed., Elsevier Publishing
Company, Amsterdam, 1959.
8. S. Stene, Ark. Kemi Mineral Geol., 18H, № 18 (1944).
9. P. L. Auer, C. S. Gardner, U. S. Atomic Energy Comm., MTA-7
(1953); Ind. Eng. Chem., 16, 39 (1954).
10. J. Bochinski, M. Smutz, F. H. Sped ding, Ind. Eng. Chem., 50,
157 (1958).
11. E. L. Koerner, M. Smutz, H. A. Wilhelm, U. S. Atomic Energy
Comm, ISC-802 (1956); Chem. Eng. Progr., 54, 9, 63 (1958).
12. H. С Peterson, G. H. Beyer, A. I. Ch. E. Journ., 2, 38 (1956).
13. E. L. Compere, A. L. Ryland, Ind. Eng. Chem., 46, 24 (1954).
14. E. G. Scheibel, Ind. Eng. Chem., 46, 43 (1954).
15. D. F. Peppard, M. A. Peppard, Ind. Eng. Chem., 46, 34 (1954).
16. E. G. Scheibel, Ind. Eng. Chem., 44, 2942 (1952).
17. B. V. С op Ian, J. K. Davidson, E. L. Zebroski, U. S. Atomic
Energy Comm., AECU-2639 (1953); пат. США 2646346, 1953; Chem. Eng.
Progr., 50, 403 (1954).
18. A. C. Schafer, J. H. Holms, Chem. Eng. Progr., 52, 201 (1956).
19. H. W. Alter, J. W. Codding, A. S. Jennings, U. S. Atomic Energy
Comm., KAPL-961 (1953); Ind. Eng. Chem., 26, 1357 (1954).
20. J. L. Bloom, U. S. Atomic Energy Comm., UCRL-4529 (1955); Chem.
Eng. Progr. Symp. Ser., 52, № 19, 183 (1956).
21. M. W. Davis, T. E. Hicks, T. Vermeulen, Chem. Eng. Progr., 50,
188 (1954); U. S. Atomic Energy Comm., ANL-4266 (1948).
22. W. G. Matters, E. E. Winter, L. G. Cor net t, R. С Cairns,
В. М. Mitchel, Atomic Energy of Canada, Ltd., CRCE-980 (1960).
23. L. К n a p p, R. Schoenherr, J. В a r g h u s e n, M. Smutz, U. S.
Atomic Energy Comm., UC-4 (1958); Ind. Eng. Chem., 51, 639 (1959).
24. G. P. Wall, Atomic Energy Research Establ. (G. Brit), Rept. CE/R 1730
(1955).
25. M. R. Fenske, R. B. Long, пат. США 2667407, 1954 г.; Chem. Eng.
Progr., 51, 194 (1955).
26. D. E. Weiss, Ind. Chemist, 31, 230 (1955).
27. M. Verzele, F. A 1 de r we i r e 1 d t, Nature, 174, 702 (1954).
28. F. A. Von Metzsch, Chem.-Ingr.-Tech., 31, 262 (1959).
29. J. A. Barker, A. F. Beech am, Austral. J. Chem., 13, 1 (1960).
30. H. A. Wilhelm, R. A. Foos, U. S. Atomic Energy Comm., ISC-458
(1954); Ind. Eng. Chem., 51, 633 (1959).
31. G. H. Morrison, H. Freiser, Solvent Extraction in Analytical
Chemistry, John Wiley and Sons, Inc., New York, 1957.
32. E. H e с k e r, Verteilungsverfahren im Laboratorium, Verlag Chemie, Wein-
heim/Bergstr., 1955.
33. E. НескегД. Allemaun, Angew. Chem., 66, 557 (1954),
440
34. L. С Craig et al, Anal. Chem., 21, 500 (1949); 22, 1346 (1950); 23,
1326 (1951).
35. L. С Craig, D. Craig, in «Techiques of Organic Chemistry», A. Weiss-
berger (ed.), 2nd ed., v. HI, pt I, p. 149—332, Interscience Publishers,
Inc., New York, 1956.
36. M. V i g п е г о n, Fractionnements par Solvents, Vigot Freres, Paris, 1954.
37 E. J. Perry, W. H. Weber, Anal. Chem., 26, 498 (1954).
38. E. Hecker, Z. Naturforsch., 8B, 77 (1953).
39. P. L. Nichols, Anal. Chem., 22, 915 (1950).
40. M. С S h e p s, R. H. P u r d y, L. L. E n g e 1, J. L. О п с 1 e y, J. Biol.
Chem., 235, 3033 (1960).
41. H. L. Meltz er, J. Biol. Chem., 233, 1327 (1958).
42. R. E. Treybal, Ind. Eng. Chem., 47, 2435 (1955).
43. H. R. С Pratt, Chem. Eng. Sci., 3, 189 (1954).
СМЕСИТЕЛЬНО-ОТСТОЙНЫЕ
ЭКСТРАКТОРЫ
ОБЩИЕ ПОЛОЖЕНИЯ
If ак только ограниченно смешивающиеся
жидкие фазы, не находящиеся в
равновесии, приводятся во взаимодействие друг
с другом, начинается самопроизвольная
диффузия веществ (входящих в состав
жидкостей), стремящаяся привести систему в
равновесие. На этом основано разделение
веществ экстракцией. Осуществляя
промышленный процесс
экстракции,необходимо создать условия, способствующие
повышению скорости процесса, чтобы
экономические затраты были наименьшими.
Скорость массообмена между двумя
жидкими фазами описывается уравнением
N = КАЬс
где N— скорость массообмена;
К — общий коэффициент массопере-
дачи;
А — межфазовая поверхность;
Ас — разность концентраций (движущая
сила процесса).
С возрастанием каждой величины,
стоящей в правой части уравнения, скорость
экстракции повышается. Процесс
экстракции ускоряется, например, при
диспергировании одной жидкости в среде другой
жидкости, так как при этом увеличивается
межфазовая поверхность, через которую
происходит перенос вещества.
Коэффициент массопередачи
определяется диффузионными сопротивлениями
сплошной и дисперсной фаз, а также
возможным сопротивлением на поверхности
раздела фаз и сопротивлениями,
обусловленными медленной химической реакцией.
Диффузионные сопротивления фаз
являются причиной возрастания К с увеличением
448
уровня турбулентности в каждой фазе. Повышение уровня
турбулентности способствует переносу вещества в результате
турбулентной диффузии, которая является более быстрым процессом,
чем молекулярная диффузия.
Конечные и начальные разности концентраций Ас
определяются материальным балансом процесса. Промежуточные
значения движущей силы процесса необходимо поддерживать на
максимально возможном уровне с помощью аппаратуры
соответствующей конструкции. При заданной скорости
массопередачи объем аппаратуры должен быть достаточным для
обеспечения необходимого времени пребывания фаз. В общем случае
идеалом является наибольшая скорость массопередачи в
наименьшей по размерам аппаратуре. Однако минимально
возможный размер аппаратуры не всегда экономически оптимален.
Многочисленные виды экстракционных аппаратов можно
разбить на две основные группы.
1. Экстракторы, состоящие из отдельных ступеней, в
которых жидкости смешиваются; при этом происходит экстракция,
затем жидкости разделяются и отдельно выводятся из ступени
(смесительно-отстойные экстракторы). Экстракционные
аппараты этого типа рассмотрены ниже. Для осуществления
многоступенчатых процессов, описанных в главах VI и VII,
отдельные ступени необходимо соединять в виде каскадов.
2. Экстракторы, в которых жидкости, движущиеся
противотоком по отношению друг к другу, непрерывно взаимодействуют.
При этом отдельный аппарат может быть эквивалентен такому
числу ступеней, какое необходимо или практически возможно.
Экстракторы,этого типа рассмотрены в главе XI.
При разработке экстракционного процесса нужно выбрать
не только тип экстракционного аппарата (из указанных двух
групп), но и наиболее экономичный размер его и определить
оптимальные условия работы экстрактора. Эти вопросы
рассмотрены в главе XII.
Ступень смесительно-отстойного экстрактора состоит из
одного или нескольких аппаратов, в которых исходные жидкости
перемешиваются, стремясь достигнуть состояния равновесия
(стадия смешения). Образующиеся продукты механически
отделяются друг от друга (стадия отстаивания) и выводятся из
ступени. Процесс можно проводить периодически или
непрерывно. В периодическом процессе один и тот же аппарат служит
одновременно для смешения и отстаивания. В непрерывном же
процессе обычно (хотя и не всегда) смешение и отстаивание
производят в разных аппаратах. Поэтому экстракторы такого
типа называются смесительно-отстойными. При перемешивании
443
одна из жидкостей, как правило, диспергируется на мелкие
капли (дисперсная фаза), распределенные в другой жидкости
(сплошная фаза). Дисперсной фазой может быть как легкая,
так и тяжелая фаза, а также экстрагент (экстракт) и раствор,
подлежащий экстрагированию (рафинат).
В большей части процессов экстракции, имеющих
промышленное значение, для разделения требуется больше одной
ступени. Любое число ступеней можно соединить в виде каскада
с перекрестным или чаще с противоточным движением контак-
тируемых жидкостей (см. главы VI и VII).
Достоинства смесительно-отстойных экстракторов. Смеси-
тельно-отстойные экстракторы являются старейшими
экстракционными аппаратами. Несмотря на то что многие экстракторы
колонного типа (см. главу XI) были сконструированы с целью
создать аппараты более совершенные, чем смесительно-отстой-
ные экстракторы, последние обладают рядом достоинств,
обеспечивающих до сих пор их широкое применение в
промышленности. Эти достоинства рассмотрены ниже.
1. По сравнению с противоточными экстракторами
непрерывно-контактного типа (в которых фазы движутся обычно под
действием сил тяжести) смеситёльно-отстойные экстракторы
характеризуются тем, что степень дисперсности или интенсивность
турбулентного движения, достигаемая в результате
механического перемешивания, не ограничена явлением «захлебывания»,
хотя и в этих экстракторах следует избегать чересчур тонкого
диспергирования, приводящего к образованию стабильных
эмульсий.
2. Смеситёльно-отстойные экстракторы могут работать при
любом соотношении фаз, которое в смесителе можно
поддерживать независимо от соотношения потоков в противоточном
каскаде в целом.
3. Экстракторы этого типа менее других чувствительны к
присутствию взвешенных твердых частиц в жидкости.
4. Обычно применяемые горизонтальные
смеситёльно-отстойные экстракторы пригодны для работы в производственных
помещениях малой высоты.
5. К горизонтальному смесительно-отстойному экстрактору
в случае необходимости всегда можно добавлять
дополнительные ступени.
^ 6. После остановки непрерывнодействующих смесительно-
отстойных экстракторов отбор продуктов можно начинать
немедленно, так как стационарные концентрации фаз
сохраняются. Это невозможно в случае колонных экстракторов, при
остановке которых жидкости распределяются под действием сил
тяжести вдоль всей высоты колонны. Поэтому при пуске
возникает необходимость в возврате (рециркуляции) целевых
продуктов до достижения установившегося состояния. Данное пре-
444
имущество смесительно-отстойных экстракторов особенно важно
в мелких производствах, когда непрерывная работа аппаратуры
в течение суток или семидневная работа в неделю экономически
нерентабельна.
7. Почти всегда можно достигнуть высокой степени
приближения к равновесию (высокой эффективности ступени), что
облегчает расчет и гарантирует необходимую эффективность
аппарата.
8. Возможен достаточно надежный переход от малых
(модельных) экстракторов к промышленным, по крайней мере для
аппаратов простейших конструкций.
9. В тех процессах, в которых остановка всего производства
нежелательна, обычно предусматривается установка запасного
оборудования на случай аварии. Механические колонные
экстракторы для обеспечения непрерывной работы необходимо
дублировать, что приводит к большим затратам. При
использовании смесительно-отстойного каскада достаточно иметь лишь
одну дополнительную ступень.
Недостатки смесительно-отстойных экстракторов. Такие
экстракторы имеют определенные недостатки, устранение которых
и являлось целью создания колонных аппаратов. Эти
недостатки заключаются в следующем:
1. Обычные горизонтальные смеситёльно-отстойные
экстракторы занимают большую площадь.
2. Необходимость в перемешивающем устройстве с приводом
для каждой ступени часто является причиной относительно
больших капитальных затрат и эксплуатационных расходов.
3. При наличии радиоактивности обрабатываемых жидкостей
усложняется эксплуатация смесительно-отстойных аппаратов.
Если при а- и р-радиоактивности обычно можно применять
механическое перемешивание, то при интенсивном у-излучении
использование такого перемешивания затрудняет эксплуатацию
оборудования.
4. Обычно необходимо перекачивание из ступени в ступень
одной, а иногда и обеих жидкостей, хотя в принципе можно
организовать движение фаз под действием сил тяжести.
Объем отстойников часто велик, что может приводить не
только к большим затратам на оборудование, но и к
значительным расходам, связанным со стоимостью растворителя (экстр-
агента). Нельзя, однако, заранее утверждать, что объем
смесительно-отстойного экстрактора или его стоимость будет больше
или меньше объема или стоимости эквивалентной
экстракционной колонны loro же назначения. Это зависит от эффективности
ступени каждого из аппаратов. Механические экстракционные
колонны будут иметь в общем случае относительно меньшие
объемы.
44S
LR D[
Xg Xg X% Xt
Концентрация В ршринате, мол. дали
Рис. 217. Способы выражения эффективности ступени:
/ — равновесная линия; 2 — рабочая линия; / — непрерывный процесс (одна ступень);
// — периодический процесс.
Методы оценки работы экстрактора (эффективность
ступени). Ступень экстрактора называется «идеальной» или
«теоретической», если выходящие из нее жидкости находятся в
химическом равновесии. Степень приближения к равновесию,
достигаемая в действительности, называется эффективностью
ступени. Ее можно выражать различными способами.
Зависимости между различными способами выражения
эффективности ступени и коэффициентами массопередачи легко
получить для разбавленных растворов и несмешивающихся
растворителей, т. е. для случаев, когда количество каждой фазы
при экстракции не изменяется. Обратимся к рис. 217. Если
начальные концентрации (в мол. долях) рафината и экстракта
равны соответственно xY и у\, а конечные концентрации — х2 и У2,
то при скоростях L моль • сект1 • м~2 для непрерывного процесса
(или при количествах жидкостей, равных L моль для
периодического процесса) количество экстрагированного вещества
составляет:
Nb
■■ LR (*, — х2) = LE (У2 — j/j)
(X,l)
Это выражение является уравнением рабочей линии процесса
(линия АВ на рис. 217). Наклон рабочей линии равен —LR/LE-
Если бы жидкости пришли в равновесие, их концентрации на
выходе из ступени равнялись бы хе и уе и соответствовали
точке С на кривой равновесия. Количество экстрагированного
вещества в этом случае составило бы:
ЬЛХ1 — хе)= LE{ye — yi)
(Х,2)
446
Следовательно, эффективность ступени можно выразить
следующим образом1:
„ NE Х\—х2 у2 — У\ АВ
Ne x\—xe Уе — У1 АС
(Х,3)
Эффективность ступени по Мэрфри2 является мерой
приближения одной жидкости к равновесию с другой жидкостью при
действительной конечной концентрации последней. Так, если
экстракт выходит с концентрацией у2, в то время как конечной
концентрации рафината х2 соответствует равновесная
концентрация у\, эффективность ступени по Мэрфри, считая по фазе
экстракта, выражается зависимостью
У2 — У1 DF
p _ У1 — У\ _ "' /у 4ч
ЕМЕ ' -"S= <X'4)
Аналогично эффективность по Мэрфри, считая по фазе
рафината, равна:
EMD = ?^4 = 4A (X,5)
'MR'
хЛ—х0 О А
Оба метода оценки эффективности не зависят.от способа
выражения концентраций.
Для смесительно-отстойного экстрактора, состоящего из
нескольких ступеней, общая эффективность Е0 равна отношению
числа идеальных (теоретических) ступеней к числу
действительных ступеней, необходимых для достижения данной степени
извлечения *:
Для данной ступени Е, ЕМе и Emr не равны друг другу и не
равны Е0, если ступень является частью смесительно-отстойного
каскада. Зависимости между этими величинами можно получить
только в том случае, если равновесная линия — прямая (т. е.
при постоянном коэффициенте распределения, см. табл. 16 на
стр. 448).
При одновременном экстрагировании нескольких веществ,
например при экстракции двумя растворителями, эффективность
ступени для каждого вещества может быть различной. В
тройных системах типа системы, показанной на рис. 105, изменения
концентраций компонентов (рабочую линию) можно
приближенно определить, поворачивая прямой угол около точки М на этом
рисунке и отмечая точки пересечения с пунктирными линиями,
выражающими концентрации в массе жидкости. Практическая
* При полном отстаивании в каждой ступени. Влияние уноса из ступени
& ступень учитывается уравнениями (X, 53) и (X, 54).
447
Зависимости между различными способами выражения
эффективности ступени
Таблица 16
Характеристика процесса
Зависимости
Одна ступень
Периодический или
непрерывный процесс
£жя(1 + <0 Емр(\ + г)
1 + *ЕМЕ ■ * + EMR
Е = 1 _ e-NOEQ+*) = j _ g-N0R (e+l)/e
е£ е£,
£..„ = -
ME
MR »-£+l 1+£Д(И(в-1)
Л1Д
F =
МЕК
Е Е.
Периодический процесс
ОЕ
е(1_£) + 1 -£жя(1-е) + е
— 1п(1 — £) 9 /£ai,e /С^лив
1 + е
К,
N
OR'
ОЕ "Е • Е
— eln(l — Е) 9 _ K'pavQ K^avQ
1+е —
9.
Кг
Непрерывный процесс
(в отсутствие
продольного перемешивания)
Непрерывный процесс
(полное
перемешивание в фазе рафината)
Непрерывный процесс
(полное
перемешивание в фазе экстракта)
Непрерывный процесс
(полное
перемешивание в обеих фазах)
OR "r • r
— In (1 — Е) К'ечН KjAH
N =
ОЕ 1_|_8
V,
лг =
OR
Е ■ Е
= — eln(l — Е) _ К'паН KRaH
Noe = -^-Eme) = NorI*
ME
nor = -^^-emr) = *Noe
= \_e-KRaH'VR
E.
MR e
N
ME
OE \—E
ME
"or \ — e
MR
448
Продолжение табл. 16
Характеристика процесса
Каскад ступеней
Перекрестный ток *
Противоток *
Зависимость
P i lg(l+e-e£)
0 lg(e+l)
lg[l+£^(e-l)] 1е[1+£жд(1/е-1)]
0 lge lgl/e
,gf !+e-£ 1
gLl+e(l-£)J
0 lge
* См. сноску на стр. 447
трудность заключается в том, что положения этих кривых
неизвестны, так что к. п. д. ступени для различных веществ в
общем случае нельзя связать друг с другом.
Зная Е, ЕМе или EMR отдельных ступеней, можно
графически определить необходимое число реальных ступеней смеси-
тельно-отстойного экстрактора (рис. 218). Для этого строят
кроме равновесной и рабочей линий так называемую псевдо-
Концентрация В рафшате, мол. доли
Рис. 218. Определение действительного числа ступеней противоточного
смесительно-отстойного каскада:
^—равновесная линия; // — псевдоравновесная линия; /// — рабочая линия каскада
(наклон LRjLEy, IV — рабочая лнния одной ступени [наклон (— LRjLE^.
29 Р. Е. Трейбал
449
fet t
Уг
x
dx
dy
3 f
i
X3
равновесную линию, связывающую
действительные концентрации на выходе из
ступеней. Например, точка В этой кривой
(см. рис. 218) получена из соотношений:
£ =
ВА
~СА
Е =JL
ME Dp
Е =™-
MR ан
Т
У,
Рис. 219. Одна
ступень в непрерывном
процессе (режим
полного вытеснения).
Найдя ряд таких точек для различных
участков рабочей линии с учетом (если это
необходимо) изменения эффективности
ступени и построив по ним кривую, легко
определить число реальных ступеней.
В табл. 16 приведены зависимости
между Е0 и эффективностью одной ступени
в случае прямой линии равновесия и
одинаковой эффективности всех ступеней
каскада для противоточной экстракции и
экстракции с перекрестным током.
Коэффициенты массопередачи и число единиц переноса.
Связь между эффективностью ступени и коэффициентом
массопередачи или числом единиц переноса можно выразить в
простой форме только в том случае, если коэффициент
распределения является величиной постоянной.
1. Непрерывный процесс. Прежде всего необходимо сделать
определенное допущение о характере изменения концентраций
в ступени. Предположим, что аппарат работает в режиме
идеального вытеснения и продольное перемешивание отсутствует.
В этом случае (рис. 219) жидкости движутся в одном
направлении от входа к выходу и на длине пути dH изменения
концентраций фаз составляют соответственно dx и dy. Тогда из
уравнений (VIII, 43) и (VIII,45) следует:
NE = КЕасЕ> ср_ (/ - у)ср Н = KRacRt ср (х - л:*)ср Н =
= К'Еа (у* - y)QpH = K'Ra (x — х*)срН
Для тех же условий имеем:
dy У2 — У1 КЕаН К'ЕаН
'ое-
(У*-У),
(Х,7)
(Х,8)
ср.
N.
OR
? dx jKj — х2
— J х — х* ~ (х — х*) ~
KRaH
K'RaH
(Х,9)
В этих уравнениях средняя движущая сила — средняя
логарифмическая из начальной и конечной величин движущей силы.
Решая совместно уравнения (X, 7) — (X, 9) и уравнения (Х,3) —
450
(X, 5), определяющие эффективность ступени, получаем
зависимости, приведенные в табл. 16 (см. стр. 448).
В аппаратах с мешалками часто одна из жидкостей (или обе)
идеально перемешивается; в полностью перемешанной фазе
концентрации постоянны по всей ступени. Например (рис. 219),
если экстракт полностью перемешан, то его концентрация во
всех точках ступени равна у2. В таком случае
(Х,10)
(X, 11)
1а0~ЕмЯ) (X, 12)
Аналогично можно вывести зависимости для других случаев:
полностью перемешанного рафината или полностью
перемешанных экстракта и рафината; эти зависимости приведены в
табл. 16.
2. Периодический процесс. Пусть в сосуде объемом v
находятся ЬЕ моль экстракта и LR моль рафината. Время
пребывания их в сосуде составляет 0. Тогда
dNE LPdy , , .
-Ж=-ЧГ=«Ху-у) (Х.13)
При постоянном коэффициенте распределения, используя
уравнение (X, I), получаем
NE = LE (y2 - ух) = K'EavQ (у* - </)ср. (X, 14)
Совместное решение этого уравнения с уравнениями для
эффективности ступени дает зависимости, приведенные в табл.16.
Последние можно получить и другим путем, с помощью
понятия «времени единицы переноса», которое определяется
следующими уравнениями 3:
гг dy Kpavd 9
"°*-/т*=7—ЕГ~ v (Х,15)
Ух
Р dx K'Dav% 9
"о*= J Т^ = -Т^- = ^ (Х,16)
Классификация аппаратуры. Оборудование смесительно-от-
стойных экстракторов можно классифицировать следующим
образом:
dx
*
х — х2
хх = х2 = y2l m = const
LRdx = K'Ra (x — x2\ dH
- K'RaH lnfl *l~xA
LR \ ATj — X2 1
29»
461
/. Смесители
А. Сосуды с перемешиванием
1. Механические
2. Пневматические
Б. Проточные, или трубные смесители
1.. С механическим перемешиванием
2. Без перемешивания
/Л Отстойники
А. Немеханическне
1. Гравитационные
2. Центробежные (циклоны)
Б. Механические (центрифуги)
///. Оборудование для улучшения отстаивания
1. Коагуляторы
2. Разделительные мембраны
3. Электростатические коагуляторы
В принципе любой смеситель с отстойником могут
обеспечить одну полную ступень в непрерывном процессе. В
периодическом процессе смешение и отстаивание производят обычнр в
одном аппарате.
АППАРАТЫ С МЕШАЛКАМИ
Процесс перемешивания можно осуществлять механическими
мешалками с вращательным или возвратно-поступательным
движением, с помощью газа, а также покачиванием или
встряхиванием всего аппарата. В процессах жидкостной экстракции
применяют в основном различные вращающиеся мешалки; реже
используют пневматическое перемешивание. Встряхивание имеет
значение только для небольших лабораторных аппаратов.
В дальнейшем будут рассмотрены вопросы, связанные с
определением наилучших форм и размеров аппарата, выбором
способа перемешивания, конструкции внутренних устройств, а
также с величиной потребляемой энергии, которую необходимо
затратить для достижения наибольших скоростей экстракции
при наименьших расходах. Однако почти все, что известно об
аппаратах с мешалками, получено в результате изучения
процессов, не относящихся к жидкостной экстракции (процессов
перемешивания растворимых жидкостей или мелкоизмельченных
твердых частиц, взвешенных в жидкости). Поэтому результаты
рассматриваемых ниже исследований (за исключением данных,
которые, как будет указано, относятся к перемешиванию
двухфазных жидких смесей) следует использовать применительно к
процессам жидкостной экстракции с большой осторожностью.
Смесители. Обычно смеситель представляет собой
вертикальный цилиндр с гладкой внутренней поверхностью (иногда с
отражательными перегородками на стенках) и предпочтительно
сферическим днищем. Менее эффективны плоскодонные смеси-
482
Рис. 220. Устройство аппара- Рис. 221. Секцнонирован-
тов с мешалками: ный смеситель для не-
а — смеситель с вертикальными отра- прерывного процесса:
жательнымн перегородками; б —сме- / — турбина; 2 —перегородка;
ситель с закрытой турбиииой мешал- 3 —вращающаяся тарелка,
кой; / — отражательная перегородка;
2 —турбина; 3 — статор.
тели прямоугольного поперечного сечения, в которых могут
образовываться застойные зоны4. Тем не менее для
обеспечения возможно большей экономичности конструкции некоторые
смесительно-отстойные экстракторы (ящичного типа) имеют
поперечное сечение прямоугольной формы (см. рис. 256).
Вопрос о минимальном отношении высоты жидкости в смесителе
к его диаметру, необходимом для хорошей циркуляции
жидкостей, подробно рассмотрен в работе Бизеля с сотр. 5Ду_чщие^е-
зультатьидетигаютсяв тех случаях, когда высота столба жид- '--
кости в аппарате ^>авна его диаметру или несколько больше.
В периодическом процессе объем аппарата равен объему
загрузки плюс объем свободного пространства над зеркалом
жидкости (высота свободного пространства обычно не меньше 0,3 м
в открытых аппаратах, но в малых смесителях может быть
меньше). В непрерывном процессе объем аппарата, занимаемый
жидкостью, равен v = QQ, где 0 — время пребывания,
определяемое скоростью экстракции и ожидаемой эффективностью
ступени. Перемешивание в непрерывных условиях можно
осуществлять как в аппаратах со свободным объемом над
поверхностью перемешиваемой жидкости, так и в полностью
заполненных аппаратах.
Примеры конструкций аппаратов для перемешивания
(смесителей с мешалками) показаны на рис. 220 и 221. Для
перемешивания чаще всего используют мешалки, изображенные на
рис. 222. Обычно их крепят на вертикальном валу, проходящем
через крышку аппарата.
483
Мешалки. Пропеллерные, турбинные, плосколопастные
мешалки и различные модификации мешалок этих типов обычно
крепят на вертикальном валу, установленном вдоль оси
аппарата. Пропеллерные мешалки, укрепленные на валу,
проходящем через боковую стенку смесителя (применяемые для
перемешивания взаимно растворимых жидкостей в крупных
аппаратах), в жидкостной экстракции не используют. Пропеллерные
мешалки, создающие осевые потоки жидкости, применяют для
перемешивания маловязких жидкостей, причем желательно
устанавливать их таким образом, чтобы они создавали потоки,
направленные вниз. Турбинные и плосколопастные мешалки в
аппаратах с отражательными перегородками (рис. 220, а)
создают радиальные потоки. В отсутствие отражательных
перегородок эти мешалки вызывают вращательное движение жидкости
вокруг центрального вала.
В жидкостной экстракции наиболее широко применяют
открытые турбинные мешалки различных видов, в частности турбины
а б в
г д
Рис. 222. Мешалки:
а —пропеллер типа морского вннта; б — центробежная турбина; в —турбина с наклонными
лопастями; г — плосколопастиая турбнна; д — плосколопастная дисковая турбина.
4S4 ,
с плоскими радиальными лопастями, турбины с лопастями,
изогнутыми в сторону, противоположную направлению вращения,
и спиральные турбины. Известны также турбинные мешалки
со стреловидными лопастями. Однако применять их для
перемешивания в системах жидкость — жидкость не рекомендуется.
Расположение мешалки в смесителе. Обычно мешалки, за
исключением пропеллерных мешалок в аппаратах без
перегородок, устанавливают на вертикальном валу, проходящем по
оси аппарата. Мешалки устанавливают на расстоянии от днища
смесителя, равном диаметру мешалки, хотя имеются указания6,
что при непрерывной экстракции в смесителе с отражательными
перегородками наиболее целесообразно устанавливать мешалки
на расстоянии от днища, равном двум третям высоты столба
жидкости в аппарате.
При непрерывной экстракции в аппаратах без перегородок
расположение мешалки по высоте аппарата имеет
относительно малое значение. Однако следует отметить, что этому
вопросу применительно к процессам жидкостной экстракции до
сих пор уделялось мало внимания. В периодическом процессе
перемешивания положение мешалки оказывает влияние на то,
какая из жидкостей будет диспергироваться (см. ниже).
Воронкообразование при перемешивании. В случае
перемешивания жидкости мешалкой, установленной на центральном
валу аппарата без отражательных перегородок, и при наличии
в аппарате свободной поверхности жидкости (т. е. когда
жидкость не полностью заполняет аппарат) после достижения
некоторого числа оборотов мешалки в перемешиваемой жидкости
образуется центральная воронка7. Образования воронки не
происходит только при перемешивании очень вязких жидкостей
(>20 000 сп), которые очень редко встречаются в процессах
жидкостной экстракции. Образование воронки приводит к
засасыванию мешалкой воздуха и перемешиванию его с
жидкостью, что нежелательно при экстракции и сопровождается
падением эффективности перемешивания.
Как неоднократно отмечалось в литературе (см., например,
статью Раштона и Костича8), в условиях образования воронки
невозможно достичь геометрического и кинематического
подобия при перемешивании данной жидкости в двух аппаратах
разных размеров. Это затрудняет моделирование, т. е.
выяснение условий перенесения данных, полученных для смесителей
небольшого размера, на крупные аппараты.
Геометрическое подобие аппаратов с перемешиванием
достигается при равенстве отношений их сходственных размеров. Для
геометрически подобных систем кинематическое подобие можег
быть достигнуто при геометрическом подобии структуры
потоков, в том числе ламинарных и турбулентных линий тока, и
характеризуется соответственно простыми соотношениями для
характеристик массопередачи. Это в свою очередь требует
соответствия отношений сил, действующих в двух системах, или
динамического подобия9.
При наличии воронки структура потоков зависит как от
критерия Рейнольдса (Re=ZVp/\x), так и от критерия Фруда
(Fr = V2/Zg). Следовательно, в двух геометрически подобных
аппаратах при перемешивании одной и той же жидкости (т. е.
при одинаковой кинематической вязкости) скорости находятся
в отношении Vi/V2=Z2/Z1 при равенстве критериев Рейнольдса
и в отношении V\/V2= (Zi/Z2)°>5 при равенстве критериев Фруда.
Таким образом, равенства обоих критериев для одной и той же
перемешиваемой жидкости достичь не удается.
Предотвращение образования воронки. Образования
воронки можно избежать с помощью одного из указанных ниже
методов.
1. Эксцентричное расположение пропеллера. Воронка не
образуется, если вал с укрепленным на нем пропеллером
установить под углом и сбоку от оси аппарата4'8. Такую
установку мешалки применяют главным образом в смесителях малых
размеров.
2. Применение перегородок. Отражательные перегородки
представляют собой узкие плоские пластины, устанавливаемые
вдоль стенок аппарата, как показано на рис. 220, а. Перегородки
предотвращают образование воронки. Их можно приваривать
или прикреплять другими способами непосредственно к стенке
аппарата. В некоторых случаях, особенно при наличии в
перемешиваемой жидкости твердых примесей, которые могут
накапливаться за перегородками, а также при перемешивании в
стеклянных или пластмассовых аппаратах для упрощения
конструкции перегородки можно устанавливать на некотором
расстоянии, равном 12—40 мм от стенок. Четыре симметрично
расположенные у стенок вертикальные перегородки с шириной,
равной Vio или Vi2 диаметра аппарата, достаточны для
устранения вращения жидкости как единого целого в аппарате и
предотвращения "образования воронки; при этом установка
дополнительных перегородок или увеличение ширины перегородок
нецелесообразно 10.
Обычно вертикальные отражательные перегородки имеют
высоту, равную высоте столба жидкости в аппарате. Однако, по
данным Нагата11, изучавшего периодический процесс
перемешивания взаимно нерастворимых жидкостей, отражательные
перегородки следует устанавливать на некотором расстоянии от
стенки аппарата и погружать в жидкость на глубину, равную
половине диаметра мешалки.
Применяют также кольцевые перегородки, состоящие из ряда
лопастей, укрепленных в кольце, расположенном на одном
уровне с турбиной и образующем статор закрытой турбинной
m
мешалки (рис. 220,6). При этом достигается более высокая
степень локальной турбулентности и вблизи мешалки создаются
более значительные срезывающие усилия, чем в случае
установки в аппарате отражательных вертикальных перегородок.
В связи с этим не следует использовать кольцевые перегородки
при перемешивании жидкостей, склонных к образованию
стабильных эмульсий.
Установка отражательных перегородок в аппаратах с
турбинными или лопастными мешалками приводит к изменению
характера движения потоков перемешиваемой жидкости:
вращательное движение ее сменяется радиальным. При этом у
стенок аппарата жидкость движется вверх и вниз от плоскости
мешалки. В результате достигается большая однородность
перемешиваемой жидкости, чем при отсутствии перегородок.
Вертикальное движение жидкости особенно желательно при
периодической экстракции, так как оно препятствует расслоению
жидких фаз. Однако при непрерывной экстракции такое движение
способствует продольному перемешиванию, которое, вообще
говоря, нежелательно.
3. Полное заполнение аппарата жидкостью. Образование
воронки исключается, если отсутствует свободная поверхность
жидкости, т. е. при полном заполнении аппарата. В этом случае
можно достигнуть как геометрического, так и кинематического
подобия при моделировании процесса для одной и той же
системы12 и в то же время уменьшить влияние продольного
перемешивания в непрерывном процессе6.
Движение жидкостей через смеситель. При перемешивании
в непрерывном процессе, проводимом в аппаратах, подобных
показанному на рис. 220, обе жидкости могут поступать в
смеситель снизу и выходить через штуцер (не показанный на
рисунке) в верхней части аппарата. При перемешивании жидкости
и газа лучше вводить газ вблизи лопастей мешалки. Возможно,
что и при жидкостной экстракции имеет смысл вводить таким
образом одну из жидкостей, по крайней мере в аппаратах с
перегородками, однако этот вопрос до сих пор не
исследовался.
Жидкости можно выводить из смесителя также через
штуцер, расположенный в боковой стенке аппарата на уровне
мешалки (турбинного типа). Напор, сообщаемый мешалкой ра-
диально движущейся жидкости, можно использовать для
перемещения жидкости в отстойник. Особенно целесообразно
располагать выходной штуцер сразу за вертикальной перегородкой
(считая по направлению потока). В таком случае одна из
жидкостей может поступать в смеситель в верхней части аппарата,
другая — снизу. Устанавливая входные штуцера перед
перегородкой, можно получить перепад давления, достаточный для
транспортирования жидкостей в аппарат из соседней ступени.
457
Как правило, полное перемешивание жидкостей (см.
уравнения (X, 11) и (X, 12) и зависимости, приведенные в табл. 16]
уменьшает движущую силу при непрерывном процессе.
Желательным является поршневой поток, приблизиться к которому
можно при секционировании аппарата, как показано на рис. 221.
В аппарате, изображенном на рисунке, обе жидкости поступают
в аппарат снизу и удаляются из его верхней части. При полном
перемешивании жидкости в вертикальном направлении в
каждой секции аппарата характер движения потока тем ближе
к поршневому, чем больше число секций13.
Конструктивные детали смесителей. В большей части
смесителей (за исключением аппаратов очень большой высоты) не
обязательно устанавливать опорные и промежуточные
подшипники для предотвращения чрезмерной вибрации и биения вала
мешалки. В случае необходимости на мешалке можно
установить стабилизаторы.
В аппаратах, работающих под избыточным давлением, в
месте прохода вала мешалки сквозь крышку (или стецку)
аппарата нужно устанавливать сальники с набивкой или
механические уплотнения. Обычно не допускают соприкосновения
сальника с жидкостью, оставляя некоторое свободное пространство
в верхней части аппарата. Корпус сальника лучше помещать
снаружи аппарата (рис. 223, а).
В закрытых аппаратах, работающих при полном
заполнении и под атмосферным давлением, нет необходимости
устанавливать сальник. В них вал мешалки может свободно
проходить через штуцер в крышке аппарата (рис. 223,6); при
этом испарение летучих жидкостей можно предотвратить при
помощи жидкостного затвора (рис. 223,0).
Привод мешалок обычно осуществляется от
электродвигателя. Небольшие мешалки можно присоединять непосредственно
к электродвигателю, работающему при 1150, 1725 и 3600 об/мин.
Для мешалок, промышленных аппаратов такие числа оборотов
Рис. 223. Устройства для ввода вала в аппарат:
а —сальник; б ~открытая втулка; в —гидравлический затвор.
458
слишком велики. Поэтому обычно число оборотов редуцируют,
чаще всего с помощью зубчатого редуктора, соединенного
непосредственно с валом электродвигателя. Иногда применяют
ременные или цепные передачи. В некоторых случаях для
приводов мешалок используют бесступенчатые редукторы. Применяют
также пневмоприводы, пригодные для мощностей, не
превышающих 1,5 квт.
В промышленной практике используют переносные мешалки
с приводом, предназначенные для открытых резервуаров; эти
мешалки крепят непосредственно к корпусу аппарата и
снабжают электродвигателями мощностью от долей киловатта до
4 квт. Мощности электродвигателей для стационарно
устанавливаемых мешалок колеблются от долей киловатта до ~400 квт.
В большей части случаев каждую ступень многоступенчатых
экстракторов снабжают индивидуальным приводом. Лишь
иногда, главным образом для небольших многоступенчатых сме-
сительно-отстойных экстракторов, используют
трансмиссионный привод.
Для создания направленного движения жидкости к мешалке
коаксиально с турбинными и пропеллерными мешалками внутри
аппарата устанавливают вертикальные цилиндры-диффузоры14,
как показано, например, на рис. 261 (стр. 511).
Содержимое смесителя нагревается или охлаждается с
помощью пара или воды, направляемых в рубашку аппарата.
Поверхность теплообмена в этом случае относительно невелика.
Большую поверхность теплообмена можно получить, помещая
около стенок аппарата прямые трубы или змеевики, причем
прямые вертикальные трубы могут одновременно служить
отражательными перегородками. Внутренние устройства смесителей
и их влияние на величину энергии, потребляемой мешалкой,
описаны в литературе4. Данные о теплообмене в смесителях, за
единственным исключением15, имеются только для однофазных
жидких систем.
Выбор дисперсной фазы. Обычно трудно, хотя и возможно,
диспергировать жидкость, составляющую более 75% общего
объема жидкой смеси.
Периодический процесс. В смесителях с отражательными
перегородками жидкость, в которую до начала перемешивания
погружена мешалка, при перемешивании, как правило,
оказывается сплошной фазой 12>1в'17. По наблюдениям Лэйти 1г, при
расположении мешалки на уровне межфазовой поверхности и
при равных объемах перемешиваемых жидкостей вода является
сплошной фазой в случае перемешивания с вязким моторным
маслом и становится дисперсной фазой при перемешивании с
керосином.
В опытах Роджера 1в> ", проводившихся в тех же условиях,
•при малой скорости вращения мешалки диспергировалась орга-
459
ническая фаза. Однако при большой скорости происходило
обращение фаз, которое наступало быстрее при больших
величинах отношения Ар/рс- При Лр/рс = 0,6 было невозможно
диспергировать в воде органическую фазу мешалкой, установленной на
межфазовой поверхности. При диспергировании воды в опытах
Роджера во многих случаях образовывались двойные эмульсии
(в крупных каплях дисперсной фазы заключены мелкие
капельки сплошной фазы). В других работах образование двойных
эмульсий наблюдалось при больших величинах отношения
|Wm*c 18- Явление обращения фаз с изменением числа оборотов
мешалки отмечалось и для аппаратов без отражательных
перегородок 19.
Непрерывный процесс. В непрерывном процессе любую фазу
можно сделать дисперсной, если ее объемная доля не
превышает указанного выше предела. Для этого следует заполнить
аппарат сплошной фазой, включить мешалку и затем вводить
жидкость, которую необходимо диспергировать.
Равномерность перемешивания. В данном случае под
равномерностью перемешивания подразумевается постоянство
соотношения сплошной и дисперсной фаз в разных частях аппарата,
а не постоянство размеров диспергированных частиц.
Аппараты без перегородок (периодический процесс). По
данным Нагата20, в случае перемешивания мешалкой с четырьмя
плоскими лопастями (с?м = Г/3) в аппарате без отражательных
перегородок минимальное число оборотов мешалки, при
превышении которого исчезают слои перемешиваемых жидкостей,
можно определить по уравнению
4,68 /цс \о,ш /Ар\о,2б
Миллер и Манн19 изучали перемешивание различными
мешалками воды и органических жидкостей (систем с высоким
межфазовым натяжением) в сосудах без отражательных
перегородок. В условиях опытов происходило образование воронки.
Однородность перемешивания определялась посредством
отбора проб из различных частей аппарата и характеризовалась
индексом перемешивания —средним отношением (в %)
объемной доли фазы, находящейся в пробе в меньшем количестве, к
объемной доле той же фазы во всем аппарате (индекс
перемешивания при полной равномерности перемешивания равен
100%). Хотя полученные значения индекса перемешивания
несколько отличались для мешалок различных типов,
приближенно можно считать, что диспергирование, близкое к
равномерному, наступает при вводе мешалкой в жидкости энергии,
равной 0,2—0,4 кет на 1 м3 объема жидкости. Некоторые опытные
данные, полученные в этой работе, приведены на рис. 224; из
460
3
I
U0
го
1
Т,м
о 0J+5
о 0,3
ь0/5
^
-rf-
г/
/Л
1
-о
них следует, что энергия, вво- too
димая на единицу объема пе- ^
ремешиваемой жидкости, оче- з? л/7
видно, может служить основой <§
для моделирования мешалок.
Более общая корреляция
для цилиндрических аппаратов
со сферическим днищем была
предложена И. С. Павлушен-
ко и А. В. Янишевским 2I.
Однако получаемые по ней
значения минимальных чисел
оборотов и мощностей,
необходимых для равномерного
перемешивания, не согласуются с
указанным выше выводом,
следующим из работы Миллера и
Манна, и представляются
заниженными.
Аппараты с отражательными перегородками (непрерывный
процесс). При периодической экстракции доля объема,
занимаемого дисперсной фазой, в среднем равна объемной доле
дисперсной фазы в загрузке аппарата. Это равенство не соблюдается в
непрерывном процессе, если количество энергии, вводимой
мешалкой, недостаточно велико.
4# во wo гоо шо юоо
„ Мощность мешалки, вт/м3
Рис. 224. Степень однородности,
достигаемая при перемешивании
(система керосин — вода;
плосколопастная мешалка; фд = 0,5;
<</ж/7- = 0,333)'».
si
op
| 0.6
Vy
уЬ,9гмЗ/ч /
/ о,игмз/ч
===-
/
/
DICI
r\ 1
IT
0.1
0ft 1,0 4 !0 U0 !00
Мощность мешалки, Вт
Рис. 225. Удерживающая способность непрерывно действующего
смесителя с отражательными перегородками [жидкости движутся
прямотоком снизу вверх; открытая турбинная мешалка с плоскими лопастями-
данные Сиволда, см. Ind. Eng. Chem., 53, 597 (1961)].
Г=Я=0,Зл; dM = 0,\SM; Q = 0,4-0,9 м'/ч; KD/KC = 0,33_1,85; Л-снстема кероснн
(дисперсная фаза) —водный раствор СаС12 (р_ = 1340; Др = 532; 0 = 38,3 • Ю-3; ц. =
==4,78 • Ю-3); В-система керосин (днслерсиая фаза)-водный раствор СаС12
(Рс = 1170; Др = 367; 0 = 36,8-10 3; цс = 1,73 ■ КГ8); С-керосии (дисперсная фаза)-
вода(рс = 1000. Др=191; а = 39,6 - 10~3; цс = 0,91 . ИГ8); D-вода (сплошная фаза) -
смесь нзобутанола и керосина Л}с = 990; Др=179; 0 = 13,9 • Ю-3; ц. = 1,14 . 10_3Y
В—изобутанол (дисперсная фаза) — вода(р_ = 968; Др —102; о = 4-10-3; р. =1,14. 10_3\
461
На рис. 225 приведены данные об удерживающей
способности, полученные в ааппарате диаметром 300 мм с
перегородками (высота аппарата равнялась его диаметру) при
перемешивании плосколопа|Стной открытой турбинной мешалкой
диаметром 150 мм в непрерывном процессе22-23. В опытах
диспергировалась легкая фа»за. Так как опыты проводили только в
аппарате одного размйра и лишь с одной мешалкой, то не было
предпринято попыткиiполучить корреляцию результатов опытов
в виде зависимости ме>жду безразмерными комплексами величин.
Макдональд и Пирует24 нашли, что для корреляции
полученных ими данных о времени смешения красителя с гомогенной
жидкостью к энергии, вводимой мешалкой, должна быть
прибавлена кинетическая энергия поступающих в аппарат потоков
жидкостей (также отшесенная к единице объема). Для системы
с малым межфазовыш натяжением, показанной на рис. 225,
кривые, полученные /для различных объемных скоростей, не
совпадают друг с другом при учете кинетической энергии
потоков.
Если мощность Р, [Потребляемую мешалкой, относить к
объемной скорости Q потооков (P/Q), то при коррелировании
опытных данных происходит разделение кривых в зависимости от
объемных скоростей сфаз для систем с высоким межфазовым
натяжением23, хотя в более ранней работе25 указывалось на
возможность коррелящии опытных данных таким способом.
Интересно отметить, чтто число оборотов, рассчитанное по
уравнению (X, 17) для периодически работающих аппаратов без
перегородок и систем (С высоким межфазовым натяжением,
приблизительно соответстгвует точке начала подъема кривых на
рис. 225. Если учесть/, что опытные данные, приведенные на
этом рисунке, получеты в аппарате объемом 22,2 л, то кривая
на рис. 224 почти совгпадет с кривой на рис. 225 для системы
керосин — вода.
Степень дисперсности (размер капель и межфазовая
поверхность). Эти вопросы «изучались в ряде теоретических
исследований, результаты котеорых подробно изложены в обзорных
работах26-28. Поэтому в данном разделе проблема определения
размера капель и межфазовой поверхности рассмотрена очень
кратко.
Максимальный разкмер капель, устойчивых в турбулентном
потоке, определяется отношением динамического давления
жидкости, стремящегося разрушить каплю, к противостоящим ему
силам межфазового иаатяжения. Если размер капли велик по
сравнению с внутренними масштабом турбулентности, то это
отношение выражается ыледугощим образом:
482
Выражение (X, 18) определяет критическое значение
критерия Вебера для капли. Величина WeHp. зависит от_уровня
турбулентности и отношения вязкости фаз. Величина V2 представляет
собой среднее значение квадрата изменения скорости в
турбулентном потоке на расстоянии, соответствующем размеру капли.
Можно показать, что в условиях изотропной однородной
турбулентности
V2=C,/—) (X, 19)
где P/vpM — энергия, вводимая на единицу массы жидкости.
Из уравнений (X, 18) и (X, 19) следует:
rf =
/We \o.e/ а \°.4vpM\0A
Ы-) Ы W (Х'20)
Как будет показано ниже, для мешалок, работающих при
высоких значениях критерия Рейнольдса в аппаратах с
отражательными перегородками, критерий мощности Я0?= Я/рv,jV3af5И
является величиной постоянной. Вводя в уравнение (X,20)
критерий Вебера для мешалок (We^ = flr^/V2pc/a) и объединяя все
постоянные величины в новую константу С2, получим:
Г г/0'4
d= Lf, n2 (X.21)
Wp°>6//0'2
Мелкие капли имеют тенденцию коалесцировать.
Рассматривая отношение кинетической энергии капель к энергии адгезии
и рассуждая аналогичным образом, Шиннар27'28 получил
следующее выражение для минимального размера капель:
с (С1*
Wejjja "
где С3 включает, в числе других величин, выражение для
энергии адгезии.
При интенсивном перемешивании можно полагать, что капли
постоянно коалесцируют и редиспергируются, результатом чего
является образование некоторого распределения капель по
размерам. Средний размер отражает состояние динамического
равновесия между явлениями распада капель и их коалесценции.
При этом распад капель доминирует в разбавленных эмульсиях,
а их коалесценция — в концентрированных. Кроме того,
известно, что скорость жидкости в сосуде с мешалкой изменяется от
точки к точке, будучи наибольшей в непосредственной близости
°т лопасти мешалки29-31. Следовательно, можно ожидать
изменения размера капель в объеме смесителя. Такие изменения
463
размера капель действительно наблюдались Э2>33.
Существование коалесценции и редиспергирования также было
экспериментально доказано33-35.
Экспериментально удельную межфазовую поверхность а
определяют обычно методами светорассеяния и
фотографирования. Для расчета межфазовой поверхности используют также
измерения интенсивности света, излучаемого сцинцилляторами,
активирующимися при прохождении через межфазовую
поверхность а- и р-частиц радиоактивного распада36. В качестве
среднего размера капель принимают диаметр сферы, у которой
отношение поверхности к объему равно этому отношению для
всей дисперсной фазы, в смесителе:
d = —j- (X.23)
Рассматриваемые далее опытные данные получены в
условиях периодического процесса и в отсутствие массообмена.
1. Открытые турбинные мешалки с шестью плоскими
лопастями (dM = 50—300 мм; сплошная фаза — вода; дисперсная
фаза — органические жидкости; <£D = 0.5; геометрически
подобные смесители диаметром от 150 до 460 мм)*6-".
Найдено, что межфазовая поверхность зависит от времени
осаждения эмульсии после прекращения перемешивания,
причем эту величину нельзя выразить в виде функции от других
известных переменных. Предложены две корреляции, из
которых одна более точная. Однако так как в нее входит время
отстаивания, эта корреляция имеет, очевидно, меньшую
практическую ценность. Другая корреляция пригодна для жидких
смесей, время отстаивания которых находится в пределах 0,2—
3 мин, и выражается уравнением
WeV5!-!^2-) е е (Х,24)
Т
Влияние числа оборотов, которое входит в WeM, близко
соответствует зависимости, выражаемой уравнением (X, 22).
Поэтому можно считать, что для довольно концентрированных
эмульсий (с которыми проводилось данное исследование)
фактором, определяющим размер капель, является их коалесценция.
2. Лопастные мешалки с четырьмя плоскими лопастями
(с?д!=170—340 мм; вода и органические жидкости; </>d=0,2—0,4;
аппараты с отражательными перегородками диаметрами 250—
500 мм)37. Для области аппарата, прилегающей к краю лопастей
мешалки, где межфазовая поверхность наибольшая, получена
зависимость
7»p(We^ 25,4^5 (We;)°-s
а~ *мП*о) = ^ (Х'25)
464
Функцию от фв (представленную авторами исследования
графически) можно в первом приближении характеризовать
зависимостью /=2,83<^5> которая приводит к конечному
выражению уравнения (X,25). Так как для геометрически подобных
аппаратов величина dM пропорциональна и'!з, это уравнение
хорошо согласуется с уравнением (X, 21).
В других опытах, проводившихся в том же смесителе как
при наличии перегородок, так и без них38, удельная
межфазовая поверхность была найдена пропорциональной
Nd^M <t>Dexp (—2 фр ). Коэффициент пропорциональности
зависел от физических свойств системы, а также от числа и ширины
перегородок. В общем случае поверхность контакта фаз,
образующаяся в смесителе с перегородками, была для данного
соотношения размеров мешалки приблизительно такой же, как и в
смесителе без перегородок при одинаковой энергии,
потребляемой мешалкой.
3. Плосколопастные мешалки с четырьмя лопастями и шести-
лопастные открытые турбинные мешалки (с?м = 60—250 мм;
органические жидкости диспергировались в воде; фв=0—0,20;
сосуд диаметром 180—380 мм, с перегородками) 39. Получено
следующее выражение для удельной межфазовой поверхности:
а = ^ (Х.26)
Здесь С=\+3,75ф0 для плосколопастных мешалок с
четырьмя лопастями {dMIT = 2lz); C=l+9 фв для шестилопастных
турбинных мешалок (dM/T = 7з). Это выражение также близко
соответствует уравнению (X, 21).
4. Мешалки различных типов (органические жидкости
диспергировались в воде, сосуды диаметром 200—40Ю мм с
отражательными перегородками)40:
CWe^RefcVrf84
а = м м и (Х) 2?)
Здесь С = 29,5 для шестилопастных турбинных мешалок;
С== 18,65 для мешалок с двумя плоскими вертикальными
лопастями; С= 13,65 для мешалок с двумя наклонными лопастями
(наклон лопастей вниз под углом 45°) и С= 13,85 для
пропеллерных мешалок. Уравнение (X, 27) также близко к
уравнению (X, 21).
5. Трехлопастные пропеллерные мешалки (dM—50—150 мм,
Фо =0,075—0,4, цилиндрические аппараты со сферическим
днищем, Т=200—300 лш)18:
30 Р. Е. Трейбал 465
Учитывая трудность экспериментального определения
межфазовой поверхности, можно считать, что приведенные
корреляции хорошо согласуются друг с другом при малых <j>D-
Возможность применения этих результатов, полученных при
периодическом процессе перемешивания, к непрерывному процессу не
установлена. Не исследован также вопрос о влиянии массообмена
на размер капель.
Расход энергии при перемешивании. Имеется большое
количество данных о расходе энергии при перемешивании
однофазных жидкостей. Наиболее обширные сведения приведены Раш-
тоном с сотр.8. Результаты опытов для аппаратов с
отражательными перегородками обычно обрабатывают графически в
виде зависимости критерия мощности р0 = pjpj\fid\j от
критерия Рейнольдса для мешалок (Ке.и = dMNp/[i). Для аппаратов
без отражательных перегородок со свободной поверхностью
жидкости (в условиях образования воронки) необходимо учитывать
дополнительный параметр — критерий Фруда для мешалок
{FrM — d.MN2lg). Энергия, потребляемая мешалкой при данном
числе оборотов, зависит не только от физических свойств
жидкости, но и от геометрической конфигурации мешалки, ее
расположения, соотношения размеров мешалки и аппарата,
внутренних устройств последнего и др.
Экспериментальные данные для двухфазных жидкостей
очень ограниченны. Для аппаратов без отражательных
перегородок, в которых возможно образование воронки, наиболее
подробное исследование выполнено Миллером и Манном19.
Однако при жидкостной экстракции образование воронки
нежелательно. Значительно больший интерес для процессов
экстракции представляют приводимые ниже данные, полученные
в условиях, когда воронка при перемешивании отсутствует.
/. Аппараты с отражательными перегородками. Опытные
данные для одно- и двухфазных систем можно описать с
помощью единой корреляции, если плотности и вязкости
двухфазных жидких смесей рассчитывать по уравнениям12:
Рс-.=РС*С + Рв*В <Х'29>
_^Л _1^ЛЛ (Х,30)
см" фс \ Н + »с)
Обработанные таким образом опытные данные для
турбинной мешалки показаны на рис. 226. При этом не было
обнаружено заметного влияния на расход энергии скорости протекания
жидкости через смеситель. Эти результаты и данные других
исследователей41 изображены в тех же координатах на рис. 227.
Каждая кривая относится как к однофазным, так и двухфазным
466
8
lot
^
JI
гоо
^•т^*"*^
600
„-!#«*$
юоо гооо воао юиоо
щ
bfip&sia
it 0000
Jffi»
*^
too
Рис. 226. Мощность, потребляемая открытыми турбинными мешалками
с шестью плоскими лопастями в аппарате, снабженном отражательными
перегородками, при перемешивании одно- и двухфазных систем [данные
для однофазных жидкостей нанесены в виде сплошной кривой; точки
относятся к двухфазным системам; Laity, Т г е у b а 1, A. I. Ch. E. Journ.,
3, 176 (1957)].
системам. В опытах с плосколопастными мешалками (четыре
лопасти)38 критерий мощности был найден постоянным и
равным Р0 = 19 W/dM при значениях ReM>6700 [плотность
рассчитывалась по уравнению (X, 29)]. До тех пор, пока не будет
накоплено достаточного количества данных, мощность,
потребляемую мешалками при перемешивании двухфазных систем,
предлагается рассчитывать по данным для однофазных систем8,
используя уравнения (X, 29) и (X, 30).
2. Аппараты без отражательных перегородок, при отсутствии
свободной поверхности жидкости (воронка не образуется).
Имеющиеся экспериментальные данные описываются единой
корреляцией с данными для однофазных жидкостей без исполь-
0,6
—-г_
ZZZ.3
_:_4
10*
/0'*
Re'
Ю5
Рис. 227. Мощность, потребляемая при перемешивании одно- и
двухфазных жидкостей [Ind. Eng. Chem., 53, 597 (1961)]:
1 — открытые турбинные мешалки с шестью плоскими лопастями в аппарате,
снабженном отражательными перегородками (Лейти и Трейбал); 2 —открытые турбинные мешалки
со стреловидными лопастями в аппарате, снабженном отражательными перегородками
(Олней и Карлсон); 3 —закрытые спиральные турбинные мешалки с кольцевым статором
в аппарате без отражательных перегородок (Олней и Карлсон); 4 — открытые турбинные
мешалки с шестью плоскими лопастями при полном заполнении жидкостью смесителя
без отражательных перегородок (Лейтн и Трейбал).
30*
467
зования критерия Фруда, если плотность рассчитывать по
уравнению (X, 29), а вязкость —по следующим уравнениям12:
"~=-^(1+^) <*->0'4) (Х-3"
'---£-('--&£-) <*-<0'4> №а>
где |д„, и \х0 — соответственно вязкости водной и органической
фаз.
Эти выражения для средней вязкости носят приближенный
характер и после накопления достаточного количества данных
должны быть уточнены. В данной работе обнаружено некоторое
влияние скорости движения жидкости через смеситель, но оно
несущественно. Зависимость критерия мощности от критерия
Рейнольдса для турбин, полученная в этих условиях, приведена
на рис. 227, причем величины Re^ рассчитывали по значениям
вязкости, определенным с помощью уравнений (X, 31) и (X, 32).
При перемешивании одно- и двухфазных жидкостей плосколо-
настными мешалками с четырьмя лопастями38 критерий
мощности при высоких значениях критерия Рейнольдса (>6700)
меняется мало и равен Po = 6WldM.
Скорости массообмена. До сих пор отсутствуют исследования,
в которых бы определяли коэффициенты массоотдачи при
перемешивании двухфазных систем й аппаратах с мешалками. Для
их нахождения приходится прибегать к косвенным данным.
Сплошная фаза. Экспериментальные данные об
исследовании массообмена в сплошной фазе при перемешивании систем
жидкость — жидкость отсутствуют. Приблизительную
информацию о величинах коэффициентов массоотдачи в сплошной фазе
можно получить на основе данных о перемешивании систем
твердое тело — жидкость.
Поверхность капель подвижна в отличие от неподвижной
поверхности твердых частиц, и коэффициенты массоотдачи в
сплошной фазе для капель больше, чем для твердых частиц
(см. главу V).
Из исследований, проведенных в аппаратах с
отражательными перегородками, следует отметить работу Баркера42.
В этой работе изучалось растворение в периодических условиях
кристаллических твердых веществ в различных жидкостях при
перемешивании плосколопастными турбинными мешалками.
Опыты проводились в цилиндрических сосудах, имевших
размеры: Т= 150—750 мм, Т=Н; dM,'T = 0,25—0,67; <j>D изменялось
от 0,005 до 0,232. Полученные значения коэффициентов
массоотдачи можно выразить эмпирическим уравнением
lg kQ = 0,8 W-03 (lg Re — 3,504) — (1,68г//з + 4,833) (X, 33)
468
Уравнение (X, 33) наиболее точно описывает данные,
полученные Баркером; с некоторым приближением результаты этого
исследования можно представить также в виде критериального
уравнения
k Т
Sh = <L_ = 0i052 Re0-833 Sc?;5 (X, 34)
Dc
Влияние формы и размера частиц на коэффициент
массоотдачи (что согласуется с наблюдениями других
исследователей) в этой работе не было обнаружено. Однако последние
данные43 указывают на то, что упомянутые факторы могут влиять
на массообмен в случае растворения очень малых частиц.
Размер капель при экстракции обычно не меньше 50—60 мк,
а при высоком межфазовом натяжении он превышает эту
величину в десять раз и более. Для частиц такого и большего
размеров44 приведенные выше уравнения хорошо согласуются со
всеми опытными данными. Хотя в уравнение (X, 34) входит
коэффициент диффузии, а в уравнении (X, 33) он отсутствует, оба
уравнения можно применять для описания экспериментальных
данных. Как было показано23'42, коэффициент диффузии Dc,
входящий в критерии Шервуда и Шмидта, в данном и других
подобных случаях сокращается из-за изменения величины Dc
с изменением отношения цс/рс- Влияние Dc на kc, если оно и
существует в аппаратах с интенсивным перемешиванием, пока
не установлено. Данные Хамфри и Ван Несса45, полученные
в условиях непрерывного движения жидкости, хорошо
согласуются с уравнениями (X, 33) и (X, 34) для условий
периодического перемешивания.
Для определения энергии, потребляемой при перемешивании
эмульсий, пользуются критерием Рейнольдса для мешалок,
рассчитанным на основе средних плотности и вязкости, однако
неизвестно, необходимо ли вести расчет таким же образом при
применении уравнений (X, 33) и (X, 34) к жидким дисперсным
системам.
Имеется значительное количество опытных данных о массо-
обмене от твердых частиц для аппаратов без отражательных
перегородок, в которых возможно образование воронки. По
этой причине они не представляют интереса для экстракции и
здесь рассматриваться не будут. Некоторые данные42,
полученные для полностью заполненных сосудов без перегородок, в
которых воронка не образуется, показывают, что коэффициент
массоотдачи имеет для данной системы те же значения 23, что
и в сосудах с перегородками при одинаковых величинах P/v.
Величина P/v имеет важное значение как критерий
интенсивности массопередачи, что было установлено теоретически и
часто подтверждалось экспериментально. Поэтому в первом
приближении можно рекомендовать применять уравнения
469
(X, 33) и (X, 34) при одинаковом расходе энергии (вводимой на
единицу объема перемешиваемой жидкости) для мешалок и
аппаратов, не испытывавшихся при выводе этих уравнений.
Дисперсная фаза. Опытные данные о массоотдаче в
двухфазных жидких системах отсутствуют. Можно, однако (см.
главу V), рассматривать капли как циркулирующие сферы с
эффективным коэффициентом диффузии Dd, изменяющимся от
DD до 2,5DD в зависимости от степени внутренней циркуляции.
Измеряя скорость растворения о-толуидина в воде в аппарате
с мешалкой, Нагата 46 пришел к выводу, что с увеличением
числа оборотов мешалки общая скорость процесса увеличивается
главным образом в результате увеличения межфазовой
поверхности, в то время как DD даже уменьшается; оба эти явления,
как установлено, обусловлены уменьшением размера капель.
Эффективность ступени. В капле дисперсной фазы,
находящейся внутри сплошной фазы в аппарате с мешалкой,
происходит процесс нестационарной диффузии растворенного вещества.
Гробер47 дал решение аналогичной задачи при теплообмене.
Он рассчитал степень приближения к температурному
равновесию твердой сферы, погруженной в жидкость с постоянной
температурой при известных времени экспозиции, диаметре
сферы, ее теплопроводности, а также коэффициенте теплоотдачи
в сплошной фазе. При подстановке в уравнение Гробера
соответствующих величин для массообмена (концентраций вместо
температур и т. д.) можно получить зависимость для
определения степени приближения к равновесию дисперсной фазы25.
Эта зависимость дана на рис. 228, где приведены эффективности
ступени по Мэрфри, определяемые по дисперсной фазе
независимо от того, является ли она экстрактом или рафинатом.
График построен на основе следующих допущений:
1. Состав сплошной фазы одинаков в объеме аппарата. Следовательно,
в случае непрерывного процесса должно происходить полное перемешивание
в сплошной фазе. Корректность такого допущения была подтверждена при
исследовании перемешивания твердых гранул в жидкости с помощью
впрыскивания красителя в сплошную фазу 48. По-видимому, полное перемешивание
достигается также в системах жидкость — жидкость в условиях как
периодического, так и непрерывного процесса (по крайней мере в аппаратах с
отражательными перегородками).
2. Дисперсная фаза находится в виде капель одинакового диаметра,
среднюю величину которого можно рассчитать, исходя из поверхности
контакта фаз.
3. Капли не подвергаются многократной коалесценции и редиспергиро-
ванию. Это явление нужно учитывать (за неимением лучшего метода)
введением некоторого поправочного множителя к коэффициенту молекулярной
диффузии DD для получения значения эффективного коэффициента диффузии
DD. Этот коэффициент должен отражать также влияние внутренней
циркуляции в капле.
470
0,1 0,2 Ofr 0,6 .1
и в to го w во юо
/nkcdp/£I^
Рис. 228. К определению эффективности ступени; m = cc/cD [T г е у b a l,
A. I. Ch. E. Journ., 4, 202 (1958)].
4. Решение, приведенное на рис. 228, очевидно, не учитывает степень
экстракции, происходящей при образовании эмульсии в начале
периодического процесса и ее осаждении в конце процесса, а также степень
экстракции в непрерывном процессе на входе и выходе жидкостей (так называемые
концевые эффекты).
5. В непрерывном процессе время пребывания капель в аппарате
предполагается постоянным и равным среднему времени пребывания Q = v<(/>D/QD.
6. Химические реакции и сопротивление массообмену на поверхности
раздела фаз не учитываются.
Пример Х-1. Аппарат диаметром и высотой, равными 300 мм,
снабженный отражательными перегородками, предназначен для непрерывной
экстракции бензойной кислоты из воды толуолом. Жидкости движутся прямотоком
снизу вверх и полностью заполняют аппарат. Расход водной фазы
составляет 5,08 мг1ч, а органической 0,50 мг/ч. Перемешивание производится шести-
лопастной турбинной мешалкой диаметром 150 мм. Определить к. п. д.
аппарата (эффективность ступени) при 200 об/мин мешалки.
Решение. Физические свойства системы: плотность водного раствора
998 кг/м3; вязкость 0,95 • 10_3 н-сек/м2; плотность толуольного раствора
865 кг/мг; вязкость 0,59-10""8 н-сек/м2; рассчитанная величина коэффициента
Диффузии бензойной кислоты в толуоле 1,68-10-9 м2/сек; межфазовое
натяжение 22-10-8 н/м; коэффициент распределения бензойной кислоты
(концентрация в толуоле/концентрация в воде) равен 20,8.
Объем аппарата и = 0,022 мг. При данном объемном соотношении фаз
Диспергироваться будет толуол. По условию QD = 0,50 мъ/ч, что составляет
9,1% от общего количества жидкости. Принимаем предварительно, что
задержка (объемная доля) дисперсной фазы в аппарате Фо = 0,091. Диаметр
мешалки dM = 0,15 м; N=200/60=3,33 об/сек.
471
По уравнению (X, 29) находим:
рсм = 998 • 0,909 + 865 • 0,091 = 986 кг/м3
Согласно уравнению (X, 30), имеем:
0,95-Ю-3 Г, . 1,5-0,59-Ю-3. 0,091
^см. = '
L 0,51
0,909 L 0,59-10" " + 0,95-10"
4f^PcM 0,152-3,33-986
= 1,1-10 н-сек/м2
Re' = м ■ '"'• = — '"_""" =67200
Нем. 1,1 • 10 3
Из рис. 227 (кривая /) следует, что Р0 = 5,8. Отсюда Р=Р0РСМ dbMNz=
=5,8-986- (0,15)5(3,33)3=15,9 вт,
Как видно из рис. 225 (кривые на рисунке относятся к данному
аппарату), Фв при этом числе оборотов действительно равно 0,091.
Коэффициент массоотдачи в сплошной фазе рассчитываем по уравнению
(Х,33): Re =(0,15)2-3,33-998/0,95 • 10"3=78000. Для и=0,0222 ж3 это
уравнение дает: &с = 0,70 • Ю"4 м/сек.
Поверхность контакта фаз рассчитаем с помощью уравнения (X, 26):
WeM = dMN2pcja = (0,15)3 (3,33)2 • 998/22 • Ю-3 = 1690
_ .100 • 0,091 • (1690)0'6 оооп з
а~ [14-9-0,091] -0,15 ~ZbW M IM
По уравнению (X, 23) имеем:
d = 6- 0,091 /2880= 1,87-10~4 .« = 0,187 мм
1>ФП 22,2- Ю-3-0,091- 3600
e = -Q7 = °^ =14'5^
m=CclcD (концентрация в воде/концеитрация в толуоле) = 1/20,8 = 0,048
Примем: D'D = 2DD = 2 • 1,68 - Ю-9 = 3,36 - Ю-9 м2 j'сек. Тогда
mkcd 0,048-0,70-10~4-1,87-10~4
2D'D 2-3,36-Ю-9
2mkcQ 2-0,048-0,70-Ю-4-14,5
= 0,093
_, — = 0,522
1,87-10 4
Из рис. 228 следует, что £MD = 0,79, или 79%. Как будет показано ниже,
при наличии концевых эффектов эффективность ступени увеличится. Кроме
того, некоторая экстракция будет происходить в отстойнике, в который
поступают жидкости из смесителя.
Для рассмотренной в примере Х-1 системы равновесное распределение
сильно сдвинуто в пользу дисперсной фазы, поэтому в сплошной фазе
сосредоточена значительно большая доля общего сопротивления массообмену, чем
в дисперсной. В связи с этим расчетная величина эффективности ступени
малочувствительна к выбору поправочного коэффициента при DD,
определяющего величину DD. Соответственно в данном случае можно ограничиться
472
учетом сопротивления только в сплошной фазе. Из табл. 16 (см. стр. 448)
при полном перемешивании в фазе рафината имеем:
ME MD e e
Полагая 0=#/(Vc +Vz>), <t>D= VD/(Vc + VD) и, пренебрегая сопротивлением
в дисперсной фазе, получаем:
тКсаН тКса% mkcaQ
VD *D <>D
Следовательно,
£ _ j e- mkcaW<t>D __ j £-0.O48-O,70-lO-4-2880-14,5/O,091 _ q jg2
MD '
Секционированные аппараты. При непрерывном движении
жидкостей через секционированный аппарат (см. рис. 221),
имеющий п одинаковых секций (эффективность каждой из
которых равна Е), в отсутствие продольного перемешивания
между соседними секциями эффективность ступени Еп можно
определить по уравнению
£я = £[Г+(1-£) + (1-£)2 + (1-£)3+...+(1-£)я-1] (Х,35)
Значение Еп больше Е и приближается к величине
эффективности ступени для идеального поршневого потока по мере
увеличения п. Уравнения для Емс, EMR или ЕМЕ для
секционированного аппарата имеют довольно сложный вид. Этими
выражениями эффективности ступени лучше пользоваться, переходя
к ним от Е или наоборот.
Пример Х-2. Определить эффективность ступени для процесса экстракции,
рассмотренного в примере Х-1, допуская, что смеситель состоит из трех
секций высотой 0,3 м каждая, а жидкости движутся через аппарат прямотоком.
Решение. Для одной секции £дп> = £дге=0,79 и e=/raVE/VR =
=20,8-0,5/5,0 = 2,08. Следовательно (см. табл. 16)
ЕМР(1+е) 0,79(1+2,08)
Р — МЕ - ! ! = 0 920
1+еЕ,,,. ~" 1+2,08-0,79 *а
1 ME ' ' '
Для трех секций по уравнению (X, 35) получаем:
Е3 = 0,920 [1 + (1 — 920) + (1 — 920)2] = 0,9995
Соответствующая величина Emd=Eme для трехсекционного аппарата
(см. табл. 16) равна:
F 0 9995
р — е - = 1^ууо g4
£лю~сл«е(1— £)+1 2,08(1—0,9995)+ 1 '
т. е. эффективность ступени составляет 99,84%.
Определение эффективности ступени в непрерывном процессе
по опытным данным периодической экстракции. Опытные
данные, полученные при периодической экстракции, как будет
показано ниже, можно описать (см. табл. 16) уравнениями:
_ Г VE 1 _ Г VRZ
= - Id+eJV^Jnep. '" (1 ~ Е) ~ ~ [ (l+e)Vw
In (1 — £) =
пер.
= —KIn(l — В) (Х.36)
473
Поэтому зависимость 1—N/Ne=l—Е от Э изображается
в полулогарифмических координатах прямыми линиями, по
наклону которых можно определить К в каждом конкретном
случае.
Для непрерывного процесса при полном перемешивании в
аппарате (см. табл. 16) имеем:
ОЕ \ V
непр.
КЕав
JME
непр.
1
N
OR
Г) -(■
" R /непр. \
Vе
*МЕ
MR
(1
■£)(1 + е)
гЕ
R /непр.
1 —£
MR
(1-£)(1 + е)
Следовательно
[КЕа (1 + е) 6/0£]непр-
непр.
(Х,37)
(X, 38)
(Х.39)
1 + \КЕа (1+8) 6/0я]непр. 1 + [KRa (1 + е) в/е*Л]непр.
Если периодический и непрерывный процессы проводят в
одинаковых аппаратах, при равных соотношениях фаз и степени
перемешивания, то
КЕау(\+г)
KRav (1 + е)
гУг
пер.
пер.
КЕа(\+г)
J непр.
У (1 + е)
80D
непр.
К
1
тс
Подставляя этот результат в уравнение (X, 39),
рывного процесса получаем:
*_ е/к _ е
(Х,40)
для непре-
(Х.41)
Это уравнение дает возможность определять эффективность
ступени, работающей в непрерывном процессе, исходя из
данных для периодической экстракции в том же аппарате.
Уравнение (X, 41) является приближенным, так как при его выводе
не учитывалось влияние непрерывного движения жидкости на
коэффициенты Keu и Kru. Кроме того, степень экстракции в
непрерывном процессе может оказаться больше расчетной
величины в результате концевых эффектов.
Пример Х-З. Хиксон и Смит' приводят следующие данные для
периодической экстракции иода из воды четыреххлористым углеродом (диаметр
аппарата 0,215 м; глубина жидкости 0,215 м; суммарный объем жидкостей
0,00709 ms, в том числе объем, занимаемый четыреххлористым углеродом,
7,09- Ю-4 1Лг\ скорость вращения мешалки 200 об/мин):
6, мин ....
N, г J2 . . . •
N/Ne = E . .
0
0
0
8
0,529
0,397
13
0,712
0,535
16
0,861
0,647
24
1,038
0,780
30
1,125
0,845
38
1,175
0,883
45
1,205
0,906
52
1,276
0,958
оо
1,330
1,000
474
Принимая, что фазы в аппарате
равномерно перемешаны, определить
эффективность ступени в непрерывном
процессе при суммарной скорости
подачи жидкостей 0,4-10"3 м3/мин в тех
же соотношениях и при той же
начальной концентрации: а) для одного
аппарата; б) для двух последовательно
соединенных аппаратов.
Решение, а) Величину ./V можно
выражать числом граммов
экстрагированного иода, причем W«= 1,330.
Значения N/Ne=E приведены в таблице
в зависимости от 6. Значения (1-Е)
в функции от В наносим на график
в полулогарифмических координатах
(рис. 229) и проводим прямую, наибо-
лее близко соответствующую экспериментальным точкам. При (1 — t)=U,l
имеем: 6 = 38,6 мин; подставляя эти значения в уравнение (X, 36), получаем
К=Ю08 сек. Для непрерывного процесса находим:
о ю го зо <w? so во
в, мин
Рис. 229. Периодическая
экстракция (к примеру Х-З).
9;
v_ = (я/4) (0,215)*-0,215 = П70 сек
Q
0,4-10" 760
По уравнению (X, 41) имеем:
й
Е
1170
- = 0,538
К + 6 ~ 1008 + 1170
вательно соединенных
Е = 0,538 [1 + (1 — 0,538)] = 0,787
б) Для двух последовательно соединенных аппаратов, согласно
уравнению (Х,35), получим:
Экспериментальные данные
об экстракции
Опубликовано лишь несколько работ, посвященных
исследованию массообмена в смесителях для систем жидкость —
жидкость. Основная трудность при проведении опытов состоит в
том, чтобы исключить массообмен в период отбора проб из
аппарата; по-видимому, экспериментальные данные большинства
исследователей включают также массообмен, происходящий при
отстаивании фаз за время отбора проб. Это затруднение можно
преодолеть, отбирая из аппарата пробы только одной фазы
через пробоотборник, изготовленный из материала,
предпочтительно смачивающего ту или иную жидкость (см. главу IX).
При рассмотрении экспериментальных данных, приводимых
ниже, предполагается, что экстракция в случае отстаивания в
пробах незначительна и ею можно пренебречь.
Периодическая экстракция. Хиксон и Смит 1 исследовали
экстракцию иода из воды четыреххлористым углеродом^ в
аппарате без перегородок при перемешивании трехлопастной
пропеллерной мешалкой, установленной по оси аппарата. В условиях
47S
100 200 UOOIOO ZOO WO 100 200 ^00
N, об/мин
а б в
Рис. 230. Периодическая экстракция иода из воды (10 объемов) четырех-
хлористым углеродом (1 объем) в геометрически подобных аппаратах
с пропеллерной мешалкой, без отражательных перегородок [dM/T = 0,5;
Н—Т; данные Хиксона и Смита, см. Ind. Eng. Chem., 41, 973 (1949)]:
а —положение пропеллера на расстоянии от дна, равном удвоенной высоте столба
экстрагента; б —то же на поверхности раздела; в —то же на расстоянии от дна, равном
половине высоты столба экстрагента.
опытов происходило образование воронки. Экспериментальные
данные наносили на график (построенный в координатах,
показанных на рис. 229), с помощью которого можно было
рассчитать коэффициенты массопередачи.
Значения коэффициентов массопередачи в зависимости от
скорости вращения мешалки приведены на рис. 230. Резкий
наклон линий на рис. 230 характерен для массообмена в системах
жидкость — жидкость. С увеличением числа оборотов мешалки
в результате усиления турбулентности возрастает не только
коэффициент массопередачи КЕ, но и поверхность контакта фаз.
Суммарное влияние этих двух явлений на массообмен
характеризуется величиной объемного коэффициента массопередачи
476
КеО- Из-за отсутствия динамического подобия вследствие
образования воронки невозможно скоррелировать данные для
аппаратов трех различных размеров. Очень малые добавки
поверхностно-активных веществ оказывают сильное влияние на
скорости экстракции, которые в этой системе связаны также с
химическим взаимодействием49.
Другие опытные данные о периодической экстракции
рассмотрены ниже в связи с исследованиями непрерывных
процессов экстракции.
Непрерывная экстракция. Наиболее подробные исследования
проводили Оверкайшер с сотр.6, изучавшие экстракцию бутил-
амина из керосина водой в смесителе непрерывного действия
'г 4 ю го ьо
Мощность, вт
б
Рис. 231. Влияние типа и размера мешалок на эффективность ступени
При непреоывной экстракции бутиламина из воды керосином [время
пребывания 1,08 мин, объемное соотношение фаз керосин — вода равно 1,57;
данные для аппаратов с четырьмя отражательными перегородками
представлены в виде сплошных кривых; пунктиром нанесены данные для
смесителей без перегородок; см. Overcashler, Klngsley, Olney,
A. I. Ch. E. Journ., 2, 525 (1956)]:
а —плосколопастные турбины; б —различные мешалки. 1, 2, 3 —плосколопастные тур-
оины; 4, 5 —пропеллеры; 6, 7 —спиральные турбины.
477
(Т = 375 мм, # = 470 мм). Жидкости двигались прямотоком
снизу вверх. Испытывался ряд мешалок как при наличии в
аппарате перегородок, так и в отсутствие последних. В последнем
случае аппарат заполнялся полностью, и, следовательно,
воронка не образовывалась.
Наиболее важные результаты этой работы представлены на
рис. 231. Приведенные в верхней части рисунка данные для
плосколопастных открытых турбинных мешалок показывают, что
в смесителе без отражательных перегородок достигается
большая эффективность ступени, приходящаяся на единицу энергии,
вводимой мешалкой, чем в аппарате с перегородками. Это,
несомненно, результат уменьшения продольного перемешивания
в отсутствие значительных вертикальных потоков, возникающих
при наличии отражательных перегородок. Такие же опыты были
проведены для других мешалок; оптимальные размеры
мешалок каждого типа приведены в нижней части рисунка. В
присутствии перегородок наилучшие результаты получены при
погружении мешалки в жидкость на глубину, соответствующую
расстоянию мешалки от днища смесителя, равному 2/3 высоты
столба жидкости в аппарате, наихудшие — когда глубина
погружения мешалки составляла 'Д высоты столба жидкости от
дна.
Для закрытых спиральных турбинных мешалок зависимость
эффективности ступени от расхода энергии на перемешивание
остается приблизительно той же, что и для открытых турбинных
мешалок в смесителе с перегородками. Для эксцентрично
установленных пропеллерных мешалок получены промежуточные
значения эффективности ступени по сравнению с эффективно-
стями, достигаемыми при том же удельном расходе энергии
пропеллерными мешалками, установленными по оси аппарата с
перегородками и без них. Опытные данные для пропеллерных,
спиральных и плосколопастных турбинных мешалок {dM —
= 100-f-254 мм) при скорости жидкостей, соответствующей
времени их пребывания в смесителе 8=1,08 мин и объемом
соотношении керосина и воды, равном 1,57, описываются
эмпирическим уравнением
0,318- \0l5(dM/T)a
£° = 1 8еЦ-р£ (Х'42)
где а = 0 для аппаратов с отражательными перегородками и
а=1,6 — для аппаратов без перегородок.
В аппарате с перегородками и аксиально установленной
пропеллерной мешалкой диаметром 203 мм было исследовано
влияние на эффективность ступени скорости жидкостей (или
времени их пребывания в смесителе). Как и следовало ожидать,
большему времени пребывания соответствует большая
эффективность. При объемном соотношении фаз, равном 1,57 (керо-
478
син/вода), опытные данные,
полученные в этих условиях,
описываются уравнением
/04-10~3Р \0-255
£*='"( х» н - 0,0425 6]
(X, 43)
Представляет интерес
проверить применимость метода
расчета с помощью графика
на рис. 228 к результатам этой
работы. Величины а и kc
можно рассчитать для
плосколопастных турбинных мешалок в
аппаратах с отражательными
перегородками. Поверхность
контакта фаз рассчитывали по
уравнениям (X, 24) и (X, 26) в
зависимости от величины
удерживающей способности, a kc —
по уравнению (X, 33).
Результаты расчета
представлены на рис. 232, где
приведены величины D'd/Dd, при которых значения эффективностей
ступени, рассчитанные по графику на рис. 228, совпадают с
опытными. Черными точками показаны величины DD/DD,
полученные для низких скоростей мешалки, при которых заметную
роль играет «входной эффект» (по данным авторов, за счет
этого эффекта достигается эффективность £' = 0,18). С
увеличением энергии, потребляемой мешалкой (при большем числе
оборотов), входной эффект имеет относительно меньшее значение.
В нижней части рис. 232 те же величины D'D/DD
изображены в функции от диаметра капель, причем темные точки
относятся к малым скоростям мешалки. Если исключить эти точки
из рассмотрения, то из графика следует, что отношение DD/ Dd<
как можно было ожидать, с возрастанием размера капель
несколько увеличивается. Ранее при обработке тех же опытных
данных было показано25, что постоянное значение отношения
D'd/Dd = 2,25 дает возможность получить расчетные величины
эффективности ступени, очень близкие к опытным.
Олней50 обработал примерно таким же способом опытные
данные для других мешалок, полученные в аппарате с
отражательными перегородками (см. рис. 231). Размер капель был
рассчитан по уравнению (X, 20) после определения величины
константы. Пренебрегая сопротивлением массообмену в
сплошной фазе, Олней нашел, что для этих мешалок отношение D'D/Dn
Мощность мешалки, Вт
4
г
i
"е? 4
г
Q6
о,и
цг
го
ио
-■■-©
*/
■
■
•
№£-
-^-й—
□
0
60
\
».
0 """""
к
д
° &м=
тар _.
о " 0,203-
4 »
-
о,гы
10
/а
Диаметр капель, м-Ю
го
4
гз
Рис. 232. Эффективный коэффициент
диффузии в дисперсной фазе
(система бутиламин — керосин — вода;
аппарат с отражательными
перегородками; открытая плосколопастная
турбинная мешалка; время
пребывания 1,08 мин; объемное
соотношение фаз керосин/вода равно 1,57).
479
1
I
о;
1
i
0,6
Oh
0,2
0,1
1
Op
Oh
0.2
0,1
E/R
0,33^
6^
^>
,s
^
<&\
У
~:Л
l^
^*
^^
^
jrr^il
e/r
6y
с
s^/X*
srfjf
A3
Лр0,зз
r^
*1 /*
^о-Усг^Ц
-if"»^
4 w3 г 4. ю* г
Расход знергии на еЗ. объема Р/ц, dw/м3
б
Рис. 233. Непрерывная экстракция бензойной кислоты водой [аппараты
с отражательными перегородками; Т = 0,146 и 0,284 м; открытая
турбинная мешалка с шестью плоскими лопастями; dM/T = 0,5; скорости
жидкостей 225—2250 кг/ч; см. F 1 у п п, Т г е у b а 1, A. I. Ch. E. Journ., 1, 324
(1955)]:
о —из толуола; б —из керосина.
изменяется от 0,5 до 2 и в среднем равно 1,0. Представляется
неясным, почему для мешалок других типов получены
величины Do/Dd меньшие, чем для турбинных.
Флин 51 экстрагировал бензойную кислоту из толуола и
керосина водой в непрерывнодействующем аппарате с
отражательными перегородками двух размеров при перемешивании
плосколопастными турбинными мешалками. Существенное
значение имел концевой эффект, особенно при больших скоростях
жидкости; поэтому корреляция опытных данных была возможна
только при внесении поправки на концевой эффект.
Эффективность ступени ЕМе, обусловленную перемешиванием,
рассчитывали, как отношение {ЁцЕ— E"me)J{\—ЕМЕ), где Е'ме—
значение эффективности, полученное в опыте, яЕМе—эффективность
при отсутствии перемешивания (при нулевой скорости мешалки).
Подобный способ учета концевых эффектов нельзя считать
совершенным. Однако расчетные данные, полученные таким
способом (рис. 233), коррелируются в функции от величины
P/Q подобно данным Оверкайшера. Аномалия, наблюдаемая у
480
кривых, соответствующих объ- /
емному соотношению фаз,
равному 3, может быть, несо- о,д
мненно, объяснена обращени- -^
ем фаз. | 0,8
Эффективности ступени, по- <§
лученные при нулевой скоро- § o,k
сти мешалки, показаны на § 0,8
рис. 234. Обрабатывать данные |-
Флина аналогично тому, как g qr
производилась обработка дан- *5.
ных Оверкайшера (рис. 232), \$ о,Ь
нецелесообразно, так как
распределение в данном случае
сильно сдвинуто в пользу
органической (дисперсной) фазы
и отношению D'DJDD можно
придать практически любое
значение. Как было показа-
E/R
ft
'
^""^-fl
^3
—в
0.S
о.зз ^
-^
^>
^р
й
7
/
—-L 1
*в
гоо воо юоо /ш шоо
Суммарный расход фаз, ке/ч
6
НО'
величина этого отноше-
Рис. 234. Эффективность (к. п. д.)
ступени при нулевой скорости
мешалки. Экстракция бензойной
ния, равная 2,25, лучше всего кислоты водой:
СООТВеТСТВуеТ ЭКСПерИМеНТаЛЬ- «-из толуола; б-из керосина (см. рис. 233).
ным данным.
Карр и Шайбель52 исследовали экстракцию в системах
уксусная кислота — метилизобутилкетон — вода" и ацетон —
ксилол— вода в одной из смесительных секций экстрактора,
показанного на рис. 296. Опыты были поставлены так, что в наса-
дочных секциях экстракции не происходило. По наблюдениям
Г*
Щ0,2
'
/
/
/
S"
гуг
з
/
/
1
и
1
13,8
Д 3,51
v 7,07
*10А
О
гоо
<*00
гоо
иоо
ш о гоо
Ц, об/мим
Рис. 235. Непрерывная экстракция в системе метилизобутилкетон
(дисперсная фаза, экстрагент) — уксусная кислота — вода [VD/VC = 1,0,
Т = 0,292; Н = 0,076; dM = 0,102 м; результаты расчета показаны в виде
сплошных кривых; условные обозначения — опытные данные Карра и
Шайбеля, см. Chem. Eng. Progr. Symp. Ser„ 50, № 10, 73 (1954)].
31 P. E. Трейбал
481
авторов, при наличии в
аппарате проволочных сеток
(см. стр. 585) движение
жидкости подобно ее
движению в смесителе с
отражательными
перегородками.
Некоторые типичные
опытные данные приведены
на рис. 235. Расчетные
кривые на этом рисунке
получены с помощью графика на
рис. 228; поверхность
контакта фаз рассчитывали по
уравнению (X, 26) и
опытным значениям <f>D, a kc —
по уравнению (X, 33),
причем отношение Do/Do было принято равным 2,25. Все опытные
данные, за исключением относящихся к самым низким
скоростям подачи жидкостей и низким числам оборотов мешалок
(когда входной эффект сказывается больше всего), хорошо
согласуются с расчетными. Следует, однако, отметить, что
значительное влияние на эффективность ступени оказывало
направление экстракции (перенос вещества к каплям или из капель).
При принятом методе расчета объяснить это явление нельзя
(см. главу V). Карр и Шайбель предложили эмпирическую
корреляцию для Каа, в которую не входят коэффициенты
диффузии и в которой Ка рассчитывают по движущей силе,
выраженной в активностях.
) "Рьон, Далей и Лоури53 экстрагировали уран из сульфатных
щелоков керосином, содержащим трибутилфосфат и ди(2-этил-
гексил)фосфорную
кислоту. Ион уранила
взаимодействует с этими
реагентами и в результате
экстрагируется. Экстракцию
проводили как
периодически, так и непрерывно
в аппаратах с
перегородками нескольких
размеров при перемешивании
плосколопастными
турбинными мешалками.
Результаты
периодических опытов
обрабатывали графически
аналогично тому, как показано на
^
/
о
Q' 02
0,3 /7,4
d„/r
05 06 07
205010U0 БЬ7 329 ZOU
Об/мин
Рис. 236. Периодическая экстракция
урана в аппарате с отражательными
перегородками [диаметр 0,3 м; открытые
турбинные мешалки с шестью плоскими
лопастями; /ге=33,4; е=33,4; 0,328 квт/м3;
см. Ryon, Daley, Lowirie, Chem.
Eng. Progr., 55, № 10, 70 (1959)].
10
l г
T=0,
•^и
^^
r,o,5~
■
С
xz****^
^T=0,15
i
□
T
0J5
b,5
Д
»
e
33,3 t
о
0
?,JJ
•
■
A
/
op
0,1 o,s oft i г и
Мощность мешалНи, Вщ/м3-Ю~^
Рис. 237. Периодическая экстракция урана
в аппаратах с отражательными
перегородками и открытыми турбинными мешалками
с плоскими лопастями (dM/T = 0,15—0,5;
данные Рьона, Дален и Лоури).
482
рис. 229. Рассчитанные таким
образом коэффициенты массо-
передачи приведены на рис. 236
и 237. Результаты
непрерывной экстракции,
обработанные для случая полного
перемешивания, показаны на рис.
238. Заслуживает внимания,
что коэффициенты массопере-
дачи не зависят от времени
пребывания жидкостей в
аппарате! Приведенные на
рисунке значения коэффицентов
массопередачи для аппарата
диаметром 0,9 м представляют
собой средние величины для
трех аппаратов, образующих
0 каскад, и соответствуют
скорости жидкости 6,9-Ю-3 мъ/сек,
времени пребывания 72 сек и
Время пребывания в отстойниках этого каскада равнялось
84 мин; в результате массообмена при отстаивании средняя
величина к. п. д. ступени возрастала до 92,6%.
г -Результаты этого исследования показывают, что
коэффициенты массопередачи в процессах периодической и
непрерывной экстракции при равной энергии, вводимой на единицу
объема аппарата, примерно одинаковы. Это подтверждает
указанную выше возможность использования опытных данных,
полученных в периодическом процессе, к непрерывному процессу
экстракции. Следует отметить, что на оси абсцисс рис. 237 и
238 отложены величины P/v, а не P/Q, как на рис. 233. Какой
из этих двух методов корреляции опытных данных лучший, пока
что установить нельзя.
При расчете эффективности описанных экстракторов с
помощью рис. 228 получаются явно завышенные значения.
По-видимому, это обусловлено влиянием медленной химической
реакции урана с реагентами в органической фазе.ГРьон53 отметил,
что влияние температуры на коэффициент массопередачи при
экстракции соответствовало в его опытах энергии активации,
равной 32- 106—40- 106 дж/кмоль. Для массообмена, не
осложненного химическим взаимодействием, эта величина близка к
20- 106 дж/кмоль, в то время как для химической реакции
наиболее вероятна величина энергии активации порядка 40Х
ХЮ6 дж/кмоль. Таким образом косвенно подтверждается, что в
данном процессе важную роль играет скорость химической
реакции. Было также отмечено, что удаление урана из экстракта
%
'
1
\
Т
0,15
OJ
0,9
е
33,3
5,33
5,33
55,5
»
о
•
д
OflU 0.1 0,2 01+ 1 4
Мощность мешалки, вт/м3-10'
Рис. 238. Непрерывная экстракция
урана в аппаратах с отражательными
перегородками и открытыми
турбинными мешалками с плоскими
лопастями (результаты опытов
приведены к dMlT = 0,333; время
пребывания 0,17—0,48 мин; данные Рьона,
Дален и Лоури).
эффективности ступени 78,9%.
31*
483
промывкой водным .раствором соды протекает с гораздо
большими скоростями, чем экстракция.
Моделирование экстракторов с мешалками. При внедрении
новых экстракционных процессов рекомендуют проводить
испытания на лабораторных моделях — аппаратах с мешалками —
по возможности с теми же растворами, с которыми будут
проводиться процессы экстракции в крупном масштабе.
Эффективность ступени нужно определять в зависимости от
возможно большего числа основных переменных (скорости
жидкостей, объемное соотношение фаз, число оборотов мешалки).
