/






































































Теги: химия химическая промышленность химические реакции
Год: 1962




Текст
ГОСУДАРСТВЕННЫЙ КОМИТЕТ СОВЕТА МИНИСТРОВ СССР
ПО ХИМИИ
МЕТОДЫ ПОЛУЧЕНИЯ
ХИМИЧЕСКИХ РЕАКТИВОВ
И ПРЕПАРАТОВ
Выпуск 4—5
ВСЕСОЮЗНЫЙ НАУЧНО-ИССЛЕДОВАТЕЛЬСКИЙ ИНСТИТУТ
ХИМИЧЕСКИХ РЕАКТИВОВ И ОСОБО ЧИСТЫХ ХИМИЧЕСКИХ ВЕЩЕСТВ
Москва — 1962
СОДЕРЖАНИЕ
Гуанидин азотнокислый. Д. Ш. Розина, Р. Л. Глобус, Т. Н. Гене-
ралова ..................................... 5
Гуанидин углекислый. В. Г. Брудзъ, Р. Л. Глобус, В. А. Иоффе,
Л. И. Грачева .........• ................... 8
Аминогуанидинбикарбоиат. Р. Л. Глобус, Р. П. Ластовский,
Д. Ш. Розина, Т. Н. Генералова ............... 11
Гуанидин роданистый. Д. Ш. Розина, Л. П. Владимирова ... 15
Гуанидин сернокислый. В. Г. Брудзъ, Р. Л. Глобус, В. А. Иоффе,
Л. И. Грачева ...................... 17
Гуанидин уксуснокислый. В. Г. Брудзъ, Р. Л. Глобус, В. А. Иоффе 18.
Гуанидин малоиовокислый кислый. В. Г. Брудзъ, Д. Ш. Розина,
Л. Т. Нестеренко .............. ............. 20
Гуанидин-алюминий сульфат. В. Г. Брудзъ, Д. Ш. Розина, Л. Т. Не-
стеренко .......................... 21
Характеристика солей гуанидина (см. таблицу-вклейку)
Дициандиамидин сернокислый. В. Г. Брудзъ, Р. Л. Глобус,
В. А. Иоффе...............'................... 23
Дициандиамидии углекислый. В. Г. Брудзъ, В. А. Иоффе, Л. И. Гра-
чева ............................ 24
Дициандиамидин двууглекислый. В. Г. Брудзъ, В. А. Иоффе,
Л. И. Грачева ........................ 26
Цианамид свинца. В. Г. Брудзъ, Р.Л. Глобус, Л. И. Грачева ... 27
Фталат свинца. В. Г. Брудзъ, Д. Ш. Розина, Л. Т. Нестеренко 30
Гексаметилендиамин уксуснокислый. В. Я. Темкина, Р. П. Ластов-
_ ский, В. Г. Брудзъ ..................... 32
Бензиламин. И. Г. Матвеев. Д. Ш. Розина ......... 33
Бензиламиноэтанол. В. Г. Брудзъ, Д. А. Драпкина.. 37
Трифеииларсип. Н. Е. Кожевникова, О. В. Иванов .. 40
Диизонитрозоацетон. Н. Е. Кожевникова, И. Д. Сапожкова 42
З-Метилпиридин. Ю. И. Чумаков ................ 44
4-Метилпиридии. К). И. Чумаков .................. 50
2,6-Диметилпиридин. Ю. И. Чумаков................ 55
N-окись пиридина. Ю. И. Чумаков ............... 59
2-Оксипиридин. Ю. И. Чумаков, 3. М. Корсакова.... 62
2-Пиридилацетат. Ю. И. Чумаков, 3. М. Корсакова.. 65
8-(п-Толуолсульфоииламино)-хинолин. В. М. Дзиомко, И. А. Краса-
вин ............................. 67
8-(Бензолсульфониламино)-хииолин. И. А. Красавин, В. М. Дзиомко 69
5-Фтор-8-меркаптохинолин и его соли. Ю. А. Банковский, 3. В. Ми-
суловина, А. Ф. Иевиньш, М. Р. Бука .......... 71
5-Хлор-8-меркаптохинолин и его соли. Ю А. Банковский, 3. В. Ма-
еуловина. Я. А. Цируле, А, Ф. Иевиньш......... 79
3
Азулен Ф, Н. Степанов, Н. А. Алданова, А. Г. Юрченко,
Н. Л. Довгань .............................................. 86
о-Сул>фобензойной кислоты дикалиевая соль. Д. Ш. Розана,
Л. Т. Нестеренко............................................ 92
о-Су льфобензойнон кислоты дихлорангидриды. Д. Ш. Розина,
Л. Т. Нестеренко.......................................... 94
о-Нитрофениларсоновая кислота. В. И. Кузнецов, Д. Ш, Розина 97
о-Аминофениларсоновая кислота. В. И. Кузнецов, Д. Ш. Розина 100
Азотол 2,4 МК. Д. Ш Розина, В. М. Дзиомко, Р. И. Розенберг 103
2-Оксибензол-(1-азо-Г)-2'-окси-3'-(2". 4"-диметилкарбоксианилидо)-
нафталин В. М. Дзиомко, Д. Ш. Розина, Р. И. Розенберг . . . 106
6-Аминотимол солянокислый. Д. Ш, Розина, Р. Г. Снятковская 109
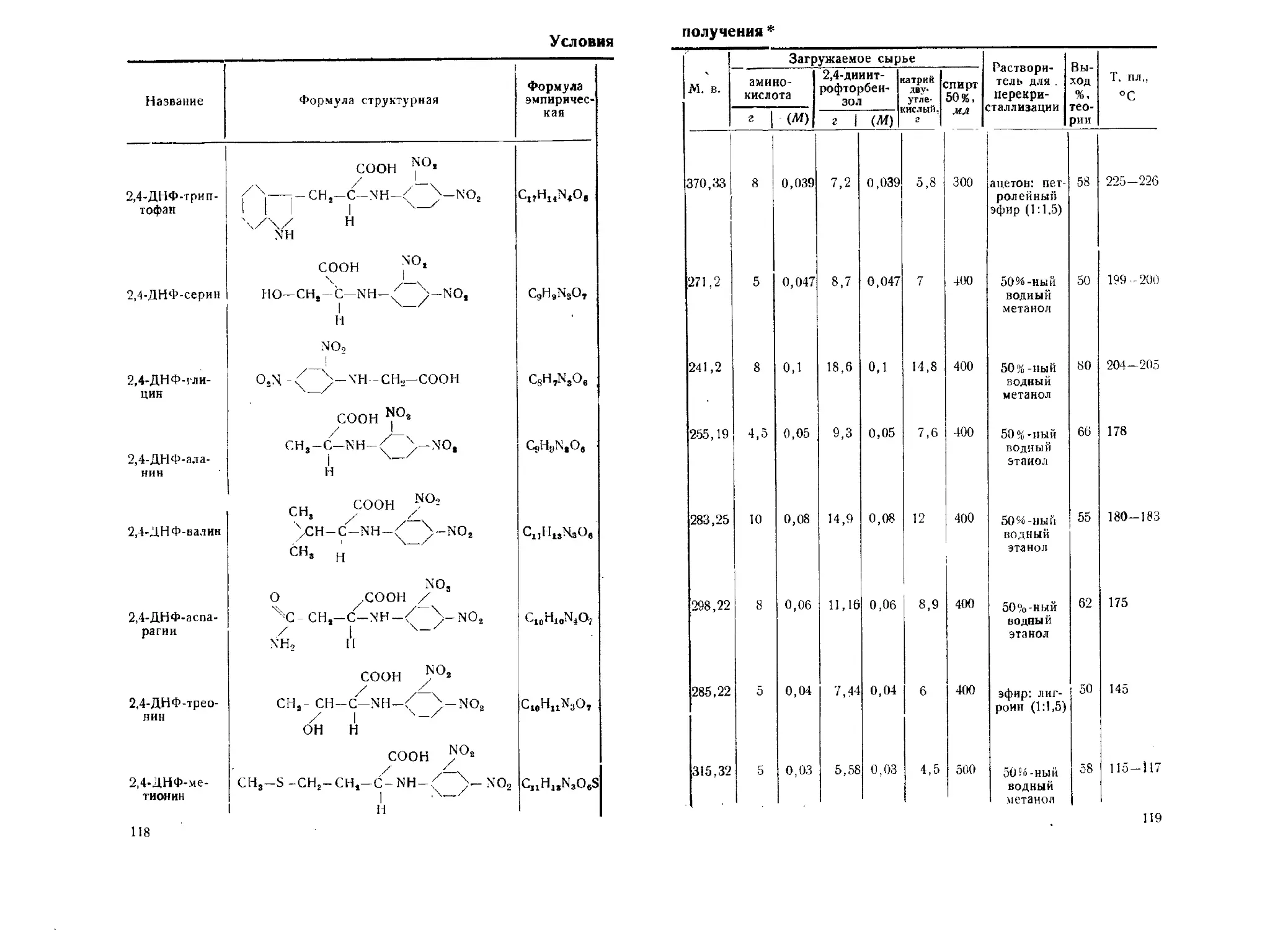
2,4-Динитрофенильные производные аминокислот. Г. Н. Кошелева,
Г. Н. Налецкая • 113
4-Метокси-4'-аминобифенил. Л. М. Литвиненко, Л. А. Перельман,
М. М. Литвиненко ............................•........... 128
4-Амино-4'-хлорбифенил. Л. М. Литвиненко, В. М. Зикранец . , . 132
2-Бром-4-нитроанилин. Л. М. Литвиненко, В. М. Зикранец .... 134
4,4'-Дитолил. Л. М. Литвиненко, Л. А. Перельман. В. М. Зикранец 135
4,4'-Дифенилдикарбоиовая кислота. Л. М. Литвиненко, Л. А. Пе-
рельман ........ • . . . ...........•............ 137
4,4'-Диннтродифенилдисульфид. Л. М. Литвиненко, Р. С. Чешко,
Л. А. Перельман.........................................• • 139
ГУАНИДИН АЗОТНОКИСЛЫЙ
(Азотнокислая соль имида мочевины)
Д. Ш. РОЗИНА, Р. Л. ГЛОБУС, Т. Н. ГЕНЕРАЛОВА
/NH2
C = NH-HNOS
XNH2
CH8O3N4 M. в. 122,09
Синтез азотнокислого гуанидина был впервые осуществлен
взаимодействием роданистого аммония с аммонием азотно-
кислым [1]. Имеется указание, что в процессе нагревания ком-
понентов произошел сильный взрыв [2]. Позднее практическое
значение приобрел способ получения азотнокислого гуанидина
сухим сплавлением дициандиамида с азотнокислым аммонием
при температуре 120—160° [3, 4, 5]. Реакция экзотермичпа, и
температура самопроизвольно повышается до 190° [6]. При
этой температуре уже возможен взрыв азотнокислого аммония
Опасность взрыва возрастает, когда азотнокислый аммоний
находится совместно с азотнокислым гуанидином и дициан-
диамидом.
Применение цианамида кальция позволяет снизить темпе-
ратуру реакции до 100° благодаря образованию аммиаката
NH4NO3 • NH3 (раствор Дивера), который резко снижает тем-
пературу плавления реакционной массы (аммиак выделяется
при реакции). Имеется указание о необходимости равномер-
ного нагревания, так как местные перегревы могут привести
к взрыву [7]. По более поздним данным, азотнокислый гуани-
дин может быть получен взаимодействием мочевины с азотно-
кислым аммонием в присутствии силикагеля (без давления)
при температуре 190—195°, с выходом 40% теории. Получен-
ный азотнокислый гуанидин не охарактеризован [8].
В основу нашего исследования был положен метод получе-
ния азотнокислого гуанидина сплавлением цианамида каль-
ция с азотнокислым аммонием.
Существенные изменения, внесенные в технологию синте-
за, обеспечили полную безопасность процесса [9]. По разрабо-
танному нами способу, цианамид кальция постепенно вносят
в предварительно полученный жидкий плав, состоящий из од-
ной трети цианамида кальция, предназначенного для загруз-
ки, азотнокислого аммония и небольшого количества воды.
Такой способ проведения синтеза позволяет нагревать реакци-
онную смесь в гомогенной среде при непрерывном перемеши-
вании, что практически исключает местные перегревы и обес-
печивает безопасность процесса. Безопасность метода под
тверждена в производственной практике.
СИНТЕЗ АЗОТНОКИСЛОГО ГУАНИДИНА
C=N
I +2NH4NOs —> C=N
N=Ca | 4-Ca(NO3)a-f-2NH3
NH8
C=N / NH2
I +NH4NO3 —> C=NH HNO3
nh2 \nh„
Суммарно:
C-N
/NH2
+3NH4NOs---->C=NH-HNO3+Ca(NO3)2+2NH.,
4NH2
Характеристика основного сырья
Цианамид кальция, технический, I сорт, ГОСТ 1780—56.
Аммиачная селитра, сорт А, ГОСТ 2—57.
Условия получения
В реактор из спецстали ЭЯ1Т емкостью 1 л (см. примеча-
ние 1), снабженный якорной мешалкой и трубкой для отвода
аммиака, вливают'53 мл воды и при температуре 95° (нагре-
вание на водяной бане) вносят в течение 10—15 минут предва-
рительно приготовленную смесь: 532 г (5,16 А1) аммиачной се-
литры и 72 г (0,5 Л1) цианамида кальция, содержащего 56%
CaCNa. Реакционную смесь нагревают 10—15 минут при 95° до
расплавления, прибавляют 0,5 мл подсолнечного масла (см.
примечание 2), перемешивают 0,5 часа и при указанной темпе-
ратуре добавляют в течение 2 часов еще 144 г (1 М) циана-
мида кальция, продолжают перемешивание еще 1 час, поддер-
6
живая температуру 90—95°, и приливают тонкой стружкой
300 мл горячей воды (90—95°).
Реакционную смесь переносят в колбу емкостью 1,5 л (см-
примечание 3), добавляют 600 мл горячей воды, 5 г активиро-
ванного угля и перемешивают 15 минут при температуре 93—
95°.
Полученный раствор азотнокислого гуанидина отфильтро-
вывают, шлам промывают три раза по 50 мл горячей (95°) во-
ды. Первую промывную воду присоединяют к основному филь-
трату (см. примечание 4). Фильтрат (~1100 мл) оставляют
па два часа при комнатной температуре, затем охлаждают в
ледяной воде до 2—3° и для более полной кристаллизации вы-
держивают при этой температуре 1 час. Выкристаллизовав-
шийся в вице белых игл азотнокислый гуанидин отфильтровы-
вают, возможно тщательнее отжимают (см. примечание 5) и
сушат при температуре 80—90° (не выше).
Выход 128—135 г азотнокислого гуанидина, содержащего
90—92% основного вещества (см. примечание 6), что состав-
ляет 65—68% теории, считая на цианамид кальция, т. пл.
205—206°, плавится в пределах не более Г. (По литератур-
ным данным, т. пл. 215° [10]).
Азотнокислый гуанидин содержит ~6% азотнокислого
кальция, который снижает его температуру плавления 11см-
примечание 7).
Перекристаллизация из воды (1:1) повышает содержание
основного вещества до 96—97%. Перекристаллизованного
азотнокислого гуанидина получают 80% от исходного.
Т. пл. 211—212°:
Примечания.
1. Синтез можно проводить в стеклянной трехгорлой колбе.
2. Нагревание реакционной смеси сопровождается довольно сильным
вспениванием; из испытанных средств, сбивающих пену, эффективным ока-
залось только подсолнечное масло.
3. Реакционную смесь приходится переносить в другой сосуд, так как
из-за маленького первоначального объема жидкости нецелесообразно уве-
личивать емкость реактора.
4. Промывные воды второй и третьей промывки используются в следую-
щей операции вместо воды для разбавления плава.
5. Влажный продукт содержит всего ~ 10% влаги. От степени отжима
в значительной мере зависит содержание основного вещества в азотнокис-
лом гуанидине.
6. Определение основного вещества в азотнокислом гуанидине. Навеску
около 0,25 г растворяют при нагревании в 25 лм дистиллированной воды.
К полученному раствору добавляют нагретый до 100° раствор из 1 г пикри-
новой кислоты в 100 мл дистиллированной воды. Массу периодически пере
мешивают в течение 1 часа, а затем оставляют в покое до следующего дня.
Выделившийся пикрат гуанидина охлаждают во льду в течение 2 часов и
отфильтровывают в тигель с пористым диом, доведенный до постоянного
веса при 100°. Остатки пикрата переносят в тигель при помощи маточного
раствора. Пикрат промывают два раза по 10 мл ледяной воды. Тигель с
пикратом высушивают при 100° до постоянного веса.
7
Содержание гуанидина азотнокислого вычисляют по формуле:
а-122,04-100
«288,16
где а—вес пикрата, г;
«-навеска, г;
122,09—мол. вес гуанидина азотнокислого;
288,16—мол. вес гуанидина пикрата.
Метод является общим для определения содержания гуанидина в его
солях.
7. Степень чистоты получаемого азотнокислою гуанидина достаточна
для большинства целей.
ЛИТЕРАТУРА
1. J. Volhard, J. prakt. Chem., 2, 21 (1874).
2. С. SchOpf, Н Klapproth, Z. angew. Chem, 49, 23 (1936).
3. Герм. пат. 527237, 1908.
4. «Синтезы органических препаратов», сб. I, стр. 176, ИЛ, 1949.
5. «Неорганические синтезы», сб. I, стр. 95, ИЛ, 1951.
6. Л. А. Кузнецов, «Производство карбида кальция», стр. 342, Гос-
химиздат (1950).
7. Н. Qockel, Z. angew. Chem., 47, 555 (1934).
8. С. И. Казарновский, Р. И. Спасская, Ж. прикл. химии, 34, 2079
(1961).
9. Д. Ш. Розина, Р. Л. Глобус, Р. П. Ластовский, А. С. Цейтленок,
П. А. Воронин и др., Авт. свид. СССР 106838, 1957.
10. Т. L. Davis, J Amer. Chem. Soc., 43, 2234 (1921).
Поступила в феврале 1962 г. ИРЕ А
ГУАНИДИН УГЛЕКИСЛЫЙ
(Углекислая соль имида мочевины)
В. Г. БРУДЗЬ, Р. Л. ГЛОБУС, В. А. ИОФФЕ, Л. И. ГРАЧЕВА
/NH2\
C=NH I • Н2СОЯ
CgHjs O3N6
М. в. 180,18
Углекислый гуанидин может быть получен из дицианди-
амида и аммиака под давлением [1], а также взаимодействием
дициандиамида с серной кислотой [2] с последующим гидроли-
зом образующегося дициандиамидина в токе углекислого га-
за [3].
Второй способ [2], технологически более простой, был поло-
жен в основу синтеза углекислого гуанидина. В условия полу-
чения [2, 3] нами были внесены изменения. Многократное упа-
ривание растворов и дробную кристаллизацию удалось заме-
нить однократным упариванием с последующим использовани-
ем маточного раствора. Исключено применение углекислого
газа в процессе гидролиза дициандиамидина, а также при
упаривании раствора углекислого гуанидина (углекислый газ
применяется только для нейтрализации избыточной щелочно-
сти); выход повышен с 68 до 80% теории.
СИНТЕЗ УГЛЕКИСЛОГО ГУАНИДИНА
/ х NHa \ / х NH2 \
21 C=NH +H2SO44-2H2O -> I C=NH IH2SO4
\ XNHCN/ \ \nHCONH2A
/ / NHa \
j C=NH I • H2SO4+Ca(OH)3------>
\ \ NHCONHjA
xNH2
-----> 2 C = NH +CaSO4 + 2H2O
\ NHCONH3
/NH„
2 C=NH +4H2O---------->
x NHCONH2
/ / NHa \
----> I C=NH i -H2CO3 + (NH4)2CO3
\ \ nh2 /2
Характеристика основного сырья
Дициандиамид, I сорт, ГОСТ 6988—54.
Известь гашеная, содержание Са(ОН)2 не менее 60%.
Условия получения
Получение сернокислого дициандиамидина. В трехгорлую
колбу, снабженную мешалкой и термометром, вносят 1,5 л во-
ды и при температуре 57—58° и перемешивании 307 г (3,35 Л1)
92%-ного дициандиамида (нагревание на водяной бане). К по-
лученному раствору добавляют тонкой струйкой в течение
15—20 минут 198 г (1,85 М) 92%-ной серной кислоты с такой
скоростью, чтобы температура не превышала 60°; по оконча-
9
нии добавления серной кислоты реакционную смесь нагрева-
ют до 95—96° и перемешивают в течение 35—45 минут, под-
держивая указанную температуру (см. примечание 1).
Получение дициандиамидина. К полученному раствору сер-
нокислого дициандиамидина, охлажденному до 20°, добавляют
в течение 10—15 минут ~ 230 г гашеной извести, содержащей
~65% Са(ОН)з, до щелочной реакции по тимолфталеину,
следя за тем, чтобы температура не превышала 40°; после че-
го реакционную смесь перемешивают еще 30 минут, периоди-
чески контролируя реакцию на щелочность, и в случае надоб-
ности добавляют известь. Полученный раствор дициандиами-
дина отфильтровывают, осадок (в основном сернокислый
кальций) промывают дважды по 150 мл горячей (~80°) воды
(см. примечание 2), присоединяя промывные воды к филь-
трату.
Получение углекислого гуанидина. Фильтрат (~1700 мл)
переносят в трехгорлую колбу, снабженную мешалкой и пря-
мым холодильником, и при перемешивании пропускают в те-
чение 30—35 минут углекислый газ до исчезновения щелочной
реакции по тимолфталеину. Нейтрализованный раствор ди-
циандиамидина нагревают на водяной бане до 98° и переме-
шивают при этой температуре в течение 4,5—5,0 часов (см.
примечание 3), совмещая нагревание с медленной отгонкой во-
ды (всего отгоняется 130—150 мл).
Образовавшийся раствор углекислого гуанидина охлажда-
ют до 10°, перемешивают при этой температуре 1 час и от-
фильтровывают от небольшого количества выделившегося нз
раствора углекислого кальция, осадок промывают 150 мл
воды.
Фильтрат вместе с промывной водой переносят в фарфоро-
вую чашку и упаривают на водяной бане до уд. в. 1,22 при
75° Упаренный раствор охлаждают до 20° и оставляют на 2
часа для кристаллизации. Выкристаллизовавшийся углекис-
лый гуанидин отфильтровывают, тщательно отжимают и су-
шат при 70—80°.
Выход 264 г с содержанием основного вещества 90,5%, что
составляет 80% теории (см. примечание 4). Маточный раст-
вор (~250 мл) присоединяют в следующей операции к раст-
вору углекислого гуанидина (см. примечание 5).
Получение чистого углекислого гуанидина. В трехгорлую
колбу, снабженную мешалкой, вливают 230 мл дистиллиро-
ванной воды и вносят при перемешивании в течение 10 минут
100 г 90%-ного углекислого гуанидина. К полученному мут-
ному раствору добавляют 10 г. активированного угля, переме-
шивают 30 минут и фильтруют. К прозрачному фильтрату при
перемешивании приливают 600 мл этилового спирта; выделив-
шийся в виде белого кристаллического осадка углекислый гуа-
10
нидин отфильтровывают, тщательно отжимают и сушат при
70°.
Получают 74 г (~80% от исходного), содержание основ-
ного вещества ~99'%.
Примечания.
1. Реакция считается законченной, когда на титрование двух контроль-
ных проб, взятых в интервале 15 минут, расходуется одинаковое количе-
ство миллилитров 0,1 и. раствора едкого натра. Для титрования берут 1 мл
реакционного раствора, который разбавляют 10 мл дистиллированной воды.
2. Конец промывки контролируют на отсутствие в промывных водах
дициандиамидина, который образует с никелем комплекс желтого цвета.
Контроль производят следующим образом:
1 г сернокислого никеля и 3 г лимонной кислоты растворяют в 15 мл
воды, добавляют 5 мл 25%-ного аммиака. 2 Л1Л полученного раствора при-
бавляют к 1 мл испытуемого раствора, в который предварительно вливают
2 мл 10%-ного едкого натра. Отсутствие желтого осадка после охлаждения
в ледяной воде указывает на конец промывки.
3. Реакция считается законченной, когда в растворе отсутствует ди-
циандиамидин (см. примечание 2),
4. Содержание основного вещества определяют пикратным методом по
ВТУ 688—52 (см. стр. 7, примечание 6).
5. Маточный раствор можно использовать 5—6 раз, после чего его
упаривают на водяной бане до густой консистенции и отфильтровывают;
осадок растворяют в воде (1,0: 1,5); полученный мутный раствор отфильт-
ровывают, а к прозрачному фильтрату добавляют двукратное количество
спирта до полного выделения углекислого гуанидина.
ЛИТЕРАТУРА
1. Т. L. Davis, J. Amer. chem. Soc., 43, 2231 (1921).
2. J. Soil, A. Stutzer, Ber., 42, 4533 (1909).
3. Герм. пат. 458437, 1928.
Поступила в феврале 19G2 г. ИРЕА
АМИНОГУАНИДИНБИКАРБОНАТ
(Гуаиилгидразии)
Р. Л. ГЛОБУС, Р. П. ЛАСТОВСКИИ, Д. Ш. РОЗИНА,
Т. Н. ГЕНЕРАЛОВА
/NH-s
С—NH -Н2СО3
xNHNH2
C2HsO3N4 М. в. 136,12
Из описанных в литературе методов получения аминогуа-
нидинбикарбоната практическое значение имеют два: восста-
новление нитрогуанидина цинковой пылью [1] и взаимодейст-
вие цианамида с гидразином или его солями с последующим
Н
превращением полученного аминогуанидина в бикарбонат
[2, 3].
Второй метод, обеспечивающий безопасность процесса и
более высокий выход, был положен в основу синтеза амино-
гуанидинбикарбоната; в качестве исходного сырья применя-
лись цианамид кальция и гидразинсульфат-
Нами установлено, что наряду с основной реакцией имеет
место полимеризация цианамида в дициандиамид. Найдены
условия (pH, температура, концентрация реагентов), подав-
ляющие реакцию полимеризации цианамида, а также усло-
вия, позволяющие почти количественно отделить дициандиа-
мид от аминогуанидинбикарбоната и получить последний в
чистом виде.
СИНТЕЗ АМИНОГУАНИДИНБИКАРБОНАТА
C=N + HsSO4 -> C=N + CaSO4
^=Са NH2
nh2
C=N + NHa + 2NH3 C = NH p (NH4)2SO4
\ I * HgSO4 \
nh2 nh2 nhnh2
nh2 nh2
C==NH + nh4hco3-» c=nh -h2co3+nh5
NHNH8 NHNH2
Характеристика основного сырья
Цианамид кальция, технический, ГОСТ 1780—56.
Гидразинсульфат, ч., ГОСТ 5841—51 (см. примечание 1).
Аммоний двууглекислый, ч., ГОСТ 3762—47.
Условия получения
Получение цианамида. В трехгорлую колбу (см. примеча-
ние 2), охлаждаемую ледяной водой и снабженную якорной
мешалкой, капельной воронкой и термометром, вливают 2,3 л
воды и при энергичном перемешивании добавляют в течение
2,0—2,5 часа 500 г (3,65 М) цианамида кальция, содержащего
20,4% связанного азота (см. примечание 3) и ~800 мл 50%-
ного раствора серной кислоты (уд. вес 1,4). Цианамид каль-
ция вносят порциями (~10 г) и одновременно добавляют из
капельной воронки тонкой струйкой серную кислоту, следя за
тем, чтобы температура не превышала 25°. Очередную пор-
12
цию цианамида кальция вносят, когда реакция становится
слабощелочной по фенолфталеиновой бумажке- По окончании
загрузки цианамида кальция продолжают прибавлять раствор
серной кислоты до исчезновения щелочной реакции по брилли-
антовой желтой бумажке и до отсутствия кислой реак-
ции по конго. Массу перемешивают 10 минут и отфильтровы-
вают от шлама (см. примечание 4); шлам по возможности
тщательнее отжимают и промывают 4 раза по 250 мл холодной
воды, присоединяя промывные воды к основному фильтрату,
затем еще 1 л воды, используя эту промывную воду вместо
воды для следующей операции. К фильтрату (2,3—2,5 л), со-
держащему цианамид, осторожно добавляют при перемешива-
нии 1%-ный раствор серной кислоты до pH 4,0—4,2 (~70 мл)
(см. примечание 5). Полученный раствор цианамида, содер-
жащий 35—37 г связанного азота в литре, упаривают на водя-
ной бане при 100—150 мм остаточного давления (темп. 70—
75°) до достижения концентрации, соответствующей 100 г свя-
занного азота в литре. Объем отогнанной воды ~ 1700 мл (см.
примечание 6). Упаренный раствор цианамида отфильтровы-
вают от небольшого количества сернокислого кальция, выде-
лившегося в процессе упаривания.
Получают 800—820 мл почти бесцветного раствора циан-
амида, содержащего 100—102 г связанного азота в литре (уд.
вес 1,022), что составляет 80—82% теории.
Получение аминогуанидина- В трехгорлую колбу (см. при-
мечание 2) емкостью 1,5 л, снабженную якорной мешалкой,
воздушным холодильником и трубкой для ввода аммиака,
вливают полученный раствор цианамида (2,9 М) и при пе-
ремешивании добавляют 177,0 г (1,35 М) 99%-ного гидразин-
сульфата. В полученную суспензию пропускают —10 минут
газообразный аммиак со скоростью 40—50 мл в 'минуту до
слабощелочной реакции по фенолфталеиновой бумажке; при
этом температура постепенно повышается до 70—75° и реак-
ционная смесь становится прозрачной. По окончании пропу-
скания аммиака возможно быстрее повышают температуру до
83—85° (нагреванием на водяной бане) и продолжают переме-
шивание в течение 5 минут; реакция должна остаться слабо-
щелочной по фенолфталеину, в противном случае снова про-
пускают аммиак. К полученному раствору аминогуанидина
добавляют 3 г активированного угля, перемешивают 10 минут
и отфильтровывают; осадок на фильтре промывают 40 мл хо-
лодной воды, присоединяя промывную воду к основному филь-
трату. Объем -~900 Д!.г (см. примечание 7).
Получение аминогуанидинбикарбоната. Раствор аминогуа
нидина переносят в колбу, добавляют 400 мл воды и нагре-
вают на водяной бане до 60°. При указанной температуре до-
бавляют при перемешивании в течение 5 минут 240 г (2 /И)
13
двууглекислого аммония, продолжают перемешивание в тече-
ние 20 минут и охлаждают до 40°. Выделившийся в виде бе-
лого кристаллического осадка аминогуанидинбикарбонат от-
фильтровывают, промывают 550—600 мл холодной воды до
отсутствия в промывных водах иона SCh2- (проба с хлористым
барием) и сушат при 35—40° (не выше) (см. примечание 8).
Получают 156—160 г амипоГуаиидинбикарбоната, содер-
жащего 97—98% основного вещества, что составляет 83—85%
теории, считая на гидразинсульфат (см. примечание 7, 9).
Найдено И: С 17,53; Н 5,73
C2H8O,N4. Вычислено %: С 17,65; Н 5,88
Маточный раствор с промывными водами используется
для получения дициандиамида (см. примечание 9).
Примечания.
1. Возможно применение гидразинсульфата технического.
2. Синтез можно проводить в реакторе из нержавеющей стали марки
ЭЯ1Т.
3. Азот определяется по ГОСТ па цианамид кальция.
4. Шлам содержит, в основном, сернокислый кальций, а также нера-
створимые в воде примеси, сопутствующие цианамиду кальция.
5. Если сразу не удается достигнуть нужного значения pH, его регу-
лируют добавлением 0,2—0,3 г цианамида кальция либо 2—3 капель серной
кислс-ы. В качестве индикатора применяется метиловый оранжевый. При
значении pH 4,0—4,2 раствор цианамида устойчив и может храниться
~ 14 дней.
6. Для правильного учета отогнанной воды приемник следует держать
погруженным в ледяную воду.
7. Аминогуанидинбнкарбонат количественно титруется нитритом нат-
рия (см. ВТУ РУ 901—53). Однако этот способ нельзя использовать для
титрования растворов, получающихся непосредственно в результате реак-
ции, так как всегда расходуется повышенное количество нитрита натрия,
по-виднмому, за счет примесей.
8. При температуре выше 35—40° аминогуанидинбикарбонат частично
разлагается и приобретает окраску от бледно-розовой до оранжевой. Целе-
сообразно, по возможности, использовать его в сыром виде.
9. Маточный раствор (~1 л) упаривают па водяной бане до уд. в.
1,23—1,24 (объем ~ 330 мл). Упаренный раствор охлаждают в ледяной во-
де в течение 3 часов. Выкристаллизовавшийся дициандиамид отфильтро-
вывают. Получают 30—32 г.
После перекристаллизации из воды (1:1) получают 25—26 г чистого
дициандиамида, т. пл. 207—208°. (Литературные данные 209,5° [4]).
ЛИТЕРАТУРА
1. J. Thiele, Liebigs Ann. Chem.. 270, 23 (1892).
2. Герм. пат. 730331, 1943.
3. Герм. пат. 689191, 1940, Chem. Abstrs., 35, 3650 (1941).
4. «Неорганические синтезы», сб. 3, стр. 41, ИЛ, 1952.
Поступила в феврале 1962 г.
И РЕ А
ГУАНИДИН РОДАНИСТЫЙ
Д. Ш. РОЗИНА, Л. П. ВЛАДИМИРОВА
XNH,
C=NH •HCNS
\ NHS
C,H,N4S.
M. в. 118,17
Практический интерес представляет способ получения ро-
данистого гуанидина взаимодействием дициандиамида с рода-
нистым аммонием. Одновременно образуется тиоаммелин (1].
Образование последнего значительно снижается, если прово-
дить реакцию под током аммиака (см. схему синтеза), что
приводит к повышению качества и выхода роданистого гуа-
нидина.,
СИНТЕЗ РОДАНИСТОГО ГУАНИДИНА
/ NH2 / nh2
C=NH + 2NH4CNS ----ч. 2C=NH -HCNS
x NHCN x NH2
Одновременно образуется тиоаммелин по схеме;
NH4CNS NH3 + HCNS.
xnh2 xn=cnh,x
C=NH + HCNS--------> HS-C N
\nhcn ^N-CNhZ
Характеристика основного сырья
Дициандиамид, технический, ГОСТ 6988—54.
Роданистый аммоний, ч., ГОСТ 3768—47.
Условия получения
В трехгорлую колбу емкостью 500 мл (см. примечание 1)
вносят предварительно приготовленную смесь из 135 г (1,5 М\
93,5%-ного дициандиамида с 236 г (3,04 Л1) 98%-ного родани-
стого аммония и постепенно нагревают на глицериновой бане
до плавления (~105°), следя за тем, чтобы разница между
температурой реакционной смеси и бани не превышала 10°. Как
только реакционная смесь расплавится, начинают пропускать
аммиак (со скоростью ~ 100 мл в минуту) и при температуре
125—130° перемешивают 4 часа. (После 2 часов нагревания
добавляют 3 г роданистого аммония). По истечении указанно-
15
го времени реакционной смеси дают охладиться до 90°, пре-
кращают подачу аммиака и, не выключая мешалки, вливают
тонкой струйкой 100 мл горячей воды (~ 90“). После 5 минут
перемешивания реакционную смесь переносят в стакан и Для
полного ее растворения добавляют еще 350 мл горячей воды.
Полученный раствор роданистого гуанидина ’упаривают в фар-
форовой чашке на водяной бане до уд. в. 1,150—1,155 при 60°,
добавляют 5 г активированного угля, охлаждают до 20° (для
выделения тиоаммелина) и отфильтровывают.
Фильтрат упаривают на водяной бане до !уд. в. 1,225—1,228
при .60° и оставляют для кристаллизации на 12 часов. Выкри-
сталлизовавшийся роданистый гуанидин отфильтровывают и
тщательно отжимают.
Получают 200—210 г роданистого гуанидина, содержащего
~10% влаги. Т. пл. НО—112° (высушена средняя проба) и
около 200 мл маточного раствора (I).
Полученный роданистый гуанидин растворяют при темпе-
ратуре 45—50° в 200 мл дистиллированной воды, раствор ох-
лаждают до 20°, оставляют на i2 часов и отфильтровывают от
небольшого количества тиоаммелина (~2 г). Фильтрат упа-
ривают на водяной бане до уд. в- 1,185—1,190 при 60°, остав-
ляют для кристаллизации на 12 часов; выкристаллизовавший-
ся роданистый гуанидин отфильтровывают, тщательно отжи-
мают и сушат при 70—75°.
Получают 140—142 г с содержанием основного вещества
97,0—98,0% (см. примечание 2), что составляет 39% теории,
считая на дициандиамид. Т. пл. 115—116°; 116—117°. (По ли-
тературным данным 118° [2]). Полученный маточный раствор
(~80 мл) вместе с маточным раствором (I) ~200 мл упари-
вают до уд. в. 1,185—1,190 при 60°. Получают дополнительно
25—30 г полноценного продукта (7—8% теории).
Общий выход 167—170 г (45—46% теории).
Примечания.
1 Колба снабжена мешалкой, термометром, трубкой для ввода и труб-
кой для отвода аммиака.
2 . Содержание основного вещества определяют по гуанидину (пикрат-
ным методом) и роданистоводородной кислоте (титрованием по Фольгар-
ду). Разница в результате — 0,2—0,3%. (см. ВТУ РУ 961—53).
ЛИТЕРА Т У Р А
1. J. S. Blair, /. М. Braham, J. Amer. Chetn. Soc., 44, 2348 (1922).
2. L. Volhard, J. Prakt. Chem. f2], 9, 15 (1874).
Поступила в феврале 1962 г. ИРЕА
ГУАНИДИН СЕРНОКИСЛЫЙ
(Сернокислая соль имида мочевины)
В. Г. БРУДЗЬ, Р. Л. ГЛОБУС, В. А ИОФФЕ, Л. И. ГРАЧЕВА
//Ж \
C=NH |-Нг5О4 0,5 Н3О
\xnh2 /2
C2HI2NeO4S-0,5H2O М. в. 225,25
По литературным данным, синтез сернокислого гуанидина
может быть осуществлен взаимодействием дициандиамида
с серной кислотой с последующим гидролизом образовавшего-
ся дициандиамидина [1].
Для получения сернокислого гуанидина могут быть исполь-
зованы также другие соли гуанидина. Наиболее целесообраз-
но исходить из гуанидина углекислого, который легко гидро-
лизуется и при взаимодействии с серной кислотой образует
сернокислый гуанидин;
/xNH2 \ //NH, \
C=NH I • H2CO3+H2SO4 -> I C=NH j H2SO4+CO2+H2O
xxh2 /2 \xnh2 A
Характеристика основного сырья
Гуанидин углекислый, технический, ВТУ 688—52.
Условия получения
В трехгорлую колбу, снабженную мешалкой и термомет-
ром, вливают 400 мл воды и при перемешивании добавляют
100 г (0,96 М) 92 %-ной серной кислоты, а затем постепенно в
течение 1 часа вносят 200 г (1 7И) 90 %-кого углекислого гуа-
нидина; реакцию доводят до нейтральной по бромтимоловому
синему (pH 7) (см. примечание 1). Реакционную смесь нагре-
вают на водяной бане до 75—80° и при этой температуре про-
должают перемешивание в течение 15 минут. Реакция должна
остаться нейтральной.