В каждом опыте необходимо рассчитывать также
удерживающую способность по дисперсной фазе. В отсутствие химической
реакции полученные данные можно использовать для расчета
эффективного коэффициента диффузии DD. Последний может
несколько меняться с расчетным размером капли в зависимости
от характера циркуляции в каплях, присутствия поверхностно-
активных веществ и пр. Эффективный коэффициент диффузии
не должен, однако, зависеть от размеров аппарата, что имеет
существенное значение для перехода от модельных аппаратов
к аппаратам большого размера (см. также гл. XII).
Перемешивание воздухом. Матерс и Уинтер54'55 описали
конструкцию смесительно-отстойного экстрактора, в котором
перемешивание осуществляется с помощью эрлифта. Воздух
подводится через штуцер в днище смесителя и распределяется
через сопло, над которым находится открытая вертикальная
труба эрлифта, погруженная в жидкость на 2/з высоты столба
жидкости в аппарате. Жидкость поступает в трубу снизу и
движется прямотоком с воздухом.
Экспериментальные данные, характеризующие работу такого
аппарата, ограниченны. В смесителе объемом 0,005 ж3
экстрагировали уксусную кислоту из воды метилизобутилкетоном при
суммарной объемной скорости жидкостей 0,0104 м3/мин и
скорости подачи воздуха 8,5 • Ю-3 м3/мин. Средняя эффективность
ступени, состоящей из смесителя и отстойника, имеющего объем
0,01 м3, равнялась EMr = 0,93 при затрате энергии на подачу
воздуха 0,745 вт (P/Q = 4300 дж/м3). Эта величина сравнима с
расходом энергии в смесителях с механическими мешалками при
такой же степени экстракции. Испытанная система жидкость —
жидкость обладает малым межфазовым натяжением, поэтому
легко диспергируется и соответственно требует малого расхода
энергии на перемешивание. Для окончательной оценки этого
аппарата необходимы данные, относящиеся к более трудно
диспергируемым системам.
Перемешивание воздухом удобно тем, что в аппарате
отсутствуют движущиеся части, соприкасающиеся с рабочими
жидкостями, которые могут быть радиоактивными или вызывать кор-
484
розию. Недостаток пневматического перемешивания —
испарение экстрагента, который нужно регенерировать, и в случае
обработки радиоактивных жидкостей необходимость тщательной
очистки выходящего воздуха.
ПРОТОЧНЫЕ СМЕСИТЕЛИ
Проточные смесители представляют собой устройства для
диспергирования одной жидкости в другой при их движении
через аппарат в определенном объемном соотношении.
Диспергирование осуществляется за счет кинетической энергии жидко-
*сти. Эти устройства можно применять только для непрерывного
или полунепрерывного процесса (когда одна жидкость
непрерывно движется, а другая непрерывно рециркулирует).
Поточные смесители отличаются от непрерывнодействующих
аппаратов с мешалками малым объемом и, следовательно, очень
малым временем пребывания жидкости. Если необходимо, время
пребывания можно увеличить, устанавливая за смесителем
отстойник или применяя соединительный трубопровод.
Смесители данного типа используют лишь в тех случаях,
когда диспергирование происходит легко и быстро устанавливается
равновесие, например для жидкостей с низким межфазовым
натяжением и малой вязкостью. Стоимость проточных смесителей
сравнительно невелика. Они находят широкое применение для
очистки легких нефтяных дистиллятов. В этом процессе
отношение количества экстрагента к количеству обрабатываемой
жидкости невелико.
Проточные смесители по конструкции устройства для
диспергирования и смешения фаз можно разделить на четыре
основных типа:
1. Форсунки или другие устройства, работающие по
принципу введения с большой скоростью одной жидкости в другую
для лучшего их перемешивания.
2. Инжекторы, в которых одна жидкость увлекается
потоком другой, что и приводит к их перемешиванию.
3. Диафрагмы и сопла, перемешивание и диспергирование в
которых осуществляется вследствие высокой степени
турбулентности, создающейся в потоке движущихся жидкостей.
4. Устройства, в которых перемешивание производится как
вследствие движения жидкостей, так и за счет механической
анергии.
Известно много разновидностей устройств последнего типа;
ниже рассмотрены лишь наиболее'важные из них. Для всех
разновидностей смесителей такого типа экспериментальные
данные очень скудны. Каличевский и Коуб56 подробно рассмотрели
Применение подобных смесителей в процессах очистки
нефтепродуктов,
488
Форсунки. Форсунки обычно наименее пригод-
^<^ ны для перемешивания взаимно нерастворимых
\\ жидкостей, хотя их успешно применяют для
смешения газов. Использование форсунок
ограничено системами с малой разностью плотностей
фаз и невысоким межфазовым натяжением, в
которых более энергичное перемешивание может
вызвать образование стабильных эмульсий.
В простейшем виде форсуночный смеситель
состоит из труб, соединенных в форме буквы Y.
Каждая жидкость подается насосом в одну из
расположенных под углом труб смесителя, и
затем они движутся совместно через общий
патрубок. Действие простейшей форсунки (рис.239)
улучшается, если использовать ее в сочетании со
смесительными соплами или диафрагмами,
описываемыми ниже.
Смеситель, представленный на рис. 240,
можно отнести к устройствам аналогичного типа;
он оказался весьма эффективным57 при
предварительном смешении масла и экстрагента в
процессе очистки смазочных масел по способу
«Дуо-Сол». Экстрагент поступает в большое
сопло по оси смесителя и проходит через
отверстия со скоростью 5—6 м/сек. Масло подается
через штуцер с противоположного конца
смесителя и, двигаясь по спирали вдоль
перфорированного конуса, тщательно перемешивается с
экстрагентом. Смесь выходит через верхний штуцер
горизонтального корпуса смесителя.
Трайс испытал58 струйный смеситель, состоящий из
цилиндрического корпуса с внутренней металлической тарелкой, о
которую ударялись жидкости, прокачиваемые насосом через
аппарат. Исследовались аппараты диаметрами 100 и 150 мм,
причем высота аппарата равнялась его диаметру. Размер
Рис. 239.
Форсунка в
колонке с диафраг-
менными
смесителями (The Du-
rlcon Co).
Смесь
Масло
Перфорации
Рис. 240. Смеситель масла и экстрагента в «Дуо-Сол-процессе» (Мах
В. Miller and Co., Inc.).
486
и
Рис. 241. Инжекторный смеситель (Hampton, пат. США 2091709):
/ — тяжелая фаза; Я —легкая фаза; III — эмульсия из смесителя в отстойник.
капель определяли методом светорассеяния. Была установлена
зависимость критерия Рейнольдса dVnpc/vc от величины
(ТУ0рс/цс)(Т]/20рс/а)у'ф0^ для двух систем. Найдено, что
коэффициент массоотдачи в сплошной фазе (определявшийся для
двухкомпонентных систем) можно рассчитать по уравнению
k£_
-. 0,03 D c
\ VC
PcDc)
(X, 44)
Вследствие того, что величина l/Dc изменяется
пропорционально Sc0'5, истинное влияние коэффициента диффузии на kc
в этой работе не было установлено23. Другая исследованная
разновидность струйного смесителя m представляла собой
вертикальный цилиндр диаметром 100 мм, в котором воду
распыляли в толуоле. Высота корпуса была достаточной для того,
чтобы фазы могли отстаиваться и порознь выводиться из
смесителя. По существу этот смеситель является смесительно-от-
стойным аппаратом, в котором перемешивание осуществляется
за счет энергии струи. При экстракции бензойной кислоты из
толуола наилучшие результаты (£Ме=0,92) получены с
распылительной трубкой диаметром 3,1 мм при суммарном расходе
7,6- Ю'3 м31мин. Расход энергии в смесителе имел
промежуточную величину между расходом энергии в смесителе с
механическим перемешиванием и в описанном выше смесителе с
пневматическим перемешиванием.
Инжекторы. Эти устройства работают по принципу действия
широко известного парового инжектора. Жидкость, подаваемая
в инжектор насосом, вызывает движение другой жидкости и
смешивается с ней. Одна из конструкций, в которой инжектор
помещен в отстойную камеру ступени экстрактора, показана на
рис. 241, а. Камера заполнена двумя взаимно нерастворимыми
жидкостями (после отстаивания смеси), поверхность раздела
487
между ними расположена приблизительно посредине отстойной
камеры. Тяжелая фаза из предыдущей ступени подается
насосом в инжектор и засасывает легкую фазу из отстойника.
Струйный экстрактор аналогичной конструкции показан на рис. 241,6,
причем в этом случае смеситель расположен вне отстойника.
Другую конструкцию струйного экстрактора (рис. 242)
применяют в полунепрерывном процессе для промывки масла.
Близкий по устройству инжекторный смеситель описан Торнтоном60.
Кафаров6I исследовал экстракцию бензойной и уксусной
кислот из четыреххлористого углерода водой в очень малых
инжекторах (длина около 75 мм, внутренний диаметр отверстия
сопла примерно 2,5 мм). Достигалась эффективность ступени от
Е = 0,8 до £=1,0 при расходах жидкостей приблизительно от
0,06 до 0,1 м3\ч. Расход энергии был относительно невысок,
величина P/Q изменялась в пределах 1400—40 000 дж/м3. Эти
инжекторы были применены для промышленной экстракции
антибиотиков62.
Сопла и диафрагмы. Смесительные сопла представляют
собой сравнительно простые устройства, которые можно
устанавливать непосредственно в трубопроводах (рис. 243). Обе
жидкости подаются одновременно в сопло или ряд последовательно
установленных сопел; качество их перемешивания зависит от
степени турбулентности, возникающей вследствие перепада
давления в жидкости. Работа диафрагменных смесителей
(рис. 244) основана на том же принципе, и они получили очень
широкое распространение в
процессах очистки легких
нефтяных погонов.
Применяющиеся для
очистки нефтепродуктов диафраг-
менные смесители обычно
состоят из 20—30 тарелок с
отверстиями (диафрагм),
установленных в трубопроводе на
расстоянии ~300 мм друг от
друга. Однако конструкция
этих смесителей не
стандартизована. При удалении
меркаптанов из бензина (процесс
«Дуалеэр») в сутки
перерабатывается около 6200 м3
бензина и экстрагирующего
раствора 63. При высокой
эффективности ступени падение
давление. 242. Инжекторный смеситель ния составляет 0,7 ат (7 X
(Ayr ее, пат США 2531547): {Qi н/мЯ) ^ *
1,11 — масло; 111 — растворитель: /V —эмуль- ' ' J '
сия. ответствует очень малому рас-
488
£П
Рис. 243.
Смесительные сопла(The
Durlcon Co).
Рис. 244. Диафрагменные
смесители (на рис. 244, б тарелки
сдвинуты относительно друг
друга на 90°).
ходу энергии P/Q = 68 дж/м3. Стоимость сопел низка, и это
удешевляет процесс экстракции.
Была измерена поверхность контакта фаз, образующаяся
при прохождении через диафрагму, установленную в смесителе,
двух несмешивающихся жидкостей64'65. Из-за коалесценции
капель поверхность контакта быстро уменьшается с возрастанием
расстояния от диафрагмы. Для системы керосин — вода в
трубе диаметром 25 мм на расстоянии 5,6 м от диафрагмы
поверхность контакта фаз была равна примерно одной десятой ее
исходной величины и оставалась далее постоянной на
расстоянии не менее 9 м. Для труб диаметром 12—50 мм при <f>D —
= 0,02—0,20 и расходах жидкостей (3,8—68) • Ю-3 м3/мин на
расстоянии 178 мм от диафрагмы удельная поверхность
фазового контакта описывается уравнением
2,68
-0,158
Z(*»r(№) №-
-,0,117
0О,878
(Х,45)
Вентили можно рассматривать как регулируемые
диафрагменные смесители. Наиболее подробные сведения о вентилях
как смесительных устройствах приведены в работе Симкина и
Олней66, В их опытах бутиламин экстрагировали из керосина
489
водой при пропускании смеси через вентиль с проходом 25 мм,
причем образующуюся эмульсию направляли в циклон, где
происходило разделение фаз. Это исследование было в основном
посвящено изучению циклона, и авторы приводят лишь весьма
ограниченные данные, касающиеся экстракции.
Кроме того, следует учитывать, что процесс экстракции
происходил одновременно в вентиле и в циклоне, и трудно
установить, какая степень экстракции достигалась только в самом
вентиле. При объемном соотношении керосина и воды 3 : 1
эффективность ступени изменялась от £0 = 0,95 до £0 = 0,633 в
зависимости от суммарной объемной скорости жидкостей,
изменявшейся в пределах (38—76) ■ 10~3 м3/мин. Падение давления в
вентиле соответствовало расходу энергии P/Q=1400—7300 дж/м3
жидкости. Для сравнения отметим (рис. 231), что расход
мощности для этой же системы в сосудах с мешалками при тех же
значениях эффективности ступени составляет 83 000—3900 дж/м3.
При достаточно тонком диспергировании, обеспечивающем
высокие скорости массопередачи, разделение фаз в циклоне было
недостаточным, однако можно использовать более эффективные
отстойники.
Лекок и Черчилль67 исследовали смеситель,
представляющий собой трубу (диаметром 12 мм и длиной 152 мм) с
насадкой из стеклянных шариков диаметром 3 мм, через которую
контактируемые жидкости прокачивали параллельным током,
а полученную смесь направляли в отстойник. Это устройство
можно рассматривать как смеситель диафрагменного типа. Для
системы бутанол (дисперсная фаза, расход 14—100 м3/м2ч) —
вода (сплошная фаза, расход 29,3—156,2 м3/м2ч) средние
величины коэффициентов массоотдачи были равны:
kca = 0,000066 L%5LD (X, 46)
kDa = 0,000117 Z.£75I%75 (X,47)
где L—объемные скорости фаз на входе в смеситель, кмольХ
Хм~2 • сект1.
Эти величины коэффициентов массоотдачи при
исследованных объемных скоростях фаз соответствуют значениям
эффективности ступени, близким к 100%. В опытах практически весь
массообмен происходил в самом смесителе.
Насосы. В качестве смесителей довольно широко применяют
насосы. Обычно жидкости, которые нужно перемешать,
засасывают в насос, где происходит их смешение под действием
рабочего колеса насоса; одновременно смесь жидкостей
перекачивают в отстойник. Такие аппараты использовали при экстракции
фенолов из подсмольных вод коксохимических производств
легкими нефтепродуктами68-69. Однако интенсивные срезывающие
усилия, возникающие в насосе, приводили к образованию в дан-
49Q
ной системе стойких эмульсий. По-
• этому на современных заводах
применяют другие экстракторы.
Эффективность ступени, равная 100%,
была достигнута при экстракции
урана из пульпы, получаемой при
выщелачивании ураносодержащих
руд70; использование в этом
процессе в качестве экстрагента гекса-
на вместо более вязкого керосина
позволяло повысить
производительность экстрактора71.
Другой тип проточного
смесителя с механическим
перемешиванием показан на рис. 245. Этот
аппарат широко применяли в
нефтяной, фармацевтической и других
отраслях промышленности.
Смесители с использованием
ультразвука. При экстракции метанола
в системе толуол — метанол — вода
в обычном распылительном
смесителе было показано72, что применение ультразвука повышает
эффективность ступени. Однако расход энергии, необходимый
для значительного увеличения эффективности, оказался очень
большим.
Рис. 245. Механический
смеситель (Chase, пат. США
2183859).
ЭМУЛЬСИИ И ДИСПЕРСИИ
При перемешивании взаимно нерастворимых жидкостей
образуется эмульсия, или дисперсия одной жидкости в среде
другой жидкости, являющейся сплошной фазой. Нестабильные
эмульсии, в которых капли дисперсной фазы относительно
велики, обычно называют дисперсиями. Принято различать
эмульсии типа «вода в масле» * или «масло в воде» в зависимости от
того, какая фаза (водная или органическая) образует
дисперсную фазу. Хотя в экстракционном процессе водная фаза может
отсутствовать, такое обозначение удобно для описания
дисперсной системы. Условия, определяющие образование эмульсий того
или другого типа, служили предметом многочисленных
исследований. Некоторые результаты, относящиеся к аппаратам с
мешалками, были приведены выше.
Поскольку сферы одинакового диаметра при предельно
плотной упаковке занимают примерно 74% общего объема дисперсии,
* Здесь и ниже под «маслом» подразумевается любая органическая фаза,
нерастворимая в воде, — Доп. ред.
491
f
долгое время считали, что эмульсии с содержанием
дисперсной фазы, превышающим 74 объемн. %, не могут существовать.
Таким образом, при содержании воды от 0 до 26% должны
получаться эмульсии «вода в масле», а при содержании воды 74—
100%—эмульсии «масло в воде»; при содержании дисперсной
фазы от 26 до 74% возможно образование эмульсий того и
другого типов.
На приведенные соотношения, однако, оказывают влияние
помимо плотности упаковки частиц и другие факторы, поэтому
были получены эмульсии с содержанием дисперсной фазы,
значительно большим 74%. Это объясняется тем, что капли
дисперсной фазы, как правило, неодинаковы по размерам. Кроме
того, высоковязкие жидкости склонны образовывать сплошную
фазу.
Большую роль играет присутствие эмульгирующих
поверхностно-активных веществ. Обычно жидкость, в которой
растворен эмульгирующий реагент, стремится образовать сплошную
фазу. Лучшая смачиваемость материала аппарата или его
внутренних устройств одной из жидкостей благоприятствует тому,
чтобы эта жидкость являлась сплошной фазой. В присутствии
твердых примесей, которые лучше смачиваются одной из
жидкостей, последняя, вероятнее всего, образует сплошную фазу. В
отсутствие эмульгаторов эмульсии типа «масло в воде» легче
образуются при низких объемных отношениях масла к воде, и,
наоборот, эмульсии типа «вода в масле» легче получаются при
низких объемных отношениях воды к маслу. Объемное
отношение фаз 74 : 26 безусловно заслуживает внимания, так как
замечено, что эмульсии часто образуют два слоя, в одном из которых
фазы находятся в этом объемном соотношении.
Обращение фаз — переход от одного типа эмульсии к
другому — происходит в том случае, когда эмульсия данного типа
в заданных условиях менее устойчива. Обращение фаз может
быть вызвано разбавлением эмульсии дисперсной фазой, причем
обращение фаз не всегда наступает при объемном соотношении
фаз 74 : 26. Сильное влияние на обращение фаз оказывает
присутствие в эмульсии твердых примесей и поверхностно-активных
веществ. В некоторых дисперсиях, образованных в сосудах с
мешалками, обращение фаз наступает при увеличении числа
оборотов мешалки. Это связано, по-видимому, с
наблюдавшимся, но труднообъяснимым явлением повышения скорости
отстаивания эмульсий, получаемых при большем числе оборотов
мешалки.
Иногда образуются двойные эмульсии, в которых сплошная
фаза одновременно распределена в виде мелких капелек,
заключенных внутри крупных капель другой фазы. Разбавляя
пробу эмульсии, можно определить ее тип: эмульсия легко
смешивается с избытком сплошной фазы и с трудом — с добавляемой
492
жидкостью, образующей дисперсную фазу. Электропроводное^
эмульсий, в которых вода является сплошной фазой, обычно
выше, чем в эмульсиях типа «вода в масле». Более подробные
сведения по этим вопросам можно получить в обзорных
работах 73> 74.
Стабильность эмульсий. С точки зрения жидкостной
экстракции стабильность эмульсий является их наиболее важным
свойством, так как фазы необходимо разделять на каждой ступени
экстракции. Стабильность эмульсий обусловливается
сопротивлением, оказываемым коалесценции капель дисперсной фазы.
Для распада дисперсии на отдельные фазы необходимо, чтобы
последовательно протекали следующие явления: а)
столкновения капель, б) их флоккуляция, или агломерация, в) коалес-
ценция капель.
а) Столкновения капель. Для очень малых
диспергированных частиц число столкновений можно рассчитывать, как для
броуновского движения. В дисперсиях, представляющих
интерес для жидкостной экстракции, капли имеют диаметр порядка
100 мк или более. Поэтому происходит относительно быстрое
осаждение капель и при движении они весьма часто
встречаются друг с другом. Скорость осаждения единичных капель
определяется уравнениями (V.71) — (V,76). Хотя эти уравнения и
нельзя применять для условий стесненного осаждения, однако
они указывают на то, что процесс отстаивания происходит тем
быстрее, чем меньше вязкость сплошной фазы и чем больше
разность плотностей фаз.
б) Агломерация капель. Не все столкновения капель
эффективны и приводят в конечном итоге к разрушению дисперсии.
Если все столкновения окажутся неэффективными, то эмульсия
будет стабильной. Стабильные эмульсии обычно
характеризуются малым размером капель, порядка 1 —1,5 мк и менее.
Однако и некоторые другие факторы в значительной степени
могут повысить стабильность эмульсии, предотвращая
агломерацию частиц.
Две чистые жидкости редко образуют стабильные
эмульсии — почти всегда необходимо присутствие третьего вещества.
Это, конечно, всегда происходит в экстракционных процессах.
Третье вещество обычно понижает межфазовое натяжение;
между стабильностью эмульсии и степенью понижения
межфазового натяжения существует по крайней мере качественная связь.
Особенно сильно понижают межфазовое натяжение и
повышают стабильность эмульсий поверхностно-активные вещества,
адсорбирующиеся на поверхности раздела жидкость —
жидкость. Присутствие твердых частиц способствует повышению
стабильности эмульсий. Если контактный угол между твердыми
частицами и двумя жидкостями имеет конечную величину, то
твердые частицы накапливаются на поверхности раздела; при
493
этом большая часть объема каждой частицы располагается в
сплошной фазе. Это может предотвращать агломерацию.
В присутствии электролитов капли вследствие адсорбции
ионов на поверхности раздела могут приобретать заряд.
Отталкивание одинаково заряженных капель препятствует агломерации.
Анионы обычно лучше растворимы в органических жидкостях,
чем катионы, и если диспергирована органическая фаза, то
капли заряжаются отрицательно. Заряженная капля находится в
окружении неподвижного диффузного слоя ионов
противоположного заряда. Когда такой комплекс движется относительно
неподвижной сплошной фазы, на поверхности сдвига возникает
потенциал, так называемый дзета-потенциал. Последний
является важным фактором, препятствующим агломерации75.
в) Коалесценция капель. Для слияния двух или более
агломерированных капель необходим разрыв пленки, окружающей
капли. Межфазовое натяжение является движущей силой этого
процесса, и для жидкостей, находящихся в равновесии, чем
выше межфазовое натяжение, тем больше скорость коалесценции.
В случае сильно вязкой сплошной фазы в связи с увеличением
времени, необходимого для утоньшения и разрыва пленки
между каплями, коалесценция замедляется. Температура, которая
влияет как на вязкость, так и на межфазовое натяжение, имеет
важное значение 76. Распределение времени коалесценции
агломерированных капель, по-видимому, беспорядочно77'78. При
коалесценции капель по крайней мере на плоской поверхности
возможно образование мелких вторичных капель, что
замедляет общую скорость процесса.
Если жидкости не находятся в равновесии, то изменение
межфазового натяжения при экстракции между каплями и
окружающей средой сказывается на скорости коалесценции34. При
наличии около капли градиента межфазового натяжения
жидкость будет перемещаться из области низкого натяжения в
область высокого. Так, если капли содержат гораздо больше
растворенного вещества, чем сплошная фаза, небольшое
количество жидкости, находящейся между каплями, при экстракции
достигнет равновесия гораздо быстрее, чем в массе жидкости,
окружающей пару капель. Если эта разность концентраций
приводит к понижению межфазового натяжения в области
между каплями, то жидкость будет перемещаться из данной
области. При этом разделяющая капли пленка становится тоньше и
коалесценция облегчается. Обратное явление наблюдается,
когда градиент межфазового натяжения таков, что движение
жидкости направлено в область между каплями. Следовательно,
направление экстракции (к каплям или из капель) может
сильно влиять на скорость коалесценции.
Нестабильные эмульсии. Мейсснер и Чертоу79 описали
поведение нестабильных дисперсий сразу же после прекращения
494
перемешивания. По прекращении перемешивания смесь
жидкостей распадается в результате осаждения и коалесценции на
два слоя (первичный распад эмульсии); этот процесс протекает
быстро, если вязкость сплошной фазы невелика. В течение
указанного периода в эмульсии можно различить три слоя: легкой
жидкости в верхней части аппарата, тяжелой фазы в его нижней
части и между ними — промежуточный слой еще не
распавшейся эмульсии. Этот период считается законченным, когда
промежуточный слой исчезает, а верхний и нижний слои жидкости
соприкасаются друг с другом, образуя ясно выраженную
границу раздела. Оба слоя могут оказаться прозрачными, но, как
правило, один из них, а иногда и оба являются мутными и
содержат в туманообразном состоянии небольшое количество
другой фазы. Мутность в конце концов исчезает, и оба слоя
становятся прозрачными (вторичный распад эмульсии).
Обычно процесс вторичного распада протекает очень
медленно. Чаще всего фаза, которая находится в большем
количестве, после первичного распада эмульсии оказывается мутной,
а другая фаза — прозрачной; такое явление наблюдается чаще
всего при больших объемных соотношениях фаз. При объемном
соотношении, большем трех (что соответствует, как отмечалось
выше, максимальной плотности упаковки сфер), слой фазы,
находящейся в меньшем количестве, обычно после первичного
отстаивания совершенно прозрачен.
Отстойники и прочее оборудование
Первичный распад дисперсий, как правило, протекает очень
быстро, и для полного разделения фаз достаточен очень
небольшой промежуток времени. В периодических процессах смесь
чаще всего отстаивается в смесителе. Для разделения фаз в
непрерывном процессе смесь направляют в отдельную емкость или
другой аппарат.
Гравитационные отстойники. Простейший гравитационный
отстойник, или декантер, чаще всего представляет собой
горизонтальный аппарат, подобный
показанному на рис. 246.
Чтобы предупредить возмущение
жидкости струей эмульсии,
поступающей в аппарат, против /
входного штуцера устанавли- ~*
вают перфорированную
отбойную перегородку с круглыми
отверстиями или щелями.
Основной объем отстойника
используют для создания лами
Рис. 246. Гравитационный отстойник:
/ — дисперсия; // —легкая фаза; /// — тяже-
лая фаза; IV — вентиль для периодического
нарнОГО ДВИЖеНИЯ ЖИДКОСТИ, удаления загрязнений с поверхности раздела.
495
Рис. 247. Отстойник с наклонными
перегородками [В и г t i s, К i г k b r i d e,
Trans. Am. Inst. Chem. Engrs., 42, 413
(1946)]:
/ — дисперсия; // — легкая фаза; /// — тяжелая
фаза.
ai Перегородка при котором возникают ус-
П /iiiVK ловия, способствующие уско-
Х^-У>ч\ рению отстаивания. Через
отводной кран,
расположенный на противоположной по
ходу жидкости стороне
аппарата, периодически
удаляют твердые частицы,
собирающиеся на поверхности
раздела фаз даже в том
случае, если поступающие
жидкости кажутся
совершенно прозрачными.
Тяжелая фаза выходит из аппарата через трубопровод с устройством
для разрыва сифона, предупреждающим полное опорожнение
отстойника. Отвод осуществляется на уровне, определяющем
положение поверхности раздела в аппарате в соответствии с
уравнением
6pr-f Дрг = арг + (с — в) рл + Дрл (Х.48)
Размеры а, Ь и с показаны на рис. 246, а величины Ар
представляют собой потери давления в соответствующих
трубопроводах.
Для более точного контроля за положением поверхности
раздела необходимо установить на трубопроводе (для выхода
тяжелой фазы в точке D) регулирующий вентиль и уровнемер;
применение устройства для разрыва сифона в этом случае не
требуется. С увеличением скорости поступления исходной смеси
слой неотстоявшейся эмульсии, распространяющийся обычно на
всю длину отстойника, увеличивается по толщине до тех пор,
пока наконец эмульсия не начинает попадать в жидкости,
покидающие отстойник.
Конструкции отстойников не стандартизованы, и в настоящее
время имеются сотни различных модификаций простейшего
гравитационного отстойника. Вместо горизонтальных аппаратов в
некоторых случаях применяют вертикальные отстойники, причем
считается предпочтительным устанавливать последние под
некоторым углом к горизонту. Смесь на отстаивание обычно
вводят на уровне раздела фаз. Часто для направления потоков в
отстойнике устанавливают перегородки, которые иногда
используют как отбойные. С помощью пакета наклонных
параллельных перегородок в отстойнике (рис. 247) достигается
ламинарное движение жидкости и уменьшается высота осаждения
капель, что также способствует ускорению отстаивания.
На рис. 248 показан отстойник оригинальной конструкции,
применяемый для очистки смазочных масел в процессе «Дуо-
Сол». Поступающая эмульсия обтекает пучок коротких патруб-
496
ков, служащих для распределения жидкости, и движется, как
показано на рисунке, по узкому горизонтальному каналу
прямоугольного сечения, расположенному вдоль оси аппарата. Так
как высота канала мала, отстаивание фактически заканчивается
за время прохождения эмульсии по каналу. Отстоявшиеся
жидкости выходят либо в конце канала, либо через отверстия в его
стенках. Тяжелая фаза движется затем в пространстве под
каналом, легкая — в пространстве над ним. Дополнительно
отстоявшаяся жидкость может перетекать из одного пространства
в другое по наклонным коротким патрубкам, расположенным
в начале канала. Жидкости отводят через штуцера, перед
которыми в корпусе устанавливают успокоительные перегородки,
как показано на рисунке.
Применяют и другие, гораздо более сложные конструкции
отстойников. Однако в настоящее время наблюдается тенденция
использовать наиболее простые и компактные конструкции этих
аппаратов с тем, чтобы уменьшить их стоимость. В
нефтехимической промышленности большая часть отстойников рассчитана
на время пребывания от получаса до одного часа. При наличии
в отстойнике перегородок, обеспечивающих ламинарное
движение жидкостей и малую высоту осаждения, это время можно
сократить до 5—10 мин, если вязкость сплошной фазы невелика
и не наблюдается аномальных явлений, связанных с коалес-
ценцией.
Время окончательного отстаивания после первичного
распада дисперсии, как правило, значительно; поэтому не следует
Г/о А-А
Рис. 248. Отстойник, применяемый в «Дуо-Сол- .роцессе» (Мах В. Miller
and Co., Inc.):
/ — дисперсия; // — легкая фаза; /// — тяжелая фаза.
32 Р. Е. Трейбал 467
стремиться к достижению
полного отстаивания дисперсии в
самом отстойнике, если за
отстойником следуют другие ступени
многоступенчатого экстрактора. Унос
небольших количеств одной фазы
с другой приводит лишь к
небольшому уменьшению
эффективности ступени [см. уравнение
(X, 53)]. Конечные же продукты,
выходящие из каскада, должны
быть свободны от уносимой
жидкости, так как иначе теряется
экстрагент.
До сих пор не разработано
удовлетворительных
теоретических методов расчета наилучшей
формы и размеров отстойника
для разделения эмульсии с
заданными свойствами, поэтому в
каждом конкретном случае
приходится проводить
предварительные опыты. Отстаивание одной
системы жидкость — жидкость
было подробно исследовано Рьоном
и др.53. В этой работе изучали
отстаивание смеси водных
растворов урана и керосина,
содержащего алкилфосфаты, в вертикальных гравитационных
отстойниках цилиндрического сечения. Результаты исследования
приведены на рис. 249 и, по-видимому, отражают общий
характер зависимости высоты слоя неотстоявшейся эмульсии от
удельной нагрузки отстойника, которую можно получить и для других
систем жидкость — жидкость.
Те же исследователи рекомендуют, чтобы площадь сечения
отстойника в плане была вдвое больше соответствующей
площади сечения, при которой толщина слоя неотстоявшейся
эмульсии для заданной производительности отстойника равна высоте
последнего. В опытах с указанной выше системой для
разделения эмульсии было достаточно времени осаждения, равного
1,4 мин. Библиография по вопросам отстаивания приведена в
обзоре Ингерсола80. •
Процесс извлечения урана из щелоков с помощью экстраген-
тов, содержащих углеводороды, можно провести значительно
экономичнее, если не осветлять раствор перед экстракцией, так
как в этом случае отпадает необходимость в установке
сгустителей и фильтров. Чтобы избежать образования стабильных
&
I
8
I
0,6
0,2
о,'
0,06
ори
0,02
0.01
III ,
о/ д^
»2 л.в
аЗ *7
ш4 од
— fi'f
1 L
if
п
г— i
/
i
!
/V
Л'2
*3
1
06 1
4 6 e io го
й Фиктивная скорость
водной фазы, мз-м~г-ч~'
Рис. 249. Непрерывное
гравитационное отстаивание смеси
водный раствор урана — керосин,
содержащий алкилфосфаты:
/ — эмульсии типа «вода в масле»:
У —диаметр отстойника 0,152 м; объемное
соотношение водной и органической
фаз 1 : 1; 2-0,152 м, 1 : 2; 3—0,55 м, 1 :1;
4— 0,56 м, 1:2; 5 —1,22 м, 1 : 1; 6— 1,22м.
1 :2; 7-2,14 м, 1:2; «-4,88 м; 1: 1.
// — эмульсия типа «масло в воде»:
/ — 0,152 м, 4 : 1 и 1 : 1; 2—0,56 jk, 4 : 1 и
1 : 1; 3-1,22 м, 4 : 1 н 2 : 1.
498
эмульсий при экстракции из таких пульп,
необходимо использовать органическую фазу в качестве
сплошной; это достигается с помощью
рециркуляции больших количеств экстрагента, направляемых
из отстойника обратно в смеситель70. Особенно
важно в данном случае предотвратить потери
экстрагента в результате уноса его с пульпой при
высоком содержании в последней твердых частиц. Эту
задачу можно по крайней мере частично решить
разбавлением исходной суспензии водой и
добавкой органических сульфонатов, чтобы изменить
смачивающие свойства твердых частиц81.
Циклоны. Основное достоинство циклонов —
очень малое время пребывания в них разделяемых
жидкостей (при удовлетворительной работе
циклона— порядка нескольких секунд). Это дает
экономический эффект вследствие компактности
аппаратуры и приводит к уменьшению необходимого объ- *""
ема экстрагента. Кроме того, в некоторых экс- рис. 250.
тракционных процессах уменьшение времени пре- Гидроциклон
бЫВаНИЯ ВажНО С ТеХНОЛОГИЧеСКОЙ ТОЧКИ ЗреНИЯ. /-дисперсия;
Например, малое время пребывания желательно дУкт;ерш-н1ж-
при экстракции различных антибиотиков, так как ннй продл".
в условиях экстракции быстро ухудшается
качество продукта. В процессах экстракции из смесей с высокой
радиоактивностью при коротком времени пребывания
уменьшается воздействие последней на экстрагент; кроме того, при
малом времени пребывания облегчается решение вопросов,
связанных с критическими объемами радиоактивных жидкостей
в аппаратуре.
Типичная конструкция циклона показана на рис. 250.
Разделение фаз в циклоне происходит под действием центробежных
сил. Легкая фаза удаляется из верхней части аппарата
(верхний продукт), а тяжелая — из нижней части (нижний продукт).
Соотношение потоков, выходящих из циклона, можно
поддерживать не обязательно равным соотношению фаз в
поступающей смеси, регулируя это соотношение с помощью вентилей на
выходных трубопроводах. Эффективность разделения в циклоне
определяется уравнением 6в-82-га
Р ^л. верх.
'т. верх.
<?л.
(Х,49)
При полном разделении фаз Q„. верх. = <2л. см. и QT. верХ.=0,
откуда Z;S=1,0, и это требует, чтобы соотношение выходящих
потоков было равно соотношению фаз в питании. Однако
поддержание такого соотношения выходящих потоков еще не
гарантирует получения Es=lfl. в отсутствие разделения
32* 499
Ул. верх. /Qt . верх. — Чл. см. см. и Eg— 0. При низком
соотношении выходящих потоков можно получить чистую легкую фазу в
верхнем продукте или чистую тяжелую фазу в нижнем
продукте; однако значение £s при этом будет невысоким. Как
правило, величина £s максимальна при некотором промежуточном
соотношении выходящих потоков. При больших скоростях
жидкостей вследствие их турбулентного движения в циклоне в нем
происходит распад капель и повторное перемешивание; в этих
условиях величина £s стремится к максимуму при соотношении
выходящих потоков, равном единице 66>84.
Исследуя процесс отстаивания в циклоне, Тип и Вудс83 не
смогли получить удовлетворительного разделения смесей изобу-
танола с водой; Es приблизительно было равно 0,1.
В опытах Симкина и Олней66 величина Es достигала 0,91
(для смесей воды с керосином или более вязким светлым
маслом), но ни одна фаза конечного продукта не была чистой.
В одной серии опытов эффективность разделения
соответствовала £8=0,71, когда средний диаметр капель в смеси был
равен 1,56 мм, и снижалась до £8=0,095 при размере капель
0,73 мм. Как уже указывалось (см. стр. 490), в условиях
эффективной экстракции (мелкие капли) разделение фаз было
плохим.
Установленные оптимальные соотношения геометрических
размеров циклона (рис. 250), по-видимому, зависят от свойств
взаимодействующих жидкостей. Аналогичные результаты
получил Хитчон84, испытывавший циклоны небольшого размера.
Полного разделения фаз можно достигнуть в двух последовательно
соединенных циклонах. В первом циклоне отбирают сверху
чистую легкую фазу (верхний продукт), а нижний продукт подают
во второй циклон. Из последнего отбирают снизу чистую
тяжелую фазу, а верхний продукт рециркулирует — направляется
в первый циклон84'85, причем рециркуляция должна
осуществляться с помощью насоса.
Центрифуги. Быстрое разделение жидкостей под действием
центробежных сил достигается в центрифугах даже при очень
малой разности плотностей фаз. Применяют вертикальные
трубчатые сверхцентрифуги, у которых барабан вращается со
скоростью 15 000 об/мин или более, и жидкостные сепараторы.
Вертикальные сверхцентр:ифуги вследствие сравнительной простоты
и легкости очистки используют чаще86. Жидкостные сепараторы
с коническими тарелками применяют обычно в тех случаях,
когда легкая фаза является дисперсной; эти аппараты могут
работать под давлением87..
Трубчатые сверхцентрифуги и жидкостные сепараторы
можно применять для выделения твердой фазы, когда, например,
обработке подвергают концентрированный шлам. В результате
отделения твердых примесей облегчается агломерация и коалес-
500
ценция дисперсий, стабилизированных в присутствии твердых
частиц. Центрифуги — сравнительно дорогие машины. Для
экстракции их применяют только в тех случаях, когда необходимо
малое время пребывания контактирующих жидкостей в
аппарате или когда затруднительно разделение фаз другим способом.
Ускорение коалесценции. Добавление избытка дисперсной
фазы к сплошной часто приводит к быстрой коалесценции,
особенно если разбавление соответствует отношению дисперсной и
сплошной фазы 3 : 1 (значение соотношения фаз 3 : 1
подчеркивалось ранее, стр. 492). Бикерман считает74, что для эмульсий
типа «вода в масле» ускорение коалесценции объясняется
присутствием растворимых в органической фазе эмульгаторов,
которые способствуют тому, чтобы органическая фаза стала
сплошной. При разбавлении до соотношения свыше 3 : 1 обычно
происходит обращение фаз, и получаемая эмульсия типа «масло
в воде» уже не является стабильной.
Описанный метод коалесценции исследовали Мейсснер и Чер-
тоу79. В этой работе было показано, что после первичного
распада эмульсии небольшое количество другой фазы, которое
имеется в находящейся в избытке фазе, может быстрее коалес-
цировать и осаждаться при добавлении приблизительно четырех
объемов дисперсной фазы. При этом, однако, избыточная фаза
после вторичного отстаивания остается мутной.
Изучение большого числа двух- и трехкомпонентных смесей
показало, что для всех смесей, за исключением смесей
глицерина с нитробензолом, коалесценция этого остаточного
количества фазы, эмульгированной в избыточной фазе, не происходит,
если дисперсная фаза неполярна. Если же дисперсная фаза
представляет собой полярное соединение, то наблюдается
коалесценция. Так, бензол, в котором в виде мути находятся капли
воды, можно осветлить после перемешивания с избытком воды.
Рис. 251. Первичное и вторичное отстаивание жидких двухфазных смесей:
1 — смеситель; 2, 3 —отстойники; / — смесь жидкостей А —В; Я —чистая фаза В;
// — жидкость А после первичного отстаивания, содержащая во взвешенном состоянии
фазу В; IV — жидкость В после первичного отстаивания, содержащая во взвешенном
состоянии фазу А; V — чистая фаза А.
601
В то же .время вода, содержащая капельки бензола, не
осветляется при добавлении последнего. Если добавлять к эмульсии
хлористоводородную кислоту, т. е. распределить между фазами
полярное соединение, то становится возможным осветление
обеих фаз. Аналогично можно разделить эмульсии нитробензола
и воды.
Для отстаивания смесей, в которых дисперсная фаза по-
лярна, Мейсснер и Чертоу предложили схему процесса,
приведенную на рис. 251. В отстойнике / производится первичный
распад дисперсии веществ А и В. Чистая фаза В выводится, а
мутный слой А перемешивается с избытком фазы В. При
окончательном разделении эмульсии в отстойнике 2 образуются
чистая фаза А и мутный слой В, который подвергается
рециркуляции. Возможны и другие варианты проведения такого процесса
разделения, но во всех случаях необходимы большие количества
рециркулирующих жидкостей.
Устройства для ускорения отстаивания. Для ускорения
разделения фаз используют коагуляторы, разделительные
мембраны и электростатические коагуляторы. Кроме того, можно
добавлять реагенты, облегчающие разрушение стабильных
эмульсий, при образовании которых применение жидкостной
экстракции в промышленном масштабе становится, как правило,
невозможным.
Коагуляторы. Мелкие капли тонких дисперсий можно
подвергнуть коалесценции, пропуская дисперсию через коагулятор;
получающиеся при этом крупные капли отстаиваются быстрее.
В коагуляторах эмульсию пропускают через слой пористого или
волокнистого материала, ускоряющего коалесценцию. При
движении эмульсии через такой слой разрываются вязкие пленки,
окружающие капли и препятствующие коалесценции. Вещество
капли должно лучше смачивать твердую поверхность пористого
материала. Поэтому капли прилипают к ней и, коалесцируя,
укрупняются. Крупные капли выносятся затем потоком
сплошной фазы.
В коагуляторах обычно используют пористые материалы с
большим отношением поверхности к объему, обеспечивающие
небольшие проходы для воздействия на весь объем дисперсии.
Необходимо также, чтобы материал обладал низким
гидравлическим сопротивлением и предпочтительно смачивался
дисперсной фазой. Кроме того, материал должен быть механически
прочным, чтобы противостоять возникающему перепаду
давления, и химически инертным по отношению к движущимся
жидкостям.
Силиконовые материалы обычно гидрофильны и могут
применяться в тех случаях, когда дисперсной фазой является вода.
Уголь, органические пластические массы, ткани с покрытием из
пластмасс и тому подобные материалы обычно обладают гидро-
502
фобными свойствами (отталкивают воду и смачиваются
неводными жидкостями) и используются при диспергировании
органической фазы. Применяют также слои гранулированных
твердых м-атериалов, такие, как песок или диатомовая земля,
древесная, стальная, медная или стеклянная стружка,
стекловолокно, специальные пластмассы, а также полиэтиленовые и
полипропиленовые пленки. Возможно эффективное применение в
коагуляторах обычных насадок для колонн, например колец Ра-
шига. В одной из работ использовали кольца из
полипропилена, плавающие в гравитационном отстойнике на поверхности
раздела водной и органической фаз.
Наиболее подробные характеристики подобных
коагуляторов приведены в работах Буртиса и Киркбрайда 88, а также Хей-
са и др.89. В их опытах дисперсию соленой воды в нефтяных
маслах пропускали через слой стекловолокна, имевший
небольшую толщину. Степень разделения была неодинаковой для
различных систем жидкость — жидкость; лучшие результаты
достигались при фиктивных скоростях жидкостей от 0,076 до
0,3 м/мин. Для коагуляции эмульсий применяли и другие
материалы, не выпускаемые промышленностью в больших
количествах 90.
Разделительные мембраны. Если размер капилляров
пористого вещества очень мал, то, несмотря на способность
жидкости, хорошо смачивающей твердую поверхность, легко
проходить через капилляры, прочные пленки, разделяющие фазы,
могут блокировать капилляры и не пропускать жидкость, не
смачивающую твердую поверхность. При достаточном давлении
возможно разрушение пленок, что позволит жидкости, не
смачивающей твердую поверхность, проходить через капилляры;
регулированием давления, в соответствии с размером пор и
межфазовым натяжением, можно обеспечить разделение фаз.
Сила, оказывающая сопротивление растяжению пленки
между фазами на входе в цилиндрический капилляр диаметром
d0, равна произведению периметра капилляра и межфазового
натяжения о. Сила, стремящаяся разорвать пленку, равна
произведению поперечного сечения капилляра и перепада давления
Др. При равновесии
ndi.
nd0o = —^- Ар (X, 50)
Например, если дисперсию бензол — вода с межфазовым
натяжением 35 дин/см пропускать через пористый фарфор,
который хорошо смачивается водой (и был предварительно смочен
ею), то при максимальном размере пор, равном 2 мк,
равновесный перепад давления, согласно уравнению (X,50), составит
0,7 ат (7 • 104 HJM2). Следовательно, если перепад давления под^
Держивать меньшим, то вода будет проходить по капиллярам, а
603
fJ7
■ Сепаратор
t=gw
Коагулятор
L-
.ОТ
=c
4l
"X
бензол задерживаться, т. е.
будет достигнуто разделение
фаз. Соответственно этому
через слой гидрофобного
вещества будет проходить бензол и
не будет проходить вода.
Разделительные мембраны такого
типа изготавливаются из
различных материалов, таких, как
фарфор или бумага,
пропитанная пластмассой, причем по-
следняя может быть гидро-
в1в фильной и гидрофобной90.
На практике перед подачей
на разделительную мембрану
дисперсию обычно сначала
пропускают через коагулятор,
чтобы предварительно
осуществить отстаивание основного
количества дисперсной фазы
для уменьшения нагрузки на
мембрану. На рис. 252
показан аппарат, применяющийся
для разделения нестабильных
дисперсий и работающий на
основе указанных выше
принципов. Эмульсия проходит вначале через пористый слой, в
котором в результате коалесценции дисперсной фазы
происходит частичное отстаивание фаз. Затем тяжелая фаза проходит
через слой пористого твердого материала с очень малым
размером пор, предварительно обработанный таким образом,
чтобы он лучше смачивался тяжелой фазой.
Рис. 252. Коагулятор с мембранным
сепаратором (Selas Corp. of America):
/ — дисперсия; // — легкая фаза; /// —
тяжелая фаза.
Подерхносто
раздела
Рис. 253. Сепаратор для отделения воды от жидкого топлива (Warner —
Lewis Co. Division, Fram Corp.):
/ — коагулятор диспергированных частиц тяжелой фазы; 2 —разделительная мембрана,
проницаемая для легкой (сплошной) фазы; 3 — воздушник; / — дисперсия; // — легкая фаза;
/// — тяжелая фаза.
Б04
Капли дисперсной фазы не могут пройти через поры, они
коалесцируют, и дисперсная фаза поднимается в верхнюю часть
аппарата. Такие аппараты рассчитаны на производительность
0,6—6,8 м3/ч, причем в них достигается полное разделение
фаз.
Другой аппарат для удаления диспергированных капелек
воды из бензина или керосина показан на рис. 253. В нем
достигается практически полное удаление воды из органической
фазы. Аппараты такого типа имеют производительность от 68
до 300 м3/ч, причем для проведения процесса обезвоживания
бензина или керосина необходима гидрофобная разделяющая
мембрана. Известны также другие конструкции аппаратов
данного типа.
Электростатические коагуляторы. Если дисперсию воды в
непроводящей сплошной фазе, стабилизированную ионами,
поместить в электрическое поле, то капли поляризуются и
располагаются в цепочки вдоль силовых линий поля91. Это приводит к
увеличению числа столкновений капель и, следовательно, к
коалесценции. Данный метод широко применяют для обессолива-
ния эмульсий, образованных сырыми нефтепродуктами и
рассолом, а также в других промышленных экстракционных
процессах92. Кроме того, электрические методы используют в
процессах экстракции урана для уменьшения уноса водной фазы
экстрактом 93.
Коалесценцию обычно производят в переменном
электрическом поле напряженностью 80—160 кв/м. При быстром
движении капелек в таком поле увеличивается число столкновений.
Реже применяют постоянный ток значительно меньшего
напряжения. Эмульсию пропускают через отстойник, в котором
установлено два или более электродов. Один из них соединен с
корпусом аппарата, другой находится в движущейся жидкости и
изолирован от стенок аппарата.
Унос и эффективность ступени. При неполном разделении
(отстаивании) дисперсии часть одной жидкой фазы уносится
другой фазой; при этом, если рассматриваемая ступень
экстракции является частью противоточного каскада, уносимая
жидкость движется в направлении, обратном нормальному
направлению движения. В случае трудно отстаивающихся систем унос
неизбежен и приводит к уменьшению общей эффективности
каскада.
Вляние уноса на общую эффективность каскада изучал Слей-
хер94. Он проводил математический анализ для противоточной
экстракции в случае применимости уравнений Кремзера
(уравнения (VI, 125) — (VI, 128)] и принятия следующих допущений:
унос каждой фазы постоянен и одинаков для всех ступеней;
фазы, выходящие с концов каскада, отстаиваются полностью и
не содержат уноса;
605
в каждом смесителе происходит полное перемешивание
обеих фаз;
коэффициенты массопередачи одинаковы для всех ступеней;
массообменом в отстойниках можно пренебречь.
Величину уноса выражали следующим образом:
ед — количество рафината, уносимое экстрактом из каждой
ступени, отнесенное к количеству поступающего в
каскад питания;
еЕ — количество экстракта, уносимое рафинатом из каждой
ступени, отнесенное к количеству поступающего в
каскад экстрагента.
Была получена довольно сложная зависимость, которую
решали на цифровой вычислительной машине для следующего
интервала переменных: число ступеней 3—10; фактор экстракции
е = 0,25—4,0; к. п. д. ступени по Мэрфри, рассчитанный по рафи-
нату, £мн=0,23—1,0; величины уноса ен и вЕ от 0 до 0,8.
Результаты расчета с точностью до 5% описываются
уравнением
«, ig (l/«) + («„ — 1) lg Л
пп lg (1/в)
(Х,51)
где пг — предполагаемое число реальных ступеней в отсутствие
уноса;
пге — число ступеней при наличии уноса.
Величина Я, равна:
х = , р м*К Г , Г (X. 52)
Последнее уравнение точно для £мн=1,0. Если унос
отсутствует, то общая эффективность E0 = nlnr выражается
уравнением, приведенным в табл. 16 (в функции от EMR). Умножая
это уравнение на уравнение (X, 51) и производя
перегруппировку членов, получаем:
„ _ 'g d/g) ( « Л , , ,Y „,
n"==-iiT-l^-1j + 1 (X'53)
Для частного случая е=1,0 имеем:
«„ = -g5- [1 + Ет (ей + ее)] - Emr{zr + гЕ) (X, 54)
MR
Общая эффективность при наличии уноса Е0е равна п/пгг.
Другой анализ проблемы уноса выполнен для случая, когда
величина уноса мала, а в отстойнике устанавливается равновесие
между увлекаемыми каплями и уносящей их жидкостью. В
пределах допустимых е влияние уноса на пГ незначительно. Из
уравнения (X, 53) получаются во всяком случае завышенные
значения пг&.
506
Для иллюстраций степени влияния уноса на общую эффективность сме-
сительно-отстойного каскада рассмотрим случай экстракции, требующий
применения пяти теоретических ступеней при факторе экстракции е =2,0 и
эффективности каждой ступени £MH = 0,90. В отсутствие уноса (табл. 16,
стр. 447) имеем:
Е lg[l+/^ (l/«-D Oo73
и пТ—5/0,873=5,80, т. е. необходимое число реальных ступеней равно шести.
Число необходимых реальных ступеней при наличии уноса, согласно
уравнению (X, 53), будет равно:
ER
0
0
0,065
0,021
0
0,387
0,129
гЕ
0
0,026
0
0,021
0,194
0
0,129
5,8
птг
= 6,0 = лг
6,0
6,0
6,0
7,0
7,0
7,0
Некоторая непрозрачность сплошной фазы, наблюдаемая после
первичного отстаивания, обычно обусловлена очень незначительным количеством
унесенной дисперсной фазы. Из рассмотренного примера становится
очевидным, что лишь чрезвычайно плохое разделение фаз в отстойниках может
вызвать необходимость увеличения числа реальных ступеней смесительно-от-
стойного экстрактора. Поэтому увеличение продолжительности пребывания
эмульсии в отстойниках сверх времени отстаивания, нужного для первичного
осветления рафината и экстракта, нецелесообразно. Конечные продукты,
выходящие из смесительно-отстойного каскада, должны быть, однако, полиостью
отстоявшимися.
СМЕСИТЕЛЬНО-ОТСТОЙНЫЕ
ЭКСТРАКТОРЫ В ПРОМЫШЛЕННОСТИ
Смесительно-отстойный экстрактор включает в себя
несколько ступеней, соединенных между собой таким образом, чтобы
обеспечить прямоточное или противоточное движение
жидкостей. Каждая ступень состоит из смесителя и отстойника, но
может включать в себя также дополнительное оборудование,
например коагуляторы. Типичная схема устройства смесительно-
отстойного экстрактора показана на рис. 254; возможны,
конечно, и другие типы устройств для смешения и разделения фаз, а
также другое расположение аппаратов. Так, например, при
ограниченной производственной площади отстойники можно уста>
навливать один над другим, а смесители и насосы — на уровне
пола69. Число различных вариантов конструкций смесителей-
отстойников очень велико. Ниже рассмотрены только некоторые
507
ш
~*<
Стуавнь 1
.'
Ступень <?
з^ УС
Ступень 3
в
итстиинаН
Рис. 254. Трехступенчатая противоточная экстракционная установка:
/, 4, 7 — смесители; 2, 5, 8 — коагуляторы; 5, 6, 9 —отстойники; / — исходная смесь;
// — экстрагент; /// — конечный экстракт; IV — конечный рафинат.
наиболее важные и оригинальные схемы устройства смеситель-
но-отстойных экстракторов, нашедших промышленное
применение.
В экстракторе Холлей — Мотта95'98 (рис. 255) устранена
необходимость в насосах для перекачки жидкостей. Устройство
одной ступени этого экстрактора показано на рис. 255, а.
Дисперсия перетекает в отстойник по средней трубе, и отстоявшиеся
фазы могут рециркулировать, т. е. возвращаться в смеситель.
Таким образом, соотношение фаз в смесителе можно
поддерживать независимо от
соотношения поступающих в него
потоков. Это особенно
важно в тех случаях, когда
последнее слишком мало или
когда желательно (для
лучшего отстаивания), чтобы
одна фаза была сплошной,
и это невозможно
осуществить при данном объемном
соотношении фаз без
рециркуляции. Рециркуляция
должна приводить к усилению
продольного перемешивания
в ступени: однако обычно
в смесителе достигается
практически полное
перемешивание фаз, поэтому
эффективность ступени
уменьшается мало.
Для уменьшения числа
Y^N
Ш— -=Л Ь^г
Рис. 255. Экстрактор Холлей-Мотта:
в —одна ступень в разрезе:
/ — /// — легкая фаза; II—IV— тяжелая фаза.
б—четырехступенчатая противоточная установка: TDv6onDOBOHOR ЧКГТПЯКтпгу
/-исходная смесь; //-экстрагент; ///-конеч- ^ » лл_„ dKLlpdKlop
ный экстракт; /V —конечный рафинат.
Холлей — Мотта имеет
508
Рнс. 256. Смесительно-отстойный экстрактор [Coplan, Davidson,
Zebroski, Chem. Eng. Progr., 50, 403 [1954)]:
/ — тяжелая фаза; // — легкая фаза; ///, /V —переток для тяжелой фазы; V —переток
для легкой фазы.
плане вид, показанный на рис. 255, б. Противоточное движение
жидкостей по каскаду осуществляется только под действием сил
тяжести, и поэтому происходит постепенное изменение уровней
в отстойниках. Выше указывалось на желательность
использования всасывающего действия мешалок, которое можно
усилить при соответствующем расположении перегородок в
смесителе.
Экстракторы Холлей — Мотта97'98 рассчитаны на очень
большую производительность, например 60 м3/ч. Конструкция
Холлей— Мотта получила дальнейшее развитие в экстракторах
ящичного типа, разработанных для экстракции из
радиоактивных растворов, хотя идея, заложенная в конструкции этих
аппаратов, уже давно известна ".
Рис. 257. Смесительно-отстойный экстрактор (рис. 256) в разрезе [Chem.
Eng. Progr., 50, 403 (1954)]:
я. ф, —легкая фаза; т. ф. — тяжелая фаза.
609
ш п
Рис. 258. Экстрактор Керра-Мак-Ги для извлечения урана:
/ — смеситель; 2 — отстойник; / — исходный водный раствор; //, ///— раф.шат; IV,
V—экстракт; VI — экстрагеит; V// —воздух.
Ящичные экстракторы занимают относительно небольшую
производственную площадь, промежуточные трубопроводы в
них отсутствуют. Одна из конструкций ящичного экстрактора
(«Pump-Mix» или KAPL-extractor 10°) показана на рис. 256 и 257.
В этом экстракторе смесители и отстойники имеют общие
плоские стенки. Мешалки работают как насосы, засасывая
жидкость через полый вал и выбрасывая ее через турбинку самой
мешалки. В главе IX указывалось, что такие экстракторы
используют в качестве лабораторных (модельных) аппаратов,
однако они находят также применение в промышленном
масштабе. Конструктивные детали аппаратов и мешалок описаны
в литературе100-102. Получены некоторые характеристики этих
аппаратов при изучении теплообмена между двумя
жидкостями103'104. Существует много конструкций ящичных
экстракторов; их описание можнс найти в обширной литературе,
посвященной технологии атомного горючего 105.
Более экономичный экстрактор представлен на рис. 258. На
рисунке схематически показаны две ступени четырехступенчатых
экстракторов, применяемых
для извлечения урана на
заводе «Kerr-McGee Oil
Industries» в Шипроке (Нью-
Мексико) 106'107. Внутри
цилиндрических деревянных
отстойников (диаметром
4,9 м и высотой 2,1 м)
находятся смесители из
нержавеющей стали
размерами 1,2x1,2 м. В экстрактор
поступает 22,7 м3/ч
обогащенного ураном раствора
после выщелачивания и
Рис. 259. Экстрактор Керра-Мак-Ги для
реэкстракции урана:
/ — смеситель; 2 — отстойник; / — экстракт из
экстрактора; //-реэкстрагирующий водный 4,5 М3/ч рЭСТВОРЭ ЭЛКИлФоС
пягтгзпп: /// — экстпягеит на чкгтпякнию vnau»- ' ' г r ~
раствор; /// — экстрагеит на экстракцию ураиа;
IV— водный раствор урана.
фатов в керосине.
510
■В
®Х—п
Рис. 260. Экстрактор Керра-
Мак-Ги для извлечения
ванадия (вид в плане):
/—водная фаза; // —органическая
фаза.
Перемешивание осуществляется
турбинной мешалкой диаметром
0,46 м, делающей 150—200 об/мин.
В каждой ступени предусмотрена
возможность рециркуляции
отстоявшегося экстракта в смеситель.
Ступени экстрактора установлены
на расстоянии 0,3 м по высоте друг
от друга для того, чтобы водная
фаза перемещалась под действием
сил тяжести; противотоком к ней
с помощью эрлифта движется
экстракт.
Уран и другие металлы
выделяют из экстракта обработкой
водным раствором соды в
двухступенчатом смесительно-отстойном экстракторе, одна из ступеней
которого показана на рис. 259. Для удаления небольших
количеств гидроокисей титана и железа, осаждающихся из раствора,
отстойники имеют конические днища. На рис. 260 показан в
плане аппарат для экстракции ванадия, установленный на том
же заводе. Внутри емкости диаметром 9,8 м размещены четыре
ступени смесительно-отстойного каскада.
Непрерывнодействующий противоточный экстрактор «Витро»
(завод в Солт-Лэйк-Сити, Юта), предназначенный для
извлечения урана, интересен тем, что смешение и отстаивание
производят в одном аппарате (рис. 261)108-109. Каждая из четырех
ступеней экстрактора
представляет собой
емкость диаметром 6 м.
В экстракторе
перерабатывают 100 м3/ч
водного раствора. Несмотря на
то что соотношение
потоков водной и
органической фаз равно шести,
органическая фаза
является сплошной. Около
мешалки объемное
соотношение органической и
водной фаз равно трем.
Это достигается
поддержанием поверхности
раздела ниже уровня
мешалки и рециркуляцией
органической фазы через
верх диффузора, поме-
Рис. 261. Экстрактор Витро для
извлечения урана:
/, /// — органическая фаза; //, /V —водная фаза.
ill
щенного над мешалкой. Исходный раствор и экстрагент подают
непосредственно в диффузор и дисперсию направляют вниз с
помощью колпака, окружающего мешалку. Вертикальные
перегородки, при наличии которых возникали бы восходящие потоки
жидкости, ухудшающие отстаивание, отсутствуют.
Перемешивание осуществляется гуммированной мешалкой «Турбо-миксер»
диаметром 460 мм, делающей 130 об/мин; привод мешалки — от
электродвигателя мощностью 1,5 кет.
Описание других конструкций смесительно-отстойных
экстракторов и показатели работы некоторых экстракционных
установок приведены в литературе "•105,110- ш. В большей части
промышленных установок эффективность ступени превышает 80%.
а в наиболее совершенных из них она приближается к 100%.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
а—удельная поверхность контакта фаз, м2/м3.
С0—постоянная диафрагмы.
Сц С2; С3; С4 — константы.
с — концентрация, кмоль/м3.
D — коэффициент диффузии, м2/сек.
D' — эффективный коэффициент диффузии, м2/сек.
dM—диаметр мешалки, м.
d0—диаметр отверстия диафрагмы, м.
d— диаметр капли, м.
dm — диаметр трубопровода, м.
Е— эффективность ступени, равная отношению
количества экстрагированного вещества к его
количеству в условиях равновесия.
EMD— эффективность ступени по Мэрфри,
рассчитанная по дисперсной фазе.
ЕМЕ — эффективность ступени по Мэрфри,
рассчитанная по фазе экстракта.
EMR — эффективность ступени по Мэрфри,
рассчитанная по фазе рафината.
Е0 — общая эффективность ступени смесительно-от-
стойного каскада в отсутствие уноса (Е0 — п/пг).
Е0г — общая эффективность ступени смесительно-от-
стойного каскада при наличии уноса (Е0=
= п[Пгг).
Es — степень разделения фаз в отстойнике или
циклоне.
Н — длина или высота ступени, м.
К—константа в уравнении (X, 36).
КЕ, KR — общий коэффициент массопередачи, кмольх
Хсек~1-м-2 {кмоль/м3).
№
Ке, Kr — общий коэффициент массопередачи, кмольх
Хсек~х-мт2 (мол. доли).
k — коэффициент массоотдачи, кмоль • сек~1 X
Хм'2 (кмоль/мъ).
L — количество жидкости, загруженной в
периодически действующий аппарат, кмоль;
мольная массовая скорость, кмоль • сек~х • м~2
(для непрерывного процесса).
m = cE/cR (при равновесии) —коэффициент
распределения.
т'= у/х (при равновесии)—коэффициент
распределения.
N — скорость мешалки, об/сек.
NE — количество экстрагированного вещества,
кмоль (периодический процесс); скорость
экстракции, кмоль/сек на 1 м2 поперечного
сечения экстрактора (непрерывный процесс).
Fr = dMN2/g—критерий Фруда для мешалок.
P0 = P/pCjV3d^—критерий мощности.
Р'а = Р /рсм NMSM— критерий мощности.
Re = d2MNpc /\ic—критерий Рейнольдса для мешалок.
R е'=с^1Л^рсм / цсм —критерий Рейнольдса для мешалок.
Sc = \i/pD - критерий Шмидта.
Sh = kT/D — критерий Шервуда.
N0E — общее число единиц переноса,
рассчитанное по экстракту.
Nor — общее число единиц переноса,
рассчитанное по рафинату.
We = <fyV2pc/a—критерий Вебера для мешалок.
We' = d3ylN2pcst /о— критерий Вебера для мешалок.
WeKpllT. — критическое значение критерия Вебера для
капель [уравнение (X, 18)].
п — число теоретических ступеней; число секций
в аппарате [уравнение (X, 35)].
пг—число реальных ступеней в отсутствие
уноса.
пгг — число реальных ступеней при наличии уноса.
Р — мощность, вт.
Ар — разность давлений, н/м2.
Q—объемная скорость потока, м3/сек.
Т—-диаметр аппарата, м.
V — фиктивная скорость, м3/сек на 1 м2
поперечного сечения экстрактора.
v — объем жидкости, ж3.
W — ширина лопасти мешалки, м.
х — концентрация в рафинате, мол. доли.
33 Р. Е. Трейбал $13
у — концентрация в экстракте, мол. доли.
z—расстояние, м.
г—фактор экстракции (ml/E/V^ = m'LE/Z^).
еЕ—количество экстракта, уносимого рафинатом (в долях от
общего количества).
er — количество рафината, уносимого экстрактом (в долях от
общего количества поступающего на экстракцию
раствора).
О — время пребывания, сек.
QOE — время общей единицы переноса, рассчитанной по
экстракту, сек.
^о.? ~время общей единицы переноса, рассчитанной порафинату,
сек.
^ — величина, определяемая по уравнению (X, 52).
[1 — вязкость, кг • м~1 • сек'1.
Мсм.—вязкость смеси, /сг-лг1 • се/с-1 [уравнения (X, 30) — (X, 32].
р — плотность, кг/м3.
Рем. — плотность смеси, кг/м3.
Ар—разность плотностей, кг/м3.
а—межфазовое натяжение, н/м.
ф — объемная доля жидкости.
Индексы
л — легкая жидкость.
ср. — средняя логарифмическая величина,
т — тяжелая жидкость.
С—сплошная фаза.
D — дисперсная фаза.
е — равновесная величина.
Е— экстракт.
F — исходный раствор.
п—номер ступени или секции.
о — органическая жидкость.
R — рафинат.
S — экстрагент.
w — водная фаза.
1—начальная величина.
2 — конечная величина.
*—равновесная концентрация (верхний индекс).
ЛИТЕРАТУРА
LA. W. Hixson, I. M. Smith, Ind. Eng. Chem., 41, 973 (1949).
2. E. V. Murphree, Ind. Eng. Chem., 17, 747 (1925).
3. A. W. Hixson, T. B. Drew, K. L. Knox, Chem. Eng. Progr., 50, 592
(1954).
4 E. S. В i s s e 11, H. С H e s s e, H. J. E v e r e 11, J. H. R u s h t о n, Chem.
Eng. Progr., 43, 649 (1947),
514
5. E. S. Bissell, H. J. Everett, J. H. Rushton, Chem. Met. Eng., S3,
118 (1946).
6. R. H. О v e г с a s h i e г, Н. А. К i n g s 1 e y, R. В. О 1 n e y, A. I. Ch. E.
Journ., 2, 529 (1956).
7. J. H. R u s li t о n, J. Y. О 1 d s h u e, Chem. Eng. Progr., 49, 161, 267
(1953).
8. J. H. Rushton, E. W. С о s t i с h, H. J. Everett, Chem. Eng. Progr.,
46, 395, 467 (1950).
9. R. E. Johnstone, M. W. Thring, Pilot Plants, Models, and Scale-
Up Methods in Chemical Engineering, McGraw-Hill Book Company, Inc.,
New York, 1957.
10. D. E. Mack, A. E. К г о 11, Chem. Eng. Progr., 44, 180 (1948).
11. S. N a g a t a, T. Yokoyama, M. Honjyo, Chem. Eng. (Japan), 15,
49 (1951).
12. D. S. Laity, R. E. Treybal, A. I. Ch. E. Journ., 3, 176 (1957).
13. H. Hof m ann. Chem. Eng. Sci., 8, 113 (1958).
14. J. H. Rushton, Chem. Eng. Progr., 43, 649 (1947).
15. G. H. Gumming s, A. S. West, Ind. Eng. Chem., 42, 2303 (1950).
16. W. A. Rodger, U. S. Atomic Energy Commision, ANL-5575 (1956).
17. W. A. Rodger, V. G. Trice, J. H. Rushton, Chem. Eng. Progr., 52,
515 (1956).
18. И. С. Павлушенко, А. В. Янишевский, ЖПХ, 32, 1495 (1959).
19. S. A. Miller, С. A. Mann, Trans. Am. Inst. Chem. Engrs., 40, 709
(1944).
20. S. N a g a t a, N. Y о s h i о k a, T. Yokoyama, D. T e r a m о t o, Trans.
Soc. Chem. Engrs. (Japan), 8, 43 (1950).
21. И. С. Павлушенко, А. В. Янишевский, ЖПХ, 31, 1348 (1958)
22. М. S ее w a 1 d, M. Ch. E. Thesis, New York Univercity, 1960.
23. R. E. Treybal, Ind. Eng. Chem., 53, 597 (1961).
24. R. W. Mac don aid, E. L. P i r e t, Chem. Eng. Progr., 47, 363 (1951).
25. R. E. Treybal, A. I. Ch. E. Journ., 4, 202 (1958); 6, 5M (I960).
26. J. O. H i n z e, A. I. Ch. E. Journ., 1, 289 (1955).
27. R. S h i п п a r, J. Fluid Mech., 10, pt. 2, 259 (1961).
28. R. S hinnar, J. M. Church, Ind. Eng. Chem., 52, 253 (1960); 53, 479
(1961).
29. S. Nagata, K. Yamamoto, K. Hashimoto, Y.Naruse, Mem
Fac. Eng. Kyoto Univ., 21, pt. 3, 260 (1959); 22, pt. 1, 68 (1960).
30. K- W. Norwood, A. B. M e t z n e r, A. I. Ch. E. Journ., 6, 432 (1960).
31. J. P. Sachs, J. H. Rushton, Chem. Eng. Progr., 50, 597 (1954).
32. H. S e t z e r, D. Eng. Sci. Thesis, New York Univercity, 1962.
33. J. H. Vanderveen, U. S. Atomic Energy Comm., UCRL-8733 (1960).
34. H. Groothius, F. J. Zuiderweg, Chem. Eng. Sci., 12, 288 (1960).
35. A. J. Madden, G. L. Damerell, A. I. Ch. E. Journ., 8, 233 (1962).
36. G. V. Chester, J. S. Newman, U. S. Atomic Energy Comm.,
. . ORNL-3018 (1961).
37. T. Vermeulen, G. M. Williams, G. E. L a n g 1 о i s, Chem. Eng.
Progr., 51, 85F (1955).
38. H. E. Re a, T. Vermeulen, U. S. Atomic Energy Comm., UCRL-2123
(1953).
39. P. H. С alder ba nk, Trans. Inst. Chem. Engrs (London), 36, 443 (1958)
40. В. В. К а ф а р о в, Б. М. Б а б а н о в, ЖПХ, 32, 789 (1959).
41. R. В. Olney, G. J. Carlson, Chem. Eng. Progr., 43, 473 (1947).
42. J. J. Barker, R. E. T г е у b о 1, A. I. Ch. E. Journ., 6, 289 (1960).
43. P. H a r r i о 11, A. I. Ch. E. Journ., 8, 93 (1962).
44. A. I. Johnson, С J. Huang, A. I. Ch. E. Journ., 2, 412 (1956).
45. D. W. Humphrey, H. С Van Ness, A. I. Ch. E. Journ., 3, 283
(1957).
46. S. Nagata, I. Yamoguchi, Kagaku Kogaku, 24, 726, 742 (1960).
47. H. Grober, Z. Ver. deut. Ing., 69, 705 (1925).
33*
SIS
48. R. V. Mat tern, О. В i 1 о u s, E. L. P i r e t, A. I. Ch. E. Journ., 3, 497
(1957).
49. A. Holm, S. G. Terjersen, Chem. Eng. Sci., 4, 265 (1955).
50. R. B. Olney, A. I. Ch. E. Journ., 7, 348 (1961).
51. A. W. Flynn, R. E. Treybal, A. I. Ch. E. Journ., 1, 324 (1955).
52. А. К a r r, E. G. S с h e i b e 1, Chem. Eng. Progr. Symp. Ser., 50, № 10,
73 (1954).
53. A. D. R у о n, F. L. Daley, R. S. Lowrie, U. S. Atomic Energy Comm.,
ORNL-2951 (1960); Chem. Eng. Progr., 55, № 10, 70 (1959).
54. W. G. Mathers, L. С. С о г п е 11, E. E. Winter, Atomic Energy of
Canada, Ltd., CRCE-790 (1959), AECL-913 (1959).
55. W. G. Mathers, E. E. Winter, Can. J. Chem. Eng., 37, 99 (1959).
56. V. А. К a 1 i с h e v s к у, K.. Kobe, Petroleum Refining with Chemicals,
D. Van Nostrand Company, Inc., New York, 1956.
57. M. B. Miller (мл.), Max B. Miller a. Co., частное сообщение, 1950.
58. V. G. Trice, U. S. Atomic Energy Comm., ANL-5741 (1957).
59. T. R. Johnstone, С W. Robinson, N. E p s t e i n, Can. J. Chem.
Eng., 39, 1 (1956).
60. J. D. T h о r n t о n, Nuclear Eng., 1, 156, 204 (1956).
61. В. В. Кафаров, С. А. Жуковская, ЖПХ, 31, 376 (1958).
62. С. А. Жуковская, Л. А. А н е н к о в а, И. Д. Бойко, Мед. пром., 13,
№ 5, 26 (1959).
63. В. F. Greek, С. A. Duval, V. A. Kalichevsky, Ind. Eng. Chem.,
49 1938 (1957)
64. J.' A. M с D о n о u g h, W. J. T о m m e, С D. Holland, A. I. Ch. E.
Journ., 6, 615 (1960).
65. "L. S. Scott, W. B. Hayes, С D. Holland, A. I. Ch. E. Journ., 4,
346 (1958).
66. D. J. Simkin, R. B. Olney, A. I. Ch. E. Journ., 2, 545 (1956).
67. J. A. Le acock, S. W. Churchill, A. I. Ch. E. Journ., 7, 196 (1961).
68. W. E. С a r b о п e, Sewage a. Ind. Wastes, 22, 200 (1950).
69. H. A. G о 11 m a r, Ind. Eng. Chem., 39, 596 (1947).
70. K. G. Shaw, R. S. Long, Chem. Eng., 64, № 11, 217 (1957).
71. H. E. Thayer, Proc. 2nd U. N. Jntern. Conf. Peaceful Uses Atomic
Energy, Geneva, 44, 22 (1958).
72. H. A. Woo die, F. С Vilbrandt, A. I. Ch. E. Journ., 6, 296 (1960).
73. P. В е с h e r, Emulsious, Theory and Practice, Reinhold Publisching
Corporation, New York, 1957.
74. J. J. Bikerman, Surface Chemistry, 2nd ed., Academic Press Inc., New
York, 1958.
75. M. Van den Temple, Rec. trav. chim., 71, 419, 433, 442 (1953).
76. T. Gilespie, E. K. R i d e a 1, Trans. Farad. Soc, 52, 173 (1956).
77. G. E. Charles, S. G. Mason, Colloid Sci., 15, 105, 236 (1960).
78. H. G. К on песке, Z. physik. Chem., 211, 208 (1959).
79. H. P. Meissner, В. С h e r t о w, Ind. Eng. Chem., 38, 856 (1946).
80. A. I. I n g e r s о 11, Petroleum Refiner, 30, № 6, 106 (1951).
81. J. B. Byrne, U. S. Atomic Evergy Comm., DOW-146 (1956).
82. K. Rietema, Chem. Eng. Sci., 2, 73 (1953).
83. J. B. T e p e, W.K.Wood s, U. S. Atomic Energy Comm., AECD-2864
(1943).
84. J. W. Hitchon, Atomic Energy Research Establ. (G. Brit.), CE/R-2777
(1959).
85. J. С. В re see, U. S. Atomic Energy Comm., CF-56-3-171 (1956).
86. M. E. O'K. Trowbridge. Brit. Chem. Eng., 4, 29 (1959).
87. F. E. S u 11 i v a n, Chem. Eng. Progr., 52, 83 (1956).
88. T. A. Burtis, С G. К i г к b r i d e, Trans. Am. Inst. Chem. Engrs., 42,
413 (1946).
89. J. G. Hayes, L. A. Hayes, H. S. Wood, Chem. Eng. Progr., 45, 235
(1949).
SIB
90 G. V. Jordan, Trans. Am. Soc. Mech. Engrs., 77, 393 (1955).
91 С A. R. P e a r с e, Brit. J. Appl. Phys., 5, 136 (1954).
92 R. J- Phillips, H. G. H a p i e r, Petrol Refiner, 38, № 2, 151 (1959).
93 W. С P h i 1 о о п, А. Н. К n e b e 1, J. A. S о u к u p, P. Fain, U. S.
Atomic Energy Comm., MCW-1461 (1961).
94. С A. Sleicher, A. I. Ch. E. Journ., 6, 529 (1960).
95 Anglo-Persian Oil Co., Ltd, A. E. Hoi ley, О. Е. М о 11, брит. пат.
321200, 1929 г.
96. A. E. H о 11 e у, О. Е. М о 11, пат. США 1953651, 1934 г.
97 D. F. Murdoch, М. С и с к п е у, Trans. Inst. Chem. Engrs (London),
24, 90 (1946).
98. E. Thorn to n, J. Inst. Petrol. Technol., 19, 957 (1933).
99. V. S. Morello, N. P о f f e nb e r g e r, Ind. Eng. Chem., 42, 1021 (1950).
100. B. V. С о p 1 a n, J. K. Davidson, E. L. Z e b г о s к i, пат. США
2646346, 1953 г.
101. E. R. Graef, S. P. Foster, Chem. Eng. Progr., 52, 293 (1956).
102. F. Roberts, B. T. Bell, Ind. Chem., 32, 221 (1956); Trans. Inst. Chem.
Engrs. (London), 35, 6 (1957).
103. A. T. Da v is, T. J. Col ven, A. I. Ch. E. Journ. 7, 72 (1961).
104. W. J. Motel, T. J. Colven, U. S. Atomic Energy Comm., DP-130
(1955); DP-254 (1957).
105. J. F. Flagg (ed), Chemical Processing of Reactor Fuels, Academic
Press, Inc., New York, 1961.
106. W. С Hazen, A. V. Henrickson, Mining Eng., 9, 994 (1957).
107. J. E. Quinn, Deco Trefoil, Denver Equipment Co. Bull., M4-B90 (1957).
108. K. Black, J. Koslov, Chem. Eng. Progr. Symp. Ser., 55, № 22, 105
(1959).
109. L. D. Lash, Mining Eng., 10, 1161 (1958).
110. M. W. Davis, T. E. Hicks, T. V e r m e и 1 e n, Chem. Eng. Progr., 50,
188 (1954).
111. R. E. Treybal, The Chemical Engineers' Handbook, 4th ed., R. H. Perry,
S. D. Kirkpatrick, С. Н. Chilton (ed.), Sect. 21, McGraw-Hill Book
Company, Inc., New York, 1963.
ЭКСТРАКТОРЫ С НЕПРЕРЫВНЫМ
(ДИФФЕРЕНЦИАЛЬНЫМ) КОНТАКТОМ
Аппараты, в которых взаимно
нерастворимые жидкости движутся противотоком
и непрерывно взаимодействуют друг с
другом (без периодического отстаивания и
физической сепарации), можно
сконструировать для получения практически любого
числа теоретических ступеней разделения.
В таких аппаратах возможен также
параллельный ток жидкостей, однако при этом
в них может быть достигнута максимально
лишь одна теоретическая ступень.
Аппараты с прямоточным движением жидкостей
относятся к категории так называемых
проточных смесителей (см. главу X).
В большей части случаев эффект
разделения должен соответствовать нескольким
теоретическим ступеням, поэтому на
практике, как правило, применяют аппараты с
противоточным движением контактируемых
жидкостей.
ОБЩИЕ ХАРАКТЕРИСТИКИ
Противоточное движение
контактируемых жидкостей осуществляется под
действием разности их плотностей. Если
жидкости движутся в поле силы тяжести,
экстракторы представляют собой вертикальные
колонны. Легкая фаза поступает в колонну
снизу и движется вверх, а тяжелая фаза
подается сверху и движется вниз
противотоком к легкой фазе. Если-движение
жидкостей осуществляется в поле
центробежных сил (возникающих при вращении
жидкостей в экстракторах), то оно направлено
радиально по отношению к оси аппарата,
которую можно расположить
горизонтально или вертикально.
Поперечное сечение экстрактора. При
противоточном движении жидкостей для
618
-каждой скорости течения одной жидкости существует
максимально возможная скорость другой жидкости. Величина
максимальной скорости зависит от физических свойств жидкостей,
К конструкции экстрактора и от способа организации
противотока (в поле сил тяжести или в поле центробежных сил). Если
при данной скорости одной жидкости скорость другой превысит
некоторое максимальное значение, аппарат не сможет пропу-
f стить заданное количество жидкостей и «захлебнется». В таких
случаях, чтобы избежать «захлебывания», необходимо
увеличить поперечное сечение аппарата.
Высота экстрактора. Время контакта двух жидкостей
определяет длину пути контакта или высоту вертикальной колонны,
которая зависит от степени и скорости экстракции и не зависит
(если пренебречь вторичными эффектами, связанными с
переходом к аппарату крупных размеров, — моделированием) от
общего количества перерабатываемых жидкостей. Скорость массо-
I передачи определяется коэффициентом массопередачи или
'высотой единицы переноса. Общую высоту, необходимую для
достижения заданной степени разделения, находят методами,
описанными ранее (см. главу VIII). Высоту аппарата можно
установить также с помощью высоты, эквивалентной теоретической
ступени разделения (ВЭТС). Иногда, в зависимости от
конструкции аппарата, некоторый его участок можно рассматривать
как реальную ступень и по эффективности ступени судить о
разделительной способности аппарата. В этих случаях применяют
методы расчета высоты аппарата, описанные в главах VI и VII.
Дисперсная и сплошная фазы. Для ускорения массопередачи
при увеличении поверхности контакта фаз одну жидкую фазу
распределяют в виде тонких пленок или мелких капель в объеме
другой (сплошной) фазы. В качестве дисперсной фазы
выбирают легкую либо тяжелую фазу, экстрагент или исходный
раствор (экстракт или рафинат). Для описания работы экстрак-
W ционного аппарата более важное значение имеют кинетические
Т характеристики, отнесенные к дисперсной или сплошной фазе
V, (например, Кт>а и HWd или Кса и HWc), чем к фазам экстракта
ф и рафината.
•% При проектировании экстракционной аппаратуры почти все-
■Ц' гда необходимо установить, какую фазу следует диспергировать.
?, Обычно считают, что целесообразнее диспергировать фазу, имею-
15 Щую большую объемную скорость, так как при данном размере
>' Капель в этом случае будет достигнута большая поверхность
*f Контакта фаз. Однако при определенных условиях желательно
Г Делать исключения из указанного правила:
1. Если объемное соотношение потоков фаз существенно от-
- личается от единицы, то в аппаратах, в которых возможно об-
, • ратное перемешивание, предпочтительно диспергировать фазу,
', движущуюся с меньшей объемной скоростью. При этом
"й 619
уменьшается степень обратного перемешивания. К аппаратам,
в которых желательно уменьшать продольное перемешивание
таким способом, относятся распылительные и насадочные
колонны, а также некоторые колонные аппараты с механическим
перемешиванием фаз, если в аппаратах этих типов обе
жидкие фазы движутся противотоком на относительно большом
пути. В таких аппаратах, как экстракторы с ситчатыми
тарелками, продольное перемешивание сравнительно невелико.
2. Дисперсная фаза находится в экстракторе обычно в
меньшем количестве, чем сплошная. Поэтому для снижения затрат
и уменьшения пожароопасности целесообразно диспергировать
более дорогую или легче воспламеняющуюся жидкость.
3. Как было отмечено в главе V, скорость массопередачи
часто зависит от направления массопередачи (эффект Маран-
гони). В данной главе показано, что от выбора
направления массопередачи зависит также объемная скорость,
соответствующая «захлебыванию» аппарата. Поэтому в некоторых
случаях выбор дисперсной фазы зависит в большей степени от
направления массопередачи, чем от относительных количеств
жидкостей.
4. В некоторых аппаратах конструкционный материал может
предпочтительно смачиваться одной из жидких фаз.
Производительность и скорость массопередачи в этом случае зависят от
того, какая жидкая фаза дисперсная. Иногда, когда объемы и
физические свойства фаз в результате экстракции значительно
изменяются по высоте аппарата, целесообразно диспергировать
обе фазы, поддерживая уровень раздела фаз в средней части
экстрактора.
Классификация аппаратов. Аппараты для непрерывного
контактирования можно классифицировать1 по признаку
усложнения конструкции их внутренних устройств. Аппараты различных
типов рассмотрены в последовательности, соответствующей
классификации, приведенной ниже:
I. Гравитационные экстракторы
А. Аппараты без механического перемешивания фаз
1. Колонны с орошаемыми стенками (пленочные колонны) и аппараты
аналогичного типа.
2. Распылительные колонны.
3. Колонны с перегородками.
4. Насадочные колонны.
5. Колонны с перфорированными тарелками (ситчатые колонны).
Б. Аппараты с механическим перемешиванием фаз
1. Колонны с вращающимися перемешивающими устройствами,
2. Пульсационные колонны:
) с пульсацией жидкостей;
) с вибрирующими тарелками.
II. Центробежные экстракторы
520
Аппараты некоторых конструкций соответствуют
одновременно признакам нескольких аппаратов различных типов,
указанных выше.
Классификацию экстракторов, несколько отличную от
приведенной, дал Пратт 2.
Конструкцию внутренних устройств аппаратов, особенно
гравитационных, обычно усложняют, чтобы увеличить скорость
экстракции и, следовательно, уменьшить необходимую высоту
аппарата, но часто указанная цель достигается в результате
снижения допустимой производительности на единицу площади
поперечного сечения экстрактора. Предложено и описано в
литературе большое число экстракционных аппаратов различных
конструкций. Здесь рассмотрены только те из них,
которые получили широкое практическое применение. Аппараты
прочих типов описаны Праттом2, а также Морелло и Поффен-
бергером 3.
Характеристики экстракционных аппаратов. Несмотря на то
что многие экстракционные аппараты эксплуатируются уже
весьма продолжительное время и широко распространены в
промышленности, сведения о них ограничены. Это объясняется
в некоторой степени тем, что эксплуатационные характеристики
экстракторов зависят от очень большого числа переменных,
Известно, что скорость процесса экстракции является
функцией характеристик системы жидкость — жидкость, условий
проведения процесса, а также типа аппарата.
Ниже приведены основные факторы, влияющие на скорость
экстракции:
1. Система жидкость — жидкость
а) Химическое сродство и соответствующие физические свойства
системы. К этой категории факторов относится также наличие в
жидкостях поверхностно-активных веществ, присутствие в них твердых взвесей
и т. п.
■б) Концентрация экстрагируемого компонента, влияющая на физические
свойства системы.
в) Направление массопередачи (перенос вещества из водной фазы в
органическую, из сплошной фазы в дисперсную или в противоположном
направлении).
г) Суммарная скорость движения жидкостей.
Д) Соотношение потоков жидкостей.
е) Выбор дисперсной фазы.
2. Аппаратура
а) Конструкция аппарата, под которой подразумевают не только общую
характеристику конструкции (например, насадочная колонна или колонна
с ситчатыми тарелками), но и особенности деталей (например, размеры и
форма иасадки, размеры и число отверстий на тарелке).
б) Способ и интенсивность механического перемешивания (вращательное или
возвратно-поступательное, интенсивное или малоинтенсивное).
521
в) Материал аппарата, от характеристик которого зависит относительная
смачиваемость его жидкостями.
г) Высота экстрактора и концевые эффекты.
д) Диаметр экстрактора и степень продольного перемешивания.
Два последних фактора [пункты г) ид)] заслуживают
специального рассмотрения. Концевые эффекты характеризуются
количеством проэкстрагироваеного вещества или скоростью
экстракции на вводе дисперсной или сплошной фазы в колонну.
Если скорость массопередачи измеряют в экстракторе
небольшой высоты, значительная доля величины скорости экстракции,
определенной в опыте, обусловлена концевым эффектом.
Полученные значения скорости экстракции нельзя использовать для
расчета аппаратов (имеющих большую высоту), в которых
влияние концевого эффекта не столь велико.
Под продольным перемешиванием понимают циркуляцию
жидкостей в вертикальном направлении (обратное
перемешивание) ^приводящую к перемещению легкой жидкости вниз, а
тяжелой— вверх (т. е. в направлении, противоположном основному
направлению движения потоков), а также поперечную
неравномерность потоков. Влияние продольного перемешивания
сказывается на уменьшение средней движущей силы процесса
экстракции и, следовательно, скорости массопередачи. Обратное
перемешивание сильно возрастает с увеличением отношения
диаметра экстрактора к его длине и, возможно, с увеличением
только диаметра экстрактора. Следовательно, если использовать
опытные данные о скорости массопередачи, полученные в
аппаратах небольшого диаметра, для проектирования экстракторов
больших размеров без учета поправки на продольное
перемешивание, то спроектированный таким образом аппарат не
обеспечит необходимой степени извлечения. Интенсивность
продольного перемешивания различна для экстракторов разных
конструкций, и в ряде случаев проблема продольного
перемешивания имеет существенное значение.
К сожалению, почти все известные опытные данные
получены ори применении малых колонн, диаметр которых редко
превышает 150 мм, а высота обычно не больше 1—2 м, причем в
большей части работ не учитывается влияние продольного
перемешивания или концевого эффекта на скорость массопередачи.
Поэтому предлагаемые расчетные зависимости следует
применять с большой осторожностью для расчета промышленных
аппаратов. Эти зависимости можно использовать для
предварительной оценки первоначальной стоимости оборудования, его
пригодности для конкретного процесса, а также для выявления
относительной значимости различных факторов и степени их
влияния на скорость экстракции.
Производительность экстракторов или предельные нагрузки,
соответствующие «захлебыванию» экстракторов, также зависят
522
н8 основном от переменных, приведенных выше. Кроме того, на
^производительность экстракторов влияет наличие переноса рас-
Ццределяемого компонента из фазы в фазу. Большая часть дан-
ряых о' захлебывании получена при отсутствии массопередачи
Ъаежду фазами. Поэтому количественные соотношения,
рассматриваемые ниже, следует применять для расчета скоростей «за-
рцлебывания» лишь в первом приближении. Результаты расчета
^необходимо корректировать на основе данных испытаний
пилотных установок.
Прежде чем перейти к описанию типовых конструкций
?экстракторов, рассмотрим некоторые процессы, общие для
Ькстракторов многих типов. К таким (процессам относятся:
образование капель, экстракция в период образования и «оалесцен-
Цции капель, обратное перемешивание.
Образование капель. Если диспергируемую жидкость вводят
|через отверстие или сопло в другую жидкость, в которой она
^нерастворима (в сплошную фазу), то процесс образования
капель сильно зависит от конструкции отверстия и смачиваемо-
*сти материала аппарата дисперсной фазой.
Когда материал перфорированной плоской тарелки
предпочтительно смачивается дисперсной фазой и скорости последней
^в отверстиях тарелки малы, диспергируемая жидкость
растекается по поверхности тарелки, образуя слой на стороне,
обращенной к оплошной фазе. Этот слой занимает площадь,-в
несколько раз большую площади сечения отверстия. От слоя от-
Jf рываются капли со скоростью, зависящей от скорости подачи
дисперсной фазы. Обычно капли велики и размер их не поддает-
-ся регулированию4'5-8. Если сильно увеличить скорость
дисперсной фазы, то картина образования капель будет аналогична
той, которая наблюдается при диспергировании фазы, не
смачивающей материал тарелки.
Если материал тарелки не смачивается дисперсной фазой
* или если капли образуются с помощью сопел, обращенных выход-
\ ными отверстиями к сплошной фазе (особенно при наличии у со-
* /«пел скошенных острых кромок), то размер образующихся капель
•Iможно регулировать. При малых скоростях жидкости в отверстии
ЬКапли периодически образуются на кромках отверстий, отры-
"■"ч-ваются и падают (или поднимаются) в слой сплошной фазы.
, ' При увеличении скорости дисперсная фаза образует струю,
Которая суживается (давая «шейку») и на некотором
расстоянии от поверхности тарелки распадается на капли. Если
скорости в отверстии сопла достигают значений порядка 0,1 м/сек,
то образующиеся капли имеют примерно одинаковый размер.
В случае дальнейшего увеличения скорости в сопле длина струи
1 Достигает максимума и затем струя распадается на капли. При
, этом получаются капли все более различающихся размеров, и,
:г по-видимому, в дальнейшем образование мелких «распылен-
w 523
I
ных» капель происходит непосредственно на кромках отверстия
сопла5-7. Такая картина образования капель наблюдается в
том случае, когда диспергируется жидкость, не смачивающая
материал тарелки.
Для улучшения каплеобразования можно рекомендовать
устанавливать на тарелках направляющие сопла или выштампо-
вывать сопла (для уменьшения части сопла, выступающей над
поверхностью тарелки) в самой тарелке, как показано на
рис. 262. Если предполагают использовать тарелку со
сверленными отверстиями, то следует также учитывать смачиваемость
жидкостью материала тарелки.
Многие металлы не обладают заметно гидрофобными или
гидрофильными свойствами; поэтому перфорированные
тарелки, изготовленные из таких материалов, в большей части
случаев работают удовлетворительно, хотя тарелки с выштампо-
ванными соплами или отверстиями более предпочтительны.
Гидрофобными материалами являются сплав железа,
молибдена и меди («Тгопсап») 8, а также большая часть пластмасс,
особенно полиэтилен, полипропилен и фторопласты. Поэтому
перечисленные материалы не следует применять при
диспергировании органических жидкостей.
Для определения диаметра капель, образующихся в
отверстиях и соплах при относительно «ебольших скоростях
дисперсной фазы и предпочтительном смачивании материала сплошной
фазой, предложено несколько корреляций 5>6-9.10. Наиболее
пригодно (при размерах отверстия сопла не более 8 мм) уравнение
Хайворта5:
у>рУо_7^°1о 179 4124s5Vc279
Др ' Ар Др1
Wvp+iMJ^-7#!^ + lnJLlu^r_ (XU)
С помощью этого уравнения можно рассчитать объем капель,
образующихся при скоростях жидкости в отверстии сопла не'
более 0,1 м/сек (когда капли имеют примерно постоянный
размер), а также объемы наибольших капель при скоростях, не
превышающих 0,3 м/сек (Кейт и Хиксон 7 использовали данное
уравнение для определения среднего размера капель при
скоростях выше этого предела). На рис. 263 представлено
графическое решение уравнения (XI, 1), полученное при условии, что
капли имеют сферическую
форму. Движение сплошной
g\ фазы противотоком к дис-
ШШ Ш1ХШ персной обычно не влияет
Направление движения на величину капель7. Си-
дисперсной фазы мес6 показал, что уравнение
(XI, 1) не применимо для
Рис. 262. Тарелка с выштампованными сопел с отверстиями диа-
отверстиями. метром больше 12 мм.
524
0,003 0,005 a,oi цог ооз ops oj о,г о,з о,5 /
Ойп _ Un Ул ' Uc'
5,гв—+и,зв-ю5-9—9-—fp-—
AJ) Ар ''°
Рис. 263. Размеры капель, образующихся при движении взаимно
нерастворимых жидкостей через отверстия тарелок и сопел, выполненных из
материала, не смачиваемого дисперсной фазой.
Наличие массопередачи в период образования капель
оказывает влияние на их размер.
Межфазовая поверхность, образованная каплями,
возникающими при распаде струи, имеет явно выраженный максимум,
соответствующий определенному значению скорости в
отверстии сопла {Vom)- Христиансен11 на основе теоретического и
экспериментального изучения распада струй органических
жидкостей в воде вывел зависимость V0m от физических свойств
бинарных систем:
Vom = *» (^У (-0,5137 РрТо,4719рс Г <*. *>
Отношение диаметров струи и сопла dj/d0 в этом уравнении
определяют из эмпирических соотношений:
625
при d0IVolg!\p < 0,785
-<2- = 0,485 -£_- +1
dJ L(a/^Ap)°'5J
при dQjYWg^P > 0,785
d„ l,61d„
(о/гДр)'
0,5
-0,12
(XI, За)
(XI, 36)
Средний диаметр капель определяют из равенства dp =
= 2dj.
При наличии третьего компонента, даже в том случае, когда
фазы находятся в равновесии, в уравнение (XI, 2) вместо
значения граничного натяжения бинарной системы Ог необходимо
ввести величину «эффективного» граничного натяжения тройной
системы Озе, являющуюся средней между значениями аг и
граничного натяжения а3 равновесной тройной системы. Величина
(ог — Озе)/(о2 — Оз) примерно равна единице для растворов,
содержащих небольшие молекулы органических веществ, и
снижается до 0,4 при наличии поверхностно-активных веществ.
Влияние массопередачи на размер капель в последнем случае
неизвестно. Симес12 указывает также, что размер капель
зависит от вязкости сплошной фазы, однако количественно эта
зависимость с достаточной точностью не установлена.
80
U0
Z*
J{
и
/
I3
~^>
г
а
а
а
I
I
"о 0,5 I 1,5
Высота колонны, м
Рис. 264. Экстракция уксусной кислоты из единичных капель
(см. таблицу):
13,14
111
1
2
3
4
Общая
скорость
экстракции
определялась
скоростями
в периоды
1, 2, 3
1, 2, 3
1, 2, 3
1, 2
Экстр агент
Метил изобутилкетон
Бензол
Изопропиловый
спирт
Изопропиловый
спирт
Дисперсная фаза
Метил изобутилкетон
Бензол
Вода
Вода
Диаметр
2,8-3,0
3,8-4,0
4,5
4,5
626
Экстракция при образовании и коалесценции капель. Шервуд
с сотр.13, по-видимому, впервые показали, что значительная
часть общего количества экстрагируемого компонента
извлекается в периоды образования капель и их коалесценции (при
слиянии их с объемом диспергируемой фазы).
На рис. 264 приведены опытные данные Шервуда 13 и Лих-
та14 об экстракции из единичных капель в период образования
капли (период /), движения в сплошной фазе (период 2) и
коалесценции (период 3) на границе раздела фаз,находящейся
у конца аппарата, противоположного вводу в него дисперсной
фазы. Определяя степень экстракции, достигаемую в аппаратах
.различной высоты, и экстраполируя полученные результаты на
нулевую высоту (см. рис. 264), можно оценить величину
концевых эффектов (периоды / и 3).
Лихт 15 и Джонсон 16>17 подробно рассмотрели методы
обработки опытных данных об экстракции, применяемые для
определения величины концевых эффектов. При этом оказалось, что
ни один метод не позволяет точно найти величину концевого
эффекта. Наибольшая ошибка может наблюдаться для
процесса экстракции в период образования капель.
Опытные данные об экстракции в период образования капель
весьма противоречивы вследствие трудностей, связанных с
получением надежных результатов. Лихт15, модифицируя допущение
Хигби (ом. главу V), предположил, что межфазовую
поверхность мож'но упрощенно рассматривать, как плоскую. Принимая
далее, что жидкость в капле полностью перемешана, вследствие
чего основное сопротивление массопередаче сосредоточено в
сплошной фазе, Лихт получил следующее уравнение,
описывающее экстракцию из капли:
2,90 (Dc6 )0,5
^,„ач.-С0.кон. = -^Г (CD,na4.-mCc) (XI- 4)
Скорость экстракции в период образования капли
определяют с помощью уравнения
vp (СР, нач. СР, кон.) ^ ,х. сч
5-1 = KDf(cDj нач,— тсс) (А1, О)
где Ар — средняя поверхность капли за время ее образования 0/.
Сравнивая уравнения (XI, 4) и (XI, 5), получим:
2,90t> (D_6,)0'5
к =—/; с /; (XI, 6)
Если предположить, что капля за время образования
сохраняет сферическую форму, то среднюю поверхность ее можно
627
найти из следующего уравнения
ь
Г 4nr2dQ
А = ° (XI, 7)
где г — радиус капли в период времени 0.
Пусть за единицу времени объем капли возрастает на
постоянную величину vplQf. Тогда в любой момент объем капли
составляет
^=4^3 да8)
О/ О
Дифференцируя уравнение (XI, 8) и подставляя производную
в уравнение (XI, 7), а также принимая, что vp = nds/6, получим18:
Л, = -|*4 (Х1'9)
_3
5
Подстановка уравнения (XI, 9) в уравнение (XI, 6) дает
«~-^ФГ
где
По данным Гарнера и Скелланда 19, экспериментальные
значения коэффициентов массопередачи больше рассчитанных по
уравнению (XI, 10) в среднем в 1,5 раза. Гарнер и Скелланд ис- |
пользовали уравнение (V, 52) для сплошной фазы и предполо^- |
жили, что внутри капли жидкость неподвижна и перенос в ней
происходит только в результате молекулярной диффузии. При
этом оказалось, что опытные значения коэффициентов
массопередачи в 7—12 раз больше рассчитанных согласно принятой
модели и в 2,1 раза больше рассчитанных по уравнению (V, 52).
Опытные значения примерно в 2 раза отличались 18 от
рассчитанных по уравнению (XI, 10).
Гарнер и Хэл20 сделали допущение, что общий коэффициент
массопередачи KDf равен коэффициенту KD для движущейся
капли, но, учитывая зависимость (XI, 9), следует вводить в
расчет лишь 0,6 от времени образования капли. Это допущение еле-
дует, однако, считать произвольным. Хертис21 также использо- I
вал 'гипотезу Хигби, но при этом предположил, что основное со- I
противление массопередаче сосредоточено внутри капли (это
предположение принял также Джонсон17). Однако опытные
данные Хертиса не соответствовали его модели. По-видимому,
сопротивление массопередаче в капле очень невелико, и
уравнение (XI, 10) должно давать в обычных условиях
удовлетворительные результаты. Влияние же поверхностно-активных
веществ, межфазовой турбулентности и других подобных явлений
I
528
(см. главу V) на скорость массопередачи.пока еще установить
нельзя. Для определения скорости массопередачи в период
образования капель вследствие отсутствия более точных
расчетных зависимостей рекомендуется применять уравнение (XI,10).
Систематизированные опытные данные о величине концевого
эффекта при коалесценции капель пока отсутствуют. На основе
имеющихся ограниченных сведений по этому вопросу Коулсон 18
сделал допущение, что коэффициент КтЛ в период коалесценции
капель по порядку величины равен коэффициенту массопередачи
в период образования капель.
Продольное перемешивание. Степень продольного
перемешивания очень сильно зависит от конструкции аппарата и
условий проведения процесса. Влияние продольного перемешивания
на необходимую высоту экстрактора можно рассчитать, если
известно общее уравнение, описывающее явление продольного
перемешивания.
Было предпринято 22> 23>24 несколько попыток решения этой
проблемы выражением эффекта продольного перемешивания с
помощью коэффициента турбулентной диффузии Е, который
можно ввести в критерий Pe'=VH/E. В качестве
определяющего линейного размера в критерий Ре' входит высота Н
аппарата. Для поршневого движения £ = 0 и Ре'=оо. Наоборот, в
аппарате идеального перемешивания Ре' = 0. Поскольку
явление продольного перемешивания можно наблюдать как в фазе
экстракта, так и в фазе рафината, обычно учитывают два
критерия Ре', определяемые для каждой фазы.
Продольное перемешивание вызывается турбулентной, или
вихревой, диффузией вещества вдоль оси экстрактора и
радиальной диффузией, обусловленной неравномерным распределением
скоростей по поперечному сечению аппарата (поперечная
неравномерность). Продольное перемешивание в дисперсной фазе
осложняется явлениями коалесценции и редиспергирования
капель. Сляйхер предложил24 описывать общий эффект
продольного перемешивания и поперечной неравномерности с помощью
одного коэффициента турбулентной диффузии, и такой подход
к решению проблемы, по-видимому, наиболее удобен.
Соответствующую систему уравнений решали24 на счетной машине.
В результате было получено эмпирическое уравнение:
( <0#'поршн. _ -поршн. РеЯРеЯ
ton
Здесь
Z = аРе^ + ЬРе'Е + с (Ре^Ре^)0-5 - d {Ре'Е + Pe'Rf5 +
+ е{Р^-Ре'Е)е-Ш/И<°«
(XI, И)
(XI, 12)
34 Р. Е. Трейбал
528
В уравнениях (XI, 11) и (XI, 12):
^паршн.—необходимая высота экстрактора для заданной
степени экстракции при идеальном противотоке;
И — необходимая высота экстрактора для достижения той
же степени экстракции при наличии продольного
перемешивания.
Из приведенных уравнений отношение Hnovmn]H можно
определить с наибольшей ошибкой, равной 10%, при следующих
значениях переменных: е = 0,25—10; Ре' = 2—55 (нижний предел
соответствует почти полному перемешиванию в аппарате, а
верхний предел — поршневому режиму движения) и NtoR =
= Я/Я(ол=1—60. Постоянные, входящие в уравнение (XI, 12),
зависят от величины фактора экстракции е, и их можно
определить24 с помощью табл. 17.
Таблица П
Значения постоянных величин в уравнении (XI, 12)
1/«
од
0,2
0,3
0,4
0,6
0,8
1,0
1,5
2,0
3,0
4,0
а
0,43
0,47
0,495
0,515
0,55
0,58
0,61
0,675
0,73
0,80
0,845
ь
0,15
0,21
0,27
0,32
0,425
0,52
0,61
0,81
1,00
1,38
2,00
с
0,31
0,485
0,625
0,75
0,975
1,16
1,31
1,61
1,85
2,12
2,25
d
0,41
0,59
0,73
0,85
1,07
1,25
1,42
1,78
2,10
2,45
2,65
е
—0,305
-0,29
—0,255
-0,22
-0,15
—0,075
0
0,18
0,35
0,64
0,865
Л
0,073
0,085
0,094
0,100
0,115
0,125
0,135
0,155
0,172
0,201
0,225
АППАРАТЫ С ОРОШАЕМЫМИ СТЕНКАМИ
(ПЛЕНОЧНЫЕ АППАРАТЫ)
В колоннах с орошаемыми стенками тонкая пленка одной
жидкости (обычно более тяжелой) стекает по внутренней
поверхности вертикальной трубы небольшого диаметра; по
остальному сечению трубы противотоком к движению пленки движется
другая жидкость. Колонны с орошаемыми стенками применяют
в лабораторных исследованиях, так как в них легко определять
величину поверхности контакта между фазами, что облегчает
изучение механизма массопередачи. К сожалению, полученные
на этих аппаратах результаты весьма противоречивы. Например,
по данным одних исследователей коэффициент массопередачи
630
в данной фазе зависит только от скорости этой фазы25-28-34;
по другим данным29 коэффициент массопередачи в фазе,
являющейся ядром потока, зависит от скорости другой фазы, но
коэффициент массопередачи в пленке не зависит от скорости в
ядре потока; наконец, отмечается 30, что скорости обеих
жидкостей влияют на коэффициенты массопередачи в каждой фазе.
Представляет интерес работа Мурдоха 27, в которой
исследовали процесс массопередачи, осложненный сольватацией
распределяемого вещества. Сольватацию можно рассматривать как
дополнительное недиффузионное сопротивление.
Вариантом колонны с орошаемыми стенками является
колонна, в которой одна жидкость течет по насадке, образующей
пирамиду из поставленных вертикально друг на друга и
скрепленных дисков28' 31. Стабильная работа таких устройств весьма
затруднена вследствие того, что пленка жидкости, обтекающая
диски, срывается с их поверхности; поэтому экстракторы такой
конструкции не пригодны для практических целей.
Другой разновидностью пленочного экстрактора является
труба, установленная в положении, близком к
горизонтальному, причем более тяжелая жидкость движется через нижнюю
половину площади поперечного сечения трубы, а легкая — через
верхнюю половину того же сечения32-33.
РАСПЫЛИТЕЛЬНЫЕ КОЛОННЫ
Распылительные колонны — простейшие экстракторы, в
которых диспергируется одна из жидкостей. Как видно из рис. 265
распылительные экстракторы представляют собой полые
колонны с устройствами для ввода
и вывода жидкостей. Вслед- г \ш \ш \ш
ствие отсутствия каких бы то ~
ни было внутренних устройств П
в активной зоне колонны
(между распределителями
жидкостей) сплошная фаза в ней
может свободно циркулировать
в вертикальном направлении
(продольное перемешивание); ^
это приводит к снижению
скорости массопередачи в
результате уменьшения
действительной движущей силы процесса.
Условия проведения
экстракции ухудшаются при
увеличении отношения диаметра и вы-
1
^CJ
в
Рис. 265. Распылительные колонны:
1 — уровень раздела фаз; 2— слой легкой
фазы; 3 — гидрозатвор; 4 — место установки
СОТЫ КОЛОННЫ. В расПЫЛИТеЛЬ- веитнля для регулирования уровня раздела
„„ т. фаз; /, /// —легкая жидкость; И. IV — тяже-
ных колоннах даже малых диа- * ' ' '
лая жидкость.
34*
631
метров (несколько сантиметров) весьма трудно достичь
степени разделения, соответствующей нескольким единицам переноса.
Вследствие отсутствия в рабочей зоне распылительных
экстракторов внутренних устройств эти аппараты применяют для
обработки жидкостей, содержащих твердые взвеси. Например,
в распылительных экстракторах цирконий и гафний разделяют
экстракцией из их растворов в тиоциановой кислоте, причем в
указанных растворах возможно образование твердых
полимеров тиоциановой кислоты.
Применяемая экстракционная установка35 состоит из
распылительных колонн диаметром 100 мм. Вследствие того, что
требуемая высота распылительной колонны была слишком велика
(30,5 м для экстракции и 91,5 м для отмывки), было
установлено несколько колонн меньшей высоты, соединенных
последовательно. В другом процессе распылительная колонна высотой
6,1 ж и диаметром 450 мм обеспечивала36 эффект разделения,
соответствующий примерно лишь одной теоретической ступени
разделения. Когда необходимо использовать распылительную
колонну с большой площадью поперечного сечения, для
уменьшения эффекта продольного перемешивания целесообразно
установить параллельно несколько колонн меньшего сечения.
Несмотря на недостатки распылительных колонн, изучение
их характеристик полезно, так как позволяет описать многие
общие характеристики работы экстракционных колонн.
Рассмотрим работу колонны, представленной на рис.265, а,
в которой диспергируется легкая жидкость. Тяжелая жидкость,
поступающая через распределитель сверху колонны, занимает
почти весь объем аппарата и в виде сплошной фазы движется
вниз колонны. Тяжелая жидкость, покидая колонну, проходит
через гидрозатвор 3, который обеспечивает полное заполнение
аппарата. Легкая жидкость вводится снизу колонны и с
помощью диспергатора распределяется в виде мелких капель.
Капли легкой фазы движутся через колонну в результате разности
плотностей фаз. На верху колонны капли сливаются и образуют
слой легкой фазы 2, которая отводится из верхней части
колонны. Давление тяжелой жидкости в гидрозатворе 3 (статическое
давление + давление на преодоление сил трения) должно
уравновешиваться давлением столба легкой и тяжелой жидкости в
колонне; соответственно этому будет устанавливаться
положение уровня раздела фаз / в колонне.
Уменьшение высоты гидрозатвора приводит к понижению
уровня раздела фаз / (как показано на рис. 265, б), и при
некотором положении гидрозатвора уровень раздела фаз
перемещается в нижнюю часть колонны. При этом в колонне
диспергируется тяжелая фаза. Кроме того, высота гидрозатвора
может быть такой, что уровень раздела фаз будет находиться в
средней части аппарата (см. рис, 265, в). Такой способ регули-
532
ровання положения уровня раздела фаз часто используют в
лабораторных и пилотных установках.
В промышленных экстракторах положение уровня раздела
фаз целесообразнее регулировать с помощью вентиля,
устанавливаемого в точке 4 трубопровода для отвода тяжелой фазы.
Этот вентиль может работать автоматически, будучи связанным
с датчиком, контролирующим положение уровня раздела фаз.
При таком методе регулирования уровня раздела фаз отпадает
необходимость в установке гидрозатвора. На рис. 265 приведены
схемы распылительных колонн для тех случаев, когда вся
колонна работает как колонна исчерпывания или колонна
обогащения; если питание вводится посредине колонны, то в этом
месте устанавливают распределитель, подобный тем, которые
показаны на рис. 265 в верхней и нижней частях колонны.
Рассмотрим работу колонны, показанной на рис. 265, а.
Пусть скорость тяжелой фазы постоянна, а скорость легкой
фазы постепенно увеличивается. При низких скоростях легкой
жидкости капли диспергируемой фазы равномерно образуются
на выходе из распределителя, свободно поднимаются вверх и
сливаются в верхней части колонны, где создается слой легкой
фазы.
По мере увеличения скорости подачи легкой жидкости
частота образования капель возрастает и капли движутся по
колонне в стесненных условиях; объемная доля дисперсной фазы
в аппарате (удерживающая способность аппарата)
увеличивается. С возрастанием скорости легкой фазы гидравлическое
сопротивление трубы для вывода ее из колонны будет
увеличиваться, соответственно уровень раздела фаз должен понижаться.
Чтобы сохранить прежнее положение уровня, нужно либо
увеличить высоту гидрозатвора 3, либо немного прикрыть вентиль,
установленный в точке 4.
Увеличение удерживающей способности по дисперсной фазе
уменьшает поперечное сечение, доступное для прохода сплошной
фазы, что приводит к значительным локальным скоростям
последней. При дальнейшем возрастании скорости легкой фазы и
соответственно объемной доли ее, удерживаемой в аппарате,
происходит еще большее увеличение скорости сплошной фазы,
вызывающее хаотическое движение капель. При этом в
аппарате могут возникать крупномасштабные вихревые токи
жидкости. В стеклянной колонне в этих условиях можно
наблюдать рециркуляцию (подъем и падение) капель. Распределитель
также уменьшает свободное сечение для прохода дисперсной
фазы.
Стеснение для движения капель, возникающее в этом месте
аппарата, может привести к слиянию капель в крупные глобулы,
которые могут увлекаться в нижнюю часть аппарата. При этом
задержка легкой фазы в аппарате резко возрастает, и большие
№
количества легкой жидкости уносятся через трубопровод для
отвода тяжелой фазы. Колонна оказывается заполненной легкой
жидкостью, которая в этих условиях часто становится сплошной
фазой (наступает захлебывание колонны), при этом
дальнейшая нормальная работа колонны становится невозможной.
Аналогичное явление наблюдается, как правило, и в тех
случаях, когда скорость легкой жидкости постоянна, а скорость
тяжелой увеличивается, хотя при очень низких скоростях легкой
жидкости еще до наступления захлебывания колонны может
наблюдаться значительный унос дисперсной фазы (легкой
жидкости). Унос легкой жидкости обусловливается высокой скоростью
сплошной фазы. Для каждой скорости течения легкой жидкости
существует определенная скорость тяжелой, при превышении
которой невозможна нормальная работа распылительной колонны.
Большую часть указанных выше причин, ускоряющих
захлебывание распылительной колонны, можно устранить, если при
ее конструировании предусмотреть плавный, с
минимальными возмущениями ввод сплошной фазы
и по возможности устранить в ней стеснение
поперечного сечения потоков. Этим требованиям
отвечает конструкция распылительной колонны,
предложенная Элджиным и Блендингом37'38. Колонна
схематично показана на рис. 266 и предназначена
для диспергирования легкой жидкости. Тяжелая
жидкость вводится сверху колонны таким
образом, чтобы не возникало высоких локальных
скоростей этой жидкости (сплошной фазы). Снизу
колонна расширяется под углом к вертикали, обычно
не превышающим 16°, что вызывает постепенное
уменьшение скорости сплошной фазы. Кольцевое
пространство между стенками расширяющейся
части колонны и распределителем должно быть
достаточным для того, чтобы скорость сплошной
фазы составляла 0,2—0,9 величины ее скорости в
корпусе колонны; при этом на выходе из
распределителя капли образуются в условиях
невозмущенного потока.
Захлебывание колонны такой конструкции
характеризуется возрастанием задержки дисперсной
фазы при уносе массы капель в нижнюю
воронкообразную часть колонны. Если дисперсной фазой
является тяжелая жидкость, колонну
устанавливают расширяющейся частью кверху. Когда
скорость коалесценции дисперсной фазы невелика,
применяют выносные отстойники, как показано на
/-легкая жид- рис. 267. На уровне границы раздела фаз из отстой-
кость; //-тяже- J г r r т
ника отводят скапливающиеся здесь загрязнения
ш
т \ t
л i п
Рис. 266.
Распылительная колонна
Элджина
(пат. США
2364892,
1944 г.):
кость:
лая жидкость
534
(шлам), препятствующие
коалесценции.
Удерживающая способность
по дисперсной фазе. Опытным
путем установлено, что
задержка дисперсной фазы, или
удерживающая способность (УС),
колонны при захлебывании
существенно отличается от
удерживающей способности до
наступления захлебывания.
Элджин с сотр. показали39-40,
что при скоростях потоков ниже
точки захлебывания отношение
скорости скольжения Vs к ско-
А С
U
ш
Рис. 267. Колонна с выносным
отстойником:
^ / — экстрактор; 2 — отстойник; / — тяжелая
рОСТИ СВОбОДНОГО ОСаЖдеНИЯ Vt жидкость (сплошная фаза); // — тяжелая
апиииииг.!* oiictuiiu r. uon,-, „d,*^.- жидкость из отстойника; /// — легкая
еДИНИЧНОИ ЧаСТИЦЫ В НеПОДВИЖ- жидкость из отстойника.
ной среде является однозначной
функцией задержки дисперсной фазы для любых дискретных
систем при движении составляющих их фаз в вертикальном
направлении (системы газ — твердое тело, жидкость — твердое
тело и жидкость — жидкость). Скорость скольжения
представляет собой истинную линейную относительную скорость фаз и
при противотоке фаз определяется из соотношения
V.-Z*.
(XI, 13)
где VD и Vc — фиктивные скорости фаз, т. е. скорости фаз,
отнесенные к полному сечению колонны.
Те же авторы нашли, что корреляцию Ценца41, полученную
для процесса псевдоожижения твердых частиц, можно
использовать для определения зависимости между отношением Vs/Vt
и фц. Эта корреляция представлена на рис. 268, где скорости
скольжения Vs определяют по соответствующему значению
удерживающей способности, а V, находят как Vs при </>!>=0.
Чтобы рассчитать скорость скольжения Vs для системы
жидкость— жидкость, значение скорости осаждения Vt вычисляют
из уравнений (V, 75) или (V, 76), а также с помощью графика
(см. рис. 100).
Удерживающую способность по дисперсной фазе,
соответствующую точке захлебывания, можно рассчитать из уравнений,
полученных Торнтоном 42, который ввел понятие
«характеристической скорости» Vk, определяемой из уравнения
v,= Mi-*D) =
(XI, 14)
535
0,0001
oj г зи вв/ г зи вею г зи ввюо
dp,
Рис, 268. Корреляция Ценца для псевдоожиженных твердых частиц 41
Торнтон предположил, что VK=Vt, однако даже в случае
несовпадения значений этих величин можно допустить, что
уравнение (XI, 14) пригодно для определения задержки при любых
режимах работы аппарата.
Величина VK действительно очень близка к величине Vt, но
наиболее точно уравнение (XI, 14) отвечает условиям
захлебывания. При захлебывании дУс1дфо=0 и из уравнения (XI, 14)
получаем:
Vdf = Wk*dfO-*df)
Аналогично dVD/d tD=0, откуда
VCF=VK(1- 2<t>DF) 0 - <>DF?
(XI, 15)
(XI, 16)
636
Исключая из уравнений (XI, 15) и (XI, 16)
характеристическую скорость VK, получают следующее уравнение для
определения задержки при захлебывании:
ф -KVnF/VCF)2 + WDF/VCFr-WnF/VrP
4(1 —V IV \
\ DPI CF>
(XI, 17)
Из уравнения (XI, 17) следует, что удерживающая
способность по дисперсной фазе при захлебывании не зависит от
размера капель и физических свойств жидкостей. Максимальная
величина задержки в распылительной колонне </-DF = 0,5
соответствует ее захлебыванию при VCF = Q.
Захлебывание. На основании уравнений (XI, 15) и (XI, 16)
можно заключить, что если величина VDF и, следовательно,' фОР
стремится к нулю, то величина VCF становится равной v'K=Vt.
Аналогично, когда VCF стремится к нулю и <f>DF стремится к
значению 0,5, величина VDF становится равной VK/4= Vt/4.
Следовательно, зависимость между скоростями фаз при
захлебывании имеет вид42, соответствующий рис. 269.
Скорости фаз при захлебывании в первом приближении
можно определить из уравнения (XI, 14), в котором VK
принимают равным Vt; однако, со статистической точки зрения,
лучшей является корреляция Минарда и Джонсона 43, которая'после
незначительного корректирования входящих в оригинальное
уравнение констант (для его упрощения) приводится к виду
У = 0,39 Др0'28
CF [0,216^ГО5р*5 + 0,2Ш°г;тР°65 (VD/Vcf5f (X1,18)
В действительности скорости фаз при захлебывании,
получаемые по уравнению (XI, 18), соответствуют уносу дисперсной
.фазы из конической части колонны Элджина. Поэтому
вычисленные при расчете по этому уравнению скорости фаз на 7—
10% выше скоростей фаз, наблюдаемых при захлебывании
обычной распылительной колонны. Кроме того, уравнение (XI, 18)
получено на основе опытов с
бинарными системами при
отсутствии перехода
распределяемого компонента из фазы
в фазу и не отражает влияния
конструкции и распределителя,
а также некоторых других
факторов «а предел
захлебывания. Поэтому при
проектировании рекомендуется
принимать величину нагрузки, не
превышающую" 40% от оассчи- ^ис-^у-(-оотношения между ско-
тянттй JX ™ /0 Л-УТо? ростями фаз при захлебывании рас-
шании по уравнению (Л1, 1»). пылительных колонн.
Рис. 269. Соотношения между ско-
537
1
\ О,»
af 0,6
02
1
0,8
0,8
£ О/
0,2
d a
•
to
A^
o\
V
3
a \ a
□
•
4 •
\
N
D
• N,
.3
ч
\
•
•
V
•
9 -1
•s»
■Bt7 ■
'Щ
2
•
2
•
V
*$/ 0,2 0,4 0,6 0,3 I. 2 4 6
LD/LC
Рис. 270. Значения высот единицы переноса в фазах для
распылительной колонны 46 (см. таблицу):
Номер
зависимости
1
2
3
4
Дисперсная
фаза
Изомасляиый
альдегид
Вода
З-Пеитанол
Вода
Сплошная фаза
Вода
З-Пентанол
Вода
Изомасляиый
альдегид
"tc
0,755 (Z.C/Z.D)°.54
0,64 (ic/iD)°'83
0,2 (ic/iD)0'96
HtD
-
°.3(£d/£C)0'114
0,426 (£D/Z.C)0.115
0,76
Теоретическое исследование УС и захлебывания
распылительных колонн приведено в работе Хармати44, который
рассматривал влияние особенностей конструкции колонн, подобных
изображенной на рис. 266, на указанные выше
гидродинамические факторы.
638
Массопередача. Имеются опытные данные о массопередаче
в распылительных колоннах диаметром, не превышающим
150 мм. Эти данные относятся к системам с третьим
компонентом, распределяемым между двумя жидкостями (тройные
системы), и к системам двух частично растворимых друг в друге
жидкостей, из которых одна не насыщена (бинарные системы).
В опытах с тройными системами можно определить только
общие для обеих фаз величины, характеризующие скорость массо-
передачи (общий коэффициент массопередачи или общую
высоту единицы переноса). В опытах же с бинарными системами,
когда массопередача может протекать лишь в одной фазе,
можно определять частные (для каждой фазы) значения
коэффициентов массопередачи или высот единиц переноса.
На рис. 270 представлены опытные данные Ладда45,
полученные при изучении массопередачи на бинарных системах.
Результаты этих опытов типичны: #(D почти не зависит от
соотношения или скоростей фаз, а Htc уменьшается с возрастанием
LD/LC или VD/Vc- Причины, обусловливающие такой характер
изменения Ht в фазах, рассмотрены ниже.
Из числа предложенных корреляций по массоотдаче в
сплошной фазе можно рекомендовать корреляцию Руби и Эл-
джина46, в которую авторы ввели поправку на концевой эффект.
В работе показано хорошее совпадение результатов опытов для
систем жидкость — жидкость со многими опытными данными,
полученными другими исследователями для систем жидкость —
твердое тело.
В наиболее простой аналитической форме корреляция Руби
и Элджина имеет вид:
Чтобы сравнить это уравнение с уравнением (V, 88) для
массоотдачи от единичной капли, представим уравнение (XI, 19)
в виде:
Shc = -&- = 0,725 Ре0'57 Sc5°'15(l -<t>D) (XI, 20)
где в диффузионный критерий Пекле входит скорость
скольжения V,.
Некоторое отличие значений коэффициентов массоотдачи,
получаемых по сравниваемым уравнениям, объясняется
различием условий переноса от системы движущихся капель и
единичной капли. Хертис21 скоррелировал свои опытные данные в
форме уравнения (V, 88) со средним значением коэффициента,
равным примерно 0,8. Полученные им величины коэффициентов
539
массоотдачи kc, включающие также концевой эффект, имеют
промежуточные значения сравнительно с величинами тех же
коэффициентов, вычисляемыми из уравнений (V, 88) и (XI, 19).
Коэффициенты массоотдачи в дисперсной фазе,
по-видимому, должны в меньшей степени зависеть от взаимного влияния
капель, чем коэффициенты массоотдачи в сплошной фазе. Для
крупных капель, в частности для капель, размер которых
больше определяемого из уравнения (V, 74), рекомендуется
пользоваться уравнением (V,83), в которое вместо Vt подставляют
скорость скольжения V&.
Для капель малых размеров можно использовать уравнение
(V, 81). Задержку дисперсной фазы (УС) можно определять
методами, описанными выше, а межфазовую поверхность
рассчитывать с помощью уравнения
л = -т^ (XI, 21)
Up
Чтобы проиллюстрировать корректность рекомендуемых уравнений
массоотдачи, используем их для расчета коэффициентов массопередачи по резуль-
20
— 10
Ус
о 3,6
о 7,3
А 11,4
v15f
<&ч^Ъ
V ССШ
V
О О
10
(Jjp--
6
12
Рис. 271. Сравнение опытных и рассчитанных значений коэффициентов
массоотдачи в распылительной колонне. Система метилизобутилкарбинол
(дисперсная фаза) — вода (сплошная фаза)47.
640
татам опытов Смита и Бекмана 47, проводившихся в распылительной колонне
диаметром 100 мм с соплами диаметром 2,7 мм. Дисперсной фазой являлся
метилизобутилкарбинол, сплошной — вода.
Фиктивные скорости фаз в колонне составляли: Vc = 3,66—16,2 и VD =
= 0,915—10,4 м3 • м~2 • ч-У.
Для определения диаметра капель был использован график, приведенный
на рис. 263.
В указанных пределах изменения скоростей дисперсной фазы диаметр
капель изменяется весьма мало. По уравнению (V, 74) можно установить, что
переходный размер капли меньше размера капель, образовывавшихся при
условиях опытов. Поэтому скорость осаждения рассчитывали с помощью
уравнения (V, 76), причем для заданных условий она была постоянная и составляла
Vt = 0,357 м/ч.
УС колонны по дисперсной фазе определяли с помощью графика (рис.268),
а соотношение Vs/Vt — по методу Элджина (т. е. по способу, аналогичному
приводимому ниже, см. пример XI-1). Межфазовую поверхность рассчитывали
по уравнению (XI, 21).
Вследствие того что размер капель изменялся в очень небольших
пределах, а скорость осаждения оставалась постоянной, УС и поверхность контакта
фаз в колонне практически не зависели от Vc и изменялись лишь при
изменении VD.
Этот вывод применим к большей части распылительных колонн при
нормальных условиях их работы. Для расчета kD применяли уравнение (V, 83)
(при замене в нем Vt на V8), а для расчета kc—уравнение (XI, 19).
Рассчитанные коэффициенты массоотдачи оказались не зависящими от скорости Vc-
Результаты расчетов приведены ниже;
vD
м3.м~2-ч-1
0,915
4,57
7,51
10,4
d
р
мм
4,66
5,45
5,25
4,9
*D
м/ч
0,28
0,27
0,259
0,25
kDa
•Г'
0,60
3,72
6,55
9,90
*С
м\ч
0,452
0,412
0,387
0,389
кса
ч '
0,92
5,7
9,8
15,3
Сравнение опытных и рассчитанных значений коэффициентов массоотдачи
показано на рис. 271, из которого видно, что они хорошо согласуются друг с
другом. Из рисунка следует также, что величины kca несколько зависят от Vc
(при расчете не было выявлено).
Значения коэффициентов масоотдачи kD приблизительно постоянны, а
межфазовая поверхность а зависит практически только от VD.
Отсюда следует, что высота единицы переноса HtD = VDkDa также
примерно постоянна.
Коэффициенты массоотдачи kc тоже близки к постоянной величине.
Поэтому высота единицы переноса Htc = Vc/kca приблизительно пропорциональна
отношению VclVD, или (Vd/Vc)'1. Это видно также из приведенных на
рис. 270 данных, относящихся к другим системам жидкость — жидкость.
541
Когда в массопередаче участвует третий компонент, можно
ожидать, что значения общих коэффициентов массопередачи
KdCI или Кф будут определяться в основном величиной
межфазовой поверхности, и, следовательно, они будут зависеть
главным образом от УС по дисперсной фазе. Обычно Htoc
уменьшается, a HtoD увеличивается при возрастании отношения
VDIVc.
Скорость изменения общей высоты единицы переноса по
любой фазе зависит от относительного влияния на ее величину
частных единиц переноса в каждой фазе.
Обычно общую высоту единицы переноса для
распылительных экстракторов и колонных экстракторов других типов
разлагают на частные (фазовые) высоты единицы переноса с
помощью уравнений (VIII,49) и (VIII, 50), представленных в
виде:
toe ~Htc~
Н.
mVD <d
(XI, 22)
Н
tOD
■ Н
ID
mVr
V,
Н
tc
(XI, 23)
Строя график зависимости Htoc от Vc/mVD или HtOD от
mVD/Vc в арифметических координатах, обычно получают
прямую линию, которую затем используют для корреляции
опытных данных. Однако ни наклон этой линии, ни отрезок,
отсекаемый ею на оси ординат,нельзя считать мерой величин Н1С и
Ню, так как часто, например, отрезок, отсекаемый на оси
ординат, является отрицательным, что
с D лишено физического смысла.
Возникающие в распылительной
колонне вертикальные
циркуляционные токи не только снижают
эффективность колонны, но и вызывают
трудности в интерпретации опытных
данных. Отбором проб жидкостей по
высоте экстрактора можно получить
профили концентраций, подобные
изображенным на рис. 272 сплош-
и , ными линиями. Если в колонне пре-
А " обладает поршневое движение сплош-
Нонцентрацця ной фазы (т. е. обратное перемеши-
распределпемоео континента вание полностью отсутствует), то
профиль концентраций изображается
Рис. 272. Профили концентра- На рис. 272 пунктирной линией DB.
ции в распылительной колонне: Если действ/тельная концентрация
/ — сплошная фаза; // — дисперсная г
фаза. распределяемого компонента в
•Q
t
*
■а:
ft
t-'
о
<о
о
СО
1
1
С"\
J
I
1
/
Сс
л7
/ *
/ /
/
/
/
/
542
сплошной фазе близка к постоянной, это отражает наличие
обратного перемешивания, интенсивность которого должна
увеличиваться с уменьшением скорости сплошной фазы48. Хертис21
наблюдал почти постоянный профиль концентрации для
сплошной фазы в исследованной им распылительной колонне.
Профиль концентраций для дисперсной фазы показывает57,
что продольное перемешивание в ней незначительно. Поэтому,
несмотря на то, что концентрации фаз на концах колонны (в
точках А, В, С и D на рис. 272) можно "рассчитать из уравнений
материального баланса, концентрации в сплошной фазе не
следуют обычным рабочим линиям, получающимся из
материальных балансов при условии поршневого движения сплошной
фазы в колонне. Значения высот единицы переноса Ню,
рассчитанные на основе действительных концентраций, будут меньше (и
точнее), чем рассчитанные для поршневого движения сплошной
фазы в колонне.
Резкое изменение концентраций в сплошной фазе «а входе в
колонну обусловливается не столько концевым эффектом,
вызванным коалесценцией дисперсной фазы (значение которого
невелико), сколько разбавлением сплошной фазы поступающей
жидкостью в результате продольного перемешивания49. Джиан-
коплис50'51 привел данные о значениях высот распылительной
колонны, эквивалентных теоретической ступени изменения
концентраций (ВЭТС), однако величины ВЭТС не поддаются
обобщениям, поскольку зависят от свойств системы и условий
проведения опыта. Было показано ^ 53, что в случае работы
распылительной колонны при относительно высоких скоростях фаз,
когда рециркуляция жидкости уменьшается, а УС и
межфазовая поверхность велики, можно получить относительно высокие
значения коэффициентов массопередачи.
Для определения общей скорости массопередачи в тройных
системах рекомендуют пользоваться методом, описанным выше
для бинарных систем (частично взаимно растворимых
жидкостей). В этом случае влияние всех явлений, связанных с
переносом через поверхность раздела и ее турбулентностью,
различием в скоростях массопередачи при противоположных
направлениях процесса и т. п., нельзя определить.
Распылительные колонны используют также как
теплообменники, позволяющие осуществлять теплопередачу при
непосредственном контакте между двумя жидкостями в отсутствие
разделяющей их металлической поверхности 53~56. Значения Ню
для теплопередачи, по-видимому, следуют тем же
закономерностям, что и Ню для массопередачи, и могут быть определены
на основе применения широко известной аналогии между тепло-
и массопереносом. При этом в уравнение массопередачи
вместо критерия Шмидта следует подставлять критерий Прандтля
и вместо критерия Шервуда — критерий Нуссельта.
643
Пример Х1-1. Определить размеры распылительной колонны для
извлечения 60% всей содержащейся уксусной кислоты из 4%-ного раствора ее в
воде (сс =0,667 кмоль/м3) метилизобутилкетоном, который не содержит
кислоты. Расходы фаз: 0,57 м3/ч водного раствора кислоты и 1,14 м3/ч кетона;
температура 25° С.
Решение. Для получения большей поверхности раздела фаз (допуская
возможность значительного обратного перемешивания в сплошной фазе)
целесообразно диспергировать ту фазу, которая имеется в избытке.
Концентрация кислоты в водном растворе после экстракции должна быть
1,6% (сс = 0,267 кмоль/м3), а концентрация кислоты в кетоне cD =
= (0,667—0,267) • 0,57/1,14 = 0,2 -кмоль/м3. Можно считать, что в пределах
указанных изменений концентраций физические свойства жидкостей остаются
неизменными и равными; для водной фазы |АС = 1 сяз( 10~3 «• се/с/ж2); Рс =
= 1000 кг/м3; для фазы кетона HD = 0,57 с/гз(0,57 • 10~3 н ■ сек/м2); р = 804
кг/м3. Коэффициент распределения т = c*D[cc = 0,545; а = 9,1 дн/см
(9,1 - Ю-3 н/м). Коэффициент диффузии для уксусной кислоты в воде Dc —
= 3,6- Ю-6 м2/ч (Ю-9 м2\сек).
Распределитель дисперсной фазы. Принимаем штампованные отверстия
диаметром rfo = 3,19 мм. По уравнению (XI, 2) скорость течения жидкости в
отверстии 0,0433 м/сек. При этой скорости потребуется чрезмерное число
отверстий в распределителе, поэтому принимаем скорость жидкости в
отверстии равной Vo=0,l м/сек. Абсцисса графика на рис. 263 при этом равна
0,0514, параметр 0,44; тогда rfP = 6,4 мм. Объемная скорость движения
жидкости в отверстии составит л • 0,003192 • 0,1 • 3600/4 = 0,00288 м3/ч, откуда
определяем необходимое число отверстий: 1,14/0,00288=396.
Диаметр колонны. Из уравнения (XI, 18) находим скорость сплошной
фазы при захлебывании. По условию VD/VC=2,0 и VCf = 1,2 • Ю-2 м/сек.
Принимаем рабочие скорости фаз Vc=0,4 • 1,2 • 10_2 = 4,8- 10~3 м/сек и
Vd = 2 • 4,8- 10~3 = 9,6 • 10~3 м/сек. Тогда поперечное сечение колонны составит
0.57/3600 • 4,8 • 10~3 = 0,033 ж2; ему соответствует диаметр колонны, равный
200 мм.
Скорость осаждения капли. По уравнению (V, 73) Р = 39,6-107 и
согласно уравнению (V, 74) величина rfPf = 3,58 мм. Так как dp>pt, используем
уравнение (V, 76), из которого Vi = 0,106 м/сек.
Удерживающая способность аппарата по дисперсной фазе. Пользуясь
графиком на рис. 268, находим, что его абсцисса для заданных условий
равна 87,0, а знаменатель ординаты составляет 49,5. Из рис. 268 определяем:
<t>D
0
0,1
0,2
0,3
Ордината
14,3
9,7
7,9
5,1
Vs(1-4>d)
м/сек
0,197
0,133
0,115
0,07
vs
(для твердых
частиц)
м/сек
0,1965 = Vt
0,148
0,12
0,1
VslVt
1,0
0,754
0,611
0,51
vs
(для жидких
чя'-тчц)
м/сек
0,106
0,08*
0,0648
0,054
* 0,754x0,106 = 0,08.
Уравнение (XI, 13) в этом случае имеет вид:
_ 9,6-10~3 , 4,8- 1(Г3
544
Решая это уравнение в пределах значений, приведенных в первом и
последнем столбцах таблицы, получим Vs»7,2 • 10~2 м/сек и 0В = О,15.
Экстракция в период образования капель. Объем капли vp = nd3/6 —
= 1,37 - 10-7 ж3. Через одно отверстие протекает 0,00288 м3/ч жидкости;
соответственно 9, = 1,37- 10-7/2,88- 10-3 = 4,8 • 10~5 ч. Из уравнения (XI, 10) имеем
Kd/ = 0,406 кмоль ■ ж-2 •«-' • Дсч. Из уравнения (XI, 9) находим: Av —
= 7,7- Ю-5 ж2.
Следовательно, для всех отверстий KofA = 0,406- 7,71 • 10~5 • 396=
= 1,24-Ю-2 кмоль ■ ч'1 • Д<г'. Применим уравнение (XI, 5): экстрагируется
кислоты 1,24 - 102 • [0,545 - 0,265 — 0]=1,79-10~3 кмоль/ч. Поэтому
концентрация кислоты в дисперсной фазе в плоскости сечения колонны,
соответствующей завершению концевого эффекта (на входе дисперсной фазы) cD =
= 1,79 • 10~3/1,14= 1,57- 10~3 кмоль/м3; концентрация кислоты в сплошной фазе
составит сс = 0,265+1,79- 10~3/1,14 = 0,2666 кмоль/м3.
Экстракция в период коалесценции капель. Примем KDA = 0,44 (см. выше).
Количество проэкстрагированной кислоты: 1,24 • 10"2 • [0,545 • 0,662 — 0,198] =
= 2,0 * 10_3 кмоль/ч. Концентрация кислоты в дисперсной фазе до концевого
эффекта cD = 0,198—(2,0- 10 3)/1,14 = 0,196 кмоль/м3; концентрация кислоты
после концевого эффекта сс = 0,662—(2,0 • 10_3)/0,57=0,658 кмоль/м3. Как
следует из приведенных расчетов, концевые эффекты в условиях данной
задачи относительно невелики.
Экстракция в средней части колонны. Определим межфазовую
поверхность' а = 60в/с?Р = 140 м2/м3. Для сплошной фазы из уравнения (XI, 19)
имеем: &с = 0,207 кмоль ■ м'2 • ч'1 Ас'1 и kca=29,Q кмоль -м3 ■ ч'1 Ас1.
Для дисперсной фазы из уравнения (V, 83) при замене Vt на Vs
находим:
ч-'-Дс"1
kD =
0,62
Для раз
кмоль ■ м~2
1
Кса
бавленных ра
н - V«
tOR KRa
mVE
VR
ч~х ■ Sc
1 +
1 k a = 87,0 кмоль ■ л
1 _ 1 ,
kca mk a 29 ~
Kca-
створов:
vc
Kca
mVD
VC
= 18,0 = KRa
4,82- 10" 3-
18,0
0,545 • 9,64
1,21 • 10
1
0,545 •
3600
10~3
-3
87,0
- 0 9€
- 1 0
Отметим, что в этом случае m = cD/cc = cE/cR. Для разбавленных
растворов (как в нашем случае) расчет высоты колонны в поршневом режиме
можно провести с использованием рис. 196 при замене xR на cR и т. д.
Так
cD, — cmlm 0,267 — 1,57 • 10~3/0,545
-М Ш.— = = 0,402
CR\ ~~ CE2lm °'667 — !>57 • Ю_3/0,545
Из рис. 196 находим:
N,OR = 5-35 Япоршн. = N<ORH,OR = ^ • °'965 = ^3 М
Если сплошная фаза (рафинат) в колонне полностью перемешана, то
Ре^ = 0. Примем Ре^ = 2; будем считать также, что режим движения
экстракта (дисперсной фазы) в основном поршневой (Ре£ = со) и что Ре£ = 55.
При е=1,09 из табл. 17 получаем а = 0,597; 6 = 0,572 и т. д. Подставляя эти
35 Р. Е. Трейбал
545
значения в уравнения (XI, 11) и (XI, 12), методом последовательного
приближения находим Я=2,3 м.
Отметим, что если при той же степени продольного перемешивания
увеличим Я вдвое (до 4,6 м), из уравнения (XI, 11) определяем ЯПОршп./Я=
=0,389 и ЯПОршн. = 1,8 м. Следовательно, величина NtOR = 1,8/0,965= 1,86.
Тогда из рис. 196 получим ординату, равную 0,35 и соответствующую степени
извлечения примерно 65% от всего количества уксусной кислоты в исходном
растворе.
\—Ш
5-
КОЛОННЫ С ПЕРЕГОРОДКАМИ
Колонны с перегородками имеют цилиндрический корпус,
внутри которого расположены горизонтальные перегородки, не
перекрывающие всего сечения колонны и служащие для
направления движения жидкостей. Типичными и простейшими
аппаратами этого типа являются экстракторы, показанные на
рис. 273.
Такие колонны изготавливают также с перегородками типа
диск — кольцо. В этом случае кольца с центральными
отверстиями, прикрепленные по периферии к обечайке колонны,
чередуются с круглыми дисками, которые расположены по оси
аппарата и поддерживаются
вертикальным штоком или
тягами, соединенными с
обечайкой аппарата. Расстояние
между перегородками обычно
составляет 100—150 мм.
Перегородки в колоннах
устанавливают для того,
чтобы уменьшить циркуляцию
жидкости в вертикальном
направлении, возникающую в
полой колонне, и в то же время
увеличить продолжительность
пребывания в колонне
дисперсной фазы. Кроме того, частая
коалесценция и редиспергиро-
вание одной из жидкостей
ускоряют процесс массопере-
дачи.
Колонны с перегородками
применяют очень давно, в
частности в процессах экстракции
уксусной кислоты из подсмоль-
ных вод и растворов,
используемых в производстве
ацетатного шелка; реже их
применяют в некоторых процессах
очистки нефтей. Краткие сведе-
■^ЕЗЯ3
^-—- У
—■£
-Ш
г—I
Рис. 273. Колонны с перегородками
(Vulcan Capper and Supply Co.):
a — направление пртоков от стенки к стенке,
б —то же от центра к центру; /, // —
тяжелая жидкость; ///, IV — легкая жидкость.
ш
цич об установках с такими колоннами приведены в обзоре Мо-
релло и Поффенбергера3. Данные для проектирования колонн
с перегородками в литературе отсутствуют.
НАСАДОЧНЫЕ КОЛОННЫ
Корпус экстракционной колонны заполняют насадкой,
которая уменьшает продольное перемешивание и способствует
£$ столкновению и разрушению капель дисперсной фазы, что при-
% водит к возрастанию скорости массопередачи. В большей части
4 й£ на садочных колонн используют стандартные насадки промыш-
JiF ленного изготовления, широко применяемые в колоннах для
Ж* обработки систем газ — жидкость, например кольца Рашига,
g кольца Лессинга и Паля, а также седла Берля и «Инталокс»,
беспорядочно засыпанные в колонну. На некоторых старых
заводах, перерабатывающих побочные продукты коксования,
раньше использовали также деревянные хордовые насадки; в
% настоящее время они вытесняются более совершенными
насадками, в частности регулярными металлическими насадками в
виде растянутых металлических сеток.
Насадка, уменьшая свободное сечение для прохода
жидкости, снижает удельную производительность насадочной колонны
(производительность на единицу поперечного сечения аппарата)
по сравнению с удельной производительностью распылительной
колонны. Кроме того, насадочные колонны не пригодны для
Ш обработки жидкостей, содержащих твердые примеси. В наса-
^ дочных колоннах можно достигнуть степени извлечения, соот-
§". ветствующей нескольким теоретическим ступеням разделения.
Поэтому данные аппараты используют почти во всех промыш-
I- ленных экстракционных процессах.
■ Для успешного применения стандартной насадки необходимо
К.*- соблюдать некоторые условия. Наиболее важное значение имеет
Ч правильный выбор материала насадки. Она должна предпо-
Г чтительно смачиваться сплошной фазой, при этом устраняется
; возможность нежелательной коалесценции капель внутри на-
л^еадки. Если насадка предпочтительно смачивается жидкостью,
^ являющейся дисперсной фазой, то это приводит к растеканию
& Жидкости вдоль поверхности насадки и уменьшению межфазо-
£'вой поверхности. Керамическая и фарфоровая насадки почти
v всегда лучше смачиваются водной фазой, чем органической;
» угольная и пластмассовая насадки лучше омачиваются органи-
-■ ческой фазой. При использовании металлических насадок
целесообразно провести предварительное испытание их
смачиваемости. В любом случае желательно, чтобы та жидкость, которая
- предпочтительно смачивает насадку, была сплошной фазой. Все
, количественные выражения, включающие характеристики
насадки, приводимые ниже, применимы исключительно в тех слу-
85*
647
новках, так как колонны
этих установок нужно заполонять насадкой того же размера
(и типа), что и промышленные колонны, в которых по
экономическим соображениям применяют насадку относительно
больших размеров. Дальнейшее рассмотрение влияния размера
насадки на работу «асадочных колонн приведено в разделе
«Удерживающая способность по дисперсной фазе» (стр. 549).
Чтобы предотвратить разрушение нерегулярной насадки при
загрузке навалом, ее засыпают в колонну, предварительно
заполненную водой. Насадка, загруженная таким образом, при
работе колонны неизбежно будет оседать, и ее объем должен
соответственно изменяться. Чтобы быстро достичь
неизменяемого объема насадки, последнюю энергично продувают
воздухом, пропуская его снизу вверх по колонне, заполненной водой.
Желательно также, чтобы сопла, используемые для
диспергирования одной из фаз, входили вглубь слоя насадки не
менее чем на 25—50 мм 38> 58>59, так как капли, образуемые в слое
светлой сплошной фазы, неизбежно сталкиваются друг с
другом и коалесцируют до достижения насадки. Капли,
образующиеся на перфорированной тарелке и смачивающие материал
тарелки, вследствие коалесценции имеют большие размеры.
Если для насадочной колонны используют обечайку колонны
Элджина (рис. 266), то необходимо, чтобы распределитель для
дисперсной фазы находился в слое насадки; насадку
загружают на решетку из металлических колосников, которые
располагают на возможно большем расстоянии друг от друга.
Однако расширения корпуса на концах насадочных экстракционных
колонн не имеют такого важного значения, как в
распылительных колоннах 58„ В колоннах с цилиндрическим корпусом уста-
648
навливают опорную колосниковую решетку для слоя
насадки или металлическую тарелку, подобную показанной на
рис. 274.
Для уменьшения продольного перемешивания и каналообра-
зования в колонне на расстоянии 3—4,5 м друг от друга
устанавливают перераспределители потоков. Схема установки
такого перераспределителя показана на рис. 274. Тяжелая
(сплошная) фаза проходит через сливные патрубки тарелки, а
легкая (дисперсная) фаза собирается под тарелкой в виде
сплошного слоя и вновь диспергируется в слое насадки,
находящемся на тарелке. Конструкция перераспределительных
тарелок приведена в разделе «Колонны с перфорированными
(ситчатыми) тарелками» (стр. 562). Аналогичные конструкции
используют для диспергирования тяжелой фазы и начального
распределения той или иной фазы на конце колонны.
На выходе дисперсной фазы из колонны необходимо иметь
в последней некоторый свободный объем (без насадки), в
котором коалесцируют капли и образуется слой дисперсной'фазы,
а также поддерживается граница раздела фаз (см. рис. 265).
В этой секции можно установить регулятор уровня и
необходимо предусмотреть устройство для периодического удаления
накапливающихся здесь загрязнений. В некоторых случаях
устанавливают дополнительно выносной отстойник, как показано
на рис. 267.
Удерживающая способность по дисперсной фазе. Объем
дисперсной фазы, удерживаемой в насадочной колонне, условно
делят на несколько частей, определение которых возможно при
использовании различных способов измерения УС. Если
одновременно прекратить подачу жидкостей в колонну и измерить
объем отстоявшейся дисперсной фазы, получают так
называемую «нормальную», или свободную, УС, которая определяется
как доля свободного объема насадки. Кроме того, существует
еще добавочный объем дисперсной фазы (удерживаемый в
насадке) , который не осаждается из насадки и называется
«перманентной» УС60'61. С помощью измерений радиоактивности62
было показано, что объемы дисперсной фазы, соответствующие
как перманентной, так и нормальной УС, во время работы
колонны, по-видимому, находятся в движении, причем
перманентная УС оказывает влияние на величину общей активной
поверхности контакта фаз, хотя степень ее влияния количественно
определить нельзя. Для УС насадочной колонны характерно
наличие небольшого гистерезиса, т. е, величина УС зависит от
того, увеличивается или уменьшается скорость движения
дисперсной фазы перед замером УС. В литературе опубликованы
лишь ограниченные сведения о величине обшей УС 6!>62.
Подробное изучение УС и связанных с ней явлений имеет
очень важное значение, так как позволяет определить поверх-
649
ность массообмена; подробные исследования выполнили Пратт
с сотр.58'60'63'64. Ниже приведены результаты этих работ.
Для каждой бинарной системы жидкость — жидкость
существует критический размер насадки dFC, определяемый для
стандартных насадок (колец и седел) по уравнению
^с = 2.42(дУ0'5 (XL 24)
Если размер насадки больше критического, то
характеристический средний размер капель дисперсной фазы dp почти не
зависит от размера и формы насадки и слабо зависит от
скоростей фаз. Для насадки, размер которой меньше критического,
величина капель возрастает, а для насадки, имеющей
критический размер, диаметр капель сильно зависит от скорости фаз.
В насадках малых размеров капли, очевидно, задерживаются
в промежутках между насадочными телами и имеют
возможность продвигаться лишь под действием толчков со стороны
других капель; эти капли коалесцируют, и их размер
увеличивается. Критический размер насадки равен примерно 12 мм;
его следует рассчитывать по уравнению (XI,24). Влиянием
размера насадки можно объяснить наблюдения Балларда и Пи-
рета 65, которые отмечали много необычных гидродинамических
явлений при работе с насадками малых размеров. Для
практического применения рекомендуют насадку, размер которой
больше dFc-
Если размер насадки больше критического, то при низких
скоростях дисперсной фазы Vd величина УС возрастает почти
линейно с возрастанием l/D до достижения первой «переходной»
точки, которая зависит от Ус и является аналогом точки под-
висания для систем газ — жидкость. При дальнейшем
увеличении Vd значение УС по дисперсной фазе возрастает резче и
капли дисперсной фазы начинают быстро коалесцировать
(достигается вторая переходная точка). Незначительное дальнейшее
возрастание VD не вызывает заметного увеличения <f>D; при этом
может произойти обращение фаз (дисперсная фаза становится
сплошной, а сплошная — дисперсной). Такой режим работы
колонны завершается захлебыванием последней.
Величину УС по дисперсной фазе вплоть до первой
переходной точки можно рассчитать из уравнения:
vr^+-T^ry.0-*D) <х'-25>
где 8 — свободный объем насадки;
VK— «характеристическая» скорость, определяемая как
предельная средняя скорость капли при Vc = 0 и малых
значениях VD.
550
0.1
г з ^ 5 7 ю
0,2 0.30^0,507 I
., „_r , сие CL_r .._._. , „_
(график не применим при диаметре колони Т < 0,075 м или dF < dFCffi.
Рис. 275. Характеристические скорости капель в различных насадках
Если в уравнении (XI, 25) вместо свободной УС ввести
величину общей УС, то, по-видимому, вместо относительной
скорости Vr найдем скорость скольжения Vs. Дифференцируя
уравнение (XI, 25) и приравнивая производные дУс/дфп и
дУв/дфю нулю, получим уравнение (XI, 17), из которого
определяют предельную УС, соответствующую переходной точке.
Эта точка отвечает скоростям фаз, составляющим 50—70% от
скоростей захлебывания. При расчете колонн рекомендуют
принимать значения УС в пределах 0,15—0,2, причем нижний
предел при малых значениях отношения VD/Vc (менее 0,5).
Характеристическая скорость капель при режимах вплоть
до первой переходной точки была определена для различных
насадок (колец размером до 37,5 мм и седел до 25 мм) по
измеренным величинам <f>D с помощью графиков, построенных на
основе уравнения (XI, 25). Была получена корреляция для
характеристической скорости в зависимости от физических
свойств систем. По графику на рис. 275, построенному в
координатах, предложенных Праттом 63, можно рассчитать величину
VK с точностью ±10%. Средний диаметр капель при dF>dFC
фактически не зависит от размера насадки и изменяется для
жидкостей, находящихся в равновесии (т. е. взаимно
насыщенных), следующим образом:
dp0VK^D
(XI, 26)
Где dp0 — диаметр капли при скорости потока, равной нулю.
651
Корреляция опытных данных имеет вид:
-^-2—= 1.42/^— (XI, 27)
V-c \V-cg
Следовательно
Дра3 ч'0'475
,= 1,42
Р-сё
Уравнение (XI,28) представлено в приближенном виде в
результате округления показателя степени и некоторого
преобразования входящих в уравнение безразмерных комплексов.
Если две жидкие фазы взаимно ненасыщены и если
происходит перенос вещества из дисперсной органической фазы в
сплошную водную, то размер капли dp будет больше, но
степень его увеличения в настоящее время установить нельзя. Если
же перенос распределяемого компонента происходит в обратном
направлении, размер капли не изменяется.
Если образующиеся «а выходе из распределителя капли
имеют размер, меньший стабильного в данных условиях, то
при прохождении через насадку размер капель постепенно
увеличивается (в результате коалесценции) до устойчивой
величины; при этом может наступить преждевременное захлебывание
колонны. Поэтому целесообразнее диспергировать фазу таким
образом, чтобы образующиеся капли имели первоначальный
размер несколько больший того, который получается расчетом
по уравнению (XI, 28); при этом капли быстро (на расстоянии
около 1 м от распределителя) разрушаются, достигая
характерного размера. Диаметры сопел распределителя необходимо
выбирать в соответствии с вышеизложенным.
Межфазовую поверхность в колонне, связанную со
свободной удерживающей способностью, определяют из уравнения:
a=-d-^ = irf- (XI-29)
ар "/;0*к
Второе выражение для а получено подстановкой уравнения
(XI, 26) в первое выражение. Второе выражение для а не
содержит фв в явном виде. Отсюда можно предположить, что
увеличение межфазовой поверхности, которое возможно при
возрастании <f>D в результате увеличения Vc, по-видимому,
компенсируется увеличением размера капель; в результате
межфазовая поверхность остается примерно постоянной вплоть до
точки подвисэния. Межфазовая поверхность, через которую
происходит массопередача, вероятно, несколько больше
определяемой из уравнения (XI, 29); эта поверхность зависит от общей
удерживающей способности, и поэтому ее трудно определить.
562
'v
s
\
S
10"г
5 Ю12 5 10*2
5 10°
Рис. 276. Корреляция по
захлебыванию насадочных колонн 67.
Захлебывание. Изучению
захлебывания посвящено
много исследований, и получен ряд
эмпирических и
полуэмпирических корреляций для
определения скоростей фаз,
соответствующих точке
захлебывания. Однако при пользовании
этими уравнениями следует
соблюдать большую
осторожность.
Большая часть корреляций
опубликована в виде
графиков, координаты которых
представляют собой очень сложные
функции от физических свойств
жидкостей и скоростей
потоков. Опытные точки по
захлебыванию, наносимые на графики, ложатся вблизи
корреляционный кривой, причем отклонения их (выражаемые обычно
в долях от величин, наносимых на координатные оси)
составляют несколько процентов. В большей части случаев
координаты содержат одноименные величины. Поэтому определяемые
таким образом отклонения нельзя считать мерой корректности
корреляций для описания захлебывания колонны; при
сравнении рассчитанных и опытных скоростей фаз, соответствующих
захлебыванию, отклонения достигают, часто, нескольких сот
процентов.
Статистическое изучение всех опубликованных данных
путем сравнения опытных и рассчитанных по известным
корреляциям скоростей захлебывания 66 показало, что корреляция Кроу-
форда и Уилки67 дает наилучшее совпадение рассчитанных и
опытных величин скоростей захлебывания, полученных в
исследовании этих авторов, а корреляция Хоффинга и Локкарта68
точнее других описывает все опубликованные опытные данные.
Корреляция Кроуфорда и Уилки приведена на рис. 276.
Корреляцию Хоффинга и Локкарта можно представить в виде:
V,
С-Др'
,0,625
водн., р
.0,275 0,125 0,10 0,125 j„/n7W62S/ / 0,12\0,838
Коэффициент С определяется по уравнениям:
при Кводн./Корг. = 0,005 —1,4
С = 7,35-10-^(Кводн./Корг.У
0,564
при Кводн./Уорг.= 1,4—100
C = 9,74-10~J(KB
,,/ ..0,295
(XI, 30)
(XI, 31)
(XI, 32)
563
Во всех опытах Хоффинга и Локкарта насадка
предпочтительнее смачивалась водной фазой.
При захлебывании всегда происходит некоторое обращение
(инверсия) фаз; поэтому для захлебывания не имеет значения,
какая фаза диспергируется, что в некоторой степени отражено
на рис. 276 (величины VD и Vc входят в корреляцию Кроуфор-
да и Уил,ки в одинаковой степени).
Величина а' в уравнении (XI, 30) является межфазовым
натяжением на границе водной фазы и воздуха; она была введена
в это уравнение для того, чтобы распространить его также на
системы газ — жидкость. Обе корреляции по захлебыванию
получены на основе обработки результатов опытов с жидкостями,
находящимися в равновесии. В связи с тем что количественно
влияние массопереноса на скорость захлебывания до сих пор
не установлено, при конструировании насадочных колонн
следует принимать скорости фаз равными не более 50—60% от
скоростей, соответствующих захлебыванию колонны.
Для данного номинального размера и типа насадки
плотность упаковки насадочных тел и, следовательно, величины
aF и 8 зависят от материала, из которого изготовлена насадка
(металл, керамика или пластмасса), способа заполнения ко-
Таблица 18
Характеристики
Номинальный
размер *
1/4" (6,3) **
3/8" (9,5) **
1/2" (12,7)**
5/8" (15,9) **
3/4" (19,0)
1"(25,4)
1 1/4" (31,8)
1 1/2" (38,1)
2" (50,8)
насадок
Керамические
кольца Рашига***
ш
0,53
0,59
0,57
0,67
0,74
0,69
0,71
0,73
683
492
328
295
292
171
184
65,6
СО
SS
S
СМ
СО
а
526
232
194
169
131
102
96,6
93,1
Угольные
кольца
Рашига
СО
0,53
0,72
0,73
0,67
0,73
а.
683
425
209
97
72,7
Керамические
седла
Берля ***
СО
0,72
0,71
0,76
0,76
376
622
301
114,5
СО
S3
о"
ш
472
169
107
64
Керамические седла
Ииталокс 4*
ш
0,75
0,78
0,77
0,775
0,81
0,79
ч.
982
622
324
255
195
118
Металличе
ские кольца
Паля <*
ш
0,902
0,934
0,94
0,94
5"
430
217
157
120
* В скобках указан номинальный размер в мм.
** У этих насадок размер иасадочиых тел может быть меньше &fq. Прежде чем
использовать эти насадки, следует провести проверку по уравнению (XI,24).
*** Величины, рекомендуемые Хоффингом и Лохкарточ для подстановки в уравнение (XI,30),
** Производства "U. S. Stonware Company" (Akron, Ohio).
654
лонны насадкой и от усадки последней при работе колонны.
Наиболее точно указанные выше характеристики насадок
можно определить, замерив их 'Непосредственно на производстве.
Если же такие замеры сделать нельзя, то при расчетах можно
пользоваться значениями а¥ и 8, приведенными в табл. 18.
Массопередача. Анализ массопередачи и получение
надежных характеристик массообмена, необходимых для
проектирования экстракционных аппаратов, невозможно без
предварительной оценки степени продольного перемешивания в этих
аппаратах. Продольное перемешивание в насадочных колоннах
значительно менее интенсивно, чем в распылительных
колоннах 57.
Однако количественные данные о продольном
перемешивании в насадочных колоннах (величинах диффузионного
критерия Пекле Ре', характеризующего продольное перемешивание)
весьма ограниченны. Соответствующие данные получены
только для сферических и кольцевых насадок на системах
керосин— вода и минеральное масло — вода22'69. Эти данные
недостаточны для анализа массопередачи и использования их при
проектировании насадочных колонн. Исследования позволяют
сделать лишь следующие качественные выводы: как и
следовало ожидать, критерий Ре'с увеличивается с возрастанием Vc
и уменьшается с увеличением Vd. Критерий Ре^ уменьшается
с увеличением Vc и уменьшением Vd (для дисперсной фазы, не
смачивающей насадку) или с возрастанием Vd (для дисперсной
фазы, смачивающей насадку).
Для определения истинной поверхности контакта фаз, через
которую происходит массопередача, необходимо также иметь
более подробные сведения о величине активной УС по
дисперсной фазе в насадочных колоннах. Однако такие данные пока
отсутствуют.
Для изучения массопередачи в каждой фазе опыты часто
проводят в системах с двумя частично растворимыми
жидкостями при отсутствии третьего (распределяемого)
компонента 45,47,60.70^ Типичные опытные данные приведены на рис. 277.
Пренебрегая для упрощения поправкой на отсутствие третьего
компонента и подставляя в общее выражение единицы переноса
значение а [по уравнению (XI,29)], получим71:
„ VD _ dp0VK
to kDa 6kD
„ _ vc _ d,ov«vc
"tc- kca &kcVD
Из этих уравнений следует, что Hw почти не зависит от
отношения Vc/Vd, a lite прямо пропорционально ему. Смит47
(XI, 33)
(XI, 34)
ббб
2
s '
*~ 0,6
0,2
в
&
О
О
В
R D
Г
/
R u
О
J
J*tc
z
A
/ „
в °
L H
Htn
oj 0,2 оь 0,6op 1 г
U 6
Рнс. 277. Значения высот единиц переноса в фазах для насадочных
колонн (см. таблицу).
Кривая
Дисперсная фаза
Сплошная фаза
Насадка
t-D
г-с
Литература (номер
ссылки)
/
Изобутиловый альдегид
Вода
Кольца диаметром 9,5 мм
1 820-4 700
3 330-10 350
45
2
Изобутаиол
Вода
Кольца диаметром 6,4 мм
1 220-10 150
1 500-8 700
70
показал, что HtD для данной системы во всех опытах остается
почти постоянной величиной, a Htc = M(VcIVD)n, причем
показатель степени п изменяется в пределах 0,54—0,98. Попытки
обобщить опытные данные для HtD не увенчались успехом;
были получены лишь эмпирические зависимости для М и п.
Вероятно, обобщенных корреляций не удалось получить
вследствие того, что из величин ВЕП, на основе которых делались
попытки обобщить опытные данные, не выделяли слагаемые
общих значений ВЕП, обусловленные продольным
перемешиванием; поэтому указанные значения ВЕП были выше
действительных.
По данным Вермюлена72, действительные значения ВЕП
могут быть на 15% ниже кажущихся, т. е. наблюдаемых в
опытах. По Пратту2'60, величина kc/Vr для сплошной фазы
является функцией комплекса (dpWpc/Vc)™- При подстановке в
зависимость между этими величинами выражения Vr из уравнения
(XI, 25) и выражения dv по уравнению (XI, 26) и при введении
эмпирической поправки на влияние размера насадки получают
уравнение
*c<40'95-7'87^
vc
-5,3.10-(^с J'5
ч
VC*D 1
vd{1-+d)\
1,5
(XI, 35)
668
Это уравнение приближенно учитывает продольное
перемешивание.
Уравнение (XI, 35) применимо для расчета коэффициентов
массопередачи в сплошной фазе при направлении массопере-
дачи из водной фазы в органическую; при противоположном
направлении массопередачи уравнение (XI,35) дает
заниженные значения kc.
Это вызвано, по-видимому, наличием межфазовой
турбулентности, а также влиянием третьего компонента на размер
капли. Вермюлен72 использовал аналогичное уравнение, в
которое входит критерий Шмидта, и высказал предположение,
что экспериментальные данные о массопередаче (с учетом
поправки на продольное перемешивание) можно применять для
приближенного расчета размеров капель с целью
использования их в корреляциях по массопередаче. Джонс и Бекманн 73
использовали для обработки опытных данных о массопередаче
уравнение (V, 88).
Уравнение (XI, 19) достаточно хорошо соответствует
опытным данным о массопередаче в потоке, движущемся через слой
твердых частиц и через систему капель. Поэтому указанное
уравнение можно рекомендовать (при замене Vs на Vr) для
расчета массопередачи в сплошной фазе. Для расчета
массопередачи в дисперсной фазе применимо уравнение (V, 83) при
условии, что размер капель больше их переходного размера,
отвечающего скорости осаждения, и при замене Vt на Vr; для
более мелких капель рекомендуют пользоваться
уравнением' (V.81).
Чтобы проиллюстрировать пригодность рекомендуемых уравнений по
массопередаче, используем для их проверки опытные данные Смита и Бекман-
на47, полученные авторами на системе метилнзобутилкарбинол (дисперсная
фаза) — вода в колонне с насадкой из колец '/г" (12,7 мм).
По уравнению (XI, 27) величина с?Р0= 1,72 - 10~3 ж; из уравнений (XI, 25),
(XI, 28) и рис. 275 находим dp, VK, Фю и VT, а из уравнения (XI, 29)—
величину а. Используем для расчета kc уравнение (XI, 19), заменив в нем V,
на Vr, и для расчета kD — уравнение (V, 83), заменив в нем Vt на VT.
Результаты расчетов приведены ниже:
м/ч
0,915
0,915
10,1
1Г.1
м/ч
3,66
16,2
3,66
16,2
"р
м
1,83-Ю-3
2,05 -10~3
2,08 -10"~3
2,5 -Ю-3
Фо
0,0С97
0,0108
0,119
0,139
м/ч
164,7
170,2
170,0
175,1
Vr
м/ч
162,2
168,4
150,0
151,0
а
м'/м>
19,1
19,0
207,2
2С0.3
kDa
ч-1
2,45
2,53
24,5
23,8
kca
ч'1
7,89
7,60
69,0
61,1
Сравнение опытных и рассчитанных значений объемных коэффициентов
массоотдачи приведено на графиках рис. 278. Опытные значения kDa очень
657
во
щ
го
о
30
%го
1
□ а
о б
V 6
9°
/
//
4
о
§
^2
о
D
п
V
«и-
3
т^
$г^
в
Q
>л
D
4 5
VD, м/ч
W
близки к расчетным, однако '
расчетные величины kco, значительно
больше опытных при высоких значениях
Vd- Кроме того, зависимость
рассчитанных величин kca от Vc обратна
аналогичной зависимости для
опытных величин kca. В связи с тем что
влияние продольного перемешивания
на массопередачу сильнее выражено
при малых значениях Vc, опытные
значения kca, не скорректированные
с учетом продольного перемешивания,
как и следует ожидать, меньше
рассчитанных при малых значениях Vc-
С помощью этих данных, принимая
рассчитанные значения dp и Фп
отвечающими действительности, можно
определить значение критерия Рес-
Опытные (и расчетные) значения
коэффициентов массоотдачи в наса-
дочных колоннах значительно выше,
чем в распылительных экстракторах
(рис. 271).
В рекомендованных
уравнениях для расчета
экстракционных аппаратов не отраже-
фаза) — вода *7:
a-Vc = 3,66 л> • мт2 ■ «-';
6 — Vq = 8,1 м> ■ м~2 -ч-1;
e — Vc = \6,2 л» ■ мт2 ■ ч"1;
1 — рассчитанная кривая для
Кс = 3,66 л3 • м~2 • ч-1; 2—рассчитанная
кривая для Vq=\6,2 м* ■ м~2 • ч~'; 3 —
рассчитанная кривая для Vq = 81 мг • м~2 • ч~К
Рис, 278. Сравнение опытных и
рассчитанных коэффициентов массо-
отдачи для колонны с насадкой из но влияния межфазовой турбу
колец диаметром 12,7 мм. Система т j r j
метилизобутилкарбинол (дисперсная лентности И сопротивления на
границе раздела фаз —
явлений, которые в настоящее
время еще не поддаются учету.
Кроме того, рекомендуемые
уравнения получены при
помощи обработки результатов
опытов, проведенных на
бинарных системах, т. е. при
растворении одной жидкости в другой, в которой первая частично
растворима. Все количество дисперсной фазы, задерживаемое
в колонне в этом случае, по-видимому, представляет собой
активную УС, т. е. участвует в массопередаче. При наличии
третьего (распределяемого) компонента некоторое количество
капель непрерывно задерживается в насадке, причем в них
быстро достигается состояние равновесия с окружающей
жидкостью; вследствие этого поверхность таких капель
нельзя рассматривать как поверхность, участвующую в
массопередаче.
До тех пор пока эти проблемы не будут решены,
рекомендуют применять указанные выше зависимости для получения
корреляций при обработке результатов испытаний опытных
установок, принимая во внимание ненадежность абсолютных
значений констант, входящих в указанные уравнения.
668
If
Имеется много опытных даганых об экстракции с участием
третьего компонента, распределяемого между фазами. Большая
часть этих данных получена на колоннах очень малых
диаметров с насадкой, размер которой либо слишком мал, либо
слишком велик по сравнению с размерами применяемых
колонн. Наиболее подробное исследование насадочных
экстракционных колонн, лишенное указанных выше недостатков,
проведено Лейбзоном и Бекманном74. Опытные данные этих
авторов весьма полезны для дальнейших обобщений.
Лейбзон и Бекманн проводили опыты в распылительных и
насадочных колоннах диаметрами 76, 102 и 150 мм, применяя
насадку из иеглазурированных фарфоровых колец Рашига.
В этих колоннах изучалась массопередача при переходе диэтил-
амина из воды (сплошная фаза) в толуол. Большая часть
опытных данных указанных авторов, скоррелированных «а
основе уравнения (XI, 22), приведена на рис. 279.
II
го
15
10
/
/
ш
!
Аг
3
4/
/ 5
/ У
10
/б 7
>
9^
10
тУв mV£ £
Рис. 279. Опытные данные 7i по экстракции диэтиламина из воды
(сплошная фаза) толуолом в распылительных колоннах с насадкой из колгц
Рашига (/,2 —распылительные колонны; 3—10 — насадочные колонны):
1-й 152 мм; 2 — d 0,76 мм; 3 — d 152 мм; насадка 25,4 мм; 4 — d 76 мм; насадка 19,0 мм;
5—d 152 мм; насадка 19,0 лл; 6 — d 152 мм; насадка 15,9 мм; 7 — d!5 мм; насадка 12,7 мм;
в—d 152 лл; насадка 12,7 .«л; 9 — d 152 мм; насадка 9,5 мм; 10 — d 76 мм, d 102 мм,
й 152 мм; насадка 6,3 мм (d — диаметр).
669
На этом рисунке нанесены не все опытные данные, но
результаты всех других опытов в координатах рис. 279 также
ложатся на прямые линии.
Полученные в опытах Лейбзона и Бекманна высоты единицы
переноса имели очень большие значения. Следует отметить,
однако, что опыты проводили на системе с большим межфазовым
натяжением, а для подобных систем насадочные колонны
обычно мало эффективны. Кроме того, опыты проводили при
значениях фактора экстракции, отличных от 8=1 —1,5. Учитывая это,
можно считать, что полученные опытные значения ВЕП не
слишком велики.
Для насадки размером '/У (12,7 мм) и более диаметр
капель не зависел от размера насадки, а для насадки 'Д" (6,3 мм)
существенно увеличивался.
Критический размер насадки в опытах был 3/s" (9,5 мм);
для данной системы межфазовое натяжение а = 25 дн/см; по
уравнению (XI, 24) критический размер насадки dFC=\0,7 мм.
Для распылительной колонны более низкие значения НЮс были
получены на колонне меньших размеров вследствие уменьшения
продольного перемешивания с увеличением отношения Н/Т. Для
колонн с насадкой lU", для которых T/dF>8, во всех
случаях не обнаружено влияние диаметра насадки на величины
Нюс-
Однако для насадок больших размеров НЮс увеличивается
с уменьшением диаметра колонны или со снижением плотности
упаковки насадки.
В колоннах очень больших диаметров влияние продольного
перемешивания может приводить к противоположным
результатам. Все опытные данные получены при применении слоев
насадки высотой 1100 мм. Лишь в опытах на колонне
диаметром 102 мм с насадкой '/г" высота слоя насадки составляла
1850 мм; при этом величина НЮс возрастала примерно на 10%,
что указывало на наличие концевого эффекта.
Пример XI-2. Определить диаметр насадочной колонны и величину ВЕП
при экстракции диэтиламина из разбавленного водного раствора толуолом
при температуре t=2TC. Расход исходного раствора 1,7 ж3/ч, расход толуола
2,72 ж3/ч.
Физические свойства водного раствора диэтиламина: вязкость 0,87 спз
(0,87- Ю-3 кг -ж4 • сек4); плотность 984 кг/ж3; коэффициент диффузии
диэтиламина в воде —4,82-10"6 м2/ч (1,34-Ю-9 м2/сек); поверхностное
натяжение на границе с воздухом 65 дн/см (65-10""3 н/м). Физические свойства
раствора диэтиламина в толуоле: вязкость 0,55 спз (0,55-10"3 кг • мч • сек4);
плотность 856 кг/ж3; коэффициент диффузии диэтиламина в толуоле
1,07 • 10"5 м21ч (2,98-Ю-9 л2/сек); межфазовое натяжение 25 дн/см
(25- 10~3 н/м), причем коэффициент диффузии и межфазовое натяжение
определены расчетом; коэффициент распределения
1 чк (кмоль диэтиламина/л3 воды)
(кмоль диэтиламина/ж3 толуола)
560
Решение. Расчет проводим для случая, когда толуол является
Дисперсной фазой. В конце примера указаны также результаты расчета, полученные
при диспергировании раствора диэтиламина.
Размер насадки. По уравнению (XI, 24) находим g?fc = 0,0107 ж, или
10,7 мм. Соответственно этому выбираем в качестве насадки керамические
кольца Рашига (предпочтительно смачиваемые водной фазой) размером 1"
(25,4 мм). По табл. 18 (стр. 554) имеем: а^ = 171 м21м3 и е = 0,69 мъ/м3.
Захлебывание, а) Воспользуемся графиком ( см. рис. 276). В нашем случае
ордината по расчету равна 0,103; соответственно находим абсциссу, равную
140. Отсюда (V% -4- V%5Ff = 75,5. Учитывая, что Vdf/Vcf = 2,72/1,7=1,6,
получим Vcf — 14,75 м/ч и VDf = 22 ж/ч.
б) Применим уравнение (XI, 30), подставляя в него значение
(а?/е1'2)°'838=102 из табл. 18. По этому уравнению Уводн. f = Vcf — 10,64 м/ч
и Vdf=17 м/ч. При расчетах возможны расхождения в результатах
определения Vcf и Vdf по уравнению (XI, 30) и графику (см. рис. 276).
Диаметр колонны и УС. Примем #d = 0,10 (если принять более высокое
значение УС, то оно будет соответствовать гидродинамическим условиям,
слишком близким к точке захлебывания колонны). Подставляя в уравнение
(XI, 25) значение Vc=VD/lfi, получим VK/VD=17,22. По уравнению (XI, 28)
находим dj, = 0,0049 ж; по уравнению (V, 73) величина Р= 21,2- 109. Ордината
графика (см. рис. 100) равна 56,5, абсцисса 16. Отсюда Vt= l&ii,cP°,i5/dPpc =
= 0,1015 м/сек.
Из рис. 275 имеем: абсцисса равна 0,781, ордината 0,50. Поскольку
величиной е~23-6т можно пренебречь, принимаем VK/0,1015=0,50. Отсюда VK =
= 0,051 м/сек; Vd = VK/17,22 = 2,95 • 10"8 ж/сек= 10,6 м/ч. Полученная скорость
дисперсной фазы составляет 62% от скорости захлебывания, рассчитанной с
помощью рис. 276 [см. пункт а) расчета] и 45% от скорости захлебывания,
рассчитанной выше [см. пункт б)] по уравнению (XI,30). Поэтому
принимаем VD = 10,6 м/ч и Vc = 10,6/1,6=6,65 м/ч. Поперечное сечение колонны
1,7/6,6=0,256 ж2, соответственно ее диаметр 570 мм.
Распределитель дисперсной фазы. Воспользуемся рис. 263, приняв V0 =
= 0,0915 м/сек. При диаметре отверстия сопла 6,35 мм величина d0 = 0,00635 м
и диаметр капли 10 мм. При диаметре отверстия сопла 3,18 мм диаметр
капли составит 7,65 мм. Полученные значения диаметра капли больше ее
критического размера для данной насадки (с?р = 4,88 мм); поэтому можно
использовать сопло с отверстием любого из двух диаметров. Сопло с
отверстием диаметром 6,35 мм меньше подвержено засорению, чем сопло
диаметром 3,18 мм, и поэтому выбираем его для данной колонны. Поперечное
сечение отверстия сопла равно 3,16- 10"5 ж2, а необходимое число сопел 2,72/3,16 X
X 10"5'0,0915-3600 = 262.
Удельная межфазовая поверхность. Согласно уравнению (XI, 29)
а= 6-0,69-01 = 85
4,88-Ю-3
Массопередача в дисперсной фазе. В данном случае ордината на рис. 100
имеет значение более низкое, чем 70, соответственно размер образующихся
капель меньше критического. По уравнению (V, 81) находим:
k 17,9-1,07-10— = 0)039з КМоль-м-*-ч-1-Ьс-у
D 4,88 • 10~3
kQa = 0,0393 • 85 = 3,36 кмоль ■ м~г ■ ч~х -Ac-1
Vn 10,6
tD k a 3,36
36 Р. Е. Трейбал 581
Массопередача в сплошной фазе. Имеем Vr=VK(l —Фв) = 1&5 м/ч.
Применим уравнение (XI, 19), заменяя V, на Vr. Находим: ^а^гРс/Ис = 255;
Ис/Рс®С= ^^' *с= "'232. Следовательно, величина kca будет равна
19,7 кмоль • мг3 • ч'1 • Асг1 и
Vr 6,65
Н.„ = -г±- = -г?г=- = 0,338 м
'С k„a 19,7
Массопередача в обеих фазах. Имеем:
И — И _| <- tD
пюс ntc^r ту^
где от =-У-=-—- = 0,735.
сс 1,36
Отсюда Vc/mVD=l/e = 0,848 и Я,ос =0,338 + 0,848 -3,18 = 3,0 ж.
Полученное значение #10с довольно близко к величине И toe по рис.279
(2,6 ж), хотя последняя найдена без учета влияния продольного
перемешивания на высоту колонны. Поправка на продольное перемешивание привела
бы к еще большему несоответствию сравниваемых величин Нюс-
Ниже приведены результаты расчета для случая, когда дисперсной фазой
является водный раствор диэтиламина (rfFC = 10,7 мм). В качестве насадки
выбираем угольные (предпочтительно смачиваемые толуолом) кольца Ра-
шига размером 1" (25,4 мм). Скорости захлебывания (см. рис. 276):
1/cf=25,0 м]ч и Vdf = 15,5 м\ч. Для сопоставления с результатами, получен-
ными в первом варианте расчета, примем рабочие скорости, равными 45% от
скоростей захлебывания: Vc = 11,25 м\ч и VD = 6,97 ж/ч. Диаметр колонны
570 мм.
УС по дисперсной фазе для данных скоростей потоков рассчитана
методом последовательного приближения: 0d = O,O66; rfp = 4,8 мм; 1/к=174 ж/ч;
Vr= 163 ж/ч; а = 59,6 м2/м3; £ва=1,09 кмоль • м'ъ ■ ч'1 • Ас'1; Я<в = 6,46 м;
£са=23,5 кмоль ■ мгъ ■ ч~' ■ Ас1; #1С=0,48 м.
Для сравнения с расчетом, проведенным ранее, определяем общую
высоту единицы переноса по фазе рафината, причем в данном случае #(0D =
= 6,87 ж. Если продольное перемешивание в этом случае не является
значительно меньшим, чем при работе колонны по первому варианту, что мало
вероятно, то следует отдать предпочтение первому варианту (диспергирование
толуола).
КОЛОННЫ С ПЕРФОРИРОВАННЫМИ
(СИТЧАТЫМИ) ТАРЕЛКАМИ
В колоннах этого типа одна из жидких фаз многократно
диспергируется и коалесцирует, протекая через установленные
в колонне тарелки, в которых имеется большое число малых
сверленых или штампованных отверстий. Скорость экстракции
при этом возрастает вследствие многократного редиспергирова-
ния и по существу — вследствие увеличения числа концевых
эффектов. Однако, вероятно, наиболее важным преимуществом
аппаратов этого типа является уменьшение в них обратного
перемешивания в результате секционирования колонны.
Простейшая и наиболее ранняя конструкция тарелок для
таких колонн — сегментные перфорированные тарелки,
напоминающие соответствующие глухие перегородки в колоннах на
562
рис. 273. Тарелки современных конструкций снабжены более
совершенными устройствами для перелива сплошной фазы с
тарелки на тарелку.
На рис. 280 показана ситчатая колонна, предложенная Хар-
рингтоном76. В этой колонне сплошная фаза течет вдоль
перфорированной горизонтальной тарелки и перетекает с тарелки на
тарелку через переливные устройства сегментного сечения.
Вертикальные пластины переливного устройства нижней своей
частью погружены в слой сплошной фазы. В пространстве
между переливным устройством и тарелкой скапливается слой
дисперсной фазы, возникающий в результате каолесценции
капель, образующихся при прохождении дисперсной фазы через
отверстия предыдущей тарелки. На рнс
лонна, предназначенная для
диспергирования легкой жидкости, однако если эту
колонну перевернуть, ее можно
использовать для диспергирования тяжелой
жидкости. Кроме того, при
соответствующем расположении переливов в одной
части колонны можно диспергировать
легкую жидкость, а в другой — тяжелую.
Колонны этого типа диаметрами до 3,7 м
очень широко используются в процессах
нефтеочистки и в нефтехимической
промышленности.
Известно очень много конструкций
экстракторов с ситчатыми тарелками;
■ниже описаны лишь некоторые из этих
конструкций. Обычной модификацией
ситчатых тарелок для колонн больших
диаметров являются тарелки, на
которых осуществляется направленный поток
сплошной фазы: от центра к периферии
и от периферии к центру на смежных
тарелках (аналогично тарелкам с
направленным движением потоков в
ректификационных колоннах).
На схеме (рис. 281) показана
тарелка, очищаемая от загрязнений,
собирающихся на поверхности раздела фаз в
межтарелочнам пространстве, с помощью
специальных приспособлений. Установка
таких приспособлений снижает
эффективность тарелки. Вместо подобных
«сливных» устройств для периодического
удаления загрязнений с поверхности
раздела фаз можно использовать отводные
. 280 изображена ко
Рис. 280. Экстрактор с
перфорированными
тарелками для
диспергирования легкой фазы:
/ — основная поверхность
раздела; 2 — граница коалесцен-
цни дисперсной фазы: 3 —
перфорированная тарелка; 4 —
слив; /, 111 — тяжелая фаза;
//, IV — легкая фаза.
36*
563
Рис. 281. Конструкция
разъемной
перфорированной тарелки [Ind. Eng. a б
Chem., 44, 2253 (1952)1: D oao r л.
, \ /1 рис 282. Схемы конструкции перфорированных
/ — съемная половина тарелки „ г; ^ . r^ г г
(отверстия 0 4,75 мм, шаг тарелок для диспергирования обеих жидких фаз:
15,8 мм); 2 — отвод загрязне- а — тарелка Кэслера и Нельсона (пат. США 2609276, 1952 г j;
ний; 3—положение поверх- б —тарелка Пэкн (пат. США 2610108, 1952 г.); 1 —
поверхности раздела фаз в межта- ность раздела фаз; 2—перфорированные части тарелок;
релочном пространстве. //—тяжелая жидкость; L — легкая жидкость.
краны, установленные «а обечайке колонны. Широко
применяют также колонны (рис. 282), в которых обе фазы
диспергируются на каждой тарелке. В колоннах очень больших
диаметров, используемых в «Udex-процессе» (см. главу. XIII),
используют тарелки77, на которых сплошную фазу направляют
из одного межтарелочного пространства в другое через
большое число переливных труб, установленных по поперечному
сечению тарелки. Кромки переливных труб выступают над
плоскостью тарелки, а не находятся на одном уровне с ней, как
показано на рис. 280. Обычно в переливные устройства (для
облегчения коалесценции капель, уносимых со сплошной
фазой) помещают насадку. Успешно применяются колонны78.79,
в которых перфорированные тарелки заменены сетками из
тонкой проволоки.
Вертикально расположенные перфорированные тарелки в
экстракционных колоннах обычно работают неудовлетворительно80,
та'к ка,к имеющийся гидростатический напор обычно
недостаточен для образования в перфорациях горизонтальных
равномерных струй дисперсной фазы. По этой же причине в процес-
664
сах экстрации неэффективны обычные
колпачковые тарелки, применяемые в
процессах ректификации 8|. Вместе с тем
в процессах нефтепереработки и других
процессах успешно используют 82>83'84
колонну Коха (рис. 283) с вертикальными
перфорированными тарелками. Тяжелая
жидкость в колонне перемещается вниз
по переливам а и через отверстия б.
Легкая жидкость, перемещаясь вверх,
входит в отверстие в и проходит через
пространства г, увлекая тяжелую
жидкость вдоль вертикальных пластин д.
Проходя последовательно через ряд
вертикальных перфорированных тарелок,
тяжелая жидкость многократно
диспергируется. Окончательная коалесценция
капель и разделение фаз происходят в
сливном устройстве е.
Конструирование тарелок и их
производительность. В литературе
опубликованы лишь методы расчета и
конструирования ситчатых тарелок наиболее
простой конструкции (рис. 280). Поэтому
мы ограничимся рассмотрением методов
расчета тарелок толыко этого типа.
Требования к конструкции тарелки, связанные с
необходимостью свободного истечения дисперсной фазы из отверстий
тарелки, освещены в литературе80'85. Материал тарелки должен
либо предпочтительно смачиваться сплошной фазой, либо
отверстия в тарелке должны быть выштампованы аналогично
показанным на рис. 262. Если дисперсная фаза смачивает
материал тарелки, в которой имеются сверленые отверстия, то
размер образующихся в отверстиях капель будет велик, что
приведет к уменьшению межфазовой поверхности и снижению
скорости массопередачи.
Отверстия обычно имеют диаметр 3—6 мм и располагаются
в вершинах правильных треугольников или квадратов с шагом
12—19 мм. Отверстия меньших диаметров предпочтительнее для
систем с высоким межфазовым натяжением. Скорость жидкости
в отверстии должна быть такой, чтобы после истечения из
отверстий происходило дробление струй на мелкие капли на
небольшом расстоянии от поверхности тарелки. Чтобы
удовлетворить этому требованию, линейные скорости жидкости в
отверстиях должны быть 0,15—0,3 м/сек. Более точно определить
величину скорости жидкости в отверстиях тарелки можно с
помощью уравнения (XI, 2). На противоположных концах
Рис. 283. Колонна Коха
(Koch Engineering Co,
Inc.):
Л // — тяжелая жидкость;
Hi, IV — легкая жидкость.
565
соседних тарелок устанавливают переливы, причем площадь
тарелки, примыкающая к переливу и соответствующая
расстоянию от перелива 25—50 мм, не перфорируется.
Переливы лучше устанавливать заподлицо с плоскостью
тарелки без выступающих кромок, как у переливов ситчатых
тарелок ректификационных колонн. Площадь поперечного сечения
переливов должна быть такой, чтобы линейная скорость
оплошной фазы в них не превышала окорости осаждения некоторой
малой капли дисперсной фазы (например, капли диаметром
0,8—1,6 мм).
В противном случае рециркуляция уносимой дисперсной
фазы вблизи тарелок может явиться причиной захлебывания
аппарата.
Высота перелива должна быть такой, чтобы его края
выходили за пределы слоя дисперсной фазы. Площадь для прохода
сплошной фазы между кромкой перелива и тарелкой, к
которой он обращен, должна быть больше поперечного сечения
перелива.
Расстояния между тарелками обычно принимают равными
150—600 мм.
Как показано ниже, скорость массопередачи на тарелке
увеличивается, а производительность снижается при
уменьшении расстояния между тарелками. Для колонн больших
размеров, где может оказаться необходимой периодическая чистка
тарелок, минимальное расстояние между тарелками принимают
равным 400—450 мм, чтобы можно было устанавливать люки
или регарды.
По экономическим соображениям, вместо применения
многочисленных люков и регард можно выполнять тарелки
колонны съемными; при этом число люков в колонне можно
значительно сократить.