Образовавшийся раствор сернокислого гуанидина охлаж-
дают до 20° и отфильтровывают от небольшого осадка, пос-
ледний промывают 50 мл воды. Фильтрат вместе с промывной
водой (525—550 мл) упаривают в течение 3—4 часов на водя-
ной бане при периодическом перемешивании до получения
густой кристаллической массы; после охлаждения до 20°, вы-
делившийся сернокислый гуанидин отфильтровывают, тща-
тельно отжимают и сушат при температуре 35—40° (не выше).
- Зак. 623 17
Выход 208 г с содержанием основного вещества 93%, что
составляет 86% теории (см. примечания 2 и 3).
Получение чистого сернокислого гуанидина. 50 г ~93%-
ного сернокислого гуанидина растворяют при перемешивании
в 50 мл дистиллированной воды. К полученному мутному ра-
створу добавляют 2,0 г активированного угля, продолжают
перемешивание в течение 30 минут и фильтруют. К фильтра-
ту приливают при перемешивании 300 мл этилового спирта,
происходит расслоение жидкости, которую энергично переме-
шивают 3 часа- Выделившийся в виде белого кристаллическо-
го осадка сернокислый гуанидин отфильтровывают, отжимают
и сушат. Получают 40 г с содержанием основного вещества
~ 99%. (~85% от исходного).
Примечания.
1. pH регулируют добавлением либо углекислого гуанидина, либо сен-
ной кислоты. Контроль производят следующим образом: 0,1 мл раствора
разбавляют в пробирке дистиллированной водой до 2 мл. От добавления
двух капель бромтимолового синего раствор должен окраситься в зеленый
цвет.
2. Содержание основного вещества определяют пикратным методом по
ВТУ РУ 640—52 (см. стр. 7).
3. Маточный раствор (~ 75 мл) присоединяют в следующей операции
к раствору сернокислого гуанидина.
ЛИТЕРАТУРА
1. J. SOU, A. Stutter, Вег., 42, 4533 (1909).
Поступила в феврале 1962 г. ИРЕА
ГУАНИДИН УКСУСНОКИСЛЫЙ
(Уксуснокислая соль имида мочевины)
В. Г. БРУДЗЬ, Р. Л. ГЛОБУС, В. А. ИОФФЕ
xnh2
C = NH -СНзСООН
XNH,
CaH9N3O2 М. в. 119,13
Гуанидин уксуснокислый может быть получен взаимодей-
ствием водного раствора углекислого гуанидина с уксусной
кислотой [1],
18
Нами уточнены условия получения уксуснокислого гуани-
дина, а также условия его выделения из раствора:
/ /NHA
C = NHj -Н2СО3 + 2СН3СООН -
XNHa4
/NH2
-> 2 С = NH-CH9COOH + СО, 4- Н,0
xNH2
Характеристика основного сырья
Гуанидин углекислый, ч., ВТУ РУ 688—52.
Кислота уксусная, 98%-ная, ГОСТ 61—51.
Условия получения
В 160 мл дистиллированной воды растворяют 100 г
(0,55 Л1) углекислого гуанидина при температуре 50—60°. К
полученному раствору постепенно добавляют уксусную кис-
лоту до нейтральной реакции по бромтимоловому синему
(слегка зеленое окрашивание. pH 7). Расходуется около 70 г
(1,13 М) уксусной кислоты. При достижении нейтральной ре-
акции полученный раствор уксуснокислого гуанидина от-
фильтровывают от взвешенных частиц. Фильтрат упаривают
в фарфоровой чашке на водяной-бане до образования значи-
тельного количества кристаллов, охлаждают до 15° и остав-
ляют на 2—3 часа для дальнейшей кристаллизации. Выкри-
сталлизовавшийся уксуснокислый гуанидин отфильтровывают,
тщательно отжимают, промывают 10 мл холодной воды (10°)
сушат при 40—50°. Получают 48 г.
Маточный раствор вместе с промывной водой (~90 мл)
упаривают до появления кристаллов, охлаждают до 10° и
оставляют на 2—3 часа. Выкристаллизовавшийся уксуснокис-
лый гуанидин отфильтровывают, по возможности лучше от-
жимают и сушат. Получают еще 40 г.
Общий выход 88 г уксуснокислого гуанидина с содержа-
нием основного вещества 95—96%, что составляет 66% теории
1см. примечание). Второй маточный раствор утилизации
не подлежит.
.Примета пи е.
Содержание основного вещества определяется пикратным методом,
г о ВТУ РУ 2064—54 (см. стр. 7)..
ЛИТЕРАТУРА
1- .4. А. Рябанш, Ж- общ. химии. 22, 541 (1952).
Поступила в феврале 1962 г. ИГ’ЕА
ГУАНИДИН МАЛОНОВОКИСЛЫЙ кислый
(Кислая малоновокислая соль имида мочевины)
В. Г. БРУДЗЬ, Д. Ш. РОЗИНА, Л. Т НЕСТЕРЕНКО
c4h6o4n3
nh2 соон
C=NH • СН2
nh2 соон
М. в. 163,13
Способ получения и свойства гуанидина малоновокислого
в литературе не описаны. Нами он получен по аналогии с не-
которыми другими солями гуанидина (см. стр. 19) взаимо-
действием углекислого гуанидина с малоновой кислотой.
NH2
C=NH
\н2
СООН
•Н2С0з + 2СН2
СООН
nh2 соон
-*2C=NH-CH2 +С02 + Н20.
\н, 'соон
Характеристика основного сырья
Гуанидин углекислый, ч„ ВТУ РУ 688—52.
Малоновая кислота, ч., ВТУ РУ 418—51.
Условия получения
В трехгорлой колбе, помещенной в водяную баню, раство-
ряют при перемешивании 182 г (1 А4) 99%-ного углекислого
гуанидина в 560 мл дистиллированной воды при 55—60°. В
полученный раствор при указанной температуре вносят в тече-
ние 1 часа 208—210 г (2,02 Af) малоновой кислоты до слабо-
кислой реакции по конго и продолжают при той же темпера-
туре перемешивать 1 час. Реакция должна остаться слабокис-
лой, в противном случае добавляют малоновую кислоту. К об-
разовавшемуся раствору малоновокислого гуанидина добав-
ляют 5 г активированного угля, перемешивают 15 минут и от-
фильтровывают. Фильтрат охлаждают до 20° и оставляют для
кристаллизации на 5 часов. Выкристаллизовавшийся в виде
20
длинных белых игл малоповокислый гуанидин отфильтровы-
вают и тщательно отжимают (получают влажного 150—160 г).
Маточный раствор (650—670 мл) переносят частями, по ме-
ре упаривания, в фарфоровую чашку емкостью ~200 мл и
упаривают на водяной бане до уд. в. 1,190—1,195 при 70° (см.
примечание 1); упаренному раствору дают охладиться до 15—
20° и оставляют на 5 часов для кристаллизации. Выделивший-
ся малоновокислый гуанидин отфильтровывают, хорошо от-
жимают (получают 145—150 г), присоединяют к полученно-
му из основного фильтрата и сушат при 90—95° до постоянно-
го веса.
Выход 239—247 г с содержанием основного вещества
99,2—99,8% (см. примечание 2), что составляет 73—76% тео-
рии. Т. пл. 150,3—150,8° Нами установлено, что температура
плавления малоновокислого гуанидина 151,0—151,5° (см. при-
мечание 3).
Примечания.
1. Удельный вес раствора определяют денсиметром в цилиндре, погру-
женном в горячую воду.
2. Содержание основного вещества определяют пикратным методом
(см. стр. 7).
Вторично полученный маточный раствор (~ 140 мл) упаривают на во
дяной бане до появления пленки. После охлаждения до 20° выделившийся
кристаллический осадок отфильтровывают, отжимают и перекристаллизо-
вывают из спирта (1:7) в присутствии угля. Получают еще 20—22 г
(~6% теории) полноценного продукта.
3. Температура плавления малоновокпелого гуанидина после перекри-
сталлизации из спирта 151,0—151,5"; последующие две перекристаллизации
не изменили ее.
Поступила в феврале /962 г. ИРЕА
ГУАНИДИН-АЛЮМИНИИ СУЛЬФАТ
. В. Г. БРУДЗЬ, Д. Ш. РОЗИНА, Л. Т. НЕСТЕРЕНКО
(nh2 \
C=NH -HA1(SO4)2-6H2O
\’Н2 /
CH6O8N3S2A1.6H2O М. в. 387,29
Гуанидин-алюминий сульфат получен взаимодействием
сернокислого гуанидина с сернокислым алюминием [1]. Нами
воспроизведена литературная пропись и уточнены условия вы-
деления препарата.
21
/ nh2 \
I C=NH -HsSO4-0,5HsO+Al2(SOt)3.18H2O -*
\ nh2 Л
nh2
-> 2C = NH • HAI(SO4)s.6H2O
XNH3
Характеристика основного сырья
Гуанидин сернокислый, ВТУ РУ 540—52.
Алюминий сернокислый, ГОСТ 3758—47.
Условия получения
В трехгорлую колбу, снабженную мешалкой, вливают
920 мл воды и при перемешивании добавляют 90 г (0,4 М)
тщательно растертого сернокислого гуанидина, В полученный
раствор вносят в течение 3—5 минут 266,5 г (0,4 М) сернокис-
лого алюминия, также тщательно растертого, и продолжают
перемешивание 5 минут. К образовавшемуся раствору гуани-
дин-алюминий сульфата добавляют 5 г активированного угля,
перемешивают 15 минут и отфильтровывают. Фильтрат упа-
ривают в фарфоровой чашке на водяной бане до уд. в. 1,28—
1,29 при 75°, упаренному раствору дают охладиться до 20° и
оставляют на 4 часа для кристаллизации. Выкристаллизовав-
шийся гуанидин-алюминий сульфат отфильтровывают и тща-
тельно отжимают. Получают 144—148 г (влажного).
Маточный раствор (260—270 мл) после обработки активи-
рованным углем (2 г) упаривают снова в условиях, принятых
для основного раствора. Получают 94—98 г влажного. Гуани-
дин-алюминий сульфат, полученный из маточного раствора,
перемешивают с полученным из основного раствора и сушат
при 90—95° до постоянного веса.
Выход 228—232 г (73—75% теории).
Найдено %: Alj(SO4)3 49,3
CH4O8N,S»A1-6H,O. Вычислено %: Al^SOj), 49,6.
Л ИТ ЕРАЗ УРА
1. И. С. Рез, Л. А. Варфоломеева, Рост кристаллов, сб. 2, стр. 126, Из.1.
АН СССР (1959).
Поступила в феврале 1962 г. ИРЕА
ДИЦИАНДИАМИДИН СЕРНОКИСЛЫЙ
В. Г. БРУДЗЬ, Р. Л. ГЛОБУС, В. А. ИОФФЕ
f / NHa \
C=NH ]-H2SOr2H2O
\ \ nhconh2/2
C4HJ8O8N8S . м. в. 338,27
. Дициандиамидин сернокислый получают взаимодействием
дициандиамида с серной кислотой [1]. Нами проверены и уточ-
нены литературные данные.
СИНТЕЗ СЕРНОКИСЛОГО ДИЦИАНДИАМИДИНА
/ nh2
2 C=NH + H2SO4 + 4Н2О
4 NHCN
/ /NH, \
IC=NH j-H8SOr2H2O
\ 4 NHCONHa/2
Характеристика основного сырья
Дициандиамид, I сорт, технический, ГОСТ 6988—54.
Условия получения
В трехгорлую колбу, снабженную мешалкой, вливают
540 мл воды и постепенно, при перемешивании, 180 г (1,7 М)
92%-нон серной кислоты. К раствор5г серной кислоты добав-
ляют в течение 10—15 минут 300 г (3,3 М) 93%-ного дициан-
диамида. Реакционную смесь разбавляют водой до объема
1.5 л и нагревают на водяной бане до исчезновения осадка.
Образовавшийся раствор сернокислого дициандиамидина
отфильтровывают от незначительного количества взвешенных
примесей, охлаждают до 20° и оставляют на 2 часа для кри-
сталлизации- Выкристаллизовавшийся серпокислый дициап-
Диамидин отфильтровывают, хорошо отжимают, промывают
Дважды, расходуя по 50 мл холодной воды, и сушат при 70—
^0°. Получают 360 г.
Маточный раствор упаривают на водяной бане до образо-
вания густой кристаллической массы (примерно до половины
объема), охлаждают до 20°. Выделившийся кристаллический
осадок отфильтровывают и растворяют в 800 мл воды; к ра-
створу добавляют 4 г активированного угля, перемешивают
Ю—15 минут и отфильтровывают от угля.
23
Полученный прозрачный, бесцветный раствор сернокислого
дициандиамидина упаривают на водяной бане до образования
густой кристаллической массы и охлаждают до 20°. Сернокис-
лый дициандиамидин отфильтровывают, промывают дважды
по 25 мл воды и сушат. Получают 154 г.
Общий выход 514 г (90% теории).
ЛИТЕРАТУРА
1. J. Soil, A. Stutzer, Вег., 42, 4533 (1909).
Поступила в феврале 1962 г.
ИРЕА
ДИЦИАНДИАМИДИН УГЛЕКИСЛЫЙ
В. Г. БРУДЗЬ, В. А. ИОФФЕ, Л. И. ГРАЧЕВА
/ х NH, \
C=NH I -Н2СО3
\ \ NHCONH/S
C6HuObN8 М. В. 266,23
Согласно литературным данным, углекислый дициандиами-
дин может быть получен только в виде водного раствора при
взаимодействии сернокислого дицианлиамидина с углекислым
барием fl, 2].
Нам удалось выделить углекислый дициандиамидип в кри-
сталлическом виде.
СИНТЕЗ УГЛЕКИСЛОГО ДИЦИАНДИАМИДИНА
//Nil, \
C=NH -H2SO4-2H2O + ВаСО3--------->
\ х NHCONH J2
/ / NH2 \
----Jc=NH -Н2СО3 + BaSO4 + 2H2O
\ x'NHCONH,/2
z NH„ \ / NH2
C==NH .H3CO3+CO5+H4O ->2C=NH HaCOf
x nhconhA xNHCONH2
24
/NHa
2C=NH -H2CO3-----------*
XNHCONH,
/ / NH2 \
-> C=NH -H2CO3+CO24-H2O
\ 4 nhconh2/2
Характеристика основного сырья
Дициандиамидин сернокислый, содержание дициандиами-
дина 59,0—61,0% (см. примечание !)•
Барий, углекислый, ч., ГОСТ 4158—48.
Условия получения
В трехгорлую колбу, снабженную мешалкой, вливают
400 мл воды, при перемешивании добавляют 76 г (0,22 М) сер-
нокислого дициандиамидина и 58,8 г (0,21 Л1) углекислого ба-
рия. Реакционную смесь продолжают перемешивать в течение
3 часов. Полученный раствор углекислого дициандиамидина
отфильтровывают от образовавшегося сернокислого бария, ко-
торый после тщательного отжима промывают дважды по
50 мл воды. С целью очистки фильтрат вместе с промывными
водами переносят в колбу и пропускают в течение 4 часов
сильный ток углекислого Газа. Образовавшийся двууглекислый
дициандиамидин отфильтровывают, тщательно отжимают и
промывают 50 мл воды, а затем (отдельно) дважды по 25 мл
спирта (см. примечание 2).
В маточный раствор вместе с промывной водой пропускают
углекислый газ в течение 2 часов. Выделившийся дициан-
тиамидин двууглекислый отфильтровывают, возможно лучше
отжимают, промывают 25 мл воды и дважды по 15 мл спирта
п присоединяют к полученному из основного раствора. (Всего
получают ~50 г).
В трехгорлую колбу, снабженную обратным холодильни-
ком, мешалкой и термометром, вливают 80 мл абсолютного
спирта и при перемешивании вносят полученный двууглекис-
лый дициандиамидин. Реакционную смесь нагревают медлен-
но на водяной бане; при 50° начинается выделение углекисло-
го газа, которое заканчивается при температуре кипения спир-
та, при этом осадок в колбе растворяется. Образовавшийся
прозрачный раствор углекислого дициандиамидина охлажда-
ют ледяной водой до 2—3°. При энергичном встряхивании вы-
деляется кристаллический осадок углекислого дициандиами-
Дина, который отфильтровывают и сушат в эксикаторе до по-
стоянного веса.
25
Выход 18 г, что составляет ~31% теории (см. примеча-
ние 3).
Найдено %: С 23,38; Н 5,58; N 44,45
CBHuO»Ng. Вычислено %: С 22,56; Н 5,20; N 42,10.
Найдено %: дицианднамидина 77,48
CsHiiOjjNg. Вычислено И: дицианднамидина 76,70.
Примечания.,
1. Дициандиамидам образует с никелем вну'рикомп.чексиую соль, мера
створимую в воде. На этом свойстве основано его'количественное опреде
ление.
2. Полученный двууглекислый дициандиамндин хранят в эксикаторе до
получения 2-ой порции из маточного раствора.
3. В маточный раствор пропускают ток углекислого газа в течение 1 ча-
са. Выделяется 9—10 г двууглекислого дициандиамидина. Хранить угле-
кислый дициаидиамидин следует в герметичной стеклянной таре, так как ои
жадно поглощает углекислый газ и переходит в двууглекислый.
ЛИТЕРАТУРА
1. J. Haag, Liebigs Ann. Chem. 122, 25 (1862).
2. E. Baumann, Ber., 7, 1766 (1874).
Поступала в феврале 1962 г.
ИРЕА
ДИЦИАНДИАМИДИН ДВУУГЛЕКИСЛЫЙ
В. Г. БРУДЗЬ, В. А. ИОФФЕ, Л. И. ГРАЧЕВА
/NH3
C=NH -Н3СО3
NIICONH,
C3H8O4N4
М. в. 164,12
Дициандиамидин двууглекислый получают пропусканием
углекислого газа в раствор углекислого дицианднамидина
(см. стр. 25).
/xnh2 \ /NH2
C = NH .H2COS+CO2+H,O 2C = NH -Н2СО8
\ xNHCONH2 /2 XNHCONH.,
Условия получения
В прозрачный раствор углекислого дициандиамидина _(см
стр. 25), охлажденный до комнатной температуры, пропуска-
26
ют ток углекислого газа в течение 3 часов. Образовавшийся
0 виде белого кристаллического осадка двууглекислый ди-
циандиамидин отфильтровывают, отжимают, промывают
50 мл воды, два раза по 25 мл спирта и сушат в эксикаторе до
постоянного веса.
В маточный раствор, к которому присоединена промывная
вода, пропускают в течение 2 часов ток углекислого газа. Вы-
делившийся двууглекислый дициандиамидин отфильтровыва-
ют, отжимают, промывают 25 мл воды и два раза по 15 мл
спирта и сушат-
Общий выход 40 г, что составляет 55% теории.
Найдено %: С 24,1; Н 4,65; N 33,00
CgHgOjNj. Вычислено %: С 25,5; Н .4,30; N 34,35
Найдено %: дицианднамидина 77,48.
C3H8O4N4. Вычислено %; дицианднамидина 76,60.
Поступила в феврале 1962 г.
ИРЕА
ЦИАНАМИД СВИНЦА
В. Г. БРУДЗЬ, Р. Л. ГЛОБУС. Л. И. ГРАЧЕВА
Pb = N—C-N
PbCN» М. в. 247,24
По имеющимся в литературе данным, цианамид свинца
получают из технического цианамида кальция [2, 3, 4, 5, 6] или
из раствора цианамида кальция при осаждении его уксусно-
кислым свинцом [1, 7]. Имеются также указания на возмож-
ность получения цианамида свинца сплавлением солей РЬСЬ
или РЬВга с NaCN при температуре ниже точки плавления ис-
ходных галоидных солей свинца [8]. В указанных литератур-
ных источниках по синтезу цианамида свинца отсутствуют
ганные, определяющие проведение процесса: нет указаний на
pH раствора при осаждении цианамида свинца, не определял-
ся насыпной вес получаемого продукта (кроме [8]).
Проведенная нами работа показала, что наиболее целесо-
образным методом получения цианамида свинца является
взаимодействие растворов азотнокислого свинца с водными
растворами цианамида или его солей, получаемых из циана
чпда кальция [9]. Другие способы, описанные в литературе,
имеют только препаративное значение.
Кроме того, нами установлено, что растворимость цианами-
да свинца в воде составляет 0,1 г/л при 20° и что наиболее
2?
благоприятными условиями для получения цианамида свинца
с наименьшим насыпным весом (0,44—0,6 г/см3) при взаимо-
действии 5—10%-ных растворов цианамида с азотнокислым
свинцом являются: проведение гидролиза цианамида кальция
водой в соотношении компонентов 1 :5; избыток кислой соли
цианамида кальция от теоретического ~10%; осаждение ци-
анамида свинца из реакционного раствора при pH 7,6—8,0.
СИНТЕЗ ЦИАНАМИДА СВИНЦА
2CaCNa-f-2HaO -> Ca(HCNa)2-|-Ca (QH)S
Ca(HCNa)2 + 2Pb (NO3)a + 2NHs ->
-> 2PbCNa+Ca (NOs)2+2NH4NOs
Характеристика основного сырья
Цианамид кальция, I сорт, ГОСТ 1780—51 (азота не менее
19%).
Азотнокислый свинец, ч., ГОСТ 4236—48 (азота не менее
98,5%; можно работать и на техническом продукте).
Аммиачная вода, I сорт, ГОСТ 9—40 (25% NH3).
Условия получения
Получение водного раствора кислой соли цианамида каль-
ция. В реактор из нержавеющей стали емкостью 3 л, снабжен-
ный мешалкой и термометром, загружают 1,25 л промывной
воды (см. примечание 1) при 18° (содержащей 1,5% Ca^HCNah
от предыдущей операции получения водного раствора кислой
соли цианамида кальция).
При энергичном перемешивании в реактор загружают в те-
чение 5 минут небольшими порциями 225 г цианамида кальция
(с содержанием 18,78% азота). Температура реакционной мас-
сы поднимается до 30°, и при этой температуре ее перемеши-
вают 30 минут. После этого отфильтровывают реакционную
массу от шлама, содержащего в основном гидроокись каль-
ция. Количество фильтрата, содержащего 10,47% Ca(HCNa)2,
составляет 907 г, pH раствора 11,0—11,5. Шлам на воронке
промывают при взмучивании ~1,1 л воды. Отфильтровывают
промывные воды (1,1 л) и .используют их в последующих опе-
рациях гидролиза цианамида кальция. Содержание Са (НС№2)з
в промывной воде составляет 1,49%. Количество шлама со-
ставляет 275 г с содержанием в нем 0,84% азота.
Приготовление раствора азотнокислого свинца. В фарфо-
ровый бачок емкостью 6 л, снабженный мешалкой, загружа-
ют 469,1 г азотнокислого свинца и 4,2 л воды. Реакционную
массу перемешивают в течение 30 минут при комнатной тем-
пературе до. полного растворения соли.
28
Очистка водного раствора кислой соли цианамида кальция
от сернистых соединений. В двухлитровый стеклянный стакан
с мешалкой загружают 907,5 г (10%-ный избыток от теорети-
ческого) водного раствора кислой соли цианамида кальция.
При перемешивании отдельными порциями приливают 100 мл
10%-ного раствора азотнокислого свинца в течение 40—60 ми-
нут, проверяя полноту осаждения серы (см. примечание 2).
Черный осадок сернистого свинца отфильтровывают. Получа-
ют 920 мл фильтрата. Осадок на воронке промывают 50 мл
воды. ПромывнЫе воды присоединяют к основному филь-
трату. „
Получают 29 г сырого или 15 г сухого сернистого свинца.
Получение цианамида свинца. В фарфоровый бачок ем-
костью 6 л, снабженный мешалкой, загружают 970 мл водного
раствора кислой соли цианамида кальция, освобожденного от
сернистых соединений (см. примечание 2). При перемешива-
нии приливают 4582 г (4211 мл) 10%-ного водного раствора
азотнокислого свинца в течение 5 минут. Выпадает бледно-кре-
мовый осадок, который постепенно желтеет и далее приобретает
зеленовато-желтую окраску. К реакционной массе при переме-
шивании приливают 75 мл 25%-ного аммиака. Осадок жел-
теет- При pH 7,6—8,0 достигается полное осаждение цианами-
да свинца. Осадок цианамида свинца отфильтровывают. По-
лучают сырого цианамида свинца 580 г и 4870 мл фильтрата.
Осадок цианамида свинца промывают на воронке 2 л дистил-
лированной воды до отрицательной реакции на иоиы NO3 и
Са (см. примечание 3). Промытый цианамид свинца сушат при
105° в течение 8 часов.
Получают 330,8 г сухого цианамида свинца, что составля-
ет 94,1% теории, считая на 100%-ный азотнокислый свинец.
Найдено %: РЬ 83, 59; 83, 61; N 11,08; 11,25,
РЬСМ,. Вычислено %:. РЬ 83,8; 1411,34.
Примечания.
I. Дли получения водного раствора кислой кальциевой соли цианамида
проводят гидролиз цианамида кальция пятикратным количеством воды при
30° с последующим отделением шлама гидрата окисн кальция. Промывные
воды полностью расходуются при повторных операциях разложения циана-
мида кальция.
2. Полноту осаждения серы проверяют добавлением к отфильтрован-
ному реакционному раствору соли азотнокислого свинца. Отсутствие бурого
осадка указывает на полноту осаждения.
3. Пробу на ион МОз проводят с дифениламином: не должно появлять-
ся синего окрашивания. Пробу на ион Са проводят с раствором щавелево-
кислого аммония: не должно появляться белого осадка.
ЛИТЕРАТУРА
1. Е. Drechsel, J, prakt. Chem. [2], 8, 330 ('873).
2. Г. И. Шелинский, Обмен опытом лакокрасочной промышленности,
№ 3, 19 (1940). •
29
3. G. Guainazzi, Pitture e Vernict, 2, 87 (1946).
4. Итал. пат. 428147, 1947.
5. Франц, пат. 966349, 1950.
6. Пат. ФРГ, 8196S8, 1951.
7. Пат. ФРГ, 803351, 1951.
8. Perret, Biechler, Bull. Soc. ind. Mulhouse, 102, 58 ('936)
9, В. Г. Брудзь. P. Л. Глобус, Л. И. Грачева, A. M. Грозовская, Хим.
пром-сть 6, 32 (1957).
Поступала в феврале 1962 г. ИРЕА
ФТАЛАТ СВИНЦА
(Средняя свинцовая соль фталевой кислоты)
В. Г. БРУДЗЬ, Д. Ш. РОЗИНА, Л. Т. НЕСТЕРЕНКО
<\-СООх
/РЬ
'х у-СОО/
C8H4O4Pb М. в. 371,31
Фталат свинка получают взаимодействием солей свинца с
солями фталевой кислоты [1]. По значительно более поздним
данным, фталат свинца может быть получен с количественным
выходом при непосредственном взаимодействии раствора фта-
левой кислоты со свинцовым глетом [2]. Этот метод имеет пре-
имущество по сравнению с первым, так как исключает две
трудоемкие операции: превращение свинцового глета и фта-
левой кислоты в соответствующие соли, а также обеспечивает
более высокий выход- Приняв за основу данные [2], нами были
внесены в условия получения фталата свинца весьма сущест-
венные изменения [3]. Использован непосредственно фталевый
ангидрид вместо насыщенного горячего раствора фталевой
кислоты, что значительно упростило технологию процесса,
применен незначительный избыток фталевого ангидрида
(1 %) • что обеспечило полное использование свинцового гле-
тр (непрореагировавший глет неизбежно попадает во фталат
свинца); улучшены и некоторые другие условия синтеза
30
СИНТЕЗ ФТАЛАТА СВИНЦА
.СО. ,соон
С6н / ^0 + н20 -> CGH4(
хсоон
,соон .соо
С,Н4( +РЬО->СвН4< ^Рь + Нго
соон хсоо7
Характеристика основного сырья
Свинцовый глет, ГОСТ 5539—50.
Фталевый ангидрид, дистиллированный, I сорт, ГОС?
7119—54.
Условия получения
В четырехгорлую колбу, снабженную мешалкой, доходящей
почти до дна (см. примечание 1), обратным холодильником,
термометром и загрузочным отверстием, вносят 800 мл воды и
при перемешивании (120—130 оборотов/минуту) 61,4 г (0,272
М) 99%-ного свинцового глета. К полученной суспензии до-
бавляют в течение 4 часов при температуре 85—90° (водяная
баня) ~40,8 г (0,275 М) фталевого ангидрида до кислой ре-
акции по конго, не исчезающей в течение 15 минут (см. приме-
чание 2), после чего перемешивание продолжают при указан-
ной температуре еще 0,5 часа; образовавшийся фталат свинца
отфильтровывают, тщательно отжимают и промывают горя-
чей дистиллированной водой (75—80°), по 50 мл в один при-
ем, до нейтральной реакции промывных вод и до отсутствия
иона свинца (см. примечание 3). Расходуется ~300 мл воды.
Полученный фталат свинца сушат при 105° до постоянного
веса.
Выход 97—98 г с содержанием основного вещества 99,7—
100%, что составляет 97—98% теории (см. примечание 4).
Найдено %: РЬ 55,85; 55,90.
С8Н4О4РЬ. Вычислено %: РЬ 55,80.
Примечания.
1, Конструкция мешалки должна обеспечить подъем тяжелого свин-
цового глста со дна колбы, а также очень хорошее перемешивание всей
реакционной массы.
2. Обычно расходуется все количество фталевого ангидрида, иногда
остается 0,1—0,2 г. Избыток фталевого ангидрида приводит к образованию
кислой соли.
3. Конец промывки контролируется следующим образом: а) к 10 мл
промывных вод прибавляют 1—2 капли раствора фенолового красного.
Желтый цвет испытуемого раствора должен, перейти в розовый при добав-
лении 2 капель 0,1 и. раствора едкого натра; б) к 5 мл промывных вод до-
бавляют несколько капель 5%-ного раствора йодистого калия; отсутствие
желтого кристаллического осадка йодистого свинца указывает на конец про-
чьшки.
4. Содержание основного вещества определяют по РТУ 616—60.
31
ЛИТЕРАТУРА
I. Carlus, Liebigs Ann. Chem, 148, 166 (1869).
2. Пат. США 2412784, 1946.
3. В. Г, Брудзъ, Д. Ш. Розина, Л. Т. Нестеренко, Труды ИРЕА, вып. 22,
стр. 139, Госхимиздат, 1958.
Поступила е мирте 1962 г.
ИРЕА
ГЕКСАМЕТИЛЕНДИАМИН УКСУСНОКИСЛЫЙ
В. Я. ТЕМКИНА, Р, 77. ЛАСТОВСКИЙ, В. Г. БРУДЗЬ
NH2(CH2)eNH2 • 2 СН3СООН
C10Ha4O4N2 М. в. 236,32.
В производстве гексаметилендиамин уксуснокислый полу-
чают взаимодействием чистого свежеперегнанного гексамети-
лепдиамина с уксусной кислотой в среде бензола с выхо-
дом 81 % теоретического.
Нами предложено получать гексаметилендиамин уксусно-
кислый в среде технического п-бутилового спирта, что позво-
лило применять технический гексаметилеидиамин и обеспечи-
ло большую безопасность процесса. Выход при этом состав-
ляет 89% теории.
СИНТЕЗ ГЕКСАМЕТИЛЕНДИАМИНА УКСУСНОКИСЛОГО
NH2(CH2)eNH2+2CH3COOH -> NH2(CH2)6NH2-2CH8COOH
Характеристика основного сырья
Гексаметилендиамин, технический, ТУ 3161—53.
Уксусная кислота, техническая, I сорт, ГОСТ 7077—54.
н-Бутиловый спирт, технический, ГОСТ 5208—50.
Условия получения
В колбу емкостью 0,25 л, снабженную обратным холодиль-
ником, термометром, мешалкой и капельной воронкой, поме-
щают 100 мл н-оутилового спирта и 22,5 г (0,19 М) мелкоиз-
мельченного гексаметилеидиамина. Смесь размешивают при
температуре ~20° в течение 0,5 часа до полного растворения.
32
К полученному раствору добавляют по каплям 25 мл (0,21 М)
уксусной кислоты с такой скоростью, чтобы температура реак-
ционной смеси не превышала 70°. При этом постепенно обра-
зуется объемистый осадок уксуснокислой соли гексаметилен-
диамина. По окончании прибавления кислоты реакционную
смесь охлаждают до 20° при размешивании в течение 1 часа.
Осадок отфильтровывают, промывают 20 мл н-бутилового
спирта, тщательно отжимают и сушат при 50°.
Получают -40 с гсксаметилендиамина уксуснокислого (89%
теории). Содержание основного вещества 98% (определено
методом потенциометрического титрования).
Найдено %: N 11.9; [2,0.
C1oH24O4N2, Вычислено %: N 11, 86.
Примечание.
н-Бутнловый спирт может быть многократно (8— 10 раз) использован
дтя последующих операций после предварительной обработки активирован-
ным углем при нагревании. Возможно применение метилового и этилового
спиртов.
Поступила в апреле 1962г. ИРЕА
БЕНЗИЛАМИН
(cij-Аминот олуол)
И. Г. МАТВЕЕВ. Д. Ш. ГОЗИНА
CH2NHa
CjHgN
М.в.107,15
По литературным данным, бензиламин получают взаимо-
действием хлористого бензила с спиртовым раствором аммиа-
ка (одновременно получаются ди- и трибензиламины [1, 2] ка-
талитическим гидрированием бензонитрила, бензальдоксима и
некоторых других подобных соединений [3, 4], превращением
а_мида фенилуксусной кислоты в амин по реакции Гофмана
[о], взаимодействием бензальдегида с формиатом натрия по ре-
Зак. G22 33
акции Лсйкарта [6], разложением двойной соли хлористого
бензила с уротропином [7]).
Последний метод, представляющий практический интерес,
был положен в основу синтеза бензиламина.
Нами внесены изменения во все стадии процесса. Исполь-
зован органический растворитель (бензол или хлороформ) для
извлечения бензиламина из водного раствора, что значительно
повысило его выход и качество. В стадии получения соляно-
кислого бензиламина промежуточная трехкратная отгонка
азеотропной смеси диэтилформаль—спирт—вода, с добавле-
нием каждый раз спирта и соляной кислоты, заменена одно-
кратной отгонкой по окончании реакции.
Выход двойной соли хлористого бензила с уротропином
значительно повышен, в основном за счет увеличения продол-
жительности нагревания реагентов.
СИНТЕЗ БЕНЗИЛАМИНА
СН2С1 СН2С1
/ \ СНС1, / \
| | 4- (CH2)6N4--> | | • (CHa)8N4
СН2С1
\
[ | • (CH?)eN4 + 12 С2Н8ОН + 3 НС1 -
CH2NH,’HC1
I
| -|-6CH2(OC2H6)2 + 3NH4C1.
CHjNH^-HCI CH2NH2
| | + NaOH _ | 4- NaCl + H2O
Характеристика основного сырья
Хлористый бензил, ч., ТУ МХП 50—49.
Уротропин, фармакопейный, ГОСТ Ф—VIII, 7/4.
Хлороформ, ГОСТ Ф—VIII, 119.
,34
Условия получения
Получение двойной соли хлористого бензила с уротропи-
ном. В трехгорлую колбу емкостью 1,5 л, снабженную мешал-
кой с глицериновым затвором и обратным холодильником (8
шариков), вливают 720 мл хлороформа, нагревают на водяной
бане до 50° и добавляют при перемешивании 138,6 г (0,99 Л1)
гексаметилентетраамина (уротропина) и через 10 минут —
126 г (I М) хлористого бензила. Реакционную смесь, не пре-
кращая перемешивания, нагревают 2 часа при 50°, а затем
5 часов при температуре кипения жидкости, после чего остав-
ляют на 12 часов при комнатной температуре- Выделившуюся
двойную соль хлористого бензила с уротропином отфильтро-
вывают, промывают дважды по 100 мл хлороформа и сушат
при 25—30°.
Выход 248—250 г, что составляет ~90% теории (см. при-
мечание 1).
Найдено %: N 20,36; 20,40.
Ci,H10N4. Вычислено %: N 21,01.
Получение солянокислого бензиламина. В трехгорлую кол-
бу емкостью 3 л, снабженную мешалкой и обратным холодиль-
ником, вливают 900 мл спирта, нагревают на водяной бане до
50° и вносят при перемешивании 248 250 г (0,93 М) двойной
соли хлористого бензила с уротропином и через 5 минут
350 мл 36 %-ной соляной кислоты. Реакционную смесь кипятят
3 часа при перемешивании, затем охлаждают до 50°, добавля-
ют 200 мл спирта. 80 мл концентрированной соляной кислоты,
продолжают кипятить при перемешивании еще 3 часа и остав-
ляют на 12 часов при комнатной температуре для более пол-
ного выделения хлористого аммония, образовавшегося в ре-
зультате реакции. Хлористый аммоний отфильтровывают, от-
жимают и промывают дважды по 50 мл спиота, присоединяя
его к основному фильтрату. Фильтрат (~ 1,6 л), содержащий
раствор солянокислого бензиламина, переносят частями (через
капельную воронку) в колбу емкостью 700 мл и отгоняют на
водяной бане азеотропную смесь диэтилформаль—спирт—во
да; температуру бани постепенно повышают до кипения, и ког-
да па кипящей водяной бане прекращается отгон жидкости
(всего отгоняется 1,10—1.15 .?), к остатку в колбе добавляют
5 г активированного угля и периодически перемешивают в те-
чение 15 минут; уголь отфильтровывают и промывают 50 мл
спирта, присоединяя его к фильтрату. Фильтрат охлаждают
в ледяной воде до 5—10° и оставляют при этой тсмпсоатуро
на 2 часа для кристаллизации. Выкристаллизовавшийся со-
лянокислый бензиламин отфильтровывают и отжимают (I).
Маточный раствор (500—550 мл) упаривают в фарфоровой
чашке на водяной бане до 100—120 мл и охлаждают до 10—
з~
35
15°; образовавшуюся густую кристаллическую массу отфильт-
ровывают и тщательно отжимают (II). Вторично полученный
маточный раствор снова упаривают до появления пленки, ох-
лаждают и отфильтровывают (III).
Солянокислый бензиламин I, II, III перемешивают и сушат
при 60—65°.
Выход 137—143 г (см- примечание 2).
Получение бензиламина. В трехгорлую колбу емкостью
500 мл, снабженную мешалкой и обратным холодильником,
наливают 140 мл 40%-ного раствора едкого натра и при пере-
мешивании постепенно вносят в течение 15—20 минут полу-
ченный солянокислый бензиламин. После 10—15 минут пере-
мешивания происходит расслоение жидкости. Верхний слой,
содержащий бензиламин, отсифонивают при помощи вакуума
(стеклянный сифон) в делительную воронку.
Из остатка в колбе бензиламин экстрагируют хлороформом
(200 мл); после энергичного перемешивания в течение 15 ми-
нут реакционную смесь отфильтровывают через фильтр из
стеклянной ткани; осадок промывают дважды по 25 мл хлоро-
форма, присоединяя его к основному фильтрату. Фильтрат —
хлороформенный раствор бензиламина — присоединяют к бен-
зиламину, уже находящемуся в делительной воронке, и прсле
энергичного встряхивания и непродолжительного (~5 минут)
отстоя отделяют нижний слой хлороформа, в который в ос-
новном переходит весь бензиламин. Верхний водный слой сно-
ва встряхивают несколько раз с 100 мл хлороформа. Хлоро-
форм отделяют и вместе с основным хлороформенным раство-
ром бензиламина сушат в течение 10 часов едким натром
(~40 г в виде сплавленных гранул). Хлороформ отгоняют на
водяной бапе (см. примечание 3), когда температура бани до-
стигает кипения, практически весь хлороформ уже отогнан,
после чего охлаждают баню до 60°, включают вакуум и при
76—78°/15—16 мм перегоняется весь бензиламин.