Глубину слоя дисперсной фазы, образующейся на тарелке,
определяют по перепаду давления, необходимому для
движения жидкостей. Увеличение скорости каждой из фаз или
усиление рециркуляции дисперсной фазы вблизи тарелки приводит к
увеличению высоты слоя скоалесцировавшей дисперсной фазы,
пока высота слоя не станет равной высоте перелива и слой
дисперсной фазы не достигнет следующей тарелки. При этом
наступит захлебывание колонны. Учитывая такую возможность,
при проектировании колонны необходимо соблюдать условие,
чтобы глубина слоя дисперсной фазы на тарелке была
сравнительно небольшой.
Общая высота h слоя определяется как сумма напоров
сплошной и дисперсной фаз:
h=hc + hD (XI, 36)
566
Для дисперсной фазы:
hD = h0 + ha (XI, 37)
где Л о — напор, необходимый для истечения жидкости из
отверстий тарелки;
ha—напор, затрачиваемый на преодоление сил межфазо-
вого натяжения.
Изучение литературных данных80'86-88 показывает, что h0
можно определить из уравнения истечения через отверстие,
принимая коэффициент истечения равным 0,67:
ho = 2* (0,67)* Ар (Х1,38)
где S0 — площадь поперечного сечения всех отверстий;
S — площадь поперечного сечения колонны (без учета
сечения переливов).
Величину ha, которую целесообразно учитывать лишь при
малых скоростях дисперсной фазы, рассчитывают по уравнению:
6(1 (XI, 39)
dpbpg
где dp—диаметр капли при скорости жидкости в отверстии
V'0 = 0,03 м/сек.
При скоростях V'0 > 0,3 м/сек величиной hj можно
пренебречь и hD~h0.
Уравнение (XI, 39) получено на основе следующих
соображений.
Избыточное давление, необходимое для преодоления сил межфазового
натяжения (выраженное высотой столба жидкости), определяется для
сферической капли из уравнения
. 4а
а~ db9g
Если вместо d подставить диаметр отверстия do, то из этого уравнения
получим значение ha, необходимое для начала истечения [см. уравнение
(X, 50)], но при движении с постоянной скоростью более правильна
подстановка в уравнение диаметра dp капли. При низких скоростях дисперсной
фазы капли образуются периодически, поэтому в уравнение необходимо
ввести средний во времени размер капли88. Предполагая, что при постоянной
скорости потока объем капли увеличивается со временем линейно и что капля
представляет собой правильную сферу, размер капли в момент времени 9
можно вычислить с помощью уравнения
1рв
--(£)'"
667
Соответственно среднюю во времени величину h0 определяют следующим
образом:
6/
4а 69
К
J dp%^g ^ /iQfy,,_ 6а
ар г/ж,/,бв=_:
\fdp Apg J V Э I dp
6/ —0 Q/dpApg J V e I dpApg
где б — знак дифференциала.
Опытным путем было установлено, что при пользовании уравнением
(XI, 37), в котором h0 находят из уравнения (XI, 38), величину ha лучше
рассчитывать из уравнения (XI, 39), используя значение dp, соответствующее
низким скоростям потока в отверстии (например, 1/о=0,03 м/сек). Кроме
того, найдено, что при скоростях в отверстии Vo>0,3 ж/сек, когда образуются
струи, величиной ha можно пренебречь и принять hD = h0. Имеются другие
способы расчета87,88 ho, в которых используют значения коэффициентов
истечения, изменяющиеся с изменением скорости потока.
Величина напора hc, необходимого для движения сплошной
фазы, складывается из потерь напора на трение в переливе
(которым обычно можно пренебречь) и на местные
сопротивления — при сужении и расширении потока на входе в перелив и
выходе из него и при двукратном резком изменении
направления движения жидкости.
Суммарно коэффициент сопротивления движению жидкости
через перелив равен 4,5; поэтому общий напор выражается
зависимостью 86
4,5 V2dpc
hc=^gir (XI-4°)
где Vd — линейная скорость сплошной фазы в переливе.
Массопередача. Опытные данные о массопередаче на сит-
чатых тарелках для данной системы жидкость — жидкость
можно обработать в соответствии с уравнениями (XI, 22) и
(XI, 23) построением графиков зависимости Ht0 от фактора
экстракции.
На таких графиках обычно получают прямые линии;однако
углы наклона этих линий и отрезки, отсекаемые ими на осях
ординат, как и при расчете других экстракторов, не могут
служить мерой Htc и HtD.
Другим способом оценки интенсивности массопередачи
является выражение ее через эффективность ступени, при этом
за 100% принимается эффективность теоретической ступени
разделения.
Для систем с относительно низким коэффициентом
межфазового натяжения диаметр отверстий и их суммарная
площадь сечения оказывают относительно небольшое влияние
568
5s?
« 30
I
t
на эффективность тарел- 50
ки (рис. 284), однако
площадь сечения
отверстия тарелки существенно
влияет на
производительность экстрактора. Для
систем с высоким
межфазовым натяжением
уменьшение размера капли,
вызванное уменьшением !Ц 4-0
диаметра отверстий, су- *"
щественно увеличивает
скорость массопередачи 90.
С увеличением
расстояния между тарелками
повышается эффективность
ступени в результате
более длительного времени
контакта жидкостей в
пространстве между
ступенями (рис. 285),
причем в большей части
случаев величина ВЭТС уве-
/ г
/
30
10 15 20
Vjj-скорость эфира, м/ч
б
Рис. 284. Влияние диаметра отверстия (а)
и свободного сечения тарелки (б) на
эффективность колонны с перфорированными
тарелками 88. Экстракция уксусной кислоты
из воды этиловым эфиром (дисперсная
фаза) при KD/KC = 2,38:
/ — диаметр отверстий dQ = 5,l мм; 2 — d0 = 2,88 мм;
3 —d =1,6 мм; 4 — площадь свободного
сечения 5,05%; 5 — 3,34%; 6—1,77%.
личивается.
Обычно эффективность ступени значительно выше для
систем с низким межфазовым натяжением (так же, как для наса-
дочных и распылительных колонн) и может достигать значений
0,5 и больше. Влияние межфазового натяжения на
эффективность ступени в первом приближении можно оценить по рис. 286,
0,25 Q5
Расстояние
между тарелками, м
Рис. 285. Влияние расстояния между
тарелками на эффективность
ступени80. Система
этилацетат—уксусная кислота — вода (сплошная фаза).
^=1.
30
20
"з
10
1
\
10
го зо 40
Мешфазное
натяжение, дин/см
Рис. 286. Влияние межфазового
натяжения на эффективность ступени
(расстояние между тарелками 150 мм;
VniVQ = 1; дисперсная фаза —
органическая; данные Гарнера85, Мэй-
филда 80, Моултона 91 и Роу92).
хотя для уточнения степени влияния а на эффективность
необходимо учесть также влияние коэффициента распределения и
суммарной скорости потоков фаз.
В колоннах с ситчатыми тарелками до сих пор не
определяли (см. стр. 555) коэффициенты массоотдачи в каждой фазе.
Вследствие отсутствия таких данных можно рекомендовать
рассчитывать коэффициенты массопередачи по способу,
предложенному Коулсоном18 для распылительных колонн. По Коулсону
допускается, что концентрация сплошной фазы между
смежными тарелками постоянна, соответственно движущая сила
процесса при образовании, коалесценции и движении капель
одинакова. На основе этих предпосылок расчетное уравнение для
объемного коэффициента массопередачи имеет вид:
KDa=2S^-h)^KOr-r (XL 41)
Первый член правой части уравнения (XI, 41) выражает
скорость массопередачи в периоды образования и коалесценции
капель, а второй — в период движения капель. Сомножителем
Kdi в первом члене правой части уравнения является суммарная
поверхность капель, приходящаяся на единицу активного
объема ступени.
Коэффициент массопередачи связан с так называемой
локальной эффективностью тарелки по Мэрфри, показывающей
степень приближения к равновесию (со сплошной фазой) в
каждой данной точке на тарелке:
KDa(Hc-h)
EMDp=l-e V° (XI. 42)
В колоннах малых диаметров обратное перемешивание
сплошной фазы на каждой тарелке, вероятно, приводит к
выравниванию концентрации сплошной фазы до ее конечного
значения (хотя обратное перемешивание между тарелками
отсутствует). В этом случае общая эффективность ступени по
Мэрфри EMD должна быть равна Emd ■ Для колонн больших
диаметров, если допустить, что обратного перемешивания на
тарелке не возникает2, эффективность ступени можно
определить на основе аналогии с одним из выражений эффективности
ступени ректификационных колонн93. В этом случае:
mVDEMDjVc ,
EMD = ± -1 (XL43)
"vd/vc
Так же как для насадочной и распылительной колонн, при
расчете эффективности ступени нельзя учесть влияния межфазо-
570
вой турбулентности и сопротивления массопереносу на
поверхности раздела фаз.
Все опытные данные о массопередаче, за исключением
результатов отдельных наблюдений3, получены на колоннах,
имеющих диаметры от 50 до 220 мм и расстояния между
тарелками ОТ 75 ДО 610 ММ 8°. 85,88,90-92, 94-97_ gce эти данные
удовлетворительно описываются эмпирической формулой:
5,63 • 10
■Зи0,5
№Г
(XI, 44)
Сходимость расчетных и опытных данных (рис. 287) была
бы, несомненно, лучшей, если бы общая эффективность ступени
выражалась по Мэрфри (EMD), однако опубликованные
результаты исследований не содержат данных, достаточных для
расчета Emd- Опытные значения межфазового натяжения для
изучавшихся систем приводятся редко, поэтому величины а
определялись расчетным путем.
Пример XI-3. Определить размеры тарелок и эффективность ступени
колонны с ситчатыми тарелками для условий примера XI-2. Объемные
скорости водного раствора диэтиламина 6,8 .и3/4, толуола 10,9 ма/ч.
Решение. В качестве дисперсной фазы выбираем толуол, так как
объемный расход толуола больше объемного расхода раствора диэтиламина.
Отверстия тарелок. Межфазовое натяжение системы велико, поэтому
принимаем диаметр отверстий тарелок равным do = 3,18 мм. Комплекс
0.8
J
I
% 0.1
!
q&i
о °
о
/
о
„Я
0 °а/
о
J
ё
о
ffib
фй*
i°
О
"о
о*
>
о
8
°8
$&
0° О
oJ$
О о
о io
о
v £»-
Опытная эффективность ЕП
Ш
Рис. 287. Сравнение опытных и рассчитанных из уравнения (XI, 44)
данных по массопередаче для колонн малых размеров с перфорированными
тарелками.
571
^o/(cr/gAp)D'5=0,707. Из уравнения (XI, За) находим dV = 2,55 мм. По
уравнению (XI, 2) при значениях ст = 2,5 ■ 10~2 н\м определяем V0= Vom = 650 м[ч
(0,18 м/сек). Суммарная площадь отверстцй на тарелке S0= 10,9/650 =
= 0,0168 ж2. Сечение одного отверстия 7,9" 10-6 м2. Число отверстий N0 =
= 1,68 • 10~2/7,9 ■ 10_6 = 2130. Если отверстия разместить в вершинах
равносторонних треугольников со сторонами 12,7 мм, то общая площадь
перфорированной части тарелки S = 0,298 м2. Средний размер капли dp=2dj = 5,\ мм.
Переливы. Скорость водной фазы в переливе должна соответствовать
скорости осаждения капель толуола диаметром 0,8 мм. Из уравнения (V, 75)
имеем: 1/,= 1/^ = 120 м/ч. Площадь поперечного сечения перелива 6,8/120 =
= 0,0566 м2.
Диаметр колонны. Площадь перфорированной части тарелки и двух
переливов суммарно составляет 0,298 + 2 • 0,0566=0,411 м2. Если принять, что у
переливов имеется неперфорированная часть поверхности тарелки шириной
25 мм, а на периферии тарелки — кольцевая поверхность шириной 50 мм,
также лишенная перфораций и предназначенная для крепления тарелки к
обечайке колонны, то общая площадь сечения колонны будет равна 0,55 м2.
Этой площади сечения соответствует диаметр колонны 0,84 м.
Высота слоя легкой жидкости под тарелкой. В данном случае S0/S =
= 0,0168/0,298 = 0,0562. По уравнению (XI, 38) величина Ло = 0,025 м.
По графику (см. рис. 263) при скорости в отверстии Ко = 0,0305 и/сек
абсцисса равна 0,0611, параметр 10,75 pDV0/Ap = 0,0677 и, следовательно,
dp=7,B мм. По уравнению (XI, 39) величина Лст= 15,7 мм. С помощью
уравнения (X, 37) определяем Лг> = 41 мм и, пользуясь уравнением (X, 40), находим
Лс = 1,98 мм. Тогда общая высота слоя жидкости под тарелкой по уравнению
(XI, 36) Л=43 мм.
Расстояние между тарелками. Примем расстояние между тарелками
равным 406 мм (Нс=0,406 м). При таком расстоянии между тарелками на
обечайке колонны можно установить люки для чистки. Высоту перелива
принимаем равной 254 мм.
УС по дисперсной фазе. Слой дисперсной фазы под тарелкой не
учитывается. Величину Vd определим по всей площади поперечного сечения
колонны (без учета площади перелива). Тогда VD= 10,9/(0,55 — 0,0566) =
= 22,1 м/ч. Поскольку сплошная фаза движется вблизи тарелки в
горизонтальном направлении, применим уравнение (XI, 13) при 1/с = 0.
Следовательно, У»=У„0с=22,1 м/ч. По уравнению (V, 73) имеем: Р=21,2-109. Из
рис. 100 при dp = 5,l мм находим: ордината 62,0; абсцисса 17,5; У(=395 м/ч.
По рис. 268 определяем, что абсцисса равна 66,5. Значения Vs<t>D для
различных 0г> приведены ниже (две первые колонки получены по данным
рис. 268):
ф0
0
од
0,2
0,3
Ордината
(рис. 268)
12,0
7,8
5,7
4Д
м/ч
492
320
233,5
168,0
Vs
(для твердых
тел)
м/ч
492
356
292
240
VslV'
1,0
0,723
0,594
0,488
(для жидкости)
м/ч
395
285
235
193
V,*D
0
28,5
47
58
Методом графической интерполяции при У,Фг> = 22,1 м/ч определяем
<fe = 0,08. Из уравнения (XI, 21) имеем: аг = 6-0,08/0,0051 «94 м2/м\
Массопередача в межтарелочном пространстве Из уравнения (XI, 19)
находим 6С=0,292 кмоль ■ м-2- ч~1 ■ Дс"1. Поскольку ордината рис. 100
меньше 70, то диаметср капли меньше критического. Следовательно, из уравнения
572
(V, 81) получаем: Яг> = 0,0375 кмоль ■ >г2 • ч-1 • Дс-1 Из уравнения (V, 60)
имеем: l/KD/> 1/0,0375 + 0,735/0,292; KDf =0,0343 кмоль ■ м~2 ■ ч~' • Дс"1; Къгаг^
= 0,0343 • 94 = 3,22 кмоль ■ лг3 • ч"1 • Дс"1.
Массопередача в период образования капель и коалесценции. Объем
капли nd'/б. Объемная скорость жидкости в перфорации nd0V0lA. Сле-
довательно, 9^ = Andpj§nd0V0 = 1,36- Ю-5 ч. Из уравнения (XI, 10)
получаем: /Сг>/ = 0,653 кмоль ■ м~2 • ч~1 ■ Дс-1. По уравнению (XI, 9) находим:
Лр=4,9-10-5 м2. Величина 2KDtAvN0/S(Hс — h) =0,763 кмоль ■ м~3 ■ ч-> ■ Дс-1.
Из уравнения (XI, 41) находим: /d,a = 3,99 кмоль • м~3 • ч~1 ■ Дс-1.
Эффективность ступени. Из уравнения (XI, 42) получаем: Е =0,064.
Если фаза водного раствора полностью перемешана на каждой тарелке, то
EMD = Е . Если же водный раствор имеет поршневой режим движения в
межтарельчатом пространстве, то из уравнения (XI, 43) определим Emd =
=0,0665. В нашем случае оба значения £мг> отличаются друг от друга на
величину, меньшую ошибки расчета. Обратимся к табл. 16 (см. стр. 448).
Поскольку Emd = EMe, to
lg
Е„ =
1+Е
MD
№-о:
о mV
lg
откуда £о = 0,069 при £AfD = 0,064 и £о = 0,072 при £мг> = 0,0665. Как и
следует ожидать для систем с высоким межфазным натяжением, эффективность
ступени оказалась невелика. (Примером для системы с низким межфазовым
натяжением является задача XI, 2, стр. 681.)
Сравнительная оценка. Определим фиктивные скорости фаз,
приходящиеся на все поперечное сечение колонны: Vd= 10,9/0,55= 19,8 м/ч и V'с =
= 6,8/0,55=12,4 м/ч. Условия задачи почти полностью соответствуют условиям
опытов D\ и D7 в исследовании Гарнера95, которые были проведены с той
же системой жидкость — жидкость на колонне диаметром 100 мм.
Расстояние между тарелками в опытах составляло 152 мм, диаметр отверстий 3,18 мм.
Полученная в этих опытах эффективность тарелки соответственно равнялась
0,091 и 0,065. Интересно также сравнить колонну с перфорированными
тарелками, рассчитанную в этом примере, с насадочнои колонной, рассчитанной в
примере XI-2. Для этого определим Htoc для колонны с перфорированными
тарелками:
Vr Vr 12,4
toe - Кса ~ mKDa ~~ 0,735 • 3,99 _ ''
В примере XI-2 для насадочнои колонны получили #(Ос = 3,0 м. Однако
этот результат нуждается в поправке на продольное перемешивание, которое
увеличивает эффективное значение Htoc, в то время как в колонне с
перфорированными тарелками продольное перемешивание отсутствует. Кроме того,
для обработки того же количества жидкостей в колонне с ситчатыми
тарелками необходимо поперечное сечение аппарата в два раза меньшее, чем у
насадочнои колонны.
ЭКСТРАКТОРЫ С МЕХАНИЧЕСКИМ
ПЕРЕМЕШИВАНИЕМ ФАЗ
К аппаратам этого типа относятся экстракторы, в которых
перемешивание фаз (при движении последних через аппарат)
осуществляется либо при непосредственном воздействии
механически движущихся внутренних устройств, либо гидравлически
573
передается и сообщается всему объему жидкостей в
аппарате.
Противоточные колонны с перемешиванием без внутренних
механических перемешивающих устройств эффективны для
обработки систем газ — жидкость, когда относительно высокое
межфазовое натяжение в этих системах преодолевается
вследствие сравнительно большой разности плотностей контактируе-
мых фаз, имеющей величину порядка 800 кг/м3. Разность
плотностей фаз в системах газ — жидкость достаточна для
сообщения жидкостям энергии, необходимой для диспергирования
одной фазы в другой.
В системах жидкость — жидкость, где разность плотностей
фаз имеет значение порядка одной десятой от указанной выше
величины, для получения той же степени диспергирования,
особенно при высоком межфазовом натяжении, нельзя обойтись без
введения дополнительной энергии извне. В аппаратах,
описанных выше, почти невозможно достичь низких значений высоты
единицы переноса (ВЕП) или высоты эквивалентной
теоретической ступени (ВЭТС) для систем с высоким межфазовым
натяжением. Для таких систем необходимо применять
экстракторы с механическим перемешиванием фаз.
РОТОРНО-КОЛЬЦЕВЫЕ ЭКСТРАКТОРЫ
Один из таких экстракторов98-99, схематично показанный на
рис. 288, представляет собой неподвижный внешний цилиндр, в
котором вращается концентрически расположенный внутренний
цилиндр. Взаимодействие жидкостей происходит в кольцевом
пространстве между цилиндрами.
Роторно-кольцевой экстрактор является, вероятно, наиболее
простым из всех экстракторов с механическим перемешиванием
фаз. Он представляет интерес для применения в атомной
промышленности вследствие простоты устройства и возможности
обеспечения кратковременного контакта фаз. Такие требования
предъявляются к аппарату, используемому в процессах
экстракции из растворов с высокой радиоактивностью для
уменьшения потерь экстракта. Сведения о применении
экстракторов этого типа в установках промышленного масштаба
отсутствуют.
При вращении ротора со скоростью, большей критической, в
роторно-кольцевом экстракторе образуются вихри с осями,
направленными тангенциально к вращающемуся цилиндру и
перпендикулярно к его оси100. Диспергируемая жидкость
увлекается вихрями и разрушается ими с образованием мелких капель101.
При этом необходимо, чтобы материал стенок, ограничивающих
кольцевое пространство, предпочтительно смачивался сплошной
фазой 98.
574
Торнтон 102 проводил опыты на роторно-кольцевых колоннах
диаметрами от 50 до 150 мм с кольцевым зазором 6,5—25,4 мм
и отношением диаметров колонны и ротора Tjdi=\,2—2,0. Было
показано, что в отсутствие распределяемого компонента и при
диспергировании фазы, не смачивающей материал колонны, УС
роторно-кольцевых колонн описывается с помощью уравнения
(XI, 14), причем входящую в это уравнение характеристическую
скорость капли определяют из уравнения:
При соблюдении тех же условий величину УС при
захлебывании можно найти по уравнению (XI, 17). Обычно за рабочую
скорость вращения ротора принимают скорость, составляющую
50% от скорости при захлебывании. Если распределяемый
компонент переходит из дисперсной органической фазы в водную,
то скорости при захлебывании увеличиваются в 2—3 раза по
сравнению со скоростями при захлебывании, полученными для
систем, находящихся в равновесии. Если массопередача
происходит в обратном направлении, то скорости захлебывания
уменьшаются на 10%. Для системы вода — ацетон — органический
растворитель при скоростях вращения ротора,
составляющих 50% от скорости захлебывания, Ф
скорости массопередачи описываются
уравнением (XI, 22). При этом величина Htc постоянна
и равна 0,067 м, а значения Hw определяют из
уравнения:
/7W2 \-0,74 , у.* 2,31
н
tD
Коэффициент С в уравнении (XI, 46)
является переменной величиной. В случае применения
в качестве экстрагента бутилацетата величина С
равна 0,32 и 0,44; соответственно для толуола
величина С составляет 0,44 и 0,57. Меньшие
значения С получены при направлении
массопередачи в органическую фазу, большие — при
обратном направлении массопередачи.
Опытным путем установлено, что скорость
массопередачи быстро уменьшается при
увеличении диаметра колонны и отношения диаметра
колонны к диаметру ротора T/d{, причем
основное сопротивление массопередаче было
сосредоточено в дисперсной фазе. Высказано
предположение ,02, что уравнение типа уравнения (XI, 46)
можно применять в качестве исходного уравне-
Ъ"
N
X
1—17
з —7
£=>**
Рис. 288.
торно-кольцевой экстрактор
(схема):
/ — неподвижная
гильза дле
предотвращения
возмущения границы
раздела фаз; 2—
ротор; 3 — кольцевое
пространство; I —
легкая жидкость;
// — тяжелая
жидкость.
675
ния для корреляции других опытных данных, но в таком виде
его нельзя использовать для расчетов роторно-кольцевых
колонн, предназначенных для работы на других системах.
Дэвис98 применил подобные колонны для системы водный
раствор азотной кислоты — уран — раствор трибутилфосфата в
керосине. Его полуэмпирическое уравнение для ВЭТС,
полученное на данной системе, по-видимому, нельзя распространить на
другие системы в связи с тем, что испытанная система обладает
необычно высоким сопротивлением массопередаче (вероятно,
вследствие наличия химической реакции, см. главу X). На
колоннах малых размеров можно получить значения ВЭТС
порядка 65 мм; на лабораторных колоннах очень малых размеров
достигали103 значений ВЭТС, равных 20 мм.
QMQsg^'
РОТОРНО-ДИСКОВЫЕ ЭКСТРАКТОРЫ |РДЭ)
Схема роторно-дискового экстрактора105 представлена на
рис. 289. К обечайке колонны прикреплены горизонтальные
«кольца статора» — кольцеобразные перегородки, разделяющие
экстрактор на ряд секций небольшого объема, имеющих высоту
Нс каждая. В центре каждой секции на валу расположены
гладкие круглые диски, которые при вращении перемешивают
жидкости в колонне. Диаметр диока d{ меньше
диаметра отверстия кольца статора ds.
Предпочтительны следующие соотношения
размеров внутренних устройств экстрактора: T/di=
= 1,5—3,0 и Т/Нс = 2—8; однако эти соотно-
? шения можно изменять в соответствии с
условиями конкретного процесса.
, На рис. 289 приведена схема экстрактора,
в котором диспергируется легкая жидкость
(граница раздела фаз находится вверху
аппарата). Однако тот же экстрактор можно
использовать для диспергирования тяжелой
жидкости.
Роторно-дисковые экстракторы
первоначально применяли для очистки нефтяных
^^-JL^-^ д/ смазочных масел фурфуролом; в настоящее
^:=^~*" время эти аппараты получили широкое
распространение во многих процессах
нефтеперерабатывающей промышленности
(включая процессы деасфальтизации нефтей
пропаном, очистки бензинов от меркаптанов), в
процессах разделения металлов, а также в
коксохимической, пищевой промышленности и
в промышленности основного органического
синтеза.
Рис. 289. Роторно-
дисковый
экстрактор (РДЭ):
/ — привод с
переменной скоростью
вращения; 2 — граница
раздела фаз; 3 — кольцо
статора; 4 — диск ро
тора; /, /// — легкая
жидкость; II, IV —
тяжелая жидкость.
576
'5Г
=3
£
с
1
3
s-
5
04,
0,2
OJ
0,06
О,0Ь
орг
т
т
k io.
'\,
^
^= = :
_^_
Л..--1
г 4 бв /я4 г 4 бвю3 г ь вею8
Критерий Рейнальдса для мешалок, Re
Рис. 290. Энергия, затрачиваемая на
перемешивание в роторно-дисковом
экстракторе 106.
Колонны
лабораторных размеров обычно
имеют диаметр 100—150 мм.
Диаметр наиболее
крупной промышленной
колонны — более 3 м.
Энергия,
затрачивавшая на перемешивание.
Энергию, потребляемую
в роторно-диоковых
экстракторах 106, определяли
на колоннах диаметрами
от 50 мм до 2,25 м с
соотношениями размеров внутренних устройств: T(di=l,6—2,2;
77ds=l,26—1,4; Г/#с = 4,1—8,8. Полученные ограниченные
опытные давдные представлены на графике (рис. 290). При этом не
определяли величину энергии, затрачиваемой на
перемешивание, при изменении соотношений размеров внутренних устройств
или размеров колонны; неизвестно также, вводились ли в
расчет средние величины физических свойств жидкостей при
высоких значениях УС. На оси ординат графика (см. рис. 290)
отложены величины потребляемой энергии, приходящейся на одну
секцию экстрактора; энергия, расходуемая в многосекционном
экстракторе, прямо пропорциональна числу секций.
УС и захлебывание. Имеется несколько эмпирических
корреляций для определения производительности экстрактора,
соответствующей захлебыванию106-108. Однако лучшим следует
считать метод расчета 109-и1, основанный на концепции
«характеристической скорости», рассмотренной выше в связи с анализом
гидродинамических характеристик экстракторов других типов.
Было показано, что уравнение (XI, 14) описывает опытные
данные по УС роторно-дисковых экстракторов, а по уравнению
(XI, 17) можно определять нагрузку при захлебывании этих
аппаратов. Однако109 в случае, когда T/(ds—dj)>24, величину
Vc, полученную по уравнению (XI, 14), следует умножать на 2,1.
Корректность уравнения (XI, 14) подтверждена в опытах на
экстракторах диаметрами 75, 150 и 1100 мм. На основании опытов,
проведенных на колоннах диаметрами 75 и 150 мм,
предложено 109, но определять характеристическую скорость Ук в
уравнении (XI, 14) в отсутствие массопередачи из уравнения:
Var /ApV'.9/ g \ / ^ \2'3 / ^г V'9/Ч V'6
-^=°-012Ы ЬиЬг) Ьг) (т-) (Х1-47)
Коэффициент 0,012 в уравнении (XI, 47) необходимо
заменять на 0,0225 при Tj(ds—di)^24. Логсдайл и0 нашел, что
направление распределения ацетона между толуолом (или бутил-
ацетатом) и водой оказывает существенное влияние на величину
37 Р. Е. Трейбал
577
VK; Стрэнд111 не обнаружил этого эффекта. Расхождения в
результатах опытов указанных авторов можно объяснить
присутствием незначительных количеств поверхностно-активных
примесей в веществах, с которыми работал один из
исследователей, в то время как другой проводил опыты с чистыми
веществами.
Стрэнд с сотр. ш определяли изменение величины УС по
высоте и диаметру роторно-дискового экстрактора. Оказалось, что
при диспергировании легкой фазы значение УС (обозначаемой в
дальнейшем fD) в зависимости от высоты колонны проходит
через максимум и остается величиной постоянной по
поперечному сечению экстрактора, возрастая лишь вблизи вала ротора.
Те же авторы считают методически правильным при проведении
лабораторных исследований УС определять величины <f-D в
точке между di/2 и ds/2 (считая от оси колонны) и в точке,
соответствующей максимуму зависимости ф0 от высоты колонны.
Средняя по всей колонне величина фц> мало отличается от
локального значения fD, найденного в этой точке.
Для расчета характеристик роторно-дисковых экстракторов,
особенно для моделирования, предложено ш связывать
характеристическую скорость VK по уравнению (XI, 14) со скоростью
осаждения Vt капли с помощью соотношения
VK=CRVt (XI, 48)
в котором коэффициент CR численно равен одному из трех
приводимых ниже выражений, имеющему наименьшее значение:
\2
№ -ДО
0,5
Соотношение между диаметром капли и скоростью ее
осаждения Vt (см. главу V) позволяет, таким образом, рассчитать
диаметр капли из данных об УС. Диаметр капли связывается
с величиной энергии, вводимой вращающимся диском в единицу
массы перемешиваемых жидкостей, с помощью уравнения типа
(X, 20):
/ а \<>,б/ пНгТ2 \о,4
*'-cUr) Ыч) <х,'<9>
По уравнению (XI, 49) рассчитывают значения С. Такой
подход позволяет применительно к какой-либо одной системе, для
которой коэффициент С должен быть постоянным, получить на
графике зависимости скорости осаждения Vt (полученной из
опытных данных об УС) от HcT2/PoN3di кривую, общую вблизи
точки захлебывания для колонн всех размеров. В первом при-
578
ближении это подтвердилось
в опытах на колоннах
диаметрами от 150 до 1050 мм.
Кривая должна быть
получена экспериментально из
данных по УС для данной
системы жидкость —
жидкость, так как величина С
может изменяться примерно
в 6 раз в зависимости от
наличия массопередачи, ее
влияния на физические
свойства жидкостей, коалесцен-
цию капель и т. п.
Массопередача. До сих
пор не опубликованы опыт- Рис- 291- Данные о массопередаче в ро-
ныр лян'ныр о окопостях торно-дисковых экстракторах при экст-
. L^tJl „,!.. ° „„ ™?„°„ Л, ракции бутиламина в системе керосин —
„„.„ „„,.,. „„„ ,„„„,, бутиламин — вода106:
1-Vd = 7J5m/4; Vc = 24 м/ч; 2 — VD= 15,45 м/ч;
Vc = 2i~Ml4.
la &
Si i-2
^ I 009
H p
§f 0.06
55
§.§ 003
§s
0
10
1
Д t
0 0/
I
M
I
\
S#
r?^
\l-
Oj
г
I*
-
m
Ю'
10'■
N3dj5
НсТг'
£/v.
массопередачи для двухком-
понентных систем, с
помощью которых можно
определять коэффициенты
массоотдачи в фазах. Известны лишь разрозненные данные о
массопередаче для колонн очень малых размеров 112-115 и
результаты двух более систематических исследований 106' ио.
Эмпирические корреляции, сходные с показанной на рис. 291,
приведены в нескольких указанных выше работах. С помощью
этих корреляций возможна по крайней мере частичная
обработка опытных данных о массопередаче в зависимости от
величины энергии, вводимой на единицу массы смеси жидкостей,
находящейся в аппарате. Предложена также и0 эмпирическая
зависимость для определения Htoc. Однако подобные
зависимости, полученные на основе общих коэффициентов массопередачи,
нельзя считать обобщенными.
Наиболее полный анализ опытных данных о массопередаче,
проведенный для выяснения возможностей моделирования
роторно-дисковых экстракторов, выполнен Стрэндом ш. Опытные
данные о продольном перемешивании при d{N/V>30 для
колонн диаметром 150 и 1050 мм были обработаны с помощью
уравнений:
И
(1-*р)Е _
НгРе'г
VrH,
' С"С
■. 0,5 +0,09(1-^)(^) $f [(^)2- (т-)1 <Х1'*»
Н
ФВЕ
37*
vDnc
;O,5+O,O90D
dtN
mm
v2
(XI, 51)
57a
Определяемые из этих уравнений значения критерия Ре'
использовали в уравнениях (XI, 11) и (XI, 12). Опытные значения
общих коэффициентов массопередачи после введения поправки
на продольное перемешивание [по уравнениям (XI, 50) и
(XI, 51)] сравнивали с расчетными, полученными на основе
коэффициентов массоотдачи. Диаметр капель рассчитывали с
помощью уравнения (XI, 48) и соотношения между Vt и dv по
экспериментально определенным значениям УС, а коэффициенты
массоотдачи — по одному из следующих двух способов:
для ка'пель без внутренней циркуляции
2я2£>
3rf„
[из уравнения (V, 79)]
kc = 0,001 Vg
для капель с внутренней циркуляцией
0,00375 V с
2я2£>г
1+-
Ит
3rf„
[из модифицированного уравнения (V, 83)]
A^f-T
[из уравнения (V, 88)]
Экспериментальные и рассчитанные значения
коэффициентов массопередачи сравнивали графически (рис. 292). Опытные
точки располагаются между значениями коэффициентов
массопередачи, рассчитанных при наличии циркуляции в каплях и
без нее, причем положение этих точек на графике зависит от
свойств системы, направления массопередачи, возможного
присутствия следов загрязнений и т. п.; вместе с тем опытные
величины коэффициентов массопередачи практически не зависят от
размера экстрактора. Таким образом, опытные данные,
полученные на экстракторах малых размеров, обработанные
описанным способом, можно с достаточной точностью использовать
9. 20
I
1
£
^
{
^
0°
о ЧФо
*-■
%9#
.«
• •
- 1
• Ф 105 м —
/г
-
§ ю3 ши /о5
i? Критерий Пекле для каплифе=с[р¥3/вл)
/О6
Рис. 292. Сравнение данных о массопередаче при экстракции ацетона
из толуола (дисперсная фаза) водой в РДЭ (0 0,15 м а 0 1,05 л)111:
1 — кривая для капель с циркуляцией; 2—кривая для капель без циркуляции.
580
для расчета характеристик экстракторов больших размеров (для
той жесисте.мы жидкость — жидкость). Для надежного перехода
к экстракторам больших размеров следует предусмотреть
возможность некоторого изменения в них скорости вращения
ротора.
Пример XI-4. Этот пример приведен для иллюстрации применения
предложенных корреляций. Будем считать, что опытные данные на рис. 292
получены только при испытании экстрактора диаметром 150 мм.
Определить высоту роторно-дискового экстрактора, предназначенного для
экстракции 80% ацетона из разбавленного раствора его в толуоле водой, по
приведенным ниже данным. Раствор ацетона в толуоле (дисперсная фаза):
цв = 0,59 спз (0,59-Ю-3 н- сек/м?); pD = 846 кг/ж3; DD = 9,65 • 10"6 м2/ч
(2,68- 10"9 м2/сек); расход 14,2 м3/ч.
Вода (сплошная фаза): Цс = 1 спз (Ю-3 н-сек/м2); рс = 985 кг/ж3;
Dc = 3,46-Ю-6 м2 (0,962- Ю-9 м2/сек); расход 11,3 м3/ч; начальная
концентрация ацетона 0%.
Межфазовое натяжение 32 дн/см (32 • Ю-3 н/м); коэффициент
распределения cd/cc = 0,58.
Размеры экстрактора: Г=1,05 м; ds = 0,715 м; di = 0,525 м; Яс=0,37 м.
Скорость вращения ротора принимается равной 75% от скорости при
захлебывании.
Решение
УС, скорость ротора, размер капель. Поперечное сечение экстрактора
Я-1,052/4 = 0,87 м2\ следовательно, Vc = 11,36/0,87= 13,1 м/ч и VD= 14,2/0,87 =
= 16,3 м/ч. Удерживающая способность экстрактора при захлебывании по
уравнению (XI, 17) составляет: <£»f = 0,35. Из уравнения (XI, 14) при
^DF = 0,35 получим Ук=Ю0 м/ч. С помощью уравнения (XI, 47) определим
скорость ротора при захлебывании: N= 191 об/мин.
Рабочая скорость ротора 0,75-191 = 143 об/мин, соответственно по
уравнению (XI, 47) находим: Ук=177 м/ч. Из уравнения (XI, 14) имеем: 0D=O,11;
Vs=157 м/ч.
Наименьшее значение CR равно (ds/T)2 = 0,446. Пользуясь уравнением
(XI, 48), определим V< = 397 м/ч; скорость капли, имеющей размер больше
критического, по уравнению (V, 76) составляет 427 м/ч. Следовательно, для
определения размера капли применимо уравнение (V, 75). При К( = 397 м/ч
величина rfp = 4,3- 10~3 м.
Коэффициенты массопередачи. Для капель без внутренней циркуляции
применим рекомендации Стрэнда:
А„ = nj D =0,0148 кмоль-м-2- ч~1-Ас-1
D Ыр
kQ = 0,001 V$ = 0,158 кмоль ■ м~2 * ч~х -Ас~1
1 1 , m 1 . 0,58
K-=Aj+A^"= Щ48 +0Л58 ~°>014 кмом ' м" * «"' • Дс
Критерий Пекле для капли dpV8/DD = 70 000. Из рис. 292 находим:
Действительное значение Кп
Значение Кп, рассчитанное для капли без циркуляции
действительное значение Кп
= 2
~~ 0,014
откуда /Сп = 0,028 кмоль • м 2 • ч'1 • Ac1; a=&<t>D/dp=\bA м2/м3; /CDa=0,028 • 154=
■=4,3 клюль • лг3 • ч-1 • Ас-1;Я,оо = Я(ол = ^»//Соа=16,3/4,3 = 3,8 м дяя
поршневого режима течения.
S81
Из графика, приведенного на рис. 196, при степени извлечения 80%
ордината равна 0,2. Фактор экстракции е= Vc/mVD= 13,1/0,58 • 16,3= 1,38.
Следовательно, NtOR=2,7 для поршневого режима течения и #поршн.=
= Я,ои-ЛГ,ов=318-2,7=10,Зл1.
Продольное перемешивание. Влияние продольного перемешивания нужно
определять методом последовательного приближения, так как уравнения
(XI, 11), (XI,50) и (XI,51) содержат неизвестную величину Я. Согласно
уравнению (XI, 50), величина Ре^ = Ре^ = 0.437Я. Из уравнения (XI, 51)
имеем: PeD = Ре^ = 1.273Я. По данным табл. 17, при 1/е=0,725 находим
а=0,569; 6 = 0,484 и т. д.
Окончательно получим Я=12,2 м; при этом Ре£=17,5; Ре„ = 51,0, по
уравнению (XI, 2) находим: Z=82,3; Яюд=4,5л.
Высота смесительной зоны экстрактора равна 12,2 м (число секций 33).
Общая высота аппарата складывается из высоты смесительной зоны и высот
отстойных зон.
ЭКСТРАКТОРЫ ТИПА «MIXCO»
Экстрактор «Mixco», известный также под названием
экстрактора Олдшу — Рештона 116, схематически показан на
рис. 293. Он состоит из ряда секций,
каждая из которых образована горизонталь-
ными кольцами статора (соединенными
2U
S ZJ
^ 18
«а
',5
1.2
6
V
5
г\Г^
V
F
~"f
==Т1
_F
— 1
F
100 300 500 700
Скорость Вращения мешалка, об/мин
Рис. 294. Данные об экстракции уксусной
кислоты в системе изобутилкетон
(дисперсная фаза)—вода (сплошная фаза).
Экстрактор «Mixco» (7= 150 мм, F — точка
захлебывания; см. таблицу)116:
Рис. 293. Схема
экстрактора
«Mixco»:
/ — перегородка; 2 —
перфорированный
распределитель фазы;
3—мешалку 4 —
горизонтальные
перегородки; /, /// — тяжелая
жидкость; //, IV —
легкая жщкость.
Номер
кривой
1
2
3
4
5
6
di
мм
50,8
50,8
50,8
50,8
50,8
76,3
",
мм
54,2
54,2
54,2
82,5
82,5
82,5
vD
м/н
4,64
8,16
8,16
11,6
8,16
4,64
vc
м/ч
2,16
8,9
5,52
5,4
3,9
2,16
582
между собой четырьмя
вертикальными пристенными отражательными
перегородками), и центрального
вала с плосколопастными турбинными
мешалками. Этот аппарат можио
рассматривать как систему
последовательно соединенных смесителей
с отражательными перегородками.
Предпочтительны следующие
соотношения размеров аппарата: Нс =
= 0,5 Г; ds>di.
Аппарат такой конструкции
можно использовать не только для
проведения процессов противоточной
экстракции, но и для обработки
жидкостей в присутствии твердых
веществ и газов при прямотоке
реагирующих веществ (см. главу X).
Несмотря на то что эти аппараты
получили промышленное
применение, в литературе опубликованы
лишь характеристики экстракторов
«Mixco» небольших диаметров.
На рис. 294 приведены опытные данные Олдшу116 для систем
с низким межфазовым натяжением (около 2—5 дн/см). Из
рис. 294 видно, что для каждого исследованного соотношения
размеров аппарата имеется оптимальное число оборотов
мешалки, различное для отличающихся по свойствам систем
жидкость— жидкость. В случае уменьшения диаметра отверстий
колец статора значения ВЭТС снижаются при одновременном
уменьшении предельно допустимой производительности
экстрактора, что, вероятно, вызвано уменьшением продольного
перемешивания между ступенями.
При экстракции урана (трудноэкстрагируемая система,
обладающая высоким межфазовым натяжением, массопередача в
которой осложнена химической реакцией), по данным Дик-
стры и7, наблюдалась оптимальная скорость мешалки (рис. 295).
Густисон118 предложил корреляцию для эффективности
ступени при экстракции урана от фактора экстракции, который
значительно меняет свою величину по высоте аппарата в результате
изменения коэффициента распределения (более высокому
значению е соответствует более низкая величина ЕМТ>).
В том же исследовании было установлено, что
эффективность ступени (секции) EMD при переходе от колонны
диаметром 150 мм к колонне диаметром 300 мм сохраняется в случае
постоянства свойств системы, массовых скоростей фаз,
энергии, вводимой в единицу объема обрабатываемых жидкостей, и
со
15
1,2
0,9
0.6
о.з
1
\
1
3
F
F.2
ч^_
F
100
14-0
180 220
Скорость вращения
мешалки, об/мин
Рис. 295. Данные о массопере-
даче при экстракции U02(N03)2
из водных растворов (3,5 н)
азотной кислоты (сплошная
фаза) 20 % -ным раствором
трибутилфосфата в V arsol'e
(7" =127 мм; d; = 63,9 мм;
d„ = 76 мм; Н' = 100 мм;
F — точка захлебывания)117:
l — VD = Vc = №,5 м/ч; 2— 10,5 м/ч;
3 — 6 м/ч.
583
при геометрическом подобии аппаратов; при этом допускалось,
что сплошная фаза в каждой секции полностью перемешана.
Если в этих условиях отношение KtAIVd можно считать
примерно постоянным, то из табл. 16 получим:
Кпа _1п(1 —£ л _ 1пП—£ \
D __ V MDI) V MD2' /VI г-пч
__ _ (XI, 52)
откуда
**М-1-0-*Ло.)ЯсЯ/ЯС1 (XL 53)
Индексы 2 и 1 в этих уравнениях относятся соответственно к
большему и меньшему аппаратам. Уравнение (XI, 53) отражает
закономерность возрастания эффективности ступени при
моделировании, которая была подтверждена опытными данными.
Однако такой метод моделирования по существу эмпирический;
более надежное проектирование этих аппаратов должно
основываться на подробных данных о продольном перемешивании,
характеристических скоростях капель и т. п.
Нагата i'9-ш провел широкое исследование других типов
экстракторов с механическим перемешиванием фаз. В этих
аппаратах мешалки располагались эксцентрично по отношению к оси
колонны и жидкости перемещались с одной секции на другую
через переточные трубки небольших диаметров (площадь
поперечного сечения каждой трубки составляла не более 3,3% от
площади поперечного сечения экстрактора).
В экстракторах такой конструкции уменьшалось обратное
перемешивание, но одновременно снижалась предельно
допустимая производительность экстрактора. Уменьшение
производительности было весьма значительным, что ограничивает
возможности промышленного применения аппаратов этого типа.
Гарнер 122 установил по оси колонны с ситчатыми тарелками
(диаметр колонны 100 мм) вал с мешалками. Мешалки были
расположены настолько близко к плоскости тарелок, что могли
разрушать капли дисперсной фазы в момент их образования на
тарелке. Применяя систему диэтиламин — вода (дисперсная
фаза) — метилизобутилкетон, отличающуюся низким значением
межфазового натяжения, при умеренных скоростях вращения
ротора Гарнер существенно увеличил эффективность ступени.
ЭКСТРАКТОРЫ ШАЙБЕЛЯ
Известны экстракторы Шайбеля двух типов. Аппарат более
ранней конструкции123 (рис. 296)—один из первых колонных
аппаратов с механическим перемешиванием, получивший
широкое применение. Он состоит из чередующихся смесительных и
отстойных секций. Перемешивание в смесительных секциях
производят с помощью мешалок, укрепленных на центральном валу.
584
Отстойные зоны заполнены плетеной сеткой с крупными
ячейками (свободный объем сетки составляет 97—98%) типа той,
которую используют в сепараторах для уменьшения уноса в
системах газ — жидкость. Относительные высоты смесительной и
отстойной зон можно менять в зависимости от условий
конкретного процесса. Модель экстрактора Шайбеля диаметром 25 мм
широко применяли в практике лабораторных исследований
процесса экстракции.
Аппараты этого типа
весьма растространеяы, однако
имеются ограниченные
сведения о характеристиках
подобных экстракторов
больших размеров. Типичные
опытные кривые,
полученные 124 при испытании
экстрактора диаметром 290 мм,
показаны на рис. 297. Для
подобных аппаратов также
существует оптимальная
скорость вращения ротора;
$>
*-ёч
сь
С!
5;
J-
<h
С;
CJ
1
41
>
emu
^
s
s
->-
u,
<o
1
'.1
memo
M
1
^
11=
Число
смеси
U
1,2
1
0,8
06
O'i
n?
4
1
1
/5
Vs
u^~~
- с
7
_~~4
2
4
N. /
\
3
0 ZOO kOO 600 800
Скорость Вращения
мешалки, об/мин
Рис. 297. Данные о массопередаче, полученные
на колонне Шайбеля ранней конструкции (Т =
= 290 мм; di = 100 мм; высота смесительной
секции — 75 мм; высота насадочной секции —
228 мм; обозначения к таблице: С — сплошная
фаза; £ — экстрагент: МИБК —
метилизобутилкетон) 124:
Рис. 296.
Первоначальная конструкция
колонны Шайбеля:
1 — вращающийся вал;
2—насадка из свернутой
проволочной сетки; 3 —турбинная
мешалка; /, III — легкая
жидкость; // — исходный
раствор (если применяется
фракционная экстракция); IV,
v—тяжелая жидкость.
Номер
кривой
1
2
3
4
5
6
1
Система
МИБК (С) —вода (£) —уксусная
кислота
МИБК — вода (С, Е) — уксусная
кислота
МИБК (С, Е) — вода — уксусная
кислота
МИБК (С) — вода {Е) — уксусная
кислота
МИБК (С, Е) — вода — уксусная
кислота
МИБК — вода (С, Е) — уксусная
кислота
Ксилол — вода (С, Е) — ацетон
Ксилол (£) — вола (С) — ацетон
Ксилол (С) — вода (£) — ацетон
Ксилол (С, Е) — вода — ацетон
VD
м/ч
12,7
12,7
7,07
7,9 i
6,75
7,9
6,45
VC
м/ч
12,7
12,7
7,07
5,27
6,45
5,27
6,75
585
-=ч
-ЕЭ-1
h^3_
h^-
h^L
эффективность колонны
зависит от направления мас-
сопередачи и от того, какая
фаза является дисперсной.
Диспергирование той или
иной фазы влияет не только
на величину межфазовой
поверхности, но, вероятно, и
на смачиваемость ситчатой
насадки. В главе X были
рассмотрены опытные
данные, полученные для
единичной смесительной
секции аппарата 125.
Более новая конструкция
экстрактора Шайбеля 126- 127,
показанная на рис. 298, в настоящее время «ашла эффективное
применение при диаметрах аппарата, превышающих 300 мм.
В аппаратах этой 'конструкции мешалки заключены в
пространство, ограниченное неподвижными кольцевыми перегородками,
поддерживаемыми с помощью тяг (на рисунке не показаны);
секции образуются кольцами статора, закрепленными на
обечайке колонны.
Рис. 298. Более поздняя конструкция
экстрактора Шайбеля:
/ — перегородки, ограничивающие секции;
2 —внутренние кольцевые перегородки.
is!
Й * Э
е g §
о «s р
СО К -Q
3°*
10
1
У
*—
и
/■
\
--/^
3
■^г
5 10г г 5 7Ю3 Z 5 7 /0*
Рис. 299. Опытные данные 126 о массопередаче для экстрактора Шайбеля
новой конструкции (7" = 287 мм; di = 100 мм; высота насадочной
секции равна высоте смесительной секции и составляет 50 мм; МИБК —
мети лизобутилкетон)'26:
Номер
кривой
2
3
4
Система
МИБК —вода —уксусная кислота
МИБК — вода —уксусная кислота
Ксилол — вода —уксусная кислота
Ксилол —вода —уксусная кислота
Ксилол — вода — фенол
МИБК—вода —уксусная кислота
Конструкция *
а
а
а
б
м/ч
19,0
7,63
15,8
* Конструкция а —с чередующимися смесительными и насадочными секциями;
конструкция б — без насадочных секций; мешалки установлены через одну секцию или в каждой
секции .
¥
В колоннах диаметром менее 900 мм, используемых для
систем с низкими межфазовым натяжением и вязкостью,
смесительные секции, представленные на рис. 298, могут чередоваться
с отстойными, заполненными проволочной сеткой. Для систем
с высокими межфазовым натяжением и вязкостью более
экономична конструкция, изображенная на рис. 298. Аппараты этого
типа должны иметь диаметр не менее 75 мм; в промышленности
Применяют экстракторы Шайбеля размером 2100 мм и более.
Типичные данные126 о массопередаче, выраженные в виде
зависимости от энергии, приходящейся на единицу объема
жидкостей, протекающих через секцию, представлены на рис. 299;
эти данные нельзя использовать для других систем жидкость —
жидкость. При критериях Рейнольдса для мешалки,
соответствующих турбулентному режиму, критерий мощности Ро=1,85.
ПУЛЬСАЦИОННЫЕ ЭКСТРАКТОРЫ
В пульсационных экстракторах жидкостям, движущимся
через аппарат, сообщаются быстрые возвратно-поступательные
колебания относительно малой амплитуды. Общий принцип
действия пульсационных экстракторов предложен Ван Дийком128,
который описал два способа сообщения пульсаций жидкостям.
По первому способу используют возвратно-поступательное
движение по вертикали горизонтальных перфорированных
тарелок, осуществляемое с помощью механических приспособлений;
при этом на основное движение жидкостей через колонну
накладываются пульсации, вызываемые вибрирующими тарелками.
По другому способу внутренние устройства экстракторов остаются
неподвижными, а пульсации сообщаются жидкости механизмом,
находящимся вне колонны. Колебательные движения от
механизма гидравлически передаются жидкостям в колонне.
. За несколькими исключениями (см. раздел «Экстракторы с
вибрирующими тарелками», стр. 596), до сих пор мало сделано
в разработке первого предложения Ван Динка. Однако второе
предложение нашло широкое применение в атомной технике —
процессах экстракции металлов из радиоактивных растворов.
В этих процессах механическое перемешивание (каким-либо
способом) необходимо для уменьшения чрезмерно большой высоты
экстракторов и соответственно для снижения расходов на
защиту обслуживающего персонала от радиоактивности.
Одновременно необходимо избегать установки в аппаратах
подшипников, сальников и других устройств, подверженных
механическому износу и коррозии, так как ремонт этих узлов
обслуживающим персоналом во многих случаях с точки зрения
техники безопасности невозможен. Идеальными для подобных
условий можно считать экстракторы, в которых пульсации
генерируются с помощью приспособлений, находящихся вне опасной
587
J
Воздух
'<вт
а
б
и
в
Рис. 300. Схемы пульсаторов.
зоны, причем пульсатор и
экстрактор связаны между собой
гидравлически. Практическое
использование подобных
аппаратов, по-видимому,
ограничено процессами экстракции,
применяемыми в атомной технике,
вследствие относительно
больших затрат энергии на
генерацию пульсаций.
Пульсационные
экстракторы с гидравлической связью
между аппаратом и генератором пульсаций можно разделить
на две группы: обычные (распылительные, насадочные,
экстракторы с перфорированными тарелками и т. д.), в которых на
основ'ное движение жидкостей через аппарат накладываются
пульсации, и аппараты с ситчатыми тарелками специальной
конструкции, используемыми только в пульсационных эстрак-
торах.
Конструкции пульсаторов. Пульсатор обычной конструкции
(рис. 300, а) представляет собой поршневой бесклапанный насос
или аналогичный специально сконструированный механизм.
Такой простой по устройству пульсатор обеспечивает легкое
регулирование амплитуды пульсаций (за счет изменения длины
хода поршня) и частоты пульсаций (с помощью коробки
скоростей на приводе).
Пульсации могут передаваться также через сильфоны
(рис. 300,6), изготовляемые из гибкого металла или
пластмассы и получающие возвратно-поступательное движение от
механического (гидравлического) привода или же от электронного
датчика. Вместо сильфона можно использовать гибкие
мембраны, или диафрагмы. Продолжительность службы сильфонов-
пульсаторов с гидравлическим приводом может составлять
30 млн. циклов и более129. Такой пульсатор можно
присоединять непосредственно к корпусу экстрактора или соединять с
последним трубой, как показано на рис. 300, а.
Обе конструкции пульсаторов (рис. 300, а и 300, б) имеют
тот недостаток, что их механизм находится в непосредственном
контакте с обрабатываемыми в экстракторе жидкостями,
которые могут быть сильно агрессивными; кроме того, пульсаторы
такого типа могут подвергаться воздействию кавитации.
Пульсатор с воздушной подушкой130, находящейся между
рабочими жидкостями в экстракторе и пульсатором, свободен от
указанного недостатка (рис. 300, в). Однако при работе этого
пульсатора затруднено точное определение амплитуды
пульсаций; кроме того, расход энергии на генерацию пульсаций
относительно высок. Чаще всего пульсаторы конструируют для по-
588
лучения синусоидальных колебаний, но применяют также
колебания пилообразной и прямоугольной форм.
Различают частоту пульсаций f, характеризующую скорость
сообщения пульсаций жидкости (цикл/ед. времени), и
амплитуду пульсаций w — линейное расстояние между крайними
положениями уровня жидкости в экстракторе (но не в пульсаторе)
за цикл. Амплитуду можно определить так же, как половину
указанного выше расстояния, по аналогии с терминологией,
принятой при изучении гармонических колебаний (при чтении
литературы на это следует обращать внимание). Объемная пульса-
ционная скорость — объемная скорость движения жидкости при
пульсировании, представляющая собой произведение амплитуды,
частоты и площади поперечного сечения экстрактора.
Расход энергии. Джелус 129 вывел уравнение для
определения расхода энергии на пульсирование жидкости в экстракторе.
Пульсатор должен воздействовать на жидкость с силой,
необходимой для преодоления статического давления столба
жидкости в колонне над уровнем установки пульсатора, инерции
жидкости и силы трения при движении жидкости. Он получил
выражение для мгновенного значения энергии, затрачиваемой
на пульсирование в колоннах с ситчатыми тарелками и
сетчатой насадкой:
( I pDzSt\ d2y
pt = s<{(PMH-Pf,z)g + {PMH + ^r)lv +
+-
Np(sysl-l)pM ,St \2 1/
o,36 ^{Sp I P"JV
'dy
Д
dy
(XI, 54)
где у — расстояние, на которое перемещается жидкость при
пульсировании.
Для простых гармонических колебаний (синусоидальные
пульсации):
у = -^- sin 2я/9
-|§-=|-(2nf)cos2nf9
Ц- = -^(2я/)281п2я/9
(XI, 55)
Для пульсатора, показанного на рис. 300, а, в этих
уравнениях величину перемещения жидкости в экстракторе связывают
с длиной штока и радиусом кривошипа.
Сравнение опытных данных, полученных на колонне
диаметром 600 мм со слоем сетчатой насадки высотой 12,6 м, с
данными, рассчитанными по приведенным уравнениям,
представлено на рис. 301. Пульсатор должен обеспечивать макси-
589
мум необходимой энергии.
Если его оборудовать
маховиком, который играет роль
аккумулятора энергии, то
рабочая энергия, затрачиваемая на
один цикл, определится как
/ PtdQ/Q. Эта величина
значительно меньше максимального
расхода энергии. Кроме
маховика можно применять ,31
последовательное соединение
двух колонн, в которых
пульсации, генерируемые одним и
тем же пульсатором,сдвинуты
по фазе на угол 180°.
Экономия энергии достигается
также при использовании
каскадного расположения пульсаци-
онных колонн
(последовательная установка колонн
небольшой высоты, работающих при
этом уменьшается статическое
Джелус рассчитал также давление в пульсаторе. Давление
можно вычислить только приближенно, поскольку при расчете
нужно принимать произвольно значение поправочного
коэффициента, учитывающего трение жидкости. Максимальное
давление всегда совпадает по фазе с максимальной потребляемой
энергией. Знание величин давлений, возникающих в пульсаторе,
очень важно, так как от величины давления зависит прочность
пульсатора. Если минимальное значение давления оказывается
меньше давления паров жидкости, находящейся в контакте с
пульсатором, то возникает кавитация и пульсирование
становится неэффективным. Вильяме ш провел подобный расчет и
установил, что фактором, определяющим возникновение кавитации,
является парциальное давление растворенного в жидкости
воздуха.
Распылительные, насадочные и ситчатые экстракторы
обычной конструкции с применением пульсаций. Биллербек 134
исследовал пульсационную распылительную колонну небольших
размеров (диаметр 38 мм). При амплитуде пульсаций 11 мм и
частоте 400 цикл/мин УС по дисперсной фазе и скорость массо-
передачи увеличивались (причем последняя возрастала
примерно на 30%) по сравнению со значениями УС и соответственно
скорости массопередачи в отсутствие пульсаций. Широтзука135
приводит дополнительные опытные данные для распылительных
пульсационных колонн небольших размеров. Можно считать, что
1.5
I
-3
V
>*з
у
*г
"\
90 180 870 360 ^50
Угол смещения Л, градусы
Рис. 301. Мощность, затрачиваемая
на пульсирование жидкости в колонне
(диаметр 600 мм; высота 12,6 м\
частота пульсации 93,6 цикл/мин;
амплитуда 12,7 мм)129:
7 —опытная кривая; 2 — расчетная кривая.
противотоке фаз), так как при
TQO
590
пульсационные экстракторы такого типа не представляют
интереса для промышленности.
В пульсационных насадочных колоннах можно использовать,
по-видимому, насадки любых типов. Насадка в виде колец или
седел, загруженная в экстрактор навалом, имеет тенденцию под
действием пульсаций ориентироваться, что может привести к
каналообразованию, т. е. неравномерному распределению
жидкости по сечению слоя насадки. Поэтому Торнтон136
рекомендовал применять вместо обычной насадки
металлическую, расположенную слоями, причем соседние слои
насадки должны располагаться под прямым углом относительно друг
друга.
Опубликованные в литературе опытные данные относятся
к насадочным колоннам очень малых размеров, и их нельзя
надежно применять для проектирования колонн промышленного
масштаба.
Фейк137 проводил опыты на колонне диаметром 38 мм с
насадкой из малых колец и седел при наличии и отсутствии
пульсаций. В процессе экстракции бензойной кислоты (система
вода — бензойная кислота — толуол) величина ВЕП снижалась
с 4,1 м (в отсутствие пульсаций) до 0,183 м (при пульсациях
частотой 400 цикл/мин и амплитудой 4,75 мм). Для систем с
низким межфазовьш натяжением эффект использования
пульсаций менее значителен. Эффективность экстракции возрастает
в основном вследствие уменьшения
размера капель и соответствующего
увеличения УС по дисперсной фазе и
межфазовой поверхности при
одновременном уменьшении
производительности экстрактора. Можно считать
установленным, что величина Ню
уменьшается с возрастанием амплитуды
пульсации и проходит через минимум
с увеличением частоты пульсаций (см.,
например, литературу138'139). Однако
эта общая закономерность
проявляется различно в зависимости от свойств
системы жидкость — жидкость и
размеров экстракторов. По имеющимся
опытным данным пока что нельзя
сделать обобщающих выводов о
количественном влиянии пульсаций на
скорость массопередачи и
производительность экстракторов. Единственные
опубликованные данные о промышленном D m п
„„ «г рис 302. Пульсационныи экс-
Применении ЭТИХ экстракторов 14° от- трактор с ситчатыми тарел-
Носятся к колонне диаметром 215 мм ками.
Тяжелая
жидкость
I
t
<ч
~jt.
Тяжелая
Жидкость^
(
Ь"
Легкая
жидкость
Сйтиатые
'тарелки
^пульсатору
Легкая
жидкость
591
с насадкой из фторопластовых колец Рашига, которую
использовали для очистки плутония от продуктов распада.
В лабораторном экстракторе с ситчатыми тарелками
обычной конструкции наблюдалось141 заметное увеличение скорости
экстракции при применении пульсаций.
Пульсационные экстракторы с ситчатыми тарелками. Схема
устройства экстрактора такого типа показана на рис. 302. В
колонне установлены горизонтальные ситчатые тарелки, у
которых в отличие от обычных ситчатых тарелок отсутствуют
переливы. В типичных малых лабораторных аппаратах диаметр
отверстий тарелок 2,5 мм и расстояние между тарелками 25 мм.
В экстракторах больших размеров диаметр отверстий
составляет обычно 3,2—6,5 мм и расстояние между тарелками равно
50 мм. Суммарная площадь отверстий тарелок может
достигать 20—25% от площади поперечного сечения колонны.
Частота пульсаций колеблется от 30 до 250 цикл/мин, амплитуда
пульсаций — от 6 до 25 мм (последнее значение
предпочтительно для экстракторов больших размеров).
Опубликованы сведения 142 о применении экстракторов
этого типа диаметром 860 мм; однако большая часть
используемых в атомной технике аппаратов имеет значительно
меньшие размеры из-за необходимости учитывать величины
«критических» объемов перерабатываемых растворов. В ситчатых
пульсационных экстракторах возможна также экстракция из
растворов, содержащих примеси твердых частиц.
Ситчатые пульсационные экстракторы работают в
вертикальном положении, однако их горизонтальное расположение имеет
ряд преимуществ. На одном из испытанных горизонтальных
экстракторов с ситчатыми тарелками модифицированной
конструкции была достигнута мз высокая эффективность массопе-
редачи, равная эффективности вертикального аппарата.
Гидродинамика. Подробное исследование гидродинамических
характеристик пульсационных ситчатых экстракторов
проведено в работе144. Одна из типичных полученных зависимостей
показана на рис. 303.
Во многих случаях при обработке систем с высоким
межфазовым натяжением размеры отверстий тарелок оказываются
настолько малыми, что препятствуют движению жидкостей до
тех пор, пока объемная пульсационная скорость не станет
равной суммарной объемной скорости потоков:
Vc+VD = wf (XI, 56)
Соответственно в области / (см. рис. 303) экстрактор
захлебывается вследствие недостаточной интенсивности пульсации.
Эдварде145 показал, что для систем с очень низким межфазовым
натяжением, когда дисперсная фаза может проходить через от-
592
По уравнению (и, 5б)
Частота пульсации
Рис. 303. Характеристики
пульсационного экстрактора
с ситчатыми тарелками
при фиксированной
амплитуде 144.
серстия тарелок под действием
статического давления столба этой
жидкости на тарелке, иногда могут быть
достигнуты скорости потоков более
высокие, чем определяемые по уравне- ^
нию (XI, 56). ^
В области 2 в пространстве между +
тарелками появляются раздельные ^
слои жидкостей (в период той части
цикла, когда жидкости отстаиваются
после импульса). Во время пульсаций,
направленных вверх, легкая жидкость
«продавливается» сквозь отверстия
тарелки и образует капли,
поднимающиеся к расположенной выше тарелке.
Во время пульсаций, направленных
вниз, аналогично ведет себя
тяжелая жидкость. В этих условиях
экстрактор работает гидродинамически устойчиво, но скорости
массопередачи низкие. Область 3 соответствует довольно
равномерному диспергированию; в течение всего цикла образуются
мелкие капли, которые распределяются в сплошной фазе. В этом
режиме достигаются наибольшие скорости массопередачи.
В области 4 наблюдаются нерегулярная коалесценция капель
и периодическая инверсия фаз. Скорости экстракции при этом
режиме работы экстрактора низкие. Дальнейшее увеличение
частоты пульсаций приводит к захлебыванию экстрактора
вследствие образования стойкой эмульсии (область 5). Переход от
одного режима работы аппарата к другому происходит
постепенно и непрерывно, а форма областей, показанных на рис. 303,
может значительно изменяться в зависимости от свойств
системы жидкость — жидкость и конструкции аппарата.
Желательно, чтобы поверхность тарелок (для улучшения
условий образования капель и редиспергирования) смачивалась
сплошной фазой. В атомной технике обычно используют тарелки
из нержавеющей стали, смачиваемость поверхности которых с
течением времени может изменяться. В этом случае
целесообразно применять тарелки с выштампованными отверстиями
(рис.262),
Ричардсон140 показал, что на таких тарелках можно достичь
производительности на 50—100% большей, чем
производительность на тарелках со сверлеными отверстиями; при этом ВЕП
на тарелках со штампованными отверстиями составляет 70%
от величин ВЕП для тарелок со сверлеными отверстиями.
Тарелки из пластмасс предпочтительно смачиваются
органическими жидкостями и поэтому могут использоваться, когда
38 Р. Е. Трейбал
693
дисперсной фазой является вода.В
колоннах тарелки из пластмассы можно
чередовать со стальными тарелками
(расстояние между тарелками около
0,3 м) с тем, чтобы намеренно
вызывать коалесценцию капель и
предотвращать образование стабиль-
8 ной эмульсии. Для этой же цели
используют «двухслойные» тарелки,
которые изготавливают из металла,
покрытого с одной стороны
пластмассой, и устанавливают таким образом,
чтобы пластмасса была обращена в
сторону входа органической жидкости
в колоину.
В колоннах диаметром 0,6 м и
более происходит интенсивное канало-
образование, снижающее скорость
массопередачи. Для улучшения массо-
передачи в колонне на расстояниях
0,9—1,2 м друг от друга помещают перераспределительные «жа-
люзийные» тарелки144'156. Схема такой тарелки показана на
рис. 304; прорези с направляющими наклонными козырьками
(«жалюзи») и вертикальные короткие перегородки,
расположенные на одной или обеих сторонах тарелки, сообщают жидкости
вращательное движение, в результате чего происходит
перераспределение потоков по сечению аппарата. При установке
перераспределительных тарелок уменьшается число обычных ситча-
тых тарелок в колонне.
Показано 147> 154>155, что при гидродинамических режимах,
соответствующих областям 3—5 (см. рис. 303) для колонн
диаметром, не превышающим по крайней мере 0,3 м, УС пуль-
сационных ситчатых экстракторов можно определить по
уравнению (XI, 14). Значение VK в отсутствие массопередачи рас~
считывают из уравнения
Рис. 304. «Жалюзийная»
тарелка для
перераспределения потоков 144:
1 — вертикальные перегородки;
2 — жалюзи; 3 — сечения А —А и
В-В.
_^с =06/Рс«Л°'24 МоРсМ°'9УсП _
О ' \^с) [ & J [Ире3] [»с
W'/^V-VAP
Рс
(XI, 57)
где i|) — максимальная удельная энергия, расходуемая на
трение при движении жидкости через тарелку.
Для синусоидальных пульсаций:
2С20Н
(XI, 58)
594
В уравнении (XI, 58) коэффициент расхода в отверстии
можно принять равным Со=0,б. Если число тарелок велико, то
H/Np практически равно расстоянию между тарелками.
Скорости жидкостей при захлебывании (для области 5) можно
рассчитать из уравнений (XI, 15) — (XI, 17). Если в колонне
происходит экстракция ацетона из толуола (дисперсная фаза)
водой, то величины VK возрастают в 2,6 раза по сравнению с
рассчитанными по уравнению (XI, 57). Смут146 предложил
следующую эмпирическую корреляцию опытных данных о
захлебывании пульсационных ситчатых экстракторов с
синусоидальными пульсациями:
V 4 / Uca3j Ub
Уравнение Смута дает среднюю ошибку, составляющую
приблизительно 20°/р.
Массопередача. Имеется значительное число работ по
исследованию скорости массопередачи в ситчатых пульсационных
экстракторах (главным образом в области атомной техники),
причем исследования проводили, в основном, на колоннах
малых размеров (диаметром 25 мм). Достаточно подробный
перечень этих работ приведен в статьях Бэбба с сотр.,42'146.
Обобщая опубликованные данные, можно сделать вывод, что
значения ВЕП довольно велики в области 2 (см. рис. 303), но
быстро уменьшаются с возрастанием частоты пульсаций до
значений, соответствующих области 3; при работе экстрактора в
режиме, соответствующем области 3, ВЕП остается величиной
почти постоянной и возрастает при переходе к режиму,
отвечающему области 4 на рис. 303. При экстракции урана или
плутония обычно Я(О=0,3—0,6 м; при экстракции продуктов распада
f/fo = 0,6—1,2 мш. Значения Ht0 зависят от физических свойств
систем, скоростей потоков и конструкции аппарата. Торн-
тон 130'147 показал, что для данной системы (в отсутствие
химической реакции) опытные данные о массопередаче можно
описать уравнением
Н1ПЛп /£2Др
Vc V ^сРс / \vd
mcOp в ЦР 1S.\ =c
0,5
M.c#Ap
,3 2
М1-*в) Рс
(XJ.60)
где С и у — постоянные, определяемые из опытов на колоннах
малых диаметров.
Величину VK рассчитывают из уравнения (XI, 14) при
использовании данных по УС, получаемых при проведении опытов по
массопередаче. Частота и амплитуда пульсаций (/ и ш) влияют
695
38*
на скорость массопередачи через величину VK. Результаты
опытов по массопередаче можно экстраполировать на колонны
больших диаметров (при одинаковой конструкции тарелок малой
и большой колонн) на основании соотношения
нюс2 = нюсУМ{Т7-т'] (XI. еь
Индексы 2 и 1 в уравнении (XI, 61) относятся соответственно
к большой и малой колоннам. Важно учитывать также
смачиваемость материала тарелки148; поэтому тарелки большой и
малой колонн необходимо изготовлять из одного и того же
материала. Имеется также эмпирическая корреляция146 для
опытных значений НЮс, однако для определения скорости
массопередачи рекомендуют применять метод, описанный выше.
Продольное перемешивание, влияние которого учитывается
наличием экспоненциального члена в уравнении (XI,61),
изучал Map142 на колонне диаметром 50 мм. Для этой колонны
при условии, что сплошной фазой является вода, коэффициент
турбулентной диффузии, используемый при расчете критерия
Ре^, определяют из уравнения
Е = 0 17 / »с У'45 /усА°.зо/дрсА0.42
^cdo ' Wvc4 \ *с I \V?c }
Экстракторы с вибрирующими тарелками. Экстракторы этой
конструкции исследованы относительно мало. В экстракторе
Карра тарелки с большой суммарной площадью отверстий
(составляющей около 63% от всего сечения колонны) укреплены
на аксиально расположенном штоке, которому сообщается
возвратно поступательное движение. В колонне Карра можно
применять тарелки из отходов штамповочного производста149. При
возвратно-поступательном движении штока и тарелок жидкости
перемешиваются. В колонне диаметром 76 мм были получены
значения ВЭТС от 0,1 до 0,225 м. Вариант колонны Карра
предложен Исааком 16°. Описан также упрощенный вариант
конструкции этой колонны ш.
Экстрактор Фенске и Лонга152,153 в некоторых отношениях
является смесителыю-отстойным. Он состоит из ряда секций,
расположенных вертикально. В каждой секции происходит
перемешивание (вследствие возвратно-поступательного движения
тарелки) и отстаивание жидкостей. Этот экстрактор применяют
в лабораторных условиях для изучения процессов экстракции,
поскольку эффективность ступени такого аппарата составляет,
как правило, 100% (см. главу IX). Несмотря на то что
предложен метод моделирования этих аппаратов, о промышленном
применении их сведений не имеется.
598
ЦЕНТРОБЕЖНЫЕ ЭКСТРАКТОРЫ
При малой разности плотностей фаз внутренняя энергия
потоков, как было показано ранее, оказывается недостаточной для
Диспергирования одной жидкости в другой; поэтому при
экстракции в контактирующие жидкости вводится
дополнительная энергия в результате их механического перемешивания. Тем
•не менее в обычных экстракторах (с механичеоким
перемешиванием) противоточное движение жидкостей обусловлено раз-
зюстью плотностей фаз, и в таких аппаратах невозможно
достичь больших скоростей потоков. Замена разности плотностей
Нраз как движущей силы противоточадго движения жидкостей
^центробежной силой (в несколько тысяч раз превышающей силу
:"тяжести) в быстро вращающихся машинах обеспечивает
высокие скорости движения жидкостей через аппарат и
соответственно уменьшает необходимый объем экстрактора.
?• При увеличении скоростей и уменьшении объема
экстрактора не только уменьшается потребность в производственной
площади и кубатуре здания, но и снижаются количества
находящихся в аппарате дорогостоящих растворителей. В
непрерывных процессах экстракции быстро достигаются стационарное
'состояние и необходимая конечная концентрация рафината. Во
sМногих случаях, в частности при экстрации антибиотиков,
процесс протекает в условиях, при которых возможна химическая
деструкция продукта (например, при низких рН), и в связи с
I этим желательно, чтобы время пребывания продукта в аппарате
i было возможно меньшим. Для достижения последней цели
центробежные экстракторы незаменимы.
| К недостаткам центробежных экстракторов относятся их
высокая стоимость, а также значительные затраты на
эксплуатацию и ремонт, вызванные сложностью конструкции этих
аппаратов. Указанные недостатки центробежных экстракторов
Приводят к тому, что экстракторы других типов в обычных
процессах экстракции становятся конкурентоспособными с
центробежными.
Экстракторы Подбильняка. Эти аппараты, бесспорно, яв-
..ляются наиболее важными представителями экстракторов
центробежного типа (рис. 305). Ротор аппарата, приводимый во
^вращение от горизонтального вала, — основная часть экстрак-
'тора; он представляет собой цилиндрический барабан, внутрен-
4Нее устройство которого может быть различным. В ранних
моделях157 экстракторов Подбильняка применяли ленту, навитую
\в виде спирали (всего около 30 витков) и образующую каналы
IПрямоугольного сечения для прохода жидкостей. В этих
каналах жидкости движутся противотоком и приходят в тесный кон-
i Такт друг с другом 158.
697
Рис. 305. Центробежный экстрактор Под-
бильняка (Podbielniak, Inc.).
1 — ротор; 2— кожух ротора; 3 — вращающийся
вал; 4 — станина; /, /К — легкая жидкость; //,
/// — тяжелая жидкость.
В экстракторе последних
моделей установлены пер~
форированные
концентрические цилиндры с
отверстиями или щелями, служащими
для прохода обеих
жидкостей. Внутренняя
конструкция экстрактора обычно
изменяется в зависимости от
особенностей процесса, для
осуществления которого
предназначена
проектируемая машина. Жидкости
вводят в экстрактор через
вращающийся вал, снабженный
механическими сальниками;
легкую жидкость подают к
периферии ротора,
тяжелую— к оси экстрактора.
При быстром вращении
ротора (число оборотов составляет несколько тысяч в минуту
и зависит от диаметра ротора) фазы движутся противотоком
в радиальном направлении, экстракт и рафинат вытекают с
противоположных концов вала экстрактора. Изменением
давления на выходе легкой жидкости 159 перемещают основную
границу раздела фаз внутри ротора в соответствии с тем, какую
фазу необходимо диспергировать. Из-за большой
центробежной силы в экстракторе возникает значительное давление,
равное примерно 1,5—12 ат. Центробежные экстракторы
характеризуются очень малой величиной УС. Например ', в
машине, перерабатывающей 3400 л/мин, общий объем жидкости
составляет всего лишь 900 л.- Имеются машины лабораторных
размеров (производительностью менее 4,5 л/мин) и
промышленные аппараты больших размеров и производительностей
(3400 л/мин) с приводом мощностью до 15 кет.
Вследствие очень малой УС экстракторы Подбильняка
применяли первоначально в фармацевтической промышленности,
особенно при экстракции пенициллина, хлоромицетина и других
антибиотиков. Впоследствии эти машины стали использовать
почти во всех процессах экстракции: при очистке растительных
масел 160'161, обесфеноливания аммиачных вод коксохимической
промышленности162, процессах очистки нефтепродуктов 163 и
при экстракции урана.
Жидкости с содержанием мелких твердых частиц также
можно обрабатывать на центробежных машинах, хотя при этом
могут возникать некоторые трудности. Обычно полагают, что
твердые частицы выводятся из экстрактора с одной из жидко-
598
;j
4
1J.
стей, однако некоторое количество твердого вещества
неизбежно аккумулируется внутри аппарата. Это может привести к
неравномерности нагрузки и дебалансу вращающихся частей.
При этом машину необходимо остановить и промыть.
На лабораторной модели диаметром 460 мм и шириной
50 мм при 5000 об/мин в опытах с системой изоамиловый
спирт — вода — борная кислота Бэрсон 164 получил от двух до
восьми теоретических ступеней. Число достигаемых
теоретических ступеней увеличивается с возрастанием соотношения
легкой и тяжелой фаз, однако при этом изменяется положение
уровня раздела фаз. При постоянных объемных скоростях фаз
число ступеней почти не зависит от скорости вращения (в
умеренных пределах). Аналогичные результаты получил Якобсон ,59.
Александр 165 на той же модели экстрактора, испытывая
систему с низким межфазовым натяжением (уксусная кислота —
вода—метилизобутилкетон), получил 12,5 ступеней. Андерсон
сообщил ,66, что при экстракции пенициллина и хлоромицетина с
содержанием твердого вещества в питании 10—15% он получил
две теоретические ступени.
В центробежных экстракторах можно получить различное
число теоретических ступеней в зависимости от свойств
обрабатываемой системы и условий проведения процесса, а также от
конструкции внутренних устройств, так как эти экстракторы
промышленных размеров большей частью конструируют спе-
Цг. циально для данного производ-
? ства.
* В фармацевтической про-
I мышленности, где технологиче-
■i окие схемы производства и но-
'г менклатура выпускаемых
продуктов быстро меняются,
центробежные экстракторы
могут устанавливаться
параллельно (для .увеличения
производительности) или последо-
- вательно (для увеличения
числа ступеней), что позволяет
легче приспосабливаться к
изменениям технологического
процесса. Фракционная
экстракция (осуществляемая при
введении питания в центр
каскада) может проводиться на
двух машинах: одна машина
предназначается для экстрак- Рис ^ Центробежный экстрактор
*, ции, другая — для окончатель- Лувеста (Centrico, Inc.):
t- НОЙ ОТМЫВ'КИ. / — легкая жидкость; // — тяжелая жидкость.
599
Экстрактор Лувеста (Центривеста). Этот экстрактор,
показанный на рис. 306, имеет ротор, вращающийся вокруг
вертикальной оси. Экстрактор является усовершенствованной
конструкцией машины, первоначально предложенной Коутором ,67.
Аппарат состоит из трех ступеней; скорость вращения 3800 об/мин;
производительность 5,9 м3/мин,68' 169. Эти аппараты более
широко распространены в Европе, чем в США, и применяются
главным образом в фармацевтической промышленности.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
Ар — средняя поверхность капли во время ее
образования, м2.
а — удельная межфазовая поверхность, м2/м3
активного объема экстрактора (без отстойных
зон).
аР—удельная поверхность насадки, м2/м3 объема
насадки.
С— постоянная.
CR—безразмерный коэффициент в уравнении
(XI, 48).
с— концентрация, кмоль/м3.
Ас — разность концентраций, кмоль/м3.
D—коэффициент молекулярной диффузии, м2/сек.
dP — диаметр насадки, м.
dFC — критический диаметр насадки, м.
dt~~ диаметр ротора или мешалки, м.
dj — диаметр струи жидкости, м.
d0— диаметр диафрагмы или сопла, м.
dp — средний диаметр капли, м.
dpM — диаметр капли, соответствующий максималь-
ной межфазовой поверхности, м.
dp0 — диаметр капли в насадочной колонне при
скорости потоков, равной нулю, м.
ds — диаметр кольцевой перегородки (отверстия
кольца статора), м.
Е— коэффициент турбулентной диффузии, м2/сек.
EMD— коэффициент эффективности ступени по Мэр-
фри.
Emdp — локальный (точечный) коэффициент
эффективности ступени по Мэрфри.
Е0 — общая эффективность ступени.
/ — частота пульсации, цикл/сек.
g— ускорение силы тяжести, м/сек2.
Н— активная высота экстрактора (высота секции,
заполненной насадкой, и т. п.), м.
600
Нс—высота секции или расстояние между
тарелками, м.
ВЭТС — высота, эквивалентная теоретической
ступени, м.
HtC — высота единицы переноса по сплошной
фазе, м.
HtD — высота единицы переноса по дисперсной
фазе, м.
Нюс—общая высота единицы переноса по
сплошной фазе, м.
Ht0D — общая высота единицы переноса по
дисперсной фазе, м.
h — глубина слоя дисперсной фазы на ситчатой
тарелке, м.
К— общий коэффициент массопередачи,
кмоль • м~2 • ч-1 (Ас=\)'1.
Ка — общий объемный коэффициент
массопередачи, кмоль • м'3 • ч~1 • (Ас — I)-1.
k— коэффициент массоотдачи,
кмоль • м~2 • ч_,(Ас= I)-1.
L — фиктивная массовая скорость, кг/ч на 1 м2
поперечного сечения колонны.
т = cD/cc — коэффициент распределения.
т! = cE/cR — коэффициент распределения.
./V — скорость вращения, об/сек.
N0 — число сопел, диафрагм или отверстий.
Np— число тарелок.
Ре — dpVs/D —критерий Пекле для молекулярной диффузии
в капле.
Ре' = Vfi/E — критерий Пекле для продольного
перемешивания.
Po==P/pc7V3^ —критерий мощности.
Re = d2.Np/n — критерий Рейнольдса для мешалок.
Sc = \i/pD — критерий Шмидта.
Р — энергия, вводимая в секцию, вт.
Pt— общая энергия, вт.
Q — общая объемная скорость, м3/сек.
г — радиус, м.
S — площадь поперечного сечения колонны (без
площади переливов), м2.
S0—суммарная площадь поперечного сечения
отверстий, м2.
Sp — площадь поперечного сечения пульсопрово-
да, м2.
St — общая площадь поперечного сечения
колонны, м2,
601
Т — диаметр колоины, м.
t — толщина тарелки, м.
V — удельная нагрузка, м3/ч на 1 м2 поперечного
сечения колонны.
Vd — окорость в переливе, м/сек.
VK—характеристическая скорость капель, м/сек.
Vo> V'o—скорости в отверстии тарелки или сопла,
м/сек.
V0M—скорость в отверстии тарелки или сопла,
соответствующая максимальной межфазовой
поверхности, м/сек.
VT — относительная скорость, м/сек.
Vs— скорость скольжения, м/сек.
V,— скорость осаждения, м/сек.
vp — объем капли, м3.
vT— объем экстрактора, м3.
w—амплитуда пульсаций, равная расстоянию
между крайними положениями жидкости в
колонне при пульсировании, м.
у — смещение жидкости, м.
z—длина пульсопровода, м.
VEm'/VR — фактор экстракции; для насадочных
экстракторов— свободный объем насадки, м3/м3.
у — постоянная.
О — время, сек.
X — угол, градусы,
ц — вязкость, кг • лг1 • се/с-1.
р — плотность, кг/м3.
Ар—разность плотностей, кг/м3.
рм—средняя плотность жидкости в экстракторе,
кг/м3.
рр — плотность жидкости в пульсопроводе.
а— межфазовое натяжение, кг/сек2.
а'—межфазовое натяжение раствора на границе
с воздухом, кг/сек2.
а2 — межфазовое натяжение для бинарной жидкой
смеси при равновесии, кг/сек2.
а3—межфазовое натяжение для тройной жидкой
смеси при равновесии, кг/сек2.
оЗЕ—эффективное межфазовое натяжение для
тройной жидкой омеси, кг/сек2.
Фр — удерживающая способность экстрактора по
дисперсной фазе, доля от свободного объема
экстрактора,
ф—максимальная энергия трения (на единицу
массы жидкости) при движении жидкости
через отверстия тарелок, м2/сек3.
Индексы
водн. — водная фаза.
С—сплошная фаза.
С1 — в период коалесценции.
D — дисперсная фаза.
Е — экстракт.
F — захлебывание.
f — в период образования капли,
кон. — конечная,
нач. — начальная.
О — в отверстии,
орг. — органическая фаза,
поршн. — поршневое (без обратного перемешивания) течение.
/? — рафияат.
г—во время подъема или падения капли; относительная
величина.
а—обусловлено поверхностным натяжением.
ЛИТЕРАТУРА
Общие вопросы
1. R. Е. Treybal, The Chemical Engineers' Handbook, 4th ed., R. H.
Perry, S. D. Kirkpatrick, С. Н. Chilton (Eds.), McGraw-Hill Book Company,
Inc., New York, 1963.
2 H. R. С Pratt, Ind. Chem., 30, 437, 475, 597 (1954); 31, 63, 505, 552
(1955).
3. V. S. Morel lo, N. P о f f e n b e r ge r, Ind. Eng. Chem., 42, 1021 (1950).
Образование капель
4. R. H. В u с h a n a n, Australian J. Appl. Sci., 3, 233 (1952).
5. С. В. Hay worth, R. E. Treybal, Ind. Eng. Chem., 42, 1174 (1950).
6. W. S iemes, Chem. Ing. Techn., 28, 727 (1956).
7. F. W. Keith, A. N. Hixson, Ind. Eng. Chem., 47, 258 (1955).
8. E. E. Ayres, Trans. Am. Inst. Chem. Engrs., 22, 9 (1929).
9. H. R. Null, H. F. Johnson, A. I. Ch. E. Journ., 4, 273 (1958).
10. K. U e у a m a, Kagaku Kogaku, 21, 766 (1957).
11. R. M Christiansen, A. N. Hixson, Ind. Eng, Chem., 49, 1017
(1957).
12. W Siemes, J. F. Kauf f man, Chem. Ing. Techn., 29, 32 (1957).
Экстракция в период
образования капель
13. Т. К. Sherwood, J. E. Evans, J. V. A. L о n g с о г, Ind. Eng. Chem.,
31, 1144 (1939); Trans. Am. Inst. Chem. Engrs., 35, 597 (1939).
14. W. Licht, J. В. С onway, Ind. Eng. Chem., 42, 1151 (1950).
15.. W. Licht, W. F. Pan sing, Ind. Eng. Chem., 45, 1885 (1953).
16. A. I. Johnson, A. E. Hamielec, A. Gold Ing, Can. J. Chem. Eng.,
36, 221 (1958).
603
17. A. I. Johnson, A. E. H a m i e 1 e c, A. I. Ch. E. Journ., 6, 145 (1960).
18. J. M Couison, S. J. Skinner, Chem. Eng. Sci„ 1, 197 (1952).
19. F. H. Garner, A. H. P. S к e 11 a n d, Ind. Eng. Chem., 46, 1225 (1954).
20. F. H. Garner, A. R. Hall, Chem. Eng. Sci., 2, 157 (1953).
21. P. M. Heertjes, W. A. H о 1 v e, H. T a 1 s m a, Chem. Eng. Sci., 3,
122 (1954).
Продольное перемешивание (теория)
22. А. К. McMullen, Т. M i у a u с h i, Т. V e r m u e 1 e n, U. S. Atomic
Energy Comm. UCRL-3911, Suppl. (1958).
23. T. Mi у a uc hi, U. S. Atomic Energy Comm. UCRL-3911 (1957).
24. С A. S lei cher, A. I. Ch. E. Journ., 5, 145 (1959).
Аппараты с орошаемыми стенками
(пленочные аппараты]
25. D. S. Brinsmade, H. Bliss, Trans. Am. Inst. Chem. Engrs., 39, 679
(1943).
26. R. F a 11 a h, T. G. Hunter, A. W. Nash, J. Soc. Chem. Ind., 53, 369T
(1934); 54, 49T (1935).
27. R. Murdoch, H. R. C. Pratt, Trans. Inst. Chem. Engrs. (London),
31, 307 (1953).
28. R. E. W a r n e r, Chem. Eng. Sci., 3, 161 (1954).
29. E. W. Comings, S. W. В г i g g s, Trans. Am. Inst. Chem. Engrs., 38,
143 (1942).
30. R. E. Treybal, L. T. Work, Trans. Am. Inst. Chem. Engrs., 38, 203
(1942).
31. R. Pass i no, A. R. G i о n a, Chim. e ind. (Milan), 42, 1077 (1960).
32.0. Be r gel in, F. J. Lockhart, G. G. Brown, Trans. Am. Inst.
Chem. Engrs., 39, 173 (1943).
33. N. F. Murphy, J. E. L a s t о v i с a, A. E. S k r z e c, A. 1. Ch. E. Journ.,
2, 451 (1956).
34. L. C. Strang, T. G. Hunter, A. W. N a s h, J. Soc. Chem. Ind., 56,
50T (1937).
Распылительные колонны
35. W. A Stickney, U. S. Bur. Mines Rept. Invest., 5499 (1959).
36. Т. О. Dee, Trans. Inst. Chem. Engrs. (London), 29, 147 (1951).
37. F. H. В1 a n d i n g, J. С Elgin, Trans. Am. Inst. Chem. Engrs., 38,
305 (1942).
38. J. С E I g i n, пат. США 2364892, 1944 г.
39. В. О. Beyaert, L. L a p i d u s, J. С Elgin, A. I. Ch. E. Journ., 7,
46 (1961).
40. R. E. C. Weaver, L. L a p i d u s, J. С Elgin, A. I. Ch. E. Journ., 5,
533 (1959).
41. F. A. Zenz, Petrol. Refiner, 36, № 8, 147 (1957).
42. J. D. Torn ton, Chem. a. Ind., 1954, 1581; Chem. Eng. Sci., 5, 201
(1956).
43. G. W. Minard, A. I. Johnson, Chem. Eng. Progr., 48, № 2, 62
(1952).
44. T. Harma ty, Acta Techn. Acad. Sci. Hung., 12, 209 (1955).
45. G. S. Laddha, J. M. Smith, Chem. Eng. Progr., 46, 195 (1950).
46. С L. Ruby, J. C. Elgin, Chem. Eng. Progr. Symp. Ser., 51, № 16, 17
(1955).
47. G. С Smith, R. B. Beckmann, A. I. Ch. E. Journ., 4, 180 (1958).
48. S. D. Cavers, J. E. Ewanchyna, Can. J. Chem. Eng., 35, 113 (1957).
49. M. L. N e w m a n n, Ind. Eng. Chem., 44, 2457 (1952).
50. R. M. К re a ger, С J. G e a n k о р 1 i s, Ind. Eng. Chem., 45, 2156 (1953).
604"
51. H. J. Vogt, C. J. Geankoplis, Ind. Eng. Chem., 46, 1763 (1954).
52. J. F. Fleming, H. F. Johnson, Chem. Eng. Progr., 49, 497 (1953).
53. A. 1. Johnson, G. W. Minard, C.-J. Huang, J. H. H a n s u 1 d,
V. M. McNamara, A. I. Ch. E. Journ., 3, 101 (1957).
54. L. Garwin, B. D. Smith, Chem. Eng. Progr., 49, 591 (1953).
55. H. Rosenthal, M. Ch. E. Thesis, New York Univers., 1949.
56. T. Wo о dwa r d, Chem. Eng. Progr., 57, № 1, 52 (1961).
57. T. E. Gier, J. О. Н о u g e n, Ind. Eng. Chem., 45, 1362 (1953).
Насадочные колонны
58. F. R. Dell, H. R. С Pratt, Trans. Inst. Chem. Engrs (London), 29,
89 (1951).
59. S. F u j i t а, К. Т о d о k о r o, E. T a n i z a w a, Chem. Eng. (Japan),
15, 164 (1951).
60. R. G а у 1 о r, H. R. С. Р r a 11, Trans. Inst. Chem. Engrs. (London) 29,
110 (1951); 31, 70, 78 (1953); 35, 267 (1957).
61. С E. Wicks, R. B. Beckmann, A. I. Ch. E. Journ., 1, 426 (1955).
62. S. E. Markas, R. B. Beckmann, A. I. Ch. E. Journ., 3, 223 (1957).
63. R. G а у 1 о r, N. W. Roberts, H. R. C. Pratt, Trans. Inst. Chem.
Engrs. (London), 31, 57 (1953).
64. J. B. Lewis, I. Jones, H. R. C. Pratt, Trans. Inst. Chem. Engrs.
(London), 29, 126 (1951).
65. J. H. Ballard, E. L. Piret, Ind. Eng. Chem., 42, 1088 (1950).
66. H. L. С h i n, M. Ch. E. Thesis, New York Univers., 1956.
67. J. W Crawford, С R. W i 1 k e, Chem. Eng. Progr., 47, 423 (1951).
68. E. H. Hoffing, F. J. Lockhart, Chem. Eng. Progr., 50, 94 (1954).
69. G. L. Jacques, J. E. Cotter, T. Vermeulen, U. S. Atomic Energy
Comm UCRL-8658 (1959).
70. A. P. Colburn, D. G. Welsh, Trans. Am. Inst. Chem. Engrs., 38, 179
(1942).
71. H. R. С Pratt, A. S. White, Chem. a. Ind., 1952, 358.
72. T. Vermeulen, N. N. Li, J. S. Moon, Т. М i у а и с h i, paper
presented to Am. Inst. Chem. Engrs., New Orleans, February, 1961.
73. L. E. Johns. R. B. Beckmann, paper presented to Am. Inst. Chem.
Engrs, New Orleans, February, 1961.
74. I. Leibson, R. B. Beckmann, Chem. Eng. Progr., 49, 405 (1953).
75. G. L. Jacques, T. Vermeulen, U. S. Atomic Energy Comm.
. . UCRL-8029 (1957).
Колонны с перфорированными
(ситчатыми] тарелками
76. P. J. Harrington, пат. США 1943822, 1934 г.; пат. СССР 21725,
1941 г.
77. G. R. Grunewald, F. J. Pierce, пат. США 2647855-6, 1953 г.; Chem.
Eng., 60, № 11, 346 (1953).
78. I. W. Hump h er ey, Ind. Eng. Chem., 35, 1062 (1943).
79. D. A. L i s t e г, пат. США 2054432, 1936 г..
80. F. D. Mayfield, W. L. Church, Ind. Eng. Chem., 44, 2253 (1952).
81. M. С Rogers, E. W. T h i e 1 e, Ind. Eng. Chem., 29, 529 (1937).
82. F. D. Fugua, Petrol. Proc, 3, 1050 (1948).
83. H. A. Kjellman, Sewage a. Ind. Wastes, 27, 854 (1955).
84. F. С Koch, пат. США 2401569, 1946 г.
85. F. H. Garner, S. R. M. Ellis, J. W. Hill, A. I. Ch. E. Journ., 1,
185 (1955); Trans. Inst. Chem. Engrs. (London), 34, 223 (1956).
86. R. Bussolari, S. Schiff, R. E. Treybal, Ind. Eng. Chem., 45,
2413 (1953).
87. С J. Major, R. R. H e r t z о g, Chem. Eng. Progr., 51, № l, 17 (1955).
605
88. С. Pyle, A. P. Colburn, H. R. Duffey, Ind. Eng. Chem., 42, 1042
(1950).
89. S. Fujita, E. T a n i z a w а, С. К а п g, Chem. Eng. (Japan), 17, 111
(1953).
90. S. K. N an d i, S. K. Gh osh, J. Indian Chem. Soc, Ind. a. News Ed., 13,
93, 103 (1950).
91. R. W. Moult on, J. E. Walkey, Trans. Am. Inst. Chem. Engrs., 40,
695 (1944).
92. S. B. Row, J. H. Koffolt, J. R. W i t h г о w, Trans. Am. Inst. Chem.
Engrs., 37, 559 (1941).
93. W.R.Lewi s, Jr., Ind. Eng. Chem., 28, 399 (1936).
94. J. Alert on, B. O. Strom, R. E. Treybal, Trans. Am. Inst. Chem.
Engrs., 39, 361 (1943).
95. F. H. Garner, S. R. M. Ellis, D. W. Fosbury, Trans. Inst. Chem.
Engrs. (London), 31,348 (1953).
96. W. M. Goldberger, R. F. В e n e n a t i, Ind. Eng. Chem., 51, 641
(1959).
97. R. E. Treabal, F. E. Dumoulin, Ind. Eng. Chem., 34, 709 (1942).
Роторно-кольцевые экстракторы
98. M. W. Davis, J. E. Weber, Ind. Eng. Chem., 52, 929 (1960).
99. R. W. S pence, R. J. Street on, пат. США 2742348, 1956 г.
100. G. I. Taylor, Phil. Trans. Roy. Soc, 223A, 289 (1923).
101. R. S p e n с e, R. J. W. S t г е с t о n, Atomic Energy Research Establ.
(G. Brit.) AERE-C/R-933 (1952).
102. J. D. Thornton, H. R. С Pratt, Trans. Inst. Chem. Engrs.
(London), 31, 289 (1953); Atomic Energy Research Establ. (G. Brit.)
AERE-C/R-906 (1953).
103. J. F. Short, G. H. Twigg, Ind. Eng. Chem., 43, 2932 (1951).
104. R. L. M а у с о с к, пат. США 2474807, 1949 г.
Роторно-днсковые экстракторы
105. G. H. Reman et al., Proc. 3rd World Petrol. Congr., Hague, Sect., Ill,
121 (1951); пат. США 2601674, 1952 г.; 2729545, 1956 г.; 2912310, 1959 г.
106. G. H. Reman, R. В. Olney, Chem. Eng. Progr., 51, 141 (1955)v
107. G. H. Re m an, J. R. Van de V u s s e, Genie chim., 74, 106 (1955).
108. G. H. Reman, Joint Symp., The Scaling-up of chemical Plant and
Processes, London, 1957, p. 26.
109. E. Y. Kung, R. B. Beckmann, A. I. Ch. E. Journ, 7, 177(1961).
110. D. H. Logsdail, J. D. Thornton, H. R. С Pratt, Trans. Inst.
Chem. Engrs. (London), 35, 301 (1957).
111. С P. Strand, R. B. Olney, G. H. Acker man, A. I. Ch. E. Journ,
8, 252 (1962).
112. V. Kalab, K- V n u k о v a, J. R e p i s, Chem. listy, 51, 1249 (1957).
113. T. Takahashi, R. Suzuki, T. Yasunaga, Nenryo Kyokaishi, 37,
547 (1958).
114. H. Vermijs, H. Kramers, Chem. Eng. Sci, 3, 55 (1954).
115. Z Ziolkowski, J. Naumowicz, Chem. Stosowana, 2, 457 (1958),
Экстракторы типа «Mlxco»
116. J. Y. Oldshue, J. H. Rusliton, Chem. Eng. Progr, 48, 297 (1952).
117. J. Dykstra, B. H. Thompson, R. J. С louse, Ind. Eng. Chem, 50.
161 (1958).
118. R A Gustison, R. E. Treybal, R. С. С а р р s, Chem. Eng. Progr.
Symp. Ser.
119. W. Eguchi, S. N a gat а, Т. Мог iy a, Kagaku Kogaku, 22, 483 (1958).
606
120. S. Nagata, W. Eguchi, Mem. Fac. Eng. Kyoto Univ., 19, 102(1957).
121. S. Nagata, W. Eguchi, T. Yokoyama, K. Maeda, Chem. Eng.
(Japan), 17, 20 (1953); 20, 2 (1956).
122. F. H. Garner, S. R. M. Ellis, J. H. Hughes, Dechema Monograph,
32, 199 (1959).
Экстракторы Шайбеля
123. E. G. Scheibel, Chem. Eng. Progr, 44, 681 (1948); пат. США 2493265,
1950 г.
124. E. G. Scheibel, A. E. К а г r, Ind. Chem, 42, 1048 (1950).
125. A. E. Karr, E. G. Scheibel, Chem. Eng. Progr. Symp. Ser, 50, № 10,
73 (1954).
126. E. G. S с h e i b e 1, A. I. Ch. E. Journ, 2, 74 (1956).
127. E. G. Scheibel, пат. США 2850362, 1958 г.; Брит. пат. 791025, 1958 г.
Пульсационные •кстракторы
128. W. J. D. V а п D i j с к, пат. США 2011186, 1935 г.
129. А. С. Jealous, H. F. Johnson, Ind. Eng. Chem, 47, 1159 (1955).
130. J. D. Thornton, Chem. Eng. Progr. Symp. Ser, 50, № 13, 39 (1954);
Nuclear Eng, 1, 204 (1956); пат. США 2818324, 1957 г.
131. W. L. Griffith, G. R. J a s п е у, Н. Т. Т u p p e r, U. S. Atomic
Energy Comm. AECD-3440 (1952).
132. A. C. Jealous, E. Lieberman, Chem. Eng. Progr., 52, 366 (1956).
133. J. A. Williams, D. J. Little, Trans. Inst. Chem. Engrs. (London),
32, 174 (1954).
134. С J. Billerbeck, J. F a r q u h a r, R. С Re id, J. С Breese,
A. S. Hoffman, Ind. Eng. Chem, 48, 183 (1956).
135. T. S h i г о t s u к a, N. Honda, H. О у a, Kagaku Kogaku, 22, 187 (1958).
136. J. D.Thornton, Brit. Chem. Eng, 3, 247 (1958).
137. G. Feick, H. M. Anderson, Ind. Eng. Chem, 44, 404 (1952).
138. W. A. Chantry, R. L. von Berg, H. F. Wiegandt, Ind. Eng.
Chem, 47, 1153 (1955).
139. А. С r i с o, Genie chim, 73, 57 (1955).
140. G. L. Richardson, paper presented to Am. Inst. Chem. Engrs, New
Orleans, February, 1961.
141 W. M. Goldberger, R. F. Benenati, Ind. Eng. Chem, 51, 641
(1959).
142. B. W. Mar, A. L. В abb, Ind. Eng. Chem, 51, 1011 (1959).
143. D H. L о g s d a i 1, J. D. T h о г п t о n, J. Nuc. Energy, B: Reactor Tech-
nol, 1, 15 (1959).
144. G. Sege, F. W. W о о d f i e 1 d, Chem. Eng. Progr, 50, 396 (1954).
145. R. B. Edwards, G. H. Beyer, A. I. Ch. E. Journ, 2, 148 (1956).
146. L. D. Smoot, B. W. Mar, A. L. В a b b, Ind. Eng. Chem, 51, 1005
(1959).
147. D. H. Logsdail, J. D. Thornton, Trans. Inst. Chem. Engrs.
(London), 35, 331 (1957).
148. R. H. Sobotik, D. M. H i m m e 1Ы a u, A. I. Ch. E. Journ, 6, 619
(1960).
149. A. E. Karr, A. I. Ch. E. Journ, 5, 446 (1959).
150. N I sa ac, R. L. DeWi tt e, A. I. Ch. E. Journ, 4, 498 (1958); Dechema
Monog, 32, 218 (1959).
151 A Guyer, A. Cuyer Jr., К. М a u 1 i, Helv. chim. acta, 38, 790, 955 (1955).
152. J. A. Carver, J. R. Felix, С H. Holder, R. B. Long, пат. США
2775543 1956 г.
153. M. R. Fensk'e, R. В. Long, пат. США 2667407, 1954 г.; Chem. Eng
Progr, 51, 194 (1955); Ind. Eng. Chem, 53, 791 (1961).
607
154 D. De fives, J Durandei, Y. L. G 1 a d e 1, Rev. Inst, tranc. petrole,
11, 231 (1956). '
155. J. D Thornton, Trans. Inst. Chem. Engrs. (London), 35, 316 (1957).
156. F. W. Wood fie Id, G. S e g e, Chem. Eng. Progr. Symp. Ser., 50, № 13,
14 (1954). " ■-'
Центробежные экстракторы
157. W. J. Podbielniak, пат. США 2044996, 1935 г.; 2670132, 1954 г и др.
158. W. J. P о d b i e 1 п i a k, Chem. Eng. Progr., 49, 252 (1953).
159. F. M. Jacobson, G. H. Beyer, A. 1. Ch. E. Journ., 2, 283 (1956).
160. H. R. Kaiser, С M. Doyle, J. Am. Oil Chem. Soc, 37, 4 (I960).
161. W. J. Podbielniak, A. M. Garvin, H. R. Kaiser, J Am Oil
Chem. Soc, 36, 238 (1959).
162. H. R. Kaiser, Sewage a. Ind. Wastes, 27, 311 (1955).
163. С M. Doule, E. R a u с h, Petrol. Engr., 27, № 5, C49 (1955).
164. N. В arson, G. H. Beyer, Chem. Eng. Progr., 49, 243 (1953).
165. M. Alexandre, P. Gentilini, Rev. Inst, franc, petrole, 11, 389
(1956).
166. D. W. Anderson, E. F. L a u, Chem. Eng. Progr. 51, 507 (1955)
167. С. С о u t о г, пат. США 2036924, 1936 г.
168. H. Eisenlohr, Ind. Chem., 27, 271 (1951); Chem. Ing. Techn., 23, 12
(1951).
169. H. Eisenlohn, A. Scharlan, Pharm. Ind., 17, 207 (1955), -
НЕКОТОРЫЕ ВОПРОСЫ ЭКОНОМИКИ
ПРОЦЕССОВ ЭКСТРАКЦИИ
Вопросы полного технико-экономического
обоснования различных химических
производств, включающих также процессы
жидкостной экстракции, подробно
рассмотрены в литературе (см. например1-2). Эта
большая и сложная проблема лишь в
кратких чертах изложена ниже.
После того как общая эффективность и
экономическая целесообразность того или
иного проектируемого процесса
установлена (что часто достаточно выяснить в первом
приближении), возникает необходимость в
подробном технико-экономическом расчете
отдельных стадий процесса для
определения оптимальных условий его проведения.
Применительно к экономике процесса
экстракции и экстракторов эти вопросы также
достаточно сложны.
Обычно для определения оптимальных
условий экстракционных процессов и
наилучшего их аппаратурного оформления
необходимо большое число различных
технико-экономических расчетов. Однако в
большинстве можно при разумных допущениях
сравнительно просто приближенно найти
оптимальные условия процесса. Подробный
расчет технико-экономических показателей
процесса в условиях, близких к
определенным в приближенном расчете, позволяет
значительно быстрее найти действительно
оптимальные условия проведения процесса.
ПРИМЕНЕНИЕ ЭКСТРАКЦИИ
ДЛЯ ИЗВЛЕЧЕНИЯ РАСТВОРЕННОГО
ВЕЩЕСТВА
Жидкостную экстракцию чаще всего
применяют для выделения определенного
вещества из раствора. Схема простейшего
экстракционного процесса, предназначенного
39 Р. Е Трейбал
для этого, показана на рис. 307. Для упрощения задачи
рассмотрим процесс экстракции без флегмы при малой взаимной
растворимости экстрагента и растворителя исходного раствора.
Стоимость экстракционного процесса в значительной степени
определяется выбором экстрагента и типом экстрактора
(смеситель-отстойник, насадочная колонна и др.).
Задача отыскания оптимальных условий осуществления
экстракционного процесса значительно усложняется, если
одновременно проводить подбор аппарата, экстрагента и
технологических показателей процесса. Гораздо проще установить
наилучшие условия проведения процесса для экстрактора данного типа
и данного экстрагента. Повторяя расчет для экстракторов
других типов и различных экстрагентов, применение которых
возможно в данном процессе, можно найти оптимальный вариант
его осуществления.
Если объемный расход исходного раствора и концентрация
извлекаемого вещества в нем известны, а также выбраны экстр-
агент и тип экстрактора, то годовая стоимость экстракционного
процесса определяется суммой следующих затрат3:
C7, = I-fll + llI + lV + V-r-VI (XII, 1)
где Ст — полная годовая стоимость экстракционного процесса;
I — годовая стоимость экстрактора;
II — годовая стоимость неизвлеченяого при экстракции
вещества;
III — годовая стоимость процесса регенерации экстрагента
из экстракта;
IV — годовая стоимость процесса регенерации экстрагента
из рафината;
V — годовая стоимость потерь экстрагента;
VI — годовая стоимость трудовых затрат.
Каждая из перечисленных статей расходов определяется
указанными ниже факторами, подлежащими оптимизации для
определения минимальной общей стоимости процесса экстракции:
Статья
расхода,
Показатель процесса зависящая
от данного
показателя
1. Конструкция экстрактора I
2. Концентрация извлекаемого вещества в рафинате, с„ I, II, III
3. Концентрация извлекаемого вещества в рециркули-
рующем экстрагенте, с„ I, III
4. Объемное соотношение экстрагента и исходного
раствора, RE I, III, V
5. Концентрация экстрагента в конечном продукте
экстракции (экстракте) III, V
6. Концентрация экстрагента в конечном рафинате , . IV, V
610
7. ФлегмОвое число при регенерации экстрагента из
экстракта дистилляцией, или число ступеней при
регенерации растворителя экстракцией, или
соответствующая им величина, если применяют какой-
либо другой метод регенерации экстрагента .... III
8. Флегмовое число при регенерации экстрагента из
рафината дистилляцией или соответствующая ему
величина при регенерации экстрагента из рафината
другими методами IV
Годовую стоимость трудовых затрат принимают постоянной,
не зависящей от этих факторов. Оптимальная концентрация в
рафинате cR обычно очень незначительна, даже при извлечении
относительно малоценных веществ, поэтому количество
экстрагента, содержащееся в выходящем из экстрактора рафинате,
практически определяется растворимостью экстрагента и
растворителя исходного раствора. Эту величину, если экстрагент
выбран, при проектировании изменить нельзя. Следовательно,
оптимальные значения факторов, приведенных под номерами 6
и 8, можно находить отдельно, методами, разработанными для
процессов дистилляции.
Концентрация экстрагента в конечном продукте экстракции
определяется требованиями, предъявляемыми к качеству
продукта, и, как правило, очень мала. В дальнейшем будем
предполагать, что потери экстрагента с конечным продуктом и с ра-
финатом (после регенерации экстрагента) пренебрежимо малы
Для нахождения минимальной общей стоимости процесса
необходимо представить каждую из статей расхода в
уравнении (XII, 1) в виде алгебраической функции от определяющих
ее переменных, взять частные
производные от величины Ст
по каждой из этих переменных
и приравнять их нулю.
Совместное решение получающихся
в результате уравнений даст
возможность найти
оптимальные величины переменных.
В некоторых случаях,
однако, к процессу экстракции
предъявляются
дополнительные требования, поэтому такой
метод нахождения оптимума
оказывается не совсем
удовлетворительным. Так, при
извлечении урана жидкостной
экстракцией (или другими
методами) из щелоков количество
неизвлеченного урана не
должно превышать определенной
Ш
г~
,Ж
\Ш
I Пи
* 1—«—' с
Рис. 307. Общая схема процесса
экстракции для выделения вещества
из раствора:
/ — экстрактор; 2 — колонна регенерации
растворителя из экстракта; 3 —колонна
регенерации растворителя нз рафината; / —
исходная смесь; // — экстрагент;/// — экстракт;
IV— рафинат; V — свежий экстрагент;
VI — регенерированный экстрагент; VII —
извлеченное вещество (конечный продукт
процесса); VIII — рафинат после регенерации
экстрагента.
39*
611
величины, которая может быть ниже оптимальной с
экономической точки зрения. Аналогичная ситуация часто возникает при
экстракции некоторых вредных примесей, например при
очистке от фенолов сточных вод коксохимических заводов. Влияние
таких ограничений на определение экономически оптимальных
условий проведения процесса рассмотрено Хаппелем '.
Если для данной системы зависимость числа ступеней от
объемного соотношения фаз RE можно определить только
графическими расчетами на фазовой диаграмме (т. е. нельзя
выразить алгебраически), то следует найти отдельно оптимальные
значения возможно большего числа других факторов,
определить на основе этих значений графически число ступеней и
построением зависимости Ст от RE определить оптимальное
объемное соотношение фаз, соответствующее минимальной
стоимости процесса. Однако даже в подобных случаях часто можно
предварительно произвести приближенный расчет на основе
упрощающих допущений. Знание примерных оптимальных
величин объемного соотношения RE и числа ступеней п в
значительной степени упрощает дальнейшие, более точные расчеты.
Уравнения, приводимые ниже, получены при следующих
допущениях:
Экстрагент и растворитель исходного раствора практически
нерастворимы друг в друге.
Коэффициент распределения постоянен.
Экстрактор представляет собой аппарат ступенчатого типа:
смесительно-отстойный каскад, тарельчатая колонна и др., т. е.
аппарат такого типа, эффективность которого можно выразить
через эффективность ступени.
Общая эффективность ступени экстрактора Е0 постоянна.
Потери экстрагента с конечным продуктом и рафинатом
после регенерации пренебрежимо малы.
Если эти допущения не отвечают действительности, то
результаты расчета можно рассматривать только как грубо при- i
ближенные.
Оптимальные условия экстракционного процесса при
регенерации экстрагента из экстракта дистилляцией. Кроме
перечисленных выше допущений далее предполагаем, что
относительная летучесть растворителя и экстрагируемого вещества
постоянна. Метод нахождения оптимальных условий регенерации
экстрагента из экстракта рассмотрен Хаппелем '.
I. Годовая стоимость экстрактора. Стоимость экстрактора
составляет:
1--|-^- (XII, 2)
СОЕ
где число теоретических ступеней пЕ определяется уравнением
(VI, 126). Если пользоваться объемными концентрациями и объ-
612
емными скоростями фаз, число теоретических ступеней можно
рассчитать по уравнению
п
(CF-CSlmE\lx 1__)
\cR-cs/mE)\ mERE)
i
Е lg mpR
E E (XII, 3)
E SE
в котором произведение mERE представляет собой фактор
экстракции.
Годовые расходы на одну ступень рассчитываются с
помощью выражения:
СЕ = CVE [Т + Ь)Е + (РА + РР) WH <ХП' 4>
Величина CVE выражает стоимость основного оборудования,
к которому относятся, например, смесители, отстойники,
насосы, перемешивающие устройства. Если стоимость
вспомогательного оборудования (инструменты, трубопроводы и пр.)
равна р—1, то сумма 'капитальных затрат на одну ступень
выражается величиной CVEp. Приблизительные значения величины р
можно найти в литературе '• 3>4.
В уравнение (XII, 4) введены обозначения: Y — срок
службы оборудования; Ь — эксплуатационные расходы (в долях от
стоимости основного оборудования); Ра + Рр— мощность
мешалок и насосов (если таковые имеются); W — стоимость энергии
и Н — время работы оборудования в течение года.
II. Стоимость неэкстрагированного вещества. Если
экстрагент и растворитель исходного раствора практически взаимно
нерастворимы, то количество неэкстрагированного вещества,
теряемого с рафинатом, равно qFcR. При цене F руб/кг годовая
стоимость неэкстрагированного вещества составит
II = FqEcRH (XII, 5)
III. Стоимость регенерации экстрагента из экстракта.
Годовая стоимость процесса дистилляции определяется
следующим выражением '■3:
ш_ CDnD°Q+r)WY+b)n
EDGD
, C.O(l-f r)(p[Y + b)n
_, h -r >\fi т iD +ChcO0+r)H (XII, 6)
Первое слагаемое правой части уравнения (XII, 6)
выражает стоимость дистилляционной аппаратуры, второе
слагаемое—стоимость теплообменной аппаратуры и, наконец, третье
слагаемое — стоимость нагревающих и охлаждающих агентов.
Уравнение (XII, 6) можно переписать в виде:
III = АВ (XII, 7)
613
Здесь
В=0(1 + г) (XII, 9)
Число теоретических ступеней nD в этих уравнениях принято
равным:
"0 = V"0,mln (ХИ,Ю)
Здесь «о, min— число ступеней при полном возврате флегмы,
величину у при оптимальном флегмовом числе можно
определить, например, по методу Джиллилэнда 5; обычно она близка
к 2,5 '. Величина По, mm определяется в свою очередь известной
зависимостью Фенске, которую можно написать в виде:
■fe) , Ч1^) ,
т i 1 - (XII.lt)
-.— lg a ' lga \ > /
Рассмотрим случай, когда экстрагент является легколетучим
компонентом экстракта. Концентрация экстрагента в кубовом
остатке, представляющем собой в датаном случае конечный
продукт экстракционного процесса, определяется требованиями,
предъявляемыми к качеству последнего, и во всяком случае
должна быть небольшой величиной. Поэтому можно написать
lg
xd \
-хр)
const (XII, 12)
D, rain lg a
Поскольку потери экстрагента с кубовым остатком
практически отсутствуют
хо _ (<fPRiPs/Ms)/0 psME
\-xD (qpREcslME)jO csMs
9sme
(XII, 13)
c„M„
»„ „,„ = r^-^—I- const (XII, 14)
D, rain In a
Тот же результат можно получить3, если легколетучим
компонентом является экстрагируемое вещество.
Количество получаемого дистиллята, если экстрагент
является легколетучим компонентом экстракта и потери его с
конечным продуктом пренебрежимо малы, составляет:
0 = М^_ (XiU5)
ms
614
Если количество экстрагируемого
компонента (кмоль/ч), поступающего на
дистилляцию экстракта, составляет
и_ 4f(cf-°r)
Мг
(хн, 16) У
то мольная доля экстрагента в
экстракте выражается уравнением
О
XE9PPS
~—ц + и- ums+re<jfPs (ХП'17)
При этом учитывается, что
концентрация cR обычно пренебрежимо мала
по сравнению с cF.
На рис. 308 показана диаграмма
процесса ректификации экстракта. Если
относительная летучесть равна а, то
/ г
</ = •
(XII, 18)
Рис. 308. Ректификации
экстракта (в случае,
когда экстрагент является
легколетучим
компонентом):
/ — линия равновесия; 2 — рз
бочая линия при минимальном
флегмовом числе.
l-fz(a—I)
и уравйение рабочей линии при минимальном флегмовом числе
принимает вид:
Лп1п _ I — У
_Ь
\—г
''rain — '
l+rmIn-T^ <XII'19>
Решая совместно уравнения (XII, 18) и (XII, 19), получаем:
1 (XII, 20)
(XII, 21)
равный
г (а— 1)
Действительное флегмовое число
Г = P/"mln
избытка флегмы, обычно
где р — коэффициент
1,05—1,30!.
Уравнения (XII, 15) — (XII, 21) дают возможность определить
величину В.
В случае, когда экстрагент является легколетучим
компонентом, имеем:
В-
<">+->-^(ч-^г)+^г <*»■->
Если легколетучий компонент — экстрагируемое вещество, то
B_0(l+r).y(l+_iT)+JS^. (XII.23)
IV. Стоимость процесса регенерации экстрагента из рафи-
ната. Как указывалось выше, расходы на регенерацию
экстрагента из рафината можно найти независимо от других величин,
характеризующих процесс экстракции. Эта статья расходов не
615
влияет на оптимизацию всего процесса в целом. Для
определения стоимости регенерации растворителя из рафината
дистилляцией можно рекомендовать методы, предложенные Хап-
пелем '
V. Стоимость потерь экстрагвнта. Для смесительно-отстой-
ного экстрактора потери экстрагвнта можно принять
пропорциональными его объемному расходу и числу ступеней:
у= nBlFRElSH (XII, 24)
ЕОЕ
где / — доля потерь экстрагвнта, приходящаяся на одну
ступень, от общей величины его расхода.
Для колонных экстракторов потери экстрагвнта можно
принимать пропорциональными только его объемному расходу:
V = qpRB!'SH (XII, 25)
где /' —потери экстрагвнта, доли от его расхода.
Влияние концентрации cR экстрагируемого вещества в
рафинате. Концентрация сн экстрагируемого вещества в рафинате
влияет (см. стр. 610) на величину расходов I, II, III [уравнение
(XII, 1)]. Стоимость регенерации экстрагента из экстракта
(III) зависит в некоторой степени от cR, так как с изменением
сн меняется и концентрация в экстракте. Однако концентрация
экстрагируемого вещества в рафинате обычно настолько мала,
что даже при изменении ее в несколько раз величина III
практически не меняется. Поэтому можно считать, что концентрация
в рафинате сказывается только на статьях расходов I и II.
Следовательно
dcR dcR ^ dcR EQE dcR ^ f
Подставляя величину пЕ из уравнения (XII, 3), получаем:
СЕ /1-VW
сЕ (l-VmE<
ЕОе\ lUmERE
X
.°Б ' " " ' (gf-Vmg) =Fq н
l(CF- CslmE) (] - 4mERE)KCR - CsimE) + l/mERE] (CR ~ C A)2 F
(XII, 27)
Влияние концентрации cs экстрагированного вещества в
регенерированном экстрагенте. Согласно предыдущему (см.
стр. 610), имеем:
i^ = JL+^- = 0 (XII, 28)
dcs dcs ^ dcs
Если взаимная растворимость экстрагента и растворителя
исходного раствора мала, то количество экстрагента, получае-
616
мое при регенерации его из рафината, невелико. Вместе с тем
в промышленных условиях потери экстрагента и, следовательно,
количество свежего экстрагента, вводимого в процесс, также
незначительны. Поэтому можно принять, что величина cs равна
концентрации экстрагируемого вещества в экстрагенте,
выходящем из колонны ректификации экстракта. Тогда уравнение
(XII, 28) принимает вид:
дСт Св дпв Cny(p/Y + b)nB dnD min
-rJ-=-FrM-^-~+ L r §^- = 0 (XII, 29)
dcs Eoe dcs Ed°d dcs
Подставляя соответствующие величины из уравнений (XII, 3)
н (XII, 14), получаем:
СЕ (l~llmERE),,
ЕОе\ lnmERE Г
у [(CF-CslmE)-{CR-CSlmE)\
mE[(CF~ Cs/mE)(l -llmERE)l(CR- Cs/mE)+VmERE}(CR- CSlmE?
D D J , =0 (XII, 30)
CSED°D]na
Если принять, что в уравнении (XII, 30) величина
(сд—cglmE) мала по сравнению с cF—cs/mE, то совместное
решение уравнений (XII, 27) и (XII, 30) дает возможность
определить оптимальную величину св:
mEyCD{PIY+b)DB
-S, опт. ED0DFqpH In a
Соответственно оптимальная величина
(XII, 31)
CR, опт. VF "■ S, опт.
= (С„ — С
К, ипт.
где
/тв) (1 "**") 0 ± Y7) + -^ (хц, 32)
_ 1£м
(CF-CS, опт>я) F1PHE0E(mERE- l) 1П mERE
J^+TT— ,, 4g„ „к* ,„ о _,ч,я, о (Х».33)
При mERE>] (что обычно наблюдается) следует брать
отрицательное значение У^.Если фактор экстракции меньше
единицы, то необходимо брать + \ J. Подставляя полученные
уравнения в уравнение (XII, 3), находим оптимальное число
теоретических ступеней:
_ ig [1-2/(1 ±/7)] .
"Я. опт. ~ W^R~E ( ' }
Число теоретических ступеней, вычисленное по уравнению
(XII, 34), будет оптимальным при данном объемном
соотношении фаз. В общем случае для нахождения числа теоретических
617
Ступеней, соответствующих наименьшим суммарным расходам
Ст, в уравнение (XII, 34) следует подставлять оптимальное
значение Re-
В частном случае, когда оптимальное значение тЕ^Е=1,0, исходя из
уравнения (VI, 128), получаем:
R, ОПТ.
CE{cP—cSi0mJmE)
10,5
£оЛЯ
CS, опт.
п
Е, опт.
\сР -cs,omJmE)EOEFqFH
10.5
— 1
(XII, 35)
(XII, 36)
Величину Cs, оит. определяют, как и ранее, по уравнению (XII, 31).
Из уравнений (XII, 10), (XII, 14) *, (XII,31) и (XII,32)
можно найти минимальное число теоретических тарелок,
необходимое для ректификации экстракта:
■2,3psMEEDGDFqpHklga
MsmEyCD(p/Y + b)DB
lg
D, mln
Iga
— 1
(XII, 37)
В этом уравнении k — мольное отношение количества
экстрагируемого вещества к количеству экстрагента в конечном
продукте. Следует отметить, что Яг>, min практически не зависит
от Re-
Влияние объемного соотношения фаз RE. Для нахождения
оптимальной величины RE имеем уравнение (см. стр. 610)
дСт ЦпЕСЕ/Е0Е) | dill
dV
dRr
dRr
дЯ^
dRr
= 0
(XII, 38)
В общем случае Е0Е и СЕ зависят от RE\ поэтому взять
первую производную в уравнении (XII, 38) формальным
дифференцированием не представляется возможным. Оптимальное
значение RE лучше определять графически, изображая зависимость
.суммы расходов (I + III + V), рассчитанных при оптимальных
значениях пЕ, cR и cs, в функции от RE. Положение минимума
на этой кривой определяет RE,om.- Приблизительное
аналитическое решение можно получить, вводя дополнительные
упрощения. Для смесительно-отстойных экстракторов Е0е обычно
находят в пределах 0,9—1,0, причем оно мало зависит от RE.
Поэтому можно принять, что Е0е — величина постоянная (для
экстракторов колонного типа величину Е0Е следует определять
подбором). Известно3, что величина энергетических затрат для
смесительно-отстойных аппаратов мала по сравнению с общей
годовой стоимостью оборудования. Это справедливо и для
других экстракторов с вводом энергии извне, за исключением, ве-
Независимо от того, какой компонент легколетучий.
616
роятно, пульсационных колонн. Поэтому величина Св в
уравнении (XII,4) приблизительно равна CVE(p/Y+b)E. Стоимость
технологического оборудования, включая и экстракторы,
обычно можно представить в виде степенной функции от
производительности3- 4:
CVE = KE4Al+RE)g <XI1'39)
Если известна стоимость ступени экстракции CYE\ при
некотором объемном соотношении фаз RE, то при произвольном RB
величина СЕ выражается уравнением
cE=cVEwr+b)E-
Ce\CveICve\
(XII, 40)
Следует отметить, что если определять число ступеней по
уравнению (XII, 34), то при умеренных изменениях RE
произведение nERE остается почти постоянным3. Этого не происходит,
если пЕ рассчитывать по уравнению Кремзера при постоянных
cR и св.
При таких упрощениях можно определить первый член
правой части уравнения (XII, 38):
1 ЧпеСе)^ -пе\йе\Се\
%е dRB EOE(l+Rmf
i + (i-g) у
(^я+1)1-^4
(XII, 41)
Выше уже было отмечено, что величина nDt mln мало меняется
при объемных соотношениях фаз, имеющих практическое
значение. Можно считать поэтому, что величина А также мало
зависит от RE и ее можно рассчитать [с помощью уравнений
(XII, 22), (XII, 23), (XII, 37) и (XII, 8)] при некотором
произвольном значении объемного соотношения. Исходя из этого и
обозначая А через Аи можно определить второй член в правой
части уравнения (XII, 38):
dill Л^ р^е
"ЖГ = М„(а-1)
(XII, 42)
Если легколетучим компонентом экстракта является экстр-
агент, то велична е = а + р—1; если же легколетучий
компонент— экстрагируемое вещество, то е=1. Так как произведение
nERE принято величиной постоянной, производная d\/dRE равна
нулю для смесительно-отстойных экстракторов; для колонных
аппаратов d\ldR^ = qFl'SH. Подставляя полученные значения в
уравнение (XII, 38), находим выражение для RE,om-'-
l+(l-g) Яр on
ХЕов(*т + 1)е
ХЕ
ОЕ
\^Е, опт. Т1) *Е. опт.
nElRElCEl
nElRElf<E<lf(P/Y+b)E
(XII, 43)
619
В уравнение (XII,43) введены обозначения:
X:
х =
(для смесителей-отстойников)
(XII, 44)
Ms(a— 1)
A.q p e
М I n ~^~ Qf^SH (для колонных аппаратов) (XII, 45)
Решение уравнений (XII, 43) — (XII, 45) относительно RE,our.
упрощается, если пользоваться графиком на рис. 309, который
можно применять для g" = 0,3—0,8. Так как при выводе
уравнения (XII, 43) применяли целый ряд допущений, результаты,
получаемые с помощью этого уравнения, необходимо уточнять в
последующих расчетах. В большей части случаев общая
стоимость процесса экстракции очень мало, изменяется в
зависимости от объемного соотношения фаз вблизи от Re, опт.-
При определении оптимальных условий проведения
экстракционных процессов рекомендуется вести упрощенный расчет в
следующей последовательности:
!. Выбор объемного соотношения фаз REi. Обычно
оптимальная величина фактора экстракции находится в пределах 1—2.
2. Расчет эффективности ступени Е0Е, размеров и стоимости
одной ступени экстракционной аппаратуры при выбранном
значении REi.
3. Определение величин В, [уравнение (XII, 22) или (XII, 23)1,
«d, min [уравнение (XII, 37)] и Л: [уравнение (XII, 8)] при
выбранном объемном соотношении
фаз Rel
4. Расчет величины /4
[уравнение (XII, 33)]. В большей
части случаев членом cs, опт./тЕ
можно пренебречь; его можно
рассчитать по уравнению
(XII.31).
5. Определение nEi по
уравнению (XII, 34).
6. Определение RE, опт.
(рис. 309). Если Re, от.
окажется сильно отличным от REi,
то следует повторить расчет
при новом значении RE-
7. Определение
оптимальных величин nE, cr и са на
основе полученного выше
оптимального объемного соотноше-
IU
7
5
3
с:
0,5
0,2
т
^>Ч
.:: Чч...
ч
Ofil QObQI Oft 1 4 Г О 4/7 100
nE RE CE
ч ч Lt
Рис. 309. Приблизительное
определение оптимального объемного
соотношения экстрагента н исходного раст- , r /vn oi\
вора [Treybal, A. I. E. Ch. Journ., ния фаз [уравнения (XII, 31) —-
5,474(1959)]. У" 'V,T ',CI"'V,T ,Cl1
(XII, 34) или (XII, 35) и (XII, 36)].
620
Ё
J
qF \Ш
-/-
п
■ш
ж
Рис. 310. Общая схема процесса экстракции, служащего для выделения
вещества из раствора, в случае регенерации экстрагента вторичной
экстракцией:
/ — основная экстракция; 2 —вторичная экстракция; /—исходная смесь; // —экстрагент;
/// — свежий экстрагент; IV — экстракт; V — рафииат; VI — экстрагеит вторичной
экстракции; VII — конечный продукт процесса.
Другой порядок упрощенного расчета оптимальных условий
экстракции предложен Шайбелем6.
Оптимальные условия процесса экстракции при регенерации
растворителя (экстрагента) из экстракта экстракционными
методами. Схема такого экстракционного процесса показана на
рис. 310. Обычно начальная концентрация экстрагируемого
вещества в растворителе, служащем экстрагентом в процессе
регенерации, равна нулю, а конечная концентрация ср
фиксирована требованиями, предъявляемыми к конечному продукту, т. е.
не подлежит оптимизации. Кроме того, в дальнейшем при
рассмотрении стадии регенерации экстрагента будут приниматься
те же основные допущения, которые были приняты выше для
процесса экстракции, в том числе постоянство фактора
экстракции msRs-
Коэффциент распределения тв для вторичной экстракции
рассматривается равным отношению концентрации извлекаемого
вещества в растворителе, служащем экстрагентом в процессе
регенерации, к его концентрации в экстракте основной
экстракции при равновесии, a RB — соответственно равным объемному
^соотношению расходов экстрагентов на стадиях регенерации и
основной экстракции. В этом случае статьи расходов I, II, IV и
N\ определяются так же, как в процессе экстракции с
регенерацией экстрагента дистилляцией.
Стоимость процесса регенерации растворителя (экстрагента)
экстракта. При регенерации растворителя экстракцией имеем:
я„С,
III =
из
Здесь
'OS
lg
{ msRs ) msRs J
lg msRs
cs = cvs(-Y + b}s + (PA + pP)sWH
(XII, 46)
(XII, 47)
(XII, 48)
621
Величину (p/Y+b) можно считать одинаковой для основной
экстракции и для стадии регенерации экстрагента. На основе
материального баланса имеем:
Rs =
CF-°R
CPRE
R,
Следовательно
s
Ig
•1
У
CF~CR CP
CSRE Csm
\ms(CF~CR)'
[ CPRE \
S .
Ig
' l [ CF Cp\
.Cs\RE ms I.
\e - s F
g с R
LP^E
(XII, 49)
(XII, 50)
(XII, 51)
Таким образом, стоимость регенерации растворителя
(экстрагента) определяется материальным балансом процесса, если
известны оптимальные условия основной экстракции.
Стоимость потерь экстрагента. На основании уравнений
(XII, 24) и (XII, 25) стоимость потерь экстрагента составит:
для смесительно-отстойных экстракторов
ОЕ
OS
qpRElSH
для колонных экстракторов
V=qpREl,SH
(XII, 52)
(XII, 53)
Оптимальные значения величин cs, nE и ns. Для
определения оптимальной концентрации в рециркулирующем экстрагенте
cs таким же образом, что и ранее (см. стр. 617), можно
вывести уравнение
_ csmE
CS, опт = РдрНЕ05 In [ms {cp - cR)l cpRE] (X"'54)
Величиной cR в уравнении (XII, 54) часто можно пренебречь.
Оптимальную концентрацию в рафинате сн, опт. и оптимальное
число ступеней пЕ, опт. определяют с помощью уравнений
(XII, 32) — (XII, 34). В эти уравнения, однако, надо подставлять
значение cs, опт-, рассчитанное по уравнению (XII, 54).
Указанные уравнения совместно с уравнением (XII,51) для
определения ns, опт. (соответствующего оптимальным значениям cs и сн)
дают возможность рассчитать наивыгоднейшие величины cs, Cn
пЕ и ns при условии, что используемая в расчетах величина Re
также оптимальна. При любом другом объемном соотношении
фаз результаты расчета будут соответствовать наименьшим
общим расходам при данном RE.
622
Оптимальное объемное соотношение экстрагента и исходного
раствора RE. Как указывалось выше (см. стр. 618),
оптимальное значение RE определяется уравнением (XII, 38). Однако если
можно выразить входящие в него величины с помощью
упрощенных зависимостей, то даже конечные уравнения в данном случае
трудно представить в простой форме. Поэтому оптимальное
объемное соотношение фаз лучше находить графически,
изображая зависимость суммы расходов (I + III + V) в функции от RE
и определяя положение минимума. Для расчета стоимости
одной ступени процесса регенерации растворителя (экстрагента)
экстракцией можно пользоваться уравнением того же вида, что
и уравнение (XII, 39):
Ce-Ke\1f(1+Re)\s{t + ь)е (ХП, 55)
is
Cs = KsllsO+Rs)lS[T + b) = *.
/?„
> CR
Т+',
I IE
(XII, 55)
Поскольку конструкционный материал, из которого
изготовлены аппараты, применяемые на стадиях регенерации и
основной экстракции, может быть различным, величины КЕ и Ks, а
также g могут иметь разные значения для процессов
регенерации и основной экстракции. Уравнения (XII, 55) и (XII, 56)
можно использовать для расчета статей расходов I и III.
Влияние конструкции экстрактора. В литературе имеется
весьма ограниченное количество данных по сравнительной
оценке влияния конструкции экстрактора на экономику
экстракционного процесса. Данные о стоимости экстракторов также очень
/
0.8
ОМ
0,2
О
4,
с^
V
7
\
/
/
\s=0,5 |
1
0,2
J L
0,3 О/, 0,6 0,8 I
Диаметр смесителя Т, м
oft- о,в 1 г и в ю го so
Относительная объемная скорость
Рис. 311. Моделирование аппаратов с мешалками при постоянном
объемном соотношении экстрагента и исходного раствора s = 0 5 [Treybal,
А. 1. Ch. E. Journ., 5, 474 (1959)].
623
скудны. В большей части случаев, особенно для экстракторов
специальных конструкций (см. главу XI), необходимые
сведения можно получить только от заводов-изготовителей. Лишь
стоимость смесительно-отстойных экстракторов простейших
конструкций можно установить по опубликованным данным4 или
из перечней цен на оборудование, помещаемых в некоторых
технических журналах (например, в «Chemical Engineering»).
Представляет интерес исследование экономики простого сме-
сительно-отстойного экстрактора3, предпринятое для
определения оптимального метода перехода от лабораторных моделей к
аппаратам промышленных размеров. Обычно аппараты с
мешалками моделируют по принципу равенства времени
пребывания жидкостей в аппарате и энергии, вводимой мешалкой в
единицу объема жидкости, равного в данном случае объему
аппарата (при наличии геометрического подобия натуры и модели).
Если диаметры большого и малого аппаратов равны
соответственно 7\ и Г2, то условие моделирования можно выразить
равенством
Т. / п. \s i p. \s
-№=№
(XII, 57)
где при условии моделирования, указанном выше, s—l/3.
На рис. 311—313 приведены кривые зависимости эффектив-.
ности ступени от размера аппарата, рассчитанные на основе
уравнений, приведенных в главе X для случая, когда объемная
доля дисперсной фазы в аппарате составляет около 0,5. Кривые
построены для значений s, равных 0,5; 0,333 и 0,25. Первое
значение s соответствует моделированию по равным массовым
скоростям жидкостей.
Uj
1
цо
0,и
0,ч
0,2
0
у
'^
/ / /
\/
/
11
/
/
/
М.л?|
~\
о,г о,з о,и о,б о,з /
Диаметр смесителя Т,м
1__1 I I J 1 L
о,г 0,5 1 г 5 ш го so too гоо воо
Относительная объемная скорость
Рис. 312. Моделирование аппаратов с мешалками при постоянном
объемном соотношении экстрагента и исходного раствора s = 0,333 [Т г е у b a I,
A. I. Ch. E. Joum., 5, 474 (1959)].
624
0,2 0,3 Oft 0,6 0,8 1 1,5 -
Диаметр смесителя Г, м
i i i i i i 1 i i i i i ' '
oj о,г о,и 1 г 4 /о го ьо юогоошюоо
Относительная объемная скорость
Рис. 313. Моделирование аппаратов с мешалками при постоянном
объемном соотношении экстрагента и исходного раствора s — 0,25 [Т г е у b a I
A. I. Ch. E. Journ., 5, 474 (1959)].
Рассмотрим применение графика на рис. 311 для моделиро-
■; вания. Пусть на основе экспериментальных данных известно,
что при суммарной объемной скорости фаз 4,25 м3/ч в смесителе
'■. диаметром 0,428 м обеспечивается эффективность ступени
EMD=0,25.
На графике величинам ЕМВ=0,2Б и 7=0,428 м соответствует
\ точка С. При этом относительная скорость жидкостей
равна 1,95. При моделировании по уравнению (XII, 57) на
производительность 42,5 м3/ч при s=0,5 относительная объемная
скорость равна 10- 1,95=19,5 и ожидаемая эффективность ступени
EMD=0,94 (последняя определяется точкой D на кривой,
проходящей через точку С).
Диаметр аппарата для новой производительности (42,5 мя/ч)
? должен составлять 1,34 м. Аналогичным образом можно
производить моделирование с помощью графиков, приведенных на
■рис. 312 и 313. В рассматриваемой работе показано, что с
экономической точки зрения выгоднее принимать величину s в
^уравнении (XII, 57) равной примерно 0,25 (вместо '/з, как
^обычно), особенно при относительно высоких значениях эффек-
Зтивностей ступени смесительно-отстойного экстрактора и при
J Ьеличине фактора экстракции, близкого к единице (в области
ч оптимума). Часто экономически целесообразно иметь несколько
t большее число ступеней меньшей эффективности.
| По данным 1958 г. были установлены затраты на основное оборудова-
; вне экстракционной установки — стальной смеситель диаметром 0,457 м и
; отстойник (рассчитанный на время осаждения 10 мин), перемешивающее
./ устройство (мешалка, вал, опоры, взрывобезопасный двигатель, редуктор
I и пр.) мощностью 13,3 вт1(м31ч). Суммарная скорость фаз 2,83 мг\ч. Эти
затраты без учета насосов определяются зависимостью
С = 730 (Q/2,83)0'5205-1-0'365
40 Р. Е. Трейбал
625
При наличии одного насоса, приходящегося на каждую ступень, затраты
определяются уравнением:
Су = 1000 (Q/2,83)°'468i'+0'332
Таким образом, величина g в уравнении (XII, 39) при s = 0,25 равна
соответственно 0,50 и 0,45. Хотя коэффициенты в этих выражениях изменяются со
временем, величина g, по-видимому, остается близкой к постоянной.
Экстракция с флегмой. При экстракции с флегмой
необходимо найти кроме рассмотренных величин оптимальное флегмовое
число. В большей части случаев применение флегмы оправдано
для систем, в которых взаимная растворимость экстрагента н
исходного раствора зависит от концентрации распределяемого
вещества. Поэтому сделанные выше допущения в данном
случае неприменимы. Для определения оптимальных условий
экстракции с флегмой необходимо прибегать к детальному технико-
экономическому расчету, так как упрощенные методы
нахождения оптимума пока отсутствуют.
ПРИМЕНЕНИЕ ЭКСТРАКЦИИ
ДЛЯ РАЗДЕЛЕНИЯ ВЕЩЕСТВ
(ЭКСТРАКЦИЯ С ДВУМЯ ЭКСТРАГЕНТАМИ)
Нахождение оптимальных условий даже в простейших
случаях экстракции с двумя экстрагентами, как видно из гл. VII,
очень сложная задача. Общий метод нахождения оптимальных
условий не разработан. Применением флегмы, как видно из
рис. 188—190, можно намного уменьшить необходимое число
ступеней. Скорости фаз при этом изменяются незначительно
вследствие малого изменения максимальной суммарной
скорости разделяемых веществ. Последнее приводит в свою очередь
к небольшому изменению производительности, если флегмовые
числа не очень велики. Учитывая это и пренебрегая влиянием
флегмового числа на стоимость регенерации экстрагента, Шай-
бель пришел к выводу6, что оптимальным флегмовым числом
при экстракции с двумя экстрагентами является такое флегмовое
число, при котором концентрации разделяемых веществ на
ступени питания и на соответствующих концах каскада одинаковы.
Шайбель отмечает, что увеличение расходов на регенерацию
экстрагента приводит к уменьшению оптимального флегмового
числа и при значительном возрастании этих расходов
экстракция с флегмой становится экономически нецелесообразной.
Пример. Экстракцией изопропнловым эфиром (экстрагент)
предполагается извлекать из 10%-ного раствора в воде органическое вещество,
стоимость которого составляет 30 коп/кг. Чистота продукта должна
соответствовать 99,9 мол. %. Расход исходного раствора 5,66 м3/ч, его плотность
993 кг/л3. Процесс проводят в экстракторе с ситчатыми тарелками при
расстоянии между тарелками, равном 0,3 м.
Для предварительной оценки общей эффективности тарелки допускается
возможность применения уравнения (XI, 44). Стоимость экстракционной ко-
626
лонны (в рублях на одну тарелку) можно принимать равной 1100 а0-7, где
а — площадь поперечного сечения тарелки (в мг). Допустимая нагрузка
Vd+Vc = 30,5 м3-м2-ч~', где VD и Vc — фиктивные скорости дисперсной н
сплошной фаз. Диспергироваться должна та фаза, расход которой больше.
Коэффициент распределения тЕ = 2,0, межфазовое натяжение 13 • Ю-3 н/м.
Молекулярный вес извлекаемого вещества 60. Стоимость экстрагента 20 коп/кг.
Регенерацию экстрагента предполагают проводить в тарельчатой
ректификационной колонне. Стоимость ректификационной колонны в расчете на
одну тарелку равна 484 руб/м2; к. п. д. тарелки 80%. Экстрагент является
легколетучим компонентом экстракта, причем его относительная летучесть
равна 2,5. Допустимая скорость паров в ректификационной колонне
73,4 кмоль • м2 • ч'1. Коэффициент избытка флегмы 1,25, при этом число
теоретических тарелок в 2,5 раза больше минимального.
Нагрузка куба ректификационной колонны по пару составляет
0,490 кмоль • м'2 • т1, стоимость 1 м2 поверхности теплообменников 14,5 руб.,
стоимость пара и хладоагентов 2 коп/кмоль дистиллята. Установка должна
работать 7200 ч в год; срок службы оборудования 3 года. Стоимость
монтажа оборудования, инструментов, трубопроводов и пр. принимается равной
1,3 стоимости оборудования, эксплуатационные расходы — 0,1 стоимости
оборудования. (Для простоты эти величины приняты одинаковыми для
экстракционного и ректификационного оборудования.). Потери экстрагента
(в данном случае очень летучего) принимаются равными 0,05% от его
расхода.
Определить оптимальные условия проведения процесса.
Решение. По условию примера заданными являются следующие
величины (единицы измерения совпадают с приведенными в списке обозначений):
^ = 5,66
тЕ = 2,0
ps = 745
Ms = 102
Лая = 60
F = 0,3O
S= 147
CD = 484
GD = 73,4
£D = 0,8
G = 0,490
CA = 14,5
P = 1,25
Y=2,5
С =0,02
he
V = 0,0005
H = 7200
p = 2,3
K=3
ft = 0,1
о = 2,5
(p/Y+ ft) = 0,867
^=100
Поскольку эффективность ступени экстрактора в значительной мере
зависит от объемного соотношения фаз, определение оптимального объемного
соотношения с помощью графика на рис. 309 является грубо приближенным.
Поэтому окончательную величину RE определяем по минимуму на кривой
зависимости суммарных расходов от RE.
В качестве первого приближения примем REi = \,Q. Фазу рафнната
считаем дисперсной. Тогда mERE1 = 2,0 • 1,0 = 2,0; Vd/Vc = 1,0. Согласно
уравнению (XI, 44), величина £015 = 0,240. Суммарный расход жидкостей равен
(jf(Re\ + 1) = 11,32 Л13/ч; площадь поперечного сечения экстрактора при
Vr> + Vc = 30,5 составляет 0,37 м2. Следовательно
Св, = 1100(0,37)0'7 • 0,867 = 495 руб/год
Из уравнения (XII, 16), пренебрегая величиной сн, находим 11 =
=9,41 кмоль/ч Далее рассчитываем: Bi = 76,7; ft = 9,995/5= 1,999 [уравнение
(XII, 22)]; nD mm= 15,50 [уравнение (XII, 37)]; Л, = 447 [уравнение (XII, 8)];
cs = 0,243 [уравнение (XII, 31)]; 7=1,0090 [уравнение (XII, 33)]; ен=0,223
[уравнение (XII, 32)]; пЕ = 7,8 [уравнение (XII, 34)]. При RE~l,0 найденные
условия проведения процесса обеспечивают наименьшие суммарные расходы.
Поскольку экстрагент является легколетучим компонентом экстракта,
е = 2,75. Величина X по уравнению (XII, 45) равна 37 000; далее с помощью
рис. 309 определяем /?Е = 0,56 (это значение соответствует абсциссе на
рисунке, равной 3,3). Так как полученная величина RE значительно отличается
от величины RE, принятой вначале, повторяем расчет, принимая RE\ = 0,5;
в результате получаем /?Е=0,65. Отсюда можно сделать вывод, что опти-
40*
627
мальное значение RE близко к 0,7. Как отмечалось, вследствие зависимости
величины Еое от объемного соотношения фаз определить точно
оптимальную величину Re по рис. 309 не представляется возможным. Поэтому
рассчитываем суммарные расходы при /?в=0,8 и /?Е =0,6:
RE
mERE
ЕОЕ
СЕ
в
nD,mln
А
cs
CR
J
ПЕ
I (руб/год)
II
III
V
ст—iv—vi
1,0
2,0
0,240
495
76,7
15,5
447 -
0,243
0,223
1,0090
7,8
16 100
2 730
34 400
3C00
56230
0,8
1,6
0,263
459
61,6
15,65
447
0,200
0,466
1,01024
10,2
17 800
5 700
27 6С0
2 400
53 500
0,6
1,2
0,297
423
46,5
15,70
447
0.Г5О
0,824
1,1468
17,5
25 000
10 700
20 800
1800
58300
Минимум величины (Ст—IV—VI) соответствует значению #£ = 0,75. Статьи
расходов IV и VI являются постоянными и могут не учитываться при
определении оптимального объемного соотношения.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
А—величина, определяемая уравнением (XII,8).
В—величина, определяемая уравнениями (ХП,9),
(XII, 22) и (XII, 23).
Ъ — годовые эксплуатационные расходы (в долях от
стоимости оборудования).
CD— стоимость [без учета расходов на установку (монтаж)]
одной ректификационной тарелки, руб[м2 тарелки.
СЕ - годовая стоимость реальной экстракционной ступени,
включая стоимость оборудования и эксплуатационные
расходы, руб/год.
Ch — стоимость [без учета расходов на установку
(монтаж)] теплообменной аппаратуры, руб/м2 поверхности
теплообмена.
С>,с— стоимость пара и хладоагентов, руб/кмоль дистиллята.
Cs— годовая стоимость ступени вторичной экстракции (при
регенерации экстрагента экстракцией), включая
стоимость оборудования и эксплуатационные расходы,
руб/год.
626
Ст—полная годовая стоимость экстракционного процесса,
руб/год.
Cv — стоимость [без учета расходов на установку (монтаж)]
основного экстракционного оборудования, руб/ступень.
сЕ — концентрация экстрагируемого вещества в экстракте,
кг/м3.
сР — концентрация экстрагируемого вещества в исходном
растворе, кг/м3.
сР — конечная концентрация экстрагируемого вещества в
растворителе (экстрагенте) при регенерации
экстрагента экстракцией, кг/м3.
Сд—концентрация экстрагируемого вещества в рафинате,
кг/л3.
cs — концентрация экстрагируемого вещества в рециркули-
рующем экстрагенте, кг/м3.
ED—общий к. п. д. тарелки ректификационной колонны.
EMD—эффективность ступени по Мэрфри, рассчитанная по
дисперсной фазе.
Еое~ общая эффективность ступени экстракции.
Eos — общая эффективность ступени экстракции на стадии
регенерации экстрагента.
F — стоимость экстрагируемого вещества, руб/кг.
0D — допустимая скорость паров в ректификационной
колонне, КМОЛЬ • Ч'1 -М'2.
Оп — нагрузка теплообменного аппарата по пару,
КМОЛЬ ■ Ч~1 • М'2.
Я— константа.
Н— время работы оборудования, ч/год.
J—величина, определяемая уравнением (XII,33).
КЕ, Ks — константы, определяемые уравнениями (XII, 39),
(XII,55) и (XII,56).
k — конечная степень чистоты (кмоль экстрагируемого
вещества после ректификации/клюль экстрагента).
/ — потери экстрагента на одну реальную ступень (в
долях от его расхода).
V — потери экстрагента (в долях от его расхода) в
колонном экстракторе.
МЕ - молекулярный вес экстрагируемого вещества.
Ms — молекулярный вес экстрагента.
шЕ — коэффициент распределения для процесса экстракции
(концентрация в экстракте/концентрация в
рафинате при равновесии).
ms — коэффициент распределения для процесса регенерации
экстрагента экстракцией (концентрация в экстракте
на стадии регенерации экстрагента/концентрация в
экстракте основной экстракции при равновесии).
tiD — число теоретических ступеней ректификации.
629
пЕ—число теоретических ступеней экстракции.
ns — число теоретических ступеней в процессе
регенерации экстрагента экстракцией.
О — расход дистиллята, кмоль/ч.
Р — энергия, вводимая мешалкой в жидкость,
вт/ступень.
РА — мощность двигателя мешалки, вт/ступень.
РР — мощность двигателей насосов, вт/ступень.
р — стоимость инструментов, трубопроводов и
монтажа (в долях от стоимости основного
оборудования) + 1.
Q — суммарный расход жидкости, м3/ч.
qF—расход исходной смеси, м3/ч.
qs — расход экстрагента, м3/ч.
РЕ — объемное соотношение экстрагента и
исходного раствора.
Ps — объемное соотношение экстрагента на
стадии регенерации экстрагента экстракцией
и экстрагента основной экстракции.
г — флегмовое число при ректификации, кмоль
флегмы/оюль дистиллята.
S—стоимость экстрагента, руб/м3.
s—фактор моделирования [уравнение (XII, 57)].
Т—диаметр аппарата, м.
U — скорость экстракции, кмоль вещества, экс-
страгированного в ч.
W ~ стоимость энергии, руб • вт1 • ч-1.
X — величина, определяемая уравнениями
(XII, 44) и (XII, 45).
xD — мольная доля экстрагента в дистилляте.
xw — мольная доля экстрагента в кубовом
остатке.
Y — срок службы оборудования, годы.
у— мольная доля экстрагента в парах.
z ■— мольная доля экстрагента в жидкости,
стекающей по колонне.
VI—статьи расходов в экстракционном
процессе, руб/год.
а — относительная летучесть.
Р = f/fmto-
Y= «d/«D, mln-
e = a + p—1, если экстрагент является
легколетучим компонентом экстракта.
е=1, если легколетучим компонентом
экстракта является экстрагируемое вещество.
ps — плотность экстрагента, кг/м3.
Индексы
D — относится к ректификации.
Е—относится к экстракции.
tnin — минимальный.
опт. — оптимальный.
S — относится к процессу регенерации экстрагента
экстракцией.
ЛИТЕРАТУРА
1 Happel J., Chemical Process Economics, John Wiley and Sons, Inc., New
York, 1958.
2. Schweyer H. E., Process Engineering Economics, McGraw-Hill Book
Company, Inc., New York, 1955.
3. Trey bal R. E„ A. I. Ch. E. Journ., 5, 474 (1959).
4. Aries R. S., Newton R. D., Chemical Engineering Cost Estimation,
McGraw-Hill Book Company, Inc., New York, 1955.
5 G i 11 i 1 a n d E. R., Ind. Eng. Chem., 32, 1220 (1940).
6. Scheibel E. G., Ind. Eng. Chem., 47, 2290 (1955).
ЭКСТРАКЦИЯ В ПРОМЫШЛЕННОСТИ
D связи с интенсивным развитием химиче-
ской техники и разнообразием
получаемых продуктов за последние годы
(особенно в период после второй мировой войны)
значительно возросла роль процессов
разделения. Это послужило причиной быстрого
проникновения жидкостной экстракции во
многие отрасли химической и
нефтехимической промышленности. Ниже будут кратко
рассмотрены экстракционные процессы,
применяемые в промышленности, и указаны
проблемы, для решения которых можно
использовать жидкостную экстракцию.
Промышленные экстракционные процессы
многочисленны, многие из них подробно
описаны в литературе. Поэтому детальные схемы
этих процессов здесь не рассматриваются.
В связи с тем что описанию некоторых
процессов посвящены целые монографии, в
этой главе, в отличие от предыдущих,
литературные ссылки по ходу изложения
приводиться не будут. Вместо этого в конце
главы помещен перечень основных работ,
которые могут более подробно ознакомить
читателя с промышленными
экстракционными процессами.
ЭКСТРАКЦИЯ В ПРОЦЕССАХ
НЕФТЕХИМИЧЕСКОГО СИНТЕЗА
Несмотря на то что процессы
жидкостной экстракции давно используют для
лабораторных и промышленных целей,
первое применение действительно в крупном
масштабе они получили в
нефтеперерабатывающей промышленности. В этой отрасли
промышленности эксплуатируют
значительное число крупнейших экстракционных
аппаратов, применяют разнообразные
экстракционные процессы, а объемы сырья,
перерабатываемого с помощью жидкостной
632
экстракции, больше, чем в любой другой отрасли
промышленности.
Нефть представляет собой сложную смесь многих
соединений, главным образом углеводородов различных типов и не-
i большого количества соединений серы, азота и кислорода. После
первоначальной дистилляции ряд фракций нефти,
различающихся по температурам кипения, перерабатывают в товарные
продукты: бензин и другие топлива, смазочные материалы,
твердые парафины и битумы. В ходе дистилляции и
дальнейшей переработки химическая природа содержащихся в нефти
соединений часто претерпевает изменения.
Перегонка нефти не приводит к разделению соединений по
их химической природе, так как в одну и ту же температурную
фракцию попадают вещества с разными химическими и
физическими свойствами (плотностью, вязкостью). Поэтому издавна
применяют химические методы разделения нефтяных
углеводородов, например отделение ненасыщенных соединений с
помощью серной кислоты. Продукты, получаемые при
взаимодействии указанных веществ с подобными реагентами, часто не
находят сбыта, что приводит к значительным потерям нефтяного
сырья. Поиски методов регенерации реагентов и путей
использования продуктов реакции во многих случаях являются
трудной задачей.
При разделении нефтяного сырья жидкостной экстракцией
избирательно извлекают, обычно без химического
взаимодействия, те или иные соединения. Очевидным преимуществом
экстракции является возможность достаточно полной регенерации
растворителей и извлеченных ими веществ. Достоинства приме-
, нения жидкостной экстракции в этой области известны давно:
J промышленная экстракция керосина была применена еще в
1909 г. в Румынии (процесс Эделеану), а использование си-
| вушных масел для экстракции керосина было предложено еще
в 1863 г. Однако большая часть экстракционных процессов в
нефтеперерабатывающей промышленности разработана в
последние годы.
Большая часть экстракционных процессов, применяемых в
промышленности нефтехимического синтеза, предназначена для
разделения углеводородов, входящих в состав легких погонов
нефти и фракций смазочных масел (очистка масел), или для
удаления сернистых соединений из нефтепродуктов. Существует
также несколько процессов другого назначения.
Смазочные масла. Углеводороды, находящиеся во фракциях
нефти, предназначенных для получения смазочных масел, не
идентифицированы полностью. Их относительные количества
зависят от источника сырья. Во фракции смазочных масел
содержатся следующие соединения:
633
1. Насыщенные углеводороды нормального и изостроения
общей формулы СпЙ2,г+2. Депарафинизированные масла
содержат, по-видимому, мало чисто парафиновых углеводородов
нормального строения.
2. Олефины или ненасыщенные углеводороды (нормальной
и разветвленной структуры) общей формулы СпНгп и С„Н2п-2.
Олефины отсутствуют в исходном сырье, но могут получаться в
результате крекинга при перегонке.
3. Нафтены — циклические соединения без двойных связей
с алкильными заместителями. Общая формула этих соединений
СпНгп-а, где а — число циклов.
4. Ароматические углеводороды — ненасыщенные
циклические соединения, примером которых могут служить бензол,
нафталин, антрацен. Неочищенные смазочные масла содержат
ароматические углеводороды общих формул СпНгп-2 и СпН2п-4
(в легких фракциях) и С„Н2п-8 (в более тяжелых фракциях).
Могут присутствовать также соединения общих формул CnH2n-io
и СпНгп-18-
5. Битумы и смолистые соединения неизвестного строения.
Углеводородные смолы летучи и могут перегоняться. Битумы
нелетучи.
Соединения, содержащиеся в маслах, обычно имеют сложное
строение и включают в себя углеводородные структуры всех
основных типов. Последние приведены выше в порядке
уменьшения отношения содержания водорода к количеству углерода.
Как правило, с уменьшением этого отношения свойства
смазочных масел ухудшаются вследствие неблагоприятного характера
температурной кривой вязкости и понижения химической
стабильности.
Различные органические растворители, применяемые или
пригодные для очистки смазочных масел, разделяют на
экстрагирующие и осадительные в зависимости от того, растворяют
они или, наоборот, не растворяют нежелательные соединения.
Например, такие растворители, как анилин и фурфурол,
селективно экстрагируют ароматические и нафтеновые углеводороды.
Селективность этих экстрагентов, рассчитанная на основе
критических температур растворения, для ароматических
углеводородов в шесть раз выше, чем для нафтенов (по отношению к
предельным соединениям). Экстракция этими растворителями
приводит к накоплению нафтеновых и парафиновых
углеводородов в рафинате, а ароматических — в экстракте.
Осадительные растворители, такие как пропан и другие
легкие углеводороды, алифатические спирты, вытесняют из
раствора (высаживают) асфальтовые вещества —их селективность
проявляется, скорее, в связи с величиной молекулярных весов,
а не с химической природной разделяемых веществ. Все раство
634
рители проявляют в определенной степени как экстрактивные,
так и осадительные свойства.
До применения экстракции в процессах очистки смазочных
масел широко применяли и отчасти применяют сейчас
(главным образом для получения светлых продуктов из масляных
фракций) концентрированную серную кислоту. Действие серной
кислоты основано как на химическом взаимодействии, так и на
растворении. Экстрагируемые кислотой вещества нельзя, однако,
регенерировать. В результате возникают трудности с
утилизацией отходов производства.
Экстрактивная очистка масел предназначена для удаления
тех составных частей масла, которые образуют при его
использовании осадок и нагар или обусловливают нежелательную
зависимость вязкости от температуры. Так как содержание
индивидуальных компонентов в маслах трудно определить, для
оценки состава масел пользуются условными величинами
(индексом вязкости, вязкостно-весовой постоянной), отражающими
степень ароматичности масел. Эти величины, являющиеся
аддитивными свойствами масел, используют также для изображения
фазовых равновесий и расчета необходимого числа ступеней
экстракции, хотя подобные способы расчета в общем случае
нельзя рекомендовать (см. главу VII). Экстракционные
процессы применительно к сложным смесям лучше рассчитывать по
методам, описанным в главе IX.
Число избирательных растворителей, предложенных и
запатентованных для экстракции смазочных масел, очень велико,
однако практическое применение нашли лишь некоторые из них.
Эти растворители являются лучшими с точки зрения их
стоимости, селективности, физических свойств и других требований,
предъявляемых к экстрагентам и рассмотренных в главе IV.
Смазочные масла очищают экстракцией одним (или смесью
растворителей) или двумя растворителями (фракционная, или
дробная экстракция).
Нитробензол. Этот растворитель обладает высокой
селективностью и хорошо растворим в смазочных маслах. Применяется
при сравнительно низкой температуре (10° С) и малом
объемном соотношении растворителя и исходного раствора,
изменяющемся в пределах 0,5—2. При наличии твердых парафинов
экстракцию необходимо проводить в аппаратах смесительно-отстой-
ного типа. Нитробензол сильно ядовит; именно этим, вероятно,
объясняется его ограниченное применение в качестве экстраген-
та (по имеющимся данным, нитробензол используют для
очистки масел только на одном заводе).
Хлорекс (р,р-дихлорэтиловый эфир). Хлорекс весьма
эффективен как растворитель для очистки масел из
пенсильванских нефтей, поскольку растворимость их в хлорексе очень
высока. Для удаления растворителя из экстракта и рафината
635
применяют перегонку с водяным паром. При этом в результате
гидролиза образуется хлористоводородная кислота, что
приводит к затруднениям, связанным с сильной коррозией
аппаратуры. Хлорекс используют для очистки масел только на
нескольких предприятиях.
Фурфурол. Один из наиболее широко распространенных
растворителей, применяемых для экстрактивной очистки масел.
Фурфурол используют при сравнительно высокой температуре,
равной 65—120° С, что позволяет производить экстракцию из
относительно вязких и парафинистых фракций, не опасаясь
забивания аппаратуры, даже в насадочных колоннах. Фурфурол
легко окисляется на воздухе и полимеризуется. Для уменьшения
потерь растворителя его обычно хранят в резервуарах с
инертной газовой подушкой, а поступающий на экстракцию раствор
подвергают деаэрации.
Фенол. Этот распространенный растворитель также
используют при высокой температуре (около 93°С), что дает
возможность очищать масла с большой вязкостью и высоким
содержанием парафинов.
Чаще всего процесс очистки проводят в экстракторах с сит-
чатыми тарелками. Температура плавления чистого фенола
довольно высока (40,9° С). Однако в присутствии воды,
применяющейся в качестве флегмы для повышения селективности, и
даже мельчайших количеств экстрагируемого материала точка
плавления фенола понижается настолько, что всякая опасность
забивания аппаратуры отпадает.
Пропан представляет собой осадительный растворитель,
вытесняющий из раствора асфальты. На большей части
нефтеперерабатывающих заводов пропан является легко доступным
и дешевым сырьем. Применяется обычно при температурах
несколько ниже 120° С и при довольно высоком объемном
соотношении растворителя и исходного раствора. Вследствие
высокой летучести пропан легко регенерируется. Экстракт (т. е.
масло, из которого удалены асфальтены) можно охлаждать в
результате испарения пропана для предварительного удаления
твердых парафинов.
S02 — бензол. Сама по себе двуокись серы является
слишком селективным растворителем по отношению к ароматическим
углеводородам для получения масел с высоким индексом
вязкости, так как она почти не извлекает нафтеновых углеводородов.
Смесь S02 и бензола (обычно в количестве 15—25%)
представляет собой более подходящий растворитель. Однако этот
смешанный растворитель не получил широкого распространения.
«Дуо-Сол-процесс». Так называется процесс экстракции
масел двумя растворителями. Экстрагентами служат пропан,
извлекающий соединения парафинового и нафтенового рядов, и
смесь, состоящая из 40% фенола и 60% крезолов («Селекто»),
636
обладающая селективными свойствами по отношению к
ароматическим и асфальтовым соединениям. Таким образом, в этом
процессе одновременно удаляют асфальтены и производят
обычную' экстрактивную очистку масел.
При экстракции масел по способу «Дуо-Сол» помимо
основного процесса разделения углеводородов происходит также
некоторая очистка масла от сернистых соединений.
Независимо от применяемых растворителей при очистке
масел достигается примерно одинаковый выход продукта (рафи-
ната с данным индексом вязкости), хотя для каждого из них
необходимо свое число ступеней и объемное соотношение
растворителя и исходного раствора.
Для очистки смазочных масел применяют аппараты
различных типов: смесители-отстойники, колонные и центробежные
экстракторы. Эффективность их обычно эквивалентна трем-восьми
теоретическим ступеням. При экстракции одним растворителем
в аппарат в качестве флегмы может подаваться часть
экстракта. Однако чаще применяется внутреннее орошение, причем
флегму получают с помощью создания определенного градиента
температуры по высоте экстрактора. При наличии
температурного градиента в результате уменьшения взаимной
растворимости масло выделяется из экстракта и переходит в фазу ра-
фината. Если растворителем служит фенол, который заметно
растворяется в воде, то ее можно подавать в аппарат в виде
флегмы (см. главу VI).
Последовательность экстракции и других операций при
очистке масел следует выбирать на основании экономических
соображений, так как независимо от последовательности
отдельных процессов можно получить одни и те же конечные
продукты. Твердые парафины (если они имеются в исходном сырье)
можно отделять как до экстракции, так и после нее.
При экстракции парафины концентрируются в рафинате, и,
следовательно, температура застывания очищенного масла
будет выше, чем у исходного. Если твердые парафины отделяют
до экстракции, точка застывания поступающего на экстракцию
масла должна быть ниже, чем необходимо для конечного
продукта; вследствие этого отделение парафинов затрудняется.
В случае если отделение парафинов следует за экстракцией,
масло, теряющееся с парафинами, является наиболее ценным
по своим свойствам, а экстракция затрудняется из-за осаждения
твердых парафинов при температуре процесса.
Смазочные масла после очистки экстракцией перегоняют для
получения легких смазочных материалов или очищают от
битумов (для получения более тяжелых масел). Очистку смазочных
масел заканчивают фильтрацией их через активированную глину.
Легкие нефтяные дистилляты. Экстракцию широко
применяют для разделения углеводородов легких погонов нефти.
637
Процесс Эделеану. В этом процессе экстракцию впервые
применили в промышленном масштабе на
нефтеперерабатывающих заводах. Процесс Эделеану первоначально предназначался
для извлечения ароматических углеводородов из керосина
(растворителем служила двуокись серы), чтобы улучшить его
качества как осветительного топлива. Селективность двуокиси серы
очень высока. Керосин экстрагируют обычно при температурах
от —12 до —6,7° С; для более тяжелых дистиллятов требуется
температура несколько выше. Одновременно с ароматикой
извлекаются также сернистые соединения. Чтобы предотвратить
образование твердых гидратов с двуокисью серы, до
поступления на экстракцию сырье освобождают от воды (обычно
осушку ведут хлористым кальцием).
На старых заводах растворитель регенерировали в
многоступенчатом испарителе. В современной промышленной
практике для той же цели используют обычную ректификацию при
более высоких температурах (при слишком высокой
температуре возможна реакция между двуокисью серы и поступающим
с сырьем сероводородом, сопровождающаяся выделением
свободной серы).
Процесс Эделеану можно применять также для удаления
ароматических соединений из дизельного топлива, так как
присутствие ароматических углеводородов ухудшает
воспламеняемость топлив. Для этого можно использовать также экстракцию
фурфуролом, которую в крупных масштабах применяют при
обработке продуктов, образующихся в результате крекинга
газойля прямой перегонки нефти.
Выделение ароматических углеводородов. Двуокись серы
применяют также для выделения ароматических углеводородов
из легких нефтепродуктов, богатых этими углеводородами. Так,
из продуктов каталитического гидроформинга получают бензол,
толуол и ксилол высокой чистоты. При помощи экстракции
двуокисью серы можно получать и высокооктановый бензин;
образующийся при этом рафинат, богатый парафинами, можно
использовать в качестве реактивного топлива.
Обычно вместе с ароматическими соединениями
экстрагируется некоторое количество парафиновых и нафтеновых
углеводородов (около 25%). Вследствие близости температур
кипения последние невозможно отделить от ароматических
углеводородов дистилляцией. Обрабатывая экстракт парафинистым
керосином с соответствующими пределами температур кипения,
можно заместить содержащиеся в экстракте неароматические
соединения углеводородами с более высокими температурами
кипения. Таким образом, становится возможным выделить
чистую ароматическую фракцию (из экстракта после регенерации
S02), которую ректификацией можно разделить на
индивидуальные углеводороды.
638
Интересно отметить, что для экстракции толуола из богатых
ароматическими углеводородами продуктов гидроформинга
можно применять воду. Однако гораздо более удобным
растворителем, который может конкурировать с S02, является диэтилен-
гликоль, используемый в процессе «Юдекс». Селективность ди-
этиленгликоля по отношению к ароматическим соединениям
увеличивается при добавлении 5—12% воды. Этот растворитель,
получающий все более широкое распространение, применяют
под давлением при относительно высоких температурах
(порядка 163° С). Так как диэтиленгликоль имеет высокую
температуру кипения, регенерация его из экстракта происходит почти
без потерь; при этом теряется, скорее, некоторое количество
углеводородов.
Процесс выделения ароматических соединений из экстракта
и регенерация экстрагента при экстракции диэтиленгликолем
производятся очень просто и представляют собой по существу
экстрактивную дистилляцию. Содержащиеся в экстракте
низшие предельные углеводороды, которые являются наиболее
легколетучей частью смеси, поднимаются вверх в виде паров.
Ароматические соединения отбирают в средней части колонны.
Низшие предельные углеводороды используют для орошения
экстрактора, чтобы заместить извлеченные высокомолекулярные
парафины (подобно тому, как при экстракции ароматических
углеводородов двуокисью серы). Для окончательного выделения
диэтиленгликоля рафинат и экстракт обычно промывают водой.
Олефины также экстрагируются диэтиленгликолем. Пропуская
экстракт (после регенерации экстрагента) через
активированную глину, вызывают их полимеризацию.
Процессы извлечения ароматических соединений из
нефтяного сырья получили настолько широкое развитие, что в
настоящее время основным источником ароматических
углеводородов стала нефтехимия, а не коксохимическая
промышленность. Кроме экстракции для выделения ароматических
углеводородов можно применять адсорбцию на активированном си-
ликагеле.
Бутадиен. Производство синтетических каучуков из
углеводородов С4 стало одной из важных отраслей химической
промышленности. Дистилляционные методы оказались в данном
случае непригодными, поэтому бутадиен из смеси углеводородов
С4 выделяют главным образом жидкостной экстракцией.
Бутадиен получают каталитической дегидрогенизацией бутилена.
Реакционную смесь экстрагируют медноаммиачным ацетатным
раствором, с которым бутадиен образует нестойкое комплексное
соединение. После выделения из экстракта бутадиен
используют для получения синтетического каучука, являющегося
продуктом сополимеризации бутадиена со стиролом. Бутадиен
можно выделять также методом экстрактивной дистилляции.
Ш
Сероочистка. Соединения серы присутствуют в нефти в виде
сероводорода (H2S), сероуглерода (CS2), меркаптанов и тиофе-
нолов (RSH), тиоэфиров (RCR'), полисульфидов, производных
тиофена и тиофана (гетероциклические соединения) и,
возможно, других соединений (R и R' обозначают алкильные группы),
Сернистые соединения могут присутствовать в количестве от
нескольких десятых процента (пенсильванская и среднеконтинен-
тальная американские нефти) до 3—4% (месторождения
Калифорнии и Мексики) и даже до 7—8% (иракская нефть).
Примеси серы в нефти нежелательны. Сероводород и
меркаптаны в присутствии воды вызывают коррозию аппаратуры.
Меркаптаны, кроме того, имеют неприятный запах, придают
нежелательную окраску легким нефтепродуктам и снижают их
стабильность. Сернистые соединения ухудшают
антидетонационные и антиокислительные свойства бензина, в частности,
снижают приемистость к тетраэтилсвинцу. При сгорании
соединения серы образуют продукты окисления, обладающие
коррозионными свойствами. По этим причинам сернистые соединения
стараются удалить, хотя бы частично, или перевести в другие,
менее вредные соединения.
Очистку от сероводорода чаще всего осуществляют
экстракцией водными растворами щелочи; при этом извлекают и тио-
фенолы. Отработанный раствор обычно не используют.
Растворы этаноламинов, фосфата калия и др., применяющиеся для
извлечения сернистых соединений из газов, можно использовать
и для жидкостной экстракции. Преимущество этих экстрагентов
заключается в том, что их можно регенерировать отпаркой
экстракта.
После удаления H2S бензины для улучшения запаха можно
подвергать дальнейшей обработке, превращая находящиеся в
них меркаптаны в меркаптиды. Последние, однако, в остальных
отношениях обладают такими же отрицательными свойствами,
как и меркаптаны. Другой способ заключается в экстракции
меркаптанов водной щелочью. Для ее регенерации меркаптаны
можно отгонять с паром. Одновременно экстрагируются
присутствующие в бензине органические кислоты ароматического и
нафтенового рядов. Они накапливаются до равновесной
концентрации в рециркулирующей щелочи. В присутствии этих
кислот коэффициенты распределения меркаптанов значительно
возрастают.
В процессе «Сольютайзер» экстрагентом служит крепкий
раствор гидроокиси калия, в котором для увеличения
коэффициентов распределения меркаптанов растворен изобутират
калия. В аналогичном процессе «Меркапсол» применяют
гидроокись натрия с добавкой нафтеновых кислот и крезолов.
Разновидностью этих процессов является процесс «Дуалэер», в
котором используют раствор едкого кали, содержащий крезилаты
(НО
калия. Меркаптаны удаляют из экстракта отпаркой.
Необходимую концентрацию щелочи поддерживают контактированием
экстрагента (перед повторным применением) с водным
раствором КОН (с которым крезилатный раствор не смешивается).
В процессе «Юнисол» к раствору едкого натра добавляют
метанол, который растворяется в бензине. Поэтому его после
экстракции повторно экстрагируют водной щелочью для
извлечения метанола. Экстрагенты регенерируют отпаркой.
Безводный фтористый водород, сам по себе или в смеси с
трехфтористым бором, также может служить растворителем для
извлечения сернистых соединений.
Другие экстракционные процессы. В
нефтеперерабатывающей промышленности экстракцию можно применять также для
извлечения нафтеновых кислот из углеводородных масел
некоторыми растворителями, например водными растворами низших
спиртов. Изопропиловый спирт используют для экстракции
нефтяных сульфонатов из масел, обработанных концентрированной
серной кислотой или олеумом.
ЭКСТРАКЦИЯ В ТЕХНОЛОГИИ ЖИРОВ,
МАСЕЛ И РОДСТВЕННЫХ ПРОЦЕССОВ
Жиры и масла как животного, так и растительного
происхождения представляют собой преимущественно эфиры глицерина
и жирных кислот общей формулы H2C(OOR/)—HC(OOR")—
—H2C(OOR'"). Углеводородные радикалы R', R" и R"' могут
быть насыщенными или ненасыщенными и содержат обычно от
8 до 32 атомов углерода. Масла и жиры любого вида содержат
большое количество разнообразных жирных кислот.
Различные жиры и масла отличаются друг от друга по типу,
числу и распределению жирных кислот, входящих в молекулы
глицеридов. В большей части случаев, особенно в маслах
растительного происхождения, жирные кислоты распределяются
между молекулами глицеридов относительно равномерно. Кроме
глицеридов в жирах и маслах, по крайней мере неочищенных,
содержатся небольшие количества свободных жирных кислот,
образующихся при частичном гидролизе триглицеридов, фосфа-
тидов (триглицеридов, в которых одна из жирных кислот
замещена сложным эфиром фосфорной кислоты, например летицин
или кефалин), а также стерины, витамины, углеводы, кароти-
ноидные пигменты, белки, токоферолы и другие вещества
неустановленного строения.
После соответствующей обработки жиры и масла применяют
для получения весьма разнообразных продуктов: пищевых
товаров, мыла, красок, чернил, различных производных жирных
кислот, смазочных материалов, косметических и медицинских
средств. При переработке жиров экстракционные процессы
4J Р. Е- Тргйбад
§41
используют для выделения примесей — жирных кислот или
родственных соединений и, кроме того, в процессе гидролиза жиров.
Содержащиеся в жирах и маслах свободные жирные кислоты
можно отделить промывкой водным раствором щелочи или соды
(при этом образуются водорастворимые мыла) с последующим
разделением фаз центрифугированием. Другой способ
заключается в проведении всего процесса (экстракции и разделения
фаз) в центробежных экстракторах. Спирты, например изопро-
пиловый спирт, экстрагируют кислоты без химического
взаимодействия. Однако широкого применения метод экстракции
спиртами не получил.
Очистка масел и жиров пропаном. Промышленная очистка
масел и жиров с помощью пропана известна под названием «Со-
лексол-процесса». Критические температуры растворения
различных компонентов масел в пропане, а также их растворимости
при высоких температурах в достаточной мере отличаются друг
от друга; вследствие этого их можно частично разделить.
Постепенное повышение температуры системы масло —
пропан приводит к последовательному растворению различных
групп компонентов масла, которые могут извлекаться одна за
другой в порядке усложнения молекулярной структуры или
увеличения молекулярного веса. Например, при разделении жира
сардины, поддерживая соответствующий температурный
градиент между последовательно соединенными экстракторами,
получают обесцвеченные фракции жира с различной степенью
ненасыщенности и витаминный концентрат. Подобно этому в
процессе очистки сырого соевого масла можно получить
обесцвеченное и не содержащее свободных жирных кислот масло или,
кроме того, выделить фракцию с высоким йодным числом и
пищевую фракцию. Таким же образом можно обесцвечивать и
фракционировать льняное масло, животные жиры и другие
продукты.
Масла можно рафинировать также более старыми
способами, заключающимися в щелочной промывке и обесцвечивании
адсорбционными или химическими методами. Экстрактивная
очистка, однако, обладает большей гибкостью и обеспечивает
более высокий выход и лучшее качество конечного продукта.
Очистка масел и жиров фурфуролом. Фурфурол селективно
экстрагирует глицериды с большей степенью ненасыщенности и,
кроме того, свободные жирные кислоты и более сложные
соединения, такие, как фосфатиды и токоферолы. Экстракция
фурфуролом дает возможность, например, получать высыхающие
масла, пригодные для призводства красок и лаков, из
«невысыхающих» масел типа соевого. К сожалению, глицериды этих масел
имеют смешанную природу и жидкостной экстракцией нельзя
полностью разделить насыщенные и ненасыщенные эфиры
глицерина.
R42
В зависимости от того, какое масло поступает на
экстракцию — сырое или предварительно обработанное щелочью, фур-
фурольную очистку соевого масла можно выполнять в двух
вариантах. Масло, предварительно обработанное щелочью,
экстрагируют фурфуролом в противоточном процессе с возвратом
экстракта в качестве флегмы; при этом в экстракт переходят
составляющие масла с относительно высокой степенью
ненасыщенности.
При экстракции сырого масла в экстракт переходят
свободные жирные кислоты, неомыляемые соединения и
антиокислители, содержащиеся как в масле, так и в фурфуроле. В
присутствии этих соединений свойства рафинированного масла как
сырья для получения красок ухудшаются. Поэтому фурфуроль-
ный экстракт вторично экстрагируют легкими нефтепродуктами
в другом экстракторе. При этом извлекаются глицериды, а
свободные жирные кислоты, антиокислители и другие
нежелательные примеси остаются в рафинате.
Рафинаты с обеих стадий экстракции можно использовать
для получения гидрированных жиров. Экстракты могут служить
сырьем для получения лаков. Побочными продуктами являются
стерины, токоферолы и свободные жирные кислоты. Экстракцию
фурфуролом можно применять также для выделения из
льняного масла фракции с высокой степенью ненасыщенности и для
выделения витаминов из жира морских рыб. Свободные жирные
кислоты можно разделять экстракцией двумя растворителями —
фурфуролом и нафтой. Моно-, ди- и триглицериды также
можно разделять фракционной (дробной) экстракцией, используя
в качестве растворителей водный раствор этилового спирта и
гептан.
Расщепление жиров. Гидролиз жиров или масел водой
производят для получения глицерина и жирных кислот. Жирные
кислоты можно затем нейтрализовать щелочью для получения
мыла или переработать другим способом. Хотя процесс
расщепления жиров лишь частично можно характеризовать как
экстракционный, технология его проведения и аппаратурное
оформление аналогичны применяемым в экстракционных
установках.
В начале процесса, когда вода взаимодействует с негидроли-
зованным маслом, реакция является гетерогенной (так как
растворимость их друг в друге очень мала) и протекает медленно.
По мере выделения жирных кислот в свободном виде
растворимость воды в масле увеличивается и реакция продолжается в
основном в органической фазе. Скорость ее значительно
увеличивается, особенно при повышенных температурах.
Образующийся глицерин переходит в водную фазу. Устанавливающееся
равновесие не зависит от температуры, что обусловлено
отсутствием заметного теплового эффекта реакции.
41*
643
Соответственно конечная степень расщепления жиров в
периодическом процессе зависит лишь от соотношения воды и
масла и повышается с увеличением количества воды, поскольку
при этом снижается концентрация глицерина. Гидролиз тригли-
церидов протекает постепенно, вследствие этого частично
расщепленные жиры содержат значительные количества моно- и
диглицеридов.
В современных установках расщепления жиров масло и
вода, из которых предварительно удален воздух, взаимодействуют
в противоточном процессе при 230—290° С и давлениях 50—
55 ат. Водный раствор, выходящий с низа колонны, содержит
10—25% глицерина, не загрязненного катализатором или
солями (в отличие от глицерина, получаемого по старым схемам
процесса). С верха колонны отбирают жирные кислоты в
количестве 97—99% от общего количества их, полученного в
результате гидролиза.
Смола и талловое масло. Неочищенная древесная смола,
получаемая перегонкой с паром или обработкой легкими
нефтепродуктами (лигроином), отличается от живичной канифоли
темным цветом. Смола плохо обесцвечивается при
использовании ее в производстве мыла, для прокатывания бумаги и
других целей. В неочищенной смоле, как живичной, так и
получаемой перегонкой с паром, содержится 80—90% абиетиновой
кислоты и ее изомеров, а также еще более высокоароматизиро-
ванные и окисленные смолистые вещества.
В процессе экстрактивной очистки сырую смолу растворяют
в легких нефтепродуктах типа бензина до получения раствора
концентрацией около 15%- Из этого раствора смолу
экстрагируют фурфуролом для извлечения нежелательных примесей.
После выпаривания растворителя и отпарки рафината получают
слегка окрашенный продукт, который практически во всех
случаях может заменить более дорогую живичную канифоль. В
качестве растворителя можно также применять, как в процессе
«Солексол», пропан.
Талловое масло представляет собой смесь 38—58%
смоляных кислот (в основном абиетиновая кислота), 54—36%
ненасыщенных жирных кислот (с 18 атомами углерода в молекуле)
и некоторого количества (5—10%) более сложных соединений,
таких, как стерины. Это масло является дешевым побочным
продуктом в сульфитном процессе получения бумаги. После
сернокислотной очистки или вакуум-дистилляции талловое масло
используют в производстве большого количества различных
продуктов.
Ценность масла в значительной степени повысилась бы, если
бы имелась возможность отделить смоляные кислоты от
жирных. Этой задаче было посвящено большое количество
исследований, и запатентовано много различных методов разделения.
644
Наиболее обещающий метод — экстракция непосредственно тал-
лового масла пропаном или экстракция предварительно
метилированного масла фурфуролом. В последнем случае метиловые
эфиры жирных кислот отделяются от неэтерифицированных
смоляных кислот.
ЭКСТРАКЦИЯ В КОКСОХИМИЧЕСКОЙ
ПРОМЫШЛЕННОСТИ
Жидкостная экстракция находит применение при
переработке некоторых побочных продуктов коксохимического
производства.
Парогазовые продукты коксования охлаждают водой;
получаемая так называемая газовая вода содержит соли аммония
(угольной, родановой, серной и других кислот) и различные
органические соединения: одноатомные и многоатомные фенолы,
пиридиновые основания, органические кислоты, ненасыщенные
углеводороды и другие соединения. Состав этих вод сильно
изменяется в зависимости от вида угля, используемого для
коксования, особенностей технологии процесса и режима его
проведения.
В прошлом эти воды после извлечения аммиака сбрасывали
в водоемы. Фенолы, попадающие таким путем в водоемы,
оказывают вредное действие на рыбу и даже при очень небольших
концентрациях (10 частей на миллион) влияют на вкус воды.
При хлорировании воды могут образовываться хлорфенолы,
которые придают воде неприятный вкус уже при концентрации
2 части на миллион. При отсутствии специальных установок для
очистки сточных вод все это может привести к весьма серьезным
затруднениям при получении питьевой воды. Поэтому фенолы
из сточных вод извлекают экстракцией или другими методами.
Общее содержание фенолов в сточных водах процессов
коксования изменяется от 0,5 до 1,5 г/л и достигает 12 г/л в
процессах полукоксования; при этом наибольшую долю составляют
одноатомные фенолы.
Собственно фенол в большей части случаев представляет
собой наиболее трудноэкстрагируемый компонент из фенолов,
присутствующих в сточных водах. Чтобы сточные воды можно было
сбрасывать, необходимо довольно полно извлечь фенолы — до
конечной концентрации, равной нескольким миллионным долям,
или еще меньшей.
• На старых заводах для экстракции применяют чаще всего
бензол и легкие нефтепродукты. После фильтрования (для
отделения твердых смолообразных веществ) сточные воды
экстрагируют в больших, заполненных коксом или хордовой насадкой
колоннах или в трехступенчатых смесительноотстойных
экстракторах. В последних для смешения фаз обычно используют
645
центробежные насосы. При этом жидкости довольно энергично
эмульгируются, что требует применения отстойников достаточно
крупных размеров.
Обесфеноленные сточные воды (рафинат) содержат
определенное количество растворенного экстрагента, который на
американских коксохимических предприятиях в значительной
степени регенерируют при последующей дистилляции. Легкие
нефтепродукты экстрагируют фенол, крезолы, высшие фенолы и,
кроме того, небольшие количества пиридина, нафталина и
нейтральных соединений.
Для выделения бензола и фенолов экстракт смешивают с
раствором щелочи, при этом фенолы экстрагируются в виде
фенолятов натрия. По мере накопления фенолов щелочь
периодически перекачивают в нейтрализатор, в котором отгоняется с
паром бензол и при подкислении серной кислотой освобождаются
фенолы. Образующийся сульфат натрия является отходом
производства. Регенерированный экстрагент отстаивается от
шлама и возвращается в экстрактор. Периодически этот экстрагент
необходимо подвергать дистилляции.
Недостаточно полное извлечение фенолов при экстракции их
легкими нефтепродуктами, склонность системы к
эмульгированию и связанная с этим необходимость иметь большие объемы
растворителя в отстойниках привели к тому, что на большей
части новых заводов используют другие экстрагенты. Применяют
сравнительно легколетучие растворители, которые можно
отогнать от экстрагированных фенолов: бутилацетат, «феносоль-
ван» (бутилацетат, содержащий другие эфиры), диизопропило-
вый эфир, метилизобутилкетон, а также смесь легких
нефтепродуктов с бензином каталитического крекинга. Для увеличения
коэффициентов распределения используют также высококипя-
щие нефтепродукты с добавкой хинолина или других смоляных
оснований и трикрезилфосфата.
Недостаток указанных растворителей, обеспечивающих
высокие коэффициенты распределения и позволяющих почти
полностью извлечь фенолы, заключается в том, что они
экстрагируют также большие количества других соединений, которые
можно отделить от фенолов, только превратив последние в
феноляты. Этот процесс дорогостоящий, и фенол, как побочный
продукт коксохимического производства, оказывается
неспособным конкурировать с высококачественным синтетическим
фенолом. Кроме того, перечисленные растворители в большей части
случаев дороже легких нефтепродуктов, и потери их необходимо
свести к минимуму.
Выделение фенолов из сточных вод является также важной
проблемой для предприятий нефтеперерабатывающей
промышленности и некоторых заводов пластмасс.