Выход 73—75 г с содержанием основного вещества (см.
примечание 4) 98,0—99,5%, что составляет 68—70% теории.
Примечания.
1. Маточный раствор хранят в закрытом сосуде; через 6—7 суток из
него выделяется 7—9 г двойной соли хлористого бензила с уротропином
(3—4% теории).
Вторично полученный маточный раствор используют вместо хлорофор-
ма в следующей операции.
2. Выход солянокислого бензиламина в процентах от теории вычислить
нельзя, так как он содержит хлористый аммоний, выделяющийся в процес-
се упаривания раствора.
3. Отгонку производят из колбы для вакуум-перегонки емкостью
200 мл с дефлегматором высотой 300 мм, перенося раствор частями через
капельную воронку, присоединенную к колбе.
Отогнанный хлороформ используется для экстрагирования бензиламннг
в следующей операции.
4. Содержание основного вещества определяют по ВТУ МХП 2808—51.
36
ЛИТЕРАТУРА
1. А. Т. Mason, J. Chem. Soc., 63, 1313 (1893).
2. Н. Limpricht, Liebigs Ann. Chem., 144, 305 (1867).
3. C. F. Winans, H. Adkins, J. Amer. Chem. Soc., 54, 306 (1932).
4. C. F. Winans, J. Amnier. Chem. Soc., 61, 3566 (1939).
5. A. W, Hoffman, Ber., 18. 2738 (1885).
6. K- Levis, J, Chem. Soc., 1950, 2249.
7. M. Delepine Bull. Soc. chim. (3). 17, 292 (1899).
Поступила в марте ig$2 г. ИРЕЛ
БЕНЗИЛАМИНОЭТАНОЛ
(М-(2-Оксиэтил) -бензиламин)
Я. Г. БРУДЗЬ, Д. А. ДРАГПДША
C6H5CH2NH-CH2CH,OH
C0H13ON М. в. 151,21
В литературе описан ряд методов получения бензиламино-
этанола- Наибольшее практическое значение имеет прямое ал-
килирование моноэтаноламниа хлористым бензилом [1, 2] и
синтез из бензиламина и окиси этилена [3—5] или этилен.хлор-
гидрина [6, 7].
Нами проверен метод получения бензиламиноэтапола из
хлористого бензила и моноэтаноламина и установлены опти-
мальные условия проведения синтеза [8],
СИНТЕЗ БЕНЗИЛАМИНОЭТАНОЛА
C6HbCH2CH-H2NCH2CH?OH------> C6H,CH2NH-CHaCH2OH
—НС1
В качестве побочного продукта получается третичный
аминдибензиламиноэтанол.
СеН5СН2С1 + C0H5CH2NH-CH3CH2OH —
-> (C6H5CH2)8N-CHsCH»OH
Характеристика основного сырья
Хлористый бензил, технический, 1-го сорта, ТУ МХП
588—49.
Моноэтаноламин, технический, ТУ МХП 2277—50.
37
Едкий натр, технический, ТУ 2263—43, 15%-ный раствор.
Хлороформ, технический, ГОСТ 1539—42.
Условия получения
Обезвоживание моноэтаноламина. проводят в круглодонной
двухгорлой колбе емкостью 5 л, снабженной насадочной ко-
лонкой длиной 500 мм и диаметром 38 мм. Насадка — стек-
лянная трубка длиной 40—42 мм и диаметром 38—40 мм. Ко-
лонка имеет головку полной конденсации с краном для ре-
гулирования скорости отбора дистиллята. Весь прибор соби-
рается на шлифах, так как резиновые пробки разрушаются
этаноламином. Обогрев колбы осуществляется электричеством
или газом через масляную баню.
В колбу загружают 3,75 кг моноэтаноламина с содержани-
ем воды 14—25% и включают обогрев при закрытом кране го-
ловки колонки. При температуре жидкости 130—140° в голов-
ке появляются пары воды. После 5—10-минутной работы ко-
лонки «на себя» начинают отбор дистиллата, для чего откры-
вают кран на головке- Скорость отбора дистиллата, регули-
руемая с помощью крана, устанавливается равной 150—
170 мл)час. При этом необходимо вести нагревание так, чтобы
скорость отбора дистиллата была примерно равной скорости
стекания флегмы.
В процессе перегонки температура жидкости, постепенно
повышаясь, достигает 171 —173°. Температура в парах (98—
98,5°) остается постоянной до конца отгонки воды, после чего
она резко повышается. Как только температура в парах до-
стигнет 125—130°, кран перекрывают и выключают обогрев
(см. примечание 1). Продолжительность отгонки 5—6 часов.
При загрузке технического моноэтаноламина с 20%-ным
содержанием воды отгоняются около 750 мл воды и 2,8—2,9 кг
обезвоженного этаноламина с общим содержанием аминов, в
пересчете на моноэтаноламин 96—97,5% (см. примечание 2).
Получение бензиламиноэтанола. В круглодопиую колбу ем-
костью 0,6 л, снабженную однолопастной якорной мешалкой,
термометром, капельной воронкой, обратным холодильником
(см. примечание 3) и помещенную в глицериновую или масля-
ную баню, загружают 315—320 г (~5 Л1) 96—97,5%-ного
обезвоженного моноэтаноламина, включают обогрев и мешал-
ку. Хлористый бензил (126,5 г) прибавляют в течение 4 часов
при температуре 105—120°, затем дают 3-часовую выдержку
при той же температуре и при перемешивании. Образуется
однородная вязкая жидкость желто-бурого цвета.
Реакционную массу переносят в делительную воронку и
нейтрализуют 235 мл 15%-ного раствора едкого натра- Едкий
натр вносят порциями по 20—30 мл до появления отчетливой
щелочной реакции по тиазоловой бумажке (pH 11,6—13,6).
38
Образуются два слоя, из которых верхний в основном состоит
из бензиламиноэтанола; последний извлекают хлороформом
(см. примечание 4). Экстракцию проводят двумя порциями по
100 мл хлороформа. Хлороформенный раствор подвергают
разгонке, а из водного слоя регенерируют моноэтаноламин
(см. примечание 5). Отгонку хлороформа и вакуум-перегонку
бензиламиноэтанола производят из круглодонной колбы ем-
костью 0,5 л, помещенной в масляную баню. Колба снабже-
на насадочным дефлегматором длиной 250—300 мм, диамет-
ром 35 мм; насадка представляет собой стеклянные кольца
длиной и диаметром около 5 мм. Хлороформ отгоняют при
нормальном давлении. Основное его количество перегоняется
при температуре бани 80—90°. По мере замедления отгонки
температура бани постепенно повышается до 180°.
По окончании отгонки хлороформа обогрев выключают, к
холодильнику присоединяют с помощью форштосса («паук»)
два приемника и перегоняют в вакууме бензил аминоэтанол. В
процессе перегонки отбирают две фракции- Первая фракция,
состоящая в основном из моноэтаноламина, отгоняется при
температуре в парах 71—74°/9—10 мм, затем температура до-
вольно быстро повышается; по достижении 142°/9—10 мм ме-
няют приемник и отбирают бензиламиноэтанол в пределах
температур 142 — 146°/9—10 мм.
Выход бензиламиноэтанола 105—115 г, что составляет
70—75% теории, считая на хлористый бензил.
Примечания.
1. Если подъем температуры в парах наступает не скачкообразно
температура жидкости ниже 170', кран перекрывают, не прекращая нагре-
ва. Колонке дают некоторое время поработать «на себя»; после того как
температура в парах снопа станет ниже 100°, возобновляют отбор дистил-
лата, ио с меньшей скоростью.
2. Содержание аминов определяют титрованием навески этаноламина
0,1 н. соляной кислотой с индикатором метиловым оранжевым.
3. Прибор собирается на резиновых пробках.
4. Верхний слой, ввиду значительного содержания в нем воды и моно-
этаноламина, непосредственно не отделяют, а извлекают хлороформом. При
экстракции хлороформом бензиламиноэтанол находится в нижнем слое, л
верхний слой 500 мл) представляет собой насыщенный хлористым нат-
рием водный раствор моноэтаноламина Регенерированный хлороформ ис-
пользуется в последующих операциях.
Для экстракции реакционного бензиламиноэтанола можно применят:,
также бензол.
5. Регенерацию моноэтаноламина проводят в круглодонной двухгор-
лой колбе емкостью 750 мл, помещенной в масляную баню, Колба снаб-
жается однолопастпой мешалкой с глицериновым затвором. В другое горд )
колбы вставляют отводную трубку, к котооой присоединяют прямой холо-
дильник. В колбу загружают 500 мл водного слоя, иолу ценного после экст-
ракции хловофоомом бензиламиноэтанола из реакционного раствора и, прн
работающей мешалке, полностью отгоняют всю жидкость. В колбе остаетс-т
осадок хлористого натрия. В результате отгопкн получают около 45(1 г 40—
45%-ного водного раствора моноэтанолатлина, от которого затем отгоняют
воду, как описано выше. Получают около 200 г обезвоженного моиоэтано-
ламина с содержанием основного вещества 96—97%. Всего, вместе с пред-
39
гоном, полученным гри разгонке бензиламиноэтанола. pi генерируется 220—
280 г мопоэтаполамтиа. Таким образом, выход бензиламиноэтанола по мо-
ноэтаноламину, с учетом регенерированного, составляет около 50% теории
ЛИТЕРАТУРА
1. P. Ruinpf, R. Kwass, Bull. Soc. chiin France, [5] 10, 347 (1943).
2. IF Wilson, J, Cbem. Soc., 1952, 3527.
3. S. Gabriel, R. Stelzner, Bcr. 29. 2382 (1896).
4. F. B. bains,
neval, J. Amer. Cbem. Soc., 47, 1981 (1925).
5. E. Wedekind E. Bruch. Liebigs Ann. chem., 471, 73 (1929).
6. Пат. США 2140259; Chem. Zbl, I, 4123 (1939).
7. W. F.'tCockbum, A. F. Me Kay, J. Organ. Chem., 18, 321 (1953).
8. В. Г. Бррдзь, Д. А. Драпкина. Труды ИРЕ4, вып. 22, стр. 142. Гос-
Химиздат, 1958.
Поступила в феврале 1962 г.
Cbem. Soc., 1952, 3527.
/?. A. Brewster, I. L. Malm, О. IF. Miller, R. B. Ma-
ИРЕА
C]fiH15As
ТРИФЕНИЛАРСИН
Н. Е. КОЖЕВНИКОВА, О. В. ИВАНОВ
С6Н6х
>s-C6H6
С,
М. в. 306,24
Трифениларсин получают действием металлического нат-
рия на смесь треххлористого мышьяка с бромбензолом в среде
серного эфира [1], бензола или ксилола [2], а также разложе-
нием двойной хлорцинковой соли хлористого фенилдиазония
цинковой пылью в присутствии треххлористого мышьяка [3, 4,
5, 6].
Трифениларсин представляет интерес в качестве исходного
продукта для синтеза некоторых экстрагентов и мышьяксодер-
жащих органических соединений [7].
Проверена и подтверждена литературная методика [5] по-
лучения трифениларсина разложением двойной хлорцинко-
вой соли хлористого фенилдиазония цинковой пылью в при-
сутствии треххлористого мышьяка.
40
СИНТЕЗ ТРИФЕНИЛАРСИНА
CeH5NH2 + 2НС14- NaNO2 — C6H5N2C1 - NaCl -|- 2FW
2CeH6N2Cl + ZnCl8 -* (CeH5N3Cl)rZnCl2
3(C8H-N2Cl)2-ZnCl2+2AsCl3^6Zn -> 2(CeH5)2As+9ZnCl24 6Na
Характеристика основного сырья
Мышьяк треххлористый, технический, с г. кип. 130°.
Цинковая пыль, техническая, ТУ ГАПУ 420—54.
Анилин, ч., ГОСТ 5819—51.
Цинк хлористый, ч„ ГОСТ 4329—48.
Условия получения
Получение двойной хлорцинковой соли хлористого фенил-
диазония. В фарфоровый стакан емкостью 6 л. снабженный
мешалкой, вносят 1 л соляной кислоты уд. в. 1,18 и 400 мл
воды и затем прибавляют 300 г анилина. Суспензию соляно-
кислого анилина при перемешивании охлаждают до 2° прибав-
лением 600 а измельченного льда. При этой температуре мед-
ленно приливают раствор нитрита натрия (220 г в 250 мл во-
ды), избыток нитрита натрия проверяют на йодкрахмальной
бумажке. Во время диазотирования постепенно прибавляют
еще 600 г измельченного льда с таким расчетом, чтобы темпе-
ратура не превышала 5°. Охлажденный раствор соли фенил-
диазония быстро отфильтровывают от незначительного осад-
ка и снова помещают в тот же сосуд. Одновременно растворя-
ют 280 г хлористого цинка в 120 мл разбавленной соляной кис-
лоты (1 : 1), смешивают с 400 г льда и медленно приливают к
энергично перемешиваемому раствору соли диазония. Выпа-
дает розовый осадок двойной хлорцинковой соли хлористого
фенилдиазония. Для того чтобы реакция прошла полностью,
перемешивание продолжают еще в течение 30 минут. Осадок
отфильтровывают, тщательно отжимают, промывают двумя
порциями сухого ацетона (см. примечание 1) по 250 мл и су-
шат на воздухе при температуре около 20°.
Выход двойной хлорцинковой соли хлористого фепнлдиа-
зония 50.0—550 г.
Получение трифениларсина. В круглодонную колбу ем-
костью 2 л, снабженную мешалкой и помещенную в баню с
охладительной смесью (лед с водой), загружают 460 г двой-
ной хлорцинковой соли хлористого фенилдиазония и раствор
138 г треххлористого мышьяка в 1,1 л сухого ацетона, высу-
шенного над хлористым кальцием- Реакционную смесь энер-
гично перемешивают и в течение 3 часов малыми порциями к
ней прибавляют цинковую пыль (167 а). Температура не дол-
жна подниматься выше 5°. По окончании загрузки цинковой
41
пыли выключают мешалку и выдерживают 12 часов. Получен-
ный раствор трифениларсина в ацетоне отфильтровывают.
Ацетон отгоняют на водяной бане. Оставшееся масло перено-
сят в стакан и обрабатывают его при перемешивании 153 мл
разбавленной (1:1) соляной кислоты. Масло быстро закри-
сталлизовывается в красно-коричневую массу. Получают
160 г трифениларсина (т. пл. 57°), его дважды перекристалли-
зовывают из метилового спирта (по 150 мл) и оставляют для
кристаллизации при 2° па 12 часов.
Выход трифениларсина 130 г, что составляет 44% теории,
считая на треххлористый мышьяк (см. примечание 2). Т. пл.
трифениларсина 61°, что соответствует литературным дан-
ным [5]-
Примечания.
1. Присутствие даже следов влаги в двойной хлорцинковой соли хло-
ристого фснилдиазония значительно снижает выход трифениларсина и
приводит к образованию смолистых побочных продуктов. Необходимо при-
менять ацетон, высушенный над хлористым кальцием.
2. Данные авторов [5] об уменьшении выхода с увеличением масштаба
загрузок подтвердились. Уменьшение загрузки в 4 раза позволяет полу-
чить трифениларсин с выходом 60—70% теории.
ЛИТЕРАТУРА
1. A. Michaelis, Liebigs Ann. Chem., 321, 160 (1902).
2. Pope, Turner. J. Chem. Soc., 117, 1447 (1920).
3. A. H. Несмеянов, ЖРФХО, 61, 1393 (1929).
4. A. H. Nesmejanow. Ber„ 62. 1010 (1929).
5. W. E. Hanby, W. A. Waters, J. Chem. Soc . 1946, 1029.
6. О. А. Реутов, Ю. Г. Еундель, Ж. обш. химии, 25.23, 24 (1955).
7. D. Jyon, F. Mann, J. Chem. Soc.. 1942, 666. .
Поступила в марте 1962 г. ИРЕА
ДИИЗОНИТРОЗОАЦЕТОН
Н. Е. КОЖЕВНИКОВА, Н. Д. САПОЖКОВА
c3h4o3n2
сн-со-сн
II li
NOH NOH
М. в. 116,08
Диизонитрозоацетон предложен в качестве реактива для
определения малых количеств галоидфосфорорганических сое-
динений, ангидридов кислот и ацилирующих средств [1].
42
По литературным данным, диизонитрозоацетон получают
действием азотистой кислоты или ее соли на ацетондикарбоно-
вую кислоту [2, 3]. В свою очередь, ацетондикарбоновую кисло-
ту получают взаимодействием лимонной кислоты с серной кис-
лотой, содержащей 20% свободного серного ангидрида [4, 5, 6,
7, 8].
Нами проверен и уточнен метод получения диизонитрозо-
ацетона [2] действием нитрита натрия на ацетондикарбоновую
кислоту или его мононатриевую соль.
СИНТЕЗ ДИИЗОНИТРОЗОАЦЕТОНА
H2SO4
НООС-СН2-СО-СН2-СООН+2NaNO2 —>
-* СН-СО-СН %-Na2CO3 % СО. + Н2О
II II
NOH NOH
Характеристика основного сырья
Ацетондикарбоновая кислота, содержащая до 20% серной
кислоты, свежеприготовленная.
Нитрит натрия, ч., ГОСТ 4197—48.
Азотная кислота, ч., ГОСТ 4461—48.
Условия получения
Получение диизонитрозоацетона. В круглодонную колбу
емкостью 5 л с мешалкой, помешенную в баню с ледяной
водой, вносят 740 г свежеприготовленной (см. примечание 1)
ацетондикарбоновой кислоты [4], содержащей до 20% сер-
ной кислоты, и 1,5 .? воды, охлаждают до 4" и неболь-
шими порциями при размешивании прибавляют насыщенный
раствор нитрита натрия (620 г нитрита натрия в 650 мл воды)
при температуре, не превышающей 6°. Наблюдается вспенива-
ние и выделение осадка. Внимание! (см. примечание 2).
После прибавления всего раствора нитрита натрия реакцион-
ную массу охлаждают до 0° и приливают небольшими порция-
ми 250 мл азотной кислоты (уд. в. 1,15). При этом также наб-
людается сильное вспенивание и дополнительное выделение
осадка (см. примечание 2). Реакционную массу охлаждают
(лед с солью) до минус ~5° в течение 10 минут, осадок быст-
ро отсасывают и тщательно промывают ледяной водой до сла-
бокислой реакции промывных вод.
Получают 215 г технического диизонитрозоацетона, его
трижды перекристаллизовывают из горячей (60—70°) воды с
активированным углем и сушат в эксикаторе над фосфорным
ангидридом. Получают 190 г сухого диизонитрозоацетона с
т. пл..136°, что соответствует литературным данным [8].
43
Выход составляет 41% теории, считая на ацетондикарбоно-
вую кислоту.
Получение мононатриевой соли диизонитрозоацетона. В
трехгорлую колбу емкостью 0,5 л, снабженную мешалкой,
ртутным затвором и холодильником с хлоркальциевой труб-
кой, загружают 75 мл абсолютного этилового спирта и к нему
медленно прибавляют 1,66 г мелконарезанного металлическо-
го натрия. После того как натрий полностью прореагирует, к
полученному раствору этилата натрия, при сильном переме-
шивании, приливают раствор из 5 г диизонитрозоацетата в
85 мл абсолютного этилового спирта. Выделяется оранжевый
или желтый осадок, который немедленно отфильтровывают,
сушат и перекристаллизовывают из водно-спиртовой смеси
(1 :4). С этой целью 6 г мононатриевой соли при нагревании
растворяют в 30 мл дистиллированной воды и туда же прибав-
ляют 120 мл этилового спирта. Выделившиеся кристаллы от-
фильтровывают, промывают этиловым спиртом и сушат в ва-
куум-эксикаторе над фосфорным ангидридом. Получают 4 г
мононатриевой соли диизонитрозоацетона.
Примечания.
1. АцетондикарбоноваЯ кислота легко разлагается на воздухе.
2. Работу необходимо вести под хорошо действующей тягой, так как
реакция сопровождается побочными процессами с выделением синильной
кислоты.
ЛИТЕРАТУРА
!. 5. Sass, W. D. Ludemann, В. Witten, V. Fischer et al, Analyt. Chem.,
29, 1346 (1957).
2. H. Pechmann, K. Wehsarg, Ber., 19, 2465 (1886).
3. K. Koessler, M. Hankl. Analyt. Chem., 40, 1717 (1918).
4. „Синтезы органических препаратов', Сб. 1, ИЛ., стр.70, 1949.
5. Willstdtter, Pfannenstiebl, Liebigs Ann. Chem,, 422, 5 (1921).
6. H. Pechmann. Ber., 17, 2543 (1884).
7. Ingold, Nickolis, J. Chem. Soc., 121, 1642 (1922).
8 «Препаративная органическая химия», стр. 743, Госхимиздат, 1959.
Поступила в марте 1962 г. ИРЕА
3-МЕТИЛПИРИДИН
(|3-Пиколин)
Ю. И. ЧУМАКОВ
сн3
(
1/
CSH,N
44
М. в. 93,12
З-Метилпиридин является одним из труднодоступных в чи-
стом виде гомологов пиридина. Он используется как исходный
продукт в синтезах никотиновой кислоты, никотинамида, нико-
типнитрила, высших 3-алкилпиридинов и ряда других соеди-
нений класса пиридина.
Обычным источником получения 3-метилпиридина являет-
ся «р-пиколиновая фракция» пиридиновых оснований, кипя-
щая около 144° и содержащая, кроме 3-метилпиридина, 4-ме-
тилпиридин и 2,6-диметилпиридин- Разделению «р-пиколипо-
вой фракции» и выделению в чистом виде ее компонентов пос-
вящено свыше ста работ, однако только немногие из предло-
женных способов позволяют получать достаточно индивиду-
альные соединения (см. обзоры литературы [1, 2]).
Нами было найдено, что 3-метилпиридин с содержанием
основного вещества 99% и выше может быть получен по мето-
дике, использующей избирательное взаимодействие 3-мегилпи-
ридина с солями меди — сульфатом [3, 5] и хлоридом [4, 6].
Вначале выделяют из «[З-пиколиновой фракции» концентрат
3-метилпиридина через комплекс с сульфатом меди [5], затем
его окончательно очищают через комплекс с хлористой медью
[6]. Контроль за содержанием получаемого 3-метилпиридина
производился по температуре кристаллизации, поскольку
это — наиболее надежный критерий чистоты [7, 8].
Попытки выделить чистый 3-метилпиридин в одну стадию
через комплекс с сернокислой медью [3] или с хлористой
медью [4] приводят к получению лишь концентрата с содержа-
нием 90—93%.
ВЫДЕЛЕНИЕ КОНЦЕНТРАТА 3-МЕТИЛПИРИДИНА
( сн3
«р-пиколиковая фракция»
C11SO4 H0SO4—
СНз
/СНз
CuSO4-2| | — 2
N
I
~!“2| 4
N
/2H,SO4
/х/СНз /\/СНз
CuSO4•2 | I % 2NaOH -> 2 | +
V N
+ CuO + Na2SO4 -% H2O
45
Характеристика основного сырья
«р-Пиколиновая фракция», ЧМТУ 3454—53.
Сернокислая медь, ГОСТ 4165—48.
Хлористая медь, ГОСТ 4164—48.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешалкой,
пропущенной через обратный холодильник, термометром и
капельной воронкой, помещают 200 мл воды и 277,5 г серно-
кислой меди (соли CuSO4 • 5Н2О (см. примечание 1). При пе-
ремешивании из капельной воронки постепенно прибавляют
80 мл концентрированной серной кислоты. Температура под-
нимается до 50—65°. Затем суспензию сернокислой меди в ра-
створе серной кислоты нагревают при перемешивании до 85° и
при этой температуре начинают прибавлять 600 г технической
«|3-пиколиновой фракции» (см. примечание 2). По окончании
прибавления, которое занимает 30—40 минут, реакционную
массу доводят до кипения (97—100° в колбе) и кипятят с об-
ратным холодильником в течение 15—20 минут. Затем охлаж-
дают при перемешивании до 15—20° (см. примечание 3). При
этой температуре реакционную массу размешивают 1—2 часа.
Выпавший осадок отфильтровывают под вакуумом на воронке
Бюхнера с полотняным фильтром, тщательно отжимая комп-
лекс на фильтре. Отключают вакуум и осадок на фильтре
пропитывают 100 мл воды (этой же водой предварительно
обмывают два раза порциями по 50 мл колбу, в которой про-
водилась реакция), разминая осадок на фильтре шпателем.
Дают постоять 20—30 минут, после чего фильтруют, тщатель-
но отжимая осадок на фильтре. В колбу, в которой проводи-
лась реакция комплексообразования, наливают 300 мл холод-
ной воды (10—15°) и переносят сюда же отжатый комплекс с
фильтра. Перемешивают в течение 20—30 минут, затем фильт-
руют на воронке Бюхнера. В результате получают около 250 г
влажной комплексной соли 3-метилпиридина и сернокислой
меди, имеющей ярко-голубой цвет (см. примечание 4).
Для перегонки с водяным паром в двухлитровую колбу
помещают 300 мл воды и 50—60 г твердого едкого натра. Сю-
да же вносят полученную комплексную соль и перегоняют с
водяным паром до полного отсутствия пиридинового основа-
ния в погоне. Получают 1000—1200 мл водного раствора 3-ме-
тилпириднна с содержанием около 10% (см. примечание 5).
Его обрабатывают при охлаждении едким натром из расчета
20 г на каждые 100 мл раствора. Выделившийся верхний слой
3-метилпиридина отделяют на делительной воронке от нижне-'
го водно-щелочного слоя и высушивают твердым едким нат-
ром, взятым в количестве 1 часть на 5 частей основания при
46
кипячении с обратным холодильником в течение 4 часов. По-
лучают в среднем 120 г технического продукта, содержащего
90—93% 3-метилпиридина (остальное — 4-метилпиридин, пи-
ридин и другие гомологи). Его без дополнительной обработки
используют для дальнейшей очистки через комплексное соеди-
нение с хлористой медью (см. примечание 6).
-ОКОНЧАТЕЛЬНАЯ ОЧИСТКА 3-МЕТИЛПИРИДИНА
/\/СНз ,/\/СНз
61 | -f- Cu2Cl2 —> CuaCla • 61 I
/х/сн3 2NaOH /ч/сн3
CuaCl2-6| I -------> 61 I +Cu2O + 2NaCl-rH2O
N N
Условия очистки
В трехгорлую колбу емкостью 250 мл, снабженную обрат-
ным холодильником, мешалкой, термометром и трубкой для
пропускания сернистого газа (см. примечание 7), помещают
80 мл смеси спирта и воды (1:1 по объему). При перемешива-
нии к суспензии 20 г хлористой меди в водно-спиртовом раст-
воре из капельной воронки быстро прибавляют 67 г техниче-
ского 3-метилпиридина, полученного, как описано выше. Затем
реакционную массу, не прекращая перемешивания и изредка
пропуская сернистый газ, постепенно охлаждают до темпера-
туры 15—20”. При этой температуре продолжают перемешива-
ние в течение получаса. Образовавшийся комплекс 3-метилпи-
ридина с хлористой медью отфильтровывают на воронке Бюх-
нера и промывают в 30 мл водно-спиртовой смеси (1 : 1). За-
тем его переносят в 70 мл водно-спиртовой смеси и перемеши-
вают в течение 10—15 минут, изредка пропуская сернистый
газ. Фильтруют, тщательно отжимая комплекс на фильтре
Получают 80 г влажного ярко-желтого комплексного соедине-
ния, имеющего состав Сп2С12 • 6C6H7N.
Для перегонки с водяным паром в колбу на 500 мл поме-
щают 50 мл воды, 20 г твердого едкого натра и сюда же вносят
полученное комплексное соединение, после чего перегоняют с
водяным паром до отсутствия пиридинового основания в пого-
не. Получают около 400 мл водного раствора 3-метилпиридина.
Его обрабатывают при перемешивании и охлаждении 80 г ед-
кого натра. Выделившийся верхний слой 3-метилпиридина от-
деляют, дополнительно высушивают гранулированным едким
47
кали (20 г на 100 мл основания) при кипячении с обратным
холодильником.
Высушенное основание перегоняют на ректификационной
колонке, эквивалентной 18—20 теоретическим тарелкам, с
флегмовым числом 15:1 (см. примечание 8). Получают в
среднем 2,4 г предгона с т. кип. до 143,5", 43,4 г основной фрак-
ции с температурой кипения 143,5—144° при 756 мм и 2,6 г
кубового остатка. Температура кристаллизации основной
фракции равна— 18,6 -н — 19,0", что соответствует 99,0%-ной
чистоте. Температура плавления пикрата 150—151,5° (испр.'
(литературные данные [7] 149—150° (см- примечание 9 и 10).
Выход составляет 48,0% теоретического, считая, что в исход-
ной «р-пиколиновой фракции» содержится 27% 3-метилпири-
дина или 12,9% (весовых), считая на «0-пвколиновую фрак-
цию».
Примечания.
1. Можно использовать технический медный купорос, нс содержащий
примесей железа. Последние ухудшают фильтрацию комплекса.
2. В реакцию брались обычные промышленные образцы «Р-пнко.зиновон
фракции». Эти образцы имеют сравнительно широкий интервал температур
кипения (138—146°) и, кроме 3-. 4-метилпиридина и 2,6-диметилпиридип.1,
содержат заметные количества 2-метилпиридпна, 2,4-диметилпиридина и
даже пиридина. В некоторых образцах «р-пиколииовой фракции» было об
наружено присутствие значительных количеств нейтральных масел (по-ви-
димому, непредельные углеводороды). Примерный состав технической
«₽-пиколиновой фракции»: 3-метилпиридина 25—30%, 4-метилпиридина 25-
30%, 2,6-диметилпирпдина 15—20%, пиридина, 2-метилпиридина и 2,4-дч.
метилпиридииа 15—25%.
3. При температуре 40—60° иногда происходит внезапное застывание
реакционной массы в густую кашу и мешалка останавливается. Это бывает
при плохом размешивании, когда мешалка не размешивает реакционную
массу у стенок колбы.
4. Промывку следует производить, строго согласно описанию, так как
именно такая промывка позволяет лучше всего отмыть комплексную соль от
гомологичных пиридиновых оснований с минимальными потерями за счет
растворимости комплексной соли. Если исходная «|3-пиколииовая фракция»
содержит нейтральные масла непредельного характера, то последние в ус-
ловиях реакции сильно осмоляются, и комплексная соль имеет грязно-голу-
бой цвет. Это не влияет на выход и качество З-метилпиридина.
5. Проба на присутствие пиридинового основания в днетнллате заклю-
чается в том, что к 5—7 мл дистиллата в пробирке прибавляют 2—3 г твер-
дого едкого натра. Появление верхнего маслянистого слоя или капель го-
ворит о присутствии пиридинового основания. Процентное содержание вод-
ного 3-метилпиридина определяют титрованием 0,1 н. раствором соляной
или серной кислоты в присутствии метилового красного или метилового
оранжевого. Более точные результаты могут быть получены потенциометри-
ческим титрованием.
6. Полученный 3-метилпиридин может быть применен для многих це-
лей без дополнительной очистки. В частности, окисление его перманганатом
калия дает никотиновую кислоту с т. пл. 235—236° и выходом 60—65%.
Повторная очистка через комплекс с сернокислой медью мало повы-
шает содержание З-.метилпиридина. Гораздо более избирательно это пири-
диновое основание взаимодействует с хлористой медью, через продукт при-
соединения с которой удается получить высокоочищенный 3-метилпиридцщ
48
7. Сернистый газ пропускается в реакционную массу для предупреж-
дения окисления хлористой меди в хлорную. Примесь хлорной меди весьма
вредна, так как комплекс 4-метилпириднна с хлорной медью более устойчив,
чем комплекс с нею 3-метилпиридина. Появление хлорной меди в реак-
ционной массе легко наблюдается по ярко-синей окраске раствора. Допу-
стим лишь голубоватый илн зеленоватый цвет раствора над осадком комп-
лекса. Вместе с тем, вреден и избыток сернистого газа, так как это приво-
дит к образованию труднофильтруемого комплекса.
8. Ректификация производилась на обычной ректификационной колонке
с высотой ректифицирующей части 40 см, внутренним диаметром 8—10 ли.
заполненной трехгранными спиралями из нихромовой проволоки (диамет-
ром 0,2 зилО размером 1,5—2 мм (насадка Левина), снабженной головкой
полной конденсации обычной конструкции. Подобная ректификация совер-
шенно необходима, так как только она позволяет освободиться от следов
пиридина, который содержится в исходной «8-пиколиновой фракции» в ко-
личестве до 5% и заметно соосаждается с 3-метилпиридином при комплек-
сообразовании с солями меди.
9. Определение температуры кристаллизации производилось в пробир-
ке длиной 15 см со стандартным шлифом № 14,5, снабженной термометром
с ценой деления 0,1 °|, пропущенным через резиновую трубку, надетую на
короткий шлиф-керн, закрывающий пробирку. Тем самым ограничивался
доступ влаги воздуха, следы которой могут исказить результаты анализа
(сухой 3-метилпиридин весьма гигроскопичен). Охлаждение пробирки про-
изводилось с помощью твердой углекислоты и ацетона. Необходимо иметь
в виду, что 3-метилпирнднн склонен легко переохлаждаться, поэтому охла-
ждение следует вести медленно, потирая стенку пробирки концом термо-
метра.
10. Наиболее строгим критерием чистоты является температура кри-
сталлизации 3-метилпиридина, поскольку показатели преломления и удель-
ные веса 3- и 4-метилпиридинов весьма близки. Температура плавления
пикрата 3-метнлпиридина может служить достаточно надежной характе-
ристикой чистоты 3-метилпиридина только в том случае, когда получение
пикрата производится в присутствии 10—15%-ного избытка пикриновой
кислоты в смеси спирт — эфир.
Согласно литературным данным [7, 9] чистый 3-метилпиридин имеет
следующие константы: т. кип. 143,14° при 760 мм, т. пл. —18,2°, Пд
1,50232, 0,95650, т. пл. пикрата 149—150°.
ЛИТЕРАТУРА
1. А. Дирихс, Р. Дубинка, «Фенолы и основания из углей», стр. 414—
426, Гостоптехиздат, 1958.
2. Ю. И. Чумаков, дисс., «Исследование в области разделения пириди-
новых оснований», Москва, Институт элементоорганических соединений,
АН СССР, 1959, стр. 6.
3. Пат. США 2489884. 1949; Chem. Abstrs. 44, 2734 (1950).
4. Пат. ГДР 6013, 1951.
5. П. А. Гангрский, Е. В. Швецова, Ю. И. Чумаков. Авт. свид. СССР
106570, 1957.
6. Ю. И. Чумаков, П. А. Гангрский. Авт. свид. СССР 128021, 1960.
7. Е. A. Coulson, I. f. [ones, j. Soc. Chem. lnd.; 65, 169 (1946).
8. D. P. Biddiscombe, E. A. Coulson., /[. Handley., E. F. G. Herin-
gton J. Chem. Soc., 1954, 1957,
9. Англ. пат. 598036, 1948; Chem. Abstrs., 42, 4614 (1948).
Поступила в феврале !962 г. Киевск. ордена Ленина политеха, ин-т (КИИ)
4 Зак. (22
4-МЕТИЛПИРИДИН
(у-Пиколии)
Ю. И. ЧУМАКОВ
СН3
1
N
C6H7N М. в. 93,12
4-Метилпиридин является промежуточным продуктом в
синтезах 4-винилпиридина (мономер), изоникотиновой кисло-
ты, изоникотиннитрила, изоникотинамида, высших 4-алкилпи-
ридинов, 4-пиридилкарбинола, 4-пиридинальдегида и других
4-замещенных производных пиридина.
Единственным существенным источником 4-метилпиридина
является «р-пиколиновая фракция» пиридиновых оснований.
Синтетические методы получения, в частности полный синтез
4-метилпиридина, исходя из кротонового альдегида и алкил-
виниловых эфиров [1], не имеют препаративного значения из-
за сложности, многостадийности и невысокого полного вы-
хода.
Для выделения из «р-пнколнновой фракции» и очистки 4-
метилпиридина использовались те же методы, что и для 3-ме-
тилпиридина (см. обзоры, стр. 45).
Для выделения только 4-метилпиридина применялись:
комплексообразование с солями платины [2], хлористым каль-
цием [3, 4J, кристаллизация в спиртовых растворах оксалата
[5—7] и др. способы.
Нами было найдено, что выделить из «р-пиколиновой фрак-
ции» в одну стадию чистый 4-метилпиридин не удается ни од-
ним из упомянутых способов, и показано, что для получения
чистого 4-метилпиридина вполне достаточна двухстадийная
очистка. Вначале выделяют концентрат 4-метилпиридина че-
рез комплекс с хлоридами кобальта [8], никеля [9] или желе-
за [10]- Полученное таким путем 85—93%-ное основание, со-
держащее в качестве примесей, главным образом 3-метилпи-
ридин и пиридин, подвергают окончательной очистке через
продукт присоединения с хлористым кальцием [3], который бе-
рут в количестве, недостаточном против необходимого по урав-
нению реакции (см. ниже). В результате получают 4-метцл-
пиридин с содержанием около 98%.
Прямое выделение 4-метилпиридина из «р-пиколнновой
фракции» через комплекс с хлористым кальцием, согласно па-
тенту [3], приводит примерно к 90%-ному основанию; необхо-
50
дима повторная очистка таким же путем. Между тем, выделе-
ние концентрата 4-метилпиридина через комплексы с хлори-
дами кобальта, никеля или железа (двухвалентного) значи-
тельно удобнее, поскольку процесс протекает в минимальных
объемах и не требует органических растворителей.
Степень чистоты получаемого 4-метилпиридина контроли-
ровалась по температуре кристаллизации [11, 12] и по темпе-
ратуре плавления пикрата.
4 I
I
ВЫДЕЛЕНИЕ КОНЦЕНТРАТА 4-МЕТИЛПИРИДИНА
4-СоС1а+4НС1 ->
«Р-пиколиновая фракция»
СН3
А /\/СНз
-CoClr4| | + 4 । । 4-4 | |
'А ХСН3
•НС1
СН3
СоС12-4 A 4-2NaOH -» 4
А
СНз
|А 4-Co(OH)3 + 2NaCl
\/
N
Характеристика основного сырья
«р-Пиколиновая фракция», ЧМТУ 3454—53.
Хлористый кобальт, ГОСТ 4525—48.
Хлористый кальций, ГОСТ 4141—48.
Условия получения
В трехгорлую колбу емкостью 1 л, снабженную мешалкой,
капельной воронкой, обратным холодильником и термометром,
помещают 110 мл концентрированной соляной кислоты (d 1,18;
46,8 г 100% НС1) и прибавляют 64,5? хлористого кобальта (со-
ли СоСЬ -бНаО). Затем из капельной воронки постепенно при-
бавляют 330 г (350 мл) технической «р-пиколиновой фракции»
(см. примечание 1). За счет тепла, выделяющегося при нейт-
4* 51
рализации соляной кислоты, температура быстро поднимается
до 93—100°, и прибавление ведут с такой скоростью, чтобы
она держалась на этом уровне (см. примечание 2). После при-
бавления пиридиновых оснований реакционную массу, разме
шивая, быстро доводят до интенсивного кипения и сразу после
этого начинают постепенно охлаждать до 10—15°. При этой
температуре реакционную массу размешивают в течение полу-
часа. Выпавший осадок комплексной соли отфильтровывают
на воронке Бюхнера с. полотняным фильтром, тщательно отжи-
мая. Отключают вакуум и осадок на фильтре смачивают 50 м'
холодной воды (5—10°). Этой же водой предварительно обмы-
вают колбу, в которой проводили реакцию. Разминают шпате-
лем, дают постоять 10—15 минут, после чего фильтруют, отжи-
мая досуха. Осадок переносят обратно в колбу и размешива-
ют со 100 мл холодной воды в течение 20—30 минут, после чего
снова фильтруют (см. примечание 3). Получают около 125 г
влажного хлорида тетра-4-метилпиридинкобальта, имеющего
серовато-розовый цвет. Его вносят при охлаждении в 100 мл
20%-ного водного раствора едкого натра и перегоняют с па-
ром до отсутствия 4-метилпиридина в дистиллате (см. приме-
чание 4). Получают около 350 мл водного раствора 4-метил-
пиридина, который при перемешивании и охлаждении обраба-
тывают твердым едким натром (из расчета 20 г на 100 мл
раствора). Выделившееся основание 4-метилпириднна отделя-
ют от нижнего щелочного слоя и определяют процентное со-
держание полученного продукта (см. примечание 5). Получа-
ют в среднем около 80 г концентрата 4-метилпиридина (72 г
сухого продукта). Содержание 4-метилпиридина в безводном
продукте составляет 87—93% (остальное — пиридин, 3-метил-
пиридин и другие компоненты «р-пиколиновой фракции») (см.
примечание 6). Его без дополнительной обработки используют
для окончательной очистки через комплекс с хлористым каль-
цием.
ОКОНЧАТЕЛЬНАЯ ОЧИСТКА 4-МЕТИЛПИРИДИНА
сн3 сн3
СаС1,-2 -'г +
\Х N
N
52
СНз сн3
I I
СаС13.2 1/х1 -> 2 + СаС12.
\/ \/
N N
Условия очистки
В трехгорлую колбу емкостью 1 л, снабженную водоотде-
лителем, соединенным с обратным холодильником (см. приме-
чание 7), мешалкой со ртутным или масляным затвором и тер-
мометром, помещают 200 мл бензола, 160 г концентрата 4-ме-
тилпиридина, полученного, как описано выше (144 г сухой сме-
си оснований), и 147 мл (196 г) 35%-ного профильтрованного
раствора хлористого кальция (d 1,336), 68,7 г 100%-ной соли
СаС12 (см. примечание 8).
Производят азеотропное обезвоживание, нагревая при пе-
ремешивании содержимое колбы на масляной бане и время от
времени сливая воду из водоотделителя в мерный цилиндр.
После того как выделится примерно 2/3 рассчитанного количе-
ства воды, начинает выпадать белый кристаллический осадок
комплекса хлористого кальция и 4-метилпиридина. Обезво-
живание продолжают до полного прекращения отделения во-
ды (см. примечание 9). Содержимое колбы охлаждают до
комнатной температуры и затем фильтруют на воронке Бюх-
нера, тщательно отжимая комплекс иа фильтре. Отсоединяют
вакуум и пропитывают осадок на фильтре 100 мл бензола, раз-
миная шпателем. Отфильтровывают и вновь повторяют про-
мывку, на этот раз 100 мл этилового эфира или низкокипяще-
го петролейного эфира. Получают около 160 г почти сухого
белого кристаллического продукта присоединения 4-метилпи-
ридина и хлористого кальция- Его переносят в колбу для пе-
регонки с паром, прибавляют 40—60 мл воды и перегоняют
с водяным паром до отсутствия в дистиллате основания (см.
примечание 4). Получают 600—700 мл водного раствора 4-ме-
тилпиридина, содержащего около 80 г чистого основания. Его
выделяют обработкой едким натром (из расчета 20 г щелочи
па 100 мл дистиллата) и высушивают твердым едким натром
при кипячении (1 часть щелочи на 5 частей основания) в тече-
ние 4—6 часов.
Высушенное основание 4-метилпиридина подвергают пере-
гонке на ректификационной колонке, эквивалентной 18—20
теоретическим тарелкам с флегмовым числом не менее 1:15.
Получают в среднем 6,0 г предгона с темп. кип. до 143,5",
59,6 г основной фракции с темп. кип. 143,5—144'7758 мм и
8,2 г кубового остатка. Температура плавления очищенного та-
ким образом 4-метилпиридина равна 2,4—2,6°, что соответст-
53
вует 97,8—98,1%-ной чистоте. Температура плавления пикра-
та, полученного из этого основания, составляет 167—168°
(испр.) (см. примечание 9). Выход 4-метилпиридина состав-
ляет 33,5% от теоретически возможного, считая, что исходная
«|3-пиколиновая фракция» содержит 27% этого основания или
9,2% (весовых), считая на «р-пиколиновую фракцию» (см-
примечание 10).
Примечания.
1. О составе применявшейся «Р-пиколиновой фракции» см. примеча-
ние 2 на стр. 48 в статье «З-Метилпиридии». Для выделения 4-метилпири-
дина могут быть использованы маточники после извлечения 3-метилпири-
дина из «р-пиколиновой фракции» с помощью сернокислой меди. Для этого
их насыщают твердым едким натром при охлаждении и перегоняют с водя-
ным паром. Из дистиллата смесь пиридиновых оснований извлекают обыч-
ным образом. Содержание 4-метилпиридина в полученной смеси пиридино
вых оснований выше, чем в «Р-пиколиновой фракции» (примерно 40%) и,
в соответствии с этим^ производится пересчет необходимых количеств хло-
ристого кобальта и соляной кислоты.
Вместо хлористого кобальта с тем же результатом могут быть взяты
эквимолекулярные количества хлористого никеля [9] или хлористого железа
[10]. В последнем случае реакция проводится в токе инертного газа (азота)
с целью предупреждения окисления соли двухвалентного железа.
2. В начале прибавления идет энергичная реакция нейтрализации со-
ляной кислоты пиридиновыми основаниями. При этом происходит сильное
дымообразование (хлоргидраты оснований), поэтому реакцию лучше прово-
дить под тягой.
3. Рекомендуется проводить промывку, строго придерживаясь приве-
денного описания (см. примечание 4, стр. 48).
4. О пробе на отсутствие пиридиновых оснований в дистилляте (см.
примечание 5, стр. 48).
5. Содержание 4-метилпиридина определяют алкалиметрически (см.
примечание 5, стр. 48).
6. Полученный концентрат 4-метилпиридина может быть без дополни-
тельной очистки использован для получения изоникотиновой кислоты окис-
лением перманганатом калия или для синтеза 4-оксиметильиых производ-
ных (полупродукты при получении 4-винилпиридина и изоникотиновой кис-
лоты).
7. Использовался водоотделитель конструкции, описанной в статье [13],
позволяющий периодически сливать выделившуюся воду.
8. Хлористый кальций берется в количестве около 90% от необходи-
мого по уравнению реакции комплексообразования этой соли с 4-метилпи-
ридииом.’Раствор хлористого кальция может быть взят практически любой
концентрации, однако целесообразнее использовать как можно более кон-
центрированные растворы этой соли.
9. Об определении температуры кристаллизации и получении пикратов
4-меТилпиридина (см. примечание 9, стр. 49).
10. Выход 4-метилпнридина может быть существенно повышен путем
использования бензольного маточника после отделения комплекса с хло-
ристым кальцием, в котором содержится примерно столько же 4-метилпи-
ридица.
Согласно наиболее достоверным литературным данным., 4-метилпирп-
дии имеет следующие константы: т. кип. 145,36’ при 760 мм, г. пл. 3,65°,
Лр 1,50144, </420 0,95478 [11], т. пл. пикрата 167—168° [12].
ЛИТЕРАТУРА
1. Пат. США 2748130, 1953; Chem. Abstrs., 50, 11369 (1956).
2. A. Ladenburg, Liebigs Ann. Chem., 247, 1—49 (1888).
54
3. Пат. США 2336502, 1943; Chem. Abstrs., 38, 3298 (1944).
4. Пат. США 2404167, 1946; Chem. Abstrs., 40, 6506 (1947).
5. Пат. США 2728771, 1955; Chem. Abstrs., 50, 10796 (1956).
6. Пат. США, 2818411, 1957; Chem. Abstrs., 52. 4964 (1958).
7. Чехосл. пат. 85823, 1956, Р.Ж.Хим., 1958, № 20, 68395.
8. Ю. И. Чумаков, Авт. свид. СССР 115571, 1958.
9. Ю. И. Чумаков, Авт. свид. СССР 105811, 1957.
10. Ю. И. Чумаков, И. И. Чумакова, Авт. свид. 106597, 1957.
11. D. Р„ Blddiscom.be, Е. A. Coulson, R. Handley, Е. F. G. Herington,
J, Chem. Soc., 1954, 1957.
12. E. Л. Coulson, J. I. Jones, J. Soc. Chem. Ind., 65, 169 (1946).
13. G. Black, E. Depp, В. B. Corson, J. Organ. Chem., 14, 14 (1949).
Поступила в феврале 1962 г. КП И
2,6-ДИМЕТИЛПИРИДИН
(2,6-Лутидин)
c7h9n
Ю. И. ЧУМАКОВ
М I!. 107,15
2,6-Диметилпиридин используется как исходный продукт
в синтезе 2-метил-6-винилпиридина, 2,6-пиридиндикарбоновой
кислоты 2,6-диоксиметилпиридина (мономеры), 2-метил-6-пи-
ридинальдегида, 2,6-диметил-З-нитропиридина и других сое-
динений. Он находит применение в бумажной хроматографии,
а также используется в качестве дегидрогалогенирующего
средства.
Основным источником получения 2,6-диметилпиридина яв-
ляется «₽-пиколиновая фракция». Известный полный синтез
2,6-диметилпиридина из ацетоуксусного эфира [1, 2] мало удо-
бен вследствие многостадийности и невысоких выходов.
Для выделения в чистом виде 2,6-диметилпиридина из «р-
пиколииовой фракции» использовались методы, общие с при-
менявшимися для 3-метилпиридина (см. ссылки, стр. 45). Для
выделения только 2,6-диметилпиридина предлагалось приме-
нять: кристаллизацию в спиртовом растворе фталата [3], комп-
лексообразование загрязняющих 2,6-диметилпиридин примесей
(3- и 4-метилпириднны) с фтористым бором [4], осаждение
55
продуктов присоединения с мочевиной [5, 6], тиомочевиной [7],
осаждение в виде соли имида бензолсульфокислоты [8], а так-
же другие способы.
При проверке описанных в литературе способов выделения
и очистки 2,6-диметилпиридина мы нашли, что наиболее удо-
бен и дает надежные результаты способ, использующий комп-
лексообразование с мочевиной и впервые описанный в патен-
те [5].
В приводимой ниже методике использован способ [5] с не-
которыми уточнениями и изменениями, имевшими целью упро-
стить работу и обеспечить воспроизводимость результатов
Очистка 2,6-диметилпиридина производилась в две стадии: вы-
деление концентрата основания через продукт присоединения
с мочевиной и последующая очистка переосаждемием такого
же комплекса из воды. Полученный 2,6-диметилпиридип имел
содержание более чем 99%. Характерно, что подобная очистка
в наших опытах дала результаты, не худшие, чем в том случае,
когда 2,6-диметилпиридин после очистки через комплекс с мо-
чевиной подвергался дополнительной семикратной очистке
фракционированным вымораживанием [9].
Контроль за чистотой 2,6-диметилпиридина производился
по температуре кристаллизации, что позволяло одновременно
оценить и его процентное содержание [10, 11], а также по тем-
пературе плавления пикрата.
Следует подчеркнуть важность для получения воспроизво-
димых результатов тщательного высушивания получаемого
основания и проведения ректификации на эффективной ко-
лонке.
ПОЛУЧЕНИЕ 2,6-ДИМЕТИЛПИРИДИНА
H2Nv
^С=О+Н2О ->
H,N
,8-пнколиновая фракция’
Iх Х| • (NH2)3CO-H2O +
НСзХ СН3
сн3
сн3 I
+ ' I +1 'I
\ / \ /
N N
56
I I . (NH2)2CO-H2O
HsC'^'V XCH3
|/X| + CO(NH2)g + HtO
H3CX к/^СН,
Характеристика основного сырья
«|3-Пиколиновая фракция», ЧМТУ 3454—48.
Мочевина, ГОСТ 6691—53.
Условия получения
В колбу емкостью 2 л, снабженную термометром, мешал-
кой и обратным холодильником, вливают 400 мл воды, нагре-
той до кипения, прибавляют 500 г мочевины и при перемеши-
вании нагревают до 90—95°. Затем, при непрерывном переме-
шивании, струей прибавляют 500 г (525 мл} технической «£-
пиколиновой фракции» (см. примечание 1). Реакционной мас-
се дают постепенно и при перемешивании охлаждаться до
комнатной температуры (см. примечание 2), затем еще 1 час
перемешивают при температуре 0-г-+5° (охлаждение водой
со льдом).
Выпавший комплекс 2,6-диметилпиридина и мочевины от-
фильтровывают, отжимают и промывают два раза по 50 мл
холодной (0-:- +5°) воды. Получают 320 г влажного комплек-
са 2,6-диметилпиридина с мочевиной. Его вносят в 200 мл во-
ды и при перемешивании нагревают до полного разложения
и появления двух несмешивающихся слоев (90—95° в реакци-
онной массе), затем охлаждают, как описано выше. Пере-
осажденный комплекс отфильтровывают и два раза промы-
вают порциями по 25 мл холодней (0-:- +5°) воды. Получают
в среднем 167 г влажного продукта, который переносят в круг-
лодонную колбу емкостью 0,5 л и перегоняют с водяным па-
ром до исчезновения пиридинового основания в дистиллатс
(см- примечание 3). Получают 400 мл водного раствора 2,6-
диметилпиридина, содержащего 62,0 г основания (см. приме-
чание 4).
Дистиллят при охлаждении обрабатывают твердым едким
натром в количестве около 20 г щелочи на каждые 100 мл ра-
створа. Выделившееся основание отделяют от нижнего водно-
щелочного слоя и дополнительно обрабатывают твердым ед-
ким натром из расчета примерно 1 часть щелочи на 5 частей
основания при кипячении с обратным холодильником в тече-
57
ние 4—6 часов. Охлаждают и фильтрованием отделяют сухое
основание от щелочи.
Полученный продукт подвергают ректификации на колон-
ке, эквивалентной 18—20 теоретическим тарелкам с флегмо-
вым числом не менее 1:15. Получают в среднем около 3,0 г
предгона с температурой кипения до 143,57752 мм, 52,8 г
фракции с температурой кипения 143,5—1447752 мм, пред-
ставляющей чистый 2,6-диметилпиридин, а также 4,1 г кубово-
го остатка (см. примечание 5). Полученный 2,6-диметилпири-
дин имеет температуру плавления —6,7—: 6,9°, что соответ-
ствует 98,1—99,1 %-ному содержанию (см. примечание 6).
Пикрат основания имеет температуру плавления 163—164°
(испр.). Согласно наиболее достоверным литературным дан-
ным, 2,6-диметилпиридин имеет следующие константы: темпе-
ратура кипения 144,05° при 760 мм, температура плавления
—6,20°, 1,49767, d.-2,i 0,92257 [11], пикрат имеет температу-
ру плавления 163—164° [10].
Из маточников после второго осаждения комплекса может
быть получено дополнительное количество чистого 2,6-дкме-
тилпиридина. Для этого маточники перегоняют с водяным па-
ром до отсутствия основания в дистиллате. Из этого дистил
лата обычным образом, обработкой едким натром, выделяют
технический 2,6-диметилпиридин, который затем вводят в ре-
акцию комплексообразования, согласно выше приведенному
описанию (см. примечание 7). Произведя осаждение и про-
мывку комплекса, его разлагают перегонкой с паром и после
выделения, описанного выше, высушивания и ректификации
получают дополнительно 3,1 г на каждый опыт чистого 2,6-ди-
метилпиридина с температурой плавления —6,9°. Полный вы-
ход составляет 70% теоретического, считая, что исходная «|3-
пиколиновая фракция» содержит 15% 2,6-диметилйиридкпа,
или 10,5% (весовых), считая на «|3-пиколиновую фракцию»
(см. примечание 8).
Примечания.
1. О составе применяемой «р-ппколиновой фракции» см. примечание 2,
стр. 49. ,
2. Горячий концентрированный раствор мочевины и «₽-пиколиновой
фракции» в воде разделяется на два слоя, из которых верхний содержит,
главным образом, пиридиновые основания, а нижний — мочевину. Переме-
шивание необходимо, чтобы создать благоприятные условия для кристалли-
зации комплекса.
3. О пробе на отсутствие пиридиновых оснований в дистиллате см. при-
мечание 5, стр. 48.
4. Содержание 2,6-диметилпиридина определяют алкалиметрически см.
примечание 5, стр. 48.
5. Применялась ректификационная колонка, описанная в примечании 8,
стр. 49.
6. Об определении температуры плавления пиридиновых оснований см.
примечание 9, стр. 49.
7. Пнрнднповое основание, мочевина и вода берутся в тех же соот-
ношениях.
58
|тр. 47 и
их насы-
водяным
обычным
8. Для выделения 2,6-диметилпиридина могут быть использованы ма-
точники после извлечения 3-метилпиридина из «р-пиколиновой фргкции» с
помощью сернокислой меди, или аналогичные маточники после стделения
4-метилпиридина через комплекс с хлористым кобальтом (см.
стр. 51), или после отделения обоих этих оснований. Для этого
щают твердым едким натром при охлаждении и перегоняют с
паром. Из днстиллата смесь пиридиновых оснований извлекают
образом (насыщение едким натром и т. д. — см. выше в методике). Выде-
ленная смесь пиридиновых оснований, которая содержит уже зн;чительи;>
больше 2,6-диметилпиридина, чем исходная «р-пиколиновая фракция», вво-
дится в реакцию комплексообразования с мочевиной, причем соо-
реагирующих компонентов берутся точно такие же, как и в случ
исходят из «р-пиколиновоп фракции».
ЛИТЕРАТУРА
1. «Синтезы органических препаратов», Сб. 2, стр. 214, ИЛ,
1949.
2. М. П. Опарина, А. Б. Карасина, Б. П. Смирнов, Ж. прикл. ;
965 (1953).
3. А. М. Григоровский, 3. М. Кимен, Авт. свид. СССР 64586,
4. И. С. Brown. S. Johnson, Н. Podall, J. Amer. Chem. Soc.,
(1954).
5. Пат. США 2295606, 1943; Chem. Abstrs., 38, 493 (1944).
ношения
ге, когда
Москва,
:имин, 11,
1945.
76, 5556
6. И. Л. Липлавк, Е. П. Болитер, Ж. прнкл. химия, 24, 191 (1951).
7. Пат. США 2325880, 1943; Chem. Abstrs., 38, 493 (1944).
8. Н. Н. Дыханов, Авт. свид. СССР 104875, 1956.
9. А. 17. Поздеева, Е. М. Гепштейн, Ж. общ. химии, 22, 2065 11952).
10. Е. A. Coulson, J. 1. Jones, J. Soc. Chem. Ind., 65, 169 (1946).
11. D. P. Biddlscombe, E. A. Coulson, R. Handly, E. E. G, Herington,
J. Chem. Soc., 1954, 1957.
Поступила в феврале 1962 г.
КПП
C5H6NO
N-ОКИСЬ ПИРИДИНА
( Пиридин- N -оксид)
Ю. И. ЧУМАКОВ
М. а. 95,10
Известны несколько способов получения N-окиси
на. Чаще всего используются два способа. Первый,
женный Отиаи, заключается в воздействии на пирид
30%-ным пергидролем в присутствии уксусной кисл
Выходы высоки, однако способ имеет недостаток — не
пириди-
предло-
ин 27—
ОТЫ [1].
обходи-
мость работать с большими объемами (большой избыток уксус-
ной кислоты), а также большая длительность процесса (12
59
часов и более). И. А. Красавиным [2] было найдено, что при
получении N-окиси пиридина по этому способу расход уксус-
ной кислоты может быть уменьшен в 5 раз- Однако при этом
существенно снижается выход N-окиси пиридина (до 70% вме-
сто 96%). Второй способ заключается в воздействии на пири-
дин 40%-ной надуксусной кислоты; выходы также высоки (до
90%), но используется опасная в обращении надуксусная кис-
лота. Синтез занимает мало времени (около 1 часа вместо
12—15 часов по упомянутым выше способам), и реакция про-
текает в небольших объемах [3].
Другие способы получения N-окиси пиридина, в частности
действием на пиридин надбензойной [4] или мононадфталевой
[5] кислотой, практического значения не имеют, поскольку мо-
лекулярный вес этих надкислот значительно выше, чем у над
уксусной, и в то же время значительно более сложным являет-
ся выделение N-окиси пиридина из реакционной массы.
Нами было найдено, что N-окись пиридина может быть по-
лучена действием на смесь пиридина и 27—30 %-ной перекиси
водорода уксусным ангидридом. При этом температура быст-
ро поднимается за счет тепла реакции и поддерживается в ин-
тервале температур 70—75°. Преимуществом такого метода
получения N-окиси пиридина является то, что реакция проте-
кает в малых объемах за короткий срок (0,5—1,5 часа), и вме-
сто довольно опасной в обращении концентрированной надук-
сусной кислоты используются перекись водорода и уксусный
ангидрид. По-видимому, в качестве промежуточного продукта
реакции образуется надуксусная кислота, которая сразу же
превращается в N-окись пиридина, причем само пиридиновое
основание катализирует реакцию образования надуксусной
кислоты из пергидроля и уксусного ангидрида (известно, что
в отсутствие катализирующих добавок серной кислоты или др.
веществ смесь уксусного ангидрида и пергидроля длительное
время остается без изменений, не образуя надуксусной кис
лоты.
Выделение N-окиси пиридина производится обычным обра-
зом— перегонкой в вакууме. Выход практически количествен-
ный (95%). Способ применим для получения N-оксидов не
только пиридина, но также N-Оксидов 2-, 3- или 4-метилпири-
динов, изомерных диметилпиридинов, хинолина, никотиновой
кислоты и других пиридиновых оснований.
СИНТЕЗ N-ОКИСИ ПИРИДИНА
/ \ сн3—cf I I
I i+H2Oa+ Х° 'n7 + сн3соон
NX СН,—с / ф . СН3СООН
о о
60
I i II
\/ \/ + СНзСООН
N N
4 -CHgCOOH I
о' о
Характеристика основного сырья
Пиридин, ГОСТ 2747—44-
Уксусный ангидрид, ГОСТ 787—55.
Перекись водорода, медицинская (27—30%-ная).
Условия получения
К 241,5 мл (237,3 г 3 М) пиридина (см. примечание 1), по-
мещенного в трехгорлую колбу емкостью 2 л, снабженную ме-
шалкой, пропущенной через шариковый холодильник, термо-
метром и капельной воронкой, при- перемешивании прибавля-
ют 363 мл (399 г) 27,5%-ного пергидроля (3,24 Л4), причем тем-
пература поднимается до 30—35°, затем в течение 1—1,5 часа
прибавляют 306 мл 1330 г 3,24 Л1) уксусного ангидрида, под-
держивая температуру в реакционной массе в интервале
70—85°. После прибавления уксусного ангидрида реакцион-
ную массу выдерживают в течение 1 часа на кипящей водяной
бане, затем по каплям прибавляют 10 мл 40%-ного водного фор-
малина (см. примечание 2) и продолжают нагревание на ки-
пящей водяной бане еще в течение 1 часа. Полученный уксус-
нокислый раствор N-окиси пиридина упаривают на кипящей
водяной бане в вакууме 20—60 мм. Внимание! (см. примеча-
ние 3). Остаток перегоняют при 1—3 мм в вакууме от масля-
ного насоса (см. примечание 4). Вначале постепенно отгоняет-
ся уксусная кислота, которую отдает при термической диссо-
циации ацетат N-окиси пиридина, затем перегоняется N-окись
пиридина. Собирают предгон с температурой кипения до
10471 мм и основную фракцию с температурой кипения 104—
10871 мм, представляющую собой N-окись пиридина (см.
примечание 5).
Выход N-окиси пиридина 240,0 г (84% теоретического)-
Температура кристаллизации 66—68°. Температура плавления
пикрата 180—181,5° (испр.). Из объединенных предгонов и ку-
бовых остатков получают дополнительно на каждый опыт по
26,2 г (11% теоретического) N-окиси пиридина с теми же кон-
стантами. Общин выход N-окиси пиридина 95% теоретическо-
го (см. примечание 6).
Примечания.
1. В реакцию вводился обычный сухой пиридин с температурой кипе-
ния 114—115°.
2. Формальдегид прибавляют для разрушения избытка перекиси водо-
рода, не вступившей в реакцию.
61
3. Внимание! Прежде чем начать перегонку в вакууме, необходимо
убедиться в отсутствии непрорсагировавшей перекиси водорода, для чего
производят анализ с йодистым калием и крахмалом. В отсутствие переки-
сей раствор не изменит окраску.
4. В случае, если используется масляный вакуум-насос, между прибо-
ром для перегонки и вакуумным насосом необходимо включить две ловуш-
ки, охлаждаемые смесью метанол — твердая углекислота емкостью каждая
не менее 100—150 мл. Ловушки должны иметь широкие вводные трубки,
иначе возможна закупорка их закристаллизовавшейся уксусной кислотой.
В ловушках собирается уксусная кислота, образующаяся при термической
диссоциации N-окиси пиридина.
5. Прибор должен быть пригоден для перегонки в вакууме затвердеваю-
щих веществ. При проверке синтеза использовался прибор, состоящий из
круглодонной колбы емкостью 1 л, соединенной иа шлифе с насадкой
Кляйзена^ имеющей длинный отвод (50 см), и «паука» с приемниками. Пе-
регонка в вакууме может быть произведена и при разрежении 15—20 мн
(от водоструйного насоса). При этом не происходит сколько-нибудь замет-
ного снижения выхода.
6. Приведенные данные получены иа десяти контрольных опытах. Спо-
соб проверялся более ста раз и была подтверждена полная безопасность
его при условии соблюдения описанных выше требований.
ЛИТЕРАТУРА
1. Е. Ochiai, J. Organ. Chem, 18, 531 (1953).
2. И. А. Красавин, Химические реактивы и препараты, вып. 1, стр. 83,
ИРЕА, 1960.
3. «Синтезы органических препаратов», сб. 5, стр. 56, ИЛ, 1954.
4. J. Melsenheimer, Вег., 59, 1а4о (1326)
5. В. Bobranski et al, Вег, 71, 2385 (193а).
Поступила в феврале 196 2 г. КПП
2-ОКСИПИРИДИН
(а-Пиридон)
CsH6ON
Ю. И. ЧУМАКОВ, 3. М. КОРСАКОВА
\Гон
М. в. 95, 10
Обычным и наиболее часто употребляемым методом полу-
чения 2-оксипиридина до последнего времени было диазотиро-
вание 2-аминопиридина, впервые предложенное Чичибабиным
62
[1]. Недостатком способа является трудность выделения в чи-
стом виде 2-оксипиридина из реакционной массы. Другой спо-
соб использует циклизацию яблочной кислоты в кумалино-
вую, превращение ее в 2-окси-5-пиридинкарбоновую кислоту
(6-оксиникотиновую) и декарбоксилирование последней в 2-
оксипиридин [2, 3]. Недостатком этого способа является много-
стадийность и невысокий выход. 2-Оксипиридин получается
также и при декарбоксилировании 6-оксихинолиновой кислоты
[5—7]. Он может быть получен при пропускании паров пириди-
на над порошком едкого кали при 300—320° или при нагрева-
нии 2-хлорпиридипа с едким натром при 175° [4].
Благодаря развитию в последние годы химии N-окиси пи-
ридина стал вполне доступным продуктом 2-пиридилацетат
[9] (см. также стр. 65). Последний легко омыляется в 2-окси-
пиридин. В литературе упоминается об омылении 2-пиридил-
ацетата 10%-ным раствором соляной кислоты. Однако в этом
случае является сложным выделение из реакционной массы
2-оксипиридина вследствие легкой растворимости его в воде.
Нами было найдено, что 2-пиридилацетат легко вступает в
реакцию переэтерификации со спиртами (метиловым, этило-
вым, н-бутиловым и др.), давая 2-оксипиридин и соответст-
вующий алкилацетат. Выходы при этом близки к количествен-
ному. Весьма существенным является то, что получаемый 2-ок-
сипиридин без дополнительной очистки свободен от уксусной
кислоты и воды в противоположность способу, использующе-
му гидролиз пиридилаиетата в водных растворах [9]. Предла-
гаемый способ не описан в литературе и позволяет сравнитель-
но простым путем, исходя из вполне доступного пиридина по-
лучить в три стадии 2-оксипиридин с общим выходом не-
сколько более 50% теоретически возможного.
2-Оксипиридин является исходным продуктом в синтезе
производных ряда пиридина (2-окси-5-пиридинкарбоновой
кислоты, 2-хлор-5-пиридинкарбоновой кислоты и др.).
СИНТЕЗ 2-ОКСИПИРИДИНА
|/Х| + н-С4Н,,ОН |ХХ| + н-С4НеОСОСН3
Х/ХОСОСН3 Y\H
Характеристика основного сырья
2-Пиридилацетат, см. стр. 65.
н-Бутиловый спирт, ГОСТ 6006—51.
Петролейный эфир, т. кип. 60—80°.
Бензол, ГОСТ 5955—51.
ИЗ
Условия получения
В круглодонную колбу емкостью 500 ли, снабженную об-
ратным холодильником, защищенным хлоркальциевой труб-
кой, заполненной натронной известью, помещают 300мл (243 г,
3,3 М) н-бутилового спирта и 50 г (0,362 М) 2-пиридилацетата
(см. примечание !)• Реакционную массу кипятят на масляной
бане в течение 6 часов с обратным холодильником. Заменяют
обратный холодильник на нисходящий и отгоняют смесь н-бу-
тилового спирта и бутилацетата вначале при атмосферном
давлении, затем в вакууме при 10—50 мм (водоструйный на-
сос). Остаток в колбе, представляющий собой технический 2-
оксипиридин, при охлаждении сразу же закристаллизовывает-
ся. Его смешивают с 20 мл петролейного эфира и отфильтро-
вывают. Осадок на фильтре промывают 2 раза по 10 мл пет-
ролейного эфира, после чего высушивают на воздухе. Полу-
чают 32,9 г технического 2-оксипиридина в виде слегка окра-
шенного кристаллического порошка. Выход 95,5% (см. приме-
чание 3). Т. пл. 103—105°. Продукт достаточно чист для боль-
шинства целей. В случае необходимости его очищают пере-
кристаллизацией из бензола, которого берут 10 мл на каждые
10 г 2-оксипиридина. Выход перекристаллизованного продукта
27 г (78,5% теоретически возможного). Т. пл. 107—108°
(испо.). По литературным данным, 2-оксипиридин имеет т. пл.
106—107°.
Примечания.
1. В реакцию вводился свежеперегнанный 2-пйридилацетат, полученный
взаимодействием N-окиси пиридина с уксусным ангидридом (см. стр. 65)
с т. кип. 78—85,5°/2 мм. 2-Пиридилацетат, хранившийся в течение года,
при переэтерификации с н-бутиловым спиртом дал сильно окрашенный
2-оксипиридни с пониженным выходом.
2. 2-Оксипиридин необходимо хранить в темноте, так как на свету он
постепенно темнеет.
3. Приведен средний выход по 16 опытам.
ЛИТЕРАТУРА
1. А. Е. Чичибабин, М. Д. Рязанцева, ЖРФХО, 47, 1571 (1915).
2. Н. V- Pechmann, О. Baltzer, Вег., 24, 3145 (1891).
3. Н. Weidel, Н. Strache, Monatsh. Chem., 7, 297 (1886).
4. А. Е. Чичибабин, Вег., 56, 1879 (1923). Герм. пат. 406208, 1924.
5. W. Koenigs, G. Koerner, Вег., 16, 2152 (1883).
6. 1Г. Koenigs, R. Oeigl, Вег., 17, 589 (1884).
7. A. Feer, W. Koenigs, Вег., 19, 2432 (1886).
8. Англ. пат. 288628; Chem. Abstrs., 23, 607, 1929.
9. Masa Katada, J. Pharmac. Soc. Japan, 67, 51 (1947); C’nem. Abstrs.,
45, 5167 (1951).
Поступила в феврале 1962 г. КПП
2-ПИРИДИЛАЦЕТАТ
(2-Ацетоксипиридин)
Ю. И. ЧУМАКОВ, 3. М. КОРСАКОВА
N ОСОСНз
С,Н,ОгК’
М. в. 137,14
2-Пиридилацетат впервые был получен Чичибабиным и
Соковым действием хлористого ацетила на безводную натрие-
вую соль 2-оксипиридина [1]. Было также найдено, что это сое-
динение может быть получено действием кипящего уксусного
ангидрида на N-окись пиридина [2]. В литературе отсутствуют
описания деталей этого синтеза. Так, в реферате упомянутой
японской работы [2] приведены слишком краткие данные, не
только недостаточные для воспроизведения синтеза, но и оши-
бочные (вероятно, ошибки референта). В частности, 2-пири-
дилацетату приписывается т. пл. 104° и т. кип- 150—160е при
0,09 мм. На самом деле 2-пиридилацетат является жидкостью
с температурой кипения НО—112° при 10 мм (см., например,
[1]).
При проверке способа получения 2-пиридилацетата из N-
окиси пиридина действием на него уксусного ангидрида мы
нашли, что лучшие выходы получаются при использовании
большого избытка уксусного ангидрида и нагревании в течение
5 часов. 2-Пиридилацетат легко выделяется из реакционной
массы перегонкой в вакууме после отгонки уксусного ангид-
рида.
2-Пиридилацетат является исходным продуктом для полу-
чения 2-оксипиридина и других производных ряда пиридина
СИНТЕЗ 2-ПИРИДИЛАЦЕТАТА
|/Х| + (СН3С0),0 -> Г Х|
\,z 'ОСОСНз + СН3СООН
N N
!
О
Характеристика основного сырья
N-окись пиридина, см. стр. 59.
Уксусный ангидрид, ГОСТ 787—55.
5 Зак. 622 65
Условия получения
В колбу емкостью 1 л, снабженную обратным холодильни-
ком, защищенным хлоркальциевой трубкой (все соединения
на шлифах), помещают 500 мл (540 г, 5,45 ЛГ) перегнанного
уксусного ангидрида, к которому прибавляют 95 г (1 Л1) N-
окиси пиридина (перегнанной, т. пл. 65—66° в запаянном ка-
пилляре)- Реакционную массу нагревают на масляной бане до
спокойного кипения уксусного ангидрида (150—155° в бане) и
продолжают кипятить с обратным холодильником в течение
5 часов. Заменяют обратный холодильник на нисходя-
щий и возможно полнее отгоняют уксусную кислоту и избыток
уксусного ангидрида (всего отгоняется около 450 мл, темпера-
тура в бане к концу отгонки около 170°). Остаток перегоняют
в вакууме (см. примечание 1), собирая фракции:
1 фракция — т. кип. до 78°/2 мм,
2 фракция — т. кип. 78—85,5°/2 мм.
В колбе остается значительное количество кубового остатка.
Выход 2-пиридилацетата составляет 53,8% теоретического
(см. примечание 2).
Примечания.
1. При перегонке в вакууме необходимо использовать ловушку доста-
точной емкости, охлаждаемую сухим льдом в метаноле или ацетоне. Пере-
гонка может быть произведена также п при вакууме 10—30 мм (от водо-
струйного пасоеа).
2. Приведен средний выход по 25 опытам, без учета 2-пиридилацетата,
содержащегося в предгонах. Из объединенных предгонов повторной пере-
гонкой может быть выделено дополнительно 3% выхода. Общий выход со-
ставляет 78,4 г, или 56,8% теоретического. При повторной перегонке 2-пи-
ридилацетат имеет т. кип. 83—85°/2 мм, n,0D 1,,5050, d^0 1,1468 (литера-
турные данные [1]: т. кип. ПО—112°/10 мм, 1,1475). Бесцветная или
слегка окрашенная жидкость с резким запахом. Легко разлагается спирта-
ми и водой. Хранить лучше в запаянной ампуле.
ЛИТЕРАТУРА
1. А. Е. Чичибабин, П. Г. Соков, Вег., 58, 2650 (1925).
2. Masa Kaiada. J. Pfiarm ас. Soc. Japan, 67, 51 (1947); Chem. Abstrs.
45, 5167, 8529 (1951).
Поступила в феврале 1962 г.
КПП
8-(п -ТОЛУОЛСУЛЬФОНИЛАМИНО)-ХИНОЛИН
(8-и-Тосиламинохинолии)
В. М. ДЗИОМКО, И. А. КРАСАВИН
CieH14N2O..S
М. в. 298,37
8-(п-Толуолсульфопиламино)-хинолин предложен ВНИИ
химических реактивов [1] для флуоресцентного определения
малых количеств цинка и кадмия. Только цинк и кадмий в
водном растворе при pH 8,0—8,3 вызывают флуоресценцию зе-
леного цвета. Чувствительность реакции, равная 0,1 мкг Zn2h
или Cd 2+ в 5 мл водного раствора, может быть увеличена в
несколько раз посредством применения экстракции. Важно,
что А13+даже в больших количествах (500 мкг в 5 мл) не вы-
зывает флуоресценции раствора. Только Си, Со, Ni и Те, при-
сутствуя в равных с определяемыми катионами количествах,
мешают реакции вследствие гашения флуоресценции.
В литературе описано получение этого вещества пои вза-
имодействии 8-аминохинолина с «-толуолсульфохлоридом в
различных условиях [‘2—4].
Впервые было кратко сообщено о проведении реакции пу-
тем нагревания эквимолекулярных количеств исходных ве-
ществ в этиловом спирте в присутствии ацетата натрия [2]. Бо-
лее подробно описаны синтезы 8-(л-толуолсульфониламино) -
хинолина, осуществленные нагреванием тех же вешсств в сре-
де пиридина при 40° в течение 12 часов [3] или добавлением
20%-ного раствора едкого натра к их раствору в ацетоне при
15° (выход неочищенного вещества 67,8%) [4].
Нами разработана методика получения 8-(л-толуолсульфо-
ниламино)-хинолина, пригодного для флуоресцентного анали-
за. Наилучшие результаты получены при проведении реакции
в среде пиридина и применении небольшого избытка (5—10%)
д-толуолсульфохлорида.