84В
Извлечение фенолов из каменноугольной смолы. Фенолы
можно выделять из каменноугольной смолы также щелочным
методом. Этот процесс, однако, является дорогим. Другим
способом может служить экстракция смолы водным раствором
метилового или этилового спирта (процесс «Метасольван») или
водным триэтиленгликолем. Кроме того, можно применять
экстракцию двумя растворителями — водным метанолом и гекса-
ном. С помощью этих двух растворителей можно отделять
фенолы друг от друга.
Данный процесс разработан для выделения фенолов, крезо-
лов и крезиловых кислот из смол полукоксования. Его можно
применять и для очистки отработанной щелочи после щелочной
промывки бензина каталитического крекинга. Аналогичным
образом можно обрабатывать буроугольные смолы. Для
экстракции фенолов изучены и предложены также другие растворители.
Ароматические углеводороды. Бензол, толуол и ксилолы
являются традиционными побочными продуктами коксохимической
промышленности. В сыром виде они содержат примеси
предельных и непредельных углеводородов, а также сернистых
соединений, например тиофена. Прежде ароматические углеводороды
очищали обработкой их серной кислотой, нейтрализацией
щелочью, промывкой водой и дистилляцией. Однако некоторые
парафиновые углеводороды трудно или даже невозможно отделить
дистилляционными методами. Для конкуренции с
высокочистыми ароматическими соединениями, получаемыми из нефтяного
сырья, на новых коксохимических заводах производят
селективное каталитическое гидрирование неароматических
непредельных углеводородов и сернистых соединений. После
отделения сероводорода смесь углеводородов экстрагируют водным
диэтиленгликолем (процесс «Юдекс», стр. 639). В результате
получают экстракт, содержащий ароматические углеводороды
высокой степени чистоты.
В отличие от нефтеперерабатывающих заводов, где на
экстракцию поступает сырье, содержащее 25—55% ароматических
углеводородов, в данном случае содержание ароматических
соединений в исходной смеси углеводородов достигает 95% и
более. Поэтому желательно селективно извлекать парафины, а не
ароматические углеводороды. Однако пригодные для этой цели
перфторуглероды (см. главу III) в настоящее время слишком
дороги.
ЭКСТРАКЦИЯ В ФАРМАЦЕВТИЧЕСКОЙ
ПРОМЫШЛЕННОСТИ
На фармацевтических заводах синтезируют или выделяют
из природных веществ тысячи сложных органических
препаратов, применяющихся в качестве лекарств, Большая часть
?47
фармацевтических препаратов чувствительна к повышенным
температурам, и их нельзя подвергать дистилляции; поэтому в
фармацевтических производствах широко используют жидкостную
экстракцию. В большей части случаев приходится разделять
небольшие количества смесей, в связи с этим довольно часто
применяют периодическую экстракцию. При выделении небольших
количеств веществ из больших объемов разбавленных растворов
возникает необходимость в крупных экстракционных установках.
Ниже будет рассмотрено лишь несколько типичных примеров
применения экстракции в фармацевтической промышленности.
Антибиотики представляют собой побочные продукты
жизнедеятельности некоторых микроорганизмов, способные замедлять
рост или вызывать гибель других микроорганизмов. Из
антибиотиков наибольшее применение находит пенициллин, являющийся
смесью нескольких веществ близкого строения.
Пенициллин получают ферментативным путем из кукурузы
и лактозы в крупных аппаратах (емкостью 190 м3 и более).
Первоначально пенициллин выделяли адсорбцией на
активированном угле, однако в настоящее время для этого применяют
исключительно жидкостную экстракцию. Реакционную смесь
подкисляют до рН = 2—2,5 (что способствует экстракции) и
экстрагируют растворителем, чаще всего бутил- или амилацетатом.
При обработке экстракта фосфатным буферным раствором
(рН = 6,8—7,5) пенициллин выделяется из раствора. Повторяя
экстракцию и обработку фосфатным буферным раствором
несколько раз, получают концентрированный и очищенный
продукт. После этого пенициллин вымораживают.
Наиболее важная стадия выделения пенициллина — его
экстракция из кислых растворов, так как при низких рН
пенициллин быстро разрушается. Поэтому необходима быстрая
экстракция при малом объеме жидкости в ступени. Идеальными в этом
отношении являются центробежные экстракторы (например,
экстрактор Подбильняка), которые обычно и применяют для этой
цели.
Среди других антибиотиков, для выделения которых
используют жидкостную экстракцию, наиболее известны ауромицин,
бацитрацин, циркулин, террамицин и стрептомицин. Несмотря
на некоторые особенности технологии выделения этих
антибиотиков, для их очистки и концентрирования применяют тот же
принцип многократного распределения между водой или
буферным раствором и органическим растворителем.
Витамины. Некоторые витамины выделяют из природных
веществ. Так, витамины А и D можно экстрагировать из масла,
выделяемого из печени рыб, а витамин Е — из растительных
масел жидким пропаном (см. раздел «Экстракция в технологии
жиров, масел и родственных процессах»). Витамин Bi2 можно
экстрагировать из отходов производства некоторых
антибиотики
ков. В процессе синтеза витамина А и некоторых витаминов из
группы В обычно также применяют жидкостную экстракцию.
Экстракцию используют также для получения многих
гормонов и алкалоидов.
ЭКСТРАКЦИЯ В ТЕХНОЛОГИИ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ
Экстракцию применяют для извлечения уксусной кислоты из
разбавленных водных растворов. Большая часть растворителей
из-за малых коэффициентов распределения с трудом
экстрагирует уксусную кислоту. Обычно используют этилацетат для
разбавленных растворов уксусной кислоты и этилацетат с добавкой
бензола (чтобы улучшить селективность и уменьшить количество
экстрагируемой воды) для более концентрированных
растворов. При больших концентрациях в качестве экстрагента можно
применять также изопропилацетат. Если содержание уксусной
кислоты превышает 35%. то для ее выделения экономичнее
использовать азеотропную перегонку. В качестве экстрагентов
уксусной кислоты применяли также диэтиловый и диизопропи-
ловый эфиры. Высокие коэффициенты распределения
обеспечивают третичные амиды высших жирных кислот, однако
маловероятно, чтобы их использовали в качестве экстрагентов на
практике.
Муравьиная кислота, образующая с водой азеотропную смесь,
экстрагируется из водных растворов еще труднее. Наилучшими
растворителями оказались тетрагидрофуран (коэффициент
распределения около двух), циклогексанон и пропилформиат
(коэффициент распределения около единицы). Эти экстрагенты,
однако, дороги. Более доступные растворители (например,
алифатические кетоны) обеспечивают достаточные величины
коэффициентов распределения только в присутствии солей в водном
растворе. Метилэтилкетон применяют для экстракции
муравьиной и уксусной кислот из сульфитных отходов бумажной
промышленности.
Аконитовую кислоту можно экстрагировать из патоки этил-
ацетатом, метилэтилкетоном или метилизобутилкетоном.
Молочная кислота хорошо экстрагируется из воды этилацетатом или
кетонами, особенно в присутствии добавок солей. Если нужно
получить эфиры этой кислоты, то лучше всего экстрагировать
ее спиртами.
В процессах окисления природного газа, гидрогенизации
окиси углерода в присутствии олефинов или без них (процессы
Фишера — Тропша, «Синтол», оксосинтез и др.) получают большие
количества кислородсодержащих соединений. В этих процессах
обычно образуются водный и масляный слои, содержащие
кислоты, альдегиды, кетоны и спирты.
649
Вещества, находящиеся в водном слое, можно разделить
дистилляцией или экстракцией, используя в качестве экстрагента,
например, изофорон. Для выделения альдегидов и кетонов из
углеводородного раствора применима экстракция водными
растворами бисульфитов, с которыми они образуют продукты
присоединения. Бисульфитные аддукты разлагаются при перегонке
с паром. Спирты можно экстрагировать диэтиленгликолем или
водными растворами низших спиртов, особенно в присутствии
гидротропных агентов (например, солей жирных кислот или
органических сульфокислот).
Экстракцию изопропиловым эфиром чаще всего применяют
для дезодорации и отделения кислот из неочищенного водного
раствора формальдегида.
Глицерин плохо экстрагируется из воды: при обычной
физической экстракции, по-видимому, наибольшие коэффициенты
распределения достигаются в случае извлечения глицерина
высшими спиртами. В присутствии кислот глицерин хорошо
экстрагируется альдегидами и кетонами в результате химической
реакции, приводящей к образованию ацеталей.
В производстве синтетического фенола по методу Рашига
бензол окисляется в фенол через стадию образования
хлорбензола. В этом процессе сырой хлорбензол экстрагируют
щелочью для удаления НС1 и бензолом извлекают фенол из
сточных вод. В процессе «Доу» хлорбензол гидролизуют щелочью.
Раствор фенолята натрия перед выделением фенолов
экстрагируют хлорбензолом для удаления дифениловых и высших эфи-
ров. Экстракцию используют в этом процессе также для
некоторых других целей.
Ряд веществ, которые можно выделить экстракцией,
содержится в сульфитных сточных водах бумажного производства.
Из окисленных стоков толуолом или ксилолом экстрагируют
ванилин. Из тех же растворов метилэтилкетоном извлекают
муравьиную и уксусную кислоты.
ЭКСТРАКЦИЯ В ТЕХНОЛОГИИ
НЕОРГАНИЧЕСКИХ ВЕЩЕСТВ
Применение экстракции в технологии неорганических
веществ (исключая металлургию) сравнительно ограничено. Это
обусловлено тем, что одним из несмешивающихся растворителей
в экстракции чаще всего является вода, другим — органический
растворитель. Растворимость же неорганических веществ в
органической среде обычно невелика. Тем не менее жидкостная
экстракция получила распространение в некоторых важных
процессах разделения этой области технологии.
Получаемый электролизом 50%-ный раствор гидроокиси
натрия содержит примеси хлорида (около 1%) и хлората (около
650
Л
0,08%) натрия. Эти загрязнения экстрагируют жидким
аммиаком, который сравнительно мало растворим в крепкой щелочи.
Вследствие относительно высокой летучести аммиак легко
регенерируют из экстракта и рафината выпариванием.
Экстракцию применяют для выделения перекиси водорода из
реакционной смеси при получении ее новейшим
«автоокислительным» способом. В этом процессе 2-этилантрахинон
каталитически гидрируется в подходящей среде (нониловом спирте или
алкилзамещенных циклогексанола) в соответствующее
производное антрагидрохинона. При окислении последнего воздухом
получают снова антрахинон и Н202 в органическом
растворителе. Перекись экстрагируют обессоленной водой в очень
крупных экстракторах, чаще всего в колоннах с ситчатыми
тарелками или в насадочных колоннах. Экстракт, содержащий около
20% перекиси водорода, для извлечения следов растворителя
экстрагируют каким-либо углеводородом, который удаляют
продувкой воздухом. После этого раствор Н202 укрепляют
дистилляцией до концентрации, равной приблизительно 90%.
Производство фосфорной кислоты по новому «мокрому»
способу, позволяющему получать высококачественный продукт,
основано на реакции между соляной кислотой и фосфатной рудой.
Образующуюся фосфорную кислоту экстрагируют бутиловым
или амиловым спиртом. Полученный экстракт промывают водой
для извлечения НС1 и выпаривают для удаления растворителя
и остатков НС1. Бутанолом экстрагируют фосфорную кислоту
также из растворов, полученных сернокислотной обработкой
фосфоритов. Экстрагент можно регенерировать и другими
методами: извлекать фосфорную кислоту из экстракта водой или
экстрагировать растворитель каким-либо углеводородом.
Метиловый эфир борной кислоты (не являющийся, правда,
чисто неорганическим продуктом) получают при реакции
борной кислоты с метанолом. Отделение эфира от избытка
метанола, с которым он образует азеотропную смесь, осуществляют
экстракцией углеводородами. Борную кислоту можно также
извлекать из водных растворов высшими спиртами. Однако вряд
ли этот метод может приобрести промышленное значение.
Пригодные для производства полупроводников
высококачественные силиконы, получаемые из тетрахлорсилана, можно
очищать от следов металлов экстракцией дымящей серной кислотой.
Большое внимание привлекает возможность применения
экстракции для получения питьевой воды из морской воды и
рассолов. Эта проблема интенсивно изучалась, однако еще не
достигла стадии промышленной реализации. Предложен процесс
экстракции воды аминами, например метиламином или этилбу-
тиламином. При нагревании экстракт разделяется на два слоя—
обогащенный водой и обогащенный амином, из которых
выделяются конечные продукты.
6S1
Необходимо доказать конкурентоспособность этого метода с
другими (выпариванием, ионным обменом и т. д.). Кроме того,
проведение такого процесса вызывает следующие
принципиальные возражения: вода экстрагируется из рассола амином
вследствие того, что активность ее в последнем меньше, чем в
рассоле. Это значит, в свою очередь, что теоретически работа
выделения воды из раствора в аминах должна быть больше
работы, необходимой для выделения ее из рассола. Поэтому
желательно экстрагировать из морской воды или рассолов соли,
получая при этом воду в качестве рафината. Однако подходящего
для этого растворителя не найдено.
Известно, что жидкостную экстракцию используют для
разделения некоторых изотопов, хотя, по-видимому, практического
применения такие процессы не получили. Так, критические
температуры растворения D20 и Н20 с фенолом и 3-пиколином
достаточно отличаются друг от друга, и их смеси можно
анализировать на содержание ЬгО по критическим температурам
растворения. Это указывает на возможность разделения D20 и
Н20 экстракцией, однако селективность оказывается слишком
малой.
Разделение D20 и Н20 возможно в результате обменной
реакции между дейтерием и водородом при взаимодействии воды
с несмешивающимся с ней меркаптаном. Хотя этот способ не
является экстракцией в обычном смысле, для его осуществления
применимы основные методы жидкостной экстракции. В
качестве еще одного примера разделения изотопов экстракцией
можно указать на возможность разделения 10В и ИВ
фракционной экстракцией борной кислоты водой и изоамиловым спиртом.
ЭКСТРАКЦИЯ В ПРОЦЕССАХ
РАЗДЕЛЕНИЯ МЕТАЛЛОВ*
Жидкостная экстракция металлов или их соединений
известна давно. В металлургических процессах примеси распределяют-
* Более подробные сведения о применении жидкостной экстракции в
атомной технике приводятся в специальной литературе. См., например,
Материалы Международной конференции по мирному использованию атомной
энергии, Женева, 1955, т. 8, «Технология производства материалов,
используемых в атомной промышленности», Металлургиздат, 1958; Труды второй
Международной конференции по использованию атомной энергии, Женева, 1958,
Избранные доклады иностранных ученых, т. 7, «Технология атомного сырья»,
Атомиздат, 1959; Мартин Ф. С, Майлс Дж. Л., Химическая переработка
ядерного топлива, перев. под ред. Галкина Н. П., Металлургиздат, 1961;
Переработка ядерного горючего, под ред. Столера С. и Ричардса Р., сокр.
перевод под ред. Зефирова А. П., Атомиздат, 1964; Пушленков М. Ф„
Зильберман Б. Я., Переработка ядерного горючего (обзор II симпозиума
Организации Европейского экономического сотрудничества и развития, ОЕСД
в Брюсселе, 1963 г.), Атомная техника за рубежом, № 2, 31 (1966).—
Доп. ред.
652
1 ся между расплавленными металлами и шлаком. Эти процессы
. хорошо изучены и описаны. В дальнейшем изложении мы
ограничимся рассмотрением процессов выделения металлов из вод-
' ных растворов.
Общие принципы. В большинстве случаев для экстракции
металлов органическими растворителями необходимо разрушить
координационные связи между молекулами воды и ионами ме-
i таллов. Для растворителей с низкой диэлектрической
проницаемостью это возможно в результате образования
электрически нейтрального соединения с экстрагирующим растворителем
или находящимся в нем комплексообразующим агентом. Все
возможные подобные взаимодействия трудно рассмотреть.
Приводимая ниже приближенная их классификация окажется
полезной при дальнейшем рассмотрении проблемы.
1. Образование хелатов. Связанная координационной связью
вода замещается хелатным комплексом, представляющим собой
ковалентное соединение, растворимое в органических
растворителях. Такие комплексы образуют ацетилацетон, диметилглиок-
сим, 8-хинолин, купферон, теноилтрифторацетон и многие
другие соединения. Образованные металлами и этими веществами
; соединения растворимы в углеводородах и их хлорпроизводных,
;, причем коэффициенты распределения указанных соединений
между водой и органическими растворителями могут иметь
значения, достигающие нескольких тысяч. Хелатные комплексы ши-
• роко используют в аналитической химии, однако подобные
методы разделения очень дороги, и их нельзя применять в
промышленном масштабе.
2. Образование других комплексных соединений. Уран,
например, легко экстрагируется в виде комплекса U02 (N03)2X4,
где X — растворитель, которым может служить диэтиловый
эфир.
3. Образование соединений с ионной связью. В этом случае
положительный и отрицательный ионы образуют нейтральные
молекулы, экстрагируемые органическими растворителями. Так,
U02X6+ экстрагируется при образовании ионной связи с 2NO;T-
: Растворителем X может быть в данном случае бутиловый спирт.
В практическом отношении последний случай представляет
собой наибольший интерес. Чаще всего экстрагируются
следующие соединения:
а) Галоиды. В большей части случаев эти соединения
экстрагируются кислородсодержащими растворителями.
б) Т и о ц и а н а т ы. Для экстракции тиоцианатов также
применяют кислородсодержащие растворители (эфиры, кетоны
и др.).
в) Нитраты. Кислородсодержащие растворители
экстрагируют также нитраты. Чаще всего нитраты и тиоцианаты
экстрагируют трибутилфосфатом (ТБФ)., например в виде комплекса
6S3
и02(Ы03)2ТБФ. Такие соединения экстрагирует сам ТБФ,
однако для уменьшения вязкости последний часто разбавляют
каким-либо углеводородом. Комплекс U02(N03)2TBO легко
экстрагируют подобной смесью.
г) Перхлораты. Эти соединения также экстрагируют
кислородсодержащими растворителями.
д) Соли алкилфосфорных кислот, например ди-
(2-этилгексил)фосфорной кислоты. Эти реагенты обычно
разбавляют неполярным растворителем, чаще всего
углеводородами. Экстракцию таких соединений часто называют «жидким
ионным обменом», с которым она действительно сходна. Так, уран
из водного раствора экстрагируют раствором алкилфосфорной
кислоты в керосине по следующей реакции:
U02S04 + 2(RO)2P(0)OH r+±. H2S04 + [(RO)2P(0)0]2U02
(вода) (керосин) (вода) (керосин)
где R — алкильная группа.
е) Соединения окиси т р и а л к и л ф о сф и н а
(например, окись н-октилфосфина). Эти реагенты обычно
разбавляют углеводородами, которые экстрагируют образующийся
комплекс металла.
ж) Амины, обычно высшие (триоктил-, трилаурил-, метил-
диоктиламины), применяют в смеси с углеводородами.
з) Соли некоторых карбокислот (бензоаты, ок-
салаты, салицилаты) экстрагируют металлы в смеси с
кислородсодержащими растворителями.
и) Соли некоторых гетерополярных соедине-
н и й (например, вольфрамфосфорные кислоты) также
экстрагируют кислородсодержащими растворителями. Разделять в
такой форме два металла трудно вследствие малой
селективности. Это обусловлено тем, что в таких больших молекулах могут
присутствовать и соэкстрагироваться оба металла.
Не следует полагать, что рассмотренные выше соединения
любых металлов всегда способны экстрагироваться. Так, хлорид
лития экстрагируется высшими спиртами, а хлорид натрия не
экстрагируется. Большое значение имеют среда (рН) и
валентность металла. Например, ион окисного (Fe3+) железа легко
экстрагируется в виде хлорида, тиоцианата, алкилфосфата и
других солей, в то время как ион закисного железа (Fe2+) не
экстрагируется. Вообще с точки зрения возможности экстракции
Fe2+ и Fe3+ ведут себя как два различных элемента. Большое
влияние оказывает также присутствие высаливающих агентов.
Подбирая рН и валентность металла, часто можно разделить
очень трудноразделимые элементы. Экономически выгодно
регенерировать экстрагент изменением направления экстракции, что
происходит во многих случаях в результате изменения рН или
валентности металла.
654
Способность образовывать экстрагируемые комплексные
соединения в наибольшей степени характерна для переходных
металлов, но при соответствующих условиях почти любой металл
можно отделить от других жидкостной экстракцией, если,
конечно, отвлечься от экономической стороны такого разделения.
До настоящего времени только наиболее дорогие металлы
выделяются или концентрируются в промышленном масштабе с
помощью жидкостной экстракции.
Стимулом для применения жидкостной экстракции в
процессах разделения металлов в крупном масштабе первоначально
послужили потребности атомной промышленности. По-видимому,
впервые экстракцию использовали для очистки урана (уранил-
нитрат экстрагировали эфиром) перед разделением изотопов на
ранней стадии развития атомной техники.
Экстракцию применяют в атомной промышленности для
решения двух проблем: а) очистки ядерного топлива и
конструкционных металлов при выделении их из руд; б) регенерации
облученного ядерного топлива. Теперь экстракцию используют
также для разделения металлов и в различных других областях,
не связанных с атомной техникой.
Извлечение урана. Для применения урана в качестве
источника атомной энергии его нужно отделить от других
металлов, содержащихся в урановой руде. Такие методы
разделения, как флотация, в данном случае оказываются
неудовлетворительными. Обычно уран выщелачивают из мелко
измельченной руды. Иногда выщелачивание производят после термической
обработки, проводимой для разложения солей с тем, чтобы
перевести в растворимое состояние находящийся в руде
ванадий.
Выщелачивание ведут кислотами (серной, азотной, соляной
или их смесями) или раствором соды в зависимости от
природы руды. Раствор после выщелачивания содержит уран,
концентрация которого относительно низка, и другие металлы.
Уран можно выделять химическими способами, ионным обменом
или жидкостной экстракцией. Особенности процесса выделения
в значительной степени определяются природой исходной руды.
г Для экстракции урана из щелоков чаще всего применяют
трибутилфосфат, додецил- или ди(2-этилгексил) фосфорную
кислоту («Далекс-процесс»). В качестве разбавителя обычно ис-
црльзуют керосин. При выделении урана из фосфорнокислых
растворов, получаемых из фосфатных руд, применяют октил- или
децилпирофосфорную кислоту в керосине. Раствор после
выщелачивания можно восстановить S02 или металлическим железом
для перевода железа в неэкстрагируемое состояние; рН
раствора устанавливается на уровне, благоприятствующем экстракции
урана. После извлечения урана, изменяя кислотность, можно
экстрагировать ванадий из рафината тем же растворителем. Из
655
экстрактов уран и ванадий выделяют с помощью
концентрированных растворов НС1, HF или соды.
Экстракцию чаще всего проводят в смесительно-отстойных
аппаратах; при переработке фосфатного сырья после смешения
фаз их"разделяют на центрифугах. На одном из заводов
применяют также центробежные экстракторы Подбильняка.
Экстракция позволяет повысить концентрацию урана в растворе в
несколько сот раз, причем при обработке в трех-пяти ступенях
степень извлечения урана достигает 99Д%.,и более, ОбычнсГуран
экстрагируют из осветленных щелоков. Иногда, однако,
производят экстракцию из неосветленных растворов. В настоящее
время исследуется возможность применения аминов для
экстракции урана («Амекс-процесс»).
Дальнейшую очистку.урана производят экстракцией уранил-
нитрата трибутилфосфатом, разбавленным керосином (при вы-
деленййурана из экстракта азотной кислотой). Для повышения
эффективности ступени керосин можно заменять менее вязкими
углеводородами, например-тексан'ом. На" некоторых заводах
применяют экстракцйю~"Миловым"Тфиром или
метилизобутилкетоном п£и_лабавлении нитрата аммония в качестве
высаливающего средства. Реэкстракцй'юосуществля'ют"водой. В результате
получают уран высокой степени чистоты, который затем
перерабатывают на металл или в случае необходимости разделения
изотопов — на гексафторид.
Регенерация облученного ядерного горючего. Облученное
ядерное горючее можно перерабатывать для регенерации
неиспользованного топлива и отделения продуктов распада.
Известно несколько способов регенерации ядерного горючего; в
некоторых из них используют жидкостную экстракцию. По одному
способу расплавленный облученный уран экстрагируют несмеши-
вающимися с ним жидкими металлами (медью, серебром) или
расплавами солей. Однако этот способ едва ли найдет
промышленное применение. Другой способ заключается в «мокрой»
переработке отработанного ядерного горючего — экстракции его
из водных растворов, причем известно несколько вариантов
регенерации ядерного топлива этим способом.
«Редокс-процесс». В этом процессе кислый раствор
отработанного ядерного горючего окисляют, чтобы перевести плутоний
в форму Ри6+ (которая легче экстрагируется), после чего
нитраты уранила и плутонила экстрагируют метилизобутилкетоном
или дибутилкарбитолом. Другие продукты распада,
перешедшие в экстракт, отмывают водой. Затем экстракт обрабаты-,
вают водным раствором, содержащим восстановитель,
переводящий плутоний в Ри3+, и высаливатель (например, нитрат
алюминия), препятствующий реэкстракции урана из органической
фазы, в то время как плутоний выделяется из нее. Уран
извлекают из органического растворителя отдельно. Как уран, так и
656
рлутоний многократно экстрагируют и реэкстрагируют для
дальнейшей очистки. В конце концов выделяют уран, плутоний и
Другие продукты распада. Описанный процесс можно несколько
видоизменить в зависимости от вида обработанного ядерного
горючего.
«Пурекс-процесс». В этом процессе в качестве растворителя
Применяют трибутилфосфат в керосине. Это дает возможность
использовать в качестве высаливателя крепкую азотную
кислоту, в среде которой метилизобутилкетон не устойчив. В
основном данный процесс аналогичен «Редокс-процессу», но
отличается от него в деталях. Например, четырехвалентный плутоний
легче экстрагируется трибутилфосфатом, чем шестивалентный.
«Торекс-процесс». Этот процесс предназначен для разделения
урана, тория и продуктов распада, получающихся при
облучении тория в реакторе-размножителе. Экстрагентом служит
раствор трибутилфосфата в парафинистом керосине (в присутствии
ароматических углеводородов возможно образование третьей
фазы). В качестве высаливателя используют нитрат алюминия.
Разделение достигается при троекратном проведении операций
экстракции, промывки и реэкстракции.
Продукты распада. Продукты распада содержат ряд
металлов, из которых с помощью жидкостной экстракции выделяют
редкоземельные и некоторые другие элементы. Полученные ис-
; кусственным путем трансурановые элементы также очищают
экстракцией.
Извлечение тория. Получаемый из монацитовых песков
торий обычно отделяют от попутных редкоземельных элементов
химическими методами. Концентрат тория в дальнейшем
очищают от примесей других металлов экстракцией. Торий
извлекают эфиром, раствором трибутилфосфата в керосине в виде
нитрата, амиловым спиртом в виде тиоцианата или, наконец,
v высшими аминами.
Разделение циркония и гафния. Вследствие коррозионной
! стойкости и малого поперечного сечения для поглощения тепло-
{ вых нейтронов цирконий имеет важное значение в производстве
! ядерной энергии. Гафний, естественный спутник циркония,
обладает большим поперечным сечением и в связи с этим служит
отличным замедлителем ядерных реакций. Для использования
каждого из этих металлов их необходимо разделить. Цирконий
и гафний настолько близки друг к другу в химическом
отношении, что в течение ста лет после открытия циркония гафний не
был в нем обнаружен, хотя и содержался во всех образцах
циркония.
Разделить гафний и цирконий химическими методами
чрезвычайно трудно. Для разделения этих элементов разработано
несколько экстракционных процессов. Наиболее важен в про-
42 Р. Е. Трейбал
6S7
мышленном отношении процесс дробной экстракции в виде тио-
цианатов из солянокислого раствора метилизобутилкетоном.
Гафний и значительное количество циркония переходят в
органический растворитель, а в рафинате остается цирконий, не
содержащий гафния. Рафинат обрабатывают кетоном для
удаления тиоцианата. Обогащенный гафнием экстракт промывают
соляной кислотой для удаления циркония, затем серной
кислотой извлекают гафний и водным раствором аммиака — тиоциа-
наты. Цирконий и гафний можно разделять также с помощью
трибутилфосфата.
Разделение тантала и ниобия. В индивидуальном виде
тантал и ниобий получили применение в химическом
машиностроении как коррозионностойкие материалы. Их используют
также для изготовления конденсаторов и выпрямителей, а ниобий
нашел некоторое применение в тепловых ядерных реакторах
Тантал и ниобий встречаются совместно и в химическом
отношении очень близки друг другу. До недавнего времени
единственным промышленным методом их разделения служил
классический метод Маринака, предложенный .еще в 1866 г. Этот
малопроизводительный метод основан на дробной
кристаллизации калиевых дифторидов этих металлов.
По одному из современных методов разделения тантала и
ниобия их окислы переводят в плавиковокислый раствор, и оба
металла экстрагируются метилизобутилкетоном. Сопутствующие
металлы, ниобий и тантал, выделяют дифференцированно из
экстракта промывкой разбавленной серной кислотой различной
концентрации. В последней стадии металлы осаждают
аммиаком. Для получения очень чистых металлов необходимо
сравнительно немного ступеней экстракции.
Разделение 'Других металлов. Хотя сведения о
промышленном разделении других металлов с помощью жидкостной
экстракции отсутствуют, представляется возможным экономически
выгодное разделение экстракцией некоторых из них. Кобальт и
никель можно разделять экстракцией триизооктиламином в виде
хлоридов или метилизобутилкетоном в виде тиоцианатов.
Разделение редкоземельных элементов возможно с помошью ал-
килфосфорных кислот или трибутилфосфатов.
Бериллий из растворов после выщелачивания экстрагируют
диалкилфосфорными кислотами в керосине. Литий можно
отделять от щелочных металлов экстракцией хлорида лития
бутиловым или амиловым спиртом. Вольфрам и молибден
можно разделить экстракцией метилизобутилкетоном из кремний-
содержащих кислых растворов. Германий и мышьяк
разделяются экстракцией хлоридов четыреххлористым углеродом.
Галлий от сопутствующих металлов можно отделить
экстракцией эфиром или трибутилфосфатом в виде хлоридов.
Ванадий извлекают из нефтяных топлив пиридином.
658
ИЗБРАННАЯ ЛИТЕРАТУРА
Экстракция в процессах
нефтехимического синтеза
1. Е. Т. Barrows, W. L. S e d d о n, Separation and Utilization of C4
Hydrocarbons, Chem. a. Ind., 1953, S57.
2 J С L. D e f i z e, On the Edeleanu Process for the Selective Extraction of
Mineral' Oils, D. B. Centne's Uitgevers-Maatschappij, N. V., Amsterdam,
1938
3. A. Ei Dunstan, A. W. Nash, В. Т. Brooks, H. Tizard (ed.), The
Science of Petroleum, v. 1—5, Oxford Univercity Press, New York, 1938—
1953
4. С A. Duval, R. T. Mai in, V. А. К a 1 i с h e v s k y, Dualayer Distillate
Fuel-Oil-Treating Process, Proc. Am. Petrol. Inst., Sect. Ill, 34, 321, 325
(1956). , „ ,
5 A W Francis, W. H. King, in «The Chemistry of Petroleum
Hydrocarbons», B. T. Brooks, С E. Boord, S. S. Kurtz, L. Schermling (ed.),v. I,
Reinhold Publishing Corporation, New York, 1954, p. 197—240.
6 G С Gester Solvent Extraction in the Petroleum Industry, Advances
in Chem. Ser., 5, 177 (1951). _гоч
7 H W Grote The Udex Process, Chem. Eng. Progr., 54, № 8, 43 (1958);
Proc. Am. Petrol. Inst., Sect. Ill, 39, 248 (1959).
8 V A Kalichevsky, Refining of Lubricating Oils, Petrol. Eng., 28,
№ 2, СИ; № 3, C70; № 5, C20 (1956). .
9. V. A. Kalichevsky, K. A. Kobe, Petroleum Refining with Chemicals,
D Van Nostrand Company, Inc., Princeton, New York, 1956.
10 К A Kobe J. J McKetta (ed.), Advances in Petroleum Chemistry
and Refining,' v. 1 (1958); v. 2 (1959), Interscience Publishers, Inc., New
York.
11 Modern Refining Processes, Oil a. Gas J., 50, № 45, 187 (1952).
12. W. F. Wilkinson, J. R. G h u b 1 i k i a n, P. О b e r g f e 11, The Sulfur
Dioxide Process, Chem. Eng. Progr., 49, 57 (1953).
Экстракция в технологии жиров,
масел и родственных процессах
13 Н D Allen, W. A. Kline, E. A. Lawrence, С. J. A r r о w s m i t h,
С J.Marse'l, Continious Hydrolysis of Fats, Chem. Eng. Progr., 43, 459
(1947)
14. K. J. Bradley, F. H. Smith, Vegetable Oil Refining, Ind. Eng. Chem.,
15 S W Gloyer, Furans in Vegetable-Oil Refining, Ind. Eng. Chem., 40,
228 (1948). , ._, , A
16 S W Gloyer, Fractionation of Drying Oils and Acids, J. Am. Oil
Chem. Soc, 27, 462 (1950). . ^ .
17 H К Kirschenbauer, Fats and Oils, An Outline of their Chemistry
and technology, 2nd ed., Reinhold Publishing Corporation, New York, 1960.
18. L. La scary, Industrial Fat Spliting, J. Am. Oil Chem. Soc, 29, 362
(1952)
19 H J Passino, The Solexol Process, Ind. Eng. Chem., 41, 280 (1949).
20 F' J. H. Riche, A. L. S t u b b s, New Drying Oils Produced by Solvent
Segregation, J. Am. Oil Chem. Soc, 37, 8 (1954).
Экстракция в коксохимической
промышленности
21 Н R Batchel der, R. В. Fi lbert, \V. H. Mink, Processing of Low
Temperature Lignite Tar, Ind. Eng. Chem., 52, 131 (1960).
42*
6S9
22. H. S. Block, R. S. Wackier, V. E. H e n n e y, Recovery of Pure
Aromatics by Udex Extraction of Coal Tar Light Oils, Gas World 145.
520 (1957).
23. J. С McCord, Design and Operation of a Phenol Recovery Plant
Sewage a. Ind. Wast., 26, 1014 (1954).
24. M. B. N e u w о r t h, V. Hofmann, Т. Е. Kelly, Fractional Extraction
Process for Recovery of Pure Tar Acids, Ind. Eng. Chem., 43, 1689 (1951).
25. H. Weigmann, Twenty-five Years of Phenol Recovery, Qluckauf, 90, 780
(1954).
26. P. J. Wilson, J. H. Wells, in «The Chemistry of Coal Utilisation»,
H. H. Lowry (ed.), John Wiley and Sons. Inc., New York, 1945.
Экстракция в фармацевтической
промышленности
27. Е. Gaden, Biochenicals Processing, Chem. Eng., 64, № 5, 237 (1957).
28. J. J. H. Hastings, Ten Years of Penicillin Manufacture, Ind. Chem., 31,
494 (1955).
29. G. Ross, Production of Antibiotics, Brit. Chem. Eng., 3, 8 (1958).
Экстракция в технологии
органических веществ
30. L. A Agnello, W. H. Williams, Synthetical Phenol, Ind. Eng. Chem.,
52, 894 (1960).
31. P. Eagles field, В. К. Kelly, J. F. Short, Recovery of Acetic Acid
from Dilute Aqueous Solutions by Liquid-Liquid Extraction, Ind. Chem.,
29, 147, 243 (1953).
32. D. F. Othme r, Acetic Acid Recovery Methods, Chem. Eng. Progr., 54,
№ 7, 48 (1958).
33. D. G. Weather, W. А. В г i g g s, Chemical Recovery from Pulp-mill
Waste Liquor, Ind. Eng. Chem., 53, 773 (1961).
Экстракция в технологии
неорганических веществ
34. A New Process for Hydrogen Peroxide, Ind. Chem., 35, 9 (1959).
35. А. В a n i e 1, R. В 1 u m b e r g, A. S 1 о n, Phosphoric Acid by Liquid-Liquid
Extraction, Brit. Chem. Eng., 4, 227 (1959); Dechema Monograph 33,
57 (1960).
36. С S. Cronan, Chemical Route to Peroxide, Chem. Eng., 66, 118 (Apr 6,
1959).
37. D. W. Hood, R. R. Davison, The Place of Solvent Extraction in Saline
Water Conversion, Advances in Chem. Ser., № 27, 40 (1960).
38. H. C. T w i e h a u s, N. J. О h 1 e r s, Caustic Soda Purified Ъу Liquid-Liquid
Extraction, Chem. Ind., 63, 230 (1948).
Экстракция в процессах
разделения металлов
39. D. J. Bauer, А. С. Rice, J. S. Berber, Liquid-Liquid Extraction of
Rare Earth Metals, U. S. Bur. Mines Rept. Invest., № 5570 (1960).
40. F. R. Bruce, J. M. Fletcher, H. H. Human (ed.), Progress in
Nuclear Energy, Series III. Process Chemistry, v. 2, Pergamon Press, New
York, 1959.
41. F. R. Bruce, J. M. Fletcher, H. H. Human, J. J. Katz (ed.),
Process Chemistry, Progress in Nuclear Energy, Series III, v, 1, McGraw-Hill
Book Company, Inc., New York, 1956.
660
42. T. A. Butler, E. E. К e t с h e n, Solvent Extraction Separation of Cerium
and Yttrium from Other Rare Earth Fission Products, Ind. Eng. Chem.,
53, 651 (1961).
43. С. Н. Chilton, Niobium and Tantalum, Chem. Eng., 65, № 22 104,
(1958).
44. J. W. С 1 e g g, D. D. F о 1 e y, Uranium Ore Processing, Addison-Wesley
Publishing Company, Reading, Mass., 1959.
45. J. E. Flagg (ed.), Chemical Processing of Reactor Fuels, Academic Press,
Inc., New York, 1961.
46. H. M. Irving, F. J. С R о s s о 11 i, R. J. P. Williams, D. E. С h a 1-
k 1 e y, A. General Treatment of the Solvent Extraction of Inorganic
Compounds, J. Chem. Soc, 1955, 1906, 1920, 1927, 1938, 1946.
47. J. M. Jacobs, Reprocessing of Irradiated Fission Reactor Fuel and
Breeding Materials an annotated bibliography, U. S. Atomic Energy,
Comm, 1958.
48. E. L. Koerner, M. S m u t z, H. A. W i 1 h e 1 m, Separation of Niobium
and Tantalum by Liquid Extraction, U. S. Atomic Energy Comm. ISC-802
(1956); Chem. Eng. Progr., 54, № 9, 63 (1958).
49. В. И. Кузнецов, Теоретические основы жидкостной экстракции
металлов, Успехи химии, 23, 654 (1954).
50. G. L. М i 1 1 е г, Tantalum and Niobium, Metallurgy of Rarer Metals, № 6,
Academic Press, Inc., New York, 1960.
51. Proceedings of the International Conference on the Peaceful Uses of
Atomic Energy, v. 7, 8, 9, United Nations, New York, 1956.
52. Proceedings, United Nations International Conference on the Peaceful Uses
of Atomic Energy, 2nd, Geneva, 1958, v. 3, 4, 17, 28, United Nations, New
York, 1958.
53. W. A. S t i с k n e y, Zirconium-Hafnium Separation, U. S. Bur. Mines Rept.
Invest, № 5499 (1959).
54. Symposium on the Reprocessing of Irradiated Fuels Held at Brussels,
Belgium, May 20—25, 1957, U. S. Atomic Energy Comm, TID-7534, Book 1
(1957).
55. United States Atomic Energy Commision, Chemical Processing and
Equipment, McGraw-Hill Book Company, Inc., New York, 1955.
56. B. Weaver, F. А. К a p p e 1 m a n n, А. С. Т a p p, Quantity Separation
of Rare Eearths by Liquid-Liquid Extraction, J. Am. Chem. Soc, 75, 3943
(1957).
ЗАДАЧИ
Глава
Н-1. Имеются равновесные данные для системы
анилин — гептан — толуол при 0° С. [D u r a n d e t,
Glad el, Rev. Inst, franc, petrole, 9, 296 (1954)]:
бинодальная кривая
Анилин,
вес. %
Гептан,
вес. %
Толуол,
вес. %
2,2
97,8
0
97,0
3,0
0
7,0
70,6
22,4
43,9
46,0
10,1
65,8
11,0
23,2
25,3
41,4
33,3
76,6
7,1
16,3
89,7
4,1
6,2
46,8
23,2
30,0
36,4
31,2
32,4
хорды равновесия
Толуол в углеводородной
фазе, вес. %
Толуол в фазе анилина,
вес. %
0
0
5,2
3,0
13,6
6,0
20,5
8,2
26,7
10,0
а) Построить треугольную диаграмму
растворимости.
б) Построить кривую распределения.
в) Построить диаграмму равновесия в
координатах Хэнда и определить состав в критической
точке.
г) Построить диаграмму равновесия в
координатах Отмера — Тобиаса.
д) Определить весовые количества каждой фазы
и равновесные концентрации для смеси, состоящей
из 60 кг анилина, 20 кг толуола и 20 кг гептана.
П-2. С помощью метода Хэнда рассчитать
распределение ацетона между водой и трихлорэтаном
по двум известным хордам равновесия в системе
вода (А)— 1,1,2-трихлорэтан (В)—ацетон (С).
[Treybal, Weber, Daley, lnd. Eng. Chem., 38,
817 (1946)].
Равновесные концентрации приведены ниже (в
вес. %):
ХАА
0,8016
0,4170
ХВА
0,0079
0,0652
ХСА
0,1905
0,5178
ХАВ
0,0133
0,1340
хвв
0,7101
0,2626
хсв
0,2766
0,6034
662
Результаты расчета сравнить с опытными данными:
ХСА
хсв
ХСА
хсв
0,0596
0,0875
0,2692
0,3852
0,0651
0,1028
0,3085
0,4297
0,1397
0,2078
0,3573
0,4821
0,1704
0,2514
0,4090
0,5395
0,1905
0,2766
0,4605
0,5740
0,2600
0,3706
0,5178
0,6034
П-3. Коэффициент распределения муравьиной кислоты тс между метил-
изобутилкетоном (В) и водой (А) равен 0,42 при 25° С и 0,37 при 54,8° С
(концентрация муравьиной кислоты в водном растворе 0,48 моль/л) по
данным Уайтхэда и Джанкоплиса [lnd. Eng. Chem., 47, 2114 (1955)]. Рассчитать
коэффициент распределения при 35° С и определить теплоту перехода
муравьиной кислоты из воды в кетон, считая объемы растворов достаточно
большими, чтобы можно было пренебречь изменением концентрации. Ответ:
0,3995; —3,49 • 106 дж/кмоль.
11-4. З-Пиколин в водном растворе ведет себя как одноосновное
основание, диссоциируя с образованием иоиа ОН".
а) Вывести выражение для коэффициента распределения 3-пиколииа
между водой и органическим растворителем, предполагая, что
распределяются только недиссоциированные молекулы пиколина. Коэффициент
распределения выразить в функции от концентрации ионов водорода в водной
фазе Н+ и константы диссоциации Ка, где Ka = Kw/Kd; Kw — константа
диссоциации воды; Kd — константа диссоциации, выраженная через
концентрацию ионов ОН*.
б) По данным Голамбика и Орчина [J. Am. Chem. Soc, 72, 4145 (1950)],
величина /С=10~5'8. Коэффициент распределения 3-пиколина между
раствором фосфатного буфера (рН = 4) и бензолом равен 0,22 (при выражении в
аналитических концентрациях пиколина). Определить величину
коэффициента распределения для фосфатного буфера с рН = 5,4. Ответ: 4,01.
Экспериментальная величина, найденная Карром и Шайбелем [lnd. Eng. Chem., 46, 1583
(1954)], равна четырем.
11-5. Ниже приведены данные о распределении уксусной кислоты (С)
между бензолом (В) и водой (А) при 25° С:
1С:
п в'
моль/л
[С ] моль/л
0,0125
0,532
0,0165
0,634
0,0443
1,148
0,0714
1,510
0,1015
1,858
0,158
2,59
Проанализировать эти данные с точки зрения простейшего закона
распределения и поправок к нему.
11-6. а) Вывести зависимость между концентрацией Н+ в водной фазе
и коэффициентом распределения тс (в общих концентрациях) двуосновной
кислоты С между водой и органическим растворителем В как функцию тс
(коэффициента распределения недиссоциированных молекул кислоты), Kdi и
Kd2- Считать, что ассоциация в органической фазе отсутствует и тс= const.
б) Вывести уравнение для расчета концентрации ионов водорода в
водной фазе, образующихся при диссоциации двуосновной кислоты, исходя из
общей ее концентрации Ст и констант диссоциации Kd\ и Kd2-
в) При 25° С концентрации щавелевой кислоты в воде, равной
0,089 моль/л, соответствует равновесная концентрация в эфире 0,0041 моль/л.
С помощью полученных выше зависимостей рассчитать равновесные
концентрации в эфирном слое для следующих концентраций в водной фазе: 0,01025;
0,298 и 0,417 моль/л. Константы диссоциации: Kdi = 6,5 • Ю-2; /Cd2=6,l • 10-s'
Ответ: 0,000136; 0,01985; 0,0314. Опытные величины (Internal Critical Tables,
v. Ill, p. 423) соответственно равны: 0,000168; 0,0200; 0,0300.
II-7. При распределении муравьиной кислоты между этиловым эфиром
(В) и водой (А) концентрации в водной фазе 0,02 моль\л соответствует
равновесная концентрация в эфире 0,00746 моль/л (18° С). Рассчитать
равновесную концентрацию муравьиной кислоты в эфнре, если в водной фазе (при
концентрации 0,02 моль/л НСООН) присутствует 0,02 моль/л муравьинокис-
лого натрия. Константа диссоциации НСООН в водном растворе равна
1,76-Ю-4. Принять коэффициент распределения недиссоциированных молекул
кислоты постоянным. Ассоциация в органической фазе отсутствует. Ответ-
0,0081 мо Ль /л.
П-8. а) Вывести уравнения для расчета одновременного распределения
трех одноосновных органических кислот между водой и органическим
растворителем, если кислоты диссоциируют в водной фазе (диссоциации или
ассоциации в органической фазе не происходит).
б) Коэффициенты распределения тс муравьиной, уксусной и пропионо-
вой кислот между метилизобутилкетоном и водой при концентрации в
водной фазе 0,05 моль/л и /=25° С соответственно равны 0,42; 0,54; 1,9.
Константы диссоциации этих кислот соответственно равны 1,76 • 10~4; 1,75 - 10~5
и 1,4 • 10"5. Рассчитать равновесные концентрации в фазе метилизобутилке-
тона, если все три кислоты распределяются одновременно и концентрация
каждой из них в водной фазе составляет 0,05 моль/л. Принять, что
распределяются только недиссоциированные молекулы кислот. Ответ: 0,0211;
0,0274; 0,0964.
П-9. Показать математически, что метод изображения равновесных
данных для тройных систем, предложенный Смитом [Chem. Eng., 54, 123 (1947)],
применим только к тем системам, которые поддаются обработке по методу
Хэнда [J. Phys. Chem., 34, 1961 (1930)], причем в соответствии с этим
методом хорды равновесия на диаграмме занимают горизонтальное положение.
Показать также, что для данных систем хорды равновесия прн продолжении
пересекаются в одной точке,
Глава III
111—1 - Коэффициент распределения ацетона (С) между водой (А) и
бензолом (В) при бесконечном разбавлении и /=15° С равен 3,84 (в случае
выражения концентраций в мольных долях). Парциальные теплоты
растворения при бесконечном разбавлении приведены ниже для температуры 15° С:
Растворенное вещество
Ацетон
Бензол
Ацетон
Вода
Растворитель
Бензол
Ацетон
Вода
Ацетон
я АН*
дж/кмоль
растворенного вещества
1300000
1088 000
—10 880 000
—377000
664
1
Рассчитать коэффициенты распределения при 30 и 45° С, приняв теплоты
^растворения постоянными. Ответ: 30° С, 4,95; 45° С, 6,41. Опытные
значения тс, полученные Бриггсом и Комингсом [Ind. Eng. Chem., 35, 411
(1943)], соответственно равны 4,71 и 5,85.
II1-2. Определить коэффициент распределения ацетона (С) между водой
(В) и н-гексаном (А) при бесконечном разбавлении и /=25° С:
а) исходя из констант Ван-Лаара для системы ацетон—вода,
приведенных на рис. 44 (см. стр. 77), и состава азеотропной смеси (53,5 вес. %
ацетона, температура кипения 49,7° С при 760 мм рт. ст. в системе ацетон —
к-гексан). Ответ: 3,46 (при выражении концентраций в весовых долях);
б) с помощью табл. 6 (см. стр. 100). Ответ 4,25 (прн выражении
концентраций в весовых процентах). Опытная величина, по данным Трейбала и
Уондрэка [Ind. Eng. Chem., 41, 1761 (1949)], равна 3,5.
111-3. Рассчитать распределение этилового спирта между глицерином и
бензолом при обычной температуре, исходя из приводимых ниже данных.
Взаимной растворимостью глицерина и бензола пренебречь.
Давления паров над растворами этилового спирта и глицерина прн 15°С
(Internet. Critical Tables, v. Ill, p. 360):
Этиловый спирт,
вес. %
Давление, мм
pm. cm.
Этиловый спирт,
вес. %
Давление, мм
pm. cm.
2
5
22,5
22,8
5
10
27,5
24,1
5,5
10,2
33,5
25,3
9,7
14,9
44,0
26,8
10
15,2
53,0
28,5
14
18,6
67,5
29,9
16,5
19,3
100
32,5
18,3
21,0
—
—
Общее давление паров в системе этанол — бензол прн 34,8° С:
Этиловый спирт,
вес. %
Давление, мм
рт. ст.
Этиловый спирт,
вес. %
Давление, мм
рт. ст.
0
147
32,15
197
2,07
174
50,15
190
3,85
185
64,91
178
9,47
195
79,88
155
17,38
198
93,28
122
25,64
198
100
103
Результаты расчета сравнить с данными Мак-Дональда [J. Am. Chem.
Soc, 62, 3183 (1940)].
111-4. По данным Джонсона и Фрэнсиса [Ind. Eng. Chem., 46, 1662
(1954)], взаимная растворимость, в системе я-гептан (А)—диэтиленгликоль
(В) равна 0,75 и 99,93 вес.% гептана, а взаимная растворимость бензола (С)
и диэтиленгликоля (В) составляет 5,9 и 67,8 вес.% гликоля при 50° С.
Рассчитать константы Ван-Лаара и найти предельную селективность
диэтиленгликоля (при *с = 0) в отношении разделения бензола и и-гептана при 50° Г
Ответ: Рсл = 29,4 (Джонсон и Фрэнсис получили (5СЛ = 28).
666
>
111-5. Согласно Фрэнсису [Ind. Eng. Chem., 36, 1096 (1944)], критическая
температура растворения смесей триэтиленгликоля и бензола равна 22° С,
Плотность триэтиленгликоля при 25° С составляет 1,1195 г/см3.
а) Исходя из этих данных, найти параметр растворимости
триэтиленгликоля для его смесей с ароматическими углеводородами и рассчитать
критическую температуру растворения смесей триэтиленгликоля и толуола. Ответ;
103° С (по данным Фрэнсиса 90° С).
б) Определить, какова должна быть критическая температура
растворения смесей триэтиленгликоля и бензола, чтобы рассчитанная и опытная
величины критической температуры растворения смесей триэтиленгликоля и
толуола совпадали друг с другом. По данным Эль Рифаи [Riv. combust., 11,
811 (1957)], триэтиленгликоль и бензол даже при 25° С растворяются только
частично. Рассчитанная величина критической температуры растворения
очень чувствительна уже к небольшим изменениям параметра растворимости.
В результате чего еще может возникнуть расхождение между опытными и
расчетными величинами критической температуры растворения?
111-6. Необходимо рассчитать распределение изопропилового спирта (С)
между водой (А) и этилацетатом (В) и определить селективность этилаце-
тата в качестве экстрагента для извлечения изопропилового спирта из воды,
пользуясь следующими данными:
а) Система А—В: при 20° С взаимная растворимость равна 7,94 и
96,99 вес. % этилацетата (Internal Critical Tables, v III).
б) Система В—С: азеотропная смесь, содержащая 30,5 мол. % изопро-
панола, с температурой кипения, равной 74,8° С при 760 мм рт. ст.
в) Система А—С: коэффициенты активности в этой системе, рассчитанные
из данных о равновесии пар — жидкость при 760 мм рт. ст., приведены в
работе Брюньеса и Богарта [Ind. Eng. Chem., 35, 255 (1943)]:
хс
Ус
Уа
0,9319
1,012
2,963
0,9! 53
1,014
2,912
0,8872
1,022
2,741
0,8520
1,027
2,657
0,0016
11,68
1,007
0,0083
10,54
1,020
0,0136
10,54
1,008
0,02040
8,00
1,102
0,0488
8,926
1,033
0,0843
5,873
1,049
0,1232
4,531
1,067
Определить константы Ван-Лаара для каждой бинарной системы и
далее с помощью уравнений Ван-Лаара второго порядка рассчитать предельную
величину коэффициента распределения изопропилового спирта (в
концентрациях, выраженных в весовых долях) и селективность. Результаты расчета
сравнить с опытными величинами, полученными Бичем и Гласстоном [J. Chem.
Soc, 1938, 67): Х'св/х'СА = 1,2 и Р'сл = 37 при 20° С. Ответ: X'CBJX'CA =
= 1,51; Рсл = 46,2.
II1-7. Следующие данные, полученные Пенингтоном и Map видом [Ind.
Eng. Chem., 45, 1378 (1953)], можно использовать для расчета равновесия в
системе н-гептан (А)—фурфурол (В) — циклогексаи (С) при 30° С.
Критические температуры растворения в бинарных системах А—В и С—В равны
соответственно 93,8 и 66,4° С. Взаимные растворимости при 30° С: система А—В,
ХАВ = °'061; ХАА = °'940' СИСТема В~ С> ХСВ = °>154°; ХВС = 0,°66- КоЭФ'
фициент распределения циклогексана между гептаном и фурфуролом при
бесконечном разбавлении равен 10 (для концентраций в мольных долях).
а) Определить коэффициент распределения циклогексана между гептаном
и фурфуролом при бесконечном разбавлении и селективность фурфурола как
экстрагента для разделения этих углеводородов при 30° С, исходя из
величин критических температур растворения и данных табл. 9. Полученные
результаты сравнить с опытными данными. Ответ: $СА = 1,76.
666
С-Циклогексан
н-Гёптан / Фурфурол
Рис. 314. Решение задачи Ш-7 (светлые точки — опытные данные):
/ — расчетные кривые растворимости; 2 — расчетная корреляционная кривая для хорд
равновесия; 3 —опытная корреляционная кривая для хорд равновесия.
б) Повторить расчет, пользуясь данными табл. 7 (см. стр. 103). Ответ:
Рсл = 3.16-
в) Повторить расчет на основе данных о взаимной растворимости и
уравнений Ван-Лаара второго порядка для бинарных систем, считая систему
гептан — циклогексан идеальной. Ответ: рс^=1,45.
г) Применив уравнение Ван-Лаара второго порядка для тройных систем
[уравнение (111,85)], написать уравнение для коэффициентов активности в
тройной системе, определяя константы по данным о взаимной растворимости
и по данным табл. 6 (для пары гептан — циклогексан). Следуя методу,
описанному в примере 111-11, рассчитать тройную диаграмму равновесия при
30° С.
На диаграмму равновесия нанести кривые растворимости и
корреляционную линию для хорд равновесия. Результаты расчета сравнить с
данными Пеннингтона и Марвила. Указание: расчет вести при *а=0,05 и 0,10;
*в = 0,5 и 0,10 по линиям постоянных ас (ас=0,25; 0,50; 0,75). Ответ: см.
рис. 314.
Глава IV
IV-1. Необходимо сравнить расходы, связанные с регенерацией
экстрагента нз насыщенного им водного раствора рафината.
В одном случае экстрагентом служит хлороформ (относительно малая
растворимость в воде, низкая температура кипения и, следовательно,
«благоприятный» для регенерации состав «гетероазеотропной» смеси), в
другом случае — н-бутанол (заметная растворимость в воде, сравнительно
высокая температура кипения и «неблагоприятный» состав гетероазеотропной
смеси).
Схема регенерации растворителя в обоих случаях одинакова. Расход
рафината, выходящего из экстрактора, составляет по воде 2270 кг/ч (без
667
учета растворенного экстрагента). Рафинат насыщен экстрагентом и
практически не содержит экстрагируемого вещества. Необходимые для расчетов
данные приведены в таблице:
Растворимость в воде при 25° С, вес. %
Температура кипения гетероазеотропной смеси
при 1 am, °C
Содержание:
экстрагента в парах гетероазеотропной
смеси, вес. %
экстрагента в дистилляте, вес. %
экстрагента в равновесных жидких фазах
при 37,8° С, вес. %
Стоимость экстрагента, pyff/кг
Экстр
хлороформ
0,75
56,2
97,2
95,0
99,35
0,77
0,36
агент
и-бутанол
6,54
92,8
57,5
55,0
78,80
6,70
0,32
а) Рассчитать годовую стоимость потерь растворителей с рафина-
том, если последние не регенерировать, в производстве при работе 24 ч в
день и 300 дней в году. Ответ: 43 800 руб. (хлороформ); 363 000 руб.
(бутанол).
б) Экстрагент регенерируют дистилляцией в колонне, работающей при
атмосферном давлении. Рафинат подают на верхнюю тарелку колонны при
^=51,7° С, куб колонны обогревают острым паром, имеющим абсолютное
давление 1,35 ат. Выходящие с верха колонны пары (состав их не
соответствует точно азеотропному составу, так как для этого потребовалось бы
бесконечно большое число тарелок) конденсируются (охлаждающая вода
поступает в конденсатор с температурой 26,7° С и выходит при ^=43,3° С)
и охлаждаются до 37,8° С. После отстаивания водный слой возвращают в
колонну, а органический слой выводят из процесса в качестве
регенерированного растворителя (экстракт). Тепло кубового остатка используют для
подогрева рафииата в теплообменнике до 51,7° С. Стоимость пара 1 руб. 45 коп.
за тонну, стоимость охлаждающей воды 2,4 коп. за кубометр. Определить
годовую сумму расходов за пар, охлаждающую воду и стоимость потерь
экстрагента, приняв, что удается регенерировать 99,5% экстрагента. Тепло-
тами растворения воды и экстрагента пренебречь. Температуру паров
дистиллята принять равной температуре кипения гетероазеотропной смеси. Ответ-
2430 руб. (хлороформ), 7200 руб. (бутаиол).
Глава V
V-1. Рассчитать и сравнить расчетные значения коэффициентов
диффузии с опытными данными (Internat. Critical Tables, v. V) для следующих
веществ:
а) н-Бутанол в разбавленном водном растворе (15°С).
б) Глицерин в разбавленном водном растворе (10, 15 и 20° С).
в) Фурфурол в разбавленном метанольном растворе (15°С).
г) Бромоформ в разбавленном растворе этилового спирта (20°С).
д) Пропиловый спирт в разбавленном бензольном растворе (15° С).
е) Ацетон в разбавленном растворе бромоформа (20° С)..
ж) Азотная кислота в разбавленном водном растворе (20° С).
з) Сульфат калия в разбавленном водном растворе (20°С).
666
V-2. Коэффициент диффузий хлорида натрий в воде при концентрации
0,05 г-экв/л и температуре 5° С равен 0,89-Ю-9 м21сек. Рассчитать
коэффициент диффузии NaCl в воде при этой концентрации и температурах 10, 15,
20, 25 и 30° С. Результаты расчета сравнить с опытными величинами
(Internal Critical Tables, v. V).
V-3. Рассчитать коэффициент диффузии гидроокиси натрия в воде в
функции от концентрации (вплоть до концентрации, соответствующей 2 н.
раствору) при 15° С и сравнить с опытными данными (Internat. Critical
Tables, v. V).
V-4. Капля толуола диаметром 3 мм движется в разбавленном водном
растворе диэтиламина (температура 20°С). Физические свойства жидкостей:
ц0 = 0,6 • Ю-3; цс = 1,0 • 10~3 к • сек/м2; рд = 866; рс = 1000 кг/ж3;
межфазовое натяжение а=35- Ю-3 н/м. Коэффициент распределения m — Сн2о/сс7н8 =
= 1,95.
а) Рассчитать скорость подъема капли. Сравнить с соответствующей
скоростью подъема (осаждения) твердой сферы того же диаметра и плотности.
Ответ: для жидкой сферы vt = 0,0855 м/сек.
б) Рассчитать общий коэффициент массопередачи Kd: 1) предполагая, что
капля ведет себя как жесткая сфера; 2) на основе уравнения Кронига —
Бринка и пенитрационной теории; 3) на основе модели Хандлоса — Барона
и пенитрационной теории; 4) на основе модели Хаидлоса — Барона и
корреляции Елзинга — Банчеро. Результаты сравнить с опытными данными Гар-
нера и Хейла [J. Appl. Chem., 5, 653 (1955)]: и,=0,084 м/сек; Кв =
=4,6-10"5 м/сек. Ответ. Кп=(1) 0,402- lO"5; (2) 1,142 - 10s; (3) 12,7- 10s;
(4) 6,38- 10"5 м/сек.
V-5. Коэффициент распределения муравьиной кислоты между
метилизобутилкетоном (В) и водой (А) при концентрации муравьиной кислоты в
воде 0,48 моль/л равен 0,42 при 25° С и 0,37 при 54,8° С.
а) Вывести уравнение для температурной зависимости коэффициента
распределения в форме [g m=f(l[T) и рассчитать теплоту перехода муравьиной
кислоты из воды в кетон. Ответ: —3 457 000 дж/кмоль.
б) Рассчитать мгновенную скорость экстракции муравьиной кислоты из
воды метилизобутилкетоном и концентрации на межфазовой поверхности в
ячейке с перемешиванием (см. рис. 96, стр. 200). Концентрация муравьиной
кислоты в воде равна 1 моль/л, в метилизобутилкетоне — нулю. Принять, что
процесс массопередачи протекает изотермически при 35° С. Коэффициенты
массоотдачи в каждой фазе равны 2,78-Ю-5 м/сек. Ответ: N=0,795 X
X Ю"5 кмоль/м2 сек; cWi=0,714; cKi=0,286 моль/л.
в) Оценить возможное влияние изменения температуры на межфазовой
поверхности на скорость экстракции, считая применимой аналогию Чилтона—
Кольбурна между тепло- и массообменом:
/ - h Pr2'» = / - Sc5'«
где h — коэффициент теплоотдачи;
C„ — теплоемкость;
p — плотность;
v— скорость;
Рг — критерий Прандтля;
Sc— критерий Шмидта.
Определить температуру и концентрации на поверхности и скорость
массопередачи на основе следующих даииых: коэффициенты массоотдачи в
каждой фазе равны 2,78 • 10-5 м/сек; температуры в массе жидкости 35° С;
концентрация в воде 1 моль/л, в метилэтилкетоне — нуль. В водной фазе
Sc= 335- Рг=5,0; в фазе метилизобутилкетона Sc=600; Pr=9,0; Ср =
=0,6 кал ■ г-' • град4 (2,51 • 103 дж ■ кг~1 • град'1; р = 820 кг /ж3). Ответ:
температура на поверхности 39°С; #=0,783 • 10"5 кмоль/м? • сек; cwi = 0,7\8;
Скг = 0,283 КМОЛЬ/М3.
669
Глава VI
VI-1. Отношение доли вещества, экстрагирующейся в п ступенях при
перекрестном токе, к соответствующей величине для бесконечного числа
ступеней при одинаковом суммарном расходе растворителя можно назвать
«относительной эффективностью» экстракции. Экстрагент содержит у кг
растворенного вещества на кг растворителя.
а) Показать, что зависимость «относительной эффективности» от
суммарного расхода экстрагента при постоянном числе ступеней проходит через
минимум;
б) рассчитать минимальную величину относительной эффективности для
одной ступени. Ответ: 77,1%.
VI-2. Смесь, состоящую из А кг растворителя с концентрацией xF кг\кг А
растворенного вещества, предполагают экстрагировать экстрагентом В
(содержащим ys кг/кг В растворенного вещества) в двух ступенях экстракции при
перекрестном токе. Компоненты А я В взаимно нерастворимы, коэффициент
распределения постоянен. Принимая, что стоимость регенерации 1 кг
экстрагента В равна стоимости X кг экстрагируемого вещества, определить
оптимальное количество экстрагента и его распределение по ступеням. Прочие
затраты считать постоянными. (Указание: найти максимум величины
стоимости экстрагируемого вещества за вычетом стоимости экстрагента.)
VI-3. Распределение бензойной кислоты между водой (Л) и бензолом (В)
в области малых концентраций при 25° С описывается уравнением (см
пример II-3, стр. 56): '
(/ = 915x1,91
где у я х — концентрации бензойной кислоты соответственно в бензоле и
воде в моль/л.
а) Необходимо понизить концентрацию бензойной кислоты в водном
растворе с 0,01 до 0,001 моль/л с экстракцией ее чистым бензолом в двух
ступенях при перекрестном токе. Определить максимальную концентрацию
бензойной кислоты, которую можно получить в суммарном экстракте, и
расходы экстрагента в каждой ступени. Ответ: 0,0055 моль/л- Bi=0 683- B,=
■=0,955 ж3/ж3 А. ' '
б) Какой состав имел бы экстракт, если бы этот процесс проводили по
схеме дифференциальной экстракции? Ответ: 0,01588 моль/л.
VI-4. Необходимо извлечь экстракцией бензолом 98% 1,4-диоксана из
20%-ного раствора его в воде, поступающего на экстракцию в количестве
450 кг/ч. Предполагая, что процесс проводят по схеме противоточной
экстракции, рассчитать:
а) минимальный расход экстрагента (ответ; 32,2 кг/ч);
б) число ступеней, необходимое для заданного извлечения диоксана, если
расход экстрагента в 1,2 раза больше минимального (ответ: 14,3);
в) расход экстрагента, достаточный для достижения заданной степени
извлечения в пяти ступенях экстракции (ответ: 63,0 кг/ч). Рассчитать также
расход экстрагента, необходимый для осуществления данного разделения в
пяти ступенях экстракции с перекрестным током при равномерном
распределении экстрагента по ступеням (ответ: 202 кг/ч).
Данные о распределении 1,4-диоксана (С) между бензолом (В) и
водой (А) при 25° С приведены ниже [В е г n d t, L у п с h, J. Am. Chem Soc,
66, 282 (1944)]:
XCA . . 0,051 0,189 0,252
XCB . . 0,052 0,225 0,32
При концентрации диоксана менее 25% бензол и вода практические не
смешиваются.
VI-5. В противоточном экстракторе, эффективность которого эквивалентна
пЕ теоретическим ступеням, смесь, поступающая в количестве А кг/ч с кои-
670
центрацией xF кг экстрагируемого вещества/кг Л, экстрагируется до
концентрации Хп- Расход экстрагента равен В кг/ч; его начальная концентрация
составляет ys кг экстрагируемого вещества/кг В. Коэффициент
распределения гпЕ постоянен. Экстракт поступает в другой противоточный экстрактор
с эффективностью ns теоретических ступеней, в котором регенерируют
растворитель (экстрагент) В до концентрации экстрагируемого вещества в нем,
равной ys- Избирательный растворитель, подаваемый на вторичную
экстракцию, не содержит экстрагируемого вещества; его расход равен S кг/ч. Если
z — конечная концентрация в растворителе S, то коэффициент распределения
для вторичной экстракции (также постоянная величина) будет равен ms = z/y
(при равновесии). Растворители принимаются взаимно нерастворимыми.
а) Доказать, что
*п ___ (4g-0(e5-l) + («g-I)(Es)(«gS-l)
хр (4£-i)(es-i)+(4£+1-i)(Es)G?-i)
где еЕ=тЕВ/Л и Es = msS/B.
б) Показать', что при фиксированных значениях xF, xn и конечной
концентрации г в растворителе S имеется минимум общего числа ступеней.
Определить для этого случая скорость рециркуляции экстрагента и число
ступеней в каждом каскаде.
VI-6. Водный раствор после сернокислотного выщелачивания урана,
содержащий 1,0 кг ТЛзОв на 1 ж3, необходимо экстрагировать до конечной
концентрации 0,002 кг U308 на 1 ж3 с помощью 0,1 М раствора додецилфосфор-
ной кислоты в керосине. Реэкстракцию урана из органического экстракта
производят 10 н. водным раствором НС1. Конечная концентрация в
солянокислом растворе должна составлять 50 кг/м3. Несмотря на то что объемная
производительность аппаратуры на стадии реэкстракции меньше, чем в
процессе основной экстракции, стоимость одной ступени из-за конструктивных
особенностей примерно одинакова. В данном процессе, кроме того,
необходима рециркуляция растворителя в каждой ступени. По этим причинам
можно принять, что размер (и, следовательно, стоимость) одной ступени не
зависит от производительности. Равновесные данные приведены ниже
[Black, Koslov, Chem. Eng. Progr. Symp. Ser., 55, № 22 (1959)]:
Для экстракции
X
У
0
0
0,002
0,3
0,01
1,0
0,038
2,5
0,09
5,0
0,12
6,0
0,20
8,0
0,41
10,0
0,60
10,65
0,80
11,05
1,00
11,30
Для реэкстракции
У
г
0
0
0,26
3,57
1,00
30,0
2,0
67,2
где х, у и г—концентрации (кг U308 на 1 ж3) соответственно в исходном
растворе (после выщелачивания), в органическом растворителе и водном
растворе НС1. Растворимость водных и органического растворов незначительна,
а изменениями объемов можно пренебречь. Определить расход экстрагента,
концентрацию U308 в нем после реэкстракции и минимальное суммарное
число теоретических ступеней. Ответ: 0,2 ж3 экстрагента/ж3 исходного раствора
(после выщелачивания); 0,25 кг/м3; 3,0 ступени основной экстракции; 2,5
ступени реэкстракции.
671
VI-7. Исходный раствор (питание) подают на экстракцию в первую
ступень противоточного экстрактора, эквивалентного п теоретическим ступеням.
Экстрагент S подают в ступень п; исходный раствор — в ступень 1.
Экстракт Е\ выходит из первой ступени, рафииат Rn — из ступени п. Рафинат
Rm после ступени т (считая от ступени, в которую вводят исходную смесь)
частично выводят из процесса в количестве К. Остальную часть RK подают
в ступень (/л+1) и подвергают дальнейшему экстрагированию.
Получить зависимости, необходимые для расчета числа теоретических
ступеней, и изобразить схему процесса на фазовой диаграмме для системы
типа I. Буквы F\ E\; S, RK и К и др. обозначают объемные скорости
соответствующих потоков (в кг/ч) и составы последних на фазовой диаграмме.
VI-8. Рассчитать процесс экстракции уксусной кислоты из водного
раствора изопропиловым эфиром. Равновесные данные для этой системы при
20° С приведены в таблице (концентрация в вес.%):
уксусная
кислота
0,69
1,41
2,89
6,42
13,30
25,50
36,70
44,30
46,40
Водный слой
вода
98,1
97,1
95,5
91,7
84,4
71,1
58,9
45,1
37,1
нзопропиловый
эфир
1,2
1,5
1,6
1,9
2,3
3,4
4,4
10,6
16,5
Органический слой
уксусная
кислота
0,18
0,37
0,79
1,93
4,82
11,40
21,6
31,1
36,2
вода
0,5
0,7
0,8
1,0
1,9
3,9
6,9
10,8
15,1
нзопропиловый
эфир
99,3
98,9
98,4
97,1
93,3
84,7
71,5
58,1
45,7
Исходный раствор состоит из 20% уксусной кислоты и 80% воды.
Конечный рафинат должен содержать 1% уксусной кислоты. Считать, что
экстрагент представляет собой чистый нзопропиловый эфир.
а) 450 кг исходной смеси экстрагируется в перекрестном токе таким
же количеством растворителя (в каждой ступени).
1. Определить необходимое число ступеней. Ответ: 10.
2. Определить общий расход экстрагента. Ответ: 4500 кг.
3. Рассчитать общее весовое количество получающегося экстракта и
концентрацию в нем уксусной кислоты. Ответ: 1,87%.
4. Составить полный материальный баланс процесса и материальный
баланс по уксусной кислоте.
б) Сравнить расход экстрагента, концентрацию уксусной кислоты в
экстракте и количество экстрагированной уксусной кислоты, полученные выше
[пункт а)], с соответствующими величинами для процесса дифференциальной
экстракции. Ответ: 3870 кг экстрагента.
в) Определить расход экстрагента, необходимый для проведения данного
разделения в одной ступени. Ответ: 32 400 кг.
г) Исходный раствор, расход которого составляет 450 кг/ч,
предполагают экстрагировать в противоточном экстракторе.
1. Определить минимальный расход экстрагента. Ответ: 1000 кг/ч.
2. Рассчитать число ступеней, необходимое для данного разделения, если
расход экстрагента составляет 1570 кг/ч. Определить также весовые
количества и составы экстракта и рафината. Ответ: 6,3 теоретической ступени.
д) Сравнить расход тепла при разделении исходной смеси дистилляцией
и экстракцией для процесса по пункту г).
872
1. Рассчитать расход тепла на ректификацию водного раствора уксусной
кислоты. Данные о равновесии пар — жидкость имеются в «The Chem. Eng.
Handbook». Принять концентрацию уксусной кислоты в дистилляте равной
1% (что примерно соответствует принятому составу рафината при
экстракции). Коэффициент избытка флегмы принять равным 1,5. При определении
расхода тепла учитывать только скрытые теплоты испарения. Ответ:
4,04- 109 дж/ч.
2. Рассчитать расход тепла в процессе экстракции [пункт г)], приняв
следующую схему регенерации экстрагента. Экстракт поступает в
ректификационную колонну /, кубовый остаток из которой, состоящий только из
изопропилового эфира и уксусной кислоты, разделяется в ректификационной
колонне 2 на чистый нзопропиловый эфир (дистиллят) и уксусную кислоту
(кубовый остаток). Коэффициент избытка флегмы в колонне 2 принять
равным 1,5.
Дистиллят из ректификационной колонны /, представляющий собой азео-
тропную смесь изопропилового спирта и воды, при конденсации образует
два жидких слоя. Органический слой используют в качестве флегмы, а
водный слой вместе с рафинатом (после экстракции) подают в
ректификационную колонну 3.
Кубовый остаток из колонны 3, почти не содержащий экстрагента,
выбрасывают. После конденсации дистиллята из колонны 3, представляющего
собой также азеотропную смесь изопропилого эфира и воды, водный слой
используют в виде флегмы, а органический слой соединяют с дистиллятом из
колонны 2 и возвращают в экстрактор. Для расчета принять, что уксусная
кислота не содержится в дистилляте ни одной из ректификационных колонн
и что тепло расходуется лишь в количествах, соответствующих внутренним
теплотам испарения (некоторая часть потребности в тепле обеспечивается
в результате рекуперации последнего).
Гетероазеотропная смесь воды и изопропилового эфира при нормальном
давлении кипит при 62,2° С и содержит 4,5% воды. Взаимная растворимость
изопропилового эфира и воды при температуре кипения азеотропнои смеси
составляет 1,0% (эфир в воде) и 0,8% (вода в эфире).
Растворы изопропилового эфира и уксусной кислоты можно
рассматривать при расчете расхода тепла в колонне 2 как идеальные. Ответ: 0,927 X
X 109 дж/ч.
е) Выгодно ли, если бы это было возможно, проводить процесс
экстракции с возвратом экстракта, получая экстракт, содержащий 30% уксусной
кислоты?
VI-9. Равновесные данные (в вес. %) для системы н-гептан—циклогек-
сан—диметилформамид (ДМФ) при .20° С [Durandet, Glade 1, Rev. Inst,
franc, petrole, 11, 811 (1956)]:
н-Гептан
99,0
80,8
63,3
46,7
30,7
15,2
0
Цнклогексан
0
18,2
35,5
. 51,5
67,0
81,8
94,5
ДМФ
1,0
1,0
1,2
"оо
2,3
3,0
5,5
н-Гептан
9,0
8,2
7,0
5,5
3,8
2,0
0
Циклогексан
0
2,4
5,7
9,0
12,5
16,0
19,8
ДМФ
91,0
89,4
87,3
85„5
83,7
82,0
80,2
Диметилформамид предполагают использовать в качестве экстрагента
в процессе противоточной экстракции для разделения смеси н-гептана и
43 Р- Е. Трейбал
673
циклогексана (1 : 1) на фракции, содержащие соответственно 10 и 90% цик-
логексаиа и не содержащие экстрагента. Поступающий в процесс экстрагент
ие содержит углеводородов.
а) Представить равновесные данные в виде, удобном для построения
диаграммы Енеке, и рассчитать селективность для каждой хорды равновесия.
б) Исходя из значения селективности при концентрации, равной
концентрации исходного раствора, рассчитать минимальное флегмовое число для
экстракта по уравнению (VI, 193) и сравнить полученную величину с
рассчитанной графически. Ответ: 4,44; 4,41.
в) Исходя из величины средней селективности, рассчитать по уравнению
(VI, 188) минимальное количество необходимых теоретических ступеней и
сравнить результаты с величиной, полученной графически расчетом. Ответ;
11,6; 11,8.
г) С помощью рис. 157 (см. стр. 301) определить флегмовое число,
необходимое для проведения данного разделения в 20 теоретических ступенях.
Полученное значение флегмового числа использовать для расчета числа
ступеней графически (в данном случае N для экстрактов не является постоянной
величиной). Ответ: 6,05; 25.
д) Рассчитать расход растворителя [на 100 кг исходной смеси для
флегмового числа, найденного по пункту г)]. Ответ: 1586 кг.
VI-10. В таблице приведены составы равновесных растворов (в мольных
долях) в системе н-гексан (Л)—метилциклопентаи (С)—анилин (В) [D а г-
went, W i n с 1 е г, J. Phys. Chem., 47, 442 (1943)]:
Температура
25
45
н-Гексан
0,930
0,838
0,736
0,589*
0,480
0,278
0,136
0,075
0
0,860
0,814
0,716
0,525
0,307
0,298
0,150
Метилциклопентаи
о,
0,090
0,187
0,326
0,430
0,618
0,744
0,795
0,856
0
0,046
0,140
0,301
0,438
0,440
0,350
н-Гексан
0,082
0,076
0,073
0,060
0,050
0,037
0,028
0,007
0
0,138
0,137
0,130
0,128
0,121
0,120
0,150
Метилциклопентаи
0
0,016
0,022
0,055
0,070 *
0,117
0,160 *
0,209
0,244
0
0,009
0,035 *
0,095 *
0,208
0,211
0,350
Округленные величины.
а) Построить (пользуясь большой линейкой и остро отточенным
карандашом) для обеих температур фазовые диаграммы в прямоугольных
координатах и кривые распределения.
б) При экстракции смеси н-гексана (50 мол. %) и метилциклопеитана
чистым анилином получают экстракт, содержащий 90 мол. %
метилциклопеитана (без учета содержания в нем экстрагента), и насыщенный анилином
рафинат, содержащий 10 мол. % метилциклопеитана (без учета экстрагента).
Процесс ведут с возвратом экстракта (не содержащего экстрагента) в виде
874
флегмы. Определить точку Q
иа рабочей линии укрепляющей
части колонны,
соответствующую Хс =0,025 мол. доли.
Каковы флегмовое число, расход
экстрагента на 100 кмоль
исходного раствора (питания) и
необходимое количество
теоретических ступеней? Процесс
проводят при 25° С. Ответ:
17 теоретических ступеней.
в) Процесс, описанный в /г
пункте б), ведут в несколько —*
измененных условиях:
исходная смесь поступает с
температурой 45° С; укрепляющая
часть экстракционной колонны
работает при 25° С, а
исчерпывающая— при 45° С.
Рассчитать расход экстрагента на 100
кмоль исходной смеси и
необходимое число теоретических
ступеней. Ответ: 1165 кмоль,
19 теоретических ступеней.
VI-i i. Необходимо
разделить смесь н-бутанола и ме-
тил-н-бутилкетоиа. Эта
система образует (пат. США
2500329) азеотропную смесь.
Поэтому для разделения
данной смеси вместо
ректификации можно применять
экстракцию водой. Данные о равновесии в системе метил-н-бутилкетон (Л) — вода
(В) и н-бутанол (С) при 38° С опубликованы Джонсом и Мак-Кантсом [Ind.
Eng. Chem., 46, 1965 (1954)]. Возможная схема процесса показана на рис. 315.
Обозначения потоков соответствуют обозначениям на эскизе фазовой
диаграммы. Так как нет данных о существовании тройной азеотропной смеси в этой
системе, можно принять следующие допущения:
1. Ректификация экстракта Et дает дистиллят Vb представляющий собой
азеотропную смесь бутанола и воды. После конденсации дистиллята водный
слой К используют в качестве флегмы в ректификационной колонне, а бута-
нольный слой подают частично (Ro) в виде флегмы в экстрактор. Остальную
часть (К'о) подвергают дальнейшей ректификации, получая дистиллят Уг
(того же состава, что и Vi) и продукт процесса Ре — бутанол. Можно
считать, что кубовый остаток SE не содержит бутанола.
2. При ректификации рафииата Rn в качестве дистиллята образуется
азеотропная смесь кетоиа и воды Т. После конденсации дистиллята
органический слой / возвращают в ректификационную колонну в виде флегмы, а
водный слой SR направляют в емкость для экстрагента. Кубовый остаток Pr —
продукт процесса (метил-н-бутилкетои), можно считать, ие содержит воды.
Состав азеотропиых смесей: V=42,5 вес.% воды; Г=30 вес.% воды.
Насыщенные растворы К; Ro; 1 и SR имеют температуру 38° С. Расход
исходной смеси (50% кетона, 50% бутанола) составляет 450 кг/ч. Определить
число теоретических ступеней и весовые расходы всех потоков (в кг/ч),
обозначенных на схеме, при следующих условиях: состав конечного экстракта
р_2% воды, 98% бутанола; состав конечного рафината —2% бутанола,
98% кетона; состав экстракта Е\ — 95% бутанола, 5% кетона (не считая
воды); флегмовое число Ro/Ре=2,0. Все составы даны в вес.% Указание:
J T SR ;_
Рис. 315. Схема процесса (к задаче VI-11).
1 — экстрактор; 2, 3, 4 — ректификационные колонны;
5, 6 — отстойники; 7 —хранилище растворителя.
43*
675
расчет следует вести графически в координатах, построенных без учета
содержания экстрагента (по диаграмме Енеке и с помощью кривой
распределения, изображенной в этих координатах). Обратить внимание на то, что R0
и РЕ имеют неодинаковый состав. Число теоретических ступеней следует
определять лишь после того, как рассчитаны составы и весовые расходы всех
потоков. Ответ: 6,3 теоретической ступени.
Глава VII
VII-I. Необходимо разделить смесь пропан-1,1-дикарбоновой (В)
(50 вес.%) и пропан-1,3-дикарбоновой (С) кислот. Для этого смесь кислот
растворяют в насыщенном водой этилацетате и экстрагируют в
перекрестном токе насыщенной этилацетатом водой (А). Коэффициенты распределения
равны: тв=2,48 и тс = 0,75 (т=концентрация в воде/концентрация в этил-
ацетате, кг растворенного вещества на 1 кг растворителя).
а) При каком объемном соотношении растворителей (экстрагентов)
достигается максимальная степень разделения в одной ступени? Найти
относительные количества обеих кислот, содержащихся в каждой фазе. Ответ: ЛД)=0,734.
б) Какое объемное соотношение растворителей в каждой ступени
необходимо для симметричного разделения кислот между рафинатом и
суммарным экстрактом в пяти ступенях экстракции при перекрестном токе и
равномерном распределении экстрагента по ступеням? Найти относительное
содержание обеих кислот в экстракте и рафинате. Ответ: Л//Э = 0,1046.
в) Какое объемное соотношение растворителей необходимо для
достижения максимальной степени разделения кислот в пяти ступенях экстракции
при перекрестном токе? Найти относительное содержание кислот в экстракте
и рафинате. Ответ: Л//Э = 0,145.
г) Раствор смесн кислот в этилацетате экстрагируют в одной ступени
водой при объемном соотношении, соответствующем максимальной степени
разделения. Водный экстракт экстрагируют еще раз этилацетатом также при
объемном соотношении, отвечающем максимальной степени разделения.
Растворы в этилацетате после двух операций экстракции соединяются.
Рассчитать общую степень разделения, а также выход и степень чистоты пропан-1,1-
дикарбоиовой кислоты в получающемся водном растворе. Какое число
ступеней потребовалось бы для получения такого же выхода и той же степени
чистоты компонента В в суммарном водном экстракте, если разделение
осуществлять экстракцией водой в перекрестном токе (при равномерном
распределении воды по ступеням)? Ответ: рв — рс = 0,290; 54 ступени.
VII-2. Доказать, что при экстракции несмешивающимися растворителями
(экстрагентами) в перекрестном токе наибольшая степень разделения двух
веществ (коэффициенты распределения их постоянны н не зависят друг от
друга) достигается при равномерном распределении экстрагента по ступеням.
VI1-3. В таблице приведены данные о .распределении ж- и л-крезолов
между бензолом и 0,25 М раствором фосфатного буфера (рН=11,0) при 25° С
[Compere, Ryland, Ind. Eng. Chem., 45, 1692 (1953)]:
Ум или Уп
хм
хп
0
0
0
0,005
0,003
0,0045
0,010
0,0068
0,0097
0,015
0,0115
0,0164
0,020
0,0175
0,0250
0,0250
0,0250
0,036 *
0,0336
0,040 *
* Округленные величины.
Здесь х и у — концентрации (в кмоль/м3 или в моль/л) соответственно в
бензоле и буферном растворе. Считать, что экстрагенты взаимно
нерастворимы и компоненты не влияют на распределение друг друга.
а) Эквимолекулярную смесь крезолов растворяют в бензоле до
концентрации 0,04 жоль/л каждого из них. Раствор экстрагируют в трех ступенях
676
экстракции при перекрестном токе буферным раствором, содержащим
0,005 моль/л л-крезола и 0,002 моль/л ж-крезола при равных объемах
органической и водной фаз в каждой ступени. Какое разделение достигается?
Определить относительное содержание крезолов в экстракте и рафинате. Ответ:
?„-?.„ =0,145.
б) Определить относительное содержание крезолов в экстракте и
рафинате в случае дифференциальной экстракции в условиях, приводящих к тому
же выходу n-крезола в рафинате, что и по пункту а) (при тех же составах
исходной смеси и экстрагента). Сравнить расход экстрагента с
соответствующей величиной, полученной по пункту а). Ответ: q —qM = 0,1637; /1(/D = 2,42.
VII-4. Эквимолекулярную смесь ж- и n-крезолов растворяют в бензоле до
концентрации 0,04 моль/л каждого и экстрагируют в противоточном процессе
водным раствором фосфатного буфера (рН=11,0) при 25° С (данные о
равновесии приведены в задаче VII-3). Буферный раствор не содержит крезолов.
Необходимо извлечь 90% ж-крезола из бензольного раствора.
а) Найти минимальное объемное соотношение расторителей
(экстрагентов), необходимое для достижения заданной степени извлечения. Ответ:
1,071 ж3 буферного раствора на 1 м3 бензола.
б) Найти необходимое число теоретических ступеней при объемном
соотношении экстрагента и исходного раствора 1,15 и относительное содержание
крезолов в экстракте. Ответ: 5 теоретических ступеней; 54,5% ж-крезола.
VII-5. Показать, что для фракционной экстракции без флегмы
(начальная концентрация разделяемых веществ в экстрагентах равна нулю,
исходная смесь поступает в виде водного раствора с концентрацией DF,
AF=0) при постоянных и независимых коэффициентах распределения
объемные соотношения фаз в случае, когда рабочие линии пересекаются в точке
пересечения равновесной кривой и линии x=xF, определяются соотношениями:
ID\ pBmc-pcmB ID'\ _ тд(1-pc)-mc(l-pB)
\ALm Рв-Рс \AL™ Рв-Рс
Показать также, что такие объемные соотношения соответствуют
бесконечному числу теоретических ступеней разделения.
VII-6. Щавелевую и сукциновую кислоты разделяют экстракцией двумя
экстрагентами: водой и н-амиловым спиртом. Равновесные данные (в г/л)
приведены в таблице [A s s е 1 i п, Comings, Ind. Eng. Chem., 42, 1198 (1950)]:
Щавелевая
кислота в воде
20
40
60
0
30
50
0
30
50
0
30
50
Сукцнновая
кислота в воде
0—50
0—50
0—50
20
20
20
40
40
40
60
60
60
Щавелевая
кислота в амиловом
спирте
6
13
21
Сукциновая
кислота в амиловом
спирте
12,5
12,0
11,0
25,0
23,5
22,5
36,5
35,0
33,5
677
Исходная смесь представляет собой водный раствор, содержащий
50 кг/м3 каждой кислоты. Начальная концентрация кислот в экстрагентах
(вода и н-амнловый спирт) равна нулю. Чистота разделения каждой кислоты
должна составлять 90%. Экстрагенты можно считать взаимно
нерастворимыми.
а) Найти предельные величины расхода экстрагентов (в м3/л13 исходного
раствора). Так как линии равновесия близки к прямым, для расчета можно
пользоваться уравнениями (VII, 71) и (VII, 72) при условии, что
коэффициенты распределения взяты при концентрации исходного раствора. Ответ: 4,60 л3
н-амилового спирта и 1,418 м3 воды на 1 м3 исходного раствора.
б) Найти расходы экстрагентов (в ж3/ж3 исходного раствора),
необходимое число теоретических ступеней и место ступени питания в случае, если
процесс экстракции ведут без флегмы, при объемном соотношении водной и
органической фаз, равном 0,47 в экстракционной части колонны и 0,36 в
промывной части колонны. Ответ: 13,5 теоретической ступени.
в) Определить необходимое число теоретических ступеней и место
ступени питания, если процесс экстракции проводят при том же расходе экстр-
агентов, что и в пункте б), но с использованием флегмы при флегмовом
числе, равном 0,5 в ступени 1, и флегмовом числе, равном 2 в ступени I1
(флегмовое число принято равным отношению количества извлеченного
вещества, возвращаемого в виде флегмы, к количеству извлеченного вещества,
выводимого из процесса). Ответ: 9,6 теоретической ступени.
VII-7. Смесь, состоящая из 40 вес.% пропан-1,1-днкарбоновой кислоты
(В) и 60 вес.% пропан-1,3-дикарбоновой кислоты (С), разделяют
экстракцией двумя взаимно насыщенными растворителями (экстрагентами); этилаце-
татом (А) и водой (D); mB=2,4 и тс = 0,75. Значения коэффициентов
распределения приведены для концентраций, выраженных в кг растворенного
вещества на 1 кг растворителя. Считать, что коэффициенты распределения
постоянны и не зависят друг от друга в пределах концентраций в обеих
фазах, не превышающих 10 вес. %.
а) Смесь кислот разделяют в противоточном процессе экстракцией с
двумя растворителями (экстрагентами); при этом получены следующие
результаты: в одной фазе относительное содержание кислот составляет 95% В
и 5% С, в другой —5% В и 95% С.
1. Определить объемное соотношение экстрагентов, положение ступени
питания и полное число ступеней для всех шести случаев, приведенных в
табл. 14 (см. стр. 369) при экстракции без флегмы.
2. Определить минимальное число ступеней, необходимое для достижения
заданного разделения при экстракции без флегмы. Ответ: 9,13.
3. Сравнить полученное значение минимального числа ступеней с
соответствующей величиной, рассчитанной по эмпирической формуле Шай-
беля.
4. Найти расход экстрагента (в кг/100 кг исходной смеси), при котором
заданная степень разделения достигается при числе ступеней, полученном в
пункте 2). Ответ: 1266 кг А; 1770 кг D.
5. Найти число ступеней, необходимое для данного процесса разделения
при объемном соотношении, полученном в пункте 1, в случае проведения
процесса с полным возвратом флегмы. Ответ: 5,07.
6. Найти необходимое при заданном разделении число ступеней для
процесса экстракции с флегмой при первоначальном объемном соотношении.
Флегмовые числа одинаковы на обоих концах колонны и равны величинам,
соответствующим приведенным в табл. 14 (стр. 369), и, кроме того, для
случая, когда а/(/— г)=2,5 и 10. Для каждого г рассчитать расход экстрагента на
100 кг исходной смеси.
б) Определить относительное содержание разделяемых кислот в экстракте
и рафинате, если процесс экстракции с двумя экстрагентами ведут в
противоточном каскаде, состоящем из 12 ступеней, без флегмы. Исходную смесь
подают в пятую ступень (считая от ввода экстрагента А), Объемное
соотношение А : D равно единице. Ответ: 89,4 и 0,895% В.
676
Глава VIII
Задачи к главе VIII приведены вместе с задачами к главе XI.
Глава IX
IX-1. В процессе лабораторной имитации четырех ступеней противоточиой
экстракции для более быстрого получения стационарного состава рафината
исходную смесь добавляют с более высокой концентрацией, чем концентрация
исходной смеси в стационарных условиях, определяемая по уравнениям
(IX, 10) и (IX, 11).
Сравнить число циклов при факторе экстракции, равном единице, которое
необходимо для достижения степени приближения к стационарным условиям
Qtlqs, равной 0,9, с количеством циклов, которое потребовалось бы для
достижения стационарного состояния при использовании концентрации исходной
смеси, равной ее концентрации в стационарном состоянии. Ответ:
соответственно четыре и шесть циклов.
IX-2. Показать, что в процессе лабораторной имитации непрерывной
экстракции двумя растворителями (экстрагентами) с подачей питания в
среднюю ступень при постоянных коэффициентах распределения и объемном
соотношении, подобранном таким образом, чтобы два вещества разделялись
симметрично, соотношение этих веществ в конечных продуктах каждого
цикла постоянно (независимо от номера цикла) и равно их соотношению в
стационарных условиях.
IX-3. Необходимо установить возможную степень разделения смеси
компонентов В и С (50% каждого) с помощью экстракции двумя экстрагентами
A и D. Компоненты В и С — трудноразделимые изомеры (их селективность
близка к единице).
Для получения более надежных данных, чем результаты однократной
экстракции, проведено пять циклов процесса лабораторной имитации каскада из
15 ступеней непрерывной экстракции двумя экстрагентами с подачей питания
в среднюю ступень. В каждой операции использовали равные объемы экстр-
агентов; исходную смесь добавляли в количестве 100 г. После пяти циклов в
фазе экстрагента А было найдено 4,16 г разделяемой смеси, содержащей
74,0% В и 26,0% С.
Определить относительное содержание компонентов В и С в фазе
экстрагента А, которое должно быть получено в прогивоточиом каскаде, состоящем
из 15 ступеней, с подачей питания в среднюю ступень при объемном
соотношении экстрагентов, соответствующем симметричному разделению. Ответ:
69,6% В и 30,4% С.
IX-4. Показать, что коэффициент распределения можно определить на
основе данных «противоточиого распределения» по Крэгу с помощью
уравнения
е = wmax — 1 0,5 (<+?) — wmax
*—«max р («max —1)(* — «max)
где 8 — фактор экстракции;
mmax—номер трубки, в которой концентрация максимальна;
t — число циклов.