5* 67
Получение 8-(и -толуолсульфониламино)-хинолина
so2ci / \ /\
А Ах/1
I 1 |+| | + C6H5N — ] N + C6H6N НС1
\/ NH
|N| |
nh2 CH, so,
I
CH3
В колбе емкостью 250 мл с мешалкой (см. примечание 1)
растворяют 14,4 г (0,1 Л1) 8-аминохинслина в 50 мл свежепе-
регианнот'о пиридина и добавляют по каплям за 1 —1,5 часа
раствор 21,0 г (0,11 А4) n-юлуолсульфохлорида (см. примеча-
ние 2) в 50 мл такого же пиридина при размешивании и ох-
лаждении на водяной бане. Продолжают перемешивание еще
2—3 часа, а затем ошавляют до следующего дня при комнат-
ной температуре. На другой день поднимают температуру ба-
ни за 1 час до 100° и выдерживают на кипящей водяной бане
1 —1,5 часа. Охладив реакционную жидкость, выливают 2—
3 мл ее при энергичном размешивании в колбу с 1000 мл ди-
стиллированной воды (см примечание 3). Когда выпавшее
вначале масло закристаллизуется, медленно выливают на воду
остальную часть реакционной массы (см. примечание 4). Об-
разовавшуюся суспензию размешивают иа холоду еще 1 час,
затем отсасывают осадок, промывают его водой на фильтре до
исчезновения запаха пиридина и отжимают. Влажный осадок
переносят в 1,5—2-литровую колбу, содержащую раствор
13,6 г едкого кали в 800 мл дистиллированной воды, и нагре-
вают до кипения (см. примечание 5). Горячий раствор филь-
труют с отсасыванием от незначительного количества нера-
створимых примесей, фильтрат переносят в чистую колбу и
подогревают до полного растворения выпавших кристаллов
калиевой соли 8-(п-толуолсульфониламино)-хинолина. К горя-
чему прозрачному раствору приливают при размешивании
10%-ный раствор уксусной кислоты в дистиллированной воде.
Осаждение заканчивается при pH 6—6,5, для чего нужно око
ло 130 мл раствора кислоты. Суспензии дают охладиться, бе-
лый осадок отсасывают, промывают дистиллированной водой
и высушивают на воздухе.
Вещество растворяют в 2500 мл кипящего этилового спир-
та (см. примечание 6), горячий раствор фильтруют с отсасы-
ванием, к фильтрату добавляют 2500 мл дважды перегнанной
дистиллированной воды (см. примечание 3) и вновь подогре-
68
вают до полного растворения. При медленной кристаллизации
в течение ночи выделяются бесцветные иглы, их отсасывают и
сушат на воздухе.
Выход 21,5—22,5 г (72—75% теоретического). Т. пл. 153,5—
154,5° (см. примечание 7). Чувствительность к цинку в водном
буферном растворе [1] не хуже 0,1 мкг в 5 мл.
Примечания.
1. Вся аппаратура должна быть чистой и совершенно свободной от
следов посторонних веществ, так как даже незначительные примеси могут
резко ухудшить флуоресцентные свойства продукта. Совершенно недопу-
стимо попадание в продукт даже следов соединений цинка, кадмия, меди,
железа, никеля и кобальта. Нельзя прикасаться к продукту металлическими
предметами (шпатели и т. п.).
2. Применялись л-толуолсульфохлорид и пиридин квалификации ч., ед-
кое кали и уксусная кислота — х. ч. Пиридин и спирт перед употреблением
перегонялись.
3. Во всех операциях данного синтеза применялась дистиллированная
вода, а при перекристаллизации — дважды перегнанная пода (вторая пере-
гонка—в цельностеклянном приборе).
4. Если реакционную массу разбавить водой быстро, то вместо светло-
серой суспензии образуются крупные комки, которые труднее промывать,
5. Чтобы ускорить растворение, полезно раздавить крупные комки
стеклянной палочкой. Обычно остается лишь очень незначительное количе-
ство нерастворимого остатка.
6. Удобнее перекристаллизовать продукт в несколько приемов, беря по
100 мл спирта на 1 г вещества и добавляя затем равный объем воды.
7. По литературным данным, т. пл. 153° (из ацетона) [4], 154° (из раз-
бавленного метилового спирта) [3], 154—156° [2],
ЛИТЕРАТУРА
1. Е. А. Божевольнов, В. А1. Дзиомко, В. Серебрякова. Авт. свид.
Г Г ГР 190020 1959
2 A. Kaufmann, О. Zeller, Вег., 50, 1620 (1917).
3. W. Langenbeck, R. JUttemann, F. Hdlrung, Liebigs Ann. Chem., 499,
201 (19.32). v,
4. E. Cerkovnikov, P. Tomaslc, Archlv Kern., 19, 38 (1947); Chem. Ab-
strs., 42, 7724 h (1948).
Поступила в августе 1961 г. ИРЕА
8-(БЕНЗОЛСУЛЬФОНИЛАМИНО)-ХИНОЛИН
И. А. КРАСАВИН, В. М. ДЗИОМКО
CisHijNaOjS
М. в. 284,34.
69
8-(Бензолеульфониламино) -хинолин может применяться
вместо 8-(пара-толуолсульфониламино)-хинолина при флуо-
ресцентном определении малых количеств цинка и кадмия по
той же методике [1]. Для определения кадмия предпочтитель-
нее использовать 8-(бензолеульфониламино)-хинолин, так как
чувствительность его к кадмию в 10 раз выше, чем к цинку, и
достигает 0,005 мкг Cd 2+ в 5 мл раствора.
В литературе имеется краткое сообщение [2] о получении
8-(бензолеульфониламино)-хинолина путем постепенного при-
бавления пиридинового раствора бензолсульфохлорида к ра-
створу эквивалентного количества 8-аминохинолина в пириди-
не. Смесь была оставлена на ночь, затем продукт был осажден
водой и перекристаллизован из спирта- Выход не указан.
Нами разработан метод получения 8-(бензолсульфонила-
мино)-хинолина, пригодного для флуоресцентного анализа. Ус-
ловия синтеза и очистки мало отличаются от применяемых для
получения 8- (пара-толуолсульфониламино)-хинолина.
Получение 8-(бензолсульфониламино)-хинолина
SO2C1 I I I
1 х / \ /
/\/\ I N
| | | + | | + C6H5N - NH +C6H5N-HC1
' 'I N Х / SO,
NH2 I
В колбе емкостью 250 мл с мешалкой (см. примечание 1)
растворяют 14,4 г (0,1 Л1) 8-аминохинолина в 50 мл свежепере-
гнанного пиридина и добавляют по каплям за 1 —1,5 часа ра-
створ 19,4 г (0,11 Л4) бензолсульфохлорида (см. примечание
2), в 50 мл пиридина при размешивании и охлаждении на во-
дяной бане. Перемешивают еще 2—3 часа и оставляют до сле-
дующего дня при комнатной температуре. Затем нагревают
баню за 1 час до 100° и выдерживают при кипении бани еще
1 час. Охладив содержимое колбы, выливают его при сильном
размешивании в колбу с 1000 мл дистиллированной воды, раз-
мешивают 1 час, осадок отсасывают, промывают несколько
раз водой и отжимают. Осадок нагревают с раствором 13,6 г
едкого кали в 660 мл дистиллированной воды до растворения
и горячую жидкость профильтровывают. Фильтрат нагревают
70
до растворения выделившихся кристаллов, и к горячему проз:
рачному раствору приливают при размешивании примерно
120 мл 10%-ной уксусной кислоты до pH 6—6,5. После ох-
лаждения белый осадок отсасывают, промывают его дистилли-
рованной водой и высушивают на воздухе- Сухое вещество ра-
створяют в 900 мл перегнанного этилового спирта, фильтру-
ют и к фильтрату добавляют 900 мл дважды дистиллирован-
ной воды. Подогревают до растворения выпавшего осадка и
оставляют на 15—20 часов. Бесцветные кристаллы отсасыва-
ют и сушат на воздухе, защищая их от пыли.
Выход 25—25,6 г (88—90% теории), т. пл. 132,5—133° (см.
примечание 3).
Примечания.
1. Требования к чистоте аппаратуры, к степени очистки исходных и
вспомогательных веществ и дистиллированной воде те же; что и при полу-
чении 8-(п-толуолсульфоииламино)-хинолина (см. примечания 1—3 на
стр. 69).
2. Применялся беизолсульфохлорид квалификации ч„ перегнанный не-
посредственно перед употреблением.
3. По литературным данным, т. пл. 133,5° [2].
ЛИТЕРАТУРА
1. Е. А. Божевольнов, В. М. Дзиомко, Г. В. Серебрякова, Авт. свид.
СССР 120029, 1959.
2. W. И. Mills, J. О. Breckenridge, Chem. Soc., 1932, 2209.
Поступила в августе 1961 г. ИРЕА
5-ФТОР 8-МЕРКАПТОХИНОЛИН И ЕГО СОЛИ
Ю. А. БАНКОВСКИЙ. 3. В. МИСУЛОВИНА, А. Ф. ИЕВИНЬШ,
М. Р. БУКА
F
I
/\ /\
-1, 1
I N
SH
I
М. в. 179,22
C3H6NSF
В данном сообщении приводится синтез не описанного в ли-
тературе 5-фтор-8-меркаптохинолина по схеме, ранее приме-
ненной для получения 8-меркаптохинолина [1, 2], З-бром-8-
71
меркаптохинолина [3], 6-бром-8-меркаптохинолина [4], 5-бром-
8-меркаптохинолина [2], 5-хлор-8-меркаптохинолина [5] и 6-
хлор-8-меркаптохинолииа [6]:
F
I
С,Н6СОС1 /\/\ НС1
-------->1 I I
МаОН \ / \ /
I N
SCOCeH6
SnCl2
•2Н2О
НС1-Н3О
5-Фторхинолин получен по методу, разработанному Баль-
цем и Шиманом для синтеза ароматических фторпроизводных
через борфториддиазониевую соль Хавкинса [7, 8]. Получение
72
5-хинолиндиазоиийборфторида и его разложение до 5-фторхи-
нолина произведено по методике Роэ [8]. Однако, по-видимому,
вследствие недостаточной чистоты имевшегося у нас 5-амино-
хинолина выходы 5-фторхинолина были несколько меньшими
и колебались от 37 до 55%. Роэ и Хавкине получали 5-фторхи-
нолин с выходом 59% по отношению к 5-аминохинолину.
Нами установлено, что разложение небольших порций бор-
фториддиазониевой соли дает большие выходы 5-фторхино-
лина.
5-Фтор-8-меркаптохинолин представляет собой красно-ко-
ричневатую маслообразную жидкость, нерастворимую в воде
и хорошо растворимую в органических растворителях. Вслед-
ствие наличия в молекуле меркаптогруппы, обладающей от-
четливо выраженным кислым характером, и основного азота
5-фтор-8-меркаптохинолин обладает амфотерными свойства-
ми— он растворим в щелочах и кислотах. Водные растворы
5-фтор-8-меркаптохинолина, особенно щелочные, легко окис-
ляются кислородом воздуха в 8,8'- (5,5'-дифтор) -дихинолил-
дисульфид:
F F F
IN N | | N
SH S--S
В чистом состоянии 8,8'-(5,5'-дифтор)-дихинолилдисульфид
может быть получен окислением щелочного раствора 5-фтор-8-
меркаптохиполипа перекисью водорода или KaFe(CN)6. Хоро-
шо растворим в хлороформе, из которого после осаждения
этанолом выпадают слегка желтоватые кристаллы с т. пл.
237—238°. Нерастворим в воде и щелочах, растворим в кисло-
тах.
Легкая окисляемость 5-фтор-8-меркаптохинолина не поз-
воляет хранить его длительное время. Однако легко могут
быть получены совершенно устойчивые соли щелочных метал-
лов, например натриевая соль:
F
I
I I I-2HA
I N
SNa
которая в закрытых сосудах может храниться неограниченное
время. При температуре 310° натриевая соль разлагается без
73
плавления. В ультрафиолете имеет интенсивную желто-зеле-
ную флуоресценцию. Хорошо растворима в воде с образова-
нием желтого или желто-оранжевого раствора, растворима в
этаноле и метаноле, нерастворима в несмешивающихся с во
дой органических растворителях.
5-Фтор-8-меркаптохинолин образует соли также с сильны-
ми кислотами, однако эти соли легко гидролизуются водой с
выделением кислот и маслообразного 5-фтор-8-меркаптохино-
лина, вследствие чего на воздухе они постепенно окисляются
в 8,8'-(5,5'-дифтор)-дихинолилдисульфид. Получен хлоргид-
рат 5-фтор-8-меркаптохинолина в виде желтых пластинчатых
кристаллов:
F
Г |XXj.HCi.H2o
I N
SH
При нагревании до 80—85° разлагается.
Условия получения
Получение 5-хинолиндиазонийборфторида. Растворяют 8,4 г
5-аминохкнслина [9] в 75 мл тетрафторборной кислоты (ко-
торую получают медленным прибавлением 61 г борной кисло-
ты к 200 мл 40%-ной фтористоводородной кислоты в плати-
новой чашке), охлаждают до 0° или ниже и приливают раст-
вор 4,2 г NaNO2 в 5 мл воды. Выпавшие красно-коричневые
кристаллы отсасывают на нутче, промывают на фильтре 20 мл
охлажденной до 0° фторборной кислоты, двумя (по 25 мл)
порциями этанола (0°) и затем тремя (по 25 мл) порциями су-
хого эфира. Выход 20 г (98%).
Получение 5-фторхинолина. В круглодонную колбу поме-
щают 20 г 5-хинолиндиазонийборфторида, приливают 50 м\
сухого толуола и осторожно нагревают на плитке до начала
разложения, затем нагревание прекращают. Дальнейшее раз-
ложение борфтордиазониевой соли идет самопроизвольно.
Когда реакция прекращается, колбу опять осторожно нагре-
вают до полного разложения борфтордиазониевой соли. Пос-
ле охлаждения толуол осторожно сливают и к твердому остат-
ку прибавляют 50 мл 10%-ной соляной кислоты, остаток то-
луола отгоняют с водяным паром. Когда весь толуол отогнан,
содержимое колбы подщелачивают 20%-ным раствором NaOH
и отгоняют с водяным паром 5-фторхино.тин. Дистиллат экст-
рагируют 100 мл эфира- Эфирный слой отделяют и сушат над
безводным Na2SO4 в течение ночи. После отгонки эфира полу-
74
чают 6 г 5-фторхинолина в виде светло-желтого масла, 47%
теоретически возможного по отношению к 5-аминохинолину.
Температура кипения 112° при 15 мм. Т. пл. пикрата 198—200°,
что соответствует литературным данным.
Получение 5-фторхинолин-8-сульфокислоты. Приливают
небольшими порциями 10 г 5-фторхинолина к 50 мл 60%-ного
олеума в 100-лы колбе со шлифом, охлаждаемой смесью льда
и соли. Охлаждение необходимо во избежание потерь SO3. К
колбе присоединяют обратный холодильник и нагревают на
водяной бане в течение 8 часов. После охлаждения содержи-
мое колбы выливают на 200 г льда. Через 20—30 минут начи-
нает выпадать белый кристаллический осадок 5-фтор-8-хино-
липсульфокислоты, который через несколько часов отсасыва-
ют, промывают небольшим количеством ледяной воды и высу-
шивают на воздухе.
Выход 10,7 г 5-фтор-8-хинолинсульфокислоты (69%).
Высушенная при 180° 5-фтор-8-хинолинсульфокислота пла-
вится при 340—342°.
Найдено %: N 6,05; S 14,01; С 47,24; Н 2,80.
C9H5NFSO3H. Вычислено %: N 6,16; S 14,17; С 47,57; Н 2,64.
Получение натриевой соли 5-фтор-8-хинолинсулъфокисло-
ты. Полученную сульфокислоту суспендируют в 20 мл воды и
нейтрализуют 20%-ным раствором NaOH до pH 7—8. После
охлаждения раствора частично выпадает дигидрат натриевой
соли: C9H5Oo,NSFNa • 2Н2О.
Раствор натриевой соли выпаривают досуха, затем сушат
в течение ночи при 180°, после чего тщательно измельчают до
состояния пудры и опять сушат несколько часов при 180° для
полного освобождения от кристаллизационной воды.
Найдено %: N 4,86; S 11,33.
CeH6NF SO8Na-2H2O. Вычислено %: N 4,91; S 11,21.
Получение 5-фтор-8-хинолинсульфохлорида. Смешивают
10 г высушенной и тщательно измельченной натриевой еоли
5-фтор-8-хинолинсульфокислоты с 8,3 г измельченного PCU в
100-.И.4 стакане и нагревают на масляной бане. При температу-
ре ~80—90° начинается реакция, смесь разжижается от об-
разующегося POCI3. Расплав выливают в фарфоровую ступку,
распределяют тонким слоем по поверхности ступки и во время
затвердевания расплав измельчают. После полного остывания
твердую, по возможности хорошо измельченную массу обраба-
тывают 100 мл ледяной воды и продолжают измельчение под
слоем воды в течение нескольких минут для выщелачивания
NaCl и разложения РОС13. Осадок отфильтровывают и отжи-
мают на нутче. После перекристаллизации из эфира — бес-
цветные иглы. Т. пл. 156°.
Найдено %: N 5,86; S 13,24; С1 14,37.
C8H6NFSO2CI. Вычислено %; N 5,72; S 13,11; С1 14,46.
75
Восстановление 5-фтор-8-хинолинсульфохлорида. Получен-
ный в предыдущей стадии и отжатый на путче влажный
5-фтор-8-хинолинсульфохлорид немедленно подвергают вос-
становлению, так как влажный сульфохлорид при стоянии раз-
лагается-
Сульфохлорид растворяют в 50 мл охлажденной в ледяной
ванне концентрированной соляной кислоты, и этот раствор не-
медленно вливают топкой струей при перемешивании в зара-
нее приготовленный раствор 32 г хлористого олова SnCl2-2H2O
в 125 мл концентрированной соляной кислоты. Смесь разогре-
вается, и вскоре выпадает лимонно-желтый осадок хлороло-
вянной соли 5-фтор-8-меркаптохинолина. После охлаждения
осадок отфильтровывают на нутче, промывают разбавленной
соляной кислотой (1:1) и затем водой.
Получение 5-фтор-8-(5-бензоил)-меркаптохинолина. К по-
лученной в предыдущей стадии хлороловянной соли 5-фтор-8-
меркаптохинолина прибавляют немного воды и хорошо разме-
шивают до образования жидкой каши, затем приливаю,
180 мл 5%-ного раствора NaOH и слегка нагревают. Хлор-
оловянная соль растворяется с образованием желтого мутного
раствора, который разбавляют водой до 400 мл, по возмож-
ности быстро охлаждают в ледяной ванне и фильтруют с отса-
сыванием. К полученному светло-желтому, постепенно мутне-
ющему раствору (от образующегося дисульфида), помещен-
ному в литровую бутыль с притертой пробкой, приливают не-
большими порциями (по 0,5—1 мл) хлористого бензоила и ин-
тенсивно встряхивают в течение нескольких минут. Прибавле-
ние хлористого бензоила и встряхивание производят до обес-
цвечивания раствора. Перед каждым прибавлением хлористо-
го бензоила контролируют pH раствора по универсальной ин-
дикаторной бумажке. Значение pH не должно быть ниже 10,
в противном случае может выпасть оловянная кислота, что
уменьшает выход 5-фтор-8-меркаптохинолина. В этом случае
приливают некоторое количество 15%-ного раствора щелочи.
5-Фтор-8-(S-бензоил)-меркаптохинолин выпадает в виде
светло-желтого зернистого осадка, который отсасывают, про-
мывают водой и высушивают на воздухе. Выход неперекри-
сталлпзованного 5-фтор-8-(S-бензоил)-меркаптохинолина 7 г.
или 60%, считая на натриевую соль 5-фторхинолин-8-сульфо-
кислоты. Перекристаллизованная соль из спирта имеет т. пл.
103°
Найдено %: X 4,97; S 11,28.
C9H6NFSCOCeH5. Вычислено %: N 4,94; S 11,32.
Получение б-фтор-в-меркаптохинолина. К 7 г неперекри-
сталлизованпого 5-фтор-8-(S-бензоил)-меркаптохинолина, по-
мещенного в круглодонную колбу со шлифом, приливают
80 мл концентрированной соляной кислоты х. ч. и кипятят с
обратным холодильником 1 час. Бензоильное производное ра-
76
створяется с образованием желтого раствора. После охлажде-
ния отфильтровывают выкристаллизовавшуюся бензойную
кислоту. Фильтрат в стакане разбавляют водой до 700 мл, при-
бавляют 100 г льда и нейтрализуют, по возможности, хорошо
охлажденным 12%-ным раствором NaOH х. ч., до pH 9. Во
время нейтрализации раствор мутнеет, и выпадает 5-фтор-8-
меркаптохинолин в виде капель, которые вскоре осаждаются
в виде винно-красиого масла на дне стакана. Желтый мутный
раствор, по возможности полностью, осторожно переливают в
другой стакан. К оставшемуся в первом стакане маслу при-
бавляют 10 мл 12%-ного раствора NaOH, интенсивно переме-
шивают некоторое время, разбавляют водой до 100 мл и обра-
зовавшийся желтый раствор присоединяют к раствору в пер-
вом стакане. На дне стакана остается нерастворяющаяся в
NaOH смолообразная масса, представляющая собой, в основ-
ном, соли 5-фтор-8-меркаптохинолина с тяжелыми металлами,
содержащимися в исходных реактивах. Желтый раствор объ-
емом около 1 л оставляют на 1 час (при этом с частично об-
разующимся дисульфидом полностью соосаждаются тяжелые
металлы) и отсасывают через самый плотный двойной фильтр.
Фильтрат нейтрализуют до pH 4 соляной кислотой ос. ч. или
перегнанной, марки х. ч. Выделившийся 5-фтор-8-меркаптохи-
нолин экстрагируют в делительной воронке тремя (по 40 мл)
порциями гексана. Гексановый экстракт высушивают в тече-
ние ночи безводным сульфатом натрия (5 г), не содержащим
следов тяжелых металлов. После отгонки гексана в вакууме
(без капилляра) получают 3,4 а 5-фтор-8-меркаптохинолина
в виде красно-кооичневатого масла, или 27%, считая на
5-фторхинолин.
Найдено %: N 7,76; S 17,73.
C,HeNSF. Вычислено %: N 7,81; S 17,85
Получение натриевой соли 5-фтор-8-меркаптохинолина. Ра-
створяют 3,4 г 5-фтор-8-меркаптохинолииа в 52 мл очищенного
этанола, приливают 11 мл дистиллированной воды и погружа-
ют в кипящую водяную баню на 5—6 минут. К горячему ра-
створу приливают по каплям спиртовой раствор этилата нат-
рия (1 а металлического натрия растворяют в 20 мл очищен-
ного этанола и перед употреблением раствор фильтруют) до
перехода окраски из темно-красной в лимонно-желтую. На дне
стакана появляется лимонно-желтый кристаллический осадок
натриевой соли 5-фтор-8-меркаптохииолина. Стакан погружа-
ют в кипящую водяную баню прогревают в течение 5—6 ми-
нут и; если появляется оранжевая окраска, опять приливают
этилат натрия до перехода окраски в лимонно-желтую. Горя-
чий раствор быстро отсасывают через неплотный стеклянный
фильтр. Получают 1.6 г натриевой соли, которую промывают
на фильтре этанольно-эфирной смесью (1:1) и высушивают
77
на воздухе. Из фильтрата при охлаждении льдом осаждается
еще около 1,7 г натриевой соли, которую отфильтровывают,
промывают, как описано выше, и высушивают иа воздухе- Пос-
ледняя порция натриевой соли имеет высокую степень чисто-
ты и пригодна для спектрофотометрических исследований.
Найдено %: N 5,93; S 13.63; С 45,57; Н 3,89.
C9H5NFS\a-2H2O Вычислено %: N 5,90; 8 13,67; С 45,56; Н 3,82.
Содержание кристаллизационной воды определено по поте-
ре веса в процессе высушивания при 120°.
Получение хлоргидрата-5-фтор-8-меркаптохинолина. К 0,1 г
5-фтор-8-меркаптохинолина прибавляют 2 капли концентриро-
ванной соляной кислоты. Через 2—3 минуты при охлаждении
льдом из желтого раствора выпадают желтые пластинчатые
кристаллы хлоргидрата, который отфильтровывают, промыва-
ют на фильтре эфиром и высушивают на воздухе.
Найдено %: N 6.08; S 13,95; С 47,38; Н 3,92
CeH6NSF-НС1-Н2О. Вычислено %: N 6,26; S 14,33; С 48,34; Н 4,05
Получение 8,8’-(5,5'-дифтор)-дихинолилдисульфида. Раст-
воряют 0,5 г натриевой соли 5-фтор-8-меркаптохинолина в 10 мл
1%-ного раствора NaOH и по каплям приливают при переме-
шивании 30%-пый Н2О2 до обесцвечивания раствора. Выпада-
ет белый творожистый осадок, который отсасывают на нутче,
промывают водой и высушивают на воздухе. Для получения
в кристаллическом состоянии растворяют 8,8'-(5,5'-дифтор) -
дихинолилдисульфид в 3 мл хлороформа, фильтруют через не-
плотный фильтр и приливают 10 мл этанола. Постепенно
осаждаются слегка желтоватые кристаллы с т. пл. 237—238л.
Найдено %: N 7,76; S 18,03.
CisHjdN-.SjF,. Вычислено %: N 7,85; S 17,95.
ЛИТЕРАТУРА
1. A. Edinger, Вег., 41, 938 (1908).
2. Ю. А. Банковский, Изв. АН Латв. ССР, 1952, № 12, 12/.
3. Ю. А. Банковский, Е М. Шварц, А. Ф. Иевиньш, Изв. АН Латв. ССР,
1958, № 2» 115.
4. Ю. А. Банковский, Е. Ф. Лобанова, /К- общ. химии, 28, 2857 (1958,.
5. Ю. А. Банковский, 3. В. Мисуловина, Я. А. Цируле, А. Ф. Иевиньш.
Изв. АН Латв. ССР, (в печати).
6. Ю. А. Банковский, Л. П, Залукаев, Р. Ф. Платниекс, Изв. АН
Латв. ССР, 1958, № 4, 95.
7. G. Balz, G. Shlemann, Ber., 69, 1186 (1927).
8. «Органические реакции». Сб. 5, 1951.
9. Dikshoorn, Recueil trav. chtm. 48, 147 (1929).
Поступила в сентябре 7967 г.
Институт химии АН Латв. ССР-
5-ХЛОР-8-МЕРКАПТОХИНОЛИН И ЕГО СОЛИ
Ю. А. БАНКОВСКИЙ, 3. В. МИСУЛОВИНА, Я. А. ЦИРУЛЕ,
А. Ф. ИБ.ВИНЬШ
C9H6NSC1
М. в. 195,67
В ряде работ показано, что 8-меркаптохинолин (тиооксин)
представляет собой ценный аналитический реактив для опре-
деления многих металлов- Значительный интерес представ-
ляют также галогенопроизводные 8-меркаптохииолина. В дан-
ной работе излагается синтез не описанного в литературе
5-хлор-8-меркаптохииолина по схеме, ранее примененной для
получения 8-меркаптохинолина [1, 2, 3], 5-бром-8-меркаптохи-
нолина [1], 6-бром-8-меркаптохииолина [4], З-бром-8-меркапто-
хинолина [5] и 6-хлор-8-меркаптохинолииа [6]:
С1 С1 С1
/Х/\ so. /\/Х NaOH /\/\ РС15
I I I—>1 I I------>1 । I-----
\,'\/ х/х/
N |N IN
SO3H SOsNa
SnCl2
C,H5COC1
NaOH ”
Cl
I
/ \/\ HC1
I N
SCOCcH6
2HaO
Cl
I
bl-HCi
|Z NX
SH
79
В противоположность 8-меркаптохинолину (тиооксину),
представляющему собой при комнатной температуре в безвод-
ном состоянии маслообразную, интенсивно синюю жидкость,
легко присоединяющую две молекулы кристаллизационной во-
ды с образованием ярко-красного дигидрата C9H7NS-2H2G,
5-хлор-8-меркаптохииолин выпадает из водных растворов н
виде бесцветных кристаллов. Таким образом, наличие гало-
гена в пятом положении хинолинового ядра (в пара-положе-
нии к меркаптогруппе) приводит к потере способности образо-
вывать устойчивый кристаллогидрат. По-видимому, вследст-
вие этого 5-хлор-8-меркаптохиполип значительно менее раст-
ворим в воде в сравнении с 8-меркаптохинолином. Однако
5-хлор-8-меркаптохинолин хорошо растворим в различных ор-
ганических растворителях. При испарении гексановых раство-
ров образуются слегка зеленоватые кристаллы с т. пл. 64—
65°. К окислительному действию кислорода воздуха они значи-
тельно более устойчивы, чем 8-меркаптохинолин, и в закрытых
сосудах могут храниться длительное время. Определение кис-
лотной константы диссоциации показывает, что меркаптогруп-
па в 5-хлор-8-меркаптохинолине вследствие негативирующего
влияния галогена обладает более кислым характером, чем в
8-меркаптохинолине. В связи с этим 5-хлор-8-меркаптохино-
лин, как и 8-меркаптохинолин, образует устойчивые соли со
щелочными металлами. Получены литиевая, натриевая, ка-
лиевая, рубидиевая и цезиевая соли общей формулы:
С1
I I |-2Н2О,
I N
SMe
где Me — Li, Na, К, Rb, Cs.
Соли щелочных металлов представляют собой лимонно-жел-
тые кристаллы, устойчивые при хранении в сухой атмосфере.
Растворимы в воде и в смешивающихся с водой органических
растворителях с образованием желтой окраски. Водные раст-
воры имеют щелочную реакцию вследствие гидролиза и срав-
нительно быстро окисляются с выпадением белого осадка —
8,8'- (5,5'-дихлор) -дихинолилдисульфида:
80
который представляет собой бесцветное кристаллическое ве-
щество с т. пл. 209°- Некоторыми восстановителями, например
фосфорноватистой кислотой в солянокислых растворах, он вос-
станавливается при нагревании в 5-хлор-8-меркаптохинолин:
Дисульфид нерастворим в воде и растворах щелочей, раство-
рим в кислотах, хлороформе, бензоле, мало растворим в аце-
тоне, четыреххлористом углероде, спиртах и почти нераство-
рим в алифатических углеводородах. Нерастворимость
8,8'- (5,5'-дихлор)-дихинолилдисульфида в гексане может быть
применена для очистки 5-хлор-8-меркаптохинолина, который в
гексане хорошо растворим. Это очень существенно для полу-
чения высокочистого реактива, так как выделяющийся из вод-
ной среды в процессе синтеза 5-хлор-8-меркаптохинолин всег-
да содержит некоторое количество 8,8/-(5,5'-дихлор)-дихино-
лилдисульфида. 5-Хлор-8-меркаптохинолин образует соли так-
же с сильными кислотами, однако, вследствие значительной
ослабленности основных свойств азота, соли 5-хлор-8-меркап-
тохинолина с кислотами еще менее устойчивы, чем соли 8-мер-
каптохинолина. Они очень легко гидролизуются водой и мо
гут быть выделены в кристаллическом состоянии из безвод-
ных растворителей. Например, хлоргидрат 5-хлор-8-меркапто-
хинолина CgHeNS-HCI получен пропусканием сухого HCI в
раствор 5-хлор-8-меркаптохинолина в абсолютном эфире.
5-Хлор-8-меркаптохииолин и его впутрикомплексные соли
с металлами в настоящее время изучаются.
Условия получения
Получение 5-хлор-8-хинолинсульфокислоты и ее натриевой
соли. В круглодонную колбу со шлифом прибавляют 12 г
5-хлорхинолина [7] постепенно небольшими порциями в тече-
ние 25 минут при перемешивании к 30 мл 55—65%-ного олеу-
ма, не допуская значительного повышения температуры олеу-
ма и потери SO3. Смесь 2 часа нагревают на масляной бане с
обратным холодильником при 150°. После охлаждения содер-
жимое колбы выливают в 100 г льда и через 3—4 часа от-
фильтровывают на нутче выкристаллизовавшуюся 5-хлор-8-
хннолинсульфокислоту почти белого цвета. Осадок на фильт-
ре промывают двумя порциями воды (по 50 мл) и высушива-
ют на воздухе.
6 Зак. 622 81
Выход 14,7 г (84% теории)
Найдено %: С 41,56; Н 3,22; N 5,45; S 12,13;
С1 13,69.
C0HeOsNSCl-H,O Вычислено %: С 41,31; Н 3,08; N 5,35; S 12,25;
С1 13,54.
Полученную 5-хлор-8-хинолинсульфокислоту нейтрализуют
в фарфоровой чашке 20%-ным раствором NaOH до нейтраль-
ной реакции. Раствор натриевой соли осторожно выпаривают
на плитке почти досуха и затем высушивают в сушильном шка-
фу при 160° в течение 8—10 часов. При охлаждении выпа-
дают кристаллы натриевой соли 5-хлор-8-хииолинсульфокнс-
лоты.
Найдено %: N 5,05; S 11,28; С| 12,52; С 37,88;
Н 2,64
С9Н6О,№С1На Н,0 Вычислено %: N 4,93; S 11,30; С1 12,49; С 38,10;
Н 2,48.
Получение 5-хлор-8-хинолинсулъфохлорида. Высушенную,
как описано выше, при 160° (для освобождения от кристалли-
зационной воды) натриевую соль сульфокислоты тщательно
измельчают в фарфоровой ступке до состояния пудры и хоро-
шо смешивают с равным по весу количеством порошкообраз-
ного РС1(5. Полученную смесь нагревают в стакане емкостью
150—200 мл, погруженном в масляную баню.’ При достижении
температуры масла 100—110° происходит разжижение реак-
ционной смеси. При периодическом перемешивании поднима-
ют температуру масляной бани до 140°С и выдерживают эту
температуру, пока совершенно жидкий расплав не станет за-
стывать (вследствие выпаривания РОС1з), на что обычно тре-
буется 40—50 минут. Затем содержимое стакана выливают в
большую фарфоровую ступку и распределяют тонким слоем
по поверхности ступки. При охлаждении во время затвердева-
ния расплава его измельчают, после полного охлаждения за-
ливают 100 мл воды со льдом и продолжают измельчение под
слоем воды (для выщелачивания NaCl и разложения РОС13).
Через 5—10 минут (оставлять под слоем воды на несколько
часов сульфохлорид нельзя, так как он разлагается до сульфо-
кислоты) желтоватый 5-хлор-8-хинолинсульфохлорид отсасы-
вают на нутче, промывают водой и тщательно отжимают. Вы-
сушенный в вакууме и перекристаллизованный из эфира суль-
фохлорид имеет следующий состав:
Найдено %: N 5,45; S 12,46; С1 26,87. . .
С9Н5О2К5С1, Вычислено %: N 5,34; S 12,21; CI 27,06.
Восстановление 5-хлор-8-хинолинсульфохлорида. Получен-
ный в предыдущей стадии влажный сульфохлорид без пере-
кристаллизации немедленно подвергают восстановлению хло-
ристым оловом. 33 г.влажного, но тщательно отжатого на нут-
че сульфохлорида растворяют в 130 мл концентрированной
82
НС1 и немедленно вливают этот раствор тонкой струей в зара-
нее приготовленный раствор SnCl2-2H2O в 200 мл концентриро-
ванной НС1- Во время сливания смесь непрерывно взбалтыва-
ют. Смесь разогревается, и выпадает желтый кристаллический
осадок хлороловянной соли, точный состав которой нами не
установлен, но данные анализа ее ближе всего согласуются
с формулой, данной в схеме синтеза,. После охлаждения хлор-
оловянную соль отфильтровывают на нутче и промывают раз-
бавленной соляной кислотой (1:1), а затем водой. Выход
~40 г влажной хлороловянной соли.
Получение 5-хлор-8-(8-бензоил)-меркаптохинолина. Выде-
лить из хлороловянной соли чистый 5-хлор-8-меркаптохинолин
прямым путем невозможно. Это можно осуществить через бен-
зоильное производное. К полученной в предыдущей стадии
хлороловянной соли приливают 200 мл 15%-ного раствора
NaOH, тщательно размешивают, нагревают до кипения, раз-
бавляют водой до 1 л и фильтруют. К полученному прозрачно-
му светло-желтому, постепенно мутнеющему (от образующе-
гося дисульфида) раствору, находящемуся в двухлитровой
банке с притертой пробкой, прибавляют небольшими порциями
(2—Змл) хлористый бензоил и после каждого прибавления по
возможности энергично встряхивают в течение нескольких ми-
нут до исчезновения капелек хлористого бензоила. Прибавле-
ние хлористого бензоила и встряхивание производят до обес-
цвечивания раствора. При этом после каждого прибавления
хлористого бензоила контролируют по универсальной индика-
торной бумажке pH раствора, которое не должно быть ниже
10. В противном случае приливают несколько миллилитров
15°/о-ного раствора NaOH. Если pH раствора падает до 6—7,
выпадает оловянная кислота, что уменьшает выход чистого
5-хлор-8-меркаптохинолина. Бензоильное производное выпада-
ет в виде светло-желтого зернистого осадка, который отсасы-
вают на нутче, промывают водой и высушивают на воздухе.
Перекристаллизованный из спирта продукт имеет температуру
плавления 98°.
Найдено %: N 4,88; S 10,60; С1 11,98
CieHt0ONSCl. Вычислено %: N 4,70: S 10,67; С1 11,82.
Выделение 8-хлор-8-меркаптохинолина и его очистка. В
крутлодонной колбе емкостью 200 мл с пришлифованным об-
ратным холодильником кипятят 10 г бензоильного производно-
го с 75 мл концентрированной соляной кислоты в течение 1,5—
2 часов до полного растворения. При этом образуется светло-
желтый прозрачный раствор, из которого после охлаждения
выпадают кристаллы бензойной кислоты, окрашенные в жел-
тый цвет. Кристаллы отсасывают на нутче и фильтрат раз-
бавляют насыщенным раствором ацетата натрия или карбона-
та натрия до pH 5. 5-Хлор-8-меркаптохинолин выпадает в ви-
6* 83
це белых иголочек, которые отсасывают на нутче и промывают
водой. Для очистки 5-хлор-8-меркаптохинолина от примесей
металлов применяют разработанный Лукшей и Шевчуком ме-
тод соосаждения [8]. Осадок 5-хлор-8-меркаптохинолина пере-
носят в литровый стакан, приливают 20 мл 5%-ного раствора
NaOH и разбавляют водой до 800—1000 мл; pH раствора дол-
жно быть около 9. Остается небольшой нерастворимый осадок
серого или буроватого цвета. Через 1 час с образующимся
при окислении кислородом воздуха 8,8'-(5,5'-дихлор)-дихино-
лилдисульфидом соосаждаются 5-хлор-8-меркаптохинолинаты
всех тяжелых металлов. Раствор отсасывают через самый
плотный двойной фильтр. Фильтрат нейтрализуют соляной
кислотой ос. ч. или перегнанной х. ч. до pH 5 по универсальной
индикаторной бумажке. Выкристаллизовавшийся 5-хлор-8-
меркаптохинолин отсасывают на нутче, промывают несколько
раз дистиллированной водой и высушивают в вакуумном шка-
фу при комнатной температуре. Полученный препарат совер-
шенно свободен от тяжелых металлов и пригоден для анали-
тических работ. Однако он содержит некоторое количество ди
сульфида. Освободить 5-хлор-8-меркаптохинолин от дисуль-
фида можно следующим образом. В противоположность
5-хлор-8-меркаптохинолину, который очень хорошо растворим
в алифатических углеводородах, 8,8'- (5,5'-дихлор) -дихинолил-
дисульфид растворим, например, в гексане очень мало. Вы-
сушенный в вакууме препарат (3—4 г) растворяют в 30 мл
гексана, нерастворившийся кристаллический осадок (дисуль-
фид) отфильтровывают и фильтрат выпаривают в токе возду-
ха (около вентиляционной трубы в вытяжном шкафу) при
комнатной температуре. После испарения гексана остаются
слегка зеленоватые кристаллы 5-хлор-8-меркаптохинолина.
Очищенный этим методом 5-хлор-8-меркаптохиполип имеет
температуру плавления 64—65°.
Найдено %: N 7,20; S 16,54; С1 17,96
C0HeNSCl. Вычислено W: N 7,15; S 16,39; С1 18,12.
Соли 5-хлор-8-меркаптохинолина.
Растворяют при нагревании 0,15 г реактива в 1,5—2 мл
этанола. К образовавшемуся темно-красному раствору прили-
вают по каплям насыщенный этанольный раствор NaOH, КОН,
RbOH, CsOH или насыщенный водный раствор LiOH до пере-
хода окраски из темно-красной в чисто-желтую. При охлажде-
нии льдом вскоре выпадают лимонно-желтые кристаллические
осадки солей 5-хлор-8-меркаптохинолина с упомянутыми ме-
таллами. Кристаллы отсасывают, промывают небольшим коли-
чеством охлажденного спирта и высушивают на воздухе.
5-Хлор-8-меркаптохинолинат лития.
Найдено И: С45.30; Н 3,93; N 6,00; S 1348; CI 15,13.
CoHsNClSlJ-SH.O. Вычислено %: С 45,48; Н 3,82; N5.89; S 13,49; CI 14,92.
84
5-Хлор-8-меркаптохинолинат натрия.
Найдено %: N5,46; S 12,42; С1 13,80; Накт1,50.
C9HsNClSNa.2H2O. Вычислено %: N5.51; S 12,63; CI 13,97; Накт1,57.
5-Хлор-8-меркаптохинолинат калия.
Найдено %: C40.I5; Н 3,23; N 5,12; S'-2.08; СИЗ,31-
C9HsNC1SK-2Н2О. Вычислено %: С 40,06; Н 3,36; N 5,19; S 11,89; С1 13,14-
5-Хлор-8-меркаптохчнолинат рубидия.
Найдено %; С 34,12; Н 2,88; N4.54; S 9,84; CI 11,45.
C0H5NClSRb-2H2O. Вычислено %: С 34,18;_Н 2,87; N 4,43; S 10,14; Cl 11,53.
5-Хлор-8-меркаптохинолинат цезия.
Найдено %: С 29,97; Н 2,52; N 3,96; S 9,10; С19.76.
C8H6NCISCs-2H2O. Вычислено %: С29.72; Н 2,80; N3,85; S 8,81; Cl 9,75.
Хлористоводородная соль. В раствор 0,1 г 5-хлор-8-мер-
каптохинолина в 20 мл абсолютного эфира пропускают,
сухой НС1. Выпадают желтые кристаллики с т. пл. 174 —
-175°.
Найдено %: N6.0I; S 13,81; CI 30,19.
C0HjNSCI.HCl. Вычислено %: N6,03; S 13,82; С130,58.
8,8'-(5,5'-дихлор)-дихинолилдисульфид. Растворяют 1 г
5-хлор-8-меркаптохинолина в 200 мл 0,5%-ного раствора
NaOH. Желтый раствор фильтруют через плотный фильтр, к
фильтрату приливают по каплям перекись водорода до обес-
цвечивания. Немедленно образуется белый осадок 8,8'-(5,5'-
тихлор-)-дихинолилдисульфида, который отфильтровывают на
нутче, промывают несколькими порциями воды и высушивают
в сушильном шкафу. Для получения в кристаллическом со-
стоянии препарат растворяют в 5 мл хлороформа и, если ра-
створ мутный, фильтруют. К фильтрату приливают 50 мл эта
иола или метанола. Через короткое время выкристаллизовы-
вается слегка желтоватый 8,8'-(5,5'-дихлор)-дихинолилдисуль-
фид, который отфильтровывают, промывают эфиром и высу-
шивают на воздухе. Т. пл. 209°.
Найдено %: С54.95; Н 2,66; N 7,33; S 16,30; С1 18,42.
C1SH1ON?S2C1,. Вычислено И; С 55,53; Н 2,58; N7.I9; S 16,47; CI 18,21.
ЛИТЕРАТУРА
I. A. Edinger. Вег., 41, 938 (1908).
2. Ю. А. Банковский, Изв. АН Латв. ССР. 1952, № 12, 127.
3. Ю. А. Банковский. А. Ф. Иевиньш, Э. А. Лукша, Ж. общ. химии 28,
2273 (1958).
4. Ю. А. Банковский, 3. Ф. Лобанова, Ж- общ. химии, 28, 2857 (1958).
5. Ю. А. Банковский, Е. Л1. Шварц, А. Ф. Иевиныи. Изв. АН Латв.
ССР, 1958, № 2, 115.
6. Ю. А. Банковский, Л. П. Залукаев и Р. Ф. Платниекс, Изв. АН Латв.
ССР, 1958, № 4, 95.
7. J. Freiidt, Monatshefte fur Chemie, 8, 583 (1887).
8. И. Шевчук, Э. Лукша, Изв. АН Латв. ССР, 1961, № 2, 127.
Поступила в сентябре 1961 г.
Институт Химии АН Латв. ССР
85
АЗУЛЕН
Ф. Н. СТЕПАНОВ. Н. А. АЛДАНОВА, А. Г. ЮРЧЕНКО, Н. Л. ДОВГАНЬ'
С10Н8 М. в. 128,16
Азулен стал сравнительно доступным препаратом с 1955
года, когда Кёниг и Рёзлер [1] с. одной стороны, и Гафнер [2]
с другой, независимо друг от друга, нашлн, что фульвен, полу-
ченный конденсацией 1-1\’-метиланилинопентадиен-1,3-аля-5
(альдегид Цинке) с цнклопентадиеном в присутствии щело-
чи, при температуре выше 150° переходит в азулен с отщепле-
нием N-метиланилина. Этот изящный метод сделал также до-
ступными и азулены, алкилированные в пятичленном и семи-
членном кольцах.
Замыкание фульвена в азулен по Гафнеру производится в
среде бензидина, нагретого до 250—270°, при этом фульвен
вносится в реакционную смесь в виде таблеток в бензидине.
Токсичность бензидина и неудобный способ прибавления твер-
дого реагента небольшими порциями в реакционный сосуд,
нагретый до высокой температуры, вызвали необходимость1
отыскать более удовлетворительный метод проведения этой
реакции. Мы испытали следующие варианты метода: замыка-
ние фульвена в азулен в высоком вакууме без растворителя,
в октадициламине, в метилоктадециламине, в дифениламине,
в хинолине, в N-изоамиланилине, в метилстеариламине и' в
триэтаноламине. Последний оказался наиболее пригодным,
так как он жидкий при комнатной температуре и нетоксичен.
При этом выход азулена не ниже, а часто выше, чем при про-
ведении реакции в бензидине.
Полный синтез включает в себя получение следующих ве-
ществ:
1) 2,4-динитрохлорфенилат пиридиния,
2) хлоргидрат N.N-Диметилдианила глутаконового альде-
гида (красная соль Цинке),
3) 1-М-метиланилинопентадиен-1,3-аль-5 (альдегид Цин-
ке) ,
4) 1 -циклопентадиенилидено-5-метиланилинопентадиен-
2,4(фульвен).
5) азулен.
Прежде мало доступный азулен служит исходным вещест-
вом для получения ряда производных, благодаря чему стано-
вится возможным изучение этого интересного класса органиче-
ских соединений.
86
СИНТЕЗ 2,4-ДИНИТРОХЛОРФЕНИЛАТА ПИРИДИНИЯ
O2NXXX/ZNO2 /X 0,N
+ IJ -
no2
Характеристика основного сырья
2,4-Динитрохлорбензол, ГОСТ 626—54.
Пиридин, ГОСТ 2747—44.
Ацетон, ГОСТ 2603—51,
Условия получения
Растворяют 203 г (1 Af) технического 2,4-динитрохлорбен-
зола в 350 мл сухого ацетона и приливают к раствору 98 г
(1,25 7И) сухого пиридина. Смесь нагревают на водяной бане
с обратным холодильником в течение 15 часов. Образовав-
шуюся густую светло-желтую кристаллическую массу хорошо
отсасывают и промывают три раза ацетоном (порциями по
70 мл) и сушат на воздухе.
Получают 270 г соли пиридиния (95,7% теоретического) в
виде светло-желтых блестящих иголок с т. пл. 180—200°.
СИНТЕЗ КРАСНОЙ СОЛИ ЦИНКЕ
ci- + 2 I
сн3
сн3
,N —СН=СН СН=СН— CH=N +
С1~
I +
NH.
no2
I
no2
87
Характеристика основного сырья
2,4-Динитрохлорфенилат пиридиния.
Монометил а нилин, РУ 678—52.
Этиловый спирт, ГОСТ 8314—57.
Ацетон, ГОСТ 2603—51.
Условия получения
Растворяют 225 г (0,8 Л1) 2,4-динитрохлорфенилата пири-
диния при нагревании в 400 мл этилового спирта и к образо-
вавшемуся раствору приливают 170 мл (1,6 А4) свежепере-
гнанного мопометиланилина. Кипятят реакционную массу с
обратным холодильником 4 часа на водяной бане. Полученно-
му темно-красному раствору дают остыть, после чего его мед-
ленно при непрерывном размешивании выливают в 2 л 5%-ной
соляной кислоты. Выделившийся объемистый желто-красный
осадок оставляют стоять на ночь, затем отсасывают, промыва-
ют два раза 5%-ной соляной кислотой, хорошо отжимают и
сушат полученную пасту на воздухе. Сырой продукт является
смесью хлоргидрата N-диметилдианила глутаконового альде-
гида и динитроанилина. Для отделения от последнего раст-
воряют продукт в 800 мл ацетона при кипячении. По
охлаждении из раствора выделяется хлоргидрат N-диметил-
дианила глутаконового альдегида в виде красивых блестящих
красных игл. Кристаллы отсасывают, промывают трижды
порциями по 50 мл холодного ацетона и сушат на воздухе.
Получают 213—223 г красной соли Цинке (85—89% тео-
ретического) с т. пл. 122—123°.
СИНТЕЗ АЛЬДЕГИДА ЦИНКЕ
СН3 СН3
I I
/N-CH=CH - СН=СН -CH=N + х,
сГ /I
NaOH
СН3
I
N-CH=CH—СН=СН-С(
н +
NHCHg
4- NaCl
88
Характеристика основного сырья
Красная соль Цинке.
Метиловый спирт, ГОСТ 6995—54.
Бензол, ГОСТ 5955—51.
Диэтиловый эфир, ГОСТ 6265—52.
Едкий натр, ч.
Условия получения
Растворяют 156 г (0,5 Л1) красной соли Цинке в 300 мл ме-
тилового спирта и к холодному раствору осторожно, неболь-
шими порциями при непрерывном размешивании прибавляют
2О°/о-ный раствор едкою натра, тщательно контролируя pH
среды. Цвет раствора при подщелачивании изменяется от
красного до желтовато-коричневого. Прибавление щелочи пре-
кращают по достижении pH 7 (по универсальной индикатор-
ной бумажке (см, примечание 1), после чего прибавляют к ра-
створу 600 мл воды. Выделившееся красно-коричневое масло
экстрагируют 150 мл бензола. Бензольный раствор сушат суль-
фатом натрия и отгоняют растворитель в вакууме. Маслооб-
разный остаток растворяют при нагревании в трехкратном по
объему количестве диэтилового эфира. Раствору дают охла-
диться до комнатной температуры, при внесении затравки из
него моментально выпадает альдегид Цинке, образуя густую
кашицу желтых игольчатых кристаллов. Осадок отсасывают,
дважды промывают на фильтре по 30 мл холодного диэтило-
вого эфира. Получают 66,3 г вещества (71% теоретического) с
т. пл. 77,0—78,5°. Альдегид Цинке постепенно полимеризует-
ся, поэтому долго хранить его не следует.
СИНТЕЗ ФУЛЬВЕНА
сн=сн 0
) >СН,+ )с-СИ=сн-сн-сн-nxxx №0С1Н1
СН-=СН и . I \------>
СН = СН СНз
- I )-c=ch-ch=ch-ch=ch-n4XX
СН = СН |Ч-Н2о
Характеристика основного сырья
Альдегид Цинке-
Дициклопентадиен,технический.
Этиловый спирт, ГОСТ 8314—57.
89
Натрий металлический,. ГОСТ 3273—55.
Уксусная кислота, ГОСТ 61—51.
Условия получения
Растворяют 56,1 г (0,3 Л4) альдегида Цинке при нагрева-
нии в 120 мл абсолютного этилового спирта и оставляют мед-
ленно остывать (см. примечание 2). К охлажденному раство-
ру альдегида прибавляют 21,6 г (0,3 Л1) свежеперегнанного
циклопептадиепа (см. примечание 3) и смесь быстро при энер-
гичном размешивании и небольшом охлаждении приливают
к приготовленному заранее раствору этилата натрия. Послед-
ний готовят растворением 6,9 г (0,3 М) металлического натрия
в 125 мл абсолютного этилового спирта. Реакционную массу
выдерживают при периодическом размешивании 30 минут при
комнатной температуре, а затем охлаждают льдом. Выпавший
при этом фульвен отсасывают, промывают два раза порциями,
по 30 мл холодного абсолютного этилового спирта, хорошо от-
жимают. Осадок переносят в 150 мл 3%-ной уксусной кисло-
ты, размешивают, снова отсасывают, промывают снова водой
до нейтральной реакции и сушат на воздухе. Некрнсталлизо-
ванный продукт представляет собой порошок кирпично-крас-
ного цвета. Получают 57,8 г фульвена (82% теоретического).
Для последующей операции—замыкания фульвена в азулен—>
применяется обычно неочищенный препарат. Фульвен можно
перекристаллизовать из петролейного эфира (фракция 60—
80°), для этого на каждый грамм фульвена берут 20 мл петро-
лейного эфира. Перекристаллизованный фульвен представляет
собой красивые разветвленные красные иголки с т. пл. 112—
112,5°. Фульвен с течением времени полимеризуется, поэтому
хранить его долго нельзя.
ЗАМЫКАНИЕ ФУЛЬВЕНА В АЗУЛЕН
СН=СН СНз
| ^С=СН— CH = CH-CH=CH-N\/\
СН=СН I I ->
Характеристика основного сырья
Фульвен.
Триэтаноламин, ВТУ ЕУ 3—54.
Петролейный эфир (т. кип. 60—80°).
Окись алюминия для хроматографии, ТУ МХП 2962—54.
90
Условия получения
В трехгорлую колбу из стекла пирекс емкостью 1 л, снаб-
женную барботером для подачи перегретого пара, капельной
воронкой, термометром и нисходящим водяным холодильни-
ком помещают 400 мл перегнанного триэтаноламина (т- кип.
190—210°/15 мм) и нагревают его до 250—260", после чего по-
дают в колбу с небольшой скоростью перегретый до 270—280°
водяной пар. Пар перегревают в стеклянном или медном зме-
евике, имеющем на выходе штуцер для замера температуры
(см. примечание 4), приемником служит колба Вюрца ем-
костью 1 л (охлаждаемая водой), к отводу которой присоеди-
нен обратный холодильник. По достижении постоянства режи-
ма (температура, скорость пара) начинают прибавлять не-
большими порциями раствор фульвена (5 г, 0,021 Л4) в 100 мл
триэтаноламина (см. примечание 5). Образующийся азулен
отгоняется с водяным паром, покрывая стенки приемника тем-
но-синим плотным слоем. После того как весь фульвен при-
бавлен, пар пропускают еще. в течение получаса, затем дистил-
лят подвергают перегонке с водяным паром. При этом азулен
отгоняется с первыми порциями погона. Экстрагируют продукт
петролейным эфиром, промывают петролейную вытяжку два-
жды 5—7%-ной соляной кислотой, затем водой, сушат суль-
фатом натрия, отгоняют большую часть растворителя с елоч-
ным дефлегматором и переносят раствор на колонку с окисью
алюминия. Азулен легко вымывается петролейным эфиром,
незначительные примеси остаются в верхней части колонки.
После полного удаления растворителя в вакууме азулен суб-
лимируют при атмосферном давлении.
Получают 1,38 г продукта с т- пл. 98° в виде синих листоч-
ков, что составляет 50,7% теоретического (см. примечание 6).
По литературным данным, температура плавления азулена
98 -99°.
Примечания.
1. Контроль pH среды должен быть особенно тщательным, так как
изменение окраски индикаторной бумажки становится заметным лишь при
значениях pH, близких к 7, а увеличение pH до 7,5 резко (в 1,5—2 раза)
снижает выход альдегида.
2. При быстром охлаждении альдегид Цинке выкристаллизовывается.
Иногда кристаллизация происходит и при медленном охлаждении, в этом
случае циклопеитадиен можно смешивать с чуть теплым раствором аль-
дегида.
3. Получение циклопситадиеиа из дицик.топеитадиена см. [3].
4. Измерение температуры пара лучше производить термопарой, т. к.
термометры при этом часто лопаются.
5. Раствор фульвена в триэтаноламине заливают в капельную вороику
небольшими порциями, чтобы предохранить его от длительного нагревания
и полимеризации. Отвод капельной воронки должен быть покороче, чтобы
раствор фульвена стекал в реакционную массу по возможности скорей. Ко-
личество триэтаиоламииа, вносимого в реакционную колбу, остается равным
400 мл и в том случае, если в реакцию вводится в 12 раз большее количесг-
91
во фульвена. Частично отгоняющийся триэтаноламин в ходе синтеза попол-
няется за счет вносимого с фульвеном.
6. Триэтаноламин, использовавшийся в синтезе, можно применять по
вторпо, подвергнув его предварительно перегонке в вакууме.
ЛИТЕРАТУРА
1. W. KOnig, Н. ROslez, Naturwissenschaften, 42, 211 (1955).
2. К. Hafner, Liebigs Ann. Chem., 606, 79 (1957).
3. Ю. К. Юрьев. «Практические работы по органической химии», вып.
1 и 2, стр. 347. Изд-во. МГУ, 1961.
Поступила в феврале 1962 г.
кпи
О -СУЛЬФОБЕНЗОЙНОЙ КИСЛОТЫ ДИКАЛИЕВАЯ
СОЛЬ
Д. Ш. РОЗИНА, Л. Т. НЕСТЕРЕНКО
COOK
I
/V-SO3K
C,H4O5SKs Z M. в. 278,36
Дикалиевую соль о-сульфобензойной кислоты * получают
взаимодействием о-сульфобензойной кислоты с едким кали [1].
Нами уточнены условия ее получения, выделения и очистки,
ПОЛУЧЕНИЕ ДИКАЛИЕВОЙ СОЛИ
соон COOK
!
|/X|-SO3H.3H2O+2KOH -> \ VS°3K-H2O
COOK COOK
I I
|/X| S°3K.2H2O |/4j-SO3K
* В дальнейшем для краткости мы будем называть дикалиевую соль
о-сульфобензойной кислоты «дикалпевая соль».
92
Характеристика основного сырья
о-Сульфобензойпая кислота, ч., ВТУ МХП 2833—53.
Кали едкое, ГОСТ 4203—48.
Условия получения
В 600 мл воды растворяют при нагревании (50°) 205 г
(0,8 А4) о-сульфобензойной кислоты. Полученный раствор упа-
ривают в колбе на асбестированпой сетке в присутствии акти-
вированного угля (10 г) до половины первоначального объе-
ма и для полного удаления окислов азота (см. примечание 1)
снова добавляют 300 мл воды и упаривают До уд. в. 1,250 при
45°. К отфильтрованному раствору добавляют 38—40%-ный
раствор едкого кали (170—180 мл) до слабощелочной реакции
по фенолфталеиновой бумажке, обрабатывают активирован-
ным углем (Ю г), упаривают до объема — 100 мл и охлаждают
в ледяной воде до 5—7°. Образовавшуюся густую кристалли-
ческую массу дикалиевой соли отфильтровывают, возможно
тщательнее отжимают, промывают 3 раза по 20 мл спирта и
сушат при 110°
Получают 120—123 г дикалиевой соли, содержащей одну
молекулу кристаллизационной воды. Для получения безводной
соли ее сушат при 140° до постоянного веса-
Выход НО—113 г с содержанием основного вещества 99,7—
99,8%, что составляет 49—51% теории (см. примечание 2).
Маточный раствор ( — 60 мл) упаривают на водяной бане поч-
ти досуха, отфильтровывают и по возможности тщательнее
отжимают. Спирт, полученный после промывки дикалиевой
соли, отгоняют; остаток в колбе вместе с дикалиевой солью,
полученной после упаривания маточного раствора ( — 80 г),
подвергают перекристаллизации из 78%-ного спирта (1:7) в
присутствии активированного угля (3 г).
Получают 45 г (—20% теории).
Общий выход составляет —70% теории.
Примечания.
1. о-Сульфобеизойная кислота получается окислением дитиосалицило-
вой кислоты крепкой азотной кислотой и всегда содержит окислы азота.
2. Содержание основного вещества определяют ионообменным методом
с применением катионита в Н-форме (КУ-1 или КУ-2) с последующим тит-
рованием 0,1 н. раствором едкого натра.
ЛИТЕРАТУРА
1. Remsen, Dohme, Amer. Chem. J., 11, 332 (1889).
Поступила в марте 1962 г.
ИРЕ Л
0-СУЛЬФОБЕНЗОЙ НОЙ КИСЛОТЫ ДИХЛОРАНГИД-
РИДЫ
(Эвтектическая смесь изомеров)
Д. III. РОЗИНА, Л. Т. НЕСТЕРЕНКО
СОС1
|XX|-SO2C1
СС12
хо
']—so2
C7H4O3SC12 М. в. 239,02
Известны два дихлорангидрида о-сульфобензойной кисло-
ты *; симметричного и лактонного строения; один из них имеет
т. пл. 79°, другой т. пл. 40°. В литературе нет единого мнения
по вопросу о том, какой из них имеет симметричное и какой
лактонное строение [1, 2].
Нами показано, что дихлорангидрнд с т. пл. 40° имеет сим-
метричное, а с т. пл. 79°—лактонное строение [3]. Оба дихлор-
ангидрида образуют эвтектическую смесь с т. пл. 21° [4].
Описанные в литературе способы получения смеси изомер-
ных дихлорангидридов основаны на взаимодействии пятихло-
ристого фосфора с солями о-сульфобензойной кислоты [2, 5, 6J.
Однако при воспроизведении условий [2, 5, 6] дихлорангид-
риды получить не удалось.
Нами установлено, что реакция успешно протекает в при-
сутствии хлорокиси фосфора при дробной чередующейся за-
грузке реагентов.
Для извлечения дихлорангидридов из реакционной смеси
использован органический растворитель (бензол), что при пос-
ледующей перегонке бензольного раствора позволило,вопреки
литературным данным [1,- 5], получить изомерную смесь ди-
хлорангидридов в чистом виде.
СИНТЕЗ ДИХЛОРАНГИДРИДОВ о-СУЛЬФОБЕНЗОЙНОЙ КИСЛОТЫ
COOK
POCI,
+ 4PCU------->
СОС1
I
/ SO2C1
* В дальнейшем дихлорангидрнд о-сульфобензойной кислоты для
краткости мы будем называть «дихлорангидрнд».
94
СС1а
I 'о
+ I р502 4- 4 Р0С13 + 4 КС1
Характеристика основного сырья
Дикалиевая соль о-сульфобензойной кислоты, содержание
основного вещества 99,5—100% (см. стр. 92).
Пятихлористый фосфор, ТУ НКХП 386—41.
Хлорокись фосфора, ВТУ У 166—51.
Условия получения
В колбу (см. примечание 1), помещенную в глицериновую
баню, постепенно вносят 51 г хлорокиси фосфора и при пере-
мешивании 70 г тщательно растертого пятихлористого фосфо-
ра и 15 а дикалиевой соли о-сульфобепзойной кислоты- После
10 минут перемешивания добавляют еще 74 г пятихлористого
фосфора.
Реакция начинается самопроизвольно, и температура повы-
шается до 40—45°, после чего реакционную смесь нагревают
до 85—90° и при этой температуре вносят в течение 1 часа
68,5 г дикалиевой соли. (Всего загружают 83,5 г (0,30 Л4) ди-
калиевой соли о-сульфобензойной кислоты и 144 г (0,69 Л4)
пятихлористого фосфора). По окончании загрузки дикалиевой
соли перемешивание продолжают 3 часа при температуре ки-
пения (115—117°). Затем реакционной смеси дают охладиться
до 60° и экстрагируют полученные дихлорангидриды 75 мл су-
хого бензола; после 15 минут перемешивания выключают ме-
шалку и оставляют реакционную смесь для отстоя на 2 часа.
Верхний слой, содержащий раствор дихлорангидридов и хлор-
окиси фосфора в бензоле, осторожно сливают, а остаток в кол-
бе отфильтровывают, осадок (в основном хлористый калий)
промывают три раза по 15 мл сухого бензола, присоединят
промывной бензол к общему фильтрату. Фильтрат, вместе со
слитым слоем бензольного раствора, переносят частями, че-
рез капельную вОронку, в колбу для вакуум-перегонки ем-
костью 100 мл, и при 45—50 мм отгоняют на водяной бане
смесь бензола и хлорокиси фосфора (см. примечание 2); оста-
ток в колбе перегоняют на масляной бане при 15—20 мм
(предварительно удаляют холодильник, присоединив колбу
непосредственно к приемникам). После небольшого предгопа
(5—6 г) температура паров быстро повышается и при 165—
95
170° переходят дихлорапгидриды, в виде бесцветной масляни-
стой жидкости (см. примечание 3).
Выход 52—54 г, что составляет 72,5—75,3% теории.
Найдено %-. С1 (в боковой цепи) 29,52; 29,80.
C^jOjSCIj. Вычислено %: С1 (в боковой цепи) 29,66.
Количество общего хлора всегда совпадало с количеством
хлора в боковой цепи, что показывает на отсутствие хлора в
ядре (см. примечания 4, 5). При гидролизе дихлорангидридов
(см. примечание 6) получается с количественным выходом
о-сульфобензойная кислота с т. пл. 67—68° (литературные
данные 68—69° [7], 70° [8]).
Примечания.
I. Трехгорлая колба емкостью 250 мл снабжена мешалкой с глицери-
новым затвором, шариковым холодильником, конец которого защищен хлоо-
кальцневой трубкой, и отверстием с стеклянной пробкой для загрузки реа-
гентов; все соединения должны быть на шлифах.
2. Для отгонки бензола и хлорокиси фосфора целесообразно использо-
вать водоструйный насос, а нс масляный, так как пары этих жидкостей
могут попасть в масло.
3. Маслянистая жидкость обычно при храпении полностью или частич-
но закристаллизовывается. Влага воздуха разлагает дихлорангидриды, по-
этому их следует хранить в герметичной стеклянной таре или в запаянных
ампулах.
4. Хлор в боковой цепи определяют после гидролиза точной навески
0,5 и. раствором едкого натра. Хлор в ядре определяют по Степанову, но
этиловый спирт заменяют бутиловым.
5. Отсутствие хлора в ядре свидетельствует о термической стойкости
обоих дихлорангидридов в процессе перегонки при 20 мм (т. кип. 165—
170°), по [1 и 5] дихлорангидрид с т. пл. 79’ в указанных условиях разла-
гается иа л-хлорбеизоилхлорид и сернистый газ).
Следует также отметить, что дихлорангидрид с т. пл. 79° выделен
нами в чистом виде из смеси дихлорангидридов, полученных после пере-
топки.
6. Гидролиз следует проводить водой в отсутствие щелочи (1 г в 50 ли
дистиллированной воды) во избежание образования минеральной соли, ко-
торая препятствует учету выхода и определению температуры плавления
получаемой о-сульфобеизойной кислоты.
ЛИТЕРАТУРА
1. R. List, М. Stein, Вег., 31, 1648 (18ОД).
2. J. Scheiber, М, К not he, Вег., 45, 2252 (1912).
3. Д. Ш. Розина, Л. Т. Нестеренко, Ю. И. Вайнштейн, Ж. общ. химии,
28, 2878 (1958).
4. Holmes, Amer. Chem. J.. 25, 202 (1901).
5. P. Fritsch, Ber., 29. 2298 (1896).
6. W. R. Orndorff, R. Corvel, J. Amer. Chem. Soc., 48, 981 (1926).
7. Remsen, Dohme, Amer. Chem .1., 11, 332 (1889).
8. Taverne, Recueil trav, chim, 25, 52 (1906).
Поступила в марте 1962 г.
ИРЕА
0-НИТРОФЕНИЛАРСОНОВАЯ КИСЛОТА
В. И. КУЗНЕЦОВ, Д. Ш. РОЗИНА
NO2
/ ^-AsO3H2
C6H6O5NAs М. в. 247,03
о-Нитрофениларсоновая кислота была впервые получена
Бартом взаимодействием хлористого о-нитрофенилдиазония
с арсенитом натрия [1, 2]. В качестве катализатора этой реак-
ции предложен медный купорос [3]. Дальнейшие исследования
не привели к существенным изменениям метода Барта [4, 5, 6].
Приведенный выход о-нитрофениларсоновой кислоты колеб-
лется от 65% [6] до 93% теории [2]. В последнем случае произ-
водилось многократное упаривание маточного раствора. Арсе-
нирование рекомендуется проводить в слабощелочной среде
[2, 4]; однако имеется указание, что для соединений, содержа-
щих отрицательные заместители, наиболее благоприятна кис-
лая среда [5, 7].
Нами установлено, что арсенирование хлористого о-нитро-
фенилдиазония при pH 5,0—5,2 приводит к очень низкому
выходу (~15% теории). Оптимальное значение pH для дан-
ной реакции находится в интервале 7—9 (выход ~80%) [81.
В отличие от [2, 4, 6J, нейтрализация избыточной кислоты в ра-
створе хлористого о-нитрофенилдиазония производилась угле-
кислым натрием до рН~5. Исследованы также температура
арсенирования, концентрация применяемых растворов, поря-
док загрузки реагентов. Внесенные изменения и уточнения
обеспечили высокий стабильный выход и чистоту о-нитрофе-
ниларсоновой кислоты.
СИНТЕЗ о-НИТРОФЕНИЛАРСОНОВОЙ КИСЛОТЫ
no2 no2
I I
|X^-NH2-i-NaNO24-2HCl -* ( X|_N+sN+NaCI + 2H,0
\ / V/
7 Зак. en? 97
no2
I Cl
/\_ m+—n
2 | | + As2O8 + 6NaOH -»
N03
/ONa
-* 2 | |-As^gNa +2NaCl+3H2O+2N2
NOa NO2
1 xONa 1 /ОН
/ |— As^-ONa . /x.—As^-OH „
| | 4-2HC1-* | | 4q + 2NaCl
Характеристика основного сырья
о-Нитроанилин технический, ТУ 1550—57-
Мышьяковистый ангидрид, рафинированный, Ост НКТП—
5884
132 '
Условия получения
Диазотирование о-нитроанилина. В фарфоровый стакан на-
ливают 150 мл воды, 150 мл 34 %-ной соляной кислоты (уд. в.
1,18) и при температуре 90—95° вносят 56,0 г (0,40 М)
98,5%-пого о-нитроанилина. Полученный раствор солянокисло-
го о-нитроанилина переносят в стеклянную банку емкостью 1 л.
снабженную мешалкой рамного типа, и охлаждают при пере-
мешивании (в бане с ледяной водой) до 5°. При этом соляно-
кислый о-нитроанилин выделяется в виде светло-желтых игл.
Когда температура снижается до 5°, добавляют 200 г мелко
раздробленного чистого льда и при хорошем перемешивании
вливают, по возможности быстрее, через трубку, опущенную в
жидкость на 1—2 см до дна, раствор 33 г (0,47 М) 97%-ного
нитрита натрия в 150 мл воды (температура повышается до
8—10°). Реакционную массу охлаждают до 3—5°, перемеши-
вают 20 минут и проверяют по йодкрахмальной бумажке на-
личие нитрита (мгновенное посинение). В случае отрицатель-
ного результата добавляют нитрит натрия.
По окончании диазотирования полученный раствор хлори-
стого о-нитрофенилдиазонияI (не должен содержать желтых
игл солянокислого, о-нитроанилина). отфильтровывают . (см.
примечание 1)? К прозрачному фильтрату желтого цвета до-<
бавляют при температуре 3—5° в течение ~ 10 минут при пе-
98
ремешивании 150 мл 10%-ного раствора углекислого натрия
до рН~5. Общий объем 800 мл.
Арсенирование. В стеклянную банку емкостью ~3 л, снаб-
женную мешалкой рамного типа, вливают заранее приготов-
ленный при 25—30° раствор арсенита натрия из 48 г (0,23 Л4)
95%-ного мышьяковистого ангидрида в 10 мл 17%-ного едко-
го натра и раствор из 4 г медного купороса в 25 мл
25%-ного аммиака. К раствору арсенита при температуре 0—
2° (наружное охлаждение льдом с поваренной солью) добав-
ляют при энергичном перемешивании из капельной воронки
в течение 1 часа (см. примечание 1) полученный раствор хло-
ристого о-нитрофенилдиазония со скоростью ~2 мл в минуту.
Спустя ~10 минут реакция становится слабощелочной по фе-
нолфталеиновой бумажке, после чего одновременно с раство-
ром диазония начинают добавлять из другой капельной ворон-
ки с такой же скоростью 10%-ный раствор едкого натра, сле-
дя за тем, чтобы реакция была сильнощелочной по бриллиан-
товой желтой и слабощелочной или нейтральной по фенолфта-
леиновой бумажке. (Реакция контролируется каждые 3—5 ми-
нут). Расходуется ~ 600 .ил раствора едкого натра (см. приме-
чание 2). По окончании добавления раствора хлористого
о-нитрофенилдиазония реакционную массу продолжают пере-
мешивать 1 час при температуре 0—5° и отфильтровывают от
небольшого осадка. Фильтрат — раствор натриевой соли о-нит-
рофениларсоновой кислоты (~ 1600 мл) упаривают в фарфоро.
вой чашке на водяной бане до объема 400—500 мл. К упарен-
ному раствору добавляют при температуре 65—70° 10 г акти-
вированного угля, перемешивают периодически от руки в те-
чение 15 минут, нагревают до 95° и отфильтровывают; уголь
промывают 100 мл горячей воды (~90э), присоединяя про-
мывную воду к фильтрату.
Превращение натриевой соли о-нитрофениларсоновой кис-
лоты в свободную кислоту. Фильтрат (~ 1600 мл) переносят
в круглодоипую колбу, снабженную мешалкой, нагревают на
водяной бане до 75° и добавляют из капельной воронки тонкой
струйкой при перемешивании ~200 мл соляной кислоты (уд.
в. 1,18) до сильнокислой реакции по конго. По мере добавле-
ния кислоты о-нитрофениларсоновая кислота выделяется в ви-
де бледно-желтых игл. Реакционной массе дают охладиться до
50°, затем снижают температуру до 10° и оставляют при этой
температуре на 1 час. о-Нитрофениларсоновую кислоту от-
фильтровывают, тщательно отжимают, промывают водой до
исчезновения С1~ в промывных водах (проба с азотнокислым
серебром) и сушат при 60—65°.
Выход 83—84 г, что составляет 84—85% теории (см. при-
мечание 3).
Найдено %: As 30,0; 30,30.
C6HeO5NAs. Вычислено %: As 30,36 (см. примечание 4).
7 > 99
Примечания.
1. Раствор хлористого о-нитрофепилдиазопия следует по возможности
охлаждать ледяной водой.
2. Процесс арсенирования сопровождается ценообразованием (выде-
ление азота); при добавлении 1—2 мл эфира или 2—3 капель подсолнечно-
го масла пена сразу спадает.
3. Маточный раствор содержит 2—3 г о-нитрофенпларсоновой кислоты
и не подлежит утилизации.
4. Мышьяк определяется по ТУ МХП 2808—51.
ЛИТЕРАТУРА
1. Н. Bart, Герм. пат. 250264, 1912; Fr. X, 1254.
2. И. Bart, Liebigs Ann. Chem., 429, 92, 106 (1922).
3. J. Kalb, Liebigs Ann. Chem., 423, 39 (1921).
4. H. Schmidt, Liebigs Ann. Chem., 421, 171 (1920).
5. L. Benda, J. prakt. Chem., 75, 95 (1917).
6. N. Jacobs, Al. Heidelberg, J. Rolf, J. Amer. Chem. Soc., 40, 1580
(1918).
7. P. Karrer., Ber, 48, 1061 (1915).
8. Д. Ш. Розина, Ж. прикл. химии, 23, 211 (1950).
Поступила в марте 1962 г. ИРЕА
0-АМИНОФЕНИЛАРСОНОВАЯ КИСЛОТА
В. И. КУЗНЕЦОВ, Д. Ш. РОЗИНА
nh2
I
CeH8O3NAs М. в. 217,04
о-Аминофениларсоновую кислоту получают восстановлени-
ем о-нитрофениларсоновой кислоты (см. стр. 91). В качестве
восстановителей предложены амальгама натрия [1], закисные
соли железа [2, 3, 4] и металлическое железо [5].
При воспроизведении условий [5] о-аминофениларсоновая
кислота получалась с низким выходом и низкой температу-
рой плавления. В пропись [5] нами внесены весьма существен-
ные изменения [6].
1) Вместо хлорного железа использован хлористый натрий,
в присутствии которого реакция протекает при pH 7,2—7,8, что
100
обеспечило селективность восстановления (не затрагивается
арсоновая группа).
2) Применена чугунная стружка, просеянная через сито в
80 мет.
3) Восстановление проводят при интенсивном кипении, а
не при 70°.
4) Увеличено количество едкого натра при разложении об-
разовавшейся железной соли о-аминофениларсоновой кисло-
ты, что обеспечило полное ее превращение в натриевую.
5) Выделен побочный продукт неполного восстановления
о-нитрофениларсоновой кислоты, который охарактеризован
нами как азобензол о-2,2'-диарсоновая кислота. (Она пол-
ностью адсорбируется шламом).
СИНТЕЗ о-АМИНОФЕНИЛАРСОНОВОИ КИСЛОТЫ
no2 nh2
I
4 Г
-AsO3H2 NaCl
+ 9Fe+4H2O
AsO,H2
' +3Fe3O.
Характеристика основного сырья
о-Нитрофениларсоновая кислота, ТУ МХП 2808—51.
Чугунные опилки, чистые, без ржавчины, просеянные через
сито в 80 меш.
Условия получения
В круглодонную колбу емкостью 2 л, снабженную якорной
мешалкой (см. примечание 1), доходящей почти до дна, об-
ратным холодильником и загрузочным отверстием, вносят 1 л
воды и при перемешивании 50 г хлористого натрия. Получен-
ный раствор хлористого натрия нагревают на глицериновой
бане до кипения и при энергичном перемешивании загружают
200 г чугунных опилок (весь процесс следует проводить при
интенсивном кипении и энергичном перемешивании). Спустя
30 минут, к образовавшейся суспензии добавляют в течение
2 часов 100 г (0,4 Л4) о-нитрофениларсоновой кислоты (см.
примечание 2), после чего реакционную смесь выдерживают
2 часа; по окончании выдержки добавляют 200 мл 17%-ного
раствора едкого натра, кипятят еще 0,5 часа и оставляют на
10—15 минут без перемешивания для отстоя шлама. Горячий
раствор (~90°) полученной натриевой соли о-аминофенилар-
соновой кислоты осторожно, стараясь не взмучивать, отфиль-
тровывают, шлам промывают сначала на воронке 200 мл, а
затем при перемешивании в 300 мл горячего (90°) 1%-ного ра-
101
створа едкого натра, присоединяя промывные воды к фильт-
рату. К фильтрату (~1,5 л), окрашенному в желтый цвет,
добавляют ~ 100 мл соляной кислоты (уд. в. 1,18) до кислой ре-
акции по конго (см. примечание 3), 10 г активированного уг-
ля, перемешивают 15 минут и отфильтровывают; уголь промы-
вают 50 мл воды, присоединяя промывную воду к фильтрату.