Сравнить это выражение с уравнениями, полученными Никольсом [Anal,
Chem 22, 915 (1950)] и Хекером [Z. Naturforsch., 8B, 77 (1953), уравнения
(5) и (6)].
IX-5. Два грамма твердой смеси исследовали методом «противоточиого
распределения» по Крэгу в аппарате, состоящем из 24 трубок, при равных
объемах воды и органического растворителя в каждой трубке. Остаток после
выпаривания жидкостей из каждой трубки взвешивали. Результаты
помещены в таблице,
679
Номер трубки
т.
1
2
3
4
5
6
7
8
9
10
11
12
Вес смеси
г
0,0023
0,0104
0,0251
0,0413
0,0599
0,0878
0,1288
0„1736
0,2030
0,2018
0,1723
0,1315
23!
(т— I)!(24 — т)\
1
23
253
1771
8 855
33 649
100 947
245157
490 314
817190
1 144 066
1 352 078
Номер трубки
т
24
23
22
21
20
19
18
17
16
15
14
13
Вес смеси
г
0,0001
0,0012
0,0063
0,0196
0,0446
0,0773
0,1051
0,1178
0,1108
0,0953
0,0870
0,0987
Какие выводы о составе исследуемой смеси можно сделать на
основании этих данных? Для облегчения вычислений в таблице приведены
значения (t—\)\1(т—i)Ut— т)\, которые симметричны относительно трубки,
находящейся в середине аппарата, т. е. указанные величины одинаковы,
например, для т—5 и /л=20. Ответ; смесь состоит из трех веществ,
содержащихся в количестве 1,20; 0,65 и 0,15 г.
Глава X
Х-1. Определить размеры аппарата с мешалкой и отражательными
перегородками, предназначенного для непрерывной экстракции диэтиламина из
разбавленных растворов в керосине (мол. вес 180) водой при температуре
25° С. Суммарная производительность 0,850 м3/ч при объемном соотношении
воды к керосину, равном 5: 1. Время пребывания должно составлять 15 сек.
Высота аппарата равна его диаметру. Перемешивание осуществляется
плосколопастной турбинной мешалкой с шестью лопастями диаметром, равным
Г/3, при числе оборотов, достаточном для поддержания соотношения фаз в
аппарате, равном 5: 1. Физические свойства фаз следующие: вязкость
раствора в керосине 2,5 спз (2,5- Ю-3 н-сек/м2); плотность 900 кг/ж3; вязкость
водного раствора 0,89 спз (0,89 • 10~3 н-сек/м2); плотность 997 кг/м3; межфазовое
натяжение 25 - 10^3 н\м. Коэффициент распределения (концентрация в во
де/концентрация в керосине) равен 0,3. Определить также эффективность
ступени и расход энергии на перемешивание. Ответ. Г = 0,356 м; Р=36 вт;
Emd = Em е= 1.
Х-2. Определить расход энергии, необходимой для образования в диа-
фрагменном смесителе той же межфазовой поверхности, что и в аппарате с
мешалкой в задаче Х-1. Отношение диаметра диафрагмы к диаметру трубы
равно 0,5. Дисперсная фаза — керосин. Ответ: 79,6 вт.
Х-3. Пересчитать эффективность ступени для аппарата с мешалкой в
задаче Х-1 для температуры 50° С при той же производительности и
одинаковой интенсивности перемешивания. Предполагая, что зависимость
коэффициента массопередачи от температуры в интервале температур от 25 до 50° С
выражается уравнением Аррениуса, рассчитать «энергию активации» для
скорости экстракции. При 50° С физические константы фаз имеют следующие
значения: вязкость керосинового раствора 0,7 спз (0,7- 10~3 н-сек/м2);
плотность 880 кг/м3; вязкость водного раствора 0,55 спз (0,55 • 10_3 н-сек/м2);
плотность 988 кг[м3; межфазовое натяжение 20 • 10"3 н/м. Коэффициент рас-
630
пределения (концентрация в воде/концентрацня в керосине) равен 0,15.
Ответ: 12,8 • 106 дж/кмоль.
Х-4. Предполагая, что в каждой секции происхбдит полное
перемешивание и отсутствует продольное перемешивание между секциями, доказать
справедливость уравнения для эффективности ступени прямоточного
секционированного смесителя, состоящего из п секций:
Еп = Е 2 (1 - E)J
J-о
где NE/Ne — эффективность ступени одной секции.
Глава XI
XI-1. Рассчитать насадочную колонну для экстракции уксусной кислоты
из водного раствора с концентрацией 4%. Экстрагент — метилизобутилкетон
с начальной концентрацией кислоты, равной нулю. Степень извлечения
уксусной кислоты 90%. Производительность колонны должна составлять 2,83 м3]ч
водного раствора и 5,66 м3/ч метилизобутилкетона. Температура 25° С.
Физические свойства фаз следующие: вязкость водного раствора 1 спз
(1-Ю-3 н-сек/м2); плотность 1000 кг/м3; коэффициент диффузии уксусной
кислоты в воде 1 • Ю-9 м2/сек; вязкость метилизобутилкетона 0,57 спз
(0,57-Ю-3 н-сек/м2), плотность 800 кг/м3; коэффициент диффузии уксусной
кислоты в метилизобутилкетоне 1,29-Ю-9 м2/сек. Коэффициент распределения
(концентрация в кетоне/концентрация в воде) равен 0,545. Межфазовое
натяжение 9,1 • Ю-3 н/м. Ответ; ответ не может быть однозначным. Для
угольных колец Рашига диаметром 25 мм в случае диспергирования воды расчет
(без учета продольного перемешивания) дает величину диаметра колонны,
равную 0,49, и высоту насадки, равную 30 м. Высота насадки должна быть
меньше, если диспергировать метилизобутилкетон, а также если в качестве
насадки использовать керамические кольца.
XI-2. Спроектировать экстрактор с ситчатыми тарелками для системы,
описанной в задаче XI-1, но вдвое большей производительности (5,66 м3/ч
водного раствора; 11,32 м3/ч органического растворителя). Дисперсная фаза—
метилизобутилкетон. Ответ: ответ не может быть однозначным. Если выбрать
диаметр отверстий в тарелках равным 6 мм и расстояние между тарелками
0,3 м, то диаметр колонны будет равен 0,9 м; EMD—0,252; Е0 = 0,263.
Потребуется 25 тарелок, что соответствует высоте рабочей зоны, равной 7,2 м.
XI-3. По опытным данным Розенталя (диссертация, Нью-Йоркский
университет, 1949), изучавшего теплообмен в распылительной колонне диаметром
0,14 м и высотой 3,3 м между толуолом (дисперсная фаза) и горячей водой
(сплошная фаза), высоты единицы переноса (без учета продольного
перемешивания) равны: Нюс—1,89 (Vc/Vd)1,04 и Я(Ос = 0,762 м. Введем
обозначения:
н Lccc _„/Г dtc
нюс = -тг-н1) j—r;
"too иа -"/} tc-tD
где L — фиктивная массовая скорость, кг • м'2 • сект1;
С — теплоемкость, дж • кет1 - град'1;
U—общий коэффициент теплопередачи, вт • м'2 • град'1;
t— температура, ° С.
Толуол диспергировали с помощью 13 сопел внутренним диаметром 7 мм.
Значения высот единицы переноса получены в интервале скоростей:
Vd = 3,6-10-3 —21.0-10-3 м/сек; У с = 12,7 • Ю'3 — 33- 10"3 м\сек.
661
Используя аналогию между тепло- и массообменом [принимая критерий
Нуссельта hd[kT эквивалентным критерию Шервуда и критерий Прандтля
cft-lkr эквивалентным критерию Шмидта (где кт—теплопроводность и А —
коэффициент теплоотдачи)], рассчитать высоты единицы переноса для
теплообмена из кинетических зависимостей для скорости массооб.мена в
распылительных колоннах при Vc = Vd = 8,5- Ю-3 м/сек. Результаты расчета сравнить с
приведенными опытными данными. Оценить также влияния продольного
перемешивания в данном случае. Температуру принять равной 25° С. Ответ-. Ht0c =
= 2,35 ж; Я,ов = 0,823 ж.
XI-4. Определить расход энергии для экстрактора, рассмотренного в
примере XI-4 (см. стр. 581).
XI-5. Определить размеры пульсационной колонны с ситчатыми тарелками,
предназначаемой для извлечения ацетона из разбавленного раствора его в
толуоле водой. Степень извлечения ацетона 80%; начальная концентрация
ацетона в воде равна нулю. Дисперсная фаза — толуол (ц.п = 0,592 • Ю~3 кг X
ХМ'1-сек-1; pD = 860 кг/ж3; расход 7,08 мъ/ч). Сплошная фаза — вода (ц.с =
= 1-10-3 кг • ж-'• сек-1; рс = 998 кг/ж3). Межфазовое натяжение 32-Ю-3 н/м;
коэффициент распределения cc/cD=0,58. Отверстия в тарелках диаметром
3,5 мм расположены по вершинам треугольников; суммарное свободное
сечение отверстий на, тарелке 25%, расстояние между тарелками 0,51 м.
Пульсации — синусоидальные; частота 160 циклов в минуту; амплитуда 25 мм.
Производительность колонны должна составлять 75% от нагрузки захлебывания.
Логсдайл и Торнтон [Trans. Inst. Chem. Engrs. (London), 35, 316, 331
(1957)] нашли, что Vk для данной системы в 2,6 раза больше величины,
определяемой из уравнения (XI, 57). Для колонны диаметром 76 мм константы
в уравнении (XI, 60) равны: С=2270 и ^=1/6. Рассчитать также расход
энергии на создание пульсаций (максимальную потребляемую мощность),
предполагая, что пульсатор непосредственно присоединен к колонне. (Условия задачи
совпадают с условиями примера XI-4, за исключением того, что скорости фаз
приняты в два раза меньшими). Ответ: диаметр колонны 0,58 ж; 114 тарелок.
Высота колонны вместе с тремя распределительными тарелками и
отстойными зонами 7,3 ж. Максимальный расход энергии 5,15 кет.
Глава XII
ХП-1. Несмотря на частичную смешиваемость с водой, метилэтилкетон
трудно выделять из разбавленных водных растворов перегонкой вследствие
образования азеотропной смеси (в данном случае не гетероазеотропной).
Поэтому иногда предлагалось извлекать метилэтилкетон (МЭК) экстракцией.
В частности, Смит и Л а Бон [Ind. Eng. Chem., 44, 2740 (1952)] предложили
применять в качестве избирательного растворителя для извлечения МЭК
2-метилфуран.
Нужно установить ориентировочные оптимальные условия и необходимое
число ступеней для процесса извлечения метилэтилкетона экстракцией 2-ме-
тилфураном, исходя из следующих данных. Исходная смесь: 9000 кг/ч.
водного раствора, содержащего 5 вес. % МЭК. Из экстракта МЭК выделяют
перегонкой. Степень чистоты метилэтилкетона должна составлять 99,95 мол. %.
Температура кипения МЭК равна 79,6° С (при 760 мм рт. ст.); стоимость
0,31 руб/кг.
Растворитель: 2-метнлфуран, температура кипения 63,7° С (при
760 мм рт. ст.); плотность 911 кг/м3 (при 25° С); растворимость в воде
0,2 вес.%; молекулярный вес 112. С водой 2-метилфуран образует гетероазео-
тропную смесь, имеющую при 740 мм рт. ст. температуру кипения 57,3° С и
состав 86 мол. % 2-метилфурана. Относительная летучесть МЭК и 2-метилфу-
рана 1,68 (близка к идеальной). В системе вода — 2-метилфуран — МЭК
тройной азеотропной смеси не образуется.
Данные о распределении МЭК между водой и 2-метилфураном имеются в
работе Смита и Ла Бона. Для разбавленных растворов можно принять коэф-
682
фициент распределения тЕ равным 10 (при выражении концентраций в кг/ж3)
в пользу экстр агента.
Экстракт предполагают разделять в тарельчатой ректификационной
колонне. Принять: CD = 290 руб/м2 площади тарелки; GD = 73,4 кмоль ■ Ж"2 • «г';
£0 = 0,85; GA = 0,50 кжоль • «г1 • м'2 поверхности теплообмена; С;,= 14,6 руб/м2
поверхности теплообмена; стоимость пара и хладоагентов Cftc = 0,02 руб/кмоль
дистиллята. Флегмовое число принять в 1,5 раза, а число тарелок —в 2,5 раза
больше минимального.
Установка должна работать 7200 ч в год. Стоимость монтажа,
инструментов и трубопроводов в 1,3 раза больше стоимости основного оборудования,
расходы на содержание равны 0,1 части стоимости основного оборудования!
Срок службы оборудования 3 года.
Принимая, что стоимость смесительно-отстойного экстрактора с одним
насосом на каждой ступени (т.. е. при движении одной из фаз под действием
силы тяжести) определяется зависимостью, приведенной в главе XII (см
стр. 625), найти оптимальные условия проведения процесса в смесительно-
отстойном экстракторе (вычислить объемное соотношение экстрагента и
исходного раствора, число ступеней, концентрацию МЭК в рафинате и
регенерированном экстракте, расходы на экстракцию и дистилляцию экстракта,
количество неизвлекаемого метилэтилкетона). Сделать приблизительную оценку
размеров экстрактора. Ответ: 12 ступеней; RE = 0,\5; 1 + 11 + 111 = 45 000 руб. в год.
XI1-2. Необходимо сравнить стоимость проведения процесса, описанного
в предыдущей задаче, в смесительно-отстойном каскаде и в колонном
экстракторе с ситчатыми тарелками. Суммарная плотность орошения по обеим фазам
в экстракторе с ситчатыми тарелками должна составлять 30 ж3 • лг* • «г1.
Эффективность тарелки следует определять по уравнению (XI, 44). Экстрагент
считать дисперсной фазой. Расстояние между тарелками принять равным 0,3 ж.
Стоимость колонны в расчете на одну тарелку оценивать с помощью
выражения 350 а0'7 руб., где а — поперечное сечение тарелки в ж2. Межфазовое
натяжение равно 30 • 10"3 н/м. Потери экстрагента принять постоянными, не
зависящими от его расхода. Схема регенерации экстрагента такая же, как и
в задаче ХП-1. Определить оптимальные условия проведения процесса в
экстракторе с ситчатыми тарелками. Ответ: RE = 0,l&; 1 + 11 + 111 = 72000 руб
в год.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА*
ТЕОРИЯ И ОБЩИЕ ВОПРОСЫ
ПРОЦЕССОВ ЖИДКОСТНОЙ ЭКСТРАКЦИИ
Абрамзон А. А., Островский М. В., Исследование скорости массопере-
носа осложненного химической реакцией в гетерогенных системах
жидкость—жидкость, ЖПХ, 36, № 3, 608 (1963).
Абрамзон А. А., Кияновская Ю. Л., Кремнев Л. Я., О
концентрации переносимого вещества в зоне реакции в системе жидкость—жидкость,
ЖПХ, 37, № 10, 2314 (1964).
Батунер Л. М., Фазовые диаграммы минимального расхода растворителя
при противоточной экстракции жидкостей, Труды Ленинградского химико-
фармацевтического института, вып. 11, 1960, стр. 166.
Бездель Л. С, Броунштейн Б. И., О некоторых закономерностях
процесса экстракции в системах жидкость — жидкость, ЖПХ, 33, № 2, 323
(1960).
Бездель Л. С, Железняк А. С, Броунштейн Б. И., Сопоставление
экспериментальных и расчетных величин по определению массопередачи
в каплях, сб. «Процессы жидкостной экстракции», Гостоптехиздат, 1963,
стр. 148.
Биккулов А. 3., Исследование влияния строения молекулы на свойства
избирательных растворителей, сб. «Процессы жидкостной экстракции и
хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, Изд. «Химия», 1965, стр. 347.
Бобиков П. И., Расчеты катионообменных экстракционных процессов, Сб.
«Процессы жидкостной экстракции и хемосорбции», Труды II Всесоюзного
совещания по жидкостной экстракции и хемосорбции, изд. «Химия», 1965,
стр. 90.
Бондаренко М. Ф., Гайлис А. А., Кондратьев А. А., Влияние
числа ступеней контактирования на показатели процесса экстракции,
Химия и технология топлив и масел, № 2, 12 (1962).
Бондаренко М. Ф., Гайлис А. А., Кондратьев А. А., О разделении
смесей, содержащих компоненты, неограниченно растворимые в
растворителе, сб. «Процессы жидкостной экстракции», Гостоптехиздат, 1963,
стр. 365.
Бондаренко М. Ф., Кондратьев А. А., Пути увеличения
разделяющей способности экстракционных систем, Новости нефтяной и газовой
техники, Нефтепереработка и нефтехимия, № 12, 40 (1961).
Брандт Б. Б., Перазич Л. И., Шехтман Е. Ш., О возможности
расчета барботажных реакторов, сб. «Процессы жидкостной экстракции и
хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 68.
Броунштейн Б. И., Метод расчета высоты противоточных насадочных и
тарельчатых колонн в процессах растворения однокомпонентной
диспергированной фазы, ЖПХ, 33, № 9, 2056 (1960).
* Библиография отечественной литературы и изобретений главным
образом за 1950—1965 гг. и зарубежной литературы— за 1963—1965 гг. Из
литературы, посвященной применению жидкостной экстракции в частности в
атомной энергетике и в гидрометаллургии редких и цветных металлов,
приведены ссылки лишь на ограниченное число работ. — Доп. ред.
684
^Броунштейн Б. И., Железняк А. С, Массопередача при экстракции
единичными каплями, ЖПХ, 36, № 11, 2437 (1963).
Броунштейн Б. И., Гитман И. Р., О роли нестационарной диффузии
в процессах жидкостной экстракции, сб. «Процессы жидкостной
экстракции», Гостоптехиздат, 1963, стр. 17.
Броунштейн Б. И., Железняк А. С, О пленочной теории
массопередачи, сб. «Процессы жидкостной экстракции», Гостоптехиздат 1963
стр. 39. '
Броунштейн Б. И., У с т р а й х М. А., Покорений В. Н.,
Многокомпонентная жидкостная экстракция в противоточных колоннах, сб.
«Процессы жидкостной экстракции», Гостоптехиздат, 1963, стр. 84.
Броунштейн Б. И., Метод расчета высоты противоточных насадочных и
тарельчатых колонн в процессах селективного растворения двухкомпо-
нентной диспергированной фазы, ЖПХ, 35, № 8, 1733 (1962).
Броунштейн Б. И., О выборе метода оценки эффективности
противоточных экстракционных колонн, сб. «Процессы жидкостной экстракции»,
Гостоптехиздат, 1963, стр. 53.
Броунштейн Б. И., Кинетика процесса экстракции многокомпонентных
систем в противоточных колоннах, сб. «Процессы жидкостной
экстракции», Гостоптехиздат, 1963, стр. 76.
Броунштейн Б. И., Кинетика процессов абсорбции и жидкостной
экстракции, сопровождаемых обратимой химической реакцией, Труды Гос.
института прикладной химии, вып. 49, Госхимиздат, 1962, стр. 162.
Броунштейн Б. И., Гитман И. Р., О некоторых моделях механизма
массопередачи в движущиеся капли, Труды Гос. института прикладной
химии, вып. 49, Госхимиздат, 1962, стр. 289.
Броунштейн Б. И., Железняк А. С, Определение частных
коэффициентов массопередачи в системах с переменным коэффициентом
распределения, ДАН СССР, 153, № 4, 889 (1963).
Броунштейн Б. И., Гитман И. Р., Железняк А. С, Массопередача
в сферические капли, ДАН СССР, 162, № 6, 1336 (1965).
Броунштейн Б. И., Железняк А. С, К вопросу о концевом эффекте
на входе диспергированной фазы в процессах жидкостной экстракции,
ЖПХ, 38, № 2, 423 (1965).
Броунштейн Б. И., Железняк А. С, Массопередача в капле,
сопровождаемая быстропротекающей химической реакцией, сб. «Процессы
жидкостной экстракции и хемосорбции», Труды II Всесоюзного совещания по
жидкостной экстракции и хемосорбции, изд. «Химия», 1965, стр. 81.
Броунштейн Б. И., Железняк А. С, К вопросу о применимости
формулы аддитивности фазовых сопротивлений, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по
жидкостной экстракции и хемосорбции, изд. «Химия», 1965,. стр. 49.
Булатов С. Н., Расчет скорости движения и времени пребывания фаз в
аппаратах с двухфазными потоками, Хим. пром., № 12, 887 (1964).
В ер туз а ев Е. Д., Сущность концевых эффектов, наблюдаемых в
экстракционных колоннах, Химия и технология топлив и масел № 7 8
(1964).
В о р о т и л и н В. П., Крылов В, С, Крылов В. Г., К теории экстракции
из падающей капли, Прикладная математика и механика, 29, вып 2,
343 (1965).
Г е л ь п е р_и н Н. И., В и л ь н и ц С. А., Истечение жидкостей из насадок и
отверстий малых диаметров, Труды Московского института тонкой
химической технологии им. М. В. Ломоносова, вып. 5, Госхимиздат, 1955, стр. 27.
Г е л ь п е р и н Н. И., В и л ь н и ц С. А., Диспергирование жидкостей при
истечении из насадков в воздух и жидкие среды, Труды Московского
института тонкой химической технологии им. М. В, Ломоносова, вып. 6,
Госхимиздат, 1956, стр. 111.
Гончаренко Г. К., Вилинова Л. С, Жеваго Л. П., О
противоточной экстракции, Мед. пром, СССР, № 1г 18 (1950).
665
Гончаренко Г. К., Готлинская А. П., О механизме массопередачи при
экстракции из растворов, Хим. наука и промышл., 3, № 4, 515 (1958).
Гончаренко Г. К., Готлинская А. П., О влиянии поверхностного
натяжения на размер поверхности раздела при экстракции с механическим
перемешиванием, Труды ХПИ им. В. И. Ленина, т. 18, вып. 5, изд.
Харьковского университета им. А. М. Горького, 1958, стр. 91.
Гончаренко Г. К., Седашева Е. Г., Нестеренко Г. Г., К вопросу
о движущей силе экстракции, Труды ХПИ им.. В. И. Ленина, т. 8, вып. 3,
изд. Харьковского госуниверситета им. А. М. Горького, 1958, стр. 97.
Гончаренко Г. К., Климко М. С, О механизме массоперехода при
экстракции, Труды ХПИ им. В. И. Ленина, т. 8, вып. 3, изд.
Харьковского госуниверситета им. А. М. Горького, 1958, стр. 101.
Гончаренко Г. К-, Готлинская А. П., Некоторые закономерности
процесса массопередачи при перемешивании жидкости мешалками, сб.
«Процессы жидкостной экстракции», Гостоптехиздат, 1963, стр. 222.
Гончаренко Г. К., Готлинская А. П., Зависимость времени
пребывания диспергируемой жидкости в перемешиваемом объеме от физико-
химических свойств систем, сб. «Процессы жидкостной экстракции и хе-
мосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 58.
Гончаренко Г. К., Коваленко В. И., Перемешивание в поле
центробежных сил, сб. «Процессы жидкостной экстракции и хемосорбции»,
Труды II Всесоюзного совещания по жидкостной экстракции и хемосорбции,
изд. «Химия», 1965, стр. 75.
Г у м е н ю к Н. А., Смирнов Н. И., Применение уравнения Г. К.
Дьяконова к экстракции из одиночных капель, ЖПХ, 38, № 4, 890 (1965).
Гуменюк Н. А., Смирнов Н. И., Применение уравнения Г. К.
Дьяконова к экстракции из группы капель, ЖПХ, 38, № 5, 1058 (1965).
Железняк А. С, Броунштейн Б. И., Массопередача при экстракции
единичными каплями, ЖПХ, 36, № 11, 2437 (1963).
Железняк А. С, Механизм массопередачи в колоннах с перфорированными
тарелками при наличии пульсаций, сб. «Процессы жидкостной экстракции
и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия*, 1965, стр. 140.
Железняк А. С, Броунштейн Б. И., К вопросу о концевых
эффектах на входе диспергированной фазы в процессах жидкостной
экстракции, ЖПХ, 38, № 2, 423 (1965).
Иофа Б. 3., Дакар Г. М., Экстракция комплексных кислот кислородо-
содержащими растворителями. Вычисление констант равновесия
гидролиза сурьмы (V) в растворах соляной кислоты и исследование
механизма ее экстракции ди-н-бутиловым эфиром, Радиохимия, 5, № 4, 490
(,963)- . к ж „
Каган С. 3., Разделение жидких смесей методом экстракции, сб.
«Жидкостная экстракция», Госхимиздат, 1958, стр. 127.
Каган С. 3„ К вопросу о расчете колонных экстракторов с учетом
продольного перемешивания фаз, Труды МХТИ им. Д. И. Менделеева,
вып. 17, 1964, стр. 39, 51.
Касаткин А. Г., Каган С. 3„ Тру ханов В. Г., Эмпирические
уравнения равновесного распределения для систем жидкость — жидкость, Хим.
пром., № 6, 50 (1960).
Кафаров В. В., Основные факторы микро- и макрокинетики,
определяющие межфазную турбулентность в процессах экстракции, сб. «Процессы
жидкостной экстракции», Гостоптехиздат, 1963, стр. 71.
Кишиневский М. X., Денисова Т. Б., Кинетика массоотдачи от
вращающегося диска при ламинарном режиме движения, ЖПХ, 37, № 7,
1544 (1964).
Кишиневский М. X., Мочалова Л. А., Сопротивление поверхности в
процессах абсорбции и экстракции, Сборник трудов по химии и хим.
технологии, вып. 4, Горький, 1961, стр. 885.
686
Кишиневский М. X., Корниенко Т. С, Исследование кинетики мас-
сообмена в системах жидкость — жидкость, ЖПХ, 36, № 5, 1008 (1963).
Кишиневский М. X., Корниенко Т. С, К кинетике экстракции из
капель, ЖПХ, 36, № 12. 2681 (1963).
Кишиневский М. X., Корниенко Т. С, Исследование массообмена
при движении капель динамическим методом, ЖПХ, 37, № 4, 844 (1964).
Кишиневский М. X., Корниенко Т. С, О кинетике экстракции,
осложненной химической реакцией, ЖПХ, 37, № 6, 1285 (1964).
Кишиневский М. X., Корниенко Т. С, О механизме перенеса в
системе н. гептан — толуол — диэтиленгликоль, ЖПХ, 37, № 9, 2071 (1964).
Кишиневский М. X., О механизме массо- и теплоотдачи от твердой
стенки к турбулентному потоку жидкости, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 62.
Ковалев Ю. Н., Каган С. 3., Межфазная поверхность в системах
жидкость— жидкость при механическом перемешивании, сб. «Процессы
жидкостной экстракции и хемосорбции», Труды II Всесоюзного совещания по
жидкостной экстракции и хемосорбции, изд. «Химия», 1965, стр. 43.
Коновалов В. И., Штробель В., Романков П. Г., Критериальное
уравнение захлебывания для противоточных экстракционных колонн,
ЖПХ, 34, №9, 1966 (1961).
Корниенко Т. С, Кишиневский М. X., Исследование кинетики
распределения бензойной кислоты между неполярным растворителем и водой,
ЖПХ, 36, № 6, 1238 (1963).
Корниенко Т. С., Кишиневский М. X., Исследование кинетики
распределения бензойной кислоты между бензолом и водой при экстракции
из капель, ЖПХ, 36, № 10, 2224 (1963).
Корниенко Т. С, Гандельман К. X., Исследование влияния поверх-
ностноактивных веществ на кинетику экстракции из капель, Ученые
записки кишиневского государственного Университета, 68, 34 (1964).
Крем н ев Л. Я., Абрамзон А. А., Кияновская Ю. Л., О механизме
массопереноса в гетерогенной системе жидкость — жидкость в условиях
перемешивания, ДАН СССР, 150, № 4, 836 (1963).
Кремнев Л. Я., Сквирский Л. Я-, Островский М. В.,
Абрамзон А. А., О сопротивлении массопереносу в гетерогенной системе
жидкость—жидкость, ЖПХ, 38, № И, 2496 (1965).
Кузнецов В. И., С е р я к о в а И. В., О механизме экстрагирования
элементов кислородосодержащими растворителями, сб. «Экстракция», вып. 1,
Госатомиздат, 1962, стр. 104.
Кузнецов В. И., Химизм экстракционных процессов, сб. «Экстракция»,
вып. 2, Госатомиздат, 1962, стр. 3.
Кущенко В. С, Смирнов В. И., Расчет числа теоретических тарелок
при разделении смесей методом ректификации, абсорбции и экстракции,
Изв. вузов, Нефть и газ, № 7, 65 (1962).
Левич В. Г., Крылов В. С, Воротилин В. П., К теории экстракции
из падающей капли. ДАН СССР, 160, № 6, 1358 (1965).
Левич В. Г., Крылов В. С, Воротилин В. П., К теории
нестационарной диффузии из движущейся капли, ДАН СССР, 161, № 3, 648 (1965).
Левин В. И., Квазиравновесные экстракционные процессы, сб. «Экстракция»,
вып. 1, Госатомиздат, 1962, стр. 143.
Лестева Т. М., Огородников С. К-, Железняк А. С, Подбор
экстрагентов при помощи газожидкостной хроматографии, сб. «Процессы
жидкостной экстракции и хемосорбции», Труды II Всесоюзного совеща-
. ния по жидкостной экстракции и хемосорбции, изд. «Химия», 1965,
стр. 369.
Лилеева А. К, Смирнов Н. И., Критериальное уравнение процесса
экстракции, ЖПХ, 34, № 5, 1158 (1961).
Л и л е е в а А К., С м и р н о в Н. И., Экстракция из одиночных капель ЖПХ,
34, № 6, 1361 (1961).
667
Орлов В. Ю., Жаворонков Н. М., Интенсификация массопередачи в
процессе противоточной экстракции под действием звуковых и
ультразвуковых колебаний, Журн. ВХО им. Д. И. Менделеева, 6, № 5, 595
(1961).
Плановский А. Н., Булатов С. Н., Аналитический расчет числа
действительных тарелок колонного массообменного аппарата (на примере
экстракции кофеина хлороформом), Хим. пром., № 8, 46 (1962).
Розен А. М., К теории пульсирующих экстракционных колонн, Научные
доклады высшей школы, № 5, 173 (1958).
Розен А. М., Физическая химия экстракционных равновесий, сб.
«Экстракция», вып. I, Госатомиздат, 1962, сто. 6.
Розен А. М., Об эффективности пульсирующих колонн (к общей теории
интенсифицированных противоточных экстракторов), сб.- «Экстракция»,
вып. II, Госатомиздат, 1962, стр. 300.
Розен А. М., Лапавок Л. И., Елатомцев Б. В., К вопросу о
гидравлическом моделировании противоточных аппаратов большого диаметра, Хим.
и нефт. машиностроение, № 4, 14 (1964).
Розен А. М., Беззубова А. И., Васильев В. А., Процессы экстракции
и их математическое описание. 3rd U. N. Internal. Conf. Peaceful Uses
Atom. Energy [Preprint], s. a. № 346 (1964).
Розен А. М., Агашкина Г. А., Константинова Н. А., Николо-
то в а 3. И., Решетько Ю. В., Тетер и н Э. Г., Хорхорина Л. П.,
Юркин В. Г., Экстракционные равновесия, сб. «Процессы
жидкостной экстракции и хемосорбции», Труды II Всесоюзного
совещания по жидкостной экстракции и хемосорбции, Изд. «Химия», 1965,
стр. 9.
Розен А. М., Беззубова А. И., Васильев В. А., Елатомцев Б. В.,
Лапавок Л. И., Массопередача при экстракции и моделирование
экстракционной аппаратуры, сб. «Процессы жидкостной экстракции и
хемосорбции», Труды II Всесоюзного совещания по жидкостной экстракции и
хемосорбции, изд. «Химия», 1965, стр. 99.
Самойлов О. Я., Тихомиров В. И., О молекулярно-кинетической
природе явления высаливания, сб. «Экстракция», вып. II, Госатомиздат, 1962,
стр. 34.
Скрябин А. К., Процесс экстракции, ЖФХ, 33, № 1, 74 (1959).
Соловкин А. С, Расчет констант экстракции сильных и слабых
электролитов, ЖНХ, 9, № 3, 746 (1964).
Сусанов Е. Я., Аналитический метод расчета жидкостной противоточной
экстракции, сб. «Процессы жидкостной экстракции», Гостоптехиздат, 1963,
стр. 92.
Тимошев В. Г., Петров К. А., Родионов А. В., Баландина В. В.,
Волкова А. А., Елькина А. В., Нагнибеда 3. И., О значение
строения и физического состояния молекул экстрагентов, сб. «Экстракция»,
вып. I, Госатомиздат, 1962, стр. 88.
Фомин В. В., Майоров Е. П., Кор ту шов а Л. Е., Определение числа
теоретических ступеней экстракционной колонны аналитическим методом,
сб. «Экстракция», вып. I, Госатомиздат, 1962, стр. 188.
Фролов А. Ф.. Степанова В. А., К расчету отношения растворителей в
противоточной экстракции, Ученые записки Ярославского
технологического института, Химия и хим. технология, 8, № 1, 347 (1962).
Фролов А. Ф., К расчету процесса экстракции с двумя растворителями,
Хим. машиностроение, № 6, 29 (1959).
Фролов Ю. Г., Механизм экстракции кислот и основность экстрагента,
Труды МХТИ им. Д. И. Менделеева, вып. 43, 1963, стр. 5.
Чернышев В. Н., О динамике массообмена в противоточных
экстракционных аппаратах, Автоматизация производственных процессов, № 4, 5
(1964).
Шувалов О. Н., Пуш ленков М. В., Метод расчета распределения
веществ при противоточной экстракции, Радиохимия, 3, № 6, 667 (1961),
666
А у е п R. J., W e s t w a t e r J. W., Рост капель в бинарных жидких системах,
A. I. Ch. E. Journal, 10, № 6, 885 (1964).
Bier у J. С, Boylau D. R., Моделирование динамической характеристики
экстракционной колонны для разделения жидких фаз, Industr. and Eng.
Chem. Fundament., 2, № 1, 44 (1963).
Бояджиев Л. О., О минимуме на кривой в координатах объемная
концентрация растворимых ПАВ — скорость массопередачи при абсорбции и
экстракции, Изв. Ин-та общ. и неорган, химии, Бълг. АН, 1, 41 (1963).
Dunn I., L a p i d u s L., Elgin E. С, Влияние массопередачи на противо-
точные флюидизированные системы жидкость — жидкость, А. 1. Ch. E.
Journal, 11, № 1, 158 (1965).
Елен ко в Д., Бояджиев Л., Механизм массопередачи между двумя
жидкими фазами в турбулентном потоке без коалеспенции капель, Докл.
Бълг. АН, 18, № 8, 755 (1965).
Ellis W. В., Beckraann R. В., Экстракция в системе жидкость —
жидкость, Ind. Eng. Chem., 57, № 11, 103 (1965).
Fischer W., Старые и новые способы многоступенчатого распределения,
Chem. Eng. Techn, 36, № 2, 85 (1964).
Fykyu С, Экстракция в системе жидкость — жидкость, Кагаку-кодзё, Chem.
Factory, 7, № 8, 83 (1963).
Gerster I. А.. Влияние продольного перемешивания на характеристику
экстракционных колонн, Chem. Eng. Progr., 59, № 11, 79 (1963).
Hadwiger H., Glur P. К., К математической теории аппарата Зигнера
для распределения веществ между двумя несмешивающимися
жидкостями, Experientia, 19, № 5, 270 (1963).
Хирата М., Экстрагирование в системе жидкость — жидкость. Расчет
равновесия и выбор растворителя. Кэмикару эндзиниярингу, 8, № 8, 734
(1963).
Hopff H., Lussi H., Gerspacher P., Hammer E., Суспензионная
полимеризация. Суспензионная полимеризация как модель для
исследования процессов перемешивания в двухфазных системах, Chem. Ing. Techn.,
36, № 11, 1085 (1964).
Ног ton Т. I., Fritsch T. R., Kintner R. С, Экспериментальное
определение скоростей экстракции внутри капель, Canad. J. Chem. Eng., 43,
№ 3, 143 (1965).
How art h W. I., Механизм массопередачи в сосудах с перемешиванием,
Chem. Eng. Sci., № 1, 47 (1963).
I а с k s о n R Образование и коалесценция капель и пузырьков в жидкостях,
Chem. Erigr., № 178, SE-107 (1964); [Trans. Inst. Chem. Eng., 42, № 4,
1964].
Johns L. E., В е с k m a n n R. В., Е 11 i s W. В., Теоретические основы мае-
сопереноса при экстракции в системе жидкость — жидкость, Brit. Chem.
Eng., 10, № 2, 86 (1965).
van Ipenberg К., Жидкостная экстракция солей металлов, Chem. weekbl.,
59, № 40, 540 (1963).
Jeffreys G. V., Ellis S. R. M., Анализ процессов экстракции
избирательными растворителями, CHISA, 1962, Lect. 1st Internat. Congr. Chem. Eng.
Equipm. Design and Automat., Prague, Czechosl. Acad. Sci., 1964, p. 65.
Jeffreys G. V., Hawksley I. L., Коалесценция капель жидкости в двух-
компонентных двухфазных системах, A. I. Ch. E. J., 11, № 3, 413
(1965).
К i e s s 1 i n g R. К., К вопросу об оптимальном методе расчета
многоступенчатых колонн при противоточной жидкостной экстракции, Kernenergie, 6,
№ 10, 540 (1963).
Kintner R. С, Явления в каплях, влияющие на процесс жидкостной
экстракции, Advances Chem. Eng., v. 4, Acad. Press, 1963, p. 51.
Kneule F., Nemek I., Увеличение пропускной способности
экстракционных колонн путем воздействия электрического поля, Chem.—Ing.—Techn ,
35, № 12, 851 (1963).
44 P. E. ТрейОал 669
Konnecke H., Селективная экстракция как один из основных процессов
химической технологии, Forsch. u. Forschritte, 37, № 5, 129 (1963).
Linde H., Thiessen D., Влияние гидродинамической нестабильности и
стабильности границы раздела жидких фаз на перенос вещества в
системе вода —бензол, Z. phys. Chem. (DDR), 227, № 3—4, 223 (1964).
Marchi В. D., Niedeger W. I., Массопередача от свободных капель,
Industr. Eng. Chem. Fundamentals, 4, № 2, 129 (1965).
Marondes N. G., Sawistowski H., Одновременный перенос двух
растворенных веществ через поверхность раздела жидкость — жидкость,
Chem. Eng. Sci., 1ft, № 11, 919 (1964).
Meyer К., Konnecke H. G., К вопросу об экстракции в системе
жидкость—жидкость, Z. phys. Chem. (DDR), 222, № 1—2. Ill (1963).
Моделирование экстракции при помощи аппарата Крэга, Chem. Eng. News,
43, № 16, 78, 80 (1965).
М 6 g 1 i А., Проблемы оптимизации колонн для противоточной экстракции,
Chem. Eng. Techn., 37, № 3, 210 (1965).
Molerus О., Движение частиц в пульсирующем потоке, Chem. — Ing.—
Techn., 36, № 8, 866 (1964).
М о р и т а X., Экстракция в системе жидкость — жидкость. Влияние добавок
неорганических солей, Mem. Fac. Technol. Kanazawa Univ., 3, № 3, 273
(1964).
Ми я ути Т., Экстрагирование в системе жидкость—жидкость. Теория
экстрагирования, Кэмикару эндзиниярингу, 8, № 8, 729 (1963).
de N i e L. Н., Экстракция в системе жидкость — жидкость, Chem. Proc. Eng.,
44, № 1, 20 (1963).
Oberg A. G., Экстракция в системе жидкость—жидкость, Chem. Engng., 70,
№ 15, 119 (1963).
Olander D. R., Гидродинамическая модель массопередачи в ячейке с
перемешиванием, Chem. Eng. Sci., 18, № 2, 123 (1963).
Olander D. R., R e d d у L. В., Влияние величины движущей силы
(разности концентраций) на массопередачу в системе жидкость — жидкость,
Chem. Eng. Sci., 19, № 1, 67 (1964).
О 1 n e у R., Характеристики капель, образующихся в противоточном
экстракторе, А. 1. Ch. E. Journal, 10, № 6, 827 (1964).
Paul H. I., Sleicher С. А., Максимальный размер устойчивой капли в
турбулентном потоке. Влияние диаметра трубы, Chem. Eng. Sci., 20, № 1,
57 (1965).
Popovich А. Т., Jervis R. E., Trass О., Массопередача при
образовании отдельной капли, Chem. Eng. Sci., 19, № 5, 357 (1964).
Princen H. M., Форма жидкой капли на поверхности раздела жидкость —
жидкость, J. Coll. Sci., 18, № 2, 178 (1963).
Prochazka I., Landau I., Изучение процесса экстрагирования. 1.
Обратное перемешивание и эффективность процесса непрерывной
ступенчатой противоточной экстракции, Coll. Czech. Chem. Comm., 28, № 8, 1927
(1963).
R u d d D. F., Blum E. D., Оптимальные условия проведения экстракции с
перекрестным током и рециркуляцией продукта, Chem. Eng. Sci., 17,
277 (1962).
Rosen E. M., Дискуссия по статье Rudd D. F., Blum E. D., «Оптимальные
условия проведения экстракции с перекрестным током и рециркуляцией
продукта», Chem. Eng. Sci., 19, № 12, 999 (1964).
Sawistowski H., James В. R., Влияние поверхностных явлений на
общие коэффициенты массопередачи при экстракции в системе жидкость —
жидкость, Trans. Inst. Chem. Eng., 35, № 3, 175 (1963).
Sawistowski H., G о 1 i t z G. E., Влияние поверхностных явлений на
скорость переноса вещества при экстракции в системе жидкость —
жидкость, Trans. Inst. Chem. Eng., 41, № 4, 174 (1963).
S idem an S., Фотографирование капель в жидкой среде, Chem. Eng. Sci.,
19, № 6, 426 (1964),
690
S h i m b a s h i Т., S h i b а Т., Скорость массопередачи через поверхность
раздела жидкость — жидкость, Bull. Chem. Soc. Japan, 38, № 4, 572 (1965).
S с h г о e d e r R. R., К i n t n e r R. С, Колебания капель, падающих в жидкой
среде, A. I. Ch, E. Journal, 11, № 1, 5 (1965).
S k е 1 1 а п d A. H. P., We lie к R. М., Сопротивление массопередаче внутри
капель, A. I. Ch. E. Journal, 10, № 4, 491 (1964).
Smith A. R., Caswell I. E., Larson P. P., Cavers S. D., Коалесцен-
ция капель в экстракционных колоннах для систем жидкость — жидкость,
Canad. J. Chem. Eng., 41, № 4, 150 (1963).
So are S., Precup I., Tar an С, Экстракция в системе жидкость —
жидкость, Bucuresti, Ed. techn. 1963.
S r a b о Т. Т., Lloyd W. A., Cannon M. R., Speaker S. S., Экстракция
с регулируемыми циклами (циклическая экстракция в системе жидкость —
жидкость), Chem. Eng. Progr., 60, № 1, 66 (1964).
S t a f f i n H. К., С h u I. С, Переходные характеристики непрерывной
экстракции с перемешиванием, A. I. Ch. E. Journal, 10, № 1, 98 (1964).
Stemerding S., Zuiderweg F. I., Продольное перемешивание
жидкостей и его влияние на эффективность экстракции. Приближенный метод
расчета, Chem. Engr., № 168, 155 [Trans. Inst. Chem. Eng., 41, №4 (1963)].
Treybal R. E., Рециркуляция при экстракции жидкости жидкостью, Ind.
Eng. Chem. Fundament., 3, № 3, 185 (1964).
Valentine R. S., Sather N. F., Heiderer W. I., Движение капель в
вязкой среде, Chem. Eng. Sci., 20, № 8, 719 (1965).
Ward W. J., Quinn J. А., Диффузия через поверхность раздела
жидкость— жидкость. II. Сопротивление поверхности раздела в трехкомпо-
нентных системах, A. I. Ch. E. Journal, 11, № 6, 1005 (1965).
W е 11 е k R. M., S к е 11 a n d A. H., Экстракция из единичных турбулентно
движущихся капель, A. I. Ch. E. Journal, 11, № 3, 557 (1965).
Wilburn N. Р., Математическое выражение кривых концентраций для
двухфазных непрерывно действующих противоточных экстракторов, Ind.
Eng. Chem. Fundament., 3, № 3, 189 (1964).
W о о d 1 e R. А., Аналогия между процессами дистилляции и экстракции, Ind.
Eng. Chem., 55, № 3, 16 (1963).
Z a h r a n d n i k R. L., Archer D. H., Применение принципа дискретного
максимума к последовательной экстракции с простым рециркулирующим
потоком, Ind. Eng. Chem., Fundament., 3, № 4, 384 (1964).
- Применение процессов жидкостной экстракции
в технологических процесевх
Авдей Г. М., Покорений В. Н., Ситько И. Л., Влияние ультразвука
на процесс экстракции ароматических углеводородов диэтиленгликолем,
сб. «Процессы жидкостной экстракции и хемосорбции», Труды II
Всесоюзного совещания по жидкостной экстракции и хемосорбции, изд. «Химия».
1965, стр. 168.
Авдей Г. М., Покорский В. Н., Экстракция диэтиленгликолем этилбен-
зола и ксилолов из нефтяного сырья, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 245.
Авдей Г. М., Покорский В. Н., Я блочки на М. Н., Экстракция
ароматических углеводородов сульфоланом, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 329.
Агафонов А. В., Аба ев а Б. Т., Окиншевич Н. К-, Получение сырья
для получения активных саж экстракцией селективными растворителями
газойлей каталитич. крекинга, Химия и техн. топлив и масел, № 7, 36 (1964).
Алаев Б. С, Перченко А. А., Котельников Б. П., Пономаре н-
к о И. Я., Способ разделения смесей водонерастворимых синтетических
жирных кислот и жирных спиртов, авт. свид. 137512, 25/IV 1961 г.; Бюлл.
изобр., № 8 (1961),
44*
6S1
Анненкова Л. А., Бойко И. Д., Жуковская С. А., Экстракционное
извлечение террамицнна из натнвных растворов с применением струйного
экстрактора, Медицинская пром. СССР, № 11, 43 (1961).
Барам М. И., Ласкорин Б. Н„ Экстракция плавиковой кислоты три-к-
бутилфосфатом, ЖНХ, 9, № 3, 738 (1964).
Бездель Л. С., Броунштейн Б. И., Фосфатная очистки бензинов от
сероводорода, Труды Всесоюзного научно-исследовательского института
нефтехимических процессов, вып. 5, Гостоптехиздат, 1962, стр. 205.
Бездель Л. С, Броунштейн Б. И., Ипатьев В. В., Теодор о-
в и ч В. П., Фосфатная очистка сжиженной пропан-пропиленовой фракции
от сероводорода, Труды Всесоюзного научно-исследовательского института
нефтехимических процессов, вып. 5, 1962, стр. 217.
Бездель Л. С, Броунштейн Б. И., Устранх М. А., Экстракция
ароматических углеводородов в пульсационных колонках, сб. Производство
бензола, Госхимиздат, 1962, стр. 69.
Бездель Л. С, Броунштейн Б. И., Ипатьев В. В., Экстракция H2S
из сжиженной пропан-пропиленовой фракции раствором КзР04 в противо-
точной колонне, ЖПХ, 35, № 8. 1715 (1962).
Бездель Л. С, Броунштейн Б. И., Ипатьев В. В., Теодор о-
в и ч В. П., Фосфатная очистка сжиженной пропан-пропиленовой фракции
и бензина от сероводорода, сб. «Процессы жидкостной экстракции», Гос-
технздат, 1963, стр. 352.
Белобородое В. В., Бухтарева Э. Ф, Жидкостная экстракция
бензиновых растворов растительных масел, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбцин, изд. «Химия», 1965, стр. 277.
Бондаренко М. Ф„ Кодымская Г. А., Сравнение избирательности
некоторых растворителей при разделении смеси метилнафталина и
«-декана, сб. «Процессы жидкостной экстракции и хемосорбции», Труды
II Всесоюзного совещания по жидкостной экстракции и хемосорбции, изд.
«Химия», 1965, стр. 357.
Борбат В. Ф., Бобиков И. И., Коуба Э. Ф., Халимова А. К.,
Способ извлечения палладия из растворов, содержащих платиноиды, цветные
металлы и железо, авт. свид. 165689 26/XI 1964 г. Бюлл. изобр., №20 (1964).
Борбат В. ф., Коуба Э. Ф., Анионообменная экстракция благородных
металлов хлористыми солями четвертичных аммониевых оснований, сб.
«Процессы жидкостной экстракции и хемосорбции», Труды II
Всесоюзного совещания по жидкостной экстракции и хемосорбцин, изд.
«Химия», 1965, стр. 298.
Брик А. И., Муравлев Д. А., Турскнй Ю. И., Филлипов И. В.,
Применение диизопропилового эфира для дефеноляции сточных вод в сб.
Очистка промышленных сточных вод. Труды совместной конференции
Института ВОДГЕО и Института водного хозяйства Министерства
земледелия, лесного и водного хозяйства ЧССР, Госстройиздат, 1962, стр. 147.
Бурмистров С. И., Зайцев В. Н., Способ получения алкилнитрофенолов,
авт. свид. № 160174 16/1 1964 г.; Бюлл. изобр., № 3 (1964).
Вдовенко В. М., Ковальская М. П., Шираинскнй Е. В.,
Экстракция тория нз сернокислых растворов октиламином, Радиохимия, 3, № 1,
3 (1961).
Вознесенская Е. В., Кутукова В. И., Рывкина С. Л.,
Двухступенчатая очистка фенолом маловязкой фракции сернистых нефтей, сб.
«Процессы жидкостной экстракции и хемосорбции». Труды II Всесоюз.
совещания по жидкостной экстракции и хемосорбции, изд. «Химия», 1965, стр. 282.
Гельперин Н., Крохин Н., Киселева Е., Экстракция из растворов
конденсирующимися парообразными экстрагентами, ЖПХ, 31, № 7, 1026
(1958).
Гельперин Н. И., А с с м у с М. Т., Коровин С. С, Извлечение галлия
методом жидкостной экстракции в инжекторной колонне непрерывного
действия, ЖПХ, 35, № 3, 516 (1962).
692
Гельперин Н. И., Пебалк В. Л., Розов В. Н.. Замышляев В Г.,
А с с м у с В. Г., М п л о в а н о в а И. Б., Епишева М. С, Аппаратурно-
технологическое оформление процесса экстракционной очистки никелевых
растворов от железа н меди, сб. «Процессы жидкостной экстракции и
хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд, «Химия», 1965, стр. 292.
Гончаренко Г. К., Готлннская А. П., Распределение кофеина между
водным экстрактом чайного листа и дихлорэтаном,' Медицинская пром.,
№ 1, 58 (1963).
Грибова К. И., Способ выделения акриловой кислоты, авт. свид. 156945,
25/IX 1963 г., Бюлл. изобр., № 17 (1963).
Гришин Е. Г., Наш опыт получения экстракторных эфирных масел, Масло-
бойно-жировая пром., № 5, 42 (1959).
Грищенко Н. Ф., Покорений В. Н., Яблочкина М. Н., Новые
селективные растворители для экстракции ароматических углеводородов, сб.
«Процессы жидкостной экстракции и хемосорбции», Труды II
Всесоюзного совещания по жидкостной экстракции и хемосорбции, изд. «Химия»,
1965, сгр. 319.
Губенко И. Б., К а р а с е в а А. А., Г р я з н о в а Н. Н., Френкель В. Л.,
Пещанская И. И., Применение роторно-дискового контактора для
интенсификации процесса двухступенчатой деасфальтизации гудронов
восточных нефтей, сб. «Процессы жидкостной экстракции и хемосорбции»,
Труды II Всесоюзного совещания по жидкостной экстракции и
хемосорбции, изд. «Химия», 1965, стр. 237.
Губенко И. Б., Черножуков Н. И., Исследование растворимости
компонентов гудрона в пропане в зависимости от основных параметров
процесса деасфальтизации, сб. «Процессы жидкостной экстракции н
хемосорбции», Труды II Всесоюзного совещания по жидкостной экстракции
и хемосорбции, изд. «Химия», 1965, стр. 342.
Долгих В И.. Бобиков П. И., Борбат В. Ф„ Ферберг М. Б.,
Опытная экстракционная установка по извлечению благородных металлов
из шламов, сб. «Процессы жидкостной экстракции н хемосорбции», Труды
II Всесоюзного совещания по жидкостной экстракции и хемосорбцин, изд.
«Химия», 1965, стр. 306.
Доронин В. Н., Николаев А. М., Экстракция высших спиртов в наса-
дочной колонне, Труды КХТИ, т. 29, Казань, 1960, стр. 192.
Д ы х а н о в Н. И., Б а т и й В. Г., Способ выделения чистого ацетилацетона,
авт. свид. 156544, 28/VIII 1963 г.; Бюлл. нзобр., № 16 (1963).
Железняк А. С., Броунштейн Б. И., Взаимная растворимость в
системе вода — уксусная кислота — этилацетат в интервале температур от
0 до 50° С, ЖПХ, 38, № 3, 694 (1965).
Железняк А. С, Лестева Т. М., Огородников С. К., Способ
выделения многоатомных спиртов, авт. евнд. 170941, 11/V 1964 г.; Бюлл.
изобр., № 10 (1965).
Ж е л е з н я к А. С., Броунштейн Б. И., Влияние пульсаций на
эффективность насадочиых колонн в процессе экстракции продуктов
нефтехимического синтеза, сб. «Процессы жидкостной экстракции н хемосорбцин»,
Труды II Всесоюзного совещания по жидкостной экстракции и
хемосорбции, изд. «Химия», 1965, стр. 115.
Индюков И. М., Сидорчук И. И.. Марджанов Г. М., Выделение
ароматических углеводородов из газойлей и нафты каталитического
крекинга в струйных экстракторах, сб. «Процессы жидкостной экстракции
и хемосорбцин», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, етр. 262.
Иоффе Э. Ш„ Экстракционное отделение кобальта от никеля н других
примесей с применением солей третичных аминов, сб. «Процессы
жидкостной экстракции н хемосорбции», Труды II Всесоюзного
совещания по жидкостной экстракции и хемосорбции, изд. «Химия», 1965,
стр. 364.
45 Р. Е. Трейбал
693
И х н о Н. П., Динамика нейтрализации примесей жирных кислот в
растительных жирах и маслах при одновременном распылении и растворении
образующегося мыла, сб. «Процессы жидкостной экстракции и хемосорбции»,
Труды II Всесоюзного_совещания по жидкостной экстракции и
хемосорбции, изд. «Химия», 196о, стр. 311.
Каган С. 3., Макаров Г. Н., В остри ков а В. Н., Применение пуль-
сационных экстракторов для обесфеноливания сточных вод, Газ. пром.,
№ 9, 16 (1958).
Каган С. 3., Сахарное А. В., Обесфеноливание производственных
сточных вод экстракцией диизопропиловым эфиром, Бюлл. по обмену опытом
в лакокрасочной промышленности, Госхимиздат, № 14 (1958).'
Каган С. 3., Волкова Т. С, Филлинов И. В., Аэров М. Э.,
Испытание опытно-промышленного роторно-дискового экстрактора для
обесфеноливания подсмольных вод, Fa3. пром., № 4, 13 (1962).
Каган С. 3., Стрельцова А. А., Волкова! С, Долотова В. А..
Двухступенчатая экстракция капролактама в роторно-дисковых
экстракторах, сб. «Полупродукты для синтеза полиамидов», Госхимиздат, 1963,
стр. 105.
Каган С. 3., Ковалев Ю. Н., Извлечение высших спиртов из их смесей
с углеводородами методом жидкостной экстракции, Труды МХТИ им.
Д. И. Менделеева, вып. 40, Москва, 1963, стр. 122.
Каган С. 3., Ковалев Ю. Н., Коган Ю. Б., Орлова Н. А.,
Исследование экстракции высших спиртов из их смесей с углеводородами,
Труды МХТИ им. Д. И. Менделеева, вып. 40, 1963, стр. 128.
Каган С. 3., Тру ханов В. Г., Костин П. А., Кудрявцев Е. Н.,
Применение промышленных роторно-дисковых экстракторов для
двухступенчатой экстракции капролактама, Хим. пром., № 2, 14 (1964).
Каган С. 3., Труха нов В. Г., Костин П. А., Кудрявцев Е. Н.,
Экстракция капролактама из сульфатных щелоков в роторно-дисковых
экстракторах, Хим. пром., № 3, 184 (1965).
Каплан Г. Е., Моисеев Е. Д., Дмитриева Л. П., Косточки-
н а С А., Экстракционное разделение циркония и гафния, сб.
«Экстракция», вып. I, Госатомиздат, 1962, стр. 117.
Каденская Н. И., Железняк А. С, Броунштейн Б. И., Массопере-
дача при экстракции уксусной кислоты единичными каплями этилацетата,
ШПХ, 38, №7, 1153 (1965).
Карасева А. А., Розенштейн М. 3., Якоби Ф. С, Новаков-
с к а я И. В., Исследование влияния гидродинамических факторов на
эффективность работы насадочной экстракционной фенольной колонны,
Химия и технология топлив и масел, № 7, 48 (1963).
Касаткин А. Г., Каган С. 3., Труханов В. Г., Исследование статики
экстракции капролактама органическими растворителями Хим проч.
№ 3, 42 (1961).
Касаткин А. Г., Каган С. 3., Труханов В. Г., Новый способ
выделения капролактама из производственных растворов экстракцией, Вестник
технической и экономической информации НИИТЭхим, № 3, 20 (1962).
Касаткин А. Г., Каган С. 3., Труханов В. Г., Аппаратурное
оформление извлечения капролактама методом жидкостной экстракции, сб.
«Процессы жидкостной экстракции», Гостоптехиздат. 1963, стр. 241.
Ко с тюк В. А., О разделении бета-пиколиновой фракции каменноугольной
смолы экстракцией двумя растворителями, Труды ИГИ АН СССР т 12,
изд. АН СССР, 1961, стр. 135.
Ковалев Ю. Н., Каган С. 3., Способ извлечения спиртов из смеси
углеводородов, авт. свид. 160175, 16/1 1964 г.; Бюлл. изобр., № 3 (1964).
Корпусов Г. В., Ескевич И. В., Жаров Е. П., Групповое разделение
редкоземельных элементов методом противоточной экстракции, сб.
«Экстракция», вып. I, Госатомиздат, 1962, стр. 125.
Крамаренко В. П., Оптимальные условия экстракции кокаина из водных
растворов органическими растворителями, Фармацевт, ж., № 1, 23 (1960).
694
Крючков Б. С, Серафимов Л. А., Львов С. В., Об извлечении
органических кислот методом жидкостной экстракции, Химия и технология
топлив и масел, № 7, 20 (1962).
Крючков Б. С, Серафимов Л. А., Львов С. В., Извлечение
органических кислот методом жидкостной экстракции, Химия и технология
топлив и масел, № 12, 58 (1963).
Крючков Б. С, Серафимов Л. А., Стрелец И. П., Г о л ы н е ц Ю. Ц.,
"Львов С. В., Извлечение двухосновных кислот методом жидкостной
экстракции, Химия и технология топлив и масел, № 4, 6 (1964).
Кузнецов Ф. А., Л и л е е в а А. К., Смирнов Н. И., Коэффициенты
распределения жирных кислот, Труды ЛТИ им. Ленсовета, вып. 60,
Госхимиздат, 1960, стр. 206.
Кузнецов Ф. Л, Лилеева А. К-, Смирнов Н. И., Изучение
равновесия процессов экстракции в некоторых системах, ЖПХ, 34, № 8, 1829
(1961).
Кутукова В. И., Вознесенская Е. В., Стоянович P. K-,
Экстрактор с вращающимися дисками для очистки фурфуролом масляных
фракций, Химия и технология топлив и масел, № 5, 16 (1960).
Кутукова В. И., Вознесенская Е. В., Стоянович P. K-,
Применение экстрактора с вращающимися дисками для селективной очистки
трансформаторного масла, Химия и технология топлив и масел, № 7, 30
(1960).
Левин А. И., Железняк А. С, С е м е н ю к Л. О., Извлечение
низкомолекулярных органических одноосновных кислот из водных растворов
методом экстракции, сб. «Ректификация и экстракция продуктов
органического синтеза», Госхимтехиздат, I960, стр. 84.
Ледяшева Г. Е., Дорогочинский А. 3., Деароматизацня дистиллята
бензина «галоша» диметилформамидом с применением дискового
ротационного экстрактора, сб. «Процессы жидкостной экстракции и
хемосорбции», Труды II Всесоюзного совещания по жидкостной экстракции л
хемосорбции, изд. «Химия», 1965, стр. 268.
Максименко М. 3., Галеев А. Ф., Актуганова Л. С,
Якушев Г. X., Повышение эффективности экстракционных колонн при
очистке масел, сб «Нефтепереработка и нефтехимия», ЦНИИТЭнефтегаз, № 10,
40 (1965).
Марушкин Б. К-, Берг Д. А., Сидорочева Л. В., Байдавлето-
в а Ф. Г., Экстрактивная депарафинизация дизельного топлива, Сборник
трудов Уфимского нефтяного института, вып. 3, Башкирское книжное изд.,
1960, стр. 187.
Митрофанов М. Г., Артемьева О. А., Ледяшева Г. Е„ Экстракция
ароматических углеводородов диэтиленгликолем с применением
ротационного дискового экстрактора, сб. «Производство бензола», Госхимиздат,
1962, стр. 55.
Митрофанов М. Г., Мартыненко А. Г., Бондаренко Н. И., М у-
лина Т. А., Ко но плев В. П., Радионова Е. М., Хабасаха-
лова Г. Я-, Освоение опытно-промышленной установки деароматизации
бензина «галоша» диэтиленгликолем, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 257.
Навтанович М. Л., Черняк А. С, Жидкостная экстракция металлов
кислыми амилфосфатами, Сб. научн. трудов Иркутского
научно-исследовательского института редких металлов, вып. 9, ГосгортехизДат, 1961,
стр. 140.
Николаев А., Поникаров И., Сравнительная эффективность аппаратов
по экстракции высших спиртов, Промышлеино-экономический бюллетень
Татсовнархоза, № 4, 10 (1960).
Огородников С. К., Немцев М. С, Морозова А И., Способ
выделения метакриловой кислоты из водных растворов, авт. свид. 170965,
11/V 1964 г.; Бюлл. изобр., № 10 (1965),
45*
696
Пальчиков Г. Ф., Меж л умов а Н И., Степуро С. И., Кага-
,нер Г. С, Кричко А. А., Экстракционное разделение нефтепродуктов
обводненным пиридином, сб. «Процессы жидкостной экстракции и хемо-
сорбции», Труды II Всесоюзного совещания по жидкостной экстракции и
хемосорбции, изд. «Химия», 1965, сгр. 337.
Петунина Н. И., Плюсин В. Г., Поиски экстрагента для извлечения
ванадия из кислых растворов, Труды Института химии АН СССР, вып. 7,
Уральский филиал, Химия и технология редких элементов, Свердловск,
1964, стр. 61.
Петунина Н. И., Плюснин В. Г., Экстракция ванадия из
производственных растворов, Труды Института химии АН СССР, вып. 7,
Уральский филиал, Химия и технология редких элементов, Свердловск, 1964,
стр. 67.
Плюснин В. Г., Петунина Н. И., Ивакин А. А., Способ извлечения
ванадия из сернокислых растворов экстракцией, авт. свид. 157783, 24/VII
1963 г.; Бюлл. изобр.. № 19 (1963).
По ко реки й В. Н., Итоги работ в области экстракции ароматических
углеводородов (бензола, толуола, ксилолов), сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции, изд. «Химия», 1965, стр. 32.
Пуш ко в А. А., Шмидт В. С, Шестериков В. Н., Экстракция
щавелевой кислоты три-н-октиламииом из азотнокислых растворов, Труды
МХТИ им. Д. И. Менделеева, вып. 43, 1963, стр. 12.
Р о з е н А. М., К а р п а ч е в а С. М., Медведев С. Ф., Родионов Е. П.,
Киселева Л. Ф., Массопередача при экстракции и реэкстракции ура-
нилнитрата в насадочных колоннах, сб. «Экстракция», вып. 2, Госатом-
издат, 1962, стр. 284.
Розен А. М., Воронецкая Е. В., Плотность, вязкость, поверхностное
натяжение растворов и коэффициенты диффузии веществ в системе
вода— уранилнитрат — азотная кислота — Т. Б. Ф., сб. «Экстракция», вып. 2,
Госатомиздат, 1962, стр. 199.
Розен А. М., Внутренняя циркуляция извлекаемых веществ и
технологический расчет колонны при экстракции трибутилфосфатом, Атомная
энергия, 7, вып. 3, 277 (1959).
С а к о д ы н с к и й К- И., М а л ю с о в В. А., У м н и к Н. Н.,
Жаворонков Н. М., Кар а пет ян Ш. А., Способ разделения смеси
тетрахлоралканов, авт. свид. 137505, 25/IV 1961 г.; Бюлл. изобр., № 8
(1961).
Сийрдэ Э. К., Лухакоодер Э. Т., Обесфеноливание подсмольной воды
горючего сланца с помощью высококипящей фракции кетонов,
выделенных из сланцевой подсмольной воды. Труды Таллинского
политехнического института, Серия А, № 63, вып. 2, Эстонское Гос. издательство,
1955, стр. 169".
Соков Ю. Ф., Путилова 3. Д., Применение роторно-дискового
контактора для экстракции ароматических углеводородов на промышленной
установке, сб. «Процессы жидкостной экстракции», Гостоптехиздат, 1963,
стр. 53.
Соков Ю. Ф., Путилова 3. Д., Кириллов Т. С, Экстракция бензола
из катализата платформинга на роторно-дисковых экстракторах, сб.
«Производство бензола», Госхимиздат, 1962, стр. 61.
Соков Ю. Ф., Путилова 3. Д., К а ста нос А. 3., Вакуленко А. А.,
Применение роторно-дискового контактора для экстракции ароматических
углеводородов диэтиленгликолем, Труды Башкирского
научно-исследовательского института по переработке нефти, вып. 7, изд. «Химия», 1964,
стр. 108.
Старков А. В., Львов С. В., Экстракция высших жирных спиртов из
их смесей с углеводородами. Химия и технология топлив и масел, № 5,
7 (1957).
696
Т р у х а н о в В. Г., Касаткин А. Г., Каган С. 3., Новый способ
экстракции капролактама, авт. свид. 145585, 21/Ш 1962 г.; Бюлл. изобр., № 6
(1962).
Тихомиров В. Б., Горяйнов Н. Е., Галкин Н. П., Федоров В. Д.,
Способ экстрагирования и устройство для выполнения этого способа. Авт
свид. 12!781, 18/VIII 1959 г.; Бюлл. изобр., № 16 (1959).
Турский Ю. И, Филиппов И. В., Прибор для измерения среднего
диаметра, скорости движения и степени дисперсности частичек при
экстракции жидкости в жидкость, авт. свид. 106621, 25/XI 1957 г.; Бюлл. изобр-,
№ 9 (1957).
У й с т р а х М. А., Б р о у н ш т е й н Б. И., П о к о р с к и й В. Н., Селективное
растворение диэтиленгликолем смесей толуол-к-гептан и бензол-к-гептан
в противоточных насадочных колоннах, ЖПХ, 35, № 11, 2454 (1962).
Филиппов И. В., Способ осуществления экстракционной очистки фенол-
содержащих сточных вод, авт. свид. 107619, 25/XI 1957 г.; Бюлл. изобр.,
№ 7 (1957).
Филиппов И. В., Турский Ю. И., Влияние различных факторов на
экстракцию фенолов из подсмольных вод бутилацетатом, Газ. пром., № 12,
19 (1958).
Филиппов И. В., К а р п а ч е в а С. М., Экстракция фенолов из
подсмольной воды^ органическими растворителями, Газ. пром., № 1, 25 (1958).
Филиппов И. В., Карпачева С. М., Турский Ю. И.,
Обесфеноливание сточных вод (экстракция), сб. «Очистка промышленных сточных вод»,
Труды совместного совещания Института ВОДГЕО и Министерства
энергетики и водного хозяйства ЧССР, Госстройиздат, 1960, стр. 83.
Филиппов И. В., Карпачева С. М., Испытание пульсационной наса-
дочной колонны в процессе обесфеноливания подсмольной воды, сб.
«Очистка промышленных сточных вод», Труды совместной конференции
Института ВОДГЕО и Института водного хозяйства Министерства
земледелия, лесного и водного хозяйства ЧССР, Госстройиздат, 1962, стр. 138.
Филиппов И В., Каган С. 3., Кондратьева М. И., К вопросу об
обесфеноливании сточных вод коксохимических заводов, Кокс и химия,
№ 12, 46 (1963).
Шостенко Ю. В., Измайлов Н. А, Чмель В. Д., Мушинская С. X.,
Способ экстрагирования веществ из водных растворов, авт. свид. 157333,
5/Х 1963 г.: Бюлл изобр., № 18 (1963).
Якубович И. А., Кириллов О. Д., Способ экстрагирования
растворимых веществ, авт. свид. 133464, 25/Х1 1969 г.; Бюлл. изобр., № 22 (1960).
Якубович И. Я-, Невский О. Б., Способ проведения противоточных
процессов массообмена в жидкой среде, авт. свид. 144465, 15/П 1962 г.;
Бюлл. изобр., № 3 (1962).
Baniel A., Blum berg R., Концентрирование водных растворов
посредством экстракции в системе жидкость — жидкость, Ind. Chemist, 39, № 9,
460 (1963).
С h i о Н i P, Протнвоточиая экстракция в системах соль — металл при
окислительно-восстановительных реакциях, Ind. Eng. Chem. Prozess Design
and De\el., 4, № 3, 299 (1965).
Ischimori Т., Akatsu E., Cheng Wen-pin at al., Данные по
экстракции неорганических растворителей, Japan Atomic Energy Res Inst.
Res. Rep., № 1062 (1964).
К u h n W., T h u r k a u f M., Экстракционный метод выделения компонентов
из жидкой смеси, швейц. пат. 358782. 31/1 1962 г
Matthews Т., Hutchinson H. Р., Перенос уксусной кислоты из
хлороформа в воду, Inter. J. Heat and Mass Transfer., 5, 1139 (1962).
Patt P. W., Метод непрерывного разделения смесей вешеств растворителями
с различающимися коэффициентами распределения, пат. ФРГ 1117544,
30/V 1962 г.
Renault Rh., Talmaut X., Работа пульсашюнных колонн. Экстракция
азотнокислою урана, Energie nucl., 5, № 3, 177 (1963),
697
Schuberth H., Метод расчета экстрактивных свойств тройных жидких
систем типа Sl/S2/Lo на примере системы я-СНзСООС2Н7 (я-СН3СООС4Н9) •
•Н20, Chem. Techn., 15, № 2, 92 (1963).
Ziolkowski Z., Respondek I., Pizondo I., Экстракция капролактама
трихлорэтиленом. II. Кинетика экстракции в пульсационной колонне,
Przem. Chem., 43, № 4, 224 (1964).
Конструкции экстракционной аппаратуры,
их исследование и расчет
Аэров М. Э., Каган С. 3., Волкова Т. С, Полупромышленные
испытания и вопросы моделирования роторно-дисковых экстракторов Хим
_ПР"» м> й **> fQfi4) '
Аэров М. Э,., Каган С. 3., Волкова Т. С, Никитин А. Я.,
Коэффициенты продольного перемешивания в роторно-дисковых экстракторах,
ЖПХ, 36, № 9, 1994 (1963).
Баранов Г. П., Чемезов В. А., Рогулин А. Н., К вопросу о
гидродинамике в пульсационных экстракционных установках, Труды
Всесоюзного научно-исследовательского института химического машиностроения
вып. 35,'1960, стр. 61.
Барбот С. И., Центробежный экстрактор, авт. свид. 148184, 21/VII 1962 г;
Бюлл. изобр., 3\|Ь 12 (1962)..
Везде ль Л. С, Броунштейн Б. И., Покоре кий В. Н., У й-
страх М. А., Шапиро Л. И., Сравнение эффективности насадочной,
пульсационной, роторной и инжекторной колонн для процесса экстракции
системы диэтиленгликоль — толуол — н. гептан в лабораторных условиях,
сб. «Процессы жидкостной экстракции», Гостоптехиздат, 1963, стр. 268.
Берестовой А. М., Воронец П. М., Болотников Ф. С, Смеситель-
но-отстойный экстрактор с центробежным разделением фаз, авт. свид.
136841, 25/П1 1961 г.; Бюлл. изобр., № 6 (1961).
Берестовой А. М., Ромаиков П. Г., Критериальные уравнения
массопередачи для смесительно-отстойных экстракторов, ЖПХ, 38, № 2, 319
(1965).
Берестовой А. М., Р о м а н к о в П. Г., Лютая Н. С, Исследование
процесса массопередачи в смесительио-отстойном экстракторе с
центробежным разделением фаз, сб. «Процессы жидкостной экстракции и
хемосорбции», Труды II Всесоюзного совещания по жидкостной экстракции и
хемосорбции. изд. «Химия», 1965, стр. 171.
Бобиков И. И., Гиндин Л. М., Некоторые вопросы кинетики обменно-
экстракционных процессов в приложении к расчетам аппаратуры, сб.
«Процессы жидкостной экстракции», Гостоптехиздат, 1963, стр. 291.
Болотников Ф. С, Романков П. Г., Исследование массопередачи в
наклонном вибрационном экстракторе, ЖПХ, 37, № 1, 46 (1964).
Болотников Ф. С, Романков П. Г., Критериальное уравнение
массопередачи для наклонного пульсационного экстрактора, ЖПХ, 37, № 2 310
(1964).
Бондаренко М. Ф., Шабалин Н. И., Мягкова А. И., Роторно-диско-
вый контактор с перфорированными тарелками, Изв. вузов, Нефть и газ,
№ 9, 91 (1960).
Борбат В. Ф., Баранов М. Н., Борисовский В. Ф., Бобиков И. И.,
Многоступенчатый экстрактор типа смеситель-отстойник для замедленных
процессов и труднорасслаиваюшихся жидкостей, авт. свид. 167819, 5/П
1965 г.; Бюлл. изобр., № 3 (1965).
Брайнес Я. М., Экстракция жидкости жидкостью (обзор современной
аппаратуры), Вестн. техн. и эконом, информации, № 4/16, 38 (1959)..
Броунштейн 8. И., Быкова Л. Г., Покорский В. Н., Уст-
раих М. А., Яблочкииа М. Н., Экспериментальная проверка метода
расчета высоты противоточных насадочных и тарельчатых колонн в про-
688
цессах растворения однокомпонентной диспергированной фазы, ЖПХ, 34,
№ 3, 548 (1961).
Броунштейн Б. И., Макаров В. В., Об условиях кавитации в
пульсационных колоннах, Труды Всесоюзного научно-исследовательского
института нефтехимических процессов, вып. 5, Гостоптехиздат, 1962, стр. 195.
Броунштейн Б. И., Бездель Л. С, Горенбург В. П.,
Соколова Е. А., К вопросу моделирования процессов жидкостной экстракции в
пульсационных колоннах, Труды Всесоюзного научно-исследовательского
института нефтехимических процессов, вып. 5, Гостоптехиздат, 1962,
стр. 148.
Броунштейн Б. И., Шапиро Л. П., Массопередача в пульсационной
колонне с учетом продольного перемешивания, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по
жидкостной экстракции и хемосорбции, изд. «Химия», 1965, стр. 133.
^/-Булатов С. Н., Плановский А. Н., Расчет диаметра экстракторов с
ситчатыми тарелками, Вестн. техн. и эконом, информации, № 6, 29 (1960).
Булатов С. Н., Плановский А. Н., Исследование гидродинамики
потоков в экстракционных аппаратах с ситчатыми тарелками, Вестн. техн.
и эконом, информации, № 4, 32 (1960).
Васильев В. А., Устройство для ввода легкой жидкости в экстракционную
колонну с пульсацией авт. свид. 121780, 18/VIII 1959 г.; Бюлл. изобр.,
№ 16 (1959).
Васильев В. А., Суховей В. Н., Чугреев Н. С, Массообмеииая
колонна, авт. свид. 148026, 21/VI 1962 г.; Бюлл. изобр., № 12 (1962).
Векслер Г. М., Ильгесонис И. В., Солнцев М. Я-, Пульсационное
смесительное устройство для экстрактора типа смеситель-отстойник, авт.
свид. 162819, 27/V 1964 г.; Бюлл. нзобр., № 11 (1964).
Векслер Г. М., Киприянов Ю. И., Поршневой пульсатор для
экстракционных аппаратов, авт. свид. 165674, 26/Х 1964 г.; Бюлл. изобр., № 20
(1964).
Вертузаев Е. Д., Плановский А. Н., Кинетические уравнения для
расчета эффективности ситчатых экстракционных колонн, ЖПХ, 36, № 2,
295 (1963).
Галкин Н. П., Тихомиров В. Б., Основные процессы и аппараты
технологии урана, Госатомиздат, 1961, стр. 160 ел.
Г а л е е в А. Ф., Михайлов Г. М., Гурьянов А. И., Николаев А. М.,
Роторно-вибрационный экстрактор, авт. свид. 149394, 28/VIII 1962 г.;
Бюлл. изобр., № 13 (1962).
Га леев А. Ф., Гурьянов А. И., Михайлов Г. М.,
Смесительно-отстойныи экстрактор колонного типа, авт. свид. 159491, 28/ХП 1963 г.}
Бюлл. изобр., № 1 (1964).
Г а л е е в А. Ф., Гурьянов А. И., Пул^сационный смесительно-отстойныи
экстрактор, авт." свид. 162503, 8/V 1964 г.; Бюлл. изобр., № 10 (1964).
Га лее в А. Ф., Гурьянов А. И., Михайлов Г. М., Смесительно-от»
стойный экстрактор, авт. свид. 167824, 5/11 1965 г.; Бюлл. изобр., № 3
(1965).
Гельперин Н. И., Волынец М. П., Колосова Г. М., Инжекторная
колонна для экстракционного выделения веществ, Хим. наука и пром.,
1, № 5, 560 (1956).
Гельперин Н. И., Исследование в области экстракции из растворов, сб.
«Вопросы массопередачи», Госхимиздат, 1957, стр. 45.
Гельперин Н. И., Лиакумович А. Г., Листопадов М. В.,
Экстракция из растворов в противоточной инжекторной колонне, Научные
доклады высшей школы, Химия и хим. технология, 1, № 1, 193 (1958).
Гельперин Н. И., Лиакумовнч А. Г., Экстракция из растворов, Хим,
наука и пром., 3, № 6, 725 (1958).
Гельперин Н. И., Ассмус М. Г., Инжекционные колонны для
непрерывной экстракции из растворов, Труды Московского института тонкой
химической технологии, вып. 8, Госхимиздат, 1958, стр. 88.
688
Гельперин Н. И., Кравченко И. И., Исследование экстракционной
колонны с чередующимися смесительными и насадочными секциями, Хим.
машиностроение, № 1, 28 (1959).
Гельперин Н. И., Асе м ус М. Г., Концевой эффект при жидкостной
экстракции в инжекторной колонке, Хим. пром., № 4, 269 (1961).
Гельперин Н И., Пебалк В. Л., Кузнецова М. И., Ротационная
экстракционная колонна с чередующимися и безнасадочными сепараци-
онными зонами, Журн. ВХО им. Д. И. Менделеева, 7, № 1, 114 (1962).
Гельперин Н. И., Пебалк В. Л., Ш а ш к о в а М. Н., Горизонтальный
многоступенчатый трубчатый экстрактор, Хим. пром., № 6, 427 (1962).
Гельперин Н. И., Ассмус М. Г., Массообмен в инжекторных массооб-
менных колоннах, сб. «Процессы жидкостной экстракции», Гостоптехиз-
дат, 1963, стр. 176.
Гельперин Н. И., Пебалк В. Л., Баранова 3. П., Исследование
массообмена в роторно-дисковых экстракторах, Химия и хим. технол топ-
лив и масел, № 6, 46 (1963).
Гельперин Н. И., Пебалк В. Л., Чичерина Т. Г., Исследование
экстракционной пульсационной колонны, Хим. пром., № 2, 31 (1963).
Гельперин Н. И., П е б а л к В. Л., Баранова 3. П.,
Кузнецова М. И., Чичерина Т. Г., Ш а ш к о в а М. Н., Результаты
исследования некоторых конструкций экстракционных аппаратов, в сб.
«Процессы жидкостной экстракции», Гостоптехиздат, 1963, стр. 105.
Гельперин Н. И., Неустроев С. Л., Исследование продольного
перемешивания в колонном экстракторе с вибрирующими тарелками Хнч
пром., № 5, 360 (1964).
Гельперин Н. И., Пебалк В. Л., Ч е х о м о в Ю. К-, Вертикальный
многоступенчатый экстрактор колонного типа, авт. свид. 166649 1/ХП 1964 г;
Бюлл. изобр., № 23 (1964).
Гельперин Н. И., Пебалк В. Л., Замышляев В. Г.,
Многоступенчатый ящичный экстрактор, авт. свид. 167827, 5/11 1965 г.; Бюлл. изобр.,
№ 3 (1965).
Гельперин Н. И., Пебалк В. Л., Замышляев В. Г., Ящичный
экстрактор, авт. свид. 168641, 26/11 1965 г.; Бюлл. изобр, № 5 (1965).
Гельперин Н. И., Пебалк В. Л., Шашкова Т. Г., Чичерина Т. Г.,
Горизонтальный распылительный многоступенчатый экстрактор авт свид
170909, 11/V 1964 г.; Бюлл. изобр., № 10 (1965).
Гельперин Н. И., Пебалк В. Л., Чичерина Т. Г., Шашкова М. Н„
Горизонтальный распылительный многоступенчатый экстрактор, Хим. и
нефт. машиностроение, № 9, 1 (1965).
Гельперин Н. И., Пебалк В. Л., Шашкова М. Н., Исследование
трубчатого горизонтального многосекционного экстрактора, сб. «Процессы
жидкостной экстракции и хемосорбции», Труды 11 Всесоюзного совещания
по жидкостной экстракции и хемосорбции, изд. «Химия», 1965, стр. 205.
Гельперин Н. И., Пебалк В. Л., Кузнецова М. И.,
Чичерина Т. Г., Исследование колонных экстракторов с механическим
перемешиванием фаз, сб. «Процессы жидкостной экстракции и хемосорбции»,
Труды II Всесоюзного совещания по жидкостной экстракции и
хемосорбции, изд. «Химия», 1965, стр. 213.
Гончаренко Г. К-, Готлинська А. П., Ахтирська А. А., Екстрак-
щя рщина — р1дина з перемгшуванием, Хим1чна пром., № 3, 15 (1962).
Григорьев С. М., Нейман Н. В., Мироедова Е. В.,
Бурцева В Н., Аникеев Л. С, Экстракционная колонна с насадкой из
подвижных сит, сб. «Процессы жидкостной экстракции» Гостоптехиздат
1963, стр. 204.
Григорьев С. М., Ко с тюк В. А., Бурцева В. Н., Экстракционный
аппарат, авт. свид. 175486, 9/Х 1965 г.; Бюлл. изобр., № 20 (1965)
Гурьянов А. И., Г а л е е в А. Ф., Николаев А М., Колонный
экстрактор с вибрирующими тарелками, авт. свид. 146281, 23/IV 1962 г.; Бюлл.
изобр., № 8 (1962),
Гурьянов А. И., Г а л е е в А. Ф., Роторно-дисковын пульсационный
экстрактор, авт. свид. 151303, 31/Х 1962 г.; Бюлл. изобр., № 21
(1962).
Гурьянов А. И., Га леев А. Ф., Николаев А. М., Роторно-вибрацион-
ный экстрактор для систем жидкость — жидкость, авт. свид. 151295
31/Х 1Р62 г.; Бюлл. изобр., № 21 (1962).
Гурьянов А. И., Га леев А. Ф., Некоторые вопросы гидродинамики
пульсационно-смесительного экстрактора (ПСЭ), Труды КХТИ им.
С. М. Кирова, т. 32, Казань, 1964, стр. 131
Гурьянов А. И., Га леев А. Ф., Исследование гидродинамики
пульсационно-смесительного экстрактора в системе жидкость — жидкость, сб.
«Процессы жидкостной экстракции и хемосорбции», Труды II Всесоюзного
совещания по жидкостной экстракции и хемосорбции, изд. «Химия», 1965,
стр. 197.
Гухман Л. А., Мирзаев С. Д., Визуальное наблюдение процесса
селективной очистки, Изв. вузов, Нефть и газ, № 8, 69 (1963).
Доронин В. Н., Жаворонков Н. М., Николаев А. М„ Исследование
массообмена в пульсирующей экстракционной колонне, Хим.
машиностроение, № 5, 21 (1962).
Доронин В. Н., Жаворонков Н. М., Николаев А. М, Исследование
массообмена в пульсирующей экстракционной колонне, сб. «Процессы
жидкостной экстракции», Гостоптехиздат, 1963, стр. 132.
Доронин В. Н., Николаев А. М., Исследование пульсационного
экстрактора с вращающимся потоком. Труды КХТИ им. С. М. Кирова, т. 32,
Казань, 1964, стр. 112.
Доронин В. Н., Николаев А. М., Исследование предельной нагрузки
пульсационной экстракционной колонны, Труды КХТИ им. С. М. Кирова,
т. 32, Казань, 1964, стр. 120.
Доронин В. Н., Николаев А. М., Исследование предельной нагрузки
пульсационной экстракционной колонны, Изв. вузов СССР, Химия и
химическая технология, 7, вып. 3, 497 (1964).
Железняк А. С, Броунштейн Б. И., Определение частных
коэффициентов массопередачи при экстракции в системе к-гептан —
толуол— диэтиленгликоль в пропеллерной мешалке, ЖПХ, 35, № 12, 2706
(1962).
Железняк А. С, Механизм массопередачи в колоннах с
перфорированными тарелками при наличии пульсации, сб. «Процессы жидкостной
экстракции и хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 140.
Змиевский Л. М„ Бедняк А. А., Покутнев Л. С, Колонный
экстрактор непрерывного действия, авт. свид. 150487, 11/Х 1962 г.; Бюлл.
изобр, № 19 (1962).
Каган С. 3., Аэров М. Э„ Волкова Т. С, Вострикова В. Н.,
Исследование экстракторов с механическим перемешиванием фаз (Пуль-
сацпонные экстракторы), Хим. пром., № 8, 43 (1959).
Каган С. 3., Волкова Т. С, Аэров М. Э., Исследование продольного
перемешивания в роторно-дисковых экстракторах, Хим. пром., № 12, 39
(1961); сб. «Процессы жидкостной экстракции», Гостоптехиздат, 1965,
стр. 65.
Каган С. 3., Волкова Т. С, Модификация роторно-дисковых
экстракторов, Труды МХТИ им. Д. И. Менделеева, вып. 33, 1961, стр. 124.
Каган С. 3., Обобщенные уравнения для расчета роторных экстракторов,
Труды МХТИ им. Д. И. Менделеева, вып. 33, 1961, стр. 118.
Каган С. 3., Аэров М. Э., Волкова Т. С, Изучение гидродинамики
роторно-дисковых экстракторов методами фото- и киносъемки, сб «Прбцес-
сы жидкостной экстракции», Гостоптехиздат, 1963, стр. 156
Каган С. 3., Аэров М. Э., Волкова Т. С, Труха но в В. Г., Расчет
диаметра капель в роторно-дисковых экстракторах, ЖПХ, 37, № 1, 58
(1964).
701
Каган С. 3., Аэров М. Э., Лоник В., Волкова Т. С,
Некоторые вопросы гидродинамики и массопередачи в ситчатых пульсацион-
ных экстракторах, Изв. вузов, Химия и хим. технол., 8, вып. 1, 142
(1965).
Каган С. 3., Аэров М. Э. и Волкова Т. С, Коэффициенты продольного
перемешивания в дисперсной фазе при жидкостной экстракции, ЖПХ 39,
№ 1, 88 (1966).
Каденская Н. И., Железняк А. С, Броунштейн Б. И., Исследопа-
ние массопередачи в экстракционной распылительной колонне, сб.
«Процессы химической технологии (гидродинамика, тепло- и массопередача)»,
Изд. АН СССР, 1965, стр. 215.
Касаткин А. Г., Kara и С. 3., Тру ханов В. Г., Удерживающая
способность роторно-дисковых экстракторов. ЖПХ, 35, № 9 1980
(1962).
Касаткин А. Г., Каган С. 3., Т р у х а н о в В. Г., Гидродинамические
характеристики роторно-дисковых экстракторов, Хим. пром № 3 38
(1962).
Касаткин А Г., Каган С. 3., Труханов В. Г., Кинетические
характеристики роторно-дисковых экстракторов, Труды МХТИ им. Д. И.
Менделеева, вып. 40, 1963, стр. 134.
Карасева А. А., Розен штейн М. 3., Якоби ф. е., Нова ко в-
с к а я И. В., Исследование влияния гидродинамических факторов на
эффективность работы насадочной экстракционной фенольной колонны,
Химия и технол. топлив и масел. № 7, 48 (1963).
Карпачева С. М., Васильев В. А., Дядина К. А., Испытание
экстракционной колонны с принудительным перемешиванием, Хим. пром
№ 6, 38 (1956).
Карпачева С. М., Розен А. М., Васильев В. А., Дядина К. А.,
Исследование пульсирующих насадочных экстракционных колонн, Хим.
машиностроение, №. 3, 6 (1959).
Карпачева С. М., Медведев С. Ф, Сенин П. Т., Эффективность
экстракционных насадочных и секционных колонн, Хим. машиностроение,
№ 4, 10 (1959).
Карпачева С. М., Дядина К- А., Васильев В. А., Пульсирующие
экстракторы. Гидравлика экстракционных насадочных колонн с
пульсацией, Вестник техн. и эконом, информ. научно-исследовательского ин-та
технико-эконом. исслед. Гос. комитета Совета Министров СССР по
химии, № 2 (14), 54 (1959).
Карпачева С. М., Дядина К. А., Васильев В. А., Пульсирующие
экстракторы. Эффективность пульсирующих экстракционных колонн.
Вестник техн. и эконом, информации Научно-исслед. ин-та технико-эконом.
исслед. Гос. комитета Совета Министров СССР по химии № 3 (15), 26
(1959).
Карпачева С. М., Родионов Е. П., Попова Г. М., Сравнение
эффективности экстракции на пульсирующих насадочных колоннах со сплошной
водной и органической фазами, Хим. пром., № 6, 496 (1960).
Карпачева С. М., Р о з е и А. М., Васильев В. А., Исследование ра
боты насадочной колонны с пульсацией, Хим. машиностроение, № 2 13
(1960).
V Карпачева С. М., К вопросу расчета экстракционной аппаратуры, сб.
«Экстракция», вып. 1, Госатомиздат, 1962, стр. 202.
Карпачева С. М., Родионов Е. П., Особенности распределения извле
каемых веществ промывной части экстракционно-промывных экстракторов.
Атомная энергия, 13, вып. 5, 486 (1962).
Карпачева С. М., Медведев С. Ф., Захаров Е. И, Белов Ю. А..
Влияние пульсаций на работу насадочных колонн, Хим. пром., № 2, 14
(1963).
Карпачева С. М., Захаров Е. И., Рагинский Л. С,
Муратов В. М., Пульсирующие экстракторы, Атомиздат, 1964,
7021
Карпачева С. М., Захаров Е. И., Киселева Л. Ф., Изучение
закономерностей движения дисперсной фазы в пульсирующей насадочной
колонне, ЖПХ, 37, № 12, 2668 (1964).
Карпачева С. М., Ч е м а р и н Н. Г., Бычков А. Е., Захаров Е. И.,
Девяткин В. И., Жданов Б. В., Исследование работы
пульсирующей экстракционной ситчатой колонны, Хим. и нефт. машиностроение,
№ 1, 24 (1965).
Карпачева С. М., Рагинский Л. С, Захаров Е. Н., Насадка для
пульсационных экстракционных колонн, авт. свид. 175489, 9/Х 1965 г.;
Бюлл. изобр., № 20 (1965).
Карпачева С. М., Захаров Е. И., Основные параметры пульсационных
колонн с различными насадками, сб. «Процессы жидкостной экстракции
и хемосорбции». Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 121.
Карпачева С. М., Захаров Е. И., Медведев С. Ф.,
Родионов Е. П., О коэффициенте моделирования пульсационных колонн, сб.
«Процессы жидкостной экстракции и хемосорбции», Труды II
Всесоюзного совещания по жидкостной экстракции и хемосорбции, изд. «Химия»,
1965, стр. 147.
Карпачева С. М., Муратов В. М., Рагинский Л. С,
Романов А. В., Расчет и моделирование пульсационных
смесителей-отстойников, сб. «Процессы жидкостной экстракции и хемосорбции», Труды
II Всесоюзного совещания по жидкостной экстракции и хемосорбции. изд.
«Химия», 1965, стр. 189.
Кафаров В. В., Жуковская С. А., Исследование основных
характеристик работы струйного экстрактора и сравнительная эффективность
экстракторов, ЖПХ, 31, № 3, 376 (1958).
\/ Кафаров В. В., Галкин Н. П., Тихомиров В. Б., Разработка новых
конструкций экстракционных колонн и основы теории процесса, сб.
«Экстракция», вып. 2, Госатомиздат, 1962, стр. 339.
Кафаров В. В., Фролов Б. Н., Гидродинамика струйной экстракционной
колонны, сб. «ПрЪцессы жидкостной экстракции», Гостоптехиздат, 1963,
стр. 215.'
Коновалов В. И., Романков П. Г., Вибрационный экстрактор, авт.
свид. 139653, 5/VI11 1961 г.; Бюлл. изобр., № 14 (1961).
Коновалов В. И., Романков П. Г., Исследование массопередачи и
гидродинамики в наклонном противоточном вибрационном экстракторе,
ЖПХ, 34, № 10, 2217 (1961).
Коновалов В. Е., Шкоропад Д. Е., Солодов А. И., Мен дел е-
вич А. И., Жданов В. В., Цилиндрические перегородки ротора
центробежного экстрактора, авт. свид. 138591, 8/VI 1961 г.; Бюлл. изобр., № 11
(1961).
Корпусов Г. В., Полупротивоточный камерный экстрактор типа смеситель-
отстойник, авт. свид. 148183, 21/VI 1962 г.; Бюлл. изобр., № 12 (1962).
К о т к а с Р. Э., Горизонтальный многоступенчатый экстрактор типа
смеситель-отстойник, авт. свид. 131355, 10/IX 1960 г.; Бюлл. изобр., № 17
(1960).
Кузнецов Ф. А., Смирнов Н. И., Изучение процесса экстракции в
экспериментальной распылительной колонне, ЖПХ, 34, № 9, 1954 (1961).
Кузнецов Ф. А., Смирнов Н. И., Экстракция в распылительных
колоннах, Труды ЛТИ им. Ленсовета, вып. 60, Госхимиздат, 1960, стр. 190.
Кузнецов Ф. А Смирнов Н. И, Количественные закономерности
процесса экстракции, ЖПХ, 34, № 9, 2111 (1961).