Из бесцветного фильтрата (~1,6 л), содержащего раствор
о-аминофениларсоновой кислоты, отгоняют на водяной бане
воду при 50—60 мм (см. примечание 4). В процессе отгонки
воды о-аминофениларсоновая кислота выделяется в виде бе-
лых блестящих игл; при накоплении кристаллов отгонку пре-
рывают; охлаждают колбу в ледяной воде до 5—10°, остав-
ляют при этой температуре на 1 час, о-аминофениларсоновую
кислоту отфильтровывают и тщательно отжимают. Из маточ-
ного раствора продолжают в указанных условиях отгонку до
конечного объема ~200 мл, проверяют реакцию по конго и в
случае надобности осторожно добавляют соляную кислоту. По-
лученный раствор охлаждают в ледяной воде 1 час, выкри-
сталлизовавшуюся о-аминофениларсоновую кислоту отфильт-
ровывают, отжимают и вместе с полученной из основного
фильтрата сушат при 60—65°. Выход 72—73 г с содержанием
основного вещества 94,5—95,0% по содержанию NaNC>2 и
94,0—94,5% по содержанию As (см. примечание 5), что состав-
ляет 77—78% теории. Т. пл. 152—153° (по литературным дан-
ным 153—154° [1], 153° [2]).
о-Аминофениларсоновая кислота содержит ~5% хлори-
стого натрия.
Перекристаллизация из воды, слегда подкисленной соля-
ной кислотой (по конго), повышает содержание основного ве-
щества до 98—99%.
Примечания.
1. Необходима эффективная мешалка, которая должна обеспечить
энергичное перемешивание реакционной смеси и подъем тяжелых чугунных
опилок.
2. При загрузке второй половины о-нитрофениларсоновой кислоты
происходит довольно сильное вспенивание; пена сразу спадает от до-
бавления двух капель подсолнечного масла, эмульгированного в 10 мл хо-
лодной воды.
3. Соляную кислоту следует добавлять' очень осторожно только до
появления кислой реакции по конго, избыток кислоты переводит о-амино-
фениларсоновую кислоту в солянокислую соль.
4. Целесообразно применять колбу емкостью ~ 700 мл, а раствор до-
бавлять по частям через капельную воронку!, присоединенную к колбе.
5. Содержание основного вещества определяют диазотированием о-ами-
нофениларсоновой кислоты 0,5 и. раствором нитрита натрия при 0°, мы-
шьяк— по ВТУ МГУХП 328—59.
ЛИТЕРАТУРА
1. Kaso-Kaschimo, J. Amer. Chem. Soc., 47, 2207 (1925).
2. L. Benda, Ber., 44, 3306 (1911).
3. L. Benda, Ber., 47, 1006, 1316 (1914).
102
4. И. Jacobs, Al. Heidelberg, J. Rolf, J. Amer. Chem. Soc., 40, 1580
(1918).
5. L. Kalb. Liebigs Ann. Chem., 423, 39 (1921).
6. Д. Ш. Розина, Ж. прикл. химии, 23, 211 (1950).
Поступила в марте 1962 г. ИРЕА
АЗОТОЛ 2,4 МК
(3-Карбокси(-2',4/-диметиланилидо)-2-оксинафталин)
Д. Ш. РОЗИНА, В. М. ДЗИОМКО, Р. И. РОЗЕНБЕРГ
/\/х/он
III
\/\/4CONHK /-к
Х-СН3
сн3
CI9HI7O2N М. в. 291,32
Азотол 2,4 МК в литературе не описан. Изомер этого азо
тола 3-карбокси- (2/6/-диметиланилидо) -2-оксипафталин полу-
чен конденсацией 2,6-ксилидина с 2-окси-З-нафтойной кислотой
в среде хлорбензола; в качестве конденсирующего реагента
применен треххлористый фосфор [1]. Синтез азотола 2,4 МК
осуществлен нами аналогичным способом
СИНТЕЗ АЗОТОЛА 2,4 МК
/х/х/он — C,HSC1
3 I I I +3H2N-/ х-СН3 + РС13------S
сн,
/Х/\/0Н
31 | | +Р(ОН)3 + ЗНС1
А /\conhX ~ Усн-
I
сн3
* Приведенное соединение мы назвали «азотол 2,4 МК». по аналогии
с' наименованиями других азотолов.
103
Характеристика основного сырья
2-Окси-З-нафтойная кислота, техническая, ТУ—У 750.
2,4-ксилидин, ч., ТУ МХП 197—51.
Хлорбензол, ТУ МХП 2877—54.
Треххлористый фосфор, ГОСТ 91—41.
Натрий углекислый, ч., ГОСТ 84—41.
Условия получения
Очистка 2-окси-З-нафтойной кислоты (см. примечание 1).
В трехгорлую колбу емкостью 1,5 л, снабженную мешал-
кой и термометром, вливают 1 л 5%-ного раствора углекисло-
го натрия, нагревают на водяной бане до 40—45э и добавляют
при перемешивании 10 мл 20%-ного раствора сульфита натрия
и 100 г 2-окси-З-нафтойной кислоты. Когда вся кислота пере-
ходит в раствор, вносят 5 г активированного угля, перемеши-
вают при указанной температуре 15 минут и отфильтровыва-
ют. Фильтрат, содержащий натриевую соль 2-окси-З-нафтой-
ной кислоты, переносят в колбу, нагревают до 85°, добавляют
при перемешивании ~85 мл соляной кислоты уд. в. 1,18 до
кислой реакции по конго и охлаждают до 20°. Выделившуюся
2-окси-З-нафтойную кислоту отфильтровывают, тщательно от-
жимают, промывают 50 мл ледяной воды и сушат при 70—757
Получают 90 г (90% теории) с т. пл. 217—218°.
Получение азотола 2,4 МК. В колбу (см. примечание 2),
помещенную в глицериновую баню, вливают 600 мл хлорбен-
зола (см. примечание 3), а при температуре 130° вносят при пе-
ремешивании 94 г (0,5 Л4) 2-окси-З-нафтойной кислоты.
После 15 минут перемешивания (растворение неполное)
вливают 60,5 г (~ 0,5 М) ксилидина (см. примеча-
ние 3). В полученный прозрачный раствор добавляют при
указанной температуре из капельной воронки в течение 2 ча-
сов смесь из 30 г треххлористого фосфора и 30 мл хлорбензо-
ла, после чего продолжают перемешивание при температуре
кипения жидкости в течение 10 часов. По истечении указан-
ного времени к реакционной смеси, окрашенной в оранжевый
цвет и содержащей осадок желтого цвета добавляют ~630 мл
3%-ного раствора углекислого натрия до слабощелочной ре-
акции по бриллиантовой желтой бумажке (рН~8) и из той
же колбы при перемешивании (во избежание толчков) отго-
няют с водяным паром хлорбензол и непрореагировавший
2,4-ксилидин. до получения прозрачного отгона, следя за тем,
чтобы сохранился первоначальный объем. По окончании от-
гонки реакционную смесь охлаждают до 70—75°, добавляют
4 г гидросульфита натрия, перемешивают 10 минут и проверяв
ют pH, значение которого должно быть 8,0, в противном слу-
чае добавляют несколько миллилитров 3%-ного раствора угле-
104
кислого натрия. Затем заменяют прямой холодильник обрат-
ным, кипятят реакционную смесь 1 час (см. примечание 4),
после чего азотол отфильтровывают, промывают горячей водой
до нейтральной реакции промывных вод.
Получают 240 г влажного азотола, окрашенного в оранже-
вый цвет (т. пл. 209—210°, см. примечание 5).
С целью очистки полученный азотол при 80° растворяют
при перемешивании в 500 мл 4%-ного раствора едкого натра,
к которому предварительно добавляют 50 мл 10%-ного ра-
створа гидросульфита натрия. К полученному раствору на-
триевой соли азотола добавляют 5 г активированного угля, пе-
ремешивают 15 минут, уголь отфильтровывают. К фильтрату
добавляют 500 мл воды и при температуре 20° выделяют азо-
тол добавлением 35%-ного раствора серной кислоты до кис-
лой реакции по конго. Расходуется ~65 мл.
Выделившийся азотол отфильтровывают, промывают горя-
чей водой до отсутствия SO42- в промывных водах (проба с
хлористым барием) и сушат при 70—75°.
Выход 116—120 г азотола светло-серого цвета, что состав-
ляет 80—83% теории. Т. пл. 216—217°. Нами установлено, что
температура плавления чистого азотола 2,4 МК 220—221° (см
примечание 6).
Найдено %: С 79 40; Н 6,2.
CijH17O2N. Вычислено %: 78,31; Н 5,9.
Примечания.
1. 2-Окси-З-нафтойную кислоту подвергают очистке только в том слу-
чае, если она имеет т. пл. ниже 217°.
2. Колба снабжена мешалкой, термометром, капельной воронкой и об-
ратным шариковым холодильником.
3. Хлорбензол предварительно сушат в течение не менее трех суток
прокаленным хлористым кальцием; 2,4-ксилидин — столько же времени ед-
ким натром.
4. При pHcv8 2-окси-З-нафтойная кислота образует соль и переходи'!
в раствор, в то время как азотол в тех же условиях нерастворим.
5. Температура плавления определена в отобранной средней пробе,
высушенной до постоянного веса.
6. Температура плавления была установлена после перекристаллиза-
ции последовательно из хлорбензола и толуола.
ЛИТЕРАТУРА
1 М. Schmidt, Е. Moser, Пат. США. 2621173, 1952; Chem. Abstrs, 48,
5507 (1954).
Поступила в апреле 1 962 г.
ИРЕА
2-ОКСИБЕНЗОЛ-(1-АЗО-1')-2'-ОКСИ-3'-(2",4"-ДИМЕТИЛ-
КАРБОКСИАНИЛИДО)-НАФТАЛИН
(Магов, обан)
В. М. ДЗИОМКО, Д. Ш. РОЗИНА, Р. И. РОЗЕНБЕРГ
C2gHaiO3N3
М. в. 411,44
2-Оксибензол-(1-азо-1') -2 -окси-3'- (2'’, 4"-диметнл карбокси-
анилидо)-нафталин предложен для колориметрического опре-
деления малых количеств магния [1,2] и как индикатор для
комплексометрического титрования [3]. Синтез реактива в ли-
тературе не описан.
Нами магон получен диазотированием о-аминофенола с
последующим сочетанием полученного диазооксида о-амино-
фенола с 3-карбокси-(2', 4'-диметиланилидо)-2-оксинафтали-
ном (азотолом 2,4 МК).
СИНТЕЗ МАГОНА
ОН о-----.
1 1 I
,'/X.-NHa <X-N=N
| | . + NaNOa-f-HCl—[ 4-NaCH 2H3O
| | | д-NaOH —>
\/x/xCONH-^ /-CH3
CH3
/\/\/0Na
\/\/\C0NH-^ ^-CH3
I
CH3
юе
О-----,
I J
,/ \ -N=N
ONa
ONa CONH-
4-HC1 —>
-CH34-NaCl
Характеристика основного сырья
о-Аминофенол, ч., ТУ МХП 1979—49.
Азотол 2,4 МК, т. пл. не ниже 216”.
Условия получения
Диазотирование о-аминофенола. В фарфоровый стакан
вливают 120 мл дистиллированной воды, 13 мл соляной кис-
лоты (уд. в. 1,18) и при перемешивании 10,8 г (0,1 М) о-ами-
нофенола. К полученному раствору солянокислого о-аминофе-
нола, окрашенному в коричневый цвет, добавляют 1,0 г акти-
вированного угля, перемешивают 5 минут и отфильтровывают.
Полученный бесцветный прозрачный раствор переносят в ста
кан, помещенный в баню со льдом и поваренной солью, ох-
лаждают до 2—3° и диазотируют раствором нитрита натрия из
107
7,1 г (0,1 М) в 30 мл воды, добавляя его при перемешивании
в течение 15 минут. При добавлении последних 2—3 мл нитри-
та натрия следят за реакцией по йодкрахмальной бумажке.
Диазотирование считают законченным, когда мгновенное по-
синение йодкрахмальной бумажки сохраняется в течение 10
минут. Полученный прозрачный раствор диазооксида о-ами-
нофенола перемешивают в точение 0,5 часа при температуре
2—3° и добавляют ~250 мл 5%-ного раствора углекислого
натрия до исчезновения кислой реакции по конго. Объем ра-
створа ~250 мл.
Сочетание. В трехгорлую колбу емкостью 1 л, снабжен-
ную мешалкой, капельной воронкой и термометром, вливают
160 мл 4%-ного раствора едкого натра, нагревают на водяной
бане до 70° и вносят при перемешивании 53 г (0,1 М) влажно-
го азотола 2,4 МК> содержащего 54,7% сухого вещества; через
5—10 минут азотол переходит в раствор. К полученному проз-
рачному раствору натриевой соли азотола при температуре
~20° добавляют при перемешивании тонкой струйкой полу-
ченный раствор диазооксида о-аминофенола и выдерживают
при перемешивании 4 часа. Реакционная смесь сразу окраши-
вается в фиолетовый цвет, переходящий через 15—20 минут в
темно-синий; одновременно начинается выделение красителя.
По окончании выдержки (см. примечание) выделившийся кра-
ситель отфильтровывают и промывают 50 мл дистиллирован-
ной воды; получают 56—58 г (влажного) красителя.
Очистка красителя. В трехгорлую колбу емкостью 500 мл.
снабженную мешалкой, вливают 300 мл 4%-ного раствора ед-
кого натра, нагревают на водяной бане до 80° и вносят при
перемешивании полученный влажный краситель (56—58 г),
перемешивание продолжают 1 час при указанной температу-
ре и отфильтровывают (примеси переходят в раствор). Остав-
шийся на воронке краситель промывают 20 мл горячего 4%-
ного раствора едкого натра и тщательно отжимают, после че-
го его снимают с фильтра и повторно обрабатывают горячим
раствором едкого натра. Краситель, полученный после повтор-
ной очистки, растворяют при 60° в 200 мл 4 %-кого спиртового
раствора едкого натра (растворение полное). К полученному
раствору добавляют при указанной температуре 12%-ный ра-
створ соляной кислоты до кислой реакции по конго. Выделив-
шийся краситель отфильтровывают, промывают дистиллиро-
ванной водой цо отсутствия С1_ в промывных водах (проба с
азотнокислым серебром) и сушат при 105° до постоянного
веса.
Выход 24,5—25,0 г, что составляет 60% теории, считая на
о-аминофенол. Т. пл. 246—247° или 247—248° (по литератур-
ным данным т. пл. 246—247° fl]).
Чувствительность реактива к Mg 0,1—0,2 мкг в 5 мл раствора.
Найдено И: С 73,10; 72,8; Н 4,80; 4рО.
CasHnO,V,. Вычислено %: С 73,02; Н 5,11.
Примечание.
Проба должна показывать отсутствие диазосоединения с резорцином и
наличие азотола (небольшой избыток) с диазо-/г-нитроанилином.
ЛИТЕРАТУРА
I. С. К. Mann, J Н. Joe, Aiialyt. chim. acta, 16. 155 (1957).
2. M. J. Maurice, Analyt chitn. acta, 20, 181 (1959).
3. R. H. Maier, Nature" 183, 461 (1959).
Поступила e марте 1S62 г. ИГЕ A
6-АМИНОТИМОЛ СОЛЯНОКИСЛЫЙ
Д. Ш. РОЗИНА, Р. Г. СНЯТКОВСКАЯ
CI0HlsONCl
СН3
HC1-NH, |
^р'он
СН(СН3)2
М. в. 201,67
6-Аминотимол солянокислый рекомендуется как эффек-
тивный реактив для колориметрического определения витами-
на В' (тиамина) в фармацевтических препаратах и зерновых
продуктах [I]. Описанные в литературе методы получения со-
лянокислого 6-аминотимола основаны на восстановлении
6-нитрозотимола различными восстановителями с последую-
щим превращением полученного 6-аминотимола в солянокис-
лую соль. В качестве восстановителей рекомендуются олово
[2], сероводород [3] и гидросульфит натрия в среде едкого
натра [4].
6-Нитрозотимол получают нитрозированием тимола нитри-
том натрия в водной среде [3] и в среде спирта, насыщенного
соляной кислотой [5, 6].
Нами проверены и, в основном, подтверждены литератур-
ные данные по нитрозированию тимола [5, 6]. Принят способ
ннтрозирования тимола в среде спирта, насыщенного соля-
ной кислотой, так как он обеспечивает более высокий выход и
109
лучшее качество 6-нитрозотимола. Уточнены количества при-
меняемого нитрита натрия и спирта. В качестве восстанови-
теля мы сочли целесообразным применить гидросульфит на-
трия. Однако восстановление в среде едкого натра при добав-
лении гидросульфита натрия мелкими порциями в сухом виде
[4] приводило к окислению получаемого 6-аминотимола (6-ами-
нотимол получался коричневого цвета) и к низкому выходу
Нами установлено, что реакция протекает успешно в ам
миачной среде при быстром добавлении раствора гидросуль-
фита натрия, взятого в небольшом избытке (5%) по отпоше
нию к теории.
СИНТЕЗ СОЛЯНОКИСЛОГО 6-АМЙНОТИМОЛА
сн8
I
./х. С.Н.ОН
I I +NaNO2 + HCl------------*
'он
СН(СН3)2
СНз
ON |
I | +NaCl+H3O
/7 хон
СН(СН3)2
СН3
ON |
Ч| 4- 2Na2S2O44-5NH4OH ->
СН(СН3)а
СНз
HSN |
| | + 2Na2SO3 + 2(NH4)3SO3 + 2H2O.
Xi/XONH4
СН(СНз),
-СНз
HSN . |
X|/4onh4
CH(CHs)2
+ HC1 *
СНз
H2N I
I I + NH4C1
Y^OH
CH(CH3)2
110
СНз СН3
H,N I HCl-HsN |
4 Pl +HC1- T4|
X|/40H X|/X0H
CH(CH3)2 CH(CH8)2
Характеристика основного сырья
Тимол, ТУ ГКХ 1653—61.
Натрий гидросульфит, ТУ ГХП 126—56.
Условия получения
Получение 6-нитрозотимола. В колбу (см. примечание 1),
помещенную в баню, содержащую^ лад с солью, вливают
700 мл этилового спирта, охлаждают до 0° и при перемешива-
нии вносят в течение 20—25 минут 90 г (0,60 М) хорошо ра-
стертого тимола. К полученному раствору тимола прибавляют
из капельной воронки тонкой струйкой 450 мл соляной кислоты
уд. в. 1,18, следя за тем, чтобы температура не превышала 5°.
Затем реакционную массу охлаждают до 0° и при этой темпе-
ратуре, не прекращая перемешивания, добавляют в течение
1,5 часа (со скоростью ~2 мл в минуту) раствор из 45 г
(0,64 М) 98%-ного нитрита натрия в 200 мл воды. По оконча-
нии добавления нитрита натрия реакционную массу переме-
шивают при 0° еще 0,5 часа, разбавляют 500 мл воды и пере-
носят в другую колбу, содержащую 3,1 л холодной воды
(~10°). После перемешивания в течение 0,5 часа, выделив-
шийся 6-нитрозотимол отфильтровывают, отжимают и промы-
вают водой до отсутствия в промывных водах С1~ (проба с
азотнокислым серебром).
Получают 325 г влажного 6-нитрозотимола, который, не вы.
сушивая, применяют для восстановления.
Выход в пересчете на сухой 101 —104 г, что составляет 94—
97% теории (см. примечание 2), т. пл. 156—157° (по литера-
турным данным, т. пл. 161—162° [5]).
Получение 6-аминотимола. В трехгорлую колбу емкостью
1,5 л, снабженную мешалкой, вливают 1 л 25%-ного аммиака,
нагревают на водяной бане до 35° и при перемешивании вносят
полученный 6-нитрозотимол (0,57 Л4); после полного раство-
рения 6-иитрозотимола образовавшийся раствор аммонийной
соли отфильтровывают; осадок промывают 50 мл воды. Филь-
трат вместе с промывной водой (~1,3 л) переносят в колбу
емкостью 4 л, помещенную.в водяную баню, и при слабом пе-
ремешивании (~-60 оборотов в минуту) вливают сразу све
жеприготовленный раствор из 261 г (1,2 Л4) 80%-ного гидро-
111
сульфита натрия в 1,55 л воды. Через 2—3 минуты реакцион-
ная масса светлеет, температура самопроизвольно повышает-
ся до 45—50° (образовавшаяся аммонийная соль 6-аминотимо-
ла выделяется в виде густого осадка кремового цвета), после
чего реакционную массу нагревают до 60°, при этой темпера-
туре перемешивают 0,5 часа, затем охлаждают до 20° при пе-
риодическом слабом перемешивании и добавляют 440 мл 18% -
ной соляной кислоты (уд. в. 1,09), 50 мл 25%-ного аммиака и
0,5 г гидросульфита натрия. Выделившийся 6-аминотимол от-
фильтровывают и промывают 2%-ным раствором гидросуль-
фита натрия до отсутствия С1“ в промывных водах (расходу-
ется 1,5 л) (см. примечание 3).
Получают 183 г влажного 6-аминотимола, который без вы-
сушивания превращают в солянокислую соль. Выход в пере-
счете на сухой (см. примечание 2) 77—79 г, что составляет
82—83% теории, считая на нитрозотимол.
Получение солянокислого 6-аминотимола. В трехгорлую
колбу емкостью 700 мл, помещенную в водяную баню, налива-
ют 400 мл дистиллированной воды и при температуре 90° до-
бавляют 50 мл соляной кислоты уд. в. 1,18, 0,5 г гидросульфит
та натрия и при перемешивании-—полученный 6-аминотимол.
Через 10—15 минут 6-аминотимол переходит в раствор в виде
солянокислой соли; после охлаждения раствора до 60—70° до-
бавляют 5 г активированного угля, перемешивают 15 минут и
отфильтровывают. Если фильтрат окрашен в желтый цвет, его
вторично обрабатывают 3 г угля в присутствии гидросульфита
натрия (0,5 а). Образовавшийся раствор солянокислого 6-амп-
нотимола охлаждают сначала при комнатной температуре до
40—45°, а затем ледяной водой до 5—6°.
Выделившийся солянокислый 6-аминотимол отфильтровы-
вают, промывают дистиллированной водой до нейтральной ре-
акции промывных вод по конго (расходуется ~ 100 мл) и су-
шат при 70°. Получают 70 г продукта.
Из маточного раствора (~670 мл) отгоняют при 150—
200 мм ~500 мл воды. К остатку в перегонной колбе добавля-
ют 2 г активированного угля и 0,5 г гидросульфита натрия;
горячий раствор отфильтровывают, фильтрат охлаждают до
5-—6°. Выделившийся солянокислый 6-аминотимол отфильтро-
вывают, промывают дистиллированной водой до нейтральной
реакции промывных вод по конго (~40 мл) и сушат при 70°.
Получают еще 18—20 г продукта.
Общий выход 88—90 г с содержанием основного вещества
99,0—99,5% (см. примечание 4). что составляет 75—76% тео-
рии, считая на нитрозотимол и 71—72% теории по тимолу.
Примечания.
I. Синтез проводят в трехгорлой колбе емкостью 2 л, снабженной ме-
шалкой, термометром и двурогим форштоссом, соединенным с капельной во-
ронкой и воздушным холодильником (D 9 мм, Н 70 см).
112
2. Для определения содержания влаги отбирают среднюю пробу, влаж-
ного 6-нитрозотимола и сушат при 50 до постоянного веса.
3. Во избежание окисления 6-ампнотпмола при фильтровании и про-
мывке необходимо следить, чтобы он все время был покрыт раствором гид
росульфита натрия.
1. Содержание основного вещества определяют титрованием 0,1 н.
раствором азотнокислой ртути в присутствии дифепилкарбазона по ВТУ
ЛИТЕРАТУРА
1. К. J. Hayden, Analyst, 82. 61, 690 (19.57).
2. R. Schiff, Вег., 8, 1501.(1875).
3. C. Liebermann, М. Jlinski, Вег.. 18. 3194 (1885).
4. Н. Е. Albert, j. Amer. Chem. Soc., 76, 4985 (1954).
5. A. Klagest, Ber.. 32, 1518 (1899).
6. «Синтезы органических препаратов», сб. I, стр. 378, ИЛ, 1949.
Поступила и марте 1962 <.
ИРЕА
2,4-ДИНИТРОФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ
АМИНОКИСЛОТ
(2,4-ДНФ-производные аминокислот)
Г. И. КОШЕЛЕВА, Г. И. НАЛЕЦКАЯ
2,4-Динитрофенильпые производные аминокислот находят
широкое применение в качестве свидетелей для идентификации
аминокислот при хроматографическом анализе гидролизатов
белков и пептидов. Они представляют собой желтые кристал-
лические вещества, обладающие большой светочувствитель-
ностью. Хранить их следует в темноте или в посуде из оранже-
вого стекла.
Синтез монозамещсипых 2,4-динитрофенильных производ-
ных аминокислот — лейцина, триптофана, серина, глицина,
аланина, валина, аргинина, аспарагина, треонина, метионина,
аспарагиновой кислоты, р-фенилаланина, пролина —осущест
в.тяли с выходом 50—60% теоретического по методу К. Р. Рао.
Н. А. Собера [1] и Ф. Сангера [2] в водно-спиртовой среде при
эквимолекулярном соотношении аминокислоты и 2,4-динитро-
фторбензола в присутствии бикарбоната натрия при темпера-
туре 60°.
Ди-2,4-дпнитрофеН11Л-гпстидип. лизин и тирозин, получали
этим же способом, но с использованием избытка 2,1-дпнитро-
8 из
фторбензола, а именно: 2,5 моля на 1 моль аминокислоты, со-
гласно Л. К. Рамахандрану [3].
При синтезе 2,4-динитрофенил-глугамнновой кислоты исхо-
дили не из глутаминовой кислоты, а из 2,4-дииитрофенил-глу-
тамина, который был получен общим способом [1, 2}. 2,4-Ди-
иитрофенил-глутамин гидролизовали затем до 2,4-динитрофе-
нил-глутаминовой кислоты по К. Р. Рао и Н. А. Соберу [1].
о-2,4-Динитрофепил-тирозин получали способом, описан-
ным Е. Р. Фритце и X. Цаном [4], который заключается в син-
тезе медного комплекса тирозина, в обработке.его 2,4-динитро-
фторбензолом и выделении о-2,4-динитрофенил-тирозина.
к-2,4-Дииитрофенил-лизин синтезировали по методу Р. Р.
Портера и Ф. Сангера [5] из лизина и 2,4-динитрофторбензола
через медную комплексную соль лизина.
е-Бензоил-а-2,4-динитрофенил-лизин получали из е-бензо-
ил-лизина, на который действовали 2,4-динитрофторбензолом
[6]. е-Бензоил-2,4-дннитрофенил-лизин гидролизовали смесью
соляной и уксусной кислот до а-2,4-динитрофенил-лизина [2].
Нами были уточнены условия синтеза и очистки 2,4-дини-
трофенильных производных аминокилот.
2,4-ДИНИТРОФЕНИЛ-ЛЕЙЦИН
(2,4-ДНФ-лейцин)
СН3 /СООН
хсн-сн,-сн
снз NH
I
I//
NOa
CiaHtjNgOe М. в. 29/,28
СИНТЕЗ СОЕДИНЕНИЯ
F
СН3 /СООН I NaHCO,
^СН-СН2-СН -J- |XpNO2 -------------
сн3 nh,2 х /
114
СООН
Сп3 ।
ХСН—СН2-СН %- NaF4-H»O -СО»
сн3
NH
— NO»
NO,
Характеристика основного сырья
Лейцин, ч., ВТУ МХП 4139—53.
2,4-Динитрофторбензол, ч., ТУ завода.
Натрий двууглекислый, ч.д.а., ГОСТ 4201—48.
Спирт этиловый, ректификат, ГОСТ 5962—51.
Условия получения
В двухгорлую круглодонную колбу емкостью 0,5 л, снабжен-
ную механической мешалкой и обратным холодильником, по-
мещают 13,1 г (0,1 М) лейцина, 18,6 г (0,1 А4) динитрофтор-
бензола, 15 г двууглекислого натрия и 400 мл 50°/о-ного этило-
вого спирта. Реакционную смесь-перемешивают 1,5 часа при
температуре водяной бани 60°. Спирт отгоняют в вакууме во-
доструйного насоса, а водный остаток экстрагируют 3 раза (по
15 мл) эфиром для удаления непрореагировавшего динитро-
фторбензола. Водный раствор подкисляют концентрированной
соляной кислотой до кислой.реакции по бумажке конго
(30 мл). Выделившееся масло промывают ледяной водой (3
раза по 25 ,ил). 2,4-ДНФ-лейцин перекристаллизовывают из
смеси эфир: петролейный эфир (1 : 1). Выход 17 г (60% тео-
рии). Т. пл. 118—120°.
a-N-2,4-ДИНИТРОФЕНИЛ-АРГИНИН
(а-1\1-2,4-ДНФ-аргинин)
H2N /СООН
^C-NH-(CH,)3-CH
HN "'NH
I
< \-NO.,
i J -
NO,
CnHjcNoO, М. в. 340,30
8« 115
СИНТЕЗ СОЕДИНЕНИЯ
F
СООН *
Н,К. / , I NO2 NaHCO3
JC-NH-(CHa)3-CH I J ---------*
HN \ x"
XNH2-HC1 I
NOa
HaN /СООН
\c-nh-(ch2)3-ch
NH
+ NaF+H2O+CO2
Характеристика основного сырья
Аргинин солянокислый, ч., ВТУ Роспромсовета 1199—59.
2,4-Диннтрофторбензол, ч., ТУ завода.
Натрий двууглекислый, ч.д.а., ГОСТ 4201—48.
Этиловый спирт ректификат, ГОСТ 5962—51.
Условия получения
В круглодонную колбу, снабженную механической мешал-
кой, помещают 7,2 г (0,0341 Л4) солянокислого аргинина,
3,16 г (0,017 А1) 2,4-диннтрофторбензола, 5,7 г (0,068 М) дву-
углекислого натрия и 400 мл 50%-кого этилового спирта. Ре-
акционную смесь перемешивают 2 часа при комнатной темпе-
ратуре и удаляют этанол в вакууме водоструйного насоса.
Остаток обрабатывают водой, в которой а-2,4-ДНФ-аргинин
нерастворим. Продукт отфильтровывают и промывают 50 мл
этанола и 50 мл эфира. а-2,4-ДНФ-аргиннн перекристаллизо-
вывают, растворяя в 2н. соляной кислоте и нейтрализуя ам-
миаком по универсальной индикаторной бумаге. Выход
а-2,4-ДНФ-аргинина 6 г (51% теории). Т. пл. 258—260°.
116
2,4-ДИНИТРОФЕНИЛ-ПРОИЗВОДНЫЕ ТРИПТОФАНА,
СЕРИНА, ГЛИЦИНА, АЛАНИНА, ВАЛИНА, АСПАРАГИ-
НА, ТРЕОНИНА, МЕТИОНИНА, АСПАРАГИНОВОЙ КИС-
ЛОТЫ, ГИСТИДИНА, р-ФЕНИЛАЛАНИНА, ПРОЛИНА,
ЛИЗИНА, ТИРОЗИНА
Общее уравнение реакции:
СООН
NaHCO,
R_C-NH2-t- f N0-
no2
СООН
I
-> R-C-NH +NaF+H2O4-COs;
no2
где R—аминокислотный остаток.
Характеристика основного сырья
Триптофан, ч., ВТУ МХП 3983—53.
Сернн, ч., ВТУ МХП 3722—53.
Гликоколь, ч., ГОСТ 5860—51.
Аланин, ч., ТУ М3 1388—51.
Валнн, ч„ ВТУ М3 1466—51.
Аспарагин, ч., ВТУ 36—56.
Треонин, ч., ТУ завода.
Метионин, имп.
Аспарагиновая кислота, ч., ВТУ РУ 1080—54.
Гистидин, ч., ТУ завода.
0-Фенилалании, ч., ВТУ РУ-1315-57.
2,4-Дннитрофторбензол, ч., ТУ завода.
Натрин двууглекислый, ч.д.а., ГОСТ 4201—48.
Спирт этиловый, ректификат, ГОСТ 5962—51.
117
Условия
Название Формула структурная Формула эмпиричес- кая
СООН NO.
2,4-ДНФ-трип- //. i-CH.—C-NH-/ /—NO, ClfH11N<0,
тофан Ml 1 '\/\/ H NH СООН j40*
2,4-ДНФ-серин НО-СН,- С—NH-/ \—NO, C9H9N3O,
1 н NO,
2,4-ДНФ-гли- OsX-\ /-\Н-СН2-СООН CgHyNjOg
цин СООН N°2
СН3-С—NH-/- NO,
2,4-ДНФ-ала- 1
НИН н гн СООН NO-'
2,4-ДНФ-валин /СН-С-NH-/ /—NO, сн, н NOS О СООН / СцН1#ХзОв
2,4-ДНФ-аспа- /с - CHs-C-NH-</-'/-NO2 c10H10N4o7
рагии \н, п СООН ^°2
2,4-ДНФ-трео- СН,- CH-C-NH-/-/-NO.
НИН 1 ОН Н СООН
2,4-ДНФ-ме- CH3-S -СН2-СН,—C-NH-/ /—NO,
тионин
118
получения *
М. в. Загружаемое сырье Раствори- тель для . перекри- сталлизации Вы- ход %, тео- рии Т. пл„ °C
амино- кислота 2,4-дииит- рофторбеи- зол натрий дву- угле- кислый. г спирт 50%, мл
г ! (Л4) г 1 (М)
370,33 8 0,039 7,2 0,039 5,8 300 ацетон: пет- ролейнып эфир (1:1,5) 58 225—226
271,2 5 0,047 8,7 0,047 7 400 50%-ный водный метанол 50 199 - 200
241,2 8 0,1 18,6 0,1 14,8 400 50%-иый водный метанол 80 204-205
2-55,19 4,5 0,05 9,3 0,05 7,6 400 50%-пый водный этанол 66 178
283,25 10 0,08 14,9 0,08 12 400 50% -нып водный этанол 55 180-183
298,22 8 0,06 11,16 0,06 8,9 400 50%-ный водный этанол 62 175
285,22 5 0,04 7,44 0,04 6 400 эфир: лиг- роин (1:1,5) 50 145
315,32 5 0,0.3 5,58 0,03 4,5 500 50%-ный водный метанол 58 115-117
119
Название Формула структурная Формула эмпириче- ская
2,4-ДНФ-аспа-
рагииовая
кислота
Ди-2,4-ДНФ-
ГИСТИДИН
соон
CHS c-nh-'/~’'>-no2
/ I ч—z
соон н
СООН у°!
N—С—CH»—С—NH ——NO.,
II II I Х—
НС сн н
C10HftN8Og
CisHieN,Ole
2,4-ДНФ-р-фе-
ннлаланин
CjsHuNjOe
2,4-ДНФ-<11-
пролИИ
Ди-2,4-ДНФ-<11-
лнзин
Дя-2,4-ДНФ-1-
тирозни
СО°Н NOS
СН.-СН. /
I ZN~\ >“
СН.,-СН/
HN-(CH,)4-CHCOOH
NO.
NO.
NH
— NO2
NO.
СООН
[-NO,
NO,
CijHjxNjOe
Перечисленные соединения были получены по аналогии с 2,4-ДНФ-
CigHlsNeO,,
120
Продолж. табл.
М. в. Загружаемое сырье Раствори- тель для перекри- сталлизации Вы- ход, % тео- рии Т. пл„ °C
амино- кислота 2,4-динит- рофторбен- зол натрий дву- угле- кислый, г спирт 50%. мл
г (Л4) г (Л4)
285,19 б 0,045 8,4 0,045 6,7 400 50%-ный водный этанол 50 186
487.36 8 0,04 18,6 0,1 13,44 400 50%-ный водный этанол 62 232-234
331,29 8 0,048 8,9 0,018 7 400 50%-ный водный этанол , 56 185
281,233 2 0,016 3 0,016 2,55 300 серный эфир: лёг- кий петро- лейиыйэфир (1:2,5) 75 181
400,390 5 0,027 10,04 0,054 9 350 ацетон: петролейный эфир (1:10) 76 275-277
513,391 3 0,016 5,95 0,038 2,55 300 ацетон: сериый эфир (1:10) 70 177-180
—лейцином (см. стр. 114)
121
а-МОНО-2,4-ДИНИТРОФЕНИЛ ГИСТИДИН
(а-моно-2,4-ДНФ-гистидин)
/COOHNa
N—С-СН,-СН |_'
НС CH
NH
CnHuNsOe М. в. 321,26
СИНТЕЗ СОЕДИНЕНИЯ
/СООН __
N-C-CH2-CH +F-/ X-NO2
" " 1 * * * * * 7 В * * ' \аНСО4
------>
НС CH NH, I
\ / NO,
NH
COOH
I NO
N-C-CH.-CH -
4 NaF% СО,4-Н,О
11 11 1 / X мп
НС CH ^-NO,
NH
Характеристика основного сырья
Гистидин, ч„ ТУ завода.
2,4-Динитрофторбензол, ч., ТУ завода.
Условия получения
В колбу, снабженную механической мешалкой и обратным
холодильником, помещают 8 г (0,04 Л1) гистидина и 13,4 г
(0,16 М) двууглекислого натрия в 800 мл воды. К смеси при-
бавляют при механическом перемешивании 3,72 г (0,02 Л4)
2,4-динитрофторбензола в 200 мл спирта. Реакционную смесь
перемешивают в течение 2,5 часов при температуре 60°, после
чего упаривают в вакууме до объема 50 мл и доводят pH
остатка до 6,5 концентрированной соляной кислотой. Выде-
лившийся осадок отфильтровывают с отсасыванием и перекри-
сталлизовывают из 1 л 50%-ного водного этанола. Желтые
кристаллы получают после 18 часов стояния при температуре
0°. Выход 8,2 г (50% теории). Т. пл. 275°.
122
2,4-ДИНИТРОФЕНИЛ-ГЛУТАМИНОВАЯ КИСЛОТА
(2,4-ДНФ-глутаминовая кислота)
НООС-СН,—СН,—СН СОО11
I
NH
|/Х|-NO,
Г
no2
CnHiiN3Qs М. в. 313,15
СИНТЕЗ СОЕДИНЕНИЯ
F
О СООН 1
+ / / +—NO, \тяНсо
С-СН2-СН2—СН +11 _+2Г2_>
NH2 NIL,
NO,
О СООН
----> С-СН3-СН3-СН +NaF+HsO+CO2
/ '
NH2 NH
I
/ \ —no2
no2
О СООН
ч /
C-CHo-CHj-CH +HC1+H,O-------->
/ I
NH2 NH
|/X|-NO2
no2
123
СООН
--> HQOC CH,-CH,-CH 4-NH4Cl
1
NH
V
NO,
Характеристика основного сырья
Глутамин, имп.
2,4-Дииитрофторбензол, ч., ТУ завода.
Натрий двууглекислый, ч.д.а., ГОСТ 4201—48.
Спирт, ректификат, ГОСТ 5962—51.
Условия получения
1) 2,4-ДНФ-глутамин. 2,4-ДНФ-глутамин синтезируют по
аналогии с 2,4-ДНФ-лейцином (см. стр. 114) из следующих
количеств веществ:
12 г (0,082 /И) глутамина,
15,3 г (0,082 .VI) 2,4-дииитрофторбензола,
12,2 г двууглекислого натрия,
500 мл 50%-ного этилового спирта.
Перекристаллизовывают 2,4-ДНФ-глутамин из 50%-ного
водного метилового спирта.
Выход 16,3 г (60% теории), т. пл. 186°.
2) 2,4-Динитрофенилглутаминовая кислота. 8 г (0,025 Л!)
2,4-ДНФ-глутамииа гидролизуют прн комнатной температуре
при перемешивании в течение 12 часов 10-кратным (по объе-
му) количеством 6 н. соляной кислоты и затем нагревают ре-
акционную смесь иа водяной бане до растворения продукта.
Раствор, охлажденный до комнатной температуры, помещают
на ночь в рефрижератор до полного отделения желтого вязкого
масла. Продукт отделяют, промывают водой до нейтральной
реакции по универсальной бумажке и сушат в вакуум-эксика-
торе над фосфорным ангидридом. Затем масло помещают в
холодильник, где оно затвердевает в течение 8—10 дней. Пере-
кристаллизовывают 2,4-ДНФ-глутаминовую кислоту из смеси
этилацетат; хлороформ = 1:2. Выход 5 г (62% теории), т. пл.
134°.
124
о-2,4-ДИНИТРОФЕНИЛ-ТИРОЗИН, ХЛОРГИДРАТ
(о-2,4-ДНФ-тирозин)
- \-сн3-сн-соон
< Х- / I
i nh2-hci
Cj,HuN3O7C1 NO2 M, в. 383,76
Характеристика основного сырья
Тирозин, ч., ВТУ РУ 723—52.
2,4-Динитрофторбензол, ч., ТУ завода.
Медь углекислая, ч., ТУ МХП 125—48.
Этиловый спирт, ректификат, ГОСТ 5962—51.
Натрий двууглекислый, ч.д.а., ГОСТ 4201—48.
Соляная кислота, ч., ГОСТ 3118—46.
Условия получения
В колбе, снабженной механической мешалкой и обратным
холодильником, растворяют 4 г (0,022 М) тирозина в 1,8 л во:
ды при кипении и затем добавляют при нагревании 3,6 г дву-
углекислого натрия н 1,5 г углекислой меди. В кипящий раст-
вор вносят 6 г (0,03 .44) 2,4-динитрофторбензола в 250 мл эти-
лового спирта.
Важно, чтобы динитрофеннлирование прошло при возмож-
но более высокой температуре. Реакционную смесь энергично
перемешивают в течение 10 мин, охлаждают до комнатной тем-
пературы и ставят на ночь в холодильник для того, чтобы мед-
ный комплекс полностью выпал. Отфильтрованный комплекс
растворяют в 1,2 л 2 и. соляной кислоты.
Из зеленого раствора выделяется при охлаждении хлор-
гидрат о-2,4-ДНФ-тирозина, уже почти чистый. Перекристал-
лизовывают из 2 и. соляной кислоты.
Выход 4 г (52% теории). Т. пл. 232° (разлагается).
а-2,4-ДЙНИТРОФЕНИЛ-ЛИЗИН
(а-2,4-ДНФ-лизин)
/СООН
H2N-(CH,)4-CH
NH
(/Xt-NO2
ХО2
C12H1(N4O6 M. в 312,29
125
СИНТЕЗ СОЕДИНЕНИЯ
F
CeHsCO-NH-(CH2)4-CH-COOH+z/\ Na>co»
I j l~NUS-------
nh2 x /
I
no2
-> CeH5CO-NH-(CH8),-CHCOOH + NaF 4- H2O 4- CO3
I
NH
i
^X|-NO2
NO,
CeH5CO-NH-(CH2)4-CHCOOH + HCl 4- CH3C00H4-H,0
NH
J
Q-no2
I
NO,
H4N-(CHs)4-CHCOOH 4- C6HSCOOH
HC1 NH
Пиридин i /X
——I | ]-NO2
H2N-(CH,)4-CHCOOH
NH NO2
|/X|- NO,
7
no2
Характеристика основного сырья
e-N-Бензоил-лизин, ч., ВТУ 1020—53.
24-Динитрофторбепзол, ч., ТУ завода.
126
Натрий углекислый безводный, ч., ГОСТ 83—41.
Уксусная кислота ледяная, хч., ГОСТ 61—51.
Соляная кислота, ч„ ГОСТ 3118—46.
Этиловый спирт, ректификат, ГОСТ 5962—51.
Условия получения
1) z-N-BeH30iui-a-N-2,4-динитрофенил-лизин. В четырехлит-
ровый фарфоровый стакан с механической мешалкой помеща-
ют 10 г (0,04 Л4) e-N-бензоил-лизина, 3 л воды и 14 мл соля-
ной кислоты (1 : 1). К смеси, находящейся в стакане, добавля-
ют по каплям при механическом перемешивании раствор 22 г
(0,11 М) 2,4-динитрофторбензола в 250 мл спирта. Затем к ре-
акционной смеси медленно присыпают 14 г соды в течение
3 часов и 11 г соды в течение 1 часа. На другой день содержи-
мое стакана подкисляют соляной кислотой (1:1) до кислой ре-
акции по конго, отделяют водный раствор от вязкой желтой
массы декантацией, промывают ее декантацией водой (100 мл)
и эфиром (50 мл). Растворяют продукт в горячем спирте и вы-
саживают водой. Перекристаллизовывают из 50%-ного водно-
го этанола. Выход 9 г (53% теории). Т. пл. е-М-бензоил-а-М-
-2,4-динитрофенил-лизипа 182—184°.
2) а-2,4-Динитрофенил-лизин. В круглодонной колбе с об-
ратным холодильником на масляной бане кипятят 9 а e-N-бен-
зоил-а-П-2,4-динитрофенил-лизина .18 часов со смесью 90 мл
ледяной уксусной кислоты и 90 мл концентрированной соля-
ной кислоты. Смесь упаривают досуха, встряхивают с 200 мл
воды и фильтруют с отсасыванием от неизменившихся веществ.
Раствор нейтрализуют пиридином по универсальной индика-
торной бумажке.
Выход 4 г (60% теории), т. пл. 260° (разлагается).
е-2,4-ДИНИТРОФЕНИЛ-ЛИЗИН, ХЛОРГИДРАТ
(е-2,4-ДНФ-лизин)
СООН
HN-(CH2)4-CH
* XNHj-HC1-H2O
I j-NO2
"l
no2
Cj2H19N4O7C1 M. b. 366,77
Характеристика основного сырья
Лизин солянокислый, ч., ВТУ МХП 1383—51.
2,4- Динитрофторбензол, ч., ТУ завода.
127
Натрий двууглекислый, ч.д.а., ГОСТ 4201—48.
Медь углекислая, ч., ТУ МХП 125—48.
Спирт этиловый, ректификат, ГОСТ 5962—51.
Условия получения
Растворяют в стакане 10 г (0,05 Л1) солянокислого лизииа
в 500 мл воды, доводят до кипения и к кипящему раствору до-
бавляют 7 г (0,057 М) углекислой меди. Избыток углекислой
меди отфильтровывают с отсасыванием и промывают 60 мя
воды. Фильтрат помещают в стакан и к нему при механиче-
ском перемешивании прибавляют избыток двууглекислого на-
трия (30 г) и раствор 15 г (0,08 М) 2,4-динитрофторбензола
в 600 мл этилового спирта. Реакционную смесь перемешивают
2 часа при комнатной температуре. Полученный зеленовато,-
желтый осадок отфильтровывают и промывают 150 мл воды,
100 мл этанола и 100 мл эфира. Затем его суспензируют в
100 .ил воды и прибавляют 1 н. соляную кислоту в количестве,
достаточном для полвого растворения продукта. Раствор ох-
лаждают льдом и через него пропускают сероводород до пол-
ного осаждения меди (5 минут). Прибавляют небольшое коли-
чество древесного угля, взбалтывают и смесь немедленно от-
фильтровывают с отсасыванием. Фильтрат упаривают досуха
в вакууме водоструйного насоса. Продукт перекристаллизовы-
вают из 20 %-ной соляной кислоты.
Выход 10,7 г (63% теории), т. пл. 182—183°.
ЛИТЕРАТУРА
I. К. R- Rao, Н. A. Sober, J. Amer. Chem. Soc., 76, 1328 (1954).
2. F. Sanger, Biochem. J.. 39. 507 (1945).
3. I. K- Ramachandran, W. B. Connell., Nature, 176, 931 (1955).
4. E. R. Fritze, H. Zahn, Z. analyt. Chem., 162, 414 (1958).
5. R. R. Porter, F. Sanger, Biochem.. J. 42, 287 (1948).
6. Г. А. Гаркуша, A. H. Гинсбург, Ж. общ. химии. 29, 1556 (1959).
7. М. Lederer, Chromatographic Reviews (Amsterdam), 2, 59 (1960).
Поступила в феврале- 1962 г, Институт химии природных соединений АН СССР.
4-МЕТОКСИ-4-АМИНОБИФЕНИЛ
Л. М. ЛИТВИНЕНКО, Л. А. ПЕРЕЛЬМАН, М. М. ЛИТВИНЕНКО
Н3СО - NH,
CJgHlaNO ~ М. в. 199,13.
Получение 4-метокси-4'-аминобифенила описано Ивановым
и Панайотовым [1]. Имеется также указание б синтезе этого
128
вещества у Тейбера [2, 3]. Нами детально разработана мето-
дика получения по той же схеме, что и у вышеприведенных ав-
торов, причем во все стадии внесен ряд изменений. Так, нами
предложен метод очистки промежуточного продукта 4-окси-4/-
аминобифенила, пригодный для работы с большими загрузка-
ми и дающий чистый продукт с температурой плавления
272,5—273,5°.
По Тейберу [2], омыление ацетаминоэфиров происходит
только при нагревании с кислотами или щелочами в спирто-
вом растворе под давлением. Однако автор не дает никаких
сведений, из которых можно было бы заключить, что он дейст-
вительно их получил. Иванов и Панайотов [1] омыляли аце-
тильные производные кипячением их с едким кали в абсолют-
ном спирте с последующим извлечением продукта омыления
серным эфиром. При этом методика омыления приводится для
очень небольших количеств ацетамина (0,001 М). Попытка
омылить Г г вещества по этому методу не привела нас к поло-
жительным результатам. Мы получили 4-метокси-4'-аминоби-
фенил с 90%-ным выходом при кипячении его ацетильного
производного в смеси равных объемов концентрированных со
ляной и уксусной кислот и воды. Полученный 4-метокси-4'-ами-
нобифенил мы пробовали перекристаллизовывать из ацетона,
бензола', петролейного-эфира, водного и разбавленного этано-
-ла и метанола. Лучшие результаты дает очистка при помощи
спиртов.
СИНТЕЗ 44ИЕТОКСИ-4'-АМИНОБИФЕНИЛА
HaN-NH8 -> ->
- -э-
—'насо-^-\—^_y~NHCOCH3
Характеристика основного сырья
Бензидин-основание, технический.
Нитрит натрия, ч., ГОСТ 4197—48.
Пиридин, ч., ГОСТ 2747—44.
Кислота уксусная, ледяная, ГОСТ 61—151.
Кислота уксусная• Эв^А-ная, х.ч., ГОСТ 61 —51.
Йодистый метил, ч., ТУ МХП'49—51.
Этиловый’спирт, ректификат.
9 Зак. 62? 129
Условия получения
Получение 4~окси-4'-аминобифенила. Растворяют при на-
гревании 50 г бензидина (~0,27 Л4) в смеси 700 мл воды и
80 мл (~2,7 Л!) технической соляной кислоты. Раствор ох-
лаждают до комнатной температуры и при перемешивании
добавляют еще 80 мл соляной кислоты. При этом выпадает
солянокислый бензидин в виде мелких кристаллов. Суспензию
охлаждают до 0° и вносят в нее по каплям раствор 37 г
(~0,54Л4) нитрита натрия (в пересчете за 100%-ный) в 200 л
воды. Конец диазотирования определяют по йодкрахмальной
бумаге при 20-минутной выдержке. Затем к диазораствору
приливают при перемешивании нагретый до 60° раствор, со-
стоящий из 50 г бензидина в 700 мл воды и 80 мл соляной кис-
лоты (при более низкой температуре выпадает о.садок соляно-
кислого бензидина), так, чтобы температура реакционной сме-
си не превышала +10°. Реакционную смесь оставляют на 2—3
суток, периодически перемешивая, при температуре 10—20%
Жидкость со временем темнеет, на поверхности образуется
немного легкого шлама, продукты разложения диазосоедине-
ния осаждаются в виде темного аморфного осадка, который
отделяют фильтрованием.
Для превращения образовавшегося аминодиазобифенил-
хлорида в 4-окси-4'-аминобифенил отфильтрованный диазора-
створ вносят по каплям в течение 1 часа в 2 л интенсивно ки-
пящей воды, перемешивая кипящую смесь и разбивая обра-
зующуюся на поверхности пену. По прибавлении всего диазо-
раствора смеси дают охладиться до комнатной температуры,
выпавший осадок отфильтровывают через бязь и промывают
на фильтре холодной водой, а затем трижды экстрагируют из
него 4-окси-4'-аминобифенил 1%-ным раствором едкого кали
(всего расходуется 5—6 л раствора щелочи, каждый раз ки-
пячение продолжают не менее 1 часа). Экстрактные вытяжки
объединяют и прибавляют к ним 150 мл 30—40%-ной серной
кислоты. Выпавшую сернокислую соль 4-окси-4'-аминобифе-
нила отфильтровывают и промывают водой. Для получе-
ния основания соль обрабатывают следующим образом: к ней
прибавляют небольшими порциями (во избежание сильного
вспенивания) при перемешивании приблизительно 1 литр 2%-
ного раствора соды (щелочная реакция ио лакмусовой бума-
жке) и смесь доводят до кипения. Продукт отфильтровывают,
промывают водой до отрицательной реакции на сульфат-ион и
сушат при 100—130°.
Получают около 50 г технического 4-окси-4/-аминобифени-
ла (~50% теории).
Для очистки 50 г продукта растворяют в 4 л 1%-ной соля-
ной кислоты, фильтруют через воронку для горячего фильтро-
вания, дают охладиться приблизительно до 50—60°, а затем
130
добавляют 150 мл 30%-ной серной кислоты (см. примечание).
Сернокислую соль переводят в основание так, как уже было
указано выше, и операцию очистки через соль проводят вто-
рично. После таких двух операций получают 42—45 г 4-окси-
-4'-аминобифенила с т. пл. ~270°, который перекристаллизо-
вывают из 1,2—1,3 л водного пиридина (3 объема пиридина и
4 объема воды).
Получают 25—30 г чистого оксиамина с т. пл. 272,5—273,5°.
Получение 4-окси-4'-ацетаминобифенила. В круглодонной
колбе с обратным холодильником нагревают при слабом кипе-
нии до полного растворения (нагревание продолжается 5 ча-
сов) 18,5 г (0,1 44) 4-окси-4'-аминобифенила в 95 мл ледяной
уксусной кислоты. По охлаждении из темно-лилового раство-
ра выпадают плотные кристаллы 4-окси-4'-ацетаминобифенн-
ла. Выход около 20 г (90% теории), т. пл. 222—224°.
После перекристаллизации из 150 мл уксусной кислоты по-
лучают 15—16 г вещества с т. пл. 223—225°.
Получение 4-метокси-4'-ацетаминобифенила. К раствору
3,0 г (0,05 Л!) едкого кали в 25 мл 70%-кого этилового спирта
прибавляют 11,5г (0.05 Л!) 4-окси-4'-ацетаминобифенила.
Смесь слегка нагревают до полного растворения продукта. К
охлажденному раствору добавляют 3,25 мл (около 0,05 44) йо-
дистого метила, смесь осторожно перемешивают, сосуд закры-
вают и оставляют при комнатной температуре, после чего вско-
ре начинают выделяться кристаллы. Через 6—7 суток выделен-
ные кристаллы отсасывают, промывают на фильтре 30 мл
70%-ного спирта и высушивают. Получают около 10 г продук-
та (83% теории) с температурой плавления 200—204°, который
перекристаллизовывают из 75 мл. уксусной кислоты.
Выход 4-метокси-4'-ацетаминобифенила 7—8 г, температу-
ра плавления 204—206°.
Получение 4-метокси-4'-аминобифенила. К 7,25 г (0,03 44)
4-метокси-4'-ацетаминобифенила прибавляют 210 мл смеси,
состоящей из равных объемов уксусной кислоты, соляной кис-
лоты и воды. Через 2—3 часа слабого кипения вещество пол-
ностью растворяется. По охлаждении омыленный продукт за-
стывает в виде кристаллической каши. Полученную солянокис-
лую соль 4-метокси-4'-аминобифенила отфильтровывают и об-
рабатывают при нагревании 50—70 мл разбавленного водно-
го аммиака (1 : 1). Амин отфильтровывают, промывают водой
до нейтральной реакции на ион хлора и сушат при 70—100°.
Получают 5,5 г ^-метокси-^-аминобифенила (приблизительно
90% теории) ст. пл. 145—146,5°.
После перекристаллизации из 220 мл этанола (активиро-
ванный уголь) выделяется 3,5—4 г бесцветных кристаллов
4-метбкси-4'-аминобифенила с т. пл. 147—147,5°.
Примечание.
Наша попытка получить солянокислую соль 4-окси.4'-аминобифенила в
кристаллическом виде дала положительные результаты лишь при примене-
9* 131
нии более чем 150-кратного объема растворителя. Поэтому мы к солянокис-
лому раствору 4-окси-4'-аминобифенила добавляли серную кислоту для пе-
ревода студневидной и плохо фильтрующейся солянокислой соли в серно-
кислую, которая отфильтровывается легче. Перекристаллизовывать амин
сразу через сернокислую соль нецелесообразно, так как эта соль намного
хуже растворяется в воде.
ЛИТЕРАТУРА
1. И. И. Иванов, И. М. Панайотов, Докл. АН СССР, S3, Мг 6, 1041,
(1953).
2. Е. TOuber, Frdl., 4, 1166 (№ 85988).
3. Е. Tauber, Вег., 27, 2629 (1894).
Поступала в апреле 1962 г. Харък. Гос. ун-т
им. А. М. Горького
(ХГУ)
4-АМИНО-4-ХЛОРБИФЕНИЛ
Л. М. ЛИТВИНЕНКО, В. М. ЗИКРАНЕЦ
HaN~\ /”\ /~С1
C„HMNCI “ М. в. 203,66
Из описанных в литературе способов синтеза 4-амино-4'-
хлорбифенила наиболее часто применяются два: бифенил ни-
труют азотной кислотой до 4-нитробифенила, который хлори-
руют, и полученный таким образом 4-нитро-4'-хлорбифенил за-
тем восстанавливают [1]; бензидин частично диазотируют, и в
полученном монодиазосоединении дназогруппу заменяют ато-
мом хлора по методу Зандмайера [2]. Мы уточнили методику
синтеза [2] и очистку готового продукта.
СИНТЕЗ 4-АМИНО-4'-ХЛОРБИФЕНИЛА
: NaNO,
----
V-/ НС1
CuCl —-с
— HtN-< \-N,Cl-► H2N—^-Cl
Характеристика основного сырья .
Бензидин технический, т. пл. 128—129",
Нитрит натрия, ч.1 ГОСТ 4197—48.
Медь однохлористад, свежеприготовленная. .
Спррт изопропиловый, оч., ТУ ОРУ 50-57.
- Условия получения
В 700 мл воды и 80 мл концентрированной соляной кисло-
ты растворяют прн нагревании 50 г бензидина. Раствор охла-
ждают до комнатной температуры и к нему добавляют еще
80 мл концентрированной соляной кислоты. Выпадает соляно-
кислая соль бензидина. Суспензию охлаждают до нуля и диа-
зотируют, медленно прибавляя при перемешивании раствор
37 г нитрита натрия в 200 мл воды. К диазораствору прилива-
ют при перемешивании нагретый до 60° раствор, состоящий из
50 г бензидина в 700.МЛ воды и 80 мл соляной кислоты (при
более низкой температуре выпадает осадок солянокислого бен-
зидина), так, чтобы температура не превышала +10°. Реак-
ционную смесь оставляют на три дня при температуре 10—
20°, время от времени ее перемешивая, затем отфильтровы-
вают от продуктов разложения диазосоединения. Диазораст-
вор по каплям при перемешивании прибавляют к раствору
54 г свежеприготовленной [3] однохлористой меди в 500 мл
концентрированной соляной кислоты, нагретому до 80°. Пос-
ле прибавления всего диазораствора смесь постепенно нагре-
вают до кипения. Дав смесн остыть до комнатной температу-
ры, образовавшийся осадок отфильтровывают, промывают 1 л
4%-ной соляной кислоты и сушат при 40—60°. Выход техниче-
ского продукта около 100 г. Продукт реакции экстрагируют
при кипячении несколькими порциями 7—8 %-ной соляной кис-
лоты: соответственно первый раз берут 5 л, а затем.4, 3, 2 и
1 л. При экстрагировании каждый раз кипятят по два часа.
Суммарный выход хлористоводородной соли 4-амино-4'-хлор-
бифенила 65 г.
Для получения свободного 4-амино-4'-хлорбифенила полу-
ченнуй хлористоводородную соль нагревают с 200 мл 10%-но-
го аммиака. Выход технического 4-амино-4'-хлорбифенила
55 г.
С целью получения чистого амина технический 4-амино-4'-
хлорбифенил трижды перекристаллизовывают из изопропило-
вого спирта с применением активированного угля.
Выход 32—33 г (60% теории). Т. пл. 131 —133°.
ЛИТЕРАТУРА
1. R. Belcher, A. Nutten, W. Stephen, J. Chem. Soc. 1953, 1334.
2. P. Gelmo, Ber., 39, 4175 (1906). . „
3. H. Д. Прянишников, «Практикум по органической химии». Госхим-
издат, стр. 131, 1956.
Поступала в акте 1961 г. ХГУ
2-БРОМ-4-НИТРОАНИЛИН
Л. М. ЛИТВИНЕНКО, В. М. ЗИКРАНЕЦ
OiN~\ /~NH*
Br.
CsH5OaN2Br M. в. 217,03
В литературе описан ряд методов получения 2-бром-4-ни-
троанилина: нагреванием 4-нитро-1,2-дибромбензола со спир-
товым раствором аммиака при температуре 180—190° [1]; ни-
трованием 2-бромацетайилида азотной кислотой (уд. в. 1,54)
с последующим омылением полученного продукта едким ка-
ли [1]; бромированием 4-нитробензанилида и дальнейшим омы-
лением образующегося 2-бром-4-нитробензанилида едким кали
[2]; бромированием п-нитроанилина либо в соляной [3], либо
в уксусной [4] кислотах.
В основу получения 2-бром-4-нитроанилина был взят пос-
ледний метод. Бромирование л-нитроанилина проводили в ук-
суснокислой среде, поскольку в результате экспериментальной
проверки оказалось, что при бромировании в соляной кислоте
наряду с 2-бром-4-нитроанилином образуется значительное ко-
личество 2,6-дибромбифенила, который трудно отделить от ос-
новного продукта.
СИНТЕЗ 2-БРОМ-4-НИТРОАНИЛИНА
> - х Dlj
O,N—NHj------- OjN-^ NHS + HBr
Br
Характеристика основного сырья
л-Нитроанилин технический, т. пл. 145—147°.
Кислота уксусная ледяная, ГОСТ 61—51.
Бром, ч., ГОСТ 4109—48.
Спирт изопропиловый, оч., ТУ ОРУ 50—57.
Условия получения
В колбе с обратным холодильником растворяют при нагре-
вании на кипящей водяной бане 100 г л-нитроанилина в 1,5 л
ледяной уксусной кислоты. К охлажденной до комнатной тем-
пературы смеси (выпадает мелкокристаллический осадок) при
134
механическом перемешивании по каплям прибавляют раствор
120 г (38 мл) брома в 100 мл ледяной уксусной кислоты. К
концу прибавления раствора брома температура реакционной
смеси повышается до 30—35°, осадок амина растворяется и
начинается выпадение темно-серого осадка 2-бром-4-нитроани-
лина. Реакционную смесь нагревают на кипящей водяной бане
в течение 30 минут. Затем охлаждают до комнатной темпера-
туры. Выпавший осадок отфильтровывают, промывают 1,5 л
воды и сушат.
Выход технического продукта 88 г.
Перекристаллизацию полученного 2-бром-4-нитроанилина
осуществляют путем растворения неочищенного продукта в
1 л разбавленного изопропилового спирта (1 объем спирта на
1 объем воды). Выход 78 г. Т. пл. 103—104°.
Для получения более чистого амина его перекристаллизо-
вывают еще раз из 0,6 л разбавленного изопропилового спир-
та. Выход 70 г (40% теории). Т. пл. 104—104,5°.
ЛИТЕРАТУРА
1. G. Korner. Gazz. chim. ital., 4, 372 (1874).
2. H. HUbner, Вег., 10, 1709 (1871).
3. В. Nicolet. W. Ray, J. Amer. Chem. Soc. 49, 1805 (1927).
4. 5. Nlyogy, J. Ind. Chem. Soc., 4., № 3, 398 (1927).
Поступила в июне IS6! г. ХГУ
4,4'-ДИТ0ЛИ<Л
Л. М. ЛИТВИНЕНКО, Л. А. ПЕРЕЛЬМАН, В. М. ЗИКРАНЕЦ
Н3С-
а
СиНи
М. в. 182,26
Ульман н Мейер (1, 2] получали 4,4'-дитолил с выходом
54% при взаимодействии расплавленного n-йодтолуола с ме-
таллической медью.
В реакции Ульмана природа меди является предметом мно-
гочисленных дискуссий. Широко употребляется порошкообраз-
ная медь, известная под названием медной бронзы. Ульман
применял особый вид бронзы «Naturkupfer С». Клайдерер н
Адамс [3] рекомендуют перед употреблением медную бронзу
135
обрабатывать йодом в ацетоне, а затем высушивать в вакуум^
эксикаторе.
Нами 4,4'-дитолил получен из n-йодтолуола с различными
видами меди: с химически осажденной медью [4]; с медью, ак-
тивированной по Клайдереру и Адамсу [3]; с продажным элек-
тролитическим медным порошком; с электролитической медью,
обработанной соляной кислотой в ацетоне (1 : 1). Лучшие ре-
зультаты дает медь, обработанная по последнем}' способу.
Реакция Ульмана чаще всего применима к небольшим ко-
личествам реагирующих веществ. Мы увеличили загрузку
n-йодтолуола в 5 раз по сравнению с предлагаемой автора-
ми [2], что не сказалось на выходе и качестве готового про-
дукта. .
СИНТЕЗ ‘М'-ДИТОЛИЛА
/ — х Си / — \ / — -
2Н3С-/ ---> Н3С-/ у-СН3
Характеристика основного сырья
n-йодтолуол, т. пл. 34—35°.
Медь, свежеприготовленная.
Изопропиловый спирт, оч., ТУ ОРУ 50-57.
Условия получения
В трехгорлую термостойкую колбу емкостью 250 мл, снаб-
женную термометром и воздушным обратным холодильником,
помещают 100 г /г-йодтолуола (см. примечание 1). Содержи-
мое Колбы нагревают до 210° и при этой температуре в рас-
плав при периодическом перемешивании медной лопаточкой в
течение 2 часов в 10--12 приемов вносят 100 г медного порош-
ка (см. примечание 2). По прибавлении всей меди температу-
ру в колбе постепенно (2—3 часа) поднимают до 260°, перио-
дически перемешивая реакционную массу. Реакцию считают
законченной, если в холодильнике не наблюдается конденса-
ции исходного продукта. После 15-минутной выдержки при
температуре 260° реакционную массу оставляют постепенно
охлаждаться до комнатной температуры. По охлаждении за-
стывший кристаллический плав извлекают из колбы (см. при-
мечание 3), измельчают и 4—5 раз экстрагируют при продол-
жительном кипячении изопропиловым или этиловым спиртом
(всего на экстрагирование идет 1,3 л спирта). Отфильтрован-
ные спиртовые вытяжки соединяют, обрабатывают при кипя-
чении углем (3—4 г), а затем отгоняют около 90% растворите-
ля. Из охладившегося раствора выпадает 26—28 г 4,4'-дито-
136
лила с т. пл. 118—120°. После перекристаллизации его из изо-
пропилового спирта~ (15 мл иа 1 г продукта) получают 23—
24 г (55—56% теории) вещества с т. пл. 120—121°.
Примечания.
1. В основу получения я-иодтолуела был взят метод, предложенный
Ульманом н Мейером [2].
2. Медь обрабатывают следующим образом: 100 г продажного медно-
го порошка заливают смесью, состоящей из 100 мл концентрированной со-
ляной кислоты и 100 мл ацетона, и размешивают в течение 5—1.0. минут,
затем фильтруют, тщательно промывают ацетоном до отрицательной реак-
ции на хлор-ион и сушат. Порошок лучше применять свежеобработанным,
хотя нами проверено, что активность меди мало .меняется в течение 2—
3 дней.
3. Плав, благодаря наличию в нем медйого порошка, довольно хрупок
и сравнительно легко извлекается.
ЛИТЕРАТУРА
I. F. Ullmann, J. Blelecki, Вег., 34, 2174 (1901).
2. F, Ullmann, О. Meyer, Liebigs Ann. Clieni., 332, 38,44 (1904).
3. £. Kleiderer, R. Adams, J. Amer. Chem, Soc., 55, 4219 (1933).
4. Ю. В. Карякин, И. И. Ангелов, «Чистые химические реактивы», 341,
Госхимиздат (1955).
Поступила s июне 1961 г. ХГУ
4,4'-ДИФЕНИЛДИКАРБОНОВАЯ КИСЛОТА
Л. М. ЛИТВИНЕНКО, Л. А. ПЕРЕЛЬМАН
НООС-/ ^-соон
СмН10О4 • ~ - М. в. 242,23
Описано несколько способов получения 4,4/-дифеиилдикар-
боновой кислоты из 4,4'-дитолила. Так, 4,4'-дитолил окисляли
хромовой кислотой в ледяной уксусной кислоте в одну стадию
до 4,4'-дифенилдикарбоновой кислоты [1}. В более поздних ра-
ботах указывается на двухстадийное окисление: сначала бихро-
матом и серной кислотой [2] или хромовым ангидридом [3] до
4-толилбензойной кислоты, затем действием на последнюю ще-
лочным раствором перманганата. В основу синтеза мы поло-
жили последний метод, существенно изменив и упростив его.
СИНТЕЗ 4,4'-ДИФЕН ИЛ ДИКАРБОНОВОЙ КИСЛОТЫ
. —. ,—. Na,Cr,O7
Н3С~/ \_/ \-сн3--------------->
— Нзс-^ \-COOH
137
KMnO,
HaC-/ ^-соон ----------------*
---► HOOC-^~^-/’\-COOH
Характеристика основного сырья
4,4'-Дитолил, т. пл. 118—120° (см. стр. 135).
Натрий двухромовокислый, ч., ГОСТ 4237—48.
Кислота уксусная, 98%-ная, х. ч., ГОСТ 61—51.
Калий марганцовокислый, ч., ГОСТ 4527—48.
Условия получения
В круглодонной колбе, снабженной обратным холодильни-
ком, механической мешалкой с затвором и тубусом для вне-
сения бихромата, растворяют при нагревании на кипящей во-
дяной бане 20 г 4,4'-дитолила в 450 мл уксусной кислоты. К
раствору прибавляют 5 мл концентрированной серной кислоты
и в течение двух часов вносят 62 г мелко растертого бихрома-
та натрия. По прибавлении всего бихромата содержимое кол-
бы нагревают при перемешивании в течение 24 часов. Затем
реакционную массу выливают в трехкратный объем воды. Вы-
павший из воды осадок представляет собой смесь 4-толил-бен-
зойной кислоты и 4,4':-дифенилдикарбоновой кислоты. Его
фильтруют через бязь и промывают на воронке водой до обес-
цвечивания промывных вод. Осадок с фильтра переносят в кол-
бу (см. примечание 1) и нагревают на кипящей водяной бане в
течение 2—3 часов с раствором .8 г едкого натра и 30 г перман-
ганата калия в 600 мл воды. Не вступивший в реакцию пер-
манганат разлагают добавлением 20—30 мл метанола до пол-
ного обесцвечивания раствора. Реакционную смесь фильтру-
ют и в фильтрат добавляют 30 мл концентрированной соляной
кислоты. Выпавший белый осадок фильтруют через бязь, про-,
мывают на фильтре дистиллированной водой, а затем 50 мл
горячего метанола для удаления возможных следов 4-толил-
бензойной кислоты. Получают . 16 г 4,4'-дифенилдикарбоновой
кислоты (60% теории). Для очистки кислоту 2—3 раза пере-
осаждают из ее натриевой соли. Для этого 15 г 4,4'-дифенил-
дикарбоновой кислоты растворяют при кипячении в 800 мл
1 %-ного раствора едкого натра, раствор отфильтровывают, а
затем осаждают из него продукт концентрированной соляной
кислотой и промывают на фильтре дистиллированной водой до
отрицательной реакции на ион хлора. Выход почти количест-
венный (см. примечание 2).
Примечания.
1. Если последующее окисление проводят не сразу и осадок успеет вы-
сохнуть, то его следует перед употреблением тщательно растереть.
138
2.4,4'-Дифенилдикарбоновая кислота практически нерастворима в
обычных органических растворителях, не плавится и не сублимируйся выше
300°. Все соли ее, за исключением щелочных, трудно растворимы ж воде.
Поэтому качество готового продукта определяется следующим образом:
0,25 а 4,4,-дифенилдикарбоновой кислоты растворяют в 35 мл 0,1 и. раствора
NaOH. Избыток щелочи оттитровывают 0,1 и. соляной кислотой в присутст-
вии фенолфталеина. Полученный продукт содержал 99—100% 4/Г-дифе-
нилдикарбоновой кислоты. Зольность его —0,1%.
ЛИТЕРАТУРА
I. О. Doebner, Вег., 9, 27.2 (1876).
2. М. Weiler, Вег., 32. 1о6' (1899).
3. A. Weissberger, J. Williams, Z. phys. Chem. [8] 3, 369 (1929).
Поступала в июне 1961 г.
ХГ9
4,4/-ДИНИТРОДИФЕНИЛДИСУЛЬФИД
Л. М. ЛИТВИНЕНКО; Р. С. ЧЕШКО, Л. А. ПЕРЕЛЬМАН
O,N—/ \-S-S-
/ ^-NOb
CisH8O4NjsSa
М. В. 308,34
В литературе описан ряд методов получения 4,4'-динитро-
дифенилдисульфида [1—II]. С практической стороны наиболь-
ший интерес представляет синтез этого вещества конденсаци-
ей n-нитрохлорбензола с дисульфидом натрия в спиртовой
среде [6, 7, 10]. Однако продукт при этом всегда получается
сильно загрязненным. В литературе приводится трудоемкий
метод очистки 4,4'-динитродифенилдисульфида, заключаю-
щийся в том, что его переводят в 4-нитромеркаптан, последний
переосаждают через натриевую соль и затем окисляют хлор-
ным железом [10].
Мы уточнили синтез 4,4'-дииитродифенилдисульфида и при-
менили значительно более простой способ его очистки.
СИ НТЕЗ 4,4/-Д И Н ИТРОД ИФ ЕН ИЛ ДИСУЛ ЬФ ИДА
2 OjN— / Cl+NajSa------>-
>0,4-/ \-S-S-^ Noa-|-2NaCl
139
Характеристика основного сырья ;
n-Нитрохлорбензол,: ч., ТУ МХП,2169—49.
Сернистый натрий, ч.д.а., ГОСТ' 2057—47.
Сера, ч. (серный цвет или черенковая).
Бензол, ч.д.а., ГОСТ 5955—51.
Кислота уксусная, 98%-ная, х. ч„ ГОСТ 61—51.
Условия получения
В двухлитровой трехгорлой колбе с механической мешал-
кой, капельной воронкой и обратным холодильником раство-
ряют на кипящей водяной бане 157,5 г (1 М) /г-ннтрохлорбен-
зола в 250 мл этилового спирта. К кипящему раствору в тече-
ние 30 минут прибавляют при перемешивании горячий спирто-
вой раствор дисульфида натрия (см. примечание I). По при-
бавлении всего раствора дисульфида реакционную смесь на-
гревают 3—4 часа при постоянном перемешивании. В процес-
се проведения реакции происходит выделение желтых кристал-
ликов продукта, и раствор заметно светлеет. Горячую реак-
ционную массу фильтруют, продукт переносят в стакан и от-
мывают от хлористого натрия водой. После этого осадок про-
мывают на фильтре 3—4 порциями этанола по 50 мл каждая.
Получают 100—115 г вещества (70—75% теории) с т. пл. 145—
160°. Полученный технический продукт обрабатывают при на-
гревании в течение 30—40 минут соответственно 120, 80 и
70 мл бензола. При первых двух обработках горячий бензол
декантируют, а при последней фильтруют через обогреваемую
воронку. Отмытый горячим бензолом продукт весит около 75 <?,
его т. пл. 176—179° (см. примечание 2).
После перекристаллизации 75 г вещества; из. 1,3—1,4 л ук-
сусной кислоты е добавлением активированного угля получа-
ют около 60 г (45% теории} 4Л'-дипитродифенилдисульфида с
т. пл. 181,5—182,5°. ...
Примечания. ,
1. Смешивают при-нагревании 180 а (0,75 М) кристаллического суль-
фида натрия (NaiS - ЭНоО) с 750 мл этилового спирта. Затем сюда же вно-
сят 24 г (Q.75 М) измельченной серы и кипятят в течение 1—1,5 часов до
полного растворения. В процессе.'приготовления раствора дисульфида, отде-
ляется более темный ннжний слой водного раствора дисульфида, состав-
ляющий 5—10% всего объема. Верхний слой сливают и в горячем состоя-
нии прибавляют его к кипящему раствору п-нитрохлорбензола. Раствор дн-_
сульфида натрия желательно применять сразу после его приготовления.
2. Иногда вполне достаточно двукратной обработки бензолом, чтобы
получить 4,4'-динитрофенилдисульфид с т. пл. 176—179°.
ЛИТЕРАТУРА
1. R. Leuckart, L. Lustig, J. prakt. Chemie, [2], 41, 199 (1890).
2. I. Blanksma, Recueil trav. chim., 20,127 (19Q.1),
3. E. Bamberger, E. Kraus,'Ber., 29, 282 (1896).
140
4. /?. Fuson, S. Melamed, J. Organ. Chem., 13, 690 (1948).
5. C. Willeerodt, Ber., 18, 333 (1885).
6. T. Wohlfahrt, I. prakt. chem., [2], 66, 553 (1902).
7. A. Ekbom, Ber., 35, 654 (1902).
8. K. Brand, A. Wlrslng, Ber., 45, 1763 (1912).
9. E. Fromm, I. Wittmann, Ber., 41, 2264 (1908).
10. T. Zinke, S. Lenhardt, Liebigs Ann. Chem., 400, 7 (1913).
11. H. M tiller, Chem. Zbl„ 1906, 11, 1588.
Поступила « июне 196/ г.
ХГУ