Ласкорин Б. Н., Пушленков М. Ф., Берестовой А. М.,
Смирнов В. Ф., Ще пети л ьн ико в Н. Н., Горизонтальный
смеситель-отстойник, сб. «Экстракция», вып. 2, Госатомиздат, 1962, стр. 347.
1/~ Ласкорин Б. Н., Худенев И. К., Смирнов В. Ф.. Красов В, Г.,
Методы расчета экстракционных аппаратов типа смеситель-отстойинк, сб.
«Экстракция», вып. 2, Госатимиздат, 1962, стр. 264.
703
Л а с к о р и и Б. Н., Скворцов Н. В., Якубович И. Л., К р а с о в В. Г.,
Смирнов В. Ф., Пивоваров В, Е., Кириллов О. Д., Т ю ф т и-
на М. П., Любимов В. К., Экстрактор непрерывного действия типа
смеситель-отстойник, авт. свид. 148383, 13/VII 1962 г.; Бюлл. изобр., № 13 (1962).
Ласкорин Б. Н., Якубович И. А., Зуев Г. П., Экстракторы для
извлечения урана и редких металлов из водных растворов, Атомная
энергия, 12, № 6, 503 (1962).
Ласкорин Б. Н., Смирнов В. Ф., Красов В. Г., Ротационный проти
воточный экстрактор ЭР-350 и пути повышения его эффективности, сб.
«Экстракция», вып. 2, Госатомиздат, 1962, стр. 361.
Ласкорин Б. Н., Саламатов И. И., Красов В. Г.. Смирнов В. Ф,
Центробежный трубчатый суперэкстрактор ЦСЭ-60 для экстракционного
извлечения цветных металлов, сб. «Экстракция», вып. 2, Госатомиздат,
1962, стр. 372.
Лукин В. Д., Романков П. Г., Берестовой А. М., Лютая Н. С,
Моделирование смесительно-отстойных экстракторов с центробежным
разделением фаз, ЛТИ им. Ленсовета, Научно-техн. конф., апрель 1965 г.
Тезисы докладов, изд. «Химия», 1965, стр. 120; ЖПХ, 38, № 12, 2671
(1965).
Лукнн В. Д.. Романков П. Г., Берестовой А. М., Исследование
процесса массопередачи в каскаде смесительно-отстойных экстракторов с
центробежным разделением фаз, сб. «Процессы жидк. экстракции и хе-
мосорбции», Труды II Всесоюзного совещания по жидкостной экстракции
и хемосорбции, изд. «Химия», 1965, стр. 182.
Лухакоодер Э. X., Раукас М. М., Сийрде Э. К, Об экстракции
жидкостей в аппарате, снабженном мешалкой, Труды Таллинского
политехнического института, Серия А, Химия и хим. технол., т. 7, Эстонское
гос. издательство, 1961, стр. 245.
[/ Лухакоодер Э. X., Сийрде Э. К., О расчете экстракторов, снабженных
пропеллерными мешалками, Труды Таллинского политехнического
института, Серия А, № 210, Эстонское гос. издательство, 1964, стр. 179.
Лухакоодер Э. X., Сийрде Э. К-, Аналитический расчет числа
экстракторов установки методом изменения ступеней конструкции, Труды
Таллинского политехнического института, Серия А, № 210, 1964, стр. 193.
Максименко М. 3., Галеев А. Ф., Гурьянов А. И., Акутугано-
в а Л. С, Исследование некоторых конструкций экстракционных
аппаратов на системе масло — фенол, Научно-техн. сб. ЦНИИТЭнефтегаз, вып. 6,
1964.
Марушкин Б. К., Лабораторная экстракционная колонна и метод
определения ее эффективности, Сб. трудов Уфимского нефтяного института,
вып. 3, Башкирское книжн. изд., 1960, стр. 183.
Марушкин Б. К-, Берг Т. А., Сидорочева Л. В., Байбавлето-
в а Ф. Г., Экстрактивная депарафинизация дизельного топлива, Сборник
трудов Уфимского нефтяного института, вып. 3, Башкирское книжное
издательство, 1960, стр. 187,
М е т к и и В. И., Д о м а и с к и й И. В., Соколов В. Н., Исследование
пневмодиспергирования несмешивающихся жидкостей в трубчатом газ-
лифтном аппарате ЛХТИ им. Ленсовета. Научно-техн. конф., апрель
1965 г., Тезисы докладов, изд. «Химия», 1965, стр. 129.
Мильвицкий Р. В., Способ экстрагирования в полых аппаратах
центробежного действия для систем жидкость — жидкость, авт. свид, 125191,
29/ХП 1959 г.; Бюлл. изобр., № 24 (1959).
Мильвицкий Р. В., Центробежный экстрактор, авт. свид. 135875, 28/П
1961 г.; Бюлл. изобр., № 4 (1961).
Мильвицкий Р. В., Зыков Д. Д., Караваев Н. М., Центробежный
массообменный аппарат, авт. свид. 140789, 20/1Х 1961 г.; Бюлл. изобр.,
№ 17 (1961).
Николаев А. М., П о н и к а р о в И. И., Расчет роторно-дискового
экстрактора, Промышленно-экономический бюллетень Татсовнархоза, № 7, 8 (1962).
704
Нярсп Э'. Ю.. Ш е л о v м о в В. В., Ящичный экстрактор, авт. свид. 167489,
18/1 1965 г.; Бюлл. изобр., № 2 (1965).
Пебалк В. Л., Гельперин Н. И., Шашкова М. Н., Аппарат
для экстрагирования, авт. свид. 144155, 6/Н 1962 г.; Бюлл. изобр., № 2
(1962).
Перовский А. П., Коссых В. Г., Основные характеристики пульсацион-
ных колонн, сб. «Процессы химической технологии», Изд-во «Наука», 1965,
стр. 208.
Питерских Г. П., Валашек Е. Р., Экстракция в турбулентном потоке.
Центробежные экстракторы, Хим. пром., № 1, 35 (1956); № 3, 158 (1957),
«-- П л а н о в с к и й А. Н., Булатов С. Н., Расчет разделительной камеры
колонных экстракторов с ситчатыми тарелками, Хим. машиностроение № 3,
9 (1960).
Плановский А. Н., Булатов С. Н., Расчет сливного устройства
колонных экстракторов с ситчатыми тарелками, Хим. машиностроение, № 2,
10 (1960).
Плановский А. Н., Булатов С. Н., В е р т у з а е в Е. Д., Расчет
колонных экстракторов с ситчатыми тарелками, Хим. пром., № 5, 364 (1962),
Плановский А. Н., Булатов С. Н., К вопросу об экспериментальном
определении размера капель при исследовании экстракционных колонн с
ситчатыми тарелками, Научн. докл. высшей школы, Химия и хим.
технология, № 4, 804 (1958).
Плановский А. Н., Булатов С. Н., Вертузаев Е. Д., Пример
расчета колонного экстрактора с ситчатыми тарелками (методическое
пособие), МИХМ (1961).
Поникаров И. И., Николаев А. М., Жаворонков Н. М.,
Сравнительная характеристика некоторых экстракционных аппаратов, Хим.
машиностроение, № 3, 11 (1962).
Поникаров И. И., Николаев А. М., Предельные нагрузки и массопере-
дача в роторно-дисковом экстракторе, Труды КХТИ им. С. М. Кирова,
т. 30, Казань, 1962, стр. 352.
Поникаров И. И., Николаев А. М., Фактор эффективности как
критерий сравнительной оценки экстракционных аппаратов, Труды КХТИ им.
С. М. Кирова, т. 30, Казань, 1962, стр. 360.
Поникаров И. Н., Николаев А. М., Жаворонков Н. М.,
Исследование гидродинамики и массопередачи в роторно-дисковом экстракторе
и его сравнительная эффективность, сб. «Процессы жидкостной
экстракции», Гостоптехиздат, 1963, стр. 166.
Поникаров И. И., Николаев А. М., Жаворонков Н. М,,
Исследование предельных нагрузок роторно-дискового экстрактора, Хим.
машиностроение, № 6, 16 (1963).
Попов А. А., Бурдаков Ф. А., Потапов В. П., Тихомиров В. Б.,
Смирнов М. И., Аппарат для проведения массообменных процессов
(например, экстракции), авт. свид. 147173, 21/V 1962 г.; Бюлл. изобр.,
№ 10 (1962).
Рагинский Л. С., Ш и р с к и й А. Н., Безмембранный пневматический
пульсатор для экстракционных пульсирующих колонн, Хим. пром., № 5, 414
(1960).
Рагинский Л. С, Иванов В. Д., Применение и расчет пневматических
пульсаторов с золотниково-распределительным механизмом при длинном
пульсопроводе, сб. «Процессы жидкостной экстракции и хемосорбции»,
Труды II Всесоюзного совещания по жидкостной экстракции и
хемосорбции, изд. «Химия», 1965, стр. 161.
Родионов Е. П., Пульсационная экстракционная колонна, авт. свид. 121776,
18/VIII 1959 г.; Бюлл. изобр., № 16 (1959).
Розен А. М., К а р п а ч е в а С. М.., Медведев С. Ф., Родионов Е. П.,
Киселева Л. Ф., Исследование массопередачи в насадочных колоннах
при экстракции трибутилфосфатом (экстракция и реэкстракция азотной
кислоты), Хим. пром., № 7, 627 (1959).
70S
Розен А. М., Васильев В. А., Беззубова А. И, Горшкова Г. П.,
Некоторые закономерности гидравлики и массопередачи в насадочных
колоннах с пульсацией, сб. «Экстракция», вып, 2, Госатомиздат, 1962,
стр. 320.
Розен А. М, К а р п а ч е в а С. М., Медведев С. Ф., Родионов Е. П.,
Киселева Л. Ф., Массопередача при экстракции и реэкстракции ура-
нилнитрата в насадочных колоннах, сб. «Экстракция», вып. 2,
Госатомиздат, 1962, стр. 284.
Р о щ и н В. Н., Баранов Г. П., Ч е м е з о в В. А., К вопросу о
гидродинамики в пульсационных эсктракционных установках. Труды Всес. научно-
исслед. и констр. нн-та хим. машнностр. НИИХИММАШ, вып. 35, 1960,
стр. 61.
Рощин А. Н, Чемарин Н. Г., Гидромеханический пульсатор для
экстракционных колонн, Хим. и нефт. машиностроение, Л» 2, 7 (1964).
Рутман Г. Б., Экстрактор ТФ-400, Вестник технпч. н экономич.
информации научно-исслед. ин-та технико-эконом. исслед. Гос. комитета хим.
пром. при Госплане СССР, вып. 3, 1964, стр. 21.
Сасин Э. М., Колонна для экстрагирования противоточного типа в системах
жидкость — жидкость, склонных к образованию эмульсий, авт. свид.
156526, 28/VII1 1963 г.; Бюлл. изобр., № 16 (1963).
Саламатов И. И, Векслер Г. М., Ротационный противоточный
экстрактор непрерывного действия, Хим. машиностроение, № 1, 12 (1959).
СоколовВ. Н., Р е ш а н о в А. С, О влиянии времени на дробление капель
в потоке, турбулнзованном барботирующим газом, ЖПХ, 33, № 5, 1068
(1960).
U" Соколов В. Н, Рейганов А. С, Барботажный многоступенчатый
экстрактор, авт. свид. 135876, 28/II 1961 г.; Бюлл. изобр., № 4 (1961).
Соколов В. Н., Решанов А. С, Межфазная поверхность и
относительный объем капель при диспергировании барботирующим газом, ЖПХ, 34,
№ 5, 1047 (1961).
Соколов В. Н., Многоступенчатый экстрактор, авт. свид. 159800, 14/1
1964 г.; Бюлл. изобр., № 2 (1964).
Соков Ю. Ф., Путилова 3. Д., Роторио-дисковый экстрактор, авт. свид.
142627, 28/ХП 1961 г.; Бюлл. изобр., № 22 (1961).
Соков Ю. Ф., Путилова 3. Д., Высокопроизводительный экстрактор
новой конструкции для извлечения ароматических углеводородов, сб.
«Процессы жидк. экстракции и хемосорбции», Труды 11 Всесоюзного совещания
по жидкостной экстракции и хемосорбции, изд «Химия», 1965, стр. 228.
Сол о до в Л. И, Шкоропад Д. Е.. Гидродинамические закономерности
центробежного экстрактора, Хим. машиностроение, № 6, 17 (1961).
Слободяник И. П., Метод анализа работы насадочных экстракционных
колонн, Изв. вузов, Пищевая технол., № 3, 111 (1960).
" Солнцев М. Я., Конструирование и исследование современных типов
экстракционного оборудования, сб. «Процессы жидкостной экстракции и
хемосорбции», Труды II Всесоюзного совещания по жидкостной
экстракции и хемосорбции, изд. «Химия», 1965, стр. 27.
Тихомиров В. Б., К а ф а р о в В. В., Струйная колонна для экстракции
в системах жидкость — жидкость, авт. свид. 117778, 20/П 1959 г.; Бюлл.
изобр., № 3 (1959).
Тихомиров В. Б., Яковцев И. В., Дубровский И. П.,
Горя ев В. М, Аппарат для экстракции в системе жидкость — жидкость,
авт. свид. 129188, 15/VI 1960 г.; Бюлл. изобр., № 12 (1960).
Т и х о м и р о в В. Б., Галкин Н. П., Федоров В. Д, Исследование
массообмена в экстракционной тарельчатой колонне с воздушным
перемешиванием, сб. «Экстракция», вып. 1, Госатомиздат, 1962, стр. 213.
Тихомиров В. Б., К расчету экстракционных колонн с провальными
тарелками, сб. «Экстракция», вып. 2, Госатомиздат, 1962, стр. 294.
Туляков П. М., Пульсатор для экстракционных колонн, авт. свид. 131356,
10/IX 1960 г.; Бюлл. изобр., № 17 (1960).
708
Ф и л и п п о с ъ я н ц Т. Т., Горизонтальные противоточные ротационные
экстракторы, Медицинская пром. СССР, № 10, 38 (1961).
Шабалин И. И., Бондаренко М. Ф., Контактная роторная колонна,
авт. свид. 142296, 14/ХП 1961 г.; Бюлл. изобр., № 21 (1961).
Шапиро Л. П., Б р о у н ш т е й н Б. И., Исследование продольного
перемешивания в насадочных пульсационных колоннах, сб. «Процессы
жидкостной экстракции и хемосорбции», Труды II Всесоюзного совещания по
жидкостной экстракции и хемосорбции, изд. «Химия», 1965, стр. 153.
Ш е н Фу, К а ф а р о в В. В., Гидродинамика и массопередача в
одноступенчатом струйном экстракторе, Изв. вузов, Химия и хим технология
4, № 3, 498 (1961).
Шкоропад Д. Е., Коновалов В. Е., Солодов А. И.,
Менделевия А. И., Жданов В. В., Центробежный экстрактор, авт. свид. 136714,
25/1II 1961 г.; Бюлл. изобр, № 6 (1961).
Шкоропад Д. Е, Лысковцов И. В, Центробежные жидкостные
экстракторы, Машгиз, 1962.
Штробель В., Романков И. Г., Коновалов В. И, Лютая Н. С,
Исследование гидродинамики без массопередачи и при наличии массопере
дачи в роторно-дисковом экстракторе, ЖПХ, 36, № 12, 2672 (1963).
Штробель В., Романков И. Г., Коновалов В И, Лютая Н. С,
Исследование процесса массопередачи в роторно-дисковом экстракторе,
ЖПХ, 37, № 1, 50 (1964).
Якубович И. А., К р а с о в В. Г., Пивоваров В. Е., Ультразвуковой
экстрактор непрерывного действия типа смеситель-отстойник, авт. свид
162111, 16/IV 1964 г.; Бюлл. изобр, № 9 (1964).
Billet R, Nagel О, Аппараты для дистилляции, сорбции и экстракции
(Achema, 1964 г.), Chem. — Eng. — Techn, 36, № 12, 1257 (1964) (см
также стр. 1283).
Bock L, Hesse A, Reich G., Исследование эффективности и приозводи-
тельности нового дискового противоточного экстрактора из иенского
стекла, Chem. Techn., 16, № 2, 85 (1964).
Brink A, Gericke I. I, Применение роторно-дискового экстрактора для
разделения неидеальных систем жидкость — жидкость. S. Afric. — Ind
Chemist., 18, № 11, 152 (1964).
Бояджиев Л, ЕленковД, Массопередача в системе жидкость—жидкость
при турбулентном течении. Межфазная поверхность в экстракторе типа
Вентури, Изв. ин-та общ. и неорган, химии Бълг. АН, 3, 147 (1965).
Chanandrasekaran P, Laddha G. S, Противоточный двухфазный
поток в насадочных экстракторах для систем жидкость — жидкость.
Расчет удерживающей способности в точке захлебывания и ниже ее,
Hydraulic and Fluid Mechanik, Pergamon Press, New York, 1964, p. 243.
С о о 1 e у С. R, Спиральный контактный элемент для пульсационной
экстракционной колонны, пат. США 2988429, 13/VI 1961 г.
D e g а 1 е е s a n Т. Е, Laddha G. S, Исследование УС при массопередаче
в насадочном экстракторе, J. Technol, 3, № 5, 137 (1965).
D e S m e t I. А, Устройство для непрерывной экстракции в системе
жидкость— жидкость, швейц. пат. 367154, 10/П 1961 г.
Еленков Д, Бояджиев Л, Массопередача между двумя жидкими
фазами в турбулентных условиях. I. Гидродинамика однофазного и
двухфазного потоков в трубке Вентури Изв. ин-та общ. и неорган, химии
Бълг. АН, 2, 131 (1964).
Еленков Д., Панев П, О скорости захлебывания и влияние
поверхностно-активных веществ на движение единичных капель в распылительной
экстракционной колонне, Химия и индустрия (Бълг), 35, № 5, 167 (1963)
Ellis S. R. M, Alfonso S, Каскадный экстрактор для системы
жидкость—жидкость, Brit. Chem. Eng, 8, № 4, 238 (1963).
Фукуи Сэн, Экстракция в системе жидкость — жидкость (одноступенчч
тые установки), Кагаку кодзё, Chem. Factory, 7, № 10, 88 (1963).
707
Фукуи Сэн, Экстракция в системе жидкость — жидкость
(многоступенчатые экстракционные установки). Кагаку кодзё, Chem. Factory, 7, № 11,
89 (1963).
Graham G., Hure J., Saint James R., Способ диспергирования
жидкости в виде однородных капель в другой жидкости, фр. пат. 1330251,
9/V 1962 г.
Gross H. H., Kriested W., Аппарат для контактирования жидкостей, пат.
США 3015545, 2/1 1962 г.
Gutoff E В., Продольное перемешивание в экстракционной колонне Реш-
тона, A. I. Ch. E. Journal, И, № 4, 712 (1965).
Haas R. A., Johnson H. F., Пенные колонны для противоточной
экстракции поверхностно-активных веществ в системе жидкость — жидкость,
А. I. Ch. E. Journal, 11, № 2, 319 (1965).
Habicht L., Устройство для непрерывного контактирования двух несмеши-
вающихся или частично смешивающихся жидкостей с различными
удельными весами, пат. ФРГ 1143185, 26/П 1956 г.
Hanson С, Промышленное оборудование для экстракции в системе
жидкость—жидкость, Brit. Chem. Eng., 10, № 1, 34 (1965).
L> Hanson С, К aye D. А., Принципы проектирования
высокопроизводительных смесителей-отстойников, Chem. Process Eng., 44, № 11, 651 (1963).
"Hanson С, К a ye D. А., Расчет смесителей-отстойников высокой
производительности, Chem. Process Eng., 45, № 8, 413 (1964).
v H a r m a t h у L., К i г a 1 у G., Проектирование экстрактора с вращаюшимся
диском, Mogyar Kemik Lapja, 20, № 1, 41 (1965).
Hazard R. E., Аппарат для интенсивного контактирования жидкостей, пат.
США 315934, 14/1 1960 г.
Hazlebeck D. E., Geankoplis С. I., Продольное перемешивание в
распылительной экстракционной колонне, !nd. Eng. Chem. Fundament., 2,
№ 4, 310 (1963).
Holmes D. В., Voncken R. M., Dekker I. А., Поток жидкости в
сосуде с перегородками и с турбинной мешалкой. Период циркуляции,
Chem. Eng. Sci., 19, № 3, 201 (1964).
[V о п s k е п R. M., Holmes D. В., Н а г t о g H. W., Дисперсия при
циркуляции, Chem, Eng. Sci., 19, № 3, 209 (1964)].
Хираи Э., Ник ахи К., К о мор и Т., Влияние сопел на эффективность
процесса экстракции в системе жидкость — жидкость, Канадзава дайгаку
когакубу киё, Mem. Fac. Technol. Kanazawa Univ., 3, № 1, 56 (1963).
С и р о ц у к а М., Гото Н., Экстрагирование в системе жидкость — жидкость.
Расчет экстракционных установок, Кэмикару эндзиниярингу, 8, № 8, 739
(1963).
S t a i п h о г р F. P.', L u d а 11 N., Обратное перемешивание в экстракторе с
вращающимися дисками, Trans. Inst. Chem. Eng., 42, № 5, 198 (1964).
Jackson M. L., Hoi man K. L., Grove D. В., Поверхностные эффекты
в пульсационной ситчатой колонне, A. I. Ch. E. Journal, 8, № 5, 659
(1962).
Jealous А. С, Колонны для экстракции в системе жидкость — жидкость,
пат. США 3085864, 20/IX 1956 г.
Jeffreys G. V., Jen son V. G., Исследование процесса периодической
экстракции, Brit. Chem. Eng., 10, № 5, 304 (1965).
К а т и я м а К-, Экстрактор с перфорированными тарелками, Кагаку кодзё,
Chem. Factory, 8, № 4, 21 (1964).
Кацуока Ц., Хитоми Ю., Колонна для проведения процесса экстракции
в условиях пульсирующей подачи жидкости, Кагаку кодзё, Chem. Factory,
8, № 4, 30 (1964).
Kiessling R. О., О влиянии прерывистости переноса фаз на кпд
пульсирующей колонны для жидкостной противоточной экстракции, Kernenergie,
6, № 4, 168 (1963).
К i г а 1 у G., Исследование роторно-дисковых экстракторов, Vespremi vegyipari
egyet kozl., 7, № 4, 351 (1963).
?0§ «
Kohl A. L., Аппарат для контактирования в системе жидкость — жидкость,
пат. США 3032403, 1/V 1962 г.
Коп песке Н. G., Р е с h s t e i n G., Wolf H. I., Массопередачи в
экстракционных колоннах с перемешиванием, Z. phys. Chem. (DDR), 223, № 1—2,
102 (1963).
К or pak W., Применение воздушных насосов для экстракционных установок
непрерывного действия, работающих при противотоке фаз, Nukleonika,
8, № 12, 857 (1963).
Ко ski О. Н., Пульсационная экстракционная колонна, пат. США 3108859,
15/ХП 1959 г.
Курихара Я-, Ядзима К-, Экстрагирование в системе жидкость —
жидкость. Современные экстракционные установки и их выбор, Кэмикару
эндзиниярингу, 5, № 8, 746 (1963).
Курихара Я., Непрерывнодействующий противоточный экстрактор с
наклонными трубками, яп. пат. 4602, 2/IX 1960 г.
Leisibach I., Новая экстракционная колонна с принудительным
перемешиванием Фаз для противоточной экстракции жидкости жидкостью, Chem. —
Ing. — Techn, 37, № 3, 205 (1965).
London I., Prochaska I., Souhrada F., Nekovar P., Изучение
процесса экстрагирования. П. Экстрактор с вибрирующими тарелками,
Coll Czech. Chem. Comm., 29, № 12, 3003 (1964).
Lorenz F., Экстракционные колонны для непрерывного разделения жидких
смесей, пат. ФРГ 1169893, 25/IX 1960 г.
Lowes L., Tanner M. Ch., Способ улучшения отстаивания в смесительно-
отстойных экстракторах, англ. пат. 909485, 31/Х 1962 г.
Meyer A., Schuhler С. К-, К расчету массопередачи в полых колоннах,
Chem. Techn.. 15, № 1, 10 (1963).
Miller D. N, Массопередача в сосудах с перемешиванием, контролируемая
жидкой пленкой, Ind. Eng. Chem., 56, № 10, 18 (1964).
Misek Т., Дробление капель с помощью вращающегося диска, Coll. Czech.
Chem. Comm., 28, № 2, 426 (1963).
Misek Т., Вертикальное движение капель в сосуде с перемешиванием, Coll.
Czech. Chem. Comm., 28, № 3, 570 (1963).
Misek Т., Гидродинамические характеристики экстракторов с
перемешиванием для системы жидкость — жидкость, Coll. Czech. Chem. Comm., 28,
№ 7, 1631 (1963).
Misek Т., Гидродинамические характеристики пульсационных экстракторов
для системы жидкость — жидкость, Coll. Czech. Chem. Comm., 29, № 8,
1755 (1964).
Misek Т., Расход энергии в роторно-дисковом экстракторе, Coll. Czech.
Chem. Comm., 29, № 8, 1767 (1964).
M i s e k Т., Коалесценция капель в экстракторе с перемешиванием для
системы жидкость — жидкость, Coll. Czech. Chem. Comm., 29, № 9, 2086
(1964).
Molvneux F., Экстракция в гидравлическом циклоне, Chem. Process Eng.,
43, № 10, 502 (1962).
M о р и т а X., Скорость достижения равновесного состояния на поверхности
раздела фаз для систем жидкость — жидкость. Значения коэффициентов
массоотдачи, Канадзава дайгаку когакубу киё, Mem. Fac. Technolog.
Kanazawa Univ., 3, № 1, 84 (1963).
Murali K., Rao M. R., Исследование массопередачи в экстракционных
колоннах с перфорированными тарелками, J. Chem. Eng. Data, 7, № 4, 468
(1962).
Новый экстрактор широкого назначения, Brit. Chem. Eng., 9, № 6, 356 (1964).
Опытная установка для экстракции в системе жидкость — жидкость с
изолированными смесительно-отстойными камерами, Brit. Chem. Eng., 9, № 12,
878 (1964).
Pechstein G., Konnecke H. G., Массообмен в экстракционных колоннах
с перемешиванием, Z. phys. Chem. (DDR), 225. № 5—6, 418 (1964).
46 P. E. ТреКбал 709
Rod V., Расчет экстракционных колонн с учетом продольного
перемешивания, Brit. Chetn. Engng., 9, № 5, 300 (1964).
Rod V., Расчет коэффициентов массопередачи и коэффициентов продольного
перемешивания на основе профилей концентрации, Coll. Czech. Chem.
Comtn., 30, № 11, 3822 (1965).
Противоточная экстракция в системе жидкость — жидкость (лабораторная
установка), Chem. Process (Engl.), 9, № 9, 6 (1963).
Пэн-Дин-и, Тан Чжи-лянь, Экстракционная колонна с тарелками,
имеющими вращательно-поступательное движение, Хуасюэ шипцзе,
Hauxue spijie, 18, № 6, 275 (1964).
R i g g R. Q., Churchill S. W., Поведение несмешивающихся жидкостей,
движущихся прямотоком через слои насадки, A. I. Ch. E. Journal, 10,
№ 6, 810 (1964).
Rushton J. H., N a gat a S., Rooney Т. В., Измерение коэффициентов
массопередачи в системе жидкость — жидкость при перемешивании,
A. I. Ch. E. Journal, 10, № 3, 298 (1964).
Saint-James R., Graham Q. P., Экстракция в системе жидкость —
жидкость в пульсационной колонне с диспергированием жидкости.
Исследование при повышенных частотах и однородных по диаметру каплях,
Genie chim., 91, № 2, 39 (1964).
С а то Т., Сугихара К., Та ни яма И., Характеристики пульсациониых
колонн с перфорированными тарелками, Кагаку когаку, Chem. Eng. Japan,
27, № 8, 583 (1963).
Сато Т., Сугихара К., Танияма И., Характеристики экстракционной
колонны с перфорированными тарелками и перемежающимися потоками
взаимодействущих фаз, Кагаку когаку, Chem. Eng. Japan, 28, № 3, 241
(1964).
S a win sky 1., Достижения в области противоточных экстракторов для
систем жидкость—жидкость, Magyar Kern, lapja, 18, № 1, 43 (1963).
Schmel G. А., В abb A. L., Исследование удерживающей способности сит-
чатой пульсационной колонны для экстракции из растворов, Ind. Eng.
Chem. Process Design and Dewel., 2, № 1, 36 (^63).
Schmel G. А., В abb A. L., Исследование продольного перемешивания в
пульсационной экстракционной колонне, Ind. Eng. Chem. Process Design
and Devel., 3, № 3, 210 (1964).
Subbaraya К., Влияние перемешивания воздухом на экстракцию в
системе жидкость — жидкость в насадочной колонне, Indian Chem. Engr., 5,
№ 3, Trans., 65 (1963).
Sterne rding S., Lumb E. C, Lips I., Продольное перемешивание з
роторно-дисковых экстракторах, Chem. — Ing. — Techn., 35, № 12, 844
(1963).
Stauff I., Koch E., Способ разделения смеси веществ посредством проти-
воточной экстракции в двух колоннах, пат. ФРГ 1124923, 20/1Х 1962 г.
S k а 1 а к Р., К 1 i m а А., Экстрактор для систем жидкость — жидкость, чехосл.
пат. 110568, 23/11 1962 г.
Смеситель, легко приспосабливаемый для обработки различных систем
жидкость—жидкость, Chem. Eng., 71, № 16, 60 (1964).
Spence R., Streeton R. I. W., Противоточная экстракция в системе
жидкость — жидкость в колонне с цилиндрическим ротором. Chem.
Process Eng., 44, № 10, 597 (1963).
Сироцука Т., Мураками А., Характеристики перемешивания на
перфорированных тарелках экстракционных колонн, Bull. Sci. Eng. Res. Lab.
Waseda Univ., № 26, 63 (1964).
Сироцука М., О расчете экстракционного оборудования, Кагаку кодзё,
Chem. Factory, 8, № 4, 7 (1964).
Todd D. В., Podbielniak W. I., Центробежные экстракторы, Chem. Eng
P'rogr., 61, № 5, 69 (1965).
Treybal R. E., Новый многоступенчатый жидкостный экстрактор, Chem.
Eng. Progr., 60, № 5, 77 (1964)."
719
Thornton J. D., Bony at is В. А., Процессы экстракции в системе
жидкость — жидкость, проводимые в сосудах с мешалками, Ind. Chem., 39,
№ 6, 298 (1963).
Tettamanti К., Uskert A., Nagy S., Способ и аппарат для
эффективного смешения и разделения жидкостей, предназначенные главным
образом для проведения экстракционных процессов, венг. пат. 150408, 12/VIII
1961 г.
-Westerterp К. R., Landsman P., Продольное перемешивание в ротор-
но-дисковом экстракторе. I. Кажущаяся продольная диффузия, Chem. Eng.
Sci., 17, 363 (1962).
Westerterp К. R., M e у b e r g W. H., Продольное перемешивание в ро-
торно-дпсковом экстракторе. П. Обратное перемешивание, Chem. Eng. Sci.,
17, 373 (1962).
What ley M. E., Woods W. M., Циклонный аппарат для контактирования
жидкостей, пат. США 3052361, 6/ХП 1960 г.
\V i 1 h е 1 m H. A., Andrews M. L., Экстрактор для системы жидкость —
жидкость, Ind. Eng. Chem. Process Design and Devel., 1, № 4, 305 (1962).
Zahdradnik R. L., Archer D. H., Пульсационная экстракционная
колонна, пат. США 3108859, 15/ХИ 1959 г.
Циклонный экстрактор для системы жидкость—жидкость, Chem. Eng. 71,
№ 18, 24 (1964).
С е г m a k А., Перегородка горизонтальной экстракционной колонны, чехосл.
пат. 109194, 16/VI 1962 г.
Z i о 1 к о w s к i Z., К u b i с a J., Кинетика экстракции в системе жидкость—
жидкость в пульсациониых колоннах, Chem. Stosow. B2, № 4, 393 (1965).
Цудзи X., Центробежная установка для экстрагирования, Кагаку кодзё,
Chem. Factory, 8, № 4, 36 (1964).
Э г у т и X., Экстракционная колонна с эксцентричной мешалкой, Кагаку
кодзё, Chem. Factory, 8, № 4, 15 (1964).
Яд зим а К, Экстрактор фирмы Fujiex, Chem. Factory, 8, № 4, 26 (1964).
Ямадзаки А., Экстрагирование в системе жидкость — жидкость.
Экстракционные установки типа смеситель-отстойник, Кэмикару эндзинияринп/,
8, № 8, 763 (1963).
Ямадзаки А., Экстрактор типа смеситель-отстойник, Кагаку кодзё, Chem.
Factory, 8, № 4, 44 (1964).
Я м а г у т и К., Современные экстракторы, Кагаку соти, Plant a. Process, 5,
№ 10, 38 (1963).
Я м а г у т и К., Современные экстракторы, Кагаку соти, Plant a. Process, 6,
№ 11, 34 (1964).
Ямагути Т., Андзава М., Экстракционная колонна с вращающимися
дисками, Кагаку кодзё, Chem. Factory, 8, № 4, 11 (1964).
X а я с и Т., Техническое обслуживание и эксплуатация экстракторов, Когаку
кодзё, Chem. Factory, 9, № 10, 89 (1965).
46*
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Агломерация капель 493, 494
Аддитивность ' фазовых
сопротивлений 196 ел.
Азеотропные смеси и регенерация
экстрагентов 162 ел.
Активности компонентов, системы
бинарные 73, 74, 76 ел.
тройные ПО, 111, 118, 119
Анилиновая точка 27, 127
Антибиотики, экстракция 18, 648
Антонова правило 148
Ароматические углеводороды,
экстракция 18, 638, 639, 648
Ассоциация растворителей, факторы
176
Атомные объемы .веществ 176, 177
Ацетон, экстракция 263 ел.
Бахмана координаты 47, 49
Бенедикта уравнения 75, 107
Бензойная кислота, экстракция 17, 56
Бериллий, экстракция 668
Бинодальная кривая 32
Бинарные системы см. также
Идеальные растворы без К.Т.Р. 24, 25
межфазовое натяжение 147 ел.
неидеальные 74 ел.
с верхней и нижней К.Т.Р. 22 ел.
Блокирование поверхности раздела
фаз 201, 202
Бутадиен, экстракция 639
Ванадий, экстракция 18, 659
Ван-Лаара уравнения 84, 85, 89, 90
для неидеальных растворов 95 ел.,
108
Вебера критерий подобия 194
Вентили как смесители 489, 490
Витамины, экстракция 648, 649
Водородные связи и расчет
равновесия в системах жидкость —
жидкость 127 ел.
Вольфрам и молибден, разделение
658
Воронкообразование при
перемешивании 455 ел.
Воспламеняемость экстрагентов 151
Выпаривание при регенерации экстр-
агентов 164
Высаливание 28, 38 ел., 65, 144
Высота единицы переноса 386 ел.
в экстраторах насадочных 555,
556
распылительных 538, 539
Высота экстракторов
проточных 384 ел.
с непрерывным контактом 519
Вязкость экстрагентов, 150, 172 ел.
Галлий, экстракция 658, 659
Германий и мышьяк, разделение 658
Гибса — Дюгема уравнение 80 ел.
Гидротропия 65
Гильдебранда и Скотта уравнения
130, 132, 133
Гордона уравнение 183
Грасгофа критерий подобия 194
Давление (я)
влияние на К-Т.Р. 25, 26
компонентов в неидеальных
бинарных растворах 76 ел.
паров экстрагентов 150
Данквертса теория обновления
поверхности 193, 194
Двухкомпонентные системы см.
Бинарные системы
Двухпленочная теория массопередачи
196, 198 ел.
Диаграммы
распределения 226, 227, 232, 233,
259 ел.
селективности 141, 142
фазовые 29 ел., 224 ел.
для систем бинарных 22 ел., 26
■ тройных 30 ел.
процесса регенерации
экстрагентов 154, 155, 157, 159, 160
Диафрагменные смесители 488, 489
Диспергирование, влияние на К.Т.Р.
27
Дисперсии 491, 494, 495
Диссоциативная экстракция 378, 379,
437, 438
Дифференциальная экстракция 227,
248 ел., 328 ел.
в случае несмешивающихся
растворителей 253, 254
Диффузионные сопротивления 396,
397
712
Диффузия 172
в ламинарном и турбулентном
потоках 189 ел.
и вязкость растворителей 173 ел.
конвективная 188 ел.
коэффициенты см. Коэффициенты
диффузии
молекулярная 168 ел., 185 ел.
Донауэ — Бэртела координаты 148
Дробная экстракция см.
Фракционная экстракция
«Дуалэер-процесе» 640
«Дуо-Сол-процеее» 636, 637
Единицы переноса массы 385 ел.
Емкость экстрагентов 144, 145
Енеке координаты 43, 44, 225, 226,
230 сл„ 238, 239, 291
Жидкостная экстракция см.
Экстракция
Жиры, экстрактивная очистка 64Гсл.
Загрязнения, Влияние иа К-Т.Р. 27,
28
Закои(ы)
распределения 20, 21
и диссоциация (ассициация)
молекул 52 ел.
при экстракции
дифференциальной 254
— — многоступенчатой 241 ел.
противоточной 276 ел.
Рауля 71 ел.
Стокеа 172
Фика 169, 171
Захлебывание, экстракторы
распылительные 537, 538
роторно-дисковые 577 ел.
с непрерывным контактом 522, 523
Идеальные растворы 71 ел.
активности и коэффициенты
активности 73 ел.
летучесть компонентов 73, 74
свободная энергия 82, 83
Изопикнические хорды 147
Изотермы растворимости, система
без тройной К.Т.Р. 32
с тройной К.Т.Р. 34
Инжекторные смесители 487, 488
Интерполяция и экстраполяция хорд
равновесия в тройных системах
44, 45
Йодное число как мера состава
многокомпонентных систем 66
Исчерпывание рафината при
противоточной экстракции с флегмой
285, 286
Капли
агломерация 493, 494
коалесценция см. Коалесценция
образование, экстракторы
ситчатые 573
с непрерывным контактом 523 ел.
размеры в смесительно-отстойных
экстракторах 462 ел.
с внутренней циркуляцией 2.11, 214
скорость осаждения 207 ел.
Керра-Мак-Ги экстрактор 510, 511
Коагуляторы 502, 503, Б05
Коалесценция
капель, экстракторы
смесительно-отстойиые 494
с непрерывным контактом 527 ел.
фаз в отстойниках 501, 502
Кобальт и никель, разделение 658
Колонны см. Экстракторы
Конвективная диффузия 188 ел.
Конода 22
Координаты
Бахмана 47, 49
Донауэ — Бэртела 148
Отмера — Тобиаса 47 ел.
прямоугольные см. Прямоугольные
координаты
тетраэдрические 59 ел., 310 ел.
треугольные см. Треугольные
координаты
Хэнда 48 ел.
Корреляция
Кроуфорда и Уилки 558
Минарда и Джонсона 537
по захлебыванию насадочных
экстракторов 558, 559
Руби и Элджина 539
Ценца 535, 536
Коррозионная активность
экстрагентов 150
Коха экстрактор 565
Коэффициент (ы)
активности
в растворах идеальных 74
неидеальных 74 ел., 80, 86,
87
при расчете равновесия 109 ел.
диффузии 169 ел.
и продольное перемешивание 529
— температура 174 ел., 178, 180
неэлектролитов 172 ел.
турбулентной 190, 212
электролитов 181 ел.
задержки 363 ел.
массоотдачи 191 ел.
в экстракторах насадочных 558
распылительных 539 ел.
и высота единицы переноса 387
713
Коэффициент
массопередачи
и высота проточных экстракторов
394 ел.
— эффективность ступени в сме-
сительно-отстойных
экстракторах 450
локальные 206
общие 197, 198
отвода 363 ел.
распределения 47, 57, 59
и селективность экстрагентов 143,
144
при низких концентрациях 119 ел.
— экстракции двумя
растворителями 318, 319
дифференциальной 330, 331
многоступенчатой 323 ел.
фракционной 357, 358, 362
самодиффузии 171, 172, 173, 175
Кривые
равновесия 2Э0, 291
распределения, системы
тройные 45 ел., 57
четырехкомпонентные 61, 62
растворимости 28, 29, 31, 32
селективности 141
Кристаллизация при регенерации
экстрагентов 165
Критерии подобия 194
Критические температуры
растворения (К.Т.Р.)
верхние и нижние 22 ел.
влияние давления 25, 26
— диспергирования 27
— загрязнений 27, 28
и селективность 126, 127
тройные 33, 34
Кронига — Бринка уравнение 211
Кроуфорда и Уилки корреляция 558
Крэга экстракторы 421 ел.
Лабораторная экстракция 19, 404,
405
непрерывная 406
противоточная 407 ел.
фракционная 415 ел.
Летучесть 72 ел.
Литий, экстракция 658
Лихта уравнение 527
Лувеста (Центривеста) экстрактор
599, 600
Марангони эффект 203
Маргулеса уравнения, неидеальные
растворы
бинарные 84 ел., 90, 95, 97
тройные 109 I
Масла, экстрактивная очистка 635,
641 ел.
Массопередача 195
в экстракторах
«Mixco» 583
насадочных 555 ел.
пульса циониых 595, 596
распылительных 539 ел.
роторно-дисковых 579 ел.
ситчатых 568 ел.
Шайбеля 585 ел.
— ячейке с перемешиванием 200
коэффициенты см. Коэффициенты
массопередачи
скорость в дисперсной и сплошной
фазах 210 ел.
теории 191 ел., 196, 198 ел.
Межфазовая поверхность,
экстракторы
насадочные 552
распылительные 541
смесительно-огстойные 462 ел.
Межфазовая турбулентность 203,
204
Межфазовое натяжение экстрагентов
147 ел.
Мембраны разделительные 503 ел.
«Меркапсол-процесс» 640
Метилэтилкетон, дегидратация
высаливанием 39 ел.
Методы
корреляции равновесных данных
для тройных систем 44 ел.
определения коэффициентов
диффузии 171, 172
— кривой растворимости 28, 29
Механические смесители 452, 490,
491
Мешалки 454
Минарда и Джонсона корреляция
537
Многокомпонентные системы
распределение веществ между
фазами 65, 66
расчет равновесия 122 ел.
экстракция многоступенчатая при
перекрестном токе 322 ел.
противоточная 332 ел.
— расчет 306 ел.
Многоступенчатая экстракция 227,
228
ацетона 244 ел.
в многокомпонентных системах
322 ел.
при перекрестном токе 236 ел.
противоточная см. Противоточная
экстракция
Моделирование экстракторов 484,
485, 623 ел.
714
Молекулярная диффузия 168 ел.,
185 ел.
Мольные объемы веществ 176, 177
Насадочные экстракторы 547 ел., 591
Насосы как смесители 490, 491
Неидеальные растворы
бинарные 74 ел.
тройные 106 ел.
Нернста уравнение 181
Неэлектролиты, коэффициенты
диффузии 172 ел.
Ниобий, экстракция 17
Нитроанилпны, экстракция 326 ел.
противоточная 341, 342
фракционная 376 ел.
Нитробензол как растворитель 635
Обогащение экстракта при противо-
точной экстракции с флегмой
283 ел.
Одноступенчатая экстракция,
системы
многокомпонентные 308 ел.
трехкомпонентиые 227 ел.
Оксифенолы, фракционная
экстракция 372 ел.
Отмера — Тобиаса координаты 47 ел.
Отстойники 495 ел.
Пекле критерий подобия 194
Перегонка с водяным паром при
регенерации экстрагентов 164
Перегородки отражательные в
экстракторах 456, 457, 466, 467
Перемешивание, экстракторы
смесительно-отстойные 452 ел., 466
ел., 484, 485
с непрерывным контактом 522, 529,
530
Пленочная теория массопередачи 191,
192
Пленочные экстракторы 530, 531
Поверхность раздела фаз
блокирование 201, 202
и градиенты концентраций 196
— температура 201
подвижность 203, 204
сопротивление при адсорбции ПАВ
201
Подбильняка экстракторы 597 «л.
Подвижность (и)
ионов при бесконечном
разбавлении 181 ел.
поверхности раздела фаз 202
Полное орошение при противоточной
экстракции 288, 290, 29§
Правило
Антонова 148
рычага 23, 30
Сеченова 65
фаз 20, 24, 33
Пропан как растворитель 636, 642
Противоточная экстракция 332 ел.,
390 ел.
ацетона 263 ел.
из нескольких растворов 270 ел.
лабораторная 407 ел.
периодическая 281, 436, 437
расход экстрагента 257, 258
расчет см. Расчет
с несмешивающимися
растворителями 274 ел.
— флегмой 282 ел., 290, 291, 295,
299, 343 ел., 413 ел.
фактор 277
Проточные смесители 485 ел.
Прямоугольные координаты
изображение противоточной
экстракции 260, 261, 273, 291
и равновесие в тройных системах
43, 44
«Пурекс-процесс» 657
Равновесие
в тройных системах 41 ел., 57 ел.
кривые 290, 291
расчет 70 ел.
по коэффициентам активности
109 ел.
хорда 22
Разделение металлов экстракцией
652 ел.
Распределение между фазами
законы см. Законы распределения
коэффициенты см.
Коэффициенты
кривые и коэффициенты 45 ел., 57,
61, 62
Распылительные экстракторы 531 ел.,
590, 591
Растворимость 131, 132
компонентов бинарных систем 28,
29
— и коэффициенты активности
94 ел.
кривые 28, 29, 31, 32
критическая точка 133
экстрагентов 145, 146
Растворители см. также Экстрагенты
вязкость и диффузия 173 ел.
для очистки смазочных масел
634 ел.
несмешивающиеся, экстракция
дифференциальная 253, 254
многоступенчатая 240 ел.
716
Растворители
несмешивающиеся, экстракция
противоточная 274 ел.
селективность 117, 118
факторы ассоциации 176
Растворы 390 ел.
идеальные см. Идеальные
растворы
неидеальные см. Неидеальиые
растворы
неэлектролитов 172 ел.
регулярные 99, 129, 130, 132, 133
свободная энергия 82, 83
сопряженные 22
Рауля закон 71 ел.
Рафинат 152, 224
исчерпывание при противоточной
экстракции 285, 286
флегма 286, 287
Расчет
высоты непрерывных проточных
экстракторов 384 ел.
коэффициентов активности
неидеальных бинаоны.х растворов 89
равновесия см. Равновесие
экстракции диссоциативной 378
— многоступенчатой 237, 239, 240
— одноступенчатой 233 ел.
— противоточной 259, 261, 262, 265
ел., 335
с флегмой 282 ел., 299
систем многокомпонентных 306 ел.,
379 ел.
— тройных 222 ел.
Реакционная способность и
стабильность экстрагеитов 149, 150
Регенерация
облученного ядерного горючего 656,
657
экстрагеитов 139 ел., 146
выпариванием !64
из азеотропных смесей 162 ел.
кристаллизацией 165
перегонкой с водяным паром
164
при экстракции многоступенчатой
237, 238
— — одноступенчатой 230
с флегмой 161 ел.
ректификацией 152 ел.
экономика 621, 622
Редлиха — Кистера уравнения,
неидеальные растворы
бинарные 85, 86, 90, 96, 97
тройные 109
«Редокс-процесс» 656, 657
Рейнольдса критерий подобия 194
Роторно-дисковые экстракторы
576 ел.
Роторно-кольцевые экстракторы 574
ел.
Руби и Элджина корреляция 539
Самодиффузия, коэффициенты 171 ел.,
175
Свободная энергия растворов 82, 83
Селективность 301, 302
анилина 134 ел.
для систем тройных 57, 120 ел.
— — четырехкомпонентных 62
и коэффициент распределения 143,
144
— К. Т. Р. 126, 127
экстрагеитов 117, 118, 139 ел.
Сеченова правило 65
Системы
двухкомпонентные см. Бинарные
системы
жидкость—жидкость, расчет
равновесия 70 ел.
многокомпонентные см.
Многокомпонентные системы
с двумя экстрагентами см.
Фракционная экстракция
сольютропные 32, 47
трехкомпонентные см. Трехкомпо-
нентиые системы
четырехкомпонентные см. Четырех-
компонентные системы
Ситчатые экстракторы 562 ел.
Сляйхера уравнение 529
Смесители 452 ел.
без перегородок '460, 461, 467, 468
диафрагменные 488, 489
инжекторные 487, 488
механические 490, 491
проточные 485 ел.
секционированные 473
с использованием ультразвука 491
— отражательными перегородками
456, 457, 461 ел.
типа вентилей 489, 490
— сопел 488, 489
— форсунок 486, 487
Смесительно-отстойные экстракторы
лабораторные 421
оборудование 451 ел.
эффективность, ступени 443 ел.
Смола древесная, экстракция 644
«Сольютайзер-процесс» 640
Сольютропные системы 32, 47
Сопла смесительные 488, 489
Сопротивление (я)
аддитивность 196 ел.
диффузионные 396, 397
поверхности раздела фаз при
адсорбции ПАВ 201
Сопряженные растворы 22
716
Степени свободы, экстракция
дифференциальная 328
многоступенчатая при перекрестном
токе 322, 323
противоточная 343 ел.
фракционная 346 ел.
Стоимость экстрагеитов 151
Стокса — Навье уравнение 208
Стокса — Эйнштейна уравнение 172
Ступени 224, 297, 301
в смесительно-отстойных
экстракторах 443, 445 ел.
при фракционной экстракции 350 ел.
эффективность 230
Сэзерленда уравнение 172, 174
Талловое масло, экстракция 644, 645
Тантал и ниобий, разделение 17, 658
Тарелки ситчатые 523, 562 ел., 591,
592
вибрирующие 596
жалюзийные 594
с выштамповаииыми отверстиями
524, 593
Температура 57 ел.
влияние на коэффициенты
активности 86, 87
характер физических
диаграмм в тройных системах
33 ел.
и коэффициенты диффузии 174 ел.,
178, 180
— многоступенчатая экстракция
239, 258, 259
— поверхность раздела фаз 201
— растворимость веществ 133
Теория(и)
абсолютных скоростей по Эйрингу
173 ел.
аддитивности сопротивлений 196 ел.
диффузии в жидкостях 172
массопередачи 191 ел., 196, 198 ел.
Термодинамическая консистентность
81
Тетраэдрические координаты 59 ел,
310 ел.
Токсичность экстрагеитов 151
«Торекс-процесс» 657
Торий, экстракция 657
Точка
замерзания экстрагеитов 150
критическая
в системах бинарных 24 ел.
тройных 42, 43, 48
растворимости ограниченно
смешивающихся веществ 133
Треугольные координаты 29 ел,
224 ел,
Треугольные координаты
графическая интерполяция хорд
равновесия 44, 45
изображение, экстракция
дифференциальная 248, 249
одноступенчатая 228 ел.
противоточная 255 ел.
Трехкомпонентные системы см.
Тройные системы
Тройные системы
активности 110, 111
без тройной К. Т. Р. 32, 33
влияние температуры на характер
фазовых диаграмм 33 ел.
межфазовое натяжение 148
неидеальные 106 ел.
образующие твердую фазу 37 ел.
плотность 146, 147
равновесие 41 ел.
расчет экстракции 22 ел.
сольютропные 32
с тройной К. Т. Р. 33, 34
— частично смешивающимися
жидкостями 31 ел.
Тура — Марчелло теория
массопередачи 194
Турбулентная диффузия, коэффициент
212
Турбулентность межфазовая 203, 204
Удерживающая способность,
экстракторы
насадочные 459 ел.
распылительные 535 ел.
роторно-дисковые 578 ел.
Уилки и Чена уравнение 176
Уксусная кислота, экстракция 205
из единичных капель 526, 527
метилизобутилкетоном 236, 398 ел.
этилацетатом 15, 17
Унос, влияние на эффективность
ступени в смесительно-отстойных
экстракторах 505 ел.
Уравнение(я)
Бенедикта 75, 107, 108
Ван-Лаара см. Ван-Лаара
уравнения
Гибса — Дюгема 80 ел.
Гильдебранда и Скотта 130, 132,
133
Гордона 183
Кронига — Бринка 211
Лихта 527
Маргулеса см. Маргулеса
уравнения
Нернста 181
Редлиха — Кистера см. Редлиха —
Кистера уравнения
Сляйхера 529
Т17
Уравнение(я)
Стокса — Навье 208
Стокса — Эйнштейна 172
Сэзерленда 172, 174
Уилки и Чена 176
Хайворта 524
Хандлоса и Барона 212
Христиансена 525
Шайбеля 177
Уран, экстракция 655, 656
алкилфосфорными кислотами 18
в смесительно-отстойных
экстракторах 482, 483
противоточная 279 ел.
Фаза(ы) 37 ел.
диаграммы см. Диаграммы фазовые
дисперсная и сплошная 459,
468 ел., 519, 520
скорость массопередачи 210 ел.
поверхность раздела и градиенты
концентраций 196
правило 20
Фактор
ассоциации растворителей 176
экстракции 243, 244, 277, 396, 397
Фенолы
как растворители 636
экстракция 645 ел.
Фика законы 169, 171
Флегма 626
получение охлаждением 302, 303
экстракция противоточная 343 ел.,
413 ел.
— фракционная 352, 359 ел., 368 ел.,
373, 374, 420
Флегмовое число, экстракция
противоточная 287, ел., 294, 297
фракционная 368 ел., 373, 374
Форсунки как смесители 486, 487
Фракционная экстракция 223, 307, 419
лабораторная 415 ел.
многокомпонентных систем 346 ел.
пиколинов 163
с флегмой 352, 359 ел., 420
экономика 626 ел.
Фруда критерий подобия 194
Фурфурол как растворитель 636, 642,
643
Хайворта уравнение 524
Харрингтона экстрактор 563
Хандлоса и Барона уравнение 212
Хигби теория массопередачи 192, 193
Химическая реакция
влияние на скорость экстракции
204 ел.
при регенерации экстрагента 165,
166
Хлорекс как растворитель 635, 636
Хлорнитробензолы, экстракция
одноступенчатая 320 ел.
противоточная 339 ел.
Холлей-Мотта экстрактор 508, 509
Хорды равновесия 22
для систем тройных 44 ел.
четырехкомпонентных 312, 313
Хоффинга и Локкарта корреляция
558
Христиансена уравнение 525
Хэнда координаты 45, 48 ел.
Центрифуги 500, 501
Ценца корреляция 535, 536
Циклоны 499, 500
Цирконий и гафний, разделение 657
Четырехкомпонентные системы
кривые распределения 61 ел.
состав, изображение 59 ел.
экстракция одноступенчатая 310 ел.
— противоточная 332 ел.
— — с флегмой 345, 346
— фракционная 348 ел.
Число
единиц переноса 386 ел.
йодное 66
ступеней при фракционной
экстракции 350 ел.
флегмовое см. Флегмовое число
Шайбеля
уравнение 177
экстракторы 584 ел.
Шервуда
критерий подобия 194
метод интерполяции хорд
равновесия 45
Шмидта критерий подобия 194
«Эделеану-процесс» 638
Эйринга теория абсолютных
скоростей 173 ел.
Экономика процессов экстракции
609 ел.
Экстрагенты см. также Растворители,
вводимые с питанием при
фракционной экстракции 356
взаимно нерастворимые 307, 316 ел.,
329 ел., 352 ел.
воспламеняемость 151
выбор 139 ел.
вязкость 150
давление паров 150
емкость 144, 145
коррозионная активность 150
коэффициенты диффузии .181 ел.
межфазовое натяжение 147 ел.
718
Экстрагенты
растворимость 145, 146
расход 257, 258
реакционная способность и
стабильность 149, 150
регенерация см. Регенерация
селективнос!ь_139 ел.
смешанные 306у314, 315
стоимость 151 '
ступенчатое взаимодействие с
исходной смесью 222 ел.
токсичность 151
точка замерзания 150
Экстракт 152, 224
обогащение прн противоточной
экстракции с флегмой 283 ел.
Экстрактор (ы)
без механического перемешивания
фаз 530 ел.
«Витро» 511
Керра-Мак-Ги 510, 511
Коха 565
Крэга 421 ел.
лабораторные 420 ел.
Лувеста (Центривеста) 599, 600
«Mixco» 582 ел.
моделирование 484, 485, 623 ел.
насадочные 547 ел., 591
непрерывные противоточные
384 ел.
Олдшу — Рештона 582 ел.
«Памп-Микс» 421
пленочные 530, 531
Подбильняка 597 ел.
пульсационные 587 ел.
распылительные 531 ел., 590, 591
роторио-дисковые 576 ел.
роторно-кольцевые 574 ел.
с вибрирующими тарелками 596
— выносным отстойником 535
смесительио-отстойиые см. Смеси-
тельно-отстойные экстракторы
с механическим перемешиванием
фаз 573 ел.
— непрерывным
(дифференциальным) контактом 518 ел.
— орошаемыми стенками 530, 531
— перегородками 548, 549
— перфорированными тарелками
(ситчатые) 562 ел., 591 ел.
Харрингтона 563
Холлей-Мотта 508, 509
центробежные 597 ел.
Шайбеля 584 ел.
Элджина и Блендиига 534
ящичные 421, 509, 510
Экстракция 12, 15 ел.
диссоциативная 378, 379, 437, 438
Экстракция
дифференциальная см.
Дифференциальная экстракция
лабораторная 404 ел,
многоступенчатая см.
Многоступенчатая экстракция
на пилотных и полупромышленных
установках 431 ел.
непрерывная 450, 451, 460 ел.,
477 ел.
одноступенчатая см.
Одноступенчатая экстракция
периодическая 459 сл„ 473 ел.
по Крэгу 423 ел.
применение в аналитической химии
423 ел.
— — коксохимии 645 ел.
нефтехимии 632 ел.
— — технологии жиров и масел
641 ел.
— — — неорганических веществ
650 ел.
— органических веществ
649 ел.
фармацевтической
промышленности 647 ел.
— при регенерации экстрагентов
165
противоточная см. Противоточная
экстракция
расчет см. Расчет экстракции
с единичными каплями 206 ел.
секции 353
селективная 139 ел.
симметричная 418 ел.
сопровождаемая химической реак
цией 204 ел.
способы организации процесса 227,
228
фактор 243, 244, 277, 396, 397
фракционная см. Фракционная
экстракция
экономика процесса 609 ел.
Электролиты, коэффициенты
диффузии 181 ел.
Эмульсии 491 ел.
Энергия свободная растворов 82, 83
Эффективность
разделения фаз в циклоне 499
ступени 230
в экстракторах ситчатых 568 ел.
■ смесительио-отстойных
445 ел., 470 ел., 505 ел.
«Юдекс-процесс» 639
«Юиисол-процесс» 641
Ящичные экстракторы 421, 509t 510
ИМЕННОЙ УКАЗАТЕЛЬ
ОХВАТЫВАЕТ ВСЕ ГЛАВЫ, ЗА ИСКЛЮЧЕНИЕМ
РАЗДЕЛА «ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА»,
ЦИФРЫ В СКОБКАХ УКАЗЫВАЮТ НОМЕРА ССЫЛОК
Альдерс Л. 9 (1)
Аненкова Л. А. 516 (62)
Бабанов Б. М. 515 (40)
Броунштейи В. И. И (1)
Железняк А, С. 11 (1)
Жуковская С. А. 516 (61, 62)
Захаров Е. И. 11 (3)
Зильбермаи Б. Я. 652
Зюлковский 3. 9 (2)
Карпачева С. М. 11 (3)
Ackerman G. Н. 606 (111)
Agnello L. A 660 (30)
Albright I. F. 219 (22)
Alders B. I, 68 (4)
Alders L. 69 (44, 68), 305
(21), 382 (22), 383 (34)
440 (7)
Alderwelreldt F. 440 (27)
Alerton J, 606 (94)
Alexandre M. 608 (165)
Allemaun K, 440 (33)
Allen H. D. 659 (13)
Allen K. A. 440 (3)
Alter H. W, 440 (19)
Amundsen N. R. 305 (6, 7)
Anderson D. W. 608 (166)
Anderson H. M. 607 (137)
Aries R. S. 631 (4)
Aris R. 305 (6)
Arnold G. B. 68 (28)
Arrowsmlth С J. 659 (13)
Asselin G. F. 383 (39)
Auer P. 1.. 220 (51), 440 (9)
Ayres E. E. 603 (8)
Babb A, L, 219 (12), 607
(142, 146)
Bachman I. 69 (39)
Baddour R. F. 219 (8)
Ballard J. H. 605 (65)
Banchero J. T. 221 (109)
Bancroft W. D. 69 (41)
Baniel A. 660 (35)
Barghusen J. 440 (23)
Barker J. A. 440 (29)
Barker J. J. 515 (42)
Baron T. 221 (103)
Barrows E. T. 659 (1)
Barry G. T. 440 (2)
Barson N. 608 (164)
Bartell F. E. 166 (10)
Bartels С. Е. 382 (23)
Batchelder H. R. 660 (21)
Bauer D. J. 660 (39)
Beams J. W. 68 (4)
Кафаров В. В. 515 (40), 516
(61)
Кишиневский М. X, 220 (46)
Кузнецов В. И. 661 (49)
Левич В. 220 (63)
Лысковцев И. В. 11 (2)
Майлс Дж. Л. 652
Мартии Ф. С. 652
Муратов В. М. 11 (3)
Павлушенко И. С. 515 (18,
21)
Bearman R. J. 219 (23)
Becher P. 516 (73)
Beck J. R, 221 (68)
Beckmann R. B. 604 (47), 605
(61, 62. 73, 74), 606 (102)
Beech D. G. 166 (2)
Beecham A. F. 440 (29)
Bedingtield С. Н. 219 (3)
Bell В. Т. 517 (102)
Benedict M. 138 (5)
Benenati R. F. 606 (96) 607
(141)
Berber J. S. 660 (39)
Berg R. L. 607 (138)
Berg L. 138 (32)
Bergelin O. 604 (32)
Beyaert В. О. 604 (39)
Beyer G. H. 440 (12), 607
(145). 608 (159. 164)
Bikerman J. J. 516 (74)
Billerbeck С J. 607 (134)
Bilous O. 515 (48)
Bird R. B. 219 (4)
Bissell E. S. 514 (4, 5)
Black С 138 (15)
Black K. 517 (108)
Blanding F. H. 604 (37)
Bliss H. 604 (25)
Block H. S. 660 (22)
Bloom J. L. 440 (20)
Blumberg R. 660 (35)
Bochinski J. 383 (38), 440 (10)
Bockelmann J. B. 68 (22).
138 (31)
Bogin C. D. 68 (26)
Bogusz E. 221 (94)
Booth H. S. 69 (63)
Boussinesq J. 221 (106)
Bowman С W. 221 (108)
Boye-Christensen G. 220 (65)
Bradlev K. J- 659 (14)
Braida L. 221 (97)
Brancher A. V. 69 (40)
Brancker A. V. 69 (47, 52),
382 (2. 3), 383 (31)
Памфилов А. В. 220 (46)
Паульсен И. А, 69 (37)
Пратт Г. 9 (3)
Пушлеиков М. Ф. 652
Рагинский Л. С. 11 (3)
Тарасенков Д. Н. 69 (37)
Фомин В. В. 10 (1)
фрумкин А. 220 (63)
Шкоропад И. Д. И (2)
Янишевский А. В. 515 (18,
21)
Bieese J. С. 516 (85). 607
(134)
Briggs S. W. 68 (10), 604
(29)
Briggs W. A. 660 (33)
Brink J. С 221 (101)
Brlnkley W. K. 382 (11)
Brinsmade D. S. 604 (25)
Brooks В. Т. 659 (3)
Brooks L, H. 220 (50)
Brown G. G. 305 (15), 604
(32)
Brown T. F. 166 (3)
Bruce F. R. 660 (40), 661 (41)
Buchman R. H. 603 (4)
Burtis T. A. 516 (88)
Bush M. T. 382 (14)
Bussolari R. 605 (86)
Butler E. D. 68 (29)
Butler T. A. 661 (42)
Byrne J. B. 516 (81)
Cairs R. S. 440 (22)
Calderbank P. II. 221 (102),
515 (30)
Capps R. С 606 (118)
Carbone W. F. 516 (68)
Carlson G. J. 515 (41)
Cadlson H. С 138 (7)
Carver J. A. 607 (152)
Cavers S. D. 605 (48)
Chalkley D. E. 661 (46)
■ Chang P. 219 (20), 220 (7)
Chang Y. 69 (56)
Chantry W. A. 607 (138)
Charles G. E. 516 (77)
Chem J. 68 (5)
Chester G. V. 515 (36)
Chilton С. Н. 661 (43)
Chin H, L. 605 (66)
Chong A. I. 220 (67)
Christiansen R. M. 603 (11)
Chu J. С 138 (19)
Church J. M. 515 (28)
Church W, L. 605 (80)
720
Churchill S. W. 516 (67)
Clegg J. W. 661 (44)
Clouse R. J, 606 (117)
Codding J, V. 382 (18), 440
(19)
Coghlan C. A, G. A. 68 (28)
Colburn A. P. 138 (7, 18, 22J
220 (41, 56), 403 (f, 4),
605 (70), 606 (88)
Colding A. 604 (16)
Colven T. J. 517 (103, 104)
Comings E. W. 68 (16), 383
(39), 604 (29)
Compere E. L. 382 (19), 440
(13)
Conway J. B. 603 (14)
Coplan B. V. 440 (17), 517
(100)
Cornett L. G. 440 (22). 515
(54)
Costich E. W. 514 (8)
Cotter J. E. 605 (69)
Coulson J. M, 604 (18)
Coutor С 608 (167)
Cox J. D. 68 (5)
Craig D. 441 (35)
Craig L. C. 440 (2), 441 (34,
35)
Crawford J. W, 605 (67)
Crespi M. A. 382 (10)
Crico A. 607 (139)
Griffith W. L. 607 (131)
Cronan С S. 660 (36)
Cruickhank A. J. B. 382 (4)
Cuckney M. 516 (97)
Cummings G. H. 515 (15)
Daley F. L, 515 (53)
Daley J. F. 69 (43) 166 (6)
Damerell G. L. 515 (35)
Danckwerts P. V. 220 (43),
221 (81, 83)
Darwent D. 68 (20)
Davidson J. K. 440 (17), 517
(100)
Davles T. V. 221 (77)
Davis A. T. 517 (103)
Davis M. W. 440 (21), 517
(110), 606 (98)
Davison R. R. 660 (37)
Deal С. А. 138 (24)
Dec Т. О. 604 (36)
Defives D. 608 (154)
Defize J. С L. 659 (2)
Deissler R. G. 220 (36)
Dell F. R. 605 (58)
Densen R. M. 382 (14)
Derr E. L. 138 (24)
De Wllte R. L. 607 (150)
Dixon H. 68 (4)
Doby-Ducleaux A. 69 (64)
Dodge B. F. 138 (3, 8)
Donahue D. I. 166 (10)
Doule С. М. 608 (160, 163)
Drew D. A. 68 (30), 138 (30)
Drew Т. В. 219 (3), 220 (41).
514 (3)
Drickamer H. G. 220 (52-54)
Drury J. S. 68 (10)
Duffey H. R. 606 (88)
Duffin J. H. 382 (17)
Duffy С. Н. 220 (29)
Dulllen F. A. L. 219 (7)
Dumoulin F. E. 606 (97)
Dunstan A. E. 659 (3)
Durandet J. 608 (154)
Duval С. А. 516 (63), 659 (4)
Dykstr.1 J. 606 (117)
Eaglesfleld P. 167 (15), 660
(31)
Edwards R. B. 607 (145)
Eguchl W. 606 (119), 607(120,
121)
Einstein A. 219 (14)
Eisenlohr H. 608 (168, 169)
Elgin J. С 403 (2), 604 (37-
40, 46)
Ellis S. R. M. 605 (85), 606
(95), 607 (122)
Elzinga E. R. 221 (109)
Engel L. L. 441 (40)
Epstein N. Б16 (59)
Evans J. E. 603 (13)
Evans T. W. 305 (4, 8, 10)
Everett H. J. 514 (4, 5, 8)
Everson H. E. 69 (63)
Ewanchyna J. E. 605 (48)
E~well R. H. 138 (32), 166 (13)
Eyring H. 219 (16—19, 21)
Fain P. 516 (93)
Fallah R. 604 (26)
Farllis J. G. 166 (11)
Farquhar J. 607 (134)
Feick G. 607 (137)
Felix J. R. 607 (152)
Fenske M. R. 68 (18), 166(4).
305 (3, 9), 440 (25), 607
(153)
Fick A. 219 (1)
Filbert R. B. 660 (21)
Flagg J. 517 (105), 661 (45)
Fleming J. F. 605 (52)
Fletcher J. M. 660 (40), 661
, (41)
Fluid J. 515 (27)
Flynn A. W. 515( 51)
Foley D. D. 661 (44)
Foos R. A. 440 (30)
Fosbury D. W. 606 (95)
Foster S. P. 517 (101)
Francis A. W. 68 (11—13, 21).
69 (50), 138 (28), 166 (8).
695 (5)
Frankforter G. B. 68 (23)
Frary F. С 68 (23)
Freiser H. 440 (31)
Fried V. 138 (20)
Friedland D. 138 (25)
Fugua F. D. 605 (82)
Fujihawa K. 220 (57)
Fujita S. 605 (59). 606 (89)
Fuoss R. M. 219 (2)
Oaden E. 660 (27)
Gardner С S. 440 (9)
Carner F. H. 220 (60, 61, 66),
221 (90, 96), 604 (19, 20).
605 (85), 606 (95), 607
(122)
Garvin A. M. 608 (161)
Garwin L. 166 (5), 605 (54)
Gaylor R. 605 (60, 63)
Geankoplis С J. 69 (62), 605
(51, 56)
Geddes A. L. 219 (5)
Geddes R. L. 383 (32)
Gentlllnl P. 608 (165)
Gester G. С 659 (6)
Ghosh S. K. 606 (90)
Ghubliklan J. R. 659 (12)
Gibbs 68 (1)
Gier Т. Е. 605 (57)
Gilespie T 516 (76)
Gilliland E. R. 219 (8), 305
(20), 631 (5)
Giona A. R. 604 (31)
Girifalco L. A. 166 (9)
Gladel Y. L. 608 (154)
Glasstone S. 166 (2), 219 (17)
Gloyer S. \V. 659 (15, 16)
Goldberger W. M. 606 (96),
607 (141)
Goldstein D. J. 219 (8)
Gollmar H. A. 516 (69)
Golumbic С 69 (46)
Good R. J. 166 (9)
Gordon A. R. 219 (9), 220
(34)
Gordon K. F. 220 (55, 58)
Graef E. R. 517 (101)
Grant D. M. 219 (21)
Greak B. F. 516 (63)
Griffith R. M. 221 (107)
Grittendon E. D. 167 (21)
Grote H. w: 167 (22), 659
(7)
Grothms H. 515 (34)
Grober H. 515 (47)
Grunewald G. R. 605 (77)
Gunn J. D. 382 (5)
Gustison R. A. 606 (118)
Guyer A. 607 (151)
Haas W. D. 382 (18)
Hadamard J. S. 221 (88)
Haertsch N. 382 (4)
Hala E; 138 (20)
Hale A. R. 220 (60, 66)
Hall A. K. 604 (20)
Harrier W. J. 138 (11)
Hamielec A. E. 221 (104), 604
(16, 17)
Hand D. B. 68 (27), 138 (27)
Handlos A. E. 221 (103)
Hansuld J. H. 605 (53)
Hapler H. G. 516 (92)
Happel J. 631 (1)
Harmathy T. Z. 221 (98)
Harmaty T. 604 (44)
Harned H. S. 219 (13), 220
(33)
Harrington P. J. 605 (76)
Harriott P. 515 (43)
Harrison J. M. 138 (32)
Hartley G. S. 219 (10)
Hartwlg G. M. 69 (58)
Hashimoto K. 515 (29)
Hastings J. J. H. 660 (28)
Hatta S. 221 (84)
Haydon D. A. 221 (76, 77)
Hayes J. G. 516 (89)
Hayes L. A. 516 (89)
Hayes W. B. 516 (65)
Havworth С. В. 68 (19), 166
(7). 603 (5)
Hazen W. С 517 (106)
Hecker E. 440 (32, 33), 441
(38)
Heririckson A. V. 517 (106)
Hermann J. A. 220 (67)
Hertjes P. M. 604 (21)
Hertzog R. R. 605 (87)
Hesse H. С 514 (4)
Heumann F. K. 382 (18)
Heuney V. E. 660 (22)
Hicks Т. Е. 440 (21), 517
(110)
Higbie R. 220 (42)
Hildebrand J. H. 68 (4), 138
(23. 34)
Hill A. F. 68 (7), 69 (33)
Hill J. W. 805 (85)
721
Himmelblau D. M. 607 (148)
Hinze J.- O. 515 (26)
Hltchon J. W. 516 (84)
Hixsor; A. N. 68 (30), 138
(30, 31), 166 (5), 167 (21),
603 (7, 11)
Hixson A. \V. 514 (1, 3)
Hobson R, I. 305 (12)
Hoffing E. H. 605 (68)
Hoffman A. S. 607 (134)
Hofmann H, 515 (13)
Hofmann V. 660 (24)
Hoffijzer P. J. 221 (86)
Holder С. Н. 607 (152)
Holland С D. 516 (64, 65)
Holley A. E, 516 (95, 96)
Holm A. 515 (49)
Holms J. H. 440 (18)
Holve W, A. 604 (21)
Honda N. 607 (135)
Honjyo M. 515 (11)
Hood D. W. 660 (37)
Hood G. С 69 (58)
Hopp H. F. 383 (37)
Horsley L. H. 138 (21), 167
(19)
Hougen J. O. 605 (57)
Hougen O. A. 69 (48)
Hu S. 221 (92)
Huang С J. 515 (44)
Hughes H. E. 68 (31)
Hughes J. H. 607 (122)
Humah H. H. 660 (40), 661
(41)
Humphrey D. W. 515 (45)
Humphrey J. W. 605 (78)
Hung С J. 605 (53)
Hunter T. G. 69 (40, 53), 305
(11, 13, 16), 382 (3, 4,
6), 403 (7), 604 (26. 34)
Hutchinson E. 220 (64)
Ibl N. V. 138 (8)
Ingensoll A. I. 516 (80)
Innes К. K- 219 (22)
Irving H. M. 661 (46)
Isaac N. 607 (150)
Jacobs J. M. 661 (47)
Jacobson F. M. 608 (159)
Jacques G. L. 605 (69, 75)
Janecke E. 69 (34)
Janlzen E. 440 (1)
Jasney G. R. 607 (131)
Jasunaga T. 606 (113)
Jealous A. C. 607 (129, 132)
Jennings A. S. 440 (19)
Jensen V. G. 221 (90)
Johns L. E. 605 (73)
Johnson A. I. 221 (87, 97,
104, 10S), 515 (44), 604 (16,
17, 43), 605 (53)
Johnson С. А. 138 (5)
Johnson H. F. 603 (9), 605
(52), 607 (129)
Johnson M. 221 (94)
Johnson P. A. 219 (12)
Johnstone T. R. 516 (59)
Jones С. А. 138 (18)
Jones J. 605 (64)
Jonstone R. E. 514 (9)
Jordan G. V. 516 (90)
Jost W. 219 (6)
Kaiser H. R. 608 (160—162)
Kalab V. 606 (112)
Kalichewsky V. A. 516 (56.
63), 659 (4, 8, 9)
Kang С 606 (89)
Kappelmann F. A. 661 (56)
Karr A. E. 167 (18) 221 (74).
515 (52), 607 (124, 125,
149)
Katz J. J. 661 (41)
Kauffman J. F. 603 (12)
Keith E. W. 603 (7)
Kelly В. К. 167 (15), 660 (31)
Kelly Т. Е. 660 (24)
Kennedy A. M. 221 (81)
Ketchen E. E. 661 (42)
King W. H. 659 (5)
Kingsley H. A. 514 (6)
Kincald J, F. 219 (18)
Ktntner R. С 221 (91—94)
Kirkbride C. G. 516 (88)
Kirkwood J. С 219 (23, 24)
Kirschenbauer H. K- 659 (17)
Kister A. T. 138 (10, 16)
Kjellman H. A. 605 (83)
Wee A. J. 221 (95)
Kleiman G. 382 (23)
Klevens H. B. 69 (65)
KUne W. A. 659 (13)
Klinkenberg A. 382 (24, 25)
Knapp L. 440 (23)
Knebel A. H. 516 (93)
Knox K. L. 514 (3)
Kobe K. A. 516 (56), 659 (9, 10)
Koch F. G. 605 (84)
Koerner E. L. 440 (11), 661
(48)
Koffolt J. H. 606 (92)
Kohn J. P. 68 (9)
Korchlnski I. J. B. 221 (102)
Koslov J. 517 (108)
Kovner E. L. 440 (11)
Кбппеске Н. G. 516 (78)
Kramers H. 606 (114)
Kreager R. M. 605 (50)
Kremser A. 305 (14)
Kroepelin H. 221 (69, 70)
Kroll A. E. 515 (10)
Kronlg R. Y. 221 (101)
Kung E. J. 606 (109)
Kurata F. 68 (9)
Kurchara T. 220 (57)
Kwauk M. 382 (1)
Laddha G. S. 604 (45)
Laldler K. J. 219 (17)
Laity D. S. 515 (12)
Langlols G. E. 515 (37)
Lapidus L. 305 (7), 604 (39,
40)
Lash L. D. 517 (109)
Lastovica J. E. 166 (11), 604
(33)
Lau E. F. 608 (166)
Lauwerier H. A. 382 (25)
Lawrence E. A. 659 (13)
Leacock J. A. 516 (67)
Leibson J. 605 (74)
Levy S. L. 138 (19)
Lewis G. N. 138 (1, 2)
Lewis J. B. 68 (6). 220 (44,
47). 221 (71), 605 (64)
Lewis W. K- 220 (40), 606
(93)
Ll J. С. М. 219 (20)
LI N. N. 605 (72)
Llcht W. 603 (14), 604 (15)
Lleberman E. 607 (132)
Llghtfoot E. N. 219 (4)
Llmmerman H. K- 68 (8)
Lin С S. 220 (37)
Lindland K. P. 220 (59)
Lister D. A. 605 (79)
Little D. J. 607 (133)
Liu Т. Н. 219 (11)
Lockhart F. J. 604 (32), 603
(68)
Logsdail D. H. 606 (110), 607
(143, 147)
Long R. B. 440 (25). 607 (152.
153)
Long R. S. 516 (70)
Longcor J. V. A. 603 (13)
Longtin B. 305 (1)
Lowrite R. S. 515 (53)
Lumb E. С 69 (66)
Lyons P. A. 220 (35)
Macdonald R. W. 515 (24)
Mack D. E. 515 (10)
Madden A. J. 515 (35)
Macda K. 606 (121)
Major С J. 605 (87)
Malin R. T. 659 (4)
Maloney J. O. 68 (31), 305
(19)
Mangules M. 138 (14)
Mann С. А. 515 (19)
Mar B. W. 607 (142, 146)
Marsel С J. 659 (13)
Martin A. J. F. 382 (20)
Mason S. G. 516 (77)
Mathers W. G. 440 (22), 515
(54), 516 (55)
Mathur К. В. 221 (90)
Mattern R. V. 515 (48)
Maycock R. L. 69 (58), 606
(104)
Mayfield F. D. 605 (80)
McBain J. W. 219 (11)
McCord J. С 660 (23)
McDonald H. J. 68 (25)
McDonough J. A. 516 (64)
McDowell W. J. 440 (3)
McGregor D. K. 221 (68)
McKetta J. J. 659 (10)
McMullen A. K- 604 (22)
Mcnamara V. M. 605 (53)
McNeil R. 383 (29)
Medcalf E. С 68 (15)
Meissner H. P. 68 (24), 516
(79)
Mellan I. 166 (12)
Meltzer H. L. 441 (41)
Metznev A. B. 515 (30)
Mhatre M. V. 221 (93)
Miller С. А. 515 (19)
Miller G. L. 661 (50)
Miller M. B. 516 (57)
Mlnard G. W. 602 (43), 605
(53)
Mink W. H. 660 (21)
Mlnty D. W. 383 (29)
Mitchel В. М. 440 (22)
Miyauchi T. 604 (22, 23), 605
(72)
Mole P. D. A. 382 (7)
Moon J. S. 605 (71)
Morello V. S. 517 (99), 603
Morgan E. L. 69 (61)
Morgenthaler A. C. 221 (68)
Morikawa K. 440 (6)
Morrison G. H. 440 (31)
Motel W. J. 517 (104)
Mott О. Е. 516 (95. 96)
Moulton R. W. 69 (56), 220
(37) 606 (91)
722
Murbach E. W. 220 (51)
Murdoch D. F. 516 (97)
Murdoch R. 221 (79), 604 (27)
Murphree E. V. 220 (38),
514 (2)
Murphy N. F. 166 (17), 604
(33)
Nagata S. 515 (11, 20, 29, 46),
606 (119), 607 (120, 121)
Nakaiki Y. 220 (57)
Nandi S. K. 606 (90)
Naruse Y. 515 (29)
Nash A. W. 69 (40, 53), 305
(11, 13, 16), 403 (7), 604
(26, 34), 659 (3)
Nassenstein H. 221 (72)
Naumowicz J. 606 (115)
Nernst W. 69 (45), 220 (31)
Neumann H. J. 221 (69)
Neuworth M. B. 660 (24)
Newman A. B. 221 (99)
Newman J. S. 515 (36)
Newman M. 68 (19), 166 (7),
605 (49)
Newton R. D. 631 (4)
Nichols P. L. 441 (39)
Nilsson K. 221 (85)
Norwood K. W. 515 (30)
Null H. R. 603 (9)
Obergfell P. 659 (12)
Ohlens N. J. 660 (38)
Olander D. R. 220 (28), 221
(80), 382 (16)
Oldshue J. Y. 515 (7), 606
(116)
Olney R. B. 515 (6, 41), 516
(50, 66), 606 (106, 111)
Onsager L. 219 (2)
Onsley J. L. 441 (40)
Orchih M. 69 (46)
Orfell J. B. 220 (39)
Oterode la Gandara J. L. 382
(8)
Othmer D. F. 68 (32), 69 (49).
167 (16, 17, 20), 403 (5,
6), 660 (32)
Overcashier R. H. 514 (6)
Owen В. В. 219 (13)
Oya H. 607 (135)
Pansing W. F. 604 (15)
Partington J. R. 220 (32)
Passino H. J. 659 (19)
Passino R. 604 (31)
Paul R. 138 (19)
Pearce С A. R. 516 (91)
Peppard D. F. 440 (15)
Peppard M. A. 440 (15)
Perry E. J. 441 (37)
Peterson H. G. 440 (12)
Phillips R. J. 516 (92)
Philocm W. G. 516 (93)
Pick J. 138 (20)
Pierce F. J. 605 (77)
Pierotti G. J. 138 (24)
Pigford R. L. 221 (82), 403
(8)
Pilloton R. L. 69 (38)
Piret E. L. 515 (24, 48), 605
(65)
Podbielnlak W. J. 608 (157,
158, 161)
Poettmann F. H. 383 (36)
Poffenberger N. 517 (99), 603
(3)
Pontius R. B. 219 (5)
Powell R. E. 219 (19)
Powers J. E. 382 (9)
Pratt H. R. С 221 (71, 75,
79), 441 (43), 603 (2), 604
(27), 605 (58, 60, 63. 64,
71), 606 (102, 110)
Prince R. G. H. 69 (57)
РгбП Е. 221 (70)
Purdy R. H. 441 (40)
Pyle С 606 (88)
Quinn J. E. 517 (107)
Raal J. D. 221 (87)
Ragatz R. A. 69 (48)
Randall M. 138 (1). 305 (1)
Ratci'tffe R. L. 167 (16)
Rauch E. 608 (163)
Rea H. E. 515 (38)
Reamer H. H. 220 (29)
Redlich О 138 (10. 16)
Ree T. 219 (21)
Reid R. С 138 (6), 219 (25).
607 (134)
Reman G. H. 382 (25), 606
(105—108)
Repis J. 606 (112)
Ricci J. 68 (2), 167 (14)
Rice A. C. 660 (39)
Rice О. К- 68 (3)
Rice S. A. 219 (24)
Richardson G. L. 607 (140)
Riche F. J. H. 659 (20)
Rideal E. K. 516 (76)
Rietema K. 516 (82)
Rius A. 382 (10)
Robbins T. 305 (12)
Roberts N. W. 605 (63)
Robinson С W. 516 (59)
Robinson P. A. 221 (68)
Roberts F. 517 (102)
Rodger W. A. 515 (16, 17)
Rogers M. С 383 (35)
Roges M. С 605 (81)
Rometsch R. 382 (26)
Rosenthal H. 605 (55)
Roseveare W. E. 219 (19)
Ross G. 660 (29)
Ross M. 383 (29)
Rossotti F. 661 (46)
Row S. B. 606 (92)
Rubin L. S. 138 (5)
Ruby C. L. 604 (46)
Rudd D. F. 305 (6)
Runnicles D. F. 219 (10)
Rushton J. H. 514 (4, 5, 7,
8). 515 (14, 17, 31), 606
(П6)
Ruthruff R. R. 69 (67)
Ryland A. 382 (19). 440 (13)
Ryon A. D. 515 (53)
Saal R. N. J. 305 (2)
Sachs J. P. 515 (31)
Sage В. Н. 220 (29)
Sage B. W. 220 (39)
Sato Y. 440 (2)
Scatchard G. 138 (11)
Schafer A. C. 440 (18)
Scharlan A. 608 (169)
Scheihel E. G. 138 (25). 167
(18), 220 (30), 221 (74),
382 (15), 383 (27. 33), 403
(5, 6). 440 (4, 5, 14, 16).
515 (52), 607 (123—127),
631 (6)
Schiff S. 605 (86)
Schoenborn E. M. 138 (18, 22)
Schoenherr R. 440 (23)
Schrage R. W. 220 (49)
Schreinemakers F. A. H. 68
(17)
Schubert A. E. 305 (19)
Schweyer H. E. 631 (2)
Scott E. J. 220 (52)
Scott L. S. 516 (65)
Scott R. L. 138 (23)
Scrlven L. E. 221 (78)
Searle R. 220 (58)
Seddou W. L. 659 (1)
Seewald M. 515 (22)
Sege G. 607 (144), 608 (166)
Setzer H. 515 (32)
Shaw K. G. 516 (70)
Shemilt L. W. 219 (7)
Sheps M. С 441 (40)
Sherwood Т. К- 69 (36), 138
(6), 219 (25). 220 (55).
221 (73, 82), 403 (8), 603
(13)
Shinnar R. 515 (27, 28)
Shlrotsuka T. 607 (135)
Short J. F. 167 (15), 606 (103),
660 (31)
Siemes W. 603 (6. 12)
Sigwart K. 221 (72)
Silllvan F. E. 516 (87)
Simkin D. J. 516 (66)
Slnfelt J. H. 220 (53)
Skelland A. H. P. 220 (61),
221 (96), 604 (19)
Skinner S. J. 604 (18)
Skogan V. G. 383 (35)
Skrzec A. E. 604 (33)
Sleicher С. А. 516 (94), 604
(24)
Slou A. 660 (35)
Smith A. S. 68 (14)
Smith B. D. 382 (11). 605 (54)
Smith F. H. 659 (14)
Smith G. С 604 (47)
Smith I. C. 69 (55). 382 (12)
Smith J. M. 138 (4), 514 (1),
604 (45)
Smith K. 138 (29)
Smith R. B. 383 (37)
Smoot L. D. 607 (146)
Smutz M. 383 (38), 440 (10,
11, 23), 661 (48)
Sobotik R. H. 607 (148)
Solomon E. 138 (5)
Souders M. 305 (15)
Soukup J. A. 516 (93)
Speddlng F. H. 383 (38). 440
(10)
Spells К. Е. 221 (88)
Spence R. 606 (99, 101)
Stearr A. E. 219 (18)
Steinberger R. L. 221 (105)
Stene S. 383 (28), 440 (8)
Sternling С V. 221 (78)
Stewart W. E. 219 (4)
Stlckney W. A. 604 (35). 661
(53)
Stokes С. А. 68 (24)
Strand С. Р. 606 (111)
Strang L. С 604 (34)
Strecton R. J. W. 606 (101)
Strecton R. J. 606 (99)
Strom В. О. 606 (94)
Strom J. R. 221 (91)
Stubbs A. L. 659 (20)
Sucherland G. В. В. М. 219
(15)
723
Suzuki R. 606 (113)
Swabb L. E. 69 (6i)
Swinton E. A. 383 (29)
Synge R. L. M. 382 (20)
Takahashi T. 606 (113)
Talsma H. 604 (21)
Tanizawa E. 605 (59), 606 (89)
Tapp A. C. 661 (56)
Tayion Q. I. 606 (100)
Tepe J. B. 516 (83)
Teramoto D. 515 (20)
Terjesen S. Q. 220 (59, 62,
65), 515 (49)
Tetervsky H. 138 (29)
Thaker M. S. 69 (49)
Thayer H. E. 516 (74)
Thiele E. W. 305 (17), 383
(32), 605 (81)
Thomas L. E. K- 220 (67)
Thompson B. H. 606 (117)
Thornton E. 517 (98)
Thornton J. D. 516 (60), 606
(102, 110), 607 (130, 136,
143, 147. 155)
Thring M. W. 514 (9)
Tiller F. M. 305 (5)
Timmermans J. 68 (6)
Tizard H. 659 (3)
Tobias P. E. 68 (32)
Todokoro K. 605 (59)
Tomme W. J. 516 (64)
Toor H. L. 220 (45)
Tornton J. D. 604 (42)
Trass O. 221 (108)
Treybal R. E. 68 (19), 69 (42,
43, 51), 138 (26), 166 (6,
7), 221 (95, 105), 441 (42).
515 (12, 23, 25, 42, 51),
517 (111), 603 (1, 5), 604
(30), 605 (86), 606 (94, 97,
118), 631 (3)
Treuger E. 157 (17)
Trice V. Q. 515 (17). 516 (58)
Trobwbridge M. 516 (86)
Tung L. H. 220 (52, 54)
Tupper H. T. 607 (131)
Turungulst С. Е. 138 (16)
Twiehaus H. С 660 (38)
TWigg G. H. 383 (30), 606
(103)
Ueyama K. 603 (iO)
Van de Vusse J. R. 606 (107)
Van den Temple M. 516 (75)
Vanderveen J. H. 515 (33)
Van Dijck W. J. D. 305 (2).
607 (128)
Van Krevelln D. W. 221 (86)
Van Laar J. J. 138 (13)
Van Ness H. С 138 (4. 9).
515 (45)
Varteressian K- A. 68 (18),
166 (4). 305 (3, 9)
Vermeulen T. 221 (100). 440
(21), 515 (37, 38), 517
(110), 604 (22), 6Э5 (69,
72, 75)
Vermijs H. 606 (114)
Verzele M. 440 (27)
Vlgneron M. 441 (36)
Vilbrandt F. С 516 72)
Vilium O. 138 (20)
Vitagliano V. 220 (35)
Vnukova K. 606 (112)
Vogt H. J. 605 (51)
Void R. 166 (1)
Vriens Q. N. 68 (15)
Wackier R. C. 660 (22)
Waiker С. А. 69 (59)
Waikey J. E. 606 (91)
Wall G. P. 440 (24)
Wang S. L. 138 (19)
Ward A. F. H. 220 (50)
Ward D. M. 221 (108)
Warner R. E. 604 (28)
Warshay M. 221 (94)
Washburn E. R. 166 (1)
Watanabe S. 440 (6)
Watson К. М. 69 (48)
Weather D. G. 660 (33)
Weareer B. 661 (56)
Weaver R. E. С 604 (40)
Weber H. С 221 (85)
Weber J. E. 606 (98)
Weber L. D. 69 (43), 166 (6)
Weber W. H. 441 (37)
Wehner J. F. 305 (18)
Wei J. С 221 (73)
Weigmann H. 660 (25)
Weiss D. E. 383 (20), 440 (26)
Welch L. M. 166 (13)
Weiier S. 69 (46)
Welis J. H. 660 (261
Welsh D. G. 220 (56). 605 (70)
West A. S. 515 (15)
West F. B. 220 (67), 221 (68)
Whatley M. E. 382 (21)
White A. S. 605 (71)
Whitehead К. Е. 69 (62)
Whitman W. G. 220 (47, 48)
Wicks С. Е. 605 (61)
Wiegand J. H. 69 (54), 382
(13)
Wiegandt H. F. 607 (138)
Wilcock D. F. 69 (67)
Wilhelm H. A. 440 (11, 30),
661 (48)
Wiike С R. 220 (26 27), 605
(67)
Wilkinson W. F. 659 (12)
Willard J. 68 (1)
Williams G. M. 515 (37)
Williams J. A. 607 (133)
Williams R. J. P. 66! (46)
Williams W. H. 660 (30)
Wilson P. J. 660 (26)
Winkler С. А. 68 (20)
Winson P. A. 69 (66)
Winter E. E. 440 (22), 515
(54), 516 (55)
Withrow J. R. 606 (92)
Wohl K. 138 (12>
Wood H. S. 516 (89)
Woodburn H. W. 138 (29)
Woodfield F. W. 607 (144, 156)
Woodle H. A. 516 (72)
Woods W. K. 516 (83)
Woodward T. 605 (56)
Work L. T. 604 (30)
Yamaguchi I. 515 (46)
Yamamoto K. 515 (29)
Yokoyama T. 515(11.20), 605
(121)
Yoshloka N. 515 (20)
Zebroski E. L. 440 (17). 517
(100)
Zenz F. A. 604 (41)
Ziolkowski Z. 606 (115)
Zuiderweg F. J. 515 (34)
P. ТРЕЙБАЛ
ЖИДКОСТНАЯ ЭКСТРАКЦИЯ
M.t Издательство „Химия", 1966 г.
724 с. УДК 66.061.5.
Редактор И. И. Ратманскнй
Техн. редактор В. В. Ксган
Художник Ю. И. Снгов
Корректоры: А. А. Иалкнна, И. С. Хрнпунсва
Подписано к печати 28/VII 1966 г. Формат 60x907,».
Бум. л. 22,625. Печ. л. 45,25. Уч.-нзд. л. 44,94.
Тираж 5500 экз. Цена 3 р. 48 к. Зак. 169.
Типогр. бум. № 1. Ки.-торг. инд. 3-14-2. Темплан 1966 г. № 128.
Ленинградская типография № 2 имени Евгении Соколовой
Главполиграфпрома Комитета по печати при Совете
Министров СССР. Измайловский проспект, 29.
ОПЕЧАТКИ
Стр.
208
292
335
350
351
534
541
558
559
601
609
Строка
20 сверху
13 »
23 »
19 »
5 »
Рис. 266
6 снизу
18 сверху
Рис. 278 (подрис.
подпись, последняя
строка)
Рис. 279 (подрис.
подпись)
17 сверху
16 снизу
Напечатано
Капли
Е'
со стороны 5
*;
R[
Должно быть
Крупные капли
£.
со стороны вершины S
Rv
Ry
Направление всех стрелок изменить
на противоположное
"«>= W
Ре'с
Vc = 81
(d —диаметр)
Ка
в большинстве
можно
^D=W
Ре'
Vc = 8,l
(d— диаметр колонны)
Ка
в большинстве случаен
можно
ч
3jki3 169. Р. Трейба.1